/









































































































































































































Author: Горст А.Г.




Text
Горст “Изготовление нитросоединений
В 17 главах книги дано изложение основных сведений по химии и тех-
нологии нитросоединений, сведений о физических, физико-химических, хими-
ческих, взрывчатых и токсических свойствах их и о применении этих соединений.
Книга может служить учебным пособием при прохождении курса химии
и технологии нитросоединений в химико-технологических втузах, а равно
может быть полезна работающим в соответствующей области инженерно-
техническим работникам заводов и научно-исследовательских институтов.
ОГЛАВЛЕНИЕ
Предисловие .......•................. 11
Перечень сокращенных обозначений журналов 13
Глава I
Способы введения нитрогрупп
Отличительная характеристика нитросоединений, нитроаминов и азотных
эфиров, спиртов н углеводов
1. Строение. 2. Химическая стойкость. 3. Чувствительность
к механическим воздействиям. 4. Восстановление водородом. 5. Осо-
бенности взаимодействия с серной кислотой................... 15—17
Способы введения нитрогрупп
1. Нитрование одной азотной кислотой. 2. Нитрование смесями
серной и азотной кислот. 3. Нитрование смесями азотной кислоты
с фосфорным ангидридом. 4. Нитрование азотной кислотой в смеси
с уксусной кислотой или уксусным ангидридом. 5. Нитрование азот-
ной кислотой в присутствии катализатора. 6. Практическое исполь-
зование способа нитрования в присутствии солей ртути. 7. Нитро-
вание азотными эфирами спиртов. 8. Нитрование азотнокислыми
солями. 9. Нитрование раствором нитрозилсерной кислоты в сер-
ной кислоте. 10. Нитрование окислами азота. J1- Нитрование дву-
окисью азота в присутствии серной кислоты. 12. Нитрование дву-
окисью азота в газовой фазе. 13. Окисление аминогруппы в нитро-
группу. 14. Действие окислов азота на аминосоединения 15. Замена
аминогруппы на нитрогруппу. 16., Электрохимическое нитрование. 18 35
Глава II
Нитросоединения алифатического ряда
Свойства нитросоединений алифатического ряда
1. Температура кипения и плотность нитропарафинов. 2. Хи-
мические свойства нйтропарафинов. 3. Продукты взаимодействия
углеводородов предельного характера с азотной кислотой при нитро-
вании в жидкой фазе. 4. Токсические свойства- 5. Свойстна отдель-
ных представителей ......................................... 37- 41
Применение нитросоединений алифатического ряда
Получение нитросоединений алифатического ряда .......... 42 -48
Глава III
Механизм нитрации азотной кислотой
1. Работы Виланда, Наметкина, Тронова, Михаэля и Карлсона.
2. Механизм альдолизации. 3,. Теория радикал-ионов Пфейффера и
Вицингера. 4. Возражения Хюккеля. 5. Схема Шааршмидта.-. . . 49-58
Глава IV
Основы процесса нитрации
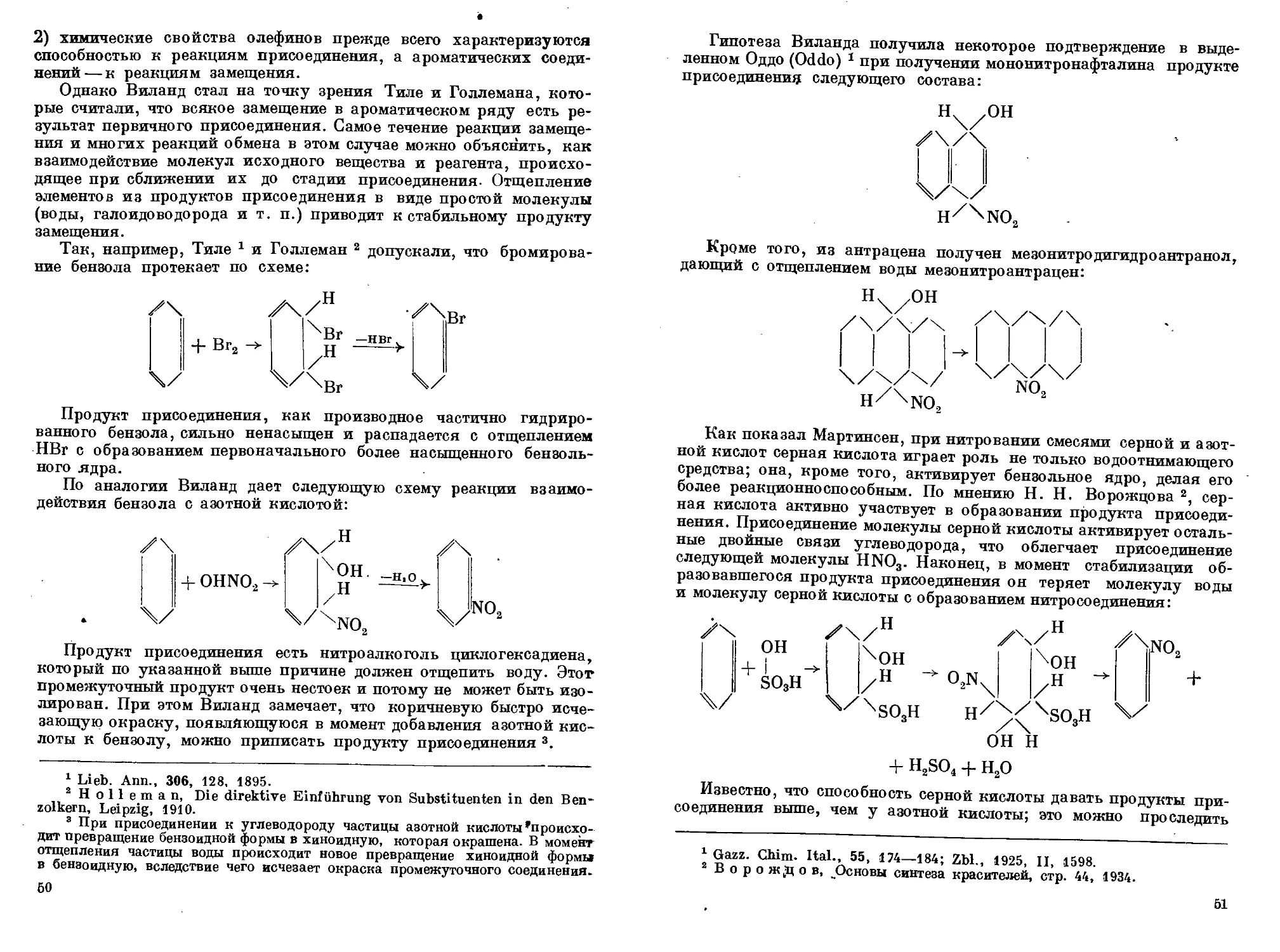
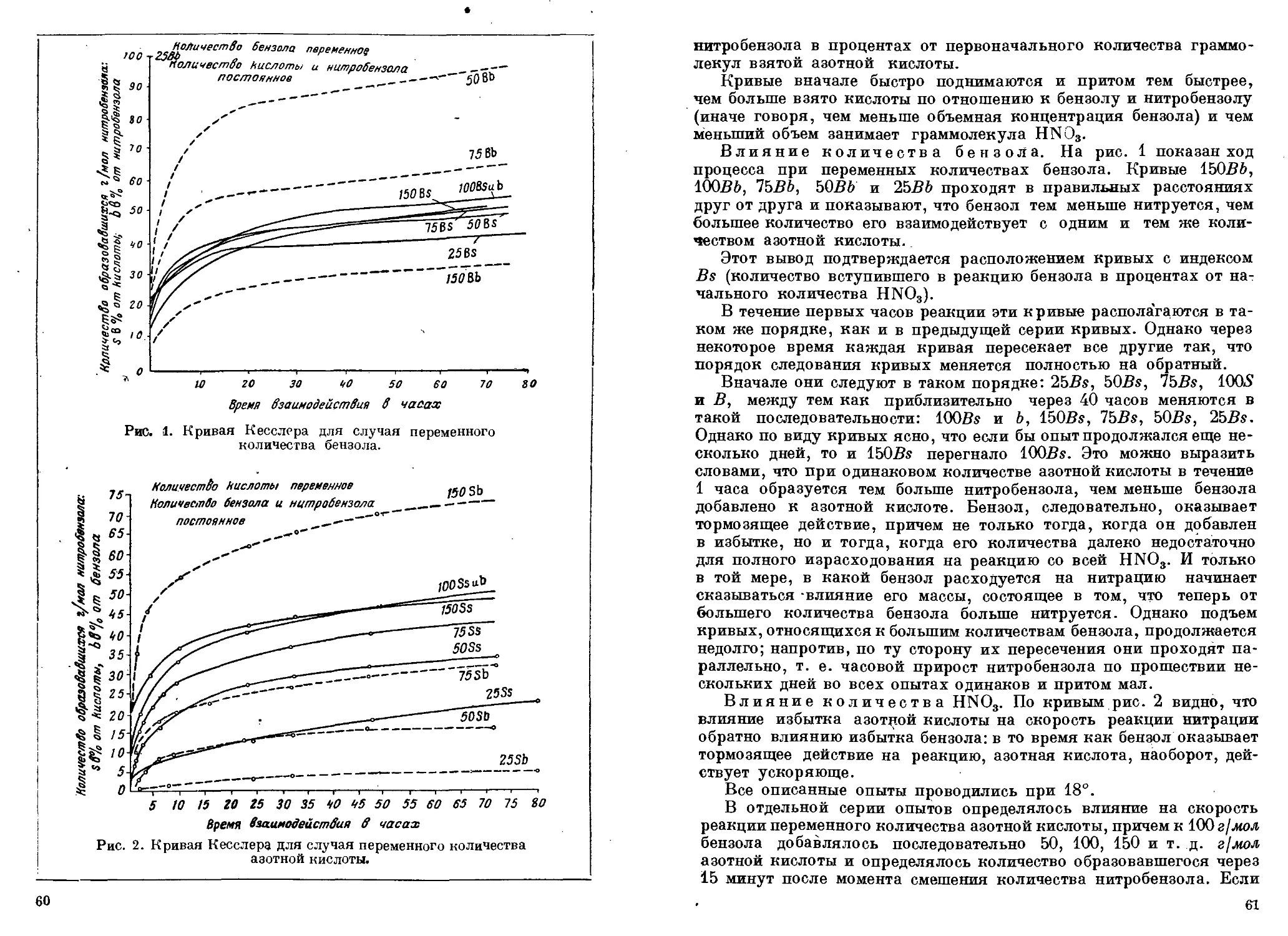
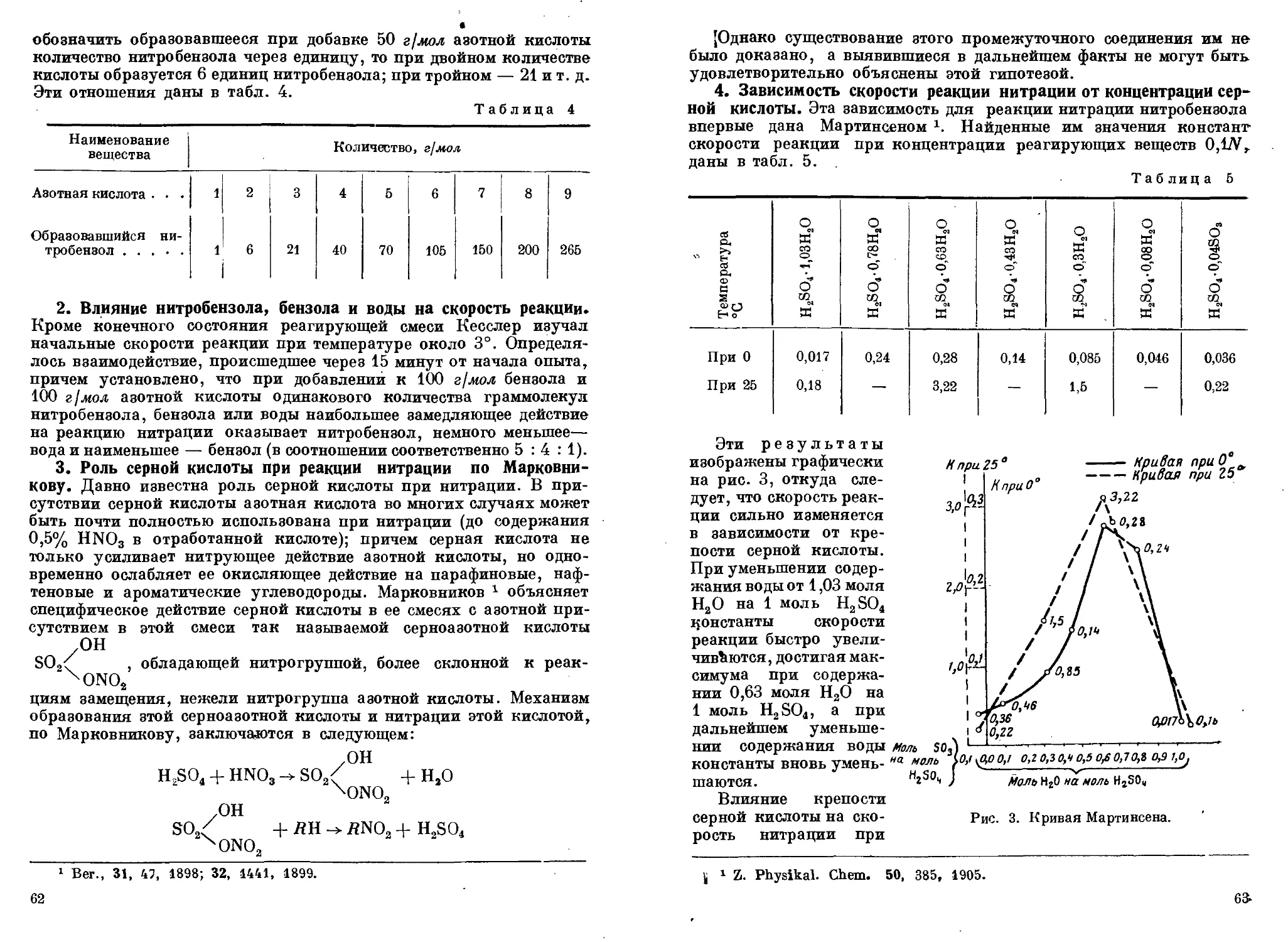
1. Влияние концентрации и количества реагирующих компо-
нентов. 2- Влияние нитробензола, бензола и воды на скорость ре-
акции. 3. Роль серной кислоты при реакции нитрации по Марков-
никову. 4. Зависимость скорости реакции нитрации от концентра-
ции серной кислоты. 5. Влияние растворителя на скорость реакции.
6. О тормозящем влиянии избытка нитробензола на скорость реак-
ции его нитрации. 7. Влияние температуры на скорость реакции
нитрации. 8. Влияние заместителей на скорость нитрации. 9. Вли-
яние окислов азота на скорость нитрации. 10. Теория нитрационных
смесей Сапожникова. И. Зависимость между составом смеси и упру-
гостью паров азотной кислоты. 12. Зависимость между составом ни-
трационной смеси и степенью нитрации клетчатки. 13. Зависимость
между составом нитрационной смеси и степенью нитрации нафталина
и толуола по Сапожникову. 14. Современное изложение теории
нитрационных смесей Сапожникова- 15- Характеристика нитра-
59-84
ционной смеси
Глава V
Влияние различных факторов на течение процесса нитрации
ароматических углеводородов
1-я нитрация
1. Приливание кислотной смеси к углеводороду. 2. Прили-
вание углеводорода к кислотной смеси. 3. Значение различных
факторов для непрерывного процесса. 4. Горячий кислотооборот . 89—95
2-я нитрация
1. Приливание смеси к мононитросоединению. 2. Приливание
моно нитросоединения к смеси. 3. Болтушка
96—97
3-я нитрация
1. Условия процесса. 2. Случаи нитрования углеводородов до
ди- и тринитросоединений в одну фазу
97—99
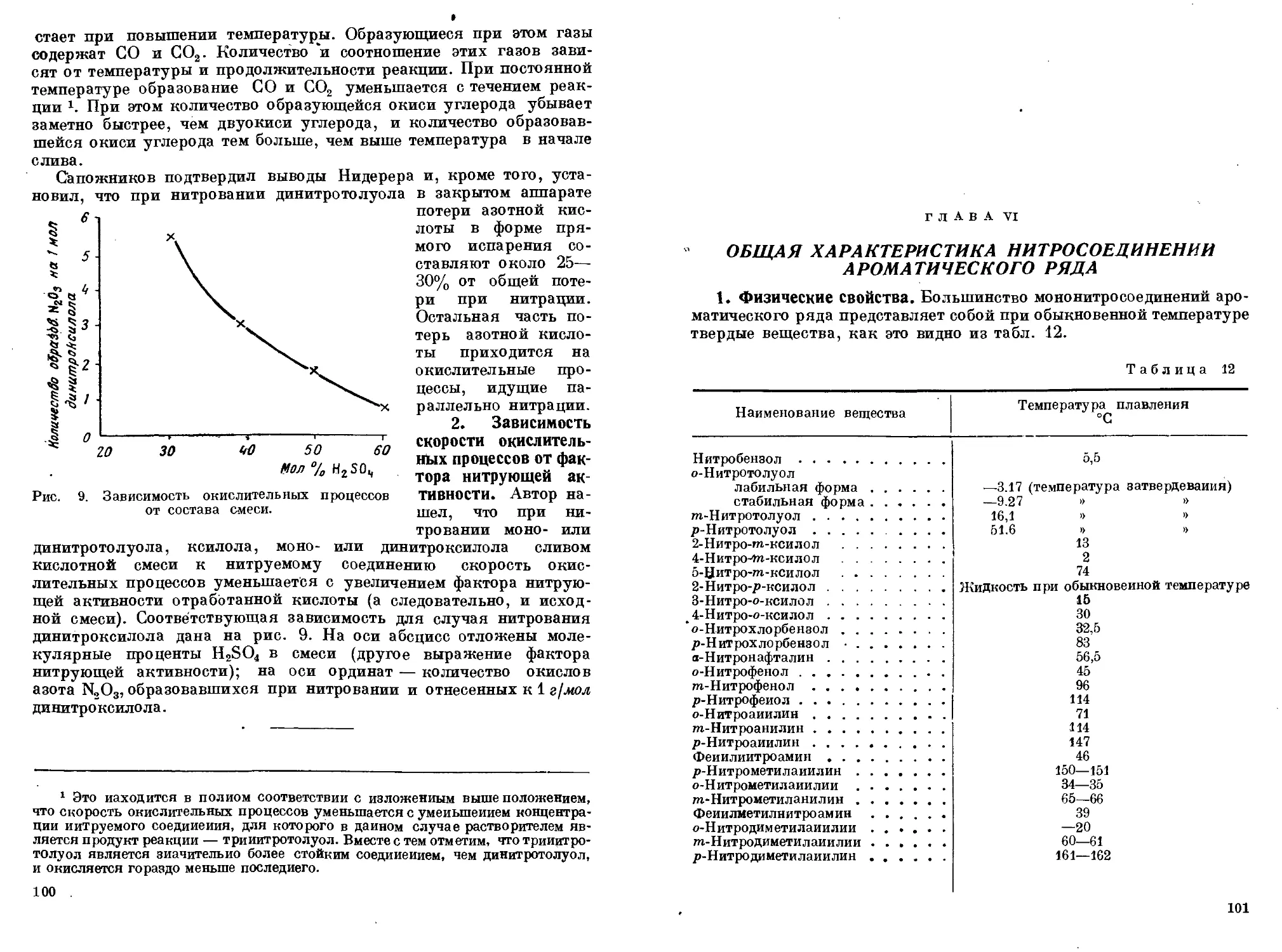
Окислительные процессы при нитрации
1. Окислительные процессы при 1-й, 2-й и 3-й нитрациях.
2. Зависимость скорости окислительных процессов от фактора ни-
99—100
трующей активности
4
Глава VI
Общая характеристика нитросоединений ароматического ряда
1. Физические свойства. 2. Химические свойства. 3. Чувстви-
тельность к удару. 4. Общий характер токсических свойств арома-
тических нитросоединений. 5. Пути отравления. 6. Теория
токсического действия ароматических нитросоединений. 7. Терапия.
8. Прогноз. 9. Применение нитросоединений ..................... 101—ПО
Глава VII
«
Классификация способов производства нитросоединений
1. По числу стадий нитрования. 2. По характеру кислотообо-
рота. 3. По цикличности процесса........................... 112—115
' Глава VIII
Нитропроизводные толуола
Толуол
1. Свойства толуола. 2. Технические условия на прием толуола
(ОСТ 464). 3. Тиофен. 4. Непредельные соединения. 5. Значение
примесей парафинов в толуоле. 6. Отличие каменноугольного то-
луола от нефтяного......................................... 116—119
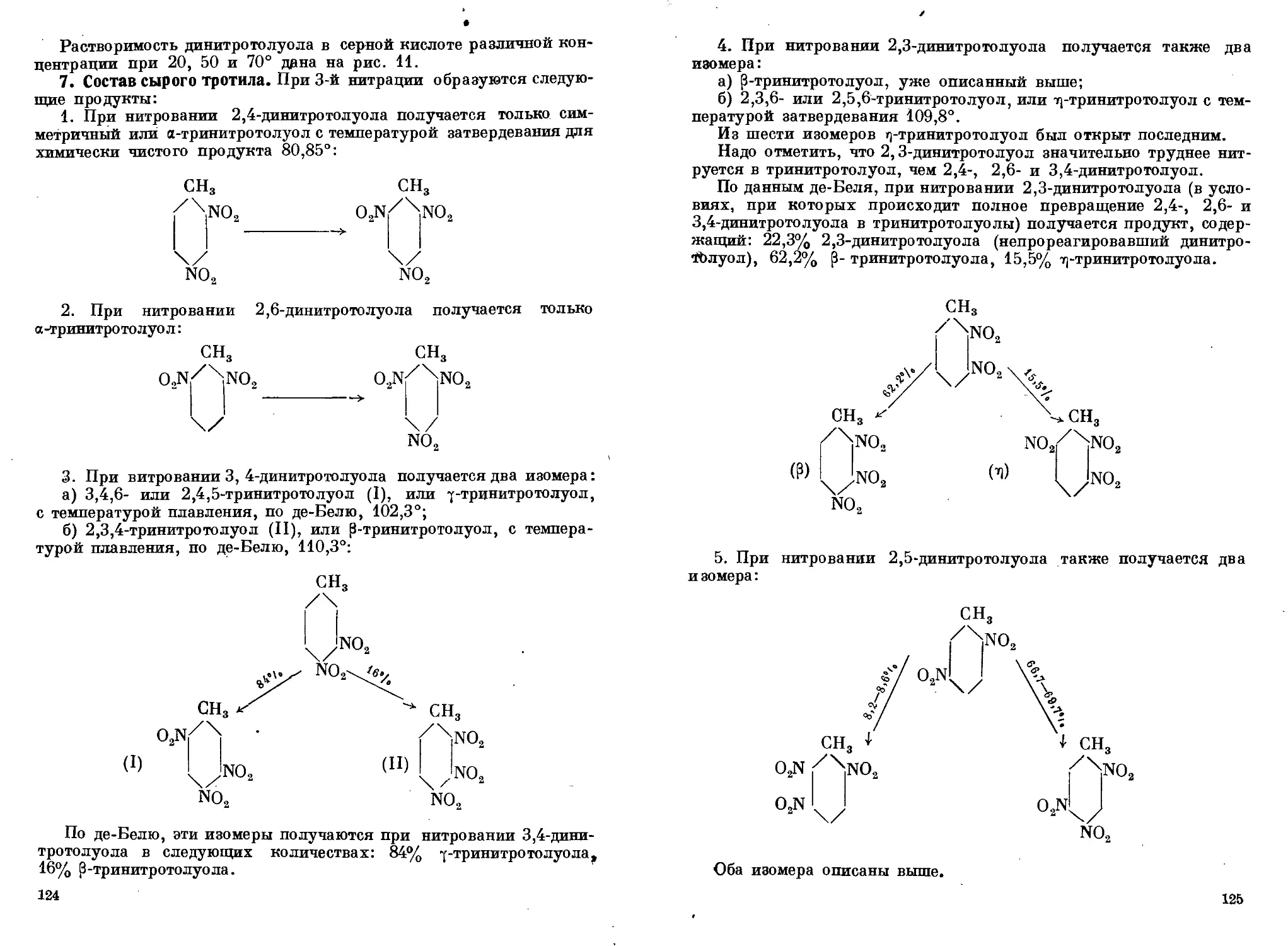
Состав продуктов 1-й нитрации толуола, их свойства и применение
1. Свойства изомеров мононитротолуола. 2. Состав продуктов
1-й нитрации толуола. 3. Технический мононитротолуол. 4. Состав
продуктов нитрации изомеров мононитротолуойа. 5. Состав про-
дуктов нитрации технического мононитротолуола. 6. Растворимость
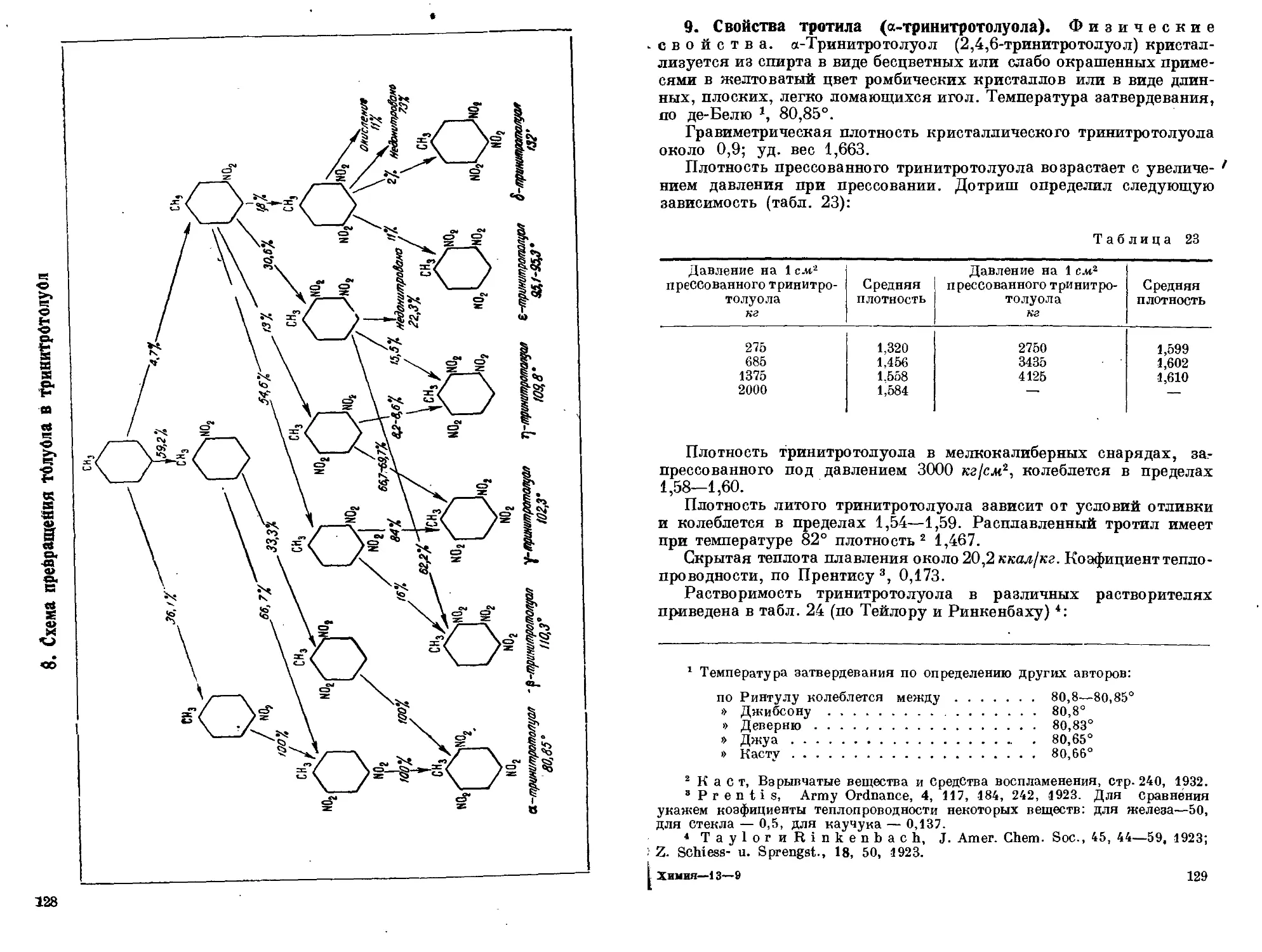
динитротолуола в кислотах. 7. Состав сырого тротила. 8. Схема
превращения толуола в тринитротолуол. 9. Свойства тротила (а-три-
нитротолуола). 10. Применение тротила. И. 2,3,4,6-Тетранитро-
толуол .................................................... 119—142
Периодические методы нитрования толуола до тринитротолуола
1. Старый способ производства тротила в три фазы. 2. Производ-
ство тринитротолуола на английских заводах во время империали-
стической войны 1914—1918 гг. 3. Итальянский способ производства
тринитротолуола с полным кислотооборотом .................. 144-155
Непрерывная нитрация
1. Аппарат Вейлер-тер-Мера. 2. Турбоаппарат Неймана, или
тутольтурбо. 3. Аппарат Куберского. 4. Английский способ непре-
рывной нитрации .......................t................... 160—162
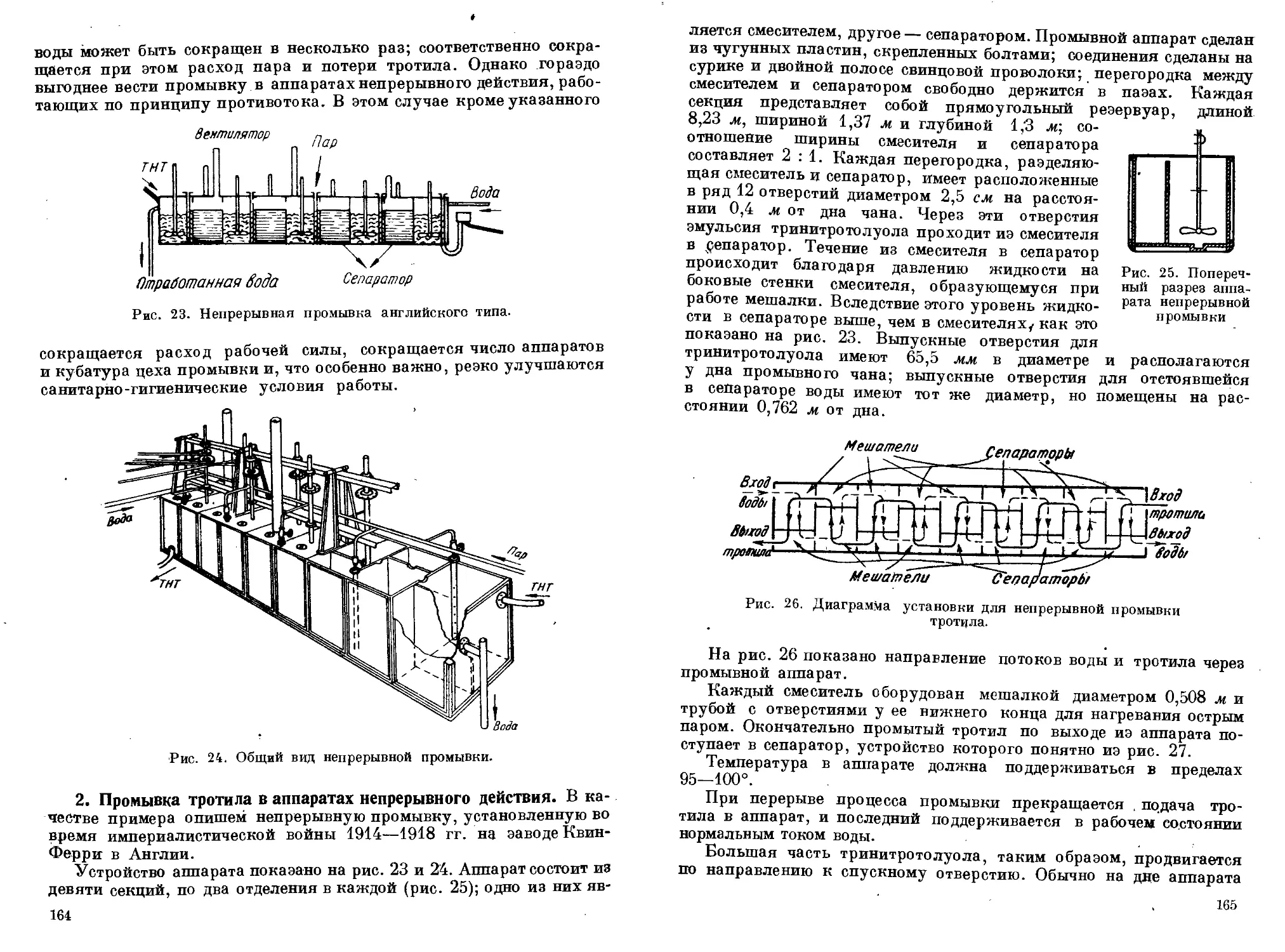
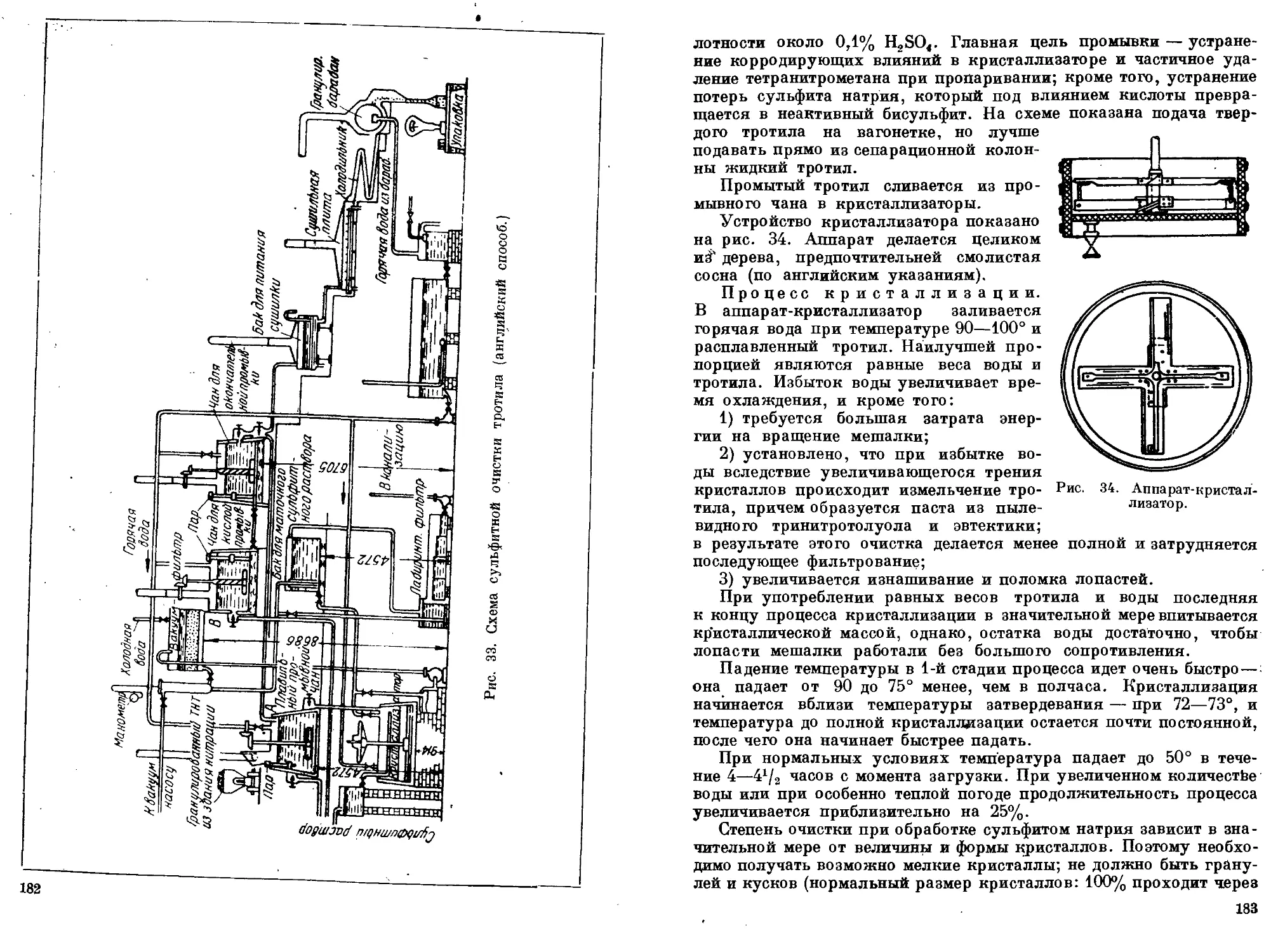
Водная промывка тротила
1. Промывка тротила в аппаратах периодического действия.
2. ПромыВка тротила в аппаратах непрерывного действия. 3. Содо-
вая промывка тротила. 4. Ликвидация промывных вод. 5. Использо-
вание загрязненного тротила. 6. Упаковка некристаллизованного
тротила. 7. Технические условия на некристаллизованный тротил.
8. Обоснование технических условий на некристаллизованный
тротил............................................'......... 163—167
Способы получения очищенного тротила
1. Перекристаллизация тротила из спирта. 2. Сульфитная
очистка тротила. 3. Другие способы очистки тринитротолуола . . 168—190
Нитрование толуол-бензина
Производство изомеров мононитротолуола
1. Применение изомеров мононитротолуола. 2. Получение изо-
меров мононитротолуола фракционированной перегонкой техни-
ческого мононитротолуола. 3. Мероприятия по технике безопас-
ности. 4. Технические условия на о-нитротолуол. 5. Получение р- *
нитротолуола вымораживанием из технического мононитротолуола.
6. Технические условия на р-нитротолуол ................. 195-200
Производство динитротолуола (травелина, порохового динитротолуола)
1. Применение динитротолуола. 2. Производство 2-,4-динитро-
толуола. 3. Технические условия на 2,4-Динитротолуол для краси-
телей. 4. Динитротолуол технический. 5. Технические требования к
динитротолуолу (техническому)............................ 200—201
Глава IX
Нитропроизводные бензола
1. Свойства бензола. 2. Токсическое действие бензола. 3. Тех-
нические •условия на бензол по ОСТ 463. 4. Свойства мононитро- ' . •
бензола и применение его. 5. Свойства динитробензола и его при-
менение. 6. Свойства тринитробензола. 1. Тетранитробензол. 8. Об-
щие-соображения о целесообразности производства и применения ди-
и тринитробензола. 9. Производство мононитробензола. 10. Произ-
водство динитробензола. 11. Получение тринитробензола.... 203—219
• ГлаваХ
Нитропроизводные ксилола
1. Исходные материалы. 2. Химические свойства изомеров кси-
лола. 3. Состав каменноугольного ксилола. 4. Состав пирогенети-
ческого ксилола. 5. Технические условия на каменноугольный кси-
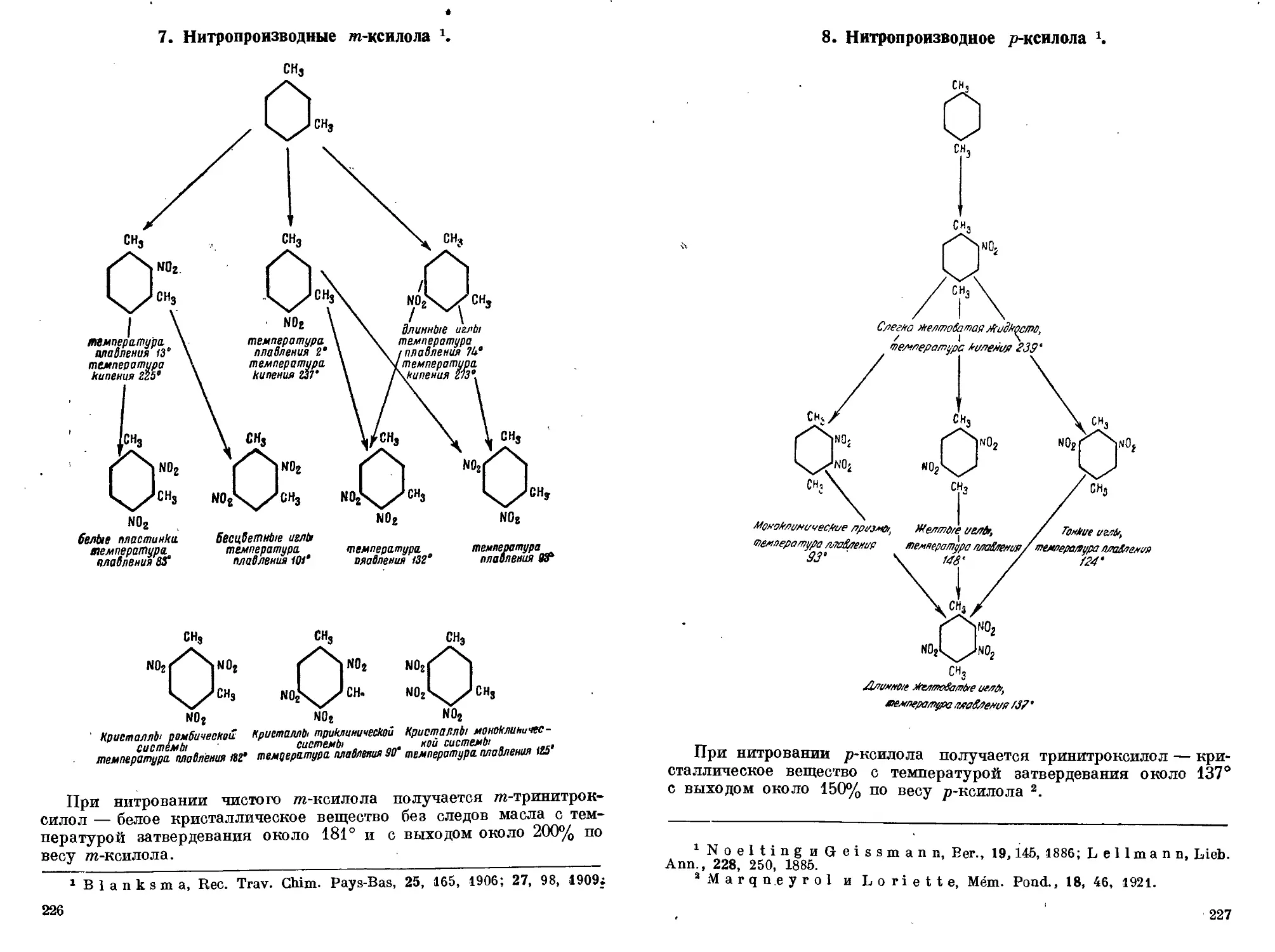
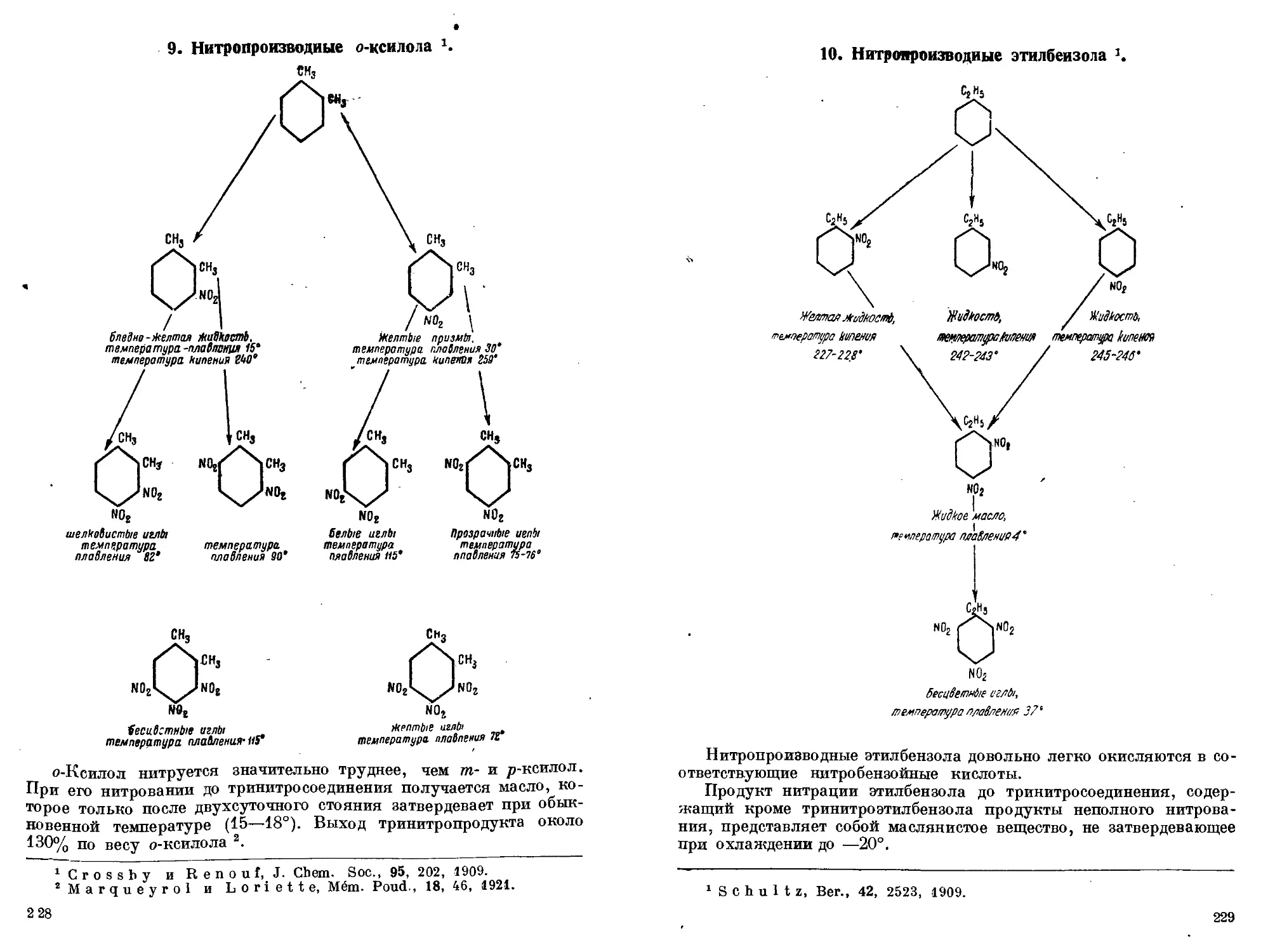
лол (ОСТ 465). 6. Выделение чистого т-ксилола. 7. Нитропроизводные
?п-ксилола. 8. Нитропроизводные р-ксилола. 9. Нитропроизводные
о-ксилола. 10. Нитропроизводные этилбензола. 11. Общее замечание
о продукте тринитрации технического ксилола. 12. Применение кси-
лила. 13. Производство ксилила. 14. Очистка ксилила. 15. Схема
технологического процесса для двухфазного способа. 16. Схема
производства ксилила. 17. Аппаратура. 18. Технические условия
на ксилил (ОСТ 3660). 19. Обоснование технических требований
к ксилилу................................................ 223—238
Глава XI
Нитропроизводные сольвент-нафты
1. Исходные материалы. Технические условия на сольвент-
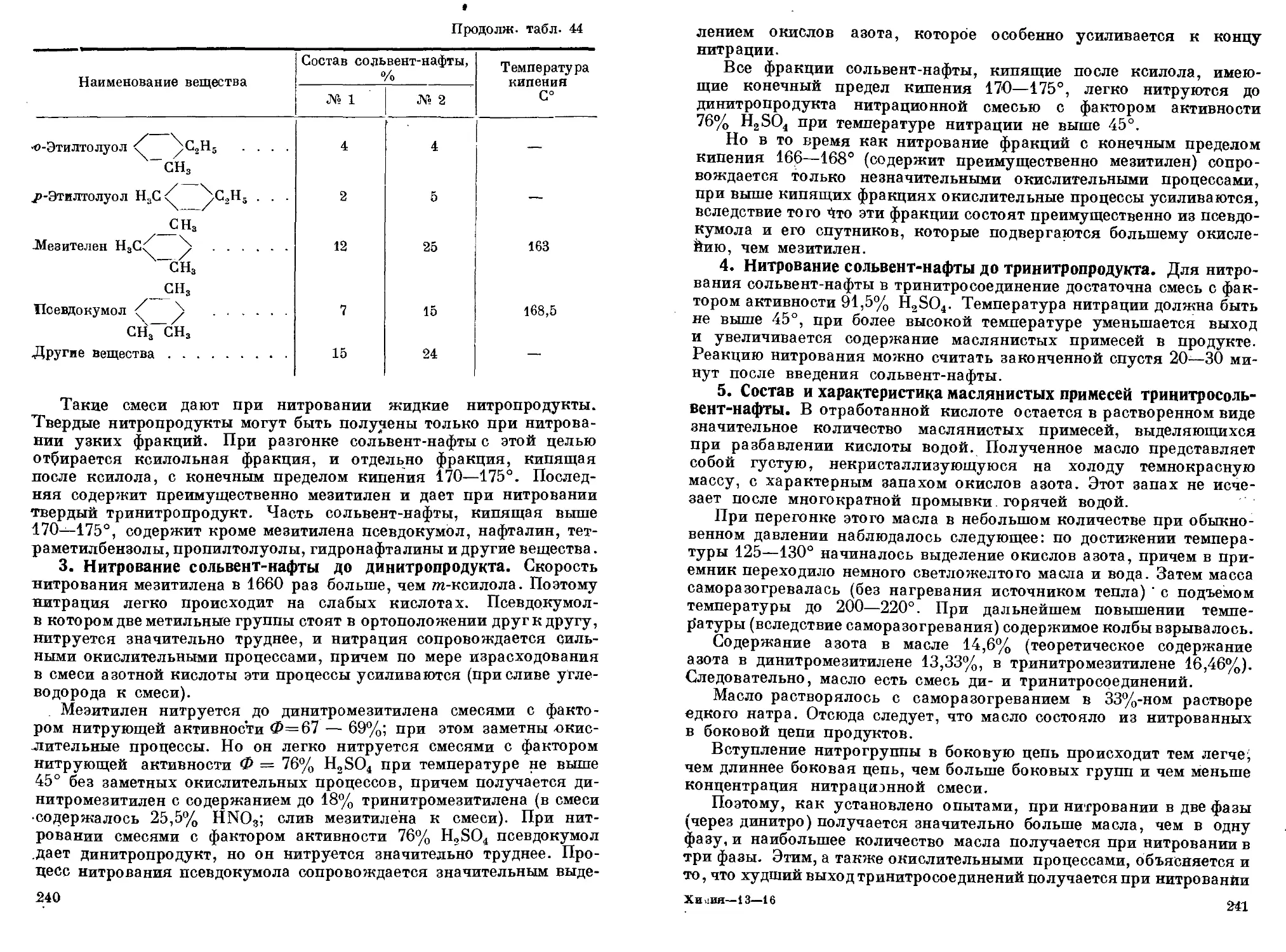
нафту. 2. Состав сольвент-нафты. 3. Нитрование сольвент-нафты до
динитропродукта. 4. Нитрование сольвент-нафты до тринитропро-
6
дукта. 5. Состав и характеристика маслянистых примесей тринитро-
сольвент-нафты. 6. Свойства нитросольвент-нафты. 7. Технические
условия на нитросольвент-нафгу 8. Применение нитросольвент-
нафты .................................................. 239—242
Глава XII
Нитропроизводные нафталина
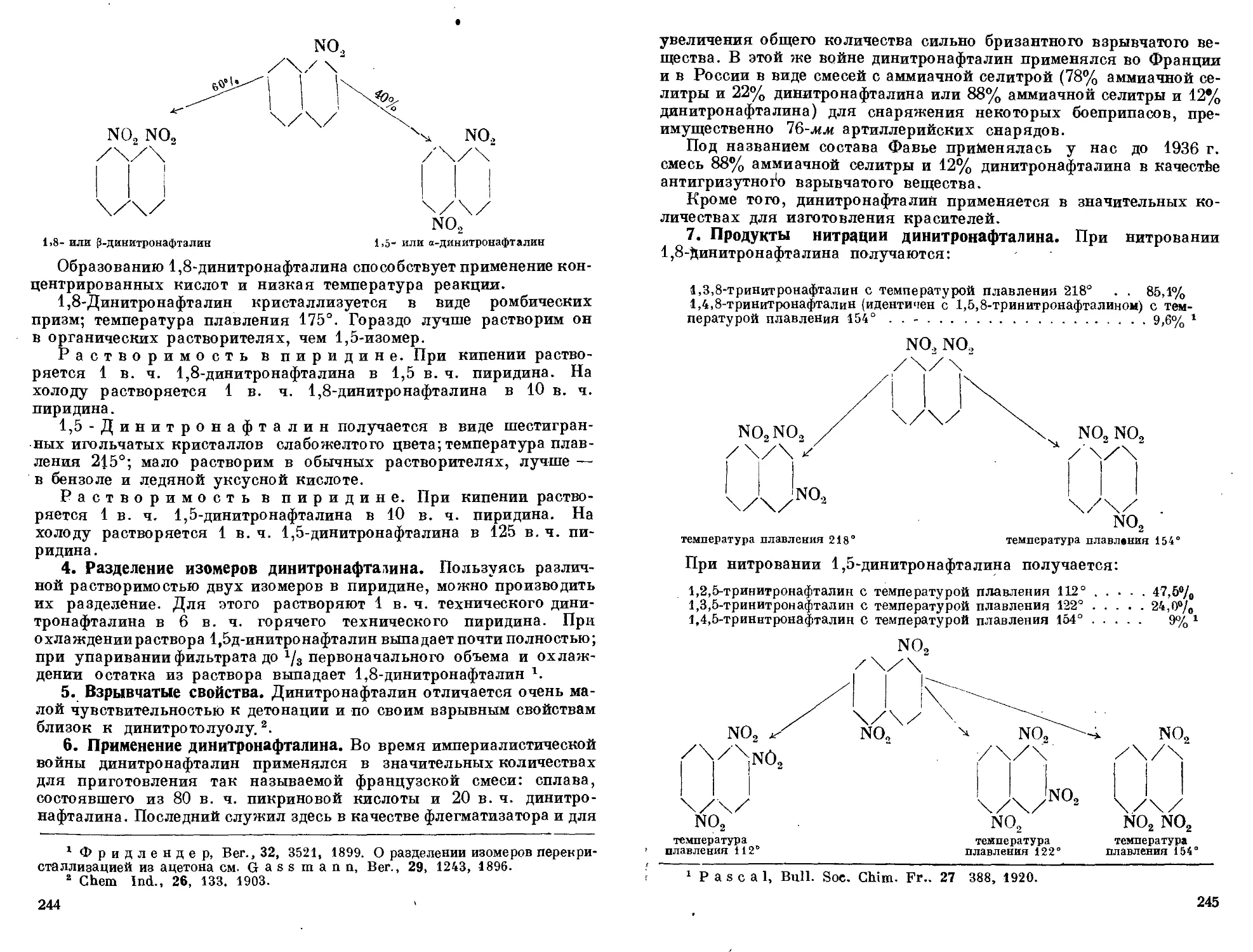
1. Технические условия на нафталин. 2. Продукты нитрации
нафталина. 3. Продукты нитрации мононитронафталина. 4. Разде-
ление изомеров динитронафталина. 5. Взрывчатые свойства. 6. При-
менение динитронафталина. 7. Продукты нитрации динитронафта-
лина. 8. Свойства тринитронафталина. 9. Свойства и применение
технического тринитронафталина. 10. Побочные реакции при нитра-
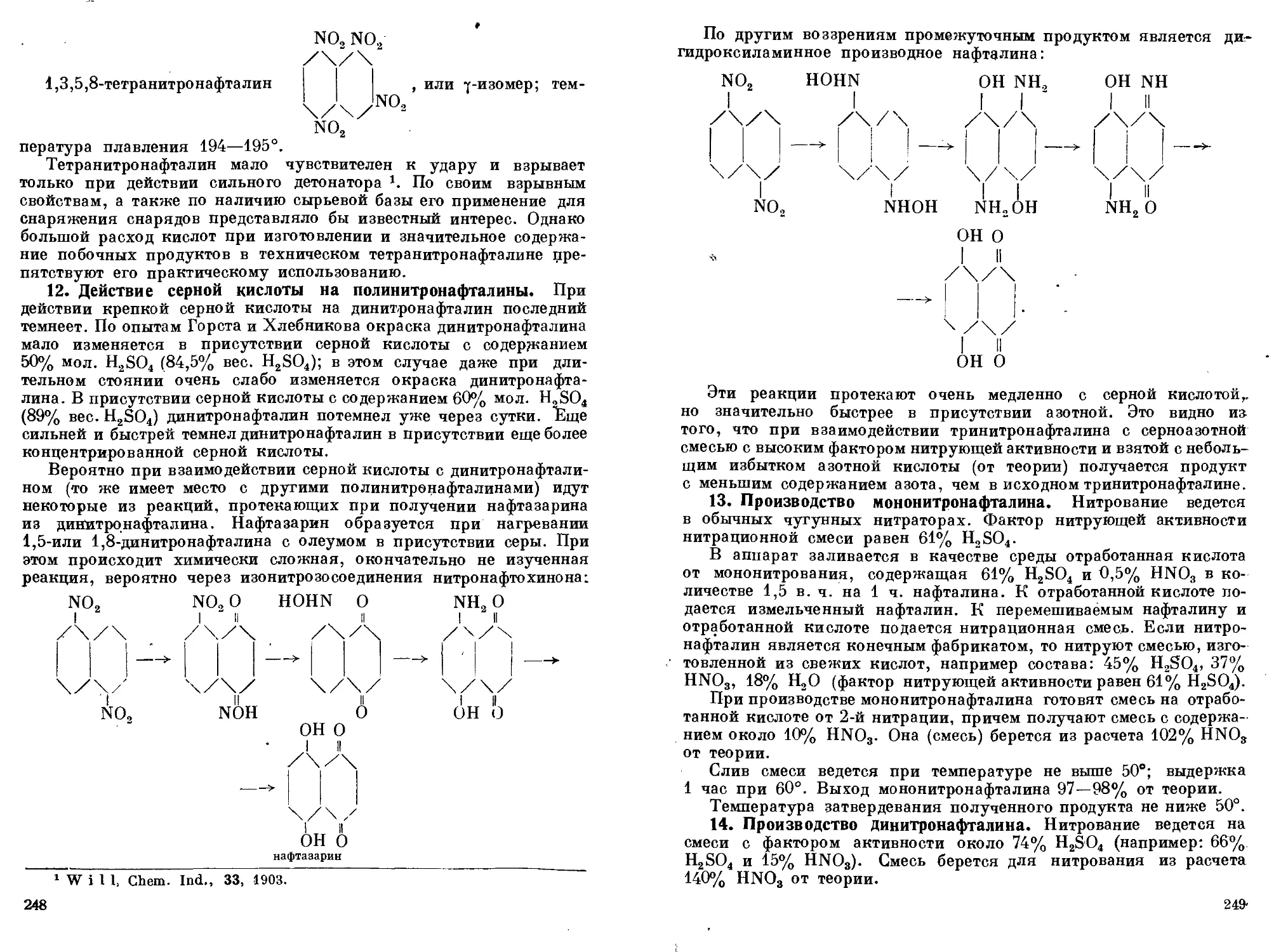
ции нафталина. И. Тетранитронафталнн. 12. Действие серной кис-
лоты на полинитронафталины. 13. Производство мононитро на фта-
лина. 14. Производство динитронафталина- 15. Технические условия
на динитронафталин (ОСТ 2940). 16. Производство тринитронафта-
лина. 17. Получение тетранитронафталина................... 243—251
Глава XIII
Нитропроизводные фенолов и их эфиров
Фенол
1. Свойства фенола. 2. Технические условия на синтетический
фенол (ОСТ 2933). 3. Технические условия на кристаллический
каменноугольный фенол (ОСТ 3296) ......................... 252—253
Продукты нитрации фенола, их свойства и применение
1. Моно нитрофенолы. 2. Строение окрашенных нитрофенолов.
3. Окраска солей нитрофенолов. 4. Причина окраски. 5. Свойства
2,4-динитрофенола. 6. Свойства 2,6-динитрофенола. 7. 2,4-динитрофе-
нолят натрия. 8. Токсические свойства динитрофенола. 9. 3,5-динитро-
фенол. 10. Свойства пикриновой кислоты. 11. Взрывчатые свойства
пикриновой кислоты. 12. физиологическое действие пикриновой
кислоты. 13. Применение пикриновой кислоты. 14. Свойства пикра-
тов. 15. Опасности, обусловленные присутствием пикратов. 16. По-
лучение пикратов. 17. Возможность образования пикратов в произ-
водственных условиях. 18. Тетранитрофенол. 19. Пентанитрофенол . 254—272
Химия процесса сульфации фенола и нитрации (и нитролиза) фенол-
сульфокислот
1. «-Сульфирования. 2. Влияние относительных количеств реаги-
рующих компонентов. 3. Температурный коэфициент реакции суль-
фапин. 4. Механизм реакции сульфации. 5. Обратимость реак-
'Ции сульфации. 6. Влияние температуры на место, занимаемое
сульфогруппой. 7. Побочные реакции при процессе сульфирования.
8. Сульфирование фенола. 9. Опыт трисульфирования фенола.
40. Свойства сульфокислот фенола. 11. Нитрование сульфопроиз-
водных фенола, 12. Нитрование чистых дисульфо-и трисульфокислот.
13. Нитрование динитрофенолсульфокислот в пикриновую кислоту.
14. Состав примесей в отработанных кислотах. 15. Опыты донитро-
вывания динитросульфокислот. 16. Реакция нитрации и реакция ни-
тролиза. 17. Скорость окисления пикриновой кислоты серноазотной
смесью .................................................... 272 —282
Периодические способы сульфирования и нитрования фенола
1. Общие сведения о способах получения пикриновой кислоты
и динитрофенола. 2. Сульфирование и нитрование фенола по фран-
цузскому способу. 3. Нитрование фенола по горшечному способу в
Англии. 4. Усовершенствованный американский способ производства
пикриновой кислоты. 5. Ход реакции нитрации и нитролиза фенол-
.сульфокислот. 6 Производство пикриновой кислоты нитрованием
"фенола крепкими кислотами. 7. Получение пикриновой кислоты по
патенту Гутенсона.............................................. 283—291
Непрерывное нитрование фенола
1. Способ Брука............................................292
Промывка пикриновой кислоты, сушка, просеивание и упаковка
1. Промывка пикриновой кислоты. 2. Сушка пикриновой- кис-
лоты. 3. Просеивание и упаковка пикриновой кислоты. 4. Выход ‘
пикриновой кислоты. 5. Технические условия на пикриновую кислоту
(ОСТ 3515). 6. Качественное определение пикриновой кислоты . . 292—296
Получение дииитрофеиола нитрованием фенола
1. Производство динитрофенола. 2. Получение динитрофенола
нитрованием фенола в растворе уксусной кислоты. 3. Получение ди-
нитрофенола нитрованием фенола азотной кислотой............ 297—298
Производство пикриновой кислоты из бензола через динитрохлорбеизол
• 4. Механизм реакции галоидирования и свойства хлорбензола и
дихлорбензолов. 2. Свойства и применение моно-и динитрохлорбензола.
3. Примеси в техническом динитрохлорбензоле. 4. Производство
хлорбензола. 5. Получение />-дихлорбензола. 6. Нитрование хлор-
бензола. 7. Техника безопасности. 8. Производство динитрохлор-
бензола в одну фазу. 9. Техника безопасности. 10. Технические
условия на динитрофенол, применявшийся во Франции для сплава
DD60/40. 11. Нитрование динитрофенола. 12. Расходные коэфици-
енты и выходы при производстве пикриновой кислоты из хлорбен-
зола. 43. Пикраминовая кислота ................................ 299—309
Производство р- и о-иитрохлорбеизола
1. Нитрование хлорбензола. 2. Сепарация кислого мононитро-
хлорбензола. 3. Нейтрализация кислого нитрохлорбензола. 4. Кри-
сталлизация мононитрохлорбензола. 5. Фуговка. 6. Разгонка эвтек-
тики. 7. Технические условия на р-нитрохлорбензол. 8. Технические
условия на о-нитрохлорбензол................................... 310—311
Нитропроизводиые анизола и фенетола
4. Свойства триниТроанизола и тринитрофенетола. 2. Получение
Ди- и тринитроанизола, ди- и тринитрофенетола. 3. Тетранитро-
анизол.............................Г..................... .... 311—312
Трииитрокрезол, или крезолит
4. Крезол. 2. Свойства три нитрокрезола. 3. Применение трини-
трокрезола. 4. Производство тринитрокрезола ................... 315—316
8
Глава XIV
Нитропроизводные ароматических аминов и их замещенных
Нитропроизводные анилина
1. Анилин. 2. Ацильные производные ароматических аминов.
3. Ароматические производные мочевины. 4. Мононитроанилины.
5. Действие окислителей на органические основания. 6. Нитрование
анилина азотной кислотой. 7. Производство />-нитроанилина. 8. Про-
изводство о-нитроанилина. 9. Частичное восстановление нитросоёди-
нений. 10. Производство т-нитроанилина. И. 2,4,6-Тринитроани-
лин, или пикрамид. 12. Отношение полинитроанилинов к диазоти-
рованию и свойства их диазониевых солей. 13. Тетранитроанилин.
14.Получение тетранитроанйлина. 15. Пентанитроанилин. 16. Нитро-
анилид (фенилнитроамин). 17. Тринитрофенилнитроамин........ 317—334
Исходные продукты для производства тетрила
1. Монометиланилин. 2. Диметиланилин. 3. Получение диметил-
анилина. 4. Технические условия на диметиланилин........... 335—338
Продукты нитрации метилаиилинов, их свойства и применение
1. Нитропроизводные монометиланилина. 2. Нитропроизводные
диметиланилина. 3. Нитрование моно- и диметиланилина азотной
кислотой. 4. Свойства тетрила. 5. Применение тетрила. 6. Тетра-
нитрофенилметилнитроамин. 7. Пентанитрофенилметилнитроамин.
8. Тринитрофенил-т-диметилдинитроамин ........................ 338—347
Растворение диметиланилина и нитрование сульфата диметиланилина
1. Схема производства тетрила. 2. Другие способы получения
тетрила....................................................... 348—353
Промывка тетрила
1. Непосредственная промывка кислого тетрила водой. 2. Про-
мывка тетрила в бензольном растворе. 3. Сушка промытого тетрила.
4. Качество промытого тетрила................................. 354 —353
Перекристаллизация тетрила
1. Растворитель для перекристаллизации тетрила. 2. Перекри-
сталлизация тетрила из ацетона. 3. Перекристаллизация тетрила из
бензола. 4. Виды брака тетрила и его исправление. 5. Сушка пере-
кристаллизованного тетрила. 6. Формирование партий, просеива-
ние, упаковка. 7. Технические условия на тетрил (ОСТ 3514).
8. Ректификация маточного ацетона. 9. Перекристаллизация тетрила из
дихлорэтана. 10. Применение нецерекристаллизованного тетрила.
11. Гексанитродифенилэтилендинитроамин, или дитетрнл. 12. Аналоги
тетрила....................................................... 356—362
Гексаиитродифениламин, или гексил
1. Свойства гексила. 2. Физиологическое действие гексила.
3. Применение гексила. 4. Получение гексила. 5. Промывка гекса-
нитродифениламина .........’.................................. 365—370
Глава XV
Соединения с двумя фенильными группами (кроме гексила)
1. Гексанитродифенил. 2. Гексанитродифенилоксид. 3. Гекса-
нитродифенилсульфид. 4. Гексанитродифенилсульфон ........ 371—374
Глава XVI
Гексоген
Строение гексаметилентетрамина
Свойства и применение гексаметилентетрамина
1. Физические свойства. 2. Химические свойства. 3. Меры пре-
досторожности при работе с гексаметилентетрамином. 4. Применение
гексаметилентетрамина. 5. Количественное определение гексамети-
лентетрамина .................................................- • 378—382
Производство гексаметилентетрамина
1. Получение гексаметилентетрамина из формалина и аммиака.
2. Другие способы получения гексаметилентетрамина .............. 383—387
Свойства гексогена
' 1. Физические свойства. 2. Химические свойства. 3. Взаимодейст-
вие гексогена с серной кислотой. 4. Химическая стойкость. 5. Взрыв-
чатые свойства гексогена. 6. Применение гексогена .......... 388—391
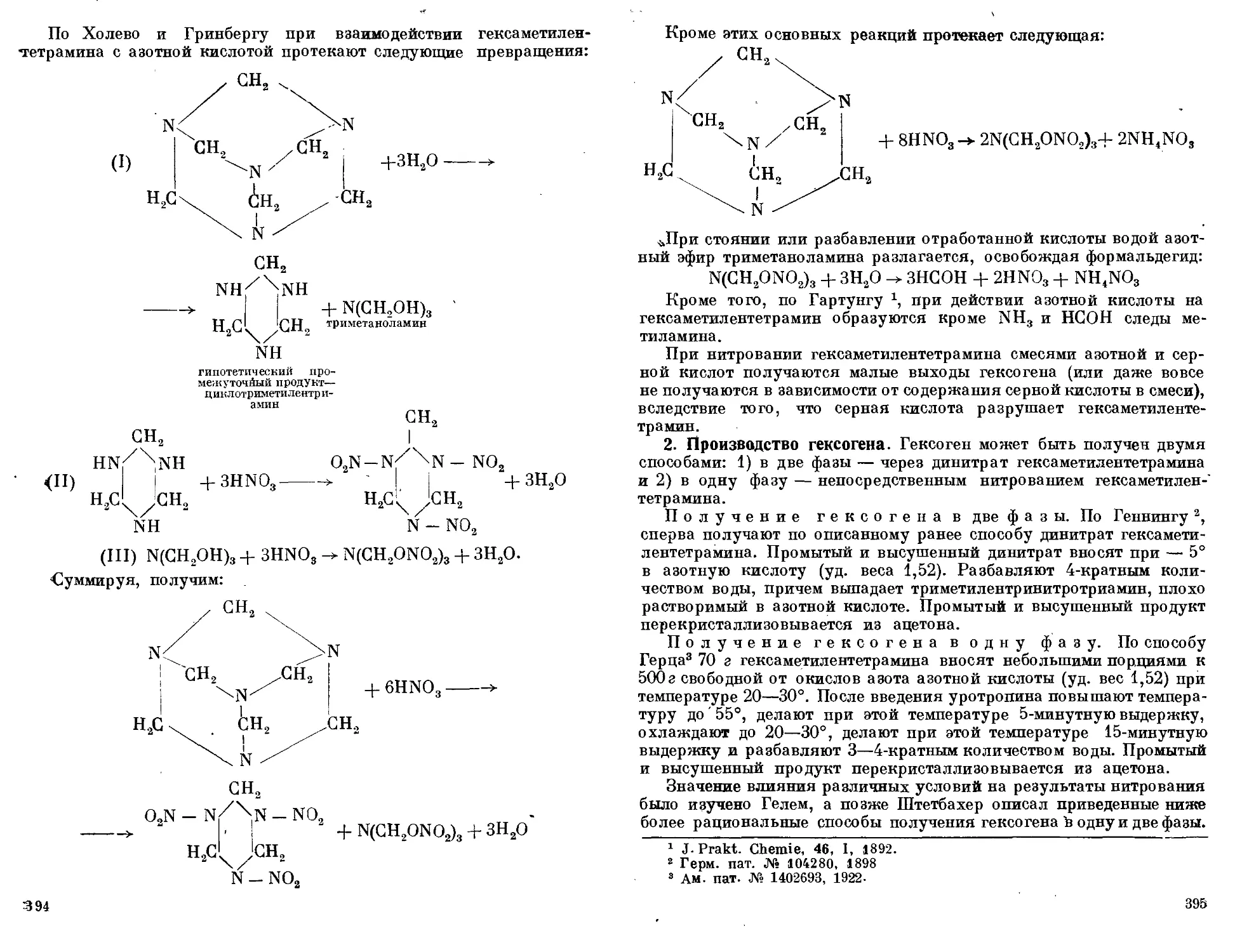
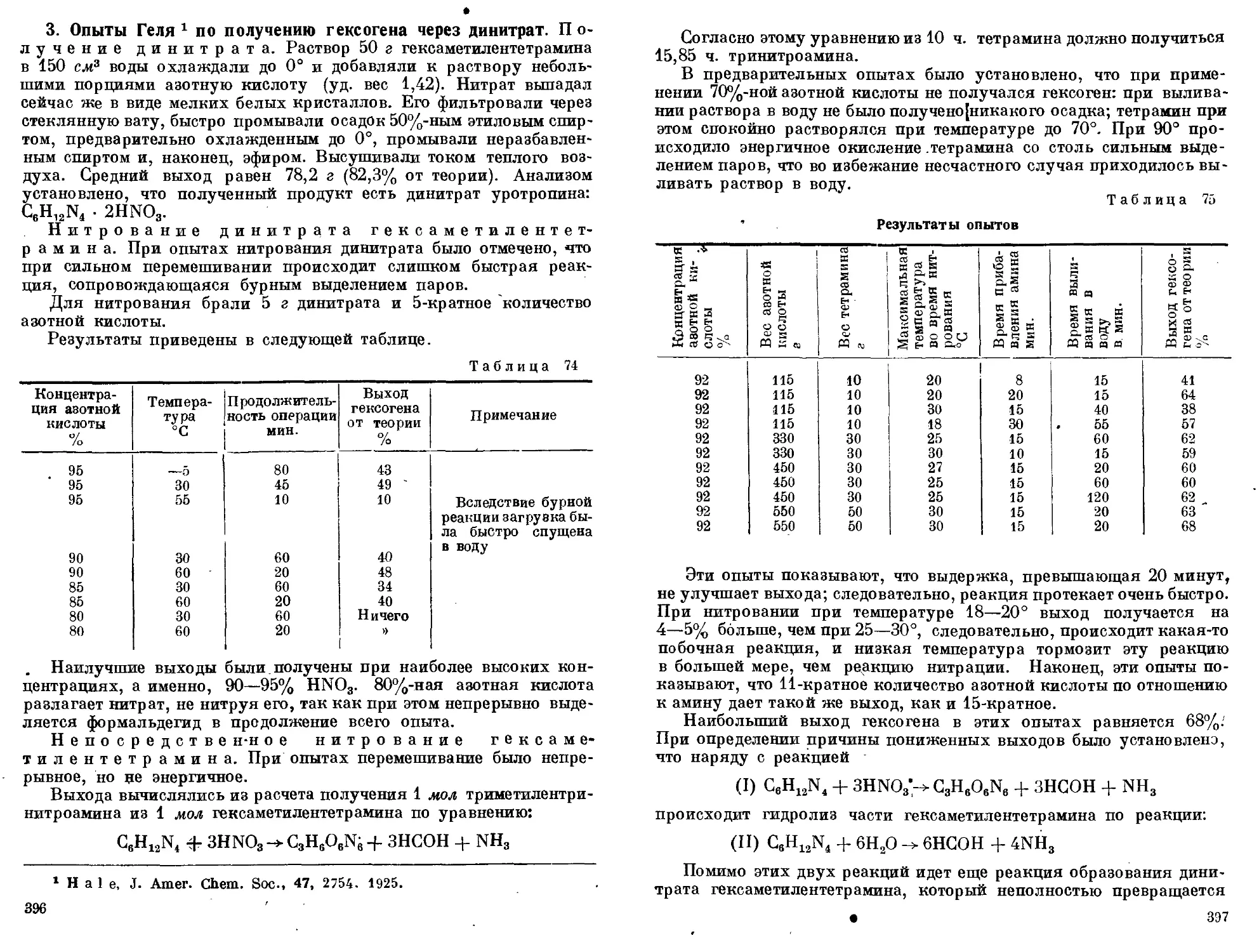
Производство гексогена
‘ 1. Течение реакции нитрации. 2. Производство гексогена.
3. Опыты Геля по получению гексогена через динитрат. 4. Получе-
ние гексогена по описанию Штетбахера- 5. Получение гексогена
в две фазы через динитрат. 6. Общее замечание о получении гексо-
гена в одну или две фазы.................................... 392—401
Глава XVII
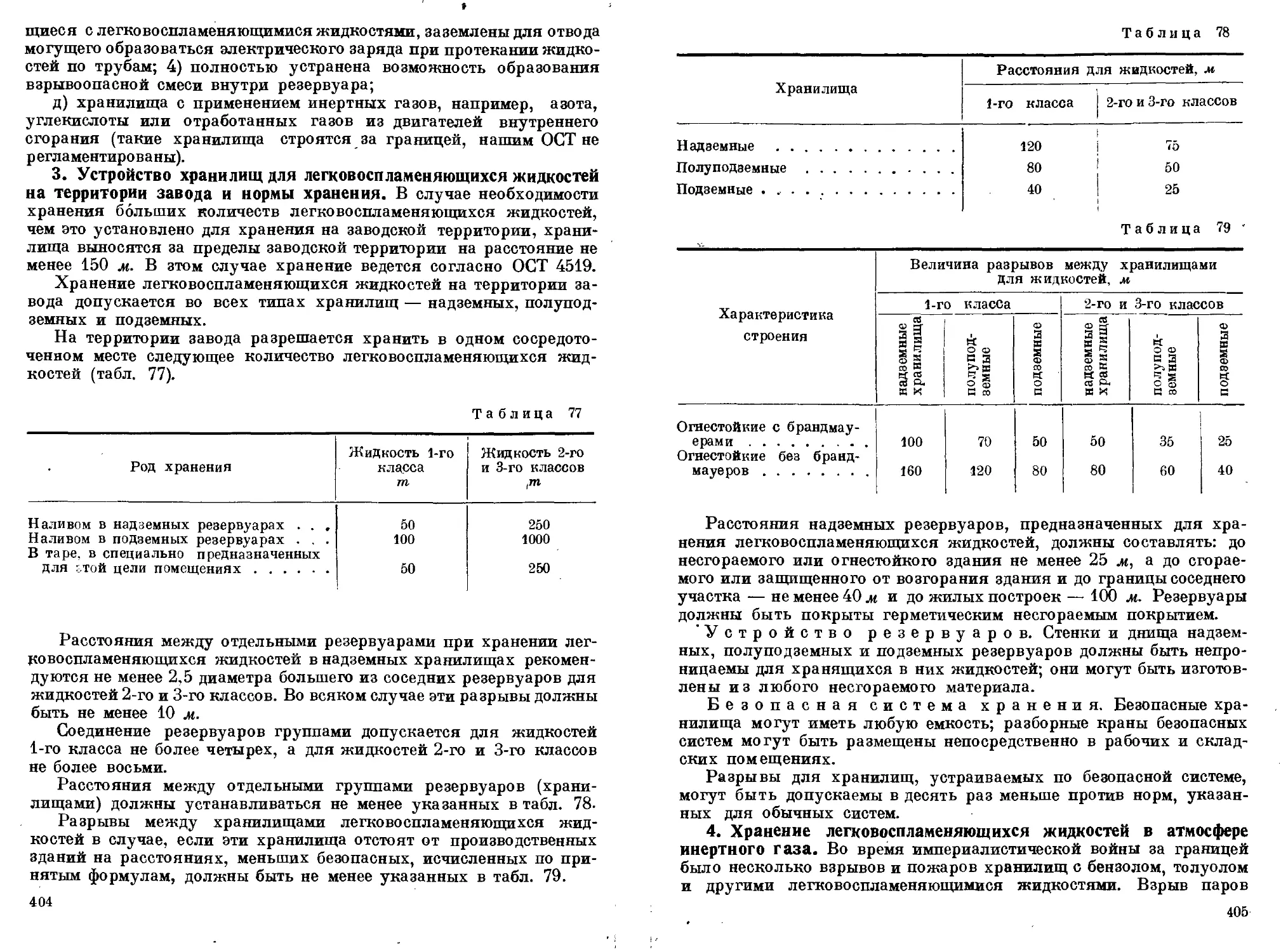
Хранение толуола и других легковоспламеняющихся жидкостей
1. Классификация легковоспламеняющихся жидкостей. 2. Клас-
сификация хранилищ для легковоспламеняющихся жидкостей.
3. Устройство хранилищ для легковоспламеняющихся жидкостей
на территории завода и нормы хранения. 4. Хранение легковоспламе-
няющихся жидкостей в атмосфере инертного газа. 5. Перевозка легко-
воспламеняющихся жидкостей. 6. Вредные пары, загрязняющие воз-
дух производственных помещений............................ 402—407
Приложение 1. Исторический очерк. 1. От открытия дымного
пороха до открытия явления детонации. 2. Развитие нитроглицери-
новых взрывчатых веществ. '3. Взрывчатые вещества военного на-
значения................................................. 408—420
Приложение 2. Кислотное хозяйство. 1. Расчет одноименных
кислот. 2. Расчет тройных смесей, приготовляемых смешением двой-
ных смесей. 3. Расчет тройных смесей, приготовляемых смешением
двойных и тройных смесей. Библиографический указатель ориги-
нальных работ по вопросу о расчете кислотных смесей. 4. Дени-
трация отработанной кислоты. 5. Утилизация потерь кислоты
при нитровании. 6. Хранение и транспортировка кислот. 7. То-
ксическое действие окислов азота и HNO3. 8. Техника безопас-
ности................................................... • 421—440
Именной указатель....................................... 441—444
Предметный указатель...................................... 445—450
ПРЕДИСЛОВИЕ
;д Автор ставил себе целью дать в настоящей книге, предназначенной
служить в качестве учебного пособия для втузов, возможно более глубокое
физико-химическое обоснование процессов нитрации и сульфации, а равно
изложить возможно полнее другие вопросы по нитросоединениям.
Поэтому в книге уделено много внимания теоретическому обоснованию
как реакций, так и условий, при которых они протекают. С этой же целью
введены главы: «Механизм реакции нитрации». «Основы процесса нитрации»,
«Влияние различных факторов на течение процесса нитрации ароматических
углеводородов», «Действие окислителей на органические основания» и ряд
других.
В главе «Способы введения нитрогрупп» наряду с основными сведениями
подробно рассмотрен вопрос о нитровании в присутствии катализаторов.
Глава «Нитропроизводные алифатического ряда» включает последние ра-
боты американских химиков. Относительно подробное изложение вопроса
о получении и свойствах нитросоединений алифатического ряда стало совер-
шенно необходимым после того, как наметилась перспектива возможного ши-
рокого применения этих нитросоединений для получения новых взрывчатых
веществ класса нитроэфиров и использования этих нитросоединений для полу-
чения ряда ценных химических продуктов.
Приведены необходимые сведения о токсических свойствах исходных и
промежуточных продуктов и нитросоединений и по технике безопасности.
Нецелесообразно приводить в книге детальные сведения, приближающиеся
к инструкциям или копирующие в известной мере эти инструкции.
Автор отдает себе отчет в том, что настоящая работа, как первый опыт
(построения руководства по химии и технологии нитросоединений, не лишена
недочетов.
Тем не менее автор надеется, что книга принесет пользу не только сту-
дентам, изучающим данный предмет, но и работникам промышленности, же-
лающим расширить свои познания в этой области.
В заключение заметим, что в нашей литературе еще не установилось ясное
отличие терминов: нитрация и нитрование, сульфация и сульфирование.
В соответствии с законами образования слов в русском языке мы приме-
няем эти термины в настоящей книге со следующими значениями: термины
нитрование, сульфирование — как обозначение действия (аппаратчика, рабо-
11
чего и пр.), а термины нитрация, сульфация—как процессы, идущие при на-
личии для их протекания подходящих условий. Поэтому мы, например, пишем:
«реакция нитрации толуола» и «методы нитрования толуола».
Глава «Кислотное хозяйство» написана совместно с инженером М. С. Гельф-
маном.
Просьба к читателям сообщить свои замечания к книге для учета их при
дальнейшей работе. *
Проф. А. Г. Горст
Москва, 23 ноября 1939 г.
ПЕРЕЧЕНЬ СОКРАЩЕННЫХ ОБОЗНАЧЕНИЙ ЖУРНАЛОВ
Сокращенное обозначение журнала
Ан. Кр. Пр.
Ж. О. X.
Ж. Пр. X.
Ж. Р. Ф. X. О.
Ж. Хим. Пр.
Укр. Хим. Журя.
X. Р. Ж.
Angew. Chem.
Army Ordnance
Вег.
Biochem. Ztschr.
Bull. Soc. Chim. Belg.
Bull: [Soc. Chim. Fr.
Chem. Ind.
Chem. Met. Eng.
Chem. News
Chem. Trade J.
Gliem. Ztg.
Chem. Ztschr.
Chim et Ind.
C. R.
Dfsch. Med. Wochschr.
Gazz. Chim. Ital.
Helv. Chim. Acta
India Rubber
Ind. Eng. Chem.
Jahresber. chem.-techn. Reichsanstalt
J. Amer. Chem. Soc.
J. Chem. Soc.
J. Chim. Phys.
J. Prakt. Chemie
J. Soc. Chem. Ind.
J. Soc. Dyers a. Colour
Полное наименование журнала
Анило-Красочная Промышленность
Журнал Общей Химии
Журнал Прикладной Химии
Журнал Русского Физико-Химического
Общества
Журнал Химической Промышленности
Украинский Химический Журнал
Химический Рефератный Журнал
Angewandte Chemie
Army Ordnance
Berichte der Deutschen Chemischen Ge-
• sellschaft
Biochemische Zeitschrift
Bulletin de la Societe Chimique de Bel-
gique
Bulletin de la Societe Chimique de
France
Die Chemische Industrie
Chemical and Metallurgical Engineering
Chemical News
Chemical Trade Journal and Chemical
Engineer
Chemiker Zeitung
Chemische Zeitschrift
Chimie et Industrie
Comptes rendus hebdomadaires des Se-
ances de 1’Academie des Sciences
Deutsche Medicinische Wochen-Schrift
Gazzetta Chimica Italiana
Helvetica iChimica (Acta
India Rubber
Industrial and Engineering Chemistry
Jahresbericht der Chemisch-Technischen
Reichsanstalt
Journal of the American Chemical So-
ciety
Journal of the Chemical Society, Lon-
don
Journal de Chimie Physique
Journal fur Praktische Chemie
Journal of the Society of Chemical In-
dustrie
Journal of the Society of Dyers and
Colour
13
KoninkJ. Akad. Wetensch.
Amsterdam
Lieb. Ann.
Man. of TNT
Mem. Artill. fr.
Мёт. Pond.
Monatsh. Chem.
Pharmac. Weekbl.
Proc. Roy. Soc.
Przeglad Artiller.
Re^- gen. mat. plast.
Rec. Trav. Chim. Pays-Bas
Riv. Artigl. Genio.
Scient. Proc. Roy. Dublin Sos.
Siiddeutsche Apoth. Ztg.
Trans- Amer. Elektrochem- Soc.
Z. Angew. Chem.
Zbl
Z. Schiess- u. Sprengst.
Z. Physikal. Chem.
Wied. Ann
Koninklijke Akademie van Wetenscha-
pen te Amsterdam, Verslag van de
gewone Vorgadering der Afdeeling Na-
turkande
Liebigs Annalen der Chemie
Technical Records of Explosives Supply
1915—1918, N 2, Manufacture of Tri-
nitrotoluene (TNT) and its Intermediate
Products, London, 1920
Memorial de Г Artillerie frangaise
Memorial des Poudres
Monatshefte fur Chemie
Pharmaceutisch Weekblad
Proceedings of the Royal Society, London.
Przeglad ArtiUerijski
Revue generale des matiers plastique
Recueil des Travaux Chimiques des Pays-
Bas
Rivista di Artigleria e Genio '
Scientific Proceedings of the Royal Dub-
lin Society.
Siiddeutsche Apotheker Zeitung
Transactions of the American Elektro-
chemical Society
Zeitschrift fur Angewandte Chemie
Chemi sches Zentralblatt
Zeitschrift fiir das gesamte Schiess-und
Sprengstoffwesen '
Zeitschrift fiir Physikalische Chemie
Wiedemanns Annalen der Chemie
ГЛАВА I
СПОСОБЫ ВВЕДЕНИЯ НИТРОГРУПП
ОТЛИЧИТЕЛЬНАЯ ХАРАКТЕРИСТИКА НИТРОСОЕДИНЕНИЙ,
НИТРОАМИНОВ И АЗОТНЫХ ЭФИРОВ СПИРТОВ И УГЛЕВОДОВ
Нитросоединения, нитроамины и азотные эфиры спиртов и угле-
водов весьма значительно различаются между собой по химиче-
скому строению, химической стойкости, чувствительности к меха-
ническим воздействиям, действию на них водорода в момент выде-
ления, взаимодействию с концентрированной серной кислотой и
по другим признакам.
1. Строение. В нитросоединениях группа NO2 связана непо-
Z0
средственно с углеродным атомом: R—С — N£ , например, в
ХО
нитробензоле:
В нитроаминах нитрогруппа связана с углеродным атомом через
/R
азот: 7?—С — , например, в фенилметилнитроамине:
XN02
Наконец, в азотных эфирах нитрогруппа связана с углеродным
Z0
атомом через кислород — С — О — N£ , например, в нитро-
Х0
глицерине:
CH2-O-NO2
ch -o-no2
ch2-o-no2
15
Как известно, наиболее прочными являются соединения, в ко-
торых нитрогруппа связана непосредственно с углеродным атомом;
менее прочны соединения, в которых нитрогруппа связана с угле-
водом черв: азот, и, наконец, наименее прочна связь нитрогруп-
пы с углеродным атомом через кислород. В полном соответствии с
-этими представлениями находятся свойства соединений, описы-
ваемых ниже.
2. Химическая стойкость. Нитросоединения являются вполне
стойкими соединениями, не способными к самопроизвольному раз-
ложению и самовозгоранию; они значительно более стойки при на-
гревании, чем азотные эфиры.
Нитроамины также не способны к самопроизвольному разложе-
нию и самовозгоранию; но они менее стойки при нагревании, не-
жели нитро соединения; например, тетрил (тринитро фенилметил-
нитроамин) начинает медленйо разлагаться уже при 130°, при 140—
180° наблюдается значительное газообразование, а при продолжи-
тельном нагревании при этой температуре происходит самовоспла-
менение.
Азотные эфиры являются химически нестойкими соединениями:
при повышенной температуре (в присутствии следов минеральной
кислоты — при обыкновенной температуре) эти вещества разла-
гаются с выделением значительного количества тепла; при (yiaro-
.приятных условиях это разложение может сопровождаться само-
возгоранием и даже взрывом.
3. Чувствительность к механическим воздействиям. Чувствг тель-
ность к механическим воздействиям (удару, трению и т. п.) у нитро-
соединений, как правило, мала. Заметно выше чувствительность у
нитро а мино в, наконец, чувствительность азотных эфиров очень
значительна.
4. Восстановление водородом. При восстановлении водородом
в момент выделения нитрогруппа в нитросоединениях восстанавли-
вается в аминогруппу х.
1 Реакция восстановления протекает по схеме:
JR — NO2 + ЗН2—» В — NH2 + 2Н2О
<
Восстановление нитросоединений протекает в несколько промежуточных
•стадий. '
При энергично протекающем восстановлении в кислой среде вначале обра-
зуется нитрозосоединение, затем производное гидроксиламина, при дальнейшем
восстановлении которого образуется амин: =. ,
R — NO2 — R — NO — R — NH(OH) — R — NH2
При восстановлении, протекающем менее энергично в щелочной среде, про-
цесс сначала идет в том же направлении: образуется нитрозосоединение и про-
изводное гидроксиламина. В присутствии щелочи чрезвычайно легко проходит
конденсация нитрозосоединения и производного гидроксиламина, причем эта
реакция протекает гораздо скорее, чем дальнейшее восстановление нитрозосо-
•единения и производного гидроксиламина; при этой конденсации образуется
16
При восстановлении нитроаминов наблюдаются некоторые осо-
бенности. При слабом восстановлении нитроаминов1 выделяется
метилгидразин; так, например, для тетрила имеем:
СН3
C6H2(NO2)3-N/no +3H2-^C6H2(NO2)3OH 4-CH3NH-—NH2
Образующийся в качестве промежуточного продукта тринитро-
фенилметилгидразин тотчас распадается по приведенной реакции,
а тринитрофенол восстанавливается с образованием аминосоеди-
нений.
При энергичном восстановлении метилгидразин восстанавли-
вается с образованием метиламина и аммиака, а пикриновая кис-
лота восстанавливается в триаминофенол:
СН3
CeH2(NO2)3 - +10H2-^C6H2(NH2)3OH+NH3+CH3NH2+7H2O
NO2 t'ri
При действии водорода в момент выделения на азотные эфиры
нитрогруппа восстанавливается до аммиака, и, как правило, реге-
нерируется исходный спирт.
5. Особенности взаимодействия с серной кислотой. Группа NO3
в нитро соединениях не вытесняется серной кислотой — реакция ни-
трации необратима. В нитро аминах нитрогруппа, связанная с угле-
родом через азот, вытесняется концентрированной серной кислотой;
например, при растворении тетрила в купоросном масле протекает
реакция:
.СН3 ZCH3
2C6H2(NO2)3N( 4- 2H5SO4 2С6Н2(N0.,)3N< +2HNO3
<NO2 ' XSO3H
В азотных эфирах нитрогруппа вытесняется серной кислотой —
реакция нитрации обратима.
азоксисоединение:
О
II
В —NO + В — NH(OH) —» В — N = N —В
азоксисоединеаие
Поэтому при щелочном восстановлении лишь небольшое количество ййиивг
Гидрокси ламина переходит в анилин, а в качестве главного продукта пмода$ф*^%
азоксибензол, который подвергается дальнейшему восстановлению/их тасти>е.сг
ном восстановлении полинитросоединений см. стр. 325): f л? /
В—N = N — В->-В — N = N — В->В—NH — NH —дй i' S I
О
азонсисоединение азосоединение гид!
’ ’Herz, Z. Schiess- u. Sprengst., 14, 101, 1919.
<имия—13—2
(/ИЯ*
17
Несмотря на отмеченное глубокое различие в строении и в свой-
ствах нитро соединений и азотных эфиров спиртов и углеводов, для
отдельных представителей азотных эфиров укоренились совершенно
неправильные названия: нитроглицерин, нитроклетчатка, нитро-
крахмал и т. п.
Ниже речь идет об истинных нитросоединениях и нитроаминах,
близких по свойствам к нитросоединениям.
СПОСОБЫ ВВЕДЕНИЯ НИТРОГРУПП
1. Нитрование одной азотной кислотой. В зависимости отксвойств
нитруемого соединения удается ввести одну, редко две и, как ис-
ключение, три нитрогруппы нитрацией одной азотной кисло-
той (нитробензол, нитротолуол, циклотриметилентринитроамин
и др.).
Нитрованием одной азотной кислотой, как правило, нельзя вы-
годно получать нитросоединения:
а) требуется большой расход азотной кислоты в связи с быст-
рым уменьшением скорости реакции нитрации вследствие выделе-
ния реакционной воды;
б) вследствие большой растворимости нитросоединений даже
в разбавленной азотной кислоте;
. в) вследствие повышенных окислительных процессов, при кото-
рых происходит разбавление азотной кислоты.
Этот способ применяется только при нитровании гексаметилен-
тетрамина.
2. Нитрование смесями серной и азотной кислот позволяет резко
снизить расход дорогой азотной кислоты; кроме того, высшие степени
нитрации, как правило, нельзя получить на чистой азотной кислоте,
и многие из них выгодно получаются при применении смесей ее
с серной кислотой. Этот вопрос будет подробно рассмотрен в сле-
дующих разделах.
3. Нитрование смесями азотной кислоты с фосфорным ангидри-
дом. Здесь фосфорный ангидрид действует, как водоотнимающее
средство:
Р2О5 + ЗН2О -> 2Н3РО4
Но так как образующаяся при соединении с водой фосфорная
кислота корродирует все материалы, применяющиеся для изготов-
ления аппаратуры, то фосфорный ангидрид на практике не исполь-
зуется для нитрования.
4. Нитрование азотной кислотой в смеси с уксусной кислотой
или уксусным ангидридом. Уксусная кислота применялась в неко-
торых случаях в качестве растворителя при реакции нитрации.
Невидимому, здесь уксусная кислота действует не специфическим
18
образом \ а как индиферентный растворитель, создающий гомоген'
ную среду 1 2.
Ортон 3 нитровал толуол в растворе уксусной кислоты (99,25%
СН3С00Н) азотной кислотой с содержанием 94—95% HNO3; он брал
300—400% азотной кислоты от теории, вел процесс при повышенной
температуре и получал выход нитротолуолов 17—18% оттеории. При
прибавлении уксусного ангидрида выход нитротолуола повышался.
Добавлением уксусного ангидрида в количестве, необходимом
для связывания всей воды (имеющейся в смеси и реакционной), до-
стигалось полное нитрование толуола с теоретическими выходами.
Вопрос нитрования в присутствии уксусной кислоты или уксус-
ного ангидрида пока представляет только теоретический интерес.
5. Нитрование азотной кислотой в присутствии катализатора.
Давно известно влияние ртути (или ее солей) на взаимодействие
между ароматическими соединениями и серной кислотой. Напри-
мер, присутствие ртути различным образом влияет на реакцию
сульфации: серная кислота приобретает свойство энергично
окислять некоторые ароматические соединения 4 (например, нафта-
лин окисляется серной кислотой в присутствии ртути во фталевую
кислоту), смещается место вступления сульфогруппы 5 * (например,
при сульфировании антрахинона серной кислотой в присутствии
ртути получается а-сульфокислота, тогда как без ртутной соли
образуется ^-сульфокислота); наконец, облегчается самая реакция
сульфирования. Все это навело на мысль об исследовании влияния
ртутных солей на реакцию нитрации.
Однако первые поставленные с этой целью опыты не дали поло-
жительных результатов ®: при нитровании толуола и нитробензола
азотной кислотой не было обнаружено влияния добавки ртутной
соли на течение реакции.
Немного позже Вольфенштейн и Бетерс 7 сделали случайное на-
блюдение, что при взаимодействии азотной кислоты с бензолом
в присутствии ртутной соли выделяются окислы азота. Заинтересо-
вавшись причиной этого явления, исследователи установили, что
при названном взаимодействии происходит окисление бензола с обра-
зованием фенола, который нитруется, причем в зависимости от усло-
вий опыта образуется моно-, ди- или тринитрофенол. Кроме того,
образуется нитробензол, количество которого зависит от условий
1 По Пикте, при смешении ледяной уксусной кислоты с дымящей азотной
кислотой образуется диацетил-о-азотная кислота (НО)3 — N — (ООССН3)2, (см.
Pictet и Gene]uand, Вег., 35, 2526, 1902). При смешении азотного и уксусного
ангидридов образуется ацетилнитрат CH3COONO2, (см. Pictet и Khotinsky,
Вег., 40,-1163, 1907).
2 Ш о р ы г и н и Соколова, Ж- Р. Ф. X. О., 62, 673, 1930, К 0-
новалов и Гуревич, Ж- Р. Ф- X. О-, 37, 537, 1905.
3 Вег., 40, 371, 1907.
‘Schmidt, Вег., 37, 66, 1904.
6Ильинский, Вег., 36, 4194, 1903.
в Holderman п, Вег., 39, 1250, 1906.
’Wolffenstein и Boters, Вег. 46, 586, 1913.
19
опыта. Они нашли, что тЬе^здие реакции зависит от концентрации
азотной кислоты. Концентрированная азотная кислота или серно-
азотная смесь давали при взаимодействии с бензолом в присутствии
ртутной соли только нитробензол; при соответствующем же раз-
бавлении азотной кислоты снижается количество образующегося
нитробензола, повышается количество образующегося нитрофенола,
или получаются только нитрофйи^ы (моно-, ди-или тринитрофенол).
Следовательно, здесь про'Цркают две конкурирующие между со-
бой . реакции: одна — непосредственно нитрующая, приводящая
прямо к образованию нитробензола; другая •— в первой стадии окис-
ляющая и только затем, во второй стадии, нитрующая, приводящая
к образованию нитрофенолов.
При образовании нитрофенолов вначале идет окисление и затем
нитрование, а не наоборот. Это доказывалось тем, что при действии
азотной кислоты в присутствии ртутной соли на нитробензол не обра-
зуется следов нитрофенолов, а нитробензол остается либо неизме-
ненным, либо переходит в динитробензол. Таким путем была выяс-
нена причина противоречия с опытами Гольдермана, который при-
менял цри нитровании толуола концентрированную азотную ки-
слоту (уд. вес 1;52) или серноазотные смеси; в этих условиях
имела место только реакция нитрации в нитротолуол, но полностью
подавлялась реакция окисления с последующей нитрацией.
Те же авторы отметили, что окислительное действие, оказывае-
мое азотной кислотой в присутствии ртути, резко отличается от ра-
нее известного окислительного действия серной кислоты на аромати-
ческие соединения в присутствии ртути: в то время как серная кислота
в присутствии ртути вызывает разрыв бензольного кольца, при дей-
ствии азотной кислоты в присутствии ртути бензольное кольцо
остается нетронутым, и в него вступает только гидроксильная группа.
В зависимости от условий процесса получается моно-, ди- или
тринитрофенол или их смеси. Например, смесь 2,4-динитрофенола
и пикриновой кислоты с очень незначительным количеством нитро-
бензола была получена при нагревании (на водяной бане и при
постоянном энергичном перемешивании) 100 г бензола, 800 г азот-
ной кислоты с содержанием 49% HNO3 и 15 г азотнокислой ртути.
При этом выделяется значительное количество окислов азота.
В дальнейших исследованиях было установлено, что выход
нитрофенола зависит от температуры и концентрации азотной кис-
лоты.
Аналогично течению реакции нитрации бензола получается ни-
тротолуол и тринитро-тп-крезол.
При нитровании бензойной кислоты азотной кислотой в при-
сутствии ртути происходит окисление и нитрация и образуется
2,4,6-тринитро-тп-оксибензойная кислота
1Wolffenstein и 'Р а аг, Вег., 46, 589, 1913.
20
соон
! он
no2
Нитрованием хлорбензола получается тринитрохлорфенол Ч
Для объяснения механизма этой реакции Блехта и Патек 1 2, ос-
новываясь на работах Димрота3, принимают, что при взаимодей-
ствии углеводородов с азотнокислой ртутью вначале образуются
соединения типа G6H5HgNO3. Эти соединения разлагаются при дей-
ствии слабой азотной кислоты, причем освобождается углеводород.
Регенерированный углеводород in statu nascendi, т. е. в очень актив-
ной форме, взаимодействует со слабой азотной кислотой, причем
происходит, с одной стороны, окисление с образованием фенола,
а с другой стороны, протекает реакция нитрации с образованием ни-
трофенола. Количественное соотношение между обоими продук-
тами реакции зависит от условий, т. е. от концентрации азотной
кислоты, температуры и пр. Чем крепче азотная кислота, тем больше
образуется нитротолуола и соответственно меньше нитрофенола.
Не только ароматические углеводороды, но и их мононитропро-
изводные способны образовывать двойные соединения с азотнокислой
ртутью4. Поэтому и здесь можно было ожидать при взаимодействии
с азотной кислотой активирования бензольного ядра.
В самом деле, опыты показали, что при этом облегчается вступ-
ление 2-й нитрогруппы, однако, не имеет места окисление, т. е.
образование соответствующего нитрофенола.
Из ряда объяснений механизма нитрации азотной кислотой
в присутствии ртутной соли наибольшего внимания заслуживает
предложенное нашим советским химиком Захаровым. В основе
предложенного Захаровым 5 объяснения механизма взаимодействия
бензола с азотной кислотой в присутствии ртути лежат следующие
соображения.
Прямое введение гидроксильной группы в молекулу бензола
или нитробензола очень трудно, но оно облегчается при ослаблении
связи водородного атома с ядром, что имеет место, например, при
разрыве двойной связи. Такой разрыв двойной связи бывает при
реакциях присоединения, которые, по мнению Голлемана, предше-
ствуют всякому процессу замещения в ароматическом ряду.
1 Davis, W а г г а 1 и др., J. Amer. Chem. Soc., 43, 594, 1921.
’BlechtanPatek, Z. Schiess- u. Sprengst., 22, 314, 1927.
* Dimroth, Ber., 32, 758, 1899. Димрот показал, что при взаимодей-
'ствии ароматических соединений с солями окиси ртути водород ароматиче-
- ского соединения замещается одновалентным остатком HgOAc : NO3HgNO3-|-
+ HCeHs -» NO3Hg.GeH6+HNO3.
4 Dimroth, Ber., 35, 2032, 1902.
‘Захаров, Ж. Хим. Пр., 4, 960, 1927 ; 5, 26, 1928; 6, 698, 1929; 8,.
30, 1931.
21
Исходя из этого, Захаров принимает, что в первой фазе реакции
взаимодействия бензола с азотной кислотой в присутствии азотно-
кислой ртути образуется продукт присоединения азотнокислой ртути
к бензолу:
/ONO,
+ и/
'ONO2
Н, O-Hg-O—NO.,
/\/ Н
-> Iх ко2
\Z
Образовавшееся при этом диеновое производное, как известно,
не является стойким к действию окислителей, и, кроме того, вообще
это соединение должно быть весьма неустойчивым. Поэтому при
одновременно протекающем процессе стабилизации и взаимодействии
с азотной кислотой протекает вторая фаза реакции:
H, O-Hg—0—NO2
X / h
+ NO2OH
ОН
II ОН
+ Hg< +hno2,
\ s ONO2
ОН
+ HNO3—j Hg(NO3)2 + Н2О
ONO2
Так как способность к реакциям замещения и соответственно
скорость этих реакций у нитрофенола значительно выше, чем у бен-
зола, то дальнейшее действие катализатора в направлении нитро-
вания осуществляется легче:
Эти две фазы реакции нитрации нитрофенола в динитрофенол
протекают настолько быстро, что изолирование промежуточного
продукта невозможно
Если крепость кислоты значительна, то взаимодействие продукта
присоединения бензола и азотнокислой ртути с азотной кислотой
22
протекает иначе, а именно, с образованием нитробензола:
Н. O-Hg—ONO,
/\/ Н
|х no2 -
+ ОН—Hg-O—no2
Отсюда понятно, почему при нитровании азотной кислотой с со-
держанием ниже 60% HNO3 количество образующегося нитробен-
зола понижается, причем тем больше, чем меньше концентрация
азотной кислоты.
В приведенной выше схеме элементы азотнокислой ртути присо-
единялись по линии одной двойной связи с образованием производ-
ного циклогексадиена
н. .гю2
Х./Н
\о-Hg—ono2
при окислении и стабилизации которого образуется нитрофенол.
Захаров же допускает возможность присоединения элементов азот-
нокислой ртути к двум двойным связям бензола с образованием
производного циклогексена:
Н 0^
/уч X /NO^/Hg
zO-NO2
+
н
О
2
н no2
Это соединение неустойчиво; в процессе стабилизации и при
одновременном взаимодействии с окислителем — слабой азотной
кислотой — протекает следующая реакция:
+ HNO2 + Hg(OH)2
23
f
Бродер 1 предлагает иной механизм реакции нитрации бензола
присутствии азотнокислой ртути. Исходя из того, что среди про-
было выделено очень нестойкое по отношению
кислоте соединение — тетранитродифенилортуть 2
^укгов нитрации
г воде и азотной
NO2 no2
это соединение промежуточным продуктом и опи-
р считает именно
^гвает процесс следующим образом:
2
+ 2HNO3
3
Дифенилортуть, по Бродеру, энергично взаимодействует с азот-
лй кислотой с образованием тетранитро дифенилортути, которая
^медленно распадается с образованием динитрофенола:
zHg4 ОН
-b4HNO3->2
ONO2
+ N2O4+ H2O
2
NO2 no2 no2
Образование нитробензола протекает по реакции:
/Hgx Hg —ONO3 no
NO
нитрофенилортуть нитрозобензол
no2
>3-^| | + hno2
Однако Захаров возражает против этой схемы, указывая, что
ричиной возникновения побочной реакции, приводящей к образо-
iDesvergneS, Chim. et Ind., 22, 451, 1929.
2 О фенилортути и ее взаимодействии с окислами азота см. В а ш Ь е г-
& г, Вег., 30, 506, 1 897.
ванию нитробензола, здесь принимается образование N2O4; в таком
случае следовало бы ожидать, что под конец процесса, когда повы-
шается содержание окислов азота в растворе, реакция должна
сдвигаться в сторону образования нитробензола; однако этого не
наблюдается. И даже при искусственном увеличении количества
N2O4 в растворе не происходит повышения выхода нитробензола.
Холево и Эйтингон 1 нитровали ти-ксилол азотной кислотой с содер-
жанием 50,2% HNO3 ПРИ 75° и получили 4-нитро-ти-ксилол и 4-нит-
розо-3-метилбензойную кислоту. Они установили, что роль ртути
при нитровании m-ксилол а иная, чем при нитровании в подобных же
условиях бензола. Это отличие обусловлено способностью ксилола
легко превращаться в мононитрокарбоновые кислоты, которые
в дальнейшем не дают возможности ртути проявить свое каталити-
ческое действие. Карбоксильная группа противодействует вступле-
нию в ядро гидроксилов.
Отметим, что изложенное толкование причины отсутствия ката-
литического действия в присутствии карбоксильной группы не вы-
зывает возражений; но остается совершенно неясным, почему соль
ртути не взаимодействует с m-ксилолом. Равным образом оно не по-
ясняет, почему Е. И. Орлов 2 при кипячении в течение 6 часов
в колбе с обратным холодильником технического ксилола и тп-кси-
лола с 45—50%-ной азотной кислотой в присутствии 2% ртути по-
лучил триоксинитродикарбоновые кислоты, из которых одна имела
строение:
СООН
он^рн
o2n„ Jcooh
2 \z
он
Известно, что в отсутствии ртути это соединение не получается
при условиях опыта Е. И. Орлова.
6. Практическое использование способа нитрования в присут-
ствии солей ртути. Захаров показал, что при повторном использова-
нии отработанной кислоты увеличивается и скорость реакции и
выход динитрофенола. Он объясняет это тем, что катализатор на-
ходится при этом в более активном состоянии. Например, при нит-
ровании свежеприготовленными реакционными растворами с содер-
жанием 59—60% HNO3 при 30%-ном содержании катализатора и
температуре 35—40° выход динитрофенола в среднем не превышает
40%. При вторичном использовании отработанной кислоты с сохра-
нением без изменения остальных условий выход динитрофенола
увеличивается до 50—55%. При последовательном использовании
отработанной кислоты в третий, четвертый и т. д. раз выходы дини-
‘Холево, и Эйтингон, Ж. Пр. X., 5, 612, 1932.
2 Укр. Хим. Журн., II, 370, 1926.
25
трофенола достигали 65, 75 и 80%. Следовательно, создается воз-
можность технического применения этой реакции.
Качество продукта. В зависимости от условий реак-
ции получается либо ди-, либо тринитрофенол, либо, наконец, их
•смесь.
По данным Деверня \ можно получить чистый тринитрофенол,
однако, с низким выходом.
Для производства красок требуется чистый 2,4-динитрофенол.
Захаров показал, что при нитровании бензола в присутствии ртути
при определенных условиях (температура 20—30°) можно получить
чистый 2,4-динитрофенол, не содержащий ни 2,4 изомера, ни пикри-
новой кислоты.
При многократном использовании отработанной кислоты с до-
бавлением концентрированной азотной кислоты до первоначаль-
ного значения и при содержании в кислоте 5—10% катализатора
реакция нитрации заканчивается в 20—25 часов. Количество ката-
лизатора не влияет на качество и выход продукта, но увеличивает
скорость процесса.
По указанию Браднера 1 2, описанный способ не получил практи-
ческого применения вследствие низких выходов продукта, медлен-
ного хода реакции и образования значительных количеств нитробен-
зола. Однако результаты получаются гораздо лучше, если к реак-
ционной смеси добавить уксусную или иную жирную кислоту (про-
пионовую или масляную). Уксусную кислоту достаточно вводить
в количестве около 10% от объема реакционной массы. По советскому
патенту № 11045 для получения динитрофенола пропускают через
нагретый до 40° раствор азотнокислой ртути в азотной кислоте
насыщенный парами бензола воздух или газ, например, получаемые
при окислении аммиака нитрозные газы; последние находятся в не-
прерывной циркуляции и многократно используются.
В заключение отметим недавнее появление французского па-
тента № 821767 (1937), согласно которому катализаторами реак-
ции нитрации могут служить соединения хрома, вольфрама, молиб-
дена, тантала, ниобия, ванадия, галлия и индия. По утверждению
патента, применением одного из этих катализаторов можно легко,
с малым расходом крепких кислот, получить тетранитронафталин
{и даже неизвестные до сих пор пента- и гексанитронафталины) и
другие высоконитрованные углеводороды. Едва ли обещания этого
патента оправдаются, но они свидетельствуют о настойчивой по-
требности в катализаторе, реакции нитрации.
Мы уже отметили, что соль ртути не только приводит к окисле-
нию бензольного ядра с образованием оксисоединения, но и является
катализатором самой реакции нитрации.
Вопрос увеличения скорости реакции нитрации применением
катализатора представляет большой практический интерес; решение
1 Desvergnes, Chim. et Ind., 22, 451, 1929.
2 Am. пат. № 1723761, Ж. Хим. Пр., 838, 1930.
26
этой задачи позволило бы заметно упростить технологический про-
цесс, сократить расход*крепких кислот на нитрацию. Это, может
быть, сделало бы возможным производство ряда веществ, как, на-
пример, тринитробензола, тринитрохлорбензола, тетранитронафта-
лина и др., которые в данное время вследствие потребного для их
производства чрезмерно большого количества крепких кислот не
могут производиться в значительных количествах.
Несмотря, однако, на такое большое практическое значение этого
вопроса, не найден еще ни один пригодный для применения в про-
изводственных условиях катализатор; и даже наиболее изученный
вопрос нитрования бензола азотной кислотой в присутствии азотно-
кислой ртути еще не вышел из стадии лабораторного изучения.
Основные причины, затрудняющие использование ртутного ка-
тализатора, следующие: 1) необходимость значительного расхода
ценной, для большинства стран дефицитной, ртути; 2) трудность
регенерации ртути; 3) некоторое загрязнение пикриновой кислоты
солью ртути, что нежелательно.
7. Нитрование азотными эфирами спиртов. Бедкер 1 нашел,
что этилнитрат энергично нитрует бензол в присутствии хлористого
алюминия. Эта реакция подробнее изучена ‘Троновым и Сибгатул-
линым, установившими, что эквимолекулярная смесь бензола и
этилнитрата в присутствии 0,5 мол А1С13 при 48-часовом стоянии
при обыкновенной температуре дает выход нитробензола от теории
50%. Реакция имеет только теоретическое значение.
8. Нитрование азотнокислыми солями. Впервые Герлянд в 1854 г.
получил нитробензойную кислоту нитрованием бензойной кислоты
смесью азотнокислого калия и концентрированной серной кислоты.
После-этого различные исследователи неоднократно пользовались
азотнокислыми солями для нитрации.
Нитрование смесями аммиачной селитры с серной кислотой по-
дробнее изучалось профессором А. В. Степановым, который приме-
нял раствор высушенной аммиачной селитры в купоросном масле.
Он указывает в своих работах на следующие преимущества при
применении нитратов: 1) удобство обращения с аммиачной селитрой
по сравнению с азотной кислотой; 2) удобство дозировки; 3) отсут-
ствие выделения окислов азота, а следовательно, малая окисляемость;
4) получаемые продукты свободны от смолистых примесей. Приме-
нение этого Аетода в производстве тормозится из-за отсутствия
целесоббразного способа использования отработанной кислоты, со-
держащей в растворе аммиачную селитру, сернокислый аммоний
и нитропродукты 2.
Вопрос нитрования легко гидролизующимися азотнокислыми
солями Al, Bi, Hg, Ag имеет только теоретическое значение. Пре-
1 Boedtker, Bull. Soc. Chim. Fr., 3, 726, 1908.
2 Безуспешные попытки применения натриевой и аммиачной селитры для
нитрования делались во время империалистической войны 1914—1918 гг.
в Англии (см. Man. of TNT, стр. 11).
27
имущественное применение в лабораторной практике имела соль
алюминия A1(NO3)3 • 9Н20. Эта соль плавится без разложения при
73°; при более высокой температуре начинается ее гидролиз и при
140° — полный распад на гидрат глинозема и азотную кислоту:
Al (N О3)3- 9Н2О А1 (ОН)3 + 3HNO3 + 6Н2О
Поэтому можно вести нитрование одной солью в отсутствии
серной или другой кислоты.
Образующаяся в результате гидролиза азотнокислого алюминия
азотная кислота нитрует; например, Наметкин успешно 'нитровал
гексаметилен.
Менке 1 применял для нитрования смесь уксусного ангидрида
с нитратами Fe’" и других металлов.
Применяя смеси уксусного ангидрида с азотнокислым железом,
он получил при нитровании фенола (при 70—85°) тринитрофенол,
при нитровании хлорбензола — р- нитрохлорбензол, при нитрова-
нии толуола — о-нитротолуол.
Преимуществом этого нитрующего средства является то, что
здесь наряду с каталитическим ускорением процесса получается
почти теоретический выход одного лишь из изомерных нитросоеди-
нений, причем реакция не сопровождается окислением или осмоле-
нием.
Нитрующее действие смеси уксусной кислоты с нитратами 2
значительно слабее смесей с уксусным ангидридом. И в этом случае
получаются хорошие выходы чистых изомеров, например, получен
почти чистый о-нитрофенол.
Бахарах 3, продолжавший работу Менке, нашел, что в растворе
уксусного ангидрида и уксусной кислоты всего быстрее действует
нитрат меди, затем железо. Активным реагентом здесь является ди-
ацетил-о-азотная кислота, т. е. слабое нитрующее средство 4.
Дешевый и простой способ технического получения чистых изо-
меров имел бы заметное практическое значение для красочной про-
мышленности.
9. Нитрование раствором нитрозилсерной кислоты в серной кис-
лоте. Нитрозилсерная кислота реагирует и как нитро со единение
и как нитрозосоединение; поэтому предполагается существование
двух таутомерных форм, находящихся друг с другом в состоянии
подвижного равновесия:
N02-SO„-OH NO—О—SO2OH
При взаимодействии нитрозилсерной кислоты в растворе серной
кислоты с диметиланилином образуется как нитрозо-, так и нитро-
диметил анилин (см. следующую таблицу на стр. 29).
'Menke, Rec. Trav. Chim. Pays-Bas, 44, 141—149, 1925.
2 Menke, Rec. Trav. Chim. Pays-Bas, 44, 2 6 9, 2 70, 1927.
3 Bacharach, J. Amer. Chem. Soc., 49, 1522, 1927.
4 Ber., 64, 2136, 1931.
28
Наименование вещества Образуется от теории, %
при 10—15° при 28—30°
р-Нитродиметиланилин 8,3 42,8
р-Нитрозодиметиланилин .... 71,5 39,3
При повышении температуры понижается количество образую-
щегося нитрозосоединения, что указывает на соответственно проис-
ходящее изменение состояния подвижного равновесия Ч
Как видно, нитро зил серная кислота является очень слабым ни-
трующим агентом, ибо она слабо реагирует даже с таким легко
нитрующимся соединением, как диметиланилин, образуя при этом
только моно нитросоединения.
10. уитрование окислами азота. Этот вопрос приобрел актуаль-
ное значение со времени установки производства синтетической
азотной кислоты. При этом производстве мы*получаем промежуточ-
ный продукт N2O4, утилизация которого для целей нитрования
представляла бы преимущество вследствие исключения конечной
фазы технологического процесса азотного завода (превращения окис-
лов азота в азотную кислоту), которая довольно сложна и связана
co значительными материальными за-
тратами.
Двуокись азота NO2 является не-
насыщенным соединением, вследствие
чего она имеет свойство реагировать
с присоединением. Это свойство выра-
жается уже в ее способности образовы-
вать димерное соединение N2O4, где
ассоциированы 2 мол NO2.
По данным Натансона 1 2, в двуокиси
•азота содержится следующее количество
NO2 при разных температурах (табл.1).
Ряд исследователей (особенно же Виланд) показал, что 2 мол
NO2 присоединяются подобно хлору к ненасыщенным алифати-
ческим углеводородам с образованием соответствующих динитро-
соединений:
Таблица 1
Температура, °C no2 %
26,7 20,0
49,6 40,0
70,0 65,6
80,6 76,6
111,3 92,7
135,0 98,7
С = С
— С —С —
no2 no2
no2 no2
Виланд3 нагревал бензол с двуокисью азота в запаянной трубке
при 80°, при этом образовалось незначительное количество нитро-
1 Biehringer и В orsum, Вег. 49, 1402, 1916.
2 Natans о n. Wied. Ann., 24, 454, 1885.
2 Wieland, Вег., 54, 1776, 1921.
29
бензола, а в качестве главного продукта образовался тринитробен-
зол, некоторое количество пикриновой кислоты и продукты разру-
шения ароматического ядра: СО, щавелевая кислота и др. Кроме
того, было установлено, что нитробензол не реагирует с двуокисью
азота.
Поэтому Виланд принимал, что к бензолу сразу присоединяется
6 мол NO2 с образованием нестойкого промежуточного продукта—
гексанитроциклогексана; последний сейчас же распадается на три-
нитробензол и 3HNO2:
+6NO
Твердый нафталин легко реагирует с NO2 и дает а-нитронафта-
лин. Фенолы реагируют очень быстро даже при охлаждении (в рас-
творе смеси бензола с бензином).
Шааршмидт применил в качестве катализаторов А1С13 и FeCl3;
например, он пропускал NO2 при обыкновенной температуре в хо-
рошо перемешиваемую смесь ароматического углеводорода с А1С13,
причем А1С13 постепенно растворялся с образованием устойчивых
комплексов типа 2А1С13 • 3(СвН6 • N2O4).
С хлорбензолом реакция идет таким образом:
2А1С13 + ЗСвН5С1 2А1С13 • ЗСвН5С1 >-
-±^> 2А1С13 • 3(СвН5С1 • N2OJ +н^ 3C6H4C1NO2 + 3HNO2
Вода разлагает комплекс, причем образуется остаток
CeH5Cl-N2O4, который тотчас же разлагается на CeH4ClNO2+HNO2
Нитрация хлорбензола приводит постоянно к ^-изомеру.
По мнению Шааршмидтд, нитрование двуокисью азота в‘ при-
сутствии А1С13 (или FeCl3) является характерным примером реак-
ций типа Фриделя и Крафтса. А1С13 или FeCl3, присоединяясь к мо-
лекуле бензола, сообщают непредельный характер двойным связям
его ядра и тем самым активируют его.
Двуокись азота присоединяется в виде двойного ангидрида азот-
ной и азотистой кислот NO • ONO2 к молекуле бензола, активиро-
ванной присоединением А1С13 или F*eCl3, образуя нитронитрит р- и
о-дигидробензола:
30
нч no2 н zno2
/\ н\><
| | и 0N0Z I
h/^ONO
Эти промежуточные соединения отщепляют молекулу азотистой
кислоты с образованием моно нитросоединения.
Титов 1 провел исследование реакции нитрапии ароматических
углеводородов двуокисью азота в присутствии А1С13, причем пришелг
к иным выводам о механизме этой реакции, чем Шааршмидт.
Титов установил следующее:
1) переход хлористого алюминия в раствор происходит при
действии двуокиси азота в количестве, меньшем в три раза, чем
это требуется для образования комплекса .Шааршмидта;
2) при дальнейшем прибавлении двуокиси азота реакция про-
текает значительно менее энергично;
3) нитросоединение может быть выделено из продукта реакции
(промежуточного продукта) без добавки воды — простым нагрева-
нием.
На основе этих наблюдений Титов считает, что реакция проте-
кает в несколько фаз, которые определяются фазами координацион-
ного насыщения хлористого алюминия.
Первая фаза реакции, протекающая наиболее энергично и соот-
ветствующая переходу А1С13 в раствор, выражается предположи-
тельно-следующим образом:
ЯН +N2O4 + 2А1С13-> ЯМ)2 -А1С13 + А1С12(ОН) NOGI^±
Z?NO2 - А1С1,(ОН) + А1С13 • NOG1
Следующие фазы реакции, связанные с менее энергично проте-
кающими фазами координационного насыщения А1С13, схемати-
чески выражаются следующим образом:
ЯГЮ2 • А1С13 4- N2O4 4- ЯН А1С12 • (ОН) (2ЯХО2) (NOC1)
A1C13(OH)NOC1 4- N2O4 4- ЯН А1С1(ОН)2 • #NO2(2NOCI)
При нагреваний эти комплексы разлагаются с выделением нитро-
соединений и хлористого нитрозила, что было подтверждено опытом.
И. Нитрование двуокисью азота в присутствии серной кислоты.
Первое исследование этого процесса произведено Пинком2. Он
прибавлял углеводород к раствору N2O4 в серной кислоте. При
этих условиях в реакцию вступала лишь половина всего коли-
чества N2O4:
CeHe + N2O4 + H2SO4—»C6H5NO2 + SO2(OH) (ONO3) + H2O
1 ж. о. X-, 7, 591, 1937.
2 J. Amer. Chem. Soc., 49, 2536, 1927.
31
При ^г01й методе нитрования наблюдались взрывы реакционных
<5месе^тге 1 производил нитрование путем прибавления двуокиси
b fC смеси нитруемого вещества и серной кислоты, причем уста-
аз°та чт0 нитрование бензола протекает нацело при 15—20° при
ловиЛ^И, что концентрация отработанной кислоты не ниже 60,7%.
Услов мер, он приводит такой состав отработанной кислоты: 60,7%
J}a®₽* 17,9% NHSO5 и 20,8% Н2О.
2 л 4рак0> если отнести процент H2SO4 только к сумме H2SO4 +
то найдем, что отработанная кислота содержит 74,5% H2SO4,
' о# и определяется нитрующая сила кислоты.
котор ге наблюдал во время нитрации при добавлении к отработан-
ь ^слотам избытка углеводорода (бензола, толуола, ксилола)
®?м рание цветных соединений. Последние имеют характер моле-
°оРа ^ых комплексных соединений и сильно окрашены.
кУляр0зол, по Баттге, образует соединения состава СвН6 • 2NHSO5;
^пазование происходит только в присутствии серной кислоты.
- ° Jj-jjafl, кроме того, во внимание структурную аналогию между
1 Рн^^цлсерной кислотой ONOSO2OH и тетраокисью азота ONONO2
® ТР одилнитрат), а также учитывая состав промежуточных соеди-
1НИТР полученных Шааршмидтом при реакции NO2 с бензольными
нени о’дородами в присутствии хлористого алюминия, Баттге выра-
^глев акцию взаимодействия бензола с окислами азота в присутстви
<ерно$ кислоты в следующем виде:
е₽Н‘ бвНв + 2NO2 + H2SO4 -> ONOCeHeNO2i5^->CeH6NO2 +
4- nhso5 + H2O
„ приписывает серной кислоте роль активатора, способствую-
u присоединению нитрозилнитратной формы двуокиси азота
ono№2 к двойной связи-
U1 ТтСГ0® и Барышникова 2 указывают, что можно вполне удовлетво-
рило предвидеть влияние различных факторов в процессе нитро-
Рите ароматических соединений двуокисью азота в присутствии
В кислоты и наглядно описать явления, допустив, что нитрова-
серно оиЗВОДИТСЯ исключительно азотной кислотой, образующейся
НИе взаимодействии:
N2O4 4- H2SO4 HNO3 + NHSO5
и рас0>1атРивая /этот nP°4eCc в связи с равновесными реакциями:
2NHSO5 + Н2О N2O3 + 2H2SO4
2HNO3 + N2O3^± 2N2O4 + H2O
H2SO4 • nH2O + H2O H2SO4 • (n + 1)H2O
, cattegay, Bull. Soc. Chim., Fr-, 43, 109, 1928.
3 О. X., VI, 1801, 1936.
32
Схему реакции нитрации с этой точки зрения следует изобразить
в следующем виде:
7?H+N2O4+tiH2SO4 • /пН2О—»7?NO24-NHSO5+(ra—l)H2SO4(zn+l)H2O
Для достижения использования N2O4, близкого к теории, необ-
ходимо наличие под конец реакции отношения между свободной
серной кислотой и водой не ниже некоторого минимального значе-
ния, характерного для каждого соединения. При увеличении кон-,
центрации N2O4 или удалении из сферы реакции N203 степень полез-
ного использования серной кислоты может быть повышена, как это
видно из приведенных уравнений.
По данным Титова и Барышниковой, в случае реакции с дву-
окисью азота требуется строгое соблюдение выработанных условий
проведения процесса. Отступление от этих условий легко приводит
к резкому усилению побочных реакций нитруемого соединения со
всеми составляющими системами (N2O4, N203, HNO3, NHSO5 и,мате-
риала аппаратов).
При опытах Титова и Барышниковой имело мрсто взрывоподоб-
ное разложение реакционной массы после окончания основной реак-
ции. Причиной взрыва послужило попадание ртути из ртутного
затвора мешалки. Специальные опыты показали, что прибавка
окиси ртути вызывает довольно энергичную реакцию между бензо-
лом и нитрозилсерной кислотой.
Наличие хлора в молекуле хлорбензола в сильнейшей степени
увеличивает стойкость бензольного ядра и предохраняет от вступ-
ления хлорбензола в побочные реакции даже в очень жестких усло-
виях — при повышенной температуре и большой концентрации
N2O4, H2SO4. Прибавка ртути также не производит заметного эф-
фекта.
12. Нитрование двуокисью азота в газовой фазе. Шорыгин и
Топчиев 1 нитровали толуол двуокисью азота в газовой фазе. При
этом исходили из воззрений П. П. Шорыгина 2, согласно которым
толуол может существовать в двух таутомерных формах (равновесие
сильно сдвинуто влево):
Вторая, более богатая энергией форма образуется, вероятно,
в сравнительно незначительном количестве при притоке энергии
извне — нагревании или облучении ультрафиолетовыми лучами.
1П. П. ШорыгиниА. В. Топчиев, Нитрование углеводородов
двуокисью азота в газовой фазе, Ж. О. X., V, 549, 1935.
767 ^Эз'б^' Ш ° Р Ы Г И Н’ ° таУ'гомеРных формах толуола, Ж- Р- Ф- X. О., 58,
Химия—13—3
33
В, этих условиях реагирующие молекулы (группы NO2) могут, сле-
довательно, присоединяться не только к двойным связям ядра, но
и к двойной связи (1 = 7):
н +2NO2->
XN02
уН
+ HNO2
V/\H
В результате же присоединения NO2 к двойным связям ядра
должны образоваться одновременно мононитротолуолы.
При температуре 14—15° и облучении ультрафиолетовыми лу-
чами в реакцию вступает около 55% взятого толуола, причем полу-
чается фенил нитрометан и мо но нитро толуол (в отношении 5 : 27—28).
Образования динитротолуола и бензойной кислоты не наблюдалось.
При повышении температуры до 58—60° выход фенилнитрометана
остается Примерно тем же, мононитротолуолы же образуются в зна-
чительно меньшем количестве.
Без облучения в реакцию вступает меньшее количество толуола,
но относительные количества фенилнитрометана и мононитротолуо-
лов при этом не изменяются существенным образом.
При опытах с бензолом ультрафиолетовые лучи не оказали ни-
какого влияния на ход реакции: выход нитробензола при облучении
и б.ез облучения был одинаков.
При взаимодействии двуокиси азота с тиофеном реакция идет
и без облучения чрезвычайно энергично с осмолением, причем обра-
зуется мононитротиофен (температура плавления 44°) и динитро-
тиофен (температура плавления 51—52°).
