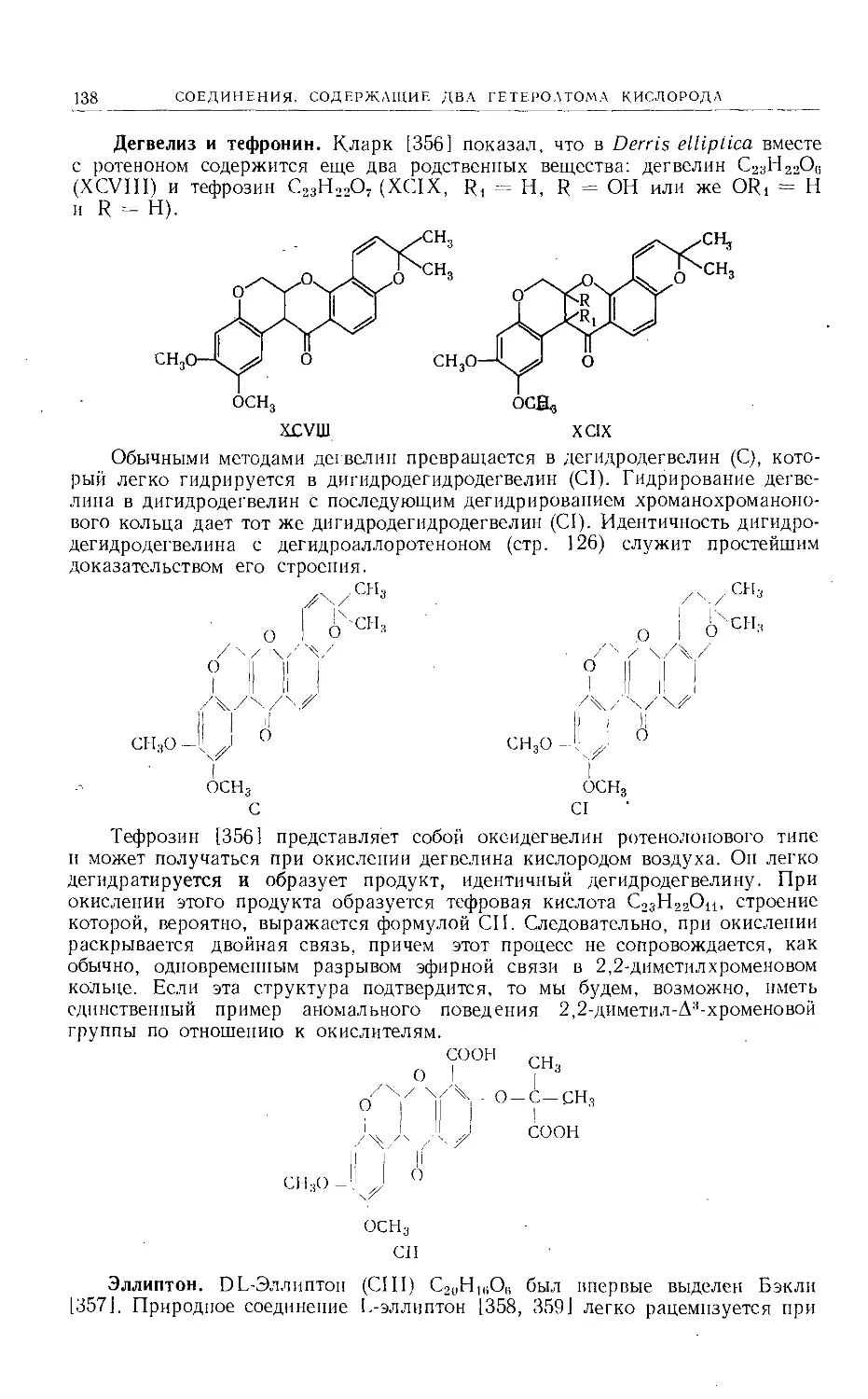
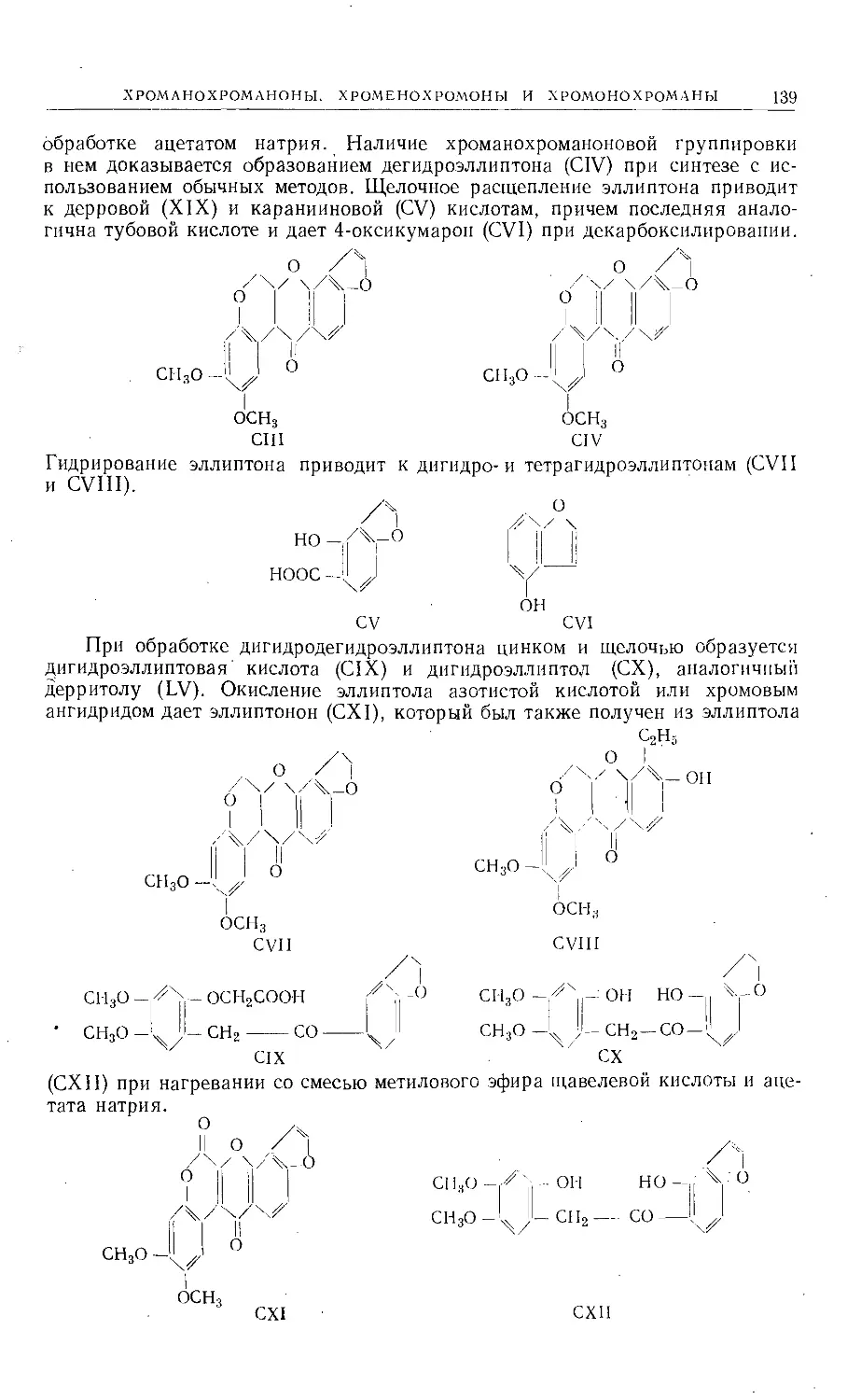
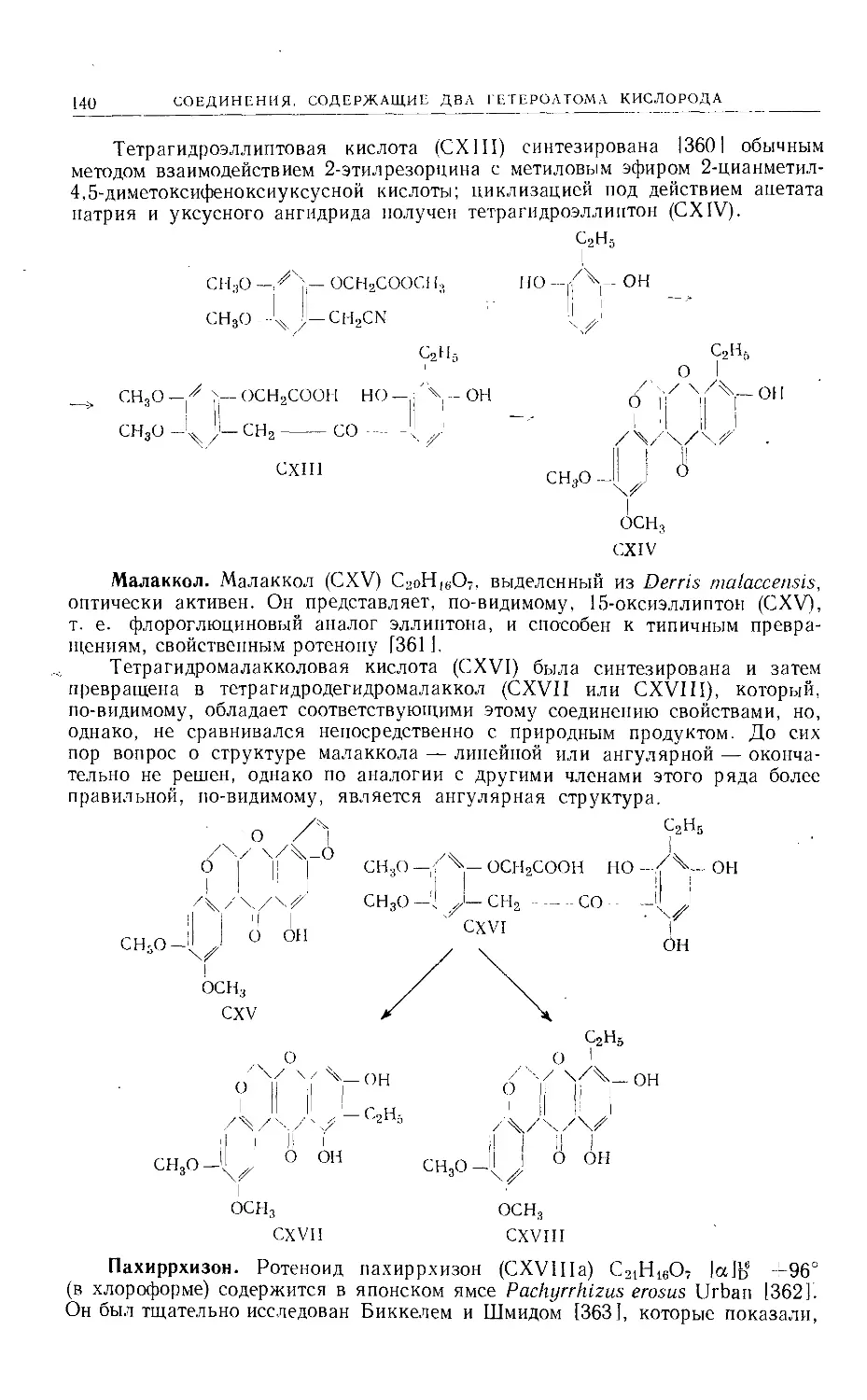
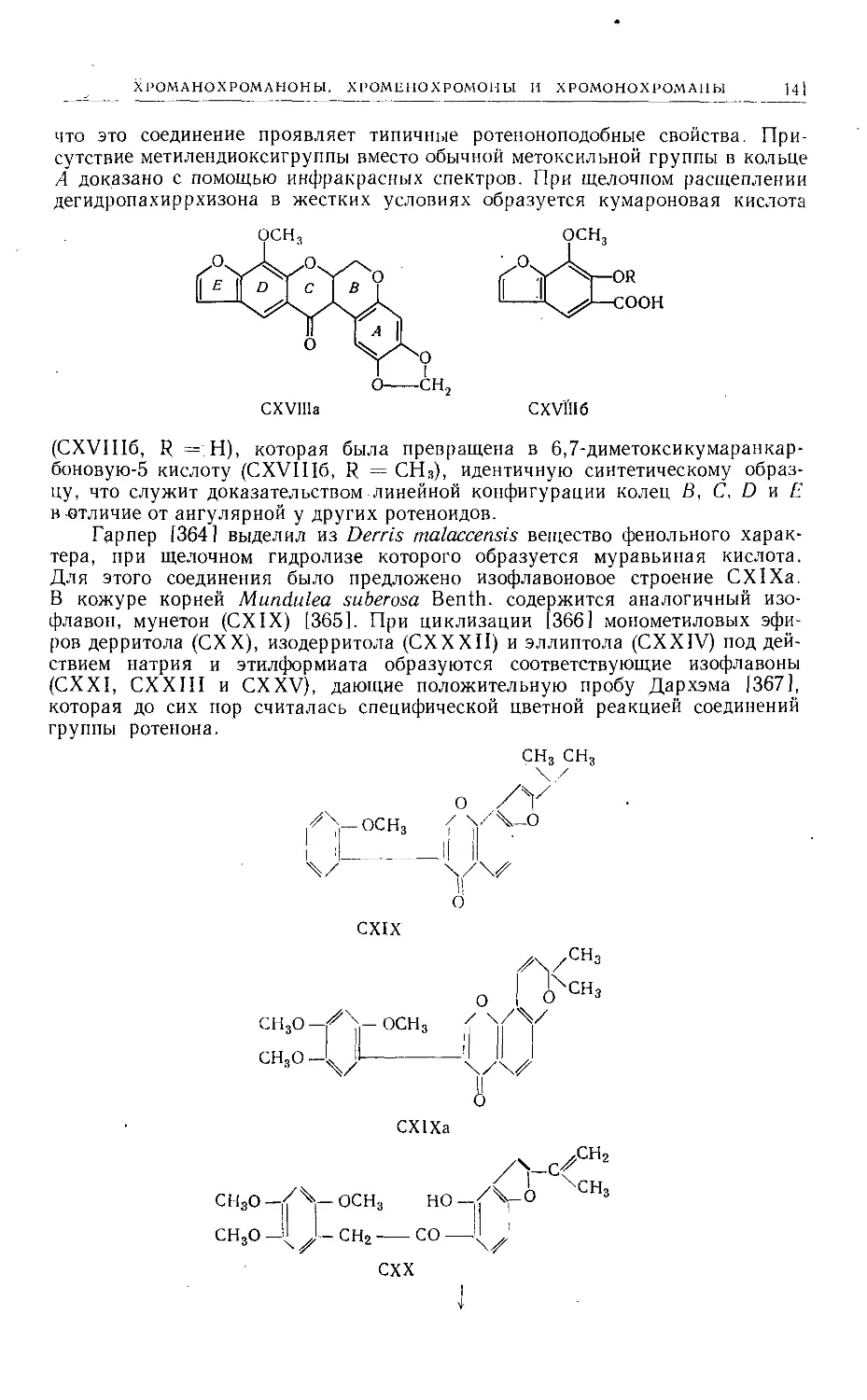
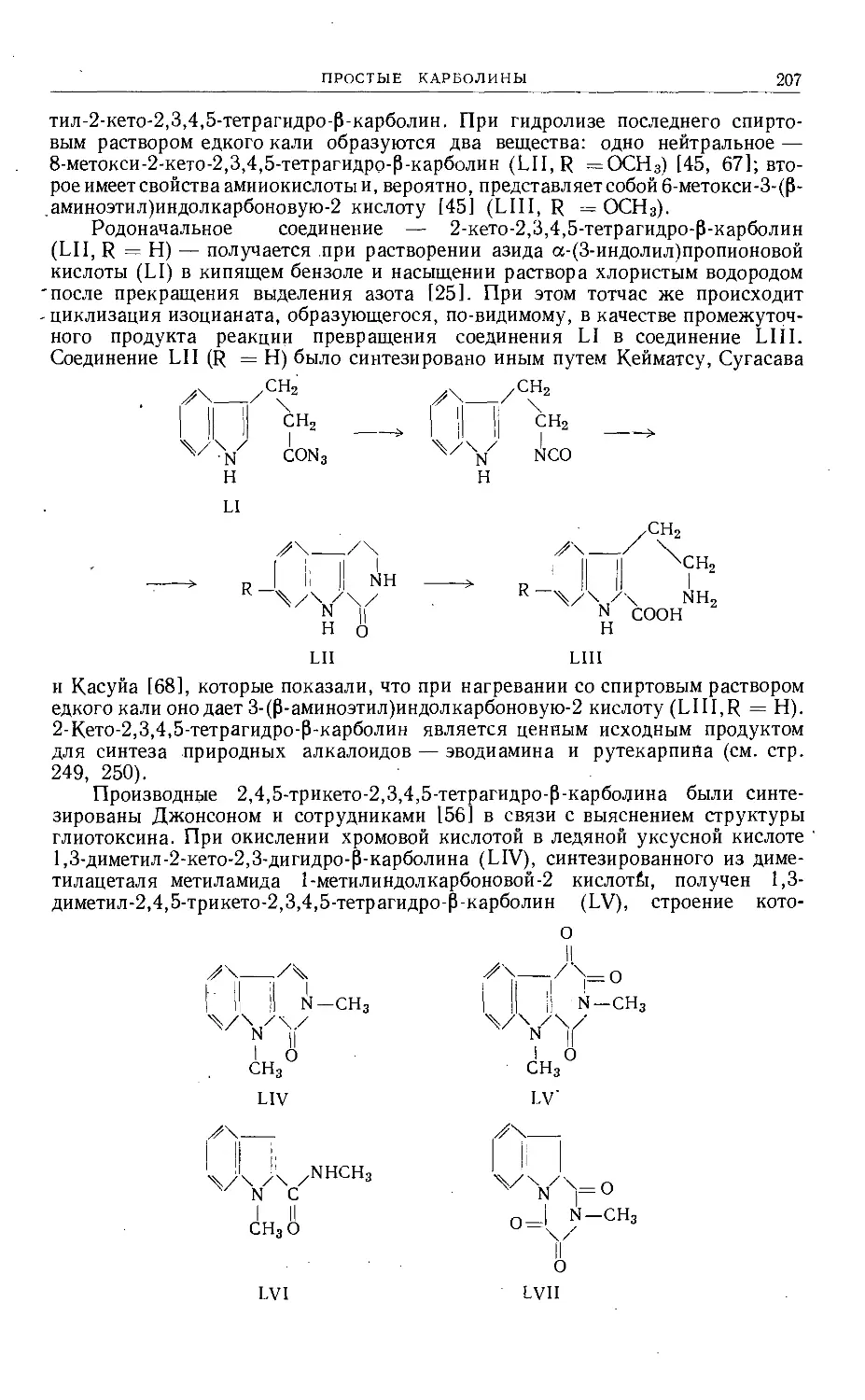
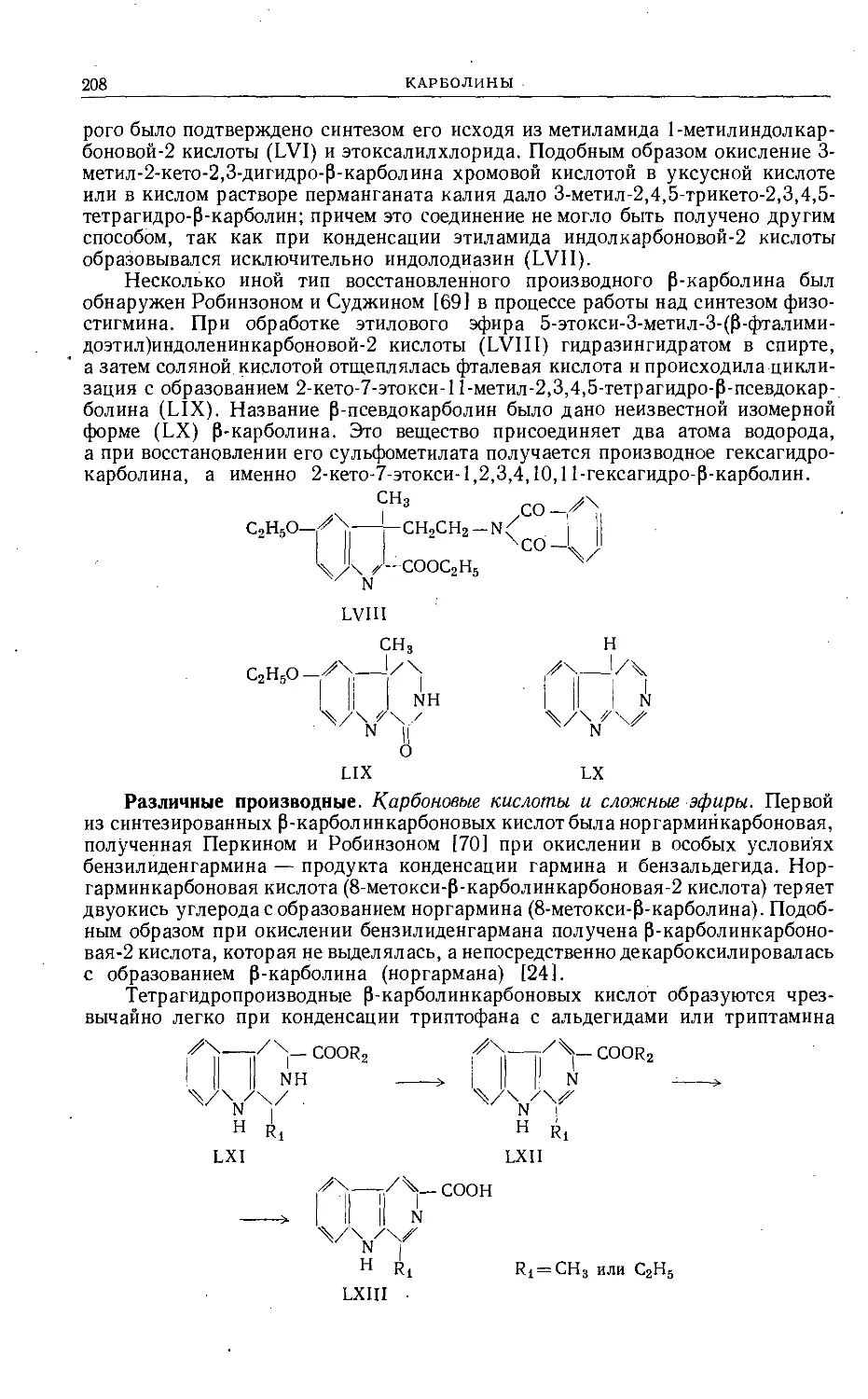
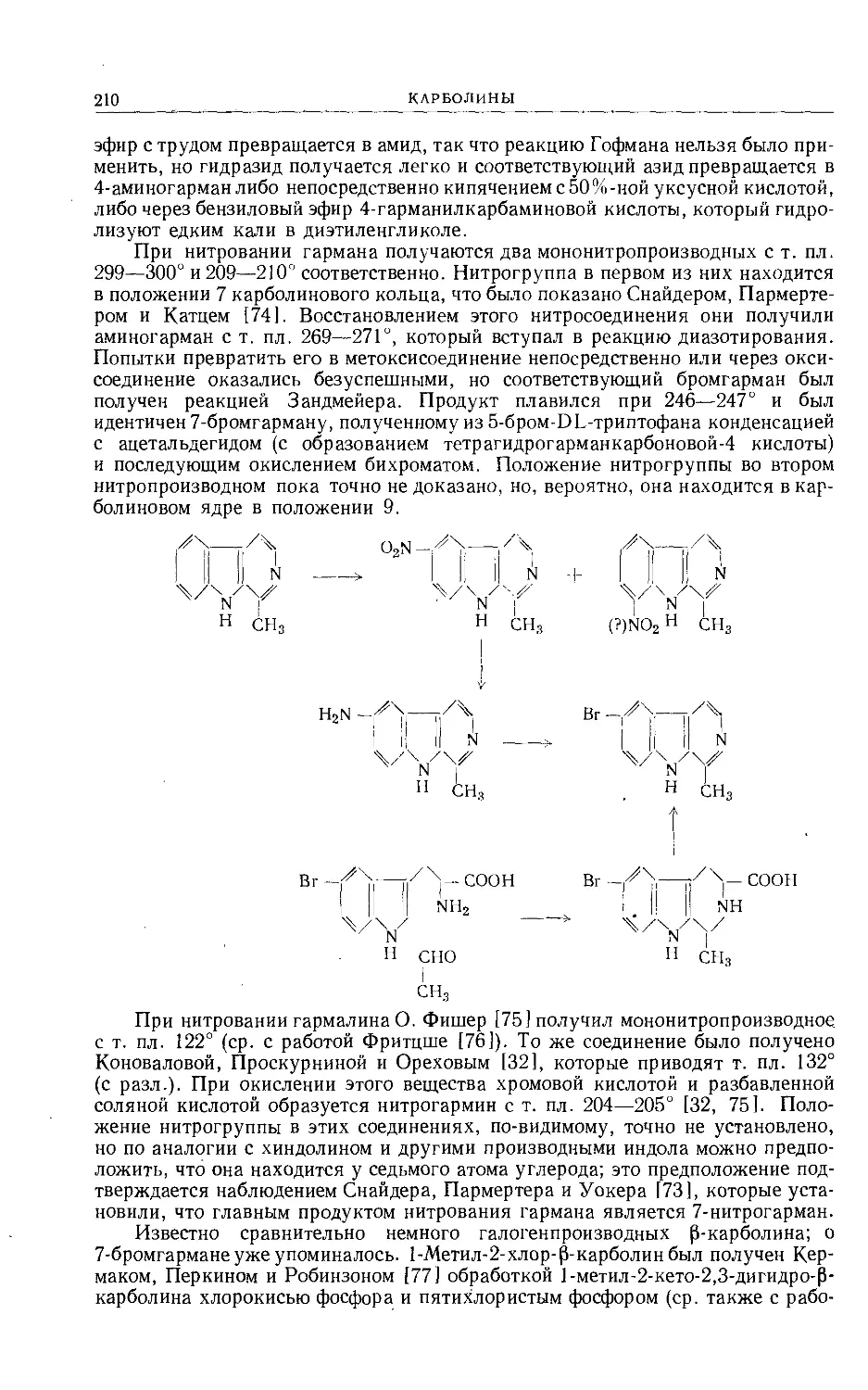
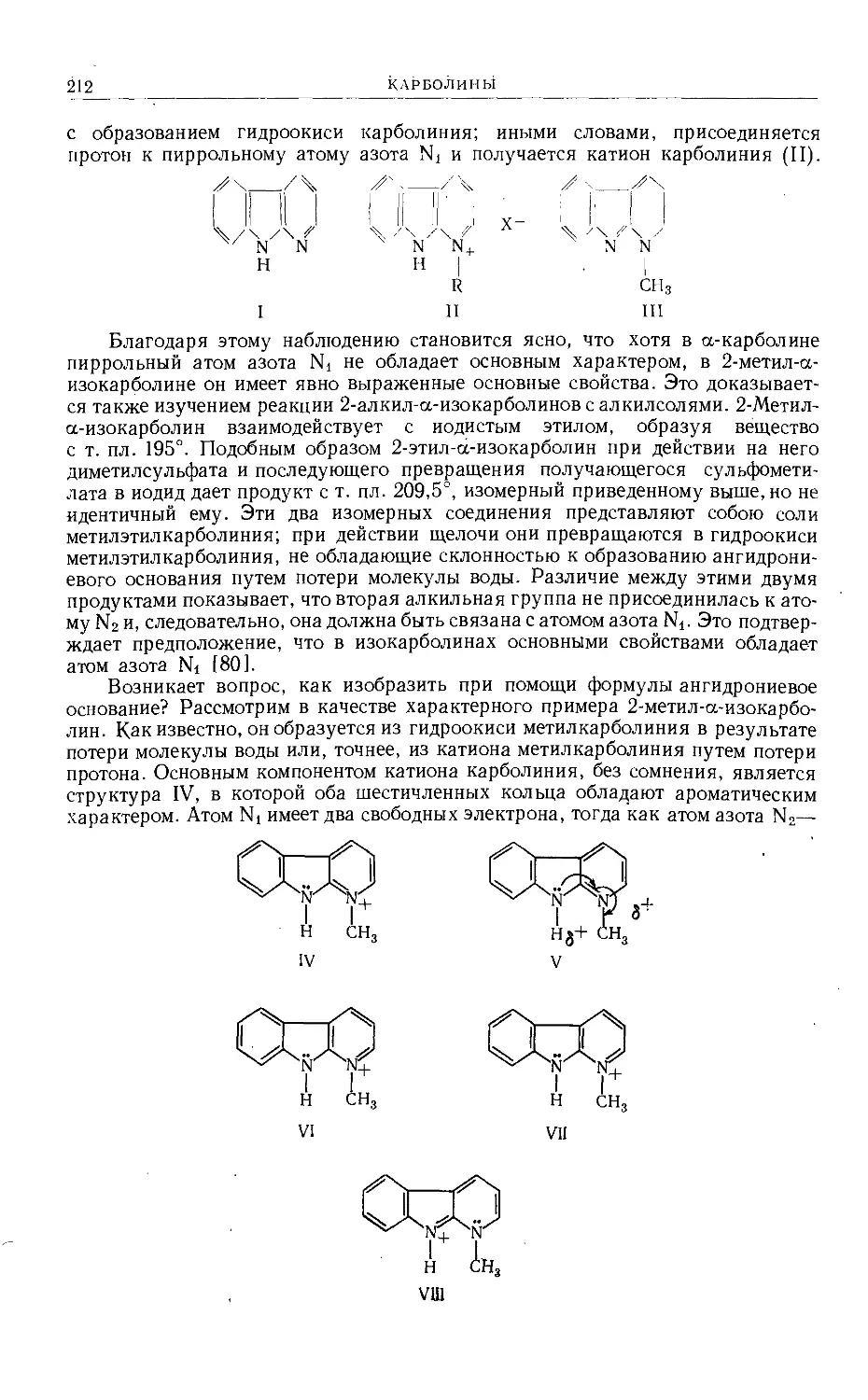
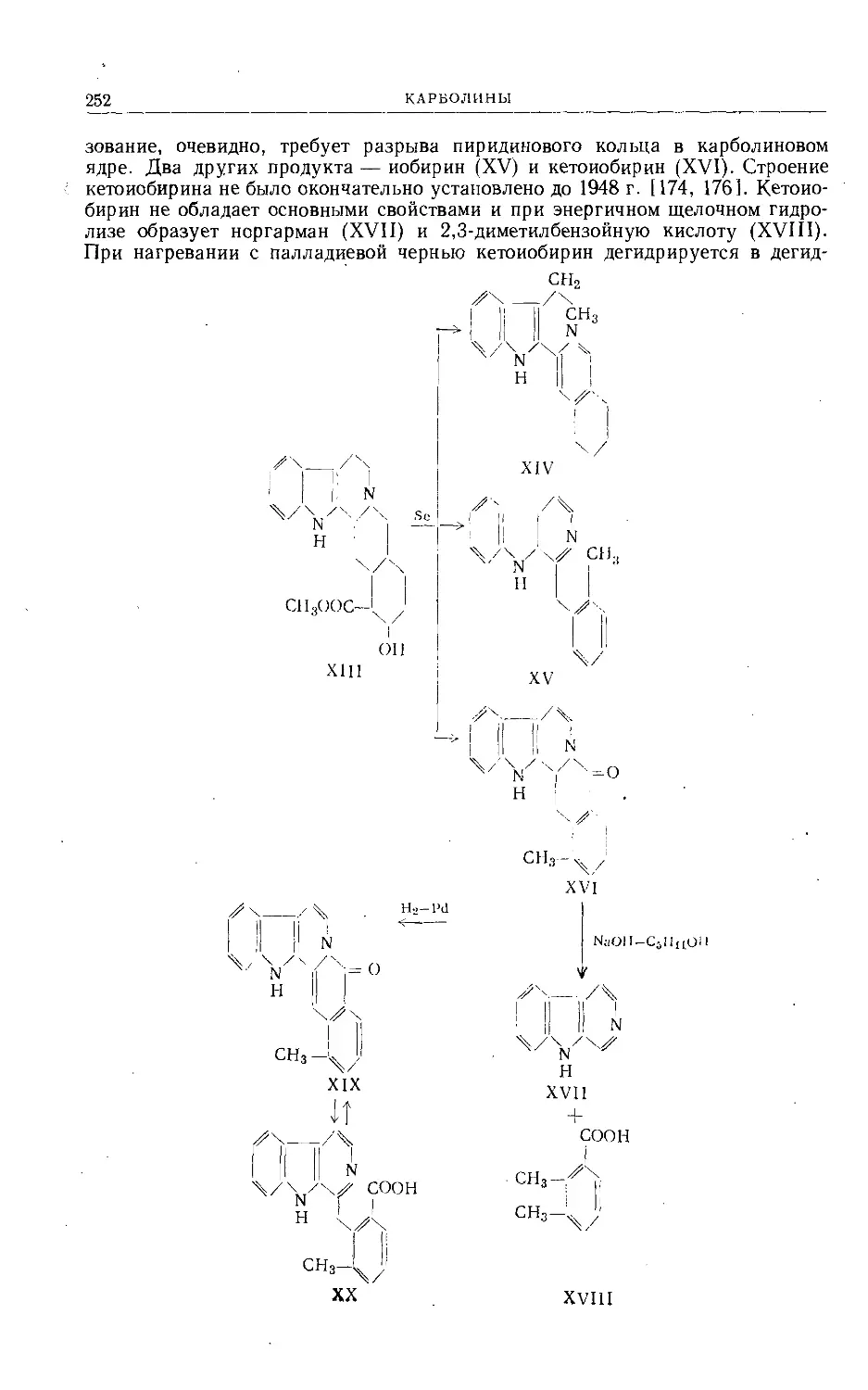
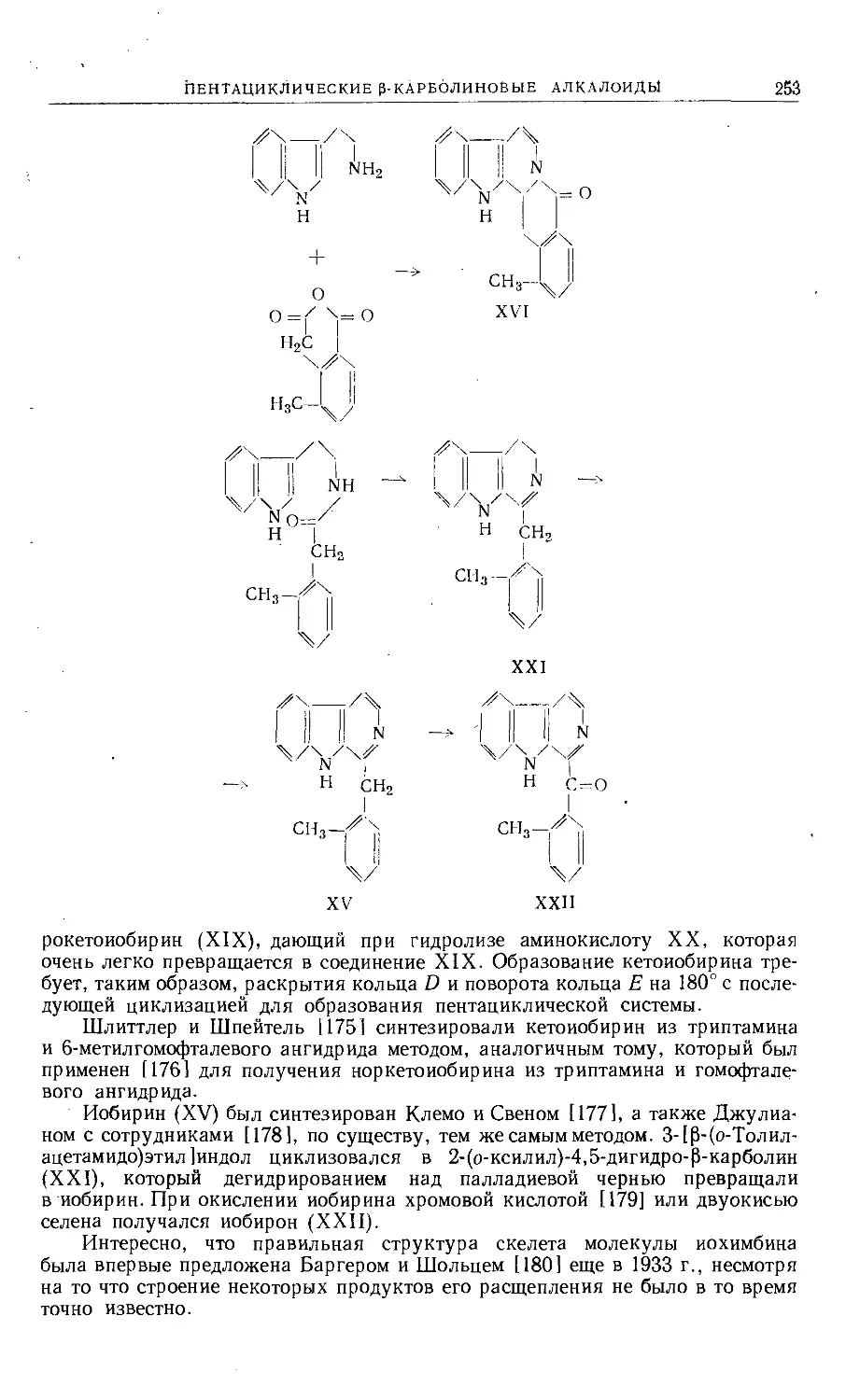
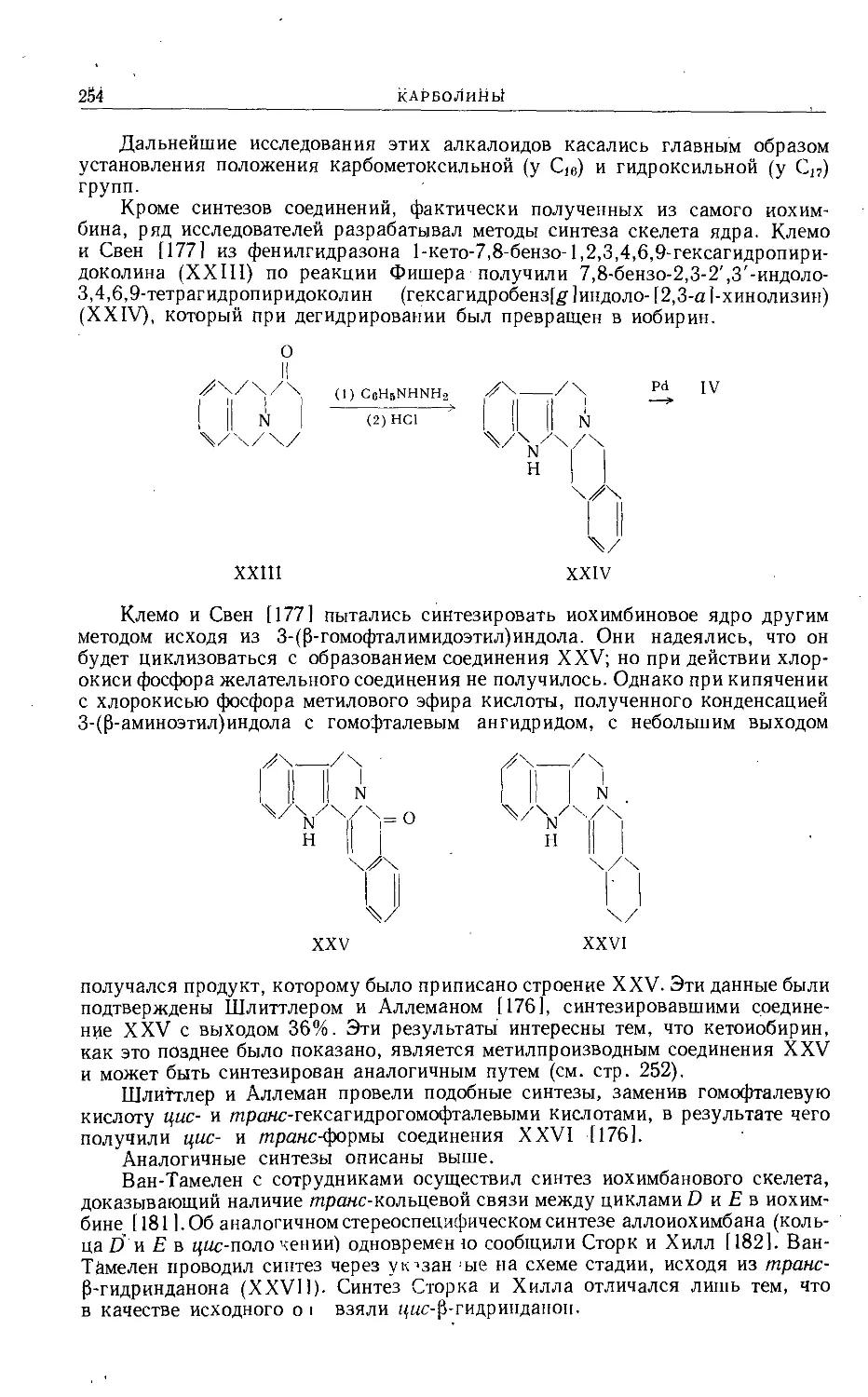
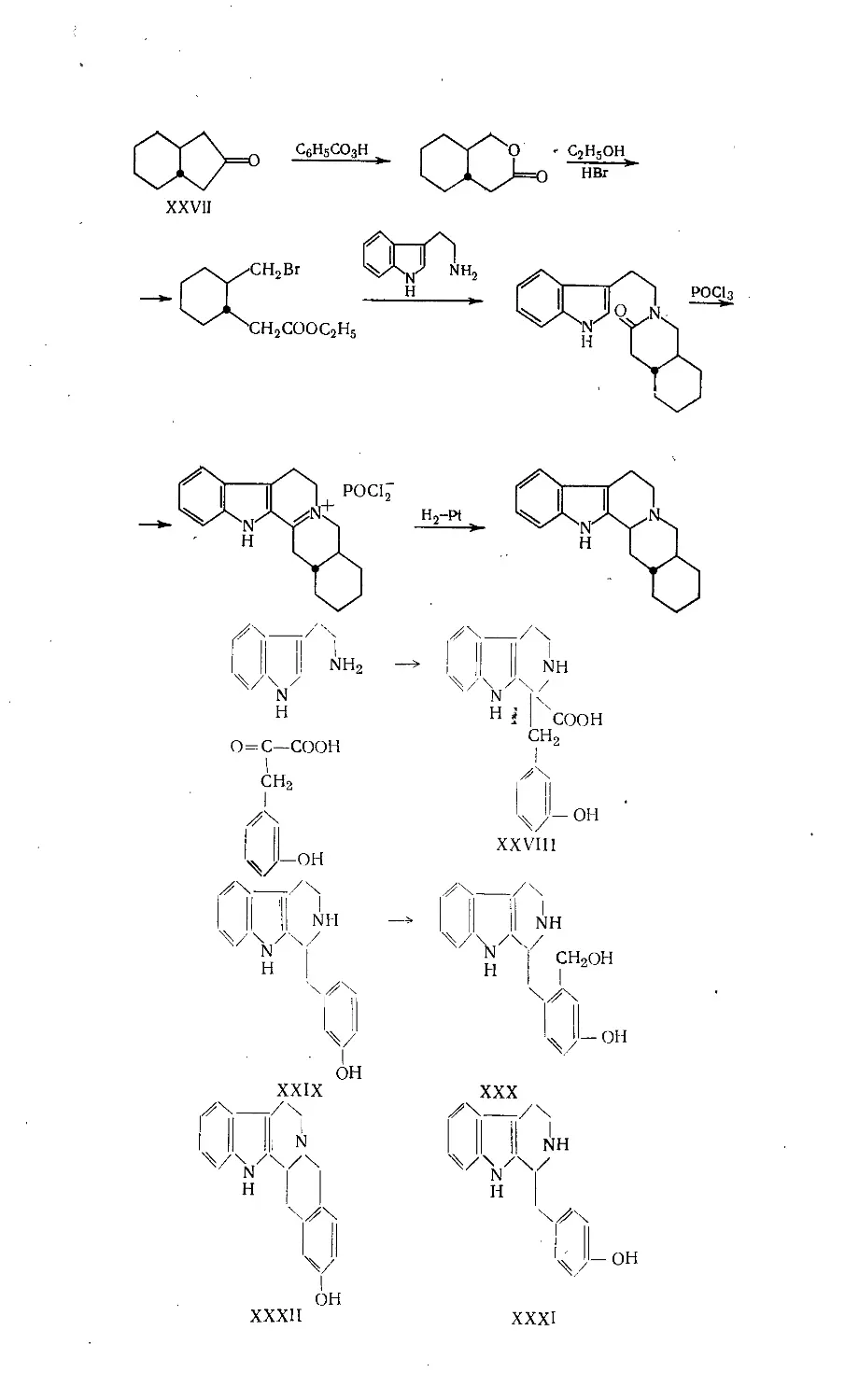
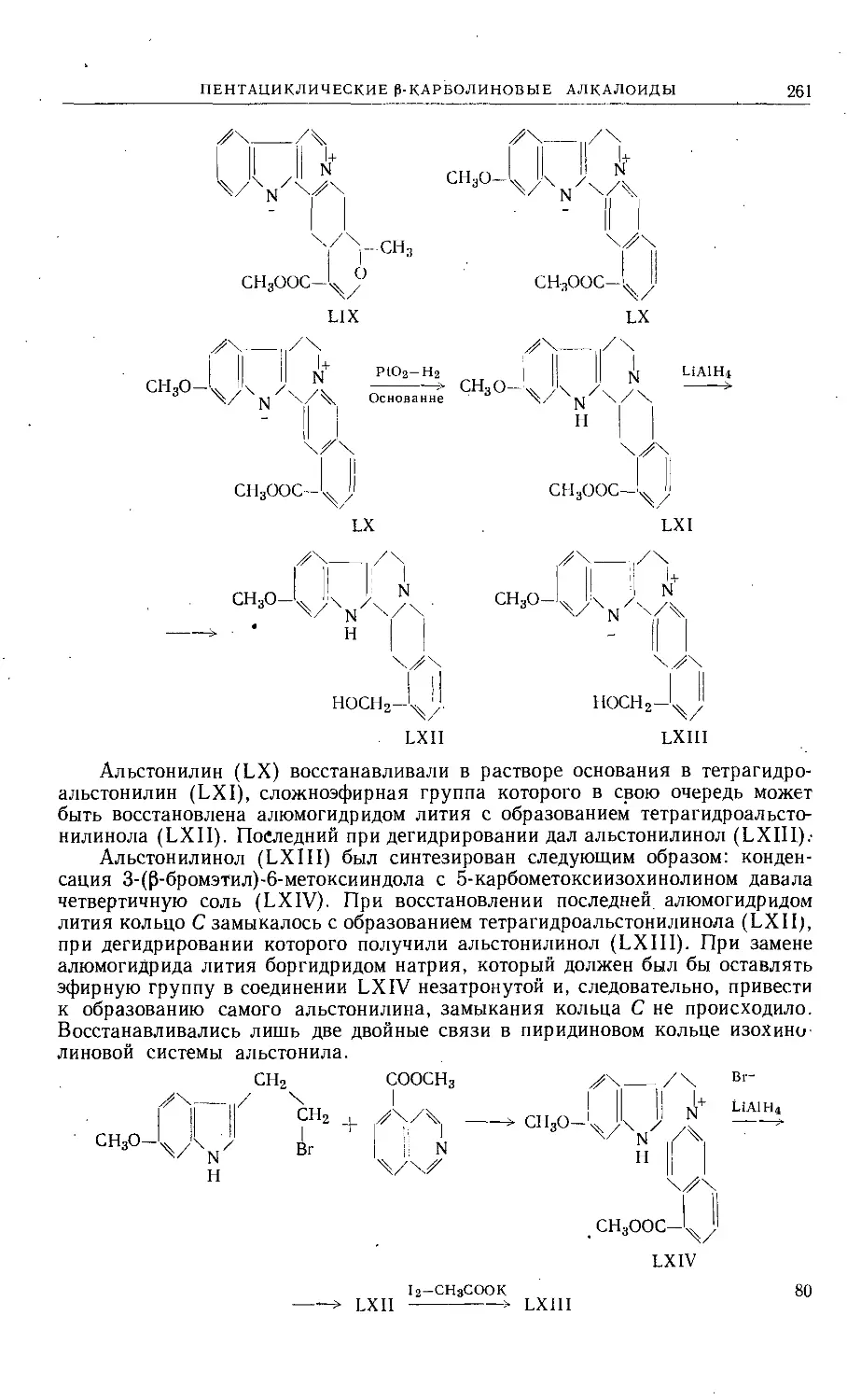
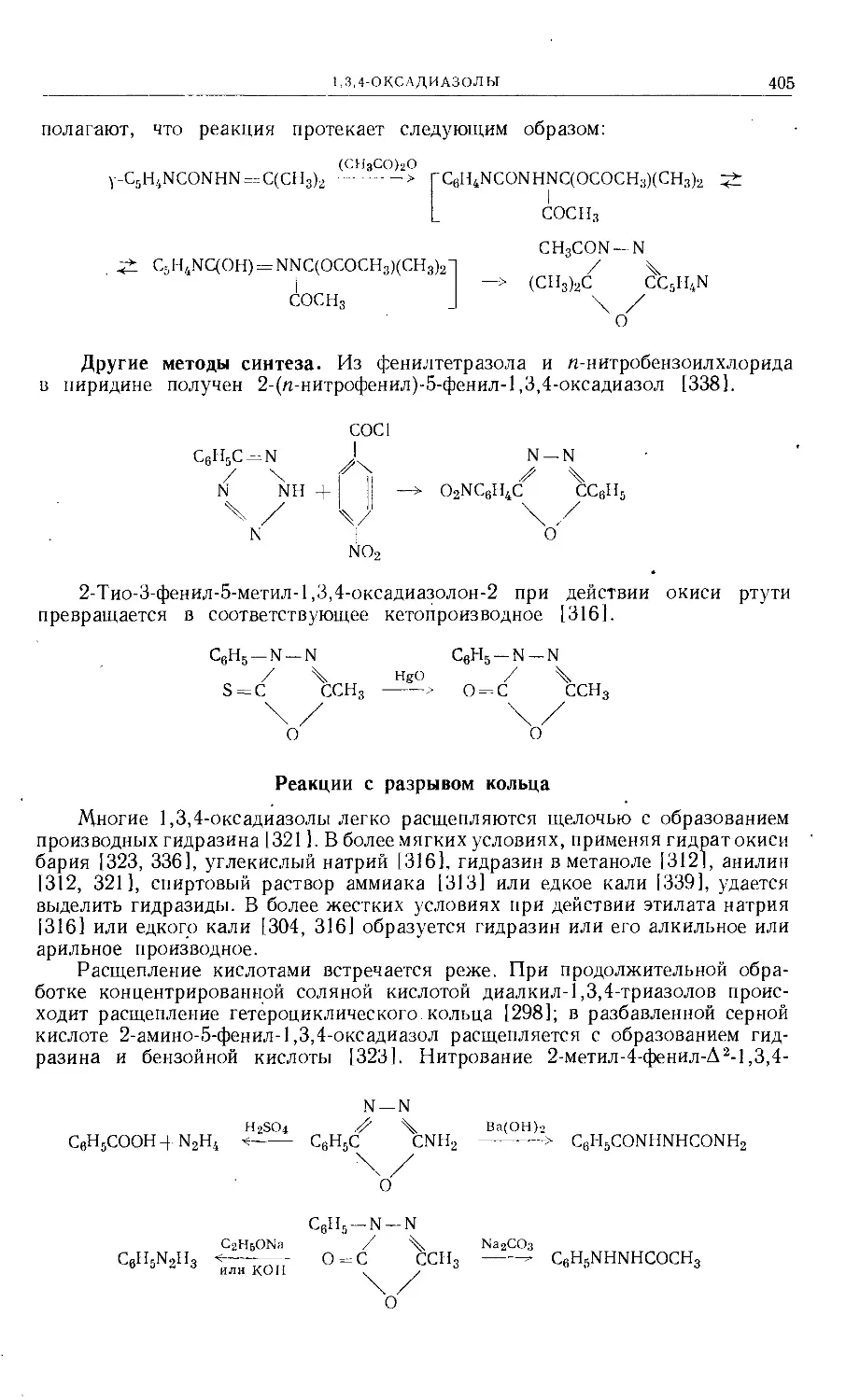
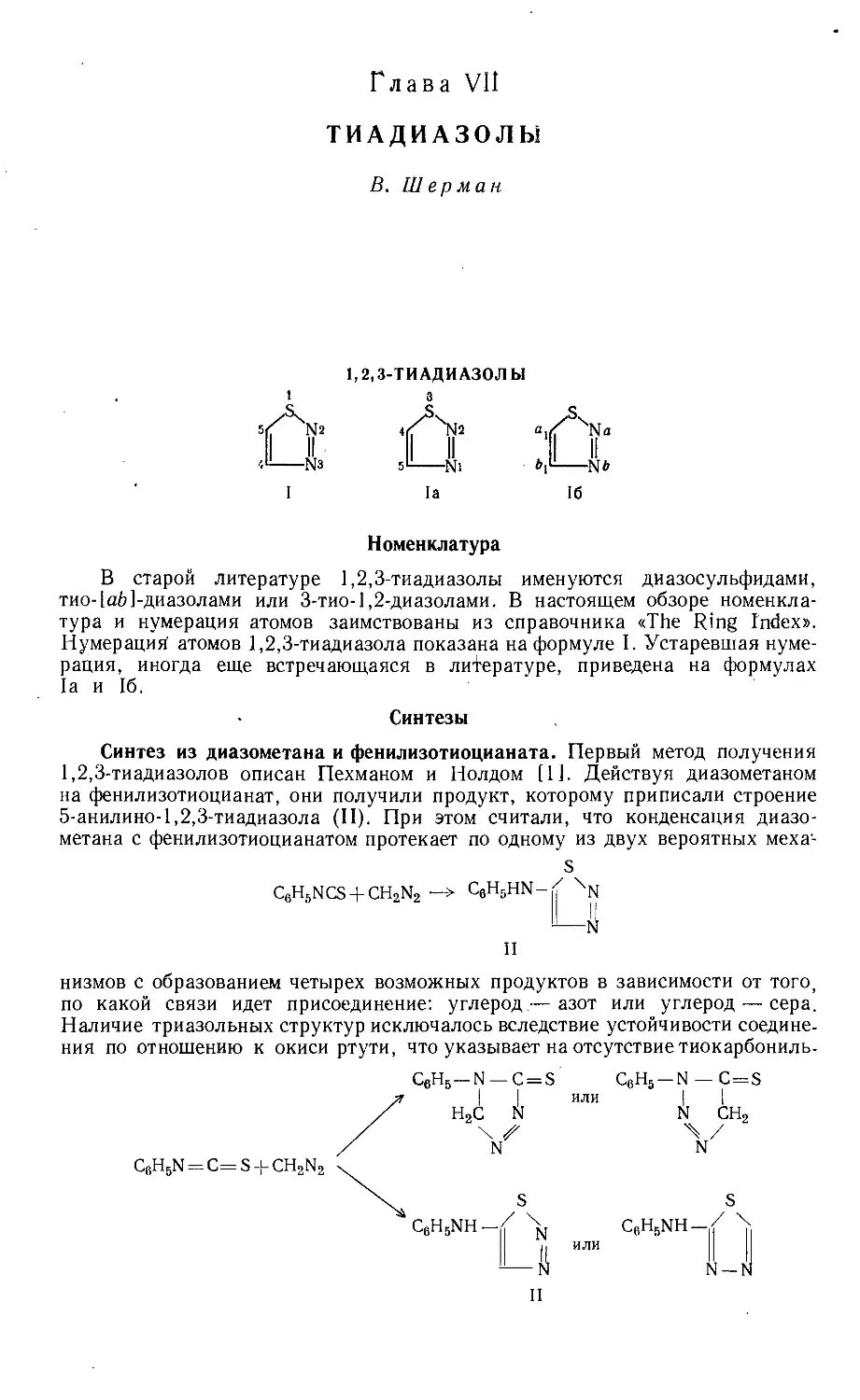
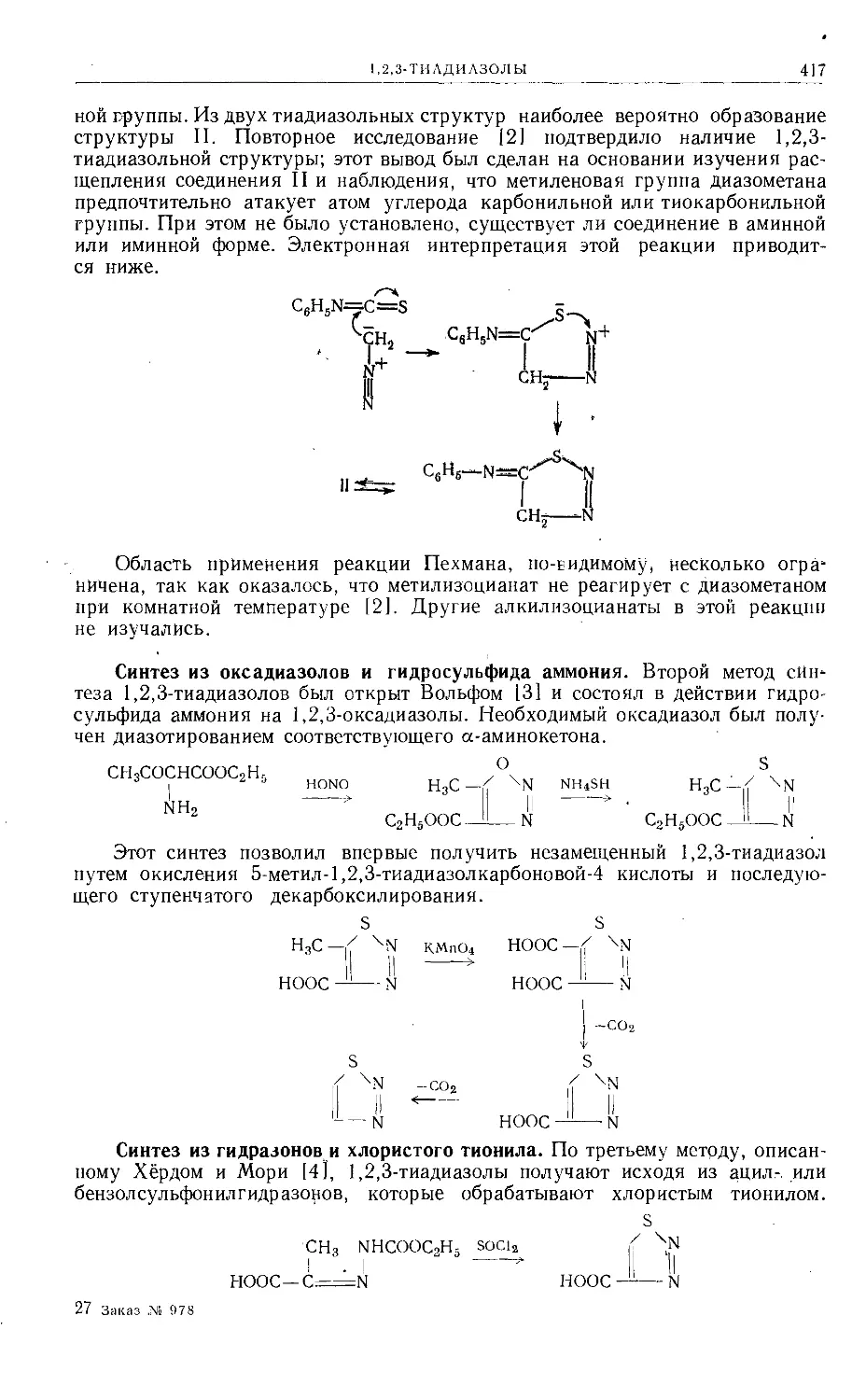
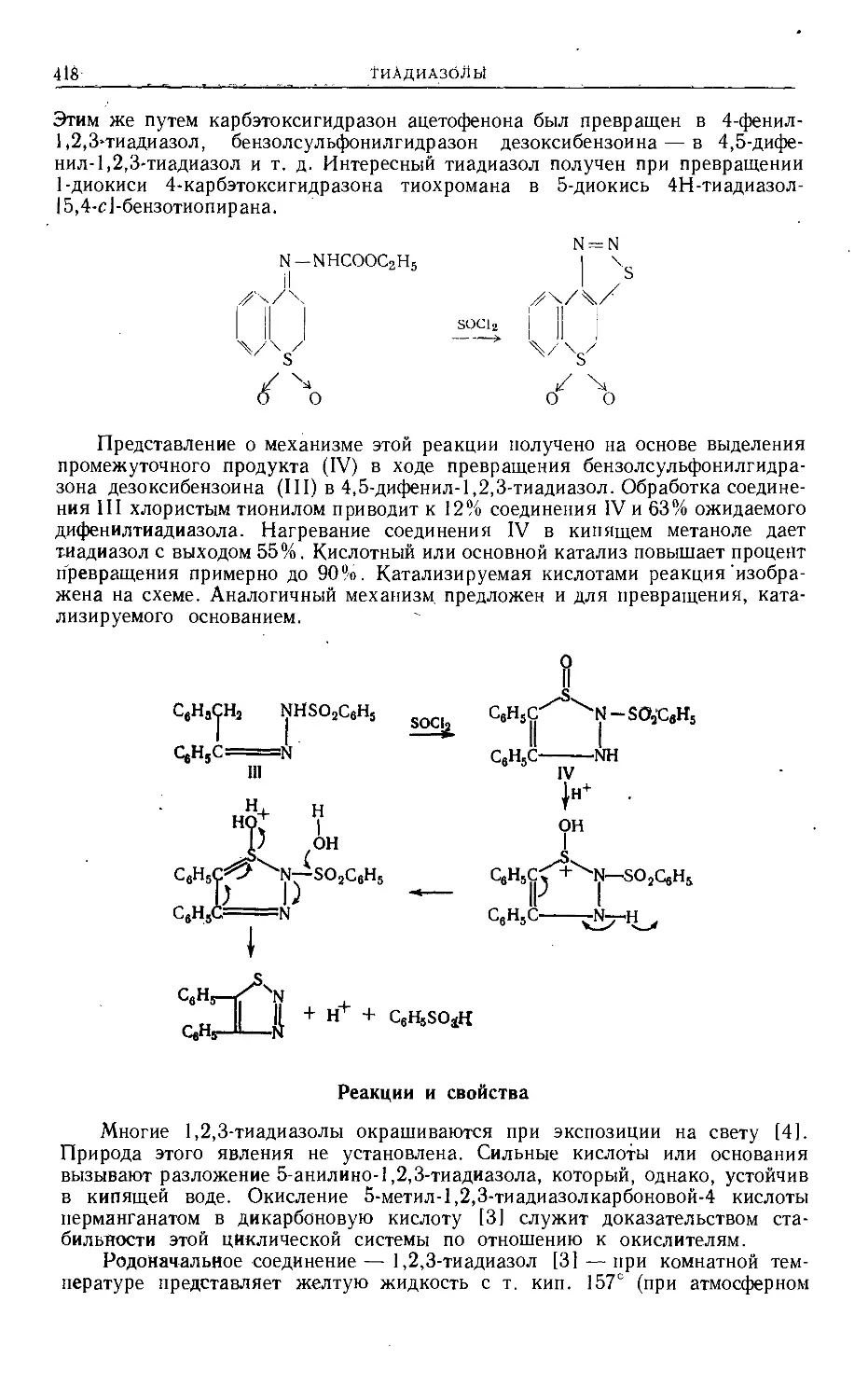
/














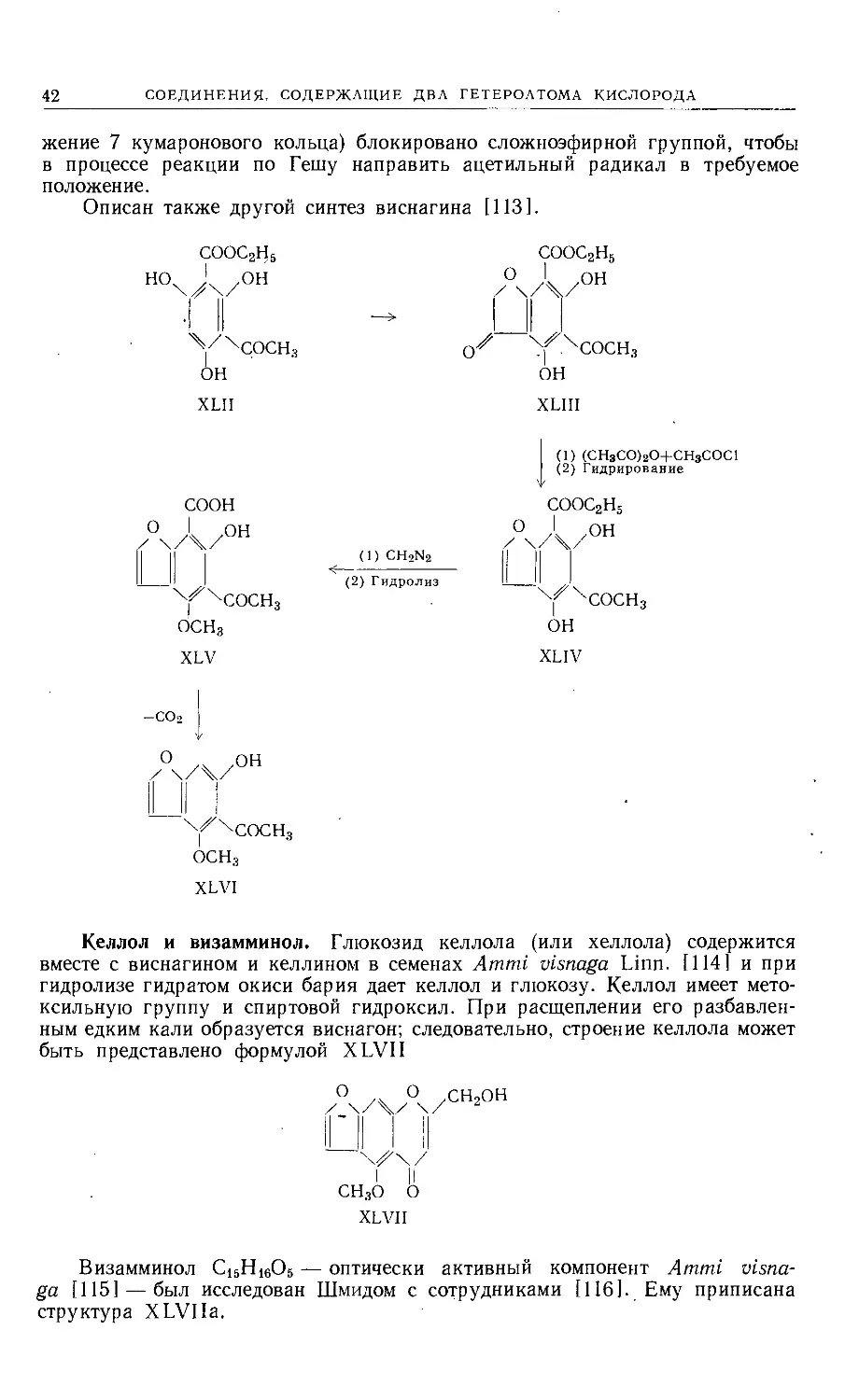
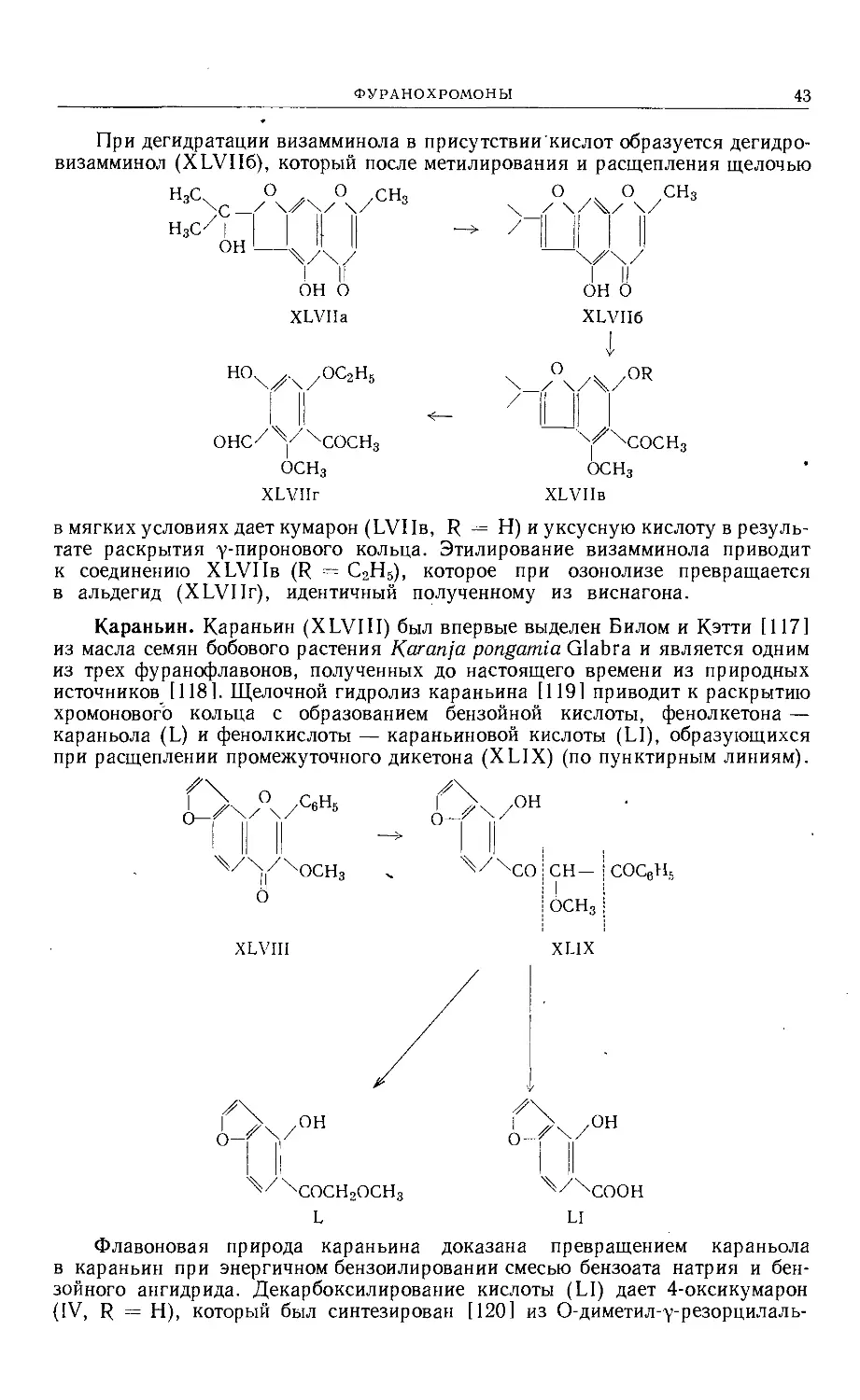
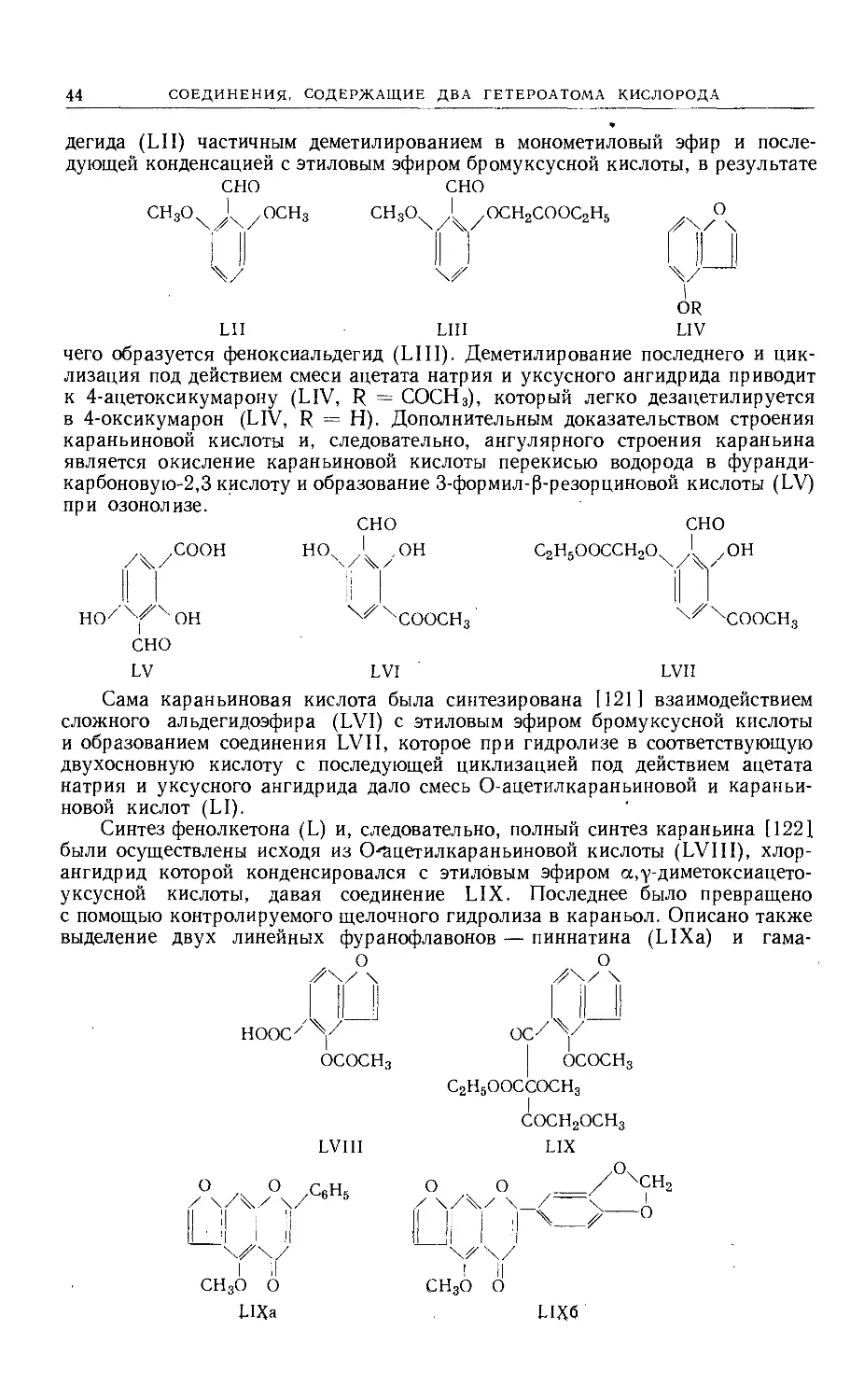
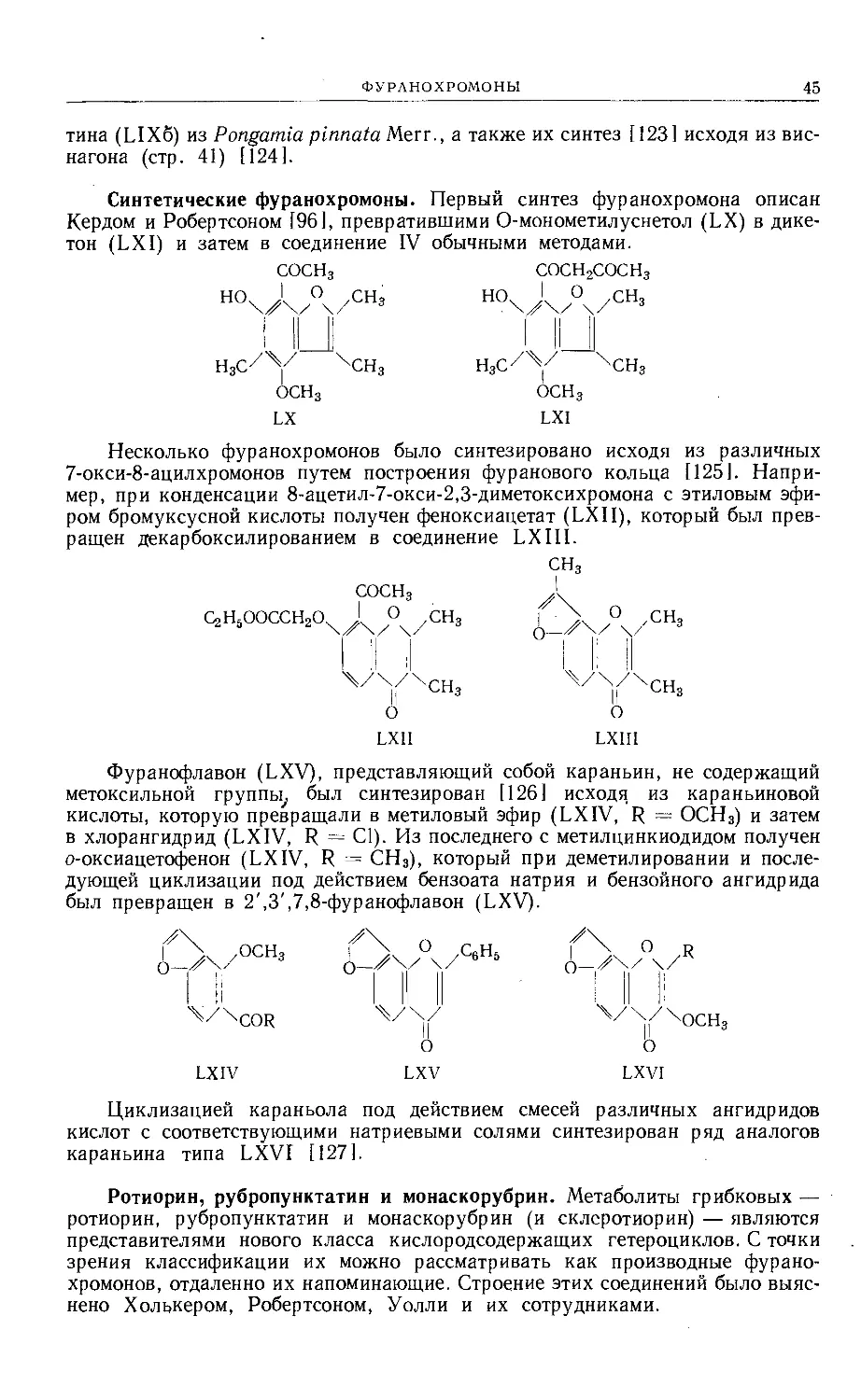
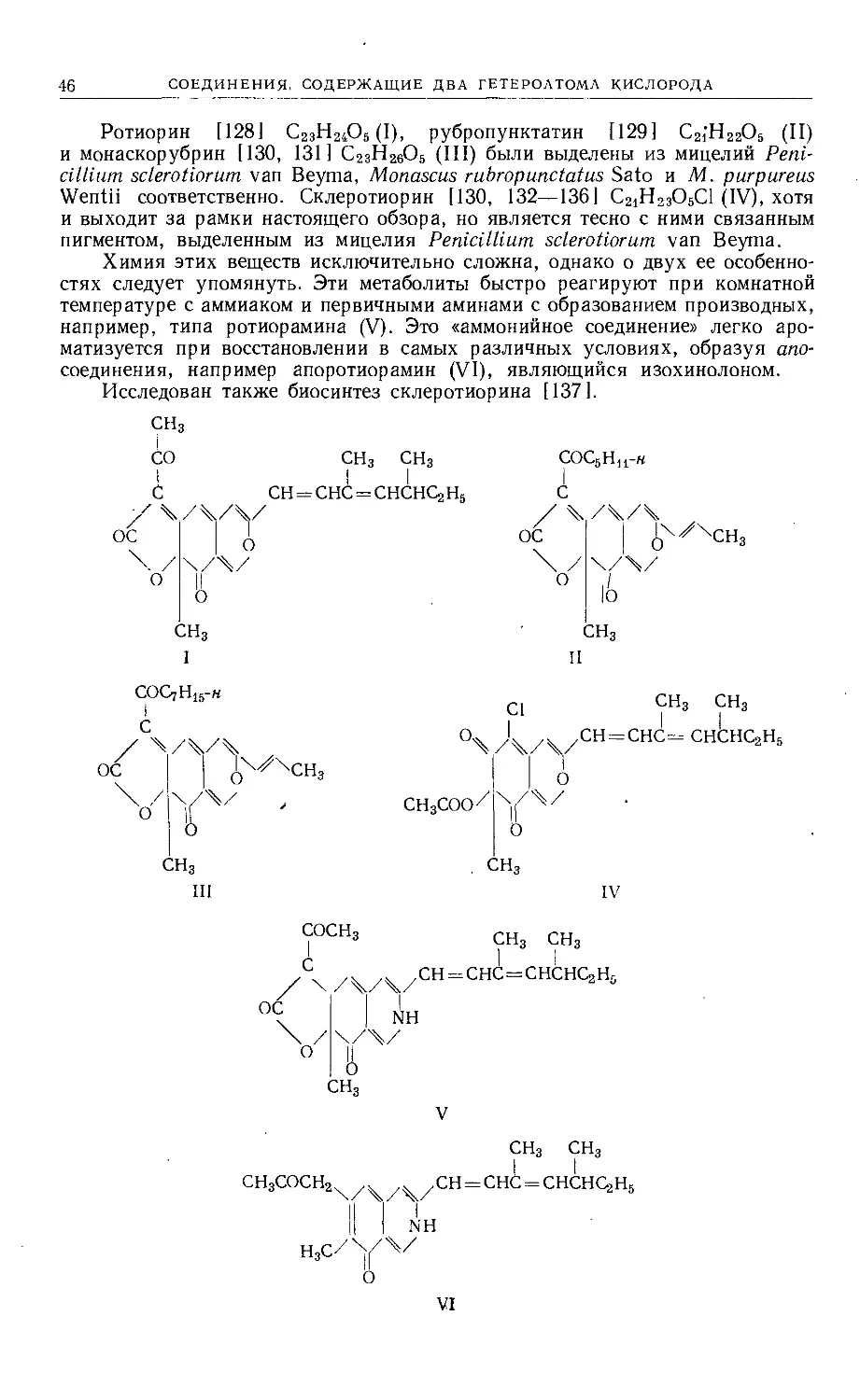
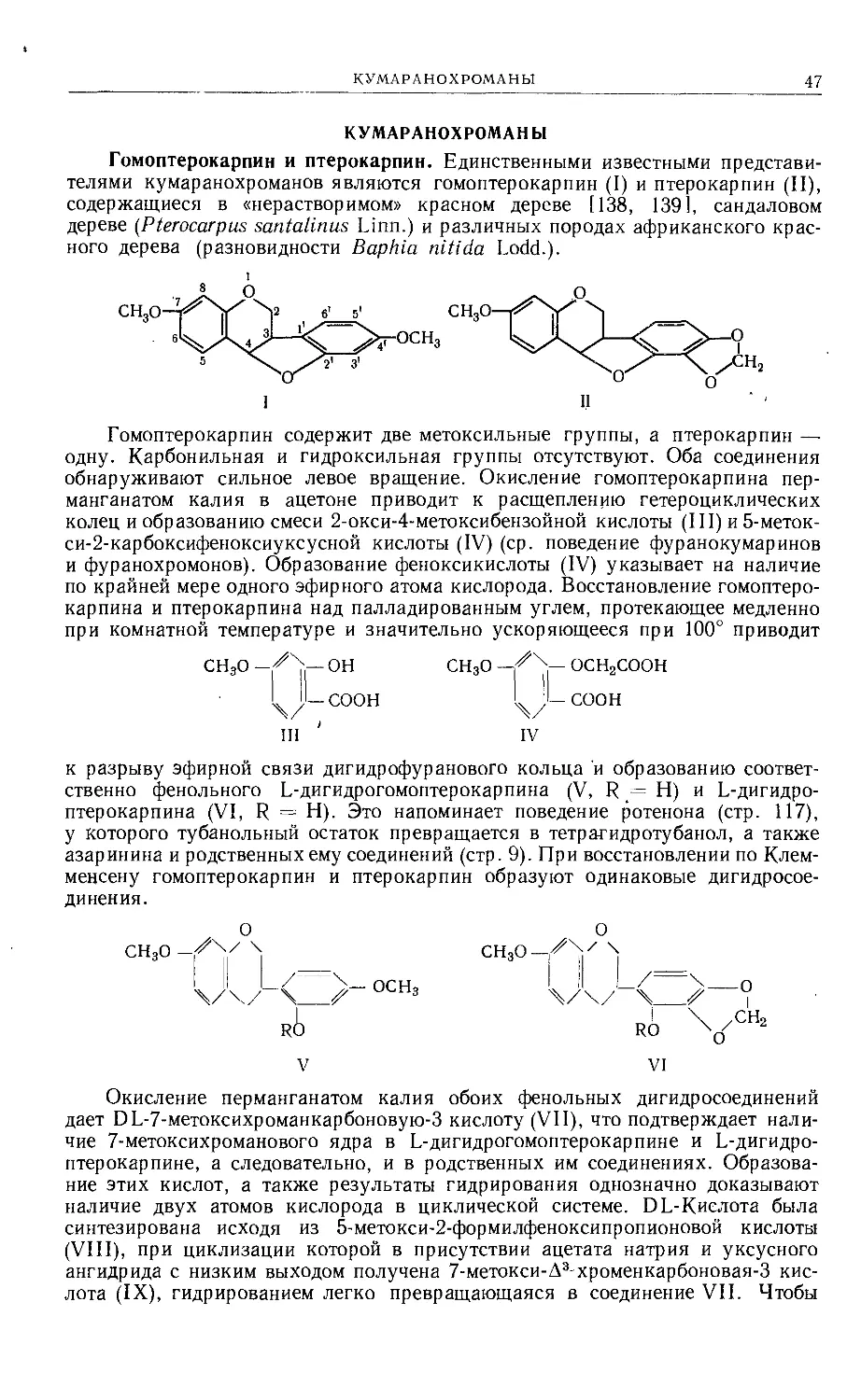
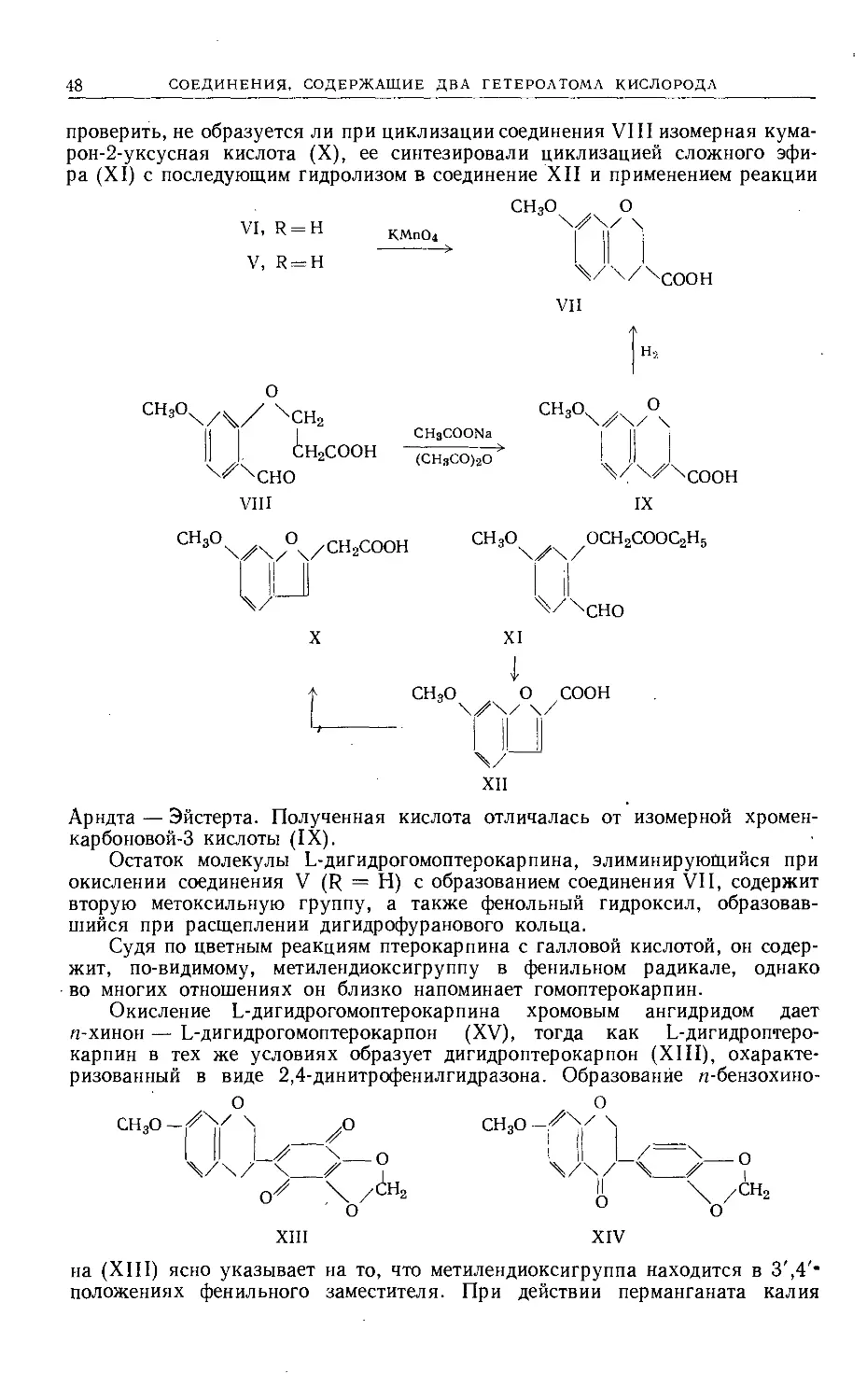
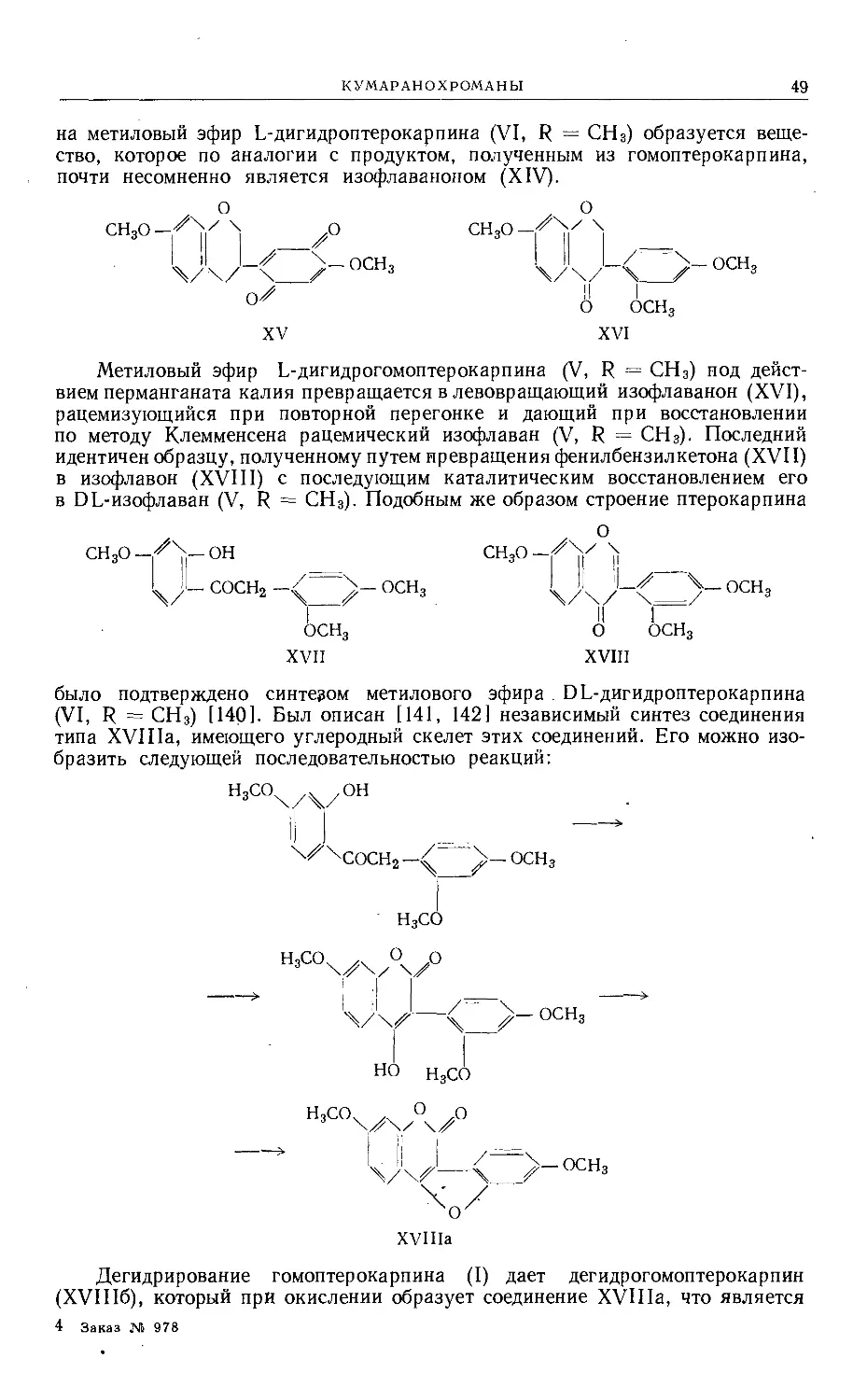
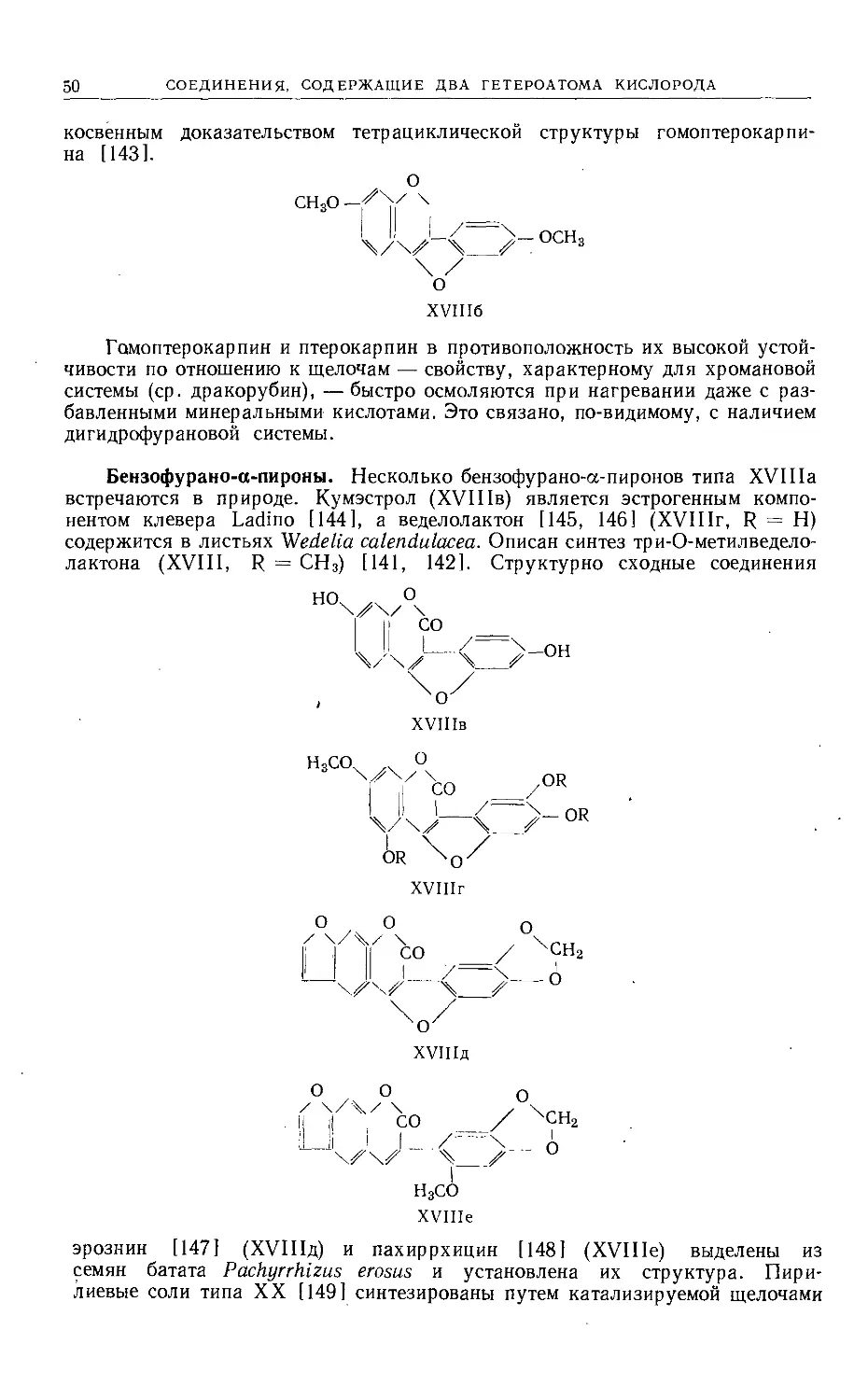
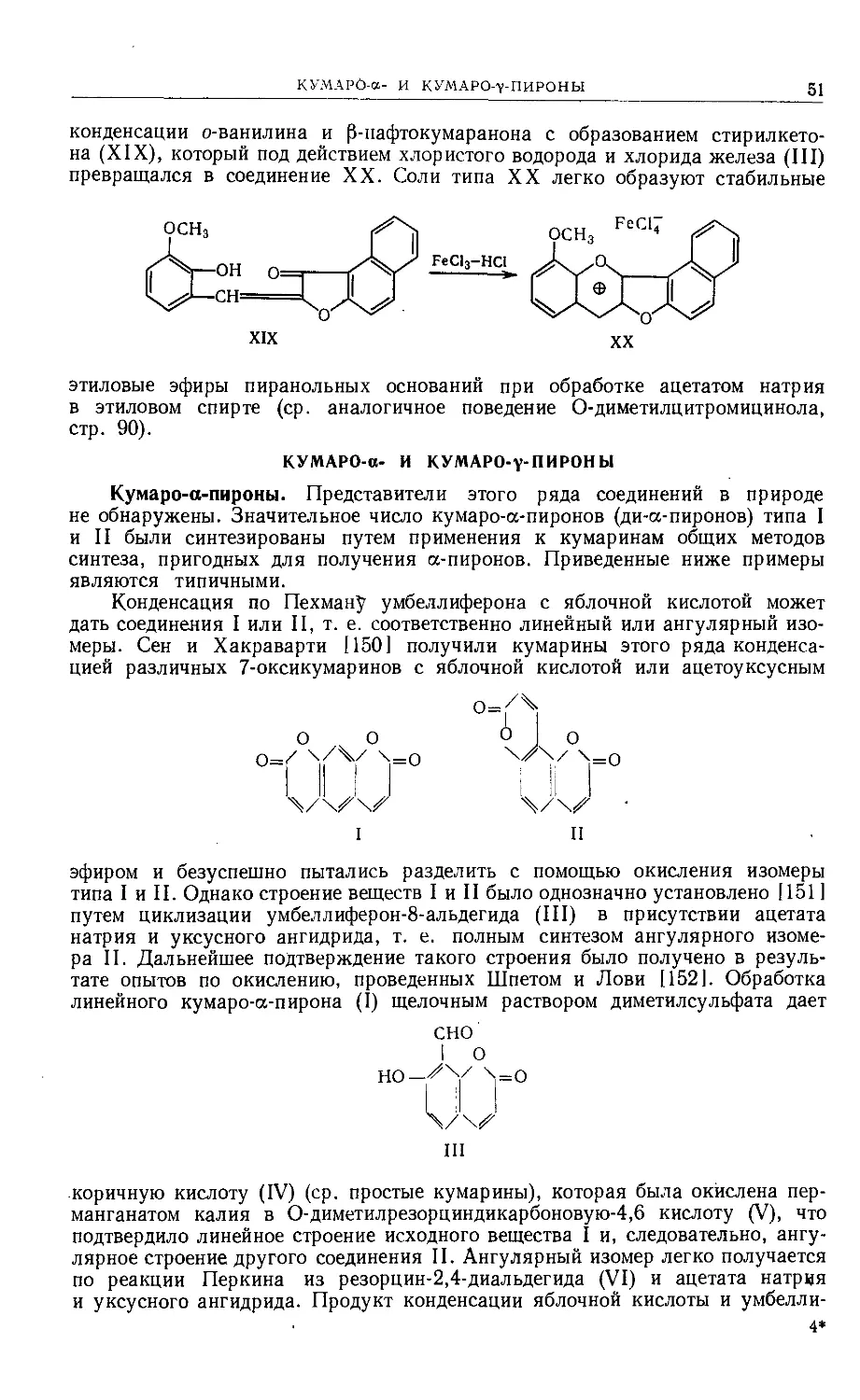
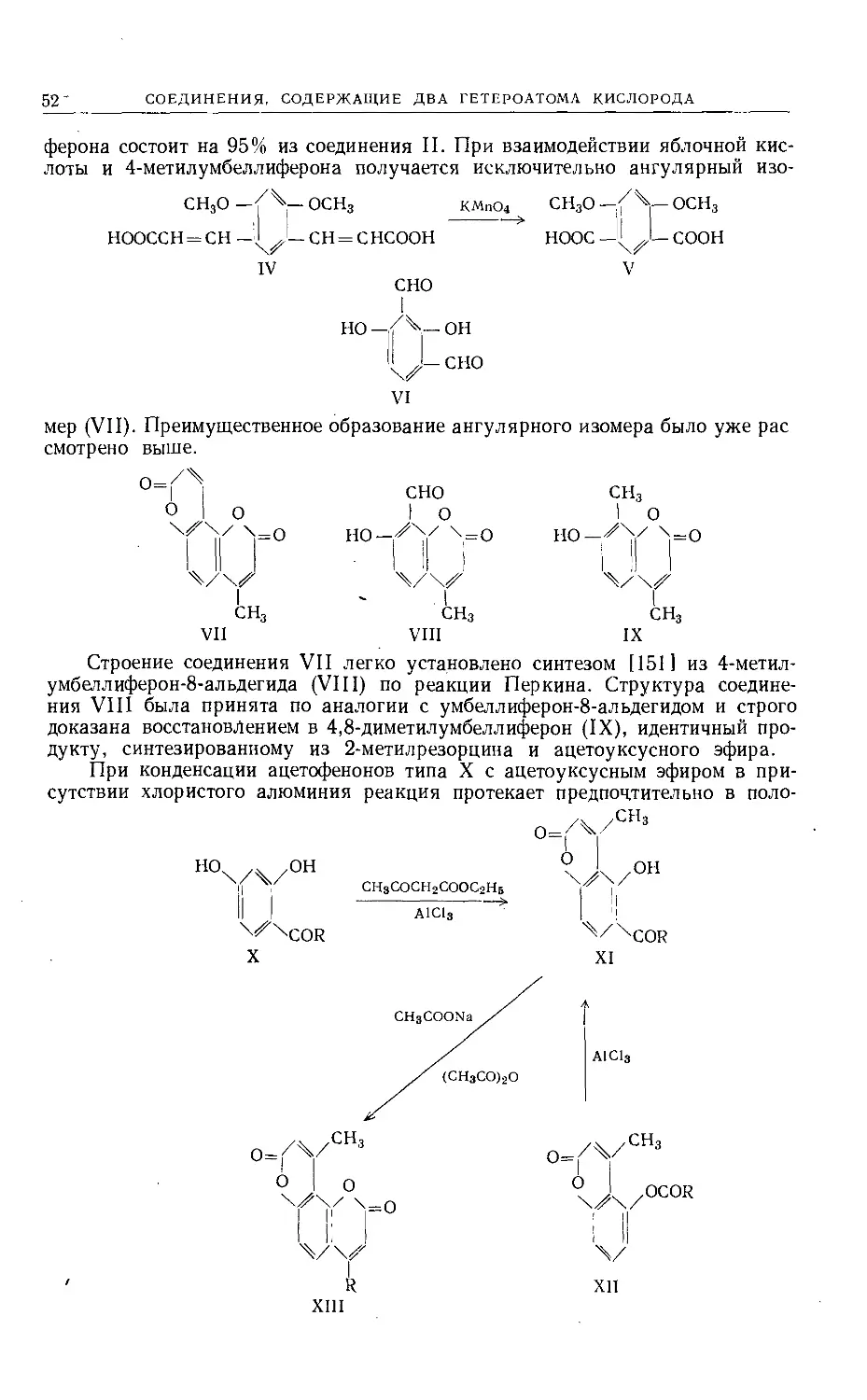
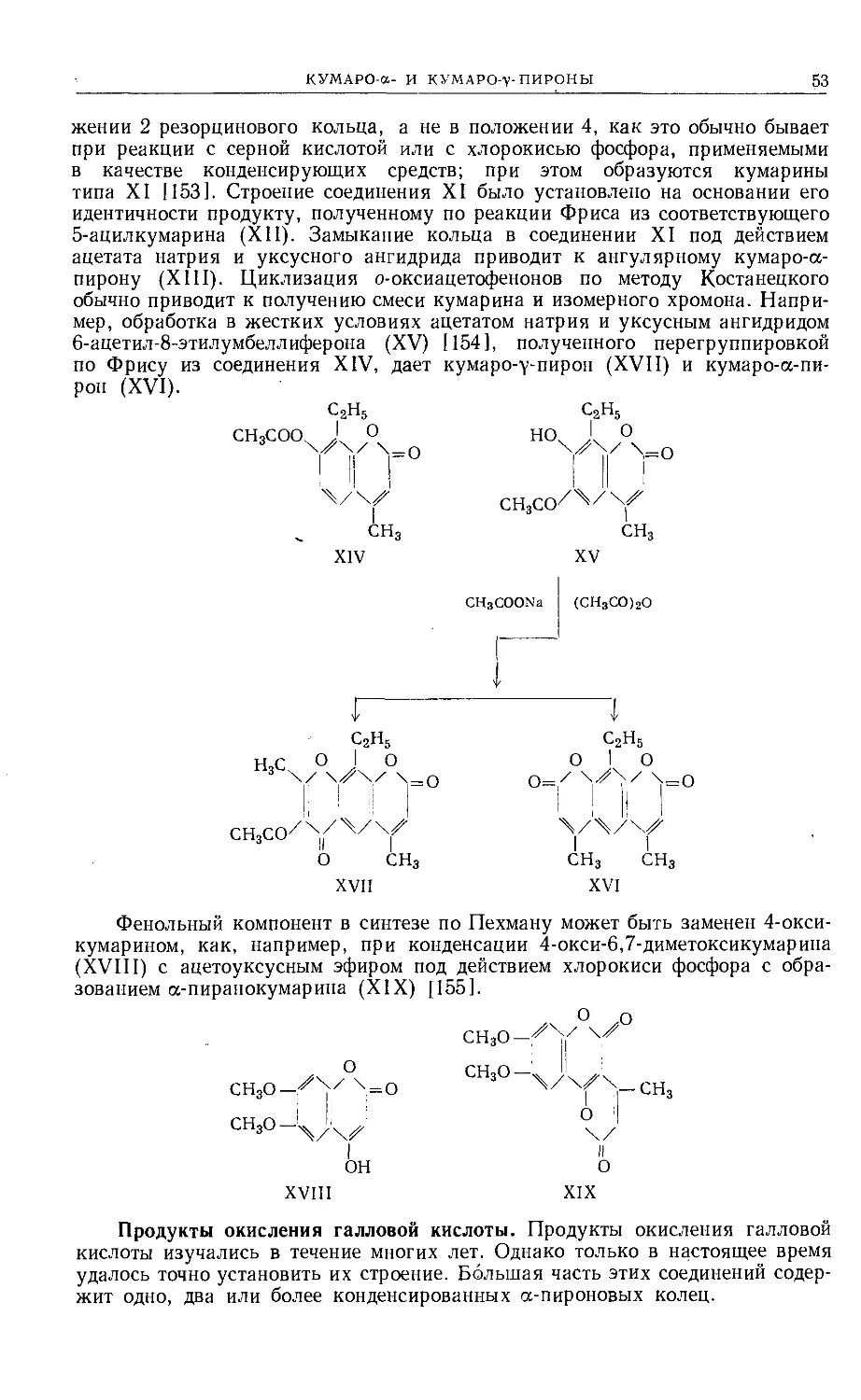
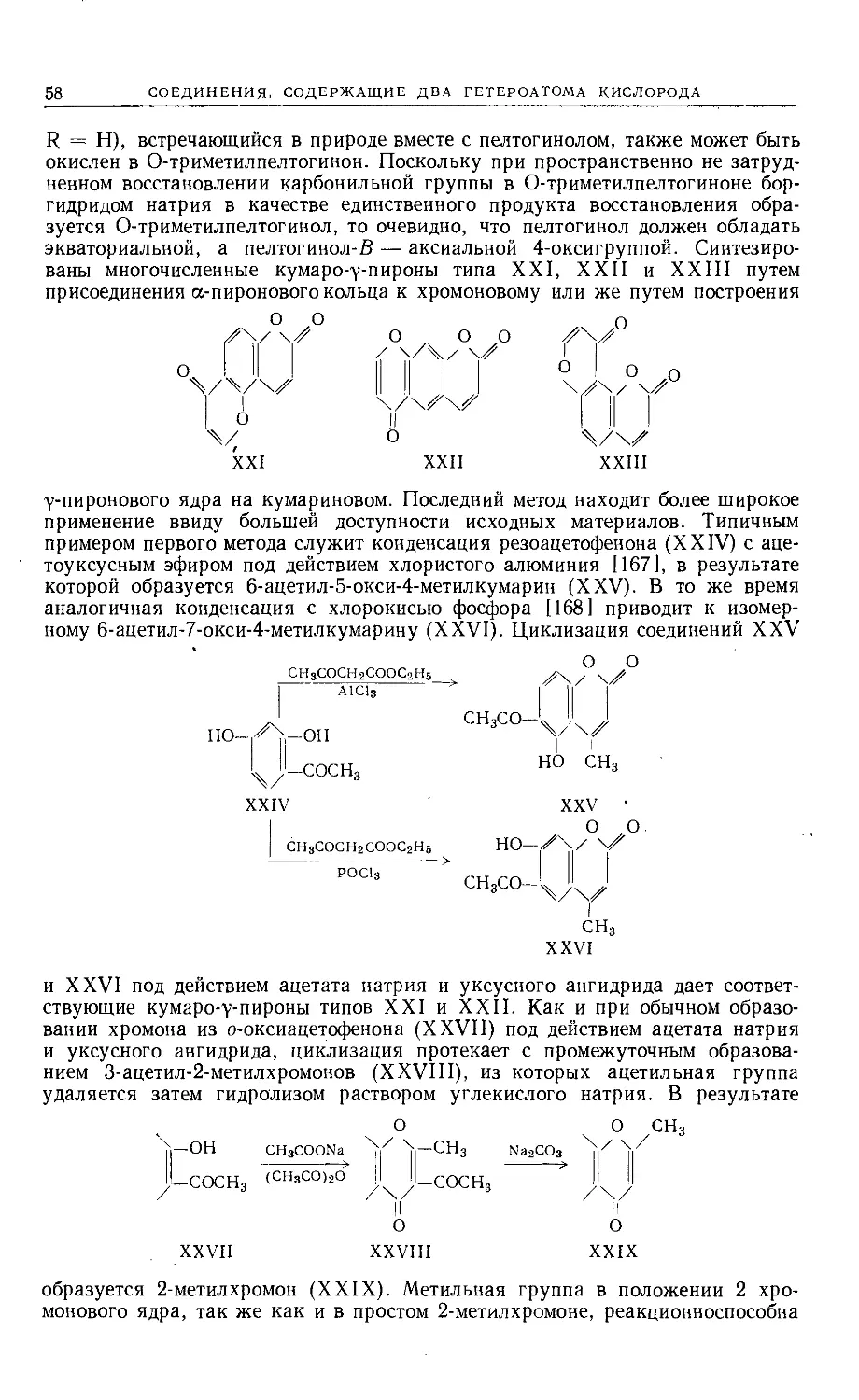
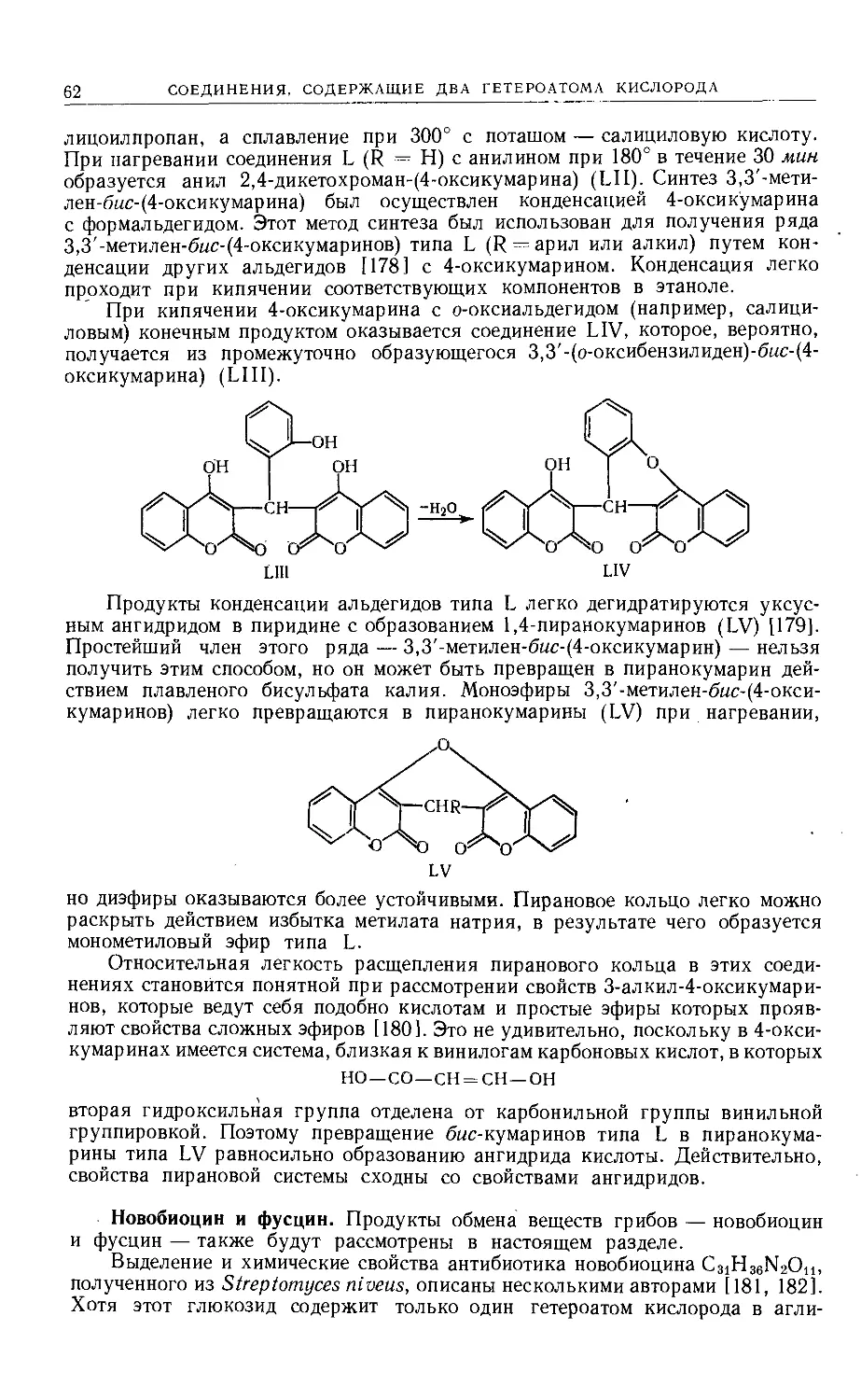
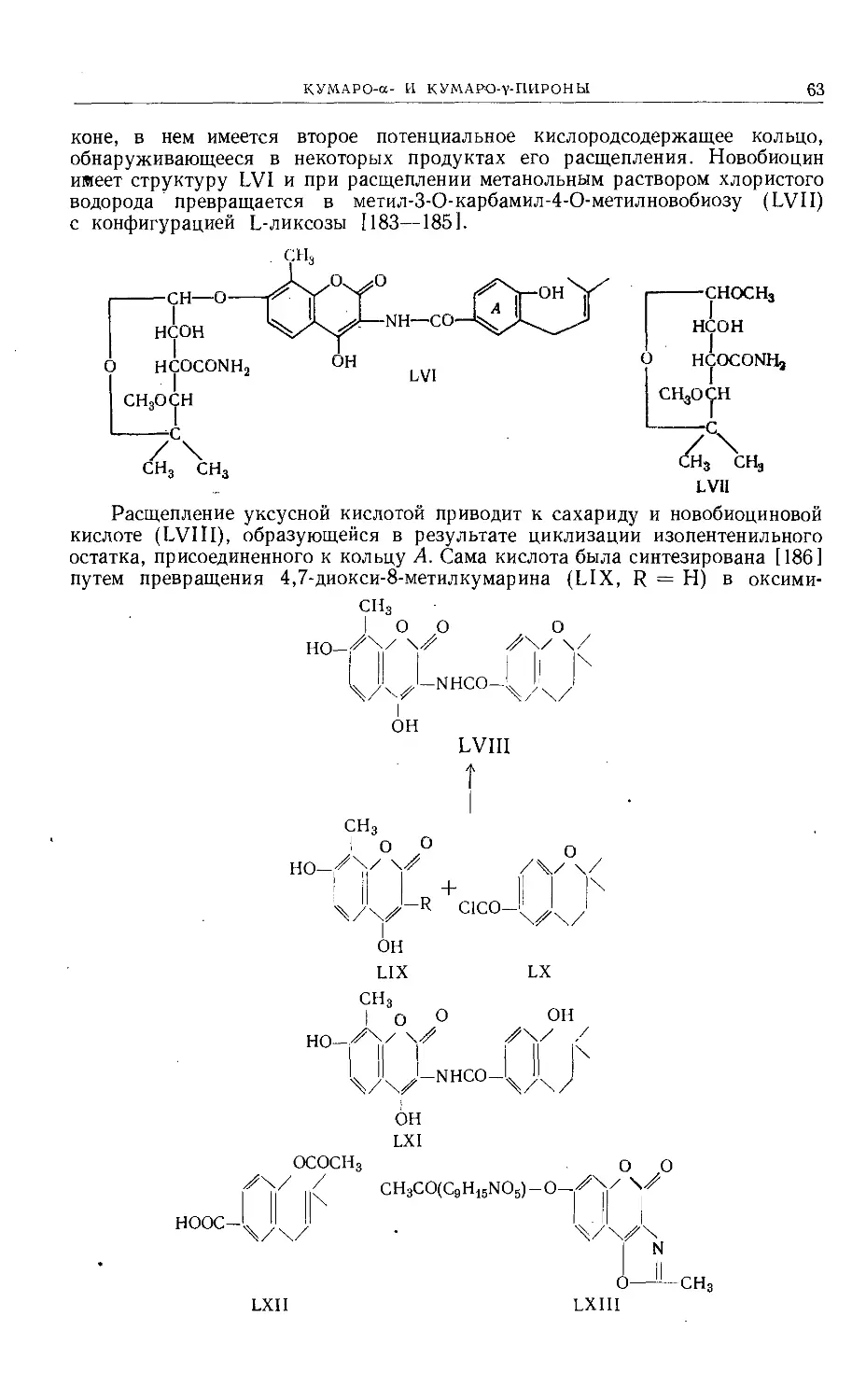
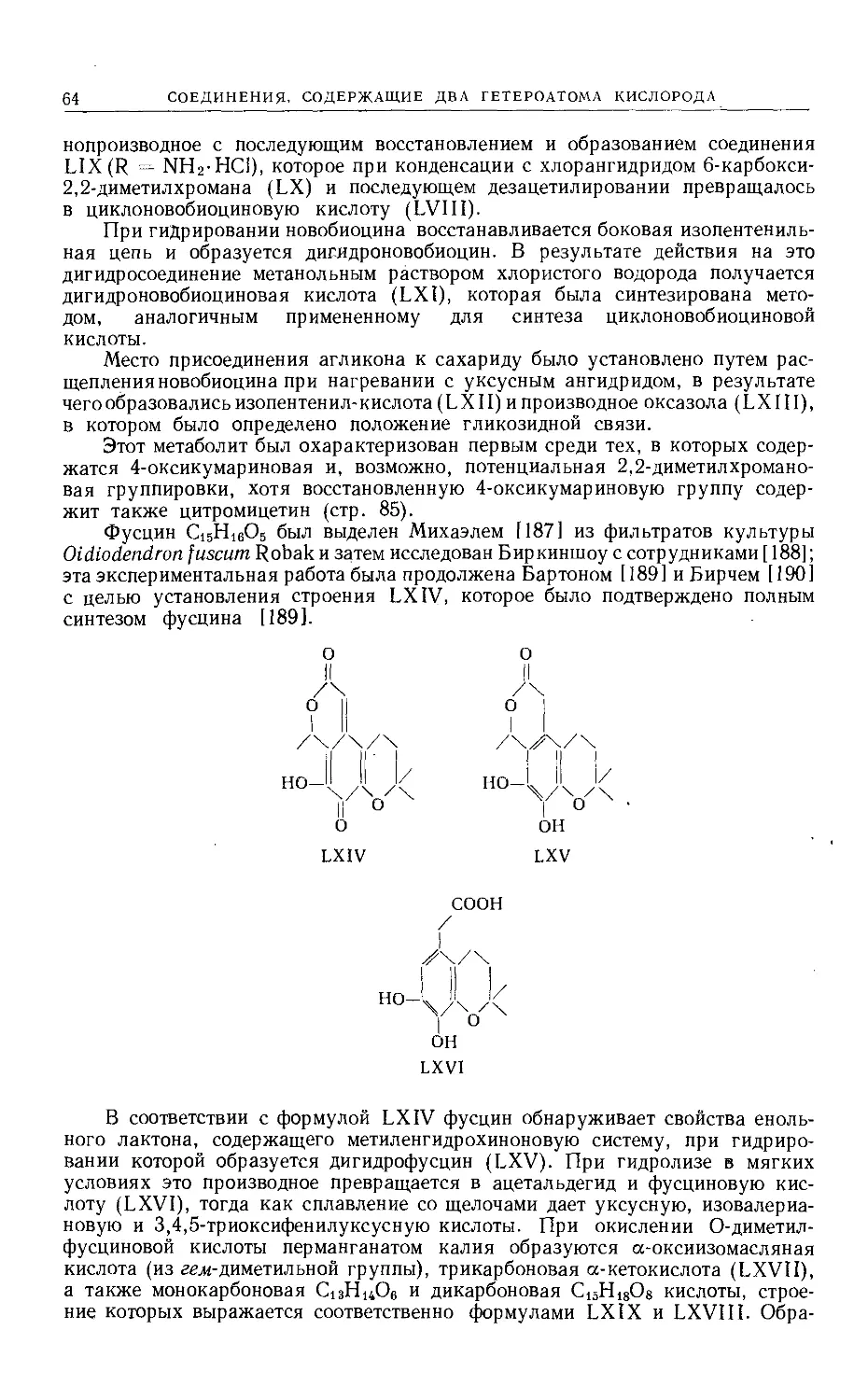
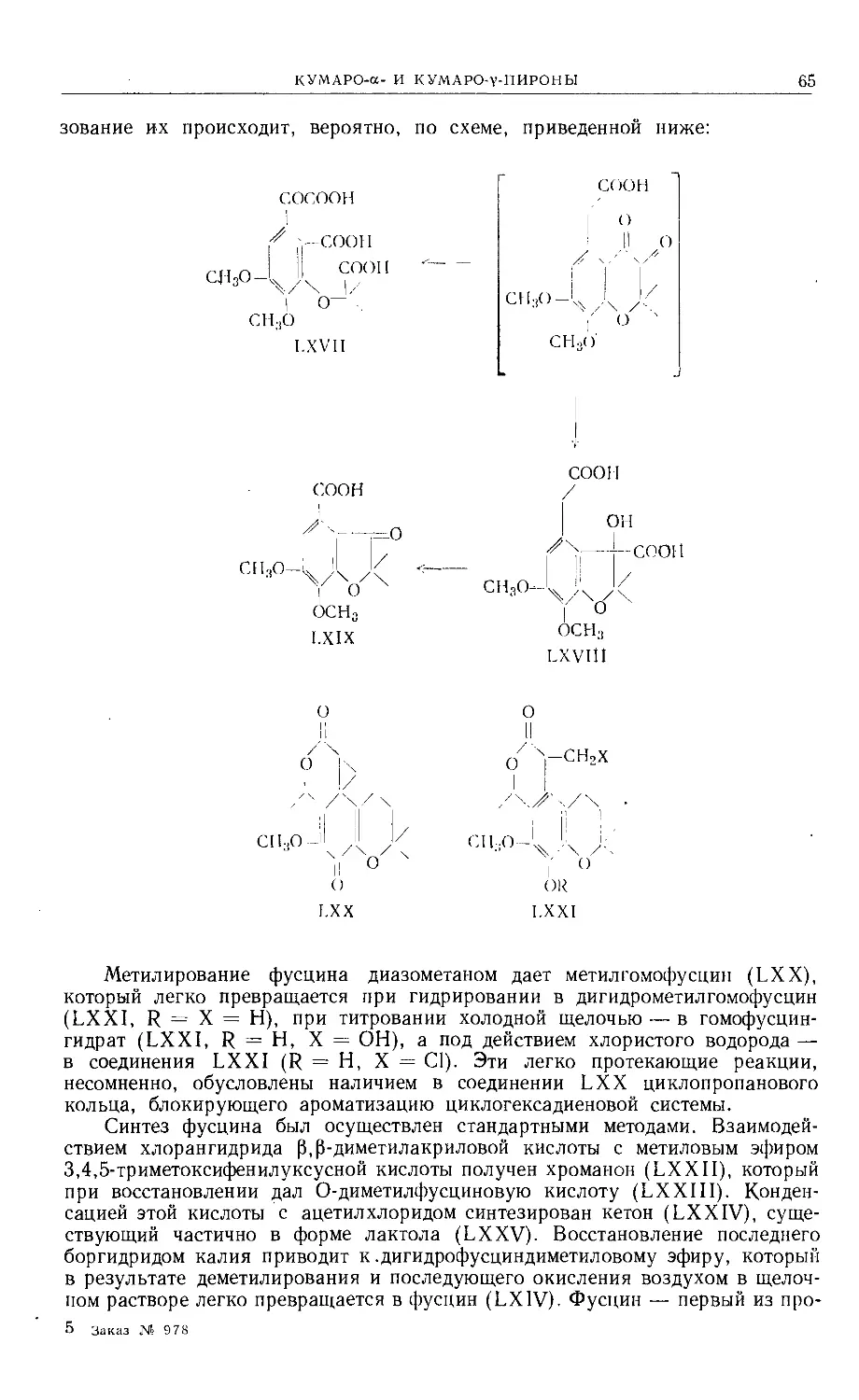
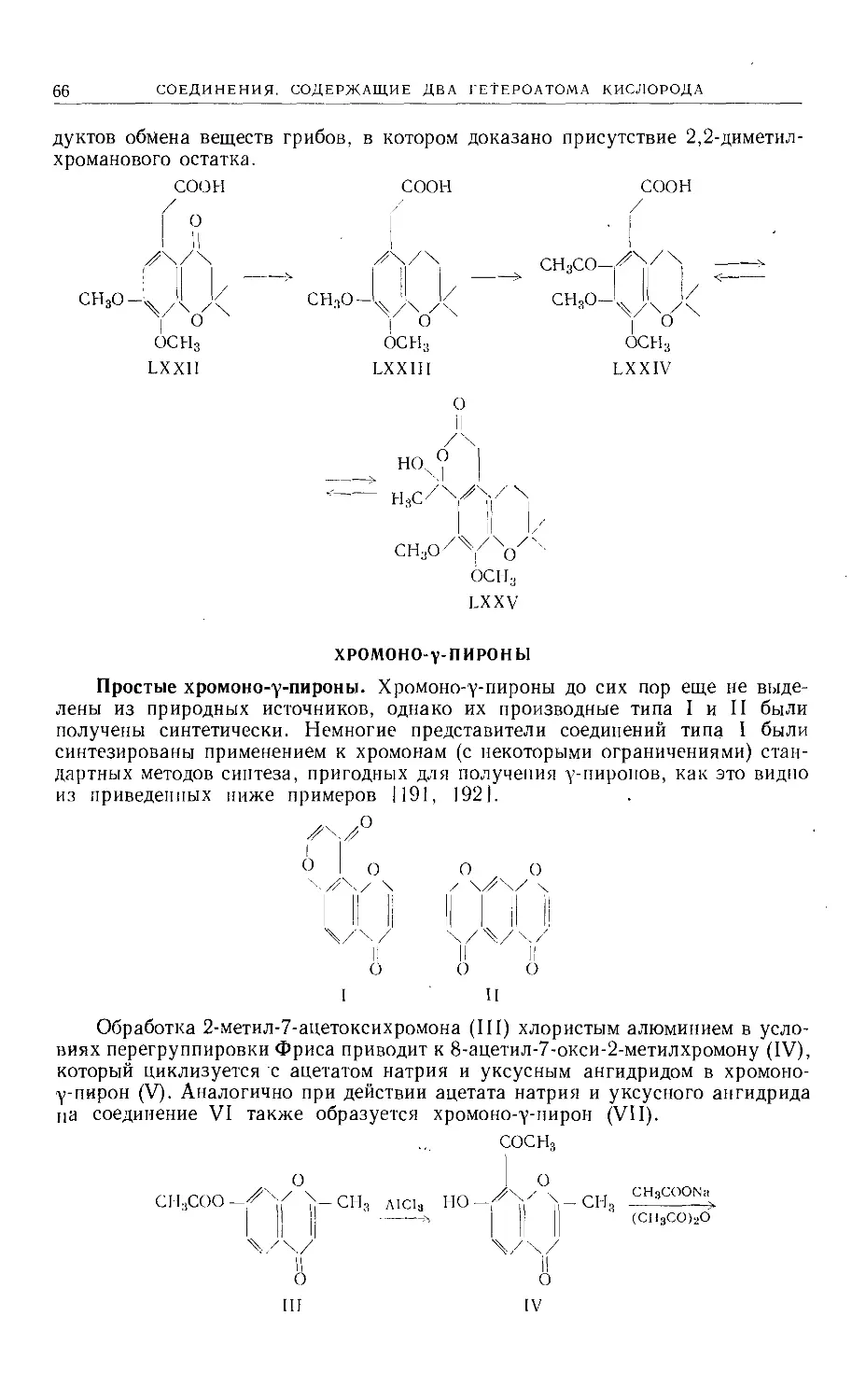
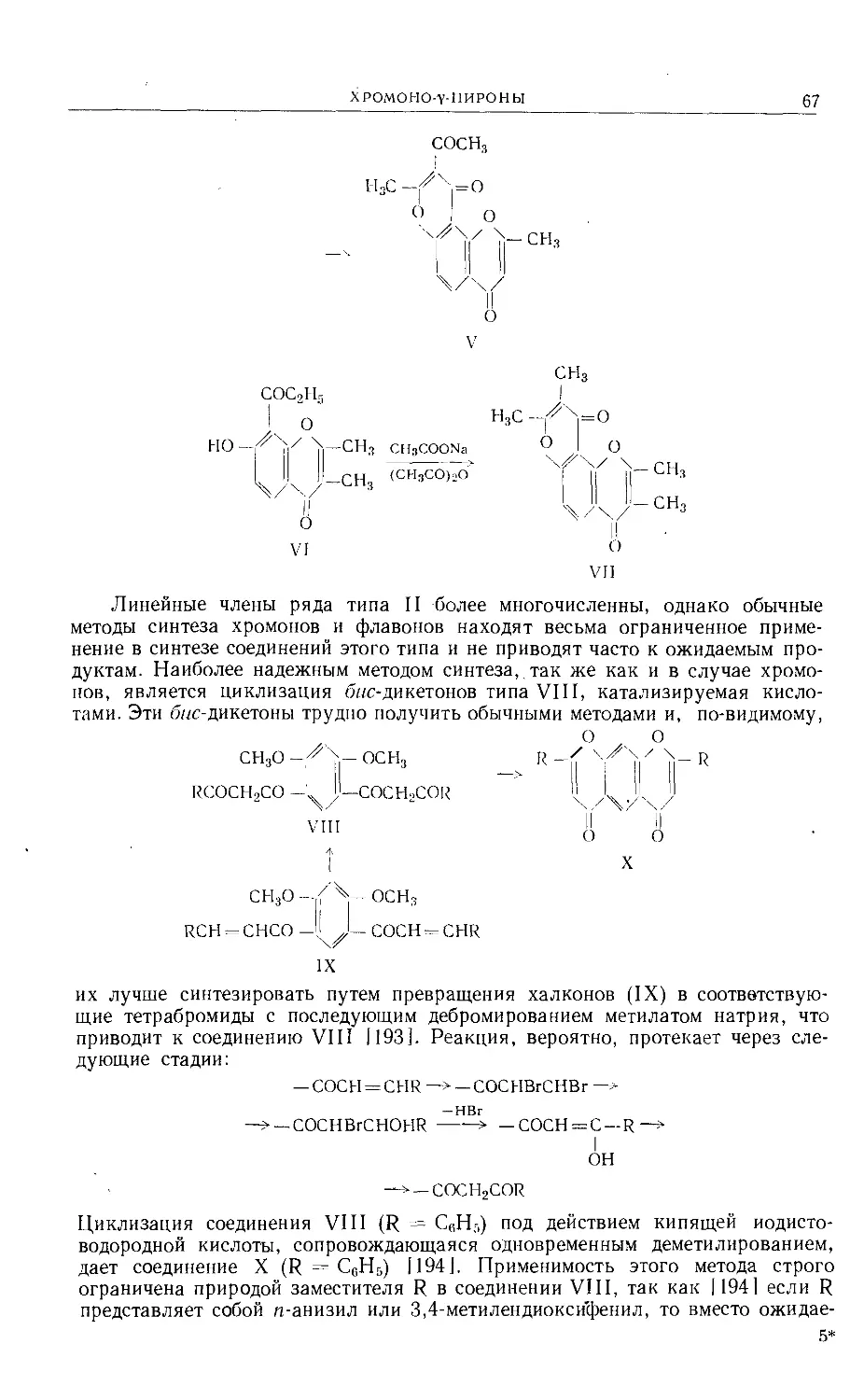
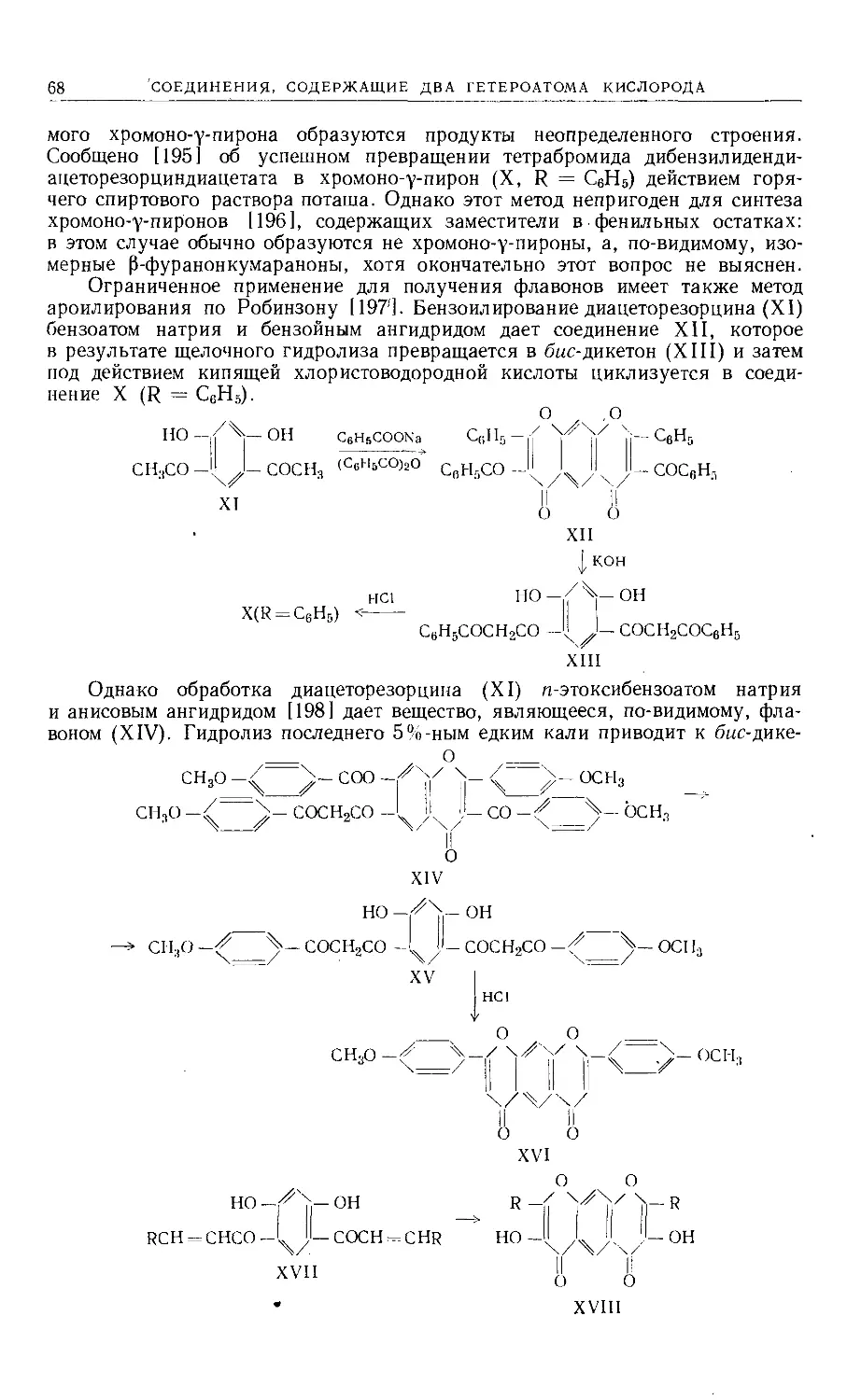
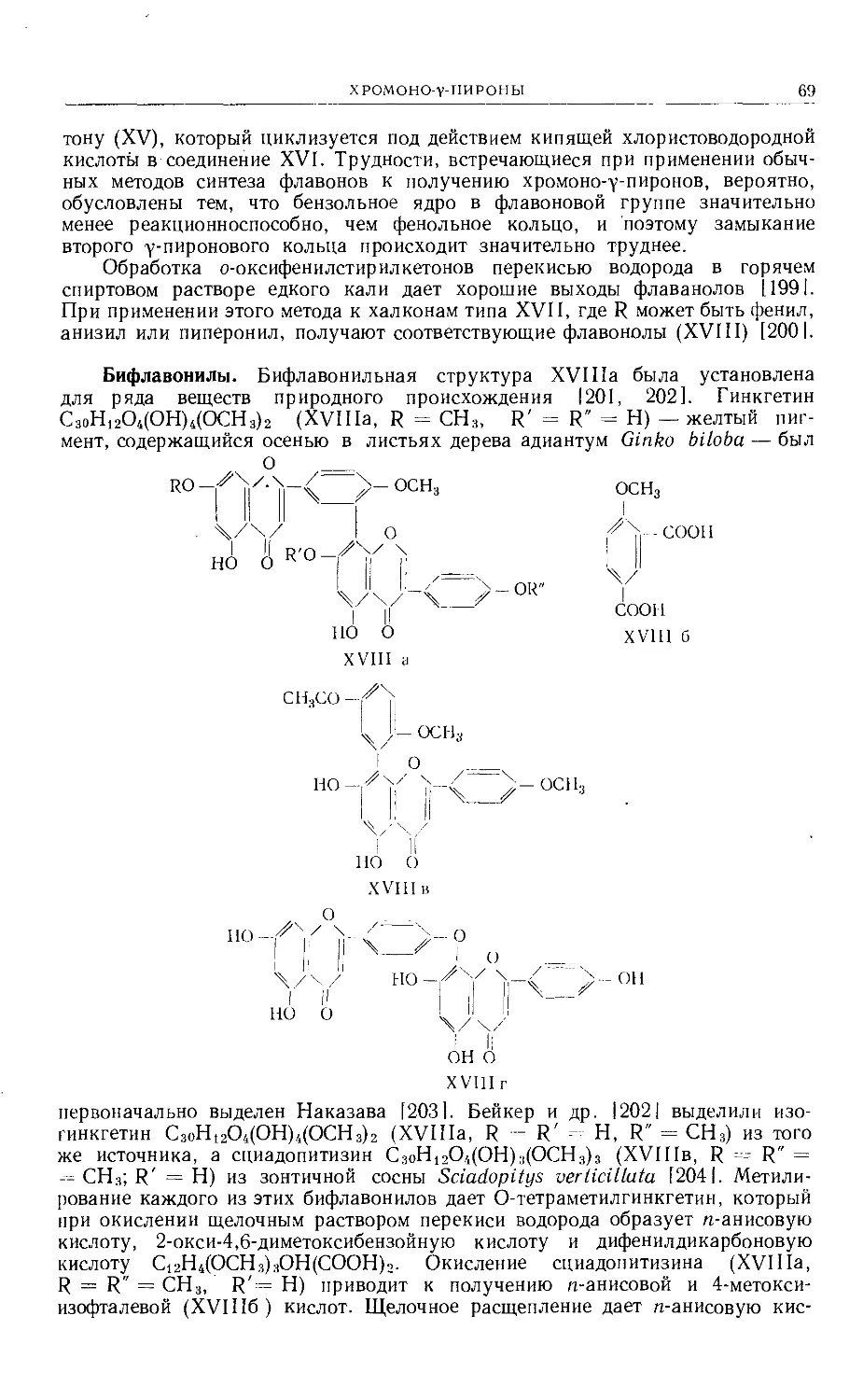


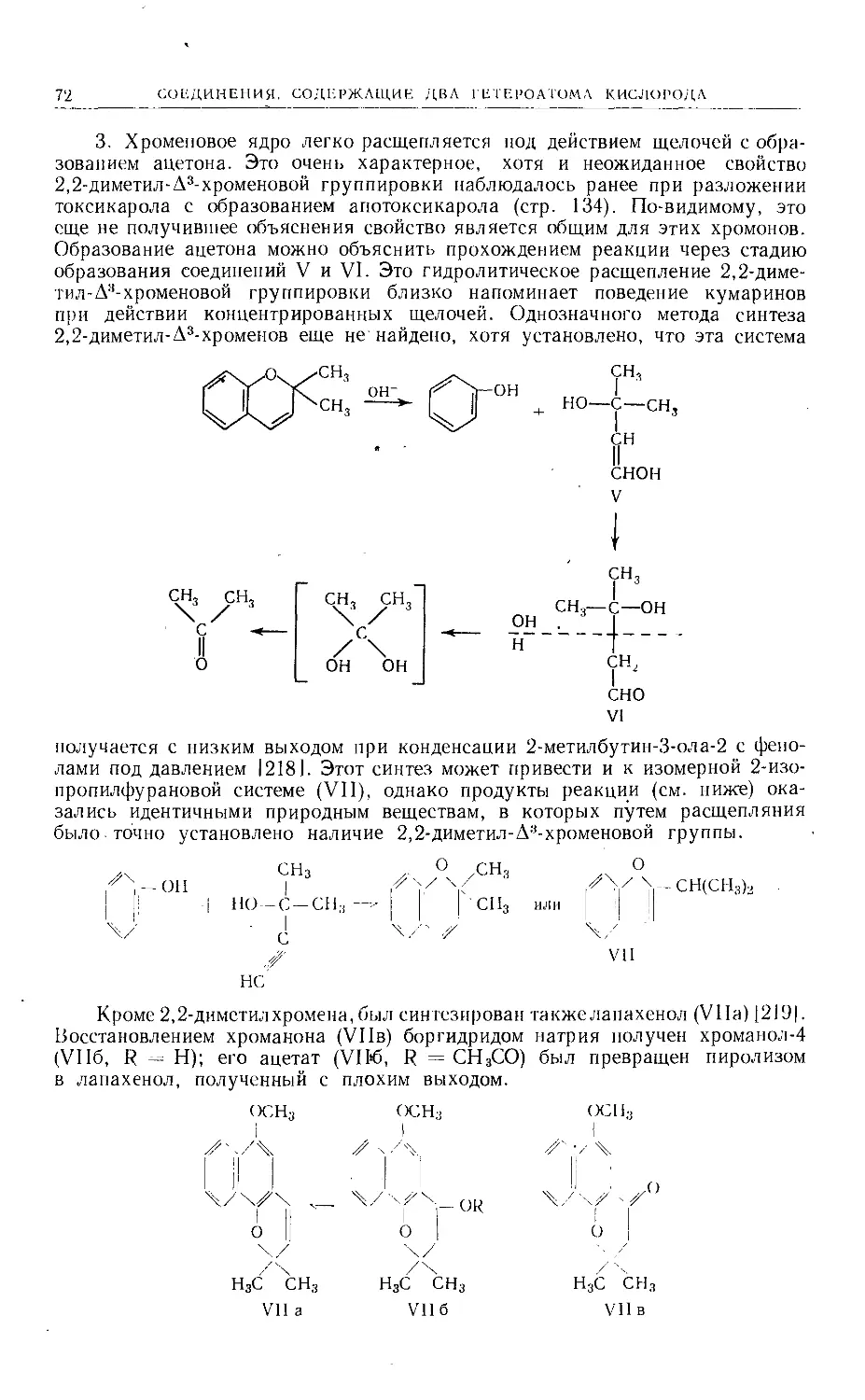
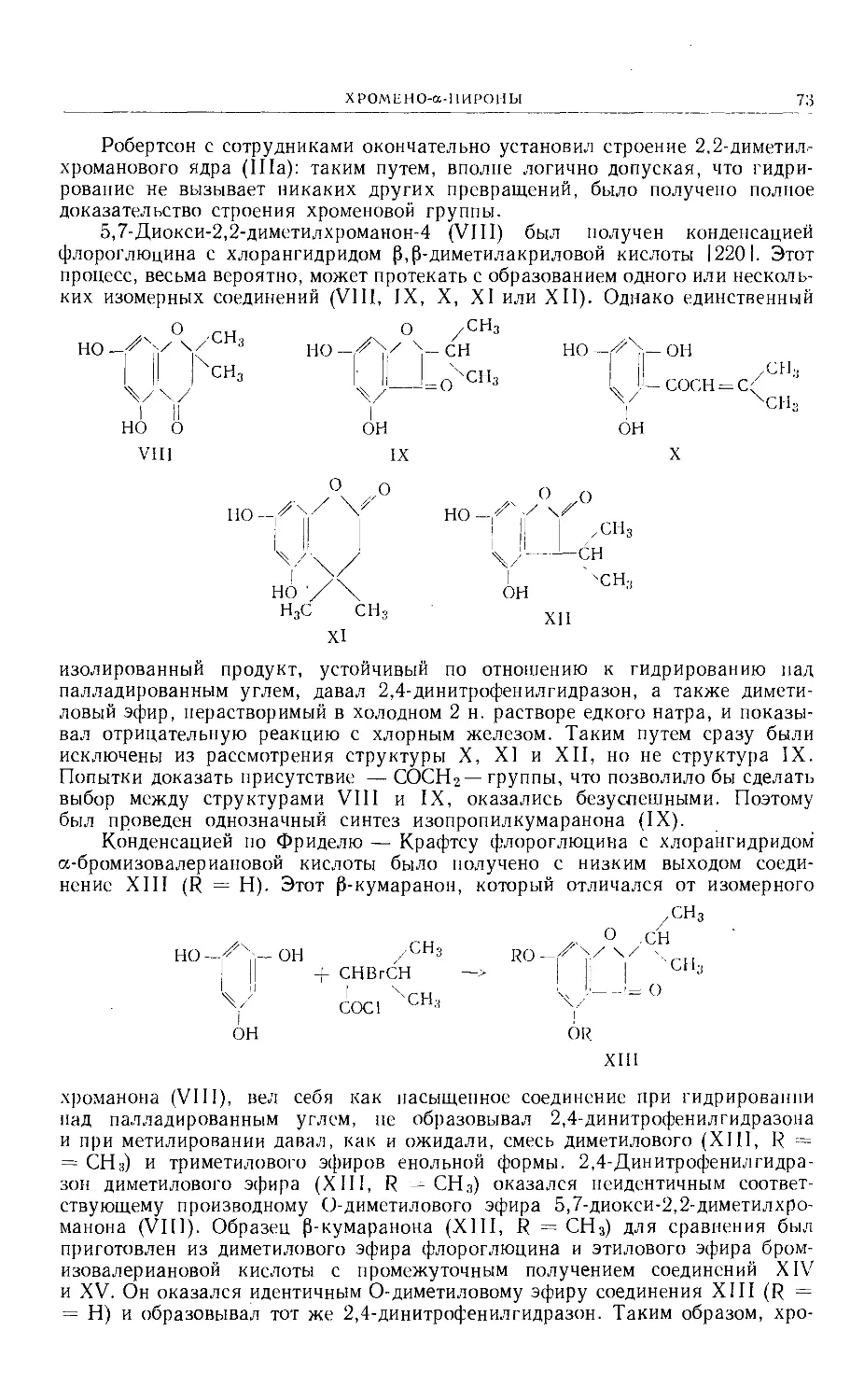
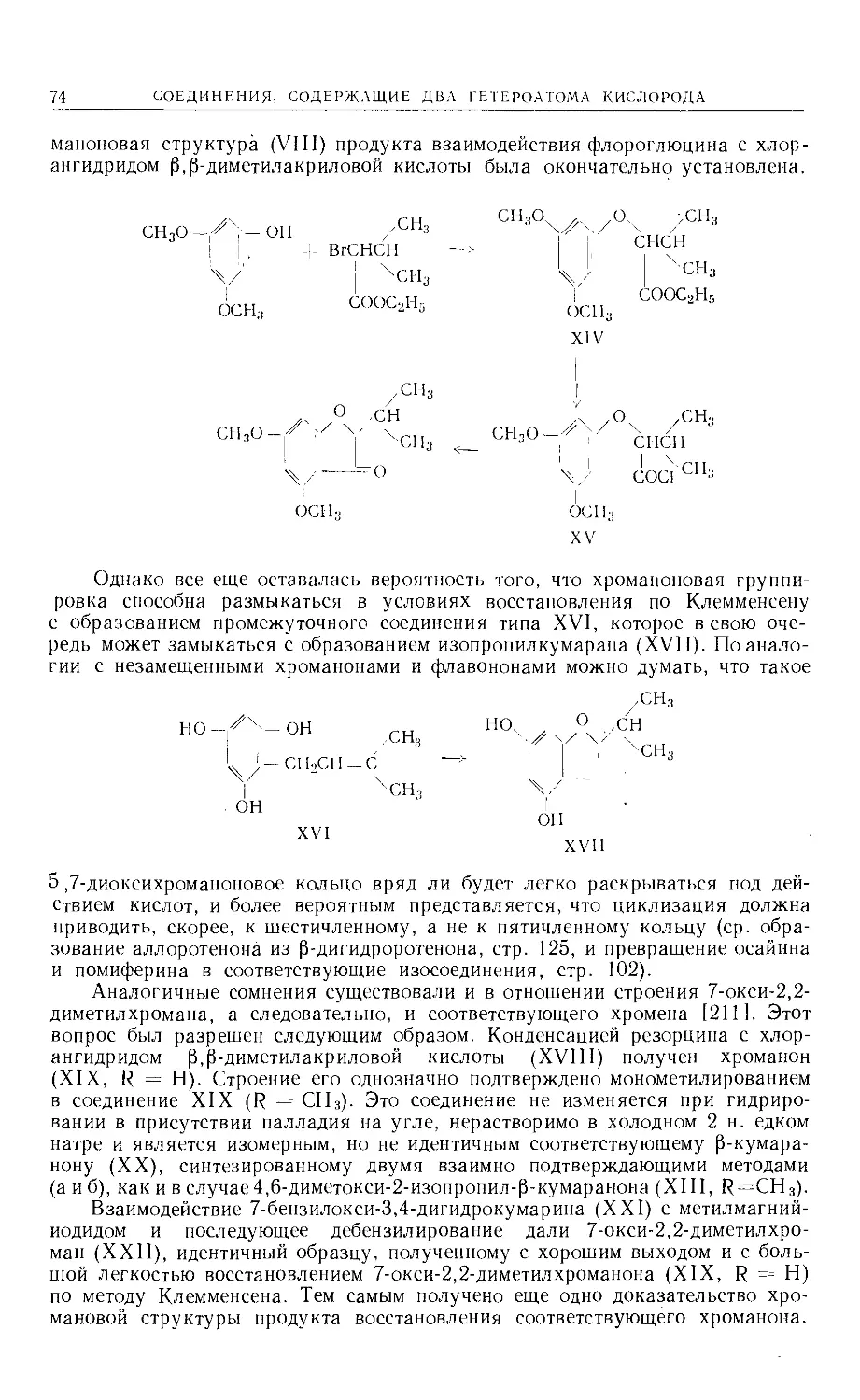
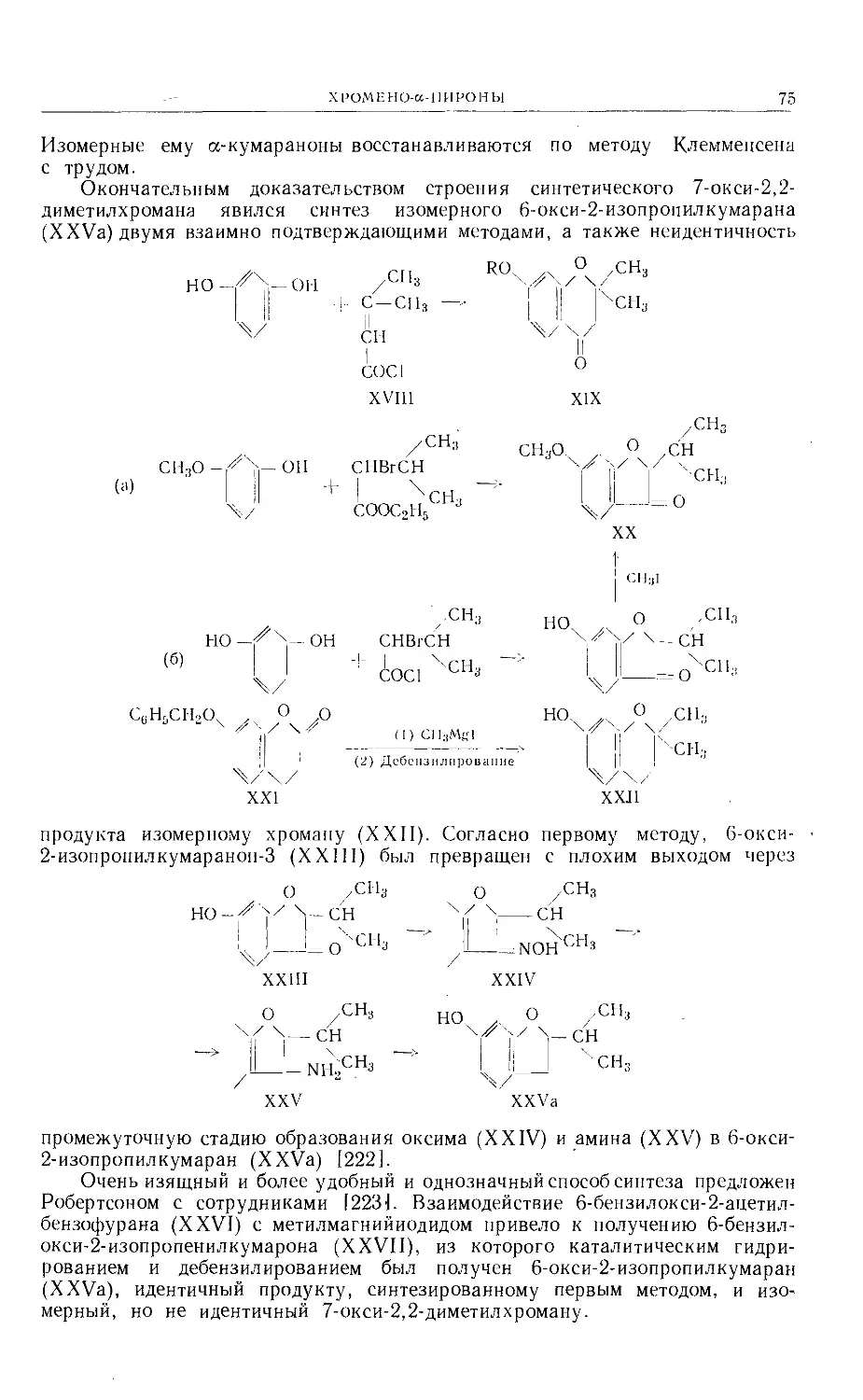
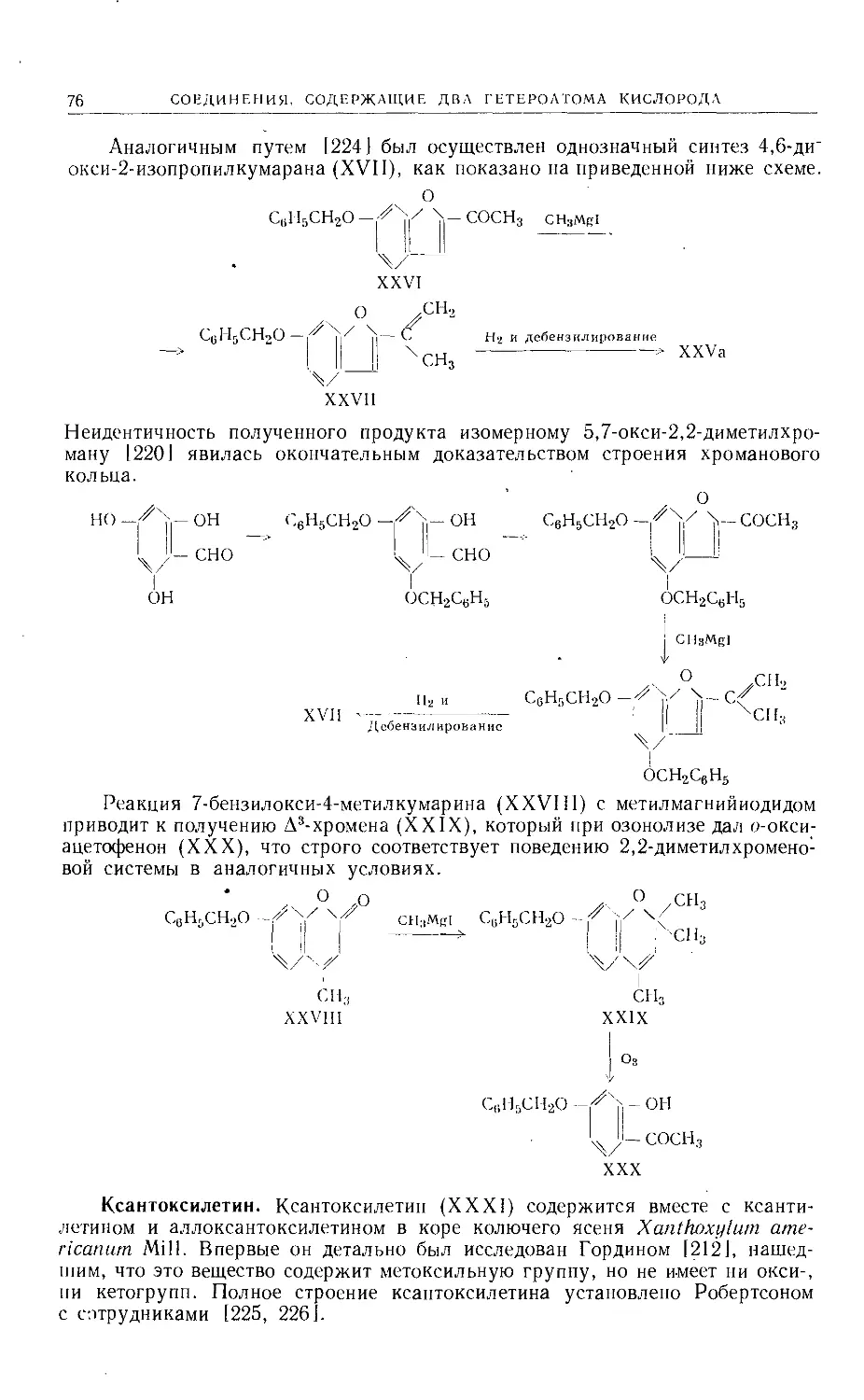
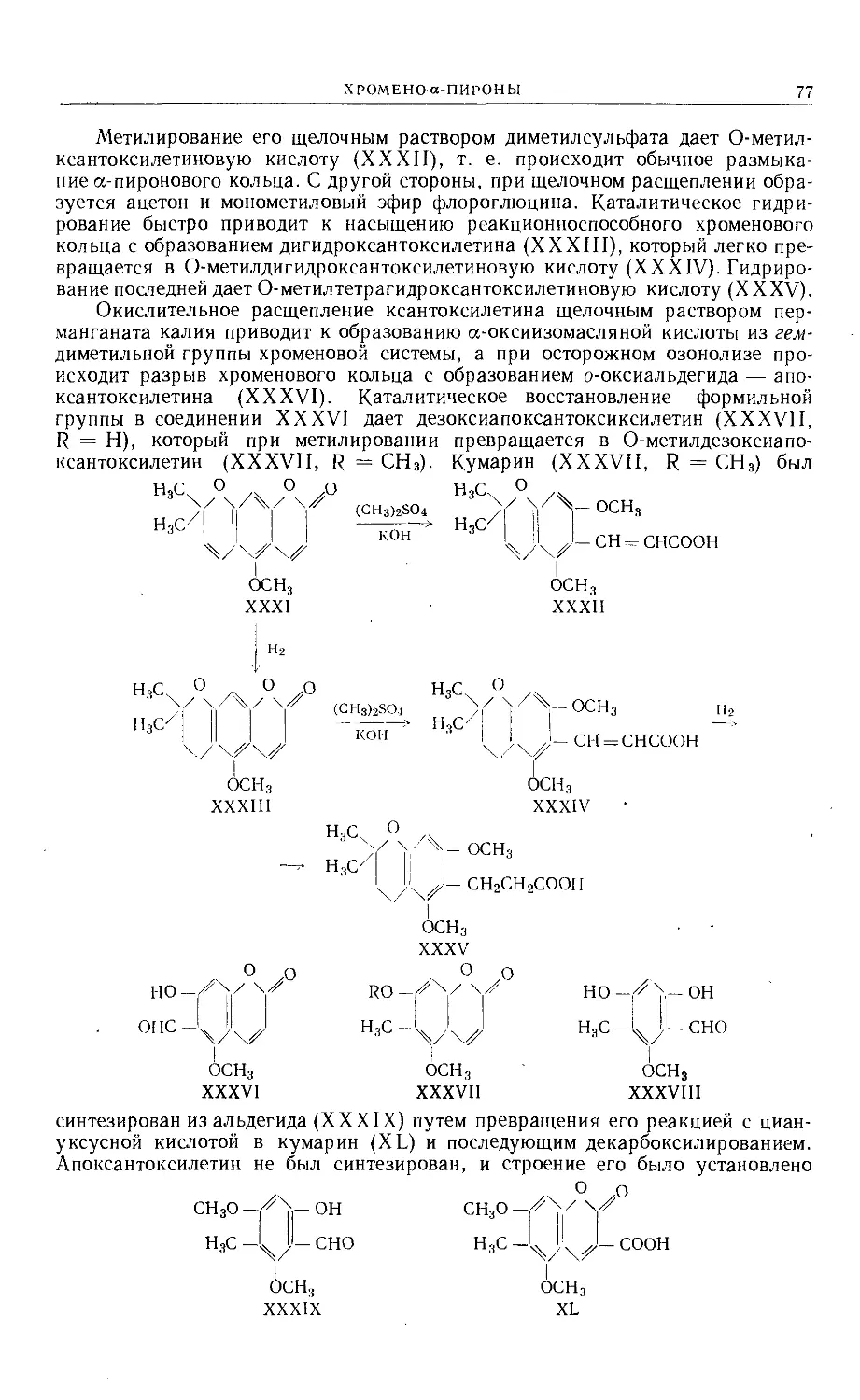

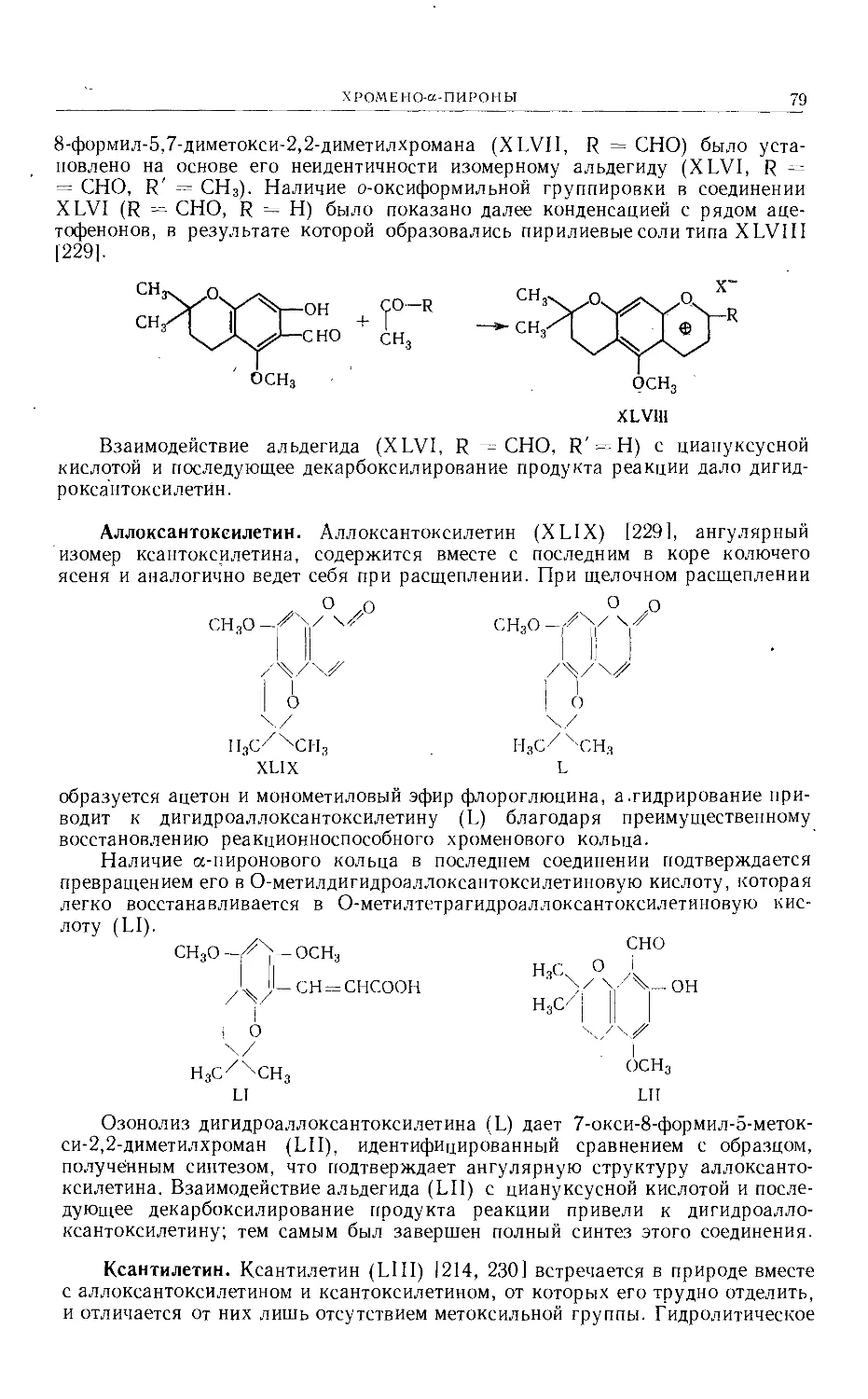
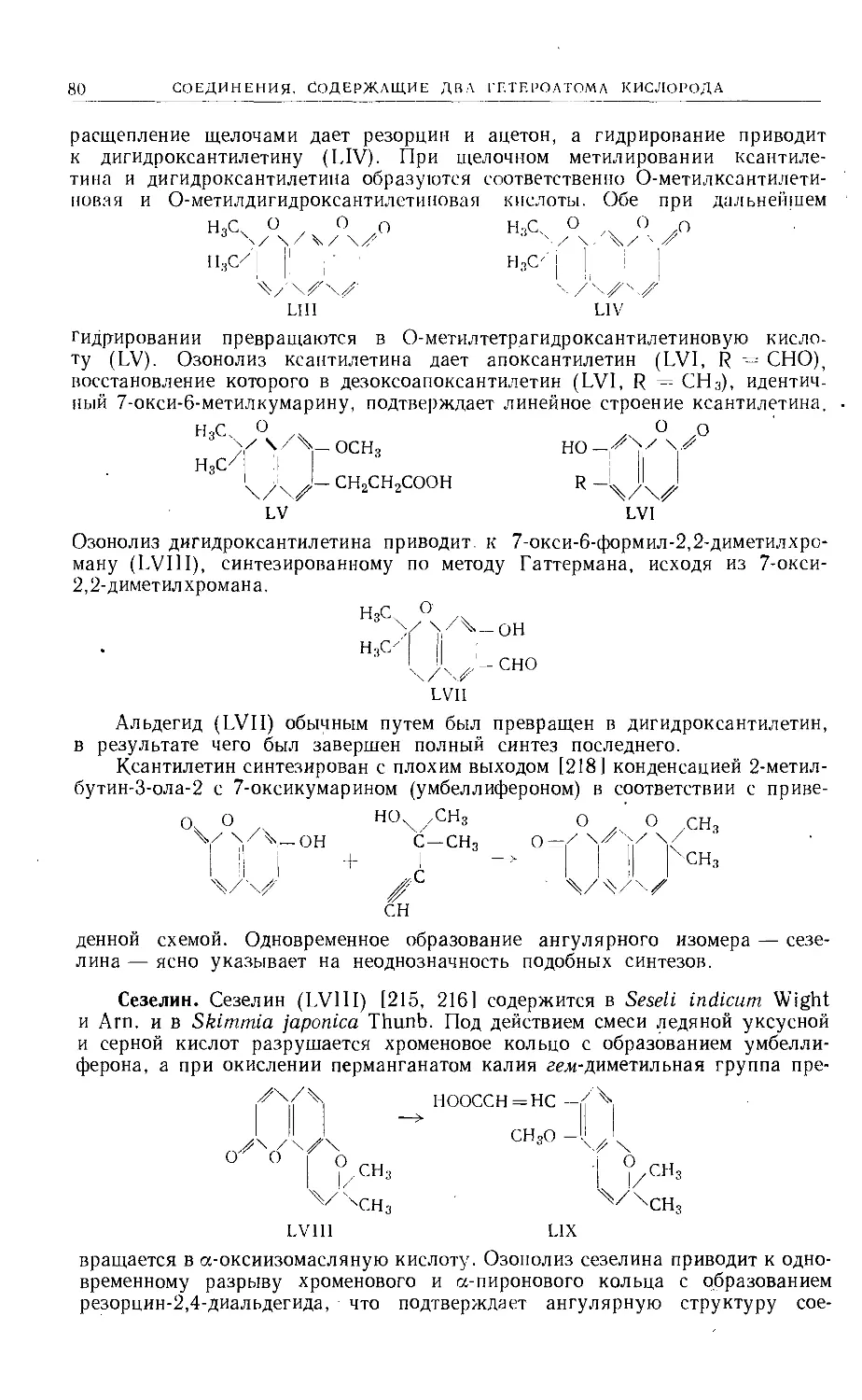


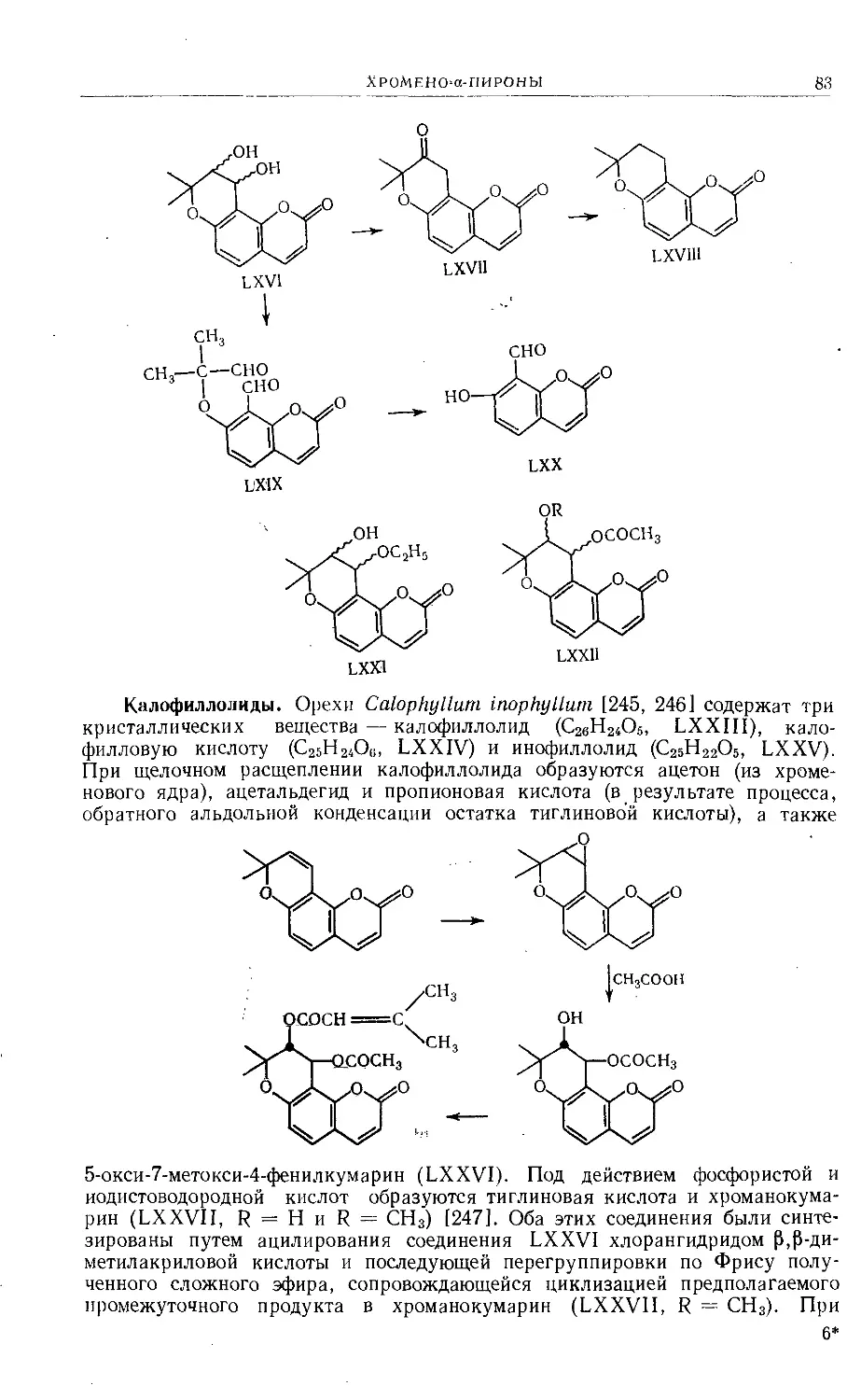
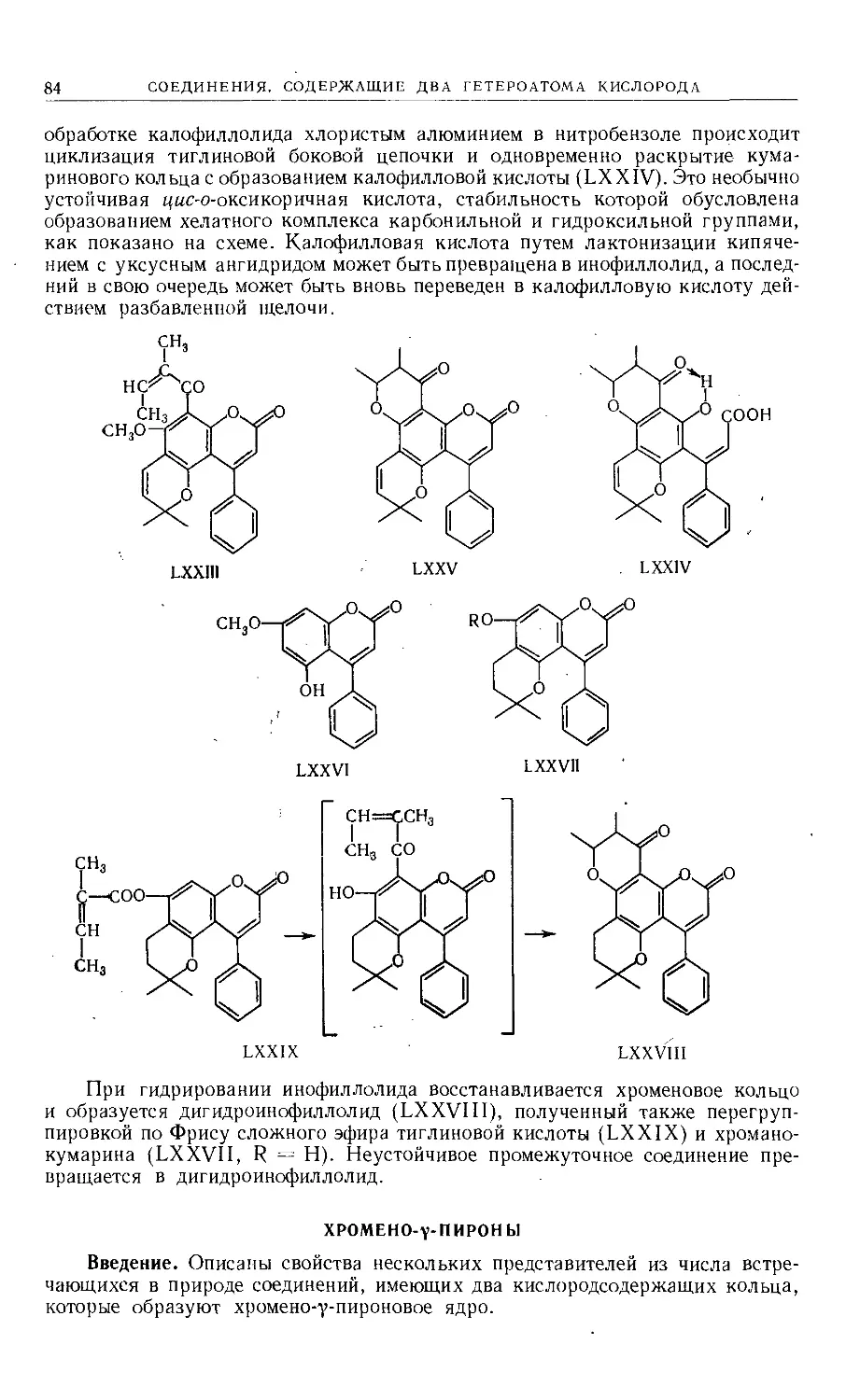

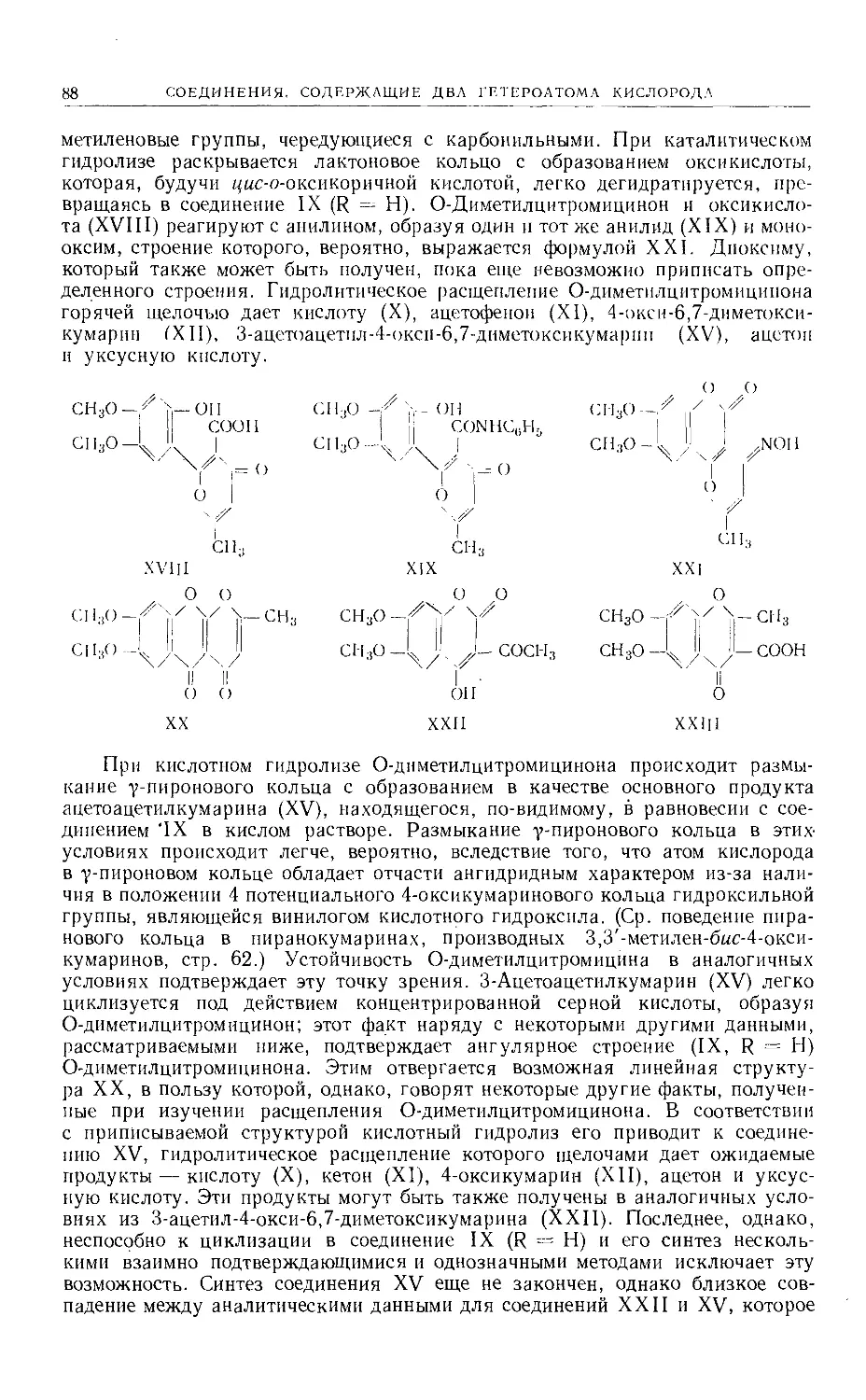
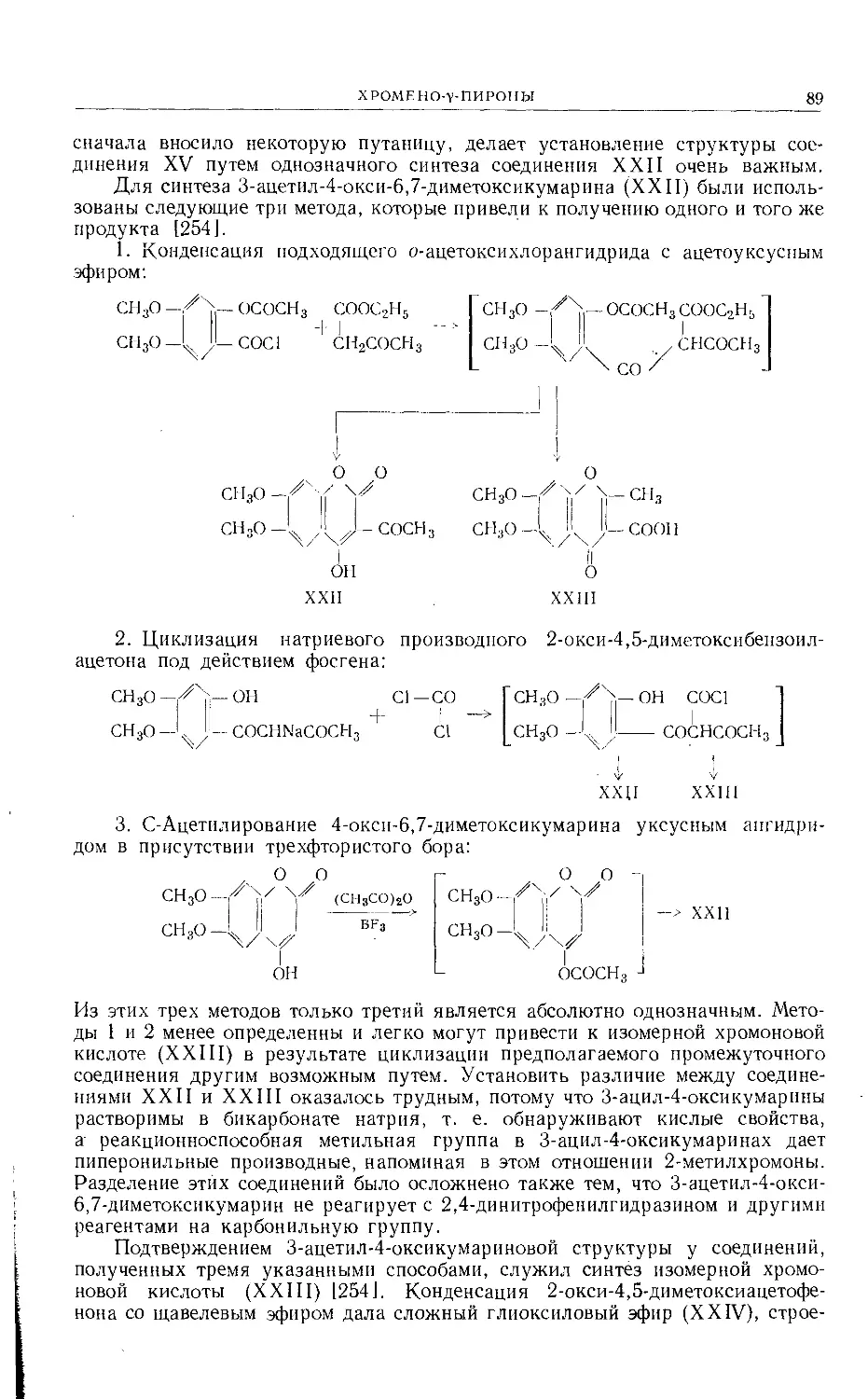
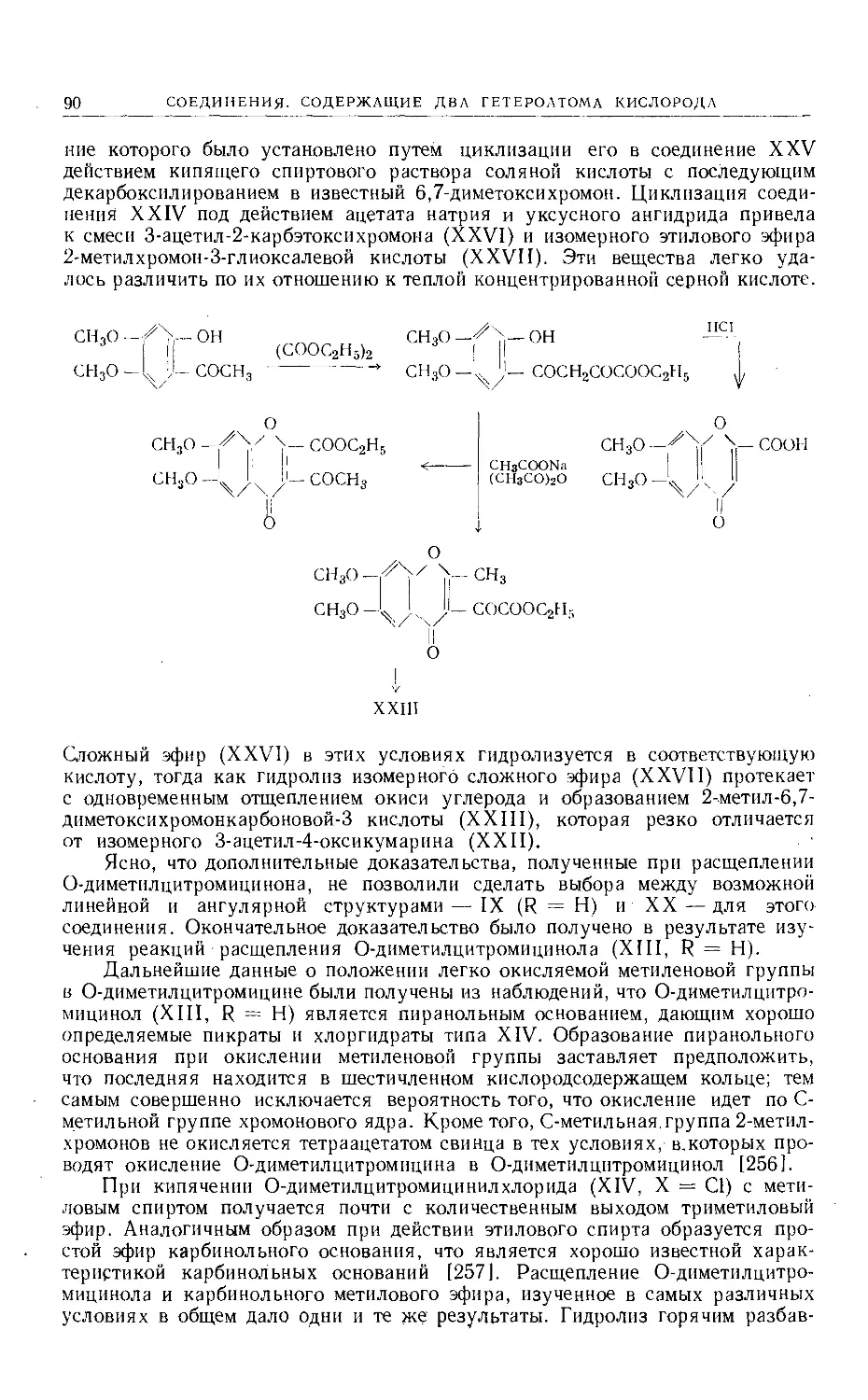
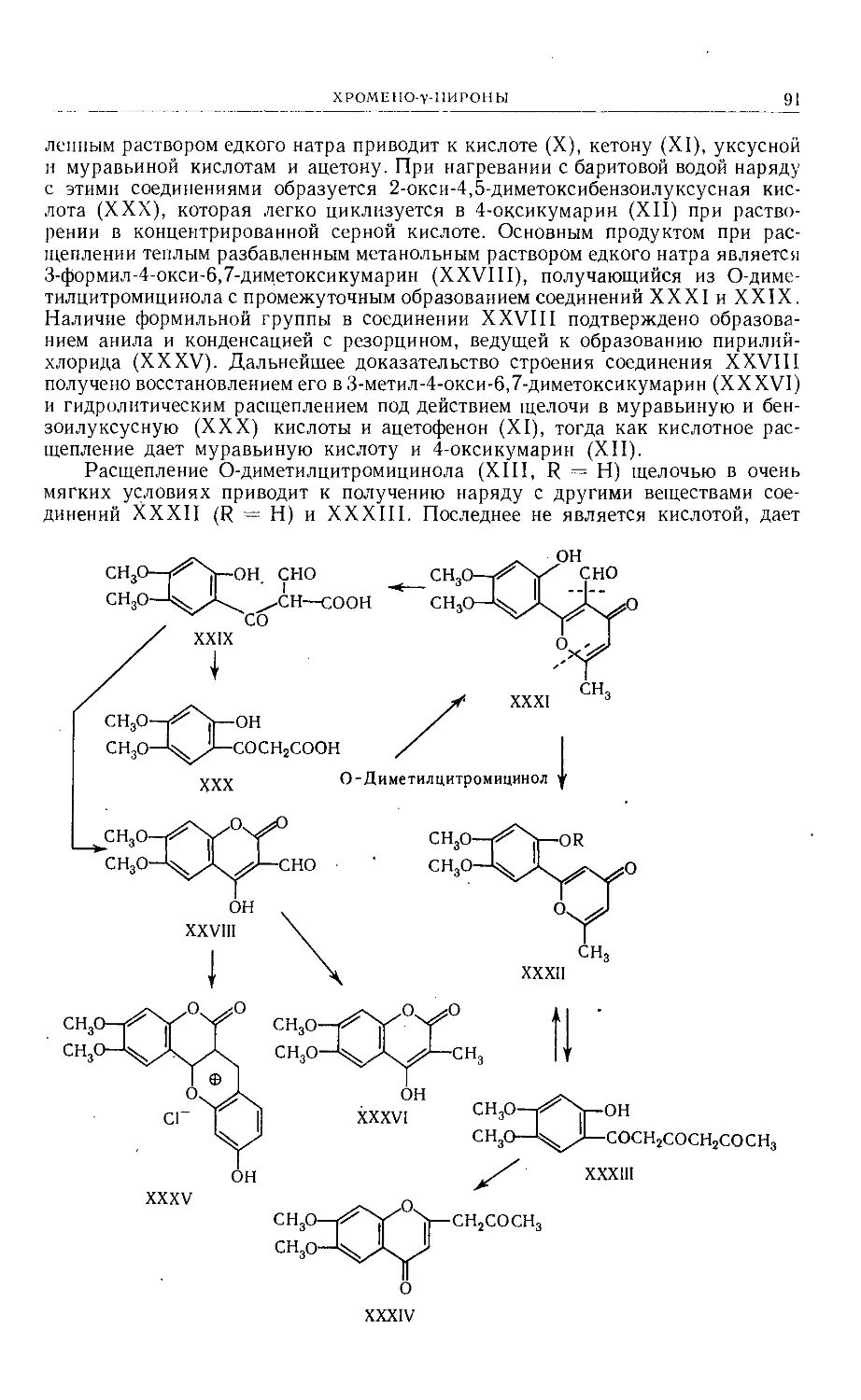






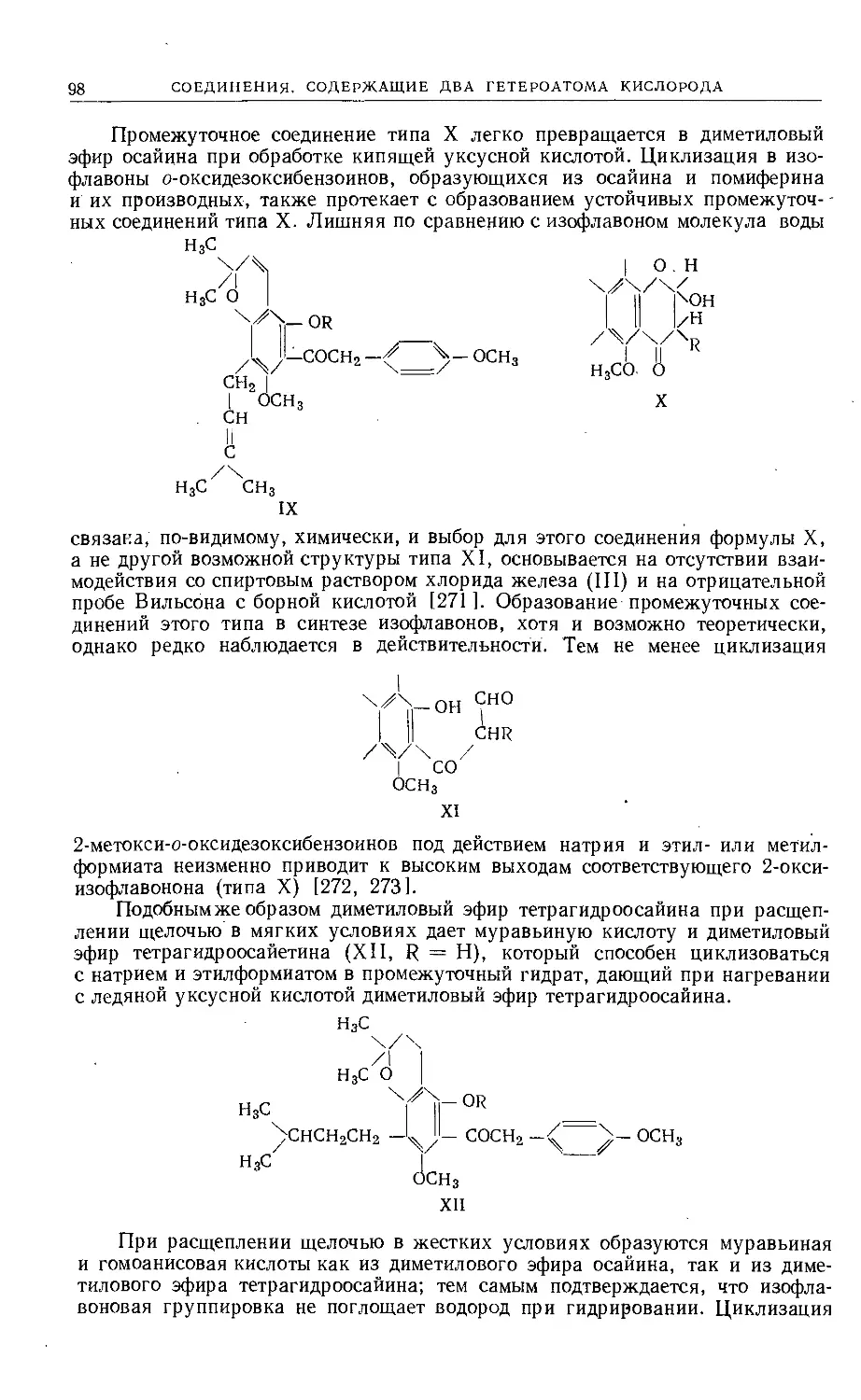
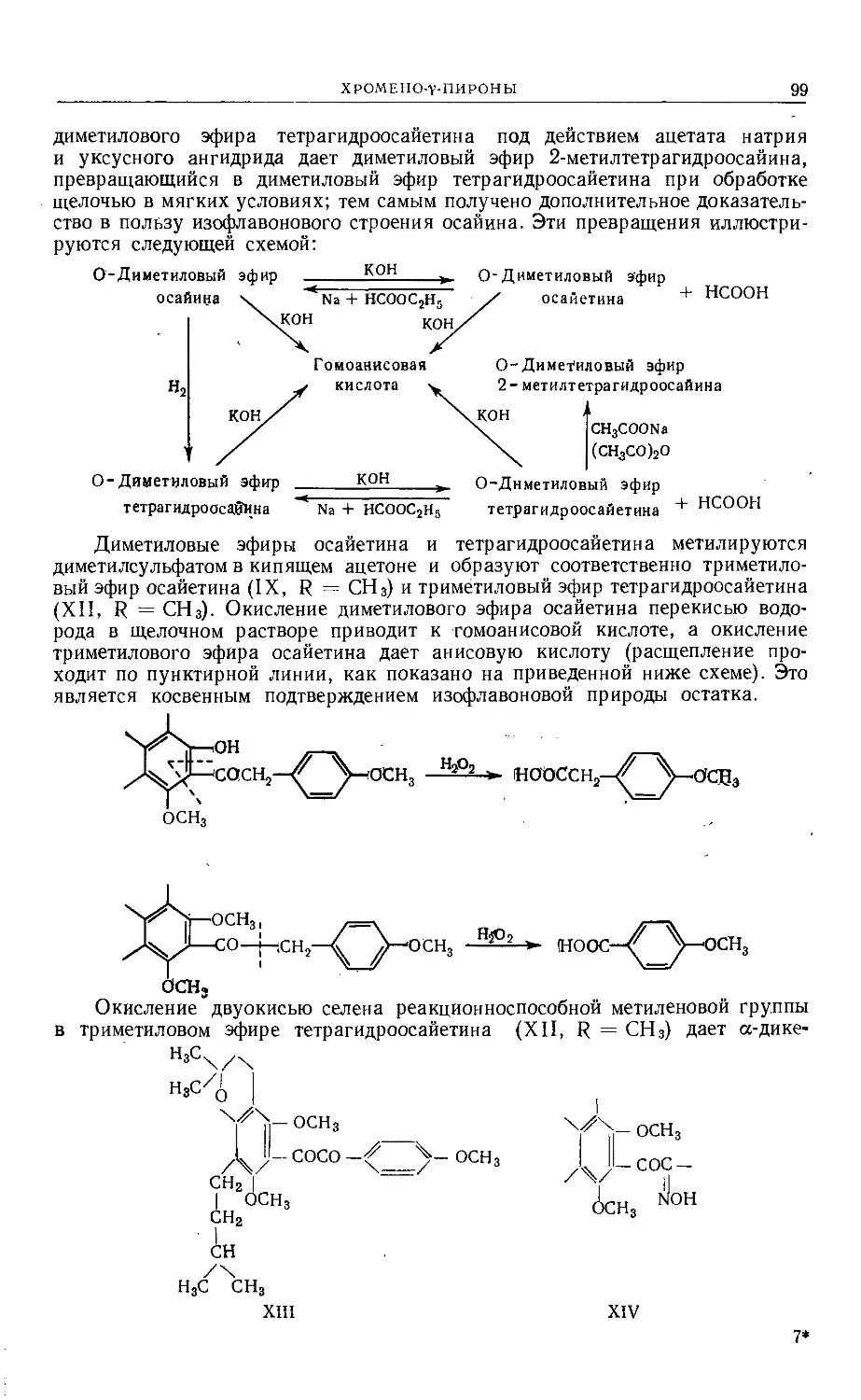
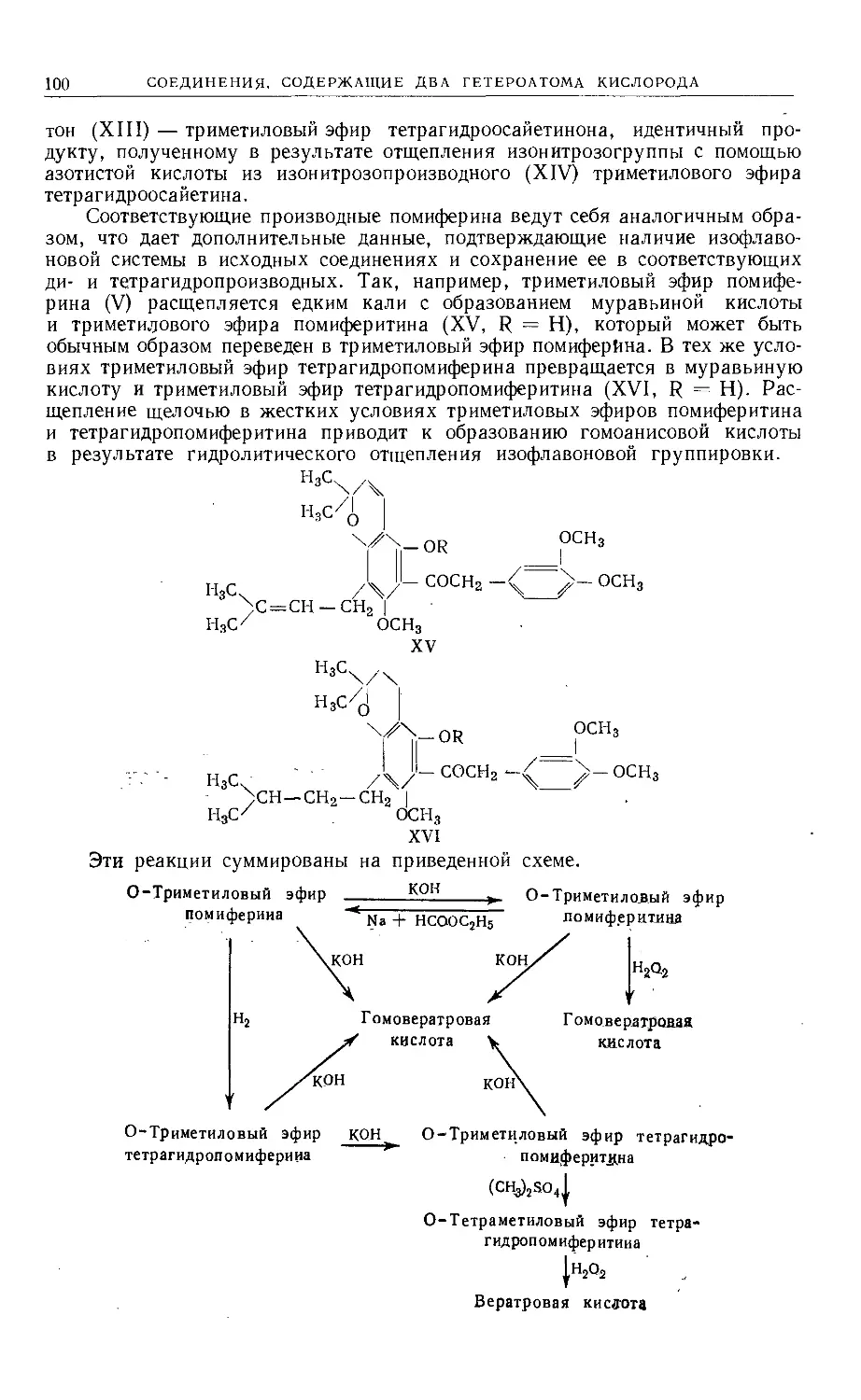
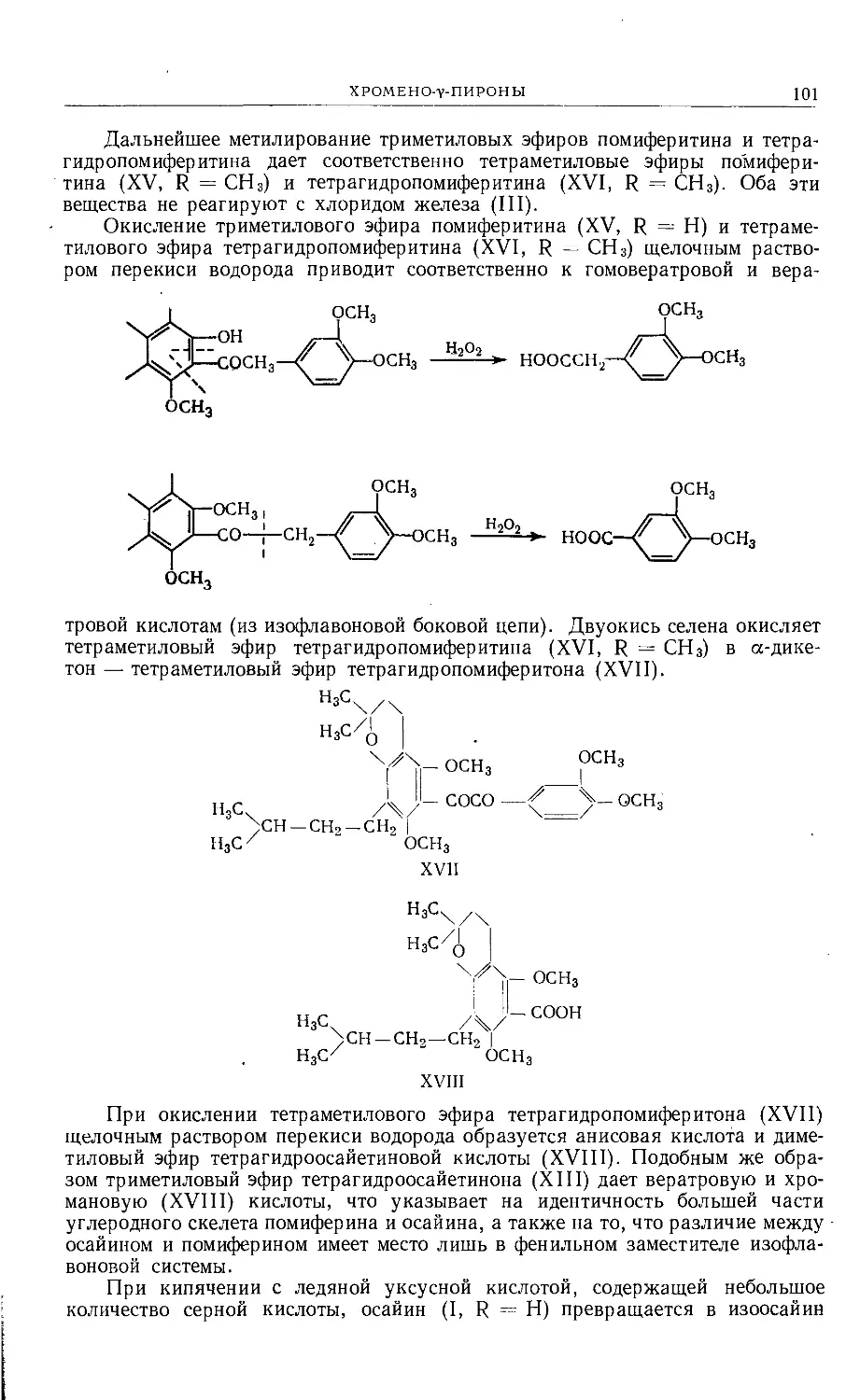
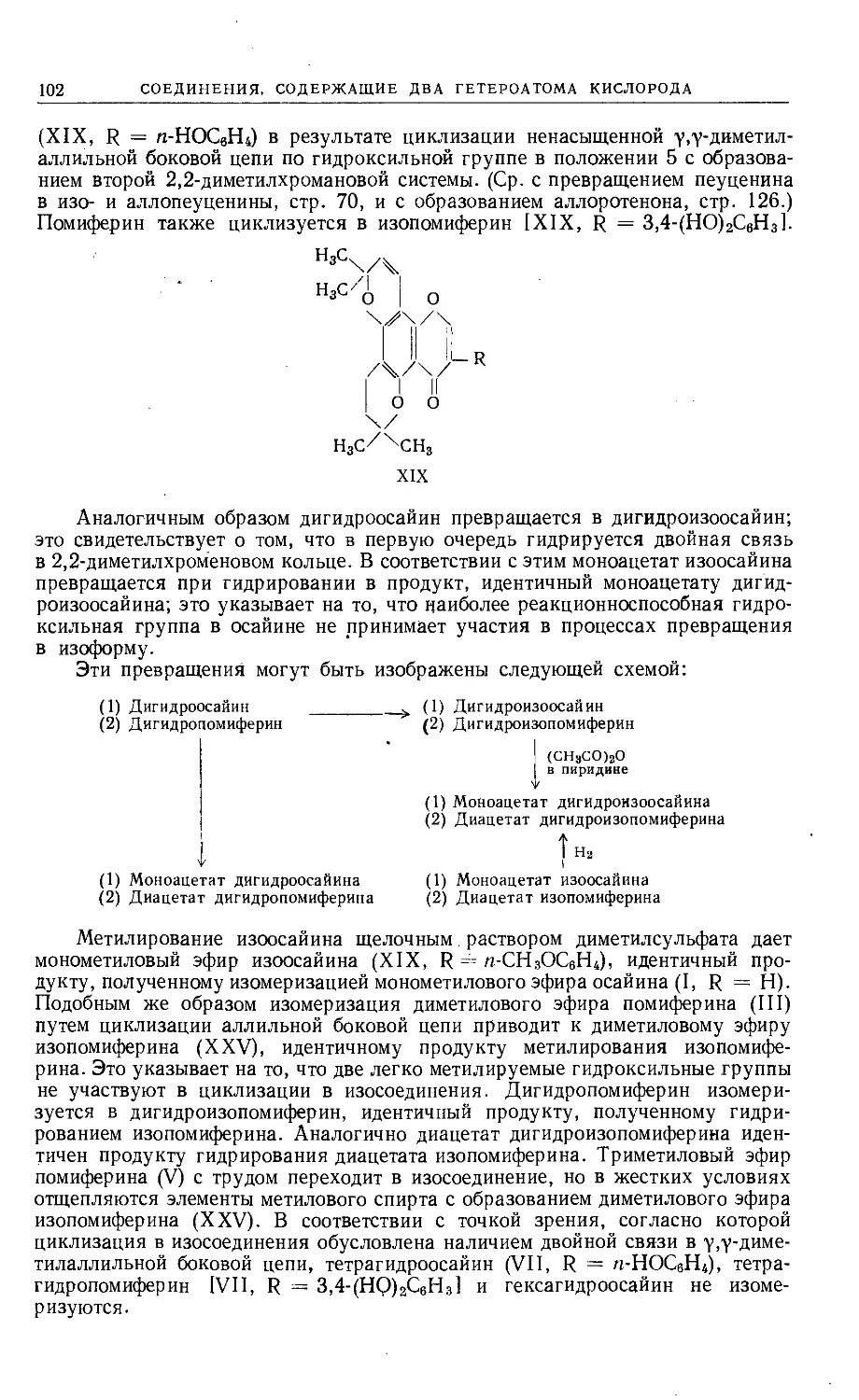

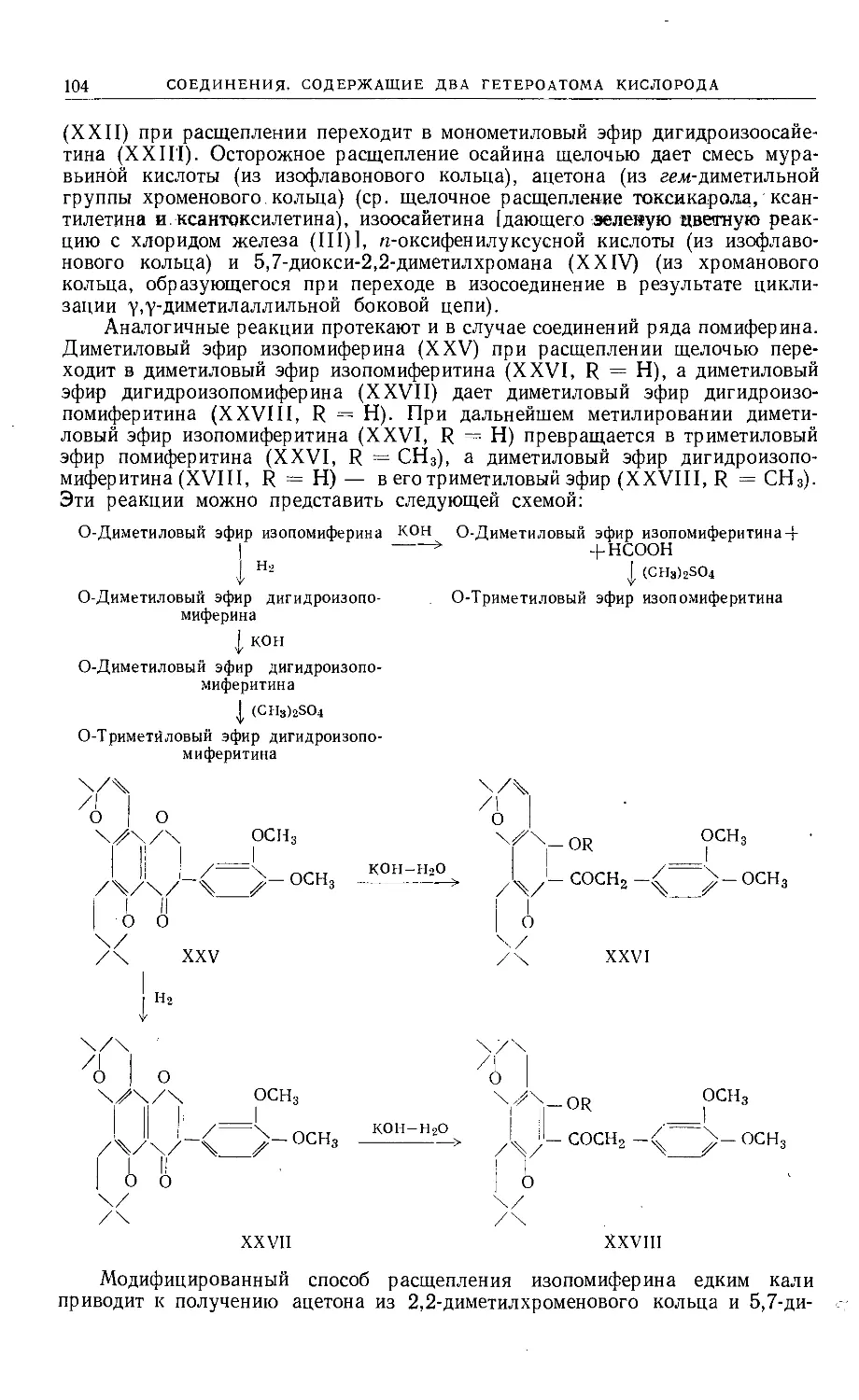

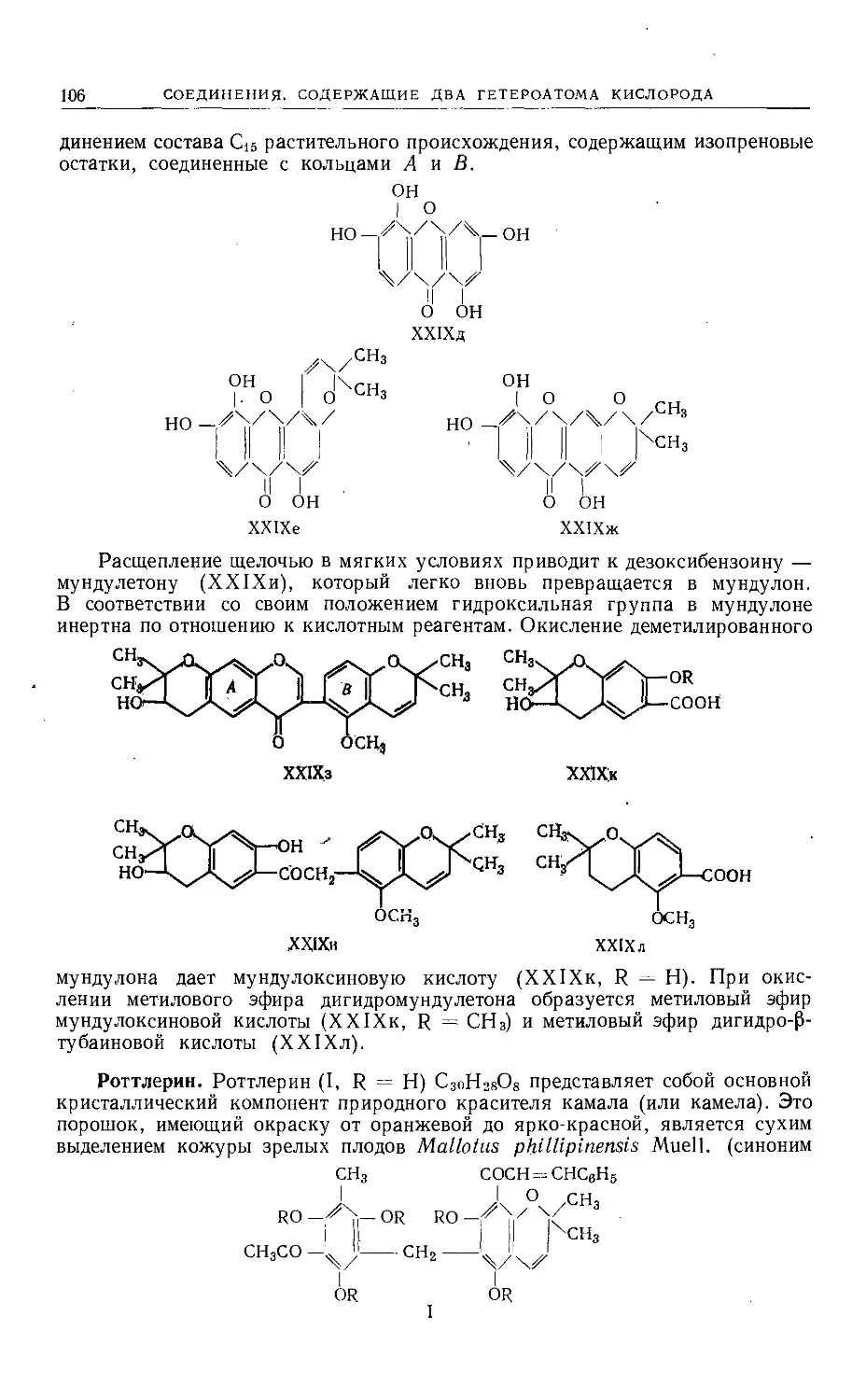




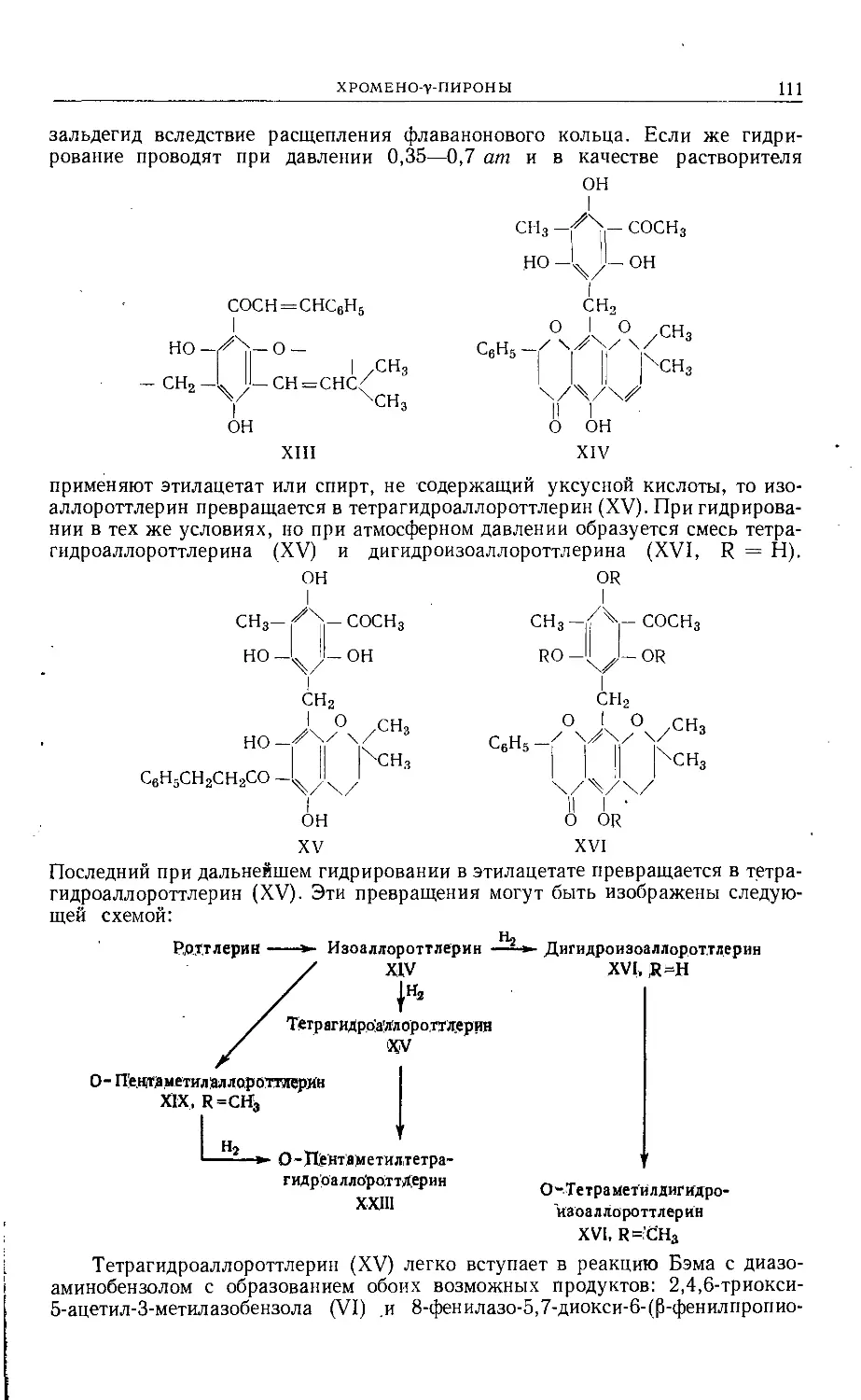

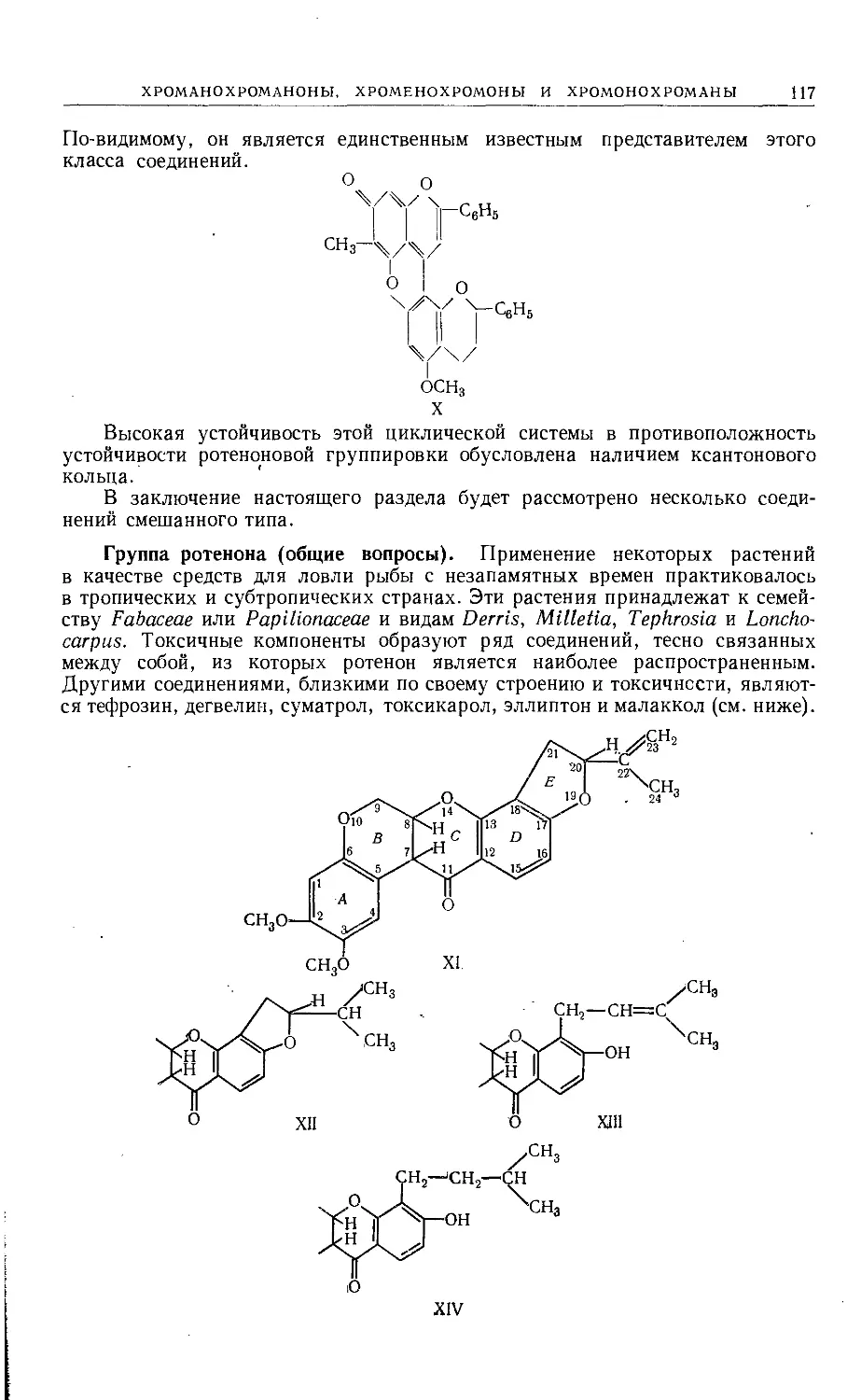


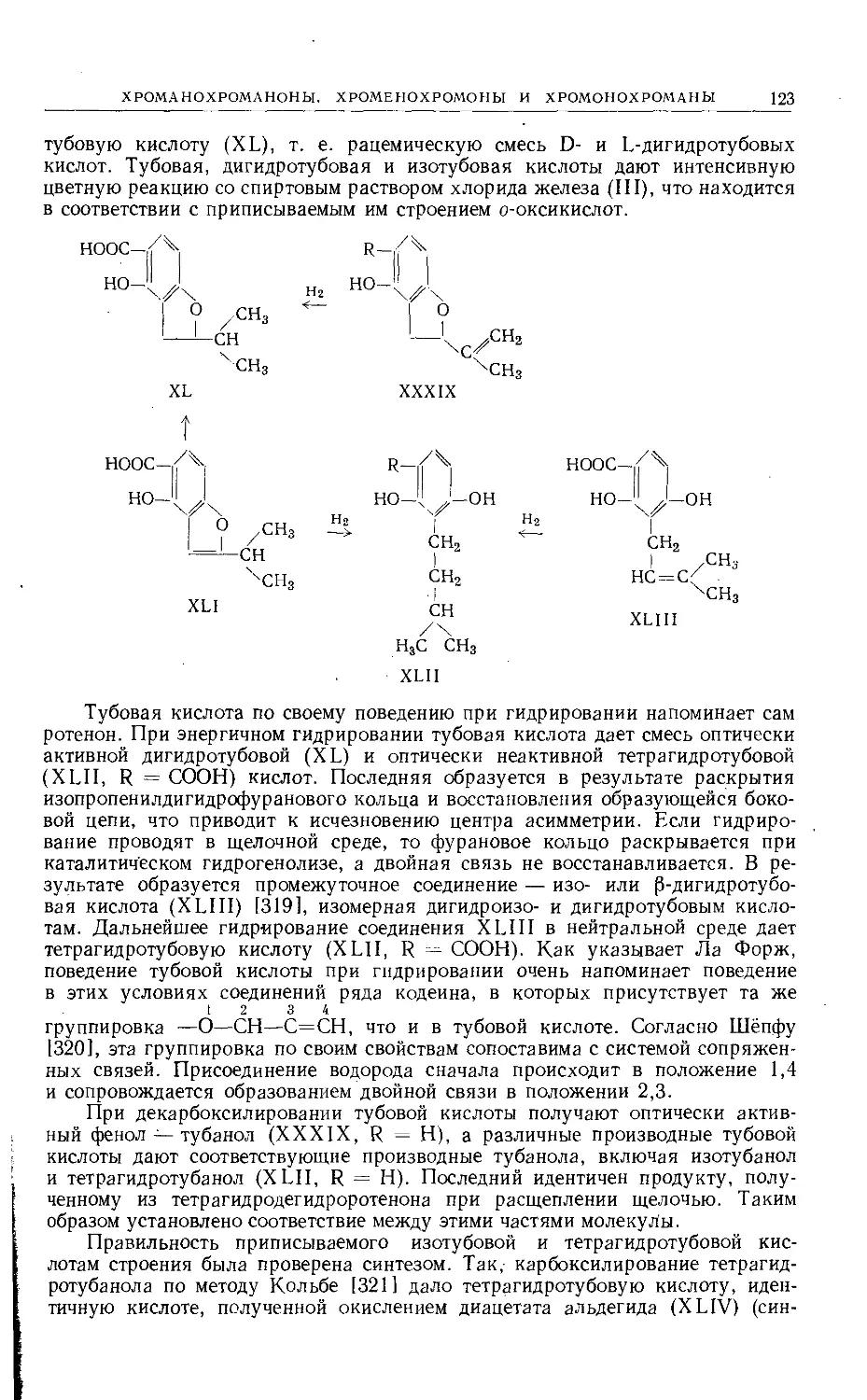
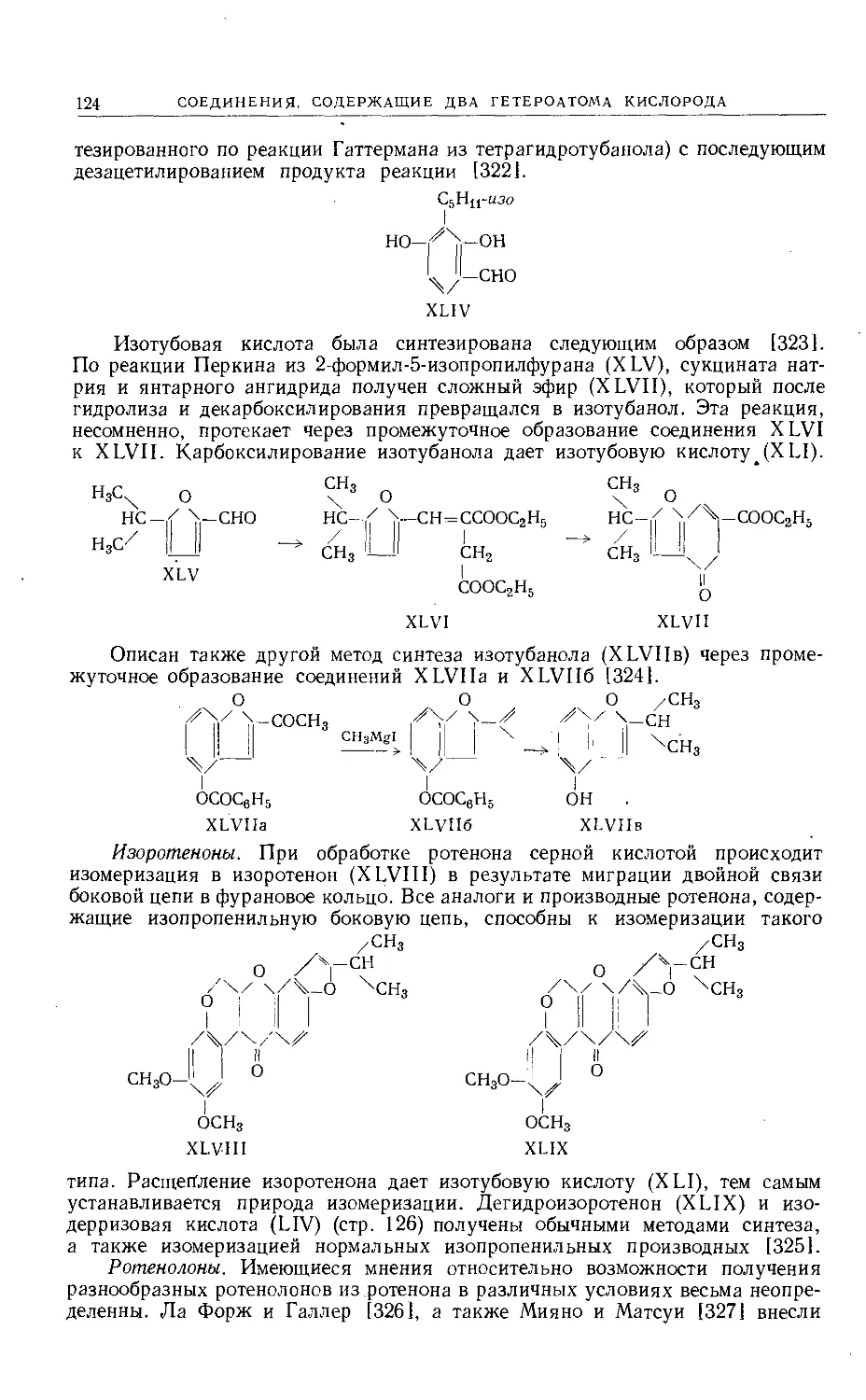
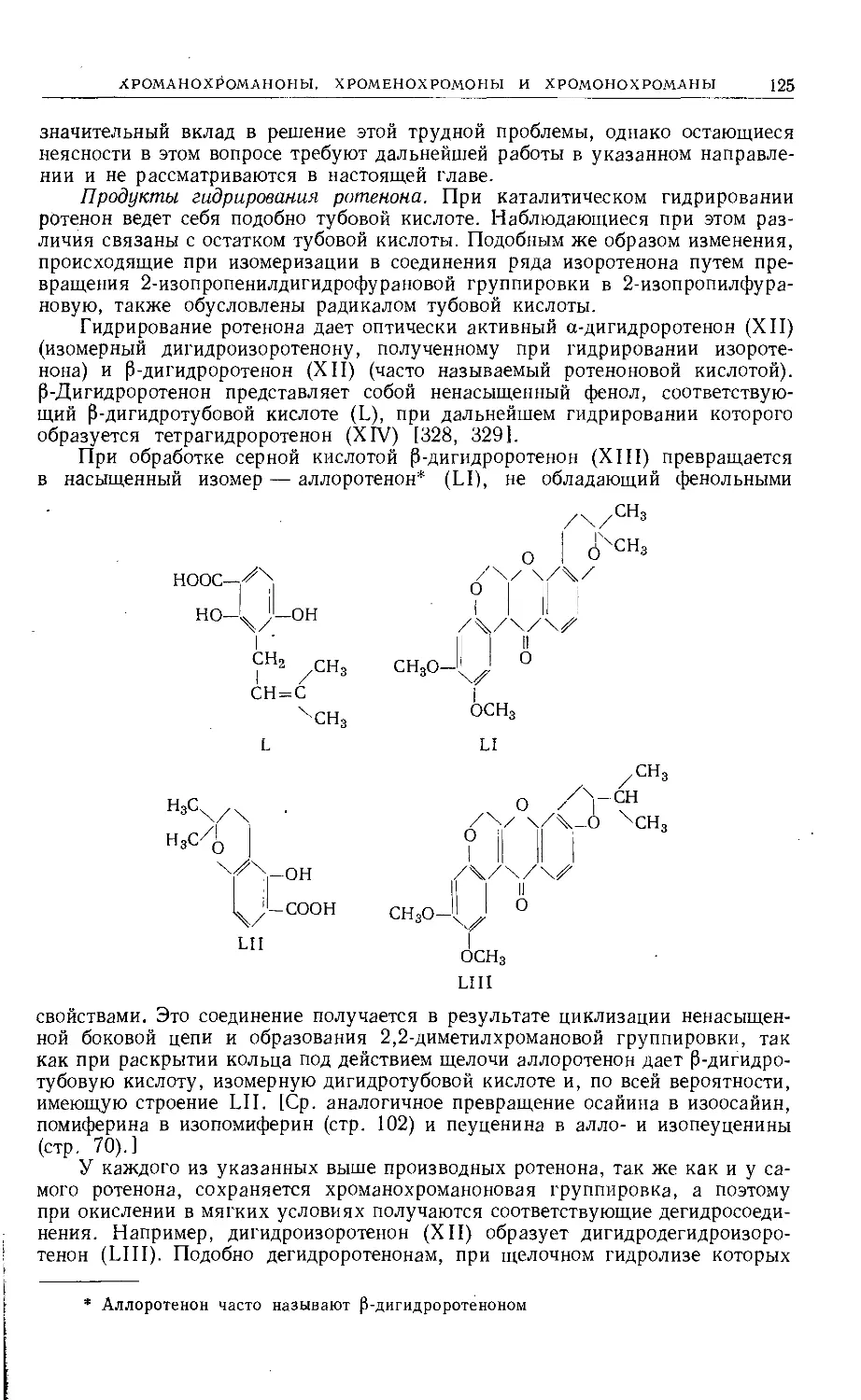
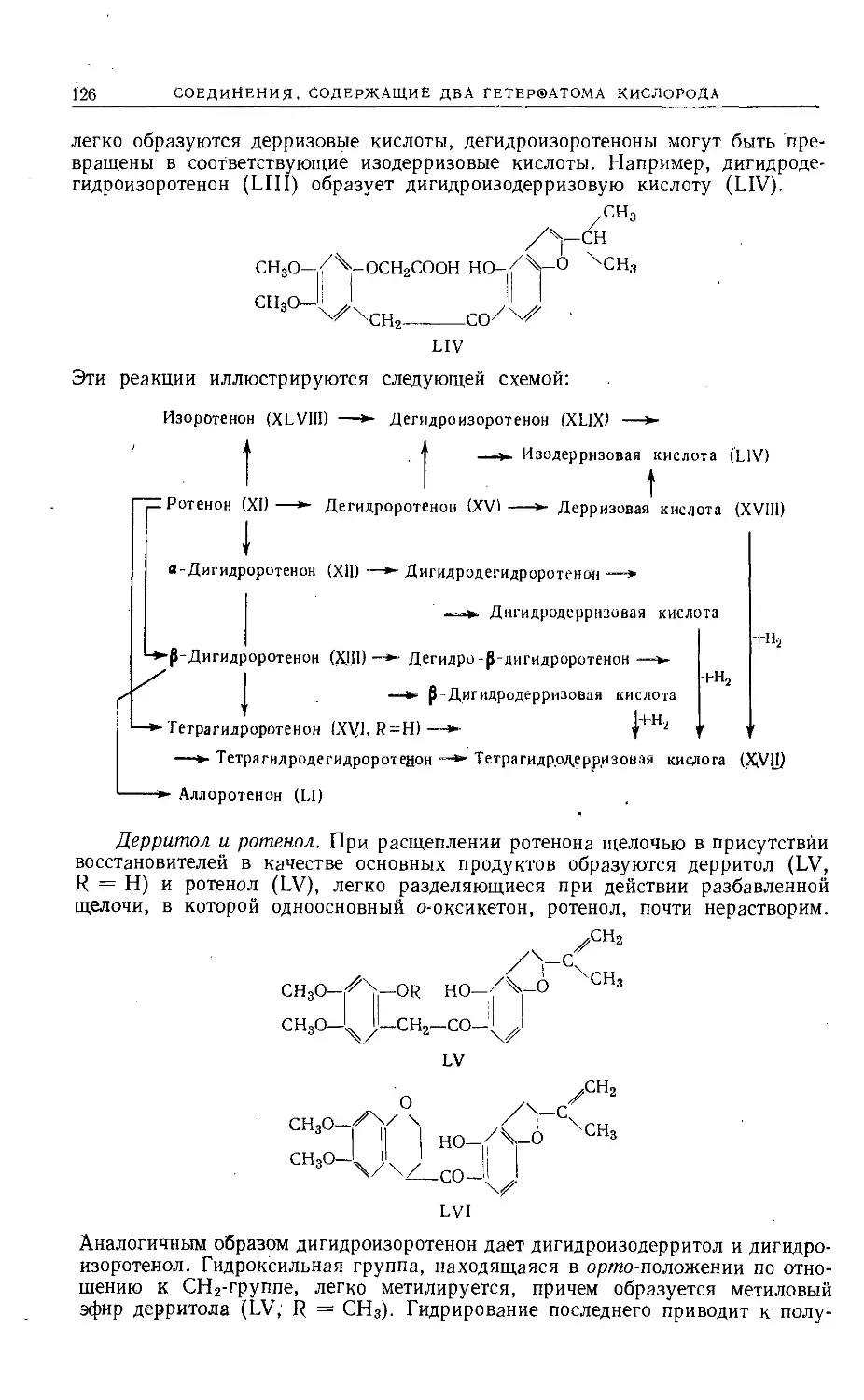
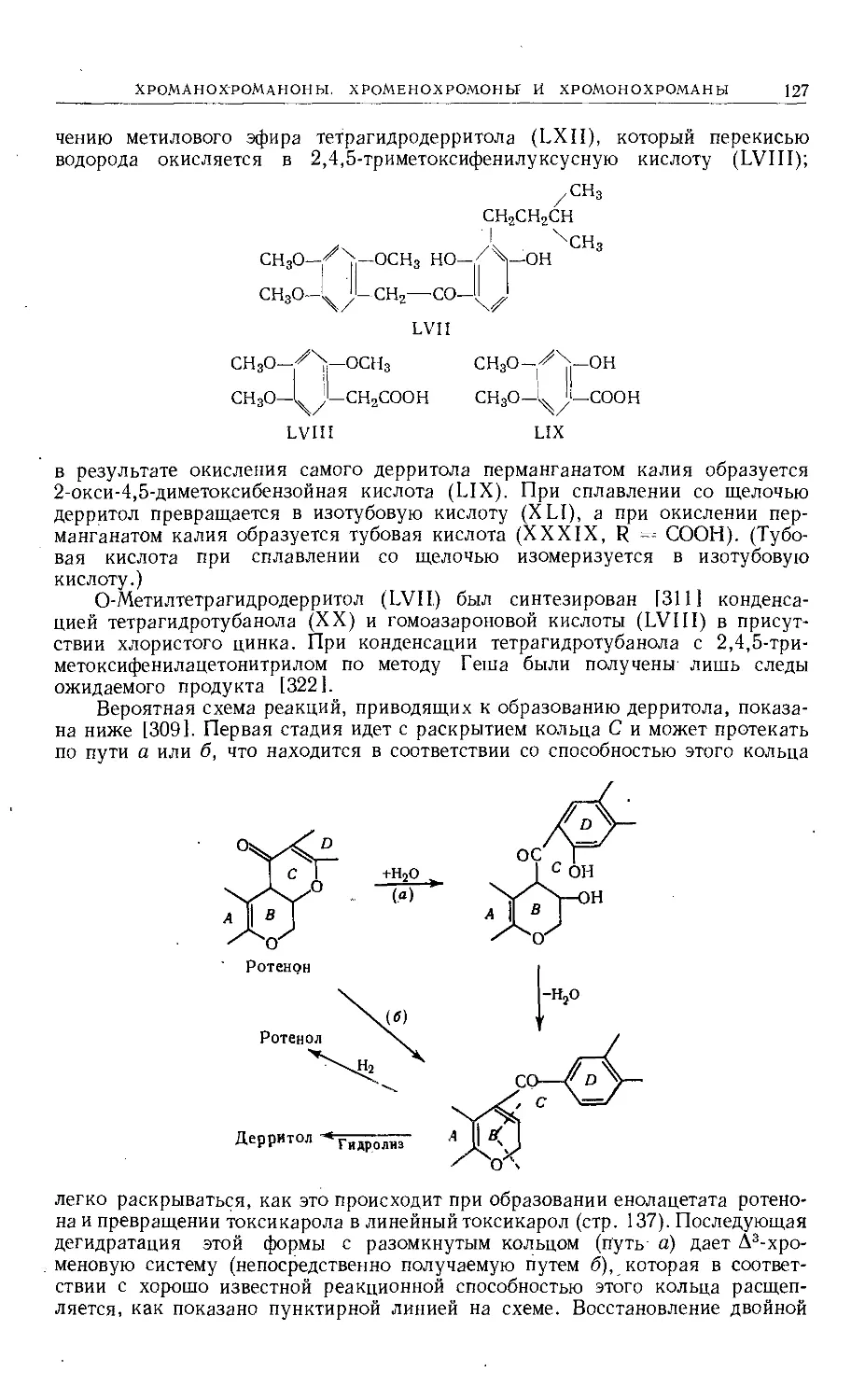
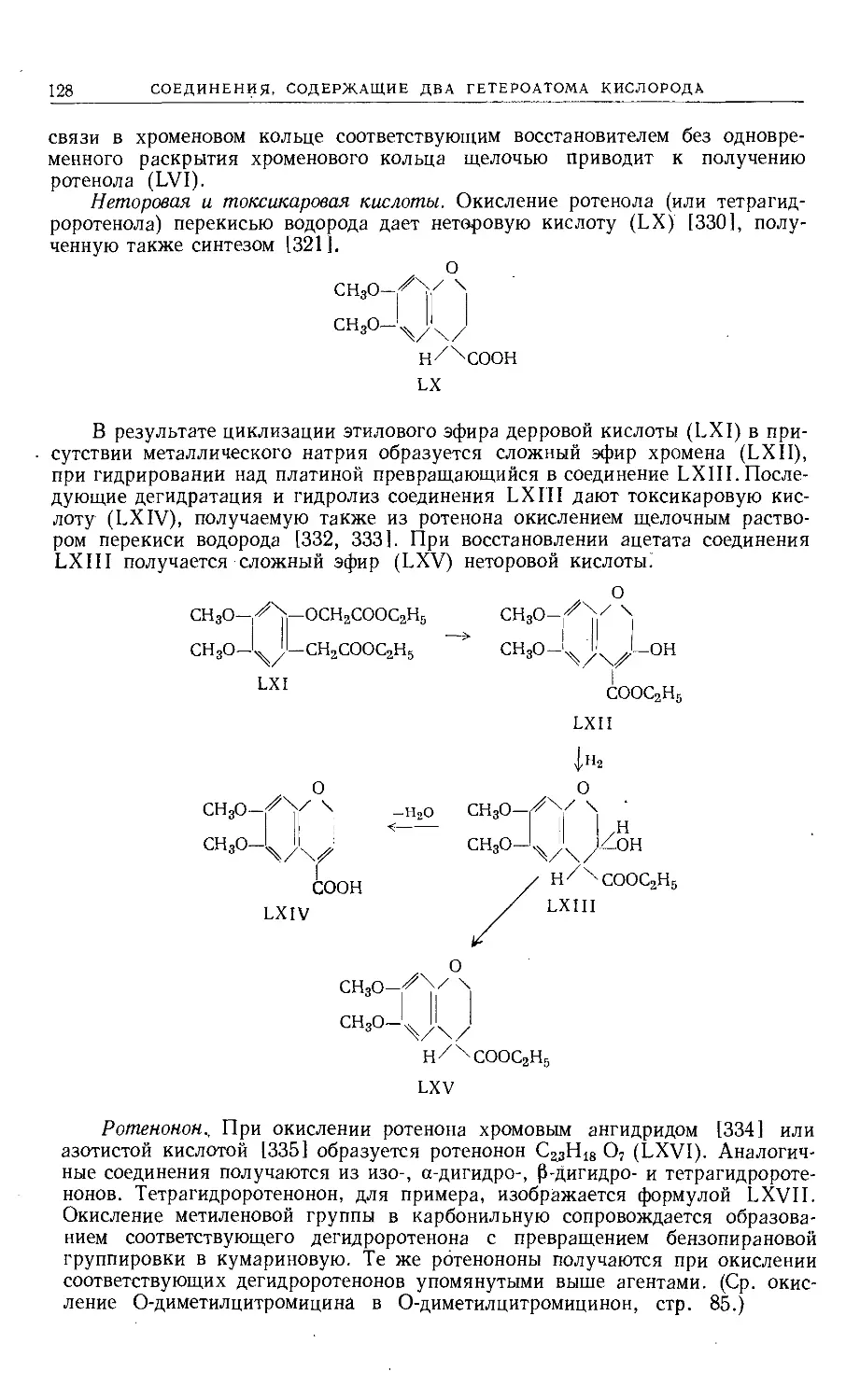
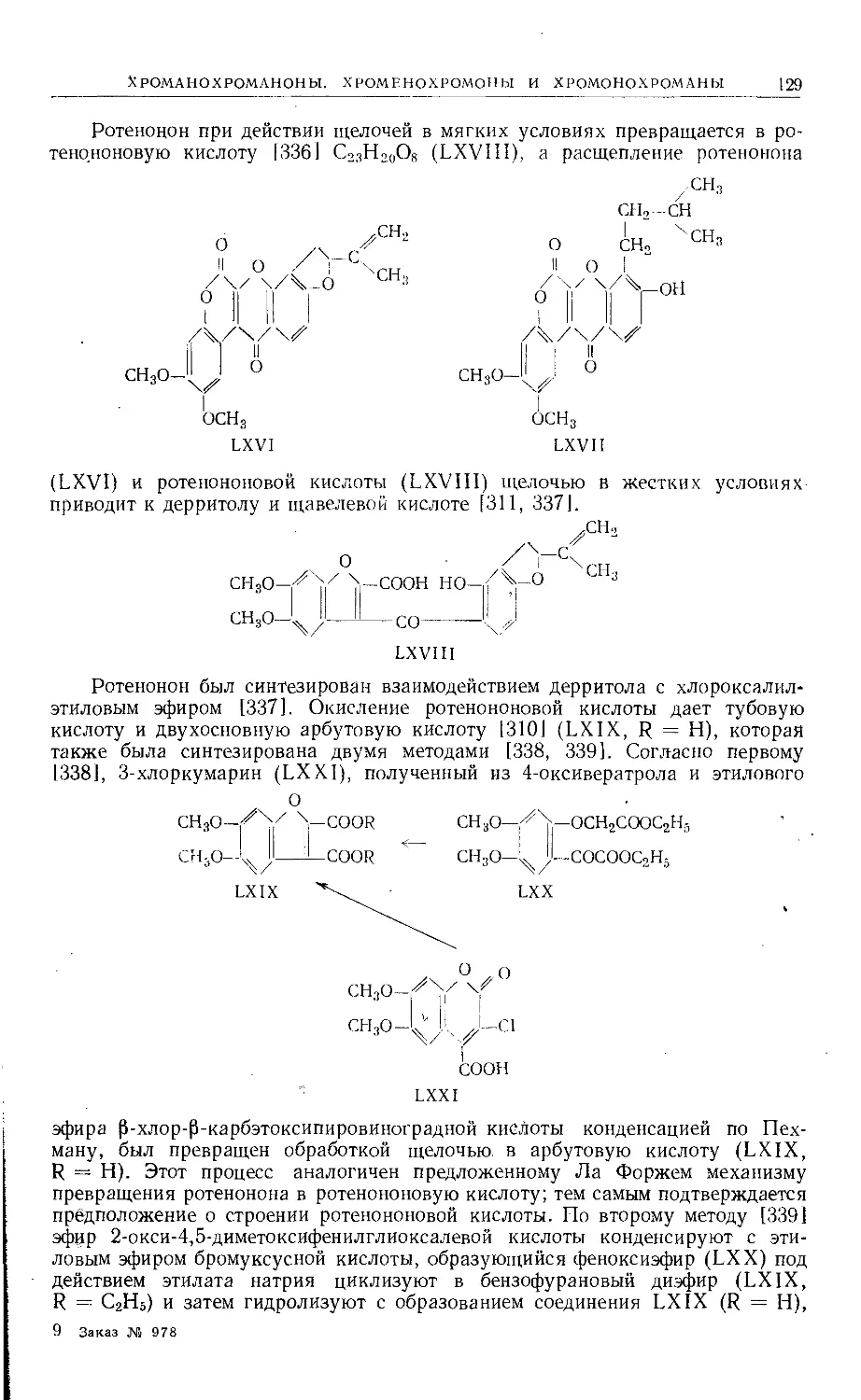


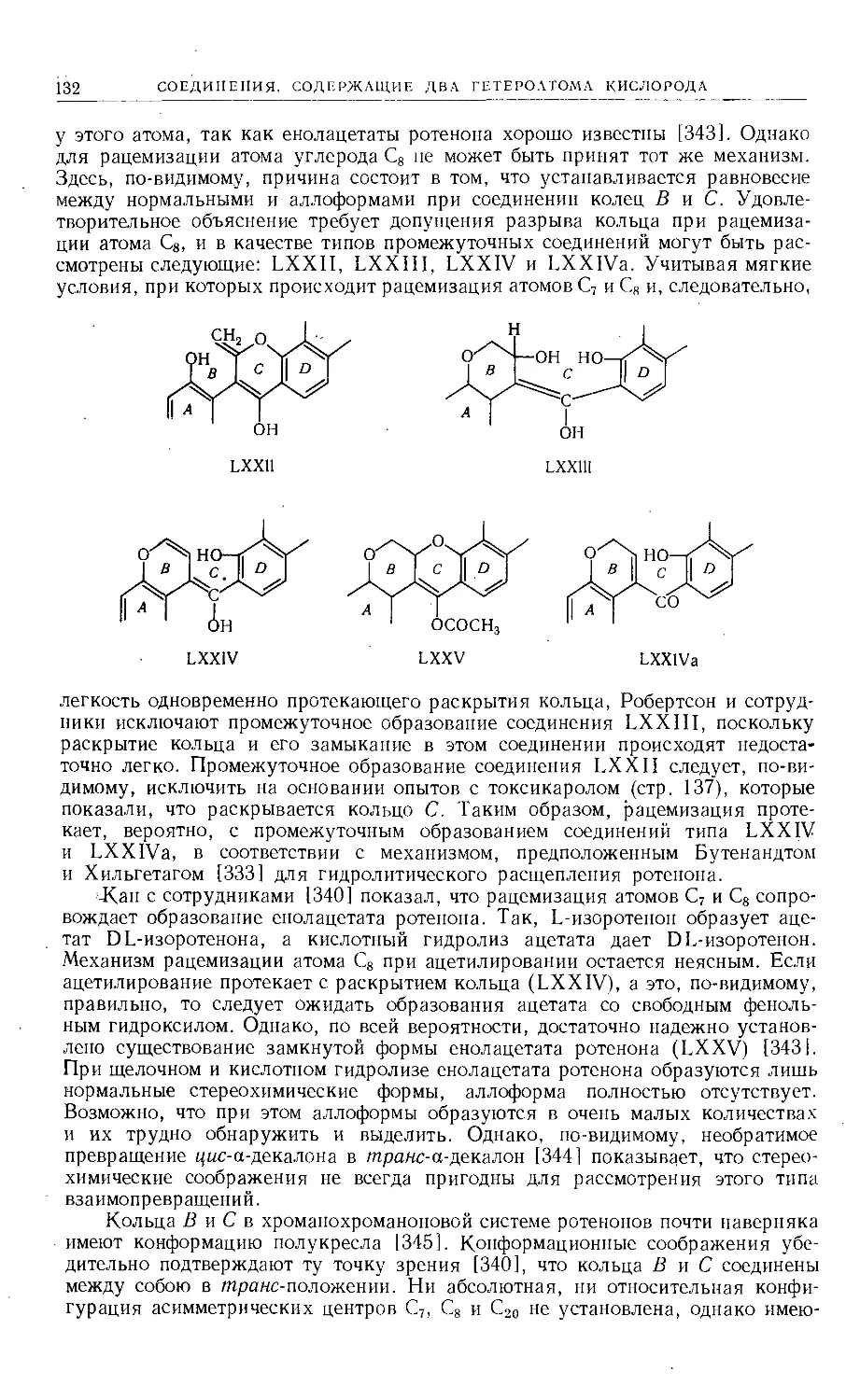

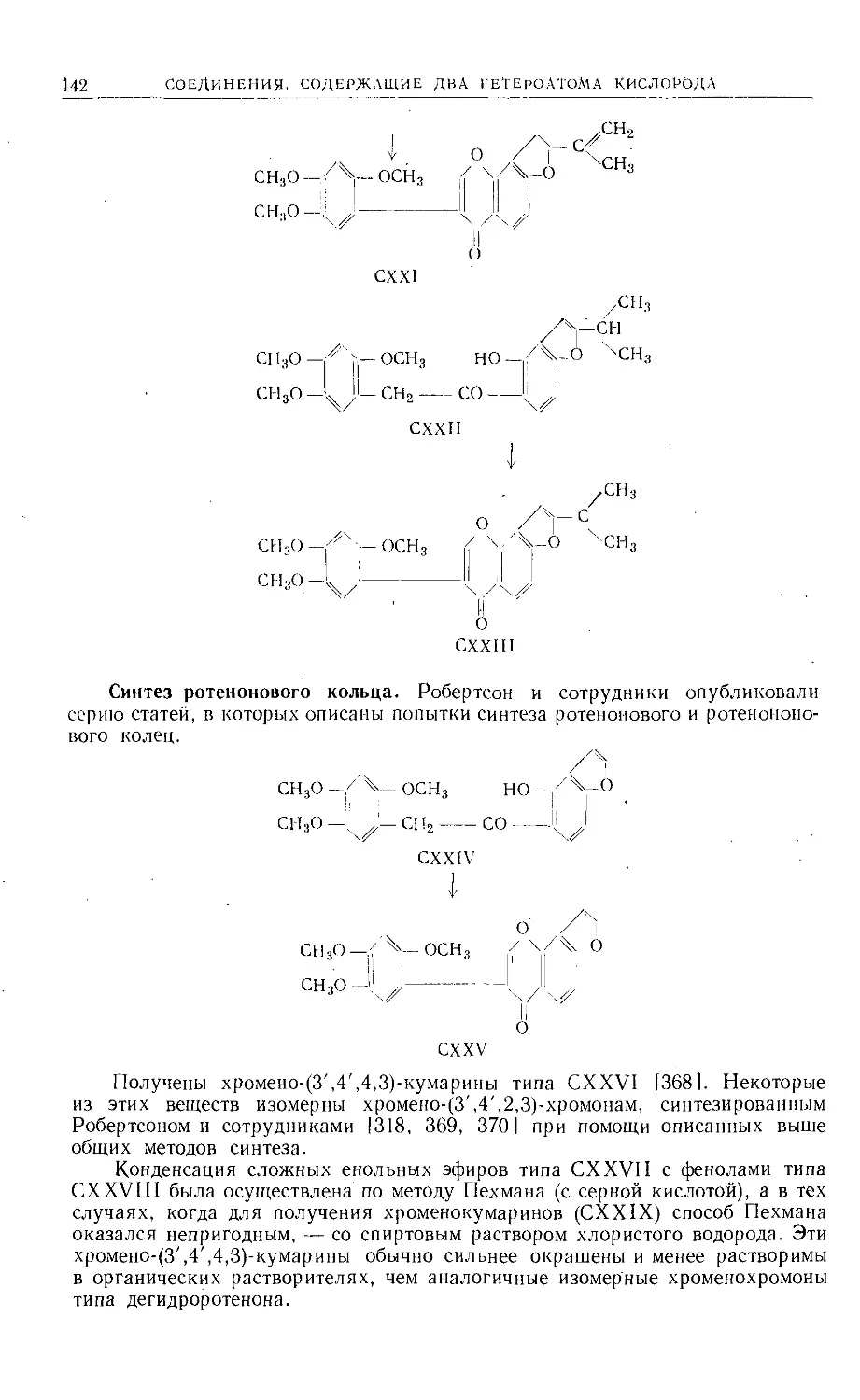
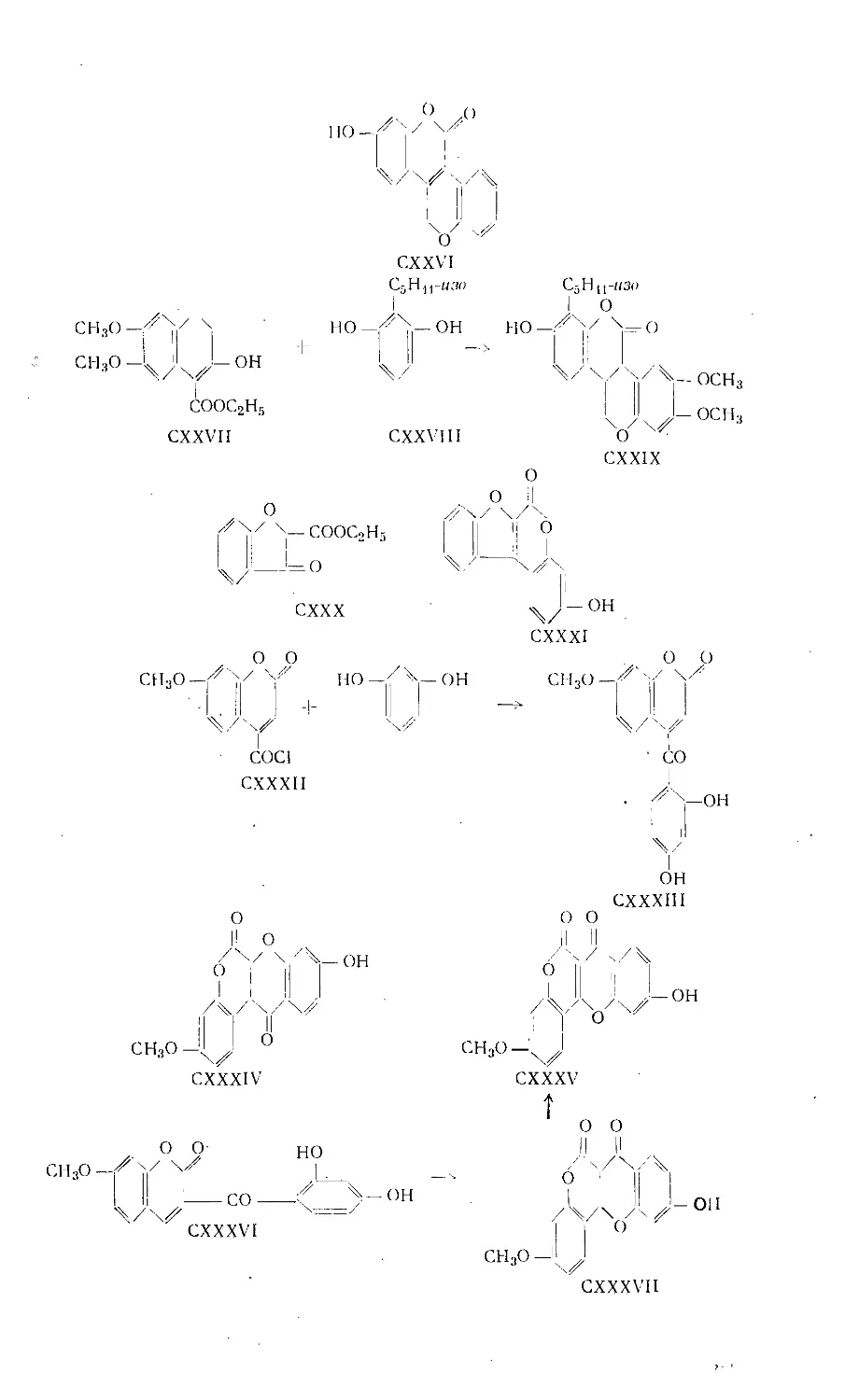
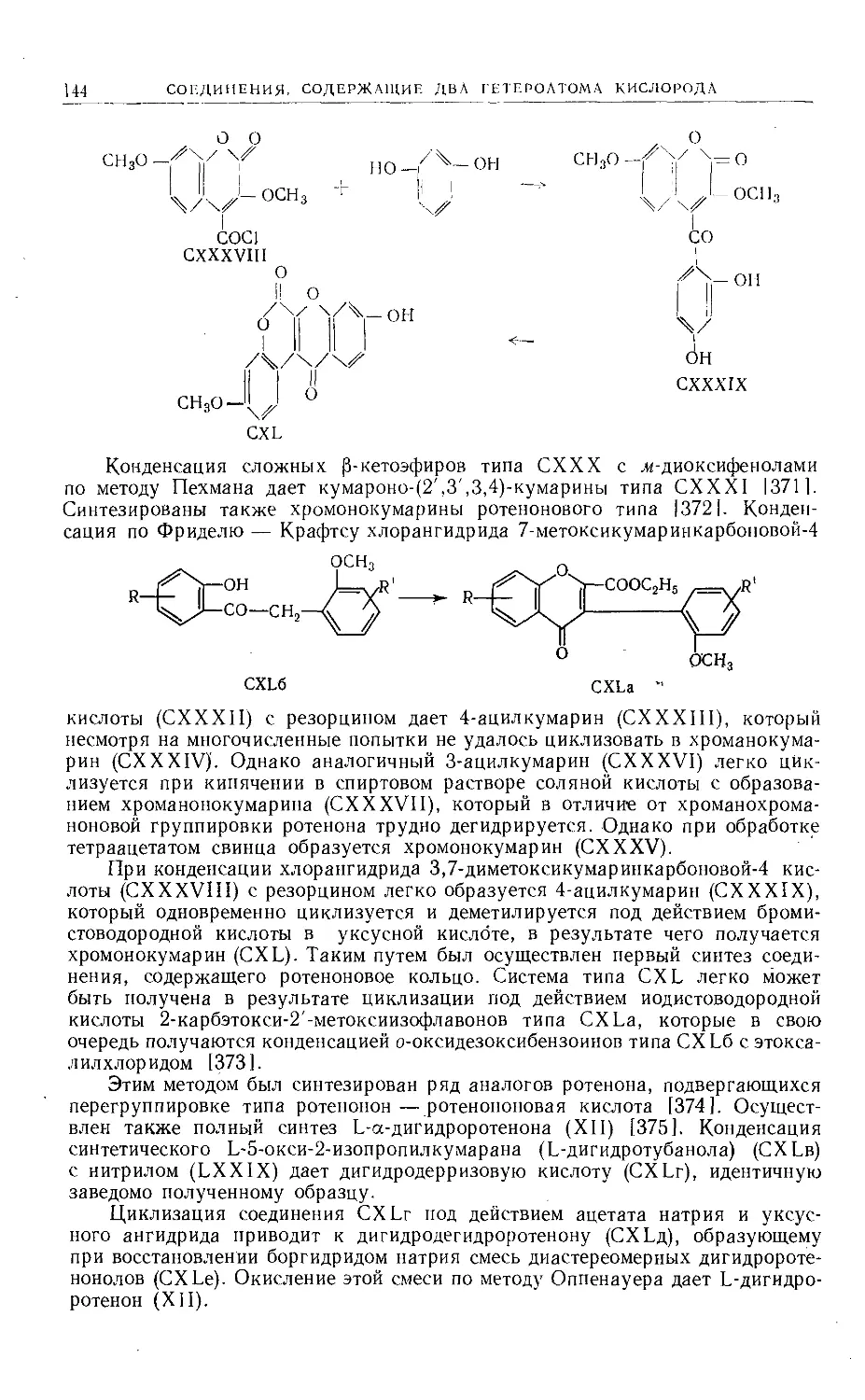
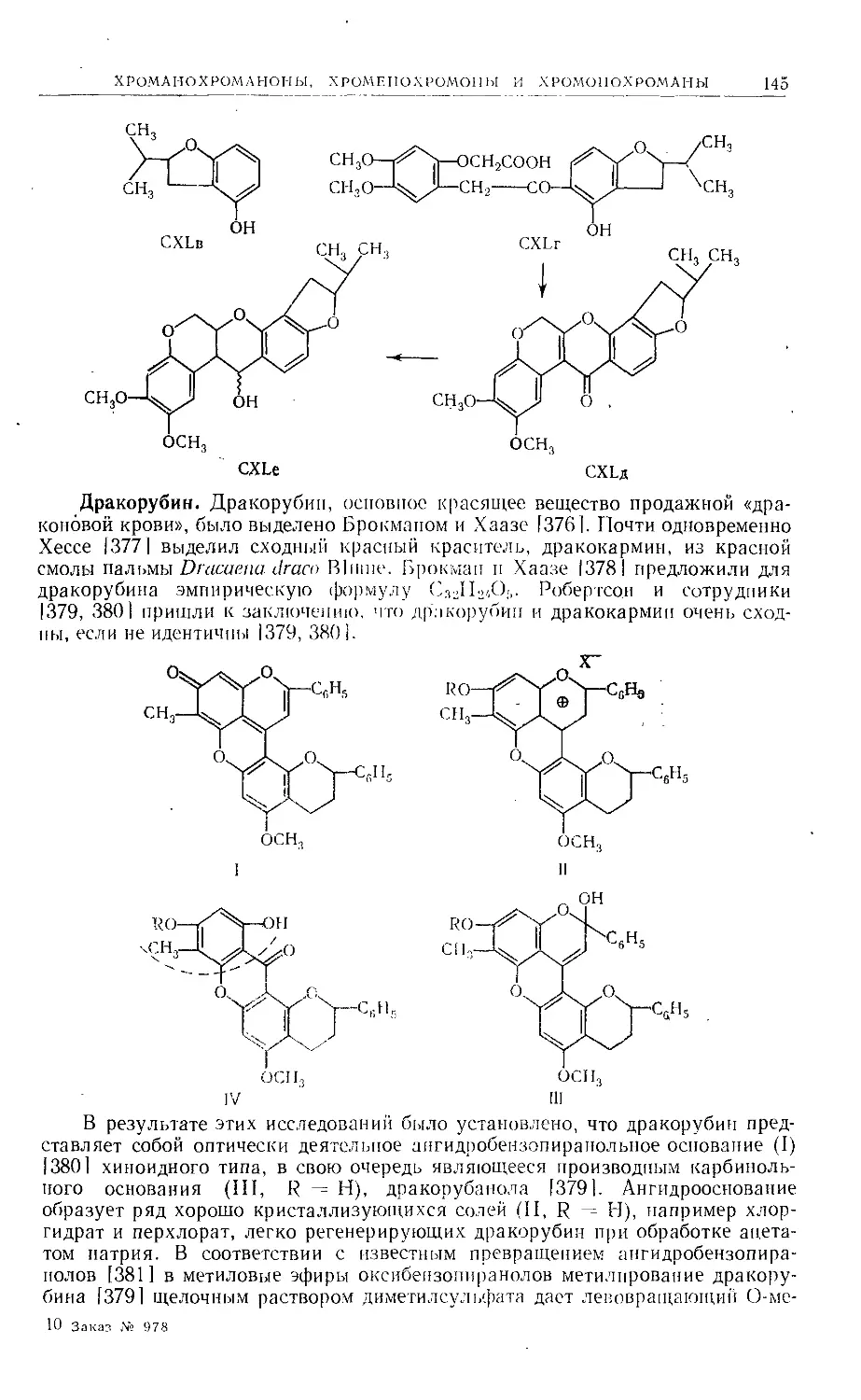









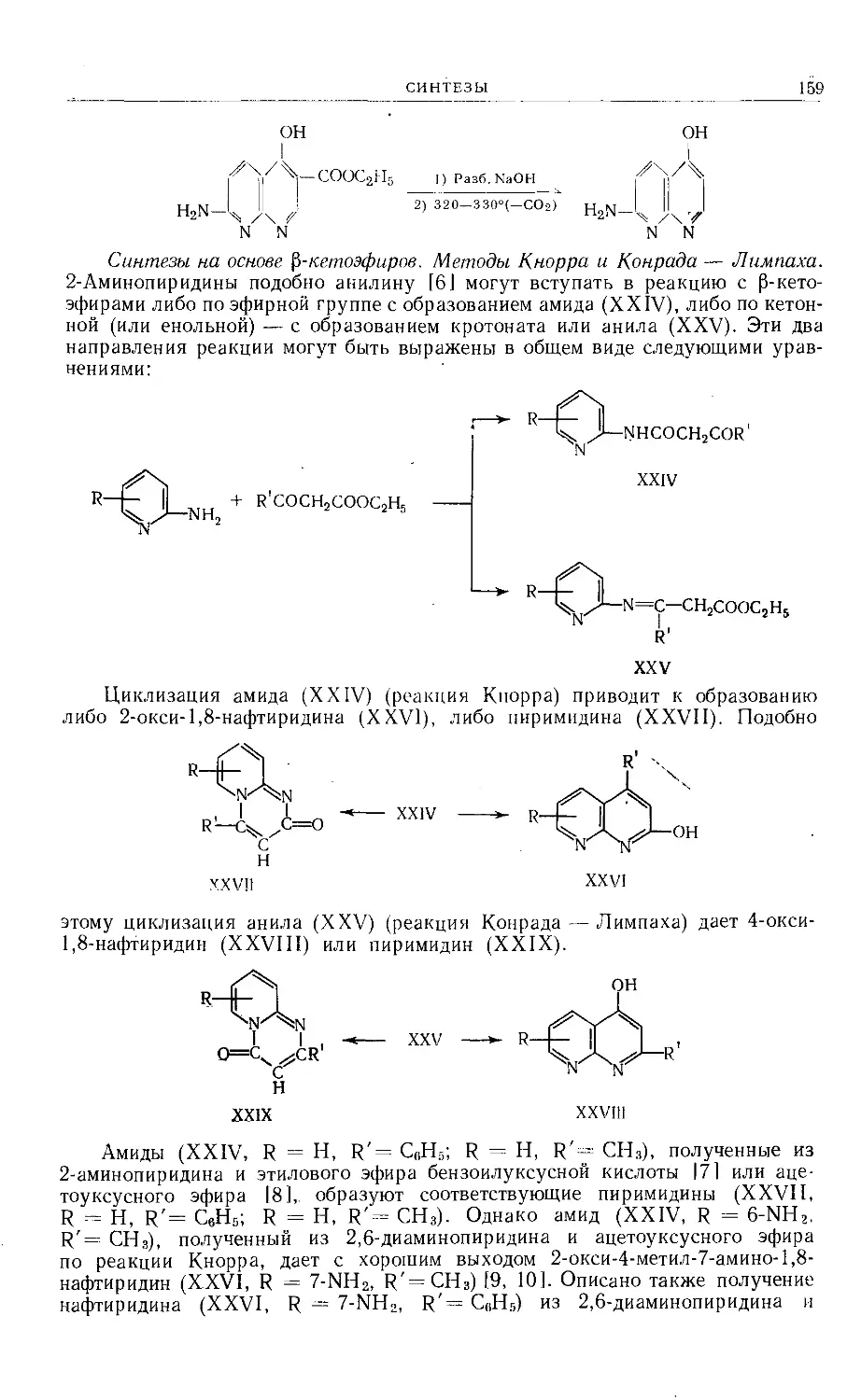
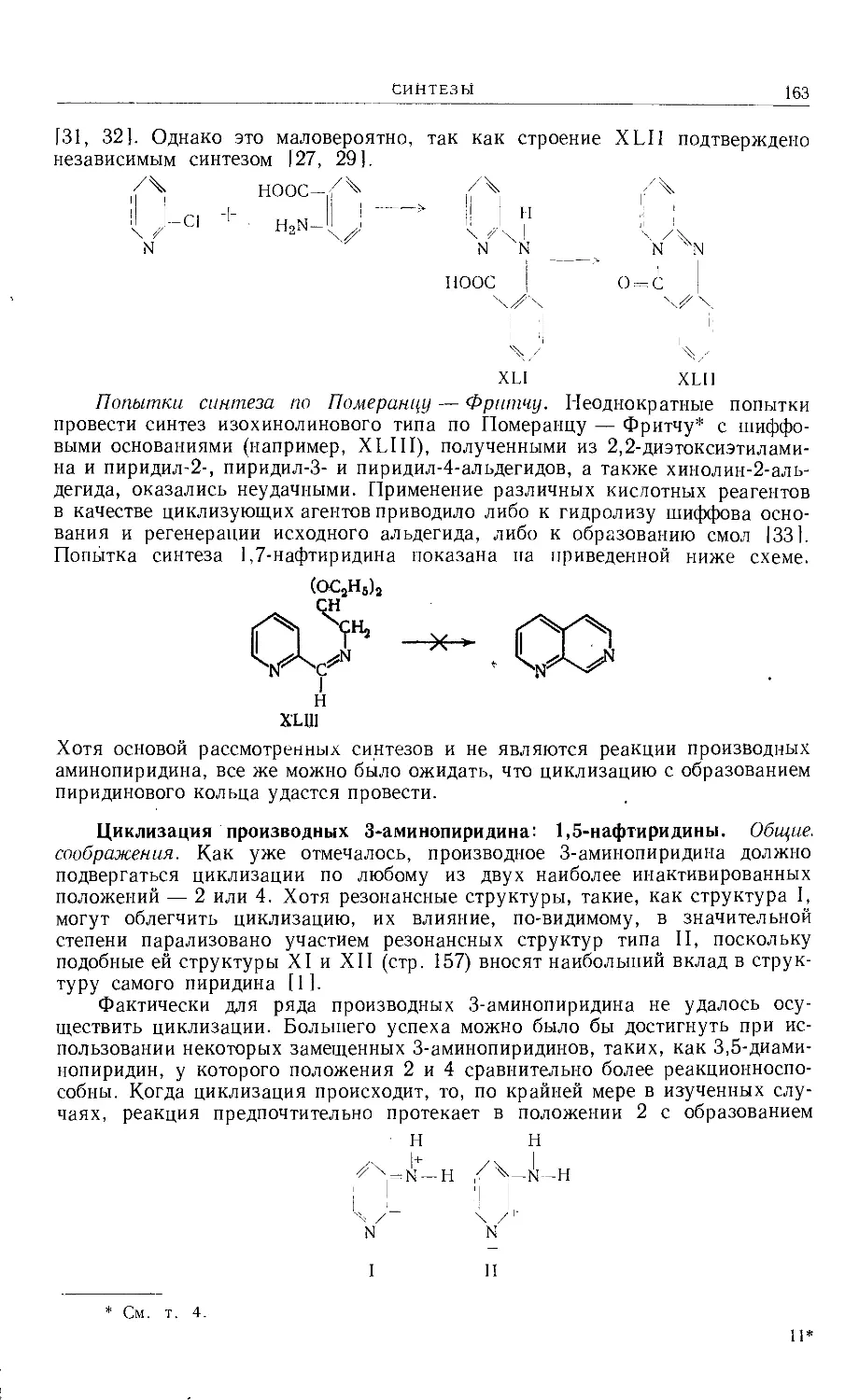

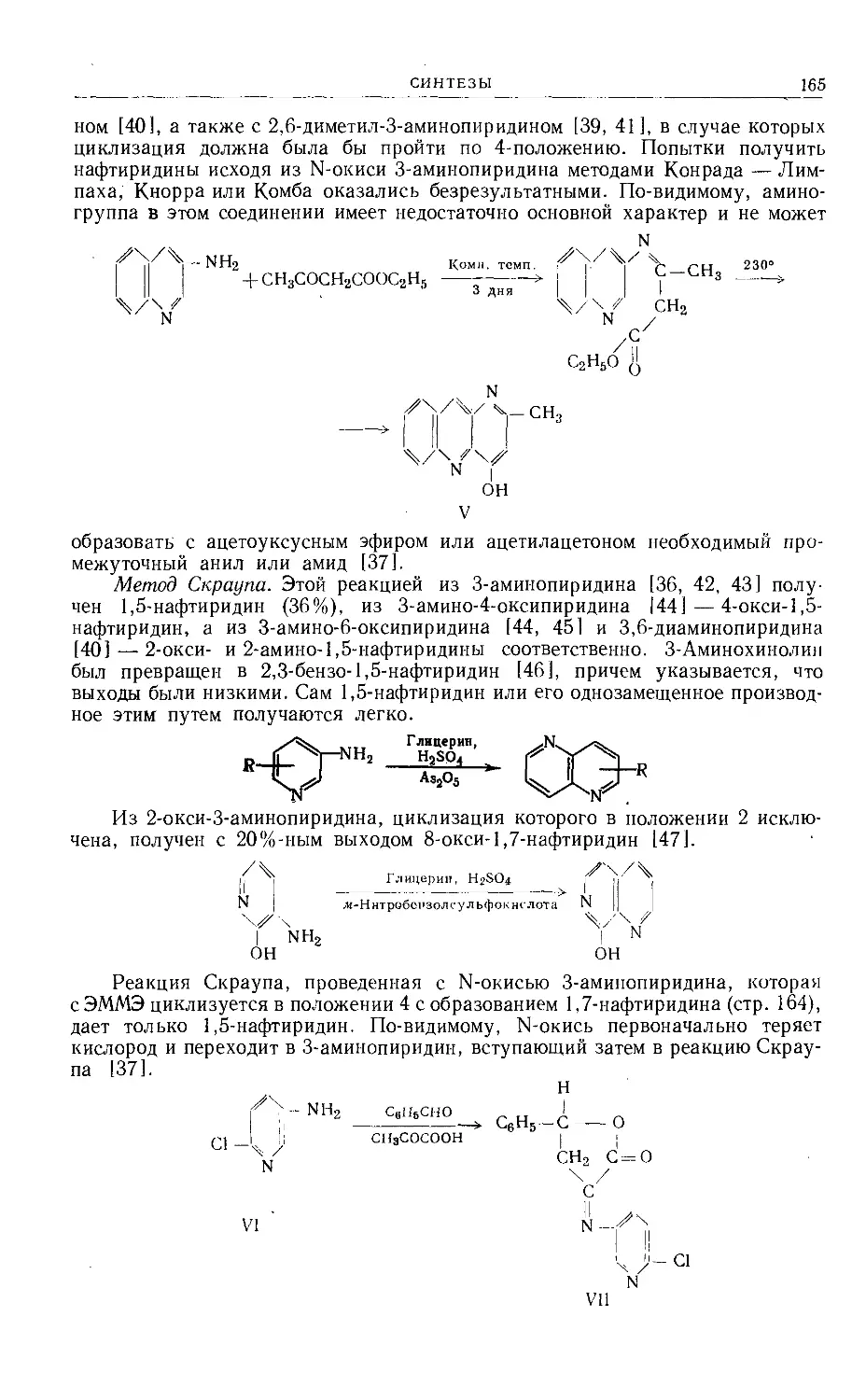
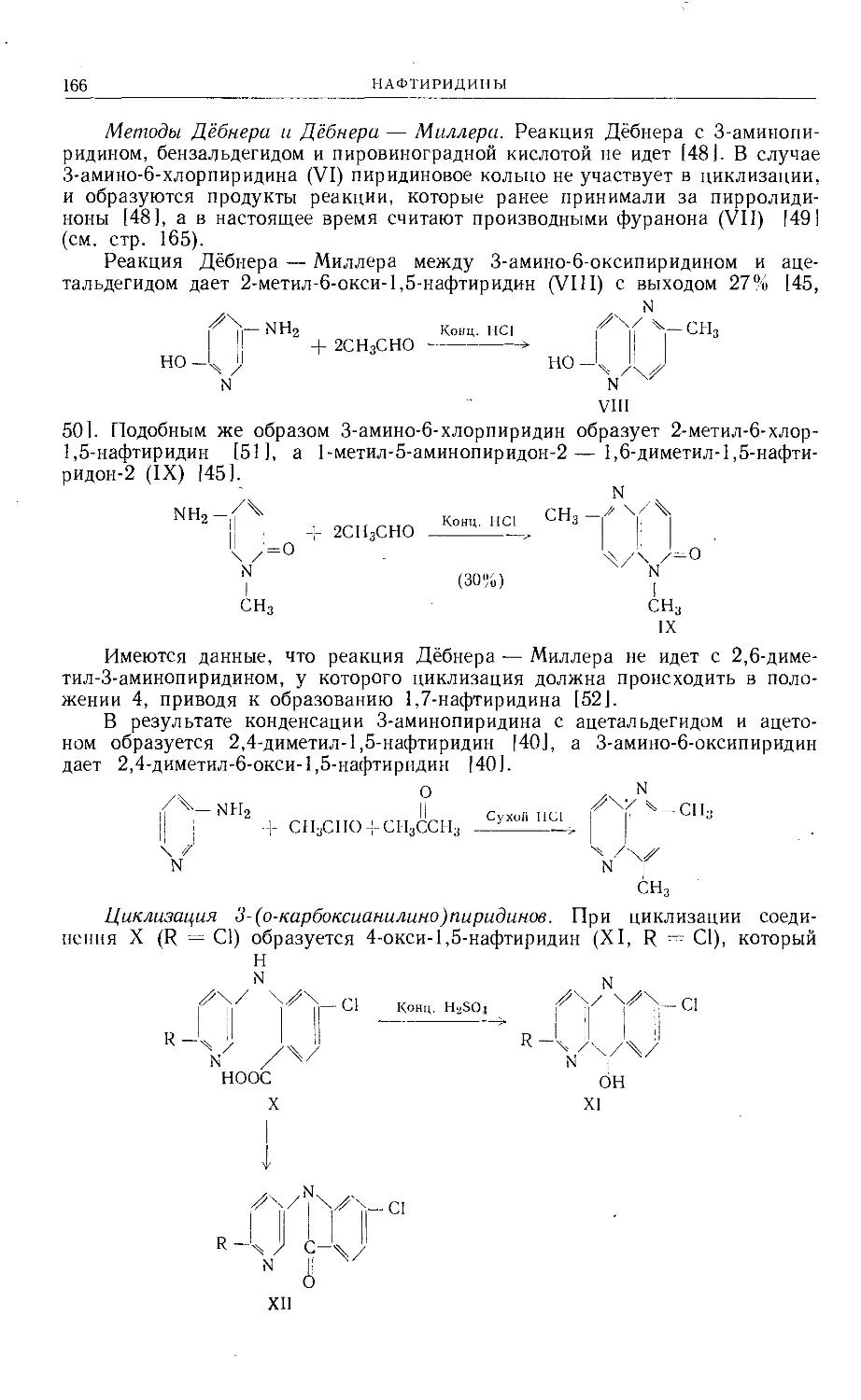

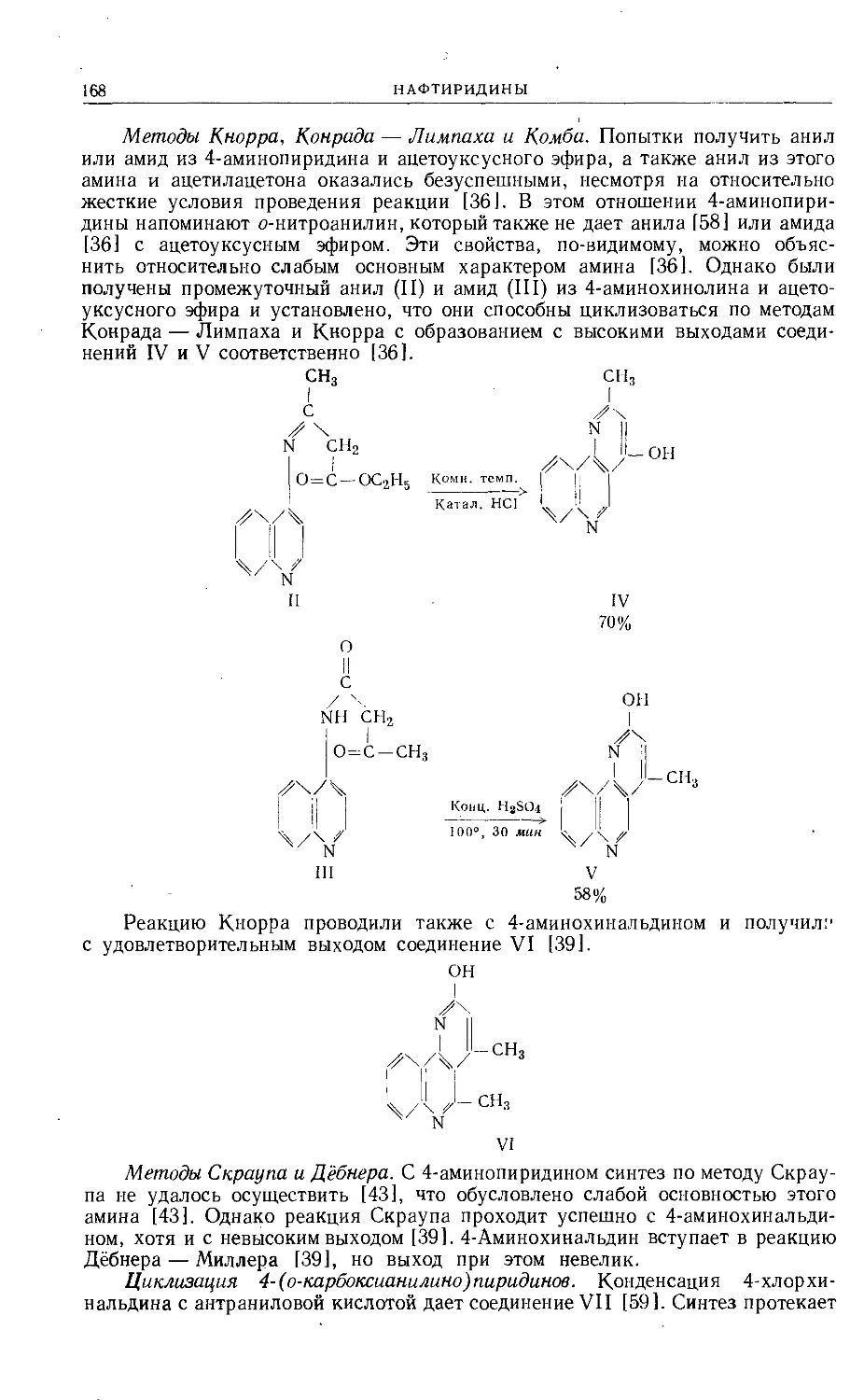



















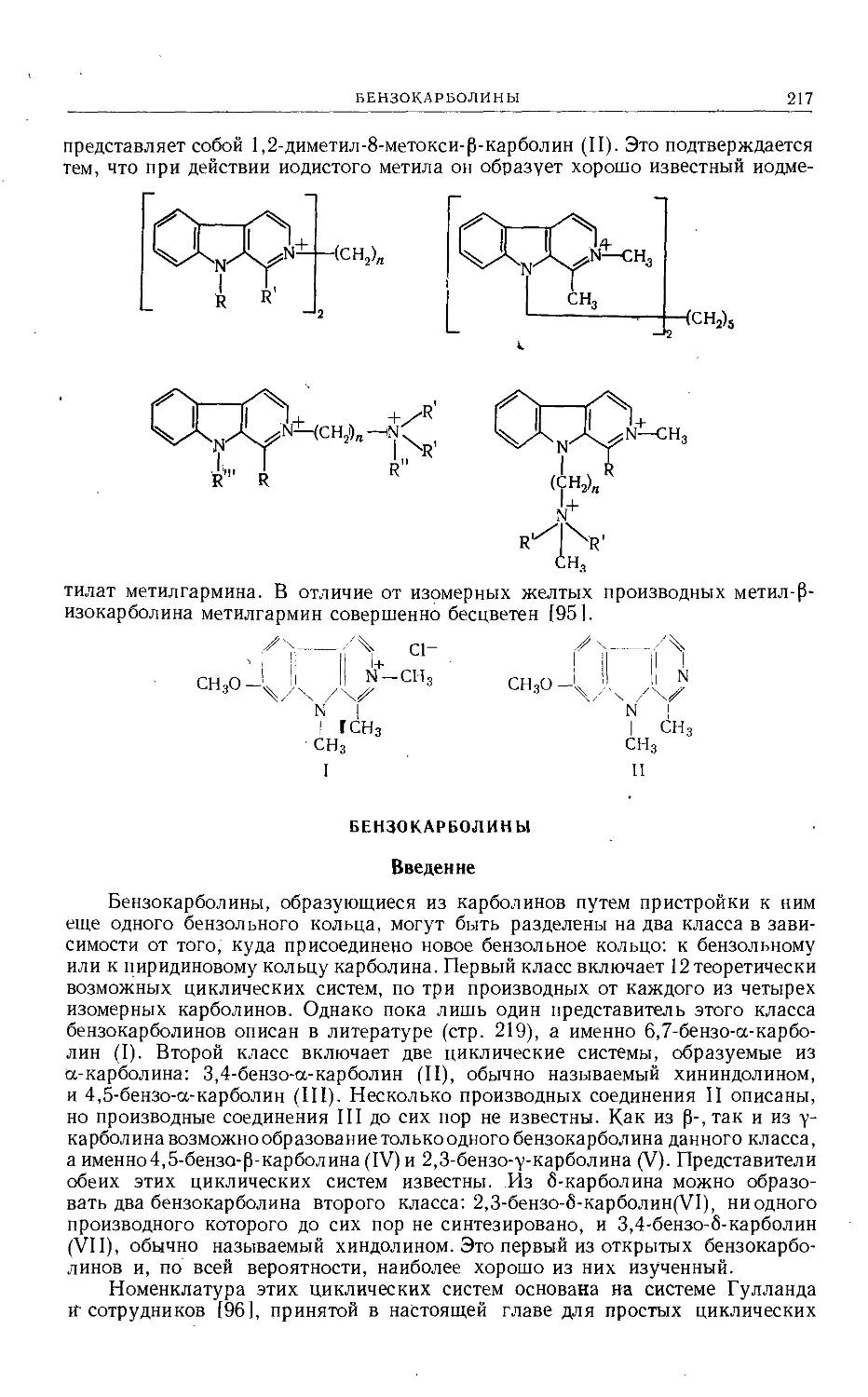
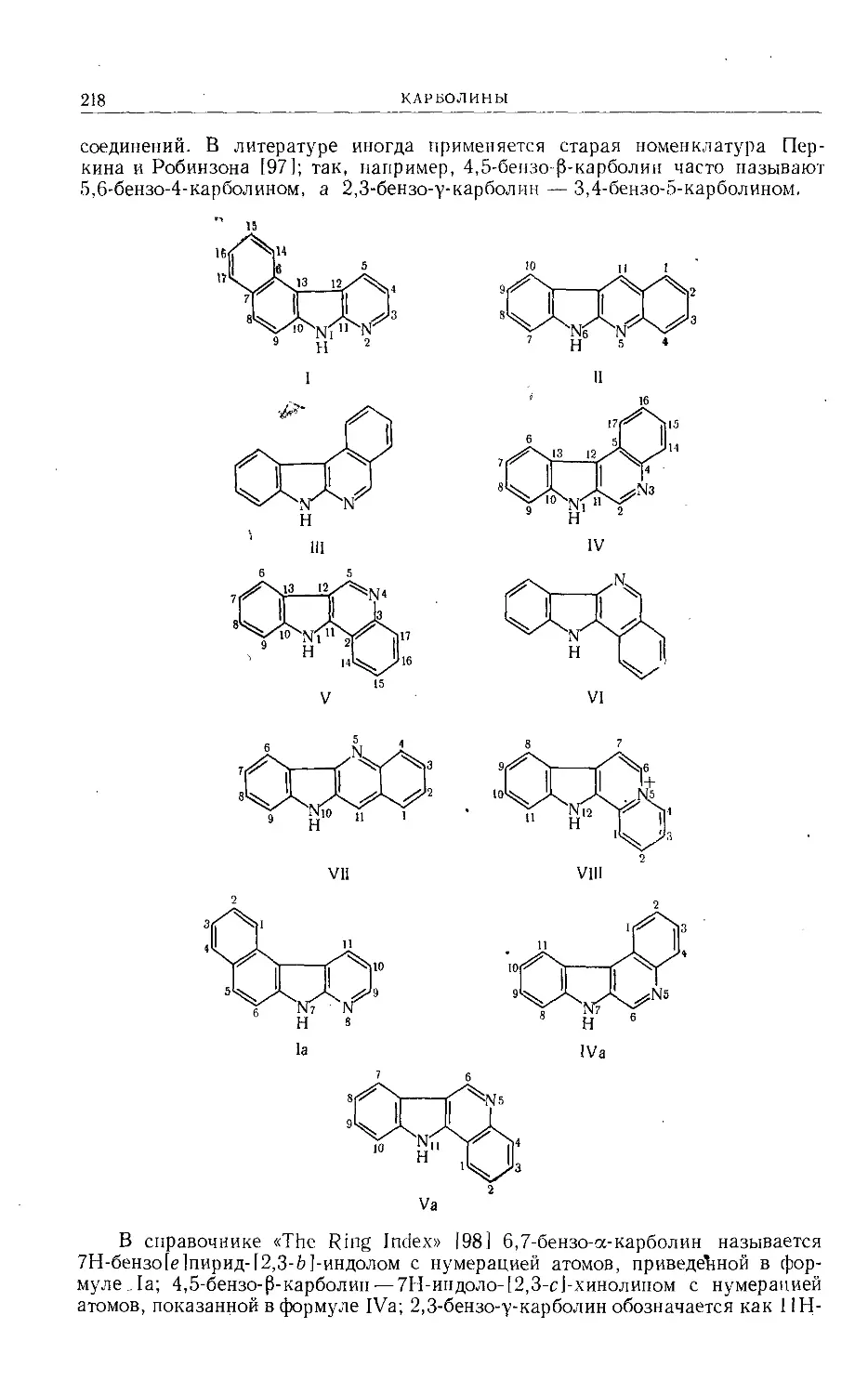

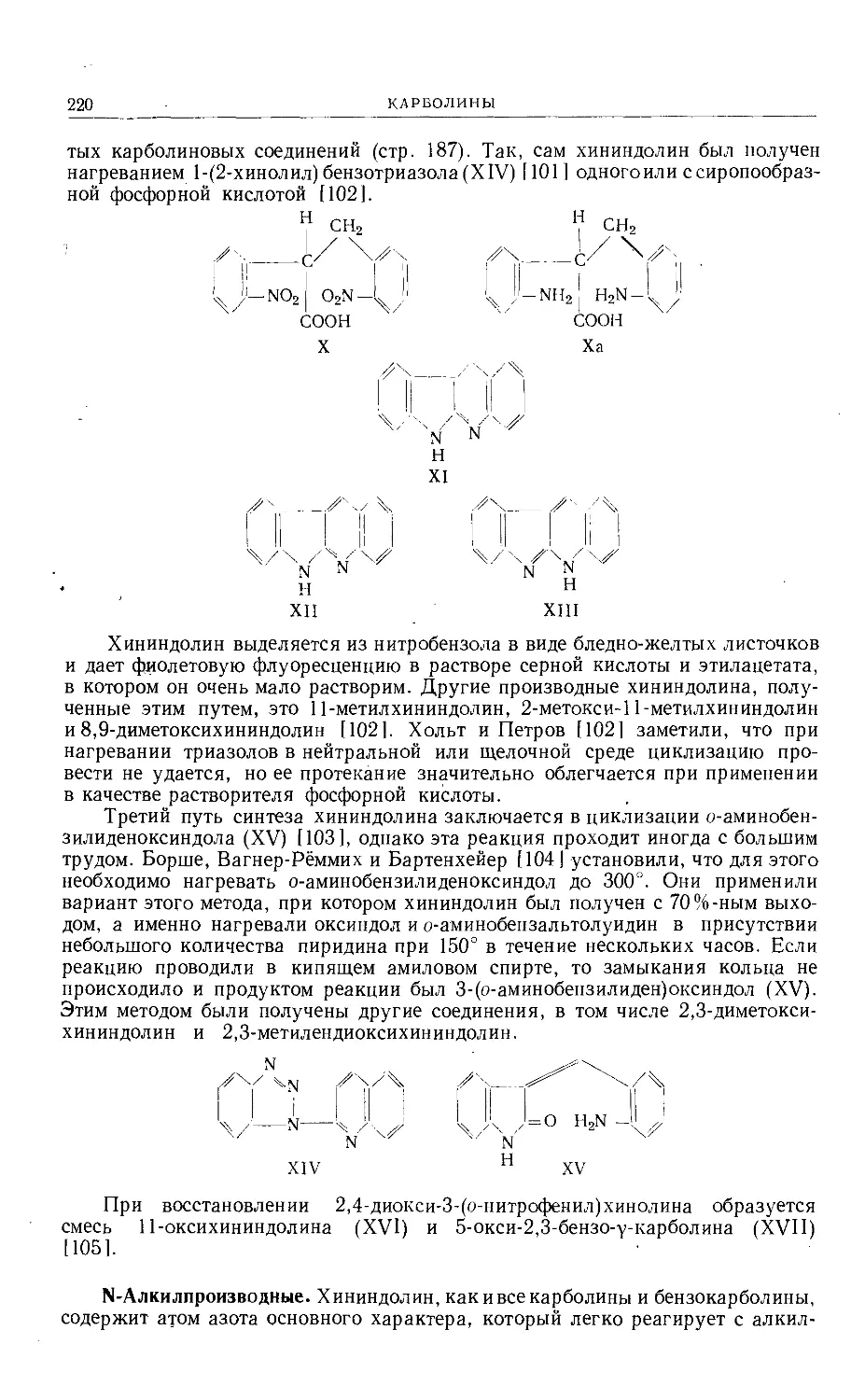


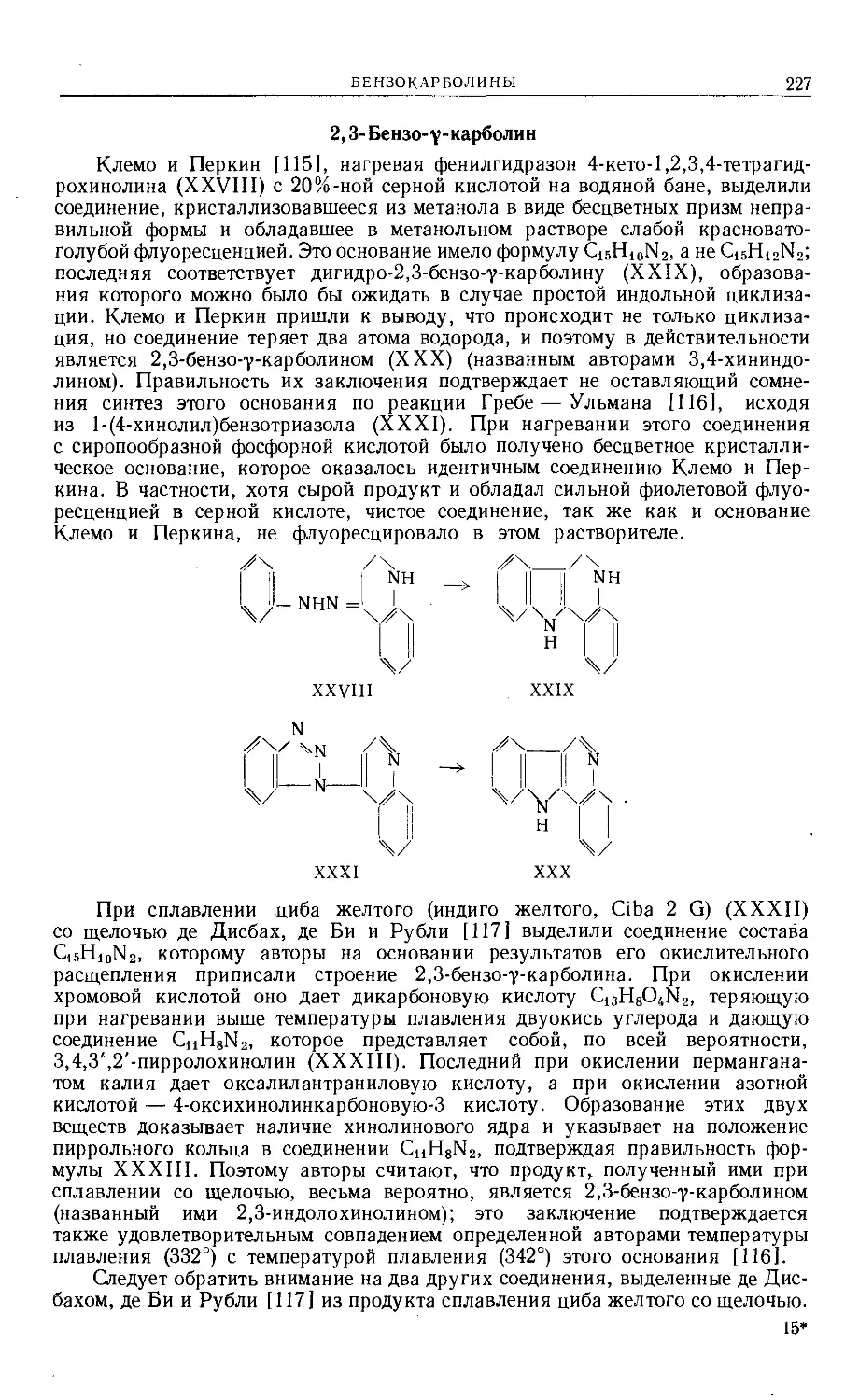
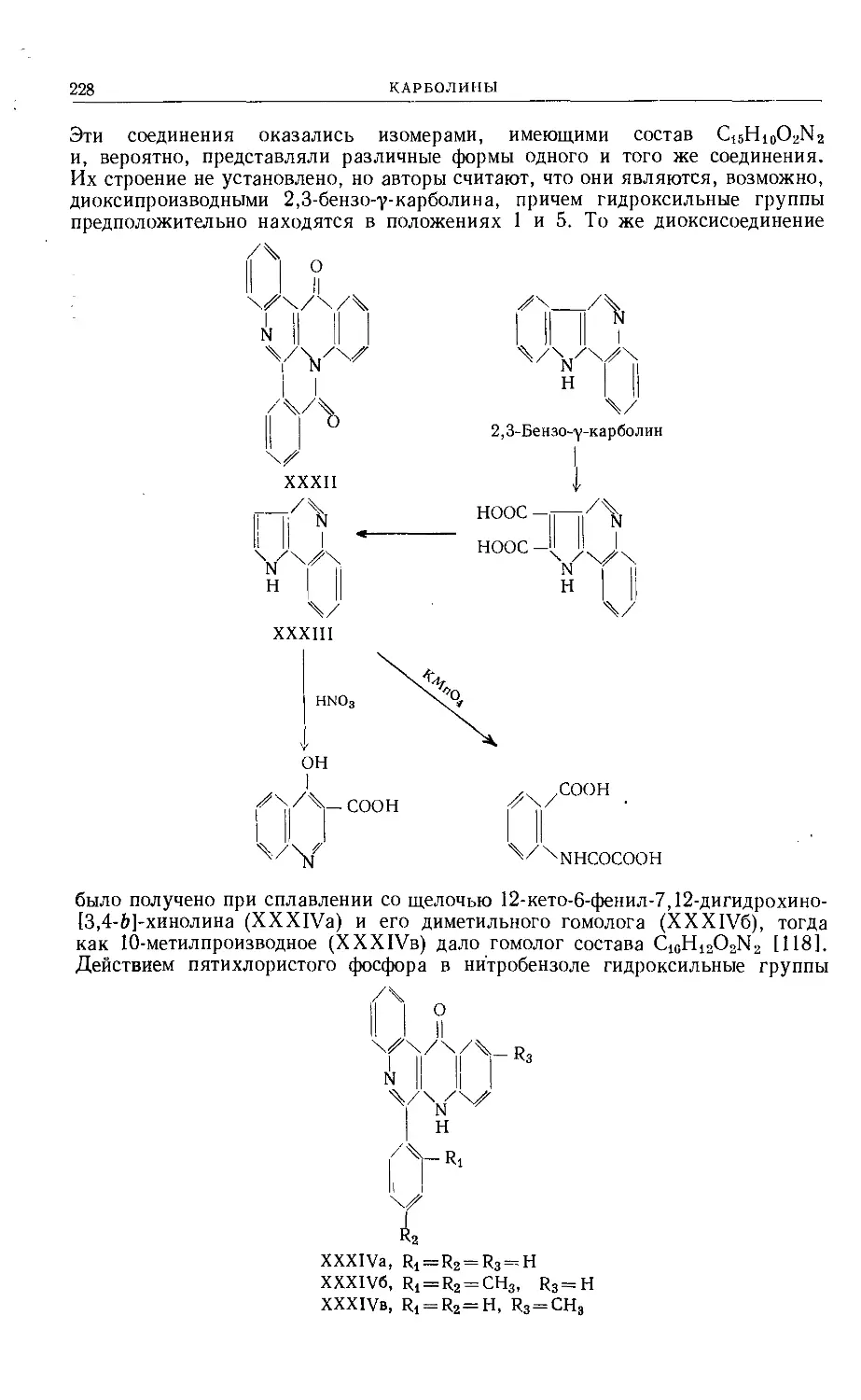
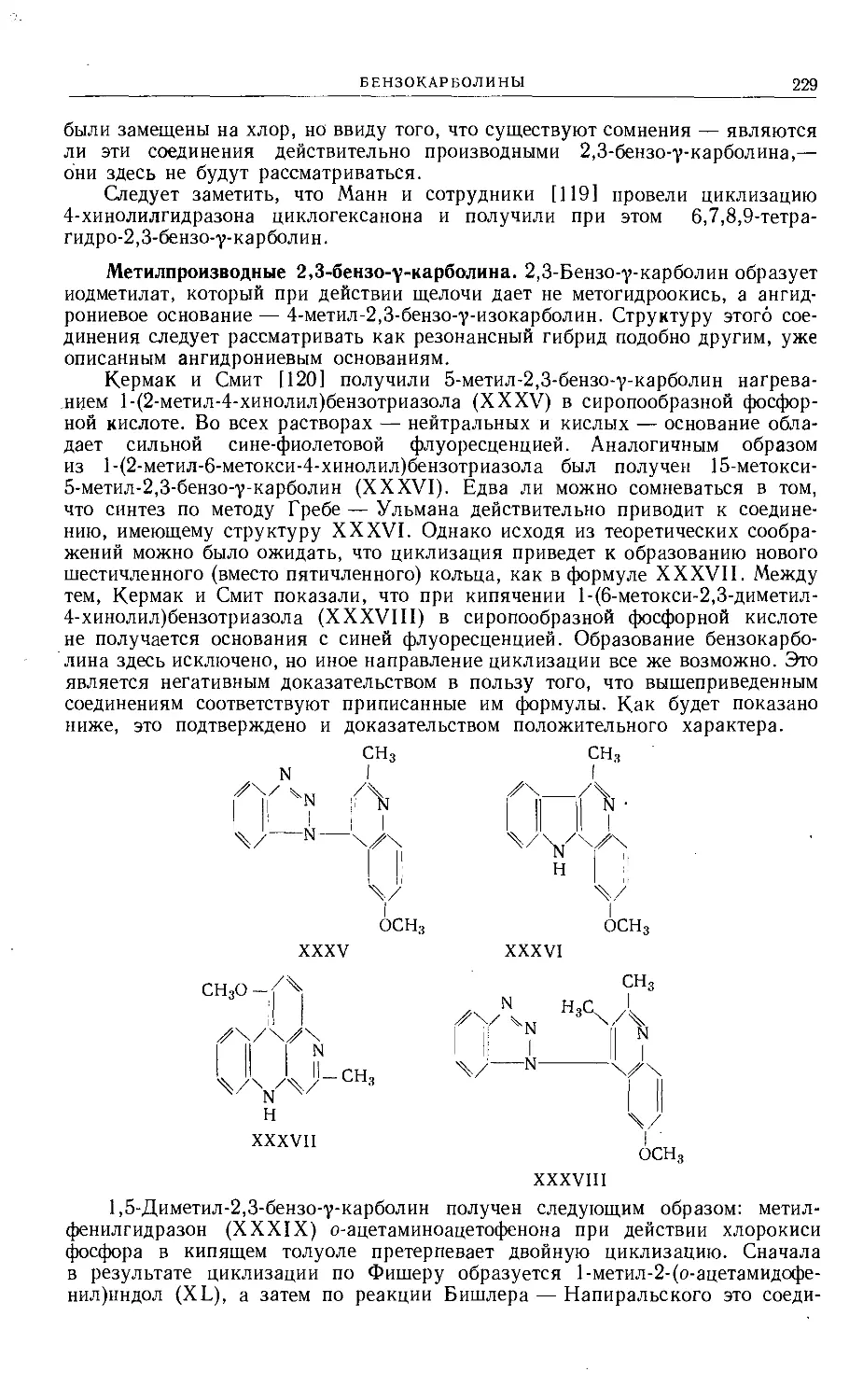
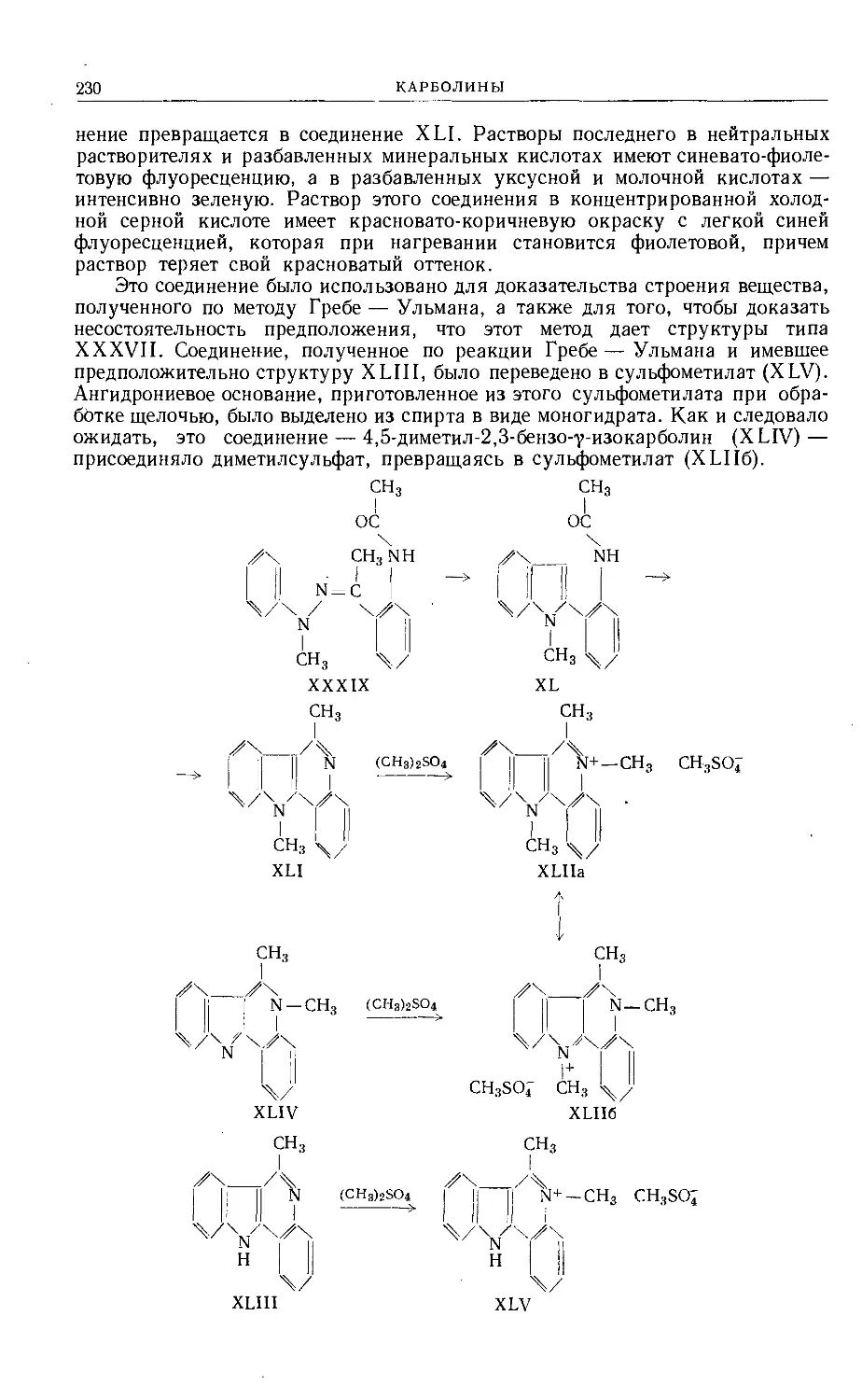





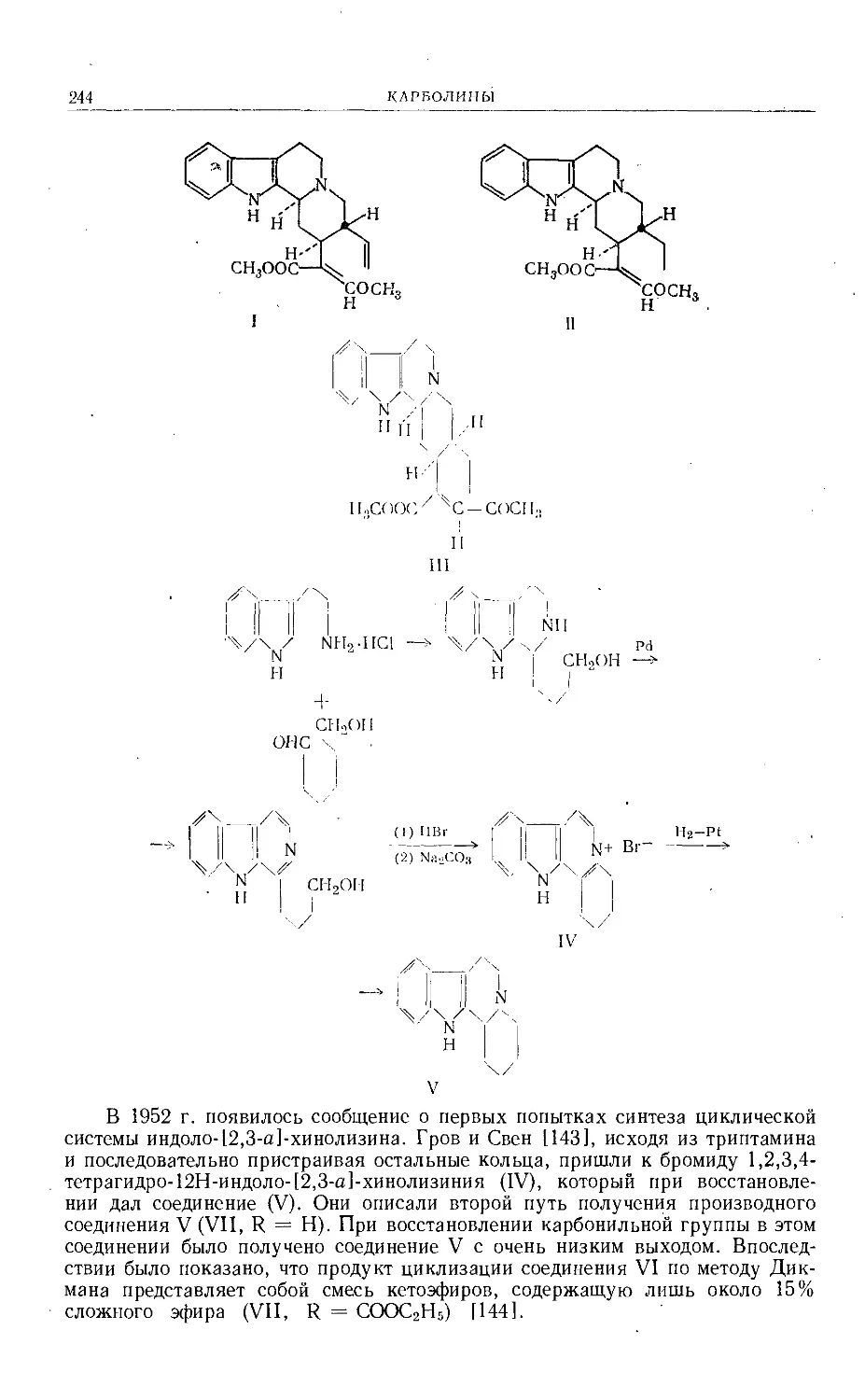
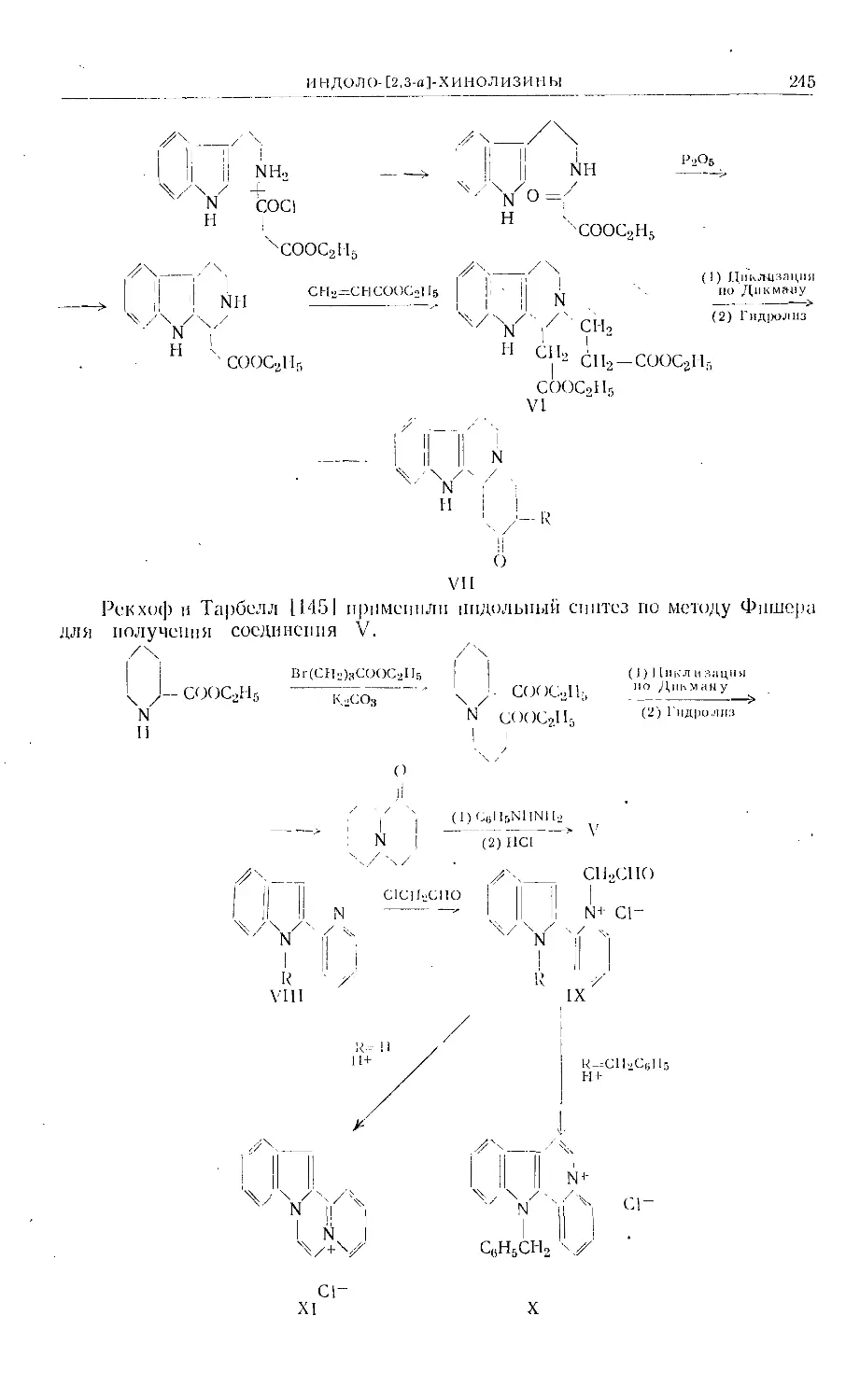
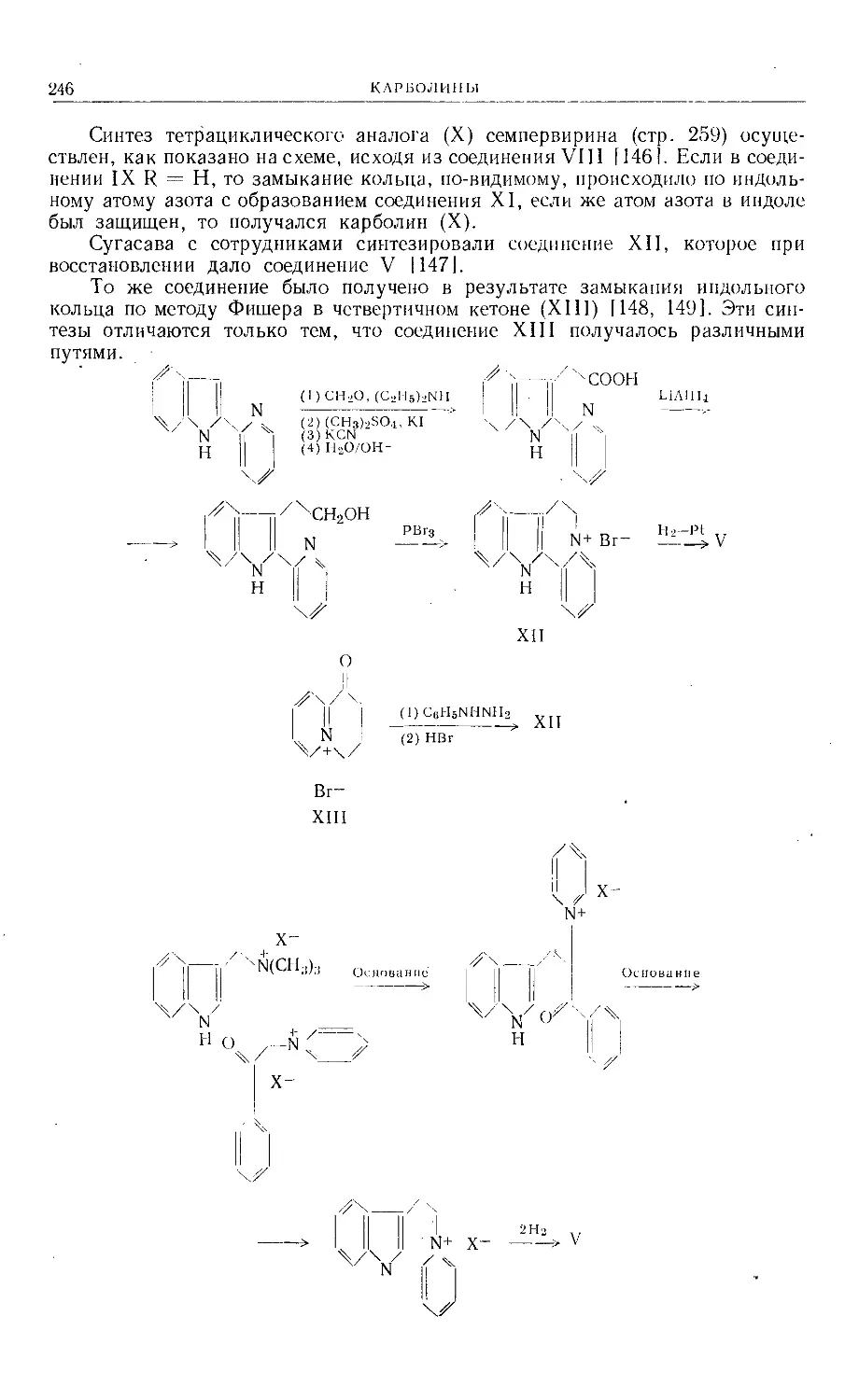
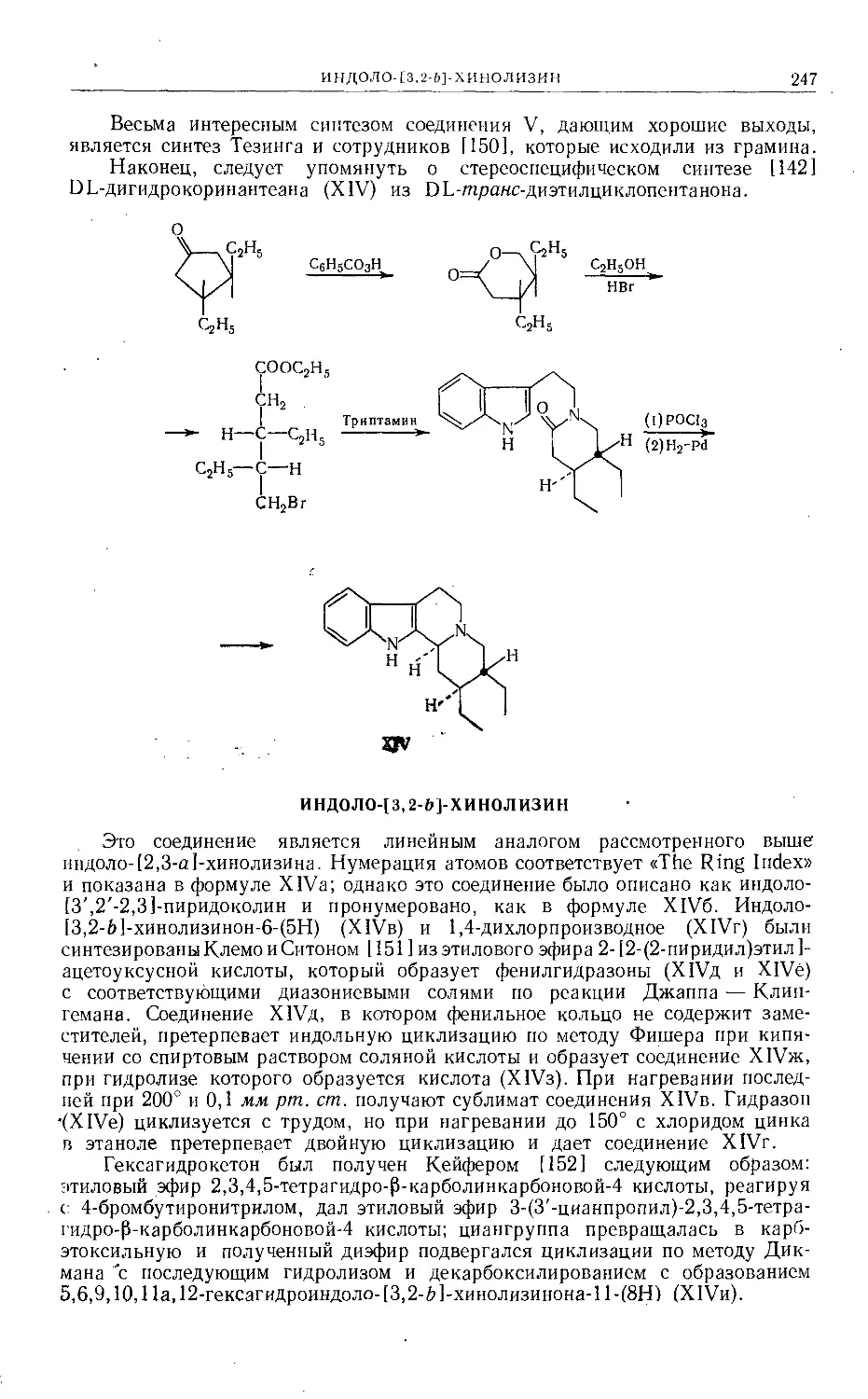

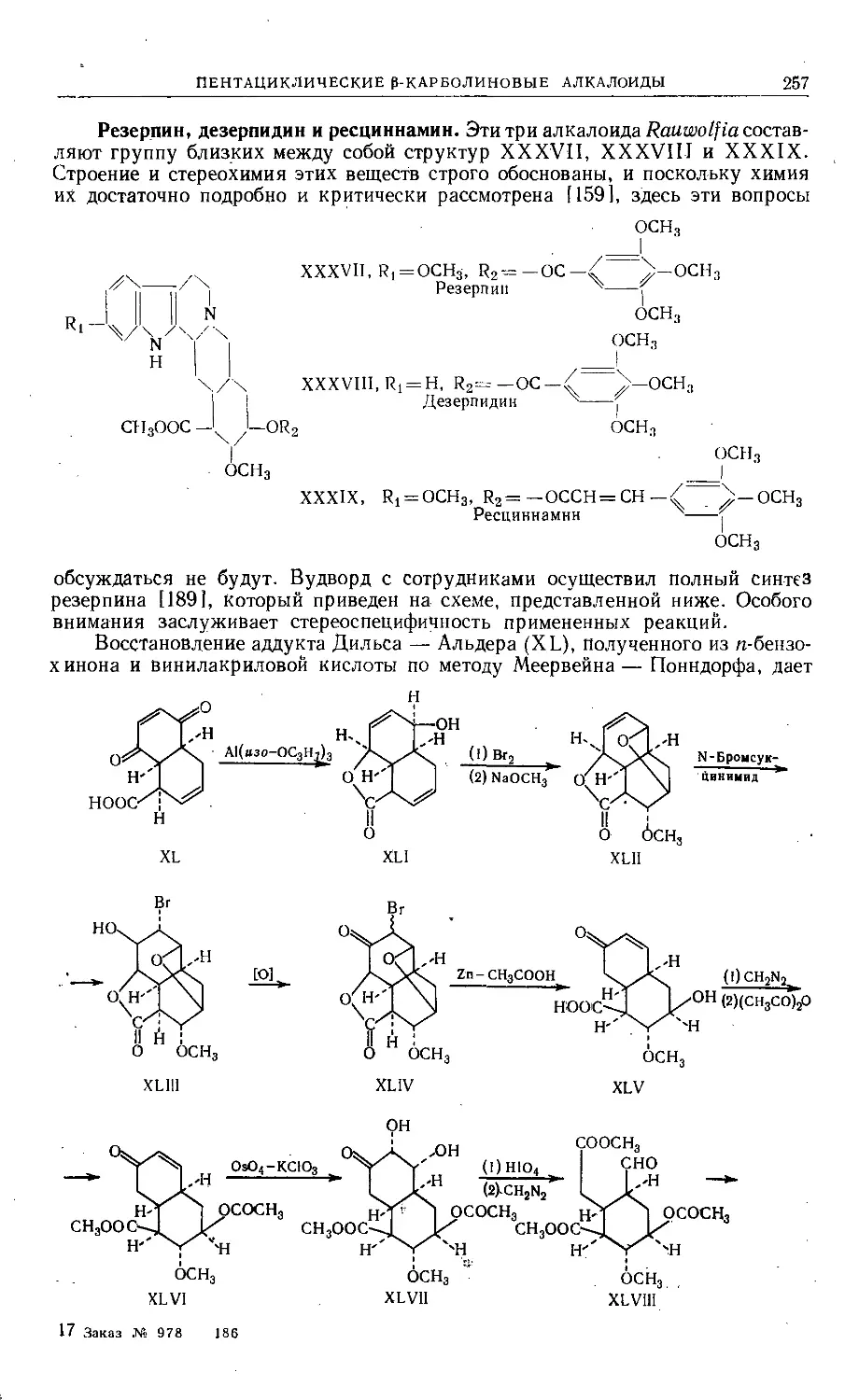
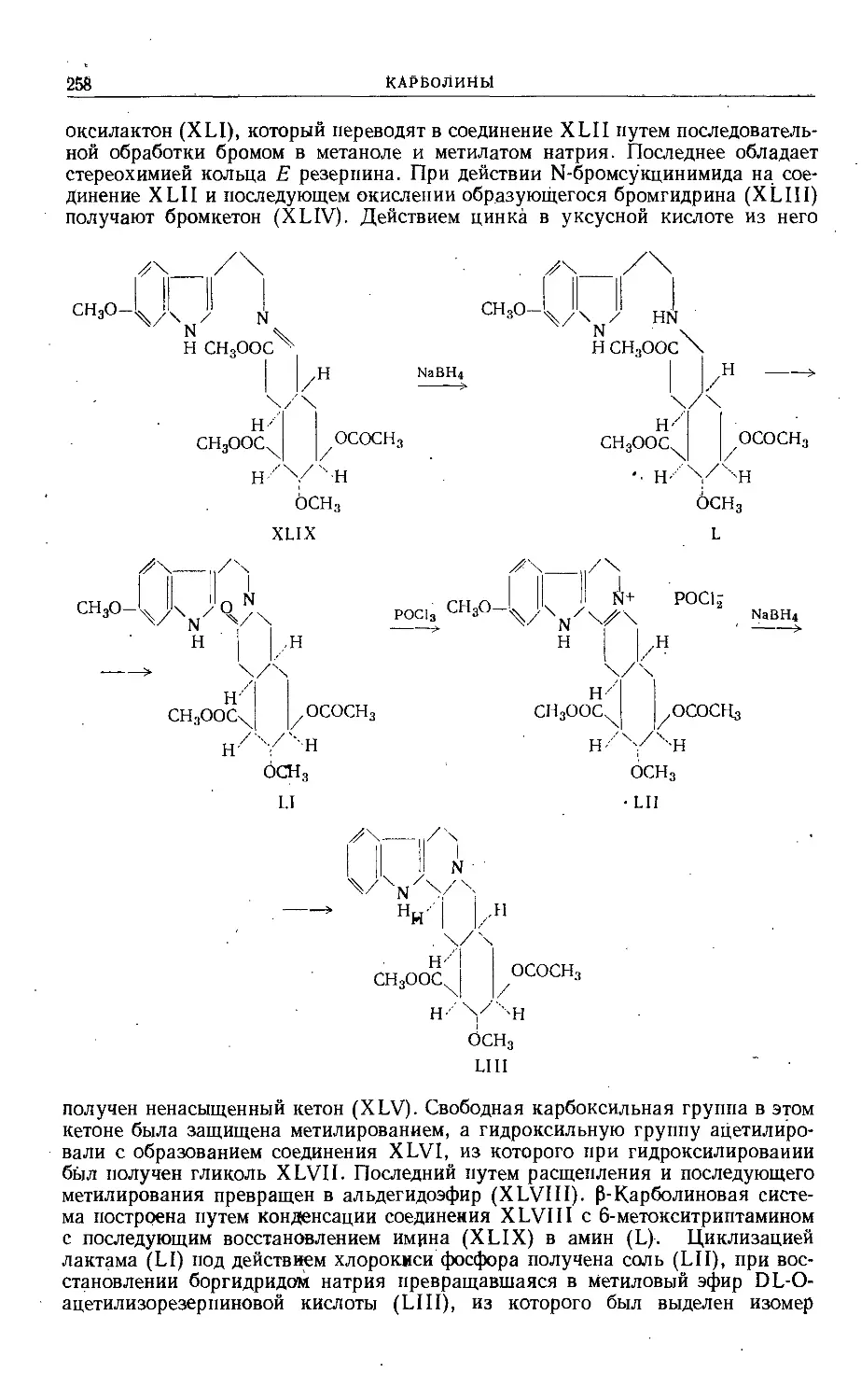
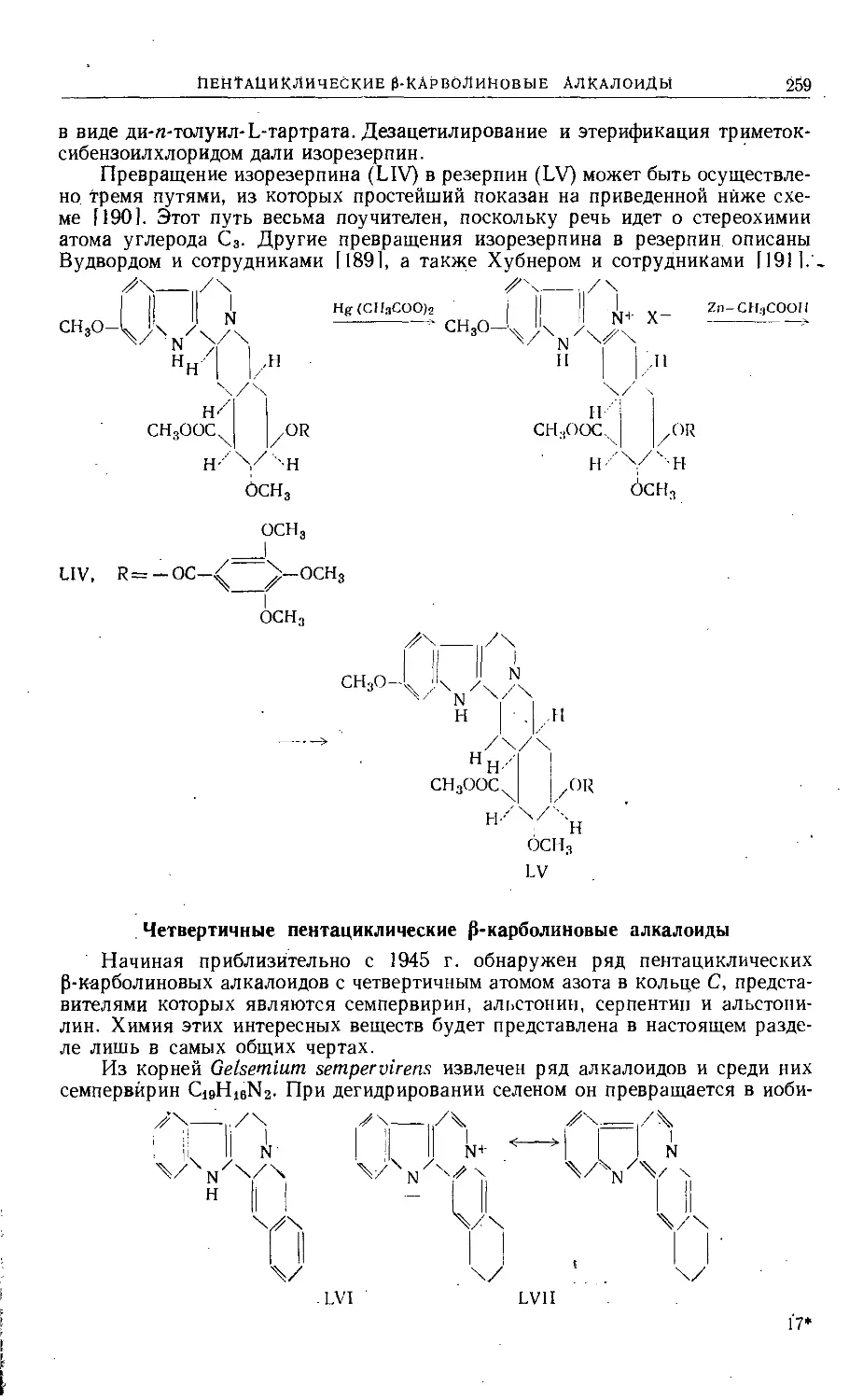
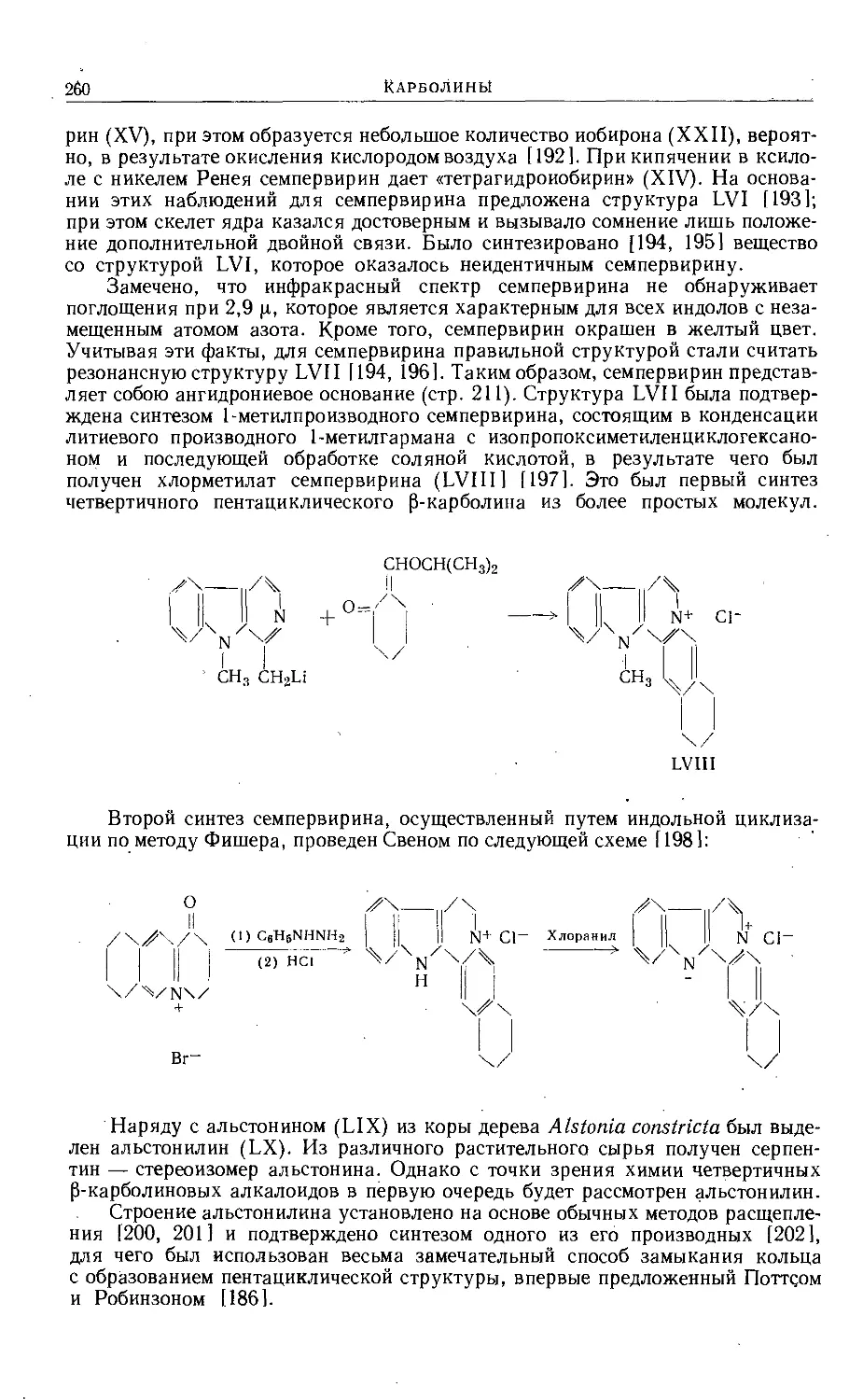








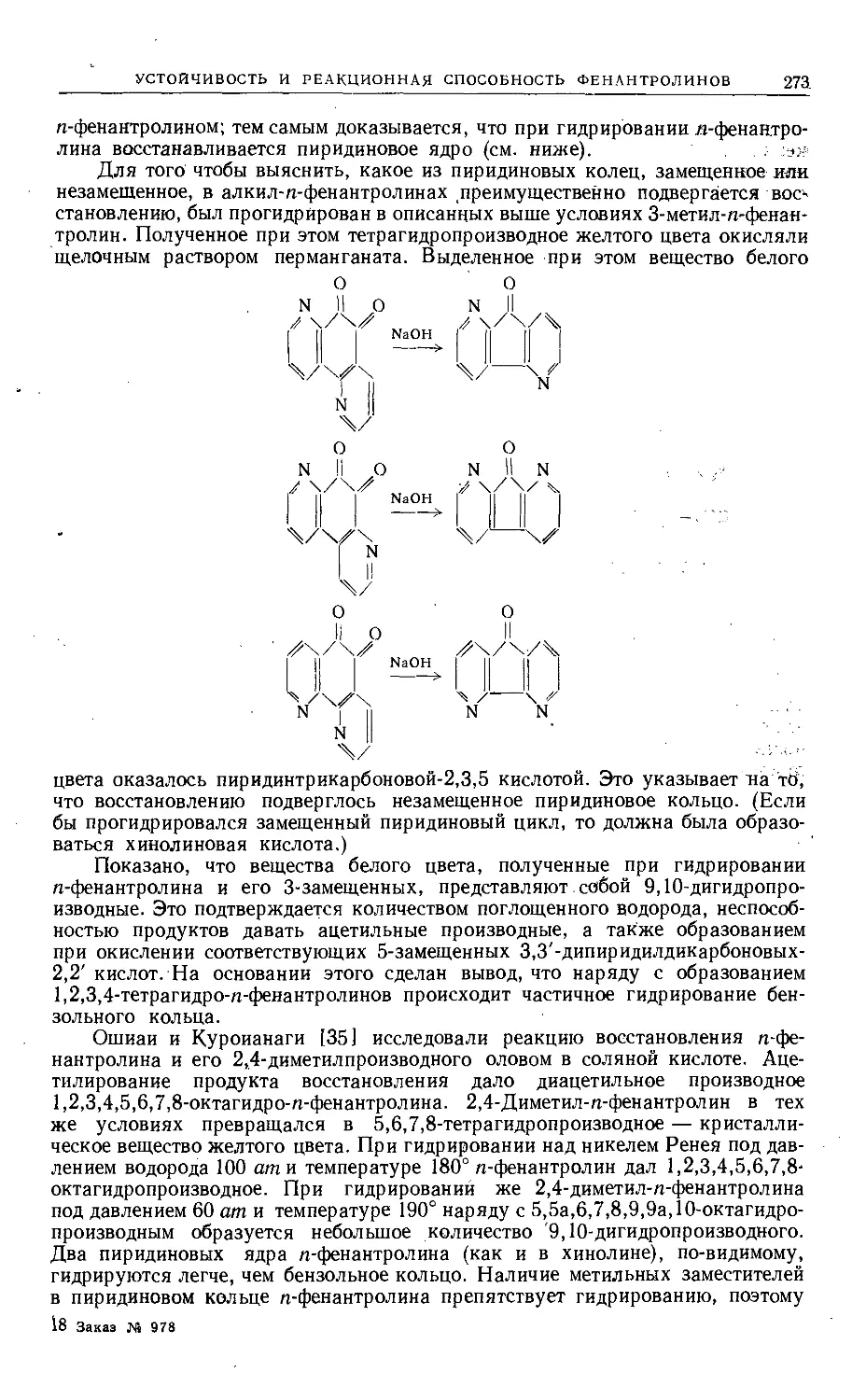







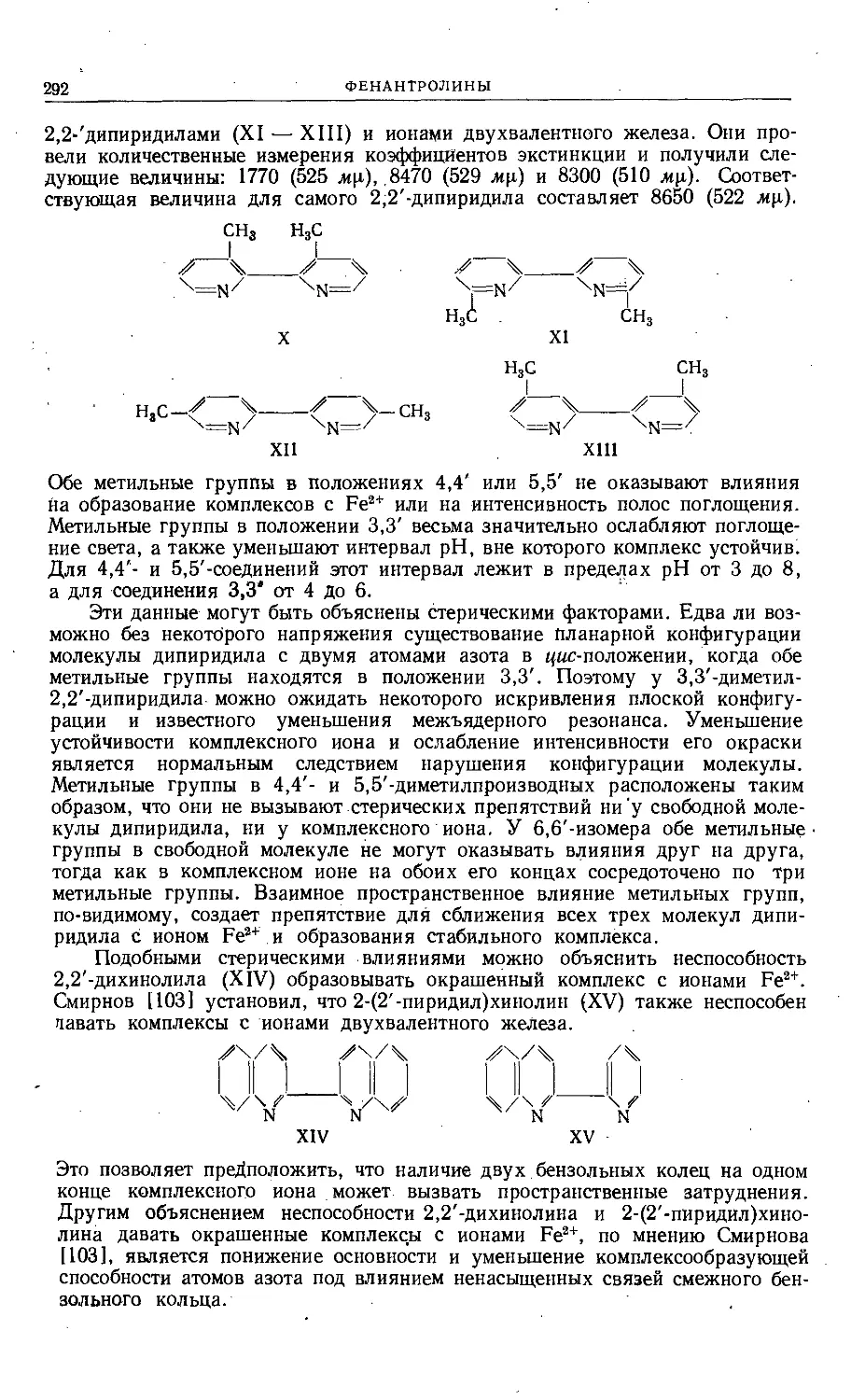




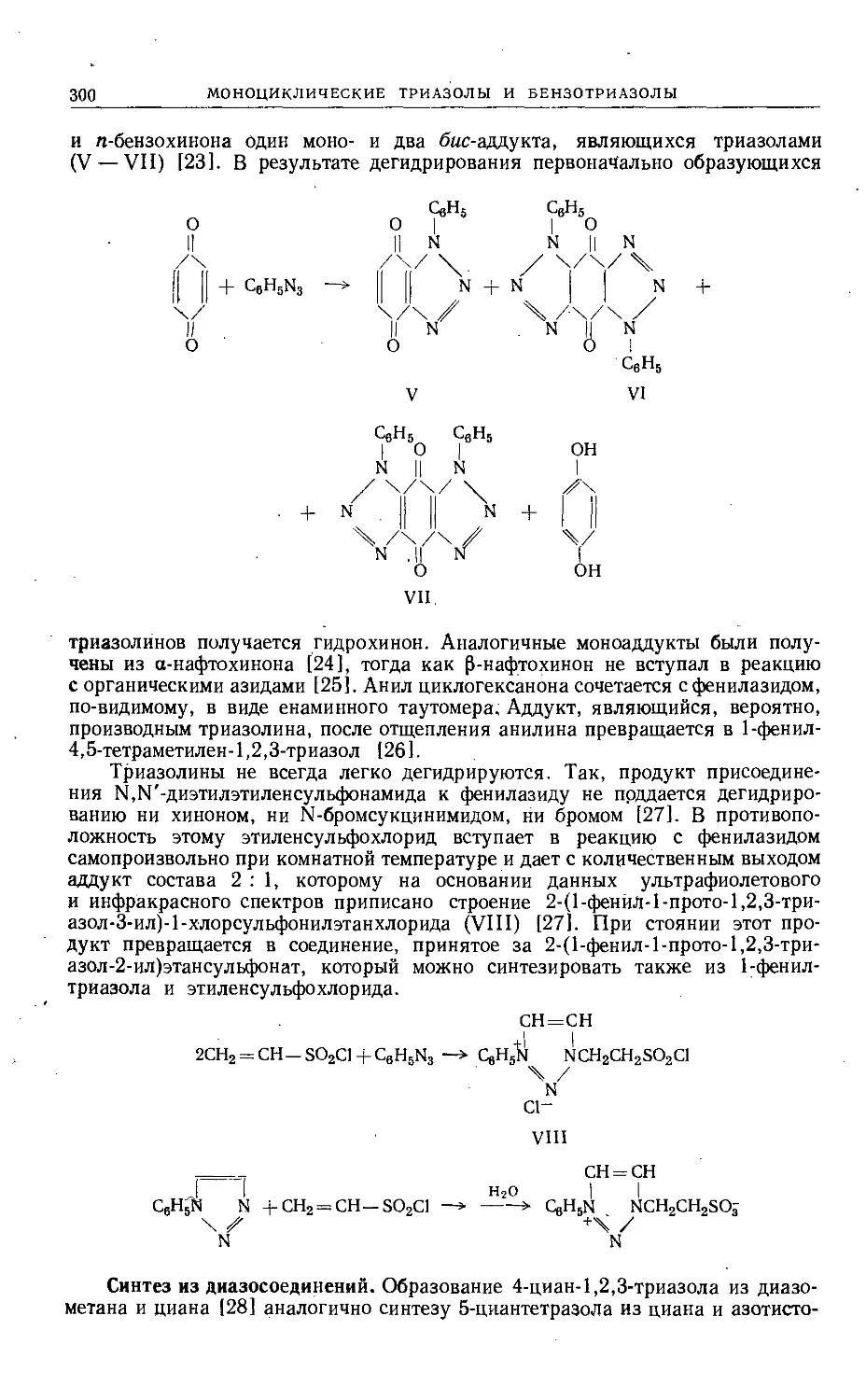




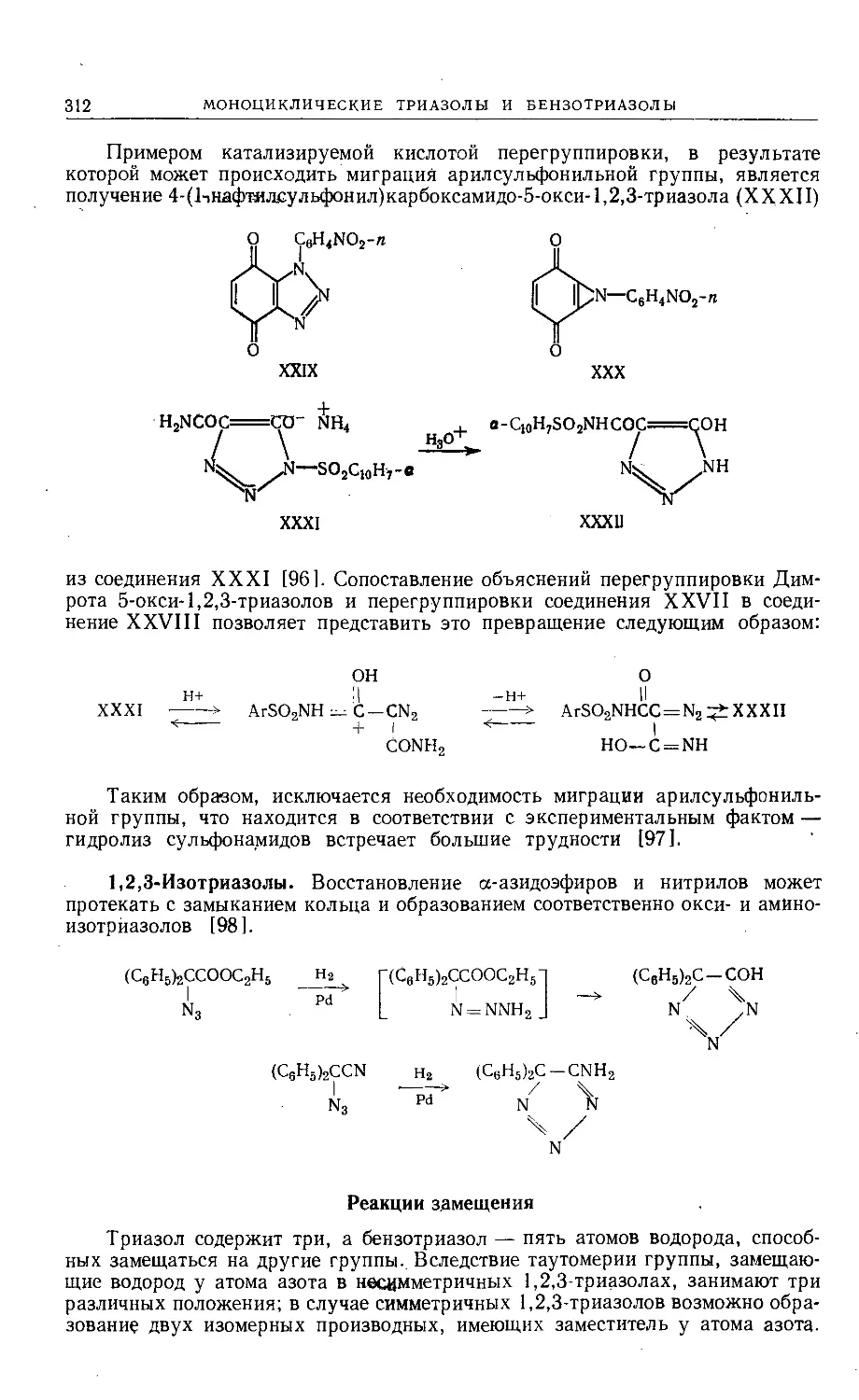
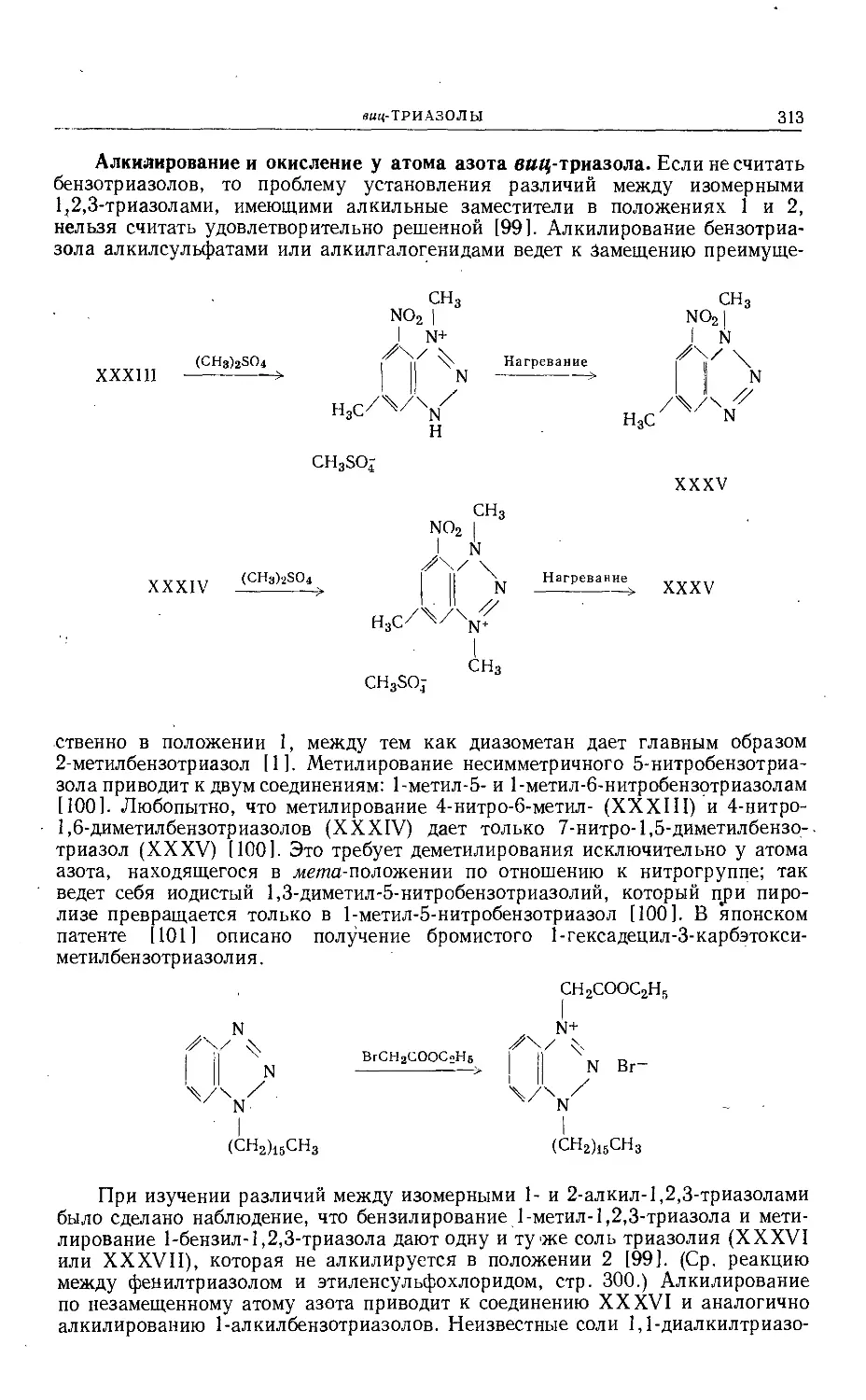


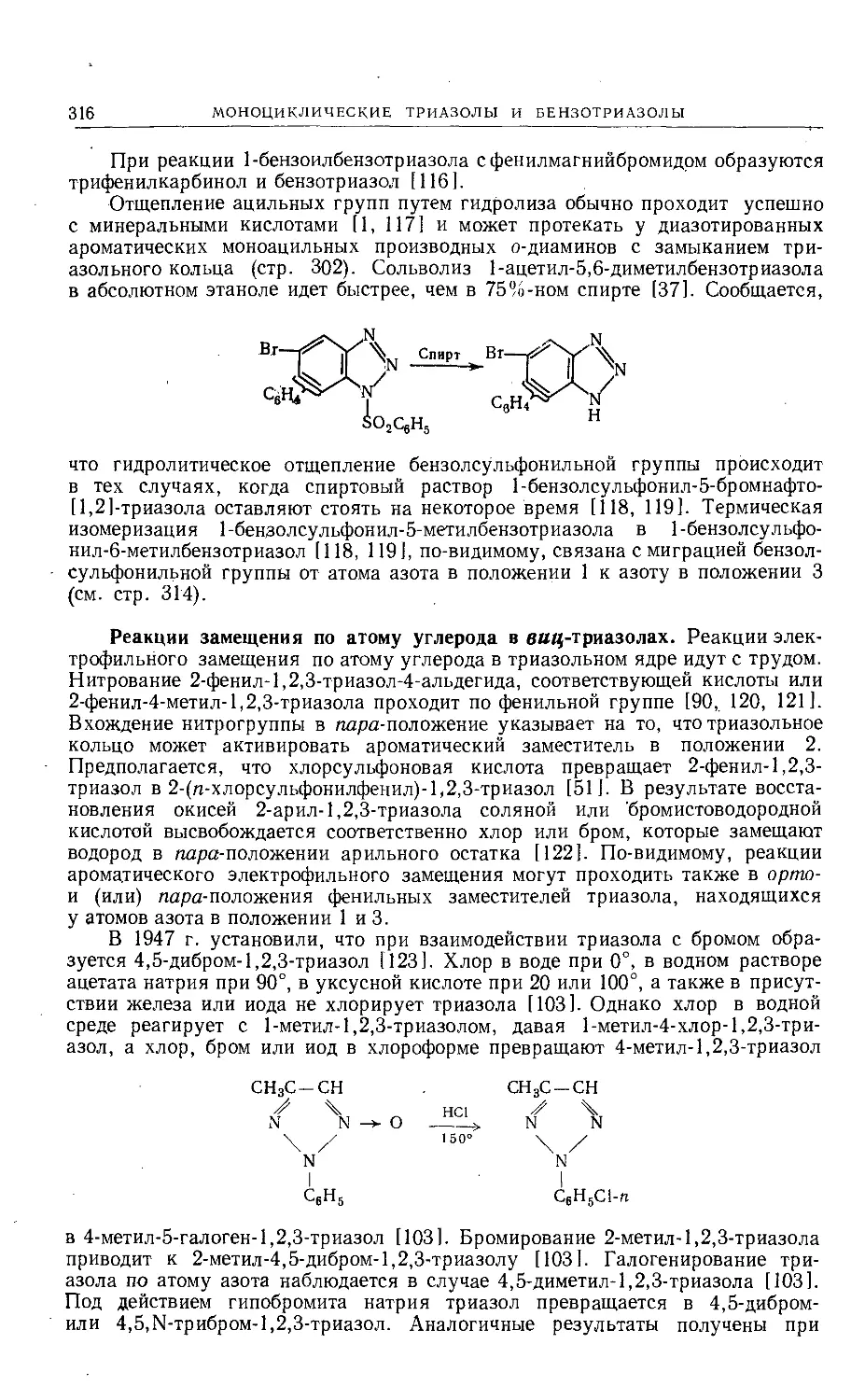

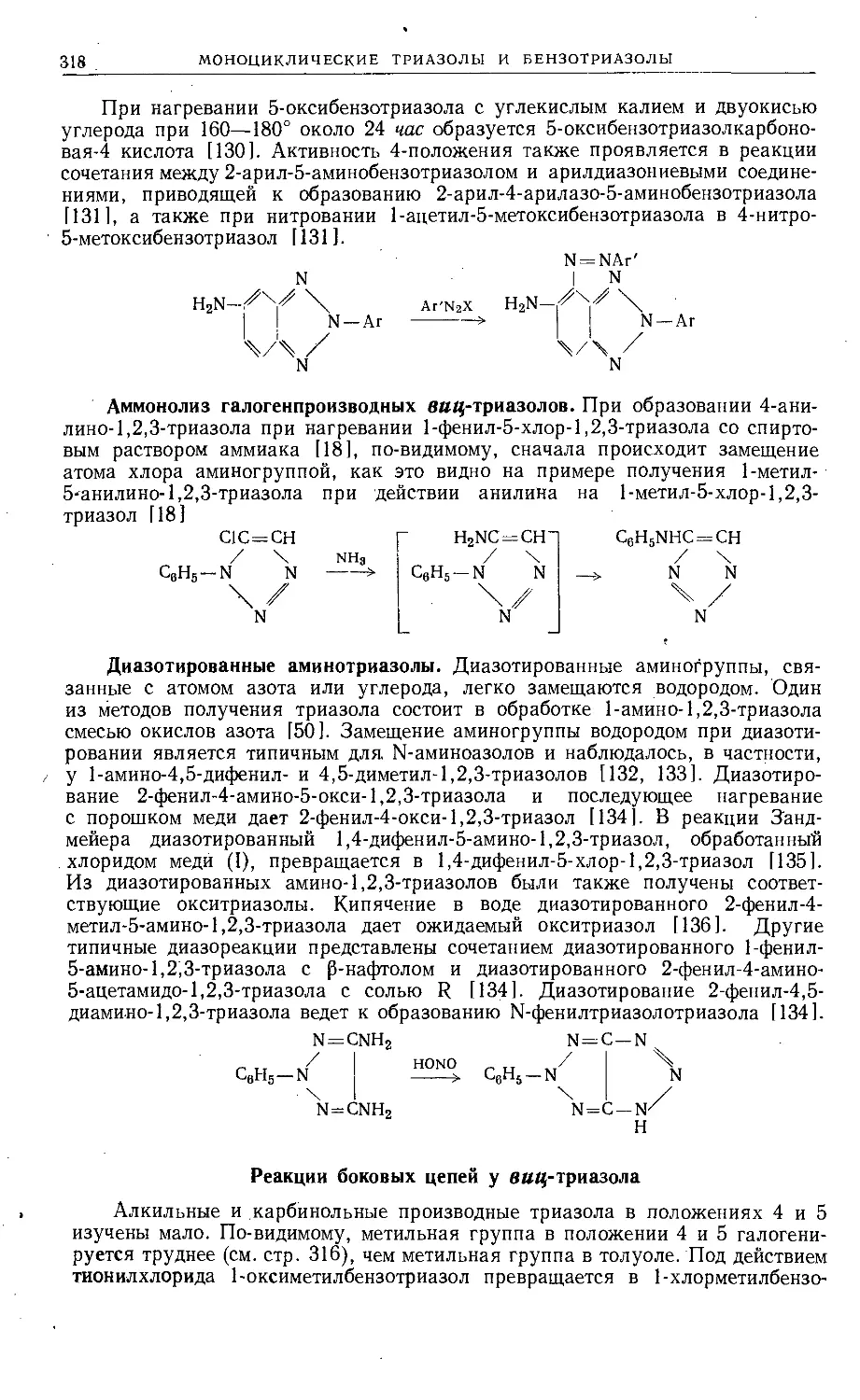





















































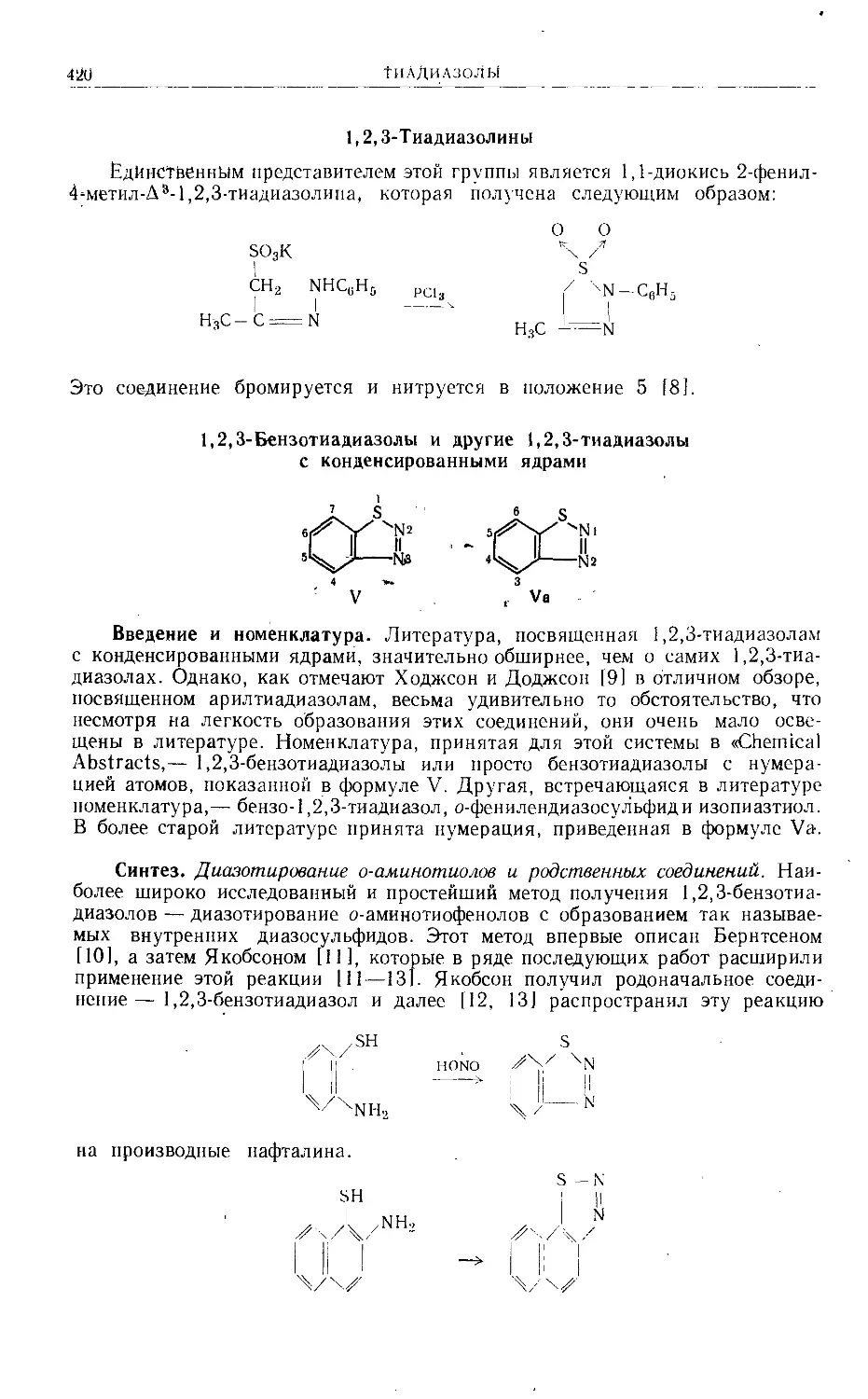
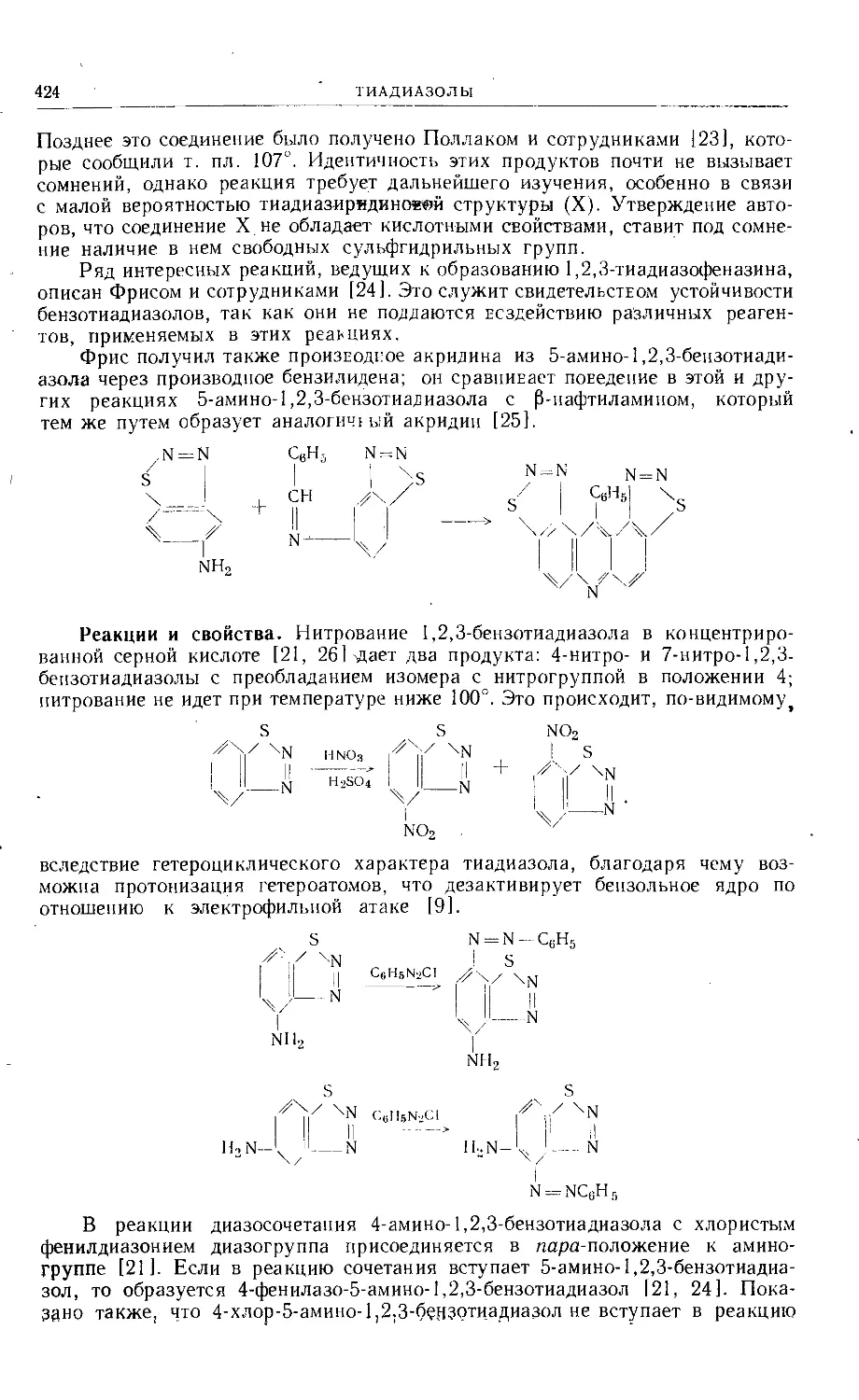