13. Окисление аминогруппы в нитрогруппу. В лабораторной
практике при синтезе некоторых соединений окисляют аминогруппу
ароматического основания в соответствующее нитрозо со единение
кисцотой Каро. При дальнейшей обработке нитрозосоединений кон-
центрированной азотной кислотой они окисляются в нитрогруппы.
Таким путем, например, Брэди и Вильямс 1 получили из 4,5-
и 3,4-динитро-о-толуидинов 2,4,5- и 2,3, 4-тринитротолуолы.
Прюдом 2 окислил анилин в нитробензол кипячением его с хлор-
ной известью, а Бамбергер 3 достиг того же окислением анилина
марганцевокалиевой солью..
Витт и Конечный 4 для получения р-динитробензола окисляли
р-нитроанилин персульфатом аммония при 45° в присутствии азот-
нокислого серебра как катализатора. Выше 45° персульфат аммо-
ния начинает самопроизвольно разлагаться со значительным разо-
*Br adv и W i Hi ams, J. Chem. Soc., 117, 1137, 1920.
2 Prudnomme, Ber., 25, 947.
3 Bamberger и Meimberg, Ber., 26, 469, 1893.
4 W i t t и Kopetschni, Ber., 45, 1134, 1912.
34
гревом, обусловливающим обугливание р-нитроанилина; поэтому
при температуре выше 45° выход динитробензола заметно умень-
шается.
14. Действие окис лов азота на аминосоединения. При действии
окислов азота на растворы ароматических аминов в разбавлен-
ной соляной кислоте 1 при 0° происходит диазотирование и нитро-
вание.
Например, анилин дает при такой обработке:
NH3 ОН
A Ano2
I , I I
I ! ! |
NO, ’
15. Замена аминогруппы на нитрогруппу. Такая замена произ-
водится, по Зандмееру * 2 3, смешением нейтрального раствора диазо-
ний-нитрата или диазоний-сульфата (полученного диазотированием
соответствующего амино соединения) с эквивалентным количеством
нитрата натрия и с последующим смешением со взмученной порош-
кообразной закисью меди.
При этой реакции вначале образуется диазоний-нитрит:
R - N = N • ONO2 + NO2Na R N = N • ONO + NO3Na
который после выделения азота и перегруппировки остатка азотистой
кислоты ONO в нитрогруппу NO2 превращается в нитросоедине-
ние:
AN = N • ONO -> 7?NO2 % N2
Этот способ находит себе практическое применение, например,
для приготовления не получаемого прямым нитрованием р-нитронаф-
талина, или, например, для лабораторного получения чистого р-ди-
нитробензола из легко получаемого р-нитроанилина.
. 16. Электрохимическое нитрование. Способ электрохимического
нитрования впервые предложил Триллер ®. Он указал, что, приме-
няя азотную кислоту с содержанием 52% HNO3, предварительно
подогретую до 80°, можно получить с хорошим выходом чистый
а-нитронафталин, не содержащий динитронафталина (это важно
в случае применения мононитронафталина для изготовления красок);
применяя же 65%-ную азотную кислоту, можно получить динитро-
нафталин.
Фихтер и Плюсе 4 проверили патент Триллера, причем прово-
дили параллельные опыты нитрования при одинаковых условиях
'Varma и Krishnamurti, Zbl, 1927, I, 1433; R i n k е s, Rec.
Trav. Chim. Pays-Bas, 46, 506, 1927.
2Sandmeyer, Ber., 20, 1494, 1887. Улучшенный метод см.
Hantzsch и Blagden, Ber., 33, 2544, 1900.
3 Герм. пат. № 100417, 1897.
4Fi cht er и Pliiss, Helv. Chim. Acta, 15, 236, 1931.
35
«
концентрации и температуры электрохимическим нитрованием и
обыкновенным способом. Они пришли к выводу, что увеличение
выхода а-нитронафталина при электрохимическом нитровании обус-
ловливается не повышением концентрации HNO3 в анодном про-
странстве, а исключительно ростом температуры реакционной смеси,
вызываемым прохождением электрического тока через эту смесь.
Кэлген и Вильсон 1 тоже изучали электрохимическое нитрова-
ние нафталина. Они констатировали, что концентрация HNO3
в анодном пространстве во время электролиза повышаемся, причем
это повышение тем сильнее, чем больше сила тока. По их мнению,
на аноде создается пленка высококонцентрированной азотной кис-
лоты, которая обусловливает повышенный нитрующий эффект.
Кэрк и Брэдт 2 проводили опыты электрохимического нитрова-
ния толуола серноазотной смесью и нашли, что в наиболее благо-
приятных условиях при электрохимическом нитровании можно по-
лучить повышение выхода на 11% по сравнению с обычным методом
нитрования.
Таким образом все имеющиеся данные по этому вопросу показы-
вают, что при электрохимическом нитровании не имеет места активи-
рование бензольной молекулы током, как это иногда предполагали,
и не получается существенных выгод по сравнению с обычным мето-
дом нитрования.
* G alha п е и W i Is о n, Trans. Amer. Elektrochem. Soc., 63, 1933.
• Kirk и Bradt, Trans. Amer. Elektrochem. Soc., 67, 1935.
ГЛАВА II
НИТРОСОЕДИНЕНИЯ АЛИФАТИЧЕСКОГО РЯДА
СВОЙСТВА НИТРОСОЕДИНЕНИЙ АЛИФАТИЧЕСКОГО РЯДА
Мононитросоединения предельного ряда (нитрометан, нитроэтан
и т. п.) представляют собой бесцветные жидкости, плохо раствори-
мые в воде.
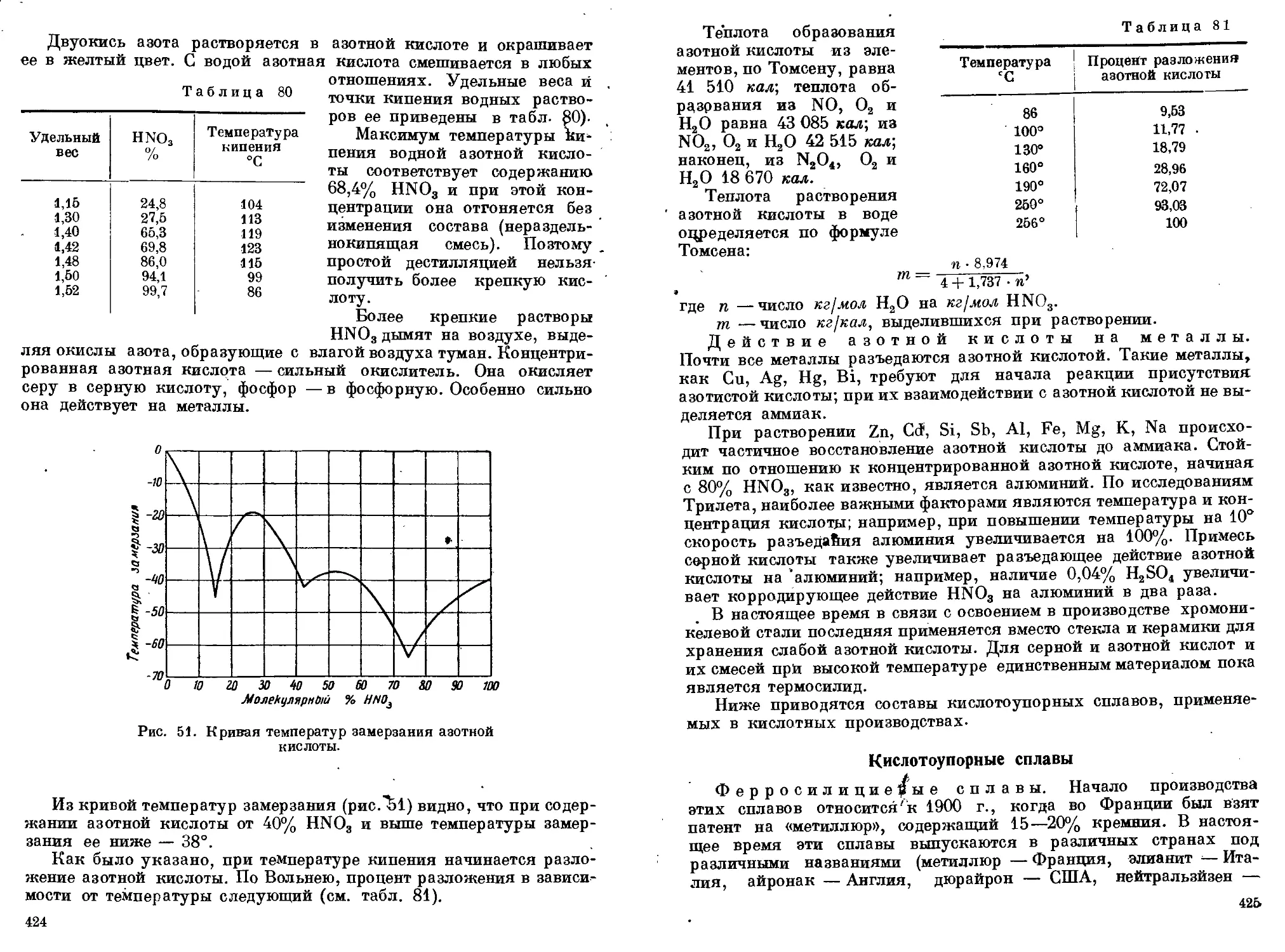
1. Температура кипения и плотность нитропарафинов видны из
табл. 2.
Таблица 2
Наименование вещества Температура кипения °C Плотность
Нитрометан 101 1,382(20720°)
Нитроэтан 116 1,0461(25725°)
1-Нитропропан 131 1,0023(25°)
2-Нитропропан 118 1,024(0°)
1-Нитробутан 151 —
139 0,9877
2-Метил-1-нитропропан 140.5 0,9625(25°/25°)
2-Метил-2-нитропропан 126,4* 0,9501 (28°/4°)
Нитропарафины имеют слабый, неприятный запах, напоминаю-
щий запах хлороформа. Перегоняются без разложения. Однако
для получения их в более устойчивой форме следует применять
перегонку их под разрежением.
2. Химические свойства нитропарафинов. Нитропарафины яв-
ляются весьма реакционноспособными соединениями.
При восстановлении в кислой среде получаются амины. Это вос-
становление может быть успешно проведено при помощи самых разно-
образных восстановителей (сернистого аммония, олова и соляной
кислоты, водорода и платины и т. п.):
CnH2n+1NO2 + ЗН2 ->CnH2n+1NH2 + 2Щ0
* Температура плавленид 25,6°.
37
Этот способ (предложен Зининым) не приобрел в химии жирного
ряда того большого практического значения, которое он имеет в аро-
матическом ряду, потому что нитро со единения жирного ряда менее
доступны, чем соответствующие им соединения ароматического
ряда.
При восстановлении в щелочной среде из первичных нитросоеди-
нений получаются альдегиды, а из вторичных — кетоны.
Конденсация альдегидов с первичными и вторичными нитропа-
рафинами дает в зависимости от условий либо нитроалкоголи, либо
нитроолефины:
I
снон
KCH2NO2 + 2К1СНО R-С—NO.,
КНСО3 ।
снон
I
R
ЯСНМ)2 + Н.сНОв присутствии д_ C-NO,4-H„O
ZnCl2 у 2 2
CH-Rx
Восстановление нитроспиртов в кислой среде ведет к получению
аминоспиртов.
При гидролизе в присутствии концентрированной соляной кис-
лоты 1 первичные нитропарафины образуют жирные кислоты и соля-
нокислый гидроксиламин:
/?CH2NO2 + НС1 + Н2О ЯСООН + NH,OH • НС1
В нитросоединениях, у которых при углеродном атоме, связан-
ном с нитрогруппой, находится один или несколько водородных
атомов, при действии щелочей один из этих водородных атомов
способен замещаться металлами, причем этот водородный атом
свою связь с углеродом меняет на связь с кислородом:
CH3CH2n/°+ NaOH -> СН3СН = n/°
X) XONa
Таким образом из настоящих нейтральных нитросоединений об-
разуются изонитросоедипения (ациформа). При выделении из со-
лей минеральными кислотами они более или менее быстро превра-
щаются снова в настоящие нейтральные соединения (псевдокис-
лоты).
1 О гидролизе в присутствии 85%-ной серной кислоты см. Lippincott
и Hass, Ind. Eng. Ghem., 31, 118—120, 1939.
38
Известны такие характерные реакции для отличия первичных,
вторичных и третичных нитросоединений: к щелочному раствору
нитро со единения добавляется сначала NaNO2, а затем серная кис-
лота. При этом происходит следующее:
а) первичные нитросоединения образуют нитро-
ловые кислоты:
СН3СН? CH3C = NOH
| .+'1 О NOH > | + Н2О
no. no.
метилнитроловая »
кислота
Нитроловые кислоты хорошо кристаллизуются, но непрочны;
растворяются в щелочах с кроваво-красным окрашиванием, образуя
при этом металлическое производное;
б) вторичные нитросоединения образуют псевдо-
нитролы:
Д2-С —Н 4-HONO ^-C —NO
I I
no2 no2
Псевдонитролы — бесцветные, твердые соединения; при растворе-
нии или плавлении они принимают интенсивный синий цвет;
в) третичные нитросоединения при этом не
взаимодействуют.
3. Продукты взаимодействия углеводородов предельного харак-
тера с азотной кислотой при нитровании в жидкой фазе1. Процесс
образования третичных нитро соединений отличается от процесса
образования вторичных и первичных нитро со единений. Это раз-
личие сказывается на механизме образования воды в том и другом
случае. В первом случае она образуется за счет третичного водорода
и гидроксильной группы азотной кислоты, во втором случае от азот-
ной кислоты берется только кислород, оба же водорода — от угле-
водорода.
Эти соотношения можно выразить так:
I I ,
/?2-С- Н+OH NO2^ в2 — c-no2+ н2о
7?3 к
третичное
иитросоедпнение
I ........Г
Я2 —С-^Н+ )N-OH->7?2—G = NOOH4-H2O
"j.. изонитросоединение
H
1 Наметкин, Ж. Р. Ф. X- О., 40, 184, 1908; €2, 581, 691, 1910.
39
I , ' >
Таким образом в первой фазе взаимодействия углеводорода пре-
дельного характера с азотной кислотой образуется изонитросоеди-
нение; это изонитросоединение тотчас же превращается либо в ста-
бильную форму, либо окисляется через альдегид или кетон (смотря
по характеру нитросоединения) в соответствующие карбоновые
кислоты. Образование альдегида или кетона протекает по реакции
Нефа:
2ЯСН = NOOH 2ЯСОН + N20 + Н30
27^0 = NOOH 2Rt — С = О + N,0 + Н.,0 .
I I
я2 я2
Сказанное о взаимодействии углеводорода с азотной кислотой можно
представить в виде следующей схемы:
углеводород+азотная кислота
I
изонитросоединение
I \
нитросоединение альдегид (кетон)
I
карбоновая кислота
Наметкин сделал из своих экспериментальных данных вывод
что «...образование нитропродукта при действии азотной кислоты
на углеводороды предельного характера (а может быть и на другие
соединения) есть лишь первая фаза реакции, а не особое ее направ-
ление».
Заметим, что изложенная схема действия азотной кислоты на
углеводороды предельного характера может служить иллюстрацией
закона постепенных стадий реакций, в силу которого 1 при всех
процессах не сразу достигается наиболее устойчивое состояние,
но сначала наступает ближайшее или из числа возможных состоя-
ний наименее устойчивое. Очевидно образование изонитро со едине-
ний в нашем случае есть стадия не только ближайшая, но и наиме-
нее устойчивая в данных условиях.
4. Токсические свойства. Вследствие присутствия нитрогруппы
можно ожидать наличия токсических свойств у нитропарафинов.
Однако опыты показали, что эта токсичность незначительна.
Нитрометан менее ядовит, чем 1-бутанол 2. Предварительные
опыты с образцами нитропропана показали, что он менее ядовит,
чем пары бензола, аммиака, сероуглерода, четыреххлористого угле-
рода, нитробензола. Поэтому американские исследователи нахо-
дят возможным применять эти нитросоединения в умеренных коли-
чествах в лаках3.
1 Оствальд, Основы неорганической химии, 224, 1914.
2 1-бутанол, или нормальный первичный бутиловый спирт, применяется
в качестве растворителя в лакокрасочной промышленности.
’Hass, Hodge и Vanderbilt, Ind. Eng. Chem., 28, 339, 1936.
40
5. Свойства отдельных представителей. Нитрометан —
жидкость, со своеобразным запахом; кипит при 102°, уд. вес 1,13-
Его пары вызывают головные боли, аналогично нитроглицерину,
но более слабые и менее продолжительные.
2-метил-2-нитропропан обладает исключительным
свойством понижать температуру затвердевания при растворении
1 моля в 1 кг растворителя на 27 °(соответствующая величина для
воды равна 1,86°).
Тетранитрометан представляет собой бесцветное масло;
затвердевает при 3°. Пары его вызывают резь в глазах, кашель,
и действуют удушающе. При 126° под обыкновенным давлением
оц кипит без разложения. Уд. вес D13 = 1,65. Сильно летуч при
обыкновенной температуре. Не горюч, практически невзрывчатый,,
но в смесях с углеводородами дает сильные взрывчатые вещества
типа веществ Шпренгеля. Чувствительность его смесей к удару зна-
чительно выше, чем у нитроглицерина. % -
Четвертая нитрогруппа в нем очень подвижна, вследствие чего'
тетранитрометан может быть применен для нитрования. Например,
при взаимодействии с р-крезолом в пиридиновом растворе он дает
нитрокрезол:
СН3
/\
।
Vno>
он
с образованием нитроформа. Формула его строения, по Шмидту
(NO2)3C — О — NO (нитрит тринитрометилового спирта). Эта фор-
мула подтвердилась при исследовании кристаллов тетранитрометана
рентгеновыми лучами 2.
Практическому применению тетранитрометана мешает его лету-
честь и высокая стоимость.
’Гексанитроэтан — белые кристаллы с температурой
плавления 142° (плавится без разложения), мало летуч. Его хими-
ческая стойкость мала: даже свежеприготовленный и обработанный,
суспендированным в воде мелом для удаления кислот он показы-
вает на лакмус кислую реакцию. С дифениламином и централитом
реагирует очень энергично. Мало чувствителен к удару и трению.
При падении груза в 2 кг с высоты в 1 м на гексанитроэтан послед-
ний взрывает. При испытании в бомбе Трауцля дает расширение
180 см3 от капсюля-детонатора № 8.
Может быть применен подобно тетранитрометану в качестве
нитрующего агента.
Гексанитроэтан не имеет практического применения прежде
всего вследствие высокой стоимости.
1 Smidt, Вег., 52, 400, 1919.
’Mark, Вег., 59, 2 989, 1926.
41
ПРИМЕНЕНИЕ НИТРОСОЕДИНЕНИЙ АЛИФАТИЧЕСКОГО РЯДА
Трудность получения нитросоединений жирного ряда до сих
пор была основным препятствием, мешавшим широкому примене-
нию этих веществ.
Больше всего привлекал внимание тетранитрометан,
который в смеси с жидкими углеводородами образует взрывчатые
вещества большой силы, могущие представить практический интерес.
Гексанитроэтан — твердое взрывчатое вещество — мало
изучен вследствие высокой стоимости.
Нитрометан представляет известный практический интерес
вследствие способности конденсироваться с альдегидами с образова-
нием спиртов, пригодных для получения сильных взрывчатых веществ,
например, нитроизобутилглицеринтринитрата. Но и нитрометан еще
мало доступен вследствие высокой стоимости его производства.
В последнее время в ряде американских работ намечается новый
способ получения нитропарафинов — путем нитрации газообраз-
ных парафинов при повышенной температуре. Эти работы имеют
целью получение дешевых нитропарафинов для применения их в са-
мых разнообразных областях:.
1) в качестве растворителей для нитро- и ацетилцеллюлозы
в смеси с другими растворителями, например со спиртом; для этой
щели, повидимому, пригодны нитропарафины, имеющие от одного
до четырех углеродных атомов; при большем числе углеродных
атомов нитропарафины недостаточно летучи;
2) в качестве добавки к топливу для дизелей с целью пониже-
ния температуры его воспламенения; это важно для таких сортов
топлива, которые не могут применяться для дизелей вследствие
слишком высокой температуры воспламенения;
3) для органических синтезов.
Как видно из сказанного о нитропарафинах, они могут служить
материалом для получения ряда практически важных химических
•соединений, а равно могут применяться для важных технических
щелей.
ПОЛУЧЕНИЕ НИТРОСОЕДИНЕНИЙ АЛИФАТИЧЕСКОГО РЯДА.
Реакция В. Мейера. При действии азотистокислого
серебра на иодистый алкил получаются нитро соединения (наряду
с образованием некоторого количества азотистокислых эфиров —
сложных эфиров азотистой кислоты):
CnH2n-f-iJ 4* AgNO2 —> Сп Hgn+fNOg 4* AgJ
параллельно идет процесс:
СцНгп > 1J + AgONO —» CnH2n+1ONO + AgJ
1 Описывается видоизмененный способ Кольбе (см. Steinkopf и
Kirchhoff, Вег., 42, 3438, 1909).
42
Получение нитрометана по Кольбе. Взаимо-
действием едкого натра 1 с монохлоруксусной кислотой получают ее
натриевую соль:
(I) СН2С1СООН + NaOH ->CH2ClCOONa + Н2О
К полученной соли добавляют теоретическое количество нитрита
натрия:
(II) CH2ClCOONa + 2NaNO2 -> CH2NO2COONa -f- NaCl
При нагревании образуется нитрометан:
(III) CH2NO2COONa + Н2О -> NO2CH3 + NaHCO3
Выход нитрометана составляет 50% от теории (по хлоруксус-
ной кислоте). Было высказано предположение, что причиной пло-
хого выхода является разложение бикарбонатом образовавшегося
нитрометана. Поэтому заменяют едкий натр гашеной известью или
свежеосажденным углекислым кальцием, для того чтобы при реак-
ции получить вместо растворимого в воде бикарбоната* натрия не-
растворимый углекислый кальций. Однако оказалось, что выход
от этого не повысился; но все же этот способ выгоднее для произ-
водства (мел дешевле едкого натра).
Некоторые улучшения в этот процесс внесены в 1930 г. китай-
скими химиками Вангом и Чао-Лун-Тзенгом х. Эти исследователи на-
блюдали, что даже при тщательной нейтрализации натриевой соли
перед добавкой нитрита остающаяся после нагревания и удале-
ния нитрометана жидкость все же показывает щелочную реакцию
по лакмусу. Поэтому можно думать, что эта возникающая во время
взаимодействия щелочная реакция и является причиной низких вы-
ходов. Нейтрализация же образующейся щелочи возможна добавле-
нием к реакционной смеси слабой кислоты. И действительно, оказа-
лось, что при добавлении борной кислоты выход повышается до 58%.
По мнению этих авторов, получившийся при реакции (III) бикарбо-
нат взаимодействует с солью хлоруксусной кислоты с образованием
соли оксиуксусной кислоты:
CH2ClCOONa + NaHC03-> HOCH2COONa + NaCl + CO2
Эта реакция частично подавляется добавлением борной кис-
лоты; последняя при взаимодействии с NaHCO3 превращается в бор-
нонатриевую соль и углекислоту.
Получение тетранитрометана. Тетранитрометан
был получен Шишковым в 1857 г. Взаимодействием уксусного ан-
гидрида с крепкой азотной кислотой 2 образуется тетранитрометан:
4(СН3СО)2О + 4HNO3 —- C(NO2)4 +СО2 + 7СН3СООН
Реакция протекает в течение нескольких суток при обыкновен-
ной температуре. При повышенной температуре реакция принимает
’Wang и Chao - Lu n - Т s е ng, Zbl 1, 1938, 55.
2 Герм. пат. № 224057, 1908; Berger, С. R., 151, 813,1910; С h a t e way,
J. Amer. Chem. Soc., 97, 2099 1910.
43
бурный характер. Повидимому, вначале образуется мононитросоеди-
нение, а затем постепенно замещаются остальные атомы метильной
группы. После этого происходит гидролиз, и образовавшаяся три-
нитроуксусная кислота медленно разлагается на СО2 и нитроформ,
который превращается азотной кислотой в тетранитрометан.
Выход — до 80% от теории.
По другому способу тетранитрометан получается нагреванием
нитробензола со смесью равных количеств азотной кислоты и
37%-ного олеума \ причем смесь берется из такого расчета, чтобы
количество азотной кислоты превышало в девять раз количество
нитробензола. Часть нитробензола нитруется при этом в тринитро-
бензол.
Выход тетранитрометана составляет 50% по весу нитробензола.
Этот способ деструктивной нитрации был не-
давно проверен 1 2 с целью определения выходов как из нитробен-
зола, так и из других ароматических нитросоединений. При этом
было найдено, что симметричный тринитробензол и симметричный
тринитротолуол очень стойки к действию на них серноазотной смеси
с высоким содержанием SO3. Только тротил-сырец дает следы тетра-
нитрометана (очевидно за счет окисления и нитрации примесей).
Равным образом при испытании моно- и динитротолуолов не образо-
вался тетранитрометан (они превращаются в тринитротолуол), при-
чем нитрование мононитротолуола сопровождается окислением, за-
висящим от температуры и концентрации SO3 в смеси.
Наибольшие выходы тетранитрометана получены при примене-
нии динитробензола, мононитробензола и нитронафталина; наибо-
лее экономичен динитробензол.
Во всех случаях выход низок по отношению к азотной кислоте.
Около 60% взятой в реакцию азотной кислоты находится в остаю-
щейся после реакции крепкой смеси, около 20% собирается в виде
слабой азотной кислоты и 20% расходуется на реакцию и теряется
безвозвратно.
Получение гексанитроэтана3. К раствору бром-
пикрина в метиловом спирте добавляют водный раствор цианистого
калия и нитрита калия. После получасового перемешивания при
температуре не выше 30° отфильтровывают осадок — тетранитро-
этанкалий. Обработкой последнего серноазотной смесью получают
гексанитроэтан:
(I) [CeH,(NO2)3O]2Ca % 10Вг2 6CNO2Br3 + 2Н2О + СаВг2
пикрат кальция ч
(П) 2CNO2Br3 % KCN _> КВг % BrCN + GNO2 - Br2
бромпикрин |
CNO2 — Br2
1 Герм. пат. № 184220.
2 Me К i e, J. Amer. Chem. Soc., 44, 430, 1923.
3 Ber., 31, 647, 1898; 35, 4289, 1902; 47, 961, 1914-
44
(III) CNO2Br2 | + 2KNO2 -> CNO2Br2 C(NO2),Br | ' + 2KBr C(NO2),Br
(IV) C(NO2)2Br 2KGN | 4- или - C(NO2),Br 2KNO3 C(NO2)2K 2BrGN | -г или C(NO2)2K 2BrNO, тетранитро- этанкалий
(V) C(NO2)2K + 2KNO3 C-(NO2)3
C(NO2)2Kb п₽исутствии h’s0? c - (NO2)3
Подобно тетранитрометану гексанитроэтан обладает подвижной
нитрогруппой, вследствие чего он может быть применен для нитро-
вания; при его взаимодействии с р-крезолом в пиридиновоэфирном
растворе получен тн-нитро-р-крезол.
Нитрование азотнойкислотой. Нитрогруппа легче всего-
Т?2
I
замещает водород третичного углеродного атома Rt — С — Н, труд-
нее — вторичного — С — Н, еще труднее — первичного R — С — Н-
| I
Н Н
Впервые прямая нитрация предельных углеводородов была осу-
ществлена М. И. Коноваловым \ При этом он пользовался азотной
кислотой уд. веса приблизительно 1,07—1,1. Например, при нитро-
вании 2-метилбутана он получил 2-метил-2-нитробутан:
(СН3)2СНСН2СН3 + HNO3 -> (GH3)2GNOaCH2CH3 % Н2О
или при нагревании диизоамила (GH3)2GHGH2CH2CH2GH2CH(GH3)2
с азотной кислотой уд. веса 1,075 (13% HNO3) — 1,1(17%
HNO3) образуются моно-и динитро соединения диизоамила. При на-
гревании же с азотной кислотой уд. веса 1,38 (61% HNO3) диизо-
амил энергично реагирует, но образуется очень мало нитросоедине-
ний. При этом взаимодействии идет сильное окисление. В продук-
тах нитрации найден альдегид.
Шорыгин и Соколова * 2 нашли, что при нитровании толуола сла-
бой азотной кислотой уд. веса 1,12 по способу Коновалова (нагре-
ванием в запаянных трубках на кипящей водяной бане в течение
'Коновалов, Ж. Р- ф. X; О-, 33, 393, 1901; О нитровании анетола
и р-крезола см. Allsop и Kenner, J. Chem. Soc., 123, 2313, 1923.
2 Ж. Р- Ф- X. О., 62, 673, 1930.
45
48 часов) образуется 3—5% фенилнитрометана (от теории) вместо
указанного Коноваловым выхода 16—17%. Поэтому эти авторы при-
бегли к нитрованию азотной кислотой, разбавленной не водой,
а уксусной кислотой, благодаря чему создавались гораздо более бла-
гоприятные условия для реакции: она шла в гомогенной среде, так
как в безводной уксусной кислоте растворяются и толуол и азот-
ная кислота. При нагревании реакционной смеси (10 см3 толуола,
10,7 см3 азотной кислоты уд. веса 1,50 и 29,3 см3 ледяйой уксус-
ной кислоты) в открытых сосудах при 110° в течение 3’/2 часов полу-
чается 9,6% от теории фенилнитрометана, считая на весь взя-
тый толуол, или 14,1% от теории, считая на толуол, вошедший в
реакцию.
Нитрование парообразных парафинов. В последние
годы (начиная с 1936 г.) американскими химиками опубликовано
несколько работ, посвященных вопросу нитрования насыщенных
углеводородов (метан, этан, пропан, бутан, пентан и некоторые
изомеры последних трех). Нитрование углеводородов, содержащих
меньше пяти атомов углерода, раньше никем не изучалось; между
тем соответствующие нитросоединения могут представлять значи-
тельный интерес в случае нахождения дешевого способа их произ-
водства вследствие наличия больших количеств дешевых простей-
ших парафиновых углеводородов и возможности широкого при-
менения таких нитропродуктов.
Мы уже видели, что нитрование предельных углеводородов
в жидкой фазе затруднено тем, что углеводороды и азотная кислота
взаимно нерастворимы; вместе с тем получающийся нитропродукт
лучше растворим в азотной кислоте, вследствие чего он подвергается
дальнейшему нитрованию и окислению.
Проведение реакции в парообразной фазе представляет опреде-
ленные преимущества, так как реагенты могут быть немедленно
смешаны в любых пропорциях; эта реакция протекает менее, чем
в 1 минуту, и почти не дает полинитросоединений.
Сущность способа нитрации в парообразной фазе заключается
в том, что пары углеводорода пропускаются через горячую концен-
трированную азотную кислоту, после чего смесь паров углеводо-
рода и азотной кислоты проходит через реакционный аппарат, имею-
щий температуру до 420°.
Известно, что азотная кислота полностью разлагается при тем-
пературе 250° и выше на окись азота, двуокись азота, воду и кис-
лород. Однако оказалось, что при высоких температурах опыта ре-
акция нитрации протекает с такой скоростью, что азотная кислота
не успевает разложиться.
Нитрование простейших парафиновых углеводородов при тем-
пературе 420° дает более или менее сложные продукты (в зависи-
мости от углеводорода). Результаты анализа продуктов нитрации
приведены в табл. 3.
В опытах молярное отношение углеводорода к азотной кислоте
было равно 2:1.
46
Т аблица'3
Эти данные показывают следующее: 1) при условиях опыта не
произошло взаимодействия между метаном и азотной кислотой;
2) обнаруживается замечательный факт, что при нитровании пара-
финовых углеводородов получались нитропроизводные более низ-
ких углеводородов, чем исходные вещества; 3) в продуктах нитрации:
изобутана (и только в одном этом случае!) получен ацетон.
Механизм реакции нитрации при образований
низших гомологов. Вначале было высказано следующее пред-
положение В сопровождающие реакцию нитрации сильные окис-
лительные процессы приводят к превращению парафинов в жирные
кислоты. Последние нитруются в a-положении и, отщепляя угле-
кислоту, превращаются в низшие парафины. Было отмечено, Что к
таким же результатам могут привести и другие реакции, например,
взаимодействие свободного алкильного радикала с молекулой дву-
окиси азота.
Однако МакКлари и Дигиринг 1 2 показали прямыми опытами,
что при обработке уксусной, пропионовой и масляной кислот азот-
ной кислотой при температуре 400—410° не образуются соответ-
ствующие нитропарафины; следовательно, жирные кислоты не яв-
ляются промежуточными продуктами при нитрации парафинов.
Поэтому отпадает приведенное выше первое предположение о .ме-
ханизме нитрации.
На основании ряда опытов МакКлэри и Дигиринг пришли к сле-
дующему представлению о механизме взаимодействия между пара-
финовыми углеводородами и азотной кислотой.
В реакции участвуют свободные радикалы, которые могут обра-
зоваться при высокой температуре реакции при окислении угле-
водорода азотной кислотой или при термическом разложении ка-
кого-либо нестойкого промежуточного соединения. Эти радикалы
1 Hass, Hodge и Vanderbilt, Ind. Eng. Chem., 28, 339, 1936.
2 M с С 1 а г у и Degering, Ind. Eng. Chem., 30, 64, 1938.
47
в свою очередь могут вступить в любую присущую свободному
- (радикалу в данных условиях реакцию, а именно:
1) реагировать с азотной кислотой
R • Н -ф- окислитель —> R 4* X
свободный
радикал
R + HONO2 7?NO2 + ОН
НН + ОН -> R 4- Н2О и т. д.
2) разложиться с образованием свободного олефина и свободного
^радикала более низкого молекулярного веса, который, в свою оче-
jpeflb, может подвергаться любой возможной реакции своего предше-
ственника :
R —> R' 4* олефин
3) соединяться с молекулой нитрующего агента с образованием
.нитропарафина и активной частицы:
R 4- HONO„ -> ANO, 4- ОН
Д'Н 4- НО -> R' 4- Н2О
R' 4- HONO2 -> fl'NO2 4- ОН и т. д.
4) столкнуться с молекулой исходного вещества и начать цеп-
ную реакцию:
R 4- НН -> ЯН 4- R
шли
Я'4-НН^Я'Н4-Я
5) соединиться с другим свободным радикалом, образуя алкан:
R + R'^RR'
6) окисляться в двуокись углерода и воду или какие-либо окис-
ленные промежуточные продукты.
Продукты окисления, получающиеся при реакции, можно объяс-
нить, если допустить: 1) непосредственное окисление исходного угле-
рода; 2) окисление нитропарафинов; 3) окисление свободных ради-
калов, продуктов распада и олефинов.
Гэсс и Петерсон 1 проверили теорию свободных радикалов в при-
менении ее к реакции нитрации. Они нашли в продуктах реакции
нитрования нитро пентана следующие вещества:
нитрометан ............... 1,1%
нитроэтан.................. 7,19%
1-нитропропан .............13,85%
1-нитробутан.............12,5%
1-нитропентан...............21,6%
2-нитропентан...............20,8%
3-нитропентан...............23,0%
Сюда вошли все соединения, которые можно построить на осно-
вании теории свободных радикалов путем прибавления нитрогруппы
к свободным радикалам, получающимся из пентана при потере во-
дородного атома или путем разрыва любой связи углерода с угле-
водом; этим подтверждается правильность теории радикалов.
1 Н ass и Patterson, Ind. Eng. Chem., 30, 67, 1938.
ГЛАВА Ш
МЕХАНИЗМ НИТРАЦИИ АЗОТНОЙ КИСЛОТОЙ
1. Работы Виланда, Наметкина, Тронова, Михаэля и Карлсона.
Виланд показал, что при взаимодействии этилена с кислотной смесью
протекает наряду с реакцией образования этилнитратнитрита сле-
дующая реакция:
СН2 ОН СН2ОН
И +1 -> I
СН2 NO3 СН^О2
Образовавшийся нитроэтиловый алкоголь этерифицируется из-
бытком HNO3, и образуется соответствующий азотный эфир.
В другом опыте Виланд и Кан 1 получили нитросоединение —
продукт замещения водорода исходного углеводорода на нитро-
группу. Механизм этой реакции они толковали следующим образом:
СН3 СН
С ОН
CH NO2
СН3
изоамилен
сн3 сн3
Хсон
> I
chno2
сн3
нитроизоамиловый
спирт
сн3 сн3
"с7
II
cno2
I
сн3
нитроизоамилен
Наряду с нитроизоамиленом выделен азотный эфир нитроизо-
амилового спирта.
В то время как нитроэтилнитрат и другие продукты присоеди-
нения алифатического ряда удалось выделить и идентифицировать,
аналогичные соединения для углеводородов ароматического ряда
выделить не удалось.
Перенесение этих представлений о механизме реакции нитрации
на ароматический ряд встречало затруднения, заключавшиеся в раз-
личии между олефиновыми и ароматическими углеводородами. Эти
различия таковы: 1) характерная для бензола и части его произ-
водных малая скорость реакции по сравнению с олефиновым рядом;
1Wieland и Kahn, Вег., 54, 1770, 1921.
Химия- 1з—4
49
2) химические свойства олефинов прежде всего характеризуются
способностью к реакциям присоединения, а ароматических соеди-
нений— к реакциям замещения.
Однако Виланд стал на точку зрения Тиле и Голлемана, кото-
рые считали, что всякое замещение в ароматическом ряду есть ре-
зультат первичного присоединения. Самое течение реакции замеще-
ния и многих реакций обмена в этом случае можно объяснить, как
взаимодействие молекул исходного вещества и реагента, происхо-
дящее при сближении их до стадии присоединения. Отщепление
элементов из продуктов присоединения в виде простой молекулы
(воды, галоидоводорода и т. п.) приводит к стабильному продукту
замещения.
Так, например, Тиле 1 и Голлеман 2 допускали, что бромирова-
ние бензола протекает по схеме:
—НВг v
,Н ------►
Продукт присоединения, как производное частично гидриро-
ванного бензола, сильно ненасыщен и распадается с отщеплением
НВг с образованием первоначального более насыщенного бензоль-
ного ядра.
По аналогии Виланд дает следующую схему реакции взаимо-
действия бензола с азотной кислотой:
н
+ OHNO2
ОН. _Н1о
н -------
• ^/xno2
Продукт присоединения есть нитроалкоголь циклогексадиена,
который по указанной выше причине должен отщепить воду. Этот
промежуточный продукт очень нестоек и потому не может быть изо-
лирован. При этом Виланд замечает, что коричневую быстро исче-
зающую окраску, появляющуюся в момент добавления азотной кис-
лоты к бензолу, можно приписать продукту присоединения 3.
1 Lieb. Ann., 306, 128, 1895.
2 Holleman, Die direktive Einfiihrung von Substituenten in den Ben-
zolkern, Leipzig, 1910.
3 При присоединении к углеводороду частицы азотной кислоты’происхо-
дит превращение бензоидной формы в хиноидную, которая окрашена. В момент
отщепления частицы воды происходит новое превращение хиноидной формы
в бензоидную, вследствие чего исчезает окраска промежуточного соединения.
60
Гипотеза Виланда получила некоторое подтверждение в выде-
ленном Оддо (Oddo) 1 при получении мононитронафталина продукте
присоединение следующего состава:
Н^/ОН
/\Х
Ч/Х/
HZ^NO3
Кроме того, из антрацена получен мезонитродигидроантранол,
дающий с отщеплением воды мезонитроантрацен:
Как показал Мартинсен, при нитровании смесями серной и азот-
ной кислот серная кислота играет роль не только водоотнимающего
средства; она, кроме того, активирует бензольное ядро, делая его
более реакционноспособным. По мнению Н. Н. Ворожцова 2, сер-
ная кислота активно участвует в образовании продукта присоеди-
нения. Присоединение молекулы серной кислоты активирует осталь-
ные двойные связи углеводорода, что облегчает присоединение
следующей молекулы HNO3. Наконец, в момент стабилизации об-
разовавшегося продукта присоединения он теряет молекулу воды
и молекулу серной кислоты с образованием нитросоединения:
+ H2SO4 Н2О
Известно, что способность серной кислоты давать продукты при-
соединения выше, чем у азотной кислоты; это можно проследить
1 Gazz. Chim. Hal., 55, 174—184; Zbl., 1925, II, 1598.
2 Ворожцов, Основы синтеза красителей, стр. 44, 1934.
51
иа следующем примере. Установлено, что окрашенные продукты при-
соединения серной кислоты образуются у тех производных 4,4'-диалк-
оксидифенила
AlkO^ / — \ /ОА1к
которые не дают продуктов присоединения с азотной кислотой,
между тем как все соединения, присоединяющие азотную кислоту,
присоединяют и серную кислоту 1.
Образование промежуточного соединения по приведенным схе-
мам делает вероятным допущение, что стабилизация его из дигидро-
бензольного в бензольное соединение может итти по двум путям.
Первый путь, рассмотренный выше, сопровождается отщеплением
воды и приводит к нитрозамещенному; второй — сопряжен с отщеп-
лением элементов азотистой кислоты, в результате чего получается
фенол (особенно в условиях недостатка водоотнимающего реагента) 2:
.Такое направление при реакции jc азотной кислотой констати-
ровано особенно в условиях отклонения процесса нитрования (по
температуре, концентрации нитрующего реагента) от нормы 3.
Наметкин и Забродина 4 указывают, что схема Виланда имела бы
большую вероятность, если бы против нее нельзя было привести
одного весьма существенного возражения: нитроалкоголь, образую-
щийся согласно этой схеме, является одним из производных цикло-
гексадиена, характерной особенностью которых является неустой-
чивость и склонность к разного рода превращениям (реакции окис-
ления, уплотнения и т. п.). Ввиду этого образование такого соеди-
нения с последующим превращением лишь в одном направлении —
в сторону образования ароматических нитроуглеводородов и без
побочных реакций, столь естественных для непредельного соедине-
ния в среде серноазотной смеси, — представляется маловероятным.
Поэтому эти авторы (и еще раньше Тронов) дают другую схему
образования промежуточного соединения, допустив, что ве элементы
1 А 1 р h е п, Zbl., 1932, 1, 675; Rec. Trav. Chim. Pays-Bas, 50, 1111, 1931.
2 Ворожцов, Основы синтеза красителей, стр. 45, 1934.
’Armstrong и Rositer, Вег., 24, 720, 1891; Noelting и
For el, Ber., 18, 2670, 1885; Noelting и Pick, Вег., 21, 3158, 1888;
Battegay, Bull. Soc. Chim. Fr., 43, 109, 1928.
4 Ж. P. ф. X. O., 57, 92, 1925.
52
азотной кислоты присоединяются к бензолу, а наоборот, элементы
бензола (Н и радикал СвН5) присоединяются к азотной кислоте:
Z° Z0
CeH. + N=o -+СвН5 —N-OH GeH5NO2
хон \он
При таком допущении промежуточный продукт присоединения
бензола к азотной кислоте сохраняет ароматический характер.
Аналогично для углеводородов ряда циклогексана:
Z z°
CjIIjH-N O -»C,HU — N—ОН
циклогексан \qjj \qjj
CeH10 = NOOH
изонитроциклогексан
Это толкование реакции Тронов и Сибгатуллин1 распространяют
и на непредельные соединения:
СН2 хО СП, СН2
II + N^O -+ || “ О ||
СН2 \он CHN^-OH ghno2
Хон
Нитроэтиловый эфир образуется при дальнейшей реакции при-
соединения азотной кислоты:
GHO CH3ONO,
|| “ + hono2 -> I
chno2 gh2no2
Тронов и Сибгатуллин указали на то, что эта схема процесса так-
же не может объяснить всех фактов.По этой схеме следовало бы ожи-
дать взаимодействия между углеводородами и эфирами азотной кис-
лоты, так как замена в азотной кислоте водорода углеродным остат-
ком не должна была бы оказать большого влияния ни на распад
молекулы HNO3 на NO2 и ОН, ни на разрыв двойной связи N с О.
Однако ни бензол, ни его гомологи (ксилол и псевдокумол), ни даже
очень легко нитрующийся анизол при стоянии с этилнитратом в те-
чение 4—5 лет не претерпевали никакого изменения.
Учитывая указание Гирсбаха и Кесслера, что при взаимодей-
ствии бензола с азотной кислотой участвуют 2 мол HNO3, Тронов
высказал предположение, что водород HN03 притягивается к угле-
роду бензольного ядра; этим расшатывается связь между углеро-
дом и водородом, стоявшим при нем раньше- Водород теперь легко
1 Ж. Р. Ф. X. О., 62, 2267, 1930.
53
отщепляется, и происходит присоединение образовавшегося ком-
плекса к новой молекуле кислоты:
н
уО
.Nf-OH
+HNO3,
'-'HNO3 ±hn?s.
HNO3 —HNQ»—н.о
2
Согласно этому предположению одна молекула HNO3 участвует
в реакции (в качестве комплексообразователя) своим водородным
атомом, другая — двойной связью. Поэтому можно было ожидать,
что ароматические углеводороды будут нитроваться и эфирами
азотной кислоты, если вместе с ними взять вещество, обладающее
достаточной способностью образовать комплексы. Это подтвержда-
лось тем, что, по данным Бедкера \ этилнитрат энергично нитрует
бензол в присутствии хлористого алюминия.
Опыты Тронова и Сибгатуллина подтвердили, что А1С13, FeCl3
и некоторые другие положительные комплексообразователи акти-
вируют реакцию нитрования этилнитратом, причем образуется зна-
чительное количество нитробензола. В этом случае молекула А1С13
играет ту же роль, как вторая молекула HNO3 при нитровании азот-
ной кислотой:
Z°
N^-OH
ХОС2Н6
А1С13
H
+ AlGl3->
А1С13 +C.H.ONO,
+ С2Н6ОН +А1С13
Однако позже, учитывая указания, что образование комплекса
хлористого алюминия с бензолом не доказано 1 2, Тронов отказался
от предположения о возможности образования первичного ком-
плекса между А1С13 и бензольными углеводородами. Он пришел
к выводу, что 2 мол HNO3 соединяются в комплекс, который затем
уже реагирует с бензолом:
2HNO3 -> НОN = О... HONO2 + СвН„ ->
О
-> НО - NO • HONO2 C„H6NO2 + Н2О + HNO3
но/\свн5
1 Boedtker, Bull. Soc. Chim. Fr., 3, 726, 1908.
2 Б. H. Меншуткин, Известия Политехнического института, 12, 1,
1909; Olivier, Rec. Trav. Chim. Pays-Bas, 33, 149, 1914.
64
Михаэль и Карлсон 1 нашли, что в противоположность мнению
Виланда азотная кислота не присоединяется к этилену или аа-ди-
фенилэтилену. По данным этих авторов, в условиях опытов Виланда*
происходило разложение азотной кислоты, и получившиеся окислы
азота взаимодействовали с непредельными углеводородами с обра-
зованием соответствующих нитро со единений. Поэтому заключение
Виланда, что азотная кислота непосредственно присоединяется как
ОН + N02 к этиленовым группам, неприемлемо, и предполагаемая
Виландом аналогия при взаимодействии бензола и азотной кислоты
не имеет экспериментального основания.
Отвергая механизм нитрации азотной кислоты, предложенный
Виландом, Михаэль и Карлсон остановились на механизме нитрации,
^аналогичном предложенному Наметкиным и Троновым и описанному
Михаэлем 2 еще в 1896 г.
Они указывают, что кислород азотной кислоты, связанный с азо-
том двумя валентностями, должен иметь гораздо больше свободной
химической энергии, чем частично гидрогенизированный кислород
гидроксила, а также большее химическое сродство к водороду и
поэтому легче может оказать то действие, которое необходимо, чтобы
отделить в бензольной молекуле водород от углерода. Доминирую-
щими движущими силами в азотной кислоте является химический
потенциал кислорода для водорода и химический потенциал азота
для арила. С этой точки зрения нитрация бензола протекает следую-
щим образом:
GeHe + HONO2 -> CeH5NO(OH)2 -> CeH5NO2 + Н20
В итоге Михаэль причисляет реакцию нитрации к реакциям
альдолизации, охватывающим соединения, содержащие негидрокси-
лированные кислородные атомы.
2. Механизм альдолизации. Механизм альдолизации может быть
схематично изображен следующим образом:
Z0
/\N<-OH y\NO2
1-N-OH->
+ Ha0
О
Сравните эту схему с механизмом образования альдоля из 2 мол
ацетальдегида:
сн3с/ + СН2С=О -> СН3С
^0 |
Т Н
Н
1 Michael и Karlson, J. Amer. Chem. Soc., 57, 1268, 1935.
2 Ber., 29, 1795, 1896.
55
Альдольная конденсация происходит таким образом, что один
из водородных атомов, связанных с углеродом, стоящим рядом
с альдегидной группой, перемещается к кислороду другой альдегид-
ной молекулы, а две освободившиеся валентности углеродных ато-
мов взаимно насыщаются. Получающиеся при этом оксиальдегиды
очень часто претерпевают дальнейшие превращения,, разлагаясь
при этом на ненасыщенный альдегид и воду:
СН3СНОНСН2СОН -> СН3СН = снсно + н2о
кротоновый
альдегид
3. Теория радикал-ионов Пфейффера и Вицингера. Пфейффер
и Вицингер 1 на основании своих работ дали отличную от всех
предыдущих новую схему механизма реакции замещения непредель-
ных и ароматических соединений.
Исследуя бромирование асимметричных дизамещенных дифенил-
этиленов, они наблюдали при взаимодействии брома с этими дизаме-
щенными (в отличие от дифенилэтилена) кратковременное темнофи-
олетовое окрашивание. Им удалось выделить темносиний проме-
жуточный продукт, дающий, по их мнению, это окрашивание, —
темносиний пербромид. Они дали следующую схему бромирования
дизамещенного дифенилэтилена:
[(GH3)2N<^ = С = СН2 + 2Вг2
-> {[(CH3)2N/ >] С - СН2Вг) Вг3 ->
неустойчивый промежуточный
продукт солеобразного ха-
рактера — темносиний пер-
бромид
-> [ (CH3)2N<^ •>] С = СНВг + НВг + Вг2
Пфейффер и Вицингер распространяют эту схему замещения
и на другие соединения, допуская, что и здесь образуются анало-
гично построенные промежуточные продукты, быстро распадаю-
щиеся, которые поэтому не могут быть выделены.
Перенося эти представления о механизме замещения на гало-
идирование бензольных углеводородов, можно легко объяснить
каталитическое действие FeCl3, SbCl3, SbBr5 и т. д. (так называемых
не рено сите л ей). Последние облегчают бензольным углеводородам
образование комплексов солеобразного характера с галоидами.
Схема реакции галоидирования, по Пфейфферу и Вицингеру,
изображается следующим образом:
1 Pfeiffer и Wizinger, Lieb. Ann., 461, 132, 1929; J. |Prakt,
Chemie, 129, 129, 1931.
56
+ Br2 + SbBr5->
4- SbBr5 4- НВг
Вицингер и Балих 1 находят подтверждение теории радикал-
истов в выделенном и идентифицированном ими солеобразном темно-
окрашенном продукте взаимодействия асимметричного ^иарилзаме-
щенного дифенилэтилена с тетранитрометаном. При обработке этого
продукта раствором едкого кали ими было получено нитрозамещен-
ное этого производного дифенилэтилена.
Течение этой реакции они изображают следующей схемой:
С = СН2 4-NO2 —C(NO2)3
[(CH3)2n
темноголубое соединение
солеобразного характера
в присутствии КОН
[С(НО2)3Г------------------>
I
в присутствии кон [(CHg)2N / \] с = CHNO2 4- К+ [C(NO2)3]~4- Н2О
По аналогии авторы дают следующую схему нитрования бензола
азотной кислотой:
промежуточный неустой-
чивый продукт
+ Н20
4. Возражения Хюккеля* Хюккель 2 считает, что образование
промежуточных солеобразных продуктов для бензольных углево-
'Wizinger и Bahlich, J. Inaug. Dissert. Bonn, 1933.
2 H iickel, Z. Physikal. Chem., 35, 163, 1937.
57
до родов гораздо менее вероятно, чем для асимметричных диарил за-
мещенных этилена, так как согласно теории перевод ароматического
состояния связей в состояние радикал-иона (CeHe(NO2)]г должно
потребовать гораздо большего количества энергии, чем превращение
в радикал-ионы диарилзамещенных этилена, уже близко подходя-
щих к трифенилметану. На основании этого Хюккель считает такой
механизм замещения мало вероятным.
5. Схема Шааршмидта. Шааршмидт дает следующую схему
механизма реакции нитрации в присутствии серной кислоты.
Серная кислота отнимает воду у азотной кислоты и образует азот-
ный ангидрид:
2HN03 + H2SO4^±N2O5 + H2SO4 • Н2О
Азотный ангидрид образует затем с углеводородом продукт при-
соединения; при разложении этого промежуточного продукта обра-
зуется нитросоединение и молекула азотной кислоты
Мы дальше увидим, что по работам Ганча в серноазотных смесях
нет азотного ангидрида; это говорит против теории Шааршмидта.
ГЛАВА IV
ОСНОВЫ ПРОЦЕССА НИТРАЦИИ
1. Влияние концентрации и количества реагирующих компонен-
тов. Первые опыты систематического изучения процесса нитрации
азотной кислотой бензола, толуола и других ароматических углево-
дородов провел Шпиндлер Он показал, что этот процесс проте-
кает очень быстро в течение первого дня взаимодействия, а после
этого резко замедляется; он установил, что добавление воды к азот-
ной кислоте резко замедляет реакцию; наконец, он впервые показал,
что процесс нитрации ароматических углеводородов является
необратимым.
Кесслер 1 2 под руководством Л. Мейера изучал влияние на про-
цесс нитрации бензола концентрации и относительных количеств
бензола, азотной кислоты и нитробензола. Он применял для своих
опытов смеси этих трех веществ, составленные таким образом, чтобы
при переходе от одной серии опытов к другой изменялась только
одна составная часть.
Для нормального опыта было взято 100 г/мол бензола, 100 г/мол
азотной кислоты 3 и 100 г/мол нитробензола, а в последующих
опытах изменяли количество одного из компонентов, сохраняя ко-
личества остальных двух постоянными.
Ход нитрации, по данным Кесслера, изображен графически на
рис. 1 и 2. Пунктирные линии с индексом ВЬ показывают коли-
чество вступившего в реакцию бензола в процентах от первоначаль-
ного количества бензола; сплошные линии с индексом Bs показы-
вают то же в процентах от первоначального количества HNO3. Ли-
ния lOOBs и Ь дана для обеих величин. Числа перед буквами Bs,
ВЬ и т. д. обозначают число граммолекул переменного вещества
на 100 г/мол каждого из двух остальных. Например, обозначение
IbBs показывает, что в этой серии опытов брали 75 г/мол бензола,
100 г/мол азотной кислоты и 100 г/мол нитробензола, а самая кри-
вая 7bBs показывает количество граммолекул образовавшегося
1 Вег., 25, 1251, 1883.
2 Z. Physikal. Chem., II, 676, 1888.
• Для опыта брали азотную кислоту с содержанием 100% HNO2.
59
Рис. 1. Кривая Кесслера для случая переменного
количества бензола.
5 10 !5 20 гб 30 35 W *5 50 55 60 65 70 75 ВО
Время взаимодействия в часах
Рис. 2. Кривая Кесслера Для случая переменного количества
азотной кислоты.
60
нитробензола в процентах от первоначального количества граммо-
лекул взятой азотной кислоты.
Кривые вначале быстро поднимаются и притом тем быстрее,
чем больше взято кислоты по отношению к бензолу и нитробензолу
(иначе говоря, чем меньше объемная концентрация бензола) и чем
меньший объем занимает граммолекула HNO3.
Влияние количества бензола. На рис. 1 показан ход
процесса при переменных количествах бензола. Кривые 150В&,
100В&, 1ЬВЬ, ЬОВЬ и 25В b проходят в правильных расстояниях
друг от друга и показывают, что бензол тем меньше нитруется, чем
большее количество его взаимодействует с одним и тем же коли-
чеством азотной кислоты.
Этот вывод подтверждается расположением кривых с индексом
Bs (количество вступившего в реакцию бензола в процентах от на-
чального количества HNO3).
В течение первых часов реакции эти кривые располагаются в та-
ком же порядке, как и в предыдущей серии кривых. Однако через
некоторое время каждая кривая пересекает все другие так, что
порядок следования кривых меняется полностью на обратный.
Вначале они следуют в таком порядке: 25.Bs, 508$, 75Bs, 1005
и В, между тем как приблизительно через 40 часов меняются в
такой последовательности: 1008$ и b, 1508$, 75Bs, 5OBs, 258$.
Однако по виду кривых ясно, что если бы опыт продолжался еще не-
сколько дней, то и 1508$ перегнало 1008$. Это можно выразить
словами, что при одинаковом количестве азотной кислоты в течение
1 часа образуется тем больше нитробензола, чем меньше бензола
добавлено к азотной кислоте. Бензол, следовательно, оказывает
тормозящее действие, причем не только тогда, когда он добавлен
в избытке, но и тогда, когда его количества далеко недостаточно
для полного израсходования на реакцию со всей HNO3. И только
в той мере, в какой бензол расходуется на нитрацию начинает
сказываться -влияние его массы, состоящее в том, что теперь от
большего количества бензола больше нитруется. Однако подъем
кривых, относящихся к большим количествам бензола, продолжается
недолго; напротив, по ту сторону их пересечения они проходят па-
раллельно, т. е. часовой прирост нитробензола по прошествии не-
скольких дней во всех опытах одинаков и притом мал.
Влияние количества HNO3. По кривым рис. 2 видно, что
влияние избытка азотной кислоты на скорость реакции нитрации
обратно влиянию избытка бензола: в то время как бензол оказывает
тормозящее действие на реакцию, азотная кислота, наоборот, дей-
ствует ускоряюще.
Все описанные опыты проводились при 18°.
В отдельной серии опытов определялось влияние на скорость
реакции переменного количества азотной кислоты, причем к 100г/мол
бензола добавлялось последовательно 50, 100, 150 и т. д. г/мол
азотной кислоты и определялось количество образовавшегося через
15 минут после момента смешения количества нитробензола. Если
61
обозначить образовавшееся при добавке 50 г {мол азотной кислоты
количество нитробензола через единицу, то при двойном количестве
кислоты образуется 6 единиц нитробензола; при тройном — 21 и т. д.
Эти отношения даны в табл. 4.
Таблица 4
2. Влияние нитробензола, бензола и воды на скорость реакции.
Кроме конечного состояния реагирующей смеси Кесслер изучал
начальные скорости реакции при температуре около 3°. Определя-
лось взаимодействие, происшедшее через 15 минут от начала опыта,
причем установлено, что при добавлений к 100 г/мол бензола и
100 г/мол азотной кислоты одинакового количества граммолекул
нитробензола, бензола или воды наибольшее замедляющее действие
на реакцию нитрации оказывает нитробензол, немного меньшее—
вода и наименьшее — бензол (в соотношении соответственно 5 : 4 : 1).
3. Роль серной кислоты при реакции нитрации по Марковни-
кову. Давно известна роль серной кислоты при нитрации. В при-
сутствии серной кислоты азотная кислота во многих случаях может
быть почти полностью использована при нитрации (до содержания
0,5% HNO3 в отработанной кислоте); причем серная кислота не
только усиливает нитрующее действие азотной кислоты, но одно-
временно ослабляет ее окисляющее действие на парафиновые, наф-
теновые и ароматические углеводороды. Марковников 1 объясняет
специфическое действие серной кислоты в ее смесях с азотной при-
этой смеси так называемой серноазотной кислоты
, обладающей нитрогруппой, более склонной к реак-
сутствием в
.ОН
SO2<
xONO2
циям замещения, нежели нитрогруппа азотной кислоты. Механизм
образования этой серноазотной кислоты и нитрации этой кислотой,
по Марковникову, заключаются в следующем:
/ОН
H2SO4 + HNO3 -> SO2< + Н2О
XONO,
/ОН
SO2< + ЛН -+ flNO2 + H2SO4
xono2
1 Ber., 31, 47, 4898; 32, 1441, 1899.
62
{Однако существование этого промежуточного соединения им не
было доказано, а выявившиеся в дальнейшем факты не могут быть
удовлетворительно объяснены этой гипотезой.
4. Зависимость скорости реакции нитрации от концентрации сер-
ной кислоты. Эта зависимость для реакции нитрации нитробензола
впервые дана Мартинсеном х. Найденные им значения констант-
скорости реакции при концентрации реагирующих веществ 0,1Л\
даны в табл. 5.
Таблица б
сб си S си ф с S Е4 о О Я СО О_ О с© 04 Я О я оо о О с© Я о Я СО О o' О со 04 Я О еч Я СО тН о” О с© 04 Я о Я СО о" О с©1 к1 О Я ОО О о” О с© еч я о § О' О с© еч Я
При 0 0,017 0,24 0,28 0,14 0,085 0,046 0,036
При 26 0,18 — 3,22 — 1,6 — 0,22
К при. 25
Эти результаты
изображены графически
на рис. 3, откуда сле-
дует, что скорость реак-
ции сильно изменяется
в зависимости от кре-
пости серной кислоты.
Приуменьшении содер-
жания воды от 1,03 моля
Н2О на 1 моль H2SO4
цонстанты скорости
реакции быстро увели-
чиваются, достигая мак-
симума при содержа-
нии 0,63 моля Н2О на
1 моль H2SO4, а при
дальнейшем уменьше-
нии содержания воды моль
константы вновь умень- "а моль
шаются. нг301'
Влияние крепости
серной кислоты на ско-
рость нитрации при
3,0^
----- Кривая при 0е
-----Привал при 25
р 3,22
Ь0,29
1л 2
2,0 р-
К при О
0,35
0,36
0,22
О0!7Ъ\О,1ь
50,} 1——---- —'—’—’—’—’—
'0,< \РОО,Г 0,20,30,4 0,5 0,6 0,70,3 0,9 1,0j
Моль Нг0 на моль H2S0w
Рис. 3. Кривая Мартинсена.
К 1 Z. Physikal. Chem. 50, 385, 1905.
63-
температуре 25° для разных соединений видно из табл, b (Мартин-
сена х).
Т аблица 6
Наименование вещества К для H2SO4-0,3H2O К для H2SO4 К для H2SO4-0,3H2O К для Н28О4
4,6-Динитро-цг-ксилол 0,0040 0,0014 2 86
jj-Хлорнитробензол 0,18 0,050 3.60
m-Хлорнитробензол 0,39 0,14 2,78
-о-Хлорнитробензол 7,15 2,18 3,28
Нитробензол 1,50 0,37 4.05
2,4-Динитроанизол ....... 0,17 0,053 3,21
2,4-Динитрофенол 0,85 0,39 2,18
Бензольсульфокислота Около 26,0 2,3 Около 11,30
Бензойная кислота » 100,0 Около 5,4 » 18,50
Указанные в таблице вещества распределяются на две группы,
из которых первая характеризуется тем, что ее скорость реакции
увеличивается приблизительно втрое при переходе от моногидрата
серной кислоты к кислоте состава H2SO4 • 0,ЗН20 (95% Н2ЗО4);для
второй группы веществ скорость реакции при этих условиях значи-
тельно превосходит трехкратную.
.К первой группе относятся нитропроизводные бензольных угле-
водородов и хлорнитропроизводные; ко второй группе относятся
органические кислоты, т. е. карбоксильные и сульфопроизводные.
Эти данные опровергают и поныне еще широко распространен-
ное мнение, что нитрация протекает тем быстрее, чем крепче сер-
ная кислота; наоборот, скорость нитрации убывает в пределах
определенной концентрации серной кислоты с повышением этой
концентрации.
5. Влияние растворителя на скорость реакции. Относительно
причины этого явления, по мнению Мартинсена, наиболее вероятно
предположение, что растворитель как среда обусловливает измене-
ние скорости нитрации, а различные степени разбавления серной
кислоты приходится рассматривать как изменение среды, в которой
идет реакция.
По вопросу о значении среды для скорости химических реакций
Н. А. Меншуткин 2 говорит следующее:
«...Скорость химической реакции может меняться в широких
пределах под влиянием растворителя.
Реакция не может быть отделена от среды, в которой она проте-
кает. Б константу скорости реакции входит определенным коэфи-
щиентом растворител ».
1 Z. Physikal. Chem., 59, 605, 1907.
s Z. Physikal. Chem., 6, 41, 1890; Ж. P. Ф. X. O., 22, 203—404, 1890.
'64
«...Проведенные опыты привели к заключению, что при изуче-
нии химического процесса неизбежно надо иметь в виду свойства
среды, в которой протекает процесс. Медленные реакции могут
быстрее проходить в целесообразно выбранном растворителе; на-
оборот, в энергично протекающих реакциях можно, если надо, при-
менением, например, углеводородов предельного ряда в качестве
растворителя, ослабить процесс».
«...Настоящее исследование показывает, что химически индифе-
рентный растворитель имеет большое влияние на скорость хими-
ческих процессов как обратимых так и необратимых. Это свидетель-
ствует о том, что между так называемыми индиферентными раство-
рителями и растворенным веществом происходит определенное кон-
тактное действие, имеющее следствием, что растворы одного и
того же вещества, в зависимости от состава и структуры индиферент-
ного растворителя, показывают различные свойства в отношении
изменения скорости химического процесса, протекающего в таких
растворах».
«...Процесс растворения двух смешивающихся жидких соедине-
ний есть первая фаза того процесса, который в дальнейших фазах
является химическим процессом».
К сожалению, в настоящее время нет еще теории влияния рас-
творителя на скорость реакции, впервые установленной Меншут-
киным.
Однако можно допустить каталитическую роль растворителя,
которая подтверждается тем, что изменение его сильно влияет на
теплоту активации реакции в растворе.
Мухин и Зильберфарб 1 установили для реакции образования
четырех замещенных аммонийных солей в различных растворителях
и в смесях этих растворителей, что в растворителе, в котором реак-
ция идет быстрее, теплота активации меньше.
Исследованиями Лауэра и Ода 2 при нитровании антрахинона
в серной кислоте найдено, что максимум скорости нитрации антрахи-
нона как азотной кислотой, так и калиевой селитрой лежит прибли-
зительно при 89% H2SO4. Теплоты активации нитрации азотной кис-
лотой и калиевой селитрой в растворе серной кислоты с содержа-
нием 87—95,6% H2SO4 примерно одинаковы и равны приблизи-
тельно 21 650 кал-, теплота же активации при нитрации в растворе
моногидрата серной кислоты значительно ниже. Но константы дей-
ствия нитрации в растворе серной кислоты с различным содержа-
нием воды очень различаются между собой. На основании этих
данных Лауэр и Ода объясняют различную скорость реакции нитра-
ции влиянием концентрации серной кислоты на величину константы
действия.
Эти же авторы рассчитали величины теплот активации и кон-
стант действия из данных Мартинсена (стр. 63), относящихся к
нитрации нитробензола в серной кислоте; причем результаты, по
1 Укр. Хим. Жури., IV, 327,
’Lauer и Oda, J. Prakt. Chemie, 144, 176, 1936.
Химия—13—5
65
мнению авторов, находятся в полном соответствии с результа-
Таблица 7 тами, полученными при нитровании антрахи-
Концентрация H2SO4 % Теплота активации кал Константа действия нона. Результаты нитрации нитробензола в растворе серной кислоты даны в
84,0 89,6 95,6 100,0 15 600 15 900 18 500 10 600 3,1 -1О10 1,3-1012 5,1-1о13 1,1-10’ табл. 7. Заметим, что эти чи- сла не находятся в пол- ном соответствии с вы- водами самих авторов;
сравнение констант дей-
ствия для двух концентраций кислот с содержанием 89,6% и
95,0% H2SO4 привело бы к выводу, что в 95%-ной кислоте скорость
реакции нитрации нитробензола должна была бы быть наибольшей,
тогда как Мартинсен определил наибольшую скорость реакции
в серной кислоте с содержанием около 89,5% H2SO4.
Изложенное указывает на неправильность распространенного
мнения, что серная кислота является только водоотнимающим сред-
ством.
Из имеющихся данных следует, что серная кислота при нитрации
является не только водоотнимающим средством, но и играет роль
активатора бензольного ядра \
,6. О тормозящем влиянии избытка нитробензола на скорость
реакции его нитрации. Гезерингтон и Массон 1 2 изучали процесс
нитрации нитробензола нитрующей смесью в гетерогенной системе
и установили тормозящее влияние избытка нитробензола на ско-
рость реакции нитрации нитробензола. Это явление автор объяс-
няет следующим образом. При смешении нитробензола с нитрующей
смесью образуются два слоя: органический слой и неор-
ганический. По заключению авторов, реакция нитрации про-
текает в неорганическом слое, и, следовательно, скорость нитрации
зависит от концентрации серной кислоты в неорганическом слое.
Концентрация серной кислоты в неорганическом слое снижается
вследствие того, что органическая фаза растворяет серную кислоту.
Это происходит вследствие образования соединения серной кислоты
с нитробензолом состава: G6H5NO2 • H2SO4, изолированного Массо-
ном3 и имеющего температуру затвердевания 11,6°.
Еще Шербулье 4 показал, что нитрогруппа обладает слабыми
основными свойствами; он выделил в кристаллическом виде серно-
кислую соль C6H5NO2 • H2SO4, которой он придает строение
[GeHeNOa • HJ+ [HSO4]—. Соответственно этому он наблюдал, что-
1 См. об этом также Battegay, Bull. Soc. Chim. Fr., 43, 109. 1928.
2 J. Chem. Soc. 105, 1933.
3 J. Chem. Soc., II, 3200, 1931.
* Helv. Chim. Acta, 6, 281, 1923; Zbl, 1923, I, 1491.
66
электропроводность серной кислоты заметно увеличивается от рас-
творения в ней нитробензола или нитротолуола, но уменьшается при
прибавлении динитробензола, который уже совершенно лишен ос-
новных свойств. Очевидно, 2-я нитрогруппа сводит на-нет основные
свойства 1-й нитрогруппы.
Указание Гезерингтона и Массона дает для одного частного слу-
чая объяснение тормозящему влиянию избытка нитробензола на
процесс нитрации. Однако против этого толкования можно при-
вести ряд доводов; в частности, при некоторых концентрациях
серной кислоты надо бы ожидать ускоряющего влияния нитро-
бензола на процесс нитрации, но это не установлено опытом.
7. Влияние температуры на скорость реакции нитрации. Влия-
ние температуры на скорость нитрации для интервала 0 и 25° дано
выше. В табл. 8 приведены эти данные для ряда других веществ и
для интервала в 10°, т. е. 25 и 35°.
Таблица 8
Наименование вещества К при 35° К при 25° К при 35° К при 25°
Для Н 2SO4-0,3 Н2О
4,6-Динитро-ж-ксилол ..... 0,013 0,004 3,25
р-Хлорнитробензол 0,47 0,18 2,61
т-Хлорнитробензол 1,23 0,39 3,15
Для Н28О4
4,6-Динитро-ш-ксилол 0,0045 0,0014 3,21
р-Хлорнитробензол 0,16 0,05 3,20
т-Хлорнитробензол 0,38 0,14 2,71
Как видно из табл. 8, температурный коэфициент нитрации
равен приблизительно трем, т. е. выше, чем для большинства хими-
цеск I реакций.
8. Влияние заместителей на скорость нитрации х. В табл. 9
приведены константы скорости нитрации для ряда веществ в одной
и той же среде — серной кислоте состава H2SO4 • 0,ЗН20 (95%
H2so4).
Таблица 9
Наименование вещества К при 25° К при 35° К при 46°
Нитробензол 1,5 —
т- Динитробензол 0 —- -—
Бензолсульфокислота 26 — —
m-Нитробензолсульфокислота . . . 0 — —
Бензойная кислота . . >100 — —
1 Z. Physikal. Chem., 59, 605, 1907.
67
Продол ж. табл. 9
Наименование вещества К при 25° К при 35° К при 46°
0,0005
0.00008
0,00008
0,00013
о-Нитробензойная кислота........
m-Нитробензойная кислота . . . .
р-Нитробензойная кислота........
о-, т- и р-Нитротолуолы . . . . .
2,4-Динитротолуол ..............
4,6-Динитро-т-ксилол............
2,4-Динитромезитилен............
о-Хлорнитробензол...............
т-Хлорнитробензол...............
р-Хлорнитробензол...............
2, 4-Динитроанизол..............
2, 4-Динитрофенетол.............
2, 4-Динитрофенол...............
Фенилуксусная кислота ......
Этиловый эфир фенилуксусной
кислоты ......................
а-Нитронафталин...............•
0,00006*
0,000009*
0,000009*
оо
0,004
6,65
7,16
0,39
0,18
0,17
0,65
0,85
Около 1.3
оо
оо
0,000043*
0,013
1,23
0,47
Из данных этой таблицы следует:
1. Нитрогруппа тормозяще действует на процесс нитрации. Это
вытекает из того, что нитро бензойные кислоты не только реагируют
много медленнее, но и нитробензолсульфоновая кислота вообще не
реагирует там, где сульфокислота очень быстро нитруется. Из кон-
стант бензойной и нитробензойной кислот можно высчитать действие
ощной нитро группы; находим, что нитрогруппа, введенная в бензой-
ную кислоту в ортоположении к карбоксильной группе, замедляет
скорость нитрации приблизительно в 0,2 • 107 раз, а в мета- или
пара положении — в 107 раз.
2. Карбоксильная группа тормозит нитрацию; из сравнения
данных для нитробензола и нитробензойной кислоты находим, что
карбоксильная группа, стоящая в ортоположении к нитрогруппе,
замедляет реакцию в 0,3 • 105 раза, а стоящая в мета- или парапо-
ложении замедляет в 1,5 • 105 раза. Если сопоставить эти числа с по-
лученными для нитрогруппы и притом так, чтобы сравнивалось
ортоположение с орто положением, мета- и параположение с мета-
и параположением, то получим, что нитрогруппа приблизительно
в 70 раз сильнее тормозит,’ чем карбоксильная; это значение можно
найти и прямым сравнением между бензойной кислотой и нитро-
бензолом.
3. Тормозящее действие сульфогруппы больше, чем карбоксиль-
ной, но меньше нитрогруппы. Действие сульфогруппы в 4 раза
* Величины, помеченные звездочками (*), вычислены из значений констант,
экспериментально полученных при другой температуре, в предположении, что
скорость реакции повышается втрое на каждые 10°.
68
больше карбоксильной, а действие нитрогруппы в 17 раз больше
сульфогруппы.
4. Из сравнения констант для нитробензола и нитрохлорбензола
видно, что хлор в орто положении ускоряет процесс, а в мета- и
параположении действует тормозяще.
5. Метильная группа ускоряет реакцию, ибо нитротолуолы реа-
гируют с бесконечно большой скоростью Вступление 2-й метиль-
ной группы в метаположение к уже имеющейся увеличивает ско-
рость нитрации в 300 раз, а вступление 3-й — тоже в метаполо-
жение — ускоряет реакцию в 1660 раз.
6. Метоксильная группа оказывает еще более ускоряющее влия-
ние, чем метильная, что видно из сравнения константы 2,4-динитро-
айизола с константой динитротолуола.
7. Этоксильная группа действует благоприятней, чем метоксиль-
ная группа.
8. Гидроксильная группа наиболее ускоряет реакцию нитрации.
Гидроксильная, метоксильная и этоксильная группы, каждая
в отдельности, действуют сильнее, чем две метильные группы, так
как динитрофенол и его алкильные эфиры обладают большей кон-
стантой нитрации, чем динитро-m-к сило л.
На основании изложенного по характеру влияния на скорость
нитрации можно установить следующий ряд заместителей:
NO2 > SO3H > СООН > Cl < GH} < (СН3)2 < ОСН3 < ОС2Н3 < ОН
Здесь хлор поставлен посредине, потому что он в одном случае
ускоряет, а в другом тормозит нитрацию. Влево от хлора расположены
группы, тормозящие реакцию, вправо — ускоряющие реакцию.
Чем дальше группа удалена от хлора, тем сильнее она тормозит
или ускоряет реакцию.
Мы уже видели, что на скорость реакции в сильнейшей степени
влияет среда, в которой реакция протекает, причем влияние среды
на скорость реакции разных веществ неодинаково. Поэтому приве-
денный ряд заместителей Мартинсена безусловно справедлив лишь
для случая, когда средой является серная кислота. При другой же
среде не только скорость реакции будет иная, но можно ожидать
и иного расположения заместителей — по силе их влияния на ско-
рость реакции нитрации.
Тронов и Бер 1 2 изучали влияние заместителей на скорость нитра-
ции замещенных бензола в растворе нитробензола (2 моля) 1 моль
HNO3 при комнатной температуре, причем полученные ими ряды,
как и надо было ожидать, отличаются от ряда Мартинсена.
При этом характерно, что распределение заместителей в ряду
зависит еще и от стадии реакции, определяемой авторами по коли-
честву израсходованной азотной кислоты.
1 Практически это значит, что при концентрации серной кислоты
H2SO4 • 0,ЗН2О (95% весовых H2SO4) в условиях опыта Мартинсена реакция
протекает настолько быстро, что ее скорость не удается определить.
2 Ж. Р. Ф. X. О., 62, 2337, 1930.
69
Ряд для момента израсходования 5% HNO3:
NO2 > С2Н5СО >С N > GNGHa > C6HSCH2 > СН3 > Н <
< С2Н5 < Вг < GH2C1 < Gl < CH3GO
Ряд для момента израсходования 15% HNO3:
...CeH5CH2> СН3> C2HS> Н < Вг <G1 < СН2С1 < СН3СО
Ряд для момента израсходования 25% HNO3:
G1 > Вг > СН3> Н <С2Н5< ... <СН3СО
В этих рядах центральное место занимает водород (Н), отвечаю-
щий незамещенному бензолу; вправо от него стоят заместители,
ускоряющие реакцию, влево — замедляющие. В этих рядах приме-
чательно в частности то, что GH3GO — группа метаориентирую-
щая — наиболее ускоряет нитрование. Заслуживает внимания по-
ложение отдельных заместителей в различных рядах.
Заместитель С2Н5, ускорявший реакцию вначале, далее замед-
ляет, затем снова ускоряет; галоиды сначала ускоряют, затем за-
медляют; метильная группа замедляет во всех случаях; 2-я, и осо-
бенно 3-я, метильная группа влияет противоположно 1-й группе,
заметно ускоряя нитрование. о-Ксилол нитруется в 1,6—1,9 раза,
пг-ксилол — в 4,5—4,9 и р-ксилол в 5,7—10,5 раза быстрее толуола.
Мезитилен и псевдокумол реагируют еще во много раз скорее.
9. Влияние окислов азота на скорость нитрации. Клеменс и
Экль 1 установили, что процесс нитрации фенола в эфирном рас-
творе есть положительный аутокаталитический процесс, и самая
скорость нитрации зависит от концентрации окислов азота, соот-
ветственно HNO2 в азотной кислоте.
Чисто аутокаталитический процесс нитрации в эфире выражается
в том, что чистая азотная кислота, свободная от двуокиси азота и
азотистой кислоты, не нитрует. Двуокись азота вызывает как процесс
нитрации, так и окислительное действие азотной кислоты по отно-
шению к бензольному производному.
Эрнол 2, нашел, что реакция нитрации фенола в спиртовом рас-
творе аутокаталитична; причиной этого является образование HNO2
в побочной реакции. Реакцию можно остановить добавлением
небольшого количества гидразина и его производных, подобно
тому, как можно остановить растворение металлов в азотной ки-
слоте прибавлением мочевйны.
По Вейбелю 3, фенол в водном растворе крайне легко нитруется.
Даже «с 12V раствором HNO3 можно провести нитрование в конеч-
ное время. Однако условием для гладкого течения реакции являет-
ся присутствие азотистой кислоты. Если этого нет, требуется пред-
1 Monatsh. Chem., 39, 641, 1918; J. Chem. Soc., 116, I. 122.
2 Arnall, J. Chem. Soc., 123, II, Д111, 1923; 124, 811—816, 1924.
3 Veibel, Ber., 63, 1577, 1930.
70
варительный индукционный период, в течение которо.го тв не иссле-
дованной точно реакции имеет место образование'Незначительных
количеств HNO2. у
Карташев 1 подтвердил своими опытами зайлючение прежних ис-
следователей о каталитическом влиянии окислов азота, причем он
пришел к выводу, что процесс нитрации фенола протекает следую-
щим образом: азотистая кислота реагирует с фенолом с образова-
нием нитрозофенола (хиноноксима), а этот последний окисляется
азотной кислотой в нитрофенол. Следовательно, процесс протекает
по следующей схеме:
(I) hno,->hno2 + o
.ОН
(II) CeHsOH + NOOH ->С6Н /
\NO
О
А ЛОН
.он
+0-C’H‘<n0!
Правильность этой схемы подтверждена Карташевым следую-
щими данными:
1) в первый момент действия азотной кислоты на фенол в слабых
водных растворах реакция Либермана дает синее окрашивание,
что указывает на наличие среди продуктов реакции нитрозо-
фенолов;
2) в начавшейся реакции нитрации дальнейшее возникновение
азотистой кислоты может происходить за счет окисления нитрозо-
фенолов в том случае, если они окисляются легче, чем фенол; это
подтвердилось тем, что прибавка к фенолу нитрозофенола снижает
необходимую для начала реакции температуру;
3) по схеме 1-й фазы реакции необходимо возникновение нитро-
зофенолов, легче окисляющихся, чем фенол; поэтому окислитель-
ные процессы, сопровождающие нитрование, могут протекать не за
счет фенола, а за счет нитро- или нитрозофенолов. Опытами было
установлено, что при действии слабой азотной кислоты нитрофенолы
не способны окисляться, — они претерпевают только дальнейшее
нитрование. Таким образом, по Карташеву, процесс нитрации
является совокупностью параллельно протекающих реакций нитро-
зирования и окисления.
1 Ж. Р. Ф. X. о., 59, 819, 883, 1927; 62, 385, 2129, 1930.
71
Мартинсен 1 нашел, что с применением серной кислоты в качестве
растворителя процесс нитрования не зависит от количества раство-
ренных в серной кислоте окислов азота.
Александров и Штамм 2 нашли, что при применении для
нитрования нафталина нитрующей смеси из химически чистых ма-
териалов, не содержащих HNO2, взаимодействие долго не начина-
лось, и только по истечении некоторого времени возникала бур-
ная реакция. При применении же обыкновенной нитрующей смеси
реакция начиналась в момент пуска первых капель этой смеси в смесь
нафталина с 61%-ной серной кислотой.
10. Теория нитрационных смесей Сапожникова. Теория Сапож-
никова 3 основана на следующих положениях и фактах:
1- Еще Вьеллем было установлено, что азотная кислота е
содержанием 78% HNO3, соответствующая составу HNO3 • Н2О,
почти не обладает нитрующей способностью по отношению
к клетчатке.
С другой стороны, при пересчете Сапожниковым состава ни-
трационных смесей, при которых нитрующая способность падает
до минимума, на молекулярные стехиометрические соотношения,
оказалось, что эти составы соответствуют формуле:
HNO3 • Н2О + п (H2SO4 • 2Н2О)
2- Опыт показал, что наилучшими для нитрации клечатки яв-
ляются смеси, содержащие H2SO4 и HNO3 в отношении приблизи-
тельно 3 : 1. При уменьшении содержания воды в таких смесях
степень нитрации клетчатки вначале повышается, а затем, с опре-
деленного значения содержания воды, падает. Еще в 1901 г- Лунге
и Беби показали, что наивысшая степень нитрации клетчатки полу-
чается при смесях, содержащих 11—12% Н2О.
3. На основании экспериментального исследования физико-хи-
мических свойств тройных смесей4, состоящих из H2SO4, HN03
и Н2О, Сапожников пришел к заключению, что в них имеет место
обратимый процесс:
HNO3nH2O + H2SO4-> HNO3(n—x)H2O + H2SO4tH2O
Предел этого процесса при возрастании содержания в смеси
совпадает с определенным молекулярным отношением состав-
ных частей рассматриваемой системы, и когда она получает состав:
HNO^H2O + a;H2SO4 HNO3 + z(H2SO4H2O)
то азотная кислота переходит в состояние свободного моногидрата.
1 Z. Physikal. Chem. 50, 385, 1905.
3 Ж. Хим. Пр., VI, 127 5, 1929.
3 Ж. Р. Ф. X. О., 41, 1712, 1909.
4 По вопросу о свойствах серноазотных смесей см. Сапожников,
Ж. Р. Ф. X. О-, 35, 1098, 1903; 36, 518, 669, 1904; 37, 374, 1905; Машкин,
Нитрация клетчатки, 1928; David, J. Soc. Chem. Ind., 41, 1922; Кон-
Д p а ц к и й, Труды Каз.-хим.-тех. института, 7, 1937.
72 • '
It. Изучая упругость паров азотной кислоты в смесях ее с сер-
ной кислотой и водой, Сапожников нашел, что по мере прибавле
ния к разбавленной азотной кислоте безводной серной кислоты
упругость пара растет и достигает максимума в момент образования
в смеси состава, отвечающего формуле:
HN03 + H2SO4H2O
Одновременно с возрастанием упругости паров азотной кислоты
растет и нитрационная способность смеси, достигая максимума одно-
временно с упругостью паров азотной кислоты, т. е. при прибавлении
серной кислоты происходит обезвоживание азотной, вследствие
чего и упругость пара возрастает.
С дальнейшим прибавлением серной кислоты упругость пара
падает, однако, не пропорционально падению концентрации без-
водной азотной кислоты в смеси (закон Генри), а несколько быстрее,
т. е. так, как это имело бы место при исчезновении части азотной
кислоты. Сапожников объяснял это явление тем, что при действии
безводной серной кислоты на азотную происходит частичное образо -
вание N2O5. Одновременно с падением упругости паров азотной
кислоты падает нитрационная способность смеси по отношению
к клетчатке.
На основании этих данных Сапожников предложил свою теорию
нитрационных смесей.
Основой теории является положение, что степень нитрации
клетчатки находится в прямой зависимости от состояния азотной
кислоты в смеси (в свободном виде или в виде того или иного ги-
драта), а это ее состояние характеризуется в значительной мере
величиной давления паров азотной кислоты в данной смеси.
Когда количество воды в нитрационной смеси недостаточно для
образования гидрата серной кислоты H2SO4H2O, то негидратирован-
ная серная кислота отщепляет воду от азотной кислоты, переводя
ее в N2O5. Вследствие этого концентрация реакционноспособной
азотной кислоты уменьшается, и нитрационная способность смеси
падает.
Дегидратирующее влияние серной кислоты по отношению к азот-
ной прекращается с момента, когда количество воды достаточно
для образования гидрата H2SO4H2O, и в этой смеси вся азотная
кислота присутствует в виде свободного моногидрата HNO3. При
этом составе смеси азотная кислота обладает максимальной упру-
гостью паров и максимальной нитрационной способностью, обеспе-
чивающей наивысшую степень нитрации клетчатки.
При дальнейшем увеличении количества воды последняя идет ча-
стично на образование второго гидрата серной кислоты H2SO4-2H2O,.
а частично на образование гидрата азотной кислоты HNO3H2O,.
и, следовательно, количество реакционно способной азотной кислоты
уменьшается, падая до нуля, между тем как количества воды до-
статочно для полного перевода всей серной кислоты в H2SO4 2Н2О
и всей азотной кислоты в HNO3H2O.
73
11. Зависимость между составом смеси и упругостью паров
азотной кислоты. Кроме изложенной теории Сапожников дал зави-
симость между составом Ьмеси и упругостью азотной кислоты, изо-
браженную им графически. Графически же он изобразил и зависи-
мость между составом нитрационной смеси и степенью нитрации
клетчатки.
Рис. 4. Диаграмма Сапожникова.
Для этой цели Сапожников воспользовался треугольником
Гиббса.
На вершинах треугольника (рис. 4) располагаются компоненты
смеси H2SO4, HNOj и Н2О. На сторонах треугольника распола-
гаются соответственные двойные смеси. Каждая точка внутри тре-
угольника, как известно, выражает состав той или другой тройной
системы.
Составы смесей выражены в молекулярных процентах.
Сплошные кривые соответствуют смесям, имеющим одно и то же
давление паров азотной кислоты.
Как видно из диаграммы, давление паров HNO3 падает быстро
по линиям HNO3 — Н2О и HNO3 — H2SO4 по мере уменьшения
74
в смесях относительного содержания, азотной кислоты, и притом
по первой линии значительно скорее, чем по второй. Общей
причиной этого является уменьшение концентрации азотной
кислоты; по линии HNO3-H3O (смеси азотной кислоты с водой),
как мы выше видели, по мере увеличения содержания воды обра-
зуются гидраты азотной кислоты; соответственно уменьшается кон-
центрация свободного моногидрата HNO3, вследствие чего
здесь имеет место быстрое падение упругости паров азотной
кислоты.
Уменьшение упругости паров азотной кислоты по линии HNO3—
H2SO4 при следовании от точки HNO3, по мнению Сапожникова,
объясняется как уменьшением концентрации всей Содержащейся
» смеси HNO3, так и частичной дегидратацией HNO3 при увеличе-
нии концентрации серной кислоты.
При рассмотрении диаграммы от точки HNO3 • Н3О по прохо-
дящей через эту точку высоте треугольника мы увидим все отмечен-
ные выше закономерности: сначала наблюдается увеличение упру-
гости паров азотной кислоты до встречи с точкой, лежащей на пря-
мой HNO3 • Н2О — H2SO4, в которой вся содержащаяся в смеси
азотная кисйота находится в виде моногидрата HNO3. При даль-
нейшем продвижении по этой линии упругость паров азотной кис-
лоты понижается вследствие уменьшения содержания HNO3 в смеси
и вследствие уменьшения концентрации свободного моногидрата
в смеси из-за дегидратирующего действия негидратированной
серной кислоты на азотную кислоту.
12. Зависимость между составом нитрационной смеси и сте-
пенью нитрации клетчатки. При нанесении на указанную диаграмму
(рис. 4) составов нитрационных смесей (выраженных, разумеется,
в молекулярных процентах) Сапожников обнаружил, что смеси,
на которых получаются определенные степени нитрации клетчатки
(например 10- или 11-азотная нитроклетчатки), располагаются в
определенных областях площади треугольника, причем наблюдаются
следующие закономерности.
• Область высоконитрованного нерастворимого пироксилина огра-
ничивается двумя линиями, из которых одна проходит близко к ли-
нии равномолекулярных отношений H2SO4 и Н2О, а другая —
очень близко к линии тройных смесей, в которых отношение числа
молекул серной и азотной кислот равно 2 : 1 и сохраняется почти
постоянным.
Значение первой линии заключается в том, что, начиная с этого
ряда смесей, вправо азотная кислота находится все время в состоя-
нии безводного моногидрата HNO3, что обеспечивает возможность
получения наиболее высокой степени нитрации клетчатки.
Значение нижней линии заключается в том, что при дальнейшем
увеличении относительного содержания серной кислоты начинает
приобретать все большее значение процесс разложения образую-
щихся азотных эфиров клетчатки серной кислотой, что вызывает
сперва понижение степени нитрации, а при еще большем количестве
75
серной кислоты в смесях —полное разложение нитроклетчатки
или невозможность самого процесса нитрации.
Рассматривая продукты нитрации, полученные на смесях, рас-
положенных близко в области нерастворимой нитроклетчатки, но
содержащих несколько большее количество воды или серной кис-
лоты, можно выделить район смесей, наиболее благоприятных для
получения пироколлодия (с числом нитрогрупп, близким к 10).
При дальнейшем продвижении в сторону увеличения относи-
тельного содержания воды и серной кислоты в смесях получаются
все более и более низкие степени нитрации с содержанием от (NO2)9
до (NO2)6. Но причины этого понижения степени нитрации в том
и другом направлении различны.
В направлении увеличения содержания воды в смесях азотная
кислота утрачивает состояние свободного моногидрата HNO3, со-
единяясь с водой в более или менее сложные гидраты, причем спо-
собность к нитрации будет сохраняться в этих смесях только до
тех пор, пока азотная кислота не перейдет в состояние гидрата
HNO3 • Н2О.
В сторону же увеличения относительного содержания серной
кислоты должен развиваться постепенно процесс денитрации, пока
количество серной кислоты не достигнет того предела, за которым
далее никакой процесс нитрации уже не будет в состоянии проис-
ходить.
По этим двум пределам, где, видимо, прекращается возмож-
ность образования каких бы то ни было степеней нитрации, про-
ведена кривая (NO2)6, которая отделяет на диаграмме целую полосу
нитрационных смесей, необходимых для получения всех степеней
нитрации от (NO2)9 до (NO2)6.
При изучении части диаграммы, ограниченной кривой (NO2)11?
надо обратить внимание еще на следующее. Для смесей, очень бед-
ных водой, т. е. очень близких к линии HNO3 — H2SO4, степень
нитрации ни разу не достигает (NO2)U и изменяется в пределах
от (N02)io,7- Это обстоятельство, по мнению Сапожникова, тесно
связано с тем, что безводная серная кислота значительно сильнее
понижает давление паров азотной кислоты, чем та же кислота
с некоторой примесью воды.
В присутствии безводной серной кислоты происходит дегидра-
тация азотной. Вследствие этого количество реакционноспособной
азотной кислоты уменьшается, и нитрационная способность смеси
падает. Поэтому концы кривой (1ЧО2)пне доведены до линии HNO3—
H2SO4.
12-азотная нитроклетчатка C24H28O8(NO3)12 на нитрационных
смесях, содержащих серную кислоту, невидимому, вообще не может
быть получена, вероятно, по той причине, что во всех смесях
с максимальной нитрующей способностью мы все же имеем дело
с подвижным равновесием:
HNO3+ n(H2SO4 Н2О) HNO3+(n—1)(H,SO4 • H,Oj+H2O +H2SO4
76
и выделяющиеся молекулы воды и серной кислоты производят
частичное омыление высоконитрованной нитроклетчатки.
В подтверждение этого можно указать на работы ряда исследо-
вателей, которые показали, что если нитрацию клетчатки произво-
дить на смесях, содержащих в качестве водосвязывающих веществ
вместо серной кислоты фосфорный, азотный или уксусный ангидрид,
т. е. на смесях, которые заведомо не содержат воды и в которых
в то же время вся азотная кислота, повидимому, находится в «сво-
бодном» реакционно способном состоянии, то действительно удается
получить нитроклетчатки, в которых содержание азота доходит
до 14 и 14,1%, что уже очень близко подходит к 12-азотной нитро-
клетчатке C24H28OS(NO3)12 (теоретическое содержание азота 14,14%),
Особенно, если учесть, что клетчатка не есть химически чистое
вещество.
13. Зависимость между составом нитрационной смеси и сте-
пенью нитрации нафталина и толуола по Сапожникову Ч Патар
сделал большое исследование по нитрации нафталина 1 2; А. В. Са-
пожников обработал материалы Патара и построил по ним диаграмму
(рис. 5).
Рис 5. Диаграмма Сапожникова для нитропроизводных
нафталина.
Относительно этой диаграммы отметим следующее. При иссле-
довании физических свойств тройных смесей из серной и азотной
1 Ж. Р. Ф. X- О.Д4, 1712, 1909.
2 Условия опытов Патара: 10 г нафталина вносилось в 300 г кислотной
смеси; смесь нагревалась до 115° и при этой температуре производилась часо-
вая выдержка.
77
кислот и воды Сапожников установил непрерывный характер изме-
нения всех свойств (упругость паров, электропроводность, удельные
леса). Этому непрерывному изменению свойств вполне отвечает
форма установленной Сапожниковым зависимости между степенью
нитрации клетчатки и составом нитрационной смеси. Естественно
было ожидать, что и зависимость между степенью нитрации нафта-
лина и составом нитрующей смеси выразится также в форме непре-
рывной функции, т. е. соответствующая кривая должна быть не-
прерывной, без скачков. На самом же деле, если мы ограничим
кривыми области моно-, ди-, три- и тетранитронафталина, то полу-
чим кривые, форма которых меняется скачкообразно. Это вызывает
большое сомнение в правильности установленной Сапожниковым
зависимости.
Еще ярче это сказывается на приведенной в работе, выполненной
под руководством Сапожникова, кривой зависимости степени нитра-
ции толуола от состава кислотной смеси (работа Костевича).
Для установления этой зависимости было сделано 44 опыта ни-
трации толуола в следующих условиях:
1) количество нитруемого толуола в одну реакцию 0,5 кг,
2) количество смеси бралось из расчета на 1 мол толуола
1,1 мол азотной кислоты (моногидрата);
3) кислота вводилась в толуол;
4) время нитрации 3 часа;
5) температура нитрации: в течение первого часа сливалось 45%
веса смеси при температуре 35°; в течение второго часа сливалось
35% веса смеси при температуре 50°; в течение (первой половины)
третьего часа вливался остаток кислоты при температуре 60°; по
окончании слива делалась выдержка при температуре 60° в течение
получаса.
Результаты опытов нанесены на диаграмму (рис. 6), на основа-
нии которой сделаны следующие выводы:
1) в части диаграммы, приближающейся к чистой воде, начиная
с содержания ее, соответствующего кривой давления паров около
5 мм, нитрация толуола совершенно отсутствует;
2) в части диаграммы, приближающейся к чистой серной кис-
лоте, уже при содержании ее в нитрационной смеси в количестве
около 65% серной кислоты обнаруживается осмоление продуктов
нитрации с выделением окислов азота и притом в большей степени
в смесях безводных;
3) кислотные смеси с содержанием серной кислоты от 0 до 35%
дают возможность получить чистый мононитротолуол независимо
от относительной пропорции азотной кислоты и воды;
4) кислотные смеси с содержанием 35—55% серной кислоты
(опять независимо от относительного содержания азотной кислоты
и воды) образуют промежуточную зону, когда получается смесь
моно- и динитротолуолов;
5) кислотные смеси, расположенные в довольно узком простран-
стве диаграммы с содержанием 55—65% серной кислоты, соответ-
78
ствуют образованию динитротолуола и, невидимому, совершенно
определенного его изомера, а именно 2 : 4.
Таким образом, если для случая нитрации нафталина мы гово-
рили о кривых, форма которых изменяется скачкообразно, то в диа-
грамме, относящейся к случаю нитрования толуола, нет даже от-
дельных участков в форме кривой.
HN03
Рис. 6. Диаграмма Сапожникова для нитропроизводных толуола.
Автор указал в 1932 г., что причиной отмеченного различия
кривых является то, что Сапожников наносил на диаграмму
первоначальный состав смеси, тогда как степень нитрации соедине-
ния определяется не начальным, а конечным составом. На диаграмму
надо очевидно наносить тот предельный состав, на котором реакция
взаимодействия между нитруемым соединением и кислотной смесью
при условиях опыта практически прекращается (подробнее об этом
см. стр. 84).
Проведенные автором с сотрудниками исследования установили
зависимость между степенью нитрации толуола и составом смеси,
показанную на рис. 7.
Совершенно аналогичного характера зависимость для случая
нитрации нитробензола в динитробензол установили Гезерингтон
и Массон 1 (рис. 8). Эти авторы называют составы кислот, лежащих
на кривых, конечными точками, или предельными
составами, имея в виду, что это точки кажущегося, а не абсо-
лютного прекращения реакции.
1 J. Chem., Soc., 105, 1933.
79
»
Рис. 7. Кривая зависимости степени нитрации толуола от состава
нитрационной смеси. Вправо от правой кривой лежит область
тринитрации; вправо от левой кривой — область диниитрации.
Рис. 8. Кривая Гезерингтона и Массона.
80
14. Современное изложение теории нитрационных смесей Сапож-
никова. Работы Сапожникова по изучению свойств смесей серной
и азотной кислот и воды выполнены в начале XX столетия.
В последующее время установлена ошибочность мнения Сапож-
никова об образовании азотного ангидрида в смесях крепких азот-
ной и серной кислот; кроме того, значительно изменились наши
взгляды на строение кислот.
Овозможности образования азотного ангидрида
в смесях азотной и серной кислот. Как изложено выше,
по мнению Сапожникова, в смесях, бедных водой, т. е. очень близко
к линии HNO3 — H2SO4 (рис. 4), происходит дегидратация азот-
ной кислоты с образованием азотного ангидрида.
. Против этого утверждения говорит следующее:
1) азотный ангидрид кипит при 45° и скорее должен бы повы-
шать, а не понижать упругость паров азотной кислоты;
2) еще в 1898 г. Хойтсема получил нитроклетчатку с содержанием
13,86% азота нитрацией клетчатки смесью азотной кислоты и азот-
ного ангидрида (11-азотная нитроклетчатка содержит 13,4% азота).
В последующие годы неоднократно бывала получена нитроклет-
чатка с весьма высоким содержанием азота на смесях азотной кислоты
с азотным ангидридом. Отсюда следует, что азотный ангидрид даже
способствует получению наивысших степеней нитрации клетчатки;
3) в новейшее время Ганч показал, что спектр поглощения рас-
твора азотного ангидрида в серной кислоте отличается от спектра
поглощения смеси безводных азотной и серной кислот. Следова-
тельно, в этой смеси нет азотного ангидрида.
Таким образом уменьшение упругости паров азотной кислоты
и ослабление нитрующей способности в очень крепких нитра-
ционных смесях не могут быть объяснены образованием азотного
ангидрида.
Ст роение кислот по Ганчу1. До Ганча азотная
кислота, ее соли и эфиры описывались как гидроксильные соединения:
HONO2; Me0NO2; CnH2n_iON02
Однако эти формулы не могут объяснить существующей фунда-
ментальной разницы между электролитами и неэлектролитами и,
следовательно, между водным раствором азотной кислоты и креп-
кой кислотой, а также между солями и эфирами азотной кислоты.
Это удалось только помощью координационной теории Вернера
и его теории комплексных соединений и ионогенной связи. Согласно
этой теории только эфиры отвечают обычной структурной форму-
ле. Соли же напротив — комплексной формуле с комплексным
анионом.
1 Вег., 58, 941, 1925.
Химия—13—6
81
Так как азотная кислота является в водном растворе сильным
электролитом, то ей придали комплексную формулу с ионогенносвя-
занным водородом: ^ONgjMe; [0Nq] Н.
Однако по данным оптического анализа эта комплексная формула
не оправдывается для высоко концентрированной азотной кислоты
и ее растворов в неионизированных средах. На основании своих
исследований Ганч пришел к заключению, что безводная азотная
кислота отвечает структурной формуле: ОН — Nf , а не комплекс-
но
ному соединению Вернера [NO3]H. Поэтому она является псевдо-
азотной кислотой.
Наряду с этим Ганч нашел, что в разбавленной азотной кислоте,
вопреки мнению Вернера, нет свободной азотной кислоты и ее сво-
бодных Н-ионов; вода, по Ганчу, функционирует по отношению
к кислотам не как растворитель, а в качестве слабого основного ан-
гидрида, и образует путем присоединения к гидроксильному водо-
роду псевдокислоты соль гидроксония (отвечающую солям аммо-
ния) Ч Следовательно, азотная кислота существует в водном рас-
творе в виде нитрата гидроксония формы:
[H3O]NO3 или [H...(HaO)n]NO3
Кроме того, Ганч установил, что в крепкой азотной кислоте и ее
смесях с крепкой серной образуются солеобразные соединения —
нитрониевые соли:
O2NOH + Н . SO4H ^[NO(OH)J[SO4H]
O2NOH 4- 2Н • SO4H -> [N(OH)3][SO4H]3
O2NOH + O2NOH [NO(OH)2] [N03]
O2NOH + 2O2NOH [N(OH)3] [NO3]2
1 В позднейших своих работах Ганч обобщил свои выводы и признал не-
правильным общепринятое До этого предположение о существовании в водных
растворах кислот свободных водородных ионов. По его мнению, при растворе-
нии кислот в воде имеет место не только физический процесс растворения, но
и химическое взаимодействие, при котором вода присоединяется к гидроксиль-
ному водороду кислот подобно аммиаку (но как гораздо более слабое основание)
с образованием подобных аммонийным гидроксониевых солей.
В соответствии с этим новым взглядом на строение кислот, растворенных
в воде, Ганч определяет силу кислоты (меру кислотности, по другому
его выражению) наклонностью ее к образованию соли, а не степенью диссоциа-
ции кислоты.
Получаемое по этому признаку расположение кислот по их силе значи-
тельно отличается от прежнего расположения их по степени диссоциации в
водных растворах, а именно:
Расположение по наклонности к образованию соли
НСЮ4 > J?'SO3H > HJ > НВг > НС1 > HNO3
Расположение по степени диссоциации в водных растворах
НС1О4 > HJ > НВг > HNO3 > НС1 > H2SO4
82
Безводная азотная кислота представляет собой, по Ганчу, раствор
нитроний-нитрата в псевдоазотной кислоте. Например, в 99,6%-ной
азотной кислоте при 0° содержится около 80% OHNO2 и около 20%
[N(OH)3](NO3)2. В крепкой азотной кислоте до момента достиже-
ния состава HNO3 • Н2О между нитрониевой солью и псевдоформой
существует состояние подвижного равновесия:
3O2NOH^[N(OH)3][NO3]2
Нитроний-нитрат нацело гидролизован в присутствии 1 мол
Н2О. Для некоторых концентраций, определено следующее содержа-
ние псевдоазотной кислоты:
Таблица 10
——— Общий процент HNOS Нормаль- ность азотной кислоты Количество молекулН2О на 1 мол HNO3 Процент псевдокис- лоты от об- щего коли- чества HNO3 Процент недис- социированной истинной ки- слоты от об- щего количе- ства HNO3 Процент ионов истин- ной кислоты от общего количества HNO3
77,3 18 1,0 ' 70 25 О
48,3 10 3,75 50 32 18
31,6 6 7,5 2 60 38
При увеличении концентрации выше 78% HNO3 происходит
быстрое нарастание процентного содержания псевдоазотной кислоты.
В 9%-ном растворе азотной кислоты можно обнаружить присут-
ствие псевдокислоты.
Новое изложение теории Сапожнико в а.
Фармер 1 впервые изложил теорию нитрационных смесей в свете
указанных представлений о строении кислот. При добавлении к вод-
ной азотной кислоте серной кислоты последняя отнимает воду
от гидроксониевой соли азотной кислоты, и увеличивается содержа-
ние псевдоазотной кислоты. По достижении определенного коли-
чества серной кислоты азотная целиком переходит в псевдосостоя-
ние. При дальнейшем увеличении количества серной кислоты и
уменьшении содержания воды в смеси образуется сульфат нитро-
ния и соответственно уменьшается количество псевдоазотной кис-
лоты.
Таким образом псевдоазотная кислота, содержание которой
в смеси обусловливает нитрующую способность смеси, находится
в меньшем количестве в безводных или почти безводных серноазот-
ных смесях, чем в смесях с некоторым содержанием воды. Наоборот,
при увеличении концентрации смеси кислот содержание псевдо-
азотной кислоты повышается до максимума, а при дальнейшем уве-
личении концентрации (согласно предыдущему) количество псевдо-
кислоты падает.
1 J. Soc. Chem. Ind., 76 Т., 1931.
83
В заключение заметим, что соображения, основанные на обра-
зовании нитрониевых солей, верны, когда реакция происходит при
низкой температуре. Эти соли настолько непрочны, что при более
высокой температуре они могут образоваться лишь в очень незначи-
тельном количестве х.
15. Характеристика нитрационной смеси. Сапожников, как мы
видели, характеризовал смеси по их начальному составу. Такая
характеристика допустима для случая нитрации клетчатки, так как
здебь начальный состав смеси относительно мало отличается от
состава отработанной кислоты (содержание азотной кислоты умень-
шается на 1—2% мол).
Иное имеет место при нитровании ароматических соединений.
Например, для нитрования толуола применяется смесь состава:
54% H2SO4, 28% HNO3, 18% Н2О и получается отработанная
кислота состава: 69% H2SO4, 0,5% HNO3, 30% Н3О-
Так как этому составу отработанной кислоты отвечает большое
количество нитрационных смесей, то очевидно, что смесь надо ха-
рактеризовать по составу отработанной кислоты, при котором пре-
кращается реакция нитрации (точнее говоря, при этой концентра-
ции скорость реакции так мала, что она не имеет практического
значения).
Очень удобно для оценки состава смеси пользоваться содержа-
нием серной кислоты в вычисленном составе отработанной кислоты,
получающейся в предположении превращения всей азотной кислоты
смеси в воду. Например, для смеси 54% H2SO4, 28% HNO3, 18%
Н2О вычисляется содержание серной кислоты в отработанной кис- \
лоте равным 70%. ' J
Активный состав отработанной кислоты.
Отработанная кислота, как правило, содержит кроме серной и
азотной кислот и воды еще растворенные органические вещества
(преимущественно нитропродукты) и нитро зил серную кислоту. По-
этому введено понятие об активном составе отрабо-
танной кислоты, под которым разумеют тройную смесь из
содержащихся в отработанной кислоте серной и азотной кислот
и воды (tz е. состав отработанной кислоты без примесей).
Концентрация серной кислоты в смеси (в активном составе отра-
ботанной кислоты или же исходной смеси), получающаяся при пре-
вращении всей азотной кислоты в эквимолекулярное количество
воды, носит название фактора нитрующей актив-
ности (принято обозначать буквой Ф) отработанной кислоты или
исходной смеси.
Вывод формулы для вычисления фактора
нитрующей активности. Обозначим через:
1 Дальнейшее развитие теории нитрационных смесей на основе представле-
ний Ганча о строении кислот см. Lauer и Oda, J. Prakt. Chemie, 144, 176,
1936.
84
S — процентное содержание H2SO4 в смеси;
N — процентное содержание HNO3 в смеси;
S' — процентное содержание серной кислоты в отработанной
кислоте, когда вся азотная кислота вынитрована (т. е. це-
ликом заменена эквивалентным количеством воды по реак-
ции С6Н6 4- HNO3 C6H5NO2 + Н2О).
При нитровании на 1 ч. HNO3 выделяется 6g = 0,286 г Н2О;
следовательно, вес кислотной смеси уменьшится на 1—0,286 =
= 0,714 в. ч., считая на 1 в. ч. HNO3, а на N в. ч. HNO3, нахо-
дящихся в 100 ч. смеси, уменьшение веса составит IV- 0,714 в. ч.
Поэтому концентрация серной кислоты в отработанной кислоте S'
определится по формуле:
о, S 100
100 — N • 0,714'
При делении числителя и знаменателя на 0,714 имеем:
____________________________140 $4
° — 140 — N'
Это и есть формула для определения фактора нитрующей актив-
ности;
140— N
Иногда дают формулу в другом виде:
ф---------------------------5.
1-А
140
Если необходимо вычислить полный состав отработанной кио-
лоты, т. е. содержание в ней серной и азотной кислот и воды, то
при этом предполагают, что реагирует количество HNO3, эквива-
лентное количеству нитруемого соединения, взаимодействующего
.со смесью. Если количество смеси взято так, что содержание HNO3
в ней больше теоретически необходимого для реакции, то в отрабо-
танной кислоте останется некоторое количество азотной кислоты.
В этом случае состав отработанной кислоты может быть вычислен
следующим образом.
Обозначим:
>S’ — процент H2SO4
TV — процентHN03 в смеси;
S' — процент H2SO4
TV' — процент HN03
Выражение для S' и N':
™ s • юо
° 100 — 0,714 ’
N, = (N-.x) 100
100 — 0,714’
в смеси;
в отработанной кислоте;
в отработанной кислоте;
(1)
(2)
где х — количество HNO3, вступившее в реакцию.
Значение х определяем следующим образом.
Пусть на а г нитруемого вещества с молекулярным весом М
взято т г смеси. Тогда на 100 г смеси придется 100 а- г нитруе-
мо а , п W
мого вещества, или г!мол. При взаимодействии этого коли-
чества граммолекул нитруемого вещества вступит в реакцию
100 • а • 63 ..„л
—т—м— г HNO3; следовательно,
63 • 100
х = --
т М
Вставляя это значение х в выражения (1) и (2), легко вычис-
лим значения S' и N'.
Приведенные расчеты фактора нитрующей активности и пол-
ного состава отработанной кислоты относятся к идеальному случаю,
когда реакция нитрации не сопровождается побочными процессами
(окисление, осмоление), дающими выделение окислов азота. Окислы
азота не только сами по себе являются балластом, но и связывают
серную кислоту в нитрозилсерную кислоту, которая в свою очередь
также является балластом.
Согласно данному выше определению фактора нитрующей ак-
тивности отработанной кислоты при его вычислении надо исходить
из ее активного состава, т. е. не учитывать содержания нитрозил-
серной кислоты и растворенных нитропродуктов.
Пример расчета - фактора нитрующей ак-
тивности отработанной кислоты. Определим
фактор нитрующей активности для среднего состава отработанной
кислоты, получающейся при производстве пикриновой кислоты из
фенола на крепких кислотах.
Состав отработанных кислот (см. стр. 290):
H,SO4................... 81%
HNO3.....................2,5%
N,O3.................... 3,5%
пикриновой кислоты .... 3,5%
Н2О..................... 9,5%
(мы не учитываем содержания в отработанной кислоте щавелевой и
динитрофенолсульфокислоты). Образовавшиеся в результате побоч-
ных реакций окислы азота- взаимодействуют с серной кислотой и
дают нитрозилсерную кислоту:
N2O3 + 2H2SO4 ->2NHSO5 + Н,0
Из этого уравнения видно, что окислы азота двояко влияют на со-
став кислотной смеси: связывая серную кислоту в нитрозилсерную,
они уменьшают фактор нитрующей активности; выделяя воду при
взаимодействии с серной кислотой, они еще больше уменьшают этот
фактор.
86
При взаимодействии 3,5 в. ч. N2O3, содержащихся в заданном
нам составе отработанной кислоты, с серной кислотой образуется
11,7 в. ч. нитро зил сер ной кислоты и 0,83 в. ч. воды. Так как на
образование этих веществ по приведенной реакции расходуется
9,03 в. ч. серной кислоты, то истинный состав заданной нам отра-
ботанной кислоты будет следующий:
H2SO4 ........................ 81—9.03=71,97%
HNO3........................... 2,50%
NHSOs.......................... 11,7%
пикриновой кислоты ............ 3,5%
Н2О........................... 10,33%
Итого . • . -100% '
Активный состав этой кислоты:
H2SO4..................... 71,97 в. ч.
HNO3 ...................... 2,5 » »
НО........................ 10,33 » »
Итого . • . 84,80 » »
Фактор нитрующей активности зтой же кислоты:
ф -______________________71,97____= 86 7°/
84,80 — 2,5 • 0,714 ои’
Об измен е-нии фактора нитрующей актив-
ности при нитрации. Так как реакция нитрации сопро-
вождается, как правило, процессами окисления, то фактор нитрую-
щей активности кислотной смеси понижается во время этого процесса.
При первой и второй нитрации толуола понижение фактора нитрую-
щей активности незначительно. При нитровании ксилола в динитро-
ксилол фактор изменяется приблизительно на 1%. При нитровании
динитротолуола в тринитротолуол понижение фактора составляет
4-5%.
Гроггинс предложил характеризовать смеси отношением коли-
чества серной кислоты в нитрационной смеси к количеству воды
в смеси, сложенной с количеством воды, образующейся из содержа-
щейся в смеси азотной кислоты к концу нитрации.
Это отношение он назвал «дегидратирующим значением серной
кислоты». В нашей химической литературе иногда пользуются этим
отношением, которое чаще называют «коэфициентом дегидратации»
или лучше, по предложению проф. Н. Н. Ворожцова, «коэфициентом
водоотнятия». Например, для названной выше смеси 54% H2SO4,
28% HN03, 18% Н2О коэфициент водоотнятия равен:
Введенное Сапожниковым определение состава смеси в молеку-
лярных процентах вместо весовых представляет большое удобство
87
*
при научных работах, так как в этом случае при изменении состава
смеси при нитровании содержание в ней серной кислоты остается
величиной постоянной (если пренебречь окислительными процес-
сами и потерей азотной кислоты испарением).
Поэтому легко и удобно определить состав смеси в любой момент
процесса нитрования по треугольной диаграмме. Кроме того, в этом
случае при нанесении на диаграмму молекулярных составов кислот
отдельные зависимости получаются в виде удобных для научных
выводов и практического использования кривых.
ГЛАВА V
.ВЛИЯНИЕ РАЗЛИЧНЫХ ФАКТОРОВ НА ТЕЧЕНИЕ ПРО-
ЦЕССА НИТРАЦИИ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
1-Я НИТРАЦИЯ
1. Приливание кислотной смеси к углеводороду. В аппарат по-
дается все количество углеводорода, предназначенное к нитрованию
в одну операцию, и затем постепенно, небольшой струей сливается
кислотная смесь. Особенностью этого порядка смешения компонен-
тов является наличие во время нитрации избытка нитруемого углево-
дорода. Наибольший избыток последнего имеется вначале, затем он
уменьшается по мере прибавления кислотной смеси и вступления
соответствующей части углеводорода в реакцию с азотной кислотой.
Лишь к концу прибавления смеси в реакционной массе оказывается
небольшой избыток азотной кислоты (против теоретически необхо-
димого для превращения всего оставшегося количества углеводо-
рода в нитросоединение).
В применяемых в промышленности для нитрования углеводоро-
дов кислотных смесях скорость реакций настолько велика, что кон-
центрация азотной кислоты в смеси в момент смешения компонен-
тов (серноазотной смеси и углеводорода) почти мгновенно падает
•до содержания 0,6% и меньше.
Это наглядно видно из табл. 11 — результатов опытного нитро-
вания толуола в заводском аппарате смесью состава 55% H2SO4,
31% HN03 и 14% Н2О (фактор нитрующей активности равен
71% H2SOJ.
Таким образом содержание HN03 в реагирующей кислоте колеб-
лется в пределах от 0,25 до 0,65%. Вероятно и эти незначитель-
ные колебания, а равно вышедшая в одном случае из отмеченных
пределов величина 1,75% HN03, объясняются недостаточным пере-
мешиванием, присущим чугунному аппарату старой конструкции,
в котором производился опыт.
Следовательно, в реакционной массе содержится кроме обра-
зовавшегося нитроуглеводорода избыток непрореагировавшего угле-
водорода, незначительное количество азотной кислоты, серная кис-
лота и нитрозилсерная кислота (образовавшаяся за счет окислов
89-
Таблица 11
Время, протек- шее от начала слива смеси час- HNO3 в кисло- те, взятой из аппарата °/ /о Количество подан- ной в аппарат смеси (от общего количества смеси, подлежащего сливу) %
1 1,75 5
2 0,35 10
3 0,55 20
4 0,25 30
б 0,46 50
6 0,45 75
7 0,65 95
8 0,3 100
азота, внесенных в смесь с азотной кислотой; при приготовлении
«меси некоторое количество окислов азота образуется за счет окис-
лительных процессов; наконец, значительное количество окислов
азота внесено с отработанной кислотой от 2-й или 3-й нитрации,
примененной для приготовления смеси).
Как показал Батге, в такой смеси образуется соединение
типа (С6Н6)П • (NHSO5)nl (H2SO4)n2. Весьма вероятно, что это
соединение представляет собой производное диенового ряда. Такие
соединения, как известно, легко окисляются и осмоляются. Опы-
том’ установлено, что осмоление и окисление идут очень мед-
ленно при обыкновенной температуре, но ускоряются при повыше-
нии температуры. Отсюда следует, что при описываемом способе
смешения компонентов надо вести процесс слива кислотной смеси
при низких температурах.
Поэтому при нитровании, например, толуола начинают слив
смеси при температуре не выше 15—18°, а затем медленно повы-
шают температуру со скоростью 2° в час. Ведение процесса при
более высокой температуре или при большей скорости подъема
температуры дает сильные процессы окисления или осмоления,
в результате чего может получиться негодный для дальнейшего
использования мононитропродукт.
По мере сливания кислотной смеси можно постепенно, как ука-
зано, повышать температуру до 40° и затем вести слив остального
количества смеси при той же температуре (40°). Отсутствие побоч-
ных процессов в этом случае может быть объяснено умеряющим
влиянием растворителя — образовавшегося мононитротолуола —
на скорость реакции окисления и осмоления. Очевидно, что скорость
этих побочных реакций тем меньше, чем меньше концентрация то-
луола в мононитротолуольном растворе, т. е., чем больше образо-
валось мононитротолуола.
Кроме температуры на скорость процессов осмоления и окисле-
ния влияет концентрация серной кислоты; чем выше фактор нитрую-
-90
щей активности реагирующей кислотной смеси, тем выше скорость
реакции осмоления углеводорода и параллельно протекающей реак-
ции окисления. Поэтому при описываемом способе смешения компо-
нентов не следует применять кислотные смеси с фактором нитрующей
активности выше 70 — 72°/0 H2SO4 в случае нитрования толуола.
При понижении концентрации серной кислоты в смеси (фактор
нитрующей активности ниже 68,5% H2SO4) резко падает скорость
процессов осмоления, но повышается скорость окислительных ре-
акций. По этой причине не следует применять при нитровании то-
луола смеси с фактором нитрующей активности ниже 68,5% H2SO4
(такие смеси, кроме того, экономически невыгодно применять, по-
тому, что в них остается много азотной кислоты).
ь На основании сказанного становится понятным изображенный
на рис- 15 температурный режим, установленный для нитрования
толуола на основании многолетней практики.
Влияние строения углеводорода на скорость оки-
сления или осмоления. Кроме указанных фйкторов на ско-
рость осмоления и окисления влияет строение углеводорода. Наиболее
устойчивым является бензол; гомологи бензола менее стойки, причем
стойкость тем меньше, чем больше в них заместителей; при одинаковом
числе заместителей углеводород тем легче окисляется, чем длиннее
углеродная цепь заместителя. Например, этилбензол окисляется
легче, чем толуол. При одинаковом числе заместителей скорость оки-
сления зависит от относительного положения заместителей: легче всего
окисляются углеводороды с заместителями, находящимися в орто-
положении друг к другу, труднее — в параположении, и наиболее
стойкими являются углеводороды, в которых заместители располо-
жены в метаположении друг к другу. Соответственно этому легче
всего окисляется о-ксилол, труднее — р-ксилол и труднее всего
нг-ксилол. Из двух находящихся в сольвент-нафте триметилбензолов
легче всего окисляется псевдокумол:
сн3
Ч/СН8
сн3
в котором одна метильная группа стоит в ортоположении и в пара-
положении к двум другим метильным группам. Гораздо труднее
окисляется мезитилен:
СН3
сн3’х^сн3
в котором все три группы стоят в метаположении друг к другу.
91
. .. Заметим, что такое влияние положения заместителей наблю-
дается не только у углеводородов — гомологов бензола, — но и
у фенолов. Например, в то время как сульфокислота ти-крезола легко
(без значительных окислительных процессов) может быть нитрова-
нием азотной кислотой превращена в тринитро-пг-крезол, сульфо-
кислоты о- и р-крезола в тех же самых условиях окисляются нацело
в щавелевую кислоту.
Алкоксильные группы влияют сильнее, чем алкильные, на спо-
собность ароматического соединения окисляться. Например, в то
время как толуол нитруется со сравнительно незначительными по-
бочными процессами окисления и осмоления при начальной темпе-
ратуре слива 15—18°, анизол легко окисляется и осмоляется при
нитровании его уже при температуре около 0°.
Испарение углеводорода.В описываемом способе нитрова-
ния создается большая поверхность испарения углеводорода вследст-
вие загрузки в аппарат в начале операции всего количества угле-
водорода. Наибольшие потери углеводорода из-за испарения имеют
место в начале слива смеси, когда в аппарате находится чистый
углеводород. По мере слива в аппарат нитрующей смеси и соот-
ветственно по мере увеличения содержания мононитросоединения
в смеси его с углеводородом упругость паров последнего понижается
и уменьшается количество испаряющегося в единицу времени угле-
водорода.
Производительность аппарата. Важное значение имеет
для производства производительность нитратора. Эта производи-
тельность мала при описываемом способе нитрования по следующим
причинам:
1) при низкой начальной температуре слива нитрующей смеси
и малой скорости подъема температуры при дальнейшем сливе тем-
пературный перепад между реагирующей массой и охлаждающей
водой очень мал, поэтому отнятие тепла от реагирующей массы идет
очень медленно;
2) так как в начале операции нитрования в аппарате находится
незначительная масса толуола, то на ее нагревание расходуется
мало, тепла, выделяющегося при реакции;
3) относительно мала поверхность охлаждения в начальный пе-
риод операции, так как находящийся в аппарате толуол занимает
незначительный объем сравнительно с рабочим объемом аппарата.
.По этим причинам при нитровании 1 т углеводорода (например
толуола) в чугунных аппаратах старой конструкции одна операция
длится от 16 часов (зимой) до 28 (летом, при теплой охлаждаю-
щей речной воде).
2. Приливание углеводорода к кислотной смеси. В этом случае
вначале в аппарат подается все количество кислотной смеси, необ-
ходимое для нитрования назначенного на одну операцию количества
углеводорода, и затем уже постепенно сливается углеводород. Осо-
бенностью этого случая является наличие в реагирующей массе
во все время нитрования избытка азотной кислоты; наибольший
92
избыток имеется в начале слива углеводорода; избыток уменьшается
по мере слива углеводорода и достигает наименьшего значения
к концу нитрования.
При этих условиях трудно допустить возможность образования
комплекса Батге, так как углеводород при соприкосновении с кис-
лотной смесью тотчас пронитровывается в прочное мононитросоеди-
нение, которое не осмоляется и не окисляется ни кислотной
смесью, ни отработанной кислотой в условиях 1-й нитрации.
Если же допустить, что некоторая часть углеводорода успевает
образовать комплекс Батге, то этот комплекс в следующий момент,
взаимодействуя с азотной кислотой, также превратится в моно-
нитросоединение. Поэтому при таком способе смешения компонен-
тов отпадает возможность осмоления, и слив можно вести при
значительно более высоких температурах, нежели в случае слива
кислотной смеси к углеводороду. По указанным причинам и
окислительные процессы протекают здесь значительно слабее, чем
при сливе смеси к углеводороду.
Практически указанные влияния выражаются в том, что, в то
время как по первому варианту можно допускать при сливе кис-
лоты повышение температуры только со скоростью 2° в час, при
работе по второму варианту температуру можно повышать со ско-
ростью 3° и больше в 5 минут. И вместе с тем в последнем случае
качество продукта значительно выше вследствие меньшего содержа-
ния продуктов осмоления. Это легко заметить по внешнему виду про-
дукта: в то время как при работе по первому способу получается
темнокоричневый продукт, во втором случае получается светлый,
очень слабо окрашенный мононитропродукт.
Образование динитросодинения. Второй особенностью
этого варианта является то, что в первый период слива реак-
ция протекает в кислотной смеси с высоким содержанием азотной
кислоты; поэтому здесь образуется повышенное количество динитро-
соединения — до 10% и больше. При работе по первому способу
получаются десятые доли процента динитропродукта.
В тех случаях, когда получаемое мононитро соединение является
промежуточным продуктом при получении ди- или тринитро соеди-
нения, частичная нитрация до динитро соединения при 1-й нитрации
является выгодной, ибо она сокращает расход более крепких кислот
на 2-ю нитрацию.
В тех же случаях, когда получаемое мононитросоединение пред-
назначается для дестилляции с целью выделения чистых изомеров
(например, о- и р-нитротолуола из технического моно нитро толуола)
или непосредственно для восстановления (нитробензол -> анилин),
присутствие значительных количеств динитро соединения является
недопустимым: в первом случае это вызывает закупорку тарелей
дестилляционных колонн; во втором случае (например при восста-
новлении нитробензола) получаются наряду с анилином фенилен-
диамины, ухудшающие качество красок, получаемых из анилина.
Поэтому для этих последних целей нельзя готовить мо но нитро соеди-
93
*
нения путем слива при нитровании углеводорода к кислотной смеси,
а надо вести процесс по первому варианту — сливом кислотной
смеси к углеводороду.
Исп арёние углеводорода. Так как при описываемом спо-
собе смешения компонентов в реакционной массе может быть только
ничтожное количество толуола и он может испаряться только
с весьма незначительной поверхности струи углеводорода, то здесь
потери углеводорода испарением должны быть весьма незначи-
тельны. Это сказывается на некотором повышении выхода нитро-
продукта сравнительно со случаем слива смеси к углеводороду.
Производительность аппарата. Производительность
нитратора при описываемом способе смешения компонентов резко по-
вышается по сравнению с первым вариантом по следующим причинам:
1) вследствие большой скорости подъема температуры при нит-
ровании быстро достигается большой температурный перепад, по-
этому отнятие тепла от реакционного аппарата идет очень быстро;
2) так как масса подаваемой до начала нитрования кислоты пре-
восходит в 3—6 раз массу нитруемого углеводорода и, кроме того,
температура в начале операции быстро поднимается до требуемого
предела, то на нагревание реакционной массы расходуется значи-
тельное количество тепла; кроме того, кислотная смесь занимает
значительно больший объем, нежели нитруемый углеводород, соот-
ветственно чему поверхность охлаждения здесь значительно больше,
чем при работе по первому варианту.
По всем изложенным причинам в этом случае отнятие тепла от
реагирующей массы протекает быстрее, чем при работе по первому
варианту. Поэтому продолжительность одной операции нитрования
сокращается весьма значительно, соответственно с чем увеличи-
вается производительность нитратора.
3. Значение различных факторов для непрерывного процесса.
а) Для случая параллельного тока взаимо-
действующих жидкостей («п р я м о т о к а»). В этом
случае в аппарат одновременно подаются компоненты в количестве,
потребном для полного пронитровывания углеводорода, т. е. про-
цесс ведется при небольшом избытке азотной кислоты. Поэтому
здесь полностью приложимо все сказанное о случае слива углево-
дорода к нитрующей смеси. Особенностью же непрерывного про-
цесса является кратковременное пребывание реагирующих веществ
в аппарате (в десятки раз меньшее время, чем при периодическом
процессе), что также сказйвается на уменьшении окислительных
процессов. Все это вместе взятое позволяет вести непрерывный
процесс 1-й нитрации при высокой температуре (40—50° и даже
выше). При такой температуре достигается большой температурный
перепад между реагирующей массой и охлаждающей водой, бла-
годаря чему увеличивается производительность аппарата и вместе
с тем получается продукт высокого качества и с хорошим выходом.
б) .Для случая противотока. Здесь могут быть са-
мые разнообразные условия в зависимости от конструкции аппарата
94
и способа работы на нем. Мы рассмотрим идеальный случай верти-
кального аппарата, в котором реагирующие кдмпоненты движутся
в двух противоположных направлениях: кислотная смесь — сверху
вниз и нитруемый углеводород — снизу вверх (типом такого аппа-
рата может служить описанный на стр. 162 аппарат Куберскогог
однако, по нашему мнению, далеко не идеальный). В этом случае
можно вести процесс двояким способом:
1-й способ: аппарат заполняется органической фазой, в ко-
торой внизу находится чистый углеводород. По мере продвижения:
кверху повышается содержание нитро соединения; вверху же ухо-
дит пронитрованный нитропродукт. Сверху поступает кислотная
смесь. Содержание азотной кислоты в смеси постепенно снижается:
цр мере прохождения через аппарат, и при выходе из аппарата
становится минимальным, при котором практически прекращается
ее взаимодействие с углеводородом. Однако условия для реакции
нитрации, если применить выводы Кесслера, не являются наиболее
благоприятными.
2-й способ. Аппарат заполняется кислотной смесью и че-
рез нее снизу пропускается нитруемый углеводород. Очевидно
здесь создаются во всех отношениях наиболее благоприятные усло-
вия для 'течения реакции нитрации, так как в наибольшей мере
устраняются побочные процессы. Сюда полностью применимо все
сказанное для случая слива углеводорода к смеси при периоди-
ческом процессе.
С теоретической точки зрения это наиболее совершенный способ
нитрации, но его использованию мешают серьезные конструктивные
затруднения.