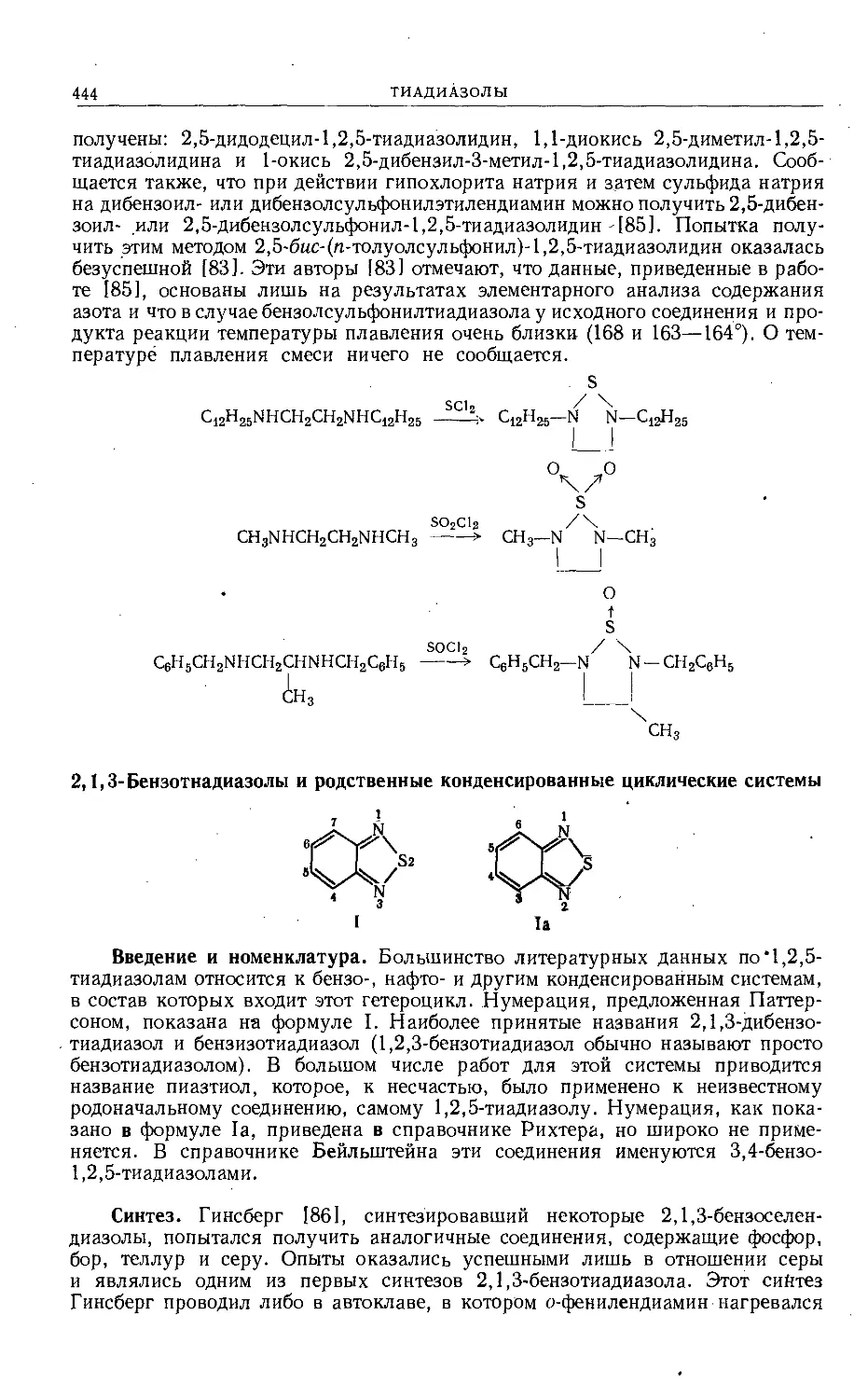



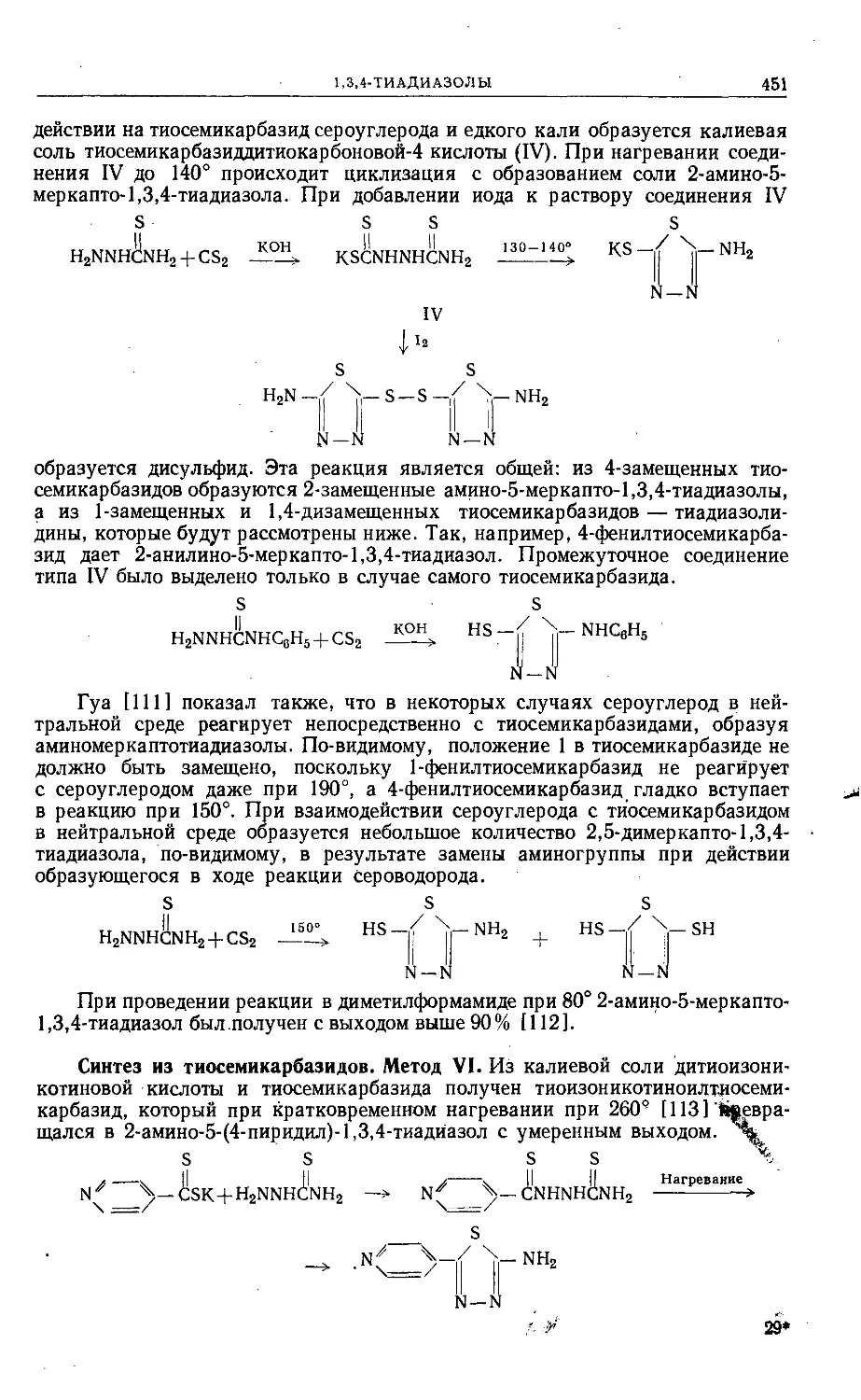
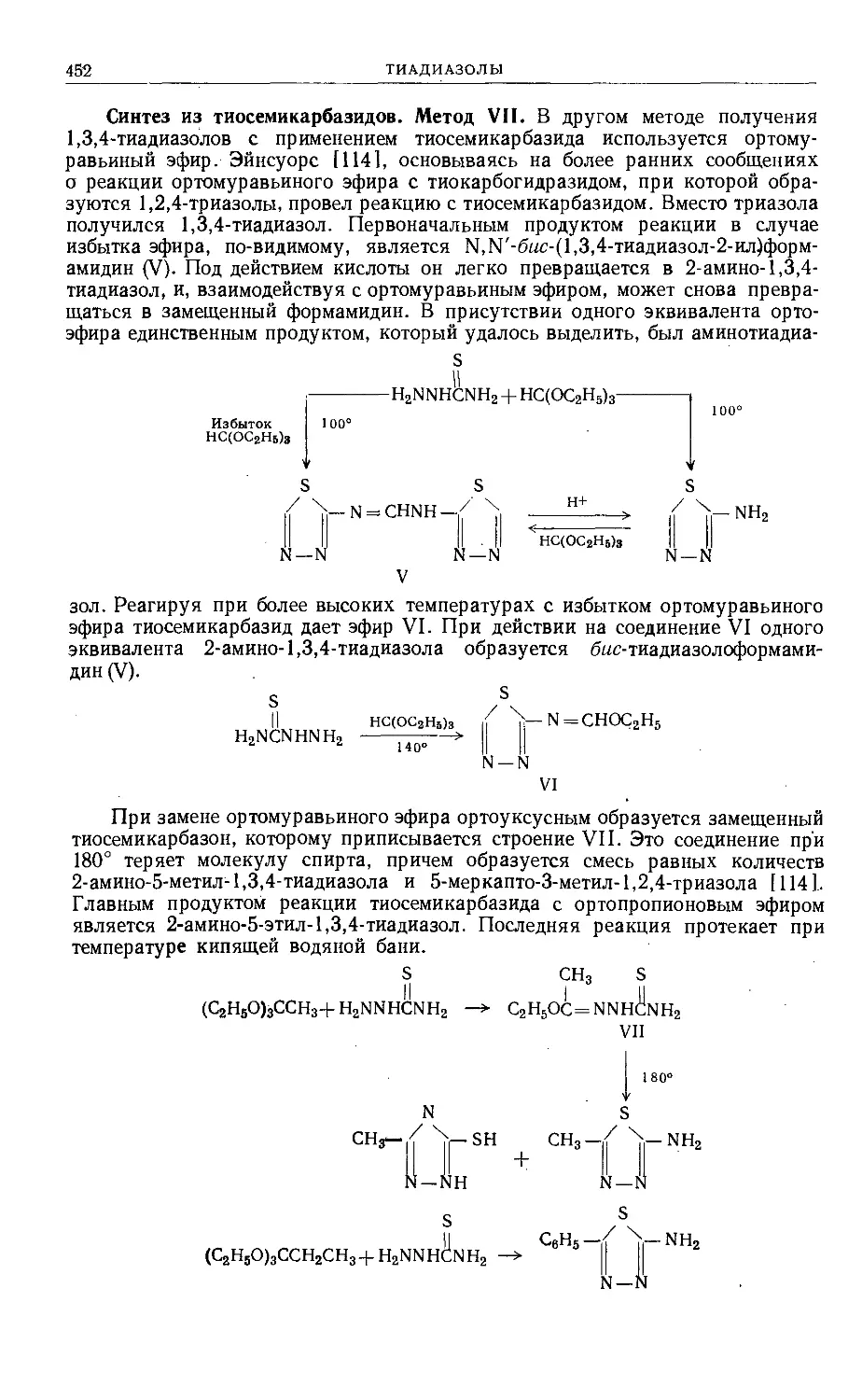
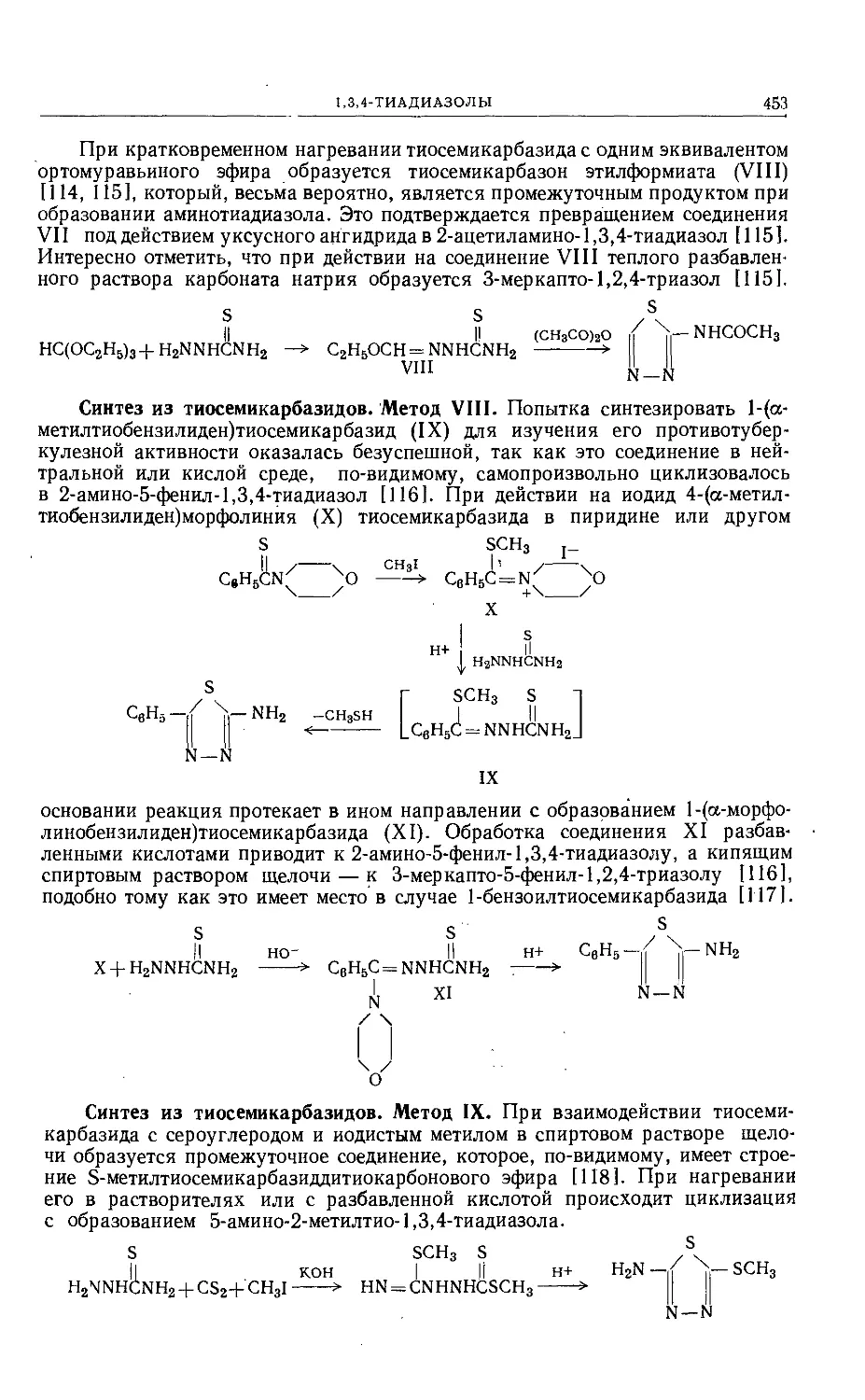
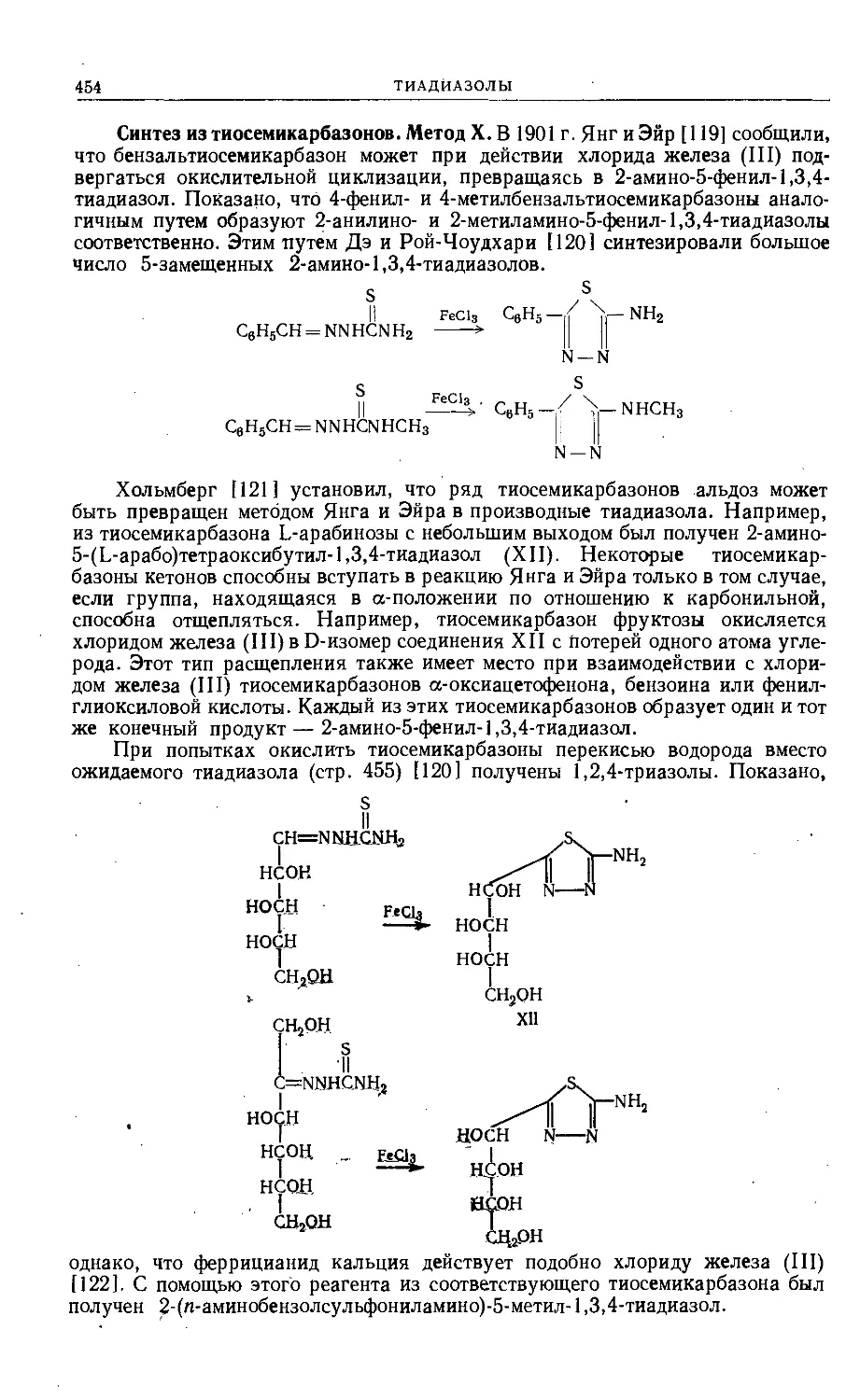
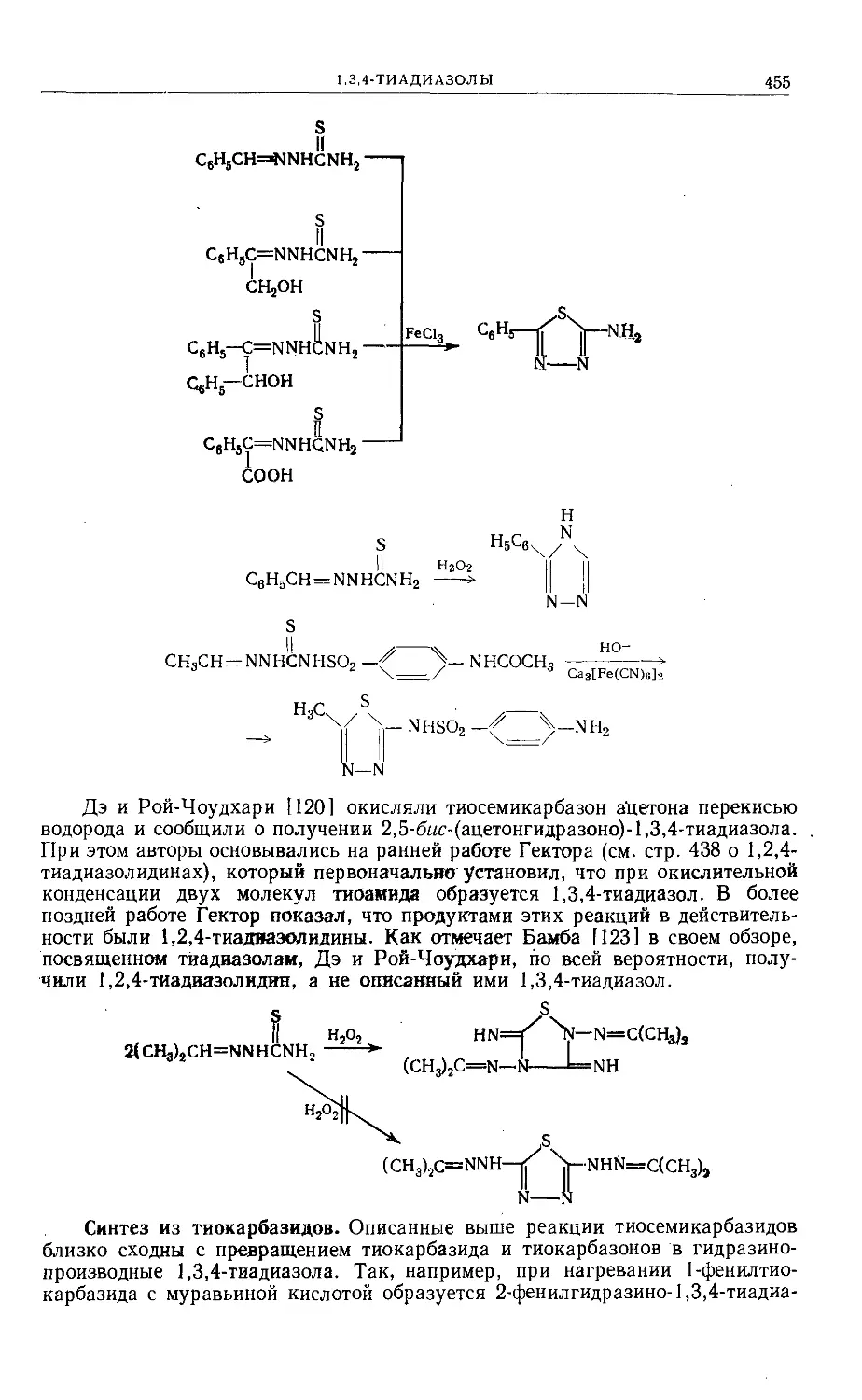

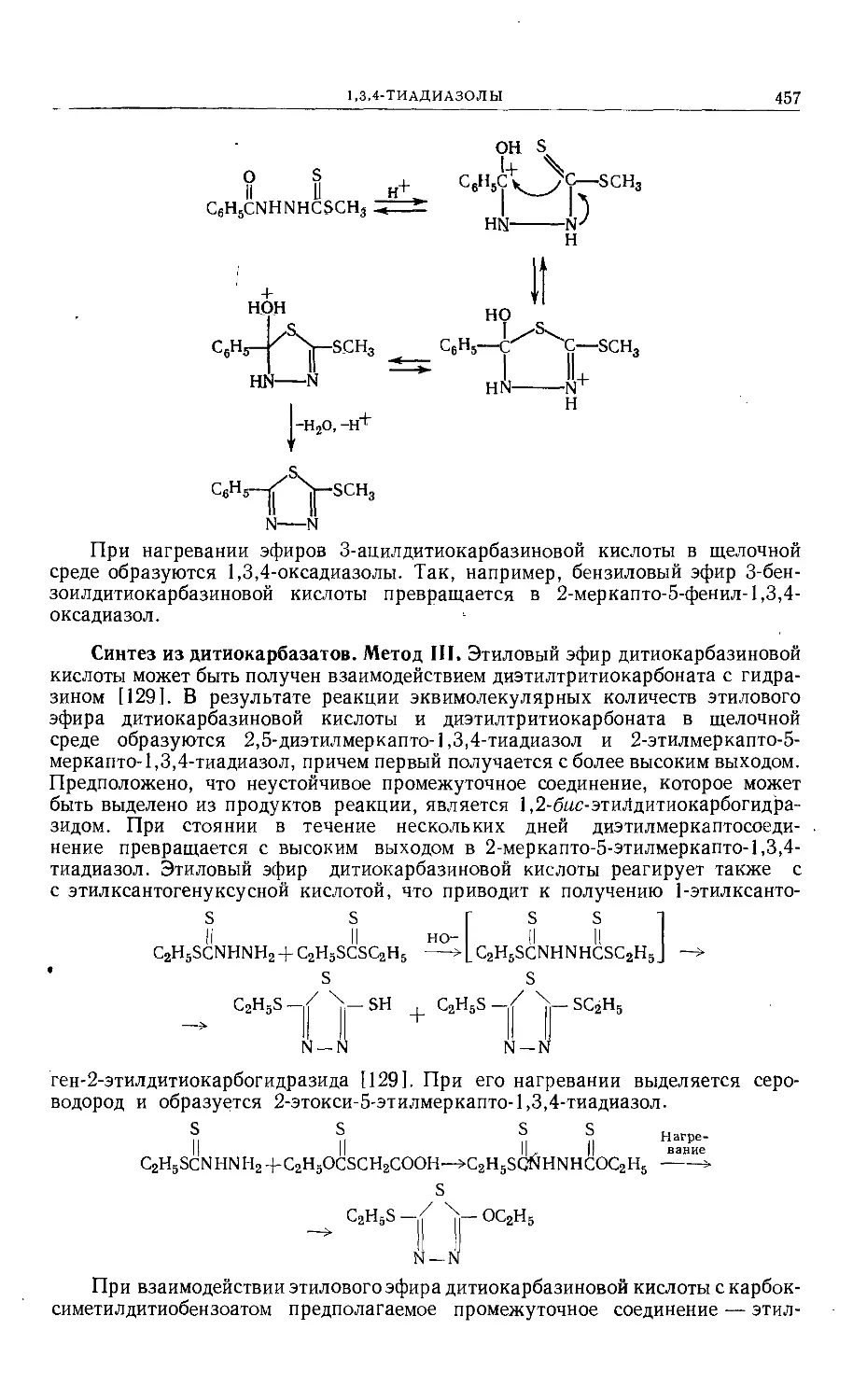

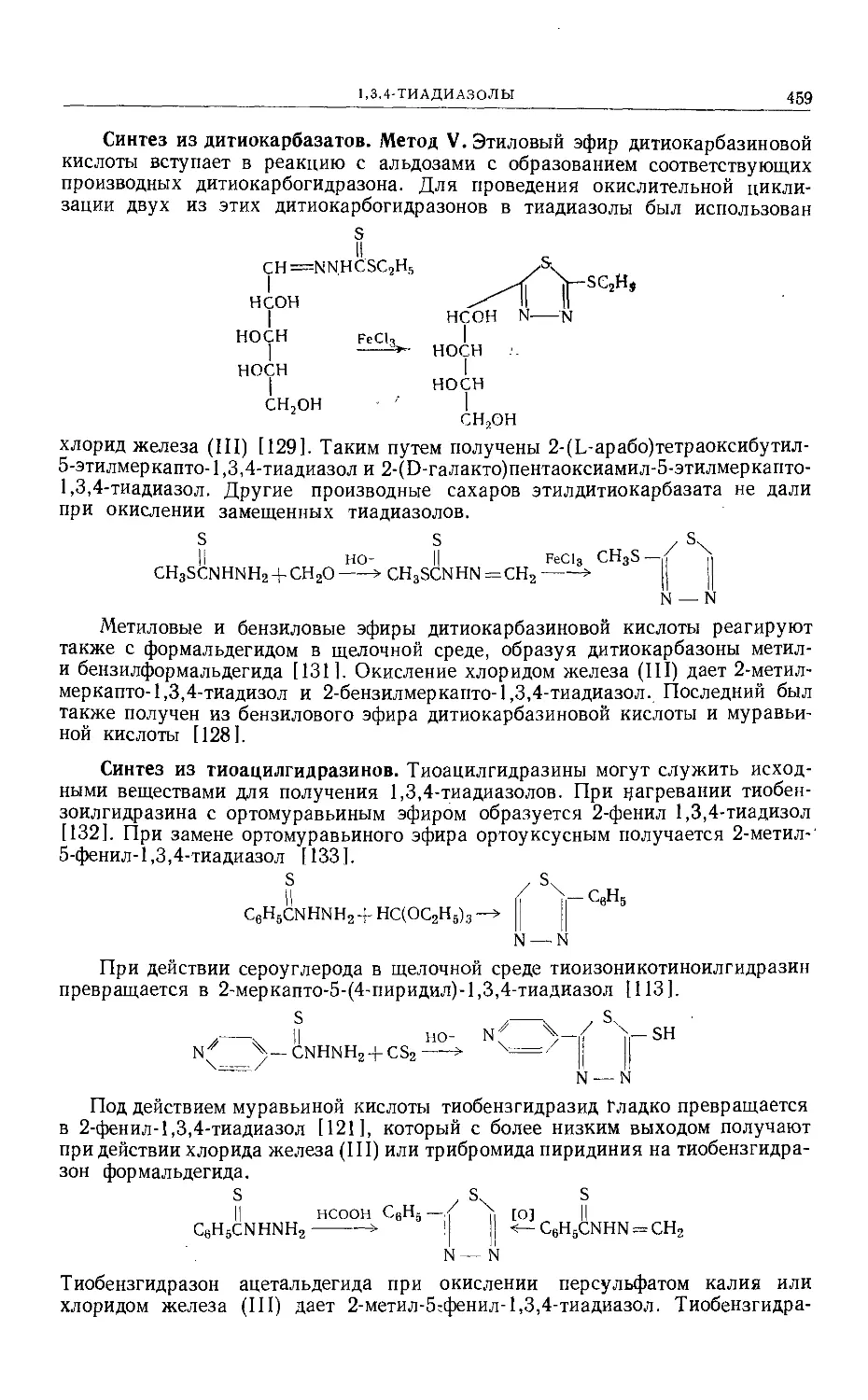

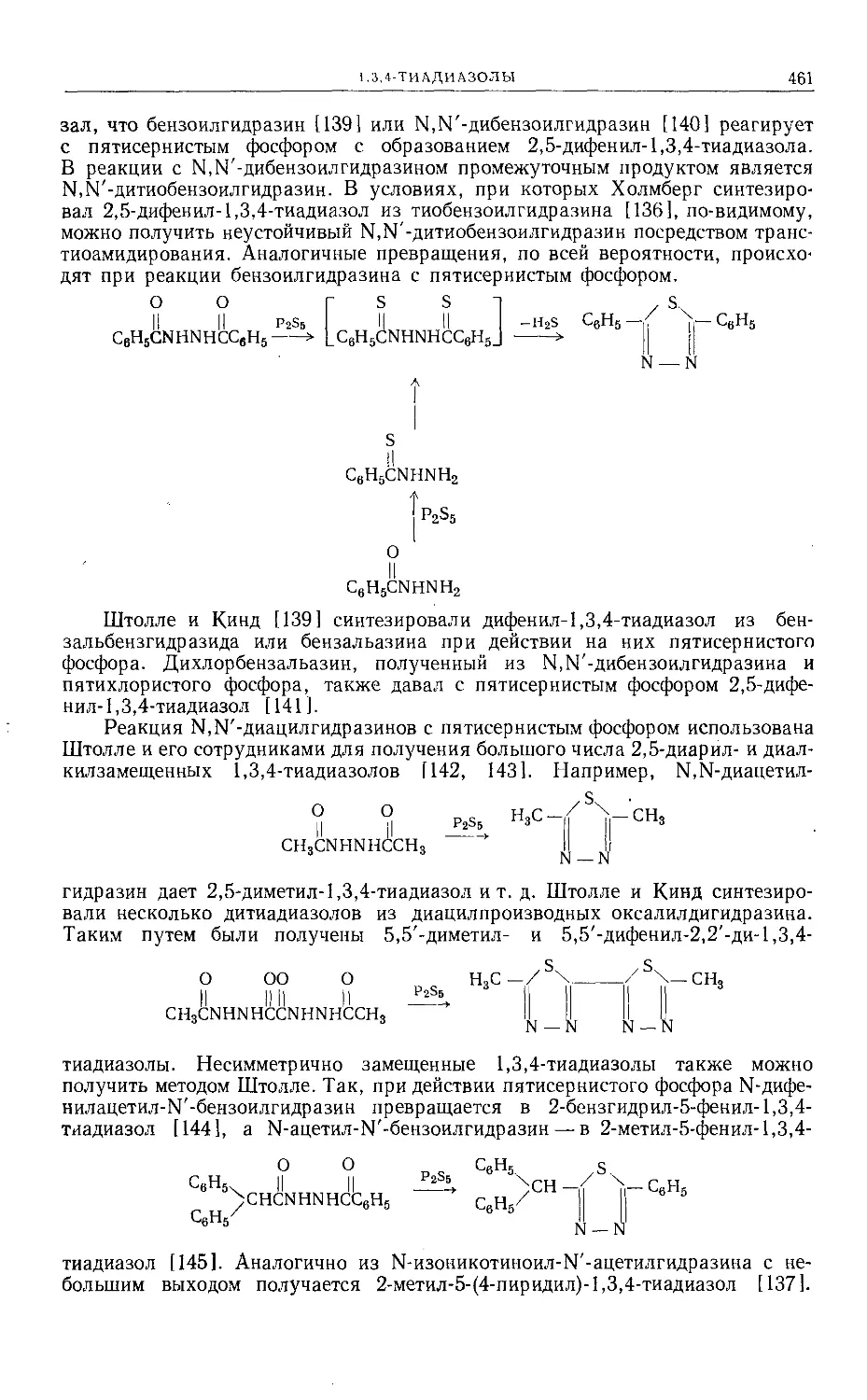

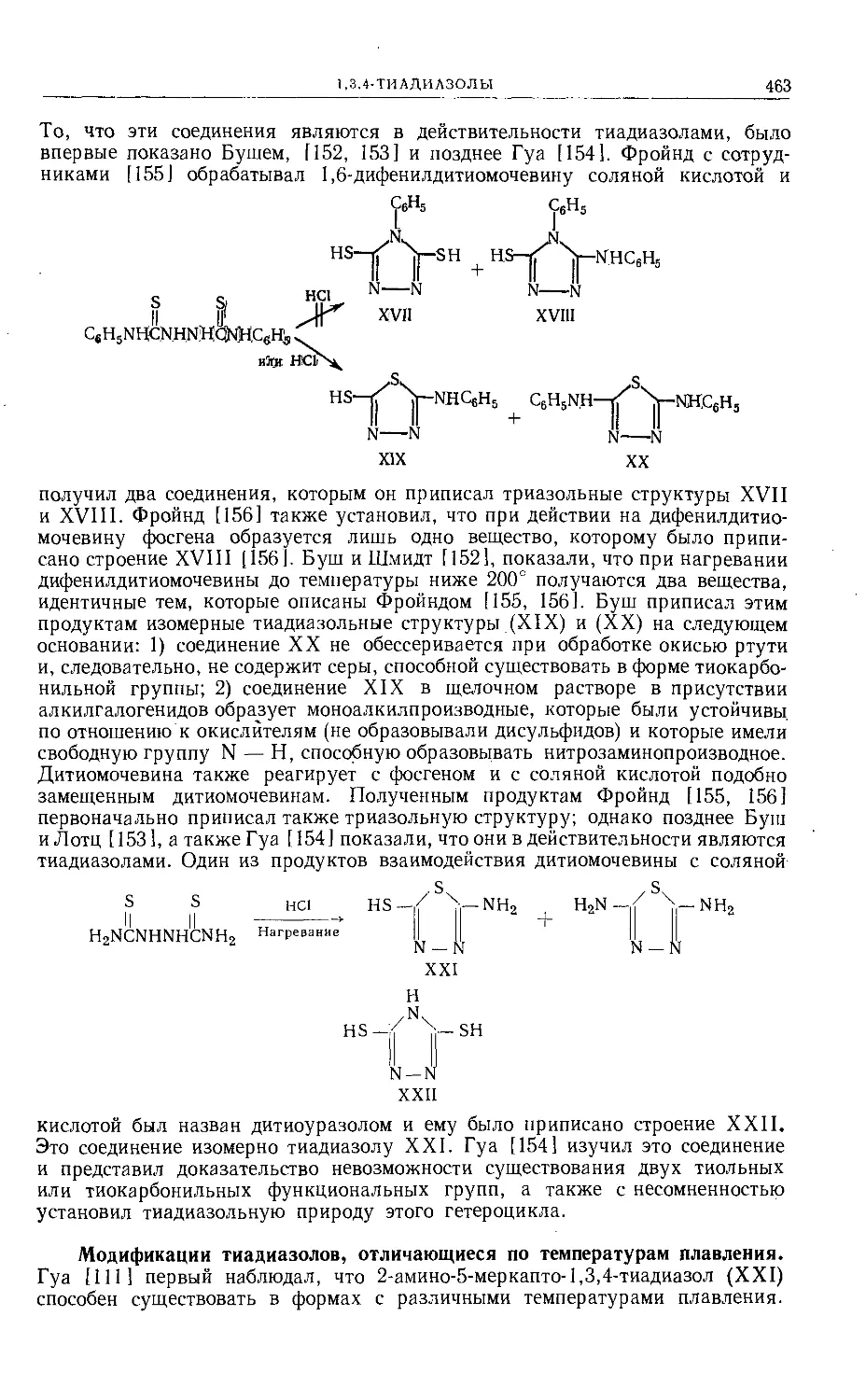



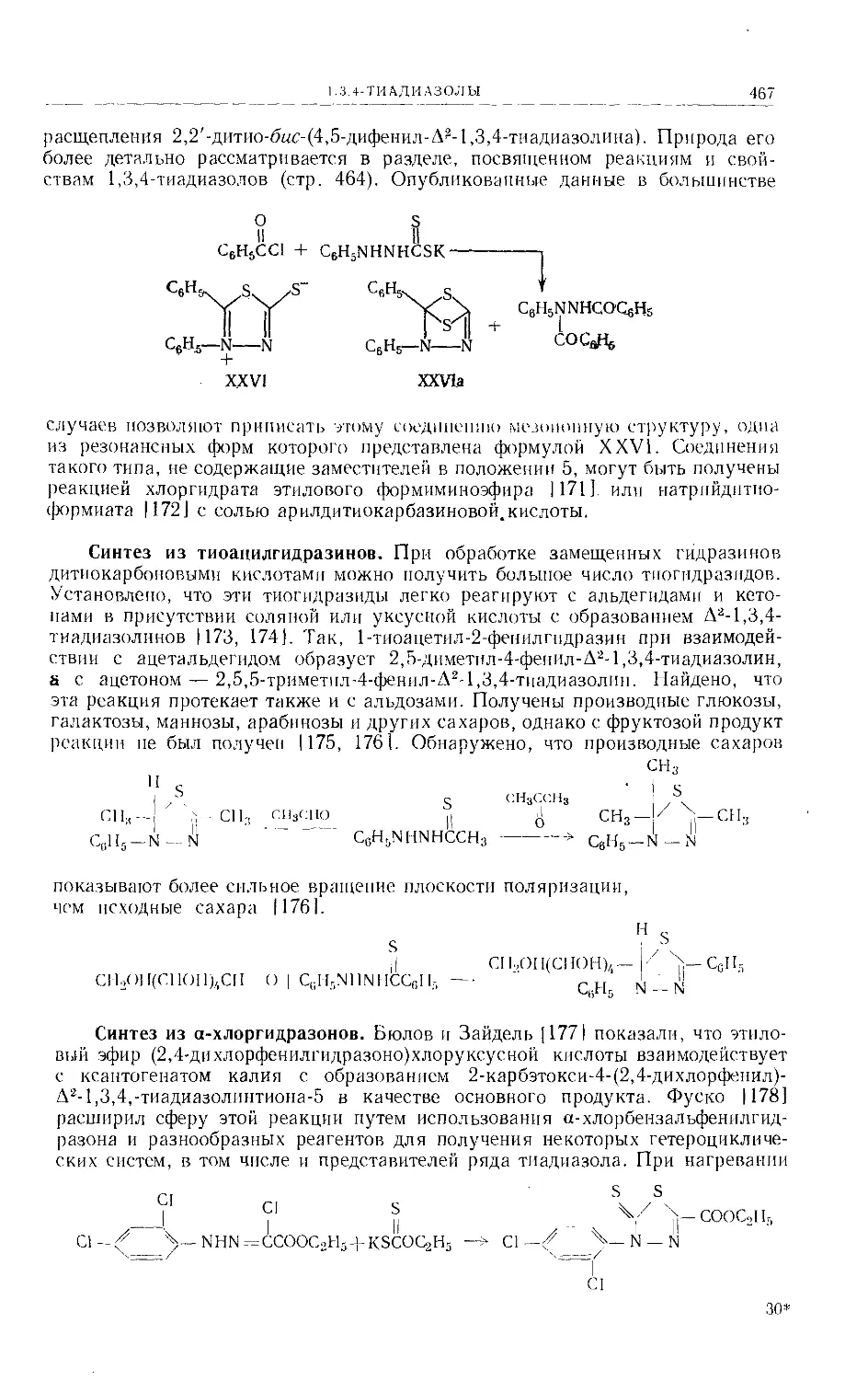




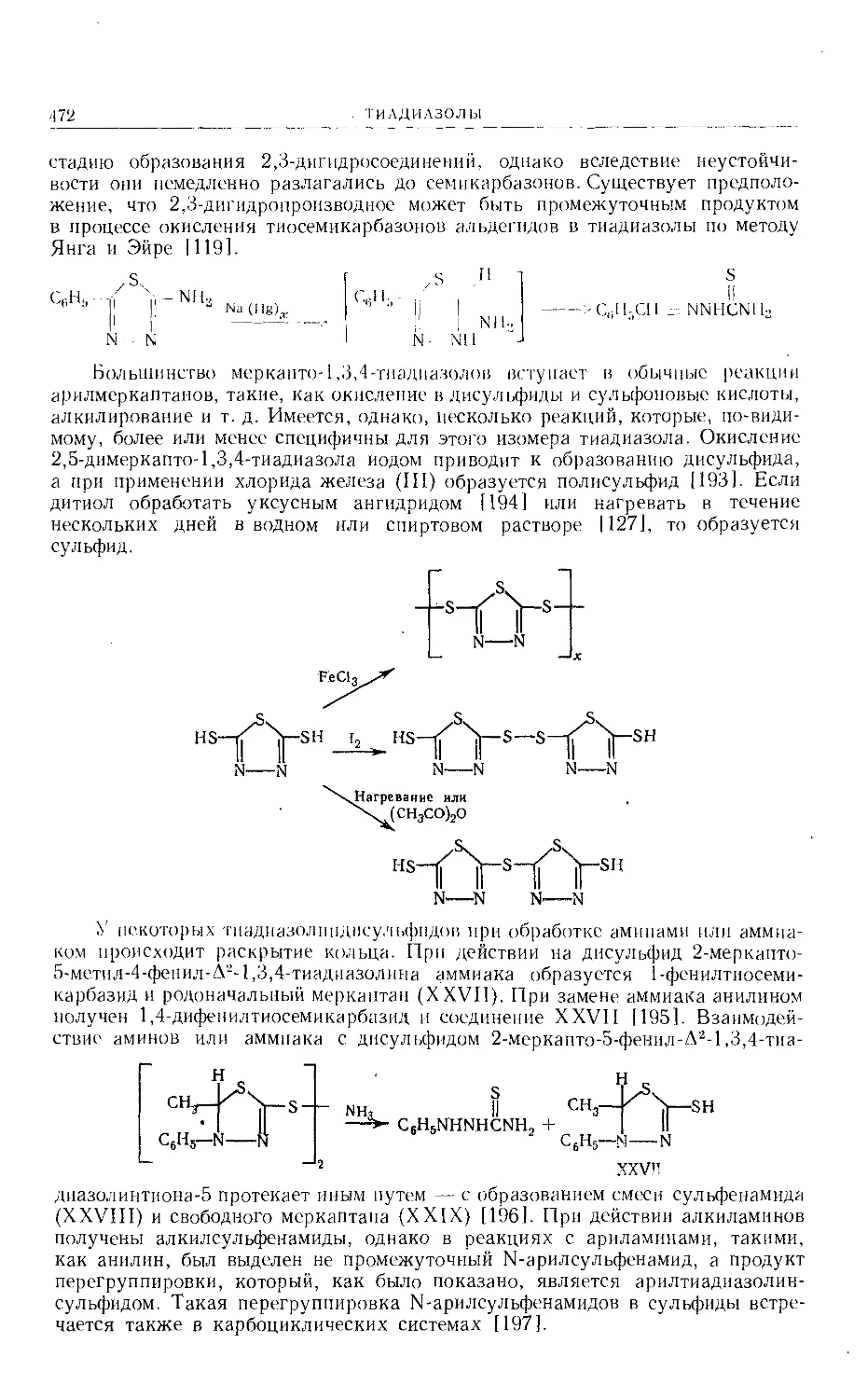
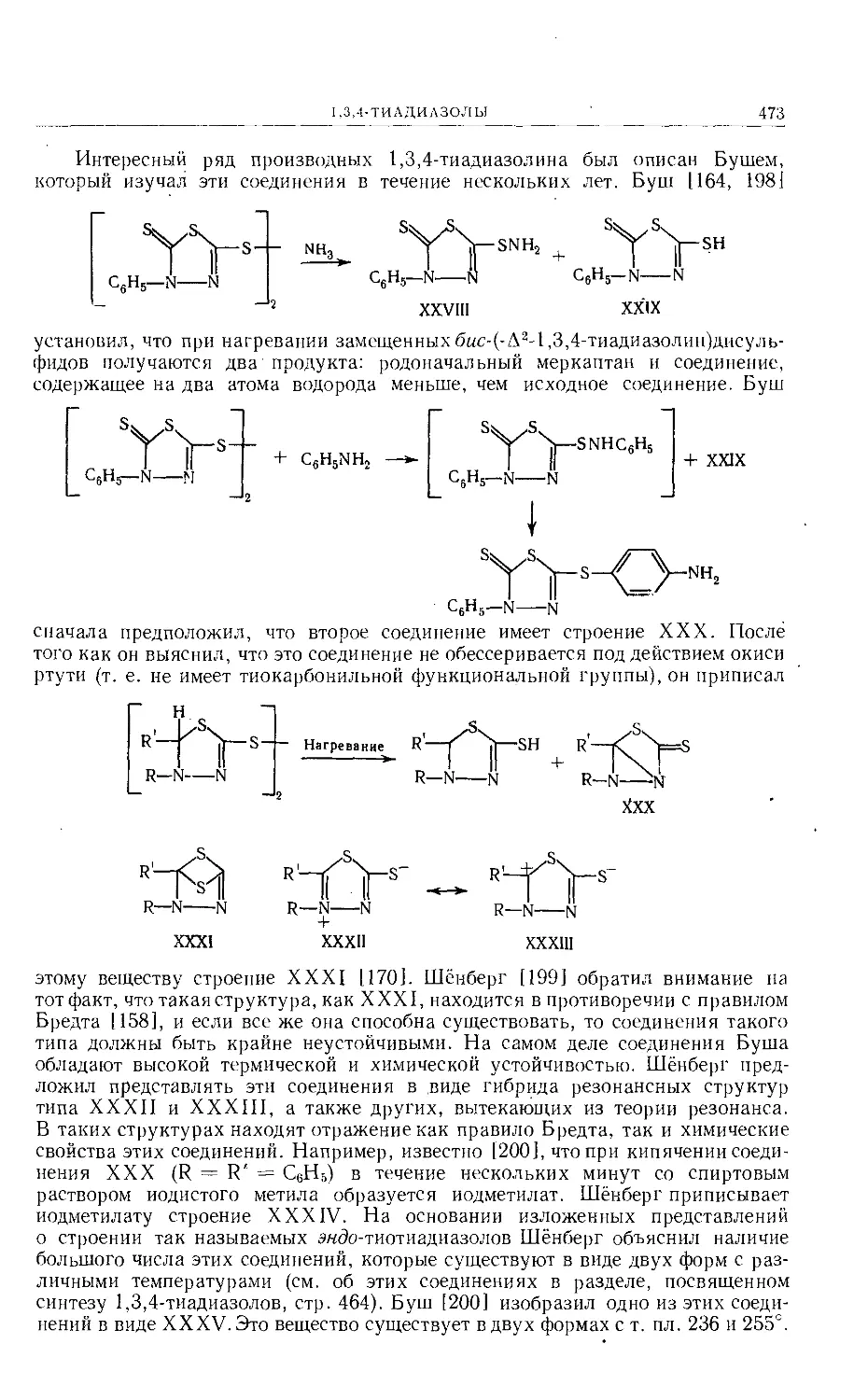



