4. Горячий кислотооборот. В целях экономии кислот при произ-
водстве тринитро- или динитро соединений используют отработан-
ные кислоты от высших степеней нитрации для приготовления из
них смесей для низших степеней нитрации. Например, используют
отработанные кислоты (частично или целиком) на приготовление
смесей для 2-й нитрации, а отработанные кислоты от 2-й нитрации—
для приготовления смесей для 1-й нитрации. В этом случае тре-
буется предварительная очистка отработанных кислот от раство-
римых и взвешенных в них нитропродуктов, что достигается раз-
бавлением их водой и длительным отстаиванием — до 20 дней и
более. Особенно тщательное отстаивание требуется в случае приме-
нения отработанной кислоты при приготовлении смеси для 1-й нит-
рации. Это необходимо потому, что (в случае прилива кислотной
смеси к углеводороду) при нитровании углеводородов, например
толуола, нитрующей смесью, приготовленной на недостаточно от-
стоявшейся отработанной кислоте, наблюдались повышенные про-
цессы окисления и осмоления; иногда происходило настолько силь-
ное осмоление, что получался негодный для дальнейшего использо-
вания продукт. Причина такого сильного осмоления заключалась
в том, что в смеси присутствовали кислые соединения, которые легко
взаимодействуют с толуолом в присутствии отработанной кислоты,.
95
•
«одержавшей лишь очень незначительное количество азотной кис-
лоты. Процесс осмоления сопровождался повышенными окислитель-
ными реакциями с выделением значительных количеств окислов азота.
Описанные явления имели место тогда, когда к углеводороду,
например толуолу, добавляли нитрующую смесь.
Иначе протекает процесс в случае сливания углеводорода, на-
пример толуола, к нитрующей смеси.
В этом случае в смеси всегда имеется избыток азотной кислоты,
наибольший в начале слива, и поэтому в момент соприкосновения
со смесью углеводорода последний мгновенно пронитровывается
до мононитросоединения, а моно нитросоединение не взаимодей-
ствует с кислыми органическими соединениями, присутствующими
в смеси, и не окисляется азотной кислотой (т. е. не осмоляется).
Аналогичные явления наблюдаются и при 2-й нитрации. Здесь
причиной осмоления и окисления являются недонитрованные угле-
водороды, содержащиеся в количестве до 1—3% в мононитросоеди-
.нении. В этом случае также имеют место процессы окисления и осмо-
ления в первый период слива нитрующей смеси к мононитросоеди-
нению, а при обратном сливе мононитросоединения к нитрующей
смеси эти процессы не протекают.
Из сказанного понцрно, что когда нитрование ведется путем
слива смеси к нитруемому соединению, эти смеси должны изготов-
ляться на тщательно отстоявшихся отработанных кислотах. Наобо-
рот, когда сливают нитруемое соединение к кислотной смеси, то эта
смесь может изготовляться и на неотстоявшихся кислотах. Прак-
тически в последнем случае поступают так: отработанные кислоты
после отделения (сепарации) нитросоединения направляют (даже
беэ предварительного охлаждения) в нитратор, где их охлаждают;
добавляют в аппарат необходимое количество азотной кислоты
или меланжа; вновь охлаждают до необходимой начальной темпе-
ратуры слива и начинают нитрование сливом к смеси нитруемого
компонента.
Такой способ работы называется горячим к и с л от о -
оборотом.
Преимущества горячего кислотооборота:
1) отпадает необходимость предварительного длительного от-
стаивания отработанных кислот; при старом способе эти кислоты от-
стаивались, как указано, до 20 и более дней, для чего требовалось
значительное количество цистерн (или колонн для отстаивания) и
специальное -их обслуживание;
2) отпадает необходимость занимать аппаратуру в мастерской
мешки и обслуживать ее, а также не требуются специальные ци-
стерны для хранения кислотных смесей.
2-Я НИТРАЦИЯ
1. Приливание смеси к мононитросоединению. Так как в техни-
ческом мононитросоединении содержится некоторое количество угле-
водорода (1—3%), то в начале слива вследствие присутствия этого
96 ’
углеводорода могут иметь место процессы осмоления и окисления,
о которых говорилось выше. Во избежание этих нежелательных
реакций необходимо начинать слив смеси при обыкновенной тем-
пературе .
Так как после вступления (при мононитрации) нитрогруппы
ароматическое ядро очень укрепляется (nocjje этого оно менее спо-
собно к реакциям осмоления и окисления, чем исходный углеводо-
род), то процесс нитрования можно вести с гораздо более быстрым
подъемом температуры, чем при 1-й нитрации (при введении 1-й ни-
трогруппы), а именно, со скоростью 2° в 5 минут; как мы выше
видели, при 1-й нитрации и при таком же порядке слива компонен-
тов температура поднимается со скоростью только 2° в 1 час.
* И здесь постепенное повышение температуры по мере слива смеси
основано на понижении окислительных процессов, имеющем место
при увеличении количества динитро со единения, т. е. по мере умень-
шения концентрации нитруемого мононитро соединения до динитро-
соединения.
Принятый температурный режим для этого случая изображен
на рис. 18.
2. Приливание мононитросоединения к смеси. Из предыдущего
ясно, что в этом случае можно начинать слив моно нитро соединения
при более высокой температуре) и вести этот слив с большей ско-
ростью подъема температуры, не боясь осмоления или окисления
углеводорода, как это имеет место при сливе кислотной смеси к угле-
водороду.
3. Болтушка. Болтушкой называют на наших заводах процесс
экстрагирования растворенных в отработанной кислоте нитропро-
дуктов. Одновременно здесь используется содержащаяся в отрабо-
танной кислоте азотная кислота, которая взаимодействует при
болтушке с недонитрованным углеводородом или мононитросоеди-
нением.
Так как для экстрагирования применяют обычно мононитросо-
единение (например, мононитротолуол при производстве ди- или
тринитротолуола) или хлорбензол (при производстве динитрохлор-
бензола), то здесь применимо все сказанное о нитровании углево-
дорода или мононитросоединения кислотной смесью, приготовлен-
ной на отработанной кислоте; а именно, нельзя приливать отра-
ботанную кислоту к мононитросоединению (всегда содержащему
некоторое количество недонитрованного углеводорода) или хлор-
бензолу во избежание осмоления и окисления. Обязательно надо
сливать мононитро соединение или хлорбензол к отработанной кис-
лоте.
3-я НИТРАЦИЯ
1. Условия процесса. При установлении условий процесса
3-й нитрации надо исходить из следующих соображений.
Вследствие тормозящего реакцию нитрации влияния двух ни-
трогрупп необходимо не только увеличение фактора активности
Химия—13—7 97
*
Кислотной смеси, но и повышенная температура при смешении ди-
нитротолуола с кислотной смесью. В'то время как при 1-й и 2-й
нитрациях углеводородов можно начать слив при температуре
около 15°, реакция нитрации динитротолуола в тринитротолуол
(а равно некоторых других динитросоединений) практически не
идет при обыкновенной температуре. Взаимодействие между кис-
лотной смесью и динитротолуолом при температуре 58—60° ста-
новится настолько значительным, что приходится вести охлажде-
ние, чтобы удержать температуру реакционной массы на заданном
уровне. При этом, однако, значительное количество добавляемого
компонента не успевает прореагировать, и потому происходит его
накопление. Когда же по окончании смешения компонентов подни-
мают температуру для выдержки, то по достижении 70° начинается
энергичное взаимодействие, в результате которого иногда начи-
нается катастрофический подъем температуры, и его не удается
остановить даже максимальным открыванием кранов для охлажде-
ния. Температура поднимается до 160°, происходят сильные окис-
лительные процессы, сопровождающиеся выделением газообраз-
ных продуктов. Из-за этого наблюдается кажущееся кипение реак-
ционной массы, ее подъем и в результате выливание из аппарата.
При смешении вылившейся массы с водой (всегда стоящей на полу,
поливаемом водой согласно установленным правилам) происходит
разложение нитрозилсерной кислоты с обильным выделением окис-
лов азота, делающих недопустимым пребывание в помещении без
противогаза. Помимо этого такой подъем, температуры не может
быть допущен потому, что нет уверенности, что при некоторых
случайно сложившихся неизвестных нам условиях бурно протекаю-
щий при такой высокой температуре процесс не может перейти
во взрыв.
Поэтому необходимо вести процесс 3-й нитрации таким образом,
чтобы температура реакционной массы ни в один момент не могла
превысить заданною предельную температуру, признаваемую без-
опасной.
Это может быть достигнуто двумя путями:
1. Ведением слива компонентов с такой скоростью и при такой
температуре, чтобы смешанные компоненты успели в период слива
почти нацело прореагировать. Опытным путем установлено, что
такое взаимодействие имеет место при температуре 72°, при кото-
рой, следовательно, не происходит накапливания подаваемого ком-
понента. Слив кислотной смеси производится здесь в пределах тем-
пературного интервала 72—78°. Верхний предел установлен с целью
ограничения окислительных реакций, имеющих место при сливе
компонентов. Реакции зти тем больше, чем выше температура. По
окончании слива смеси делают выдержку при 110—115° для до-
нитровывания не успевшего прореагировать динитротолуола. Та-
кая выдержка необходима потому, что к концу нитрации понижается
концентрация динитротолуола в образовавшемся тринитротолуоле
(действующему аналогично тормозящему влиянию нитробензола на
98
реакцию нитрации бензола в опытах Кесслера или динитробензола на
реакцию нитрации нитробензола в опытах Гезерингтона и Массона).
2. В целях рационализации процесса 3-й нитрации может быть
желательным вести процесс при более низкой температуре и слив
компонентов производить с большей скоростью, чем зто установ-
лено по предыдущему режиму.
Это может быть допущено при условии, что при полном, притом
мгновенном, взаимодействии динитротолуола и кислотной смеси,
имеющихся в любой момент в аппарате, температура (теоретически
подсчитанная) реакционной смеси не превысит определенного за-
данного предела. В качестве такого предела у нас принята темпе-
ратура 120°.
4 2. Случаи нитрования углеводородов до ди- и тринитросоединений
в одну фазу. Иногда применяют (или применяли раньше) нитрование
в одну фазу до ди- и тринитросоединения, например, нитрование то-
луола в динитротолуол или ксилола в ди- или тринитроксилол,
сольвент-нафты до ди- или тринитросольвент-нафты и т. п.
На основании ранее сказанного ясно, что если приливать кислот-
ную смеСь к нитруемому углеводороду, то вследствие высокой кон-
центрации серной кислоты в смеси при недостатке в ней (в реаги-
рующей смеси) азотной кислоты будет иметь место сильное осмоле-
ние. Это явление не будет иметь места при обратном сливе.
Поэтому нитрование углеводорода до ди- или тринитросоедине-
ния в одну фазу можно и должно вести только сливом углеводорода
к смеси.
ОКИСЛИТЕЛЬНЫЕ ПРОЦЕССЫ ПРИ НИТРАЦИИ
1. Окислительные процессы при 1-й, 2-й и 3-й нитрациях. При
1-й и 2-й нитрациях окислительные процессы весьма незначительны.
Это видно из того, что при нитровании бензола или толуола можно
получить выход нитробензола или нитротолуола 98—99% от тео-
. рии; выход динитробензола или динитротолуола по мононитросоеди-
нению составляет 96—97% от -теории.
Но гораздо более значительные окислительные процессы имеют
место при нитровании динитро со единения до тринитросоединенйя.
Они протекают со значительным газообразованием. Уже Гайзерман 1
заметил, что при нитровании динитротолуола идет значительное
газообразование, которое очень умеренно при нитровании чистого
динитротолуола, однако, сильнее при нитровании технического
динитротолуола.
По Нидереру 2, при нитровании технического динитротолуола
в тринитротолуол при температурах ниже 75° газообразование не-
значительно; оно заметно увеличивается около 76° и быстро возра-
1 Z. Angew. Chem. 4, 611, 1891.
2 Z. Schiess u. Sprengst, 27, 217 (1932).
99
»
стает при повышении температуры. Образующиеся при этом газы
содержат СО и СО2. Количество *и соотношение этих газов зави-
сят от температуры и продолжительности реакции. При постоянной
температуре образование СО и СО2 уменьшается с течением реак-
ции х. При этом количество образующейся окиси углерода убывает
заметно быстрее, чем двуокиси углерода, и количество образовав-
шейся окиси углерода тем больше, чем выше температура в начале
слива.
Сапожников подтвердил выводы Нидерера и, кроме того, уста-
новил, что при нитровании динитротолуола
в закрытом аппарате
потери азотной кис-
лоты в форме пря-
мого испарения со-
ставляют около 25—
30% от общей поте-
ри при нитрации.
Остальная часть по-
терь азотной кисло-
ты приходится на
окислительные про-
цессы, идущие па-
раллельно нитрации.
2. Зависимость
скорости окислитель-
ных процессов от фак-
тора нитрующей ак-
тивности. Автор на-
шел, что при ни-
тровании моно- или
Рис. 9. Зависимость окислительных процессов
от состава смеси.
динитротолуола, ксилола, моно- или динитроксилола сливом
кислотной смеси к нитруемому соединению скорость окис-
лительных процессов уменьшается с увеличением фактора нитрую-
щей активности отработанной кислоты (а следовательно, и исход-
ной смеси). Соответствующая зависимость для случая нитрования
динитроксилола дана на рис. 9. На оси абсцисс отложены моле-
кулярные проценты H2SO4 в смеси (другое выражение фактора
нитрующей активности); на оси ординат — количество окислов
азота N,O3, образовавшихся при нитровании и отнесенных к 1 г]мол
динитроксилола.
1 Это находится в полном соответствии с изложенным выше положением,
что скорость окислительных процессов уменьшается с уменьшением концентра-
ции нитруемого соединения, для которого в данном случае растворителем яв-
ляется продукт реакции — тринитротолуол. Вместе с тем отметим, что тринитро-
толуол является значительно более стойким соединением, чем динитротолуол,
и окисляется гораздо меньше последнего.
100 .
ГЛАВА VI
ОБЩАЯ ХАРАКТЕРИСТИКА НИТРОСОЕДИНЕНИИ
АРОМАТИЧЕСКОГО РЯДА
1. Физические свойства. Большинство моно нитросоединений аро-
матического ряда представляет собой при обыкновенной температуре
твердые вещества, как это видно из табл. 12.
Таблица 12
Наименование вещества
Температура плавления
°C
Нитробензол....................
o-Нитро толуол
лабильная форма ................
стабильная форма .........
ш-Нитротолуол .................
р-Нитротолуол.......... . . . .
2-Нитро-т-ксилол ..............
4-Нитро-ш-ксилол ..............
5-Цитро-т-кСилол ..............
2-Нитро-р-ксилол...............
З-Нитро-о-ксилол...............
4-Нитро-о-ксилол...............
о-Нитрохлорбензол..............
р-Нитрохлорбензол •............
а-Нитронафталин................
о-Нитрофенол...................
ш-Нитрофенол ..................
р-Нитрофеиол ..................
о-Нитроаиилин..................
ш-Нитроанилин .................
р-Нитроаиилин .................
Феиилиитроамин .... ...........
р-Нитрометилаиилин.............
о-Нитрометилаиилии.............
т-Нитрометиланилин.............
Феиилметилнитроамин ...........
о-Нитродиметилаиилии...........
ш-Нитродиметилаиилии...........
р-Нитродиметилаиилин...........
5,5
—3.17 (температура затвердевания)
—9.27 » »
16,1 » »
51.6 » »
13
2
74
Жидкость при обыкновенной температуре
16
30
32,5
83
56,5
45
96
114
71
114
147
46
150—151
34—35
65—66
39
—20
60—61
161—162
101
Большинство мононитросоединений перегоняется в вакууме
с очень незначительным осмолением, чем пользуются в технике
для получения некоторых чистых изомеров (о-нитротолуола, о-нитро-
хлорбензола и др.). Некоторые о-изомеры, например, о-нитрофенол,
о-нитроанилин, легко перегоняются с водяным паром, тогда как
соответствующие ^-изомеры перегоняются очень слабо; этим поль-
зуются в лабораторной практике для получения некоторых чистых
изомеров, а иногда пользуются и в заводской практике, например
для получения о-нитрофенола.
Динитросоединения перегоняются в вакууме, а также с водя-
ным паром, но значительно труднее мононитросоединений.
Тринитросоединения не перегоняются в вакууме, а некоторые
из них, хотя и с большим трудом, перегоняются с водяными парами.
Последнее имеет практическое значение при очистке отработанных
кислот от нитропродуктов при денитрации.
2. Химические свойства. Ароматические нитро соединения пред-
ставляют собой третичные нитро со единения, т. е. в них нитро-
группы соединены с углеродными атомами, не связанными с ато-
мами водорода; поэтому они в отличие от первичных или вторичных
алифатических нитросоединений не могут образовать щелочных
солей путем перегруппировки в обладающие кислотными свой-
ствами изо нитро соединения. Однако ароматические нитросоединения
способны присоединять как алкоголяты щелочных металлов, так
и щелочи. При этом происходит превращение бензоидной формы
в хиноидную и образуются окрашенные соли, строение которых
может быть выражено следующей формулой:
Н/ХОС2Н5
Эти соли характеризуются резко повышенной чувствительностью
к механическим воздействиям и нагреванию по сравнению с ней-
тральными нитросоединениями А
Нитросоединения обладают сильно выраженной способностью
к образованию продуктов присоединения. Эта способность усили-
вается при повышении числа нитрогрупп в ядре. Так, например,
симметричный тринитробензол соединяется как с ненасыщенными,
так и с ароматическими углеводородами, например, со стильбеном
1 Более подробные сведения о металлических производных ароматических
нитросоединений изложены в главе «Свойства тротила» (см. стр. 131—135).
102
и гексаметилбензолом, причем в результате присоединения полу-
чаются темноокрашенные двойные молекулы:
CeH3(NO2)3 • СвН5СН = СН • CeH5 C6H3(NO2)3 • Ce(CH3)e
тринитробензолстильбен тринитробензолгексаметилбензол
Нитрогруппа ароматических соединений способна активировать
как замещайщие группы бензольного ядра, находящиеся по отно-
шению к ней в орто- и параположениях, так и водородные атомы,
находящиеся в этих же положениях. Характер этого активирую-
щего действия может быть различный. Рассмотрим несколько при-
меров.
х» а) Активирующее действие нитрогруппы на другую нитро группу,
находящуюся в орто- или параположении к первой, выражается
в ослаблении связи этой нитрогруппы с ядром. Поэтому она легко
может быть замещена другими группами. Так, например, нитро-
бензол не реагирует при нагревании ни с аммиаком, ни с алкоголя-
том натрия; в о-динитробензоле и его ^-изомере (но не в т-изомере)
нитрогруппа может быть легко заменена аминогруппой или алк-
оксильной группой:
,nh2 zno2
\ + NH3 /----х +CH3ONa
^no2 < \no2----------------->
OCH3
zno2
Аналогично в этих изомерах замещается та же нитро группа
(подобно всем «подвижным» нитрогруппам) на сульфогруппу при
взаимодействии их с водным раствором сульфита натрия:
NO, SO3Na
\ " + Na,SOa /----ч
^NO2--------> / \NO2 + NaNO2
Это активирующее действие нитрогруппы усиливается при на-
коплении числа нитрогрупп в ядре. Соответственно этому не только
облегчается замещение одной из нитрогрупп в нитросоединениях
бензола, стоящих в орто- или параположении друг к другу в при-
сутствии третьей нитро группы (в двух несимметричных тринитро-
бензолах), но и в симметричном тринитробензоле проявляется спо-
собность одной из нитрогрупп к замещению, например, при кипя-
чении его с раствором алкоголята:
NO,
NO,
/NO,
У + NaNO2
ОСН3
Активирующее действие нитрогруппы в отношении других ни-
тро групп уменьшается со вступлением в ядро электроцоло-
103
жительных заместителей и увеличивается со вступлением
в ядро электроо трицательных заместителей. Сооб-
разно с этим действие это уменьшается при переходе слева направо
в ряду Мартинсена (стр. 69) и увеличивается для перехода в обрат-
ном направлении.
б) Активирующее действие нитрогруппы на хлор (выражающееся
также в ослаблении связи хлора с ядром) легко проследить на при-
мере хлорбензола и его нитропроизводных. В то время как в хлор-
бензоле замена хлора на гидроксил при действии едкого натра мо-
жет быть достигнута только при высокой температуре с применением
катализатора [например, по американскому патенту № 1868140
(1932) при нагревании хлорбензола с едким натром в автоклаве
при температуре 300—375° в присутствии меди], эта замена заметно
облегчена в нитрохлорбензоле и очень легко протекает в динитро-
хлорбензоле. Присутствие двух нитрогрупп уже настолько активи-
рует Cl-заместитель, что замена хлора легко идет, притом почти
количественно, при обработке динитрохлорбензола при 100° слабым
водным раствором едкого натра при избытке последнего в 2—3%
от теоретического количества. При этом можно установить общее
положение, что подвижность атома хлора (как и вообще всех гало-
идных атомов) усиливается под влиянием нитрогрупп, находящихся
по отношению к нему в орто- или параположениях.
в) С точки зрения пояснения процессов, протекающих при осмо-
лении, часто наблюдающемся (и особенно легко воспроизводимом
при .специально подобранных условиях) при нитровании аромати-
ческих соединений, интересно отметить реакцию взаимодействия
бензальдегида с динитротолуолом. В то время как получение стиль-
бена путем конденсации толуола с бензальдегидом не удается, эта
конденсация проходит легко с о-о- и о-р-динитротолуолом, так как
водородные атомы метильной группы обладают в этих соединениях
большой подвижностью и способны вступать в реакцию с бензальде-
гидом. В качестве конденсирующего средства здесь применяют
алкоголят натрия:
NO2
O2n/ ^СНз + ОСн/ \--------------->
/NO2
----->O2N/' ‘>сн=сн/ у + н,о
г) Прямое введение гидроксильной группы в молекулу бензола
или нитробензола при помощи процессов окисления очень трудно.
Оно хорошо удается с тп-динитробензолом и еще лучше с симметрич-
ным тринитробензолом, благодаря активирующему влиянию нитро-
групп на водородный атом, стоящий в орто- или параположении
к ним:
104
NO.,
^OH
NO3
no2 no.,
\ окисление /-------\
/----------->O2N/ Noh
no2 no2
Поэтому тринитробензол легко и нацело окисляется в тринитрофе-
нЬл при действии железосинеродистым калием 1 K3Fe(CN)6.
3. Чувствительность к удару. Химическое строение вещества и
характер заместителей влияют на взрывчатые свойства. Ясно, что
количество внутренней энергии возрастает с увеличением числа
кислородсодержащих групп (нитро-, или сульфогрупп, или других)-
и уменьшается при вступлении групп, не содержащих кислорода
(алкильные, аминогруппы и др.).
Влияние природы и числа заместителей на чувствительность
нитро соединения к удару впервые изучено Велером и Венцельбер-
гом 2. Хотя выводы этих авторов не могут быть признаны оконча-
тельными, однако, они несомненно правильно наметили общие за-
кономерности в этом вопросе.
Чувствительность к удару у ароматическйх нитро со единений
возрастает с увеличением числа заместителей в ядре. При этом
влияние группы СН3 слабее влияния других групп (ОН, С1, Вг)..
Повышение чувствительности к удару при введении заместите-
лей сйидетельствует об ослаблении устойчивости бензольного ядра.
Ослаблением этой устойчивости облегчается распад молекулы.
Обнаруживается замечательный факт, что бризантность, по
Гессу, и чувствительность к детонации располагаются в порядке,
обратном чувствительности к удару. Наиболее бризантен тринитро-
ббнзол, его же чувствительность к детонации наибольшая; и вместе
с тем для его взрыва требуется максимальная работа удара при испы-
тании на копре. С другой стороны, тринитромезитилен, наименее
бризантный, наиболее чувствителен к удару (работа удара прибли-
зительно равна той, которая требуется для тринитроксилола, тогда
как более бризантный тротил является менее чувствительным к удару,
чем ксилил или тринитромезитилен).
Молекула тринитробензола, следовательно, является наиболее
устойчивой по отношению к удару; введение метильной группы (три-
нитротолуол) уменьшает прочность молекулы; поэтому энергия,
необходимая для разрушения молекулы тринитротолуола, меньше,
чем для тринитробензола. Введение второй метильной группы (три-
1 Н е р р, Lieb. Ann., 215, 344, 1882.
2W6hlern Wenzelberg, Z. Angew; Chem. 46,173 1933; Mem-
ArtiU- fr., 14, 467, 1935.
10b
нитроксилол) вызывает новое ослабление устойчивости бензольного
ядра, соответственно требуется еще меньшая работа удара. Введение
третьей метильной группы, по данным Велера и Венцельберга, не
усиливает этого эффекта Ч
Влияние заместителей на чувствительность к удару видно из
опытных данных (табл. 13), в которых мерилом чувствительности
к УДаРУ принята работа удара в кг/см2.
Таблица 13
Взрывчатое вещество
Число заме-
стителей
Работа удара
кг!смг
Влияние метильной группы
Динитробензол 2 19,5
Динитротолуол 3 18,9
Динитроксилол • • 4 14,6
Динитромезитилен 5 13,8
Тринитробензол 3 12,1
Тринитротолуол 4 11,4
Тринитроксилол- ... 5 5,7
Тринитромезитилен 6 5,9
Влияние гидроксильной группы
Динитробензол ..." 2 19,5
Динитрофенол 3 12,7
Динитрорезорцин 4 10,3
Тринитробензол 3 12,1
Тринитрофенол 4 8,2
Тринитромезитилен 5 4,0
Влияние хлора
.Динитробензол...........................
Хлординитробензол........................
Дихлординитробензол......................
Тринитробензол...........................
Тринитрохлорбензол.......................
Влияние нитрогруппы
.Динитробензол..........................I
Тринитробензол..........................I
Динитротолуол...........................|
Тринитротолуол..........................।
Динитроксилол ...........................
Динитромезитилен ........................
Тринитроксилол ..........................
Тринитромезитилен........................
Динитрофенол ............ , ...........
Тринитрофенол ...........................
Динитро резорцин.........................
Тринитрорезорцин . ......................
Динитрохлорбензол .......................
Тринитрохлорбензол.......................
19,5
12,0
10,2
12,1
11,3
19,5
12,1
18,9
11,4
14,6
13.8
5,7
5,9
12,7
8.2
10,3
4,0
12.0
11,3
1 Повидимому, чувствительность к удару тринитромезитилена немного
больше, чем у тринитроксилола, хотя ее труднее обнаружить в условиях опыта
названных авторов. Но эту разницу легко установить для смесей с аммиачной
селитрой, показывающих, что чувствительность таких смесей с тринитромези-
тиленом выше, чем с кСилилОм.
106
Производные бензола независимо от вида заместителей при оди-
наковом числе их требуют приблизительно одинаковой работы,
которая с увеличением числа заместителей уменьшается. Это видно
из табл. 14.
Таблица 14
Взрывчатое вещество j Число заме- стителей Работа удара кг/см*
Дивитрохлорбензол 3 12.0
Дрнитроанилин 3 12,6
Динитрофенол 3 12,7
Динитроанизол 3 13,0
Тринитробензол 3 12,1
Динитрохлорбензол 4 10,2
Динитрорезорцин 4 10,3
'Гринитроанилин 4 10,4
Тринитротолуол . . . 4 11,4
Из приведенных данных видно, что первая метильная группа
оказывает небольшое влияние на чувствительность к удару, вто-
рая — более сильное, третья — опять небольшое. Первая гидро-
ксильная группа сильно повышает чувствительность к удару анало-
гично третьей нитрогруппе; вторая гидроксильная группа повышает
эту чувствительность также довольно сильно. Первый хлорзамести-
тель повышает чувствительность динитробензола аналогично нитро-
группе, второй хлорзаместитель — еще сильнее. Одинаково дей-
ствуют группы NO2, NH, и ОСН3. Влияние метильной группы
меньше других, что особенно заметно в случае динитро- и тринитро-
крезолов при сравнении их с соответствующими нитрофенолами.
Были проделаны опыты с целью выяснения влияния относитель-
ного положения заместителей в бензольном ядре на чувствительность
к удару. Однако при этом существенного различия найдено не было.
4. Общий характер токсических свойств ароматических нитро-
соединений. Ароматические нитросоединения действуют отравляю-
щим образом на центральную нервную систему и на кровь. На
центральную нервную систему действуют сначала раздражающе:
вызывают головные боли, падение температуры, временами судороги,
(иногда сводит конечности), расстройство чувствительной сферы,
расстройство зрения; в дальнейшем вызывают расстройство в сер-
дечно-сосудистой системе: кровяное давление падает, кровеносные
сосуды расширяются, образуются застойные явления во внутрен-
них органах, главным образом, в печени и легких. Наблюдаются
кровоизлияния под кожу, поверхностное разрушение эпидермиса.
Одновременно наблюдается изменение в картине крови.
Ароматические нитросоединения тормозяще действуют на крове-
творные органы, вследствие чего развивается анемия со значитель-
107
«
яым разрушением красных кровяных шариков и с явлением дегене-
рации белых. Вследствие этого понижается поступление кислорода
в организм и правильное его распределение. В результате нару-
шается деятельность сердца, нервно-мозговые функции и дыхание.
Степень действия отдельных нитропроиз-
водных неодинакова. В общем можно сказать, что то-
ксичность уменьшается с повышением числа нитро групп, а при одном
и том же числе нитрогрупп отравляющее действие уменьшается с по-
вышением числа метильных групп.
Ядовитее всех нитропроизводные бензола, среди них в особен-
ности динитробензол.
Динитробензол действует раздражающе на центральную нервную
систему: появляется усталость, головные боли, цианоз (синюшное
окрашивание слизистых оболочек и конечностей), неспокойный сон,
аппетит теряется. При тяжелых отравлениях наблюдается затруднен-
ное дыхание, конвульсии, расстройство зрения, расстройство же-
лудка, желтуха. Такие тяжелые отравления в большинстве смер-
тельны.
Отравление нитротолуолом вызывает лихорадочное состояние,
усталость, одышку, опухание печени, сыпь на коже.
Динитрохлорбензол и гексил, попадая на кожу, вызывают сыпь,
а иногда и язвы.
Характерным признаком отравления ароматическими нитросо-
единениями является цианоз (синюшная окраска) и желтушная
окраска роговицы глаз, обусловленные преимущественно разру-
шением красных кровяных телец.
5. Пути отравления. Отравление идет, главным образом, через
кожу и дыхательные органы. Отравление нитросоединениями идет
через кожу вследствие резорбции (всасывания), даже тогда, когда
кожа не повреждена, а только загрязнена. Так как яд поступает
медленно, то отравление имеет хронический характер. Отравление
через дыхательные пути происходит при вдыхании паров или пыли
(например, при сушке, просеивании и упаковке).
В практических условиях работы большое значение имеют физи-
ческие свойства веществ, например, летучесть; поэтому чаще всего
имеет место отравление легко летучими нитросоединениями, напри-
мер, при дестилляции мононитротолуола.
Вредное влияние нитро со единений на организм зависит в неко-
торой степени от количества проникшего в организм вещества, а еще
в большей мере от сопротивляемости данного организма.
Особенно вредно влияет отравление на лиц, систематически
пьющих спиртные напитки. Малоустойчивы молодые, плохо упи-
танные организмы, еще менее устойчивы женщины.
Иммунитета за редкими исключениями не наблюдается; наобо-
рот, лица, перенесшие отравление, делаются менее устойчивыми.
6. Теория токсического действия ароматических нитросоедине-
ний. По Гейбнеру, как у нитро-, так и у аминосоединений на мозг
действует фенильная группа.
108
Действие на кровь объясняется следующим образом. В орга-
низме человека образуется, например, из нитробензола /,-нитрозо-
фенол. Последний окисляет гемоглобин в метгемоглобин \ а самый
р-нитрозофенол при этом благодаря восстановлению превращается
в хинонимин, вызывающий также образование метгемоглобина. Из
анилина в организме образуется сначала р-аминофенол, окисляемый
оксигемоглобином в хинонимин; последний, как мы видели, является
причиной образования метгемоглобина.
Аналогичные процессы происходят при действии всех нитро- и
аминосоединений.
Позднее было высказано предположение, что активным проме-
жуточным продуктом при действии нитро- и аминосоединений
является p-фенилгидроксиламин, который под влиянием оксигемо-
глобина окисляется в азоксибензол. При этом кислород активи-
руется и превращает гемоглобин в метгемоглобин. Различная чув-
ствительность у людей зависит вероятно также и от весьма сильных
индивидуальных колебаний газообмена. На основании этой теории,
например, различие в токсичности динитробензолов объясняется
различной скоростью, с которой они подвергаются восстановлению
в организме. Точно так же сравнительно незначительная токсич-
ность тринитробензола и особенно симметричного тринитротолуола
объясняется трудностью восстановления их в тканях.
7. Терапия. При слабом отравлении необходима перемена
одежды, ванна, тщательное мытье волос. При острых отравлениях
(но не очень тяжелых) — вдыхание кислорода.
В тяжелых случаях отравления — кровопускание, вливание фи-
зиологического раствора, искусственное дыхание, клизма из физио-
логического раствора.
При упадке сердечной деятельности рекомендуется холод на
сердце, раздражение кожи, переливание крови, возбуждающие
средства (камфора, кофеин).
При хроническом отравлении: свежий воздух, хорошее пита-
ние, покой и средства, усиливающие кровообращение (гимнастика,
Душ)-
8. Прогноз. При отравлении нитросоединениями выздоровление
наступает медленно.
1 Гемоглобин—красное красящее вещество крови. Он легко поглощает
кислород, главным образом, в легких, причем переходит в оксигемоглобин.
Последний, в свою очередь, легко отдает кислород клеткам тканей при их омы-
вании кровью, причем вновь регенерируется гемоглобин. Это процесс посто-
янно повторяется в организме, обеспечивая перенос кислорода тканям из
воздуха.
При действии некоторых веществ, например, р - нитрозофенола, хиноними-
на и Др-, образуется более прочное соединение гемоглобина с кислородом,
метгемоглобин, не способный отдавать кислород тканям, и из которого лишь
медленно регенерируется опять гемоглобин. Поэтому при образовании мет-
гемоглобина соответствующее количество гемоглобина временно прекращает
Свои функции переносчика кислорода, и уменьшается питание организма
кровью; это вызывает кислородное голодание, свидетельствующее об отравле-
нии организма.
109
9. Применение нитросоединений. Широкое практическое значе-
ние приобрели только ароматические нитро соединения; причина
этого заключается, главным образом, в легкости их получения и
в доступности сырья для их изготовления. В то время как арома-
тические нитро со единения легко получаются при действии серно-
азотных смесей на углеводороды или их производные, нитросоеди-
нения жирного ряда получаются с большим трудом, притом срав-
нительно дорогими методами.
Наряду с этим большое значение для того или иного практи-
ческого применения имеют и свойства соединений. Ароматические
нитро со единения характеризуются значительной физической и хи-
мической стойкостью при большой силе взрыва, что делает их
весьма пригодными для применения в качестве взрывчатых веществ
для снаряжения боеприпасов и для других целей. Кроме того, они
служат и для изготовления промежуточных продуктов в фабрика-
ции самых разнообразных красителей. Нитросоединения жирного
ряда характеризуются пониженной химической и физической стой-
костью (она значительно ниже, чем у ароматических соединений);
вместе с тем они не находят применения для изготовления кра-
сителей.
Большой интерес представляет собой тетранитрометан; он обра-
зует очень сильные взрывчатые смеси с жидкими горючими; однако
вследствие его дороговизны, большой летучести и ядовитости он
не находит себе практического применения.
В настоящее время из числа неароматических нитросоединений
применяется только гексоген (тринитроциклотриметилентриамин),
повидимому, гетероциклическое соединение, но его структурная
формула не может быть признана окончательно установленной.
Ароматические моно нитро соединения применяются в ограничен-
ных количествах в качестве примесей ко взрывчатым тринитросо-
единениям (например, мононитронафталин в сплаве с пикриновой
кислотой для флегматизации последней). Нитробензол применялся
раньше в значительных количествах в смесях типа река-рок или
белого пороха Винера (с бертолетовой солью). Моно нитро со едине-
ния не обладают взрывчатыми свойствами и должны рассматри-
ваться как легко сгорающие примеси в смесях с окислителями, на-
пример в хлоратитах. Горючесть мононитросоединений может быть
повышена введением сульфогруппы (например, предложенные
Фойгтом щелочные соли нцтрокрезол-, нитрофенол- и нитронафталин-
сульфокислот в смеси с калиевой или натриевой селитрой образуют
смеси, взрывающиеся непосредственно от бикфордова шнура без
капсюля).
Значительные количества мононитросоединений (нитробензол,
о-и />-нитротолуол, изомеры хлорнитробензола и т. п.) производятся
для изготовления полупродуктов анилокрасочного производства.
Динитросоединения обладают взрывчатыми свойствами, однако,
их восприимчивость к детонации мала, вследствие чего они требуют
сильного детонатора. Это твердые вещества; они применяются в зна-
110
чительных количествах во взрывчатых смесях. Вследствие указан-
ной малой восприимчивости к детонации и невзрывчатости их при
пожаре динитросоединения разрешается перевозить по железной
дороге в некоторых странах наравне с невзрывчатыми грузами.
И только у тринитро соединений, в которых в одном бензольном
ядре содержится три нитрогруппы, взрывчатые свойства столь
ясно выражены, что они могут применяться непосредственно в ка-
честве взрывчатых веществ.
В тетранитросоединениях (тетранитроанилин, тетранитрофенол:
и т. п.) нитро группы связаны менее прочно с ароматическим ядром;
они менее стойки при высоких температурах, 4-я нитрогруппа
цргко отщепляется, иногда при обыкновенной температуре.
Тетранитросоединения более чувствительны к механическим воз-
действиям, чем тринитросоединения. По этим причинам тетрани-
тросоединения за очень редким исключением не находят себе
применения.
ГЛАВА VII
КЛАССИФИКАЦИЯ СПОСОБОВ ПРОИЗВОДСТВА НИТРО-
СОЕДИНЕНИЙ
Каждый способ производства полинитросоединений характери-
зуется следующими основными признаками: 1) числом стадий нитро-
вания; 2) характером кислотооборота; 3) цикличностью процесса.
На основе этих признаков примем следующую классификацию
способов производства нитросоединений:
1. По числу стадий нитрования: 1) способ в одну фазу; 2) способ
в две фазы через мононитросоединение; 3)способ в две фазы через дини-
тросоединение; 4) способ в три фазы; 5) способ в четыре и более фаз.
1. При ведении процесса в одну фазу реакция идет по урав-
нению (на примере нитрования толуола):
CeH3CH3 + 3HNO3 -> CeH2CH3(NO2)3 + ЗН2О
В этом случае вся отработанная кислота получается состава,
необходимого для вступления 3-й нитрогруппы в ядро, т. е. ма-
ксимальной концентрации. Расход кислот наибольший; причем для
составления кислотных смесей требуются исходные кислоты наи-
высшей концентрации (олеум и концентрированная азотная кис-
лота). В большом количестве концентрированных отработанных
кислот остается в растворе значительное количество нитросоедине-
ния, вследствие чего понижается выход конечного продукта.
Поэтому этот способ экономически невыгоден. В частности, для
производства тротила он никогда не применялся.
2. При способе в две фазы через мононитросоединение протекают
следующие две реакции (на примере получения тринитротолуола):
CeH5CH3 + HNO3 CeH4CH3NO2 + Н2О
CeH4CH3NO2 + 2HNO3 ->CeH2CH3(NO2)3 + 2Н2О
В этом способе при введении 1-й нитрогруппы пользуются наи-
более слабой кислотной смесью (например, при получении моно-
нитротолуола или мононитроксилола фактор нитрующей актив-
ности равен 69—71), а крепкая кислота, отвечающая смеси 3-й нит-
рации (фактор нитрующей активности равен 93—94), расходуется
на введение только двух нитрогрупп в бензольное ядро. Поэтому
112
этот способ выгоднее предыдущего; он применялся раньше, но
в ограниченных размерах, на некоторых заграничных заводах.
3. Выгоднее вести производство в две фазы через динитросоедине-
ние, например, для случая нитрования толуола в тринитротолуол:
CeH5CH3 + 2HNO3 ->CeH3CH3(NO2)2 + 2Н2О
CeH3CH3(NO2)2 + HNO3 -> CeH2CH3(NO2)8 + H20
В этом случае крепкая смесь с фактором нитрующей активности,
равным 93—94% H2SO4, расходуется только для введения одной
нитрогруппы во 2-й фазе, а введение двух первых нитрогрупп
производится на менее крепкой смеси с фактором нитрующей актив-
ности, равным 80—82% H2SO4.
Этот способ применялся прежде широко в практике некоторых
заграничных заводов.
4. Еще выгоднее способ в три фазы, при котором крепкая смесь
расходуется на введение только 3-й нитрогруппы в ядро. Менее
крепкая смесь расходуется на введение только 2-й нитрогруппы,
и, наконец, наименее крепкая, следовательно, наиболее дешевая
смесь расходуется на введение 1-й нитрогруппы в ядро.
В этом способе расход кислот наименьший. На приготовление
смесей для 1-й и 2-й фаз могут применяться более слабые исход-
ные кислоты. Наименьшее количество нитро соединений остается
в растворенном виде в отработанных кислотах, а потому этот спо-
соб является экономически наиболее выгодным из изложенных че-
тырех вариантов.
Этот способ преимущественно применялся в производстве три-
нитротолуола.
5. При дальнейшем увеличении числа стадий нитрования расход
крепких кислот еще больше снижается. Однако с увеличением числа
стадий усложняется оборудование завода и увеличивается расход
рабочей силы на обслуживание этого оборудования, что связано
с удорожанием строительства и с увеличением эксплоатационных
расходов. Но при удачном оформлении технологического процесса
и удачной конструкции аппаратов число стадий нитрования может
быть с выгодой значительно увеличено. Примером удачного реше-
ния проблемы может служить английская многокамерная система,
•описанная ниже (стр. 162), в которой процесс 2-й и 3-й нитра-
ций разбит на семь стадий, в результате чего не только сокращается
расход кислот, но оказывается Возможным обойтись без применения
олеума. Кроме того, при этом способе расход рабочей силы не больше,
л даже меньше, чем при нитровании в три стадии.
При каждом из описанных способов процесс можно вести со сле-
дующими вариациями слива компонентов:
а) слив кислотной смеси к нитруемому соединению;
б) слив нитруемого соединения к кислотной смеси;
в) одновременный слив компонентов.
Влияние порядка слива компонентов мы рассмотрели в гл. V.
Химия—13—8 113
f
В отношении степени нитрации в отдельных стадиях различают:
а) ведение процесса с соблюдением точных молекулярных ста-
дий нитрации, т, е. при 1-й нитрации нитруют весь углеводород
(разумеется, за исключением трудно нитрующихся к концу опера-
ции небольших количеств углеводорода — около 1%) до монони-
тросоединий; при 2-й нитрации нитруют мононитросоединение до
динитросоединения (и здесь к концу операции в продукте остается
около 1% мононитросоединения, трудно нитрующегося при малой
концентрации кислоты к концу нитрации);
б) ведение процесса без соблюдения точных молекулярных ста-
дий нитрации. Этот способ был впервые применен в Англии во время
империалистической войны 1914—1918 гг. при нитровании толуола
и характеризуется следующими особенностями.
В 1-й фазе нитрации превращают только половину толуола в мо-
нонитротолуол. Это делается с целью максимального извлечения
азотной кислоты и мононитротолуола из отработанной кислоты от
1-й фазы нитрации. Так как в полученном продукте на 1 мол толуола
приходилось 1/2 группы NO2, то англичане обозначили полученный
продукт символом г/2 НТ, где число перед буквами НТ обозначает
число нитрогрупп, приходящееся на одну молекулу исходного то-
луола. Во 2-й фазе нитрации ведут процесс так, чтобы полученный
нитропродукт был жидким при температурах в пределах 30—35°.
Это достигается при смесях нитропродуктов, содержащих 50—75%
динитротолуола и соответственно 50—25% мононитротолуола. Так
как в таких смесях на 1 мол пронитрованного толуола приходилось
приблизительно от 1г/2 до 13/4 нитрогрупп, то ее обозначали сим-
волами I1 /2 НТ или 13/4 НТ.
Такая низкоплавкая смесь нитропродуктов получалась во
2-й фазе нитрации для удобства отделения нитропродукта от отра-
ботанной кислоты. Так как сепарация производилась при относи-
тельно низкой температуре, то в отработанной кислоте оставалось
в растворенном виде значительно меньше нитропродукта, чем при
обычной сепарации оставалось полностью пронитрованного веще-
ства •—динитротолуола, имеющего температуру плавления 56—58°
(температура затвердевания 54—56°).
Таким образом процесс производства тротила при описываемом
способе состоит из следующих ф аз:
Т-,——т—V2HT------------ИЧ.-^ЛНТ-----------»ЗНТ
(толуол) ' 2 ’ «4
2. По характеру кислотооборота способы производства нитро-
соединений подразделяются на следующие: 1) способ без примене-
ния кислотооборота; 2) способ с неполным кислотооборотом; 3) спо-
соб с полным кислотооборотом.
Производство тротила без применения кислотооборота имело
место в самый ранний период; при этом все три кислотных смеси—
для 1-й, 2-й и 3-й фаз нитрации — готовились на свежих кисло-
тах. Впоследствии начали применять часть отработанной кислоты
114
3-й фазы для приготовления смеси для 2-й фазы. Еще позже стали
применять отработанные кислоты (часть 3-й и часть 2-й отрабо-
танной кислоты) также и для приготовления смеси для 1-й фазы.
При таком неполном кислотообороте снизился расход
свежих кислот
Наконец, приблизительно со времени империалистической
войны 1914—1918 гг. стали применять полный ки с лото-
оборот — наиболее выгодный способ, обладающий следующими
преимуществами:
1) уменьшается общий расход серной кислоты;
2) уменьшается расход азотной кислоты вследствие более пол-
ного использования азотной кислоты, остающейся в отработанных
кислотах;
3) увеличивается выход тринитротолуола вследствие экстрак-
ции растворенных в отработанных кислотах нитропродуктов" от
предыдущей фазы нитрации;
4) облегчается регенерация отработанных кислот при денитра-
ции, так как они получаются в виде наиболее слабой отработан-
ной кислоты от 1-й фазы нитрации с содержанием 60—70% кислоты.
В этой отработанной кислоте содержится минимальное количество
нитросоединений (причем легко возгоняющихся мононитросоедине-
ний), азотной и нитрозилсерной кислот; последняя, как известно,
разлагается при денитрации тем легче, чем слабее серная кислота,
в которой она растворена. •
3. По цикличности процесса различают: 1) периодические спо-
собы нитрования, 2) непрерывные способы нитрования.
Периодический способ, по которому впервые получались нитро-
соединения, начиная от вначале установленного способа Гайзер-
мана без кислотооборота, постепенно совершенствовался до способа
с полным кислотооборотом.
Однако наиболее совершенными являются непрерывные процессы,
наиболее безопасные, требующие наименьших капиталовложений,
наименьшего расхода рабочей силы, дающие наибольший выход ко-
нечного продукта при минимальном расходе кислот и являющиеся'
в итоге наиболее экономичными.
ГЛАВА VIII
НИТРОПРОИЗВОДНЫЕ ТОЛУОЛА
ТОЛУОЛ
1. Свойства толуола. Толуол представляет собой бесцветную
подвижную жидкость со специфическим запахом.
Приводим некоторые константы для химически чистого толуола
по Тимермансу и Мартину 1 *: температура кипения 110,8° при нор-
мальном давлении; температура затвердевания—95,0°; плотность
О° = 0,88545; D” = 0,86697. Изменение плотности, соответствую-
щее 1° в интервале от 0 до 30°, — 0,00925.
2. Технические условия на прием толуола (ОСТ 464).
• Таблица 15
Показатели Каменноугольный толуол Нефтяной толуол
Удельный вес 0,870±0,002 при 15° 0,863-0,870 при 15°
Пределы кипения при 780 мм\
начало кипения 109° 108,5°
конец кипения 111° 111°
Степень окраски серной кислоты по шкале Кремера и Шпилькера 95% перегоняется в пределах 1° —
не более 0,5 0,5
Бромное число не более 0,8 г 0,4 г
(на 100 см3 4 толуола) (на 100 см3 толуола)
Хамелеоновая проба1 —— не менее 2 минут
Суль ] ируемость серной кислотой
с 5% SO3 — не менее 96%
по объему
1 Температура’кипения и удельные веса толуола указаны по данным Между-
народного бюро физико-химических эталонов в Брюсселе. По этим данным
прежние литературные указания ошибочны, если толуол для испытания не
подвергался предварительно очистке через кристаллическое соединение (р-то-
луиДин или р-толуолсульфокислоту). См. Timmermanse A. Mar-
tin, J. Chim- Phys., 23, 733, 1926; цитировано по Stettbacher,
Chem. Ztg., Fortschrittberichte, 1—20, 1929.
4 Эта проба в настоящее время не применяется.
116
Раньше в толуоле определялось еще содержание тиофена и тио-
толена; в настоящее время эта проба исключена из технических
условий из-за малого содержания этих веществ, не имеющего прак-
тического значения.
НС —СН
II II
3. Тиофен, НС СН, представляет собой бесцветную жид-
У'
кость, кипящую при 84°. Обладает слабым, мало характерным за-
пахом; уд. вес 1,062 при 23°. а-Метилтиофен (температура кипения
413°) и р-метилтиофен (температура кипения 114°), называемые
тиотоленами, сопровождают каменноугольный толуол. Диметилти-
офен или тиоксен сопровождают каменноугольный ксилол.
Реакция тиофена и его производных обладает большим сходством
с реакциями ароматических соединений. Химически это сходство
проявляется в устойчивости тиофена к реакциям окисления (несмотря
на присутствие атома двухвалентной серы), в трудности протека-
ния реакции присоединения, в способности легко нитроваться,
сульфироваться и т. д.
В большинстве случаев реакции замещения протекают для тио-
фена значительно легче, чем для бензола; например, при обработке
серной кислотой тиофен сульфируется гораздо легче бензола. Креп-
кая серная кислота разрушает тиофен и его гомологи. Однако раз-
бавленные растворы их гладко сульфируются.
При нитровании тиофена азотной кислотой в смеси с уксусным
ангидридом получается с хорошим выходом а-нитротиофен. При
нитровании нитротиофена серноазотной смесью получается дини-
тротиофен (температура плавления 520)1.
Тротил, ксилил и другие взрывчатые вещества, содержащие ни-
тропроизводные тиофена и его гомологов, окрашиваются в желто-
красноватый цвет, который все более и более углубляется при дей-
ствии света.
Характерной реакцией на присутствие тиофена в углеводородах
является голубая окраска (образование индофенина), вызываемая
действием сернокислого раствора изатина.
4. Непредельные соединения* Как в каменноугольном, так и
в нефтяном бензоле и толуоле содержатся непредельные соединения.
Количество непредельных соединений определяется по бромному
числу, т. е. по количеству граммов брома, потребного для их свя-
зывания, и по пробе с серной кислотой. При взаимодействии сер-
ной кислоты с примесями, содержащимися в бензоле, толуоле и
других углеводородах, происходят следующие реакции:
1. Присоединение серной кислоты к непре-
дельным соединениям с образованием сернокислых
1 В a tasi ni an, J. Amer. Chem. Soc., 50, 2748, 1928.
117
эфиров:
R - CH
ОН
R -CH2
R— CHOSO2OH
2
2. По лимеризация непредельных соедине-
Z\____________________________CH Z\_____CH
ний. В частности инден, | || || и кумарон | |1
\Z\/GH \/\zLH
СН2 О
осмоляются с выделением большого количества тепла и образова-
нием кумароновой смолы.
3. Образование продуктов конденсации. Не-
предельные углеводороды легко конденсируются с бензолом и его
гомологами в присутствии серной кислоты, причем образуются го-
мологи бензола предельного характера, например, с амиленом обра-
зуется амилбензо л:
свнв + с5н10-+свн5санп
Аналогично стирол СвН5СН = СН2 конденсируется с гомоло-
гами бензола в высокомолекулярные углеводороды.
В присутствии непредельных соединений при смешении бензола
(толуола, ксилола и других погонов) с серной кислотой происхо-
дит окрашивание кислоты от слабожелтого цвета до темного — в за-
висимости от характера и содержания этих примесей.
На этом основана следующая проба с серной кислотой. В мер-
ный цилиндр емкостью около 25 мл (с притертой пробкой) вводят
5 мл «химически чистой» или «чистой» серной кислоты уд. веса 1,84
(см. ОСТ 361), 5 мл исследуемого бензола (толуола, ксилола), сильно
взбалтывают в течение 5 минут и после 2-минутного отстаивания
сравнивают окраску серной кислоты с окраской образцового рас-
твора бихромата калия.
Сравнение ведут в сосудах одинакового диаметра. Степень
окраски обозначают соответствующим номером шкалы образцовых
растворов бихромата, отвечающих определенному количеству грам-
мов К2Сг2О, на 1 л 50%-ной «химически чистой» или «чистой»
серной кислоты.
Образцовые растворы приготовляют путем растворения в 1 л
50%-ной «химически чистой» или «чистой» серной кислоты следую-
щего количества химически чистого бихромата калия в граммах:
®,1, 0,2,.0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1,5, 2,3, 4.
5. Значение примесей парафинов в толуоле. Толуол, получаемый
на коксобензольных заводах, почти совершенно свободен от пара-
финов. В очень редких случаях в толуоле из коксовых печей содер-
жится до 0,5% парафинов. Однако толуол, получавшийся в Англии
в период империалистической войны 1914—1918 гг. из газовой
смолы, особенно тех заводов, которые пользовались вертикальными
ретортами, содержал до 4—5% и больше парафинов. Такой то-
118
луол смешивали с большим количеством толуола, не содержавшего
парафинов, для уменьшения содержания последних.
По английским данным при содержании парафинов от 4 до 5%
промывка полученного мононитротолуола от кислот с последующей
отгонкой от него водяным паром парафиновых углеводородов (по-
добно тому, как это производилось при получении мононитротолуола
из тблуол-бензина) является невыгодной, так как стоимость этих
операций почти равна стоимости добавочной азотной кислоты, не-
обходимой для получения из такого толуола конечного продукта
удовлетворительного качества.
Однако в случае толуола, содержавшего более чем 5% парафи-
новых углеводородов, выгодней процесс, употребляемый для толуол-
,бензина, а именно, отмывка продукта 1-й нитрации от кислот и от-
гонка парафинов водяным паром Ч ч
6. Отличие каменноугольного толуола от нефтяного. Каменно-
угольный толуол содержит следующие примеси: 1) непредельные
соединения; 2) фенолы; 3) сернисты^ соединения — сероуглерод и
тиофен. В нефтяном толуоле, получаемом при пиролизе нефти, нет
сернистых соединений.
Но при пиролизе нефти или ее погонов образуется наряду с аро-
матическими углеводородами большое количество как непредельных
соединений, так и бензинов, имеющих температуру кипения, весьма
близкую к температуре кипения толуола. Эти продукты остаются
в толуоле после дестилляции в большем или меньшем количестве
Поэтому методы испытания каменноугольного и нефтяного толуола
должны быть различны и, конечно, не могут быть ограничены одйой
только перегонкой с последующей поверкой на степень очистки,
как это имеет место при приеме каменноугольного толуола. По тех-
ническим условиям на каменноугольный толуол нефтегазовый за-
воды могли бы сдать различные смеси бензинов с толуолом.
Поэтому в технические условия на нефтяной толуол введено
добавочное требование, чтобы сульфируемость толуола была не
меньше 96% (по объему).
СОСТАВ ПРОДУКТОВ 1-Й НИТРАЦИИ ТОЛУОЛА, ИХ
СВОЙСТВА И ПРИМЕНЕНИЕ
1. Свойства изомеров мононитротолуола. При нитрации толуола
образуются три теоретически возможных изомера: о-, т- и р-нитро-
толуолы.
«-Нитротолуол представляет собой маслянистое вещество соло-
менно-желтого цвета с запахом горького миндаля, с температурой
кипения 221,3°, по Тимермансу, с температурой затвердевания, по
де-Белю 2, для лабильной формы (^-форма) 3,17°, для стабильной
формы (а-форма) 9,27° и с плотностью при 0° 1,1775, при 15°
1,168 и при 23° 1,163.
1 Man. of TNT.
! De Beule, Bull. Soc. Chim. Belg., 42, 27, 1933.
119
m-Нитротолуол — жидкость желтоватого цвета с температурой
кипения, по Тимермансу, 232,7°, с температурой затвердевания, по
де-Белю, 16,1° и с плотностью при 22° 1,168.
^-Нитротолуол — бесцветные кристаллы с температурой кипе-
ния, по де-Белю и Тимермансу, 238,4°, с температурой затвердева-
ния, по де-Белю, 51,6°.
2. Состав продуктов 1-й нитрации толуола. Состав технического
мононитротолуола представляет значительный практический инте-
рес, так как продукты нитрации содержащегося в нем т-нитрото-
луола ухудшают качество тринитротолуола. Кроме того, относи-
тельное содержание изомеров мононитротолуола имеет большое
значение при выделении из технического продукта изомеров о- и
р-нитротолуолов, применяемых для изготовления красок и фарма-
цевтических препаратов.
По вопросу о содержании изомеров в техническом мононитрото-
луоле имеются значительные расхождения у различных авторов.
По Нельтингу и Форелю, при нитровании толуола одной азотной
кислотой получается около 66% р-нитротолуола; наоборот, при ни-
тровании смесями серной и азотной кислот получается около 60%
о-нитро толуола.
По Голлеману, при нитровании толуола, серноазотной смесью
при разных температурах получается следующий состав мононитро-
толуола (табл. 16).
Таблица 16
Температура нитрации °C o-H итротолуол % р-Нцтротолуол О/ /о m-Н итротолуол %
—30 55,6 41,7 2,7
0 56,0 49,9 3,1
+30 56,9 39,9 3,2
+60 57,5 38,5 4,6
Джибсон с сотрудниками 1 нашли, что мононитротолуол, полу-
ченный при обычных условиях нитрования, содержит 61—63%
о-нитротолуола, 33,5 — 32% р-нитротолуола, 4,5 — 4,2% m-нитро-
толуола.
По данным тех же авторов, при нитровании толуола одной азот-
ной кислотой мононитротолуола содержалось:
Таблица 16а
Температура нитрации °C о-Нитротолуол % р -Н итрото луо л [m-Н итротолуол
% %
40 60,7 35 4,3
0 60,3 37,2 2,5
‘Gibson, Duckham and Fairbairn, J, Chem. Soc., 121, 27, 1922.
120
Наиболее надежным надо считать данные де-Беля (табл. 17).
Таблица 17
Условия нитрации о-Нитротолуол % р-Н итротолуол о/ /о m-Н итротолуол %
Одной азотной кислотой при 20° 58,4 37,4 4,2
Смесью серной и азотной кислот при 30—50° .... 59,2 36,1 4,7
Из приведенных данных видно, что:
1. Результаты нитрования толуола одной азотной кислотой,,
полученные Нельтингом и Форелем, неправильны. При применении
одной азотной кислоты вмес-
то серноазотной смеси сни-
жается содержание о-изоме-
ра приблизительно на 1% и
на столько же увеличивает-
ся содержание р-изомера,
что не может иметь практи-
ческого значения. Содержа-
ние ?п-изомера остается без
перемен.
2. При понижении темпе-
ратуры в случае нитрования
серноазотной смесью незна-
чительно понижается содер-
жание о- и тп-изомера и со-
ответственно . повышается
содержание р-йзомера. Это
влияние также не имеет
практического значения.
3. Технический мононит-
ротолуол. Полученный по
описанному способу моно-
Рис. 10. Растворимость мононитрото-
луола в серной кислоте при 20°.
нитротолуол представляет
собой жидкость, окрашенную примесями (побочными продуктами)
в красно-коричневый цвет.
В воде почти не растворяется, растворяется в серной и азотной
кислотах и в большинстве органических растворителей.
Растворимость мононитротолуола в серной кислоте приведена
в табл. 18.
Растворимость мононитротолуола в серной кислоте различной
концентрации 1 дана на рис. 10.
1 Man of TNT.
121
Таблица IB
Концентрация I Растворимость
% I при 20° | при 60°
60 0,04 0,08
75 0,50 0,81
80 1,56 2,09
83 2,73 12,28
88 19,50 20,14
90 33,20 33,90
4. Состав продуктов нитрации изомеров мононитротолуола. При
нитровании о-нитротолуола получаются:
.2,4- Динитротолуол (I) кристаллизуется в виде моно-
клинических иго л со слегка желтоватым оттенком. Температура
затвердевания химически чистого продукта, по Штельцнеру, 70,5°.
Кипит с разложением при 300° при нормальном давлении.
2,6- Динитротолуол (II) кристаллизуется в виде слегка
желтоватых игол-
Температура затвердевания, по Джибсону, 64—65°.
При нитровании о-нитротолуола получается 66,6% 2,4-изомера
и 33,3% 2,6-изомера.
Де-Бель показал, что при нитровании о-нитротолуола не обра-
зуется 2,5-динитротолуола вопреки утверждению ряда авторов.
При нитровании ти-нитротолуола получаются:
122
1) 3,4- динитротолуол
по де-Белю, 58,5°;
2) 2,3- динитротолуол
по де-Белю, 59,23°,
3) 2,5- динитротолуол
по де-Белю, 50,25°;
4) 3,5- динитротолуол
по де-Белю, 92,1°.
По де-Белю, при нитровании
четырех динитротолуолов в следующих соотношениях (см. табл. 19).
При нитровании p-нитро-
толуола получается только
2,4-динитротолуол, описан-
ный выше:
100'Л
(I); температура затвердевания,
(II); температура затвердевания,
(III); температура затвердевания,
(IV); температура затвердевания
n-нитротолуола получается смесь
Таблица .19
Наименование вещества О/ /о
3,4-Динитротолуол 54,6
2,3-Динитротолуол 30,6
2,6-Динитротолуол 13,0
3,5-Динитротолуол .... . 1,8
5. Состав продуктов нитрации технического мононитротолуола.
На основании приведенных данных вычисляется состав (табл. 20)
продуктов нитрации технического мононитротолуола (без учета со-
державшихся в мо но нитро соединении примесей).
6. Растворимость динитротолуола в кислотах. Растворимость
динитротолуола в 90%-ной серной кислоте (табл. 21).
Рис. 11. Растворимость динит-
ротолуола в серной кислоте.
2,4-Динитротолуол .
2,6-Динитротолуол .
3,4-Динитротолуол .
2,3-Динитротолуол .
2,б-Динитротолуол .
3,6-Динитротолуол .
Таблица 20
Наименование вещества %
76,1 1
19,8 |
2,26
1,23
0,54
0,08
95,9
4,1
Таблица 21
Температура, °C Растворимость, %
20 8,5
60 16,8
70 20,0
90 20,6
123
Растворимость динитротолуола в серной кислоте различной кон-
центрации при 20, 50 и 70° дана на рис. 11.
7. Состав сырого тротила. При 3-й нитрации образуются следую-
щие продукты:
1. При нитровании 2,4-динитротолуола получается только сим-
метричный или а-тринитротолуол с температурой затвердевания для
химически чистого продукта 80,85°:
СН3 СН3
/XNO2 O2N/4jNO2
Xno2 \о2
2. При нитровании 2,6-динитротолуола получается только
а -тринитро то луо л :
СН3 сн3
C^n/^NO, o2nz\no2
3. При витровании 3, 4-динитротолуола получается два изомера:
а) 3,4,6- или 2,4,5-тринитротолуол (I), или 7-тринитротолуол,
с температурой плавления, по де-Белю, 102,3°;
б) 2,3,4-тринитротолуол (II), или ^-тринитротолуол, с темпера-
турой плавления, по де-Белю, 110,3°:
СН3
По де-Белю, эти изомеры получаются при нитровании 3,4-дини-
тротолуола в следующих количествах: 84% у-тринитротолуола,
16% [3-тринитротолуола.
124
kt. При нитровании 2,3-динитротолуола получается также два
изомера:
а) ^-тринитротолуол, уже описанный выше;
б) 2,3,6- или 2,5,6-тринитротолуол, или ^-тринитротолуол с тем-
пературой затвердевания 109,8°.
Из шести изомеров ^-тринитротолуол был открыт последним.
Надо отметить, что 2,3-динитротолуол значительно труднее нит-
руется в тринитротолуол, чем 2,4-, 2,6- и 3,4-динитротолуол.
По данным де-Беля, при нитровании 2,3-динитротолуола (в усло-
виях, при которых происходит полное превращение 2,4-, 2,6- и
3,4-динитротолуола в тринитротолуолы) получается продукт, содер-
жащий: 22,3% 2,3-динитротолуола (непрореагировавший динитро-
:Й)луол), 62,2% р- тринитротолуола, 15,5% ^-тринитротолуола.
(?)
сн3
/X|NO2
^J.NO2
no2
no2zxno,
(7|)
5. При нитровании 2,5-динитротолуола также получается два
и зомера:
Оба изомера описаны выше.
125
Как и 2,3-динитротолуол, 2,5-динитротолуол очень трудно нит-
руется.
Состав продуктов нитрации 2,5-динитротолуола, по де-Белю,
колеблется в пределах: 21,7—25,1% 2-5-динитротолуола (непрореаги-
ровавший динитротолуол), 66,7—69,7% у-тринитротолуола, 8,2—
8,6% ^-тринитротолуола.
6. Нитрование 3,5-динитротолуола представляет особый интерес.
Билль 1 обрабатывал 3,5-динитротолуол смесями различных; кон-
центраций и вел реакцию при температуре 95—100° в течение не-
скольких дней. Однако в продукте реакции он нашел только 1,3,5-
динитробензойную кислоту наряду с неизмененным динитротолуолом.
Маркероль, Келер и Жовине 2 отметили в работе, проведенной
в 1916 г., что 3,5-динитротолуол при различных условиях нитрации
серноазотной смесью не изменяется или же подвергается разру-
шению.
Де-Бель нашел, что при нитровании 3,5-динитротолуола более
крепкой серной кислотой, чем обычно применяемая при нитровании
динитротолуола (соответствовала смесц: 86% H2SO4, 14% HNO3),
происходит частичная нитрация 3,5-динитротолуола. Во время ни-
трации наблюдается выделение пузырьков газа вследствие окис-
ления нитро производно го. При одном из опытов во время первой
половины нитрации произошло окисление, и температура поднялась
до 150°.
При нитрации получено: 73% 3,5-динитротолуола (непрореаги-
ровавший динитротолуол), 11% 2,3,5-тринитротолуола (е-тринитро-
толуол)3, 2% 3,4,5-тринитротолуола (8-тринитротолуол)4, 14%
потерь вследствие окисления:
Таким образом из всех изомеров динитротолуола труднее всего
нитруется 3,5-динитротолуол.
1 W i 1 1, Вег., 47, 704, 1914.
2 Marqueyrol, Kohler и Jovinet, Mem. Poud., 18, 66, 1921.
3 По де-Белю, температура плавления 95,1—95°.
4 Температура плавления 132°.
126
Из приведенных данных де-Бель определил следующий состав
промытого тринитротолуола при условии полного превращения
2, 4- и 2,6-динитротолуолов в а-тринитротолуол:
Таблица 22
Наименование вещества 0/ /0
а-Тринитротолуол 95,1
у-Тринитротолуол 2,69
[J-Тринитротолуол
^-Тринитротолуол 0,29 4,0
Е-Тринитротолуол 0,009
В-Тринитротолуол 0,002
2,3-Динитротолуол ..... 0,33 )
2,6-Динитротолуол 0,16 } 0,64
З,б-Динитротолуол 0,06 J •
Промытый производственный тротил содержит кроме указанных
следующие примеси: до 2,5% динитротолуола (2,4- и 2,6-динитро-
толуола, остающихся недонитрованными), тетранйтрометан в очень
малых количествах х, очень малые количества динитробензола и
тринитроксилола (получены от нитрации небольших количеств
бензола и ксилола, содержавшихся в исходном толуоле), очень
малые количества тринитробензола (получен при горячей промыв-
ке из тринитро бензойной кислоты, образующейся При окислении
тротила во время нитрации динитротолуола) и, наконец, окрашен-
ные соединения фенольного характера.
По Шмиту 1 2, в тротиле могут быть динитрооксибензойные кис-
лоты, образующиеся при горячей промывке из несимметричных
тринитробензойных кислот. В то время как при кипячении водного
раствора симметричной тринитробензойной кислоты выделяется СО2
и образуется тринитробензол (см. стр. 221), при нагревании водного
раствора несимметричных тринитробензойных кислот происходит
гидролиз с образованием динитрооксибензойных кислот:
СООН
СООН
z\no2
он\/
no2
СООН
^Xx'|NO2 + Hj0
\/N°2
no2
СООН
7 >NO2
* \/он
no2
1 Об образовании тетранитрометана при взаимодействии нитропроизводных
толуола см. М с К i е, J. Soc. Chem. Ind., 44, 430, 1923.
2 Schmitt, Мёт. Pond., 37, 131, 1937.
127
8. Схема превращения тблубла в тринитротолуол
128
9. Свойства тротила (а-тринитротолуола). Физические
свойства. а-Тринитротолуол (2,4,6-тринитротолуол) кристал-
лизуется из спирта в виде бесцветных или слабо окрашенных приме-
сями в желтоватый цвет ромбических кристаллов или в виде длин-
ных, плоских, легко ломающихся игол. Температура затвердевания,
по де-Белю 80,85°.
Гравиметрическая плотность кристаллического тринитротолуола
около 0,9; уд. вес 1,663.
Плотность прессованного тринитротолуола возрастает с увеличе- 1
нием давления при прессовании. Дотриш определил следующую
зависимость (табл. 23):
Таблица 23
Давление на 1 см2 прессованного тринитро- толуола кг Средняя плотность Давление на 1 см2 прессованного тринитро- толуола кг Средняя ПЛОТНОСТЬ
275 1,320 2750 1,599
685 1.456 3435 1,602
1375 1.558 4125 1,610
2000 1,584 — —
Плотность тринитротолуола в мелкокалиберных снарядах, за-
прессованного под давлением 3000 кг1см2^ колеблется в пределах
1,58—1,60.
Плотность литого тринитротолуола зависит от условий отливки
и колеблется в пределах 1,54—1,59. Расплавленный тротил имеет
при температуре 82° плотность 2 1,467.
Скрытая теплота плавления около 20,2 ккал/кг. Коэффициент тепло-
проводности, по Прентису 3, 0,173.
Растворимость тринитротолуола в различных растворителях
приведена в табл. 24 (по Тейлору и Ринкенбаху) 4:
1 Температура затвердевания по определению других авторов:
по Ринтулу колеблется между.............. 80,8—80,85°
» Джибсону ............................. 80,8°
» Деверню............................... 80,83°
» Джуа.................................. 80,65°
» Касту................................. 80,66°
2 Каст, Взрывчатые вещества и Средства воспламенения, стр-240, 1932.
8 Prentis, Army Ordnance, 4, 117, 184, 242, 1923. Для Сравнения
укажем козфициенты теплопроводности некоторых веществ: для железа—50,
для Стекла — 0,5, для каучука — 0,137.
4 Т а у 1 or и R i n k enb а с h, J. Amer. Chem. Soc., 45, 44—59, 1923;
Z. Schiess- u. Sprengst-, 18, 50, 1923.
Химия—13—9
129
Таблица 24
Темпера- тура °C В 100 мл растворителя растворяется тринитротолуола, г
вода 95%'Ный спирт толуол бензол эфир ацетон серо- углерод
При 15 0,012 1.07 45 50 2.85 92 0,35
» 20 0,0130 1.23 55 67 3,29 100 0,48
» 30 0,0175 1.80 84 ИЗ 4.56 156 0,85
» 45 — — —. — — — 2,02
» 60 0,0675 8.30 367 478 —. 600 —
» 65 — — — —. — 843 —
» 75 0,0975 19.50 1685 2028 — — —
» 100 0,1475 — —• — — — —.
Растворимость тротила в маточных растворах отличается незна-
чительно от таковой в чистом спирте. Тротил мало растворим в ли-
гроине, петролейном эфире, бензине.
Растворимость тротила в серной кислоте и в серноазотных смесях
приведена в табл. 25 и 26.
Таблица 25
Растворимость тринитротолуола в серной кислоте, %
Темпера- тура '°C Концентрация кислоты
70% 75% 80% 85% 90% 95% юо%
При 0 0.3 0,4 0.6 2,0 3.5 13,0
» 10 — 0.3 0,45 0,75 2,2 4.0 13,5
» 20 — 03 0,50 0,85 2,5 4,8 15.0
>> 25 — 0,32 0,55 0,95 2,6 5.2 15,5
» 30 — 0 35 0,60 1.0 2,7 6,0 16,5
» 40 0.2 0.40 0.65 1,3 3,0 7.0 18,0
» 50 0.2 0,45 0.70 1,7 3,5 8.5 21,0
» 60 0.22 0.50 1,0 2,3 5,2 11.0 24.8
» 70 0.35 0,7 1,6 3,3 7,0 13,5 29.0
» 80 0,6 1,3 2,4 4.8 10,0 18.0 36,5
Химические свойства. Подобно пикриновой кислоте,
нитробензолу и другим нитросоединениям тротил образует с анили-
ном, ароматическими углеводородами и другими соединениями про-
дукты присоединения, предлагавшиеся в качестве взрывчатых
веществ.
Тротил не взаимодействует с металлами. С щелочами и алкотоля-
тами щелочных металлов реагирует, образуя металлические произ-
водные — тротилаты.
130
Таблица 26
Растворимость тринитротолуола в серноазотных смесях
Состав серноазотной смеси % Растворимс сть
ILSO., HNO, Н2О при 20° при 50° при 70°
60 0 40 0,20 0.52 0,70
80 0 20 0,59 1.25 2.07
90 0 10 2.55 4.70 7,63
60 1 39 0.22 0,41 0.62
80 1 19 0.55 1,08 1,68
90 1 9 1,85 4,35 7,49
60 5 35 0,25 0,55 1.23
80 5 15 0,73 1,48 1,85
90 5 5 1,76 4,49 7,53
Ганч и Киссель1 обстоятельно исследовали тспрос о взаимодей-
ствии алкоголятов и спиртовых растворов щелочей. Они указали,
что на основании имеющихся данных весьма вероятно, что образо-
вание солей первичных и вторичных нитросоединений при взаимодей-
ствии последних с едким натром или алкоголятом натрия протекает
по следующей схеме:
Z0
R • СЕЛО. 4- NaOH -> R - CH, Z<-ONa-------->
хон
неустойчивый промежуточ-
ный продукт
Z0
------> ДСП = N< + Н2О
XONa
.0
R - CH,NO, + C0HsONa -> R - CH2 - N<ONa---------->
\OC2H5
неустойчивых! промежуточ-
ный продукт
------= 4-CJL0H
^ONa -
Здесь первоначально образуется неустойчивый промежуточный
продукт присоединения едкого натра, соответственно алкоголята
натрия, к нитро соединению. При последующей стабилизации этого
продукта образуется соль изонитросоединения и освобождается
вода или спирт.
1 Hantzsch и Kissel., Вег., 32, 3137, 1899.
131
Третичные нитросоедирения не могут превратиться в изонитро-
соли, так как у них недостает группы СН—N02, или, иначе говоря,
недостает атома водорода, необходимого для превращения нитро-
группы в изонитро группу.
Взамен этого, по Ганчу и Кисселю, у третичных нитросоедине-
ний может иметь место солеобразование, состоящее в прямом при-
соединении едкого натра или аЛкоголята натрия к нитрогруппе.
Образующийся продукт присоединения вследствие отсутствия во-
дородного атома не может ангидризироваться в изонитросоль, сохра-
няется таковым в форме соли и может быть в некоторых случаях
даже изолирован в виде свободной кислоты. По этим соображениям
можно предположить, что ароматические нитросоединения могут
образовать продукты присоединения алкоголятов состава:
у°
R -N<ONa
ХОС2Н5
Соответственно изложенному мы имели бы для тринитротолуола;
CeH,(CH3) (NO2)3 + СН3ОК С6Н2(С нз) (N0,)2 - OK
\осн3
тринитротолуол - метилат-
калий или динитротолуол-
эфирнокислый калий
Мейзенгеймер 1 установил новый взгляд на строение продуктов
присоединения алкоголятов к полинитросоединениям. Так как при
действии алкоголятов на почти бесцветный тринитробензол полу-
чается глубоко окрашенное вещество, то Мейзенгеймер предполо-
жил, что при этом бензоидная форма ядра переходит в хиноидную,
и полученное соединение следует рассматривать как соль хиноло-
нитрокислоты :
нх/осн3
02N|/X N02
Nf
Ч)К
Мейзенгеймер приводит результаты исследования продуктов вза-
имодействия алкоголятов с нитроантраценом и этилата и изобути-
лата калия с тринитроанизолом. Анализ полученных темнокрасных
1 Meisenheimer, Ann. der Chemie, 323, 205, 1902.
132
соединений подтвердил, что они являются продуктами присоедине-
ния алкоголятов к нитросоединениям. При действии разбавленной
серной кислоты на спиртовой раствор калиевой соли тринитроани-
зола регенерируется соответствующее нитросоединение.
Позже Ганч 1 присоединился к выводам Мейзенгеймера и ука-
зал, что появление хиноидного строения очень хорошо объясняет
причину превращения под влиянием алкоголятов и щелочей почти
бесцветных полинитросоединений в сильно окрашенные вещества,
причем хромофором здесь является группа =
хОМе
Стефанович выделил продукты присоединения 1, 2 и 3 мол ал-
ко голята.
Принимая хиноидное строение продуктов присоединения к три-
нитросоединениям 1 мол алкоголята, данное Мейзенгеймером, Сте-
фанович 2 предложил следующее строение для продуктов присоеди-
нения 2 и 3 мол алкоголята к а-тринитротолуолу:
Н5С2О. .СН3
Н5С2ОЧ ,СН3
Охх X .0
о
хок
ос2н5
ок
КСИ
Н
н5с2о/\/Хчн
4 ок
О 2Н5
хок
Стефанович установил, что не только а-тринитротолуол, но и
1,2,4-динитротолуол, 2,4,6-тринитро-лг-ксилол и 1,3,5-трини-
тробензол в абсолютной толуолалкогольной среде присоединяют
к молекуле нитро соединения число молекул алкоголятов щелочных
металлов, равное числу нитрогрупп в веществе. Для тетрила были
изолированы продукты присоединения 1 и 2 мол и для одного ами-
лата калия 3 мол алкоголята; более 3 мол алкоголята не удалось
присоединить к тетрилу.
Производное аммония удалось изолировать только для а-три-
нитротолуола; повидимому, оно представляет продукт присоеди-
нения к нитро соединению 1 лол гипотетического алкоголята аммония
(или 1 мол аммиака и 1 мол алкоголя). Металлические производ-
ные — продукты присоединения алкоголятов — аморфные, глубоко
окрашенные порошки разнообразных оттенков — от темнокоричне-
вого до карминово-красного.
При действии на а-тринитротолуол щелочей в присутствии окисли-
телей получается гексанитродибензил (температура плавления 212°).
1 Вег., 42, 2119, 1909.
2 Стефанович, Металлические производные ароматических полини-
тросоединений, ВТА, 1928 .
р- и у-тринитротолуолы при обработке едкими или углекислыми
щелочами в присутствии или отсутствии алкоголята очень легко
обменивают одну нитрогруппу на гидроксил или алкоксильную
группу и образуют соли или эфиры динитрокрезолов:
СН3
O,NZ X',N0,
1 Г
NO,
при обработке едкими
или углекислыми щелочами
в присутствии окислителя
сн3
zL при обработке едкими
' |NO., или углекислыми щелочами
х Jno2
no2
сн3
|ZXjNO2
NO2
сн3
при обработке едкими
или углекислыми щелочами
Механизм реакции между щелочами и нитро со единениями выяс-
нен только отчасти. Кописаров 1 представляет его в виде следующей
схемы:
CeH2(NO2)3CH3
Ф
LH.,CeH,(N0,)3 CH.,CeH2(N3.,), _ Q
. " 6 ' конденсация , - в -/3 конденса-
CH2CeH,(NO2)2NO CH2CeH2(NO,_)2N комплекс-
| ‘ "|| ное азо-
। * соедине-
CH.,CeH„(NO,).,N = О Н"е
I +
CH,CeH,(NO,)3 !
СНСвН2(Ю2)^О - - - I
|| -----------> конденсация в азокисоединение-!
CHC6H2(NO2),NO
1 Kopisarow, Chem. News, 112, 283, 1915.
134
При действии воды и разбавленных кислот на металлические
производные тринитробензола и тринитро фенилметилнитрамина
регенерируется соответствующее нитро соединение.
Совершенно иначе действует вода и разбавленные кислоты на ме-
таллические производные тринитротолуола и тринитроксилола: по-
лучаются темнобурые, почти черные, вещества, описанные Ганчем,
Кописаровым и др. как сложного состава неоднородные продукты
интрамолекулярного окисления и конденсации, которые эти авторы
считают производными дибензила и стильбена.
По чувствительности к удару выделенные Стефановичем метал-
лические производные нитро соединений близки к гремучей ртути
(производные тротила, тринитробензола, тетрила) и только произ-
водные тринитроксилола близки в этом отношении к азиду свинца.
К трению эти аморфные вещества мало чувствительны. Темпера-
тура вспышки для некоторых металлических производных а-трини-
тротолуола определена около 50° (например для тротилата аммония).
Повидимому, тротилаты образуются при непосредственном сплав-
лении щелочей и некоторых солей щелочных металлов с а-тринитро-
толуолом. По Кописарову, тринитротолуол, нагретый с небольшим
количеством едкого кали, взрывается при 160°.
По опытам Солонина такой сплав взрывается уже при нагрева-
нии до 100°.
При смешении расплавленного тротила с калиевой селитрой
Солонина обнаружил сильное побурение сплава около 160°. После
часового нагревания такой бурый сплав показал резко повышен-
ную чувствительность к удару и пониженную температуру вспышки
около 160°.
В 1916 г. на одном русском заводе произошел большой взрыв
при смешении расплавленного тротила с калиевой селитрой. Этот
сплав применялся для снаряжения снарядов. По данным обследо-
вания причиной взрыва могло быть образование металлического
производного тротила. Вероятнее, однако, что этот взрыв был резуль-
татом диверсии.
Действие света на тринитротолуол. Под дей-
ствием света тринитротолуол буреет с поверхности. Вопрос о при-
чинах этого побурения и о его практическом значении для качества
взрывчатых веществ неоднократно изучался различными исследо-
вателями.
Краутц и Турек 1 пришли к выводу, что под действием солнеч-
ного света на а-тринитротолуол последний окисляется в пикрино-
вую и тринитро бензойную кислоты, повышающие его чувствитель-
ность к удару. Более опасны соли этих кислот, могущие образо-
ваться при соприкосновении их с металлами.
Последующие исследователи, однако, отвергли эти выводы.
Шульц и Гангули 2 на основании своих исследований пришли
‘KrautzHTurek, . Schiess-u. Sprengst., 20, 49—58, 1925.
2 Schultz и Ganguli, Вег., 58, 702, 1925.
135
*
к выводу, что при действии света происходит фотоизомеризация
тринитротолуола, механизм которой обоснован ими следующим
образом.
Продукты, получающиеся при действии света на тротил, пред-
ставляют собой коричневый краситель, растворимый в воде и плавя-
щийся выше 280°: этот краситель состоит из двух соединений, из
которых одно легко растворимо в ацетоне. Одно из этих соединений
в твердом состоянии окрашено в коричневый цвет, другое — в черный.
Эти два соединения имеют одинаковую с тротилом формулу,
с.ледовательно, представляют собой продукты фотоизомеризацци тро-
тила. Они дают при ацетилировании одно и то же ацетильное произ-
водное; в ацетоином растворе происходит постепенный переход
коричневого соединения в черное; следовательно, они представляют
собой две таутомерные формы.
Обе формы образуют при кипячении с уксусным ангидридом ди-
ацетильное производное, следовательно, оба изомера имеют по два
гидроксильных остатка.
Так как оба таутомера образуются фотоизомеризацией 2,4,6-три-
нитротолуола, то два атома кислорода, необходимые для образова-
ния гидроксильных групп, могут происходить только от кислорода
нитрогрупп.
Перемещение кислородного атома нитрогруппы вследствие осве-
щения о-нитроароматических соединений наблюдалось уже раньше.
Чамичан и Зильбер 1 показали, что о-нитробензойный альдегид под
влиянием света переходит почти количественно в о-нитробензойную
кислоту. Позже был установлен ряд других аналогичных реакций 2.
Все эти исследования показали, что светочувствительна только
нитрогруппа, находящаяся в ортоположении по отношению к боко-
вой цепи.
Следовательно, в 2,4,6-тринитротолуоле под влиянием света
могут изменять свое положение только два кислородных атома
двух нитрогрупп, стоящих в ортоположении по отношению к
метильной группе.
Так как в 2,4,6-тринитротолуоле свободны положения 3 и 5,
то можно допустить, что обе гидроксильные группы образуются или
в 3,5-положениях, или же одна гидроксильная группа образуется
в ядре, а другая — в боковой цепи. Следовательно, оба таутомера
могут иметь только следующие конфигурации:
сн3-
ONf^NO
(!)
ОШ
ОН
no2
СН2ОН
ON^NO
или (II)
no2
1 Ci amici ап и Silber, Вег. 34, 2040, 1901.
’CohnuFriedlander, 35, 1925, 1902; SachsHSchiiller,
Вег., 36, 962, 3392, 1903; 37, 1870, 3425, 1904.
136
Конфигурация (I) представляет собой нитродинитрозоорсин.
Однако ни один из таутомеров, образующихся при освещении
2,4,6-тринитротолуола, не дает характерной реакции нитронитро-
зоорсинов — зеленого осадка с раствором FeSO4. Кроме того, фор-
мула (I) не объясняет происхождения двух таутомерных форм. По-
этому наше соединение может иметь только формулу (II). При этой
формуле возможна таутомерия вследствие перемещения атома водо-
рода, находящегося в ядре гидроксильной группы, в соседнюю
или противоположную нитрозогруппу с образованием о- или
р-хиноноксима:
СН,ОН
ON./^NOH
\/=°
no2
о-хиноноксйм
СН2ОН
on/4no
\/он
no2
неустойчивая
форма
СН2ОН
HON=/4jNO
°
N02
р-хиноноксим
Оба таутомера являются хиноноксимами. Это подтверждается
тем, что оба эти соединения являются красными красителями с оди-
наковым спектром поглощения; этого могло бы не быть, если бы
один из таутомеров имел энольную формулу, а другой — кетонную
(хиноноксима).
На основании изложенных данных можно вывести заключение,
что реакция фотоизомеризации 2,4,6-тринитротолуола протекает
по следующей схеме:
Н2СН^-| ОНСНОН НОСНОН
°2г*/\чоо on//'\no on<\no
2,4,6-тринитро- промежуточный наименее устойчивый
толусл продукт продукт
Вследствие стабилизирующего влияния двух находящихся в орто-
положении нитро зогрупп неустойчивая форма не превращается
в альдегид с отщеплением воды, но один кислородный атом пере-
ходит в ядро и становится в свободное место — в метаположение
к метильной группе. Полученный при этом продукт является также
неустойчивым: водород переходит к нитрозогруппе с образованием
двух хиноноксимов: о-хиноноксима или /^-хиноноксима — конечных
продуктов фотоизомеризации тринитротолуола под влиянием света.
Павлик 1 изучал спектры поглощения а-тринитротолуола, при-
чем подтвердил выводы Шульца и Гангули о том, что при действии
света на тротил образуются оксимы. При этом было установлено,
1 Pawlik, Chim. et Ind., 245, 29, 6 bis, 1933.
137
»
что такое же влияние на спектры поглощения, как и свет, оказывает
длительное нагревание тротила, а также действие на него сульфита
и щелочи. Поэтому Павлик пришел к заключению, что вещества,
получающиеся при действии света, высоких температур иди щело-
чей на тротил, представляют одинаковые химические соединения —
оксимы.
Орлов выделил из тротила продукт воздействия на него света.
Это вещество темно коричнево го цвета. Эмпирический состав и мо-
лекулярный вес выделенного продукта одинаковы с таковыми же
тротила. Температура вспышки около 230°; чувствительность к удару
значительно превосходит чувствительность тетрила (76% взрывов).
Чувствительность тротила к нагреванию.
Температура вспышки тротила около 300°; вспышка сопровождается
пламенем и копотью, но не производит взрыва; взрыв происходит
только в закрытой оболочке, например, при нагревании в прочной,
хорошо запаянной стеклянной трубке.
Некоторые примеси понижают температуру вспышки тротила.
Например, примесь высохшей олифы понижает температуру вспышки
до 248°; примесь синтетической смолы — идитола — в количестве
10% понижает температуру вспышки до 250°, примесь 50% этой же
смолы понижает температуру вспышки до 220°.
По данным Верола, при нагревании а-тринитротолуола до 150°
не наблюдается разложения. При нагревании до 180—200° происхо-
дит частичное разложение, вследствие чего значительно понижается
его температура затвердевания.
Орлов определил следующие данные о влиянии нагревания на
тринитротолуол (табл. 27).
Таблица 27
Темпе рату pa бани °C Время нагревания 'час. Температура затвердевания °C Чувствитель- ность к удару °/ /о Температура вспышки °C
До опыта 80.3 10 295 — 300
120 200 80.2 15 295 — 300
135 210 80.0 12 295 — 300
15J 50 79.0 Через 20 часов темная окраска, смолистый вид
150 100 76.4 4 280 — 285
165 50 80.0 - -
185 Через. 93 часа тротил в пробирке обуглился
Восприимчивость к детонации. Восприимчи-
вость к детонации тротила меньше, чем у тетрила и пикриновой кис-
лоты, и больше тринитроксилола, как это видно из табл. 28 величин
предельных инициирующих зарядов гремучей ртути и азида свинца \
'Koester, Untersuchungen fiber die sogenannte Grenzladungen von Ini-
tialsprengstoffen, 1929.
138
Таблица 28
Наименование вещества Предельный инициирующий заряд, г
гремучей ртути азида свинца
Тетрил 0,36 0,03
Пикриновая кислота 0,36 0,05
Тринитротолуол . ....... 0,38 0,15
Тринитроксилол Около 0,62 Около 0,45
Таблица 29
Плотность Скорость детонации^ Ai/сек
0.81 4400
0.94 4700
1.31 5800
1.40 6200
1.50 6500
1.54 6700
1.59 6700
1,61 6700
Эти данные относятся к прессованному тротилу. Восприимчи-
вость к детонации литого тринитротолуола значительно меньше,
чем прессованного. Для взрывания литого тротила необходим про-
межуточный детонатор, тогда как для взрывания прессованного
тротила достаточен капсюль-детонатор.
Скорость детонации тротила сильно меняется в зависимости от
плотности, что видно из табл. 29
для прессованного тротила (по
Касту). Расширение в бомбе Тра-
уцля 280 мл.
Чувствительность
тротила к удару. При
употребительной у нас пробе ис-
пытания чувствительности к уда-
ру (вес груза 10 кг, высота паде-
ния 25 см, навеска 0,03 г) при
испытании тротила получается
4—12% взрывов, причем не наблю-
дается заметной разницы между
тротилом-сырцом и очищенным.
Свойства изомеров тринитротолуола и их
влияние на качество тротила. Изомеры тринитро-
толуола отличаются по своим химическим и физико-химическим
свойствам, но почти тождественны между собой и с а-тринитро-
толуолом по взрывчатым свойствам. Удельные веса их близки к 1,62;;
температура вспышки 290—310°; в бомбе Трауцля они дают почти
одинаковое расширение и на копре обнаруживают почти одинаковую
чувствительность к удару.
Присутствие их в тротиле нежелательно по следующим основным,
причинам:
1. Вместе с а-тринитротолуолом, продуктами неполной нитра-
ции и посторонними примесями они образуют низкоплавкую массу,,
обусловливающую так называемую маслянистость тротила.
2. В присутствии изомеров понижается температура затверде-
вания тротила, причем нельзя удобными для производства мето-
дами установить, что причиной такого понижения температуры
затвердевания являются только эти изомеры, а не какие-нибудь.
другие примеси.
139>
*
В остальном нет никаких оснований считать изомеры а-три-
нитротолуола опасными примесями в тринитротолуоле.
10. Применение тротила. Требования к качеству тро-
тила, применяемого для снаряжения артиллерий-
ских снарядов. Рассмотрим тротил с точки зрения требований,
обусловленных характером службы боеприпасов.
Основные требования, которым должны удовлетворять взрыв-
чатые вещества, применяемые для снаряжения артиллерийских
снарядов, следующие:
1) неизменность при хранении — химическая и физическая
стойкость; 2) малая чувствительность к удару и другим механи-
ческим воздействиям; 3) максимальное фугасное и бризантное дей-
ствие.
1. Неизменность при хранении — химическая и
‘физическая стойкость. Еще Гайзерман 1 нашел, что перекристалли-
зованный тротил является химически стойким, но в некристаллизо-
ванном тротиле, т. е. тротиле-сырце, очищенном промывкой во-
дой и слабым содовым раствором, спустя короткое время хранения
при 50° наблюдались следы азотистой кислоты (по пробе Абеля
при 70—75°). Это очевидно обусловлено присутствием в тротиле
малостойких примесей.
Высказывалось мнение о том, что при хранении некристалли-
зованного тротила в нем происходят процессы, в результате кото-
рые увеличивается содержание низкоплавких примесей, т. е. так
называемого тротилового масла. Однако это мнение не доказано
прямым опытом или строгим наблюдением.
В начале 1917 г. впервые было отмечено массовое явление выте-
кания из снарядов маслообразного вещества, получившего у нас на-
звание «тротилового масла». Это явление наблюдалось и в других
странах, участвовавших в империалистической войне 2.
Вытекание масла из разрывного заряда сопровождается следую-
щими явлениями:
а) Разрыхление взрывчатого вещества; оно делается менее плот-
ным, рыхлым, ноздреватым. Если масло вытекло через дно (напри-
мер через нарезы ввинтного дна), то сделался более рыхлым слой,
прилегающий к дну. В результате этого возможно смещение заряда
при выстреле, воспламенение заряда от удара и преждевременный
взрыв.
б) При корпусах снарядов с ввинтным дном тротиловое масло,
несущее в растворенном виде тротил, остается в нарезах; при этом
взрыв может быть при ударе (в момент выстрела) металлических
поверхностей, между которыми масло заключено в нарезах снаряда;
или же может быть воспламенение пороховыми газами — передачей
-огня по маслу, заключенному в нарезах дна снаряда (как по сто-
пину).
1 Н aussermann, Z. Angew. Chem., 4, 508, 661, 1891.
2 Army Ordnance, 5, 611, 612, 1924; Przeglad Artiller., 3, 381—384, 1925.
U40
в) Тротиловое масло, выделяющееся из тротила, иногда соби-
рается в гнезде под взрывателем (или детонатором). В этом случае,
если детонатор не включен в металлическую герметически закры-
тую оболочку (таковы, например, английские тротиловые детона-
торы в шелковых мешочках), то он пропитывается маслом, отчего
теряет восприимчивость к детонации; в результате бывают непол-
ные взрывы или отказы. Но иногда наблюдалось и проникновение
масла во взрыватель, где оно попадало в капсюль-детонатор ц
флегматизировало гремучую ртуть, что также имело следствием
неполные взрывы или отказы.
Таким образом некристаллизованный тротил (тротил-сырец) яв -
ляется веществом физически нестойким. В результате этой нестой-
кости могут быть преждевременные разрывы (в канале орудия или
чаще всего перед дулом орудия), неполные взрывы или даже отказы
при стрельбе.
2. Чувствительность к удару. Как правило, не-
кристаллизованный тротил менее чувствителен к удару, чем тротил
кристаллизованный. Однако попадаются образцы, даже в одной и
той же валовой партии, которые обладают несколько повышенной
чувствительностью к удару.
3. Максимальное фугасное или бризант-
ное действие. При испытании в броневой камере снарядов,
снаряженных некристаллизованным тротилом, получается меньшее
осколочное действие, чем у снаряженных кристаллизованным тро-
тилом. Это, разумеется, зависит от меньшей чувствительности к
детонации некристаллизованного тротила сравнительно с кристалли-
зованным.
Равным образом опыты, поставленные для проверки фугасного
действия снарядов, показали, что здесь некристализованный
тротил значительно уступает кристаллизованному.
Если к изложенному добавить, что тротил, перекристаллизо-
ванный из спирта, имеющий температуру затвердевания не ниже
80°, является химически и физически стойким веществом, мало
чувствительным к удару и другим механическим воздействиям,
обладает хорошей восприимчивостью к детонации, то ясно требова-
ние, существующее во всех странах, о снаряжении снарядов в мир-
ное время тротилом, перекристаллизованным из спирта, или очи-
щенного другим способом, но равного по качеству перекристалли-
зованному из спирта.
Для изготовления аммонитов обычно применяется тротил некри-
сталлизованный и даже тротиловое масло и другие отходы тротило-
вого производства.
Применение тротила. Тротил применяется во мно-
гих странах для снаряжения всех видов боеприпасов: артиллерий-
ских снарядов, авиабомб, морских мин, для изготовления подрыв-
ных патронов и др. Во время империалистической войны 1914—
1918 гг. тротил применялся в огромных количествах в смесях с ам-
миачной селитрой для снаряжения многих видов боеприпасов (амма-
141
толы да/40 и 83/20; здесь числитель показывает сЬдержание аммиач-
ной селитры, а знаменатель — содержание тротила). В весьма зна-
чительных количествах тротил расходуется на изготовление аммо-
нитов для промышленных взрывных работ: такие аммониты дешевы
и в то же время безопасны в применении.
Наконец, тротил применяется для изготовления капсюлей-дето-
наторов (толовые капсюли), детонирующих шнуров и промежуточ-
ных детонаторов (для последней цели широко применялся раньше
в Англии).
Широкое применение тротила в качестве взрывчатого вещества
обусловлено его малой чувствительностью к механическим воздей-
ствиям, большой бризантностью, химической нейтральностью, срав-
нительной дешевизной и наличием широкой сырьевой базы (в стра-
нах с развитой коксохимической или нефтегазовой промышлен-
ностью). Все эти причины ставят тротил в положение первокласс-
ного по качеству и значению взрывчатого вещества.
11. 2,3,4,б-Тетранитротолуол. Получен Голлеманом1 по сле-
.дующей схеме:
+HNO»
СН3
O2N^NO2
^/ОСНз
NO.,
+NH3 ;
При нагревании метилового эфира m-крезола с азотной кислотой
*(уд. вес 1,52, 99,6% HNO3)2 получается его тринитропроизводное,
представляющее собой бесцветные кристаллы с температурой плав-
ления 92°.
Для получения тринитротолуидина из метилового эфира три-
нитрокрезола раствор последнего в N растворе аммиака в метило-
вом спирте нагревают на водяной бане. Реакция проходит быстро и
с хорошим выходом.
Замещение группы NH2 может быть произведено, как ниже опи-
сано для пикрамида (стр. 209), однако, здесь выход мал. Напротив,
при окислении группы NH2 помощью персульфата получается очень
удовлетворительный выход. Для окисления смешивают предвари-
тельно приготовленные растворы тринитротолуидина и отдельно
1 Holleman, Rec. Trav. Chim. Pays-Bas, 49, 501, 1930.
2 Blanksma, Rec. Trav. Chim. Paxs-Bas, 21, 331, 1902.
142
персульфата натрия в купоросном масле. Смешение ведется без
охлаждения, причем температура поднимается до 100°. Получен-
ный продукт очищается перекристаллизацией из 65%-ной азотной
кислоты или лучше из хлороформа (больше выход и выше качество
продукта).
Тетранитротолуол можно получить также по способу Борше:
нагреванием метилового эфира тринитро-т-крезола со спиртовым
раствором гидроксиламина получают З-гидроксиламино-2,4,6-три-
нитротолуол; при обработке последнего горячей азотной кислотой
получается тетранитротолуол.
В 2,3,4,6-тетранитротолуоле нитрогруппы занимают те же
места, как и в описываемом ниже 2,3,4,6-тетранитробензоле. По-
этому на этих двух веществах можно проследить влияние метиль-
ной группы на свойства соединения.
При вступлении метильной группы сильно возрастает прочность
связи нитрогруппы с ядром. В то время как тетранитробензол
сообщает желтую окраску (вследствие образования пикриновой кис-
лоты) холодной воде через полчаса, тетранитротолуол показывает
эту реакцию только через день. Тетранитробензол разлагается во-
дой при 80° в несколько минут, между тем как тетранитротолуол
при суточном нагревании с водой при этой температуре разла-
гается только на 57%. Тетранитробензол разлагается азотной кис-
лотой, а тетранитротолуол можно перекристаллизовать из 65%-ной
азотной кислоты.
Чувствительность к удару, измеренная высотой падения груза
весом в 2 кг, равна для тетранитробензола 25 см, для тетранитро-
толуола 50 см.
Таким образом вступление метильной группы значительно уве-
личило прочность связи 4-й нитро группы с ядром. Кроме того,
в противоречии с выводами Велера и Венцельберга вступление ме-
тильной группы увеличило прочность ядра, так как на взрыв тетра-
нитротолуола требуется вдвое больше энергии удара, чем на взрыв
тетранитробензола.
Наконец, работоспособность тетранитротолуола, равная 327 см3,
заметно меньше, чем у тетранитробензола (447 см3), что объясняется
худшим кислородным балансом и соответственно меньшей теплотой
взрывчатого разложения.
ПЕРИОДИЧЕСКИЕ МЕТОДЫ НИТРОВАНИЯ ТОЛУОЛА ДО
ТРИНИТРОТОЛУОЛА
Мы познакомимся с основными особенностями периодических
методов нитрования, а равно основными этапами их развития на
трех примерах: 1) старого способа производства тротила в три фазы,
2) способа завода Квин-Ферри, установленного в Англии во время
империалистической войны 1914—1918 гг. и 3) итальянского спо-
соба с полным кислотооборотом.
143
1. Старый способ производства тротила в три фазы. Схему про-
изводства тротила см. на рис. 12.
Толуол доставляется на завод в железнодорожных цистернах,
из которых перекачивается в железные цистерны 1 для хранения.
Отсюда толуол подается по трубам в мерники 2, а из последних
в нитратор 4. Необходимое для нитрования количество нитрацион-
ной смеси подается из специального мерника 3. По окончании
1-й нитрации содержимое аппарата отсасывается помощью вакуум-
насоса (не показан на схеме) в подъемник 5, из которого затем
передавливается для отстаивания и сепарации в колонну 6. Между
подъемником 5 и вакуум-насосом установлен нейтрализационный
аппарат (не показан на схеме) для нейтрализации кислых паров из
воздуха перед его поступлением в вакуум-насос. Отстоявшийся
в колонне 6 мононитротолуол подается в специальный мерник 8,
откуда поступает в аппарат-болтушку 10, где перемешивается с от-
работанной кислотой 2-й нитрации с целью зкстрагирования раство-
ренного в последней динитротолуола. Из аппарата-болтушки’ его
содержимое засасывается в подъемник 11 для передачи в сепаратор
12, из которого болтаный мононитротолуол поступает через мер-
ник 15 в аппарат для 2-й нитрации 17. По окончании 2-й нитрации
содержимое нитратора через подъемник 18 перегоняется в колонну
сепаратора 79; отстоявшийся здесь динитротолуол помощью подъем-
ника 21 подается в мерник 22, обогреваемый паром, и отсюда по-
дается на 3-ю нитрацию в аппарат 24. Отстоявшаяся же отработан-
ная кислота от 2-й нитрации подается из колонны 19 помощью
подъемника 20 в названный выше аппарат-болтушку; отработан-
ная кислота 2-й нитрации поступает после её обработки мононитро-
толуолом в цистерну, откуда расходуется на изготовление смеси для
1-й нитрации.
По окончании 3-й нитрации содержимое нитратора 24 поступает
в сепаратор 26, из которого отсепарированный тротил поступает
в промывной чан, а отработанная кислота — в отстойную колонну
27. Промытый до нейтральной фракции тротил разливается в лотки
(сковородки) 30, где он застывает.
По затвердевании тротила его выламывают из лотков, разбивают
и укупоривают в деревянные ящики.
Промывные воды от промывки кислого тротила проходят через
ловушки 31 и затем поступают в канализацию.
Часть отработанных кислот 3-й нитрации после небольшого
разбавления водой и надлежащего отстоя поступает на освежение
для получения нитрационных смесей для 1-й и 2-й нитраций, а избы-
ток разбавляется водой до содержания 70—72% H2SO4 для выделе-
ния растворенного тротила и поступает дальше на денитрацию.
Равным образом избыток 2-й отработанной кислоты, не используе-
мый на составление смеси для 1-й нитрации, разбавляется водой для
выделения растворенных нитропродуктов, после чего поступает на
денитрацию.
144
26
Рис. 12. Схема производства тротила по старому способу.
/—цистерна д ля толуола; 2—мерник для толуола; 3—мерник для 1-й нитросмеси;
4—нитратор 1-й нитрации; 5—подъемник для продуктов 1-й нитрации; «—колонна
для сепарации; 7—подъемник для отработанной кислоты 1-й нитрации; «—мерник
для сырого мононитротолуола; 9—мерник для отработанной кислоты 2-й нитрации;
10—аппарат для «болтушки»; 11—подъемник для продуктов «болтушки»; 12—сепара-
тор для продуктов «болтушки»; 13—подъемник для болтаной отработанной кислоты
2-й нитрации; 14—отстойная колонна для болтаной отработанной кислоты 2-й нит-
рации; 15—мерник для болтаного мононитротолуола; /«—мерник для 2-й нитро-
смеси; /7—нитратор 2-й нитрации; 18—подъемник для продуктов 2-й нитрации;
19—колонна для сепарации; 20—подъемник для отработанной кислоты 2-й нитрации;
21—подъемник для динитротолуола; 22 — мерник для динитротолуола; 23—мерник
для 3-й нитросмеси; 24—нитратор 3-й нитрации; 25—подъемник для продуктов 3-й
нитрации; 2в—сепаратор для продуктов 3-й нитрации; 27—отстойная колонна для
отработанной кислоты 3-й нитрации; 28— подъемник д ля отработанной кислоты 3-й
нитрации; 29—чан для промывки тринитротолуола; 30—лоток для тринитротолуола;
31—ловушка для промывных вод.
Химия—13—10
145
Рис. 13. Нитратор ста-
рой Конструкций.’
Юписание производства. Нитратор. Для ни-
трования ароматических углеводородов применяются чугунные ап-
параты. Устройство и основные размеры аппарата старой системы
показаны на рис. 13.
Аппарат имеет двойные стенки, образующие рубашку для охла-
ждения или нагревания содержимого аппарата. Наружный котел
делается из обыкновенного литейного чугуна (иногда делается же-
лезный клепаный); внутренний котел, являющийся собственно ре-
акционным сосудом, делается из кремнистого чугуна во избежание
быстрого его разъедания кислотами. Внут-
ренний котел снабжен крышкой, закре-
пляющейся на болтах. Для герметичности
между крышкой и телом нитратора поме-
щается прокладка из асбеста или свинца.
На оси аппарата подвешен вал с насажен-
ными на него винтообразными лопастями,
служащими для перемешивания. Мешалка
делает около 120 об/мин. На валу ставится
маслоуловитель для масла, стекающего из
подшипников, во избежание попадания масла
в аппарат и в реакционную массу. В каче-
стве простейшего маслоуловителя может
служить круговая тарель около вала. Для
добавочного охлаждения внутри аппарата
поставлен кольцевой холодильник, стенки
которого одновременно служат также на-
правляющими поверхностями для жидкости,
движущейся во время работы мешателя.
Междустенные пространства аппарата и
кольцевого цилиндра соединены между со-
бой, и через них последовательно проходит
содержимого аппарата (или пар при нагре-
вода при охлаждении
ваний).
Аппарат снабжен трубами для заливки в аппарат кислот и ни-
труемого вещества, для впуска сжатого воздуха, вентиляционной
трубой для выпуска газов, трубой для выдачи продукта нитрации
(продуктовая труба), трубой для подачи воды или пара в междустен-
ное пространство и выпуска воды из кольцевого цилиндра, термо-
метром, который во избежание поломки движущейся массой жид-
кости окружен железным футляром. Последний для лучшей пере-
дачи тепла термометру либо заполняется купоросным маслом, либо
делается в виде трубки, открытой с нижнего конца, которая запол-
няется жидкостью, находящейся в нитраторе.
Термометры должны быть снабжены паспортом, содержащим
поправки показаний термометра. Для удобства работы у аппарата
на соответствующем расстоянии от крышки нитратора, на которой
находятся приборы для управления аппаратом во время процесса
нитрования, устраивается железный помост. Внутренний котел
146
Рис. 14. Нитратор американского
типа с «пальцами».
нитратора, мешалка и вал быстрее всего разъедаются отработан-
ными кислотами 1-й нитрации толуола, содержащими около 68,5%
H2SO4. Поэтому нитраторы 1-й нитрации быстрее других выходят
из строя.
Часто бывает выгодно с целью увеличения поверхности охлажде-
ния ставить взамен внутреннего
кольцевого холодильника свинцо-
вый или стальной змеевик.
На рис. 14 изображен нитра-
тор американского типа с рифле-
ными стенками и 7—9 внутрен-
ними вертикальными трубками
(пальцами) для охлаждения. Ко-
тел окружен железной рубашкой.
Нитратор снабжен мешалкой про-
пеллерного типа, окруженной на-
правляющим жидкость стаканом.
Пропеллер делает 250—300 об/мин.
Недостатком аппарата являет-
ся трудность отливки чугунного
котла с рифлеными стенками. Пре-
имуществом его является большая
охлаждающая поверхность и энер-
гичная циркуляция жидкости.
Кроме того, в дне нитратора сде-
лано углубление, в которое захо-
дит конец продуктовой трубы; это
позволяет при разгрузке нитра-
тора полностью освободить его от
реакционной массы.
1-я нитрация. Толуол
подается из цистерны в мерник
1-й нитрации помощью центробеж-
ного насоса или насоса системы
Вортингтон. Мерник для толуола
представляет собой цилиндриче-
ский бак из клепаного железа.
Сбоку мерника установлена пере-
полнительная трубка для автома-
тического отмеривания потребного
количества толуола. Толуол заливается в мерник до переполнитель-
ной трубки, по которой избыток его выливается через воронку в
трубу для обратного спуска в подъемник. Отмеренное таким образом
количество толуола подается в нитратор. Способ отмеривания то-
луола по объему имеет тот недостаток, что при нем в зависимости от
температуры наружного воздуха отмериваются разные весовые ко-
личества толуола. Поэтому рекомендуется отмеривать не по объему,
а по весу.
147
Нитрационная смесь отмеривается в мернике такой же кон-
струкции.
В крышке мерника имеются два отверстия: одно для подачи смеси,
другое — для прохождения воздуха при загрузке или разгрузке
мерника. В днище имеется отверстие для выпуска содержимого мер-
ника.
Нитрационная смесь первой нитрации содержит 54—55% H2SO4
и 27—28% HNO3.
Количество смеси берется из расчета, чтобы количество азот-
ной кислоты в ней составляло 103% от теоретического. При расчете
количества смеси учитывается содержание бензина в толуоле.
Процесс 1-й нитрации. В междустенное простран-
ство нитратора и во внутреннее кольцо пускают холодную воду для
предварительного охлаждения аппарата, после чего загружают
в него 1000 кз толуола, пускают мешалку и по охлаждении аппа-
рата до 15—25° начинают подачу в него нитрационной смеси из мер-
ника. Железная труба, по которой подается нитрационная смесь,
снабжена смотровым фонарем, служащим для наблюдения за вели-
чиной струи кислоты, регулируемой вентилем. Расход смеси опреде-
ляется по шкале мерного стекла мерника и записывается через
каждые 30 минут в нитрационный листок.
Во время нитрации люк аппарата не закрывается наглухо мас-
сивной чугунной крышкой, а лишь прикрывается легкой алюми-
ниевой крышкой (то же при 2-й и 3-й нитрациях, а также при ни-
трации других соединений, как-то: бензола, ксилола и т. п.). Замена
чугунной крышки алюминиевой произведена с целью обезопасить
процесс нитрации: в случае неожиданного подъема температуры
в аппарате и разложения нитропродукта алюминиевая крышка легко
сбрасывается при повышении давления в аппарате.
После слива толуола пускают мешалку и охлаждают содержимое
аппарата до температуры 15—18°. Слив кислот производится таким
образом, чтобы происходил плавный подъем температуры. В пер-
вые 2 часа температура должна подниматься не более, чем на 4°
в час, а в каждый последующий час (до достижения температуры 38°)
по 2° в час; начиная с 38°—1° в час. Вся кислота должна быть
спущена при температуре не выше 40°.
Изменение температуры во время процесса нитрации изображено
на диаграмме (рис. 15).
. По окончании слива в течение 40 минут продолжается перемеши-
вание при охлаждении, причем температура немного падает, вслед-
ствие того что теплота реакции при незначительном количестве ни-
трующегося в этой фазе толуола меньше количества тепла, отнимае-
мого охлаждающей водой. После этого выключают охлаждение и,
если нужно, паром в течение 10 минут поднимают температуру до
50° и делают при этой температуре часовую выдержку.
Проверяют конец реакции исследованием полученного продукта
в лаборатории. Моно нитросоединение должно содержать не выше 1%
толуола (отгон до 150°) и иметь удельный вес не менее 1,17.
148
В случае, если мононитротолуол содержит недонитрованного
толуола больше 1%, нитратор охлаждают до 40° и в аппарат до-
бавляют обычным порядком соответствующее количество смеси (из
расчета 103% теории по количеству недонитрованного толуола,
вновь поднимают температуру до 50° и делают получасовую вы-
держку. Вновь отбирают пробу для анализа.
Продолжительность операции нитрования зависит от темпера-
туры охлаждающей воды: зимой достигает 12—13 часов, летом же
при температуре охлаждающей воды 20—25° затягивается до (30—
36 часов.
гоооео 60 60 60 SO 60 60 60 60 60 № 60 60 60 60 60 60 60 миидты
12 3 4 5 6 7 8 9 10 П 12 13 10 15 16 П № часы
Рис. 15. Температурная диаграмма нитрации толуола в моно-
нитротолуол.
Слив смеси продолжается около 9 часов, причем в первые 41/2 часа
удается слить % смеси, а в остальные часа — 2/3 смеси.
При 1-й нитрации наблюдаются следующие ненормальности.
1. При быстром приливании кислот и слабом охлаждении может
иметь место подъем температуры до 60° и выше. При этом умень-
шается выход вследствие испарения толуола и ухудшается качество
вследствие происходящего при этом окисления и осмоления про-
дукта.
2. Если нитрационная смесь приготовлена на плохо отстояв-
шихся отработанных кислотах, то при повышенных температурах
резко усиливаются процессы окисления и осмоления. При этом
образуется пена, иногда настолько большая, что масса частью вы-
ливается через люк аппарата на площадку. Для прекращения вспе-
нивания обычно приходится останавливать на 2—3 часа спуск кис-
лоты, не прекращая перемешивания.
Сепарация. По окончании выдержки содержимое аппарата
передается через подъемник в моновую колонну или в подъемник-
сепаратор (рис. 16), а в междустенное пространство нитратора пус-
кается холодная вода для охлаждения аппарата.
149
Сепарация основана на разности удельных весов мононитрото-
луола (около 1,17) и отработанной кислоты (около 1,6) и дости-
гается простым отстаиванием в течение 20—30 минут.
Сепарационные колонны (рис. 17) склепаны из котельного
железа, опаяны внутри рольным свинцом; устанавливаются на
кирпичном фундаменте. На верху колонны
устроены мостики, чтобы можно было осмо-
треть внутренность колонны.
Подъемник-сепаратор представляет собой
сосуд из кислотоупорного чугуна с кониче-
ским днищем, до которого доходит отводя-
щая труба. По окончании от-
Рис. 17. Сепараци-
онная колонна.
стаивания, заканчивающегося
через 20—30 минут, в подъем-
ник подается сжатый воздух.
Вначале из подъемника выго-
няется нижний слой — кисло-
та, которая направляется в от-
стойные колонны для дополни-
тельного отстоя взвешенных
нитропродуктов. Моно нитрото-
луол, появление которого на-
блюдается в фонарь, поставлен-
ный на отводящей трубе, на-
правляется в болтушку с отра-
Рис. 16 Подъ-
емник-сепара-
тор.
ботАнной кислотой 2-й нитрации, после чего нитропродукт посту-
пает на 2-ю нитрацию.
Выход мононитротолуола составляет 1,42—1,43 (около 95% от
теории), тогда как теоретический выход равен 1,49 на 1 в. ч. толуола.
Причинами потерь являются:
1) растворение нитропродуктов в отработанной кислоте до 1%;
2) частичное испарение толуола в нитраторе во время нитрации;
3) окислительные процессы.
Мононитротолуол из сепарационной колонны спускают в мер-
ник для сырого мононитротолуола, а отработанные кислоты через
подъемник перегоняют в отстойную колонну, в которой происходит
дополнительный отстой кислот. Выделяющийся здесь мононитро-
толуол периодически спускают в мерник и передают на болтушку,
а отработанную кислоту отправляют на денитрацию.
Средний состав отработанной кислоты следующий: 68,5% H2SO4,
0,5% HNO3, 1,5% N2O3, 0,7—0,8% нитропродуктов; уд. вес кис-
лоты 1,62.
2-я нитрация ведется в нитраторе такого же устройства
и размеров, как и 1-я нитрация.
Состав нитрационной смеси для 2-й нитрации: 67% H2SO4,
23% HNO3, 10% Н2О.
Количество смеси берется из расчета, чтобы количество азотной
кислоты в ней составляло 111% от теории.
150
Процесс 2-й нитрации. В охлаждающую рубашку и
внутреннее кольцо нитратора пускают воду, охлаждают аппарат
до 20—25°, загружают в него 1200 кг болтаного мононитропро-
дукта, одновременно пускают мешалку и начинают прилив нитра-
ционной смеси из мерника. Приливание регулируется таким обра-
зом, чтобы температура в аппарате повышалась не более как на 2°
в 5 минут. Вся кислота должна быть прилита при температуре не
выше 80°. Изменение температуры во время процесса нитрации пока-
зано на температурной диаграмме (рис. 18).
По окончании слива кислоты поддерживают температуру 80°
в течение 1 часа, после чего нагревают паром в течение часа до 105°
и делают часовую выдержку при этой температуре.
VI
Рис. 18. Температурная диаграмма 2-й нитрации.
Конец операции контролируется по температуре затвердевания
кислого динитропродукта. Если она ниже 52°, то охлаждают аппа-
рат до 70—80°, добавляют еще нитрационной смеси и вновь подни-
мают температуру до 105°, производят получасовую выдержку и
вновь испытывают полученный продукт. Обычно это бывает при не-
правильной дозировке смеси или при неправильном ведении про-
цесса нитрации. Например, если во время нитрации происходят
температурные скачки, то улетучивается повышенное количество
азотной кислоты.
Иногда пониженная температура затвердевания кислого дини-
тротолуола получается при ошибочной подаче избыточного коли-
чества смеси вследствие растворения азотной кислоты в динитро-
толуоле. В этом случае динитротолуол сильно пахнет азотной кис-
лотой. Дабы не терять азотную кислоту в отработанной кислоте,
в аппарат после его охлаждения до 70—80° подают незначительными
порциями (по 20—30 кг) моно нитротолуол, поднимают температуру
до 105°, делают выдержку, затем вновь испытывают полученный ди-
нитротолуол.
151
Пониженная температура может получиться при наличии в ди-
йи^ротолуоле значительного количества тринитротолуола. В этом
случае такой продукт без предварительного исправления направ-
ляется на 3-ю нитрацию.
Наконец, пониженная температура может быть вызвана наличием
посторонних примесей, продуктов окисления и осмоления, являю-
щихся результатом неправильно проведенных 1-й и 2-й нит-
раций.
В этом случае нельзя улучшить качества продукта, и аппарат
разгружают, а динитротолуол перерабатывают отдельно от осталь-
ной массы кондиционного динитротолуола, чтобы не загрязнять всего
продукта.
Операция 2-й нитрации протекает наиболее ровно, редко при ней
бывают какие-нибудь ненормальности.
По окончании нитрации содержимое аппарата отсасывают в подъ-
емник и отсюда передают в сепарационную колонну 2-й нитрации
или в железный сепаратор, устроенный аналогично сепаратору
1-й нитрации, но с рубашкой для обогрева паром. При значитель-
ной разнице удельных весов динитротолуола (1,34) и отработанной
кислоты (1,7) практически полное их разделение происходит в 25—
30 минут.
Отделившийся на верху сепарационной колонны динитротолуол
сливается в мерник, обогреваемый паром, и из последнего подается
затем на 3-ю нитрацию.
Выход динитротолуола составляет 1,26 ч. динитротолуола на
1 ч. мононитротолуола (против теоретических 1,32).
Болтушка. В отработанной кислоте от 2-й нитрации
остается значительное количество растворенного и взвешенного ди-
нитротолуола, всего до 4% и до 2,5% HNO3. Поэтому она после
отстаивания обрабатывается мононитротолуолом, который экстра-
гирует из нее взвешенный и растворенный в ней динитропродукт.
Одновременно мононитротолуол и находящийся в нем толуол нит-
руются содержащейся в обработанной смеси азотной кислотой.
Эта операция выполняется следующим образом: отстоявшаяся
в сепарационной колонне отработанная кислота от 2-й нитрации
подается в аппарат, снабженный мешалкой и рубашкой для нагре-
вания. Обычно для этой цели применяют обыкновенный нитрацион-
ный аппарат. Загрузку берут равной количеству отработанных кис-
лот, получающихся от одной нитрации, охлаждают до 30—40°, за-
гружают еще около 50 кг слабой азотной кислоты из конденсацион-
ной установки. Приливают при температуре не выше 35° мононитро-
продукта в количестве 1000 кг тонкой струей; затем нагревают до
55° (по 2° в 5 минут). По достижении 55° делают часовую вы-
держку, охлаждают до 40° и перегоняют содержимое аппарата
в сепаратор.
Отработанные кислоты содержат после болтушки: 0,8% HNO3,
0,7—0,8% нитропродуктов. Удельный вес мононитротолуола подни-
мается от 1,172 до 1,218.
162
Сравнение данных растворимости моно- и динитротолуола пока-
зывает, что растворимость динитротолуола меньше мононитрото-
луола, а именно, в 80%-ной серной кислоте при 25° растворяется
1,56% моно- и 0,9% динитротолуола.
Для чего, в таком случае, применяется описанная выше бол-
тушка?
При применении болтушки достигается следующее:
1. Содержание динитротолуола в отработанной кислоте повы-
шено, главным образом, за счет взвешенного в кислоте динитрото-
луола, легко растворяющегося в мононитротолуоле при бол-
тушке.
2. Растворимость нитропродуктов в отработанной кислоте падает
^следствие уменьшения в ней содержания азотной кислоты с 2,5
до 0,8%, а в случае добавления слабой азотной кислоты
при болтушке — еще и за счет уменьшения концентрации серной
кислоты.
3. Экономически выгоднее терять в отработанной кислоте (если
она поступает на денитрацию) моно нитротолуол, а не динитротолуол.
Кроме того, значительно легче денитровать отработанную кислоту,
содержащую в растворе моно-, а не динитротолуол.
4. Используется содержащаяся в отработанной кислоте азотная
кислота, как указано выше.
5. Основное значение болтушки заключается в том, что она
позволяет охлаждать кислоты и передавать их уже охлажденными.
Не болтаные горячие кислоты, например от 2-й нитрации,
содержат в растворенном виде значительное количество нитропро-
дуктов, которые выкристаллизовываются при охлаждении и заби-
вают трубопроводы и аппараты.
3-я нитрация ведется в нитраторе такого же устройства,
как 1-я и 2-я, но без внутреннего кольца.
Состав нитрационной смеси при этом: 82 — 83% H2SO4 и 17,5—
18,5% HNO3.
Количество нитрационной смеси берется из расчета, чтобы в смеси:
содержалось 164% HNO3 от теории (или 570 кг моногидрата HNOa
на 1000 кг динитротолуола).
Загрузка динитротолуола может быть произведена двумя спо-
собами:
1) через мерник;
2) непосредственно из сепарационной колонны в нитратор. В по-
следнем случае в нитраторе предварительно замеряется измеритель-
ной линейкой мертвый остаток тротила с отработанной кислотой
(то количество, которое не может быть отсосано из аппарата вслед-
, ствие того, что продуктовая труба не доходит до дна нитра-
, тора).
Процесс 3-й нитрации. В аппарат подается из мер-
! ника 1000 кг динитротолуола и затем нитрационная смесь. Прили-
[ вание смеси и охлаждение регулируются таким образом, чтобы
| температура в аппарате держалась в пределах 72—77°. Слив про-
153
•
должается 31/2 часа. Большая скорость слива не допускается, вслед-
ствие того что в этом случае при температуре слива динитротолуол
не успевает пронитроваться, а при дальнейшем повышении темпе-
ратуры по окончании слива начнется бурная реакция со значитель-
ным подъемом температуры, с большим газообразованием и выбра-
сыванием реагирующей массы из аппарата. Слив регулируется та-
ким образом, чтобы в течение первых 2 часов было слито не более
1/3, а в оставшиеся 1г/5—2 часа — остальные 2/3 назначенного ко-
личества смеси.
После слива всей смеси наблюдают за подъемом температуры,
рёгулируя его охлаждением водой или нагреванием паром (так, чтобы
температура в первые 30 минут поднималась не быстрее 1° в 5 ми-
нут, а затем не более 2° в 5 минут до достижения 115°). При этой
температуре делают часовую выдержку.
По окончании выдержки останавливают мешалку на 15 минут
и отбирают пробу для определения температуры затвердевания кис-
лого тринитротолуола.
Если тротил предназначен для последующей сульфитной очистки,
то температура затвердевания влажного тротила (под водой)
должна быть не меньше 74,2 или 77,5° в пересчете на сухой
продукт Ч
По окончании нитрации содержимое нитратора отсасывают
в подъемник, а отсюда перегоняют сжатым воздухом в так называе-
мую тротиловую колонну. Во избежание затвердевания тротила при
перегонке продукта нитрации в тротиловую колонну все проходимые
им сосуды и трубы делаются с кожухом для обогревания паром.
Для уменьшения расхода пара следует сперва прогнать через си-
стему горячую кислоту для обогрева сосудов и труб, а затем уже
тротил.
Тротиловая сепарационная колонна устроена подобно выше-
описанной моновой колонне, отличаясь от нее большими размерами
и тем, что в верхней части колонны приделаны патрубки с клепа-
ными жолобами, обложенными внутри свинцом, для спуска тротила.
Кроме того, в верхней части колонны поставлены свинцовые зме-
евики для нагревания кислоты и тротила в случае понижения тем-
пературы или охлаждения содержимого колонны и затвердевания
тротила.
В колонне тротил обычно отстаивается 20—30 минут, после чего
его спускают по жолобу в.промывные чаны, а отработанная кислота
разбавляется водой.
Горячая отработанная кислота содержит до 14% растворенного
тротила. Если она предназначена для приготовления смеси для
2-й нитрации, то она разбавляется в сепарационной колонне до со-
держания 82% H2SO4; если же она поступает для приготовления
1 Температура затвердевания влажного тротила в присутствии избытка
воды, т. е. под водой, на 3,3° меньше, чем у высушенного тротила.
154
смеси для 1-й нитрации, то ее разбавляют до 74—П°/о HgSO4. После
этого отработанные кислоты переливаются в отстойные колонны, где
они отстаиваются в течение 10—20 дней; затем они поступают либо
на изготовление новых нитрационных смесей, либо на денит-
рацию.
Описанный старый способ производства тротила применялся дли-
тельное время на многих заводах. Способы работы отдельных заводов
отличались только в отдельных деталях, сохраняя основные приемы
процесса.
В послевоенное время опубликовано несколько способов произ-
водства тротила, которые внесли ряд существенных изменений в преж-
нцй процесс.
2. Производство тринитротолуола на английских заводах во время
империалистической войны 1914 —1918 гг. х. Английские заводы
отказались от точных молекулярных стадий нитрации до моно- и
динитротолуола как промежуточных стадий при получении трини-
тротолуола. С целью максимального извлечения азотной кислоты
из отработанной кислоты в 1-ю стадию превращали только поло-
вину толуола в нитротолуол. Для удобства отделения динитрото-
лубла от отработанной кислоты при температуре около 30—35°
(необходимой для обеспечения максимальной полноты отделения)
нужно, чтобы нитропродукт при зтих температурах был жидким.
Это достигалось частичным нитрованием мононитротолуола до ди-
нитротолуола (см. стр. 114).
На заводах, изготовлявших тринитротолуол из толуол-бензина;
в 1-й фазе получали моно нитротолуол.
Производство тринитротолуола на заводе
Квин-Ферри. 1-я фаза (супердетолуация). В 1-й фазе гото-
вился мононитротолуол из каменноугольного или нефтяного толуола.
Мононитротолуол поступал на промывку (болтушку) отработанной
кислотой 2-й нитрации.
' Эта операция называется супердетолуацией. Ее целью
является экстракция растворенного тринитротолуола и использо-
вание непрореагировавшей азотной кислоты для нитрации моно нитро-
толуола в динитротолуол (а в случае мононитротолуола из каменно-
угольного толуола также для нитрации ненитрованного толуола
в мононитротолуол), осветление отработанной кислоты и приведение
ее в состояние, годное для денитрации.
Болтаный мононитротолуол нитровался дальше смесью, изго-
товлявшейся на всей отработанной кислоте 3-й нитрации с добав-
лением 50—55%-ной азотной кислоты из конденсационных установок
и промывной воды из чана предварительной промывки тринитрото-
луола.
Эта операция называлась детолуацией, но она по су-
ществу являлась 2-й нитрацией, при которой получался PL—
13/4НТ.
1 Man. of TNT, стр. 4.
155
Схема производства тринитротолуола на заводе Квин-Ферри
11/г-1®/4-НТ + кислотная смесь для 3-й нитрации
кислый тринитротолуол отработанная кислота
I 3-й нитрации
промывка отработанная кислота 3-й
I нитрации-р5О°/о-ная азот-
I на я кислота 4-
кислая npoiMbiB-
ная вода
тринитротолуол
кислая промывная вода +
болтаный мононитротолуол
(детолуация или 2-я нитра-
ция)
толуол + кислая смесь
I
отработанная. мононитротолуол
кислота
I I
ф Ф
I1/,-I3/, НТ отработанная
кислота +
-|- мононигротолуол
^супердетолуа'ция или Сол-
тушка)
на денитрацию
1
Ф
болтаное моно
на детолуацию
I
Ф
отработанная
кислота
I
Ф
на денитра-
цию
Производство тринитротолуола идет в три фазы: толуол->НТ->
Р/г-Р/^НТ ЗНТ.
1-я нитрация ведется, как при старом способе, описан-
ном выше.
2-я нитрация (детолуация). Отработанную кислоту сли-
вают в нитратор (по английскому обозначению — детолуатор) при
температуре около 80°; пускают мешалку, дают охлаждение и по-
дают последовательно мононитротолуол, слабую азотную кислоту
и воду из чана первоначальной промывки. При предварительной
промывке получается промывная вода, содержащая 14% HNO3;
здесь регенерируется около 90% всей растворенной в тринитрото-
луоле азотной кислоты, что составляет около 65 кг HNO3 на 1 тп
тринитротолуола.
166
.Слив мононитротолуола ведется при температуре 75—80°; при
этой же температуре делается получасовая выдержка.
После этого реакционную массу охлаждают до 50° и добав-
ляют воды с целью уменьшения растворимости нитропродуктов
в отработанной кислоте.
Температуры затвердевания полученного промытого сухого про-
дукта колебались в пределах 24—44,5° и соответственные пределы
уд. весов 1,31—1,39.
Супердетолуация (болтушка) необходима тогда, когда отрабо-
танная кислота 2-й нитрации не используется для 1-й нитрации,
а поступает на денитрацию.
Отработанная кислота 2-й нитрации (детолуации), разбавленная
во^ой, содержит до 1—2% нитропродукта, состоящего из мононитро-
толуола и динитротолуола. Эта кислота растворяет мононитротолуол
в количестве около 0,3%. Остальное количество нитропродуктов
содержится во взвешенном состоянии.
Супердетолуация производилась таким образом, что одновременно
в аппарат подавались при температуре около 50° отработанная кис-
лота и моно нитротолуол. После слива мононитропродукта добавляли
воду для разбавления отработанной кислоты до 70%. Наконец,
охлаждали до 20—35° при перемешивании.
Английские источники указывают, что если отработанная кислота
подана раньше мононитротолуола, то требуется больше времени на
экстрагирование растворенных и взвешенных нитропродуктов, чем
при их параллельном сливе.
После супердетолуации в отработанной кислоте остается около
0,4—0,5% мононитротолуола в растворенном (0,3%) и эмульгиро-
ванном (0,1—0,2%) виде и незначительные количества HNO3.
Полученный после операции нитропродукт поступает на 3-ю ни-
трацию, а отработанная кислота — на денитрацию.
3-я нитрация. Состав смеси в этом случае таков: 80%
H2SO4, 18% HNO3 и 2% Н2О.
Содержание воды в смеси обусловлено недостатком олеума и,
слёдовательно, необходимостью изготовления смесей с применением
значительного количества купоросного масла.
В аппарат загружается смесь, а динитротолуол подается к смеси
при температуре ниже 66°. Обычно после слива динитротолуола не-
обходимо непродолжительное нагревание для достижения темпера-
туры 66°. При 66° начинается реакция, и температуре загрузки
дают повышаться на 3° в 10 минут до 90° и затем на 5° в 10 ми-
нут до 100°.
При 100° делают выдержку до получения желательной темпера-
туры затвердевания. Для контроля отбирают пробу на испыта-
ние и с определенного момента это испытание повторяют каждые
полчаса.
Затем реакционную массу охлаждают до 90° и добавляют воду
для уменьшения растворимости тринитротолуола в отработанной
кислоте.
157
Рйс. 19. Диаграмма распределения азота
при производстве тротила в процентах от
азота, содержавшегося в исходной селитре.
После подачи воды массу перемешивают в течение 5 минут и пе-
редают затем содержимое аппарата в сепаратор.
Особенности английского способа. Отмечаем следующие
особенности английского способа по сравнению со старым способом.
1) отказ от точных молекулярных стадий нитрации и замена их
промежуточными;
2) использование всей отработанной кислоты 3-й нитрации для
2-й нитрации;
3) применение для изготовления 2-й смеси слабой азотной кислоты
(50%-ной) и промывной
воды от предварительной
промывки тротила;
4) прилив динитрото-
луола к смеси при 3-й
нитрации;
5) прилив динитрото-
луола к смеси при тем-
пературе ниже 66° с по-
следующим (после слива
всего динитротолуола) ее
повышением до 100°.
Составление ба-
ланса на заводе
Квин-Ферри. Кроме
обычного контроля про-
изводства на зтом заводе
систематически составлял-
ся баланс толуола, сер-
ной и азотной кислот с точ-
ным распределением и
правильным указанием различных источников потерь материалов.
Как пример такого баланса приводится диаграмма (рис. 19),
показывающая распределение азота в процессе за период 28 дней
(1918 г.). Составление подобного рода диаграмм требует очень тща-
тельной системы контроля установки и точной проверки результа-
тов. Только в этом случае такие записи имеют реальную ценность.
При подобном ведении работы на заводе Квин-Ферри были обна-
ружены и устранены некоторые невыясненные источники потерь,
в результате чего удалось понизить расходные козфициенты.
3. Итальянский способ производства тринитротолуола с полным
кислотооборотом. Процесс построен таким образом, что вся отрабо-
танная кислота 3-й нитрации поступает на изготовление смеси
для 2-й нитрации, а вся отработанная кислота 2-й нитрации —
на 1-ю.
1-я нитрация. Состав смеси при зтом: 66% H,SO4, 6%
HNO3, 2,6% N2O3, 25,4% Н2О.
В нитратор загружают 6000 кг смеси и при 15° начинают.при-
лив толуола или толуол-бензина с содержанием 500 кг толуола.
158
К концу прилива температура должна быть 40°; делают часовую
выдержку, массу охлаждают до 25° и сепарируют.
2-я нитрация. В нитратор загружают отработанную кис-
лоту от 3-й нитрации и 40%-ную азотную кислоту (из абсорбционной
установки мастерской нитрации и денитрации).
Таким образом получают 7680 кг смеси состава: 73% H2SO4r
7% HNO3, 2,7% N3O3, 17,3% Н2О.
Затем смесь охлаждают до 50° и к ней приливают (при 50°)
1000 кг мононитротолуола. Подъем температуры: 1° в 5 минут.
К концу слива температура достигает 75°. После этого производяг
нагрев до 100° и делают часовую выдержку. Потом охлаждают до
66° и сепарируют.
* Полученная отработанная кислота поступает целиком на изго-
товление смеси для 1-й нитрации. Кислотооборот, при котором смесь
готовится в нитраторе путем смешения неохлажденной отработанной
кислоты с азотной (или меланжем), получил у нас название горя-
чего кислотооборот а.
3-я нитрация. Применяется смесь состава: 83—85%
H2SO4 и 16,5—17,5% HNO3. Смесь приливают к динитротолуолу
при температуре 80—85°, затем делают часовую выдержку при 110°.
После сепарации вся отработанная кислота от 3-й ниграции по-
ступает на изготовление смеси для 2-й нитрации.
Особенности итальянского способа нитрации
сравнительно с описанным старым способом и английским:
1) применение полного кислотооборота;
2) применение так называемого горячего кислотооборота;
3) слив толуола и мононитротолуола к смеси (обратный слив);
4) отметим, что приливание динитротолуола к смеси при высо-
кой температуре 80—85° не является новым; но при зтой температуре
идут повышенные окислительные процессы, и потому лучше вести
слив при температурах 72—77°, при которых окислительные про-
цессы меньше, а скорость реакции достаточна.
НЕПРЕРЫВНАЯ НИТРАЦИЯ
Аппараты непрерывного действия несравненно выгоднее аппара-
тов периодического действия как с точки зрения экономики, так и
техники безопасности. Энергичная разработка вопроса непрерыв-
ной нитрации началась за границей после известного взрыва боль-
шой установки для нитрации бензола в Руммельсбурге около Бер-
лина в феврале 1914 г., при котором были убитые и раненые. Этот
взрыв произошел, повидимому, оттого, что рабочий, не пустив ме-
шалку, открыл кран для подачикислотной смеси. Кислотная смесь,,
подававшаяся к бензолу, садилась при зтом на дно аппарата, и
когда рабочий через некоторое время пустил мешалку, то одновре-
менно вступило в реакцию большое количество бензола. Вследствие
быстрого выделения при зтом большого количества тепла произошло
сильное разогревание, и бензол закипел вместе с кислотной смесью.
Образовавшееся большое количество паров бензола, окислов азота
16»
I
Рис. 20. Аппарат Вейлер-тер-Мера.
л
ж азотной кислоты не могло достаточно быстро удалиться из аппарата
через узкое отверстие вытяжной трубы. Развившимся в результате
итого давлением с аппарата была сорвана тяжелая чу унная крышка,
-закрывавшая люк. Искра, образовавшаяся при падении крышки,
воспламенила смесь паров бензола, нитробензола, окислов азота
и воздуха.
Из большого числа различных систем непрерывно действующих
нитрационных аппаратов, судя по литературным данным, получили
практическое применение следующие: 1) аппарат Вейлер-тер-Мера
2) тутольтурбо; 3) аппарат Ку-
берского; 4) английский сек-
ционный аппарат.
1. Аппарат Вейлер-тер-Мера
(рис. 20) состоит из цилиндра,
снабженного рубашкой для
охлаждения. В нижнем конце
он имеет 2 штуцера для пода-
чи бензола и кислотной смеси,
а наверху на крышке, — вы-
пускной штуцер. Мешалка со-
стоит из вертикального вала,
на который насажено 6 винтов.
Аппарат разделен двумя
неподвижыми дырчатыми ди-
сками на 3 камеры, которые,
в свою очередь, делятся попо-
лам тремя подвижными диска-
ми, насаженными на вал ме-
шалки. Реагирующие жидкости
поступают из расположенных
выше баков (с очень точно рабо-
тающими регулировочными приспособлениями^ эквивалентных коли-
чествах в нижнюю камеру аппарата и здесь тесно смешиваются. Ско-
рость подачи бензола и кислотной смеси, поверхность охлаждения
и количество охлаждающей воды подбираются таким образом, чтобы
в нижней камере постоянно удерживалась оптимальная темпера-
тура 50°. Реакционная смесь должна пройти в аппарате длинный
путь, обусловленный наличием неподвижных и подвижных дисков.
Наконец, она выходит наверху аппарата в виде готового продукта,
состоящего из эмульсий отработанной кислоты и нитробензола.
Этот продукт поступает в присоединенные к нитратору сборники,
также снабженные мешалками, где производится отбор пробы, и там
же в случае надобности могут быть сделаны небольшие поправки.
Из описания видно, что аппарат постоянно наполнен жидкостью
ж никогда не содержит больших количеств бензола вместе с боль-
шим же количеством кислотной смеси; поэтому процесс можно в любой
момент прекратить и возобновить снова. При остановке нитробензол
собирается над отработанной кислотой. При возобновлении работы
160
в нижней части быстро происходит необходимый подъем температуры
и непрерывный процесс продолжается дальше нормальным образом.
Аппарат имеет объем около 3 м3. В час в него подается около
300 кг бензола и соответствующее количество нитрационной смеси.
Для полного окончания реакции продукт должен находиться
в аппарате около 4 часов.
Производительность аппарата около 9 т нитробензола
2. Турбоаппарат Неймана, или ту-
тольтурбо (рис. 21) состоит из сосуда
А с двойными стенками и вращаю-
щегося полого цилиндра В. Последний
имеет такие размеры, что между ним и
восудом А остается только небольшой
промежуток, образующий реакционное
пространство. Нитруемое вещество и
кислотная смесь подаются снизу по
двум трубам и после окончания реак-
ции вытекают вверху нитратора у С.
Находящийся во время процесса нит-
рации в движении вращающийся ци-
линдр В имеет на наружной стенке
лопаточки а, проходящие между лопа-
точками Ь, насаженными на внутренних
(неподвижных) стенках нитратора.
Помощью этих лопаточек происходит
энергичное перемешивание. Охлажде-
ние производится помощью наружной
Рис. 21. Аппарат Неймана,
или тутольтурбо.
рубашки аппарата. В первый период
эксплоатации этого аппарата охлажде-
ние производилось при участии вра-
щающегося цилиндра, в кольцевое пространство которого (коль-
цевое пространство образовано между стенками вращающегося ба-
рабана и концентрически вставленного в него второго барабана)
пропускалась вода для охлаждения. Однако охлаждение помощью
вращающегося барабана оказалось излишним.
Емкость этого аппарата 180 л. По словам Наума, в таком аппа-
рате можно пронитровать в сутки около 4500 кг толуола и получить
соответственно около 9000 кг динитротолуола.
В технической литературе иногда приводятся колонны Кубер-
ского, достоинство которых якобы заключается в том, что в них вовсе
отсутствуют мешалки.
3. Аппарат Куберского (рис. 22). Исходные материалы — бензол
или толуол и кислотная смесь — непрерывно притекают в нитра-
ционную колонну, причем бензол, поступающий снизу в колонну
по трубе а, заполняет ее, все более и более нитруясь по мере
продвижения вверх, и выходит по верхней выходной трубе с.
Напротив, кислотная смесь вступает в колонну сверху b и вслед-
ствие более высокого удельного веса тонет в бензоле, проходит через
Химия—13—11
161
Рис. 22. Аппарат Куберского.
а и b—впускные трубы, сид —выпускные
трубы продукта и отработанной кислоты;
вода для охлаждения входит в змеевик d
через е и выходит через f. г
парфорйрованные перегородки в мелко распределенном виде. Та-
ким образом здесь достигается реакция между опускающейся кислот-
ной смесью и поднимающимся бен-
золом по принципу противотока.
Отработанная кислота собирает-
ся на дне колонны и уходит по
трубе д.
В нитрационной колонне ус-
троены змеевики для охлаждения
водой. Вода входит в змеевик dче-
рез е и выходит через /. Обе реа-
гирующие жидкости — бензол и
кислотная смесь — проходят одна
сквозь другую в совершенном про-
тивотоке, так что подходящая
к основанию отработанная кислот-
ная смесь постоянно соприкасается
со свежим бензолом, а полученный
нитробензол в верхней части ко-
лонны обрабатывается свежей кис-
лотной смесью. Ход реакции на-
блюдают по плотности нитробен-
зола, которая определяется по
ареометру, подвешенному в пере-
ливе. По показанию этого арео-
метра устанавливается или регу-
лируется относительное количе-
ство бензола и кислоты.
4. Английский способ непрерыв-
ной нитрации. Процесс, установ-
ленный в 1917 г. на заводе в Ольд-
бюри, основан на принципе про-
тивотока и аналогичен ниже опи-
санной непрерывной промывке
завода Квин-’Ферри (стр. 164).
Крепкая серная кислота и моно-
нитротолуол поступают с противо-
положных концов установки и вы-
ходят соответственно в форме три-
нитротолуола и отработанной
кислоты. Азотная кислота посту-
установки.
пает в промежуточных фазах
Установка состоит из нескольких пар чугунных сосудов. Один
сосуд каждой пары служит смесителем, а другой — отстойником:
и в то же время сепаратором. Из отстойника (сепаратора) нитропро-
дукт проходит в следующий смеситель, кислота же движется в обрат'-
ном направлении, тоже в следующий смеситель. Смесители снаб-i
жены змеевиками для регулирования температуры.
162
ВОДНАЯ ПРОМЫВКА ТРОТИЛА
После отделения от отработанных кислот тротил подвергается
промывке водой для удаления кислот до содержания их не более 1
0,1% для некристаллизованного тротила (т. е. для продукта, не
подвергающегося дальнейшей очистке и предназначенного, напри-
мер, для изготовления аммонитов). Если тротил предназначается
для перекристаллизации, то он должен быть отмыт до содержания
кислотности не более 0,02% (соответственно требованию по кислот-
ности для перекристаллизованного тротила) и не более 0,05%, в
случае если он предназначен для очистки раствором сульфита
натрия.
^Водная промывка может быть произведена в аппаратах как пе-
риодического, так и непрерывного действия.
1. Промывка тротила в аппаратах периодического действия.
Промывка тротила производится в деревянных чанах емкостью
около 1,3 ль3, выложенных внутри рольным свинцом толщиной
6 мм. На уровне дна (или выше на 5 ель, чтобы не спускать с жид-
ким тротилом отстоявшиеся примеси, например песок) находится
выпускное отверстие для тротила, а на высоте 1/3 от дна нахо-
дится второе выпускное отверстие для спуска промывной воды.
Внутри чана расположена свинцовая труба, оканчивающаяся на
дне чана специальным змеевиком с большим числом отверстий для
подачи пара или воздуха для перемешивания.
В пустой промывной чан наливают 500—1000 кг жидкого тро-
тила, затем горячую воду (вес воды приблизительно равен весу тро-
тила) и пускают пар, которым вода нагревается до кипения.
Для более энергичного неремешивания подают сжатый воздух
в течение 3—5 минут после начала кипения. После 10—15-минут-
ного отстаивания спускают воду и вновь добавляют к жидкому тро-
тилу горячую воду. Подобным образом промывку повторяют 6—
8 раз, до тех пор, пока промывная вода не покажет нейтральную
реакцию на лакмус.
Процесс промывки тротила (от момента начала подачи кислого
тротила до момента выпуска промытого продукта из аппарата) про-
должается около 7 часов.
! После спуска последней воды с промытого тротила его выпускают
через нижний кран промывочного бака помощью шланга в желез-
ные противни. Вместимость противня — около 16 кг тротила. Он за-
стывает в противнях через 6—12 часов в зависимости от температуры
воздуха. При его промывке вместе с водяными парами выделяются
ркислы азота (в начале подачи воды к кислому тротилу) и раздражаю-
щие глаза пары тетранитрометана. Окислы азота и тетранитрометан
Ьчень ядовиты, поэтому выделяющиеся при промывке пары следует
Тщательно отводить вытяжной вентиляцией.
‘ При промывке тротила без водооборота расход воды составляет
В л3 и более на 1 т тротила. С применением водооборота расход
1 В пересчете на серную кислоту.
163
воды может быть сокращен в несколько раз; соответственно сокра-
щается при этом расход пара и потери тротила. Однако гораздо
выгоднее вести промывку в аппаратах непрерывного действия, рабо-
тающих по принципу противотока. В этом случае кроме указанного
Рис. 23. Непрерывная промывка английского типа.
сокращается расход рабочей силы, сокращается число аппаратов
и кубатура цеха промывки и, что особенно важно, резко улучшаются
санитарно-гигиенические условия работы.
Вода
Рис. 24. Общий вид непрерывной промывки.
2. Промывка тротила в аппаратах непрерывного действия. В ка-
честве примера опишем непрерывную промывку, установленную во
время империалистической войны 1914—1918 гг. на заводе Квин-
Ферри в Англии.
Устройство аппарата показано на рис. 23 и 24. Аппарат состоит из
девяти секций, по два отделения в каждой (рис. 25); одно из них яв-
164
ляется смесителем, другое — сепаратором. Промывной аппарат сделан
из чугунных пластин, скрепленных болтами; соединения сделаны на
сурике и двойной полосе свинцовой проволоки; перегородка между
смесителем и сепаратором свободно держится в пазах. Каждая
секция представляет собой прямоугольный резервуар, длиной
8,23 м, шириной 1,37 м и глубиной 1,3 м; со-
отношение ширины смесителя и сепаратора
составляет 2:1. Каждая перегородка, разделяю-
щая смеситель и сепаратор, имеет расположенные
в ряд 12 отверстий диаметром 2,5 см на расстоя-
нии 0,4 м от дна чана. Через эти отверстия
эмульсия тринитротолуола проходит из смесителя
в сепаратор. Течение из смесителя в сепаратор
Рис. 25. Попереч-
ный разрез аппа-
рата непрерывной
промывки
происходит благодаря давлению жидкости на
боковые стенки смесителя, образующемуся при
работе мешалки. Вследствие этого уровень жидко-
сти в сепараторе выше, чем в смесителях^ как это
показано на рис. 23. Выпускные отверстия для
тринитротолуола имеют 65,5 мм в диаметре и располагаются
у дна промывного чана; выпускные отверстия для отстоявшейся
в сепараторе воды имеют тот же диаметр, но помещены на рас
стоянии 0,762 м от дна.
Рис. 26. Диаграмма установки для непрерывной промывки
тротила.
На рис. 26 показано направление потоков воды и тротила через
промывной аппарат.
Каждый смеситель оборудован мешалкой диаметром 0,508 м и
трубой с отверстиями у ее нижнего конца для нагревания острым
паром. Окончательно промытый тротил по выходе из аппарата по-
ступает в сепаратор, устройство которого понятно из рис. 27.
Температура в аппарате должна поддерживаться в пределах
95—100°.
При перерыве процесса промывки прекращается . подача тро-
тила в аппарат, и последний поддерживается в рабочем состоянии
нормальным током воды.
Большая часть тринитротолуола, таким образом, продвигается
по направлению к спускному отверстию. Обычно на дне аппарата
165
остается слой тринитротолуола толщиной 2,5 см. Для спуска его
открывают свинцовые заглушки, которые закрывают сточные отвер-
стия на дне отдельных секций.
При спуске промывного аппарата стоки вновь закрываются
свинцовыми заглушками и промывные аппараты наполняются холод-
ной водой. Затем подают пар через паровые нагревательные трубы,
и в то же время начинается приток горячей воды. Когда темпера-
тура воды по всей промывной установке достигнет 95°, пускаются
ТНТ в непрерывную
сушильню
Рис. 27. Сепаратор.
мешалки, и начинают подачу тринитротолуола, вначале медленно,
затем скорость постепенно увеличивается до нормальной. Произво-
дительность описанной установки около 1,5 m тротила в час.
Расход воды на 1 и промытого тротила составляет приблизи-
тельно 2,5 т.
3. Содовая промывка тротила В прежнее время на некоторых
заводах Западной Европы применяли промывку тротила содовым
раствором с целью ускорения отмывки кислот. Однако при такой
промывке имели место случаи воспламенения тротила, вероятно,
вследствие образования тротилатов натрия. Поэтому не следует
допускать промывку тротила содовым раствором.
4. Ликвидация промывных вод. Высаживание тро-
тила. Тяжелым бременем являются для завода красно-коричне-
вые кислые промывные воды, собирающиеся от промывки тротила.
В них содержится до 4% тротила в суспендированном виде и неко-
торое количество растворенного тротила, который по охлаждении
воды выделяется в виде тонкой и рыхлой массы. Для улавливания
этого тротила сточные воды спускаются в бетонные или дере-
вянные отстойники (ловушки), в которых они охлаждаются. При
166
медленном прохождении через ловушки тротил осаждается и соби-
рается в них. Тротил периодически извлекается из ловушек, про-
мывается вместе с кислым тротилом, а отстоявшаяся и прошедшая
через отстойники вода спускается в канализацию.
Нейтрализация отстоявшихсяпромывных
вод. Промывные воды, прошедшие отстойники, содержат в рас-
творенном виде еще незначительное количество тротила и других при-
месей — нитросоединений, а потому являются чрезвычайно ядови-
тыми. Кроме того, они содержат небольшое количество кислот, ко-
торые также заражают почву и водоемы. Поэтому на некоторых
заводах сначала нейтрализуют эти воды, а затем пропускают через
песочные фильтры. Для этой цели к промывным водам добавляют
до нейтральной (или слабо щелочной) реакции известковое молоко
и энергично перемешивают чугунной пропеллерной мешалкой. Ней-
трализованная сточная вода вновь спускается в систему отстойни-
ков, после которых пропускается через два песочных фильтра,
откуда направляется в реку.
5. Использование загрязненного тротила. Если собранный из
ловушек или иных мест тротил сильно загрязнен и не может быть
поэтому пущен на промывку вместе со свежими порциями кислого
тротила, то он может быть очищен следующим способом. Тротил
расплавляют и сливают в железные формы, суженные книзу. При
зтом образуется два слоя: верхний слой состоит из грязи и прочих
примесей. После охлаждения тротила с него снимают слой грязи,
который сжигают. Остальная масса тротила, освобожденная от ме-
ханических примесей, направляется в промывные чаны вместе с
кислым тротилом.
6. Упаковка декристаллизованного тротила. Если тротил пред-
назначен для отправки в декристаллизованном виде, то его после
водной промывки спускают в противни. После остывания в про-
тивнях его выламывают из них, разбивают деревянными лопат-
ками и упаковывают в деревянные ящики по 45 и 80 кг в каж-
дом для отправки на склад.
-7. Технические условия на декристаллизованный тротил. 1. Тро-
тил декристаллизованный может иметь вид или зерен или кусков
весом до 0,5 кг, причем он не должен заключать в себе посторон-
них примесей, видимых на-глаз, не должен иметь признаков под-
мочки и не должен быть темнее эталона.
2. Температура его затвердевания должна быть не ниже 76°.
3. Влажность и летучесть тротила должны быть не более 0,1%.
4. Кислотность его в переводе на серную кислоту (азотной кис-
лоты допускаются только следы) не более 0,1%.
5. Нерастворимых в бензоле примесей не более 0,15%.
Валовая партия тротила должна быть не менее 10 т. Исключе-
ние составляет последняя партия, предъявляемая в конце наряда,
вес которой должен быть не менее 3 т.
8. Обоснование технических условий на декристаллизованный
тротил. Некристаллизованный тротил применяется для производ-
ит
ства аммонитов. Норма для температуры затвердевания 76° уста-
новлена потому, что она легко достигается при производстве тро-
тила, а приготовленный на таком тротиле аммонит удовлетворяет
установленным для него техническим требованиям. Норма влаж-
ности 0,1% установлена с целью уменьшения количества влаги,
вносимой тротилом в аммонит; при повышении содержания влаж-
ности в тротиле до 0,3—0,5% соответственно повышается содержа-
ние влажности в выпускаемых с аммонитовых заводов аммонитах,
что вредно отражается на качестве последних. Кислотность 0,1%
установлена потому, что она достигается без затруднений при водной
промывке; с другой стороны, при таком содержании кислоты не
наблюдалось какого-либо отрицательного влияния ее на качество
аммонита; при повышенной кислотности, например, возможно ее
действие на бумажную гильзу аммонитовых патронов, в результате
чего эти гильзы становятся ломкими и, следовательно, легко разру-
шаются.
Норма нерастворимых примесей предусматривает тот предел
содержания их, который легко достигается при производстве тро-
тила и при котором получают нормальное качество аммонитов.
СПОСОБЫ ПОЛУЧЕНИЯ ОЧИЩЕННОГО ТРОТИЛА
Для получения очищенного тротила применяют три основных
способа:
1) перекристаллизация его из спирта;
2) сульфитная очистка тротила;
3) кристаллизация его из отработанной кислоты.
Кроме того, предлагались и некоторое время применялись
следующие способы:
1) перекристаллизация из других растворителей: мононитро-
толуола, четыреххлористого углерода, бензола и пр.;
2) промывка спиртом, четыреххлористым углеродом и другими
растворителями;
3) очистка промывкой гипохлоритом;
4) очистка мононитротолуола от m-иитротолуола или динитро-
толуола от продуктов денитрации ^-нитротолуола.
1. Перекристаллизация тротила из спирта. Схема произ-
водства (рис. 28). Отвешенный на весах тротил засыпается
в растворитель 3, в который из мерников 1 и 2 предварительно
залито необходимое количество спирта или спиртотолуольной
смеси. Массу подогревают до 75° и по растворении тротила рас-
твор передавливается сжатым воздухом через фильтр-пресс 9 или
другой фильтрующий прибор в кристаллизатор 4. По окончании
процесса кристаллизации содержимое кристаллизатора сливается
в центрифугу 5 для отжимки маточного раствора. Иногда содер-
жимое кристаллизатора сливают в вакуум-воронку, где пред-
168
варительно отсасывается маточный раствор до содержания в тро-
тиле 12—15% спирта, после чего переводят тротил в центрифугу
и фугуют. Тритил выгребается из центрифуги в ящик 6, а маточный
раствор поступает в отстойники 7 для охлаждения и выделения из~
него взвешенного и частично растворенного тротила.
Рис. 28. Схема очистки тротила кристаллизацией.
7—мерник для маточного спирта; 2—мерник для чистого спирта;
растворитель; 4—кристаллизатор; 5—центрифуга; 6—ящик для кри-
сталлизованного тринитротолуола; ~—отстойник для маточного спирта;
S—подъемник; 5—фильтр-пресс.
Описание производства. Аппарат для растворе-
ния тротила в спирте (рис. 29) представляет собой горизонтальный,
цилиндрический котел, имеющий в нижней половине рубашку для
нагревания. Сверху к аппарату присоединены две трубы: одна—для.
заливки спирта, вторая — для
выхода газов. Он имеет дв а люка
для загрузки кускового троти-
ла, закрывающиеся привинчи-
ваюримися крышками с рези-
новыми прокладками. Внутри
цилиндра проходит горизон-
тальный вал с лопастями, слу-
жащими для перемешивания
содержимого аппарата. Вал ме-
шалки делает 20—30 об/мин.
Емкость аппарата — около
4,5—5 м9- ,
Рис. 2 9. Аппарат для растворения'
тротила.
В аппарат заливается спирт, а если работают на спиртотолуоль-
ной смеси, то заливается и толуол. Пускают в ход мешалку и загру-
жают тротил из такого расчета: 900 кг тротила на 240 ведер спирта *'
1 Температура кипения спирта 78°; D*° = 0,804 (при содержании 94%.
С2Н5ОН). Граница взрывчатости смеси паров спирта с воздухом 3,5—19%, или
при содержании в 1 *3 воздуха 67—364 в спирта. При 20° и „760 мм давле-
ния в 1 л3 воздуха при полном насыщении содержится 5,9% (объемных) спирта
(Иордан, Химическая технология растворителей, стр. 338, ОНТИ, 1934).
169
или 1000 кг тротила на 280 ведер спиртотолуольной смеси. Со-
держание толуола в смеси 2—3%.
После загрузки тротила закрывают люк аппарата, пускают пар
в его рубашку и нагревают до 75—78°. При этой температуре выдер-
живают 20—25 минут.
При температуре кипения 100 в. ч. спирта растворяют 22 в. ч.
тротила. Принимая D™ спирта = 0,804, определим вес одного
ведра спирта: 12,3 л • 0,804 = 8,889 8,9 кг. Следовательно, вес
340 ведер спирта равен
3346 кг. Это количество
спирта растворяет 33,6 X
X 22,1=743 кг, или 82,5%
от загруженного тротила
(растворимость тротила
в спирте при 80° равна
22,1 в. ч. тринитротолуо-
ла в 100 в. ч. спирта).
Аналогично приняв вес
280 кг спиртотолуольной
смеси равным 2770 кг (при
расчете по плотности спир-
та), мы определим (без
учета увеличения раство-
римости в присутствии
Рис. 30. Аппарат-кристаллизатор. 2—,3% толуола), что при
температуре кипения спир-
та растворяется 27,7 • 22,1 = 612 кг тротила, или 61,2% от
загруженного тротила. Остальное его количество остается в жид-
кости в виде раствора спирта в тротиле, из которого при охлаж-
дении и перемешивании кристаллизуется тротил.
По окончании выдержки раствор передавливается через фильт-
рующие приборы (фильтр-пресс или приборы упрощенного типа)
в кристаллизатор. Перед началом передавливания содержимого ап-
парата продуктовая линия прогревается паром; после перегонки
раствора производится новая продувка продуктовой линии паром
для очистки ее от осевшего там тротила. Средняя продолжительность
операции растворения с загрузкой и выгрузкой длится 1г/2
часа.
Кристаллизатор (рис. 30) представляет собой желез-
ный цилиндрический котел’, снабженный у дна и с боной рубашкой
для охлаждения, рамочной мешалкой на вертикальном валу для
перемешивания содержимого. Мешалка делает 24—27 об/мин.
Температурный режим при кристаллиза-
ции. Охлаждать раствор следует медленно, с целью получения
крупных кристаллов; изомеры ди- и тринитротолуола и другие при-
меси отлагаются на поверхности крупных кристаллов чистого три-
нитротолуола и легко удаляются при промывке спиртом.
170
Приводим два варианта режима охлаждения:
1-й вариант. В кристалли-
затор подается раствор 240 кг три-
нитротолуола в спирте. Температура
раствора в момент окончания загруз-
ки около 70°.
1) 1х/2 часа выдерживают без
охлаждения. Температура падает
медленно на 5° (с 70 до 65°);
2) после этого охлаждение регу-
лируется так, чтобы падение темпе-
ратуры в аппарате не превышало 3°
за.ч15 минут;
3) по достижении установленной
температуры выгружают содержи-
мое аппарата в вакуум-воронку.
2-йвариант. В кристаллиза-
тор подается раствор 500 кг трини-
тротолуола в спиртотолуольной сме-
си. В момент окончания загрузки
температура раствора около 70°:
1) после загрузки кристаллиза-
тор охлаждают водой до 45°. В этот
период на стенках аппарата нара-
стает корка толщиной 0,5 мм',
2) по достижении 45° выпускают
воду из рубашки и нагреванием па-
ром поднимают температуру на 5°
(до 50°);
3) по достижении 50° опять дают
охлаждение до установленной тем-
пературы аппарата; выгружают в
вакуум-воронку или центрифугу.
Установленная конечная температура зависит от оборота спирта.
Иногда ее приходится менять и в зависимости от качества сырого
тротила. С повышением оборота спирта, т. е. при увеличенном
содержании примеси в спирте, надо выше держать конечную темпе-
ратуру. При плохом качестве исходного сырого тротила приходится
иногда держать конечную температуру выше установленной нормы.
Можно указать следующие величины конечных температур:
для нолевого оборота ................ 25°
» 1-го маточного................... 27°
» 2-го » 28—30°
» 3-го » 30—32°
Средняя продолжительность операции кристаллизации с загруз-
кой и выгрузкой по 1-му варианту 6 часов, по 2-му варианту 7 часов.
Отжимка. Отжимка отработанного спирта от тротила ве-
дется на центрифугах. Для повышения производительности центри-
фуг и уменьшения потерь спирта иногда отжимают предварительно
на вакуум-воронке.
Центрифуга имеет обычное устройство, отличаясь лишь тем,
что для безопасности работы корзина делается латунной или медной;
кроме того, она герметически закрывается во время работы во избе-
жание потерь спирта из-за испарения при фуговке.
Диаметр корзины 1 м, высота 0,5 м. На боковых ее стенках
имеются отверстия диаметром в 5 “мм. На внутренней поверхности
корзины укладывается три слоя сеток: 1-я, ближайшая к корзине,—
с отверстиями 5x5 мм, 2-я, шелковая или медная,— с 3600 от-
верстиями на 1 см2 и 3-я, предохранительная, — из листовой
меди, пробивная. Вместимость центрифуги 100—160 кг отжатого
тротила с 4—5% спирта.
Загрузка центрифуги производится самотеком из кристаллиза-
тора; в случае предварительного отсоса на вакуум-воронке тротил
загружается из воронки в центрифугу алюминиевыми или медными
совками.
171
Вначале центрифугу пускают на тихий ход — около 500 об/мин-
Через 5 минут переводят ее на 1000 об/йин. Это делается с той
целью, чтобы в начальный период (еще при большом содержании
жидкости) не спрессовался тринитротолуол, что сильно затрудняет
фильтрование и увеличивает продолжительность фуговки.
Через 10 минут после начала фуговки производят спиртовую
промывку тротила для удаления маслянистых примесей с поверх-
ности кристаллов и удаления остатков маточного раствора. Подачу
спирта для промывки делают либо на ходу центрифуги, через отвер-
стие в крышке центрифуги, либо останавливают центрифугу,
подают спирт, размешивают тротил совком и вновь пускают центри-
фугу. Дозировка промывного спирта зависит от оборота маточного
спирта, а отчасти и от качества сырого тринитротолуола. Например:
для нолевого оборота................. 1 ведро спирта на центрифугу
» 1-го маточного .............. 2 ведра » » »
» 2-го » 3 » >> » »
> > 3-го » 4 » » » »
При плохом качестве сырого тротила приходится брать в каж-
дом случае на 1 ведро больше, чем здесь показано. Перед подачей
в центрифугу спирта его нагревают до 40°.
После подачи спирта производится отжимка при 1000 об/мин.,
а затем выгрузка тротила в деревянный ящик-ларь при центрифуге
с нижней выгрузкой. Продолжительность операции фуговки с
загрузкой и выгрузкой 50 минут.
Тротил упаковывают в деревянные ящики или брезентовые
мешки и отправляют на сушку. При этом формируют частные пар-
тии из тротила, получаемого от одной загрузки в растворитель.
Следовательно, вес одной частной партии составляет около 800—
900 кг тротила.
Собирание маточных и промывных спир-
тов. Стекающий из вакуум-воронок и центрифуг маточный спирт
несет с собой некоторое количество тротила во взвешенном виде;
часть растворенного тротила выпадает при охлаждении. Этот тро-
тил улавливается в ловушках, состоящих из ряда секций; из послед-
ней секции маточный спирт подается в мерник, находящийся на
площадке над растворителями. Остаток, не вмещающийся в мернике,
качается в бак для 3-го маточного спирта. Остаток всех маточных
спиртов (вместе с 3-м) составляет 3-й маточный, а 4-й маточный по-
ступает на ректификацию. Иногда работают на четырех маточных
и очень редко на пяти. В этом случае, разумеется, слив остатков
производится к соответствующему последнему маточному.
По другому методу работы собирают соответствующие маточные
спирты в сборники и периодически работают только на одном каком-
нибудь маточном, например, 1-м, 2-м и т. д. Этот способ лучше,
ибо маточные спирты предварительно отстаиваются, следовательно,
очищаются и получаются более однородные валовые партии тро-
тила из частных партий, изготовленных на одном и том же номере
маточного спирта.
172
Спи р то о бо р о т. Применением спиртооборота достигается
уменьшение расхода чистого спирта, потерь спирта и тротила и умень-
шение потребной мощности установки для ректификации спирта.
Таблица 30
Загрузка спирта при работе без добавки толуола
Наименование маточного спирта Загрузка промы- того тринитро- толуола кг Загрузка чистого спирта в ведрах Загрузка маточ- ного Спирта в ведрах
Нвлевой 900 340
1-й маточный .... 900 40 300
2-й » .... 900 60 280
3-й » .... 900 80 260
К загрузке чистого спирта засчитывается израсходованный при
промывке на центрифуге «промывной спирт», который смешивается
с соответствующим маточным спиртом.
Таблица 31
Загрузка спирта и толуола при работе на спиртотолуольной смеси
Наименование маточного спирта Загрузка про- мытого три- нитротолуола кг Загрузка Спирта в ведрах Загрузка ма- точного Спирта в ведрах Загрузка толуола в ведрах
Нолевой 1 1000 260 — 20
1-й маточный.... 1000 60 215 5
2-й » .... 1000 100 170 10
3-й » . . . • ; юоо i 150 115 15
Часть толуола подается в виде толуольной фракции, получаю-
щейся при ректификации маточного спирта и содержащей 40—50%
толуола и 60—50% спирта.
Потери спирта в обоих способах одинаковы и составляют около
2000° на 1 т кристаллизованного тротила. Преимущество от при-
менения спиртотолуольной смеси —увеличение производительности
установки; отрицательная ее сторона—потери толуола при перекри-
сталлизации и ядовитость его паров, вредных для здоровья рабочих,
что дает себя особенно чувствовать в летнее время.
Потери тротила при его перекристаллизации составляют 12%,
из них: около 10% остается в маточном растворе и около 2%
теряется при отжимке, сушке и упаковке.
Сушка тротила. Прежде сушка перекристаллизованного
из спирта тротила производилась в камерных сушилках; теперь
для этой цели применяются пневматические сушилки.
173
Пневматическая сушил к а (рис. 31) состоит из ка-
лорифера А, вентилятора В, трубы с засыпной воронкой С, сушиль-
ной трубы Z), циклонов Е и F и пыльной камеры G-
Засасываемый снаружи воздух проходит через калорифер А,
где нагревается паром до 68—72° и гонится вентилятором В по
трубе D, в которой находится засыпная воронка С для подачи
Рис. 31. Пневматическая сушилка.
влажного тротила. Схема этой трубы показана в увеличенном мас-
штабе на отдельном рисунке (рис. 32). Диаметр трубы сужается
около воронки. Скорость воздуха в широкой части трубы 15—
20 м/сек, а в суженной — значительно больше, благодаря чему
создается разрежение (по принци-
Т пу эжектора), которым тротил заса-
<. \ сывается в трубу и гонится возду-
s' хом в циклоны Е и F, где 1 из-за
потери скорости воздуха кристаллы
Рис. 32. Деталь трубы с засып- тротила оседают на дно циклона и
ной воронкой. п0 трубам ссыпаются в ларь Н,
откуда тротил вручную выгру-
жается в ящики или мешки. Мелкие частицы тротила в виде
пыли проходят в верхнюю пыльную камеру, в которой натянут ряд
рыболовных сеток с ячейками в 1 см2 (или мешковины), на кото-
рых оседает унесенная сюда тротиловая пыль. Эта пыль по мере
ее накопления стряхивается с сеток и сметается в циклон F, откуда
она ссыпается в ларь Н. Освобожденный от частиц тротила воздух
выходит через трубу I наружу.
Скорость воздуха при выходе из жолоба в 1-й циклон около
3—4 м/сек, а при выходе из 2-го циклона — около 1 м/сек.
174
При сушке сохраняют частичные партии, сформированные в цехе
кристаллизации тротила. В зависимости от размеров производитель-
ность пневматической сушилки достигает 20 т тротила в сутки
и больше.
Анализ частных партий, просеивание и
упаковка. От частных партий тротила отбираются пробы для
анализа. Тротил отправляется в упаковочное помещение, где он
просеивается. Однако во избежание пыления лучше производить про-
сеивание до сушки. В этом случае просеивание производится череа
сито с ячейками 4x4 мм. При этом получают 6—10% по весу
тротила комков, корок и пр., которые либо вновь перекристалли-
зовывают из спирта, либо применяют для производства аммони-
тов4: Упаковка производится в деревянные ящики вместимостью
45 и 80 кг тротила. Ящики внутри обкладывают пергаментной или
оберточной бумагой.
Из кондиционных частных партий формируются валовые партии.
Технические требования ккристаллизован-
номутротилу.
1. Мелкий, кристаллический порошок, светложелтого цвета, не
заключает видимых на-глаз посторонних примесей; должен пред-
ставлять собой совершенно однородную массу ине носить явных при-
знаков подмочки. При просеивании взятой пробы через проволоч-
ное сито со стороной ячеек в 3 мм тротил должен проходить без.
остатка.
2. Температура затвердевания не ниже 80,2°.
3. Влажности и летучих веществ не более 0,075%.
4. Кислотность в переводе на серную кислоту (азотной кислоты
допускаются только следы) не более 0,01%.
5. Нерастворимых в бензоле примесей не более 0,15%.
6. Содержание золы не более 0,1°/0.
7. Маслянистость не более, чем у тротила, принятого за эталон.
Обоснование требований технических ус-
ловий. Тротил, применяемый для ответственных изделий, должен
быть возможно высокой степени чистоты, что обеспечивает наилуч-
шую восприимчивость к детонации, минимальною чувствительность
к удару, максимальную физическую стойкость.
В частности: 1) температура затвердевания является основным
критерием чистоты продукта, 2) влажность понижает восприимчи-
вость к детонации тротила и вызывает вспенивание при плавке тро-
тила; 3) кислотность вызывает коррозию металлических стенок изде-
лия. Образующиеся окислы метила могут при длительном хранении
в присутствии аммиачной селитры (в амматоле) привести к образова-
нию опасного тротилата аммония; 4) нерастворимые примеси могут
(в зависимости от их природы) увеличивать чувствительность к удару;
в этом отношении особенно опасен песок; 5) о значении маслянистости
говорится выше (см. гл. VIII, стр. 140, Применение тротила).
Ректификация отработанного спирта. Ма-
точный спирт отстаивается в железных колонках не менее 5 суток.
175
При этом выкристаллизовывается нитропродукт с температурой за-
твердевания около 60°, содержащий значительное количество дини-
тротолуола; зтот продукт носит название осадочного тре-
ти л а и пригоден для изготовления аммонитов. Осадочного тротила
получается около 2% по весу тротила-сырца. Ректификация спирта
ведется в обычных дестил л яцио иных установках, применяемых на
спиртовых заводах.
На английских заводах обнаружили в парах спирта окислы
азота, поэтому к спирту перед перегонкой прибавлялось немного мо-
чевины для разрушения этих окислов. По мнению английских хи-
миков, окислы азота образуются из этийнитрита, получающегося
при взаимодействии этилового спирта с изомерами тротила. В при-
сутствии окислов азота происходит значительная коррозия холо-
дильников, требующая их частого ремонта.
Тротиловое масло. При ректификации спирта остается
смесь нитросоединений, состоящая из некоторого количества a-три-
нитротолуола и примесей, содержавшихся в тротиле-сырце, а именно:
изомеры тринитротолуола (главным образом ^-изомер), динитротолуол
.и названные ранее малые количества тетранитрометана, тринитробен-
зола и др. а-Тринитротолуол находится в тротиловом масле в коли-
честве, отвечающем его растворимости в холодном маточном спирте.
Состав тротилового масла не является постоянным, а изменяется
в некоторых пределах, зависящих от производственных процессов
(от условий нитрации, от условий предварительного отстаивания
и разбавления водой маточных спиртов, так как при этом выделяются
разные количества «осадочного тротила» и, следовательно, меняется
состав остающихся в растворе нитропродуктов).
Содержание компонентов колеблется в следующих пределах:
тринитротолуола 25—40%, динитротолуола 70—50%; мононитро-
толуола и других примесей 5—10%.
Физические свойства. Тротиловое масло получается
при ректификации спирта в виде густой темнобурой жидкости.
При охлаждении и стоянии этой жидкости из нее выделяются кристал-
лические вещества, образующие с жидкой фазой сильно маслянистую
густую массу, с температурой плавления, колеблющейся для раз-
личных образцов в пределах 25—‘60°. Из этой массы в теплое время
года вытекает более или менее значительное количество маслянистой
жидкости.
Взрывчатые с,в о й с т в а. Тротиловое масло в виде жид-
кости, густой массы или сплошной затвердевшей массы очень мало
чувствительно к детонации и не взрывается даже при применении
мощного капсюля. Порошкообразный продукт (полученный, напри-
мер, измельчением замороженного масла) детонирует с большим тру-
.дом от мощного капсюля.
Применение тротилового масла. Тротиловое
масло не может применяться в чистом виде в качестве взрывчатого
вещества, но оно вполне пригодно для изготовления аммо-
нитов.
176
По ОСТ 40099 допущен к применению аммонит № 3-ТМ следую-
щего состава: 85% аммиачной селитры, 13% тротилового масла,
2% древесной муки; вследствие непостоянства состава тротилового
масла аммонит № 3-ТМ допущен только для открытых работ.
У нас были разработаны трудно замерзающие динамиты на основе
тротилового масла. Однако примесь тротилового масла, даже взя-
тая в значительном количестве, не обеспечивает необходимого в ус-
ловиях наших северных районов понижения температуры замерза-
ния динамита; кроме того, при высоком содержании в таких дина-
митах тротилового масла они имели отрицательный кислородный
баланс, и поэтому продукты взрыва содержали большой процент
ядовитых газов. Такие динамиты не могли применяться для подзем-
ных работ. Наконец, при содержании тротилового масла динамиты
обладали пониженной химической стойкостью.
По этим причинам такие динамиты не нашли промышленного
применения.
Из тротилового масла можно выделить 2,3,4-тринитротолуол.
Для этой цели отделяют кристаллы от масла и обрабатывают их сер-
ной кислотой. При этом удаляются динитротолуол и другие при-
меси, и получается чистый 2,3,4-тринитротолуол, применяющийся
в Англии для получения красок.
2. Сульфитная очистка тротила. Историческая справ-
к а. Давно известно, что если в ароматическом нитро со единении две
нитрогруппы находятся в орто- или параположении относительно
друг друга, то одна из них является подвижной; например, при дей-
ствии едкого натра легко идут следующие реакции:
NO. NO2
/x,no2 z\qh
+ NaOH--------> | % NaNO2
NO, NO.
+ NaOH-------> | % NaNO3
Xno2 Xoh
Лаубенгеймер 1 впервые показал, что при обработке 3,4-динитро-
хлорбензола водным раствором сульфита натрия подвижная нитро-
группа замещается сульфогруппой. По данным исследований фран-
цузских химиков, проведенных во время империалистической войны
1914—1918 гг., эта реакция является, повидимому, общей для всех
полинитросоединений с подвижной нитрогруппой. Легко реагируют
с водным раствором сульфита натрия асимметричные тринитрото-
1 Laubenheimer, Вег., 16, 597, 1883.
Химия—13—12
177
луолы с образованием легко растворимых в воде сульфонатов. Суль-
фит натрия легко реагирует с р- и о-динитробензолом, с 3,4-динитро-
хлорбензолом, но труднее реагирует с соответствующими динитро-
соединениями толуола, в которых подвижность 2-й нитро группы ослаб-
лена благодаря присутствию метила. При применении сульфита
натрия для очистки тротила были получены теми же французскими
исследователями настолько хорошие результаты, что способ сульфит-
ной очистки стал широко использоваться при производстве очищен-
ного тротила вместо перекристаллизации из спирта.
Вскоре этот способ стали применять в других странах: в Англии
(1916 г.), в США, в Италии и др.
Первоначально применявшийся во Франции процесс сульфитной
очистки заключался в том, что тротил, отлитый в виде болванок,
измельчался, а затем промывался водным раствором сульфита нат-
рия, получавшимся от производства синтетического фенола.
Большим недостатком этого способа являлась операция измельчения
тротила.
В 1917 г. в Англии процесс этот видоизменили следующим обра-
зом. Сырой тротил, как и раньше, подвергался предварительной про-
мывке для удаления кислот. После этого жидкий тротил медленно
охлаждался под водой при постоянном перемешивании. Таким пу-
тем получалась влажная масса мелких, но хорошо образованных
кристаллов чистого тротила. Примеси, как и при получении круп-
ных кристаллов, концентрируются на поверхности кристаллов. После
соответствующего охлаждения массы добавляли при продолжаю-
щемся перемешивании раствор сульфита натрия. Продолжали пе-
ремешивание в течение получаса, а затем отсасывали темнокрасный
раствор и промывали тротил водой. Для обеспечения полного уда-
ления щелочей расплавленный тротил промывали слабым раство-
ром серной кислоты. При сульфитной промывке достигалось повы-
шение температуры затвердевания 1 на 2,2°.
Реакции, протекающие при сульфитной
очистке2. При сульфитной очистке тротила протекают следую-
щие химические реакции:
1. я-Тринитротолуол образует мало стойкий в слабых сульфит-
ных растворах (меньше 5% Na2SO3) продукт присоединения, окра-
шенный в яркокрасный цвет. При подкислении раствора это соеди-
нение разрушается и выделяется неизмененный тротил.
Растворимость этого продукта присоединения в водных раство-
рах сульфита натрия при обыкновенной температуре такова:
3%-ный раствор сульфита натрия растворяет . . 0,3% а-тринитротолуола
6*/0 » » » » » . . 0,6% »
12% >> » » » » . . 2,3% »
1 Man. of TNT.
’ Muraour, Bull. Soc. Chim. Fr., 35, 367—379, 1924; Army Ord-
nance, 5, 507—511, 1924; Z. Schiess- u. Sprengst., 19, 177, 178, 1924.
178
При разбавлении раствора этот продукт присоединения разру-
шается, и соответствующее количество тротила выпадает в осадок.
2. При взаимодействии сульфита натрия с нессиметричными три-
нитротолуолами нитрогруппа, стоящая в метаположении к метиль-
ной и в ортоположении к другой нитрогруппе, замещается сульфо-
группой:
СН3
I
o2n/\
xJsO3Na
NO,
+ NaNO2
3. С тетранитрометаном эта реакция протекает энергичнее, чем
с несимметричными тринитротолуолами и сопровождается выделе-
нием тепла:
C(NO2)4 + Na,SO3 C(NO2)3SO3Na + NaNO2
C(NO2)3SO3Na + H,0 -> CH(NO2)3 + NaHSO4
4. С динитротолуолами сульфит натрия не реагирует.
'Месир 1 показал, что при обработке раствором сульфита натрия
синтетических смесей а-тринитротолуола с 2,4-динитротолуолом не
происходит никакого взаимодействия. Однако если эти смеси вклю-
чают еще и несимметричные тринитротолуолы, то при обработке их
раствором сульфита натрия вместе с несимметричными тринитрото-
луолами в раствор переходит и 2,4-динитротолуол. Месир не дал.
объяснения причины этого явления.
По данным того же автора, симметричный тринитротолуол может
быть количественно освобожден промывкой раствором сульфита нат-
рия от примесей 3,4-динитротолуола и 2,3-динитротолуола.
5. Сульфит натрия не реагирует с »г-динитробензолом и трини-
тро -т-ксилолом, но с несимметричными тринитро-да-ксилолами
сульфит натрия реагирует аналогично реакции с несимметричными
тринитротолуолами.
'б. С тринитробензолом сульфит натрия образует продукт при-
соединения, легко растворяющийся в разбавленном растворе суль-
фита. При длительном стоянии это вещество переходит в сульфосо-
единение. То же происходит при длительном стоянии продукта при-
соединения а-тринитротолуола с сульфитом натрия.
7. Окрашенные продукты окисления фенольного характера, со-
держащиеся в сыром тротиле, легко растворяются в разбавленных
водных растворах сульфита натрия (вероятно с образованием раство-
римых в воде фенолятов). Этим обусловлено происходящее при суль-
фитной очистке обесцвечивание тротила.
1 М е с i г,’ Chim. et Ind., 6 bis, 925, 1933.
179
Механизм взаимодействия сульфита нат-
рия с нитросоединениями. По Бреди, Гиветсону и
Клейну1, механизм реакции взаимодействия нитро со единений
с сульфитом натрия можно рассматривать двояко в зависимости
от того, принять или не принять гипотезу об асимметричном строе-
нии сульфитов.
При допущении асимметричного строения сульфита реакция мо-
жет быть выражена простой схемой:
zNa
7?NOo + S02<-------> 7?SO,ONa + NaNO2
xONa
Если же принять симметричное строение сульфитов, то можно
объяснить реакцию образованием промежуточного продукта по из-
вестной схеме2:
7?NO2 R /ONa / rjq + 0 = S\ ’° = S~ONa ‘ xONa \UPIa ONa » >S< + NaNCL CK xONa
Свойства натриевых солей динитро сульфокис лот.
Бреди с сотрудниками выделил продукты взаимодействия р-и у-три-
нитротолуолов с сульфитом натрия. Это — кристаллические соеди-
нения, растворимые в воде и спирте и нерастворимые в эфире и
бензоле. При нагревании они дают сильную вспышку. С трудом
реагируют с водным и спиртовым растворами аммиака (с замеще-
нием сульфогруппы на аминогруппу), но легко взаимодействуют
с метиламином, с образованием соответствующих метилтолуидинов.
Приготовление сульфитного раствора3. Во
время войны на английских заводах применялся сульфит, полу-
чавшийся в виде отхода при производстве синтетического фенола.
Средний состав этого продукта 73,8% Na2SO37H2O, 1,83% Na2CO3,
0,46% NaOH, 1,54% нерастворимых примесей, остальное — сульфат
натрия, гидрат окиси железа, сульфит кальция, вода и др.
Полученный после растворения мутный раствор перекачивался
в ряд отстойников, из которых он после 24-часового отстаивания
поступал в расходный резервуар. Употребляемая концентрация
равна 10—15% Na2SO3. Более высокой концентрации избегают из
опасения кристаллизации в трубопроводах в холодную погоду.
Так как щелочь, особенно едкая, значительно повышает раство-
римость тринитротолуола и таким образом повышает потери его,
то присутствие щелочи в сульфите нежелательно. Было испробовано
1 Brady, Hewetson, Klein, J. Chem. Soc., 125, 240, 4924.
2 Эфраим, Неорганическая химия, ч. II, стр. 31—32, Госхимиадат, 1933.
3 Man. of TNT., стр. 97.
180
частичное подкисление раствора серной кислотой, после чего ана-
лизы показали, что щелочность значительно понижалась без одно-
временного действия на сульфит.
Кроме того, промышленность изготовляет сульфит натрия взаимо-
действием сернистого газа с раствором соды, причем получается
(после упаривания и кристаллизации) продукт состава Na2SO3 • 7Н2О.
Технические условия на технический
сульфит натрия (кристаллический) (ОСТ 3511). Сульфит
натрия представляет собой продукт, кристаллизующийся с 7 мол
воды. Он должен удовлетворять следующим требованиям:
1) цвет кристаллов должен быть белым или слегка желтоватым;
2) содержание сернокислого натрия Na2SO3 • 7Н2О не менее
§4,7% или сернистого ангидрида SO2 не менее 21,5%;
3) содержание углекислого натрия Na2CO3 • 10Н2О не более 4,5%;
4) содержание железа в пересчете на FeO не более 0,1%;
5) нерастворимого в воде остатка должно быть не более 0,1%;
6) сульфит натрия должен быть рассыпчатым и неслипающимся.
Упаковка. Сульфит натрия упаковывают в деревянные
бочки (сухотарки) или ящики. Предельный вес нетто: для бочек
100 кг, для ящиков 50 кг. На каждой упаковке должно быть обо-
значено название треста, завода, продукта, номер партии, вес нетто
и брутто и ОСТ 3511.
Растворимость сульфита натрия в воде1
(дана в процентах к раствору, т. е. р выражено в граммах безводного
вещества в 100 г раствора)
i° = 0 10 20 30 33,4 40 50 60 70 80 90 100'
р = 12,5 16,0 20,7 26,1 28,0 27,0 25,7 24,5 23,5 22,6 21,7 21,2
Схема установки сульфитной промывки
завода Квин-Ферри2 (рис. 33). Гранулированный .после
предварительной промывки тротил подвозится в вагонетках к про-
мывному чану сульфитной мастерской. После промывки этого тро-
тила в промывном чане он передается в деревянный чан-кристалли-
затор, в котором производится кристаллизация тротила под водой.
В этом же чане после кристаллизации производится промывка суль-
фитным раствором. По окончании промывки содержимое кристалли-
затора помощью центробежного насоса подается на вакуум-воронку,
где отсасывается маточный сульфитный раствор и производится
промывка водой до полного удаления маточного раствора.
Тротил из фильтра передаемся в чан для кислой промывки, а от-
сюда в чан для окончательной промывки. Окончательно промытый
тротил поступает по трубопроводу в бак для питания сушилки,
а отсюда в сушильную плиту. Высушенный тротил получается
в виде чешуек на специальном барабане.
Описан и-е процесса сульфитной промыв-
к и. Тротил подвергается перед кристаллизацией промывке до кис-
1 Foerster, Brosche, Norberg-Schulz, Physikal.
Chem. 110, 435, 1924. По «Спутнику Химика», II. 361, ОНТИ, 4936.
2 Man of TNT
181
Маноме/м колодная Горячая
xl Soda "-fl П — дпдп
182
лотности около 0,1% H2SOV Главная цель промывки — устране-
ние корродирующих влияний в кристаллизаторе и частичное уда-
ление тетранитрометана при пропаривании; кроме того, устранение
потерь сульфита натрия, который под влиянием кислоты превра-
щается в неактивный бисульфит. На схеме показана подача твер-
дого тротила на вагонетке, но лучше
подавать прямо из сепарационной колон-
ны жидкий тротил.
тротил сливается из про-
мывного чана в кристаллизаторы.
Устройство кристаллизатора показано
на рис. 34. Аппарат делается целиком
ий' дерева, предпочтительней смолистая
сосна (по английским указаниям).
Процесс кристаллизации.
В аппарат-кристаллизатор заливается
горячая вода при температуре 90—100° и
расплавленный тротил. Наилучшей про-
порцией являются равные веса воды и
тротила. Избыток воды увеличивает вре-
мя охлаждения, и кроме того:
1) требуется большая затрата энер-
гии на вращение мешалки;
2) установлено, что при избытке во-
ды вследствие увеличивающегося трения
кристаллов происходит измельчение тро-
тила, причем образуется паста из пыле-
видного тринитротолуола и эвтектики;
в результате этого очистка делается менее полной и затрудняется
последующее фильтрование;
3) увеличивается изнашивание и поломка лопастей.
При употреблении равных весов тротила и воды последняя
к концу процесса кристаллизации в значительной мере впитывается
кристаллической массой, однако, остатка воды достаточно, чтобы
лопасти мешалки работали без большого сопротивления.
Падение температуры в 1-й стадии процесса идет очень быстро—•
она падает от 90 до 75° менее, чем в полчаса. Кристаллизация
начинается вблизи температуры затвердевания — при 72—73°, и
температура до полной кристаллизации остается почти постоянной,
после чего она начинает быстрее падать.
При нормальных условиях температура падает до 50° в тече-
ние 4—4х/2 часов с момента загрузки. При увеличенном количестЬе
воды или при особенно теплой погоде продолжительность процесса
увеличивается приблизительно на 25%.
Степень очистки при обработке сульфитом натрия зависит в зна-
чительной мере от величины и формы кристаллов. Поэтому необхо-
димо получать возможно мелкие кристаллы; не должно быть грану-
лей и кусков (нормальный размер кристаллов: 100% проходит через
183
*
сито со 100 отверстиями на 1 см2 и 80% проходит через сито с 400
отверстиями на 1 см2).
Если в процессе кристаллизации образовались сростки кристал-
лов тринитротолуола, то они не будут в достаточной мере очищаться
раствором сульфита. Таким образом маленькие частички окажутся
чище, чем большие куски. Это доказывается тем, что если обработан-
ный сульфитом продукт просеять, то тринитротолуол, прошедший
сквозь сито, будет лучшего качества, чем остальная масса. Разница
в температуре затвердевания получается около 0,3°.
Промывка тротила раствором сульфита
натрия. В результате исследований, произведенных на англий-
ском заводе во время империалистической войны 1914—1918 гг.,
определены следующие оптимальные условия для процесса промывки:
1) температура реакции 40—45°; при более высоких температу-
рах действие сульфита натрия на а-тринитротолуол быстро увеличи-
вается при пониженных температурах действие сульфита на изо-
меры становится очень медленным;
2) количество сульфита по отношению к тринитротолуолу 3,5—4%;
3) концентрация реагирующего раствора 3—4%;
4) продолжительность взаимодействия раствора с тротилом —
полчаса.
Теоретически для полного удаления изомеров необходимо меньше
2,5% сульфита натрия; следовательно, при 3,5—4% его избыток
против теории составляет 40—60%.
Для ускорения процесса вели сульфитную промывку при 50%
причем продолжительность процесса кристаллизации составляла
5 часов, которые складывались из следующих стадий:
Кристаллизация и охлаждение ............. 4 часа
вливание сульфита и обработка им ... . 40 мин.
загрузка и выгрузка чана................ 20 »
Сульфитный раствор подавался в кристаллизатор при помощи
перфорированной трубы, расположенной над кристаллизатором. По
окончании подачи раствора сульфита продолжают перемешивание
в течение получаса, после чего открывается спускной вентиль и со-
держимое кристаллизатора спускается в сборник для кристалли-
ческой массы, находящейся внизу, и затем перекачивается на фильтр
при помощи центробежного насоса. В случае надобности разбавляют
густую кристаллическую массу путем прибавления маточного рас-
твора от предыдущей загрузки. Перекачка массы идет хорошо при
добавлении 3 в. ч. маточного раствора к 2 ч. кристаллического
тротила. Во избежание попадания расплавленного тротила (во время
загрузки) в спускную трубу и ее закупорки спускное отверстие за-
1 Барбье пришел к близким выводам; он указал, что минимальные потери
тротила имеют место, если обрабатывать раствором сульфита натрия не более
1 часа при температуре ниже 40°, а по окончании реакции тотчас же разбавить
равным объемом воды. Barbier е, Mem. Poud., 26, 294, 1934/1935.
184
полняется влажной кристаллической массой от предыдущей загрузки-
Это оказалось лучше, чем заполнение сухими кристаллами или гра-
нулированным тринитротолуолом.
На работу мешалки расходуется энергии около 4—5 л. с. Однако
мощность мотора должна быть больше вследствие большого сопро-
тивления осевшей кристаллической массы при пуске мешалки после-
случайных остановок.
Фильтр (вакуум-воронка). Для удаления маточного
раствора и окончательной отмывки растворенных сульфонатов слу-
жит фильтр.
В случае недостаточной отмывки сульфонатов при последую-
щей кислой промывке тротила образуются сульфоновые кислоты,
которые, повидимому, оказывают эмульгирующее действие на смесь-
воды с тротилом, что обусловливает неполадки процесса.
Вакуум создается вакуум-насосом, соединенным с фильтром воз-
душной линией- Раствор попадает с фильтра в спускную торичел-
лиеву трубу, снабженную вакуум-камерой, которая служит брызго-
уловителем; сверху этой камеры подведена вакуум-линия. Ниж-
ний конец спускной трубы опущен в промежуточный сосуд, из ко-
торого маточный раствор сливается в лабиринт. Длина спускной
трубы около 7,6 м. и практически можно создавать разрежение
в 50 см рт. ст. без опасения, что раствор поднимется в спускную
трубу и попадет через вакуум-линию в насос. Однако во избежание
повреждения насоса при случайном засасывании раствора следует
ставит s мокрый вакуум-насос.
Процесс фильтрации. После передачи массы из кри-
сталлизатора отсасывают маточный раствор, затем тротил промы-
вают до удаления следов красного маточного раствора. Промывку
ведут водой, нагретой до температуры 60—65°. При этом одновре-
менно удаляется эвтектика, образуемая динитротолуолом с троти-
лом, повышается температура затвердевания тротила и уменьшается
его маслянистость. По окончании промывки тротил выгребают
в чан для кислой промывки, расположенный непосредственно под
вакуум-воронкой. Маточные растворы и промывные воды проходят
через лабиринт, где при охлаждении выделяется растворенный тро-
тил и осаждается механически увлеченный; последний собирается
и возвращается на промывку. Общая продолжительность обработки
продукта на фильтре около46 часов.
Образование нитродиазотолуолсульфо-
кислоты при подкислении маточных суль-
фитных вод минеральными кислотами1. Суль-
фитные воды содержат натриевую соль динитро толуолсульфокислоты,
нитрит натрия и сульфит натрия. При подкислении этих вод обра-
зуются NaHS03 и S02, которые восстанавливают динитротолуол-
сульфокислоты; при этом образуются нитроамидосульфокислоты^
1 В a ti k, Chim. et Ind., 29, 6 bis, 952—959, 1933 .
18»
которые в избытке ^минеральных кислот и NaNO2 диазотируются
с образованием нитро диазотолуолсульфэкислэ ты:
СН3
+ Na,SO,
GH3
o.,n,/x,
| + so,
'x/'SO3Na
no2
или NaHSO»
+ SO, или NaHSOs
SO3Na
NH2
4- HNO,____
в прие. H,SO, *
CH3
o2n/x-
I
I^SOjNa
N : N • SO4H
Нитродиазотолуолсульфокислота при нагревании до 130—140°
взрывает с большой силой; иногда она взрывает при 75°. Воспламе-
няется она от легкого удара между железными предметами. Обра-
зование этой кислоты при производстве тринитротолуола является
очень опасным. Поэтому маточные и сульфитные промывные воды
следует удалять таким образом, чтобы они не смешивались с водами,
содержащими свободные минеральные кислоты.
Ненормальности при фильтровании1. Орга-
нические кислоты, содержащиеся в тротиле, поступающем в кристал-
лизаторы, растворяют железо (железных частей аппарата). При при-
бавлении сульфита железо осаждается в виде гидрата окиси. Боль-
шая часть этого гидрата остается в коллоидальном растворе;
частью же он осаждается на кристаллах тротила и смывается на филь-
трующую ткань, забивая ее. Эта ткань быстро становится непро-
ницаемой (часто после двух загрузок). Поэтому необходимо приме-
нять грубые сорта ткани для фильтров.
Устройство фильтрующей части. На дне
*фильтра покоится на 5-см опорах перфорированное железное осно-
вание в виде четырех квадрантов; поверх него лежит фильтрующая
ткань, укрепленная по краям фильтра тросом. Поверх троса лежит
алюминиевая пластинка с большим числом отверстий диаметром
около 0,65 см и, наконец, — грубая проволочная сетка с такими же
отверстиями.
Кисла я п р о мывка. Чтобы обеспечить полное удаление
щелочи, могущей остаться после сульфитной очистки и последующей
водной промывки, а также из опасения, что при промывке тротила
раствором сульфита натрия могли образоваться тротилаты, тротил
подвергается добавочной кислой промывке.
В чан для кислой промывки подается горячая вода и серная
кислота из расчета, чтобы получить концентрацию около 0,1% H2SO4.
1 По английским данным, ив книги Man. of TNT. •
186
В горячую подкисленную воду загружается лопатами содержи-
мое фильтра-, причем массу одновременно перемешивают и подают
в нее пар.
По расплавлении ее перемешивают еще несколько минут, отстаи-
вают и спускают кислую воду. Затем чан наполняют горячей водой
и спускают тротил в чан для окончательной водной промывки.
В чан кислой промывки подают (к оставшейся там воде) необходимое
количество серной кислоты для промывки новой загрузки тротила.
Ненормальности при промывке. При описан-
ном способе кислой промывки не достигается полное удаление гид-
рата окиси железа. Кристаллы тротила загружают в чан в форме
влажных, слипшихся кусков, которые при попадании в кипящую
воду начинают расплавляться (от внешней своей поверхности). При
таких условиях плавления тротила частицы гидрата окиси железа
смешиваются с расплавленным тринитротолуолом, обволакиваются
пленкой жидкого тринитротолуола и становятся недосягаемыми для
действия кислой воды. Гидрат окиси железа является при этом
прекрасным эмульгирующим агентом, вследствие чего в чан окон-
чательной промывки вместе с тротилом будет попадать кислая вода,
а суспендированный тротил будет уходить из этого чана вместе с про-
мывной водой в лабиринты.
От гидрата окиси железа можно полностью освободиться, если
загружать кристаллы с фильтра в воду, имеющую температуру,
меньшую температуры плавления тринитротолуола. В этом случае
гидрат окиси железа полностью растворяется в кислоте. Конечный
продукт получался светлый и вполне устранялось эмульгирование.
Однако этот способ имел два недостатка, вследствие чего его скоро
оставили: 1) процесс кислой промывки требовал почти вдвое больше
времени, чем при промывке плавленого тротила, и, таким образом,
уменьшалась производительность установки; 2) мешалки быстро
снашивались от трения о кристаллы.
Поэтому вернулись к прежнему методу, заменив железные ло-
пасти мешалки деревянными. Однако вследствие невозможности
полного устранения из аппарата железных частей в тротиле всегда
оставалось некоторое количество гидрата окиси железа.
Окончательная водная промывка. Если опе-
рация кислой промывки была проведена правильно, то первая про-
мывная вода (при окончательной промывке) оказывается нейтраль-
ной. В противном случае эту операцию продолжают до достижения
необходимой степени промывки (например, меньше 0,02% по H2SO4).
Промытый тротил поступает в сушилку.
Промывная вода из чана окончательной промывки возвращается
на промывку кислого тринитротолуола (до кристаллизации под
водой).
Повышение температуры затвердевания
при сульфитной промывке. Среднее повышение тем-
пературы затвердевания при сульфитной промывке на английских
заводах равно 2,2°. Этим определялся нижний предел температуры
187
затвердевания тротила, допускавшегося к сульфитной очистке,
а именно, 74,2° для влажного тринитротолуола. В значение для
температуры затвердевания влажного тринитротолуола вносилась
поправка1 в 3,7° для получения температуры затвердевания сухого
тринитротолуола. При таком тринитротолуоле после обработки
сульфитом натрия и сушки получался следующий продукт:
температура затвердевания влажного тринитротолуола 74,2°
поправка на влажность......................... 3,7°
повышение температуры затвердевания при сульфит-
ной промывке.................................. 2,2°
температура затвердевания очищенного и высушен-
ного тринитротолуола........................... 80,1°
Потери при сульфитной промывке. При суль-
фитной промывке удаляются изомеры, содержащиеся в количестве
около 4,2% и, кроме того, теряется около 4% а-тринитротолуола.
Потери а-тринитротолуола происходят за счет взаимодействия его
с сульфитом натрия, растворяясь в маточном растворе, а частью
за счет механически уносимого с маточным раствором и промывными
водами тринитротолуола.
Сушка и фильтрация. Тринитротолуол, поступающий
из промывки в сушилку, содержит небольшое количество осадка,
состоящего из сернокислого свинца, песка, побочных продуктов и
волокон дерева от промывных чанов. Кроме того, при 85° он содер-
жит 1,1% растворенной воды и больше 0,5% воды в виде эмульсии;
эмульгированная вода легко испаряется при температуре выше 100°.
Тяжелые примеси осаждаются в баке для питания сушилки.
В нем скопляется осадок, от которого необходимо очищать его раз
в 4 месяца.
Для очистки от примесей, не осаждающихся во время сушки,
необходимо фильтровать тротил. Опыт показал, что трудно филь-
тровать сырой тринитротолуол, содержащий воду в эмульгирован-
ном виде, так как при этом фильтр из тонкой сетки быстро заби-
вался. Однако значительно легче идет процесс после того,кактро-
тил пройдет через сушилку. Сетка делается из фосфористой бронзы;
стальные сетки или сетка из луженой стали непригодны, так как они
ржавеют.
Для очистки сушилки от осадка ее освобождают насколько воз-
можно от тринитротолуола и охлаждают. Затем снимают крышку
и осадок отбивают помощью зубила и молотка, сделанных из соот-
ветствующего твердого дерева; в конце пропаривают струей пара.
Удаленный осадок сжигается.
Непрерывная сушка (рис. 35). Из чана окончатель-
ной промывки тротил поступает в сборник 7, служащий одновре-
менно питательным баком для сушильной плиты 9. В этом сборнике
тротил, поступающий сюда с температурой около 85°, нагревается
паровым змеевиком до температуры 103—106°. Здесь испаряется
значительная часть механически увлеченной и эмульгированной во ды.
1 Ср. с указаниями на стр. 154.
188
! Из сборника тротил перетекает в сушильный жолоб, вдоль кото-
рого он протекает тонкой струей (толщина слоя около 12 мм). Жо-
! лоб обогревается помощью паровой рубашки. Высушенный тротил
проходит через ряд охлаждающих желобов, где он охлаждается воз-
духом до 85° и затем поступает на аппарат — барабан — для по-
лучения его в форме чешуек.
Рис. 35. Испаритель и сушильная плита.
Получение тринитротолуола в виде че-
шуек. Для получения тринитротолуола в виде чешуек служит
медный барабан, диаметром около 0,6 м и длиной около 0,9 м.
Толщина его стенок около 18 мм. Барабан укреплен на цапфах;
через одну из цапф поступает охлаждающая вода; уходит она из
барабана через другую цапфу, расположенную так, что барабан
всегда наполнен водой наполовину. Барабан делает 5—10 об/мин.
Он погружен в ванну с жидким тротилом; ванна снабжена рубашкой
для нагревания паром в случае застывания тротила. Барабан при
вращении смачивается слоем тротила, который застывает при одном
обороте барабана; затвердевший слой тротила срезается по всей
длине барабана медным ножом.
Весь аппарат закрыт легкой крышкой из листового железа с при-
соединенной вытяжной трубой; внизу расположен жолоб, через
который тринитротолуол подается на весы.
Утилизация маточных сульфитных щело-
ков. В английской литературе 1 предлагается вести обработку
маточных сульфитных щелоков при температуре 70° метиламином:
сн3
ySOjNa
4- NH2GH3
GH3
o„n/\no2
+ NaHSO3
1 'NHGH3
1 Man. of TNT.
189
При нитровании полученных динитрометилтолуидинов легко
образуется тринитротолилметилнитроамин:
СН3
O2N[/\,NO2
^JnHCH3
+ HNO3
сн3
I /"N°2
no2 3
Гонелл и Робинсон 1 получили из ^-тринитротолуола натрие-
вую соль 2,4-динитротолуол-З-сульфокислоты, восстановлением ко-
торой получили пг-толилендиамин-3-сульфонат натрия, кристалли-
зующийся из воды в виде блестящих пластинок. Это соединение
представляет интерес как исходный продукт для изготовления кра-
сителей. При этом все оттенки получаются более красными, чем
у производных 2,4-диаминотолуолсульфокислоты.
При окислении 2,4-динитротолуол-З-сульфоната натрия перман-
ганатом калия в присутствии соды получается 2,4-динитро-З-сулъфо-
бензойная кислота, на основе которой могут быть получены азокра-
сители.
3. Другие способы очистки тринитротолуола. Промывка
гипохлоритом натрия и буферными раство-
рами, содержащими сульфит натрия. Описанный
способ очистки тринитротолуола промывкой растворами сульфита
натрия включает громоздкую операцию кристаллизации под водой,
сравнительно длительную промывку и операцию плавления трини-
тротолуола. Поэтому неоднократно делались попытки вести очистку
продукта в плавленом состоянии. Однако при обработке тринитро-
толуола при температуре выше температуры его плавления раство-
рами сульфита встретились большие затруднения, так как при этих
условиях сульфит действует на а-тринитротолуол подобно едким
щелочам с образованием продуктов конденсации и осмоленияг
(Месир выделил гексанитро дибензил).
В Англии во время империалистической войны 1914—1918гг. был
разработан и применялся на одном заводе способ, состоявший в обра-
ботке плавленого тринитротолуола раствором гипохлорита натрия.
Гипохлорит вводили во время промывки горячей водой. Недостат-
ком этого способа является то, что таким путем можно повысить
температуру затвердевания тринитротолуола только на 1—1,5°,
т. е. меньше, чем при обработке раствором сульфита натрия.
Химические реакции, протекающие между несимметричными
тринитротолуолами и гипохлоритом натрия, до сих пор не уста-
новлены. Очевидно некоторые примеси окисляются. Полученный
при этом тротил почти бесцветен, и содержание в нем органических
примёсей, нерастворимых в бензоле, очень мало. Этот способ имел
1 Gornall nRobinson, J. Chem. Soc., 1983, 1926.
’ Muraour, Bull. Soc. Chim. Fr., 35, 367, 1924; M e c i r, Chim. et
Ind., 29, 952, 1933.
190
лишь кратковременное применение во время империалистической:
войны 1914—1918 гг. и непригоден для получения тротила высокого
качества.
Английский патент № 382222 предлагает так называемые бу-
ферные растворы, содержащие сульфит натрия. В этих растворах
щелочные свойства горячих сульфитных растворов понижаются пу-
тем добавления:
1) первичных или вторичных фосфор но натриевых солей;
2) уксусной кислоты с уксуснокислым натрием;
3) буры с борной кислотой.
Заметим, однако, что если бы по этому патенту и удалось устра-
нись окисляющее и осмоляющее действие сульфита натрия при
обработке тринитротолуола горячими сульфитными растворами, то
все же качество тринитротолуола, полученного по этому способу,,
будет хуже, чем при способе, описанном выше, так как здесь отпа-
дает частичная очистка тринитротолуола от динитротолуола, проис-
ходящая при промывке тротила на вакуум-воронке после сульфит-
ной промывки.
Кристаллизация тротила из отработанной
кислоты. Этот метод был впервые предложен и применен в Ита- .
лии- Он состоит в следующем.
Содержимое нитратора по окончании 3-й нитрации передается
в кристаллизатор. Последний представляет собой чугунный аппа-
рат, отличающийся от обычно применяющегося нитратора только
тем, что снабжен внизу выпускной трубой для выпуска содержи-
мого по окончании процесса кристаллизации на вакуум-во ронку;
Подобно нитратору кристаллизатор снабжен рубашкой для охлажде-
ния, мешалкой, термометром, вытяжной трубой, люком и т. д.
После загрузки кристаллизатора пускается в ход мешалка и
охлаждение. Процесс кристаллизации начинается по достижении
температуры около 75—77° и держится на этом уровне в течения
всего периода кристаллизации тротила (за счет теплоты его кристал-
лизации). По окончании кристаллизации продолжают охлаждение
до 30—35° и затем спускают содержимое кристаллизатора на ва-
куум-во ро нку.
Получение продукта высокого качества основано здесь на сле-
дующем. При перемешивании расплавленного тринитротолуола с от-
работанной кислотой примеси (изомеры, динитротолуол и др.) рас-
пределяются между слоем тринитротолуола и слоем кислоты. При
охлаждении а-тринитротолуол кристаллизуется из жидкого трини-
тротолуола, причем эвтектика обогащается примесями, которые
перераспределяются между эвтектикой и отработанной кислотой
(так как твердый а-тринитротолуол перестает быть растворителем
для примесей). Этот процесс продолжается по мере кристаллизации
а-тринитротолуола, причем органическая часть, растворенная в_ртра-
ботанной кислоте, все больше обогащается примесями.
Вакуум-воронка представляет собой железный клепаный, внутри
гомогенно освинцованный сосуд. Фильтрующим слоем здесь служит
191
(гравий или кварц (дйаметр кусочков кварца 3—4 мм). Кварц уло-
жен между двумя железными, гомогенно освинцованными решетками.
Здесь отсасывают на вакуум-воронке отработанную кислоту,
промывают разбавленными отработанными кислотами убывающей
концентрации, например, с содержанием 75% H2SO4, затем 55%
H2SO4. Такая промывка ведется с целью предупредить оплавление
кристаллов вследствие разогревания, которое могло бы иметь место
в случае непосредственной промывки водой на вакуум-воронке
после отсасывания крепкой отработанной кислоты. Кроме того,
_при этом вытесняются отработанные кислоты вместе с растворен-
ными в них низкоплавкими примесями. Эти примеси в случае их
выделения при непосредственном разбавлении водой понизили бы
температуру затвердевания тротила и увеличили бы его масля-
нистость.
По окончании отсасывания 55%-ной кислоты начинают промывку
холодной водой.
Установлено, что на вакуум-воронке затруднительно промыть
тротил до кислотности 0,02% и ниже по серной кислоте. Поэтому
'Содержимое вакуум-воронки переводится в промывной чан, где тро-
тил расплавляется и промывается до необходимой степени кислот-
ности.
Качество тротила, полученного по этому способу, ниже, чем по-
лученного перекристаллизацией из спирта или сульфитной очист-
кой. Для получения тротила нормального качества кристаллический
продукт после промывки водой (без расплавления тротила) подвер-
гается дополнительной сульфитной очистке с применением вдвое
или втрое меньшего количества сульфита натрия, чем при сульфит-
ной промывке тротила-сырца. После этого тротил промывается го-
рячей водой и сушится в расплавленном виде.
Недостатком этого способа является накопление низкоплавких
примесей в отработанной кислоте, что приводит к ухудшению ка-
чества тринитротолуола в случае применения полного кислотообо-
рота, поэтому необходимо отстаивание отработанной кислоты до ее
пуска на изготовление кислотных смесей. Следовательно, здесь
Невозможен горячий кислотооборот; кроме того, имеет место силь-
ная коррозия трубопроводов и аппаратуры.
Был предложен способ перекристаллизации тринитро то луе ла
из серной кислоты, который не получил практического приме-
нения.
Промывка тротийа ч е т ы р е х х л о р и ст ы м угле-
родом. В США во время империалистической войны 1914—1918 гг.
.для повышения качества тротила его промывали четыреххлористым
^углеродом и отсасывали жидкость на вакуум-воронке.
Промывка тротила спиртом. На одном заводе
*США промывали тротил полуторным весовым количеством спирта;
затем отсасывали промывной спирт на вакуум-воронке; наконец,
.вытесняли оставшееся количество спирта водой (0,9 в. ч. воды на
1 в. ч. тринитротолуола). Выход очищенного тринитротолуола — 90%.
192
При промывке тротила четыреххлористым углеродом или спиртом
необходимо предварительное его измельчение.
Перекристаллизация тротила из бензола.
На одном английском завода тротил перекристаллизовывался из бен-
зола — 3-й маточный поступал на дестилляцию. Выход тротила
мало отличался от выхода при перекристаллизации его из спирта.
Перекристаллизация изо-нитротолуола \
По словам Штетбахера, в обыкновенной аппаратуре для перекри-
сталлизации тротила можно перекристаллизовать его из о-нитрото-
луола в 4—5 раз больше, чем из спирта, причем с повышенным вы-
ходом.
2000 кг тротила нагревают при 80° с 7000 кг о-нитротолуола.
Раствор фильтруется, охлаждается в кристаллизаторах до темпе-
ратуры не ниже 16° (процесс кристаллизации ведется быстрее,
чем при спиртовом способе). Затем отсасывают на нутче, фугуют,
промывают на центрифуге небольшим количеством холодного спирта
для удаления остатков о-нитротолуола.
Преимуществом этого способа является то, что содержавшиеся
в тротиле примеси динитротолуола вновь нитруются, так как маточ-
ные растворы поступают для нитрации до ди- и тринитротолуола.
Недостатком этого способа является то, что побочные продукты
(несимметричные тринитротолуолы, продукты осмоления и др.) не
удаляются из цикла, а накапливаются, в- результате чего должно
ухудшаться качество получаемого тротила и после определенного
числа операций очистка должна даже совсем прекратиться.
Способ итальянского завода в Ченчжио.
Вследствие высокого акциза на спирт в Италии там еще в довоенное
время изыскивались способы получения тротила высокого качества
без перекристаллизации его из спирта.
По разработанному на заводе в Ченчжио способу динитротолуол
промывали водой и разливали в лотки. После застывания его выни-
мали из лотков, разбивали и переносили в отделение с температурой
40—50°, где укладывали на решетку; при этой температуре расплав-
лялись примеси, образовывавшие с динитротолуолом эвтектику,
которая стекала в поставленные внизу сосуды. На решетках оставался
продукт с температурой плавления 62—66°.
При этом способе получали из 1 кг толуола 1,1 кг твердого
и 0,6 кг жидкого динитротолуола.
Твердый продукт нитровался в тринитротолуол, причем полу-
чался продукт, удовлетворяющий техническим условиям на кристал-
лизованный тринитротолуол. Жидкий динитротолуол продавали
в другие страны, где спирт был дешевле; там его нитровали, а полу-
ченный тротил перекристаллизовывали из спирта.
Описанный способ разделения динитротолуола громоздок и имеет
кустарный характер. Лучше произвести разделение промывкой ди-
нитротолуола водой на вакуум-воронке или на центрифуге.
1 Штетбахера Пороха и взрывчатые вещества, стр. 391, 1936.
Химия—13—13 193
Выделение m-изомера из мононитротолуола.
Во время империалистической войны 1914—1918 гг. в Германии вос-
пользовались на некоторых заводах дестилляционными установками,
на которых в мирное время получали изомеры о-, т- и р-нитротолуола,
для очистки мононитротолуола от пг-нитротолуола. При нитровании
о-или р-изомера получается тринитротолуол с температурой затвер-
девания 1 около 79°. Однако и в Германии этот способ не нашел ши-
рокого применения. Недостатки способа: 1) заметные потери моно-
нитротолуола при дестилляции, 2) значительное удорожание про-
дукта.
Фуговка тринитротолуола с примесью фе-
нола. Примеси тринитротолуола дают эвтектику, которая частично
может быть отмыта центрифугированием измельченного тринитро-
толуола в струе нагретой (до температуры ниже температуры пла-
вления тринитротолуола) воды. Лучшие результаты получались
проведением процесса в две фазы при различных температурах.
Процесс был усовершенствован применением фенола: тринитро-
толуол смешивался с фенолом и медленно охлаждался. При этом
фенол образует эвтектику с примесями тринитротолуола. Эвтектика
отжималась на центрифуге, а тротил промывался затем во-
дой для полного удаления фенола. Получался тротил с температу-
рой затвердевания 80°.
НИТРОВАНИЕ Т0ЛУ0Л-БЕНЗИНА
Выше уже было сказано (стр. 119), что при содержании в толуоле
более 5% парафиновых углеводородов сильно увеличивается расход
кислот при получении из него тринитротолуола и ухудшается
качество продукта.
Толуол-бензин в зависимости от природы нефти, из которой он
получен, содержит от 60 до 90% бензина. Так как эти смеси по
своему составу близки к нераздельно кипящим смесям, то выделение
из них чистого толуола дестилляцией невозможно. Для получения
тротила из толуол-бензинов поступают следующим образом.
Нитруют толуол-бензин обычной нитрационной смесью, беря
количество азотной кислоты по содержанию толуола в обрабатывае-
мой порции толуол-бензина (на английских заводах берут 105%
HN03 от теории, а не 103%, как при нитровании каменноуголь-
ного толуола). Эта смесь практически не реагирует с бензином.
По окончании процесса нитрования реакционная масса состоит из
отработанной кислоты и смеси (раствора) моно нитротолуола с бен-
зином. Обычным способом отделяют органическую фазу от отрабо-
танной кислоты, промывают продукт водой для полного удаления
кислот и затем передают его в перегонный куб, где отгоняется бензин—
сначала глухим, а под конец острым паром. Так как при отгонке
бензина паром перегоняется некоторое количество мононитротолуола,
то во избежание значительных потерь последнего должно быть по-
1 Schwarfe, Die Techiik im Weltkriege, стр. 108, Berlin, 1920.
194
дано возможно меньше острого пара. В кубе остается свободный от
бензина (или содержащий минимальное его количество, не имеющее
заметного значения для последующих стадий нитрования) моно-
нитротолуол, который поступает в следующую фазу нитрации.
Отогнанный бензин может быть использован после очистки
в качестве моторного топлива.
ПРОИЗВОДСТВО ИЗОМЕРОВ МОНОНИТРОТОЛУОЛА
1. Применение изомеров мононитротолуола, о-и р-нитротолуолы
широко применяются для производства красок и фармацевтических
препаратов.
Основное применение о-нитротолуол находит для производства
о-толуидина и толидина:
о-толуидин
толидин
р-Нитротолуол применяется для получения р-толуидина и ди-
амино стильбендисульфокислоты:
р-Толуидин
СН—=сн
диаминостильбендисульфовдслота
о-Т олуидин — маслянистая жидкость (температура плавле-
ния —21° и —16°); применяется в производстве целого ряда азо-
красителей, при производстве синего сернистого красителя, сафра-
нина и др.
р-Толуидин — фисталлическое вещество (температура
плавления 43°); применяется в производстве m-нитро-р-толуидина.
Так как выделение вг-нитротолуола из технического мононитро-
толуола весьма трудно, то выгоднее получать его в чистом виде из
р-толуидина по схеме:
СН3 СН3
/\ /\
1 > х Jno2
NHCOCH3 NHCOCH3
195
При восстановлении /«-нитротолуола получается /«-толуидин,-
который подобно другим изомерам применяется в технике при про-
изводстве различных красителей.
2. Получение изомеров мононитротолуола фракционированной
перегонкой технического мононитротолуола. Разделение изомеров
мононитротолуола производится в технике фракционированной пе-
регонкой в вакууме.
Производство мононитротолуола для д е-
стилляции. В мононитротолуоле, поступающем на дестилля-
цию, не должно быть примеси динитротолуола; последний, перего-
няясь вместе с мононитротолуолом, раньше всего конденсируется
в дестилляционной колонке. Там он выделяется в кристаллическом
виде и закупоривает тарели, что приводит к ухудшению качества
продукта и понижению производительности дестилляционной ко-
лонки.
Для приготовления мононитрэтолуола с минимальным содержа-
нием динитротолуола пользуются следующим способом.
Приготовляют на свежих кислотах (нэ допускается применение
отработанных кислот 2-й и 3-й нитраций) кислотную смесь, приме-
нявшуюся при старом способе (состава 54%. H2SO15 29% HNO3,
17% Н,0 или, еще лучшэ, с меньшим содержанием азотной кислоты
с фактором нитрующей активности Ф = 68—70%). Кислотную
смесь дозируют из расчета, чтобы азотной кислоты было около 95%
от теоретического количества. Эту смесь приливают к толуолу (от-
нюдь не наоборот!) при температурном режиме, установленном для
старого способа, но без выдержки при 50°. Охлаждают массу до 35°
и направляют содержимое аппарата на сепарацию.
При таких условиях в мононитротолуоле остается от 5 до 10%
толуола.
Отсепарированный мононитротолуол подвергается тщательной
промывке для удаления кислот. Присутствие даже незначительных
количеств кислоты приводит к быстрому разрушению дестилляци-
онной установки. Поэтому мононитротолуол подвергают водной про-
мывке, затем содовой и еще раз водной; допускается содержание не
более 0,001% кислоты в пересчете на серную кислоту.
Дестилляция мононитротолуола. Известно,
что при перегонке под обыкновенным давлением вследствие высокой
температуры кипения изомеров мононитротолуола происходит силь-
ное осмоление; кроме того, если бы даже можно было пренебречь
этим осмолением, то потребовалась бы установка специальных паро-
вых котлов высокого давления. Во избежание этого ведут перегонку
в вакууме.
В табл. 32, (стр. 197) приведены температуры кипения изомеров
мононитротолуола при разных давлениях.
Дестйлляционный аппарат, служащий для разделения изомеров,
состоит из перегонного куба, обогреваемого паром, ректификацион-
ной колонки и трубчатого конденсатора. Колонка имеет высоту около
12 м, снабжается тарелями и колпачками. Вверху колонка закан-
196
Таблица 32
Наименование вещества Температура, °C
при 760 мм при 100 мм при 40 мм
о-Нитротолуол 221,3 147,7 122,7
т-Нитротолуол 232,7 158,3 134,4
р-Нитротолуол 238,4 163,6 137,9
чивается дефлегматором высотой 1,3 м. По выходе из дефлегматора
пары конденсируются в трубчатом конденсаторе, откуда сконденси-
ровавшаяся жидкость поступает в приемник.
Процесс перегонки ведется следующим образом. Устанавливают
вакуум в дестилляционной системе, нагревают перегонный куб паром
с давлением около 2 ат (температура греющего пара около 120°).
При этом перегоняется так называемая головка, состоящая из не-
прониТрованного толуола. После отгонки толуола давление пара по-
вышают до 7 ат (температура греющего пара около 164°) и ведут
некоторое время перегонку при этом давлении. К концу перегонки
повышают давление пара до 10 ат.
Вначале отбирают около 30% по весу загруженного моно нитро-
толуола. Эта фракция представляет собой о-нитротолуол с не-
значительным содержанием р-изомера (обычно до 2%, при плохой
работе колонки до 6%). После этого отбирают промежуточную фрак-
цию, состоящую из о-изомера (с содержанием 8—9% р-изомера)
всего около 20% по весу загрузки мононитротолуола. Эта фракция
возвращается на вторичную перегонку для выделения чистого о-изо-
мера.
Остаток, состоящий из смеси о-, р- и незначительного количества
иг-нитротолуола (всего около 45—50% по весу загрузки мононитро-
толуола), поступает в железные ящики-кристаллизаторы, где при
охлаждении выкристаллизовывается р-нитротолуол. Последний вы-
деляется в количестве около 55% по весу кубового остатка (или
около 25% по весу начальной загрузки мононитротолуола). р-Изомер
отжимается на вакуум-воронке или, лучше, на центрифуге, а ма-
точный раствор поступает вновь на ректификацию.
Даже при описанной перегонке при пониженных температурах
происходит частичное осмоление мононитротолуола; продукты осмо-
ления собираются н маточном растворе. При перегонке последнего
произошла бы частичная закупорка тарелей дестилляционной ко-
лонки. Поэтому маточный раствор не пускают непосредственно на
дестилляцию, а предварительно очищают его перегонкой с водяным
паром. Очищенный таким образом продукт дестиллируют вместе
со свежим мононитротолуолом, а остаток после перегонки с водяным
паром, представляющий собой смесь смолы и кокса, получающийся
в количестве около 5% от маточного раствора, сжигается.
197
3. Мероприятия по технике безопасности. При перегонке моно-
иитротолуола могут иметь место процессы разложения, сопровож-
дающиеся повышением давления. Во избежание аварии котел для
перегонки снабжают предохранительным приспособлением из тон-
кой алюминиевой пластинки, имеющей форму хомутика. Под этой
пластинкой ставится острие, предназначенное для того, чтобы про-
бить пластинку, если в аппарате образуется давление.
Работающим в дестилляционном отделении надо помнить, что
пары мононитротолуола чрезвычайно ядовиты; они вызывают тяже-
лые сердечные заболевания. Поэтому нельзя допускать неплот-
ностей в аппаратуре, через которые в помещение могли бы проник-
нуть пары мононитротолуола, не разливать в помещении моно-
нитротолуол и не пачкать руки мононитротолуолом.
Ориентировочная схема производства изомеров мононитротолуола
100 в. ч. технического мононитротолуола
4
б в. ч.
головки
I
___________I__________
30 в. ч.о-нитротолу-
ола с содержанием до
2% р-нитротолуола
20 в. ч. o-нитротолуола 45 в. ч. ос-
с содержанием 8—9% татка, смесь р-
р-изомера (промежу- и «-изомеров
точная фракция)
на нитрацию
кондиционный про-
на охлаждение в
к ристаллизаторы
I . I
на вторичную разгонку Ф
дукт отжим на ва-
на склад
куум-воронке или
лучше фуговка
I
I
25 в. ч. кондиционного
р-изомера (55% по весу
остатка от дёстилляции)
' I
I
на склад
I
Ф
маточный раствор
Ф
перегонка с во-
дяным паром
смесь о- и р-изомеров
I
Ф
дестилляция вместе со
свежим мононитрото-
луолом
смола и кокс
(5% по весу ма-
точного раствора)
на сжигание
При неудовлетворительном качестве 1-й фракции ее смешивают
с промежуточной фракцией и дестиллируют. При этой операции по-
лучают около 50% кондиционного продукта по весу перегоняемой
смеси.
4. Технические условия на о-нитротолуол.
Показатели -Л i 1-й сорт 2-й сорт
1. Внешний вид............
Прозрачная маслообразная жидкость
от светложелтого до светлокоричне-
вого цвета, без видимых на-глаз при-
месей
2. Температура кипения:
90% объема при давлении
760 мм должно отгоняться
в пределах ...............
3. Содержание динитротолуола .
4. Содержание углеводородов
не более . . ........
5. Содержание р-нитротолуола
не более............. . . . .
6. Содержание влаги не более . .
218 — 223° 217—223,5°
Отсутствие
0,1% 0,1%
1% 2%
0,1% 0,1%
Упаковка. о-Нитротолуол отгружают в чистых железнодорожных
цистернах или железных бочках.
5. Получение р-нитротолуола вымораживанием из технического
мононитротолуола. Потребности промышленности в р-нитротолуоле
значительно выше, чем в о-нитротолуоле, но количество последнего,
получаемое при дестилляции моно нитротолуола, значительно пре-
вышает количество получаемого при этом р-изомера.
Поэтому пользуются для получения р-изомера более простым и
дешевым способом — вымораживанием р-изомера из технического
мононитротолуола путем охлаждения последнего до —20°.
В зтом случае производство упрощается еще благодаря тому,
что не требуется приготовления мононитротолуола по описанному
специальному способу, а пригоден обычный мононитротолуол,
приготовленный по старому способу, либо в аппарате для непрерыв-
ного нитрования.
При охлаждении технического мононитротолуола до темпера-
туры —20° и? него выкристаллизовывается около 14% р-нитро-
толуола, считая по весу загрузки. Кристаллическая масса отжи-
мается от маточного раствора на центрифуге. Качество полученного
таким способом р-изомера выше, нежели полученного при дестил-
ляции (при охлаждении кубовых остатков), так как в последнем
случае продукт заметно загрязнен смолистыми примесями, образо-
вавшимися при перегонке.
199
6. Технические условия на р-нитротолуо i.
Показатели
1-й сорт
2-й сорт
1. Внешний вид...............
2. Температура застывания су-
хого продукта не ниже . . .
3. Содержание Динитротолуола .
4. Содержание углеводородов . .
5. Содержание влаги не более
6. Содержание примесей, не рас-
творимых в толуоле, не более . .
Кристаллы от светлосерого до светло-
желтого цвета
50° । 49°
Отсутствие
Отсутствие
1% | 1%
0Д% ! 0 1%
Упаковка. р-Нитротолуол отпускается в чистых железны'- барабанах
или Деревянных бочках. Вес нетто для барабанов 100 кг, для бочек 300 кг.
ПРОИЗВОДСТВО ДИНИТРОТОЛУОЛА
(травелина, порохового динитротолуола)
1. Применение динитротолуола. 2,4-динитротолуол применяется
для изготовления сернистого оранжевого красителя и для получе-
ния лг-толуилендиамина
Н3С
-NH2
который является важным промежуточным продуктом для получе-
ния различных красителей.
Кроме того, динитротолуол применяется под названием траве-
лина в качестве пластификатора при изготовлении нитроглицери-
новых порохов.
2. Производство 2,4-динитротолуола. 2,4-динитротолуол можно
получить двумя способами: 1) из технического динитротолуола пу-
тем фракционированного плавления и удаления низкоплавких фрак-
ций; 2) нитрованием р-нцтротолуола.
П р о и з в о дство 2,4-д инитротолуола из тех-
нического динитротолуола. Нитрованием толуола
получают динитротолуол, температура затвердевания должна быть
для получения хорошего выхода о-, р-изомера не ниже 55°.
По окончании нитрации отстоявшийся над отработанной кисло-
той динитротолуол сжатым воздухом передавливается в аппарат-
гранулятор. Гранулятор представляет соOoiPJlepевянный чан, снаб-
женный деревянной мешалкой, делающей'30 об/мин. В аппарат
2 00
предварительно загружается холодная вода и при работающей
мешалке подается динитротолуол; одновременно непрерывно посту-
пает из водопровода холодная вода. При этом динитротолуол затвер-
девает в виде гранулей. •
Полученный гранулированный динитротолуол поступает на цен-
трифугу. Центрифуги применяются с медной корзиной с двойным
фильтром из сукна и полотна; число оборотов от 750 до 950 в ми-
нуту- ЗагрузК'а в зависимости от диаметра корзины от 120 до 160 кг'
гранулированного динитротолуола. Продукт размещается ровным
слоем в центрифуге.
Во время работы центрифуги на продукт через разбрызгиватель
спускается вода, предварительно нагретая до 27—32°. Теплая вода,
пфоходя через слой продукта, нагревает последний и уходит через,
спускное отверстие кожуха центрифуги, увлекая с собой жидкую
фракцию нитропродукта, носящую название низкоплава.
После получасовой промывки водой, имеющей температуру 27—
32°, начинают промывку динитротолуола водой, нагретой до темпе-
ратуры 37—40° и ведут эту промывку также в течение получаса.
Наконец, его отжимают от воды и низкоплавкой фракции в течение-
15 минут.
Центрифугирование считается законченным, когда температура
затвердевания динитротолуола достигнет 64,5° и выше.
Выход 2,4-динитротолуола 62% от теории.
Производство 2,4 - динитротолуола из р - ни-
тротолуола. Нитруют р-нитротолуол, имеющий температуру
затвердевания не ниже 48°. Для нитрования применяется такая кис-
лотная смесь, чтобы отработанная кислота содержала около 80%.
H2SO4; смесь берется из расчета, чтобы избыток азотной кислоты
составлял 2% от теории. Содержание HN(X в отработанной кислоте
около 0,5%.
Полученный 2,4-динитротолуол гранулируется, как описано-
в предыдущем случае.
Выход 91,3% от теории по р-нитротолуолу-
3. Технические условия на 2,4-динитротолуол для красителей.
Температура затвердевания не меньше 64,5°. Содержание влажности
не больше 20%. Содержание о-р-изомера не меньше 96%.
4. Динигротолуол технический. Кроме травелина для различ'
ных технических целей изготовляется технический продукт, полу-
чаемый после 2-й нитрации и промывки водой.
5. Технические требования к динитротолуолу (техническому).
1. .Технический динитротолуол представляет плавленые куски
весом до 0,5 кг, желтого цвета и не должен включать посторонних
видимых на-глаз примесей.
2. Влажность не более 1,5%;
3. Содержание моно нитро толуола не более 1,0%;
4. Содержание нерастворимых в бензоле примесей не бо-
лее 0,1%.
201
5. Содержание золы не более 0,1%.
6. В том числе песка не более 0,05%.
7. Содержание кислоты в пересчете на серную кислоту не более
0,005%.
8. Температура затвердевания сухого промытого динитротолуола
не ниже 50° и не выше 56°.
9. При дальнейшей нитрации динитротолуола должен полу-
читься тринитротолуол с температурой затвердевания не ниже 76°.
ГЛАВА IX
НИТРОПРОИЗВОДНЫЕ БЕНЗОЛА
’ 1. Свойства бензола. Химически чистый бензол имеет темпера-
туру кипения 80,4°; температура его затвердевания 1 5,5°; уд. вес
В° = 0,89996; D‘° = 0,87895. Он очень мало растворим в воде
(при 23° 0,061 ч. в 100 ч. воды); вода в бензоле также мало рас-
творима (0,20 ч. в 100 ч. воды).
Каменноугольный бензол содержит сернистые соединения: иногда
следы сероуглерода (температура кипения 47°), почти всегда тиофен
(температура кипения которого 84°); поэтому его нельзя нацело
удалить из бензола перегонкой. Содержание тиофена в бензоле
доходит до 0,5%.
Присутствие тиофена в бензоле определяется при помощи индо-
фениновой реакции В. Мейера, основанной на том, что при взаимо-
действии тиофена с изатином в присутствии концентрированной сер-
ной кислоты образуется индофенин, окрашивающий жидкость
в голубой цвет.
В нефтяном бензоле не содержится ни сероуглерода, ни тиофена.
При действии озона бензол образует кроме небольшого количества
фенола продукт присоединения озона — бензолтриозонид:
Это вещество белого цвета, легко разлагающееся при действии воды,
1 Температура затвердевания и удельные веса приведены по данным Ме-
ждународного бюро физико-химических эталонов, опубликованным в статье
Timmermans и Martin, J. Chim. Phys. 23, 733, 1926 (см. Stett-
bacher, Chem. Zig, Fortschrittberichte, 1—20, 1929). По данным Международ-
ного бюро при поглощении влаги ив воздуха температура затвердевания
бензола понижается на 0,1°.
203
Кислот или щелочей; при быстром нагревании вспыхивает при 50° т
очень чувствительно к удару и трению.
Оксозон О4 образует с бензолом аналогичный предыдущему
продукт присоединения — бензолтриоксозонид, еще более чувстви-
тельный к удару и трению, чем нитроглицерин.
Оба эти вещества практически неприменимы вследствие большой
чувствительности к механическим воздействиям и химической не-
стойкости.
2. Токсическое действие бензола. Бензол является сильным
ядом. При вдыхании незначительных количеств бензола появляется
головокружение, головная боль, шум в ушах и рвота. При вды-
хании значительного количества отравления могут быть настолько
сильными, что кончаются смертью.
3. Технические условия на бензол по ОСТ 463
1. Цвет и прозрачность: бесцветен, прозрачен.
2. Удельный вес при 15° 0,880—0,885.
3. Испаряемость: испаряется без остатка.
4. Пределы кипения при барометрическом давлении 760 .мм:
начало перегонки не ниже 78,5°, конец перегонки не выше 80,6°.
В пределах 1° должно перегоняться не менее 95% по объему.
5. Степень очистки: окраска с серной кислотой по шкале Кремера
и Шпилькера не выше 0,5. Бромное число не выше 0,6 г брома на
100 мл бензола.
Нефтяной бензол содержит до 5% бензинов, но не содержит фено-
лов й сернистых соединений (сероуглерода и тиофена).
Присутствие бензинов (D=0,868) влияет на удельный вес и пре-
делы кипения, поэтому существуют особые технические требования
на нефтяной бензол.
4. Свойства мононитробензола и применение его. Мононитро-
бензол (мирбановое масло) представляет собой жидкость с запахом
горького миндаля. Температура кипения 208°; температура плав-
ления 5,5°; уд. вес 1,204 (при 20°); ядовит.
Прежде применялся в составах типа Шпренгеля (река-рок, со-
стоит из 79% бертолетовой соли и 21% мононитробензола). В на-
стоящее время применяется преимущественно для приготовления
полупродуктов для красителей.
5. Свойства динитробензола и его применение. Температуры
плавления изомеров (по Вилеру): т-динитробензола 1 89,9° о-изо-
мера 117° и р-изомера 172°.
Динитробензол легко летуч с парами воды и легко возгоняется.
В кислой среде динитробензол мало реакционноспособен.
Метилат натрия или калия, растворимый в абсолютном метило-
вом спирте, взаимодействует с раствором о- и р-динитробензола
1 По М с CamishHSalathe, температура плавления гга-динитробен-
зола 89,57° (см. J. Amer. Chem. Soc., 50, 1785, 1928.
204
в абсолютном метиловом спирте, образуя количественно соответ-
ствующие нитроанизолы, например:
/X,NO2
+ CH3ONa
\/NO2
+ NaNO2
В этих условиях тп-динитробензол не реагирует с метилатом натрия,
вследствие чего эта реакция может применяться для количествен-
ного определения о-, р -изомеров в смеси трех изомеров.
Однако в присутствии влаги частично реагирует и тп-динитро-
бензол, например, при применении для описанной реакции рас-
Tbopa едкого натра в метиловом спирте (вследствие выделения воды
при образовании метилата: NaOH 4-СН3ОН -> CH3ONa + Н2О)
или при применении водного метилового спирта. При взаимодей-
ствии с аммиаком одна нитрогруппа в о- или р-динитробензоле может
быть замещена аминогруппой. В этих изомерах одна нитрогруппа
легко замещается на сульфогруппу при взаимодействии их с вод-
ным раствором сульфита натрия:
/ >О2
Na2SO3
\^NO2
+ NaNO2
^SO3Na
Этой реакцией пользуются в производстве для удаления о- и р-изо-
меров из технического динитробензола для получения чистого тп-Ди-
нитробензола.
Перекристаллизованный, свободно насыпанный динитробензол
детонирует от капсюля с 1,5 г гремучей ртути; прессованный под
давлением 290 кг/см2 с кубической плотностью 1,29, он детони-
рует от капсюля с 3 г гремучей ртути; наконец, прессованный под
давлением 585 кг/ем2 с кубической плотностью 1,44, он не дето-
нирует о г 3-г капсюля. Литой динитробензол и в виде литого заряда
и измельченный в порошок значительно менее восприимчив к дето-
нации,чем мелкокристаллический продукт, перекристаллизованный
из растворителя.
Динитробензол — ядовитое вещество. При работе с ним имели
место многочисленные случаи отравления рабочих 2. Поэтому надо
принимать меры предосторожности: работать под вентиляционными
шкафами, защищать нос и рот фильтрами для пыли, работать в пер-
чатках и пр.
Делались попытки применения динитробензола для снаряже-
ния снарядов. Он не может применяться в смеси с тротилом, так как
эти смеси обладают слишком низкими температурами плавления.
1 Z. Schiess- u. Sprengst-, 13, 245—247, 268, 269, 1918; Dtsch. Med.
Wochschr., 45, 1273, 1274, 1919; Z. Schiess- u. Sprengst., 18, 14—16, 1923.
205
Применение чистого динитробензола для снаряжения снарядов
затруднительно вследствие его пониженной чувствительности к де-
тонации; оно возможно лишь при условии применения мощного
детонатора; это несколько усложняет процесс снаряжения.
Например, в Германии во время империалистической войны
1914—1918 гг. снаряжение производилось следующим образом: за-
ливали в снаряд динитробензол; образовавшуюся при остывании
усадочную раковину заполняли тротилом, служившим детонатором
для взрывания заряда динитробензола.
Кроме того, динитробензол раньше применяли для изготовления
бронебойного состава (95% тротила, 4% нафталина и 1% динитро-
бензола). Беллит — смесь аммиачной селитры с динитробензолом —
применяли раньше (до империалистической войны) в качестве про-
мышленного взрывчатого вещества. Однако вследствие частых от-
равлений при изготовлении беллита он теперь не применяется.
Во время империалистической войны смеси динитробензола с ам-
миачной селитрой применялись широко в Германии для снаряжения
различных боеприпасов.
Значительные количества динитробензола применяются для из-
готовления тга-нитро анилина и иг-фенилендиамина — полупродуктов
красочного производства.
6. Свойства тринитробензола. Тринитробензол кристаллизуется
в виде белых листочков (из спирта) или в виде пластинок ромби-
ческой формы. Температура его затвердевания 122,5°; плотность
может быть доведена прессованием до 1.67,
'У тринитробензола в высокой степени сказывается активирую-
щее влияние нитро групп на соседние группы и атомы, отмеченное
в главе «Общие свойства нитро соединений» (стр. 103):
1- С метилатом и этилатом щелочей он дает продукт присоединения
О = N - ОК
а
О2Н^ JNO,
NO2 /\
осн3
o2n/\no2
+ СНзОК
2. При действии разбавленного раствора едкого натра одна
нитро группа замещается группой — ONa:
ONa
+ NaOH
O,NX NO2
O2Nx/NO2
3. Еще легче проходит замещение нитрогруппы на гидроксиль-
ную группу у несимметричных тринитробензолов; для этого здесь
206
достаточно кипячения с водным раствором соды:
NO2 ONa
2
+ Na3CO3
замещается на аминогруппу:
NH2
zxno2
no2 no2
При нагревании несимметричных тринитротолуолов с водным
раствором аммиака нитрогруппа
NO2
* <У*о2
| + NHs
no2 no2
Еще легче проходит замена той же нитрогруппы при взаимодей-
ствии с водным раствором сульфита натрия:
NO, SO3Na
!Z4NO, zzno2
+ NaNO2
+ Na,S0a
no2 NO,
4. Три нитрогруппы, находящиеся в ядре бензола, настолько
активируют водородный атом, что тринитробензол уже при дей-
ствии слабого окислителя К3 [Fe (CN)6J окисляется в пикриновую,
кислоту.'
no2
N02
Лон
I
no2 o,n
При восстановлении тринитробензола получается триамино-
бензол.
Взрывчатые свойства. Чувствительность к удару
(табл. 33):
0,N
!NO2
Таблица 33
Наименование вещества
Высота падения груза, вызывающая
детонацию одного из двух образцов
при весе груза 2 кг, см
Тринитробензол.................
Тротил ........................
Пикриновая кислота ............
Тетрил.........................
150
100
50
50
207
Расширение в бомбе
Трауцля при взрыве
15 г взрывчатого ве-
щества (см. табл. 34).
Скорость детонации
близка к 7000 м/сек.
По Дотришу, мощ-
ность тринитробензола
превышает
пикриновой
на 5%.
1,2,3,5 -тетранитро-
Таблица 34
Наименование вешества Расширение в бомбе Трауцля СМ3
Тринитротолуол 459
Пикриновая кислота . . . 469
Тринитробенвол 480
Свойства
таковую
кислоты
7. Тетранитробензол.
•бензола. 1,2,3,5-тетранитробензол получен Голлеманом в виде
«ветложелтых кристаллов с температурой плавления 129—130°.
Он плавится без разложения и не разлагается при нагревании при
•температуре плавления в течение 6 часов. Нерастворим в воде;
легко растворяется в кипящем бензоле и дихлорэтане. Разлагается
при действии жидкостей, содержащих кислород (спирт, ацетон,
эфир и т. п.), вследствие чего может быть очищен перекристаллиза-
цией только из углеводородов и галоидопроизводных углеводородов.
При смачивании его водой сначала не наблюдается изменения
окраски; но через полчаса вода становится слегка желтой вследствие
взаимодействия тетранитробензола с водой с образованием пикри-
новой кислоты, окрашивающей воду; через неделю 3,3% вещества
разлагается на пикриновую кислоту и окислы азота. Скорость раз-
ложения сильно увеличивается при повышении температуры; при
температуре кипения воды вещество в несколько минут превращается
нацело в пикриновую кислоту. При хранении в лаборатории в тече-
ние 9 месяцев наблюдалось незначительное разложение поверх-
ностного слоя вещества. При хранении на сухом воздухе вещество
не показывает следов разложения после годового хранения.
Водный раствор щелочи разлагает тетранитробензол на пикраты
л нитриты; при растворении в растворе метилата натрия в абсолют-
ном метиловом спирте
при обыкновенной тем-
пературе образуется
нитрит натрия и три-
нитро анизол.
Тетранитробензол ко-
личественно превраща-
ется в пикрамид при
действии водного рас-
твора аммиака.
Его чувствитель-
ность к удару опреде-
лялась в приборе Ка-
юта; результаты см. в табл. 35 (вес груза равен 2 кг).
Отсюда видно, что у тетранитробензола чувствительность к удару
Таблица 35
Наименование вещества Высота падения груза см
Тетранитробензол .... 25
Пикриновая кислота . . . 83
Тринитротолуол 130
Тетрил 60
70% тетранитробензола+ + 30% тротила 60
"208
значительно больше, чем у применяемых взрывчатых веществ и даже
при добавлении к нему 30% тротила его чувствительность к удару
не меньше, чем тетрила.
Работоспособность его, определенная в бомбе Трауцля при на-
веске Ю г, равна:
Таблица 36
Наименование вещества 1-й опыт 2-й опыт Среднее По отноше- нию к тро- тилу °/ /о
Тетранитробензол 433 460,5 447 161
Пикриновая кислота ....... 316,5 335,5 325 117
Тринитротолуол 70% тетранитробензола + 30% тро- 267,5 286,5 277 100
тила — 340 123 123
Следовательно, по работоспособности тетранитробензол на 61%
превосходит тротил: однако флегматизированный тротилом состав
не имеет преимуществ перед тетрилом.
Получение тетранитробензола. По Голле-
м а н у х, тетранитробензол получается пропусканием двуокиси
азота через суспендированный в 65%-ной азотной кислоте пикра-
мид.^ По мнению Голлемана, при этом протекают следующие
реакции:
NH2 N = N • NO2 NO2
O2N.//Z\NO2 OoN/^NO, O^/^jNOa
NO2 no2 no2
При взаимодействии пикрамида с окислами азота образуется
азотистокислый диазотринитробензол, который разлагается с обра-
зованием тетранитробензола и выделением окислов азота.
Очистка полученного по этому способу тетранитробензола про-
изводится перекристаллизацией его из бензола. Бензольный рас-
твор предварительно обрабатывают серной кислотой для извлечения
примесей и промывают водой до нейтральной реакции.
Выход чистого тетранитробензола составляет 69% от теории.
По способу Борше при нагревании на водяной бане
спиртового раствора тринитрофенетола с гидроксиламином обра-
зуется тринитрофенилгидроксиламин,
NHOH
O2n/4NO2
Xno2
1 Holleman, Rec. Trav. Chim. Pays-Bas, 49, 112, 1930.
Химия—13—14
209
При нагревании на водяной бане полученного продукта с кон-
центрированной азотной кислотой образуется 1,2,3,5-тетранитро-
бензол.
По Борше и Феске1 2, 3-гидроксиламино-4,6-динитро-
1-феноксибензол реагирует с гидроксиламином:
H5c6o/44nhoh
O2N^/NO2
nhoh/^nhoh
O2N\/NO2
При нагревании на водяной бане полученного продукта с кон-
центрированной азотной кислотой он окисляется в 1,2,4,5-тетра-
нитробензол:
O.2N/XNO2
o2nX/no2
8. Общие соображения о целесообразности производства и при-
менения ди- и тринитробензола. Бензол получается в значительно
больших количествах, чем толуол и ксилол, как это видно из сле-
дующих данных (табл. 37).
Таблица 37
Относительное количество ароматических углеводородов, получающихся
из 1 т сухого угля3
Наименование вещества В Донбассе и Кемерово кг В новых быстро- ходных печах Донбасса кг
Бензола 2 4.8
Толуола 1 0,6—0,7
Ксилола 0.45 0,25
Фенола 0,12 0,1
Креэолов и ксилолов .... 0,36 0,18
Нафталина 1,2 (только для Донбасса) 1.5 2,6
(только Для Кемерово)
1 В orsch е и F esk е, Вег., 56, 1494, 1923; 4,6-дифениловый эфир ди-
нитрорезорцина получается при нагревании на водяной бане 4,6-динитро-1,3-ди-
хлорбенвола с фенолом и металлическим натрием. При нагревании Спиртового
раствора полученного продукта с гидроксиламином образуется:
NHOH' рС,Н5
o2n^Jno2
2 Н. Н. Ворожцов, Основы синтеза промежуточных продуктов и
красителей, стр. 18, 1934.
210
Выход продуктов при пиролизе нефти, или нефтяных погонов,
по весу исходного сырья таков: 6,6% бензола, 4,4% толуола, 9,8%
со львент-нафты.
Из этих сравнительных данных видно огромное значение
бензола, как сырьевой базы для получения взрывчатых веществ.
Бензола получается приблизительно в 2 раза больше, чем
толуола на старых печах и в 7—8 раз больше на новых быстроход-
ных печах.
Обычно из него производится пикриновая кислота и только
ничтожная часть используется для получения динитробензола. Ис-
пользование бензола для производства динитро бензола было бы
значительно выгоднее, чем производство пикриновой кислоты, как
:Ло видно из следующего сопоставления:
1) выход пикриновой кислоты по бензолу незначителен; он со-
ставляет лишь около 54% от теории. Выход динитробензола состав-
ляет 80—85% от теории;
2) производство динитробензола проще и стоимость его значи-
тельно меньше пикриновой кислоты;
3) динитробензол — нейтральное вещество, не взаимодействующее
с аммиачной селитрой, а потому может быть использован для сме-
сей типа амматола; это серьезнейшее преимущество динитробензола.
По этим причинам вопрос получения динитробензола имеет
большое практическое значение. Наряду с этим не следует упускать
из виду серьезные преимущества пикриновой кислоты в отношении
се взрывчатых свойств: она чувствительнее к детонации. В то время
как плавленая пикриновая кислота взрывается от З-з гремуче-
ртутного капсюля, для взрывания динитробензола требуется мощ-
ный промежуточный детонатор.
По работоспособности и бризантности пикриновая кислота зна-
чительно превосходит динитробензол. Поэтому вопрос о предпочтении
того или другого из этих веществ не может быть решен однозначно:
очевидно, во многих случаях окажется правильным применение
динитробензола (преимущественно в виде его смесей с аммиачной
селитрой). В ряде других случаев окажется, вероятно, более целе-
сообразным применение пикриновой кислоты.
Наконец, следует отметить вопрос о производстве и применении
тринитробензола.
Тринитробензол в качестве взрывчатого вещества также пред-
ставляет большой практический интерес. При сравнении пикриновой
кислоты с тринитробензолом мы должны отметить следующие серьез-
ные преимущества последнего:
1) тринитробензол менее чувствителен к механическим воздей-
ствиям, чем пикриновая кислота и даже чем тротил; поэтому он
мог бы применяться с успехом для снаряжения ряда ответственных
боеприпасов, где вопрос пониженной чувствительности к удару при
большой силе имеет особое значение;
2) чувствительность к детонации у тринитробензола выше, чем
у пикриновой кислоты;
211
3) работоспособность его выше, чем у пикриновой кислоты;
4) тринитробензол — нейтральное вещество, не взаимодей-
ствует с аммиачной селитрой, а потому может быть использован
для смесей типа амматолов.
По этим причинам вопрос нитрации динитробензола в тринитро-
бензол привлекает к себе в последнее время внимание ряда иссле-
дователей.
9. Производство мононитробензола. Зависимость между составом
нитрационной смеси и степенью нитрации бензола изучена Гроггин-
сом х. В своих опытах он делал выдержку при 60°. При этом он уста-
новил, что наилучшей смесью для нитрования бензола, является
смесь, имеющая коэфициент дегидратации 3,6 и состав: 60% H2SO4,
32% HNO3, 8% Н2О.
Фактор нитрующей активности этой смеси равен 78. При этом
Гроггинс указывает, что смесь состава: 54% H2SO4, 38% HNO3,
8% Н2О (фактор нитрующей активности этой смеси равен 75) часто
приводит к нитробензолу со значительным содержанием бензола.
В настоящее время применяют смесь состава: 58—60% H2SO4,
30—32% HNO3, остальное Н2О.
Смесь приливают к бензолу при температуре до 40—50°; вы-
держка — 1 час при 60°.
По окончании выдержки и охлаждения до 30—35° реакционная
масса переводится в сепаратор, где отстаивается не менее 30 минут.
Отстоявшийся нитробензол промывается водой и затем содовым
раствором.
Выход нитробензола 98% от теории или 154,5 ч. на 100 в. ч.
бензола.
Часто берут смесь, соответствующую содержанию около 74—
75% H2SO4 в отработанной кислоте, с целью устранения образо-
вания динитробензола. В этом случае, несмотря на то, что берут
102% HNO3 от теории, получают несколько процентов недонитро-
ванного бензола. После промывки полученного нитробензола отде-
ляют непрореагировавший бензол — отгонкой острым паром — до
начала перегонки масла с удельным весом, большим единицы. Это
масло отделяют от воды и направляют на нитрацию.
Если исходный бензол содержит тиофен, то при нитровании
образуется динитротиофен, присутствие которого можно обнаружить
по красному окрашиванию, получаемому после обработки поташем
спиртового раствора нитробензола.
Нитрование бензола и р и повышенной тем-
пературе. При нитровании бензола (так же, как при нитрова-
нии толуола по старому способу, см. стр. 148) процесс проводится,
начиная от обыкновенной температуры с медленным постепенным
подъемом ее до 50°. При этой температуре заканчивают слив смеси
в бензол.
‘Groggins, Anilin and its derivatives, New-York, 1924-
212
Благодаря малому температурному перепаду, особенно в первый
период слива смеси, значительно увеличивается продолжительность
процесса нитрации.
Такой температурный режим установлен, как мы видели раньше,
во избежание образования продуктов осмоления и окисления; кроме
того, при таком режиме почти устраняется образование динитробен-
зола.
С целью уменьшения продолжительности нитрования Малярев-
ский1 изучил нитрование бензола при повышенной температуре,
причем установил, что в обычного типа нитраторах можно без ощу-
тительного ущерба для качества конечного продукта повысить тем-
пературу нитрования до 80°. Одновременно, разумеется, сокра-
тцается расход энергии, воды для охлаждения и рабочей силы; со-
кращается также расход серной кислоты, так как можно снизить
концентрацию отработанной кислоты. Для избежания частичной
перенитрации при выдержке и для полного использования исход-
ного бензола необходимо энергичное и совершенное перемешивание.
На английских заводах, по Гроггинсу, охлаждение во время
нитрации производится не водой, а рассолом из холодильных уста-
новок. Кроме того, слив нитрационной смеси к бензолу ведется
в зависимости от степени совершенства перемешивания при темпе-
ратурах 24 или 60°. Для удобства регулирования .температуры
в аппарат заливается перед началом нитрации отработанная кис-
лота в количестве 2 ч. на 2,6 ч. бензола. По словам Гроггинса,
этим процесс ускоряется на много часов.
Технические условия на мононитробен-
з о л (ОСТ 467):
1. Мононитробензол должен быть прозрачен и не содержать
взмученных механических примесей.
2. Уд. вес при 15° 1,207—1,208.
3. Реакция нейтральная или слабо щелочная.
4. Содержание воды: не более 0,5% (допускается до 1,5%).
5. Содержание бензола: не более 1%.
6. Пределы кипения нитробензола (предварительно высушенного
прокаленным поташем) должны заключаться в интервале 8°, отсчи-
тываемых вверх от начальных точек кипения 205—208°.
10. Производство динитробензола. По данным Мартинсена о за-
висимости скорости нитрации нитробензола от крепости серной кис-
лоты, видно, что максимальная скорость имеет место при составе
кислоты H2SO4 • 0,63 мол Н2О или при содержании 89,5% H2SO4
в отработанной кислоте. На самом деле современные смеси, приме-
няемые для нитрации мононитробензола, близки к этому со-
ставу.
В начале XX столетия динитробензол получался, по Кайзеру,
следующим образом2.
1 Ж. X. П., IV, 399, 1927-
’Ullman, Enz. der.techn. Chem. т. II, стр 275, 1928.
213
К 175 в. ч. моно нитробензола добавлялась в течение 5 часов
смесь из 1350 кг серной кислоты (уд. вес 1,84) и 450 кг азотной
кислоты с содержанием 86% HNO3. Температура поднималась
в течение первых 1г/2 часов до 115° и эту температуру держали до
конца слива.
При смешении указанных кислот получается смесь состава:
71,5% H2SO4, 17% HNO3, 11,5% Н2О, что соответствует содержа-
нию 81,5% H2SO4 в отработанной кислоте; количество HNO3 — 430%
от теории. Большой расход кислот по этому способу, повидимому,
объясняется: 1) недостаточной концентрацией серной кислоты
в смеси, 2) высокой температурой слива, при которой должно те-
ряться повышенное количество HNO3 испарением и должны иметь
место повышенные окислительные процессы, 3) применением смеси
с большим содержанием воды (11,5% Н2О), вследствие чего неиз-
бежно повышается количество отработанной кислоты.
Гроггинс рекомендует для получения динитробензола смесь со-,
става: 80% H2SO4, 18% HNO3, 2% Н2О.
Коэфициент дегидратации равен 7 (фактор нитрующей активности
равен 91,5%), избыток HNO3 равен 10%.
Заводский способ получения динитробензо л а.
В аппарат подается 4600 кг нитробензола и 4500 кг серной кислоты
с содержанием 98% H2SO4. К этой смеси постепенно прибавляют в те-
чение 8—10 часов смесь состава: 27,5% H2SO4, 68% HNO3, 4,5% Н2О.
Избыток HNO3 — 10% от теории.
При сливе поднимают температуру с 40 до 76°. По окончании
слива нагревают содержимое аппарата до 90° и при этой темпера-
туре делают 2-часовую выдержку. Отработанная кислота содер-
жит в среднем: 87% H2SO4, 4% HNO3.
По охлаждении до 80° отделяют динитробензол, цромывают его
горячей водой, а затем содой.
Продукт сушится в расплавленном состоянии при нагревании
глухим паром до 120° с одновременной продувкой воздухом. Полу-
ченный таким образом продукт не должен пахнуть нитробензолом и
должен иметь температуру плавления не меньше 81°.
На 100 в. ч. мононитробензола получается 132 в. ч. сырого ди-
нитробензола (96,5% от теории).
Состав сырого динитробензола. Сырой дини-
тробензол содержит кроме указанного ранее количества изомеров
динитробензола очень малые количества пикриновой и стифниновой
кислот 2.
Кроме того, содержатся следы красящих веществ.
1 По Bennet и Joule (J. Chem. Soc., 1816, 1938), при нитрации
бензола образуется 0,03% динитрофенола, а при нитрации нитробензола — от
0,5 до 6,5% стифниновой кислоты в зависимости от условий нитрации. К сожа-
лению, в опытах этих авторов условия нитрации нитробензола сильно отлича-
лись от обычных заводских условий нитрации нитробензола, вследствие чего
по ним нельзя сделать вывода о количественном содержании этих побочных
продуктов в техническом динитробензоле.
214
Влияние условий нитрования на состав
динитробензола. Для практических целей как при при-
менении в качестве взрывчатого вещества, так и при применении
для изготовления красок необходимо иметь т- динитробензол
с возможно малым содержанием о- и р-изомеров. Поэтому изуча-
лись условия получения прямым нитрованием продукта с возможно
высоким содержанием т- динитробензола.
Уже давно 1 было установлено, что состав кислотной смеси мало
влияет на соотношение между изомерами, но большое влияние ока-
зывает температура нитрации.
Вилер 2 недавно вновь изучал влияние температуры нитрации
на соотношение между изомерами, причем получил следующие дан-
ные (табл. 38):
Таблица 38
Температура нитрации °C Температура плавления по- лученного Ди- нитробензо ла °C Состав динитробенвола, % Процент />-изо- мера, если сумму о-ивомер 4- р-изо- мер приравнять к 100%
т- о- Р-
124—129 79,9 85,7 13,9 0,4 3
108—113 81,2 87,6 •10,2 2,2 18
90—100 80,0 87,0 7,8 1,2 9
65—75 82,1 89,2 8,7 2,1 19
65—69 83,0 89,3 7,7 3,0 28
25—29 85,0 92,6 5,0 2,4 32
—5 до 4-5 86,8 94,7 3,5 1,8 34
—17 до —10 86,9 95,1 2,5 2,4 49
Таким образом при низких температурах можно получить тех-
нический динитробензол с содержанием 5—6% о- и р-изомеров. Как
указывает Вилер, при нитровании при низких температурах не
образуются красящие вещества, загрязняющие обычно динитробен-
зол, полученный нитрованием при высоких температурах.
Недостатком способа нитрования при низких температурах яв-
ляется необходимость применения для охлаждения холодильной
установки, увеличение продолжительности нитрации и повышенный
расход олеума.
Получение очищенного динитробензола
(с высоким содержанием т - д и н и т р о б е н з о ла).
В последнее время разработан следующий способ получения высоко-
качественного m-динитробензола с малым содержанием о- и р-изо-
меров.
1 Holleman, Вег., 39, 1715, 1906; L о Ь г у de Bruyn, Rec.
Trav. Chim. Pays-Bas, 19, 79, 1909.
2 W у 1 e r, Helv. Chim Acta, 15, 23, 1932.
215
Отделенный в сепарационной колонке от отработанной кислоты
динитробензол направляется на гранулирование; с этой целью он
сливается в чан с энергично перемешиваемой холодной водой; одно-
временно с динитробензолом из водопровода подается холодная
вода.
Гранулированный продукт подается на центрифугу (где он от-
жимается, и промывается холодной водой для удаления кислотно-
сти), откуда его смывают в аппарат для сульфитной промывки.
Очистка динитробензола ведется по способу О. М. Голосенко Ч
В авторском свидетельстве О. М. Голосенко указано на давно
известные и описанные в литературе наблюдения, что при действии
солей сернистой кислоты все три изомера динитробензола вступают
в реакцию с образованием растворимых в воде сульфокислот. Это
взаимодействие происходит при почти совпадающих условиях.
Это совпадение условий реакции для всех трех изомеров не поз-
воляло использовать реакцию с солями сернистой кислоты в технике
для очистки сырого динитробензола от о- и р-изомеров.
При изучении условий взаимодействия трех изомеров динитро-
бензола с сульфитом натрия О. М. Голосенко установила, что все
они легко и почти количественно реагируют с солями сернистой
кислоты уже при 79°. Однако, если вести процесс при 65—70° при
энергичном перемешивании и брать сульфит натрия в количестве,
отвечающем содержанию о- и р-изомеров в смеси, то реакция вза-
имодействия сульфита натрия с о- и р-динитро бензолом протекает
количественно, причем заканчивается через 2—3 часа; т-изомер
в Этих условиях не реагирует с сульфитом натрия.
По указанию автора, после 3—4-кратной промывки реакционной
массы водой получается продукт с количественным выходом т-изо-
мера с температурой плавления 89,5—90°, почти свободный ото- и
р-изомера.