Text
Больше химической литературы на .
vk.com/chemzone и vrww.cnemzone.injо
More chemistry books you can find on
our webresources vk.cpm/chemzone
and www.chemzone.info
vk.com/chemzone
ИЗДАТЕЛЬСТВО
«МИР »
HETEROCYCLIC
COMPOUNDS
Volume 7
Edited by
ROBERT. C. ELDERFIELD
NEW YORK
196 1
ГЕТЕРОЦИКЛИЧЕСКИЕ
СОЕДИНЕНИЯ
Том 7
Под редакцией
Р. ЭЛЬДЕРФИЛЬДА
Перевод с английского
В. А. ГЕТЛИНГА и В. В. ЩЕКИНА
Под редакцией
доктора хим. наук В. Г. ЯШУНСКОГО
ИЗДАТЕЛЬСТВО «МИР»
Москва 1965
Книга представляет собой очередной том серии «Гетероциклические
соединения», издаваемой с 1953 г. и предназначенной для широкого круга
химиков-органиков (в книгу включены только первые 7 глав американского
издания).
Подобно предыдущим томам, книга составлена по публикациям, выхо-
дящим в свет на английском, немецком, французском, японском и других
языках. Помещен большой обзор, основательно и систематически освещающий
химию соединений, содержащих атомы кислорода в двух конденсированных
гетероциклах. В круг рассматриваемых объектов входят многие природные
соединения, в том числе и фуранокумарины, среди которых найдены эффек-
тивные инсектициды.
Редакция литературы по химии
ПРЕДИСЛОВИЕ ЭЛЬДЕРФИЛЬДА
К РУССКОМУ ИЗДАНИЮ
Мне доставляет большое удовольствие написать предисловие к русскому
изданию седьмого тома «Гетероциклических соединений».
Когда в 1949 г. была начата публикация этого издания, то предпола-
галось выпустить не более трех томов критического, но не энциклопеди-
ческого характера. В дальнейшем стало ясно, что уложиться в такой объем
не удастся. В настоящее время число томов этой серии достигло семи
и готовится к изданию восьмой том. Однако по возможности сохранен
первоначальный замысел — дать критическую оценку значительного и все
возрастающего числа работ, посвященных химии гетероциклических соеди-
нений, этого важного раздела органической химии. Читатель не найдет
в этом труде перечисления всех тех гетероциклических соединений, описание
которых имеется в соответствующих изданиях. Мы постарались лишь про-
иллюстрировать основные положения путем тщательного подбора примеров.
При издании настоящего тома снова возникла проблема номенклатуры,
которая, насколько возможно, отвечает принятой в Chemical Abstracts.
При применении другой номенклатуры это всегда оговаривается. Двойные
связи в кольце обозначены на всех структурных формулах, но атомы водо-
рода кольца не приводятся, за исключением тех случаев, когда это вызвано
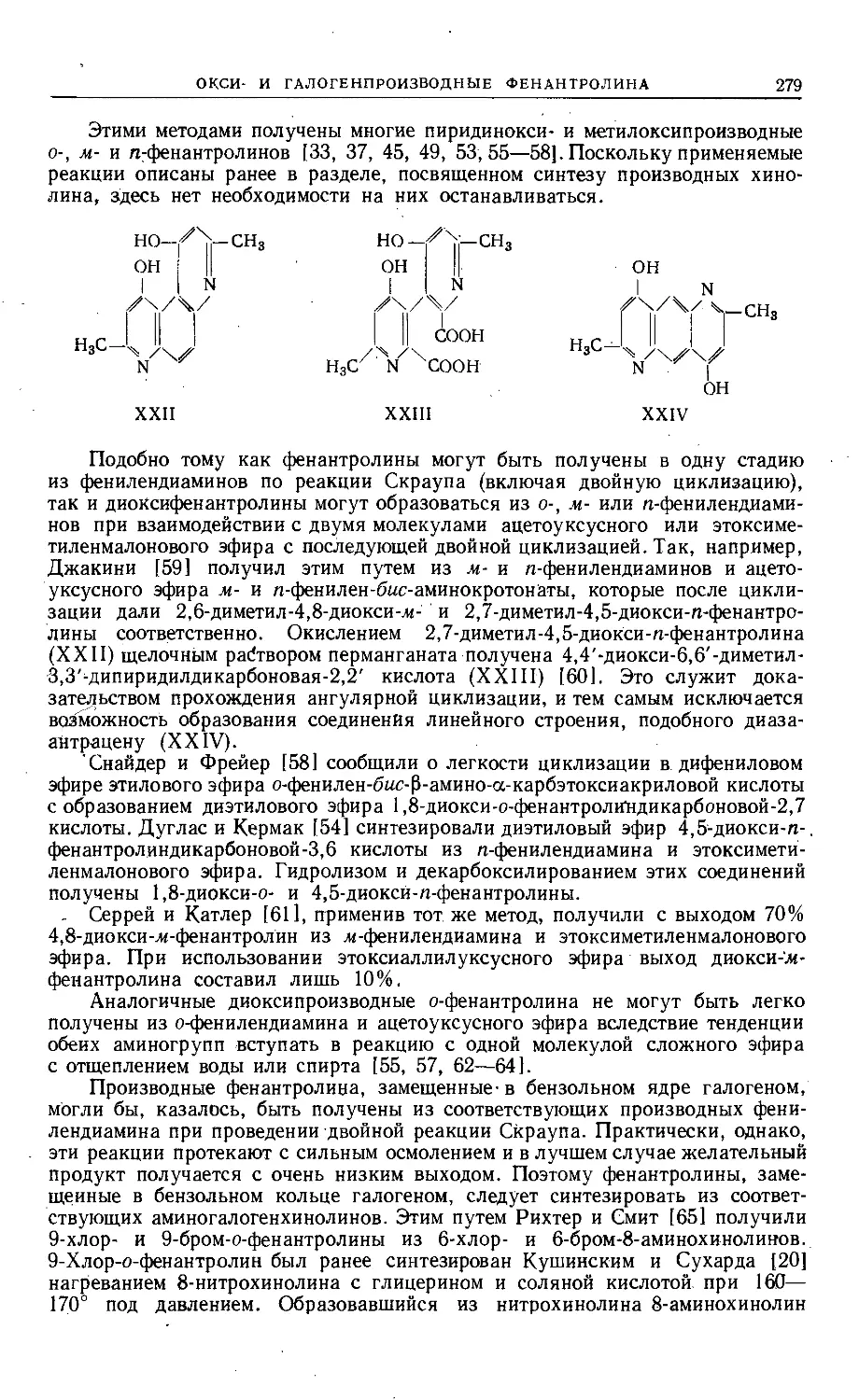
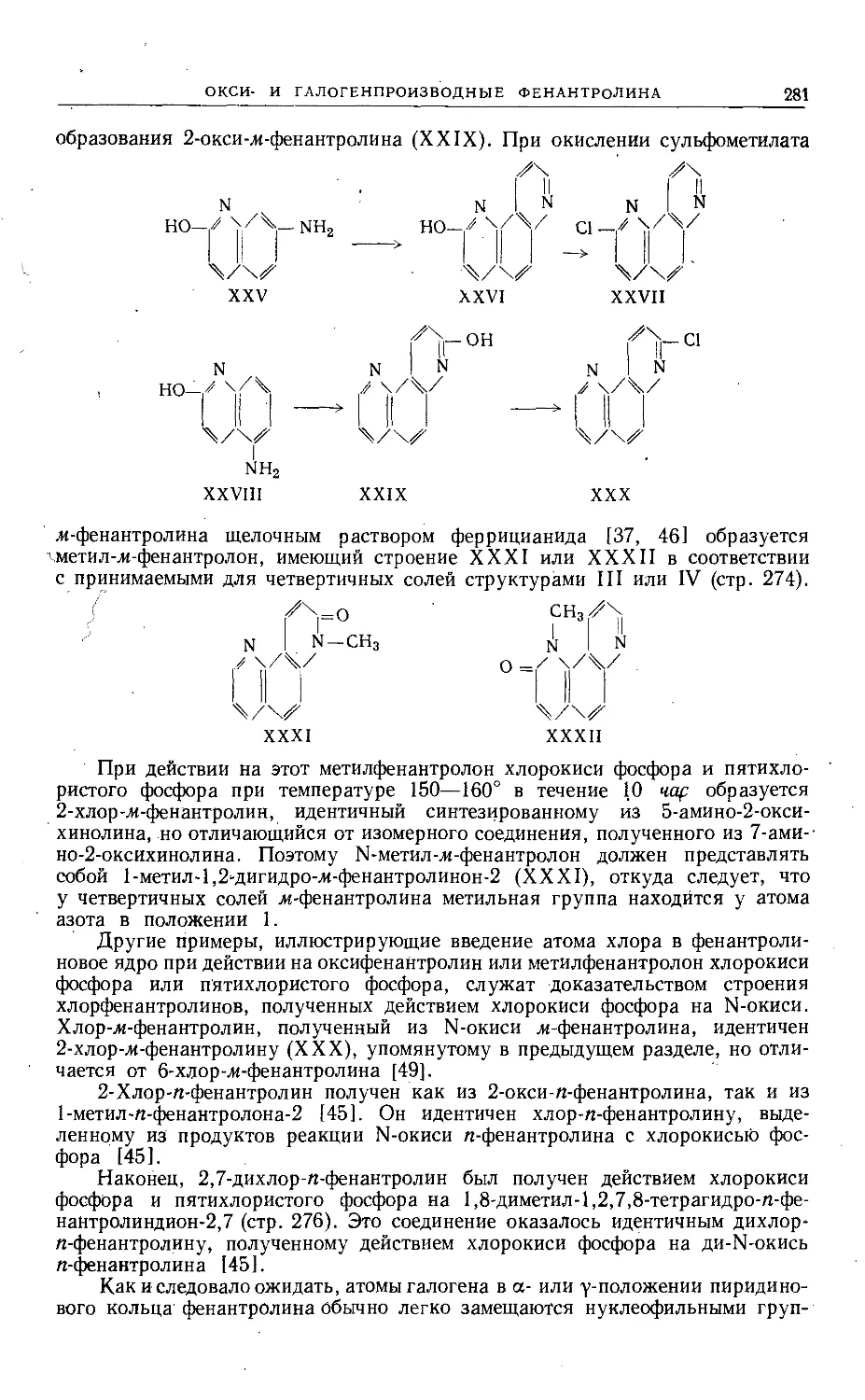
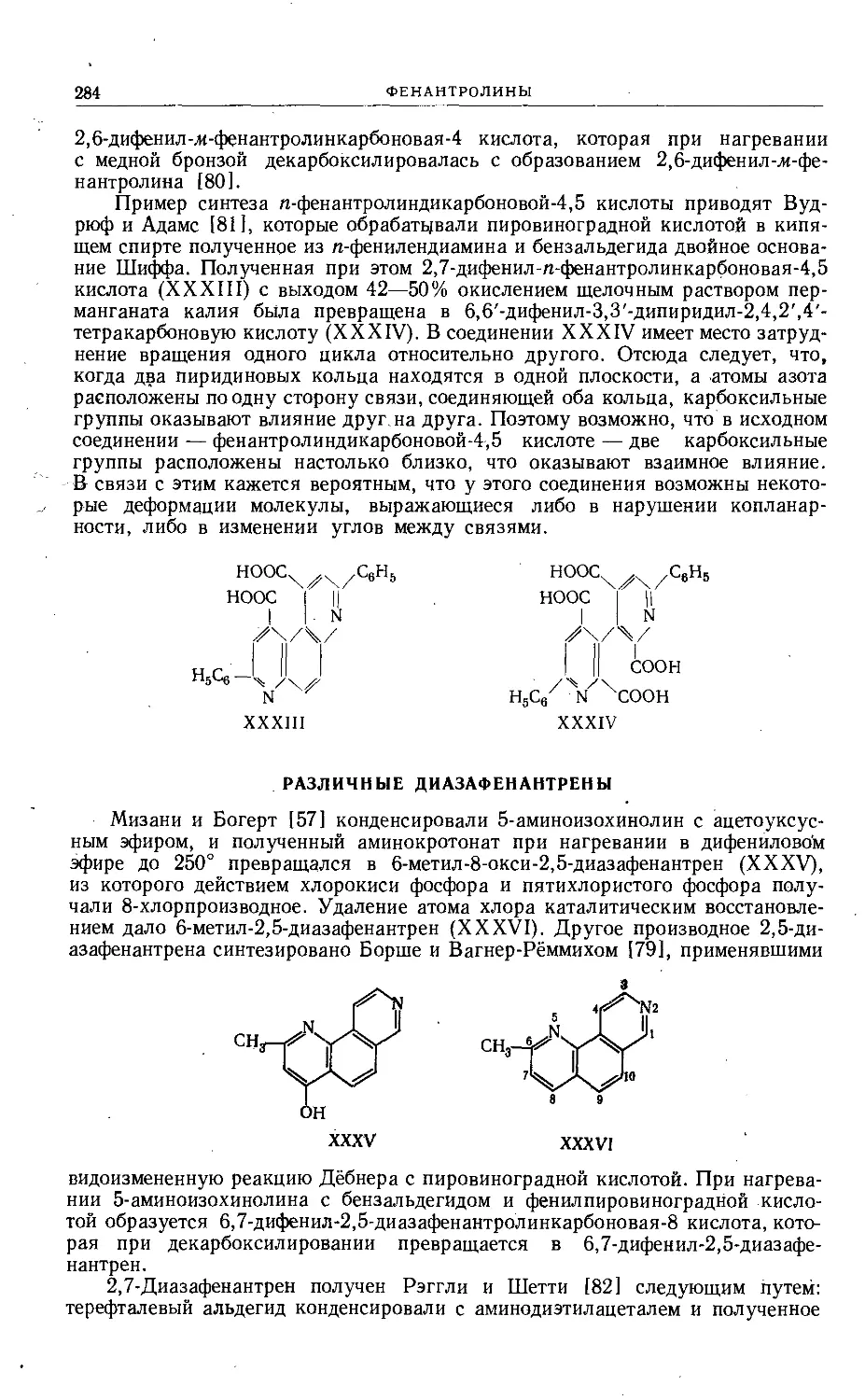
требованиями наглядности.
Мне приятно поблагодарить всех тех, кто способствовал выходу в свет
этого тома. Я благодарен также моей жене за неоценимую помощь при чте-
нии корректур и подготовке указателя. Мы очень ценим, что русские коллеги
признали это издание заслуживающим перевода, и искренне надеемся, что
наше сотрудничество будет способствовать лучшему взаимопониманию.
Ann Arbor, Michigan,
январь 1964 г.
Роберт Эльдерфильд
Глава I
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
В РАЗЛИЧНЫХ КОЛЬЦАХ
У. Б. Валли
ВВЕДЕНИЕ
В настоящей главе рассматриваются гетероциклические соединения,
содержащие лишь кислород в качестве гетероатома и имеющие два (или более)
таких атомов. Этот класс соединений включает большое количество природных
веществ, в том числе и обладающих биологической активностью. Разрозненные
сведения, имеющиеся в отношении соединений этой группы, сведены воедино
в настоящей главе, где рассматриваются синтез и расщепление как природных,
так и синтетических Представителей этого ряда.
Рассматриваемые соединения по возможности расположены в порядке
возрастания сложности молекулы; соединения с пятичленными гетероциклами
предшествуют шестичленным гетероциклическим системам, хотя в некоторых
случаях имеются вынужденные отклонения от этого порядка.
ФУРАНОФУРАНЫ. БИСФУРАНОИДНЫЕ ЛИГНАНЫ
Многие фуранофураны встречаются в природе и образуют группу бисфу-
раноидных лигнанов, в свою очередь состоящую из двух подгрупп: одна из них
включает соединения, содержащие метилендиоксигруппы в 3-, 4-, 3'- и 4'-
положениях, а другая — соединения, имеющие в тех же положениях метоксиль-
ные или гидроксильные группы. К первому классу относятся сезамин (I)
[1, 2] и сезамолин (II) [3], содержащийся в семенах сезама и в сезамовом
масле, а также изосезамин [4], L-сезамин [5] и азаринин [6] (I) (эпи-Ь-сеза-
мин), первоначально названный ксантоксилом S [7], который встречается
во многих растениях, произрастающих в Азии, и в коре колючего ясеня
Xanthoxylum americanum Mill.
Второй класс включает пинорезинол [8] (III, R = ОН, R' = СН3О),
входящий в состав смолистых выделений различных хвойных, и эвдесмин [9 ]
(III, R = ОН, R' = СНзО), выделенный из камеди эвкалипта.
Деметиленирование сезамина, сопровождающееся метилированием, дало
возможность установить определенную связь между обеими группами соеди-
нений. Например, D-сезамин был превращен таким путем в оптический антипод
эвдесмина, О-диметиловый эфир D-пинорезинола [10]. Аналогичным образом
L-азаринин был превращен в L-эвдесмин.
Этим соединениям посвящено большое число работ (см. библиографию
в обзоре Карнмальм и др. [3]). В настоящей главе приведены только данные,
характеризующие конечные стадии определения их строения.
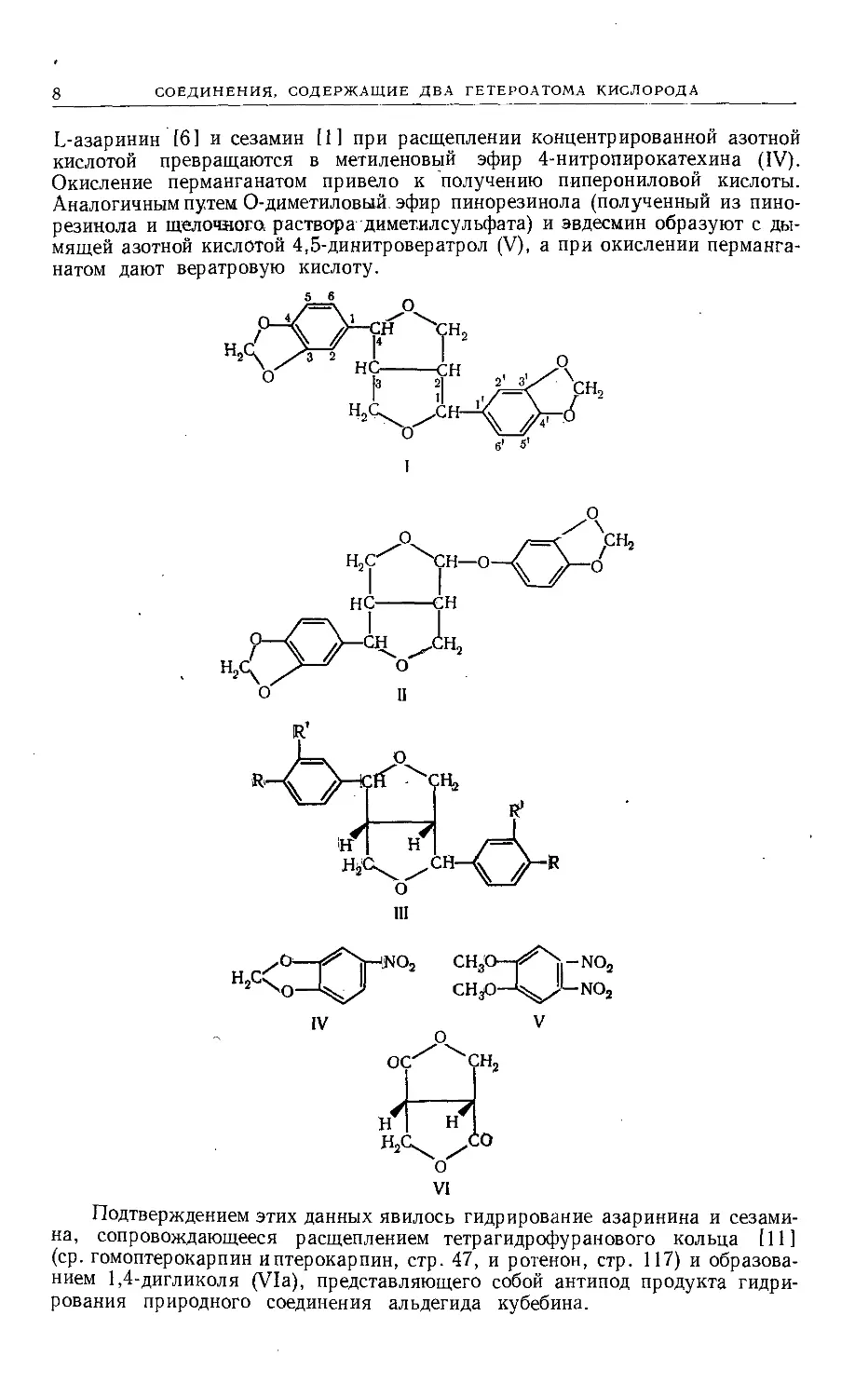
Общие черты структуры этих веществ были установлены: а) окислением,
в результате которого образуется пиперониловая или вератровая кислота
совместно с ди-у-лактоном а,|3-бпс-(оксиметил)янтарной кислоты (VI),
и б) расщеплением азотной кислотой, иногда с предварительным бромирова-
нием, в результате чего образуются нитро- или бромнитрофенолы. Например,
8
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
L-азаринин [6] и сезамин [1] при расщеплении концентрированной азотной
кислотой превращаются в метиленовый эфир 4-нитропирокатехина (IV).
Окисление перманганатом привело к получению пиперониловой кислоты.
Аналогичным путем О-диметиловый эфир пинорезинола (полученный из пино-
резинола и щелочного раствора диметилсульфата) и эвдесмин образуют с ды-
мящей азотной кислотой 4,5-динитровератрол (V), а при окислении перманга-
натом дают вератровую кислоту.
VI
Подтверждением этих данных явилось гидрирование азаринина и сезами-
на, сопровождающееся расщеплением тетрагидрофуранового кольца [11]
(ср. гомоптерокарпин иптерокарпин, стр. 47, и ротенон, стр. 117) и образова-
нием 1,4-дигликоля (Via), представляющего собой антипод продукта гидри-
рования природного соединения альдегида кубебина.
ФУРАНОФУРАНЫ. БИСФУРАНОИДНЫЕ ЛИГМАНЫ
9
Основным препятствием на пути к полному выяснению строения этих
веществ служит наличие у них четырех асимметрических центров (атомы
углерода в положении 1,2, 3 и 4 в формулах I и II), в связи с чем возникают
стереохимические проблемы такого же порядка, как и в случае биотина (т. I,
стр. 199).
При кипячении со спиртовым раствором хлористого водорода природный
сезамин частично превращается в изосезамин, что считают результатом эпиме-
ризации асимметрических центров 1 или 4. Эта реакция, будучи обратимой,
приводит к образованию равновесной смеси сезамина и изосезамина независи-
мо от того, какое вещество служит исходным. Азаринин является оптическим
антиподом изосезамина и при обработке кипящим спиртовым раствором хло-
ристого водорода образует подобным же образом равновесную смесь изосеза-
мина и L-сезамина [6]. Схематически это превращение изображено ниже.
О-Диметиловый эфир D-пинорезинола
D-Сезамин
[a]D + 68° RCH2
f^+Hd !
rt 1’2 I
Изосезамин------> CH ------
la]D H- 122° |
CH2OH
Эвдесмин
[a]D—68°
L-Азаринин
CH2R [<x]d — 122c
I ' П +Hcl
I +H 2
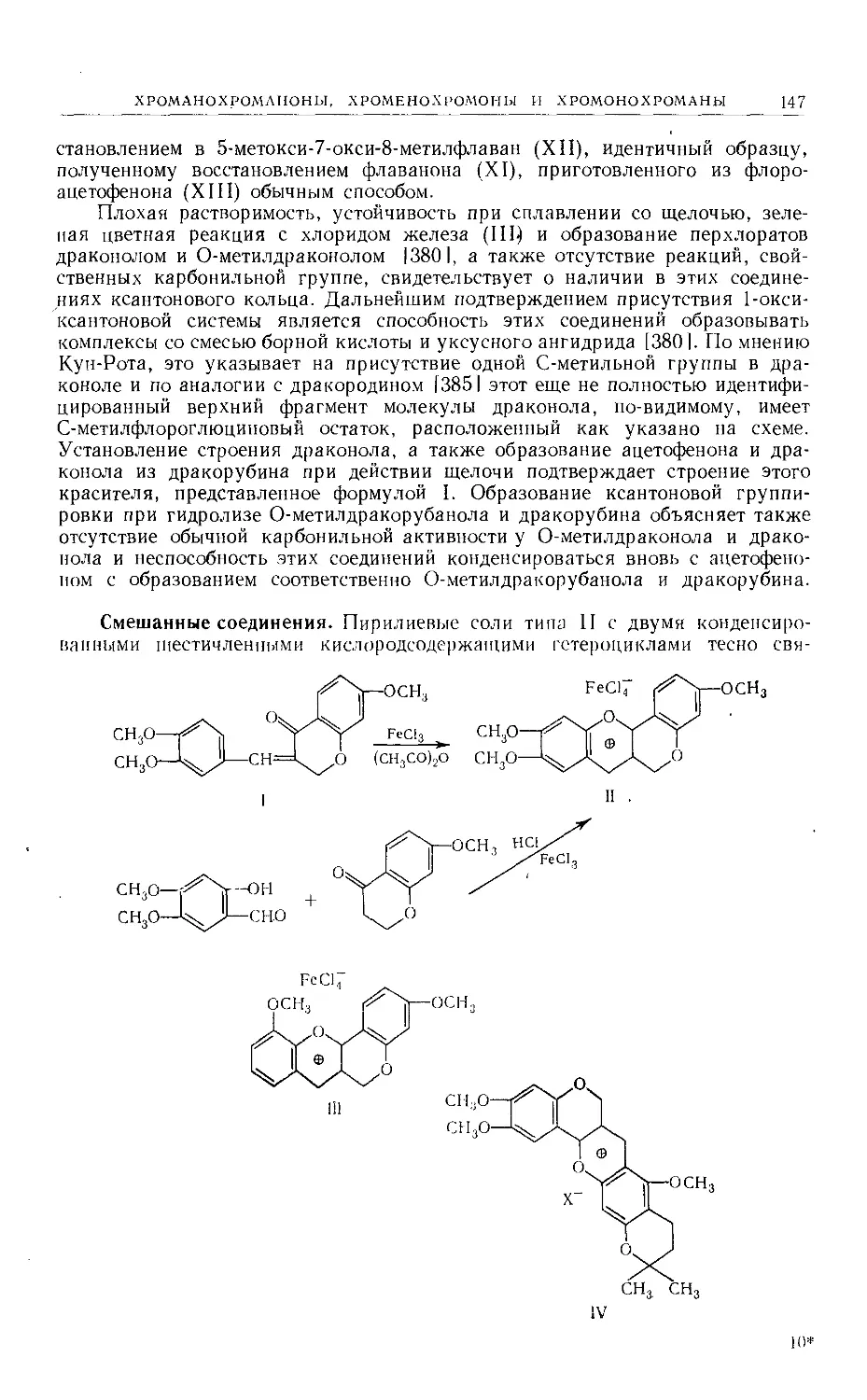
CH <-----L-Сезамин
| (изоазаринин)
CH2OH [a]p —68°
Via
Лактон (VI), получаемый окислением сезамолина, сезамина и пиноре-
зинола, оптически активен и поэтому должен быть г^нс-лактоном 112], что
находится в соответствии с общим выводом о слишком большом напряжении,
препятствующем существованию транс-лактона 15, 12].
Следовательно, строение этих бисфураноидов можно представить форму-
лами VI6, VIb или VIr.
VI6
R
VIb
Эрдтман [8] и Грипенберг [13] показали, что пинорезинол и эвдесмин
«симметричны» и поэтому должны быть представлены структурами VI6 и VIb,
тогда как эпипинорезинол (VIr) не является «симметричным» [13].
10
.СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Указанные соотношения приведены ниже. Сезамолин, как было показано,
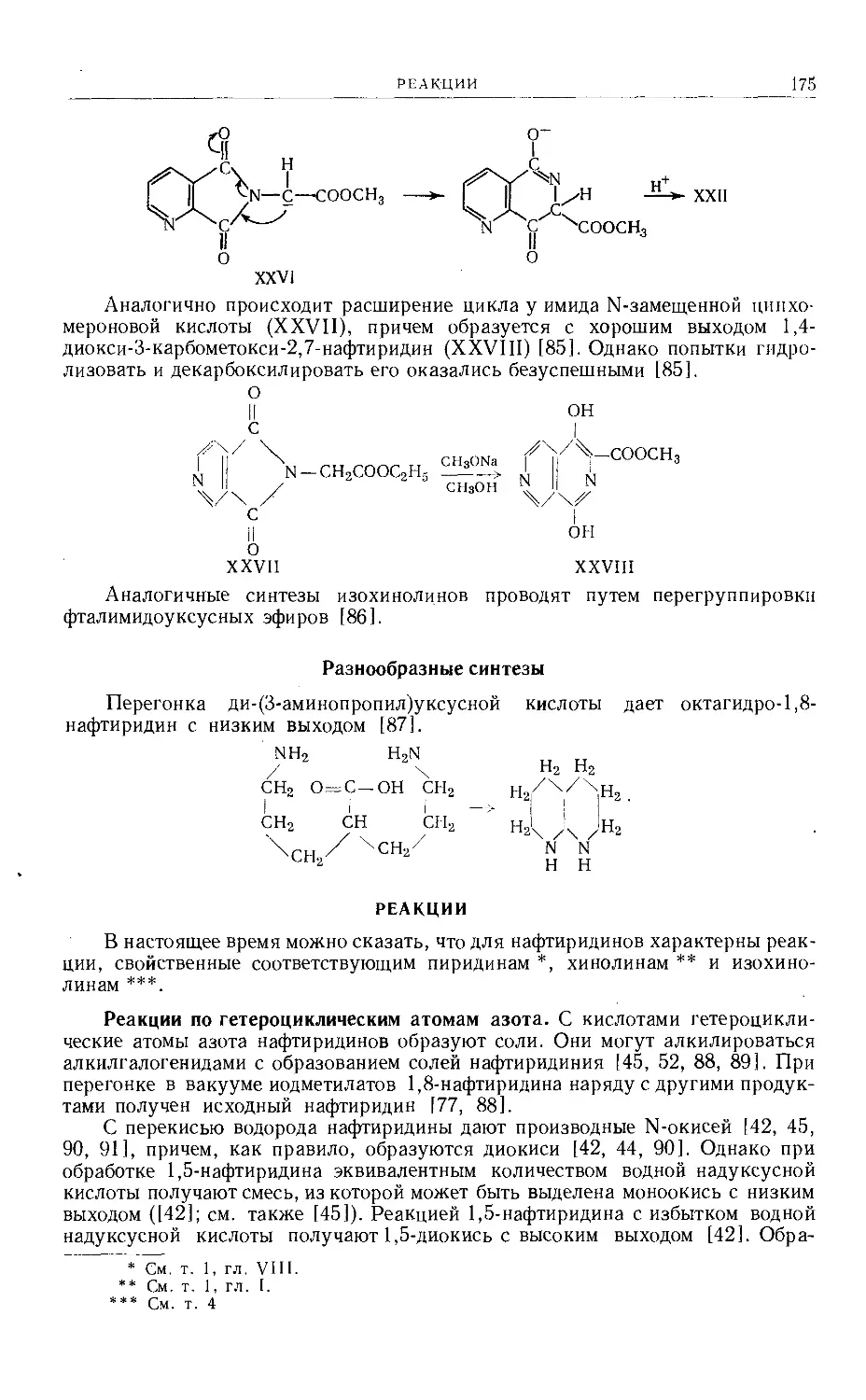
имеет ацетальную структуру (II) [3—5], афарагол идентичен DL-сезамину [3].
«Симметричные» «Несимметричные»
L-Сезамин , > L-Азаринин
L-Эвдесмин ~ L-Эпиэвдесмин
D-Сезамин ~ > D-Изосезамин
Диметиловый эфир D-пииорезинола ( > Диметиловый эфир D-эпипинорезинола
Синтез DL-сезамина, а следовательно, и DL-азаринина, в который первый
легко может быть эпимеризован, был осуществлен [14] исходя из диэтилового
эфира а,а'-дипиперонилянтарной кислоты (У1д). Это вещество существует
в кристаллической и жидкой модификациях. Восстановление последней
алюмогидридом лития приводит к тетраоксисоединению (Vie), а последую-
щее замыкание цикла с помощью разбавленной соляной кислоты дает DL-
сезамин.
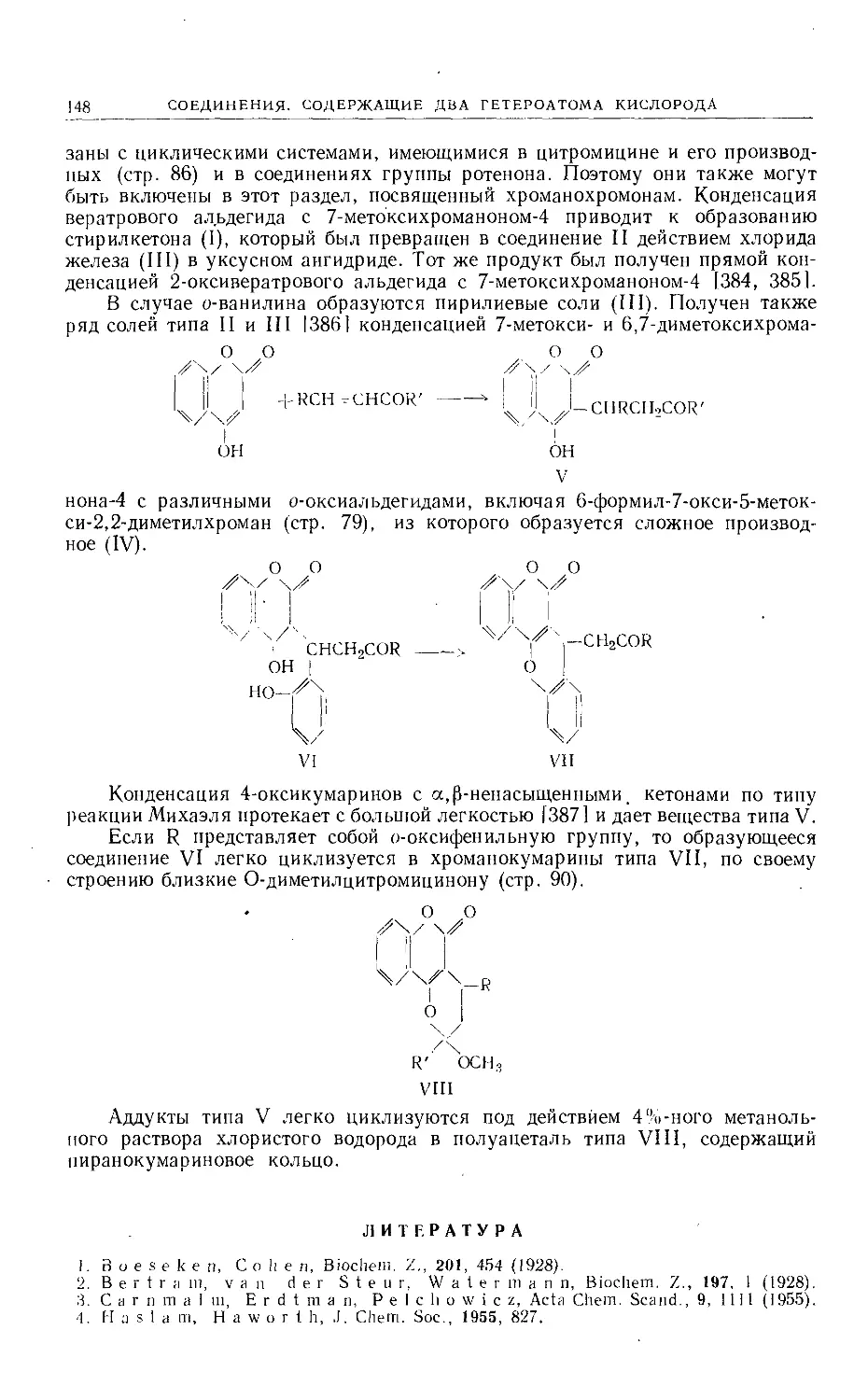
RCOCHCOOC2H5
I
RCOCHCOOC2H5
У1д
R£H-CH-CH2OH
он ,он
/СН-СН
НОСН2 XR
Vie
Фуранобензофураны. Известно сравнительно немного представителей
этого класса соединений, содержащих два фурановых кольца, сконденсиро-
ванных с бензольным ядром. Большинство из них синтезировано конденсацией
бензоинов с двухатомными фенолами [15—18]. Взаимодействием бензоина
и резорцина в присутствии концентрированной серной, кислоты получена
смесь соединений VII, VIII и IX.
VII
nt в сн:
0 0
VIII
CrOg
с6н5сохуч/сосвн5
! »
но/^/чон
X
Свн5
-С6н5
СгОз
C6H5CO/Y\COC6H5
он
IX
ФУРАНОФУРАНЫ. БИСФУРАНОИДНЫЕ ЛИГНАНЫ
11
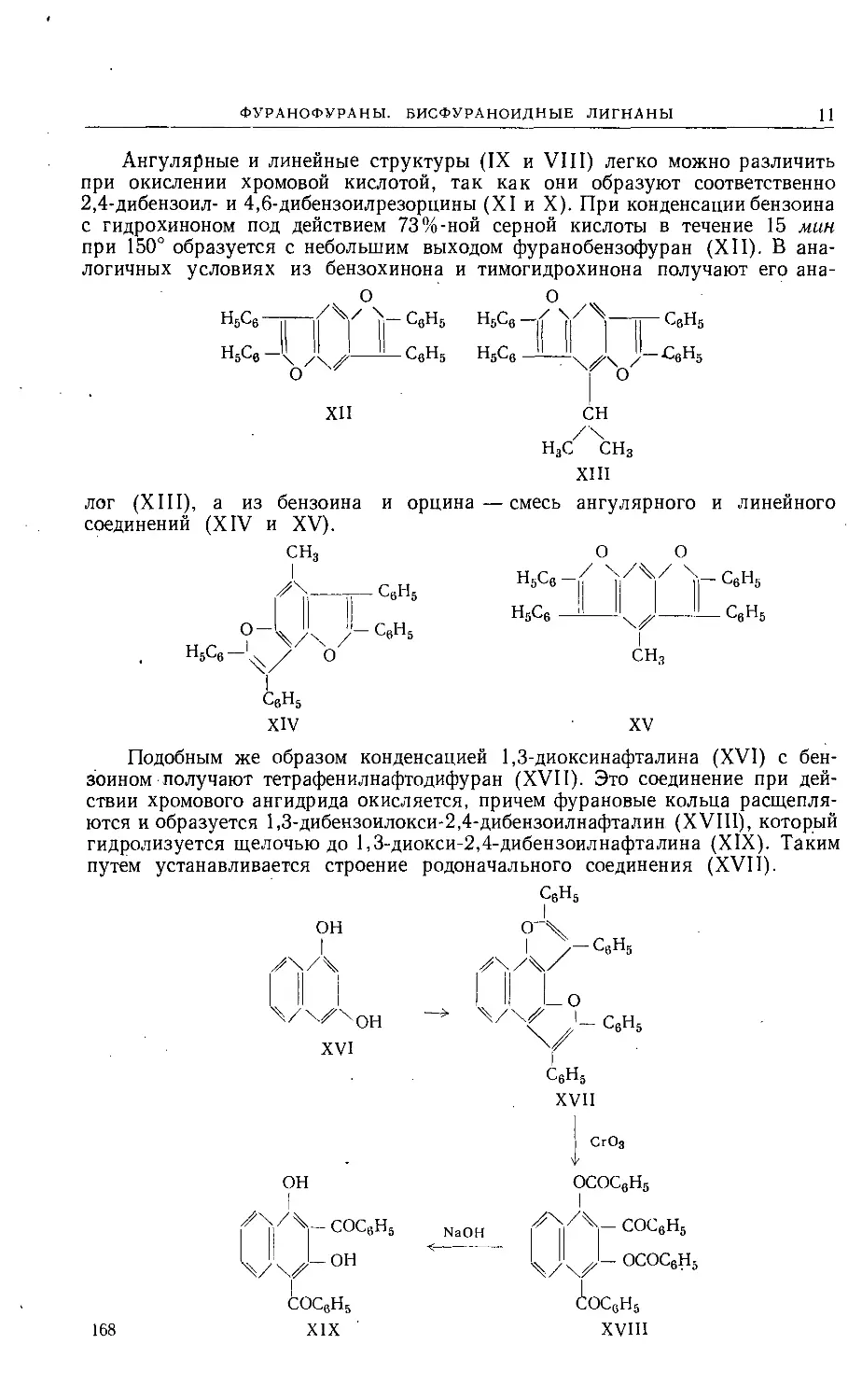
Ангулярные и линейные структуры (IX и VIII) легко можно различить
при окислении хромовой кислотой, так как они образуют соответственно
2,4-дибензоил- и 4,6-дибензоилрезорцины (XI и X). При конденсации бензоина
с гидрохиноном под действием 73%-ной серной кислоты в течение 15 мин
при 150° образуется с небольшим выходом фуранобензофуран (XII). В ана-
логичных условиях из бензохинона и тимогидрохинона получают его ана-
Н3С/ 'сНз
XIII
лог (XIII), а из бензоина и орцина — смесь ангулярного и линейного
соединений (XIV и XV).
СН3
о.Г|м1?
нг Y \/\/“СвН5
. Н5С6 — о
С8Н5
XIV
о о
н5св~/ у у у свн6
н5с6 J---\-----L С6Н6
I
СН3
XV
Подобным же образом конденсацией 1,3-диоксинафталина (XVI) с бен-
зоином получают тетрафенилнафтодифуран (XVII). Это соединение при дей-
ствии хромового ангидрида окисляется, причем фурановые кольца расщепля-
ются и образуется 1,3-дибензоилокси-2,4-дибензоилнафталин (XVIII), который
гидролизуется щелочью до 1,3-диокси-2,4-дибензоилнафталина (XIX). Таким
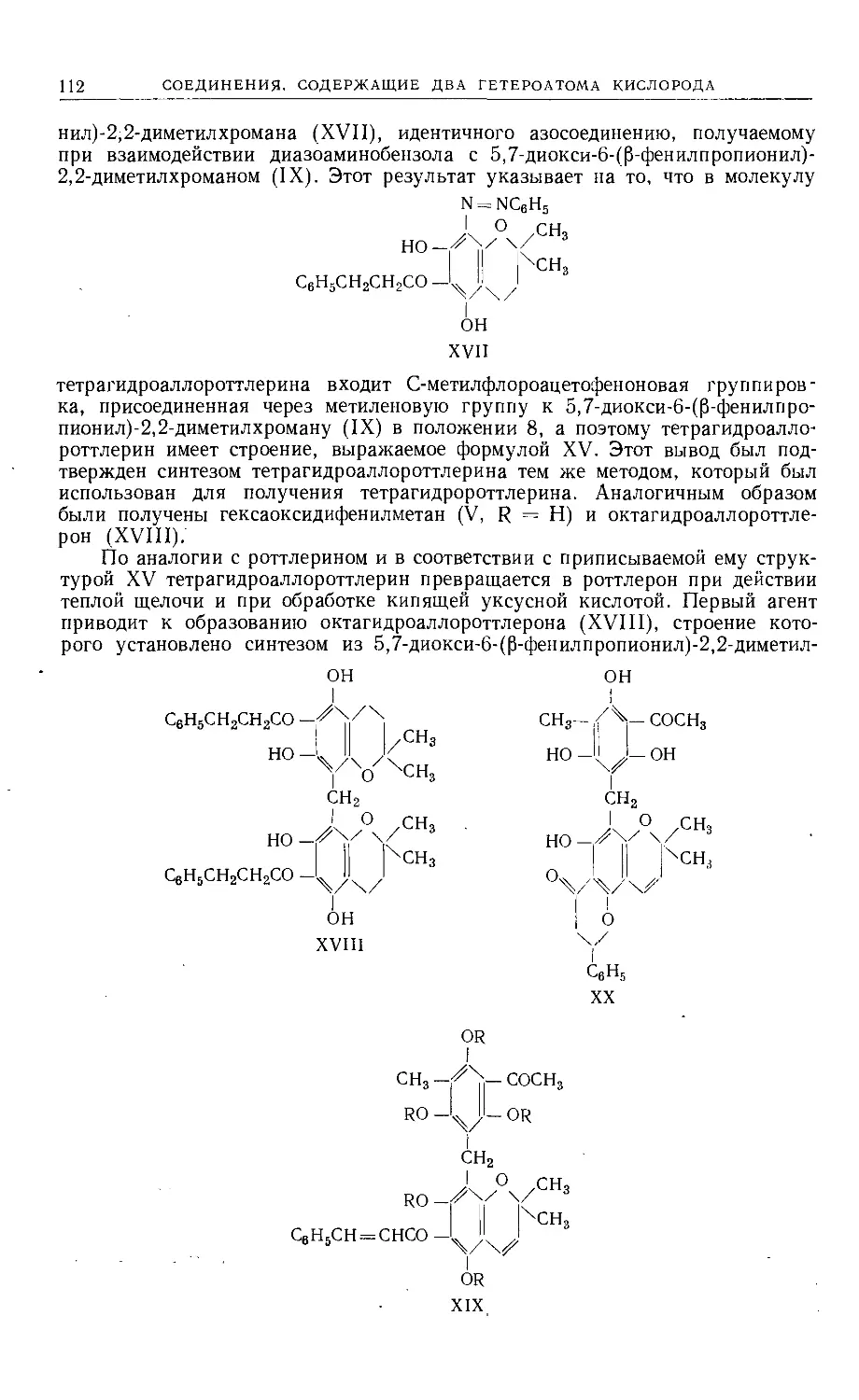
путем устанавливается строение родоначального соединения (XVII).
С6Н5
I
ОН 0%
I I /-с6н5
I II I I II Lo
\/\z\Oh '-ед
XVI
С6н5
XVII
i ^г<^3
ОН ОСОС6Н5
I I
^\/Ч СОС6Н8 NaOH А|/Ч|- С0СвН5
U \ Аон ' U U-ососвН5
ioceH5 ioceH5
168
XIX XVIII
12
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
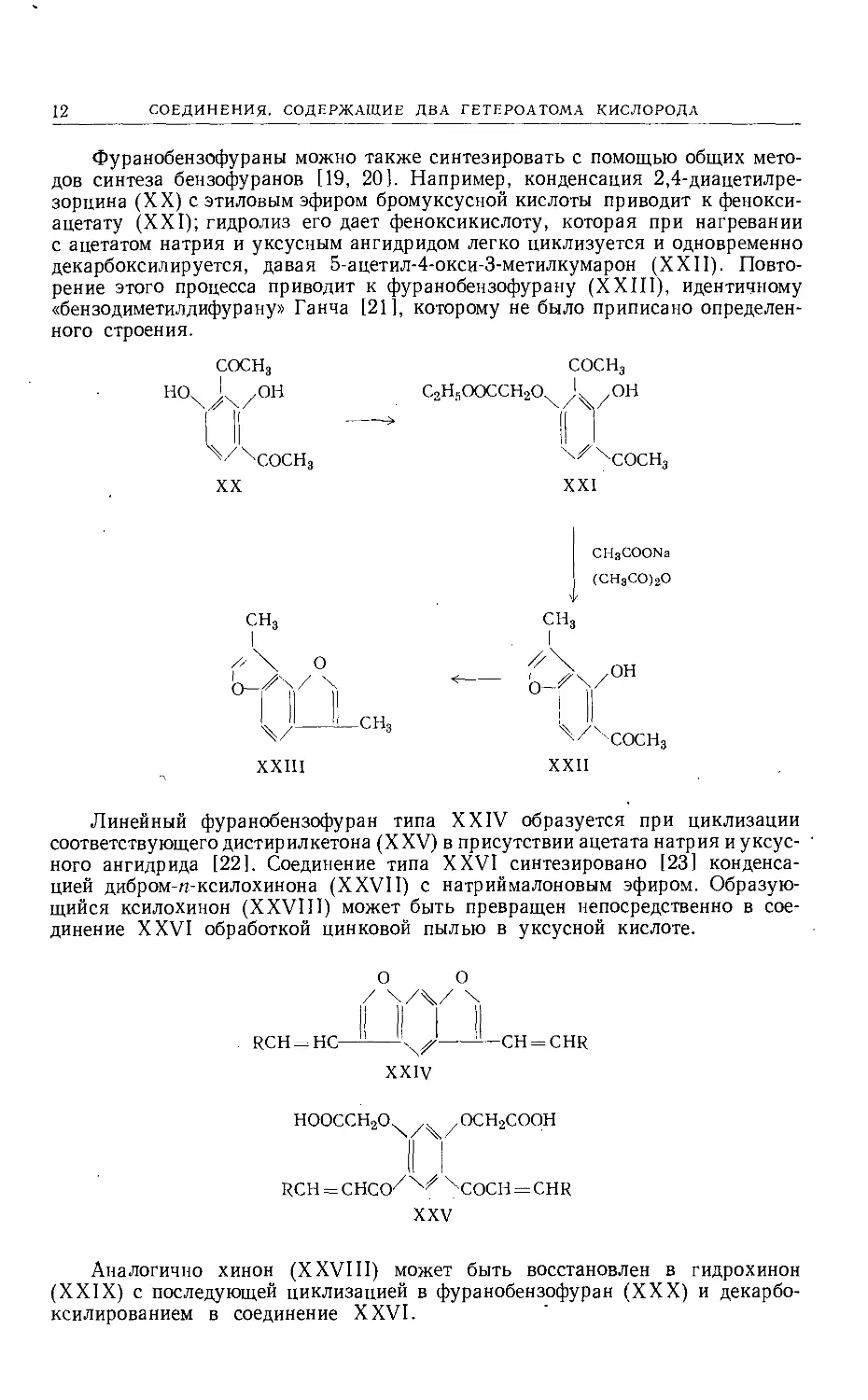
Фуранобензофураны можно также синтезировать с помощью общих мето-
дов синтеза бензофуранов [19, 20]. Например, конденсация 2,4-диацетилре-
зорцина (XX) с этиловым эфиром бромуксусной кислоты приводит к фенокси-
ацетату (XXI); гидролиз его дает феноксикислоту, которая при нагревании
с ацетатом натрия и уксусным ангидридом легко циклизуется и одновременно
декарбоксилируется, давая 5-ацетил-4-окси-3-метилкумарон (XXII). Повто-
рение этого процесса приводит к фуранобензофурану (XXIII), идентичному
«бензодиметилдифурану» Ганча [21], которому не было приписано определен-
ного строения.
СОСН3
С2Н5ООССН2ОХ / / он
Х^ХСОСН3
XXI
CH3COONa
(CH3CO)2O
XXIII
СН3
I
^/^СОСНз
XXII
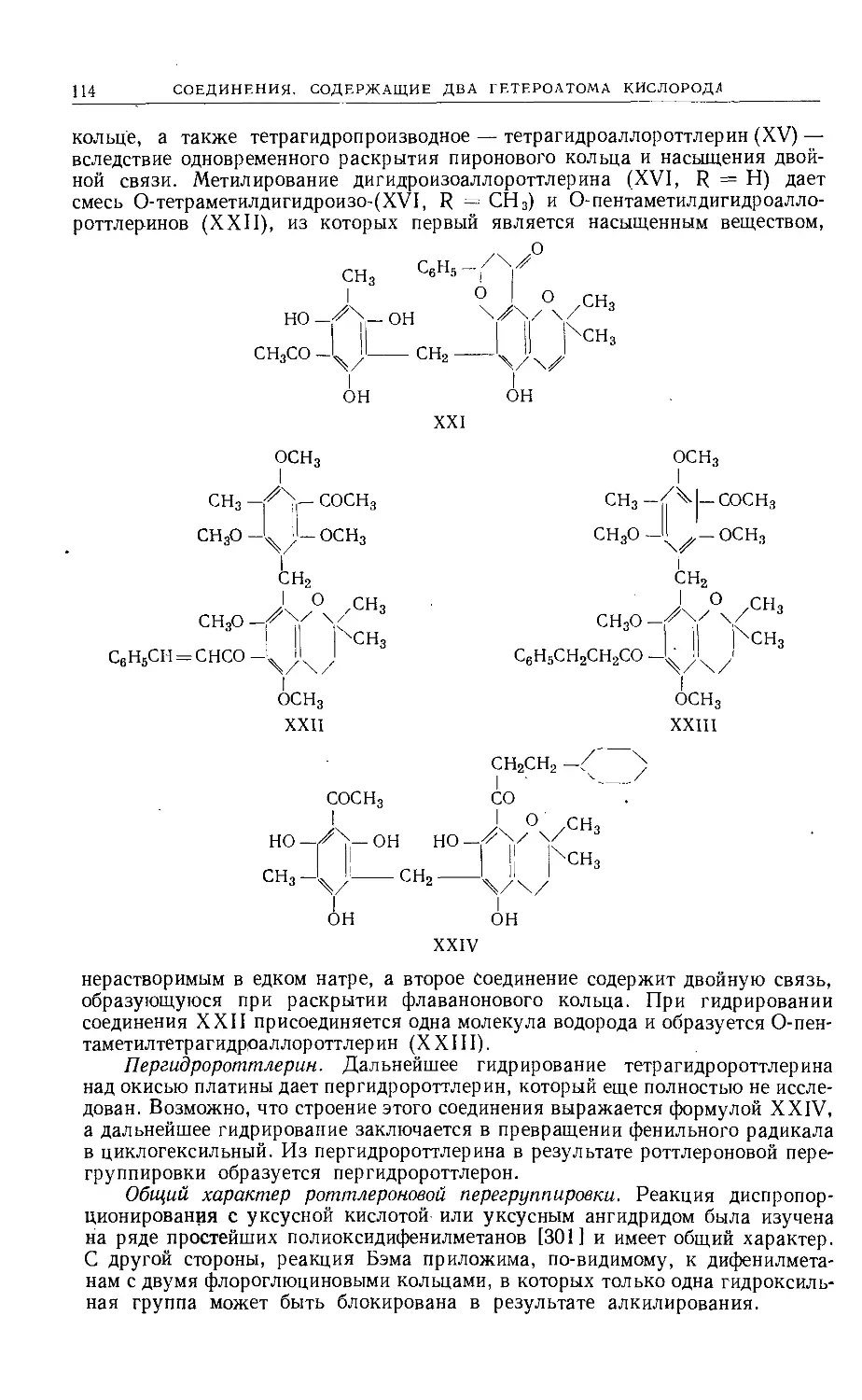
Линейный фуранобензофуран типа XXIV образуется при циклизации
соответствующего дистирилкетона (XXV) в присутствии ацетата натрия и уксус-
ного ангидрида [22]. Соединение типа XXVI синтезировано [23] конденса-
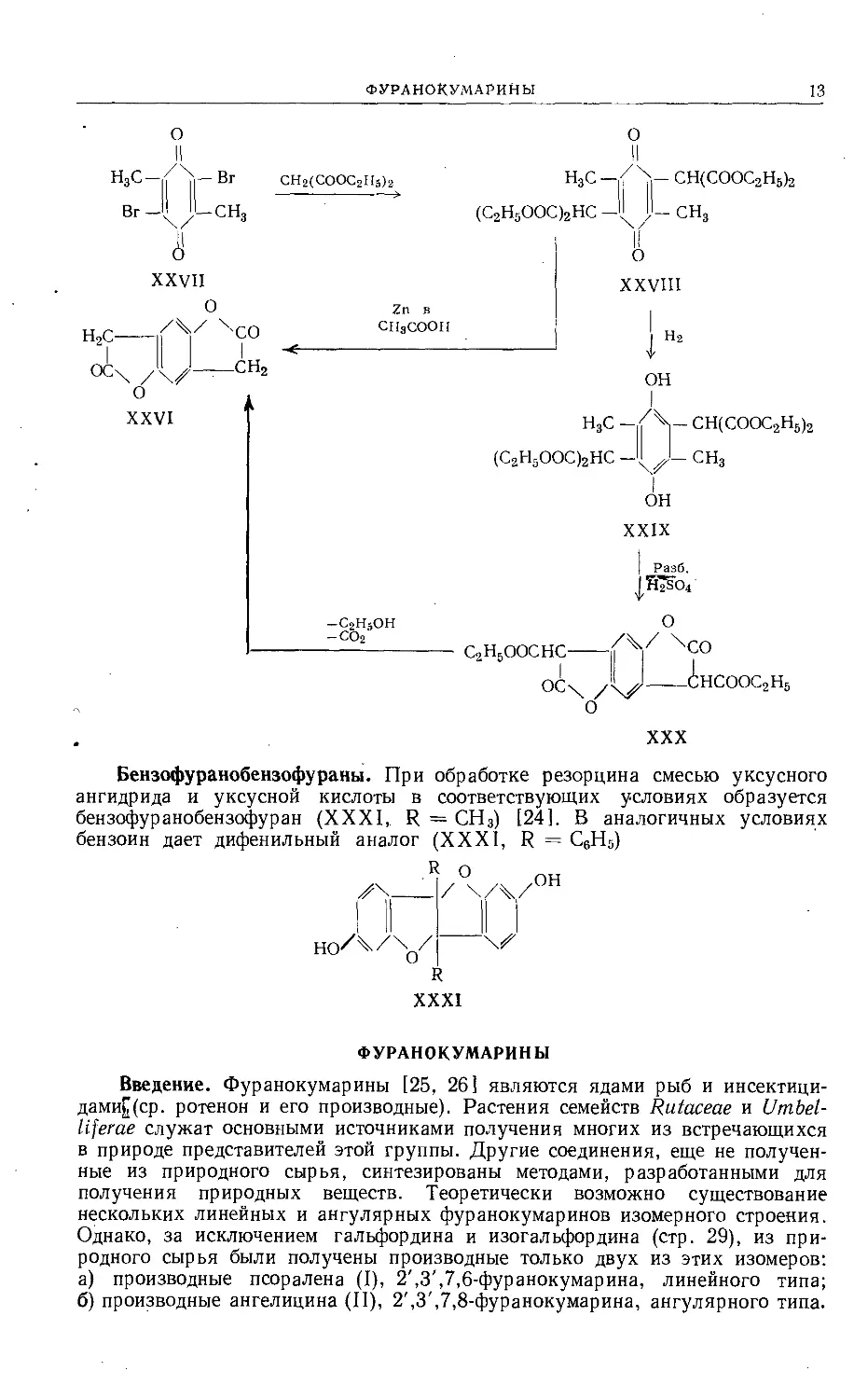
цией дибром-ц-ксилохинона (XXVII) с натриймалоновым эфиром. Образую-
щийся ксилохинон (XXVIII) может быть превращен непосредственно в сое-
динение XXVI обработкой цинковой пылью в уксусной кислоте.
О О
/ \/\/ \
RCH = НС— J----LH = CHR
XXIV
НООССН2ОХ „ /ОСН2СООН
Г|
RCH = СНСО/Х^ ' COCH = CHR
XXV
Аналогично хинон (XXVIII) может быть восстановлен в гидрохинон
(XXIX) с последующей циклизацией в фуранобензофуран (XXX) и декарбо-
ксилированием в соединение XXVI.
ФУРАНОКУМАРИНЫ
13
О
II
B3C lj/xj'l
Br —Ml—СН3
XXVII
о
H2i -fY41°
ОС\ /U—сн2
0
XXVI
СН2(СООС2Н5)2
о
II
Н3С —СН(СООС2Н6)2
(С2Н5ООС)2НС JL СН3
О
XXVIII
Zn в
СН3СООН
-С2Н5ОН
-со2
ОН
Н3С -|А- СН(СООС2Н6)2
(С2Н5ООС)2НС СН3
I
он
XXIX
| Разб.
^Ti^SO4
О
С2Н6ООСНС-----М/ ХСО
ОС\ /U--------^НСООС2Н6
о
XXX
Бензофуранобензофураны. При обработке резорцина смесью уксусного
ангидрида и уксусной кислоты в соответствующих условиях образуется
бензофуранобензофуран (XXXI, R = СН3) [24]. В аналогичных условиях
бензоин дает дифенильный аналог (XXXI, R = С8Н5)
XXXI
ФУРАНОКУМАРИНЫ
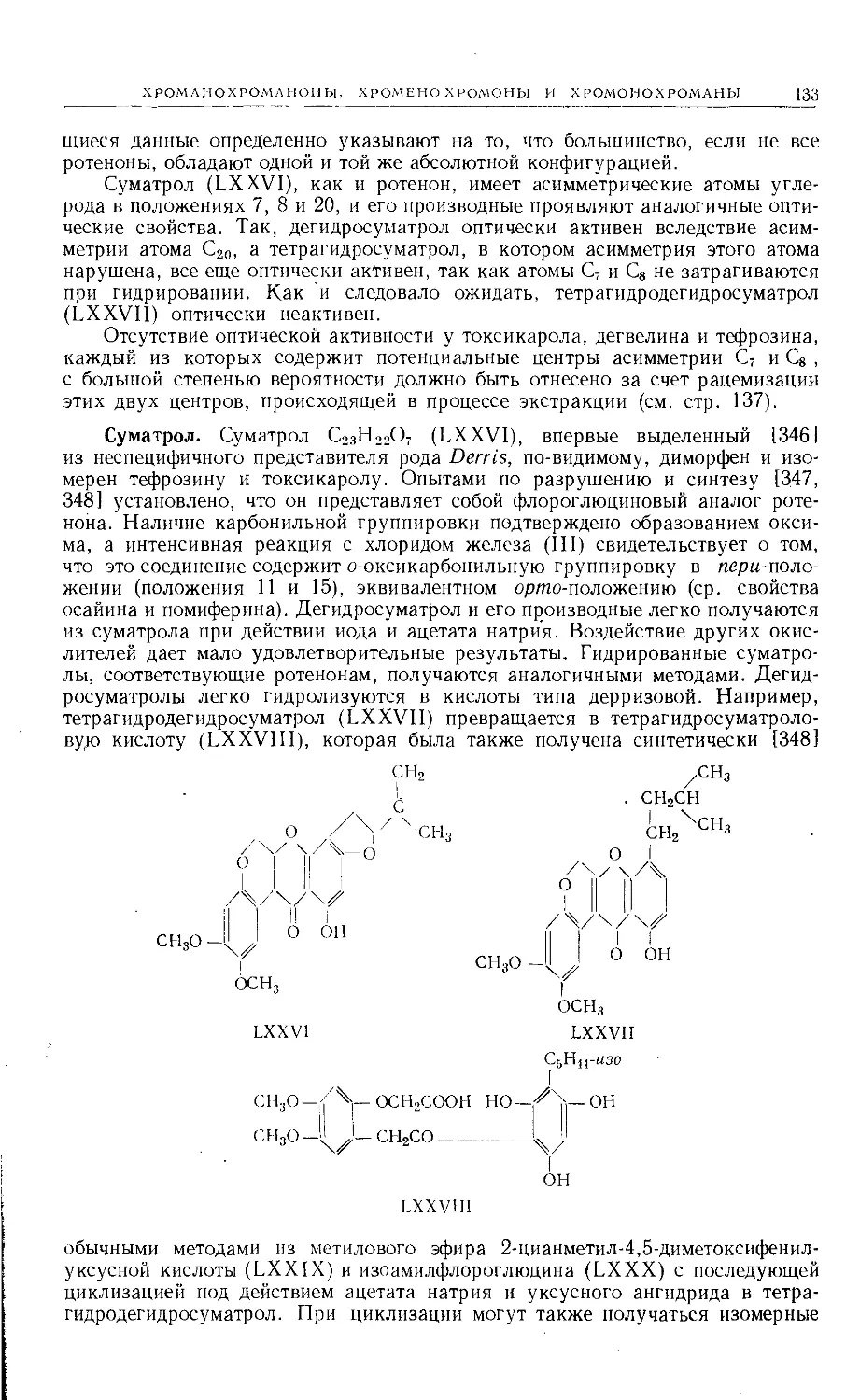
Введение. Фуранокумарины [25, 26] являются ядами рыб и инсектици-
дами[(ср. ротенон и его производные). Растения семейств Rutaceae и Umbel-
liferae служат основными источниками получения многих из встречающихся
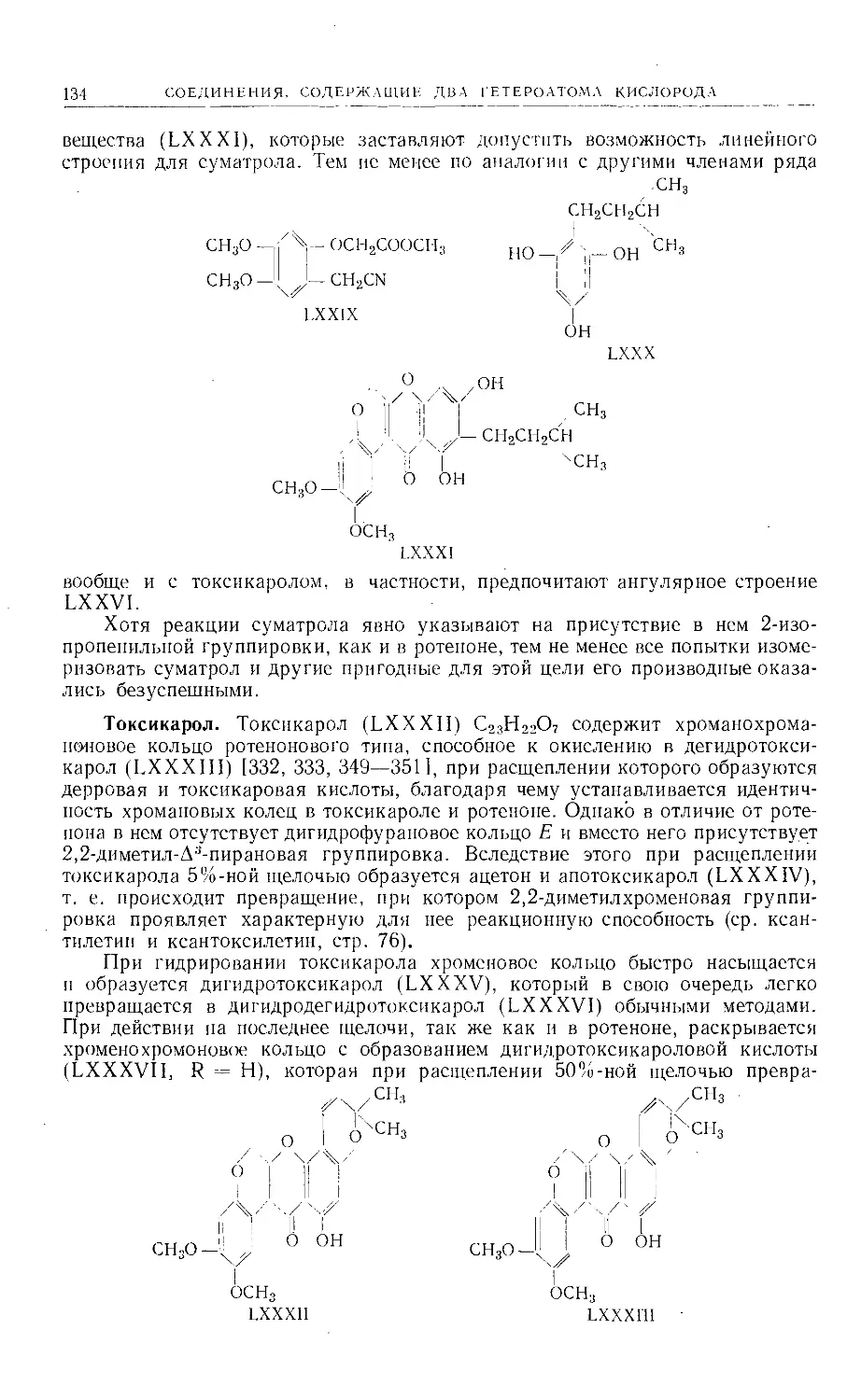
в природе представителей этой группы. Другие соединения, еще не получен-
ные из природного сырья, синтезированы методами, разработанными для
получения природных веществ. Теоретически возможно существование
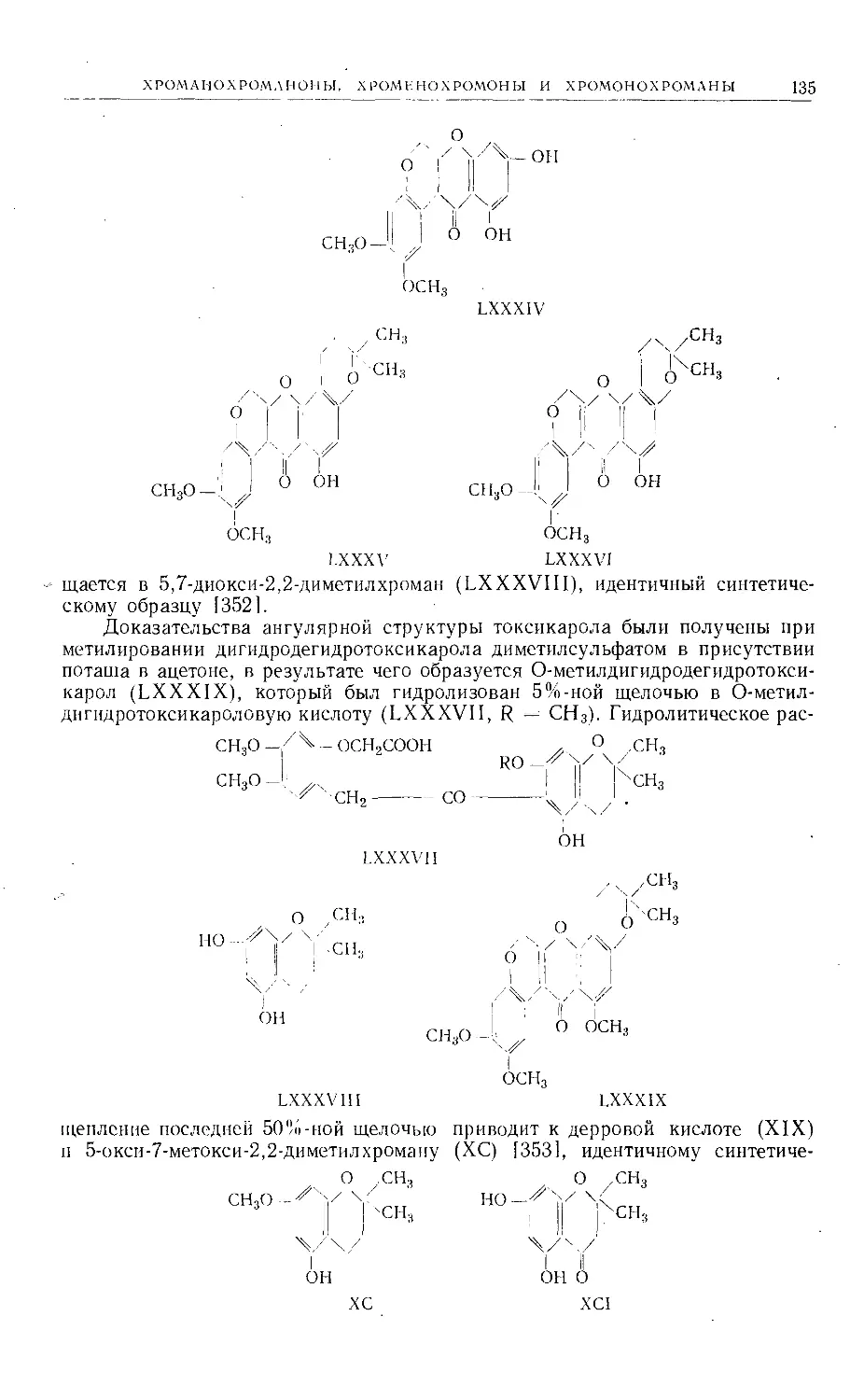
нескольких линейных и ангулярных фуранокумаринов изомерного строения.
Однако, за исключением гальфордина и изогальфордина (стр. 29), из при-
родного сырья были получены производные только двух из этих изомеров:
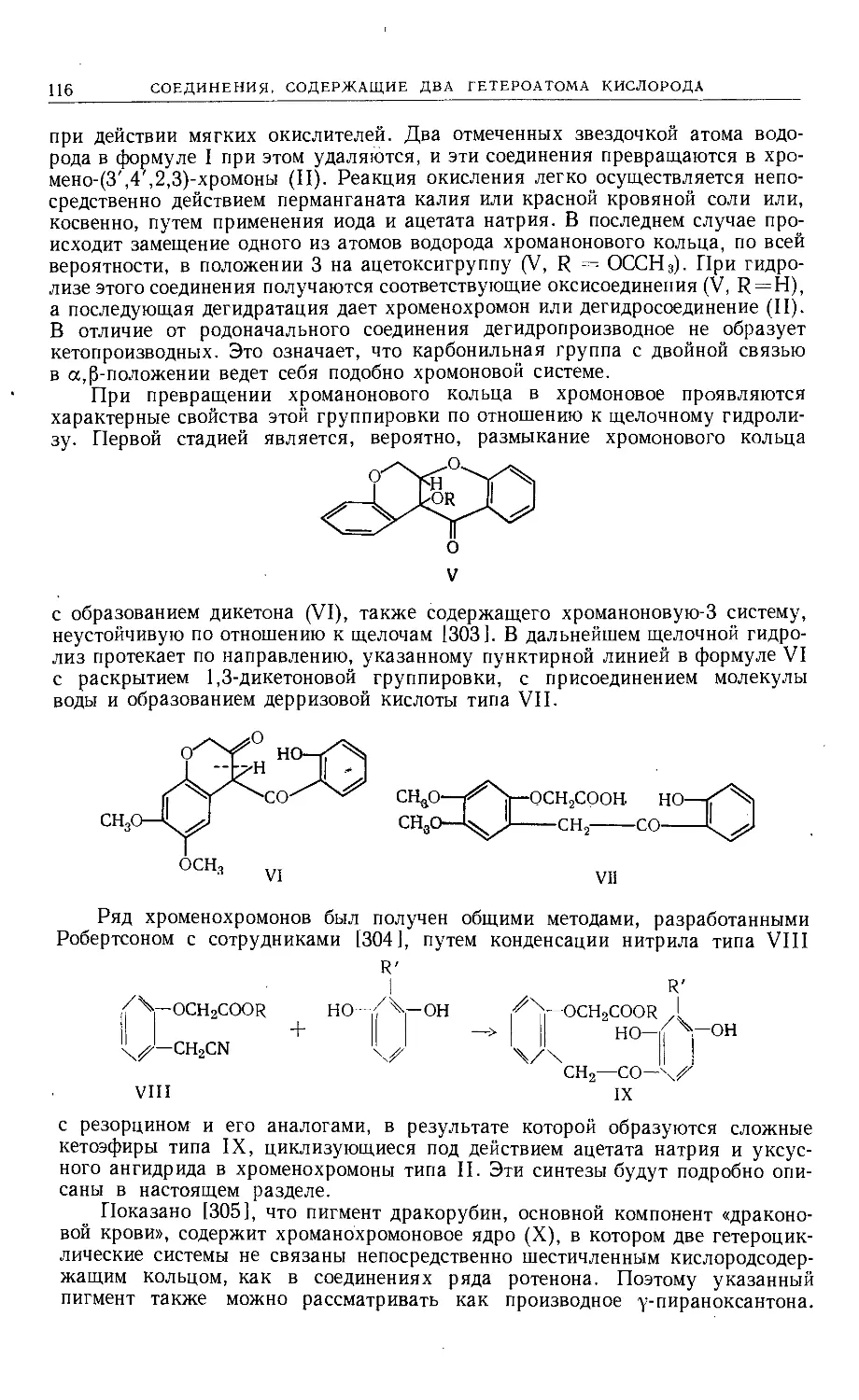
а) производные псоралена (I), 2',3',7,6-фуранокумарина, линейного типа;
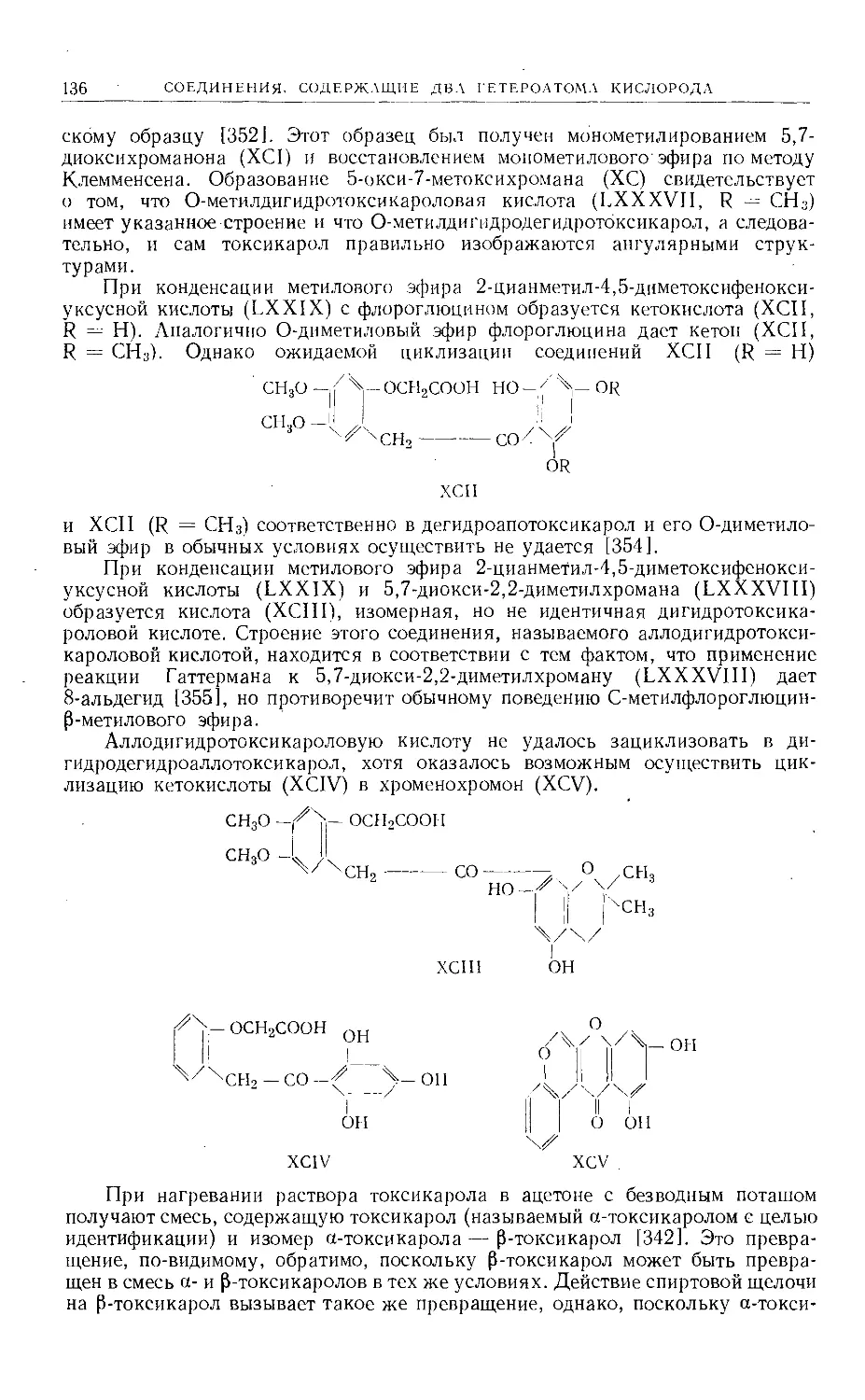
б) производные ангелицина (II), 2',3',7,8-фуранокумарина, ангулярного типа.
14 СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Также были синтезированы фуранокумарины типа III, однако представители
других возможных типов неизвестны.
Расщепление. Основными методами расщепления, применяемыми для
выяснения строения фуранокумаринов, являются следующие: 1. Гидрирова-
ние, обычно с последующим окислением. 2. Окисление. 3. Щелочное мети-
лирование. 4. Сплавление со щелочью.
1. При восстановлении фуранокумаринов амальгамой натрия образуется
дигидрокоричные кислоты типа IV, тогда как каталитическое гидрирование
в присутствии платины или палладия приводит к быстрому насыщению свя-
зи 4',5' фуранового кольца, после которого проходит медленное восстановле-
ние а-пиронового кольца с образованием 3,4,4',5'-тетрагидрофуранокумари-
нов (тип V). Раскрытие лактонного кольца при растворении в 5%-ном едком
кали сопровождается образованием о-оксикоричной кислоты, которая легко
гидрируется над палладием; свободные о-оксидигидрокоричные кислоты
(тип IV), полученные любым методом, могут быть превращены в соответствую-
щие 3,4-дигидрокумарины возгонкой в вакууме или нагреванием выше темпе-
ратуры плавления. Дигидро- и тетрагидрофуранокумарины легко дегидри-
руются в фуранокумарины при нагревании с палладиевой чернью [271.
2. В результате окисления перманганатом калия разрушается концевой'
гетероцикл и образуется замещенная бензойная кислота из центрального
бензольного кольца, а также жирные кислоты из ненасыщенных боковых
цепей. Разрыв гетероциклов происходит также и при озонолизе; при этом
из бензольного кольца образуется о-оксикарбонильный остаток и одновремен-
но получаются кетоны и альдегиды из ненасыщённых боковых цепей. Пере-
кись водорода в противоположность перманганату калия расщепляет а-пиро-
новое и бензольное кольца, не затрагивая фурановое; продукты окисления
выделяются в виде производных фурандикарбоновых-2,3 кислот. Окисление
3,4-дигидрофуранокумаринов азотной кислотой в течение нескольких дней при
комнатной температуре приводит к образованию янтарной кислоты в результате
расщепления а-пиронового кольца. Следует заметить, что атомы углерода
обеих карбоксильных групп в фурандикарбоновой-2,3 кислоте и атом углеро-
да одной из карбоксильных групп в янтарной кислоте происходят из цен-
трального бензольного ядра.
3. При метилировании фуранокумаринов с помощью щелочного раствора
диметилсульфата [28] раскрывается кумариновое кольцо и образуется мети-
ловый эфир о-метоксикоричной кислоты. Последний в результате гидролиза
до коричной кислоты и последующего окисления перманганатом калия дает
о-метоксибензойную кислоту с одновременным расщеплением фуранового
кольца. Прежде чем применять эти методы деструктивного окисления, необ-
ходимо метилировать оксифуранокумарины диазометаном.
ФУРАНОКУМАРИНЫ
15
-° ч ,осн3
О СО У
II I кон ||
/х=^ /Х'СН = СНСООСН3
| (1) Гидролиз
(2) КМпО<
Ч/ОСН3
^/^СООН
4. При сплавлении фуранокумаринов со щелочами разрушаются конце-
вые кислородсодержащие кольца, в результате чего образуются фенолы
и фенолкарбоновые кислоты (из центрального бензольного кольца).-
Известные фуранокумариновые эфиры (за исключением метиловых эфи-
ров) весьма чувствительны к кислотам И легко расщепляются на фенольный
компонент и спирт при действии ледяной уксусной кислоты, содержащей
несколько капель концентрированной серной кислоты.
Синтез. Синтез многочисленных природных и большого числа не встре-
чающихся в природе фуранокумаринов был осуществлен сочетанием несколь-
ких методов, обычно применяемых для получения кумаринов и кумаронов.
Возможны два основных пути.
1. Присоединение соответствующим способом а-пироновой системы
к о-оксиальдегидобензофурану дает продукты с четко определенным строе-
нием. Часто применяют конденсацию по Пехману Р-кетокислот с соответ-
ствующими фенолкумаранами с последующим дегидрированием [27 ] образую-
щихся 4',5'-дигидрофуранокумаринов. Однако этот метод нужно применять
с осторожностью, потому что в результате реакции могут образоваться изо-
мерные хромоны или смесь хромона и кумарина. Строение получающихся
при этом кумаринов также неопределенно, поэтому метод синтеза не является
однозначным.
2. Пристройку фурановой системы к о-оксиальдегидо- или о-оксикетоку-
марину можно осуществить обычным образом действием феноксикислоты. Для
этой цели наиболее пригодны различные о-оксиацилкумарины, которые дают
4'-замещенные фуранокумарины. Однако о-оксиальдегидокумарины трудно
доступны, вследствие чего возможности получения таким путем фуранокума-
ринов, не замещенных в положении 4', весьма ограниченны.
Производные псоралена. Псорален (I), известный также под названием
фикусина, содержится в Ficus carica Linn, и Psoraleacorylifolia Linn. Обработка
его щелочным раствором диметилсульфата и последующее окисление образую-
щейся р-(6-метоксибензофурано)-5-акриловой кислоты (VI) перманганатом
калия приводит к резорциндикарбоновой-4,6 кислоте (VII), идентифициро-
ванной в виде тетраметилового эфира (VIII) [29], чем доказывается ее линей-
ное строение.
° . .осн, НО\/Ч/ОН сн,0 осн,
от LJ о
----ч^\сн=снсоон НООС/Х^ЧСООН сн3оос/\^\соосн3
VI VII VIII
Псорален был синтезирован Шпетом [29] следующим образом: 6-оксибен-
зофуран (IX) был прогидрирован в кумаран (X), а затем подвергнут по методу
Пехмана конденсации с яблочной кислотой в присутствии серной кислоты,
16
Соединения, содержащие два гетероатома кислорода
в результате чего образовался главным образом 4',5'-дигидропсорален (Х1а).
Дегидрированием над палладием на угле получен псорален (I) с примесью
следов ангелицина (II). Превращение соединения IX в X до образования
кумаринового кольца является необходимым вследствие чувствительности
незамещенных бензофуранов к минеральным кислотам. Кумараны более
О
НгС/ VS- 0Н
। *
н2с-—сно
XI
О
Яблочная
кислота
4*
H2SO4
Pd при
I <-------
о о
Нг| YY “
НгС----Y\^
Xia
устойчивы к кислотам. Этот синтез не является однозначным. Робертсон
с сотрудниками [30] описал другой, строго определенный путь синтеза
псоралена.
Применение метода синтеза альдегидов по Гаттерману к 6-оксикумарану
(X) приводит к альдегиду (XI), который был превращен в кумарин’ (дигидро-
псорален) (Х1а) конденсацией с циануксусной кислотой с последующим гид-
ролизом и декарбоксилированием. Дегидрирование соединения Х1а дает
псорален (I). Строение альдегида (XI) было установлено восстановлением
6-окси-5-метилкумарана (XVI), идентичного образцу, приготовленному в соот-
ветствии со схемой XII—*- XVI.
В связи с гидрированием кумаронов в кумараны следует заметить, что
каталитическое гидрирование а,р-незамещенных бензофуранов проходит очень
легко, но наличие карбоксильной, карбометоксильной или карбэтоксильной
группы в a-положении сильно затрудняет присоединение водорода к фурано-
вому кольцу [31]. Скелет псоралена имеется в значительном числе природных
фуранокумаринов, отличающихся от псоралена только наличием гидроксиль-
ных или алкоксильных групп в положениях 5 и 8.
НО.
ОН
но. .. .он
Н3С/Х^ХСНО
С6Н6СН2О
Н3С
XII XIII XIV
H0\Z\/°\CH
I II 1
А/------ СН2
Н3СХ Х/
XVI
„ С6Н6СН2ОХ ,, ,ОСН2СООН
(1) Циклизация ь ° Х/\/ 2
А-СОг I
(3) Гидрирование Н I
Н3С/Х^ХСНО
XV
Бергаптол (XVII, R = Н), 5-оксипсорален, содержится в плодах Citrus
bergamia Risso. Определение его строения является заслугой главным обра-
зом Померанца [32], Томса [33] и, в меньшей степени, Шпета [34].
ФУРАНОКУМАРИНЫ
17
Метилирование бергаптола дает метиловый эфир, бергаптен (XVII,
R = СН3). Гераниловый эфир (XVII, R — геранил) — бергамоттин (назы-
ваемый также бергаптином)— содержится в бергамотовом масле. При нагрева-
нии в высоком вакууме или же при обработке смесью ледяной уксусной и сер-
ной кислот бергамоттин расщепляется, образуя бергаптол и гераниловый
спирт [35].
При сплавлении бергаптола с поташом разрушается концевое кольцо
с образованием флороглюцина, а нитрование метилового эфира (бергаптена)
в ледяной уксусной кислоте дает мононитробергаптен. При восстановлении
последнего в амин с последующим окислением бихроматом калия в серной
кислоте образуется хинон, идентичный ксантотоксинхинону (стр. 19).
О О
Il Т п°
----
OR
XVII
Наличие фуранового кольца в бергаптоле, а также в аллобергаптоле
(XVIII, R = Н), обладающем ангулярной структурой, доказано окислением
с помощью перекиси водорода в фурандикарбоновую-2,3 кислоту. Синтез
Шпета [36], дающий смесь бергаптола (XVII, R = Н) и аллобергаптола
(XVIII, R = Н), не является однозначным. Исходным веществом служит
3,4,6-триацетоксикумаран (XXII), приготовленный исходя из соединения XIX.
Смесь, содержащая главным образом аллобергаптол (XVIII, R = Н), при
действии диазометана дает аллобергаптен (XVIII, R = СН3) вместе с неболь-
шим количеством бергаптола (XVII, R = Н), получающимся в результате
ОН СНзСОО
XVIII XIX XX
СНзСОО.
12
СНОСОСНз
н2
ОСОСНз
XXII
(CH3CO)2O CH3COCI
V
СНзСОО 0
у и7 хсн
' И
---------СОСОСНз
I
ОСОСНз
XXI
о
конденсации 3,4,6-триацетоксикумарана (XXII) с натрийформилуксусным
эфиром в присутствии этилата натрия. Следует отметить, что, когда два воз-
можных орто-положения могут принимать участие в замыкании цикла с об-
разованием линейной и ангулярной структур, получается почти исключитель-
но ангулярный изомер. Этот эффект особенно заметен, когда соединение
содержит флороглюциновый остаток. Положение 7 в бензофуране и положе-
ние 8 в кумарине более реакционноспособны, чем соответствующие положе-
ния 5 и 6. Превосходным доказательством этого [37] является образование
исключительно 7-формилпроизводного при применении реакции Гаттермана
2 Заказ № 978
18
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
к а-замёщенным бензофуранам, производным флороглюцина (замещение
в положение 5 полностью отсутствует). Робертсон описал [38] однозначный
рациональный синтез бергаптена из апоксантоксилетина (XXIII) [39] (стр. 77).
Конденсация соединения (XXIII) с этиловым эфиром бромуксусной кислоты
в кипящем ацетоне, содержащем поташ, дает 6-формил-5-метокси-7-карбэток-
симетоксикумарин (XXIV). Под действием ацетата натрия и уксусного ангид-
рида соответствующая кислота превращается в бергаптен.
НО. .. О С2Н6ООССН2ОЧ z. о
I II ?° III )°
ОСН3 ОСН3
XXIII XXIV
Робертсон с сотрудниками [31 ] описал также рациональный синтез алло-
бергаптена. Сложный феноксиэфир (XXV) циклизуется в присутствии этилата
натрия с образованием этилового эфира 6-бензилокси-4-метоксикумаронкар-
боновой-2 кислоты (XXVI, R = СвН6СН2), который над палладиевым ката-
лизатором в присутствии водорода дебензилируется с образованием этилового
эфира 6-окси-4-метоксикумаронкарбоновой-2 кислоты (XXVI, R = Н). Из него
по методу Гаттермана получен альдегид (XXVII), строение которого подтвер-
ждено восстановлением и метилированием в известный этиловый эфир 4,6-
диметокси-7-метилкумаронкарбоновой-2 кислоты (XXVIII). Декарбоксили-
рование альдегида (XXVII) приводит к 7-формил-6-окси-4-метоксикумарону
СвН6СН2Оч/. /ОСНгСООСгНз
III
^^сно
осн3
XXV
сно
СООС2Н6
XXVI
ОСН3
XXVII
СНз
ch3o4J4/ox/cooc2h5
I
ОСНз
XXVIII
сно
ОСНз
XXIX
(XXXI), который при конденсации с циануксусной кислотой дает аллобер-
гаптен (XVIII, R — СН3). Следует отметить, что наличие карбометоксильной
группы в a-положении препятствует восстановлению фуранового кольца при
каталитическом гидрировании соединения XXVI (R = С6Н5СН2) в соединение
XXVI (R = Н) и соединения XXVII в соответствующий монометиловый
ФУРАНОКУМАРИНЫ
19
эфир, соединения XXVIII. Метилирование бергаптола щелочным раствором
диметилсульфата дает г{«с-р-(4-окси-6-метоксибензофурано)-5-акриловую кис-
XXX XXXI
лоту (XXX), которая превращается в транс-форму при нагревании до 100°
в течение 2,5 час с 5%-ным едким кали. При подкислении образуется изо-
бергаптен (XXXI) [40].
Ксантотоксин (XXXII) (ксантотоксолметиловый эфир, или 8-метокси-
псорален) содержится в семенах Fagara xanthoxyloid.es Lam. Строение этого
соединения определено Томсом [33, 41], получившим при сплавлении его
с едким кали пирогаллолкарбоновую-4 кислоту и пирогаллол. При нитро-
вании ксантотоксина образуется мононитросоединение, изомерное, но не иден-
тичное мононитропроизводному бергаптена и легко превращающееся при
ОСН3
О | О
XXXII
окислении хромовым ангидридом соответствующего амина в бергаптенхинон
(стр. 17). Синтез был осуществлен Шпетом [42, 43] исходя из кумаранона
(XXXIII), который был прогидрирован в кумаран (XXXIV), а последний был
превращен реакцией Пехмана с яблочной кислотой в 8-окси-4',5'-дигидро-'
6,7,3',2'-фуранокумарин (XXXV). Метилирование соединения XXXV с после-
дующим дегидрированием над палладием на угле дает ксантотоксин (XXXII)
(ср. синтез бергаптена). Ксантотоксин через промежуточную стадию полу-
чения 5-нитроксантотоксина [44] был превращен в изопимпинеллин (стр. 23).
XXXIII
XXXIV
Яблочная
кислота
H2S.O4
Ф
он
(1) ch2n2
XXXII <------------------
(2) Дегидрирование
XXXV
2*
20
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Соотношение между бергаптолом и ксантотоксолом может быть выражено
следующей схемой:
Бергаптен
Бергаптенхинон
Ксантотоксин
А
I
Бергаптол
Изопимпииеллин
ОН
Ксантотоксол
Биакангелицин (XXXVI, R = R' = Н), содержащийся в корнях Angelica
glabra Makino, образует кристаллы светло-желтого цвета, [a Jd 24,62° (в пири-
дине) [45]. При его ацетилировании образуется диацетат (XXXVI, R = R' =
= СНзСО). На присутствие фуранового кольца указывает образование фуран-
дикарбоновой-2,3 кислоты при окислении перекисью водорода. Восстановление
амальгамой натрия с последующей перегонкой продукта реакции приводит
/СН3
OCH2CHORC-CH3
I
осн3
XXXVI
XOR'
OR
II I II I
---\/\Z
I
OCH3
XXXVII
к 3,4-дигидробиакангелицину, при окислении которого азотной кислотой
образуется янтарная кислота. Разложение биакангелицина в ледяной уксусной
кислоте, содержащей несколько капель концентрированной серной кислоты,
приводит к фенолу (XXXVII, R = Н), из которого при метилировании диазо-
метаном образуется изопимпииеллин (XXXVII, R = СН3) (стр. 23). Дегидра-
тация пятиокисью фосфора в толуоле а-дигликолевой группировки в биакан-
гелицине дает ангидробиакангелицин (XXXVIII). При окислении перманга-
натом калия биакангелицин отщепляет боковую, цепь в виде а-оксиизомасля-
ной кислоты, тогда как окисление хромовым ангидридом приводит к бергап-
тенхинону и биакангелициновой кислоте (XXXIX). Последняя в свою очередь
была синтезирована из 5-метокси-8-оксипсоралена и метилового эфира
хлоруксусной кислоты. 5-Метокси-8-оксипсорален был получен нитрованием
ФУРАНОКУМАРИНЫ
21
бергаптена в 5-метокси-8-нитропсорален (XL) с последующим восстановле-
нием нитросоединения и диазотированием обычным образом.
/СН3
ОСН2СОСН
II I II ?°
\/w
осн3
XXXVIII
no2
I
осн3
XL
ОСН2СООН
О I о
I
ОСН3
XXXIX
Биакангеликол (XLI) [45], [aId 34,77° (в пиридине), содержится вместе
с биакангелицином в корнях Angelica glabra Makino. Несмотря на название,
биакангеликол не имеет свободных гидроксильных групп. Восстановление
/СН3
ОСН2СН-С(
\ / ХСН3
о
о
сн2
О Хснс/
I о он
сн3
СН3
О
ОСН3
XLI
/ 'у4)7
\^ХСН = СНСООН
6сн3
XLII
а-пиронового кольца амальгамой натрия приводит к соответствующей о-окси-
дигидрокоричной кислоте, при окислении которой азотной кислотой образует-
ся янтарная кислота. Окисление биакангеликола хромовым ангидридом дает
бергаптенхинон наряду с биакангелициновой кислотой (XXXIX). При окис-
лении перекисью водорода образуется фурандикарбоновая-2,3 кислота, а под
действием перманганата калия боковая цепь отщепляется, в виде а-оксимасля-
ной кислоты. Отщепление эфирной боковой цепи под действием смеси уксус-
ной и серной кислот дает 5-метокси-8-оксипсорален, который в свою очередь
может быть превращен в изопимпинеллин при метилировании диазометаном.
Эпоксидная группа в боковой цепи размыкается при действии ацетата
натрия и уксусного ангидрида с образованием биакангелициндиацетата
и может быть непосредственно прогидратирована продолжительным нагрева-
нием с 1%-ным водным раствором щавелевой кислоты, в результате чего
образуется биакангелицин. При кипячении толуольного раствора биакангели-
кола с фосфорным ангидридом биакангеликол изомеризуется в ангидробиакан-
гелицин (XXXVIII). При нагревании до 100° в течение 1 час с 10%-ным спир-
товым раствором поташа биакангеликол превращается, вероятно, через про-
межуточную стадию образования биакангелицина в изобиакангеликол,
который, по-видимому, имеет строение XLII.
Императорин (XLIII) (мармелозин, или у,у-диметилаллиловый эфир
ксантотоксола) содержится главным образом в плодах Aegle marmelos Linn.
[46—48]. При его каталитическом гидрировании легко получается гексагидро-
императорин, при окислении которого азотной кислотой образуется янтарная
кислота (из а-пиронового кольца). При действии перекиси водорода на импе-
раторин образуется фурандикарбоновая-2,3 кислота, а при окислении перман-
ганатом калия — а-оксиизомасляная кислота (за счет боковой цепи). Окисление
хромовым ангидридом приводит к получению ацетона также за счет окисле-
ния боковой цепи. Расщепление с помощью смеси ледяной уксусной и серной
кислот дает ксантотоксол (XLIV) и у,у-диметилаллиловый спирт (XLV),
22
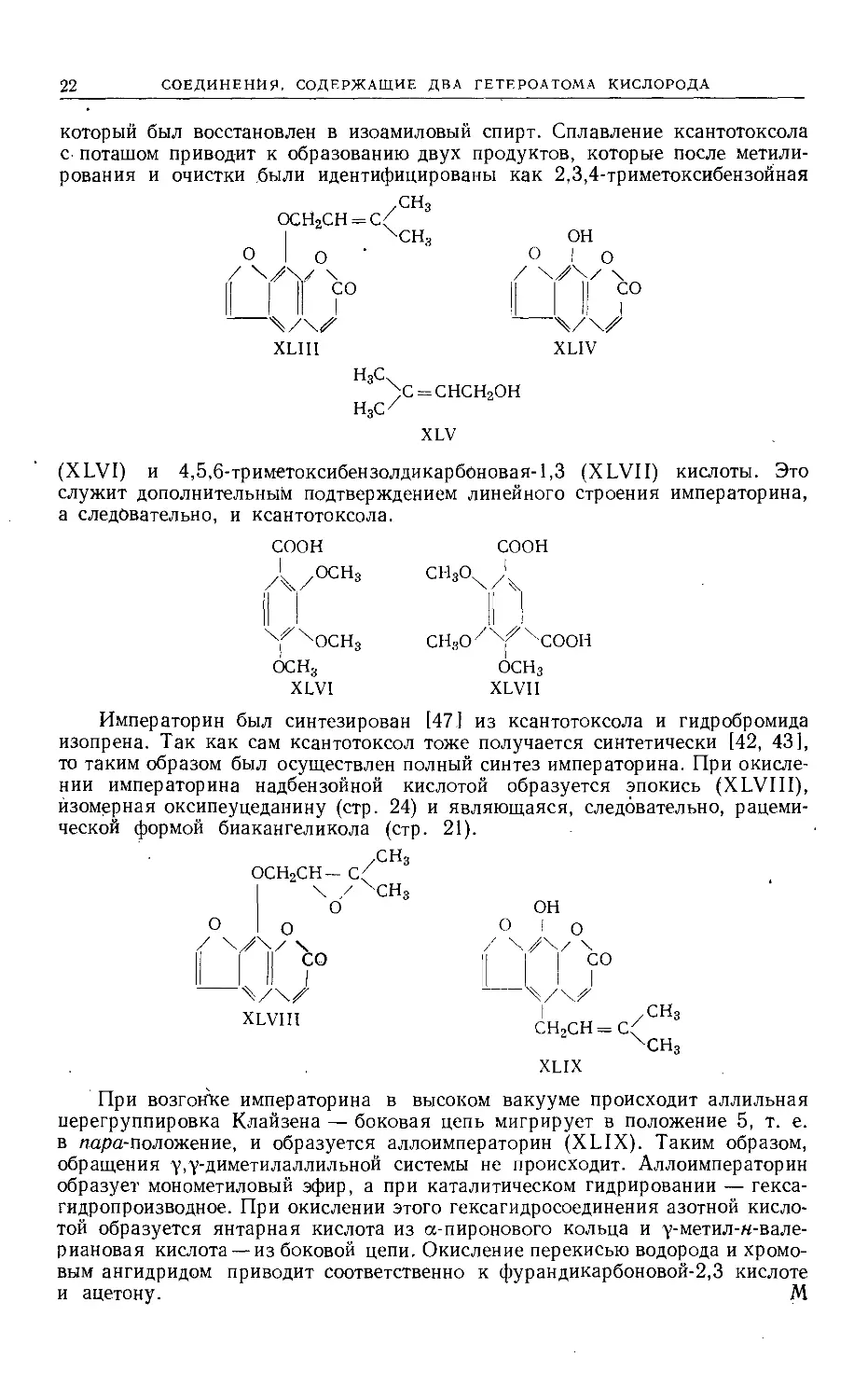
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
который был восстановлен в изоамиловый спирт. Сплавление ксантотоксола
с поташом
рования и
приводит к образованию двух продуктов, которые после метили-
очистки были идентифицированы как 2,3,4-триметоксибензойная
, /СН3
ОСН2СН = С<
| ХСН3
О | о
X
I Н со
ОН
I О
X
II СО
‘ (XLVI) и
XLIII
н3сх
)С = СНСН2ОН
XLIV
XLV
4,5,6-триметоксибензолдикарбоновая-1,3 (X LVII)
служит дополнительным подтверждением линейного строения
а следовательно, и ксантотоксола.
СООН
/I /°СН3
кислоты. Это
императорина,
СООН
СН3О. '
СН3О/Х^ХСООН
ОСНз
XLVH
[47] из ксантотоксола и
ОСНз
XLVI
Императорин был синтезирован
изопрена. Так как сам ксантотоксол тоже получается синтетически [42, 43],
то таким образом был осуществлен полный синтез императорина. При окисле-
нии императорина надбензойной кислотой образуется эпокись (XLVIII),
изомерная оксипеуцеданину (стр. 24) и являющаяся, следовательно, рацеми-
ческой формой биакангеликола (стр. 21).
СН3
ОСН2СН— С(
\ / ХСН3
о
о
' 'со
гидробромида
XLVIH
он
о ! о
/ \/\/ X
il I I (°
----\/-Х^
I
сн2сн = с
СН3
СН3
XLIX
О
3
При возгонке императорина в высоком вакууме происходит аллильная
перегруппировка Клайзена — боковая цепь мигрирует в положение 5, т. е.
в пара-положение, и образуется аллоимператорин (XLIX). Таким образом,
обращения у,у-диметилаллильной системы не происходит. Аллоимператорин
образует монометиловый эфир, а при каталитическом гидрировании — гекса-
гидропроизводное. При окислении этого гексагидросоединения азотной кисло-
той образуется янтарная кислота из а-пиронового кольца и у-метил-«-вале-
риановая кислота — из боковой цепи. Окисление перекисью водорода и хромо-
вым ангидридом приводит соответственно к фурандикарбоновой-2,3 кислоте
и ацетону. М
ФУРАНОКУМАРИНЫ
23
же, как ксантотоксол к бергаптолу.
Изоимператор ин (L) (у,у-диметилаллиловый эфир бергаптола) содержится
в корневищах Peucedanum ostruthium Koch, (синоним Imperatoria ostruthium
Linn.) и относится к императорину так
0 О
/ \
II СО
ОСН2СН
/СН3
чсн3
L
Образование фурандикарбоновой-2,3 кислоты [49, 50] при окислении пере-
кисью водорода указывает на наличие в молекуле (L) незамещенного фурано-
вого кольца. О строении эфирной боковой цепи свидетельствует образование
ацетона при окислении хромовым ангидридом и присоединение кислорода
к двойной связи при действии надбензойной кислоты с образованием эпокси-
соединения, идентичного оксипеуцеданину (стр. 24). Гидрирование дает гек-
сагидроизоимператор ин, при окислении которого азотной кислотой образуется
янтарная киблота из а-пиронового кольца и у-метил-«-валериановая кислота
из боковой цепи. При действии смеси уксусной и серной кислот получается
бергаптол и у,у-диметилаллиловый спирт.
Изоимператорин был синтезирован конденсацией бергаптола с гидробро-
мидом изопрена (ср. синтез императорина); поскольку сам бергаптол также
получен синтетически, был осуществлен, таким образом, полный синтез изо-
императорина.
Изопимпииеллин (LI) представляет собой 5,8-диметоксипсорален и содер-
жится [44] в корневищах Pimpinella saxifraga Linn. Образование фуран-
дикарбоновой-2,3 кислоты при окислении перекисью_ водорода свидетель-
ствует о наличии незамещенного —
ОСН3
О | о
/ \
II I СО
фуранового кольца. Присутствие сс-пироно-
ОСН3
А А /ОСНз
ОСН3
LII
осн3
LI
вого кольца и связь между пимпинеллином и изопимпинеллином подтвер-
ждаются результатами метилирования последнего щелочным раствором диме-
тилсульфата, в результате чего образуется р-(4,6,7-триметокси-5-бензофурано)-
акриловая кислота (LII), идентичная продукту, получающемуся по той же
реакции из пимпинеллина. Полный синтез изопимпинеллина [51 ] был осу-
ществлен исходя из бергаптола окислением его хромовым ангидридом в бер-
гаптохинон с последующим восстановлением и метилированием.
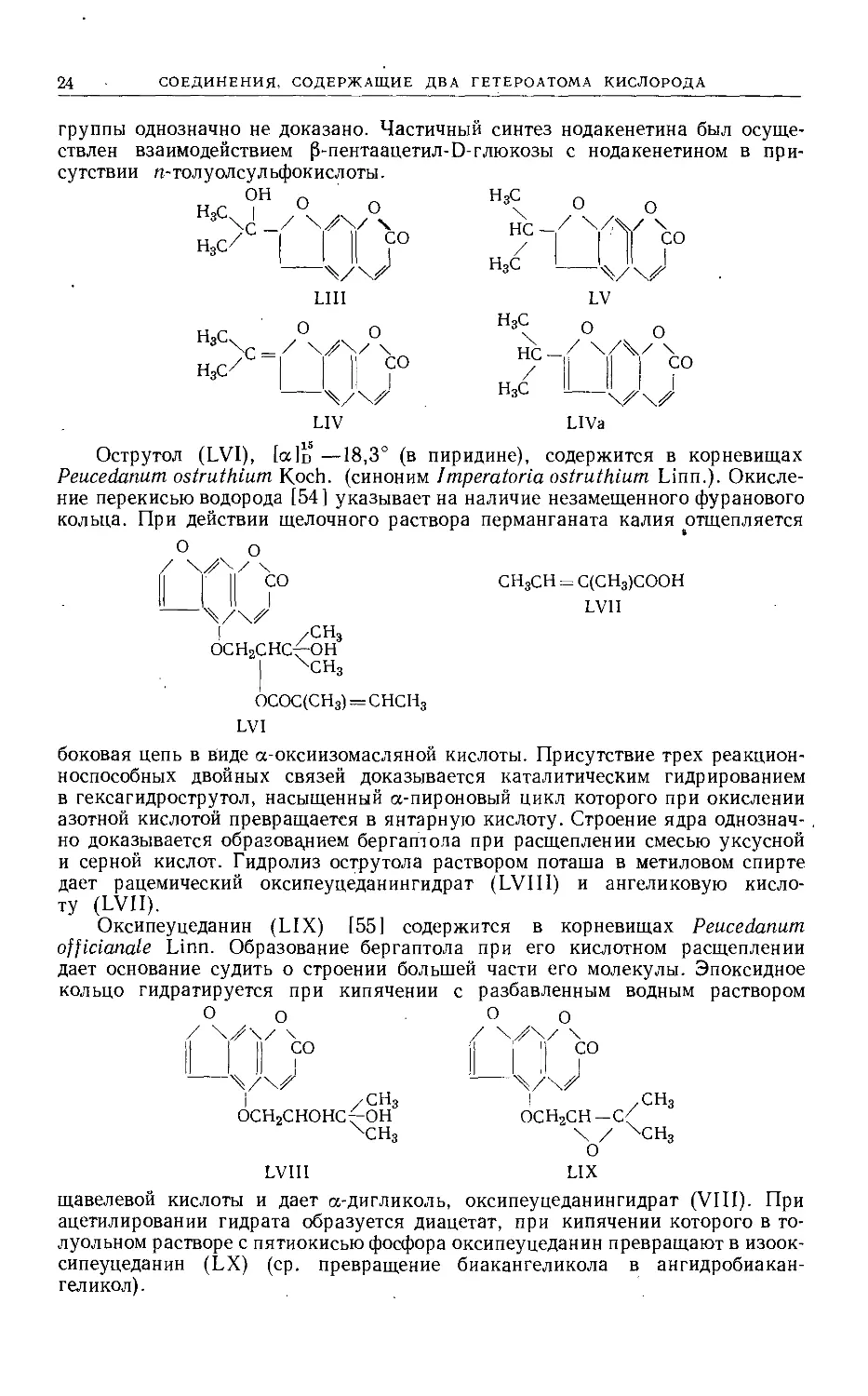
Нодакенетин (LIII), [ссЬ — 22,4°, содержится в виде глюкозида, нода-
кенина С2оН2909-Н20, в Peucedanum decursivum Maxim. [52, 53]. Перегонка
нодакенетина с пятиокисью фосфора в высоком вакууме дает ангидронода-
кенетин, образующийся при дегидратации третичного спирта, строение кото-
рого может быть выражено формулой LIV или, более вероятно, LIVa. Гидри-
рование соединений LIV приводит к тетрагидронодакенетину, идентичному
дезоксидигидроореоселону (LV) (стр. 25). При окислении щелочным раствором
перманганата калия из изопропилиденовой группы фуранового кольца обра-
зуется ацетон. При помощи этих реакций установлена наиболее вероятная
структура нодакенетина, а именно L1II, хотя положение гидроксильной
24
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
группы однозначно не доказано. Частичный синтез нодакенетина был осуще-
ствлен взаимодействием (З-пентаацетил-D-глюкозы с нодакенетином в при-
сутствии п-толуолсульфокислоты.
LIV LIVa
пиридине), содержится в корневищах
Острутол (LVI), [сс]ц —18,3° (в
Peucedanum ostruthium Koch, (синоним Imperatoria ostruthium Linn.). Окисле-
ние перекисью водорода [54] указывает на наличие незамещенного фуранового
кольца. При действии щелочного раствора перманганата калия отщепляется
О о
/ \
II Г СО
СН3СН = С(СН3)СООН
LV1I
осн2снсф-он
ОСОС(СН3) = СНСН3
LVI
боковая цепь в Виде а-оксиизомасляной кислоты. Присутствие трех реакцион-
носпособных двойных связей доказывается каталитическим гидрированием
в гексагидрострутол, насыщенный а-пироновый цикл которого при окислении
азотной кислотой превращается в янтарную кислоту. Строение ядра однознач- .
но доказывается образованием бергатола при расщеплении смесью уксусной
и серной кислот. Гидролиз острутола раствором поташа в метиловом спирте
дает рацемический оксипеуцеданингидрат (LVIII) и ангеликовую кисло-
ту (LVII).
Оксипеуцеданин (LIX) [55] содержится в корневищах Peucedanum
officianale Linn. Образование бергаптола при его кислотном расщеплении
дает основание судить о строении большей части его молекулы. Эпоксидное
кольцо гидратируется при кипячении
О о
/ \^\/ \
II I II СО
с разбавленным водным раствором
О о
/ \У\/\
II 'I СО
I /СН3
ОСН2СНОНС^ОН
I /СН3
ОСН2СН—С(
\ / ХСН3
о
LIX
оксипеуцеданингидрат (VIII). При
LVIII
щавелевой кислоты и дает а-дигликоль,
ацетилировании гидрата образуется диацетат, при кипячении которого в то-
луольном растворе с пятиокисью фосфора оксипеуцеданин превращают в изоок-
сипеуцеданин (LX) (ср. превращение биакангеликола в ангидробиакан-
геликол).
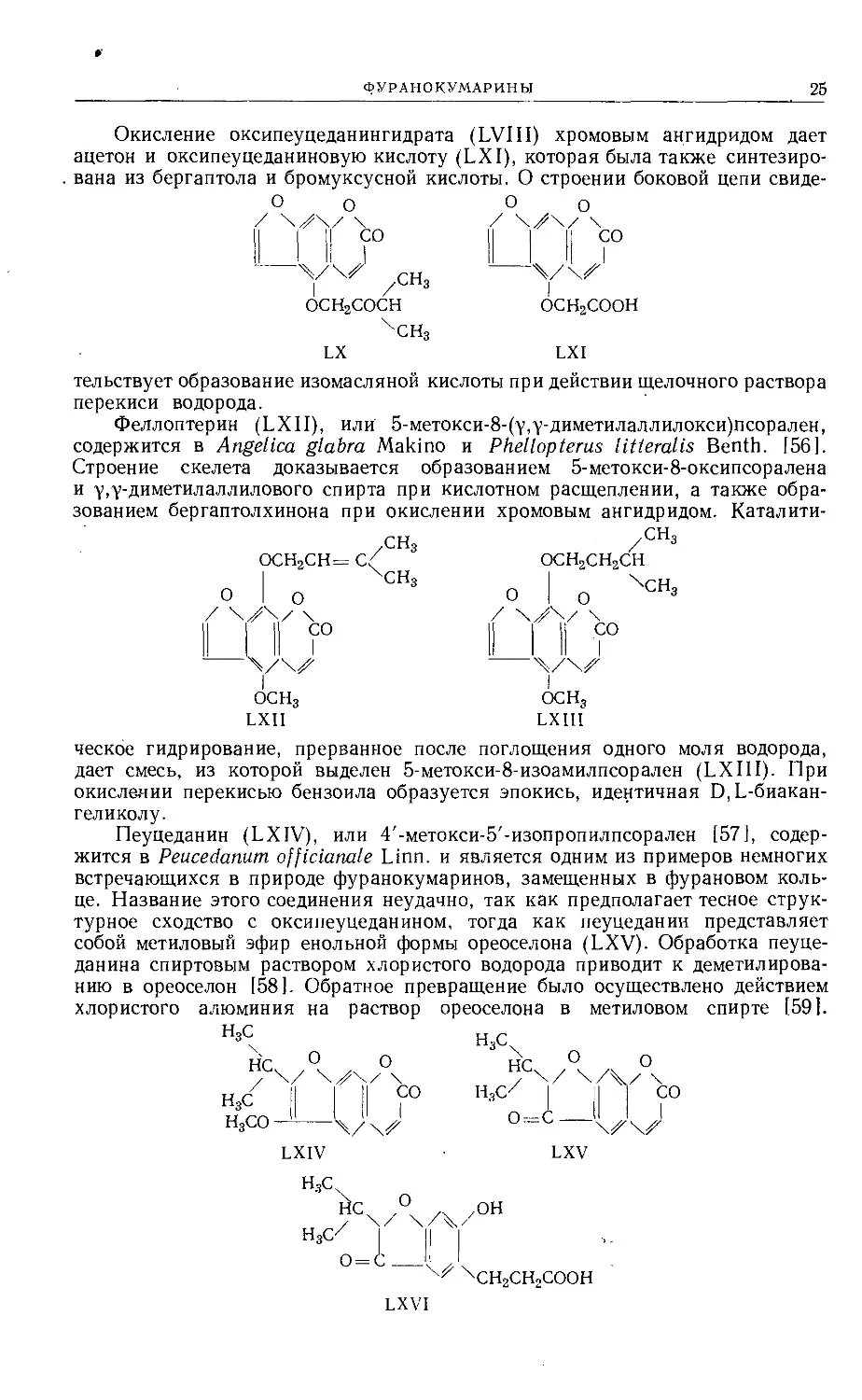
ФУРАНОКУМАРИНЫ
25
Окисление оксипеуцеданингидрата (LVIII) хромовым ангидридом дает
ацетон и оксипеуцеданиновую кислоту (LXI), которая была также синтезиро-
. вана из бергаптола и бромуксусной кислоты. О строении боковой цепи свиде-
О о О о
ОСН2СОСН ОСН2СООН
чсн3
LX LXI
тельствует образование изомасляной кислоты при действии щелочного раствора
перекиси водорода.
Феллоптерин (LXII), или 5-метокси-8-(у,у-диметилаллилокси)псорален,
содержится в Angelica glabra Makino и Phellopterus litteralis Benth. [56].
Строение скелета доказывается образованием 5-метокси-8-оксипсоралена
и у,у-диметилаллилового спирта при кислотном расщеплении, а также обра-
зованием бергаптолхинона при окислении хромовым ангидридом. Каталити-
СН3 /СНз
----- - / ОСН2СН2СН
I ХСН3
О I о
LXII LXIII
ческое гидрирование, прерванное после поглощения одного моля водорода,
дает смесь, из которой выделен 5-метокси-8-изоамилпсорален (LXIII). При
окислении перекисью бензоила образуется эпокись, идентичная П,Ь-биакан-
геликолу.
Пеуцеданин (LXIV), или 4'-метокси-5'-изопропилпсорален [57], содер-
жится в Peucedanum officianale Linn, и является одним из примеров немногих
встречающихся в природе фуранокумаринов, замещенных в фурановом коль-
це. Название этого соединения неудачно, так как предполагает тесное струк-
турное сходство с оксипеуцеданином, тогда как пеуцеданин представляет
собой метиловый эфир енольной формы ореоселона (LXV). Обработка пеуце-
данина спиртовым раствором хлористого водорода приводит к деметилирова-
нию в ореоселон [58]. Обратное превращение было осуществлено действием
хлористого алюминия на раствор ореоселона в метиловом спирте [59].
LXIV LXV
Н,СХ
ЙСХ О .. .он
H,cUj I
ХСН2СН2СООН
LXVI
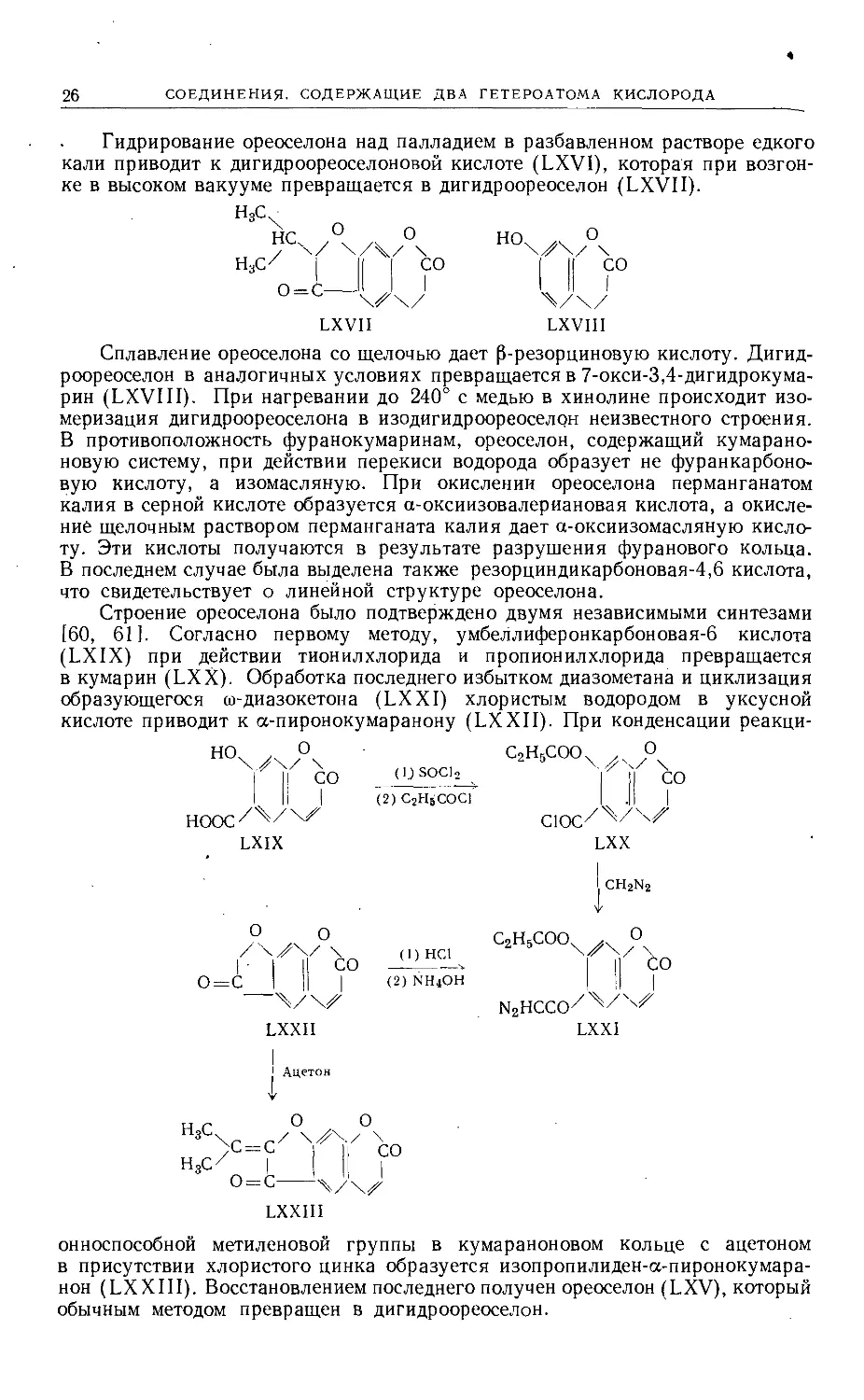
26
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Гидрирование ореоселона над палладием в разбавленном растворе едкого
кали приводит к дигидроореоселоновой кислоте (LXVI), которая при возгон-
ке в высоком вакууме превращается в дигидроореоселон (LXVII).
LXVII
LXVIII
Сплавление ореоселона со щелочью дает [3-резорциновую кислоту. Дигид-
роореоселон в аналогичных условиях превращается в 7-окси-3,4-дигидрокума-
рин (LXVIII). При нагревании до 240° с медью в хинолине происходит изо-
меризация дигидроОреоселона в изодигидроореоселон неизвестного строения.
В противоположность фуранокумаринам, ореоселон, содержащий кумарано-
новую систему, при действии перекиси водорода образует не фуранкарбоно-
вую кислоту, а изомасляную. При окислении ореоселона перманганатом
калия в серной кислоте образуется а-оксиизовалериановая кислота, а окисле-
ние щелочным раствором перманганата калия дает а-оксиизомасляную кисло-
ту. Эти кислоты получаются в результате разрушения фуранового кольца.
В последнем случае была выделена также резорциндикарбоновая-4,6 кислота,
что свидетельствует о линейной структуре ореоселона.
Строение ореоселона было подтверждено двумя независимыми синтезами
[60, 61]. Согласно первому методу, умбеллиферонкарбоновая-6 кислота
(LXIX) при действии тионилхлорида и пропионилхлорида превращается
в кумарин (LXX). Обработка последнего избытком диазометана и циклизация
образующегося ю-диазокетона (LXXI) хлористым водородом в уксусной
кислоте приводит к а-пиронокумаранону (LXXII). При конденсации реакци-
I II i°
HOOC'/'^/xZ
LXIX
(IJ SOCl-2
(2) C2H5COC1
(I) HC1
(2) NH4OH
LXXII
1 Ацетон
С2Н5СООХ Д
1.11)°
C10C/'^/4iZ
LXX
ch2N2
c,Hscoow о
III ?
n2hcco/'^/
LXXI
О о
г \
о= I I! “
0 = с---
LXXIII
онноспособной метиленовой группы в кумараноновом кольце с ацетоном
в присутствии хлористого цинка образуется изопропилиден-а-пиронокумара-
нон (LXXIII). Восстановлением последнего получен ореоселон (LXV), который
обычным методом превращен в дигидроореоселон.
ФУРАНОКУМАРИНЫ
27
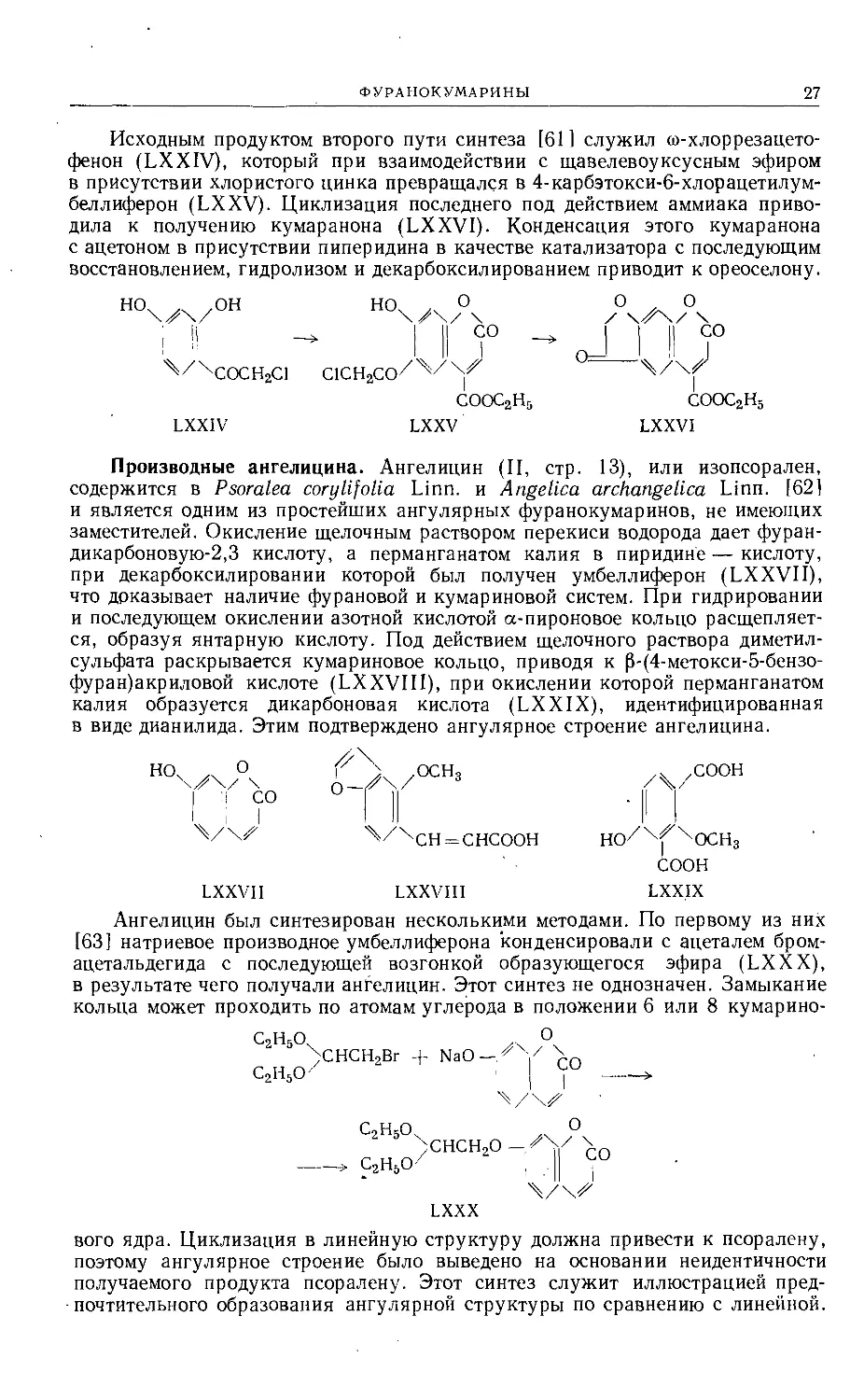
Исходным продуктом второго пути синтеза [61] служил со-хлоррезацето-
фенон (LXXIV), который при взаимодействии с щавелевоуксусным эфиром
в присутствии хлористого цинка превращался в 4-карбэтокси-6-хлорацетилум-
беллиферон (LXXV). Циклизация последнего под действием аммиака приво-
дила к получению кумаранона (LXXVI). Конденсация этого кумаранона
с ацетоном в присутствии пиперидина в качестве катализатора с последующим
восстановлением, гидролизом и декарбоксилированием приводит к ореоселону.
НО\Л/°Н
I II ? ’
^/ХСОСН2С1 ClCHaCo/^/4^
СООС2Н5
LXXIV
LXXV
Производные ангелицина. Ангелицин (II, стр. 13), или изопсорален,
содержится в Psoralea corylifolia Linn, и Angelica archangelica Linn. [62]
и является одним из простейших ангулярных фуранокумаринов, не имеющих
заместителей. Окисление щелочным раствором перекиси водорода дает фуран-
дикарбоновую-2,3 кислоту, а перманганатом калия в пиридине — кислоту,
при декарбоксилировании которой был получен умбеллиферон (LXXVII),
что доказывает наличие фурановой и кумариновой систем. При гидрировании
и последующем окислении азотной кислотой а-пироновое кольцо расщепляет-
ся, образуя янтарную кислоту. Под действием щелочного раствора диметил-
сульфата раскрывается кумариновое кольцо, приводя к р-(4-метокси-5-бензо-
фуран)акриловой кислоте (LXXVIII), при окислении которой перманганатом
калия образуется дикарбоновая кислота (LXXIX), идентифицированная
в виде дианилида. Этим подтверждено ангулярное строение ангелицина.
НО
LXXVII
LXXVIII
ч ZCOOH
|| I
но/х^хосн3
СООН
LXXIX
Ангелицин был синтезирован несколькими методами. По первому из них
[63] натриевое производное умбеллиферона конденсировали с ацеталем бром-
ацетальдегида с последующей возгонкой образующегося эфира (LXXX),
в результате чего получали ангелицин. Этот синтез не однозначен. Замыкание
кольца может проходить по атомам углерода в положении 6 или 8 кумарино-
С2Н5ОЧ /ч 0
)CHCH2Br + NaO—Z
с2н5О' ' I --------->
С2Н5О /Ок
;СНСН2О \
----> С2Нйо/ ’ II I
LXXX
вого ядра. Циклизация в линейную структуру должна привести к псоралену,
поэтому ангулярное строение было выведено на основании неидентичности
получаемого продукта псоралену. Этот синтез служит иллюстрацией пред-
почтительного образования ангулярной структуры по сравнению с линейной.
28
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Второй синтез Шпета [64] также неоднозначен и осуществлен путем
превращения умбеллиферона под действием гексаметилентетрамина в ледяной
уксусной кислоте в умбеллиферон-8-альдегид (LXXXI) [65]. При конденса-
ции натриевого производного (LXXXI) с этиловым эфиром иодуксусной
СНО
LXXXI
СНО
с2н5ооссн2оч J о
I II ?
LXXXII
кислоты образуется 7-карбэтоксиметоксиумбеллиферон-8-альдегид (LXXXII),
который в свою очередь гидролизуется, а затем циклизуется с одновременным
декарбоксилированием, превращаясь в ангелицин (II). Строение альдегида
(LXXXI) установлено [66] восстановлением его в продукт, идентичный
7-окси-8-метилкумарину, приготовленному из 2-метилрезорцина конденса-
цией по Пехману.
Третий синтез ангелицина был проведен Лимайе [67] исходя из караниола.
Караниол (LXXXIII), полученный расщеплением караньина (стр. 43), был
превращен в 5-формил-4-оксикумарон (LXXXIV) по методу Реймера —
Тимана (чувствительность незамещенного фуранового кольца к действию
минеральных кислот исключала возможность применения реакции Гаттер-
мана). Последующая конденсация по Перкину приводила к ангелицину.
В связи с синтезом альдегида (LXXXIV) нельзя не учитывать некоторых
отклонений в поведении кумаронов в условиях получения альдегидов по ме-
тоду Гаттермана. Бензофураны, не замещенные в а- и p-положениях, очень
чувствительны по отношению к минеральным кислотам. Однако наличие
карбоксильной группы в «-положении в фурановом кольце стабилизирует
всю систему и делает возможным проведение синтеза Гаттермана [34]. Альде-
гидная группа в этом случае входит в положение 7 бензольной системы, если
эта последняя является производной флороглюцина. После гидролиза защитная
группа легко удаляется декарбоксилированием. Наличие метильной группы
в а- и p-положениях также стабилизирует систему. Если метильная группа
находится в p-положении, то формильная группа вступает в фурановое кольцо
в «-положение по отношению к бензольному кольцу. Если же бензофурановая
система такова, что замещение возможно в обоих кольцах, и если желательно
ввести формильную группу в бензольное кольцо, то a-положение в фурановом
цикле должно быть блокировано, например карбоксильной группой.
LXXXIII LXXXIV
Пимпинеллин (LXXXV), или 5,6-диметоксиангелицин, содержится
в корневищах Pimpinella saxifraga Linn, и Heracleum sphondyliutn Linn. [44]
LXXXV
_/X/ra-
I II
сн3с/ у 'CH=CHCOOH
OCHg
LXXXVI
ФУРАНОКУМАРИНЫ
29
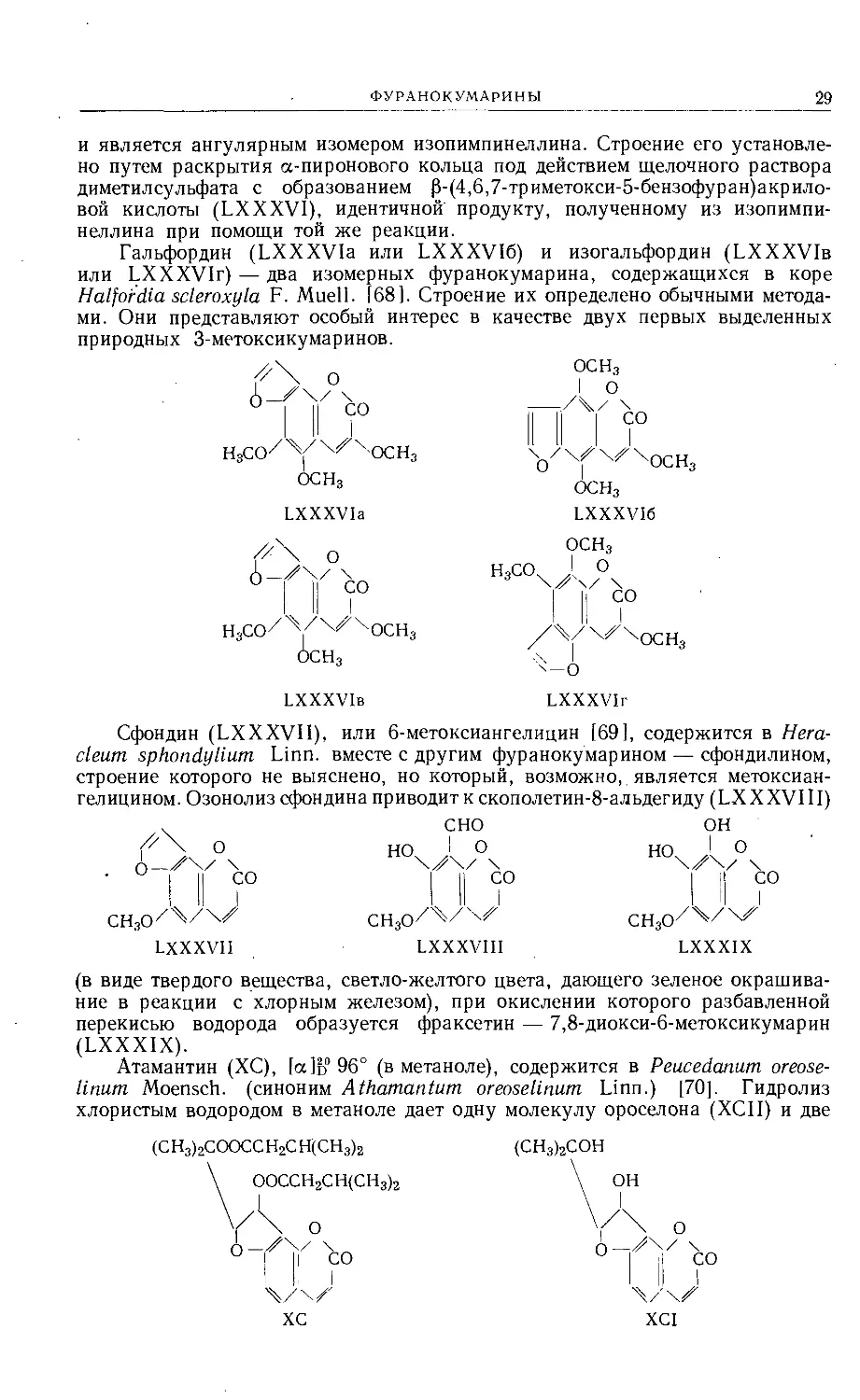
и является ангулярным изомером изопимпинеллина. Строение его установле-
но путем раскрытия а-пиронового кольца под действием щелочного раствора
диметилсульфата с образованием р-(4,6,7-триметокси-5-бензофуран)акрило-
вой кислоты (LXXXV1), идентичной продукту, полученному из изопимпи-
неллина при помощи той же реакции.
Гальфордин (LXXXVIa или LXXXVI6) и изогальфордин (LXXXVIb
или LXXXVIr) — два изомерных фуранокумарина, содержащихся в коре
Halfordia scleroxyla F. Muell. 168]. Строение их определено обычными метода-
ми. Они представляют особый интерес в качестве двух первых выделенных
природных 3-метоксикумаринов.
ОСН3
LXXXVIa
ОСН3
ОСН3
LXXXVI6
О
*~Г' ~
НзСО/ХЧ./Х^ЧОСНз
ОСНз
LXXXVIb
ОСН3
Н3СО. । о
I II ?
'^хосн3
LXXXVIr
Сфондин (LXXXVII), или 6-метоксиангелицин [69], содержится в Нега-
cleum sphondylium Linn, вместе с другим фуранокумарином — сфондилином,
строение которого не выяснено, но который, возможно, является метоксиан-
гелицином. Озонолиз сфондина приводит к скополетин-8-альдегиду (LX XXVIII)
СНзО7^74^
LXXXVII
С НО
I II ?
СНзО7^7
LXXXVIII
ОН
H0\zkz\
I II )°
СНзО7^747,
LXXXIX
(в виде твердого вещества, светло-желтого цвета, дающего зеленое окрашива-
ние в реакции с хлорным железом), при окислении которого разбавленной
перекисью водорода образуется фраксетин — 7,8-диокси-6-метоксикумарин
(LXXXIX).
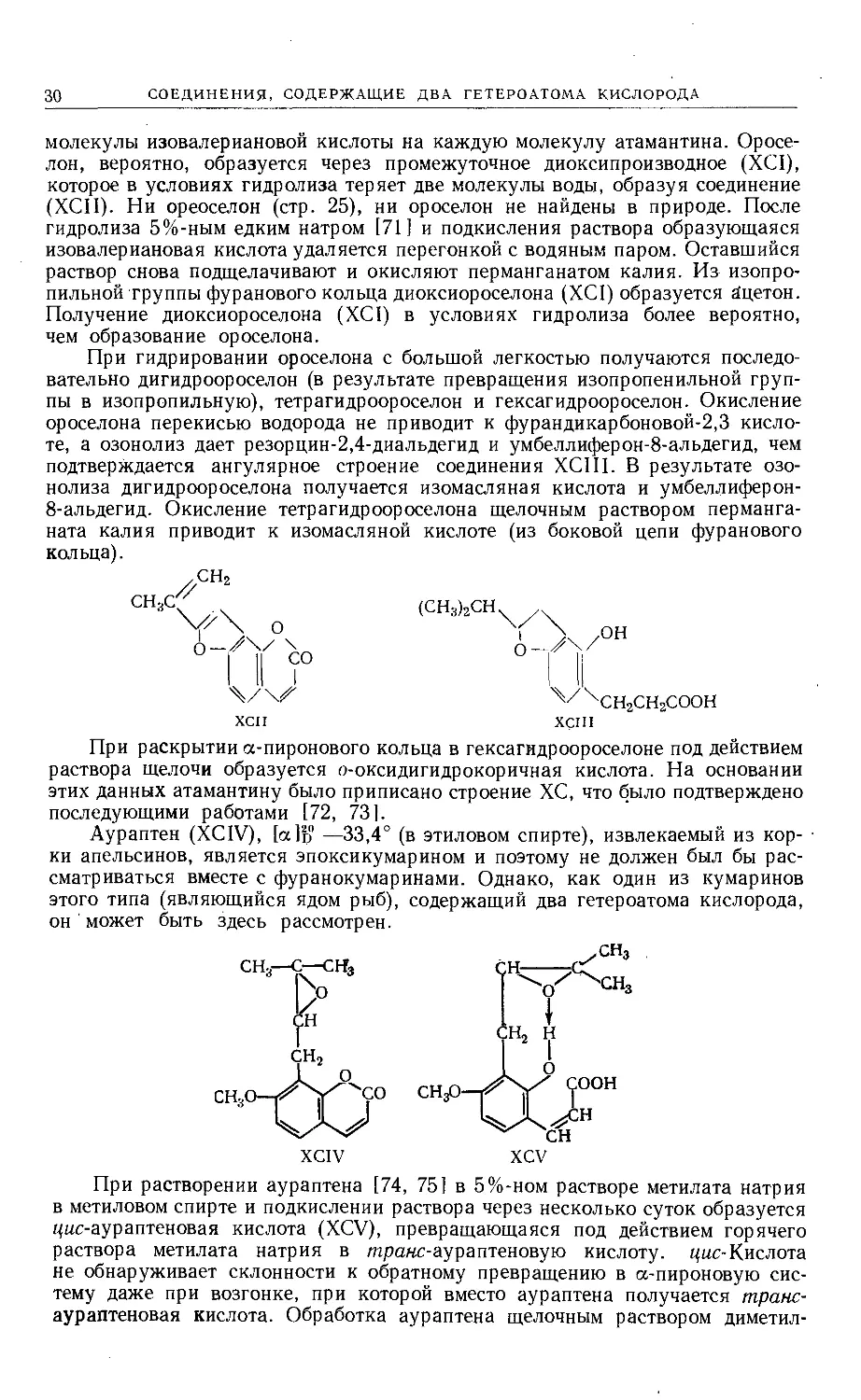
Атамантин (ХС), [а]Ь° 96° (в метаноле), содержится в Peucedanum oreose-
linum Moensch. (синоним Athamantum oreoselinutn Linn.) [70]. Гидролиз
хлористым водородом в метаноле дает одну молекулу ороселона (XCII) и две
(СН3)2СОН
(СН3)2СООССН2СН(СН3)2
ХС
XCI
30
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
молекулы изовалериановой кислоты на каждую молекулу атамантина. Оросе-
лон, вероятно, образуется через промежуточное диоксипроизводное (XCI),
которое в условиях гидролиза теряет две молекулы воды, образуя соединение
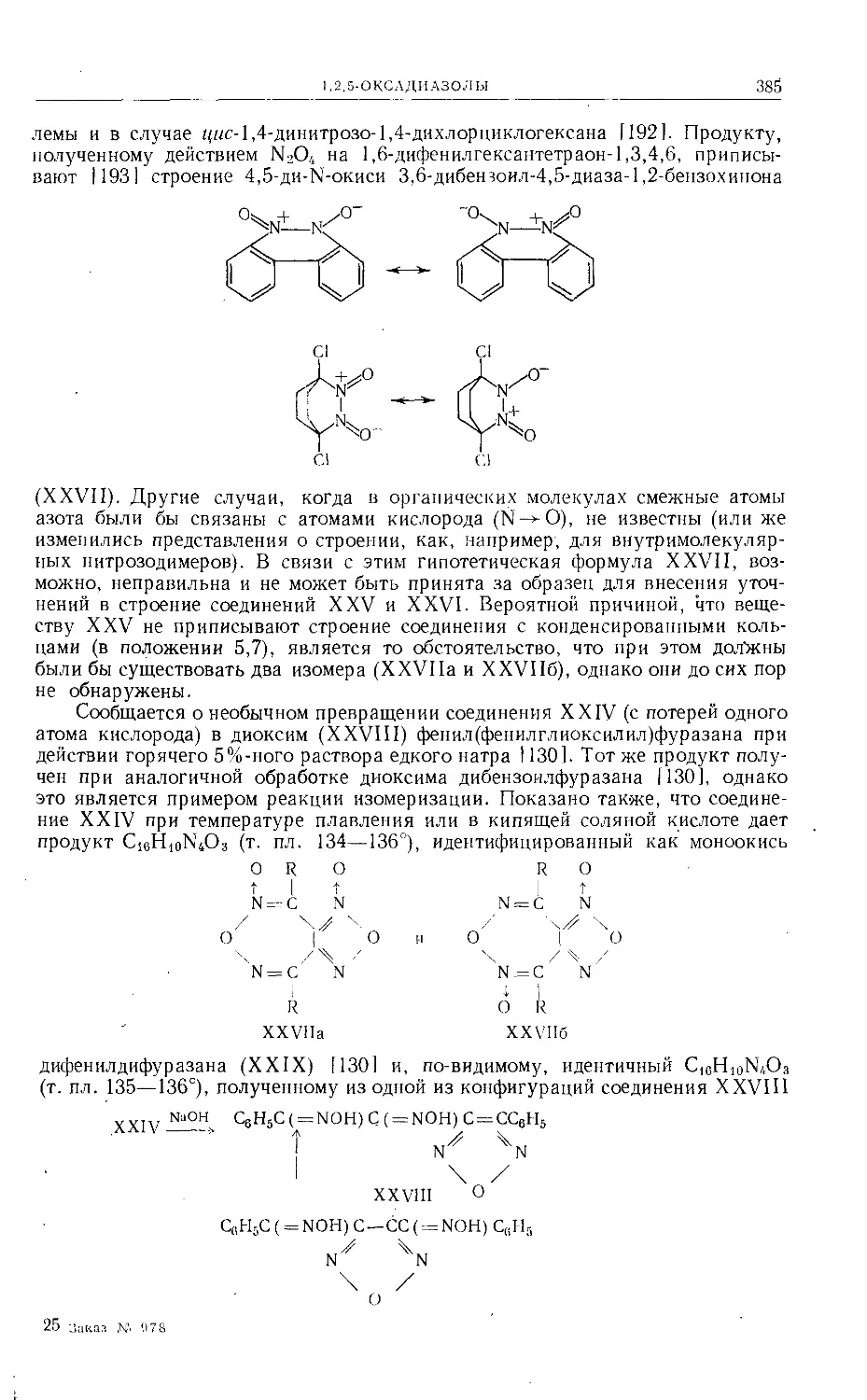
(ХСП). Ни ореоселон (стр. 25), ни ороселон не найдены в природе. После
гидролиза 5%-ным едким натром [71] и подкисления раствора образующаяся
изовалериановая кислота удаляется перегонкой с водяным паром. Оставшийся
раствор снова подщелачивают и окисляют перманганатом калия. Из изопро-
пильной группы фуранового кольца диоксиороселона (XCI) образуется ацетон.
Получение диоксиороселона (XCI) в условиях гидролиза более вероятно,
чем образование ороселона.
При гидрировании ороселона с большой легкостью получаются последо-
вательно дигидроороселон (в результате превращения изопропенильной груп-
пы в изопропильную), тетрагидроороселон и гексагидроороселон. Окисление
ороселона перекисью водорода не приводит к фурандикарбоновой-2,3 кисло-
те, а озонолиз дает резорцин-2,4-диальдегид и умбеллиферон-8-альдегид, чем
подтверждается ангулярное строение соединения ХСШ. В результате озо-
нолиза дигидроороселона получается изомасляная кислота и умбелдиферон-
8-альдегид. Окисление тетрагидроороселона щелочным раствором перманга-
ната калия приводит к изомасляной кислоте (из боковой цепи фуранового
кольца).
хсп хеш
При раскрытии а-пиронового кольца в гексагидроороселоне под действием
раствора щелочи образуется о-оксидигидрокоричная кислота. На основании
этих данных атамантину было приписано строение ХС, что было подтверждено
последующими работами [72, 73].
Аураптен (XCIV), [а]р —33,4° (в этиловом спирте), извлекаемый из кор-
ки апельсинов, является эпоксикумарином и поэтому не должен был бы рас-
сматриваться вместе с фуранокумаринами. Однако, как один из кумаринов
этого типа (являющийся ядом рыб), содержащий два гетероатома кислорода,
он ’ может быть здесь рассмотрен.
XCIV
XCV
При растворении аураптена [74, 75] в 5%-ном растворе метилата натрия
в метиловом спирте и подкислении раствора через несколько суток образуется
цис-аураптеновая кислота (XCV), превращающаяся под действием горячего
раствора метилата натрия в трояс-аураптеновую кислоту. цис-Кислота
не обнаруживает склонности к обратному превращению в а-пироновую сис-
тему даже при возгонке, при которой вместо аураптена получается транс-
аураптеновая кислота. Обработка аураптена щелочным раствором диметил-
ФУРАНОКУМАРИНЫ
31
сульфата по методу Кантера и Робертсона [28] дает о-оксикоричную кислоту,
а не сложный эфир о-метоксикоричной кислоты или о-метоксикоричную кисло-
ту. Однако при действии диазометана на свободную кислоту образуется
сложный эфир транс-кислоты. Попытки прометилировать фенольную гидрок-
сильную группу оказались безуспешными.
Гидрирование цис- и транс-аураптеновых кислот приводит к одной и той
же дигидроаураптеновой кислоте, идентичной продукту, полученному восста-
новлением аураптена в разбавленной щелочи. Дигидроаураптеновая кислота
возгоняется без изменения, не обнаруживая склонности к превращению в ди-
гидрокумарин, обычной для о-оксидигидрокоричных кислот. Это необычное
поведение кумаринового ядра аураптена трудно объяснимо, и объяснить его
стерическим влиянием заместителя большого размера в положении 8, по-
видимому, невозможно, так как остенол (XCVI) содержит такой же замести-
тель в том же положении и все же ведет себя нормально [76]. Эти особенности
дигидроаураптеновой кислоты напоминают о-оксикоричные кислоты, произ-
водные кумаринов, содержащие отрицательные (электрофильные) замести-
тели [77—79], и могут быть также объяснены наличием водородной связи,
как показано в формуле XCV [80].
СН3 /Снз
СН2СН = С/ СН2СОСН
НО. I О СН3 н0 I о \Сн3
\Z\z \ \^\/ \ 3
I [ ? i II г
\/\Z \/\Z
XCVI XCVII
При кипячении в течение 4 час с 20%-ной серной кислотой аураптен
изомеризуется в оптически неактивный изоаураптен (XCVII), что напоминает
превращение биакангеликола в ангидробиакангелицин (стр. 20). Гидратация
эпоксидного кольца нагреванием с разбавленной щавелевой кислотой при-
водит к а-дигликолю (XCVIII), превращающемуся в изоаураптен при кипя-
чении гликоля с 20%-ной серной кислотой. Дальнейшим доказательством
наличия эпоксидного кольца в аураптене является присоединение диметил-
амина [81] с образованием аминоспирта.
------------------с/----------------> -с/
/\ / \ HN(CH3)2 /у | \
О---------------------------ОН N(CH3)2
Окисление аураптена хромовым ангидридом дает ацетон и 7-метоксику-
марин-8-уксусную кислоту, идентичную образцу, полученному из остола [82].
При действии на аураптенгидрат [а-дигликоль (XCVIII)] тетраацетата свинца
образуется ацетон и 7-метоксикумарин-8-ацетальдегид (XCIX), дающий при
обработке окисью серебра 7-метоксикумарин-8-уксусную кислоту [83], иден-
тичную образующейся при окислении аураптена хромовым ангидридом. Эта
кислота может быть декарбоксилирована в 7-метокси-8-метилкумарин.
Н3С СН3
С—ОН
Н-С-ОН
I
сн2
сно
сн2
XCVIH
XCIX
32
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
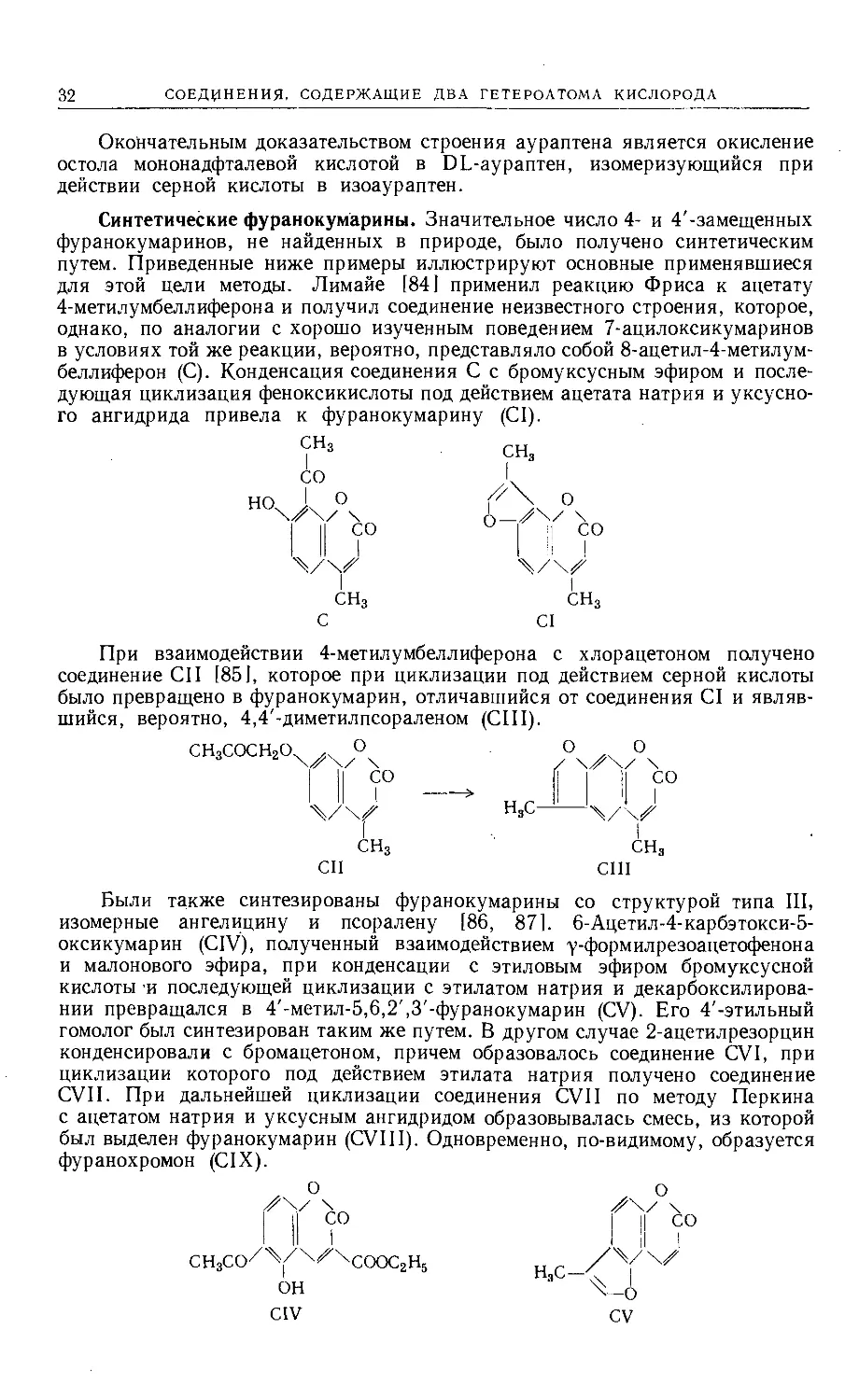
Окончательным доказательством строения аураптена является окисление
остола мононадфталевой кислотой в DL-аураптен, изомеризующийся при
действии серной кислоты в изоаураптен.
Синтетические фуранокумарины. Значительное число 4- и 4'-замещенных
фуранокумаринов, не найденных в природе, было получено синтетическим
путем. Приведенные ниже примеры иллюстрируют основные применявшиеся
для этой цели методы. Лимайе [84] применил реакцию Фриса к ацетату
4-метилумбеллиферона и получил соединение неизвестного строения, которое,
однако, по аналогии с хорошо изученным поведением 7-ацилоксикумаринов
в условиях той же реакции, вероятно, представляло собой 8-ацетил-4-метилум-
беллиферон (С). Конденсация соединения С с бромуксусным эфиром и после-
дующая циклизация феноксикислоты под действием ацетата натрия и уксусно-
го ангидрида привела к фуранокумарину (CI).
СН3
СО
СН
I
сн3
При взаимодействии 4-метилумбеллиферона с хлорацетоном получено
соединение СП [85], которое при циклизации под действием серной кислоты
было превращено в фуранокумарин, отличавшийся от соединения CI и являв-
шийся, вероятно, 4,4'-диметилпсораленом (СШ).
СН3СОСН2О. О 0 0
II ? —> нс II I !|
\/\Z НзС----\/\^
I .. I
сн3 сн3
СП СШ
Были также синтезированы фуранокумарины со структурой типа III,
изомерные ангелицину и псоралену [86, 87]. 6-Ацетил-4-карбэтокси-5-
оксикумарин (CIV), полученный взаимодействием у-формилрезоацетофенона
и малонового эфира, при конденсации с этиловым эфиром бромуксусной
кислоты и последующей циклизации с этилатом натрия и декарбоксилирова-
нии превращался в 4'-метил-5,6,2',3'-фуранокумарин (CV). Его 4'-этильный
гомолог был синтезирован таким же путем. В другом случае 2-ацетилрезорцин
конденсировали с бромацетоном, причем образовалось соединение CVI, при
циклизации которого под действием этилата натрия получено соединение
CVII. При дальнейшей циклизации соединения CVII по методу Перкина
с ацетатом натрия и уксусным ангидридом образовывалась смесь, из которой
был выделен фуранокумарин (CVIII). Одновременно, по-видимому, образуется
фуранохромон (CIX).
СН3СО/‘^|/Ч^чСООС2Н5
он
CIV
CV
ФУРАНОКУМАРИНЫ
33
СОСН3.
О=С —сн3
CVI
СОСНз
но
CVII
СН3
СОСН3
Н>С\А
| со
CH3COONa
(СН3СО)2О
СНз
СТХ
CH3COONa (СН3СО)2О
CVIII
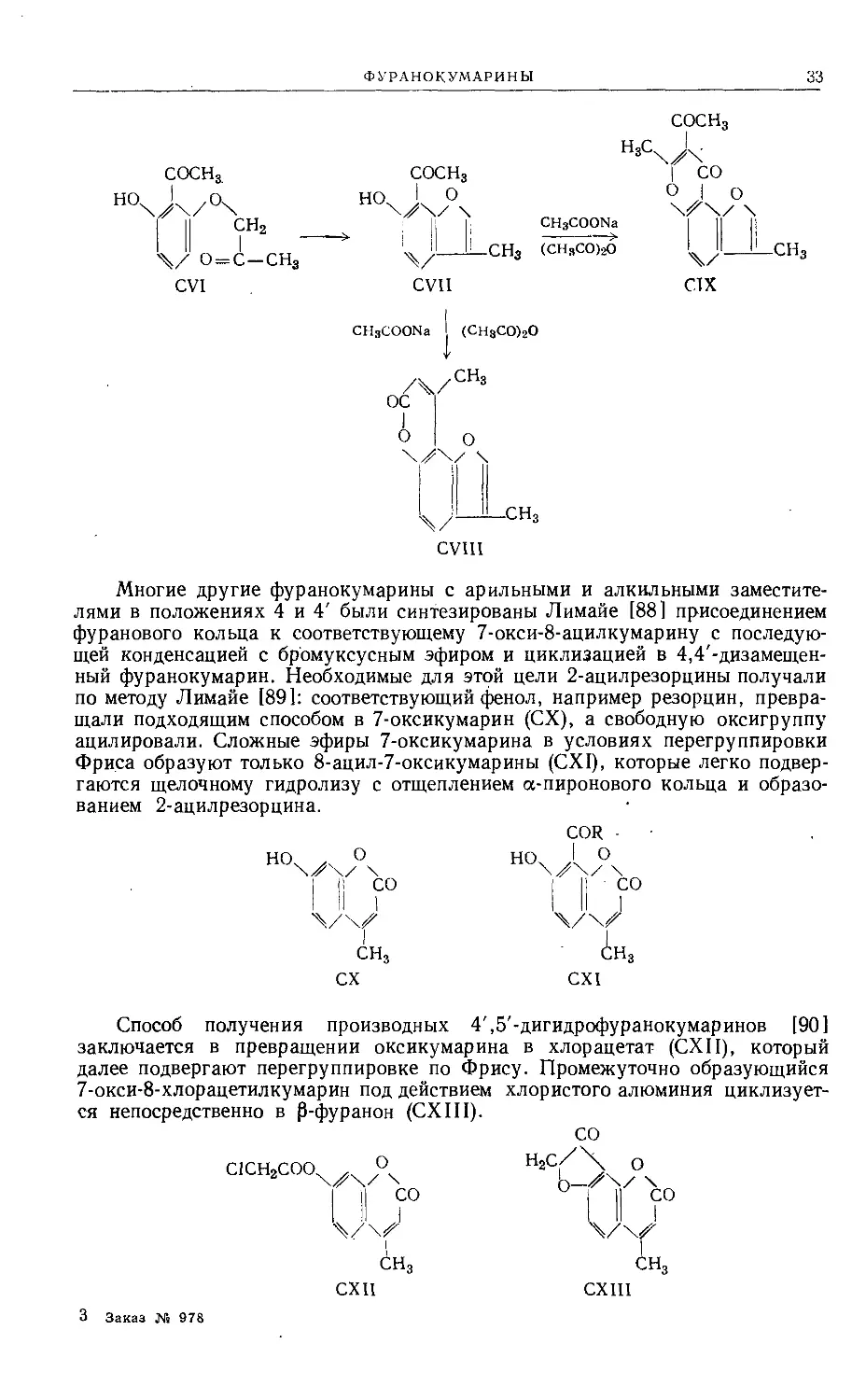
Многие другие фуранокумарины с арильными и алкильными заместите-
лями в положениях 4 и 4' были синтезированы Лимайе [88] присоединением
фуранового кольца к соответствующему 7-окси-8-ацилкумарину с последую-
щей конденсацией с брбмуксусным эфиром и циклизацией в 4,4'-дизамещен-
ный фуранокумарин. Необходимые для этой цели 2-ацилрезорцины получали
по методу Лимайе [891: соответствующий фенол, например резорцин, превра-
щали подходящим способом в 7-оксикумарин (СХ), а свободную оксигруппу
ацилировали. Сложные эфиры 7-оксикумарина в условиях перегруппировки
Фриса образуют только 8-ацил-7-оксикумарины (CXI), которые легко подвер-
гаются щелочному гидролизу с отщеплением а-пиронового кольца и образо-
ванием 2-ацилрезорцина.
COR -
СХ CXI
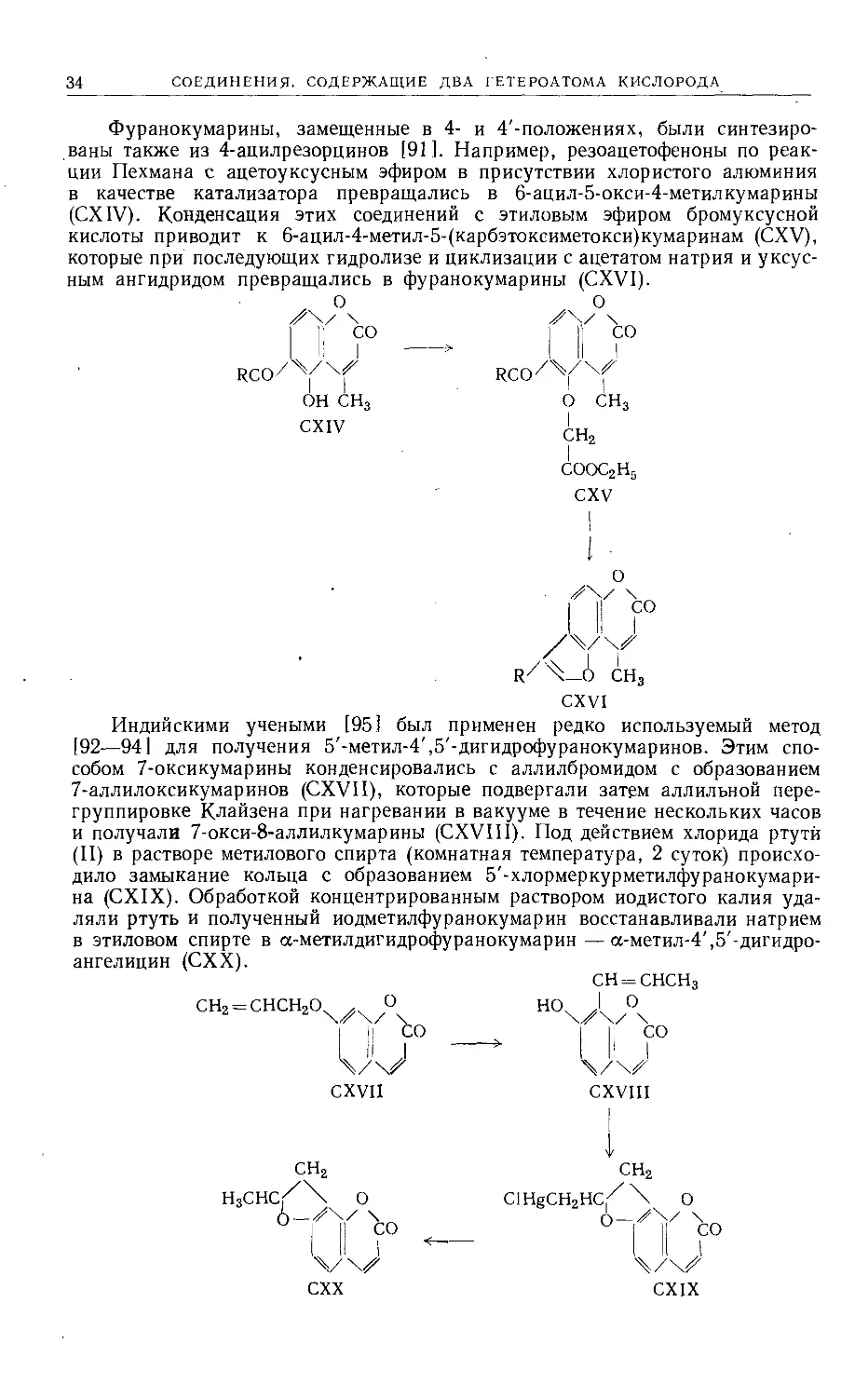
Способ получения производных 4',5'-дигидрофуранокумаринов [90]
заключается в превращении оксикумарина в хлорацетат (СХП), который
далее подвергают перегруппировке по Фрису. Промежуточно образующийся
7-окси-8-хлорацетилкумарин под действием хлористого алюминия циклизует-
ся непосредственно в |3-фурайон (СХШ).
СО ссн,соох \ Н!СОчД I s СО % || со 1 1 СН3 СН3 СХП CXIII
3 Заказ № 978
34
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Фуранокумарины, замещенные в 4- и 4'-положениях, были синтезиро-
ваны также из 4-ацилрезорцинов [91]. Например, резоацетофеноны по реак-
ции Пехмана с ацетоуксусным эфиром в присутствии хлористого алюминия
в качестве катализатора превращались в 6-ацил-5-окси-4-метилкумарины
(CXIV). Конденсация этих соединений с этиловым эфиром бромуксусной
кислоты приводит к 6-ацил-4-метил-5-(карбэтоксиметокси)кумаринам (CXV),
которые при последующих гидролизе и циклизации с ацетатом натрия и уксус-
ным ангидридом превращались в фуранокумарины (CXVI).
О О
А\/ \ /\/ \
I I! ?° I II ?
ОН сн3 о сн3
CXIV сн2
СООС2Н8
CXV
CXVI
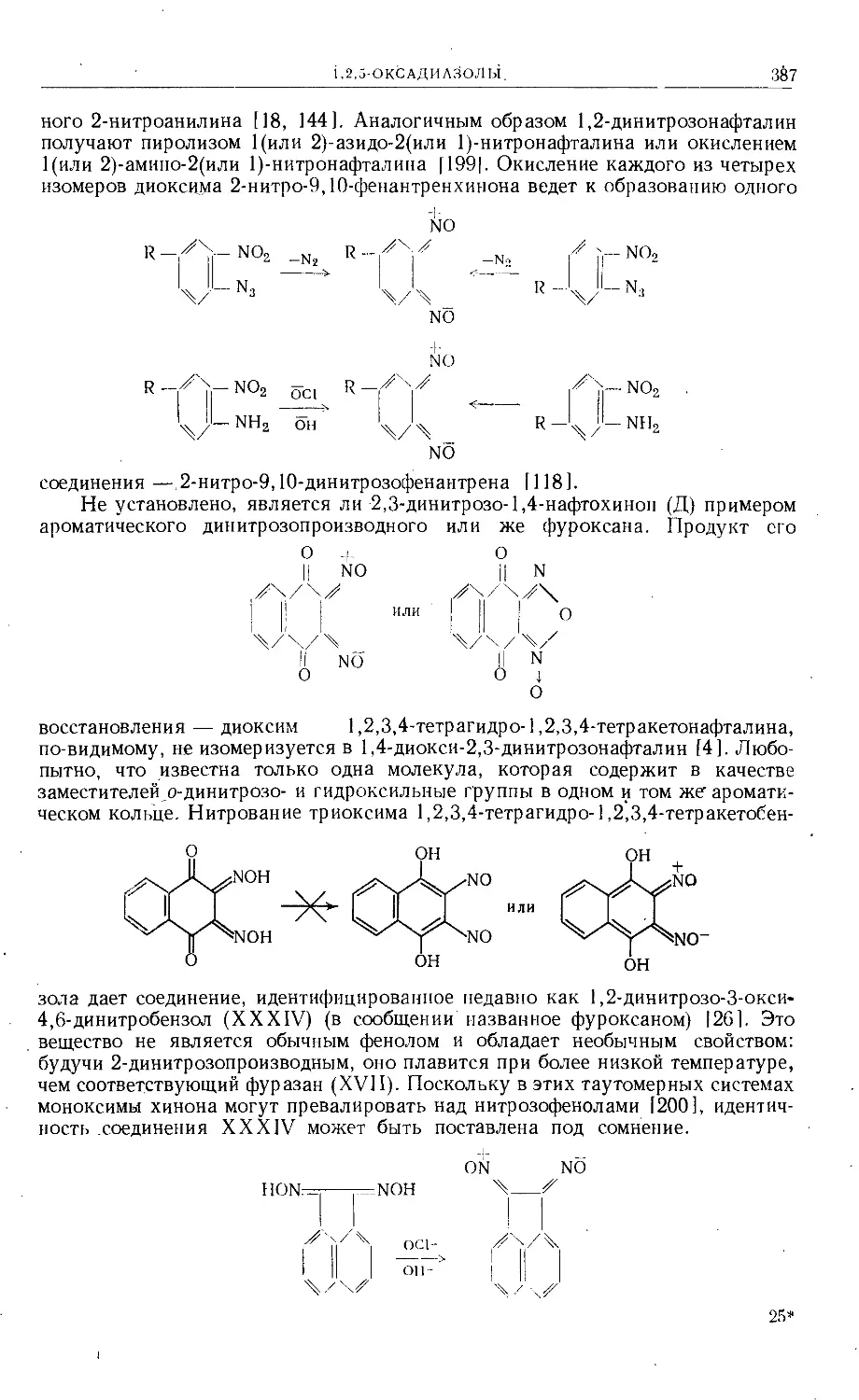
Индийскими учеными [95] был применен редко используемый метод
[92—94] для получения 5'-метил-4',5'-дигидрофуранокумаринов. Этим спо-
собом 7-оксикумарины конденсировались с аллилбромидом с образованием
7-аллилоксикумаринов (CXVII), которые подвергали затем аллильной пере-
группировке Клайзена при нагревании в вакууме в течение нескольких часов
и получали 7-окси-8-аллилкумарины (CXVIII). Под действием хлорида ртути
(II) в растворе метилового спирта (комнатная температура, 2 суток) происхо-
дило замыкание кольца с образованием 5'-хлормеркурметилфуранокумари-
на (CXIX). Обработкой концентрированным раствором йодистого калия уда-
ляли ртуть и полученный иодметилфуранокумарин восстанавливали натрием
в этиловом спирте в а-метилдигидрофуранокумарин — а-метил-4',5'-дигидро-
ангелицин (СХХ).
СН = СНСН3
снг-снснго^хд НС\АА
I II СО I |; СО
I li । —" И ।
Ч/\А \/\А
CXVII CXVIII
сн2
СХХ
сн2
ClHgCHzHC./X о
О — А\/ \
I II 7
CXIX
ФУРАНОХРОМОНЫ
35
ФУРАНОХРОМОНЫ
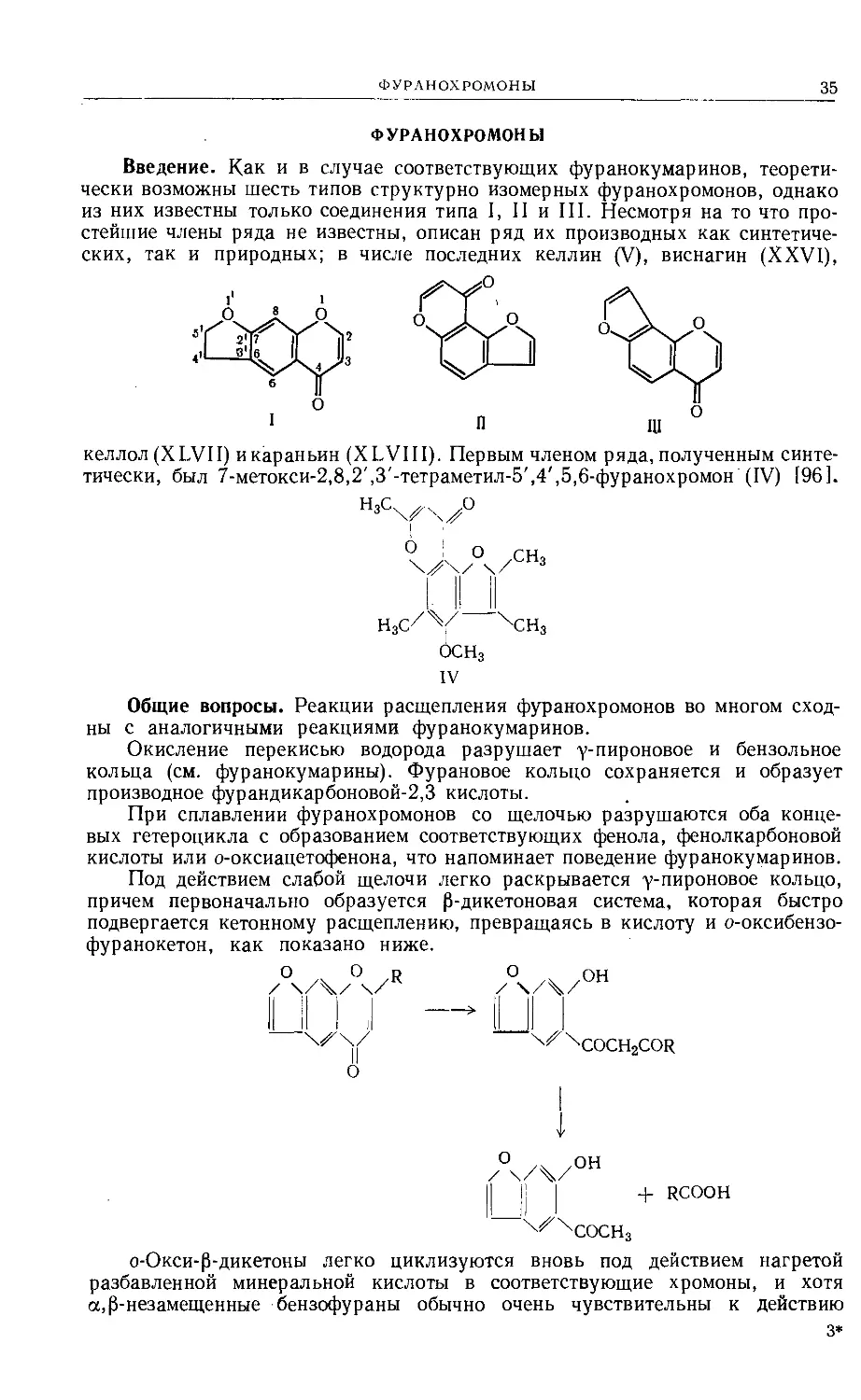
Введение. Как и в случае соответствующих фуранокумаринов, теорети-
чески возможны шесть типов структурно изомерных фуранохромонов, однако
из них известны только соединения типа I, II и III. Несмотря на то что про-
стейшие члены ряда не известны, описан ряд их производных как синтетиче-
ских, так и природных; в числе последних келлин (V), виснагин (XXVI),
келлол(ХЬУП) икараньин (XLVIII). Первым членом ряда, полученным синте-
тически, был 7-метокси-2,8,2',3'-тетраметил-5',4',5,6-фуранохромон (IV) [96].
Общие вопросы. Реакции расщепления фуранохромонов во многом сход-
ны с аналогичными реакциями фуранокумаринов.
Окисление перекисью водорода разрушает у-пироновое и бензольное
кольца (см. фуранокумарины). Фурановое кольцо сохраняется и образует
производное фурандикарбоновой-2,3 кислоты.
При сплавлении фуранохромонов со щелочью разрушаются оба конце-
вых гетероцикла с образованием соответствующих фенола, фенолкарбоновой
кислоты или о-оксиацетофенона, что напоминает поведение фуранокумаринов.
Под действием слабой щелочи легко раскрывается у-пироновое кольцо,
причем первоначально образуется 0-дикетоновая система, которая быстро
подвергается кетонному расщеплению, превращаясь в кислоту и о-оксибензо-
фуранокетон, как показано ниже.
ОН
COCH2COR
ОН
СОСН3
+ RCOOH
о-Окси-0-дикетоны легко циклизуются вновь под действием нагретой
разбавленной минеральной кислоты в соответствующие хромоны, и хотя
а,р-незамещенные бензофураны обычно очень чувствительны к действию
з*
36
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
сильных минеральных кислот, «.^-незамещенные фуранохромоны обнаружи-
вают значительную, а в некоторых случаях даже исключительную стабиль-
ность по отношению к этим реагентам вообще и к иодистоводородной кислоте,
в частности (см. ниже).
Подобно фуранокумаринам, фуранохромоны могут быть получены комби-
нацией тех же методов, которые применяются для синтеза хромонов и бензо-
фуранов. Существуют два основных пути: 1) фурановое кольцо может быть
пристроено к уже существующему хромону или 2) у-пироновое кольцо присое-
диняется путем циклизации к бензофурану. Второй метод обладает, по-види-
мому, большими возможностями, поскольку необходимые о-оксибензофуран-
кетоны гораздо более доступны, чем о-оксиформил- и о-оксикетохромоны,
из которых строится фурановое кольцо.
Лишь небольшое число фуранохромонов выделено из природного сырья,
большинство же получено синтетически. В первую очередь будут рассмотрены
встречающиеся в природе представители указанного класса соединений.
Келлин. Келлин (V), содержащийся в семенах Ammi visnaga Linn. 1971,
использовался как антиспазматическое и сосудорасширяющее средство [98].
Первоначально этому соединению ошибочно приписывали кумароновую
структуру [99], однако аналитическое исследование Шпета [97] в сочетании
с синтезами Кларка и Робертсона [100], а также Бэкстера, Рэмеджа и Тим-
сона [101] с несомненностью установили строение, соответствующее фор-
муле V. Окисление келлина перекисью водорода дает фурандикарбоно-
вую-2,3 кислоту, а обработка слабой щелочью приводит к размыканию хро-
монового кольца с последующим расщеплением первоначально образую-
щегося р-дикетона, в результате чего образуется уксусная кислота и кел-
линон (VI, R = Н). Ацетилирование последнего соединения в мягких усло-
виях дает ацетат (VI, R = СН8СО), однако обработка теми же реагентами
в более жестких условиях .приводит к 3-ацетилкеллину (VII), из которого
действием нагретого раство'ра карбоната натрия получают келлин [97].
ОСН3
СН3
СОСН3
VII
VIII
Этилирование келлинона (VI, R = Н) щелочью и диэтилсульфатом дает
о-этилкеллинон (VI, R = С2Н8), представляющий собой бесцветную жидкость
и охарактеризованный в виде семикарбазона. При окислении келлинона
дымящей азотной кислотой образуется хинон (VIII).
При озонолизе этилового эфира келлинона (VI, R = С2Н8) фурановое
кольцо размыкается и образуется о-оксиальдегид (IX), дающий при этили-
ровании 3,6-диметокси-2,4-диэтокси-5-формилацетофенон (X). Окисление
последнего перманганатом калия приводит к кислоте (XI), которая при декар-
боксилировании превращается в 3,6-диметокси-2,4-диэтоксиацетофенон (XII),
идентифицированный путем синтеза, чем окончательно было доказано линей-
ное строение келлина. Будучи производным 2-метилхромона, келлин кон-
денсируется с ароматическими альдегидами с образованием стирилпроиз-
водных.
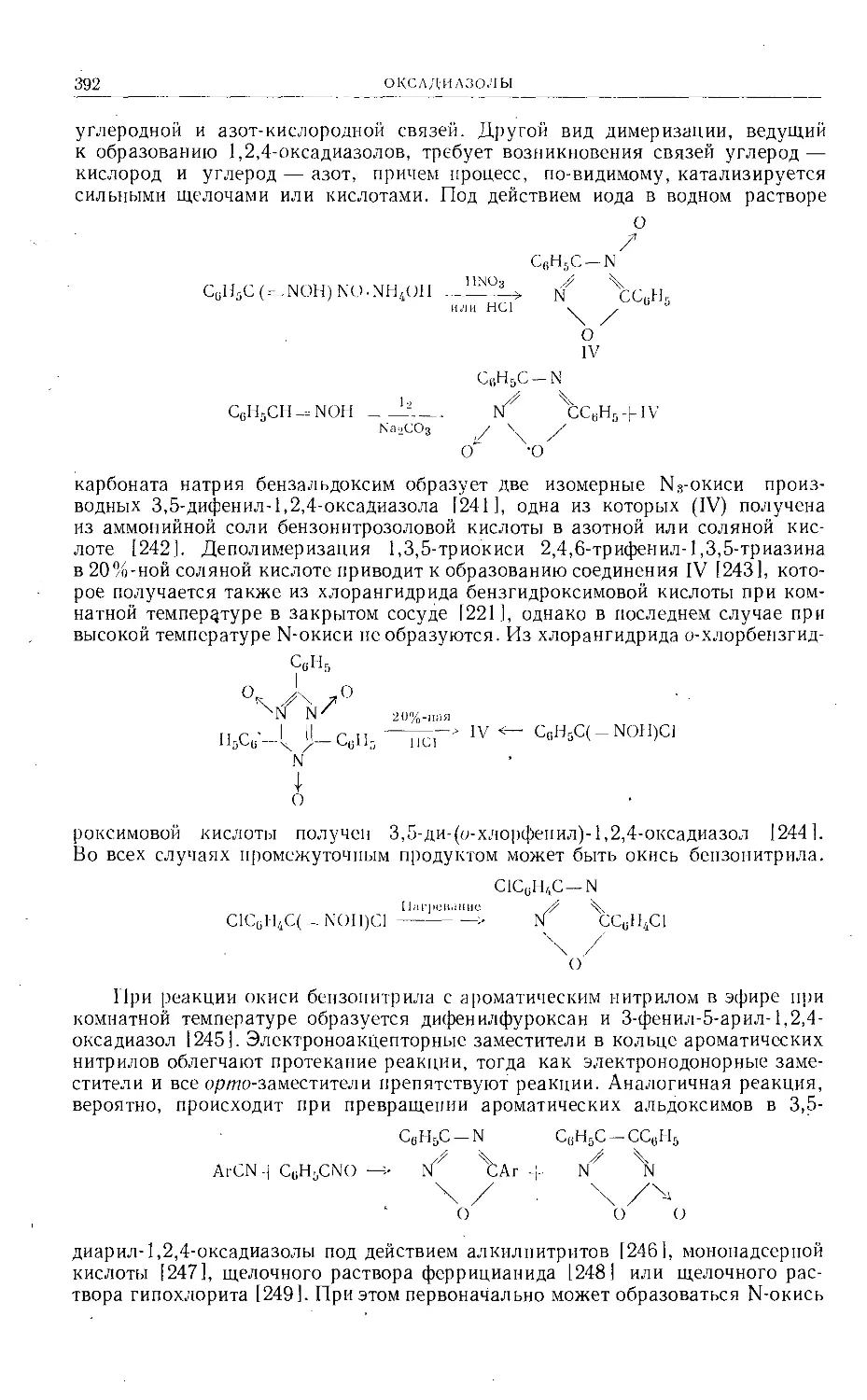
Келлин был синтезирован четырьмя группами исследователей [100—103].
Метод Кларка и Робертсона [100] заключается в приложении реакции Гаттер-
ФУРАНОХРОМОНЫ
37
мана к 2,5-диметоксирезорцину (XIII), в результате чего образуется альдегид (XIV, R = Н), превращаемый бензилированием в эфир (XIV, R = C6HSCH2),
ОСНз ОСНз
НОХ Л/0С!Н’ (C2Hs)2SO4 С2Н5ОХ JX/OC2H5 1 и
ОНС' II 1 ' > /чучсоснз онс/^/хсосн3
ОСНз ОСНз
IX X
! КМпО4
ОСНз ОСНз
СгН5Оч А/0СгН> 1 с2що 1ос2н5 II
"Y^COCHj HOOC/4j^4COCH3
ОСНз ОСНз
XII XI
строение которого было доказано положительной реакцией с хлоридом желе-
за (III) и превращением в соединение XV (R = С6Н5СН2) обычным мето-
дом. Каталитическое дебензилирование последнего приводит к получению
ОСНз ОСНз ОСНз
но\Л/он Il 1 RO. 1 /ОН 1 1 RO J О ЙОД 1 II li
II 1 1! 1 у\сно 1 1! li. \/ 1
ОСНз ОСНз ОСНз
XIII XIV XV
соединения XV (R = Н) (наличие карбэтоксильной группы предотвращает
восстановление фуранового кольца), а при конденсации с хлористым ацети-
лом в условиях реакции Фриделя — Крафтса соединение XV (R = Н) обра-
зует кислоту (XVI), декарбоксилирующуюся с образованием синтетического
ОСН3
° /\ /0Н
ноос “LII
V^coch,
I J
0CH3
XVI
0CH3
он
COCH2COCH3
ОСНз
XVII
кетона — келлинона, идентичного природному продукту. Конденсация син-
тетического келлинона с этилацетатом приводит к дикетону (XVII), легко
циклизующемуся под действием кислот в келлин.
Получение 2-метилхромонов из о-оксиацетофенонов через стадию про-
межуточного образования дикетонов позволяет избежать применения высо-
ких температур и образования 3-ацилхромонов, которые получаются в резуль-
тате хорошо известного процесса ароилирования по Робинзону; поэтому
этот метод синтеза является более надежным.
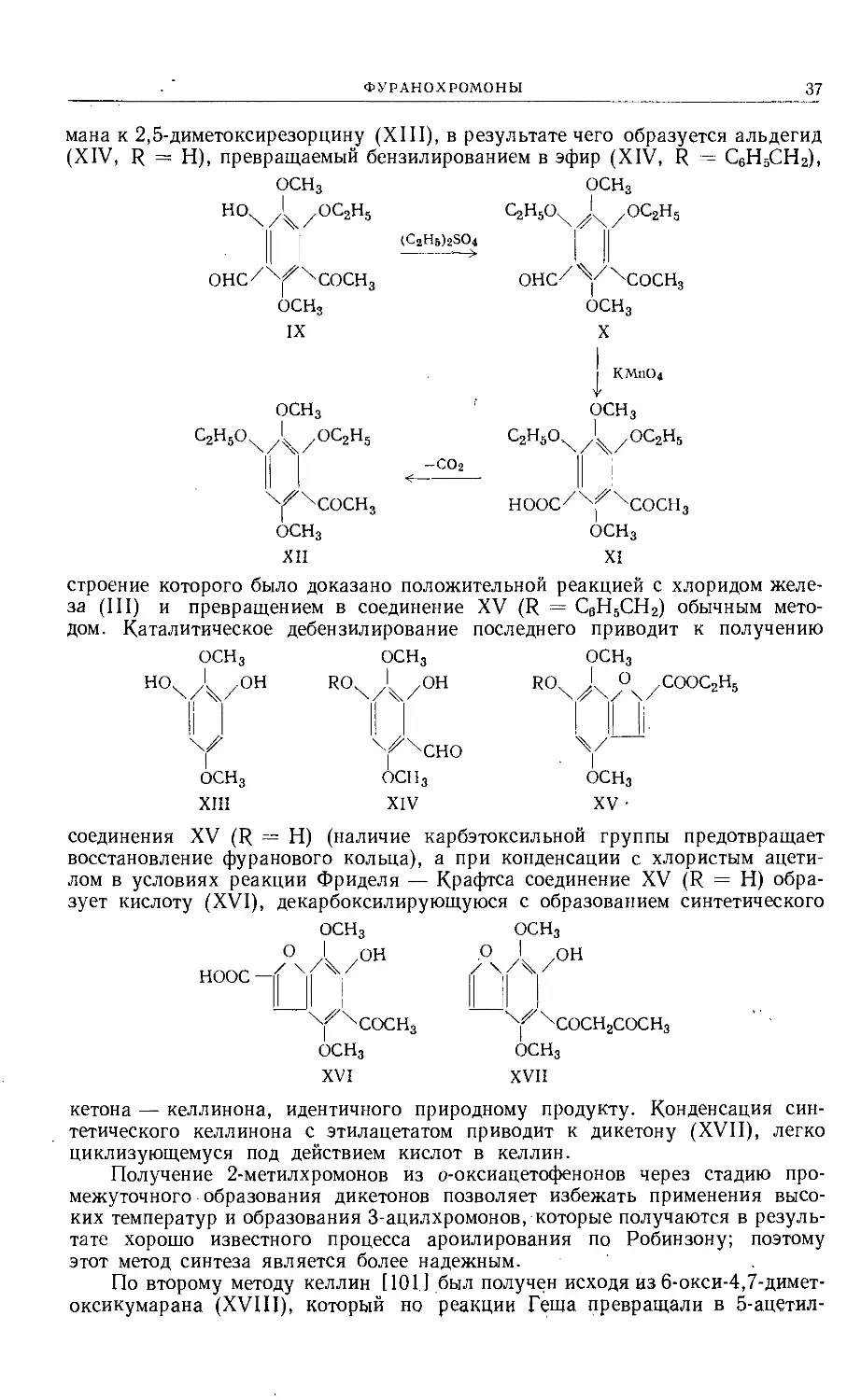
По второму методу келлин [101] был получен исходя из 6-окси-4,7-димет-
оксикумарана (XVIII), который но реакции Геша превращали в 5-ацетил-
38
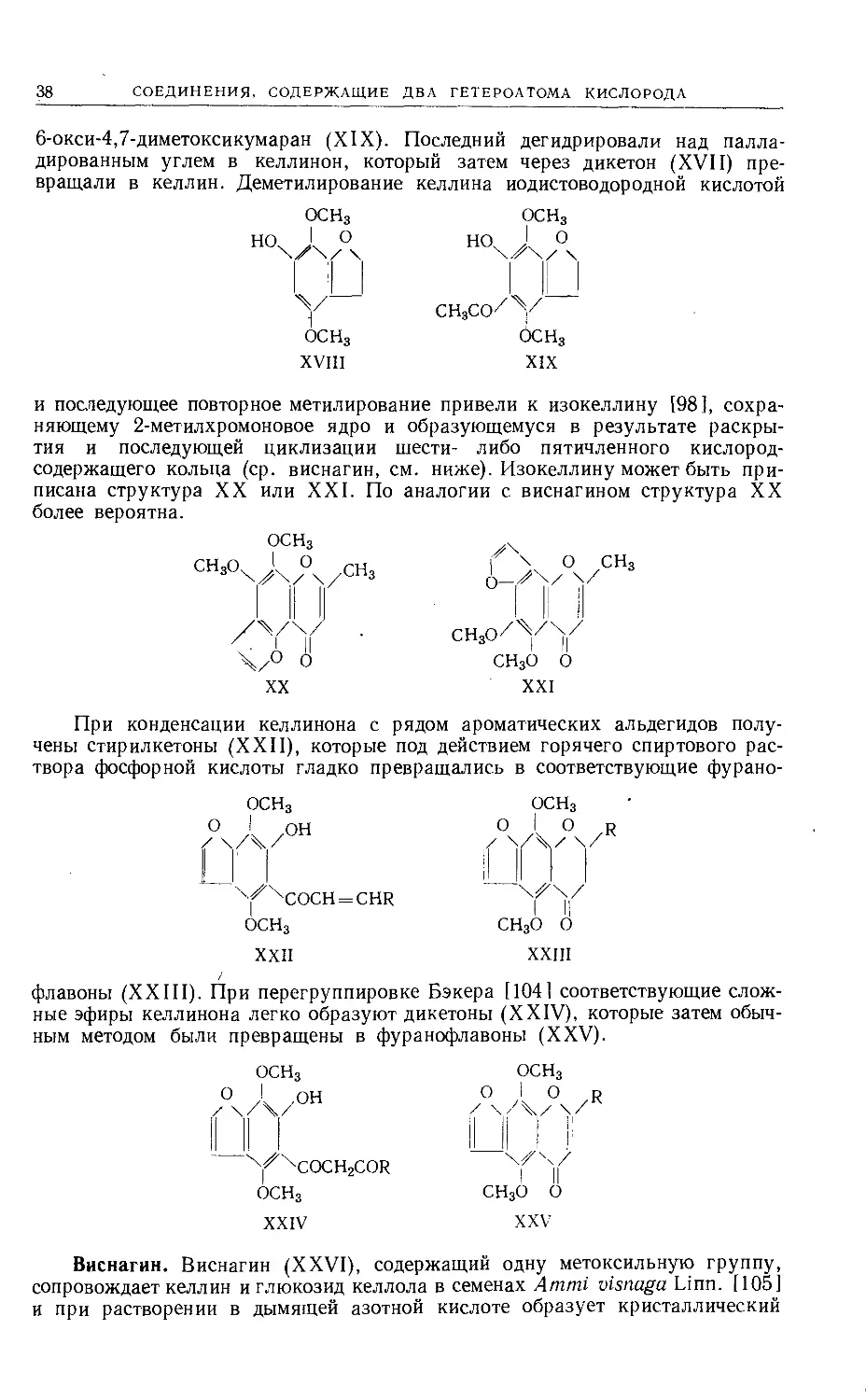
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
6-окси-4,7-диметоксикумаран (XIX). Последний дегидрировали над палла-
дированным углем в келлинон, который затем через дикетон (XVII) пре-
вращали в келлин. Деметилирование келлина иодистоводородной кислотой
ОСН3
ОСН3
XVIII
ОСН3
ОСН3
XIX
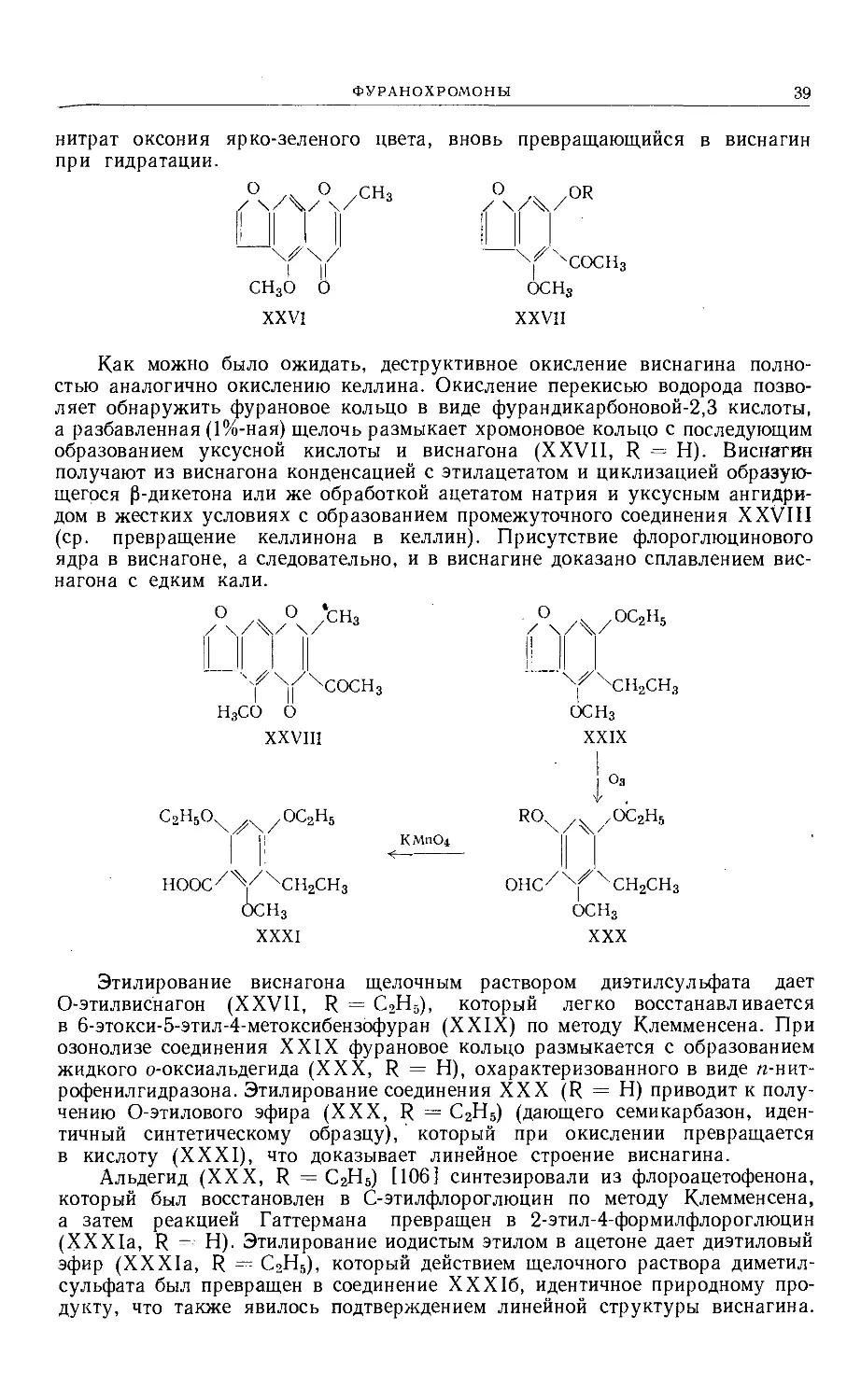
и последующее повторное метилирование привели к изокеллину [98], сохра-
няющему 2-метилхромоновое ядро и образующемуся в результате раскры-
тия и последующей циклизации шести- либо пятичленного кислород-
содержащего кольца (ср. виснагин, см. ниже). Изокеллину может быть при-
писана структура XX или XXI. По аналогии с виснагином структура XX
более вероятна.
ОСНз
XX
СН3О О
XXI
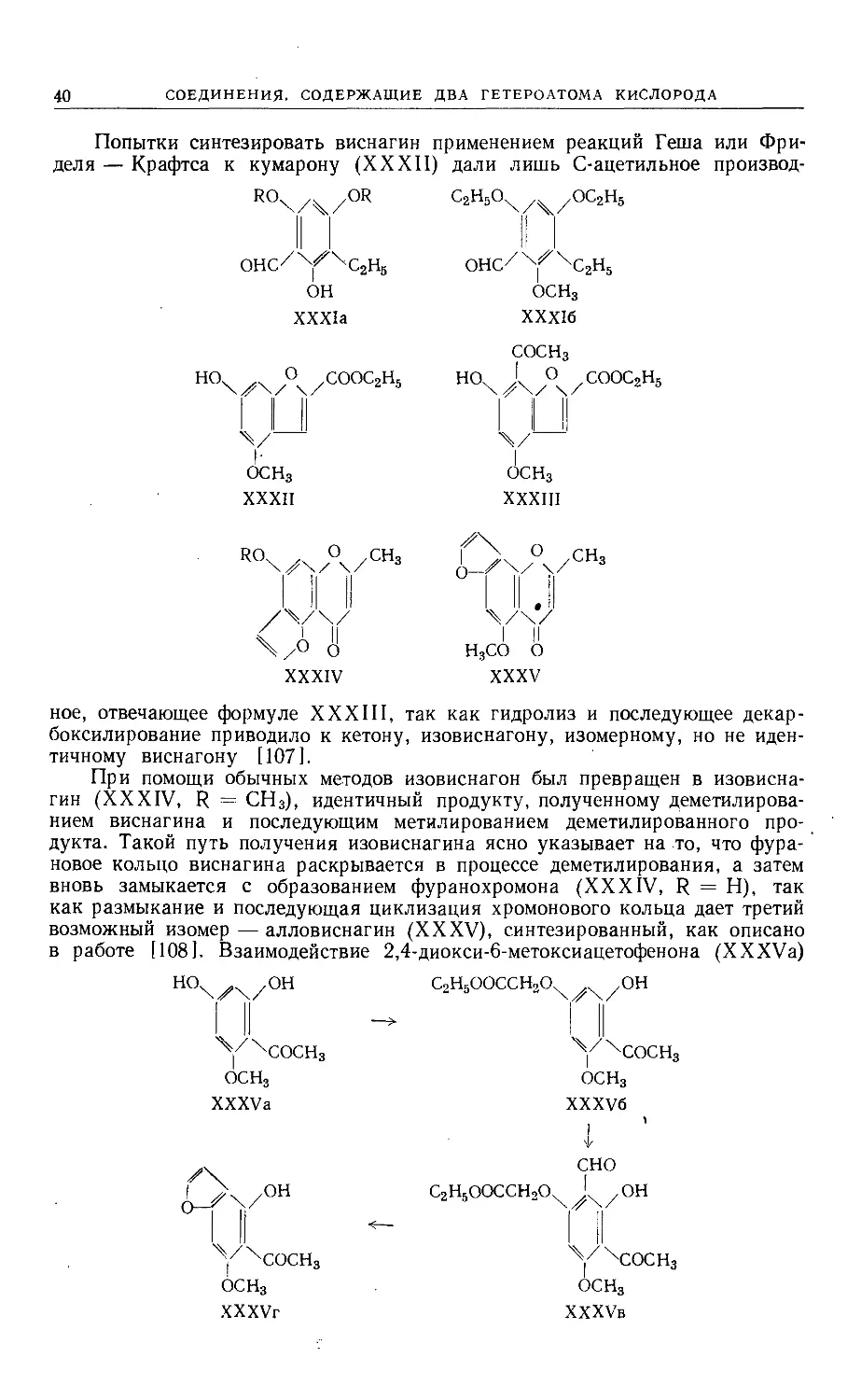
При конденсации келлинона с рядом ароматических альдегидов полу-
чены стирилкетоны (XXII), которые под действием горячего спиртового рас-
твора фосфорной кислоты гладко превращались в соответствующие фурано-
ОСН3
ДА/ОН
LI' I
X^4COCH = CHR
ОСН3
XXII
ОСНз
СН3О О
XXIII
флавоны (XXIII). При перегруппировке Бэкера [104] соответствующие слож-
ные эфиры келлинона легко образуют дикетоны (XXIV), которые затем обыч-
ным методом были превращены в фуранофлавоны (XXV).
ОСН3
ЛА/0Н
IIJI I
''^'COCH2COR
ОСНз
XXIV
ОСНз
О I О R
/ уу \/к
\/\/
1 II
СН3О о
XXV
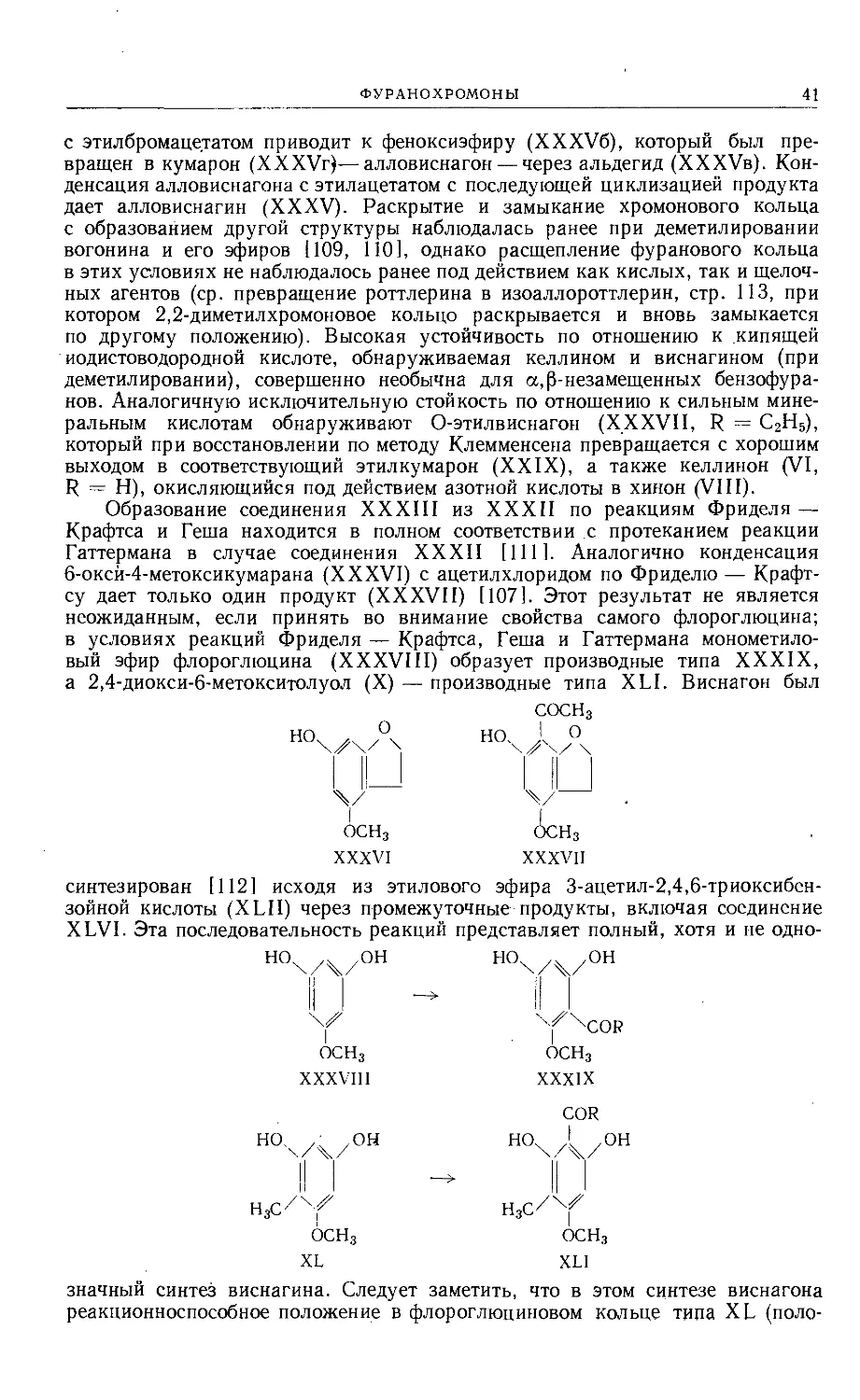
Виснагин. Виснагин (XXVI), содержащий одну метоксильную группу,
сопровождает келлин и глюкозид келлола в семенах Ammi visnaga Linn. [105]
и при растворении в дымящей азотной кислоте образует кристаллический
ФУРАНОХРОМОНЫ
39
нитрат оксония ярко-зеленого цвета, вновь превращающийся в виснагин
при гидратации.
I
СН3О
XXVI
'^'СОСНз
осн3
XXVII
Как можно было ожидать, деструктивное окисление виснагина полно-
стью аналогично окислению келлина. Окисление перекисью водорода позво-
ляет обнаружить фурановое кольцо в виде фурандикарбоновой-2,3 кислоты,
а разбавленная (1%-ная) щелочь размыкает хромоновое кольцо с последующим
образованием уксусной кислоты и виснагона (XXVII, R = Н). Виснагин
получают из виснагона конденсацией с этилацетатом и циклизацией образую-
щегося р-дикетона или же обработкой ацетатом натрия и уксусным ангидри-
дом в жестких условиях с образованием промежуточного соединения XXVIII
(ср. превращение келлинона в келлин). Присутствие флороглюцинового
ядра в виснагоне, а следовательно, и в виснагине доказано сплавлением вис-
нагона с едким кали.
Y^^COCHj
Н3СО О
XXVIII
с2н5охух/ос2н5
НООс/^/^СНгСНз
ОСН3
КМпОа
<------
XXXI
х^хсн2сн3
ОСНз
XXIX
ь.
RO^/ч ZOC2H5
III
ОНС/Х^ХСН2СН3
ОСНз
XXX
Этилирование виснагона щелочным раствором диэтилсульфата дает
О-этилвиснагон (XXVII, R = C2HS), который легко восстанавливается
в 6-этокси-5-этил-4-метоксибензофуран (XXIX) по методу Клемменсена. При
озонолизе соединения XXIX фурановое кольцо размыкается с образованием
жидкого о-оксиальдегида (XXX, R = Н), охарактеризованного в виде л-нит-
рофенилгидразона. Этилирование соединения XXX (R = Н) приводит к полу-
чению О-этилового эфира (XXX, R = С2Н5) (дающего семикарбазон, иден-
тичный синтетическому образцу), который при окислении превращается
в кислоту (XXXI), что доказывает линейное строение виснагина.
Альдегид (XXX, R = С2Н5) [106] синтезировали из флороацетофенона,
который был восстановлен в С-этилфлороглюцин по методу Клемменсена,
а затем реакцией Гаттермана превращен в 2-этил-4-формилфлороглюцин
(XXXIa, R =- Н). Этилирование иодистым этилом в ацетоне дает диэтиловый
эфир (XXXIa, R = С2Н5), который действием щелочного раствора диметил-
сульфата был превращен в соединение XXXI6, идентичное природному про-
дукту, что также явилось подтверждением линейной структуры виснагина.
40
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Попытки синтезировать виснагин применением реакций Геша или Фри-
деля — Крафтса к кумарону (XXXII) дали лишь С-ацетильное производ-
RO. .OR
\/\/
ОНС/Х^ЧС2Н5
ОН
XXXIa
С2Н5О. /. /ОС2Н5
\/ч/
II)
онс/у ^С2Н5
ОСНз
XXXI6
НО^^Д/СООСгЩ
ОСН3
XXXII
I
ОСНз
СООС2Н5
XXXIII
XXXV
ное, отвечающее формуле XXXIII, так как гидролиз и последующее декар-
боксилирование приводило к кетону, изовиснагону, изомерному, но не иден-
тичному виснагону [107].
При помощи обычных методов изовиснагон был превращен в изовисна-
гин (XXXIV, R = СНз), идентичный продукту, полученному деметилирова-
нием виснагина и последующим метилированием деметилированного про-
дукта. Такой путь получения изовиснагина ясно указывает на то, что фура-
новое кольцо виснагина раскрывается в процессе деметилирования, а затем
вновь замыкается с образованием фуранохромона (XXXIV, R = Н), так
как размыкание и последующая циклизация хромонового кольца дает третий
возможный изомер—алловиснагин (XXXV), синтезированный, как описано
в работе [108]. Взаимодействие 2,4-диокси-6-метоксиацетофенона (XXXVa)
ОН
С ОСН3
ОСН3
XXXVa
'Y'XCOCH3
ОСНз
XXXVr
C,HSOOCCH,O4 .. /ОН
“ s “ \у\/
III
у хсосн3
ОСНз
XXXV6
сно
C2H5OOCCH,O. I /ОН
III
Y ЧСОСН3
ОСНз
XXXVB
ФУРАНОХРОМОНЫ
41
с этилбромацетатом приводит к феноксиэфиру (XXXV6), который был пре-
вращен в кумарон (XXXVr)—алловиснагон — через альдегид (XXXVb). Кон-
денсация алловиснагона с этилацетатом с последующей циклизацией продукта
дает алловиснагин (XXXV). Раскрытие и замыкание хромонового кольца
с образованием другой структуры наблюдалась ранее при деметилировании
вогонина и его эфиров [109, 110], однако расщепление фуранового кольца
в этих условиях не наблюдалось ранее под действием как кислых, так и щелоч-
ных агентов (ср. превращение роттлерина в изоаллороттлерин, стр. 113, при
котором 2,2-диметилхромоновое кольцо раскрывается и вновь замыкается
по другому положению). Высокая устойчивость по отношению к кипящей
иодистоводородной кислоте, обнаруживаемая келлином и виснагином (при
деметилировании), совершенно необычна для а,р-незамещенных бензофура-
нов. Аналогичную исключительную стойкость по отношению к сильным мине-
ральным кислотам обнаруживают О-этилвиснагон (XXXVII, R = С2Н5),
который при восстановлении по методу Клемменсена превращается с хорошим
выходом в соответствующий этилкумарон (XXIX), а также келлинон (VI,
R = Н), окисляющийся под действием азотной кислоты в хинон (VIII).
Образование соединения XXXIII из XXXII по реакциям Фриделя —
Крафтса и Геша находится в полном соответствии с протеканием реакции
Гаттермана в случае соединения XXXII [111]. Аналогично конденсация
6-оксй-4-метоксикумарана (XXXVI) с ацетил хлор и дом по Фриделю — Крафт-
су дает только один продукт (XXXVII) [107]. Этот результат не является
неожиданным, если принять во внимание свойства самого флороглюцина;
в условиях реакций Фриделя — Крафтса, Геша и Гаттермана монометило-
вый эфир флороглюцина (XXXVIII) образует производные типа XXXIX,
а 2,4-диокси-6-метокситолуол (X) — производные типа XLI. Виснагон был
СОСН3
ОСН3 ОСН3
XXXVI XXXVII
синтезирован [112] исходя из этилового эфира 3-ацетил-2,4,6-триоксибен-
зойной кислоты (XLII) через промежуточные продукты, включая соединение
XLVI. Эта последовательность реакций представляет полный, хотя и не одно-
н°\/\/0Н
I
ОСНз
XXXVIII
H°.x/WOH
Н3С/Х/
ОСНз
XL
но
ОСНз
XXXIX
COR
ОСНз
XLI
значный синтез виснагина. Следует заметить, что в этом синтезе виснагона
реакционноспособное положение в флороглюциновом кольце типа XL (поло-
42
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
жение 7 кумаронового кольца) блокировано сложноэфирной группой, чтобы
в процессе реакции по Гешу направить ацетильный радикал в требуемое
положение.
Описан также другой синтез виснагина [113].
СООС2Н6
Н() ' .ОН
I II
^/хсосн3
он
XLII
О'
СООС2Н6
0 X /ОН
Y•хсосн3
он
XLIII
(1) (СН3СО)2О+СН3СОС1
(2) Гидрирование
соон
/сосн3
I 3
ОСНз
(1) ch2n2
(2) Гидролиз
СООС2Н6
Х^ХСОСН3
он
XLIV
XLV
-со2
,^ СОСН3
I 3
ОСН3
XLVI
Келлол и визамминол. Глюкозид келлола (или хеллола) содержится
вместе с виснагином и келлином в семенах Ammi visnaga Linn. [114] и при
гидролизе гидратом окиси бария дает келлол и глюкозу. Келлол имеет мето-
ксильную группу и спиртовой гидроксил. При расщеплении его разбавлен-
ным едким кали образуется виснагон; следовательно, строение келлола может
быть представлено формулой XLVII
0 /чЧ 0 .СН2ОН
СН3О о
XLVII
Визамминол С16Н1вО6 — оптически активный компонент Ammi visna-
ga [115] — был исследован Шмидом с сотрудниками [116]. Ему приписана
структура XLVIIa.
ФУРАНОХРОМОНЫ
43
При дегидратации визамминола в присутствии кислот образуется дегидро-
визамминол (XLVII6), который после метилирования и расщепления щелочью
XLVIIa
нохух/ос2н6
ОНС'/'У'ЧСОСН3
ОСНз
XLVIIr
XLVII6
z Ul
Ч|^ХСОСН3
ОСНз
XLVIIb
в мягких условиях дает кумарон (LVIIb, R = Н) и уксусную кислоту в резуль-
тате раскрытия у-пиронового кольца. Этилирование визамминола приводит
к соединению XLVIIb (R = С2Н5), которое при озонолизе превращается
в альдегид (XLVIIr), идентичный полученному из виснагона.
Караньин. Караньин (XLVIII) был впервые выделен Билом и Кэтти [117]
из масла семян бобового растения Karanja pongamia Glabra и является одним
из трех фуранофлавонов, полученных до настоящего времени из природных
источников [118]. Щелочной гидролиз караньина [119] приводит к раскрытию
хромонового кольца с образованием бензойной кислоты, фенолкетона —
караньола (L) и фенолкислоты — караньиновой кислоты (LI), образующихся
при расщеплении промежуточного дикетона (XLIX) (по пунктирным линиям).
^/^/^ОСНз
О
XL1X
L LI
Флавоновая природа караньина доказана превращением караньола
в караньин при энергичном бензоилировании смесью бензоата натрия и бен-
зойного ангидрида. Декарбоксилирование кислоты (LI) дает 4-оксикумарон
(IV, R = Н), который был синтезирован [120] из О-диметил-у-резорцилаль-
44
соединения, содержащие два гетероатома кислорода
дегида (LII) частичным деметилированием в монометиловый эфир и после-
дующей конденсацией с этиловым эфиром бромуксусной кислоты, в результате
СНО СНО
CH3OXJX/OCH3 СН3Ох/1^/ОСН2СООС2Н6 ухД
OR
LII LIII LIV
чего образуется феноксиальдегид (LIII). Деметилирование последнего и цик-
лизация под действием смеси ацетата натрия и уксусного ангидрида приводит
к 4-ацетоксикумарону (LIV, R = СОСН3), который легко дезацетилируется
в 4-оксикумарон (LIV, R = Н). Дополнительным доказательством строения
караньиновой кислоты и, следовательно, ангулярного строения караньина
является окисление караньиновой кислоты перекисью водорода в фуранди-
карбоновую-2,3 кислоту и образование 3-формил-р-резорциновой кислоты (LV)
при озонолизе.
СНО СНО
z 7С00Н НОХ ,ОН С2Н6ООССН2ОХ/' он
.и xi ) Т II1
HOZ х он хсоосн3 хсоосн3
СНО
LV LVI ' LVII
Сама караньиновая кислота была синтезирована [121] взаимодействием
сложного альдегидоэфира (LVI) с этиловым эфиром бромуксусной кислоты
и образованием соединения LVII, которое при гидролизе в соответствующую
двухосновную кислоту с последующей циклизацией под действием ацетата
натрия и уксусного ангидрида дало смесь О-ацетилкараньиновой и караньи-
новой кислот (LI).
Синтез фенолкетона (L) и, следовательно, полный синтез караньина [1221
были осуществлены исходя из О-щцетилкараньиновой кислоты (LVIII), хлор-
ангидрид которой конденсировался с этиловым эфиром а,у-диметоксиацето-
уксусной кислоты, давая соединение LIX. Последнее было превращено
с помощью контролируемого щелочного гидролиза в караньол. Описано также
выделение двух линейных фуранофлавонов — пиннатина (LIXa) и гама-
О y\z \ 1 и НОСКУ ОСОСН3 LVIII z°\/\z°\/CeHs 111 i II \У\/ 1 II сн3о о LIXa О y\z \ 1 II л ос^ ососн3 С2Н6ООССОСН3 1 СОСН2ОСН3 LIX .0. 0 0 / Хсн2 Z \/\Z \_/ \ 1 UJ.J 1 II СН3О 0 L1X6
ФУРАНОХРОМОНЫ
45
тина (ЫХб) из Pongamia pinnata Merr., а также их синтез [123] исходя из вис-
нагона (стр. 41) [124].
Синтетические фуранохромоны. Первый синтез фуранохромона описан
Кердом и Робертсоном [96], превратившими О-монометилуснетол (LX) в дике-
тон (LXI) и затем в соединение IV обычными методами. СОСН3 СОСН2СОСН3 H0\Az°z/CH> H0\Az°z/CH- I IIJ 1 IIJ Нзс/у7 XCH3 H3C'/^'/ хсн3 осн3 осн3 LX LXI
Несколько фуранохромонов было синтезировано исходя из различных
7-окси-8-ацилхромонов путем построения фуранового кольца [125]. Напри-
мер, при конденсации 8-ацетил-7-окси-2,3-диметоксихромона с этиловым эфи-
ром бромуксусной кислоты получен феноксиацетат (LXII), который был прев-
ращен декарбоксилированием в соединение LXIII.
СН3
С°СН3 J
С2Н5ООССН2Оч I 0 .СН3 I \ ,° /СН3
2 5 2 \Z\/ \/ А_А\/ \/ d
I a j и л
II ^п3 || ^пз
О о
LXII LXIII
Фуранофлавон (LXV), представляющий собой караньин, не содержащий
метоксильной группы^, был синтезирован [126] исходя из караньиновой
кислоты, которую превращали в метиловый эфир (LXIV, R = ОСН3) и затем
в хлорангидрид (LXIV, R = С1). Из последнего с метилцинкиодидом получен
о-оксиацетофенон (LXIV, R = СН3), который при деметилировании и после-
дующей циклизации под действием бензоата натрия и бензойного ангидрида
был превращен в 2',3',7,8-фуранофлавон (LXV).
ОСН3
COR
LXIV
Циклизацией караньола под действием смесей различных ангидридов
кислот с соответствующими натриевыми солями синтезирован ряд аналогов
караньина типа LXVI [127].
Ротиорин, рубропунктатин и монаскорубрин. Метаболиты грибковых —
ротиорин, рубропунктатин и монаскорубрин (и склеротиорин) — являются
представителями нового класса кислородсодержащих гетероциклов. С точки
зрения классификации их можно рассматривать как производные фурано-
хромонов, отдаленно их напоминающие. Строение этих соединений было выяс-
нено Холькером, Робертсоном, Уолли и их сотрудниками.
46
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Ротиорин [128] С23Н24О6(1), рубропунктатин [129] С21‘Н22О6 (II)
и монаскорубрин [130, 131] С2зНгвО6 (III) были выделены из мицелий Peni-
cillium sclerotiorum van Beyma, Monascus rubropunctatus Sato и M. purpureas
Wentii соответственно. Склеротиорин [130, 132—136] C21H23O6C1 (IV), хотя
и выходит за рамки настоящего обзора, но является тесно с ними связанным
пигментом, выделенным из мицелия Penicillium sclerotiorum van Beyma.
Химия этих веществ исключительно сложна, однако о двух ее особенно-
стях следует упомянуть. Эти метаболиты быстро реагируют при комнатной
температуре с аммиаком и первичными аминами с образованием производных,
например, типа ротиорамина (V). Это «аммонийное соединение» легко аро-
матизуется при восстановлении в самых различных условиях, образуя апо-
соединения, например апоротиорамин (VI), являющийся изохинолоном.
Исследован также биосинтез склеротиорина [137].
СОС7Н16-к
с
СОС6Н41-к
С
сн3
II
сн3
СНзСОо/
сн3
сн3 сн3
I I
сн=снс= СНСНС2Н6
сн3 сн3
СН = СНС=СНСНС2НЙ
СН3
V
сн3 сн3
СН3СОСН2х/. . /СН^Н^СНСНСгНб
Il I NH
HgC/Y'47
о
VI
КУМАРАНОХРОМАНЫ
47
КУМАРАНОХРОМАН Ы
Гомоптерокарпин и птерокарпин. Единственными известными представи-
телями кумаранохроманов являются гомоптерокарпин (I) и птерокарпин (II),
содержащиеся в «нерастворимом» красном дереве [138, 139], сандаловом
дереве (Pterocarpus santalinus Linn.) и различных породах африканского крас-
ного дерева (разновидности Baphia nitida Lodd.).
Гомоптерокарпин содержит две метоксильные группы, а птерокарпин —
одну. Карбонильная и гидроксильная группы отсутствуют. Оба соединения
обнаруживают сильное левое вращение. Окисление гомоптерокарпина пер-
манганатом калия в ацетоне приводит к расщеплению гетероциклических
колец и образованию смеси 2-окси-4-метоксибензойной кислоты (I II) и 5-меток-
си-2-карбоксифеноксиуксусной кислоты (IV) (ср. поведение фуранокумаринов
и фуранохромонов). Образование феноксикислоты (IV) указывает на наличие
по крайней мере одного эфирного атома кислорода. Восстановление гомоптеро-
карпина и птерокарпина над палладированным углем, протекающее медленно
при комнатной температуре и значительно ускоряющееся при 100° приводит
СН3О ОН
соон
III '
сн3о —осн2соон
JL соон
IV
к разрыву эфирной связи дигидрофуранового кольца и образованию соответ-
ственно фенольного L-дигидрогомоптерокарпина (V, R = Н) и L-дигидро-
птерокарпина (VI, R = Н). Это напоминает поведение ротенона (стр. 117),
у которого тубанольный остаток превращается в тетрагидротубанол, а также
азаринина и родственных ему соединений (стр. 9). При восстановлении по Клем-
менсену гомоптерокарпин и птерокарпин образуют одинаковые дигидросое-
динения.
Окисление перманганатом калия обоих фенольных дигидросоединений
дает DL-7-метоксихроманкарбоновую-З кислоту (VII), что подтверждает нали-
чие 7-метоксихроманового ядра в L-дигидрогомоптерокарпине и L-дигидро-
птерокарпине, а следовательно, и в родственных им соединениях. Образова-
ние этих кислот, а также результаты гидрирования однозначно доказывают
наличие двух атомов кислорода в циклической системе. DL-Киелота была
синтезирована исходя из 5-метокси-2-формилфеноксипропионовой кислоты
(VIII), при циклизации которой в присутствии ацетата натрия и уксусного
ангидрида с низким выходом получена 7-метокси-А3-хроменкарбоновая-3 кис-
лота (IX), гидрированием легко превращающаяся в соединение VII. Чтобы
48
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
проверить, не образуется ли при циклизации соединения VIII изомерная кума-
рон-2-уксусная кислота (X), ее синтезировали циклизацией сложного эфи-
ра (XI) с последующим гидролизом в соединение XII и применением реакции
VI, R = H
V, R = H
KMnOi
------>
VII
CH3COONa
---------->
(СНЯСО)2О
СН3О. о
3\У\/\
'^/W'VcOOH
СНз°\/\/ чсн2
|| |. (1;н2соон
х^чсно
VIII
СН3О ОСН2СООС2Н5
Арндта — Эйстерта. Полученная кислота отличалась от изомерной хромен-
карбоновой-3 кислоты (IX).
Остаток молекулы L-дигидрогомоптерокарпина, элиминирующийся при
окислении соединения V (R = Н) с образованием соединения VII, содержит
вторую метоксильную группу, а также фенольный гидроксил, образовав-
шийся при расщеплении дигидрофуранового кольца.
Судя по цветным реакциям птерокарпина с галловой кислотой, он содер-
жит, по-видимому, метилендиоксигруппу в фенильном радикале, однако
во многих отношениях он близко напоминает гомоптерокарпин.
Окисление L-дигидрогомоптерокарпина хромовым ангидридом дает
/г-хинон — L-дигидрогомоптерокарпон (XV), тогда как L-дигидроптеро-
карпин в тех же условиях образует дигидроптерокарпон (XIII), охаракте-
ризованный в виде 2,4-динитрофенилгидразона. Образование /г-бензохино-
О
XIII
XIV
на (XIII) ясно указывает на то, что метилендиоксигруппа находится в 3',4'°
положениях фенильного заместителя. При действии перманганата калия
КУМАРАНОХРОМАНЫ
49
на метиловый эфир L-дигидроптерокарпина (VI, R = СН3) образуется веще-
ство, которое по аналогии с продуктом, полученным из гомоптерокарпина,
почти несомненно является изофлаваноном (XIV).
XV XVI
Метиловый эфир L-дигидрогомоптерокарпина (V, R = СН3) под дейст-
вием перманганата калия превращается в левовращающий изофлаванон (XVI),
рацемизующийся при повторной перегонке и дающий при восстановлении
по методу Клемменсена рацемический изофлаван (V, R = СН3). Последний
идентичен образцу, полученному путем превращения фенилбензилкетона (XVII)
в изофлавон (XVIII) с последующим каталитическим восстановлением его
в DL-изофлаван (V, R = СН3). Подобным же образом строение птерокарпина
СН3О-
— ОН
— СОСН2
осн3
XVII
XVIII
было подтверждено синтезом метилового эфира . DL-дигидроптерокарпина
(VI, R = СН3) [140]. Был описан [141, 142] независимый синтез соединения
типа XVIПа, имеющего углеродный скелет этих соединений. Его можно изо-
бразить следующей последовательностью реакций:
НзС0\/\/0Н
il I
\^\сосн2 -/ ОСНз
н3со
' i I /—v
<_>- 0СНз
НО Н3со
XVUIa
Дегидрирование гомоптерокарпина (I) дает дегидрогомоптерокарпин
(XVII16), который при окислении образует соединение XVIПа, что является
4 Заказ К» 978
50
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
косвенным доказательством тетрациклической структуры гомоптерокарпи-
на [143].
О
СН3О—\
\/\ОСНз
XVIII6
Гомоптерокарпин и птерокарпин в противоположность их высокой устой-
чивости по отношению к щелочам — свойству, характерному для хромановой
системы (ср. дракорубин), —быстро осмоляются при нагревании даже с раз-
бавленными минеральными кислотами. Это связано, по-видимому, с наличием
дигидрофурановой системы.
Бензофурано-а-пироны. Несколько бензофурано-а-пиронов типа XVI Па
встречаются в природе. Кумэстрол (XVIIIb) является эстрогенным компо-
нентом клевера Ladino [144], а веделолактон [145, 146] (XVIIIr, R = Н)
содержится в листьях VJedelia calendulacea. Описан синтез три-О-метилведело-
лактона (XVIII, R = СН3) [141, 142]. Структурно сходные соединения
XVIIIb
XVIIIr
Н3СО
XVIIIe
эрознин [147] (XVIПд) и пахиррхицин [148] (XVIIIe) выделены из
семян батата Pachyrrhizus erosus и установлена их структура. Пири-
лиевые соли типа XX [149] синтезированы путем катализируемой щелочами
КУМАРО-а- И КУМАРО-у-ПИРОНЫ
51
конденсации о-ванилина и p-нафтокумаранона с образованием стирилкето-
на (XIX), который под действием хлористого водорода и хлорида железа (III)
превращался в соединение XX. Соли типа XX легко образуют стабильные
XIX
XX
этиловые эфиры пиранольных оснований при обработке ацетатом натрия
в этиловом спирте (ср. аналогичное поведение О-диметилцитромицинола,
стр. 90).
КУМАРО-а- И КУМАРО-у-ПИРОНЫ
Кумаро-а-пироны. Представители этого ряда соединений в природе
не обнаружены. Значительное число кумаро-а-пиронов (ди-а-пиронов) типа I
и II были синтезированы путем применения к кумаринам общих методов
синтеза, пригодных для получения а-пиронов. Приведенные ниже примеры
являются типичными.
Конденсация по Пехману умбеллиферона с яблочной кислотой может
дать соединения I или II, т. е. соответственно линейный или ангулярный изо-
меры. Сен и Хакраварти [150] получили кумарины этого ряда конденса-
цией различных 7-оксикумаринов с яблочной кислотой или ацетоуксусным
эфиром и безуспешно пытались разделить с помощью окисления изомеры
типа I и II. Однако строение веществ I и II было однозначно установлено 1151 ]
путем циклизации умбеллиферон-8-альдегида (III) в присутствии ацетата
натрия и уксусного ангидрида, т. е. полным синтезом ангулярного изоме-
ра II. Дальнейшее подтверждение такого строения было получено в резуль-
тате опытов по окислению, проведенных Шпетом и Лови 1152]. Обработка
линейного кумаро-а-пирона (I) щелочным раствором диметилсульфата дает
СНО
I О
НО— ^=0
III
коричную кислоту (IV) (ср. простые кумарины), которая была окислена пер-
манганатом калия в О-диметилрезорциндикарбоновую-4,6 кислоту (V), что
подтвердило линейное строение исходного вещества I и, следовательно, ангу-
лярное строение другого соединения II. Ангулярный изомер легко получается
по реакции Перкина из резорцин-2,4-диальдегида (VI) и ацетата натрия
и уксусного ангидрида. Продукт конденсации яблочной кислоты и умбелли-
4*
52*
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
ферона состоит на 95% из соединения II. При взаимодействии яблочной кис-
лоты и 4-метилумбеллиферона получается исключительно ангулярный изо-
СН3О ОСНз КМПО4 СН3О—/4 ОСНз
нооссн=сн IV сн=снсоон сно но — / Ч— он 'U-ch° VI ноос JI L V СООН
мер (VII). Преимущественное образование ангулярного изомера было уже рас
смотрено выше.
СН3
VII
СНО
I О
\=О
\/\Z
. I
СНз
VIII
СНз
I О
I
сн3
IX
Строение соединения VII легко установлено синтезом [151] из 4-метил-
умбеллиферон-8-альдегида (VIII) по реакции Перкина. Структура соедине-
ния VIII была принята по аналогии с умбеллиферон-8-альдегидом и строго
доказана восстановлением в 4,8-диметилумбеллиферон (IX), идентичный про-
дукту, синтезированному из 2-метилрезорцина и ацетоуксусного эфира.
При конденсации ацетофенонов типа X с ацетоуксусным эфиром в при-
сутствии хлористого алюминия реакция протекает предпочтительно в поло-
XIII
КУМАРО-а- И КУМАРО-у-ПИРОНЫ
53
жении 2 резорцинового кольца, а не в положении 4, как это обычно бывает
при реакции с серной кислотой или с хлорокисью фосфора, применяемыми
в качестве конденсирующих средств; при этом образуются кумарины
типа XI [1531. Строение соединения XI было установлено на основании его
идентичности продукту, полученному по реакции Фриса из соответствующего
5-ацилкумарина (XII). Замыкание кольца в соединении XI под действием
ацетата натрия и уксусного ангидрида приводит к ангуляркому кумаро-а-
пирону (XIII). Циклизация о-оксиацетофенонов по методу Костанецкого
обычно приводит к получению смеси кумарина и изомерного хромона. Напри-
мер, обработка в жестких условиях ацетатом натрия и уксусным ангидридом
6-ацетил-8-этилумбеллиферона (XV) [154], полученного перегруппировкой
по Фрису из соединения XIV, дает кумаро-у-пирон (XVII) и кумаро-а-пи-
рон (XVI).
С2Н5
сн3соо
II г
X/XZ
I
сн3
но
СН3СО
С2Н5
I о
/\/\Z
I
СН3
XIV XV
CHsCOONa (СН3С0)2О
___________
С2Н5
XVI
XVII
Фенольный компонент в синтезе по Пехману может быть заменен 4-окси-
кумарином, как, например, при конденсации 4-окси-6,7-диметоксикумарина
(XVIII) с ацетоуксусным эфиром под действием хлорокиси фосфора с обра-
зованием а-пиранокумарина (XIX) [155].
СН3О—х=о
I
ОН
о о
СНзО-.-^'у
CH>°~JUx_CH
I I '
О 1
II
о
XVIII XIX
Продукты окисления галловой кислоты. Продукты окисления галловой
кислоты изучались в течение многих лет. Однако только в настоящее время
удалось точно установить их строение. Большая часть этих соединений содер-
жит одно, два или более конденсированных а-пироновых колец.
54
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Эллаговая кислота известна уже по крайней мере 130 лет как компонент
дубильных веществ. Она может быть получена синтетически окислением
галловой кислоты мышьяковой кислотой. Строение ее было выяснено глав-
ным образом Герцигом и Поллаком [156].
О О
II II
О
XIXa XIX6
Окисление щелочного раствора галловой кислоты воздухом дает галло-
флавин (XIXa) [157, 158], строение которого было установлено Хэуорсом
и сотрудниками [159, 160] на основании изучения изогаллофлавина (XIX6,
R = Н), полученного из галлофлавина действием щелочи. Щелочное расщеп-
ление в жестких условиях декарбоксилированного тетраметилизогаллофла-
вина приводит к муравьиной кислоте и фталиду (Х1Хв), как это показано
на приведенной ниже схеме.
Положение карбоксильной группы в изогаллофлавине, который по лег-
кости декарбоксилирования напоминает продукт, чувствительный к кисло-
там, в противоположность устойчивому к кислотам изогаллофлавину было
установлено путем превращения триметилизогаллата (XIX6, R = Н) в про-
изводное Х1Хд через соединение Х1Хг. Строение 2-(«-пропил)фурана Х1Хд
было доказано путем расщепления щелочью, в результате чего образуются
соединение XIХв и метил-н-пропилкетон. В соответствии с установленной
для изогаллофлавина структурой (XIX6, R = Н) галлофлавин имеет строе-
ние XIXa. Переход галлофлавина в изогаллофлавин протекает по хорошо
известному пути превращения 3-метокси-, 3-окси- и 3-галогенкумаринов
в 2-карбоксибензофураны, например, так же как перегруппировка ротенона
в ротеноновую кислоту (стр. 129).
Тетраметилизогаллофлавин
н3соху./соон
II!---сн-со
нзсо/у о/ ।
ОСНз \ I
сн=сн
Н3СО, ,х .соон
A J—снон
н3со/У I
ОСНз СО
СН2СНО
О+НСООН
С ОСН3
Х1Хв
КУМАРО-а- И КУМАРО-у-ПИРОНЫ
55
Триметилизогаллат
1) РС15
2) Cd (С2Н6)2
H>cow\A
I II. ?
НзС0/уу\
Н3СО О\ X
I
СОСН2СН3
Х1Хг
о
НзСо/^/^Х
н3со
I
сн2сн2сн3
Х1Хд
Другим продуктом окисления галловой кислоты мышьяковой кислотой
в серной кислоте является флавогаллол, гексаоксифенол С21Н8О12 (XIXe) [1611.
О
ОН ОСН3
Одно из лактонных колец в флавогаллоле очень неустойчиво и легко
раскрывается (давая флавогалловую кислоту) в таких условиях, при которых
не затрагиваются кумарины. Это реакционноспособное кольцо, несомненно,
56
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
является кольцом А в соединении XIХе, так как из-за стерических препят-
ствий оно не обладает плоской конфигурацией. При энергичном метилиро-
вании, сопровождаемом гидролизом, флавогаллол превращается в двухоснов-
ную кислоту (XIХж, R'= Н), в которой расположенная в центре сложно-
эфирная группа пространственно труднодоступна и вследствие этого устой-
чива по отношению к гидролизу. Поэтому флавогаллол ведет себя как анги-
дрооснование, производное катиона с плоской структурой типа Х1Хз.
Синтез флавогаллола был осуществлен [162] конденсацией по Ульману
сложных бромэфиров (Х1Хи, Х1Хк), сопровождаемой одновременным деме-
тилированием и циклизацией промежуточного соединения Х1Хж (R = СН3).
«Дегидродиэллаговая кислота» (XI Хл) образуется в результате автоконден-
сации сложного эфира (Х1Хк).
СООСН3
НзСС/^/^ОСНз
ОСН3
Х1Хи
Вг
н3соч^х/соосн3
I |1 ОСНз
.. . Ах A Jx /ОСНз
Н3СО 1 II
СН3ООС/Х/'ОСН3
Х1Хк
II
он
он
\/\/\
он
он
Х1Хл
СНз
СО I
/ \/\
О-Дигиталоза-Н-фукоза - 0 0 Г I
I НА
z\/\/v
I 'I I о
। со
но
Х1Хм
Гликозидный антибиотик — чатрейзин [163] имеет строение, подобное
строению эллаговой кислоты с конденсированными «-пироновыми кольцами
(XI Хм).
КУМАРО-а- И КУМАРО-у-ПИРОНЫ
57
Кумаро-у-пироны. Единственно известными природными представите-
лями этого ряда являются О-диметилцитромицинон (XX, R = Н), метил-О-
диметилцитромицетинон (XX, R = СООСН3) (см. цитромицетин, стр. 85)
и дистемонантин (ХХа) Ci7H10C)9, содержащиеся в древесине Distemonanthus
, О О
сн3о -/V V
CH3°A/vJ\^°
R О |
I
СН3
XX
benthamianus. Хромоно-(3',2',3,4)-изокумариновая структура была установлена
путем расщепления щелочью [165] пентаметилового эфира, в результате чего
образовались гемипиновая кислота (ХХб) и ю-метоксиацетофенон (ХХв).
Соответствующая обработка пентаэтилового эфира позволила установить поло-
жение метоксильной группы. Хотя сам дистемонантин не синтезирован, сое-
динения такого типа были получены, например, ацилированием ю-метокси-
резоацетофенона 2-метоксикарбонилбензоилхлоридом. Полученный продукт
СН3°
ХХа
ОСН3
I
/\-ОСН3
J.'- СООН
I
СООН
ХХб
СН3О -/ V- он
СН3О JLcOCH2OCH3
I
ОСН3
ХХв
подвергался перегруппировке Бекера — Венкатарамана и было получено
соединение ХХг, которое при деметилировании дало хромоноизокумарин ХХд.
ХХг
I
СООСН3
ХХд
Лейкоантоцианидин, пелтогинол [166] (XX, R = Н), содержащийся в древесине
Peltogyne porphyrocardia, является производным флавандиола-3,4 с восстанов-
ленным дистемонантиновым кольцом. Окисление О-триметилпелтогинола
(ХХе, R = СНз) дает О-триметилпелтогинон (ХХж). Пелтогинол-В (ХХе,
ХХе
ХХж
58
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
R = Н), встречающийся в природе вместе с пелтогинолом, также может быть
окислен в О-триметилпелтогинон. Поскольку при пространственно не затруд-
ненном восстановлении карбонильной группы в О-триметилпелтогиноне бор-
гидридом натрия в качестве единственного продукта восстановления обра-
зуется О-триметилпелтогинол, то очевидно, что пелтогинол должен обладать
экваториальной, а пелтогинол-В — аксиальной 4-оксигруппой. Синтезиро-
ваны многочисленные кумаро-у-пироны типа XXI, XXII и XXIII путем
присоединения а-пиронового кольца к хромоновому или же путем построения
О О
XXI
XXII
А
у-пиронового ядра на кумариновом. Последний метод находит более широкое
применение ввиду большей доступности исходных материалов. Типичным
примером первого метода служит конденсация резоацетофенона (XXIV) с аце-
тоуксусным эфиром под действием хлористого алюминия [167], в результате
которой образуется 6-ацетил-5-окси-4-метилкумарин (XXV). В то же время
аналогичная конденсация с хлорокисью фосфора 1168] приводит к изомер-
ному 6-ацетил-7-окси-4-метилкумарину (XXVI). Циклизация соединений XXV
СН3С0СН2С00С2Н5
| Aids
ho-Z^.-oh
И1-сосн3
XXIV XXV
° °
СН3СОСН2СООС2Н5 но—Z\z \Z
рш^ * сн,со I II I
сн3
XXVI
и XXVI под действием ацетата натрия и уксусного ангидрида дает соответ-
ствующие кумаро-у-пироны типов XXI и XXII. Как и при обычном образо-
вании хромона из о-оксиацетофенона (XXVII) под действием ацетата натрия
и уксусного ангидрида, циклизация протекает с промежуточным образова-
нием З-ацетил-2-метилхромонов (XXVIII), из которых ацетильная группа
удаляется затем гидролизом раствором углекислого натрия. В результате
CH3COONa
(СНзСО)2О
II н
о о
XXVIII XXIX
>|-°Н
J-COCHg
XXVII
образуется 2-метилхромон (XXIX). Метильная группа в положении 2 хро-
монового ядра, так же как и в простом 2-метилхромоне, реакционноспособна
КУМАРО-ос- И КУМАРО-у-ПИРОНЫ
59
и может вступать в конденсацию с ароматическими альдегидами с образова-
нием соответствующих стирилпроизводных.
Аналогично при конденсации 6-бромрезоацетофенона (XXX) [169, 170]
с ацетоуксусным эфиром в присутствии хлористого алюминия образуется
Br
CH3CO-
OH CH3
XXXI
CH3COONa
(СН3СО)2О
HO-/Y-OH
Br-I^ J-COCHg
CHSCOCH2COOC2H5
Aicis
XXX
Br
I О дэ
. 0 I IH
I A L
СНзСО-!^ СНз
I
CH3
XXXII
кумарин (XXXI), который циклизуется при действии ацетата натрия и уксус-
ного ангидрида в кумаро-у-пирон (XXXII). Аналогичным путем 5-этилрезо-
ацетофенон (XXXIII) дает кумаро-у-пирон (XXXV), причем в качестве про-
межуточного продукта образуется 6-ацетил-8-этил-5-окси-4-метилкумарин
(XXXIV). Подобная конденсация орцинола и ацетоуксусного эфира [171]
С2Н5
I О '
XX/ \=о
НО—| ц—ОН СН3СОСН2СООС2Н5 I II | CH3COON3
С2Н5-Ц1-СОСН3 --------> СНзСО-^1^ (СНзСО)2о
XXXIII он сн3
XXXIV
сн3со-^z° СНз
сн3
XXXV
приводит к 5-окси-4,7-диметилкумарину (XXXVI). Этерификация соедине-
ния XXXVI бромистым аллилом дает аллиловый эфир (XXXVII), пре-
терпевающий при нагревании перегруппировку Клайзена с одновременным
замыканием кольца и образованием хроманокумарина (XXVIII). Кумаро-у-
пироны типа XXIII легко доступны при получении по схеме XXXIX XLI
с применением метода циклизации по Костанецкому 8-ацил-7-оксикумаринов,
60
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
XXXVI
о о
H3c-|^Y
\/\Z
I сн3
ОСН2СН = СН2
XXXVII
XXXVIII
полученных перегрупировкой Фриса из сложных эфиров 7-оксикумаринов.
Кумарины не вступают в реакцию кетонного синтеза по Гешу и поэтому тре-
буемые для этого синтеза о-оксикетоны не могут быть получены этим способом.
X 0 0
RCOO—
I II I
\/\z
I
сн3
COR
I О О
I II I
\/\/
I
сн3
CH3COONa
-------—>
(СН3СО)20
R
Н3С | О
о I о О
\Z\z\Z
XXXIX
XL
\/\/
сн3
XLI
Присоединение а-пиронового кольца к хромоновому ядру менее привле-
кательно по сравнению с только что описанным методом,, поскольку оксихро-
моны не вступают в реакцию конденсации по Пехману, в реакцию синтеза
кетонов по Фриделю— Крафтсу и в реакцию синтеза альдегидов по Гаттер-
ману. Однако формильная группа может быть введена, хотя и с плохим выхо-
дом, в положение 8 оксихромона действием гексаметилентетрамина в ледя-
ной уксусной кислоте. Получающиеся 7-окси-8-формилхромоны могут быть
превращены в кумарины при помощи стандартных методов, таких, например,
как конденсация по Перкину или же с худшим результатом действием циан-
уксусной кислоты.
Строение хромон-8-альдегидов доказано их восстановлением в 8-метил-
хромоны, идентичные соответствующим хромонам, синтезированным из 2-метил-
резорцина.
Получен также ряд кумаро-у-пиронов типа XXI [172]. Например, 7-окси-
3-метоксифлавон (XLII, R = Н) дает альдегид (XLII, R = СНО), превра-
щающийся в результате конденсации по Перкину в у-пиронокумарин
(XLIII).
О—
R и-1
I о о ! о
но-/у >-с6н5 Yy ус6н5
MJLoch, UJL ОСН,
<4 и
XLII
XLIII
КУМАРО-а- И КУМАРО-у-ПИРОНЫ
61
Аналогичным образом из 7-окси-3-метокси-2-метилхромона (XLIV, R = Н)
получают альдегид (XLIV, R = СНО) и затем циклизуют его в кумаро-у-
пирон (XLV). Путем конденсации 8-формил-7-окси-3-метоксифлавонов типа
О—
R I
I О о I о
но-А/ у СН3 у СНз
-yNI-оснз уМЬоснз
II II
о о
XLIV XLV
(XLIX) и замещенных умбеллиферон-8-альдегидов типа XLVIII с о-окси-
ацетофенонами в стандартных условиях был синтезирован также ряд пири-
лиевых солей типа XLVI и XLVII, аналогичных хромоно-а- и хромоно-у-
пиронам [173].
XLVlll XL1X
1,4-Пиранокумарины. Производные 3,3/-метилен-биб’-(4-оксикумаринов).
Установлено, что вещество, вызывающее геморрагическое заболевание круп-
ного рогатого скота и содержащееся в клевере, является 3,3'-метилен-б«с-
(4-оксикумарином) (L, R = Н) [174—177], который образует диметиловый
эфир по двум кислым енольным гидроксильным группам. Расщепление сое-
динения L (R = Н) 10%-ным едким кали при 100° дает дикетон (LI), 1,3-диса-
ОООО
I II I I II I п л
—СОСН2СН2СН2СО—
он он
L
LI
О О
Z\z \Zz
uu
6н
L1I
62
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
лицоилпропан, а сплавление при 300° с поташом — салициловую кислоту.
При нагревании соединения L (R = Н) с анилином при 180° в течение 30 мин
образуется анил 2,4-дикетохроман-(4-оксикумарина) (LII). Синтез 3,3'-мети-
лен-бис-(4-оксикумарина) был осуществлен конденсацией 4-оксикумарина
с формальдегидом. Этот метод синтеза был использован для получения ряда
3,3'-метилен-б«с-(4-оксикумаринов) типа L (R = арил или алкил) путем кон-
денсации других альдегидов [178] с 4-оксикумарином. Конденсация легко
проходит при кипячении соответствующих компонентов в этаноле.
При кипячении 4-оксикумарина с о-оксиальдегидом (например, салици-
ловым) конечным продуктом оказывается соединение LIV, которое, вероятно,
получается из промежуточно образующегося 3,3'-(о-оксибензилиден)-бис-(4-
оксикумарина) (LIII).
LII1
Продукты конденсации альдегидов типа L легко дегидратируются уксус-
ным ангидридом в пиридине с образованием 1,4-пиранокумаринов (LV) [179].
Простейший член этого ряда — 3,3'-метилен-б«с-(4-оксикумарин) — нельзя
получить этим способом, но он может быть превращен в пиранокумарин дей-
ствием плавленого бисульфата калия. Моноэфиры 3,3'-метилен-б«с-(4-окси-
кумаринов) легко превращаются в пиранокумарины (LV) при нагревании,
но диэфиры оказываются более устойчивыми. Пирановое кольцо легко можно
раскрыть действием избытка метилата натрия, в результате чего образуется
монометиловый эфир типа L.
Относительная легкость расщепления пиранового кольца в этих соеди-
нениях становится понятной при рассмотрении свойств З-алкил-4-оксикумари-
нов, которые ведут себя подобно кислотам и простые эфиры которых прояв-
ляют свойства сложных эфиров [180]. Это не удивительно, поскольку в 4-окси-
кумаринах имеется система, близкая к винилогам карбоновых кислот, в которых
но—со—сн = сн—он
вторая гидроксильная группа отделена от карбонильной группы винильной
группировкой. Поэтому превращение б«с-кумаринов типа L в пиранокума-
рины типа LV равносильно образованию ангидрида кислоты. Действительно,
свойства пирановой системы сходны со свойствами ангидридов.
Новобиоцин и фусцин. Продукты обмена веществ грибов — новобиоцин
и фусцин — также будут рассмотрены в настоящем разделе.
Выделение и химические свойства антибиотика новобиоцина C31H36N20n,
полученного из Streptomyces niveus, описаны несколькими авторами [181, 182].
Хотя этот глюкозид содержит только один гетероатом кислорода в агли-
КУМАРО-а- И KVMAPO-Y-ПИРОНЫ
63
коне, в нем имеется второе потенциальное кислородсодержащее кольцо,
обнаруживающееся в некоторых продуктах его расщепления. Новобиоцин
имеет структуру LVI и при расщеплении метанольным раствором хлористого
водорода превращается в метил-З-О-карбамил-4-О-метилновобиозу (LVII)
с конфигурацией L-ликсозы [183—185].
СН3 сн3
-----СНОСНз
неон
о HCOCONH,
СН3О^Н
-----С^
СНз СНз
LVII
Расщепление уксусной кислотой приводит к сахариду и новобиоциновой
кислоте (LVIII), образующейся в результате циклизации изопентенильного
остатка, присоединенного к кольцу А. Сама кислота была синтезирована [186]
путем превращения 4,7-диокси-8-метилкумарина (LIX, R = Н) в оксими-
LXI
ОСОСНз о О
LXII LXIII
64
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
нопроизводное с последующим восстановлением и образованием соединения
LIX(R = NH2-HCI), которое при конденсации с хлорангидридом 6-карбокси-
2,2-диметилхромана (LX) и последующем дезацетилировании превращалось
в циклоновобиоциновую кислоту (LVIII).
При гидрировании новобиоцина восстанавливается боковая изопентениль-
ная цепь и образуется диглдроновобиоцин. В результате действия на это
дигидросоединение метанольным раствором хлористого водорода получается
дигидроновобиоциновая кислота (LXI), которая была синтезирована мето-
дом, аналогичным примененному для синтеза циклоновобиоциновой
кислоты.
Место присоединения агликона к сахариду было установлено путем рас-
щепления новобиоцина при нагревании с уксусным ангидридом, в результате
чего образовались изопентенил-кислота (L X11) и производное оксазола (LX 111),
в котором было определено положение гликозидной связи.
Этот метаболит был охарактеризован первым среди тех, в которых содер-
жатся 4-оксикумариновая и, возможно, потенциальная 2,2-диметилхромано-
вая группировки, хотя восстановленную 4-оксикумариновую группу содер-
жит также цитромицетин (стр. 85).
Фусцин Ci5Hi6O5 был выделен Михаэлем [187] из фильтратов культуры
Oidiodendron fuscum Robak и затем исследован Биркиншоу с сотрудниками [ 188];
эта экспериментальная работа была продолжена Бартоном [189] и Бирчем [ 190 ]
с целью установления строения LXIV> которое было подтверждено полным
синтезом фусцина [189].
LXIV
СООН
LXVI
В соответствии с формулой LXIV фусцин обнаруживает свойства еноль-
ного лактона, содержащего метиленгидрохиноновую систему, при гидриро-
вании которой образуется дигидрофусцин (LXV). При гидролизе в мягких
условиях это производное превращается в ацетальдегид и фусциновую кис-
лоту (LXVI), тогда как сплавление со щелочами дает уксусную, изовалериа-
новую и 3,4,5-триоксифенилуксусную кислоты. При окислении О-диметил-
фусциновой кислоты перманганатом калия образуются а-оксиизомасляная
кислота (из гсщ-диметильной группы), трикарбоновая а-кетокислота (LXVII),
а также монокарбоновая С1зН14О6 и дикарбоновая Ci5Hi80s кислоты, строе-
ние которых выражается соответственно ф°РмУлами LXIX и LXVIII. Обра-
КУМАРО-а- И КУМАРО-у-ПИРОНЫ
65
зование их происходит, вероятно, по схеме, приведенной ниже:
cocoon
f '-соон
CH3oJJ4
Y o-z,
сн;!о
LXVII
соон
соон
ОСНз
LXIX
соон
СН3О-
соон
LXVIII
LXX
LXXI
Метилирование фусцина диазометаном дает метилгомофусцин (LXX),
который легко превращается при гидрировании в дигидрометилгомофусцин
(LXXI, R = X = Н), при титровании холодной щелочью — в гомофусцин-
гидрат (LXXI, R = Н, X = ОН), а под действием хлористого водорода —
в соединения LXXI (R = Н, X = С1). Эти легко протекающие реакции,
несомненно, обусловлены наличием в соединении LXX циклопропанового
кольца, блокирующего ароматизацию циклогексадиеновой системы.
Синтез фусцина был осуществлен стандартными методами. Взаимодей-
ствием хлорангидрида р,|3-диметилакриловой кислоты с метиловым эфиром
3,4,5-триметоксифенилуксусной кислоты получен хроманон (LXXII), который
при восстановлении дал О-диметилфусциновую кислоту (LXXIII). Конден-
сацией этой кислоты с ацетил хлоридом синтезирован кетон (LXXIV), суще-
ствующий частично в форме лактола (LXXV). Восстановление последнего
боргидридом калия приводит к .дигидрофусциндиметиловому эфиру, который
в результате деметилирования и последующего окисления воздухом в щелоч-
ном растворе легко превращается в фусцин (LX1V). Фусцин — первый из про-
5 Заказ М 978
66
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
дуктов обмена веществ грибов, в котором доказано присутствие 2,2-диметил-
хроманового остатка.
СООН СООН СООН
LXXII LXXIJI LXXIV
ноу '|
Нзс/
сн3о/ухо/х
ОСН3
LXXV
ХРОМОНО-у-ПИРОНЫ
Простые хромоно-у-пироны. Хромоно-у-пироны до сих пор еще не выде-
лены из природных источников, однако их производные типа I и II были
получены синтетически. Немногие представители соединений типа I были
синтезированы применением к хромонам (с некоторыми ограничениями) стан-
дартных методов синтеза, пригодных для получения у-пиропов, как это видно
из приведенных ниже примеров 1191, 1921.
Обработка 2-метил-7-ацетоксихромона (III) хлористым алюминием в усло-
виях перегруппировки Фриса приводит к 8-ацетил-7-окси-2-метилхромону (IV),
который циклизуется с ацетатом натрия и уксусным ангидридом в хромоно-
у-пирон (V). Аналогично при действии ацетата натрия и уксусного ангидрида
на соединение VI также образуется хромоно-у-пирон (VII).
СОСН3
о I о
Ггт гмп /'''•/ \ си чс. \ ru CH3COONa
CJiyCOO । .I i( CH3 AlClj HO ( I, 11 OHo--------->
I || || , I [| || lai.coi.o
Ч/\/ \/\/
II II
о о
III IV
XPOMOHO-Y-ПИРОНЫ
67
COCHg
I
I-I3C-^X,=O
о I о
\________рц
I !l I! ’
\/\/
II
о
V
CH3
COC2H5 I
I 0 H3C -A=O
НО-// VCH.3 CH3COONa ° | O
^jLVCHgVW V/ VCH3
? vy~ra>
VI о
VII
Линейные члены ряда типа II более многочисленны, однако обычные
методы синтеза хромонов и флавонов находят весьма ограниченное приме-
нение в синтезе соединений этого типа и не приводят часто к ожидаемым про-
дуктам. Наиболее надежным методом синтеза, так же как и в случае хромо-
нов, является циклизация бис-цикетонов типа VIII, катализируемая кисло-
тами. Эти бис-дикетоны трудно получить обычными методами и, по-видимому,
/- 0 / 0
СН3О -/ / ОСНз -V / \ / / R
RCOCH2CO/COCH2COR \/\/\ /
VUl If V
А
I X
СН3О / V ОСНз
RCH = СНСО -/J— COCH = CHR
IX
их лучше синтезировать путем превращения халконов (IX) в соответствую-
щие тетрабромиды с последующим дебромированием метилатом натрия, что
приводит к соединению VIII 1193]. Реакция, вероятно, протекает через сле-
дующие стадии:
- COCH = CHR —> - СОСНВгСНВг —-
-НВг
—» — COCHBrCHOHR---> -СОСН = С —R—>
I
ОН
—coch2cor
Циклизация соединения VIII (R = СвН5) под действием кипящей иодисто-
водородной кислоты, сопровождающаяся одновременным деметилированием,
дает соединение X (R = С6Н5) 1194]. Применимость этого метода строго
ограничена природой заместителя R в соединении VIII, так как 1194] если R
представляет собой и-анизил или 3,4-метилендиокси'фенил, то вместо ожидае-
5*
68
'соединения, содержащие два гетероатома кислорода
мого хромоно-у-пирона образуются продукты неопределенного строения.
Сообщено [195] об успешном превращении тетрабромида дибензилиденди-
ацеторезорциндиацетата в хромоно-у-пирон (X, R = С6Н5) действием горя-
чего спиртового раствора поташа. Однако этот метод непригоден для синтеза
хромоно-у-пиронов [196], содержащих заместители в фенильных остатках:
в этом случае обычно образуются не хромоно-у-пироны, а, по-видимому, изо-
мерные p-фуранонкумараноны, хотя окончательно этот вопрос не выяснен.
Ограниченное применение для получения флавонов имеет также метод
ароилирования по Робинзону [197']. Бензоилирование диацеторезорцина (XI)
бензоатом натрия и бензойным ангидридом дает соединение XII, которое
в результате щелочного гидролиза превращается в бис-дикетон (XIII) и затем
под действием кипящей хлористоводородной кислоты циклизуется в соеди-
нение X (R = С6Н5).
О ,0
НО /Ч-ОН CeHsCOONa СВН5-/
СН3СО СОСН3 (CeH5c°)2° CRH5CO сосйн5
XI II
Л| о о
XII
I кон
НС1 НО -/ОН
X(R = CeH5) <--
С0Н5СОСН2СО СОСН2СОС6Н5
XIII
Однако обработка диацеторезорцина (XI) и-этоксибензоатом натрия
и анисовым ангидридом [198] дает вещество, являющееся, по-видимому, фла-
воном (XIV). Гидролиз последнего 5%-ным едким кали приводит к бпс-дике-
XIV
—> СН3О
НО ОН
Y СОСН2СО -МL СОСН2СО _______________Y ОСНз
XV
НС1
СН3О
ОСНз
НО ОН
RCH = СНСО - Y J— СОСН - CHR
XVII
XVIII
ХРОМОНО-у-ПИРОНЫ
69
тону (XV), который циклизуется под действием кипящей хлористоводородной
кислоты в соединение XVI. Трудности, встречающиеся при применении обыч-
ных методов синтеза флавонов к получению хромоно-у-пиронов, вероятно,
обусловлены тем, что бензольное ядро в флавоновой группе значительно
менее реакционноспособно, чем фенольное кольцо, и поэтому замыкание
второго у-пиронового кольца происходит значительно труднее.
Обработка о-оксифенилстирилкетонов перекисью водорода в горячем
спиртовом растворе едкого кали дает хорошие выходы флаванолов 11991.
При применении этого метода к халконам типа XVII, где R может быть фенил,
анизил или пиперонил, получают соответствующие флавонолы (XVIII) [2001.
Бифлавонилы. Бифлавонильная структура XVI Па была установлена
для ряда веществ природного происхождения 1201, 2021. Гинкгетин
СзоН1204(ОН)4(ОСН3)2 (XVIIIa, R = СНз, R' = R" = Н) — желтый пиг-
мент, содержащийся осенью в листьях дерева адиантум Ginko biloba — был
XVIII а
ОСН3
!
СООН
Ч/
I
СООН
XV1I1 б
ОСН3
XVIII в
XVIII г
первоначально выделен Наказава [203]. Бейкер и др. 1202[ выделили изо-
гинкгетин СзоН1204(ОН)4(ОСН3)2 (XVIIIa, R == R' = Н, R" = СН3) из того
же источника, а сциадопитизин С3оН1204(ОН)3(ОСНз)з (XVIIIb, R — R" =
--= СН3; R' = Н) из зонтичной сосны Sciadopitys verticillata [204]. Метили-
рование каждого из этих бифлавонилов дает О-тетраметилгинкгетин, который
при окислении щелочным раствором перекиси водорода образует м-анисовую
кислоту, 2-окси-4,6-диметоксибензойную кислоту и дифенилдикарбоновую
кислоту Ci2H4(OCH3)3OH(COOH)2. Окисление сциадопитизина (XVIIIa,
R = R" = СН3, R'= Н) приводит к получению и-анисовой и 4-метокси-
изофталевой (XVII16) кислот. Щелочное расщепление дает и-анисовую кис-
70
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОЛТОМА КИСЛОРОДА
лоту, 2,6-диокси-4-метоксиацетофенон и кетон (XVIПа). На основании этих
данных и других соображений [201, 2021 было установлено строение гинкге-
тина, изогинкгетина и сциадопитизина 1205].
Карийон и Савада [206] показали, что кайафлавон C3oHt204(OH)3(OCH3)3
и сотетсуфлавон С3оН1204(ОН)5ОСН3 являются бифлавонилами, близко род-
ственными гинкгетину, так как при метилировании они дают тетраметиловый
эфир гинкгетина. Бейкер с сотрудниками [202] определил строение кайа-
флавона (XVII la, R = Н, R' — R" == СН3) и сотетсуфлавонa (XVIIIa,
R = R' = R" = Н).
Хинокифлавон из листьев Chamaecyparis obtusa [2071, вероятно, также
имеет строение бифлавона (XVIIIг).
Синтезирован также ряд других бифлавонилов 12081.
Пейценин (XIX, R = Н) [2091, содержащийся в корневищах Peuce-
danum. ostruthium Koch., не имеет метоксильных групп и имеет только один
гетероатом кислорода, однако некоторые из продуктов его превращения
являются хромано-у-пиронами (дигидрохромоно-у-пиронами). При метили-
ровании диазометаном легко образуется монометиловый эфир пейценина
(XIX, R = СН3), однако вторая гидроксильная группа, экранированная
двумя соседними карбонильными группами (ср. осайин и помиферин), мети-
лируется с трудом. Ацетилирование монометилового эфира в жестких усло-
виях с помощью ацетата натрия и уксусного ангидрида дает моноацетат моно-
метилового эфира. При гидрировании пейценина восстанавливается двойная
связь в боковой цепи с образованием дигидропейценина, который, как и исход-
ное соединение, легко образует монометиловый, но не диметиловый эфир.
Пейценин при кипячении в растворе ледяной уксусной кислоты, содержа-
щей небольшое количество серной кислоты, легко превращается в смесь двух
изомерных 2,2-диметилхромано-у-пиронов — аллопейценин (XXI) и изопей-
ценин (XX) — при циклизации ненасыщенной у,у-диметилаллильной боковой
цепи. (Ср. аналогичное превращение осайина и помиферина в изоосайип и изо-
помиферин соответственно, стр. 102.)
Циклизация монометилового эфира пейценина (XIX, R = СН3) в этих
условиях дает в качестве единственного продукта монометиловый эфир алло-
пейценина, чем доказывается ангулярное строение аллопейценина (XXI)
и, следовательно, линейное строение изопейценина (XX).
Озонолиз пейценина дает ацетон, образующийся из у,у-диметилаллиль-
ной боковой цепи, а щелочное расщепление приводит к получению уксусной
кислоты из хромонового кольца и 5,7-диокси-2,2-диметилхрома:на. Последний
образуется в результате циклизации боковой цепи в процессе обработки
ХРОМЕНО-а-НИ РОНЫ
71
щелочью (ср. поведение осайина и помиферина). Отсутствие подобной группи-
ровки в самом пейценине легко доказывается тем, что при щелочном расщеп-
лении дигидропейценина, не способного к циклизации, образуется только
уксусная кислота и изоамилфлороглюцин.
Метиловый эфир аллопейценина был синтезирован 12101 конденсацией
8-ацетил-7-окси-5-метокси-2,2-диметилхромана (XXII) с этилацетатом и после-
дующей циклизацией образующегося дикетона.
ХРОМЕНО-а-П ИРОН Ы
Введение. Простейшие представители этого класса (I и II) и соответствую-
щие им ангулярные формы не известны. Первым выделенным представителем
этого класса был ксантоксилетин из Xanthoxylum americanutn Mill. [211, 2121
5
I П
Затем из этого же источника были изолированы один из ангулярвых изоме-
ров—аллоксантоксилетин [213i и родоначальное соединение ряда—ксан-
тилетин 1214]. Из природных источников был выделен также 1215, 216] сезе-
лин, а в орехах Calophyllum inophyllum обнаружены три более сложных пред-
ставителя этого класса [217].
В этих соединениях сс-пироновое кольцо ведет себя подобно простым
кумаринам, а двойная связь трудно гидрируется без предварительного пре-
вращения в коричную кислоту.
2,2- Диметил-А3-хроменовое ядро. 2,2-Диметил-А3-хроменовое ядро (III),
содержащееся в соединениях этого класса, имеет несколько характерных
особенностей и специфических свойств. В связи с трудностями, возникающими
при окончательном доказательстве строения, и ввиду наличия этой группи-
ровки во многих природных веществах представляется уместным рассмотреть
химию этой важной циклической системы, прежде чем перейти к рассмотре-
нию поведения веществ, в которых она содержится.
2,2- Диметил-А3-хроменовое ядро характеризуется некоторыми реак-
циями, свойственными высокореакционноспособной двойной связи.
1. Гидрирование протекает очень быстро с полным превращением хро-
мена (III) в хроман (Ша), прежде чем значительно более устойчивая двойная
связь в a-пироновом кольце хромено-а-пироновой системы подвергнется воз-
действию водорода.
2. Хроменовое ядро легко расщепляется под действием окислителей.
Аномальные результаты получены при озонолизе, при котором удаляется
гем-диметильная группировка с одновременным разрывом двойной связи
в положении 3,4 и эфирной связи с образованием о-оксиальдегида, вместо
того чтобы окисление протекало только у двойной связи с образованием сое-
динения типа IV, как этого следовало бы ожидать. Окисление такими аген-
тами, как перманганат калия, также приводит к расщеплению обеих эфир-
ных связей и двойной связи с образованием a-оксиизомасляной кислоты.
Д\/°Х/СНЗ Г;Л/СНз °C сн3
I || |ХСНз | || ХСН3 | |) ХСООН
Vxz Ч/\/ Ч/хСоон (СНО)
(СНО)
III III а IV
72
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМЛ КИСЛОРОДА
3. Хроменовое ядро легко расщепляется под действием щелочей с обра-
зованием ацетона. Это очень характерное, хотя и неожиданное свойство
2,2-диметил-Д3-хроменовой группировки наблюдалось ранее при разложении
токсикарола с образованием апотоксикарола (стр. 134). По-видимому, это
еще не получившее объяснения свойство является общим для этих хромонов.
Образование ацетона можно объяснить прохождением реакции через стадию
образования соединений V и VI. Это гидролитическое расщепление 2,2-диме-
тил-Д3-хроменовой группировки близко напоминает поведение кумаринов
при действии концентрированных щелочей. Однозначного метода синтеза
2,2-диметил-Д3-хроменов еще не найдено, хотя установлено, что эта система
получается с низким выходом при конденсации 2-метилбутин-3-ола-2 с фено-
лами под давлением 12181. Этот синтез может привести и к изомерной 2-изо-
пропилфурановой системе (VII), однако продукты реакции (см. ниже) ока-
зались идентичными природным веществам, в которых путем расщепляния
было точно установлено наличие 2,2-диметил-Д3-хроменовой группы.
СН3
।
НО--С-СН-,
. I
с
НС
о
/V >,-сн(сн3)2
или ) I
VII
Кроме 2,2-диметилхромена, был синтезирован также лапахенол (Vila) [2191.
Восстановлением хроманона (VI 1в) боргидридом натрия получен хроманол-4
(VII6, R Н); его ацетат (VII>6, R = СН3СО) был превращен пиролизом
в лапахенол, полученный с плохим выходом.
ОСН-;
I
НзС7 СН3
Н3с СНз
VII в
Vila VII6
X POM Е Н О-a- П И РО Г! Ы
Робертсон с сотрудниками окончательно установил строение 2,2-диметил-
хроманового ядра (Illa): таким путем, вполне логично допуская, что гидри-
рование не вызывает никаких других превращений, было получено полное
доказательство строения хроменовой группы.
5,7-Диокси-2,2-диметилхроманон-4 (VIII) был получен конденсацией
флороглюцина с хлорангидридом р,0-диметилакриловой кислоты 12201. Этот
процесс, весьма вероятно, может протекать с образованием одного или несколь-
ких изомерных соединений (VIII, IX, X, XI или XII). Однако единственный
изолированный продукт, устойчивый по отношению к гидрированию над
палладированным углем, давал 2,4-динитрофенилгидразон, а также димети-
ловый эфир, нерастворимый в холодном 2 н. растворе едкого натра, и показы-
вал отрицательную реакцию с хлорным железом. Таким путем сразу были
исключены из рассмотрения структуры X, XI и XII, но не структура IX.
Попытки доказать присутствие — СОСН2— группы, что позволило бы сделать
выбор между структурами VIII и IX, оказались безуспешными. Поэтому
был проведен однозначный синтез изопропилкумаранона (IX).
Конденсацией по Фриделю — Крафтсу флороглюцина с хлорангидридом
a-бромизовалериановой кислоты было получено с низким выходом соеди-
нение XIII (R = Н). Этот p-кумаранон, который отличался от изомерного
/СН3
г\ .У, ,
НО-.^'.-ОН /СНз
I || + СНВгСН
СОС1 СНз
он
OR
XIII
хроманона (VIII), вел себя как насыщенное соединение при гидрировании
над палладированным углем, не образовывал 2,4-динитрофенилгидразона
и при метилировании давал, как и ожидали, смесь диметилового (XIII, R =
= СНЙ) и триметилового эфиров енольной формы. 2,4-Динитрофенилгидра-
зон диметилового эфира (XIII, R — СН3) оказался неидентичным соответ-
ствующему производному О-диметилового эфира 5,7-диокси-2,2-диметилхро-
манона (VIII). Образец p-кумаранона (XIII, R = СН3) для сравнения был
приготовлен из диметилового эфира флороглюцина и этилового эфира бром-
изовалериановой кислоты с промежуточным получением соединений XIV
и XV. Он оказался идентичным О-диметиловому эфиру соединения XIII (R =
= Н) и образовывал тот же 2,4-динитрофенилгидразон. Таким образом, хро-
74
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
малоновая структура (VIII) продукта взаимодействия флороглюцина с хлор-
ангидридом р,р-диметилакриловой кислоты была окончательно установлена.
сн3о W ! \/ ! ОСН;, он /СНз ВгСНСН ХСН3 СООС2Н5 СНз°\^./° 1 !' "Ч/ ()С11з XIV „ /СНз снсн ХСНз COOC2HS
СН3О „СН3 о ОН ' 1 ХСНз 1 1 /О СН3О-^Х/ \ /сн- снсн
\./—"° Ч ' COCI СНз
ОСНз ОСНз
XV
/СН3
НО ° . ,сн
' У У Х/ ХСН3
он
XVII
Однако все еще оставалась вероятность того, что хроманоповая группи-
ровка способна размыкаться в условиях восстановления по Клемменсену
с образованием промежуточного соединения типа XVI, которое в свою оче-
редь может замыкаться с образованием изопропилкумарана (XVII). По анало-
гии с незамещенными хроманонами и флавононами можно думать, что такое
но-р-он СНз
р-СН2СН-С
i ХСН3
он
XVI
5 ,7-диоксихроманоновое кольцо вряд ли будет легко раскрываться под дей-
ствием кислот, и более вероятным представляется, что циклизация должна
приводить, скорее, к шестичленному, а не к пятичленному кольцу (ср. обра-
зование аллоротенона из p-дигидроротенона, стр. 125, и превращение осайина
и помиферина в соответствующие изосоединения, стр. 102).
Аналогичные сомнения существовали и в отношении строения 7-окси-2,2-
диметилхромана, а следовательно, и соответствующего хромена [2111. Этот
вопрос был разрешен следующим образом. Конденсацией резорцина с хлор-
ангидридом р,р-диметилакриловой кислоты (XVIII) получен хроманон
(XIX, R = Н). Строение его однозначно подтверждено монометилированием
в соединение XIX (R =-- СН3). Это соединение не изменяется при гидриро-
вании в присутствии палладия на угле, нерастворимо в холодном 2 н. едком
натре и является изомерным, но не идентичным соответствующему р-кумара-
нону (XX), синтезированному двумя взаимно подтверждающими методами
(а и б), как и в случае4,6-диметокси-2-изопропил-|3-кумаранона (XIII, R=CH3).
Взаимодействие 7-бензилокси-3,4-дигидрокумарина (XXI) с метилмагний-
иодидом и последующее дебензилирование дали 7-окси-2,2-диметилхро-
ман (XXII), идентичный образцу, полученному с хорошим выходом и с боль-
шой легкостью восстановлением 7-окси-2,2-диметилхроманона (XIX, R == Н)
по методу Клемменсена. Тем самым получено еще одно доказательство хро-
мановой структуры продукта восстановления соответствующего хроманона.
ХРОМЕНО-а-ПИРОНЫ
75
Изомерные ему а-кумараноны восстанавливаются по методу Клемменсена
с трудом.
Окончательным доказательством строения синтетического 7-окси-2,2-
диметилхромана явился синтез изомерного 6-окси-2-изопропилкумарана
(XXVa) двумя взаимно подтверждающими методами, а также неидентичность
но ои
/СН3
г С-СНз
il
сн
I
COCI
XVI11
/СНз
СНВгСН
I ^СНз
СООС2Н5
НО — он
|«| I j
X/
СоН^НгО^ ° у)
XXI
,СН3
СНВгСН
(2ОС1 Х'СНз
(1) Cl
(2) Дебензилирование
ХХД1
продукта изомерному хромапу (XXII). Согласно первому методу, 6-окси-
2-изопропилкумараноп-З (XXIII) был превращен с плохим выходом через
О /СН3 о /СН3
но — X > / \— сн х >— сн
Р \---L О ХСНз J-J NOH СНз
XXIII XXIV
О /СН3 НО О /СНа
—сн р у сн
L NHCH3 i I Хснз
XXV XXVa
промежуточную стадию образования оксима (XXIV) и амина (XXV) в 6-окси-
2-изопропилкумаран (XXVa) [222].
Очень изящный и более удобный и однозначный способ синтеза предложен
Робертсоном с сотрудниками [223]. Взаимодействие 6-бензилокси-2-ацетил-
бензофурана (XXVI) с метилмагнийиодидом привело к получению 6-бензил-
окси-2-изопропенилкумарона (XXVII), из которого каталитическим гидри-
рованием и дебензилированием был получен 6-окси-2-изопропилкумаран
(XXVa), идентичный продукту, синтезированному первым методом, и изо-
мерный, но не идентичный 7-окси-2,2-диметилхроману.
76
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Аналогичным путем [2241 был осуществлен однозначный синтез 4,6-ди"
окси-2-изопропилкумарана (XVII), как показано на приведенной ниже схеме.
О
СВН5СН2О —СОСН3 CH3Mgi
XXVI
О уСН2
С0Н5СН2О—V — С Ня и дебензилирование
I II II ХСН3---------------------™
\/
XXVII
Неидентичность полученного продукта изомерному 5,7-окси-2,2-диметилхро-
ману [2201 явилась окончательным доказательством строения хроманового
кольца.
О
но ОН С6Н6СН2О -|^|- ОН СВН6СН2О V У- СОСНз
1^ J- СНО NL СНО ----L!
! \ !
ОН ОСН2С6Н5 ОСН2СвН5
j СН3МК1
я
п2 И СВН5СН2О у У- (У
XVII --------------- ' ХСНз
Деоензилированис || ||
\/
бсн2с6н5
Реакция 7-бензилокси-4-метилкумарина (XXVIII) с метилмагнийиодидом
приводит к получению Д3-хромена (XXIX), который при озонолизе дал о-окси-
ацетофенои (XXX), что строго соответствует поведению 2,2-диметилхромено-
вой системы в аналогичных условиях.
* О О о сн3
СВНЬСН2ОY V сн;1мй1 свн5сн2о-Я>/Y
! il i ---------------------* ! I! гси»
VV Ч/\У
CH-, ch3
XXVIII XXIX
O3
V
CBH5CH2O - OH
Y1- соси.,
XXX
Ксантоксилетин. Ксантоксилетин (XXXI) содержится вместе с ксанти-
летином и аллоксантоксилетином в коре колючего ясеня Xanthoxylum ате-
ricanum Mill. Впервые он детально был исследован Гордином [2121, нашед-
шим, что это вещество содержит метоксильную группу, но не имеет ни окси-,
пи кетогрупп. Полное строение ксантоксилетина установлено Робертсоном
с сотрудниками [225, 2261.
X РОМЕНО-а-ПИРОН Ы
77
Метилирование его щелочным раствором диметилсульфата дает О-метил-
ксантоксилетиновую кислоту (XXXII), т. е. происходит обычное размыка-
ние а-пиронового кольца. С другой стороны, при щелочном расщеплении обра-
зуется ацетон и монометиловый эфир флороглюцина. Каталитическое гидри-
рование быстро приводит к насыщению реакционноспособного хроменового
кольца с образованием дигидроксантоксилетина (XXXIII), который легко пре-
вращается в О-метилдигидроксантоксилетиновую кислоту (XXXIV). Гидриро-
вание последней дает О-метилтетрагидроксантоксилетиновую кислоту (XXXV).
Окислительное расщепление ксантоксилетина щелочным раствором пер-
манганата калия приводит к образованию а-оксиизомасляной кислоты из гем-
диметильной группы хроменовой системы, а при осторожном озонолизе про-
исходит разрыв хроменового кольца с образованием о-оксиальдегида — апо-
ксантоксилетина (XXXVI). Каталитическое восстановление формильной
группы в соединении XXXVI дает дезоксиапоксантоксиксилетин (XXXVII,
R = Н), который при метилировании превращается в О-метилдезоксиапо-
ксантоксилетин (XXXVII, R = СН3). Кумарин (XXXVII, R = СН3) был
(CH3)2SO4
кон
— осн3
-СН=.СНСООН
осн3
XXXII
(CH3)2SOj
кон
Н3С /°х /<к
У у ОСН3
Нзс/
( сн=снсоон
ОСНз
XXXIV
I
ОСНз
XXXVI
Н3С ° /К
У у V оснз
НзС' I I
сн2сн2сооп
I
ОСНз
XXXV
.. О о z.
RO -У У у НО -у у он
н3с -у У Н3с -у- СНО
! I
ОСНз ОСН3
XXXVII XXXVIII
синтезирован из альдегида (XXXIX) путем превращения его реакцией с циан-
уксусной кислотой в кумарин (XL) и последующим декарбоксилированием.
Апоксантоксилетин не был синтезирован, и строение его было установлено
О Q
СН3О -А- он сн3о У у У
Н3сДу-СНО Н3С _у1У-СООН
ОСН., ОСНз
XXXIX XL
78
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
косвенно следующим образом. 7-Этокси-5-метокси-8-метилкумарин (XLI,
R Н) был приготовлен из альдегида (XLII) через кислоту (XLI, R =
СН3 СНз
I О О
С2Н5О - / V с2н5о он
II I ' р 111 с но
к ч/ н
ОСНз ОСНз
XLI XLII
= СООН). Обработка этого кумарина щелочным раствором диметилсульфата
дала 2,6-диметокси-4-этокси-3-метилкоричную кислоту (XLIII), идентичную
кислоте, полученной в аналогичных условиях из О-этилдезоксиапоксантокси-
летина. Строение этого эфира должно отвечать формуле XLIV, поскольку
СНз
I
С2Н5О -/Д-ОСН,
>
сн=снсоон
ОСНз
XLIII
ОСНз
XLIV
соединение, имеющее другое возможное строение,— 5-этокси-7-метокси-6-ме-
тилкумарин — при обработке щелочным раствором диметилсульфата должно
превращаться в 4,6-диметокси-2-этокси-3-метилкоричную кислоту (XLV). Сле-
довательно, строение апоксантоксилетина выражается формулой XXXVI,
СНз
4 НзС\ /Я,
СН3О ОСНз V > z4.- OR'
НзС'
4./- CII = CHCOOII ч R
ОС2Н5 ОСНз
XLV XLVI
из которой ясно следует линейное строение ксантоксилетина. Дальнейшее
доказательство строения апоксантоксилетина, а следовательно, и ксантокси-
летина было получено благодаря синтезу дезоксиапоксантоксилетина [2271
и этилового эфира (XLIV) исходя из 2,4-диокси-5-метил-6-метоксибензальде-
гида (XXXVIII) [2281 обычными методами.
Наличие 2,2-диметилхроменового ядра в ксантоксилетине было также
доказано озонолизом дигидроксантоксилетина)(ХХХШ), в результате кото-
рого образовался о-оксиальдегид; поскольку ксантоксилетин имеет линейное
строение (XXXI), то строение получающегося о-оксиальдегида должно отве-
чать формуле XLVI (R = СНО, R' = Н). Метилирование последнего дает
соединение XLVI (R = СООН, R' = СН3), которое окисляется в кислоту
(XLVI, R = СООН, R' = СНз) и затем декарбоксилируется в 5,7-диметокси-
2,2-диметилхроман (XLVII, R = Н). Последний был идентифицирован пре-
вращением в альдегид по методу Гаттермана (XLVII, R = СНО). Строение
R
Н3СЧ ° J
4 / ' z 0СН3
НзС-О
OCII3
XI.VI!
ХРОМЕ НО-а-ПИРОНЫ
79
8-формил-5,7-диметокси-2,2-диметилхромана (XLVII, R = СНО) было уста-
новлено на основе его неидентичности изомерному альдегиду (XLVI, R =
= СНО, R' = СН3). Наличие о-оксиформильной группировки в соединении
XLVI (R = СНО, R = Н) было показано далее конденсацией с рядом аце-
тофенонов, в результате которой образовались пирилиевые соли типа XLVIII
[229].
XLV111
Взаимодействие альдегида (XLVI, R СНО, R' = H) с циануксусной
кислотой и последующее декарбоксилирование продукта реакции дало дигид-
роксантоксилетйн.
Аллоксантоксилетин. Аллоксантоксилетин (XLIX) [229], ангулярный
изомер ксантоксилетина, содержится вместе с последним в коре колючего
ясеня и аналогично ведет себя при расщеплении. При щелочном расщеплении
О о ° О
сн3о сн3о
/\/\/' /\/\^
К U
ПзС/^СНз Н3С/чСНз
XLIX L
образуется ацетон и монометиловый эфир флороглюцина, а .гидрирование при-
водит к дигидроаллоксантоксилетину (L) благодаря преимущественному
восстановлению реакционноспособного хроменового кольца.
Наличие a-пиронового кольца в последнем соединении подтверждается
превращением его в О-метилдигидроаллоксантоксилетиновую кислоту, которая
легко восстанавливается в О-метилтетрагидроаллоксантоксилетиновую кис-
лоту (LI).
СН3О ОСН3 СНО
н3с О !
У-СН = СНСООН VVV-OH
Y н,<у ||
! О \/xY
\/ . I
Н3С/ХСН3 0СНз
LI LII
Озонолиз дигидроаллоксантоксилетина (L) дает 7-окси-8-формил-5-меток-
си-2,2-диметилхроман (LII), идентифицированный сравнением с образцом,
полученным синтезом, что подтверждает ангулярную структуру аллоксанто-
ксилетина. Взаимодействие альдегида (LII) с циануксусной кислотой и после-
дующее декарбоксилирование продукта реакции привели к дигидроалло-
ксантоксилетину; тем самым был завершен полный синтез этого соединения.
Ксантилетин. Ксантилетин (LIII) [214, 230] встречается в природе вместе
с аллоксантоксилетином и ксантоксилетином, от которых его трудно отделить,
и отличается от них лишь отсутствием метоксильной группы. Гидролитическое
80
соединения. Содержащие два гетеролтомл кислорода
расщепление щелочами дает резорцин и ацетон, а гидрирование приводит
к дигидроксантилетину (LIV). При щелочном метилировании ксантиле-
тина и дигидроксантилетина образуются соответственно О-метилксантилети-
новая и О-метилдигидроксантилетиновая кислоты. Обе при дальнейшем
Н3<\ Н3СЧ Ох
НзИ j' ' H3cq ) ! |
4/\Z\Z' VW
LIII LIV
гидрировании превращаются в О-метилтетрагидроксантилетиновую кисло-
ту (LV). Озонолиз ксантилетина дает апоксантилетин (LVI, R - СНО),
восстановление которого в дезоксоапоксантилетин (LVI, R = СН3), идентич-
ный 7-окси-6-метилкумарину, подтверждает линейное строение ксантилетина.
-ОСН3
- СН2СН2СООН
LV
LVI
Озонолиз дигидроксантилетина приводит к 7-окси-6-формил-2,2-диметилхро-
ману (LVIII), синтезированному по методу Гаттермана, исходя из 7-окси-
2,2-диметилхромана.
Н3С °
Н3С
/\z'-CHO
LVII
Альдегид (LVII) обычным путем был превращен в дигидроксантилетин,
в результате чего был завершен полный синтез последнего.
Ксантилетин синтезирован с плохим выходом [218] конденсацией 2-метил-
бутин-З-ола-2 с 7-оксикумарином (умбеллифероном) в соответствии с приве-
СН
денной схемой. Одновременное образование ангулярного изомера — сезе-
лина — ясно указывает на неоднозначность подобных синтезов.
Сезелин. Сезелин (LVIII) [215, 216] содержится в Seseli indicum Wight
и Агп. и в Skimmia japonica Thunb. Под действием смеси ледяной уксусной
и серной кислот разрушается хроменовое кольцо с образованием умбелли-
ферона, а при окислении перманганатом калия гещ-диметильная группа пре-
нооссн=нс
СНоО
LVIII LIX
вращается в а-оксиизомасляную кислоту. Озонолиз сезелина приводит к одно-
временному разрыву хроменового и а-пиронового кольца с образованием
резорцин-2,4-диальдегида, что подтверждает ангулярную структуру сое-
ХРОМЕ НО-а-ПИРОНЫ
81
динения LVIII. При щелочном метилировании сезелина образуется корич-
ная кислота (LIX).
Сезелин был получен конденсацией умбеллиферона и 2-метилбутин-3-ола-2.
Уже упоминалось (стр. 80) о конкурирующем образовании линейного сое-
динения — ксантилетина — и этот факт вместе с синтезом дигидроксантиле-
тина из умбеллиферона и изопрена [231] является дальнейшей иллюстрацией
двойственного характера этого метода синтеза.
Лувангетин. Лувангетин (LX) [232] содержится в плодах Luvanga scan-
dens Наш. вместе с изопимпинеллином, ксантотоксином и ксантилетином.
По своему химическому поведению лувангетин сходен с ксантоксилетином,
ОСН3
LX
от которого он отличается только положением метоксильной группы, являясь
производным пирогаллола, тогда как ксантоксилетин представляет собой
производное флороглюцина. Гидрирование дает последовательно дигидролу-
вангетин (LXI) и тетрагидролувангетин (LX1I).
LXII
Окисление азотной кислотой обоих — ди- п тетрагидросоединений приво-
дит к янтарной кислоте, а при осторожном озонолизе лувайгетина преимуще-
ственно расщепляется хроменовое кольцо с образованием 6-формил-8-меток-
сиумбеллиферона (LXIV, R = СНО), идентичного соединению, полученному
при озонолизе ксантотоксина (стр. 19). Окисление перманганатом калия
в щелочном растворе дает а-оксиизомасляную кислоту из гем-диметилыюй
группы хроменового ядра.
Лувангетин был синтезирован следующим образом (2331. Пирогаллол
метилировали в 2-метиловый эфир и по методу Гаттермана превращали в альде-
гид (LXIII). Циклизация альдегида по Перкину давала 8-метоксиумбеллифе-
рон (LXIV, R = Н), который превращался в лувангетин с очень низким
выходом при конденсации с 2-метилбутин-3-олом-2 в запаянной трубке. Сле-
дует отметить, что наличие 2,2-диметилхроманового кольца в дигидросезе-
сно
II I
НО он
I
ОСНз
LXIII
ОСН3
I О о
НО—
н J« I
к \/\Z
LXIV
лине и дигидролувангетине, по-видимому, не было отмечено, и большая часть
имеющихся аналитических и синтетических данных вполне совместима с фура-
нокумариновой структурой типа LXV. По аналогии с ксантилетином, ксанто-
6 Заказ .\и 9 78
82
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
ксилетином и т. и. для этих двух соединений наиболее вероятной является
хроменокумариновая структура. Работа Шмида (см. ниже), посвященная
выяснению строения виснагина, подтверждает эту точку зрения.
изСх о О о
НС__/ \^\/
Н,с/ ММ
\/\М
LXV
Представляют интерес биогенетические соотношения между соединениями,
содержащими а-изопропилфурановую группировку, такими, как эйпарин,
подакенетин и ротенон, и соединениями, содержащими 2,2-диметилхромено-
вую группировку,— роттлерином, ксантоксилетином, ксантилетином, токси-
каролом, дегвелином и пигментами маклюры оранжевой. Концевые фурано-
вое и хроменовое кислородсодержащие кольца в каждом типе соединений тео-
ретически могут быть построены путем присоединения изопреновой группы
к фенольной системе. Только различием в месте присоединения потенциаль-
ного гетероатома определяется образование пяти- или шестичленного кольца.
Эта точка зрения более полно рассмотрена Уолли [234].
Виснаганы. Кроме описанных выше кристаллических производных фура-
иохромонов (стр. 36), из семян Ammi visnaga Linn, путем разделения аморф-
ной фракции [235—238], названной виснаганом, были выделены три других
вещества, не обладающие этим строением: самидин С21Н22О7, дигидросамидин
С21Н24О7 и виснадин С21Н24О7. Эти соединения (виснаганы) были охарактери-
зованы Шмидом и его сотрудниками [239—241], а впоследствии Смитом и дру-
гими авторами [242, 243] как производные 2,2-диметилхроменокумаринов.
Гидролиз самидина, дигидросамидина и виснадина дает уксусную кислоту
и один и тот же келлактон (LXVI), а также соответственно сенециевую, изо-
валериановую и а-метилмасляную кислоты. Строение келдактона установлено
дегидратацией его в хромонокумарин (LXVII), сопровождающейся восстанов-
лением в дигидросезелии (LXVIII). Окисление а-дигликольной части иодной
кислотой дает диальдегид (LX1X), легко гидролизующийся в умбеллифе-
рон-8-альдегид (LXX), который образуется вместе с муравьиной кислотой
и ацетоном. При обработке горячим спиртовым раствором щелочи виснаганы
образуют сенециевую, изовалериановую и L-a-метилмасляную кислоты наряду
с обычными продуктами расщепления — уксусной кислотой и продуктом
этанолиза, соединением LXX1. Эту реакцию представляют как отщепление
О-алкильной группы бензилацетата с последующим образованием этилового
эфира [244]. На этом основании строение самидина, дигидросамидипа и вис-
падина соответственно выражается формулами
/ ZCH3\ ( /СНз\
I.XXII R = CO—СН = С/ , LXXII R = COCH2CH
\ 'СНд/ у хсн3/
и LXXII /R = COCHCHCH3\ .
1
\ СН3 /
Хотя стереохимия келлактона еще неполностью выяснена, вполне вероятно,
что a-дигликольная группировка в нем имеет цис-конфигурацию [241].
Синтез DL-m/шнс-самидина из сезелина был осуществлен по схеме, при-
веденной на стр. 83 [241].
ХРОМЕЙО-а-ПИРОНЫ
83
Калофиллолиды. Орехи Calophyllum inophyllum [245, 2461 содержат три
кристаллических вещества — калофиллолид (С2вН24О5, LXXIII), кало-
филловую кислоту (С25Н24О0, LXXIV) и инофиллолид (С25Н22О5, LXXV).
При щелочном расщеплении калофиллолида образуются ацетон (из хроме-
нового ядра), ацетальдегид и пропионовая кислота (в результате процесса,
обратного альдольной конденсации остатка тиглиновои кислоты), а также
5-окси-7-метокси-4-фенилкумарин (LXXVI). Под действием фосфористой и
иодистоводородной кислот образуются тиглиновая кислота и хроманокума-
рин (LXXVII, R = Н и R = СН3) [247]. Оба этих соединения были синте-
зированы путем ацилирования соединения LXXVI хлорангидридом р,р-ди-
метилакриловой кислоты и последующей перегруппировки по Фрису полу-
ченного сложного эфира, сопровождающейся циклизацией предполагаемого
промежуточного продукта в хроманокумарин (LXXVII, R = СН3). При
6*
84
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
обработке калофиллолида хлористым алюминием в нитробензоле происходит
циклизация тиглиновой боковой цепочки и одновременно раскрытие кума-
ринового кольца с образованием калофилловой кислоты (LXXIV). Это необычно
устойчивая ^uc-о-оксикоричная кислота, стабильность которой обусловлена
образованием хелатного комплекса карбонильной и гидроксильной группами,
как показано на схеме. Калофилловая кислота путем лактонизации кипяче-
нием с уксусным ангидридом может быть превращена в инофиллолид, а послед-
ний в свою очередь может быть вновь переведен в калофилловую кислоту дей-
ствием разбавленной щелочи.
LXXIX
LXXV1II
При гидрировании инофиллолида восстанавливается хроменовое кольцо
и образуется дигидроинофиллолид (LXXVIII), полученный также перегруп-
пировкой по Фрису сложного эфира тиглиновой кислоты (LXXIX) и хромано-
кумарина (LXXVII, R = Н). Неустойчивое промежуточное соединение пре-
вращается в дигидроинофиллолид.
ХРОМЕНО-у-ПИРОН Ы
Введение. Описаны свойства нескольких представителей из числа встре-
чающихся в природе соединений, имеющих два кислородсодержащих кольца,
которые образуют хромено-у-пироновое ядро.
X POME HO-Y-ПИ РОНЫ
85
Цитромицетин (I, R СООН) — продукт метаболизма различных видов
Citrotnyces — единственный из известных представителей этого ряда, имею-
щий ангулярное строение, содержит два конденсированных шестичленных
кислородсодержащих цикла. К нему тесно примыкаютпирилиевые соли типа II
(стр. 147), являющиеся синтетическими продуктами. Ангидрофульвиновая
кислота (1а), легко получаемая из продуктов обмена веществ грибов, и фуль-
виновая кислота (16) обладают линейной структурой.
Ямаицин, якарейбин и пигменты маклюры оранжевой — осайин и поми-
ферин — содержат хромено-у-пироновый скелет типа III. Вообще говоря,
осайин и помиферин проявляют в реакциях характерные свойства 2,2-диме-
тил-Д3-хроменовой и у-пироновой систем, тогда как свойства колец цитро-
мицетина и фульвиновой кислоты довольно специфичны. Свойства и реакции
дигидроротенонов и родственных им веществ (Ша), содержащих «обращенное»
цитромицетиновое ядро, более подробно будут рассмотрены в разделе, посвя-
щенном хроманохроманонам (стр. 115), которые являются единственными
синтетическими представителями этого ряда.
В заключение речь пойдет о химии роттлерина, включение которого
в обзор является вполне уместным, поскольку многие из продуктов его пре-
вращения представляют собой хромено-у-дигидропироны, хотя сам роттле-
рин содержит только 2,2-диметил-Д3-хроменовую систему.
Цитромицетин, фульвиновая кислота. Цитромицетин (I, R = СООН)
С14Н10О7 — продукт обмена веществ грибов, обладающий кислотными свой-
ствами, впервые был выделен Хезерингтоном и Райстриком [2481 из мета-
болитной жидкости некоторых образцов Cytrotnyces, выросших на питатель-
ной среде, содержащей либо глюкозу, либо глицерин в качестве источника
углерода. После детального исследования с целью нахождения оптимальных
условий образования цитромицетина, указанные авторы начали изучение
О
СН3О — \
CH3o4Jl !WO
R О |
I
CH3
IV
«6 СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ. ДВА ГГ.ТЕРОАТОМЛ КИСЛОРОДА
этого вещества и его производных, показав, что оно является диоксикислотой,
образующей диацетат, простой диметиловый эфир и диметиловый эфир слож-
ного метилового эфира (IV, R — СООСН3). При нагревании с разбавленной
серной кислотой цитромицетин декарбоксилируется в двухатомный фенол
цитромицин (1, R Н), образующий диацетат и диметиловый эфир. Те же
авторы 1248] показали, что цитромицетин содержит оксихинольный цикл,
а метил-О-диметилцитромицетин (IV, R = СООСН3) при щелочном гидро-
лизе образует диметоксиоксифталевую кислоту, являющуюся, вероятно,
3-окси-5,6-диметоксифталевой кислотой (V). Хезерингтон и Райстрик выде-
лили также продукт щелочного расщепления, который, по их предположе-
нию, представлял собой либо 4-окси-6,7-диметокси-3-метилкумарин (VI), либо
же изомерный 3-окси-6,7-диметокси-2-метилхромон (VII).
сн3о - А|- он
сн3осоон
I
соон
V
О О о
сн3о-/у у сн3о-^у\-си3
сн3о.уу J-СНз CH3O-NNI-OH
I II
он о
VI VII
о
СП3О - У |Х у СОСН3
ch3o-L 1—Lo
VIII
Вслед за этим, Робертсон, Уолли и их сотрудники 1249—255] обстоя-
тельно исследовали химию и строение цитромицетина и подтвердили наличие
в нем скелета 2,4,5-триоксифталевой кислоты, а также приписали ему струк-
туру V [249] на основании синтеза с применением обычных методов. Далее
было показано, что вторичный продукт щелочного расщепления метил-О-диме-
тилцитромицетина не идентичен полученным в этих условиях соединениям VI
и VII или же изомерному 5,6-диметокси-2-ацетилкумаранону-3 (VIII) [250]
и является, по всей вероятности, случайным образованием.
На основании исследований Робертсона и Уолли цитромицетину при- '
писана структура 1 (R = СООН), а цитромицину — формула I (R = Н).
Для исследования продуктов расщепления был выбран О-диметилцитро-
мицнн. Это соединение обладает сильными основными свойствами и дает ряд
солей — бромгидрат, хлоргидрат, HFeCl4, а также имеет один активный атом
водорода и одну связанную с атомом углерода метильную группу. Это сое-
динение не содержит реакционноспособной двойной связи и не дает произ-
водных с обычными реагентами на гидроксильную и карбонильную группы.
Метильная группа, связанная с атомом углерода, конденсируется с пиперо-
налем в присутствии этилата калия с образованием стирилпроизводного (XVI)
(ср. аналогичное поведение 2-метилхромонов). Окисление О-диметилцитро-
мицина хромовой кислотой дает лактон (IX, R = Н) — О-диметилцитроми-
цинон. Последнее соединение образуется вместе с промежуточным продуктом
окисления — О-диметилцитромицинолом (XIII, R = Н) в том случае, если
окисление проводится озоном. Дальнейшее окисление О-диметилцитро-
мицинола (XIII, R = H) озоном или хромовой кислотой приводит к О-диме-
тилцитромицинону. При окислении О-диметилцитромицина тетраацетатом
свинца с хорошим выходом образуется только О-диметилцитромицинол.
Выводы о строении О-диметилцитромицина и, следовательно, цитроми-
цина и цитромицетина почти полностью основываются на реакциях О-диме-
тилцитромицинона и О-диметилцитромицинола; наиболее полное понимание
сложной химии этого соединения было получено из рассмотрения реакций
обоих этих веществ.
ХРОМЕНО-у-ПИРОНЫ
87
При исследовании методом Куна — Рота найдено, что О-диметилцитро-
мицинон (IX, R = Н) содержит С-метильную группу. Окисление пиперо-
нилиденового производного (ХУ1).О-диметилцитромицина дает стирилпроиз-
водное 0-диметилцИтромицин.она (XVII). Таким образом, С-метильная группа
в у-пироновом кольце не является той метиленовой группой, которая при
окислении О-диметилцитромицина дает О-диметилцитромицинол (XIII, R = И)
ОН
XV
п О-диметилцитромицинон (IX, R = Н). Характер расщепления О-диметил-
цитромицинона определяется его структурой, которая, за исключением бен-
зольного кольца, включает действительно существующие или потенциальные
88
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
метиленовые группы, чередующиеся с карбонильными. При каталитическом
гидролизе раскрывается лактоновое кольцо с образованием оксикислоты,
которая, будучи ^uc-о-оксикоричной кислотой, легко дегидратируется, пре-
вращаясь в соединение IX (R — Н). О-Диметилцитромицинон и оксикисло-
та (XVIII) реагируют с анилином, образуя один и тот же анилид (XIX) и моно-
оксим, строение которого, вероятно, выражается формулой XXI. Дпоксиму,
который также может быть получен, пока еще невозможно приписать опре-
деленного строения. Гидролитическое расщепление О-диметилцитромиципона
горячей щелочью дает кислоту (X), ацетофенон (XI), 4-окси-6,7-диметокси-
кумарин (XII), 3-ацетоацет11л-4-оксп-6,7-диметоксикумар11п (XV), ацетон
и уксусную кислоту.
СН3()
сп3о
ххш
При кислотном гидролизе О-днметилцитромицинона происходит размы-
кание у-пиронового кольца с образованием в качестве основного продукта
ацетоацетилкумарина (XV), находящегося, по-видимому, в равновесии с сое-
динением ‘IX в кислом растворе. Размыкание у-пиронового кольца в этих-
условиях происходит легче, вероятно, вследствие того, что атом кислорода
в у-пироновом кольце обладает отчасти ангидридным характером из-за нали-
чия в положении 4 потенциального 4-оксикумаринового кольца гидроксильной
группы, являющейся винилогом кислотного гидроксила. (Ср. поведение пира-
нового кольца в пиранокумаринах, производных 3,3'-метилен-бш?-4-окси-
кумаринов, стр. 62.) Устойчивость О-диметилцитромицина в аналогичных
условиях подтверждает эту точку зрения. З-Ацетоацетилкумарин (XV) легко
циклизуется под действием концентрированной серной кислоты, образуя
О-диметилцитромнцИнон; этот факт наряду с некоторыми другими данными,
рассматриваемыми ниже, подтверждает ангулярное строение (IX, R = Н)
О-диметилцитромицинона. Этим отвергается возможная линейная структу-
ра XX, в пользу которой, однако, говорят некоторые другие факты, получен-
ные при изучении расщепления О-диметилцитромицинона. В соответствии
с приписываемой структурой кислотный гидролиз его приводит к соедине-
нию XV, гидролитическое расщепление которого щелочами дает ожидаемые
продукты — кислоту (X), кетон (XI), 4-оксикумарин (XII), ацетон и уксус-
ную кислоту. Эти продукты могут быть также получены в аналогичных усло-
виях из 3-ацетил-4-окси-6,7-диметоксикумарина (XXII). Последнее, однако,
неспособно к циклизации в соединение IX (R = Н) и его синтез несколь-
кими взаимно подтверждающимися и однозначными методами исключает эту
возможность. Синтез соединения XV еще не закончен, однако близкое сов-
падение между аналитическими данными для соединений XXII и XV, которое
X POME HO-Y-ПИ РОНЫ
89
сначала вносило некоторую путаницу, делает установление структуры сое-
динения XV путем однозначного синтеза соединения XXII очень важным.
Для синтеза 3-ацетил-4-окси-6,7-диметоксикумарина (XXII) были исполь-
зованы следующие три метода, которые привели к получению одного и того же
продукта [254].
1. Конденсация подходящего о-ацетоксихлорангидрида с ацетоуксусным
эфиром:
СН3О ОСОСН3 СООС2Н5
-1-1
СН3О-^—СОС1 СН2СОСН3
о о
сн3о у у у
СН3О -MyL СОСНз
I
он
XXII
СН3О—
СН3О -
УОСОСНд СООС2Н5
ч (I СНСОСНз
ч/ \ СО /
XXIII
2. Циклизация натриевого производного 2-окси-4,5-диметоксибензоил-
ацетона под действием фосгена:
СН3О—ОН С1-СО Гсн,о ОН СОС1
СН3О —У - 00CHNaC0CH3 Cl СН3О —j-----------СОСНСОСН3
ХХЦ XXIII
3. C-Ацетилирование 4-окси-6,7-диметоксикумарина уксусным ангидри-
дом в присутствии трехфтористого бора:
О О
СН3О— /У у (сн3со)г0
fH->0_I I БРз
I
OH
CH3O
XXII
CH3O
Из этих трех методов только третий является абсолютно однозначным. Мето-
ды 1 и 2 менее определенны и легко могут привести к изомерной хромоновой
кислоте (XXIII) в результате циклизации предполагаемого промежуточного
соединения другим возможным путем. Установить различие между соедине-
ниями XXII и XXIII оказалось трудным, потому что З-ацил-4-оксикумарины
растворимы в бикарбонате натрия, т. е. обнаруживают кислые свойства,
а реакционноспособная метильная группа в З-ацил-4-оксикумаринах дает
пиперонильные производные, напоминая в этом отношении 2-метилхромоны.
Разделение этих соединений было осложнено также тем, что З-ацетил-4-окси-
6,7-диметоксикумарин не реагирует с 2,4-динитрофенилгидразином и другими
реагентами на карбонильную группу.
Подтверждением З-ацетил-4-оксикумариновой структуры у соединений,
полученных тремя указанными способами, служил синтез изомерной хромо-
новой кислоты (XXIII) [254]. Конденсация 2-окси-4,5-диметоксиацетофе-
нона со щавелевым эфиром дала сложный глиоксиловый эфир (XXIV), строе-
90
СОЕДИНЕНИЯ- СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
ние которого было установлено путем циклизации его в соединение XXV
действием кипящего спиртового раствора соляной кислоты с последующим
декарбоксилированием в известный 6,7-диметоксихромон. Циклизация соеди-
нения XXIV под действием ацетата натрия и уксусного ангидрида привела
к смеси З-ацетил-2-карбэтоксихромона (XXVI) и изомерного этилового эфира
2-метилхромоп-З-глиоксалевой кислоты (XXVII). Эти вещества легко уда-
лось различить по их отношению к теплой концентрированной серной кислоте.
сн3о -А- он
СН3О сосн3
(СООС2Н5)2
СН3О -А- ОН
СН3О — I I’— СОСН2СОСООС2Н5
сн3о -/у у СООС2Н5
сн3о У у У сосн3
CHsCOONa
(СН3СО)2О
о
сн3о—у/ Y- соон
cco-’J!,)
о
о
сн3о-/у у сн3
СН3О У / сосоос2н5
"||
о
I
XXIII
Сложный эфир (XXVI) в этих условиях гидролизуется в соответствующую
кислоту, тогда как гидролиз изомерного сложного эфира (XXVII) протекает
с одновременным отщеплением окиси углерода и образованием 2-метил-6,7-
диметоксихромонкарбоновой-3 кислоты (XXIII), которая резко отличается
от изомерного З-ацетил-4-оксикумарина (XXII).
Ясно, что дополнительные доказательства, полученные при расщеплении
О-диметилцитромицинона, не позволили сделать выбора между возможной
линейной и ангулярной структурами — IX (R = Н) и XX —для этого
соединения. Окончательное доказательство было получено в результате изу-
чения реакций расщепления О-диметилцитромицинола (XIII, R = Н).
Дальнейшие данные о положении легко окисляемой метиленовой группы
в О-диметилцитромицине были получены из наблюдений, что О-диметилцитро-
мицинол (XIII, R = Н) является пиранольным основанием, дающим хорошо
определяемые пикраты и хлоргидраты типа XIV. Образование пиранольного
основания при окислении метиленовой группы заставляет предположить,
что последняя находится в шестичленном кислородсодержащем кольце; тем
самым совершенно исключается вероятность того, что окисление идет по С-
метильной группе хромонового ядра. Кроме того, С-метильная.группа 2-метил-
хромонов не окисляется тетраацетатом свинца в тех условиях, в.которых про-
водят окисление О-диметилцитромицина в О-диметилцитромицинол [256].
При кипячении О-диметилцитромицинилхлорида (XIV, X = С1) с мети-
ловым спиртом получается почти с количественным выходом триметиловый
эфир. Аналогичным образом при действии этилового спирта образуется про-
стой эфир карбинольного основания, что является хорошо известной харак-
теристикой карбинольных оснований [257]. Расщепление О-диметилцитро-
мицинола и карбинольного метилового эфира, изученное в самых различных
условиях в общем дало одни и те же результаты. Гидролиз горячим разбав-
ХРОМЕНО-Т-ПИРОНЫ
91
ленным раствором едкого натра приводит к кислоте (X), кетону (XI), уксусной
и муравьиной кислотам и ацетону. При нагревании с баритовой водой наряду
с этими соединениями образуется 2-окси-4,5-диметоксибензоилуксусная кис-
лота (XXX), которая легко циклизуется в 4-оксикумарин (XII) при раство-
рении в концентрированной серной кислоте. Основным продуктом при рас-
щеплении теплым разбавленным метанольным раствором едкого натра является
3-формил-4-окси-6,7-диметоксикумарин (XXVIII), получающийся из О-диме-
тилцитромицинола с промежуточным образованием соединений XXXI и XXIX.
Наличие формильной группы в соединении XXVIII подтверждено образова-
нием анила и конденсацией с резорцином, ведущей к образованию пирилий-
хлорида (XXXV). Дальнейшее доказательство строения соединения XXVIII
получено восстановлением его в 3-метил-4-окси-6,7-диметоксикумарин (XXXVI)
и гидролитическим расщеплением под действием щелочи в муравьиную и бен-
зоилуксусную (XXX) кислоты и ацетофенон (XI), тогда как кислотное рас-
щепление дает муравьиную кислоту и 4-оксикумарин (XII).
Расщепление О-диметилцитромицинола (XIII, R = Н) щелочью в очень
мягких условиях приводит к получению наряду с другими веществами сое-
динений XXXII (R = Н) и XXXIII. Последнее не является кислотой, дает
XXXIV
92 СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
цветную реакцию со спиртовым раствором хлорида железа (III) и образует
производное изоксазола, что указывает на наличие 1,3-дикарбонильной груп-
пировки. Расщепление щелочью дает ацетон, ацетофенон (XI) и 4-оксикума-
рин (XII), образующийся при циклизации фрагмента бензоилуксусной кис-
лоты (XXX). Растворение в концентрированной серной кислоте привело
к образованию 2-ацетонилхромона (XXXIV).
Другое соединение (XXXII, R = Н) в соответствии с его формулой
не дает реакции с хлоридом железа (III), не образует карбонильных производ-
ных и, таким образом, легко отличается от трикетона и ацетонилхромона
(XXXIV), которому оно изомерно. Это соединение может образоваться из
О-диметилцитромицинола с промежуточным получением соединения XXXI
и с последующим отщеплением формильной группы. Ацетилирование трике-
тона (XXXIII) дает ацетат (XXXII, R = СН3СО), идентичный ацетату
у-пирона (XXXII, R = Н), а дезацетилирование ацетатов, полученных раз-
личными способами, приводит к одному и тому же у-пирону (XXXII, R = Н).
Эти превращения О-диметилцитромицинола и характер продуктов его
расщепления в сочетании с образованием муравьиной кислоты и 3-формил-
4-оксикумарина (XXVIII), а также реакции О-диметилцитромицинона, дают
полное основание считать, что молекулы О-диметилцитромицина, а следова-
тельно, и цитромицина имеют ангулярное строение IV (R = Н) и I (R = Н).
Дальнейшее подтверждение наличия у О-диметилцитромицинона у-пироно-
кумариновой структуры было получено при конденсации 4-окси-6,7-диметок -
сикумарина и ацетоуксусного эфира, в результате чего был получен а-пиро-
нокумарин (XXXVII). Он оказался изомерным (но не идентичным) О-диме-
тилцитромицинону и легко от него отличался благодаря нерастворимости
в кислотах, неспособности образовывать соли, а также наличию у него свойств,
присущих а-пиронокумаринам.'
XXXV11
Подтверждение аннулярного строения этих веществ синтетическим путем
получено [258] конденсацией 6,7-диметоксихроманона (XXXVIII) с ацетил-
ацетоном (XXXIX) под действием хлорокиси фосфора, в результате чего обра-
зуется ппранольное основание (XL), идентичное продукту, полученному при
СН3О-
СН3о-
XXXVIII
сн3
I
со
С Но
I
со
I
сн3
о
XXXIX XL
действии метилмагнийиодида на О-диметилцитромицин. Окисления метилено-
вой группы в соединениях типа О-диметилцитромицина ранее не наблюда-
лось, однако реакционноспособность метиленовой группы в ядре обращенного
типа, содержащемся в ротеноне (стр. 129), хорошо известна. Озонолиз про-
текает, по-видимому, однозначно, и, вероятно, по свободнорадикальному
ХРОМЕНО-Т-ПИРОНЫ
93
механизму, что подтверждается окислением О-диметилцитромицина тетра-
ацетатом свинца. Для последней реакции свободнорадикальный механизм
надежно установлен.
СН3О -fy - он
СН3О-^- СООН
соосн3
XLI
СН3О J -СОСН,СОСН3
СН3ООС он
XLIII
XLII
Кажется весьма вероятным, что цитромицетин отличается от цитромицина
только наличием карбоксильной группы, что подтверждается образованием
кислого сложного эфира фталевой кислоты (XLI) при окислении метилового
эфира О-диметилцитромицетина (IV, R = СООСНз) перманганатом калия
и опытами по щелочному расщеплению. Тем не менее не исключена вероят-
ность более глубоких изменений при декарбоксилировании. Однако в рабо-
те [259], посвященной выяснению этого вопроса, показано, что формулы
IV (R = СООСНз) для метилового эфира О-диметилцитромицетина и, следо-
вательно, I (R = СООН) для цитромицетина правильны.
Окисление метилового эфира О-диметилцитромицетина озоном затрудни-
тельно, но окисление тетраацетатом свинца приводит к О-дидоетилцитромице-
тинолу (XIII, R = СООСН3), который далее может быть превращен в метило-
вый эфир О-диметилцитромицетинона (IX, R = СООСНз) действием хро-
мовой кислоты. Последний можно получить непосредственно из соединения
IV (R = СООСН3) при действии хромовой кислоты. Реакционноспособная
С-метильная группа в у-пироновом кольце образует стирильное производное,
окисляющееся хромовой кислотой в стирильное производное метилового
эфира О-диметилцитромицинона, что указывает на неидентичность С-метиль-
ной группы и метиленовой группы в О-диметилцитромицетине, реакционно-
способной по отношению к окислителям.
Гидратация соединения IX (R = СООСН3) дает неустойчивую ццс-о-окси-
коричную . кислоту (XLII, R = ОН), образующую анилид (XLII, R =
= NHC6Hj), идентичный анилиду, полученному непосредственно из соеди-
нения IX (R — СООСН3). Кислотный гидролиз метилового эфира О-диме-
тилцитромицетинона приводит к размыканию концевого у-пиронового кольца
с образованием З-ацетоацетил-4-оксикумарина (XLIII), легко дегидратирую-
щегося в исходное соединение.
Изомерные гидраты XV и XVIII, производные О-диметилцитромицина,
и XLIII, и XLII (R = ОН), производные метилового эфира О-диметилцитро-
мицетина, не трудно отличить друг от друга. Кислоты [XVIII и XLII (R =