Технические требования к динитробензолу
Показатели 1-й сорт | 2-й сорт
1. Внешний вид 0 Мелкий кристаллический порошок или сплавленные небольшие гранули желтого цвета. Динитробензол не дол- жен заключать посторонних примесей, видимых на-глаз. При просеивании взятой пробы через проволочное сито со стороной ячеек в 3 мм кристалли- ческий динитробензол должен прохо- дить без остатка (См. продолжение^
1 Авторское свидетельство на изобретениие № 40343, 1934.
216
Показатели 1-й сорт | 2-й сорт 1
I
2. Влажность и летучие вещества Не более 0.05% Не более 0,1%
3. Температура затвердевания . . Не ниже 88° Не ниже 81.5®
4. Кислотность в переводе на серную кислоту (азотной допуска-
ются только следы) 5. Нерастворимого в бензоле Не более 0,02% Не более 0,02%
остатка Не более 0,15% Не более 0,15%
'*» 6. вг-Изомера Не менее 95% Не менее 95%
7. Солей натрия . • Не более следов Не более следов
8. Маслянистость: по эталону для кристаллизованного из спирта балла 2
тротила • . Не выше балла 2 Не выше или 3.
Технические условия на динитробензол для приготовления
m-нитроанилина и т-фенилендиамина
1.. Содержание т-динитробензола............не меньше 98%
2. Температура затвердевания...............не ниже 89°
3. Содержание о- и р-изомеров..............не больше 1%
Обоснование технических требований к д и н и т р о-
бензолу. Взрывчатые свойства о- и р-динитробензола практически
не отличаются от таковых m-динитробензола, и с этой точки зрения
их присутствие не отразилось бы отрицательно на качестве динитро-
бензола. Но их присутствие влияет на физические свойства динитро-
бензола, изготовляемых из него смешением с другими нитросоедине-
ниями сплавов, а также смесей с аммиачной селитрой. Вследствие
образования низкоплавкой эвтектики здесь возможна течь из снаря-
женных этими составами боеприпасов; соответственно и здесь при-
менимо целиком сказанное по вопросу о влиянии изомеров на ка-
чество тротила. Поэтому установлены минимальные значения допу-
стимых температур затвердевания, содержания т-динитробензола»
а также введена проба на маслянистость.
Натриевые соли могут присутствовать либо в виде сульфита нат-
рия (при щелочной реакции) или нитрита натрия (при нейтраль-
ной реакции). Присутствие щелочи недопустимо вследствие возмож-
ного взаимодействия с нитросоединениями при длительном хранении
динитробензола или его сплавов с другими полинитросоединениями..
Нитрит, как нестойкая соль, при хранении разлагается с выделе-
нием окислов азота, — что может отрицательно повлиять на хими-
ческую стойкость взрывчатого вещества.
Остальные требования технических условий обеспечивают опре-
деленную степень чистоты продукта.
21?
Обоснование требований технических
условий на динитробензол для анилинокра-
«очной промышленности. При содержании о- и р-ди-
нитробензола в продукте, применяемом для приготовления тп-нп-
троанилина или m-фенилен диамина, уменьшается выход как аминов,
так и красителей. Например, при применении сырого (неочищен-
ного) динитробензола, содержащего до 15% о- и р-изомерд, выход
да-нитроанилина уменьшается на 30—35%. При применении т-фе-
нилендиамина (полученного из неочищенного динитробензола), со-
держащего небольшие примеси о- и р-изомеров, выход красителя
•сильно уменьшается (например, выход анил-коричневого ДЗГ па-
дает на 30%, анил-черного Е — на 10%).
Поэтому особенно строги требования к качеству т-динитробен-
зола, применяемого для целей анилинокрасочной промышленности.
Определение содержания m-изомера в ди-
нитробензоле. Метод определения содержания т-динитро-
бензола основан на том, что m-фенилендиамин сочетается с диазо-
бензолом в уксуснокислой среде:
ю- и р-фенилендиамины в тех же условиях не реагируют с диазо-
бензолом. Определение ведется следующим образом. Навеску около
5 г высушенного динитробензола растворяют при подогревании на
водяной бане в 100 сл3 этилового спирта в колбе Эрленмейера ем-
костью на 500 сл3, закрытой пробкой с длинной вертикальной труб-
кой в качестве обратного холодильника.
По растворении тп-динитробензола' добавляют 50 ел3 соляной
кислоты (уд. веса 1,19), после чего небольшими порциями добав-
ляют цинковую пыль.
Реакцию следует вести не слишком бурно. Когда раствор почти
•обесцвечен, сразу прибавляют несколько большую порцию цинко-
вой пыли; обесцветившийся при этом раствор быстро фильтруется
через воронку Бюхнера; фильтр промывают водой до исчезновения
реакции на хлор с азотнокислым серебром. Фильтрат переливают
в мерную колбу на 500 сл3 и разбавляют водой до метки.
В толстостенный стакан емкостью 500 сл3 берут пипеткой 50 сл3
полученного раствора, нейтрализуют содой свободную минераль-
218
ную кислоту, добавляют 100 см3 20%-ного раствора уксуснокислого
натрия, 5 слг! 10%-ного раствора уксусной кислоты и помещают
в лед для охлаждения до температуры 0—5°.
Нитруют 1 2/20Лг раствором хлористого фенилдиазония до появле-
ния реакции с Н-кислотой. Содержание m-динитробензола рассчиты-
вают по формуле:
n М • X • 500 • 100 • 100
и — 1000.20 • 50 • а (100 — 6)’
где D — лг-динитро бензол в сухом продукте, %;
-У — израсходованный раствор фенилдиазония, см3;
«, Ь — влажность динитробензола;
а — навеска динитробензола.
11. Получение тринитробензола. Нитрование дини-
тробензола. Проф. Солонина в 1911 г. разработал следую-
щий способ получения тринитробензола.
* • — - ~ - - С- —------— — _ _•* » ттЛ ттттг-t Л птлгт О Л -» Л г»/~» rtiTTA
Опечатки
Cmp. Строка Напечатано Должно быть По чьей, вине
218 19 сверху Т Z II z\ / N=NCl f l авт. и ред.
219 4 сверху Нитруют Титруют авт.
А. Г. Горст, Химия и технология нитросоединений. Зак. № 13
то при сохранении прочих условий нитрования получается про-
дукт, содержащий 30% тринитробензола и 70% динитробензола.
Наконец, Редклиф и Подлит 3 нашли, что если брать 1200%
HNO3 от теории, то в продукте нитрации содержится 70% трини-
тробензола и 30% динитробензола.
Во всех этих опытах смеси содержали от 82 до 87% мол H2SO4
(96,6—97,5% вес. H3SO4 в отработанной кислоте).
Очевидно, что при таком большом расходе крепких кислот и при
малых выходах тринитробензола промышленное получение трини-
тробензола нитрованием динитробензола неэкономично.
Хороший выход может быть получен только при нитровании под
давлением, что сложно и опасно.
1 Drummond, J. Soc. Chem. Ind., 41, 338—340, 1922.
2 R adcli 11 e и P ol 1 i 11, J. Soc. Chem. Ind., 40, 45—48, 1921.
219
*
Обоснование требований технических
условий на динитробензол для анилинокра-
«оч но й промышленности. При содержании о- и р-ди-
нитробензола в продукте, применяемом для приготовления т-нп-
троанилина или m-фенилендиамина, уменьшается выход как аминов,
так и красителей. Например, при применении сырого (неочищен-
ного) динитробензола, содержащего до 15% о- и р-изомерд, выход
m-нитроанилина уменьшается на 30—35%. При применении тп-фе-
нилендиамина (полученного из неочищенного динитробензола), со-
держащего небольшие примеси о- и р-изомеров, выход красителя
•сильно уменьшается (например, выход анил-коричневого ДЗГ па-
дает на 30%, анил-черного Е — на 10%).
Поэтому особенно строги требования к качеству т-динитробен-
зола, применяемого для целей анилинокрасочной промышленности.
Определение содержания т - и з о м е р а в ди-
нитробензоле. Метод определения содержания т-динитро-
бензола основан на том, что m-фенилендиамин сочетается с диазо-
ЙРНЯГ»ТТПА.Т п тгтгрттоггпхгттлттл-Д- л-плттл.
v g динитрооензола растворяют при подогревании на
водяной бане в 100 ел3 этилового спирта в колбе Эрленмейера ем-
костью на 500 см3, закрытой пробкой с длинной вертикальной труб-
кой в качестве обратного холодильника.
По растворении m-динитробензола добавляют 50 см3 соляной
кислоты (уд. веса 1,19), после чего небольшими порциями добав-
ляют цинковую пыль. '
Реакцию следует вести не слишком бурно. Когда раствор почти
•обесцвечен, сразу прибавляют несколько большую порцию цинко-
вой пыли; обесцветившийся при этом раствор быстро фильтруется
через воронку Бюхнера; фильтр промывают водой до исчезновения
реакции на хлор с азотнокислым серебром. Фильтрат переливают
в мерную колбу на 500 сл3 и разбавляют водой до метки.
В толстостенный стакан емкостью 500 сл3 берут пипеткой 50 см3
полученного раствора, нейтрализуют содой свободную минераль-
218
ную кислоту, добавляют 100 сл3 20%-ного раствора уксуснокислого
натрия, 5 см3 10%-ного раствора уксусной кислоты и помещают
в лед для охлаждения до температуры 0—5°.
Нитруют Х/2(Д раствором хлористого фенилдиазония до появле-
ния реакции с Н-кислотой. Содержание m-динитробензола рассчиты-
вают по формуле:
п М • X • 500 • 100 • 100
и — 1000 • 20 • 50 • а (100 — 6)’
где D — m-динитробензол в сухом продукте, %;
X — израсходованный раствор фенилдиазония, см3;
* Ь — влажность динитробензола;
а — навеска динитробензола.
11. Получение тринитробензола. Нитрование дини-
тробензола. Проф. Солонина в 1911 г. разработал следую-
щий способ получения тринитробензола.
В колбу с обратным холодильником наливается 30 г азотной
кислоты (уд. вес 1,5) и 7,6 г олеума, содержащего 45 50% SO3;
затем всыпается 10 г m-динитробензола. Смесь сильно взбалтывается
и нагревается сначала при 85° в течение 8 часов, затем 16 часов
при 120°. После этого берется проба, и если окажется заметное ко-
личество динитробензола, то смесь продолжают нагревать при 140—
150° до исчезновения динитробензола. При этом происходит силь-
ное окисление динитробензола и до 40% его окисляется в тетра-
нитрометан .
Содержимое колбы разбавляется толченым льдом, затем осто-
рожно холодной водой; тринитробензол отделяется, промывается и
высушивается.
Опыты Друммонда 1 показали, что при применении крепких
смесей и 400% HNO3 от теории при 3—6-часовой выдержке при
130° получается продукт, содержащий около 20% тринитробен-
зола и 80% динитробензола. Если взять 700% HNO3 от теории,
тог при сохранении прочих условий нитрования получается про-
дукт, содержащий 30% тринитробензола и 70% динитробензола.
Наконец, Редклиф и Поллит 2 нашли, что если брать 1200%
HNO3 от теории, то в продукте нитрации содержится 70% трини-
тробензола и 30% динитробензола.
Во всех этих опытах смеси содержали от 82 до 87% мол H2SO4
(96,6—97,5% вес. H2SO4 в отработанной кислоте).
Очевидно, что при таком большом расходе крепких кислот и при
малых выходах тринитробензола промышленное получение трини-
тробензола нитрованием динитробензола неэкономично.
Хороший выход может быть получен только при нитровании под
давлением, что сложно и опасно.
1 Drummond, J. Soc. Chem- Ind., 41, 338—340, 1922.
2 R ad cli 11 e и P о 1 li 11, J. Soc. Chem. Ind., 40, 45—48, 1921.
219
t
Получение тринитробензола восстанов-
лением хлористого пикрила. Получение
тринитрохлорбензола. Тринитрохлорбензол полу-
чается легче тринитробензола. По Франкланду и Гарнеру1, он
получается следующим образом: 100 ч. динитрохлорбензола рас-
творяют в 750 ч. моногидрата серной кислоты и добавляют 125 ч.
азотной кислоты с содержанием 94% HN03 (323% от теории),
нагревают при 130° в течение 12 часов. При этом выход тринит-
рохлорбензола составляет 85% от теории; температура его за-
твердевания 76° (температура затвердевания химически чистого ве-
щества 81,66° по Деверню).
Восстановление хлористого пикрила. В 1901 г.
Ульман и Белецкий 2 опубликовали работу по вопросу о синтезе
соединений дифенильного ряда. Они получали эти соединения пу-
тем взаимодействия соответствующих галоидных соединений с по-
рошкообразной медью (см. о получении гексанитро дифенила,
стр. 371). При реакции получения гексанитродифенила эти авторы
пользовались для достижения спокойной реакции в качестве разба-
вителя нитробензолом. При одном опыте конденсации они приме-
няли в качестве разбавляющего средства толуол, причем из про-
дукта реакции изолировали кроме гексанитродифенила незначитель-
ное количество тринитробензола. Однако при. дальнейшей работе
авторам не удалось найти условия, ведущие к образованию трини-
тробензола, но они указали, что, повидимсму, для образования
•тринитробензола необходима влажность..
Условия образования тринитробензола были указаны в патенте
Мейера 1909 г. 3. Описанный в этом патенте способ основан на том,
что тринитрохлорбензол легко восстанавливается в тринитробензол
при обработке его порошкообразной медью в нейтральном раствори-
теле в присутствии воды. В качестве растворителей можно приме-
нять спирты (метиловый, этиловый, амиловый), ацетон, эфир, бензол
и пр. Вместо меди можно брать латунь, цинк, железо, алюминий,
магний.
При взаимодействии протекает следующая реакция:
С1
o2n/\no2 o2n/4xno2
2 J J + 3Cu'+ H2O-----------> 2 | + Cu2C12 + CuO
no2 no2
Для этой реакции 100 ч. хлористого пикрила взмучивают в 100 ч.
95%-ного спирта и прибавляют 32 ч. меди. Смесь нагревают в тече-
ние 2 часов на водяной бане с обратным холодильником. Продукт ре-
акции выпадает в виде блестящих чешуек. Выход сырого продукта
1 Frankland и Garner, J. Soc. Chem. Ind-, 39, 257, 1920-
2 Вег-, 34, 2179, 1901
3 Герм. пат. № 234726, 1909.
220
составляет 68% от теории. Этот метод получения тринитробензола
не имеет промышленного применения из-за трудности получения
хлористого пикрила и малого выхода тринитробензола.
Окисление 2, 4, 6-тринитротолуола в три-
нитробензойную кислоту и последующее по-
лучение тринитробензола.
а) Окисление тринитротолуола хромовой
смесью. По Келеру \ в чугунном аппарате растворяют 12 кг
тротила в 200 кг купоросного масла при 80°, охлаждают до 40°
и при этой темцературе прибавляют небольшими порциями 20 кг
бихромата в течение 3 часов. После этого оставляют в покое на 5 ча-
сов; затем разбавляют водой и промывают. Полученную тринитро-
бензойную кислоту растворяют в 180 л воды, нагревают в течение
5—6 часов. При этом выделяется углекислота и образуется трини-
тробензол 1 2. Выход — 8 кг, т. е. 71% от теории; выход мал и спо-
соб дорог.
б) Окисление тринитротолуола азотнойки-
с л о т о й (серноазотной смесью). Этот способ основан на следую-
щей реакции:
СН3
no2z4xno2
+ 6HNO3
СООН
o2nzzno.,
| + 6NO2 + 4Н2О
NO2
no2
Полученную тринитробензойную кислоту превращают в трини-
тробензол кипячением с водой.
Условия реакции окисления по Келеру:
а) дозировка:
тринитротолуол .......................... 14.
азотная кислота с содержанием 90 — 92% HNO3 5 »
93%-ная серная кислота................... 10 »
Тротил растворяют в 4 ч. серной кислоты. Остальные 6 ч. сер-
ной кислоты смешивают с 5 ч. азотной и смесь понемногу приливают
к раствору тротила;
б) наилучшая температура для окисления — около 190°;
в) продолжительность операции — около 40 часов.
После отделения тринитро бензойной кислоты остается отрабо-
танная кислота состава: 84% H2SO4, 2% HNO3, 1% NO2, 13%
Н2О примеси.
1 Desvergnes, Chim. et Ind., 25, 3—17; 291—306, 1931.
2 О разложении симметричной тринитробензойной кислоты в растворе воды
и других растворителей см. Moelwyn-HughesHHinshelwood,
Proc. Roy. Soc., A, 131, 186, 1931.
О взаимодействии несимметричных тринитробензойных кислот с водой
см. стр. 127.
При поверке этого способа на заводе в Сен-Шама (Франция;
произошел сильный взрыв.
При изучении причин взрыва было установлено следующее:
1) 4 ч. 94%-ной серной кислоты недостаточно для растворения
1 ч. тротила при 190°. Для этого необходимо 6 ч. кислоты;
2) при вдувании небольшого количества воздуха при 209—210°
происходит разложение тротила со взрывом;
3) без доступа воздуха удается повысить температуру в аппа-
рате до 230—235°, причем разложения не происходит.
Из сказанного следует, что последний метод непригоден для про-
мышленного применения вследствие его опасности. Пригоден только
метод окисления бихроматом.
Получение тринитробензола из тринитро-
т - к си л о л а. При окислении 2,4,6-тринитро-нг-ксилола хромо-
вой смесью получается тринитроизофталевая кислота:
СН3 СООН
ojn/^no, o2n/\no,
it л it I 4
-----*
^/'СНз ^усоон
no2 no2
температура плавле-
ния 192°
При нагревании выше температуры плавления кислота теряет
2 мол углекислоты и переходит в 1,3,5-тринитробензол.
Нагревание тринитросоединений при такой высокой температуре
небезопасно.
ГЛАВА X
НИТРОПРОИЗВОДНЫЕ КСИЛОЛА
ч»1. Исходные материалы. о-Ксилол как и другие изомеры, бес-
цветная жидкость; температура кипения 141°; температура плавле-
ния 27°, уд. вес 0,863.
т - К си л о л : температура кипения 139°; температура плав-
ления 54°, уд. вес 0,862 (£>’“ = 0,865 по Голлеману).
р-Ксилол: температура кипения 1 136°; температура плав-
ления 15°, уд- вес- 0,861-
Этилбензол: температура кипения 136°; температура
плавления 94°, уд. вес 0,867 (ZT° — 0,867 по Голлеману).
2. Химические свойства изомеров ксилола. Из трех изомерных .
ксилолов тп-ксилол при действии концентрированной серной кис-
лоты легче всего переходит в сульфокислоту. Это различие между
ксилолами стоит в полном соответствии с направляющим влиянием
боковых цепей — при реакции образования сульфокислот. Так как
углеводородные радикалы направляют группу SO3H в орто- или
параположение, то переход нг-ксилола в сульфокислоты
протекает под согласным влиянием обеих метильных групп; в слу-
чае же р- или о-ксилола сульфогруппа должна занять метаположе-
ние по отношению к одной из метильных групп:
СН3 СН3
Z\SO3H /Ч,СН3
\ И \ '
СН3 SO3H
1 По данным Международного бюро физико-химических эталонов в Брюс-
селе, температура кипения р-ксилола 138,40°; уд. вес Dt = 0,86100 (см.
J. Chim. Phys. 23, 733, 1926); Stettbacher, Chem. Ztg., Fortschritt-
berichte, 1—20, 1929.
2 23
Это последнее обстоятельство и является задерживающим факто-
ром при сульфировании о- и р-ксилола и, следовательно, причи-
ной более трудной растворимости этих углеводородов в концентри-
рованной серной кислоте сравнительно с т-ксилолом.
По данным Кижнера и Венделыптейна \ при взбалтывании в те-
чение получаса 8,6 в. ч. р-ксилола с 22 в. ч. серной кислоты (уд.
вес 1,84) растворилось 64% углеводорода; после 2-часового взбал-
тывания в раствор перешло 68% углеводорода. При растворении
наблюдается слабое разогревание смеси. При 100° (при указанном
соотношении реагентов) р-ксилол вполне растворяется в серной кис-
лоте. *
В отличие от р-ксилола т-ксилол (при том же соотношении
с серной кислотой) сполна и с сильным разогреванием растворяется
в серной кислоте.
Слабая азотная кислота (30% HNO3) при 100° окисляет о- и
р-ксилол в соответствующие толуиловые кислоты, остается без из-
менения т-ксилол.
Явление более легкого окисления о- и р-ксилола сравнительно
с тп-ксилолом можно объяснить, по данным М. И. Коновалова 2,
тем, что процесс окисления ароматических углеводородов связан
с нитрованием боковых цепей при действии разбавленной азотной
кислоты (см. стр. 39—40), а большая стойкость т-ксилола по отно-
шению к азотной кислоте зависит от того, что этот углеводород в
боковую цепь нитруется труднее сравнительно с о- и р-ксилолом.
Смесь хромовой и серной кислоты окисляет о-ксилол до СО2 и
Н2О,а т- и р-ксилолы до фталевых кислот.
3. Состав каменноугольного ксилола.
Таблица 39
Наименование вещества По данным НИИ % По данным Централь- ной лаборатории Ко- ксобензола %
да-Ксилол 66 65—70
р-Ксилол 20 12—15
•о-Ксилол ’. . . 10 5—8
•Этилбензол 4 8—10
Парафины • 1-2
1 Ж. Р. Ф. X. О., 62, 1, 1925 .
2 Ж. Р. ф. X. О., 37, 531, 1905 .
224
4. Состав пирогенетического ксилола
Таблица 40
Наименование вещества По данным НИИ °/ /° По данным Справоч- ника 1
т -Ксилол 29-31 30—45
о-Ксилол ЭтиЙбензол . . 8—11 23—27 21—23 20—47 15—25
Бензины 13—15 8—10
5. Технические условия на каменноугольный ксилол (ОСТ 465).
Цвет и прозрачность...................... бесцветен и прозрачен
Уд. вес при 15°.......................... 0,867—0,002
Испаряемость..............•.............. без остатка
Пределы кипения при давлении 760 мм:
Начало перегонки......................... 136,5°
Конец перегонки..........................141,5°
В пределах 4,5° перегоняется............. не менее 95% по объему
Степень очистки: окраска серной кислотой
по шкале Кремера и Шпилькера .... не более 3,0.
н Я
6. Выделение чистого m-ксилола. Добавляют ксилол к купо-
росному маслу. Температура поднимается сама собой до 45°; вы-
держка 2 часа при 50°. Образовавшиеся сульфопроизводные о-
лг-ксилолов* извлекаются водой; отделяются р-ксилол, этилбензол
парафины.
Сульфокислоты 2 m-ксилола разлагаются паром при 130°.
Сульфокислоты о-ксилола разлагаются паром при 160°.
Водный раствор сульфопроизводных ксилола нагревают в аппа-
ратах из ферросилиция; при этом регенерируются углеводороды:
с6н3 (СН3)2 SO3H + Н2 о С6Н 4 (СН3)2 + н2 so4
Ведя это разложение последовательно при 130°, а затем при
160°, получают оба изомера.
1 Справочник по производству взрывчатых веществ, под ред. Лебедева,
стр. 86, 1934.
2 О разделении сульфокислот, основанном на различной растворимости
их кальциевых солей см. Patterson и Sommerville, J. Chem.
Soc., 125, 2488, 1925; о разделении вымораживанием см. Fischer,
Z.t. Elektrochemie, 16, 161, 1910.
Химия—13—15 225
7. Нитропроизводные m-ксилола *.
N0z NOj N02
Кристаллы ромбической Кристаллы триклинической Кристаллы моноклиничес-
системЬ/ системы ной системы
температура плавления Я? температура плавления SO температура плавления tti
При нитровании чистого m-ксилола получается лг-тринитрок-
силол — белое кристаллическое вещество без следов масла с тем-
пературой затвердевания около 181° и с выходом около 200% по
весу т-ксилола.
1 Blanksma, Rec. Trav. Chim. Pays-Bas, 25, 165, 1906; 27, 98, 1909a
226
8. Нитропроизводное р-ксилола *.
вемпе/хмура ма&яешя /3?'
При нитровании р-ксилола получается тринитроксилол — кри-
сталлическое вещество с температурой затвердевания около 137°
с выходом около 150% по весу р-ксилола 1 2.
1 Noelting nGeissmann, Вег., 19, 145, 1886; Lellmann, Lieb.
Ann., 228, 250, 1885.
2 Marqneyrol и Loriette, Mem. Pond., 18, 46, 1921.
227
9. Нитропроизводиые о-ксилола
шелковистые иглЫ
температура
плавления 82*
температура,
плавления 90'
ВелЫе иелЫ
температура
плавления 115'
Прозрачные иелЫ
температура
плавления п-76°
CHS
ОГИ,
N0g
NOg
(есивстнЫе иелЫ
температура плавления-НГ
Сн3
I t0”3
NoJ 'NOg
NOg
Желтые иглЫ (
температура плавления Л
о-Ксилол нитруется значительно труднее, чем т- и р-ксилол.
При его нитровании до тринитро соединения получается масло, ко-
торое только после двухсуточного стояния затвердевает при обык-
новенной температуре (15—18°). Выход тринитропродукта около
130% по весу о-ксилола 1 2.
1 Crossby и Renouf, J. Chem. Soc., 95, 202, 1909.
2Marqueyrol и Loriette, Mem. Poud., 18, 46, 1921.
2 28
10. Нитропроизводиые этилбензола 3.
no2
бесцветнее игМн,
температура п/>овлеш№ 37"
Нитропроизводиые этилбензола довольно легко окисляются в со-
ответствующие нитро бензойные кислоты.
Продукт нитрации этилбензола до тринитросоединения, содер-
жащий кроме тринитроэтилбензола продукты неполного нитрова-
ния, представляет собой маслянистое вещество, не затвердевающее
при охлаждении до —20°.
1 Schultz, Вег., 42, 2523, 1909.
229
11. Общее замечание о продукте тринитрации технического кси-
лола. Нитрованием технического ксилола нельзя получить немасля-
нистого продукта; количество масла зависит от содержания о-кси-
лола, этилбензола в исходном ксилоле и динитроксилолов в про-
дукте нитрации. Так как в техническом ксилоле относительное со-
держание т- и р-ксилолов, дающих при нитрации кристаллические
тринитропродукты, колеблется в значительных пределах, то темпе-
ратура затвердевания тринитропродукта не является достаточным
критерием для оценки качества этого продукта.
Свойства тринитроксилола (ксилила). Тех-
нический тринитроксилол, или ксилил, представляет собой мелко-
кристаллическое вещество белого или слегка желтоватого цвета.
Его гравиметрическая плотность 0,6.
Температура затвердевания продукта, полученного в одну фазу
без обработки его горячей водой на центрифуге, 160—165°. Темпе-
ратура затвердевания очищенного ксилила 170—176°.
Растворимость тринитроксилолов в 100 см3 95°-ного спирта:
Таблица 41
Наименование вещества Количество тринитроксилола в 100 см3 95%-ного спирта, г
при 20° при 15°
2,4,6-Тринитро-тп-ксилол 0,039 0,024
2,4,5-Тринитро-иг-ксилол 1,88 —
4,5,6-Тринитро-тп-кСилол 1,205 —
Тринитро-р-ксилол — 0,325
3,4,6-Тринитро-о-ксилол • — 1,159
3,4,5-Тринитро-о-ксилол — 0,874
Растворимость тринитро-тп-ксилола в спиртобензольных смесях1
Таблица 42 (для определения растворимости взят образец ксилила с температурой затвер- девания 180,5°) (см. табл. 42).
Объемное соотношение спирта и бензола Растворимость ксилила %
спирта бензола при 20° при 8°
0,5 1,0 1,5 2,0 строению фановичу, растворе. 1 1 1 1 тротилато тринитрокс тогда как 0,71 ’0,45 0,29 0,2 в (Мейзенг ;илол не об ТрОТИЛ В Э1 алкоголятов на три- 0,32 нитроксилол образу- 0,24 ются продукты при- — соединения, строение которых аналогично эймер, Стефанович). Но, по Сте- эазует солей в аммиачно-спиртовом "их условиях образует весьма опас-
1 Справочник по производству взрывчатых веществ, под ред. Лебедева,
стр. Из, Госхимтехиздат, 1934-
230
ные при обращении тротилаты аммония. Температура вспышки
ксилила 330°.
Бризантное и фугасное действие ксилила зависят мере от содержания в нем маслянистых примесей, этих примесей кси- лил по бризантному в значительно# Очищенный от Таблица 43
и фугасному дейст- вию немного слабее тротила, как это вид- но из следующей таб- Взрывчатые вещества Обжатие свинцовых столбиков мм Расширение в бомбе Трауцля см3
лицы (табл. 43): В книге Венена 1 #риводятся следую- щие данные для кси- Тротил Ксилил 12,5 12,0 285 270
лила: плотность при
прессовании колеблется от 1,4 до 1,58; скорость детонации для
запрессованного под давлением 320 кг/см2 6250 м[сек; для за-
прессованного под давлением 1280 кг/см2 6600 м/сек. Для смеси
77% аммиачной селитры и 23% сырого ксилила при плотности
1,47 скорость детонации равна 5700 м/сек, а при плотности 1,68
скорость детонации равна 6075 м/сек.
12. Применение ксилила. Ксилил применялся во время империа-
листической войны в России в смеси с тротилом и аммиачной селит-
рой для снаряжения снарядов. Состав смеси: 50% аммиачной се-
литры, 37,5% тротила и 12,5% ксилила. Смесь состава 82% аммиач-
ной селитры и 18% ксилила применялась для снаряжения мин и
ручных гранат.
По ОСТ 40099 допущен для подземных работ аммонит следую-
щего состава: 88% аммиачной селитры и 12% ксилила.
13. Производство ксилила. Как мы видели из работ Мартинсена,
при введении 2-й метильной группы в ядро скорость реакции нитра-
ции значительно увеличивается. Скорость реакции нитрации иг-кси-
лола в 300 раз больше, чем толуола. Поэтому и смеси для нитрова-
ния ксилола могут быть значительно слабее, чем для нитрования
толуола. Этим же обусловлено то, что ксилол можно даже в завод-
ских условиях нитровать в тринитроксилол в одну фазу, в то время
как для толуола нитрование в одну фазу никогда и нигде не про-
изводилось, вследствие того, что это сопряжено с огромным расхо-
дом кислот, а потому неэкономично, не говоря о других трудностях
и невыгодах нитрования толуола в тринитротолуол в одну фазу.
Во время империалистической войны в России производили
ксилил нитрованием ксилола в одну фазу; в это же время был разра-
ботан более экономичный способ Филиппова в две фазы, через моно-
нитроксилол. Этот способ, однако, не был освоен. Позже (1927—
1928 гг.) был разработан способ нитрования в две фазы, но через
1 Венен, Бюрло и Лекорше, Пороха и взрывчатые вещества,
370—371, ОНТИ, 1936.
231
динитроксилол. Французы нитровали ксилол во время войны
1914—1918 гг. в три фазы. Все эти способы ниже кратко описы-
ваются .
Производство ксилила в одну фазу по спо-
собу Солонина. Схема производства. Кислотная
смесь загружается в нитратор обычного типа и к ней приливается
из мерника ксилол. По окончании нитрации содержимое нитратора,
имеющее вид тонкой кашицы, подается через подъемник в вакуум-
воронку, где отсасывается отработанная кислота и производится
предварительная промывка холодной водой. Отсюда ксилил спус-
кается по жолобу в промывной чан для окончательной отмывки
кислот. Промытый ксилил отжимается на центрифуге и направляется
на сушку.
Отработанная кислота поступает из вакуум-воронки в ловушку,
а отсюда в отстойную колонну, где разбавляется водой до 72% H2SO4
для высаживания растворенных нитропродуктов. Оставшаяся отра-
ботанная кислота поступает на денитрацию. Промывные воды из ва-
куум-воронки, из промывного чана и из центрифуги проходят через
ловушку в канализацию.
Описание производства. На Штеровском заводе во
время империалистической войны применялась смесь состава: 79%,
H2SO4, 18% HNO3, 3% Н2О, причем брали 13,5 в. ч. этой смеси
на 1 в.ч. ксилила или 143% HNO3 от теории.
На заводе Кроте в целях экономии серной кислоты применялась
смесь состава: 75% H2SO4, 22% HNO3, 3% Н2О.
. Избыток азотной кислоты брали здесь 45,2% от теории. Прилив
ксилола к смеси и выдержку вели при температуре не выше 35°.
Позже был установлен следующий режим. Начало слива кси-
лола при обыкновенной температуре. По достижении 30—40° ведут
слив основной массы ксилола при этой температуре, а к концу слива
поднимают температуру до 65—70°. В это время содержимое аппа-
рата сильно вспучивается, пенится, и температура поднимается за
счет выделяющейся теплоты реакции до 90°. После прекращения
вспучивания и пенения нагревают содержимое нитратора до 105°
и делают при этой температуре часовую выдержку. Состав отрабо-
танных кислот при смесях Штеровского завода: 81—82,5% H2SO4,
0,8—1,5% Н NO3, 3—5% Н2О, 3—4,5% нитропродуктов.
При уменьшении концентрации серной кислоты (против состава
Штеровского завода) выход ксилила немного уменьшается, а про-
дукт получается более мдслянистый, вследствие большего содержа-
ния низших степеней нитрации. Получался более мелкий кристалл,
плохо фильтрующийся и плохо отстаивающийся.
Большое значение имеет скорость слива ксилола; при очень
быстром сливе получается худший продукт, чем при более мед-
ленном.
По окончании нитрации содержимое нитратора передается на ва-
куум-воронку. Последняя имеет съемную железную освинцованную
решетку, на которую укладывается свинцовая сетка с отверстиями
232
не более 1 мм. Эта сетка собственно и служит фильтром. Спущен-
ный на вакуум-воронку ксилил с отработанной кислотой отстаивается
2—3 часа, затем дается вакуум.
При форме и размерах вакуум-воронки, применявшейся на Ште-
ровском заводе (цилиндрический свинцовый бак высотой 1,4 м, диа-
метром 1,2 м) полная фильтрация шла 7—9 часов, а для ускоре-
ния производства отсасывалось лишь 30% отработанной кислоты.
На фильтре делалась одна-две промывки водой, после чего кси-
лил переводился в промывной чан, где он промывался до кислот-
ности ниже 0,1% по H2SO4, для чего требовалось две холодных и
три горячих промывки. Промытый ксилил отжимался в течение 30 ми-
нут на центрифуге, после чего направлялся с влажностью 13—18%,
в сушку. Температура в камерной сушилке держалась вначале 45—
50°, в конце сушки 80°; продолжительность сушки 24 часа; темпе-
ратура затвердевания полученного ксилила около 163—165°.
Способ Филиппова. Во время империалистической
войны Филипповым был разработан способ получения тринитрокси-
лола в две фазы через мононитроксилол.
Для нитрования ксилола до мононитроксилола можно приме-
нять более слабые нитрационные смеси, чем для нитрования толуола.
Однако так как более слабые кислоты сильнее разъедают нитраторьц
то Филиппов остановился на том же составе смеси, который приме-
няется для мо но нитрации толуола.
Слив смеси к ксилолу ведется при температуре не выше 35—40%
выдержка 2%2 часа, при тех же температурах, в крайнем случае
не выше 45°.
При температуре выше 50° в чугунной или железной аппара-
туре получаются черные отработанные кислоты и нитропродукты.
Нитропродукт в этом случае труднее отделяется от отработанной
кислоты, а отсепарированный мононитроксилол плохо нитруется
в тринитроксилол; это не наблюдалось, если нитрация проводилась
при температуре ниже 50°. Для контроля полноты 1-й нитрации
Филиппов рекомендует перегонять моно нитропродукт с равным
объемом спирта, собирая перегон в воду. При этом спирт раство-
ряется в воде, а ксилол всплывает поверх воды.
Нитрование моно нитроксило л а в тринитроксилол ведется на
смеси, рекомендованной Солонина. Избыток HNO3 21,5% от тео-
рии. Слив смеси к мононитропродукту ведется при температуре
60—65°; после слива делается часовая выдержка при НО—115°.
По окончании приливания кислот непрерывное размешивание
прекращается; ведется периодическое размешивание исключительно
с целью осадить в кислоту массу, вспучившуюся в виде шапки. По
словам автора, продукт получается кристаллическим только при
условии отсутствия непрерывного перемешивания, причем он лучше
пронитровывается, легче отделяется от кислоты и имеет высшую
температуру затвердевания. При непрерывном же размешивании
образовавшийся вначале твердый продукт сбивается в комки, неви-
димому, из-за присутствия плохо нитрующихся продуктов, вслед-
233
*
ствие чего нитрация задерживается и не всегда доходит до
конца.
Получение в две фазы, но через динитроксилол.
Получение динитроксилола. Для динитрации ксилола при-
меняется смесь следующего состава: 65% H2SO4, 20% HNO3,
15% Н2О, соответствующая 76% H2SO4 в отработанной кислоте.
Избыток HNO3 равняется 10% от теории.
Процесс нитрации идет спокойнее, если пользоваться смесями,
содержащими не более 20% HNO3.
Начальная температура нитрации не выше 25°; во время слива
смеси температуру поднимают до 35—40°. После слива смеси вы-
держка при 100° в течение 1г/2 часов.
Если выдержку производить при 80°, то получается динитрокси-
лол с более низкой температурой затвердевания, с большим содержа-
нием жидких нитропроизводных.
Если динитроксилол будет фракционироваться, как ниже опи-
сано, то надо делать выдержку при 100°; если же он поступит непо-
средственно на тринитрование без фракционирования, то достаточна
выдержка при 80°.
Выход динитро ксилола 1,74 по весу ксилола, или 94% от тео-
рии. Температура затвердевания промытого водой динитроксилола
около 32°.
Для дальнейшей нитрации в тринитроксилол полученный ди-
нитроксилол подвергается разделению на твердый и жидкий про-
дукты промывкой водой, нагретой до 20—22°, на центрифуге или ва-
куум-воронке. При этом низкоплавкие примеси (эвтектика) выде-
ляются вместе с водой, и остается динитроксилол с температурой за-
твердевания около 38°. Таким образом отделяется около 20%
масла.
Динитроксилол, получающийся после нитрации и промывки в виде
жидкой массы, очень медленно затвердевает, иногда только после
суточного стояния. Отделенный жидкий динитропродукт содержит
значительное количество динитроэтилбензола, который при после-
дующей нитрации частично переходит в тринитро соединение.
Эти нитропроизводиые этилбензола с продуктами нитрации
о-ксилола являются основной причиной маслянистости ксилила,
поэтому их удаление из динитроксилола обеспечивает получение
немаслянистого ксилила.
Нитрование динитроксилола. Для нитрования динитро-
ксилола применяют смесь' 80% H2SO4, 18% HNO3, 2% Н2О, избы-
ток HNO3 равен 80% от теории.
Слив смеси производится при температуре 50—55°; выдержка
при НО—120°.
Нитрованием двух полученных фракций динитроксилола полу-
чают из высокоплавкого динитроксилола 1,47—1,54 ч. по весу кси-
лола немаслянистого ксилила с температурой затвердевания около
170°; из низкоплавкого динитроксилола — 0,256 ч. по весу кси-
234
лола маслянистого ксилила, с температурой затвердевания 160—164°,
вполне пригодного для изготовления аммонитов.
Получение тринитроксилола в три фазы. Этот метод
применялся во время империалистической войны во Франции.
Для 1-й нитрации берется кислотная смесь, близкая по составу
к применяемой для мононитрации толуола: 56% H2SO4, 28% HNO3,
10% Н2О. HNO3 берется избыток — 3% от теоретического. Процесс
нитрования ведут при температуре не выше 35°. Выход мононитро -
продукта 92% от теории.
Для 2-й нитрации берется смесь: 74% H2SO4, 11% HNO3, 15%
Н2О, отвечающая содержанию 80,5% H2SO4 в отработанной кислоте.
, Слив ведется при 35—50°; выдержка в течение 1 часа при 85°.
Выход динитроксилола 96% от теории, считая на мононитроксилол.
Для 3-й нитрации берется смесь с фактором нитрующей актив-
ности 82,6—84,3%.
Приливание кислотной смеси к динитроксилолу начинают при
60—70°. К концу слива смеси температуру поднимают до 80—85°;
выдержка при 95—100° или 110 — 120°. Выход тринитроксилола
составляет 74% от количества взятого динитро ксило ла.
Полученный по этому способу ксилил после отмывки кислот про-
мывается на холоду 5-кратным количеством спирта. При этом на
100 ч. сырого ксилила получается около 75 ч. очищенного.
14. Очистка ксилила. Маслянистые примеси в ксилиле значи-
тельно уменьшают восприимчивость его к детонации. Во время импе-
риалистической войны при хранении снарядов, снаряженных спла-
вом тротила с ксилилом, из них вытекала маслянистая жидкость,
что приводило к нарушению физической структуры заряда, как это
имело место со снарядами, снаряженными некристаллизованным
тротилом.
Как мы видели выше, во Франции промывали ксилил 5-кратным
количеством спирта, причем в спирте растворялось около 25% по
весу ксилила. В спирте хорошо растворимы маслянистые (низко-
плавкие) примеси и очень мало растворим тринитро-т-ксилол. Боль-
шой расход спирта препятствует широкому применению этого спо-
соба.
Применению предложенного для этого способа получения кси-
лила высокого качества путем фракционированной кристаллизации
динитроксилола мешает длительная кристаллизация динитроксилола
и усложнение технологического процесса.
При промывке ксилила на Центрифуге горячей водой удается
удалить значительное количество маслянистых примесей, причем
заметно поднимается температура затвердевания ксилила (до 174—
176°). Однако при этом все же не получается необходимая степень
очистки.
Были проведены опыты очистки ксилила промывкой водным рас-
твором сульфита натрия. Мюраур 1 нашел, что при действии водного
1 Muraour, Mem. Poud., 18, 1921.
235
раствора сульфита натрия на тринитропроизводные о- и р-ксилола
образуются растворимые в воде сульфосоли.
Лабораторные опыты показали, что при обработке технического
ксилила 2,5—5%-ным раствором сульфита натрия при 60°в течение
3 часов при соотношении веса ксилила к весу раствора 1 : 2 значи-
тельно уменьшается его маслянистость; температура затвердевания
поднимается с 162—163° до 168°. Однако при этом остается еще
некоторая маслянистость, превышающая допустимые нормы.
При увеличении количества сульфитного раствора (отношение
веса ксилила к весу раствора равно 1 : 3) повышается температура
затвердевания ксилила, уменьшается его маслянистость и увеличи-
ваются потери.
При промывке ксилила 7—9%-вым раствором сульфита натрия
при 70° в течение 4 часов получался немаслянистый ксилил с тем-
пературой затвердевания до 174,6°. Однако потери увеличивались
до 15—24%.
Вследствие таких больших потерь, неизбежных при получении
ксилила высокого качества, этот способ не нашел практического при-
менения.
15. Схема технологического процесса для двухфазного способа.
Из описанных способов получения ксилила наиболее выгодным яв-
ляется способ в две нитрации через динитроксилол.
Наименьший же расход кислот имеет место при трехфазном про-
цессе. Однако экономия на кислотах по сравнению с двухфазным мето-
дом не оправдывает расхода на добавочное оборудование. Из двух
способов ведения двухфазного процесса—через моно- или динитро кси-
лол — второй требует меньшего расхода кислот, так как крепкие
кислоты расходуются только на нитрование ди- в тринитроксилол,
тогда как при нитровании нитроксилола в тринитроксилол необхо-
димо дозировать смесь из расчета на введение двух нитрогрупп.
Подобно описанному ранее итальянскому методу получения тро-
тила и при производстве ксилила выгодно применять способ с пол-
ным кислотооборотом.
16. Схема производства ксилила. В отделении динитрации кси-
лол нитруется в динитроксилол на смеси, составленной из кислоты
3-й нитрации и азотной кислоты. Отсюда содержимое аппарата по-
ступает в сепаратор. Отработанная кислота поступает в колонку
для дополнительного отстаивания, а динитропродукт — в аппарат
3-й нитрации. По окончании 3-й нитрации содержимое аппарата
спускается в центрифугу. Отработанная кислота передается в аппа-
рат динитрации ксилола, а ксилил промывается в промывных чанах;
отсюда он переводится на центрифугу, где промывается горячей
водой для улучшения его качества (отмывка низкоплавких приме-
сей), после чего ксилил поступает на сушку.
17. Аппаратура. Нитратор имеет устройство обычных нитрато-
ров для тротила. Удобно оборудовать аппараты с нижней выгруз-
кой во избежание закупорки труб твердым высокоплавким продук-
том, имеющей место при обычных нитрационных аппаратах, в кото-
236
рых реакционная масса выдается засасыванием или под давле-
нием. Сепаратор такой же, как и для динитротолуола.
Центрифуга может быть железная, если на ней не производится
водная промывка, и из нержавеющей стали V4A, если производится
водная промывка. При отжиме от отработанной кислоты в ксилиле
остается 20—30% кислоты, которую затем приходится отмывать
водой.
Рис. 36. Чан для горячей промывки
ксилила.
Промывные таны — деревянные, внутри освинцованИые (рис. 36).
К тану подведена холодная и горячая вода, сжатый воздух для пе-
ремешивания и жолоб, по которому ксилил поступает из центри-
фуги. У дна чана имеется выпускная труба для передачи ксилила
с водой на центрифугу.
Водная центрифуга. По достижении в промывных ча-
нах установленной нормы кислотности (0,1—0,2% H2SO4) ксилил
передается для отжимки на центрифугу. Условия работы центри-
фуги такие же, как раньше указывалось при отжимке тротила от
маточного спирта: малое число оборотов при наполнении (200—
237
t
240 об/мин.) и окончательная отжимка на полном ходу (800 —
900 об/мин.).
Ксилил имеет после отжимки 15—18% воды, с которой направ-
ляют его на сушку.
Сушилки — камерные или туннельные. Сушка
в них производится при температуре 60—70°.
18. Технические условия на ксилил (ОСТ 3660).
Показатели 1-й сорт 2-й сорт
1. Внешний вид.............
2. Цвет.......................
3. Температура затвердевания
не ниже................. ....
4. Влажности и летучих веществ
не более .......................
5. Нерастворимого в бензоле
остатка не более . . . .
6. Кислотность (в пересчете на
H2SO4) не более................
7. ’ Маслянистость...........
Мелкокристаллический порошок, без
механических примесей, видимых на-
глаз.
Белый или слегка желтоватый
163° 160°
0,1% 0,2%
0,2% 0,5%
0Д% 0,1%
Не более эталона
19. Обоснование технических требований к ксилилу. Ограничен
ние содержания влажности вызывается тем, что она понижает вос-
приимчивость к детонации и другие взрывчатые качества ксилила
и изготовленного на нем аммонита. В последнем повышение влаж-
ности увеличивает его слеживаемость и ухудшает его взрывные
свойства.
Повышенное содержание минеральных примесей, особенно
песка, повышает чувствительность ксилила к механическим воздей-
ствиям. При повышенном содержании минеральных кислот в ксилиле
происходит коррозия металлических стенок (например снаряда).
Маслянистые примеси сильно понижают чувствительность ксилила
к детонации и другие его взрывные свойства.
238
ГЛАВА XI
НИТРОПРОИЗВОДИЫЕ СОЛЬВЕНТ-НАФТЫ
I. Исходные материалы. Технические условия на
. /ОСТ 6396 \
сольвент-нафту (нкт^йг)
Показатели Сольвент-нафта очищенная № 1 Сольвент-нафта очищенная № 2
1. Удельный вес при 15° .... 0,87 ± 0,01 0,89 ± 0,02
2. Цвет Бесцветная, допу- Бесцветная или
скается слабое по- желтение слабожелтая, до- пускается потем- нение или покра- снение
3. Начало кипения не ниже . . . Отгоняется не менее 90% при 120° 135°
температуре не выше Степень очистки (окраска с сер- ной кислотой по шкале Кремера— 160° 180°
Шпилькера) не более X 3,0 Не нормируется
2. Состав сольвент-нафты. Сольвент-нафты № 1 и 2 представ-
ляют собой смеси значительного количества углеводородов и имеют
приблизительно следующий состав (табл. 44)
Таблица 44
Наименование вещества Состав сольвент-нафты °/ /о Температура кипения °C
№ 1 № 2
Этилбензол / )с2н5 5 2 136
m-Ксилол \ снГ >сн3 55 25 139
239
Продолж- табл. 44
Наименование вещества Состав сольвент-нафты, % Температура кипения С°
№ 1 № 2
о-Этилтолуол \с2Н5 .... ~сн3 4 - 4 —
у>-Этилтолуол Н3С<^ уС2Н5 . . . сн3 2 5 —
-Мезителен Н3С\ > СН3 сн3 12 25 163
Псевдокумол / сщсн, 7 15 168,5
Другие вещества 16 24 —
Такие смеси дают при нитровании жидкие нитропродукты.
Твердые нитропродукты могут быть получены только при нитрова-
нии узких фракций. При разгонке сольвент-нафты с этой целью
отбирается ксилольная фракция, и отдельно фракция, кипящая
после ксилола, с конечным пределом кипения 170—175°. Послед-
няя содержит преимущественно мезитилен и дает при нитровании
твердый тринитропродукт. Часть сольвент-нафты, кипящая выше
170—175°, содержит кроме мезитилена псевдокумол, нафталин, тет-
раметилбензолы, пропилтолуолы, гидронафталины и другие вещества.
3. Нитрование сольвент-нафты до динитропродукта. Скорость
нитрования мезитилена в 1660 раз больше, чем m-ксилола. Поэтому
нитрация легко происходит на слабых кислотах. Псевдокумол-
в котором две метильные группы стоят в ортоположении друг к другу,
нитруется значительно труднее, и нитрация сопровождается силь-
ными окислительными процессами, причем по мере израсходования
в смеси азотной кислоты эти процессы усиливаются (при сливе угле-
водорода к смеси).
. Мезитилен нитруется до динитромезитилена смесями с факто-
ром нитрующей активности Ф=67 — 69%; при этом заметны окис-
лительные процессы. Но он легко нитруется смесями с фактором
нитрующей активности Ф — 76% H2SO4 при температуре не выше
45° без заметных окислительных процессов, причем получается ди-
нитромезитилен с содержанием до 18% тринитромезитилена (в смеси
•содержалось 25,5% HNO3; слив мезитилена к смеси). При нит-
ровании смесями с фактором активности 76% H2SO4 псевдокумол
дает динитропродукт, но он нитруется значительно труднее. Про-
цесс нитрования псевдокумола сопровождается значительным выде-
240
лением окислов азота, которое особенно усиливается к концу
нитрации.
Все фракции сольвент-нафты, кипящие после ксилола, имею-
щие конечный предел кипения 170—175°, легко нитруются до
динитропродукта нитрационной смесью с фактором активности
76% H2SO4 при температуре нитрации не выше 45°.
Но в то время как нитрование фракций с конечным пределом
кипения 166—168° (содержит преимущественно мезитилен) сопро-
вождается только незначительными окислительными процессами,
при выше кипящих фракциях окислительные процессы усиливаются,
вследствие того что эти фракции состоят преимущественно из псевдо-
кумола и его спутников, которые подвергаются большему окисле-
Йию, чем мезитилен.
4. Нитрование сольвент-нафты до тринитропродукта. Для нитро-
вания сольвент-нафты в тринитросоединение достаточна смесь с фак-
тором активности 91,5% H2SO4. Температура нитрации должна быть
не выше 45°, при более высокой температуре уменьшается выход
и увеличивается содержание маслянистых примесей в продукте.
Реакцию нитрования можно считать законченной спустя 20—30 ми-
нут после введения сольвент-нафты.
5. Состав и характеристика маслянистых примесей тринитросоль-
вент-нафты. В отработанной кислоте остается в растворенном виде
значительное количество маслянистых примесей, выделяющихся
при разбавлении кислоты водой. Полученное масло представляет
собой густую, некристаллизующуюся на холоду темнокрасную
массу, с характерным запахом окислов азота. Этот запах не исче-
зает после многократной промывки горячей водой.
При перегонке этого масла в небольшом количестве при обыкно-
венном давлении наблюдалось следующее: по достижении темпера-
туры 125—130° начиналось выделение окислов азота, причем в при-
емник переходило немного светложелтого масла и вода. Затем масса
саморазогревалась (без нагревания источником тепла) ' с подъемом
температуры до 200—220°. При дальнейшем повышении темпе-
ратуры (вследствие саморазогревания) содержимое колбы взрывалось.
Содержание азота в масле 14,6% (теоретическое содержание
азота в динитромезитилене 13,33%, в тринитромезитилене 16,46%).
Следовательно, масло есть смесь ди- и тринитросоединений.
Масло растворялось с саморазогреванием в 33%-ном растворе
едкого натра. Отсюда следует, что масло состояло из нитрованных
в боковой цепи продуктов.
Вступление нитрогруппы в боковую цепь происходит тем легче'
чем длиннее боковая цепь, чем больше боковых групп и чем меньше
концентрация нитрационной смеси.
Поэтому, как установлено опытами, при нитровании в две фазы
(через динитро) получается значительно больше масла, чем в одну
фазу, и наибольшее количество масла получается при нитровании в
три фазы. Этим, а также окислительными процессами, объясняется и
то, что худший выход тринитросоединений получается при нитрованйи
Хи.:ия—13—16 241
в три фазы, более высокий — в две и наилучший — при нитровании
в одну фазу.
6. Свойства нитросольвент-нафты. При температуре в 40° и выше
нитро сольвент-нафта имеет вид густой жидкости от желтого до
темно коричнево го цвета. При нормальной температуре нитросоль-
вент-нафта представляет собой неоднородную смесь жидкой массы
с кристаллическим веществом.
При содержании азота от 13 до 15% (по техническим условиям)
нитро сольвент-нафта состоит из смеси ди- и тринитросоединений
с небольшим содержанием мононитросоединений Ч
7. Технические условия на нитросольвент-нафту
1. Содержание влажности и летучих веществ . не более 1,5*/,
2. » нерастворимого в бензоле ос-
татка .................................. не более 0,2%
3. Кислотность (при пересчете на серную кис-
лоту) .......................................не более 0,02%
4 Стойкость, по Абелю, при 75°............. не менее 20 мин.
5. Содержание азота ........................ от 13 до 15%
6. Отсутствие механических примесей
8. Применение нитросольвент-нафты. Нитро сольвент-нафта при-
менялась у нас с 1932 г. для
Таблица 46 изготовления нитроглицериновых
тт . Наименование компонентов %
Нитроглицерин .... 8
Нитросольвент-нафта . 5
Коллодионный хлопок . 0.2
Мел . 0.5
Древесная мука .... 3
Аммиачная селитра . . 83,3
взрывчатых веществ с о л ь ве-
ли т о в следующего состава
(табл. 45):
Сольвениты применялись до
1936 г. для промышленных взрыв-
ных работ и допущены к приме-
нению во всех подземных работах
за исключением шахт, опасных
по газу и пыли.
С 1936 г. сольвениты не изго-
товляются и взамен их применяется менее опасный в обращении
зерненый динафталит.
1 О нитропроизводных сольвент-нафты см. Холево, Ж- Пр. Хим., 3, 251—
254, 1930.
ГЛАВА XII
НИТРОПРОИЗВОДНЫЕ НАФТАЛИНА
1. Технические условия на нафталин. Для нитрования приме-
няют кристаллический нафталин 1-го сорта по ОСТ 277, удовлетво-
ряющий следующим техническим условиям:
1. Внешний вид ясно кристаллический.
2. Температура застывания не ниже 79,6°.
3. Реакция с серной кислотой: при нагревании с серной кисло-
той нафталин может давать окраску, по интенсивности и оттенку
не превышающую цвета контрольных растворов, содержащих в 1 л
воды 0,012 г КМпО4 или 0,01 г КМпО4 + 0,15 г К2Сг2О7.
4. Реакция с азотной кислотой: при стоянии над азотной кисло-
той в эксикаторе в течение получаса не должно происходить по.жел-
тения нафталина.
5. Содержание золы — не более 0,05%.
Нафталин, поступающий для нитрования в виде литых кусков,
предварительно измельчается в дезинтеграторе.
2. Продукты нитрации нафталина. При мононитрации нафта-
лина получается почти исключительно а-нитронафталин; ^-нитро-
нафталин получается либо в виде следов, либо совсем не получается.
Температура плавления химически чистого а-нитронафталина 1
56;5°.
Желтый цвет технического мононитронафталина обусловлен при-
сутствием динитронафтола (около 1%).
Мононитронафталин кристаллизуется в виде игол; легко раство-
рим в сероуглероде. В концентрированной серной кислоте он рас-
творяется, образуя раствор с интенсивной темнокрасной окра-
ской.
3. Продукты нитрации мононитронафталина. При нитровании
а-нитронафталина получается смесь изомеров динитронафталина,
среди которых преобладают 1,8- или [З-динитронафталин (60%) и 1,5-
или а-динитронафталин (40%).:
1 Е n z и Pfister, Helv. Chim. Acta, 13, 194, 1930.
243
1,8- или 3-динитронафталин
1,5- или а-динитронафталин
Образованию 1,8-динитронафталина способствует применение кон-
центрированных кислот и низкая температура реакции.
1,8-Динитронафталин кристаллизуется в виде ромбических
призм; температура плавления 175°. Гораздо лучше растворим он
в органических растворителях, чем 1,5-изомер.
Растворимость в пиридине. При кипении раство-
ряется 1 в. ч. 1,8-динитронафталина в 1,5 в. ч. пиридина. На
холоду растворяется 1 в. ч. 1,8-динитронафталина в 10 в. ч.
пиридина.
1,5- Динитронафталин получается в виде шестигран-
ных игольчатых кристаллов слабожелтого цвета; температура плав-
ления 215°; мало растворим в обычных растворителях, лучше —
в бензоле и ледяной уксусной кислоте.
Растворимость в пиридине. При кипении раство-
ряется 1 в. ч. 1,5-динитронафталина в 10 в. ч. пиридина. На
холоду растворяется 1 в. ч. 1,5-динитронафталина в 125 в. ч. пи-
ридина .
4. Разделение изомеров динитронафтазина. Пользуясь различ-
ной растворимостью двух изомеров в пиридине, можно производить
их разделение. Для этого растворяют 1 в. ч. технического дини-
тронафталина в 6 в. ч. горячего технического пиридина. При
охлаждениираствора 1,5д-инитронафталин выпадает почти полностью;
при упаривании фильтрата до х/3 первоначального объема и охлаж-
дении остатка из раствора выпадает 1,8-динитронафталин Ч
5. Взрывчатые свойства. Динитронафталин отличается очень ма-
лой чувствительностью к детонации и по своим взрывным свойствам
близок к динитротолуолу.1 2.
6. Применение динитронафталина. Во время империалистической
войны динитронафталин применялся в значительных количествах
для приготовления так называемой французской смеси: сплава,
состоявшего из 80 в. ч. пикриновой кислоты и 20 в. ч. динитро-
нафталина. Последний служил здесь в качестве флегматизатора и для
1 Фридлендер, Вег., 32, 3521, 1899. О разделении изомеров переври
сталлизацией из ацетона см. Gass шапп, Вег., 29, 1243, 1896.
2 Chem Ind., 26, 133. 1903.
244
увеличения общего количества сильно бризантного взрывчатого ве-
щества. В этой же войне динитронафталин применялся во Франции
и в России в виде смесей с аммиачной селитрой (78% аммиачной се-
литры и 22% динитронафталина или 88% аммиачной селитры и 12%
динитронафталина) для снаряжения некоторых боеприпасов, пре-
имущественно артиллерийских снарядов.
Под названием состава Фавье применялась у нас до 1936 г.
смесь 88% аммиачной селитры и 12% динитронафталина в качестве
антигризутно^о взрывчатого вещества.
Кроме того, динитронафталин применяется в значительных ко-
личествах для изготовления красителей.
7. Продукты нитрации динитронафталина. При нитровании
1,8-Динитронафталина получаются:
1,3,8-тринитронафталин с температурой плавления 218° . . 85,1%
1,4,8-тринитронафталин (идентичен с 1,5,8-тринитронафталином) с тем-
пературой плавления 154° . . -................................9,6% 1
При нитровании 1,5-динитронафталина получается:
1,2,5-тринитронафталин с температурой плавления 112°..........47,5%
1,3,5-тринитронафталин с температурой плавления 122°..........24,0%
1,4,5-триннтронафталин с температурой плавления 154°............ 9% 1
температура
плавления 112°
температура
плавления 122°
температура
плавления 154°
1 Pascal, Bull. Soc. Chim. Fr.. 27 388, 1920.
245
8. Свойства тринитронафталина. Тринитронафталин растворим
в ацетоне, хлороформе и других органических растворителях.
Водный раствор бисульфита натрия растворяет тринитро нафта-
лин на холоду без изменения, а при нагревании переводит в нитро-
аминонафталинсульфокислоты. Сульфит натрия реагирует со всеми
изомерами тринитронафталина, не действуя, однако, на динитрона-
фталин, чем пользуются при количественном их разделении \
9. Свойства и применение технического тринитронафталина. Во
время империалистической войны 1914—1918 гг. тринитронафталин
изготовлялся в Германии с содержанием 14,0—14,2% азота. Он
получался в виде гранулей; температура плавления в пределах
145—155°; температура вспышки 345—355°.
Скорость детонации для указанного продукта найдена в зависи-
мости от условий опыта:
1) в железной трубе с толщиной стенки, равной 2 мм, с диамет-
ром 38 мм; труба снаряжена гранулированным продуктом, грави-
метрическая плотность 0,6—0,7; при испытании применен промежу-
точный детонатор. Скорость детонации найдена равной 2400—
2500 м/сек;
2) для прессованных шашек с плотностью 1,4 без оболочки при
применении тетрилового капсюля-детонатора скорость детонации
определена равной 5650 м/сек;
3) при применении в предыдущем случае промежуточного дето-
натора из прессованной пикриновой кислоты скорость детонации
равна 5400—6000 м/сек.
Эти большие скорости детонации для слабонитрованного про-
дукта (нетрудно видеть, что применявшийся в Германии продукт был
по своему составу ближе к динитронафталину, теоретическое содер-
1 Теплоты горения и образования некоторых нитропроизводных нафта-
лина (по В adosche, Bull. Soc. Chim. Fr. [5] 4, 3, 559, 1937:
Таблица 46
Наименование вещества Молекулярные теплоты горения ккал при; Теплоты образования ккал
постоянном объеме ПОСТОЯННОМ давлении
Нафталин 1230,33 1229,17 —11,57
1-Нитронафталин 1189,95 1190,09 — 6,67
1,5-Динитронафталин 1153,48 1152,61 — 3,36
1,8-Динитронафталин 1154,74 1153,87 — 4,62
1,3,8-Трииитронафталин 1119,75 1117,86 — 2,79
1,4,5-Трииитронвфталин 1122,72 1120,83 — 5,76
Из этой таблицы видно, что при превращении 1,5-динитронафталина в 1,4,5-
тринитронафталин происходит не выделение, а поглощение теплоты в количестве,
равном 2,4 ккал. Во всех остальных случаях замена атома водорода на нитро-
группу сопровождается выделением тепла, т. е. является реакцией экзотерми-
ческой. При этом количество выделяемого тепла уменьшается по мере пере-
хода ко все более и более замещенным молекулам.
246
жание азота в котором равно 12,9%, чем к тринитронафталину,
в котором теоретическое содержание азота равно 15,98%) неожи-
данны, так как при испытании в бомбе Трауцля он дает расшире-
ние всего лишь 140—150 сл3, что свидетельствует о неполноте взрыва
в последнем случае.
При испытании взрыванием в отдельных ящиках в количестве
от 15 до 25 кг было установлено, что при применении сильных про-
межуточных зарядов тринитронафталин указанного качества регу-
лярно детонирует Ч
По своим взрывчатым свойствам (чувствительности к детонации,
фугасному действию и бризантности) динитро нафталин сходен с ди-
нитротолуолом, тринитронафталин — с динитробензолом и тетра-
* нитронафталин — с тринитротолуолом.3.
Тринитронафталин применялся в Германии во время империали-
стической войны в виде сплава, состоявшего из 66% тротила и 33%
тринитронафталина, под названием три-тринала для снаряжения
снарядов, а чистый продукт — в прессованном виде с сильным
детонатором из пикриновой кислоты для снаряжения мелкокалибер-
ных снарядов.
Во Франции он применяется для изготовления антигризутных
взрывчатых веществ — гризунафталитов.
10. Побочные реакции при нитрации нафталина. Нитрация нафта-
лина сопровождается частичным окислением его с образованием
нитробензойной и нитрофталевой кислот. Незначительное количество
нафталина окисляется в нафтол, который превращается в динитро-
нафтол при мононитрации нафталина.
Кроме того, образуются буроватые вещества, растворимые в ще-
лочах и щелочных углекислых солях; эти вещества очень нестойки;
они разлагаются при нагревании, вспучиваясь и воспламеняясь3.
11. Тетранитронафталин. При нитровании 1,8-динитронафта-
лина получается преимущественно 1,3,6,8-тетранитронафталин
, или p-изомер; температура плавления 200°.
N02
При нитровании 1,5-динитронафталина получаются преимуще-
ственно следующие два изомера:
1,2,5,8-тетранитронафта лин
no2 no2
'\/\no2
, или 8-изомер; при
нагревании он разлагается ниже температуры плавления.___________________
1 Jahresber. chem.-techn. Reichsanstalt, VIII, 119, 1930.
2 Chem. Ind., 26, 133, 1903.
3 Паскаль, Взрывчатые вещества, пороха и боевые газы, стр. 116, Гос1
химтехиздат, 1932.
247
1,3,5,8-тетранитронафталин
NO, NO2
Z\/\
, или 7-изомер;
тем-
NO2
пература плавления 194—195°.
Тетранитронафталин мало чувствителен к удару и взрывает
только при действии сильного детонатора *. По своим взрывным
свойствам, а также по наличию сырьевой базы его применение для
снаряжения снарядов представляло бы известный интерес. Однако
большой расход кислот при изготовлении и значительное содержа-
ние побочных продуктов в техническом тетранитронафталине пре-
пятствуют его практическому использованию.
12. Действие серной кислоты на полинитронафталины. При
действии крепкой серной кислоты на динитронафталин последний
темнеет. По опытам Горста и Хлебникова окраска динитронафталина
мало изменяется в присутствии серной кислоты с содержанием
50% мол. H2SO4 (84,5% вес. H2SO4); в этом случае даже при дли-
тельном стоянии очень слабо изменяется окраска динитронафта-
лина. В присутствии серной кислоты с содержанием 60% мол. H2SO4
(89% вес. H2SO4) динитронафталин потемнел уже через сутки. Еще
сильней и быстрей темнел динитронафталин в присутствии еще более
концентрированной серной кислоты.
Вероятно при взаимодействии серной кислоты с динитронафтали-
ном (то же имеет место с другими полинитронафталинами) идут
некоторые из реакций, протекающих при получении нафтазарина
из динитронафталина. Нафтазарин образуется при нагревании
1,5-или 1,8-динитронафталина с олеумом в присутствии серы. При
этом происходит химически сложная, окончательно не изученная
реакция, вероятно через изонитрозо соединения нитронафто хинона:
NO2 О HOHN О
ОН О
I II
нафтазарин
‘Will, Chem. Ind., 33, 1903.
248
По другим воззрениям промежуточным продуктом является ди-
гидроксиламинное производное нафталина:
ОН О
Эти реакции протекают очень медленно с серной кислотойг
но значительно быстрее в присутствии азотной. Это видно и»
того, что при взаимодействии тринитронафталина с серноазотной
смесью с высоким фактором нитрующей активности и взятой с неболь-
щим избытком азотной кислоты (от теории) получается продукт
с меньшим содержанием азота, чем в исходном тринитронафталине.
13. Производство мононитронафталина. Нитрование ведется
в обычных чугунных нитраторах. Фактор нитрующей активности
нитрационной смеси равен 61% H,SO4.
В аппарат заливается в качестве среды отработанная кислота
от мононитрования, содержащая 61% H2SO4 и 0,5% HN03 в ко-
личестве 1,5 в. ч. на 1 ч. нафталина. К отработанной кислоте по-
дается измельченный нафталин. К перемешиваемым нафталину и
отработанной кислоте подается нитрационная смесь. Если нитро-
нафталин является конечным фабрикатом, то нитруют смесью, изго-
товленной из свежих кислот, например состава: 45% H2SO4, 37%
HN03, 18% Н20 (фактор нитрующей активности равен 61% H2SO4).
При производстве мононитронафталина готовят смесь на отрабо-
танной кислоте от 2-й нитрации, причем получают смесь с содержа-
нием около 10% HNO3. Она (смесь) берется из расчета 102% HNOa
от теории.
Слив смеси ведется при температуре не выше 50°; выдержка
1 час при 60°. Выход мононитронафталина 97—98% от теории.
Температура затвердевания полученного продукта не ниже 50°.
14. Производство динитронафталина. Нитрование ведется на
смеси с фактором активности около 74% H2SO4 (например: 66%
H2SO4 и 15% HNO3). Смесь берется для нитрования из расчета
140% HNO3 от теории.
249-
Слив жидкого монопродукта производится при температуре
20—40°.
Выдержка 1 час при 50°.
Содержимое нитратора спускается на центрифугу, сделанную из
V4A, где отжимается отработанная кислота и производится предва-
рительная промывка небольшим количеством воды. После этого
.динитронафталин передается в промывные чаны для окончательной
отмывки кислот.
Отработанная кислота от 2-й нитрации содержит около 2,5%
HNO3, поэтому в отжатом от кислот динитронафталине остается
заметное количество азотной кислоты. С целью ее использования
на французских заводах 1 отделенный от отработанной кислоты ди-
нитронафталин промывают отработанной кислотой от 1-й нитрации
и используют промывную кислоту для приготовления нитрационной
«меси для 1-й нитрации.
15. Технические условия на динитронафталин (ОСТ 2940).
Показатели 1-й сорт 2-й сорт 3-й сорт
1. Внешний вид Динитронафталин должен быть со- вершенно однородным, порошкообраз-
2. Температура затвердевания ным или гра жать видимь примесей нулированныл' lx для глаза и не содер- посторонних
не ниже ИЗ. Влажность и летучие веще- 150° 150° 150°
ства не более 4. Нерастворимых веществ в 0,5% 0,5% 0,5%
смеси ацетона и ксилола не более 0,2% 0,3% 0,5%
5. Золы не более В том числе: 0.2% 0,2% 0,3%
Кремнезема SiO2 не более .... 0,05% 0.05% 0,05%
Свинца не более Не допускается Следы 0,03%
Железа 6. Кислотность (по Н28О4) не Следы Следы Не определено
«олее 0,1% 0,1% 0,2%
Примечания. 1. Определение кремнезема не производится, если про-
цент зольности не выше 0,05.
2. Количественное определение свинца в 3-м сорте динитронафталина не
производится, если зольность не превышает 0,03%.
'Паскаль, Взрывчатые вещества, пороха, боевые газы, Госхимтехиэ-
дат, стр. 119, 1932.
-250
16. Производство тринитронафталина. Трини т р о н а ф т а-
л и н получается во Франции нитрованием мононитро нафталина,
причем применяют смесь состава г: 55% H2SO4, 40% HNO3, 5% Н2О.
Фактор активности 77% H2SO4.
Этой смеси берут 470 кг на 100 кг мононитронафталина (256%HNO3
от теории) и добавляют к ней 800 кг отработанной кислоты от три-
нитронафталина. В нитратор подается 1000 кг отработанной кис-
лоты, к которой одновременно вливают при нитрации расплавленный
мононитронафталин и кислотную смесь.
Слив ведется при температуре около 55°; при этой же температуре
делается выдержка 2 часа.
Выход 120% по весу мононитронафталина.
17. Получение тетранитронафталина. Одной из наиболее выгод-
ных из числа опубликованных в химической литературе смесей для
получения тетранитронафталина является рекомендованная Пата-
ром смесь состава: 64,5% H2SO4, 29% HNO3 и 6.5% Н2О.
Этой смеси берется 100 в. ч. на 7 в. ч. динитронафталина, т. е.
почти 15 в. ч. смеси на 1 в. ч. динитронафталина. Уже из этих
данных ясно, что этот способ не может иметь промышленного
значения.
При получении тетранитронафталина образуются в наибольшем
количестве побочные продукты, в том числе нестойкие, разлагаю-
щиеся при нагревании; вследствие их присутствия получающийся
при нитровании тетранитронафталин обладает низкой химической
стойкостью.
1 В е н е н, Бюрло иЛекорше, Пороха и взрывчатые вещества,
стр. 376, ОНТИ, 1936.
ГЛАВА XIII
НИТРОПРОИЗВОДНЫЕ ФЕНОЛОВ И ИХ ЭФИРОВ
ФЕНОЛ
Рис. 37. Диаграмма раствори-
мости воды в феноле и фенола
в воде.
1. Свойства фенола. Химически чистый фенол кристаллизуется
в бесцветных ромбических иглах; температура плавления, по Бе-
галю, 42,5—43°, температура кипения, по Кальбауыу 1 * (Kahlbaum),
181,5°; уд. вес, по Коппу (Корр), при
32,9° 1,0597; уд. вес по Андриенцу
(Andrienz), при 40° 1,05433; уд. вес,
по Андриенцу, при 50° 1,04663.
Растворимость воды в феноле и фе-
нола в воде растет вначале почти про-
порционально температуре, и, начиная
с 68,3°, фенол и вода смешиваются во
всех отношениях (критическая темпе-
ратура растворения). Критический ра-
. створ содержит в этих условиях 33%
фенола. Взаимная растворимость фе-
нола и воды изображена на рис. 37.
При длительном соприкосновении с
воздухом фенол, окисляясь, принимает
красноватую окраску, а поглощая вла-
гу из воздуха, —расплывается. Легко
растворяется в растворе едкого кали
или натра с образованием фенолятов:
СвН5ОН + NaOH ->C6H5ONa Ц- Н2О
но не растворяется в растворе соды.
Фенол разъедает кожу и вызывает язвы. Принятый внутрь, дей-
ствует, как сильный яд. Противоядием служит известковый сахарат,
и если предполагают, что фенол проник в кишечник, то дают серно-
кислый натрий (глауберова соль).
Для количественного определения фенола приливают к водному
раствору, содержащему около 0,1 г фенола, 100 см31Н раствора
1 Разные авторы определили температуру кипения фенола в пределах от
178,9° до 184,1°.
252
брома в едком натре и 5 см3 крепкой соляной кислоты. Смесь взбал-
тывают и через 15 минут определяют избыток брома добавлением
Ю см3 раствора йодистого калия и титрованием 10%-ным гипосуль-
фитом.
2. Технические условия на синтетический фенол (ОСТ 2933).
Показатели 1-й сорт 2-й сорт
1. Внешний вид Бесцветный Бесцветный или
кристаллический слабо розовый
сплав кристаллический
сплав
2- Температура затвердевания
не ниже . 39° 36°
3. Нелетучих веществ не более 0,1% 0,1%
4. Растворимость в воде после
прибавления едкого натра Полная Полная
5. Растворимость в дестиллиро-
ванной воде в отношении 1:5. . . Полная без мути Полная без мути
Для производства пикриновой кислоты допускается только 1-й сорт
фенола. При нитровании фенола 2-го сорта кислота получается пони-
женного качества.
3. Технические условия на кристаллический каменноугольный
фенол (ОСТ 3296)
Показатели Нормы
1. Цвет и внешний вид . 2. Температура затвердевания не ниже 3. Температура затвердевания йосле просушки не ниже 4. Растворимость в воде 5. Остаток после выпаривания не более 6. Реакция Бесцветная розовая или красноватая кристаллическая масса 37° 40.5° Раствор фенола в 15 в. ч. воды при 15° должен быть прозрачным1 0,1% Нейтральная
Упаковка: железные оцинкованные барабаны емкостью 5, 10, 25, 100, 150
и 300 кг.
Каменноугольный фенол пригоден для производства пикрино-
вой кислоты; однако синтетический фенол чище (содержит меньше
крезолов или вовсе их не содержит). Поэтому следует предпочесть
синтетический фенол, который дает лучшего качества пикриновую
кислоту, причем расход азотной кислоты при его нитровании меньше.
1 Указание на отсутствие крезола.
253
t
ПРОДУКТЫ НИТРАЦИИ ФЕНОЛА, ИХ СВОЙСТВА
И ПРИМЕНЕНИЕ
1. Мононитрофенолы. При нитровании фенола азотной кислотой
получается смесь о- и р-нитрофенолов. Они могут быть разделены
при помощи перегонки с водяным паром, так как с парами воды
перегоняется лишь о-изомер. ,
тп-Н итрофенол, не образующийся при прямом нитрова-
нии фенола, получают из m-нитроанилина через диазосоединение;
он не окрашен, без запаха, не перегоняется с водяным паром; тем-
пература его плавления 96°. тп-Нитрофенол не имеет практического
значения.
о-Н итрофенол окрашен в желтый цвет; температура плав-
ления его равна 45°; он обладает резким запахом; применяется в ка-
честве исходного материала для получения о-аминофенола, о-нитро-
анизола, о-анизидина, о-дианизидина
H2n/ \ / >nh2
ОСН3 осн3
и сернистых красителей.
р-Нитрофенол имеет температуру плавления 114°. Соединение
это бесцветно, но раствор его в щелочи окрашен в желтый цвет.
Применяется для получения фенетидина (фенетидинами называются
этиловые эфиры аминофенолов):
C6H4(NH2)(OC2H5)
о- и р-нитрофенолы могут быть получены нитрованием фенола; при
этом образуется тем больше р-изомера, чем ниже температура
2. Строение окрашенных нитрофенолов. Уже давно было отме-
чено, что о- и р-нитрофенолы так сильно отличаются по своим свой-
ствам, что это отличие не могло быть объяснено одним лишь относи-
тельным расположением заместителей в молекуле, а именно, орто-
и параположением нитрогруппы относительно гидроксильной группы.
В самом деле: о-нитрофенол летуч с водяными парами, окра-
шен в интенсивный желтый цвет, имеет характерный, напоминаю-
щий нитробензол, запах; р-нитрофенол — без цвета, запаха и не
перегоняется с водяными парами. Армстронг 1 2 впервые указал, что
такие отличия не могут быть отнесены к простой изомерии положе-
ния и что следует о-нитрофенолу придать хиноидную формулу:
О
‘ 'I О
n/
| хон
т. е. хинон-о-нитроксима.
1 Гольдштейн, Ж. Р.Ф. Х..О., 10, 352, 1878; Pictet, С. R., 116,
817, 1893.
2 Armstrong, Proc. Chem. Soc., 101, 1892.
В подтверждение этого взгляда Армстронг указал, что метиловые
эфиры обоих изомеров не окрашены и в одинаковой степени летучи
с парами воды, т. е. в этом случае изомерия обусловлена только
различием в относительном положении заместителей (изомерия по-
ложения), но нет изомерии, обуслбвленной различием структуры
ядра. Окраска металлических производных р-нитрофенола объяс-
няется изменением структуры ядра при образовании соли (образова-
ние хиноидной формы).
Однако Армстронг не привел доказательств правильности своего-
мнения о хиноидной природе окрашенных нитро фенолятов. Эти дока-
зательства были даны позже Ганчем, который показал, что многие
бесцветные или почти бесцветные соединения, образующие интен-
сивно окрашенные соли, являются псевдо кислотами.
Применяя эти взгляды к нитрофенолам, Ганч 1 пришел к выводу,
/ОН
что истинные нитрофенолы С6Н4<^ бесцветны, а ацинитрофе-
XNO2
нолы СвН4< окрашены. Правильность этой точки зрения:
-NO ОН
была доказана не только физико-химическими методами, но чисто
химически, а именно, открытием двух рядов эфиров нитрофенЬлов:
истинного нитрофенола и ацинитрофенола:
/ОСПН2П 4
О6Н4ч
xno2
эфир истинного нитро-
фенола бесцветный
сн^°
^NOOCnH2n + 1
эфир ациформы нитрофе-
нола окрашенный
При замене алкильного остатка СпН2п+1 на водород получаются
соединения, окрашенные соответственно так же, как и их эфиры.
Таутомерную форму можно определить по окраске. Бесцветные,
или имеющие только слабый желтый оттенок, нитрофенолы являются
в твердом состоянии (почти) полностью истинными нитросоедине-
ниями. Другие нитрофенолы, например, о-нитрофенол, более окра-
шены, но никогда столь интенсивно, как их соли. Это, по Ганчу,
твердые растворы малых количеств окрашенных ацинитрофенолов
в больших количествах истинных нитрофенолов, между которыми:
существует состояние подвижного равновесия:
с,н4
С6Н
XNOOH
Состояние этого равновесия так же, как и для равновесия в жид-
кости, зависит от температуры: при повышении температуры окраска
делается интенсивней, следовательно, равновесие перемещается
в сторону лабильной, при обыкновенной температуре — хиноидной
формы; при охлаждении происходит обратное.
1 Hantzsch, Вег., 39, 1084, 1906
255
Очень часто наблюдается, что при увеличении числа нитро групп
® ядре окраска (вопреки первоначальным представлениям хромо-
сферной теории) не делается более глубокой, а наоборот, становится
светлей. Например, при введении в желтый о-нитрофенол 2-й нитро-
группы получается бесцветный динитрофенол. По мнению Ганча,
влияние вступления новой нитрогруппы на окраску можно объяс-
нить тем, что эта нитрогруппа, как и всякий новый заместитель,
несколько изменяет свойства молекулы и, в частности, может влиять
ла тенденцию к перегруппировке
,NO2 .NOOH
Аг^----------> Аг^
хон ХО
как в смысле усиления ее, так и ослабления. Согласно хромофорной
теории хромофор одним своим присутствием сообщает бесцветной
молекуле окраску; но это действие хромофора проявляется или уси-
ливается при посредстве ауксохромной, например, гидроксильной
группы. Тот факт, что соли нитрофенолов всегда окрашены много
интенсивнее, чем свободные соединения, объяснялся по хромофорной
теории тем, что ауксохромная природа гидроксильной группы про-
является только путем солеобразования.
Согласно изложенной теории перегруппировки Ганча, развитой
и доказанной им на примере многих псевдокислот, специфический
способ действия, присущий ауксохромам в содержащих хромофор
производных бензола, объясняется возникновением благодаря им
хиноидных форм. Например, появление интенсивной окраски при
введении ауксохромов (ОН или NH2) в нитробензол объясняется
образованием не самых продуктов замещения, но возникающих при
введении этих заместителей (путем перегруппировки) веществ с хино-
идным строением.
По теории перегруппировки в солях нитрофенолов благодаря
положительному щелочному металлу почти полностью восстанов-
лена ациформа, соответственно стремлению металла связаться
с отрицательно заряженным местом молекулы. Таким образом равно-
весие у «водородных» (т. е. таких, в которых соответствующий во-
дородный атом не замещен металлом) соединений смещено почти
полностью в сторону фенольной формы х:
.ОН ZO
Аг <-----------AtQ
xNO2 xNOOH
свободные нитрофенолы
ONa ,0
А т» ' _ у Д т» Д'
XN02 ^NOONa
соли нитрофенолов
1 Возражения против теории Ганча в пользу хромофорной теории см. К а п £•
m а п п, Вег., 433, 237, 1923; возражения против статьи Кауфман а—
см. Hantzsch, Lieb. Ann., 436, 321, 1924.
256
3. Окраска солей нитрофенолов *. При изучении окрашенных
эфиров и солей нитрофенолов было отмечено, что, в то время как
эфиры окрашены в красный цвет, щелочные соли соответствующего
нитрофенола большей частью желтые. Это привело к предположению
о существовании двух изомерных солей нитрофенола: одной желтой
и другой красной, так как красные эфиры должны отвечать красным
щелочным солям.
Ганч объясняет существование двух рядов окрашенных солей
пространственной изомерией, подобно тому как различная окраска
двух рядов диазосульфонатов объясняется стереоизомерией:
Аг • N Аг • N
11 II
MeO3S • N N • SO3Me
синсоль» темнее . антисоль светлее
окрашена окрашена
Для установления стереоформул, построенных аналогично
диазосульфонатам, Ганч отходит от обычной дикетонной фор-
мулы хинона и производит их (стереоформулы) от перекисной
О
формы: I
XNOOMe
В соответствии с этим Ганч формулирует строение производных
нитрофенола следующим образом:
.OR
6hZ
бесцветное соединение
(эфир)
С6Н4 - о
NO2Me
красная соль
С6Н4 - о
MeO2N
желтая соль
С
В пользу такого пространственного расположения в красной и
желтой соли говорит следующее:
1) можно допустить, что красные соли соответствуют более темным
синсолям 1 2 диазосульфонатов, а желтые, более светлые,—антисолям
этих сульфонатов;
2) красные эфиры нитрофенолов, построенные аналогично крас-
ным солям, очень легко превращаются самопроизвольно в бесцвет-
ный истинный эфир. Это говорит о том, что метильная группа в крас-
ных эфирах, а следовательно, и металл в красных солях, находятся
по соседству к первоначальному, ставшему хиноидным при солеобра-
зовании, фенольному кислородному атому3:
1 Hantzsch, Вег., 40, 330, 1907.
2 В настоящем случае син- и антиположения определяются положением
относительно кислородного атома, а не арила.
3 Дальнейшее развитие взглядов Гаича по вопросу о зависимости между
цветом и строением Нитросоединений см. Hantzsch, Lieb. Ann., 492, 65,
1931; X. Р. Ж-, 4, 7. 453, 1932.
Химия—13—17
Ar-----О Ar — О Ar — OCH3
\ /<ОМе----------> \/ ОСН3-----------> \
NO NOZ NO2
нрасьая соль красный эфир бесцветный эфир
4. Причина окраски. Теория хромофоров как в ее первоначаль-
ном виде, так и в видоизменении Ганча констатирует наблюденную
связь между определенным строением молекулы и окраской, но она
не объясняет, почему бензоидные и хиноидные формы нитропроиз-
водных фенола (и вообще замещенных бензола) неодинаково абсор-
бируют различно окрашенные составные части солнечного света.
Объяснение это может быть дано на основе современных физи-
ческих представлений о взаимодействии между световыми лучами и
валентными электронами.
Согласно этим представлениям все световые волны являются но-
сителями энергии; при прохождении через кристалл световые лучи
рассеиваются в разных направлениях. Когда период волны равен
свободным периодам электронов, то эти электроны приводятся в силь-
ную вибрацию и поглощают энергию. Таким образом те волны, ко-
торые имеют периоды, одинаковые с периодами электронов, погло-
щаются, и происходит абсорбция света. Другие световые волны, не
находящиеся в периоде с электронами, не поглощаются, но частично
отражаются с поверхности кристалла, обусловливая определенную
его окраску. Поглощение света по описанной схеме производится
валентными электронами и зависит от состояния этих электронов
(энергетический уровень электронов, прочность связи электронов).
Электроны, вызывающие поглощение световых волн, должны
находиться уже на сравнительно высоком энергетическом уровне
(слабо связанные, «разрыхленные» электроны).
Из спектров поглощения неорганических атомных ионов известно,
что слабо связанные электроны, поглощение которых сильно сме-
щено в длинноволновую область спектра,появляются у всех тех ионов,
у которых электронные оболочки не имеют законченной конфигура-
ции благородных газов. Сюда относятся в первую очередь ионы пе-
реходных элементов, как, например, Fe, Fe, Си, Си, Ru и ионы
редких земель. Наоборот, ионы с заполненными оболочками благо-
родного газа, как, например, К, Na, Mg, Са, F', Cl', поглощают,
только начиная с границы кварцевой ультрафиолетовой области;
наибольшая прозрачность наблюдается у самих благородных газов.
Среди органических молекул ненарушенную конфигурацию бла-
городного газа в смысле октетной теории Льюиса в первую очередь
н н н
имеют насыщенные углеводороды, например, пропан Н : С : С : С : Н;
Н Н Н
эти углеводороды поглотают свет в наиболее коротковолновой об-
ласти.
268
Нарушение такой симметричной октетной конфигурации, иными
словами, появление слабо связанных электронов, может происходит
как вследствие недостачи электронов, например, у свободных ради-
Н : : : Н
калов R : С : Я, или появления двойных связей, С С , так
R Н: : : Н
и вследствие соединения между собой атомов, имеющих различное
Н
сродство к электрону, например, Н : С : J: (при перетягивании
Н
электронов к атому с большим сродством к электрону симмет-
рий нарушается, что обнаруживается также и по появлению диполь-
ного момента). Особенно сильное смещение мы имеем тогда, когда
обе последние причины действуют совместно, например, при появлё-
нии хромофорных групп.
Таким образом появление хромофорных групп связано с нару-
шением конфигурации благородного газа, при этом появляются ла-
бильные (слабо связанные) электроны, поглощающие энергию волн
определенного периода и тем обусловливающие появление опреде-
ленной окраски вещества.
Относительная степень ослабления связи (разрыхления) таких
лабильных электронов не является постоянной, но зависит от различ-
ных влияний (введение хромофора в молекулу и др.). По имеющимся
наблюдениям при повышении лабильности электронов вещество по-
глощает более длинные волны. Эта теория вместе с хромофорной
позволяет дать следующее более вероятное объяснение окраски пи-
кратов аммония.
Пикрат аммония состоит из двух хромоизомеров: бензоид бесцве-
тен, а хиноид имеет окраску от желтого до красного. Бензоидный
изомер либо имеет мало лабильных электронов, либо вовсе их не имеет,
поэтому он не абсорбирует свет,т.е. бесцветен. Хиноидный изомер
имеет лабильные электроны, свободный период которых совпадает
с периодом коротких световых волн; более длинные волны поэтому
не абсорбируются, но отражаются. Степень разрыхления лабильных
электронов в пикрате аммония зависит до некоторой степени от ряда
влияний: температуры, щелочности, концентрации; соответственно
изменяется окраска. В зависимости от этих влияний получаются
желтые, оранжевые, или красные кристаллы, а отдельные кристаллы
могут быть внутри красными, а снаружи желтыми и наоборот, что на
самом деле наблюдается при получении кристаллов.
5. Свойства 2,4" динитрофено ла» 2,4-динитрофенол окрашен в жел-
товатый цвет. Температура его затвердевания 112,5° (по Деверню),
удельный вес при 24° 1,683; перегоняется с водяным паром. Раство-
римость в воде г: 1. ч. динитрофенола в 197 ч. воды при 18°;
1 ч. динитрофенола в 21 ч. воды при 100°.
Бром реагирует с ним с образованием 2,4-динитро-6-бромфенола
1 Grunner, J. Prakt. Chemie, 1, 102—122; 2, 225—228.
259
С иодом он образует 2,4-динитро-6-иодофенол. При нагревании
его до 175° с крепким раствором аммиака образуется 2,4-динитро-
анилин. При введении по каплям раствора цианистого калия в на-
гретый до 60° раствор 2,4-динитрофенола выделяется аммиак, и по
охлаждении осаждается порошкообразная кристаллическая соль,
которая является калиевой солью яг-пурпуровой кислоты:
o,n/\nhoh
xZ0H
Это темнокрасное вещество с металлическим блеском; растворяется
в воде и спирте с весьма интенсивным багрово-красным окрашива-
нием. Применяется для изготовления сернистых красок и 2,4-диамино-
фенола (проявителя).
6. Свойства 2,6-динитрофенола. Температура его затвердевания
61,52° (по Деверню). Он перегоняется с парами воды, но в мень-
шей степени, чем 2,4-динитрофенол.
Растворимость его в 100 ч. воды следующая: при 15° 0,0315;
при 50° 0,5121; при 100° 1,22.
7. 2,4-Динитрофенолят натрия. Подобно другим динитрофеноля-
там щелочей динитрофенолят натрия образуется, очень легко при
действии едких или углекислых щелочей на динитрофенол. 2,4-Дини-
трофенолят натрия представляет собой светложелтые шелковистые
иглы. Растворимость в воде: при 85° 27,4%, при 23° 4,4%.
8. Токсические свойства динитрофенола. Случаи отравления на
производстве динитрофенолом приписывались вначале содержа-
щимся в нем примесям. Однако позже было доказано, что все эти
примеси значительно менее ядовиты, чем самый 2,4-динитро-
фенол.
Он действует отравляюще независимо от того, каким путем про-
ник в животный организм: введением ли в вену, или под кожу, че-
рез дыхательные и пищеварительные пути, или же при смазывании
кожи.
Отравляющая его доза равна около 0,01 г на 1 кг животного
организма. Динитрофенол, как и динитро хлор бен зол, усиливает
процессы физиологического окисления и вызывает сильную лихо-
радку, сопровождающуюся иногда смертью.
Хроническое отравление динитрофенолом сопровождается увели-
ченным обменом веществ и вызывает иногда чрезмерное выделение
мочи, богатой азотистыми и фосфорнокислыми веществами; хрони-
ческое отравление динитрофенолом может вызвать повреждение пе-
чени и почек. На степень отравляющего действия динитрофенола
{как и многих других твердых нитросоединений) оказывают косвен-
ное влияние летучие примеси. Эти примеси, повидимому, способ-
260
ствуют улетучиванию динитрофенола и тем самым увеличивают со-
держание ядовитых веществ в окружающей атмосфере.
Констатирована возможность привыкания к динитрофенолу (Де-
вернь).
9. 3,5-Динитрофенол, бесцветные кристаллы с температурой плав-
ления 122°. Практического применения не имеет. Получается, по
Лобри де-Брюину по схеме:
O,N
NO2
ОСН3
он
| CH3ONa
;no2 O2nI4/'nO2
при омылении
* O^/'NO,
^Нагревают 1,3,5-тринитробензол с раствором едкого натра
в метиловом спирте. Образуется метиловый эфир 3,5-динитрофенола.
Для омыления этого эфира Лобри де-Брюин нагревал его при тем-
пературе 180° с концентрированной соляной кислотой. Однако при
таком способе омыления получается пониженный выход.
Количественный выход 3,5-динитрофенола получается, по Ганчу2,
при нагревании эфира при 130° с концентрированной серной кис-
лотой.
10. Свойства пикриновой кислоты. Физические свой-
ства. Пикриновая кислота получается в обычных производствен-
ных условиях в виде пластинчатых кристаллов желтого или светло-
желтого цвета. Из крепкой соляной кислоты выкристаллизовывается
почти бесцветной.
Она окрашивает шелк й шерсть в желтый цвет. 1 в. ч. пикрино-
вой кислоты окрашивает 100 000 в. ч. воды в отчетливо видимый
желтый цвет. Температура ее плавления 3 122,5°; температура за-
твердевания 3 121,9°; уд. вес пикриновой кислоты 1,813 (Rudorf);
уд. вес плавленой пикриновой кислоты при 124° 1,589; гравиметри-
ческая плотность кристаллической пикриновой кислоты 0,9—1,0.
Плотность, получающаяся при прессовании (по Дотришу):
•Давление в ат 275 685 1375 2000 2750 3435 4125
Плотность 1,315 1,480 1,615 1,672 1,714 1,731 1,740
Однако в производстве в целях безопасности не переходят давле-
ния 2000 кг, что отвечает, по Дотришу, плотности 1,672 (в произ-
водственных условиях получается 1,63). Скрытая теплота плавле-
ния пикриновой кислоты 20,4 ккал/кг.
Химические свойства. Кислотные функции фенола
увеличиваются от вступления в него нитро групп, причем тем больше,
чем больше нитрогрупп в него входит. В то время как фенол не дей-
ствует на лакмус, пикриновая кислота окрашивают его в красной
цвет.
1 L ob г у d е Bruyn, Rec. Trav. Chim. Pays-Bas, 9, 209, 1890.
2 Hantzsch, Ber., 40, 341, 1907 .
’Munroe и Howell, Z. Schiess- u. Sprengst., 18, 87, 1923.
261
*
Пикриновая кис-
Таблица 47
Растворимость пикриновой кислоты в воде
(по Финдлею)
лота разлагает кар-
бонаты, образуя пик-
Темпера- тура °C Раствори- мость в 100 г воды г Темпера- тура °C Раствори- мость в 100 г воды г раты, в то время как СО2 разлагает феноляты, образуя карбонаты.
0 ! 1,05 5 : 1,07 10 ' 1,10 15 1,16 20 1.22 25 1,37 30 1,55 Растворимость пикрине кислоте разне 40 i 1,98 50 2.53 60 ; 3.17 70 1 3,89 80 \ 4.66 90 5.49 100 1 6.33 Таблица 48 »вой кислоты в серной >й концентрации1 Сухая пикрино- вая кислота не дей- ствует на металлы, но в присутствии вла- ги довольно энергич- но вступает с ними в реакцию, образуя пикраты. По ско- рости их взаимодей- ствия с пикриновой кислотой металлы можно расположить
Содержание ТТ ап Содержание пикриновой кислоты в 100 г раствора, г в ряд: Zn, Fe, Си, Al, Sn; из них пер-
% при 18° при 50° при 80° вые три реагируют довольно быстро, алюминий медлен-
0 2,3 4.7 10.0 18,0 . 25.5 50,5 69,7 87.9 97,4 100,0 Растворимост! в 1,184 0.230 0,142 0.091 0.079 0,092 0.429 0.928 2.461 7,532 10.180 Т а б л и пикриново! спирте 2,389 0,692 0,368 0.265 0,214 0,230 0.645 1.424 5.826 12,785 16,230 ц а 48а кислоты нее, а олово не ре- 1 940 агирует. Легче, чем с ме- 0J27 таллами, пикриновая 0,561 кислота реагирует с 0,587 иХ окислами, гидра- 1,104 г 2 2оз тами окислов и со- 7,’бЮ лями; она разлагает 24,020 в присутствии влаги 25,860 некоторые нитраты и хлораты. Подобно другим тринитро соединениям пикриновая кислота образует с ароматически- ми и особенно хорошо с поли- ядерными углеводородами трудно растворимые и хорошо кристал-
Температура °C Растворимость в 100 з спирта 0
20 78 реакция ирг g 23 единения. С нафталином образова- fir9„ ние молекулярного соединения ’ С10Н8 C6H2(NO2)3OH идет коли- чественно, вследствие чего эта [меняется для количественного определения нафта-
1 Techn. Rec., Synthetic phenol and picric acid, London, 1921.
262
лина. Многие орга- нические основания образуют с пикрино- вой кислотой краси- вые труднораствори- мые пикраты. Например, акри- Таблица 486 Растворимость пикриновой кислоты в бензоле (по Финдлею)
Темпера- тура °C Раствори- мость в 100 г бензола г Темпера- тура °C Раствори- мость в 100 г бензола г
криновой КИСЛОТОЙ g акридинпикрат, пло- ю хо растворимый в во- 15 де, бензоле и спир- ТС- Эта реакция при- меняется для количественного oi В аналитической практике пр 3,70 5,37 7,29 9.56 12,66 аределения и работе с 35 45 55 65 75 пикриново пикриново 21.38 33.57 50,67 71,31 96,7 й кислоты. й кислотой
играет большую роль другое органическое основание — нитрон, с
которым пикриновая кислота образует нерастворимое в воде соеди-
нение.
Нитрон образуется из трифениламиногуанидина при нагревании
его с муравьиной кислотой:
H.C6HN—N
I
н - с< + h5c6hn. I
он >с
h5c6hn/
трифениламиногуанидин
С6Н5 - N—------------N
/NC6H3 |
СН( > с
хж6н/
нитрон
Нитрон образует желтые листочки; плавится при 189°. Ацетат
его легко растворим в воде; нитрат и пикрат практически нерас-
творимы. На этом основано применение нитрона для количественного
определения нитратного и пикратного ионов; реакция обычно ве-
дется в уксуснокислом растворе.
. 11. Взрывчатые свойства пикриновой кислоты. Чувствитель-
ность к удару пикриновой кислоты — около 32% взрывов при
весе груза в 10 кг и высоте падения 25 см (при тех же условиях
тротил дает 4—8% взрывов). Чувствительность к удару пикрино-
вой кислоты является предельной для взрывчатых веществ, при-
меняемых для снаряжения артиллерийских снарядов. Во время
империалистической войны в России и Франции чистой пикриновой
кислотой снаряжали только 15-мм (соответственно 3-дюймовые)
артиллерийские снаряды, а снаряды более крупного калибра сна-
ряжались флегматизированным составом (с динитронафталином или
динитрофенолом), значительно менее чувствительным к удару.
Примесь кремнезема значительно увеличивает чувствительность
пикриновой кислоты к удару. Ниже приводятся результаты испыта-
ния на копре при весе груза в 10 кг и высоте его падения
25 см.
263
♦
Таблица 49 Температура ее
Содержание кремнезе- Число Полные вспышки 300—310°.
мистого песка в пикри- До 100 кг она
новой кислоте % испытаний ' о/ /о сгорает без взрыва на воздухе. Но при
0 25 25 сгорании больших
0.25 25 60 количеств часто на-
0.5 25 100 блюдались взрывы.
При быстром на-
гревании до 300° в замкнутой оболочке взрывает.
При выжигании в снаряженных снарядах, уложенных в шта-
бели, пикриновая кислота сгорает без взрыва, если диаметр открытого
отверстия в снаряде не меньше диаметра разрывного заряда. В про-
тивном случае горение пикриновой кислоты большей частью сопро-
вождается детонацией выжигаемого штабеля1. Например, при вы-
жигании штабелей из 76- или 45-лмг английских мелинитовых снаря-
дов, имеющих узкое очко, диаметр которого значительно меньше
диаметра разрывного заряда, количество детонаций составляет 75—
80% от всего количества выжигаемых штабелей. Штабели из анало-
гичных тротиловых или амматоловых снарядов, как правило, выго-
рают без взрыва.
Восприимчивость к детонации зависит от давления, при котором
запрессован заряд, и соответственно от плотности: при давлении
1500 кг/см2 и плотности 1,58 для взрыва необходим капсюль-де-
тонатор с 0,4 г гремучей ртути; при давлении 2900 кг/см2 и плот-
ности 1,68 для взрыва необходим капсюль-детонатор с 0,68 г гре-
мучей ртути; для литой пикриновой кислоты необходим капсюль-
детонатор с 3 г гремучей ртути. Расширение в бомбе Трауцля 305 лш;
бризантность, по Гессу, 16 мм.
Скорость детонации в детонирующем шнуре с оловянной оболоч-
кой около 7000 м/сек.
По Дотришу, скорость детонации при плотности 1,74 и сильном
капсюле около 7645 м/сек.
Для порошкообразной пикриновой кислоты, спрессованной в за-
рядах до плотности 1,5,— до 5000 м/сек.
12. Физиологическое действие пикриновой кислоты. Пикрино-
вая кислота имеет горький вкус; она представляет собой слабый яд.
При попадании в дыхательные пути она их сильно разъедает.
При продолжительном действии поражает легкие и почки; вы-
зывает различные кожные болезни: экземы, чирья, нарывы и т. п.
Пикриновая кислота имеет неприятную особенность — окраши-
вать белки глаз в желтый цвет, если попадает в организм даже
в очень маленьких количествах. Кроме того, она очень интенсивно
окрашивает кожу человека в желтый цвет, причем это окрашивание
очень стойкое и исчезает только через большой промежуток времени.
1 А. Г. Горст, Несчастные случаи при разрядке огнеприпасов в 1925 г.
Разрядка и уничтожение огнеприпасов, стр. 97, АУ РККА, 1927.
264
13. Применение пикриновой кислоты. До империалистической
войны пикриновая кислота заливалась в снаряды в значительных
количествах в чистом виде. Однако это представляет ряд неудобств.
Чувствительность пикриновой кислоты велика для ныне принятых
начальных скоростей снарядов при выстреле, и ее необходимо по-
нижать добавлением флегматизаторов. Заливку чистой пикриновой
кислоты приходится вести при температуре около 130° для получе-
ния хорошего и плотного заряда; но при этой температуре процесс
становится более опасным, так как вместе с возрастанием темпера-
туры увеличивается чувствительность этого вещества к механи-
ческим воздействиям. Наконец, выделяющиеся из нагретой расплав-
ленной массы пары очень вредны для здоровья. Поэтому едва ли
чистая пикриновая кислота применяется ныне для заливки снаря-
дов. Обыкновенно ее применяют в смеси с флегматизирующими и
понижающими температуру плавления ароматическими нитросоеди-
нениями, реже в смеси с парафином или вазелином.
В Англии применялась под названием лиддита смесь следующего
состава: 87% пикриновой кислоты, 10% динитробензола, 3% вазе-
лина или 90% пикриновой кислоты и 10% вазелина.
Во Франции до и во время империалистической войны приме-
нялся для снаряжения снарядов крезилит 60/40, т. е. содержав-
ший 60% пикриновой кислоты и 40% тринитрокрезола (температура
плавления около 85°). Однако после введения способа Брежа для
улавливания паров летучих растворителей (особенно в пороховом
производстве) крезол почти целиком расходовался на установках
Брежа. В связи с этим взамен крезилита была введена новая смесь.
DD-60/40, содержавшая 60% пикриновой кислоты и 40% динитро-
фенола (температура плавления около 85°). Однако приготовление
этой смеси потребовало особых приемов вследствие большой ядови-
тости динитрофенола: необходимо было вести мешку так, чтобы устра-
нялось попадание пыли смешиваемых веществ в помещение мешки.
Кроме того, во Франции применялись следующие смеси:
MDN: 80% пикриновой кислоты и 20% динитронафталина (тем-
пература плавления 100—110°).
МР: 88% пикриновой кислоты, 12%, парафина.
Смесь с мононитронафталином: 90% пикриновой кислоты, 10%
мононитронафталина.
В России чистая пикриновая кислота применялась для сна-
ряжения 76-лгм снарядов. Для снаряжения снарядов среднего
калибра (122- и 152-л«л«) она применялась исключительно в виде
флегматизированного состава, состоявшего из 80% пикриновой кис-
лоты и 20% динитронафталина. Этот состав был заимствован из Фран-
ции, вследствие чего применялся у нас под названием «французской
смеси». Он применялся также для снаряжения авиабомб. В сравни-
тельно меньшем количестве применялся для снаряжения авиабомб
состав из 90% пикриновой кислоты и 10% мононитронафталина.
14. Свойства пикратов. Чувствительность к удару..
Наиболее чувствительны соли тяжелых металлов, несмотря на тог
265
что они содержат кристаллизационную воду (даже высушенные
при 100°).
Наиболее чувствителен к удару пикрат свинца; за ним в порядке
убывающей чувствительности следуют Fe, Си, Ag, щелочноземель-
ные металлы, щелочные металлы1.
Чувствительность пикратов к удару в значительной мере зависит
от содержания кристаллизационной воды, как это отчетливо видно
из следующей таблицы 2:
Таблица 50
Чувствительность к удару и температура взрыва солей пикриновой кислоты и
динитрофенола
Взрывчатые вещества Кристаллиза- ционная вода Температура сушки ~ • и CTt ео ЗБ тг о Температура вспышки | °C Примечание
инимальная высот; щения груза в 2 к вызывающая по гера: ней мере один взрг :у из Г см
или вспышг испытаний, ।
ё
1 2 3 4 5 6
Г ремучая ртуть . Тетрил Тротил Пикриновая ки- слота Пикрат аммония. Пикрат натрия . Динитрофенолят натрия .... Пикрат меди .. . Пикрат цинка . . Пикрат алюминия Пикрат закисного железа .... Пикрат окисного железа .... Безводный Н2О Безводный н2о Безводный ЗН2О Безводный 6Н2О Безводный 10Н2О 2Н2О 8Н2О Высушен над серной кисло- той в вакуум- эксикаторе Н2О О* О ОО (О о 5» СП I I III О О О О1 О U' СОСОСДОСДСО О О 1 1 III оооо О О ООООООООО оо 5,1 20,3 35,6 36,6 43,2 43,2 38,1 40,6 .38,1 48,3 30,5 86,4 30,5 91,4 40,6 40,6 91,4 35,6 91,4 20,3 17.8 15 2 210 260 470 320 320 360 370 300 310 360 310 295 Температура вспы- шки определялась, как температура, необходипая для того, чтобы вы- звать в течение 5 секунд вспышку или взрыв несколь- ких млг взрывча- того вешества, по- мешенного в тон- костенный медный капсюль, погру- женный на опреде- ленную глубину в баню из расплав- ленного металла. Опыты произво- дились через каж- дые 5°
1 Kast, Z. Schiess- и Sprengst., 6, 7, 31, 1911.
2 Hopper, J. Franklin Institute, 225, 219, 1938.
266
Из этой таблицы видно, что более гидратированные модифика-
ции менее чувствительны к удару, чем менее гидратированные; без-
водные модификации наиболее чувствительны.
Часть гидратной воды удаляется при нагревании до 80—100°,
но для удаления всей кристаллизационной воды и получения, таким
образом, наиболее чувствительных форм требуется длительное на-
гревание при более высокой температуре, а именно при 130—150°.
В свою очередь безводные модификации быстро поглощают влагу
из воздуха и вследствие этого становятся менее чувствительными.
Пикраты натрия, меди, цинка, алюминия, закисного и окисного
железа, содержащие кристаллизационную воду, полученные кри-
сталлизацией из воды с последующей сушкой, менее чувстви-
тельны к удару, чем основные военные взрывчатые вещества—
тротил, пикриновая Кислота и тетрил. Чувствительность к удару
пикрата аммония меньше тротила.
Более чувствительны, чем пикриновая кислота, обезвоженные
при высокой температуре пикраты меди, цинка и окисного железа;
наоборот, безводные пикраты натрия и алюминия и динитрофенолят
натрия менее чувствительны, чем пикриновая кислота.
Пикрат закисного железа обладает чувствительностью такого же
порядка, как и пикриновая кислота.
Пикрат окисного железа, высушенный при 100°, по своей чув-
ствительности несколько превосходит перекристаллизованный тетрил.
Следовательно, как общее правило, чувствительные безводные
модификации получаются лишь путем предварительного нагревания
пикратов при 100° и выше.
Для сравнения чувствительности к удару наиболее опасного пи-
крата свинца рассмотрим следующие данные.
Таблица 51
Наименование вещества Взрыв с высоты (см) при весе груза
2 кг 10 кг
Нитроглицерин 15 3
Гремучий студень 10 4
Пироксилин 10 4
Пикрат свинца, безводный 10 8
Отсюда следует, что наиболее чувствительный безводный пикрат
свинца обладает чувствительностью к удару, равной таковой же
пироксилина и гремучего студня. Для практической оценки этого по-
рядка чувствительности отметим, что сухой пироксилин применяется
только при производстве динамита, причем предписаны строгие
правила предосторожности при обращении с ним. Особенно опасна
сухая пироксилиновая пыль. Гремучий студень — самое опасное в
обращении и применении промышленное взрывчатое вещество —
267
*
давно не применяется у нас вследствие большого числа несчастных
случаев, имевших место при обращении с ним и при его применении.
Температуры вспышки некоторых пикра-
тов, по данным Гоппера, приведены в табл. 50; в следующей таб-
лице даны температуры вспышки, определенные другими исследо-
вателями:
Таблица 52
Температура вспышки пикратов
Наименование вещества Температура вспышки
по Солонина °C По Касту °C
Пикрат калия 335 335—339
» натрия — 316—321
» бария 340 335—340
» кальция — 331—335
» закиси железа 300 256—261
» цинка . 315 340—342
» меди 310 279—280
» свинца 260 285—287
Температуры вспышки пикратов, приведенные в табл.’50, ко-
леблются в пределах от 295° для пикрата окисного железа, до 360°
для пикратов натрия и алюминия. При параллельных опытах тем-
пература вспышки пикриновой кислоты определена равной 320°, а
тетрила 260°.
Такого же порядка значения температуры вспышки для пикратов
щелочноземельных металлов и свинца находим в табл. 52. При
этом температура вспышки наиболее чувствительного к нагреванию
пикрата свинца не ниже, чем тетрила.
Чувствительность к трению. Заметную чувстви-
тельность обнаруживают при испытании на трение в фарфоровой
ступке пикраты свинца, калия и безводного пикрата бария; за ними
следуют пикраты кальция и серебра. Остальные пикраты ведут себя
при этом испытании индиферентно.
15. Опасности, обусловленные присутствием пикратов. Присут-
ствие пикратов в пикриновой кислоте служило причиной взрыва.
Неоднократно бывали объяснявшиеся присутствием пикратов
взрывы пикриновой кислоты во время пожаров. Взрыв происходил
при падении в горячую расплавленную массу пикриновой кислоты
частей обрушивавшегося здания. В этом случае высокая чувстви-
тельность пикратов (чаще железа или свинца) обусловлена, во-пер-
вых, обезвоживанием при высокой температуре сплава и, во-вторых,
еще более резким повышением чувствительности при той же высокой
температуре.
Кроме того, пикраты способны при воспламенении или нагрева-
нии до высокой температуры взрываться, тогда как чистая пикри-
268
новая кислота легко сгорает. Эта особенность свойственна уже
смесям пикриновой кислоты с окислами металлов.
В статистике несчастных случаев иногда без достаточного осно-
вания приводятся и такие, в которых заранее исключается при-
сутствие больших количеств пикратов. Здесь допускали, что неболь-
шие количества пикратов, мелко распределенные в массе пикрино-
вой кислоты, действовали как детонатор. Однако это допущение
мало вероятно. Во-первых, инициирующее действие даже самого
сильного пикрата — пикрата свинца — далеко уступает действию
инициирующих взрывчатых веществ, например, гремучей ртути;
во-вторых, инициирующее действие пикратов можно допустить
только тогда, когда они сосредоточены в отдельных местах и нахо-
дятся в соприкосновении с главной массой пикриновой кислоты.
Там, где эти условия не имели места, трудно объяснить возникно-
вение взрыва действием пикратов.
В производственных мастерских, не по правилам построенных,
где не соблюдаются необходимые правила чистоты, пикраты могут
образоваться и собраться на больших поверхностях в тонком слое,
например, на полу, на стенах и трубопроводах. В этом случае соз-
дается определенная опасность, поскольку эти соли легко воспла-
меняются от удара и трения и лучше распространяют огонь в тон-
ком слое, чем маленькие количества сравнительно трудно воспламе-
няющейся пикриновой кислоты. В действительности это свойство
пикратов, особенно пикрата железа, во многих случаях было при-
чиной пожара.
Пикраты могут образоваться при взаимодействии пикриновой
кислоты с щелочноземельными основаниями и солями, содержа-
щимися в цементных полах мастерских.
Во время империалистической войны, когда в России в мастерских
пикриновых цехов полы делались цементные, бывали случаи вспышки
таких пикратов при хождении и при ремонте полов.
Опасность образования пикратов устраняется в производстве
(а также в снаряжательных мастерских) тщательным соблюдением
чистоты, соответствующей окраской стен и труб, применением ас-
фальтовых полов, возможным исключением металлов из строитель-
ных конструкций.
В практике возникает иногда вопрос о допустимости перевозки
пикриновой кислоты с содержанием незначительных количеств
пикрата железа или свинца. Очевидно, нужны специальные условия,
в частности, резкое местное повышение концентрации пикратов,
для того чтобы можно было говорить об опасности, создаваемой
лрисутствием пикратов для перевоз'ки или хранения. Для обычных же
случаев, встречающихся на практике, страх перед пикратами можно
считать в значительной степени преувеличенным.
16. Получение пикратов. Пикраты щелочных и щелочноземель-
ных металлов получаются легко при взаимодействии пикриновой
кислоты с углекислыми солями этих металлов, по уравнению:
2C6H2(NO,)3OH + Ме2СО3 -> 2C6H2(NO2)3OMe + Н2О + СО2
269
Таблица 53
Растворимость пикрата калия в воде
Температура °C Растворимость в 100 г воды г
0 0,0029
15 0,0049
100 7,14
Пикраты железа, ртути, цинка, свинца’
меди, алюминия и никеля легко получаются при дей-
ствии сернокислой соли металла на водный раствор пикрата бария:
[C6H2(NO2)3O]2Ba + MeSO4 -> [C6H2(NO2)3O]Me + BaSO4
Пикрат калия получается добавлением поташа к горя-
чему раствору пикриновой кислоты. Нагревание производится до
прекращения выделения угольной
кислоты.
По охлаждении раствора вы-
кристаллизовывается пикрат ка-
лия в виде длинных золотисто-
желтых призм с металлическим
блеском. При осторожном нагрева-
нии пикрат калия окрашивается в
красно-оранжевый цвет, но при
охлаждении к нему возвращается
первоначальный цвет.
Пикрат аммония полу-
чается насыщением водного рас-
твора пикриновой кислоты аммиаком. В зависимости от условий
получения пикрата аммония получается либо желтая, либо крас-
ная модификация, причем1:
1) красный пикрат аммония получается при избыточном содер-
жании аммиака при повышенной температуре и при перекристал-
лизации желтого пикрата аммония из водного раствора аммиака;
2) при перекристаллизации красного пикрата аммония из вод-
ного раствора пикриновой кислоты получается желтый пикрат;
3) при рассмотрении красных кристаллов пикрата аммония под
микроскопом заметно, что только наружная часть кристаллов окра-
шена в красный цвет, внутри же кристаллы имеют желтый цвет.
По американскому способу пикрат аммония получается при рас-
творении 1 в. ч. пикриновой кислоты в 10—12 в. ч. воды и прибав-
лении аммиака после нагревания раствора до полной нейтрализации.
П рименение пикрата аммония. Вследствие малой чув-
ствительности пикрата аммония к удару и трению он применяется
в США с 1900 г. для снаряжения бронебойных снарядов. Пикрат
аммония не плавится при нагревании, а взрывается при темпера-
туре 300—320°.
17. Возможность образования пикратов в производственных
условиях. Несмотря на то, что в кислотах, в которых получается
в производстве пикриновая кислота, и с которыми она соприкасается
до момента промывки, содержатся соли металлов, например, соли
свинца и железа, тем не менее здесь пикраты не образуются, так
как серная кислота вытесняет пикриновую кислоту из ее металли-
ческих солей, а не наоборот.
1 Hale, Army Ordnance, 6, 39—41, 1925 .
270
Пикраты в пикриновой кислоте образуются в производстве при
взаимодействии ее с солями, содержащимися в воде, оставшейся
в пикриновой кислоте после фуговки (или отсасывания на вакуум-
воронке). Скорость взаимодействия увеличивается при просушке
пикриновой кислоты при 50—60°. Легче получаются соединения
Са и Fe, так как они находятся в воде в виде основных солей
(они реагируют с пикриновой кислотой легче, чем средние соли).
18. Тетранитрофенол. Тетранитрофенол впервые был получен
Нитцки и Буркхардом 1 обработкой дихиноилтриоксима азотной
кислотой. Дихиноилтриоксим, существующий в двух таутомерных
формах, получается при взаимодействии динитрорезорцина с гидро-
ксиламином:
ОН
Z^NO,
, | " + NH,OH
k/0H
no2
О N - ОН
II II
/X|=NOH-------> |/4|=0
1= NOH <----NOH
II 'I
NOH NOH
При взаимодействии 1 ч. дихиноилтриоксима с 4 ч. азотной
кислоты (с содержанием 50% HNO3) образуется тетранитрофенол..
Наиболее вероятная его формула:
ОН
2
VN°2
no.
Тетранитрофенол легко растворяется в воде и из нее очень-
трудно кристаллизуется. Температура плавления его 130°; темпе-
ратура вспышки 130°.
Бланксма 2 получил тетранптрофенол прямым нитрованием,
щ-нитрофенола. Он обработал 5 г ///.-нитрофенола смесью 12 г азот-
ной кислоты уд. веса 1,52 (99% HNO3) и 15 г крепкой серной
кислоты с выдержкой при 50° в течение 15 минут.
Получено вещество, представляющее собой 1,2,3,4,6-тетрани-
трофенол,
OH
'NO,
NO,
1 Burkhard, Вег, 30, 184, 1897.
2 Blanksma, Rec. Trav. Chim. Pays-Bas, 21, 256, 1902 .
271
Однако это вещество обладает иными свойствами, чем получен-
ное Нитцки и Буркхардом. Оно имеет температуру плавления 140°
и при этой температуре спокойно переходит в жидкость, а при даль-
нейшем нагревании медленно разлагается с выделением газов.
Тетранитрофенол — вещество нестойкое. Нитрогруппа, нахо-
дящаяся в положении 3, легко подвижна и очень склонна к заме-
щению; при действии воды она замещается гидроксильной группой,
образуя тринитрорезорцин; с метилатом и этилатом натрия заме-
щается группами ОСН3 и ОС2Н5. При обработке спиртовым рас-
твором аммиака нитрогруппа замещается аминогруппой.
Стойкость тетранитрофенола значительно повышается при его
тщательной очистке. Для такого очищенного продукта температура
взрыва определена1 равной 245—251°.
19. Пентанитрофенол получен Бланксма нитрованием 3,5-дини-
трофенола 2. 2 г динитрофенола обрабатывались при охлаждении
смесью, составленной из 6 г азотной кислоты уд. веса 1,52 (99%
HNO3) и 8 г купоросного масла.
Пентанитрофенол плавится с разложением при 190°.
Обе нитрогруппы, стоящие в метаположении к гидроксильной
группе, являются нестойкими и способны легко замещаться; напри-
мер, при взаимодействии с водой или со спиртовым раствором ам-
миака.
Тетра- и пентанитрофенолы не могут иметь практического при-
менения вследствие их нестойкости.
ХИМИЯ ПРОЦЕССА СУЛЬФАЦИИ ФЕНОЛА
И НИТРАЦИИ (И НИТРОЛИЗА) ФЕНОЛСУЛЬФОКИСЛОТ
1. г.-Сульфирования. При замещении водородного атома в аро-
матическом ядре (бензоле, толуоле и др..) гидроксилом получаются
вещества, которые не только легче нитруются, чем углеводород, но
и легче окисляются и осмоляются.
При введении в молекулу фенола сильно электроотрицательных
групп NO2, SO3H повышается сопротивляемость молекулы окисле-
нию, причем эта сопротивляемость растет с возрастанием числа
электроотрицательных групп. Поэтому с целью получения про-
дукта с хорошим выходом и высокого качества вводят в фенол одну
или несколько сульфогрупп сульфированием и нитруют полученный
сульфопродукт. Процесс .сульфирования согласно уравнению реак-
ции:
R • Н + H2SO4-> R SO3H + Н2О
сопровождается выделением реакционной воды, разбавляющей сер-
ную кислоту. При наличии избытка сульфируемого вещества по
мере течения реакции достигается такая концентрация серной кис-
1 Rec. Trav. Chim. Pays-Bas, 39, 162, 1920.
2 Rec. Trav. Chim. Pays-Bas, 21, 201, 1902.
лоты, при которой сульфирование при данной температуре прекра-
щается.
Этот предел концентрации серной кислоты, выраженный в про-
центах SO3, при котором сульфирование данного вещества при дан-
ной температуре прекращается (строго говоря, при этой концентра-
ции скорость сульфирования настолько мала, что практически
мы можем считать ее равной нулю), называется it данного сульфиро-
вания.
Величина it-сульфирования зависит от природы сульфируемого
углеводорода, от имеющихся в нем заместителей и температуры
сульфирования.
Так, например, моно сульфирование бензола при 100° останавли-
вается, когда содержание SO3 в серной кислоте достигает 66,4%,
т. е. it-сульфирования бензола равен 66,4.
к для моносульфирования нафталина имеет следующие значе-
ния: при 55—60° около 56% SO3; при 160° около 52%.
к для дисульфирования нафталина: при 10° около 82% SO3;
при 80—90° около 80,8%; при 160° около 66,5%.
it для трисульфирования нафталина: при 160° около 79,8% SO3.
Нитробензол и нитротолуол имеют it для моносульфирования,
близкий к 82% SO3 (олеум с 2% свободного SO3).
Следовательно, сильно электроотрицательные заместители (NO2,
SO3H) затрудняют вступление сульфогруппы в ядро; соответственно
с этим повышается значение it для сульфирования.
Зная значение it для данного сульфирования, можно легко
определить количество серной кислоты, необходимое для сульфиро-
вания1.
Если обозначим через X количество рерной кислоты с содержа-
нием а°/0 SO3, необходимое для сульфирования 1 моля органиче-
ского вещества, и примем во внимание, что на сульфирование 1 моля
органического вещества будет израсходован 1 моль (равный 80), то
можем написать:
-у- Л
100
= 80 + (Х-80)^.
Отсюда:
v _ 80 (100 — те)
(а—те) -
Чем выше начальная концентрация серной кислоты а, тем больше
знаменатель этой дроби и, следовательно, тем меньшее количество
серной кислоты потребуется для сульфирования.
При применении для сульфирования серного ангидрида по при-
веденному уравнению X = 80, т. е. для сульфирования требуется
теоретическое количество серного ангидрида.
1 Court ot и Bonnet, С. R., 182, 855, 1926.
Химия—13—18
273
Для любого количества Р кг сульфируемого соединения с моле-
кулярным весом М количество потребной для сульфирования кис-
лоты X будет равно:
у _ 80(100 —л) Р
~ (а—л)Л/~’
2. Влияние относительных количеств реагирующих компонентов.
Подобно установленному Кесслером влиянию относительных коли-
честв нитруемого вещества и азотной кислоты на количество образо-
вавшегося нитропродукта при сульфировании увеличение коли-
чества сульфируемого вещества также тормозит реакцию; напротив,
с увеличением количества серной кислоты увеличивается коли-
чество образовавшегося сульфосоединения. Константа скорости ре-
акции сульфации тем выше, чем выше концентрация серной кис-
лоты (разумеется, при прочих равных условиях). Следовательно,
скорость реакции уменьшается во время процесса сульфирования
вследствие выделения реакционной воды, уменьшающей концентра-
цию серной кислоты.
3. Температурный коэфициент 1 реакции сульфации равен
2—2,5 (для р-нитротолуола). Константа скорости нитрации при-
близительно в 1000 раз более, чем у реакции сульфации и тем-
пературный коэфициент выше (около 3).
4. Механизм реакции сульфации 2. Подобно описанному ранее
механизму нитрации азотной кислотой реакция сульфации идет
через стадию присоединения серной кислоты к молекуле аромати-
ческого соединения. Этот продукт присоединения неустойчив, при
его’ стабилизации выделяется 1 мол воды и образуется сульфосо-
единение, например:
5. Обратимость реакции сульфации. Реакция сульфации обра-
тима:
7?Н + H2SO4^zz± 7?SO3H + Н2О
В реакционной смеси устанавливается равновесие между7 прямой
реакцией сульфирования .и обратной реакцией гидролиза.
6. Влияние температуры на место, занимаемое сульфогруппой.
Еще Кекуле (1869 г.) показал, что при нагревании о-фенолсульфо-
кислоты при 100—110° она превращается в р-фенолсульфокислоту.
1 Сведения о скоростях реакции сульфации взяты из книги Н. Н. Ворож-
цова (Основы синтеза промежуточных продуктов и красителей, стр. 73, Гос-
химтехиздат, 1934).
2 См. там же.
274
Это обстоятельство используется техникой, так как р-фенолсульфо-
кислота легче нитруется, чемо-изомер. При сульфировании фенола
на холоду купоросным маслом образуется небольшое количество
о-фенолсульфокислоты; при последующей выдержке при температуре
100—110° образовавшаяся вначале о-сульфокислота превращается
в р-изомер.
Аналогичные явления наблюдаются и в других случаях. Напри-
мер, при мо но сульфировании нафталина (при низкой температуре
35—60°) получается в качестве главного продукта а-сульфокислота;
та же реакционная смесь образует при высокой температуре (160°)
преимущественно ^-сульфокислоту. Равным образом, если сначала
получить а-сульфокислоту и затем, не выделяя продукта, нагреть
до** 160°, то по мере нагревания заметно уменьшится количество
«-сульфокислоты и возрастет количество ^-изомера1.
Здесь, как и во многих аналогичных случаях сульфирования,
имеются равновесные системы из нескольких компонентов, и каж-
дой температуре при достаточной выдержке отвечают строго опре-
деленные соотношения между количеством отдельных компонентов.
Почти всегда вызываемый повышением температуры сдвиг равнове-
сия в пользу одного продукта объясняется участием образующейся
при сульфировании воды, которая, несмотря на присутствие избы-
точной серной кислоты в реакционной массе, вызывает отщепление
сульфогруппы от молекулы сульфокислоты с образованием (по при-
веденному уравнению) серной кислоты и первоначального органи-
ческого соединения. Последнее образует при повышенной темпера-
туре другой сульфоизомер с более устойчивой по отношению к гид-
ролизу сульфогруппой 2, например:
SO3H
7. Побочные реакции при процессе сульфирования. При повы-
шенных температурах процесс сульфирования может сопровождаться
побочными реакциями — окислением органического вещества, обра-
зованием сульфонов, конденсациями.
Образование сульфонов происходит или из 2 мол сульфокислоты
по уравнению 3:
2HSO3H = T?SO2H + H2SO4
или (более вероятно) из 1 мол сульфокислоты и несульфированного
вещества:
#SO3H + ЯН = T?SO2T? + Н2О
1 Euwes, Rec. Trav. Chim. Pays-Bas, 28, 298, 1909.
2 H. H. Ворожцов, Основы синтеза красителей и полупродуктов,
стр. 76, Госхимтехиздат, 1934.
8 Fonque и Lacroix, Zbl., 1923, 1, 1120.
275
Таблица 54
Концентрация серной кислоты °/ /о Количество фенола, превращенное в фе- нолдисульфокислоту %
92 62
93 66
94 71
96 82
97 91
100 100
Таблица 55
Продолжи- Количество фенола,
тельность превращенного в фе-
нагревания нолдисульфокислоту
час. %
v4 48
х/2 65
1 66
2 68
4 68.5
Образованию сульфонов содействует высокая температура, зна-
чительная концентрация сульфирующего агента (серная кислота)
при наличии сульфокислоты и непросульфированного вещества1.
8. Сульфирование фенола. По Обермиллеру2, при нагревании
1 в. ч. фенола с 1,5 в. ч. моногидрата серной кислоты при 45—50°
получается фенолмоносульфокислоты в количестве 98% от теории.
Отношение о- к р-изомеру равно 1 : 35,5. Образованию о-сульфо-
кислоты способствует малая концентрация и низкая температура.
При сульфировании фенола с большим избытком серной кислоты
и при повышенной температуре образуется дисульфокислота. Напри-
мер, при 6—8-часовом нагревании при 90—100° 1 ч. фенола с 4 ч.
95%-ной серной кислоты получено около 65—70% от теории фенол-
2,4-дисульфокислоты. По Обермиллеру, температура выше 100°
благоприятствует образованию сульфонов.
Данные Маркероля и его сотрудников
о сульфировании фенола. По данным Маркероля, ко-
личество полученной при суль-
фировании фенолдисульфокислоты
зависит от следующих факторов:
1) концентрации серной кис-
лоты;
2) продолжительности процес-
са сульфирования;
3) температуры сульфирования.
Ниже приводятся опытные дан-
ные Маркероля.
Для опытов брали постоянно
100 в. ч. фенола и 500 в. ч. сер-
ной кислоты.
Влияние концентра-
ции- В табл. 54 дано количество
фенола, превращенное в фенолди-
сульфокислоту после часового на-
гревания при 100°.
Из этой таблицы видно, что
количество фенола, превращенное
в фенолдисульфокислоту, растет с
увеличением концентрации серной
кислоты, причем его рост быстрее
роста концентрации кислоты.
тельности сульфирова-
Х)° с серной кислотой, содержащей
Влияние температуры. Опыты велись с 93%-еой серной
кислотой при температуре 75 и 150° (см. табл. 56).
Влияние продолжи
ния. Опыты проведены при 1(
93% H2SO4 (см. табл. 55).
1 О получении сульфонов см. Mayer, Lieb. Ann., 433, 327, 1923.
3 О b е rm i 1 1 е г, Вег., 40, 3623, 1907; 41, 696, 1908.
276
Из приведенных
данных легко опреде- —
лить наилучшие усло-
вия для получения
наибольшего коли-
чества фенолдисуль- —
фокислоты: темпера-
тура сульфирования
около 100°, продол-
жительность нагрева-
ния (выдержка), при
этой температуре около
назревание реакционной
Таблица 56
Темпера- тура °C Продолжитель- ность нагревания час. Количество фенола, превращенного в фе- нолдисульфо кислоту о/ /о
75 V2 51
75 1 59
150 50
150 V» 66
150 1 66,5
30 минут (до 1 часа). Бесполезно вести
смеси (выдержку) в течение нескольких
%1сов. Это увеличение времени нагревания даже нежелательно
по разным причинам; например, после продолжительного • на-
гревания полученный сульфофенол много легче кристаллизуется;
вследствие этого слив фенолдисульфокислоты может быть затруднен.
9. Опыт трисульфирования фенола. При нагревании фенола с
10-кратным количеством 20%-ного олеума при 120° в течение 3 часов
получена смесь ди- и фенолтрисульфокислоты с преобладающим
содержанием последней. При этом не получено ощутимых коли-
честв сульфонов или их сульфопроизводных.
10. Свойства сульфокислот фенола. Сульфокислоты относятся
к наименее изученным веществам органической химии. Их изучение
чрезвычайно затрудняется тем, что они не имеют определенной тем-
пературы кипения и характерной температуры плавления.
Для разведения сульфокислот пользуются различной раствори-
мостью их солей в воде. Очистка их производится путем многократ-
ной перекристаллизации из воды.
Соли моносульфокислот фенола — белые кристаллы; на воздухе,
и особенно в растворе, они окрашиваются в коричневый или красно-
ватый цвет. Все свободные сульфокислоты хорошо растворимы в
в воде; они являются сильными кислотами и обычно образуют два
типа солей:
1) при замещении водорода сульфогруппы металлом;
2) при замещении металлом водорода гидроксильной группы.
Сульфо группа легко нейтрализуется даже'карбонатами. Гидро-
ксильная группа медленно и неполностью нейтрализуется карбона-
тами; при увеличении числа сульфогрупп в молекуле скорость
нейтрализации гидроксильной группы увеличивается. Соли сульфо-
групп дают весьма слабокислую реакцию с синим лакмусом; соли
гидроксильной группы гидролизуют и переводят красный лакмус
в синий.
Кислые свойства сульфокислот фенола увеличиваются в такой
последовательности: ?и-,р-,о-фенолди сульфокислота, а способность
к гидролизу уменьшается в таком же порядке.
Молекулярная электропроводность растет регулярно от моно-
к трисульфокислоте фенола.
277
11. Нитрование сульфопроизводных фенола. До империалистиче-
ской войны 1914—-1918 гг. пикриновая кислота получалась нитрова-
нием моносульфофенола, содержавшего до 7% дисульфофенола. Та-
кой сульфофенол получался сульфированием фенола 5в. ч. купорос-
ного масла. И только во время войны (1915 г.) в Германии и Англии
было установлено производство пикриновой кислоты нитрованием
сульфопродукта, не содержавшего фенолмоносульфокислоты, а пре-
имущественно фенолдисульфокислоты и некоторое количество фенол-
трисульфокислоты. При этом заметно повысился выход пикриновой
кислоты.
Влияние состава сульфопродукта на выход пикриновой кислоты
изучалось во Франции Маркеролем с сотрудниками; результаты
этих опытов приводятся ниже.
Влияние состава продукта сульфации фо-
нола на выход пикриновой кислоты. Проведены
сравнительные опыты нитрования фенолсульфо кислоты, содержа-
щего 93% моносульфо- и 7% фенолдисульфокислоты и фенолсуль-
фокислоты с содержанием 100% фенолдисульфокислоты.
Фенолсульфокислота с 7% фенолди сульфо кислоты приготовлена
сульфированием фенола при температуре 50—75° 5-кратным коли-
чеством серной кислоты (с 92% H2SO4). Фенолдисульфокислота (100%
дисульфо-) получена сульфированием фенола 5-кратным количеством
моногидрата серной кислоты при температуре 100° и при продол-
жительности реакции в 30 минут.
Нитрование проведено в условиях французского способа, сливом
сульфофенола, полученного от сульфирования 100 в. ч. фенола и
смеси 400 в. ч. азотной кислоты 28° Вё (39% HNO3)h 300 в. ч. нат-
риевой селитры. Опыты нитрования были поставлены с нагреванием
реакционной массы до 112 и до 122°.
___ Таблица 57
Количество фенол- дпсульфокислоты г Выход пикрино- вой кислоты на 100 в. ч. исход- ного фенола в. ч. Содержание дини- тросульфокислоты в отработанной кислоте в пересче- те на пикриновую кислоту г Общий выход пи- криновой кислоты на 100 в. ч. исход- ного фенола
Нит рование при температуре до 112°
7 180 8 188
7 179 И 190 ,
100 191 ' 6 197
100 194 5 199
Нитрование при температуре до 122°
7 175 9 184
7 176 10 186
100 195 6 201
100 194 4 198
Из этих данных видно, что выход пикриновой кислоты зависит
от содержания фенолмоно- и дисульфокислот в продукте сульфи-
278
рования фенола. Он заметно выше для фено л дисульфо кислоты,
чем для фенолмоносульфокислоты. Повышение температуры от 112°
до 122° не увеличило выхода пикриновой кислоты.
При нитровании сульфопродукта, полученного сульфированием
фенола 10-кратным количеством олеума, проведенном в указанных
условиях французского способа и при температуре 112—115° (к концу
слива сульфо-), получен выход 215—220% пикриновой кислоты по
весу фенола.
12. Нитрование чистых дисульфо- и трисульфокислот. Нитрова-
ние велось азотной кислотой крепостью 40° Вё (62% HNO3 при тем-
пературе 110°):
Таблица 68
•ь
Наименование вещества Выход пикрино- вой кислоты в пересчете на 100 в. ч. фенола °/ /о Выход от теории %
Фенолсульфокислота . . . 208 — 210 86
Трисульфокислота .... 220 — 221 90—91
Таким образом приходим к выводу, что выход пикриновой кис-
лоты растет регулярно с возрастанием числа сульфогрупп в фенол-
сульфокислоте. Это объясняется тем что с возрастанием числа
сульфогрупп растет сопротивляемость молекулы фенола окислению.
13. Нитрование динитрофенолсульфокислот в пикриновую кис-
лоту. Из приведенных данных видно, что для повышения выхода
пикриновой кислоты надо донитровать оставшиеся в отработанной
кислоте в растворенном виде продукты — динитро сульфокислоты
фенола. Эти недонитрованные продукты не только уменьшают вы-
ход пикриновой кислоты, но из-за их частичного растворения в пи-
криновой кислоте, из которой они не вымываются нацело при вод-
ной промывке, ухудшается ее качество и получается повышенный
процент брака (вследствие нерастворимости динитрофенолсульфо-
кислоты в бензоле получается повышенное содержание нерас-
творимых в бензоле примесей).
14. Состав примесей в отработанных кислотах. По данным Мар-
кероля, значительная часть динитросульфокислот фенола содер-
жится в отработанной кислоте, полученной при нитровании по фран-
цузскому способу, в форме 2,4-динитрофенол-6-сульфокислоты фе-
нола:
ОН
!
Xno2
279
t
которая получается очевидно при неполном нитровании фенол-2,4-
или 2,6-дисульфокислот, потому что в фенолмоносульфокислоте
сульфогруппа находится в положении 4.
Кроме того, в тех же отработанных кислотах находится некото-
рое количество 2,6-динитрофенол-4-сульфокислоты фенола:
ОН
O,N/4NO2
XSO3H
которая получается при неполном нитровании фенол-4-сульфокис-
лоты, а может быть также фенолдисульфокислоты.
Иногда в случае недостаточного количества азотной кислоты
к концу нитрования или в случае недостаточного перемешивания
во время нитрования 1 находят также мононитрофенолсульфокис-
лоты.
В отработанных кислотах определена щавелевая кислота в коли-
честве около 8,7 г (СООН)2 2Н2О на 100 г взятого для нитрования
фенола.
Все эти данные о примесях в отработанных кислотах относятся
к французскому способу. О содержании и составе этих примесей
в отработанных кислотах, получаемых при производстве пикрино-
вой кислоты на крепких кислотах (немецкий и английский способ),
в литературе данных не имеется. Но известно, что количество обра-
зующейся щавелевой кислоты тем меньше, чем больше сульфогрупп
введено в ядро и чем выше концентрация серной кислоты, в которой
протекает реакция 2.
15. Опыты донитровывания динитросульфокислот. Маркероль
нагревал 2 г динитрофенолсульфоната натрия с 10 см3 азотной
кислоты крепостью 40° Вё (62% HNO3) или 14,8 см3 азотной кис-
лоты 31,5° Вё (44,4% HNO3), что отвечает каждый раз 8,5 г HNO3.
Полученные результаты приведены в табл. 59 (стр. 281).
Во второй серии опытов Маркероль приготовил нитрационные
смеси добавлением к серной кислоте с содержанием 86,3% H2SO4
азотной кислоты в таком количестве, чтобы в 1 л кислоты содержа-
лось 50 г и в другом образце 25 г HNO3. К 100 см3 приготовлен-
ной таким образом кислоты добавлялись 2 г динитросульфоната.
Остальные условия опыта и результаты приведены в табл. 60 (стр. 281).
Из этих данных следует, что превращение 2,4-динитрофенолсуль-
фокислоты в пикриновую идет труднее, чем динитро-2, 6-фенолсуль-
фо кислоты-4.
1 Надо помнить, что здесь идет речь о нитровании в горшках по француз-
скому способу, где не было механического перемешивания.
2 King, J. Chem- Soc., 119, 2105, 1921.
280
Таблица 69
Положение сульфогруппы Содержание в 1 л реакцион- ной смеси г । Температура °C Продолжи- тельность на- гревания час. Выход % Содержание в 1 л реакцион- ной смеси г Температура °C Продолжи- тельность на- гревания час. Выход %
4 6 4 6 4 6 4 6 50 50 50 50 50 50 50 50 115 115 110 110 100 100 90 90 2 2 2 2 6 6 12 12 96 92 96 75 92 61 82 36 25 25 25 25 25 25 25 25 115 115 ПО ПО 100 100 90 90 3 3 3 5 6 6 8 8 92 51 88 48- 51 36- 41 19
Таблица 60
Нитрование с азотной кислотой с содержанием 62% HNO3 Нитрование с азотной кислотой с содержанием 44,4% HNOa
Положение сульфогруппы Температура 1 t °C Продолжи- тельность нагревания час. Выход пикри- новой кислоты от теории % Положение сульфогруппы Температура Г Продолжи- I тельность нагревания час. ' 1 Выход пикри- новой кислоты от теории %
4 6 4 6 4 6 4 • 6 105 105 100 100 90 90 80 80 2 2 2 2 2 2 2 2 96 96 96 80 96 76 82 70 4 6 4 6 4 6 4 6 по по 105 105 1 100 100 90 90 2 2 2 2 2 2 2 2 96 96 96 48 94 29 89 17
Это объясняет, почему в отработанных кислотах содержится пре-
имущественно динитро-2,4-$енолосульфокислота-6. Этим, неви-
димому, объясняется и давно сделанное наблюдение, что фенол-
4-сульфокислота легче превращается в пикриновую, чем фенол-
2-сульфокислота.
16. Реакция нитрации и реакция нитролиза. При взаимодействии
фенолсульфокислот с азотной кислотой для получения пикрино-
вой кислоты имеют место два вида реакции: 1) реакция нитра-
ции ЛН + HNO3—» 7?NO2 Н2О и 2) реакция нитролиза:
7?SO3H HNO3—» Z?NO2 +H2SO4.
Условия, наиболее благоприятные для течения этих реакций,,
неодинаковы. Наибольшая скорость реакции нитрации достигается
при высоком факторе нитрующей активности, который точно не-
281
определен, но находится в пределах 80—90% H2SO4, вероятно, ближе
ж 90%; наибольшая же скорость реакции нитролиза достигается
при значительно меньшем факторе, вероятно, ниже 70% H2SO4.
Поэтому, например, образовавшаяся в первой стадии реакции
2-нитрофенол-4-сульфокислота может дальше реагировать по двум
направлениям:
ОН
В случае (I) идет реакция нитрации и получается 2,6-динитро?
«фенол-4-сульфокислота, а в случае (II) идет нитролиз с образова-
нием 2-4-динитрофенола.
При высокой концентрации серной кислоты, в которой проте-
кает взаимодействие, преобладает реакция (I); наоборот, при низ-
кой-концентрации ее преобладает реакция нитролиза1.
Эти зависимости имеют заметное практическое значение вслед-
ствие того, что динитрофенол превращается в пикриновую кислоту
только при нитровании его концентрированной серноазотной
смесью, тогда как динитрофенолсульфо кислота, наоборот, легко
превращается в пикриновую кислоту при взаимодействии в слабой
серной кислоте. Тем не менее эти зависимости не учитываются
в производстве пикриновой кислоты по французскому способу,
а равно при производстве пикриновой кислоты на крепких кислотах
по немецкому способу; но они играют заметную роль на англий-
ских и американских заводах.
1'7. Скорость окисления пикриновой кислоты серноазотной смесью.
Теммик2 определил скорость окисления пикриновой кислоты при
разных температурах серноазотной смесью, следующего состава:
14,7% H2SO4, 22,6% HNO„ 62,7% Н2О.
В 100 г этой смеси растворялось а г (см. табл. 61) пикрино-
вой кислоты; раствор нагревался в течение определенного времени
при определенной температуре и по окончании опыта устанавли-
вали количество оставшейся в кислоте пикриновой кислоты.
‘King, J. Chem. Soc., 119, 2105, 4921.
’ Н ammick, J- Soc. Chem. Ind., 40, 26, 1921
282
Таблица 61
Результаты приведены в табл. 61.
Температура 116° ± 1 106° + 1 96° ± 1
Содержание пикриновой кислоты в 100 см3 смеси а = 0,809 а = 1,198 а ~ 1,813
Продолжительность нагревания а—х* а—х а—х
2 дня 0.490 0,920 1,657
3 >> . 0,368 —- 1,644
4 » 0,291 0,838 1,600
Вывод. Даже при сравнительно высоких температурах ско-
рость разложения пикриновой кислоты серноазотной смесью на-
столько мала, что не может иметь практического влияния на ее вы-
ход. При этом надо учесть, что смесь Геммика содержала небольшой
процент серной кислоты и большой процент азотной по сравнению
с отработанной кислотой, получаемой при производстве пикриновой
кислоты, т. е. должна была окислять сильнее, чем смесь, в кото-
рой протекает реакция в производственных условиях.
ПЕРИОДИЧЕСКИЕ СПОСОБЫ СУЛЬФИРОВАНИЯ
И НИТРОВАНИЯ ФЕНОЛА
1. Общие сведения о способах получения пикриновой кислоты
и динитрофенола. Пикриновая кислота производится двумя спо-
собами:
1) из фенола нитрованием предварительно сульфированного
фенола;
2) из бензола через динитрохлорбензол.
Основным, притом наиболее старым и простым, является способ
производства из фенола. Второй способ получения через динитро-
хлорбензол, впервые примененный англичанами во время англо-
бурской войны, широко применялся во время империалистической
войны 1914—1918 гг., так как производство по этому способу мож-
но было быстро и без больших капитальных затрат установить на
красочных заводах, изготовлявших динитрохлорбензол для произ-
водства красок.
Динитрофенол получается тремя способами:
1) из динитро хлорбензола — этим способом получают наиболь-
шие количества динитрофенола;
2) нитрованием слабой азотной кислотой фенола, сульфирован-
ного меньшим количеством купоросного масла, чем при приготовле-
нии из него пикриновой кислоты французским способом;
* Через х обозначено количество окислившейся при опыте пикриновой
кислоты; следовательно, а—х есть количество оставшейся в растворе после
Опыта неокисленной пикриновой кислоты.
283
3) нитрованием фенола, растворенного в инертном растворителе.
Последние два способа применялись во время империалистической
войны во Франции.
2. Сульфирование и нитрование фенола по французскому способу.
Во время империалистической войны пикриновая кислота готовилась
в России из фенола по французскому способу, незначительно видо-
измененному.
Применялись следующие исходные материалы: фенол, купорос-
ное масло с содержанием около 92% H2SO4, натриевая селитра, азот-
ная кислота крепостью 31°
Рис. 38. Нитрационный горшок (француз-
ский способ).
Вё (43,5% HNO3).
В случае поступления на .
завод более крепкой азотной
кислоты она разбавлялась
слабой азотной кислотой или
водой.
Сульфирование.
Для сульфирования брали
на 1 ч. фенола 5 ч. купо-
росного масла.
На деревянном помосте
был установлен чугунный
эмалированный котел, снаб-
женный змеевиком (или ру-
башкой) для обогрева для
плавки фенола. Расплавлен-
ный фенол спускался самоте-
ком в расположенные ниже
чугунные эмалированные
аппараты с мешалкой, куда
предварительно наливалось
купоросное масло. Реакция
шла со значительным выде-
лением тепла, вследствие
чего температура поднималась до 50 — 75° (на других заводах
сульфирование фенола производилось в кислотоупорных гончарных
горнах, куда сперва заливалось отвешенное количество фенола и
к последнему из мерника добавлялось купоросное масло. Содержи-
мое горшков перемешивалось вручную стеклянной палкой или
алюминиевой трубкой. Иногда горшки окружали изоляцией из ма-
териала, не проводящего тепло, для достижения более высокой
температуры при сульфировании и медленного охлаждения).
Нитрование. Готовый раствор фенолсульфокислоты само-
теком спускался в нитрационные горшки (рис. 38).
Нитрационные горшки были керамиковые. Устройство показано
на чертеже. Горшок закрывался отъемной крышкой, имеющей четыре
отверстия: 1) для вливания фенолсульфокислоты, 2) для трубки,
подающей сжатый воздух (для перемешивания), 3) для термометра,
284
4) для удаления из горшка газов (воздуха, подаваемого для переме-
шивания, и окислов азота). Горшки устанавливались на деревянных
вагонетках, где они вставлялись в деревянные ящики. Промежутки
между стенками горшка и ящика заливались цементом х. В одну
операцию в горшке нитровали 22,5 кг фенола.
В горшок предварительно подавались 3 ч. натриевой селитры
и 4х/2 ч. азотной кислоты (с содержанием 43,5% HNO3) на 1 ч.
нитруемого фенола. После этого горшок закрывали крышкой и под-
возили в нитрационное отделение, где через большое отверстие
в крышке и посредством перекидного колена его соединяли с систе-
мой керамиковых баллонов и конденсационной башней для конден-
сации азотной кислоты.
*В одно отверстие вставляли стеклянную трубку для перемешива-
ния сжатым воздухом, в другое — термометр, в третье — воронку
для слива фенолсульфокислоты. Все соединительные щели замазы-
вали смесью жидкого стекла с асбестом. Слив фенолсульфокислоты
вели постепенно, наблюдая по термометру за температурой, таким
образом, чтобы к концу слива температура поднялась до 115°.
По окончании слива фенолсульфокислоты пропускали воздух
до тех пор, пока температура не опускалась до 105° (это перемеши-
вание необходимо для получения хороших кристаллов, иначе полу-
чалась ноздреватая масса), после чего вывозили горшки в особое
помещение — отстойную, — где оставляли их в покое на 2 часа;
затем в них заливали 65 л холодной воды и оставляли в покое
на 12—16 часов для выделения кристаллов пикриновой кислоты.
Пикриновая кислота собиралась на поверхности жидкости в виде
объемистой кристаллической массы светло желто го цвета.
3. Нитрование фенолапо горшечному £пособу в Англии. В отличие
от французского способа здесь не применялась для нитрования фе-
но лсульфокислоты натриевая селитра, а только азотная кислота
(с содержанием 65% HNO3); последняя подавалась к сульфофе-
нолу (по французскому способу — наоборот).
В начале ее слива к фенолсульфокислоте наблюдалось значи-
тельное повышение температуры, вследствие чего происходило по-
вышенное окисление и осмоление; в этом случае выход и качество
пикриновой кислоты понижались.
Во избежание этого было введено разбавление фенолсульфокис-
лоты водой до удельного веса приблизительно 1,35.
Такое разбавление было удачно использовано для удаления
сернокислого свинца, который содержится в растворенном виде
в концентрированной серной кислоте, но выпадает из раствора при
разбавлении этой кислоты. После разбавления производилось фильт-
рование сульфофенола через песочный фильтр или простое отстаивание
1 Это необходимо было потому, что стенки котлов сильно разрушались от
кислот и сравнительно высокой температуры нитрации и часто растрескивались,
причем содержимое котлов при отсутствии цементной одежды разливалось
и обжигало рабочих. Средняя служба горшков доходила лишь до 30 Операций
нитрования.
285
еульфофенола в течение 2—3 часов. Такая очистка фенолсульфо-
кислоты обеспечивала получение пикриновой кислоты с содержа-
нием не больше 0,02% золы.
Другой особенностью английского способа является применение
железного сульфуратора, снабженного механической мешалкой
стальным змеевиком для обогрева. Выдержка велась при 70° в те-
чение 4 часов.
На одном заводе был установлен иной порядок разбавления
сульфофенола. При этом исходили из следующих соображений. При
реакции нитрования фенолдисульфокислоты (по английским данным
образуется в количестве около 70% фенолдисульфокислоты при суль-
фировании фенола при 100°) сначала замещается нитрогруппой-атом
водорода и образуется мононитрофенолдисульфокислота. Здесь идет
непосредственная реакция нитрования. 2-я и 3-я нитрогруппы вво-
дятся путем замещения сульфогруппы нитролизом. Отсюда следует,
что выход пикриновой кислоты может быть улучшен, если 1-я нитро-
группа вводится в среду крепких кислот; 2-я и 3-я фазы реакции
нитрации в целях облегчения отщепления сульфогрупп протекают
легче в менее крепких растворах.
В этом случае ведут процесс следующим образом.
Сначала добавляют к неразбавленному раствору фенолсульфо-
кислот при температуре 35—50° 1,25 моля HNO3 (на 1 мол фе-
нола) в виде 65%-ной азотной кислоты. После этого смесь разбав-
ляют равным количеством воды, снова подают азотную кислоту в
количестве 2,75 моля HNO3 и делают выдержку 6—8 часов.
4. Усовершенствованный американский способ производства пи-
криновой кислоты. Во время империалистической войны в Америке
было проведено много исследовательских работ с целью повышения
выхода пикриновой кислоты при ее производстве. При этом выяв-
лены следующие условия \ при которых выход составляет 220%.
Загрузка: 302,2 кг фенола, 605 кг (93% H2SO4) серной кислоты;
выдержка при 95—98° 6 часов; при этом получается продукт, со-
держащий 80% фенол-4-сульфокислоты и 20% фенол-2,4-дисуль-
фо кислоты.
Сульфосмесь разбавляют 737,5 л воды; для этой цели обычно
применяют промывные воды. Содержание фенола в разбавленной
сульфосмеси 18,4%.
Подачу азотной кислоты ведут в две фазы. 1-я фаза: в аппарат
подается теоретически необходимое для введения одной нитрогруппы
количество азотной кислоты; начало слива ведут при 30°, к концу
слива поднимают температуру до 52°. Для нитрования применяют
смесь состава 70% HNO3, 10% H2SO4, 20% Н2О.
Азотная кислота приливается к фенолсульфо кислоте с той целью,
чтобы в 1-й фазе образовалось только мононитросоединение, а не ди-
нитросоединение.
1 Olsen и Goldst ei n, Ind. Eng. Chem., 16, 66—71, 1924.
286
По окончании 1-й фазы поднимают температуру до 60°, начи-
нают слив остальной части кислотной смеси; температура повы-
шается в течение первого часа слива до 110—115°. Всего подают
за обе фазы 1250 кг смеси (2,75 кг азотной кислоты на 1 кг фе-
нола).
При таком ведении процесса не происходит выделения обычно
наблюдающихся бурых паров окислов азота; нитрование заканчи-
вается, когда красный цвет кислоты, обусловленный образованием
нитрофенолсульфокислоты, переходит в нормальный желтый цвет
пикриновой кислоты.
Вес промытой пикриновой кислоты в пересчете на сухой вес со-
ставляет 667,6 кг, или 220% по весу фенола.
Отработанная кислота, содержащая 1,3% HNO3, разбавляется
двойным количеством (по весу) воды (промывной) и раствор кипя-
тится в течение 5 часов. После охлаждения собирают осевшую пикри-
новую кислоту, которая составляет 15% по весу фенола. Потери
пикриновой кислоты в промывных водах и в растворе отработанной
кислоты равны 1% по весу фенола. Следовательно, из теоретически
возможных 243 кг на 100 кг фенола получено 235 кг; 1 кг потерян
в промывных водах и отработанных кислотах — всего 236 кг; оки-
слено в щавелевую кислоту и потеряно в процессе производства
(пролито, распылено и т. п.) 7 кг.
5. Ход реакции нитрации и нитролиза фенолсульфокислот. Если
в смеси имеется фенол-4-сульфокислота, то в 1-й фазе нитрации
нитрогруппа вступает в ортоположение, образуя 2-нитрофенол-4-
сульфо кисло ту. В следующей фазе сульфогруппа в положении 4 за-
мещается нитрогруппой и образуется 2,4-динитрофенол. Эту стадию
легко заметить при лабораторном получении пикриновой кислоты
(если сульфирование производилось купоросным маслом, а не олеу-
мом) по выделению вначале темнокоричневых кристаллов динитро-
фенола. По мере нитрации эти кристаллы расплавляются благодаря
частичной нитрации до пикриновой кислоты, которая образует с ди-
нитрофенолом сплав, температура плавления которого ниже темпе-
ратуры реакционной массы.
• При продолжающейся нитрации выделяются кристаллы пикри-
новой кислоты с постепенным и полным исчезновением расплавлен-
ной массы.
При реакции нитрации фенол-2,4-дисульфокислоты вначале
1-я нитрогруппа вступает в незанятое ортоположение, образуя
6-нитрофенол-2,4-дисульфокислоту. Во второй фазе сульфогруппа
в положении 4 замещается нитрогруппой; образуется 2,4-динитро-
фе но л-6-сульфо кислота.
Это вещество легко превращается в пикриновую кислоту при 100^
в присутствии слабой кислотной смеси.
Динитрофенол никогда не наблюдается в качестве промежуточ-
ного продукта при нитровании фенолдисульфокислоты.
6. Производство пикриновой кислоты нитрованием фенола креп-
кими кислотами. Общие сведения. Приведенные данные
287
нитрования фенола горшечным способом широко применялись во
время империалистической войны во многих странах, потому что
«ни не требовали сложного оборудования, а работа была сравни-
тельно проста, не требовала квалифицированных рабочих: необхо-
димая квалификация быстро приобреталась новыми рабочими.
Все же эти способы имели серьезнейший недостаток. Это были
типичные кустарные способы, громоздкие, малопроизводительные.
При больших расходных коэфициентах по кислотам и фенолуи соот-
ветственно малых выходах пикриновой кислоты, при большом расходе
рабочей силы, они были неэкономичны. Нитрование велось в кера-
миковых горшках, не допускавших искусственного охлаждения
или подогрева, что не позволяло регулировать температуру про-
цесса. Наконец, применение слабых кислот не позволяло применять
металлическую аппаратуру с механическими мешалками.
В целях разработки более совершенного метода нитрования
были произведены исследования условий протекания реакции суль-
фирования и нитрования, приведшие к новому способу нитрования
фенола крепкими кислотами в металлической аппаратуре.
Нитрование фенола крепкими кислотамипо
немецкому способу. В основе этого способа лежат сле-
дующие ранее обоснованные положения:
1. При нитровании смеси фенолдисульфо- и трисульфокислот
получается лучший выход пикриновой кислоты вследствие меньшей
окисляемости.
2. При нитровании крепкими кислотами можно вести процессы
сульфирования и нитрования в обычных больших чугунных аппа-
ратах нитрационных заводов. Благодаря этому производство по срав-
нению с французским способом становится более компактным, умень-
шаются площади производственных зданий и расход рабочей силы,
легче рационализировать отдельные стадии процесса, что привело
к сокращению потерь, т. е. к повышению выхода. Наконец, при
новом способе значительно сокращается расход азотной кислоты,
а отработанная кислота может быть подвергнута денитрации.
По сообщению Каста1, производство пикриновой кислоты нитро-
ванием фенола крепкими кислотами было установлено в Германии
во время империалистической войны. Этот способ описывается2
следующим образом: к 94 г фенола добавляют при температуре
ниже 90° 400 г олеума (с содержанием 20% SO3), и нагревают
5 часов при 90—100° для образования ди сульфокислоты. Добавляют
"200 ч. серной кислоты уд. веса 1,84 при температуре ниже 50°.
После этого приливают 8СУ ч. (1 моль) азотной кислоты уд. веса
1,46 (80% HNO3). Затем добавляют 80 ч. этой кислоты при темпе-
ратуре 60—80° и, наконец, 100 ч. той же кислоты при темпера-
туре выше 80°. Выход 200—205% по весу фенола.
1 Каст, Взрывчатые вещества и средства воспламенения, стр. 211, 212,
Госхимтехиздат, 1932.
2 Герм. пат. № 298021, 1915.
288
Описание технологического процесса ни-
трования фенола на крепких кислотах. Суль-
фирование фенола. Барабан с фенолом очищают от при-
липшей к его наружным стенкам грязи, вскрывают и вносят в же-
лезный обогреваемый паром плавильный аппарат. Расплавленный
фенол помощью железного мерника отмеривается и подается в аппа-
рат-сульфуратор. Сульфуратор представляет собой железный или
чугунный аппарат, с рубашкой для нагрева или охлаждения, с ме-
ханической мешалкой, термометром, люком и трубой для удаления
газов.
На 1 ч. фенола берется 4,25 ч. олеума с содержанием 20% SO3.
После смешения компонентов массу нагревают до 100° и делают
при этой температуре выдержку. По окончании выдержки охлаж-
дает, добавляют 2 ч. купоросного масла на 1 ч. фенола (1 ч. купо-
росного масла можно без ущерба для производства заменить отрабо-
танной кислотой), охлаждают до 35° и переводят в сборник
сульфосмеси.
Основное назначение добавляемого купоросного масла (или отра-
ботанной кислоты) — разбавление раствора фенол- ди- и трисульфо-
кислот для предупреждения их кристаллизации.
Нитрова ние. Применение чистой азотной кислоты для ни-
трования, как это рекомендуется в приведенном патенте, потребо-
вало бы применения алюминиевого оборудования для подачи и отме-
ривания кислоты. Для возможности применения обычного желез-
ного оборудования к азотной кислоте добавляют некоторое коли-
чество серной кислоты. Например, применяют смесь состава: 80%
H2SO4, 10% HNO3, 10% Н2О. Нитрование ведется в чугунных аппа-
ратах обычного типа; для ускорения процесса рекомендуется ста-
вить змеевики для охлаждения. Загрузка сульфосмеси в зависи-
мости от объема нитратора в среднем около 350—380 кг сульфи-
рованного фенола. Загрузка азотной кислоты берется из расчета
125% от теории.
Смесь охлаждают до 40° и начинают приливание азотной кис-
лоты. Приливание ее производится в три приема: 1) первый моль азот-
ной кислоты (из расчета на моль фенола) приливают с такой ско-
ростью, чтобы температура поднималась на 2° в 5 минут; к концу
слива температура достигает 60°. Этот период отвечает мононитра-
ции фенола. Затем добавляется второй моль в пределах 60—80°,
при этом происходит динитрация фенола. Наконец, остаток кис-
лоты (1,75 моля азотной кислоты) приливается при температуре
80—100°. По окончании слива массу подогревают до 100° и при
этой температуре делается часовая выдержка.
По окончании выдержки продукты охлаждают до 35° и переда-
вливают в напорный бак, находящийся в отделении промывки и слу-
жащий для питания центрифуги. Напорный бак представляет собой
железный цилиндрический сосуд с конусообразным дном, закан-
чивающимся трубой с краном, через которую масса спускается
в центрифугу.
Химия—13—19
•269
Отжим от кислот. Для отжима от отработанной кислоты
масса из напорного бака спускается на центрифугу, приготовлен-
ную из V4A. Центрифугу загружают пикриновой кислотой около
200 кг (в пересчете на сухую). В отжатой пикриновой кислоте
остается около 10% серной кислоты при отжиме при 700 об/мин.
и диаметре корзинки 1200 мм. Отработанная кислота содержит 80—
82% H2SO4, 2,5—3% HNO3, 3—4% N2O3, 3—3,5% пикриновой
кислоты.
Отработанная кислота отстаивается в отстойных цистернах, где
из нее выделяется некоторое количество пикриновой кислоты.
Английский способ нитрования фенола
крепкими кислотами. Как при нитровании фенола гор-
шечным способом, так и при нитровании крепкими кислотами в Ан-
глии для сульфирования фенола применяли 1 купоросное масло.
Это было основано на том, что при проведенных в полузаводском
масштабе опытах сульфирования фенола олеумом получили пикри-
новую кислоту худшего качества, чем при сульфировании купо-
росным маслом; предполагали, что причиной является образование
сульфонов при сульфировании. Соотношение между купоросным мас-
лом и фенолом 1 : 3,5—5; но при этом было отмечено, что чем
больше серной кислоты брали для сульфирования, тем лучше
выход 2.
Особенности английского способа нитро-
вания фенола на крепких кислотах (проверен
на полузаводекой установке). При сульфировании брали на 1 ч.
фенола 3—5 в. ч., купоросного масла. Выдержка при 100° в тече-
ние 2 часов. Сульфосмесь охлаждалась до обыкновенной темпера-
туры, после чего начинался прилив кислотной смеси состава:
57,5% H2SO4, 38,5% HNO3, 4% Н2О; половина смеси добавлялась
при температуре ниже 50°; при превышении этой температуры пони-
жался выход пикриновой кислоты. Отмечено, что в стадии моно-
нитрования прилив смеси вызывает заметную тенденцию к повыше-
нию температуры, что приводит к окислению, а после введения
первой четверти всего количества кислоты остальная кислота может
быть прилита быстрее.
Если при приливании первой четверти происходит подъем тем-
пературы выше 50°, то имеется опасность, что содержимое нитра-
тора может быть выброшено. После прилива первой половины кис-
лотной смеси массу нагревают паром до 65°; приливают три четверти
второй половины смеси, причем температура должна повышаться
1 Здесь описываются английские методы работы периода империалистиче-
ской войны 1914—1918 гг.; изложено по Technical Records, Synthetic phenol
and picric acid, 1918.
2 По нашему мнению, ссылка на образование сульфонов является мало об-
основанной. Известно, что во время империалистической войны в Англии был
большой недостаток олеума, недостаточно было и купоросного масла. Недостат-
ком олеума надо объяснить применение купоросного масла для сульфирования
. фенола, а также применение его по отношению к фенолу в меньшей пропорции,
чем 5 к 1.
290
со скоростью 1° в каждые 3 минуты. Оставшуюся после этого кис-
лотную смесь приливают в течение 35—40 минут, причем темпера-
тура должна подняться от 80 до 115°.
По достижении 115° оставляют содержимое нитратора стоять
в покое для охлаждения до 100°, после чего охлаждают водой до 25°.
Температура выдержки 115° обосновывалась следующим образом.
Если кончать процесс ниже 115°, то полученная пикриновая кис-
лота часто содержит недонитрованный продукт с пониженной тем-
пературой затвердевания. Если же конечную температуру поднять
выше 115°, то пикриновая кислота вследствие начинающегося про-
цесса плавления имеет тенденцию к образованию гранулей.
В первом случае продукт чрезвычайно медленно фильтруется,
цо втором случае его невозможно отмыть от минеральных кислот.
Содержимое нитратора по окончании операции выгружалось на
асбестовый фильтр, отработанная кислота отсасывалась, после чего
пикриновая кислота промывалась небольшим количеством серной
кислоты (с содержанием 75% H2SO4). Этим путем вытеснялась вся
азотная кислота из межкристальной жидкости, чем облегчается по-
следующая отмывка кислот. Если не применяется промывка серной
кислотой, то при загрузке пикриновой кислоты водой происходит
обильное выделение окислов азота.
Состав отработанной кислоты, %:
H2SO4 (включая динитрофенолсульфокислоты) 73,0
HNO3...................................... 0,65
HNO2...................................... 3,58
пикриновая кислота ......................... 4,2
прочие органические соединения ............ 2,02
вода ...................................... 16,55
Итого . . - 100
7. Получение пикриновой кислоты по патенту Гутенсонах. Спо-
соб имеет целью избегнуть предварительного сульфирования фе-
нола. Для уменьшения скорости окисления и осмоления фенола по-
следний растворяют в парафиновом или минеральном масле, не
взаимодействующем с серной и азотной кислотами или их смесью,
в отношении 1 в. ч. фенола на 4 в. ч. парафинового масла. Этот
раствор сливается в смесь серной и азотной кислот, составленной
из 1’в. ч. серной и 8 в. ч. азотной. Смесь берется из расчета 3,5—
4 в. ч. азотной кислоты на 1 в. ч. фенола. Рекомендуется прили-
вать сначала половину фенольного раствора, а вторую половину
добавлять на второй день.
Пикриновая кислота выделяется в кристаллическом виде, а па-
рафиновое масло может служить опять для новой нитрации.
Способ не имеет заводского применения.
1 Герм. пат. № 126197, 1900
291
НЕПРЕРЫВНОЕ НИТРОВАНИЕ ФЕНОЛА
Рис. 39. Аппарат Брука.
вые патрубки, через которые в аппарат
1. Способ Брука. Аппарат-нитратор Брука состоит из ванны,
образованной из поставленных на силикатно-асбестовой замазке
керамиковых кирпичей. Длина аппарата 25 м, ширина 0,60 м и
глубина 0,50 м, толщина стенки около 20 мм (рис. 39).
Ванна покрыта керамиковыми плитами со смотровыми отвер-
стиями. На расстоянии 1 м друг от друга по всей длине аппарата
расположены керамико-
вые трубы для отвода воз-
духа с кислыми парами;
между каждыми двумя
такими трубами помеща-
ются глиняные паровые
трубы, спускающиеся до
дна для нагревания смеси
острым паром. На рас-
стоянии 9 м друг от дру-
га поставлены алюминие-
к протекающей жидкости
подается азотная кислота.
Оба конца ванны открыты, с одного конца имеется секция для
смешивания (длина 2 м), отделенная от остальной ванны кирпичной
перегородкой, которая не доходит на 0,10 м до верха ванны; на дне
ванны имеется дырчатая спираль для подачи пара.
Фенол сульфируется сначала в отдельном аппарате, а затем
вводится в первую секцию этого аппарата, где к нему прибавляется
горячая отработанная кислота. Смешанные здесь фенолсульфокис-
лота и отработанная кислота непрерывно переливаются через вер-
тикальную перегородку и текут дальше, встречая по пути азотную
кислоту, непрерывно подаваемую через указанные алюминиевые
патрубки. Здесь нитрация идет постепенно при средней температуре
102°. Реакционная масса, прошедшая через весь аппарат, перетекает
в аппараты-кристаллизаторы, где выкристаллизовывается пикри-
новая кислота.
Для сульфирования фенола брали 2 моля H2SO4 в виде купоросг
ного масла на 1 мольфенола, сульфосмесь разбавлялась водой до уд.
веса 1,36. В 1-й секции к сульфофенолу добавляли отработанной
кислоты из расчета 4 моля H2SO4 на 1 моль фенола.
Фенолсульфокислота проходила через аппарат в течение 5 часов
ПРОМЫВКА ПИКРИНОВОЙ кислоты, СУШКА,
ПРОСЕИВАНИЕ И УПАКОВКА
1. Промывка пикриновой кислоты. По французскому способу
в отстойной горшки охлаждаются до 60°, после чего перевозятся
в промывочную мастерскую. При охлаждении ниже 60° вместе
с пикриновой кислотой выкристаллизовывается большое количество
бисульфата, трудно отмываемого от нее.
292
Отработанные кислоты сливаются посредством сифонов, а пикри-
новая кислота выгружается деревянными ковшами в деревянные
промывные чаны. Последние имеют дырчатое дно и ниже — второе,
сплошное. Отверстия в верхнем дне имеют форму усеченного конуса,
обращенного большим основанием вниз. Диаметр верхнего основа-
ния конуса 0,5 мм. При такой форме отверстий они задерживают
воду, не закрываясь при разбухании намокшего дерева, что часто
бывает с цилиндрическими отверстиями.
Пикриновая кислота промывается 4—5 раз (а иногда и больше)
для отмывки от нее кислот и бисульфата.
Промывные воды спускаются по желобу в систему осадочных
чанов, где к ним добавляются отработанные кислоты в таком коли-
честве, чтобы концентрация серной кислоты была не меньше 10%.
Это необходимо для максимального уменьшения потерь пикри-
новой кислоты в промывных водах, как это видно из табл. 48, на
стр. 262.
Собирающаяся на поверхности жидкости в осадочных чанах и на
дне последних пикриновая кислота выгребается черпаками и снова
промывается. Отстоявшиеся промывные воды после выделения из
них увлеченнойщз промывных баков или выделившейся при разбав-
лении отработанных кислот пикриновой кислоты поступают в кана-
лизацию.
Промытая пикриновая кислота отжимается на центрифуге и
отправляется в сушилку.
Промывка пикриновой кислоты на англий-
ских заводах. Особенностью этого способа являлось то, что
для достижения большего однообразия качества окончательного про-
дукта вся пикриновая кислота перед промывкой пропускалась
через вальцовую мельницу *, состоявшую из рифленых вальцов,
расположенных попарно (одна пара под другой). Верхняя пара
имела промежуток между вальцами от 25 до 28 мм, а нижняя пара
вальцов прилегала друг к другу вплотную.
Промывка пикриновой кислоты при про-
изводстве ее на крепких кислотах. При фран-
цузском способе производилась 4—5-кратная промывка пикрино-
вой кислоты, причем каждый раз брали чистую воду. При этом
расходовалось от 5 до 15 ч. воды на промывку 1 ч. пикриновой
кислоты. Таков же был расход воды на промывку пикриновой кис-
лоты во время империалистической войны по описываемому ниже
способу получения ее через динитрохлорбензол. При этом имели
место значительные потери пикриновой кислоты, уносившейся в
промывных водах в растворенном и взвешенном виде.
Поэтому была разработана противоточная промывка, которая
выполняется по следующей схеме (рис. 40).
1 Возможно, что это имело целью разрушение сростков кристаллов, что
должно было облегчить промывку.
293
Из напорного бака *4 реакционная масса подается в центри-
фугу В. После окончания фуговки пикриновая кислота выгру-
жается в декантер I, куда предварительно наливается кислая вода,
отжатая на центрифуге С, в количестве 4 ч. на 1 ч. пикриновой
кислоты. После 5-минутного перемешивания воздухом содержимое
декантера 1 при помощи насоса перекачивается в декантер 2, распо-
ложенный на уровне напорных резервуаров. В декантере 2 дают
Рис. 40. Схема фуговки и противоточной промывки пикриновой
кислоты.
массе отстояться; кристаллы пикриновой кислоты оседают на дно;
сливают кислую воду помощью крана через верхнюю сливную трубку,
проходящую через стенку декантера. Эта вода проходит через серию
ловушек для выделения механически увлеченной пикриновой кис-
лоты и из них в канализацию. Из декантера таким образом сливается
около 3/4 всей находящейся в нем вместе с пикриновой кислотой
воды.
После этого в декантер 2 добавляют чистой воды из расчета 2 ч.
ее на 1 ч. пикриновой кислоты, перемешивают в течение несколь-
ких минут воздухом и спускают на водную центрифугу. После от-
жима от второй воды на центрифугу подают еще чистой воды в ко-
личестве, равном весу пикриновой кислоты; воды смешивают и по-
дают в декантер 1 для промывки новой порции кислой пикриновой
кислоты.
При этом способе промывки на 1 т пикриновой кислоты расхо-
дуется только 3 т воды, что заметно сказывается на уменьшении
потерь пикриновой кислоты при промывке х.
1 В Англии запрещается спускать в каналы и реки промывные воды, содер-
жащие минеральные кислоты или пикриновую кислоту. Нейтрализация мине-
ральных кислот производится известью в колодцах (см. стр. 167). Был разрабо-
тан Способ восстановления пикриновой кислоты в промывных водах железом.
О восстановлении нитросоединений железом см. Lyons и Smith, Вег.. 60,173,1927-
294
2. Сушка пикриновой кислоты. Отжатая на водной центрифуге
пикриновая кислота с содержанием 6—7% влаги поступает в сушку.
Для сушки могут применяться следующие типы сушилок:
а) Камерные сушилки, наименее экономичные. Про-
должительность сушки 16 часов при 60°. Применялись во время им-
периалистической войны; ныне мало употребительны.
б) Туннельные сушилки. Пикриновая кислота насы-
пается на деревянные лотки с доньями из тонкой фанеры. Лотки
устанавливаются на вагонетках, которые вводятся в канал сушилки
и медленно движутся по каналу. Навстречу вагонеткам через канал
протягивается при помощи вентилятора нагретый до 60° воздух.
Перед поступлением в вентилятор воздух проходит через влажный
фильтр (во избежание попадания взрывчатой пыли в вентилятор).
Продолжительность сушки пикриновой кислоты около 5 часов.
в) Барабанные сушилки состоят из вращающегося
деревянного барабана, закрытого с обоих концов неподвижными ко-
жухами. Пикриновая кислота непрерывно подается алюминиевым
шнеком в барабан. При вращении барабана продукт пересыпается
по наклонным полкам внутри барабана, непрерывно подвигаясь впе-
ред. Навстречу (или попутно) движется нагретый до 60° воздух.
Высушенный продукт с другого конца высыпается в приемный
бункер, откуда по короткому холщевому рукаву ссыпается в подстав-
ленные ящики.
Осушающий воздух засасывается вентилятором извне, пропу-
скается через фильтр и нагнетается в паровой калорифер, где и на-
гревается; затем по жестяному луженому трубопроводу подводится
в барабан. Пройдя барабан, воздух, насыщенный пылью пикриновой
кислоты, направляется в рукавный фильтр, где освобождается от
пикриновой кислоты, и через трубу удаляется в атмосферу.
г) Пневматическая сушилка по типу применяю-
щейся для сушки тротила может с успехом применяться для сушки
пикриновой кислоты.
3. Просеивание и упаковка пикриновой кислоты. Высушенную
пикриновую кислоту перевозят в отделение упаковки, где она про-
сеивается через медное сито с отверстиями 3X3 еж2 для удаления
случайно попавших посторонних тел и кусочков (из слипшихся кри-
сталлов) пикриновой кислоты. Ввиду вредности для здоровья рабо-
чих пыли, получающейся при просеивании сухой пикриновой кис-
лоты, рекомендуется делать просеивание влажного продукта (до
сушки).
Просушенную и просеянную пикриновую кислоту упаковывают
в ящики, которые делаются из обрезных досок на шипах; внутри
ящики выкладываются бумагой. На крышке и стенках ящика
делаются установленные ОСТ надписи.
4. Выход пикриновой кислоты при ее производстве француз-
ским и английским способами колебался в пределах 160—190% по
весу фенола — в среднем около 175%, тогда как теоретически из
100 в. ч. фенола должно получиться 243,6 в. ч. пикриновой кислоты.
295
Выход пикриновой кислоты при нитровании фенола на креп-
ких кислотах с применением олеума для сульфирования составлял
около 200% по весу фенола.
Причинами пониженного выхода пикриновой кислоты при ее
производстве по горшечному способу являются: 1) повышенное
окисление фенолсульфокислот при нитровании сравнительно с ди-
сульфокислотами, 2) недонитровывание части сульфокислот, остаю-
щихся в отработанных кислотах в виде динитрофенолсульфокислот,
3) потери продукта в условиях кустарного производства, в част-
ности, при промывке пикриновой кислоты.
На качество пикриновой кислоты при описанном способе влияют:
1. На температуру затвердевания — содержание динитрофенола,
чистота фенола, т. е. содержание в нем крезола и других органи-
ческих примесей.
2. На количество золы, в особенности песка, — содержание их
в селитре, примененной для нитрования (по французскому способу),
кроме того, сернокислый свинец, осаждающийся при разбавлении
купоросного масла, примененного для сульфирования.
3. На количество нерастворимых в бензоле примесей — получе-
ние при нитровании динитросульфокислоты фенола, которая частично
растворяется в пикриновой кислоте и трудно от нее отмывается.
5. Технические условия на пикриновую кислоту (ОСТ 3515):
Показатели Нормы
1. Внешний вид для всех сортов . 2. Цвет Кристаллический порошок без механи- ческих примесей, видимых на глаз Светложелтый
3. Температура затвердевания не ниже . , 119.5° 119,5°- 119°
4. Влажность и летучие веще- ства не более .... 0,5% 0,5% 0,5%
б. Нерастворимых в бензоле примесей не более 0,2% 0,3% 1,0%
В том числе: а) пикратов Не допу- Следы Не более
б) кремнезема не более скается 0.05% 0,05% 0.03% 0,1%
6. Серной кислоты не более . . . 0.1% 0,2% о.з%
7. Галоидных соединений > . . . Следы Следы Следы
Упаковка: деревянные ящики или бочки с толщиной стенок не менее 1,5 см.
6. Качественное определение пикриновой кислоты. Для определе-
ния малых количеств пикриновой кислоты пользуются тем, что при
взаимодействии пикрата натрия с цианистым калием образуется
изопурпуровая кислота *, причем раствор окрашивается в красный
‘Grignard, C.R., 142, 545, 1906; 145. 1115, 1907; Kling, Zbl,
1921, II, Ю09.
296
цвет. Для этой цели берут в пробирке 5 мл исследуемой жидкости,
нейтрализуют раствором едкого натра, прибавляют 0,5 мл свеже-
приготовленного 5%-ного раствора цианистого калия и нагревают
до кипения.
В присутствии пикриновой кислоты или динитрофенола получают
окрашивание от розового цвета до смородинно-красного.
Чувствительность реакции — около 1/50 000 для тринитрофенола
и около 1/20 000 для динитрофенола.
Структура изопурпуровой кислоты твердо еще не установлена.
Ей приписывается одна из следующих формул:
ONa
O2N
ONa
+ 3HCN —
ONa
O.N./^NfCN),
’ I или
no2
O2NZXNH2
+ HCNO 4- H2O
NC\/GN
no2
ПОЛУЧЕНИЕ ДИНИТРОФЕНОЛА НИТРОВАНИЕМ ФЕНОЛА
Во Франции во время империалистической войны получали ди-
нитрофенол нитрованием фенола. Введение этого метода было вы-
звано стремлением изъять из употребления хлорбензол, получавшийся
из Америки.
1. Производство динитрофенола, а) Сульфировали фе-
но л в аппарате из листового железа, снабженном мешалкой и зме-
евиком для нагревания. Загружали 840 кг фенола, нагревали до
•80—85°, после чего приливали 1200 кг 93%-ной серной кислоты
(1,43 ч. серной кислоты или 1,33 ч. моногидрата H2SO4 на 1 ч.
фенола). Температура поднималась сначала до 120°, потом, по исте-
чении 4 часов, снижалась до 110° и спустя 6 часов — до 100°.
Сульфофенол в количестве, соответствующем 280 кг фенола, на-
правлялся в мерник, в котором находилось 220 л воды.
б) Нитрование сульфофенола производилось в ап-
паратах из листового железа, выложенных внутри кислотоупорным
кирпичом.
Процесс проводился в 3 фазы.
1-я фаза: получение мононитрофенолсульфокислоты.
В аппарате растворяли 336 кг натриевой селитры в 448 кг воды
и прибавляли сульфофенол. Температура возрастала быстро до 100°.
2-я фаза: превращение мононитрофенолсульфо кислоты в
1,2, 4-динитрофенол и 1,2,6-динитрофенолсулнфокислоту.
297
В нитратор добавляли 225 кг воды и 270 кг натриевой селитры.
Затем добавляли постепенно 400 кг серной кислоты 60° Вё. При
W° реакция становилась весьма интенсивной и температура подни-
малась до 103—104°. На поверхности раствора постепенно соби-
рался жидкий 1,2,4-динитрофенол. По охлаждении до 101° при-
бавляли треть селитры, потребной для 3-й фазы, чем облегчалось
выделение динитрофенола на поверхности раствора.
Динитрофенол снимали медным черпаком и направляли на про-
мывку.
3-я фаза: превращение динитрофенолсульфокислоты в тринитро-
феиол. В нитратор добавляли небольшими порциями 440 кг се-
литры. После этого постепенно сливали 744 кг серной кислоты с со-
держанием 82% H,SO4. При сливе температура поднималась с 80°
до НО—112°. По окончании слива оставляли в покое на 7 часов,
поле чего прибавляли 800 л воды. Дальше поступали, как изло-
жено в описании получения пикриновой кислоты французским спо-
собом.
2. Получение динитрофенола нитрованием фенола в растворе
уксусной кислоты. Этот способ подобно способу Гутенсона основан на
том, что: 1) скорость окисления азотной кислотой фенола, раство-
ренного в уксусной кислоте, значительно меньше, чем нерастворенного
фенола; 2) динитрофенол, образующийся при нитровании, не раство-
рим в 65%-ной и более слабой уксусной кислоте и поэтому он легко и
удобно отделяется от отработанной кислоты. Способ Келера состоите
следующем: 100 г фенола растворяют в 360 см3 66—67%-ной уксусной
кислоты и полученный раствор вводят постепенно в азотную кислоту
40° Вё (62% HNO3) при температуре ниже 20°. Выход динитро-
фенола 70—75% от теории. Способ не находит промышленного приме-
нения вследствие дефицитности и высокой цены уксусной кислоты.
3. Получение динитрофенола нитрованием фенола азотной кис-
лотой. В аппарат загружают 1500 кг азотной кислоты 20° Вё (27%
HNO3), нагревают до 25—30° и вводят нагретую до 55—60° эмуль-
сию из 100 кг фенола и 50 кг воды. Температура быстро подни-
мается, но нельзя допускать подъема ее выше 82°.
Выход динитрофенола 155% от веса фенола (или 80% от тео-
рии). Содержание азота в динитрофеноле 15% (теоретическое содер-
жание 15,2%).
Эти данные о нитровании фенола азотной кислотой взяты у Де-
верня Ч Однако вызывает сомнение возможность получения такого
высокого выхода динитрофенола при нитровании одной азотной кис-
лотой. Во-первых, легко подсчитать, что при расходе согласно тео-
рии 2 мол. HNO3 на 1 мол фенола концентрация HNO3 в отрабо-
танной кислоте составит около 13,5%. Едва ли при такой концентра-
ции возможно нитрование фенола до динитрофенола даже при вы-
держке при температуре до 82°. Кроме того, по данным Маркероля
и Лориетта, выход динитрофенола увеличивается при добавлении
1 Desvergnes, Chim. et. Ind., 26, 507—520; 1271—1281, 1931.
298
к азотной кислоте серной кислоты; тем не менее эти авторы полу-
чили выход динитрофенола только 138,5% по весу фенола, при
следующих условиях Ч
К смеси 632 г серной кислоты с содержанием 67% H2SO4 и 360 г
’ азотной кислоты с содержанием 52,8% HNO3 (состав смеси: 42,7%
H2SO4, 19,10% HNO3, 38,2% Н2О; Ф 49,5% H2SO4; количество
HNO3 в смеси равно 150% от теории) добавляется раствор 94 ч.
фенола в 30 ч. воды.
Температура слива не выше 30°, по окончании слива температура
поднимается до 90—100°. Динитрофенол получается в виде мелких
светложелтых кристаллов 2,4-динитрофенола с температурой пла-
вления 101—103°.
ПРОИЗВОДСТВО ПИКРИНОВОЙ КИСЛОТЫ ИЗ БЕНЗОЛА
ЧЕРЕЗ ДИНИТРОХЛОРБЕНЗОЛ
Это производство состоит из следующих основных операций:
1) производство хлорбензола; 2) нитрование хлорбензола; 3) омыле-
ние динитрохлорбензола; 4) нейтрализация динитрофенолята; 5) ни-
трование динитрофенола; 6) промывка и сушка пикриновой кислоты.
1. Механизм реакции галоидирования и свойства хлорбензола
и дихлорбензолов. Хлорбензол легко образуется при действии хлора
на бензол в присутствии так называемых переносчиков га-
лоида, т. е. веществ, каталитически ускоряющих процесс. В ка-
честве такого переносчика в заводской практике применяют исклю-
чительно железо, как наиболее дешевый и легко доступный материал
(опилки, куски листового железа и т. п.).
По теории радикал-ионов Пфейффера роль переносчиков гало-
ида, в присутствии которых идет замещение водорода бензольного
ядра, заключается в том, что они образуют промежуточные про-
дукты, представляющие собой галоидные соли:
СН
сн/^сн
СН1
СН
СН
[С1АГ---->
-Ь сь А
СНС1
СН
сн^^сн
СН
.сн
СС1
+ НС1 + А
где А — переносчик галоида (ср. стр. 56 и 57).
1 М а г q u еу го1 и Loriette, Bull. Soc. Chim. Fr., 25, 370—375,
1919.
299
Свойства хлорбензола. Температура кипения 132°.
В то время как галоидопроизводные углеводородов жирного ряда
являются очень реакционноспособными соединениями (их галоидные
атомы обладают большой подвижностью и могут вступать в различные
реакции обмена), ароматические галоидопроизводные, галоид кото-
рых стоит в бензольном ядре, обладают другим характером; связь
галоида с ядром здесь очень прочна и замещение галоида идет с боль-
шим трудом. Так, например, хлорбензол реагирует с аммиаком
в автоклаве лишь при нагревании до 180—200° в присутствии солей
меди или медного порошка; концентрированные водные растворы
щелочей отщепляют хлор от хлорбензола лишь при температуре
около 1 300°. Прочность связи галоида с ядром уменьшается заметно
при вступлении в ядро так называемых «отрицательных групп» 2,
например, NO,, СООН и др. При этом особенно сильно активируют
атом хлора и делают его подвижным группы, стоящие к нему в
орто- или параполо-
жении. Увеличение
числа нитрогрупп в
орто- и параположе-
ниях к хлору осо-
бенно сильно увели-
чивает реакционную
способность послед-
него 3.
условиях всегда по-
количество дихлор-
Наименование вещества Температура кипения °C Температура плавления °C
о-Дихлорбензол 4 . 180,3
р- Дихлорбензол 173 53
При хлорировании бензола в заводских
лучается наряду с хлорбензолом некоторое
бензола, преимущественно о- и р-дихлорбензола (см. табл.).
2. Свойства и применение моно- и динитрохлорбензола. Свой-
ства мононитрохлорбензола. р-Нитро хлор бензол —
твердое вещество. Плавится при температуре 83°.
1 В углеводородах жирного ряда наблюдается прочная связь галоидного
атома с углеродом в тех случаях, когда галоид стоит у двойной связи, например,
в СН3СН = СНС1. В хлорбензоле галоид тоже связан с не вполне насыщенным
атомом углерода, и потому можно считать, что он тоже стоит у двойной связи:
IC1
В этом отношении отмечается, следовательно, определенная аналогия в
свойствах галоидозамещенных жирного и ароматического рядов.
2 Группы NO2, SO3H, СООН с точки зрения ионно-электронной теории
являются положительными группами. Мы, однако, сохраняем здесь издавна
укоренившееся в органической химии наименование «отрицательные группы»,
которым подчеркивается, что эти группы придают углеводородному остатку
кислые свойства.
3 J. Schmidt, Uber den Einfluss der Kernsubstitution auf die Reakti-
onsfahigkeit aromatischer Verbindungen, Stuttgart, 1902.
4 О физических свойствах о-дихлорбензола см. К а р з у э л, Ind. Eng.
Chem., 7, 1928 (Ж. Хим. Пр., 21—22, 1928).
300
о-Нитрохлорбензол —твердое вещество. Плавится при темпе-
ратуре 32,5°.
Свойства динитрохлорбензола. Химически чи-
стый 1,2,4-динитрохлорбензол представляет собой почти бесцветные
кристаллы с температурой затвердевания 50,1° (по Деверню); раство-
римость его в 100 ч. воды (по Деверню):
при 15° ........ 0,00018
» 50°.............0,0410
» 100°............0,1590
1,2,4-Динитрохлорбензол легко омыляется едкими щелочами,
образуя 1,2,4-динитрофеноляты. •
При омылении в присутствии спиртов дает соответственно 1,2,4-
динитрофенетол, 1,2,4-динитроанизол и т. п.
Применение динитрохлорбензола. Во время импери-
алистической войны во Франции пытались применять динитрохлор-
бензол в виде сплавов с пикриновой кислотой (например, 60%
пикриновой кислоты и 40% динитро хлорбензола), однако, такие
сплавы эксудировали («текли» — по нашей терминологии). Даже
очищенный динитро хлорбензол с температурой затвердевания 49°
давал в сплаве указанного состава 17,9% эксудата после 16 часов
хранения при температуре 50°.
2,4-динитро-1-хлорбензол применяется для изготовления целого
ряда сернистых красок. Он конденсируется с большим числом соеди-
нений ароматического ряда, а пцлучаемые продукты конденсации
восстанавливаются сернистым натрием и серой.
Омылением динитрохлорбензола получают динитрофенолят, пе-
рерабатываемый далее в пикриновую кислоту (по методу, описан-
ному в следующей главе).
3. Примеси в техническом динитрохлорбензоле. По данным Де-
верня1, в техническом динитрохлорбензэле содержатся следующие
примеси:
1) мононитрохлорбензол — о-, р- и немного т-йзомеров (содер-
жится несколько процентов);
2) продукты нитрации m-нитрохлорбензола, количество которых
зависит от количества последнего, увеличивавшегося с повыше-
нием температуры мононитрации, как это видно по следующим кос-
венным данным: при температуре 1-й нитрации 30—36° температура
получающегося динитрохлорбензола 47,4°,упри 70° 46,2°, при 115—
120° 45,5°; при существующих условиях нитрации содержание этих
продуктов составляет несколько процентов;
3) m-динитробензол, получаемый от динитрации примеси бензола,
содержащейся в хлорбензоле (содержится около 0,1%);
4) динитродихлорбензол, получаемый при нитровании дихлор-
бензола;
1 Девернь, 2,4-Динитрофенол, его производство, свойства,применение,
Сборник переводных статей «Взрывчатые вещества», 3, 67.
301
ci/\oh
5) хлорнитрофенол ; это вещество образуется, по дан-
\ю2
ным Деверня, во время нитрации благодаря тому, что одна из нитро-
/\/N°2
СГ ।
групп хлоро-о-динитробензола восстанавливается, затем
\/\no2
диазотируется, образуя диазопроизводное, гидролиз которого дает
хлорнитробензол х.
Кроме того, хлорнитрофенол может образоваться во время омы-
ления едким натром 3,4-динитро-1-хлорбензола по реакции:
NO2
l/C1
+ NaOH
ОН
O2N|/X|
+ NaNO,
\/C1
Ядовитость получаемого из динитрохлорбензола динитрофенола
увеличивается при содержании в нем мононитро хлорбензола и хлор-
нитрофенола. ,
4. Производство хлорбензола. В аппарат, устройство которого
понятно из чертежа (рис. 41), загружают около 4 м3 бензола и.
пропускают хлор при температуре 30—32° в течение 40 часов. По-
лученный после этого продукт имеет уд. вес около 1,0 и состав:
хлорбензола 40—65%, бензола 25—55%, полихлоридов 3—8%.
1 Мы считаем более вероятным образование хлорфенола при действии азо-
тистой кислоты на хлорбензол, аналогично взаимодействию HNO2 с толуолом:
• СН3 СН3 СН3
| | + hno2----
N = N ОН ОН
(см. статью Титова и Барышциковой, Ж. О. X., 68, 1801, 1936).
С1 С1 С1
| + hno2
N = N • ОН ОН
Заметим, что вследствие ускоряющего реакцию нитрации влияния гидроксиль-
ной группы хлорфенол должен пронитроваться до динитрохлорфенола, а не"
хлорнитрофенола, как это утверждает Девернь.
302
Рис. 41. Абсорбер.
I—труба для подачи хлора; 2—внутреннее
охлаждение; 3—наружное охлаждение;
а и 6—полки для катализатора.
После превращения более 50% бензола в хлорбензол усиливается
образование дихлорбензола. Образующийся хлористый водород от-
водится через обратный холодильник в орошаемую водой керамико-
вую башню, где получается соляная кислота крепостью до 22° В&
(уд. вес 1,18 или 35,4% НС1). Полученный продукт хлорирования
нейтрализуется кальцинированной содой для удаления хлороводо-
рода и хлористого железа х. Эта операция производится в чугунном
резервуаре, снабженном механи-
ческой мешалкой. По окончании
перемешивания хлорбензола с до-
бавленной содой отстаивают и вы-
пускают продукт в бак — хра-
нилище хлорированного бензола.
Хлорбензол выпускают из ней-
трализатора через трубу, погру-
женную в него уширяющимся
концом, на который надет колпак
из проволоки, обтянутой плотной
бумажной тканью и холстом, яв-
ляющимся фильтром.
Нейтрализованный, отделен-
ный от осадка продукт поступает
в дестилляционный аппарат для
отгонки хлорбензола. Отгоняю-
щийся здесь бензол с примесью
хлорбензола возвращается в аб-
сорбер для хлорирования. Про-
межуточная фракция, содержа-
щая около 50% бензола и 50%
хлорбензола, добавляется при
разгонке к следующей порции
сырого хлорбензола.
Выход хлорбензола составляет
79% от теории. Выход дихлор-
бензола равен 14% от веса взятого для хлорирования бензола.
При охлаждении дихлорбензола выкристаллизовывается р-ди-
хлорбензол, который применяется для дезинфекции и для производ-
ства красок.
НЕПРЕРЫВНОЕ ХЛОРИРОВАНИЕ БЕНЗОЛА
При хлорировании бензола скорость реакции между хлором и
бензолом и между хлором и хлорбензолом отличается очень мало.
1 Хлористое железо необходимо удалить потому, что оно при нагревании
с водяным паром образует хлороводород:
3FeCl2 + 4Н2О ->Fe3O4 + 6НС1 + Н2О,
который Сильно корродирует дестилляционную установку.
303-
Поэтому при хлорировании бензола в производственных усло-
виях получается до 15% и больше полихлоридов наряду с основным
пр о д ук том—моно хл ор б ензол ом.
. Понижение образования дихлорбензола может быть достигнуто
разными путями:
1) ведением хлорирования в условиях, при которых заметно
увеличивается разница между скоростью вступления первого и
второго атома хлора в бензольное ядро;
2) ведением процесса хлорирования таким образом, чтобы обра-
зовавшийся хлорбензол возможно меньше подвергался действию
новых количеств хлора. Это может быть достигнуто применением
непрерывного хлорирования.
Непрерывное хлорирование дает ряд преимуществ; уменьшение
объема аппаратуры, автоматизация
процесса и др.; поэтому этот спо-
соб подробно изучен и проверен с
Рис. 41а. Аппарат для непрерыв-
ного хлорирования.
положительными результатами в за-
водских условиях.
На рис. 41а показан аппарат для
непрерывного хлорирования. Этот
аппарат представляет собой чугун-
ную колонку, собранную из несколь-
ких царг. На чугунные решетки,
делящие внутренний объем аппарата
на несколько отделений, загружают
катализатор. Бензол поступает свер-
ху через ороситель, хлор—-через
барбатер. Катализатор загружают
через специальные лазы, имеющиеся
у каждой царги.
Охлаждение производится оро-
шением водой внешней поверх-
ности колонны, для чего охлаждаю-
щая вода подводится по трубе а; дальше вода проходит через
кольцевые оросители Ь, стекает по внешней поверхности стенок
царг и собирается в кольцевые желоба с, откуда отводится по
трубе d.
Колонну заполняют сначала бензолом, а затем подают непре-
рывно хлор через барбатер е и бензол через ороситель g.
Хлор и бензол движутся навстречу друг другу (по принципу
противотока). Образующиеся газообразные продукты удаляются
из верхней части колонны, а хлорированная жидкость вытекает
из нижней.
Процесс регулируется скоростью подачи хлора и бензола и
отвода прореагировавшей жидкости и контролируется измерением
температуры во всех царгах колонны1.
1 По вопросу о непрерывном хлорировании бензола см. Ворожцов,
Зильберман и Григорьев, Ж. Пр. X., 8, 872, 1935.
304
5. Получение р-дихлорбензола. Полихлориды от производства
хлорбензола содержат 60% р-дихлорбензола, 35% о-дихлорбензола
и 5% хлорбензола.
Для получения р-дихлорбензола сливают полихлориды при 40°
небольшой струей на струю холодной (водопроводной) воды. При
этом выкристаллизовывается р-дихлорбензол. Смесь поступает на
нутч-фильтр, где отжимают р-дихлорбензол от эвтектики до дости-
жения температуры плавления р-дихлорбензола 45°. После этого про-
мывают на нутч-фильтре водой с температурой 25—30°.
По достижении точки плавления 51—52° р-дихлорбензол упако-
вывают в оцинкованные барабаны.
. Жидкие полихлориды применяются для уничтожения сусликов
(лучше и удобнее сероуглерода) и могут применяться для понижения
точки затвердевания отравляющих веществ в снарядах.
6. Нитрование хлорбензола. Производство динитро-
хлорбензола на заводе в Сан-Фонс во Фран-
ции. Производство велось в две фазы: 1) получение моно нитрохлор-
бензола, 2) нитрование полученного моно- в динитро хлорбензол.
Получение мононитрохлорбензола (1-я
нитрация). Смесь для 1-й нитрации приготовлялась из отрабо-
танной кислоты от 2-й нитрации'(или серной кислоты) и азотной
кислоты и имела состав: 65,0% H2SO4, 18,2% HNO3, 2,3% NO3,
14,5% Н3О.
Эта смесь соответствует 77% H2SO4 в отработанной кислоте (без
учета того, что часть серной кислоты связывается NO2 в нитрозил-
серную кислоту).
Слив смеси к хлорбензолу производится в пределах 30—34°,
выдержка при температуре 60° в течение 20 минут. Мононитрохлор-
бензол сепарируется и передается в аппарат 2-й нитрации.
Состав отработанной кислоты: 75,0% H2SO4, 0,3% HNO3, 2,2%
NO3, 22,5% Н2О 4-нитротела.
2-я нитрация. К мононитрохлорбензолу добавляется при
температуре 75° смесь состава: 71,5% H3SO4, 21,5% HNO3,
7;о% н2о.
Эта смесь соответствует содержанию 84,5% H2SO4 в отработанной
кислоте. Азотная кислота берется из расчета 124,5% от теории.
Выдержка 45 минут при 95°.
Затем охлаждается до 75—80° и передается в бак, в котором
температура реакционной массы поддерживается между 50 и 60°
до исчезновения эмульсии. Если эмульсия не исчезает, то нагревают
до 80° и дают остыть. По исчезновении эмульсии производят сепа-
рацию.
Выход динитрохлорбензола 172%, по весу хлорбензола, или
95,5% от теории.
7. Техника безопасности. Соприкосновение с динитрохлорбензо-
.лом вызывает экзему, а ожоги кислотой, содержащей этот продукт,
весьма опасны. Такие ожоги необходимо промывать теплым, а потом
касколько возможно горячим раствором соды.
Химия—13—20 305
Полы в цехах, где ведется работа с динитрохлорбензолом, должны
содержаться влажными во избежание пыления.
Некоторые предосторожности в производ-
стве. По данным Деверня \ на французском заводе в Сан-Фонс
произошел взрыв резервуара с кислотной смесью, приготовленной
на отработанной кислоте. Взрыв сопровождался длинным светя-
щимся пламенем. По данным расследования при этом, повидимому,
имели место: 1) взрыв, 2) воспламенение плававшего на поверхности
кислотной смеси динитро хлорбензола. Повидимому, взорвалась смесь
окислов азота, воздуха и водорода; последний мог образоваться от
действия слабых кислот на стенки аппарата. Слабая же кислота могла
образоваться при конденсации воды в трубопроводе со сжатым воз-
духом.
Воспламенение указанной взрывчатой газовой смеси могло быть
вызвано воспламенением упавшего в кислоту кусочка каучука от
пробки. В связи с этим Девернь рекомендует следующие предосто-
рожности в производстве динитро хлорбензола.
1) совершенно избегать каучуковых соединений;
2) установить конденсационный горшок на трубопроводе для сжа-
того воздуха;
3) очистить насколько возможно полно отработанную кислоту от
динитро хлорбензола.
8. Производство динитрохлорбензола в одну фазу * 2. Нитрование-
хлорбензола производилось смесью состава: 70% H2SO4 и 30%
HNO3, соответствующей содержанию 89% H2SO4 в отработанной
кислоте. Нитрование производилось с теоретическим количеством
азотной кислоты. Выдержка при 85°. Получался выход 170% по
весу хлорбензола, температура затвердевания динитрохлорбензола
была между 46,5—47°.
Другой способ производства динитро-
хлорбензола. Для нитрования применяют смесь с фактором
нитрующей активности 83% H2SO4 (например, смесь следующего
состава: 65,7 ± 0,5% H2SO4, 28 ± 0,5% HNO3, около 0,8% NO3,
5,5 ± 0,5% Н2О. Смесь дозируют из расчета 245% HNO3 от теории.
К смеси при работающей мешалке и полном охлаждении при 16—20°
начинают спуск хлорбензола. Температуру равномерно поднимают
до 40° и поддерживают на этом уровне до конца слива; лишь при
спуске последней порции хлорбензола поднимают температуру до 45°.
По окончании слива реакционную массу нагревают до 105—1100,
и делают выдержку до достижения температуры затвердевания не
ниже 47° (для кислого продукта).
Экстрагирование отработанной кислоты.
Отсепарированная отработанная кислота имеет состав: 79,5—82,0%
H2SO4, 0,5—0,8% HNO3, 0,5—1,0% NO2, 16,8—19,5% Н2О, 0,5% ни-
тротел.
‘Девернь, 2,4-Динитрофенол, его производство, свойства и применение^
Сборник переводных статей «Взрывчатые вещества», 3, 71, Химиздат, 1933..
2 Этот способ применялся во время войны в Германии.
308
Так как эта отработанная кислота поступает на денитрацию, то
предварительно производится обработка ее хлорбензолом для извле-
чения нитропродуктов и азотной кислоты. Для этой цели в обычный
чугунный нитратор подают отработанную кислоту, охлаждают до
45° и сливают хлорбензол так, чтобы температура к концу прилива
не превышала 55—60°. При этой температуре дается 20-минутная вы-
держка, после чего масса передается на делительную воронку. <<Экс-
трагированный» хлорбензол подается на нитрацию, а отработанная
кислота на денитрацию.
Промывка динитрохлорбензола. Промывка ди-
нитрохлорбензола производится в промывных чанах, аналогичных
применяющимся для промывки тротила при периодическом способе
промывки. 4 раза промывается водой при температуре 55—80°.
При 5-й промывке добавляется сода для ускорения отмывки
кислот.
Как при выборе конструкции промывных аппаратов, так и при
ведении процесса промывки надо внимательно учитывать сильную
ядовитость динитро хлорбензола. Поэтому следует вести промывку
в закрытых аппаратах, из которых пары не могут попасть в атмосферу
рабочих помещений.
В случае, если динитрохлорбензол выпускается с завода как го-
товый фабрикат, продукт после промывки поступает в сушилку-от-
стойник, где он подогревается свинцовым змеевиком до 70—80°.
Нагретый продукт отстаивается и сливается в железные бочки для
отправки потребителю.
Омыление динитрохлорбензола. Омыление дини-
трохлорбензола производится в железных клепаных котлах с рубаш-
кой для охлаждения, со змеевиком для нагрева острым паром и ме-
шалкой. На 100 в. ч. динитрохлорбензола берется 42,5 ч. едкого
натра в виде 40%-ного водного раствора.
В котел загружают едкий натр, разбавляют водой до содержа-
ния не более 1С% NaOH; воду предварительно нагревают до
75—80° и загружают потребное количество динитро хлорбензола.
При этом происходит разогревание и температура повышается
до 93—95°. При этой температуре делают выдержку в течение
15—20 минут. Омыление считается законченным, если при по-
верке анализом динитрохлорбензол отсутствует, а содержание
едкого натра в растворе равно 0,4—0,8%. При наличии в реак-
ционной массе неомыленного динитрохлорбензола наряду с нормаль-
ным или повышенным содержанием едкого натра продолжают
выдержку еще 15—20 минут, при пониженном — добавляют рассчи-
танное количество щелочи. Наконец, при повышенном содержании
щелочи и отсутствии динитрохлорбензола прибавляют в омылитель
рассчитанное количество динитро хлорбензола. По получении удов-
летворительного результата анализа омыление считается закончен-
ным, и полученный динитрофенолят передавливают в аппарат для
его разложения. Тотчас после выдавливания фенолята в омылитель
заливают воду в количестве, достаточном для того, чтобы покрыть
307
змеевики. Это делается во избежание воспламенения на змеевиках
смеси сухого фенолята со щелочью.
Разложение динитрофенолята натрия. Разложение
динитрофенолята ведется в деревянном чане, снабженном деревян-
ной рамочной мешалкой и свинцовым змеевиком для охлаждения.
В чан заливают воду и приблизительно 40%-ной серной кисло-
ты, необходимой для нейтрализации фенолята. Количество воды бе-
рется из расчета, чтобы получить сильно разбавленный раствор сер-
ной кислоты. После этого в чан подают динитрофенолят, к концу слива
которого температура в разлагателе должна быть 60—70°. После по-
дачи всего фенолята добавляют серную кислоту до кислой реакции
на конго, поддерживая температуру 60— 70°. По окончании подачи
серной кислоты отбирают пробу для анализа на присутствие нераз-
ложенного фенолята и на кислотность. Фенолят должен отсутство-
вать, а кислотность должна быть в пределах 0,25—0,8%.
При удовлетворительных результатах анализа массу охлаждают
до 40—50°, включив водяное охлаждение. Дальнейшее охлажде-
ние до 30—35° достигается добавлением в аппарат холодной воды;
затем спускают содержимое разлагателя по свинцовому трубопро-
воду на центрифугу.
Фуговка. Центрифуга состоит из стального барабана, покры-
того эбонитом. Кожух железный, внутри освинцованный. Барабан
может быть и медный. Фильтрующий материал — сукно.
Динитрофенол отжимается до содержания 10—20% влажности
и .отправляется на упаковку.
Если динитрофенол предназначен для дальнейшей нитрации,
то содержание влажности после фуговки должно быть минималь-
ным достижимым на центрифуге.
Маточные растворы, уносящие с собой динитрофенол во взвешен-
ном состоянии и отчасти растворенном виде, направляются в отстой-
ники-ло ушки, где собираются значительные количества динитро-
фенола, а отсюда они идут через колодцы в канализацию. Выход
динитрофенола от хлорбензола — 91% от теории (по Венену, от
90 до 95%).
9. Техника безопасности. Девернь рекомендует следующие меры:
1) насколько возможно избегать соприкосновения с динитрохлор-
бензолом;
2) конденсировать все улетучивающиеся при омылении пары;
сильно вентилировать помещение, держать закрытыми чаны для под-
кисления и снабдить их’вытяжными трубами;
3) не допускать попадания динитрофенолята натрия на паропро-
вод, так как динитрофенолят бурно разлагается в присутствии из-
бытка едкого натра (если капнуть каплю едкого натра на нагретый
до 200° динитрофенолят, то он быстро и полностью сгорает); вслед-
ствие этого необходимо тщательно промывать омылители после
каждой операции;
4) обслуживающий персонал должен часто менять белье и каж-
дый день принимать душ.
308
10. Технические условия на динитрофенол, применявшийся во
Франции для сплава DD-60/40. 1) температура затвердевания не
меньше 110°; 2) кислотность в пересчете на H2SO4 не более 0,1%;
3) содержание растворимого свинца не больше 0,0004%; 4) общее
содержание золы не больше 0,2%.
11. Нитрование динитрофенола. В нитратор подается отработан-
ная кислота в количестве 2,5 в. ч. и 1 в. ч. динитрофенола; содер-
жимое нитратора перемешивается для взмучивания динитрофенола
в кислоте и сливается нитрационная смесь состава х: 70% H2SO4,
28% HNO3 и 2% Н2О. Количество смеси берется из расчета, чтобы
в ней содержалось 125% потребной по теории HNO3. Загрузка
динитрофенола около 750 кг.
Начало слива смеси при температуре 50°; постепенно повышают
температуру и заканчивают слив при температуре не выше 80°.
Нагревают до 110° и делают при этой температуре 2-часовую
выдержку.
Весь остальной технологический процесс выполняется так же,
как зто описано для процесса получения пикриновой кислоты из
фенола на крепких кислотах.
12. Расходные коэфициенты и выходы при производстве пикри-
новой кислоты из хлорбензола1 2.
Расходные коэфициенты
На 1 т хлорбензола
бензола........................ 0,878 т
хлора......................... 0,850 »
соды кальцинированной ..... 0,051 »
олеума для сушки ............. 0,494 >>
На 1 т динитрофенола
хлорбензола.................... 0,69 »
олеума........................ 0,76 »
86%-ной азотной кислоты....... 0,9 >>
85%-ного каустика............. 0,72 »
На 1 т пикриновой кислоты из бензола
олеума........................2,6 т
азотной кислоты............... 1,32 >>
бензола....................... 0,61 >>
соды.......................... 0,031 >>
100%-ного хлора............... 0,54 »
85%-ного каустика............. 0,68 »
олеума для сушки хлора........ 0,095 >>
Выходы на разных операциях
1 т бензола дает 1,05 т. хлорбензола, или 71,5% от теории.
1 т хлорбензола дает 1,73 т динитрохлорбензола, или 96% от теории.
1 т хлорбензола дает 1,73 • 0,86=1,49 т ди нитрофенола, или 91% от теории-
1 т бензола дает 1,03 • 1,49 =1,53 т динитрофенола, или 64% от теории.
1 т динитрофенола дает 1,05 т пикриновой кислоты, или 84% от теории.
1 т бензола дает 1,60 т пикриновой кислоты, или 54% от теории.
1 Смесь содержит 56,3% мол. H2SO4 и соответствует содержанию 87,5%
H2SO4 в отработанной Кислоте.
2 Эти данные взяты из материалов времен империалистической войны.
309
t
13. Пикраминовая кислота. Пикраминовая кислота применяется
для приготовления красок и диазодинитрофенола. Получается ча-
стичным восстановлением пикриновой кислоты.
Производство пикраминовой кислоты. Растворяют
1 ч. пикриновой кислоты в 8,5 ч. воды и к раствору добавляют пор-
циями кальцинированную соду до щелочной реакции. Для полноты
реакции нагревают массу до 75°. В том же аппарате (обычно деревян-
ный чан с деревянной рамочной мешалкой и железным змеевиком)
ведется восстановление, для чего охлаждают массу до 45° и при пе-
ремешивании подают раствор дисульфида. Температура должна
быть при сливе не выше 50°. Делают часовую выдержку. При этом
идет реакция:
ONa
o3n/\no2
+ Na2S2 + H2O
ONa
O2N/XN
nh2
4- Na2S2O3
NO2
no2
Для уменьшения растворимости пикрата к раствору добавляют
поваренной соли из расчета 1,75 в. ч. на 1 в. ч. взятой для реак-
ции пикриновой кислоты. Массу охлаждают до 25° и спускают
на нутч-фильтр. Путч-фильтр представляет собой деревянный чан
с дёревянным ложным дном. Фильтрующей поверхностью служит
бельтинг.
Вследствие большого объема жидкости ведут фильтрование в не-
сколько приемов: наполняют до краев нутч-фильтр, перемешивают
в чане реакционную массу, дают ей отстояться и включают вакуум.
Отфильтровав а/3 объема 1-й порции, выключают вакуум и вновь
заполняют нутч-фильтр из чана. Это повторяют до спуска всей массы
из чана. Последний отжим ведут до возможно полного удаления жид-
кости. После этого наливают в нутч-фильтр насыщенный раствор
поваренной соли, тщательно все перемешивают (веслом или лучше
опускающейся мешалкой), дают отстояться и, наконец, включают
вакуум для отсасывания рассола. Таким образом промывают еще
два раза.
Промытый продукт переводят для разложения в деревянный чан
с рамочной мешалкой и свинцовым змеевиком. Сюда заливают воду
примерно в 5-кратном количестве к пикраминовокислому натрию,
перемешивают и нагревают до 65°; при этой температуре прибав-
ляют серную (или соляную) кислоту до слабокислой реакции. Делают
часовую выдержку и затем оставляют в покое на 8 часов для кристал-
лизации пикраминовой кислоты.
После этого, предварительно перемешав массу, спускают ее на
нутч; фильтруют через шинельное сукно. После удаления маточного
раствора три раза промывают ее водой.
310
ПРОИЗВОДСТВО Р- И о-НИТРОХЛОРБЕНЗОЛА
1. Нитрование хлорбензола. Процесс нитрования ведется таким
образом, чтобы образовалось минимальное количество динитрохлор-
бензола, присутствие которого вызывает закупорку в дестилляцион-
ной установке и, кроме того, ухудшает качество краски. Поэтому
ведут: 1) прилив смеси к хлорбензолу; 2) слив смеси при температуре
не выше 35°, а выдержку делают при температуре не выше 45°.
Нитрование ведется в обыкновенном чугунном нитраторе. Фактор
нитрующей активности смеси — 75% (например, 63,3—65,4% H2SO4,
18—22% HNO3). Избыток азотной кислоты от теории 2,5%.
Начало слива смеси производится при температуре 18—20°, при-
чем вода для охлаждения подается в рубашку после начала подъема
температуры (за счет теплоты реакции). При преждевременном
охлаждении может иметь место переохлаждение хлорбензола, что
в свою очередь вызовет последующий перегрев при вступлении боль-
шого количества нитросмеси в реакцию и в результате — выброс из
нитратора.
Присутствие в молекуле заместителя — хлора — понижает ре-
акционноспособность молекулы при ее взаимодействии с окислите-
лем, поэтому температуру повышают с 18 до 35° в течение 1 часа.
Эту температуру поддерживают до конца слива и лишь при послед-
ней порции смеси поднимают температуру до 45° и делают при этой
температуре 45-минутную выдержку.
Температура плавления получаемого нитрохлорбензола колеб-
лется в пределах 53—57°. Содержание HNO3 в отработанной кис-
лоте должно быть не больше 1%.
По окончании выдержки содержимое нитратора нагревают до
60—65° (для расплавления нитрохлорбензола) и передают в дели-
тельную воронку, предварительно нагретую до 65—70°.
2. Сепарация кислого мононитрохлорбензола. Верхние царги се-
парационной колонны обогреваются паровым змеевиком для преду-
преждения охлаждения и соответственно затвердевания нитропро-
дукта. Отделенный от кислоты нитропродукт поступает в нейтрали-
затор.
3. Нейтрализация кислого нитрохлорбензола. В чугунный ней-
трализатор, снабженный рубашкой для обогрева и пропеллерной ме-
шалкой (80 об/мин.), загружают предварительно раствор соды (5—
10% Na2CO3), затем подают кислый нитрохлорбензол. После 10-ми-
нутного перемешивания поверяют реакцию по конго. Если реакция
кислая, то добавляют содовый раствор до исчезновения кислотности
и делают 2-часовую выдержку при температуре 60°. По окончании
выдержки передают нитропродукт и часть раствора сульфата из ней-
трализатора в кристаллизатор, предварительно пропарив материаль-
ную линию.
4. Кристаллизация мононитрохлорбензола. Полученный моно-
нитрохлорбензол содержит 33% о-изомера и 67% р-изомера. При
охлаждении происходит кристаллизация р-изомера.
311
Кристаллизатор представляет собой чугунный аппарат с рубаш-
кой для охлаждения и рамочной мешалкой (15 об/мин.). Охлаждают
рубашкой и водой, подаваемой непосредственно в нижнюю часть
аппрата. Вода проходит через нитропродукт, поступает в верхнюю
часть аппарата и оттуда, пройдя ловушку, поступает в канализацию.
Таким образом охлаждают нитропродукт до 16—17°, делают вы-
держку 6 часов и спускают содержимое кристаллизатора на центри-
фугу.
5. Фуговка. Фильтрующий слой — тонкое полотно, покрытое
медной сеткой. Для защиты медной сетки от механических поврежде-
ний при разгрузке центрифуги она покрыта пробивной сеткой из
кровельного железа (или листовой меди).
Продукт, поданный из кристаллизатора, отжимают в течение
5—10 минут, причем вместе с водой отжимается эвтектика, и затем
для более полной отмывки эвтектики (на 2-й скорости) его промы-
вают водой с температурой 45—50°. Промывку прекращают по до-
стижении температуры 81,5° (для о-нитрохлорбензола). Отделив-
шаяся эвтектика, содержащая 60% о-изомера и 4С% /^-изомера, по-
ступает на разгонную колонну.
6. Разгонка эвтектики. Эвтектика, имеющая температуру плавле-
ния 14,5—20°, подается в куб разгонной колонны.
Дестилляционный аппарат состоит из следующих частей: 1) куба,
снабженного паровым змеевиком для обогревания жидкости; 2) ко-
лонны, состоящей из трех царг, заполненных кольцами Рашига;
3) дефлегматора; 4) холодильника; 5) приемников для погона.
В кубе создается вакуум 700—720 .ил< рт. ст., в колонне 720—
730 мм.
В куб разгонной колонны загружают 15 000—17 000 л эвтектики.
Вначале отгоняется вода, затем хлорбензол и, наконец, р-нитро-
хлорбензол. Разница между температурами кипения обоих изомеров
очень небольшая: о-изомера 245,5° (при 760 мм рт. ст.; р-изомера
238,5° (при 760 мм рт. ст.).
Вследствие малой разницы между температурами кипения обоих
изомеров требуется мощная колонна для разгонки. В начале идет
«головка» (вода, хлорбензол), затем р-нитрохлорбензол, промежуточ-
ная фракция и, наконец, «хвост» погона — о-изомер.
Когда вся ортофракция отгонится, выключают вакуум и спускают
кубовые остатки в бочки.
Вся операция разгонки длится 54—77 часов. Каждая из фрак-
ций — пара- и ортопродукта — содержит некоторое количество эв-
тектики, для удаления которой эти фракции передаются в кристалли-
заторы, а отсюда на центрифугу.
7. Технические условия на р-нитрохлорбензол:
1-й сорт 2-й сорт 3-й сорт
Температура затвердевания не меньше 81,5° 81,2°, 81,0°
Содержание влаги не больше 6%.
8. Технические условия на о-нитрохлорбензол: температура за-
твердевания не меньше 31,5°; содержание влаги не больше 8%.
312
НИТРОПРОИЗВОДНЫЕ АНИЗОЛА И ФЕНЕТОЛА
1. Свойства тринитроанизола и тринитрофенетола. Плотность ли-
того тринитроанизола 1,6—1,62.
Температура плавления химически чистого тринитроанизола 71е*
(технического 65—68°), тринитрофенетола 81°. Бризантность три-
нитрофенетола меньше, чем тротила, а бризантность тринитроани-
зола больше, чем тротила, но меньше, чем пикриновой кислоты.
Скорость 1 детонации тринитроанизола 7500 м)сгк.
Чувствительность к удару при весе груза в 2 кг: тринитроанизол
не взрывает при высоте 195 см; при высоте 200 см тринитро анизол:
дал 50% взрывов полных, 25% взрывов неполных и 25% отказов.
Д1ри том же весе груза чистая пикриновая кислота дала взрывы
при высоте 100 см.
По этим данным тринитроанизол занимает промежуточное место
между пикриновой кислотой и тротилом по фугасному действию,,
будучи менее чувствительным к удару, чем пикриновая кислота.
Преимущество тринитроанизола и тринитрофенетола перед пикри-
новой кислотой — их нейтральность, что должно бы позволить
удобно применять их в смесях с аммиачной селитрой. Однако эти
смеси не оправдали себя во время первой империалистической войны,
когда они применялись в Германии. Оба эфира (легче тринитрофене-
тол) постепенно гидролизуются в присутствии влаги с образованием
пикриновой кислоты и соответствующего спирта.
2. Получение ди- и тринитроанизола, ди- и тринитрофенетола.
Медленно вливают раствор едкого натра в смесь динитрохлорбензола
и метилового спирта:
C6H3C1(NO2)2 + NaOH + СН3ОН ->C6H3(NO,)2OCH3 +NaCl +Н2О
Во время реакции температура поднимается от 18—20° до 45°.
Выход динитроанизола 94% от теории. Нитрование динитроанизола
производится так же, как динитрофенола.
Ди- и тринитро фенетол приготовляются вливанием раствора ед-
кого натра в смесь динитро хлорбензола и этилового спирта и получен-
ный динитрофенетол нитруют — все аналогично получению ди- и три-
нитроанизола.
3. Тетранитроанизол. Тетранитроанизол получается, по Бланкс-
ма 2, нитрованием симметричного динитроанизола, получаемого, по
Лобри де-Брюину3, обработкой симметричного тринитробензола
в растворе метилового алкоголя с метилатом натрия.
При нагревании 5 г симметричного динитроанизола в течение
2 часов со смесью 15 см3 азотной кислоты (уд. вес 1,44) и 15 см?
серной кислоты получается тринитроанизол. Вследствие трудности
вступления нитрогруппы (между двумя нитрогруппами) 3-я нитро-
1 Венен, Бюрло и Лекорше, стр. 401, ОНТИ, 1936.
2 Blanksma, Rec- Trav. Chim. Pavs-Bas, 9, 208, 1890.
3 Сообщения Академии наук Амстердама От 28 марта 1903 г.
313:
группа становится при нитровании в ортоположение по отношению
к метоксильной группе. Эта нитрогруппа вместе с тем должна быть
подвижной вследствие совместного влияния на нее двух нитрогрупп,
из коих одна стоит в ортоположении, а другая в параположении к
введенной 3-й нитрогруппе. Поэтому полученный 2,3,5-тринитро-
анизол легко реагирует с раствором метилата натрия в метиловом
спирте, с аммиаком (при обработке спиртового раствора тринитроани-
зола с водным раствором аммиака), с метиламином и другими
веществами:
ОСН3
ОСН3 |
| I NO2 -(.раствор CH.ONa I ОСН3
--------?--->
в метиловом-„
O„N—J—NO, спирте O,N I — NO,
\/ 2 \/
или при обра-
ботке с водным
раствором амми-
ака
------------------>
температура
плавления 104°
осн3
o2n!\Z-no2
или с метил-
амином
ОСНз
/\- NHCH3
O2N— -NO,
При нитровании 2,3,5-тринитроанизола серноазотной смесью
получается 2,3,5,6-тетранитроанизол, причем образуются две мо-
дификации: одна с температурой плавления 112°, другая 154°. При
перекристаллизации полученного при нитровании и промытого про-
дукта из спирта получаются обе модификации. Вещества с температу-
рой затвердевания 112° легко переходят в модификацию с темпера-
турой плавления 154° посредством перекристаллизации из бензола.
В 2,4,5,6-тетранитроанизоле более подвижными являются две
нитрогруппы, стоящие в метаположении к метоксильной группе.
При обработке тетранитро анизола раствором метилата натрия в ме-
тиловом спирте получается:
ОСН3
I ОСН3
O3N j/ZVX'j NO2 + СН ONa O2N NO2
O2N Ж НзСО^/- och3
Температура плавления 112 или 154°.
По Клессену1 при нитровании m-нитро анизола получается
2,3,4,6-тетранитроанизол. Тетранитроанизолы обладают высокой
бризантностью; однако они недостаточно стойки, притом очень до-
роги, вследствие чего и не находят себе применения.
1 Герм. пат. №238655, 209466, 1914.
314
ТРИНИТРОКРЕЗОЛ, ИЛИ КРЕЗОЛИТ
1. Крезол. Крезол, получающийся только при пирогеретических
процессах (коксование, полукоксование, газовое производство
и т. п.), состоит из трех изомеров: о-крезола с температурой кипе-
ния 182° (по Джуа) \ m-крезола с температурой кипения 200° (по
Джуа) и р-крезола с температурой кипения 199,5° (по Джуа).
Вследствие близости температур кипения т- и р-крезола перегон-
кой можно отделить только о-крезол.
Обыкновенный технический крезол содержит в среднем: 40%
о-крезола, 35% тп-крезола, 25% р-крезола.
После отгонки ортокрезола получается смесь, содержащая 60%
щ-крезола и 40% р-крезола 1 2. Как о-крезол, так и р-крезол не дают
при нитровании тринитронроизводных (можно получить моно- и
динитропроизводные при нитровании, но они при дальнейшем ни-
тровании окисляются азотной кислотой в щавелевую кислоту); и
только тп-крезол дает при нитровании тринитро-тп-крезол.
Вследствие этого производство тринитрокрезола связано со зна-
чительным расходом азотной и серной кислот.
2. Свойства тринитрокрезола. Химически чистый тринитро-т-
крезол плавится при температуре 105—106°, технический трини-
трокрезол плавится при температуре около 100°. Он плохо раство-
рим в холодной воде (1 : 449), немного лучше в горячей (1 : 123).
Образует соли с металлами, но труднее, чем пикриновая кислота.
Все его соли (за исключением аммониевых и основных алюминие-
вых солей) чувствительней к удару, чем тринитрокрезол.
Изменение чувствительности разных солей следует тому же по-
рядку, как и пикраты, а именно: наиболее чувствительны соли тяже-
лых металлов, а из них — соль свинца; затем следует железо, медь,
серебро, щелочноземельные металлы, щелочные металлы.
К трению наиболее чувствительны соли свинца и калия; затем
следуют соли кальция, серебра и бария.
Остальные соли (подобно тринитрокрезолу) ведут себя при испы-
тании в фарфоровой ступке индиферентно.
3. Применение тринитрокрезола. Во Франции прежде (до обо-
рудования установок Брежа) применялся сплав пикриновой кис-
лоты с тринитрокрезолом под названием крезилита. Например,
смесь из 40% пикриновой кислоты и 60% тринитрокрезола (крези-
лит 60/40) плавится при 85°, делается пластичной при 65—70°,
что является ценным свойством этого сплава при снаряжении, так
как позволяет легко получить плотный заряд.
4. Производство тринитрокрезола. Производство тринитро кре-
зола сходно с таковым пикриновой кислоты. Выход тринитро крезола
получается около 110% по весу взятого крезола. Малый выход объяс-
няется окислением р-крезола.
1 Температуры плавления: о-крезола 31°, р-крезола 36°, т-крезол— жид-
кость при обыкновенной температуре
2 Способ определения т.-крезолов в смеси см. Z. Angew. Chem., 759,1900.
315
По данным Эскалеса 1 при нитровании чистого m-крезола полу-
чается выход 150%; теоретический же выход — 220% (считая на
100 в. ч. исходного т-крезола).
С целью уменьшения расхода азотной кислоты на окисление
р-крезола -и крепкой серной кислоты, необходимой для связывания
реакционной воды, предлагались методы выделения р-крезола обра-
боткой крезола олеумом. При этом р-изомер дает кристаллическое
сульфосоединение, слаборастворимое в кислоте, m-изомер дает рас-
творимое в кислоте сульфо соединение. Затем отделяют на центрифуге
или вакуум-воронке кристаллическое соединение. При нитровании
раствора получают тринитро-т-крезол с хорошим выходом; из кри-
сталлического сульфо соединения можно нитрованием получить ди-
нитрокрезол, или гидролизом с водяным паром выделить р-крезсл.
1 Escales, Nitrosprengstoffe, 1914.
ГЛАВА XIV
НИТРОПРОИЗВОДНЫЕ АРОМАТИЧЕСКИХ АМИНОВ
И ИХ ЗАМЕЩЕННЫХ
НИТРОПРОИЗВОДИЫЕ АНИЛИНА
1. Анилин, являющийся исходным продуктом для получения
некоторых нитро анилинов, представляет собой бесцветное, трудно
растворимое в воде масло, со слабым ароматическим запахом, кипящее
при 182°; температура плавления 6°. При действии воздуха анилин
быстро темнеет. Вдыхание значительных количеств анилиновых па-
ров может вызвать явления отравления (головокружение, состояние
возбуждения). Для открытия анилина имеется ряд цветных реак-
ций: раствор хлорной извести окрашивает водный раствор анилина
в фиолетовый цвет, а двухромовокислый калий и серная кислота
окрашивают его сначала в красный, а затем в синий цвет.
При нагревании анилиновой соли с метанолом в автоклаве полу-
чаются моно- и диметиланилин; причем в зависимости от относитель-
ного количества взаимодействующих веществ, от давления и от
температуры реакции образуется больше или меньше моно- или
диметиланилина:
G6H5NH„ • НХ + СН3ОН -> G6H5NHGH3 • НХ + Н2О
C6H5NH2 • НХ + 2GH3OH ->GeH5N(GH3)2 • НХ + 2Н2О
При нагревании равномолекулярных количеств анилина и соля-
нокислого анилина в автоклаве при 200—230° образуется дифенил-
амин:
G6H5NH2 • НС1 + С6Н5 — NH2 -> С6Н5 — NH — С6Н5 + NH4G1
Аминогруппа, так же как и гидроксил, облегчает хлорирование
ароматического ядра; ароматические амины, как правило, чрезвы-
чайно легко вступают в реакцию с галоидами. Для проведения про-
стого хлорирования реакцию необходимо вести с ацетильными про-
изводными данного амина. При хлорировании ацетанилида обра-
зуется наряду с некоторым количеством о-хлоранилида, главным
образом, р-хлоранилид; при дальнейшем действии хлора получается
2,4-дихлор-, а затем 2,4,6-трихлоранилид.
317
Атомы хлора, находящиеся в орто- и параположениях к амино-
группе, сильно ослабляют основные свойства аминов, но хлор в ме-
таположении оказывает лишь незначительное влияние на основ-
ность амина.
Сульфирование ароматического ядра затрудняется аминогруп-
пой.
Анилин-р-сульфокислота образуется при нагревании анилина
с концентрированной серной кислотой до 200°. Реакция протекает
с образованием промежуточного продукта — бензолсульфаминовой
кислоты, которая перегруппировывается затем в сульфаниловую
кислоту 1:
N;
NH2
NH,
I I + H2SO4
SO3H
сульфаниловая кислота
бензолсульфаминовая
кислота
Анилин-т-сульфокислота, или метаниловая кислота, получается
из нитробензола путем сульфирования и последующего восстанов-
ления.
Труднее всего получается о-изомер; исходным материалом для
его получения является р-броманилин, который сначала сульфи-
руют, а затем восстановлением удаляют из полученной сульфокис-
лоты бром:
NH2
NHa
SO3H
NH2
'XSO3H
Br
Br
анилин-о-сульфокислота
Все попытки сульфировать нитроанилин и его замещенные (в ами-
ногруппе) дали отрицательные результаты 2.
Подобно тому как аммиак реагирует с альдегидами с отщепле-
нием воды и с образованием многочисленных аминопроизводных,
так и амины реагируют с альдегидами с отщеплением воды. Ани-
лин легко реагирует с формальдегидом с образованием красивого,
с шелковистым блеском вещества — анги дроформальдегиданилина 3:
c6ii5n.h7; OCH2-*C6H5N = СН2 + Н2О
. ангидроформаль-
дегиданилин
1 О перегруппировке аналогичного характера см. стр. 333—334.
2 Wi tt и W i tt е, Ber., 41, 3090, 1908.
2 Tollens, Ber., 17, 653, 1884.
318
При восстановлении этого соединения 1 цинковой пылью в кон-
центрированном водном растворе едкого натра получают мономе-
тиланилин; выход составляет до 55% от теории.
2. Ацильные производные ароматических аминов. Первичные и
вторичные ароматические амины могут быть ацилированы, т. е.
один водородный атом может быть заменен на различные кислотные
остатки. Для этой цели нагревают амины с безводными кислотами
или обрабатывают их хлорангидридами или же ангидридами кис-
лот:
C6H5NH24-HOOCH->C6H5NH -^СОН 4-Н2О
форманилид
CeH5NH2 + (СН3СО)2О ->C6H5NH - СОСНз + СНзСООН
ацетанилид
2C6H5NH2 + С1СОС6Н5 C6H5NHCOC6H5 + C6H5NH2HC1
б енз ои лани лид
Ацилированные амины нейтральны и не растворяются ни в вод-
ных растворах щелочей, ни в кислотах; при длительном нагрева-
нии как щелочи, так и кислоты омыляют ацильные производные
аминов.
Омыление проходит легче в кислой среде, чем в щелочной.
Водород, связанный с атомом азота ацилированного амина, мо-
жет быть в безводной среде заменен на атом щелочного металла.
Получающиеся таким образом натриевые или калиевые соединения
C6H5NNa—СОСН3 снова полностью разлагаются при действии
воды.
При действии алкилирующих средств на эти металлические со-
единения происходит замещение металла на алкил:
,Na .СН3
C6H5N< + CH3J -> C6H5N( 4- NaJ
XCOCH3 XCOCH3
3. Ароматические производные мочевины получаются при взаимо-
действии аминов с фосгеном или арилизоцианатом:
/С1 h2nc6h5 ЛНС6Н5
СО< + ----> со<
ХС1 h2nc6h5 xnhc6h5
ЛНС6Н5
C6H5N = с = о + h,nc6h4ch3 -> со<
XNHC6H4CH3
4. Мононитроанилины. Положение, занимаемое нитрогруппой по-
отношению к аминогруппе, зависит от способа нитрования и от рода,
исходного материала.
1 Герм. пат. № 75854 и фр- пат. № 212506.
О побочных реакциях и механизме реакции см. Frankland, Chal-
lenger и Nicholls, J. Chem- Soc., 115, 198, 1919-
31»
При нитровании азотной кислотой анилинсульфата в растворе
концентрированной серной кислоты получаются в приблизительно
равных количествах т- и р-нитро анилины.
При нитровании ацилированного анилина (например, форм- или
ацетанилида) образуется наряду с небольшим количеством о-нитро-
соединения, главным образом, р-нитроацетанилид:
и небольшое количество о-нитроацетанилида
;Лсосн3
( 4,NO„
I I '
о- и р-нитроанилин могут быть легко получены путем нагревания
с аммиаком соответствующих изомеров мононитрохлорбензола, так
как в этих соединениях ослаблена связь атомов хлора с ядром вслед-
ствие присутствия нитрогруппы.
Общие сведения о свойствах мононитро-
а н и л и н о в. Нитрогруппы, находящиеся по отношению
к NH2-rpynne в орто- и параположениях, ослабляют основные
•свойства аминов, причем особенно сильно сказывается в этом отно-
шении влияние заместителя в ортоположении; однако т-нитроани-
лин обладает почти такой же основностью, как анилин. Различная
основность нитроанилинов может быть использована при их разде-
лении.
Аминогруппа в о- и р-нитроанилине легко может быть заме-
щена на гидроксил при нагревании этих соединений с щелочами.
Нитрогруппа ослабляет также связь аминогруппы с ядром, причем
аналогично другим подобным случаям (например, в случае замести-
теля хлора) эта связь тем больше ослабляется, чем больше нитро-
групп находится в ядре. Поэтому гидролитическое отщепление
аминогруппы протекает особенно легко в случае 2,4,6-тринитро-
анилина (пикрамида).
Мононитроанилины представляют собой хорошо кристаллизую-
щиеся вещества желтого цвета, мало растворимые в воде, хорошо—
в спирте.
320
о Н ит р о а ни л и н, оранжево-желтые иглы, температура
плавления 71°.
m-Нитроанилин, желтые кристаллы, температура плав-
ления 114°. Применяется для получения некоторых азокрасок и
т-фенилендиамина.
р - Нитро анилин, желтые кристаллы, температура плав-
ления 147°. Применяется для получения красителей и р-фенилен-
диамина.
5. Действие окислителей на органические основания. Многие ор-
ганические основания легко окисляются в щелочном или нейтраль-
ном растворе, но стойки в кислом растворе. Это можно объяснить
тем, что они, обладая основными свойствами и соединяясь с кисло-
тами в продукты присоединения, являются ненасыщенными и, как
таковые, легко окисляются. При растворении этих оснований в ми-
неральных кислотах образуются соли, причем ненасыщенный 3-атом-
ный азот оснований переходит в насыщенный 5-атомный1 азот
соли.
Более сильные основания лучше могут быть защищены от окис-
ляющего действия путем переведения их в соли; например, сернокис-
лые соли этиламина на холоду не восстанавливают КМпО4, между
тем как соли более слабых оснований как гидроксиламина, гидра-
зина, значительно легче окисляются.
В ароматических соединениях реакционно способный арил дей-
ствует совместно с ненасыщенным азотом, чем обусловливается лег-
кая разлагаемость ненасыщенных ароматических оснований, как,
например, анилина и его замещенных. Ароматические основания
значительно слабее, чем алифатические; это, например, выражается
в том, что соли ароматических оснований в водном растворе большей
частью распадаются на свободное основание и свободную кислоту.
Поэтому, хотя и можно затруднить окисление ароматических осно-
ваний нейтрализацией их минеральными кислотами, но не удается
затруднить в такой мере, как в более сильных алифатических. Лучше
всего защищает от окисления концентрированная серная кислота или
дымящая соляная кислота2 (разумеется, последняя только в тех слу-
чаях, когда окислителем является не азотная кислота).
Ароматические амины легко реагируют с азотной и азотистой
кислотами с образованием нитро- и нитрозопроизводных; поэтому
они применяются в качестве стабилизаторов азотнокислых эфиров,
входящих в состав бездымных порохов.
1 На степень насыщения атома азота влияют у отдельных оснований сила
основания и кислоты. Если азот в индиферентных веществах вообще не соеди-
няется с кислотами, то он насыщен трехатомно. Ароматические аминыхаракте-
ризуются значительно более слабой основностью, чем амины жирного
РяДа. *
2 В производстве нитрогоединений ароматических аминов последние перед
нитрованием либо нейтрализуются крепкой серной кислотой, либо произво-
дится предварительное ацилирование аминов. В обоих случаях сильно умень-
шается наклонность молекулы н^окислению или осмолению.
Химия—13—21 321
При алкилировании в аминогруппе анилина прочность соедине-
ния увеличивается: при взаимодействии с азотной кислотой легче
всего окисляется и осмоляется анилин, прочнее моно метил анилин
и наиболее прочным является диметиланилин. Этим объясняются
пониженные выходы тетрила при нитровании монометиланилина
сравнительно с диметиланилином.
6. Нитрование анилина азотной кислотой. Гюбнер показал х, что
незамещенный в аминогруппе анилин можно нитровать в том слу-
чае, если его вводить в реакцию с азотной кислотой в виде раствора
в концентрированной серной кислоте. Реакцию ведут таким обра-
зом, что сернокислый раствор анилина по каплям подают при силь-
ном охлаждении к серноазотной смеси, содержащей теоретическое
количество азотной кислоты, необходимое для введения одной
нитро группы.
При этом получается значительное количество р- и т-нитроани-
лина 1 2 и меньшие количества о-нитроанилина.
Известно, что аминогруппа ориентирует в орто- и параположе-
ния. Образование же большого количества тге-нитроанилина указы-
вает на то, что группа — NH2H9SO4 обладает в значительной мере
метаориентирующим действием, причем было установлено, что
это действие усиливается при увеличении концентрации и коли-
чества серной кислоты, в которой растворен анилин. Кроме того,
и при нитровании замещенных анилина (ацетиланилида, диметил-
анилина) при применении больших количеств концентрированной
серной кислоты наблюдается тенденция ко вступлению нитрогруппы
в метаположение к аминогруппе 3.
При нитровании анилина в присутствии слабой кислоты, а именно,
ледяной уксусной кислоты, и при применении уксусного ангидрида
в качестве водоотнимающего средства образуется 75% о-нитроани-
лина и 25% д-нитроанилина.
7. Производство р-нитроанилина. Так как аминогруппа сооб-
щает молекуле анилина способность к легкому окислению и осмоле-
нию при взаимодействии с азотной кислотой, то ее приходится пе-
ред нитрованием «защищать». Это достигается очень удобно, если
вместо анилина подвергают нитрованию ацетанилид или форманилид
(последний дешевле, поэтому предпочтительней); после нитрова-
ния ацильную группу удаляют омылением.
Н
Форманилид C6H6N<f представляет собой нейтраль-
. ХСОН
ное вещество с температурой плавления около 43—45°.
1 Н u b п е г, Вег., 10, 1716, 1877.
2 По W i 11 и W i t t e (Ber., 41, 3090, 1908), при нитровании раствора
анилина в концентрированной серной кислоте до 45% его превращается
в m-нитроанилин.
3 W i 11 и [Hermann, Вег., 39, 3901, 1906.
322
форманилид передается в расплавленном виде для растворения
в верной кислоте в нитратор, куда предварительно загружается мо-
ногидрат серной кислоты. Процесс растворения ведется при темпе-
ратуре не выше 5°. При более высокой Температуре происходит
разложение форманилида (омыление) на анилин и муравьиную кис-
лоту, а присутствие анилина отражается неблагоприятно как на
течении процесса нитрования, так и на качестве конечного продукта.
По окончании растворения охлаждают массу в нитраторе до—1°
и при этой температуре начинают подачу азотной кислоты, регули-
руя скорость слива таким образом, чтобы температура реагирую-
щей массы не поднималась выше —1°; при более высокой темпера-
туре образуются смолистые примеси, загрязняющие продукт. Кроме
того, увеличивается количество образующегося о-нитроанилина,
что также понижает качество р-нитроанилина. По окончании подачи
азотной кислоты делают 30-минутную выдержку при температуре
не выше нуля.
По окончании процесса нитрования в нитраторе получают р-ни-
троформанилид, растворенный в серной кислоте. -
Способ выделения р-нитроформанилида из его раствора в серной
кислоте основан на уменьшении его растворимости при разбавлении
этой кислоты. Разбавляют полученный при нитровании раствор
большим количеством воды при охлаждении. Температура при этом
не должна подниматься выше 4°, так как при более высокой
температуре р-нитроформанилид омыляется с выделением р-нитро-
анилина по реакции:
б присутствии сер-
ной кислоты
+ нсоон
р-Нитроанилин образует с серной кислотой растворимую серно-
кислую соль
NH2 • H„SO4
которая останется в растворенном виде в маточном растворе при
фильтровании нитроформанилида, что понизит выходы р-нитро-
анилина.
При разбавлении раствора р-нитроформанилид выпадает в виде
мелкокристаллического осадка бледножелтого цвета.
323
Так как и при пониженной температуре кислотное омыление
продолжает итти, хотя и медленно, то надо время операции разбав-
ления уменьшить в максимальной мере и тотчас по окончании про-
цесса разбавления отфильтровать выделившийся продукт, промывая
водой до нейтральной реакции.
К полученному продукту добавляют воды, тщательно перемеши-
вают и посредством сжатого воздуха передают в омылитель.
Омыление р-нитроформанилида можно вести и одной водой при
нагревании; процесс ускоряется в присутствии едкой щелочи.
Кроме того, при отсутствии щелочи освобождающиеся р-нитро-
анилин и муравьиная кислота образуют растворимую в воде соль:
NH2 •НСООН
NO2
(муравьинокислый р-нитроанилин).
В присутствии едкого натра эта соль разлагается с образованием
муравьинокислого натрия и из раствора выпадает малорастворимый
в воде р-нитроанилин.
Омыление ведется в 3%-ном растворе едкого натра при темпе-
ратуре 80°.
По окончании процесса омыления содержимое аппарата охлаж-
дают при работающей мешалке до 45°, оставляют в покое в течение
20—25 минут и спускают раствор, содержащий избыток щелочи,
муравьинокислый натрий и о-нитроанилин. Так как о-нитроанилин
сильнее растворим в воде, чем р-нитроанилин, то повторной
промывкой продукта водой можно очистить продукт от этого
изомера.
Обычно после слива маточного раствора массу промывают еще
один раз водой и сифонируют при температуре 40°.
После этого переводят продукт на центрифугу, промывают его
водой и отжимают.
Производство р-н и т р о а н ил и н а из р-н итрохлорбен-
зо ла. Это производство основано на замене хлора аминогруппой
по реакции:
Cl
NH2
Расплавленный в специальном плавильном котле р-нитрохлор-
бензол передавливают в стальной автоклав, подают нашатырный
спирт, герметизируют автоклав и нагревают паром помощью ру-
324
башки. По достижении в автоклаве 100° пускают в ход мешалку и
массу нагревают до 175—180°; при этой температуре дают выдержку
в течение 12 часов.
После спуска давления в автоклаве до атмосферного и охлажде-
ния до 60° выдавливают массу на друк-фильтр, где сырой р-нитро-
анилин отжимают от маточника (раствора хлористого аммония) и
промывают холодной водой.
Выход от теории по нитро хлорбензолу до 95%.
8. Производство о-нитроанилина. Аммонолиз о-нитрохлорбен-
зола производится аналогично описанному для р-нитроанилина.
После спуска давления в автоклаве и охлаждения до 18—20° пере-
давливают реакционную массу на нутч-фильтр, где ее отжимают
от маточника, промывают водой, вновь отжимают и выгружают
в деревянные бочки.
Выход от теории по о-нитрохлорбензолу 80—85%.
Готовый продукт содержит 98—99% о-нитроанилина.
9. Частичное восстановление нитросоединений. Известно, что
при восстановлении полинитро соединений в щелочной среде сер-
нистыми щелочными металлами восстанавливается лишь одна ни-
трогруппа до аминогруппы. В качестве восстановителей применяют,
главным образом, дешевые и легко доступные соли: сернистый нат-
рий Na„S, сульфгидрат натрия NaHS и полисульфиды натрия Na^,
Na2S3, Na^, Na2S5.
Реакция между нитросоединениями и сернистым натрием идет
по уравнению:
4Z?N02 4- 6Na,S 4- 7Н,0 4Z?NH„ 4- 3Na2S,O, + 6NaOH
it I 4 1 4 4 1 4 4 0*
Здесь в процессе реакции выделяется щелочь, и потому щелоч-
ность раствора повышается.
При применении полисульфидов восстановительный процесс не
сопровождается выделением щелочи:
#NO2 + Na2S2 + Н20ЯГШ2 + Na2S,O3
• 4*44*4 4*440
дисульфид тиосульфат
натрия натрия
При восстановлении полисульфидом с большим содержанием
серы, чем в дисульфиде натрия, процесс восстановления сопровож-
дается одновременным выделением серы:
7?NO2 + Na2Ss + Н2О -> 7?NH, + 3S + Na2S2O3
пентасульфид
натрия
Так как при наличии в среде щелочи процесс восстановления
может привести к получению значительных количеств нитроазокси-
соединений, то целесообразно применение восстановителей, при
которых процесс восстановления не сопровождается выделением
щелочи.
325
Реакцией частичного восстановления полинитро соединений поль-
зуются для получения m-нитроанилина и пикраминовой кис-
лоты х.
10. Производство ///-нитроанилина. Производство ш-нитроани-
лина состоит из следующих операций:
Приготовление полисульфида ведется путем
растворения серы в водном растворе сернистого натрия при темпе-
ратуре до 95°; эти вещества берут в соотношении, отвечающем по-
лисульфиду состава Na2S2,5:
2Na2S + 3S -> 2Na2S2>5
Необходимо иметь в виду, что сернистый натр, попадая на кожу,
вызывает тяжелое кожное заболевание; поэтому надо разбивку и
загрузку сернистого натра вести обязательно в закрытых очках и
в рукавицах.
Восстановление m-динитробензола проте-
кает по реакции:
NO, NO2
2| I + 2NaaS2,5 + 2Н2О-->2| i + 2Na,S2O3+S
x^/NO, \/NH2
ведется в чугунном или железном аппарате с мешалкой и паровой
рубашкой (или паровым барбатером). В аппарат загружают воду и
динитробензол. Вода берется в 20—30-кратном количестве по весу
динитробензола. Ввиду сильной ядовитости динитробензола и вы-
зываемых им тяжелых кожных заболеваний работу с ним необхо-
димо вести в перчатках, очках и респираторе. Нагревают массу
при 93° до полного расплавления динитробензола. При работающей
мешалке приливают нагретый до 75—80° раствор полисульфида
так, чтобы температура массы поднималась постепенно и равно-
мерно до 91° к концу слива. По окончании прилива полисульфи-
дов массу поверяют на отсутствие динитробензола. В случае нали-
чия невосстановленного динитробензола делают выдержку в течение
15 минут при температуре 94—96°.
После этого нейтрализуют раствор при температуре 93—96° до-
бавлением бисульфита, делают 10-минутную выдержку и опреде-
ляют pH реакционной массы. Реакция должна быть нейтральной или
слабокислой, но ни в коем случае не щелочной.
Затем поднимают температуру до 98—99° и выдавливают массу
сжатым воздухом на друк-фильтр для освобождения от смол. Фильт-
рующий материал — бельтинг 2.
1 С obenz 1, Chem. Ztg, 37, 299, 1913 .
8 Бельтинг — толстое бумажное полотно.
326
Во избежание кристаллизации m-нитроанилина материальные
линии и друк-фильтр предварительно обогревают паром в течение
Ю—15 минут.
Кристаллизация. В деревянный освинцованный чан,
снабженный деревянной мешалкой и свинцовым или железным змее-
виком, загружают хорошо промытый лед в крупных кусках. На
лед подают из друк-фильтра раствор ш-нитроанилина. Как только
лед растает настолько, что станет возможным пустить в ход
мешалку, начинают перемешивание содержимого чана. По дости-
жении температуры 40—45° начинают охлаждать рассолом, а по
достижении 20—25° спускают в центрифугу.
Центрифугирование. В центрифугу со стальным,
гуммированным барабаном (фильтрующий материал—бельтинг) при
400 об/мин. спускают из кристаллизатора раствор с кристаллами
тге-нитроанилина, промывают холодной водой, отжимают при,
1400 об/мин. и выгружают в бумажные мешки, а затем упаковы-
вают в бочки или фанерные барабаны. Выход около 70—77% от
теории по динитробензолу.
Перекристаллизация. Для очистки нестандартного
продукта, а также для получения высококачественного продукта,
не содержащего нерастворимых в соляной кислоте примесей, при-
меняется перекристаллизация.
Для этого в редуктор заливают горячую воду (48 л на 1 кг про-
дукта), нагревают до 98—99° и при этой температуре размешивают
в течение 1 часа. Затем, не прекращая размешивания, выдавливают
раствор на друк-фильтр.
Выход перекристаллизованного продукта от взятого на пере-
кристаллизацию 80%.
2, 4-Динитроанилин,—желтые кристаллы, температура плавления
176°.
11. 2,4,6-Тринитроанилин, или пикрамид. 2,4,6-Тринитроанилин
(пикрамид), температура плавления 192—-195°.
Так как при нитровании раствора анилина в большом количе-
стве серной кислоты получается до 45% тл-нитро анилина, который
не может быть превращен в пикрамид, то для получения последи^о^
исходят из о- или /?-нитроанилина. о-Нитроанилин pearnpyej^Uejr-- ..
гичнее, чем /?-цитроанилин. У
Пикрамид получают по следующему способу Витта и рйоте ;
видоизмененному Голлеманом 1 2. -о
414 в. ч. (3 моля) р-нитроанилина растворяют в 4140 BViipQSptJ
ной кислоты с содержанием 95% H2SO4, отдельно готовят
606 в. ч. калиевой селитры в 4140 в. ч. серной кислоты (95%
охлаждают до 0° до выделения из него значительного количества
KHSO4, после этого начинают сливать к нему раствор /?-нитроани-
лина, поддерживая температуру в пределах 0—5° и сильно переме-
шивая. После слива раствора нитроанилина реакционную массу
оставляют в покое. На следующий день, охладив массу ниже 0°,
1 Wi tt и Wi И е, Вег,. 41, 3090, 1908.
2 Holleman Rec. Trav. Chim. Pays-Bas, 49, 112, 1930.
327
добавляют при механическом перемешивании 4,5 л насыщенного
раствора соли, охлажденной предварительно до 10°, при этом вы-
деляется много соляной кислоты.
Пикрамид выделяется на поверхность в виде мелких кристаллов.
Его отжимают на центрифуге и промывают водой. Выход 510 г
(74,5% от теории). Пикрамид имеет температуру плавления 192—
195°, т. е. он почти химически чист.
Пониженный выход обусловлен частичным гидролизом (за счет
реакционной воды), при котором образуется пикриновая кислота
и аммиак. Кроме того, образуется некоторое количество тринитро-
фенилнитро амина.
Если исходить не из анилина, а из ацетанилида, и, тем пода-
вить промежуточное образование т-нитросоединения, то можно
легко получить любое количество пикрамида прямым нитрованием
ацетанилида (или форманилида). По Голлеману, в этом случае выход,
ацетилпикрамида составляет 78,3% от теории.
12. Отношение полинитроанилинов к диазотированию и свойства
их диазониевых солей. Динитроанилины диазотируются с трудом
и дают при этом плохие выходы солей диазония. Это обусловлено
тем, что основность этих соединений сильно ослаблена имеющи-
мися в ядре отрицательными группами, вследствие чего соли их
отчасти или полностью гидролизуются в водном растворе. В отли-
чие от обычных методов диазотирования в этих случаях ведут реак-
цию, растворяя амин в концентрированной серной кислоте и внося
в раствор твердый азотистокислый калий или раствор нитрозил- /
серной кислоты в концентрированной серной кислоте.
По Витту, растворяют амин в концентрированной азотной кис-
лоте и вносят затем эквивалентное количество восстановителя (на-
пример, пиросульфита калия); этот последний восстанавливает со-
ответствующее количество азотной кислоты до азотистой кислоты,
которая и диазотирует имеющийся в растворе амин. После разбав-
ления реакционной смеси льдом получают водный раствор соли ди-
азония.
До самого последнего времени считали, что пикрамид и анало-
гичные ему по ли нитро амины вообще не способны диазотироваться.
Однако Мисслин установил, что если растворить пикрамид в ледя-
ной уксусной кислоте и прибавить к раствору на холоду по каплям:
раствор нитрозилсерной кислоты, то и этот амин диазотируется.
Полученный таким образом раствор нельзя, однако, разбавлять
водой, но следует потреблять для дальнейших реакций (сочетаний),,
так как вода почти моментально действует на это соединение, от-
щепляя от него одну из нитрогрупп:
N2 • SO4H N2 • SO4H
O2n/\)H
I I +H2O—> I I +HNO2.
XNO3 XNO2
328
„ Такое взаимодействие с водой обусловлено тем, что диазогруппа’
действует разрыхляющим и отщепляющим образом на заместители,.
Находящиеся по отношению к ней в орто положении.
13. Тетра нитроанилин. Способ получения и применения тетра-
Нитроанилина в качестве взрывчатого вещества впервые опублико-
ван Флюршеймом в 1910 г. Этим веществом много и серьезно за-
нимались, однако, оно не нашло себе практического применения.
В США во время империалистической войны был построен завод для
производства тетранитроанилина, однако, ввиду окончания к этому
времени войны завод успел выпустить лишь небольшую партию
этого продукта.
Свойства тетранитроанилина. Тетранитроани-
лин образует желтые кристаллы. Плавится с частичным разложе-
нием при температуре 217—220°.
Его уд. вес 1,867; при прессовании достигается плотность 1,68г
как у тетрила. Не растворяется в воде и плохо растворяется в обыч-
ных органических растворителях. Может быть перекристаллизован
из нитроксилола.
Гигроскопичность его меньше, чем у тетрила: при специально
поставленных опытах тетранитроанилин поглотил в неделю 0,12%.
Н2О при относительной влажности воздуха 85%; тетрил поглотил
в неделю 0,30% Н2О при той же относительной влажности воздуха.
Перекристаллизованные продукты поглотили при тех же условиях
еще меньше влажности: тетранитроанилин 0,023%; тетрил 0,024%.
Вода при обыкновенной температуре действует очень медленно
на тетрзнитроанилин. Кипящая вода действует очень энергично,
превращая его в тринитро-тя-аминофенол. Даже при 50° вода дей-
ствует довольно заметно. При растворении же его в сыром ацетоне
образуется 2,4,6-тринитроаминофенол; то же при нагревании его
с водой. При нагревании с этиловым спиртом образуется тринитро-
аминофенетол.
При нагревании тетранитроанилина с метиловым спиртом обра-
зуется тринитроаминоанизол.
• Нитрогруппа, стоящая в метаположении к аминогруппе, мгно-
венно и полностью удаляется при обыкновенной температуре в вод-
ном ацетоновом растворе уксуснокислого натрия, причем образуется
тринитроаминофенол. Последний превращается в тринитро резор-
цин при взаимодействии с водным раствором едкого кали. Этими
реакциями было установлено строение тетранитроанилина х:
NH2
при взаимодейст-
1'<и„вии е раствором
| CHgCOONa
'\JnO2
NO,
в присутствии
водного рас-
твора едкого
кали
он
o2n/\no2
!
1 /он
no2
1Fliirscheim и Simon, Proc. Chem. Soc., London, 26, 81, 1910.
32»
Легко можно приготовить продукт, выдерживающий пробу Абеля
при 71° в течение 1 часа.
При испытании нагреванием при 100 и 120° в вакууме полу-
чены результаты не хуже, чем для тетрила т: температура вспышки
222—233° по Касту, 222° по Флюршейму (температура вспышки
тетпила 186° ппи па-
раллельном испыта- нии Флюршейма). Чув ствительно сть к удару при испыта- нии на копре при весе груза 30 аз (см. табл): Как видно, чувст- Высота падения груза
вещества 16 см 30 см
Тетранитроанилин . . . Тетрил 0% взрыва 40% » 20% взрыва 60% »
вительность к удару
тетранитро анилина значительно меньше, чем тетрила.
Расширение в бомбе Трауцля, по данным Флюршейма, при на-
веске в 10 г взрывчатого вещества и капсюле-детонаторе № 8:
тетранитроанилин............... 430 см3
тетрил......................... 375 »
пикриновая кислота ............ 297 »
тротил......................... 254 »
Чувствительность к детонации равна таковой же тетрила.
По мнению Солонина, подробно изучавшего свойства тетранитро-
анилина, последний может заменить тетрил и в мирной и в военной
промышленности.
Применение тетранитроанилина 1 2. Тетрани-
троанилин изучался в США во время империалистической войны
с целью использования его для изготовления детонаторов. Там счи-
тали, что способность тетранитроанилина реагировать не умень-
шает его ценности как военного взрывчатого вещества, так как он
должен быть сухим при снаряжении им детонаторов, и, кроме того,
он негигроскопичен. Поэтому он был одобрен в США как замена
тетрила и для его производства был построен упомянутый ранее
завод.
При систематическом изучении в течение ряда лет стойкости
изготовленного на этом заводе тетранитроанилина, хранившегося
на складе в виде 400-г патронов, были получены следующие дан-
ные (табл. 63).
Эти данные указывают на такие изменения, происшедшие после
D-летнего хранения тетранитроанилина: 1) понижение температуры
плавления на 5,90°; 2) повышение влажности с 0,25 до 0,45%;
3) повышение .кислотности с 0,02 до 1,12%; 4) чувствительность
к детонации заметно понизилась; 5) стойкость понизилась настолько,
что пришлось изъять тетранитроанилин из склада и уничтожить
его.
1Fliirscheim, Z. Schiess. u. Sprengst., 8, 185, 1913.
2 Ingraham, Army Ordnance, 11, 59, 1930.
330
Таблица 63
се ss в « ь? Песочная проба
X о Ф о ф ПО ля Испытание « С Ф
3 К _S О н S Ф ® к я к g ф 2 2 о- >-< ф Я! Е- S rt а* О- ф 2 * в « в S М н rt О X С м ф ж ф ф ss* в < iQ ь ‘О *5 'о нагреванием при 135° ЕГ S ф £-8 песка, I ;шего ж э 30 ме
к s и О" Я « К св и О Я о о “ ь*
к « S ® ЧЦ Е-1 С О с в vp О ® 0х- о Q.O.S S и я я Ф И Л Ф д S ® Я О Q
1921 0,02 210,0 0,25 72 0,18
1923 0,05 208,4 0,21 72 Взрыва не было 0,18 42,3
1928 через 5 часов
0,59 204,1 0,35 6 — 0,24 40,9
1929 1,12 204,4 0,45 1 Взрыв через 0,24 38,1
1 минуту
Из сказанного следует, что для практического использования
тетранитроанилина необходимо найти условия, устраняющие воз-
можность взаимодействия его с водой. Так как высушивание его
до содержания 0,02% влажности не представляет большого затруд-
нения, то, быть может, этим путем удастся решить задачу полу-
чения стойкого, пригодного для практического применения тетра-
нитроанилина.
14. Получение тетранитроанилина. Получение тетра-
нитроанилина по Флюршейму. Растворяют 1 в. ч.
иг-нитроанилина, 3 в. ч. KNO3 (или соответствующие количества
NaNO3 или NH4NO3) в 36 в. ч. серной кислоты (уд. вес 1,84) при
температуре 68—70°. После начала реакции охлаждают, чтобы тем-
пература не поднималась выше 100°. По окончании реакции нагре-
вают еще несколько минут при 100°; затем охлаждают и отфильтро-
вывают образовавшийся продукт. Выход тетранитроанилина 130—
140% по весу нитроанилина (теоретический выход, считая по весу
нитроанилина, равен 204%), т. е. около 70% от теории.