= ОН)] легко растворимы в растворе бикарбоната натрия и в минеральных
кислотах, благодаря солеобразующим свойствам у-пйронового кольца, тогда
как 3-ацетоацетильные производные (LXV и LVIII) растворимы в бикарбо-
нате натрия лишь с трудом, не растворяются в минеральных кислотах, но обра-
зуют карбонильные производные.
94
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Щелочное расщепление метилового эфира О-диметилцитромицинола, кото-
рый, будучи пиранольным основанием, дает хорошо определяемые пикрат, хлор-
гидрат и хлорплатинат, приводит к образованию муравьиной кислоты в соот-
ветствии с его ангулярным строением XIII (R = СООСН3). Основным про-
дуктом щелочного расщепления метилового эфира О-диметилцитромицетинона
является вещество состава С13Н12О6, которому первоначально приписывали
строение XLIV. Оно образуется в результате циклизации промежуточного
соединения XLV. Однако структуру индандиона-1,3 окончательно доказать, что это вещество имеет (XLVI) не удалось [260]. • СН3О— у-ОН
О CH3O-/V \-сн3 CH3°~4JxzO СН3О-^\ он у/
СНз°ЛЛ/ 1 II НООС О XLIV СНЭО -\J-I- СОСН2СОСНЭ 6—СН । (Z 1 СООН СОСНд XLV XLVI
Фульвиновая кислота С14Н12О8 была впервые выделена Райстриком
и сотрудниками как метаболит Penicillium griseofulvum Dierckx [261] и интен-
сивно исследовалась Дином с сотрудниками [260], приписавшими ей строе-
ние XLVII (R = Н). В соответствии с таким строением — полуацеталя,
I II
ноос о
XLVII
ноос о
XLVIII
находящегося в «замаскированном» сопряжении с а, (3-ненасыщен ной карбо-
нильной группой, фульвиновая кислота легко дегидратируется в ангидро-
фульвиновую кислоту (XLVIII). Катализируемое кислотами и основаниями
присоединение элементов воды или спиртов к ангидрофульвиновой кислоте
снова дает фульвиновую кислоту (XLVII, R = Н) или ее соответствующие
производные (XLVII, R = алкил).
Метилирование фульвиновой кислоты приводит к метиловому эфиру
О-диметилангидрофульвиновой кислоты, в котором С-метильная группа в кон-
цевом кольце обладает повышенной реакционной способностью по отношению
к ароматическим альдегидам и к образованию стирильных производных. Мети-
ловый эфир О-диметилангидрофульвиновой кислоты окисляется во фтале-
которая получается из метилового эфира
СН2ОН —> Фульвиновая кислота
1
СО —СН
I
СОСН2СОСН3
вую кислоту (XLI), идентичную той,
НО ОН
Цитромицетин <—
НО— I II
XLIX
О
сн3о-/у \/Ч_сн3
СН,0_' II II
СН3ООС О °
L
з
LI
СН3ООС о
ХРОМЕНО-Т-ПИРОНЫ
95
О-диметилцитромицетина (стр. 93), и гидролизуется щелочами в 2-ацетил-
индандион-1,3 (XLVI), идентичный полученному из метилового эфира О-диме-
тилцитромицетинона. Таким образом, индандион-1,3, несомненно, образуется
случайно в результате самопроизвольной циклизации промежуточного сое-
динения XLV так же, как это происходит и в случае цитромицетина 12601.
Хотя ни один из продуктов расщепления фульвиновой кислоты и ее производных
не содержит хромоновой группировки, тем не менее свойства фульвиновой
кислоты, а также и цитромицетина (стр. 93) полностью подтверждают при-
писанную ей структуру XLVII (R = Н).
Хотя фульвиновая и ангидрофульвиновая кислоты не обнаруживают
свойств карбонильных соединений, тем не менее метиловый эфир О-диметил-
ангидрофульвиновой кислоты с гидроксиламином образует N-окись пири-
дина (L), получающуюся также из соединения LI при окислении воздухом
(ср. работу Роджерса [262]).
Примечательно, что и цитромицетин и ангидрофульвиновая кислота
(а следовательно, и фульвиновая кислота) могут быть получены исходя из одно-
го и того же гипотетического промежуточного соединения XLIX.
Осайин, помиферин и родственные соединения. Желтые пигменты осайин
(I, R = Н)С25Н24О5 и помиферин (I, R = ОН) С26Н24Ов [263—270] содер-
жатся в маклюре оранжевой Maclura pomifera Raf. Изофлавоновое ядро в этих
соединениях сконденсировано с 2,2-диметил-А3-пирановым кольцом, и, кроме
того, имеется другой потенциальный 2,2-диметилпирановый цикл. Они являют-
ся немногими представителями природных веществ, имеющих подобное строе-
ние, и их сопоставление с калофиллолидами (стр. 83) представляет большой
интерес. Эти соединения не содержат метоксильной и реакционноспособной
карбонильной групп, оптически неактивны и не дают аддуктов с малеиновым
ангидридом.
Обработка диазометаном ' или щелочным раствором диметилсульфата
в мягких условиях приводит к метилированию оксигрупп только в фенильном
радикале с образованием монометилового эфира осайина (II) и диметилового
эфира помиферина (III). Оба эти вещества дают зеленую цветную реакцию
со спиртовым раствором хлорида железа (III). Положение гидроксильной
и карбонильной групп в положениях 5 и 4 эквивалентно их орто-положению.
Эта эквивалентность выявляется по устойчивости к дальнейшему метилиро-
ванию монометилового эфира осайина и диметилового эфира помиферина,
которые только в жестких условиях при применении диметилсульфата в кипя-
щем ацетоне или йодистого метила в горячем ацетоне дают соответственно
диметиловый эфир осайина (IV) и триметиловый эфир помиферина (V).
\ ° \ °
0СНз 0СНз
I II I II
но О СН3О О
II IV
96
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
ОСНз
_ J
ОСНз
ш
\ / \ ОСНз
U! S-осн’
I II
СН3О о
На наличие одной реакционноспособной гидроксильной группы в осайине
и двух в помиферине указывает также легкое образование моноацетата осай-
ина и диацетата помиферина при обработке уксусным ангидридом в пиридине
на холоду. Ацетилирование осайина или его моноацетата в жестких условиях
ацетатом натрия и уксусным ангидридом дает диацетат осайина, а помиферин
или его диацетат при подобной обработке образуют триацетат. В тех же усло-
виях нереакционноспособная гидроксильная группа в монометиловом эфире
осайина и в диметиловом эфире помиферина может быть проацетилирована
с образованием ацетата О-монометилового эфира осайина и ацетата О-диме-
тнлпомиферина. Эти превращения можно представить в виде следующей схемы:
Помиферин (CH3)2SO4^ О-Диметиловый эфир помиферина
Осайин на холоду О-Монометиловый эфир осайина
(CH3)2SO4 I (CH3)2SO4
в кипящем в кипящем
ацетоне I ацетоне
О-Триметиловый эфир помиферина
О-Диметиловый эфир осайина
CH3COONa, (СН3СО)2О
V
Ацетат О-диметилового эфира помиферина
Ацетат О-монометилового эфира осайина
Окисление диметилового эфира осайина и триметилового эфира поми-
ферина перекисью водорода в щелочной среде дает соответственно анисовую
и вератровую кислоты из изофлавонового кольца молекулы; при этом про-
исходит раскрытие гетероциклических колец (ср. свойства фурано-а- и фура-
но-у-пиронов).
Н3С Н3С
/|/Х| /Г ।
Н3С О I о Н3с о I о
\^\/\ \^\/ \
но о ™2но о
с сн
/\ /\
Н3С сн3 Н3С сн3
VI VII
VIII
ХРОМЕНО-Т-ПИРОНЫ
97
При гидрировании осайина в этиловом спирте под платиновым катали-
затором при прекращении реакции после поглощения одной молекулы водо-
рода в результате восстановления хроменового кольца образуется дигидро-
осайин (VI, R = п-НОС6Н4). Следовательно, реакционная способность двой-
ной связи в 2,2-диметилхроменовом кольце даже выше, чем реакционная спо-
собность двойной связи в у,у-диметилаллильной боковой цепи. Дальнейшее
гидрирование осайина или дигидроосайина приводит к тетрагидроосайину
(VII, R = п-НОС6Н4), в котором хроменовое кольцо превратилось в хро-
мановое, а у,у-диметилаллильная боковая цепь также восстановлена. Конеч-
ным продуктом гидрирования является гексагидроосайин. Последний пред-
ставляет собой, по-видимому, изофлаванон (VIII), хотя сравнительно большая
легкость частичного восстановления изофлавонового кольца в изофлавононовое
довольно поразительна и, вероятно, обусловлена активирующим влиянием
других заместителей.
Различие в реакционной способности двух гидроксильных групп, наблю-
даемое в случае осайина, характерно и для продуктов его восстановления:
в пиридине и уксусном ангидриде образуются соответственно моноацетаты
дигидроосайина, тетрагидроосайина и гексагидроосайина из ди-, тетра- и гекса-
гидроосайина. В жестких условиях ацетилирования ацетатом натрия и уксус-
ным ангидридом ди-, тетра- и гексагидроосайины или их моноацетаты пре-
вращаются соответственно в диацетаты дигидро-, тетрагидро- и гексагидро-
осайинов. Эти превращения иллюстрируются следующей схемой:
н2
Осайин----------> (1) Днгидроосайин
(2) Тетрагидроосайин
CH3COONa
(СН3СО)2б"(3) Гексагндроосаинн
I (СН3СО)2О,
w ф пиридин
(1) Диацетат дигидроосайина
п „ CHgCOONa
(2) Диацетат тетрагидроосаиина <=—.----
(СН3СО)2О
(3) Диацетат гексагидроосайина
(1) Моноацетат дигидроосайина
(2) Моноацетат тетрагидроосайина
(3) Моноацетат гексагидроосайина
Помиферин при гидрировании ведет себя аналогично осайину, образуя
первоначально дигидропомиферин (в результате восстановления хроменового
кольца) [VI, R = 3,4-(НО)2С6Н3], дающий ди- и триацетаты. При дальней-
шем поглощении водорода восстанавливается боковая аллильная цепь и обра-
зуется тетрагидропомиферин [VII, R = 3,4-(ОН)2С6Н3], из которого приго-
товлены ди- и триацетаты. Помиферин проявляет обычные свойства изофла-
вонов и не дает гексагидропроизводных.
При расщеплении диметилового эфира осайина (IV) щелочью в мягких
условиях образуется одна молекула муравьиной кислоты и одна молекула
о-оксикетона (IX, R = Н) (диметилового эфира осайетина); в этом про-
является изофлавоновый характер осайина. Расщепление диметилового эфира
осайина (IV) или же диметилового эфира осайетина (IX, R = Н) щелочью
в более .жестких условиях дает гомоанисовую кислоту в результате отщепле-
ния от центрального, полностью замещенного флороглюцинового ядра п-меток-
сифенацильного остатка из фенильной группы изофлавона. (Ср. аналогичное
отщепление 7-ацетильной группы от усниновой кислоты и ее производных.)
На наличие о-оксидезоксибензоиновой группировки в диметиловом эфире
осайетина указывает его легкая циклизация под действием натрия и этил-
формиата в исходный изофлавон через промежуточную стадию образова-
ния гидрата, строение которого выражается, по-видимому, формулой
X (R = п-СНзОСвНО.
7 Заказ № 978
98
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Промежуточное соединение типа X легко превращается в диметиловый
эфир осайина при обработке кипящей уксусной кислотой. Циклизация в изо-
флавоны о-оксидезоксибензоинов, образующихся из осайина и помиферина
и их производных, также протекает с образованием устойчивых промежуточ- -
ных соединений типа X. Лишняя по сравнению с изофлавоном молекула воды
Н3С
н3с
О . Н
'V
|\ОН
—сосн2 —
сн2 I
I ОСН3
сн
з
Н3СО. О
X
С
Н3С СН3
IX
связана, по-видимому, химически, и выбор для этого соединения формулы X,
а не другой возможной структуры типа XI, основывается на отсутствии взаи-
модействия со спиртовым раствором хлорида железа (III) и на отрицательной
пробе Вильсона сборной кислотой [271]. Образование промежуточных сое-
динений этого типа в синтезе изофлавонов, хотя и возможно теоретически,
однако редко наблюдается в действительности. Тем не менее циклизация
СНО
I I CHR
/\/\ /
| со
осн3
XI
2-метокси-о-оксидезоксибензоинов под действием натрия и этил- или метил-
формиата неизменно приводит к высоким выходам соответствующего 2-окси-
изофлавонона (типа X) [272, 2731.
Подобным же образом диметиловый эфир тетрагидроосайина при расщеп-
лении щелочью в мягких условиях дает муравьиную кислоту и диметиловый
эфир тетрагидроосайетина (XII, R = Н), который способен циклизоваться
с натрием и этилформиатом в промежуточный гидрат, дающий при нагревании
с ледяной уксусной кислотой диметиловый эфир тетрагидроосайина.
Н3С
Н3С
Н3С I =
>СНСН2СН2 —сосн2 осн.
НзСх I “
н3
XII
При расщеплении щелочью в жестких условиях образуются муравьиная
и гомоанисовая кислоты как из диметилового эфира осайина, так и из диме-
тилового эфира тетрагидроосайина; тем самым подтверждается, что изофла-
воновая группировка не поглощает водород при гидрировании. Циклизация
ХРОМЕНО-Т-ПИРОНЫ
99
диметилового эфира тетрагидроосайетина под действием ацетата натрия
и уксусного ангидрида дает диметиловый эфир 2-метилтетрагидроосайина,
превращающийся в диметиловый эфир тетрагидроосайетина при обработке
щелочью в мягких условиях; тем самым получено дополнительное доказатель-
ство в пользу изофлавонового строения осайина. Эти превращения иллюстри-
руются следующей схемой:
О-Диметиловый эфир
осайица
кон
Н2
О-Диметиловый эфир
Na + НСООС2Н5 / осайетина + НС°ОН
КОН/
кон
Гомоанисовая
кислота
О-ДимеТиловый эфир
2-метилтетрагидроосайина
КОН
О-Диметиловый эфир
тетрагидроосадЯна
КОН
Na + НСООС2Н5
CH3COONa
(СН3СО)2О
О-Днметиловый эфир
тетрагидроосайетина + НСООН
Диметиловые эфиры осайетина и тетрагидроосайетина метилируются
диметилсульфатом в кипящем ацетоне и образуют соответственно триметило-
вый эфир осайетина (IX, R = СН3) и триметиловый эфир тетрагидроосайетина
(XII, R = СН3). Окисление диметилового эфира осайетина перекисью водо-
рода в щелочном растворе приводит к гомоанисовой кислоте, а окисление
триметилового эфира осайетина дает анисовую кислоту (расщепление про-
ходит по пунктирной линии, как показано на приведенной ниже схеме). Это
является косвенным подтверждением изофлавоновой природы остатка.
ОСН3
ОСН.
Окисление двуокисью селена реакционноспособной метиленовой группы
в триметиловом эфире тетрагидроосайетина (XII, R = СНз) Дает а-дике-
НзС\/\
сн
н3с/Хсн3
XIII
XIV
7*
100
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
тон (XIII) — триметиловый эфир тетрагидроосайетинона, идентичный про-
дукту, полученному в результате отщепления изонитрозогруппы с помощью
азотистой кислоты из изонитрозопроизводного (XIV) триметилового эфира
тетрагидроосайетина.
Соответствующие производные помиферина ведут себя аналогичным обра-
зом, что дает дополнительные данные, подтверждающие наличие изофлаво-
новой системы в исходных соединениях и сохранение ее в соответствующих
ди- и тетрагидропроизводных. Так, например, триметиловый эфир помифе-
рина (V) расщепляется едким кали с образованием муравьиной кислоты
и триметидового эфира помиферитина (XV, R = Н), который может быть
обычным образом переведен в триметиловый эфир помиферйна. В тех же усло-
виях триметиловый эфир тетрагидропомиферина превращается в муравьиную
кислоту и триметиловый эфир тетрагидропомиферитина (XVI, R = Н). Рас-
щепление щелочью в жестких условиях триметиловых эфиров помиферитина
и тетрагидропомиферитина приводит к образованию гомоанисовой кислоты
в результате гидролитического отщепления изофлавоновой группировки.
НзС\/\
Н3С<1 |
Н3С. /\/— С0СН2 —ОСН3
)С = СН — СН2 |
H3CZ ОСНз
XV
нзс\/\
НзС/j |
Х^Х___пр ОСН3
| II /=1
н,с - ДЛ соси, ОСН,
)СН—СН2—СН2 I
H3CZ ОСНз
XVI
Эти реакции суммированы на приведенной схеме.
О-Триметиловый
помиферииа
эфир --------522----О-Триметиловый эфир
Na + нсоос2н5 помиферитина
Гомовератроиая
кислота
О-Триметиловый эфир КОН
тетрагидропомиферина
О-Триметиловый эфир тетрагидро-
помиферитина
(CH^SO^I
О-Тетраметиловый эфир тетра-
гид ропомиферитииа
|н2О2
Вератровая кислота
XPOMEHO-y-ПИРОНЫ
101
Дальнейшее метилирование триметиловых эфиров помиферитина и тетра-
гидропомиферитина дает соответственно тетраметиловые эфиры помифери-
тина (XV, R = СНз) и тетрагидропомиферитина (XVI, R = СН3). Оба эти
вещества не реагируют с хлоридом железа (III).
Окисление триметилового эфира помиферитина (XV, R = Н) и тетраме-
тилового эфира тетрагидропомиферитина (XVI, R = СН3) щелочным раство-
ром перекиси водорода приводит соответственно к гомовератровой и вера-
тровой кислотам (из изофлавоновой боковой цепи). Двуокись селена окисляет
тетраметиловый эфир тетрагидропомиферитина (XVI, R = СН3) в а-дике-
тон — тетраметиловый эфир тетрагидропомиферитона (XVII).
НзС\/\
НзС^ I
ОСНз °СНз
НзС Д J- coco —0СНз
)СН —СН2-СН2 |
Н3С7 ОСНз
XVII
НзС\/\
НзС/I |
V V ОСН3
н3с /ULc00H
>СН —СН2—СН2 I
Н3С7 ОСНз
XVIII
При окислении тетраметилового эфира тетрагидропомиферитона (XVII)
щелочным раствором перекиси водорода образуется анисовая кислота и диме-
тиловый эфир тетрагидроосайетиновой кислоты (XVIII). Подобным же обра-
зом триметиловый эфир тетрагидроосайетинона (XIII) дает вератровую и хро-
мановую (XVIII) кислоты, что указывает на идентичность большей части
углеродного скелета помиферина и осайина, а также на то, что различие между
осайином и помиферином имеет место лишь в фенильном заместителе изофла-
воновой системы.
При кипячении с ледяной уксусной кислотой, содержащей небольшое
количество серной кислоты, осайин (I, R — Н) превращается в изоосайин
102
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
(XIX, R = п-НОС8Н4) в результате циклизации ненасыщенной у,у-диметил-
аллильной боковой цепи по гидроксильной группе в положении 5 с образова-
нием второй 2,2-диметилхромановой системы. (Ср. с превращением пеуценина
в изо- и аллопеуценины, стр. 70, и с образованием аллоротенона, стр. 126.)
Помиферин также циклизуется в изопомиферин [XIX, R = 3,4-(НО)2СвН3].
Н*С\/Ч.
' ' H3c/i | о
\У\/\
I и lr
/ч/\/ к
I 1 11
| о о
н3с/чсн3
XIX
Аналогичным образом дигидроосайин превращается в дигидроизоосайин;
это свидетельствует о том, что в первую очередь гидрируется двойная связь
в 2,2-диметилхроменовом кольце. В соответствии с этим моноацетат изоосайина
превращается при гидрировании в продукт, идентичный моноацетату дигид-
роизоосайина; это указывает на то, что наиболее реакционноспособная гидро-
ксильная группа в осайине не принимает участия в процессах превращения
в изоформу.
Эти превращения могут быть изображены следующей схемой:
(1) Дигидроосайин
(2) Дигидропомиферин
__________(1) Дигидроизоосайин
(2) Дигидроизопомиферин
(СН3СО)2О
в пиридине
(1) Моноацетат дигидроизоосайина
(2) Диацетат дигидроизопомиферина
(1) Моноацетат дигидроосайина
(2) Диацетат дигидропомиферина
(1) Моноацетат изоосайина
(2) Диацетат изопомиферина
Метилирование изоосайина щелочным раствором диметилсульфата дает
монометиловый эфир изоосайина (XIX, R = /г-СН3ОС8Н4), идентичный про-
дукту, полученному изомеризацией монометилового эфира осайина (I, R = Н).
Подобным же образом изомеризация диметилового эфира помиферина (III)
путем циклизации аллильной боковой цепи приводит к диметиловому эфиру
изопомиферина (XXV), идентичному продукту метилирования изопомифе-
рина. Это указывает на то, что две легко метилируемые гидроксильные группы
не участвуют в циклизации в изосоединения. Дигидропомиферин изомери-
зуется в дигидроизопомиферин, идентичный продукту, полученному гидри-
рованием изопомиферина. Аналогично диацетат дигидроизопомиферина иден-
тичен продукту гидрирования диацетата изопомиферина. Триметиловый эфир
помиферина (V) с трудом переходит в изосоединение, но в жестких условиях
отщепляются элементы метилового спирта с образованием диметилового эфира
изопомиферина (XXV). В соответствии с точкой зрения, согласно которой
циклизация в изосоединения обусловлена наличием двойной связи в у,у-диме-
тилаллильной боковой цепи, тетрагидроосайин (VII, R = п-НОС8Н4), тетра-
гидропомиферин [VII, R = 3,4-(HQ)2C8H3] и гексагидроосайин не изоме-
ризуются.
ХРОМЕНО-Т-ПИРОНЫ
103
Обработка осайина и его диметилового эфира горячей смесью иодисто-
водородной и уксусной кислот дает дигидроизоосайин. В тех же условиях
помиферин или его триметиловый эфир образуют дигидроизопомиферин. Вос-
становление имеющегося хроменового кольца в хромановое сопровождается
деметилированием, протекающим одновременно с циклизацией у,у-диметил-
аллильной боковой цепи и образованием второй 2,2-диметилхромановой
системы, характерной для соединений изо-ряда.
При осторожном окислении триметилового эфира помиферина получается
его эпокись (XX). Способность триметилового эфира тетрагидропомиферина,
диметилового эфира изопомиферина и монометилового эфира изоосайина
образовывать эпокиси еще раз подтверждает следующее: 1) двойная связь
в изофлавоновом кольце участвует в образовании эпокисей; 2) изофлавоно-
вая группировка не восстанавливается в тетрагидропомиферине, а следова-
тельно, сохраняется также и в тетрагидроосайине; 3) «изофлавоновая» двойная
связь не принимает участия в изомеризации.
Эпоксидная природа продуктов окисления подтверждена реакцией с кипя-
щей уксусной кислотой, содержащей иодистый калий, в результате чего выде-
лялся свободный иод. Эта реакция характерна для кетоокисей [274].
Дальнейшее окисление эпокисей триметиловых эфиров помиферина,
тетрагидропомиферина и изопомиферина, а также диметилового эфира поми-
ферина дает во всех случаях вератровую кислоту. Аналогично окисление
перекисью водорода эпокиси монометилового эфира изоосайина и мономети-
лового эфира осайина приводит к анисовой кислоте. Эти результаты показы-
вают, что 4'-гидроксильная группа в осайине и 3',4-гидроксильные группы
в помиферине не участвуют в изомеризации. Образование этих эпокисей
и восстановление изофлавонового кольца изофлавоновой системы в гекса-
гидроосайине указывает на необычайно реакционноспособную двойную связь
у этих изофлавонов в положении 2,3. Дополнительным подтверждением нали-
чия изофлавоновой группировки в осайине и помиферине, а также в соответ-
ствующих им изосоединениях служит способность соединений изо-ряда реа-
гировать со щелочью так же, как исходные соединения и их производные.
Так, монометиловый эфир изоосайина (XIX, R = п-СН3ОС8Н4) при обра-
ботке спиртовой щелочью в мягких условиях образует муравьиную кислоту
и монометиловый эфир изоосайетина (XXI, R = Н), дающий зеленую цвет-
ную реакцию с хлоридом железа (III); при его метилировании образуется
диметиловый эфир изоосайетина (XXI, R = СН3), не реагирующий с хлори-
дом железа (III). Подобным образом монометиловый эфир дигидроизоосайина
104
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
(XXII) при расщеплении переходит в монометиловый эфир дигидроизоосайе-
тина (XXIH). Осторожное расщепление осайина щелочью дает смесь мура-
вьиной кислоты (из изофлавонового кольца), ацетона (из гел«-диметильной
группы хроменового кольца) (ср. щелочное расщепление токсикарола, ксан-
тилетина и. ксантоксилетина), изоосайетина [дающего зеленую цветную реак-
цию с хлоридом железа (III) 1, n-оксифенилуксусной кислоты (из изофлаво-
нового кольца) и 5,7-диокси-2,2-диметилхромана (XXIV) (из хроманового
кольца, образующегося при переходе в изосоединение в результате цикли-
зации у,у-диметилаллильной боковой цепи).
Аналогичные реакции протекают и в случае соединений ряда помиферина.
Диметиловый эфир изопомиферина (XXV) при расщеплении щелочью пере-
ходит в диметиловый эфир изопомиферитина (XXVI, R = Н), а диметиловый
эфир дигидроизопомиферина (XXVII) дает диметиловый эфир дигидроизо-
помиферитина (XXVIII, R = Н). При дальнейшем метилировании димети-
ловый эфир изопомиферитина (XXVI, R = Н) превращается в триметиловый
эфир помиферитина (XXVI, R = СН3), а диметиловый эфир дигидроизопо-
миферитина (XVIII, R = Н) — в его триметиловый эфир (XXVIII, R = СН3).
Эти реакции можно представить следующей схемой:
О-Диметиловый эфир изопомиферина КОН О-Диметиловый эфир изопомиферитина+
| ----> +НСООН
I Нз I (CH3)2so4
О-Диметиловый эфир дигидроизопо- О-Триметиловый эфир изопомиферитина
миферина
I кон
О-Диметиловый эфир дигидроизопо-
миферитина
| (CH3)2SO4
О-Триметйловый эфир дигидроизопо-
миферитина
XXVII
кон-н2о
xxviii
Модифицированный способ расщепления изопомиферина едким кали
приводит к получению ацетона из 2,2-диметилхроменового кольца и 5,7-ди-
ХРОМЕНО-Т-ПИРОНЫ
105
окси-2,2-диметилхромана из 2,2-диметилхроманового кольца, образующегося
в процессе превращения в изоформу.
Щелочное расщепление диметилового эфира тетрагидроосайина и три-
метилового эфира тетрагидропомиферина дает небольшое количество 5-окси-
7-метокси-2,2-диметил-7-изовалерилхромана (XXIX), охарактеризованного
в виде 3,5-динитробензоата.
О синтезе этого хромана сообщалось [275]. Заслуживает внимания раз-
дельное расщепление 2,2-диметилхроменового и изофлавонового колец в осай-
ине и помиферине, а также в соответствующих их производных, которое пока-
зало, что в этих веществах, по крайней мере в отношении расщепления щело-
чами, 2,2-диметилхромановая система значительно превосходит по устойчи-
вости изофлавоновое кольцо.
Строение, приписываемое осайину и помиферину, было подтверждено
полным синтезом монометилового эфира дигидроизоосайина (XXII) и диме-
тилового эфира дигидроизопомиферина (XXVII), осуществленным Вольфро-
мом и Уилди [276].
При взаимодействии флороглюцина с у,у-диметилаллилбромидом в при-
сутствии хлористого цинка из продуктов реакции был выделен хроман (XXIХа).
Конденсация последнего с п-метоксибензилцианидом дает монометиловый
_ QLI
СН. I !|
^сн-сн2-CH2Z'Y
СНз ОСН3
XXIX
эфир дигидроизоосайетина (XXIII), который был превращен обычными мето-
дами в монометиловуй эфир дигидроизоосайина. Тем же методом был синте-
зирован диметиловый эфир дигидроизопомиферина.
Изофлавон, ямаицин (XXIX6) С22Н18О6, содержащийся в коре Piscidia
erythrina L., имеет строение, сходное со строением осайина и помиферина [277].
сн3 сн2 О
XXIX6 XXIXb XXIXt
Основные особенности его структуры были выяснены в результате образова-
ния 6-метоксипиперониловой кислоты (XXIXb) и р-тубаиновой кислоты
(XXIXr) при окислении щелочным раствором перекиси водорода.
Якарейбин С^ЬЩОв был выделен из древесины Calophyllum brasilien.se
Catnb. [278]. Под действием щелочи разрушается 2,2-диметилхроменовое
кольцо и образуется 1,3,5,6-тетраоксиксантон (ХХ1Хд), идентифицированный
синтезом. Имеющиеся данные не позволяют пока сделать выбора между двумя
возможными структурами — XXIXe и ХХ1Хж — для якарейбина, однако
исходя из общих соображений можно допустить, что наиболее вероятной
является ангулярная структура, выражаемая формулой XXIXe.
Мундулон(ХХ1Хз)С2вН26О6, выделенный [279] из коры Mundulea sericea,
был первым из найденных в природе хроманолов-3 и первым фенольным сое-
106
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
динением состава Ci5 растительного происхождения, содержащим изопреновые
остатки, соединенные с кольцами А и В.
он
I О
но —
ХХ1Хд
он
I- О
о он
XXIXe
НО 3
III II I |чсн»
II I
о он
ХХ1Хж
Расщепление щелочью в мягких условиях приводит к дезоксибензоину —
мундулетону (ХХ1Хи), который легко вновь превращается в мундулон.
В соответствии со своим положением гидроксильная группа в мундулоне
инертна по отношению к кислотным реагентам. Окисление деметилированного
мундулона дает мундулоксиновую кислоту (XXIXk, R = Н). При окис-
лении метилового эфира дигидромундулетона образуется метиловый эфир
мундулоксиновой кислоты (XXIXk, R = СН3) и метиловый эфир дигидро-р-
тубаиновой кислоты (ХХ1Хл).
Роттлерин. Роттлерин (I, R = Н) C3oH2s08 представляет собой основной
кристаллический компонент природного красителя камала (или камела). Это
порошок, имеющий окраску от оранжевой до ярко-красной, является сухим
выделением кожуры зрелых плодов Mallotus phillipinensis Muell. (синоним
сн3 сосн = снс6н5
I I О гн
RO-/\-OR RO-/V V
I I СНз
СНзСО-1^----СН2—
I I
OR OR
I
XPOMEHO-V-ПИРОНЫ
107
Rottlera tinctoria Roxb.), небольшого вечнозеленого дерева, произрастающего
в тропической Индии, Индокитае, на Яве и островах Малайского архипелага.
Камала применяется как субстантивный краситель для шелка и шерсти
(в щелочных ваннах), а в Индии еще и как антигельминтное средство. Роттле-
рин был, по-видимому, впервые выделен Андерсоном [280] (ср. Лейбе [281]),
который, однако, не сумел получить его в кристаллическом состоянии, а затем
Леркиными [282], назвавшими его маллотоксином и показавшими, что
он идентичен роттлерину Андерсона [280] (ср. работу Явейна [283]).
Это соединение служило объектом многочисленных исследований [284—
290], и хотя полученные результаты в свете наших теперешних представлений
несколько противоречивы и нет необходимости рассматривать их целиком,
все же следует отметить, что наличие в молекуле роттлерина флороглюци-
новой группы, остатка коричной кислоты [284], метилфлороглюцина и двой-
ных связей, а также его восстановительное расщепление с образованием диме-
тилфлорглюцина [286] находят свое отражение и в принятых в настоящее
время формулах.
В обширном и элегантном исследовании химии и строения 'роттлерина
Робертсон с сотрудниками [291—296] с несомненностью установили струк-
туру роттлерина и смогли определить природу тех изменений, которым спо-
собно подвергаться это хамелеоноподобное соединение (ср. Брокман и Майер
[297]).
СОСН = СНС8Н5 СОСН = СНС6Н5
Роттлерин (I, R = Н) экстрагируется из камала кипящим бензолом.
Сырой продукт очищают затем экстракцией четыреххлорйстым углеродом
и кристаллизацией из этилацетата и ацетона. Вещество имеет эмпирическую
формулу С3оН2808 и образует пентаацетат С3оН2308(СН3СО)5 (I, R = СН3СО)
СН3
I
НО -,/^i-OH
CH3CO-4^J—
СОСН2СН2С6Н5
I I
он он
III
и пентаметиловый эфир С3оН2308(ОСН3)5 (I, R = СН3). Гидролитическое
расщепление щелочью в отсутствие цинковой пыли дает уксусную, бензойную
и коричную кислоты, а также флороглюцин. В присутствии цинковой пыли
получают С-метил- и С-диметилфлороглюцины, а также р-фенилпропионовую
кислоту. Триметилфлороглюцин при этом не образуется (ср. Телле [286]).
С ацетатами фенилгидразина и семикарбазида соединение I (R = Н) дает
аморфные продукты. При обработке роттлерина водным раствором соды
получается с плохим выходом хорошо выкристаллизовывающийся продукт
роттлерон (II, R = Н) С41Н38О8, образующий О-тетраметиловый эфир
С41Н32О4(ОСН3)4 (II, R = СНз).
Аналогично тетрагидророттлерин (III) (получаемый гидрированием роттле-
рина в этилацетате в присутствии палладия на угле) с 8%-ным водным раство-
108
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
ром едкого натра дает октагидророттлерон (IV), идентичный продукту гидри-
рования роттлерона и образующий тетраацетат и тетраметиловый эфир
СОСН2СН2С6Н5 COCH2CH2C6HS
он он
IV
С41Н40О4(ОСН3)4. Эти взаимные превращения могут быть представлены сле-
дующей схемой:
н2
Роттлерин (I, R = H)----> Тетрагидророттлерин (III)
| Щелочь | Щелочь
н2
Роттлерон (II, R = H)---> Октагидророттлерон (IV)
При расщеплении тетрагидророттлерина кипящей уксусной кисло-
той получают второй продукт, производное гексаоксидифенилметана
Ci9H2o08(V, R = Н). При превращении роттлерина и тетрагидророттлерина
СН3 СН3
I I
RO OR RO -/'Vi- OR
СН3СО -М'------СН2------ML СОСН3
I I
OR OR
V
соответственно в роттлерон и октагидророттлерон образуется в небольших
количествах С-метилфлороглюцин. Наличие С-метилфлороглюциновой груп-
пировки в роттлерине подтверждено также образованием С-метилфлороацё-
тофенона, выделенного в виде 2,4,6-триокси-5-ацетил-3-метилазобензола (VI)
при расщеплении роттлерина под действием диазоаминобензола по методу
Бэма [298].
Строение роттлерина выведено исходя из структуры тетрагидророттле-
рина, также дающего 2,4,6-триокси-5-ацетил-3-метилазобензол (VI) при рас-
щеплении по методу Бэма. Роттлерон получается лишь в незначительном
количестве, однако октагидророттлерон образуется с почти количественным
выходом. Октагидророттлерон дает тетраацетат С41Н4о04(ОСОСН3)4, тетра-
метиловый эфир С41Н4о04(ОСН3)4 и диоксим C41H48O8N2.
СН3 R
I I О гн
НО он HO-^V ч/
I снз
СН3СО -n = nc6hs Ч/\/
I I
он он
VI VII
Расщепление октагидророттлерона щелочью приводит к р-фенилпро-
пионовой кислоте, 5,7-диокси-2,2-диметилхроману (VII, R = Н) и 5,7-диокси-
8-(Р-фенилпропионил)-2,2-диметилхроману (VII, R = C8HSCH2CH2CO). Пер-
воначально строение октагидророттлерона было не совсем ясно. Судя по со-
ХРОМЕНО-'у-ПИРОНЫ
109
ставу продуктов расщепления, казалось, что его правильное строение выра-
жается формулой дигидроциннамоилхромана (VIII, R — Н, или IX). Одна-
ко синтез показал [293], что два возможных дигидроциннамоилхромана не-
идентичны октагидророттлерону, откуда был сделан вывод, что октагидро-
роттлерон представляет собой дихроманилметан (IV).
СвН5СН2СН2—СО
НО _У\/°\/СНз НО —/у°\/СНз ЬоНз | Гсн3 X) СвН5СН2СН2СО ) 1 1 OR ОН VIII IX
При конденсации p-фенилпропионитрила с диоксихроманом (VII, R = Н)
по методу Геша образуется смесь кетонов (VIII, R = Н, и IX). При алки-
лировании соединения VIII (R = Н) легко образуется монобензиловый и мо-
нометиловый эфиры, нерастворимые в разбавленном едком натре и дающие
резко выраженную реакцию с хлоридом железа (III), что характерно для
о-оксикетона типа VIII (R = СН3).
Метилирование монобензилового эфира (X, R = CH2CeH5, R' = Н)
дает эфир (X, R = СН3, R' = СН2С6Н5). При дебензилировании последнего
образуется монометиловый эфир (X, R = СН3, R' = Н), легко растворимый
в разбавленном растворе едкого натра и в соответствии с приписываемой
ему структурой дающий отрицательную реакцию с хлоридом железа (III).
Строение этого соединения было в дальнейшем подтверждено конденсацией
5,7-диокси-8-ацетилхромана (XI) с бензальдегидом с образованием 8-цинна-
моилхромана (XII), при гидрировании которого было получено соединение
VIII (R = Н).
СОСН2СН2СвН5
1 II Гсн*
Ч/\/
I
OR
X
Получение 8-(|3-фенилпропионил)хромана (VIII, R = Н) этим методом
открыло простой путь синтеза октагидророттлерона. При взаимодействии
формальдегида и хромана (VII, R = C6HSCH2CH2CO) в спиртовом растворе
серной кислоты с высоким выходом получен октагидророттлерон, идентич-
ный природному продукту, что и подтвердило его диарилметановое строение.
СОСН3
СОСН = СНС6Н5
ОН
XI
ч/\/
I
он
XII
Строение дифенилметана (V, R = Н), полученного вместе с октагидро-
роттлероном при действии горячей уксусной кислоты на тетрагидророттлерин,
было установлено следующим образом. При расщеплении щелочью роттлерина
и тетрагидророттлерина до соответствующих роттлеронов обязательно обра-
зуется в небольшом количестве С-метилфлороглюцин. В соответствии с пред-
но
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
положением, что эта группировка присутствует в молекулах роттлерина
и тетрагидророттлерина, Брокман и Майер [297 ] получили азопроизводное
С-метилфлороацетофенона по реакции Бэма. Несомненно, что эта группа
имеется в гексаоксидифенилметане. Это было подтверждено превращением
этого соединения в 2,4,6-триокси-5-ацетил-3-метилазобензол (VI) по методу
Бэма, применяемому для расщепления дифенилметанов. Дальнейшим под-
тверждением этого вывода явился синтез 2,4,6,2',4',6'-гексаокси-5,5'-диаце-
тил-3,3'-диметилдифенилметана (V, R — Н) взаимодействием С-метилфлоро-
ацетофенона с формальдегидом в спиртовом растворе серной кислоты.
Что касается структуры тетрагидророттлерина, то образование соеди-
нений II и V (R = Н) может являться подтверждением его более сложного
строения, чем строение соединения III. Разложение тетрагидророттлерина
и роттлерина с образованием азопроизводного (VI) по реакции Бэма, несом-
ненно, требует данных, подтверждающих существование С-метилфлороаце-
тофеноновой группы, присоединенной к остатку молекулы через метилено-
вую группу. Превращение тетрагидророттлерина в смесь соединений II и V
(R = Н), казалось бы, может указывать на наличие в молекуле тетрагидро-
роттлерина обеих группировок. Однако результаты определения молеку-
лярных весов роттлерина, тетрагидророттлерина и их метиловых эфиров исклю-
чают эту возможность. В соответствии с этим роттлерин изображается фор-
мулой I (R = Н), а превращение его в роттлерон и соединение V (R =Н) под
действием уксусной кислоты рассматривается как реакция диспропорциони-
рования, согласно приведенной ниже схеме:
2RCH2R' CH2R2 + CH2R' (1)
В подтверждение этого было найдено [296], что при взаимодействии соеди-
нения VIII (R = Н) с метилфлороацетофеноном и формальдегидом в спир-
товом растворе серной кислоты получается тетрагидророттлерин, а также
дифенилметан (V, R = Н) и дихроманилметан (IV). Обнаружено также [296],
что реакция диспропорционирования, показанная уравнением (1), действи-
тельно является обратимой.
В соответствии с ранее приведенным строением тетрагидропроизводного,
роттлерину должна соответствовать формула I(R = Н). Это строение нахо-
дится в согласии с тем фактом, что роттлерин имеет две двойные связи и что
под действием щелочных агентов в результате обратимой реакции образуется
бензальдегид.
CeHsCHO + CH3COR C6H5CH = CHCOR
Далее, вторая двойная связь в тетрагидророттлерине должна находиться
в 2,2-диметилхромановой системе. Следовательно, роттлерин содержит 2,2-
диметил-Д3-хроменовую группировку.
Такая структура согласуется с неустойчивостью молекулы в присут-
ствии теплых кислотных или щелочных агентов. Хотя остаток о-оксикорич-
ной кислоты и способен к циклизации с образованием 2-фенилдигидро-у-
пирона (т. е. соединения флаванонового типа), однако эта реакция обратима,
а из изучения реакций гидрирования и метилирования становится ясным,
что роттлерин содержит стирилкетонную группировку.
Аллороттлерин. При продолжительном кипячении с уксусной кисло- .
той, спиртом, толуолом и другими высококипящими растворителями роттле-
рин превращается в изомерное соединение с т. пл. 182° [294, 297] —изо-
аллороттлерин (XIV), первоначально названный Брокманом и Майером изо-
роттлерином [297] (см. стр. 113).
Гидрирование изоаллороттлерина протекает более сложно, чем гидри-
рование роттлерина. Восстановление при атмосферном давлении в уксусной
кислоте приводит к насыщению хроменового кольца и образованию дигидро-
изоаллороттлерина (XVI, R = Н), дающего при кипячении со щелочью бен-
ХРОМЕНО-Т-ПИРОНЫ
111
зальдегид вследствие расщепления флаванонового кольца. Если же гидри-
рование проводят при давлении 0,35—0,7 ат и в качестве растворителя
он
I
сн3 -А,- СОСНз
НО ОН
СОСН = СНС6Н5
I
но О -
1 /СНз
СН2-1 1|-СН = СНС<
У ХСН3
он
СеНз
XIII
I
сн2
XIV
применяют этилацетат или спирт, не содержащий уксусной кислоты, то изо-
аллороттлерин превращается в тетрагидроаллороттлерин (XV). При гидрирова-
нии в тех же условиях, но при атмосферном давлении образуется смесь тетра-
гидроаллороттлерина (XV) и дигидроизоаллороттлерина (XVI, R = Н).
ОН
OR
J
СН3 -i/S- СОСНз
RO -yL OR
I
сн2
СН3-СОСНз
но ~у J.L он
I
сн2
I
он
XV XVI
Последний при дальнейшем гидрировании в этилацетате превращается в тетра-
гидроаллороттлерин (XV). Эти превращения могут быть изображены следую-
щей схемой:
Р.О„ттлерин---*- Изоаллороттлерин — Дигидроизоаллорот.тлерин
// XIV XVI.
' |Н2
Тетрагидроаллороттлерин
XV
О- Пентдметилаллороттлерин
XIX, R=CH3
н2 п „
------► О-Пентаметилтетра- ' ’
гидроаллороттхерин О-.ТетраметилДигидро-
XX® изоаллороттлерин
XVI, R = CH3
Тетрагидроаллороттлерин (XV) легко вступает в реакцию Бэма с диазо-
аминобензолом с образованием обоих возможных продуктов: 2,4,6-триокси-
5-ацетил-З-метилазобензола (VI) .и 8-фенилазо-5,7-диокси-6-(Р-фенилпропио-
112
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
нил)-2,2-диметилхромана (XVII), идентичного азосоединению, получаемому
при взаимодействии диазоаминобензола с 5,7-диокси-6-(|3-фенилпропионил)-
2,2-диметилхроманом (IX). Этот результат указывает на то, что в молекулу
N = NC6H5
। 0 ,СН3
, /\ У X / 6
С6Н5СН2СН2СО—1
3
ОН
XVII
тетрагидроаллороттлерина входит С-метилфлороацетофеноновая группиров-
ка, присоединенная через метиленовую группу к 5,7-диокси-6-(|3-фенилпро-
пионил)-2,2-диметилхроману (IX) в положении 8, а поэтому тетрагидроалло-
роттлерин имеет строение, выражаемое формулой XV. Этот вывод был под-
твержден синтезом тетрагидроаллороттлерина тем же методом, который был
использован для получения тетрагидророттлерина. Аналогичным образом
были получены гексаоксидифенилметан (V, R = Н) и октагидроаллороттле-
рон (XVIII).
По аналогии с роттлерином и в соответствии с приписываемой ему струк-
турой XV тетрагидроаллороттлерин превращается в роттлерон при действии
теплой щелочи и при обработке кипящей уксусной кислотой. Первый агент
приводит к образованию октагидроаллороттлерона (XVIII), строение кото-
рого установлено синтезом из 5,7-диокси-6-(|3-фенилпропионил)-2,2-диметил-
ОН
он
СвН5СН2СН2СО - А|/Х,
/СН3
НО— 1 II )(
СН3- ,/^j- сосн3
НО—II J— он
сн2
I О .СН3
НО —х/
I В hCH3
свн5сн2сн2со -1^х/1
I
он
XVIII
сн2
10 сн3
НО — \/
о. I II ГСН'
С6Н5
XX
OR
I
CH3-^V- соснз
RO -L J- OR
сн2
к А /снз
СвН5СН = СНСО— I
OR
XIX
ХРОМЕНО-у-ПИРОНЫ
113
хромана (IX) и формальдегида обычным путем. Хотя октагидророттлерон
и октагидроаллороттлерон очень похожи друг на друга, они заметно отли-
чаются по отношению к диазоаминобензолу в кипящем спирте. Октагидро-
аллороттлерон медленно реагирует, давая почти количественный выход азо-
производного, тогда как октагидророттлерон даже при продолжительном
кипячении с этим реактивом не вступает в реакцию. Было высказано предпо-
ложение, что это различие обусловлено высокой реакционной способностью
положения 8 в 5,7-диокси-2,2-диметилхромане, например, в отношении реак-
ций Гаттермана и Геша [295, 299], а также тем, что разрыв связи метиленовой
группы с углеродным атомом в положении 8 происходит более легко. Это
наблюдение согласуется, по-видимому, с тем фактом, что тетрагидроаллороттле-
рин (XV) при обработке горячим спиртовым раствором диазоаминобензола
дает два азосоединения (VI и XVII), тогда как тетрагидророттлерин в этих
условиях не дает азопроизводных кетохромана. Показано, что в этом ряду,
так же как и в ряду роттлерина, превращение в роттлерон в уксусной кислоте
является обратимым.
Оригинальное объяснение Брокмана и Майера [297 ] изомеризации роттле-
рина в дигидроизоаллороттлерин и обратного процесса как следствия цикли-
зации халконовой группировки с образованием соединения типа дигидро-
а-пирона (флаванона) (XXI) не было принято Робертсоном и сотрудниками.
Они выдвинули другое объяснение, подтвержденное расщеплением и синте-
зом. По их мнению, эти превращения заключаются в глубоких изменениях моле-
кулярного строения, в результате которых размыкается хроменовое кольцо
с образованием промежуточного соединения (XIII) и с последующим замы-
канием кольца в последнем таким образом, что образуется изомер — алло-
роттлерин (XIX, R = Н). Одновременно халконовая система в аллороттле-
рине циклизуется, образуя дигидро-у-пироновое кольцо, и дает изоаллороттле-
рин (XX или XIV). Робертсон и сотрудники думают, что название изороттле-
рин должно быть сохранено для еще неизвестного соединения (XXI), обра-
зующегося при циклизации халконовой системы в роттлерине. Следовательно,
теоретически возможен только один изороттлерин. С другой стороны, та же
последовательность превращений аллороттлерина (XIX, R — Н) может дать
два изомера — а- и p-изоаллороттлерины (XIV и XX). Какая из этих двух
формул соответствует изоаллороттлерину, пока не ясно. Если циклизация
халконовой системы в роттлерине предшествует раскрытию хроменового
кольца, как это предположено Сешадри [300], то вполне вероятно, что изо-
аллороттлерин имеет линейное строение XIV. Робертсон и сотрудники при-
держиваются иной точки зрения на течение реакции, поскольку очевидно,
что образование дигидро-у-пиронового кольца с блокировкой гидроксильной
группы в хроменовом кольце будет стабилизировать последнее. Поэтому
они считают, что предпочтительным будет механизм, предполагающий перво-
начальное размыкание хроменового кольца. Строение XX или XIV, припи-
сываемое изоаллороттлерину, дает рациональное объяснение свойств этого
соединения в отношении реакций гидрирования и метилирования.
Размыкание 2,2-метилХроменовой циклической системы, происходящее
при превращении роттлерина в изоаллороттлерин, ранее не наблюдалось.
Этот факт совершенно аналогичен раскрытию фуранового кольца в виснагине
и келлине, которое также происходит в кислой среде с образованием изовис-
нагина и изокеллина соответственно (стр. 38) и является дальнейшей иллю-
страцией равновесия, существующего в кислотных и щелочных средах между
открытыми и замкнутыми формами кислородсодержащих циклов. Положе-
ние равновесия и, следовательно, соотношение между циклическими и разом-
кнутыми формами в смеси зависит от pH раствора и строения молекулы.
При гидрировании изоаллороттлерин (XIV) образует дигидроизоалло-
роттлерин (XVI, R = Н) в результате насыщения двойной связи в хроменовом
8 Заказ № 978
114
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
кольце, а также тетрагидропроизводное — тетрагидроаллороттлерин (XV) —
вследствие одновременного раскрытия пиронового кольца и насыщения двой-
ной связи. Метилирование дигидроизоаллороттлерина (XVI, R = Н) дает
смесь О-тетраметилдигидроизо-(ХУ'1, R = СН3) и О-пентаметилдигидроалло-
роттлеринов (XXII), из которых первый является насыщенным веществом,
СН3 С6Н5~~
। 0 I О сн,
но_лгон YY V
СНз
сн3со --------сн2----
I I
он он
XXI
ОСНз
I
ОСНз
СНз -А- СОСНз
CH3O-A-OCH3
I
сн2
СНз-/^-|-СОСНз
СН3О Д^-ОСНз
I о
СН3О-
с6н5сн=снсо-
I
ОСНз
XXII
I
сн2
СН3О-
С6Н5СН2СН2СО —
СН3
СНз
XXIII
СН2СН2 — /
I
СОСНз со
но-А,-он НО YY°>\СНз
СНз-1 I'— СН2 I !1 I СНз
сн3 сн2 ^/ч/
I I
он он
XXIV
нерастворимым в едком натре, а второе Соединение содержит двойную связь,
образующуюся при раскрытии флаванонового кольца. При гидрировании
соединения XXII присоединяется одна молекула водорода и образуется О-пен-
таметилтетрагидроаллороттлерин (XXIII).
Пергидророттлерин. Дальнейшее гидрирование тетрагидророттлерина
над окисью платины дает пергидророттлерин, который еще полностью не иссле-
дован. Возможно, что строение этого соединения выражается формулой XXIV,
а дальнейшее гидрирование заключается в превращении фенильного радикала
в циклогексильный. Из пергидророттлерина в результате роттлероновой пере-
группировки образуется пергидророттлерон.
Общий характер роттлероновой перегруппировки. Реакция диспропор-
ционирования с уксусной кислотой или уксусным ангидридом была изучена
на ряде простейших полиоксидифенилметанов [301] и имеет общий характер.
С другой стороны, реакция Бэма приложима, по-видимому, к дифенилмета-
нам с двумя флороглюциновыми кольцами, в которых только одна гидроксиль-
ная группа может быть блокирована в результате алкилирования.
ХРОМАНОХРОМАНОНЫ, ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ
115
Мангостин. Мангостан С24Н2вО6— основной компонент латекса манго-
станового дерева Garcinia mangostana. Ранее приписывавшиеся ему строение
и формулы пересмотрены Ятесом и Стутом [302], предложившими для деметил-
и изодеметилмангостинов ксантоновую (XXV) и 2,2-диметилхромановую
(XXVI и XXVII) структуры.
z О
НО— Z \Z\—он
I II II Сн,
7 он о
I
XXV
ХРОМАНОХРОМАНОНЫ, ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ
Введение. Ряд встречающихся в природе веществ этого ряда вклю-
чает ротенон и его гомологи, содержащие хромано-(3',4';2,3)-хроманоновое
кольцо (I).
Хроманоновая система в ряду ротенона очень сходна с соответствующей
системой в группе флаванонов (III); следовательно, эти соединения могут
по существу рассматриваться как изофлаваноны, в которых равновесная
халконо-флаваноновая система может проявляться в щелочной или кислой сре-
де. Это означает, что в соответствующих условиях хроманоновое кольцо может
раскрываться, давая изомерный халкон типа IV.
IV
Хроманохроманоны, обладающие, подобно хроманонам, обычными харак-
терными для кетонов свойствами, проявляют также некоторые особенности
8*
116
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
при действии мягких окислителей. Два отмеченных звездочкой атома водо-
рода в формуле I при этом удаляются, и эти соединения превращаются в хро-
мено-(3',4',2,3)-хромоны (II). Реакция окисления легко осуществляется непо-
средственно действием перманганата калия или красной кровяной соли или,
косвенно, путем применения иода и ацетата натрия. В последнем случае про-
исходит замещение одного из атомов водорода хроманонового кольца, по всей
вероятности, в положении 3 на ацетоксигруппу (V, R = ОССН3). При гидро-
лизе этого соединения получаются соответствующие оксисоединения (V, R = H),
а последующая дегидратация дает хроменохромон или дегидросоединение (II).
В отличие от родоначального соединения дегидропроизводное не образует
кетопроизводных. Это означает, что карбонильная группа с двойной связью
в а,р-положении ведет себя подобно хромоновой системе.
При превращении хроманонового кольца в хромоновое проявляются
характерные свойства этой группировки по отношению к щелочному гидроли-
зу. Первой стадией является, вероятно, размыкание хромонового кольца
О
V
с образованием дикетона (VI), также содержащего хроманоновую-3 систему,
неустойчивую по отношению к щелочам [303]. В дальнейшем щелочной гидро-
лиз протекает по направлению, указанному пунктирной линией в формуле VI
с раскрытием 1,3-дикетоновой группировки, с присоединением молекулы
воды и образованием дерризовой кислоты типа VII.
ОСЩСООН.
—СН2---
VII
Ряд хроменохромонов был получен общими методами, разработанными
Робертсоном с сотрудниками [304], путем конденсации нитрила типа VIII
R'
I R'
OCH2COOR НО-/Ч-ОН OCHoCOOR I
LJ-chcn + If ~"l HO-A-OH
CH2CN \/\ II I
ch2—co—
VIII IX
с резорцином и его аналогами, в результате которой образуются сложные
кетоэфиры типа IX, циклизующиеся под действием ацетата натрия и уксус-
ного ангидрида в хроменохромоны типа II. Эти синтезы будут подробно опи-
саны в настоящем разделе.
Показано [305], что пигмент дракорубин, основной компонент «драконо-
вой крови», содержит хроманохромоновое ядро (X), в котором две гетероцик-
лические системы не связаны непосредственно шестичленным кислородсодер-
жащим кольцом, как в соединениях ряда ротенона. Поэтому указанный
пигмент также можно рассматривать как производное у-пираноксантона.
ХРОМАНОХРОМАНОНЫ, ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ
117
По-видимому, он является единственным известным представителем этого
класса соединений.
5
ОСН3
X
Высокая устойчивость этой циклической системы в противоположность
устойчивости ротеноновой группировки обусловлена наличием ксантонового
кольца.
В заключение настоящего раздела будет рассмотрено несколько соеди-
нений смешанного типа.
Группа ротенона (общие вопросы). Применение некоторых растений
в качестве средств для ловли рыбы с незапамятных времен практиковалось
в тропических и субтропических странах. Эти растения принадлежат к семей-
ству Fabaceae или Papilionaceae и видам Derris, Milletia, Tephrosia и Loncho-
carpus. Токсичные компоненты образуют ряд соединений, тесно связанных
между собой, из которых ротенон является наиболее распространенным.
Другими соединениями, близкими по своему строению и токсичности, являют-
ся тефрозин, дегвелин, суматрол, токсикарол, эллиптон и малаккол (см. ниже).
XIV
118
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
В результате исследования ротенона и родственных ему соединений были
получены многие продукты расщепления. Для полного познания химии этой
сложной молекулы достаточно описать лишь некоторые из этих веществ.
Для ротенона и его аналогов было подмечено два характерных факта:
1) дегидрирование хроманохроманоновой системы в хроманохромоновую
систему и 2) образование тетрагидропроизводных в результате расщепления
фуранового и дигидрофуранового колец с одновременным насыщением двой-
ной связи при гидрировании над палладиевым катализатором. Так, ротенон
(XI) присоединяет либо два атома водорода с образованием а-дигидророте-
нона (XII) или p-дигидроротенона (XIII), либо четыре атома водорода и пре-
вращается в тетрагидроротенон (XIV) [3061. Ротенон и родственные соеди-
нения, а также многие их производные в значительной степени проявляют
диморфизм, поэтому к использованию температуры плавления как критерия
чистоты и идентичности соединений этого ряда следует относиться с осторож-
ностью.
Несколько представителей ряда ротенона содержат пятичленное кисло-
родсодержащее кольцо: в ротеноне и суматроле — дигидрофурановая груп-
па, в эллиптоне, пахиррхизоне и малакколе — фурановый цикл, а в дегвелине
и токсикароле — 2,2-диметил-Д3-пирановая группировка (см. приведенную
ниже схему). Вследствие сложности систематической номенклатуры производ-
Дигидрофуранохроманохроманоны
Ротенон С23Н22О6
Суматрол С23Н22О7
Пиранохроманохроманоны
Дегвелин С23Н22О6
Тефрозин С23Н22о7
Токсикарол С23Н22О7
' Фуранохроманохроманоны
Эллиптон С20Н1вОв
Малаккол С20Н16О7
Пахиррхизон С21Н16О7
ных и продуктов расщепления соединений группы ротенона разработана номен-
клатура, основанная на получивших широкое применение «тривиальных»
названиях. Эта терминология принята и в настоящем разделе, хотя следует
учесть, что многие из названий претерпели изменения в процессе развития
химий ротенона. Поэтому некоторые названия, применяемые в настоящее
время, означают совсем не то, что в более ранних работах, а это, к сожалению,
может иногда приводить к недоразумениям.
Различным образом нумеруют также атомы углерода в соединениях ряда
ротенона. Наибольшее применение получила нумерация, показанная в фор-
муле XI, которая используется в настоящей главе. Отдельные кольца в роте-
ноне и подобных молекулах часто обозначаются различными буквами —
Д, В, С, D и Е, как это показано в формуле XI.
Ротенон. Из соединений этой группы наиболее полно исследован ее ди-
морфный представитель — ротенон (XI) С23Н22О6, т. пл. 163 и 185°. Поэтому
как типичный пример он будет описан здесь более подробно. Ротенон был
впервые выделен в 1895 г. из Robinia nicuo [3071— растения, произрастающего
во французской Гвиане. Вслед за тем он стал объектом изучения большого
числа исследователей. Наиболее выдающимися были работы Ла Форжа и со-
трудников [3081. В 1932 г. окончательно была установлена структура роте-
нона [309—312].
Оптически активный ротенон содержит две метоксильные группы, реак-
ционноспособную кетонную группу, способную к енолизации, три,неактивных
атома кислорода в гетероциклах и реакционноспособную двойную связь.
ХРОМАНОХРОМАНОНЫ, ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ 119
Хроманохроманоновая группировка в нем легко дегидрируется в хромено-
хромоновую (II) под действием мягких окислителей, например перманганата
калия и красной кровяной соли. При обработке ротенонов ацетатом натрия
XV
СН3СН3
хсн
I
сн2
сн2
ОСН3
XVI
п
СН2
СН3
ч/
с
СН3О ОСН2СООН / V
Н0-/\-0
сн3о i !'
СН3СН3
хсн
I
сн2
I
сн2
СН3О-/\- ОСН2СООН I
си.о-11 Н0-П
z gh2 —со—
XVIII
XVII
и иодом образуются различные ацетилротенолоны (V, R = СН3СО) в резуль-
тате ацетоксилирования атома углерода в положении 2, или, более вероятно,
в положении 3 хромановой системы (отмечен звездочкой в соединении I).
Гидролиз ацетатов ротенолона в рбтенолоны (V, R = Н) с последующей дегид-
ратацией дает тот же хроменохромон или дегидросоединение, которое полу-
чается из ротенона и названо дегидроротеноном (XV) [3131.
Карбонильная группа в соединениях ряда дегидроротенона лишена
обычной кетонной активности (ср. хромоны).
Карбонильная группа хроманохроманонового ядра способна к енолиза-
ции и может быть прометилирована и проацетилирована (см. стр. 129), но вслед-
ствие особенностей строения хроменохромонов (т. е. дегидроротенонов) подоб-
ная енолизация невозможна в случае дегидроротенонов.
Строение ротенона становится особенно понятным при рассмотрении
реакций тетрагидродегидроротенона (XVI, R = Н), который гидролизуется
щелочью в тетрагидродерризовую кислоту (XVII) [3081. Аналогично щелочной
гидролиз дегидроротенона (XV) дает исходную дерризовую кислоту (XVIII)
[308]. Тетрагидродерризовая кислота (XVII) — одноосновная кетокислота,
дающая интенсивную реакцию с хлоридом железа (III) в спирте вследствие
наличия карбонильной группы в орто-положении по отношению к гидроксилу.
Хроменохромоновая группировка регенерируется при кипячении с ацетатом
натрия и уксусным ангидридом [3031. Этим способом из тетрагидродерризо-
120
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
вой кислоты получают ацетат тетрагидродегидроротенона (XVI, R = СН3СО),
вероятно, с промежуточным образованием соединения типа VI.
Эта точка зрения подтверждается тем наблюдением, что в подобных же
условиях простые аналоги тетрагидродерризовой кислоты типа XVI 1а цик-
лизуются в 4-ароилхроманон-З типа XVII6 1314].
СН3 О-/\-ОСН2СООН СН3О ОСН3
I 11_сн2---со------' II
XVIIa
О
fu,O___\
• I II I
н ОСНз
I
СО —— ОСНд
XVII6
Расщепление тетрагидродерризовой кислоты горячей концентрирован-
ной щелочью дает дерровую кислоту (XIX) и 2-изоамилрезорцин (тетрагидро-
тубанол) (XX) [315].
Эти характерные реакции суммированы на приведенной ниже схеме.
Ротенон (XI) —
Дегидроротенон (XV)
+ 2Н2О | j - 2Н2О
Дерризовая кислота (XVIII)
—> Тетрагидроротенон (XIV)
Тетрагидродегидроротенон (XVI, R = H)
-2Н2° || + 2Н2О
Тетрагидродерризовая кислота (XVII)
Дерровая кислота (Х1Х)-|-Тетрагидротубанол (XX)
Строение тетрагидротубанола (XX) было установлено синтезом исходя
из 2,6-диметоксибензонитрила (XXI) [316] путем взаимодействия его с изо-
бутилмагнийбромидом с последующим восстановлением' в изовалерофенон
(XXII, R = СН3) и деметилированием образующегося изовалерилрезорци-
нового эфира (XXIII, R = СН3).
Тетрагидротубанол (XX) был также получен и другим путем [317].
Применение перегруппировки Фриса к сложному изовалериановому
эфиру (XXIV) 7-окси-4-метилкумарина приводит к 7-окси-8-изовалерил-4-
метилкумарину (XXV), дающему при расщеплении щелочью 2,6-диоксиизо-
валерофенон (XXII, R = Н). При восстановлении последнего по методу Клем-
менсена образуется тетрагидротубанол (XX).
СН3
СН2СН2СН
СН3О -/ОСН2СООН НО -[^'>1- ОН СНз
сн3о сн2соон
XIX XX
Строение двухосновной дерровой кислоты (XIX) было выяснено посред-
ством окисления и подтверждено синтезом [312]. Эта кислота, содержащая
две метоксильные группы и один эфирный атом кислорода, имеющийся также
и в ротеноне, дает при окислении перманганатом калия ближайший низший
гомолог (XXVI) — риссовую кислоту. Последняя при декарбоксилировании
превращается в декарбориссовую кислоту (XXVII, R = Н); эта кислота
ХРОМАНОХРОМАНОНЫ. ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ
121
была синтезирована конденсацией 3,4-диметоксифенола (XXVIII) с иодэтил-
ацетатом с последующим гидролизом сложного эфира (XXVII, R = С2Н5).
СН3
СН3
CN
СОСН2СН
СН2СН2СН
СН3О ОСНз
RO —OR СНз
RO —/Ч— OR СНз
XXI
XXII
XXIII
Н3С CHS
сн
сн2
Н3С
О о
со
I о °
СНСН2СОО -.
Н3С
СН3
СНз
XXV
Синтез риссовой кислоты был осуществлен исходя из 3,4-диметоксифенола
(XXVIII) через соединения XXIX, XXX и XXXI, а дерровой кислоты —
через соединения XXX, XXXII и XXXIII с применением обычных методов.
сн3о -/Ч-ОСН2СООН
XXIV
-со2
сн3о och2coor
СН3О соон
XXVI
СН3О -/Ч- ОН
СН3О он
СН3О
XXVII
СН3О ОСН2СООС2Н5
СН3О
XXVIII
сн3о —С । - сно
СН3О -I Н- СНО
СН3О -/'>1- ОСН2СООС2Н5
сн3о ОСН2СООС2Н5
СН,О-1 1'-СН=С-СО
СН3О —ч II-соон
о
XXXI
N = C---CeHs
XXXII
СНз °-А|- ОСН2СООН Н2О2
СН3О -I IL СН2СОСООН Х1Х
XXXIII
122
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
После установления строения обеих «половин» тетрагидродерризовой
кислоты можно попытаться воссоздать ее формулу. Однако следует учитывать,
что воссоединение обеих «половин» с отщеплением воды и образованием кето-
кислоты со свойствами, отвечающими тетрагидродерризовой кислоте, может
быть произведено одним из двух путей, каждый из которых приводит к раз-
личным формулам — XXXIV или XXXV.
СН3О ОСН2СООН I СН3О CH2COOH I
I ho/V-oh HO -Д-он
сн«°АЛ II I сн-°ААо гн ro II I
CH2—co—O-CH2-CO-\^
XXXIV XXXV
Выбор между этими двумя структурами был сделан в результате осу-
ществления полного синтеза тетрагидродерризовой кислоты и тетрагидроде-
гидроротенона [3181. Обработка оксима (XXXVI) пировиноградной кислоты
(XXXIII) ацетатом натрия и уксусным ангидридом дала нитрил (XXXVII,
R = Н), при этерификации которого диазометаном получен сложный метило-
вый эфир (XXXVII, R = СН3).
СН3О -/Ч||- ОСН2СООН
СН3О II- СН2ССООН
г^он
XXXVI
СН3О och2coor
CH3O-J^-CH2CN
XXXVII
Конденсация этого сложного нитрилоэфира с 2-изоамилрезорцином (XX)
(тетрагидротубанолом) по методу Геша приводит к получению сложного
метилового эфира кетокислоты (XXXIV), который способен гидролизоваться
в свободную кислоту, идентичную тетрагидродерризовой кислоте. Выделение
этого сложного эфира доказывает, что тетрагидродерризовая кислота имеет
строение XXXIV, а не другую возможную структуру XXXV. Циклизация
соединения XXXIV под действием ацетата натрия и уйсусного ангидрида
приводит к синтетическому тетрагидродегидроротенону. Следует отметить,
что эту циклизацию нельзя использовать при выборе между структурами
XXXIV и XXXV, поскольку последняя может дать хроменохромон типа
XXXVIII. Отсюда вытекает важность выделения сложного эфира, получаемо-
го конденсацией по методу Геша.
C5Hn-irw
0 1
I II II II I
сн3о-1 Д Д Д J
' v о Y
о
XXXVIII
Тубовая кислота. Молекула ротенона включает 2-изопропенилкумара-
новое ядро, и производные тубовой кислоты (XXXIX, R = СООН), содер-
жащие эту группировку, были первыми продуктами, полученными при рас-
щеплении ротенона. Это соединение, образующееся при щелочном расщеп-
лении ротенона, является оптически активной одноосновной кислотой феноль-
ного типа, содержащей реакционноспособную двойную связь, насыщение кото-
рой приводит к оптически активной дигидротубовой кислоте (XL). При сплав-
лении со щелочью или при обработке уксусной кислотой, содержащей неболь-
шое количество серной кислоты, тубовая кислота изомеризуется в оптически
неактивную изотубовую (XLI), которая с трудом гидрируется в дигидроизо-
ХРОМАНОХРОМАНОНЫ. ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ
123
тубовую кислоту (XL), т. е. рацемическую смесь D- и L-дигидротубовых
кислот. Тубовая, дигидротубовая и изотубовая кислоты дают интенсивную
цветную реакцию со спиртовым раствором хлорида железа (III), что находится
в соответствии с приписываемым им строением о-оксикислот.
НООС—,j \
н°4А н°-\А
| ? /С«з
1-— СН
х СНз
XL
НООС-/Ч
ноАА
I ° /сн»
-=!-сн
хсн3
XLI
I °
~ХС^СН2
ХСН3
XXXIX
аП
но-1; J-oh
Нг ( Н2
сн2
I
сн2
сн
Н3С СНз
XLII
hooc-ZS
но-^-он
I
сн2
I
нс=с
XLIII
/СН3
ХСНз
Тубовая кислота по своему поведению при гидрировании напоминает сам
ротенон. При энергичном гидрировании тубовая кислота дает смесь оптически
активной дигидротубовой (XL) и оптически неактивной тетрагидротубовой
(XLII, R = СООН) кислот. Последняя образуется в результате раскрытия
изопропенилдигидрофуранового кольца и восстановления образующейся боко-
вой цепи, что приводит к исчезновению центра асимметрии. Если гидриро-
вание проводят в щелочной среде, то фурановое кольцо раскрывается при
каталитическом гидрогенолизе, а двойная связь не восстанавливается. В ре-
зультате образуется промежуточное соединение — изо- или [3-дигидротубо-
вая кислота (XLIII) [319], изомерная дигидроизо- и дигидротубовым кисло-
там. Дальнейшее гидрирование соединения XLIII в нейтральной среде дает
тетрагидротубовую кислоту (XLII, R — СООН). Как указывает Ла Форж,
поведение тубовой кислоты при гидрировании очень напоминает поведение
в этих условиях соединений ряда кодеина, в которых присутствует та же
12 3 4
группировка —О—СН—С=СН, что и в тубовой кислоте. Согласно Шёпфу
1320], эта группировка по своим свойствам сопоставима с системой сопряжен-
ных связей. Присоединение водорода сначала происходит в положение 1,4
и сопровождается образованием двойной связи в положении 2,3.
При декарбоксилировании тубовой кислоты получают оптически актив-
ный фенол — тубанол (XXXIX, R = Н), а различные производные тубовой
кислоты дают соответствующие производные тубанола, включая изотубанол
и тетрагидротубанол (XLII, R = И). Последний идентичен продукту, полу-
ченному из тетрагидродегидроротенона при расщеплении щелочью. Таким
образом установлено соответствие между этими частями молекулы.
Правильность приписываемого изотубовой и тетрагидротубовой кис-
лотам строения была проверена синтезом. Так, карбоксилирование тетрагид-
ротубанола по методу Кольбе [321] дало тетрагидротубовую кислоту, иден-
тичную кислоте, полученной окислением диацетата альдегида (XLIV) (син-
124
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
тезированного по реакции Гаттермана из тетрагидротубанола) с последующим
дезацетилированием продукта реакции [322].
I
HO-/V-OH
U-ch°
XLIV
Изотубовая кислота была синтезирована следующим образом [323].
По реакции Перкина из 2-формил-5-изопропилфурана (XLV), сукцината нат-
рия и янтарного ангидрида получен сложный эфир (XLVII), который после
гидролиза и декарбоксилирования превращался в изотубанол. Эта реакция,
несомненно, протекает через промежуточное образование соединения XLVI
к XLVII. Карбоксилирование изотубанола дает изотубовую кислотуJXLI).
н3сх о
НС—/ X—СНО
н3с/ IHI
XLV
СН3
\ 0
НС—/ СН=ССООС2Н5
7 1
сн3 L.II снг
I
соосгн5
XLVI XLVII
Описан также другой метод синтеза изотубанола (XLVIIb) через проме-
жуточное образование соединений XLVI 1а и XLVII6 [324].
ОСОС8Н5 ОСОС8Н5 ОН
XLVIIa XLVII6 XLVIIb
Изоротеноны. При обработке ротенона серной кислотой происходит
изомеризация в изоротенон (XLVIII) в результате миграции двойной связи
боковой цепи в фурановое кольцо. Все аналоги и производные ротенона, содер-
жащие изопропенильную боковую цепь, способны к изомеризации такого
I I
ОСН3 ОСН3
XLVIII XLIX
типа. Расщепление изоротенона дает изотубовую кислоту (XLI), тем самым
устанавливается природа изомеризации. Дегидроизоротенон (XLIX) и изо-
дерризовая кислота (LIV) (стр. 126) получены обычными методами синтеза,
а также изомеризацией нормальных изопропенильных производных [325].
Ротенолоны. Имеющиеся мнения относительно возможности получения
разнообразных ротенолонов из ротенона в различных условиях весьма неопре-
деленны. Ла Форж и Галлер [3261, а также Мияно и Матсуи [327] внесли
ХРОМАНОХРОМАНОНЫ, ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ
125
значительный вклад в решение этой трудной проблемы, однако остающиеся
неясности в этом вопросе требуют дальнейшей работы в указанном направле-
нии и не рассматриваются в настоящей главе.
Продукты гидрирования ротенона. При каталитическом гидрировании
рртенон ведет себя подобно тубовой кислоте. Наблюдающиеся при этом раз-
личия связаны с остатком тубовой кислоты. Подобным же образом изменения,
происходящие при изомеризации в соединения ряда изоротенона путем пре-
вращения 2-изопропенилдигидрофурановой группировки в 2-изопропилфура-
новую, также обусловлены радикалом тубовой кислоты.
Гидрирование ротенона дает оптически активный а-дигидроротенон (XII)
(изомерный дигидроизоротенону, полученному при гидрировании изороте-
нона) и Р-дигидроротенон (XII) (часто называемый ротеноновой кислотой).
Р-Дигидроротенон представляет собой ненасыщенный фенол, соответствую-
щий p-дигидротубовой кислоте (L), при дальнейшем гидрировании которого
образуется тетрагидроротенон (XIV) [328, 329].
При обработке серной кислотой Р-дигидроротенон (XIII) превращается
в насыщенный изомер — аллоротенон* (LI), не обладающий фенольными
НООС—^'4.
он
/СНз
сн=с
хсн3
L
нзс\/\
Нз<4 |
Ч/Ч-ОН
Ы-соон
LII
свойствами. Это соединение получается в результате циклизации ненасыщен-
ной боковой цепи и образования 2,2-диметилхромановой группировки, так
как при раскрытии кольца под действием щелочи аллоротенон дает Р-дигидро-
тубовую кислоту, изомерную дигидротубовой кислоте и, по всей вероятности,
имеющую строение LII. [Ср. аналогичное превращение осайина в изоосайин,
помиферина в изопомиферин (стр. 102) и пеуценина в алло- и изопеуценины
(стр. 70).]
У каждого из указанных выше производных ротенона, так же как и у са-
мого ротенона, сохраняется хроманохроманоновая группировка, а поэтому
при окислении в мягких условиях получаются соответствующие дегидросоеди-
нения. Например, дигидроизоротенон (XII) образует дигидродегидроизоро-
тенон (LIII). Подобно дегидроротенонам, при щелочном гидролизе которых
* Аллоротенон часто называют 0-дигидроротеноном
126
соединения, содержащие два гетероатома кислорода
легко образуются дерризовые кислоты, дегидроизоротеноны могут быть пре-
вращены в соответствующие изодерризовые кислоты. Например, дигидроде-
гидроизоротенон (LIII) образует дигидроизодерризовую кислоту (LIV).
/СН3
//-СН
CH3O-/V-OCH2COOH H0-/V-0 хсн3
__н । II I
3 \^\сн2 со/\^
LIV
Эти реакции иллюстрируются следующей схемой:
Изоротенон (XLVIII) —► Дегидроизоротенон (XLJX) —►
(—Ротенон (XI)
Дегидроротенон (XV)
Изодерризовая кислота
(L1V)
Дерризовая кислота
(XVIII)
о-Дигидроротенон (XII) —*- ДигидродегидроротеноН
—*. Дигидродеррнзовая кислота
+Н2
+Н2
-►p-Дигидроротенон (XJII) —*- Дегидро-р-дигидроротенон —*-
I —► p-Дигидродеррнзовая кислота
Тетрагидроротенон (XVLR-H)—* у 2
—► Тетрагидродегидроротедон —*• Тетрагидродерризовая кислота (ХУП)
Аллоротенон (LI)
Дерритол и ротенол. При расщеплении ротенона щелочью в присутствии
восстановителей в качестве основных продуктов образуются дерритол (LV,
R = Н) и ротенол (LV), легко разделяющиеся при действии разбавленной
щелочи, в которой одноосновный о-оксикетон, ротенол, почти нерастворим.
их ^СН2
/ ГС\
CH3O-/V~OR H0-/V-0 СНз
ch3o-N'-ch2-co-/J
LV
СН3О—I
0 ZY
I но—/S-о
уСн2
хсн3
LVI
Аналогичным образом дигидроизоротенон дает дигидроизодерритол и дигидро-
изоротенол. Гидроксильная группа, находящаяся в орто-положении по отно-
шению к СН2-группе, легко метилируется, причем образуется метиловый
эфир дерритола (LV, R = СН3). Гидрирование последнего приводит к полу-
ХРОМАНОХРОМАНОНЫ. ХРОМЕНОХРОМОНЬГ И ХРОМОНОХРОМАНЫ
127
чению метилового эфира тетрагидродерритола (LXII), который перекисью
водорода окисляется в 2,4,5-триметоксифенилуксусную кислоту (LVIII);
/СН3
снгснгсн
/I ЧсНз
СНзО-^.-ОСНз НО-/Ч-ОН
СН3ОJ Ьснг—CO-IN
LVII
СН3О-^>,—ОСНз СНзО-^.-ОН
СН3О—I Jl—CH2COOH СН3О—СООН
LVIII LIX
в результате окисления самого дерритола перманганатом калия образуется
2-окси-4,5-диметоксибензойная кислота (LIX). При сплавлении со щелочью
дерритол превращается в изотубовую кислоту (XLI), а при окислении пер-
манганатом калия образуется тубовая кислота (XXXIX, R ~ СООН). (Тубо-
вая кислота при сплавлении со щелочью изомеризуется в изотубовую
кислоту.)
О-Метилтетрагидродерритол (LVII) был синтезирован [311] конденса-
цией тетрагидротубанола (XX) и гомоазароновой кислоты (LVIII) в присут-
ствии хлористого цинка. При конденсации тетрагидротубанола с 2,4,5-три-
метоксифенилацетонитрилом по методу Геша были получены лишь следы
ожидаемого продукта [322].
Вероятная схема реакций, приводящих к образованию дерритола, показа-
на ниже [3091. Первая стадия идет с раскрытием кольца С и может протекать
по пути а или б, что находится в соответствии со способностью этого кольца
легко раскрываться, как это происходит при образовании енолацетата ротено-
на и превращении токсикарола в линейный токсикарол (стр. 137). Последующая
дегидратация этой формы с разомкнутым кольцом (путь а) дает А3-хро-
, меновую систему (непосредственно получаемую путем б), которая в соответ-
ствии с хорошо известной реакционной способностью этого кольца расщеп-
ляется, как показано пунктирной линией на схеме. Восстановление двойной
128
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
связи в хроменовом кольце соответствующим восстановителем без одновре-
менного раскрытия хроменового кольца щелочью приводит к получению
ротенола (LVI).
Неторовая и токсикаровая кислоты. Окисление ротенола (или тетрагид-
роротенола) перекисью водорода дает неторовую кислоту (LX) [330], полу-
ченную также синтезом [321].
О
СН3О—4
CH3°-U\/
н/хсоон
LX
В результате циклизации этилового эфира дерровой кислоты (LXI) в при-
сутствии металлического натрия образуется сложный эфир хромена (LXII),
при гидрировании над платиной превращающийся в соединение LXIII. После-
дующие дегидратация и гидролиз соединения LXIII дают токсикаровую кис-
лоту (LXIV), получаемую также из ротенона окислением щелочным раство-
ром перекиси водорода [332, 333]. При восстановлении ацетата соединения
LXIII получается сложный эфир (LXV) неторовой кислоты;
CH3O-/V-ОСН2СООС2Н5
CH3O-^J— СН2СООС2Н5
LXI
о
I 1| |
сн^-ЦМ-он
СООС2Н5
LXII
о
СНзО-^у \
ch3°-UU
соон
LXIV
-н2о
сн3о-^у \
н/чСООС2Н5
LXV
Ротенонон.' При окислении ротенона хромовым ангидридом [334] или
азотистой кислотой [335] образуется ротенонон С23Н18 О7 (LXVI). Аналогич-
ные соединения получаются из изо-, а-дигидро-, [J-дигидро- и тетрагидророте-
нонов. Тетрагидроротенонон, для примера, изображается формулой LXVII.
Окисление метиленовой группы в карбонильную сопровождается образова-
нием соответствующего дегидроротенона с превращением бензопирановой
группировки в кумариновую. Те же рбтенононы получаются при окислении
соответствующих дегидроротенонов упомянутыми выше агентами. (Ср. окис-
ление О-диметилцитромицина в О-диметилцитромицинон, стр. 85.)
ХРОМАНОХРОМАНОНЫ, ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ
129
Ротенонон при действии щелочей в мягких условиях превращается в ро-
тенононовую кислоту [3361 С23Н20О8 (LXVIII), а расщепление ротенонона
/СНз
СН2--СН
О СН2 ЧсНз
и о I
° II II г
СНО II I о
СН3О—
ОСН3
LXVII
(LXVI) и ротенононовой кислоты (LXVIII) щелочью в жестких условиях
приводит к дерритолу и щавелевой кислоте [311, 337].
ZH2
^Х/°\ П Схсн.;
СН3О—у Х-СООН НО-А-°
СН3О—J------со-----\ )
LXVIII
Ротенонон был синтезирован взаимодействием дерритола с хлороксалил-
этиловым эфиром [337]. Окисление ротенононовой кислоты дает тубовую
кислоту и двухосновную арбутовую кислоту [310] (LXIX, R = Н), которая
также была синтезирована двумя методами [338, 339]. Согласно первому
[338], 3-хлоркумарин (LXXI), полученный из 4-оксивератрола и этилового
0
CHgO-.^Y х—COOR СН3О—ОСН2СООС2Н5
СН3О—-----LcoOR * CH3O-^J— СОСООС2Н5
LXIX LXX
соон
LXXI
эфира р-хлорф-карбэтоксипировиноградной кислоты конденсацией по Пех-
ману, был превращен обработкой щелочью в арбутовую кислоту (LXIX,
R = Н). Этот процесс аналогичен предложенному Ла Форжем механизму
превращения ротенонона в ротенононовую кислоту; тем самым подтверждается
предположение о строении ротенононовой кислоты. По второму методу [339]
эфир 2-окси-4,5-диметоксифенилглиоксалевой кислоты конденсируют с эти-
ловым эфиром бромуксусной кислоты, образующийся феноксиэфир (LXX) под
действием этилата натрия циклизуют в бензофурановый диэфир (LXIX,
R = С2Н5) и затем гидролизуют с образованием соединения LXIX (R = Н),
9 Заказ № 978
130 соедийеНйЯ, Содержащий Два йетёроатома кислорода
идентичного соединению, полученному при помощи первого метода, и арбу-
товой кислоте.
Оптическая активность и стереохимия ротенона и родственных ему
соединений. Оптическая активность ротенона обусловлена наличием в нем
трех асимметрических атомов углерода (7, 8 и 20 вформуле XI), один из которых
С20 находится в дигидрофурановом кольце, а два других — в положениях 7 и 8
хроманонового кольца. Это следует из того, что если асимметрия атома угле-
рода в дигидрофурановом кольце нарушена в результате изомеризации в изо-
соединение или в результате восстановления в Р-дигидро- или тетрагидроро-
тенон, то получаемые продукты все же обладают оптической активностью.
Если же хроманоновое кольцо в этих изомерных продуктах или в продуктах
гидрирования превращалось в хромоновое, то тогда такое соединение, т. е.
дигидроизоротенон или тетрагидродегидроротенон, становилось оптически
неактивным вследствие одновременного исчезновения асимметрии атомов
углерода 7 и 8 в хроманоновом кольце. Если же сначала разрушаются эти
активные центры, например при окислении ротенона в дегидроротенон, то тогда
дегидроротенон оказывается оптически активным вследствие наличия асим-
метрического атома углерода С20 в дигидрофурановом кольце. Если же этот
асимметрический центр ликвидируется, как это имеет место, например, при
превращении дегидроротенона в дегидроизоротенон или тетрагидродегидро-
ротенон, то продукт опять получается оптически неактивным. Наибольший
вклад в решение вопросов оптической активности и стереохимии соединений
группы ротенона внес Кан с сотрудниками 1340]. Стереохимическая проблема
в случае этих соединений состоит в следующем. В результате соединения колец
В и С в XI может образоваться цис- или транс-конфигурация. Хотя образо-
вание транс-конфигурации более вероятно, тем не менее окончательного-
решения по этому поводу принято не было. Одна из конфигураций, какой бы
она ни была, называется нормальной формой, а другая — аллоформой [340].
Таким образом изменение конфигурации атомов С7 или С8 влечет за собой
переход от нормальной к аллоформе. На стереохимию атома С20 указывает
наличие или же отсутствие приставки эпи-. Если эта приставка отсутствует,
то атомы водорода у С20 и С8 находятся в том же соотношении, как и в роте-
ноне. Атом углерода С7 не может служить опорным пунктом для С20 по той
причине, что, как это станет ясно из дальнейшего изложения, активность
этого центра асимметрии легко нарушается в результате енолизации.
Ротенон (XI) является левовращающим в бензоле и при окислении дает
L-дегидроротенон (XV), в котором отсутствуют центры асимметрии С7 и С8.
Отсюда следует, что левовращающим является атом углерода в положении 20.
При нагревании ротенона в уксусной кислоте с добавкой серной кислоты
происходит изомеризация его в изоротенон, который также является лево-
вращающим 1341]. Нет никаких оснований предполагать, что эта изомериза-
ция затрагивает атомы С7 и С8: нарушена асимметрия только атома углерода
С20. Следовательно, С7 и С8, рассматриваемые как единое целое, являются
левовращающими. В 'приведенной ниже таблице иллюстрируется использо-
вание различных терминов (придерживаясь номенклатуры, предложенной
Капом, по аналогии со стеринами) и поясняется их значение [3401.
Для изоротенона (XLVIII), дегвелина (XCVIII), токсикарола (LXXXII)
и других веществ, у которых центр асимметрии в кольце Е отсутствует, сущест-
вуют обычные L-, D- и DL-нормальные и L-, D- и DL-аллоформы. Дегидро-
соединения ие могут существовать в нормальной и аллоформах, поэтому
имеется только оптически неактивный дегидроизоротенон и D-, L- и DL-дегид-
роротеноны.
При растворении L-ротенона в спиртовом растворе едкого кали враща-
тельная способность его постепенно падает и через несколько часов становится
приблизительно постоянной. Полученный продукт, изомерный ротенону и на-
Хроманохроманоны. хроменохромоны и хромонохромлны
131
Номенклатура стереоизомеров ротенона
Приставка Центр асимметрии при атомах Рацемические
углерода формы
Никакой или L
D
L-эпи
D-эпи
L-алло
D-алло
L-эпиалло
D-эпиалло
Нормальный ряд
Аллоряд
DI.
’ DL-эпи
DL-алло
DL-эпиалло
зываемый мутаротеноном, имеет т. пл. 146° и [a]D —83° [340, 342]. Это же
вещество получается при нагревании L-ротенона либо с ацетатом калия в абсо-
лютном или 96%-ном спирте, либо с поташом в ацетоне, содержащем неболь-
шое количество воды. При кристаллизации мутаротенона из четыреххлористо-
го углерода приблизительно половина по весу от первоначально взятого
количества вещества выделяется в виде ротенона, содержащего кристаллиза-
ционный четыреххлористый углерод. Остаток представляет собой некристалли-
зующуюся смолу. Окисление мутаротенона щелочным раствором иода дает
почти с количественным выходом L-дегидроротенон. Это указывает на то,
что все компоненты молекулы мутаротенона имеют конфигурацию левовра-
щающего атома С20. Подобным образом серная кислота превращает мутароте-
нон с высоким выходом в DL-изоротенон. Это свидетельствует о том, что ком-
поненты смеси имеют только нормальную стереохимическую конфигурацию
атомов С7 и С8 и что смесь состоит из равных количеств стереоизомеров, имею-
щих взаимно противоположную оптическую конфигурацию атомов С7 и С8.
Эта смесь состоит наполовину из L-ротенона, имеющего асимметрические атомы.
L-(C?+ С8) и L-C20, и наполовину из вещества нормального ряда, изомерного
ротенону, имеющего асимметрические атомы D-(C74- С8) и L-C20, т. е. D-эпи-
ротенона.
Поташ в ацетоне или ацетат натрия в спирте вызывают быструю рацеми-
зацию атомов углерода С7 и С8; это наиболее удобный метод для получения
оптически неактивных изоротенона, дигидродегвелина и изодигидроротенона
из оптически активных изомеров [340]. Рацемизация С20 в D-эпиротеноне при
изомеризации дает с высоким выходом D-изоротенон, энантиоморфный L-изо-
ротенону. Это наблюдение подтверждено получением DL-изоротенона смеше-
нием равных частей D- и L-изоротенонов. Таким образом основным компонен-
том смолообразного D-эпиротенона является правовращающее соединение
с асимметрическими атомами (С7Ц- С8). Дальнейшее подтверждение этого пред-
положения о структуре D-эпиротенона получено в опытах по гидрированию.
В присутствии катализатора Адамса D-эпиротенон в этилацетате превра-
щается в D-дигидроэпиротенон, не энантиоморфный хорошо известному L-ди-
гидроротенону. При гидрировании D-эпиротенона над палладированным суль-
фатом бария в диоксане образуется фенольная смола, которая почти несом-
ненно представляет неочищенный D-P-дигидроротенон, поскольку он легко
превращается с высоким выходом в D-дигидродегвелин (D-дигидроалло-
ротенон)*.
Механизм рацемизации атомов углерода С- и С8 в соединениях группы
ротенона. Рацемизация С7 происходит, вероятно, благодаря енолизации
* Термин «алло» в данном случае не имеет отношения к конфигурации колец В и С-
9*
132 СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОЛТОМЛ КИСЛОРОДА
у этого атома, так как енолацетаты ротенона хорошо известны [343]. Однако
для рацемизации атома углерода С8 не может быть принят тот же механизм.
Здесь, по-видимому, причина состоит в том, что устанавливается равновесие
между нормальными и аллоформами при соединении колец В и С. Удовле-
творительное объяснение требует допущения разрыва кольца при рацемиза-
ции атома С8, и в качестве типов промежуточных соединений могут быть рас-
смотрены следующие: LXXII, LXXIII, LXXIV и LXXIVa. Учитывая мягкие
условия, при которых происходит рацемизация атомов С7 и С8 и, следовательно,
легкость одновременно протекающего раскрытия кольца, Робертсон и сотруд-
ники исключают промежуточное образование соединения LXXIII, поскольку
раскрытие кольца и его замыкание в этом соединении происходят недоста-
точно легко. Промежуточное образование соединения LXXII следует, по-ви-
димому, исключить на основании опытов с токсикаролом (стр. 137), которые
показали, что раскрывается кольцо С. Таким образом, рацемизация проте-
кает, вероятно, с промежуточным образованием соединений типа LXXIV
и LXXIVa, в соответствии с механизмом, предположенным Бутенандтом
и Хильгетагом [333] для гидролитического расщепления ротенона.
Кап с сотрудниками [340] показал, что рацемизация атомов С7 и С8 сопро-
вождает образование енолацетата ротенона. Так, L-изоротенон образует аце-
тат DL-изоротенона, а кислотный гидролиз ацетата дает DL-изоротенон.
Механизм рацемизации атома С8 при ацетилировании остается неясным. Если
ацетилирование протекает с раскрытием кольца (LXXIV), а это, по-видимому,
правильно, то следует ожидать образования ацетата со свободным феноль-
ным гидроксилом. Однако, по всей вероятности, достаточно надежно установ-
лено существование замкнутой формы енолацетата ротенона (LXXV) [3431.
При щелочном и кислотном гидролизе енолацетата ротенона образуются лишь
нормальные стереохимические формы, аллоформа полностью отсутствует.
Возможно, что при этом аллоформы образуются в очень малых количествах
и их трудно обнаружить и выделить. Однако, по-видимому, необратимое
превращение цис-а-декалона в транс-а-декалон [344] показывает, что стерео-
химические соображения не всегда пригодны для рассмотрения этого типа
взаимопревращений.
Кольца В и Св хроманохроманоновой системе ротенонов почти наверняка
имеют конформацию полукресла [345]. Конформационные соображения убе-
дительно подтверждают ту точку зрения [340], что кольца В и С соединены
между собою в транс-положении. Ни абсолютная, ни относительная конфи-
гурация асимметрических центров С7, С8 и С20 не установлена, однако имею-
ХРОМАНОХРОМАНОНЫ, ХРОМЕНО ХРОМОНЫ И ХРОМОНОХРОМАНЫ
133
щиеся данные определенно указывают на то, что большинство, если не все
ротеноны, обладают одной и той же абсолютной конфигурацией.
Суматрол (LXXVI), как и ротенон, имеет асимметрические атомы угле-
рода в положениях 7, 8 и 20, и его производные проявляют аналогичные опти-
ческие свойства. Так, дегидросуматрол оптически активен вследствие асим-
метрии атома С20, а тетрагидросуматрол, в котором асимметрия этого атома
нарушена, все еще оптически активен, так как атомы С7 и С8 не затрагиваются
при гидрировании. Как и следовало ожидать, тетрагидродегидросуматрол
(LXXVII) оптически неактивен.
Отсутствие оптической активности у токсикарола, дегвелина и тефрозина,
каждый из которых содержит потенциальные центры асимметрии С7 и С8 ,
с большой степенью вероятности должно быть отнесено за счет рацемизации
этих двух центров, происходящей в процессе экстракции (см. стр. 137).
Суматрол. Суматрол С23Н22О7 (LXXVI), впервые выделенный [3461
из неспецифичного представителя рода Derris, по-видимому, диморфен и изо-
мерен тефрозину и токсикаролу. Опытами по разрушению и синтезу [347,
348] установлено, что он представляет собой флороглюциновый аналог роте-
нона. Наличие карбонильной группировки подтверждено образованием окси-
ма, а интенсивная реакция с хлоридом железа (III) свидетельствует о том,
что это соединение содержит о-оксикарбонильную группировку в пери-поло-
жении (положения 11 и 15), эквивалентном орто-положению (ср. свойства
осайина и помиферина). Дегидросуматрол и его производные легко получаются
из суматрола при действии иода и ацетата натрия. Воздействие других окис-
лителей дает мало удовлетворительные результаты. Гидрированные суматро-
лы, соответствующие ротенонам, получаются аналогичными методами. Дегид-
росуматролы легко гидролизуются в кислоты типа дерризовой. Например,
тетрагидродегидросуматрол (LXXVII) превращается в тетрагидросуматроло-
вую кислоту (LXXVIH), которая была также получена синтетически [348]
СН2 ZCH3
LXXVI LXXVII
С5Ни-изо
I
CH3O — OCH2COOH HO — p 0H
CH3O J- CH2CO--------------N'
I
OH
LXXVIII
обычными методами из метилового эфира 2-цианметил-4,5-диметоксифенил-
уксусной кислоты (LXXIX) и изоамилфлороглюцина (LXXX) с последующей
циклизацией под действием ацетата натрия и уксусного ангидрида в тетра-
гидродегидросуматрол. При циклизации могут также получаться изомерные
134 СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
вещества (LXXX1), которые заставляют допустить возможность линейного
строения для суматрола. Тем не менее по аналогии с другими членами ряда
СН3
СН2СН2СН
СН3О q A- ОСН2СООСНз но _} (|„ он СН3
сн3о-11 Lch2cn i
LXXIX [
он
LXXX
.. ZOH
° К Y xf . снз
Д 11 СН2СН2СН
. V А/ 'У
осн3
LXXXI
вообще и с токсикаролом. в частности, предпочитают ангулярное строение
LXXVI.
Хотя реакции суматрола явно указывают на присутствие в нем 2-изо-
пропенильной группировки, как и в ротеноне, тем не менее все попытки изоме-
ризовать суматрол и другие пригодные для этой цели его производные оказа-
лись безуспешными.
Токсикарол. Токсикарол (LXXXII) С23Н22О7 содержит хроманохрома-
поновое кольцо ротенонового типа, способное к окислению в дегидротокси-
карол (LXXXIII) [332, 333, 349—351], при расщеплении которого образуются
дерровая и токсикаровая кислоты, благодаря чему устанавливается идентич-
ность хромановых колец в токсикароле и ротеноне. Однако в отличие от роте-
нона в нем отсутствует дигидрофурановое кольцо Е и вместо него присутствует
2,2-диметил-Д3-пирановая группировка. Вследствие этого при расщеплении
токсикарола 5%-ной щелочью образуется ацетон и апотоксикарол (LXXXIV),
т. е. происходит превращение, при котором 2,2-диметилхроменовая группи-
ровка проявляет характерную для нее реакционную способность (ср. ксан-
тилетип и ксантоксилетин, стр. 76).
При гидрировании токсикарола хроменовое кольцо быстро насыщается
и образуется дигидротоксикарол (LXXXV), который в свою очередь легко
превращается в дигидродегидротоксикарол (LXXXVI) обычными методами.
При действии па последнее щелочи, так же как и в ротеноне, раскрывается
хроменохромоновое кольцо с образованием дигидротоксикароловой кислоты
(LXXXVII, R = Н), которая при расщеплении 50%-ной щелочью превра-
ОСН, ОСН3
LXXXII LXXXIII
ХРОМАНОХРОМЛНОНЫ. ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМЛНЫ
135
ОСНз
LXXXV
LXXXVI
- щается в 5,7-диокси-2,2-диметилхроман (LXXXVIII), идентичный синтетиче-
скому образцу [352].
Доказательства ангулярной структуры токсикарола были получены при
метилировании дигидродегидротоксикарола диметилсульфатом в присутствии
поташа в ацетоне, в результате чего образуется О-метилдигидродегидротокси-
карол (LXXXIX), который был гидролизован 5%-ной щелочью в О-метил-
днгидротоксикароловую кислоту (LXXXVII, R СН3). Гидролитическое рас-
СН3О - - ОСН2СООН
он
СН3О-1'
сн2------------------со
LXXXV1I
СН3О
LXXXVIH
/ Ч/СН3
iXCH3
'll ' I
' . || i
О ОСНз
i
осн3
LXXXIX
щеплснпе последней 5016-ной щелочью
п 5-окси-7-метокси-2,2-диметилхроману
О /СН3
' Ден,
\/\/
он
хс
приводит к дерровой кислоте (XIX)
(ХС) [353], идентичному синтетиче-
I Й
ОН о
XCI
136 СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
скому образцу [352]. Этот образец был получен монометилированием 5,7-
диоксихроманона (XCI) и восстановлением монометилового’эфира по методу
Клемменсена. Образование 5-окси-7-метоксихромана (ХС) свидетельствует
о том, что О-метилдигидротоксикароловая кислота (LXXXVII, R — СН3)
имеет указанное строение и что О-метилдигидродегидротоксикарол, а следова-
тельно, и сам токсикарол правильно изображаются ангулярными струк-
турами.
При конденсации метилового эфира 2-цианметил-4,5-дцметоксифенокси-
уксусной кислоты (LXXIX) с флороглюцином образуется кетокислота (ХСП,
R =-• И). Аналогично О-диметиловый эфир флороглюцина дает кетон (ХСП,
R = СН3). Однако ожидаемой циклизации соединений ХСП (R = Н)
СН3О — /\-ОСН2СООН НО —7 OR
I г
'.in >.>./ |
OR
XCII
и ХСП (R = СНз) соответственно в дегидроапотоксикарол и его О-диметило-
вый эфир в обычных условиях осуществить не удается [354].
При конденсации метилового эфира 2-цианметил-4,5-диметоксифенокси-
уксусной кислоты (LXXIX) и 5,7-диокси-2,2-диметилхромана (LXXXVIII)
образуется кислота (ХСШ), изомерная, но не идентичная дигидротоксика-
роловой кислоте. Строение этого соединения, называемого аллодигидротокси-
кароловой кислотой, находится в соответствии с тем фактом, что применение
реакции Гаттермана к 5,7-диокси-2,2-диметилхроману (LXXXVIII) дает
8-альдегид [355], но противоречит обычному поведению С-метилфлороглюцин-
(3-метилового эфира.
Аллодигидротоксикароловую кислоту не удалось зациклизовать в ди-
гидродегидроаллотоксикарол, хотя оказалось возможным осуществить цик-
лизацию кетокислоты (XCIV) в хроменохромон (XCV).
СН3О -/ ОСН2СООН
CHjO-L
^/ХСН2--------СО------о сн3
но-/У У 3
I II Гсн»
\/\/
I
хеш он
ОСН2СООН он
I Н L
^/ХСН2 — СО-/ /-ОН
I
он
XCIV
XCV
При нагревании раствора токсикарола в ацетоне с безводным поташом
получают смесь, содержащую токсикарол (называемый а-токсикаролом е целью
идентификации) и изомер а-токсикарола — ^-токсикарол [342]. Это превра-
щение, по-видимому, обратимо, поскольку ^-токсикарол может быть превра-
щен в смесь а- и Р-токсикаролов в тех же условиях. Действие спиртовой щелочи
на ^-токсикарол вызывает такое же превращение, однако, поскольку а-токси-
ХРОМАНОХРОМЛНОНЫ, ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ
137
карол значительно меньше растворим в спирте, чем Р-токсикарол, то реакция
доходит почти до конца, так как образующийся а-токсикарол выделяется
из раствора. Это превращение нельзя объяснить какой-либо инверсией центров
асимметрии у атомов углерода в положении 7 и 8, так как в этом случае дегид-
ро-а-токсикарол и дегидро-р-токсикарол должны были бы быть идентич-
ными. Однако они различаются между собой, и истинное соотношение между
а- и ^-изомерами показано ниже.
В результате применения обычных методов из а-токсикарола (LXXXII)
получают дигидродегидро-а-токсикарол (LXXXVI), а ф-токсикарол образует
изомерный дигидродегидро-р-токсикарол. При нагревании с раствором едкого
кали эти два соединения дают одну и ту же дигидродегидротоксикароловую
кислоту (LXXXVII, R = Н). Это возможно лишь в том случае, если дигидро-
дегидро-Р-токсикарол имеет линейное строение (XCVI), а следовательно, и р-
токсикарол должен обладать соответствующей линейной структурой XCVII.
XCVII
Токсикарол, дегвелин и тефрозин, получающиеся обычной щелочной
экстракцией, оптически неактивны. Однако долгое время подозревали, что эти
вещества, находящиеся в природных источниках, оптически активны. В отно-
шении токсикарола это было подтверждено двумя группами исследователей
[340, 342], изолировавшими L-a-токсикарол, т. пл. 101—102°, [а]д —53
в бензоле, путем осторожной экстракции смолы Derris. Кану с сотрудниками
[340] удалось доказать, что это соединение действительно является L-a-токси-
каролом, а не каким-либо изомерным соединением. Так, L-a-токсикарол под
действием щелочи превращался в DL-a-токсикарол, что доказывает отсутствие
в нем суматрола, имеющего левовращающий атом углерода С20, который
может содержаться в виде примеси. При обработке спиртовым раствором
едкого кали получается смесь а- и p-токсикаролов, что дает дальнейшие дока-
зательства природы этого предшественника DL-a-токсикарола.
При растворении L-a-токсикарола в метанольном растворе едкого кали
первоначальное небольшое левое вращение быстро переходит в сильное пра-
вое вращение (от+ 300° до +321°). Если этот раствор подкислить, прежде чем
начнет уменьшаться вращение, то получается неизмененный L-a-токсикарол.
Поэтому изменение оптических свойств в данном случае следует приписать
[340] енолизации атома водорода при С7, а следовательно, атом С7 в L-a-токси-
кароле является левовращающим, а атом С8 — правовращающим, причем
левое вращение у атома С7 преобладает.
Оптическое вращение ротенона не претерпевает больших изменений
в этих условиях, что находится в соответствии с предположением, согласно
которому хроманокарбонильная группа в кольце С значительно менее спо-
собна к енолизации.
Следовательно, выделение L-a-токсикарола из щелочного раствора пока-
зывает, что рацемизация атома С7 в этих условиях не происходит, а конфигу-
рация атома С8 такова, что способствует перемещению атома водорода обратно
в его исходное (транс) положение с образованием связи нормального типа
между кольцами В и С. Это ясно указывает на то, что аллоформа токсикарола
образуется с большим трудом.
138
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Дегвелиз и тефронин. Кларк 1356] показал, что в Derris elliptica вместе
с ротеноном содержится еще два родственных вещества: дегвелин С23Н22ОВ
(XCVIII) и тефрозин С23Н22О7 (XCIX, Rt = Н, R = ОН или же ORt = Н
и R = Н).
Обычными методами дегвелин превращается в дегидродегвелин (С), кото-
рый легко гидрируется в дигидродегидродегвелин (CI). Гидрирование дегве-
лина в дигидродегвелин с последующим дегидрированием хроманохроманоно-
вого кольца дает тот же дигидродегидродегвелин (CI). Идентичность дигидро-
дегидродегвелина с дегидроаллоротеноном (стр. 126) служит простейшим
доказательством его строения.
ОСН3 ОСН3
С CI
Тефрозин [3561 представляет собой оксидегвелин ротенолопового типе
и может получаться при окислении дегвелина кислородом воздуха. Он легко
дегидратируется и образует продукт, идентичный дегидродегвелину. При
окислении этого продукта образуется тефровая кислота (КШааОц, строение
которой, вероятно, выражается формулой СП. Следовательно, при окислении
раскрывается двойная связь, причем этот процесс не сопровождается, как
обычно, одновременным разрывом эфирной связи в 2,2-диметилхроменовом
кольце. Если эта структура подтвердится, то мы будем, возможно, иметь
единственный пример аномального поведения 2,2-диметил-Д3-хроменовой
группы по отношению к окислителям.
СООН
О
О
сн
О-С—СНз
1
соон
о
ОСН3
СП
Эллиптон. DL-Эллиптоп (СШ) С20Н1вОв был впервые выделен Бэкли
[357]. Природное соединение L-эллиптон [358 , 359] легко рацемизуется при
ХРОМАНОХРОМЛНОНЫ. ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ
139
обработке ацетатом натрия. Наличие хроманохроманоновой группировки
в нем доказывается образованием дегидроэллиптона (CIV) при синтезе с ис-
пользованием обычных методов. Щелочное расщепление эллиптона приводит
к дерровой (XIX) и каранииновой (CV) кислотам, причем последняя анало-
гична тубовой кислоте и дает 4-оксикумарон (CVI) при декарбоксилировании.
О О
\/\_О /X/ \/Х-0
? I II I ? II II
/\/Х/\Х
CH3oJ[J 0 CH.3oJ[J 0
ОСНз ОСНз
CHI C1V
Гидрирование эллиптона приводит к дигидро-и тетрагидроэллиптоиам (CVII
и CVIII).
/X , О
“-fx r'iljl
ноос-у у
ОН
CV CVI
При обработке дигидродегидроэллиптона цинком и щелочью образуется
дигидроэллиптовая' кислота (CIX) и дигидроэллиптол (СХ), аналогичный
дерритолу (LV). Окисление эллиптола азотистой кислотой или хромовым
ангидридом дает эллиптонон (CXI), который был также получен из эллиптола
С2Н5
(СХП) при нагревании со смесью метилового эфира щавелевой кислоты и аце-
тата натрия.
140
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
Тетрагидроэллиптовая кислота (CXIII) синтезирована 13601 обычным
методом взаимодействием 2-этилрезорцина с метиловым эфиром 2-цианметил-
4,5-диметоксифеноксиуксусной кислоты; циклизацией под действием ацетата
натрия и уксусного ангидрида получен тетрагидроэллиптон (CXIV).
С£ 1;,0 — OCH2COOCI1,
СН3Оch2CN
СН,О ОСН2СООН НО \- ОН
СН3О -L СН2---СО----I
схш
С2Н5
I
но ОН
1| !
С2Н6
О 1
о
! ч Hi
/\/\/\^ .
II I
сн3о -!^,1 °
осн3
CXIV
Малаккол. Малаккол (CXV) С2оН)вО-, выделенный из Derris malaccensis,
оптически активен. Он представляет, по-видимому, 15-оксиэллиптон (CXV),
т. е. флороглюциновый аналог эллиптона, и способен к типичным превра-
щениям, свойственным ротенону [361].
Тетрагидромалакколовая кислота (CXVI) была синтезирована и затем
превращена в тетрагидродегидромалаккол (CXVII или CXVIII), который,
по-видимому, обладает соответствующими этому соединению свойствами, но,
однако, не сравнивался непосредственно с природным продуктом. До сих
пор вопрос о структуре малаккола — линейной или ангулярной — оконча-
тельно не решен, однако по аналогии с другими членами этого ряда более
правильной, по-видимому, является ангулярная структура.
Пахиррхизон. Ротеноид пахиррхизон (CXVlIIa) С21Н16О7 Icclfc? -L96°
(в хлороформе) содержится в японском ямсе Pachyrrhizus erosus Urban 1362].
Он был тщательно исследован Биккелем и Шмидом [363], которые показали,
ХРОМАНОХРОМАНОНЫ. ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ 14|
что это соединение проявляет типичные ротеноноподобные свойства. При-
сутствие метилендиоксигруппы вместо обычной метоксильной группы в кольце
А доказано с помощью инфракрасных спектров. При щелочном расщеплении
дегидропахиррхизона в жестких условиях образуется кумароновая кислота
CXVIIIa
CXVIII6
(CXVIII6, R — Н), которая была превращена в 6,7-диметоксикумаранкар-
боновую-5 кислоту (CXVII 16, R = СН3), идентичную синтетическому образ-
цу, что служит доказательством линейной конфигурации колец В, С, D и С
в отличие от ангулярной у других ротеноидов.
Гарпер [364] выделил из Derris malaccensis вещество фенольного харак-
тера, при щелочном гидролизе которого образуется муравьиная кислота.
Для этого соединения было предложено изофлавоновое строение CXlXa.
В кожуре корней Mundulea suberosa Benth. содержится аналогичный изо-
флавон, мунетон (CXIX) [365]. При циклизации [366] монометиловых эфи-
ров дерритола (СХХ), изодерритола (CXXXII) и эллиптола (CXXIV) под дей-
ствием натрия и этилформиата образуются соответствующие изофлавоны
(CXXI, СХХШ и CXXV), дающие положительную пробу Дархэма 1367],
которая до сих пор считалась специфической цветной реакцией соединений
группы ротенона.
CXIXa
СН3О—ZS—ОСН3 НО —
СН3О —сн2 — со
СХХ
142 Соединения, содержащие двА геТероАгоМа кислорода
II
о
CXXI
СН3
//чр сн
СНзО-.-^-ОСНз НО—I^-O хсн3
сн3о СН2 — со---------
CXXII
о
сххш
Синтез ротенонового кольца. Робертсон и сотрудники опубликовали
серию статей, в которых описаны попытки синтеза ротенонового и ротенононо-
вого колец.
СН3О ОСНз НО —Y°
СН3О-^-СП2------СО---lN
CXXIV
o'
сн3о— .< V-och3 / о
CH0JI :_________I
CH3O-
II
О
cxxv
Получены хромено-(3',4',4,3)-кумарины типа CXXVI [368]. Некоторые
из этих веществ изомерны хромено-(3',4',2,3)-хромонам, синтезированным
Робертсоном и сотрудниками 1318, 369, 370] при помощи описанных выше
общих методов синтеза.
Конденсация сложных енольных эфиров типа CXXVII с фенолами типа
CXXVIII была осуществлена по методу Пехмана (с серной кислотой), а в тех
случаях, когда для получения хроменокумаринов (CXXIX) способ Пехмана
оказался непригодным, — со спиртовым раствором хлористого водорода. Эти
хромено-(3',4',4,3)-кумарины обычно сильнее окрашены и менее растворимы
в органических растворителях, чем аналогичные изомерные хроменохромоны
типа дегидроротенона.
плхххэ
1ХХХЭ
о
Х1ХХЭ
1 (['-'J
1ЛХХЭ
о
/ \
144
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОЛТОМА КИСЛОРОДА
О О
СОС1
CXXXVIII
о
Н о
/\z \/Ч._ом
пин
СНО II I о
CrigO
CXL
СН3О—/V \=о
</V~0C11:i
I
co
CXXXIX
Конденсация сложных p-кетоэфиров типа СХХХ с л-диоксифенолами
по методу Пехмана дает кумароно-(2',3',3,4)-кумарины типа CXXXI 1371].
Синтезированы также хромонокумарины ротенонового типа 13721. Конден-
сация по Фриделю — Крафтсу хлорангидрида 7-метоксикумаринкарбоновой-4
CXL6
кислоты (СХХХП) с резорцином дает 4-ацилкумарин (СХХХШ), который
несмотря на многочисленные попытки не удалось циклизовать в хроманокума-
рин (CXXXIV). Однако аналогичный 3-ацилкумарин (CXXXVI) легко цик-
лизуется при кипячении в спиртовом растворе соляной кислоты с образова-
нием хроманонокумарина (CXXXVII), который в отличие от хроманохрома-
ноновой группировки ротенона трудно дегидрируется. Однако при обработке
тетраацетатом свинца образуется хромонокумарин (CXXXV).
При конденсации хлорангидрида 3,7-диметоксикумаринкарбоновой-4 кис-
лоты (CXXXVIII) с резорцином легко образуется 4-ацилкумарин (CXXXIX),
который одновременно циклизуется и деметилируется под действием броми-
стоводородной кислоты в уксусной кислоте, в результате чего получается
хромонокумарин (CXL). Таким путем был осуществлен первый синтез соеди-
нения, содержащего ротеноновое кольцо. Система типа CXL легко может
быть получена в результате циклизации под действием иодистоводородной
кислоты 2-карбэтокси-2'-метоксиизофлавонов типа CXLa, которые в свою
очередь получаются конденсацией о-оксидезоксибензоинов типа CXL6 с этокса-
лилхлоридом [373].
Этим методом был синтезирован ряд аналогов ротенона, подвергающихся
перегруппировке типа ротенонон — ротенононовая кислота [374]. Осущест-
влен также полный синтез L-a-дигидроротенона (XII) [375]. Конденсация
синтетического Ь-5-окси-2-изопропилкумарана (L-дигидротубанола) (CXLb)
с нитрилом (LXXIX) дает дигидродерризовую кислоту (CXLr), идентичную
заведомо полученному образцу.
Циклизация соединения CXLr под действием ацетата натрия и уксус-
ного ангидрида приводит к дигидродегидроротенону (СХЦд), образующему
при восстановлении боргидридом натрия смесь диастереомерных дигидророте-
нонолов (CXLe). Окисление этой смеси по методу Оппенауера дает L-дигидро-
ротенон (XII).
ХРОМАНОХРОМЛНОНЫ, ХРОМЕПОХРОМОНЫ И ХРОМО11ОХРОМАНЫ 145
Дракорубин. Дракорубип, основное красящее вещество продажной «дра-
коновой крови», было выделено Брокманом и Хаазе [3761. Почти одновременно
Хессе [377] выделил сходный красный краситель, дракокармин, из красной
смолы пальмы Dracaena draco Blume. Брокман и Хаазе 13781 предложили для
дракорубина эмпирическую формулу С32Н.и,О;,. Робертсон и сотрудники
1379, 3801 пришли к заключению, что дракорубин и дракокармин очень сход-
ны, если не идентичны 1379, 380 [.
В результате этих исследований было установлено, что дракорубин пред-
ставляет собой оптически деятельное ангидробензопиранольное основание (I)
13801 хиноидного типа, в свою очередь являющееся производным карбиноль-
ного основания (III, R -= Н), дракорубанола [3791. Ангидрооснование
образует ряд хорошо кристаллизующихся солей (II, R Н), например хлор-
гидрат и перхлорат, легко регенерирующих дракорубин при обработке ацета-
том натрия. В соответствии с известным превращением ангидробензопира-
полов [381] в метиловые эфиры оксибензопиранолов метилирование дракору-
бина [3791 щелочным раствором диметилсульфата даст левовращающий О-ме-
10 Заказ № 978
146
СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
тилдракорубанол (III, R = СН3), из которого легко получены соответствую-
щие соли (II, R = СН3). При щелочном расщеплении дракорубина образуется
ацетофенон и небольшое количество бензойной кислоты (образующейся из аце-
тофенона) в качестве единственных идентифицированных веществ. Окисление
щелочным раствором перекиси водорода приводит к разрыву пиранольного
Кольца и образованию оптически активного двухатомного фенола, драконола
(IV, R = Н), содержащего, так же как и дракорубин, трудно обнаруживае-
мую метоксильную группу, на что не обратили внимания немецкие исследова-
тели. Метилирование 1379] этого фенола дает О-метилдраконол (IV, R = СН3),
идентичный продукту, полученному вместе с ацетофеноном при щелочном
расщеплении О-метилдракорубанола.
Окисление дракорубина перекисью водорода или дальнейшее окисление
драконола приводит к насыщенной левовращающей драковой кислоте (V)
1382], не обладающей кетонными свойствами и дающей интенсивную фиолето-
вую цветную реакцию с хлоридом железа (III). Эта кислота, строение кото-
рой было неправильно указано Брокманом [382], была синтезирована сле-
дующим путем 1380]. 5,7-Диоксифлаванон (VI) превращали в монобензиловый
эфир (VII), затем метилировали в соединение VIII и каталитически дебензи-
лировали с образованием 7-окси-5-метоксифлаванона (IX). Последний восста-
навливали с высоким выходом, применяя модифицированный метод Клеммен-
сена, в ПБ-7-окси-5-метоксифлаван (X, R = Н). Применение реакции Гаттер-
мана к соединению X (R = Н) легко дает соединение X (R = СНО) в качестве
единственного продукта, который в свою очередь был превращен ацетили-
рованием, окислением и дезацетилированием в DL-драковую кислоту (V).
Расщепление этой DL-кислоты на оптические антиподы с помощью бруцино-
вых солей привело к получению L-драковой кислоты, идентичной во всех
отношениях природному продукту. Вероятно, она образуется из драконола
при окислительном расщеплении, как показано пунктирной линией в фор-
муле IV (R = Н). Строение альдегида (X, R — СНО) было установлено вос-
СООН
XI XII XIII
ХРОМАНОХРОМАНОНЫ, ХРОМЕНОХРОМОНЫ И ХРОМОНОХРОМАНЫ
147
становлением в 5-метокси-7-окси-8-метилфлаван (XII), идентичный образцу,
полученному восстановлением флаванона (XI), приготовленного из флоро-
ацетофенона (XIII) обычным способом.
Плохая растворимость, устойчивость при сплавлении со щелочью, зеле-
ная цветная реакция с хлоридом железа (III) и образование перхлоратов
драконолом и О-метилдраконолом 1380], а также отсутствие реакций, свой-
ственных карбонильной группе, свидетельствует о наличии в этих соедине-
ниях ксантонового кольца. Дальнейшим подтверждением присутствия 1-окси-
ксантоновой системы является способность этих соединений образовывать
комплексы со смесью борной кислоты и уксусного ангидрида [380]. По мнению
Кун-Рота, это указывает на присутствие одной С-метильной группы в дра-
коноле и по аналогии с дракородином [385] этот еще не полностью идентифи-
цированный верхний фрагмент молекулы драконола, по-видимому, имеет
С-метилфлороглюциновый остаток, расположенный как указано на схеме.
Установление строения драконола, а также образование ацетофенона и дра-
конола из дракорубина при действии щелочи подтверждает строение этого
красителя, представленное формулой I. Образование ксантоновой группи-
ровки при гидролизе О-метилдракорубанола и дракорубина объясняет также
отсутствие обычной карбонильной активности у О-метилдраконола и драко-
нола и неспособность этих соединений конденсироваться вновь с ацетофено-
ном с образованием соответственно О-метилдракорубанола и дракорубина.
Смешанные соединения. Пирилиевые соли типа II с двумя конденсиро-
ванными шестичленными кислородсодержащими гетероциклами тесно свя-
10*
148 СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
заны с циклическими системами, имеющимися в цитромицине и его производ-
ных (стр. 86) и в соединениях группы ротенона. Поэтому они также могут
быть включены в этот раздел, посвященный хроманохромонам. Конденсация
вератрового альдегида с 7-метоксихроманоном-4 приводит к образованию
стирилкетона (I), который был превращен в соединение II действием хлорида
железа (III) в уксусном ангидриде. Тот же продукт был получен прямой кон-
денсацией 2-оксивератрового альдегида с 7-метоксихроманоном-4 [384, 385].
В случае о-ванилина образуются пирилиевые соли (III). Получен также
ряд солей типа II и III 1386] конденсацией 7-метокси- и 6,7-диметоксихрома-
0 0 О О
'x/xj -i'RCH-CHCOR' 4 J_CHRCH2COR'
I ' I
он он
нона-4 с различными о-оксиальдегидами, включая 6-формил-7-окси-5-меток-
си-2,2-диметилхроман (стр. 79), из которого образуется сложное производ-
ное (IV).
VI VII
Конденсация 4-оксикумаринов с а,р-ненасыщенными. кетонами по типу
реакции Михаэля протекает с большой легкостью [387] и дает вещества типа V.
Если R представляет собой о-оксифен ильную группу, то образующееся
соединение VI легко циклизуется в хроманокумарины типа VII, по своему
строению близкие О-диметилцитромицинону (стр. 90).
R' ЧОСН3
VIII
Аддукты типа V легко циклизуются под действием 4%-ного метаноль-
ного раствора хлористого водорода в полуацеталь типа VIII, содержащий
пиранокумариновое кольцо.
ЛИТЕРАТУРА
1. Boeseken, Cohen, Biochein. Z., 201, 454 (1928).
2. Bertra in, van d e r Steur, W a I e r ill a n n, Biochem. Z., 197, 1 (1928).
3. С a r n m a I in, E r d t m a n, P e I c li о w i c z, Acta Chem. Sea nd., 9, 1111 (1955).
4. Haslam, Haworth, .1. Chem. Soc., 1955, 827.
ЛИТЕРАТУРА
149
5, В e г о z a, J. Am. Clieni. Soc., 77. 3332 (1955).
u a n g-M i и I о и, Ber., 70, 951 (1937).
S c 11 w e n g I e r. Arch. Pharin.. 277, 33 (1939).
Sveusk Kem. Tidskr.. 48, 230, 236, 250 (1936).
n, S ni i I Ii, J. Proc. Roy. Soc. N. S. Wales, 84, 449 (1914).
J. Pharin. Soc., Japan, 57. 1015 (1937).
6. H
7. 1)
8.
r d t in a n,
о b e r t s о
a к u, R i,
В r u с к h a u s e n, G e r h a r d, Ber.. 72. 830 (1939).
E r d t m a ii,
G r i
Ber
D i
D i
D i
D i
L i
Li-
H a n t s c h, Ber., 19, 2928 (1896).
A 1 g a r. Barry,' T m e Proc. Roy. Irish Acad., B41, 8 (1932).
S ш i t Ii, Nichol s, J. Am. Chein. Soc., 65, 1739 (1943).
N i ' ‘ • - - -- - — ----
P
e
P
a
S p
F о
F о
P о
33. T Ii
34. S p
35. S p
36. S p
37. Fo
Howel,
В
S
T
s
s .
W e s s e
N о
S
S
S
S
S .
W e s s e
S p
S p
S p
S p
N о „
Spath, К I a g e r, Ber., 66, 749 (1933).
Spath, К I a g e r, Schlosser, Ber.. 64, 2203 (1931).
Schmid, Ebnother, Helv. Chim. Acta, ..................
В r u с к h
К u m
Spath,
Spat'
S i. a t h,
Duff, 1
R a n g a s
L i ni a у e, Rasayanam, 1, 1 (1936).
Hegarty, Lahey, Australian J. Chein., 9, 120 (1956).
Spath, Schmid, Ber., 74, 595 (1941).
° ~ , P I a t z e r, ’S c h m i d, Ber., 73, 709 (1940).
10.
I I.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
Е
R
К
S
S
S
С
P e
о z
Gripeuberg, Acta Chem. .Scand., 1, 7) (1947).
n b e r g, Acta Chem. Scand., 2, 82 (1948).
a, S c h ' ' ’ ‘ " .... ' ’
n
и
и
и
h t e r, J. Am. Chem. Soc., 78, 1242 (1956).
r, Monatsh., 62, 263 (1933).
r, Monatsh., 66. 201 (1935).
r, Monatsh., 68, 41 (1936).
r, Monatsh., 74, 25 (1941).
С
f
f
d о
<1 о
d о
ci о
N a g a г к a r, Rasayanam, 1, 255 (1943).
Pause, Rasayanam, 1, 231 (1941).
е
е
е
е
c h
c h
in a у e,
in a у e,
S
г
f
е
е
е
e d e r 1, Nagel, J. Am. Chem. Soc., 63. 580 (1941).
a t h, Ber., A70, 83 (1937).
t h n a, S h a h, Chem. Revs., 36, I (1945).
"th, G a 1 i n о v s к y, Ber., 70, 235 (1937).
ter, R о b e r t s о n, J. Chem. Soc., 1931, 1875.
t 11 et al., Ber., 69, 1087 (1936).
ter,
ter,
m e r a
о in s,
a t h,
a t h,
a t h, '
s t e r,
а
П
а
s
s
Р а
11 о
Р й
Р а
Р
Р
Р
Р
Р
В u s Ii r a, J. Chein. Soc., 1948, 2254.
R о b e r t so и, J. Clieni. Soc., 1939, 930.
e, Monatsh., 12, 379 (1891); 14, 28 (1893).
e t с к e, Ber., 45, 3705 (1912).
b i c z e k, Ber., 70, 1253 (1937).
i и r a th, Ber., 70, 2272 (1937).
s s e 1 у, К a i и r a t h, Ber.. 70, 243 (1937).
Robertson,
Howell,
i n z
В a
К u
К a
We
Robertson, J. Clieni. Soc., 1939, 921.
Robertson, ,1. Chein. Soc., 1937, 293.
I, Robertson,
t h,
in s,
t h,
t Ii,
ш а и i a tn, .1. Chem. Soc., 19.30, 627.
1253 (1937).
g u c
" t '
t
t
t
t
а
а
а
а
а
а
а
а
а
g
S ii b r a
К u b i c z e k. Ber., 70,
Ber., 44, 3325 (1911).
P a i I e r, Ber., 69, 767
Vierhapper, Ber.,
у, К a 1 1 a b, Monatsh., 59, 161 (1932).
hi, К a w a n a ni i, Ber., 71, 344, 1428 (1938); 72, 483 (1939).
H о ' " •
H о
h et al.,
К a
D о
(1936).
70, 248 (1937).
I’,
h.
I z е n, Вег., 66, 1 137 (1933).
I z e n, Ber., 68, 1123 (1935).
Ber., 70, 1021 (1937).
h о v e c, Ber., 66, 1 146 (1933).
b г о v о 1 n y. Ber., 72, 52 (1939).
К a I I a b, Monatsh., 59, 161 (1932).
Kainrat h, Ber., 69, 2062 (1936).
T у r a y, Ber., 72, 2089 (1939).
Christian!, Ber., 66, I 150 (1933).
К I a g e r, Ber., 66, 914 (1933).
t
t
t
t
u t i, К a w a n a ni i, J. Pharin. Soc. Japan, 60. 57 (1940).
а г,
И,
В
Ebnother, Helv. Chim. Acta, 34. 1982 (1951).
a u s e ii, Hoffman n, Ber., 74, 1584 (1941).
Ram, R a y, J. Indian Chem. Soc., 23, 370 (1946).
P e s t a, Ber., 67, 853 (1934).
P a i 1 e r, Ber., 67, 1212 (1934).
P a i I e r, Ber., 68, 940 (1935).
ills, J. Chem. Soc., 1934, 1305.
w a m i, S e s h a d r i. Proc. Indian Acad. Sci., 9A, 7 (1939).
Spath,
Spath,
Schmid, Sci. Proc. Royal Dublin Soc., 27, 145 (1956).
Halpern, Wiser, Schmid, Helv. Chem. Acta, 40, 758 (1957).
P I a
Schmid, Ber.. 73, 1309 (1940).
150 СОЕДИНЕНИЯ, содержащие два гетеролтома кислорода
74. Bohme, Р i е t s с 11, Вег., 72, 774 (1939).
75. Boh in е, S с h и е i d e r, Ber., 72, 780 (1939).
76. S p a t h, В r u c k, Ber., 70, 1023 (1937).
77. M i I I e г, К i u к e I i n, Ber., 22, 1706 (1889).
78. Jordan, Thorpe, J. Chem. Soc., 1915, 387.
79. Whalley, J. Chem. Soc., 1951, 3235.
80. Crawford, Rasburn, J. Chem. Soc., 1956, 2155.
81, F о u r n e a u, T i f f e n e a u, Compt. rend., 146. 699 (1908).
82. S p a t h, P e s t a, Ber., 66, 754 (1933).
83. D e I e p i n e, Compt. rend., 149, 39 (1909).
84. L i m a у e, Ber., 65, .375 (1932).
85. Ray, S i I о о j a, V a i d, J. Chelll. Soc.. 1935, 813.
86. Shah, Sha h, J. Indian Chem. Soc., 17, 41 (1940).
87. L i m a у e, S a t h e. Rasayanain, 1, 87 (1937).
88. L i m a у e et al., Rasayanain, 1, 187 (1939).
89. L i m a у e, Ber., 67, 12 (1934).
90. Row, Seshadri, Proc. Indian Acad. Sci., 11 A. 206 (1940).
91. Chudgar, S h a h, J. Univ. Bombay, 13, Pt. 3, 15 (1944).
92. A d a m s, Roman, S p e r r v, J. Ain. Chem. Soc., 44, 1781 (1922).
93. M i 1 I s, A d a m s, J. Am. Chem. Soc., 45, 1842 (1923).
94. Несмеянов, Царевич, Ber., 68, 1476 (1935).
95. К r i s h n a s w a m у, S e s h a d r i, Proc, Indian Acad. Sci., 13Д, 43 (1941).
96. Curd, Robertson, J. Chem. Soc., 1933, 1 173.
97. S p a t h, Gruber, Ber., 71, 106 (1938).
98. A.n r e p et al., Lancet, 1, 557 (1947).
99. F a n t I, Salem, Biochem. Z., 226, 166 (1930).
100. Clarke, Robertson, J. Chem. Soc., 1949, 302.
101. В a x t e r. Ramage, T i m son, J, Chem. Soc., 1949, S30.
102. Murti, Seshadri, Proc. Indian Acad. Sci., 30, 107 (1949).
103. G e i s m a n n, H a I s a 1 1, J. Ami Chem. Soc., 73, 1280 (1951).
104. В a к er, J. Chem. Soc., 1933, 1381; 1940, 1370.
105. S p a t h, Gruber, Ber., 74, 1492 (1941).
106. Cp. Curd, R о b e r t s о n, J. Chein. Soc., 1933, 437.
107. С 1 a г к e, Glaser, R о b e r t s о n, J. Chem. Soc., 1948, 2260.
108. P h i I I i p p s, Robertson, W li a 1 1 e y, J. Chem. Soc., 1952, 4951.
109. W e s s e 1 e у et al., Monatsh., 56, 97 (1930); 60, 26 (19.32).
НО. H a t t о r i, Ber., 72, 1914 (1939).
111. F о s t e r, Howell, Robertson,.!. Chem. Soc., 1939, 930.
112. G r u b e r, Horvath, Monatsh., 80, 874 (1949).
113. Davies, Norris, J. Chem. Soc., 1950, 3195.
114. Spath, Gruber, Ber., 74, 1549 (1941).
115. S ni i t h, P u с с u, В у w a t e r, Science, 115, 520 (1952).
116. S c h rn i d, Sci. Proc. Royal Dublin Soc., 27, 145 (1956); В e и c z e, E i s e n b e. i s s,
Schmid, Helv. Chim. Acta, 39, 923 (1956).
117. В e a I, К a t t i, Chem. Zentr., 1926, H, 596; J. Am. Pharin. Assoc., 14, 1086 (1925).
118. L i m a у e, Rasayanam, 1, I (1936).
119. M a n j u n a t h, Seetharamiah, S i d d a p p a, Ber., 72, 93 (1939).
120. L i m a у e, Rasayanam., 1, I 19 (1937).
121. Foster, Robertson, J. Chem. Soc., 1948, 115.
122. Seshadri, Venkateswarlu, Proc. Indian Acad. Sci., 13A, 404 (1941).
123. Pavanaram, Row, Australian J. Chem., 9, 132 (1956).
124. Row, Australian J. Sci. Research, A5, 754 (1952).
125. Kelkar, Limaye, Rasayanam; 1, 228 (1941).
126. Manjunath, Seetharamiah, Half-Yearly J. Mysore Univ., B2, Pt. 3, 19
(1941).
127. Manjunath, Seetharamiah, Ber., 72, 97 (1939).
128. Jackman et al., J. Chem. Soc., 1958, 1825.
129. Haws et al., J. Chem. Soc., 1959, 3598.
130. Powell, Robertson, Wliallev Chem. Soc. (London) Spec. Publ. No. 5,
27 (1956).
131. Fielding et al., Tetrahedron Letters, No. 5, 24 (I960).
132. E a d e et at., J. Chem. Soc., 1957, 4913.
133. Graham et al., J. Chem. Soc., 1957, 4924-
134. Fielding et al., J. Chem. Soc., 1957, 4931.
135. Fielding et al., J. Chem. Soc., 1958, 1814.
136. Dean, Staunton, Whalley, J. Chem. Soc., 1959, 3004.
137. В irch et al., J. Chem. Soc., 1958, 4576.
138. M c G о о к i n, Robertson, Wba I lev, J. Chem. Soc., 1940, 787.
139. Spath, Schlager, Ber., 73, 1 (1940).
ЛИТЕРАТУРА
151
140. Robertson, Whalley, J. Chem. Soc., 1954, 1440.
141. В о w у e r, Robertson, Whalley,.!. Chem. Soc., 1957, 542.
142. Govindachari, N a g a r a j a n, P a r t h a s a r a t h y, J. Chem. Soc.,
1957, 548.
143. Handlord, Whalley, неопубликованные данные.
144. Emerson, В i с k о f f, J. Am. Chem. Soc., 80, 4381 (1958).
145. Govindachari, Nagarajan, P a i, J. Chem. Soc., 1956, 629.
146. Govindachari, Nagarajan, Pai, Partliasarathv.J. Chem. Soc.
1957, 545.
147. E i s e n b e i s s, Schmid, llelv. Chitn. Acta, 42, 61 (1959).
148. S i гл о n i t s c h, Frei, Schmid, Monatsh., 88, 54 (1957).
149. Ridgeway, Robinson..!. Chem. Soc., 125, 214 (1924).
150. Sen, C h a k r a v a r t i, J. Indian Chem. Soc., 6, 793 (1929).
151. R а и g a s w a in i, S e s h a d r i, Proc. Indian Acad. Sci., 6A, 112 (1937).
152. Spath, Low y, Monatsh., 71, 365 (1938).
153. Del i wala, Shah, J. Chem. Soc., 1939, 1250.
154. Limaye, G h a t e, Rasayanam, 1, 169 (1939).
155. В a d с о c k et al., ,1. Chem. Soc., 1950, 903.
156. Herzig, Pollak, Monatsh., 29, 263 (1908).
157. Bohn, G r a e b e, Ber., 30, 2327 (1897).
158. Herzig, Ann., 421, 247 (1920).
159. Haworth, McLachlan, J. Chem. Soc., 1952, 1583.
160. Grimshaw, Haworth, J. Chem. Soc., 1956, 418.
161. Bleuler, Perkin, J. Chem. Soc., 109, 529 (1916).
162. Grimshaw, H a w о r t h, J. Chem. Soc.. 1956, 4225.
163. Leach et al., J. Am. Chem. Soc., 75, 4011 (1953).
164. Simoni tsch et al., Helv. Chini. Acta, 43, 58 (I960).
165. King, King, Stokes, J. Chem. Soc., 1954, 4594.
166. Chan, Forsyth, Hassall, J. Chem. Soc., 1958, 3174.
167. S e t h n a, Shah, Shah, J. Chem. Soc., 1938, 228.
168. Desai, Hamid, Proc. Indian Acad. Sci., 6A, 185 (1937).
169. Deliwala, Shah, Proc. Indian Acad. Sci., 13A, 352 (1941).
170. Desai, Ekhlas, Proc. Indian Acad. Sci., 8A, 567 (1938).
171. К r i s h n a s w a m y, Rao, Seshadr i, Proc. Indian Acad. Sci., 19A, 5 (1944).
172- R angaswami, Seshadr i, Proc. Indian Acad. Sci., 9A, (1939).
173. Row, Seshadri, Proc. Indian Acad. Sci., 15A, 118 (1942).
174. Campbell, Link,!. Biol. Chem., 138, 21 (1941).
175. Stahmann, Huebner. Link, J.l Biol. Chetn., 138, 513 (1941).
176. Huebner, L i n k, J. Biol. Chem., 138, 529 (1941).
177. Overman et al., J. Biol. Chem., 142, 941 (1942).
178. Sullivan et al., J. Am. Chem. Soc., 65, 2288 (1943).
179. Huebner et al., J. Am. Chem. Soc., 65,2292 (1943).
180. Jones et al., J. Chem. Soc., 1949, 562.
181. Hinman et al., J. Am. Chem. Soc., 78, 1072 (1956).
182. Sh un k et al., J. Am. Chem. Soc., 78, 1770 (1956).
183. Walton et al., J. Am. Chem. Soc., 78, 5454 (1956).
184. Stammer et al., J. Am. Chem. Soc., 80, 137 (1958).
185. Spencer et al., J. Am. Chem. Soc., 80, 140 (1958).
186. Spencer et al., J. Am. Chem. Soc., 78 , 2655 (1956).
187. Michael, Biochem. J., 43, 528 (1948).
188. Birkinshaw et al., Biochem. J., 48, 67 (1951).
189. Barton, Hendrickson, Chem. & Ind. (London), 1955, 682; J. Chem. So
1956, 1028.
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
200.
201.
202.
203.
204.
205.
Birch, Chem. & Ind. (London), 1955, 682.
Wittig, Banger t, Richter, Ann., 446, 155 (1925).
Wittig, Ber., 59, 116 (1926).
Pond, Maxwell, Norman, J. Am. Chem. Soc., 21, 964 (1899).
Al gar, Hanway, Proc. Roy. Irish Acad., 42B, 9 (1934).
Ryan, O’N e i 1 1, Proc. Ro ’. Irish Acad., 32, 48 (1915).
Ryan et al- Proc. Roy. Irish Acad., 32, 167, 185, 193 (1915); 39, 432 (1930).
Allan, Robinson, J. Chem. Soc., 125, 2192 (1924).
Algar, McCarthy, Dick, Proc. Roy. Irish Acad., 41B, 155 (1932).
A 1 g a r, Flynn. Proc. Roy. Irish Acad., 42B, 1 (1934).
Algar, Hurley, Proc. Roy. Irish A-'ad., 43B, 83 (1936).
В a k e r et al., Proc. Chem. Soc., 1959, 91.
Baker, Ollis, Robinson, Proc. Chem. Soc., 1959, 269.
Nakazawa, J. Pharm. Soc. Japan, 61, 174, 228 (1941).
К a r i y/> n e, Kawano, J. Pharm. Soc. Japan, 76, 448, 451, 453, 457 (1956).
Kawano, Chem. and Ind. (London), 1959, 852.
152 СОЕДИНЕНИЯ. СОДЕРЖАЩИЕ ДВА ГЕТЕРО АТОМ А КИСЛОРОДА
206. К а г i У '> и г. S в w и (| а. J. Pharin. Soc. Japan, 78, 1010, 1013, 1016 (1958).
207. F u к u i. К a w a n o, J. Am. Cheiii. Soc., 81. 6331 (1959).
208 Chen et al.. Proc. Cheiii. Soc., 1959, 232.
209. S p a t ii, E i t c r, Ber., 74, 1851 (1911).
210. Backhouse, Roberts о n, .1. Chem. Soc., 1939. 1258.
211. Staples, Am. J. Pharni., 1829, 163.
212. Cm. Gordin, J. Am. Chem. Soc., 28. 1649 (1906).
213. Robertson, Subrn in a n i a in, J. Ch.mi. Soc., 1937, 1515.
214 Bell, R о b e r t s о n, J. Client. Soc., 1936, 1828.
215' Spath et al., Ber.. 72, 821(19.39).
216. Spath, Hillel, Ber., 72, 963 (1939).
217 Polonsky, Bull. soc. chiin. France, 1956. 91 I.
218. Spath, Hillel, Ber., 72, 2093 (1939).
219. Livingstone, Watson, J . Client. Soc... 1956, 3701.
220. Bridge, H e у e s, Robertson,.!. Chem. Soc., 1937, 279.
221. Bridge et al., J. Chem. Soc., 19.37, 1530.
222. К a m t h о ii g, Robertson, J. Chein. Soc., 1939, 933.
223. McKenzie, Robertso n, .1. Chem. Soc., 1949, 2057.
224. Robertson, Towers, неопубликованные данные.
225. Bell, Robertson, S u b r a in a n i a in, J. Client. Soc., 1936, 627.
226, Robertson, S u b r a in a n i a nt, J. Client. Soc., 1937, 286.
227. Robertson, Whalley,.). Client. Soc., 1951, 1935.
228. Robertson, W li a 1 ) e y, J. Chem. Soc., 1950, 1882.
229. Robertson, 1 s a r a s e и а, неопубликованные, данные.
230. Bell, Bridge, Ro h e r t s о n, J. Client. Soc., 1937, 15-12.
231 .Spath, M о c n i k, Ber., 70. 2276 (1937).
232 Spath et al., Ber., 73, 1361 (1940).
233. Spath, Sclnni d, Ber., 74, 193 (1941).
234. Whalley, неопубликованные данные.
235. S a in a a n. Quart. J. Pharin. and Pharmacol., 4, 14 (1931).
236. C a v a 1 1 i t o, Rockwell , .1. Org. Chein., 15, 820 (1950).
237. S in i t h, Pucci, В у w a t e r, Sfience, 115, 520 (1952).
238 Smit h. It о s a n s k v, В v w a t e r, 126th Meeting Am. Chem. Soc., Abstr.,
1954, 24N.
239. Schmid, Sci. Proc. Royal Dublin Soc., 27, 145 (1956).
240. В e и c z e, FI a 1 p e r n, S c ii m i d, Experientia, 12, 137 (1956).
241. S c 11 in i d, частное сообщение.
242. Smit h, H о s a n s k у, В у w a t e r, Chem. & Ind. (London), 1956, 718.
243. Smit h et al., J. Am. Chem. Soc., 79, 35,34 (1957).
244. 1) a v les, К e п у о n. Quart. Rev. Chem. Soc., 9, 203 (1955).
245. О г ш а и с e у - P c> t i e г, В и z a s, 1. e d e r e r. Bill I. soc. ch im. France, 1951, 5.77.
246. Polonsk у, T о ii b i a n a, Conipt, rend., 242, 2877 (1956).
247. Polonsky, Compt. rend., 242, 2961 (1956).
248. Hetherington, R a i s t r i c k, Phil. Trans. Roy. Soc. London, B220, 209 (1931).
249. M a с k e и z i e, R о b e r t s о n, J. Chem. Soc., 1949, 497.
250. J о n e s et al., J. Chem. Soc., 1949, 562.
251.
Robert so ii,
W li a 1 1 e y, J. Chem. Soc., 1949, 848.
252. Cavil!, Robertson, W h a 1 1 e y, .1. Chem. Soc., 1949, 1567.
253. Dean, R о b e r t s о n, W li a 1 1 e y. J. Chem. Soc.., 1950, 895.
254. В a d с о c k et al., J. Chem. Soc., 1950. 903.
255. C a v i 1 1, Robertson, Whalley,,!. Chem. Soc., 1950. 1031.
256. C a v i 1 1, R о b e r t s о n, W h a 1 1 e y, J. Chem. Soc., 1949, 1567.
257, Z
i g 1 e r, Ann.. 434. 34 (1923).
258. Mackenzie,
259. Robertson,
260. D e. a n et al., J.
26!. Oxford,
262. John
263. W
264. W
265. W
266. W
267. W
e
t
f
f
f
f
a 1 I
о 1 1
о 1 1
о 1 1
о 1 1
(1941).
268. W о 1 '
269. W о 1
270. W о 1
271. Wil
f
f
f
Robertson, W h a
W h a 1 1 e v, Y a t e s,
Chem. Soc., 1957, 3497.
strick, Simonar
J.
t,
e v, J. Chem. Soc., 1950, 2965.
Chem. Soc., 1951, 207,3.
s о n,
er,
г о
г о
г о
г о
m
m,
m,
m,
Biochem. J., 29, 1102 (1935).
R a i
Rogers, Trappe, J. Chem. Soc., 1956, 1093.
W о 1 f г о m, Hess, J. Am. Chein. Soc., 60, 574 (19.38).
et al., J. Am. Chem. Soc., 61, 2832 (1939).
Gregory. J. Am. Chem. Soc., 62, 651 (1940).
Mo
о
M
M
M
r g
a n, Benton, J. Am. Chem. Soc., 62, 1484 (1940).
an, В e n t о n, J. Am. Chein. Soc., 63, 422, 1248, 125.3, 3356
h a n, J. Am. Chein. Soc., 64, 308 (1942).
f f e t, J.Am. Chem. Soc., 64, 311 (1942).
m,
m,
m et al., J. Arn. Chem. Soc., 68, 406 (1946).
о
о
о
n, J. Am. Chem. Soc., 61, 2303 (1939).
272. W h a 1 1 e y, J. Chem. Soc., 1953, 3366.
r
s о
a
о
ЛИТЕРАТУРА
153
273. W h а 1 1 е у, J. Am. Chem. Soc., 75, 1059 (1953).
274. Weitz, Scheffer, Ber., 54, 2327 (1921).
275. Pierpoint, Robertson, W h a 1 1 e y, J. Chem. Soc., 1951, 3104.
276. Wolf rom, W i 1 d i, J. Am. Chem. Soc., 73, 235 (1951).
277. S t a m m, Schmid, Biichi, Helv. Chim. Acta, 41, 2006 (1958).
278. King, King, M a n n i n g, J. Chem. Soc., 1953, 3932.
279. Burrows et al., Proc. Chem. Soc., 1959, 150.
280. Anderson, Edinburgh New Phil. J., 1, 296 (1855).
281. L.e u b e, Vierteljahresschr. prakt. Phann., 9, 319 (1860).
282. Perkin, Perkin, Ber., 19, 3109 (1886).
283. J a w e i n, Ber., 20, 182 (1887).
284. P e г к i n, J. Chem. Soc., 63, 975 (1893); 67, 230 (1895); 75. 443, 829 (1899).
285. В a r t о 1 о t t i, Gazz. chim. ital., 24, I, 1 (1894); 24, II, 480 (1894).
286. Telle, Arch. Pharm., 244, 441 (1906).
287. Hermann, Arch. Pharm., 245, 572 (1907).
288. Dutt, J. Chem. Soc., 127, 2044 (1925).
289. Dutt, G о s w a n i, J. Indian Chem. Soc., 5, 21 (1928).
290. Hoffmann, F a r i, Arch. Pharm., 271, 97 (1933).
291. McGookin, Reed, Robertson, J. Chem. Soc., 1937, 748.
292. McGookin, Percival, Robertson, J. Chem. Soc., 1938, 309.
293. Backhouse, Robertson, J. Chem. Soc., 1939, 1257.
294. McGookin, Robertson, Tittensor, J. Chem. Soc., 1939, 1579, 1587.
295. Morton, Sawires, J. Chem. Soc., 1940, 1052.
296. Backhouse et al., J. Chem. Soc., 1948, 113.
297. Brock m an n, Maier, Anri., 535, 149 (1938).
298. Boehm, Ann., 318, 262 (1901).
299. ло ber tso n, Subra ma n ia m. J. Chem. Soc., 1937, 286.
300. Seshadr i, Sci. Proc. Royal Dublin Soc., 27, 84 (1956).
301. McGookin, Robertson, Simpson, J. Chem. Soc., 1951, 2021.
302. Yates, Stout, Chem. & Ind. (London), 1956, 1392; J. Am. Chem. Soc., 80, 1691
(1958).
303. Robertson, частное сообщение.
304. Robertson, J. Chem. Soc., 1933, 489.
305. Robertson, Whalley, Yates, J Chem Soc., 1950, 3117.
306. La Forge, Smith, J. Am. Chem. Soc., 51, 2574 (1929).
307. G e о f f г о y, Ann. inst. colon. Marseille, 2, 1 (1895).
308. Cm. La Forge, Haller, Smith, Chem. Revs., 12, 181 (1933)
309. La Forge, Ha 1 1 e r, J. Am. Chem. Soc., 54, 810 (1932).
310. Butenandt, McCartney, Ann., 494, 17 (1932).
311. Takei, Miyajima, Ono, Ber., 65, 1041 (1932).
312. Robertson, J. Chem. Soc., 1932, 1380.
313. L a Forge, S m i t h, J. Chem. Soc., 52, 1091 (1930).
314. Whalley, неопубликованные данные.
315. Robertson, неопубликованные данные.
316. H a 1 1 e r, J. Am. Chem. Soc., 55, 3032 (1933).
317. Robertson, S ii b r a m a n i a m, J. Chem. Soc., 1937, 278.
318. Robertson, J. Chem. Soc., 1933, 1163.
319. Takei, Miyajima, Ono, Ber., 65, 279 (1932).
320. S c h й p f, Ann., 483, 157 (1930).
321. T a k e i, К о i d e, Ber., 62, 3030 (1929).
322. Robertson, Rusby, J. Chem. Soc., 1935, 1371.
323. Reichstein, Hirt, Helv. Chim. Acta, 16, 121 (1933).
324. M i у ano, Matsu i, Bull. Agr. Chem. Soc. Japan, 23, 141 (1959).
325. Butenandt, Hildebrandt, Ann., 477, 245 (1930).
326. La Forge, Haller, J. Am. Chem. Soc., 56, 1620 (1934).
327. Miyano, Matsu i, Chem. Ber., 92, 1438 (1959).
328. H a 1 I e r, J. Am. Chem. Soc., 53, 733 (1931).
329. H a 1 1 e r, J. Am. Chem. Soc., 54, 2126 (1932).
330. Smith, La Forge, J. Am. Chem. Soc., 52, 4595 (1930).
331. Robertson, Rusby, J. Chem. Soc., 1936, 212.
332. Butenandt, Hi Igetag, Ann., 495, 172 (1932).
333. Butenandt, Hilgetag, Ann., 506, 158 (1933).
3. 34. Takei, Biochem. Z., 157, 1 (1925).
335. Butenandt, Ann., 464, 253 (1928).
336. La Forge, Smith, J. Am. Chem. Soc., 52, 1091 (1930).
337. La F о r g e, J. Am. Chem. Soc., 54, 3377 (1932).
338. Holton, Parker, Robertson, J. Chem. Soc., 1949, 2049.
339. Hargreaves, McGookin, Robertson, J. Chem. Soc., 1957, 5093.
340. Cahn, Phipers, В о a rn. J. Chem. Soc., 1938, 513.
154
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИСЛОРОДА
341. Wright, J. Am. Chem. Soc., 50, 3355 (1928).
342. Tattersfield, Martin, Ann. Appl. Biol,, 23, 899 (1936); ,1. Soc. Chein. Ind,
(London), 56, 77T, 88T (1937).
343. Smith, La Forge, J. Am. Cheni. Soc., 54, 2996 (1932).
344. H ii с к e 1, Ann., 441, 1 (1925).
345. Cm. Whalley, The Chemistry of Vegetable Tannins — A Symposium, Cambridge,
1956, editor the Society of Leather Trades’ Chemists — Croydon 1957, p. 151.
346. Cahn, В о a m, .1. Soc. Chem. Ind. (London), 54, 42T (1935).
347. Robertson, Rush y, J. Chein. Soc., 1937, 497.
,348. Kenny, Robertson, George, J. Chem. Soc., 1939, 1601.
349. Clark, J. Am. Chem. Soc., 52, 2461 (1930).
350. С 1 a r k, J. Am. Chem. Soc., 53, 2264 (1931).
351. С 1 a r k, J. Am. Chem. Soc., 54, 1600, 2537 (1932).
352. Bridge, Heyes, Robertson, J. Chem. Soc., 1937, 279.
353. George, Robertson, J. Chem. Soc., 1937, 1535.
354. Heyes, Robertson, J. Chein. Soc., 1935, 681.
355. Robertson, Subramaniam, J. Chem. Soc., 1937, 286.
356. С 1 a r k, J. Am. Chem. Soc., 53, 729, 2369 (1931).
357. В ti с к 1 e y, J. Soc. Chem. Ind. (London), 55, 285T (1936).
358. Meyer, Koolhaas, Rec. trav. chim., 58, 207, 875 (1939).
359. Harper, J. Chem. Soc., 1939, 1099, 1424.
360. H a r p e r, J. Chem. Soc., 1942, 593.
361. H a r p e r, J. Chem. Soc., 1940, 309.
362. Th. M. Meiyer, Rec. trav. chim., 65, 835 (1946).
363. Bickel, Schmid, Helv. Chim. Acta, 36, 664 (1953).
364. Harper, J. Chem. Soc., 1940, 1178.
365. D ti t t a, J. Indian Chem. Soc., 36, 165 (1959).
366. H a r p e r, J. Chem. Soc., 1942, 595.
367. Harper, J. Chem. Soc., 1939, 1099.
368. Hilton et al., J. Chem. Soc., 1936, 423.
369. Robertson, J. Chem. Soc., 1933, 489.
370. King, Robertson, J. Chem. Soc., 1935, 993.
371. King et al., J. Chem. Soc., 1948, 1672.
372. Parker, Robertson, J. Chem. Soc., 1950, 1121.
373. Baker, H a r b о r n e, О 1 1 i s, J. Clieni. Soc., 1953, 1860.
374. Whalley, Lloyd,,). Chem. Soc., 1956, 3213.
375. M a t s ti i, M i у a n o, Proc. Jap. Acad., 35, 175 (1959).
376. Brockman n, Haase, Ber., 69, 1950 (1936).
377. Hesse, Ann., 524, 14 (1936).
378. Brockman n, Haase, Ber., 70, 1733 (1937).
379. Collins et al., J. Chein. Soc., 1950, 1876.
380. Robertson, Whalley, Yates, J. Chem. Soc., 1950, 31 17.
381 .Pratt, Robinson, J. Chem. Soc., 1923, 739.
382. В г о с к ni a n n, F r e i e n s e h n e r, Ber., 77, 279 (1944).
383. Robertson, Whalley, J. Chem. Soc., 1950, 1882.
384. Perkin, Ray, Robinson, J. Chem. Soc., 1926, 950.
385. Keller, Robinson, J. Cheni. Soc., 1934, 1533.
386. Isarasena, Robertson, неопубликованные данные.
387. I к a w a, Stahmann, L i n k, J. Am. Chein. Soc., 66, 902 (1944).
Глава II
НАФТИРИДИНЫ
Л1. В е йс, Ч. Г а у з е р
ВВЕДЕНИЕ
• Соединения, содержащие два конденсированных пиридиновых кольца,
называются нафтиридинами. В зависимости от взаимного расположения ато-
мов азота нафтиридины могут быть шести типов (см. приведенную ниже схему).
Эти соединения называли также диазонафталинами, бензодиазинами и пири-
допиридинами. Нафтиридин типа 1,5 называется изонафтиридином, а типа
5 4
1,8.— 1,7- 1,6-
2,7 — копирином. Известны представители всех шести типов нафтиридинов,
однако из незамещенных описаны только 1,8- и 1,5-нафтиридины.
СИНТЕЗЫ
Методы синтеза нафтиридинов в большинстве случаев аналогичны мето-
дам получения хинолинов*. Их удобно классифицировать на два типа: тип А,
включающий реакцию циклизации с образованием пиридинового кольца,
и тип Б, когда эта реакция отсутствует. В синтезах типа А исходными вещест-
вами служат аминопиридины, а в синтезах типа Б — орто-дизамещенные
соединения. В первом случае (тип А) особое значение имеет электронодонор-
ная способность пиридинового кольца, тогда как во втором (тип Б) это свой-
ство существенного значения не имеет. Кроме того, известно несколько раз-
личных примеров синтеза, которые не относятся ни к первой, ни ко второй
группе реакций.
Синтезы типа А
Циклизации с образованием пиридинового кольца.Методы типа А пред-
ставляют видоизменения хорошо известных синтезов с применением этокси-
метиленмалонового эфира (ЭММЭ), методов Конрада — Лимпаха, Кнорра,
Комбе, Скраупа и других синтезов с использованием аминопиридина вместо
* См. т. 4, гл. II.
156
НАФТИРИДИНЫ
анилина. Аминониридин обрабатывают соответствующим карбонильным соеди-
нением, например ЭММЭ, ацетоуксусным эфиром или ацетил ацетоном с целью
получения кротоната, анила или амида, который затем циклизуют при нагре-
вании, часто в кислой среде. Подобные реакции циклизации сопровождаются
углерод-углеродной конденсацией ионного типа, причем пиридиновое кольцо
служит донором электронов, а карбоксильная группа — акцептором (см.
соединения I, III, V и VII). Из 2-аминопиридина образуется 1,8-нафтиридин
(II), из 3-аминопиридина — 1,5-(IV) или 1,7-нафтиридины (VI) и из 4-амино-
пиридина — 1,6-нафтиридин (VIII).
Таблица 1
Заместители в пиридиновом кольце, образующемся при циклизации
Карбонильное соединение
RCOCH2COOC2H5
rcoch2cooc2h6
RCOCH2COR'
С2Н5ОСН = С(СООС2Н5)2
сн2=снсно
RCHO, СНзСОСООН
СН3СН0, СН3СНО
CH^COOC^h
Метод
_________________
г
Кнорра
Конрада — Лимпаха
Комба
ЭММЭ
Скраупа
Дёбнера
Дёбнера— Миллера
Чичибабина
I
Заместители
2-ОН, 4-R
4-ОН, 2-R
2-R, 4-R'
4-ОН, 3-СООС2Н5
Нет
2-R, 4-СООН
2-СН3
2-ОН, 4-ОН
СИНТЕЗЫ
157
Эти реакции циклизации непосредственно или о одновременной дегидро-
генизацией приводят к образованию нового пиридинового кольца, которое
обычно содержит различные заместители. Характер заместителя зависит
от природы карбонильного соединения, а в некоторых случаях от применяемого
метода синтеза (табл. 1).
Вообще синтез нафтиридинов этими методами менее эффективен, чем
синтез хинолинов, что не является неожиданностью, так как гетероцикличе-
ский азот склонен оттягивать электроны из остатка пиридинового кольца,
снижая тем самым электронодонорные свойства последнего в реакциях цикли-
зации соединений I, III, V и VII по сравнению с бензольным кольцом соответ-
ствующих производных анилина (IX). Этот характер атома азота в пиридине
особенно ярко проявляется в положениях 2 и 4, как это показано в формуле X.
Действительно, соответствующие резонансные формы XI и XII с положи-
тельными зарядами в положениях 2 и 4 имеют существенное значение для
понимания строения j.самого пиридина [11.
IX
Поскольку циклизация производных 3-аминопиридина (III или V) долж-
на проходить по 2- или 4-положениям, можно ожидать, что эта реакция будет
идти с большими трудностями. Действительно, только небольшое число реак-
ций циклизации производных 3-аминопиридина протекает успешно. Цикли-
зация производных 2- или 4-аминопиридинов, по-видимому, протекает легче,
но применение этой реакции для получения нафтиридинов ограничивается
другими факторами.
Циклизация производных 2-аминопиридина: 1,8-нафтиридины. Общие
соображения. Значение 2-аминопиридинов для получения 1,8-нафтиридинов
ограничено тем, что промежуточные продукты — анил, кротонат или амид
(I) — могут циклизоваться не только в положении 3 пиридинового кольца
с образованием нафтиридина (II), но также по атому азота кольца, как это
показано в формуле XIII, что приводит к производному пиримидина (XIV).
ХШ XIV
Вообще нормальным ходом реакции является образование пиримидина.
Это не вызывает удивления, так как резонансная структура XV, которая
может рассматриваться как активированная форма, ведущая к образованию
пиримидинов, по-видимому, вносит значительный вклад в структуру произ-
водных 2-аминопиридина [2]. Электронодонорная способность гетероцикли-
ческого азота может быть дополнительно усилена благодаря таким резо-
нансным структурам, как XVI, поскольку вклад соответствующих структур
XI и XII в структуру самого пиридина значителен [1]. Резонансные струк-
туры XVII или XVIII [3], которые можно рассматривать как активированные
формы, приводящие к образованию 1,8-нафтиридинов, также могут иметь
значение, однако, как правило, реакция циклизации будет происходить только
при наличии стерических препятствий у атома азота в кольце.
158
НАФТИРИДИНЫ
Таким образом, хотя нафтиридины обычно не образуются из производных
2-аминопиридина с боковой цепью, их удается получить из производных
2,6-диаминопиридина, у которых реакция по циклическому атому азота затруд-
нена вследствие стерических препятствий [4]. Положение 3 в кольце произ-
водного 2,6-диаминопиридина, по-видимому, активируется благодаря нали-
чию резонансной структуры XIX.
Синтезы на основе этоксиметиленмалонового эфира (ЭЛ4Л4Э). Реакция
2-аминопиридина с ЭММЭ (XX), протекающая, по-видимому, с присоедине-
нием в положения 1,4 и сопровождающаяся отщеплением этоксигруппы,
приводит к образованию кротоната (XXI), который при циклизации дает
3-карбэтокси-4-окси-1,8-нафтиридин (XXII) или пиримидин (XXIII). Схемы
этих реакций приведены ниже. Нафтиридин образуется с хорошим выходом,
когда R является 6-амино-[5], 6-метил-[4] или 6-этоксигруппой [4]. Пирими-
дин [4] получается, когда R — 4-метил-, 5-метил-, 5-хлор-, 5-бром- или 4-эток-
сигруппы. Если R —-водород [4, 51, 4-хлор [4] или 6-бром [4|, то наблю-
далось лишь образование смол. Следует отметить, что два последних произ-
водных содержат относительно реакционноспособный атом галогена. Из 6-ок-
си-, 5-нитро- или 5-карбэтокси-2-аминопиридина кротонат не образуется |4|.
СООС2НЭ
ХХ111
Оба типа образующихся гетероциклов заметно отличаются друг от друга-
Нафтиридины (XXII) высокоплавки (выше 220°), нерастворимы в большин-
стве органических растворителей, за исключением пиридина, и гидролизуются
щелочами с образованием соответствующих нафтиридинкарбоновых кислот.
Пиримидины (XXIII) имеют более низкие температуры плавления (ниже 170°),
растворимы в большинстве органических растворителей и разлагаются щело-
чами с образованием исходных 2-аминопиридинов [4]. Реакцией декарбэтокси-
лирования (гидролиз и декарбоксилирование) эти нафтиридиновые эфиры,
по-видимому, могут быть превращены в 7-замещенные 4-окси-1,8-нафтири-
дины. Так, например, из 3-карбэтокси-4-окси-7-амино-1,8-нафтиридина был
получен с хорошим выходом 4-окси-7-амино-1,8-нафтиридин [51.
СИНТЕЗЫ
159
ОН ОН
Х\//СООС2Н5 |) Разб. NaOH
H2N-\ X } V320-330"(-co;r h2N-X К J
NN NN
Синтезы на основе |3-кетоэфиров. Методы Кнорра и Конрада — Лимпаха.
2-Аминопиридины подобно анилину [61 могут вступать в реакцию с р-кето-
эфирами либо по эфирной группе с образованием амида (XXIV), либо по кетон-
ной (или енольной) — с образованием кротоната или анила (XXV). Эти два
направления реакции могут быть выражены в общем виде следующими урав-
нениями:
XXV
Циклизация амида (XXIV) (реакция Кнорра) приводит к образованию
либо 2-окси-1,8-нафтиридина (XXVI), либо пиримидина (XXVII). Подобно
XXV!)
XXIV
этому циклизация анила (XXV) (реакция Конрада — Лимпаха) дает 4-окси-
1,8-нафтиридин (XXVIII) или пиримидин (XXIX).
Амиды (XXIV, R = Н, R' = CeHs; R = Н, R'== СН3), полученные из
2-аминопиридина и этилового эфира бензоилуксусной кислоты |71 или аце-
тоуксусного эфира [81, образуют соответствующие пиримидины (XXVII,
R = Н, R'= С6Н5; R = Н, R'= СН3). Однако амид (XXIV, R = 6-NH2.
R'= СН3), полученный из 2,6-диаминопиридина и ацетоуксусного эфира
по реакции Кнорра, дает с хорошим выходом 2-окси-4-метил-7-амино-1,8-
нафтиридин (XXVI, R = 7-NH2, R'=CH3) [9, 10]. Описано также получение
нафтиридина (XXVI, R = 7-NH2, R'= С6Н5) из 2,6-диаминопиридина и
60
НАФТИРИДИНЫ
С-ацетилпроизводного этилового эфира бензоилуксусной кислоты, причем
в ходе реакции ацетильная группа, по-видимому, отщеплялась fill.
Аналогично анил (XXV, R = Н, R' = СН3), полученный из 2-аминопи-
ридина и ацетоуксусного эфира, вероятно, образует пиримидин (XXIX,
R = Н, R' = СН3) [121. Однако анил (XXV, R = 6-NH2, R'= СН3), полу-
ченный из 2,6-диаминопиридина и ацетоуксусного эфира в результате реак-
ции Конрада—Лимпаха, дает с 8%-ным выходом 2-метил-4-окси-7-амино-
1,8-нафтиридин (XXVIII, R = 7-NH2, R' = СН3) 112]. Эта реакция совер-
шенно неожиданно проходила при комнатной температуре в условиях, при
которых обычно образовывался только анил, однако продолжительность реак-
ции была значительно больше, чем обычно. Кроме нафтиридина, был выделен
также дианил (XXX) с выходом 13%ь Циклизацией дианила путем кратко-
временного нагревания с обратным холодильником в кипящем теплоносителе
(даутерм А) был получен продукт, представляющий собой либо пиримидонаф-
тиридин (XXXI), либо пиридонафтиридин (XXXII) 112].
ОС2Н5
С = О
ОН
I
О = С (2CI-H
\ Z
СН
XXXI
XXXII
В результате конденсации 2,6-диаминопиридина с диэтиловым эфиром
щавелево-а-пропионовой кислоты, который может рассматриваться как р-кето-
эфир, по-видимому, образуется 1,8-нафтиридин (XXXIV) [12]. Эта реакция,
которую проводили в присутствии драйерита при продолжительном нагрева-
нии с обратным холодильником в кипящем этаноле, по-видимому, протекала
с образованием промежуточного анила (XXXIII, R = 6-NH2). Однако в резуль-
тате реакции между 2-аминопиридином и диэтиловым эфиром щавелево-а-
пропионовой кислоты [12] не удалось выделить какого-либо продукта. Из 2-
амино-6-метилпиридина не был получен ни нафтиридин, ни пиримидин, а был
выделен малеимид (XXXV) 112]. Он получается, по-видимому, в результате
реакции промежуточно образующегося анила (XXXIII, R = 6-СН3) со вто-
рой молекулой аминопиридина. Описан аналогичный малеимид, выделенный
при реакции л-хлоранилина с диэтиловым эфиром щавелево-а-пропионовой
кислоты [13].
Синтезы на основе fi-дикетонов. Метод Комби. При взаимодействии
2,6-диаминопиридина с ацетилацетоном образуется анил, который в резуль-
тате циклизации дает 2,4-диметил-7-амино-1,8-нафтиридин (XXXVI) ] 14, 151.
СИНТЕЗЫ
161
Выход (60—70%) при проведении реакции в концентрированной серной кис-
лоте значительно выше, чем в присутствии хлорида цинка Г10, 16].
+ С2Н5ООССОСН(СП3)СООС2Н5
Аналогично проведена циклизация анила, полученного из 2,6-диамино-
пиридина и несимметричного 0-дикетона — бензоилацетона [17]. Однако,
какой образовался изомер — 2-метил-4-фенил- или 2-фенил-4-метил-1,8-наф-
тиридин — окончательно не было установлено. Несмотря на то что из 2-ами-
но-6-метилпиридина и ацетилацетона анил получается легко, попытки его
циклизации остались безрезультатными [12].
Синтезы на основе малонового эфира. Метод Чичибабина. 2-Аминопиридины
реагируют с малоновым эфиром, причем образуются амиды (XXXVII), которые
затем могут быть превращены циклизацией либо в нафтиридины (XXXVIII),
либо в пиримидины (XXXIX); фактически нафтиридин удается получить
только в том случае, когда заместитель находится в положении 6. Если этими
заместителями являются амино-, ацетамидо- или этоксигруппа [18],
то с высокими выходами получаются 7-замещенные 2,4-диокси-1,8-нафти-
ридины (XXXVIII). Из 6-метилпроизводного [18] выделен только соответ-
ствующий нафтиридин с очень малым выходом. Циклизация с образованием
пиримидина (XXXIX) протекает с хорошим выходом в случае 4- и 5-метил-[ 18],
5-хлор-[18] или 5-бром-[18] и незамещенного [29] аминопиридина. С моно-
алкил- или моноарилзамещенным малоновым эфиром 2-амино-6-метилпири-
II Заказ № 978
162
НЛФТИРИДИНЫ
дин дает 2,4-диокси-7-метил-1,8-нафтиридин, замещенный соответственно в по-
ложении 3 (выход 35—50%) [20].
XXXVIII XXXIX
Попытки синтеза по Дёбнеру, Дёбнеру — Миллеру и Скраупу. Имелись
сообщения, что 2-аминопиридин вступает в реакцию Дёбнера с пировино-
градной кислотой и бензальдегидом 121 ], анисовым альдегидом [22] и другими
ароматическими альдегидами [221 с образованием 2-арил-1,8-нафтиридин-
карбоновых-4 кислот. Однако более поздние работы показали, что 1,8-наф-
тиридины, по-видимому, не были получены, а вместо них были выделены такие
соединения, как соединение XL [23]. Оно, вероятно, образуется в результате
присоединения 2-аминопиридина к n-метоксифенилпировиноградной кислоте.
Этим соединениям приписана пиримидиновая структура [24].
СН3О-^ ^-СНО + СН3СОСООН----------->
х---х 2-Аминопиридин
----->сн3о-^ ^-СН--СНСОСООН---------------->
о
----> СН3О-^ ^—СНСН2ССООН
x=z I
NH
XL
Неудачной была также попытка осуществить реакцию Дёбнера — Мил-
лера с 2-аминолепидином и паральдегидом с целью получения бензо-2,5-
диметил-1,8-нафтиридина [25].
Сообщено, что в обычных условиях реакция Скраупа с 2-аминопиридином
[26], а также с 2-аминолепидином [25] не происходит.
Попытки циклизации 2-(р-карбоксианилино)пиридинов. Попытки про-
вести синтезы различных бензо-4-окси-1,8-нафтиридинов циклизацией пири-
дина [27—291 или хинолина [30—32], замещенных в положении 2 о-карбок-
сианилиновой группой, привели к образованию только производных пири-
мидина. Поэтому взаимодействие 2-хлорпиридина с антраниловой кислотой
приводит к соединению XLII через промежуточное образование соединения
XLI [27]. Соединение XLII было также получено из 2-аминопиридина и
о-хлорбензойной кислоты [29]. В отношении некоторых соединений, полу-
ченных при этой конденсации, было высказано предположение о возможности
образования антраниловой структуры, как, например, XII (см. стр. 166)
Синтезы
163
[31, 32]. Однако это маловероятно, так как строение XLII подтверждено
независимым синтезом [27, 29].
НООС-/^
И !-----ч I I тт ।
НООС I О = С I
\Z\ \
\/
XLI XLII
Попытки синтеза по Померанцу — Фритчу. Неоднократные попытки
провести синтез изохинолинового типа по Померанцу — Фритчу* с шиффо-
выми основаниями (например, XLIII), полученными из 2,2-диэтоксиэтилами-
на и пиридил-2-, пиридил-3- и пиридил-4-альдегидов, а также хинолин-2-аль-
дегида, оказались неудачными. Применение различных кислотных реагентов
в качестве циклизующих агентов приводило либо к гидролизу шиффова осно-
вания и регенерации исходного альдегида, либо к образованию смол [33].
Попытка синтеза 1,7-нафтиридин а показана на приведенной ниже схеме.
(ОС2Н5)2
XLUJ
Хотя основой рассмотренных синтезов и не являются реакции производных
аминопиридина, все же можно было ожидать, что циклизацию с образованием
пиридинового кольца удастся провести.
Циклизация производных 3-аминопиридина: 1,5-нафтиридины. Общие,
соображения. Как уже отмечалось, производное 3-аминопиридина должно
подвергаться циклизации по любому из двух наиболее инактивированных
положений — 2 или 4. Хотя резонансные структуры, такие, как структура I,
могут облегчить циклизацию, их влияние, по-видимому, в значительной
степени парализовано участием резонансных структур типа II, поскольку
подобные ей структуры XI и XII (стр. 157) вносят наибольший вклад в струк-
туру самого пиридина [1].
Фактически для ряда производных 3-аминопиридина не удалось осу-
ществить циклизации. Большего успеха можно было бы достигнуть при ис-
пользовании некоторых замещенных 3-аминопиридинов, таких, как 3,5-диами-
нопиридин, у которого положения 2 и 4 сравнительно более реакционноспо-
собны. Когда циклизация происходит, то, по крайней мере в изученных слу-
чаях, реакция предпочтительно протекает в положении 2 с образованием
н н
I II
* См. т. 4.
11*
164
НАФТИРИДИНЫ
1,5- нафтиридина. Циклизация, несомненно, возможна и в положении 4 с обра-
зованием 1,7-нафтиридинов, если у исходных производных 3-аминопиридина
положение 2 блокировано каким-либо заместителем. В одном из сообщений
указывается, что у N-окиси 3-аминопиридина циклизация происходит пред-
почтительно в положении 4 с образованием 1,7-нафтиридина.
Метод с применением этоксиметиленмалонового эфира (ЭММЭ). Реак-
ция 3-аминопиридина с ЭММЭ дает 3-карбэтокси-4-окси-1,5-нафтиридин (III)
15, 34]. Однако удовлетворительные выходы были достигнуты только при
нагревании реагентов в кипящем теплоносителе (даутерм А) в сильно раз-
бавленном растворе. При гидролизе и последующем декарбоксилировании
соединения III с хорошим выходом образовывался 4-окси-1,5-нафтиридин
[5, 34]. Синтезы по этому методу проводились также с З-амино-6-метоксипи-
ридином [35], З-амино-6-бутоксипиридином [35] и 3-аминохинолином [36].
При реакции N-окиси 3-аминопиридина с ЭММЭ циклизация протекает
по 4-положению с образованием 1,7-нафтиридина (IV) [37]. Восстановление,
гидролиз и декарбоксилирование последнего приводят к образованию 4-окси-
1,7-нафтиридина с суммарным выходом 22% [37].
/x-nh2
I н -J-C2HSOCH--С(СООС2Нй)2 —
NZ
N N
Z\z ч. Ч
I II ( (I) Разб. NaOH I II |
> к Д^-СООС2Па (^) 320-330» *
N । N ।
ОН ОН
III
+ С2Н5ОСН = С(СООС2Н5)2 ——->
ОН ОН
1 (1) Fe, СНзСООН [
\__rQOCoHr (2) Разб. NaOH У Ч-Z \
----> I II > CUUC2H5---------------> |
O^_N I (3) Кипящий хинолин N
\/\/ Z
N Х/ N
IV
Приведенный пример циклизации указывает на способность N-окиси
пиридина претерпевать электрофильное замещение предпочтительно в поло-
жении 4 [38].
Методы Кнорра, Конрада — Лимпаха и Комба. Эти методы не дали поло-
жительных результатов с 3-аминопиридином, несмотря на то что промежуточ-
ные анилы или амиды были предварительно выделены ]36]. Однако, как сооб-
щается, реакция Конрада — Лимпаха с З-амино-6-метоксипиридином и 3-ами-
но-6-бутоксипиридином проходила успешно и был получен с 80%-ным выхо-
дом соответствующий 2-метил-4-окси-6-алкокси-1,5-нафтиридин [35]. Метод
Кнорра оказался неприменимым к 3-аминохинолинам, однако по методу
Конрада — Лимпаха из этого амина оказалось возможным получить с 40 % -
ным выходом 2-метил-4-окси-5,7-бензо-1,5-нафтиридин (V) [36].
Попытки провести реакцию Кнорра с 3-аминохинальдином и ацетоуксус-
ным эфиром были безуспешны [39]. Сообщается также о неудачной попытке
проведения реакции Комба с ацетилацетоном и З-амино-6-ацетамидопириди-
СИНТЕЗЫ
165
ном [40], а также с 2,6-диметил-З-аминопиридином [39, 41 ], в случае которых
циклизация должна была бы пройти по 4-положению. Попытки получить
нафтиридины исходя из N-окиси 3-аминопиридина методами Конрада — Лим-
паха, Кнорра или Комба оказались безрезультатными. По-видимому, амино-
группа в этом соединении имеет недостаточно основной характер и не может
+ СН3СОСН2СООС2Н5
Комн. темп.
3 дня
СН2
/с/
С2Н5О й
230°
образовать с ацетоуксусным эфиром или ацетилацетоном необходимый про-
межуточный анил или амид [37].
Метод Скраупа. Этой реакцией из 3-аминопиридина [36, 42, 43] полу-
чен 1,5-нафтиридин (36%), из З-амино-4-оксипиридина [44] — 4-окси-1,5-
нафтиридин, а из З-амино-6-оксипиридина [44, 451 и 3,6-диаминопиридина
[40]—• 2-окси- и 2-амино-1,5-нафтиридины соответственно. З-Аминохинолин
был превращен в 2,3-бензо- 1,5-нафтиридин [46], причем указывается, что
выходы были низкими. Сам 1,5-нафтиридин или его однозамещенное производ-
ное этим путем получаются легко.
Глицерин,
H2SP4
AsjOj
Из 2-окси-З-аминопиридина, циклизация которого в положении 2 исклю-
чена, получен с 20%-ным выходом 8-окси-1,7-нафтиридин [47].
I NH2
ОН
Глицерин, H2SO4
Л1-Ннтробензол сульфокислота
Реакция Скраупа, проведенная с N-окисью 3-аминопиридина, которая
сЭММЭ циклизуется в положении 4 с образованием 1,7-нафтиридина (стр. 164),
дает только 1,5-нафтиридин. По-видимому, N-окись первоначально теряет
кислород и переходит в 3-аминопиридин, вступающий затем в реакцию Скрау-
па [37].
Н
I
С6н5-с — о
сн2 с = о
Z'N-NHz С6Н6СНО
Ci —L li СН3СОСООН
N
VII
166
НАФТИРИДИНЫ
Методы Дёбнера и Дёбнера — Миллера. Реакция Дёбнера с 3-аминопи-
ридином, бензальдегидом и пировиноградной кислотой не идет [48J. В случае
З-амино-6-хлорпиридина (VI) пиридиновое кольцо не участвует в циклизации,
и образуются продукты реакции, которые ранее принимали за пирролиди-
ноны [48], а в настоящее время считают производными фуранона (VII) [49]
(см. стр. 165).
Реакция Дёбнера — Миллера между З-амино-6-оксипиридином и аце-
тальдегидом дает 2-метил-6-окси-1,5-нафтиридин (VIII) с выходом 27% [45,
NH2
+ 2СН3СНО
но Д;
N
Конц. HCI
VIII
50]. Подобным же образом З-амино-6-хлорпиридин образует 2-метил-6-хлор-
1,5-нафтиридин [51], а 1-метил-5-аминопиридон-2— 1,6-диметил-1,5-нафти-
ридон-2 (IX) [45].
nh2-
/ N
9ГЫ СИП КОНЦ’ НС1 СНЗ
2L.ri3L.rlkJ
„ /=°
N
(30%)
СНз
СН3
IX
Имеются данные, что реакция Дёбнера — Миллера не идет с 2,6-диме-
тил-3-аминопиридином, у которого циклизация должна происходить в поло-
жении 4, приводя к образованию 1,7-нафтиридина [52].
В результате конденсации 3-аминопиридина с ацетальдегидом и ацето-
ном образуется 2,4-диметил-1,5-нафтиридин [40], а З-амино-6-оксипиридин
дает 2,4-диметил-6-окси-1,5-нафтиридин [40].
О
II
CH3CIIO-|-CII3CCH3
Сухой I1CI
N ।
сн3
N
Циклизация 3-(о-карбоксианилино)пиридинов. При циклизации соеди-
нения X (R = С1) образуется 4-окси-1,5-нафтиридин (XI, R = С1), который
н
XII
СИНТЕЗЫ
167
можно превратить в трихлорпроизводное [53]. Указывается, что аналогично
из соединения X (R = Н) синтезировано соединение XI (R — Н) 154], однако
получить дихлорпроизводное из последнего не удалось [54]. Промежуточные
продукты X (R = Н и С1) были получены конденсацией 2,4-дихлорбензойной
кислоты с соответствующим 3-аминопиридином.
Петров и Стёрджен [45] изучили эту реакцию и высказали предположе-
ние, что продуктом ее является антранил (XII). Имеются сообщения, что
некоторые попытки проведения циклизации 3-(о-карбоксианилино)пиридина
и 3-(о-карбоксианилино)хинолина оказались неудачными [55, 56].
Однако описан и успешный синтез подобного типа [57]. Циклизацию
проводили, нагревая соединение X (R = алкоксигруппа) с хлорокисью фосфора.
Этим методом можно непосредственно получать 4-хлор-1,5-нафтиридины, кото-
рые могут быть превращены в 4-аминопроизводные, аналоги акридинового
препарата, хинакрина, обладающего противомалярийным действием. Сообщает-
ся, что один из таких аналогов (ХПа, азакрин) обладает ценными антималя-
рийными свойствами.
HNCH(CH2)3N(C2H5)2
I
СНз
ХПа
Циклизация производных 4-аминопиридина: 1,6-нафтиридины. Общие
соображения. Производные 4-аминопиридина подобно производным 2-амино-
пиридина могут циклизоваться по наименее дезактивированному третьему
атому углерода кольца с образованием нафтиридинов. В отличие от производ-
ных 2-аминопиридина замещенные 4-аминопиридина не могут циклизоваться
по атому азота в кольце с образованием пиримидинов. Поэтому можно было
ожидать, что 4-аминопиридины будут циклизоваться в нафтиридины не толь-
ко с большей легкостью, чем 3-аминопиридины (которые циклизуются по наи-
более дезактивированному положению), но и легче, чем производные 2-ами-
нопиридина, многие из которых циклизуются с образованием пиримидинов.
В тех случаях, когда могли быть получены промежуточные производные 4-ами-
нопиридина, это предположение подтвердилось. Однако из 4-аминопиридина
в отличие от 2- или 3-аминопиридинов не удалось получить промежуточных
продуктов обычного типа, тогда как из 4-аминохинолина они образуются
чрезвычайно легко.
Метод на основе этоксиметиленмалонового эфира (ЭММЭ). Эта реакция
проводилась с 4-аминопиридином и привела к 1,6-нафтиридину (I) с суммар-
ным выходом 68%; с 4-аминохинолином выход составил 93% [36].
+ С2Н5ОСН = С(СООС2Н5)2
i 10°
ОС2Н5
о=с
Кипячение с обратным
холодильником
------------------->
в даутерме А
он
I
/VVcooc2H5
168
НАФТИРИДИНЫ
Методы Кнорра, Конрада — Лимпаха и Комба. Попытки получить анил
или амид из 4-аминопиридина и ацетоуксусного эфира, а также анил из этого
амина и ацетилацетона оказались безуспешными, несмотря на относительно
жесткие условия проведения реакции [36]. В этом отношении 4-аминопири-
дины напоминают о-нитроанилин, который также не дает анила [58] или амида
[36] с ацетоуксусным эфиром. Эти свойства, по-видимому, можно объяс-
нить относительно слабым основным характером амина [36]. Однако были
получены промежуточный анил (II) и амид (III) из 4-аминохинолина и ацето-
уксусного эфира и установлено, что они способны циклизоваться по методам
Конрада — Лимпаха и Кнорра с образованием с высокими выходами соеди-
нений IV и V соответственно [36].
СН3
I
С
S \
N СН2
I
О = С- ОС2Н5 комн, тем п.
Катал. НС1
сн3
I
II
IV
70%
ОН
I
' Lc„,
У\/\/ 3
О
II
С
NH СН2
I
о=с—сн3
I II I Конц. H2SO4 I II 1
L Д / 100», 30 ДЛ" L Д )
N 4 N
III V
58%
Реакцию Кнорра проводили также с 4-аминохинальдином и получил:'
с удовлетворительным выходом соединение VI [39].
ОН
VI
Методы Скраупа и Дёбнера. С 4-аминопиридином синтез по методу Скрау-
па не удалось осуществить [43], что обусловлено слабой основностью этого
амина [43]. Однако реакция Скраупа проходит успешно с 4-аминохинальди-
ном, хотя и с невысоким выходом [39]. 4-Аминохинальдин вступает в реакцию
Дёбнера — Миллера [39], но выход при этом невелик.
Циклизация 4-(о-карбоксианилино)пиридинов. Конденсация 4-хлор хи-
нальдина с антраниловой кислотой дает соединение VII [59]. Синтез протекает
СИНТЕЗЫ
169
в две стадии. Нагревание реагентов в кипящем растворе уксусной кислоты
приводит к промежуточному продукту — 4-(о-карбоксианилино)хинальдину,
который затем циклизуется при кратковременном нагревании при 100° в рас-
творе концентрированной серной кислоты; о выходах не сообщается.
. П
У\/
7 I Lo„
Y A + ILN - р р/Y
k/LLc"3 noocjjl NIJ
VII
Синтезы типа Б
Циклизация opzwo-дизамещенных соединений. opmo-Дизамещенные пири-
дины, как правило, аминопиридины, имеющие структуру типа I или II, а также
opmo-дизамещенные производные бензола могут вступать в реакции циклиза-
ции с различными соединениями, в том числе содержащими активный водород,
образуя нафтиридины. Из 2,3-дизамещенных аминопиридинов со структурой I
получаются 1,8-нафтиридины, а из соединений со структурой II — 1,5-нафти-
ридины.
I II
Эти синтезы обычно основаны на применении некоторых методов получе-
ния хинолинов, в том числе методов Ниментовского, Фридлендера и Пфитцин-
гера [2 ]. В большинстве случаев синтез нового пиридинового кольца вклю-
чает две конденсации: с образованием азот-углеродной и углерод-углеродной
связей.
Азот-углеродная конденсация приводит к получению анила или амида,
а углерод-углеродная, как правило, является конденсацией альдольного или
ацильного типа. Окси нафтиридин получается в результате промежуточного
образования амида или протекания конденсации ацильного типа.
Поскольку эти методы обычно не приводят к циклизации с образованием
пиридинового кольца, они не ограничены факторами, рассмотренными при
обсуждении синтезов типа А.
Синтезы на основе о-аминопиридинкарбоновых кислот или антраниловой
кислоты. Метод Ниментовского. Метиловый эфир 2-аминоникотиновой кис-
лоты при реакции с малоновым эфиром образует 2,4-диокси-З-карбометокси-
1,8-нафтиридин (III), который в результате гидролиза и декарбоксилирования
превращается в 2,4-диокси-1,8-нафтиридин (IV). Заслуживает внимания факт
переэтерификации в процессе реакции. Сообщается о хорошем суммарном
выходе продукта [60]. Аналогичными методами из 2-амино-З-карбометокси-
хинолина и З-амино-З-карбэтоксипиридина получены с хорошими выходами
2,4-диокси-6,7-бензо-1,8-нафтиридин (V) [61 ] и 2,4-диокси-1,5-нафтирипин
(Va) [62] соответственно.
Примером другого применения этого метода служит реакция между
3-аминопиколиновой кислотой и флороглюцином с образованием соединения
VI, которое в результате окисления и последующего декарбоксилирования
переходит в 4-окси-1,5-нафтиридин [63]. Сообщается о хорошем выходе веще-
170
НАФТИРИДИНЫ
ства VI, однако выход 4-окси-1,5-нафтиридина не указывается [63]. Это соеди-
нение, по-видимому, более удобно получать синтезом с ЭММЭ (стр. 164).
/Ч СООСНч
| • +сн.да^нЛ 2*!==.
N
ОН
ОН
ЧЧХЧ- СООСНз 15%-пый води, р-р КОН ЧЧЧ'Ч
он
NZ N
III
Il ОН
N N
IV
Описан аналогичный путь образования 4-окси-1,8-нафтиридина из 2-амино-
никотиновой кислоты [64].
2 05°, 30 мин
без растворителя
VI
(1) HNO3
(2) -СО2
Антраниловая кислота и 2,4,6-триоксипиридин дают 1,6-нафтиридин
(VII), но в результате последующего окисления непрочное диоксипиридиновое
кольцо разрушается и образуются производные хинолина [65]. Синтез соеди-
ОН
ОН ОН
/Ч- соон
Ijj-NH2
II N
НО— ОН
ЧЧ/Ч/Ч
I SI I Чо„
\/\
N
VII
пения VII проходил с отличным выходом при проведении реакции в растворе
уксусной кислоты или в нейтральной водной среде, однако попытки осущест-
вить реакцию в щелочной среде или в растворе минеральной кислоты были
ОН
СООН |
,/Ч/ /Ч Ч\/Ч/ч
II I II U I I! II
\ /\Z V /\
N N N N
Н Via
СИНТЕЗЫ
171
безуспешными [65]. Интересно, что аналогичная реакция между антраниловой
кислотой и флороглюцином приводит к трем различным продуктам [66].
При нагревании 2-анилиноникотиновой кислоты с полифосфорной кисло-