По опытам Солонина и Крюкова1 нитрование
пг-нитроанилина удобнее производить азотной кислотой, причем
реакция идет спокойней. Они брали 1 в. ч. ттг-нитроанилина,
31 в. ч. азотной кислоты (уд. вес 1,5) и 36 в. ч. серной кислоты
(уд. вес 1,84).
При нитровании ттг-нитроанилина кислотной смесью сначала обра-
зуется нитрофенилнитроамин, который затем перегруппировывается
в динитро анилин.
Ван-Дуин2 указывает следующий способ получения тетра-
нитроанилина нитрованием анилина. 26 г анилина растворяют в
1 А. Солонина, Технология взрывчатых веществ, ч. V, стр. 91,
Ленинград, 1925.
2 V a n D и у n, Rec. Trav. Chim- Pays-Bas., 37, 1,111, 1917.
331
♦
700 см3 купоросного масла и при температуре 5° (с применением
охлаждающей смеси) добавляют смесь 16 см3 азотной кислоты
(уд. вес 1,49) и 80 см3 купоросного масла. После прибавления всей
смеси массу оставляют в охлаждающей смеси в течение 2 часов,
после чего прибавляют 117 г калиевой селитры при температуре
не выше 50°. Оставляют стоять на день, затем нагревают при 500,
в течение Р/2 часов и оставляют еще на день. Фильтруют, промы-
вают продукт 50%-ной серной кислотой и, наконец, водой. Выход—
26 г тетранитроанилина с температурой плавления 207°.
Очевидно, что и этот способ, при котором на 1 в. ч. анилина
расходуется 55 в. ч. купоросного масла и большое количество азот-
ной кислоты и калиевой селитры, непригоден для производства
тетранитроанилина.
15. Пентанитроанилин впервые описан в 1910 г. х. Более по-
дробные данные об условиях его получения опубликованы в 1928 г.1 2.
3,5-Динитроанилин получен восстановлением симметричного три-
нитробензола сернистым аммонием. При нитровании 3,5-динитро-
анилина, так же как и при нитровании ш-нитроанилина, образуются
нитроамины. Эти нитроамины характеризуются химической не-
стойкостью: они легко воспламеняются при благоприятных усло-
виях. Это воспламенение не происходит при нитровании ш-нитро-
анилина, так как все получающиеся при его нитровании амины
хорошо растворимы в кислотной смеси, и только к концу реакции
выделяется образовавшийся тетранитроанилин, плохо растворимый
в отработанной кислоте, химически стойкий, - не самовоспламеняю-
щийся.
.Иначе протекает процесс при нитровании 3,5-динитроанилина;
получающиеся здесь нитроамины плохо растворимы в кис-
лотной смеси; они выделяются из раствора, вследствие чего их
перегруппировка в соответствующее нитро со единение замед-
ляется.
При этом, если вести процесс с помешиванием, то суспендиро-
ванные в жидкости мелкие кристаллы соединяются в более крупные
частицы, причем они разлагаются со вспышкой. Во избежание этого
явления ведут реакцию нитрации без помешивания. В этом слу-
чае красновато-оранжевый цвет нитроаминов постепенно меняется
на желтый — пентанитроанилина. Чистый пентанитроанилин
имеет золотисто-оранжевый цвет; он плавится с разложением
при 192°.
В нем две нитрогруппы, стоящие в метаположении к аминной
группе, легко замещаются. Например, при действии соды образуется
тринитроаминорезорцинат; при действии водного аммиака легко
получается тринитротриаминбензол:
1 F 1 и г s ch ei m и Simon, Proc. Chem. Soc., London, 26, 81, 1910.
2 Fliir scheim и Holmes, J. Chem. Soc., 3041, 1928.
332
NH2
o2n z\no2
; I + Na,co,
O.,N^ Jno2
no2
NH„
NaO^^ONa
NH2
NH,
2
H2N'X/INH2
no2
* n
Вследствие высокой чувствительности к механическим воз-*
действиям и малой химической стойкости пентанитро анилин не-
пригоден для практического применения.
н,
16. Нитроанилид (фенилнитроамин) C6H5N<^ может быть
получен, по Бамбергеру, обработкой анилина при низкой темпера-
туре смесью концентрированной азотной кислоты и уксусного ан-
гидрида:
NH2
н
2
+ HONO2
+ н20
Это соединение хорошо кристаллизуется и плавится при 46°,
при 100° оно разлагается с выделением азота.
Нитроанилид довольно неустойчив и при действии света или
при нагревании, а также и при обработке кислотами перегруппиро-
вывается в нитроанилин
NO
1 Bamberger и Landsteiner, Ber , 26, 482, 1893. Перегруппировка
фенилнитроанилина в нитроамин является одним из многих примеров интрамо-
лекулярной перегруппировки, состоящей в обмене водородного атома ядра на
заместитель боковой цепи. Аналогичные перегруппировки: образование са-
лициловокислого натрия из фенолуглекислого натрия (С3Н5О • COONa —>
/ОН \ / ZH
—СвН4/ I нитрозооснований из нитрозоаминов ( C3H6N<
xCOONa / \'
/NH2\
----> С6Н4/ I фенолсульфокислоты из фенилсерной кислоты,
\NO /
/ /ОН \
C3H5OSO3H -> свн4<
\ \SO3H /
333
н
NH
2
2
При этом образуется, главным образом, о-нитроанилин и неболь-
шое количество парасоединения.
Его металлические производные отвечают двум десмотропным
формам, в виде которых может реагировать нитроанилид:
,Н
О
2
он
(I) 1
(И)
Соответствующее формуле (I) метильное производное может
быть получено из натрийпроизводного нитро анилида при действии
на него йодистого метила:
Метилнитроанилид плавится при температуре 39° и может быть
путем перегруппировки превращен в о- и р-нитрометиланилин.
При взаимодействии серебряной соли нитроанилида (II) с йоди-
стым метилом образуется метиловый эфир ациформы:
N = nX°
/\ ЧОСН3
Это соединение представляет собой желтую жидкость, очень
нестойкую; оно полностью .разлагается через несколько часов.
ХН
• 1Л ^2
17. Тринитрофенилнитроамин 02N/ \ ГЮ2.Тринитрофенилнитро-
NO,
амин представляет собой твердое кристаллическое вещество;
334
Температура плавления около 91,5°. Химическая стойкость этого*
нитроамина очень мала; он воспламеняется при нагревании между
80 и 100°. Растворяется в водных щелочах, образуя темные пурпу-
рово-красные растворы, из которых можно изолировать характер-
ные соли.
Тринитрофенилнитроамин превращается в тетрил при обработке
метилсульфатом и водной щелочью.
По Джонсу и Вильсону \ тринитрофенилнитроамин можно'по-
лучить с хорошим выходом нитрованием сульфаниловой кислоты.
Растворяют 104 г этой кислоты в 400 сл3 нагретой серной кис-
лоты (уд. вес 1,84); к этому раствору при температуре не выше 15°
приливается 300 г азотной кислоты (уд. вес 1,5). После слива кис-
лоты продолжают перемешивание при температуре не выше 20е*
в течение 24 часов. При дальнейшем стоянии смеси выпадает-
нитроамин в виде мелких кристаллов. Выход 120 г, или 75% от
теории.
Нитрация протекает медленно, и для предотвращения воспламе-
нения необходимо перемешивание.
Этот нитроамин можно также получить нитрованием пикрамида-
Для этой цели растворяют 2 г пикрамида в 18,4 г купоросного
масла и к раствору постепенно добавляют 4,2 г азотной кислоты
(уд. вес 1,5). Через 2 часа выпадают кристаллы тринитрофенил-
нитроамина.
ИСХОДНЫЕ ПРОДУКТЫ ДЛЯ ПРОИЗВОДСТВА ТЕТРИЛА
Диметиланилин, применяющийся в качестве исходного про-
дукта для получения тетрила, получается конденсацией анилина
с метиловым спиртом; при этом в зависимости от температуры реак-
ции и соотношения взаимодействующих веществ получается больше
либо моно-, либо диметиланилина:
CeH5NH2 • НХ + СН3ОН C6HSNHGH3 • НХ + Н2О
. C6H5NH2. НХ + 2СН3ОН -> С6Н5 — N(GH3)2 • НХ 4- 2Н2О
1. Монометиланилин кипит при 193°. При нагревании до 330°’
он частично перегруппировывается в р-толуидин:
NHGH. NH,
9 &
II I 1
сн3
При взаимодействии с уксусным ангидридом он образует аце-
тильное производное; с окислами азота образует нитрозамин.
‘Jones и Willson, J. Chem. Soc., 2277—2279, 1930.
335-
72. Диметиланилин. Уд. вес 0,9553; температура кипения 193,5°;
температура затвердевания 1,96°; температура плавления 1 2,07°.
Параположение к диметиламинной группе обладает в диметилани-
лине большой реакционной способностью. Так, например, при дей-
ствии на него азотистой кислоты образуется зеленый р-нитрозоди-
.метиланилин:
N(CH3)2 N(CH3)2
I >| + HONO------> | I +H2O
Аналогично диметиланилин легко реагирует с фосгеном:
/°1 ~>N(CH3), ^N(CH3)2
СО + 27 -----*С0 + 2НС1
ХС1 <Х1Х(СНз)2 X /Ц(снз)2
кетон Михлера
Диметиланилин обладает также характерной для третичных ами-
'яов способностью образовывать при действии перекиси водорода
аминоксид (диметиланилиноксид). Это соединение обладает основ-
ными свойствами и образует с хлористым водородом вещество, по
своему характеру сходное с солями аммония:
CeH5N(CH3)2 + Н2О2 ->C8H5N(CH3)2 Д- Н2О
II
О
C6H5N(CH3)2 + НС1 [C6H5N(CH3)2]C1
о он
Подобно анилину диметиланилин гораздо труднее сульфируется,
чем бензол и другие ароматические углеводороды. По Смиту, моно-
сульфокислота получается при обработке диметиланилина эквива-
лентным количеством серной кислоты при температуре 180—190°.
При этом получается диметиланилин-р-сульфокислота, представ-
ляющая собой бесцветную кристаллическую массу, плавящуюся2
с потемнением при температуре 257°.
Введение же нескольких сульфогрупп в молекулу диметилани-
лина по настоящее время не удалось.
При взаимодействии диметиланилина с 8—10-кратным коли-
чеством купоросного масла при температуре до 40° легко обра-
зуется сернокислая соль диметиланилина.
1 J о n е s, J. Soc. Dyers a. Colour, 35, 43, 1919.
2Michaelis и Godchaux, Вег., 23, 555, 1890.
33S
Эта соль имеет вид белых кристаллов, температура ее плавле-
ния 91,5°; сильно гигроскопична, хорошо растворима в серной
кислоте, воде и спирте; при действии щелочей разлагается с выде-
лением свободного диметиланилина. Как соль слабого основания
и сильной кислоты легко гидролизуется и имеет кислую реакцию.
При нагревании сухой соли свыше 130° она переходит в суль-
фокислоту:
C6H5N(CH3)2 • H2SO4->C6H4N(CH3)2SO3H + Н2О
При вступлении нитрогруппы в бензольное ядро основной ха-
рактер алкиланилинов (так же как и анилина) ослабляется. Такие
замещенные поэтому не образуют столь стойких солей, как анилин
или алкиланилин; их соли водой частично или полностью разру-
шаются, а некоторые разлагаются при хранении на воздухе.
Например, по данным Мертенса 1 2,4-динитродиметиланилин по-
глощает сухой НС1 из воздуха, а при пропускании над ним тока
сухого НС1 образует солянокислую соль C6H3(NO2)2N(CH3)2 • НС1.
Однако эта соль на воздухе полностью теряет HG1 в 2—3 дня,
а при нагревании до 70° происходит бурное выделение газа и
остается свободный динитродиметиланилин.
3. Получение диметиланилина. Диметиланилин получают путем
обработки анилина метиловым спиртом в автоклаве в присутствии
серной кислоты; кислота играет роль катализатора.
На 2,7 в. ч. анилина берут 1,8 в. ч. метилового спирта и
0,27 в. ч. серной кислоты (с содержанием 92—96% H2SO4).
Применяются стальные автоклавы с чугунными эмалированными
вкладышами, двумя эмалированными гильзами для термометров,
эмалированной трубой (для сжатого газа при передавливании),
двумя манометрами, двумя предохранительными клапанами и двумя
вентилями. После загрузки материалов автоклав закрывают и под-
нимают температуру до 200°; при зтом давление достигает при-
мерно 30 ат. По достижении температуры 200° делают выдержку,
после чего содержимое автоклава охлаждают и ведут отгон избытка
метилового спирта. Для отделения кислоты передавливают содер-
жимое автоклава в сепаратор, откуда отстоявшийся продукт
передается в нейтрализатор для нейтрализации раствором едкого
натра.
В этом котле реакционную массу нагревают в течение 5 час.
при 170° под давлением 8—10 отп; при этом идет расщепление чет-
вертичного аммонийного основания, образовавшегося в процессе
метилирования в результате присоединения диметилсульфата к ди-
метил анилину.
По окончании разложения вторично отгоняют освободившийся
метиловый спирт; затем острым паром отгоняют диметиланилин
<(1-ю фракцию — головку — и 3-ю — окрашенную — смешивают;
1 Mertens, Вег., 19, 2123, 1886.
X имин—13—2 2
337
♦
эта смесь перерабатывается отдельно); диметиланилин отстаивают
в сепараторе, отделяют от водного слоя, сушат твердым каустиком
и затем подвергают окончательной очистке перегонкой в вакууме.
4. Технические условия на диметиланилин (по стандарту
GT 27 \
Главоргхимпрома 12817/
Показатели 1-й сорт 2-й сорт 3-й сорт
Внешний вид Жидкость Светложел- того цвета Жидкость от желтого до светло- коричневого цвета Жидкость от желтого до коричне- вого цвета
Удельный вес при 20° Содержание влаги в объемных 0,955—0,960 0,955—0,965 0,955—0,965
процентах не более . Содержание аммиака Содержание монометиланилина Отсутствие Качествен- ная проба 0,1% Качествен- ная проба 0,5% 2%
не более В пределах 2° в интервале тем- ператур: для 1-го сорта 192—195° для 2-го сорта 191,5—195° для 3-го сорта 191,5—196° Должно перегоняться в объем- Отсутствие о,з% 2%
ных процентах не менее 95% 93% 90%
Упаковка и маркировка. Диметиланилин отпускают в железных
бочках; вес нетто 400—500 кг. На днищах бочек обозначают наименование
завода, название продукта, номер партии, номер бочки, вес брутто, нетто
СТ 27
и ГОХП 2817 •
ПРОДУКТЫ НИТРАЦИИ МЕТИЛ АНИ Л ИНОВ,
ИХ СВОЙСТВА И ПРИМЕНЕНИЕ
1. Нитропроизводные монометиланилина. р-Нитромонометил-
/СН3
N /
ХН
алии: / ; желто-коричневые кристаллы с фиолетовым
Х2
отблеском (из спирта); температура плавления 150—151°; хорошо
растворим в спирте и бензоле.
338
УСН3
N(
XTJ
о-Нитромонометиланилин: ; красные кристаллы с фиоле-
I I 2
товым оттенком; температура плавления 34—35°.
/СН3
N(
m-Нитромонометиланилин: /\ ; желто-красные кристаллы;
* \/no2
температура плавления 65—66°; растворим в горячей воде; легко
растворим в спирте и эфире.
СН3
2,4-Динитромонометиланилин: канареечно-желтые кри-
NO2
сталлы; температура плавления 176—178°.
СИ3
2,6-Динитромонометиланилин: O2N/\i\in > температура пла-
вления 106°.
/СН3
\ тт
2-5-Динитромонометиланилин: ’ температура плавле-
р-Нитро диметиланилин;
стальным отливом; тем-
ния 161°.
2. Нитропроизводные диметиланилина.
(XN;/ ; желтые кристаллы со
' х—Х хсн3
пература плавления .161—162°; растворим
в горячем алкоголе и
бензоле.
_____^°2 СН3
о-Нитро диметиланилин: / ; подвижное оранже-
\----7 хсн3
во-желтое масло; температура плавления 20°.
339
\ ZCH3
m-Нитро диметиланилин: 6 z^K > красные кристаллы;
4----7 СН3
температура плавления 60—61°.
2,4-Динитро диметиланилин: O2N^ z^\ » светложелтые
Х-----Z ХСН3
кристаллы; температура плавления 87°.
no2 сн3
2,6-Динитро диметиланилин:
пения 78°.
; температура плав-
/----\ /СНз
3,4-Динитродиметиланилин: O2N
NO^ СНз
плавления 174—175°,
температура
/-0-?- /СН3
2.5-Динитродиметиланилин: С ; температура плав- NO2 СНз
пения 112°.
3. Нитрование моно- и диметиланилина азотной кислотой. Мер-
тенс1 нитровал моно- и диметиланилин сильно разбавленной азот-
ной кислотой.
При нитровании монометиланилина (10 г монометиланилина рас-
творено в смеси 100 г азотной кислоты и 50 г воды и раствор
осторожно нагревался при 60°) получен тринитромонометилани-
лин. При нитровании диметиланилина (500 г диметиланилина рас-
творили при 0° в смеси 6 л азотной кислоты и 6 л воды и оста-
вили на несколько часов) получен 2,4-динитродиметиланилин.
По Ортону 2, при нитровании диалкиланилинов существенно
важно присутствие азотистой кислоты. По его мнению, здесь возмож-
но образование промежуточного продукта — нитрозосоединения,
ON • C6H4N(CH3)2, которое дальше окисляется в нитро соединение.
4. Свойстватетрияа. физические и физико-химические
свойства тетрила. Химически чистый тетрил имеет вид белых
кристаллов; температура плавления, по Касту, 131,5°; температура
затвердевания, по Касту, 128,3°, по Штетбахеру, 128,6°, по
Джуа, 128,7°.
Прессованием достигается плотность 1,68.
Хорошо растворим в бензоле, толуоле, ацетоне, дихлорэтане,
четыреххлористом углероде, азотной и уксусной кислотах; мало
растворим в спирте; растворимость в воде см. табл. 64.
1 Mertens, Вет., 19, 2123, 1886.
2 Orton, Вег, 40, 370, 1907.
340
Смесь тетрила с тротилом в отношении 1 :1 плавная при 63°.
Химические свойст-
ва тетрила. При нагревании
тетрила со слабым раствором со-
ды получается в незначительных
количествах пикрат натрия; та-
кое же разложение тетрила, хотя
и значительно слабее, происходит
при кипячении с водой Ч
* Примесь от 1 до 10% пикрата
натрия не изменяет чувствитель-
ности тетрила к удару. Примесь
10% пикрата натрия понижает
температуру плавления тетрила
Ноульса* 2, следует, что применение соды в виде слабого водного
раствора для ускорения отмывки от тетрила кислот, как это
практикуется во Франций, является вполне допустимым.
Таблица 66
Растворимость тетрила в различных растворителях
Таблица 64
Растворимость тетрила в воде
Температура °C Содержание тетрила в 100 см3 раствора г
15 0,007
30 0,017
50 0,020
80 0,051
100 0,16
на 0.6°. Отсюда, по мнению
Температура °C В 100 ем3 растворителя растворяется тетрила, г
спирт бензол ацетон дихлорэтан
17 — 4,57
20 0,563 3,55 45,82 —
40 — — — 11,46
50 ( 9,99 111,85 —
60 — —. — 27,82
80 — — — 60,48
При взаимодействии тетрила с едким кали получается пикрат
калия и метиламин:
,СН3
CeH2(NO2)3N( + 2КОН ->C6H2(NO2)3OK + CH3NH2 + KNO3
xno2
Химическая стойкость тетрила. Сырой тет-
рил, т. е. неперекристаллизованный из растворителя, обладает
весьма низкой химической стойкостью: он выдерживает пробу Абеля
лишь в течение нескольких минут, тогда как тетрил перекристалли-
зованный выдерживает эту пробу десятки минут.
Ноульс 2 считает, что причиной этой нестойкости является при-
месь — тетранитрофенилметилнитроамин, — названный им тге-ни-
тротетрилом. По данным Ноульса, тге-нитротетрил выдерживает
пробу Абеля лишь 1 минуту. При прибавлении к образцу тетрила,
выдержавшего пробу Абеля в течение 25 минут, 1% т-нитротетрила
’Van Dnyn, Rec. Trav. Chim- Pays-Bas, 37, I, 111, 1917.
2 Knowles, Ind. Eng. Chem., 12, 246, 1920.
341
это время сокращается до 15 минут, а при прибавлении 10% полу-
чается смесь, выдерживающая эту пробу только 3 минуты.
Ноульс считает пробу Абеля малоценной для определения стой-
кости тетрила и рекомендует, как более надежную, пробу Обермюл-
лера. По его определениям wz-нитротетрил дает в вакууме увеличе-
ние давления на 588 мм в 1 час, а кондиционный (т. е. очищен-
ный перекристаллизацией) тетрил дает только 20—30 мм, откуда
видно, что m-нитротетрил разлагается в 25 раз быстрее, чем чистый
тетрил.
Прибавление небольших количеств пг-нитротетрила к тетрилу
сильно увеличивает скорость разложения последнего в вакууме.
Присутствие в тетриле nz-нитротетрила было установлено на осно-
вании следующих данных.
При варке тетрила с водой с целью его стабилизации на амери-
канском заводе было замечено, что в большинстве случаев про-
мывные воды приобретали темный цвет и часто обнаруживали кис-
лотность даже в том случае, если кислотность была предварительно
уничтожена в данном образце тетрила. При выпаривании этих
вод досуха было получено бесцветное кристаллическое вещество —
тринитрометил нитроаминофенол.
По вопросу о причине образования тге-нитротетрила Ноульс до-
пускает, что некоторое количество m-нитротетрила может образо-
ваться при нитровании диметиланилина; но наряду с этим он счи-
тает более вероятным, что основным источником образования
пг-нитротетрила является примесь монометиланилина в исходном ди-
ме^ил анилине. В действительности, при поверке этого предположе-
ния было установлено, что количество образующегося пг-нитротет-
рила находится в прямой зависимости от содержания монометилани-
лина в исходном сырье.
Отношение тетрила к нагреванию. Приводим
Таблица 66 данные 1 относительно убыли веса при нагрева-
Температура Продолжительность Убыль веса нии тетрила (табл. 66):
°C нагревания % При 110° уже через
V ДИУИ MilЧИНЙЛии_Ь
50 320 дней 0,05 гичное прогрессивное
75 320 » 0,3 разложение.
97 320 » 11,5 При нагревании в
16 » 11,5 пробирке при 130° на-
чинается разложение
тетрила, сопровождающееся газообразованием и наступающее тем
раньше, чем менее чист продукт. Это влияет на результаты опре-
деления температуры затвердевания тетрила; поэтому для получе-
ния возможно точных результатов определения температуры за-
твердевания тетрила необходимо самое определение проводить воз-
можно быстрее.
1 Солонина, Технология взрывчатых веществ, V, стр. 71—72.
342
Вследствие начинающегося при температуре плавления разло-
жения тетрила он не применяется в литом виде, но он может приме-
няться в виде смесей с тротилом или другими нитросоедшу^иями.
При 145—150° газообразование уже относительно велщьри вде-
нет за собой при более продолжительном нагревании самовоспла-
менение. Температура вспышки 190—194°.
При воспламенении тетрил горит энергично и в отличие от пи-
криновой кислоты и тротила при горении не плавится.
При испытании тетрила раствором дифениламина в серной кис-
лоте он всегда дает указание на присутствие азотной кислоты, как бы
тетрил ни был очищен.
Взаимодействие тетрила с концентриро-
в'а иной серной кислотой. При действии на тетрил
серной кислоты- в присутствии металлической ртути он выделяет
нацело одну пятую часть азота в виде окиси.
По данным Солонина, в этом случае протекает следующая
реакция:
2C6H2(NO2)3NCH3NO2 + 2H2SO4 -> 2C6H2(NO2)3NCH3SO3H + 2HNO3
3HNO3 + 6Hg -> 3HgaO + 2NO + H2O
_______________3Hg2O + 3H2SO4-> 3HgSO4 + 3H2O____________
2C6H2(NO2)3NCH3NO2 + 5H2SO4 + 6Hg 2C6H2(NO2)3NC H3SO3 H +
+ 3HgSO4 + 2NO + 4H2O
При нагревании водного раствора происходит гидролиз трини-
трофенилметилсульфамина с образованием тринитрофенилметил-
амина:
C6H2(NO2)3NCH3SO3H + Н20 -^C6H2(NO2)3NHCH3 + H2SO4
Подвижность нитро группы в нитроаминах.
Мы уже отмечали явление перемещения группы SO3H в солях аро-
матических аминов (при нагревании анилина с концентрированной
серной кислотой).
Вероятно процесс протекает таким образом, что в качестве про-
межуточного продукта из сульфата анилина получается сульфами-
новая кислота, которая далее перегруппировывается в сульфокис-
лоту амина. Бамбергер 1 констатировал два перехода от^действия
концентрированной серной кислоты: при более низкой температуре
в о-сульфокислоту анилина, а при высокой температуре (180—190°)
в сульфаниловую кислоту:
/\,NH2H2SO4 |^\nHSO3H
| I I I
___ или ПРИ i/Z'4iNH3
^Jso3n 18°-190° SOsH^J
1 Bamberger, Ber., 30, 654, 1897; 30, 2274, 1897.
343
Аналогичные переходы констатированы и для других аминов.
Переход остатка сульфокислоты от азота аминогруппы в ядро
является частным случаем общей тенденции амино соединений, имею-
щих у азота электроотрицательный радикал (ОН, Cl, NO,), усту-
пать его ядру.
По Бамбергеру, при действии минеральных кислот или света
на фенилнитроамин образуется нитроанилин:
JI
nh2
2
2
при действии мине-
I ' ральных кислот или 1 * 1
\ / света \ /
Аналогично фенилметилнитроамин легко превращается в о- или
р-нитромонометиланилин:
NzCH3
/ч NO2
СН8
сн3
<>NO2
или
no2
В тетриле переход подвижной нитрогруппы, связанной с азо-
том, не происходит потому, что орто- и параместа уже заняты тремя
нитрогруппами. В этом случае эта нитрогруппа легко замещается
сульфогруппой при взаимодействии тетрила с концентрированной
серной кислотой с выделением азотной кислоты.
Этим объясняются отмеченные выше факты пониженной стой-
кости тетрила при нагревании или при взаимодействии с серной
кислотой и, в частности, возможность определения
азота в нитрометре Лунге.
Взрывчатые свойства тетрила,
ность к удару по международной пробе 40 см высоты
в 2 кг.
При испытании по русской пробе при весе груза в
падения груза 25 см получается около 60% полных взрывов. Чув-
ствительность к трению значительно выше, чем для пикриновой
кислоты.
Расширение в бомбе Трауцля 340 см3. Бризантность, по Гессу,
19 мм.
Скорость детонации, по Касту, при плотности 1,63 7200 м]сек Ч
При достигаемой прессованием максимальной плотности 1,68
тетрил детонирует еще от капсюля с 0,54 г гремучей ртути.
нитроаминного
Чувствитель-
при весе груза
10 кг и высоте
1 Для сравнения взрывчатых свойств дадим соответствующие данные для
четырех нитросоединений из табл. 67 на стр. 345.
344
5. Применение тетрила. Тетрил применяется преимущественно
для снаряжения детонаторов и капсюлей-детонаторов.
6. Тетранитрофенилметилнитроамин (метанитротетрил, пентил),,
впервые получен Ромбургом (в 1889 г.).
Его строение: V
N/CH3
O3n/XNO°2
* no2
По Ромбургу и Шеперсу \ он получается следующим образомг
10 г диметиланилина растворяют в 370 г крепкой серной кислоты.
Такое большое количество концентрированной серной кислоты
(37-кратное количество) необходимо брать для того, чтобы получить,
максимальный выход m-нитродиметиланилина при вступлении 1-й
нитрогруппы в ядро (см. стр. 322), так как только при нитрова-
нии этого изомера получается /и-нитротетрил
К охлажденному до—2° раствору приливают по каплям
смесь 12 г азотной кислоты (уд. веса 1,49) (89,6% HNO3) и 60 ?
крепкой серной кислоты, поддерживая все время температуру—2°.
После слива кислоты оставляют в покое еще на полчаса при —2°.
При этих условиях образуется, главным образом, т- и р-нитро-
диметиланилин.
Полученный раствор вносят в 150 см3 охлажденной азотной кис-
лоты (уд. веса 1,49) и оставляют в покое на неделю. Образую-
щийся пентил выкристаллизовывается из раствора, а одновременно
образовавшийся тетрил остается в растворе. При обработке пентила
раствором углекислого натрия замещается на группу ONa не
только нитрогруппа, стоящая в метаположении к нитроаминной
Таблица 67
(к стр. 344)
Наименование вещества Применяемая максимальная плотность Скорость детонации м)сек Расширение в бомбе Трауцля см3 Сжатие медных крешеров на бризанцмессере Каста мм
Тринитробензол . . 1.63 7000 300 4.1
Тринитротолуол . . 1,59 6700 285 3,6
Пикриновая кислота 1,69 7100 305 4,1
Тетрил 1,63 7200 3400 4.2
1 Rombourgh
XXII, 293, 1913.
и Schepers, Koninkl. Akad. Wetensch. Amsterdam,
345
группе, но и самая нитроаминная группа, и образуется динатровая
соль стифниновой кислоты.
Температура плавления пг-нитротетрила 146°.
При кипячении пентила с водой, метиловым или этиловым спир-
том та же нитрогруппа замещается гидроксилом, метоксильной груп-
пой или этоксильной группой:
•O2N
при кипячении с
НаО, СНаОН, СаН&ОН
N/™3
'Л NO, '
o9n< x,no2
[Я=ОН;ОСН3;ОС2Н5]
\ / °R
no2 .
7. Пентаиитрофенилметилиитроамин. Пентанитрофенилметил-
нитроамин получается по следующей схеме:
NO2
при действии (HN,),S
°2N\JnO2
! i
O2N:. zNO2
при нагревании в запаянной
трубке
с эквивалентным количе-
ством CH3J в метиловом
спирте
Две нитрогруппы, стоящие в метаположении к нитроаминной
группе, легко подвижны и при нагревании вещества с водой заме-
щаются гидроксильными группами:
• \сн3
O2N|Z
no2
при нагревании
с водой
Пентанитрофенилметилнитроамин получается в виде бесцветных-
кристаллов, плавящихся с разложением при 132°.
346
8. Тринитрофенил-7/г-диметилдинитроамин. Ромбург 1 получил
это вещество, исходя из 1,3-диметилфенилендиамина обработкой
его дымящей азотной кислотой:
и йз тетраметилфенилендиамина 2 при обработке его раствора в сер-
ной кислоте (1 объем тетраметилфенилендиамина в 2 объемах сер-
ной кислоты) азотной кислотой (уд. веса 1,48):
+ HNOa
Бланксма и Тервогт 3 получили его нагреванием при 150° в тече-
ние 5 часов nz-дихлординитробензола с 4 мол спиртового раствора
метиламина и обработкой полученного динитрометил-пг-фенилен-
диамина азотной кислотой (уд. веса 1,52):
Cl NHCH3
NO, ' NO,
А £.
/СН3
N/
I XNO,
o2n< \no2"
.CH3
\ 7 N\no
no2 °2
По своей взрывчатой силе это соединение стоит между тетри-
лом и тетранитрофенилметилнитроамином.
По данным Ван-Дуина и Леннепа 4, введение 2-й метилнитроами-
ногруппы в тетрил или 4-й нитрогруппы в ядро повышает чувстви-
тельность к механическим воздействиям полученного соединения
настолько, что новое соединение принадлежит к другим классам
1 Rombourgh, Rec. Trav. Chim. Pays-Bas, 7, 3, 1888.
2 Rec. Trav. Chim- Pays-Bas, 8, 280, 1889.
8 Blanksma и Terwogt, Rec. Trav. Chim- Pays-Bas, 21, 90, 1889.
4 Rec. Trav. Chim- Pays-Bas, 39, 145—177 1920.
347
чувствительности, чем обычно применяющиеся полинитро сое дине’
ния. Введение аминогруппы (а также алкилирование фенола)умень-
шают чувствительность к механическим воздействиям. Наконец,
подвижность нитро групп повышает чувствительность.
РАСТВОРЕНИЕ ДИМЕТИЛАНИЛИНА И НИТРОВАНИЕ
СУЛЬФАТА ДИМЕТИЛАНИЛИНА
1. Схема производства тетрила (рис. 42). В аппарат 2 подается
из мерника 1 серная кислота и к ней добавляется диметиланилин
из мерника 3 для образования диметиланилинсульфата. Раствор
последнего в серной кислоте направляется в сборник, из которого
поступает через мер-
ник в нитратор 4, ку-
да предварительно
подается азотная
кислота или смесь
азотной кислоты с
серной кислотой. По
окончании процесса
нитрования содержи-
мое нитратора пере-
дается на вакуум-
воронку или центри-
фугу (или, наконец,
в промывные ванны
для разбавления во-
дой, как это показа-
но на схеме). После
Рис. 42; Схема установки для нитрации диметил- отжатия кислоты кис-
анилина. лый тетрил поступает
в чан для промывки,
затем на вакуум-воронке или центрифуге отжимается вода. Отжатый
тетрил после сушки перекристаллизовывается из ацетона, либо
без предварительной сушки поступает на перекристаллизацию из
бензола.
Нейтрализация диметиланилина. По изло-
женным в разделе «Действие окислителей на органические основа-
ния» соображениям нельзя нитровать непосредственно диметилани-
лин, его надо предварительно обработать купоросным маслом для
получения сернокислого диметиланилина:
C6H5N(CH3)2 + H2SO4 -> C6H5N(CH3)2 • h2so4
На основании практики заводской работы считают, что наилуч-
шими условиями нейтрализации, при которых при последующем
нитровании обеспечивается наибольший выход и наилучшее качество
продукта, являются:
1) концентрация серной кислоты 92—93%;
348
2) соотношение между диметиланилином и купоросным маслом
равно 1 : 8—10;
3) температурный режим: прилив диметиланилина при 25 —40°,
выдержка при 40°.
При указанных условиях образуются сернокислый диметил-
анилин х, но не образуется сульфо диметил ^илин.
Нейтрализация. Процесс нейтрализации прежде назы-
вался неправильно «сульфированием», раствор диметиланилинсуль-
фата «сульфо» и нейтрализатор «сульфуратором». Эти названия даны
были тогда, когда считали, что при обработке диметиланилина сер-
ной кислотой образуется сульфодиметиланилин, а не диметил-
ацилинсульфат. Мы избегаем применения этих неправильных
названий.
Нейтрализатор представляет собой железный двустенный аппа-
рат с мешалкой, термометром, продуктовой трубой, трубой для при-
лива диметиланилина и серной кислоты и вентиляционной трубой.
В нейтрализатор подается серная кислота (с содержанием 92—
93% H2SO4). На 1 в. ч. диметиланилина берется 8—10 в. ч. серной
кислоты. Кислота подогревается в нейтрализаторе до 25—30°,
после чего начинают слив диметиланилина.
После подъема температуры до 35° ведут прилив остального
количества диметиланилина при температуре 35—40°. По оконча-
нии прилива делают выдержку при 40° в течение 30 минут; затем
охлаждают водой до 18° и перегоняют в хранитель.
Для контроля отбирают из нейтрализатора по окончании вы-
держки 40 см3 раствора, разбавляют 10-кратным количеством воды,
тщательно перемешивают и охлаждают. В случае содержания не-
прореагировавшего диметиланилина на поверхности водного рас-
твора всплывают масляные капли диметиланилина. Контроль дол-
жен осуществляться очень тщательно, так как при содержании
в кислоте непрореагировавшего диметиланилина, даже при очень
хорошем охлаждении нитратора, во время слива сернокислого
раствора диметиланилинсульфата к азотной кислоте получается
вспышка, которая может привести к загоранию уже образовавше-
гося нитропродукта.
Полученный раствор сернокислого диметиланилина в серной кис-
лоте имеет вид слабокоричневой прозрачной жидкости. Кислот-
ность ее по серной кислоте 80—82%.
Продолжительность операции нейтрализации при загрузке
112 кг диметиланилина составляет 5 часов.
Нитрование сернокислого диметилани-
лина. Нитратор представляет собой двухстенный железный,
внутри освинцованный котел с мешалкой, состоящей из стального
вала и крыльев, обложенных свинцом. Мешалки делают
110—120 об/мин. Нитратор оборудован нижней спускной трубкой
1 По практическим данным при нейтрализации 1 кг диметиланилина сер
ной кислотой выделяется 160 Кал.
349
с керамиковым или термосилидовым краном для спуска содержи-
мого нитратора.
Применяют также нитраторы из V2A.
Вследствие большей опасности нитрования сернокислого ди-
метиланилина, чем ароматических углеводородов или сульфофе-
нола, вначале была установлена загрузка в нитратор около 25 кг
диметиланилина. Однако опыт показал возможность загрузки до
100 кг, которая по современным воззрениям является вполне до-
пустимой и не более опасной, чем прежняя вчетверо меньшая за-
грузка.
В прежнее время применяли для нитрации азотную кислоту
(с содержанием 91—94% HN03). Такая кислота сильно корро-
дирует железную аппаратуру, трубопроводы и свинцовый котел
нитратора. Для уменьшения этой коррозии добавляют к азотной
кислоте серную кислоту с расчетом, чтобы получить смесь состава:
84—86% HNO3, 8—6% H2SO4.
При нитровании диметиланилина одновременно протекает про-
цесс окисления одной метильной группы, причем здесь возможны
две реакции: одна с восстановлением азотной кислоты до двуокиси
азота, другая с восстановлением азотной кислоты до элементарного
азота:
СН3
(I) C6H5N(CH3)2 + 10HNO3->C6H2(NO2)3 — N< +
xno2
+ CO, + 8H2O + 6NO2
,CH„
(II) 5C6H5N(CH3)2 + 26HNO3 -> 5C6H2(NO2)3N/ +
XNO2
+ 5CO2 + 28H2O + 3H2 + N2
Ha 1 в. ч. диметиланилина расходуется по уравнению (I)
5,2 в. ч. азотной кислоты, по (II) 2,7 в. ч.
Опытные данные показывают, что реакция идет одновременно
по обоим уравнениям, но, главным образом, по уравнению (I).
Например, по Лангеншейдту, при нитровании на 1 в. ч. диметил-
анилийа берется 4,7 в. ч. моногидрата азотной кислоты. Пренебре-
гая небольшим количеством азотной кислоты, не участвующей в ре-
акции (остающейся в отработанной кислоте и улетучивающейся),
легко подсчитать, что по уравнению (I) в этом случае реагирует
80% и по (II) 20% азотной кислоты.
Дозировка азатной кислоты. В прежнее время,
когда тетрил перекристаллизовывался из ацетона, требования к ка-
честву тетрила-сырца, или промытого тетрила, были относительно
мягкими (температура затвердевания нитрованного тетрила не ниже
124°) и считали наилучшим количественным соотношением HNO3h
диметиланилина 5,4: 1; при меньшем количестве азотной кислоты
наблюдалось уменьшение выхода и ухудшение качества продукта.
Однако при перекристаллизации тетрила из бензола необходимо
значительно повысить требования к качеству нитрованного тетрила,
350
так как иначе получается повышенный процент брака (температура
затвердевания нитрованного продукта должна быть не ниже 127°
и небольшой процент в пределах 126—127°).
Процесс нитрования. Процесс можно вести только
сливанием раствора сернокислого диметиланилина к азотной кислоте
(или ее смеси с серной кислотой). При обратном порядке слива
реакция сопровождается такими сильными окислительными про-
цессами, что легко происходит самовоспламенение. Реакция проте-
кает при нитровании таким образом, что вначале в ядро вступают
две нитрогруппы, затем, главным образом,выдержке, происхо-
дит окисление одной метильной группы, и после этого вступают
остальные две нитрогруппы.
По еле загрузки в аппарат нитрующей смеси пускают мешалку
и подогревают паром; по достижении 40° выключают пар; темпера-
тура в аппарате продолжает подниматься, и по достижении 45®
начинают прилив раствора сернокислого диметиланилина. Регули-
руют процесс охлаждением и скоростью прилива так, чтобы темпе-
ратура поднималась в течение 10 минут на 1,5° до достижения 50°.
Остальное количество сульфата сливают при температуре в пределах
50—52°.
При загрузке 270 кг раствора (28 кг диметиланилина) прилив
сульфата продолжается в среднем 2х/2 часа летом и около 2 часов
зимой. По окончании, слива делают часовую выдержку при
55—56°.
В начале прилива раствора сульфата содержимое нитратора имеет
обычный вид смеси жидкого нитропродукта с кислотой. К концу
слива и в начале выдержки образуется обильная пена из-за выделе-
ния углекислоты при реакции окисления метильной группы и одно-
временно образующихся окислов азота или элементарного азота по
2-й реакции. Во время выдержки пена постепенно спадает, причем
за конец реакции можно грубо принять момент по истечении 15 м -
нут после исчезновения пены.
В случае чрезмерно сильного кипения необходимо: 1) прекра-
тить дальнейший приток сульфата; 2) если температура поднимается
выше установленного предела, то пустить в паровую рубашку нитра-
тора воду для охлаждения; 3) если эти мероприятия не останавли-
вают вспенивания, то дать 0,5—1 кг кислотной смеси.
Особенно опасной является во время нитрации остановка ме-
шалки (обрыв ремня, авария на электростанции и т. п). В этом слу-
чае возможно сильное повышение температуры реакционной смеси,
результатом чего может быть выброс, самовоспламенение или взрыв.
Поэтому надо иметь на случай остановки мотора либо аварийную
паровую машину (наименее надежна), либо аккумуляторную под-
станцию, либо, наконец, иметь в нитраторе подводку для сжатого
воздуха, чтобы закончить операцию.
В случае полной остановки мешалки или опасного подъема тем-
пературы в аппарате следует содержимое его спустить в промывную
ванну с водой.
351
Указанные выше температуры нитрации не следует переходить
вследствие сохранившейся и у сульфата тенденции к окислению и
•осмолению при высоких температурах. При приливе раствора суль-
фата диметиланилина в указанных температурных пределах полу-
чается светложелтый тетрил кристаллического строения. При веде-
нии же нитрации в пределах до 65° получается тетрил зернистого
строения темножелтого цвета и с низкой температурой затвердева-
ния. В случае же подъема температуры выше 65—75° продукт
иногда осмоляется до такой степени, что содержимое нитрата пред-
ставляет собой вязкую черную тестообразную массу, затвердеваю-
щую при охлаждении. Это явление сопровождается в большинстве
случаев вспышкой.
Условия перемешивания играют при нитровании диметилани-
линсульфата большую роль, чем при нитровании ароматических
углеводородов или фенолсульфокислоты. Для обеспечения быстрого
перемешивания сульфата с кислотой необходимо, чтобы сульфат
приливался на конец крыла мешалки и, чтобы струя сливаемого
сульфата не попадала в воронку, образуемую жидкостью около
вала мешалки, так как здесь происходит очень слабое перемеши-
вание жидкости; при попадании сульфата в эту воронку обра-
зуются крупные гранули тетрила и происходит осмоление сульфата
диметиланилина.
Продолжительность операции нитрации,. начиная от момента
начала подачи кислотной смеси в аппарат и до момента окончания
разгрузки аппарата — около 51/2 часов. Отделение тетрила от от-
работанной кислоты может производиться отжимом на центрифуге,
отсасыванием отработанной кислоты на вакуум-воронке и, нако-
нец, разбавлением реакционной массы обильным количеством воды
с последующим фильтрованием через полотняный фильтр
На Шлиссельбургском заводе во время империалистической войны
реакционную массу спускали из нитратора в железную центрифугу
и после отжима кислоты тетрил направляли на промывку.
Менее удовлетворительно идет процесс отделения отработанной
кислоты отсасыванием на вакуум-воронке, которая может быть сде-
лана из железа На некоторых заграничных заводах применяют
вакуум-воронки из V4A. Наконец, наименее совершенным является
примитивный способ разбавления реакционной массы водой В этом
случае после обычного для всех способов охлаждения содержимого
аппарата до 15—25° (в зависимости от температуры охлаждающей
воды) спускают его небольшой струей в ванну, наполненную на
высоты водой при параллельном пуске струи воды. Ванна пред-
ставляет собой деревянный или железный ящик, обложенный внутри
свинцом; объем ее рассчитан таким образом, чтобы на 1 кг
нитруемого диметиланилина приходилось около 60 л емкости
ванны.
Разбавленная отработанная кислота стекает из ванн по жолобу
в систему ловушек, расположенную в крытом помещении вблизи
отделения нитрации. Ловушки представляют собой ряд последова-
352
тельно соединенных между собой деревянных ящиков, обложенных
свинцом.
Тетрил из ванн спускается на воронку с полотняным фильтром.
Оставшийся на воронке тетрил направляется на промывку.
2. Другие способы получения тетрила. Способ Б р юнел я.
Для нейтрализации диметиланилина берется на 1 в. ч. его
10 в. ч. купоросного масла. Нитрование ведется смесью состава:
47,5% H2SO4, 26,5% HNO3, 26% Н2О. Эта смесь смешивается со
значительным количеством отработанной кислоты, причем на 1 в. ч.
диметиланилина берется 66 в. ч. такой <х%си с отработанной кисло-
той. При этих условиях температура приреакции поднимается до
100°, и получают выход 92—93% от теории.
Получение тетрила в две фазы по Келеру1.
Добавлением диметиланилинсульфата к водной серноазотной смеси
получают в 1-й фазе динитродиметиланилин, который затем добав-
ляют к более крепкой серноазотной смеси, причем получают тетрил.
Получение тетрила из монометиланилина
и динитрохлорбензола. Этот способ невыгоден вслед-
ствие высокой стоимости монометиланилина. Поэтому в последнее
время ведут разработку метода получения дешевого монометилани-
лина. Девернь указывает следующий наиболее выгодный способ
получения монометиланилина.
Нагревают в автоклаве в течение 10 часов при температуре 193—
198° и под давлением 13—17 ат сухой солянокислый анилин с ме-
тиловым спиртом, взятым с избытком в 20%.
После отгонки полученного продукта водяным паром в присут-
ствии извести получают смесь следующего состава: 5,4% анилина,
70,0% монометиланилина, 24,6% диметиланилина.
Из этой смеси монометиланилин можно выделять, основываясь
на различной растворимости в воде щавелевых солей этих трех ве-
ществ. Следует, однако, заметить, что вследствие большей наклон-
ности (сравнительно с диметиланилином) монометиланилина к реак-
циям окисления и осмоления при взаимодействии его с азотной кис-
лотой при его нитровании получаются меньшие выходы, чем при
нитровании диметиланилина 2.
Получение тетрила из динитрохлорбензола и
метиламина. Метиламин может быть получен взаимодействием
формальдегида с хлористым аммонием или, что дешевле, из ледя-
ной уксусной кислоты — массового продукта лесохимической
промышленно сти.
Для получения метиламина из ледяной уксусной кислоты посту-
пают следующим образом. Действием аммиака на ледяную уксус-
ную кислоту получают уксусноаммониевую соль, при нагревании
1 Desvergnes, Mem. Poud., 3-me fasc-, 1922.
2 О применении монометиланилина для приготовления тетрила см. No-
lan, Scient. Proc. Roy. Dublin Soc., 17, 219—223, 1923; Desvergnes,
1 Chim. et Ind., 24, 785—793, 1304—1316, 1936.
L Химия—13—23 353
которой получают ацетамид. При взаимодействии ацетамида с из-
вестковым молоком выделяется газообразный метиламин, который
улавливается соляной кислотой или соответствующим растворите-
лем. По этому способу метиламин получается за границей в завод-
ском масштабе.
Однако метиламин, получающийся этими методами, слишком
дорог, вследствие чего способ получения тетрила из динитрохлор-
бензола с метиламином не находит промышленного применения.
При взаимодействии динитрохлорбензола и метиламина в при-
сутствии щелочи (чтобы не образовалась свободная НС1) получается
динитрофенилмонометиламин. Конденсация производится при тем-
пературе 65—70° добавлением 15—20% водного раствора метил-
амина к бензольному раствору динитрохлорбензола \ а нитрова-
нием полученного динитромонометиланилина при температуре 30—
40° получается тетрил:
С1
/ ^NOg + сн3хн,
\о2
+ HNO3
/СН3
N\
। XNO2
O2Nz xno.
ПРОМЫВКА ТЕТРИЛА
Отмывка тетрила от кислот может производиться двумя спосо-
бами: 1) непосредственной промывкой кислого тетрила водой; 2) про-
Рис. 43. Схема установки для промывки тетрила.
1—вода; 2—пар; 3—спуск промывной воды; 1—вакуум-
воронка; S—отстойные ванны.
мывкой водой тетрила,
предварительно раство-
ренного в бензоле.
1. Непосредственная
промывка кислого те-
трила водой произво-
дится в деревянных ча-
нах (рис. 43), снабжен-
ных мешалками, со-
стоящими из стального
вала с алюминиевым
пропеллером внизу и
алюминиевыми пере-
кладинами по середине
мешалки. Мешалка де-
лает 75 об/мин. Над
чанами ставятся колпа-
ки, соединенные с вентиляцией для отвода пара и газов, выделяю-
щихся при промывке.
1 Deseigne, Mem. Poud-, 28, 156—170, 1938.
3&4
Емкость одного чана 1350 л, рабочая емкость 1150 г (85% за-
полнения).
Тетрилу после промывных ванн нитрационного отделения дается
в промывном чане одна холодная промывка и четыре промывки при
температурах 40, 60, 80 и 90°.
Подогрев ведется подачей в жидкость острого пара через желез-
ную спираль, расположенную у дна чана. По достижении необхо-
димой температуры перемешивают 8 минут (при 1-й промывке при
обыкновенной температуре перемешивают полчаса), потом останав-
ливают на полчаса мешалку. В это время тетрил осаждается на
дно, а промывная вода спускается чер^х поставленный на соответ-
ствующей высоте кран. Затем в чан подайся новое количество воды
длй следующей промывки. После последней промывки тетрил с
частью промывной воды спускается на центрифугу для отжатия
воды; отжатый на центрифуге тетрил, содержащий 7—10% воды,
поступает на сушку.
Тетрил должен иметь после промывки кислотность не выше
0,015% (в пересчете на серную кислоту).
Указанным числом промывок часто не удается получить конди-
ционный по кислотности тетрил и приходится делать до И—14 про-
мывок.
Продолжительность одной горячей промывки около часов.
С промывными водами уносится 5—8% тетрила; поэтому их про-
водят через систему ловушек для улавливания унесенного тетрила.
Ловушки чистятся периодически и собирающийся здесь тетрил про-
мывается в промывных чанах и направляется на сушку.
Ван-Дуин указывает ], что после промывки тетрила водой его
промывают еще раз холодным спиртом. По его предположению при
этом растворяются нестойкие, нерастворимые в бензоле, побочные
продукты, так как предварительно промытый спиртом тетрил после
перекристаллизации из бензола гораздо белее и более стоек, чем
без такой промывки, а в промывном спирте легко обнаруживается
присутствие довольно большого количества этилнитрита.
2. Промывка тетрила в бензольном растворе. Описанный выше
способ промывки тетрила требует длительной обработки горячей
водой ввиду трудности отмывки кислот из кристаллического про-
дукта. Процесс значительно ускоряется при промывке тетрила,
растворенного в бензоле. Такой метод, по указанию Штетбахера 1 2,
применяется в Америке и состоит в следующем. Растворяют кислый
тетрил в бензоле в отношениях 1: 3,5—4, и полученный раствор
промывают несколько раз теплой водой; каждый раз после тщатель-
ного перемешивания сливают нижний слой промывной воды и по-
дают свежую порцию воды.
При таком способе работы отпадает необходимость в специаль-
ной мастерской промывки, так как здесь процесс промывки произ-
1 Van Du у n, Rec. Trav. Chim. Pays-Bas, 37, I, 111, 1917.
2. Z. Schiess- u. Sprengst., 7, 262, 1929.
355
водится в аппарате-растворителе; сокращается расход воды, пара,
рабочей силы и резко сокращается длительность процесса промывки;
при всем этом качество промытого тетрила лучше (меньший брак по
кислотности).
3. Сушка промытого тетрила. При перекристаллизации тетрила
из ацетона его необходимо предварительно высушить, чтобы избе-
жать сильного разбавления растворителя водой. В случае же пере-
кристаллизации тетрила из бензола предварительная его сушка яв-
ляется излишней, так как вода нерастворима в бензоле. Сушка тет-
рила производится в обычных камерных сушилках. Он рассыпается
на лотках по 8—10 кг на каждом. Лотки устанавливаются на стел-
лажах. Температура сушки 55—60°.
Продолжительность сушки 24—28 часов.
Во время империалистической войны в целях ускорения про-
цесса вместо сушки производилось обезвоживание промытого тет-
рила промывкой его спиртом после отсасывания на вакуум-воронке
воды. Промытый спиртом тетрил после отсасывания спирта поступал
непосредственно в аппарат-растворитель мастерской перекристалли-
зации.
4. Качество промытого тетрила. В сухом промытом тетриле содер-
жатся продукты неполной нитрации диметиланилина и продукты
осмоления, которые понижают его восприимчивость к детонации
и бризантность. Кроме того, образуются побочные продукты нитра-
ции, которые обусловливают пониженную химическую стойкость
тетрила. К числу таких продуктов относится описанный выше тет-
ранитрофенилметилнитроамин (так называемый <<т-нитротетрил»).
Примесь илистых веществ, попадающая из неочищенной речной
воды во время половодья, не оказывает заметного влияния на вос-
приимчивость к детонации и бризантность при содержании их до
нескольких десятых долей процента; но содержание кремнеземистого
песка (который может попасть при производстве в количестве
выше 0,05%) заметно повышает чувствительность тетрила к удару
и трению.
Поэтому тетрил необходимо очистить перекристаллизацией из
растворителя для удаления из него указанных примесей.
Наконец, пикриновая кислота, присутствующая в очень малых
количествах, не влияющих на свойства тетрила, является продук-
том гидролиза тетрила, образующимся вероятно при горячей его про-
мывке.
ПЕРЕКРИСТАЛЛИЗАЦИЯ ТЕТРИЛА
1. Растворитель для перекристаллизации тетрила. Для перекри-
сталлизации тетрила применяются два растворителя: ацетон и бен-
зол. При перекристаллизации из ацетона легче получить тетрил
высокого качества с более высокой температурой затвердевания,
в виде более крупных кристаллов; продукт обладает немного луч-
шей восприимчивостью к детонации.
356
Однако для огромного большинства изделий качество тетрила,
перекристаллизованного из бензола (так называемый бензольный тет-
рил), является вполне удовлетворительным. Вместе с тем примене-
ние в качестве растворителя бензола вместо ацетона имеет следую-
щие преимущества: 1) бензол значительно (в 6—7 раз) дешевле ацето-
на; 2) ацетон является дефицитным продуктом, тогда как бензол легче
может быть получен в потребных для производства количествах.
Свойства ацетона. Ацетон — бесцветная жидкость. Уд.
вес ее 0,818 при 0°.
Температура его кипения 56,1°; температура плавления 94°. Он
смешивается с водой во всех отношениях.
2. Перекристаллизация тетрила из ацетона. Схема уста-
новки. Высушенный тетрил поступает в аппарат-растворитель,
где растворяется в ацетоне. Раствор продавливается через фильтр-
пресс, после чего направля, в кристаллизатор. По окончании
процесса кристаллизации содержимое кристаллизатора поступает
в вакуум-воронку, а после отсасывания растворителя и промывки
на воронке спиртом тетрил поступает на сушку. Отработанный аце-
тон собирается в хранитель, из которого поступает через мерник
в дестилляционный аппарат для отгонки чистого ацетона.
Описание производства. Аппарат-растворитель —
железный клепаный, с двойными стенками для нагревания, с мешал-
кой, делающей 60 об/мин. Для улавливания образующихся во время
растворения тетрила паров растворителя они отводятся для конден-
сации в холодильники — железные цилиндрические сосуды, снаб-
женные внутри медными трубками для охлаждения водой.
В растворитель загружается тетрил и ацетон в соотношении
1: 1 при свежем, так называемом нолевом, ацетоне. В этом случае
на 64 кг тетрила берется 64 кг ацетона. Нагревают до 56—58° и
при этой температуре делают 35 минут выдержку для обеспечения
растворения всего загруженного в аппарат тетрила, после чего про-
гоняют раствор через фильтр-пресс под давлением в 1—1,2 ат,
создаваемым в аппарате-растворителе сжатым воздухом.
. Фильтр-пресс применяется чугунный, с тремя рамами и двумя
плитами. Фильтр-пресс перед прогонкой через него раствора нагре-
вается паром до 50—55° для предупреждения охлаждения раствора
и кристаллизации из него тетрила.
При загрузке маточных растворов добавляется небольшое коли-
чество свежего ацетона и спирта (последний для уменьшения
растворимости тетрила) и уменьшается загрузка тетрила согласно
табл. 68 (стр. 358).
Кристаллизации ведутся сериями, т. е. ведется последовательно
ряд операций на одноименном растворителе. Например, пять кри-
сталлизаций на свежем (нолевом) ацетоне, затем столько же на 1-м
маточном, дальше столько же на 2-м и т. д.
Продолжительность одной операции растворения, считая от мо-
мента начала наполнения до конца опорожнения, 1 ч. 15 м. Продол-
жительность одной операции фильтрации 40 мин.
357
Таблица 68
№ ацетона Загрузка, кг
тетрила свежего ацетона маточного ацетона спирта
0 64 64 __
d 40 4 60 2
2 40 4 60 9
3 35 4 60 2
4 30 4 60 .
5 25 4 60
6 25 4 60 —
Из фильтр-пресса раствор подается
Кристаллизация.
в аппарат-кристаллизатор — железный аппарат с двойными боко-
выми стенками, с мешалкой рамного типа, делающей 28 об/мин.
Аппарат снабжен внизу дна спусковой трубой с краном, на конец
трубы надевается полотняный или резиновый рукав для спуска со-
держимого кристаллизатора на вакуум-воронку.
Кристаллизатор должен быть перед загрузкой горячего раствора
нагрет до 45—50°.
После подачи раствора в кристаллизатор его оставляют в тече-
ние 40—60 минут для медленного охлаждения, т. е. без подачи
воды в рубашку. Затем производят медленный пуск воды и посте-
пенно увеличивают открытие вентиля до максимума в течение 1х/2—
2 часов. Охлаждают зимой до 10°, летом до 15°. По достижении этой
температуры спускают содержимое кристаллизатора на вакуум-
воронку.
Продолжительность одной операции кристаллизации около
2 ч. 55 м.
После отсасывания маточного раствора на вакуум-воронке про-
мывают спиртом для наилучшего вытеснения ацетона из тетрила.
Тщательная отмывка ацетона необходима потому, чтобы уменьшить
его потери, а также потому, что в присутствии ацетона при сушке
тетрила получаются темные корки, и готовый продукт приобретает
пониженную температуру затвердевания Ч
Отжатый тетрил содержит 10—12% спирта. Продолжительность
одной операции отсасывания на вакуум-воронке 50 минут.
3. Перекристаллизаций тетрила из бензола. Основным методом
перекристаллизации тетрила ныне является бензольный. Схема про-
1 По Деверню, тетрил вступает в реакцию с примесями, содержащимися
в ацетоне. При нагревании неотмытого от ацетона тетрила образуется чернова-
тое вещество с температурой плавления 121—123°; дальнейшее нагревание вновь
поднимает температуру затвердевания тетрила до 127—128°j Поэтому надо сле-
дить за тем, чтобы промытый спиртом тетрил не имел запаха ацетона. В против-
ном случае при сушке его получаются темные корки, и продукт приобретает
пониженную температуру затвердевания.
358
изводства и здесь та же, как и при ацетоином способе. Основное
отличие в устройстве аппаратуры и в ведении самого процесса заклю-
чается в следующем:
1) вследствие значительно меньшей растворимости тетрила
в бензоле соотношение между тетрилом и бензолом при загрузке
в аппарат-растворцтель равно 1 : 3,5—4. Соответственно большему
объему, занимаемому раствором, увеличены размеры соответствую-
щих аппаратов;
2) вследствие более высокой температуры кипения бензола жид-
кость нагревается в аппарате-растворителе до 70,5° (влажный бензол
кипит при температуре 70,5°), это сказывается соответственно на ре-
жиме при охлаждении в кристаллизаторе;
" 3) после отсасывания бензольного раствора от перекристалли-
зованного тетрила необходимо промыть тетрил спиртом или другой
жидкостью для удаления б_..о<лла. Это вызывается ядовитостью па-
ров бензола, в присутствии которых нельзя сушить тетрил в камер-
ных сушилках.
Качество бензола для перекристаллиза-
ции тетрила. Касаясь вопроса о перекристаллизации тетрила
из бензола, Ван-Дуин отмечает, что нельзя полагаться на продукт,
который имеется на мировом рынке в продаже под названием
«чистый бензол», так как после перекристаллизации из него почти
белый тетрил становится темнокоричневым. Бензол для перекри-
сталлизации тетрила должен удовлетворять особым требованиям
в отношении его чистоты.
4. Виды брака тетрила и его исправление: 1) пониженная тем-
пература затвердевшего нитрованного тетрила.
При перекристаллизации тетрила из ацетона такой брак исправ-
лялся некоторое время промывкой азотной кислотой или перекри-
сталлизацией из нее. Однако работа с азотной кислотой крайне
неудобна, поэтому позже перекристаллизовывали из бензола, а за-
тем вторично — из ацетона. Выгоднее донитровать такой тетрил
для получения кондиционного по температуре затвердевания нитро-
ванного тетрила;
2) при небольших отклонениях по температуре затвердевания
или кислотности перекристаллизованного тетрила удается часто
получить кондиционный продукт промывкой тетрила бензолом и
спиртом (ацетоном при ацетоином тетриле);
3) при большом содержании нерастворимых примесей — золы
в тетриле, собираемом из ловушек или всасывающих колодцев (для
промывных вод, прошедших ловушки), производится предваритель-
ная перекристаллизация из бензола;
4) тетрил, высаженный из маточных растворов (маточного аце-
тона или бензола), обладает пониженной температурой затвердева-
ния (от 116 до 121°) вследствие значительного содержания про-
дуктов неполной нитрации и смолистых примесей. Такой продукт
прежде часто сжигался. Были попытки исправить его подобно некон-
диционному нитрованному продукту промывкой азотной кислотой
359
»
или перекристаллизацией из азотной кислоты или из бензола.
Однако эти способы невыгодны; выгоднее донитровать этот продукт.
5. Сушка перекристаллизованного тетрила производится в ка-
мерных сушилках. На каждый лоток загружается около 7 кг кри-
сталлизованного тетрила. Для ускорения процесса сушки и преду-
преждения образования корок и комков необходимо периодическое
перелопачивание его на лотках. Сушка ведется при температуре не
выше 60°. Продолжительность сушки около 15 часов. Для сушки
бензольного тетрила лучше применять барабанные или пневмати-
ческие сушилки, так как в этом случае не требуется предваритель-
ная отмывка бензола дорогим спиртом, исключается систематическое
отравление рабочих бензолом, остающимся всегда в некотором ко-
личестве в тетриле после спиртовой промывки, а равно исключается
возможность образования комков и корок при сушке.
6. Формирование партий, просеивание, упаковка. Вес партии
тетрила не менее 1 т. Для получения однообразной партии необ-
ходимо тщательно перемешать порции тетрила, получаемые при от-
дельных кристаллизациях, которые отличаются друг от друга по
качеству. Для этой цели отдельные кристаллизации, давшие при
анализе удовлетворительные результаты, смешиваются. Эта мешка
удобно производится одновременно с просеиванием тетрила (для
удаления комков и крупных кусков) в сетчатом барабане, куда одно-
временно и непрерывно загружается тетрил от разных кристалли-
заций.
Более примитивный способ просеивания заключается в том, что
на. сито насыпается послойно тетрил из 10—15 кристаллизаций;
эти слои сперва грубо перемешиваются вручную, а затем оконча-
тельно перемешиваются при просеивании через сито. Сита (качаю-
щиеся) устанавливаются в деревянных ларях. Применяется медное
сито с 900 отверстиями на 1 сл2.
Тетрил упаковывается в коробки из белой жести емкостью около
8—10 кг. Вместо этих коробок можно упаковывать в брезентные ме-
шочки такой же емкости. Коробки или мешочки по 4 штуки укла-
дываются в деревянные ящики.
7. Технические условия на тетрил (ОСТ 3514).
Показатели 1-й сорт 2-й сорт 3-й сорт
1. Внешний вид для всех сортов 2. Ивет 3. Температура затвердевания не ниже 4. Влажности и летучих веществ не более 5. Нерастворимых в ацетоне при- месей не более Мелко крист: ханических - С 127,7° 0,02% 0,1% ^ллический по примесей, вит эломенно-желт 127,7° 0,02% 0,25% эошок без ме- 1мых на-глаз ый 127° 0,05% 0,25%
360
Показатели 1-й сорт 2-й сорт 3-й сорт
В том числе: золы не более 0,05% 0,05% 0,05%
кремнезема не более .... 0,015% 0,02% 0,02%
6. Кислоты в пересчете на H2SO4 не более 0,01% 0,01% 0,015%
7. Окись азота на 1 г при темпе- ратуре 0° и Давлении 760 мм рт. Ст- не менее 75 мл 75 мл 65 мл
8. Ректификация маточного ацетона. Непригодный для новых
перекристаллизаций тетрила маточный ацетон разбавляется водой
специальном баке с целью высаживания из него основной массы
растворенных нитро продуктов; воды добавляется 30% по весу аце-
тона. После перемешивания с водой раствор оставляют в покое на
1х/2—2 суток. После отстоя смесь ацетона с водой отделяется от
нитропродуктов и направляется в мастерскую ректификации.
Мастерская ректификации ацетона состоит из простейшего обо-
рудования — разгонного куба и холодильника.
Разгонный куб представляет собой железный цилиндрический
сосуд с паровой рубашкой и спускной трубой в днище куба.
При гонке собираются три фракции: 1) до 65°—чистый ацетон—
используется наравне со свежим ацетоном; 2) от 65 до 88° — смесь
ацетона со сниртом — подвергается вторичной перегонке; 3) от 80
до 90° — наиболее богатая спиртом фракция; она применяется вместо
чистого спирта для промывки тетрила на вакуум-воронке.
Из 140 кг загруженного ацетона получается 84 кг 1-й фрак-
ции, 21 кг 2-й , 7 кг 3-й и остаток 28 кг.
9. Перекристаллизация тетрила из дихлорэтана. Как при перекри-
сталлизации тротила, так и при перекристаллизации тетрила при-
менение горючих летучих растворителей вносит значительный до-
полнительный источник возможности возникновения пожара и
взрыва в производстве. Поэтому издавна стремились к замене этих
растворителей негорючими. В Америке применяют для перекристал-
лизации тетрила менее горючий дихлорэтан. Вследствие большей
растворимости тетрила в дихлорэтане, чем в бензоле, тетрил и ди-
’хлорэтан берутся в отношениях 1 : 1,7, что позволяет уменьшить
размеры аппаратуры сравнительно с бензольным способом. Нагре-
вание при растворении при 80°. Возможность применения этого ме-
тода в производстве решается в основном стоимостью дихлорэтана.
10. Применение неперекристаллизованного тетрила. Для изго-
товления некоторых изделий, например, капсюлей-детонаторов для
промышленных взрывных работ, в некоторых странах (Германия,
Швеция и др.) применяют неперекристаллизованный тетрил, но вы-
держивающий установленное время при испытании по пробе Абеля.
Очевидно здесь получается нитрованный тетрил с высокой темпера-
турой затвердевания, обладающий хорошими взрывчатыми свой-
ствами, и поверяется его химическая стойкость.
361
11. Гексаннтродифенилэтилендинитроамин, или дитетрил Ди-
фенилэтилендиамин CeHsNHCH2CH2NHCeH5 получается кипяче-
нием смеси дихлорэтана с анилином. Полученное основание легко
очищается в виде его сульфата перекристаллизацией из воды.
Диаминсульфат растворяется в концентрированной серной кис-
лоте и раствор добавляется к концентрированной азотной при тем-
пературе 30—35°; выдержка при 80°.
Октонитропроизводное
zno2 no2X
N—СН2 - СН2_'N
O2N " 2 । 2 представ-
I ! i ।
no2 no2
ляет собой бледножелтый кристаллический порошок, плавящийся
с разложением при температуре 213°. При быстром нагревании он
взрывает. При нагревании с концентрированным раствором едкого
натра разлагается с образованием пикриновой кислоты.
12. Аналоги тетрила. Преимущество тетрила при применении
его для изготовления капсюлей-детонаторов или детонаторов заклю-
чается в том, что он, будучи сравнительно дешевым и легко доступ-
ным веществом, обладает большой мощностью, необходимой для
взрыва заряда. Однако в ряде случаев требуются еще более мощные
детонаторы: это обеспечило бы большую безопасность антигризутных
взрывчатых веществ, предупредило бы в ряде случаев образование
повышенного количества ядовитых газов при применении взрывча-
тых веществ при подземных работах; это обеспечило бы возмож-
ность более глубокого суррогатирования взрывчатых веществ, при-
меняемых для снаряжения боеприпасов.
Для решения этой задачи были предложены более мощные ана-
логи тетрила, которые были подробнее изучены по инициативе гор-
ного бюро взрывчатых веществ в США 2, а именно, тринитрофе-
нилнитроамино этилнитрат и гексанитродифениламиноэтилнитрат.
Тринитрофенилнитроаминоэтилнитрат,
л е н т р и л,
/NO,
.
xch2ch2ono2,
o2n< xno2
Xno2
Bennet, J. Chem. Soc., 115, 576, 1919.
Clark, Ind. Eng. Chem-, 25, 1385—1390, 1933: 26, 554—556, 1934.
362
получается нитрованием 2,4-динитрофенилэтаноламина. 2, 4-Дини-
трофенилэтаноламин получается конденсацией 2,4-динитрохлорбен-
зола с этаноламином в спиртовом растворе в присутствии едкого
натра:
Cl HN-CH2CH2OH
|/X|N°2 <4|N02
I I + NH2CH2CH2OH + NaOH----------> | 4- NaGl + H2O
XNO2 4NO2
Динитрофенилэтаноламин получается в виде крупных оранжево -
жёлтых кристаллов с выходом 70% от теории. После повторной пе-
рекристаллизации продукт имел температуру плавления 92°. При
этой реакции получается в качестве побочного продукта тетранитро-
дифенилэтаноламин:
С1
/Ч,№2
2 | | + NH2CH2CH2OH
Xno2
ZCH2CH2OH
/хро2
Xno2
применяющийся для приготовления описанного ниже второго анаг
лога тетрила.
100 г 2,4-динитрофенилэтаноламина растворялось в 1000 г 95%-ной
серной кислоты при температуре 20—30°. Полученный раствор
вводился в 300 г азотной кислоты (с содержанием 87% HNO3) при
20—30° с выдержками при 30, 40 и 50° — при каждой температуре
по полчаса.
П е н т р и л выделялся в виде суспензии в отработанной кис-
лоте, из которой он изолировался выливанием этой кислоты в ледя-
ную воду Выход нитропродукта 90% от теории.
После отмывки кислот продукт перекристаллизовывался из
бензола и получался в виде микроскопических светложелтых кри-
сталлов.
Температура плавления продукта после повторной перекри-
сталлизации была 128°.
Пентрил выдержал испытание на химическую стойкость по между-
народной пробе нагреванием при 75° в течение 48 часов без потери
в весе. Ряд других испытаний показал, что он очень устойчив при
длительном хранении при 75° (только через 40 дней слегка измени-
лась окраска и температура плавления понизилась на 0,5°).
Чувствительность пентрила к удару немного меньше, чем тетрила
и больше пикриновой кислоты. Чувствительность к трению несколько
больше тетрила. Чувствительность его к детонации одинакова
с таковой же тетрила.
363
Фугасное действие на 16% больше* чем тетрила; бризантное дей-
ствие на 12% больше тетрила. 0,5 г пентрила, инициируемого 0,2 г
гремучей ртути, подрывает 40%-ный динамит на расстоянии на
37% дальше, чем тетриловый детонатор с весом заряда в 1,34 г (кап-
сюль-детонатор № 8).
Гексанитродифениламин о»э тилнитрат
получается по следующей схеме:
С1
/\no,
2! . I +NH2CH2CH2OH
\о2
N-CH2CHaOH
ch2ch2ono2
O9N,/'41NO2
2 2 I 2
no2 Xno2
Тетранитродифениламино этанол получается в виде побочного про-
дукта при получении динитрофенилэтаноламина. Тетранитропродукт
растворяется в 7-кратном количестве купоросного масла и раствор
приливается к 4-кратному количеству (тоже по весу тетранитропро-
дукта) азотной кислоты (с содержанием 90% HNO3) при температуре
не выше 30° и с последующей выдержкой при температуре 50°.
Выход промытого водой гексанитропродукта составляет 80% от
теории.
Свойства гексанитро дифениламиноэтилнитрата.
Очищенный путем растворения в ацетоне и высаживания из этого
раствора 95%-ным спиртом гексанитро продукт имеет вид бледно-
желтых блестящих пластинок с температурой плавления 184°. Плот-
ность 1,63. Температура воспламенения около 390°. Чувствитель-
ность к удару немного меньше, чем у пентрила и тетрила. Чув-
ствительность к детонации близка к таковой же тетрила. Сила, опре-
деленная по песочной пробе, на 3% меньше тетрила.
364
ГЕКСАНИТРОДИФЕНИЛАМИН, ИЛИ ГЕКСИЛ
Гексил
o2n/xno2 o2n/x
no2
был впервые описан в 1874 г.
NO2 . NO2
Аустеном \ получившим его нитрованием пикрил-р-нитроанилина.
В том же году его получил Гнем 1 2 нитрованием дифениламина.
Ни один из этих способов не нашел практического приме-
нения вследствие высокой стоимости получавшегося продукта.
В,'1913 г. Картер 3 /азработал нашедший практическое примене-
ние метод, основан /ый на реакции взаимодействия 2,4-динитро-
хлорбензола с анилином, в результате чего получается динитродц-
фениламин. Нитрованием последнего азотной кислотой в две стадии
получается гексанитро дифениламин. Этот метод значительно усовер-
шенствован Маршалем 4, который заменил азотную кислоту серно-
азотной смесью.
1. Свойства гексила. Гексил получается в виде желтого кри-
сталлического порошка; плавится при температуре 249° с разло-
жением; не растворим в обычных растворителях. Его можно
перекристаллизовывать из ледяной уксусной кислоты и концентри-
рованной азотной.
Вследствие присутствия 6 нитрогрупп водородный атом NH-rpyn-
пы приобрел отрицательный характер и может быть замещен ме-
таллом.
Соответствующие его соли можно получить нагреванием водных
или спиртовых растворов (или суспензий) гидроокисей металлов
или карбонатов с суспендированным в этих растворах гексанитро-
дифениламином 5.
Все соли, за исключением аммониевой и магниевой, чувствитель-
ней к удару, чем чистый гексил. Вообще ряд чувствительности (если
исключить натриевую соль) таков же, как и у соответствующих
солей пикриновой кислоты и тринитрокрезола.
Первой в этом ряду стоит наиболее чувствительная свинцовая
соль; за ней следует Си, Na, Fe, К, Са, NH4.
При практической оценке этих данных надо, однако, учесть, что
вследствие исключительно трудной растворимости гексила в воде
скорость образования солей очень замедлена.
Опасность при применении гексанитро дифениламина должна быть
поэтому много меньше, чем у несколько легче растворимого трини-
трокрезола и особенно у значительно легче растворимой пикрино-
вой кислоты.
1 Austen, Вег., 7, 1248, 1874.
’Gnehrn, Вег., 7, 1399, 1874; 9, 1245, 1876.
’Carter, Z. Schiess- u. Sprengst-, 8, 205 и 251, 1913.
* Marshall, Ind. Eng. Chem., 12, 336, 1920.
5 К a s t и L anghans, Z. Schiess-u. Sprengst-, 14, 1, 25, 1919.
365
Кроме того, надо отметить, что опасность образования солей во
время производства этого вещества существует только тогда, когда
в растворе нет минеральных кислот.
Химическая стойкость. Маршаль 1 применил для
определения химической стойкости гексила видоизмененную им
пробу Обермюллера, которая состояла в следующем. Отвешенное
количество испытываемого материала нагревалось при 120° в ва-
кууме в колбе, соединенной с манометром. Давление по манометру
отмечалось через каждый час, а увеличение давления пересчитыва-
лось на число кубиков газа, выделявшегося в течение часа при дан-
ном давлении.
При параллельном испытании по этой пробе гексила, тротила и
тетрила получены следующие результаты:
Таблица 69
Наименование вещества Температура плавления °C Стойкость, выраженная числом с.и3 газа, выде- ляющегося в 1 час на 1 г при 100 м-л давления
ГекСил 240 4,4
Тротил 77,3 3,6
Тротил 80,1 2,1
Тетрил 127.0 13,8
Тетрил . 128.2 2,6
Тетрил 129,0 1.6
Отсюда видно, что стойкость гексила меньше, но близка к стой-
кости тротила и тетрила.
По данным Штетбахера2, шведский гексил, плавящийся не
ниже 240°, выдерживает пробу Абеля при 135° не менее 1 часа и
является, следовательно, значительно более стойким, чем тетрил.
Взрывчатые свойства гексила. Гексил менее
чувствителен к удару, чем тетрил, и больше, чем пикриновая кислота.
Чувствительность к детонации. По Маршалю',
для полной детонации названных ниже взрывчатых веществ необ-
ходимы следующие минимальные количества инициирующего взрыв-
чатого вещества (смесь гремучей ртути с бертолетовой солью):
для гексила...................... 0,18 г
» тетрила....................... 0,20 »
» тетраиитроанилина............. 0,20 »
» тротила....................... 0,25 »
1 Marshall, loc. cit.
2 Штетбахер, Пороха и взрывчатые вещества, стр. 424, ОНТИ, 1936-
366
Отсюда следует, что гексил немного чувствительней к детонации,
чем тетрил и тетранитроанилин.
Бризантность по Касту:
Гексил с плотностью 1,32 ..3,32
Пикриновая кислота с плотностью
1,34.................... 2,91
Скорость детонации по Касту:
При плотности 1,58 ........ 7095 м!сек
» » 1,68 ........... 7145 »
2. Физиологическое действие гексила. О ядовитости гексила
имеются в литературе разноречивые данные.
По мнению одних авторе^, гексил — самое ядовитое из всех
взрывчатых веществ. Эти ан Ары указывают, что при работе с гекси-
лом наблюдается воспаление кожи: образуются очень болезненные
маленькие пузыри, как при ожоге; при этом эпидермис совершенно
разрушается. Болезненное состояние продолжается несколько. ме-
сяцев. Чрезвычайно вредно действует пыль гексила на слизистые
оболочки рта, носа и легких.
Однако, по мнению других авторов, вреден не гексил, а динитро-
хлорбензол, если он недостаточно тщательно отмыт; последний вы-
зывает разрушение кожи с последующим воспалительным процес-
сом нарывного характера. Эти авторы указывают, что при работе
с гексилом в течение 1х/2 лет не наблюдали ни одного случая забо-
левания, хотя в работе принимали участие люди разного возраста и
пола.
3. Применение гексила. Гексил применяется для снаряжения
морских мин в смеси с тротилом. Для приготовления этой смеси
тротил плавят и к нему добавляют при перемешивании гексил;
к 40—30 в. ч. тротила добавляется 60—70 в. ч. гексила. Та-
кой сплав носит название новита или гексанита. Плотность новита
1,64—1,70.
Во время первой империалистической войны немцы снаряжали
морские снаряды смесью тротила с гексилом, флегматизированной
небольшим количеством парафина. Такая смесь применялась для
снаряжения морских снарядов только в качестве суррогата, при
недостатке тротила.
Вследствие недостатка тетрила в США во время империалисти-
ческой войны было предпринято исследование гексила с целью
использования его в детонаторах в качестве замены тетрила. При
этом были получены положительные результаты, которые, однако,
не были реализованы вследствие окончания войны к этому времени.
4. Получение гексила. Гексил можно получить по методу, ана-
логичному способу получения тетрила из диметиланилина: раство-
рением дифениламина в серной кислоте (нейтрализация дифенил-
амина) и медленным сливанием полученного раствора к концентри-
рованной азотной кислоте. Этот способ опасен, так как при нитро-
вании легко возможно самовоспламенение и взрыв; кроме того, ди-
фениламин дорог. Поэтому при получении гексила в заводском мас-
штабе исходят из динитрохлорбензола и анилина, конденсацией
367
которых получают динитро дифениламин:
NH2HC1
Полученный динитродифениламин нитруют в две фазы для по-
лучения гексанитро дифениламина.
Получение динитродифениламина. По перво-
начально принятому способу в качестве среды служит 2,5 в.ч . спир-
та на 1 в. ч. динитро хлор бензола. На 1 мол динитро хлорбензола
берется 2 мол анилина, из которых одна идет на нейтрализацию.
К концу реакции все застывает вместе с растворителем в виде
темнокрасной массы. Затем к этой массе добавляют воды, нейтрали-
зуют мелом и промывают водой.
Получается динитро дифениламин с температурой плавления 150—
454°. Температура плавления чистого продукта 156—157°.
При получении динитро дифениламина по описанному способу
расходуется спирт; выгоднее и удобней следующий способ Ч
2 мол анилина и 1 мол динитро хлор бен зола суспендируют
в нагретой до 60° воде, взятой в 3-кратном количестве по отношению
к их общему весу. Нагревают при непрерывном перемешивании
.до 80° и поддерживают эту температуру в течение 1 часа. Обра-
зуется динитродифениламин в виде больших сростков, состоящих
из красных иголкообразных кристаллов. Для полного растворения
солянокислого анилина перемешивают еще полчаса при 80°, пере-
водят полученную массу на фильтр, промывают разбавленной со-
ляной кислотой и водой и сушат.
Выход динитро дифенила мина выше 95% от теории. Температура
плавления 148—152°.
Получение тетранитродифениламина. Ни-
трование в тетранитропродукт производится за границей в стой-
ких к температуре керамиковых горшках (из витреозиля).
На 1 в. ч. динитро дифениламина берется 8 в. ч. азотной кислоты
36° Вё (52% HNO3).
Начало подачи динитропродукта к азотной кислоте установлено
при температуре 40°. Подъем температуры к концу подачи динитро-
дифениламина не выше 90—100°. При охлаждении выпадает раство-
ренный в горячей азотной кислоте тетранитродифениламин:
-----NH ------
( 4,NO2 O2N(z\
I । г
XNO2 XNO2
1 Am. пат. № 1309580, 1919.
368
Серноазотную смесь, по Картеру, не применяют для тетрани-
трования, так как динитродифениламин плохо в ней растворяется,
а поэтому плохо нитруется.
Однако, по данным Маршаля, это мнение неправильно. Ока-
залось, что нитрование протекает успешно, если вводить сухой ди-
нитродифениламин при 70° к Зх/2— 4 в. ч. кислотной смеси, содер-
жащей от 30 до 45% HNO3 и 50—40% H2SO4, после чего сделать
выдержку при 80—90°. Затем массу охлаждают и фильтруют. Полу-
чают аморфное вещество коричневато-желтого цвета, содержащее
небольшое количество боле\ высоконитрованных продуктов, кото-
рое нитруют до гексанитр/ Соединения.
Нитрование тетранитродифениламина до
г*е ксанитродифениламина. По первоначально приня-
тому за границей способу гексанитрование производится в эмали-
рованных чугунных нитраторах или в аппаратах из V2A. На 1 в. ч.
тетранитропродукта берется 8 в. ч. азотной кислоты 48? Вё
(92% HNO3). Подача тетранитропродукта при 40°. Постепенный
подъем температуры до 90°. Потом отсасывают на вакуум-воронке,
промывают сначала крепкой, потом слабой азотной кислотой для
удаления клейких примесей и, наконец, промывают водой и сушат
при 50—60°.
По этому способу также требуются эмалированные, чугунные
или алюминиевые нитраторы и алюминиевые трубопроводы; кроме
того, велик расход концентрированной азотной кислоты.
По данным Маршаля, и на этой стадии можно применять кис-
.лотную смесь. В этом случае вводят при температуре 70° кислый
тетранитродифениламин к 3,75 в. ч. кислотной смеси состава:
40% Н2ЗО4и 60% HNO3. После подачи нитропродукта делают часо-
вую выдержку при 90°, охлаждают до комнатной температуры и
фильтруют. Получают окрашенное в желтый цвет кристаллическое
вещество, плавящееся с разложением при температуре 238,5°—•
239,5°.
При нитровании динитропродукта до тетра- и гекса со единений
сёрноазотными смесями выход составляет 86% от теории.
Состав отработанных кислот при этом следующий:
Таблица 70
Наименование вещества Отработанная кислота
от тетранитро- вания 7о от гексанитро вания о/ /0
H2SO4 ....... 60,95 40,03
HNO3 14,28 33,62
NHSO5 - 7,45 11,30
Н2О . . Органические веще- ства, растворимые 26,13 14,16
в эфире 1,07 0,89
Химия—13—24
3S9
•
Получение гексанитродифениламина ни-
трованием динитро дифениламина в одну фазу.
Можно нитровать динитродифениламин не в две, а в одну фазу1. Для
этого вносят 1 в. ч. динтродифениламина к 3,75 в. ч. кислотной смеси,
содержащей 30% H2SO4 и 70% HNO3 при температуре 70—80°. При
этом получается тетранитросоединение. Затем при температуре
80—90° осторожно прибавляют 2 ч. олеума, содержащего 107—
108% H2SO4 (около 30% свободного SO3).
Делают часовую выдержку при этой температуре. Охлаждают
и фильтруют. Получают вещество аморфного вида, с температурой
плавления 238°, с выходом 85 % от теории.
Подсчет стоимости показал, что выгоднее вести процесс в две ста-
дии. Кроме того, надо избегать получения аморфного вещества,
которое в сухом виде легко превращается в пыль и, следовательно,
представляет опасность отравления при работе с ним.
5. Промывка гексанитродифениламина. Для промывки гекса-
нитродифениламина нельзя применять щелочи, так как он образует
с ними растворимые в воде соли. Очень трудно отмыть полностью
кислоты из аморфного продукта, но кристаллическое вещество мо-
жет быть полностью отмыто от кислот 2—3-часовой промывкой ки-
пящей водой. Затем отжимают воду на центрифуге.
Г Л А В А XV
СОЕДИНЕНИЯ С ДВУМЯ ФЕНИЛЬНЫМИ ГРУППАМИ
* . (КРОМЕ ГЕКСИЛА)
Ниже приводятся краткие сведения о гексанитропроизводных
дифенила, дифенилоксида, дифенилсульфида и дифенилсульфона.
____no2 no,
1. Гексанитродифенил: O2N\ -------/NO2-
NO, NO7
Этот простейший и сильнейший представитель соединений с двумя'
фенильными группами — «удвоенный тринитробензол» — впервые
получен Ульманом и Белецким 1 в 1901 г.
Ульман и Белецкий нашли, что при взаимодействии аромати-
ческих галоидопроизводных легко получаются соединения ряда
дифенила (причем эта реакция не ограничена только применением
галоидонитросоединений, имеющих подвижный галоидный атом,
как, например, пикрилхлорид, динитро хлорбензол и др., но в нее
вступают и те ароматические галоидные производные, которые
не имеют отрицательной группы в ортоположении к галоидному
атому). И в последнем случае могут быть получены с хорошим вы-
ходом дифенильные производные, если соответствующие иодистые
соединения обработать медью.
Реакция протекает настолько энергично, что для спокойного ее
течения приходится применять разбавитель, например, нитробен-
зол или песок.
Для получения 2,2'-динитродифенила конденсируют о-нитро-
хлорбензол при температуре 200—210°, применяя в качестве раз-
бавляющей среды песок:
1 Marshall, loc. cit.
Выход продукта €0% от теории.
‘Ullman и Bielecki, Вег., 34, 2179, 1901 .
371
При получении 4,4' -динитродифенила приходится применять
р-нитроиодбензол, так как при параположении нитрогруппы к га-
лоиду очень мало ослабляется связь последнего с ядром, причем
не применяется разбавитель (нитробензол или даже песок) и реакция
протекает при 220—235°:
O2N У J 4- j/ ) NO2 + 2Сп---->
----> Q,n/ /~\ /NO2 + 2CuJ
Гораздо большей подвижностью обладает галоидный атом в ди-
нитросоединениях, и соответственно реакция идет значительно энер-
гичней. При получении 2,2',4,4'-тетранитродифенила взаимо-
действием с 2,4-динитрохлорбензолом недостаточно умеряющего
влияния песка на течениё реакции. Поэтому растворяют динитро-
хлорбензол в нитробензоле и нагревают с медью в течение 1 часа
при кипении (208°):
____NO2 __________
O2n/ ^)С1 + С1/ \nO2 + 2Cu--------------->
NQT
-----> O2N/ Х~\ >NO2 + 2CuC1
no2 no7
Выход 60% от теории.
Наконец, для получения 2,2',4,4',6,6'-гексанитродифенила не-
обходимо вести реакцию конденсации пикрилхлорида только с при-
менением разбавителя, так как при нагревании обоих веществ при
127° и в отсутствии разбавителя они реагируют со взрывом. При
применении же нитробензола в качестве растворителя реакция про-
текает вполне нормально, причем выход составляет 55% от теории:
___no2 no2
O2n/ ^С1 + С1/ ^NO2 + 2Cu ——>
no2 nd7
____no2 no2
----> O2n/ ^NO24-2CuC1
no2 N07
Гексанитро дифенил представляет собой нейтральное, стабильное
вещество. Температура плавления 230°. Температура вспышки
выше 320°. По опытам Ван-Дуина 1 гексанитродифенил несколько
более чувствителен к удару, чем тринитробензол, но значительно
меньше, чем тетрил. Он на 10% сильнее гексила.
'Van Du у n, Rec. Tray. Chim. Pays-JJas, 39, 685, 1920.
He применяется в технике вследствие высокой стоимости.
_________________________________no2 .________no2
2. Гексанитродифенилоксид: O2N\ /—О—/ /NO2
‘ NO2 NOT
получается так: m-нитрофенол растворяют в малом количестве аце-
тона и полученный раствор смешивают с крепким водным раствором
едкого натра; нагревают на водяной байе и добавляют ацетонный
раствор тринитрохлорбензола \ Затем нагревают при слабом кипе-
нии, отгоняют ацетон и извлекают уксусной кислотой тетранитроди-
фенилоксид:
____NO2 ___NO2
* O2nZ + NaO/ \------------------>
4 NO2
____________no2 __________no2
----> O2n/
NO2
Полученный продукт нитруют серноазотной смесью; выдержка
при 100°.
Температура плавления 2 гексанитродифенилоксида 269°.
По данным Вестфальского акционерного общества, запатенто-
вавшего это вещество, чувствительность его к удару мала; напротив,
по исследованиям Ван-Дуина 3, чувствительность велика.
____no2 no2
3. Гексанитродифенилсульфид4 O2N\ \ /^О2
no2 no7
, При взаимодействии 2 мол динитрохлорбензола с 2 мол гипо-
сульфита идет следующая реакция:
2C6H3(NO.2)2C1 + 2Na2S2O3 -> C6H3(NO2)2SC6H8(NO2)2 +
+ 2NaCl + Na2S3Oe.
При тех же условиях тринитрохлорбензол образует с гипосульфи-
том темную массу. Но можно получить хорошего качества продукт
с хорошим выходом, если вместо 2 мол гипосульфита ввести в реак-
цию 1 мол и нейтрализовать образующуюся при реакции серную
кислоту:
2CeH2(NO2)3Cl + Na2S2O3 + Н2О -> C6H2(NO2)3SC6H2(NO2)3 +
+ 2NaCl 4- H2SO4
1 В герм. пат. № 635185, (1936) описывается способ получения гексани-
тродифенилоксида нитрованием дифенилоксида .
2 По другим данным 227° .
3 Z. Schiess- u. Sprengst-, 17, 21, 1922.
1 Rec. Trav. Chim. Pays-Bas, 39, 157—159, 1920.
372
373
Для нейтрализации серной кислоты добавляется углекислый магний.
Тринитрохлорбензол берется в спиртовом растворе.
Надо очень точно следить за тем, чтобы не допустить в реакцию
ни малейшего избытка гипосульфита, в противном случае произой-
дет потемнение продукта и понизится выход.
Температура плавления очищенного перекристаллизацией гекса-
нитро дифенилсульфида 233—234°.
Гексанитродифенилсульфид обладает большой химической стой-
костью; по фугасному действию не отличается от гексила; чувстви-
тельность его к удару немного меньше, чем у гексила. Он не ядовит
и не вызывает заболеваний кожи.
Применялся в Германии во время империалистической войны
в смеси с тротилом (50: 50) для снаряжения авиабомб.
___no2 no2
4. Гексанитродифенилсульфон: O2NZ $ — \ /^О2
no?
В то время как тетранитродифенилсульфид окисляется в сульфон
азотной кислотой 1 или хромовой кислотой в кислом растворе, не
удается превратить гексанитродифенилсульфид в сульфон ни нагре-
ванием сульфида с азотной кислотой (уд. вес 1,52) в запаянной
трубке, при температуре до 200°, ни кипячением с безводной хро-
мовой кислотой, растворенной в уксусной кислоте 2.
Окисление дипикрилсульфида удается смесью хромовой кислоты
с концентрированной азотной кислотой'3.
Температура плавления гексанитродифенилсульфона 307°.
Чувствительность его к удару меньше сульфида, но больше,
чем тетрила. Немецкие авиабомбы снаряжались смесью 50 в. ч.
тринитротолуола с 50 в. ч. гексанитродифенилсульфона.
По словам Штетбахера, зти соединения, как и гексанитродифенил-
сульфид, применялись для снаряжения авиабомб потому, что к их
значительному взрывному действию присоединялось отравляющее
действие, вызываемое образованием окислов серы.
1 Бейльштейн и К у р б а т о в, Lieb. Ann.,197, 78., 1879. По данным этих
авторов ЗЛ^Ч'-тетранитродифенилсульфид окисляется в сульфон при на-
гревании с азотной кислотой уд. вес 1,5 при 120° в течение 6 часов в запа-
янной трубке.
2 Blanksma, Rec. Tray. Chim., Pays-Bas, 20, 425, 1901 .
’Van D u у n и van L e n n e p, Rec. Trav. Chim. Pays-Bas, 39, 160,
1920. Ср. герм. пат. № 269826, Zbl, 1914, I, 723.
Г ЛАВ А XVI
ГЕКСОГЕН
Гексоген получается нитрованием гексаметилентетрамина (уро-
тропина).
СТРОЁНИЕ ГЕКСАМЕТИЛЕНТЕТРАМИНА
Бутлеров предложил 1 следующую формулу строения гексамети-
лентетрамина:
N <гн.
В этой формуле 1 атом азота является 3-валентным и 3 атома
5-валентными.
Лозеканн 2 предложил формулу, в которой все 4 атома азота
являются 3-валентными:
,СН2 — N =СН2
N^-CH2- n = ch2
xCH2-N=CH2
Этой формулой Лозеканн пытался объяснить то, что гексаметилен-
тетрамин образует с бромистым серебром продукт присоединения.
Ею легко объяснить взаимодействие гексаметилентетрамина с азо-
тистой кислотой с образованием динитрозо пентаметилентетр амина:
NO
СН2 — N — СН2
/СН2 — N = СН2 / \
N<CH2-N = CH2 4- 2N2O3------> N------CH2-N
xch2-n=ch2 \ /
CH, — N—CH2
I
NO
Однако по данным Пуммерера и Гофмана 3, гексаметилентетрамин
не гидрируется водородом в присутствии платины и, следовательно,
не содержит двойных связей.
1 Б у т л е р о в,-Lieb. Ann., Ill, 250, 1859; 115, 322, 1866.
2 L Osekann, Chem. Ztg-, 14, 1409, 1890.
Pummerer и Hofmann, Ber., 56, 1255, 1923.
375
Трилла и Файолла 1 при восстановлении гексаметилентетрамина
наблюдали образование аминов и отсюда сделали вывод, что это со-
единение имеет строение:
N = CH2
“2'>xn = СН2
т. е. имеет состав C3HeN2.
По их мнению, образование метиламина при восстановлении ге-
ксаметилентетрамина цинковой пылью в солянокислом растворе
идет следующим образом:
ZN = CH, ,NH — СН3 NH,CH3
Н2с/ “ + 2Н,^НС/ 3 ±Н,О_>СН2О+ ‘
xn = ch2 4nh-ch3 nh2ch3
Но Грасси и Мотта 2 показали, что при восстановлении гексаме-
тилентетрамина в кислом растворе кроме метиламина получается еще
и триметиламин, вследствие чего они предпочли формулу Лозеканна.
Дуден и Шарфф 3 предположили, что в гексаметилентетрамине
атомы азота расположены в углах правильного тетраэдра, вследствие
чего они приписывают ему следующее строение:
или
Доказательством правильности этой формулы, по Дудену и
Шарффу, являются продукты расщепления гексаметилентетрамина
при его взаимодействии с азотистой кислотой и хлористым бен-
зоилом:
NO
I
N N - СССвН5
Н2С^\,СН2 н2с/хсн2
I I
ON - N1^ /IN — NO HsCeOCN / /NCCC6H 8
CH2 CH2
тринитрозотриметилентриамин трибензоилтриметилентриамин
1 Trillat и Fayollat, Bull. Soc. Chim- Fr., 3 Serie, 14, 22, 1891.
2 Grassi и M otta, Gazz. Chim. Ital., 29, 33, 1899.
3 Duden и Scharff, Lieb. Ann., 288, 218, 1895.
376
Кон 1 2 на основании результатов исследования нитро- и бромпро-
изводных гексаметилентетрамина дает ему строение метиленара-
бинозы:
СН2 = С—СН—СН—СН—СН
\/ I I II
NH NH2 NH2 NH
В этом случае динитрозопентасоединение имело бы строение:
СНО- СН - СН - СН - СН
I I II
NH NH NH NH
. II
NO NO
Формула Кона лучше всего объясняет существование продуктов
присоединения 2 и 4 атомов галоида к 1 мол гексаметилентетра-
мина и легкую отщепляемость 2 галоидных атомов от продуктов
присоединения:
СН2Вг - СВг - СН - СН - СН - СНВг
\ III
NH NH2 NH2 NHBr
CH,Br -;CBr - CH - CH - CH - CH
\ I I II
NH NH2 NH2 NH
В 1923 г. ряд авторов ? опубликовал исследования о строении
кристаллической решетки гексаметилентетрамина. По их данным
атомы азота расположены на углах тетраэдра, а углеродные атомы—
на углах октаэдра, описанного вокруг тетраэдра. Эти данные подтвер-
ждают правильность формулы Дудена и Шарффа.
Тем не менее в последний период опубликован ряд исследований
Адессандрини и Маротта 3 4, в которых они на основании изучен-
ного ими ряда химических превращений доказывают правильность
формулы Лозеканна
Мы принимаем формулу Дудена и Шарффа. Эта формула наи-
более подтверждается всеми имеющимися экспериментальными дан-
ными, а с другой стороны, реакции взаимодействия гексаметилен-
тетрамина при нитровании (как получение гексогена, так и ряд
побочных реакций) легче всего объясняются, если принять для ге-
ксаметилентетрамина формулу Дудена и Шарффа. Наконец, строение
1 Kohn, J. Р г a k t. Chemie, 56, 345, 1897.
2 Dickinson и Raymont. J. Amer. Chem. Soc., 45, 22, 1923;
Gonell и Mark, Z.PhysikaL Chem., 107, 181, 1923.
sA4essandrini и Marotta, -Graz. Chim. Ital., 59, 942, 1929.
4 А. И. Настюков в «Успехах химии» (стр. 151, т. V, 1936) говорит,
что работами Маротта и Алессандрини твердо установлена правильность фор-
мулы строения гексаметилентетрамина, предложенной ранее Лозеканном.
377
гексогена легче всего устанавливается, если принять для гексаме-
тилентетрамина формулу Дудена и Шарффа.
В этом случае гексоген, подобно гексаметилентетрамину, рассмат-
ривается как производное гипотетического циклотриметилентри-
-амина:
СН2
nh/^nh
gh2^Jgh2
NH
циклотриметилентриамин
сн2
NO2 - - NO2
1 !
CH2!x^CH2
N — NC)2
тринитроц иклотриметилентриамин
или гексоген
СВОЙСТВА И ПРИМЕНЕНИЕ ГЕКСАМЕТИЛЕНТЕТРАМИНА
1. Физические свойства. Гексаметилентетрамин представляет
собой белый кристаллический порошок без запаха, сладкий на вкус.
Стоек при хранении на воздухе. При нагревании под обыкновенным
давлением выше 100° возгоняется очень небольшое его количество
с частичным разложением, причем образуется метиламин.
В вакууме он возгоняется при 230—270° почти без разложения Г
Молекулярная теплота его сгорания при постоянном объеме
1005,85 кал 1 2, по другим измерениям 1036,9 кал3.
Растворимость гексаметилентетрамина в разных растворителях 4.
В 100 мл воды растворяется при комнатной температуре 167 г;
плотность этого раствора 1,0985.
В 100 мл абсолютного этилового спирта растворяется на холоду.2,89 г
» . . , 90»/0 . . » .... - 5,58 .
. , „ , бензола , , . „ 0,23 .
. » . ксилола п * „ . . 0,14 ,
В петролейном эфире гексаметилентетрамин вовсе не раство-
ряется. Отделенный от растворителя гексаметилентетрамин немного
летит с ним при температуре сушильного шкафа; в течение 40 часов
улетучивается 18,38% вещества.
Растворимость гексаметилентетрамина в водных растворах ам-
миака различной концентрации см. стр. 386. При взаимодействии
гексаметилентетрамина с кислотами легко отщепляется NH3 и ре-
генерируется формальдегид 5.
2. Химические свойства. Гексаметилентетрамин разлагается под
действием соляной, серной, уксусной и салициловой кислот, обра-
зуя, главным образом, формальдегид, аммиак, углекислоту и метил-
1 Tollens, Вег., 17, 653, 1884.
8 Delepine, Bull. Soc. Chim- Fr., 3 Ser’e, 15, 1199, 1896.
3 Harvey, и Baekeland, Ind. Eng. Chem. 13, 135, 1929.
* U tz, Siiddeutsche Apoth. Ztg, 59, 832, 1919.
s Tollens, Ber., 17, 653, 1889.
378
амин Ч Относительные количества образующихся продуктов раз-
ложения зависят от концентрации Применяемых кислот, темпера-
туры и продолжительности их действия.
При пропускании слабого тока N2O3 через раствор гексамети-
лентетрамина в ледяной уксусной кислоте происходит выделение
газа: образуется формальдегид, аммиак и метиламин. При пропус-
кании N2O3 через тот же раствор при повышенной температуре обра-
зуется много СО2 и метиламин.
Тренделенбург 8 установил, что скорость расщепления гекса-
метилентетрамина тем больше, чем больше концентрация водород-
ных ионов. Соответствующая зависимость видна на рис. 44
и 45.
Из кривых рис. 44 и 45 видно, что скорость реакции падает
с уменьшением концентрации водородных ионов, однако, не пропор-
ционально последней, но медленнее. И в нейтральном растворе,
следовательно, при равном содержании в растворе Н’ и ОН' -ионов
при температуре 38° разлагается еще измеримое количество гекса-
метилентетрамина .
Эти данные показывают, что упаривание нейтральных водных
растворов гексаметилентетрамина под атмосферным давлеЯием со-
провождалось бы заметными потерями гексаметилентетрамина вслед-
ствие разложения последнего. Поэтому при получении гексамети-
лентетрамина в технике, как это мы увидим на стр. 385, ведут упари-
вание в вакууме и постоянно добавляют незначительные количества
аммиака, чтобы реакция раствора была щелочная.
При взаимодействии гексаметилентетрамина с азотистой кисло-
той 3 при отсутствии избытка последней образуется преимущественно
динитро зопента метилентетр амин:
NO
N - СН2 - N • N.0 СН2 — N — СН2
СН2/\СН2 ;СН2 ------CH,-N
n-gh2-n-no \ /
СН2 —N —СН2
I
NO
формула Делепина*
формула Лозеканва
Это вещество легко разлагается горячими минеральными кис-
лотами с образованием НСОН, N2 и NHg.
1 Jschidz и Inouye, Zbl., I, 1087, 1906; Hartung, J. Prakt.
Chemie, 46, I, 1892.
2 Trendelenburg, Biochem. Ztschr., 59, 146, 1919.
’Griess и H arrow, Ber., 16. 2737, 1888; Fr. Mayer, Ber., 16,
2883’, 1888.
4 C. R., 120. 197, 1895
379
время 6 минутах
Рис. 44. Разложение гексаметилентетрамина в кислотах
разной концентрации при температуре 38°.
Кривая I: pH в начале 0,5; в конце 0,6 Кривая 6: рНвначале 3,9 ; в конце 4,0
Кривая А—процент разложенного гексаметилентетрамина через час при 38°;
кривая В — то же через 6 час. при 38°.
______________________________________________________________________________________i
380
В присутствии большого избытка азотистой кислоты образуется,
главным образом, триметилентринитрозамин:
N - NO
ON -N'^yN - NO
сн2
При нагревании с разбавленными кислотами это вещество разла-
гается с выделением НСОН и N2. Концентрированные кислоты
разлагают это соединение моментально с выделением окислов азота.
Гексаметилентетрамин представляет собой слабое основание.
Образует соли с кислотами, весьма реакционноспособен.
При взаимодействии его с азотной кислотой в зависимости
от условий реакции образуется нитрат гексаметилентетрамина
C6H12N4 • HNO3, динитрат гексаметилентетрамина CeH12N4 • 2HNO3
или гексоген C3HeNeO6.
Нитрат гексаметилентетрамина C6H12N4 • HNO3 образуется
при взаимодействии спиртового раствора гексаметилентетрамина
со спиртовым же раствором азотной кислоты. Нитрат представляет
собой кристаллическое вещество, легко растворяется в воде. Вод-
ный раствор его имеет нейтральную реакцию *.
Динитрат гексаметилентетрамина 1 2 C6HL2N4 • 2HNO3 образуется
при добавлении азотной кислоты к водному раствору гексаметилен-
тетрамина. При этом из раствора выпадает соль. Динитрат можно
также получить путем добавления твердого гексаметилентетрамина
при охлаждении к разбавленной азотной кислоте 3. Например,
100 г гексаметилентетрамина добавляют' к 258,3 г азотной кислоты
(с содержанием 34,8% HNO3) при температуре не выше 5°; после
некоторого стояния осадок отделяют и тотчас же высушивают током
воздуха. Выход составляет 180 г (теоретически 190 г).
По Геннингу4, растворяют 1 в. ч. гексаметилентетрамина
в 2 в. ч. воды при 0—5°, добавляют к этому раствору по каплям
0,9 в. ч. азотной кислоты уд. веса 1,46 (65% HNO3). Реакция
образования динитрата идет по схеме:
CeH.,N4 + 2HNO3 ->C6H12N42HNO,
Позже было установлено, что можно вести слив кислоты при тем-
пературе до 40—50° без ухудшения выхода или качества продукта.
1 Cambi ег и Brocket,Bull. Soc. Chim. Fr., 3 ser., 13, 392, 1895.
2 Moschatos и T oil ens, Lieb. Ann., 272, 273, 1892.
* Герм. пат. № 298412 от 12/VII 1916; выдан 3/VII 1919.
4 H e n n i n g, Ber., 21, 1999, 1888.
381
Из раствора выпадает белый кристаллический о.садок динитрата
гексаметилентетрамина, который отфильтровывают, промывают
50%-ным водным раствором спирта и эфиром. Выход динитрата гек-
саметилентетрамина составляет в среднем 95% от теории.
Свойства динитрата. Температура плавления дини-
трата — 165°, он легко растворим в воде; раствор имеет сильно
кислую реакцию; при стоянии он разлагается, выделяя формаль-
дегид; нерастворим в спирте, эфире, хлороформе, ацетоне, четырех-
хлористом углероде.
При взаимодействии гексаметилентетра-
мина с перекисью водорода в присутствии лимонной
или азотной кислот получается гексаметилентрипероксиддиамин:
/СН2-О-О —СН2Х
N—СН2 — О — О — СН,—N или
\СН2 - О - О - СН/
о-сн2. сн2-о
I \N-CH2-O-O-CH2-N/
О - СН/ СН2 _ 0
Это соединение предложено в качестве инициирующего взрывча-
того вещества.
3. Меры предосторожности при работе с гексаметилентетрамином.
При длительном соприкосновении с гексаметилентетрамином проис-
ходит раздражение кожи, что необходимо учитывать при работе
с этим продуктом х.
4. Применение гексаметилентетрамина. Гексаметилентетрамин
применяется для изготовления гексогена; предложен для изготовле-
ния гексам етилентрипероксиддиамина.
Он широко применяется в медицине. Вследствие его растворяю-
щего действия на мочевую кислоту и ее соли применяется при моче-
вых камнях, для лечения гриппа (входит в состав кальцекса), брон-
хита и при большом числе других заболеваний.
Гексаметилентетрамин применяется в некоторых случаях в пи-
щевой промышленности для консервирования; как дезинфицирующее
средство; для приготовления искусственных смол и пластмасс; в ре-
зиновой промышленности в качестве ускорителя при вулканизации
каучука. Наконец, гексаметилентетрамин применяется в лаборатор-
ной практике для синтеза многих аминосоединений (в частности, для
получения мо но метиламина).
' 5. Количественное определение гексаметилентетрамина. 1. Для
быстрого и надежного определения объемным методом можно поль-
зоваться пикриновой кислотой, образующей с гексаметилен-
тетрамином двойное соединение2 C6H12N4 • С6Н2ОН (NO2)3.
1 India Rubber, 71, 12, 1926.
2 Подробности о способе получения этого соединения и о его свойствах
см. Moschatos и Tollens. Lieb. Ann., 272, 275, 1892.
362
К раствору 0,15—0,20 г основания в небольшом количестве
воды добавляют 20 см3 0,057V раствора пикриновой кислоты, пе-
ремешивают 5 минут, доводят объем до 100 см3, фильтруют и опре-
деляют в 50 см3 избыточную пикриновую кислоту титрованием
0,17V раствора едкого натра в присутствии метилоранжа.
Ошибка метода составляет ± 0,3%.
2. Для быстрого определения гексаметилентетрамина его можно
разложить:
C6H12N4 + 6Н2О + 2H2SO4 -> 6НСОН + 2(NH4).2SO4
сильным кипячением с серной кислотой и с полной отгонкой фор-
мальдегида. Нагревают 0,5 г основания с 40 слс3 7V раствора
серной кислоты в течение получаса до исчезновения запаха фор-
ума ль дегида; по охлаждении разбавляют и титруют свободную сер-
ную кислоту в присутствии метилоранжа. Расчет производят по
формуле:
ct • Зо л /
—Гп7Г — % гексаметилентетрамина,
где а — количество израсходованных см3 раствора едкого натра;
А' — навеска основания 1 2 1.
ПРОИЗВОДСТВО ГЕКСАМЕТИЛЕНТЕТРАМИНА
Гексоген получается нитрованием гексаметилентетрамина (уро-
тропина).
1. Получение гексаметилентетрамина из формалина и аммиака..
Исторические сведения. В 1859 г. Бутлеров2 опу-
бликовал свое наблюдение, что при пропускании сухого аммиака
через сухой триоксиметилен образуется кристаллическое вещество,
не обладающее запахом формальдегида, с основными свойствами.
Дальнейшими исследованиями 3 он установил, что соединение
представляет собой гексаметилентетрамин и имеет формулу CgH^N.j.
В 1869 г. Гофман 4 получил гексаметилентетрамин при упари-
вании формальдегида с аммиаком.
Длительное время после этого гексаметилентетрамин получался
в ограниченных количествах, так как в то время получение концен-
трированных растворов формальдегида представляло большие за-
труднения и получавшийся продукт был очень дорог. Например,
9в 1884 г. Толленс описал способ, при котором из 1100 см3 раствора
, сырого формалина и 200 см3 10%-ного аммиака получается 7 г
' гексаметилентетрамина.
1 Об определении гексаметилентетрамина осаждением хлорной ртутью см.
Z. Anal. Chem-, 36, 44. 1897.
2 Бутлеров, Lieb. Ann., Ill, 250, 1859.
3 Lieb. Ann., 115, 322, 1866.
4 Hofmann, Ber., 2, 153, 1869 .
385
В последнем десятилетии XIX столетия гексаметилентетрамин
•стали применять в значител ных размерах в медицине с этим сов-
падает разработка более совершенных способов получения гекса-
метилентетрамина. В 1899 г. Грасси и Мотта 1 опубликовали способ
его получения, при котором обрабатывали триоксиметилен кон-
центрированным аммиачным раствором; взаимодействие протекало
с болыгим выделением тепла. Раствор упаривался на водяной бане
при постоянном добавлении аммиака и сиропообразный остаток
обрабатывался абсолютным спиртом. Выход гексаметилентетрамина
составлял 70% от теории.
По этому методу в дальнейшем стали получать гексаметилен-
тетрамин в значительных количествах, причем взамен триоксимети-
лена брали кон ентрированные (с содержанием 30—36% формаль-
дегида) растворы формалина. При смешении компонентов избегали
повышения температуры выше 20° и при этом получали почти
теоретические выходы. z
Общие сведения о получении гексаметилентет-
рамина. Образование гексаметилентетрамина идет по реакции:
6СН2О,4- 4NH4CH = (CH2)6N4 6Н2О; при соблюдении надлежащих
.условий процесс идет почти количественно. Теоретически требуется
180 в. ч. формальдегида и 80 в. ч. аммиака для изготовления 140 в. ч.
гексаметилентетрамина. Практически, однако, требуется небольшой
избыток обоих компонентов, так как имеют место незначительные
потери. Особенно надо заботиться о том, чтобы к концу реакции
в реакционной массе присутствовал свободный аммиак, так как
в противном случае идет обратный процесс — гидролиз гексамети-
лентетрамина,— что приводит к уменьшению выхода, а с другой
стороны, начинается образование триметиламина, имеющего отврати-
тельный запах и делающего продукт негодным.
Описание производства гексаметилен-
тетрамина. Подробное описание производства гексаметилен-
тетрамина в большом масштабе дает Мак Ланг 2.
Исходными материалами здесь являются 30%-ный раствор фор-
мальдегида с содержанем 1—2% метилового спирта и аммиак, свобод-
ный от пиридина.
Поступающий из аммиачных баллонов А (рис. 46) аммиак по-
.дается в аппараты В, снабженные мешалками; емкость каждого
реакционного аппарата 45 J кг 30%-но, о формальдегида. Во время
--Z*------------------------------ ЛхгтгОиспЯТПТСЯ ВО ПОЙ
реакционного аппарата 45J m 30%-hoi о формальдегида. Во время 24 ча“сов ~~ ------o*v1[)ulMn VJlliai. ирИ w_ в
подачи аммиака мешалки работают и аппараты охлаждаются водой МаточныйЧеМ ПОлУчается чистый продукт. НИе
(температура охлаждающей воды около 8°). Температура реакцион- ЛОвину собиРаюЩийся в сосудахF, упаривается на™
ной смеси во время подачи аммиака 22° и держится постоянной фугуЮт и с ₽ сталлиз°вавпшйся при этом гексаметилентетрамин
е 5 чачив. По истечении 5 часов температура поднимается Л. J • 1
теплоты реакции либо уменьшением подачи охлаждающей uuwle второго упар
F ----™ к° R “?точные растворы перерабатываются
в течение 5 часов. По истечении 5 часов температура поднимается!
(за счет ’) eilJlUlbl рсапцпи ------ г ,
воды, либо увеличением скорости подачи аммиака) до 25°. В тече-j
ние 1 часа в каждый реакционный аппарат подается в среднем 3 кг
‘GrassieMotta, Gazz. Chim. Ital., 29, 43, 1899.
2 Me Lang, Chem. Trade, J , 76, 325, 1925.
•384
аммиака. Теоретически на 1800 кг 30%-ного формальдегида (за-
грузка 4 реакционных аппарата) требуется 204 кг 100%-ного аммиака.
Применяется избыток 6 кг NH3.
По окончании подачи аммиака раствор спускается в сосуды С,
где он оставляется на 1 день. Отсюда раствор подается в вакуум-
аппараты D для упаривания. Одновременно приводят в действие
автомат (на схеме не показан), подающий в минуту 7 см? водного
раствора аммиака к упариваемому раствору.
Рис. 46. Схема производства гексаметилентетрамина.
По достижении вакуума в 72—74 см водяного столба медленно
нагревают до 30° [находящуюся в аппарате жидкость и упаривают
ее при перемешивании. Отгоняющаяся вода имеет слабощелочную
реакцию. На упаривание требуется 12—14 часов. По прошествии
приблизительно 11 часов начинает выделяться гексаметилен-
тетрамин в виде красивых белоснежных кристаллов.
• По окончании упаривания останавливают вакуум-насосы и от-
крывают воздушные краны. Теплый раствор спускают непосредствен-
но на центрифугу Е, откуда маточный раствор стекает в сосуды F.
Фугований гексаметилентетрамин сушат при 30° в течение
ПОРЛП ---~ —
Получающиеся после второго упаривания и отделения продукта
ячннр пйг.таппи отдельно. Вследствие накоп-
ления в них посторонних примесей при упаривании этих маточных
растворов не получается чистый продукт, а так называемый тёхни-
теский гексаметилентетрамин.
Переработка технического гексаметилентетрамина на чистый
ЙЮизводится следующим образом.
Ййрш—13—25
385
Концентрация $одногораст$Ора амниона
Рис. 47. Растворимость гексаметилентетрамина
в аммиачных растворах.
300 кг технического гексаметилентетрамина растворяют в 400 л
дестиллированной воды; раствор фильтруют и упаривают с добавле-
нием несколько повышенного количества аммиака (10—12 смэ в ми-
нуту). Отгоняют около 300 л воды, фугуют и получают опять
чистый гексаметилентетрамин.
Получающийся здесь маточный раствор соединяют с таковым
от второго упаривания и все вместе упаривают в вакууме при 74 см
продукт ОПЯТЬ раС-
и получают чистый
составляет приблизи-
тельно 96% от тео-
рии (по формальде-
гиду)-
Чтобы использо-
вать холод, получаю-
щийся благодаря ра-
боте расширения ам-
миака, аммиачные
баллоны соединяют
с рефрижератором
для производства
льда; при этом амми-
ачные баллоны не по-
крываются льдом,
чем понижалось бы
давление аммиака.
Часть этого льда ис-
пользуется для ох-
лаждения воды (ох-
лаждающей).
Описанный Хем-
нициусом 1 способ
получения гексаме-
тилентетрамина от-
личается от преды-
дущего следующим.
Реакционные аппараты емкостью 250 л установлены в деревян-
ных чанах с водой для охлаждения. По окончании реакции к рас-
твору, который должен иметь запах аммиака, добавляют уголь для
обесцвечивания и фильтруют. Вакуум-аппараты эмалированные.
При упаривании к жидкости добавляют немного жира для устра-
нения вспенивания. Упаренный раствор вместе с выкристаллизо-
вавшимся веществом переводят в эмалированные чаны. На другой
день сливают с помощью сифона маточный раствор, покрывающий
кристаллы, и добавляют его к новому раствору. К влажным кристал/
лам добавляют немного аммиака (на 100 кг около 300—400 г
.. --------------------------------------------------------<
1 Chemni tins. Chem. Ztg, 735, 1928.
25%-ного аммиака), фугуют, затем промывают небольшим количеством
. дестиллированной воды и сушат при 50° на матерчатых лотках.
Выход по формальдегиду составляет около 96% от теории.
В описанных способах производства гексаметилентетрамина до-
бавлялось только такое количество аммиака, чтобы раствор имел
отчетливый запах его. Если, однако, добавить к реакционной массе
значительно больше аммиака, чем требуется для полного превраще-
ния формальдегида в гексаметилентетрамин, то растворимость по-
следнего начинает убывать с повышением концентрации аммиака.
Этим путем 1 удается выделить из реакционной жидкости главную
массу гексаметилентетрамина в форме чистого кристаллического
продукта. Растворимость гексаметилентетрамина в аммиачных рас-
тнорах различных концентраций показана на рис. 47.
При практическом выполнении этого способа отделяют выделив-
шийся из раствора кристаллический продукт и высушивают. Полу-
чают чистый продукт. Из фильтрата отгоняется избыточный аммиак,
раствор упаривается и вновь насыщается аммиаком. Можно к ча-
стично упаренному раствору добавить свежий раствор формальде-
гида и после этого насытить аммиаком.
Для выделения гексаметилентетрамина из растворов можно
пользоваться также сушкой в распылительных установках 2.
2. Другие способы получения гексаметилентетрамина. При вза-
имодействии формальдегида с углекислым аммонием3 на холоду 4
образуется гексаметилентетрамин: '
/ onh4 onh4 \
4 сС О + С = О + 18НСОН -» 3CeH12N4+8CO2 + 22НгО.
\ \)н 4 NH2 /
. Реакционную смесь упаривают в вакууме, а осадок очищается
перекристаллизацией из алкоголя или возгонкой в вакууме.
। Гексаметилентетрамин образуется при взаимодействии хлори-
стого аммония с формальдегидом в присутствии щелочи 5, а также
при взаимодействии сернокислого или азотнокислого аммония с фор-
малином 6.
1 Ам. пат. № 1566820 от 25/VII 1924; выдан 22/XII 1925.
' 2 S t е w а г t, Chem. Met. Eng., 472, 1928.
3 Продажный углекислый аммоний представляет собой смесь кислой соли
р4НСО3 и карбаминовокислого аммония NH.COONH,.
г *Z. Angew. Chem., 33, 48, 1920, англ- пат. № 17693, 1897.
5 Lieb. Ann., 288, 251. 1895; С. R., 120, 557, 1895; Z. Physikal. Chem.,
, 107, 1913.
3 Z. Angew. Chem., 33, 84, 1920; Pharmac. Weekbl., 58, 1463, 1922.
386
387
В последнее время приобрел производственное значение способ
получения гексаметилентетрамина при взаимодействии аммиака
с хлористым метиленом х. Реакция идет так:
6СН2С12 + 16NH3 -> C6H12N4 • НС1 + 12NH.C1
Водный раствор аммиака с хлористым метиленом нагреваю
в автоклаве при температуре 100—120°. После отгона непрореаги-
ровавшего хлористого метилена выделяют гексаметилентетрамин
обычной кристаллизацией.
Леб 1 2 нашел, что гексаметилентетрамин образуется при дей-
ствии тихого электрического разряда на алкоголь в присутствии
аммиака или азота. При этом имеет место сложная цепь реакций:
алкоголь расщепляется на ацетальдегид и водород; альдегид рас-
щепляется на метан и окись углерода; окись углерода с водородом
образует формальдегид. Одновременно при взаимодействии азота с
водородом образуется аммиак, образующий с формальдегидом ге-
ксаметилентетрамин.
Гексаметилентетрамин получается при пропускании смеси ме-
тана, аммиака и воздуха 3 или кислорода через металлическую
трубку, нагретую до температуры 500—700°. В трубку вводится ка-
тализатор в виде фольги, проволоки или кусочков листового ме-
талла (меди, серебра, олова, никеля или их сплавов).
СВОЙСТВА ГЕКСОГЕНА
1. Физические свойства. Гексоген представляет собой белые кри-
сталлы без запаха и вкуса. В отличие от большинства нитросоедине-
ний — не ядовит; плотность 1,82 (плотность тетранитропентаз{.и-
трита 1,77); температура его плавления, по Деверню, 203,5°, раство-
римость в воде: 0,01% при комнатной температуре и 0,15%
при 100°.
1 Н б 1 a n d, Lieb. Ann., 240, 225, 1887; D е 1 е р i n е, Bull. Soc.
Chim. Fr., 3 Serie, 11, 556, 1894; герм. пат. № 406227 от 29/III 1922, вы-
дан 15/11 1924; ам^ пат. № 1566821 от 5/VII 1924, выдан 22/ХП 1925; ам.
пат. № 1655707 от 9/11 1924, выдан 12/VII 1927.
Хлористый метилен употребляется в качестве растворителя и экстраги-
рующего вещества; его получают в большом масштабе хлорированием ме-
тана.
2 L б b, Biochem. Ztschr., 20, 136, 1909.
3 Герм. пат. № 357742 от 10/IX 1919, выдан 31/VIII 1922.
Его растворимость в органических растворителях, по Деверню:
Таблицу 71
Растворители Температура °C 100 в. ч. растворителя растворяют в. ч. гексо- гена
Ацетон 20 7,413
(по. другим Данным 6,0)
31 9,0
53 17,5
96%-ный этиловый спирт 20 0 104
Метиловый спирт 21 0 187
БенЗЬл 21 0015
Толуол 18 0 032
Четыреххлористый углерод .... 18 Не растворяет
п Раст р и 20°: воримость гекс Отношение ацетон : вода 1 : 1 1 : 2 1 : 3 огена в смеси в 100 г смеси 0,29 0,07 0,04 ацетона и воды
р а с т в о римость г е к с о г ена в азотной кислоте при 20°
Концентрирован- ная HNOa,% 93 80 70 60 в 100 в. ч. кислоты 12,5 2,2 0,44 Следы
2. Химические свойства. Концентрированная серная кислота
разлагает гексоген.
В азотной кислоте он растворяется без разложения и может быть
осажден из нее простым разбавлением кислоты водой.
Не реагирует с основаниями.
3. Взаимодействие гексогена с серной кислотой1. Вернаццо
исследовал влияние концентрированной серной кислоты на гексо-
ген. По его данным, гексоген растворяется в концентрированной сер-
ной кислоте, но не растворяется в разбавленной. Тем не менее он
йе выпадает из раствора после разбавления его водой. Отсюда сле-
дует, что гексоген разлагается при растворении в концентрирован-
ной серной кислоте. По Вернаццо, взаимодействие гексогена с кон-
центрированной серной кислотой можно выразить следующей фор-
‘мулой:
> C3H6N6O6 4- 2H2SO4 -> ЗСН2О + 2SO2 (ОН) (ONO) -f- 2N2-f-H2O
t--------------------------------------------------------------
1 V era azz o, Z. Schiess- u. 'Sprengst., 31. 57, 1936.
389
388
Этой реакцией объясняется невозможность точного определения
содержания азота в гексогене в нитрометре Лунге; по этому методу
определяется только б/6 азота, содержавдегося в гексогене, причем*
и это количество определяется неточно. Эта же реакция показывает,
что образующийся при нитровании гексаметилентетрамина креп-
кими кислотными смесями азотной и серной кислот гексоген разла-
гается при его взаимодействии с концентрированной серной кисло-
той, а потому в этом случае гексогена вовсе не получается. При ни-
тровании же гексаметилентетрамина менее концентрированными
кислотными смесями, не разлагающими гексогена, получаются пони-
женные выходы гек-
согена вследствие ча-
стичного разложе-
Таблица 72
Данные определения1 pH по Деверню
(испытания проведены при температуре 132°)
Продолжи- тельность нагревания час. Гексоген Тэн Тетрил
0 6,63 6,56 6,85
1 —. 3,03 3,11
2 5,81 2,64 2,96
3 —. 2,32 —
4 — 2.22 2,89
5 5,73 Красные пары —
8 5,68 — 2,68.
ния гексаметиленте-
трамина серной кис-
лотой.
4. Химическая
стойкость. Гексоген
может больше меся-
ца храниться при
50° без разложения.
По данным Авогадро,
он выдерживает про-
бу Абеля при 60°
больше 60 часов. Ре-
зультаты определе-
ния pH приведены
в табл. 72.
Отсюда следует, что стойкость химически чистого гексогена
значительно превышает таковую тетрила.
Но стойкость технического продукта часто значительно ниже,
чем чистого 2, и разница между стойкостью гексогена и тэна в этом
случае становится значительно меньше.
По Иццо, гексоген начинает разлагаться при 200°. Температура
его вспышки 215° — выше температуры вспышки тетрила.
5. Взрывчатые свойства гексогена. Теплота взрывчатого разло-
жения гексогена, по Авогадро, 1390 кал.
Чувствительность гексогена к удару: высота падения груза при
весе в 2 кг 30—32 см (для тэна 30 см., для тетрила 40 см); чув-
ствительность гексогена к удару немного меньше, чем тэна. Расши-
рение3 в бомбе Трауцля 475 см3 (для тэна 515 и для тетрила 340).
При испытании по пробе Гесса (навеска 50 г) гексоген анало-
гично тзну дает полную деформацию столбика (тетрил дает 17,3 мм).
1 Напомним, что pH = 7 соответствует нейтральной реакции-
pH =4,6 >> слабокислой реакции
pH = 0,3 <> сильнокислой реакции
2 Izzo, L’industria Chimica, 2, 1933.
3 Naoum, Nitrocellulose, 5, 184, 1934.
390
Скорость детонации гексогена при плотности 1,70 равна
8370 м/сек (для тэна 8440 м/сек при плотности 1,62, а для тет-
рила 7200 при плотности 1,63); как видно, она немного меньше,
чем у тэна.
6. Применение гексогена. Гексоген является сильнейшим из всех
химически однородных твердых взрывчатых веществ (он уступает
только гексанитроманниту, который не применяется вследствие его
малой стойкости). Как в чистом виде, так и в виде определенных
смесей гексоген является наиболее бризантным взрывчатым веще-
ством. По сравнению с тэном обладает большим преимуществом —
большей химической стойкостью.
Он применяется для изготовления капсюлей-детонаторов и для
снаряжения некоторых боеприпасов, причем для уменьшения чув-
ствительности к удару его флегматизируют добавлением к нему па-
рафина, воска, канифоли и других веществ, а также ди- и трини-
тротолуола и других полинитросоединений.
Вследствие высокой температуры плавления гексогена он не мо-
жет быть залит, в боеприпасы, и остается один способ снаряжения
им — прессование. Однако вследствие большой чувствительности
к механическим воздействиям он может быть спрессован лишь в ко-
личестве до 10 г; при большем заряде вероятность взрыва и разру-
шительное действие при взрыве увеличиваются до недопустимых
размеров. Поэтому в настоящее время чистый гексоген (аналогично
тэну) применяется только для изготовления капсюлей-детонаторов
или зарядов диаметром до 2 см для снаряжения мелкокалиберных
гранат. Для больших зйрядов применяется флегматизированная
смесь с менее чувствительными ароматическими нитросоединениями,
например, тротилом, динитротолуолом, реже тетрилом.
Однако при флегматизации не только уменьшается чувствитель-
ность к удару, но и плотность, работоспособность и скорость де-
тонации.
В следующей таблице показано действие влияния примеси флег-
матизатора на чувствительность к удару и работоспособность тэна,
близкого по своим взрывчатым свойствам к гексогену *
Таблица 73
Чувствитель- Работоспособ-
Наименование вещества ность к удару ность по Трауцлю
см см3
Тэн, чистый 30 470
» с примесью 5% парафина 35 415
» .» » 10% » 60 340
» >> » 5% динитротолуола . . — 450
» » » 10% » .... 57 425
» » » 20% » .... — 355
1 Stettbacher, Nitrocellulose, 4, 179—184, 199—206, 1933; 5, 6—12,
1934.
39 L
Отсюда видно, что при добавлении к тэну 10% парафина или 20%
динитротолуола работоспособность смеси равна таковой же тетрила,
и нет смысла применять такую смесь.
Сказанное здесь о тэне полностью применимо к гексогену.
На шведских заводах в Бофорсе применяется смесь из 55% гексо-
гена и 45% тротила под названием боннита в виде прессованных за-
рядов.
ПРОИЗВОДСТВО ГЕКСОГЕНА
1. Течение реакции нитрации. Реакция взаимодействия гекса-
метилентетрамина с азотной кислотой протекает значительно слож-
нее, чем изученные нами реакции нитрации (замена атома водорода
на нитрогруппу) или реакция нитролиза (замена группы SO3H на
нитрогруппу). Это взаимодействие состоит из нескольких стадий:
1) выделение из молекулы гексаметилентетрамина группы атомов
N(CH2)3 и взаимодействие ее с азотной кислотой и водой; 2) вступ-
ление во вновь образовавшуюся молекулу трех нитрогрупп.