той и хлорокисью фосфора образуется соединение Via [67].
Синтез незамещенного бензо-1,6-нафтиридина (VIb) проводят следующим
образом: циклизуют полученное из анилина и 1-ацетил-З-карбэтоксипипери-
дона-4 о-дизамещенное производное пиперидина (VI6); после снятия ацетиль-
ной группы и дегидрирования образуется нафтиридон, который превращают
с помощью цинка в щелочи в дигидропроизводное и, наконец, дегидрируют
[68].
С2Н5ООС
V16
(I) НС1
(2) Pd/C
(3) Zn, NaOH
(4) Pd/C
VIb
Конденсацией 3-аминопиколиновой кислоты с метазоновой кислотой
(оксимом нитроацетальдегида) с образованием промежуточного этилиденового
производного (VIII) синтезирован З-нитро-4-окси-1,5-нафтиридин (IX), при-
чем выход не указан [44]. Этот нафтиридин был также получен нитрованием
4-окси-1,5-нафтиридина, который можно синтезировать по реакции Скраупа
[44] из З-амино-4-оксипиридина. Сообщено о безуспешной попытке осущест-
вить реакцию между 3-аминоизоникотиновой и метазоновой кислотами
[69, 70].
nh2
II ,|_соон +HON-chch2no2 ->
N
N = CHCH2NO2
COOH
N
(CH3CO)2O
CHgCOONa
IX
VIII
Синтезы с антраниловым альдегидом. Метод Фридлендера. При взаимо-
действии антранилового альдегида с 2,4,6-триоксипиридином образуется 2,3-
бензо-5,7-диокси-1,6-нафтиридин (X); выход не указан [71]. Из продуктов
реакции было выделено также вещество XI, которое, как предполагают, обра-
зуется в результате конденсации соединения X со второй молекулой антра-
нилового альдегида. В результате обработки соединения X соляной кислотой
при 215° или горячей 25%-ной щелочью диоксипиридиновое кольцо разру-
шается и образуются производные хинолина [71].
Анил n-толуидина и З-аминопиридил-4-альдегида (XII) были применены
в синтезе по Фридлендеру для получения с высокими выходами некоторых
1,7-нафтиридинов [69]. Так, например, конденсацией соединения XII с аце-
тофеноном получен 2-фенил-1,7-нафтиридин (XIII), а с циклогексаноном —
тетрагидробензо-1,7-нафтиридин (XIV) [70]. При действии на соединение XII
172
НАФТИРИДИНЫ
изонитрозоацетона образуется с 56%-ным выходом 1,7-нафтиридин-2-аль-
доксим. Анил (XII) приготовлен окислением З-нитро-4-пиколина двуокисью
ОН
V сно /А
I I' + II N ->
4/~nh2 но-IM-он
он он
XI
селена, в результате чего образуется З-нитропиридил-4-альдегид (37%) с по-
следующим получением производного п-толуидина (95%), которое, наконец,
восстанавливается сульфидом натрия (80%) [691.
XIV XII
66%
XIII
84%
Восстановление оксима (XV) сопровождается циклизацией с образова-
нием 5,8-диокси-7-карбометокси-1,6-нафтиридина (XVI) [721. Несмотря на то
что соединение XVI получается с хорошим выходом, синтез исходного оксима
(XV) сложен, поэтому соединение XVI удобнее получать методом расширения
цикла (стр. 174) [44].
ОН
х /соосн- хх А
I Л NOH | II J*
I Л il Ч /X СООСНз
N ХС — С—СООСН3 1У (
« он
XV XVI
Внутримолекулярные синтезы с образованием лактама. Лактамная цик-
лизация |3-[4-(3-аминопиридил)]акриловой кислоты (XVIII), полученной
восстановлением ее 3-нитропроизводного (XVII), дает с отличным выходом
2-окси-1,7-нафтиридин [69]. Этот синтез представляет собой видоизменение
предложенного Хиоцца способа получения 2-оксихинолина из о-нитрокорич-
СИНТЕЗЫ
173
ной кислоты [1, 73]. Промежуточный продукт (XVIII) синтезируют из 3-нит-
ро-4-пиколина окислением его двуокисью селена в 4-альдегид и последующей
конденсацией с малоновой кислотой [691.
А-сн = снсоон Fe(0H)2 сн = снсоон
^J-№2 * NJLNh2 yU-°h
XVII XVIII
Сплавление аммонийной соли Р-гомохинолиновой кислоты дает 6,8-диокси-
1,7-нафтиридин (выход 23%) [69, 74]. Этот метод аналогичен описанному син-
тезу 1,3-диоксиизохинолина из гомофталевой кислоты [75].
— СН2СООН Через стадию образования — ОН
I аммонийной солн .1
I’ II -СООН--------------------------> N
% z LUUH \
N N ।
ОН
Циклизация продуктов присоединения кетонов к четвертичным солям
никотинамида. Описан интересный синтез 1-окси-З-арил- или 1-окси-З-алкил-
2,7-нафтиридинов путем циклизации продуктов присоединения кетонов, легко
получаемых из четвертичных солей никотинамида [76, 77]. З-Фенилпроизвод-
ное синтезировано из продукта присоединения с ацетофеноном. Можно при-
менять также аддукты, полученные из хлорметилата-никотинамида, причем
метильная группа из промежуточного 2-хлорметилата нафтиридина удаляется
возгонкой при 300° в виде хлористого метила [77].
СН3
I I
С1 С1
Синтезы с отщеплением галогеноводородов. Циклизация соединения XIX
под действием спиртового раствора едкого кали дает дигидро-1,8-нафтиридин
VXNH2CH-CH2NHCH3
Вг
XIX
вг __Ч.
( II |/н 9Н’
WXCH2-Агн
I
н
(1) СНз 1
(2) КОН
XX
174
НАФТИРИДИНЫ
(XX) с низким выходом [78, 79]. Сообщается, что превращение этого продукта
в триметил-четвертичную соль и последующая реакция с едким кали дает
2-метил-6-бром-1,8-нафтиридин [75, 78].
Расширение кольца у циклических имидов. У N-замещенного циклического
имида (XXI), полученного взаимодействием этилового эфира бромуксусной
кислоты с калиевой солью хинолинимида, при нагревании с раствором мети-
лата натрия в метаноле происходит расширение кольца с образованием 5,8-
диокси-7-карбометокси-1,6-нафтиридина (XXII) (выход 50%) и изомерного 5,8-
диокси-6-карбометокси-1,7-нафтиридина (XXIII) (выход 20%) [47]. Сложные
метиловые эфиры образуются в результате переэтерификации под действием
растворителя. Основной продукт (XXII) после кратковременного нагрева-
ния в растворе иодистоводородной кислоты гидролизуется и декарбоксили-
руется с образованием 5,8-диокси-1,6-нафтиридина (XXIV) с выходом 55%
[80]. Несмотря на то что эта реакция была известна ранее [80—82], образова-
ние 1,7-нафтиридина (XXIII) первоначально не было обнаружено, возможно,
вследствие его большой растворимости в воде.
Эти же два продукта получены действием раствора метилата натрия
в метаноле на соединение XXV [83].
Расширение цикла может сопровождаться ионизацией а-водорода ими-
доэфира (XXI) с образованием аниона сложного эфира [84], который претер-
певает перегруппировку. Можно предположить, что перегруппировка прохо-
дит через стадию карбаниона XXVI, являющегося резонансной структурой
CHgONa
I!
—с—nhch2cooc2h5
-СООН
он
I
/^/^-.соОСНз
! i N
N ,
ОН
XXIII
20%
XXII % XXIII
XXV
аниона сложного эфира, в котором анионный атом углерода, реагируя с а-
карбонильной группой имидного кольца, образует соединение XXII (показано
стрелками), а с Р-карбонильной группой — соединение XXIII. 1,6-Нафтиридин
(XXII) является основным продуктом реакции, что обусловлено большей
реакционной способностью а-карбонильной группы.
РЕАКЦИИ
175
Аналогично происходит расширение цикла у имида N-замещенной цинхо-
мероновой кислоты (XXVII), причем образуется с хорошим выходом 1,4-
диокси-3-карбометокси-2,7-нафтиридин (XXVIII) [85]. Однако попытки гидро-
лизовать и декарбоксилировать его оказались безуспешными [85].
О
II ОН
С 'I
I || \-сн2соМ!н5 I || I СООСН*
с I
II ОН
о
XXVII XXVIII
Аналогичные синтезы изохинолинов проводят путем перегруппировки
фталимидоуксусных эфиров [86].
Разнообразные синтезы
Перегонка ди-(3-аминопропил)уксусной
нафтиридин с низким выходом [87].
nh2 h2n
СН2 О=С-ОН сн2
I I I
сн2 сн сн2
^СНг/ СН27
кислоты дает октагидро-1,8-
н2 Н2
Нг/^/ЛНз .
Н2\ Jh2
N N
РЕАКЦИИ
В настоящее время можно сказать, что для нафтиридинов характерны реак-
ции, свойственные соответствующим пиридинам *, хинолинам ** и изохино-
линам ***.
Реакции по гетероциклическим атомам азота. С кислотами гетероцикли-
ческие атомы азота нафтиридинов образуют соли. Они могут алкилироваться
алкилгалогенидами с образованием солей нафтиридиния [45, 52, 88, 89]. При
перегонке в вакууме иодметилатов 1,8-нафтиридина наряду с другими продук-
тами получен исходный нафтиридин [77, 88].
С перекисью водорода нафтиридины дают производные N-окисей [42, 45,
90, 91], причем, как правило, образуются диокиси [42, 44, 90]. Однако при
обработке 1,5-нафтиридина эквивалентным количеством водной надуксусной
кислоты получают смесь, из которой может быть выделена моноокись с низким
выходом ([42]; см. также [45]). Реакцией 1,5-нафтиридина с избытком водной
надуксусной кислоты получают 1,5-диокись с высоким выходом [42]. Обра-
* См. т. 1, гл. VIН.
** См. т. 1, гл. I.
*** См. т. 4
176
НАФТИРЙДИНЫ
ботка ди- и моноокисей 1,5-нафтиридина хлорокисью фосфора дает соответ-
ственно 2,6-дихлор- и 2-хлор-1,5-нафтиридины (реакция Мейзенгеймера, ср.
стр. 182).
О
Избыток водн. /
|рОС13
СН3
I
CH3CONH-\ Jx J- СН3
N N
Г I-
C2H5
НС(ОС2Н5)з
_________________
избыток (CHgC0)2O5
кипячение 30 мин с
обратным холодиль-
ником
N N
1+
с2н5
сн3
I '
CH3CONH Д А /-
N N
|+
с2н5
CHqCONH
сн3 сн3
I I
CH3CONh J'% Д J-CH=CH-CH=!4 J-NHCOCHa
N N
- !
с2н5
II
или NHCOCH3
XN
СН = СН- СН=/ 'N--C2II5 .
\ -/
I- I
сн3
III
или NHCOCH3
XN
С2Н5 —NZ \-СН = СН-СН=/ XN--C,H.
- Т '°Т
сн3 сн3
I IV
РЕАКЦИИ
177
Реакция алкилнафтиридинов. Можно было ожидать, что замещенные нафти-
ридины с алкильной группой в а- или у-положении по отношению к атому азота
в кольце будут по аналогии с соответствующими пиридинами * и хинолинами **
вступать в различные реакции конденсации типа Клайзена и Михаэля, в кото-
рых алкильная группа является донором водорода 192]. Однако, за исключе-
нием упоминаемых ниже случаев, примеры реакций такого типа не известны.
С целью получения ряда карбоцианиновых красителей из нафтиридина
были применены обычные методы синтеза. Так, например, конденсацией
1-иодэтилата 2,4-диметил-7-ацетамино-1,8-нафтиридина (I) с ортомуравьиным
эфиром в избытке уксусного ангидрида был получен карбоцианиновый краси-
тель с выходом 69% [89]. Строение этого продукта, для которого могут быть
предложены три формулы — II, III или IV, точно не определено. При проведе-
нии конденсации в щелочной среде (спирт — пиперидин, спирт — потащ или
пиридин — триэтиламин) выходы значительно ниже, около 20%. При приме-
нении ортоуксусного и ортопропионового эфиров вместо ортомуравьиного,
а также 1-иодэтилата 2,4-диметил-7-метокси-1,8-нафтиридина вместо 1-иод-
этилата 2,4-диметил-7-ацетамино-1,8:нафтиридина были получены удовлетво-
рительные результаты [89].
Реакции электрофильного замещения. Попытки нитрования 1,5-нафтири-
дина, как это можно было ожидать по аналогии с пиридином ***, оказались
безуспешными [42]. Однако наличие гидрокисла или аминогруппы облегчает
электрофильное замещение. Так, например, нитрование 2-окси-1,5-нафтиридина
даете хорошим выходом 2-окси-3-нитро-1,5-нафтиридин [44, 93], а из 4-окси-
1,5-нафтиридина получается 3-нитро-4-окси-1,5-нафтиридин [44, 94].
Аналогично при бромировании 4-окси-1,5-нафтиридина в водном растворе
с хорошим выходом образуется 3-бром-4-окси-1,5-нафтиридин [44]. По патент-
ным данным [85], хлорирование 2-амино-5,7-диметил-1,8-нафтиридина дает
с высоким выходом 3-хлор производное.
ОН ОН
N I N I
/ VS HNOg /\/\-NO2
I II I H2sot I Ii I
\ Z\ Z \./\ z
N N
Предположение, что замещающая нитрогруппа или атом галогена вступает
в положение 3 основано на аналогии с известными реакциями оксипиридинов
и оксихинолинов [44]. Строение З-нитро-4-окси-1,5-нафтиридина, получен-
ного нитрованием 4-окси-1,5-нафтиридина, твердо установлено путем синтеза
его из 3-аминопиколиновой и метазоновой кислот (см. стр. 171) [44]. Отме-
чается, однако, что при нитровании 2,7-диокси-5-метил-1,8-нафтиридина, по
всей вероятности, образуется 4-нитропроизводное [96].
Обработка 1,5-нафтиридина бромом в холодном растворе хлороформа дает
с высоким выходом соединение V [42], представляющее собой соль нафтири-
* См. т. 1, гл. VIII.
** См. т. 4, гл. I.
*** См. т. 1, гл. VIII.
12 Заказ № 978
178*
НАФТИРИДИНЫ
дилнафтиридиния. В результате реакции этой соли с водой при 100° образуется
смесь 1,5-нафтиридина и 4-окси-1,5-нафтиридина, которую можно разделить,
однако выходы не приводятся [42]. Последнее соединение было получено
Н2О
100°
также по реакции Скраупа из З-амино-4-оксипиридина (см. стр. 165) [42]
и синтезом на основе ЭММЭ из 3-аминопиридина с последующим гидролизом
и декарбоксилированием промежуточного 3-карбэтоксипроизводного [5, 34]
(см. стр. 164).
Сообщается [65, 71], что оксипроизводные 1,6-нафтиридинов (VII и X,
стр. 170, 172) в щелочной среде сочетаются с хлористым п-нитрофенилдиазонием.
Окси нафтиридины могут также вступать в реакцию с реакционноспособ-
ными альдегидами [96]. Так, например, описано, что при действии на 2,4-
диоксинафтиридин глиоксалевой кислоты образуется 3,3'-метилен-бцс-производ-
иое [97]. Это соединение было получено как аналог дикумарина.
Некоторые а-гидразинонафтиридины при действии азотистой кислоты
циклизуются с образованием производных тетразола [9, 11, 16]. Так, напри-
мер, 2,7-дигидразино-4-метил-1,8-нафтиридин (VI) превращается в соединение
VII |9]. В этой реакции гетероциклический атом азота ведет себя как донор
электронов. При окислении соединение VII переходит в тетразол (VIII) [9].
Замещение первичной аминогруппы на гидроксил. При действии азотистой
кислоты на некоторые 2-аминонафтиридины происходит замещение аминогруп-
пы на гидроксил, как это имеет место при обычных диазореакциях [11, 15, 16,
411. Так, например, из 2,7-диамино-4-метил-1,8-нафтиридина (IX) получают
с хорошим выходом 2,7-диокси-4-метил-1,8-нафтиридин (X) [9]. Данные об
успешном проведении реакций Зандмейера или Гаттермана с аминонафтири-
дином, по-видимому, отсутствуют.
Замещение гидроксильной группы на хлор или водород. Гидроксильная
группа, находящаяся в а- или у-положении молекулы нафтиридина, при
РЕАКЦИЙ
17й
обработке хлорокисью фосфора [9, 11, 15, 35, 981 или смесью хлорокиси фосфо-
ра с пятихлористым фосфором [9, 10, 45, 60, 93] может замещаться на хлор.
По-видимому, не известен случай замещения Р-гидроксильной группы на атом
галогена. Предпочтительное замещение гидроксильной группы в а-положении
видно на примере реакции 5,8-диокси-1,б-нафтиридина (XI)' с хлорокисью фос-
фора с образованием 5-хлор-8-окси-1,б-нафтиридина (XII) ([72, 99]; см. также
[49]).
СН3
I
СН3
HNO2
H2N -ч Д ,
N N
IX
ОН
но -4 Д 0H
N N
X
Cl
N
N Г
ОН
N у
ОН
XI
Сообщается о дегидроксилировании под действием цинковой пыли, однако
выходы при этом были очень низкие [59, 63]. При дегидроксилировании
у-нафтиридина цинковой пылью и едким натром выход составил 34% 1'68].
Замещение атома галогена основаниями. Атом хлора в нафтиридине, нахо-
дящийся в а- или у-положении, может замещаться под действием различных
нуклеофильных реагентов, в том числе алкоголятов металлов [14—16, 98,
100, 101], фенола [5, 45], меркаптидов металлов [101], аммиака [102], различ-
ных аминов [11, 14, 16, 45] и гидразинов [9, 11, 13, 41]. Реакция с аминами
нашла применение для синтеза аналогов некоторых антималярийных препа-
ратов [5, 53, 57, 103, 104]. Так, например, 4-хлор-1,5-нафтиридин, полученный
из соответствующего 4-оксипроизводного, при взаимодействии с 1-диэтилами-
но-4-аминопентаном, дал соединение XIII с выходом 85% [5].
CH3CH(CH2)3N(C2H5)2
I
NH
N I
Cl
N I
/ \/Ч
I II | + CH3CHCH2CH2CH2N(C2H6)2 ДД—Д
A' %,
N
XIII
Изучение реакционной способности 2,4-дихлор-1,5-нафтиридина по отно-
шению к нуклеофильным реагентам позволило установить ярко выраженное
предпочтительное замещение атома хлора в положении 2 на аммиак, гидразин
и воду (водн. соляная кислота) [62]. Однако в случае анилина и бензиламина
2,4-диаминопроизводные были получены даже в тех случаях (с анилином), ког-
да расходовалась только половина молярного эквивалента амина.
NHRj
N I
/ \,/Ч
LI J- NHRi
х/ N
R1 = CeH5, CeH5CH2
Cl
N i
// \/4
r = nh2, hnnh2, oh
12*
180
НАФТИРИДИНЫ
Для ряда 2,4-дихлор-1,8-нафтиридинов также наблюдалось, что атомы
хлора в положении 2 обладают предпочтительной реакционной способностью
по отношению к гидразину [105, 1061.
Из соответствующего 8-оксинафтиридина и диазометана был синтезиро-
ван 8-метокси-1,6-нафтиридин [84].
Другие реакции нуклеофильного замещения. Известен случай нуклеофиль-
ного замещения гидрид-иона на амид-ион при синтезе 2-амино-1,5-нафтиридина
из 1,5-нафтиридина и амида натрия [42].
При взаимодействии соединения XIV с амидом калия в жидком аммиаке
(комнатная температура, продолжительность 48 час) происходит замещение
фенильной группы с образованием 2-амино-1,7-нафтиридина (XV) с удовлетво-
рительным выводом [107].
KNI-b
XV
Реакции карбоновых кислот и их производных. Карбоновые кислоты при
нагревании легко декарбоксилируются [5, 34, 48, 60]; как уже указывалось
[11, 24, 39], эта реакция использовалась в некоторых синтезах. Декарбокси-
лированию подвергаются карбоксильные группы, находящиеся в а- и |3-поло-
жениях по отношению к атому азота в кольце. 2,4-Диокси-3-карбометокси-1,8-
нафтиридин при обработке аммиаком, а затем хлорокисью фосфора превращает-
ся в соответствующий дихлорнитрил (XVI) [100].
ОН С1
1 J
ZX/V- СООСН3 (!) NH3 1^X./S-CN
L J\)— °н <2) рос‘з L /\ J—ci
NN NN
XVI
Дегалогенирование, гидрирование и восстановление. Амино- и гидроксиль-
ная группы могут замещаться на водород через стадию дегалогенирования
(— NH2—— ОН * — Cl-^ — Н). Это очень важно для синтеза, так как
получение многих нафтиридинов связано с первоначальным образованием
амино- или оксизамещенных продуктов.
Описано несколько случаев успешного дехлорирования под действием
водорода в присутствии палладия на углекислом кальции. Однако выходы не
приводятся [10, 15, 40, 108]. Так, например, гидрирование 2,7-дихлор-4-метил-
1,8-нафтиридина (XVII) в присутствии этого катализатора дает 4-метил-1,8-
нафтиридин [15, 108]. Попытки осуществить дегалогенирование действием
водорода в присутствии катализатора — палладия на угле [15] или красным
фосфором п иодистоводородиой кислотой при 170—180е [9] привели не только
РЕАКЦИИ
181
к удалению хлора, но и, по крайней мере, к частичному восстановлению коль-
ца. При применении йодистого калия и иодистоводородной кислоты при 127
происходит гидролиз и образуется диоксипроизводное [9]. Однако сообщается
об удовлетворительном дехлорировании 1,6-нафтиридина иодистоводородной
XVII
Jлoтoй [47]. Описаны попытки дегалогенирования других хлорнафтириди-
различными путями: действием водорода над платиновым катализатором
, 99, 100] или над палладием на угле в присутствии метанольного раствора
ого кали [10, 40], с помощью порошкообразного железа и разбавленной
wHHoft кислоты [100], цинковой пыли и водно-спиртовой смеси [1001, под
действием концентрированной иодистоводородной кислоты и йодистого фосфора
[100]. Все эти попытки оказались либо безуспешными, либо приводили к вос-
становлению кольца. Отдельные попытки дегалогенирования .натрием в спирте
привели к образованию декагидропроизводных [98, 109]. В результате ката-
литического гидрирования 2,4-дихлор-1,8-нафтиридина в присутствии палла-
дия на угле получена смесь, из которой с низким выходом выделен 1,8-нафти-
ридин 199, 100).
В патенте [93] описано дегалогенирование и восстановление нитрогруппы
2-хлор-З-нитро-1,5-нафтиридина водородом над палладиевым катализатором.
Избежать при этом гидрирования ядра, по-видимому, удалось в результате
прекращения реакции на стадии, поглощения рассчитанного количества
водорода.
Некоторые нафтиридины, имеющие в одном из пиридиновых колец одну
или две метильные группы, подвергались гидрированию под высоким давле-
нием над платиной или никелем Ренея. Преимущественно гидрировалось
неметилированное пиридиновое кольцо с образованием тетрагидропроизвод:
ных [10, 16, 40, 65]. Однако восстановление этих метилнафтиридинов натрием
в спирте привело к образованию декагидро- и гексагидропроизводных, у кото-
рых были восстановлены оба кольца [40, 65]. Гидрирование 1,5-нафтиридина
над платиной или никелем Ренея дает тетрагидропроизводное [40].
Обработка щелочью толуолсульфонилгидразинного производного, полу-
ченного конденсацией н-толуолсульфонилгидразида с хлорнафтиридином,
является примером другого метода дегалогенирования [42, 62]. Этим путем
из 2-хлор-1,5-нафтиридина, а также из 2,6-дихлор-1,5-нафтиридина, получен
(без указания выхода) 1,5-нафтиридин [42].
N
Окисление. Как уже отмечалось (стр. 170), оксинафтиридины легко
окисляются [65]. Несколько неожидайно, что даже незамещенный 1,8-пафти-
182
НАФТИРИДИНЫ
ридин, по имеющимся данным, обесцвечивает холодный разбавленный водный
раствор перманганата калия 188].
Моноиодметилат 2,4-диметокси-1,8-нафтиридина (XVIII) окисляется фер-
рицианидом калия с образованием производного нафтиридона (XIX) [109].
ОСН3
I
II
к ;1 ,/С- ОСНз
N, N
I I-
СН3
K3Fe(CN)6
---------->
XVIII
ОСН3
I
i II
О- ^-ОСНз
N N
I
СНз
XIX
N-Окиси. Образование и реакции N-окисей нафтиридина изучены весьма
ограниченно [106]. При взаимодействии 1,5-нафтиридина с 30%-ной перекисью
водорода в уксусной кислоте получается Мх,М5-диокись и небольшое количест-
во моноокиси. В противоположность легко протекающему нитрованию N-окиси
пиридина с образованием 4-нитропроизводного [ПО], N-окиси 1,5-нафтиридина
в условиях, которые не ведут к разложению, совершенно не поддается нитро-
ванию. Это обусловлено дезактивирующим влиянием пиридинового кольца,
примыкающего к месту возможного замещения.
Диокись 1,5-нафтиридина при действии хлорокиси фосфора претерпевает
реакцию Мейзенгеймера и дает 2,6-дихлор-1,5-нафтиридин. Окисление этого
дихлорпроизводного перекисями ведет к образованию диокиси, однако с поте-
рей одного атома хлора.
В противоположность этому 2,4-дихлор-1,8-нафтиридин относительно
устойчив в реакциях образования N-окисей. Он дает с небольшим выходом
диокись (также с потерей одного атома хлора) и продукт, принимаемый за моно-
окись этого дихлорпроизводного (см. стр. 179).
ЛИТЕРАТУРА
1. S с h о m a k е г, Р a u I i n g, J. Am. Chem. Soc., 61, 1769 (1939).
2. S t e с к, Ewing, J. Am. Chem. Soc., 70, 3397 (1948).
3. R e m i c k, Electronic Interpretations of Organic Chemistry, 2nd ed., John Wiley and
Sons, New York, 1949, p. 372.
4. L a p p i n, J. Am. Chem. Soc., 70, 3348 (1948).
5. Adams et al., J. Am. Chem. Soc., 68, 1317 (1946).
6. Hauser, Reynolds, J. Am. Chem. Soc., 70, 2402 (1948).
7. Зейде О. A., Ber., 58, 352 (.1925).
8. C r i p p a, S c e v о 1 a, Gazz. chim. itai., 67, 327 (1937) [C. A., 32, 166 (1938)].
9. 3 e й д e O. A., Ber., 59, 2465 (1926).
10. M i у a к i, J. Pharin. Soc. Japan, 62, 26 (1942) [C. A., 45, 627 (1951)].
11. Mangini, Colonna, Boll. sci. fac. chim. ind. Bologna, 1941, 85 [C. A., 37,
3096 (1943)].
12. Hauser, W e i s s, J. Org. Chem., 14, 453 (1949).
13. Surrey, G u t 1 e r, J. Am. Chem. Soc., 68, 514 (1946).
14. Mangini, Boll. sci. fac. chim. ind. Bologna, 1940, 165 [C. A., 36, 5476 (1942)].
15. Ochiai, M i у a к i, Ber., 74, 1115(1941).
16. M a n g i n i, Colonna, Gazz. chim. ital., 73, 323 (1943) [C. A., 41, 1225 (1947)].
17. M a n g i n i, Colonna, Gazz. chim. ital., 73, 330 (1943) [C. A., 41, 1225 (1947)].
18. Lappin, Petersen, Wheeler, J. Org. Chem., 15, 377 (1950L
19. Ч и ч и б а б и н A. E., Ber., 57, 1168 (1924).
20. В u u - H о i, D e с 1 e r c q, Rec. trav. chim., 73, 377 (1954).
21. Mazza, Migliardi, Atti accad. sci. Torino, Classe sci. fis. mat. e nat., 75, 438
(1940) [C. A., 36, 5477 (1942)].
22. Migliardi, Atti accad. sci. Torino, Classe sci. fis. mat. e nat., 75, 548 (1940) [C. A.,
38, 1507 (1944)].
23. A 1 1 e n, Spangler, W e b s t e r, J. Org. Chem., 16, 17 (1951).
ЛИТЕРАТУРА
183
24. Petrow, Rewald, Sturgeon, J. Chem. Soc., 1947, 1407.
25. M a r с к w a 1 d, Ann., 279, 1 (1894).
26. Bobranski, Sucharda, Roczniki Chem., 7, 192 (1927) [C. A., 22, 777 (1928)1.
27. Spath, Kufner, Ber., 71, 1657 (1938).
28. 3 e й д e О. А., Ч e л и н ц е в В. В., ЖОХ, 7, 2314 (1937) [С. А., 32, 572 (1938)1.
29. 3 е й д е О. A., Ann., 440, 311 (1924).
30. 3 е й д е О. А., Ч е л и н ц е в В. В., ЖОХ, 7, 2318 (1937) [С. А., 32, 572 (1938)1.
31. Bose, S е n, J. Chem. Soc., 1931, 2840.
32. В а с к е b е г g, J. Chem. Soc., 1933, 390.
33. Hart, J. Chem. Soc., 1954, 4030.
34. Price, Roberts, J. Am. Chem. Soc., 68, 1204 (1946).
35. Goldberg, Theobald, Williamson, J. Chem. Soc., 1954, 2357.
36. Hauser, R e у n о 1 d s, J. Org. Chem., 15, 1224 (1950).
37. M u г г а у, Hauser, J. Org. Chem., 19 , 2008 (1954).
38. О c h i a i, J. Org. Chem., 18, 534 (1953).
39. Lions, Ritchie, J. Proc. Roy. Soc. N. S. Wales, 74, 443 (1941) [C. A., 35, 4771
(1941)1.
40. M i у a к i, J. Pharm. Soc. Japan, 62, 257 (1942) [C. A., 45, 2950 (1951)j.
41. G u 1 1 a n d, R о b i n s о n, J. Chem. Soc., 127, 1493 (1925).
42. H a r t, J. Chem: Soc., 1954, 1879.
43. Bobranski, Sucharda, Ber., 60, 1081 (1937).
44. H a r t, J. Chem. Soc., 1956, 212.
45. Petrow, Sturgeon, J. Chem. Soc., 1949, 1157:
46. S h i m i z u, J. Pharm. Soc. Japan, 64, № 10A, 46 (1944) [C. A., 45, 8525 (1951)].
47. A 1 b e r t, H a m p t о n, J. Chem. Soc., 1952, 4985.
48. W e i s s, H a u s e r, J. Am. Chem. Soc., 68, 722 (1946).
49. M e у e г, V a u g h a n, J. Org. Chem., 22, 1554, 1560, 1565 (1957).
50. Герм. пат. 507637 (1926) [Chem. Zentr., 101, II, 3084 (1930)].
51. Takahashi et al., J. Pharm. Soc. Japan, 64, Ns 7/8A, 9 (1944) [C. A., 46, 1 10 (1952)1.
52. G u 1 1 a n d, R о b i n s о n, J. Chem. Soc., 127, 1493 (1925).
53. Takahashi, Hayas e, J. Pharm. Soc. Japan, 65, № 5/6A, 9 (1945) [C. A., 45,
8530 (1951)].
54. P r i c e, Roberts,.!. Org. Chem., 11, 463 (1946).
55. Kermack, Weatherhead, J. Chem. Soc., 1942, 726.
56. P e t г о w, J. Chem. Soc., 1945, 927.
57. Besly, Goldberg, J. Chem. Soc., 1954, 2448.
58. С о f f e y, Thomson, W i 1 s о n, J. Chem. Soc., 1936, 856.
59. Backeberg, J. Chem. Soc., 1933, 390.
60. Koller, Ber., 60,. 407 (1927). Синтез 3-карбометокси-2!4-Диокси-6,7-циклогек-
сено-1,8-нафтиридина этим методом см. Dornow, Neuse, Arch. Pharm., 288, 174
(1955) [С. A., 50, 16769 (1956)].
61. К о 1 1 e r, Strang, Monatsh., 50, 144 (1928).
62. О a k e sw R у d о n, J. Chem. Soc., 1958, 204.
63. Klisiecki, Sucharda, Roczniki Chem., 7, 204 (1927) [C. A., 22, 777 (1928)].
64. Sucharda, Kosmos, 1920 [C. A., 22, 2948 (1928)].
65. von Niementowski, Sucharda, J. prakt. Chem. [2] 94, 193 (1916).
66. von Niementowski, Ber., 29, 76 (1896); 39, 385 (1906).
67. C a r b о n i, Gazz. chim. ital., 85, 1201 (1955) [C. A., 50, 9404 (1956)].
68. С о s c i a, D i c k e r m a n, J. Am. Chem. Soc., 81, 3098 (1959).
69. Baum g'a rten, Krieger. J. Am. Chem. Soc., 77, 2439 (1955).
70. В a u m g a r t e n, С о о k, J. Org. Chem., 22, 138 (1957).
71. von Niementowski, Sucharda, Ber., 52, 484 (1919).
72. О c h i a i, M i у a k i, Sato, Ber., 70, 2018 (1937).
73. C h i о z z a, Ann., 83, 117 (1852).
74. R e y-B ellet, Erlenmeyer, Helv. Chim. Acta, 39, 2106 (1956).
75. M e у e г, V i t t e n i t, Ann. chim. (Paris), [10] 17, 291 (1932).
76. Krohnke, Ellegast, Bertram, Ann., 600, 198 (1956).
77. В i r k о f e r, Kaiser, Ber., 90, 2933 (1957).
78. Ж у к о в а и др., Изв. АН СССР, ОХН, 1952, 743 [С. А., 47, 401 (1953)].
79. Гольдфарб, Кондакова, Изв. АН СССР, ОХН, 1951, 610 [С. А. 46, 8113
(1952)].
80. F е 1 s, Вег., 37, 2129 (1904).
81. О с h i a i, А г a i, J. Pharm. Soc. Japan, 59, 458 (1939) [C. A., 34, 108 (1940)].
82. О c h i a i et al., Ber., 70, 2018 (1937).
83. О c h i a i et al., J. Pharm. Soc. Japan, 65, 69 (1945) [C. A., 45, 8018 (1951)].
84. H a u s e г, К a n t о r, J. Am. Chem. Soc., 73, 1437 (1951).
85. G a b r i e 1, Colman, Ber., 35, 1358 (1902).
86. Гетероциклические соединения, т. 4, Издатинлит, М.. 1957.
87. R е i s s е г t, Вег., 27,. 979 (1894),
184
НАФТИРИДИНЫ
88. Koller, Ber., 60, 1918 (1927).
89. P a i 1 e r, Kuhn, Monatsh., 84, 85 (1953).
90. С о 1 о n n a, Runt i, Gazz. chim. ital., 82, 513 (1952) [C. A., 48, 680 (1954)].
91. Colonna, Risaliti, Boll. sci. fac. ch ini. ind. Bologna, 9, 82 (1951) [C. A., 46,
7102 (1952)].
92. Weiss, H a u s e r, J. Am. Chem. Soc., 71, 2023 (1949); 71, 2026 (1949).
93. Герм. пат. 700862 (Dec. 5, 1940) [C. A., 35, 7422 (1941)].
94. S ii s et al., Ann., 593, 91 (1955).
95. Швейц, пат. 263148 (Nov. 1, 1949) [C. A., 44, 6088 (1950)].
96. Mangini, Boll. sci. fac. chi. ind. Bologna, 1940, 165 ]C. A., 36, 5476 (1942)].
97. F u c i к et al., Chem. listy, 45, 23 (1951) [C. A., 45, 10245 (1951)].
98. К о 1 1 e r, Strang, Monatsh., 54, 952 (1929).
99. О c h i a i, M i у a к i, J. Pharm. Soc. Japan, 58, 764 (1938) [C. A., 33, 2525 (1938)].
100. Koller, Ber., 60, 1572 (1927).
101. Пат. США 2517929 (Aug. 8, 1950) [C. A., 45, 672 (1951)].
102. Англ.пат. 583109 (Dec. 9, 1946) [C. A., 42, 2403 (1948)].
103. К e r m a c k, S t о r e y, J. Chem. Soc., 1951, 1389.
104. Colonna, Gazz. chim. ital., 78, 502 (1948).
105. Dor now, von Loh, Arch. Pharm., 290, 136 (1957).
106. H у d о r n, Ph. D. Dissertation, University of Michigan, 1960.
107. Berg, P e t г о w, J. Chem. Soc., 1952, 3713.
108. M i у a к i, К a t а о к a, J. Pharm. Soc. Japan, 60, 367 (1940). [C A., 35, 1404 (1941)].
109. Koller, Kandler, Monatsh., 58, 213 (1931).
НО. О c h i a i, J. Org. Chem., 18, 534 (1953),
Глава III
КАРБОЛИНЫ
/> К. ер мак, Дж. Мак-К eh'л
ВВЕДЕНИЕ
Карболиновая циклическая система состоит из бензольного, пиррольного’
и пиридинового колец, соединенных таким образом, что пирролыюе кольцо
находится между двумя другими, присоединенными к нему через его атомы угле-
рода в положениях 2,3 и 4,5. Карболин можно рассматривать как карбазол
(том 3, гл. III), в котором одно из бензольных колец заменено пиридиновым.
Совершенно очевидно, что атом азота в пиридиновом кольце может находиться
в четырех положениях (они показаны в формулах I — IV), поэтому известны
четыре ряда карболиновых соединений.
6 5
а-Карболин
I
9-Пирид-[2,3-Ь]-индол
1а
р-Карболин
II
9-Пирид-[3,4-Ь]-индол
Па
6 5
7-Карболин
III
З-Карболин
IV
186
КАРБОЛИНЫ
Для карболиновых циклических систем применялись различные наимено-
вания и нумерация атомов. В настоящей книге принята система Гулланда
(1 — IV) [11 главным образом потому, что она позволяет избежать громоздкой
терминологии. В справочнике «The Ring Index» [21 карболины называются
пиридиндолами и атомы нумеруются, как показано в формулах 1а — IVa.
Иногда систему Гулланда модифицируют, заменяя греческие буквы а, 0, у и б,
указывающие положение основного атома азота в пиридиновом кольце, на
цифры 2, 3, 4 и 5. Так, например, а-карболин называется 2-карболином.
В системе, предложенной Перкином и Робинзоном [31, общие атомы углерода
нумеруются вместе с другими, как показано в формулах V —VIII, а четыре
циклические системы обозначаются соответственно как 3- 4-, 5- и 6-карболииы.
9 6 9 6
V VI
Можно предположить, что, кроме упомянутых выше карболиновых струк-
тур, существуют близкие им изомерные формы, у которых атом водорода, нахо-
дящийся у атома азота в положении 1, перемещается к одному из других атомов
молекулы. Некоторые из этих изомерных форм известны, но лишь в виде
алкильных или других устойчивых производных. Сюда относятся а-, 0- и у-
нзокарболины (IX — XI), алкильные производные которых получены из чет-
вертичных солей соответствующих карболинов в виде анГидрониевых основа-
ний (стр. 211 и 213).
Следует упомянуть о довольно необычном соединении L1X (стр. 208),
которое можно рассматривать как тетрагидропроизводное гипотетической
«псевдокарболиновой» структуры XII.
Кроме этих четырех карболинов, будут рассмотрены некоторые более слож-
ные системы, которые можно считать производными карболинового ядра.
Сюда относятся системы из четырех циклов, известные под названием бензо-
карболинов, а также ряд природных алкалоидов более сложной структуры.
Первая часть настоящей главы будет посвящена простым карболипам,
включая гарминовые алкалоиды; во второй части будут рассмотрены бензокар-
ПРОСТЫЕ КАРБОЛИНЫ
187
болины (и родственные им соединения); в третьей части будут описаны некото-
рые природные соединения, молекула которых содержит карболиновое ядро
как часть более сложной циклической системы.
ПРОСТЫЕ КАРБОЛИНЫ
Введение
Карболиновую циклическую систему можно рассматривать как карбазол,
в котором одна из групп — СН = бензольного кольца заменена на — N .
Поэтому химии карболинов и карбазолов свойственны некоторые общие черты.
Метод синтеза карбазолов по Гребе — Ульману (том 3, стр. 235) может быть
применен и для карболинового ряда. Он является наиболее общим методом
синтеза карболиновых производных. Все четыре простых карболина, а также
разнообразные бензокарболины были синтезированы этим способом. Как
и в карбазоле, атом азота пиррольного кольца не имеет основного характера.
Как карбазол, так и карболины не дают ни одной из характерных цветных-
реакций, свойственных пиррольным или индольным производным, не имеющим
заместителей в а- или ^-положениях. И карбазольная, и карболиновая цикли
ческие системы проявляют ярко выраженный ароматический характер и весьма
устойчивы. Соединения, не имеющие заместителей в кольце, устойчивы при
перегонке в токе водорода над цинковой пылью; карболиновое ядро обычно
обнаруживают в продуктах глубокого расщепления сложных природных осно-
ваний, содержащих карболиновый скелет.
Карболины отличаются от карбазолов тем, что обладают явно основными
свойствами благодаря наличию атома азота пиридинового кольца. Они ведут
себя как одноатомные основания и дают с минеральными кислотами хорошо
выделяемые соли. С такими реагентами, как иодистый метил или диметилсуль-
фат, они образуют также четвертичные соли. Один из наиболее интересных
вопросов в химии карболинов — связь этих четвертичных солей с получающи-
мися из них ангидрониевыми основаниями — будет рассмотрен ниже (стр. 211).
Наибольшее число проведенных до настоящего времени исследований
простых карболинов относится к производным Р-карболина. Поэтому удобнее
рассмотреть сначала производные а-, у- и 6-карболинов вместе, а затем [3-кар-
болины и, наконец, ангидрониевые основания.
а-, у- и 6-Карболины
Применение метода синтеза карбазолов Гребе — Ульмана. Синтез
а- и у-карболинов с помощью этого метода осуществляется легко. 2-(2'-Амино-
анилино)пиридин (XIII), необходимый для получения а-карболина, можно легко
приготовить из о-фенилендиамина и 2-хлор- [4] или 2-бромпиридина 15]. Обра-
ботка соединения XIII азотистой кислотой дает 1-(2-пиридил)бензотрйазол
(XIV) [4], который при энергичном нагревании с плавленным хлоридом цинка
теряет азот и переходит в а-карболин (I). Разложение бензотриазола (XIV)
проходит более гладко и с лучшими выходами в горячей сиропообразной
фосфорной кислоте [6].
а-Карболин кристаллизуется из бензола в виде бесцветных плоских игл.
Разбавленные нейтральные растворы а-карболина обладают фиолетовой флу-
оресценцией, тогда как растворы его солей не флуоресцируют (ср. с Р-карбо-
лином, стр. 195). Восстановление натрием в бутаноле приводит к образованию
3-(3'-аминопропил)индола, который был выделен в виде фталимидного производ-
ного наряду с другими продуктами реакции.
у-Карболин (III) может быть получен из о-фенилендиамина и 4-хлорпйрп-
дина при помощи такого же ряда реакций 17, 81. И в этом случае превращение
188
КАРБОЛИНЫ
триазола в карболки лучше всего проходит в горячей сиропообразной фосфор-
ной кислоте. у-Карболин образует бесцветные блестящие призматические иглы;
XIII
растворы его, нейтральные или кислые, водные или спиртовые, не флуоресци-
руют при дневном свете, но в ультрафиолетовом свете солянокислый спиртовой
раствор у-карболина дает синевато-фиолетовую флуоресценцию [7]. у-Карбо-
лип был получен Бремером [9] с высоким выходом нагреванием 1-фенилпиридо-
|3,4-с(]-«ш4-триазола (XV) в парафине при 320—350". Кристаллографические
данные для у-карболина приведены Гринвудом [10].
Нагревание
III
XV
Получение [5- и 6-карболинов методом Гребе — Ульмана более сложно
вследствие трудности синтеза 3-(2'-аминоанилино)пиридина. Последний был
получен Шпетом и Эйтером [11] нагреванием 3-бромпиридина с о-фениленди-
амином в запаянной трубке в присутствии сульфата меди и небольшого коли-
чества воды при 155е. Обработка 3-(2'-аминоанилино)пиридцна азотистой
кислотой дает 1-(3'-пиридил)бензотриазол (XVI), при нагревании которого
с хлоридом цинка в высоком вакууме в течение 15 мин при 320° получена смесь
трех компонентов. Один из них, как было показано, представлял собой 3-ани-
линопиридин; два других по анализу имели составы, соответствующие брутто-
формуле CuH8N2, причем один с т. пл. 198,5° был идентичен [3-карболину (II)
(см. стр. 192), а другой оказался неизвестным до того времени 6-карболином
(IV), т. пл. 214—215°. Смешанная проба с а-карболином (т. пл. 218°) давала
ZnCl2
320°
-NHC6H6
II
О
N
IV
Простые карболины.
189
депрессию температуры плавления нового соединения, что исключает возмож-
ность изомеризации при нагревании, необходимом для превращения триазола
в карболин. Изомерные карболины, хотя это и маловероятно, по-видимому,
образовались в результате простой миграции радикала, находящегося в а-
или у-положении пиридинового кольца.
Получение а-, у- и 6-карболинов разложением азидов. При термическом
разложении 3-(2'-азидофенил)пиридина образуется смесь а- и у-карболинов
(11 и 111) [ 12]. В противоположность этому из 2-(2'-азидофенил)пиридина 6-кар-
болин не получается. Термическое разложение в этом случае, по-видимому,
сопровождается отщеплением двух атомов водорода либо от молекулы раство-
рителя, либо от другой молекулы с образованием 2-(2'-аминофенил)пиридина.
1%-ный раствор
в декалине.
170-180°
__/К
\/'Vxz
н
II III
Получение карболинов реакций Пшорра. В условиях фенантренового
синтеза по методу Пшорра из 2-метил-(2'-аминофенил)аминопиридина (XVII)
был получен с выходом 7% 1-метил-а-карболин (XVIII). Основному продукту
реакции (84%) была приписана структура XIX [13]. Однако, если взять такие
исходные соединения, чтобы замыкание кольца происходило у бензольного,
а не у пиридинового кольца, то выход соединения XVIII увеличивается до
78% (XX — XVIII).
Разные методы. Одно из производных 6-карболина было получено Гуллан-
дом и сотрудниками [1] окислением 3,4-диоксихиндолина (стр. 243) азотной
кислотой (29 или 47%-ной). При этом образовывался трудно поддающийся
очистке студенистый продукт, из которого при нагревании с уксусной кислотой
выделялась двуокись углерода и получалась, по всей вероятности, 7-нитро-6-
карболинкарбоновая-3 кислота. Положение нитрогруппы не было точно уста-
новлено, и возможно, хотя и маловероятно, что карбоксильная группа находит-
ся в положении 4.
Йодиды 1,2,4-триалкил-у-карболиния были получены [14] из соединения
ХХа, синтезированного из 2-литий- 1-метилиндола и диалкиламиноацетонов.
При диметиламинометилировании соединения ХХа по методу Манниха полу-
чено соединение ХХб, которое при действии йодистого метила давало четвер-
тичную соль (ХХв). При кипячении водного раствора соединения ХХв обра-
190
КАРБОЛИНЫ
зуется иодид тетрагидро-у-карболиния (ХХг), который нагреванием при 220°
превращается в соль у-карболиния (ХХд). Если для реакции применяют бен-
зилэтиламиноацетон и продукт дебензилируют, то получающийся вторичный
амин (ХХе) может при действии формальдегида циклизоваться в тетрагидро-
у-карболин (ХХж) (аналогично образованию тетрагидро-Р-карбол.инов из трип-
тамина и альдегидов).
N(CH3)2
ХХа ХХб
Г N(CH3)3
ХХв ХХг
220°
ХХд
nhc2h5
Н 1
,сн2
ХХе
1з
ХХж
Гидрированные производные. При нагревании фенилгидразона 1-метилпи-
перидона-4 (XXI) с разбавленной серной кислотой происходит замыкание
индольного кольца по Фишеру (том 3, стр: 7) с образованием 4-метил-2,3,4,5-
тетрагидро-у-карболина (XXII) [15]. Несколько лучшие выходы получаются
при насыщении спиртового раствора фенилгидразона хлористым водородом] Id].
/ч /\
!| I N-CHj
X/ \N-nX'' /
R
XXI, R = H
XXIII, Н = -алкил
XXII, R = H
XXIV, К = алкил
Этот общий метод был использован для получения 1-замещенных тетрагидро-у-
карболинов из N-алкилзамещенных фенилгидразонов (XXIII и XXIV) [17].
ПРОСТЫЕ КАРБОЛИНЫ
191
Робинзону и Торнлею [7] не удалось получить аналогичный тетрагидро-
карболин из фенилгидразона триацетонамина, но при нагревании метилфенил-
гидразона триацетонамина с разбавленной серной кислотой циклизацию удалось
осуществить и был выделен 1,3,3,5,5-пентаметил-2,3,4,5-тетрагидро-у-карбо-
лин (XXV). Позднее Бёкельхайде и Айнсворт!16] синтезировали тетрагидрокар-
болин (XXVI) конденсацией фенилгидразона триацетонамина под действием
хлористого водорода в спирте [18]. Из фенилгидразона 1-метил-3,5-ди-
этилпиперидона-4 получен гексагидрокарболин (XXVII) [18]. При восста-
новлении у-карболина натрием в бутаноле образуется 2,3,4,5-тетрагидро-у-
карболин, являющийся сильным основанием [7].
н3сх/сн,
^А /А
I !! :
^/VN-N^A/Асня
R
XXV, R = CH3
!
R
XXVI, R=H
Манн, Приор и Виллкокс сообщили, что при нагревании 4-пиридилгидра-
зона циклогексанона в течение 10 мин с хлоридом цинка при 240° образуется
6,7,8,9-тетрагидро-у-карболин [19].
Р-Карболины
Введение. Из всех карболинов наиболее подробно изучены производные
Р-карболина (I). Это, несомненно, объясняется тем, что большое число алка-
лоидов являются производными Р-карболина. Первые алкалоиды, для которых
это было установлено, выделены из семян Peganum harmala, причем двумя
главными основаниями были гармин и гармалин. Затем было выяснено, что
некоторые алкалоиды, выделенные из других растений, также представляют
собой простые производные карболина: телепатин, ягеин и банистерин из Bani-
steria caapt идентичны гармину, а арабин из Araribra rubra (идентичный лоту-
рину из Symplocos racemosa) — даже более простое производное Р-карболина.
В некоторых других алкалоидах Р-карболиновая циклическая система является
частью более сложного скелета молекулы. Среди них следует упомянуть эво-
диамин и рутекарпин из Evodia rutaecarpa, иохимбин и связанные с ним
основания, а также алкалоиды различных видов Rauwolfia (резерпин и род-
ственные ему алкалоиды). Поскольку эти вещества содержат более трех
Р-карболиновых колец, они будут рассмотрены в одном из следующих разде-
лов. Так как природные соединения исчерпывающе описаны в трудах, посвящен-
ных химии алкалоидов [20], они здесь не будут рассматриваться. Однако
в настоящей главе будут освещены те разделы химии алкалоидов, которые
способствуют пониманию химии Р-карболинов.
Что касается номенклатуры, то, согласно принятой в этой главе системе,
рассматриваемые соединения будут обозначаться как производные Р-карболина
с нумерацией атомов, как указано в формуле I. Однако при пользовании лите-
ратурой читатель должен иметь в виду, что некоторые авторы применяют систе-
192
КАРБО ЛИНЫ
му нумерации, показанную формулой II и предложенную Перкином и Робин-
зоном 121], согласно которой рассматриваемая кольцевая система называется
4-карболином. В ряде случаев очень удобно описывать соединения как произ-
водные встречающихся в природе алкалоидов гармина и гармалина. Это особенно
относится к соединениям, родственным этим алкалоидам или легко из них полу-
чающимся. Г армии представляет собой 8-метокси-2-метил-Р-карболин (III), а гар-
малин—его 4,5-дигидропроизводное (IV). Алкалоид гармалол, найденный вместе
с сопровождающими его гармином и гармалином в семенах Р. harmala, представ-
ляет собой8-окси-2-метил-4,5-дигидро-Р-карболин. Гармол—это 8-оксиА2-метил
Р-карболин, а гарман—соединение, образующееся из гармина после удалени-
метоксигруппы, это—2-метил-Р-карболин. Гарман, по-видимому, не содержится
в семенах Р. harmala. Однако установлено, что арабин из Araribra rubra и
лотурин из Symplocos racemosa идентичны этому основанию [221. Показано
также, что пассифлорин, получаемый из страстоцвета, идентичен гарману [23].
Поскольку сам Р-карболин получен впервые путем удаления метильной груп-
пы из гармана, он был первоначально известен как норгарман, причем это
название до сих пор иногда употребляется.
Общие методы синтеза. Существующие методы синтеза р-карболинового
ядра удобно систематизировать следующим образом. Т
1. При применении синтеза карбозолов по методу Гребе — Ульмана
к бензотриазолу, образующемуся из 3-(2'-аминоанилино)пиридина, получается
смесь Р- и 6-карболинов (см. стр. 188).
2. При обработке диалкилацеталей амида индолилкарбоновой-2 кислоты
общей формулы V спиртовым раствором соляной кислоты может произойти
циклизация с образованием производных 2-кето-2,3-дигидро-Р-карболина
(VI) (см. стр. 201).
3. При обработке ацильных производных триптамина такими циклизующи-
ми агентами, как хлорокись или пятиокись фосфора, образуется третье кольцо
ПРОСТЫЕ КАРБОЛИНЫ
193
согласно следующей схеме:
R
Этот синтез совершенно аналогичен синтезу изохинолинов из ацильных
производных фенилэтиламина по реакции Бишлера — Напиральского (т. 4).
4. При взаимодействии триптамина с соответствующими альдегидами
в кислом или нейтральном растворе образуются производные 2,3,4,5-тетра-
гидро-Р-карболина, предположительно через шиффово основание, согласно
следующей схеме:
Аналогия этого метода с синтезом изохинолина по методу Пикте — Шпенгле-
ра очевидна (т. 4). Этот удобный синтез особенно подробно исследован
Ханом и сотрудниками, которые показали, что альдегид можно с успехом
заменить какой-либо а-кетокислотой; получающийся при этом продукт пред-
ставляет собой производное 2,3,4,5-тетрагидро-р-карболинкарбоновой-2 кисло-
ты (стр. 204).
5. В упомянутой выше реакции с альдегидом триптамин может быть заме-
нен триптофаном; в результате получается 2,3,4,5-тетрагидро-р-карболинкар-
боновая-4 кислота (стр. 209).
6. При применении реакции Курциуса к р-(3-индолил)пропионилазиду,
получающийся сначала карбимид немедленно циклизуется с образованием
2-кето-2,3,4,5-тетрагидро-р-карболина (стр. 207).
7. Производные р-карболинкарбоновой-4 кислоты получают конденсацией
2-карбэтоксииндол-З-альдегида с гиппуровой кислотой и обработкой образую-
щегося азлактона спиртовым раствором едкого кали. Одним из продуктов реак-'
ции является калиевая соль 2-кето-2,3-дигидро-р-карболинкарбоновой-4 кисло-
ты (стр. 209).
8. Другой метод с промежуточным образованием 2,3-дизамещенного
производного индола представлен синтезом гармалина, проведенным Ингом,
Манске и Робинзоном, которые получили 2-ацетил-3-(Р-фталимидоэтил)-7-меток-
сииндол путем циклизации соответствующего гидразона, синтезированного
реакцией Джаппа — Клингемана. При удалении фталевой кислоты амин
немедленно терял воду и превращался в гармалин (стр. 198).
Р-Карболин (норгарман). p-Карболин впервые был получен из гармина
путем последовательного удаления метоксильной и метильной групп. Метод,
которым из гармина получали деметоксигармин, будет описан в разделе, посвя-
щенном гарману; для удаления метильной группы ее сначала превращали
в карбоксильную (стр. 208). Получающуюся гармановую кислоту декарбокси-
лировали нагреванием в глицерине [24]. Р-Карболин (I) был получен синтети-
чески различными путями. Впервые он был получен из 1-метилиндолкарбоно-
вой-2 кислоты, которую конденсировали с диэтилацеталем аминоацетальдегида.
Циклизация образующегося при этом амида (VII) привела к 2-кето-1-метйл-2,3-
дигидро-Р-карболину (VIII) (стр. 201). При перегонке этого соединения над
цинковой пылью в токе водорода был получен не совсем чистый продукт,
13 Заказ 978
194
КАРБОЛИНЫ
представлявший, вероятно, смесь Р-карболина (I) и 1-метил-Р-карболина (IX)
[24]. Вещество, полученное после очистки, оказалось идентичным норгар-
ману, приготовленному из гармина, как описано выше. Другие производные Р-
__г ___________________________________
I II П H2NCH2CH(OC2H5)2 I |1 II
ММ1-С00Н * L CONHCH2(OC2H5)2
СН3 СН3
VII
I
карболина, такие, как 2_-кето-2,3,4,5-тетрагидро-Р-карболин [24] (стр. 207)
или 2-кето-3-метил-2,3-дигидро-Р-карболин [25] (стр. 201), при перегонке над
цинковой пылью также дают норгарман, обычно с примесью индола и других
продуктов распада [26].
Ниже кратко описаны другие методы синтеза p-карболина. Один из них
уже упоминался (стр. 192) — это применение синтеза Гребе—Ульмана к
1- -(З-пиридил)бензотриазолу, из которого получают смесь р- и 6-карболинов.
А
Pd
168
XI
ПРОСТЫЕ КАРБОЛИНЫ
195
Р-Карболин был также синтезирован исходя из 2-карбэтоксииндол-З-аль-
дегида. Путем ряда последовательных реакций, которые будут приведены ниже
(стр. 209), его превращали в 2-хлор-Р-карболинкарбоновую-4 кислоту. После
дегалогенирования и декарбоксилирования получался Р-карболин.
Конденсацией триптофана с формальдегидом может быть получена 2,3,4,5-
тетрагидро-Р-карболинкарбоновая-4 кислота (X). При окислении хромовой
кислотой она теряет 4 атома водорода и одновременно отщепляется карбоксиль-
ная группа [24]. Описаны усовершенствования первоначальной методики [27]
и предложен механизм реакции для объяснения интересного превращения
соединения X в соединение I [27].
Шпет и Ледерер [28] получили подобным образом р-карболин из трипт-
амина и формальдегида, причем промежуточное соединение — 2,3,4,5-тетра-
гидро-р-карболин (XI) — дегидрировали нагреванием с губчатым палладием.
Р-Карболин кристаллизуется в виде бесцветных игл. Его растворы в раз-
бавленных кислотах, как и аналогичные растворы многих его производных,
обладают интенсивной синей флуоресценцией. Р-Карболин образуется в резуль-
тате распада многих природных алкалоидов и благодаря своей флуоресценции
может быть легко обнаружен. Наличие его в продуктах распада обычно указы-
вает на присутствие Р-карболиновой системы в исходном алкалоиде.
При ацилировании Р-карболин образует ацильные производные по индоль-
ному, а не по пиридиновому атому азота [291.
Алкил- и алкоксипроизводные. Гарман и гармин. Как уже указывалось
(стр. 192), 2-метил-Р-карболин (гарман) (XII), хотя и не был найден вместе
с гармином и гармалийом в Р. harmala, но оказался идентичен встречающимся
в природе алкалоидам — арабику и лотурину; он был также обнаружен в про-
дуктах распада более сложных алкалоидов. Показано [30], что мелинонин F,
полученный из Strychnos melinoniana, является четвертичной солью гармана.
Гарман был получен из гармина (XIII) путем удаления метоксильной группы.
Деметилирование в гармол (XIV) может быть гладко проведено по методу
Культарда, Левина и Пимана [31] кипячением гармина с серной кислотой,
взятой в количестве 35—60 вес.%; требующееся для реакции время может
изменяться от 100 до 2 час в зависимости от концентрации кислоты. Если коли-
чество кислоты превышает 60 вес.%, то получают продукт, загрязненный
гармолсульфоновой кислотой. Коновалова, Проскурнина и Орехов [32] реко-
мендуют применять сиропообразную фосфорную кислоту (уд. вес 1,7), при
нагревании с которой при 150° гладко удаляется метильная группа и обра-
зуется гармол с выходом 78%. При применении этого метода устраняются ослож-
нения, встречающиеся при реакции с серной кислотой. Когда гармол обраба-
тывают смесью хлорида цинка с аммиаком [33], то гидроксильная группа заме-
щается аминогруппой и образуется аминогарман (XV). При удалении из этого
13*
196
КАРБОЛИНЫ
соединения аминогруппы обычными методами получают гарман, 2-метилф-
карболин.
Гарман, подобно многим другим производным p-карболина, отличается
интенсивной флуоресценцией. Очень разбавленные растворы в кислых и ней-
тральных растворителях обнаруживают яркую фиолетовую флуоресценцию.
Впервые эту флуоресценцию наблюдали Гопкинс и Коле [34], хотя в то время
они не знали истинной природы соединения, которое ее вызывало.
Гарман легко, получается из гармалана (2-метил-4,5-дигидро-Р-карболина)
(стр. 198) в результате дегидрирования, например при нагревании с губчатым
палладием при 200° [351; таким образом, синтезы гармалана, описанные ниже,
являются также синтезами гармана. Подобным образомдегидрирование 2-метил-
2,3,4,5-тетрагидро-р-карболина (тетрагидрогармана) нагреванием в водном
растворе с малеиновой кислотой и палладиевой чернью [36] дает гарман.
При окислении 2-метил-2,3,4,5-тетрагидро-р-карболинкарбоновой-4 кислоты
хромовой кислотой [24] одновременно происходит дегидрирование и декарбо-
ксилирование и образуется то же соединение. Как будет показано ниже, тетра-
гидрогарман и тетрагидрогарманкарбоновая-4 кислота очень легко получаются
конденсацией ацетальдегида с триптамином или с триптофаном соответственно.
Впервые лабораторный синтез гармана осуществили Гопкинс и Коле
в 1903 г. [34] при изучении строения недавно открытой тогда аминокислоты —
триптофана, хотя они и не установили природу синтезированного соединения.
При окислении этого соединения хлоридом железа (III) они получили два
продукта: один нейтральный — с т. пл. 195°, а другой — основание C12H10N2
с т. пл. 238°, растворы которого в разбавленной кислоте обладали, как указы-
вают авторы, очень интенсивной синей флуоресценцией. Первое из этих двух
соединений, как было показано несколько лет спустя, представляло собой индол-
3-альдегид; природа второго оставалась невыясненной до 1919 г., когда Перкин
и Робинзон [37] выдвинули веские доводы в пользу (i-карболиновой структуры
гармина и гармалина. Кроме того, этим авторам удалось показать идентичность
соединения Гопкинса и Коле с гарманом, полученным из гармина в результате
удаления метоксильной группы. Простейшее объяснение состоит в том, что
триптофан взаимодействует с альдегидом, образуя тетраГидрогарманкарбо-
новую-4 кислоту (XVI), которая окисляется с одновременным декарбоксили-
рованием и дает гарман (XII). В своей статье Гопкинс и Коле указывали, что
реакционную смесь после окисления экстрагировали эфиром для выделения
нейтрального продукта и экстракт затем оставляли стоять в течение нескольких
дней до окончательной обработки — выделения основания. Возможно, что эфир,
применяемый для экстрагирования, содержал спирт, окислявшийся при стоянии
Hi |Ггсоон
nh2
н нсо
I
сн3
сн3
XVI
до альдегида, который и реагировал с триптофаном. Такой синтез гармана, как
это сразу было оценено, доказывал, что метильная группа находится в положе-
нии 2, а не 4 (факт в то время еще не установленный экспериментально).
ПРОСТЫЕ КАРБОЛИНЫ
197 .
Алкилирование гармина, гармана и их производных алкилгалогенидами
или алкилсульфатами первоначально протекает по пиридиновому атому
азота (N3). Поскольку химия этих веществ тесно связана с проблемой получаю-
щихся из карболинов ангидрониевых оснований, они будут подробно рассмот-
рены ниже (стр. 214).
Алкилпроизводные p-карболина, в которых алкильная группа находится
у индольного атома азота, были получены из ацилпроизводных 1-алкилтрипт-
аминов [28, 38].
При взаимодействии Nz-метилтриптамина (XVII) с формальдегидом
в 0,5 н. серной кислоте образуется смесь соединений XVIII, XIX и XX, причем
происхождение этих веществ очевидно [391.
QqQ_cHi
н Н
XVII XVIII
Если алкилировать гармин путем последовательной обработки амидом
калия и алкилгалогенидом в толуоле, то алкильная группа вступает к индоль-
ному атому азота [27, 38, 40, 41].
Алкил
Замещенные в бензольном кольце метокси-p-карболины теоретически могут
быть получены из соответствующих производных пиридилбензотриазола,
но этот метод трудно осуществим на практике из-за сложности синтеза промежу-
точных продуктов. Поэтому те p-карболины с метоксигруппой в бен-
зольном кольце, которые были синтезированы, получались окислением соот-
ветствующих ди гидро- и тетрагидропроизводных (стр. 198). Так, гармин легко
был получен'из гармалина или из тетрагидрогармалина, и приведенные ниже
(стр. 199) синтезы этих соединений являются, следовательно, и синтезами гар-
мина. Среди метокси-р-карболиновых соединений, синтезированных Шпетом
и Ледерером [28], имеются 7-метокси- и 9-метокси-2-метил-Р-карболины. Дру-
гие примеры замещенных в бензольном кольце Р-карболинов будут приведены
при описании многоядерных р-карболинов.
Ряд О-длкиловых эфиров гармола был синтезирован Культхардом, Леви-
ном и Пиманом [31 ]. Общим методом получения служила обработка спиртового
раствора гармола 1 молем едкого натра и 1 молем алкил- или аралкилгалоге-
нида с последующим нагреванием смеси с обратным холодильником. Главным
продуктом реакции был О-алкиловый или О-аралкиловый эфир, который, одна-
ко, был загрязнен N-алкил- и О,М-диалкилпроизврдными. Синтезированы
были все О-алкиловые эфиры от этилового до додецилового. Представляет
интерес поведение хлоргидратов этих соединений при действии на них воды.
198
КАРБОЛИНЫ
Горячие концентрированные водные растворы бутилового эфира и следующих
за ним ближайших гомологов при охлаждении становятся желеобразными.
Начиная с н-октилового эфира и выше соли мало растворимы даже в горячей
воде, но при контакте с водой делаются желеобразными. Соль амилового эфира
и его высших гомологов постепенно гидролизуется в растворе, причем выде-
ляются кристаллы основания. Лактаты, которые растворимы значительно луч-
ше, чем хлоргидраты, желатинируются труднее.
Гидрированные производные Р-карболина. Гармалин и тетрагидрогармин.
Первые синтезированные простые соединения ряда Р-карболина были произ-
водными гармина, которые были выделены из природных веществ или легко
получались из природных алкалоидов. Гармалин — это дигидрогармин; уста-
новление его точного строения представляет определенный интерес. Гармалин
может быть окислен в гармин, но осуществить обратную реакцию непосредст-
венно не удается. Так, например, гармаллн превращается в гармин при окис-
лении хромовой кислотой [42] или азотной кислотой в спиртовом растворе
[43]; последний способ рекомендуется для получения Чистого гармина из неочи-
щенной смеси алкалоидов, выделенных из семян Р. harmala, основным компо-
нентом которой является гармалин [421. Гармалин может быть также дегид-
рирован нагреванием при 200° в присутствии губчатого палладия [35]. Для
превращения гармина в гармалин его необходимо сначала восстановить в тетра-
гидрогармин и затем перевести это основание с помощью контролируемого
окисления в гармалин [441. Гармалан, дигидрогарман, так же дегидрирова-
нием над губчатым палладием при нагревании превращается в гарман [35].
Интересный вопрос о точном положении двух дополнительных атомов водо-
рода в дигидропроизводных, таких, как гармалин, будет рассмотрен после
обсуждения химического строения некоторых метилпроизводных.
Гармалин реагирует с диметилсульфатом или иодистым метилом с обра-
зованием четвертичных солей, легко растворимых в воде и не осаждающихся
аммиаком. Однако концентрированная щелочь осаждает основание, анализ кото-
рого показывает, что его состав соответствует метилгармалину, получаемому
из гидроокиси четвертичного основания при потере элементов воды.
Метилгармалин [45], так же как и сульфометилат гармалина [24], легко
окисляется перманганатом калия с образованием нейтрального соединения
C13H14O2N2, т. пл. 227—228°, структура которого установлена синтезом и не
вызывает сомнений. Ниже (стр. 201) мы рассмотрим синтез 8-метокси-З-метил-
2-кето-2,3-дигидроф-карболина (XXI), получаемого из 6-метоксииндол-2-карб-
оксидиметилацеталилметиламида путем обработки спиртовым раствором хло-
ристого водорода. Восстановление этого соединения натрием в бутиловом
XXI XXII
XXIII
XXIV
спирте дает 3-метил-2,3,4,5-тетрагидроноргармин (XXII) с т. пл. 182°. Это
основание может быть вн<5вь превращено в нейтральное соединение ст. пл. 227—
228°, образующееся из него при окислении перманганатом калия. Обратная
ПРОСТЫЕ КАРБОЛИНЫ
199
реакция протекает при восстановлении нейтрального соединения металли-
ческим натрием в бутиловом спирте [26].
Эти наблюдения показывают, что соединение с т. пл. 227—228° должно
иметь строение XXIII и что единственной приемлемой структурой для метил-
гармалина является структура XXIV. Превращение метилгармалина в соедине-
ние с т. пл. 227—228° заключается просто в замещении метиленовой группы
атомом кислорода. Следует заметить, что идентичность синтетического метил-
тетрагидроноргармина (XXII), полученного из гармалина через соединение
XXIII, служит первым эксперимейтальным доказательством положения мето-
ксильной группы в гармине и гармалине.
При установлении точной структуры гармалина необходимо сделать выбор
между двумя возможными структурами — XXV и XXVI. То обстоятельство,
что гармалин .образует с диметилсульфатом соли, не осаждающиеся из водных
растворов аммиаком и, следовательно, сходные с четвертичными солями, заста-
вило предположить, что атом азота в пиридиновом кольце гармалина третич-
ный, и, таким образом, этот факт свидетельствует в пользу структуры XXV.
На основании этого метилгармалину следует приписать строение XXIV, кото-
рое хорошо объясняет легкость его окисления в соединение ст. пл. 228° (XXI).
С другой стороны, если гармалин имеет строение XXVI, то метилгармалину
следует приписать строение XXVII и его окисление в соединение XXI ста-
новится малопонятным. Дальнейшим подтверждением структуры XXIV слу-
жит тот факт, что гармалин и его соли легко реагируют с такими соединениями,
как синильная кислота, и что гармалин, экстрагированный из растения, полу-
чается в оптически неактивной форме и поэтому, вероятно, не содержит асим-
метрического атома углерода.1
XXV XXVI
XXVII
Позднее гармалин был синтезирован Манске, Перкином и Робинзоном
[461 с помощью ряда реакций, показанных на приведенной ниже схеме.
Последняя стадия состоит в удалении остатка фталевой кислоты из
2-ацетил-3-(Р-фталимидоэтил)-6-метоксииндола (XXVIII). Получающийся при
этом 2-ацетил-3-(Р-аминоэтил)индол (XXIX) немедленно циклизуется с потерей
молекулы воды и образует гармалин. Эти результаты лучше и естественней
согласуются с формулой XXV в качестве возможной структуры основания.
С2Н5ООССН(СН2)з— N
СОСН3
NaOOCCH(CH2)3—NHCO —
СОСН3 NaOOC —J
СН3О
| || ch2ch2ch2n
N=C
сосн3
200
КАРБОЛИНЫ
XXVIII
XXX
Манске, Перкин и Робинзон [46] аналогичным образом синтезировали
гармалан (2-метил-4,5-дигидро-Р-карболин); непосредственное образование
гармалана из 2-ацетил-3-(Р-фталимидоэтил)индола при удалении заместителя
у аминогруппы является в данном случае подтверждением предложенной для
этого основания структуры XXX.
Приведенная выше структура гармалина (формула XXV) не лишена
недостатков. Так, например, гармалин образует ацетильное производное, не
обладающее основными свойствами, что, по-видимому, говорит в пользу
формулы XXVI. Однако Нишикава, Перкин и Робинзон [45] показали, что
ацетилгармалин C15H18O2N2 может быть окислен в соединение C14H14O3N2
с т. пл. 207—208°. Очевидно, что при этом теряется СН2-группа и присоеди-
няется атом кислорода. Эта реакция, по всей вероятности, аналогична той,
которая проходит при окислении метилгармалина в соединение XXIII (стр. 198).
Это заставляет предположить, что ацетилгармалин имеет структуру XXXI
и тогда продукт окисления будет иметь строение XXXII, что хорошо согла-
суется со всеми известными фактами. Следует отметить, что и метилгармалин
и ацетилгармалин являются производными соединения XXXIII, которое мож-
но рассматривать как изогармалин, и возможно, что в определенных условиях
гармалин представляет собой равновесную смесь изомерных структур XXXIV
и XXXIII со значительным преобладанием первой. Оба изомера, вероятно,
находятся в равновесии с катионом гармалиния (XXXV), и та или иная струк-
тура образуется в зависимости от того, каким образом удаляется протон, что
приводит к молекуле основания, не имеющей заряда (ср. [44]).
Таутомерная природа гармалана подтверждается получением гармалина
[47] и 6-метокситриптамина и гармалана из триптамина или триптофана путем
взаимодействия с гликолевым альдегидом с образованием соответствующего
2-оксиметил-2,3,4,5-тетрагидро-Р-карболина, и последующей дегидратацией
под действием горячей серной или фосфорной кислоты. Синтезированная таким
путем из триптофана гармаланкарбоновая-4 кислота идентична кислоте, полу-
ченной циклизацией этилового эфира 2-ацетамино-2-карбэтокси-3-(3-индолил)-
пропионовой кислоты под действием хлорокиси фосфора, с последующим гидро-
лизом и отщеплением двуокиси углерода [47, 48]. Гармалин получается также
окислительным декарбоксилированием 2-метил-2,3,4,5-тетрагидро-р-карбо-
ПРОСТЫЕ КАРБОЛИНЫ
201
линкарбоновой-2 кислоты при кипячении ее водного раствора со свежеприго-
товленной окисью серебра.
Другой синтез гармалина, который целиком согласуется с формулой
XXXIV, предложен Шпетом и Ледерер'ом [35]. Они обрабатывали ацетильное
производное 6-метокситриптамина (XXXVI) пятиокисью фосфора в кипящем
ксилоле. Происходящая циклизация полностью аналогична синтезу дигидро-
изохинолинов по методу Бишлера — Напиральского. 0 помощью этой же
реакции из ацетилтриптамина получен гармалан.
Широкое применение синтезов типа Бишлера — Напиральского для
получения соединений 0-карболинового ряда продемонстрировано многими
исследователями. Так, например, Асахина и Осада [49] синтезировали
2-фенил- и 2-бензил-4,5-дигидро-0-карболины из соответствующих бензоил-
и фенацетилпроизводных триптамина, а Хан с сотрудниками применил эту
реакцию к амидам высших жирных кислот к триптамину, а также к бис-
триптаминовым производным некоторых алифатических двухосновных кислот
[50]. (См. также Шпет и Ледерер [28], Хан и Людевиг [51], Клемо и Сван [52],
Снайдер и Катц [53] и Джулиан и др. [54].)
Производные 2-кето-2,3-дигидро-^>-карболина. Значительное число произ-
водных 2-кето-2,3-дигидро-0-карболина было синтезировано по методу Кермака,
Перкина и Робинзона [24]. При обработке амидов общей формулы XXXVII
спиртовым раствором хлористого водорода или эфирным раствором серной
кислоты происходит циклизация с выделением двух молекул спирта. При этом
могут получаться два типа соединений в зависимости от того, будет ли ацетиль-
ная группа реагировать с атомом азота в положении 1 или с атомом углерода
в положении 3 индольного кольца. В первом случае продуктом реакции будет
индолодиазин (XXXVIII), во втором — 0-карболиновое производное (XXXIX).
Показано, что если в индольном кольце свободны оба положения — 1 и 3,
то преимущественное образование того или другого продукта реакции будет
зависеть главным образом от того, содержит ацеталь первичную или вторичную
аминогруппу. В первом случае главным продуктом будет индолодиазин; если
же аминогруппа вторичная, как, например, у метиламинодиэтилацеталя
202
КАРБОЛИНЫ
(XXXVII, Rt = H, R2 = CH3, R3 = H или алкил, R4=C2H6), то основным продук-
том реакции оказывается карболиновое производное, а в некоторых случаях
реакция идет исключительно в сторону их образования.
HC(OR4)2
'CHR,
I J
,N— R2
X
R! О
XXXVII
XXXIX
Если атомы водорода в положении 1 или 3 индольного кольца замещены,
например'метильной группой, то циклизация идет в единственно возможном
направлении, т. е. по незамещенному месту. Если же оба положения (1 и 3)
индольного кольца защищены, то не образуется вещества, похожего на кар-
болин или индолодиазин. Влияние природы заместителей видно из табл. 1.
Таблица 1
Индолодназин
Амнд (XXXVII) Карболнн (XXXIX) Индолодназин (XXXVIII) Литература (примечания)
R1 R2 Rs R1 R1 Rs Rs х R2 R3 X
Н н н С2Н5 — 1 1 Не выделено Н н — 24
Н н н С2Н5 — н н Н - н Н — 55а '
- 20% 80% .
сн3 н н С2Н5 — сн3 Н н — 24
СН3 н н с2н5 — сн3 Н н — 55
(выход 65%)
н н н с2н5 6-СН3О I Чет н Н 7-СН3О 24
н сн3 н сн3 — н сн3 н — Нет 26
н сн3 н С2Н5 — н сн3 н — Нет 55
н н сн3 С2Н6 — н н сн3 — н СН3 — 55
н сн3 сн3 С2Н5 — н сн3 сн3 — Нет 55 6
сн3 сн3 н с2н5 — сн3 сн3 н — —. 56
сн3 СНз сн3 с2н5 — сн3 сн3 сн3 — 55
(выход 62%)
н сн3 н сн3 4-СН3О н сн3 н 6-СН3О Не выделено 57
н н н сн3 5-СН3О 5-СН3О 8-СН3О
н н н С2Н5 Чет Н Н 57
н сн3 н С2Н5 5-СН3О н сн3 н 7-СН3О сн3 н 8-СН3О 57
н сн3 н сн3 6-СН3О н сн3 н 8-СН3О сн3 н 7-СН3О 26
н сн3 н сн3 7-СН3О н сн3 н 9-СН3О сн3 н 6-СН3О 57
800/6 20%
а Выделены противоточным методом.
б На основании ультрафиолетового спектра в сыром продукте индолодназин не обнаружен.
ПРОСТЫЕ КАРБОЛИНЫ
203
Так, в случае вторичного аминоацеталя, когда положения 1 и 3 в индоль-
ном ядре свободны, на соотношение количеств образующихся индолодиазина
и карболина могут влиять заместители, находящиеся в бензольном ядре.
Наличие метоксигруппы в положении 5 или 6 индольного кольца, по-видимо-
му, благоприятствует образованию индолодиазина. Если в реакции участвует
первичный аминоацеталь, то метоксильная группа не оказывает такого дейст-
вия, так как в этом случае даже при отсутствии какого-либо заместителя
в бензольном кольце карболин образуется в настолько незначительных коли-
чествах, что его даже невозможно выделить.
О 2-кето-2,3-дигидро-0-карболинкарбоновой-4 кислоте см. стр. 209.
Тетрагидро-^-карболины. Ранее наиболее известными представителями
этого типа соединений были тетрагидрогармин и N-метилтетрагидрогармин.
По мере развития химии пентациклических 0-карболинов (стр. 248) было синте-
зировано большое число других тетрагидро-0-карболинов, Тетрагидрогармин
получается восстановлением гармина или гармалина, N-метилтетрагидрогар-
мин— из метилгармина или метилгармалина [44]. То, что метилирование
вторичного основания — тетрагидрогармина дает такой же метилтетрагидро-
гармин, как и полученный из метилгармина или метилгармалина, показывает,
что в этих двух соединениях метильная группа действительно находится у ато-
ма азота пиридинового кольца.
Наблюдалась интересная реакция при нагревании соли четвертичного
основания (XL) — сульфометилата К3-метилтетрагидрогармина в метанольном
растворе едкого кали. При этом получалось соединение, имеющее эмпирическую
формулу CieH24O2N2, что соответствует замещению группы CH3SO’ метоксиль-
ной; продукт реакции представляет, вероятно, 2-(а-метокси)этил-3-(0-диметил-
аминоэтил)-8-метоксииндол (XLI). Если вместо метилового взять этиловый
спирт, то получается гомологичное основание, содержащее вместо метоксигруп-
пы этоксильную. Это доказывает, что источником метоксильной группы являет-
ся растворитель. Хотя основание наиболее вероятно, имеет строение XLI,
не исключена возможность и структуры XLII; при этом следует предположить,
что расщепление кольца происходит несколько иначе.
СН2
CH3SOj
/Чсн2
\ N (СН3)2
СНОСН3
I
сн3
XL XLI
СН2
/\
| || || СН2ОСНз
н CH-N(CH3)2
н I
сн3
XLII
Производные тетрагидро-0-карболина легко могут быть получены взаимо-
действием триптамина или триптофана с альдегидами в соответствующих усло-
виях; в реакциях с триптамином альдегиды могут быть заменены а-кетокисло-
тами. Этот тип реакции был широко изучен рядом исследователей, в том числе
Татсуи [58], Акабори и Сайто [36], Ханом с сотрудниками [51, 59—62], Дже-
кобсом и Крейгом [63], Уадсвортом и Пангборном [64], Харвеем и Робсоном
204
КАРБОЛИНЫ
[65] и Харвеем, Миллером и Робсоном [66]. Такого рода конденсации прово-
дились первоначально в присутствии кислоты, но Хан с сотрудниками [51,
59] показал, что эти реакции протекают более гладко при 37° или даже при
комнатной температуре при pH в пределах 3—7,4. Татсуи [58] нашел, что
триптамин легко конденсируется с паральдегидом, причем образуется 2-ме-
тил-2,3,4,5-тетрагидро-0-карболин (тетрагидрогарман), из которого он полу-
СН2
____/\
|. || || снсоон
\/\N / ^н2
„ сно
н г
R
сн2
\/\N / NH2
Н СО-СООН
R
чил ряд производных, в том числе 2,3-диметил-2,3,4,5-тетрагидро-0-карболин
(Мя-метилтетрагидрогарман).
Акобори и Сайто [36] аналогичным путем синтезировали тетрагидрогар-
ман из ацетальдегида и триптамина в кислом растворе. Хан и Людевиг [51]
получили тетрагидрогарман из триптамина и ацетальдегида в буферных ра-
створах при pH 5,2 и 6,2; они синтезировали также 2-бензил-2,3,4,5-тетрагиДро-
0-карболин из фенилацетальдегида и триптамина, причем лучший выход был
получен при pH 6 и температуре 25°.
В этой реакции ацетальдегид может быть заменен а-кетокислотами.
Действительно, Хан и сотрудники [59] показали, что такая видоизмененная
реакция проходит значительно легче, причем продуктами реакции оказывают-
ся производные тетрагидро-0-карболинкарбоновой-2 кислоты. Выделение дву-
окиси углерода происходит очень легко. Благодаря применению этого метода
многие производные тетрагидрокарболина стали легко доступными. Так были
получены, например, 2-метил-, 2-фенил-, 2-бензил- и 2-(3',4'-метилендиокси-
бензил)-2,3,4,5-тетрагидро-0-карболины [59].
Если триптамин обрабатывать производными фенил ацетальдегида, содер-
жащими гидроксильную или метоксигруппу, то p-карболин может и не полу-
читься. Однако при использовании соответствующих производных фенилпи-
ровиноградной кислоты реакция проходит гладко. Так, З-метокси-4-оксифе-
нилпировиноградная кйсдота, 3,4-диметокси- и 3,4,5-триметоксифенилпи-
ровиноградные кислоты дают соответственно 2-(3-метокси-4-оксибензил)-,
2-(3,4-диметоксибензил)- и 2-(3,4,5-триметоксибензил)-2,3,4,5-тетрагидро-Р-кар-
болинкарбоновые-2 кислоты I59E Реакция метилтриптамина с формальде-
гидом описана на стр. 197.
Обычно эти тетрагидро-Р-карболинкарбоновые-2 кислоты претерпевают
декарбоксилирование при попытке этерифицировать их по методу Фишера —
Шпейера. Обработка кислот хлористым водородом в горячем этаноле действи-
ПРОСТЫЕ КАРБОЛИНЫ
205
тельно представляет удобный способ получения тетрагидро-0-карболина из
соответствующей карбоновой-2 кислоты.
При действии на 2-метил-2,3,4,5-тетрагидро-0-карболинкарбоновую-2
кислоту (XLIII) хлористого водорода в метаноле получают некоторое коли-
чество сложного метилового эфира (XLIV) наряду с декарбоксилированным
основанием [59]. Хан и Ханзель [60] воспользовались этим наблюдением для
того, чтобы решить вопрос, действительно ли продукт, полученный из тр'ипт-
амина и а-кетокислоты, представляет производное тетрагидро-0-карболина
(XLIII), а не просто шиффово основание (XLV). Не подлежит сомнению, что
декарбоксилированные соединения, получаемые в результате обработки
хлористым водородом в метаноле, являются производными тетрагидро-0-
карболина, но циклизация может происходить под действием кислоты. Если
метиловый сложный эфир (XLIV) гидролизовать кипячением непродолжитель-
ное время со спиртовым раствором едкого кали, то получается небольшое коли-
чество исходной кислоты. Это указывает на то, что тетрагидро-0-карболнновая
кольцевая система, безусловно присутствующая в сложном эфире после обра-
ботки кислотой, должна была содержаться и в исходной кислоте, откуда сле-
дует, что циклизация должна проходить в мягких условиях, применявшихся
Ханом и сотрудниками для проведения этих конденсаций.
____/\
1 || NH----------> I || || NH +
%/\ /\/ \/\ /\/
N /\ N /\
нсн3 СООН Нсн3 СООСНз
XLIII XLIV
С—СООН
I
СН3
XLV
Интересно отметить, что эти конденсации протекают гладко и дают хоро-
шие выходы при «физиологических» условиях pH и температуры, но при pH
в интервале от 3 до 7 они не проходят удовлетворительно даже при' более
высоких температурах. Когда, однако, pH уменьшается до величин, близких
к 1, реакции проходят гладко как при 25°, так и при значительно более высо-
ких температурах. Действительно, при таком pH более высокая температура
представляет определенное преимущество, реакция протекает гораздо скорее
и до некоторого предела с повышением температуры- выходы увеличиваются.
Так, при конденсации триптамина с .и-оксифенилпировиноградной кислотой
выходы 2-(.м-оксибензил)-2,3,4,5-тетрагидро-Р-карболинкарбоновой-2 кислоты
при pH 1, следующие: при 25° за 24 дня — 58%; при 35° за 22 дня — 68%;
при 45° за 74 час — 65%; при 55° за 48 час — 54%; при 70° за 24 час — 36%.
При 100° за 48 час кислота получилась лишь с выходом 10%; кроме того, был
выделен с выходом 35% хлоргидрат декарбоксилированного основания.
Очевидно, водная кислота при относительно высоких температурах способст-
206
КАРБОЛИНЫ
вует декарбоксилированию. Хорошие выходы при низком pH и высокой темпе-
ратуре получаются и с другими кислотами, кроме упомянутой лг-оксифенилпи-
ровиноградной, например с 3,4-диметоксифенилпировиноградной кислотой.
Эта реакция при физиологических условиях протекает удовлетворительно,
когда кетокислота содержит вторую карбоксильную группу. Из триптамина
и а-кетоглутаровой кислоты при 25° и pH 3,8—5,8 была получена с выходом
приблизительно 50% 2-(2-карбоксиэтил)-2,3,4,5-тетрагидро-0-карболинкарбо-
новая-2 кислота (XLVI). Если это соединение обработать хлористым водо-
родом в метаноле, то оно теряет карбоксильную группу у атома углерода
в положении 2, и одновременно теряется молекула воды в результате образо-
вания другой карбоксильной группой внутреннего амида. Получающееся соеди-
нение выделяется в виде хлоргидрата, и поскольку эта соль не очень сильного
основания, то считается, что аминогруппа в пиперидиновом кольце осталась
свободной и, следовательно, в образовании амида должен участвовать азот
индольного ядра. Поэтому основание имеет строение XLVII; новое кольцо,
как это будет видно из дальнейшего, является шестичленным.
СН2
СООН
XLVI
При замене монокетокислот какой-либо а,а'-дикетодикарбоновой кислотой
происходят дальнейшие изменения. Так, при pH 1 и 45° триптамин и а,а'-
дикетопимелиновая кислота дают либо бис-тетрагидрокарболиновое основание
(XLVIII), либо тетрациклическую кислоту (XLIX), которая образуется, оче-
видно, в результате самопроизвольной циклизации промежуточного соедине-
ния L. В обоих случаях декарбоксилирование проходит у атома углерода карбо-
линового ядра в положении 2.
XLVIII
1 || II 1N11
\/\/\/ о = с—соон
N | |
н сн2 сн2
хсн2/
L
8-Метокси-2-кето-3-метил-2,3,4,5-тетрагидро-0-карболин уже упоминался
в связи с получением его путем окисления метилгармалина (стр. 197); окисле-
нием ацетилгармалина (стр. 200) получен соответствующий 8-метокси-З-аце-
ПРОСТЫЕ КАРБОЛИНЫ
207
тил-2-кето-2,3,4,5-тетрагидро-0-карболин. При гидролизе последнего спирто-
вым раствором едкого кали образуются два вещества: одно нейтральное —
8-метокси-2-кето-2,3,4,5-тетрагидро-0-карболин (LII, R =ОСН3) [45, 671; вто-
рое имеет свойства аминокислоты и, вероятно, представляет собой 6-метокси-3-(0-
,аминоэтил)индолкарбоновую-2 кислоту [45] (ЫН, R =ОСН3).
Родоначальное соединение — 2-кето-2,3,4,5-тетрагидро-0-карболин
(LII, R = Н) — получается при растворении азида а-(3-индолил)пропионовой
кислоты (LI) в кипящем бензоле и насыщении раствора хлористым водородом
'после прекращения выделения азота [25]. При этом тотчас же происходит
-циклизация изоцианата, образующегося, по-видимому, в качестве промежуточ-
ного продукта реакции превращения соединения LI в соединение LIII.
Соединение LII (R = Н) было синтезировано иным путем Кейматсу, Сугасава
LII LIII
и Касуйа [68], которые показали, что при нагревании со спиртовым раствором
едкого кали оно дает 3-(0-аминоэтил)индолкарбоновую-2 кислоту (LIII,R = Н).
2-Кето-2,3,4,5-тетрагидро-0-карболин является ценным исходным продуктом
для синтеза природных алкалоидов — эводиамина и рутекарпина (см. стр.
249, 250).
Производные 2,4,5-трикето-2,3,4,5-тетрагидро-0-карбодина были синте-
зированы Джонсоном и сотрудниками [56] в связи с выяснением структуры
глиотоксина. При окислении хромовой кислотой в ледяной уксусной кислоте
1,3-диметил-2-кето-2,3-дигидро-0-карболина (LIV), синтезированного из диме-
тилацеталя метиламида 1-метилиндолкарбоновой-2 кислотйц получен 1,3-
диметил-2,4,5-трикето-2,3,4,5-тетрагидро-0-карболин (LV), строение кото-
NHCHg
СН3О
LVI
208
КАРБОЛИНЫ
рого было подтверждено синтезом его исходя из метиламида 1-метилиндолкар-
боновой-2 кислоты (LVI) и этоксалилхлорида. Подобным образом окисление 3-
метил-2-кето-2,3-дигидро-0-карболина хромовой кислотой в уксусной кислоте
или в кислом растворе перманганата калия дало 3-метил-2,4,5-трикето-2,3,4,5-
тетрагидро-0-карболин; причем это соединение не могло быть получено другим
способом, так как при конденсации этиламида индолкарбоновой-2 кислоты
образовывался исключительно индолодиазин (LVII).
Несколько иной тип восстановленного производного 0-карболина был
обнаружен Робинзоном и Суджином [69] в процессе работы над синтезом физо-
стигмина. При обработке этилового эфира 5-этокси-3-метил-3-(0-фталими-
доэтил)индоленинкарбоновой-2 кислоты (LVIII) гидразингидратом в спирте,
а затем соляной кислотой отщеплялась фталевая кислота и происходила цикли-
зация с образованием 2-кето-7-этокси-11-метил-2,3,4,5-тетрагидро-0-псевдокар-
болина (LIX). Название 0-псевдокарболин было дано неизвестной изомерной
форме (LX) 0-карболина. Это вещество присоединяет два атома водорода,
а при восстановлении его сульфометилата получается производное гексагидро-
карболина, а именно 2-кето-7-этокси-1,2,3,4,10,11-гексагидро-0-карболин.
LVIH
LX
Различные производные. Карбоновые кислоты и сложные эфиры. Первой
из синтезированных 0-карболинкарбоновых кислот была норгарминкарбоновая,
полученная Перкином и Робинзоном [70] при окислении в особых условиях
бензилиденгармина — продукта конденсации гармина и бензальдегида. Нор-
гарминкарбоновая кислота (8-метокси-0-карболинкарбоновая-2 кислота) теряет
двуокись углерода с образованием норгармина (8-метокси-р-карболина). Подоб-
ным образом при окислении бензилиденгармана получена 0-карболинкарбоно-
вая-2 кислота, которая не выделялась, а непосредственно декарбоксилировалась
с образованием 0-карболина (норгармана) [24].
Тетрагидропроизводные 0-карболинкарбоновых кислот образуются чрез-
вычайно легко при конденсации триптофана с альдегидами или триптамина
LXI LXII
—/Ч>____соон
I 'II II N
N ।
н Rt
LXIII
Ri = CH3 или С2Н5
ПРОСТЫЕ КАРБОЛИНЫ
209
с а-кетокислотами. Об этих конденсациях уже упоминалось в связи с синтезом
различных производных тетрагидрокарболина, легко получающихся из этих
кислот в результате декарбоксилирования, поэтому нет необходимости здесь
на них останавливаться. Однако можно упомянуть работу Снайдера и сотрудни-
ков [71], которые показали, что сложные эфиры (LXI) 2,3,4,5-тетрагидро-0-
карболинкарбоновых-4 кислот можно дегидрировать либо серой в ксилоле,
либо хлоранилом в тетрахлорэтане и получать соответствующие 0-карболи-
новые сложные эфиры (LX 11), которые могут быть омылены в карбоновые кисло-
ты (LXIII).
Интересный синтез 2-кето-2,3-дигидро-0-карболинкарбоновой-4 кислоты
описан Кингом и Стиллером [72]. При гидролизе азлактона (LXIV), образую-
щегося из 2-карбэтоксииндол-З-альдегида, 10%-ным раствором едкого кали
в метиловом спирте получалась калиевая соль 2-кето-2,3-дигидро-0-карболин-
карбоновой-4 кислоты (LXVI), образовавшаяся из промежуточной 0-(2-карбок-
сииндол)-а-аминоакриловой кислоты (LXV). Маточные растворы содержали
соединение, при кислотном гидролизе которого в мягких условиях получен
метиловый эфир 2-кето-2,3-дигидро-0-карболинкарбоновой-4 кислоты. Реакции
этого соединения показали, что оно представляет собой метиловый эфир
4- (2-кето-2,3-дигидро-р-карболин)ортомуравьиной кислоты (LXVII).
.ч ,СН = С-С=О .. СН
\\\—II 1 „/° А—II /соон
I Ii II N-f —» I' II II %
^/\/ХСООС2Н5С6Н5 'Ч/\/ \ NH2
Н Н СООН
N II
н о
LXVI
LXIV LXV
СООН
LXVIII
С(ОСН3)3
LXVII
Тот же метиловый ортомуравьиный эфир получается и тогда, когда в каче-
стве исходного азлактона для синтеза брали 2-карбометоксииндол-З-альдегид.
При действии на азлактон (LXIV) спиртового раствора едкого кали, кроме
карбоксикарболина (LXVI), получается соответствующий этиловый ортому-
равьиный эфир, который легко гидролизуется кислотой в этиловый' эфир
2-кето-2,3-дигидро-0-карболинкарбоновой-4 кислоты.
Если 2-кето-2,3-дигидро-0-карболинкарбоновую-4 кислоту обработать
пятихлористым фосфором в хлорокиси фосфора, а неочищенные продукты
реакции превратить в их метиловые сложные эфиры, то получается метиловый
эфир 2-хлор-0-карболинкарбоновой-4 кислоты, а также побочный продукт
с брутто-формулой C27H20O4N4, строение которого не было выяснено. Атом гало-
гена в хлоркарболиновом сложном эфире и в свободной 2-хлоркарболинкарбо-
новой-4 кислоте (LXVIII) настолько прочно связан, что не может быть удален
каталитически. Однако при восстановлении сложного эфира иодистоводородной
кислотой и красным фосфором при 180° образуется 0-карболинкарбоновая-4
кислота, которая при декарбоксилировании дает Р-карболин [72].
Нитро-, амино- и галогенпроизводные. Аминопроизводные самого Р-карбо-
лина, по-видимому, не были получены, но различные аминогарманы описаны
в литературе. Один из них — 8-аминогарман (8-амино-2-метил-Р-карболин) —
уже упоминался как промежуточный продукт при получении гармана из гарми-
на (стр. 195). 4-Аминогарман был синтезирован Снайдером, Пармертером
и Уокером [73] из метилового эфира гарманкарбоновой-4 кислоты. Этот
14 Заказ № 978
210
КАРБОЛИНЫ
эфир с трудом превращается в амид, так что реакцию Гофмана нельзя было при-
менить, но гидразид получается легко и соответствующий азид превращается в
4-аминогарман либо непосредственно кипячением с 50 %-ной уксусной кислотой,
либо через бензиловый эфир 4-гарманилкарбаминовой кислоты, который гидро-
лизуют едким кали в диэтиленгликоле.
При нитровании гармана получаются два мононитропроизводных с т. пл.
299—300° и 209—210° соответственно. Нитрогруппа в первом из них находится
в положении 7 карболинового кольца, что было показано Снайдером, Пармерте-
ром и Катцем [74]. Восстановлением этого нитросоединения они получили
аминогарман с т. пл. 269—271°, который вступал в реакцию диазотирования.
Попытки превратить его в метоксисоединение непосредственно или через окси-
соединение оказались безуспешными, но соответствующий бромгарман был
получен реакцией Зандмейера. Продукт плавился при 246—247° и был
идентичен 7-бромгарману, полученному из 5-бром-ПЬ-триптофана конденсацией
с ацетальдегидом (с образованием тетрагидрогарманкарбоновой-4 кислоты)
и последующим окислением бихроматом. Положение нитрогруппы во втором
нитропроизводном пока точно не доказано, но, вероятно, она находится вкар-
болиновом ядре в положении 9.
При нитровании гармалина О. Фишер [75] получил мононитропроизводное
с т. пл. 122° (ср. с работой Фритцше [76]). То же соединение было получено
Коноваловой, Проскурниной и Ореховым [32], которые приводят т. пл. 132°
(с разл.). При окислении этого вещества хромовой кислотой и разбавленной
соляной кислотой образуется нитрогармин ст. пл. 204—205° [32, 75]. Поло-
жение нитрогруппы в этих соединениях, по-видимому, точно не установлено,
но по аналогии с хиндолином и другими производными индола можно предпо-
ложить, что она находится у седьмого атома углерода; это предположение под-
тверждается наблюдением Снайдера, Пармертера и Уокера [73], которые уста-
новили, что главным продуктом нитрования гармана является 7-нитрогарман.
Известно сравнительно немного галогенпроизводных (3-карболина; о
7-бромгармане уже упоминалось. 1-Метил-2-хлор-0-карболин был получен Кер-
маком, Перкином и Робинзоном [77] обработкой 1-метил-2-кето-2,3-дигидро-0-
карболина хлорокисью фосфора и пятихлористым фосфором (ср. также с рабо-
Простые карболиНЫ
211
той Кермака и Тебриха [78]). Это соединение не реагировало с иодистым метил-
магнием в кипящем изоамиловом эфире с образованием метилгармана; единст-
венным выделенным при этом соединением был 2-кето-1-метил-2,3-дигидро-р-
карболин. По-видимому, образовывались лишь следы метилгармана, о чем
можно было судить по его флуоресценции.
Атом хлора в 1-метил-2-хлор-р-карболине, по-видимому, еще в меньшей
степени способен к реакции с первичными аминами, чем атом хлора в 2-хлорпи-
ридине или 2-хлорхинолине. При нагревании этого соединения с диэтиламино-
этиламином, хотя и происходила, по-видимому, какая то реакция, но чистого
продукта выделить не удалось. Когда в реакцию вводился сульфометилат
1-метил-2-хлор-р-карболина, реакционноспособность атома хлора в положении
2 увеличивалась и удалось выделить салициловую соль образовавшегося осно-
вания, а именно дисалицилат 2-(р-диэтиламино)этиламино-1,4-диметил-р-кар-
болиния. При взаимодействии с 3-диметиламинопропиламином была получена
соль соответствующего гомолога.
Ангидрониевые основания, получаемые из карболинов
В карболиновом ряду мы встречаемся с явлением, которое хотя и наблюда-
лось у многих типов органических соединений, однако здесь оно особенно
ярко выражено, почему и послужило предметом тщательного изучения. Это
образование так называемых «ангидрониевых оснований». Все четыре карболи-
на способны давать такие основания. Ангидрониевым основаниям, образовав-
шимся из p-карболинов, уделялось значительно больше внимания, несомненно,
потому, что они встречаются в большой группе алкалоидов. Ангидрониевые
основания а- и у-каболинов несколько менее изучены, а соответствующие осно-
вания 6-карболинов, по-видимому, вовсе не исследовались.
Первые исследования ангидрониевых оснований а- и у-карболинов проведе-
ны Робинзоном и сотрудниками, что послужило прочной основой для последую-
щего глубокого изучения p-карболиновых оснований. Выводы Робинзона, каса-
ющиеся структуры этих оснований, тем более замечательны, что в то время,
когда они были сделаны, современные концепции, основанные на теории резонан-
са, были еще сравнительно мало разработаны. Армит и Робинзон [79] ввели
термин «ангидрониевое основание» для обозначения ангидропроизводных аро-
матических ониевых гидроокисей. В настоящей главе прежде всего будет уде-
лено внимание основаниям, являющимся производными а- и у-карболинов, а
затем уже будут рассмотрены p-карболиновые основания.
Образование ангидрониевых оснований лучше всего можно проследить
на конкретном примере. В а-карболине (I) атом азота пиррольного кольца
(Ni) практически не обладает основными свойствами, тогда как атом азота пири-
динового кольца (N2) сообщает молекуле свойства основания. а-Карболин
легко реагирует с диметилсульфатом, образуя сульфометилат [79, 80], с иодис-
тым метилом дает иодметилат [80], а с этилсульфатом — сульфоэтилат [80];
в этих соединениях алкильная группа присоединена к пиридиновому атому
азота (II). Если раствор сульфометилата обработать крепким едким кали,
то выделяется масло, которое затвердевает и, будучи перекристаллизованным
из ацетона, плавится при 138—139° [79, 80]. По составу это соединение являет-
ся метил-а-карболином и, очевидно, образуется из метогидроокиси в результа-
те потери молекулы воды. Метильная группа должна быть связана с атомом
азота N2,h по сравнению с^родоначальным соединением — а-к'арболином — здесь
отсутствует атом водорода у атома азота NP Ангидрониевое основание, очевид-
но, является производным не самого а-карболина, а гипотетического а-изо-
карболина (IX) (стр. 187).
2-Метил-а-изокарболин (III) растворяется в воде, причем раствор имеет
щелочную реакцию; при этом, очевидно, присоединяется молекула воды
*41
212
КАРБОЛИНЫ
с образованием гидроокиси
протон к пиррольному атому
карболиния; иными словами, присоединяется
азота Nj и получается катион карболиния (II).
I
Благодаря этому наблюдению становится ясно, что хотя в а-карболине
пиррольный атом азота Ni не обладает основным характером, в 2-метил-а-
изокарболине он имеет явно выраженные основные свойства. Это доказывает-
ся также изучением реакции 2-алкил-а-изокарболиновсалкилсолями. 2-Метил-
а-изокарболин взаимодействует с иодистым этилом, образуя вещество
с т. пл. 195°. Подобным образом 2-этил-а-изокарболин при действии на него
диметилсульфата и последующего превращения получающегося сульфомети-
лата в иодид дает продукт с т. пл. 209,5°, изомерный приведенному выше, но не
идентичный ему. Эти два изомерных соединения представляют собою соли
метилэтилкарболиния; при действии щелочи они превращаются в гидроокиси
метилэтилкарболиния, не обладающие склонностью к образованию ангидрони-
евого основания путем потери молекулы воды. Различие между этими двумя
продуктами показывает, что вторая алкильная группа не присоединилась к ато-
му N2 и, следовательно, она должна быть связана с атомом азота Nj. Это подтвер-
ждает предположение, что в изокарболинах основными свойствами обладает
атом азота Ni [80].
Возникает вопрос, как изобразить при помощи формулы ангидрониевое
основание? Рассмотрим в качестве характерного примера 2-метил-а-изокарбо-
лин. Как известно, он образуется из гидроокиси метил карболиния в результате
потери молекулы воды или, точнее, из катиона метилкарболиния путем потери
протона. Основным компонентом катиона карболиния, без сомнения, является
структура IV, в которой оба шестичленных кольца обладают ароматическим
характером. Атом Nt имеет два свободных электрона, тогда как атом азота N2—
VII
ПРОСТЫЕ карболины
213
четвертичный и свободных электронов у него нет. Отсутствие основных свойств
у атома Ni а-карболина, а также карбазола показывает, что два свободных элек-
трона в значительной мере участвуют в образовании циклической ароматичес-
кой системы. В ионе карболиния можно ожидать еще более значительного сме-
щения этих электронов в сторону пиридинового кольца с его положительно
заряженным атомом азота, и структуру иона можно частично представить эле-
ктромерными формами, какУ. Таким образом, в дополнение к различным резо-
нансным формам структуры IV, например VI и VII, можно добавить форму VIII,
где положительный заряд находится не у N2, а у атома NP Если мы отнимем
протон от IV, то получим для ангидрониевого основания формулу IX. В ней оба
шестичленных кольца обладают ароматическим характером, но у атомов Nj
и N2 имеются заряды; атом Ni имеет четыре свободных электрона и несет отрица-
тельный заряд; атом N2, не имея свободных электронов, несет положительный
заряд. Молекула является диполем, как молекула бетаина. Но если считать, что
протон удален не от IV, а от VIII, то мы получим другую структуру — X.
Здесь ни один из атомов азота не несет заряда; отсутствие локализованного заря-
да будет благоприятствовать существованию этой формы, а не дипольной (IX).
С другой стороны, у формы X пиридиновое кольцо не полностью ароматично,
что должно противодействовать ее образованию.
__/\
М' ! - 1 Г!
\ /\ /\ Z \/\ z\ /
NN NN
' Г - i
сн3 СН3
IX X
Даже в то время, когда важное значение резонанса в органических струк-
турах не оценивалось и отсутствовала интерпретация на основе квантовой
механики, Армит и Робинзон 1791 уже предполагали, что для полного изобра-
жения ангидрониевого основания, такого, как 2-метил-а-изокарболин, необ-
ходимы обе структуры IX и X и что это соединение является в некотором роде,
гибридом этих двух форм. В ангидрониевом основании существованию одной
формы благоприятствует то, что она полностью ароматична, но это преимущест-
во ослабляется тем, что молекула представляет достаточно сильный диполь.
В катионе карболиния обе формы несут положительный заряд, и здесь форма,
имеющая полностью ароматический характер, получает преобладающее зна-
чение, так как распределение зарядов не является помехой.
При наличии соответствующего катиона или катион-радикала (как,
например, Н4- или СН3) атом Nj в формуле IX благодаря своему отрицатель-
ному заряду будет притягивать молекулу реагента, с которой затем соединится.
Отсюда следует, что у такого ангидрониевого основания, как 2-метил-а-изокар-
болин, центром основных свойств будет атом NP
Образование ангидрониевого основания можно проиллюстрировать на
примере соединения из ряда у-карболинов. Так, например, при действии на
сульфометилат у-карболина (XI) большого избытка горячего концентрирован-
ного раствора едкого кали получается 4-метил-у-изокарболин [811. Строение
этого соединения так же, как и строение 2-метил-а-изокарболина, лучше все-
з
XI
XII
XIII
214
КАРБОЛИНЫ
го представить в виде резонансного гибрида двух форм — XII и XIII.
4- Метил-у-изокарболин—основание, имеющее желтую окраску в противополож-
ность бесцветному у-карболину; в безводном состоянии он плавится при 203—
204°, но очень гигроскопичен. В холодной воде этот изокарболин дает щелочную
реакцию, а в горячей воде он растворяется с образованием сильнощелочного
раствора, содержащего, без сомнения, метогидроокись у-карболина. Безвод-
ный 4-метил-у-изокарболин образует бесцветный сульфометилат, в котором вто-
рая метильная группа связана с атомом азота в положении 1. Метогидроокись,
получаемая действием едкого кали на растворы этого сульфометилата, нерас-
творима в эфире и бензоле, но растворяется в воде; сульфометилат 4-метил-у-
изокарболина резко отличается по своим свойствам от сульфометилата
у-карболина.
Образование ангидрониевых оснований в ряду p-карболинов изучено весь-
ма подробно. Химия p-карболиновых оснований будет рассмотрена в следую-
щих разделах. Практически, все, что было сказано выше, также применимо
и к этому ряду соединений. Однако следует указать на одно обстоятельство:
структура соединений,соответствующих формулам X или XII, в случае р-изокар-
болина не вполне очевидна. Мы не можем представить себе сопряженную систе-
му, расположенную по кратчайшему пути от одного атома азота к другому.
Оказалось необходимым представить незаряженную форму р-изокарболина
так, как изображено в формуле XIV. В этом случае сопряженная система меж-
ду двумя атомами азота проходит через бензольное кольцо. Обе формы струк-
туры p-изокарболина представлены формулами XIV и XV.
z\—-Л ' ---/ч
I | N-Clh | |l !| N —СПз -
XIV XV
Выводы, к которым пришел Робинзон с сотрудниками относительно хими-
ческого строения ангидрониевых производных карболина, получили веское
подтверждение в последующих работах. В частности, следует указать на
критический обзор и дальнейшее исследование p-карболинангидрониевых соеди-
нений, опубликованное Шварцем и Шлиттером [82]. Их исследование и выте-
кающие из него выводы одинаково хорошо приложимы к р-карболинангидро-
ниевым соединениям как природным, так и синтетическим независимо от слож-
ности их строения. В общем для этих соединений характерны следующие
свойства: все эти основания окрашены, начиная от интенсивно желтого цвета
до оранжево-красного. Основность этих оснований несколько больше, чем
основность аммиака (величина рК колеблется в пределах 10,4—10,6).
Спиртовые растворы оснований — желтого цвета, а растворы в абсолют-
ном ацетоне или хлороформе — оранжево-красные. Следовательно, цвет рас-
творов этих оснований зависит от полярности растворителя. Видимый и ультра-
фиолетовый спектры этих оснований в кислых и нейтральных спиртовых раство-
рах одинаковы и имеют максимумы поглощения около425, 315 и 255 мц и мини-
мумы — около 330 и 280 мр. В слабощелочной среде наблюдается характерный
сдвиг поглощения в направлении более длинных волн с максимумами около
425, 330 и 285 мц и минимумами при 350 и 300 мц.
Основания кристаллизуются из различных водных растворов в виде
гидратов, содержащих от одной до трех молекул кристаллизационной воды,
причем последнюю молекулу воды удалить из них крайне трудно.
Основания образуют соли с одним молем кислоты. Нитраты характеризу-
ются малой растворимостью. Соли имеют несколько более светлую окраску, чем
основания, и кристаллизуются с одной или двумя молекулами воды; в без-
водном состоянии сильно гигроскопичны.
ПРОСТЫЕ КАРБОЛИНЫ
215
Строение четвертичных солей пиридиния и получающихся из них псевдо-
оснований уже обсуждалось (т. 1) [83—85]. Шварц и Шлитер [821
считают, что образование p-карболиновых ангидрониевых оснований может
протекать по приведенной ниже схеме. Паолини и Марини-Беттоло [86] обра-
тили внимание на валентные состояния атомов азота в карболинах и образую-
щихся из них ангидрониевых основаниях.
Псевдооснование
(бледно-желтое)
1
Альдегидная форма
Образование ангидрониевых оснований известно и в ряду бензокарболинов;
некоторые исследования, касающиеся этого явления, будут описаны в соот-
ветствующих разделах настоящей главы (стр. 224). Обнаружено, что соедине-
ния, получаемые из простых карболинов в виде ангидрониевых оснований,
обычно имеют более желтую окраску, чем самые близкие им соединения с нор-
мальной карболиновой структурой; это распространяется и на ряд бензокарбо-
линов, но ангидрониевые основания соединений этого ряда иногда бывают
красного или пурпурного цвета. Следует ожидать, что изокарболиновые
производные будут иметь более интенсивную окраску, чем соединения типа
нормальных карболинов, если правилен высказанный выше взгляд на их
структуру, как резонансный гибрид; но специальной теории о зависимости
их цвета от структуры не выдвигалось.
Представляет интерес поведение p-карболиновых ангидрониевых оснований
при восстановлении [82]. При действии натрия в спирте или олова в соляной
кислоте гидрируется пиридиновое кольцо с образованием тетрагидропроизвод-
ных (XVI — XVII). Тот же результат получают при каталитическом гидрирова-
нии над окисью платины в щелочной среде. В противоположность этому при
восстановлении в уксуснокислом растворе затрагивается бензольное кольцо
XVIII XVI XVII
индольной части молекулы (XVI — XVIII). При восстановлении бор-
гидридом натрия бромметилат гармана восстанавливается в 2,3,4,5-тетрагидро-
гарман [87].
Интересное исследование сравнительной основности а-, р- и у-карболино-
вых ангидрониевых оснований, целиком согласующееся с ранее высказанными
216
КАРБОЛИНЫ
взглядами Робинзона, было опубликовано Греем 188]. Величины рА”„ для
ангидрониевых оснований 2-метил-а-карболина, З-метил-0-карболина и 4-метил-
у-карболина, определенные в 60%-ном этаноле при 25°, составляли 7,75,
— /ч. ____/ч
I il II rLСПз ' Н^° - I II I' N—-СН3 +Н3О+
/\Ч Ч/ Ч /
N N
Н
11,11 и 10,54 соответственно. Повышенная основность p-производного, обус-
ловленная увеличением свободной энергии при удалении протона из соли,
может быть предположительно приписана более низкой энергии стабилизации
Р-карболинангидрониевого основания. Выводы, которые можно сделать из рас-
смотрения хиноидных форм (XIX — XXI) относительно общей устойчивости
СПз
XIX
XXI
этих оснований, подтверждают эти предположения. Поскольку структура XX,
в которой имеет место нарушение резонанса бензольного кольца, является
структурой с относительно более высокой энергией, можно ожидать, что
P-основание будет энергетически менее выгодным, чем любой из его аналогов
(XIX и XXI).
N-Алкилпроизводные Р-карболина
p-Карболины, имеющие алкильные заместители у одного или обоих ато-
мов азота, могут быть получены несколькими способами.
Применение стандартного синтеза p-карболинов (стр. 195) к 1-алкилза-
мещенным триптамина приводит к 1-алкил-р-карболинаМ 189, 90].
-LRCHO
При алкилировании p-карболинов с помощью амида калия в жидком
аммиаке [91 ] или амида натрия в толуоле [92] и алкилгалогенида алкильная
группа присоединяется к атому азота индольного кольца. При действии алкил-
сульфатов или алкилиодидов на p-карболины четвертичные соли образуются
за счет атома азота пиридинового кольца.
Эта реакция была рассмотрена в разделе, посвященном ангидрониевым
основаниям (стр. 211). Применяя различные сочетания этих трех методов, Грей
и сотрудники [92 , 93] получили большое число четвертичных карболинов, из
которых некоторые имеют второй четвертичный атом азота. Ряд подобных
соединений приведен на стр. 217, некоторые из них обладают сильным гипо-
тенсивным действием.
При нагревании хлористого диметилгармина (хлористого 1,2,3-триметил-
8-метокси-р-карболиния) (I) при 290—300° и 10 мм рт. ст. выделяется хло-
ристый метил и образуется метилгармин ст. пл. 124—125°, отличающийся от
рассмотренного ранее. Как и можно было ожидать, наиболее легко будет отщеп-
ляться метильная группа от пиридинового атома азота, и новый метилгармин
БЕНЗОКАРБОЛИНЫ
217
представляет собой 1,2-диметил-8-метокси-р-карболин (II). Это подтверждается
тем, что при действии йодистого метила он образует хорошо известный иодме-
R^f^R'
тилат метилгармина. В отличие от изомерных желтых производных метил-0-
изокарболина метилгармин совершенно бесцветен [95].
N |
I Г СН3
СНз
БЕНЗОКАРБОЛИНЫ
Введение
Бензокарболины, образующиеся из карболинов путем пристройки к ним
еще одного бензольного кольца, могут быть разделены на два класса в зави-
симости от того, куда присоединено новое бензольное кольцо: к бензольному
или к пиридиновому кольцу карболина. Первый класс включает 12 теоретически
возможных циклических систем, по три производных от каждого из четырех
изомерных карболинов. Однако пока лишь один представитель этого класса
бензокарболинов описан в литературе (стр. 219), а именно 6,7-бензо-а-карбо-
лин (I). Второй класс включает две циклические системы, образуемые из
а-карболина: 3,4-бензо-а-карболин (II), обычно называемый хининдолином,
и 4,5-бензо-а-карболин (III). Несколько производных соединения II описаны,
но производные соединения III до сих пор не известны. Как из 0-,так и из у-
карболина возможно образование только одного бензокарболина данного класса,
а именно4,5-бензо-р-карболина (IV) и 2,3-бензо-у-карболина (V). Представители
обеих этих циклических систем известны. Из 6-карболина можно образо-
вать два бензокарболина второго класса: 2,3-бензо-6-карболин(У1), ни одного
производного которого до сих пор не синтезировано, и 3,4-бензо-6-карболин
(VII), обычно называемый хиндолином. Это первый из открытых бензокарбо-
линов и, по всей вероятности, наиболее хорошо из них изученный.
Номенклатура этих циклических систем основана на системе Гулланда
И-сотрудников [96], принятой в настоящей главе для простых циклических
218
КАРБОЛИНЫ
соединений. В литературе иногда применяется старая номенклатура Пер-
кина и Робинзона [97]; так, например, 4,5-бензо-|3-карболин часто называют
5,6-бензо-4-карболином, а 2,3-бензо-у-карболин — 3,4-бензо-5-карболином.
В справочнике «The Ring Index» [98] 6,7-бензо-а-карболин называется
7Н-бензо[е]пирид-[2,3-6[-индолом с нумерацией атомов, приведённой в фор-
муле. 1а; 4,5-бензо-р-карболин — 7Н-индоло-[2,3-с|-хинолином с нумерацией
атомов, показанной в формуле IVa; 2,3-бензо-у-карболин обозначается как 11Н-
БЕНЗОКАРБОЛИНЫ
219
индоло-[3,2-с]-хинолин; нумерация атомов этого соединения приведена в фор-
муле Va. Названия хининдолин и хиндолин, как и нумерация атомов, указан-
ная в формулах II и VII, приняты в справочнике «The Ring Index».
Для бензокарболинов, не являющихся производными хиндолина и хинин-
долина, будет применяться нумерация атомов, принятая для простых карболи-
нов, причем необобществленные атомы углерода в новом бензольном кольце
будут иметь номера 14, 15, 16 и 17, как показано в формулах I, IV и V. Нумера-
ция атомов хиндолина и хининдолина будет соответствовать общепринятой
в литературе и в справочнике «The Ring Index».
Природных производных бензокарболина пока не найдено, за исключени-
ем алкалоида криптолепина. Поскольку некоторые бензокарболины синтезиру-
ются легче, чем сами карболины, то они изучены довольно полно.
Некоторые производные бензокарболина удобно получать по методу Гре-
бе — Ульмана (стр. 187), но в отдельных случаях применялись и другие методы,
приведенные в соответствующих разделах.
Кроме упомянутых бензокарболинов, описано несколько производных типа
VIII, которые большей частью являются продуктами восстановления исход-
ной структуры VIII. Их называют индоло-[2,3-а]-хинолизинами и нумерация ато-
мов в них соответствует принятой в справочнике «The Ring Index».
6,7-Бензо-а-карболин
При реакции 2-хлорпиридина с 1,2-диаминонафталином образуется пири-
диламинонафтиламин; по мнению Фрика и Робинзона [99], это скорее всего
1-амино-2-(2-пиридиламино)нафталин, а не изомерное ему 2-амино-1-(2-пиридил-
амино)-п(юизводное. Полученный из этого соединения действием азотистой
кислоты 3-(2-пиридил)-нафто-[1, 2]-триазол (IX) разлагается при нагревании
с сиропообразной фосфорной кислотой, образуя 6,7-бензо-а-карболин (9,10-
бензо-3-карболин) (I).
Хининдолин
Хининдолин (3,4-бензо-а-карболин, nepu-хиндолин, индоло-2',3',2,3-хино-
лин, бензо-3-карболин) впервые получен Габриелем и Эшенбахом [100] путем
восстановления либо а,р-бис-о-нитрофенилпропионовой кислоты (X) сульфатом
железа (II) и аммиаком, либо нитрила этой кислоты спиртовым раствором сер-
нистого аммония в запаянной трубке при 100°. Вместо ожидаемого продукта
C15H12N2 (XI), получающегося в результате отщепления двух молекул воды
от диаминокарбоновой кислоты (Ха), было получено соединение формулы
CisHjoNa, которому была приписана структура XII или XIII и дано название
«хининдолин», потому что его можно рассматривать как соединенные хинолино-
вую и индольную структуры. В настоящее время установлено, что это основа-
ние имеет структуру XII; но, как будет показано ниже, производные изохинин-
долина (XIII) легко получаются в виде ангидрониевых оснований из метилатов
хининдолина.
Второй способ синтеза хининдолиновых производных состоит в применении
реакции Гребе — Ульмана, уже упоминавшейся при описании синтеза прос-
220
КАРБОЛИНЫ
тых карболиновых соединений (стр. 187). Так, сам хининдолин был получен
нагреванием 1-(2-хинолил) бензотриазола (XIV) [101] одного или с сиропообраз-
ной фосфорной кислотой [102].
СООН СООН
Ха
Хининдолин выделяется из нитробензола в виде бледно-желтых листочков
и дает фиолетовую флуоресценцию в растворе серной кислоты и этилацетата,
в котором он очень мало растворим. Другие производные хининдолина, полу-
ченные этим путем, это 11-метилхининдолин, 2-метокси-11-метилхининдолин
и 8,9-диметоксихининдолин [102]. Хольт и Петров [102] заметили, что при
нагревании триазолов в нейтральной или щелочной среде циклизацию про-
вести не удается, но ее протекание значительно облегчается при применении
в качестве растворителя фосфорной кислоты.
Третий путь синтеза хининдолина заключается в циклизации о-аминобен-
зилиденоксиндола (XV) [103], однако эта реакция проходит иногда с большим
трудом. Борше, Вагнер-Рёммих и Бартенхейер [104] установили, что для этого
необходимо нагревать о-аминобензилиденоксиндол до 300°. Они применили
вариант этого метода, при котором хининдолин был получен с 70%-ным выхо-
дом, а именно нагревали оксиндол и о-аминобензальтолуидин в присутствии
небольшого количества пиридина при 150° в течение нескольких часов. Если
реакцию проводили в кипящем амиловом спирте, то замыкания кольца не
происходило и продуктом реакции был 3-(о-аминобензилиден)оксиндол (XV).
Этим методом были получены другие соединения, в том числе 2,3-диметокси-
хининдолин и 2,3-метилендиоксихининдолин.
XV
При восстановлении 2,4-диокси-3-(о-нитрофенил)хинолина образуется
смесь 11-оксихининдолина (XVI) и 5-окси-2,3-бензо-у-карболина (XVII)
[105].
N-Алкилпроизводные. Хининдолин, как и все карболины и бензокарболины,
содержит атом азота основного характера, который легко реагирует с алкил-
БЕЙЗОКАРБОЛИНЫ
221
галогенидами или соответствующими сложными алкиловыми эфирами, образуя
5-алкилхининдолиниевые соли. Эти соли при действии щелочей превращаются
XVI XVIIj
не в гидроокиси алкилхининдолиния, а в соответствующие ангидрониевые
основания, которые можно рассматривать как производные гипотетического
изохининдолина (XIII) и аналоги ангидрониевых оснований карболинового
ряда, рассмотренных выше (стр. 211). Подобно последним, их лучше представ-
лять в виде гибридных структур: бензоидной формы с зарядами (XVIII) и сопря-
женной, небензоидной, не несущей зарядов (XIX).
XVIII
R
XIX
2,3-Метилендиокси-5-метилизохининдолин был синтезировав Армитом
и Робинзоном [106]. Действуя на б'-аминопиперонилиденоксиндол диметил-
сульфатом они получили б'-метиламинопиперонилиденоксиндольную соль
метилсерной кислоты (XX), которую затем циклизовали при помощи спирто-
вого раствора едкого кали. Получающийся 5-метил-2,3-метилендиоксиизохинин-
долин (XXI) кристаллизуется из спирта в виде лимонно-желтых игл. Раствор
в серной кислоте дает яркую синевато-фиолетовую флуоресценцию.
XX
Борше, Вагнер-Рёммиху и Бартенхейеру [104] не удалось получить
хининДолины из 3-(о-аминобензилиден)-1-метоксиндолов при нагревании
с пиперидином в различных условиях. Портер, Робинзон и Вайлер [107] также
встретили затруднения при циклизации 3-(6-аминопиперонилиден)-1-метил-
оксиндола, но они установили, что реакция может быть проведена при нагре-
вании с сульфаминовой кислотой в растворе хинолина; образующийся при этом
2,3-метилендиокси-6-метилхининдолин флуоресцирует в нейтральных и кислых
растворах.
Нитропроизводные. Габриель и Эшенбах [100] нитровали хининдолин
с целью получения мононитропрбизводного. Следовало ожидать, что хинин-
долин будет нитроваться в положении 9. Хольт и Петров [102] подтвердили
222
КАРБОЛИНЫ
это следующим образом. Они конденсировали 5-нитрооксиндол с о-нитробенз-
альдегидом и получившийся 2,5-динитробензилиденоксиндол восстанавливали
каталитическим путем в ледяной уксусной кислоте. Ацетилирование получен-
ного продукта уксусным ангидридом дало триацетилпроизводное 9-аминохи-
ниндолина. Это соединение идентично полученному из мононитропроизводного
хининдолина в результате восстановления последнего в аминосоединение
и последующего ацетилирования. Промежуточный продукт — микрокристал-
лический желтый порошок — представлял собой 9-аминохининдолин. 9-Нит-
рохининдолин дает хлорметилат, который при обработке щелочью образует
ангидрониевое основание — 9-нитро-5-метилизохининдолин.
Хининдолинкарбоновые кислоты. При кипячении индиго с водноспирто-
вым раствором едкого натра одним из продуктов оказалась карбоновая кислота
с формулой CieH10O2N2; при нагревании выше 300° она теряет двуокись углеро-
да и дает хининдолин [1081. В качестве промежуточного продукта, очевидно,
образуется хининдолинкарбоновая-11 кислота по следующей схеме:
СООН
I
Изоиндиго
СООН
I
Хининдолин
Хининдолинкарбоновая-11 кислота получается также в качестве проме-
жуточного продукта реакции при конденсации оксиндола с изатином.
4,5-Бензо-Р-карболин
Первый синтез одного из производных 4,5-бензо-р-карболина был описан
Лаусоном, Перкином и Робинзоном [109]. Основная реакция — это конденса-
ция 2,2'-диаминобензофенона с каким-либо а-галогенкетоном в соответствии
с приведенным ниже уравнением. Ввиду относительной доступности был при-
менен диаминовератрон (6,6'-диамино-3,4,3',4'-тетраметоксибензофенон). При
нагревании с ю-бромацетофеноном в растворе уксусной кислоты он давал бром-
гидрат N-фенацилдиаминовератрона. При добавлении 30%-ного водного рас-
твора едкого кали к суспензии сухого бромгидрата в спирте получали темно-
бензокарболины
223
красный раствор; после кипячения в течение 1 мин цвет его становился светло-
оранжевым и при разбавлении водой выпадали бесцветные игольчатые кристал-
лы 2-фенил-7,8,15,16-тетраметокси-4,5-бензо-р-карболина (фенилдивератро-
гармирина). Это вещество растворимо в метиловом спирте и выпадает из рас-
твора в виде призм, не растворяющихся вновь при нагревании. Растворы этого
основания в нейтральных растворителях имеют фиолетовую флуоресценцию,
а кислые желтые спиртовые растворы обладают лишь слабой зеленой флуорес-
ценцией. Раствор этого основания в серной кислоте имеет заметную зеленую
флуоресценцию.
При реакции диаминовератрона с бромацетоном в уксусной кислоте не уда-
лось выделить промежуточного соединения. Единственным полученным про-
дуктом оказался бромгидрат 2-метил-7,8,15,16-тетраметокси-4,5-бензо-
p-карболина, из которого действием водного раствора едкого кали полу-
чено свободное основание.
Горячие водные растворы этого основания при охлаждении остаются пере-
сыщенными и становятся чрезвычайно вязкими, вследствие чего даже разбавлен-
ные растворы по своему внешнему виду похожи на глицерин. При добавлении
затравки основание закристаллизовывается почти нацело и жидкость утрачи-
вает свою аномальную вязкость. Бензольные растворы этого основания обла-
дают фиолетовой флуоресценцией. Вещество является сильным основанием и на
воздухе образует углекислую соль. Растворы основания в водных кислотах
имеют бледно-зеленую флуоресценцию, а в кислых спиртовых растворах и в ле-
дяной уксусной кислоте обладают более сильной зеленовато-голубой флуорес-
ценцией. Основание образует кристаллический сульфометилат, который в рас-
творе этилового спирта имеет исключительно яркую голубовато-зеленую флу-
оресценцию.
При кипячении бромгидрата 2-метил-7,8,15,16-тетраметокси-4,5-бензо-р-
карболина с иодистоводородной кислотой (уд. вес 1,7) выделяется иодистый
метил и образуется метилтетраокси-4,5-бензо-р-карболин. Основание, выделен-
ное из водного раствора иодида добавлением ацетата натрия, легко окисляется
воздухом и трудно очищается. При окислении этого тетраоксисоединения хромо-
вой кислотой получалась кислота, превращавшаяся при перегонке над натрон-
ной известью в токе водорода в 7-метил-6-азаиндол (апогармин) (XXII) с незна-
чительным выходом.
Второй способ синтеза производных 4,5-бензо-р-карболина заключается
в получении их из ацетильных производных 3-(о-аминофенил)индола. 3-(о-
Нитрофенилиндол)карбоновая-2 кислота легко образуется при действии
спиртового раствора хлористого водорода на фенилгидразон о-нитрофенилпи-
ровиноградной кислоты, причем происходит индольная циклизация по Фише-
ру. Декарбоксилирование 3-(о-нитрофенилиндол)карбоновой-2 кислоты,
СН3
XXII
сопровождающееся восстановлением, дает 3-(о-аминофенил)индол. Аналогично
из метилфенилгидразона получают 1-метил-3-(о-аминофенил)индол, причем
в этом случае циклизация проходит несколько легче [110].
При кипячении 3-(о-аминофенил)индола с безводной муравьиной кислотой
получается 3-(о-формамидофенил)индол и следы 4,5-бензо-|3-карболина, обра-
зующегося, по-видимому, при дегидратации формамидофенилиндола под дейст-
вием муравьиной кислоты. Сырое формильное производное при обработке
хлорокисью фосфора в кипящем толуоле циклизуется с образованием 4,5-
224
КАрбоЛинЫ
бензо-0-карболина. Это соединение не вступает ни в одну из обычных реакций
индола; в кислых растворах оно обладает красивой голубовато-зеленой флуо-
Схема 1
ресценцией. При обработке З-(о-ацетамидофенил)- или 3-(о-пропионамидофе-
нил)индола хлорокисью фосфора в кипящем толуоле аналогичным образом
получены 2-метил- и 2-этил-4,5-бензо-р-карболины (схема 1, R4 = Н).
N-Метилпроизводные, метилаты и т. д. Вследствие сравнительной легкости
синтеза 1-метил-3-(о-аминофенил) индола, о котором уже сообщалось, произ-
водные 1-метил-4,5-бензо-р-карболина особенно легко доступны. Так, при
кипячении 3-(о-формамидофенил)-1-метилиндола с хлорокисью фосфора в толу-
оле получается 1-метил-4,5-бензо-р-карболин. 3-(о-Ацетамидофенил)-1-метил-
индол в тех ’же условиях дает 1,2-диметил-4,5-бензо-р-карболин (схема 1,
Ri = R2 = СН3.)
4,5-Бензо-р-карболины легко дают четвертичные соли, причем алкильная
группа присоединяется к атому азота пиридинового кольца. Таким образом
в результате действия диметилсульфата на соответствующие основания были
получены следующие метилаты [111]: сульфометилат 4,5-бензо-р-карболина,
его 1-метил-, 1,2-диметил- и 2-этил производные. Все эти сульфометилаты легко
растворимы в воде, причем растворы обладают сильной флуоресценцией, обычно
зеленоватой, голубовато-зеленой или зеленовато-желтой. С увеличением числа
метильных групп флуоресценция становится все более желтой (см. работу Кер-
мака и Слетера [111]).
Ангидрониевые основания получаются при действии щелочи на растворы
метилатов 4,5-бензо-р-карболина, не замещенных в 1-положении. Эти основания
представляют собой производные 4,5-бензо-р-изокарболина. Относительная
легкость синтеза соединений ряда 4,5-бензо-р-карболина позволяет легко
получить экспериментальные доказательства положения метильных групп
в солях четвертичных оснований.
Если водный раствор сульфометилата 4,5-бензо-р-карболина подщело-
чить, то осаждается оранжевое ангидрониевое основание. При растворении
в бензоле и обработке диметилсульфатом это 3-метил-4,5-бензО-р-изокарболи-
новое основание дает сульфометилат, идентичный сульфометилату, получен-
ному из 1-метил-4,5-бензо-р-карболина (см. выше). Так как в последнем соеди-
нении одна метильная группа, несомненно, присоединена к атому азота пир-
рольного кольца, то, следовательно, при присоединении диметилсульфата к ан-
бензокарболины
225
гидрониевому основанию, у которого метильная группа связана с атомом
азота в пиридиновом кольце, новая метильная группа присоединяется к атому
азота пиррольного ядра [111]. Этот вывод был в дальнейшем подтвержден
при проведении параллельных опытов с гомологичными 2-метилсоединениями;
сульфометилат, полученный из 2,3-диметил-4,5-бензо-0-изокарболина, оказал-
ся идентичным сульфометилату 1,2-диметил-4,5-бензо-0-карболина [111].
Производные 2-окси-4,5-бензо-0-карболина. 2-Окси-4,5-бензо-0-карболин
и его производные синтезируются особенно легко. Так, например, при восста-
новлении 3-(о-нитрофенил)индолкарбоновой-2 кислоты происходит циклизация
с образованием 2-окси-4,5-бензо-р-карболина (XXV). Это соединение, вероятно,
существует главным образом в таутомерной форме в виде 2-кето-2,3-дигидро-
4,5-бензо-р-карболина (XXIV), который можно рассматривать как внутренний
амид, получающийся в результате отщепления воды от карбоксильной и ами-
ногруппы индоламинокислоты (XXIII). Восстановление удобно проводить
цинковой пылью в 80%-ной уксусной кислоте; циклизация З-(о-аминофенил)-
индолкарбоновой-2 кислоты наступает немедленно. О легкости, с которой замы-
кание кольца происходит даже в щелочной среде, свидетельствует следующее:
при проведении восстановления при помощи сернокислого железа (II) и амми-
ака раствор оставался щелочным в течение всего опыта. В фильтрате после
отделения окиси железа продукт не был обнаружен; производное бензо-р-
карболина было получено после растворения окиси железа в разбавленной
кислоте.
I 8 I »
ZX—/V
I II В N°2 I || II NH2
L Д ^-соон L H Ji-соон
V N X/ N
H H
XXIII
2-Кето-2,3-дигидро-4,5-бензо-р-карболин при кристаллизации из пиридина
образует бесцветные прямоугольные призматические иглы. Он практически
нерастворим в большинстве органических растворителей, кроме ледяной уксус-
ной кислоты и пиридина. В уксуснокислом растворе, так же как и при кипячении
с водой, содержащей следы соляной кислоты, он дает красивую синюю флуорес-
ценцию, Превращение этого соединения в.4,5-бензо-0-карболин затруднительно,
и лишь восстановлением цинковой пылью в токе водорода удалось превратить
его в основание с небольшим выходом [110].
Нагревание 2-кето-2,3-дигидро-4,5-бензо-0-карболина при 110° с хлор-
окисью фосфора и одним молем пятихлористого фосфора приводит к получению
2-хлор-4,5-бензо-0-карболина. При освещении вольтовой дугой (но не при
дневном свете) сильно разбавленные спиртовые растворы этого соединения
дают сине-фиолетовую флуоресценцию. Аналогично 2-кето-1-метил-2,3-дигидро-
4,5-бензо-р-карболин, получаемый восстановлением 1-метил-3-(о-нитрофенил)-
15 Заказ JM5 978
226
КАРБОЛИНЫ
индолкарбоновой-2 кислоты, превращается в 2-хлор-1-метил-4,5-бензо-р-
карболин, очень сходный по своим свойствам с соединением, не имеющим метиль-
ного заместителя.
Если при реакции 2-кето-2,3-дигидро-4,5-бензо-р-карболина с хлорокисью
фосфора и пятихлористым фосфором взять больше 1 моля последнего на 1 моль
карболина, то получается дихлорбензо-p-карболин. То, что второй атом хлора
находится в карболиновом кольце в положении 7, было доказано синтезом этого
соединения из 5-хлор-3-(о-нитрофенил)индолкарбоновой-2 кислоты. Кислота
при восстановлении циклизуется обычным образом и дает 7-хлор-2-кето-
2,3-дигидро-4,5-бензо-p-карболин, который при реакции с хлорокисью фосфора
и 1 молем пятихлористого фосфора (на 1 моль карболина) дает 2,7-дихлор-4,5-
бензо-p-карболин, идентичный упомянутому выше дихлорбензо-Р-карболину
[112]. Присоединение атома хлора в пара-положение относительно группы
NH согласуется с тем, чего следовало бы ожидать на основании общих сооб-
ражений. Хлорирующее действие пятихлористого фосфора в данном случае
аналогично наблюдаемому при действии избытка пятихлористого фосфора
в хлорбензоле на некоторые дифениламинокарбоповые кислоты [113].
Как и можно было ожидать, атом хлора, находящийся в 4,5-бензо-Р-кар-
болинах в положении 2, в некоторой степени способен к реакциям с анио-
ноидными реагентами. Например, при нагревании 2-хлор-1-метил-4,5-бензо-
Р-карболина с р-диэтиламиноэтиламином при 145° образуется 2-(Р-диэтил-
аминоэтиламино)-!-метил-4,5-бензо-р-карболин. Разбавленные спиртовые раст-
воры его обладают сине-фиолетовой, а разбавленные водные растворы —
зеленовато-синей флуоресценцией. Он имеет ясно выраженный горький вкус
и даже следы его оказывают анестезирующее действие на язык. 2-(Р-Диэтил-
аминоэтиламино)-4,5-бензо-Р-карболин может быть получен подобным обра-
зом; очень разбавленные водные растворы его обладают сине-фиолетовой
флуоресценцией, исчезающей под действием кислоты [112].
2-Метил-6,7,8,9-тетрагидро-4,5-бензо-р-карболин синтезирован Робин-
зоном и Робинзоном [114] кипячением смеси 3-гидразинохинальдина, цикло-
гексанона и ледяной уксусной кислоты в течение 10 час. Образующийся в каче-
стве промежуточного соединения гидразон (XXVI), котор&й не был выделен,
претерпел индольную циклизацию по Фишеру и дал 2-метил-6,7,8,9-тетра;
гидро-4,5-бензо-р-карболин (XXVII) с общим выходом 63%. Слабый спирто-
вой раствор этого соединения имеет светло-фиолетовую флуоресценцию,
XXVI XXVII
которая становится более интенсивной при добавлении соляной кислоты.
Сульфометилат этого основания, образующий микроскопические бледно-
желтые иглы, легко растворим в воде и спирте; растворы имеют очень яркую
синевато-фиолетовую флуоресценцию. Обработка этих растворов щелочью
дает ангидрониевое основание, 2,3-диметил-6,7,8,9-тетрагидро-4,5-бензо-р-изо-
карболин, в виде длинных желтых игл; желтые растворы последнего в эфире
или бензоле обладают зеленой флуоресценцией. Это соединение образует
сульфометилат, раствор которого флуоресцирует ярко-голубым цветом. Кон-
центрированный водный раствор едкого натра осаждает метогидроокись,
нерастворимую в эфире и бензоле. Как и следовало ожидать, образования
ангидрониевого основания в этом случае не происходит.
БЕНЗОКАРБОЛИНЫ
227
2,З-Бензо-у-карболин
Клемо и Перкин [115], нагревая фенилгидразон 4-кето-1,2,3,4-тетрагид-
рохинолина (XXVIII) с 20%-ной серной кислотой на водяной бане, выделили
соединение, кристаллизовавшееся из метанола в виде бесцветных призм непра-
вильной формы и обладавшее в метанольном растворе слабой красновато-
голубой флуоресценцией. Это основание имело формулу С15Н1(^2, а не C15H12N2;
последняя соответствует дигидро-2,3-бензо-у-карболину (XXIX), образова-
ния которого можно было бы ожидать в случае простой индольной циклиза-
ции. Клемо и Перкин пришли к выводу, что происходит не только циклиза-
ция, но соединение теряет два атома водорода, и поэтому в действительности
является 2,3-бензо-у-карболином (XXX) (названным авторами 3,4-хининдо-
лином). Правильность их заключения подтверждает не оставляющий сомне-
ния синтез этого основания по реакции Гребе — Ульмана [116], исходя
из 1-(4-хинолил)бензотриазола (XXXI). При нагревании этого соединения
с сиропообразной фосфорной кислотой было получено бесцветное кристалли-
ческое основание, которое оказалось идентичным соединению Клемо и Пер-
кина. В частности, хотя сырой продукт и обладал сильной фиолетовой флуо-
ресценцией в серной кислоте, чистое соединение, так же как и основание
Клемо и Перкина, не флуоресцировало в этом растворителе.
ХХУШ
При сплавлении .циба желтого (индиго желтого, Ciba 2 G) (XXXII)
со щелочью де Дисбах, де Би и Рубли [117] выделили соединение состава
C15H10N2, которому авторы на основании результатов его окислительного
расщепления приписали строение 2,3-бензо-у-карболина. При окислении
хромовой кислотой оно дает дикарбоновую кислоту C13H8O4N2, теряющую
при нагревании выше температуры плавления двуокись углерода и дающую
соединение СцН8М2, которое представляет собой, по всей вероятности,
3,4,3',2'-пирролохинолин (XXXIII). Последний при окислении пермангана-
том калия дает оксалилантраниловую кислоту, а при окислении азотной
кислотой — 4-оксихинолинкарбоновую-З кислоту. Образование этих двух
веществ доказывает наличие хинолинового ядра и указывает на положение
пиррольного кольца в соединении CnH8N2, подтверждая правильность фор-
мулы XXXIII. Поэтому авторы считают, что продукт, полученный ими при
сплавлении со щелочью, весьма вероятно, является 2,3-бензо-у-карболином
(названный ими 2,3-индолохинолином); это заключение подтверждается
также удовлетворительным совпадением определенной авторами температуры
плавления (332°) с температурой плавления (342°) этого основания [116].
Следует обратить внимание на два других соединения, выделенные де Дис-
бахом, де Би и Рубли [117] из продукта сплавления циба желтого со щелочью.
15*
228
КАРБОЛИНЫ
Эти соединения оказались изомерами, имеющими состав C15H10O2N2
и, вероятно, представляли различные формы одного и того же соединения.
Их строение не установлено, но авторы считают, что они являются, возможно,
диоксипроизводными 2,3-бензо-у-карболина, причем гидроксильные группы
предположительно находятся в положениях 1 и 5. То же диоксисоединение
было получено при сплавлении со щелочью 12-кето-6-фенил-7,12-дигидрохино-
[3,4-6]-хинолина (XXXIVa) и его диметильного гомолога (XXXIV6), тогда
как 10-метилпроизводное (XXXIVb) дало гомолог состава C10H12O2N2 [118].
Действием пятихлористого фосфора в нитробензоле гидроксильные группы
I н
2
XXXIVa, R1 = R2 = R3 = H
XXXIV6, r1 = r2 = ch3, r3 = h
XXXIVb, R1 = R2=H, R3 = CH3
бензокарболины
229
были замещены на хлор, но ввиду того, что существуют сомнения — являются
ли эти соединения действительно производными 2,3-бензо-у-карболина,—
они здесь не будут рассматриваться.
Следует заметить, что Манн и сотрудники [119] провели циклизацию
4-хинолилгидразона циклогексанона и получили при этом 6,7,8,9-тетра-
гидро-2,3-бензо-у-карболин.
Метилпроизводные 2,3-бензо-у-карболина. 2,З-Бензо-у-карболин образует
иодметилат, который при действии щелочи дает не метогидроокись, а ангид-
рониевое основание — 4-метил-2,3-бензо-у-изокарболин. Структуру этого сое-
динения следует рассматривать как резонансный гибрид подобно другим, уже
описанным ангидрониевым основаниям.
Кермак и Смит [120] получили 5-метил-2,3-бензо-у-карболин нагрева-
нием 1-(2-метил-4-хинолил)бензотриазола (XXXV) в сиропообразной фосфор-
ной кислоте. Во всех растворах — нейтральных и кислых — основание обла-
дает сильной сине-фиолетовой флуоресценцией. Аналогичным образом
из 1-(2-метил-6-метокси-4-хинолил)бензотриазола был получен 15-метокси-
5-метил-2,3-бензо-у-карболин (XXXVI). Едва ли можно сомневаться в том,
что синтез по методу Гребе — Ульмана действительно приводит к соедине-
нию, имеющему структуру XXXVI. Однако исходя из теоретических сообра-
жений можно было ожидать, что циклизация приведет к образованию нового
шестичленного (вместо пятичленного) кольца, как в формуле XXXVII. Между
тем, Кермак и Смит показали, что при кипячении 1-(6-метокси-2,3-диметил-
4-хинолил)бензотриазола (XXXVIII) в сиропообразной фосфорной кислоте
не получается основания с синей флуоресценцией. Образование бензокарбо-
лина здесь исключено, но иное направление циклизации все же возможно. Это
является негативным доказательством в пользу того, что вышеприведенным
соединениям соответствуют приписанные им формулы. Как будет показано
ниже, это подтверждено и доказательством положительного характера.
СН3 СН3
XXXV XXXVI
ОСН3
XXXVIII
1,5-Диметил-2,3-бензо-у-карболин получен следующим образом: метил-
фенилгидразон (XXXIX) о-ацетаминоацетофенона при действии хлорокиси
фосфора в кипящем толуоле претерпевает двойную циклизацию. Сначала
в результате циклизации по Фишеру образуется 1-метил-2-(о-ацетамидофе-
нил)нндол (XL), а затем по реакции Бишлера — Напиральского это соеди-
230
КАРБОЛИНЫ
нение превращается в соединение XLI. Растворы последнего в нейтральных
растворителях и разбавленных минеральных кислотах имеют синевато-фиоле-
товую флуоресценцию, а в разбавленных уксусной и молочной кислотах —
интенсивно зеленую. Раствор этого соединения в концентрированной холод-
ной серной кислоте имеет красновато-коричневую окраску с легкой синей
флуоресценцией, которая при нагревании становится фиолетовой, причем
раствор теряет свой красноватый оттенок.
Это соединение было использовано для доказательства строения вещества,
полученного по методу Гребе — Ульмана, а также для того, чтобы доказать
несостоятельность предположения, что этот метод дает структуры типа
XXXVII. Соединение, полученное по реакции Гребе — Ульмана и имевшее
предположительно структуру XLIII, было переведено в сульфометилат (XLV).
Ангидрониевое основание, приготовленное из этого сульфометилата при обра-
ботке щелочью, было выделено из спирта в виде моногидрата. Как и следовало
ожидать, это соединение — 4,5-диметил-2,3-бензо-у-изокарболин (XLIV) —
присоединяло диметилсульфат, превращаясь в сульфометилат (XLII6).
СН3 СН3
I I
ОС ОС
XXXIX XL
СН3 СН3
I I
XLI XLIIa
СН3 СН3
XLIV XLII6
СН3 СН3
I I
XLIII XLV
бензокарболины
231
При обработке 1,5-диметил-2,3-бензо-у-карболина диметилсульфатом
получена четвертичная соль (XLIIa). Температура плавления смеси этих
двух соединений не обнаруживала заметной депрессии, т. е. они были иден-
тичны; растворимость, флуоресценция и другие физические свойства обоих
сульфометилатов полностью совпадали. Это говорит в пользу того, что про-
дукт, синтезированный по методу Гребе — Ульмана, имеет строение XXXVI,
а не XXXVII [120].
Метод, применявшийся для синтеза 1,4-диалкил-,у-карбониевых солей
(см. стр. 189), был использован для получения иодида 4-этил-10,11,12,13-
тетрагидро-1-метил-2,3-бензо-'у-карболиния (XLVa) из 2-литий-1-метилин-
дола и 2-диэтиламиноциклогексана [121]. Иодид 4,4-диэтил-2,3,4,5,10,11,12,13-
октагидро-2-окси-1-метил-2,3-бензо-у-карболиния (XLV6) был получен ана-
логичным путем. При нагревании в о-нитротолуоле до 230° он превращался
в соединение XLVa.
XLVa
XLV6
Индоло-[2,3-в]-хинолизин. Это соединение можно рассматривать как
родственное у-карболину, однако его называют 11Н-индоло-[2,3-в]-хиноли-
зином; нумерация атомов приведена в формуле XLVb. Описан синтез неко-
XLVb
XLVk
Ан, сн3
XLVe, R = H
XLVjk,R = CH2N(CH3)2
XLVa, R = CH2N(CH3)3
XLVa
232
карболины
торых его производных [121]. Эти соединения были названы хинолизино-
[2,3-в]-индолами, а нумерация атомов соответствовала формуле XLVb. При
взаимодействии 2-литий-1-метилиндола с 2-ацетилпиридином образуется
2-[1-окси-1-(2-пиридил)этил]-1-метилиндол (XLVe). Основание Манниха
(XLVjk), полученное при реакции соединения XLVe с формальдегидом >и диме-
тиламином, даете иодистым метилом четвертичный иодид (ХЬУз). Нагрева-
ние раствора соединения XLV3 в целлозольве (1,2-диэтоксиэтане) до 100°
приводит к иодиду 11,12-диметил-11Н-индоло-[2,3-в]-5-хинолизиния (XLVr).
Каталитическое восстановление соединения X LVe водородом на платине в уксус-
ной кислоте дает производное пиперидина (XLVh), которое при действии
формальдегида в уксусной кислоте циклизуется с образованием 1,2,3,4,5,6,12,
12а-октагидро-12-окси-11,12-диметил-11Н-индоло-[2,3-в]-хинолизина (LXVk);
последний при действии йодистого метила образует соль четвертичного осно-
вания (XLVn). При нагревании последнего до 250—270° отщепляется вода
и метан и образуется иодид 1,2,3,4-тетрагидро-11,12-диметил-11Н-индоло-
[2,3-в]-5-хинолизиния (XLVa).
2,3-Бензо-6-карболин. Эта циклическая система (XLVm) известна также
под названием 11Н-индоло-[3,2-с]-изохинолина (согласно справочнику «The
Ring Index») с нумерацией атомов, как указано в формулах XLVm или XLVh.
Авторы придерживаются нумерации XLVm. Производные соединения XLVm
получают циклизацией З-ациламино-2-фенилиндолов по методу Бишлера —
Напиральского [122]. Замыкание кольца проходит легче, когда атом азота
в индольном кольце замещен. Из З-ацетиламино-2 фенилиндола (XLVo) при
обработке пятиокисью фосфора в кипящем нитробензоле получен с низким
выходом один лишь 4-метил-2,3-бензо-6-карболин (XLVc). Робинзону и Торнли
[123] не удалось зациклизовать соединение XLVo при помощи треххлористого
XLVm
XLVh
XLVo, Ri=H, R2=CH3
XLVn, Rj— CgHg, R2=CH3
XLVg, R,= R2=C6H5
XLVc,R,=H, R2=CH3
XLVt, R^CgHfc R2=CH3
XLVy, Rt=R2 = C6H5
<?H3
XLVcj), R,-H
XLVx, Rj=CeH}
бензокарболины
233
фосфора в кипящем эфире. 4-Метил-1 -фенил- (XLVt) и 1,4-дифенил-2,3-бен-
зо-6-карболины (XLVy) были получены с удовлетворительными выходами
при кипячении 3-ацетиламино- (XLVn) и соответственно 3-бензоиламино-
1,2-дифенилиндола (XLVp) с пятиокисью или хлорокисью фосфора в ксилоле.
Из соединений XLVc и XLVt были приготовлены иодметилаты (ХЬУф
и X LVx). Не сообщается о попытке получить ангидрониевое основание из соеди-
нения X ЬУф, однако это соединение вводили в конденсацию с п-диметиламино-
бензальдегидом, 2-окситианафтен-З-альдегидом и нитрозосоединениями для
получения цианиновых красителей [124].
Хиндолин (3,4-бензо-6-карболин)
Хиндолин был первым из описанных в литературе производных бензо-
карболина. Его открыл Лоран [125], который, конечно, совершенно не знал
структуры соединения, с которым он имел дело. Изучая продукты распада
индиго, он обнаружил образование желтого кристаллического соединения,
названного им «флавиндином» и заметил, что этот продукт легко растворим
в аммиаке. При нагревании это соединение превращалось в бесцветное веще-
ство, которое возгонялось. По всей вероятности, этот «флавиндин» являлся
хиндолинкарбоновой-11 кислотой, а бесцветное соединение, полученное при
нагревании, представляло сам хиндолин (нумерацию атомов см. стр. 218).
Следующее упоминание о хиндолине встречается в работе Шютценбер-
гера [126], который также получил его из индиго. Он нагревал 1 ч. индиго-
тина, 2 ч. кристаллического барита, 1,5 ч. цинковой пыли и 10 ч. воды в закры-
том сосуде в течение 48 час при 180°. Сначала индиготин восстанавливался
в белое индиго, но после 48-часового нагревания голубая окраска при встря-
хивании на воздухе более не появлялась. После извлечения твердого остатка
спиртом и возгонки экстрагированного продукта в присутствии цинка
Шютценбергер получил соединение, которому он приписал формулу Ci8Hi4N2.
Он назвал его «индолин», так как считал его продуктом восстановления индиго,
содержавшим два индольных ядра. Позднейшие работы показали, что это
соединение на самом деле является хиндолином [127].
Модификацией проведенного Шютценбергером превращения индиго
в индолин служили опыты Жиро [128], который нагревал индиго со щелочным
гидросульфитом при 186—190° в течение 48 час. Из полученного продукта
он выделил кислоту, которой приписал формулу C32H24O5N4 и которую считал
идентичной «флавиндину», полученному Лораном. Эта кислота при пере-
гонке над цинковой пылью давала «индолин»; в отсутствие цинковой пыли
выход «индолина» был значительно меньше. Кислота Жиро действительно
была хиндолинкарбоновой-11 кислотой, вероятно гидратированной, и при
перегонке с цинковой пылью она декарбоксилировалась с образованием хин-
долина. Жиро ошибочно считал, что происходит восстановление, а не декар-
N
п СООН
234
карболины
боксилирование и, так же как Шютценбергер, приписывал получавшемуся
«индолину» формулу C16H14N2 вместо правильной C15Hi0N2.
Истинная природа «флавиндина» и «индолина» была выяснена работами
Нольтинга и Штейера [129] и Фихтера и Ронера [130]. Нольтинг и Штейер
сообщили, что в лабораториях «Badische Anilin und Soda-Fabrik» «флавин-
дин» был получен конденсацией индоксила с изатином в щелочном растворе
и, следовательно, он являлся хиндолинкарбоновой-11 кислотой. Образование
этой кислоты проходило, как показано на стр. 233.
Нольтинг и Штейер показали, что сам хиндолин можно получить конден-
сацией о-нитробензальдегида с индоксилкарбоновой-2 кислотой, протекаю-
щей с отщеплением двуокиси углерода и с последующим восстановлением
получающегося промежуточного соединения цинковой пылью в уксусной
кислоте, оловом в соляной кислоте или щелочным гидросульфитом; при этом
немедленно происходит циклизация. Они синтезировали хиндолин также
конденсацией индоксилкарбоновой-2 кислоты с о-аминобензальдегидом в при-
сутствии соляной кислоты.
Н Н
В этих условиях происходило и декарбоксилирование и циклизация.
Почти одновременно Фихтер и Ронер [130] пришли к таким же выводам.
Они показали, что хиндолинкарбоновая-11 кислота, полученная от «Badische
Anilin und Soda-Fabrik», вероятно синтезированная из изатина и индоксило-
вой кислоты, дает при восстановлении цинковой пылью в растворе едкого
натра и последующем окислении воздухом хиндолин. В качестве промежу-
точного продукта предположительно получается дигидрохиндолинкарбоновая
Н СООН н СООН
н
N N
БЕНЗОКАРБОЛИНЫ
235
кислота, легко отщепляющая двуокись углерода. Таким образом, подтверди-
лись и были объяснены наблюдения Жиро [128], что «флавиндин», или хиндо-
линкарбоновая кислота, может быть восстановлен в хиндолин.
За несколько лет до этого Фихтер и Берингер [131] получили хиндолин
совершенно иным путем: при действии горячего спиртового раствора едкого
натра на диэтиловый эфир ди-о-нитробензилмалоновой кислоты (I) происхо-
дила бурная реакция; раствор приобретал красную окраску, и после удале-
ния углекислого натрия и последующего концентрирования раствора выпадала
натриевая соль красного цвета. При подкислении кипящего спиртового
раствора этой соли уксусной кислотой выпадал мелкий кристаллический
порошок соединения состава ClsHi0O2N2, представлявшего собой, по всей
вероятности, оксихиндолин, возможно 10,11-диоксихиндолин (II).
При действии фенилгидразина это вещество легко теряло атом кислорода,
вероятно находившийся у атома Ni0, образуя соединение III; при более энер-
гичном восстановлении фенилгидразином или иодистоводородной кислотой
и фосфором вещество теряло оба атома кислорода и получался хиндолин.
Выше упоминалось о превращении индиго в хиндолин в опытах Шютцен-
бергера [126] и Жиро [128]. Гранмужен 1127] предложил три возможных
механизма этой реакции.
1. Разложение индиго на индоксилальдегид и антраниловую кислоту
с последующей конденсацией и замыканием кольца.
2. Расщепление индиго на индоксил и антраниловую кислоту с после-
дующей конденсацией и циклизацией в оксихиндолин и восстановлением
в хиндолин.
3. Превращение индиго в изатин и индоксил с последующей конденса-
цией и циклизацией в хиндолинкарбоновую-11 кислоту. Последнее объясне-
ние кажется наиболее вероятным (см. схему на стр. 236).
Гранмужен [132] нашел, что индиго может быть превращено в хиндолин
иным путем — через дианилиновый синий, который получается при взаимо-
действии 2 молей анилина и 1 моля индиго и имеет формулу C28H20N4 и вероят-
ную структуру IV. Это соединение при действии минеральной кислоты на его
раствор в уксусной кислоте дает желтое изомерное основание, которое при
энергичном гидролизе кислотой теряет 1 моль анилина и образует новое желтое
основание C22H16ON3 — по всей вероятности, антранилилхиндолин, так как
при сплавлении со щелочью это вещество дает антраниловую кислоту и хин-
долин. Аналогичный ряд соединений получен исходя из n-толуидина. В каче-
236
КАРБОЛИНЫ
стве конечного продукта получалось соединение, выпадающее из спирта
в виде бесцветных игл,- которому Гранмужен приписал строение 2-метил-
хиндолина (V). Строение промежуточных соединений и их превращения пока
не выяснены.
Наилучшим методом получения хиндолина, по-видимому, является непос-
редственное декарбоксилирование хиндолинкарбоновой-11 кислоты нагреванием
Н
Хиндолин
ее в парафине при 300° [133]. Такой способ декарбоксилирования более удо-
бен, чем косвенный метод Фихтера и Ронера [130], заключающийся в восста-
новлении цинковой пылью в растворе едкого натра с последующим окислением
воздухом, или модификация этого способа, предложенная Армитом и Робин-
зоном [134], которые проводили восстановление с помощью амальгамы натрия.
Хиндолинкарбоновые кислоты. Как уже сообщалось, хиндолинкарбоно-
вая-11 кислота («флавиндин») образуется при реакции индоксила с изатином
в щелочном растворе (Фихтер и Ронер [130], Нольтинг и Штейер [129]). Если
считать, что из изатина образуется изатиновая кислота, то конденсация про-
ходит по схеме, приведенной на стр. 233.
БЕНЗОКАРБОЛИНЫ
237
Предложены различные видоизменения этого метода. Армит и Робинзон
[134] применяли «технический плав индоксила», содержавший требуемое
эквивалентное количество индоксила, и, проводя реакцию в атмосфере све-
тильного газа, получали хиндолинкарбоновую-11 кислоту почти с количе-
ственным выходом. Хольт и Петров [133] применяли О-ацетилиндоксил или
О,М-диацетилиндоксил также в атмосфере светильного газа. При взаимодей-
ствии с тионилхлоридом образуется хлоргидрат хлорангидрида кислоты,
из которого легко получаются метиловый эфир и амид. Если амид подвергнуть
возгонке при 300° или кипятить с уксусным ангидридом или пятиокисью
фосфора в ксилоле, то образуется 11-цианхиндолин. Амид не удалось превра-
тить в амин по реакции Гофмана, однако последний можно получить из кис-
лоты реакцией Курииуса [133].
Бром-, нитро- и аминопроизводные хин долина. При бромировании хин-
долина Фихтер и Ронер [130] получили монобромпроизводное. Хольт и Пет-
ров [133] доказали, что при этом образуется 7-бромхиндолин (VI), осуществив
синтез его из 5-бромдиацетилиндоксила и изатина. Промежуточно образую-
щаяся 7-бромхиндолинкарбоновая-11 кислота (VII), полученная в виде аморф-
ного оранжевого порошка при бромировании хиндолинкарбоновой кислоты,
отщепляла двуокись углерода при нагревании в жидком парафине при 285°
и превращалась в 7-бромхиндолнн (см. схему на стр. 238).
При нитровании хиндолина концентрированной азотной кислотой, взятой
в избытке, получается смесь веществ . (Фихтер и Ронер [130]), которая, как
показали Хольт и Петров [133], может быть хроматографическим путем раз-
делена на два соединения. Главный продукт — 7-нитрохиндолин (VIII), в кото-
ром положение нитрогруппы доказано превращением этого соединения через
7-аминохиндолин (IX) в 7-бромхиндолин. Второе нитропроизводное, обра-
зующее великолепные алые шелковистые иглы, представляет собой 11-нитро-
хиндолин (X), так как при восстановлении оно дает 11-аминохиндолин (XI).
Строение его было доказано синтезом, исходя из хиндолинкарбоновой-11
кислоты путем превращения ее в гидразид (XII), из которого реакцией Кур-
циуса получали 11-аминохиндолин (XI).
При нитровании хиндолина в различных условиях главным продуктом
реакции всегда оказывается 7-нитропроизводное, и, если подобрать подходя-
щие условия реакции, то получаются лишь следы 11-нитросоединения. Один
из динитрохиндолинов был получен при кипячении хиндолина с концентри-
рованной азотной кислотой. Одна нитрогруппа в этом соединении находится
в положении 7, поскольку оно получается также и при нитровании 7-нитро-
хиндолина. Однако оно не образуется при нитровании 11-нитрохиидолина;
таким образом, положение второй нитрогруппы не известно. Нитрование хин-
долинкарбоновой-11 кислоты при 100° дает 7-нитрохиндолинкарбоновую-11
кислоту (XIII), при декарбоксилировании которой получают 7-нитрохин-
долин [133].
N-Алкилпроизводные. Фихтер и Берингер [131] установили, что иодме-
тилат хиндолина (XIV) при обработке едким натром в водном растворе дает
продукт темно-красного цвета, разлагающийся со вспениванием при 160°
и по составу отвечающий формуле CieHi4ON2 или C17Hi6 ON2; первая соот-
ветствует метогидроокиси, а вторая — тому же соединению, в котором гид-
роксил заменен метоксигруппой. Фихтер и Пробст [135] показали, что пра-
вильна первая формула. Это соединение при действии иодистоводородной
кислоты вновь превращается в иодметилат; оно реагирует даже с двуокисью
углерода с образованием соли хиндолиния. Армит и Робинзон [134] получили
сульфометилат хиндолина в виде оранжево-желтых игл, которые со щелочью
дали лиловато-розовый, похожий на мел осадок, являющийся, по-видимому,
гидратной формой метогидроокиси. Хольт и Петров [136] описывают основа-
хи
н
IX
« соон
VIII
XIII
V
N
__/ \/Ч
XI
бензокарболины
239
ние, полученное из хлорида 5-метилхиндолиния (хлорметилата хиндолина)
как псевдооснование темно-фиолетового цвета, легко растворимое в хлоро-
форме. Они согласны с Армитом и Робинзоном, что это соединение не теряет
СН3 СН3
XIV XV
молекулы воды и не образует ангидрониевого основания. Неоднократные
попытки удалить элементы воды из этого соединения оказались безуспеш-
ными. Как Фихтер и Берингер [131], они также считают, что соединение,
вероятно, имеет структуру XV, причем гидроксильная группа присоединена
к общему атому углерода, а не к атому Си. Главным доказательством этого
служит сделанное Фихтером и Берингером наблюдение, что при окислении
это соединение не дает 5-метил-11-кето-5,11-дигидрохиндолина.
Более поздние работы показали, что строение криптолепина соответст-
вует формуле XXVII; вопрос о гидратации будет рассмотрен ниже при описа-
нии этого алкалоида (стр. 240).
При действии на хлорид 7-нитро-5-метилхиндолиния (XVI) кипящего
раствора едкого натра в метаноле получается 7-нитро-5-метилизохиндолин
(XVII) в виде темно-красных игл. Очевидно, наличие нитрогруппы способ-
ствует выделению воды из промежуточно образующейся метогидроокиси.
С другой стороны, хлорид 7-амино-5-метилхиндолиния — желтый микро-
кристаллический порошок, полученный при восстановлении нитросоедине-
ния, не удалось перевести в ангидрониевое основание; при обработке щелочью
этот хлорид дал соединение, выделявшееся из хлороформа в виде игл цвета
синего индиго. Хольт и Петров [136] считают, что это псевдооснование (XVIII).
СН3 CI- СН3
XVI XVII
СН3
H0N
H2N -
н
XVIII
При действии на 11-аминохиндолин йодистого метила в кипящем мета-
ноле получен иодид 11-амино-5-метилхиндолиния (XIX) в виде желтых микро-
скопических игл, разлагающихся при нагревании с выделением иода. В пользу
строения, приписываемого этому соединению, свидетельствует тот факт, что
триацетильное производное 11-аминохиндолина (10-ацетил-11-диацетиламино-
хиндолин) при действии диметилсульфата дает соответствующую соль 11-амино-
5-метилхиндолиния (XXI). При гидролизе разбавленной соляной кислотой
240
КАРБОЛИНЫ
соль (XXI) превращалась в хлорид (XXII). Как хлорид, так и иодид, полу-
ченные столь различными путями, дают одно и то же алкоксипсевдооснование
СН3 СН3 СН3
XIX XX XXII
ххш
сн3
N+ CH3SO7
/ \,/\
I 11 I I
N I
I N(COCH3)2
СОСНз
XXI
и ангидрониевое основание. Так, при обработке иодметилата кипящим раст-
вором едкого натра в метаноле получен 11-амино-11-метокси-5-метил-5,11-
дигидрохиндолин (XX, R = СН3), а в этаноле — соответствующее этоксипро-
изводное. Оба эти соединения при нагревании в вакууме до 110° превращаются
в 11-имино-5-метил-5,11-дигидрохиндолин (XXIII). Такой же ряд соединений
был получен из хлорида 11-амино-5-метилхиндолиния [136].
Хиндолинкарбоновая-11 кислота слишком плохо растворима, чтобы ее
можно было непосредственно перевести в четвертичную соль, которая легко
получается из метилового эфира сульфометилата 5-метил-11-карбометоксихин-
долиния (XXIV) и при его гидролизе дает хлорид 11-карбокси-5-метилхиндо-
линия (XXV); из последнего легко можно получить цвиттер-ион (XXVI) [136]..
XXIV
СН3
N+
XXV
XXVI
Криптолепин. Единственным известным в настоящее время природным
хиндолином является весьма интересный алкалоид криптолепин, который
был выделен из корней Cryptolepis triangularis N. Е. Вг. и из Cryptolepis
sanguinolent а. Доказано, что он имеет структуру, изображенную форму-
бензокарболины
241
лой XXVII [137]. Криптолепин образует темно-фиолетовые кристаллы.
В растворе их цвет зависит от полярности растворителя и меняется от желтого
до красного. Соли его имеют желтую окраску. Как и ангидрониевое соедине-
ние, он сольватируется при перекристаллизации из гидроксилсодержащнх
растворителей.
При каталитическом гидрировании над окисью платины в метаноле или
при восстановлении гидросульфитом натрия криптолепин дает желтое дигид-
росоединение, которое легко окисляется кислородом обратно в криптолепин.
При дегидрировании селеном теряется одна метильная группа и образуется
хиндолин (XXVIII). Каталитическое восстановление хлоргидрата (XXVIII)
или
186
16 Заказ .№ 978
242
КАРБОЛИНЫ
над окисью платины дает тетрагидропроизводное (XXIX), которое при дей-
ствии йодистого метила образует тетрагидрокриптолепин (XXX), выделен-
ный в виде нитрата. Таким образом, во время дегидрирования селеном не про-
исходит глубокой деструкции углеродного скелета молекулы. При восстано-
влении криптолепина над окисью платины в уксусной кислоте получается
тетрагидропроизводное (XXXI). При действии кислот на соединение XXXI
образуются соли (XXX), идентичные получаемым действием йодистого метила
на соединение (XXIX). Каталитическое восстановление криптолепина в соля-
ной кислоте ведет к образованию октагидропроизводного (XXXII). Эти пре-
вращения наряду с другими приведены ниже.
Хотя формула XXVII отражает, по существу, структуру криптолепина,
по-видимому, невозможно получить ангидрониевое основание, свободное
от растворителя, применявшегося для кристаллизации. Армит и Робинзон
1134] показали, что имевшиеся у них различные образцы метогидроокиси
хиндолина содержали от 1,6 до 2,4 молекул воды. Последний образец соот-
ветствует свободному от спирта основанию криптолепина Ci6Hi2N2-2,5H2O,
описанному Желлером и сотрудниками {137]. При нагревании основания
криптолепина C16H12N2-2,5H2O до 120° при 0,01 мм рт. ст. в течение 5 час
был получен полугидрат Ci6Hi2N2-0,5H2O; таким образом могут быть удалены
2 молекулы воды, хотя они и удерживаются довольно прочно.
Сравнение ультрафиолетовых спектров поглощения спиртовых растворов
показало близкое сходство спектров криптолепина, его солей, иодметилатов
криптолепина и хиндолина и метогидроокиси хиндолина (метилхиндоланола).
Спектры криптолепина и метогидроокиси хиндолина, растворенных в 0,1 н.
спиртовом растворе едкого кали, были также сходны и по сравнению со спект-
рами растворов в чистом спирте обнаруживали поглощение в области более
длинных волн. Таким образом, в спирте ангидрониевое основание В отнимает
протон от растворителя, образуя ВН+; в присутствии едкого кали протон
отщепляется и вновь образуется основание В. Сильные основные свойства
и характер ультрафиолетовых спектров позволяют предполагать, что в спир-
товом растворе едкого кали ангидрониевое основание (XXVII), несомненно,
сольватировано. Выделение полугидрата находится в согласии со структурой
XXVII; два протона молекулы воды группируются у отрицательно заряжен-
ного атома азота молекулы криптолепина.
Другие производные хиндолина. При конденсации 6-аминопипероналя
с индоксилом в атмосфере светильного газа Гулланд и сотрудники [96] полу-
чили продукт, который предположительно является б'-аминопиперонилиден-
индоксилом (XXXIII). При кипячении с ледяной уксусной кислотой соеди-
нение XXXIII циклизовалось в 2,3-метилидендиоксихиндолин (XXXIV),
который в этиловом спирте дал фиолетовую, а в серной кислоте — голубую
XXXIV
XXXV
ЙНДОЛО- [2,3-а]-ХИНОЛИЗИНЫ
243
флуоресценцию. Это соединение легко окислялось раствором хромовой кис-
лоты в уксусной, однако в результате удалось выделить лишь небольшое
количество кислоты, вероятно, отвечающей формуле XXXV. 2,3-Метилеи-
диоксихиндолин при нитровании дал мононитропроизводное, по-видимому,
7-нитро-2,3-метилендиоксихиндолин. Положение нитрогруппы было уста-
новлено на основании устойчивости диоксиметиленового цикла по отношению
к горячему спиртовому раствору этилата натрия. Это свидетельствует о том,
что нитрогруппа вступает в незамещенное бензольное кольцо, как это имеет
место в случае карбазола и самого хиндолина (стр. 237).
3,4-Диоксихиндолин (XXXVI) получен из метилендиоксипроизводпого
кипячением последнего с иодистоводородной кислотой в резорцине или, лучше,
нагреванием в запаянной трубке при 180—190° с раствором едкого кали в мети-
ловом спирте. Свободное основание ввиду его нестойкости может быть полу-
N N
Ai—\ Wон П—f
Uу JU- он . ад 'U-соон
н н
XXXVI XXXVII
чено из хлоргидрата с большим трудом. При окислении соединения XXXVI
азотной кислотой получается смесь дикарбоновой и монокарбоновой кислот;
первая была превращена во вторую нагреванием с уксусной кислотой.
Анализ и свойства этой кислоты указывают на то, что она представляет собой
7-нитро-6-карболинкарбоновую-3 кислоту (XXXVII), полученную с очень
низким выходом при окислении 7-нитро-2,3-метилендиоксихиндолина 47 %-ной
азотной кислотой.
Как уже упоминалось на стр. 235, Фихтер и Берингер [131] получили
из диэтилового эфира бис-(о-нитробензил)малоновой кислоты соединение,
представлявшее собою, вероятно, 10,11-диоксихиндолин, а из него действием
фенилгидразина — монооксисоединение, по-видимому 11-оксихиндолин. Они
также синтезировали ряд производных этого соединения, включая простые
метиловые эфиры и ацетаты, а также нитропроизводное монооксихиндолина
и соответствующее аминосоединение. Они полагали, что нитрогруппа нахо-
дится в положении 7, что подтверждено позднейшими работами по нитрованию
хиндолина.
С азотистой кислотой хиндолин дает 10-нитрозохиндолин, из которого
хиндолин может быть получен кипячением со спиртовым раствором соляной
кислоты [130]. При восстановлении хиндолина оловом в соляной кислоте
образуется двухосновное дигидропроизводное, которое легко окисляется
в хиндолин. В отличие от диацетильного производного этого соединения моно-
ацетильное—обладает основным характером. Восстановление иодида метил-
хиндолиния ведет к образованию 5-метилдиоксихиндолина, который полу-
чается при метилировании дигидрохинолина [130] (ср. стр. 195).
ИНДОЛО-[2,3-а]-ХИНОЛИЗИНЫ
Некоторые алкалоиды содержат 0-карболиновое ядро как часть тетра-
циклической индоло-[2,3-а]-хинолизиновой структуры. Коринантеин (I), дигид-
рокоринантеин (II) и коринантеидин (III) были выделены из коры Pseudo-
cinchona africana A. Chev. [138, 139]. Для коринантеина Жано, Гутарель
и Прелог предложили структуру I [140]; строение II и III было установлено
Жано, Гутарелем и Шабасс-Массоно [139]. Стереохимия этих алкалоидов
была изучена благодаря двум исследованиям [141, 142].
16*
244
КАРБОЛИНЫ
В 1952 г. появилось сообщение о первых попытках синтеза циклической
системы индоло-[2,3-а]-хинолизина. Гров и Свен [143], исходя из триптамина
и последовательно пристраивая остальные кольца, пришли к бромиду 1,2,3,4-
тетрагидро-12Н-индоло-[2,3-а]-хинолизиния (IV), который при восстановле-
нии дал соединение (V). Они описали второй путь получения производного
соединения V (VII, R = Н). При восстановлении карбонильной группы в этом
соединении было получено соединение V с очень низким выходом. Впослед-
ствии было показано, что продукт циклизации соединения VI по методу Дик-
мана представляет собой смесь кетоэфиров, содержащую лишь около 15%
сложного эфира (VII, R = СООС2Н5) [144].
И НДОЛО-[2,3-о]-ХИНОЛИЗИН Ы
245
CH2=CHCOOC2|I6
NH,
4-
С0С1
I
XCOOC2II5
QU (2) Гидролиз
I 2
CH2 — СООС2МЯ
COOCoIIr,
VI
P2O6
(1) Циклизация
по Дикману
VII
для
Рекхоф и Тарбелл 1145] применили индольный синтез по методу Фишера
получения соединения V.
i' Xi
(J) ) 1икл и зация
ио Дикману
(2) Гидролиз
вг(сн=)зсоос2115 I
J-cooc2h5 ----------„ (О.(----- ч J сопель,
N ’ ' N СООСИЦ
П ! ,
\ ,•
О
(l)C(ill5NI|Nfl2
N 1 (2) на
С1-
XI X
246
КАРБОЛИНЫ
Синтез тетрациклического аналога (X) семпервирина (стр. 259) осуще-
ствлен, как показано на схеме, исходя из соединения VIII [1461. Если в соеди-
нении IX R = Н, то замыкание кольца, по-видимому, происходило по индоль-
ному атому азота с образованием соединения XI, если же атом азота в индоле
был защищен, то получался карболин (X).
Сугасава с сотрудниками синтезировали соединение XII, которое при
восстановлении дало соединение V 11471.
То же соединение было получено в результате замыкания индольного
кольца по методу Фишера в четвертичном кетоне (XIII) [148, 149]. Эти син-
тезы отличаются только тем, что соединение XIII получалось различными
путями.
(I)CH2O, (C2H5)2NH
LiAllIi
(2) (СНз)2ЬО,1, KI
(3) KCN
(4) н2о/он-
ИНДОЛ О- [3,2-6]-ХИНОЛ ИЗИ н
247
Весьма интересным синтезом соединения V, дающим хорошие выходы,
является синтез Тезинга и сотрудников f 1501, которые исходили из грамина.
Наконец, следует упомянуть о стереоспецифическом синтезе [142]
DL-дигидрокоринантеана (XIV) из DL-транс-диэтилциклопентанона.
ИНДОЛО-13,2-&J-XHH0JI изин
Это соединение является линейным аналогом рассмотренного выше
индоло-[2,3-а!-хинолизина. Нумерация атомов соответствует «The Ring Index»
и показана в формуле XlVa; однако это соединение было описано как индоло-
13', 2'-2,31-пиридоколин и пронумеровано, как в формуле XIV6. Индоло-
13,2-6 ]-хинолизинон-6-(5Н) (XIVb) и 1,4-дихлорпроизводное (XlVr) были
синтезированы Клемо и Ситоном [1511 из этилового эфира 2-[2-(2-пиридил)этил]-
ацетоуксусной кислоты, который образует фенилгидразоны (Х1Уд и XlVe)
с соответствующими диазониевыми солями по реакции Джаппа — Клип-
гемана. Соединение Х1Уд, в котором фенильное кольцо не содержит заме-
стителей, претерпевает индольную циклизацию по методу Фишера при кипя-
чении со спиртовым раствором соляной кислоты и образует соединение Х1Уж,
при гидролизе которого образуется кислота (Х1Уз). При нагревании послед-
ней при 200° и 0,1 мм рт. ст. получают сублимат соединения XIVb. Гидразон
•(XlVe) циклизуется с трудом, но при нагревании до 150° с хлоридом цинка
в этаноле претерпевает двойную циклизацию и дает соединение XIVr.
Гексагидрокетон был получен Кейфером [152] следующим образом:
этиловый эфир 2,3,4,5-тетрагидро-Р-карболинкарбоновой-4 кислоты, реагируя
с 4-бромбутиронитрилом, дал этиловый эфир 3-(3'-цианпропил)-2,3,4,5-тетра-
гидро-Р-карболинкарбоновой-4 кислоты; циангруппа превращалась в карб-
этоксильную и полученный диэфир подвергался циклизации по методу Дик-
мана 'с последующим гидролизом и декарбоксилированием с образованием
5,6,9,10,11а,12-гексагидроиндоло-[3,2-6]-хинолизинона-11-(8Н) (Х1Уи).
248
КАРБОЛИНЫ
XlVfl. Х = Н
XlVe, X = CI
XlVn
ПЕНТАЦИКЛИЧЕСКИЕ р-КАРБОЛИНОВЫЕ АЛКАЛОИДЫ
Существует довольно большая группа;алкалоидов, содержащих fj-карбо-
линовое ядро, являющееся частью пснтациклической системы. С 1950 г. иссле-
дования этих веществ приобретают все большее значение, что стимулирова-
лось появлением резерпина, очень ценного лекарственного препарата, обла-
дающего транквилизирующим и гипотенсивным действием. В числе fl-карбо-
линовых алкалоидов следует упомянуть эводиамин и рутекарпин, иохимбин
и близкие ему основания, алкалоиды Rauwolfia, алкалоиды Alstonia и многие
другие. Поскольку химии этих природных веществ посвящен ряд превосход-
ных работ [153—1611, здесь нет необходимости подробно останавливаться
на описании этих алкалоидов. Больше внимания будет обращено на химию
карболинового ядра и будут приведены доказательства наличия его в алка-
лоидах, а также описаны некоторые методы синтеза.
Эводиамин и рутекарпин
Эводиамин CieH17ON3 и рутекарпин C18H13ON3 — два алкалоида, содер-
жащиеся в сухом плоде Evodia rutaecarpa. Эводиамин может быть превращен
в рутекарпин; при нагревании с водно-спиртовым раствором соляной кислоты
он последовательно образует изоэводиамин и ангидроизоэводиамин (III);
последний при нагревании без растворителя отщепляет хлористый метил
и дегидрируется с образованием рутекарпина [162, 163]. Эводиамин при
кипячении со спиртовым раствором едкого кали дает N-метилантраниловую
кислоту и дигидроноргарман (3,4-дигидро-р-карболин). В аналогичных усло-
виях изоэводиамин C19H19O2N3 образует N-метилантраниловую кислоту
и триптамин [164]. Рутекарпин, обработанный раствором едкого кали в кипя-
щем амиловом спирте, превращается в антраниловую и 3-(Р-аминоэтил)индол-
карбоновую-2 кислоту (V) [165]. На основании этих реакций эводиамину
ПЕНТАЦИКЛИЧЕСКИЕ Р-КАРБОЛИНОВЫЕ АЛКАЛОИДЫ
249
и рутекарпину было приписано строение I и IV, а изоэводиамину — II; как
будет показано ниже, образование его требует раскрытия пиридинового кольца
Эводиамин
I КОН.
С2Н5ОН
Н
NIICH3
I
Ангндропзоэводвамил
I Сухое
| нагревание
nh2 \
I IV
—СООН Рутекарпин
в карболиновом ядре. Частичный синтез рутекарпина был впервые осущест-
влен Асахина с сотрудниками 1166, 167] исходя из 3-(0-аминоэтил)индолкар-
VI
VII
250
КАРБОЛИНЫ
боновой-2 кислоты (V), полученной, как уже указывалось, при разложении
этого алкалоида. о-Нитробензоильное производное (VI) этой аминокислоты
восстанавливали до соответствующего антранилпроизводного VII, которое
СООСНд
I
“!NYii
циклизовалось под действием хлорокиси фосфора в четыреххлористом углероде
с образованием рутекарпина (IV).
ПЕНТАЦИКЛИЧЕСКИЕ 0-КАРБОЛИНОВЫЕ АЛКАЛОИДЫ
251
Более прямой путь синтеза рутекарпина предложен Асахина, Манске
и Робинзоном [168], которые конденсировали метиловый эфир антраниловой
кислоты с 2-кето-2,3,4,5-тетрагидро-[5-карболином при кипячении в течение
4 час с треххлористым фосфором. Несколько похожий синтез был осуществлен
Ота [169], который нагревал изатойный ангидрид (VIII) с 2-кето-2,3,4,5-
тетрагидро-Р-карболином при 195° и 20 мм рт. ст. и получил рутекарпин
с хорошим выходом.
Эводиамин получен Асахина и Ота [170] из N-метилизатойного ангидрида
(IX) (приготовленного из этилхлорформиата и N-метилантраниловой кис-
лоты) и триптамина. При взаимодействии этих двух соединений образуется
3-(М-метилантраниламидоэтил)индол (X), при обработке которого ортому-
равьиным эфиром при 175—180° происходит двойная циклизация с образова-
нием DL-эводиамина.
Йохимбин и родственные алкалоиды
Йохимбин встречается в коре Johimbe вместе с рядом близких ему алка-
лоидов. Различные виды Rauwolfia являются также превосходным сырьем
для получения алкалоидов, родственных иохимбину. Из Rauwolfia serpentina
было выделено по крайней мере 20 алкалоидов, большинство которых содер-
жало {5-карболиновое ядро как часть пентациклической системы. Из других
видов Rauwolfia получено множество разнообразных алкалоидов этой группы
[159]. Ниже будут приведены типичные примеры расщепления и синтеза,
однако рассмотрение всех членов этой группы алкалоидов выходит за пределы
настоящей работы.
Родоначальный циклический скелет (XI) можно рассматривать как резуль-
тат соединения {5-карболинового и изохинолинового колец, поэтому ядро
можно назвать 2,3,2',3'-изохинолино-[5-карболином. В справочнике «The
Ring Index» [171] принято систематическое наименование для родоначального
соединения — бенз[^г]индоло-[2,3-а]-хинолизин. В процессе выяснения струк-
туры иохимбина была разработана система нумерации атомов, приведенная
в формуле XI. Нумерация, принятая в справочнике «The Ring Index», показана
в формуле XII. В настоящее время применяются обе системы. В данном обзоре
будет в основном использована система нумерации атомов, приведенная
в формуле XI. Ее преимущество заключается в соответствии с нумерацией, уже
применявшейся для [5-карболина. В некоторых случаях, когда для ясности необ-
ходимо пользоваться нумерацией по формуле XII, это будет отмечаться особо.
Благодаря наличию трех асимметрических атомов углерода в скелете
молекула XI обладает стереоизомерией; примеры всех возможных конфигу-
раций известны. Однако подробно рассматриваться эти изомеры не будут.
Возможны и известны также соединения с ненасыщенными кольцами С, D и Е,
а у некоторых алкалоидов отмечено включение дополнительного гетероатома
(кислорода) в кольцо Е.
Йохимбин. При дегидрировании иохимбина (XIII) с помощью селена
получаются три продукта [172, 173]. Одним из них является «тетрагидроиоби-
рин» — 2-[3-(5,6,7,8-тетрагидроизохинолил)]-3-этилиндол (XIV), и его обра-
252
КАРБОЛИНЫ
зование, очевидно, требует разрыва пиридинового кольца в карболиновом
ядре. Два других продукта — иобирин (XV) и кетоиобирин (XVI). Строение
кетоиобирина не было окончательно установлено до 1948 г. [174, 176]. Кетоио-
бирин не обладает основными свойствами и при энергичном щелочном гидро-
лизе образует норгарман (XVII) и 2,3-диметилбензойную кислоту (XVIII).
При нагревании с палладиевой чернью кетоиобирин дегидрируется в дегид-
XVIII
Г1ЕНТАЦИКЛИЧЕСКИЕ Р-КАРБОЛИНОВЫЕ АЛКАЛОИДЫ
253
XXI
рокетоиобирин (XIX), дающий при гидролизе аминокислоту XX, которая
очень легко превращается в соединение XIX. Образование кетоиобирина тре-
бует, таким образом, раскрытия кольца D и поворота кольца Е на 180° с после-
дующей циклизацией для образования пентациклической системы.
Шлиттлер и Шпейтель [175] синтезировали кетоиобирин из триптамина
и 6-метилгомофталевого ангидрида методом, аналогичным тому, который был
применен [176] для получения норкетоиобирина из триптамина и гомофтале-
вого ангидрида.
Иобирин (XV) был синтезирован Клемо и Свеном [177], а также Джулиа-
ном с сотрудниками [178], по существу, тем же самым методом. 3-[[5-(о-Толил-
ацетамидо)этил ]индол циклизовался в 2-(о-ксилил)-4,5-дигидро-[5-карболин
(XXI), который дегидрированием над палладиевой чернью превращали
в иобирин. При окислении иобирина хромовой кислотой [179] или двуокисью
селена получался иобирон (XXII).
Интересно, что правильная структура скелета молекулы иохимбина
была впервые предложена Бартером и Шольцем [180] еще в 1933 г., несмотря
на то что строение некоторых продуктов его расщепления не было в то время
точно известно.
264
карболиНь!
Дальнейшие исследования этих алкалоидов касались главным образом
установления положения карбометоксильной (у С)6) и гидроксильной (у CJ7)
групп.
Кроме синтезов соединений, фактически полученных из самого иохим-
бина, ряд исследователей разрабатывал методы синтеза скелета ядра. Клемо
и Свен [177] из фенилгидразона 1-кето-7,8-бензо-1,2,3,4,6,9-гексагидропири-
доколина (XXIII) по реакции Фишера получили 7,8-бензо-2,3-2',3'-индоло-
3,4,6,9-тетрагидропиридоколин (гексагидробенз[^]индоло- [2,3-а]-хинолизин)
(XXIV), который при дегидрировании был превращен в иобирин.
(1) c6h5nhnh2
(2)НС1
ХХП1
IV
Клемо и Свен [177] пытались синтезировать иохимбиновое ядро другим
методом исходя из 3-([5-гомофталимидоэтил)индола. Они надеялись, что он
будет циклизоваться с образованием соединения XXV; но при действии хлор-
окиси фосфора желательного соединения не получилось. Однако при кипячении
с хлорокисью фосфора метилового эфира кислоты, полученного конденсацией
3-ф-аминоэтил)индола с гомофталевым ангидридом, с небольшим выходом
XXV
получался продукт, которому было приписано строение XXV. Эти данные были
подтверждены Шлиттлером и Аллеманом [176], синтезировавшими соедине-
ние XXV с выходом 36%. Эти результаты интересны тем, что кетоиобирин,
как это позднее было показано, является метилпроизводным соединения XXV
и может быть синтезирован аналогичным путем (см. стр. 252).
Шлиттлер и Аллеман провели подобные синтезы, заменив гомофталевую
кислоту цис- и транс-гексагидрогомофталевыми кислотами, в результате чего
получили цис- и транс-формы соединения XXVI [176].
Аналогичные синтезы описаны выше.
Ван-Тамелен с сотрудниками осуществил синтез иохимбанового скелета,
доказывающий наличие транс-кольцевой связи между циклами D и Е в иохим-
бине [1811.06 аналогичном стереоспецифическом синтезе аллоиохимбана (коль-
ца D и Е в цас-поло кении) одновремен ю сообщили Сторк и Хилл [ 182]. Ван-
Тймелен проводил синтез через уктзан ые на схеме стадии, исходя из транс-
Р-гидринданона (XXVII). Синтез Сторка и Хилла отличался лишь тем, что
в качестве исходного о i взяли цнс-р-гидриндапон.
XXXII
XXXI
256.
карболины
Изящный синтез иохимбинового скелета был выполнен Ханом и сотруд-
никами [183], применившими общий метод получения тетрагидро-р-карболи-
новых соединений из триптамина, и производных пировиноградной кислоты,
рассмотренный на стр. 203. Конденсацией триптамина с л«-оксифенилпировино-
градной кислотой при стоянии в течение 10 дней при pH 4,2 и температуре 25°
была получена с 87%-ным выходом 2-(л«-оксибензил)-2,3,4,5-тетра<идро-р-
карболинкарбоновая-2 кислота, которая при действии соляной кислоты в мета-
нольном растворе отщепляла двуокись углерода и превращалась в 2-(л«-окси-
бензил)-2,3,4,5-тетрагидро-[5-карболин (XXIX). Из последнего при взаимодей-
ствии с формальдегидом при pH 4,4 быстро образовывалось оксиметильное
производное. Оксиметильная группа находится, вероятно, в положении 6 фе-
нильного кольца соединения XXX, а не в орто-положении к уже имеющейся
гидроксильной группе, о чем можно судить по тому, что оксибензильный пара-
изомер (XXXI) не реагирует в аналогичных условиях с формальдегидом.
При растворении хлор гидрата соединения XXX в теплой воде и обработке
его карбонатом натрия до щелочной реакции происходит циклизация; выпав-
шее кристаллическое основание имеет строение XXXII.
Полный стереоспецифический синтез самого иохимбина осуществлен
Ван-Тамеленом с сотрудниками [184].
Исключительно интересная реакция циклизации с образованием пента-
циклического p-карболинового производного описана Белло [1851, а также
Поттсом и Робинзоном [186]. Продукт конденсации 3-ф-хлорэтил)индола с изо-
хинолином (XXXIII) при восстановлении алюмогидридом лития дал пента-
циклический p-карболин (XXXIV). Механизм этой реакции до сих пор не выяс-
нен. С соответствующими солями пиридиния (XXXV) циклизация не про-
XXXIII
ходит [187], и продукт реакции, вероятно, имеет строение XXXVI. С помощью
боргидрида натрия не удается провести циклизации даже солей изохинолиния
[188]. Еще один пример этого синтеза приведен на стр. 260.
Li А! Н* или
NaBHi
XXXVI
XXXV
ПЕНТАЦИКЛИЧЕСКИЕ р-КАРБОЛИНОВЫЕ АЛКАЛОИДЫ
257
Резерпин, дезерпидин и ресциннамин. Эти три алкалоида Rauwolfia состав-
ляют группу близких между собой структур XXXVII, XXXVIII и XXXIX.
Строение и стереохимия этих веществ строго обоснованы, и поскольку химия
их достаточно подробно и критически рассмотрена [159], здесь эти вопросы
ОСН3
I
XXXVII, R1 = ОСНз, R2=-ОС-Z >-осн3
Резерпин ----1
ОСН3
ОСН3
XXXVIII, R1 = H, R2=—ОС—>—ОСНз
Дезерпидин -----1
СН3ООС —OR2 ОСН3
I ОСН3
ОСН3 |
XXXIX, Р1 = ОСН3, R2=-OCCH=CH— ^-ОСН3
Ресциннамин 4---1
ОСНз
обсуждаться не будут. Вудворд с сотрудниками осуществил полный синтез
резерпина [189], который приведен на схеме, представленной ниже. Особого
внимания заслуживает стереоспецифичность примененных реакций.
Восстановление аддукта Дильса — Альдера (XL), Полученного из «-бензо-
хинона и винилакриловой кислоты по методу Меервейна — Понндорфа, дает
N-Бромсук-
цинимид *
IO]
XL XLI XLII
17 Заказ № 978 186
258
КАРВОЛИНЫ
оксилактон (XLI), который переводят в соединение XLII путем последователь-
ной обработки бромом в метаноле и метилатом натрия. Последнее обладает
стереохимией кольца Е резерпина. При действии N-бромсу'кцин имида на сое-
динение XLII и последующем окислении образующегося бромгидрина (XLIII)
получают бромкетон (XLIV). Действием цинка в уксусной кислоте из него
XLIX
получен ненасыщенный кетон (XLV). Свободная карбоксильная группа в этом
кетоне была защищена метилированием, а гидроксильную группу ацетилиро-
вали с образованием соединения XLVI, из которого при гидроксилировании
был получен гликоль XLVII. Последний путем расщепления и последующего
метилирования превращен в альдегидоэфир (XLVIII). p-Карболиновая систе-
ма построена путем конденсации соединения XLVIII с 6-метокситриптамином
с последующим восстановлением имцна (XLIX) в амин (L). Циклизацией
лактама (LI) под действием хлорокиси фосфора получена соль (LII), при вос-
становлении боргидридом натрия превращавшаяся в метиловый эфир DL-O-
ацетилизорезерпиновой кислоты (LIII), из которого был выделен изомер
ПЕНТАЦИКЛИЧЕСКИЕ g-КАРВОЛИЙОВЫЕ АЛКАЛОИДЫ
259
в виде ди-л-толуил-Ь-тартрата. Дезацетилирование и этерификация триметок-
сибензоилхлоридом дали изорезерпин.
Превращение изорезерпина (LIV) в резерпин (LV) может быть осуществле-
но Тремя путями, из которых простейший показан на приведенной ниже схе-
ме [190]. Этот путь весьма поучителен, поскольку речь идет о стереохимии
атома углерода С3. Другие превращения изорезерпина в резерпин описаны
Вудвордом и сотрудниками [1891, а также Хубнером и сотрудниками [191[.'»
Четвертичные пентациклические p-карболиновые алкалоиды
Начиная приблизительно с 1945 г. обнаружен ряд пентациклических
Р-Карболиновых алкалоидов с четвертичным атомом азота в кольце С, предста-
вителями которых являются семпервирин, альстонин, серпентин и альстони-
лин. Химия этих интересных веществ будет представлена в настоящем разде-
ле лишь в самых общих чертах.
Из корней Gelsemium sempervirens извлечен ряд алкалоидов и среди них
семпервирин C19H16N2. При дегидрировании селеном он превращается в иоби-
1'7*
260
Карболины
рин (XV), при этом образуется небольшое количество иобирона (XXII), вероят-
но, в результате окисления кислородом воздуха [192]. При кипячении в ксило-
ле с никелем Ренея семпервирин дает «тетрагидроиобирин» (XIV). На основа-
нии этих наблюдений для семпервирина предложена структура LVI [193];
при этом скелет ядра казался достоверным и вызывало сомнение лишь положе-
ние дополнительной двойной связи. Было синтезировано [194, 195] вещество
со структурой LVI, которое оказалось неидентичным семпервирину.
Замечено, что инфракрасный спектр семпервирина не обнаруживает
поглощения при 2,9 р, которое является характерным для всех индолов с неза-
мещенным атомом азота. Кроме того, семпервирин окрашен в желтый цвет.
Учитывая эти факты, для семпервирина правильной структурой стали считать
резонансную структуру LVII [194, 196]. Таким образом, семпервирин представ-
ляет собою ангидрониевое основание (стр. 211). Структура LVII была подтвер-
ждена синтезом 1-метилпроизводного семпервирина, состоящим в конденсации
литиевого производного 1-метилгармана с изопропоксиметиленциклогексано-
ном и последующей обработке соляной кислотой, в результате чего был
получен хлорметилат семпервирина (LVIII] [197]. Это был первый синтез
четвертичного пентациклического ^-карболина из более простых молекул.
1 СН3 CH2Li
СНОСН(СН3)2
Второй синтез семпервирина, осуществленный путем индольной циклиза-
ции по методу Фишера, проведен Свеном по следующей схеме [ 198]:
Наряду с альстонином (LIX) из коры дерева Alstonia constricta был выде-
лен альстонилин (LX). Из различного растительного сырья получен серпен-
тин — стереоизомер альстонина. Однако с точки зрения химии четвертичных
[5-карболиновых алкалоидов в первую очередь будет рассмотрен альстонилин.
Строение альстонилина установлено на основе обычных методов расщепле-
ния [200, 201] и подтверждено синтезом одного из его производных [202],
для чего был использован весьма замечательный способ замыкания кольца
с образованием пентациклической структуры, впервые предложенный Поттсом
и Робинзоном [186].
ПЕНТАЦИКЛИЧЕСКИЕ Р-КАРБОЛИНОВЫЕ АЛКАЛОИДЫ
261
Альстонилин (LX) восстанавливали в растворе основания в тетрагидро-
альстонилин (LXI), сложноэфирная группа которого в свою очередь может
быть восстановлена алюмогидридом лития с образованием тетрагидроальсто-
нилинола (LXII). Последний при дегидрировании дал альстонилинол (LXIII).-
Альстонилинол (LXIII) был синтезирован следующим образом: конден-
сация 3-(Р-бромэтил)-6-метоксииндола с 5-карбометоксиизохинолином давала
четвертичную соль (LXIV). При восстановлении последней алюмогидридом
лития кольцо С замыкалось с образованием тетрагидроальстонилинола (LXII),
при дегидрировании которого получили альстонилинол (LXIII). При замене
алюмогиДрида лития боргидридом натрия, который должен был бы оставлять
эфирную группу в соединении LXIV незатронутой и, следовательно, привести
к образованию самого альстонилина, замыкания кольца С не происходило.
Восстанавливались лишь две двойные связи в пиридиновом кольце изохино
линовой системы альстонила.
-> LXII
12-СН8СООК
----------> LXIII
80
262
КАРБОЛИНЫ
Сообщается о других методах синтеза пентациклического ядра альстони-
Лина. При фишеровской индольной конденсации четвертичного кетона (LXV)
с различными арилгидразинами получены аналоги (LXVI) альстонилина,
не содержащие карбометоксигруппы 1203]. Получить этим способом соедине-
ния, содержащие карбометоксигруппу, не удалось.
ЛИТЕ РАТ УРА
1. G u 1 1 а п d et al., J. Chem. Soc., 1929, 2926.
2. Patterson, Capel 1, Walker, The Ring Index, 2nd ed., American Chemical
Society, 1960. \ ,
3. Perkin, Robinson, J. Chem. Soc., 115, 970 (1919). \
4. Lawson, Perkin, Robinsun, J. Chem. Soc., 125, 626 (1924).
5. Пат. США 2688022 [С. A., 50, 1925 (1956)].
6. Freak, Robinson, J. Chem. Soc., 1938, 2013.
7. Robinson, Thornley, J. Chem. Soc., 125, 2169 (1924).
8. G r a y, J. Am. Chem. Soc., 77, 5930 (1955).
9. Bremer, Ann., 514, 291 (1934).
10. Greenwood, Mineralogical Magazine, 20, 393 (1925); цитируется в [7J.
11. Spath, E i t e r, Ber., 74, 719 (1940).
12. S m i t h, В о у e r, J. Am. Chem. Soc., 73, 2626 (1951). Синтез и,свойства б-карболина
см. Abram ovitch et al., Can. J. Chem., 38, 2152, 2273 (1960).
13. Abramovitch, Hey, Mulley, J. Chem. Soc., 19§4, 4263.
14. К e b r 1 e, Rossie, Hoffman, Helv. Chim. Acta, 42, 907 (1959).
15. С о о k, R e e d, J. Chem. Soc., 1945, 399.
16. В о e k e 1 h e i d e, Ainsworth, J. Am. Chem. Soc., 72, 2132 (1950).
17. Horlein, Chem. Ber., 87 , 463 (1954).
18. Rosnati, Palazzo, Gazz. chim. ital., 84, 644 (1954).
19. M a n n, Prior, W i 1 1 с о x, J. Chem. Soc., U)59, 3830.
20. Henry, The Plant Alkaloids, Blakiston, Philadelphia, 4th ed., 1949; M a n s k e,
Holmes, The Alkaloids, Academic Press, New York, 1952, Vol. 2, p. 371; 1960,
Vol. 7, p. 4.
21. Perkin, Robinson, J. Chem. Soc., 115, 970 (1919).
22. Spath, Monatsh., 40, 351 (1919); 41, 401 (1920).
23. Neu, Arzneimittel Forsch., 6, 94 (1956).
24. Ker mack, Perkin, Robinson, J. Chem. Soc., 119, 1602 (1921).
25. Manske, Robinson, J. Chem. Soc., 1927, 240.
26. Kermack, Perkin, Robinson, J. Chen . Soc., 121, 1872 (1922).
27. G r a y, Spinner, C a v a 1 i t t o, J. Am. Chem. Soc., 76, 2792 (1954), и приве-
денные там ссылки.
28. S р a t h, Lederer, Ber., 63, 2102 (1930).
29. S p e i t e 1, Schlittler, Helv. Chim. Acta, 32, 863 (1949).
30. В a c h 1 i et al., Helv. Chim. Acta, 40, 1167 (1957).
31. Coulthard, Levene, Pyman, Biochem. J., 27, 727 (1933).
32. Коновалова, Проскуриина, Орехов, Arch. Pharm., 273, 156 (1935).
33. Fischer, Festschrift zum 80. Geburstag des Prinzregenten Luitpold, Erlangen, 1901
(Chem. Soc. Abstr., 1901, 80, 405).
34. H о p k i n s, С о 1 e, J. Physiol. (London), 29, 451 (1903).
15. S p a t h, Lederer, Ber., 63, 122 (1930).
36. A k a b о r i, Saito, Ber., 63, 2245 (1930).
37. P e r k i n, R о b i n s о n, J. Chem. Soc., 115, 970 (1919).
ЛИТЕРАТУРА
263
38. Н 6 г 1 е i п, Chem. Вег., 87, 463 (1954).
39. Юрашевский, ЖОХ, 24, 729 (1954).
40. A net et al., J. Chem. Soc., 1954, 1242,
41. G г a у et al., J. Am. Chem. Soc., 77, 3533 (1955). JI
42. Hasenfratz, Sutra, Compt. rend., 182, 703 (1926).
43. E л г а з н н С., Хим. фарм. пром., № 5, 270 (1933).
44. Perkin, Robinson, J. Chem. Soc., 115, 933 (1919).
45. Nishikawa, Perkin, Robinson, J. Chem. Soc., 125, 657 (1924).
46. Manske, Perkin, Robinson, J. Chem. Soc., 1927, 1.
47. Spenser, Can. J. Chem., 37, 1851 (1959).
48. Hardegger, Corrodi, Helv. Chim. Acta, 39, 984 (1956).
49. A s a h i и a, О s a d a, J. Pharm. Soc. Japan, № 534, 629 (1926).
50. H a h n, G u d j о n s, Ber., 71, 2175 (1938).
51. Hahn, Ludewig, Ber., 67, 2031 (1934).
52. С 1 e m o, S w a n, J. Chem. Soc., 1946, 617.
53. S и у d e г, К a t z, J. Am. Chem. Soc., 69, 3140 (1947).
54. J u 1 i a n et al., J. Am. Chem. Soc., 70, 180 (1948).
55. J о h n s о n et al., J. Am. Chem. Soc., 69, 2364 (1947).
56. Johnson et al., J. Am. Chem. Soc., 67, 423 (1945).
57. В 1 a i k i e, P e r k i n, J. Chem. Soc., 125, 296 (1924).
58. T a t s u i, J. Pharm. Soc. Japan, 48, 92 (1928).
59. H a h n et al., Ann., 520, 107 (1935).
60. H a h n, Hansel, Ber., 71, 2163 (1938).
61r',H a h n, Hansel, Ber., 71, 2192 (1938).
62. H a h n, W e r n e r, Ann., 520, 123 (1935).
63. J а с о b s, C r a i g, J. Biol. Chem., 113, 759 (1936).
64. Wadsworth, Pangbor ne, J. Biol. Chem., 116, 423 (1936).
65. H a r v e y, R о b s о n, J. Chem. Soc.,, 1938, 97.
66. Harvey, Miller, Robson, J. Chem. Soc., 1941, 153.
67. Barret t, Perkin, R о b i n s о n, J. Chem. Soc., 1929, 2942.
68. Keimatsu, Sugasawa, Kasuya,J. Pharm. Soc. Japan, № 558, 762 (1928).
69. Robinson, S u g i и о m e, J. Chem. Soc., 1932, 304.
70. P e r k i n, Rob i nso n, J. Chem. Soc., 101, 1778 (1912),
71. Sny der et al., J. Am. Chem. Soc., 70, 219 (1948).
72. К i n g, S t i 1 1 e r, J. Chem. Soc., 1937, 466.
73. Snyder, P ar mer ter, Walker, J. Am. Chem. Soc., 70, 237 (1948).
74. Snyder, Parmer ter, Katz, J. Am. Chem. Soc., 70, 222 (1948).
75. Fischer, Boesler, Ber., 45, 1930 (1912).
76. Fritzsche, Ann., 68, 355 (1848); 88, 329 (1853).
77. Kermack, Perkin, Robinson, J. Chem. Soc., 121/1872 (1922).
78. Kermack, Tebrich, J. Chem. Soc., 1940, 314.
79. A r m i t, R о b i n s о n, J. Chem. Soc., 127, 1604 (1925).
80. F r e a k, R о b i n s о n, J. Chem. Soc., 1938, 2013.
81. Robinson, Thornley, J. Chem. Soc., 125, 2169 (1924).
82. S c h w a r z, Schlittler, Helv. Chim. Acta, 34, 629 (1951).
83. К a u f m a n n, S t r il b i n, Ber., 44, 680 (1911).
84. H a n t z s c h, Kalb, Ber., 32, 3109 (1899).
85. G a d a m e r, Arch. Pharm., 243, 12 (1905); 246, 89 (1908).
86. Paolini, Marini-Bettolo, Nature (London), 179, 41 (1957).
87. G r a y, Spinner, C a v a 1 1 i t o, J. Am. Chem. Soc., 76, 2792 (1954).
88. G r a y, J. Am. Chem. Soc., 77, 5930 (1955).
89. A k a b о r i, Saito, Ber., 63, 2245 (1930).
90. H б r 1 e i n, Chem. Ber., 87, 463 (1954).
91. Stephen, Robinson, Nature, 162, 167 (1948); M u k h e r j i, Robinson,
Schlittler, Experientia, 5, 215 (1949).
92. Gray, Spinner, C a v a 1 1 i t o, J. Am. Chem. Soc., 76, 2792 (1954).
93. Gr a у et al., J. Am. Chem. Soc., 76, 1862 (1954); 77, 3533 (1955).
94. C a v a 1 1 i t o, Gray, O’D e 1 1, Arch, intern, pharmacodynamic, 101, 38 (1955)
O’D ell, Luna, N a p о 1 i, J. Pharmacol. Exptl. Therap., 114, 306 (1955).
95. Iyer, Rob inson, J. Chem. Soc., 1934, 1635.
96. G u 1 1 a n d et al., J. Chem. Soc., 1929, 2926.
97. P e r k i n, R о b i n s о n, J. Chem. Soc., 115, 970 (1919).
98. Patterson, Capel 1, Walker, The Ring Index, 2nd ed., American Chemical
Society, 1960.
99. F r e a k, R о b i n s о n, J. Chem. Soc., 1938, 2013.
100. Gabriel, Eschenbach, Ber., 30, 3017 (1897).
101, Perkin, Robinson, J. Chem. Soc., 115, 970 (1919).
102. Holt, P e t г о w, J. Chem. Soc., 1948, 922.
103. Kirchner, Nachr. kgl. Ges. Wiss. Gottingen, 1921, 161.
264
КАРБОЛИНЫ
'04. В о г s с h е, W a g п е г - R о е m m i с 11, В а г t h е n h е i е г, Ann., 550, 160
(1942).
105. de Diesbach, Gross, Tschannen, Helv. Chim. Acta, 34, 1050 (1951).
106. Armit, Robinson, J. Chem. Soc., 127, 1604 (1925).
107. Porter, Robinson, Wyler, J. Chem. Soc., 1941, 620.
108. Friedlander, Sander, Ber., 57, 648 (1924).
109. Lawson, Perkin, Robinson, J. Chem. Soc., 125, 626 (1924).
110. К e r m a c k, S 1 a t e r, J. Chem. Soc., 1928, 32.
111. Ker in ack, Slater, J. Chem. Soc., 1928, 789.
112. К e r in а с к, T e b r i c h, J. Chem. Soc., 1940, 314.
113. G о о d a 1 1, К e r m a c k, J. Chem. Soc., 1936, 1163.
114. Robinson, Robinson, J. Chem. Soc., 125, 832 (1924).
115. С 1 e ni о, P e г к i n, J. Chem. Soc., 125, 1612 (1924).
116. К e r m a c k, S t о r e y, J. Chem. Soc., 1950, 607.
117. de Diesbach, de Bie, Rubli, Helv. Chim. Acta, 17, 113 (1934).
118. deDiesbach, de Bie, Rubli, Helv. Chim. Acta, 20, 132 (1937).
119. M a n n, Prior, W i 1 1 с о x, J. Chem. Soc., 1959, 3830.
120. Kermack, Smith, J. Chem. Soc., 1930, 1999.
121. К e b r 1 e, Rossi, Hoffmann, Helv. Chim. Acta, 42, 907 (1959).
122. Huang-Hsinmin, Mann, J. Chem Soc., 1949, 2911.
123. ixobinson, Thornley, J. Chem. Soc., 1926, 3144.
124. G 1 a u e r t, M a n n, J. Chem. Soc., 1952, 2135; англ. пат. 684187 [С. A., 47, 6291
(1953)].
125. Laurent, Compt. rend. trav. chim., 1, 190 (1849); Ann., 72, 282 (1849); J. prakt.
Chem., [1], 47, 153 (1849).
126. Schiitzenber ger, Compt. rend., 85, 147 (1877).
127. Grandmougin, Rev. chim. ind. Paris, 41, 130 (1932).
128. G i r a u d, Compt. rend., 89, 104 (1879); 90, 1429 (1880).
129. Nolting, .S t e u e r, Ber., 43, 3512 (1910).
130. Fichte.r, Rohner, Ber., 43, 3489 (1910).
131. Fichte r, Boehringer, Ber., 39, 3932 (1906).
132. Grandmougin, Compt. rend., 174, 1175 (>1922).
133. Holt, P e t г о w, J. Chem. Soc., 1947, 607.
134. Armit, Robinson, J. Chem. Soc., 121, 836 (1922).
135. Fichte r, Probst, Ber., 40, 3478 (1907).
136. Holt, P e t г о w, J. Chem. Soc., 1948, 919.
137. Gellert, Raymond-Hamet, Schlittler, Helv. Chim. Acta, 34 , 642
(1951).
138. Henry, The Plant Alkaloids, Longmans Green and Co., Philadelphia, 4th ed., 1949.
139. Janot, Goutarel, Chabass e-M assonneau, Bull. soc. chim. France,
1953 1033
140. Janot, Goutarel, Prelog, Helv. Chim. Acta, 34, 1207 (1951).
141. Janot et al., Helv. Chim. Acta, 38, 1073 (1955).
142. Van Tamelen, Aldrich, Katz, Chem. and Ind. (London), 1956,' 793.
143. Groves, S w a n, J. Chem. Soc., 1952, 650.
144. Prasad, Swan, J. Chem. Soc., 1958, 2045.
145. Reckhow, Tarbell, J. Am. Chem. Soc., 74, 4961 (1952).
146. Bentley, Stevens, Thompson, в книге Recent Work on Naturally Occu-
ring Nitrogen Heterocyclic Compounds, Special Publication № 3, The Chemical Society,
London, 1955, p. 21.
147. Sugas.aw a, Terashim а, К а и о к a, Pharm. Bull. (Tokyo), 4, 16 (1956).
148. EIderfield et al., J. Org. Chem., 23, 435 (1958).
149. Prasad, Swan, J. Chem. Soc., 1958, 2024.
150. Th'esing et al., Angew. Chem., 68, 387 (1956); Chem. Ber., 89, 2896 (1956).
151. С 1 e m o, S e a t о n, J. Chem. Soc., 1954, 2582.
152. К e u f e r, Ann. pharm. Iran?., 8, 817 (1950).
153. Manske, Holmes, The Alkaloids, Academic Press, New York, 1952, Vol. 2,
p. 369.
154. Sumpter, Miller, Heterocyclic Compounds with Indole and Carbazole Systems,
Interscience, Publishers, New York, 1954, p. 196.
155. Boekelheide, Prelog, Progress in Org. Chem., 3, 218 (1955).
156. S a x г о n, Quart. Revs. (London), 10, 108 (1956).
157. Henry, The Plant Alkaloids, 4th ed., Longmans Green and Co., Philadelphia, 1949.
158. Chatterjee, Fortschr. Chem. org. Naturstoffe, 10, 390 (1953).
159. Woodson et al., Rauwolfia: Botany, Pharmacognosy, Chemistry and Pharmaco-
logy, Little, Brown and Co., Boston, 1957.
160. Stevens, в книге Recent Work on Naturally Occuring Nitrogen Heterocyclic Com-
pounds, Special Publication № 3, The Chemical Society, London, 1955, p. 19.
161. Battersby, Hodson, Quart. Revs. (London), 14, 77 (1960).
ЛИТЕРАТУРА
265
162. A s a h i п а, О h t a, J. Pharm. Soc. Japan, № 530, 293 (1926).
163. A s a h i n a, О h t a, J. Pharm. Soc. Japan, 49, 157 (1929).
164. A s a h i n a, J. Pharm. Soc. Japan, № 503, 1 (1924).
165. Asahina, Fujita, J. Pharm. Soc. Japan, № 476, 863 (1921).
166. Asahina et al., J. Pharm. Soc. Japan, № 543, 51 (1927); 48, 51 (1928).
167. Asahina, Ishimasa, J. Pharm. Soc. Japan, № 534, 61 (1926).
168. Asahina, Manske, Robinson, J. Chem. Soc., 1927, 1708.
169. О h t a, J. Pharm. Soc. Japan, 60, 109 (1940).
170. Asahina, О h t a, Ber., 61, 319, 869 (1928).
171. Patterson, Capel 1, Walker, The Ring Index, 2nd ed., American Chemical
Society, 1960.
172. Mendlik, Wibaut, Rec. trav. chim., 48, 191 (1929); 50, 91 (1931).
173. Wibaut, Van Gastel, Rec. trav. chim., 54, 85 (1935).
174. Woodward, Witkop, J. Am. Chem. Soc., 70, 2409 (1948).
175. Schlittler, S p e i t e 1, Helv. Chim. Acta, 31, 1199 (1948).
176. Schlittler, Alleman, Helv. Chim. Acta, 31, 128 (1948); ср.. С 1 e ni o,
Swan, Nature, 162, 693 (1948); J. Chem. Soc., 1949, 487.
177. Clemo, Swan, J. Chem. Soc., 1946, 617.
178. Julian et al., J. Am. Chem. Soc., 70, 174 (1948).
179. Scholz, Helv. Chim. Acta, 18, 923 (1935).
180. Barger, Scholz, Helv. Chim. Acta, 16, 1343 (1933).
181. V a n T a m e 1 e n, S h a m m a, J. Am. Chem. Soc., 76, 951 (1954); Van T a in e-
len, Shamma, Aldrich, ibid., 76, 4628 (1956).
182. Stork, H i 1 1, J. Am. Chem. Soc., 76, 949 (1954); 79, 495 (1957).
183. Hahn et al., Ann., 520, 107, 123 (1935).
184. Van Ta me le n et al., J. Am. Chem. Soc., 80, 5006 (1958).
185. В e 1 1 e a u, Chem. and Ind. (London), 1955, 229.
186. Potts, Robinson, J. Chem. Soc., 1955, 2675.
187. Elderfield, Fischer, Lagowski, J. Org. Chem., 22, 1376 (1957).
188. Elderfield, Fischer, J. Org. Chem., 23, 949 (1958).
189. Woodward et al., J. Am. Chem. Soc., 78, 2023, 2657 (1956); Tetrahedron, 2, 1
(1958).
190. Weisenborn, Diass i, J. Am. Chem. Soc., 78, 2022 (1956).
191. Huebner et al., Experientia, 12, 249, 250 (1956). •
192. P r e 1 о g, Helv. Chim. Acta, 31, 588 (1948).
193. Goutarel, Janot, Prelog, Experientia, 4, 24 (1948).
194. Edwards, Marion, J. Am. Chem. Soc., 71, 1694 (1949).
195. Swan, J. Chem. Soc., 1949, 1720.
196.
197.
198.
199.
200.
201.
202.
203.
Woodward, Witkop, J. Am. Chem. Soc., 71, 379 (1949).
Woodward, McLamore, J. Am. Chem. Soc., 71, 379 (1949).
Swan, J. Chem. Soc., 1958, 2038.
Обзор алкалоидов Alstonia см. Elderfield, в книге Festschrift Arthur Stoll,
Birkhauser,
E 1 d e r f i
E 1 d e r f i
E 1 d e r f i
E 1 d e r f i
Soc., 1958,
Basel, 1957, p. 353.
eld, W у t h e, J. Org. Chem., 19, 683 (1954).
eld, M с C u r d y, J. Org. Chem., 21, 295 (1956).
eld, Fischer, J. Org. Chem., 23, 332, 949 (1958).
e 1 d et al., J. Org. Chem., 23, 435 (1958); G 1 о v e'r, J о n e s, J. Chein.
1750; cp. Prasad, Swan, ibid., 1958, 2045.
Глава IV
ФЕНАНТРОЛИНЫ
13. К. ер мак, Дж. Мак-Кейл
ВВЕДЕНИЕ
Название фенантролин применяется для обозначения любой из трех гетеро-
циклических систем — I, II и III,— образующихся в результате замещения
двух групп — СН=в кольце фенантрена на — N =: одной в положении I или
4 и второй в положении 5 или 8. Эти циклические системы можно считать так-
.же 4,5-диаза- (I), 1,5-диаза- (II) и 1,8-диазафенантренами (III). Нумерация
атомов аналогична принятой для фенантрена (IV). Несмотря на то что систе-
матическая номенклатура обладает рядом достоинств, в настоящей главе
применяется широко распространенный термин фенантролин. Как будет пока-
зано ниже, все три основных соединения — I, II и III — могут быть получены
реакцией Скрауяа из соответствующих о-, м- и n-фенилендиаминов, вследствие
чего различают о-, м- и п-фенантролины.
Термин фенантролин в принципе должен был бы включать, кроме соеди-
нений I, II и«Ш, и другие диазафенантрены, однако обычно ограничивают его
применение циклическими системами I, II и III.
Диазафенантрены можно разделить на три группы: 1) с двумя атомами
азота в одном кольце; 2) с двумя атомами азота в смежных кольцах и 3) с од-
ним атомом азота в каждом из внешних колец. К числу диазафенантренов пер-
вой группы относятся бензоциннолины, бензохиназолины или бензохинокса-
лины; ко второй группе — бензонафтиридины. Соединения этих двух групп
описаны в т. 6, гл. V, VIII и X, а также в гл. II настоящей книги, поэтому ниже
речь будет идти только о диазафенантренах, принадлежащих к третьей группе.
Таких диазафенантренов десять. Три из них о-(4,5-), лс-(1,5-) и п-(1,8-)
фенантролины. Другие семь 1,6-, 1,7-, 2,5-, 2,6-, 2,7-, 3,5- и 3,6-диазафенантре-
ны. Большинство этих циклических систем относительно труднодоступны
ОБЩИЕ МЕТОДЫ СИНТЕЗА
267
и исследованы сравнительно мало. Ниже (стр. 284) будут приведены некото-
рые сведения, касающиеся этих необычных типов соединений.
Фенантролины могут рассматриваться как продукты конденсации пири-
динового ядра с ядром хинолина, вследствие чего их можно именовать хино-
пиридинами или пиридохинолинами. Так, например, соединения I, II и III
иногда называют: 7,8-3',2'-, 5,6-2',3'- или соответственно 7,8-2',3'- и 5,6-3',2'-
пиридохинолинами.
В настоящей главе, как уже говорилось, принята система нумерации,
основанная на общепринятой для фенантренов. Таким образом, атомы в цик-
лах о-фенантролина, или 4,5-диазафенантрена, нумеруются, как показано
в формуле I, м-фенантролина, или 1,5-диазафенантрена,—как в формуле II,
и n-фенантролина, или 1,8-диазафенантрена, как в формуле III. Эта нумерация
находится в соответствии с принятой у Бейлынтейна, но отличается от нуме-
рации в справочнике «The Ring Index», в котором применяется система, при-
веденная в формулах V, VI и VII, и номенклатуры— 1,10-, 1,7- и 4,7-фенантро-
лины. В Англии для м- и n-фенантролинов принята нумерация атомов, показан-
ная в формулах II и III, но для о-фенантролина чаще применяется нумерация,
как в формуле V. Нумерация, принятая в данной главе, имеет то преимущест-
во, что она применима, если соединения описываются как диазафенантрены.
Эта система может быть легко приспособлена для обозначения не только трех
типов собственно фенантролинов, но также и других и предусматривает еди-
ную систему нумерации для всего ряда.
В некоторых случаях, когда в одном из пиридиновых колец фенантролина
необходимо показать положение заместителя относительно атома азота, удобно
обозначать его греческими буквами а, 0 и у, как это часто делается в случае
аналогичных производных пиридина и хинолина.
ОБЩИЕ МЕТОДЫ СИНТЕЗА
Практически все известные производные фенантролина получают одним
из стандартных методов синтеза хинолинов, применяя их к соответствующему
производному аминохинолина с аминогруппой в бензольном кольце или фени-
лендиамина. С помощью реакции Скраупа о-, м- и n-фенантролины могут
быть синтезированы не только из о-, м- и n-фенилендиаминов, но также
и из 8-амино-, 5-амино и 6-аминохинолинов соответственно.
Вместо реакции Скраупа могут быть использованы и другие методы синтеза
хинолинов, применимые к простым первичным ароматическим аминам. В после-
дующих разделах будет также упомянут метод конденсации с ацетоуксусным
эфиром или щавелевоуксусным эфиром по Конраду — Лимпаху или до неко-
торой степени аналогичные синтезы с применением этоксиметиленмалонового
эфира. Применялись также реакции циклизации Дёбнера с использованием
пировиноградной кислоты, позволяющие, в частности, из 5-аминохинолина,
бензальдегида и пировиноградной кислоты получить 6-фенил-л-фенантролин-
карбоновую-8 кислоту [1], а из 6-аминохинолина — изомерную 2-фенил-п-
фенантролинкарбоновую-4 кислоту [2]. Рао и У.илер [3] синтезировали диэти-
ловые эфиры 4,8-диокси-2,6-дифенил-ле-фенантролиндикарбоновой-3,7 и 4,5-
диокси-2,7-дифенил-п-фенантролиндикарбоновой-3,5 кислот путем конденса-
цйи диимидохлоридов (полученных из дибензоильных производных м- и п-
268
ФЕНАНТРОЛИНЫ
фенилендиамина) с натриймалоновым эфиром. Продукты конденсации цикли-
зовали нагреванием при 200—210°. Поскольку практически все фенантролины
синтезируют, пользуясь стандартными методами получения хинолинов, здесь
Н5С2ООС СООС2Н5
н с—С6Н5
II
N N
СвН5С/ \^\/
.'I и
с-н
С2Н5ООС/ЧСООС2Н5
СООС2Н5
I
с2н5оос
I
он
подробно не будет рассматриваться механизм циклизации, так как реакции
Скраупа, Конрада — Лимпаха и др. рассмотрены в т. 4, гл. I. Однако особое
значение в связи с синтезом фенантролинов приобретает вопрос о направлении
замыкания кольца.
Уже отмечалось, что «-фенантролин может быть получен по методу Скрау-
па из*н-фенилендиамина или 6-аминохинолина. Совершенно очевидно, что вто-
рое пиридиновое кольцо может присоединяться к 6-аминохинолину (VIII)
двумя путями — с замыканием цикла либо по углеродному атому в положе-
нии 5, либо в положении 7 хинолинового кольца с образованием соответствен-
но ангулярного «-фенантролина (IX) или линейного 1,5-диазаантраКена (X).
Практически образуется только ангулярный «-фенантролин. Ниже (стр. 271)
приводится доказательство того, что продукт реакции имеет ангулярную,
а не изомерную линейную конфигурацию.
VIII XI X
Уместно напомнить, что при применении реакции Скраупа к 2,5-диметил-
/i-фенилендиамину (XI), когда могла бы образоваться линейная, а не ангуляр-
ная трициклическая система, ни одна из них фактически не обнаружена
и выделенное соединение представляет 6-амино-5,8-диметилхинолин (XII) [41.
Если реакцию Скраупа применять к 6-амино-5-хлорхинолину, то хлордиазаан-
трацен либо совсем не образуется, либо получается с небольшим выходом
и вместо него, как правило, удается выделить лишь некоторое количество
СН3 СН3
СН3 СН3
XI XII
/г-фенантролина [51. Таким образом, тенденция к ангулярной циклизации
настолько преобладает над линейной, что создающий препятствие атом хлора
удаляется из положения 5 и замыкание кольца происходит предпочтительнее
по этому месту, а не в положении 7, где нет помех для замыкания кольца»
д-, jk- й n-ФЕНАНТРОЛиИЫ
269
Эта тенденция к образованию ангулярного фенантролина аналогична
наблюдаемой у р-нафтиламина и его производных. Так, например, из р-нафтил-
амина реакцией Скраупа получают 5,6-бензохинолин; это же соединение
наряду с 8-хлор-6,7-бензохинолином (8-хлор-1-азантраценом) получается
из 1-хлор-2-нафтиламина.
При циклизации производных аминохинолина с аминогруппой в бензоль-
ном ядре, по-видимому, не имеет места исключение из правила, и если возмож-
но образование обеих систем — линейной и ангулярной, то получаются
продукты исключительно ангулярного строения. В отдельных случаях, когда
нет надежных доказательств строения полученного соединения, можно смело
утверждать, что образуется ангулярная система. Это правило, в частности,
распространяется на синтезы Скраупа и Конрада — Лимпаха.
о-, м-и и-ФЕНАНТРОЛ И Н Ы
О-Фенантролин. Впервые о-фенантролин (1,10-фенантролин, 4,5-фенантро-
лин, а-фенантролин, изофенантролин, 7,8,3',2'-пиридохинолин, дипириди-
но-3',2', 1,2,2",3",3,4-бензол) был получен Бляу реакцией Скраупа как из
о-фенилендиамина, так и из 8-аминохинолина [61. Позднее различные исследо-
ватели изучали синтез этого соединения с целью подбора условий полу-
чения его с максимальным выходом. Большинство авторов установили,
что из о-фенилендиамина о-фенантролин образуется с низким выходом. Неко-
торые авторы, в том числе Смит [7 ], указывают, что получить о-фенантро-
лин исходя из о-фенилендиамина вообще невозможно. По-видимому, некото-
рое количество фенантролина в действительности все же образуется, однако
в жестких условиях реакции Скраупа происходит столь сильное осмоление,
что выделение продукта представляет значительные трудности [8]. Тартарини
и Самайа [91 использовали способность о-фенантролина образовать комплексы
с двухвалентным ионом железа. Эти авторы обрабатывали продукт реакции
Скраупа сернокислым железом (II), что приводило к образованию железо-
о-фенантролинового комплекса, который осаждали хлоридом ртути (II). Эту
соль разлагали едким натром и свободное основание экстрагировали петролей-
ным эфиром [9]. Хэдель и Гизин [10] сообщили об усовершенствовании реак-
ции получения о-фенантролина по методу Скраупа, проводя ее в присутствии
солей меди. Из медного комплекса основание выделено с помощью сероводоро-
да. Выход продукта, полученного из о-фенилендиамина, составил 30%.
В цитируемой работе условия проведения реакции Скраупа были отно-
сительно жесткими, причем применялась концентрированная серная кислота
и, насколько это было возможно, исключалось попадание влаги. В модифици-
рованном синтезе Скраупа [11 ] применялась примерно 69%-ная серная кисло-
та и реакция проводилась в значительно менее жестких условиях; все это
уменьшало тенденцию к смолообразованию. Используя измененный метод
синтеза, Холкроу и Кермак [12] получили о-фенантролин из о-фенилендиами-
на с выходом 45% (на сырой продукт), затратив меньшие усилия, чем требо-
валось по методам Хибера и Мюльбауэра [8], а также Тартарини и Самайя [9].
Большинство авторов установило, что 8-аминохинолин в качестве исход-
ного продукта дает лучшие выходы, чем о-фенилендиамин, однако смолообра-
зование все же продолжает оставаться весьма значительным, а очистка про-
дукта представляем собойJ довольно затруднительную операцию. Кюстер
и Ролл [13] получили 6 г гидрохлорида из 20 г 8-аминохинолина. Смит и Гетц
подробно описывают проведение синтеза о-фенантролина из 8-аминохинолина
в большом масштабе; выход составлял 40% (20% в расчете на о-нитроанилин).
Эти авторы выделили также побочный продукт зеленого цвета, который,
по их мнению, является |3-(8-хинолиламино)пропионовым альдегидом [14].
270
Фенантролины
о-Фенантролин кристаллизуется из воды в виде моногидрата с т. пл. 102°;
безводный плавится при 117°; кипит без разложения выше 360°. С кислотами
он образует соли обычно состава 1:1. Бихромат и хлороплатинат имеют сле-
дующие формулы: (С12Н8К2)2-Н2Сг2О7-2Н2О и (Ci2H8N2)2-H2PtCle- 11/2(2)Н2О.
о-Фенантролин окисляется щелочным раствором перманганата в 2,2'-дипи-
ридилдикарбоновую-3,3' кислоту [7].
о-Фенантролин обладает ясно выраженной склонностью вступать в соеди-
нение с ионами различных металлов, особенно с двухвалентными металлами
переходной группы. В аналитической химии приобрел важное значение ком-
плекс с ионом двухвалентного железа, называемый ферроином. Строение и свой-
ства комплексов металлов более полно рассмотрены на стр. 287.
Кейс [15] синтезировал ряд ди- и триметил-о-фенантролинов с помощью
реакции Скраупа исходя из соответствующего 8-амйнохинолина и глицерина,
а-метилакролеиндиацетата или метилвинилкетона. Автор сообщает, что
наилучшие выходы были получены при реакции с глицерином, средние —
с а-метилакролеиндиацетатом и наименьшие — с метилвинилкетоном. Так,
например, 1,10-диметил-о-фенантролин получался из 4,5-диметил-8-амино-
хинолина и глицерина с выходом 24%. Однако синтез того же соединения
из 6-метил-8-аминохинолина и метилвинилкетона осуществить не удалось.
Кейс [16] синтезировал различные фенил-о-фенантролины, применяя
реакцию Скраупа к соответствующему фенил-8-аминохинолину и глицерину
или p-хлорпропиофенону. Автор считает это первым случаем применения
p-хлорпропиофенона в качестве второго компонента в реакции Скраупа. Несмо-
тря на то что в некоторых случаях с этим реагентом могут быть достигнуты
хорошие выходы, оказалось, что реакция не идет, если фенильная группа
находится у_атома,углерода,' соседнего с тем, который участвует в замыкании
кольца. Так, например, в результате реакции 6-фенил-8-аминохинолина с
Р-хлорпропиофеноном не удалось получить 1,10-дифенил-о-фенантролина. Эта
неудача обусловлена стерическими препятствиями.
лс-Фенантролин.Скрауп и Бортман [17] в 1882 г. синтезировали щ-фенант-
ролин, известный также под названиями фенантролин, 1,7-фенантролин,
1,5-фенантролин, 4,10-фенантролин, 5,6,3',2'- или 7,8,3',2'-пиридохинолин,
дипиридино-2'3', 1,2,2",3",3,4-бензол и 4,10-нафтизодиазин, нагреванием смеси
двойной оловянной соли щ-фенилендиамина, глицерина, м-динитробензола
и концентрированной серной кислоты. Почти одновременно с ними Ла Кост
[18] получил /{-фенантролин из щ-нитроанилина, глицерина, серной кислоты
и нитробензола наряду с небольшим количеством побочного продукта, который,
как он полагал, представлял собой 2-окси-щ-фенантролин. Впоследствии было
показано, что это 10-оксипроизводное (см. стр. 277). Время от времени различ-
ные авторы предлагали модификации синтеза Скраупа с целью повысить выход
/{-фенантролина из /{-фенилен диамина. Смит [7] получил 40—45 г дигидрата
основания из 50 г щ-фенилендиамина. При замене глицерина акролеином
At-фенантролин выделить не удалось {7].
Доказательством того, что продуктом двойной реакции Скраупа действи-
тельно является,/{-фенантролин (1,5-диазафенантрен) (XIII), а не линейный
1,8-диазаантрацен (XIV) служит тот факт, что соединение XIII может быть
\/\^
ХШ
N I N
/ \/\/
II СООН
соон
XV .
б-, м- й л-4>еНАнтроЛинЫ
271
получено из 5-аминохинолина реакцией Скраупа, а также результаты окисле-
ния основания щелочным раствором перманганата калия [7, 17]. При этом
образуется 2,3'-дипиридилдикарбоновая-3,2' кислота (XV), которая при декар-
боксилировании переходит в 2,3'-дипиридил. Такой дипиридил, безусловно,
не мог образоваться из 1,8-диазаантрацена (XIV). Кушинский и Зухарда [20]
установили, что лс-фенантролин можно получить нагреванием 7-нитрохино-
лина с глицерином и концентрированной соляной кислотой при 160—170°
в течение 20 час. Соответствующая реакция с 5-нитрохинолином дает 10-хлор-
ле-фенантролин (стр. 280).
Безводный л-фенантролин образует бесцветные таблички с т. пл. 78°,
обладающие характерным запахом; дигидрат кристаллизуется в виде бесцвет-
ных игл. С одним эквивалентом минеральных кислот основание образует
устойчивые соли. Дихлоргидрат Ci2H8N2-2HC1 нестоек, при выпаривании
водных растворов этой соли выпадает монохлоргидрат. л-Фенантролин легко
растворим в спирте, средне растворим в горячей воде и плохо — в эфире,
бензоле и петролейном эфире. Он слегка летуч с водяным паром и хорошо
растворим в разбавленных кислотах.
и-Фенантролин. n-Фенантролин, известный также под названиями 4,7-
фенантролин, 1,8-фенантролин, 5,6,2',3'-пиридохинолин, дипиридино-
2',3',1,2,3",2",3,4-бензол, псевдофенантролин, хино-м-(а-хинолин) и 4,7-наф-
тизодиазин был впервые получен Скраупом и Бортманом [21 ] нагреванием
двойной оловянной соли п-фенилендиамина с глицерином, нитробензолом
и серной кислотой. В качестве побочного продукта было выделено небольшое
количество хинолина. Позднее Смит [7] описал синтез n-фенантролина из
п-фенилендиамина (выход 60%). То же соединение с небольшим выходом наря-
ду с 6-нитрохинолином было получено Борнеманом [22], а также Клаусом
и Висом [23] при нагревании смеси м-нитроанилина, глицерина, нитробензола
и концентрированной серной кислоты. Хаскельберг [24] усовершенствовал
этот метод, в результате чего выход n-фенантролина из м-нитроанилина увели-
чился до 49%. Модификацией метода явилось применение вместо м-фениленди-
амина м-аминоазобензола. Нагревая последний с глицерином и концентриро-
ванной серной кислотой, Лельман и Липперт [25] получили м-фенантролин
наряду с некоторым количеством хинолина. Позднее Мацумура [26] рекомен-
довал этот метод, как дающий высокие выходы (около 85%) и не требующий
большого труда для синтеза промежуточных продуктов. Мацумура сделал
также любопытное наблюдение, что n-фенантролин может быть выделен с не-
большим выходом из продуктов реакции Скраупа диметилового эфира м-амино-
резорцина с 2,4-диоксиазобензолом или 2,4-диметоксиазобензолом [26].
Помимо синтеза n-фенантролина двойной реакцией Скраупа исходя из п-
фенилендиамина или другого соединения с промежуточным образованием
м-фенилендиамина, существует еще метод получения n-фенантролина из '6-ами-
нохинолина. При нагревании последнего с глицерином, мышьяковой кислотой
и концентрированной серной кислотой Кауфман и Радошевич [27] получили
n-фенантролин с количественным выходом. Представляет интерес модифика-
ция этого метода, предложенная Кушинским и Зухарда [20]: м-фенантролин
получен нагреванием смеси 6-нитрохинолина, глицерина и концентрирован-
ной соляной кислоты при 160—170° в течение 20 час.
Ни один из этих методов не дает доказательства строения соединения XVI,
так как все они не исключают возможности образования характерной для 1,5-
диазааитрацена (XVII) линейной конфигурации. Строение соединения XVI
было установлено окислением его щелочным раствором перманганата калия
[7, 27.]; при этом образуется 3,3'-дипиридилдикарбоновая-2,2' кислота (XVIII),
превращающаяся при нагревании в результате декарбоксилирования в 3,3'-
дипиридил.
272
ФЕНАНТРОЛИНЫ
XVI XVII XVIII
n-Фенантролин кристаллизуется из лигроина в виде безводных игл
с т. пл. 177°. Основание начинает возгоняться при 100°; растворим в спирте
и хлороформе. С хлоридом железа (III) в спиртовом растворе п-фенантролин
дает оранжевую окраску; образует соли как с одним, так и с двумя эквивален-
тами минеральных кислот.
(УСТОЙЧИВОСТЬ И РЕАКЦИОННАЯ СПОСОБНОСТЬ ФЕНАНТРОЛИНОВ
Заслуживает внимания устойчивость всех трех циклических систем фенан-
тролина. Они с большим трудом нитруются и очень стойки ко многим окисли-
тельным агентам. Смит и Кэгл [28] установили, что на о-фенантролин не дей-
ствует кислый раствор бихромата, нейтральный, кислый или щелочный раство-
ры перекиси водорода, подкисленный ванадат или перйодат. Линекер и Эванс
[29] нашли, что м- и м-фенантролины не окисляются хромовой кислотой, дву-
окисью селена, пятиокисью ванадия, йодноватой или иодной кислотой.
Несмотря на то что все три фенантролина могут быть окислены щелочным
раствором перманганата калия в дипиридилдикарбоновые кислоты, фенантро-
лины не удается превратить в 9,10-фенантролохиноны при действии окислите-
лей, с которыми обычно протекают реакции этого типа [28, 29]. В этом отноше-
нии фенантролины резко отличаются от фенантрена, из которого окислением
легко получить 9,10-фенантрахинон. Единственный, полученный до настоя-
щего времени 9,10-фенантролохинон выделен Смитом и Кэглом в качестве
побочного продукта при нитровании о-фенантролина в очень жестких условиях
(см. стр. 282).
Однако Дрюи и Шмидт [30] сообщили о получении- с 90%-ным выходом
.и-фенантролин-9,10-хинона при окислении 9-метокси- или 9-окси-л«-фенантро-
лина смесью дымящей азотной и серной кислот. Авторы указывают, что при
действии других окислителей этот продукт не образуется. Подобным образом
9-метокси-м-фенантролин превращается в п-фенантролин-9,10-хинон, а 9-ме-
токси-о-фенантролин — в о-фенантролин-9,10-хинон. Сообщается, что в эту
реакцию вступают фенантролины, имеющие свободную или замещенную гид-
роксильную или аминную группу в положении 9 или 10 [31].
о-Фенантролин-9,10-хинон при действии разбавленного раствора едкого
натра отщепляет окись углерода и переходит в новое азотсодержащее гетеро-
циклическое соединение — 4,5-диазафлуоренон-9 [32]. Это первая реакция,
в ходе которой из 1,2-дикетонной группировки выделялась окись углерода.
Дрюи и Шмидт [30] действием водного раствора едкого натра при 70° в тече-
ние 4 час превратили о-, м- и м-фенантролин-9,10-хиноны соответственно в 4,5-
диазафлуоренон-9, 1,5-диазафлуоренон-9 и 1,8-диазафлуоренон-9.
Сирлс и Уоррен [33] исследовали гидрирование п-ф>енантролина и его
3-замещенных. Гидрирование самого n-фенантролина в абсолютном спирте
над никелем Ренея и этилатом натрия протекает с поглощением 2 молей водо-
рода и ведет к образованию двух продуктов: более низкоплавкого соединения
белого цвета, не способного образовать ацетильное производное.^и более
высокоплавкого вещества желтого цвета, температура плавления 'которого
совпадает с температурой плавления, найденной Мацумура [34] для 1,2,3,4-
тетрагидро-м-фенантролина. Образование ацетильного производного и данные
анализа подтверждают, что желтое вещество является 1,2,3,4-тетрагидро-
УСТОЙЧИВОСТЬ И РЕАКЦИОННАЯ СПОСОБНОСТЬ ФЕНАНТРОЛИНОВ
273.
n-фенантролином; тем самым доказывается, что при гидрировании л-фенан.тро-
лина восстанавливается пиридиновое ядро (см. ниже).
Для того чтобы выяснить, какое из пиридиновых колец, замещенное или
незамещенное, в алкил-п-фенантролинах преимущественно подвергается вое*
становлению, был прогидрйрован в описанных выше условиях 3-метил-п-фенан-
тролин. Полученное при этом тетрагидропроизводное желтого цвета окисляли
щелочным раствором перманганата. Выделенное при этом вещество белого
цвета оказалось пиридинтрикарбоновой-2,3,5 кислотой. Это указывает на тб,
что восстановлению подверглось незамещенное пиридиновое кольцо. (Если
бы прогидрировался замещенный пиридиновый цикл, то должна была образо-
ваться хинолиновая кислота.)
Показано, что вещества белого цвета, полученные при гидрировании
n-фенантролина и его 3-замещенных, представляют собой 9,10-дигидропро-
изводные. Это подтверждается количеством поглощенного водорода, неспособ-
ностью продуктов давать ацетильные производные, а также образованием
при окислении соответствующих 5-замещенных 3,3'-дипиридилдикарбоновых-
2,2' кислот. На основании этого сделан вывод, что наряду с образованием
1,2,3,4-тетрагидро-п-фенантролинов происходит частичное гидрирование бен-
зольного кольца.
Ошиаи и Куроианаги [35] исследовали реакцию восстановления п-фе-
нантролина и его 2,4-диметилпроизводного оловом в соляной кислоте. Аце-
тилирование продукта восстановления дало диацетильное производное
1,2,3,4,5,6,7,8-октагидро-м-фенантролина. 2,4-Диметил-м-фенантролин в тех
же условиях превращался в 5,6,7,8-тетрагидропроизводное — кристалли-
ческое вещество желтого цвета. При гидрировании над никелем Ренея под дав-
лением водорода 100 ат и температуре 180° n-фенантролин дал 1,2,3,4,5,6,7,8-
октагидропроизводное. При гидрирований же 2,4-диметил-м-фенантролина
под давлением 60 ат и температуре 190° наряду с 5,5а,6,7,8,9,9а, 10-октагидро-
производным образуется небольшое количество '9,10-дигидропроизводного.
Два пиридиновых ядра n-фенантролина (как и в хинолине), по-видимому,
гидрируются легче, чем бензольное кольцо. Наличие метильных заместителей
в пиридиновом кольце n-фенантролина препятствует гидрированию, поэтому
18 Заказ № 978
27.4
ФЕНАНТРОЛИНЫ
предпочтительно образуется тетрагидропроизводное. Двойная связь в поло-
жении, 9,10, по-видимому, более реакционноспособна, чем соответствующая
связь в ядре хинолина, так как при гидрировании образуются 9,10-дигидро-
и 5,5а,6,7,8,9,9а, 10-октагидропроизводные. Это указывает на сходство реак-
ционной способности «-фенантролина и фенантрена.
I N
СНз-Д^-СНз
МЕТИЛАТЫ. ПРОДУКТЫ ВОССТАНОВЛЕНИЯ И ОКИСЛЕНИЯ
Вступая в реакцию с одной молекулой йодистого метила, о-, м- и «-фенан-
тролины легко образуют моноиодметилаты Cl2HgN2-CH3I [12, 27, 36]. Оба
атома азота в о- и «-фенантролинах равноценны и, следовательно, моноиодмети-
латы, несомненно, имеют строение I и II, однако у л-фенантролина теоретиче-
ски возможны две различные четвертичные соли III и IV. Экспериментально
удалось получить лишь один иодметилат или сульфометилат [37]. Исходя
из общих положений можно предположить, что он имеет строение III. Правиль-
ность этого вывода подтверждается исследованиями, результаты которых
приведены на стр. 281.
МЕТИЛАТЫ. ПРОДУКТЫ ВОССТАНОВЛЕНИЯ И ОКИСЛЕНИЯ 275
Сайкес [381 синтезировал моноиодметилаты 10-хлор-, 10-ацетамино-
и 10-бром^п-фенантролинов. Во всех случаях единственными продуктами реак-
ции были соответствующие 10-замещенные 8-иодметилаты. Позднее этот автор
[391 показал, что у моноиодметилатов «-фенантролинов, замещенных в поло-
жении 2, четвертичным становится атом азота в положении 8. Невозможность
получить какой-либо из 1-иодметилатов Сайкес объясняет стерическими
затруднениями у атома азота в положении 1, обусловленными наличием
заместителя в положении 2.
Из о- и лефенантролинов не удается получить метилатов, в которых оба
атома азота были бы четвертичными. бис-Четвертичные соли образует «-фенан-
тролин, который, например, при нагревании с большим избытком йодистого
метила в спирте дает дииодметилат Ci2H8N2-2(CH3I) [21]. Эту соль легче
получить обработкой «-фенантролина диметилсульфатом с последующим пре-
вращением дисульфометилата в дииодметилат (V) [27].
При обработке гидросульфитом натрия иодметнлаты о-, м- и «-фенантроли-
нов восстанавливаются с образованием дигидропроизводных. Первоначально
их обозначали формулами VI—VIII, что отвечало строению, приписываемому
1,2-дигидропроизводным, полученным аналогичным путем из четвертичных
солей метилпиридиния [36]. Однако Трабер и Каррер [40] показали, что полу-
ченные восстановлением с помощью гидросульфита дигидропиридины пред-
ставляют, собой 1,4-дигидропроизводные, а менее стабильные 1,2-дигидропро-
изводные, легко превращающиеся в 1,4-дигидроизомеры, образуются при
восстановлении боргидридом натрия. Иодметилат «-фенантролина при вос-
становлении боргидридом натрия дает неустойчивое дигидропроизводное,
отличающееся от продукта, полученного восстановлением гидросульфитом,
но легко в него превращающееся [41 ]. По аналогии можно считать вероятным,
что неустойчивый продукт восстановления боргидридом натрия имеет строе-
ние VIII, а стабильный продукт восстановления гидросульфитом — строение
Villa. По-видимому, дигидрометил-о- и дигидрометил-л«-фенантролинам, полу-
ченным восстановлением гидросульфитом, соответствует строение Via и Vila,
а продуктам восстановления боргидридом — строение VI и VII, однако точные
доказательства отсутствуют: Эти соединения энергично восстанавливают нитрат
серебра. 1 -Метил- 1,2-(VIII) и Пметил-1,4-дигидро-«-фенантролины (Villa)
восстанавливаются каталитически водородом над платиной с образованием
1-метил-1,2,3,4-тетрагидро-«-фенантролина (IX). Как это соединение, так
и соответствующие тетрагидро-о- и тетрагидро-л«-производные могут быть так-
же получены непосредственно каталитическим восстановлением иодметила-
тов. При восстановлении дииодметилата «-фенантролина с помощью гидросуль-
фита натрия в присутствии бикарбоната натрия образуется темно-красный
А",
| N — CH3
zx/'v
VIII
VII
Н2А
N I N —СН3
/ \/Ч/
A h
\/\z
Vila
H2A .
| N-CH3
z\/x/
Villa
18*
276
ФЕНАНТРОЛИНЫ
раствор, из которого выделить основание экстракцией эфиром невозможно.
Манометрические наблюдения показали, что на один моль дииодметилата
n-фенантролина расходуется один атом водорода [42], в результате чего,
по-видимому, образуется продукт с семихиноидной структурой, изображен-
ной формулой X [37 ] и сходный с продуктом, полученным из дииодметилата
у/у'-дипиридила [43]. О восстановлении иодметилатов производных а-метокси-
феиантролинов см. в работе Каррера и Плетчера [44].
Окисление метилатов фенантролина щелочным раствором феррици-
анида протекает так же, как окисление метилатов пиридина и хинолина
(см. т. 1и т. 4). Так, например, иодметилат n-фенантролина дает при
этом 1-метил-1,2-дигидро-м-фенантролинон-2 (XI), а дииодметилат аналогич-
ным путем окисляется в 1,8-диметил-1,2,7,8-тетрагидро-п-фенантролинди-
он-2,7 (XII) [27]. Это соединение может быть получено также окислением иод-
метилата соединения XI [45]. Четвертичные метильные соли о- и .«-фенантро-
линов дают соответствующие метил-о- и метил-лефенантролины [12, 37, 46].
I N —СН3
^\/\/ .
Вибо, Сйиерс и Оувельтьис [47 ] сообщают, что при получении м-фенан-
тролина из м-фенилендиамина методом Смита [71 они выделили побочный
продукт, идентичный полученному Мацумура [48] при восстановлении м-фе-
нантролина натрием в амиловом спирте или оловом в соляной кислоте. Это
основание, которое не может быть окислено в n-фенантролин и образующее
моноацетильное и монобензоильное производные, вероятно, представляют собой
1,2,3,4-тетрагидро-н-фенантролин.
N-ОКИСИ ФЕНАНТРОЛИНА
Поскольку у фенантролинов имеются два третичных атома азота, можно
ожидать образования моно- и ди-М-окисей. N-Окись .«-фенантролина [49]
и N-окись п-фенантрблина [45] образуются при обработке соответствующего
фенантролина надбензойной кислотой в растворе хлороформа. Так как при
обработке N-окиси .«-фенантролина хлорокисью фосфора [49] получается
2-хлор-л(-фенантролйн [37] (стр. 281), атом кислорода в исходной окиси нахо-
ОКСИ- И ГАЛОГЕНПРОИЗВОДНЫЕ ФЕНАНТРОЛИНА
277
дится у первого, а не у пятого атома азота (XIII). При действии на М’фенантро-
\/\Z
хш
лин надбензойной кислоты получена также ди-М-окись [45], которая идентич-
на соединению, синтезированному Линекером и Эвансом [29] окислением
n-фенантролина перекисью водорода в кипящей ледяной уксусной кислоте.
Те же авторы при действии перекиси водорода в уксусной кислоте на о- и .«-фе-
нантролины получили соответствующие ди-N-окиси [29].
При нагревании N-окисей хинолина с хлорокисью фосфора или с хло-
ристым сульфурилом образуются производные 2-хлор-, 4-хлор- 'и иногда
3-хлорхинолина (см. т. 4). Как уже упоминалось, .«-фенантролин при ки-
пячении с хлорокисью фосфора дает 2-хлор-ле-фенантролин; 4-хлор-.м-фенан-
тролин при этом выделить не удалось [49]. Тенденцию превращаться
в производные а-хлорфенантролина обнаруживают и другие N-окиси фенан-
тролина. Так, например, N-окись л-фенантролина дает 2-хлор-м-фенантро-
лин, а ди-N-oKHCb м-фенантролина — 2,7-дихлор-м-фенантролин [45]. Дан-
ные о строении этих хлор-.м- и хлор-м-фенантролинов, а также дихлор-м-
фенантролина будут приведены в следующем разделе.
ОКСИ- И ГАЛОГЕНПРОИЗВОДНЫЕ ФЕНАНТРОЛИНА
Оксифенантролины могут быть синтезированы: а) из соответствующего
оксисоединения реакцией Скраупа; б) из аминохинолина реакцией типа Кон-
рада — Лимпаха или Кнорра, при помощи которых удобно удалить карб-
этоксигруппу гидролизом и декарбоксилированием.
Ю-Окси-л«-фенантролин (XIV) наряду с ^-фенантролином впервые полу-
чен Ла Костом [18] из .м-нитроанилина реакцией Скраупа. По мнению автора,
соединение представляло собой 2-окси-ле-фенантролин, однако его действи-
тельное^ строение было установлено Мацумура [26], который получил его
с низкйм выходом наряду с хинолином реакцией Скраупа из 5-бензолазо-8-окси-
хинолина (XV). В опытах Мацумура 5-амино-8-оксихинолин, очевидно, обра-
зовался в качестве промежуточного продукта, и весьма вероятно, что в опы-
тах Ла Коста из л-нитроанилина первоначально образовывался 5-нитро-
хинблин, который при восстановлении превращался в оксиаминохино-
лин, изомеризующийся в 5-амино-8-оксихинолин (XVI), из которого уже был
получен 10-окси-л-фенантролин.
- Хеуорс и Сайкес [50] получили 10-окси-л-фенантролин из 5-амино-8-окси-
хинолина (XVI); другой синтез разработан Турским и Клейном [51], которые
подвергли 2,4-диаминофенол (XVII) двойной реакции Скраупа. Ю-Окси-
.м-фенантролин был получен также Гошем, Роем и Бенерджи [52] при нагре-
вании 2,4-динитрофенола (XVIII) с глицерином и серной кислотой, причем
сам динитрофенол при этом действовал как окислитель. Выход составил 50%
[52].
Оксифенантролины с гидроксильной группой в одном из двух пиридино-
вых колец могут быть синтезированы любым из двух общих методов, указан-
ных выше. Так, например, 4-окси-м-фенантролин (XX) (т. пл. 300—301°)
был получен как реакцией Скраупа из 4-окси-6-аминохинолина (XIX) [53],
так и конденсацией 6-аминохинолина (XXI) с этоксиметиленмалоновым эфиром
с последующей циклизацией в этиловый эфир 4-окси-м-фенантролинкарбоно-
278
ФЕНАНТРОЛИНЫ
вой-3 кислоты,•который затем подвергали гидролизу и декарбоксилирова-
нию [54]. Продукты циклизации имеют ангулярную структуру в обоих слу-
чаях, что доказано образованием n-фенантролина при перегонке декарбокси-
лированного продукта с цинковой пылью [53].
XIX XX
XXI
С2Н5ООС СООС2Н6
"с/ . СООСгЩ
Ч I
си но-/\
NH I N
ОКСИ- И ГАЛОГЕНПРОИЗВОДНЫЕ ФЕНАНТРОЛИНА
279
Этими методами получены многие пиридинокси- и метилоксипроизводные
о-, м- и П7фенантролинов [33, 37, 45, 49, 53,55—58]. Поскольку применяемые
реакции описаны ранее в разделе, посвященном синтезу производных хино-
лина, здесь нет необходимости на них останавливаться.
но—сн
ОН II
I N
х\/\/
| II соон
/>N/X'COOH
он
он
XXIV
Н3С
XXII XXIII
Подобно тому как фенантролины могут быть получены в одну стадию
из фенилендиаминов по реакции Скраупа (включая двойную циклизацию),
так и диоксифенантролины могут образоваться из о-, м- или м-фенилендиами-
нов при взаимодействии с двумя молекулами ацетоуксусного или этоксиме-
тиленмалонового эфира с последующей двойной циклизацией. Так, например,
Джакини [59] получил этим путем из м- и м-фенилендиаминов и ацето-
уксусного эфира м- и п-фенилен-бис-аминокротонаты, которые после цикли-
зации дали 2,6-диметил-4,8-диокси-л<- и 2,7-диметил-4,5-диокси-п-фенантро-
лины соответственно. Окислением 2,7-диметил-4,5-диокси-м-фенантролина
(XXII) щелочным раствором перманганата получена 4,4'-диокси-6,6'-диметил-
3,3'-дипиридилдикарбоновая-2,2' кислота (XXIII) [60]. Это служит дока-
зательством прохождения ангулярной циклизации, и тем самым исключается
возможность образования соединения линейного строения, подобного диаза-
айтрацену (XXIV).
"Снайдер и Фрейер [581 сообщили о легкости циклизации в дифениловом
эфире этилового эфира о-фенилен-бие-0-амино-а-карбэтоксиакриловой кислоты
с образованием диэтилового эфира 1,8-диокси-о-фенантролиНдикарбоновой-2,7
кислоты. Дуглас и Кермак [54] синтезировали диэтиловый эфир 4,5-диокси-п-,
фенантролиндикарбоновой-3,6 кислоты из м-фенилендиамина и этоксимети-
ленмалонового эфира. Гидролизом и декарбоксилированием этих соединений
получены 1,8-диокси-о- и 4,5-диокси-п-фенантролины.
- Серрей и Катлер [611, применив тот же метод, получили с выходом 70%
4,8-диокси-ле-фенантролйн из ле-фенилендиамина и этоксиметиленмалонового
эфира. При использовании этоксиаллил уксусного эфира выход диокси-ле-
фенантролина составил лишь 10%.
Аналогичные диоксипроизводные о-фенантролина не могут быть легко
получены из о-фенилендиамина и ацетоуксусного эфира вследствие тенденции
обеих аминогрупп вступать в реакцию с одной молекулой сложного эфира
с отщеплением воды или спирта [55, 57, 62—641.
Производные фенантролина, замещенные-в бензольном ядре галогеном,
могли бы, казалось, быть получены из соответствующих производных фени-
лендиамина при проведении двойной реакции Скраупа. Практически, однако,
эти реакции протекают с сильным осмолением и в лучшем случае желательный
продукт получается с очень низким выходом. Поэтому фенантролины, заме-
щенные в бензольном кольце галогеном, следует синтезировать из соответ-
ствующих аминогалогенхинолинов. Этим путем Рихтер и Смит [65] получили
9-хлор- и 9-бром-о-фенантролины из 6-хлор- и 6-бром-8-аминохинолинов.
9-Хлор-о-фенантролин был ранее синтезирован Кушинским и Сухарда [20]
нагреванием 8-нитрохинолина с глицерином и соляной кислотой при 160—
170° под давлением. Образовавшийся из нитрохинолина 8-аминохинолин
280
ФЕНАНТРОЛИНЫ
при хлорировании, по-видимому, превращается в N-хлораминохинолин, кото-
рый изомеризуется в 5-хлор-8-аминохинолин. Из этого соединения должен
образоваться тот же хлор-о-фенантролин, который получается из 6-хлор-8-
аминохииолниа. .Э'-БрамтОтфенаддеоанн синтезирован йак-^из 5^бром-8-амино-
хителвни !15!,"тажгм: из б^броиш'роизводтгого [‘651. Кейс ЛВ6Ч получил при
помощи реакции Скраупа ряд бром-о-фенантролинов с целью использования
их в качестве осадителей в аналитической химии (см. табл. 1).
Таблица 1
Бром-о-фенантролины
о-Феиантролин Применявшийся 8-аминохииолин Второй компонент Выход, %
2-Бром З-Бром Глицерин 20,4
2,10-Днбром 6-Бром а-Бромакролеиндиацетат -1,4
2,9-Днбром 3,6-Дибром Глицерин 28,2
9,10-Ди бром 5,6-Дибром » 14,1
2,9,10-Трнбром 3,5,6-Трибром » 26,9
2,7,9,10-Тетрабром 3,5,6-Трибром а-Бромакролеиндиацетат 4,3
Запатентовано [67] получение 10-хлор- и Ю-бром-ж-фенантролинов
из 4-хлор- и 4-бром-ж-фенилендиамина двойной реакцией Скраупа. Хеуорс
и Сайкес [50] подтвердили возможность такого синтеза для бромопроизводных,
но отметили низкие и неустойчивые выходы (10—30%). Уже упоминавшийся
10-бром-ж-фенантролин может быть синтезирован из 5-амино- или 5-бензолазо-
8-бромохинолина по реакции Скраупа [67]. 10-Хлор-ж-фенантролин получен
нагребанием 5-нитрохинолина при 160—170° с глицерином и концентрирован-
ной соляной кислотой [20]. Эта реакция аналогична приведенной выше для
8-нитрохинолина, причем 8-хлор-5-аминохинолин, вероятно, образуется
в Качестве промежуточного продукта [20].
9-Бром-я-фенантролин может быть синтезирован [67] двойной реакцией
Скраупа из 2-бром-п-фенилендиамина, однако Хеуорс и Сайкес [50] рекомен-
дуют исходить из 8-бром-6-аминохинолина.
Уже упоминалось, что N-окиси м- и n-фенантролинов при действии на них
хлорокиси фосфора дают хлорпроизводные, содержащие атом хлора в a-поло-
жении пиридинового кольца. Более общий метод получения производных
фенантролина с галогеном в а- или у-положении в одном из пиридиновых колец
состоит в обработке соответствующих оксипроизводных галогенокисью фос-
фора или пентагалогенидом фосфора. Этим методом Дуглас и Кермак [54] син-
тезировали 1-хлор-п-фенантролин из 1-оксипроизводного почти с количе-
ственным выходом. С другой стороны, можно использовать N-метилфенантро-
лон, однако в этом случае условия реакции обычно должны быть более жест-
кими. Применяемые при этом реакции сходны с реакциями в ряду пиридина
и хинолина, и поэтому достаточно рассмотреть один-два примера. Первый
пример служит доказательством приведенного выше утверждения, что чет-
вертичные метильные соли м-фенантролина имеют структуру III (но не IV)
(стр. 274).
7-Амино-2-оксихинолин (XXV) по реакции Скраупа дает соединение,
которое из-за его тенденции к ангулярной циклизации следует принять
за 6-окси-ж-фенантролин (XXVI). Обработка его хлорокисью фосфора и пяти-
хлористым фосфором в течение 3 час приводит к 6-хлор-ж-фенантролину
(XXVII). Изомерный 2-хлор-ж-фенантролип (XXX) получается аналогичным
путем из 5-амино-2-оксихинолина (XXVIII) через стадию промежуточного
ОКСИ- И ГАЛОГЕНПРОИЗВОДНЫЕ ФЕНАНТРОЛИНА
281
образования 2-окси-л-фенантролина (XXIX). При окислении сульфометилата
I II I II
N N I N N I N
НО—/ NH2 НО—/ V'4/ Cl— /W
I II I —> I II I I I.
\/\s \/\z
XXV XXVI XXVII
z/\_орг Г1
I Н ОН . Ц-С1
nh2
XXVIII XXIX XXX
л-фенантролина щелочным раствором феррицианида [37, 461 образуется
метил-лгфенантролон, имеющий строение XXXI или XXXII в соответствии
с принимаемыми для четвертичных солей структурами III или IV (стр. 274).
XXXI XXXII
При действии на этот метилфенантролон хлорокиси фосфора и пятихло-
ристого фосфора при температуре 150—160° в течение 10 чар образуется
2-хлор-л£-фенантролин, идентичный синтезированному из 5-амино-2-окси-
хинолина, но отличающийся от изомерного соединения, полученного из 7-ами--
но-2-оксихинолина. Поэтому М-метил-л-фенантролон должен представлять
собой 1 -метил- 1,2>дигидро-.и-фенантролинон-2 (XXXI), откуда следует, что
у четвертичных солей .«-фенантролина метильная группа находится у атома
азота в положении 1.
Другие примеры, иллюстрирующие введение атома хлора в фенантроли-
новое ядро при действии на оксифенайтролин или метилфенантролон хлорокиси
фосфора или пятихлористого фосфора, служат доказательством строения
хлорфенантролинов, полученных действием хлорокиси фосфора на N-окиси.
Хлор-л-фенантролин, полученный из N-окиси л-фенантролина, идентичен
2-хлор-лгфенантролину (XXX), упомянутому в предыдущем разделе, но отли-
чается от б-хлор-л-фенантролина [49].
2-Хлор-л-фенантролин получен как из 2-окси-л-фенантролина, так и из
1-метил-л-фенантролона-2 [45]. Он идентичен хлор-л-фенантролину, выде-
ленному из продуктов реакции N-окиси л-фенантролина с хлорокисью фос-
фора [45].
Наконец, 2,7-дихлор-л-фенантролин был получен действием хлорокиси
фосфора и пятихлористого фосфора на 1,8-диметил-1,2,7,8-тетрагидро-л-фе-
нантролиндион-2,7 (стр. 276). Это соединение оказалось идентичным дихлор-
л-фенантролину, полученному действием хлорокиси фосфора на ди-И-окись
л-фенантролина [45].
Как и следовало ожидать, атомы галогена в а- или у-положении пиридино-
вого кольца фенантролина обычно легко замещаются нуклеофильными труп-
282
ФЕНАНТРОЛИНЫ
пами, такими, как алкил О — или RNH—. Атом галогена в бензольном коль-
це или в 0-положении пиридинового кольца менее реакционноспособен и, как
правило, может быть замещен только в значительно более жестких условиях.
НИТРО- И АМИНОФЕНАНТРОЛИНЫ
Согласно Хеуорсу и Сайкесу [50], ни м-, ни n-фенантролин не нитруются
и после обработки концентрированной или дымящей азотной кислотой в при-
сутствии концентрированной серной кислоты при 20 или 100° выделяются
неизмененными. Друи и Шмидт [30] при обработке .«-фенантролина смесью
дымящей азотной и серной кислот получили с выходом 8% Ю-нитро-л-фенан-
тролин. Не удалось также получить нитрофенантролин двойной реакцией
Скраупа из 2-нитро-л-фенилендиамина или 4-нитро-л-фенилендиамина 168],
однако, согласно Коржинскому и Брайдоуну [69], Э-нитро^-фенантролин
образуется из 5-нитро-л-фенилендиамина в результате двойной реакции
Скраупа.
о-Фенантролин с определенным трудом может быть пронитрован [70]
смесью серной и дымящей азотной кислот при 100° с образованием мононитро-
производного. Холкрау и Кермак [12] доказали, что нитрогруппа вступает в по-
ложение 9 (или 10) и получили то же соединение из 5-нитро-8-аминохинолина
реакцией Скраупа [12]. Однако синтезировать это соединение из 4-нитро-о-
фенийендиамина двойной реакцией Скраупа им не удалось.
Смиту и Кэглу [28] удалось добиться выхода при нитровании о-фенантро-
лина более 90%, проводя реакцию при 165° с концентрированной азотной
кислотой (уд. вес 1,42) и 15—25%-ным олеумом. Из маточного раствора после
стояния в течение 15 суток было выделено небольшое количество желтых
пластинок, анализ и свойства которых почти с несомненностью доказывают,
что это фенантролин-9,10-хинон. Согласно Рихтеру и Смиту [65], 9-метил-
о-фенантролин нитруется легче, чем о-фенантролин, причем образуется, по-ви-
димому, 9-метил-10-нитро-о-фенантролин.
9-Амино-ж-фенантролин может быть получен восстановлением соответ-
ствующего Э-нитро-л-фенантролина хлоридом олова (II) [7Г]. Однако удобный
метод синтеза Ю-амино-л-фенантролина и 9-амино-л-фенантролина восста-.
новлением соответствующих нитропроизводных по существу отсутствует,
ввиду малой доступности последних. Эти аминосоединения образуются при
обработке соответствующих бромфенантролинов аммиаком в запаянной трубке
в присутствии меди в качестве катализатора [50, 71 ]. Ю-Амино-л-фенантролин
также получен Хеуорсом и Сайкесом нагреванием Ю-окси-л-фенантролина
с аммиаком в течение .15 час при 210—220° [50, 72].
При обработке 9-амино-п-фенантролина 0-диэтиламиноэтилхлоридом
в толуоле в присутствии амида натрия по методу Дьюара [73] получают
9-(0-диэтиламиноэтиламино)-л-фенантролин. В этих условиях удается избе-
жать образования четвертичных солей, которые получаются при присоеди-
нении диэтиламиноалкилхлорида к пиридиновым атомам азота, что происхо-
дит, очевидно, когда аминофенантролин обрабатывают непосредственно
диэтиламиноэтилхлоридом [74].
Выше отмечалось (стр. 281), что атомы галогена в а- или у-положении
пиридинового кольца фенантролина легко замещаются нуклеофильными
реагентами, и в том числе первичными аминами. Этим путем получают раз-
личные производные, главным образом диалкиламиноалкиламинопроизводные,
которые представляют интерес ввиду их возможного химиотерапевтического
значения. Типичными представителями этого класса соединений являются
следующие: 3-(2-диэтиламиноэтиламино)-о-фенантролин [12], 1-(4-диэтил-
амино-1 -метилбутиламино)-10-метокси-о-фенантролин [58 ], 1,8-бис-(4-диэтил-
амино- 1-метилбутиламино)-о-фенаитролин [58], 8-(2-диэтиламиноэтиламино)-
ФЕНАНТРОЛИНКАРБОНОВЫЕ КИСЛОТЫ И СЛОЖНЫЕ ЭФИРЫ 283
6-метил-л«-фенантролин [37], 4-(4-диэтиламино-1-метилбутиламино)-2-метил-
.и-фенантролин [37], 6-(2-диэтиламиноэтиламино)-8-метил-.и-фенантролин [37],
4-(2-диэтиламиноэтиламино)-2-метил-л-фенантролин [53], 2-(4-диэтиламино-
1-метилбутиламино)-л-фенантролин [31 ], 4-(3-диэтиламинопропиламино)-9-
хлор-п-фенантролин [54], 4-(4-диэтиламино-1-метилбутиламино)-л-фенантро-
лин [54], 1,10-ди-(3-диэтиламинопропиламино)-л-фенантролин [54], 4-(3-ди-
этиламино-2-оксипропиламино)-л1-фенантролин [61 ], 8-(3-диэтиламино-2-окси-
пропиламино)-4-окси-л(-фенантролин [61 ], 8-(2-оксиэтиламиноэтиламино)-4-
окси-л-фенантролин [61] и 4,8-бис-(3-диэтиламино-2-оксипропиламино)-л-
фенантролин [61L
ФЕНАНТРОЛИНАЛЬДЕГИДЫ
л-Фенантролин-2- и лгфенантролин-8-, а также л-фенантролин-4-альдегид
получены окислением соответствующих метилфенантролинов Двуокисью селе-
на [75]. Однако 1-метил-о-фенантролин, хотя и восстанавливал двуокись
селена до селена, но альдегид при этом выделить не удалось.
ФЕНАНТРОЛИНКАРБОНОВЫЕ КИСЛОТЫ И СЛОЖНЫЕ ЭФИРЫ
Карбоновые кислоты и сложные эфиры фенантролинов не заслуживают
подробного рассмотрения. Фенантролинкарбоновые кислоты получаются окис-
лением метилфенантролинов. Так, например, лгфенантролинкарбоновая-2
и л-фенантролинкарбоновая-10 кислоты [77] синтезированы соответственно
из 2- и Ю-метил-л-фенантролинов. Однако Рихтер и Смит [65] не смогли
окислить 9-метил-о-фенантролин в о-фенантролинкарбоновую-9 кислоту.
Сайкес [39] описал синтез л-фенантролинкарбоновой-2 кислоты броми-
рованием 2-метил-п-фенантролина с последующим гидролизом. Попытки
получить эту кислоту реакцией Рейсерта оказались безуспешными.
Большая часть известных фенантролинкарбоновых кислот содержит
карбоксильную группу в одном из пиридиновых колец. Эти кислоты полу-
чают одним из мётодов синтеза хинолинов, позволяющих ввести карбоксиль-
„ную или карбэтоксильную группу в нужное положение. На стр. 277 уже
приводились примеры получения оксифенантролинкарбоновых кислот и слож-
ных эфиров из аминохинолинрв и щавелевоуксусного, а также этоксиметилен-
малонового эфиров. Сарри и Катлер [61] показали, что динатриевая соль, полу-
ченная омылением диэтилового эфира 4,8-диокси-л-фенантролиндикарбоновой-
3,7 кислоты под действием соляной кислоты переходит в нерастворимую
мононатриевую соль. Эта соль не изменяется при действии горячей 3 н. соля-
ной кислоты. Гидролиз диэфира разбавленной соляной кислотой приводит
к образованию дикарбоновой кислоты с количественным выходом. Об исполь-
зовании метода синтеза хинолинов с пировиноградной кислотой (метода
Дёбнера) упоминалось выше (см. стр. 267). Другими примерами являются
получение б-метил-л-фенантролинкарбоновой-8 кислоты из 5-аминохинолина
[1] и 2-метил-л-фенантролинкарбоновой-4 кислоты из 6-аминохинолина [2].
Аллан [78] сообщил о синтезе 10-метил-2,6-дифенил-.и-фенантролинди-
карбоновой-4,8 кислоты из 4-амино-5-метил-7-изатинглиоксалевой кислоты
и ацетофенона путем кипячения водного, раствора основания с обратным холо-
дильником. В качестве побочного продукта при этом образуется некото-
рое количество 2-фенил-4-карбокси-5-амино-6-метилхинолин-8-глиоксалевой
кислоты.
Борше и Вагнер-Рёммих применили к аминохинолинам видоизмененную
реакцию Дёбнера с пировиноградной кислотой. Так, например, нагреванием
8-аминохинолина с бензальдегидом и фенилпировиноградной кислотой синте-
зирована 2,3-дифенил-о-фенантролинкарбоновая-1 кислота [79]; из 7-амино-
2-фенилхинолина, бензальдегида . и пировиноградной кислоты получена
284
ФЕНАНТРОЛИНЫ
2,6-дифенил-Л1-фенантролинкарбоновая-4 кислота, которая при нагревании
с медной бронзой декарбоксилировалась с образованием 2,6-дифенил-л(-фе-
нантролина [80].
Пример синтеза п-фенантролиндикарбоновой-4,5 кислоты приводят Вуд-
рюф и Адамс [81], которые обрабатывали пировиноградной кислотой в кипя-
щем спирте полученное из n-фенилендиамина и бензальдегида двойное основа-
ние Шиффа. Полученная при этом 2,7-дифенил-п-фенантролинкарбоновая-4,5
кислота (XXXIII) с выходом 42—50% окислением щелочным раствором пер-
манганата калия была превращена в 6,6'-дифенил-3,3'-дипиридил-2,4,2',4'-
тетракарбоновую кислоту (XXXIV). В соединении XXXIV имеет место затруд-
нение вращения одного цикла относительно другого. Отсюда следует, что,
когда два пиридиновых кольца находятся в одной плоскости, а атомы азота
расположены по одну сторону связи, соединяющей оба кольца, карбоксильные
группы оказывают влияние друг.на друга. Поэтому возможно, что в исходном
соединении — фенантролиндикарбоновой-4,5 кислоте — две карбоксильные
группы расположены настолько близко, что оказывают взаимное влияние.
В связи с этим кажется вероятным, что у этого соединения возможны некото-
рые деформации молекулы, выражающиеся либо в нарушении копланар-
ности, либо в изменении углов между связями.
ноосх^х/свн6
НООС I ||
XXXIII
НООС\У\/СбН5
НООС I II
I I N
^\/\/
II СООН
XXXIV
РАЗЛИЧНЫЕ ДИАЗАФЕНАНТРЕНЫ
Мизани и Богерт [57] конденсировали 5-аминоизохинолин с ацетоуксус-
ным эфиром, и полученный аминокротонат при нагревании в дифениловом
эфире до 250° превращался в 6-метил-8-окси-2,5-диазафенантрен (XXXV),
из которого действием хлорокиси фосфора и пятихлористого фосфора полу-
чали 8-хлорпроизводное. Удаление атома хлора каталитическим восстановле-
нием дало 6-метил-2,5-диазафенантрен (XXXVI). Другое производное 2,5-ди-
азафенантрена синтезировано Борше и Вагнер-Рёммихом [79], применявшими
XXXV
видоизмененную реакцию Дёбнера с пировиноградной кислотой. При нагрева-
нии 5-аминоизохинолина с бензальдегидом и фенилпировиноградной кисло-
той образуется 6,7-дифенил-2,5-диазафенантролинкарбоновая-8 кислота, кото-
рая при декарбоксилировании превращается в 6,7-дифенил-2,5-диазафе-
нантрен.
2,7-Диазафенантрен получен Рэггли и Шетти [82] следующим путем:
терефталевый альдегид конденсировали с аминодиэтилацеталем и полученное
РАЗЛИЧНЫЕ ДИАЗАФЕНАНТРЕНЫ
285
фталилиденовое соединение (XXXVII) восстанавливали в п-ди-(0,0-диэтокси-
этиламинометил)бензол (XXXVIII). При обработке 33%-ным олеумом отщеп-
ляется 4 молекулы этанола и 4 атома водорода и с небольшим выходом обра-
зуется 2,7-диазафенантрен (XXXIX).
XXXVII XXXVIII
Берг и Петров [83], применив перегруппировку Штиглица, синтезировали
2,10-диазафенантрены. Из 1,3-диметил-2-азафлуоренона-9 и фенилмагнийброми-
да получен 1,3-диметил-9-фенил-2-азафлуоренол-9 с выходом 52%. Пятихло-
ристый фосфор в толуоле превращает это соединение в 9-хлорпроизводное,
которое под действием амида калия в жидком аммиаке дает соответствующее
9-аминопроизводное. При окислении 1 н. гипохлоритом калия и последующем
подкислении соляной кислотой образуется 9-хлораминопроизводное, которое
286
ФЕНАНТРОЛИНЫ
действием безводного метилата натрия в пиридине превращают в 1,3-диметил-
9-фенил-2,10-диазафенантрен с 20%-ным выходом. Ход синтеза показан
на приведенной на стр. 285 схеме.
Хаузер и Рейнольдс [84] описали различные способы получения 3- и
4-аминохинолинов: с применением этоксиметиленмалонового эфира, а также
методов Конрада — Лимпаха и Кнорра. Первичные продукты конденсации
образовывались из обоих аминохинолинов легко (о невозможности осуще-
ствить с 4-аминопиридином первую ступень синтезов Конрада — Лимпаха
и Кнорра см. стр. 168). Циклизация промежуточных продуктов, полученных
из 4-аминохинолина, происходит легче, чем циклизация аналогичных продук-
тов из 3-аминохинолина. Так, например, из 4-аминохинолина и этоксимети-
ленмалонового эфира был получен этиловый эфир 1-окси-4,9-диазафенантрен-
карбоновой-2 кислоты (XL) с выходом 92%.
В противоположность этому выход диазаантрацена (XLI) составил всего
22%. Аналогичным образом амид, полученный из 4-аминохинолина и ацето-
уксусного эфира дал З-окси-1-метил-4,9-диазафенантрен (XLII) с выходом
58%, тогда как соответствующий амид из 3-аминохинолина не удалось зацик-
лизовать. Далее, 4-аминохинолин и ацетоуксусный эфир в присутствии ката-
литических количеств кислоты при комнатной температуре образует 1-окси-
З-метил-4,9-диазафенантрен (XLIII), между тем циклизация промежуточного
продукта, полученного из 3-аминохинолина, в соединение XLIV требует
нагревания. По аналогии с образованием 1,5-нафтиридинов из 3-аминопири-
дина (стр. 163) соединениям XLI и XLIV приписана линейная структура.
Однако, учитывая большую тенденцию к образованию ангулярных структур,
о чем уже упоминалось выше (стр. 268), может оказаться, что эти соединения
являются 1,9-диазафенантренами.
/V СООС2Н5
У\А/^0Н
ии
х N
ОН
XL1
XL
ОН
XLII
СН3
XLIII
он
XLIV
XLV
КОМПЛЕКСЫ 2,2'-ДИПИРИДИЛА И О-ФЕНАНТРОЛИНА С МЕТАЛЛАМИ
287
О возможности использования упоминавшейся выше реакции сообщает
Девис [85]. Проводя реакцию Скраупа с 4-амино-2-фенилхинолином, он полу-
чил 10-фенил-4,9-диазафенантрен (XLV):
Таким,, образом, реакция 4-амино-2-фенилхинолина о. этоксиметилен-
малоноЕым эфиром проходила несколько иначе, чем с 4-аминохинолином
у Хаузера и Рейнольдса. Ниже приведена схема реакций.
NH2
| II | + С2Н5ОСН=С(СООС2Н5)2
С°Нб
N
/СН
NH
С(СООС2Н5)2
— I If Lc«H
CH—NHX /=\
NH — СО—А
I I г n/
J сосен, iH5
H,c.J II '
Нагревание
КОМПЛЕКСЫ 2,2 -ДИПИРИДИЛА И о-ФЕНАНТРОЛИНА С МЕТАЛЛАМИ
Уже упоминалось о способности о-фенантролина образовывать комплекс-
ные соединения с ионом двухвалентного железа и ионами других металлов
[86]. С этой точки зрения о-фенантролин очень напоминает 2,2'-дипиридил,
поэтому при изучении комплексов о-фенантролина с металлами не следует
забывать и бб аналогичных соединениях дипиридила.
Как дипиридил, так и о-фенантролин образуют стабильные комплексные
соли с различными переходными металлами общей формулой М (фен)3Х2-уН2О
(где фен — о-фенантролиц). Металлы имеют координационное число 6, и, таким
образом, координационные связи полностью насыщены тремя молекулами
бидентата 2,2'-дипиридила (дипи) или о-фенантролина. Соли эти боль-
шей частью окрашены. Так, например, никельсодержащее соединение
[М1(фен)3]Вг2-7Н2О кристаллизуется в форме розовато-красных табличек,
а кобальтовая соль [Со(фен)3]С12-8Н2О образует бледно-желтые таблички;
[Ru (дипи)3]Вг2-6Н2О кристаллизуется в виде розовых листков. Наиболее
интенсивно окрашены соли двухвалентного железа, как, например,
[Ре(феи)3]Вг2-7Н2О, которая кристаллизуется в . виде призм красного
цвета.
Как дипиридильные, так и о-фенантролиновые комплексы ионов двух-
валентного железа обладают в водном растворе интенсивной красной окраской.
288
ФЕНАНТРОЛИНЫ
Это дает возможность определять следы Fe2+ при концентрациях порядка
1 ч. на 1 млн. ч. Такие ионы являются красителями, окрашивающими непро-
травленный шелк и шерсть в розово-красный цвет и обладают значительной
устойчивостью; холодные разбавленные кислоты и щелочи на них не действуют,
однако оии разрушаются более концентрированными горячими растворами.
Строение и оптическая активность комплексов с металлами. Установ-
лено, что устойчивые формы таких соединений, как дифенил или дипиридил,
состоят из двух лежащих в одной плоскости ароматических колец, причем
углы между связями близки к 120°.
2,2'-Дипиридил известен в двух устойчивых конфигурациях: с атомами
азота, расположенными по одну сторону (цис-форма, I) и по обеим сторонам
(транс-форма, II) центральной оси. Совершенно очевидно, что в образовании
комплекса участвует цис-форма, геометрически сходная с о-фенантролином
(III). Действительно, если допустить, что все валентные углы равны 120°,
Ч___Ч Ч__________________/N=\ Ч______________У Ч
\=NZ 4N=/ 4=N/ 4=N/ XN = Z
I II III
а длины всех связей одинаковы, то группа — СН = СН —, необходимая
для превращения цис-2,2'-дипиридила (I) в о-фенантролин (III), может быть
введена без нарушения конфигурации остатка молекулы дипиридила. По-ви-
димому, оба эти предположения не очень строго обоснованы, но они достаточ-
ны, чтобы показать весьма близкое сходство координационной способности
обеих молекул.
Имеется достаточно доказательств того, что шесть координационных свя-
зей вокруг атомов таких металлов, как железо или платина, расположены
октаэдрически вдоль трех общих координационных осей (IV). Если мы пред-
ставим, что все связи имеют равную длину, то концы их займут вершины пра-
вильного октаэдра. В некоторых случаях удобно принять, что октаэдры ориен-
тированы, как показано на схеме V. Оба атома азота любой координационно
связанной молекулы дипиридила или фенантролина, естественно, займут
смежные вершины. Таким образом, три органические молекулы могли бы
занять соответственно положения АА', ВВ’ и СС (Via). Совершенно очевидно,
что образующийся комплексный ион имеет элемент асимметрии и не идентичен
своему зеркальному изображению. У энантиоморфного соединения три моле-
кулы будут занимать соответственно положения. Я С', ВА’ и СВ1, как показано
КОМПЛЕКСЫ 2,2'-ДИПИРИДИ Л А И о-ФЕНАНТРОЛИНА С МЕТАЛЛАМИ
289
на схеме VI6. Отсюда следует, что если эти комплексные ионы достаточно
4 устойчивы, то они могут быть расщеплены на оптически активные формы.
В некоторых случаях это расщепление было осуществлено фракционной кри-
сталлизацией их солей с помощью оптически активной кислоты,
Так, например, Вернер [87] кристаллизацией тартрата и превращением
его в бромид получил левовращающий трис-2,2'-дипириднлбромид железа (II)
[Ре(дипи)31Вг2-6Н2О, [а I лампа Нернста = — 520°. трыс-2,2'-Дипириднлхло-
рид никеля аналогичным путем был расщеплен Морганом и Бёрсталлом
[88]: для правовращающего и левовращающего изомеров [а]”^ =+529°
и —501° ([М] = + 3735° и —3536°). Соли железа и никеля практически
теряют свою оптическую активность после 2-часового пребывания в водном
растворе. Интересно, что трыс-2,2'-дипиридилрутенийхлорид [Ки(дипи)3]Вг2-
•6Н2О, также разделенный Бёрсталлом [86] оказался значительно устойчи-
вее. Найдено, что для него [а]^ =+860° и —815° (]7И] = + 7205°
и —6828°). Эта соль сохраняет свою оптическую активность при комнатной
температуре неограниченное время не только в твердом состоянии, но ив 0,1%-
ном растворе. Оптическая активность не исчезает после перекристаллизации
из горячей воды или при кипячении водного раствора в течение нескольких
минут.
Особые свойства комплексов железа и рутения. Атом железа взаимодей-
ствует с дипиридилом и о-фенантролином не только в двухвалентном, но и в
трехвалентном состоянии. Бляу [6] установил, что темно-красный раствор
комплекса о-фенднтролина и двухвалентного железа ,[(фен)3Ре]2+, для удобства
называемого ферроином, при окислении приобретает синюю окраску, но менее
интенсивную, чем у комплекса трех валентного железа. Синий о-фенантроли-
новый комплекс трехвалентного железа [(фен)3Ре]3+ «феррин» не может быть
получен взаимодействием о-фенантролина с ионами Fes+. Согласно Бляу [6]
о-фенантролин образует плохо охарактеризованное соединение с ионами
трехвалентного железа. Гейнс, Гаммет и Вальден [90] выделили кристалличе-
ское соединение коричневого цвета, которому они приписали следующее
строение.
Если красные ионы Fe2+ и светло-синие ионы Fe3+ находятся в равнове-
сии, то, казалось, можно было бы измерить окислительно-восстановительный
потенциал. Однако провести эти измерения с дипиридильными соединениями
не удалось, по-видимому, вследствие неустойчивости образующегося комп-
лекса с ионами трехвалентного железа. Для о-фенантролин-железного комп-
лекса измерить точный окислительно-восстановительный потенциал легче;
величина его по отношению к нормальному водородному электроду равна
1,06 в [911. Вальден и Эдмондс [92] приводят слегка завышенную величину
1,14 в.
Свойства ферроина, сообщающие ему особую ценность как окислитель-
но-восстановительного индикатора в аналитической химии, следующие:
1. Относительно высокий окислительно-восстановительный потенциал вслед-
ствие фактической неспособности окисляться под действием окисляющего
реагента, до тех пор пока более легко окисляющееся Исследуемое соединение
19 Заказ М 978
290
ФЕНАНТРОЛИНЫ
практически не окислится полностью. 2. Окраска изменяется от темно-крас-
ной к светло-синей или, при малых концентрациях, от красной к бесцветной.
Ли, Кольтгофф и Леуссинг [93] измерили константы диссоциации феррои-
на и феррина и определили константу скорости реакции [Ре(фен)3]2+ =
= Fe2+ + 3(фен), равную 5-10~22, которая у феррина зависит от концентрации
присутствующей серной кислоты и изменяется от 8-10-15 в 0,05 М серной кис-
лоте до 7,5-10~21 в 8 М серной кислоте.
Рутений занимает в периодической системе положение, очень близкое
к железу, и поэтому можно было ожидать, что его комплексы с 2,2'-дипирйди-
лом и о-фенантролином будут напоминать комплексы железа и смогут приме-
няться в аналитической химии. Комплекс хлорида рутения (II) и 2,2'-дипири-
дила [Ки(дипи)3]С12-Н2О, исследованный Бёрсталлом [89], а также Штейг-
маном, Бирнбаумом и Эдмондсом [94], имеет окислительно- восстановительный
потенциал, равный 1,33 в. Концентрированные растворы окрашены в оран-
жево-красный цвет, который при окислении изменяется до зеленого. У раз-
бавленных растворов окраска изменяется от желтой до бесцветной. Относи-
тельно слабо выраженное изменение цвета отрицательно влияет на примене-
ние его в качестве индикатора окисления. Комплексы о-фенантролина с ионами
двухвалентного рутения, по-видимому, пока еще недостаточно исследованы.
Комплексы с другими металлами. Способность этих органических моле-
кул взаимодействовать с металлами не ограничена металлами переходной
группы. Пфейфер и Кристелейт (95) описали следующие соли: [Ы(фен)]С1О4,
[Ыа(фен)2]С1О4-ЗН2О, [А§(фен)2]ЫО3-Н2О, [Т1(фен)2]ЫО3-2,5Н2О и
[Са(фен)4] (С1О4)2-ЗН2О. Медь и серебро образуют комплексы как в одно-
валентном, так и в двухвалентном состоянии. Дипиридилтиоцианат меди
Си(дипи)(БСЫ)2 окрашен в светло-зеленый цвет, а после восстановления
бисульфитом натрия образует оранжево-красную медную соль Си(дипи)БСЫ
[96]. При взаимодействии двух молекул о-фенантролина с азотнокислым сереб-
ром в растворе образуется бесцветный желеобразный осадок, который под
действием персульфата аммония переходит в стабильный ди-о-фенантролин-
персульфат серебра [А§(фен.)2]Б2О8 коричневого цвета. Из раствора этой
соли в 64%-ной азотной кислоте могут быть осаждены различные соли, как,
например, перхлорат [А§(фен)2](С1О4)2 [8]. Морган и Бёрсталл [97] описали
аналогичные устойчивые серебряные комплексы с дипиридилом, а также
с трипиридилом (см. следующий раздел). Неустойчивый ион серебра стабили-
зируется при образовании комплексов с органическими молекулами.
Соединения, родственные о-фенантролину и дипиридилу. Как и следо-
вало ожидать, комплексообразующие свойства проявляются также и у ряда
простых производных о-фенантролина. Так, например, 9-хлор-, 9-бром-,
Таблица 2
Окислительно-восстановительные потенциалы комплексов металлов
Соединение Е° комплекса с
железом рутением хромом
о-Фенантролин 1,06 [98] 0,77
9-Метилфенантролин 1,02 [98]
9-Хлорфенантролин . 1,12 [98]
9-Бромфенантролин 1,12 [98]
9-Нитрофенантролин 1,25 [98]
9-Нитро-10-метилфенантролин 1,23 [98]
2,2'-Дипиридил 0,97 1,33 [94]
КОМПЛЕКСЫ 2,2'-ДИПИРИДИЛА И о-ФЕНАНТРОЛИНА С МЕТАЛЛАМИ
291
9-метил- и 9-нитро-о-фенантролины >образуют темно-красные комплексы
с ионами Fe2+. Окислительно-восстановйтельные потенциалы этих комплекс-
ных ионов приведены в табл. 2. Эти соединения исследовались с целью нахож-
дения аналитического реагента, превосходящего по своим свойствам о-фенан-
тролин. Однако, по-видимому, за исключением 9-метил- и 9-нитропроизвод-
ных, ни одно из этих соединений не представляет ценности как реагент в ана-
литической химии. 9-Нитро-о-фенантролинсульфат железа (II) известен под
названием нитроферроин. Он применяется для титрования в тех случаях,
когда полезен относительно высокий окислительно-восстановительный
потенциал.
Как и 2,2'-дипиридил, способность образовывать внутрикомплексные
соли проявляют 2,2',2"-трипиридил [2,6-бис-(2-пиридил)пиридин, трипи, VII]
и 2,2',2'',2'"-тетрапиридил [6,6'-бис-(2-пиридил)-2,2'-дипиридин, тетрапи,
VHI], которые ведут себя соответственно как тридентатные и тетрадентатные
основания. Трипиридил дает соли состава [(трипи)2М]Х2-1/Н2О или
[(трипи)2М]Х3, где М— двух- или трехвалентный элемент из числа пере-
ходных, как, например, железо (II), рутений, осмий (II), кобальт (II и III),
никель или хром. Уже были высказаны соображения, которые позволяют
с уверенностью утверждать, что в образовании комплекса участвует цис-
изомер с тремя атомами азота, расположенными по одну сторону. Комплекс-
ные ионы имеют структуру IX, у которой три смежные вершины октаэдра,
лежащие в одной плоскости, проходящей через центр, координируются
с тремя атомами азота одной молекулы трипиридила. Если это действитель-
но так, то образующийся ион не будет асимметрическим, что подтверждается
отсутствием расщепления красного рутениевого комплекса [(трипи)2Ии]2+
при кристаллизации тартрата [99].
Тетрапиридил (VIII) также образует комплексы с различными ионами
металлов и обычно ведет себя, как тетрадентатное основание. Морган и Бёр-
сталл [99] получили следующие соли: [Ее(тетрапи)15О4-4Н2О, красно-корич-
невые иглы; [Ее(тетрапи)]Вг2-2Н2О, зеленые иглы или черные ромбы;
[Со(тетрапи)1С12-2Н2О розовые листочки; [СоС12(тетрапи)]С1-ЗН20, серебри-
стые таблички; [2п(тетрапи)]С12-2Н2О, бледно-желтые листочки. Весьма
вероятно, что все четыре пиридиновых кольца лежат в одной плоскости,
а четыре атома азота связаны с расположенным в центре атомом металла
четырьмя копланарными валентными связями. Относительная жесткость
плоской структуры тетрапиридила делает вероятным некоторое искривление
координационных валентных связей металла [99].
6,6'-Диметилдипиридил (X), синтезированный Виллинком и Вибо [100],
не дает окраски с ионами двухвалентного железа [101]. Позднее Кэгл и Смит
[102] исследовали реакцию между изомерными 3,3'-, 4,4'- и 5,5*-диметил-
19*
292
ФЕНАНТРОЛИНЫ
2,2*'дипиридилами (XI — XIII) и ионами двухвалентного железа. Они про-
вели количественные измерения коэффициентов экстинкции и получили сле-
дующие величины: 1770 (525 лр), ,8470 (529 лр) и 8300 (510 лр). Соответ-
ствующая величина для самого 2,2'-дипиридила составляет 8650 (522 жр).
XII Х111
Обе метильные группы в положениях 4,4' или 5,5' не оказывают влияния
на образование комплексов с Fe2+ или на интенсивность полос поглощения.
Метильные группы в положении 3,3' весьма значительно ослабляют поглоще-
ние света, а также уменьшают интервал pH, вне которого комплекс устойчив.
Для 4,4'- и 5,5'-соединений этот интервал лежит в пределах pH от 3 до 8,
а для соединения 3,3* от 4 до 6.
Эти данные могут быть объяснены стерическими факторами. Едва ли воз-
можно без некоторого напряжения существование планарной конфигурации
молекулы дипиридила с двумя атомами азота в tfuc-положении, когда обе
метильные группы находятся в положении 3,3'. Поэтому у 3,3'-диметил-
2,2'-дипиридила можно ожидать некоторого искривления плоской конфигу-
рации и известного уменьшения межъядерного резонанса. Уменьшение
устойчивости комплексного иона и ослабление интенсивности его окраски
является нормальным следствием нарушения конфигурации молекулы.
Метильные группы в 4,4'- и 5,5'-диметилпроизводных расположены таким
образом, что они не вызывают стерических препятствий ни‘у свободной моле-
кулы дипиридила, ни у комплексного иона. У 6,6'-изомера обе метильные
группы в свободной молекуле не могут оказывать влияния друг на друга,
тогда как в комплексном ионе на обоих его концах сосредоточено по три
метильные группы. Взаимное пространственное влияние метильных групп,
по-видимому, создает препятствие для сближения всех трех молекул дипи-
ридила с ионом Fe2+ и образования стабильного комплекса.
Подобными стерическими влияниями можно объяснить неспособность
2,2'-дихинолила (XIV) образовывать окрашенный комплекс с ионами Fe2+.
Смирнов [103] установил, что 2-(2'-пиридил)хинолин (XV) также неспособен
лавать комплексы с ионами двухвалентного железа.
XIV XV
Это позволяет предположить, что наличие двух бензольных колец на одном
конце комплексного иона может вызвать пространственные затруднения.
Другим объяснением неспособности 2,2'-дихинолина и 2-(2'-пиридил)хино-
лина давать окрашенные комплексы с ионами Fe2+, по мнению Смирнова
[103], является понижение основности и уменьшение комплексообразующей
способности атомов азота под влиянием ненасыщенных связей смежного бен-
зольного кольца.
КОМПЛЕКСЫ 2,2'-ДИПИРИДИЛА И о-ФЕНАНТРОЛИНА С МЕТАЛЛАМИ
293
Согласно Бреккенриджу, Льюису и Квику 1104], 2,2'-дихинолил обра-
зует устойчивые комплексы с хлоридами меди (II) и кобальта (II) —
Ci8Hi2N2CuC12 и C18H12N2CoC12. Первый представляет собой оранжево-крас-
ный микрокристаллический порошок, а второй — блестящие серо-зеленые
листочки. С ионом одновалентной меди 2,2'-дихинолил дает интенсивную
пурпурную окраску, появление которой обусловлено, по-видимому, образо-
ванием комплекса. Выделена также не совсем чистая коричневато-зеленая
микрокристаллическая соль, вероятно, состава (C18H12N2)2CuC1. Цветная
реакция с дихинолилом весьма характерна для ионов одновалентной меди
и чувствительность ее составляет 1 : 108. Если растворы соли меди (I) в раз-
бавленной соляной кислоте, уксусной кислоте, спирте или диоксане смешать
с растворами основания, то жидкость окрашивается в темно-пурпурный цвет.
Склонность к образованию комплексов с медью очень велика, о чем можно
судить по тому, что окрашивание появляется при соприкосновении металли-
ческой меди со спиртовым раствором основания, а также при контакте в тече-
ние некоторого времени соединения меди со спиртом. Длительное экспониро-
вание на солнечном свету, кипячение в течение нескольких часов или проду-
вание через раствор воздуха в течение нескольких дней не оказывают замет-
ного действия на окраску.
Комплексы, содержащие С1, СО и NO. У большинства известных соеди-
нений координационные валентности металла все направлены к атому азота
о-фенантролина, дипиридила или любого другого органического соединения.
Исключение составляет тетрапиридил в сочетании с хлоридом кобальта.
В этом соединении [СоС12(тетрапи)1С1 четыре из шести координационных
валентностей кобальта направлены к тетрапиридилу, а две — к атомам хлора.
Другие комплексы тетрапиридила с общей формулой М(тетрапи)Х2 могут
также включать в качестве координационных компонентов атомы хлора, однако
тетраэдрическая конфигурация, по мнению Моргана и Бёрсталла [97], менее
вероятна, чем планарный координационный комплекс типа [М(тетрапи)1Х2.
Описан ряд комплексных ионов, содержащих металл, координационно
связанный не только с дипиридилом, но и с СО или NO. Так, например, Морган
и Бёрсталл показали, что калийнитрозорутенийпентахлорид KalRuNOCU]
дает с 2,2'-дипиридилом темно-зеленые кристаллы комплекса [RuNOCl3(flHnH)]
или оранжево-коричневую соль [RuNOCl(flHnH)2][RuNOCl5] в зависимости
от условий реакции.
Известны комплексы, содержащие СО и о-фенантролин, отличающиеся
от уже упомянутых тем, что в них входит не ион, а нейтральный атом металла.
Так, например, Хибер и Мюльбауэр [105] показали, что при легком нагрева-
нии пентакарбонила железа Fe(CO)5 и о-фенантролина в ацетоне при темпера-
туре 60° появляется синее окрашивание; при 65—70° выделяется СО, окрас-
ка переходит в красную и выпадает осадок желтовато-коричневого соединения
состава Ее(СО)3(фен). Подобным образом карбонил никеля Ni(CO)4 и о-фе-
нантролин в пиридине, ацетоне или безводном спирте дают относительно устой-
чивый комплекс №(СО)3(фен) в виде рубиново-красных игл, а карбонил;
кобальта Со(СО)4 и о-фенантролин в разбавленном метаноле дают темно-
коричневый комплекс Со(СО)3(фен) [1061.
К этим солям близок смешанный нитрозо-о-фенантролиновый комплекс
Ре(ЫО)2(фен), полученный Хибером и Андерсоном [107] действием о-фенан-
тролина на нитрозокарбонил железа Fe(CO)2(NO)2 в бензоле. Нитрозокарбо-
нил кобальта Co(NO)(CO)3 и о-фенантролин в пиридине дают черные с голубым
оттенком иглы Со(ЫО)(СО)(фен).
Следует отметить, что соединения Ре(ЫО)2(фен), Со(ЫО)(СО)(фен)
и №(СО)2(фен) образуют ряд, в котором число электронов и сумма атомных
номеров атомов, образующих комплекс, остаются постоянными.
294
ЛИТЕРАТУРА
. ЛИТЕРАТУРА
1. Willgerodt, von Neander, Вег., 33, 2928 (1900).
2. Willgerodt, Jablonski, Вег., 33, 2918 (1900).
3. R a о, W h e e 1 e г, J. Chem. Soc., 1938, 476.
4. Marckwal d, Ann., 274, 336 (1893).
5. Cairns, Kermack, неопубликованные данные.
' 6. Blau, Monatsh., 19, 647 (1898).
7. S m i t h, J. Am. Chem. Soc., 52, 397 (1930).
8. Hieber, Muhlbauer, Ber., 61, 2149 (1928).
9. Tartarin i, Samaja, Ann. chim. appl., 23, 353 (1933).
10. Пат. США 2535417 [С. A., 45, 4747 (1951)].
11. Англ пат. 394416 [Brit. Chem. Abstr., В, 856 (1933)].
12. Halcrow, Kermack, J. Chem. Soc., 1946, 155.
13. К ii s t e r, R о 1 1, Z. Physiol. Chem., 155, 157 (1926).
14. S m i t h, Getz, Chem. Revs., 16, 113 (1935).
15. C a se, J. Am. Chem. Soc., 70, 3994 (1948); 71, 821, 1828 (1949).
16. Case, J. Org. Chem., 16, 1541 (1951).
17. Skraup, Vortmann, Monatsh., 3, 572 (1882).
18. L a Coste, Ber., 16, 674 (1883).
19. Skraup, Vortmann, Monatsh., 5, 531 (1884).•
20. Kuczynski, Sucharda, Roczniki Chem., 16, 513 (1936).
21. Skraup, Vortmann, Monatsh., 4, 570 (1883).
22. В о r n e m a n n, Ber., 19, 2377 (1886).
23. С 1 a u s, W i s, J. prakt. Chem., [2], 38, 393 (1888).
24. H a s k e 1 b e r g, J. Am. Chem. Soc., 69, 1538 (1947).
25. L e 1 1 m a n n, Lippert, Ber., 24, 2623 (1891).
26. M a t s u m u r a, J. Am. Chem. Soc., 52, 3974 (1930).
27. Kaufmann, R a dose v i c, Ber., 42, 2612 (1909).
28. Smith, C a g 1 e, J. Org. Chem., 12, 781 (1947).
29. L i n s k e r, E v a n s, J. Am. Chem. Soc., 68, 403 (1946).
30. D r u e y, Schmidt, Helv. Chim. Acta, 33, 1080 (19o0).
31. Англ. пат. 688802 [С. A., 48, 4009 (1954)].
32. I n g 1 e t t, S m i t h, J. Am. Chem. Soc., 72, 842 (1950).
33. S e a r 1 e s, W a r r e n, J. Org. Chem., 18, 1317 (1953).
34. Matsumura, J. Am. Chem. Soc., 57, 495 (1935).
35. О ch iai, Kuroyanagi, J. Pharm. Soc. Japan, 63, 213 (1943) [C. A., 45, 5152
(1951)].
36. К ar r er, Pletscher, Manz, Helv. Chim. Acta, 30, 1146 (1947); 31, 1431 (1948).
37. Kermack, Webster, J. Chem. Soc., 1942, 213.
38. Sykes, J. Chem. Soc., 1953, 3543.
39. Sykes, J. Chem. Soc., 1956, 3087.
40. Tr aber, К a r r e r, Helv. Chim. Acta, 41, 2066 (1958).
41. Traber, Hubman, Karrer, Helv. Chim. Acta, 43, 265 (1960).
42. Karrer, Pletscher, Manz, Helv. Chim. Acta, 31, 1431 (1948).
43. Karrer et al., Helv. Chim. Acta, 21, 223 (1938).
'44. Karrer, Pletscher, Helv. Chim. Acta, 31, 786 (1948).
45. Douglas, Jacomb, Kermack, J. Chem. Soc., 1947, 1659.
46. Герм. пат. 654444.
47. Wibaut, Spiers, Out we 1 t j es, Rec. trav. chim., 56, 1219 (1937).
48. M a t s u m u r a, J. Am. Chem. Soc., 57, 495 (1935).
49. К e r m a c k, T e b r i c h, J. Chem. Soc., 1945, 375.
50. Haworth, S у k e s, J. Chem. Soc., 1944, 311.
- 51. T u r s k i, К 1 e j n, Roczniki Chem., 18, 31 (1938).
52. Ghosh, Roy, Banerjee, J. Indian Chem. Soc., 22, 219 (1945).
53. Kermack, Weatherhead, J. Chem. Soc. 1940, 1164.
54. Douglas, К e r m a c k, J. Chem. Soc., 1949, 1017.
55. Hazlewood, Hughes, Lions, J. Proc. Roy. Soc. N. S. Wales, 71, 462 (1938)
56. Burger, Bass, Fredericksen, J. Org. Chem., 9, 373 (1944).
57. M i s a n i, В о g e r t, J. Org. Chem., 10, 347 (1945).
58. S n у d e r, F r e i e r, J. Am. Chem. Soc., 68, 1320 (1946).
59. Jacini, Bertilli, Gazz. chim. ital., 69, 111, 405 (1939).
60. J a c i n i, S a 1 i n i, Gazz. chim. ital., 69, 717 (1939).
61. Surrey, Cutler, J. Am. Chem. Soc., 76, 1109 (1954).
62. H i n s b e r g, Koller, Ber., 29, 1500 (1896).
63. Sexton, J. Chem. Soc., 1942, 303.
64. В a x t e r, S p r i n g, J. Chem. Soc., 1945, 229.
65. Richter, Smith, J. Am. Chem. Soc., 66, 396 (1944).
66. Case, J. Org, Chem., 16, 941 (1951).
ЛИТЕРАТУРА
295
67. Англ. пат..454526 [Brit. Chem. Abstr., В, 1179 (1936)].
/ 68. Bulow, Man n, Ber., 30, 979 (1897).
69. Korczynski, Brydowna, Bull. soc. chim. France, [4] 37, 1483 (1925).
70. Hammett, Walden, Edmonds, J. Am. Chem. Soc., 56, 1092 (1934).
71. Англ. пат. 454525 [Brit. Chem. Abstr., B, 1179 (1936)].
72. Англ. пат. 451932 [Brit. Chem. Abstr., B, 1144 (1936)].
73. D e w a r, J. Chem. Soc., 1944, 619.
74. Jacomb, Kermack, J. Chem. Soc., 1946, 62.
75. E i f e r t, H a m i 1 t о n, J. Am. Chem. Soc., 77, 1818 (1955).
76. Ger deissen, Ber., 22, 251 (1889).
77. S к r a u p, Fischer, Monatsh., 5, 527 (1884).
78. A 11 a n, Chem. listy, 46, 224 (1952).
79. Borsch e, Wagner-Roemmich, Ann., 544, 280 (1940).
80. Borsch e, Wagner-Roemmich, Ann., 544, 287 (1940).
81. Woodruff, A d a m s, J. Am. Chem. Soc., 54, 1977 (1932).
82. R u g g 1 i, S c h e t t y, Helv. Chim. Acta, 23, 725 (1940).
83. В e r g, P e t г о w, J. Chem. Soc., 1952, 3713.
84. Hauser, Reynolds, J. Org. Chem., 15, 1224 (1950).
85. Davis, J. Chem. Soc., 1958, 828.
86. Brandt, Dwyer, Gyarfas, Chem. Revs., 54, 959 (1954).
87. Werner, Ber., 45, 433 (1912).
88. Morgan, Burstall, J. Chem. Soc., 1931, 2213.
89. Burstal 1, J. Chem. Soc., 1936, 173.
90. G a i n e s, Hammett, W a 1 d e n, J. Am. Chem. Soc., 58, 1668 (1936).
91. H u m e, К о 1 t h о f f, J. Am. Chem. Soc., 65, 1895 (1943).
92. Walden, Edmonds, Chem. Revs., 16, 81 (1935).
93. L e e, К о 1 t h о f f, L e u s s i n g, J. Am. Chem. Soc., 70, 2348 (1948).
94. Steigman, Birnbaum, Edmonds, Ind. Eng. Chem., Anal. Ed., -14, 30
(1942).
95. Pfeiffer, C h r i s t e 1 e i t, Z. anorg. u. allgem. Chem., 239, 133 (1938).
96. T a г t a r i n i, Gazz. chim. ital., 63, 597 (1933).
97. Morgan, Burstall, J. Chem. Soc., 1930, 2595; 1937, 1649.
98. S m i t h, Richter, Ind. Eng. Chem., Anal. Ed., 16, 580 (1944).
99. Morgan, В u r s t a 1 1, J. Chem. Soc., 1938, 1672.
100. W i 1 1 i n k, W i b a u t, Rec. trav. chim., 54, 275 (1935).
101. F e i g 1, Ind. Eng. Chem., Anal. Ed., 8, 406 (1936).
102. Cagle, S m i t h, J. Am. Chem. Soc., 69, 1860 (1947).
103. Smirnoff, Helv. Chim. Acta, 4, 802 (1921).
104. Breckenridge, Lewis, Quick, Can. J. Research, 17B, 258 (1939).
105. Hieber, Muhlbauer, Ber., 65, 1082 (1932).
106. Hieber, Muhlbauer, Ehmann, Ber., 65, .1090 (1932).
107. H i e b er, Anderson, Z. anorg. u. allgem. Chem., 211, 132 (1932).
Глава V
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕН30ТРИА30ЛЫ
Дж. Бойер
ВВЕДЕНИЕ
Триазолы представляют собой ароматические циклические соединения
с тремя атомами азота и двумя атомами углерода. Два возможных сочетания
этих пяти атомов дают так называемые вицинальные и симметричные триазолы.
Вицинальные триазолы в «Chemical Abstracts» именуются также— 1Н-1,2,3-
триазолы или пирро-[а&]-диазолы и 2Н-1,2,3-триазолы или nHppo-laaJ-диазо-
лы. В старой литературе симметричные триазолы иногда называли пирродиазо-
лами. Термин озотриазол относится к производным 2Н-1,2,3-триазола,
особенно к полученным из озазонов. Реже применяемая номенклатура будет
приведена при соответствующих формулах. Производные 1,2,4-изотриазолов
пока не известны, производные же 1,2,3-изотриазолов впервые описаны
в 1958 г. (стр. 312).
eutf-Триазол, или 1-,2,3-триазол:
нс=сн НС—СН Н2С—сн
/ \ ч / . ч
HN N-------> N N ------> N N
— \ / "— ч /
N N N •
I
Н
1Н-1,2,3-Триазол 2Н-1,2,3-Триазол, 1,2,3-Изотриазол
озотриазол,
1,2,5-триазол,
2,1,3-триазол
силш-Триазол, или
HN-N
1Н-1,2,4-Триазол
1,2,4-триазол:
N = N
Н2С СН
\ Z
N
4Н-1,2,4-Триазол,
1,3,4-триазол,
4,1,2-триазол,
пирродиазол
1,2,4-Изотриазол
Если считать, что таутомеры не поддаются разделению, то замещение
водорода как у вицинальных, так и у симметричных триазолов (за исключе-
нием изотриазолов) допускает образование трех однозамещенных, четырех
дизамещенных (при одинаковых заместителях), семи дизамещенных (при
различных заместителях), двух тризамещенных (при одинаковых заместите-
лях), пяти тризамещенных (при двух различных заместителях) и девяти
виц-ТРИ АЗОЛЫ
297
тризамещенных (при трех различных заместителях). В большинстве слу-
чаев это, по-видимому, точно характеризует структурную изомерию триазо-
лов. Исключение представляют таутомеры: 3-метил-5-нитрамино-1,2,4-три-
азол и 3-нитроамино-5-метил-1,2,4-триазол (стр. 350).
Для бензотриазола характерна орто-конденсация бензольного кольца
с кольцом euq-триазола. В «Chemical Abstracts» таутомерные формы родоначаль-
иого соединения обозначаются как 1Н-бензотриазол и 2Н-бензотриазол и атомы
нумеруются так, как показано в приведенной ниже формуле.
Н
1Н-Бензотриазол,
азимидобензол,
бензолазоимид,
бензизотриазол,
1,2,3-бензотриазо л
2Н-Бензотриазол,
псевдоазимидобензол,
2,1,3-бензотриазол
У бензотриазола с заместителем в положении 2 отсутствуют некоторые
хиноидные свойства и строение их, по-видимому, нельзя исчерпывающе
описать при помощи только ковалентных связей.
Химия euq-триазолов, включая бензотриазол, рассмотрена в работе [1].
виц-ТРИ АЗОЛЫ
Методы получения
Ацетиленовые соединения и азиды. Продукт присоединения фенилазида
к метиловому эфиру ацетиленди карбоновой кислоты неустановленного состава
впервые описан в 1893 г. [2—4]. Несколько лет спустя Димрот и Фестер уста-
новили, что 1-фенилтриазол получается в результате присоединения фенил-
азида к ^ацетилену [5]. По-видимому, эта общая реакция образования кольца
триазола необратима. В частности, фенилазид, присоединяясь к аддукту,
обратимо образующемуся из дивинилфульвена и метилового эфира ацетилен-
дикарбоновой кислоты, дает диметиловый эфир 1-фенилтриазолдикарбоно-
вой-4,5 кислоты (I) [6].
Монозамещенные ацетиленовые соединения дают изомерные продукты
присоединения [7, 8], одйако преимущественно получаются 1,4-, а не 1,5-диза-
мещенные триазолы [9]. Несимметричные дизамещенные ацетиленовые про-
изводные также образуют изомерные продукты присоединения, как, например,
дифенилтриазолальдегиды, полученные из фенилазида и фенилпропиолового
альдегида [10]. Алкилазиды и азотистоводородная кислота также вступают
в реакцию с ацетиленовыми соединениями. Интересный ряд триазолов получен
298
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
в результате присоединения фенил-, бензил-, н-гексил- и н-додецилазидов
110“
CeH5C = CCHO + CgHjNg —
ОНСС—N
II II +
CeH5C N
N
I
СвНв
40%
CeH5C —N
II II
ОНСС N
N
свн5
22%
к диацеталю 2-бутиндиаля-1.,4. Гидролиз аддукта приводит к соответствующим
1-замещенным 4,5-диформилтриазолам (II) [.И]. Триазолы, а также 4-заме-
щенные и 4,5-дизамещенные триазолы получены реакцией присоединения
В запаянной трубке Н3О+
(С2Н6О)2СНС = CCH(OC2H6)2 + RN3
100“
онсс=ссно
RNZ 'n
II
R=CeH5, CeH6CH2, СН3(СН2)5, CH3(CH2)tl
ацетиленовых соединений к азотистоводородной кислоте [1, 2]. Любопытно,
что монозамещенные 4-алкил-1,2,3-триазолы оставались неизвестными до
1954 г., когда они были синтезированы этим методом [12].
Ацетиленовые соединения с сопряженными связями, реагируя с азотисто-
воДородной кислотой, алкил- или арилазидами, дают ди-(1,2,3-триазолы) [13].
/ ои \
ОН ОН (СН3)2СС = С —
II / \
(СН3)2СС = СС == СС(СН3)2RN3 --* • RN N
\ XZ А
R=H, СвН5, С8Н5СН2
Оптическая активность 4,5'-бис-(1-оксициклогексил)-ди-[5,4'-(1,2,3-триазолил)]-
1, Г-диацетилуксусной кислоты является результатом пространственного за-
труднения свободного вращения двух триазольных колец [13].
ОН НО
/ ----с=с--------с=с--------/ у
. Rif Rtf XN
V V
r=ch2cooh
Алкоголяты и азиды. При нагревании фенилазида с этилатом натрия
в этиловом спирте при атмосферном давлении образуются 1-фенил-1,2,3-три-
азол, анилин, азот и едкий натр. Арилтриазолы с алкильными заместителями
в положении.4 получаются из высших первичных спиртов, тогда как в случае
алкоголятов вторичных спиртов происходит только восстановление в анилин
[14—161. Удовлетворительного объяснения этой реакции не предложено.
CH3CH2ONa + CeH5N3---> HC = CH + N2 + CeH5NH2 + NaOH
С2Н5— / \
\ /
N
виц-ТРИАЗОЛЫ
299
Соединения с активными метиленовыми группами и азиды. В результате
реакции между фенилазидом и ацетоуксусным эфиром в присутствии этилата
натрия образуется 1-фенил-4-карбалкокси-5-метил-1,2,3-триазол [17]. Анало-
гичные реакции наблюдались между арилазидами и другими соединениями,
содержащими способные к енолизации метиленовые группы [2]. Взаимодей-
ствием эфиров циануксусной кислоты с фенилазидом синтезированы 1-фенил-
5-амино-1,2,3-триазолы [18]. Продукт (III), полученный из ацетофенона
CeH5COCH3+CeH5N3
CaHsONa
CeH5C— N.
II 7
CeH5COCH2N=NC —Nz
III
и фенилазида, строение которого подтверждено независимым синтезом, содер-
жит связь N = N, появление которой не удается объяснить [19]. Некоторые
арилазиды не вступают в эту реакцию, и иногда наряду с триазолами обра-
зуются пиразолы [2]. Для аналогичных синтезов триазолов успешно при-
меняются алкил-, ацил- и сульфонилазиды [1].
В присутствии /пре/п-бутилата калия в безводном тетрагидрофуране
алкилазиды при комнатной температуре вступают в реакцию с фенилацето-
нитрилом с образованием 1-алкил-4-фенил-5-амино-1,2,3-триазолов [20].
Ацетилениды металлов и азиды. В результате присоединения реактивов
Гриньяра к азидам образуются триазеновые соли [2]. При взаимодействии
BrMgC == CMgBr-j-Br——N3-------->
С6Н4Вг-и
НС — N.
II у
n-BrCeH4NHN = NC — W
IV
гриньяровского ацетиленового комплекса с n-бромфенилазидом образуется
не только линейный бис-триазен, но и изомерное диазоаминопроизводное
триазола (IV) [21]. Аналогичная реакция фенилацетиленида натрия с избыт-
ком тозилазида приводит к натриевой соли триазолтриазена. При гидролизе
получается соответствующий триазолазид [22].
Фенилацетиленид натрия также вступает в реакцию с 0-хлорэтилазидом,
по-видимому взаимодействуя с азидогруппой [22], а не с атомом хлора.
TsN3
CeH5C = CNa-------> [СвН5С = С — N — N=N — Ts]-Na+
CeH5C=C-Na+
TsN^ 'n
TsNg
|-CeH5C = CNN = NTs-1
TsNZ N
\ /
L N J
H3O+
---->
CeH5C=CN3
TsN N
_ Na+
Олефины и азиды. Присоединение азидогруппы к олефинам ведет, как
правило, к образованию триазолинов. Вольф и Грау получили из фенилазида
300
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
и n-бензохинона Один моно- и два бис-аддукта, являющихся триазолами
(V — VII) [23]. В результате дегидрирования первоначально образующихся
триазолинов получается гидрохинон. Аналогичные моноаддукты были полу-
чены из а-нафтохинона [24], тогда как 0-нафтохинон не вступал в реакцию
с органическими азидами [25]. Анил циклогексанона сочетается с фенилазидом,
по-видимому, в виде енаминного таутомера. Аддукт, являющийся, вероятно,
производным триазолина, после отщепления анилина превращается в 1-фенил-
4,5-тетраметилен-1,2,3-триазол [26].
Триазолины не всегда легко дегидрируются. Так, продукт присоедине-
ния М,М'-диэтилэтиленсульфонамида к фенилазиду не поддается дегидриро-
ванию ни хиноном, ни N-бромсукцинимидом, ни бромом [27]. В противопо-
ложность этому этиленсульфохлорид вступает в реакцию с фенилазидом
самопроизвольно при комнатной температуре и дает с количественным выходом
аддукт состава 2:1, которому на основании данных ультрафиолетового
и инфракрасного спектров приписано строение 2-(1-фениЛ-1-прото-1,2,3-три-
азол-3-ил)-1-хлорсульфонилэтанхлорида (VIII) [27]. При стоянии этот про-
дукт превращается в соединение, принятое за 2-(1-фенил-1-прото-1,2,3-три-
азол-2-ил)этансульфонат, который можно синтезировать также из 1-фенил-
триазола и этиленсульфохлорида.
сн=сн
2СН2 = СН — SO2Cl + CeH5N3 —> СоН^ NCH2CH2SO2C1
\ /
N
С1-
VIII
_ сн = сн
J I н2о | I
C6H5N N +СН2 = СН—SO2C1 —>---------> CeHjN . NCH2CH2SOj
Синтез из диазосоединений. Образование 4-циан-1,2,3-триазола из диазо-
метана и циана [28] аналогично синтезу 5-циантетразола из циана и азотисто-
виц-ТРИ АЗОЛЫ
301
водородной кислоты. Подобно этому диазоэтан при взаимодействии с цианом
HC=CCN
-10“ I |
CH2N2 + (CN)2 —> HN N
дает 4-циан-5-метил-1,2,3-триазол [28]. В реакцию вводились другие циани-
стые соединения: метилцианугольный эфир, хлор- и бромциан, а также тозил-
ацетонитрил [1]. По-видимому, весьма сходной является реакция образова-
ния 4-фенил-1,2,3-триазола йз закиси азота, ацетонитрила и фениллития [29].
Изомеризация а-диазоамидов в 4-окси-1,2,3-триазолы, катализируемая,
как правило, щелочами, известна под названием перегруппировки Димрота
/30,31]. Сообщается [32], что диазотирование некоторых а-аминоамидов
НОС = ССООС2Н5
C2H5OOCCHCONH2
nh2-hci
может привести к образованию триазола. При подкислении солей окситриазола
вновь получаются линейные а-диазоамиды. Изучение этих превращений имело
С=СО~ К+
N2CHCONH2 —hn" \
большое значение для исследования природы таутомерии. В качестве цикли-
зующих агентов могут быть использованы другие вещества; так, метиловый
эфир 1-фенил-5-хлор-1,2,3-триазолкарбоновой-4 кислоты (IX) [18] получен
нагреванием метилдиазомалонанилида с пятихлористым фосфором.
O=CNHCeH5 pci6
С1С-
CH3OCOCN2
СН3ОСОС- nZ
\ Таутомерная перегруппировка, по-видимому, носит бимолекулярный
характер с первоначальной атакой протона по замещенному атому азота
кольца [33]. Перегруппировка 1-арил-4-карбэтокси-5-окси-1,2,3-триазолов
ROOCC==CO~ Н+ ROOCC=C-
ROOjJ—С=О
N2 NHR
замедляется электронодонорными заместителями, находящимися в пара-
положении арильных групп, применением среды, в которой триазол хорошо
растворим, а также, при добавлении некоторых солей [34].
’ Замыкание цикла, приводящее к образованию триазолов из а-диазокето-
нов, а также из соединений, содержащих аминогруппу, как, например, гидро-
302
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
ксиламин, первичные амины и гидразины, является необратимым процес-
сом [35].
СН3С=ССООС2Н5
CH3COC(=N2)COOC2H5 + RNH2 —> rn" \
Бензотриазол и многие его производные получают из диазотированного
о-фенилендиамина или из соответствующим образом замещенного производ-
ного [1]. Источником третьего атома азота в триазоле вместо азотистой кисло-
ты может служить соль диазония. Так, например, бензотриазолкарбоновая-4
кислота получена из 2,3-диаминобензойной и диазотированной сульфаниловой
кислот [36]. При наличии моноалкильных или моноарильных заместителей
у атома азота замыкание кольца приводит к образованию соответствующих
1-замещенных производных бензотриазола [1], однако сообщается, что некото-
рые моноациламины дают 1-'ацилбензотриазолы [37, 38] и что ацильная группа
N = CHR N = N
R=n-CeH4NO2
может отщепиться в процессе диазотирования [39]. Диазотирование моно-
сульфонилпроизводных ароматических о-диаминов также сопровождается
образованием триазола [40,41].
Примером редко наблюдаемого замещения аминопроизводного в положе-
нии 1 у выц-триазола является реакция, имеющая, по-видимому, общий
характер. При циклизации диазотированного 4-метил-5-амино-6-(п-нитробен-
заль)гидразинопиримидина образуется соответствующий 1-(п-нитробензаль)-
аминотриазол [42]. Аналогичным образом при диазотировании 4-окси-8-
аминоциннолица получается триазолоциннолон [43].
Реакции конденсации смежных групп. При исследовании продуктов
окисления, подученных из фенилозазонов 1,2-дикарбонильных производных,
Пехман обнаружил образование моноциклических виц-триазолов [44]. Окисле-
нием фенилозазона диметилглиоксаля (X) получен продукт, позднее иденти-
фицированный как бис-азобутен. При нагревании этого соединения в кислоте
образуется 2-фенил-4,5-диметил-1,2,3-триазол (XI).
CH3C = NNHCeH5
CH3C=NNHCeH5
[О]
CH3CN=NC6H5
CH3L = NCeH5
СН3С—ссн3
X
виц-ТРИАЗОЛЫ
303
Выделять промежуточные азопроизводные нет необходимости; при нагре-
вании бис-гидразона диацетила до 170° образуются 4,5-диметил-1,2,3-триазол
и аммиак и происходит полимеризация [45]. При пиролизе фенилозазона (X)
или монофенилгидразонмоноксима диацетила получается соединение XI
и наряду с ним анилин или вода соответственно [44, 45]. Полученное при
пиролизе фенилгидразона пировиноградной кислоты соединение XI является
продуктом распада первоначально образующегося вещества X через промежу-
точные соединения XII и XIII, которые также были выделены [45].
СН, СН,
I + J -С-^
CeH5NHN = ССООН q* c6h5nhnh = ссоо-
—> [СвН5ЫнЬн=:ССН3] —> CeH5NHN = CHCH3
XII XIII
300°
XII + XIII -> X ---> XI+CeH5NH2
Фенилозазоны или фенилгидразоноксимы 1,2-дикарбонильных соедине-
ний превращаются в триазолы при более низких температурах под действием
уксусного ангидрида, уксусной кислоты или пяти хлористого фосфора в хлоро-
форме [1]. Этим путем получено большое число разнообразных 2,4-ди- или
2,4,5-тризамещенных триазолов. Исследуя фенилозазоны сахаров, Гудсон [46]
установил, что сульфат меди в кипящей воде отщепляет анилин, в результате
чего образуются озотриазолы сахаров. Теубер [47] с помощью нитрозо-
дисульфоната калия превратил глюкозазон в глюкозотриазол. Фенилозазон
бензила не взаимодействует с этим реагентом. Получены озотриазолы сахаров
с изотопом брома в 2-(п-бромфенильном) заместителе [48].
Окиси триазола образуются при нагревании производных монофенил-
гидразонмоноксима 1,2-дикарбонильных соединений с окисью ртути или
N2O4 [1 ]. В восстановительных условиях реакции Кижнера — Вольфа а-окси-
минокетоны образуют триазолы [49]. /
CH3C = NOH СН3С-ССН3
I Желтая Hg° 0«-N \
I ИЛИ N9O4 ч ,
CH3C = NNHCeH5 \ /
N
I
j СвН5
2-Фенил-1,2,3-триазол (XV) синтезирован из а, 0-бис-(бензолазо)этилена
при действии хлорида железа (III) в разбавленной кислоте, причем исходный
продукт получают окислением фенилозазона глиоксаля (XIV) [50]. Соедине-
ние XIV при кипячении в водном растворе сульфата меди также переходит
в триазол (XV) [51]. В противоположность этому бензоилозазон глиоксаля
в щелочном растворе феррицианида калия превращается в 1-бензамидо-
HC=NNHCeH5
HC = NNHCeH5
К2СГ2О7
CH3COOH
HCN = NCeH5
HCN = NCeH5
FeCl3
' ' - J*
разб. HCI
HC —CH
I
CeH5
XV
XIV
. 1,2,3-триазол, а бензоилозазон метилглиоксаля — в 1-бензамидо-4-метил-
1,2,3-триазол и 1-бензамидо-5-метил-1,2,3-триазол [50]. Аналогично заме-
304
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛ Ы
щенные в 1-положении 1,2,3-триазолы получают из тозилозазонов димеТил-
глиоксаля действием металлического натрия в этиленгликоле [52]. Тозилоза-
зон метилфенилдикетона вступает в аналогичную реакцию, тогда как тозилоза-
зон бензила с этим реагентом дает дифенилацетилен [52].
CH3C=NNHSO2C7H7 СН3С = ССН3
| c7h7so2Nhn n
ch3c=nnhso2c7h7 (CHa0H)2 \ /
N
Имеется очень мало работ, посвященных изучению образования триазолов
путем отщепления различных аминов от озазонов. Так, от фенил-о-карбокси-
фенилозазона метилглиоксаля элиминируется анилин [53]
HC=NNHCeH4COOH
CH3C=NNHCeH5
Горячая CHgCOOH
- CeH5NH2
НС-ССН3
С6Н4СООН
Другие три'метода синтеза триазолов, вероятно, имеют отношение к мето-
дам, обсуждаемым в настоящем разделе. Так, 2-оксй-5-метилтриазолкарбо-
новая-4 кислота (XVI) получена дегидратацией триазолина, образующегося
при восстановлении этилового эфира а-оксимиио-0-нитрозоиминомаеляной
кислоты [54]. Обработка аминОиминосукцинонитрила, тетрамера цианистого
водорода [55], азотистой кислотой приводит к образованию 4,5-дициан-
1,2,3-триазола (XVII), в котором может происходить аналогичное замыкание
CH3C=NNO
I Zn
г и mJ ыпи СНзСООН*
С2Н5ООСС=NOH
Конц. HCI
120°
XVI
кольца. Третьим примером является получение 2-окси-4,5-тетраметилен-
1,2,3-триазола (XVIII) действием хлоргидрата гидроксиламина на натриевую
соль моноксима циклогександиона-1,2 [56].
NCC=CCN
NCC = NH hnO2 / \
| ----> HN N
ncchnh2 \ Z
N
XVII
о
/\у
a
NONa
3NH2OHHC1
H2O
CaCO3
H2O
Кипячение
48 час
XVIII
Некоторые типы циклизации, характерные лишь для реакций, ведущих
к образованию триазолов, сконденсированных с ароматическими кольцами,
аналогичны Другим методам, описанным в настоящем разделе. Так, например,
бензотриазол образуется при нагревании о-аминофенилгидразоиа кетомало-
виЧ-ТРИ АЗОЛЫ
305
новой кислоты с аммиачным раствором сульфата меди [57 ]. Примером цикли-
зации о-нитрогидразинопроизводных ароматических соединений служит полу-
чение 1-окси-6-нитробензотриазола (XIX) в результате реакции, когда для
открытия некоторых карбонильных соединений динитрофенилгидразином
употребляют щелочь [58]. Аналогичным образом циклизация 2,4-динитро-
5-карбоксигидразобензола приводит к соответствующей 1-окиси бензотриазола
N
НООС NHNHCeH5 CH3CQOH НООС —\
------* N-CeH5
O^-J^II-no., O2N-I J /
N
I
О
XX
(XX) [59]. При избытке гидразосоединения или при наличии некоторых дру-
гих восстановителей образуется бензотриазол вместо его N-окиси [1].
Каждое из Двух азопроизводных, полученных в результате реакции
между диазотированным анилином и л-фенилендиамином, окисляется. Образу-
ющиеся бис-триазолбензолы являются изомерами. Строение ангулярного
изомера было подтверждено независимым синтезом [60]. Структура бис-
триазолбеизола, замещенного в каждом триазольном кольце в положении 2,
до сих пор не установлена, поэтому приписывать 2-замещенным бензотриазолам
хиноидную форму, по-видимому, нет необходимости. При действии более
энергичных окислителей получаются линейные бис-триазолбензохиноны, вос-
становление которых приводит к соответствующим гидрохинонам [61 ]. В щелоч-
ной среде происходит циклизация 2,4-динитро-5-карбэтоксиметилфенилгидра-
зина с образованием 1-окси-5-карбэтоксиметил-6-нитробензотриазола, однако
20 Заказ /в 978
306
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
2,4-динитро-5-гидразинофенилгидразин в тех же условиях разлагается [62].
Сообщено о получении три(триазол)бензола [63].
Циклизация ароматических о-аминоазопроизводных происходит как
в окислительных [60], так и в восстановительных [64] условиях с образова-
c7h7so2nh-
c6h5n=n-
-NHSO2C7H,
-n=nc6h5
NagCfjOy
нием производных бензотриазола. При действии сульфида аммония на о-нитро-
азобенЗол образуется 2-фенилбензотриазол [65], однако 2-нитро-4-метилазо-
бензол при этом превращается в 1-окись 2-фенил-6-метилбензотриазола [66].
N
С2Н5ООССН2 -Z\- n2h3 ‘ С2Н5ООССН2 -/V Ч
• - N
ОгЫ-^^-ыо, ОгЫДД /
N
ОН
Замыкание триазольного цикла может происходить и при взаимодействий
азофункции с о-фенилгидразинной, азидной и тиониламинной группами [1].
Некоторые р-замещенные производные азоксибензола могут циклизоваться
с образованием бензотриазолов или их N-окисей. При нагревании о-амино-
азоксибензола в концентрированной серной кислоте происходит дегидратация
и образуется 2-фенилбензотриазол [67]. 1-Окись 2-фенилбензотриазола полу-
чена при действии сульфида аммония на о-нитроазоксибензол, щелочи на
о-оксиаминоазоксибензол и перекиси на о-аминоазобензол (возможно, через
обычный промежуточный продукт [1]).
Сужение кольца. У 2,3-дизамещенных 2,3-дигидротетразинов при нагре-
вании или под действием кислот происходит сужение кольца с образованием
_ триазола. Сообщается, что как дигидротетразины, так и изомерные им бис-азо-
N c6H5c — CCgHj
c6h5c = nnhc6h5 NaOC2H5 C6H5 — / xNC6H5 c2h5oh N
c6h5c=nnhc6h5 12 - 1, C6H5-^ ZNC6HS HCI N i. У •' N |
CeH5
олефины являются продуктами окисления озазонов. Полученный из бензил-
фенилозазона 2,3,5,6-тетрафенил-2,3-дигидро-виц-тетразин вспиртовом растворе
хлористого водорода превращается в 2,4,5-трифенил-1,2,3-триазол [68].
В работе [50] описано превращение ряда дигидротетразинов в три азолы под
действием кислот или при нагревании. Достоверное объяснение этихпревраще-
виц-ТРИАЗОЛЫ
307
ний требует дальнейших исследований.
НС = ССН3 N
(CeH5CO)2NN^ \ \ 4NCOCeH5
\ Z Н3с-1 /NCOCeHj
кт /
N N
С2Н8ОН
НС1
СН3С=СН
N
Превращения циклов. Некоторые производные изоксазола легко пре-
вращаются в триазолы. Диазотирование 4-амино-3,5-диметилизоксазола
и последующее кипячение с подкисленным раствором сульфата меди дает
4-ацетил-5-метил-1,2,3-триазол [69]. При диазотировании 3-(п-метоксианили-
но)-4-амино-5-фенилизоксазола изоксазольное кольцо не разрушается, а обра-
Н3СС—О СН3СОС = ССН3
II I / \
H2NC N . HN N
? N
сн3
зующийся продукт представляет, по-видимому, 3-(п-анизил)-6-фениЛтриазол-
[с]-изоксазол [70]. Другой пример демонстрирует термическую перегруппи-
ровку фенилгидразона З-ацетил-5-метилизоксазола с образованием 2-фенил-
4-метил-5-ацетонил-1,2,3-триазола [71]. Эта реакция напоминает образование
Свн5
C=CNH2
о <!:nh—свн4осн3
свн4осн3
триазолов из производных монофенилгидразонмоноксимов 1,2-дикарбониль-
ных соединений. Некоторые изоксазолы, как, например, XXI и XXII, не пре-
терпевают такой перегруппировки [71]. Термическая изомеризацйя 3-метил-
4-бензолазо-5-фенил изоксазола в 2-фенил-4-метил-5-бензоил-1,2,3-трйазол [72]
' CeH5C=NNHCeH5 CeH5C = NNHCeH5
I I
C = NX C = NX
I/O I/O
СвН5С = С/ HC = CZ
I I
CeH5 CeH5
XXI XXII
I
CH3
CH3C=NNHCeH5
C = NX
I >0
. HC=CZ
CH3C—ссюсвн5
CH3C = N 180° \
| )O — --> N N
CeH5N = NC=C/ /
ceH5 N
20*
308
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
может проходить аналогично. Примером менее понятного превращения изокса-
золов в триазолы является синтез 1-окиси 2-фенил-4-бензолазо-5-метилтриазо-
ла из 3,5-диметил-4-нитроизоксазола и щелочного раствора гидроокиси фенил-
диазония [73]. Предложен следующий возможный механизм реакции:
СН3С—CNO2
CeH8N2OH
СН3С—CN = NCeH5
У X
О -е- N N
Н5
Ациламинотриазолы получаются пиролизом гидразонов 3-ацил-1,2,4-
оксадиазолов [74].
Более глубокая перегруппировка имеет место при действии едкого кали,
а затем анилина на диоксим 3,4-диформилфуроксана, тетрамер гремучей
кислоты. Строение продукта реакции — (1-фенил-1,2,3-триазол-4-ил)изо-
нитрозоацетанилида — установлено путем превращения его действием тионил-
хлорида в 1-фенил-4-циан-1,2,3-триазол [75].
C6H5C = NNHC6H5
I
190°
C = N
N = C
C6HSC —CNHCOC6H5
У \
N N
NOH
HON = CHC—CCH = NOH
N^ \
(1) KOH
(2) CeH5NH2
C6H5NHCOCC = CH
NZ \-CeH5
4 /
N
Расщепление бензотриазолов. Устойчивость триазольного кольца бензо-
триазола к окислению — это свойство, которое, в частности, проявляется
при окислении перманганатом 5-метилбензотриазола в триазолдикарбоновую-
4,5 кислоту [76]. Аналогичным образом окисление перманганатом 1-окси-
6-нитробензотриазола дает 1-окси-1,2,3-триазолдикарбоновую-4,5 кислоту
(XXIII) [77].
I
ОН
ноосс=ссоон
Щелочной раствор КМпОд /
-----------------* HON N
XXIII
ноосс=ссоон
СНз-/
Алкильные заместители, находящиеся у атома азота триазола, устой-
чивы к окислению [78], а винильные [22,79] и ароматические — неустой-
чивы. Ароматический остаток с активирующим заместителем легче отщепляет-
виц-ТРИАЗОЛЫ
309
ся, чем незамещенный [80, 81].
НООСС = ССвН4СООН-о
СвН5-г/ \
\ /
N
При неокислительном расщеплении из обработанного щелочью бис-аддук-
та хинона и фенилазида получена 1-фенил-1,2,3-триазолкарбоновая-4 кисло-
та [82]. Различные хлорированные и содержащие кислород производные
СвН5О
6%-НЫЙ NaOH
---------.—>.
НООСС = СН
ди- и тетрагидробензотриазола также подвергали щелочному расщеплению,
причем получены соответствующие замещенные моноциклические триазолы [11.
N—N —СвН5
II I
О НО соон
XXIV
XXV
Сообщается о двух примерах гомоциклического сужения кольца: образо-
вание соединения XXIV в результате перегруппировки типа бензиловой
кислоты [83] и превращение диазотированного 2-фенил-4-амино-5-оксибензо-
триазола в соединение XXV [84].
Окисление и декарбоксилирование боковой цепи. Деструктивный метод
декарбоксилирования применяется весьма широко для синтеза производных
триазола с небольшим числом заместителей. Известно, что связанные с атомом
азота триазола винильные и активированные арильные группы легко окисляют-
ся; продукты реакции — карбаминовые кислоты — могут декарбоксилиро-
ваться в процессе их образования. Окисление 2,4-бис-(диаминофенил)-1,2,3-
триазсла перманганатом дает триазолкарбоновую кислоту (XXVI) [85].
310
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛ Ы
Для превращения 2-нитрофенил-1,2,3-триазолкарбоновой кислоты в соеди-
нение XXVI, по-видимому, необходимо восстановить исходное соединение
до 2-аминофенилтриазолкарбоновой кислоты, которую потом окисляют [86].
(H2N)2CeH3C-CH
КМпО4
NaOH
100°
нс=ссоон
HNZ 'n
XXVI
I
СвНз(ИН2)2
Неактивированные арильные или алкильные заместители у атома азота триа-
зола более устойчивы к окислению, чем алкильные заместители у атома углерода
триазольного кольца. Окисление перманганатом 1-бензил- [82], а также
1-фенил-5-метил-1,2,3-триазола [87] дает соответствующую 1-замещенную
триазолкарбоновую-5 кислоту.
При окислении карбинольных боковых цепей в положениях 4 и 5 могут
образоваться триазол альдегиды. При окислении фенилглюкОзотриазола крас-
ной окисью свинца (РЬ3О4) с выходом 83% получен 2-фенил-4-формил-1,2,3-
триазол [88].
Декарбоксилирование у атома углерода в триазольном цикле протекает
труднее и обычно требует нагревания примерно до 200° [1]. Типичными при-
мерами являются образование 1-этил-1,2,3-триазола из 1-этил-1,2,3-триазол-
карбоновой-5 кислоты [82], а также 1,2,3-триазол-1-илуксусной кислоты
из 4,5-дикарбокси-1,2,3-триазол-1-илуксусной кислоты [89]. Декарбокси-
лирование серебряной соли 2-(п-нитрофенил)-1,2,3-триазолкарбоновой-4 кис-
лоты протекает при 250° [90]. Карбоксильную функцию в арильных группах,
связанных с атомом азота или углерода кольца триазола, удалить довольно
трудно. При кипячении в хинолине с обратным холодильником в присутствии
меди 2-фенил-4-(о-карбоксифенил)-1,2,3-триазолкарбоновая-5 кислота пре-
вращается в 2,4-дифенилтриазол [85], тогда как 2-(п-карбоксифенил)-1,2,3-
триазолдикарбоновая-4,5 кислота при возгонке дает 2-(п-карбоксифенил)-
1,2,3-триазол [91]. 4-Карбокси-5-оксибензотриазол при кипячении в воде
теряет двуокись углерода и переходит в 5-оксибензотриазол, а изомерный
5-окси-6-карбоксибензотриазол в этих условиях не изменяется [92].
0-НООССвН4С—ССООН свн5с—сн
N N —> N N
CeH5 CeHs
НООСС- ССООН НС—сн
N N
C6H4COOH-n (iBH4COOH-n
Пиролиз 1-карбалкоксибензотриазолов полностью сходен с реакциями
декарбоксилирования. Нагревание метилового эфира бензотриазолкарбоно-
вой- 1 кислоты приводит к отщеплению двуокиси углерода и образованию
1-метил- (60%), а также 2-метилбензотриазолов (35%) [79].
Молекулярная перегруппировка, происходящая в процессе декарбокси-
лирования 1-фенил-5-амино-1,2,3-триазолкарбоновой-4 кислоты до 4-анилино-
йг/ч-ТРИАЗОЛЫ
311
1,2,3-триЗзола, рассматривается в следующем разделе.
H2NC = CCOOH
\ /
, N
-СО2
CeH5NHC=CH
HN/ "n
\
N
Изомерия положения. Димрот [18] в 1909 г. открыл термическую изомери-
зацию 1-арил-5-амино-1,2,3-триазолов. В спиртовом растворе при 150° из сое-
динения XXVII образуется равновесная смесь, состоящая на 77% из 4-карб-
этокси-5-анилино-1,2,3-триазола (XXVIII, Аг = С6Н5) (кислый) и на 23%
из 1-фенил-4-карбэтокси-5-амино-1,2,3-триазола (XXVII, Аг — С8Н5) (ней-
тральный). Количественное превращение нейтральной формы в- кислую про-
исходит в присутствии пиридина или этилата натрия. Предложенное объясне-
ние этой изомеризации предполагает первоначальное раскрытие кольца,
затем состояние промежуточного равновесия “между а-диазоацетамидинами
и далее замыкание кольца [93], т. е. близко объяснению механизма аналогич-
H2NC = ССООС2Н5
Аг—1/ \
150°
XXVII
ArNHC = CCOOC2H5
hn" \
\ z
N’
XXVIII
ной перегруппировки некоторых тетразолов [93]. Состояние промежуточного
равновесия требует, чтобы аминогруппа в 5-положении соединения XXVII
содержала по крайней мере один атом водорода. В своем оригинальном иссле-
довании Димрот -[18] установил, что 1-фенил-5-метиламино-1,2,3-триазол
при 110° перегруппировывается в 1-метил-5-анилино-1,2,3-триазол.
NH2 NH
! _ I
XXVII ArNC = CN = N ArNHC = CN = N XXVIII
+ I I
COOC2H5 COOC2H5
В кинетическом исследовании изомеризации [94] скорости превращения
XXVII XXVIII измерялись путем титрования соединения XXVIII спирто-
вым раствором едкого кали. Каталитическое действие пикриновой или хлор-
ной кислоты, эффективных при [Н+] < 10-4 М, может быть объяснено анало-
гичным механизмом, который согласуется со способностью электроноакцеп-
торных групп, находящихся в пара-положении арильного заместителя, увели-
чивать скорость реакции, в то время как электронодонорные заместители
- NH2 н+ NH2
I 7ZZZ II —ZZ
XXVII^±ArN = C — C(R)N2 ArNH —С —C(R)N2 н+
NH
II
Д". ArNHCC(R)N2 XXVIII
в пара-положении оказывают противоположное действие. Все попытки синте-
зировать соединёнйе XXVII (Ar = n-O2NCeH4) привели к образованию соеди-
нения XXVIII [94], что находится в соответствии с аналогичной реакцией
изомеризации тетразола [93]. Образование из n-нитрофенилазида и п-бензо-
хинона [95] производного азациклопропена (XXX) вместо ожидаемого триазола
(XXIX) происходит благодаря неустойчивости триазольного кольца.
312
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
Примером катализируемой кислотой перегруппировки, в результате
которой может происходить миграция арилсульфонильной группы, является
получение 4-(1-)Нафтил£ульфонил)карбоксамидо-5-окси-1,2,3-триазола (XX ХП)
H2NCOC=CO- nh4
XXX
Н3О'
—SO2Ci0H7-c
XXXI!
XXXI
из соединения XXXI [96]. Сопоставление объяснений перегруппировки Дим-
рота 5-окси-1,2,3-триазолов и перегруппировки соединения XXVII в соеди-
нение XXVIII позволяет представить это превращение следующим образом:
ОН
н+
XXXI-------> ArSO2NH С —CN2
<--- + ।
conh2
о
ArSO2NHCC=N2 X X XII
HO—C = NH
Таким образом, исключается необходимость миграции арилсульфониль-
ной группы, что находится в соответствии с экспериментальным фактом —
гидролиз сульфонамидов встречает большие трудности [97].
1,2,3-Изотриазолы. Восстановление а-азидоэфиров и нитрилов может
протекать с замыканием кольца и образованием соответственно окси- и амйно-
изотрйазолов [98].
(С8Н5)2ССООС2Н5
N3
Из
Pd
(С8Н5)2ССООС2Н5-
n=nnh2_
(C8H5)2C —COH
(C8H5)2CCN
N3
(С8Н5)2С —cnh2
NZ \
Реакции замещения
Триазол содержит три, а бензотриазол — пять атомов водорода, способ-
ных замещаться на другие группы. Вследствие таутомерии группы, замещаю-
щие водород у атома азота в несимметричных 1,2,3-триазолах, занимают три
различных положения; в случае симметричных 1,2,3-триазолов возможно обра-
зование двух изомерных производных, имеющих заместитель у атома азота.
виц-ТРИАЗОЛЫ
313
Алкилирование и окисление у атома азота ви^-триазола. Если не считать
бензотриазолов, то проблему установления различий между изомерными
1д2,3-триазолами, имеющими алкильные заместители в положениях 1 и 2,
нельзя считать удовлетворительно решенной [99]. Алкилирование бензотриа-
зола алкилсульфатами или алкилгалогенидами ведет к Замещению преимуще-
ХХХ111
(CH3)2SO4
СН3
NO2 |
I N+
Нагревание
CH3SOj
XXXV
XXXIV
(CH3)2SO4
---------->
сн3
NO2 I
сн3
CH3SOj
^рева^ XXXV
ственно в положении 1, между тем как диазометан дает главным образом
2-метилбензотриазол [1]. Метилирование несимметричного 5-нитробензотриа-
зола приводит к двум соединениям: 1-метил-5- и 1-метил-6-нитробензотриазолам
[100]. Любопытно, что метилирование 4-нитро-6-метил- (XXXIII) и 4-нитро-
1,6-диметилбензотриазолов (XXXIV) дает только 7-нитро-1,5-диметилбензо-
триазол (XXXV) [100]. Это требует деметилирования исключительно у атома
азота, находящегося в лгета-положении по отношению к нитрогруппе; так
ведет себя иодистый 1,3-диметил-5-нитробензотриазолий, который при пиро-
лизе превращается только в 1-метил-5-нитробензотриазол [100]. В японском
патенте [101] описано получение бромистого 1-гексадецил-З-карбэтокси-
метил бен зотр и азол и я.
СН2СООС2Н5
(СН2)15СН3 (СН2)15СН3
При изучении различий между изомерными 1- и 2-алкил-1,2,3-триазолами
было сделано наблюдение, что бензилирование 1-метил-1,2,3-триазола и мети-
лирование 1-бензил-1,2,3-триазола дают одну и туже соль триазолия (XXXVI
или XXXVII), которая не алкилируется в положении 2 [99]. (Ср. реакцию
между фенилтриазолом и этиленсульфохлоридом, стр. 300.) Алкилирование
по незамещенному атому азота приводит к соединению XXXVI и аналогично
алкилированию 1-алкилбензотриазолов. Неизвестные соли 1,1-диалкилтриазо-
314
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
лия по своему строению сходны с 1-окисью 1,6-диметилбензотриазола [100].
НС=СН НС = СН
/ \ / \
СН3—N • N —СН2С8Н5 «-->• СН3—N N— CH2C8HS
+ / \ // +
N N
XXXVI
нс=сн сн3
/ \ /
N N
/+\
N СН2С8Н5
XXXVII
Метил- и бензил-1,2,3-триазолы [102] получены из ацетилендикарбоновой кис-
лоты и метил- и бензилазидов соответственно с последующим декарбоксилиро-
ванием. Один из них был также получен из серебряной соли триазола и йоди-
стого метила [5]. При метилировании 1,2,3-триазола диметил сульфатом полу-
чают как 1-метилтриазол (в преобладающем количестве), так и 2-метилтриазол
[103]. Алкилирование. 2-замещенных 1,2,3-триазолов не исследовалось.
Сопряженное присоединение бензотриазола к акриловой кислоте, акри-
лонитрилу, акриламиду, кротоновой кислоте, коричному альдегиду или бен-
зальацетофенону приводит к производным 1-бензотриазола [104], идентифици-
руемым с помощью ультрафиолетового спектра (двойной максимум при 255
и 283 л/р). Подобным продуктам присоединения, полученным из 1,2,3-триазола,
приписывается по аналогии строение 1-алкилтриазола [104]. Как бензотриазол,
так и 1,2,3-триазол в реакциях с формальдегидом и аминами дает основания
Манниха с заместителями в положении 1 [105]. Из формальдегида и бензо-
триазола получен 1-оксиметилбензотриазол [105], который при действии
тионилхлорида превращается в 1-хлорметилбензотриазол. Аналогичной реак-
цией из нитробензальдегида и бензотриазола получен нитрофенилкарбинол-
[104]. а-Бензотриазоилизомасляная кислота образуется при кипячении смеси
бензотриазола, едкого натра, хлороформа и ацетона [106].
В отдельных сообщениях описывается алкилирование производных бензо-
триазола в положении 2. Смесь 1,5,6- и 2,5,6-триметилбензотриазолов получе-
на при метилировании 5,6-диметилбензотриазола диметил сульфатом и ще-
лочью [78]. При метилировании 4,5,6,7-тетрахлорбензотриазола тем же спосо-
бом получена смесь 1- и 2-метилпроизводных [107]. Акрилонитрил, кротоновая
кислота и другие подобные им соединения реагируют с 4,5,6,7-тетрахлорбензо-
триазолом не по 1-, а по 2-положению, что, по-видимому, обусловлено влиянием
стерических факторов [107].
Метилирование 1-оксибензотриазола ставит задачу изучения сравнитель-
ного алкилирования у атомов кислорода и азота. При использовании метил-
сульфата соотношение продуктов алкилирования по кислороду и по азоту
в 2Л1 щелочи составляло 1,12 для гидроокиси лития; 0,97 для едкого натра;
1,02 для едкого кали; 1,17 для гидроокиси рубидия; 1,26 для гидроокиси цезия
и 1,53 для гидроокиси метилтриэтиламмония [108].
Ацилирование у атома азота в«^-триазолов. Реакция ацилирования у
атома азота известна для бензотриазолов и неизвестна для моноциклических
1,2,3-триазолов. Ряд N-ацильных и N-сульфонильных производных 1,2,3-триа-
зола получены другими методами, например присоединением ацилазида или
сульфонилазида к ацетиленовому производному или к натриевой соли производ-
ного малонового эфира.
виц-ТРИАЗОЛЫ
315
Ацилирование бензотриазолов происходит, по-видимому, в большин-
стве случаев по атому азота в положении 1 (или 3). Из 5-метилбензотриазола
и ацетилхлорида (или уксусного ангидрида) получена смесь 1-ацетил-4-
и 1-ацетил-5-метилбензотриазолов [109]. По имеющимся данным продукт,
полученный из 5-метоксибензотриазола и уксусного ангидрида, представляет
собой 2-ацетил-5-метоксибензотриазол (т. пл. 94—96°). Его изомеры — 1-аце-
тил-5-метоксибензотриазол (т. пл. 140—142°) и 1-ацетил-б-метоксибензотриа-
зол (т. пл. 107—108°) — синтезированы диазотированием соответствующих
ацетамидоанизидинов [ПО]. При действии на 5,6-диметилбензотриазол уксус-
ного ангидрида, бензоилхлорида или бензолсульфохлорида происходит ацили-
рование у атома азота в положении 1 [111], причем нет указаний о замещении
у атома азота в положении 2. Присоединение фенилизоцианата к бензотриазо-
лу приводит к 1-фенилкарбамилбензотриазолу [112].
CONHCeHs
Дезалкилирование и дезацилирование у атома азота ea^-триазола. Реак-
ция окислительного отщепления алкильных и арильных групп от атома азота
триазола рассматривалась выше (стр. 309). При гидрировании 1-бензил-1,2,3-
триазола над палладием на угле при 175° и давлении 28—84 ат образуется
1,2,3-триазол [102]; под действием алюмогидрида лития 1-бензоилбензотриазол
превращается в бензотриазол [113]. Исключительная устойчивость кольца
СН3
I N
H2N -Xх/ \
Нг, Ni-Ренея
CeFJeN = N—'
N — С8Н5
120°
62 ат
3
N
вц/4-триазола к гидрированию обнаружена также на примере восстановления
2-фенил-4-метил-5-амино-6-бензолазобензотриазола в 2-фенил-4;метил-5,6-ди-
аминобензотриазол [64]. Восстановение сульфометилата 1-метил-2-бензил-
бензотриазолия аммиачным раствором гидросульфита натрия протекает
с количественным выходом, при этом отщепляется бензольная группа в положе-
нии 2 и образуется 1-метилбензотриазол [79]. Легкость, с которой 1-оксиме-
СН2ОН-
СН2ОН
XXXVIII
тил-5-оксибензотриазол перегруппировывается в 4-оксиметил-5-оксибензо-
триазол (XXXVIII) [114], кажется несколько неожиданной, если учесть, что
ни алюмогидрид лития, ни водород над никелем Ренея, ни палладий на угле
не действуют на 1-оксиметилбензотриазол [113]. Сообщается, что аммонолиз
1-(2,4-динитрофенил)-4-карбометокси-5-окситриазола в метаноле при 100° сопро-
вождается образованием 4-карбометокси-5-окситриазола [115].
316
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
При реакции 1-бензоилбензотриазола сфенилмагнийбромидом образуются
трифенилкарбинол и бензотриазол [116].
Отщепление ацильных групп путем гидролиза обычно проходит успешно
с минеральными кислотами [1, 117] и может протекать у диазотированных
ароматических моноацильных производных о-диаминов с замыканием три-
азольного кольца (стр. 302). Сольволиз 1-ацетил-5,6-диметилбензотриазола
в абсолютном этаноле идет быстрее, чем в 75%-ном спирте [37]. Сообщается,
что гидролитическое отщепление бензолсульфонильной группы происходит
в тех случаях, когда спиртовый раствор 1-бензолсульфонил-5-бромнафто-
[1,2]-триазола оставляют стоять на некоторое время [118, 119]. Термическая
изомеризация 1-бензолсульфонил-5-метилбензотриазола в 1-бензолсульфо-
нил-6-метилбензотриазол [118, 119], по-видимому, связана с миграцией бензол-
сульфонильной группы от атома азота в положении 1 к азоту в положении 3
(см. стр. ЗГ4).
Реакции замещения по атому углерода в в«^-триазолах. Реакции элек-
трофильного замещения по атому углерода в триазольном ядре идут с трудом.
Нитрование 2-фенил-1,2,3-триазол-4-альдегида, соответствующей кислоты или
2-фенил-4-метил-1,2,3-триазола проходит по фенильной группе [90,. 120, 121].
Вхождение нитрогруппы в пара-положение указывает на то, что триазольное
кольцо может активировать ароматический заместитель в положении 2.
Предполагается, что хлорсульфоновая кислота превращает 2-фенил-1,2,3-
триазол в 2-(п-хлорсульфонилфенил)-1,2,3-триазол [51]. В результате восста-
новления окисей 2-арил-1,2,3-триазола соляной или 'бромистоводородной
кислотой высвобождается соответственно хлор или бром, которые замещают
водород в пара-положении арильного остатка [122]. По-видимому, реакции
ароматического электрофильного замещения могут проходить также в орто-
и (или) пара-положения фенильных заместителей триазола, находящихся
у атомов азота в положении 1 и 3.
В 1947 г. установили, что при взаимодействии триазола с бромом обра-
зуется 4,5-дибром-1,2,3-триазол [123]. Хлор в воде при 0°, в водном растворе
ацетата натрия при 90°, в уксусной кислоте при 20 или 100°, а также в присут-
ствии железа или иода не хлорирует триазола [103]. Однако хлор в водной
среде реагирует с 1-метил-1,2,3-триазолом, давая 1-метил-4-хлор-1,2,3-три-
азол, а хлор, бром или иод в хлороформе превращают 4-метил-1,2,3-триазол
СН3С-СН
N N —О
I
с6н5
СН3С-СН
С6Н5С1-П
в 4-метил-5-галоген-1,2,3-триазол [103]. Бромирование 2-метил-1,2,3-триазола
приводит к 2-метил-4,5-дибром-1,2,3-триазолу [103]. Галогенирование три-
азола по атому азота наблюдается в случае 4,5-диметил-1,2,3-триазола [103].
Под действием гипобромита натрия триазол превращается в 4,5-дибром-
или 4,5,М-трибром-1,2,3-триазол. Аналогичные результаты получены при
вы«-ТРИАЗОЛ Ы
317
реакции с гипохлоритом натрия [103].
НОС = СН
HONO
——>
HOC = CNO
HONO
-СО2
НОС=ССООН
Электрофильное замещение по атому углерода триазольного кольца
облегчается присутствием электронодонорной группы, находящейся у другого
атома углерода. Нитрозирование 1-фенил-5-окси-1,2,3-триазола дает 1-фенил-
5-окси-4-нитрозо-1,2,3-триазол [124]. Этот же продукт получен из 1-фенил-
5-окси-1,2,3-триазолкарбоновой-4 кислоты [124]. Как это видно на примере
диазотированного n-толуидина, 4-окси-1,2,3-триазол сочетается с ароматиче-
скими диазониевыми соединениями, образуя 4-окси-5-арилазо-1,2,3-триазолы
[125]. Галогенирование, нитрование, сульфирование или другие реакции
электрофильного замещения в активированном триазольном кольце не изучены.
Реакции замещения в бензотриазоле. Нитрование бензотриазола и 1-метил-
бензотриазола дает соответственно 4-нитробензотриазол (31%) и 1-метил-7-
нитробензотриазол (32%) [126]. Как 1-арил-, так и 2-арилбензотриазолы
нитрируются в положение 4 (7) [127]. Сравнение реакции нитрования бензо-
триазола и других «бензазолов», приводящей к мононитропроизводным, обна-
руживает сходство с бензофуразаном (бензо-1,2,3-оксадиазолом), который
тоже нитруется в положение 4, но не с бензимидазолом, индазолом или 2,3-
диметилиндолом, которые нитруются в положение 5. Относительно получения
динитропроизводных бензотриазола данные отсутствуют. Представляет инте-
рес изучить, будет ли 5-нитробензотриазол (из диазотированного 4-нитро-1,2-
диаминобензола) нитроваться в соседнее Р(или 6)-положение (как это наблю-
далось у 5-нитробензимидазолов и 6-нитроиндазола) или же в «(или 4)-положе-
ние [128].
Хлорирование бензотриазола и 2-метилбензотриазола смесью соляной
Ф азотной кислот (царской водкой) приводит к образованию 4,5,6,7-тетрахлор-
бензотриазола (87%) и 2-метил-4,5,6,7-тетрахлорбензотриазола (50%) [107].
В этих условиях из 1-метилбензотриазола получен неидентифицированный
продукт, которому приписано строение Гметил-4,5,6-трихлорбензотриазола
[107], а из 5-метил- или 5-бромбензотриазола — 4,6,7-трихлорпроизводные
[129].
Реакции замещения в ряду 4- и 5-окси-, а также4- и 5-аминобензотриазолов
идут легче; они изучены более широко [114, 127]. Ниже приводится схема
превращения 5-аминобензотриазола в 5-(«, |3-дихлорвинил)триазол-4-илглиок-
силовую кислоту.
С12
SnCl2
Na ОН
С1 н
HNOg
I N
ci-ZV \
I [I /
</Y \
О н
С1
I
С1СН = сс= ссосоон
HN N
318
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
При нагревании 5-оксибензотриазола с углекислым калием и двуокисью
углерода при 160—180° около 24 час образуется 5-оксибензотриазолкарбоно-
вая-4 кислота [130]. Активность 4-положения также проявляется в реакции
сочетания между 2-арил-5-аминобензотриазолом и арилдиазониевыми соедине-
ниями, приводящей к образованию 2-арил-4-арилазо-5-аминобензотриазола
[131], а также при нитровании 1-ацетил-5-метоксибензотриазола в 4-нитро-
5-метоксибензотриазол [131].
N = NAr'
N |N
H2N-/4/ \ Ar'N2X H2N-/x/ \
I I N —Ar-------> N —Ar
v\ / \/\ /
N N
Аммонолиз галогенпроизводных ви^-триазолов. При образовании 4-ани-
лино-1,2,3-триазола при нагревании 1-фенил-5-хлор-1,2,3-триазола со спирто-
вым раствором аммиака [18], по-видимому, сначала происходит замещение
атома хлора аминогруппой, как это видно на примере получения 1-метил-
5*анилино-1,2,3-триазола при действии анилина на 1-метил-5-хлор-1,2,3-
триазол [18]
С1С=СН
NH3
H2NC=CH~
CeH5 —N7 ~N
CeH5NHC = CH
sf 'n
Диазотированные аминотриазолы. Диазотированные аминогруппы, свя-
занные с атомом азота или углерода, легко замещаются водородом. Один
из методов получения триазола состоит в обработке 1-амино-1,2,3-триазола
смесью окислов азота [50]. Замещение аминогруппы водородом при диазоти-
ровании является типичным для N-аминоазолов и наблюдалось, в частности,
/ у 1-амино-4,5-дифенил- и 4,5-диметил-1,2,3-триазолов [132, 133]. Диазотиро-
вание 2-фенил-4-амино-5-окси-1,2,3-триазола и последующее нагревание
с порошком меди дает 2-фенил-4-окси-1,2,3-триазол [134]. В реакции Занд-
мейера диазотированный 1,4-дифенил-5-амино-1,2,3-триазол, обработанный
хлоридом меди (I), превращается в 1,4-дифенил-5-хлор-1,2,3-триазол [135].
Из диазотированных амино-1,2,3-триазолов были также получены соответ-
ствующие окситриазолы. Кипячение в воде диазотированного 2-фенил-4-
метил-5-амино-1,2,3-триазола дает ожидаемый окситриазол [136]. Другие
типичные диазореакции представлены сочетанием диазотированного 1-фенил-
5-амино-1,2,3-триазола с р-нафтолом и диазотированного 2-фенил-4-амино-
5-ацетамидо-1,2,3-триазола с солью R [134]. Диазотирование 2-фенил-4,5-
диамино-1,2,3-триазола ведет к образованию N-фенилтриазолотриазола [134].
N = CNH2
CeHs-Z
N=CNH2
HONO
N=C—N
Реакции боковых цепей у виц-триазола
Алкильные и карбинольные производные триазола в положениях 4 и 5
изучены мало. По-видимому, метильная группа в положении 4 и 5 галогени-
руется труднее (см. стр. 316), чем метильная группа в толуоле. Под действием
тионилхлорида 1-оксиметилбензотриазол превращается в 1-хлорметилбензо-
виц-ТРИАЗОЛЫ
319
триазол, который при восстановлении алюмогидридом лития может дать
1-метилбензотриазол [113]. При получении 2-фенил-1,2,3-триазол-4-илуксусной
кислоты из 2-фенилтриазол-4-карбинола последний действием бромистоводо-
родной кислоты переводят в бромид, который, взаимодействуя с цианистым
калием, дает триазоилацетонитрил, превращающийся при гидролизе в ука-
занную кислоту [120].
Альдегиды триазола оказались ценными исходными продуктами для
получения разнообразных производных 1,2,3-триазола. 2-Фенил-1,2,3-триа-
зол-4-альдегид (XXXIX) по реакции Канницаро превращается в 2-фенил-
1,2,3-триазол-4-карбинол [121], а с реактивом Гриньяра образует ожидаемый
вторичный спирт [121 ]. Реакции конденсации соединения XXXIX с ацетофено-
ном или уксусным ангидридом ведут к образованию соответствующего а,|3-
непредельного кетона и кислоты [121]. В синтезе аминокислот по методу
Штрекера соединение XXXIX нормально реагирует со смесью цианистого
калия и хлористого аммония [120].
Из альдегидов триазола получены азлактоны. 1-Фенил-1,2,3-триазол-
4-альдегид и гиппуровая кислота дают азлактон (XL, R = С8Н5), который
переводят в замещенную гиппуровую кислоту (XLI, R = CeHs) действием
горячей щелочи с последующим восстановлением образовавшейся а,|3-непре-
дельной кислоты окисью платины. При восстановлении красным фосфором
НС = С—СН = С — С=О НС=ССН2СНСООН
rn" 'n n о rn ' n nhcoc6h5
Cells
XL XLI
HC = CCH2CHCOOH
/ \ I
RN N NH2
\.Z
. N
hc=cch2ch2nh2
rZ n
XLII
XLIII
и иодистым водородом образуется аминокислота (XLII, R = CeH5). Аналогич-
ные соединения (XL, XLI, XLII, R = CH3) синтезированы из 1-метил-1,2,3-
триазол-4-альдегида [137]. Продукт конденсации 1-фенил-1,2,3-триазол-4-
альдегида с нитрометаном восстанавливали над палладием на угле в оксим,
который в свою очередь восстановлением над окисью платины превращали
в замещенный этиламин (XLIII, R = С8Н5) [137]. Продукту конденсации
альдегида с аммиаком приписывается строение XLIV. Из 1-метил-1,2,3-триазол-
4-альдегида и родамина получен продукт конденсации, который по методу
ОН
I
11С=CCHN = СНС = СН
hc=cch2ch2nh2
CHg-Z N
XLIV
XLV
Шихана и Робинзона [10, 138] может быть превращен в замещенный оксим
пировиноградной кислоты. Из последнего пиролизом может быть получен
нитрил, который восстанавливается до амина (XLV) [137].
Диальдегид— 1-гексил-4,5-диформил-1,2,3-триазол (II, стр. 298) — при
конденсации с глиоксальбисульфитом дает 1-гексил-4,7-дикето-4,7-дигидро-
320
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
5,6-диоксибензотриазол [11].
О С8Н13
II n
CHOHSO3Na но \
I +11-^ N
CHOHSO2Na HO-'Vk /
II N
О
Следует упомянуть о двух реакциях хлорангидридов триазолкарбоновых
кислот. Хлорангидрид 2-фенил-1,2,3-триазолкарбоновой-4 кислоты, реагируя
с диазометаном, образует диазокетон, который при действии аммиака в присут-
ствии спиртового раствора азотнокислого серебра дает соответствующий три-
азоилацетамид [139]. С бромистоводородной кислотой диазокетон образует
а-бромкетон, который алкилирует метиламин. Полученный продукт может
быть восстановлен в замещенный этаноламин [139]. В другом примере хлор-
ангидрид 1-фенил-5-метил-1,2,3-триазолкарбоновой-4 кислоты действием пере-
киси натрия превращается в перекись, которая при гидролизе опять дает
кислоту [140].
Ангидрид 2-фенил-1,2,3-триазолдикарбоновой-4,5 кислоты конденсирует-
ся в присутствии серной кислоты с гидрохиноном, образуя 2-фенил-4,9-дигид-
ро-4,9-дикето-5,8-диоксинафто-[2,31-триазол [ 141 ].
О
II
C-C=N.
+ О< | )N- С8Н5
'С —C=NZ
H2SO4
ОН О
N
Из о-фенилендиамина и соответствующих о-хинонов были получены
триазолфеназины [127].
N—N —СвН5 N —N—С8Н5
|| Isl N || N
Этилацетат конденсируется в присутствии металлического натрия с 1-фе-
нил-4,5-дикарбэтокси-1,2,3-триазолом; при этом отщепляется карбэтоксиль-
ная группа и образуется 1-фенил-4-(а-карбэтоксиацетил)-1,2,3-триазол [142].
С2Н5ОСОС = ССООС2Н5 НС = ССОСН2СООС2Н5
/ \ сн3соос2н8 / \
C8H5-N N ---------------------> С6Н5—N N
\ Z \ Z
N N
Ацилоксирадикал, образующийся при пиролизе перекиси диацила 1-фе-
нил-5-метил-1,2,3-триазолкарбоновой-4 кислоты, вступает в реакцию с гало-
генопроизводными бензола, давая фениловый сложный эфир [143].
СН3С—ССОО- СН3С—ССООС8Н5
/ \ С6Н5С1 / \
C6H5N N---------------> C8H5N N
\ z \ Z
N N
виц-ТРИАЗОЛЫ
321
Выделение азота из вн^-триазольного кольца
Пиролиз 1-ацетил-1,5-дигидро-5-фенил-1,2,3-триазоло[с( [триазола в спирте
сопровождается выделением азота и образованием 2-фенил-4-ацетиламино-
1,2,3-триазола [134]. По меньшей мере в одном случае при пиролизе триазола,
по-видимому, образуется азациклопропен (стр. 311). Пиролиз 1-арилбензо-
N N
СОСНа
с2н5он
CH3CONHC—СН
триазолов при температуре, как правило, выше 350° является общим методом
получения карбазолов [144] и карболинов [145]. В качестве растворителя
для этого используется сиропообразная фосфорная кислота [146]. В аналогич-
ной реакции 1-бензилбензотриазол превращается в фенантридин [147].