По вопросу о течении и механизме этой реакции существует не-
сколько указаний в литературе.
Штетбахер коротко утверждает в своем учебнике, что в качестве
побочных продуктов реакции образуются только газы и притом
СО2 и N2. Однако на это последовало возражение, что развитие
газов происходит только при выгорании загрузки, причем выде-
ляются СО2, NO, NO2 и только незначительное количество азота.
При нормальном же течении реакции нитрования процесс протекает
без образования газов (заметно лишь незначительное образование
окислов азота).
Девернь 1 указывает, что при нитровании протекают следующие
реакции:
(I) C6H12N4 + 4HNO, ->C3H6N6O6 + ЗНСОН + NH4NO3
(II) C6H12N4 + 28HNO3 -> 6CO2 + 24NO2 + 12H2O + 4NH4NO3
(III) C6H12N4 + 6H2O 6HGOH + 4NH3
Выделяющийся по реакции (III) формальдегид реагирует, по Де-
верню, главным образом, при стоянии отработанной кислоты.
Образующийся при взаимодействии аммиака с азотистой кисло-
той азотистокислый аммоний разлагается с образованием свободного '
азота NH4NO2 -> 2Н2О 4- N2. Однако все эти р еакции Деверня ни-
чего не говорят о самом механизме реакции взаимодействия гекса-
метилентетрамина с азотной кислотой. Кроме того, вопреки утвер-
1 Desvergnes, Chim. et Ind., 28, 1038—1044, 1932 .
392
ждению Деверня, формалин при смешении с азотной кислотой вза-
имодействует сравнительно быстро и энергично, тогда как энергичные
окислительные процессы начинаются в отработанной кислоте после
ее стояния в течение нескольких часов. Эти факты лучше учиты-
ваются в механизме реакции, предложенном Герцем.
По Герцу *, при нормальном течении реакции взаимодействия
между азотной кислотой и гексаметилентетрамином происходит
расвдепление молекулы тетраметилентетрамина с образованием на-
ряду с гексогеном очень неустойчивого метилендинитрата 2. Этот
динитрат сравнительно стоек в крепкой отработанной кислоте, но
при разбавлении кислоты немедленно омыляется, причем освобож-
даются азотная кислота, формальдегид и аммиак (тотчас связывае-
мой азотной кислотой). Формальдегид обнаруживается в разбавлен-
ной кислоте по запаху и легко может быть отогнан из нейтрализо-
ванного раствора при нагревании. Наконец, при нейтрализации
аммиаком он превращается в гексаметилентетрамин.
Эти реакции могут быть выражены следующими уравнениями:
СН2
o2n-n/^.n - no2
-----> I + 3CH2(ONO2), + nh4no3 + 3H2O
н2с'ч Jch2
N - NO2
При стоянии отработанной кислоты или при ее разбавлении ме-
тилендинитрат омыляется:
CH2(ONO2)2 + 2Н2О -> СН2(ОН)2 + 2HNO3
СН2(ОН2)->СН2О-|-Н2О
1 Н е г z, Z. Schiess- u. Sprengst., 29, 162, 1934.
О метилендинитрате см. Travagli, (jazz. Chim. Ital., 68, 718—721,.
1938.
39$
По Холево и Гринбергу при взаимодействии гексаметилен-
тетрамина с азотной кислотой протекают следующие превращения:
NH
Н2С
+ N(GH2OH)3
триметанолам ин
гипотетический про-
межуточйый продукт—
циклотриметилентри-
амин
сн2
<Н) I
Н2С^ 'сн2
NH
сн2
O2N~n/\n - no2
+ 3HNO3----> ' I +зн2о
Н2С‘Ч ^сн2
N - NO2
(III) N(GH2OH)3 + 3HNO3 -> N(GH2ONO2)3 + 3H2O.
Суммируя, получим:
СН2
O,N- n/>N-NO2
N-NO2
+ N(GH2ONO2)3 + 3H2O
394
Кроме этих основных реакций протекает следующая:
-> 2N(CH2ONO2)3+ 2NH4NO3
*При стоянии или разбавлении отработанной кислоты водой азот-
ный эфир триметаноламина разлагается, освобождая формальдегид:
N(GH2ONO2)3 + ЗН2О -> 3HGOH + 2HNO3 + NH4NO3
Кроме того, по Гартунгу х, при действии азотной кислоты на
гексаметилентетрамин образуются кроме NH3 и HGOH следы ме-
тиламина.
При нитровании гексаметилентетрамина смесями азотной и сер-
ной кислот получаются малые выходы гексогена (или даже вовсе
не получаются в зависимости от содержания серной кислоты в смеси),
вследствие того, что серная кислота разрушает гексаметиленте-
трамин.
2. Производство гексогена. Гексоген может быть получен двумя
способами: 1) в две фазы — через динитрат гексаметилентетрамина
и 2) в одну фазу — непосредственным нитрованием гексаметилен-
тетрамина.
Получение гексогена в две фазы. По Геннингу 1 2,
сперва получают по описанному ранее способу динитрат гексамети-
лентетрамина. Промытый и высушенный динитрат вносят при — 5°
в азотную кислоту (уд. веса 1,52). Разбавляют 4-кратным коли-
чеством воды, причем выпадает триметилентринитротриамин, плохо
растворимый в азотной кислоте. Промытый и высушенный продукт
перекристаллизовывается из ацетона.
Получение гексогена в одну фазу. По способу
Герца3 70 г гексаметилентетрамина вносят небольшими порциями к
500г свободной от окислов азота азотной кислоты (уд. вес 1,52) при
температуре 20—30°. После введения уротропина повышают темпера-
туру до' 55°, делают при этой температуре 5-минутную выдержку,
охлаждают до 20—30°, делают при этой температуре 15-минутную
выдержку и разбавляют 3—4-кратным количеством воды. Промытый
и высушенный продукт перекристаллизовывается из ацетона.
Значение влияния различных условий на результаты нитрования
было изучено Гелем, а позже Штетбахер описал приведенные ниже
более рациональные способы получения гексогена й одну и две фазы.
1 J. Prakt. Chemie, 46, I, 1892.
2 Герм. пат. № 104280, 1898
3 Ам. пат. № 1402693, 1922-
395
3. Опыты Геля 1 по получению гексогена через динитрат. П о-
лучение динитрата. Раствор 50 г гексаметилентетрамина
в 150 сл3 воды охлаждали до 0° и добавляли к раствору неболь-
шими порциями азотную кислоту (уд. вес 1,42). Нитрат выпадал
сейчас же в виде мелких белых кристаллов. Его фильтровали через
стеклянную вату, быстро промывали осадок 50%-ным этиловым спир-
том, предварительно охлажденным до 0°, промывали неразбавлен-
ным спиртом и, наконец, эфиром. Высушивали током теплого воз-
духа. Средний выход равен 78,2 г (82,3% от теории). Анализом
установлено, что полученный продукт есть динитрат уротропина:
C6H12N4 • 2HNO3.
Нитрование динитрата гексаметилентет-
рамина. При опытах нитрования динитрата было отмечено, что
при сильном перемешивании происходит слишком быстрая реак-
ция, сопровождающаяся бурным выделением паров.
Для нитрования брали 5 г динитрата и 5-кратное количество
азотной кислоты.
Результаты приведены в следующей таблице.
Таблица 74
Концентра- ция азотной кислоты % Темпера- тура °C Продолжитель- ность операции мин. Выход гексогена от теории % Примечание
95 —5 80 43
95 30 45 49 '
95 55 10 10 Вследствие бурной реакции загрузка бы- ла быстро спущена в воду
90 30 60 40
90 60 20 48
85 30 60 34
85 60 20 40
80 30 60 Ничего
80 60 20 »
. Наилучшие выходы были получены при наиболее высоких кон-
центрациях, а именно, 90—95% HNO3. 80%-ная азотная кислота
разлагает нитрат, не нитруя его, так как при этом непрерывно выде-
ляется формальдегид в продолжение всего опыта.
Непосредстве н-н ое нитрование гексаме-
тилентетрамина. При опытах перемешивание было непре-
рывное, но не энергичное.
Выхода вычислялись из расчета получения 1 мол триметилентри-
нитроамина из 1 мол гексаметилентетрамина по уравнению:
C6H12N4 ф 3HNO3 -> C3H6O6Ng + ЗНСОН + NH3
1 Н а 1 е, J. Amer. Chem. Soc., 47, 2754. 1925.
396
Согласно этому уравнению из 10 ч. тетрамина должно получиться
15,85 ч. тринитроамина.
В предварительных опытах было установлено, что при приме-
нении 70%-ной азотной кислоты не получался гексоген: при вылива-
нии раствора в воду не было получено|никакого осадка; тетрамин при
этом спокойно растворялся при температуре до 70°. При 90° про-
исходило энергичное окисление .тетрамина со столь сильным выде-
лением паров, что во избежание несчастного случая приходилось вы-
ливать раствор в воду.
Таблица 75
Результаты опытов
Концентрация азотной ни- . слоты * % Вес азотной кислоты е Вес тетрамина г Максимальная] температура во время нит- рования °C Время приба- вления амина МИН. । Время выли- вания в воду в мин. ексо- ео рии
Выход г гена от т %
92 115 10 20 8 15 41
92 115 10 20 20 15 64
92 115 10 30 15 40 38
92 115 10 18 30 . 55 57
92 330 30 25 15 60 62
92 330 30 30 10 15 59
92 450 30 27 15 20 60
92 450 30 25 15 60 60
92 450 30 25 15 120 62
92 550 50 30 15 20 63
92 550 50 30 15 20 68
Эти опыты показывают, что выдержка, превышающая 20 минут,
не улучшает выхода; следовательно, реакция протекает очень быстро.
При нитровании при температуре 18—20° выход получается на
4—5% больше, чем при 25—30°, следовательно, происходит какая-то
побочная реакция, и низкая температура тормозит эту реакцию
в большей мере, чем реакцию нитрации. Наконец, эти опыты по-
казывают, что 11-кратное количество азотной кислоты по отношению
к амину дает такой же выход, как и 15-кратное.
Наибольший выход гексогена в этих опытах равняется 68%.'
При определении причины пониженных выходов было установленэ,
что наряду с реакцией
(I) C6H12N4 + 3HNO3’->G3HeO6N6 + ЗНСОН + NH3
происходит гидролиз части гексаметилентетрамина по реакции:
(II) G6H12N4 + 6Н2О -> 6HGOH + 4NH3
Помимо этих двух реакций идет еще реакция образования дини-
трата гексаметилентетрамина, который неполностью превращается
397
в гексоген, вследствие чего этот динитрат может быть обнаружен
в отработанной кислоте:
(III) C6H12N4 + 2HNO3->G6H13N4 • 2HNO3
' Часть выделяющегося при гидролизе аммиака взаимодействует
с азотистой кислотой с образованием азотистоаммониевой соли,
которая разлагается даже при низкой температуре:
NH4NO2z->N2 + 2Н2О
На основании приведенных данных, а равно и дополнительно
поставленных опытов Гель пришел к следующим выводам:
1) при применении кислоты относительно низкой концентрации—
до 70% IjINO3 — идет гидролиз гексаметилентетрамина, нитра-
ция почти не имеет места;
2) при более высоких концентрациях азотной кислоты проте-
кают все три реакции одновременно, причем реакция (I) превали-
рует при концентрациях 95—100% и при низких температурах.
При применении безводной азотной кислоты и при низкой темпера-
туре реакция гидролиза в начале добавления тетрамина фактически
не идет вследствие отсутствия воды. Это наиболее благоприятные
условия для получения высоких выходов гексогена. Однако близкого
к 100% выхода получить не удается вследствие того, что по мере
добавления тетрамина выделяющаяся реакционная вода обусловли-
вает реакцию гидролиза, а с другой стороны, образующийся дини-
трат не нацело превращается в гексоген.
4. Получение гексогена по описанию Штетбахера. Способ
Ш тетбахера \ По Штетбахеру, лучшие выходы получаются
при прямом нитровании гексаметилентетрамина концентрированной
азотной кислотой при условии применения 10-кратного количества
азотной кислоты.
Расчет величины избытка HNO3, отвечающего этой дозировке азот-
ной кислоты, затруднителен потому, что неизвестно точное течение
реакции между азотной кислотой и гексаметилентетрамином. Как
мы видели, на 1 мол тетрамина расходуется, по Деверню, 4 мол
HNO3, по Герцу, 10 мол HNO3, по Холево и Гринбергу, 6 мол.
Штетбахер тоже принимает 6 мол HNO3, но в правой части уравне-
ния он приводит в качестве продуктов взаимодействия кроме гексо-
гена воду, углекислоту и азот, что неправильно. Поэтому мы при-
нимаем для расчетов более приемлемую реакцию Холево и Грин-
берга:
C6H12N4 + 6HNO3 C3H6N6O6 + N(CH2ONO2)3 + ЗН2О
140,13 663,02 222,1
100 269,9 158,5 теоретически
100 + 1000 115(72,8% от практически
(избыток 270% теории)
от теории)
•> 1 S t е t t b а с h е г, Nitrocellulose, 4, 200, 1933.
398
15 г кристаллического, измельченного в порошок гексаметилен-
тетрамина вносятся при сильном перемешивании в 150 г азотной
кислоты (уд. веса 1,52).
При внесении тетрамина температура должна быть 15—20°, и
не выше 25°. Тетрамин полностью растворяется, раствор остается
прозрачным до конца внесения тетрамина. После внесения тетрами-
на сливают содержимое колбы в 4—5-кратное количество ледя-
! ной воды, причем гексоген выпадает в виде белых кристаллов. Отса-
сывают на воронке, промывают многократно водой и один раз кипя-
тят с водой, вновь отсасывают; пропаривают в автоклаве при давле-
! нии 3—3,5 ат. Благодаря этой обработке в автоклаве продолжи-
[ тельность стабилизации сокращается во много раз, ибо уже при
i 3 ат температура воды достигает 134°, вследствие чего следует
1 ускорение очистки в 50—100 разно сравнению с простым кипячением
при обыкновенном давлении. Большей частью достаточно получасо-
' вого пропаривания при 3,5 ат. Затем охлаждают, отсасывают на во-
J ронке и многократно промывают водой. После такой обработки ге-
ксоген выдерживает нагревание неделями при 100° без потери веса.
При двух опытах стабилизации нагреванием в течение 1 часа при дав-
лении в 3 ат определены потери от сырого продукта; они оказались
равными 2,73 и 2,80%.
Выход гексогена 17,3 г, или 115% по весу исходного тетрамина;
температура плавления 20*0—201°.
5. Получение гексогена в две фазы через динитрат. Получение
тетраминдинитрата протекает согласно уравнению: '
C»H12N4 -|- 2HNO3 —> C,H12N4 • 2HNO3
140,13 263,02 266,17
100 90,0 190,0 (теоретически)
100 160 174 (91,6% (практически)
(175 см3 в виде от теории)
65%-ной азотной кислоты)
100 г тетрамина растворяют в 135—140 см3 воды. Не дожидаясь
полного растворения тетрамина, начинают медленно подавать 175 см3
65%-ной азотной кислоты (уд. вес 1,40). Температура может при этом
достичь 40—50° без всякой опасности разложения. Тетрамин быстро
растворяется, но скоро начинается выделение соли — нитрата гекса-
метилентетрамина. К концу содержимое аппарата получает вид
тяжелой кашеобразной массы, которая отсасывается на воронке по
достижении 15°. Не промывая динитрат водой, сушат его на филь-
тровальной бумаге или в вакууме над серной кислотой в эксикаторе.
По удалении главной массы воды его можно сушить при повышенной
температуре — до 70°. Фильтрат применяется для растворения сле-
дующих порций гексаметилентетрамина, что повышает выход дини-
трата и уменьшает расход азотной кислоты.
Выход динитрата 195 г влажного, содержащего 21 г воды (сла-
бой азотной кислоты). Влажный динитрат пахнет сильно формальде-
гидом и немного азотной кислотой. Этот запах свойствен и воздушно-
сухому продукту.
399
При хранении 50 г воздушносухого динитрата в эксикаторе над
серной кислотой определена следующая потеря в весе:
Таблица 76
Потеря в весе Продолжительность
% хранения
0.95 5 час.
1,01 6*/2 »
1,35 22 »
1,46 3 ДНЯ
1,49 10
1,50 14 »
При хранении на открытом воздухе он увлажняется, и вследствие
этого увеличивается его вес. При высушивании потеря веса, однако,
больше содержания воды, откуда следует, что динитрат является
нестойким соединением.
При температуре 157° динитрат гексаметилентетрамина разла-
гается; нагретый в капилляре выше температуры разложения, он
энергично взрывается. При взрыве 10 г капсюлем-детонатором № 8
он действует на медную пластинку аналогично динитробензолу.
При хранении без доступа воздуха соль можно хранить годами;
но вследствие легкой отщепляемости формальдегида и слабо связан-
ной азотной кислоты следует учитывать возможность саморазогре-
вания и последующего разложения.
Нитрование динитрата гексаметилентетрамина протекает соглас-
но уравнению:
CeH12N4 • 2HN0, + 4HNO,-----------> C3HeNeOe + N(CH2ONO2)3 + ЗН2О
266,17 4-63,02 222,096
100 94,7 83,43 (теоретически)
100 420 55,3 (практически)
।соответствует 52,5 г (или на 100 г тетрамина
гексаметилентетрамина) 800 г HNOj, или избыток
от теории 196 ’/„)
Из этого расчета видно, что расход азотной кислоты при нитро-
вании динитрата гексаметилентетрамина составляет 80% сравнитель-
но с расходом при нитровании тетрамина.
В остальном условия нитрования такие же, как при нитровании
тетр амина.
Выход, по Штетбахеру, меньше, чем при нитровании гексамети-
лентетрамина, а именно:
При нитровании 100 г свободного основания . ; . 115 г гексогена
(72,8% от теории)
» >> 100 г динитрата................> . 96,8 а гексогена
(61% от теории)
400
Следовательно, по Штетбахеру, при нитровании динитрата выход
гексогена приблизительно на 16% меньше, чем при нитровании сво-
бодного основания.
Ведение нитрования при низких температурах (0—5°) не повы-
шает выхода гексогена.
6. Общее замечание о получении гексогена в одну или две фазы.
При нитровании динитрата, как это следует из сказанного на стр. 321
об окисляемости органических оснований, меньше сказывается вли-
яние концентрации и количества азотной кислоты и содержания
окислов азота в последней; кроме того, нитрование менее опасно,
и при одинаковых условиях нитрования выход больше на
5—6%, вопреки утверждению Штетбахера.
ч» Но оборудование завода значительно проще для однофазного
процесса, чем для двухфазного.
Химия—13—26
ГЛАВА XVII
ХРАНЕНИЕ ТОЛУОЛА И ДРУГИХ ЛЕГКОВОСПЛАМЕ-
НЯЮЩИХСЯ ЖИДКОСТЕЙ
1. Классификация легковоспламеняющихся жидкостей Ч Л е г-
ковоспламеняющимися называются такие жидкости,
которые легко воспламеняются при соприкосновении с пламенем,
искрой или раскаленными предметами или выделяют огнеопасные
пары, образующие при смешении с воздухом взрывчатые смеси.
Сюда относятся жидкие ароматические углеводороды (бензол, то-
луол, ксилол и т. п.), ацетон, метиловый и этиловый спирты,
нефть и ее продукты (нефтяной эфир, газолин, бензин, лигроин, ке-
росин) и др. ,
. Многие из этих жидкостей применяются в производстве нитро-
соединений и могут быть источником пожара или взрыва (смесей
паров с воздухом) как при хранении на складах, так и в производ-
ственных мастерских.
По степени пожарной опасности и опасности взрыва легковос-
пламеняющиеся жидкости делятся на классы в соответствии с
разделением жидкостей на классы по температуре вспышки паров2
при давлении 760 мм рт. ст.
1 Правила хранения легковоспламеняющихся жидкостей регламентируются
следующими ОСТ:
ОСТ 4518: Классификация хранилищ1 легковоспламеняющихся жид-
костей.
ОСТ 4519: Хранилища-склады для легковоспламеняющихся жидкостей.
ОСТ 4520: Хранилища для легковоспламеняющихся жидкостей на тер-
ритории предприятий или хозяйств.
ОСТ 4521: Склады жидкого топлива (мазута на территории предприя-
тий или хозяйств.
Эти ОСТ утверждены в 1931 г. и нуждаются в дополнениях или измене-
ниях применительно к условиям того или другого производства.
2 В следующей таблице, приводятся температуры вспышки паров летучих
веществ.
Наименование вещества Температура вспышки °C Класс пожар- ной опасно- сти, к которо- му относится Наименование вещества Темпера- тура вспы- шки °C Класс пожар- ной опасно- сти, к которо- му относится
Бензол От—17 до —19 1-й класс Метиловый спирт —1 1-й класс
Толуол 7 1-й » Этиловый спирт . 9 1-й
Ксилол . . 27,5 1-й * т-Крезол .... 86 3-й »
Сольвент-нафта. От 49 до 67 3-й » Ацетон — 18 1-й
Нафталин .... От 81 ДО 89 3-й Нитробензол • • 89,5 3-й »
Фенол . .... 94 3-й « Диметиланилин * 61 3-й »
Хлорбензол . . . 27,5 1-й »
402
С
а) к 1-му классу относятся жидкости, дающие вспышку паров
при температуре ниже 28° (например бензин, эфир, сероуглерод);
б) ко 2-му классу относятся жидкости, дающие вспышку паров
при температуре от 28 до 45° (например керосин);
в) к 3-му классу относятся жидкости, дающие вспышку при тем-
пературе от 45 до 100° (например мазут).
Очевидно, что чем ниже температура вспышки, тем жидкость
опаснее.
2. Классификация хранилищ для легковоспламеняющихся жид-
костей. По целевому назначению складские сооруже-
ния для легковоспламеняющихся и горючих Жидкостей делятся на
следующие разновидности:
' а) склады как самостоятельные предприятия или хозяйства,
предназначенные для образования запасов легковоспламеняющихся
и горючих жидкостей с целью снабжения ими какого бы то ни было
потребителя (базисные склады);
б) хранилища легковоспламеняющихся и горючих жидкостей
на территории предприятий или хозяйств, сооруженные с целью
образования емкостей, потребных для производственных нужд дан-
ного предприятия или хозяйства;
в) склады для жидкого топлива (мазут), подлежащего хранению
на территории предприятия или хозяйства.
По роду применяемых устройств сооружения
для легковоспламеняющихся жидкостей делятся на следующие:
а) надземные, к которым относятся хранилища, где днища ре-
зервуаров находятся на уровне земли или выше поверхности окру-
жающей местности, или когда наивысший возможный уровень жид-
кости в хранилище находится выше 1,5 м над уровнем земли в месте
нахождения резервуара;
б) надземные, к которым относятся все хранилища, где наивыс-
ший возможный уровень жидкостей в резервуарах не выше поверх-
ности прилегающей территории; *
в) полуподземные, т. е. частично заглубленные в земле резер-
вуары, в которых наивысший возможный уровень находится не бо-
лее, чем на 1,5 м выше прилегающей территории, при условии,
что резервуар покрыт железнрй герметической крышкой или железо-
бетонным покрытием, обсыпанным слоем земли толщиной не менее
0,5 м;
г) кроме описанных хранилищ ОСТ регламентирует склады с
безопасной системой хранения воспламеняющихся жидкостей; сюда
относятся склады, удовлетворяющие следующим общим требованиям:
1) при всякого рода порче оборудования, поломке трубопроводов,
пожаре у мест приемки и раздачи или в непосредственной близости
к хранилищу огонь не может передаваться внутрь резервуаров с
жидкостями и помещений, в которых находятся резервуары; 2) при
порче оборудования, поломке трубопроводов и пр. жидкость не мо-
жет выливаться наружу на окружающую территорию; 3) все метал-
лические части хранилища, трубопроводы и арматура, соприкасаю-
Ж
»
щиеся с легковоспламеняющимися жидкостями, заземлены для отвода
могущего образоваться электрического заряда при протекании жидко-
стей по трубам; 4) полностью устранена возможность образования
взрывоопасной смеси внутри резервуара;
д) хранилища с применением инертных газов, например, азота,
углекислоты или отработанных газов из двигателей внутреннего
сгорания (такие хранилища строятся за границей, нашим ОСТ не
регламентированы).
3. Устройство хранилищ для легковоспламеняющихся жидкостей
на территории завода и нормы хранения. В случае необходимости
хранения больших количеств легковоспламеняющихся жидкостей,
чем это установлено для хранения на заводской территории, храни-
лища выносятся за пределы заводской территории на расстояние не
менее 150 м. В зтом случае хранение ведется согласно ОСТ 4519.
Хранение легковоспламеняющихся жидкостей на территории за-
вода допускается во всех типах хранилищ — надземных, полупод-
земных и подземных.
На территории завода разрешается хранить в одном сосредото-
ченном месте следующее количество легковоспламеняющихся жид-
костей (табл. 77).
Таблица 77
Род хранения Жидкость 1-го класса т Жидкость 2-го и 3-го классов ,т
Наливом в надземных резервуарах . . , 50 250
Наливом в подземных резервуарах . . . В таре, в специально предназначенных 100 1000
для '.той цели помещениях 50 250
Расстояния между отдельными резервуарами при хранении лег-
ковоспламеняющихся жидкостей в надземных хранилищах рекомен-
дуются не менее 2,5 диаметра большего из соседних резервуаров для
жидкостей 2-го и 3-го классов. Во всяком случае эти разрывы должны
быть не менее 10 м.
Соединение резервуаров группами допускается для жидкостей
1-го класса не более четырех, а для жидкостей 2-го и 3-го классов
не более восьми.
Расстояния между отдельными группами резервуаров (храни-
лищами) должны устанавливаться не менее указанных в табл. 78.
Разрывы между хранилищами легковоспламеняющихся жид-
костей в случае, если эти хранилища отстоят от производственных
зданий на расстояниях, меньших безопасных, исчисленных по при-
нятым формулам, должны быть не менее указанных в табл. 79.
404
Таблица 78
Хранилища Расстояния для жидкостей, м
1-го класса j 2-го и 3-го классов
Надземные Полуподземные Подземные . 1 120 ! 75 80 j 50 40 | 25 Таблица 79 •
Характеристика строения Величина разрывов между хранилищами для жидкостей, м
1-го класса 2-го и 3-го классов
надземные хранилища 1 полупод- земные подземные i надземные хранилища полупод- земные подземные
Огнестойкие с брандмау- ерами . . Огнестойкие без бранд- 100 70 50 50 35 25
мауеров . 160 120 80 80 60 40
Расстояния надземных резервуаров, предназначенных для хра-
нения легковоспламеняющихся жидкостей, должны составлять: до
несгораемого или огнестойкого здания не менее 25 м, а до сгорае-
мого или защищенного от возгорания здания и до границы соседнего
участка — не менее 40 м и до жилых построек — 100 м. Резервуары
должны быть покрыты герметическим несгораемым покрытием.
Устройство резервуаров. Стенки и днища надзем-
ных, полуподземных и подземных резервуаров должны быть непро-
ницаемы для хранящихся в них жидкостей; они могут быть изготов-
лены из любого несгораемого материала.
Безопасная система хранения. Безопасные хра-
нилища могут иметь любую емкость; разборные краны безопасных
систем могут быть размещены непосредственно в рабочих и склад-
ских помещениях.
Разрывы для хранилищ, устраиваемых по безопасной системе,
могут быть допускаемы в десять раз меньше против норм, указан-
ных для обычных систем.
4. Хранение легковоспламеняющихся жидкостей в атмосфере
инертного газа. Во время империалистической войны за границей
было несколько взрывов и пожаров хранилищ с бензолом, толуолом
и другими легковоспламеняющимися жидкостями. Взрыв паров
405
f
легковоспламеняющихся жидкостей возможен только при наличии
взрывоопасной смеси их с воздухом и при действии на эту смесь
искрой (наиболее частая причина таких взрывов), пламенем или
при нагревании этой смеси до определенной температуры (это в
практических условиях хранения может быть только при пожаре).
Для предупреждения образования взрывчатых смесей паров
с воздухом было предложено заполнить пространство над легко вос-
пламеняющейся жидкостью в цистерне инертным газом взамен воз-
духа. В качестве инертного газа применяют азот, углекислоту и отра-
ботанные газы из двигателей внутреннего сгорания; чтобы воздух не
попадал в трубопровод, последний окружали вторым трубопрово-
дом, а образовавшееся при этом кольцевое пространство заполняли
тем же инертным газом. Такие хранилища значительно безопаснее
обыкновенных, недостатком же их является высокая стоимость.
5. Перевозка легковоспламеняющихся жидкостей. Перевозка
легко воспламеняющихся жидкостей производится обычно в железно-
дорожных цистернах емкостью от 13 до 45 т. Передача этих жид-
костей из железнодорожных цистерн в заводские и из заводских
Рис. 48. Схема установки для оттаивания бензола в железно-
дорожной цисте рне.
в производственные помещения производится обычно центробеж-
ными насосами, или насосами системы Вортингтон, или специаль-
ными насосами для перекачки огнеопасных жидкостей. Передачи
легковоспламеняющихся жидкостей помощью сжатого воздуха или
вакуума следует избегать вследствие опасности взрыва смесей воз-
духа с парами жидкости.
^Перевозка бензола в холодное время. Вслед-
ствие высокой температуры замерзания бензола (4-5,5°) в холодное
время железнодорожная цистерна с замерзшим бензолом подается
в тепляк, аналогичный применяемому для оттаивания цистерн
с олеумом; для оттаивания замерзшего бензола необходимо до 24—
30 часов. Перекачка бензола продолжается 24—30 часов.
Для ускорения оттаивания бензола применяется ряд методов,
из которых опишем применяющийся на заводах (рис. 48).
406
В цистерну с замерзшим бензолом опускается труба; конец ее
заключен в опущенную в цистерну трубу-рубашку, через которую
проходит пар для предварительного обогрева бензола.
Когда часть бензола расплавилась, пускают в ход поршневой на-
сос и начинают пропускать оттаявший бензол через подогреватель
обратно в цистерну, а когда весь бензол расплавится, его передают
на склад. При работе по этому способу оттаивание и перекачивание
продолжаются 4—5 часов.
6. Вредные пары, загрязняющие воздух производственных поме-
щений. Большинство паров летучих растворителей вредно для здо-
ровья’. Поэтому необходимо для предупреждения отравления рабочих
производственных помещений периодически контролировать содер-
жание вредных примесей в воздухе Ч
Законом 1 2 установлены следующие предельно допустимые кон-
центрации ядовитых газов, паров и пыли в воздухе рабочей зоны
производственных помещений:
мг/л
ацетон..............................0,2
бензол, толуол, ксилол........... 0,1
этиловый спирт ..................... 1,0
дихлорэтан.......................... 0,05
При одновременном выделении в воздух рабочих помещений па-
ров нескольких растворителей (спирта и толуола, спирта и бензола
и др.) их суммарная концентрация не должна превышать допустимой
концентрации, установленной для наиболее вредного вещества.
К сожалению, правилами не установлены предельно допустимые
концентрации для ряда паров и пылей, выделяющихся при производ-
стве нитросоединений, например, паров мононитросоединений, пы-
лей твердых нитросоединений и др.
1 Подробные указания о методах этого контроля даны в книге А. С- Жит-
ковой, Методика определения вредных газов и паров в воздухе, Оборон-
гиз, 1939.
2 См. ОСТ 90014—39: Общесоюзные санитарные нормы н правила строи-
тельного проектирования промышленных предприятий, Госстройиздат, 1939.
ПРИЛОЖЕНИЕ 1
ИСТОРИЧЕСКИЙ ОЧЕРК
(Важнейшие этапы истории взрывчатых веществ)
1. От открытия дымного пороха до открытия явления детонации.
На основании имеющихся исторических документов не представ-
ляется возможным установить не толькб имя, но и национальность
изобретателя дымного пороха; равным образом неизвестны последо-
вательные этапы, через которые проходило самое развитие пороха.
Но несомненно, что изобретение пороха не было делом рук одного
человека; напротив, имел место длительный, протекавший в течение
многих столетий процесс постепенного развития состава пороха,
начиная от первоначальных примитивных селитросодержащих соста-
вов древних, через длинный ряд промежуточных этапов, привед-
ших к собственно пороху лишь к концу XIII или началу XIV сто-
летия.
Основная составная часть дымного пороха — селитра — впервые
стала известна еще в глубокой древности в Китае, где согласно имею-
щимся указаниям изготовлялись зажигательные стрелы задолго до
появления пороха в Европе; в XI столетии нашей эры в Китае из-
готовляли ракеты, в состав которых входила селитра, сера и уголь.
Однако это были только зажигательные составы и позже составы с
реактивным действием, но не пороха, т. е. эти вещества не обладали
метательным действием.
Греческий огонь, приобревший крупное военное значение
в VII столетии нашей эры, состоял вначале из смеси серы, пека и
смолы, позже в его состав ввели селитру; но и после добавления се-
литры это был только зажигательный состав.
Для перехода от зажигательных и реактивных составов китайцев
и от греческого огня к собственно пороху требовался длительный
процесс постепенного развития этих составов. Сюда относятся: под-
бор компонентов — селитры, серы и угля; установление правиль-
ной пропорции этих компонентов; познание значения степени измель-
чения и тесноты смешения составных частей; нахождение способа
очистки селитры от большого количества примесей, без чего нельзя
получить пороха. При низком уровне науки и техники в древние
времена и при чрезвычайно медленном их развитии, естественно, тре-
бовались столетия для прохождения указанных этапов и для решения
названных основных задач. И только решение всех этих частных
408
задач привело в конце XIII или в начале XIV столетия к веществам,
обладавшим метательным действием.
Практическое значение порох приобрел только в начале XIV сто-
летия, после изобретения огнестрельного оружия.
Об историческом значении появления в Европе пороха и огне-
стрельного оружия Ф. Энгельс говорит следующее г:
«В начале XIV в. среди западноевропейских народов входит
в употребление порох, заимствованный ими у арабов и, как это
известно всякому школьнику, он произвел переворот в способе ве-
дения войн. Но введение пороха и огнестрельного оружия отнюдь,
не было делом насилия, а делом промышленного, т. е. экономи-
ческого прогресса. Промышленность остается промышленностью,,
зайята ли она производством полезных предметов или же таких,
которые служат для целей разрушения. Введение огнестрельного"
оружия повлияло революционизирующим образом не только на
самое ведение войны, но и на политические отношения господствую-
щих и угнетенных классов. Чтобы добыть огнестрельное оружие,
нужны были промышленность и деньги, а тем и другим владели
горожане. Огнестрельное оружие было поэтому с самого начала ору-
жием городов и возвышающейся монархии, которая в своей борьбе
против феодального дворянства опиралась на города. Неприступные
до тех пор каменные стены рыцарских замков не устояли перед
пушками горожан; пули бюргерских ружей пробили рыцарские
панцири».
В Московской Руси порох и артиллерийские орудия появились
впервые в княжение Дмитрия Ивановича Донского. Это отмечено
летописцем (Голицынская летопись) такими красочными словами:
«... Лета 6897 (1389) вывезли из Немец арматы на Русь и огненную
стрельбу и от того часу уразумели из них стреляти». В 1400 г. на-
чали изготовлять порох в Москве, но большие пороховые заводы были
построены при Пеуре Первом.
На протяжении почти 500 лет со времени открытия дымного по-
роха и огнестрельного оружия не было найдено ни одного нового
практически пригодного взрывчатого вещества.
В конце XVIII столетия в связи с развитием промышленного
капитализма стала ощущаться потребность в более сильном мета-
тельном средстве, нежели дымный порох. Эта потребность стала
острее в XIX столетии. Кроме того, развитие добывающей промьпп-
лейности в течение XIX столетия потребовало также более мощного
взрывчатого вещества для ведения горных работ. Наконец, во вто-
рой половине XIX столетия развитие военной техники потребовало
более сильного взрывчатого вещества для снаряжения артиллерий-
ских снарядов. В соответствии с этим мы видим, начиная с конца
XVIII столетия, ряд изысканий, имевших целью дать более мощные
метательные средства и взрывчатые вещества. Эти изыскания увен-
1 Ф. Энгельс, Антн-Дюрннг, стр. 137, 1938 .
40Э
чались успехом в середине и во второй половине XIX столетия на
основе бурно развивавшейся в это время органической химии.
Первая серьезная попытка решить эту задачу была сделана Бер-
толле в связи с открытием им в 1786 г. хлорноватой кислоты и ее
•солей.
Считая, что на основе этих солей можно приготовить более мощ-
ный порох, чем единственно известный в то время дымный порох,
Бертолле занялся приготовлением такого пороха.
В 1788 г. он поставил первый заводской опыт приготовления
пороха на основе хлорноватокалиевой соли. При обработке под бе-
гунами тройной смеси — бертоллетовой соли, серы и угля — произо-
шел взрыв, сопровождавшийся человеческими жертвами. Однако
Бертолле приготовил позднее такой порох, но при первом же его ис-
пытании стрельбой произошел разрыв орудия, также сопровождав-
шийся человеческими жертвами. После этой неудачи опыты с новым
порохом были прекращены.
В 1815 г. англичанин Игг изобрел капсюль-воспламенитель, со-
стоявший из медного колпачка, в который запрессовывался хлорат-
содержащий состав.
Гремучую ртуть, изобретенную Говардом в 1805 г., первона-
чально не применяли в ударных составах для капсюлей-воспламени-
телей вследствие ее большой чувствительности к механическим воз-
действиям; но с 1831 г. гремучая ртуть вошла в качестве существен-
ной составной части в капсюльные составы.
Значительным затруднением при ведении взрывных работ в то
время являлось отсутствие достаточно безопасных методов воспла-
менения зарядов при этих работах. Для этой цели пользовались
палочками, обмазанными пороховым тестом или медленно горящим
серным фитилем. Серный фитиль состоял из пропитанной жидкой
•серой хлопчатобумажной нити, завернутой в бумажную трубочку.
Такие способы воспламенения были и неудобны и небезопасны.
Поэтому два открытия, сильно уменьшившие опасность взрывных
работ, имели очень большое значение для развития взрывного дела:
открытие способа электровоспламенения в 1804 г. и изобретение
Бикфордом медленно горящего шнура в 1831 г.
В 1832 г. Браконо получил ряд продуктов взаимодействия кон-
центрированной азотной кислоты с крахмалом, сахаром, древесными
волокнами. Эти продукты он назвал общим именем «ксилоидинов».
В 1838 г. Пелуз рекомендовал эти вещества для изготовления пиро-
технических изделий, но и-для этой цели они были непригодны вслед-
ствие их химической нестойкости.
Решающее значение для решения поставленной задачи имело от-
крытие Шенбейном нитроклетчатки нитрованием хлопка и открытие
Собреро нитроглицерина. Об обоих этих открытиях было сообщено
в 1846 г.
Вслед за открытием Шенбейна вначале в Австрии, а затем во
Франции и в других странах были построены пироксилиновые заводы.
Но уже в 1847 г. взорвался завод в Австрии, а в последующие годы
410
имели место взрывы пироксилиновых заводов во Франции и в других
странах. Причиной этих взрывов было саморазложение пироксилина,
который долгое время не удавалось получить в стойком виде,
несмотря на производившуюся самую тщательную и сложную
промывку.
Только в 1865 г. английский химик Абель ввел измельчение пи-
роксилиновых волокон на голландерах (по типу применяющихся
в писчебумажной промышленности), в результате чего удалось от-
мыть полностью кислоту, заключавшуюся во внутренней трубчатой
полости волокон. Одновременно был введен метод контроля пирокси-
лина — испытание его химической стойкости по пробе, названной
по имени предложившего ее автора «пробой Абеля». Введение этой
пробы, а позже еще более совершенных проб, устранило опасность
взрыва от саморазложения и положило прочное начало производству
пироксилина.
Обратимся теперь к открытию Собреро — нитроглицерину. В своем
докладе о получении нитроглицерина Собреро правильно описал
взрывчатые свойства этого вещества. Но он не сумел использовать
свое открытие на практике. Считая невозможным практическое при-
менение жидкого взрывчатого вещества, он занялся изучением дру-
гого азотного эфира—гексанитроманнита, который он хотел исполь-
зовать в капсюлях-воспламенителях в качестве ударного состава.
Но для этой цели нитроманнит оказался непригодным, ибо его чув-
ствительность к удару недостаточна, да и сырьевая база крайне
ограничена.
Спустя 9 лет после опубликования работы Собреро известный
русский ученый-химик Николай Николаевич Зинин впервые- поста-
вил вопрос о применении нитроглицерина в качестве взрывчатого
вещества. В 1854 г. в связи с войной (Крымская кампания) Зинин
предложил снаряжать гранаты нитроглицерином. Артиллерист
Петрушевский разработал способ снаряжения гранат этим веще-
ством. Однако вследствие косности чиновников, находившихся в то
время у власти, из-за их безразличия к работе Зинина и Петрушев-
ского; из-за общей обстановки, обусловленной отсталостью царской
России, замечательная инициатива Зинина и Петрушевского не по-
лучила необходимой поддержки. И после происшедшего однажды
взрыва снаряженной нитроглицерином гранаты, при котором было
ранено 3 человека, опыты со снаряжением гранат нитроглицерином
были прекращены.
В это время в России жил молодой* Альфред Нобель, который
здесь вместе со своим отцом по поручению русского правительства
занимался изысканием новых взрывчатых веществ и применением
их для снаряжения морских мин.
Нобель подробно познакомился с работами проф. Зинина и Петру-
шевского, посещая в течение ряда лет их лабораторию. После отъ-
езда вместе со своим отцом к себе на родину в Швецию, Нобель
в 1859—1861 гг. успешно продолжал работы над изысканием методов
производства и применения нитроглицерина.
411
В 1862 г. был построен первый опытный нитроглицериновый
завод в Швеции, близ Стокгольма. Этот завод взорвался в 1864 г.
После этого Нобель перенес свои опыты на пароход, на Малярское
озеро, а затем, на основе результатов своих опытных работ, построил
около Гамбурга нитроглицериновый завод.
Основным затруднением с самого начала работ Зинина над вопро-
сом применения нитроглицерина в качестве взрывчатого вещества
являлось отсутствие надежного способа взрывания его.
До этого был известен лишь один способ взрывания, применяв-
шийся для пороха,—воспламенение огнем. Однако нитроглицерин
не взрывался от луча огня, например от стопина или бикфордова
шнура. Для взрывания нитроглицерина русские ученые, а по их при-
меру Нобель, применяли заряд дымного пороха, который воспла-
менялся обычным огневым способом. При некоторых своих опытах
Нобель надевал на конец бикфордова шнура капсюль-воспламени-
тель для лучшего воспламенения порохового заряда. При одном
опыте взрывания нитроглицерина таким капсюлем без промежуточ-
ного заряда из дымного пороха был получен взрыв необычайной
силы. В дальнейших опытах гильза капсюля была вытянута для
удобства надевания капсюля на бикфордов шнур и закрепления на
нем и был увеличен заряд гремучей ртути. Таким образом в 1865 г.
Нобелем был изобретен капсюль-детонатор и им же было открыто
явление детонации взрывчатых веществ — открытие, ставшее пово-
ротным пунктом в истории взрывчатых веществ и положившее начало
бурному росту бризантных взрывчатых веществ.
2. Развитие нитроглицериновых взрывчатых веществ. В 1866 г.
сделано важное изобретение гур динамита, производство кото-
рого было начато в 1867 г. До этого нитроглицерин перево-
зился в жестяных бидонах, причем либо в чистом виде, либо (в целях
уменьшения опасности при перевозке и хранении) в форме раствора
в метиловом спирте. В некоторых случаях во избежание опасной
перевозки нитроглицерин изготовляли в небольших количествах
примитивными методами вблизи места его применения. В Америке
в это время, по предложению Мовбрея, нитроглицерин перевозили
в замороженном состоянии в жестяных банках. Такой способ пере-
возки применялся в Америке в некоторых местах еще долгое время
после изобретения динамитов.
Все описанные способы перевозки нитроглицерина были свя-
заны с большими опасностями. Кроме того, и самое употребление
жидкого нитроглицерина’ сопровождалось большими неудобствами
и опасностями- Поэтому изобретение гурдинамита, порошкообраз-
ного взрывчатого вещества, которое можно было подобно дымному
пороху патронировать в бумажную оболочку и перевозить в обыч-
ной деревянной упаковке, имело большое практическое значение.
В 1867 г. шведы Олльсон и Норбин изобрели первое аммиачно-
селитренное взрывчатое вещество «аммонкрут». С целью устранить
с пути опасных конкурентов по приготовлению динамитов Нобель
закупил патент Олльсона и Норбина, но не для производства более
412
дешевого аммонкрута, а, наоборот, чтобы его не допустить. Этим
Нобель на долгое время затормозил производство и применение ам-
миачно-селитренных взрывчатых веществ.
В 1873 г. Нобель изобрел гремучий студень и желатин-динамиты;
в 1878 г.—камфорный гремучий студень, а в 1888 г. — нитроглицери-
новый порох—баллистит, состоящий из 40% нитроглицерина и 60%
коллодионного хлопка и пироксилина.
В 70-х гг. прошлого столетия в связи с увеличением добычи угля
и с углублением угольных шахт (вследствие чего ухудшается венти-
ляция и увеличивается содержание метана в атмосфере каменно-
угольных шахт) и с применением высоко бризантных взрывчатых
веществ — динамитов — участились случаи взрывов газовой атмо-
сферы в угольных шахтах; увеличился и размер сопровождавших
такие взрывы катастроф. Это побудило к организации в 1877 г. во
Франции антигризутной комиссии, специально для изучения вопроса
безопасности ведения взрывных работ в угольных шахтах. Анало-
гичные комиссии были созданы в 1879 г. в Англии и Бельгии и
в 1880 г. в Германии. В результате работы этих комиссий с 1880 г.
во Франции, а позже и в других странах началось широкое примене-
ние аммиачной селитры для изготовления антигризутных взрывча-
тых веществ, а наряду с этим аммиачно-селитренные взрывчатые
вещества стали постепенно и все шире применяться для многих
промышленных взрывных работ помимо угольных шахт.
3. Взрывчатые вещества военного назначения. В 1869 г. Броун
в Англии детонировал влажный пироксилин, что положило начало
применению его (в виде прессованных шашек или так называемого
лекального пироксилина) для* снаряжения артиллерийских снаря-
дов и мин и для подрывных работ (пироксилиновые шашки). В Рос-
сии пироксилин был принят в 1875 г. морским ведомством для
снаряжения мин, а в 1876 г. военным ведомством для снаряжения
артиллерийских снарядов.
Пикриновая кислота. Первые сведения о пикриновой
кислоте относятся к 1771 г., когда Вульф наблюдал образование
желтого красящего вещества при действии азотной кислоты на шелк.
Спустя 12 лет (1783 г.) Гауссман получил пикриновую кислоту дей-
ствием на индиго
zco4 z со,
С6Н4( >С = С/ /С6Н4
азотной кислотой, причем он дал ей название «пикриновая кислота»
аа горький вкус (по гречески «тсьхро^» — горький) и кислотный
характер.
Лоран впервые получил в 1843 г. пикриновую кислоту нитрова-
нием фенола и показал идентичность ранее полученной желтой
краски с полученным им соединением. После этого стали приготов-
лять пикриновую кислоту в больших количествах для окраски
шерсти и шелка.
413
Более 100 лет со времени открытия пикриновой кислоты оста-
вались неизвестными ее взрывчатые свойства, и во взрывной тех-
нике сначала применялись или предлагались смеси пикриновой
кислоты или ее пикратов с селитрами или другими кислородсодер-
жащими веществами.
Например, в 1868 г. Дезиньоль предложил пикрат калия как
составную часть пороха (80% KNO3, 11% угля и 9% пикрата ка-
лия), а в 1869 г. Брюжер во Франции и независимо от него Эбль
в Англии предложили пикрат аммония тоже как составную часть
пороха.
Опыты с порохом Брюжера, состоявшим из 43% калиевой се-
литры и 57% пикрата аммония, дали в 1884 г. такие хорошие ре-
зультаты, что если бы не появился бездымный порох Вьелля, то
вероятно порох Брюжера получил бы практическое применение
в военном деле.
Д этот период считали, что вследствие малого содержания кисло-
рода в пикриновой кислоте она не может применяться в чистом виде
в качестве взрывчатого вещества, а потому ее или ее соли смешивали
с солями, богатыми кислородом.
В 1873 г. Шпренгель впервые указал (на заседании Лондон-
ского химического общества) на способность пикриновой кислоты
давать сильный взрыв от действия капсюля-детонатора. Однако
она была введена для снаряжения снарядов лишь после предложе-
ния Тюрпена в 1885 г. применять чистую литую пикриновую кис-
лоту. Причем им же было указано, что для взрыва литой пикрино-
вой кислоты необходим более мощный детонатор, так называемый
промежуточный детонатор, из прессованной пикриновой кислоты,
которая в свою очередь надежно взрывается под действием капсюля-
детонатора.
Это важный этап в истории развития военных взрывчатых ве-
ществ. Впервые нашли взрывчатое вещество, которое при большой
силе и бризантности было гораздо безопаснее в хранении и приме-
нении, чем известные до этого в военной практике дымный порох и
пироксилин. Пикриновая кислота оказалась вполне пригодной для
снаряжения артиллерийских снарядов..
Уже в 1886 г. пикриновая кислота под названием мелинита была
введена для снаряжения снарядов во Франции; в Англии она вве-
дена в 1888 г. под названием лиддита, в Японии под названием «ши-
мозе», в Германии под обозначением С/88.
В России первая мастерская для производства пикриновой кис-
лоты была построена в 1896 г. на Охтенском пороховом заводе.
Этот мелинитовый завод сгорел в 1907 г. После этого пожара еще
в течение года изготовляли пикриновую кислоту под открытым не-
бом и затем прекратили это производство, так как на том же Охтен-
ском заводе было начато производство тротила.
Непредвиденные большие потребности во взрывчатых веществах,
обнаружившиеся вскоре после начала империалистической войны
414
1914—1918 гг. и недостаток толуола заставили вернуться к произ-
водству и применению пикриновой кислоты. Производство пикрино-
вой кислоты было установлено на двух заводах.
На этих заводах пикриновая кислота готовилась из фенола так
называемым французским способом. Кроме того, было установлено
производство пикриновой кислоты из бензола через динитрохлорбен-
зол на двух содовых Заводах: «Электрон» в Славянске и Любимова,
Сольвей и К° около Лисичанска (ст. Рубежная).
Во время империалистической войны в Германии и Англии был
установлен новый способ производства пикриновой кислоты из фенола
нитрованием его крепкими кислотами, что позволило применять,
обычную чугунную аппаратуру для нитрования, а это знаменовало
собо’Й переход от полукустарного французского «горшечного» способа,
к более совершенному заводскому способу.
Вскоре после введения во Франции пикриновой кислоты для
снаряжения снарядов оказалось необходимым в целях ее флегмати-
зации (уменьшения чувствительности к механическим воздействиям),,
а отчасти и для удешевления снарядов, добавлять к ней некоторое
количество динитронафталина, мононитронафталина, динитрофе-
нола или других нитро соединений. При применении динитрофенола
оказалось возможным заменить им 40% пикриновой кислоты, т. е.
применять сплав, состоящий из 60% пикриновой кислоты и 40% ди-
нитрофенола. Это потребовало во Франции производства значитель-.
ных количеств динитрофенола. В России не применялись сплавы
пикриновой кислоты с динитрофенолом, а применялась, так назы-
ваемая «французская смесь», состоящая из 80% пикриновой кис-
лоты и 20% динитро нафталина и в значительно меньших размерах
применялась во время империалистической войны смесь 90% пикри-
новой кислоты и 10% мононитронафталина.
К недостаткам пикриновой кислоты относится то, что ее нельзя
смешивать с аммиачной селитрой для образования смесей, аналогич-
ных амматолам. Возможность такого смешения с аммиачной селитрой
имела бы большое практическое значение в условиях военного вре-
мени как в целях удешевления взрывчатых веществ, так и в еще
более важных целях увеличения общего количества взрывчатых ве-
ществ. Поэтому в Америке еще до империалистической войны стали
применять для снаряжения снарядов давно предложенные под на-
званием «громобоя» (русским ученым Чельцовым в 1886 г.) или ма-
исита 1 — смеси состава: 72,5 % аммиачной селитры и 27,5% пи-
крата аммония).
Вскоре после начала применения пикриновой кислоты выяви-
лись отрицательные свойства ее: способность образования более
1 Hale, Army Ordnance, VI, 39—40; Re e, J. Frankl. Inst., 760,
1924. Маисит, запатентованный в 1886 г. в Америке, имел состав: 72—40%
аммиачной селитры и 2 8—60% нитрата аммония.
41&
"чувствительных к удару и трению пикратов, высокая температура
плавления. Все это заставило искать новые взрывчатые вещества,
лишенные этих недостатков. В качестве такого взрывчатого вещества
намечался тринитрокрезол. Последний обладал рядом преимуществ
перед пикриновой кислотой: растворимость в воде меньше, поэтому
наклонность к образованию солей меньше, чем у пикриновой кис-
лоты; соли его менее взрывчаты, — чувствительность к удару
меньше, чем у солей пикриновой кислоты.
Очень вероятно, что это вещество (в чистом виде, или в смеси
с пикриновой кислотой) приобрело бы большое значение для снаря-
жения снарядов, если бы к этому времени всеобщее внимание не
"было привлечено новым появившимся веществом — тринитрото-
луолом. Это вещество обладало крупными преимуществами перед
пикриновой кислотой: заметно меньшая чувствительность к удару,
юно нейтрально — не образует опасных солей (как позже было уста-
новлено, тринитротолуол может образовать соли, но при особых усло-
виях), не растворяется в воде, имеет низкую температуру плавле-
ния, удобную для заливки снарядов, мало ядовит, имеет слабые
красящие свойства, не имеет горького вкуса, столь тягостного при
^работе с сухой пикриновой кислотой, способен сгорать в больших
количествах без взрыва и дешев; исходное сырье — толуол — полу-
чалось легко, дешево и в больших количествах из побочных
продуктов коксового производства.
Кроме того, относительное потребление толуола в промышлен-
ности гораздо меньше, чем фенола и бензола, вследствие чего при
удовлетворении спроса на бензол и фенол толуол оказывается избы-
точным продуктом.
Тринитротолуол был впервые получен Вильбрандом в 1863 г.
В 1891 г. Гайзерман указал на его взрывчатые свойства; в том же
году он разработал технический способ получения тринитротолуола,
и вскоре производство тринитротолуола было установлено в Герма-
нии на химическом заводе Грисгейм. Изготовлявшийся здесь тротил
применялся для производства аммонитов.
Прусское военное министерство еще в 1887 г. ставило опыты
•со смесями тринитротолуола с другими веществами (окислителями),
но при этом не было получено практических результатов. В 1892 г.
<были проведены новые опыты, но с чистым тринитротолуолом.
Однако только в 1902 г. он был принят в Германии для снаряже-
ния снарядов.
Особый интерес, проявленный в Германии к тротилу, обусловлен
требованием морской артиллерии на бронебойный снаряд, для кото-
рого требовалось очень мало чувствительное к механическим воз-
действиям взрывчатое вещество, не взрывающееся в момент удара
снаряда о броню. Применение для этой цели тротила позволило наи-
лучшим образом разрешить эту задачу.
С другой стороны, появление в начале XX столетия дешевого
олеума — после постройки сернокислотных заводов, работающих по
.методу контактного окисления сернистого газа—удешевило производ-
-416
ство тринитротолуола настолько, что он мог изготовляться и приме-
няться в больших количествах.
В России производство тринитротолуола было впервые установ-
лено в 1908 г. на Шлиссельбургском заводе (первоначальная произ-
водительность цеха была 250 т тринитротолуола в год; к концу
года цех был расширен до 600 т в год).
В 1909 г. было начато производство тротила на Охтенском за-
воде, которое велось здесь до известного взрыва 1916 г. При этом
взрыве тротиловый цех был разрушен и больше не восстанавли-
вался.
Краткие сведения о развитии технологи-
ческого процесса производства тротила.
Впервые тротил изготовлялся на химическом заводе Грисгейм
в Германии по способу, разработанному и установленному на этом
заводе Гайзерманом. По этому способу толуол нитровали в три фазы
с соблюдением точных молекулярных стадий нитрации, т. е. полу-
чали в 1-й фазе мононитротолуол, во 2-й — динитротолуол и в 3-й
тринитротолуол. Нитрующие смеси для всех трех фаз готовили на
свежих кислотах.
В целях экономии кислот позже стали применять часть отрабо-
танной кислоты от 3-й нитрации для изготовления смеси
для 2-й нитрации, и часть отработанных кислот 3-й и 2-й
нитрации — для изготовления кислотной смеси для 1-й ни-
трации.
Нитрование производилось путем слива нитрационных смесей
к нитруемым соединениям при всех трех фазах.
Опыт показал, что при приготовлении смесей для 1-й и 2-й фазы
с частичным использованием отработанных кислот наблюдалось за-
метное ухудшение качества продукта (усиливались процессы окисле
ния и осмоления) в случае применения плохо или вовсе не отстояв-
шихся отработанных кислот. Для получения удовлетворительных
результатов требовалось предварительное длительное отстаивание
отработанных кислот — до 20 суток и более. Это заметно осложняло
кислотное хозяйство завода.
Английские заводы, установившие у себя впервые производство
тротила во время империалистической войны, отказались от соблю-
дения точных молекулярных стадий нитрации: с целью наилучшего
использования азотной кислоты при 1-й фазе они вели здесь процесс
так, чтобы пронитровывалось до мононитротолуола только 50% толу-
ола; во 2-й фазе они вели процесс так, чтобы продукт нитрации имел
в составе 50—75% динитротолуола и соответственно 50—25% мо-
нонитротолуола. Этим достигалось то, что продукт имел темпера-
туру плавления ниже 35°; такой продукт легко отделялся от отрабо-
танной кислоты и достигалась максимальная полнота отделения.
Другой особенностью английского способа было то, что при нем вся
отработанная кислота от 3-й нитрации применялась для изготовле-
ния смеси для 2-й, причём вместо частичного разбавления отрабо-
танной кислоты водой практиковали разбавление ее слабой азотной
Химия—13—27
417
кислотой (из конденсационной системы для отходящих из нитраторов
газов) и промывной воды от предварительной промывки
тротила (содержала несколько десятков килограммов азотной кис-
лоты). Наконец, здесь велся прилив динитротолуола к смеси, а не
наоборот.
Следующим значительным этапом в развитии тротилового про-
изводства является введение так называемого полного кислотообо-
рота. Первые сведения об зтом способе стали известны из итальян-
ских источников. В зтом способе процесс построен таким образом,
что вся отработанная кислота от 3-й нитрации поступает на изготов-
ление смеси для 2-й, а вся отработанная кислота от 2-й нитрации
поступает на изготовление смеси для 1-й. Здесь был впервые приме-
нен так называемый «горячий кислотооборот», при котором смесь
для 2-й нитрации готовилась в нитраторе подачей сюда неотстояв-
шейся, не охлажденной после сепарации тротила отработанной кис-
лоты от 3-й нитрации и азотной кислоты. При полном кислотообо-
роте оказалось возможным пользоваться для изготовления смеси
для 2-й нитрации не крепкой азотной кислотой, а слабой (напри-
мер, 40%-ной азотной кислотой). Как при 1-й, так и при 2-й нит-
рации нитруемое соединение сливалось к смеси.
В заключение отметим, что еще с 1914 г. началась усиленная раз-
работка способов непрерывной нитрации жидких ароматических
углеводородов, и несколько установок было поставлено во время
империалистической войны (тип Неймана в Германии, камерная си-
стема в Англии).
Развитие производства ксилила. Тринитро-
ксилол был впервые получен Фитигом в 1869 г. Швейцер получил
в 1883 г. при нитровании каменноугольного ксилола продукт
с температурой затвердевания в пределах 160—177° (для разных
образчиков).
Заводское производство ксилила было впервые установлено во
время империалистической войны в России и во Франции. В России
производство ксилила было установлено проф. Солонина. По этому
способу получали ксилил нитрованием ксилола в одну фазу сливом
углеводорода к кислотной смеси.
Во Франции был установлен способ в три фазы, причем полу-
ченный ксилил после отмывки кислот очищался от маслянистых
примесей промывкой . его на холоду 5-кратным количеством
спирта.
В 1916 г. Филиппов разработал способ получения ксилила
в две фазы—через мононитроксилол. Этот способ заметно выгоднее
способа проф. Солонина, однако, он не был освоен.
В 1927—1928 гг. был разработан в лабораторном масштабе
способ получения ксилила в две фазы через динитроксилол.
418
Тетрил впервые был получен Мертенсом в 1877 г. и подроб-
нее изучен Ромбургом в 1883 г. В результате исследования тетрилу
была дана следующая формула строения:
СН3
n^no2
\о2
^Взрывчатые свойства тетрила впервые изучались по инициативе
Ленце в прусском военном министерстве. Результаты этого иссле-
дования установили его непригодность для снаряжения снарядов
вследствие его большой чувствительности к механическим воздей-
ствиям. Но он оказался вполне пригодным для снаряжения детонато-
ров и капсюлей. Для этой цели он уже в 1906 г. изготовлялся в зна-
чительных количествах в Германии. В России производство тетрила
было впервые установлено в 1908 г. В 1912 г. Артиллерийское ве-
домство построило тетриловый цех на одном из заводов взрывчатых
веществ, а во время войны 1914—1918 гг. тетрил изготовлялся и на
другом заводе.
Гексоген. Учитывая опыт мировой войны, государства, ли-
шенные достаточных сырьевых ресурсов (каменного угля для произ-
водства ароматических углеводородов, жиров для производства гли-
церина и т. п.) для производства взрывчатых веществ, стремятся изы-
скать такие взрывчатые вещества, для которых сырье может быть
в неограниченных количествах изготовлено синтезом из легко до-
ступных веществ: угля, воздуха и воды. Среди ряда веществ, изу-
чавшихся под этим углом зрения, большой интерес наряду с азот-
ным эфиром пентаэритрита представляет циклотриметилентрини-
тро амин или гексоген.
. Гексоген открыт в 90-х гг. прошлого столетия, а способ его по-
лучения впервые описан Генингом в 1898 г. Техника не заинтересо-
валась тогда этим веществом, и оно было надолго забыто. Лишь во
время империалистической войны 1914—1918 гг. вернулись к изуче-
нию гексогена. Он был предложен как замена нитроглицерина в по-
рохах 1 и в качестве добавки к нитросоединениям 2 для увеличения
их взрывного действия.
Начиная с 1929 г., научные лаборатории и техника стали уси-
ленно интересоваться гексогеном, а в настоящее время он произво-
дится в значительных масштабах в ряде стран (Италия, Германия
и др.).
Хлоратные взрывчатые вещества. Причина
описанных выше неудачных опытов Бертолле с применением хлор-
1 Герм. пат. № 298539, 1916.
2 Герм. пат. № 299028, 1916; швейц, пат. Герца № 88759, 1921
419
новатокалиевой соли заключается в том, что во время производства
этих опытов не были известны ни большая чувствительность к меха-
ническим воздействиям смесей бертолетовой соли с серой и углем,
ни бризантность таких смесей, делающие их весьма опасными при
изготовлении и непригодными для применения в качестве метатель-
ных средств.
Второй этап развития в истории хлоратных взрывчатых веществ
наступил после открытия Блека (1869 г.) способности этих веществ
детонировать от капсюля-детонатора. Это позволило применить
составы, значительно менее чувствительные к лучу огня. Вслед за
этим Шпренгель предложил составы, названные его именем, сильно
понизившие опасность при производстве хлоратных взрывчатых
веществ, так как эти составы готовились простым насыщением на
местах работы патронов из хлората калия нитробензолом. Здесь
опасность изготовления и применения взрывчатых веществ на-
столько уменьшилась, что новые взрывчатые вещества стали с успе-
хом применять при ведении взрывных работ.
Наконец, третий, последний, этап наступил в конце 90-гг. про-
шлого столетия, когда стали получать дешевую бертолетову соль
электролитическим путем и когда французский химик Стрит изо-
брел новые хлоратные составы, шеддиты (это название они получили
от наименования местечка Шедд во Франции, где они изготовлялись)
со значительно пониженной чувствительностью к механическим воз-
действиям. Это понижение чувствительности достигалось добавле-
нием флегматизаторов — касторового масла, парафина и других ве-
ществ.
В настоящее время применяются как хлоратные взрывчатые ве-
щества типа Шпренгеля, так и вещества типа шеддитов.
ПРИЛОЖЕНИЕ Z
(Написано совместно с М. С. Гельфманом
КИСЛОТНОЕ хозяйство
Свойства серной кислоты
100%-ная серная кислота —моногидрат —представляет собой
маслянистую тяжелую жидкость, уд. веса 1,88 при 20°.
Технические сорта крепкой серной кислоты, применяемые на
практике: купоросное масло с содержанием 92—93% H2SO4 и олеум
(содержит до 20% свободного §О3). Технические сорта слабой
серной кислоты: камерная содержит 65% H2SO4 и гловерная или
башенная — 75% I^SO^. В последнее время на башенных установ-
ках получают непосредственно купоросное масло.
Олеум иначе называется контактной серной кислотой, так как
он получается на контактном сернокислотном заводе- Купоросное
масло обычно получается путем упаривания слабой серной кис-
лоты на концентрационных установках. Для удобства учета вне
зависимости от крепости кислоты в производстве она учитывается
в пересчете на моногидрат.
Крепость серной кислоты в заводских условиях и на практике
в первом приближении определяется посредством замера удел!-
ного веса. В сернокислотном производстве употребительны арео-
метры Боме.
.В области до содержания 90% H2SO4 ареометр представляет
собой достаточно чувствительный прибор. При более высоких кон-
центрациях, где удельный вес мало меняется с концентрацией, кре-
пость кислоты определяется анализом.
Для хранения и транспорта кислоты большое значение имеет
точка ее плавления.
На рис. 49 дана диаграмма плавкости серной кислоты • различ-
ной концентрации.
Из этой диаграммы видно, что транспортабельными в зимнее
время и, следовательно, круглый год являются кислоты: 57%-ная
камерная кислота, 68—75%-ная башенная кислота и 93%-ное купо-
росное масло (замерзают при температуре ниже 25°). Сорта олеума,
содержащие 18—20% свободного SO3, замерзают при—11°, а так
называемый высокопроцентный олеум, содержащий 65% свободного
SO3, замерзает при температуре 0,8°.
421
Рис. 49. Диаграмма плавкостей системы H2SO4—Н2О.
Рис. 50v График теплоемкости и температур кипения
растворов серной кислоты различной концентрации.
Сплошная кривая—удельная теплоемкость при 20°; пунктир-
ная—средняя теплоемкость в интервале от 0° до температуры
кипения.
422
У^елы&ге теплоемкости и температуры кипения серной кислоты
приведены? в графике рис. 50.
Теплота смешения H2SO4 и Н2О определяется по следующей
формуле Томсена:
' „ _ 17 860 • п
~ п +1,798 ’
где п — Число кг)мол Н2О, прибавляемое к 1 кг/мол H2SO4; теплоту
смешения можно определить, по Портеру, с учетом температуры,
при которой происходит смешение:
504,2-й . 0,714« —15)
Н ~~ п + 0,2013+ п + 0,062 ’
где п —количество кг Н2О на 1 кг SO3. В обоих случаях теплота
смешения получается в больших калориях.
Действие H2SO4 на металлы. При крепости серной
кислоты до 80% свинец является вполне устойчивым металлом как для
горячей, так и для холодной кислоты. Эта устойчивость обусловли-
вается образованием защитной пленки сульфата свинца, нераство-
римой в кислоте. Однако с повышением крепости кислоты раствори-
мость пленки в ней повышается, и поэтому для крепких кислот свиг
нец не стоек.
С повышением крепости кислоты с 97 до 100% H2SO4 раство-
римость сульфата свинца возрастает примерно в 20 раз и соответ-
ственно увеличивается корродирующее действие кислоты на свинец.
Коррозия зависит также от чистоты свинпа. Поэтому по ОСТ 2985
свинец должен содержать не более 0,1% примесей.
Аппаратура для концентрированной кислоты (купоросное масло)
делается обычно из чугуна; для олеума применяется железо.
Стойкость черных металлов в крепкой кислоте объясняется также
образованием защитной пленки сульфата железа, растворимость ко-
торой уменьшается с повышением крепости кислоты.
Техника безопасности. Серная кислота, особенно
крепкая, действует на вещества растительного и животного про-
исхождения обугливающим образом. При ожоге рекомендуется
не смывать водой, а стереть тряпкой и затем смыть содовым раство-
ром. Не следует лить воду в крепкую кислоту. При обслуживании
кислотной аппаратуры и ремонте ее, следует быть в очках и надевать
резиновые перчатки. При сливе кислоты из бочек и цистерн, чистке
цистерн нужно также иметь противогаз. Нужно помнить, что при
действии кислоты на железо выделяется водород; поэтому от искры
может произойти взрыв образующегося при выделении водорода гре-
мучего газа.
Свойства азотной кислоты
Азотная кислота в чистом виде представляет собой бесцветную
жидкость уд. веса 1,52, которая при —42° затвердевает и кипит при
86°.'При этой температуре она начинает медленно разлагаться по
уравнению:
2HNO, -> 2NO2 + Н2О + |о2
423
Двуокись азота растворяется в азотной кислоте и окрашивает
ее в желтый цвет. С водой азотная кислота смешивается в любых
Таблица 80
Удельный HNO3 Температура
вес % кипения °C
1,15 24,8 104
1,30 27,5 113
1,40 65,3 119
1,42 69,8 123
1,48 86,0 115
1,50 94,1 99
1,52 99,7 86
азота, образующие
с
отношениях. Удельные веса й ,
точки кипения водных раство-
ров ее приведены в табл- $0).
Максимум температуры ки-
пения водной азотной кисло-
ты соответствует содержанию
68,4% HNO3 и при этой кон-
центрации она отгоняется без
изменения состава (нераздель-
нокипящая смесь). Поэтому .
простой дестилляцией нельзя-
получить более крепкую кис-
лоту.
Более крепкие растворы
HNO3 дымят на воздухе, вы де-
влагой воздуха туман. Концентри-
ляя окислы
рованная азотная кислота — сильный окислитель. Она окисляет
серу в серную кислоту, фосфор — в фосфорную. Особенно сильно
она действует на металлы.
Рис. 51. Кривая температур замерзания азотной
кислоты.
Из кривой температур замерзания (рис.^1) видно, что при содер-
жании азотной кислоты от 40% HNO3 и выше температуры замер-
зания ее ниже — 38°.
Как было указано, при температуре кипения начинается разло-
жение азотной кислоты. По Вольнею, процент разложения в зависи-
мости от температуры следующий (см. табл. 81).
424
Теплота образования
азотной кислоты из эле-
ментов, по Томсену, равна
41 510 кал; теплота об-
разования из NO, О2 и
Н2О равна 43 085 кал; из
NO2, О2 и Н2О 42 515 кал;
наконец, из N2O4, О2 и
Н2О 18 670 кал.
Теплота растворения
азотной кислоты в воде
определяется по формуле
Томсена:
Таблица 81
Температура СС Процент разложения азотной кислоты
86 9,53
100° 11,77 -
130° 18,79
160° 28,96
190° 72,07
260° 93,03
266° 100
п 8,974
, т ~ 4 + 1,737 • п>
где п —число кг!мол Н2О на кг!мол HNO3.
т — число кг]кал, выделившихся при растворении.
Действие азотной кислоты на металлы.
Почти все металлы разъедаются азотной кислотой. Такие металлы,
как Си, Ag, Hg, Bi, требуют для начала реакции присутствия
азотистой кислоты; при их взаимодействии с азотной кислотой не вы-
деляется аммиак.
При растворении Zn, Cd, Si, Sb, Al, Fe, Mg, K, Na происхо-
дит частичное восстановление азотной кислоты до аммиака. Стой-
ким по отношению к концентрированной азотной кислоте, начиная
с 80% HNO3, как известно, является алюминий. По исследованиям
Трилета, наиболее важными факторами являются температура и кон-
центрация кислоты; например, при повышении температуры на 10°
скорость разъеда&ия алюминия увеличивается на 100%. Примесь
серной кислоты также увеличивает разъедающее действие азотной
кислоты на ’алюминий; например, наличие 0,04% H2SO4 увеличи-
вает корродирующее действие HNO3 на алюминий в два раза.
В настоящее время в связи с освоением в производстве хромони-
келевой стали последняя применяется вместо стекла и керамики для
хранения слабой азотной кислоты. Для серной и азотной кислот и
их смесей прй высокой температуре единственным материалом пока
является термосилид.
Ниже приводятся составы кислотоупорных сплавов, применяе-
мых в кислотных производствах-
Кислотоупорные сплавы
Ферросилицие^ые сплавы. Начало производства
этих сплавов относится'к 1900 г., когда во Франции был взят
патент на «метиллюр», содержащий 15—20% кремния. В настоя-
щее время эти сплавы выпускаются в различных странах под
различными названиями (метиллюр — Франция, элианит -— Ита-
лия, айронак — Англия, дюрайрон — США, нейтральзйзен —
425
♦
Германия, термоеилид — СССР. Обычно они содержат от 15 до 20%
кремния, так как при содержании ниже 14% их свойства мало
отличаются от обычного чугуна,' а при содержании выше 22%
их химическая устойчивость мало повышается, но зато резко сни-
жаются механические качества.
Приводим состав американского «дюрайрона»: 15,51%Si,
82,23% Fe, 0,66% Мп, 0,83% Сг, 0,57% Р, 0,01% S (уд. вес 6,94).
Железохромистые сплавы. Вначале эти сплавы
под названием «нержавеющей стали» применялись для противодей-
ствия атмосферному влиянию; они содержали 12—14% хрома.
Позже их стали применять в кислотном производстве, причем
для азотной кислоты применяется сталь с содержанием 17—19% Сг,
0,1% С, 0,5% Si.
В отличие от хромоникелевых сплавов железохромистые приме-
няются в том случае, где требуется также жароупорность.
Хромоникелевые сплавы. Из этих сплавов приме-
няются, главным образом, следующие четыре группы: VIM и V2A —
Германия, Anka — Ан-
глия, Calite А — США,
имеющие следующий
состав (в %) (табл. 82).
Наибольшее распро-
странение получили ста-
ли типа V2A с та-
ким средним составом:
18% Сг, 8%Ni, 0,1% С.
Они вполне устой-
чивы по отношению к
температуре и применяются для
Таблица 82
Наимено- вание примеси V1M V2A Anka Calite А
с . . . . 0,15 0,25 0,1 0,8
Сг . . . 14 20 15 15
Ni ... 1,8—2 7 10—12 35
невысокой
кислотам
при
слабым
аппаратуры и кислотопроводов.
Мешка кислот
Из рассмотрения дозировочных расходных коэфициентов и кис-
лотных модулей при получении нитросоединений можно составить
себе представление о размерах потребления и оборота кислот, кото-
рые в несколько раз превышают количество выпускаемой продук-
ции. Кроме того, даже в случае наличия кислотных производств на
месте при мастерских нитрации должны быть постоянные запасы
кислот для обеспечения бесперебойности производства, не говоря
уже о тех случаях, когда производство пользуется привозными
кислотами. Эти факторы и определяют собой размер кислотного
хозяйства.
Кислотная мастерская, таким образом, должна включать в себя:
хранилища для свежих кислот, установку по сливу кислот из желез-
нодорожных цистерн, мешатели, мерники, темпер а ционные котлы
с водяными рубашками, вакуум-установки для зарядки сифонов,
насосную станцию, дозировочные котлы, хранилища нитросмеси и
отработанной кислоты, лабораторию для контроля.
426
Постоянные и правильно организованные замеры кислот являются
существенным элементом контроля, особенно если учесть большие
количества оборачиваемых кислот и трудность обнаружения перерас-
ходов, учет которых очень важен для экономичности ведения про-
цесса, и если еще учесть, что свыше 80% стоимости исходных мате-
риалов для нитрации падает на кислоты.
При приготовлении нитрационной смеси помимо правильной
дозировки и анализа решающую роль играет тщательное перемеши-
вание для достижения однородности смеси.
Мешка воздухом, несмотря на сравнительную простоту, имеет
ряд органических недостатков: выдувание азотной кислоты, увлаж-
нение и, следовательно, разбавление смеси влагой атмосферного воз-
духа, разъедание перфорированных труб (барботеров), по которым
воздух подается в смесь, большая продолжительность процесса
благодаря различной вязкости кислот. При мешке сжатым воздухом
охлаждение не практикуется, получается значительное повышение
температуры, влекущее за собой разложение азотной кислоты, а при
наличии нитропродуктов в отработанной кислоте может произойти
и взрыв вследствие местного перегрева и воспламенения нитросоеди-
нений.
На рис. 52 приводятся основные схемы мешки кислот.
Схема мешки кислот воздухом. По этой схеме
кислоты перемешиваются в смесителе D, который для охлаждения
орошается водой. Смесь кислот перед подачей ее в сборник L охлаж-
дается дополнительно в сборнике К.
Меш ка кислотно схеме, описанной Наумом.
По этой схеме кислоты поступают из мерников N и 5 в нижнюю
часть сосуда-смесителя, снабженного термометром и змеевиком для
охлаждения. По наполнении смесителя кислота спускается в подъем-
ник D. Кислые пары уходят через трубу Т: Кислоты поступают
в смеситель в тангенциальном направлении и здесь смешиваются;
при прохождении из смесителя К в подъемник D, а также в самом
подъемнике происходит дальнейшая мешка кислот. Готовая смесь
передается по трубе MS в сборник кислот.
По словам Наума, мешка получается настолько удовлетворитель-
ной, что этот способ вполне пригоден даже для нитроглицериновых
заводов.
Схема механической мешки кислот с вер-
тикальным смесителем. Охлаждение производится
помощью охлаждающей рубашки.
Схема механической мешки кислот с гори-
зонтальным смесителем. Охлаждение в метателе из-
бегается непрерывным добавлением в цистерну холодной смеси кис-
лот, получаемой охлаждением в холодильнике К (частичная рецир-
куляция смеси). Кислота перекачивается в сборник центробежным
насосом Р по трубе NS.
Схема мешки кислот посредством цирку-
ляции. Кислоты непрерывно поступают через дозеры (не пока-
427
' Схема
мешки кислот Воздухом
Мешка кислот по
схеме Наума
[горизонталЬнЬ1й котел сласосом)
Схема механической
мешки кислот
(SepmukaobHbiu котел)
Схема мешки кислот
посреОртвом циркуляции
Рис. 52. Основные схемы мешки кислот.
428
заны на рисунке) в смеситель К, снабженный змеевиком для охла-
ждения. Применением частичной рециркуляции смеси, охлаждае-
мой дополнительно в холодильнике К', значительно увеличивается
производительность смесителя.
При точной работе кислоты отвешиваются; для бесперебойного
процесса требуется четыре мерника.
Расчет кислотных смесей
Может быть три случая расчета кислотных смесей: 1) расчет
одноименных кислот, 2) расчет разноименных кислот (тройных сме-
сей), приготовляемых смешением одноименных кислот, и, наконец,
3) расчет разноименных кислот, приготовляемых смешением трой-
ных смесей с одноименными кислотами (исправление кислот, при-
готовление тройной смеси смешением отработанной кислоты с ме-
ланжем и одноименными кислотами).
1. Расчет одноименных кислот. Примем следующие обозначения:
А — концентрация крепкой кислоты;
В —концентрация заданной кислоты;
С — концентрация слабой кислоты;
х — количество слабой кислоты, которое надо взять для получе-
ния кислоты заданной концентрации;
у — количество крепкой кислоты, потребное для той же цели;
Р —количество кислоты концентрации В, которое задано по-
лучить.
а) Приготовление кислоты заданной кон-
центрации из двух кислот: более крепкой и
более слабой, чем заданная концентрация,
т. е. А > В > С-
Очевидно:
Ау + Сх = РВ. 1
у = Р— X. ’ J W
Отсюда:
Р(А-В).
А —С ’
у = Р -X-
б) Если для
кислота, а во
разбавления берется не слабая
д а, то С = 0 и из уравнений (2) получаем:
У =
у = Р-----X.
(3)
в) Если задано не количество кислоты но-
вой концентрации, а количество Рх крепкой
кисло ты, которое требуется разбавить от концентрации А до
концентрации В, то из уравнения
АРг + Сх = (Рх + х) В
429
найдем:
т_ Л И-В)
• х- в^С
Количество кислоты концентрации В, которое здесь получим, оче-
видно равно
г) П р а в и л о «креста» (правило К о б е н ц л я).
Правило Кобенцля, очень удобное для расчетов, основано на сле-
дующем.
Подставляем в формулу у = Р — х значение х — и3
формул (2). После простых преобразований получим:
Деля друг на друга левые и правые части выражений для у и хг
получим:
у = В-С
х А — В ’
Отсюда следует, что для составления заданной кислоты концентра-
ции В надо брать кислоты с концентрацией А и С в соотношении
(В—С): (Л — В), т. е. надо взять (В — С) частей крепкой кислоты
(концентрации А) и (А —В) частей слабой кислоты (концентра-
ции С). Это правило нагляднее изображается графической схемой в
виде прямоугольника с двумя диагоналями
А
С
В— С = а
А — В = Ь
Числа, обозначающие концентрацию смешиваемых кислот, пи-
шем на левых углах прямоугольника, а заданную концентрацию—
на пересечении диагоналей. Вычитаем числа, расположенные на
одной диагонали, из большего —меньшее, и разность пишем на дру-
гом конце той же диагонали. Полученные числа дают относительные
количества кислот, которые следует смешать для получения кислоты
заданной концентрации, причем они относятся к тем кислотам, с
которыми соединены горизонталями.
Пользуясь графической схемой, мы легко подсчитаем, что для со-
ставления кислоты концентрации В надо взять В—С=а в. ч. креп-
кой кислоты и А—В=Ь в. ч. слабой кислоты, причем получим
a-j-b в. ч. смеси заданной концентрации.
д) Расчет олеума. В заводской практике иногда требуется
приготовление олеума определенной концентрации. Разбавление
производится обычно серной кислотой и реже —слабым олеумом.
Определение концентрации серной кис-
ло т ы в олеуме. Обычно концентрация олеума дается в про-
430
центах содержащегося в нем S03; для определения
жания моногидрата в олеуме (это количество моногидрата (ябШИИ
вается из содержащегося в олеуме моногидрата серной кисшмЙгЖ
моногидрата, образующегося из S03 при добавлении к последйейф
эквивалентного количества воды) исходим из того, что 100 в. ч. SO»
дают 122,5 в. ч. Поэтому, если олеум содержит р% H2SO4+
+ q°/0 SO3, то общее содержание в нем моногидрата серной кислоты:
5 = р + 1,225? = (? + ?) + 0,225? = 100 + 0,225?. (6>
Отсюда видно, что величина 0,225? представляет собой то коли-
чество воды, которое надо добавить к 100 в- ч. олеума, чтобы превра-
тить весь свободный SO3, содержащийся в нем, в моногидрат серной
кислоты.
Расчет олеума можно произвести по правилу креста, для чего
надо выразить концентрацию олеума в процентах моногидрата сер-
ной кислоты, пользуясь предыдущим расчетом.
Пример. Пусть требуется разбавить олеум с содержанием 28% свобод-
ного SO3 до содержания последнего 18%. Для разбавления служит купорос-
ное масло с содержанием 92% H„SO4. В каком соотношении надо смешать имею-
щиеся кислоты?
Определяем по формуле (6) крепость обоих сортов олеума в процентах моно-
гидрат a H»SO4:
S, = 100 + 0,226 • 28 = 106,3% H2SO4;
S3 = 100 + 0,226 • 18 = 104,05% H2SO4;
Строим схему Кобенцля.
106,3^ /12,05(104,05 — 92)
104,05
92 Х2,25(106,3 — 104,05)
Следовательно, для получения олеума заданной концентрации надо смешать
12,05 в. 1 ч. 28%-ного олеума и 2,25 в. ч. 92%-ного купоросного масла.
2. Расчет тройных смесей, приготовляемых смешением двойных1
смесей. Примем следующие обозначения:
Таблица 83
Название кислоты или смеси * Количе- ство кг Состав смесей, %
H2SO4 HNO3 Н2о Итого
Заданный состав тройной смеси . , р А В с 100
Крепкая азотная кислота X — ьа —-
Слабая азотная кислота — — —
Олеум или серная кислота .... У а2 — — —
Меланж . Z а ъ с 100
Отработанная кислота и а1 Ьг С1 100
Вода t — — — —-
1 Чистые кислоты, как содержащие только два компонента — моногидрат и
воду, — носят название двойных смесей. Тройными смесями называются смеси,
в состав которых входит три компонента: HsSO4, HNO3 и Н2О.
431
Выведем ряд формул для решения задач, чаще всего встречаю-
щихся в заводской практике.
а) Приготовить Р кг тройной смеси из серной и азотной кислот
и воды.
Из условия, что количество моногидрата серной кислоты в Р кг
смеси равно количеству того же моногидрата в х кг серной кислоты,
следует:
РА_____уа2'
100 = 100’
отсюда
Аналогично:
РА
у = —
и а2
РВ
х = т~-
Ъ2
-Очевидно :
t = Р — (»+ у).
(7)
(8)
(9)
Приготовление кислотной смеси с добавкой воды часто практи-
куется в лабораториях, реже на опытных установках; в производ-
стве же оно является совершенно недопустимым; по экономиче-
ским соображениям здесь нужно вместо воды применять слабую азот-
ную кислоту.
б) Приготовить Р кг нитрационной смеси
из серной, крепкой азотной и слабой азот-
ной кислот.
Для составления Р кг смеси с содержанием А°/о H2SO4 необхо-
димо взять
п А
у = Р— кг
аг
(Ю)
серной кислоты с содержанием а2°/0 H2SO4.
Обозначим через а ту часть азотной кислоты от величины В (кон-
центрации HNO3 в смеси), которая вводится в смесь с х кг крепкой
азотной кислоты; тогда с хх кг слабой азотной кислоты будет вве-
дена часть (В — а) той же величины.
В этом случае имеем:
х=Р-^-;
Ъ2
-Р^В-а)
-П
(Н)
Ь3 '
Очевидно у х х± = Р; вставляя значения у, х и xt из
формул (10) и (11), имеем:
Р(В-а) _
Ъ3
432
Решая это уравнение относительно а, найдем:
а 2
а -а ----------s
1_____1_ •
Ь2 Ь3
Вставляя это значение а в выражение для количества
азотной кислоты х и сделав преобразования, найдем:
РВ-(Р-у)Ь3
Ъ2-Ъ3
^Наконец, значение хх найдем из уравнения:
Xj = Р—у — х.
в) Приготовить Р кг нитрационной
из крепкой серной, слабой серной и азотной
кислот.
Аналогично предыдущему легко вывести:
крепкой
(12)
(13)
смеси
— РА — (р —-g)°3
а3 — а3 ’
где а3 —концентрация слабой серной кислоты;
У1 = Р — х — у,
где ух —количество слабой серной кислоты для изготовления Ркг
смеси.
3. Расчет тройных смесей, приготовляемых смешением двойных
и тройных смесей, а) Приготовить Р кг смеси из сер-
ной и азотнойкислот и меланжа (или отра-
ботанной кислот ы).
Составляем баланс моногидратов смешиваемых кислот:
а - а, п А
2 100 100 — Р 100’ (15)
21^+^ = ^^ <16>
Кроме того, очевидно:
Р = z + х + у. (17)
Определим из уравнения (17) значение г, подставляем его в урав-
нения (15) и (16); решением двух последних уравнений относительно
х найдем:
х = р g(a2Tha,)~Z?(--F— • (18)
(bs — b) — afrs
Химия—13—28
433
Величину z определяем из следующих соображений.
В Р кг смеси должно содержаться Р ~ кг моногидрата азотной
кислоты. С х кг азотной кислоты вводится х кг моногидрата
HNO3. Следовательно, с меланжем (отработанной кислотой) должно
вводиться кг моногидрата азотной кислоты. При со-
держании в меланже (отработанной кислоте) b%HNO3 надо взять
такой кислоты:
z = РВ ^хЬ2. (19)
Вставляя в формулу (19) значение х, вычисленное по формуле (18),
найдем значение z, т. е. количество меланжа (отработанной кис-
лоты), которое надо взять для составления Р кг кислотной смеси
заданного состава.
Определив значение х и z, легко найдем у по формуле
у - Р — х — z. (20)
б) Приготовить Р кг кислотной смеси из
серной кислоты, меланжа и отработанной
кислоты.
Аналогично предыдущему случаю составляем баланс моногидра-
тов смешиваемых кислот
а2у + az + ахи = РА;
bz + Ьг и = РВ;
у 4- z + и = Р.
Решая эти уравнения относительно у, z и «, получим:
р (A—a) (bj—Ь) —(В —Ь) (а!—а).
У (а2 —«) (&1— Ь) + Ь («1 — а) ’
2___р В (а1 аг) bt (А аг)___
— (а2 —a)(bi —b) + b(ai—а)’
„ __ р (В — Ь) (а2 а) + b (А — ах)
(а2 a)(bi Ь) + Ь (ctj — а)
(21)
(22)
(23)
(24)
(25)
(26)
БИБЛИОГРАФИЧЕСКИЙ УКАЗАТЕЛЬ ОРИГИНАЛЬНЫХ РАБОТ ПО
ВОПРОСУ О РАСЧЕТЕ КИСЛОТНЫХ СМЕСЕЙ
1. М. Gerster, Oleum-Mischungen, Chem. Ztg., 1, 3, 1887.
2. A. С 0 b e n z 1, Einfache Mischungs-Berechnung, Chem. Ztschr., 108, 954,
1910.
3. H. Mager, Mischungs-Berechnungen, Chem. Ztschr., 97, 865, 1910.
4. T. Evers-Dusseldorf, Eine leichte Rechnungsart zur Herstel-
lung einer Fliissigkeit, deren Gradstarke zwischen derjenigen zweier gegebenen
Fliissigkeiten liegt, Chem. Ztg-, 53, 476, 1910.
5. Fowler. J. Soc. Chem. Ind., 38 T., 34, 1919.
434
6. S о s ё, Rev. gen. mat. plast-, 2, 3, 6, 7, 1931; 1, 2, 1932. Русфи*
перевод см. «Нитроклетчатка и бездымный порох», Сборник перёвоДЗДк
статей, вып. 44, 1933. '
Т. Берль, Герберт и Валит, Номограммы для химической пик
мышленности, Госхимтехиздат, 1932. . -
8. С. Бесков, Номограммы для подсчета кислотных смесей, Хим-
строй, 8, 462, 1934.
9. С. Бесков, Расчет смесей кислот графическим способом, Ж. Хим.
Пр., 8, 69, 1933.
4. Денитрация отработанной кислоты. До поступления отрабо-
танной кислоты на денитрацию она подвергается отстаиванию (отра-
ботанные кислоты, содержащие ди- и тринитротолуол; однако при
современных способах производства тротила с полным кислотообо-
.ротом эта операция излишня, так как в этом случае на денитрацию
поступают только кислоты от 1-й нитрации). Отработанные кислоты
Рис. 53. Схема денитрационной установки.
Перегретый
_рг-
Отпавтатт
А
кислота
от производства пикриновой кислоты и тетрила должны быть подверг-
нуты специальной очистке во избежание осаждения возгоняющихся
нитропродуктов в верхних холодных частях башни и взрыва их.
• Содержащиеся в отработанной кислоте от 1-й нитрации толуола
нитро продукты летят вместе с парами воды и азотной кислоты и окис-
лами азота.
Как и процесс концентрации азотной кислоты (отгонкой из смеси
ее с серной), процесс денитрации состоит в том, что стекающая сверху
колонны отработанная кислота нагревается до 150—180° за счет теп-
лоты конденсации перегретого пара и разбавления серной кислоты;
при этом разлагается нитровилсерная кислота и отгоняются окислы
азота и азотная кислота и возгоняются нитросоединения. Легко окис-
ляющиеся органические примеси окисляются при этом в углекислоту
и воду.
Смесь уходящих из денитрационной башни паров и газов
направляется по трубопроводу в конденсационную систему.
Схема денитрационной установки. Отработан-
ная кислота поступает из питателя А (рис. 53) в денитрационную
435
♦
колонну В, навстречу ей у основания колонны подается перегре-
тый пар с температурой 250°; стекающая в низ колонны центриро-
ванная отработанная кислота поступает в сборник С, снабженный
змеевиком для охлаждения,и отсюда направляется на концентра-
ционную установку. Летящие из башни пары и газы направляются
по трубопроводу в воздушный холодильник И; конденсирующиеся
в первой половине холодильника при температурах выше 55—60°
вещества спускаются в сепаратор Е, где отделяются всплывающие
наверх нитропродукты, а через нижний штуцер спускается слабая
азотная кислота. Часть азотной кислоты конденсируется во второй
половине холодильника. Не конденсирующиеся в холодильнике
пары и окислы азота вместе с газами (воздухом) направляются на
абсорбционную установку.
В качестве денитрационной башни применяется термосилидовая
колонна тарельчатого типа.
Примерный состав отработанных кислот, поступающих на дени-
трацию с тротилового производства: 73% H2SO4, 0,5—0,7% HNO3,
0,5% нитротел, 4% N2O4, 21% Н2О.
При денитрации такой кислоты получается незначительное ко-
личество азотной кислоты с содержанием 15—20% HNO3. Поэтому
при таком составе отработанной кислоты ’(с малым содержанием
азотной кислоты) основным назначением денитрации является полу-
чение серной кислоты, свободной от азотной кислоты и нитротел.
Стекающая из денитрационной башни серная кислота содержит
68.-72% H2SO4.
Примерные расходные коэфициенты при денитрации
(на 1 т моногидрата в денитрированной серной кислоте)
Отработанной кислоты 1.02— 1,03 т моногидрата Серной кислоты
пара................. 0,2 — 0,3 »
воды . .,............... 10,0—15,0 -и3
электроэнергии.......... 3,0 — 5,0 квтч
Режим работы денитрационной колонны:
Давление пара перед пароперегревателем..........4,5 ат
» » » колонкой...................... 300 мм рт. ст.
Температура пара после перегревателя............ 210—250°
» кислоты, поступающей на колонку . . 30° '
Температура вытекающей денитрированной кислоты 150°
Состав вытекающей денитрированной кислоты .... H2SO4—67—70%
* N2O4—0,02—0,04%
Нитротел—0,3%
Температура газа на верху колонны............... 105°
» » после оросительного холодильника 55—60°
Разрежение на верху колонны.................. , 10 мм рт. ст.
» у эксгаустера ...........................25 мм рт- Ст.
Производительность колонны диаметром 0,85 м порядка 25—30 т
в сутки денитрированной серной кислоты, считая по моногидрату.
436
Полученная слабая денитрированная серная кислота подается
на концентрацию. Для концентрации служат аппараты «Хемико»,
Кесслера, Бюшинга, вакуум-концентраторы.
По преимуществу получили распространение аппараты «Хемпко».
Расходные коэфициенты на аппаратах «Хемико» следующие:
слабой серной кислоты......... 1,05 т
мазута ..................... 0,060 >>
воды ....................... 5,0 м3
электроэнергии .............20,0 квтч
Полученное в результате концентрации купоросное масло частью
используется для составления кислотных смесей, либо подается
для интенсификации олеумного завода, либо продается на сторону
(товарный продукт).
5. Утилизация потерь кислоты при нитровании. Количество наи-
более ценной азотной кислоты, теряющейся при нитровании, в отдель-
ных случаях достигает 20—25% всех затрат ее на производство.
Главным источником этих потерь являются отходящие газы и
промывные воды. Использование азотной кислоты из промывных
вод затрудняется чрезвычайно низкой концентрацией, которая не
превышает 1—1,5%. Поэтому утилизация этих потерь очень затруд-
нительна. Что касается азотной кислоты, уходящей с вентиляцион-
ными газами, то ее улавливание не представляет особых затрудне-
ний. Газы, отходящие из аппаратов 1-й и 2-й нитрации толуола,
состоят преимущественно из окислов азота; газы из аппаратов 3-й
нитрации содержат, кроме того, значительное количество азотной
кислоты. Простейшим способом улавливания этих газов и паров
является абсорбция их водой.
При взаимодействии окислов азота с водой и кислородом воздуха
происходят следующие реакции:
2NO + О2 = 2NO2 + 26 920 кал;
3NO2 + Н2О = 2HNO3 + NO + 32 520 кал.
Таким образом этот процесс состоит из ряда последовательных
реакций окисления NO в NO2 и абсорбции последней водой с обра-
зованием азотной кислоты.
Поэтому поглотительная установка должна состоять из ряда
последовательно поставленных башен (абсорберов), через которые
эксгаустерами протягивается газ, содержащий окислы азота. Абсор-
беры орошаются слабой азотной кислотой, причем здесь осуще-
ствляется принцип противотока, т. е. на последнюю башню подается
вода, которая постепенно закрепляется при последовательном про-
хождении через все башни, и из первых башен выдается азотная кис-
лота. Реакционное тепло уводится на оросительных холодильниках
путем охлаждения циркулирующей кислоты Ч
1 Эта поглотительная установка почти целиком соответствует обычным
абсорбционным установкам азотных заводов, подробно описываемым в учебни-
ках и учебных пособиях по азотной кислоте. Поэтому мы здесь не приводим
этого описания.
437
6. Хранение и транспортировка кислот. Хранение сер-
ной кислоты. Как видно из приведенных ранее данных о тем-
пературах замерзания серной кислоты разных концентраций, кис-
лоты с содержанием 80—85% H2SO4 и 98—100% H2SO4 замерзают
при температурах выше 0°. Поэтому избегают хранить и перевозить
серную кислоту такой концентрации. Для хранения серной кислоты
крепостью 52—55° Вё (65—70% H2SO4) применяется свинец. > Храни-
лища представляют собой обычно деревянные ящики, покрытые из-
нутри листовым свинцом. Для хранения кислоты средней концен-
трации (75% H2SO4) применяют наряду со свинцом железо. При
этом цистерны делаются из котельного железа, клепаные. Для купо-
росного масла и олеума применяется исключительно железо. Для
хранения купоросного масла пригоден также чугун, содержащий
несколько процентов кремния.
/Применяются, хотя и не получили большого распространения,
следующие материалы: андезитовый бетон (состоит из молотого ан-
дезита, жидкого стекла и кремнефтористого натрия), плавленые
естественные минералы (базальт и др.) и железобетон, облицованный
несколькими слоями кислотоупорных плиток.
Конструктивно хранилища оформляются в зависимости от ем-
кости. Хранилища для хранения до 30 т олеума или серной кислоты
делаются цилиндрической формы, горизонтальные с выпуклыми
днищами из клепаного котельного железа диаметром 2 м и длиной
10 м. Большей емкости хранилища делаются вертикальные,, пре-
имущественно с верхней выгрузкой помощью сифона.
Хранение азотной кислоты. Хранилища для кон-
центрированной азотной кислоты делаются из алюминия или желе-
зобетона, облицованного внутри кислотоупорными плитками.
Хранилища для слабой азотной кислоты делаются почти исклю-
чительно из хромоникелевой стали и реже из железобетона, обли-
цованного кислотоупорными плитками.
Транспортировка кислот. Наиболее практичной
для перевозки кислот является современная цистерна цилиндри-
ческой формы с соответствующими приспособлениями (сливные и
наливные приспособления, воздушки, предохранительные клапаны).
В отношении грузоподъемности, учитывая массовые перевозки кис-
лот, необходимо ориентироваться на большегрузные цистерны
(50—70 т), у которых помимо этого достигается более благоприят-
ное соотношение между,мертвым и полезным весом. Так, например,
если у двухосной (14—15-ти) цистерны на 1 т кислоты приходится
0,58 т тары, то у 50-тп цистерны приходится соответственно
0,47 т тары. Но основным вопросом здесь является выбор металла.
Для перевозки концентрированных серных кислот за границей
применяются кислотостойкие стали; для перевозки серной кислоты
средней концентрации (52—55% Вё)—освинцованное железо. При
отсутствии гуммированного металла или освинцованного железа
приходится пользоваться для перевозки слабых кислот стеклянной
и отчасти керамиковой тарой.
438
Для перевозки концентрированной серной кислоты у нАс ^ЙЙ^?
меняют обычные железные (типа нефтяных) цистерны, соответствуй^
щим образом оборудованные; для перевозки серной кислоты сред-
ней концентрации такие цистерны освинцовываются. \
Перевозка азотной кцслоты. Для перевозки кон-
центрированной азотной кислоты применяют алюминиевые цистерны;
для перевозки слабой кислоты применяют цистерны из хромонике-
левой стали или стеклянную тару (бутыли).
7. Токсическое действие окислов азота и HNO3. О кислы
а зо та. Раздражающий газ для легких и дыхательных путей,
разъедает ткани.
Кратковременное вдыхание небольших количеств нитрозных
г^зов вызывает только продолжающееся несколько часов легкое
кашлевое раздражение со скудным выделением мокроты.
Действие больших количеств вызывает сильное кашлевое раз-
дражение, а временами также головную боль и рвоту. После этого
часто следует видимое выздоровление; наступает период более или
менее нормального состояния длительностью 2—12 часов, редко
больше, после чего у больного появляется ясно выраженное чувство
слабости, часто озноб, тошнота, иногда рвота и другие болезненные
явления. При тяжелых отравлениях смерть наступает чаще в первые
24 часа. Индивидуальная восприимчивость к окислам азота у раз-
личных лиц неодинакова.
Хронические отравления окислами азота наблюдаются при вды-
хании небольших количеств окислов азота в течение длительного вре-
мени. Такие отравления приводят к хроническим катарам дыха-
тельных путей.
Необходима хорошая вентиляция производственного помещения
и исправная, возможно лучше герметизированная, аппаратура для
тех производственных процессов, при которых происходит выделе-
ние окислов азота.
Азотная кислота. Общий характер действия паров
азотной кислоты такой же, как и окислов азота. Азотная кислота
йдовитее всех остальных минеральных кислот.
8. Техника безопасности. По американским исследованиям, наи-
большее число несчастных случаев в кислотных хранилищах про-
исходило от образования водорода при действии кислоты на металл;
поэтому имели место разрывы цистерн от увеличения давления в
случае повышения температуры и взрывы гремучего газа при
случайном возникновении искры.
При заполнении хранилищ и цистерн считается нормальным
оставление над кислотой свободного пространства в 75—80льи;при
высоте свободного пространства в 30 мм и при повышении темпе-
ратуры на 10—15° имеет место увеличение давления до 10 ат.
Ремонт цистерн и хранилищ из-под кислоты следует производить
лишь после тщательной промывки водой и раствором щелочи, тща-
тельно освободив их (цистерны) до промывки от кислоты. Особенно
внимательно надо проверить отсутствие в хранилищах азотной
439
кислоты и окислов азота. В противном случае могут быть случаи тя-
желых отравлений включительно до смертельных.
Законом установлена предельно допустимая концентрация окис-
лов азота в воздухе рабочей зоны производственных помещений
0,005 мг/л в пересчете на N2O5.
На всех хранилищах наверху должны быть втулки, которые
необходимо время от времени открывать для удаления газов.
Следует обязательно пользоваться защитными очками при работе
с кислотой, при пуске насосов и вентиляторов, при отборе проб
кислоты, при наливе кислот в бутыли и при погрузке кислот в
вагоны.
ИМЕННОЙ УКАЗАТЕЛЬ
Александров 72
Барышникова 32, 33
§ер 69
Бутлеров 375, 383
Бюрло 231, 251, 312
Вендельштейн 224
Венен 231, 251, 312
Ворожцов Н. Н. 51, 52, 210, 274,
275
Голосенко О. М. 214
Гольдштейн 254
Горст А. Г. 264
Гуревич 19
Житкова 407
Забродина 52
Захаров 21, 22, 23, 24, 25, 26
Зильберфарб 65
Зинин Н. Н. 38, 411
Ильинский 19
Иордан 169
Карташев 71
Каст 288
Кижнер 224
Кондрацкий Б. Л. 72
Коновалов М. И. 19, 45, 46, 224
Крюков 331
Курбатов 374
Лебедев И. В. 225
Лекорше 231, 251, 313
"Марковников 63
Машкин 72
Меншуткин Б. Н. 54
Меншуткин Н- А. 64
Мухин 65
Наметкин С. С. 39, 49, 52, 55
Настюков. А. И. 377
Орлов Е. И. 25
Оствальд 40
Паскаль 250
Петрушевский 411
Сапожников А. В. 72, 73, 74, 75,
76, 78, 79, 81, 83, 84, 87
Сибгатуллин Н. X. 27, 53, 54
Соколова А. М. 19, 45
Солонина А. А. 135, 219, 232, 233,
331, 342, 418
Степанов А. в. 27
Стефанович 133, 230
Титов А. И. 31, 32, 33
Топчиев А. В. 33
Тронов 27, 49, 52, 53, 54, 55, 69
Филиппов О. Г. 231, 233
Холево Н. А. 25, 242
Чельцов 415
Шишков 43
Шорыгин П. П. 19, 33, 45
Штамм 72
Штетбахер 366
Энгельс 409
Эйтингон И. И. 25
Эфраим 180
Alessandrini (Алессандрини) 377
Alphen (Альфен) 52
Allsop (Олсоп) 45
Armstrong (Армстронг) 52, 254, 255
Arnall (Эрнолл) 70
Austen (Аустен) 365
Bacharach (Бахарах) 28
Badosche (Бадош) 247
Baekeland (Бекленд) 378
Bahlich (Балих) 57
Bamberger (Бамбергер) 24, 333,
343, 344
Barbiere (Барбье) 184
Batasinian (Бетесиниен) 116
Battegay (Батге) 32, 52
Batik (Батик) 185
Bebie (Беби) 72
Berger (Бергер) 43
Beilstein (Бейлыптейн) 374
Bennet (Беннет) 214, 362
De Beule (Де-Бель) 119, 120
Biehringer (Бирингер) 29
Bielecki (Белецкий) 220, 371
Blagden (Благден) 35
Blanksma (Бланксма) 142, 226, 271,
313, 34 7, 374
Blechta (Блехта) 21
Boedtker (Бедкер) 27, 54
Bonnet (Бонне) 273
Borsche (Борше) 209, 210
Borsum (Борзум) 29
Boters (Бетерс) 19
Bradner (Браднер) 26
441
Bradt (Бредт) 36
Brady (Бреди) 34, 180
Brochet (Броше) 381
Broders (Бродер) 24
Brosche (Броше) 181
Bruck (Брук) 292
Burkhard (Буркхард) 271, 272
Brunel (Брюнель) 353
Calhane (Келген) 36
Cambier (Камбье) 381 »
McCamish (Мак Кемиш) 204
Carter (Картер) 365, 369
Carswell (Карзуэл) 300
Chalenger (Челенджер) 319
Chao-Lun-Tseng (Чао-Лун-Тзенг) 43
Chateway (Четвей) 43
Chemnitius (Хемнициус) 386
Cherbulier (Шербулье) 66
Ciamician (Чамичан) 136
Claessen (Клессен) 314
Clark (Клерк) 362
McClary (Мак Клери) 47
Cobenzl (Кобенцль) 326
Cohn (Кон) 136
Courtot (Курто) 273
Crafts (Крафтс) 30
Crossby (Кросби) 228
David (Дэвид) 72
Davis (Дэвис) 21
Degering (Дигиринг) 47
Delepine (Делепин) 378, 388
Deseigne (Десинь) 354
Desvergnes (Девернь) 24, 26, 221,
299, 301, 305, 353, 358, 392
Dickinson (Дикинсон) 377
Dimroth (Димрот) 21
Drummond (Друммонд) 219
Duden (Дуден) 376
Ekl (Экль) 70
Enz (Энц) 243
Euwes (Эйвес) 275
Escales (Эскалес) 316
Fayollat (Файолла) 376
Feske (Феске) 210
Fichter (Фихтер) 35
Flurscheim (Флюршейм) 329, 330,
331, 332
Foerster (Ферстер) 181
Fonque (Фонк) 275
Forel (Форель) 52, 120,- 121
Frankland (Франкланд) 220, 319
Friedel (Фридель) 30
Friedlander (Фридлендер) 136, 244
Ganguli (Гангули) 135
Garner (Гарнер) 220
Gassmann (Гасман) 244
Geissmann (Гейсман) 227
Genequand (Генекан) 19
Gibson (Джибсон) 120
Giua (Джуа) 315
Gnehm (Гнем) 365
Godchaux (Годшо) 336
Goldstein (Гольдштейн) 286
Gonell (Гонелл) 377
Gornall (Гонелл) 190
Grassi (Грасси) 376, 384
Griess (Грисс) 379
Grignard (Гриньяр) 296
Groggins (Гроггинс) 87, 212, 213
Grunner (Груннер) 259
Gutenson (Гутенсон) 291, 298
Hale (Гель) 270, 396
Hall (Голл) 415
Hammick (Геммик) 282, 283
Hantzsch (Ганч) 35, 58, 81, 82, 83,
131, 132, 255, 256, 257, 258, 261
Harrow (Гарроу) 379
Hartung (Гартунг) 379, 395
Harvey (Гарви) 378
Hass (Гесс) 38, 40, 47, 48
Haubner (Гейбнер) 108
Haussermann (Гайзерман) 99, 140
Henning (Генинг) 381, 395, 419
Hepp (Гепп) 105
Herland (Герлянд) 27
Herz (Герц) 17, 393, 395
Hetherington (Геверингтон) 66, 67,
79, 80, 99
Hewetson (Геветсон) 180
Hiersbach (Гирсбах) 53
Hinshelwood (Гиншельвуд) 221
Hodge (Годж) 40, 47
Hofmann (Гофман) 375, 383
Hoitsema (Гойтзема) 81
HOland (Геланд) 388
Holdermann (Гольдерман) 19, 20
Holleman (Голлеман) 21, 50, 120,
142, 209, 215, 327
Holmes (Гольме) 332
Hopper (Гоппер) 266
Howell (Гоуэлл) 261
Hiibner (Гюбнер) 322
Hiickel (Хюккель) 57
Ingraham (Ингрегем) 330
Inouye (Инуи) 379
Ischidzu (Ишидзу) 379
Izzo (Иццо) 390
Jones (Джонс) 335, 336
Joule (Джоуль) 214
Jovinet (Жовине) 126
Kahn (Кан) 49
Karlson (Карлсон) 49, 55
Kaufmann (Кауфман) 256
Kast (Каст) 129, 266, 365
Kenner (Кеннер) 45
Kessler (Кесслер) 53, 59, 60, 62, 95,
99, 274
McKie (Мак Кай) 44, 127
King (Кинг) 280
Kirchhoff (Кирхгоф) 42
442
Kirk (Кэрк) 36
Kissel (Киссель) 131, 132
Klein (Клейн) 180
Klemenc (Клеменс) 70
Kling (Клинг) 296
Knowles (Ноульс) 341, 342
Koester (Кестер) 138
KOhler (Келер) 126, 221, 298, 353
Kohn (Кон) 377
Kopetschni (Конечный) 34
Kopisarow (Кописаров) 134, 135
Krantz (Краутц) 135
Krishnamurti (Кришнамурти) 35
Kubierschky (Куберский) .161
Lacroix (Лакруа) 275
Landsteiner (Ландштейнер) 333
Langhans (Лангганс) 365
Laubenheimer (Лаубенгеймер) 177
Lauer (Лауэер) 65, 84
Lellman (Лельман) 227
Lennep (Леннеп) 347, 374
Lippincott (Липинкот) 38
Lob (Леб) 388
Lobry de Bruyn (Лобри де-Брюин)
215, 261, 313
Loriette (Лориет) 227, 228, 298, 299
Ldsekann (Ловеканн) 375
Lunge (Лунre) 72
Lyons (Лайонс) 294
Mark (Марк) 41, 377
Marshall (Маршаль) 365, 366
Martin (Мартин) 116, 203
Martinsen (Мартинсен) 51, 63, 64,
65, 66, 69, 213
Marotta (Маротта) 377
Marqueyrol (Маркероль) 126, 227,
228, 276, 278
Masson (Массон) 66, 67, 79, 80, 99
Mayer Fr. (Майер) 379
Meyer L. (Мейер) 59
Meyer (Мейер) 220, 276
Mecir (Месир) 190
Meimberg (Меймберг) 34
Meisenheimer (Мейзенгеймер) 132,
133, 230
Menke (Менке) 28
Mertens (Мертенс) 337, 340
Michael (Михаэль) 49
Michaelis (Михаэлис) 336
Misslin (Мисслин) 328
Moelwyn-Hughes (Мелвин-Хьюз) 221
Moschatos (Мошатос) 381, 382
Motta (Мотта) 376, 384
Munroe (Мунрое) 261
Muraour (Мюраур) 178, 190, 235
Naoum (Наум) 390
Natanson (Натансон) 29
Neumann (Нейман) 161
Nickoils (Никколс) 319
Nietzki (Ницки) 271, 272
Nobel (Нобель) 411, 412, 413'
Noelting (Нельтинг) 52,
227
Nolan (Нолан) 353 , г
Norberg-Schulz (Норберг-Шульц)184j
Obermiller (Обермиллер) 276
Oda (Ода) 65, 84
Oddo (Оддо) 51
Olivier (Оливье) 54
Olsen (Олсен) 286.
Orton (Ортон) 340
Paar (Паар) 20
Pascal (Паскаль) 245
Patart (Патар) 7 7
Patek (Патек) 21
Patterson (Пэтерсон) 48, 225
Pawlik (Павлик) 137, 138
Pfeiffer (Пфейффер) 56
Pfister (Пфистер) 243
Pink (Пинк) 31
Pick (Пик) 52
Pictet (Пикте) 19, 254
Pluss (Плюс) 35
Pollitt (Подлит) 219
Prentiss (Прентисс) 129
Prudhomme (Прюдом) 34
Pummerer (Пуммерер) 375
Radkliffe (Редклиф) 219
Raymont (Реймонт) 377
Reese (Риз) 415
Renouf (Рену) 228
Rinkenbach (Ринкенбах) 129
Rinkes (Ринкес) 35
Robinson (Робинзон) 190
Rombourgh (Ромбург) 345, 347
Rositer (Розитер) 52
Sachs (Закс) 136
Salathe (Селэт) 204
Sandmeyer (Зандмеер) 35
Schaarschmidt (Шааршмидт) 30, 31,
32 58
Scharff (Шарфф) 376
Schepers (Шеперс) 345
Schmidt (Шмидт) 19, 41
Schmidt J. (Шмидт) 300
Schmitt (Шмит) 127
Schuller (Шюллер) 136
Schultz (Шульц) 135, 229
Schwarte (Шварте) 194
Silber (Зильбер) 136
Simon (Саймон) 329, 332
Smith (Смит) 294
Sommerville (Зоммервиль) 225
Spindler (Шпиндлер) 59
Steinkopf (Штейнкопф) 42
Stettbacher (Штетбахер) 116, 193»
203, 223, 355, 391, 398
Stewart (Стюарт) 387
Taylor (Тейлор) 129
Terwogt (Тервогт) 347
443-
Thiele (Тиле) 50
Timmermans (Тиммерманс) 116, 119,
120, 203
Tollens (Толленс) 318, 378, 381, 382
Travagli (Травагли) 398 i
Trendelenburg (Тренделенбург) 379
Tryllat (Трилла) 376
Tryller (Триллер) 35
Turek (Турен) 135
Ullman (Ульман) 213
Ullman (Ульман) 220, 371
Utermann (Утерман) 322
Utz (Утц) 378
Vanderbilt (Вандербилт) 40, 47
Van Duyn (Ван Дуин) 331, 341, 347,
355, 359, 372, 373, 374
Varma (Варма) 35
Veibel (Вейбель) 70
Vernazzo (Вернаццо) 389
Veroia (Верола) 138
Vielle (Вьелль) 72
Wang (Ванг) 43
Warral (Ворел) 21
Weiler-ter-Meer (Вейлер-тер-Мер) 160
Wenzelberg (Венцельберг) 105, 106,
143
Werner (Вернер) 82
Wieland (Виланд) 29, 30, 49, 50,
Will ’(Билль) 126, 248
Williams (Вильямс) 34
Willson (Вильсон) 335
Wilson (Вильсон) 36
Witt (Витт) 34, 318, 322, 327
Witte (Витте) 318, 322, 327
Wizinger (Вицйнгер) 55, 57
Wolffenstein (Вольфенштейн) 19, 20
Wohler (Велер) 105, 105, 143
Wyler (Вилер) 215
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азотная кислота, свойства 423—425
— — токсическое действие 439
Аммонит № 3—ТМ 177
Аммиачно-селитренные взрывчатые
вещества, исторические сведения
412
Ангидроформальдегиданилин 318
Анилин, свойства 317
— нитрование азотной кислотой
322
Анилин-т-сульфокислота 318
Анилин-р-сульфокислота 318
Аппарат для нитрования (см. нитра-
тор)
Аппарат для промывки тротила, пе-
риодический 163
----------непрерывный англий-
ский 164—165
Аппарат для растворения тротила 169
— кристаллизатор 170
Ароматические производные моче-
вины 319
Ацильные производные ароматиче-
ских аминов 319
Оензол, влияние концентрации азот-
• ной кислоты на скорость нитра-
ции 19
— свойства 203
— технические условия 204
— токсическое действие 204
Бензойная кислота, нитрование в
присутствии ртути 20
----- скорость нитрации 63, 67
Бензолсульфокислота, скорость ре-
акции нитрации 63, 67
Бензолтриозонид 203
Бикфордов шнур, исторические све-
дения 410
Болтушка 97, 152, 153, 155, 156, 157
Бутан, нитрование 46
Галоидирование, схема реакции 56
— механизм реакции 399
Гексаметилентетрамин, количествен-
ное определение 382
— меры предосторожности при
работе 382
— применение 382
— производство 383—387
— разные способы получения
387—388
-— растворимость в аммиачных
растворах 386
— свойства 378—382
— строение 375—378
— течение реакции нитрации 392
Гексанитродибензил 133
Гексанитролифенил 371
Гексанитродифениламин (см. гексил)
Гексанитродифениламиноэтилнитрат
364
Гексанитродифенилоксид 373
Гексанитродифенилсульфид 373
Гексанитродифенилсульфон 374
Гексанитроэтан 42
Гексанитродифенилэтилендинитро-
амин 362
Гексил, исторические сведения 365
— получение 367—370
— применение 367
— свойства 365, 366, 367
Гексоген, исторические сведения 419
— опыты получения 396—398
— применение 110, 391
— производство 395, 398—401
— свойства 388—390
Гидроксиламин солянокислый, обра-
зование при гидролизе первичных
нитропарафинов 38
Гремучая ртуть 410
Гремучий студень 413
Громобой 415
Гурдинамит 412
Денитрация отработанной кислоты
435, 436
Детолуация 155, 156, 157
Диметилаиилин, нейтрализация 348,
349
445
Диметиланилин, нитрование азотной
кислотой 340
— нитрование сернокислого 349,
350, 351, 352
— получение 337
— продукты нитрации 339
— свойства ЗЗб
— технические условия 338
Динитробензол, обоснование техни-
ческих требований 217
— обоснование технических усло-
вий 218
— получение очищенного 215, 216
— применение 205, 206, 210, 211
— примесь в тротиле 127
— производство 213—217
— свойства 204, 205
— скорость реакции нитрации 67
— состав сырого 214, 215
— техника безопасности при ра-
боте 205
— технические требования 216
— технические условия 217
Динитродиметиланилин, изомеры 340
2,2'-Динитродифенил 371
Динитродифениламин 368
Динитроксилол, изомеры 226, 227,
228
4,6-Динитрометаксилол, скорость
реакции нитрации 63, 67
Динитрометил-т-фенилендиамин 347
Динитромонометиланилин, изомеры
339
1,8-Динитронафталин 243, 244
1,5-ДинитрОнафталин 243, 244
Динитронафталин технический
— действие серной кислоты 248
— продукты нитрации 245
— производство 249, 250
— применение 244, 245
— разделение изомеров 244
— свойства 244
— технические условия 250
Динитронафтол 243
Динитрооксибензойная кислота 127
2,4-Динитротолуол 200, 201
Динитротолуол 123, 127
Динитротолуолсульфокислоты 180
2,4-Динитрофенилэтаноламин 863
2.4-Динитрофенол 259
2,6-Динитрофенол 260
3,5-Динитрофенол 261
Динитрофенол, нитрование 309
— получение 25, 297, 298
— скорость реакции нитрации 63
— техника безопасности 308
— технические условия 308
— токсические свойства 260
Динитрофенолсульфокислота, нитро-
вание 279, 280, 281
*
Дитетрил (см. гексанитродифенил
этилендинитро амин)
Динитрохлорбензол, применение 301
— примеси в техническом 301, 302
— производство в одну фазу 306
— свойства 301
— техника безопасности 305, 306
р-Динитробензол 304, 305
Железохромистые сплавы 426
Капсюль-воспламенитель 410, 412
Капсюль-детонатор' 412
Кислотооборот горячий 96
Кислоты, строение по Ганчу 81
— хранение и транспортировка
438, 439
— техника безопасности 439
Клетчатка, зависимость между со-
ставом нитрационной смеси и
степенью нитрации 75
Крезол 315
Крезолит (см. тринитрокрезол)
Ксилил, исторические сведения 418
— обоснование технических тре-
бований 238
— применение 231
— производство 231—238
— технические условия 238
Ксилоидины 410
Ксилол каменноугольный 224, 225
— пирогенетический 225
т-Ксилол, нитрование в присутствии
катализатора 25
— получение чистого 225
— свойства 223, 224
р’-Ксилол 223, 224
о-Ксилол 223, 224
Легковоспламеняющиеся жидкости,
допустимые нормы паров в произ-
водственных помещениях 407
-------классификация жидкостей
402
----- классификация хранилищ
403
----- перевозка 406
-----хранение в атмосфере инерт-
ного газа 405
---— хранилища 404
Метан, нитрование 46
Метанитротетрил (см. тетранитро-
фенилметилнитроамин)
Метилгидразин, образование при вос-
становлении нитроаминов 17
446
2-Мети л-1-нитропропан 37
2-Метил-2-нитропропан 37, 41, 45,
Мешка кислот 426—429
Механизм альдолизации 55
Механизм реакции нитрации 47, 49,
51
Монометиланилин 335, 340
Мононитроанилины 319, 320
Мононитробензол (нитробензол) 204,
212, 213
Мононитротолуол, выделение т-изо-
мера 194
— дестилляция 196
— применение изомеров 195
ч> — производство для дестилляции
196
— состав продуктов нитрации 123
— техника безопасности при де-
стилляции 198
Мононитрокс илол, изомеры 226, 227,
228
Мононитронафталин 110, 249
Моно нитрохлорбензол 300, 311
Нафтазарин 248, 249
Нафталин, зависимость между соста-
вом смеси и степенью нитрации
77
— побочные реакции при нитра-
ции 247
— продукты нитрации 243, 244
— технические условия 243
Непредельные соединения, примесь
в толуоле 117, 118
Нитратор американского типа 147
— английского типа 162
— Брука 292
— Вейлер-тер-Мера 161
— Куберского 161
— • Неймана, или тутольтурбо 161
— со змеевиком 147
Нитроанилид 333
Нитроанилины (изомеры) 322, 324,
325, 326
Нитробензол (мононитробензол), при-
менение 110
— скорость реакции нитрации 110
zn-Нитробензолсульфокислота, ско-
рость нитрации 67
Нитробутан 37
Нитроглицерин, исторические сведе-
ния 410, 411, 412
Нитродиазотолуолсульфокислота
185, 186
Нитродиметиланилин 339, 340
Нитроклетчатка, см. пироксилин
Нитролиз 281, 282, 287
Нитроловые кислоты 39
Нитромонометиланилин 338, 339
Нитрометан 37, 41, 42
Нитронафталин (изомеры) 243
Нитроолефины 38
Нитропарафины 37, 40
Нитропропан 37
Нитросоединения, классификация
способов производства 112, 113
— механизм образования вторич-
ных 39
— механизм образования первич-
ных 39
— механизм образования третич-
ных 39
— применение 110, 111
• — прогноз 109
— пути отравления 108
— • строение 15
— теория токсического действия
— ароматических 108, 109
— терапия 109
— токсические свойства 107, 108
— физические свойства 101, 102
— химические свойства 102, 103
— химическая стойкость 16
— частичное восстановление 325
— чувствительность к механиче-
ским воздействиям 16, 105,
106, 107
Нитросольвент-нафта 242
тп-Нитротолуол, применение 196
— продукты нитрации 122, 123
— свойства 120 -»
о-Нитротолуол, применение 195
— продукты нитрации 122
— • свойства 119, 120
— технические условия 199
р-Нитротолуол, получение вымора-
живанием 199
— применение 195
— продукты нитрации 122
— технические условия 200
Нитрофенолы, образование при ни-
тровании в присутствии катали-
заторов 20
— окраска солей 257
— причина окраски 258, 259
— строение окрашенных 254
тп-Нитрофенол 254
о-Нитрофенол 254
р-НитрофенОл 254
о-Нитрохлорбензол 311, 312
р-Нитрохлорбензол 311
Нитроэтан 37
”кислы азота, токсическое действие
439
Олеум 421
Отработанная кислота, активный со-
став 84
447
Парафины, примесь в толуоле 118
Пентан, нитрование 46
Пентанитроанилин 332
Пентанитрофенилметилнитроамин
346
Пентанитрофенол 272
Пентил (см. тетранитрофенилметил-
нитроамин|
Пентрил (см. тринитрофенилнитро-
аминоэтилнитрат)
Пикрамид 327
Пикраминовая кислота 310
Пикраты аммония 270
Пикраты, возможность образования
в производственных условиях 270,
271
— исторические сведения 414
— опасности, обусловленные при-
сутствием 268, 269
— получение 269, 270
— свойства 265—269
Пикрил хлористый, см. тринитрр-
хлорбензол
Пикриновая кислота, американский
способ получения 286
-----выход 295, 309
----- качественное определение
296
-----получение по английскому
способу 285, 286
— ’— получение по патенту Гутен-
сона 291
-----получение по способу Брука
292
-----получение по французскому
способу 284, 285
----применение 110, 265
------ производство из бензола че-
рез динитрохлорбензол 299
-----промывка 292—294
-----просеивание и упаковка 295
----- процесс нитрования фенола
287, 289, 290, 291
— — расходные коэфициенты при
производстве через хлорбен-
зол ЗОЭ
----- свойства 261—264
-----скорость окисления серно-
азотной смесью 282
-----сушка 295
— — технические требования 296
-----физиологическое действие
264
Пироксилин, исторические сведения
410, 411, 413
Полинитроанилины, диазотирование
328
Порох, исторические сведения 408,
413
Пропан, нитрование 46
Псевдокислоты 39
тп-Пурпуровая кислота 260
Расчет кислот 429—435
Серная кислота, свойства 421—423
— обращение 423
Сольвениты 242
Сольвент-нафта 239—241
Спирт, маточный 171, 172, 173
— промывной 172
— ректификация отработанного
175
Сульфация, влияние катализатора на
реакцию 19
— влияние различных факторов
на реакцию 276, 277
— влияние температуры на место,
занимаемое сульфогруппой при
274, 275
— механизм реакции 274
— обратимость реакции 274
— побочные процессы при реак-
ции 275
Сульфирование 272, 273
Сульфит натрия, механизм взаимо-
действия с нитросоединениями 180
— приготовление раствора 180
— растворимость в воде 181
— технические условия 181
Сульфокислота фенола, нитрование
278, 279
----- свойства 277
Супердетолуация 155, 157
•Сушилка пневматическая 174
> емпература вспышки паров летучих
веществ 402
Тетранитроанизол 313, 314
Тетранитроанилин 329—331
Тетранвдрометан, взаимодействие с
сульфитом натрия 179
— получение 43
— примесь в тротиле 127
— применение 110
— свойства 41
Тетранитробензол 208, 209
Тетранитродифенил 372
Тетранитродифениламин 368, 369
Тетранитродифенилэтаноламин 363
Тетранитронафталин 26, 247, 248,
251
Тетранитротолуол 142, 143
Тетранитрофенилметилнитроамин
345
Тетранитрофенол 271, 272
448
Тетрил, аналоги 362
— взаимодействие с алкоголятами
133, 135
— виды брака и его исправление
359
— исторические сведения 418» 419
— качество бензола для перекри-
сталлизации 359
— качество промытого 356
— перекристаллизация из ацетона
357, 358
'— перекристаллизация из бензола
358, 359
— перекристаллизация из дихлор-
этана 361
— получение в две фазы 353
— получение из динитрохлорбен-
зола и метиламина 353
— получение по способу Брюнеля
353
— применение 345, 356, 357, 361
— промывка в бензольном рас-
творе 355, 356
— промывка кристаллического
продукта 354, 355
— растворитель для перекристал-
лизации 356—357
— свойства 340—345
— сушка перекристаллизованного
360
— сушка промытого 356
— схема производства 348
— технические условия 360
Тиофен, свойства 117
Толуол 77, 116, 119
Толуол-бензин 194
Тринитроанизол 313
2,4,6-Тринитроанилин, см. пикрамид
Тринитробензойная кислота 127
Тринитробензол, взаимодействие с
алкоголятами 133, 135
• — взаимодействие с сульфитом
натрия 179
— получение 219—222
— применение 210, 211
— примесь в тротиле 127
— свойства 206, 207
Тринитроксилол (см. ксилил), взаи-
модействие с алкоголятами 133
— взаимодействие с сульфитом на-
трия 179
— изомеры 226, 227, 228
— окисление хромовой смесью 222
— примесь в тротиле 127
— свойства 230
Тринитро крезол 315
Тринитронафталин 245, 246, 250
Тринитро-тп-оксибензойная кислота,
образование при нитровании в
присутствии ртути 20
Тринитросольвент-нафта 241
Тринитротолуол (тротил), изомеры
124—127
— исторические сведения 416—418
— окисление азотной кислотой 221
— окисление хромовой смесью 221
— перекристаллизация из бензола
193
— перекристаллизация из о-нитро-
толуола 193
— примеси в неочищенном 127
— промывка буферными раство-
рами 190
— промывка гипохлоритом натрия
190
промывка спиртом 192
— промывка четыреххлористым
углеродом 192
— свойства 129—139
— фуговка с примесью фенола 194
Тринитротолуолы несимметричные,
взаимодействие с сульфитом нат-
рия 179
Тринитротолилметилнитроамин 190
Тринитрофенетол 313
Тринитрофенил-т-диметилдинитро-
амин 347
Тринитрофенилметилнитроамин, см.
тетрил
Тринитрофенилнитроамин 334
Тринитрофенилнитроаминоэтилни-
трат 362, 363
Тринитрохлорбензол 220, 372, 373
Тринитрохлорфенол, образование
при нитровании хлорбензола в
присутствии ртути 21
Тринитроциклотриметилентриамин,
см. гексоген
Тротил (тринитротолуол), кри-
сталлизация из отработанной кис-
лоты 191
— перекристаллизация из спирта
168—175
— применение 140—142
— производство 144—194
— промывка 163—166
— состав сырого 124—128
— сульфитная очистка 177—188
— технические условия на некри-
сталлизованный 167
— технические требования к пере-
кристаллизованному 175
Тротиловое масло 176, 177
У ротропин, см. гексаметилентетр а
мин
Фактор нитрующей активности 84,
85, 86, 87
Химия—13—29
449
Фенол 252, 253
Феиилнитроамин, см. нитроанилид
Ферросилициевые сплавы 425, 426
Форманилид 323, 324
Хлорнитробензолы, скорость реакции
нитрации 63, 67
Хлорноватокалиевая соль, историче-
ские сведения 410
Хромоникелевые сплавы 426
Хлоратные взрывчатые вещества,
исторические сведения 419, 420
Хлорбензол 21, 300, 302, 305
Этан, нитрование 46
Этилбензол 223, 229
Эфиры спиртов, азотные 15, 16, 17
Редактор Д. А. Катренко
Техн, редактор А. Н. Савари
Сдано в набор 31/XII 1939 г.
Подписано к печ. 26/VII 1940 г.
Автор, дог. № 607- Инд. 5-2.
Тираж 3000. Кол. печ. лист.
2874. Учетно-авт. лист. 32,11.
Форм. бум. 60x92/ls. А25118.
Зака) №13.
Цена 12 руб., переплет 2 руб.
Типография Оборонги а
Киев, Крещатик, 42