Хотя триазольное кольцо устойчиво по отношению к восстановителям,
однако под действием натрия в бутаноле 1-алкилбензотриазолы превращаются
в моноалкил-о-фенилендиамины [148]. 1,4-Диалкил-1,2,3-триазолы, устойчи-
N
СН2—
вые к расщеплению кольца действием амальгамы натрия в метаноле или цинка
в,соляной кислоте, могут быть восстановлены с хорошим выходом в амины
натрием в абсолютном спирте [9].
Кислотность eeq-триазольного кольца
Кислотность вы^-триазола, по-видимому, меньше, чем у тетразола
(К = 1,62-10“5) [149], но измерена не была. Производные, диссоциация кото-
рых происходит за счет водорода, связанного с атомом азота кольца, являются
слабыми кислотами. К их числу относятся: 4-фенил-5-(п-нитрофенил)ймино-
1,2,3-триазол, р/Са 6,60 [150]; 4-фенил-5-(м-метоксифенил)амино-1,2,3-триазол,
рКа 7,91 [150]; 4-фенил-1,2,3-триазол, рКи 7,68 [151 ]. Исключение составляет
21 Заказ № 9 78
322 МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
4,5-дициан-1,2,3-триазол, рКа2,53 [152], у которого исключительное значение
может иметь резонансная стабилизация соответствующего аниона. Триазол-
карбоновые кислоты и окситриазолы, по-видимому, более сильные кислоты,
чем аналогичные производные бензойной кислоты и фенола [151, 153].
4-Окси-вы/{-триазол титруется как одноосновная кислота [154], в то время как
окси-вы^-триазолкарбоновые кислоты титруются как двухосновные кислоты
[153]; в качестве индикатора применяется фенолфталеин. При титровании
едким натром 1,2,3-триазол-4-альдегида и 1-метил-1,2,3-триазолкарбоновой-4
кислоты требуется 1 моль щелочи, а в случае 1,2,3-триазолкарбоновой-4 кисло-
ты— 2 моля [123]. Получены продукты присоединения 1-фенил-1,2,3-три-
азолальдегида с едким кали или метилатом натрия [123]. Кислотность бензо-
триазола (рКа 8,2) [144] повышается, если атомы водорода в бензольном
кольце замещаются на хлор, как это имеет место у 5-хлорбензотриазола,
рКа 7,7 [155] и у 4,5,6,7-тетрахлорбензотриазола, рКа 5,48 [107]. Последний
образует с гидроокисью бензилтриметиламмония взаимопревращаемые соли
в молекулярных соотношениях 2:1 и 3 : 1 [107].
Основность eeq-триазольного кольца
Известен неустойчивый хлоргидрат вы^-триазола [86] и хлороплатинат
1-фенил-1,2,3-триазола [4]. Если у атомов углерода или азота триазольного
кольца имеются алкильные группы, то основность соединения возрастает [156].
Бензотриазол или 1-алкилбензотриазолы в отличие от 2-алкилбензотриазолов
реагируют с соляной кислотой или хлористым водородом в эфире [79]. Пред-
положение, что 2-алкил-вы^-триазолы обладают меньшей основностью, чем
замещенные в положении 1 изомеры, подтверждается тем, что 2-метил-1,2,3-
триазол не образует нерастворимой серебряной соли, хлоргидрата или пикрата
[1031, а 1-метилбензотриазол более растворим в азотной кислоте, чем 2-метил-
бензотриазол [107]. Бензотриазол, 5-хлор бензотриазол и 2-метилбензотриазол
в отличие от 4,5,6,7-тетрахлорбензотриазола растворимы в царской водке
[107]. 1-Бензил-1,2,3-триазолы образуют кристаллические хлоргидраты,
которые не получены для соответствующих 1-фенил-1,2,3-триазолов [91.
Соли и комплексы вы^-триазольного кольца
Применение триазолов в фотографии и аналитической химии связано
с их способностью образовать производные металлов. Как бензотриазол, так
и его 5-бромпроизводное могут применяться для количественного определения
ионов серебра в присутствии ионов меди, никеля, висмута, таллия, свинца,
кадмия, цинка, железа, кобальта и хлора [157, 158]. Изучены комплексы
бензотриазола с кобальтом [1591 и палладием [160]. 1,2,3-Триазол образует
также нерастворимую серебряную соль с азотнокислым серебром.
4- и 5-Окси-еиц-триазолы, как и другие триазолы, дают характерные
цветные реакции с хлоридом железа (III) [84].
, Сообщается о бензотриазольных производных дибензолсульфонимида.
[161], трифенилфосфина 1162] и малеинового ангидрида [163]. С малеиновым
ангидридом 1-метил-, но не 2-метилбензотриазол дает аналогичную реакцию
[163]. Некоторые полинитроароматические соединения образуют с 2-фенил-
нафтотрназолом комплексные продукты присоединения в соотношении
1 : 1 [164].
Водородные связи в eeq-триазолах
Определение молекулярных весов бензотриазолов в нафталине [165]
и бензоле [166] показало, что они представляют собой высокоассоциированные
линейные полимеры, в которых водород связан с атомом азота триазола.
йич-ТРИАЗОЛЫ
323
Между тем 1- и 2-замещенные бензотриазолы являются мономерами. Наличие
внутримолекулярной водородной связи объясняет большую летучесть, лег-
кость восстановления, более низкую кислотность и меньшую ассоциацию
4-нитробензотриазола по сравнению с 5-нитроизомером [126].
Спектры поглощения в«ч-триазолов
Ультрафиолетовые спектры поглощения виц-триазолов, включая бензо-
триазолы, изучены весьма детально [167].
Первые ультрафиолетовые спектры поглощения моноциклических виц-
триазолов были сняты в 1954 г. Родоначальное соединение показывает
максимум при 210 л/ц (log е 3,6), который смещается для 4-алкилпроизводных
к 215—216 M[i (log е 3,65) [11]. Максимум при 245 л/р, (log е 4,2) и минимум
при 218 л/р, (log е 3,6) характерны для двух соединений: 4-фенил-ви/{-триазола
и бифенила [12]. Аналогичный максимум при 243 л/р, (log е 4,0) имеется
у 1-фенил-ви/{-триазола [27], однако его иодметилат, которому приписывается
строение иодида 1-фенил-З-метилтриазолия, поглощает при 251,6 л/р, (log е 3,9)
[27]. 2-Фенил-1,2,3-триазол поглощает при 262 л/р, (log е 4,21) [167]. Поглоще-
ние в инфракрасной области для кольца вш{-триазолов лежит в интервалах
8,8—9,2 р, и 9,8—10,3 р, [12, 27].
В растворах концентрированных кислот и оснований бензотриазол погло-
щает при 274 л/р,, тогда как в слабокислых или нейтральных растворах имеется
два максимума поглощения: при 260 и 274 л/р, 1155]. Принято считать, что
для бензоидной .хромофорной группы характерно поглощение с меньшей
длиной волны, а для хиноидной — с большей [155]. Свойственный 1-алкил-
бензотриазолам двойной максимум поглощения при 255 л/р, (log е 3,81) и 283 лр,
(log е 2,68) и единственный максимум при 275 л/р, (log е 3,90), который дают
2-алкилбензотриазолы, позволяет разделять эти изомеры 1104]. Приписы-
ваемая бензотриазолу хиноидная хромофорная структура, которая обычно
фигурирует при рассмотрении 2-замещенных бензотри азолов, плохо согласует-
ся с химическими свойствами этих соединений [107].
Для бензотриазола и 1- и 2-метилбензотриазолов были изучены спектры
комбинационного рассеяния. Флуоресцирующие бензотриазолы применяются
в красителях [169].
Различные свойства
Благодаря стабильности триазольного кольца многие производные три-
азола могут перегоняться неизмененными, как, например, 1,2,3-триазол
с т. пл. 23° и т. кип. 204° (740 мм). При попытке перегонять бензотриазол
при температуре 160° (2 мм) происходит сильное разложение со взрывом
[170]. Галогенпроизводные «///{-триазола бурно взрываются при температуре
выше 260°; 1-фенил-4-метил-5-окси-1,2,3-триазол разлагается при 134°, а при
более высокой температуре взрывает; триазол карбоновые кислоты нестойки
'выше 200°.
Многие триазолы растворимы в воде и гигроскопичны; так, например,
4,5-диметил-1,2,3-триазол образует тригидрат с т. пл. 97°.
Дипольный момент eu/j-триазола равен 1,77 р, [171]; он ближе к диполь-
ному моменту пиразола (1,57 ц), а не имидазола (3,84 ц). Это позволяет
утверждать, что таутомерная структура с атомом водорода в положении 2
наиболее точно отражает строение молекулы. Дипольный момент бензотриазола
равен 4,07 ц, 1-фенилбензотриазола — 4,08 р, а , .1-метилбензотриазола —
4,16 р. Эти величины заметно отличаются от дипольного момента 2-фенил-
бензотриазола, который равен 0,97 р [171, 172].
Восстановительные потенциалы различных производных триазола имеют
следующие значения: бензотриазол-4,7-хинон (XLVI), £о = 0,526 в; 1-фенил-
21*
324
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
бензотриазол-4,7-хинон (XLVII), Ео = 0,539 в; 2-фенилбензотриазол-4,7-хинон
(XLVIII), Ео = 0,487 в. Эти данные заставляют предположить, что последнее
соединение не так легко ароматизируется [173].
Знание показателей преломления и удельных весов позволило вычислить
величины молекулярных рефракций eu^-триазолов. Вычисленная рефракция
для триазольного скелета C2N3H равна 13,11 ±0,16 [12]. При изучении
показателей преломления замещенных в положении 2 бензотриазолов
оказалось, что экзальтации этих соединений сравнимы с таковыми для 2-ал-
килиндазолов, которые могут иметь и хиноидную структуру.
Теплота сгорания бензотриазола равна 794,97 ± 0,21 ккал/моль. Эта вели-
чина позволяет подсчитать энергию резонанса, равную 57 ккал/моль [175].
Для сравнения может быть приведена теплота сгорания изомерного фенил-
азида, равная 817,4 ккал/моль [176].
XLVI
XLVII
XLVIII
Вычисленные электронные плотности у атомов азота eu^-триазола равны
1,406 (в положениях 1 и 3) и 1,356 (в положении 2) [177].
Описаны оптические кристаллографические свойства некоторых виц-
триазолов [178], а также полиморфизм бензотриазола [179].
Стирол в присутствии бензотриазола полимеризуется при 70° [180].
Образование предполагаемого промежуточного соединения [125] при
превращении 1-фенил-4-фенилазо-5-окситриазола в анилид 2-фенилтетразол-
карбоновой-5 кислоты (кипячение с ледяной уксусной кислотой) напоминает
получение триазолов из а-диазокетонов и первичных аминов (стр. 302)
НС = СОН
N N— СвН5
\ /
N
СНзСООН
CeH5N = NC-COH -
n2 riceH5_
,n=cconhc6h5
-> c8h5n/ I
XN = N
Перегруппировка 1-фенил-4-бензоилоксимино-1,2,3-триазолона-5 в 1-фе-
нилтетразолкарбоновую-5 кислоту проходит в холодном водном растворе
едкого натра [18Г].
CeH5COON=C—N
O = CZ N
NaOH
-10°
N —N
НООСС N
СбЩ
Биохимические свойства вн^-триазолов
Рост Micrococcus pyogenes aureus, Е. coli, Klebsiella pneumoniae и Serratia
marcescens в некоторой степени ингибируется 4-алкил-ви^-триазолами. Эти сое-
динения неэффективны в отношении роста саркомы 180 у мышей [12]. Некого-
симм-ТРИ АЗОЛ Ы
325
рые триазольные аналоги гистамина обнаруживают антигистаминную и анти-
ацетилхолиновую активность. eu^-Триазольное кольцо является антагонистом
имидазольного кольца в триазольных аналогах аденина и гуанина [10, 1381.
Сообщается о мидриатическом действии этаноламинных производных виц-
триазола. Аминотриазолкарбоновые кислоты обладают сосудорасширяющим
действием, а также являются ингибиторами роста [182]. Сообщается, что
в некоторых случаях бромид 1-додецил-З-бензилтриазолия обладает таким
же мутагенным действием на Chlamydomonos eugametos, как и рентгеновские
лучи [183].
При исследовании бензотриазолов как антагонистов роста оказалось,
что 4-метокси-6-нитробензотриазол угнетает простейшее одноклеточное Т. gelii,
нуждающееся в гуанине, и тормозит развитие эмбриона R. pipiens [184].
Упомянутое выше соединение — 4-окси-6-нитробензотриазол — ингибирует
деление клеток Е. coli [185]. Соли Ы,Ы'-диалкилбензотриазолия обладают
/ермицидной активностью [101].
симм-ТРИ АЗОЛЫ
Методы получения
Внутримолекулярные конденсации. Циклизация в щелочной среде. Цикли-
зация ацильных производных семикарбазидов, тиосемикарбазидов или амино-
гуанидинов в щелочных растворах представляет собой широко применяемый
метод получения сил<л<-триазолов. Исходные вещества получают ацилированием
семикарбазида, тиосемикарбазида, аминогуанидина или их производных.
Необходимые ацилсемикарбазиды или ацилтиосемикарбазиды могут быть
синтезированы также взаимодействием ацилгидразинов или тиокарбогидрази-
дов с цианатом или тиоцианатом калия [186, 187].
Известно небольшое число окситриазолов, полученных из ацилсемикар-
базидов в щелочном растворе. Гелен [186] указал, что 3-окси-5-алкил-с«лои-
триазолы получаются по этому методу с 40—50%-ным выходом, однако соот-
ветствующие 5-арилтриазолы образуются с более низкими выходами. Согласно
данным ранних работ, аналогичной реакцией из ацилированных производных
1-фенилсемикарбазида [188] получены 1-фенил-3-окси-5-алкил-с«л<л<-триазолы.
Этот метод был успешно применен для синтеза сил<л<-3,3'-диокси-5,5'-дитриазо-
ила (I) [186]. N-Циан-М'-ацилгидразины, полученные из соответствующего
гидразида или бромистого циана, также превращаются в щелочном растворе
в окситриазолы [186].
R'
RCONNHCONH2
R' = H, СеН5
кнсо3
HN —N
RC7
Ч
СОН
N-NH
кон
(H2NCONHNHCO)2------->
При получении смлои-триазолов из ацилтиосемикарбазидов применение
сильной щелочи позволяет уменьшить или совсем избежать образования
тиадиазолов [189]. Ряд 3-алкил-5-меркапто-1,2,4-триазолов (III) был получен
действием метилата натрия [190] или углекислого натрия [191] на соответ-
326
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И ЕЕНЗОТРИЛЗОЛЫ
ствующий 1-ацилтиосемикарбазид (II, R' = R" = Н) или 4-ацилтиосемикар-
базид. Изомерные 1-метил- и 4-метилпроизводные соединения (III) образуются
R' HN-N
I CHgONa 7 \
RCONHNCSNIIR" ----------•- HSC СК
II X /
N
III
аналогичным путем из 1-ацил-2-метилтиосемикарбазида (11, R' — СН3, R" Н)
или 1-ацил-4-метилтиосемикарбазида (II, R' Н, R" - СН3) соответственно
[190.]. Действием едкого натра карбэтокситиосемикарбазид (II, R =- ОС2Н;,,
R' = R" -= Н) и карбэтоксифенилтиосемикарбазид . (Il, R ; ОС2Н5,
R' = Н, R" = СеН5) можно превратить в 3-окси-5-меркапто-1,2,4-триазол
(III, R = ОН) (тиоуразол) и его 4-фенильное производное [1921. Иминотио-
уразол или его таутомер (III, R -- NH2) получен из бпс-тиомочевины
(H2NCSNH)2 [1931. Меркаптотриазол (III, R =- Н) образуется из 1-этокси-
метиленсемикарбазида (H2NCSNHN=CHOC2H5) в водном растворе угле-
кислого натрия 1194 ]; эта методика сходна с методом получения 3-меркапто-
4-амино-1,2,4-триазола из тиокарбогидразида и этилортоформиата [194].
В процессе получения тиокарбогидразида из диэтилксантата и гидразина
образуется в небольшом количестве 3-меркапто-4-амино-5-гидразино-1,2,4-три-
азол [1951. При этом может происходить внутримолекулярная конденсация
дитиосемикарбазида.
. N—N
n2h4 Ч,
4C2H5)2CS3------> (H3N2)2CS —> (H3N2CSNH)2 —> HSC c —n2h3
N
I
nh2
Циклизация фенилкарбамилтиосемикарбазида (II, R = CeH5NH, R' =
= R" = H) приводит к тиоуразолу и 4-фенилтиоуразоЛу в зависимости
от того, выделяются ли соответственно анилин или аммиак. Отмечается, что.
N —N
NaOH У Ч
XCSNHNHQ -NH)NH2. -------> HSC C-NH2
IV \ /
N
I
R
V
первая реакция преобладает 1196]. Меркаптотриазоламины (V) образуются
при циклизации в щелочной среде Ц-алкил(или арил)-М'-гуанидинотиомоче-
вин (IV, X = RNH или ArNH) или гуанидинодитиокарбаминовых кислот
jIV, X = SH) [197].
Ациламидогуанидины близки по своим свойствам к соединению IV и пре-
вращаются в соответствующие 5-амино-силг.и-триазолы при нагревании или
под действием водного раствора соды [198]. При действии хлористого бензоила
на 2-метйлгуанидин в растворе едкого натра проходит циклизация 1-бензами-
до-2-метилгуанидина, приводящая к образованию 1-метил-3-фенил-5-амино-
1,2,4-триазола, выделенного в форме бензоильного производного [199].
СН3 Н3С—N-N
CeH5CONHNC( = NH)NH2 -» CeHsCONHcf *C-CeH5
симм-ТРИ АЗОЛЫ
327
Сообщается о превращениях фенилазоацетальдоксима в присутствии
йодистого метила и метилата натрия, а также соответствующего N-метилпро-
изводного в присутствии метилата натрия в 1-фенил-3-метил-1,2,4-триазол
1200].
СН3 СН3 H5C,-N-N
CH3ONa i | CH3ONa / %.
CeH5N = NC(CH3) = NOH------> CeH5NN = CN = O----------> НС C—CH,
cH3i /
N
Замыкание цикла в кислой среде. Склонность ацилтиосемикарбазидов пре-
вращаться в присутствии кислот преимущественно в тиадиазолы, а не в три-
азолы [201] ограничивает число производных силл-три а зол а, которые могут
быть получены этим путем. В концентрированной соляной кислоте из карба-
милтиосемикарбазида (VI, R = Н) образуется тиоуразол (III, R = ОН),
однако фенилкарбамилтиосемикарбазид (VI, R = СвН5), по-видимому, пре-
вращается в 2-амино-5-окситиадиазол [201]. В концентрированной соляной
кислоте из соединения VII получен 4-аминотиоуразол [187].
RNHCONHNHCSNH2 h2nnhcsnhnhcsnh2
VI VII
Ацильные производные аминогуанидина (но не семикарбазида) 1188]
превращаются в кислой среде в силл-триазолы. Дегидратация аминогуанидин-
карбоната концентрированной серной кислотой рекомендуется как метод
получения 3-амино-1,2,4-триазола 1203]. Синтез 3-амино-5-алкил-1,2,4-три-
азолов осуществлен нагреванием аминогуанидиннитрата с карбоновой кисло-
той [204]. Дегидратация кислотой бензальдегидного производного гидразида
бензгидроксамовой кислоты ведет к образованию 3,5-дифенил*-!,2,4-триазола
[205].
CeH5 HN — N
I н+ / \ ’
c6h5ch=nnhc=noh —’• СвН5С ссвн5
ч//
М,М'-бис-Бензимидилгидразин lC(iHsC( NH)NH]3, полученный в резуль-
тате реакции двух молекул бензиминоэфира с гидразином, устойчив по отно-
шению к щелочам, однако в разбавленных кислотах или в кипящей уксусной
кислоте превращается в 3-,5-дифенил-1,2,4-триазол [206]. Это же соединение
получено из бензоилгидразида и бензонитрила в присутствии бензолсульфо-,
кислоты [207]. В качестве промежуточных соединений в этой реакции обра-
зуется М-бензоил-М'-бензимидилгидразин С8Н5С(= NH)NHNHCOCeH5.
Замыкание некоторых триазольных колец осуществляется путем дегид-
рирования. По этому методу продукты конденсации (VIII) «дицианфенил-
гидразина» с альдегидами [208], семикарбазонами или тиосемикарбазонами
CN Н5С6—N-N
I / ч
RCH = N-C = NNHCeH5 -> . RC CCN
VIII % /
N
могут быть превращены в силл-триазолы. Хлорид железа (III) окисляет про-
изводное 2-бензилсемикарбазида бензальдегида и производное 2-метилсеми-
карбазида n-анисового альдегида в соответствующие 1,3,5-тризамещенные
328
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
сылш-триазолы [209, 210]. В результате подобного окисления тиосемикарбазо-
R RN —N
I FeCl3 / Ч
ArCH = NNCONH2-------НОС САг
нов можно ожидать образования как аминотиадиазолов, так и меркаптотриазо-
лов. Алкилирование у атома серы, предшествующее окислительной циклиза-
ции, по-видимому, исключает образование тиадиазола [211]. По этому методу
из тиосемикарбазона бензальдегида, а также его S-метилпроизводного (IX,
R = С0Н5, R' = СН3) получены 3-фенил-5-метилтио-1,2,4-триазол [212]
(X, R = CeH5, R ' = СНз), а из тиосемикарбазонап-ацетиламинобензальдегида,
а также его S-бензилпроизводного (IX, R = n-CH3CONHC0H4, R' =
= С0Н5СН2) — 3-(п-ацетиламинофенил)-5-бензилтио-1,2,4-триазол (X, R =
= n-CH3CONHC0H4, R'=C0H5CH2) [213].
SR' HN — N
I Feels / 4
RCII^NN^CNH2 ---------- R'SC CR
ix 4 /
N
X
Окислительная циклизация в присутствии перекиси. Сообщается, что
окисление S-алкилтиосемикарбазонов перекисью водорода позволяет, по край-
ней мере в некоторых случаях, получать триазолы с лучшими выходами, чем
при окислении хлоридом железа (Ш) [211]. Из S-метилтиосемикарбазона
бензальдегида (IX, R = С0Нй, R ' = СН3) окислением перекисью водорода
[211] получен 3-фенил-5-метилтио-1,2,4-триазол (X, R = С0Н5, R = СН3)
с хорошим, но точно не установленным выходом. При окислении же хлоридом
железа (Ш) выход составил 53% [212]. В присутствии избытка перекиси
тиосемикарбазон бензальдегида превращается в 3-фенил-1,2,4-триазол, причем
в некоторых случаях первоначально образовавшиеся меркаптотриазолы
окисляются в соответствующие дисульфиды [211].
Термическая циклизация. Некоторые ацильные производные семикарбази-
, дов, тиосемикарбазидов и аминогуанидинов превращаются при нагревании
в симм-триазолы. Согласно одному из наиболее ранних сообщений о получении
уразола, б«с-мочевину нагревали некоторое время при 180—250° [214]. Сер-
нистый аналог (H2NCSNH)2 превращается в дитиоуразол и З-амино-5-меркап-
то-1,2,4-триазол при 210°. Аналогичным образом получают 4-арил-З-амино-
5-меркаптотриазолы [215]. Дифенилгидразидины, полученные из хлорбензани-
лида и из семикарбазида или тиосемикарбазида теряют при нагревании аммиак
-и превращаются в триазолы [216]. Отщепление метилмеркаптана от S-метило-
вого эфира фенилгуанилтиомочевины происходит с образованием фенилгуана-
зола [196]. Элиминирование метилмеркаптана может сопровождаться цикли-
СеН5 N —N
I 200» \
c0h5n=cnhnhconh2----------> нос СС0Н5
сйн5
NH NH N—N
II II // Ч
c6h5nhcnhnhcsch3 —> h2nc cnh2+ch3sh
I
с«лел£-ТРИАЗОЛ Ы
329
зацией другого типа, ведущей к образованию монотиоуразола [196]. В анало-
гичных условиях из 1-ацил-4-алкилтиосемикарбазидов образуются симм-
HN — N
/ ч
CH3SCONHNHCSNH2 —> HOC CSH + CH3SH
N
триазолы 1202]. В кипящем спирте, содержащем аминогуанидин, 1-формамидо-
N —N
200° \
RCONHNHCSNHR'---------> RC CSH
N^
А'
R = H, СН3; R' = C2H5
2-нитрогуанидин превращается в 3-нитрамино-1,2,4-триазол, выделенный
в виде аминогуанидинной соли [217]. Дифенилбензамидогуанидин дает при
HN —N
hconhnhc(nh2) = nno2 -» нс7 ^NHNO2
\ /
N
нагревании 3,4-дифенил-5-анилино-1,2,4-триазол [218] и 1-фенилтиокарбамил-
2-анилиногуанидин C8H5NHCSNHC( = NH)NHNHC8H5; при пиролизе отщеп-
ляется сероводород и образуется 1-фенил-3-амино-5-анилино-1,2,4-триазол
1219]. Дифурилгидразидин в кипящей уксусной кислоте превращается
в 3,5-дифурил-1,2,4-триазол [220], а бензоил-п-толилгидразидин при 120°
образует 3-фенил-5-(п-толил)-1,2,4-триазол [220]. Образование 3,5-динитрами-
по-1,2,4-триазола (XI) при восстановлении сероводородом неочищенного
N
азида, полученного из 1,6-динитро-З-аминогуанилбигуанидина и азотистой
кислоты или же при выделении 5-аминотетразола из образовавшегося в резуль-
тате перегруппировки азида неустойчивого тетразола, по-видимому, происхо-
дит в результате термически индуцируемых реакций.
NH NC( = NH)N3
И II
O2NNHCNHNHCNHNO2
г
HN —N
o2nnhc cnhno2
^'n^
XI
HN —N
NH NC^ j I
|| || N-N HN —N
O2NNHCNHNHCNHNO2 XI -|- h2ncZ II
N —N
Получение диалкиламино-силш-триазола нагреванием алифатического
нитрила и гидразина в железном сосуде [222], а также образование 1,3,5-три-
330
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗбЛЫ
фенил-1,2,4-триазола из фенилгидразина и бензонитрила в присутствии метал-
лического натрия [223] представляют собой реакции, в которых вероятными
N2I<4
R —CN-------
Г NH NH~|
II I!
LRCNIINIICR _
n2h4
N—N
промежуточными соединениями являются амидразоны. Нерастворимый поли-
мер, полученный из полиакрилонитрила и гидразина, содержит повторяющиеся
звенья 4-амино-1,2,4-триазолов [224]. Дегидратация ацилпроизводных co-N-
Na G6H6CN
CeH6N2H3 + CeH5CN-----> |C8H6NHNHC( = NH)C8H5---------->
H5C8 —N —N
/ 4,
—> CeH6NHN = C(C8Hs)NHC(CeH5) = NH] —•> H5C8-N CC8H5
N
фенил-С-цианформамидразона — одна из наиболее давно известных реакций,
ведущих к образованию снлш-триазолов [225]. При 200° как оксальамидразбн,
так и оксальгидразидин превращаются в 4-амино-1,2,4-триазол [226].
CN H5Ce-N-N
I / \
RCONHC=NNHCeH5 —> RC CCN
\ /
N
H2NCCOOH
II
h2nn
N —N
// \
нс сн
200°
-co2
—N2H4
h2nnhccooh
II .
h2n.n
ч Межмолекулярные конденсации. Диацилгидразины и амины. При обра-
ботке диформилгидразина (XII, R = Н) избытком аммиака в автоклаве при
200° в течение 24 час образуется с хорошим выходом 1,2,4-триазол (XIII,
R = R ' = Н) [190]. В тех же условиях смесь гидразина, аммиака и формамида
дает диформилгидразин, который в результате последовательных реакций
превращается в 1,2,4-триазол [190]. Другие алифатические и ароматические
диацилгидразины вместе с аммиаком (иногда в виде комплексов с хлоридом
цинка) применялись для получения 3,5-дизамещенных снлш-триазолов [227].
Замещенный в 4-положение триазол образуется в результате замены аммиака
гидразином [228] или первичным амином [229]; однако некоторые гетероцикли-
ческие первичные ароматические амины не вступают в эту реакцию [229].
Низкие выходы 3,4,5-трифенил-1,2,4-триазола (XIII, R =R' = С8Н6) при
получении его из дибензоилгидразина и анилина могут быть повышены до 90%,
если реакцию проводить в присутствии пятиокиси фосфора [230] или фенил-
фосфазоанилида [231]. Из N-метилдиформилгидразина и формамида получен
1-метил-1,2,4-триазол [232]. При взаимодействии бисмочевины (XII, R =
= NH2) и анилина образуется 4-фенилуразол (XIII, R = ОН, R' = СеН6)
[233]. При получении 4-амино-1,2,4-триазола (XIII, R = Н, R ' = NH2)
из окиси углерода и гидразина промежуточным соединением, по-видимому,
является диформилгидразин (XII, R = Н), который затем претерпевает
циклизацию [234]. Триазолы могут быть синтезированы из N-ациламидо-
силгл4-ТРИ АЗОЛЫ
331
дитиокарбаминовой кислоты и гидразина. Так, например, N-бензамидодитио-
N—N
R'NH2 /А \.
RCONHNHCOR-------> RC CR
XII \ /
N
R'
XIII
карбаминовая кислота и гидразин образуют 3-фенил-4-амино-5-меркапто-
1,2,4-триазол [235].
N —N
n2h4 / Ч
c6h5conhnhcs2h-------> use CC6II5
I
nh2
Дибензгидразиддихлорид [C6H5C(C1) N —]2, полученный из дибензоил -
гидразина и пятихлористого фосфора, конденсируется с замыканием кольца
с аммиаком или первичным амином. Этим путем из дихлорида дибензоил-
гидразина и соответственно аммиака, анилина и гидроксиламина синтезированы
3,5-дифенилтриазол, 3,4,5-трифенилтриазол и 3,5-дифенил-4-окситриазол 1236].
Представляет интерес возможность наличия таутомерии у последнего соеди-
N —N
HN-N
Н5С6С СС6Н5
N
ОН
нения, поскольку N-окиси с«жж-триазола, по-видимому, не известны. Анало-
гичная реакция наблюдалась для бензальазина при превращении его под дей-
ствием смеси азотной и уксусной кислот и уксусного ангидрида в бензальпро-
изводное 3,5-дифенил-4-амино-1,2,4-триазола [237]. Гидразон бензальдегида,
по-видимому, является промежуточным продуктом гидролиза.
N-N
(C6H5CH=N)2 \
(C6H3CH -.N), -> C6H5CH=-NNH2------------> Н5С6С СС6Н5
I
h5c6hc = n
Амидразоны или гидразидины и карбонильные производные. Две молекулы
бензамидразона легкд конденсируются друг с другом, образуя 3,5-дифенил-
1,2,4-триазол и соединение, которому было приписано строение 3,6-дифенил-
1,2-дигидро-1,2,4,5-тетразина 1206] (см. стр. 336). В результате аналогичной
реакции конденсации из фенилбензамидразона получен дифенилтетразин
и 3,4,5-трифенил-1,2,4-триазол 1216]. Подобного рода реакция протекает
при пиролизе гидразидов кислот [228, 238] и может иметь место при попытке
перегонки гидразида 1239]. При действии безводного гидразина на этиловый
эфир n-сульфаниламинобензойной кислоты образуется соответствующий
гидразид, две молекулы которого, конденсируясь друг с другом в присутствии
безводного гидразина, дают 3,5-ди-(п-сульфамидофенил)-4-амино-1,2,4-триазол
332
МОНОЦИК.ЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
[240]. При реакции эквимолекулярных количеств бромистого циана и гидра-
зина образуется аминогуаназол [241].
NH2 NH —NH HN —N
CCeH5-l-C6H5C
CeH5C=NNH2
ССВН5
N — N
NHCgHj
N
N —N
N — N
CeHaC -NNHZ
C6H5C
C6H,
N-N
N
ceH5
N —N
n2h4
H2NSO2CeH4COOC2Hs-----> ArCON2H3
h2nso2c6h4c
cc6h4so2nh2
N -
NH2
HN—NH
BrCN + N2H4
|NCNHNH2]
HN = C
C = NH
nh2
w-Алкил- или w-арилбензамидразоны вступают в реакцию с карбоновыми
кислотами, хлорангидридами или ангидридами кислот, давая сыл-ш-триазолы.
Так, например, 1,3-дифенил-5-метил-1,2,4-триазол (XIV, R = С6Н5, R ' = СН3)
получен из ю-фенилбензамидразона и уксусного ангидрида, а 1-метил-3,5-
дифенил-1,2,4-триазол (XIV, R = СН3, R ' = QHs) — из w-метилбензамидра-
зона и хлористого бензоила [242, 243].
RN —N
C6H5C = NNH2 + R'COX
nh2
R'C
CCeH,
N
XIV
Из аминогуанидина и карбоновых кислот синтезированы З-амино-5-алкил-
1,2,4-триазолы [1991. Аналогичная реакция, по-видимому, приводит, к обра-
зованию 3-метил-4,5-диамино-1,2,4-триазола (XV) при восстановлении нитро-
аминогуанидина цинком в уксусной кислоте или водородом над платиной
в уксусной кислоте [244]. Нитрон (XVI) легко получить аналогичной реак-
цией между трифениламиногуанидином и муравьиной кислотой [245]. Перво-
NH N-N
I [H]
H2NNCNHNO2 c—;oo7i- H2NC
CCI-I,
N
fW2
XV
начально его считали бициклическим соединением, но позднее для нитрона
было предложено цвиттерионное [246] и мезоионное [247] строение. Аналогии-
N
симм-ТРИ АЗОЛЫ
333
но могут быть охарактеризованы продукты реакции альдегидов с 1,4-диарил-
тиосемикарбазидами [245], а также с ангидридом камфорил-ф-семикарбазида
[248].
NC6H5
II
c6h5nhnhcnhc6h5
нсоон
XVI
+
С,Н,—N—N
V—NC6H5
К
С6Н5
(дополнительные
резонансные
структуры)
ArN—N
RC CNA?
(и другие тиазольные
или тиадиазольные
структуры)
Сообщается о замыкании триазольного кольца в результате реакции
между S-алкилтиосемикарбазидами и уксусным ангидридом [249], которая
напоминает образование 1-фенил-3-окси-1,2,4-триазола из 1-фенилсемикарбази-
да и муравьиной кислоты [250].
Получение линейных поли-4-амино-1,2,4-триазолов из дигидразидов дикар-
боновых кислот и гидразина «в условиях, способствующих полимеризации»-
S
• it—N NH
II || (СН3СО)2О
!( NHNHCSCH3------------->
S
CH3SC
ссн3
N —N----
H5Ce —N —N
нсоон / \
CeH5NHNHCONH2-------s- НС СОН
X/
N
по-видимому, представляет собой новое применение реакции образования
3,5-дизамещенных 4-аминотриазолов при пиролизе гидразидов кислот [251],
причем промежуточными продуктами могут быть гидразидины.
Диациламины и гидразины (реакция Эйнхорна — Бруннера). Образование
триазолов из диациламинов и гидразинов носит название реакции Эйнхорна —
Бруннера [252]. Гидразин или его монозамещенные производные вступают
в реакцию с симметричными или несимметричными диациламинами в растворе
органической кислоты, в кислом буферном растворе или в присутствии хлор-
гидрата пиридина. Более сильные кислоты вызывают понижение выходов
триазолов и соответственно увеличение количества образующихся гидразидов,
аммонийных солей и соединений неизвестного строения. Из несимметричных
334
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛь!
диациламинов изомерные триазолы не образуются [253, 254]. Согласно имею-
щимся ограниченным литературным данным, реакция не сопровождается
первоначальной амидо-гидразидной перегруппировкой.
Типичным примером является образование [253] 1,5-дифенил-1,2,4-три-
азола (XVII, R = Н, R ' = R" = СвН5) из формилбензамида и фенилгидра-
зина [252—254]. Другие триазолы (XVII) образуются при аналогичных
реакциях, когда R =СН3, R' = С2Н5, R" = С6Н5 [253]; R=CH3, R' =
= С6Н5СН=СН2, R"=C6H5 [255]; R = СН3, R' R'' -С«Н3 [253];
R =Н, R' =CeH5, R" =СН3 [242]; R =R' = СН3, R' = С6Н5 [242].
Реакция между N-ацетилуретаном и фенилгидразином приводит к 1-фенил-
3-метил-5-окси-1,2,4-триазолу (XVII, R = СН3, R' = ОН, R" = С6Н5) [242,
256]. З-Фенилтриазол получают из формилбензамида и семикарбазида [242].
R"N — N
/ \
RCONHCOR' + R"NHNH2 R'C CR
\ /
N
XVII
Известен ряд триазолов (XVII), полученных в результате взаимодействия
симметричных диациламинов с гидразинами: R= R' = СН3, R" = С6Н5 [2571;
R = R' =R" = СН3 [258]; R = R' = СН3, R" = H2NCO [259]; R = R ' -
= NH2, R" = H [214].
Диациламины могут быть заменены производными, у которых имино-
группа замещена одним или двумя карбонильными атомами кислорода.
Так, ацетил- и бензоилдициандиамиды под действием гидразина превращают-
ся соответственно в З-уреидо-5-метил- и 3-уреидо-5-фенил-1,2,4-триазолы [260].
Любопытно, что ацетилгуанилмочевина CH3CONHC(NHC) = NCONH2 не реа-
гирует с гидразином с образованием триазола [260]. По-видимому, аналогичная
реакция между углекислым гуанидином и гидразином приводит к образованию
NCN HN —N
II N2H4 / \
rconhcnh2-----> RC cnhconh2 •
4-амино-3,5-дигидразино-1,2,4-триазола [194].
Примером получения триазолов из дигуанидов является реакция между
л-оксифенилдигуанидом и гидразином [261]. Эта реакция использована для
NH NH HN —N
II II . n2h4 ' / \
HOCbH4NHCNHCNH2 -------> H2NC CNHC6H4OH-/i
получения 3,5-дпамино-1,2,4-триазола (гуаназола) [262, 263] или 1-фенил-
NH HN-N
II n2h4 / \
h2ncnhcn-------h2nc cnh2
NH Н5С6 —N —N —СН3
H2N(!nHCNH-C6H5NHNHCH3—» HN = C/ 'c=NH
N
H
сил4Л4-ТРИАЗОЛЫ
335
2-метилгуаназола из дициандиамида и гидразина или 1-фенил-2-метилгидра-
зина соответственно [263].
Один или оба атома кислорода карбонильной группы у диациламинов
могут быть замещены серой. Дитиоуразол и 1-фенилдитиоуразол образуются
в результате взаимодействия гидразина или соответственно фенилгидразина
с надтиоциановой кислотой [264]. Реакция между гидразином и бензоилизо-
тиоцианатом, по-видимому, по своему характеру близка к рассмотренным
выше реакциям [265].
RN —N
RN2H34-H2NCSNHCSoH —> hsc7 \zsh
V /
N
R = H, ОД
HN —N
n2Hi / \
C6H6CONCS-----C6H5C CSH
4/
N
Гидразиды и амиды (реакция Пеллиццари). Нагревание гидразидов
и амидов при высоких температурах в отсутствие растворителей приводит
к силш-триазолам [266] и является методом, известным как реакция Пеллиц-
цари [267]. В качестве промежуточных продуктов при этом образуются ацил-
гидразидины. Характерные для этой реакции низкие выходы связаны с боль-
шими потерями амидов вследствие их дегидратации в соответствующие нитри-
лы, которые не принимают участия в реакции Пеллиццари, ведущей к образо-
ванию триазолов. Возможность миграции ацильной группы между амидом
и гидразидом может осложнить идентификацию продукта. Эта перегруппиров-
ка не происходит до тех пор, пока при нагревании ацетамида с 1-фенил-2-про-
пионилгидразином температура не поднимается выше 210°. Ниже этой тем-
пературы единственным продуктом реакции является 1-фенил-3-этил-5-метил-
1,2,4-триазол (XVII, R = СН3, R ' = CeH5, R" = С2Н5), тогда как при более
высокой температуре образуется в существенных количествах 1-фенил-3,5-
диметил-1,2,4-триазол (XIX, R = СН3, R' = QHs) [253].
Из бензамида и 1-фенил-2-ацетилгидразина, кроме ожидаемого 1,5-дифе-
нил-3-метил-1,2,4-триазола (XVIII, R = R' = С6Н5, R" = СН3), образуется
1-фенил-3,5-диметил-1,2,4-триазол (XIX, R' = CeH5, R" = СН3) [253].
Как из бензамида и ацетилгидразина, так и из ацетамида и бензоилгидразина
получается ожидаемый 3-метил-5-фенил-1,2,4-триазол, однако во второй
реакции в результате ацильной перегруппировки образуется также 3,5-дифе-
нил-1,2,4-триазол [242]. Незамещенный 1,2,4-триазол получают из формамида
и формилгидразина, а 1-метил- или 1-фенил-1,2,4-триазол — из (]юрмамида
и соответственно из 1-формил-2-метил- или 1-формил-2-фенилгидразина [267].
Интересным примером замены амида амидином служит реакция между
R'N —N
RC7 \zR"
% /
N
XVIII
R'N-N
RCONH2-|-R'NHNHCOR / \
—> (R'')RC CR(R")
(или R''CONH2 + R'NHNHCOR") \ /
N
RCONH2-|-R'NHNHCOR" —>
R"CONH2 + R'NHNHCOR
XIX
336
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
цианацетилгидразином или формилгидразином и Ы,Ы'-дифенилформамидином.
Эта реакция ведет к образованию 3-цианметил-4-фенил-1,2,4-триазола
(R = CH2CN) или 4-фенил-1,2,4-триазола (R = Н) соответственно [268].
N-N
RCONHNH24-C6HsN = CHNHC6Hs —> Rdf "сн
CeH5
Вариацией реакции Пеллиццари является применение хлорангидрида
карбаминовой кислоты и 1-фенил-2-ацилгидразинов [R = СН3, СН3СН2,
С6Нц (циклогексил), но не C6HS] [269]. Из 2,4-дифенилтиосемикарбазида
CeH5—N —N
RCONHNHCeH5 + H2NCOCl —> ^CR
I
conh2
и хлорбензанилида получен 1,3,4-трифенил-1,5-дигидро-5-тиокето-1,2,4-три-
азол [216].
CeH5 ci с6Н6—N—N
| СвНбС=КСвНб / \
CeH5NHCSNNH2 --------------> S = C ССеН5
\'n//
I
CfiH6
Хлбргидрат аминогуанидина и мочевина дают 3-амино-5-окси-1,2,4-
триазол.
Молекулярные перегруппировки. Окси-сижж-триазолы получают пиро'
лизом гидразонов пировиноградногидроксамовых кислот (реакция Гастальди)
1270]. При этом первоначально, по-видимому, происходит перегруппировка
гидроксамовой кислоты по Лоссеню; циклизация в присутствии ангидрида
кислоты дает соответствующий эфир триазола. Примером реакции может слу-
жить получение 1-фенил-3-метил-5-окси-1,2,4-триазола (R = Н) и его этило-
вого эфира (R = С2Н5) из фенилгидразона пировиноградногидроксамовой
кислоты в пропионовом ангидриде [270].
СН3 CeH5- N— N
I (С2Н6СО)20 / \
CeH6NHN = CCONHOH--------------> ROC ССН3
Ч /
N
Поскольку 1,2-дигидро-1,2,3,4-тетразин фактически представляет собой
изомерный 4-амино-1,2,4-триазол, катализируемое основаниями сужение коль-
ца бис-диазоуксусной кислоты (дигидротетразина) приводит к 4-амино-1,2,4-
триазолдикарбоновой-3,5 и 3-амино-1,2,4-триазолкарбоновой-5 кислотам. Оче-
видно, «дифенилуразин» представляет собой изомерный N-фениламинотриазол.
а «гуаназин»—4-аминогуаназол [271 ]. Позднее была осуществлена катализируе-
мая основаниями перегруппировка 1,4-дигидро-1,4-дифенил-1,2,4,5-тетразина
Н5С6—N—N
N N —С6Н5 CoHsONa / \
I I----------------> НС CNHCeH6
H6C6_N N х /
ы
.илсж-ТРИ АЗОЛЫ
337
в 1-фенил-З-анилино-1,2,4-триазол [272]. 2-Арил-З-ароиламинохиназолоны
в водной щелочи перегруппировываются в 3,5-диарил-4-(о-карбоксиарил)-
триазолы (см. т. 6).
N N-N
I ii ।— Разб. щелочь \
| II N-NHCOC6H5 > Н6С0С сс0н5
\/\/ \ /
II N
О
С6Н4СООН-о
Бензальдегидное производное 4-аминоуразола, по-видимому, образуется
в результате протекающей в кислом растворе перегруппировки соединения XX,
сопровождающейся расширением цикла [273].
О
HN-NH
CeH5CH = NN/ XNNH2 ----°=с^ /С=О
[С 14
II I
о n=chc6h9
XX
сижж-Триазолы образуются при взаимодействии 5-замещенныХ тетраЗоЛоВ
с иминохлоридами [274]. На следующем примере показан новый путь получе-
ния полиароматических соединений и предложен вероятный механизм реак-
ции [274].
HN-N zN=N _2N2
| I + 2CoH6N = C(C1)C6H6--->
N = NZ ----- 44-NH
N—N N—N
ЛОВ
CgHs
Пиролиз 5-замещенных тетразолов может
и тетразинов 1275].
Сб^5
привести к получению триазо-
N-NH
RC^ | +R"N = C(C1)R'
XN = N
-НС1
/R'
.N — NC^,
RCT | XNR"
XN = N
338
МоноЦикЛиЧеские ТРИАЗОЛЫ И БЕЙЗоТРИАзбЛЫ
Неклассифицированные методы. Отдельные примеры получения симм-
триазолов, по-видимому, не имеют отношения к другим методам и остались
не разработанными. Полученный в одном случае из бензальазина и циановой
кислоты триазолотриазол был превращен в 1-бензил-3-фенил-5-окси-1,2,4-
триазол, идентичный продукту, полученному окислением бензальдегидного
производного 2-бензилсемикарбазида (стр. 327) [209]. Упомянутые триазоло-
триазолы, полученные замещением циановой кислоты тиоциановой кислотой
или фенилизоцианатом, не образуют в аналогичных условиях триазолов [209].
(CeH6CH = N)2
IIOCN
он н" \(1н5
кон
СеН6СН2—N—N
нос7 %свнй
Пиролиз бензилазида в ксилоле дает смесь пяти продуктов: 3,5-дифенил-
4-бензил-1,2,4-триазола, тетрафенилтетразина, дибензилбензамидина, три-
фениламина и бензилбензилиденамина. Курциус и Эрхардт сообщают об обра-
зовании третичного амина и триазола в соответствии с приведенным ниже
уравнением [276]. При проведении реакции в диметиланилине выход триазола
увеличивается [276].
N —N
9CBH5CII2N3 > CgHjC CCgHr, 2(C0HbCH2)3N-^5N2-f-4HN3
^N^
СН2С6Н5
Получение 3-меркапто-4-амино-1,2,4-триазола из тетрахлордиметилсуль-
фида и гидразина [277] напоминает некоторые другие методы получения
4-аминотриазолтиолов, как, например, реакции между тиокарбогидразидом
и этилортоформиатом, а также между диэтилксантатом и гид раз и hqm (стр. 326).
(C12CH)2S
N-N
Производное триазола —4-оксамидо-1-оксамолил-1,2,4-триазолидин-
дион-3,5 — получается при конденсации гидразида оксаминовой кислоты
с фосгеном [278]. В результате гидролиза образуется кислота, при декарбокси-
лировании которой получен 1-формил-4-формиламино-1,2,4-триазолидин-
дион-3,5. Последний образуется также при нагревании гваяцилформилгидра-
ct/.м.м-ТРИАЗОЛЫ
339
зинкарбоксилата [278].
H2NCOCON—NH
COCI 2 /' \
h2ncoconhnh2---------> o=c c=o
N11COCONH2
COOR HCON—NH HOOCCON—NH
NH —ROH O = C C = O -CO9 O = C C = O
I ----> \ / <--- \ /
NH V V
CHO I I
NHCHO NHCOCOOH
R.= C6H4OCH3-o
Реакции замещения в снзгл-триазольном кольце
В следующих разделах на ряде примеров будет показана инертность
силии-триазольного кольца. Оно устойчиво к действию фосфора и йодистого
водорода при 160° (стр. 345), к расщеплению хлористым бензоилом [279],
к восстановлению различными агентами: алюмогидридом лития (стр. 346),
водородом над платиной (стр. 332) или никелем (стр. 345), цинком в уксусной
кислоте (стр. 332) или натрием в жидком аммиаке (стр. 347). силш-Триазольное
кольцо устойчиво также к окислению перманганатом, перекисями и другими
реагентами. В концентрированной серной кислоте при 190° происходит пол-
ный распад 3-метил-5-амино-1,2,4-триазола с образованием аммиака, двуокиси
углерода, гидразина и уксусной кислоты [198].
Поскольку таутомерия допускает переход уразолов и гуаназолов в гидра- •
зопроизводные, легкое окисление этих сылци-триазолов объясняют образова-
нием соответствующих циклических азопроизводных [196, 233]. Полученные
продукты—твердые, легко разлагающиеся, темно-красного цвета, с острым запа-
хом; строение их окончательно не установлено. Удовлетворительным окисли-
телем является щелочной раствор феррицианида калия, но сообщается и об ана-
логичном действии иона на серебряную соль уразола [280]. Окисленная форма
фенилуразола в водной среде частично превращается в фенилуразол; кроме
того, образуются анилин, двуокись углерода и азот [280].
Сообщается, что прибавление небольшого количества гидросульфита
натрия во время перекристаллизации 1-замещенных гуаназолов позволяет
избежать потерь, вызванных образованием продуктов окисления [281].
Реакции электрофильного замещения у атома углерода. Реакция этого
типа наблюдается при конденсации 1,2,4-триазола и его 1-бензилпроизводного
с формальдегидом [190]. В первом случае получается З-оксиметил-1,2,4-
триазол, а в другом — либо 1-бензил-З-, либо 1-бензил-5-оксиметил-1,2,4-
триазол. Реакции галогенирования, сульфирования, алкилирования по Фри-
делю — Крафтсу и ацилирования силш-триазольного кольца не известны.
Нитрование 3-окси-1,2,4-триазола дымящей азотной кислотой дает 3-окси-
5-нитро-1,2,4-триазол [282]. В двух случаях обнаружено, что нитрование
с«жж-триазолов с фенильными группами, связанными с атомами либо углеро-
да, либо азота идет по бензольному кольцу независимо от того, несет ли три-
22*
340
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
азольное кольцо активирующие заместители [210, 282]. Появление красной
окраски [283] при смешении 3-окси-с«лии-триазола с диазобензолсульфокис-
лотой может служить указанием на прохождение реакции сочетания.
Реакции электрофильного замещения у атома азота. Эти реакции иссле-
довались более широко. Алкилирование натриевого производного триазола,
по-видимому, проходит в положении 1, поскольку в результате реакции алки-
лирования иодистым метилом и реакции конденсации диформилметилгидра-
зина с формамидом получается один и тот же продукт [232, 258]. Алкилиро-
вание в положении 1 наблюдается также при действии на 1,2,4-триазол и его
3,5-диметил- или 3,5-дифенилпроизводные либо натрия в метаноле, а затем
N—N —СН3
У \
Н3СС ссвн5
\ /
N
ch2n2
HN— N
Na
CH3OH
CH3 — N—N
H:iCC \zC0H-,
\ /
N
алкилгалогенида [190, 258], либо диазометана или диазоэтана [258]. Любо-
пытно, что в реакции с 3-фенил- или З-метил-5-фенил-1,2,4-триазолом диазо-
метан атакует главным образом вицинальный атом азота, соседний с фениль-
ным заместителем, в то время как метилирование иодистым метилом происхо-
дит преимущественно по другому вицинальному атому азота [242]. В противо-
положность этому продуктам алкилирования З-уреидо-5-фенилтриазола (XXI)
диметилсульфатом или хлористым бензилом в присутствии едкого натра при-
писывается строение 3,4,5-тризамещенных силии-триазолов [260]. Тому же
четвертичному иодиду, полученному в результате метилирования 1,3,5- или
3,4,5-триметил-1,2,4-триазола иодистым метилом, приписывается строение
N—N
N—N
СвН5С
CNHCONH2
RC1
ИЛИ
R2SO4
NaOH
У У
СсН6С CNHCONH2
I
R
С6Н5СН2
R = CH3,
N
Н
иодида 1,3,4,5-тетраметил-1,2,4-триазолия [279, 284].
Алкилирование функциональных групп в боковых цепях может проте-
кать в конкурирующих направлениях. Степень енолизации, как показывает
метилирование диазометаном, в случае 4-фенил-, 1,4-дифенил- и 1,2-диметил-
уразолов незначительна. Количественное метилирование превращает 1-фенил-
4-метилуразол в его 3-метоксипроизводное, а 1-фенил-2-метилуразол— в его
4-метилпроизводное. Метилирование атома азота ведет к образованию 1,2-диме-
тил-4-фенилуразола из 4-фенилуразола, а метилирование кислорода —
к З-метокси-4-метилтриазолону из 4-метилуразола. У уразола, его 1-фенил-
и 1-метилпроизводных енолизуется только одна амидная группа [285]. Под дей-
ствием диазометана уразол превращается в 1,4-диметил-З-метокситриазолон,
а 1,2-диметилуразол — в 1,2,4-триметилуразол [285]. Акри [286] сообщает
об образовании 1-фенил-3-метокси-4,5-дигидро-4-метил-5-оксо-1,2,4-триазола
из 1-фенилуразола и диазометана. С метилатом натрия и иодистым метилом
симм-ТРИ АЗОЛЫ
341
в метаноле 1-фенил-3-этил-5-окси-1,2,4-триазол дает соответствующее 4-метил-
производное [269]. Между тем 1-фенилуразол в аналогичных условиях пре-
вращается в 1-фенил-2-метилуразол [285], а 3-фенил-5-меркапто-1,2,4-триа-
зол — в З-фенил-5-метилтио-1,2,4-триазол [212].
С6Н5—N—N С6Н5 —N—N
/ \ ch2n2 / \
О = С СОН-------------> О-.С СОСНз
С6Н5 — N-N
нос7
CH3I
КО11
С6НЬ—-N- N
/ Ч.
О — С СС2Н;,
СН3
HN—N
HN-KN
СцЩС7 \sh
N
сн31
кон
/ Ч
С6Н5С CSCH3
Замещение в триазоле, происходящее, по-видимому, в положении 1,
обусловлено сопряженным присоединением 1,2,4-триазола к а,Р-непредсльным
карбонильным соединениям [287]. От продуктов присоединения алкоксимети-
ленмалоновых эфиров к 3-амино-5-алкил-1,2,4-триазолам (XXII) отщепляется
молекула спирта, причем у атома азота триазола остается винильная группа
[288]. Атака по атому азота в триазольном кольце является также причиной
образования продукта XXIII из соединения XXII и аллилацетоуксусного
эфира при нагревании их в нитробензоле при 160° в течение 1 час [288].
Продукт реакции — N-ацилпроизводное триазола, как сообщается, не обла-
HN—N HOOCCH2CH2-N-N
/ ч, / ч
НС СН С1Н снсоон —> ИС СП
ч / /
N N
N—NH
RC CNH2 4- ROCH = C(COOR)2
\ /
N
XXII
N—N —CH=C(COOR)2
RC^ XCNH2
\ Z
N
дает неустойчивостью, обычно характерной для этого типа соединений.
СН3 СН2СН = СН2
I I
N-N — С = С —СО —N—N
XXII
XXIII
Внутримолекулярная конденсация может
за сходными реакциями алкилирования или
сопровождать или следовать
ацилирования у атома азота
342
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И ВЕН30ТРИА30ЛЫ
силни-триазольного кольца в З-амино-1,2,4-триазоле. При взаимодействии
с этиловым эфиром Р-хлоркротоновой кислоты образуется 5-окси-7-метил-
1,2,8-триазаиндолизин (XXIV), полученный также из 2-гидразино-4-метил-
6-оксипиримидина [289]. Как сообщается, из четырех возможных изомерных
продуктов 5-окси-7-метил-1,3-8-триазаиндолизин (XXV) является единствен-
ным, полученным из 3-амино-1,2,4-триазола и ацетоуксусного эфира или кетена
[290]. Продукт конденсации, полученный из а-карбэтоксициклогексанона
N—NH
N—NH
N N
И-,С-/ \
I N
I N ?
Т (311
ОН •
нсоон
I
ОН
XXIV
N . N
СН3СОСНгС(>ОС..Н5 П3С —/ \
---------------’ I IN ZCH
| N
ОН
XXV
идентифицирован как 7-окси-5,6-тетраметилен-1,3,8(или 1,2,8)-триазаиндо-
лизин [291].
Конденсация гуаназола с этилбензоилацетатом дает гуаназопиразолон,
а с диформилгидразицом — гуаназотетразин [292]. Реакция получения гуанат
NH
II
NH
(HCONH)2
HN=C
C-..NH
NH—NH
зола из гидразина и дициандиамида (стр. 334) может сопровождаться конден-
сацией гуаназола с дициандиамидом и образованием 3,5,7-триамино-1,2,6,8-
тетразаиндолизина [262].
II h2nc \ H2N-f у \
H2NCNHCN + I N —-> NN N
HNX / Ч/\ /
ХС I с
I nh2 I
nh2 nh2
По-видимому, такое же
триазола (предположительно
неустойчивое ацетильное производное 1,2,4-
в положении 1) образуется из калиевой соли
силл-ТРИ АЗОЛЫ
343
триазола и хлористого ацетила или из триазола и уксусного ангидрида [258].
Легкость, с которой происходит отщепление ацетильной группы, наглядно
видна при образовании пикрата триазола при действии пикриновой кислоты
в хлороформе на N-ацетилтриазол, что согласуется с предпочтительным заме-
щением у атома азота (а не у атома углерода) [258]. При нагревании 1,2,4-
триазола с хлористым бензоилом до 100° в течение 3 час не было обнаружено
образования бензоилтриазола (исходный триазол был возвращен с 94%-ным
выходом) [279]. Однако под действием хлористого бензоила в пиридине соеди-
нение XXI превращается в 3-уреидо-4-бензоил-5-фенил-1,2,4-триазол [260].
Ацилирование 1-фенилгуаназола этилформиатом или формамидом идет в поло-
жении 2. Однако производные высших жирных кислот не вступают в эту реак-
цию. При взаимодействии с диэтилмалонатом образуется 1-фенил-2-карб-
этоксиацетилгуаназол [293].
CeH5—N-NH ' C6H5-N—N— СОСН2СООС2Н5
IIN = C/" 4=--NH-------> HN = C/ —NH
X'n'//
h J H
Некоторые 1,2,4-триазолы вступают в реакцию с циановой кислотой [259]
или фенилизоцианатом [294], при этом происходит замещение у атома азота
триазола и образуются производные мочевины.
HN- N H2NCO-N-N
/ \ HCNO / \
сн3с ссн3--------- СН3С ссн3
Ч / X/
N N
Замещение функциональных групп водородом. Легкость, с которой
от атома азота триазола отщепляются ацильные группы, свойственна также
и карбамильной группе. Под действием теплой воды, спирта, анилина или
семикарбазида 1-карбамил-3,5-диметил-1,2,4-триазол превращается в 3,5-
диметил-1,2,4-триазол [259]. При гидролизе 1-фенил-3-метил-4-карбамил-
5-кето-4,5-дигидро-1,2,4-триазола происходит разрыв связи азота с внеколь-
цевой карбонильной группой [269].
CeH5-N—N С6Н5 —N—N
/ \ н2о / Ч „
О = С ССН3 ----------> НОС ССН3
\ / Ч /
N N
I
conh2
Окисление активированных арильных групп, связанных с атомом азота
в силой-три азол ах, представляет собой свойство, характерное также для ана-
логичных производных вицинальных триазолов. В одном из наиболее ранних
методов получения силои-триазола 1-фенил-5-карбокси-1,2,4-триазол перво-
начально нитровался в бензольном кольце. После восстановления аминофе-
нильную группу удаляли, проводя, окисление и декарбоксилирование.
Образующийся 5-карбокси-1,2,4-триазол при температуре его плавления (137°)
превращался в триазол [295]. Окисление 1-фенил-1,2,4-триазола перманга-
натом в кислом растворе сопровождается декарбоксилированием и образова-
нием 1,2,4-триазола [267]. Алкильные группы у углеродного атома симм-
триазола, по-видимому, окисляются легче, чем неактивированные арильные
группы, связанные с атомом азота силмртриазола. Андреоччи [296] описал
получение 1-фенил-1,2,4-триазола путем окисления и декарбоксилирования
344
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
его 3-метилпроизводного. З-Карбокси-1,2,4-триазол получен окислением
3-метилтриазола [190]. Как и следовало ожидать, алкенильные заместители
у атома углерода триазола более подвержены окислению, чем алкильные
группы. Частичным доказательством строения 1-фенил-3-метил-5-стирил-1,2,4-
триазола является окисление и декарбоксилирование его в 1-фенил-З-метил-
1,2,4-триазол [255].
С6Н5—N-N
C(iH5CH -СНС'7 \сн3
\ /
N
c6h5-n-n
' / ч
НС ссн3
ч. /
N
Известны производные силои-триазолов с аминогруппами в положении
3, 4 или 5, но не 1 и не 2. Диазотирование 3- или 5-аминотриазолов протекает
нормально. Однако при действии азотистой кислоты происходит замещение
аминогруппы в положении 4 на водород [235]. Диазотирование З-метил-4,5-
диамино-1,2,4-триазола в соляной кислоте приводит к получению хлорида
3-метил-1,2,4-триазол-5-илдиазония, который при действии фосфорноватистой
кислоты превращается в 3-метил-1,2,4-триазол [244, 297]. В кипящем этаноле
диазотированный З-амино-5-карбокси-1,2,4-триазол дает 1,2,4-триазол [198].
Аналогичная реакция декарбоксилирования происходит при образовании
hno2
НС1
HN—N
CIN-C7 \-СН3
“Ч. /
N
H3PO2
------->
HN— N
/ 4
нс ссн3
\ /
N
3-хлор-, 3-бром- или 3-иод-1,2,4-триазолов из диазотированного З-амино-5-карб-
окси-1,2,4-триазола под действием соответственно соляной кислоты, бромисто-
водородной кислоты или йодистого калия [198, 282]. При обработке разбав-
ленными минеральными кислотами или уксусной кислотой в мягких условиях
соль диазония превращается в 3-окси-5-карбокси-1,2,4-триазол, который
декарбоксилируется при температуре плавления [283].
Гидроксильные или сульфгидрильные группы, связанные с атомом угле-
рода триазола, могут замещаться водородом. Открытое Пеллиццари превраще-
ние уразола в 1,2,4-триазол при действии пятисернистого фосфора [267]
было распространено позднее на получение З-фенил-1,2,4-триазола из его
5-оксипроизводного [298]. Еще более любопытны превращения 1-метил-З-
фенил-5-окси- и 1-фенил-3,4-диметил-5-кето-3,5-дигидро-1,2,4-триазолов соот-
ветственно в 3-фенил- и 1-фенил-3-метил-1,2,4-триазолы [198]. Отщепление
метильной группы напоминает элиминирование метилмеркаптана и образова-
ние 3,5-диамино-4-фенил-1,2,4-триазола при нагревании S-метилового эфира
P2S5
HN-N
HN—N
c6h5-n-n
о=с \сн3
I
СН3
c6h5-n-n
симм-ТРИ АЗОЛЫ
345
фенилгуанилтиомочевины (стр. 328). Дегидроксилирование 1-фенил-З-окси-
1,2,4-триазола осуществляется с помощью пятисернистого фосфора или путем
нагревания его серебряной соли [250].
Замещение сульфгидрильных групп водородом происходит при действии
как окислителей, так и восстановителей. Разбавленная перекись водорода
превращает З-меркапто-1,2,4-триазол и его производные в соответствующие
дисульфиды [202, 211] и(или) соответствующий десульфурированный триазол
[202, 212]. Метод обессеривания З-алкил-1,2,4-триазолтиолов-5 требует при-
менения концентрированной азотной кислоты [190]. Аналогичные результаты
достигаются применением никеля Ренея. В результате восстановительного
расщепления 5-меркаптопроизводного образуется 3-фенил-4-амино-1,2,4-три-
азол 1235].
N-N N-N
Ni- Ренея
^н2
Различные реакции. Атом галогена у углерода в силыи-триазоле менее
реакционноспособен, чем в вицинальном триазоле. При кипячении в 20%-ном
растворе едкого натра 3-иод-1,2,4-триазол не изменяется [282]. Действие
дымящей азотной кислоты на 3-бром- или 3-иод-1,2,4-триазол вызывает образо-
вание нитрата триазола, тогда как З-хлор-1,2,4-триазол не вступает в реакцию
[282]. При нагревании З-хлор-1,2,4-триазолов с фосфором и иодистым водо-
родом в течение 5 час при 160° получаются соответствующие триазолы [188].
.Образование 5-окси-7-метил-1,2,8-триазаиндолизина (XXIV) из З-бром-1,2,4-
триазола и амида ацетоуксусной кислоты в кипящем этаноле требует заме-
щения атома брома. Предположение [289], что З-амино-1,2,4-триазол является
промежуточным продуктом (см. стр. 342), находится в противоречии с инерт-
ностью галогенпроизводных силья-триазолов.
Метод получения хлоридов триазолов из окси-силш-триазолов и пяти-
хлористоРо фосфора [188] применяется редко. Замещение диазониевых групп
галогеном (стр. 344), вероятно, может быть использовано, однако со времени
публикации Тиле и Маншо метод, по-видимому, никем не применялся.
Некоторые диазониевые соединения при действии сульфаминовой кислоты
превращаются в соответствующие амины. Одним из наиболее реакционноспо-
собных в этом отношении является диазотированный 3-амино-5-карбокси-1,2,4-
триазол [299].
При действии окиси ртути на 5-тио-1,3,4-трифенил-1,2,4-триазолон сера
замещается на кислород [216].
n-n-c0h5
с6н5с
I
с6н5
HN-N
N:-N-C„H5
Hgo / \
----> с6н5с с=о
160° ь ь /
NZ
с6н5
hscZ \sh
X /
N
ceH5
346
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
Таутомерные превращения некоторых меркаптидов cruut-три азол а цвиттер-
ионного строения в аминотиадиазолы рассматривались на стр. 333. Весьма
сходным, по-видимому, является образование 2-анилино-1,3,4-тиадиазолтио-
ла-5 из 4-фенил-1,2,4-триазолдитиола-3,5, происходящее в соляной кислоте
[264].
Реакции боковой цепи
Известны реакции замещения у атома углерода, связанного с углеродом
сищщ-триазольного кольца. Карбинолы взаимодействуют с хлористым тиони-
лом, образуя а-хлоралкил-сшюи-триазолы, алкилирующие в нормальных
условиях аммиак и амины [190]. 3-Этоксиметил-1,2,4-триазол (но не 3-метокси-
метил- или 3-феноксиметил-1,2,4-триазол) при кипячении с 30%-ным раствором
бромистого водорода в уксусной кислоте гидролизуется в 3-оксиметил-1,2,4-
триазол [190].
Окисление боковых цепей сююи-триазолов с последующим декарбоксили-
рованием уже рассматривалось в качестве препаративного метода получения
некоторых производных триазола (стр. 343). По-видимому, карбинольные,
альдегидные или кетонные производные сшищ-триазола в качестве продуктов
окисления не получались. Карбоксильные или карбалкоксильные группы
в боковых цепях сшш-триазола гладко восстанавливаются до карбинольных
под действием алюмогидрида лития. Типичными примерами являются: образо-
вание 1,2,4-триазол-З-илкарбинола из этилового эфира 1,2,4-триазолкарбо-
новой-3 кислоты и 0-(1,2,4-триазол-1-ил)-этанола из а-(1,2,4-триазол-1-
ил)уксусной кислоты [190]. Однако соответствующий триазолилацетамид не
HN—N HN-N
/ Ч ЫЛ1Н4 / \
НС ССООС2Н5----------> нс ссн2он
\ Ч /
N N
НООССН2—N—N НОСН2СН2--- N-N
удалось восстановить алюмогидридом лития [190], в то время как амиды 1,2,4-
триазол-3-илуксусной кислоты превращались в соответствующие триазолэтил-
амины [190].
Щелочной раствор перманганата окисляет З-амино-1,2,4-триазол и его
5-метилпроизводное в соответствующие линейные азопроизводные (ср. стр. 343),
которые могут быть восстановлены хлоридом олова (II) до гидразосоединений
[198]. Последние легко окисляются кислородом воздуха, хлоридом железа
HN—N
НС7 \ын2
X /
N
КМпО4
HN-N HN—N
/ \ / Ч.
НС CN--NC СН
(III) или аммиачным раствором азотнокислого серебра [198]. Диазотирован-
ные амино-шщщ-триазолы по сравнению с солями фенилдиазония обнаружи-
вают определенную стабильность. Азокрасители легко могут быть получены
с помощью обычных реакций сочетания с нафтолами, нафтиламинами и дру-
гими ароматическими соединениями [198]. Одно азопроизводное получено
из ацетоуксусного эфира и диазотированного 3-амино-5-пропил-1,2,4-триазола.
Диазотированные амино-сююи-триазолы могут быть восстановлены в соответ-
ствующие гидразинотриазолы с помощью хлорида олова (II) [282]. Декарбокси-
симы-ТРИ АЗОЛЫ
347
лирование и восстановление диазотированной З-амино-1,2,4-триазолкарбоно-
вой-5 кислоты дает ожидаемый гидразин, который, реагируя с азотистой
кислотой, превращается в 3-азидо-1,2,4-триазол [282], а с бензальдегидом
дает гидразон [198].
Гидразоны образуются при конденсации альдегидов с 4-амино-1,2,4-
триазолом [300]; однако, если в положении 3(или 5) имеется также аминогруп-
па, то в результате конденсации могут образоваться триазолы с конденсиро-
ванными кольцами. Из бензила и 3-фенил-4,5-диамино-1,2,4-триазола (XXVI)
С0НЬСОСОС6Н5
h2nc \
I N
h2nn у
хс'
СбН5
XXVI
получен 3,6,7-трифенил-1,2,5,8-тетразаиндолизин [301]. Замена бензила диаце-
тилом дает аналогичный продукт [301 ]. Продукту, полученному из дибензо-
ильного производного соединения XXVI в концентрированной соляной кислоте,
приписывают строение триазолотриазола с конденсированными ядрами [301].
N N
C6H5COC1 HC1 X \
XXVI------------>-------> С6Н5С N
\ /\ /
N С
н д
сен5
Конденсация бромистого фенацила с 3-фенил-4-амино-5-меркапто-1,2,4-
триазолом приводит к 3,6-дифенил-7,8-дигидро-8-тиа-1,2,5-триазаиндоли-
зину [235]
N s N
hsX \
С6Н5СОСН2ВГ-Г I N-------------> I I N
H=NN у. с.нД N
С N С
I i
С6Н5 С6Н5
Окисление «uui-триазолтиолов в дисульфиды можно проводить с помощью
перекисей [202], брома [192] или иода [215]. Хлорангидрид 1,2,4-триазол-
сульфоновой-3 кислоты получен окислением и хлорированием 3-меркапто-
1,2,4-триазола действием соляной кислоты и избытка хлора в течение 1 час
при температуре ниже 5° [302]. Обработка более концентрированным раствором
перекиси или азотной кислотой вызывает замещение меркаптогруппы водоро-
дом (стр. 346). В противоположность этому меркапто-, алкилмеркапто- и сульфо-
группы у атома углерода в cruut-триазоле не замещаются при действии аммиака
или аминов [212, 272]. Восстановительное расщепление 3-бензилмеркапто-
5-арил-1,2,4-триазола натрием в жидком аммиаке дает соответствующий
триазолилтиол (стр. 328) [213].
Кислотность производных симм-триазола
Кислотная диссоциация силиртриазола не изучалась, однако кислотный
характер этого соединения подтверждается образованием из него мононатрие-
вой [232] и серебряной [303] солей. Аналогичные производные металлов
348
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
известны для 5-хлор-1,2,4-триазола [198], З.З'-азо-сююи-триазола [198]
и 3,5-диметил-1,2,4-триазола [259]. Кислотным характером 3,3'-азо-симм-
триазола можно объяснить его растворимость в щелочных растворах, из кото-
рых он осаждается минеральной кислотой [198].
Окси- и меркаптопроизводные сшюи-триазола — более сильные кислоты.
Уразол дает с хлоридом железа (III) красное окрашивание, разлагает карбо-
наты и окрашивает в красный цвет лакмусовую бумажку [214]. Его 1-фениль-
ное производное является двухосновной кислотой с константами диссоциации
1 -10-5 и 5-10~7 [286]. Сообщается о растворимости 3-окси-5-фенил-1,2,4-
триазола в растворах как щелочи, так и минеральных кислот [304]. Однако
3-уреидо-5-фенил-1,2,4-триазол [260] и 3,3'-диокси-5,5'-ди-сил1Л1-триазолил
[186] (соединение I на стр. 325) растворимы в щелочах и нерастворимы в вод-
ных растворах минеральных кислот. Моно- и димеркапто-1,2,4-триазолы более
сильные кислоты, чем их кислородные аналоги. Как окси-, так и меркапто-
cruut-триазолы образуют соли с металлами и в некоторых случаях (например,
при образовании соли C4O2N6Ag4 из соединения I [186]) происходит замещение
водорода триазольного кольца.
Для измерения констант диссоциации производных амино-силмх-триазолов
применялись потенциометрический и спектрофотометрический методы [305,
306]. Оба метода характеризуют З-метил-5-нитрамино-1,2,4-триазол как силь-
ную кислоту с рК 4,8, со второй константой диссоциации рК 11,30, определен-
ной спектрофотометрическим методом. Изомерный 3-нитрамино-5-метил-1,2,4-
триазол имеет рК 4,3 и 11,10. Близкие величины рК (,3,95 и 10,80) найдены
для 3-нитрамино-1,2,4-триазола, который образует стабильную аминогуаниди-
новую соль [217]. Кислотная диссоциация 3-амино-1,2,4-триазола слишком
мала, чтобы ее можно было измерить потенциометрическим методом.
Количественные измерения кислотности сшюи-триазолкарбоновых кислот
не проводились.
Основность производных силглг-триазола
Величины рассчитанной л-электронной плотности у атомов азота в симм-
триазоле не согласуются между собой [242, 307], поэтому их использование
для учета реакционной способности весьма ограничено [242].
1,2,4-Триазол является слабым основанием, его k = 2-Ю-12 [308]. Срав-
нительно низкая основность С-аминопроизводных по мере увеличения числа
алкильных заместителей в кольце [198], по-видимому, возрастает. Соли мине-
ральных кислот известны для моно-, ди- и триалкил-сшюи-триазолов, а также
арил-силш-триазолов [223,232,252, 259]. Моноокси-сшим-триазолы образуют
хлоргидраты [188], однако уразол их, по-видимому, не образует [214]. Имеют-
ся данные о слабой основности З-хлор-1,2,4-триазола [198], 1,5-дифенил-З-хлор-
1,2,4-триазола [188] и 1,5-дифенил-З-метокси-1,2,4-триазола [188]. Описаны
амфотерные свойства 1 -фенил-3,5-димеркапто-1,2,4-триазола [264] и монотио-
уразола [202]. Гуаназол — слабое основание, для которого известны как
моно-, так и дихлоргидраты [263]. Упомянутый 3-амино-1,2,4-триазол найден
в почвах, где он принимает участие в системах катионного обмена. Описан ряд
солей аминотриазола с кислотами, обладающими гормональным действием
[310].
Нитрон (стр. 332)—сильное основание и применяется как реагент для
определения нитратов, которые он количественно осаждает в виде трудно
растворимого кристаллического нитрата нитрона C20Hi6N4-HNO3 [245,
311]. Нитрон образует также соли с хлорной, иодистоводородной, тио-
циановой, хромовой, хлорноватой, азотистой и бромистоводородной кислотами.
Сообщается также о получении трифторметилацетата нитрона [312]. М
симм-ТРИАЗОЛЫ
349
Соли и комплексы симм-триазолов
Металлические производные триазолов часто описывают как характерные
осадки, но не приводится сведений ни об их элементарном составе, ни других
аналитических данных. Предполагается, что аддукты силш-триазолов, не содер-
жащих способного к замещению водорода, и некоторых неорганических солей
являются комплексными соединениями. В качестве примеров могут быть при-
ведены комплексы, полученные взаимодействием хлорида ртути (II) с 1-фенил-
3,5-диметил-1,2,4-триазолом [313], а также азотнокислого серебра с 1,3,5-
трифенил-1,2,4-триазолом [236]. При этом может происходить как замещение
водорода, так и образование комплекса: например, из 3,5-диметил-1,2,4-триазо-
ла и хлорида ртути (II) получено производное (C4H6N3)2Hg-2HgCl2 с т. пл. 242°
[259]. Эти же компоненты образуют также комплекс состава С4Н7Нз-4HgCl2
с т. пл. 185° [259]. Соли и комплексы могут быть получены из пикриновой
кислоты и многочисленных производных силш-триазола, например: 1,5-дифе-
нил-3-хлор-1,2,4-триазола [188], 3-амино-5-метил-1,2,4-триазола [198], 3-окси-
5-амино-1,2,4-триазола [282] и 1,5-дифенил-1,2,4-триазола [252, 254]. Получе-
ны такие производные с платинохлористоводородной [188, 232, 252], щавелевой
[232], азотной [232, 282], соляной и серной кислотами, а также с дибензол-
сульфонимидом [314].
Спектры поглощения си мм-триазолов
силш-Триазол [255], его алкил- и З-амино-5-алкилпроизводные прозрачны
для ультрафиолетового излучения с длиной волны выше 214 мр [199, 255, 315].
Отдельные полосы поглощения для каждого из трех фенил-, четырех дифенил-,
двух трифенил-, семи метилфенил-, пяти диметилфенил-силш-триазолов лежат
в области 224—272 мр (величина е колеблется от 300 до 29200) [255]. Близость
ультрафиолетовых спектров 1-фенил-3-окси-1,2,4-триазола (X 282 мр, е 9100)
и 1-фенил-2,3-дигидро-2-метил-З-кето-1,2,4-триазола (X 280 мр, е 7400) застав-
ляет предположить для первого триазолоновое строение [255]. Установлено
сходство спектров поглощения 3,5-диметил-1,2,4-триазола (X 222 мр, е 6900)
и 3,5-диметил-1-(1,2,4-триазол-3-ил)-1,2,4-триазола (А, 220 -мр, & 6000) [199].
Величины поглощения для 3-амино- и 3-окси-5-фенил-1,2,4-триазолов сходны.
Как катион, так и нейтральная молекула последнего обнаруживают максимум
поглощения при 260 мр; однако для нейтрального соединения е равна 8000,
тогда как у катиона е 15 000 [199].
N —N — -- С —N
\
Н3сс ССН3 N СН
\ Z \ /
N N
Н
На основании изучения ультрафиолетовых спектров 1-замещенным гуана-
золам, которые можно изобразить пятью таутомерными структурами [316], при-
писано строение 3(5)-амино-1 -арил-5(3)-имино-1,2,4-триазолонов (А и Б) [317].
RN—NH
С—NH
RN-N
RN—NH
HN = C CNH2
\Z
RN—NH
h2nc/ c—nh
RN—N
\nh2
350 МОНОЦИКЛИЧЕСКИЕ ФрИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
Спектр 1-фенилгуаназола (максимумы при 266 мц, е 10,6-103 в этаноле,
при 250 Л1Ц, е 8,4-103 в 0,01 н. соляной кислоте и при 254 Atp, е 8,9-103 в 0,01 н.
едком натре) отличается от спектра 1-фенил-2-метилгуаназола (максимумы
при 216 лщ, е 30,0-103 в этаноле, при 224 лщ, е 19,7• 103 в 0,01 н. соляной кисло-
те и при 220 мц, е 22,1 • 103 в 0,01 н. едком натре). Эти различия приписываются
наличию резонансных таутомерных и ионных структур у первого соединения
и отсутствию их у второго [317].
Для 3-нитрамино-1,2,4-триазола и изомерного 3(5)-метил-5(3)-нитрамино-
1,2,4-триазола (стр. 348) характерны максимумы поглощения при 284—288 мц
[306]. Отмечаются различия в ультрафиолетовых спектрах изомерных 4-амино-
1,2,4-триазолов и дигидротетразинов [318].
Для сююи-триазола изучены инфракрасные спектры [319]. Рассмотрение
инфракрасного спектра З-метил-5-нитрамино-1,2,4-триазола заставляет пред-
положить, что азометиновые группы триазольного кольца поглощают при
6,15 ц и что для кольца характерны максимумы при 9,11 и 9,83 ц [320]. Пред-
ставляет интерес появление у силии-триазола азометиновой полосы поглощения,
которая, по-видимому, отсутствует у вицинальных триазолов и тетразолов
[320, 321].
Разработан колориметрический метод определения 3-амино-1,2,4-триазо-
ла [322].
Различные свойства и применение
Для 1-фенил-3,5-диметил-1,2,4-триазола [323], З-метил-5-амино-1,2,4-три-
азола [315] с т. пл. 149° и 152°, а также для 1,2-диацетил-1,2,4-триазолона-5
наблюдался полиморфизм [186]. Приводится рентгеноструктурный анализ
гуаназола [211]. Сообщается, что у 1-фенил-З-анилино-1,2,4-триазола [324]
дипольный момент составляет 3,54 0,02D, а у незамещенного 1,2,4-триазо-
ла—3.17D [325].
Смолы, получаемые из производных аминотриазола и альдегидов, при-
годны в качестве пластификаторов и клеев [326]. Разнообразные аминотриазо-
лы являются ингибиторами вуалирования, а также стабилизаторами фотогра-
фических эмульсий и цветообразователей для получения фотографического
изображения желтого цвета [327]. - • .
Биохимические свойства
Гербицид амизол (3-амино-1,2,4-триазол) особенно эффективен в отношении
многолетних сорных трав, таких, как ядовитый плющ, ядовитый дуб, канад-
ский татарник, пырей обыкновенный и водные сорняки [328]. Этот амин также
сильно понижает активность каталазы печени и почек крыс (но не крови)
в течение 3 час после внутрибрюшинного или внутривенного введения [329].
Изучена зависимость гистаминной активности аоюи-тр'иазоилэтиламинов
и имидазоилэтиламинов от их структуры. Соединения XXVII, XXVIII
и XXIX активны, в то время, как XXX и XXXI неактивны [190].
У гипертонических собак 1-метил-5-ф-аминоэтил)-1,2,4-триазол (XXIX)
вызывает снижение кровяного давления, однако после одного или двух приемов
животные становятся нечувствительными к этому препарату [190]. З-ф-Амино-
этил)-1,2,4-триазол вызывает длительное понижение кровяного давления
у млекопитающих. Увеличение или уменьшение длины, а также разветвлен-
СН3 —N—X N—N — СН3
/ \ Z \
НС CCH2CH2NHo нс cch2ch2nh2
\ / \ /'
N N
XXVII, Х = СН XXIX
XXVIII, X = N
Литература
351
N—X
н</ \ch2ch2nh2
CH3
XXX, х = сн
XXXI, X = N
иости боковых цепей уменьшает или сводит на нет фармакологическую актив-
ность [190].
Некоторые водорастворимые алкил- и арил-сшилс-триазолы могут инги-
бировать активность холинэстеразы [330].
ЛИТЕРАТУРА
1. Benson Saveli, Chem. Revs., 46, 1 (1950).
2. Boyer, Canter, Chem. Revs., 54, 1 (1954).
3. Michael, J. prakt. Chem., [2] 48, 94 (1893).
4. Michael, Luehn, Higbee, Am. Chem. J,, 20, 377 (1898).
5. Dimroth, Fester, Ber., 43, 2219 (1910).
6. Alder, Trimborn, Ann., 566, 58 (1950).
7. Mugnani, Grunanger, Atti accad. nazl. Lincei, Rend., Classe sci. fis., mat.
e nat., 14, 95 (1953) [C. A., 48, 2684 (1954)].
8. Kirmse, Horner, Ann., 614, 1 (1958).
9. Moulin, Helv. Chim. Acta, 35, 167 (1952).
10. Sheehan, Robinson, J. Am. Chem. Soc., 73, 1207 (1951).
11. Henkel, Weygand, Ber., 76, 812 (1943).
12. H a r t z e 1, В e n s о n, J. Am. Chem. Soc., 76, 667 (1954).
13. Dornow, Rombusch, Chem. Ber., 91, 1841, 1851 (1958).
14. В e г t h o, Ber., 58, 859 (1925).
15. В e г t h o, J. prakt. Chem., [2] 120, 89 (1929).
16: В e r t h o, Holder, J. prakt. Chem., [2] 119, 173 (1928).
17. Dimroth, Ber., 35, 1029 (1902).
18. Dimroth, Ann., 364, 183, 212 (1909).
19. Dimroth, Frisoni, Marshall, Ber., 39, 3920 (1906).
20. L i e b e r, Rao, R a j kumar, J. Org. Chem., 24, 134 (1959).
21. К 1 e i n f e 1 1 e r, Bonig, J. prakt. Chem., [2] 132, 175 (1932).
22. Boyer et al., J. Org. Chem., 23, 1051 (1958).
23. Wolff, Grau, Ann., 394, 68 (1912).
24. Chattaway, Parkes, J. Chem. Soc., 127, 1307 (1925).
25. F i e s e r, Hartwell, J. Am. Chem. Soc., 57, 1479 (1935).
26. Alder, Stein, Friedrichsen, Ann., 501, 1 (1933).
27. Rondest vedt, Chang, J. Am. Chem. Soc., 77, 6532 (1955).
28. Peratone r, Az z а г e 1 To, Gazz. chim. ital, 38, I, 76 (1908).
2.9. Meier, Chem. Ber., 86, 1483 (1953).
30. Curtius, Thompson, Ber., 39, 4141 (1906).
31. Dimroth, Ann., 373, 349 (1910); 377, 131 (1910).
32. Cook, Heilbron, Hunter, J. Chem. Soc., 1949, 1443.
33. Brown, H a m m i c k, J. Chem. Soc., 1947, 1384.
34. L e f f 1 e r, L i u, J. Am. Chem. Soc., 78, 1949 (1956).
35. Wolff, Ann., 394, 49 (1912).
36. Griess, Ber., 15, 2199 (1882).
37. Benson, Hartzell, Saveli, J. Am. Chem. Soc., 74, 4917 (1952).
38. Belcher, Sykes, Ta tlow, J. Chem. Soc., 1954, 4159.
39. Sanni e, Compt. rend., 233, 1494 (1951).
40. S 1 a t e r, J. Chem. Soc., 1932, 2196.
41. Красовицкий, Кочергина, ДАН СССР, 86, 1121 (1952).
42. Taylor, 135th American Chemical Society National Meeting, Boston, April 5—10,
1959, Abstracts,of Papers, p. 8N.
43. Schofield, Theobald, J. Chem. Soc., 1949, 2404.
44. v. P e c h m a n n, Ber., 21, 2751 (1888); v. Pechmann, Wehsarg, Ber,, 21,
2992 (1888).
45. Boyer, Morgan, J. Am. Chem. Soc., 80, 3012 (1958).
352
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
46. Н а п n, Н u d so п, J. Am. Chem. Soc., 66, 735 (1944); Haskins, Hann, Hud-
s о n, ibid., 69, 1050 (1947).
47. T e u b e r, J e 1 1 i n e k, Chem. Ber., 85, 95 (1952).
48. Weygand, Grisebach, Schmeiser, Angew. Chem., 63, 27 (1951).
49. Rapoport, C h e n, J. Org. Chem., 25, 313 (1960).
50. Stolle, Ber., 59, 1742 (1926).
51. R i e b s о m e r, J. Org. Chem., 13, 815 (1948).
52. Bamford, S t e v e n s, J. Chem. Soc., 1952, 4735.
53. Kliegl, Schmalenbach, Ber., 56, 1517 (1923).
54. E u 1 e r, Euler, Ber., 36, 4256 (1903); 37, 49 (1904).
55. Hinkel, Richards, Thomas, J. Chem. Soc., 1937, 1432.
56. Banks, P f 1 a s t e r e r, J. Org. Chem., 18, 267 (1953).
57. F r i e s, Franke, Bruns, Ann., 511, 241 (1934).
58. To wer s, Mortimer, Nature, 174, 1189 (1954).
59. G о 1 d s t e i n, S t a m m e, Helv. Chim. Acta, 35, 1470 (1952).
60. Schmidt, Hagenbocker, Ber., 54, 2191, 2201 (1921).
61. M u z i 1, Allan, Chem. listy, 52, 486 (1958) [C. A., 53, 4173 (1959)].
62. В о r s c h e, Ber., 54, 669 (1921).
63. Ch m a t a 1, Allan, M u i. i k, Chem. listy, 52, 948 (1958) [C. A., 53, 5251 (1959)].
64. M u2 i k, A 1 1 a n, Chem. listy, 48, 221 (1954) [C. A., 49, 2426 (1955)].
65. G r a n d m о u g i n, J. prakt. Chem., [2] 76, 134 (1907).
66. Bamburger, Hubner, Ber., 36, 3822 (1903).
67. C u s m a n o, Gazz. chim. ital., 51, II, 71 (1921).
68. Spasov, Elenkov, Robev, Bulgar. Acad. Nauk., Otdel Geol.-geograf. i Khim.
Nauk., Izvest. Khim. Inst., 1, 229 (1951) [C. A., 47, 2153 (1953)].
69. Q u i 1 i с о, M u s a n t e, Gazz. chim. ital., 71, 327 (1941).
70. W i e 1 a n d, G m e 1 i n, R о s e e u, Ann., 375, 300 (1910).
71. Ajello, Cusmano, Gazz. chim. ital., 70, 770 (1940); Ajello, Tornetta,
ibid., 77, 332 (1947).
72. W i t t i g, В e n g e r t, Kleiner, Ber., 61, 1140 (1928).
73. Q ui 1 ico, M u s a n t e, Gazz. chim. ital., 72, 399 (1942).
74. Gr a man tier i, Gazz. ital., 65, 102 (1935).
75. Wieland, Frank, Kitasato, Ann., 475, 42 (1929).
76. В 1 a d i n, Ber., 26, 545, 2736 (1893).
77. Cur t i us, M a у e r, J. prakt Chem., [2] 76, 369 (1907).
78. P 1 a u t, J. Am. Chem. Soc., 76, 5801 (1954).
79. Krollpfeiffer, Potz, Rosenberg, Ber., 71, 596 (1938).
80. E 1 b s et al., J. prakt. Chem., [2] 108, 209 (1924).
81. Charrier, Beretta, Gazz. chim. ital., 56, 191 (1926).
82. Wolff, Ann., 394, 23, 59, 68 (1912).
83. R assow, R u 1 k e, J. prakt. Chem., [2] 65, 107 (1902).
84. Sus et al., Ann., 579, 133 (1953).
85. G h i g i, P о z z i - В a 1 b i, Gazz. chim. ital., 71, 228 (1941).
86. В a 1 t z e r, v. P e c h m a n n, Ann., 262, 302 (1891).
87. Dimroth, Ber., 35, 1031 (1902).
88: V a г g h a, R e m e n у i, J. Chem. Soc., 1951, 1068.
89. C u r t i u s, К 1 a v e h n, J. prakt Chem., [2] 125, 498 (1930).
90. Bishop, Science, 117, 715 (1953).
91. J о n a s, v. P e c h m a n n, Ann., 262, 277 (1891).
92. S с a 1 e r a, A d a m s, J. Am. Chem. Soc., 75, 715 (1953).
93. H e n r y, Finnegan, L i e b e r, J. Am. Chem. Soc., 76, 88 (1954).
94. Brown, H a m m i c k, H e r i t a g e, J. Chem. Soc., 1953, 3820.
95. Car о n n a, Palazzo, Gazz. chim. ital., 82, 292 (1952).
96. C u r t i u s, J. prakt. Chem., [2] 125,303 (1930).
97. Snyder, Heckert, J. Am. Chem. Soc., 74, 2006 (1952); Snyder, Geller
ibid., 74, 4864 (1952).
98. Hohenlohe-Oehringen, Monatsh., 89, 557, 562, 588, 597 (1958).
99. W i 1 e у, M о f f a t, J. Am. Chem. Soc., 77, 1703 (1955).
100. Brady, Reynolds, J. Chem. Soc., 1928, 193; 1930, 2667.
.101. Япон. пат. 158894 [С. A., 43, 7971 (1949)].
102. Wiley, Hussung, Moffat, J. Org. Chem., 21, 190 (1956).
103. Huttel, Welzel, Ann., 593, 207 (1955).
104. Wiley et al., J. Am. Chem. Soc., 76, 4933 (1954).
105. L i c a r i et al., J. Am. Chem. Soc., 77, 5386 (1955): Burckhalter, Stephens,
Hall, ibid., 74, 3868 (1952).
106. Galimberti, Def r ancesch i, Gazz. chim. ital., 77, 431 (1947).
107. Wiley, Hussung, Moffat, J. Am. Chem. Soc., 77 , 5105 (1955).
108. Brady, Jakobvits, J. Chem. Soc., 1950, 767.
109. Morgan, Mickelthwait, J. Chem. Soc., 103, 1391 (1913).
ЛИТЕРАТУРА
353
ПО. Фельдман, Усовская, ЖОХ, 18, 1699 (1948).
111. Benson, Hartzel, Saveli, J. Am. Chem. Soc., 74, 4917 (1952).
112. Henry, D e h n, J. Am. Chem. Soc., 71, 2297 (1949).
113, Gaylord, J. Am. Chem. Soc., 76, 285 (1954).
114. Fries, Guterbock, Kuhn, Ann., 511, 213 (1934).
115. D i m г о t h, A i c k e 1 i n, Ber., 39, 4390 (1906).
116. Mustafa, Asker, H i s h m a t, J. Am. Chem. Soc., 77, 5127 (1955).
117. G i 1 1 e s p i e, E n g e 1 m a n, Graff, J. Am. Chem. Soc., 78, 1651 (1956).
118. M о r g a n, G о d d e n, J. Chem. Soc., 97, 1702 (1910).
119. M о г g a n, S c h a r f f, J. Chem. Soc., 105, 120 (1914).
120. R iebsomer, Stauffer, J. Org. Chem,, 16, 1643 (1951).
121. R iebsomer, Su m re 1 1, J. Org. Chem., 13, 807 (1948).
122. P о n z i o, Gazz. chim. ital., 29, 280 (1899).
123. Hiittel, Gebhardt, Ann., 558, 34 (1947).
124. Dimroth, Taub, Ber., 39, 3912 (1906).
125. Dimroth, Aickelin, Ber., 39, 4390 (1906); Curtius, Thompson,
Ber., 39, 3403 (1906); Pederson, Acta Chem. Scand., 12, 1236 (1958).
126. M i 1 1 e r, W a g n e r, J. Am. Chem. Soc,, 76, 1847 (1954).
127. Fries, Roth, Ann., 389, 318 (1912); Fries, Empson, Ann., 389, 358 (1912);
Fries, Noll, Ann., 389, 367 (1912); Fries, Guterbock, Kuhn, Ann.,
511,213 (1934); Z i n c k e, Ann., 311, 276 (1900).
128. Schofield, Quart. Rev, (London), 4, 388 (1950).
129. Zincke, Arzberger, Ann., 249, 270 (1888).
130. Пат. США 2700669 [C.A., 49, 6614 (1955)].
131. Пат. США 2462405 [С. А., 44, 341 (1950)].
132. V. Pechmann, Bauer, Ber,, 42, 659 (1909).
133. S t о 1 1ё, M и п с h, К i n d, J. prakt. Chem., [2], 70, 437 (1904).
134. Thiele, Schleussner, Ann., 295, 129 (1897).
135. Dimroth, Letsche, Ber., 35, 4054 (1902).
136, Bamberger, de Gruyter, Ber., 26, 2783 (1893); Jagerspacher, Ber.,
28, 1283 (1895).
137. H ii t t e 1 et al., Ann., 585, 115 (1954).
138. Sheehan, Robinson, J. Am. Chem. Soc., 71, 1436 (1949).
139. Stein, D’A n t о n i o, Farmaco (Pavia) Ed. sci., 10, 235 (1955) [C. A., 50, 4924
(1956)].
140. Cooper, J. Chem. Soc., 1952, 2408.
141, Beretta, Gazz. chim. ital., 55, 788 (1925).
142. Borsch e, Hahn, Wagner- Roemmisch, Ann., 554, 15 (1943).
143. F о r d, M с К a y, J. Chem. Soc., 1957, 4620; 1958 1290, 1294.
144. Campbell, Barclay, Chem. Revs., 40, 359 (1947); Plant, Powell,
J. Chem. Soc., 1947, 937; Allen, Suschitzky, ibid., 1953, 3845.
145. Lawson, Perkin, Robinson, J. Chem. Soc., 125, 626 (1924).
146. Ker mack, Storey, J. Chem. See,, 1950, 607.
147. Gibson, J. Chem. Soc., 1956, 1076.
148. S t e t t e r, Chem. Ber., 86 , 69 (1953).
149. Mihina, Herbst, J. Org. Chem., 15, 1082 (1950); Oliveri-Mandala,
Gazz. chim. ital., 44, 175 (1914).
150. Lieber et al., J. Org. Chem., 23, 1916 (1958).
151. Oliveri-Mandala, Gazz. chim. ital., 46, 308 (1916).
152. Taylor, Can. J. Research, 20B, 161 (1942).
153. Dimroth, Ann., 338, 154 (1905).
154. Curtius, Bockmuhl, Ber., 43, 2441 (1910).
155. F a g e 1, E w i n g, J. Am. Chem. Soc., 73, 4360 (1951).
156, D e d i c h e n, Ber., 39, 1831 (1906).
157, Тарасевич, Вестник МГУ, 3, № 10, 161 (1948).
158. Cheng, Anal. Chem., 26, 1038 (1954).
159, Cambi, Canonic a, Sironi, Atti accad. nazl. Lincei, Rend., Classe sci. fis.,
mat. e nat., 18, 583 (1955).
160. W i 1 s о n, W i 1 s о n, J. Am. Chem. Soc., 77, 6204 (1955); Anal. Chem., 28, 93
(1956).
161. Runge, Engelbrecht, Franke, Chem. Ber., 88, 533 (1955).
162. Horner, Kliipfel, Ann., 591, 69 (1955).
163. Ghigi, Rocchi, Gazz. chim. ital., 85, 183 (1955).
164. Po.zzo-Balbi, Ann. chim. (Rome), 43, 222 (1953).
165, Heafield, Hunter, J. Chem. Soc., 1942, 420.
166. White, Kilpatrick, J. Phys. Chem., 59, 1044 (1955).
167. Dal Monte, Mangini, Montanari, Gazz. chim. ital., 88, 1035 (1958);
Dal Monte et al., ibid., 88, 977 (1958).
168. К о h 1 г a u sch, S e k a, Ber., 73, 162 (1940).
23 Заказ Ka 978
354 МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
169. Англ. пат. 726119 [С. А., 49, 15253 (1955)]; пат. США 2684966 [С. А., 48, 14227
(1954)].
170. Chem. Eng. News. 34, 2450 (1956).
171. Jensen, Friediger, Kgl. Danske Videnskab. Selskab, Mat.-fys. Medd., 20,
№ 20, 1 (1943) [C. A., 39, 2068 (1945)].
172. L e Fevre, Liddicoet, J. Chem. Soc., 1951, 2743.
173. F i e s e r, Ames, J. Am. Chem. Soc., 49, 2604 (1927); Fieser, Martin, ibid.,
57, 1840 (1935).
174. v. Auwers Ber., 71, 604 (1938).
175. Fagley, Klein, Albrecht, J. Am, Chem. Soc., 75, 3104 (1953); F a g 1 e y,
частное сообщение.
176. R о t h, M fl 1 1 e r, Ber., 62, 1190 (1929).
177. Basu, Proc. Natl. Inst. Sci. India, 21A, 173 (1955) [C. A., 50, 6175 (1956)].
178. Castle, Mikrochim. Acta, 1953, 196.
179. К о f 1 e r, Mikrochemie ver. Mikrochim. Acta, 34, 15 (1948).
180. Англ. пат. 684489 [С. A., 47, 7258 (1953)].
181. Dimroth, Dienstbach, Ber., 41, 4055 (1908).
182. Пат. США 2714110 [C. A., 50,12116 (1956)].
183. Jerchel, FIAT Rev. German Sci. Biochemistry Pt. I, 59 (1939—1946) [C. A., 42,
5059 (1948)].
184. Gillespie, Engelman, Graff, J. Am. Chem. Soc., 76, 3531 (1954).
185. Loveless, Spoerl, Weisman,J. Bacteriol., 68, 637 (1954).
186. G e h 1 e n, Ann., 563, 185 (1949); 577, 237 (1952).
187. G u h a, De, J. Indian Chem. Soc., 1, 141 (1924).
188. С 1 e v e, Ber., 29, 2671 (1896); W i d m a n, Ber., 29, 1946 (1896); W i d m a n,
С 1 e v e, Ber., 31, 378 (1898).
189. Arndt, Bielich, Ber., 56, 2276 (1923); Girard, Compt. rend., 225, 458 (1947).
190. Ainsworth, Jones, J. Am. Chem. Soc., 75, 4915 (1953); 76, 5651 (1954); 77,
621, 1538 (1955).
191. Wojahn, Arch. Pharm,, 285, 122 (1952).
192. Fromm, Nehring, Ber., 56, 1374 (1923).
193. G u h a , J. Am. Chem. Soc., 45, 1036 (1923).
194. К a n a о k a, J. Pharm. Soc. Japan, 75, 1149 (1955) [C. A., 50, 5647 (1956)]; S t о 1 1 ё,
Bowles, Ber., 31, 1101 (1908).
195. Audrieth, Scott, Kippur, J. Org. Chem., 19, 733 (1954).
196. Arndt, M i 1 d e, Ber., 54, 2089 (1921); Arndt, Mil de, Tschenscher,
Ber., 55, 341 (1922); Arndt, Tschenscher, Ber., 56, 1984 (1923).
197. Англ, пат. 741228 [С. A., 50, 9913 (1956)]; англ. пат. 736568 [С. А., 50, 13097 (1956)].
198. Thiele, Hei denreich, Ber., 26, 2598 (1893); Thiele, Manchot, Ann.,
303, 52 (1898).
199. Atkinson et al., J. Chem. Soc., 1954, 4508.
200. Bamberger, Frei, Ber., 35, 746 (1902).
201. M i c h a e 1, J. prakt. Chem., [2] 60, 292 (1899).
202. Freund, Ber., 29, 2483 (1896); Freund, Schander, Ber., 29, 2506 (1896).
203. Allen, Bell, Org. Syntheses, 26, 11 (1946).
204. Biemann, Bretschneider, Monatsh., 89, 603 (1958).
205. Wieland, Ber., 42, 4199 (1909).
206. Pinner, Ber., 26, 2130 (1893); 27, 985 (1894); Ann., 297, 223, 249 (1897).
207. P о t t s, J. Chem. Soc., 1954, 3461.
208. В 1 a d i n, Ber., 25, 183 (1892).
209. В a i 1 e у, M о о г e, J. An . Chem. Soc., 39, 279 (1917); Bailey, McPherson,
ibid., 39, 1322 (1917).
210. Backer, Mulder, Rec. trav. chim., 44, 1113 (1925).
211. De, Chakravorty, J. Indian Chem. Soc., 7, 875 (1930).
212. Hoggarth, J. Chem. Soc., 1949, 1160.
213. Duschinsky, Gainer, J. Am. Chem. Soc., 73, 4464 (1951).
214. Pellizzari, Cuneo, Gazz. chim. ital., 24, 506 (1894).
215. Guha, Mehta, J. Indian Inst. Sci., 21A, 41 (1938) [C. A., 33, 598 (1939)].
216. Busch, Schneider, J. prakt. Chem., ]2] 89, 310 (1914).
217. Henry, DeVries, Boschan, J. Am. Chem. Soc., 77, 5693 (1955).
218. Busch, Brandt, J. prakt. Chem., [2] 74, 533 (1906).
219. Fromm, Vetter, Ann., 356, 178 (1907).
220. P i n n e г, С a г o, Ber., 27, 3273 (1894); Ber., 28, 465 (1895).
221. Henry, Skolnik, Smith, J. Am. Chem. Soc., 75, 955 (1953).
222, Oda, T a n i m о t o, J. Chem. Sc. Japan, Ind. Chem. Sect., 55, 595 (1952).
223. Engelhardt, J. prakt. Chem., [2] 54, 143 (1896).
224. Sonner skog, Acta Chem. Scand., 12, 1241 (1958).
225. В 1 a d i n, Ber., 18, 1544 (1885).
226. Rat z, Schroeder, J. Org. Chem., 23, 1931 (1958).
ЛИТЕРАТУРА
355
227. S t о 1 1 ё, Вег., 32, 796.(1899).
228. Herbst, Garrison, J. Org. Chem., 18, 872 (1953).
229. Wiley, Наг t, J. Org. Chem., 18, 1368 (1953).
230, В u s c h, J. prakt. Chem., [2] 89, 552 (1914).
231. Klingsberg, J. Org. Chem., 231, 1086 (1958).
232. Pellizzari, Soldi, Gazz. chim. ital., 35, 373 (1905).
233. Thiele, Strange, Ann., 283, 41 (1894); d’A г с a n g e 1 o, Rev. fac. cienc.
quim. Univ. nacl. La Plata, 18, 81 (1943) [C. A., 41, 948 (1947)].
234. Buckley, Ray, J. Chem. Soc., 1949, 1156.
235. Hoggarth, J. Chem. Soc., 1952, 4811.
236. S t о 1 1 ё, T h 6 m a, J. prakt. Chem., [2] 73, 288 (1906),
237. M i у a t a к e, J. Pharm. Soc. Japan, 72, 1486 (1952).
238. Pellizzari, Atti reale accad. Lincei, 8, I, 330 (1899); Ruheman, Stap-
le t о n, J. Chem. Soc., 75, 1131 (1899).
239. Smith, Organic Reactions, 3, 367 (1946).
240. Schrader, J. prakt. Chem., [2] 95, 312 (1917).
241. Migrdichian, The Chemistry of Organic Cyanogen Compounds, Reinhold Pub-
lishing Corp., New York, 1947, p. 104.
242. Atkinson, Polya, J. Chem. Soc., 1954, 3319; J. Am. Chem Soc., 75, 1471 (1953).
243. R i e d, M u 1 1 e r, Chem. Ber., 85, 470 (1952); Jerchel, Kuhn, Ann., 568, 185
(1950); Jerchel, Fischer, Ann., 574, 85 (1951).
244. Lieber et al., J. Org. Chem., 18, 218 (1953).
245. Busch, Ber., 38, 856 (1905); J. prakt. Chem., [2] 67, 201 (1903).
246. Warren, J. Chem. Soc., 1938, 1100.
247. Baker, Ollis, Poole, J. Chem. Soc., 1950, 1545.
248. Forster, F-i e r z, J. Chem. Soc., 87, 722 (1905).
249. Beyer, Kreutzberger, Reese, Chem. Ber., 84, 478 (1951),
250. W i d m a n, Ber., 26, 2612 (1893).
251. Англ. пат. 682399 [С. A., 47, 4132 (1953)].
252. Einhorn, Bischkopff, Szelinski, Ann., 343, 227 (1905); Brunner,
Ber., 47, 2671 (1914).
253. Atkinson, Polya, J. Chem. Soc., 1952, 3418.
254. Thompson, J. Am. Chem. Soc., 73, 5914 (1951).
255. Atkinson, Parkes, Polya, J. Chem. Soc., 1954, 4256.
256. Ghosh, Betrabet, J. Indian Chem. Soc., 7, 899 (1930).
257. К о m z a k, Polya, J. Appl. Chem., 2, 666 (1952); Brunner, Monatsh., 36,
509 (1915).
258. Atkinson, Polya, J. Chem. Soc., 1954, 141.
259. Brunner, Medwit h, Monatsh., 47, 741 (1926); Brunner, ibid., 36, 509
(1915).
260. Kaiser, Peters, J. Org. Chem., 18, 196 (1953); пат. США 2399598 [С. A., 40,
4227 (1946)].
261. Пат. США 2406591 [С. А., 41, 488 (1947)].
262. Kaiser, Peters, Wystrach, J. Org. Chem., 18, 1610 (1953); Pelliz-
zari, Gazz. chim. ital., 24, 481 (1894).
263. Pellizzari, L’Orosi, 17, 143, 185 (1893).
264. Guha, Janniah, J. Indian Inst. Sci., 21A, 60 (1938); Fromm, Schneider,
Ann., 348, 174 (1906).
265. Hoggarth, J. Chem. Soc., 1949, 1160.
266. S i 1 b e r r a d, J. Chem. Soc., 77, 1190 (1900).
267. Pellizzari, Gazz. chim. ital., 24, 222 (1894); 41, 20 (1911); Atti reale accad.
Lincei, 10 297 (1901).
268. Papin i, Checchi, Ridi, Gazz. chim. ital., 84, 769 (1954).
269. Rupe, Labhart, Ber., 22, 233 (1900); R u p e, M e t z, Ber., 36, 1092 (1903).
270. G a s t a 1 d i, Gazz. chim. ital., 53, 629, 635 (1923).
271. В ii 1 о w, Ber., 39, 4106 (1906); Curtius, Darapsky, Muller, Ber., 41,
3161 (1908).
272. В а к e г, О 1 1 i s, P о о 1 e, J. Chem. Soc., 1950, 3389.
273. D i e 1 s, G r u b e, Ber., 53, 854 (1920).
274. Huisgen, Sauer, Seidel, Chem. & Ind. (London), 1958, 1114.
275. Losse n, Statius, Ann., 298, 97 (1897).
276. Curtius, Ehrhardt, Ber., 55, 1559 (1922).
277. Feichtinger, Z. Naturforsch., 3b, 377 (1948),
278. Rat z, Schroeder, J. Org. Chem., 23, 2017 (1958); Diels, Ber., 47, 2183 (1914).
279. Atkinson, Polya, Chem. & Ind. (London), 1954, 462.
280. S t о 1 1 e, Ber., 45, 273 (1912).
281. Steck, Brundage, Fletcher, J. Am. Chem. Soc., 80, 3929 (1958).
282. Manchot, Noll, Ann., 343, 9 (1905); В 1 a d i n, Ber., 25, 741 (1892).
283. Manchot, Ber., 31, 2444 (1898).
23*
356
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫ И БЕНЗОТРИАЗОЛЫ
284. Duffin, Kendall, Waddington, Chem & Ind. (London), 1954, 1458.
285. Arndt, Loewe, T ar 1 a n - A к on, Rev. fac. sci. univ. Istanbul, 13A, 127
(1948) [C. A., 42, 8190 (1948)].
286. A c r e e, Am. Chem. J., 38, 1 (1907).
287. Wiley et al., J. Am. Chem. Soc., 77, 2572 (1955).
288. Пат. США 2476549 [С. A., 44, 66 (1950)]; пат. США 2444605 [С. А., 42, 7178 (1948)].
289. Birr, Werner, Chem. Ber., 86, 1401 (1953).
290. Allen et al., 135th National American Chemical Society Meeting, Boston, April
5—10, 1959, Abstracts of Papers, p. 78-0.
291. Cook, Gentles, Tucker, Rec. trav. chim., 69, 343 (1950).
292. Papin i, Checchi, Gazz. chim. ital.,-82, 735 (1952).
293. Papin i, Manganelli, Gazz. chim. ital., 80, 855 (1950).
294. H e n r y, D e h n, J. Am. Chem. Soc., 71, 2297 (1949).
295. В 1 a d i n, Ber., 25, 741 (1892).
296. Andreocci, Ber., 25, 225 (1892).
297. Henry, Finnegan, J. Am. Chem. Soc., 76, 290 (1954).
298. You n g, J. Chem. Soc., 87, 625 (1905).
299. Grimmel, Morgan, J. Am. Chem. Soc., 70, 1750 (1948).
300. Stolid, Ber., 39, 826 (1906); Ruhemann, Ber., 39, 1228 (1906); N о z о e,
I t о, M a t s u i, Proc. Japan. Acad., 30, 313 (1954) [C. A., 50, 972 (1956);]; S a z a к i,
Pharm. Bull. (Japan), 2, 123 (1954) [C. A., 50, 972 (1956)].
301. Hoggarth, J. Chem. Soc., 1950, 614.
302. Пат. США 2554816 [С. A., 46, 3087 (1952)].
303. S t r a i n, J. Am. Chem. Soc., 49, 1566, 1995 (1927).
304. Y о u n g, W i t h a m, J. Chem. Soc., 77, 224 (1900).
305. Lieber, Patinkin, Tao, J. Am. Chem. Soc'., 73, 1792 (1951).
306. D e V r i e s, G a n t z, J. Am. Chem. Soc., 76, 1008 (1954).
307. Basu, Proc. Natl. Inst. Sci. India, 21A, 173 (1955) [C. A., 50, 6175 (1956)].
308. D e d i c h e n, Ber., 39, 1849 (1906).
309. S u n d, J. Agr. Food Chem., 4, 57 (1956).
310. Leaper, Bishop, Research Correspondence, Suppl. to Research (London). 8,
№ 8, S40 (1955) [C. A., 50, 7783 (1956)].
311. Gut bier, Z. angew. Chem., 18, 494 (1905).
312. Haszeldine, Leedham, J. Chem. Soc., 1952, 3483.
313. Komzak, Polya, J. Appl. Chem., 2, 666 (1952).
314. Runge, Engelbrecht, Franke, Chem. Ber., 88, 533 (1955).
315. Goerdeler, Wember, Worach, Chem. Ber., 87, 57 (1954).
316. Stolid, Dietrich, J. prakt. Chem., [2] 139, 193 (1934).
317. Steck, Nachod, J. Am. Chem. Soc., 79, 4411 (1957).
318. Grammaticakis, Compt. rend., 241, 1049 (1955).
319. Otting, Chem. Ber., 89, 2887 (1956).
320. Lieber, Levering, Patterson, Anal. Chem., 23, 1594 (1951).
321. Boyer, Borger s, Wolford, J. Am. Chem Soc., 79, 678 (1957).
322. S u n d, J. Agr. Food Chem., 4, 57 (1956).
323. Branstatter, Grimm, Mikrochim. Acta, 1956, 1175 [C. A., 50, 9093 (1956)].
324. Edgerley, Sutton, J. Chem. Soc., 1950, 3394.
325. Jensen, Friedriger, KgL Danske Videnskab, Mat.-fys. Medd., 20, № 20,
1 (1943) [C. A., 39, 2068 (1945)].
326. Англ. пат. 583367 [С. A., 41, 2607 (1947)]; пат. США 2406591 [С. А., 41, 488 (1947)];
пат. США 2723275 [С. А., 50, 10135 (1956)].
327. Пат. США 2444605 [С. А., 42, 7178 (1948)]; пат. США 2449225 [С. А., 43, 52 (1949)];
англ. пат. 741228 [С. А., 50, 9913 (1956)]; пат. США 2406654 [С. А., 41, 42, (1947)].
328. Т a f uro, Beatty, Guest, Proc. Northeast. Weed Control Conf., 1955, 31
[C. A., 49, 6527 (1955)]
329. Heim, Appelman, Pyfrom, Am. J. Physiol., 186, 19 (1956) [C. A., 50,
15665 (1956)].
330. Polya, Nature, 176, 1175 (1955).
Глава VI
ОКСАДИАЗОЛЫ
Дж. Бойер
ВВЕДЕНИЕ
Каждое из четырех оксадиазол иных колец состоит из двух атомов углеро-
да, двух атомов азота и одного атома кислорода. Приведенные под каждой
формулой названия применяются для обозначения производных неизвестных
незамещенных гетероциклов.
1,2,3-Оксадиазол,
фуро[аЬ]диазол,
диазоксид
НС—СН
N—СН
1,2,4-Оксадиазол,
фуро[аЬ]диазол,
азоксим
N—N
1,2,5-Оксадиазол,
фуро[аа1]диазол, фуразан,
азоксазол
1,3,4-Оксадиазол,
фуро[661]диазол, биазол,
оксибиазол, оксадиазол
В настоящей главе материал расположен по степени важности соединений,
вследствие чего сначала рассматриваются 1,2,5- и 1,2,4-оксадиазолы. Боль-
шинство производных 1,2,3-оксадиазола можно представить как изомерные
а-диазокарбонильные соединения и поэтому они здесь не будут рассмотрены.
Природные соединения с оксади азольными кольцами не известны. Это
обстоятельство, а также отсутствие исследований в области промышленного
применения этих гетероциклов является причиной того, что ряд проблем,
касающихся их строения, реакций и свойств, остается нерешенным.
1,2,5-ОКСАДИАЗОЛЫ
Получение фуразанов
Дегидратация глиоксимов. Фуразаны — внутримолекулярные ангидриды
глиоксимов — получены при нагревании глиоксимов с водой [1], водным
раствором аммиака или едким натром [2] и, реже, с мочевиной [3], уксусным
[4] или янтарным ангидридом [5], а также с хлорокисью фосфора [6]. Приме-
нение других кислотных дегидратирующих агентов, по-видимому, безрезуль-
татно [5], а действие хлорокиси фосфора на глиоксимы вызывает образование
1,2,4-оксадиазолов [7]. Другие методы включают обработку а- или |3-бензил-
358
ОКСАДИАЗОЛЫ
диоксима сернокислой медью [8], а также длительное экспонирование а-бен-
зилдиоксима в разбавленной щелочи на солнце в условиях тропиков [9].
Фуразаны могут быть получены при действии щелочи [10, 11] на сложные
диэфиры глиоксима или при перегонке их с паром [12]. В некоторых случаях
фуразаны образуются при взаимодействии гидроксиламина с 1,2-дикарбониль-
ными соединениями [13]. Ариламиноглиоксимы, по-видимому, существуют
в виде двух структурных изомеров, один из которых образует сложный диэфир
диоксима, а другой — изомерное O.N-диацильное производное. В присутствии
едкого натра сложный диэфир диоксима дегидратируется с образованием
1,2,4-оксадиазола, тогда как изомер теряет молекулу карбоновой кислоты,
что приводит к образованию арилациламинофуразана [14].
nh4oh СН3С—ССН3
ИЛИ У к
NaOH Янтарный ангидрид
[СН3С( = NOH)]2 — > <------------- [CH3C( = NOH)]2
о
//°\/c=NOH\
Olli )
NH2CONH2, 80°
или H2O, 1 65°
сн3
I
CH3CO2N = CC—cnh2
СН3С( = NOH)C( = NOH)C( = NOH)NH2
(CH3CO)2O
25°
СбЩС---CCgHg
C6H5Q = NOH )C( = NOH)NH2
POCIa
(CeH5C = NOCOCH3)2
KOH
C6H5C-CCeH3
l/ *N
C6H5C( = NOCOR)C—N
NaOH
:6h5c(=nocor)C(=nocor)nh2 —->
— H2O
C6H5C—CNHCOR
NaOH
C6H5C( = NOCOR)C(=NCOR )NHOH
— KUUUrl
Попытки получить три- и тетраметиленфуразаны из соответствующих
алициклических глиоксимов действием мочевины [3], уксусного или янтар-
ного ангидрида, гидроокиси аммония или едкого натра оказались безуспеш-
ными [15]. Однако сообщается, что дегидратация диоксима (I) в фуразан (II)
проходит в присутствии аммиака при 160° [16]. Это является единственным
известным примером образования фуразанового (не считая фуроксанов) кольца,
1,2,5-ОКСАДИАЗОЛЫ
359
конденсированного с неароматическим пятичленным циклом. Тетрабромтетра-
метиленфуразан (IV) получен из бензофуразана (III) и брома [17].
(СН3)2 n
(CH3)Y | О
(СН3)2
(СН3)2
NOH
NOH
NH4OH
------->
160°
II
Перегонка с паром щелочного раствора диоксима о-бензохинона [18]
используется в качестве метода получения бензофуразана (III). Все известные
производные, содержащие фуразановое кольцо, конденсированное с арома-
тическим кольцом, могут быть получены дегидратацией соответствующих
диоксимов о-хинонов. Дегидратация иногда происходит самопроизвольно [19],
либо при хранении продукта при комнатной температуре [18], либо при нагре-
вании до температуры плавления [20]. Диоксимы о-хинонов могут быть дегид-
ратированы в фуразаны уксусным ангидридом [18, 21 ], фенилизоцианатом [22],
Вг
.NOH /ч N I N
-Н2О I^Y \ Вг2 Вг— \
II —ч I > BJ I >
у»
Вг
III IV
абсолютным спиртом (при 130— 150°) [23], серной кислотой [24], а также хло-
ристым ацетилом или хлористым бензоилом со щелочью [24, 25]. При дегид-
ратации уксусным ангидридом тетраоКсим (V) превращается (ступенчато)
в ангулярный трициклический фуразан (VI) [26], а диоксим (VII) тетракето-
тетрагидронафталина — в фуразан (VIII) [4]
NOH N N
-НОН \ -НОН \
II L > —* .1 L /°
HON^Y^NOH HON^Y N N
NOH NOH I ||
O—N
V VI
О О
УХ/хУ1^ (CHCOljO
I и । (UHgUO)2U I и 1 \
I II I CH3COONa' I || I Z°
^Y^NOH '4/\/ N
О 0
VII VIII
Изомерные иминопроизводные моноксима Р-нафтохинона превращаются
при окислении в тот же фуразан [27]. Образуется ли при этом в качестве про-
межуточного продукта диоксим, установлено не было. Окисление 1-нитрозо-
\/\Z
X = NOH, Y=NH
X = NH, Y = NOH
Щелочной раствор
------------------->
КзРе(СЫ)в нлн NaOCl
Щелочной раствор
KsFe(CN)e нли NaOCl
A=NO,:. B=NH2
A = NH2j B=NO
360
оксадиАзолы
2-амино- и 1-амино-2-нитрозонафталинов гипохлоритом или щелочным раство-
ром феррицианида приводит к тому же фуразану [27, 28]. В аналогичных
условиях щелочной раствор гипохлорита количественно окисляет о-нитрозо-
ацетанилид (о-нитрозоанилин неизвестен) в бензофуразан128].
При превращении монооксима метилглиоксаля [29] в метиламинофура-
зан действием щелочного раствора гидроксиламина, по-видимому, происходит
CH3COCH = NOH
h2noh
NaOH
СН3С—CNH2
как аминирование, так и дегидратирование диоксима. Об аналогичном
превращении оксима фенилглиоксаля (изонитрозоацетофенона) в фениламино-
фуразан сообщалось ранее [30].
Превращения циклов. У некоторых ацилизооксазолов при действии щелоч-
ного раствора гидроксиламина происходит раскрытие цикла с последующим
СН3СОС-СН
h2noh
NOH NOH
II II
C6H5CCH2C
NOH
II
ссн3
C6H5CCH2C—ссн3
II к.
HON N N
образованием фуразана [31]. Так, З-бензоил-5-фенил-1,2,4-оксадиазол пре-
вращается при действии гидроксиламина в З-бензамидо-4-фенилфуразан
(IX) [32]. Образующийся стабильный оксим исходного соединения перехо-
С6Н5С—CNHCOC6H5
N N
С6Н5СОС—N
h2noh
60°~*
IX
дит при температуре плавления в соединение IX [32]. При действии гидро-
ксиламина изонитрозопирролы также превращаются в фуразаны [33],
СН3
I
Н3С—г—= NOH h2noh СН3С—ССНССН3
[ ----> // \ II
Н3с— СН3 N' N NOH
N \ /
О
Различные методы. Сообщается о превращении перицианиловой кислоты
в теплом хлористом тиониле в изоцианат цианфуразана [34].
HON NO2H
II II
NCC —CCH = NOH
NCC—CNCO
При взаимодействии ацетилена с азотной кислотой в присутствии ацетона
[35] наряду с изоксазолами получаются фуразаны, одним из представителей
которых является фуразандинитроловая кислота [36].
1,2,5-ОКСАДИАЗОЛЫ
361
HNO3
С2Н2------>
NOH NOH
II II
o2ncc~ ccno2
При образовании дианилинофуразана из фенилтиоцианата и гидроксил-
амина [37], по-видимому, имеют место дегидрирование, обессеривание и дегид-
ратация.
c6h5nhc—cnhc6h5
h2noh
C6H5CNS------>
Представляет интерес способ получения 5-метоксибензофуразана
из 2-нитро-4-метоксиацетанилида путем обработки этилкарбаматом и пяти-
окисью фосфора в кипящем ксилоле [38].
NHCOCH3 н2мсоос2н5
СНзО-I^J-NO2
р2о6
СН3О -1
В случае многих, но не всех фуроксанов при действии пятихлористого
фосфора происходит отщепление кислорода с образованием соответствующих
фуразанов (см. стр. 370).
Получение фуроксанов
Дегидрирование глиоксимов. Фуроксаны представляют собой N-окиси
фуразанов. Превращение глиоксимов в фуроксаны осуществляется с помощью
щелочных растворов феррицианидов, окислов азота, галогецов или щелочных
растворов гипогалогенитов [35]. Диоксим бензила при действии щелочного
раствора феррицианида [1, 40], хлора в этаноле или бензоле [39], щелочного
раствора гипохлорита [41] или N2O3 [42] превращается в дифенилфуроксан
(X), который получается также (но в виде следов) из диоксима и N2O4 [39].
Из диалкилглиоксимов и N2O4 соответствующие диалкилфуроксаны полу-
чаются с хорошим выходом [39], поэтому метод рекомендуется для получения
диметилфуроксана. Диоксим циклогександиона-1,2 (ниоксим) при действии
N2O4 нитруется и(или) окисляется, образуя адипиновую кислоту [39]. Тетра-
метиленфуроксан получают из ниоксима и гипохлорита натрия [39]. Для подоб-
ных превращений был также использован иод в присутствии ацетата ртути
в неводной среде [431.
С6Н5С—ССвН5
(C6H5C=NOH)2
о
X
362
ОКСАДИАЗОЛЫ
Несимметричные глиоксимы окисляются в изомерные фуроксаны.
Из у- и 6-модификаций диоксима n-метоксибензила получены оба возможных
фенил-п-анизилфуроксана [44]. Каждая из двух других модификаций диок-
сима образует смеси фуроксанов. Аналогичным образом получены изомерные
фуроксаны из диоксимов метиларилдикетонов [45]. Метилфенилглиоксим и фер-
С6Н5С---: СС6Н4ОСН3 С6Н5С—СС6Н4ОСН3
N fi _______________> Sf •
он он \о
С6Н5С-----СС6Н4ОСН3
II • II
N N
С6Н5С-СС6Н4ОСН3
но' но"
рицианид калия дают метилфенилфуроксан [46]. Фенилфурбксан получают
из фенилглиоксальдиоксима и N2O4 [47] или пятиокиси азота [48].
По-видимому, окисление феррицианидом предшествует замыканию кольца,
так как попытки осуществить непосредственное окисление фуразана в фурок-
сан оказались безуспешными. Это согласуется с наблюдением, что в щелочном
растворе а-аци-нитрооксимы дегидратируются с образованием фуроксанов
[49, 50]. При смешении глиоксимов и галогена, гипогалогенида или N2O4
RC—CR'
RC —CR' -NaOH \
II || -----> N Nx
HON NO2Na \ /4
\о/ О
может появиться синяя окраска [39]. Это считают доказательством образова-
ния промежуточного гелг-галогеннитрозо- или гелг-нцтронитрозопроизводного.
Отщепление галогена, галогеноводорода, N2O4 или азотистой кислоты приво-
дит к образованию фуроксана.
(RC = NOH)2
NaOCl или CI2
или N2O4
(RQX)NO)2
или
RC(X)NO
RC=NOH
—х2
----
или
-НХ
RC-CR
Х = С1 или NO2
Попытки получить триметиленфуроксан из диоксима циклопентандиона-1,2
с помощью перечисленных выше реагентов оказались безуспешными [15].
В противоположность этому диоксим камфорхинона превращается в один из двух
ожидаемых изомерных фуроксанов (XI) [15, 51], что является единственным
доказательством образования конденсированной системы, состоящей из пяти-
членного и фуроксанового колец.
XI
Фуроксаны из окисей нитрилов. Окись бензонитрила впервые получена
в 1894 г. [52]. Полагают, что во многих реакциях те или другие окиси нитрилов
образуются в качестве промежуточных продуктов. Окиси нитрилов димери-
1,2,5-ОКСАДИАЗОЛЫ
363
зуются в фуроксаны (N-окиси 1,2,5-оксадиазолов), N-окиси азоксимов (N-окиси
1,2,4-оксадиазолов) или 1,4-диокса-2,5-диазины, а также тримеризуются
с образованием 1,3,5-триокисей сылглг-триазина. Окиси нитрилов являются,
по-видимому, промежуточными продуктами в каждом из следующих четырех
синтезов фуроксанов.
Альдоксимы и окислы азота. Ароматические альдоксимы превращаются
в соответствующие диарилфуроксаны при действии N2O4 [53], трехокиси азота
[42], окислов азота [54, 55] или амилнитрита [56, 57]. Применение алкилнит-
ритов может привести к образованию 1,2,4-оксадиазолов. Ароматические
альдоксимы одновременно нитруются и окисляются N2O4 [58]. При более
высоких температурах нитрование дает более высокий выход нитроловых
кислот, которые, по-видимому, теряют элементы азотистой кислоты с образо-
ванием окисей нитрилов. Димеризация ведет к образованию фуроксанов.
Алифатические нитроловые кислоты более стабильны, чем ароматические или
ацилнитроловые кислоты, поэтому в мягких условиях они не могут превра-
щаться в диалкилфуроксаны.
RC-CR NO2
\ -hno2 I
N N <— RCNO <-----------RC —NOH
I n2o4 • I
RCH = NOH <-------RCH = NOH
Превращение альдоксимов в фуроксаны не начинается с окисления в аци-
нитроалканы. Вследствие этого такие окислители, как щелочной раствор
гипохлорита, щелочной раствор феррицианида или кислота Каро, при взаимо-
действии с альдоксимами дают другие продукты.
Окисление и декарбоксилирование а-оксиминокарбоновых кислот. Дифенил-
фуроксан получают из а-изонитрозофенилуксусной кислоты действием N2O4
[59] или иода в растворе соды [60]. а-Изонитрозо-а-алкилуксусные кислоты
в этих условиях превращаются в соответствующие нитроловые кислоты [61].
C6H5C( = NOH)COOH CeH6(C2N2O2)C6Hs
N2O4
RC( = NOH)COOH-------> RC( = NOH)NO2
Метилкетоны и азотная кислота. Голлеман [62] открыл превращение
метиларилкетонов в фуроксаны действием азотной кислоты. Из ацетофенона
получены дибензоилфуроксан (XII) [63] и бцс-бензоилформальдоксимино-
фуроксан (XIII) [64]. Эти же соединения синтезированы действием на метил-
фенилкарбинол [65] и 1,6-дифенилгексантетраон-1,3,4,6 [66] азотной кислоты,
а также из хлорангидрида бензоилгидроксамовой кислоты и уксуснокислого
О О
II II
С6Н5С СС6Н5
I I
С6Н5СОС — ССОС6Н5 C6H5COON = СС — СС= NOOCC6H5
hno3 У \ У \
С6Н5СОСН3 —:> N N + N . N
rlUlNU
N 0 О 0
XII XIII
натрия [67]. Очень широко изучалась реакция с ацетофеноном [64, 65, 68, 69].
То, что сначала Происходит нитрозирование по метильной группе, согла-
суется с наблюдениями, что не содержащая окислов азотная кислота не пре-
вращает ацетофенон ни в соединение XII, ни в XIII. Эти продукты получены
364
ОКСАДИАЗОЛЫ
также из изонитрозоацетофенона или w-нитроацетофенона при действии
не содержащей окислов азотной кислоты [68]. Предложена схема вероятного
превращения изонитрозоацетофенона в окись бензоилнитрила, которая затем
димеризуется с образованием дифенилфуроксана [69]. Попытки получить моно-
мерную окись бензоилнитрила из бромистого бензоила и гремучего серебра
HONO HNO3
СвН5СОСН3 ----> CeH5COCH=NOH------->
NO2
I
CeH5COC-NOH
-hno2
-----> CeH5COCNO
-> XII
привели к образованию бензоилмочевины [70]. Найдено также, что при диме-
ризации окиси бензоилнитрила может образовываться новая окись нитрила,
которая, димеризуясь, дает соединение XIII [64].
C6H5COCNO ---C6H5CONCCOC6He -*-*
CNO
С6Н.С—ON=CCOC6H5 —C6HSCOON—CCOCeHs
*CNO CNO
Аналогичные дизамещенные фуроксаны получены из а-ацетилфурана [71]
и р-ацетилтианафтена [72]. Превращение ацетонилацетона вфуроксан, по-види-
мому, идет по-стадийно в соответствии с приведенной ниже схемой [73].
О наличии продуктов, строение которых подобно строению соединения XIII,
при этом не сообщается.
(СН3СОСН2)2
hno2
HNO3
~CH3COC = NOH
_СН3СОСН2
COCNO~1
c4h4no—сос - ССО — C4H4NO
—► хш
При действии смеси 100%-ной азотной кислоты и азотнокислого аммония
ацетоксим превращается в 3,4-диацетилфуроксан (74], который может быть
получен также из а-оксиминоацетона и N2O4 [75].
(CH3)2C = NOH
hno3
СН3СОС-ССОСН3
N2O4
:--- СН3СОСН = NOH
Тетрамер гремучей кислоты по своему строению близок к соединению
XIII и образуется при димеризации окиси нитрила формгидроксамовой кис-
лоты, являющейся в свою очередь димером гремучей кислоты (76)
HON = СНС - ССН = NOH
HONC HON = CHCNO
Хлорангидриды. гидроксамовых кислот. Нитроловая и нитрозоловая кис-
лоты. Некоторые ароматические окиси нитрилов получены' из соответствую-
1,2,5-ОКСАДИАЗОЛЫ
365
щих галогенангидридов гидроксимовых кислот или нитроловых кислот. Окись
бензонитрила (т. пл. ,15°) получается из хлорангидрида бензгидроксимовой
кислоты при действии водного раствора карбоната натрия при 0° [52] или
при самопроизвольном разложении фенилнитроловой кислоты [77].
Cl NO2
I I
C6H6C = NOH -> c6h5cno «— C6H5C = NOH
Окись бензонитрила при комнатной температуре в течение 1 час димери-
зуется в дифенилфуроксан. Эта реакция ускоряется в присутствии щелочи
[78] и, по-видимому, необратима, по крайней мере при низких температурах.
В щелочной среде образуется также другой димер — 4-окись 3,5-дифенил-
1,2,4-оксидиазола — и тример [79]. Тример легко деполимеризуется и ведет
себя в реакциях как мономер. В ксилоле при 110° окись бензонитрила диме-
ризуется в дифенилфуроксан и перегруппировывается в фенилизоцианат [80].
О С6Н5
t I
N-C С6Н5С —N—* О
Щелочь Ч X Ч
X C6H5CNO--------------> CeH5C N—>0+ N С-С6Н5+Х
W \о/
О свн5
Многие превращения хлорангидридов гидроксимовых кислот или нитро-
ловых кислот в фуроксаны, по-видимому, протекают с промежуточным обра-
зованием окисей нитрилов. Дифенилфуроксан получен при обработке хлоран-
гидрида бензгидроксимовой кислоты водным раствором углекислого натрия,
азотнокислого серебра или м- или n-нитробензоата серебра [52, 81]. Хлор-
ангидриды м- или n-нитробензгидроксимовой кислоты при нагревании с раст-
вором едкого кали превращаются в ожидаемые фуроксаны [82]. Из хлорангид-
ридов ароматических гидроксимовых кислот и аммиака или аминов образуются
как фуроксаны, так и амидоксимы [83].
Cl N(CH3)2
/\-C = NOH (CH3)2nh ArC=NOH + ArC-CAr
u! —( к
I X / о
no2 о
Ранее уже упоминалось, что окиси ацилнитрилов могут образоваться
в качестве промежуточных продуктов в некоторых реакциях получения фурок-
санов. Щавелевый эфир окиси нитрила, полученный из этилового эфира бром-
уксусной кислоты и азотнокислого серебра [84], по-видимому, является про-
межуточным продуктом при превращении как хлорангидрида карбэтоксигид-
роксимовой кислоты, так и этилового эфира динитроуксусной кислоты в
в дикарбэтоксифуроксан [78, 85—87]; он образуется также при действии
азотной кислоты на изонитрозоацетоуксусный эфир [88]. При температуре
—16° этиловый эфир бромуксусной кислоты и нитрит натрия реагируют
hno3 O2NCCOOC2H5 _hno2
CH3COCCOOC2H5
II
NOH
ONCCOOC^Hj
С2Н5ООСС — ССООС2Н5
г/
0
с диметилформамидом, образуя дикарбэтоксифуроксан [89]. Хлорангидриды
алифатических гидроксимовых кислот (но не алифатические нитроловые кис-
366
ОКСАДИАЗОЛЫ
лоты) могут быть превращены в фуроксаны [87, 90]. Окиси алкилнитрилов
не известны, за исключением окиси трифенилметилнитрила, полученной
из хлористого трифенилметила и гремучего серебра [91]. Однако этот метод
не является общим и, как правило, ведет к образованию изоцианатов.
С1 (п-С1С6Н4)2СНС-ССН(СвН4С1-п)2
I Na2CO3 \
(n-ClCeH4)2CHC = NOH----> N N
\/ЧО
CeH5C( = NOH)C - СС( = NOH)CeH5
5%-ный Na2CO3 \
C6H5C( = NOH)C( = NOH)I---------> N N
Хлорангидрид хлоргидроксимовой и бромангидрид бромгидроксимовой
кислот превращаются при действии аммиака или аминов в соответствующие
дигалогенфуроксаны с высоким выходом [92, 93].
Окись бензонитрила считают возможным промежуточным продуктом
при образовании дифенилфуроксана из толуола, хлора и окиси азота [94],
а также из бромистого бензила и нитрита натрия [88].
С1
С6Н6СН3
С12
NO
_CeH5C = NOH
c6h5cno
X
NaNOa
CeH6CH2Br —C6H5CH2NO2
c6h5chno2
bJO
C6H5CNO
—> X
Серебряная соль фенилнитрозоловой кислоты при взаимодействии с иодом
дает дифенилфуроксан [95].
NOAg i2
II —> X
CeH5CNO
Алкены или алкины и азотистый ангидрид. Некоторые псевдонитрозиты,
продукты присоединения азотистого ангидрида к олефинам, могут изомеризо-
ваться в соответствующие оксимы нитрокетонов. При обработке разбавленного'
щелочного раствора а-нитрооксима холодной соляной кислотой наряду с дру-
гими продуктами образуется фуроксан и при этом в одной модификации, если
структура имеет изомерные формы [50, 96]. Нитрометилкетоксим не дает
RC-CR'
RCH—CHRr RC----------CR' \
II II II —» N N
NO NO2 NOH NO2H \
О о
О
R = 3,4-CH2 С6Н3 —, 4-СН3ОС6Н4 —, 3,4-(СН3О)2С6Н3—; R' = CH3
^о7
фуроксана, вместо него получается соответствующий нитрометилкетон [50].
Образование метилнитрофуроксана из пропилена и азотноватого или азоти-
стого ангидрида, возможно, протекает через стадию циклизации а-оксимино-
нитроловой кислоты [97]
СН3С—CNO2
NOH NOH NOH у \
II -> || || —^ N N
CH3CH = CH2 —> CH3CCH2NO2 ch3c—cno2 \ /4
о о
или изомер
1,2,5-ОКСАДИ АЗОЛЫ
367
Азотистый ангидрид может быть получен in situ при действии на нитриты
металлов кислоты [11, 98—100]. Коричный альдегид под действием окислов
азота дает смесь продуктов, содержащую фенилнитрофуроксан [101].
CeH5C- CNO2
СвН5СН=СНСНО •—> N или изомер
Фенилфуроксан получен из фен ил ацетилен а и азотистого ангидрида [102].
Соли гремучей кислоты и галогены. Ртутные и серебряные соли гремучей
кислоты при действии хлора, брома или иода образуют дигалогенфурок-
саны [103].
ХС-СХ
У \
MONC + Х2 -> N N
ХЧЧ
M = Ag или 1/2Hg2+
Х = С1, Вг или I
Различные методы. Бром в серной кислоте окисляет бензоиламиноглиок-
сим или его таутомер в бензоиламинофуроксан, что отличает его от таких реа-
гентов, как N2O4, щелочной раствор гипохлорита и азотная кислота, которые
C6H5COC( = NOH)C(==NOH)NH2 C6H5CO(C2N2O2)NH2
[или C6H5COC( = NOH)C( = NH)NHOH]
не обладают этим действием [11, 104]. Дифенилфуроксан образуется при дей-
ствии на бензилмеркурхлорид азотной кислоты или при реакции дибензил-
ртути с азотноватым ангидридом, взятым в недостаточном количестве [105].
Азид-ион быстро атакует как цис,- так и транс-1,2-динитроолефины [106],
причем азот выделяется по мере образования диалкилфуроксана. Из 2,3-динит-
рогексена-2 получен З-пропил-4-метилфуроксан, как и ожидали, в одной из его
RC__CR'
RC = CR' NaN3 \
| |--------> N N
o2n no2 \ /\
о о
R = R' = CH3, С2Н5, С6Н6
R = CH3; R' = n-C3H7
двух структурных модификаций. Эта реакция напоминает образование о-динит-
розоароматических производных из соответствующих о-азидонитросоединений,
который будут рассмотрены в следующем разделе.
о-Д и и и трозоароматические производи ые
Разработано несколько методов введения в ароматические соединения
о-динитрозогрупп, из которых наиболее часто применяются следующие:
1) пиролиз или фотолиз о-нитроарилазидов; 2) окисление о-нитроариламинов
щелочным раствором гипохлорита; 3) окисление диоксимов о-хинонов.
Удобным методом получения о-динитрозобензола является пиролиз
о-нитрофенилазида [107—109]; он протекает с удовлетворительной скоростью
при температуре ниже 65° [110]. Отрицательная величина энергии активации
указывает на образование циклического переходного состояния (циклического
активированного комплекса) [НО]. При облучении [111] 2-азидо-З-нитро-
368
ОКСАДИАЗОЛЫ
и 2-азидо-3,5-динитробифенилов ртутной лампой мощностью 100 вт образуются
соответствующие динитрозобифенилпроизводные с выходами 52 и 85%. При
пиролизе выходы составляют соответственно 55 и 83%. Пиролитический метод
распространен широко и, по-видимому, применим ко всем о-нитроарилазидам
R R +
I I NO
_м
I II Нагревание I
R' —no2 или hv R' —
NO-
R = R'=H или R = CeH5; R'=H или NO2
Пиролиз 8-нитротетразолпиридина, по всей вероятности, включает перво-
начальную изомеризацию его в 2-азидо-З-нитропиридин [112, 113].
N = N
Окисление о-нитроанилина щелочным раствором гипохлорита — наиболее
удобный метод получения о-динитрозобензола [114, 115]. Аналогичным спо-
собом получены 4-хлор-, 4-метил-, 3,5-диметил- и 4-метоксипроизводные
1,2-динитрозобензола [115, 116]. Окисление щелочным раствором гипохло-
рита 3,3'-динитро-4,4'-диаминобифенила дает ожидаемый тетранитрозоби-
фенил [115]. Тем же путем получают 1,2-динитрозонафталин из соответст-
вующих нитронафтиламинов [28]. Гипогалогениты в нейтральных растворах
окисляют о-нитроариламины в о,о'-динитроазопроизводные [115]. В щелочном
растворе может происходить замещение некоторых групп, находящихся
в пара-положении к аминогруппе. При действии щелочного гипохлорита
в метаноле на 2,4-динитроанилин образуется 1,2-динитрозо-4-хлор-5-метокси-
бензол [115].
__ми Cl__
I II NaOCl C1 | |
O2N -L IL NO2 nToh^ CH3O -I L
В водных растворах щелочной гипохлорит полностью разрушает бен-
зольное кольцо в 2,4-динитроанилине, 2-нитро-1,4-диаминобензоле, 1-амино-
2-нитро-4-ацетаминобензоле и 4-амино-З-нитробензолсульфокислоте [115].
Сообщается об окислении 5-хлор-, 5-метил- и 5-метоксипроизводных 2-нитро-
анилина под действием фенилиодозоацетата в бензоле в соответствующие
производные 1,2-динитрозобензола [117]. Если бензол заменить уксусной
кислотой, то получаются производные азобензола.
Предпочтительным способом получения о-динитрозопроизводных арома-
тических соединений с конденсированными ядрами часто может служить
окисление диоксимов о-хинонов. Корев [40] в 1886 г. обнаружил о-динитрозо-
производные в продуктах окисления диоксима Р-нафтохинона в 1,2-динитро-
зонафталин с помощью щелочного раствора феррицианида. Несколько лет
спустя о-динитрозобензол был получен из диоксима о-бензохинона действием
щелочного раствора феррицианида или азотной кислоты [107]. Однако этот
метод в общем не очень удобен для получения моноциклических ароматических
о-динитрозопроизводных, так как сами диоксимы в большинстве случаев
(если не считать продукты их восстановления) не известны. Каждый из четырех
геометрических изомеров диоксима 2-нитрофенантренхинона окисляется хло-
1.2,5-ОКСАДИАЗОЛЫ
369
ром в водном растворе этанола или концентрированной азотной кислотой
с образованием 2-нитро-9,10-динитрозофенантрена, который также получается
при действии на диоксим фенантренхинона азотной кислоты [118].
Br Вг
J ы I ЙО
Вг—\ КОН Х^Х
О----->
Br —1
Br j
О
XIV
'Ч/'Чью-
Вг
XV
По другому методу тетрабромтетраметиленфуроксан (XIV) дегидроброми-
руется в щелочи с образованием 1,2-динитрозо-3,6-дибромбензола (XV) [17, 119],
который был идентифицирован восстановлением в 1,2-диамино-3,6-дибром-
бензол [119].
С6Н5 +
I NO
N2m N
XII-----»
N
СГ,Н5
В заключение упомянем метод, заключающийся в обработке диацилфурок-
сана гидразином; этим способом из дибензоилфуроксана (XII) получен
3,6-дифенил-4,5-динитрозопириДазин [66, 120].
Реакции фуразанов, фуроксанов и о-динитрозоароматических производных
Окисление и восстановление. Фуразаны не удается одностадийным окисле-
нием превратить в фуроксаны, и сообщение, что фуразан был окислен азотной
кислотой в фуроксан, по-видимому, не соответствует действительности [121]..
Устойчивость к окислению кольцевого атома азота, находящегося
по соседству с атомом кислорода в гетероциклических соединениях, по-види-
мому, является общим правилом.
Фуроксаны также не поддаются окислительному расщеплению; их пре-
вращения в соответствующие нитронитрозо- или динитропроизводные не изве-
стны. В противоположность этому о-динитрозобензол окисляется в о-динитро-
бензол под действием надтрифторуксусной кислоты [122].
Алкильные боковые цепи окисляются обычными способами. Диметилфура-
зан с помощью перманганата калия может быть по-стадийно превращен
в метилфуразанкарбоновую и фуразандикарбоновую кислоты [2]. Следует
отметить, что фуроксанкарбоновые кислоты не образуются при окислении
соответствующих алкилфуроксанов. Гидролиз озонида, полученного из дифе-
нилфуроксана [123], дает бензойную кислоту.
Н3СС —ССН3 НООСС —ссн3
КМпО< КМпО4
N N-----------> N N ------------->
НООСС — ссоон
N
N
О
\oz ХО
Восстановление фуразанов может привести к образованию диаминов или
углеводородов. При действии цинка в соляной кислоте 4-нитробензофуразан
переходит в 1,2,3-триаминобензол [18, 124]. Каталитическое восстановление
одного фуразанового кольца в ангулярной бис-фуразанбензоле дает 4,5-диами-
24 Заказ № 978
370
оКсАДиАзолЫ
нобензофуразан [125]. При температуре выше 200° фосфор с иодистым водоро-
дом превращают дифенилфуразан в дифенилэтан.
Восстановление фуроксанов изучалось довольно подробно. Многие фур'о-
ксаны непосредственно превращаются в фуразаны, теряя кислород при
обработке пятихлористым фосфором. Типичными примерами являются
дифенилфуроксан (R = R' = С6Н5) и метил-п-анизилфуроксан (R или
р, Н1
230°
СвН5С—СС6Н5
С6Н5СН2СН2С6Н5
R' = п-СН3ОСвН4 и R или R' = СН3) [126, 127]. Сообщается, что один из изо-
меров фенилметилфуроксана не реагирует с пятихлористым фосфором, тогда
как другой превращается при этом в соответствующий метилфенилфуразан
[128]. о-Динитрозоароматические производные не дают фуразанов при дей-
ствии РС15 [26, 127, 129].
RC —CR' RC — CR'
Ччо// ° Хчо//
При восстановлении фуроксанов цинком в уксусной кислоте получаются
диоксимы [11, 47, 51, 52, 54, 130, 131]. При этом, по-видимому, первоначально
образуются омфи-изомеры [44], которые далее перегруппировываются в диок-
симы с более устойчивой конфигурацией или дегидратируются в фуразаны.
Восстановление дибензоилфуроксана (XII, стр. 363)цинком в уксусной кислоте
дает с хорошим выходом дибензоилэтан [132]. Не известно, является ли диок-
сим в этом случае промежуточным продуктом. Олово или хлорид олова (II)
Zn
XII ----—> СвН5СОСН2СН2СОС6Н5
СНзСООН ° z z ь 5
в соляной кислоте восстанавливают фуроксаны в фуразаны, причем диоксимы,
по-видимому, являются промежуточными продуктами. Дибензоилфуразан
образуется при длительной обработке дибензоилфуроксана хлоридом олова
RC-GR'
'онон
RC—CR'
(II) и хлористым водородом в уксусной кислоте [133]. Алкиларилфуроксаны
также превращаются в фуразаны [11], однако фенилнитрофуроксан с хлоридом
олова (II) дает фениламинофуроксан [101]. Циангруппа в фенилцианфурок-
сане нереакционноспособна по отношению к хлориду олова (II) и хлористому
i ,2,5-ОКСАДИАЗОЛЫ
371
водороду в уксусной кислоте в условиях образования фенилцианфуразана
[134]. о-Фенилендиамин образуется при восстановлении о-динитрозобензола
оловом в соляной кислоте [108]. Восстановление 1,3-динитро- и 1,2-динитро-
4,5-динитрозобензолов дает соответствующие тетрамины, а 1,2-динитрозо-
нафталин— 1,2-диаминонафталин [19].
Каталитическое восстановление фуроксанов мало изучено. Фуроксан,
полученный при действии гипогалогенида на диоксим камфорхинона, при вос-
становлении над платиной дает один из известных (по-видимому, а.ифи-кон-
фигурации) диоксимов камфорхинона [135]. Метилнитрофуроксан в уксусной
кислоте восстанавливается над платиной в соответствующий метиламино-
фуроксан [97].
Йодистый водород восстанавливает о-динитрозоароматические производ-
ные при температуре ниже 80° и не восстанавливает фуроксаны. При исполь-
зовании недостаточного количества реагента 1,2-динитрозо-4-азидо-5-нитробен-
зол восстанавливается в 1,2-диамино-4-азидо-5-нитробензол, а при избытке
реагента образуется 2,4,5-триаминонитробензол [136]. о-Динитрозобензол
восстанавливается по этому методу в диоксим о-бензохинона [137]. Диметил-
и дифенилфуроксаны не вступают в реакцию с иодистоводородной кислотой при
кипячении; между тем иодистый водород с красным фосфором при 190° превра-
щает дифенилфуроксан в дифенилфуразан.
Щелочные или аминные растворы гидроксиламина восстанавливают
о-динитрозоароматические производные в диоксимы соответствующих хинонов,
однако не восстанавливают фуроксанов. Этим методом, например, получены
H2N —— NH2 Избыток
02n J- NH2 Н!
N3 YYNH2
OoN -Y’-NHz
/ч /NO
n3 у
O2N —I L
^NO-
диоксимы 1,2-бензохинона и его 4-хлор-, 3-метил-, 4-метил- и 4-хлор-5-метокси-
производных {18, 81, 115]. При образовании диоксима 3-аза-4-амино-5-метил-
бензохинона-1,2 в результате восстановления 2,3-динитрозо-5-метилпиридина
в диэтиламине происходит аминирование [137].
/\ /NO
H3c-|^Y
V^NO-
ii2noh
r2nh
/\ /NOH
H3C-/V
H2N Д /I
N wH
Гидразингидрат восстанавливает о-динитрозобензол в о-фенилендиамин,
а 4-метил-1,2-динитрозобензол — в 3,4-диаминотолуол, однако 1,2-динитро-
зонафталин при этом образует соответствующий фуразан [138]. При действии
сероводорода в спиртовом растворе аммиака [18] или сульфида натрия в раз-
бавленном спирте [112, 139] ароматические динитрозопроизводные превра-
щаются в диамины. Боргидрид натрия восстанавливает как о-динитрозоаро-
матические соединения, так и фуроксаны в диоксимы. Выход диоксима о-бен-
зохинона составляет 54%, а диоксима бензила — 44% [122].
В случае дизамещенных фуразанов и фуроксанов при действии алюмо-
гидрида лития происходит восстановительное расщепление кольца с образо-
ванием первичных аминов [501. Каждый из двух изомерных фенилметилфурок-
свнов дает этиламин и бензиламин. Тетраметиленфуроксан восстанавливается
24*
372
ОКСАДИАЗОЛЫ
в гексаметилендиамин. Алюмогидрид лития не вызывает разрыва углерод-
углеродной связи у о-динитрозобензола, 1,2-динитрозоаценафтена или фенант-
рен-9, 10-фуразана, каждый из которых восстанавливается в соответствующий
О
/ \./\с—ССН3
Н2С I II Ч 1ЛА1Щ
2 \ II N N--------------------* СН2О2СвНзСН2Ь1Н2 + СНзСН2Ь1Н2
о Ч'/ \ / Ч
о о
ароматический диамин. Предполагается [50], что распад фуроксана является
первичной реакцией, далее следует восстановление нитрила и аниона первич-
ного нитросоединения.
RC —CR'
Z \ н-
N N ------------> RCN + R'CH = NOJ
'о О
Весьма любопытна реакция восстановления о-динитрозобензола в о-нитро-
анилин под действием меди или ее солей в кислом растворе. Предполагается,
что образуется промежуточная содержащая медь хелатная соль диоксима
о-бензохинона и что в этих условиях диоксим изомеризуется в о-нитроанилин
/ V NO2
L nh2
[140]. Наблюдалось также полное восстановление натриевой соли бйсульфит-
ного производного 1,2-динитрозонафталин-6-сульфокислоты в разбавленном
растворе карбоната натрия в двунатриевую соль 1-амино-2-нитронафталин-
4,6-дисульфокислоты [141]. Те же вещества в кипящем 12,5%-ном растворе
карбоната натрия дают соответствующий нитронафтолдисульфонат, а в 15%-ном
растворе едкого натра при комнатной температуре — соответствующий п-нафто-
хинондисульфонат в виде диоксима [141].
NaO3S-
NO
•NaHSO3
1%-ный р-р NasCOg
NHg
SO3Na
Диоксимы диацилфуразанов и фуроксанов окисляются N2O4, пермангана-
том, гипохлоритом или азотной кислотой [142, 143]. Строение продуктов, рас-
сматриваемых соответственно как производные фуразана и фуроксана, у кото-
рых пятичленное фуразановое кольцо сконденсировано с восьмичленным коль-
цом диоксадиазина, точно не установлено.
Действие кислот. Устойчивость фуразанового кольца в растворе кислот
наглядно проявляется в ряде примеров: дифенилфуразан устойчив к действию
соляной кислоты при 200° [1441; при нитровании метил-п-анизилфуразана [11]
1.2,5-ОКСАДИАЗОЛЫ
373
получается мононитропроизводное, а дифенилфуразана [1]—динитропроиз-
водное. Дибромфуроксан растворяется и остается неизмененным в дымящей
Н3СС—СС6Н4ОСН3-И Н3СС — CC6H3(NO2)OCH3
С6Н5С — CCgHtj ОгМСвЩС—CC6H4NO2
/ \ ONOSO^
азотной кислоте, но при длительной обработке концентрированной соляной
кислотой при 100° расщепляется, давая щавелевую и бромистоводородиую
кислоты, гидроксиламин и аммиак [145].
В противоположность этому сообщается, что З-фенил-4-дезилфуразан
в кипящей соляной кислоте обратимо превращается в 3-бензоил-4,5-дифенил-
изоксазол [1461.
С6Н5
С6Н5С—С^НСОС6Н5 C6HSC-CCOC6H5
n —c6H5cf \
\ / -H2NOH /
о о
Диазотирование З-амино-4-метилфуразана приводит к образованию триа-
зена [29].
H3CC-CNH2 Н3СС—CN = NNHC-CCH3
При нитровании бензофуразана образуется 4-нитропроизводное, динитро-
NO2
производное при этом не обнаруживается. Для превращения фуразана (XVI)
в 4,6-динитро-7-оксибензофуразан (XVII) — единственный известный пример
бензофуразана с гидроксильным заместителем — необходимо проведение гид-
О ОН
NOCOCH3 NO2
XVI XVII
N—О N-0
XVIII
374
ОКСАДИАЗОЛЫ
ролиза, окисления и нитрования [26]. Нафтофуразан (XVIII) сульфируется
в положении 4 [141 ].
Ни фенил-, ни дифенилфуроксан не изменяются под действием кипящей
концентрированной соляной кислоты [126, 144]. Метил-(3,4-диметоксифенил)-
CH3(C2N2O2)C6H3(OCH3)2 CH3(C2N2O2)C6H2(OCH»)2NO2
фуроксан легко нитруется в бензольное кольцо [141]. Сообщается, что алкил-
арилфуроксаны претерпевают взаимное превращение при кипячении с пропио-
новой кислотой [127].
о-Динитрозобензол образует 3-мононитро- и 3,5-динитропроизводные
[124]. При нитровании 1,2-динитрозо-4-нитробензола получен 1,2-дйнитрозо-
4,5-динитробензол [124]. Этот пример образования динитропроизводных
со смежными нитрогруппами напоминает нитрование 5-нитробензимидазола
и 6-нитроиндазола, которые превращались в соответствующие 5,6-динитро-
производные [147].
Действие щелочей. Под действием щелочных растворов легко происходит
расщепление однозамещенных фуразановых и фуроксановых колец. Фенил-
фуразан (XIX) изомеризуется в а-изонитрозофенилацетонитрил [12]. Диалкил-,
диарил- или алкиларилфуразаны более устойчивы к действию щелочей. Фена-
NaOH
C6HSC=NOH
CN
С6Н5С—СН
XIX
цилфенилфуразан в концентрированном спиртовом растворе едкого кали пре-
вращается в метилфенилфуразан [148]. Дифенилфуразан не изменяется под
действием горячей щелочи [1], однако сообщается, что метил-п-анизилфура-
зан разлагается спиртовым раствором едкого кали с образованием и-анисовой
кислоты [100]. Моно- и диацилфуразаны в щелочной среде превращаются
C6HsC-CCH2COCeHs с6н5с—ссн3
-У КОН к/
N N . ---> N N
V 'Хо'/
RCOC-CCOR
Холодный р-р NaOH
RCOC—СН
О
RCOCCN
II
NOH
в а-изонитрозо-а-ацилацетонитрилы [149]. Метилбензоилфуразан претерпе-
вает щелочной гидролиз с образованием бензойной кислоты и аммиака [149].
Аминоацилфуразаны изомеризуются в щелочной среде в 1,2,4-оксадиазолы
1,2.5-ОКСАДИАЗОЛЫ
375
[1501. Бензофуразан более устойчив к действию щелочей; обсуждалась даже
возможность получения его из диоксима о-бензохинона путем щелочной пере-
гонки с паром.
С6НЙСОС — CNHCOC6HS nccnhcoc6h5 ncc —n
X \ KOH H 4
N N _ —> NOH _» N CC6H6
\ / ROH \ /
О О
Фенилфуроксан неустойчив в щелочных растворах (см. стр. 384). Дифенил-
фуроксан при действии метанольного раствора едкого кали дает бензойную
кислоту [151]. Дизамещенные фуроксаны с двумя алкильными группами или
одной алкильной и второй арильной также неустойчивы по отношению к щело-
чам. Показано, что один из двух изомеров фенилметилфуроксана в присут-
ствии этилата натрия превращается в 3-фенил-4-амино-5-оксиизоксазол (или
в его иминный таутомер); аналогичным образом метилбензилфуроксан обра-
зует 3-бензил-4-амино-5-оксиизоксазол [153]. Предложен механизм этого пре-
вращения [152].
с н ом» H2NC —ССН2С6Н5
CH3(C2N2O2)CH2C6H5 _1Л----, II II
HOC N
Карбинол — бцс-оксибензилфуроксан — в водном растворе едкого натра
образует бензальдегид [122].
с6н5сн(он)с—ссн(он)с6нй —2L. с6нйсно
^'о^ 'о
Нуклеофильное замещение электроноакцепторньгх заместителей у атомов
углерода фуроксана происходит без разрыва кольца. В .спиртовом растворе
едкого кали фенилнитро- и фенилхлорфуроксаиы превращаются в соответст-
вующий фенилалкоксифуроксан [101, 154]. Аналогично в разбавленном водном
растворе щелочи образуется феиилоксифуроксан [101]. Дибромфуроксан
C6HS(C2N2O2)OH ДДгДД c6hs(C2n2o2)X c6h5(C2N2o2)or
NaOH ROH
X = NOZ, Cl
с аминами дает диоксимы оксамидов, которые при дегидратации могут пре-
вращаться в амидоксимы [103]. Обработка дибензоилфуроксана слабой щелочью
приводит к бензойной [69] и бензоилметазоновой [71] кислотам; при взаимо-
Br(C2N2O2)Br [R2NC(=NOH)]2—> R2N(C2N2O2)NR2
действии с аминами образуются амидоксимы [155]. Амидоксим, получающийся
при действии фенилгидразина, дегидратируется в изоксазол, который в теплой
C6H5CO(C2N2O2)COC6Hs —C6HsCOCCH2NO2
II
NOH
уксусной кислоте дает З-бензоил-5-фенилгидразин-1,2,4-оксадиазол [1561.
Аналогичные продукты получены из других диароилфуроксанов и фенил-
гидразина [71, 72]. По-видимому, продукт с брутто-формулой C6H7N3O2
[62], полученный из дибензоилфуроксана и аммиака, является 3-беизоил-
376
ОКСАДИАЗОЛЫ
5-амино-1,2,4-оксадиазолом [157], а
лом [104].
не З-бензоил-4-амино-1,2,5-оксадиазо-
С6Н5СОС-ССОС6НЬ
N N\j
°
C6H5N2H3
— Cel IsCONKNUCgHs
-1ОО
C0H5COC( = NOH)C(-=NOH)NHNHC6H5----
C6H5C=CNO
о cnhnhc6h5
n=cnhnhc6h5
CHgCOOH I I
------=> СсН5СОС о
z z
N
Диэтиловый эфир фуроксандикарбоновой кислоты в растворе едкого
бария омыляется, и бариевая соль, которую удалось выделить, легко пре-
терпевает гидролитический разрыв кольца [158].
Ва(ОН)2
С2Н5ООС (C2N2O2)COOC2H5------Ba2+[OOC(C2N2O2)COO12- —-
NOH
HOOCCCONHOH
Изоцианиловая кислота — диоксим диформилфуроксана и тетрамер
гремучей кислоты — изомеризуется в крепкой щелочи в эрмтро-цианило-
вую кислоту [159]; в растворе аммиака изомеризация ведет к получению
nepu-цианиловой кислоты через промежуточное образование эпм-цианиловой
кислоты [159]. Производное оксима метилбензоилфуроксана в разбавленном
HON = CHC — CCH-NOH ONC-CCH=NOH
НО(/ \
'o 'b
| NH4OH H
v
NCC( = NOH)C( = NOH) C (= NOH)OH —> NCC( = NOH)C(=NO2H)CH = NOH
растворе едкого натра при 0° претерпевает реакцию, аналогичную превраще-
нию изоцианиловой кислоты в эпм-цианиловую кислоту; образуется натриевая
соль <щ«-нитробензилметилфуразана, которая изомеризуется в разбавленной
серной кислоте [1601.
NaOH H2SO4
CH3(C2N2O2)CC6H5-----> CH3(C2N2O2)CC6H5----->
II °° II
NOH NO2Na
CH3(C2N2O) CHC6H5
I
no2
о-Динитрозоароматические производные легко вступают в реакции с осно-
ваниями. о-Динитрозобензол в метанольном растворе едкого кали превра-
щается в бензофуразан [17], по-видимому, с промежуточным образованием
диоксима о-бензохинона. Указывается, что применение щелочного раствора
гидроксиламина позволяет приостановить реакцию на стадии диоксима.
^/^NO-
сн3ок
СН3ОН
NOH-
NOH..
1,2,5-ОКСАДИАЗОЛЫ
377
Как 3-нитро-, так и 3,5-динитро-1,2-динитрозобензолы реагируют с водными
растворами едких кали и натра, а также с карбонатами натрия или калия
[115, 124]. При этом могут быть выделены металлические соли 3,5-динитро-
1,2-динитрозобензола, которые при подкислении превращаются в исходное
соединение. Образование соли, по-видимому, происходит в результате заме-
щения протона в положении 4 ионом металла, так как 1,2-динитрозо-3,5-
динитро-4-метилбензол не образует соли [116]. Калиевая соль представляет
некоторый интерес как взрывчатое вещество. При продолжительном воздей-
ствии щелочи она распадается, при этом появляется запах аммиака. Попытки
КНСОз
(C6HN4O6)-K+
проалкилировать калиевую соль хлористым или подпетым метилом в жидком
аммиаке оказались безуспешными [116].
Как 1,2-динитрозо-3,5-динитро-, так и 1,2-динитрозо-4,5-динитробензол
вступают в реакцию присоединения с анилином [124]. Во втором случае обра-
зуются красные иглы 1,2-динитрозо-3,6-дианилино-4,5-динитробензола, тогда
NO NC6H5
/V II
! | + c6h5nh2 c6h5nh—
'xJLnhC6H5
II
nc6h5
как в первом выделен неидентифицированный продукт присоединения одной
молекулы анилина. При 150—160° о-динитрозобензол взаимодействует с ани-
лином, образуя дианил дианилино-п-бензохинона [161].
Различные реакции. Фуроксаны легко реагируют с реактивами Гриньяра,
тогда как фуразаны, по-видимому, инертны. Пиперонилметилфуроксан с фенил-
магнийбромидом дает пиперонилнитрил, ацетофенон, неидентифицированный
кетон и нитропроизводное, которое было принято за аци-l-нитро- 1-фенилэтан
[162]. С метилмагнийиодидом получают пиперонилнитрил и пиперонилметил-
кетон [135]. Метил-п-метоксифенилфуроксан с фенилмагнийбромидом дает
п-метоксибензонитрил [135] и неустойчивое соединение [135], являющееся,
вероятно, аци-\-нитро- 1-фенилэтаном [50]; при гидролизе образуется ацето-
фенон [135].
RMgx ArCN + RC=NO2H
Ar(C2N2O2)CH3 —|
CH3
rc=no2h ho
I __RCOCH3
CH3
Единственным удобным способом синтеза 2-анилинотрифениламина
является реакция между бензофуразаном и фениллитием. При объяснении
механизма этой реакции предположили, что наибольший вклад в гибридную
структуру бензофуразана (предполагается, что она принадлежит к мезоион-
ным [164]), вносит структура с противоположно заряженными атомами азота;
378
ОКСАДИАЗОЛЫ
при этом реакция с фениллитием может идти, как показано на схеме:
.Li
N N
СбН— ХоCfiH5Li ГИг"N^CeH^Li ceHsLi
U\ / > ~ * U-N(C6H5)OLi
N ' N
I
C6H5
N(C6H5)Li .^\-NHC6H5
NLn(C6H5)2 ^J-N(C6Hs)2
Имеется указание, что у дибензоилфуроксана под действием уксусного
ангидрида происходит разрыв кольца [165].
О
C6H5CO(C2N2O2)COC6HS C6HsCOC( = NOCOCH3)C( = NOCOCH3)COC6HS
Как бензофуразан, так и о-динитрозобензол присоединяют бром, образуя
соответственно тетрабромтетраметиленфуразан (IV) и тетрабромтетраметилен-
фуроксан [17].
Вг
NO । N
в, Вг-/УХ
I I ^11°
^NO~ Br“YV
ВГ I
О
Наблюдалось образование комплексов ароматических углеводородов
с 1,2-динитрозо-3,5-ди нитробензолом, 1,2,3,4-тетранитрозо-5-нитробензолом,
гексанитрозобензолом и 1,2-динитрозо-4,5-динитробензолом [166].
Физические свойства
В литературе имеются данные о цироскопических и кристаллографических
константах, теплотах сгорания, дипольных моментах и спектрах комбина-
ционного рассеяния света 1,2,4-, 1,2,5- и 1,3,4-оксадиазолов, содержащих как
алкильные, так и арильные заместители [167]. Теплота сгорания фуразана,
как правило, на 20 ккал выше, чем теплота сгорания соответствующего фурок-
сана [168]. Для некоторых метиларилфуроксанов наблюдалась разница в теп-
лотах сгорания в 8—10 ккал у двух изомерных форм [168].
Исследование поглощения в инфракрасной области спектра для этих
изомерных циклов позволяет считать полосу 1380 см'1 [169], которая имеется
в инфракрасных спектрах пиррола и фурана, характеристической для пяти-
членного кольца. Полосу в инфракрасном спектре фуразана в области 1570 слГ1
также относят к пятичленному циклу. Полоса поглощения при 1030 см'1,
найденная в спектрах 1,2,4- и 1,3,4-оксадиазолов (но не у фуразанов), отвечает
колебаниям связи С — О. Связь N — О [167] в фуразанах характеризуется
поглощением в области 1430—1385 см'1 (ср. с фуроксанами). Известны неко-
торые данные о поглощении бензофуразана в области спектра 6,0—7,0 р [116].
Диметилфуразан дает сильное непрерывное поглощение в ультрафиолето-
вой области loge 0,67—2,85 при X 2409—2280 А [180],
1.2,5-ОКСАДИАЗОЛЫ
379
Инфракрасные спектры фуроксанов характеризуются наличием семи
полос поглощения: 1625—1600 (С = N), 1475—1410 (дублет О-—N-> О),
1360—1300 (N — О), 1190—1150, 1030—1000 и.890—840 (фуроксановое кольцо)
и 950—900 см"1 (диароилфуроксаны) [171].
Поглощение в инфракрасной области четырех о-динитрозоароматических
производных незначительно отличается от поглощения фуроксанов [172].
Диалкил-, диарил- и диацилфуроксаны поглощают в ультрафиолетовой
области 255—285 мр, а о-динитрозоароматические соединения — в области
350—410 му, [172].
Дифенилфуразан перегоняется с частичным разложением и дает бензо-
нитрил, фенилизоцианат и 3,5-дифенил-1,2,4-оксадиазол [1].
Сообщаются цироскопические константы и дипольные моменты для
некоторых о-динитрозоароматических производных [173]. Теплота сгорания
о-динитрозобензола равна 752 ккал, тогда как аналогичная константа для
бензофуразана составляет 753 ккал [174].
Проблема строения фуроксана
Исследование структур Виланда— Вернера. Фуроксаны и 1,2-динитро-
зопроизводные при исследовании их строения оказались тесно связанными друг
с другом вследствие внутримолекулярной ассоциации нитрозогрупп. В раз-
личное время для этих соединений предлагалась и признавалась та или иная
структура от А до М.
В настоящее время структурная формула Виланда — Вернера (Е) наи-
более широко принята для моноциклических дизамещенных фуроксанов
[96, 175], как предложил их называть Виланд [96]. Признание существования
сходных по структуре изомерных фуроксанов [44, 123] (ср. стр. 381) исклю-
О
NZ О
II N
Б
N
о чсод5
I I
о СС6Н5
N
Корев, 1886 Корев, 1886
Ильинский, 1886 Ильинский, 1886
Корев, 1886
Г
Нельтинг, Коон, 1894
Цинке, Шварц, 1894
Д
Цинке, Оссенбек, 1894
Виланд, 1903
Вериер, 1904
(и изомер)
Ж
Виланд, 1907
380
ОКСАДИАЗОЛЫ
Бойер и др., 1955
чает рассмотрение таких симметричных молекул, как диоксадиазин (В) [4, 40]
и эпоксипроизводное (К) [18, 176], однако это не относится к геометрически
изомерным динитрозоолефинам (Л) [177] или бициклическому оксазирану
(Ж) [126]. Данные инфракрасных спектров для изомерных фенилметилфурок-
санов допускают для каждого из них структурную формулу Виланда — Вер-
нера (Е) [50, 1781.
Потеря кислорода фуроксанами при действии пятихлористого фосфора —
свойство, которое не распространяется на известные динитрозосоединения
и наличие которого нельзя ожидать у неизвестных цис- и mpawc-динитрозооле-
финов (Л). Структура Ж (оксазиран) не отражает устойчивости фуроксана,
так как показано, что оксазираны подвергаются необратимому пиролизу
в нитроны даже при такой низкой температуре, как 80° [179].
° t
свн5сн-\с(сн3)3 -гр-!!аГ^> с6н5сн = nc(ch3)3
Фуроксановые (но не фуразановые) соединения легко вступают в реакцию
с озоном и, по-видимому, той стороной молекулы, где находится экзоцикличе-
ский атом кислорода [123]. При действии на озониды холодного разбавленного
раствора едкого натра образующаяся кислота сначала содержит заместитель
у атома углерода фуроксана, смежного со связью N —О [123].
С6Н5С-СС6Н4ОСН3
l/ \ СН3ОС6Н4СООН + Остаток
\ / Ж)-О3
О
. Кипячение „ ,
Остаток-------.. СвН5СООН
NaOH
C6HsC-CCeH4OCH3
,N С6Н5СООН -P Остаток
o3-oz \ /
о
Остаток CH3OC6H4COOH
NaOH
Деполимеризация дифенилфуроксана в окись бензонитрила обнаружена
совершенно независимо при перегруппировке последнего в фенилизоцианат
[80, 144]. При термической изомеризации арилметилфуроксанов при темпе-
С6Н5С-СС6Н5
г/ \ _22. c6h5cno c6h5nco
200° 110°
О
i ,2,5-ОКСАДИАЗОЛЫ
38 i
-ратуре, близкой к 140°, деполимеризации не происходит, так как симметрич-
ные фуроксаны, по-видимому, не образуются [127]. Атомы кислорода должны
стать равноценными в переходном состоянии, возможно существующем в виде
активированного динитрозопроизводного.
'ArC—CCHg
II II
N N+
“О О
АгС—ССНз
О
АгС—ССН31
II II
+N N
О О-
ArC—CCHg
По крайней мере в одном случае был получен только один из ожидаемых
двух изомерных фуроксанов. При окислении гипохлоритом каждого из четы-
рех геометрически изомерных диоксимов камфорхинона получается один
и тот же фуроксан [135] (XI), который при восстановлении [135] вновь дает
один из четырех диоксимов, по-видимому, в виде алк/ш-формы. Положение
внекольцевого атома кислорода в фуроксане не известно, и можно предпола-
гать, что с точки зрения пространственных факторов, он удален от метильной
группы, находящейся у мостика.
Монозамещенные фуроксаны. Кроме а- (т. пл. 111—112° [177]) и 0-форм
(т. пл. 108° [180]), полученных из изомерных диоксимов фенилглиоксаля,
фенилфуроксан (XX) описан как изомерная окись изонитрозофенилацетонит-
рила (XXI, т. пл. 112—113° с разл.) [177]. Неидентифицированный изомер
(т. пл. 96—97° с разл.) образуется из хлорангидрида бензгидроксамовой кис-
лоты и гремучего серебра [70].
CeH5C = NOH
CNO
XXI
а-Форма 6-Форма
XX
С6Н5С( = NOH)C1 C8HeN2O2 .
Сообщается об изомеризации [126, 177] a-формы соединения XX в орга-
нических растворителях: уксусной кислоте, бензойном ангидриде, а также
в водном растворе карбоната натрия. Продукт, представляющий окись изо-
нитрозофенилацетонитрила (XXI), и a-форма соединения XX изомеризуются
при кипячении с пропионовой кислотой, по-видимому, в фенилокси-1,2,4-
С6Н5С—СОН
а-ХХ —> XXI —> [CeH5C( = NOH)C( = NOH)OH] \
XXIII \ /
о
XXII
c6h5c-n
1/ сон
HOC—N
~Н2° [XXIII]
ХХШа ХХШб
оксадиазол (ХХШа или б), а не в фенилоксифуразан [177] (XXII). Из пред-
полагаемого промежуточного продукта (XXIII) путем дегидратации, сопро-
вождаемой перегруппировками Бекмана и Лоссеня [71 ], могут быть получены
изомерные фенилокси-1,2,4-оксадиазолы (ХХШа и б); между тем циклизация
при дегидратации без перегруппировки, если бы она произошла, дала бы
382
ОКблДиАзолЫ
соединение XXII. Структурные формулы, приписываемые как оксифураза-
нам, так и окси-1,2,4-оксадиазолам, очевидно, не могут основываться на полу-
чении этих соединений из глиоксимов.
Однозначного метода синтеза каждого из этих трех продуктов (XXII,
ХХШа и б) не разработано, однако существуют два метода, из которых
любой кажется бесспорным, но они не дают совпадающих результатов, о чем
речь будет ниже. По-видимому, надежный метод синтеза метилового
(т. пл. 58—59°)и этилового (т. пл. 49—50°) эфиров соединения ХХШб раз-
работали Янг и Джонсон [181]. Он основан на взаимодействии диалкилбен-
зоилимидотиокарбонатов и гидроксиламина. Предполагаемая структура этих
CeH5CON = COR h2noh CgH5CON = COR _н2о ROC—N
SR HONH r/ CC8II5
О
ХХШв
R=CHg, С2Н5
сложных эфиров (ХХШв) подтверждается их способностью восстанавли-
ваться в бензоилмочевину при действии цинка в уксусной кислоте [181].
Строение, приписываемое З-окси-5-фенил-1,2,4-оксадиазолу (ХХШб,
т. пл. 201—202°), полученному из соответствующего хлорида и разбавленного
раствора щелочи [6], основывается теперь на получении его метилового
ХХШв CeH5CONHCONH2
СН3С00Н
(т. пл. 58—59°) и этилового (т. пл. 47—48°) эфиров при действии на серебря-
ную соль фенола соответствующего ал кил галогенида или алкилсульфата [6],
что не согласуется с идентификацией этих соединений как эфиров фенилок-
сифуразана (XXII), не известных до этого. Основываясь на упомянутой иден-
С1С—N
c6h5c(=noh)C(=noh)ci / YceH5 —2^ ХхШб
ХХШг \ /
о
тификации соединения ХХШб, соответствующий хлорид, полученный из глиок-
сима (ХХШг) и хлорокиси фосфора, представляет собой З-хлор-5-фенил-
1,2,4-оксадиазол (т. пл. 37—38°), а не фенилхлорфуразан, как указывалось
ранее [6] на основании неверного утверждения, что хлорокись фосфора всегда
переводит глиоксимы в фуразаны. Обработка глиоксима (ХХШг) пятихлори-
стым фосфором, по-видимому, вызывает перегруппировку и образование
З-фенил-5-хлор-1,2,4-оксадиазола (т. пл. 5°), а не изомерного З-хлор-5-фенил-
1,2,4-оксадиазола, как это предполагалось ранее [6], или дегидратацию в неиз-
вестный фенилхлорфуразан. Хлорид при действии соответствующих алкого-
лятов натрия превращается в метиловый (т. пл. 26°) и этиловый (т. пл. 64°)
CeH5C- N
ХХШг —^ci ХХШа
О
эфиры [6, 181 ],. которые разлагаются иодистым водородом с образованием
вещества с брутто-формулой C8H6N2O2 (т. пл. 175—176° [6]), которое может
быть либо соединением XXIIIa, что вп?лне вероятно, либо фенилоксифураза-
ном (XXII) [183]. Однако последнее было отвергнуто [182] и предпочтение
было отдано формуле XXII16 на основании того, что это соединение с анили-
ном при 200° образует Ы,Ы'-дифенилмочевину. Идентификация вещества
1,£.5-6кСАДИАЗОЛЫ
383
состава CsHeN2O2 (т. пл. 175—176°) [184] осложняется еще тем, что описаны
метиловый (т. пл. 116° [184]) и этиловый (т. пл. 35—36° [185]) эфиры 3-фенил-
5-окси-1,2,4-оксадиазола (XXIПа). Для некоторых из этих производных
замещение может происходить по атому азота, а не кислорода.
Второй синтез, который, по-видимому, является однозначным, разработан
Фальком в 1885 г. [186]. Обработка бензамидоксима этиловым хлоругольным
эфиром и последующий гидролиз дают продукт ст. пл. 201—203°, идентифи-
цированный как З-фенил-5-окси-1,2,4-оксадиазол (ХХШа) [187]. Это предпо-
ложение подтверждено получением таутомера соединения (ХХШа (т. пл. 198°),
из хлорангидрида бензгидроксимовой кислоты и цианата калия [188]. Ранее
СвН5С(.= NOH)NH2 CeH5C( = NOCOOC2H5)NH2 —> XXIII (т. пл. 201-203°)
CeH5C-NH
CeH5C( = NOH)C1 2£22 т/ СО ДагРевание CeH5Q = NOH)NH2-H2CO3
это соединение было получено пиролизом карбоната бензамидоксима по методу
Фалька [189]. Избежать неожиданной молекулярной перегруппировки на одной
из стадий и получить неизмененное ядро фенил-1,2,4-оксадиазола при приме-
нении синтезов Янга — Джонсона и Фалька не удается. Ниже будут при-
ведены данные, показывающие, что метод Фалька не всегда может быть одно-
значным (стр. 391).
По-видимому, аутентичный образец фенилоксифуразана (XXII) не был
получен, и имеющиеся данные лучше всего подтверждают, что в указанных
условиях происходит изомеризация и a-форма соединения XX превращается
в изомер XXIII (т. пл. 201—202°), а не в соединение XXII. Наблюдалась также
изомеризация a-формы соединения XX в 0-форму, которая проходит при кипя-
чении в ксилоле в течение 1 час [177]. 0-Изомер при кипячении с пропионовой
кислотой превращается в соединение XXIII (т. пл. 201—202°) [177].
Влажные амины, реактивы Гриньяра, ангидриды кислот и минеральные
кислоты [177] также дают одни и те же продукты при взаимодействии как
с a-формой соединения XX, так и с соединением XXI. С аминами в отсутствие
влаги реагирует соединение XXI, а a-форма соединения XX не вступает
в реакцию; соединение XXI взаимодействует с водными растворами галогено-
водородов при комнатной температуре, однако в случае а-изомера XX тре-
буется более высокая температура; хлорокись фосфора вступает в реакцию
с соединением XXI и не реагирует с a-формой соединения XX (сообщается,
что пятихлористый фосфор переводит а-изомер в З-хлор-5-фенил-1,2,4-окса-
диазол [190]); цинк в уксусной кислоте дает различные продукты восстановле-
ния, и лишь соединение XXI при этом димеризуется.
а-ХХ или XXI -2221. CeH5C( = NOH)C( = NR)NHOH
а-ХХ или XXI 2222^. Л2^ CeH5C( = NOH)C( = NOH)CH3
а-ХХ или XXI 2222222 [CeH5C(=NOCOCH3)CNO]2
а-ХХ или XXI -(СбН5222!2» СвН5С = NOCOCeH5
I 4-
С( = NOCOCeH5)OCOCeH5
+ C6H5C( = NOH)C( = NOCOCeH5)OCOC0H5
CeH5C-CNHCOC6H5
а-ХХ или XXI JЛ5_22> Z \ + CeH5C( = NOH)C(--= NO2H)H
9f)0/ ,ная НС1
а-ХХ или XXI —------Л С6Н5СООН + H2NOH
384
ОКСАДИАЗОЛЫ
В инфракрасном спектре а-фенилфуроксана с т. пл. 108° (по-видимому,
нечистого) наблюдались полосы поглощения в области 2229 (слабая) и 3225 смГ1,
относящиеся к гидроксильным группам в нитрилах и оксимах, что указывает
на присутствие соединения XXI. Аналогичные полосы (сильное поглощение
группы С s N) характерны также для изонитрозофенилацетонитрила [178],
однако в спектре p-фенилфуроксана эти полосы отсутствуют [178].
а-ХХ ___Zn..> С6Н5( = NOH)C( = NOH)H
CH3COOH
XXI
Zn C6H5C=NOH
-----—> I
CH3COOH CN
C6H5C( = NOH)C—CC( = NOH)C6H5
XXI
8 0°
CeHs
XXIV
В присутствии этилата натрия p-фенилфуроксан изомеризуется в аци-
фенилнитроацетонитрил [177], причем предполагается, что механизм реакции
аналогичен превращению а-изомера соединения XX в соединение XXI.
p-xxJ^0!^. c6h5c=no2h
CN
Р-Фенилфуроксан и магнийметилиодид дают продукт присоединения, при
гидролизе которого образуются фенилацетат и аммиак [177].
р-хх СНзМеА» 2^2» С6Н5ОСОСН3+NH3
При восстановлении фенилфуроксана (не охарактеризованного) алюмо-
гидридом лития был получен диаминоэтилбензол [50]. По аналогии с реакцией
Гриньяра, можно предполагать, что для реакции был взят а-изомер.
XX (вероятно, а)—22 С6Н5СН (NH2) CH2NH2
Продукты превращения диоксимов диацилфуроксанов и диацилфуразанов.
При окислении диоксима (XXIV) дибензоилфуроксана образуется твердое
вещество желтого цвета CieHioN404 (т. пл. 162—168°, с разл.), которое оши-
бочно было принято за циклическую перекись с конденсированными циклами
(XXV) [67, 90, 177]. Аналогичным образом диоксим дибензоилфуразана дает
желтый продукт С^Ню^Оз (т. пл. 171—172°, с разл.), который идентифици-
ровали как перекись (XXVI) [142]. Особую трудность в каждом из этих
примеров представляет установление строения внутримолекулярной ассоциа-
ции 1,4-динитрозопроизводных. В качестве примера внутримолекулярной
ассоциации нитрозогрупп с образованием связи азот — азот предложен
о,о'-динитрозодифенил [191]. По-видимому, подобные представления прием-
С6Н5 О
с6н5
XXV
СОС6Н5
N^=°
=0
V
СОС6Н5
XXVI
XXVII
1,2,5-ОКСЛДИАЗОЛЫ
385
лемы и в случае цис- 1,4-динитрозо-1,4-дихлорциклогексана [192]. Продукту,
полученному действием N2O4 на 1,6-дифенилгексантетраон-1,3,4,6, приписы-
вают 1193] строение 4,5-ди-№-окиси 3,6-дибензоил-4,5-диаза-1,2-бензохинона
(XXVII). Другие случаи, когда в органических молекулах смежные атомы
азота были бы связаны с атомами кислорода (N-*O), не известны (или же
изменились представления о строении, как, например, для внутримолекуляр-
ных нитрозодимеров). В связи с этим гипотетическая формула XXVII, воз-
можно, неправильна и не может быть принята за образец для внесения уточ-
нений в строение соединений XXV и XXVI. Вероятной причиной, что веще-
ству XXV не приписывают строение соединения с конденсированными коль-
цами (в положении 5,7), является то обстоятельство, что при этом должны
были бы существовать два изомера (XXVIIa и XXVII6), однако они до сих пор
не обнаружены.
Сообщается о необычном превращении соединения XXIV (с потерей одного
атома кислорода) в диоксим (XXVIII) фенил(фенилглиоксилил)фуразана при
действии горячего 5%-ного раствора едкого натра 1130]. Тот же продукт полу-
чен при аналогичной обработке диоксима дибензоилфуразана [130], однако
это является примером реакции изомеризации. Показано также, что соедине-
ние XXIV при температуре плавления или в кипящей соляной кислоте дает
продукт CieH10N4O3 (т. пл. 134—136°), идентифицированный как моноокись
О R О
t | t
N = C N
/ 4
О I О и
'n = C/ \
।
R
R О
I t
N = C N
О R
XXVII6
XXVIIa
дифенилдифуразана (XXIX) [130] и, по-видимому, идентичный C16Hi0N4O3
(т. пл. 135—136°), полученному из одной из конфигураций соединения XXVIII
XVJV NaOH C6H5C( = NOH)C( = NOH)C = CC6H6
N
N
XXVIII 0
C0H5C( = NOH)C—CC( = NOH) C(iH5
О
25 Зака?. W 0 7 8
386
ОКСАДИАЗОЛЫ
[133]. Изомер C16H10N4O3 (т. пл. 131—132°) получен окислением другой
конфигурации соединения XXVIII. Диоксим (XXVIII) при дегидратировании
дает дифенилдифуразан C|6H|0N4O2 (XXX, т. пл. 135—136°) [133].
Свн5 (C2n2o2) (C2N2O) сен5 с6н5 (C2n2O) (C2n2O) с6н5
XXIX XXX
Диоксим диацетилфуроксана при окислении N2O4 образует твердое веще-
ство желтого цвета CeHeN4O4 (XXXI) с т. пл. 187° (с разл.), которому была
приписана структура перекиси, аналогичная структуре XXV [194]. Окисле-
ние диоксима диацетилфуроксана азотной кислотой дает неидентифицирован-
пый продукт (XXXII) C6H6N4O5 [195], а обработка щелочью — твердое веще-
ство желтого цвета C6H0N4O3, (XXXIII, т. пл. 188—190°), которому приписы-
вается строение соединения с конденсированными в положении 5,7 кольцами.
Структурные формулы линейных полиариальных соединений (XXIX
и XXX), по-видимому, правильны, однако производные с конденсированными
кольцами (XXV, XXVI, XXXI и XXXIII) должны быть повторно исследованы.
СН3 О
I t
N = C N
/ \
О ( О
\ / \ /
N~C N
I
СН3
XXXIII
о-Динитрозоароматические производные. Бледно-желтому (бесцветному
после тщательной очистки) о-динитрозобензолу было приписано строение бен-
зофуроксана (3, стр, 379), после того как он был описан Грином и Рове в 1912 г.
[115]. В следующем году эти авторы предложили [18] структуру эпоксипроиз-
водного (И) с целью объяснить симметрию половин группы N2O2. Это важное
свойство о-динитрозобензола допускает наличие двух, но не четырех изомер-
ных монозамещенных производных. Хиноидные свойства как бензофуразона,
так и о-динитрозобензола, проявляющиеся в реакциях присоединения брома,
едва ли могут быть объяснены структурами, характеризующимися наличием
бензоидного кольца, т. е. эпоксипроизводным (И) или о-динитробензолом (Г).
Уже сообщалось о невозможности существования 1,2,4,5-тетранитрозобензола
[116], так как одновременная внутримолекулярная ассоциация этих двух
пар смежных нитрозогрупп, очевидно, не может иметь места. Это указывает
на нестабильность бензоидных форм, таких, как Г и И, по сравнению с о-хиноид-
ными формами ароматических о-динитрозопроизводных. В противополож-
ность этому молекулы желтого 1,2,3,4-тетранитрозо-5-нитробензола [116]
и гексанитрозобензола [197] стабильны и могут иметь хиноидную форму.
Сообщается, что 1,2-динитрозо-4,5-диаминобензол [136, 198] обладает меньшей
устойчивостью.
Требование, чтобы о-динитрозобензол имел симметричное расположение
нитрозогрупп и одновременно хиноидную форму, привело к мысли о резонанс-
ном гибриде (М), в котором большую роль играют ионные хиноидные и бен-
зоидные структуры [140]. Такое представление о строении о-динитрозобензола
говорит о сходстве многих его химических и физических свойств со свойствами
п-динитрозобензола и несходстве с л-динитрозобензолом, как, например, в отно-
шении восстановления в диоксимы бензохинонов.
Неоднократно была показана равноценность в молекуле 1,2-динитрозо-
бензола положений 4 и 5 с целью их использования для замещений. Пиролиз
4- и 5-замещенных 2-нитрофенилазида ведет к получению того же продукта,
который образуется при окислении щелочным гипохлоритом 4- или 5-замещен-
1,2,5-ОКСАДИЛЗОЛЫ.
387
ного 2-нитроанилина [18, 144]. Аналогичным образом 1,2-динитрозонафталин
получают пиролизом Цили 2)-азидо-2(или 1)-нитронафталина или окислением
Цили 2)-амино-2(или 1)-нитронафталина [ 199|. Окисление каждого из четырех
изомеров диоксима 2-нитро-9,10-фенантренхинона ведет к образованию одного
соединения — 2-нитро-9,10-динитрозофенантрена [118].
Не установлено, является ли 2,3-динитрозо- 1,4-нафтохиноп (Д) примером
ароматического динитрозопроизводного или же фуроксана. Продукт его
О Ч-
Н NO
N
восстановления — диоксим 1,2,3,4-тетрагидро-1,2,3,4-тетракетонафталина,
по-видимому, не изомеризуется в 1,4-диокси-2,3-динитрозонафталин [4]. Любо-
пытно, что известна только одна молекула, которая содержит в качестве
заместителей о-динитрозо- и гидроксильные группы в одном и том же аромати-
ческом кольце. Нитрование триоксима 1,2,3,4-тетрагидро-1,2,3,4-тетракетобен-
зола дает соединение, идентифицированное недавно как 1,2-динитрозо-З-окси-
4,6-динитробензол (XXXIV) (в сообщении названное фуроксаном) [26]. Это
вещество не является обычным фенолом и обладает необычным свойством:
будучи 2-динитрозопроизводным, оно плавится при более низкой температуре,
чем соответствующий фуразан (XVII). Поскольку в этих таутомерных системах
моноксимы хинона могут превалировать над нитрозофенолами [200], идентич-
ность соединения XXXIV может быть поставлена под сомнение.
ON NO
HONr=---r=NOH
25*
388
окслдилзолы
Для фуразанового кольца несвойственно образование конденсированных
систем с другими пятичленными циклами. Попытки дегидрировать диоксим
аценафтенхинона в его ангидрид --- фуразан [15] — оказались безуспешными.
Правда, щелочным гипохлоритом удалось окислить этот диоксим [201 ]. Однако
полученному при этом продукту, вероятно, неправильно было приписано строе-
ние фуроксана [201]; его следует рассматривать как другой пример арома-
тического о-динитрозопроизводного.
1,2,4-ОКСАДИАЗОЛЫ
Методы получения
Пиролиз амидоксимов и их сложных эфиров. Бензамидоксим при 170
теряет аммиак, азот и закись азота и при этом образуются бензамид, бензо-
нитрил, 3,5-дифенил-1,2,4-триазол, 2,4,6-трифенил-1,3,5-триазин и 3,5-дифе-
нил-1,2,4-оксадиазол (I) [202]. Нагревание бензамидоксима в низкомолеку-
лярных жирных кислотах [203], а также обработка азотистой кислотой [2041
или хлором [205] в этаноле, также служат методами получения соединения I.
Более общим методом получения некоторых дизамещенных 1,2,4-окса-
диазолов оказался пиролиз амидоксимов сложных эфиров; при этом замести-
тели могут оказаться одинаковыми или разными. Сложный о-толуиловый
эфир о-толиламидоксима при 180° дает 3,5-ди-(о-толил)-!,2,4-оксадиазол [2061,
а ацетат п-бромбензамидоксима — 3-(п-бромфенил)-5-метил-1,2,4-оксадиазол
C6H5C( = NOH)NH2
Нагревание
ИЛИ С12«
ИЛИ HNO2
C6H6C-N
/ СС6Н5
[207]. Этим методом получены другие 3,5-дизамещенные 1,2,4-оксадиазолы:
3-(3-НОСеН4)-5-СН3-[208], 3-(а- или ₽-С10Н7)-5-СН3-[209, 210], 3-(4-СН3ОСвН4)-
5-СН3- или 3-(4-СН3ОС6Н4)-5-С6Н5- [212] и 3-[2,4-С6Н3(СН3)г]-5-С6Н5- 12121.
С7Н7С ( == NOCOC7H7) nh2
BrC6H4C — N
ВгСвН4С (= NOCOCH3) nh2
В 3-положение могут быть введены алкильные заместители. Нагревание ацетата
бензилгидроксамовой кислоты в водной среде дает З-бензил-5-метил-1,2,4-окса-
диазол [213]; соответствующий карбинол получают аналогичной обработкой
ацетата амидоксима миндальной кислоты [214]. Из бензоата амидоксима корич-
с6н5сн2с —n
CgH5CH2C(==NOCOCH3)NH2 »^рева™ч iZ \сн3
o'/
ной кислоты получен 3-стирил-5-фенил-1,2,4-оксадиазол [215]. Монобензоат (II)
в соляной кислоте дает соответствующий З-ароил-5-фенил-1,2,4-оксадиазол,
тогда как в разбавленном растворе едкого натра реакция ведет к образованию
1,2.4-ОКСЛДИЛЗОЛЫ
389
производного оксима [216]. В некоторых других случаях аналогичная дегид-
ратация происходит в присутствии серной кислоты [209, 217].
NOH
II
С7Н7СС —N Na0H C7H7C-^NOH НС1 С7Н7СОС—N
У Ч ' <----------- I -----> J Ч
N СС6Н5 H2NC = NOCOCeH5 N CC(iH5
'''о'7 Хо/
II
Выделение эфира амидоксима не всегда необходимо. Во многих случаях
оксадиазол с лучшими выходами получают при нагревании смеси амидоксима
с соответствующим ангидридом кислоты или хлористым ацилом. Производные
бензамидоксима в янтарном ангидриде дают соответствующую |3-(3-арил-1,2,4-
оксадиазол-5-ил)пропионовую кислоту [218]. Взаимодействуя с уксусным,
О
II
СН2С
ArC( —NOH) NH2 + О
СН2С
il
О
ArC-N
Ч Ч
N ССН2СН2СООН
o'
пропионовым или масляным ангидридом, бензамидоксим образует соответ-
ствующие З-фенил-5-алкил-1,2,4-оксадиазолы [203, 204]. Большое число раз-
личных замещенных в кольце производных бензамидоксима обрабатывали
аналогичным путем уксусным ангидридом, хлористым ацетилом или хлори-
стым бензоилом [207, 208, 209, 211, 219, 220]. Однозначный синтез 3-амино-
5-фенил-1,2,4-оксадиазола (III, т. пл. 164°) осуществляется путем обработки
бромгидрата диоксигуанидина хлористым бензоилом и бикарбонатом натрия
[221, 222]. Тот же продукт получается при гидролизе 1\т,О-дибензоилокси-
гуанидина [222].
H2NC —NOCOCgH5 H2NC-N c6h5coci ’ HON = CNH2
1XIHCOC6H6 " NX 5CCliH5 N11011
111
Производное 5-ацетопил-1,2,4-оксадиазола получено действием ацетоук-
сусного эфира на п-толиламидоксим. 5-Галогенметильпые производные синте-
зируют, используя галогепацетилхлорид [220, 223]. Применялись и дру-
гие ацилирующие агенты: например, бензотрихлорид и бензойная кис-
лота [204].
Алифатические амидоксимы обнаруживают аналогичные реакции. При
взаимодействии амидоксима миндальной кислоты с уксусным ангидридом
[214 I, ацетамидоксима с хлористым бензоилом [224 | и моноамидоксима малоно-
вой кислоты с бензойной кислотой [2251 получаются ожидаемые оксадиазолы.
N —С —ч
I \ \
[H2NC(=^NOH)J2----> / Н3СС N
\ Л
U-I2NC(=-NOH)j2Cll2
390
ОКСАДИ АЗОЛЫ
Тот же продукт — З-метил-5-фенил-1,2,4-оксадиазол— образуется в соот-
ветствии с последними двумя схемами, причем вторая реакция сопровож-
дается декарбоксилированием. В уксусном ангидриде как диаминоглиоксим,
так и диамидоксим малоновой кислоты дают бис-1,2,4-оксадиазолы [226].
В противоположность этому 1,3-диамиио-1,2,3-триоксиминопропан в уксусном
H2NC- СС (= NOCOCH3) NH2
II2NC( NOII) С (— NOH)C( = NOH)NH2 • ••
''''o''
ангидриде дает фуразан, тогда как 1,4-диамино-1,2,3,4-тетраоксиминобутап
в холодном уксусном ангидриде в реакцию не вступает [226].
Циклизация моноксимов диациламидов. Имидохлорид дибензамида дает
с гидроксиламином 3,5-дифенил-1,2,4-оксадиазол [227]. Вероятным проме-
С6Н5СС1
II
NCOC6H6
h2noh
"C6H5C = NOH -
NIICOCGH5_
c6h5c-n
О
жуточным соединением является оксим дибензамида.
Аналогичный промежуточный продукт, по-видимому, образуется при
получении по методу Янга — Джонсона [228] (ср. стр. 382) З-анилино-5-фенил-
1,2,4-оксадиазола, который идентифицирован восстановлением в фенилбен-
зоилгуанидин. Этот метод был использован для синтеза 5-окси-1,2,4-оксадиа-
CuHgNHCSCH,
' II
NCOCBII5
HgNOH
---->
“C6H6NHC=NOH ’
NHCOCBIIS_
Cyl lgNl IC - N
\ Zn
N CC6II5--------------> CyH^NHC ( — NH) NIICOCaHr,
\ J C1I3COO11 ' ) ь э
(J
C2HbOOCNHCSCH, Nnn C.,Hr,OOCNHC— N
О a [ |.>NO11 « 0
il -----> X
NCOOC2H;, N COII
зол-3-илуретана [229]. В синтезе З-уреидо-5-замещепных 1,2,4-оксадиазолов
исходя из дициандиамидов и гидроксиламина предполагаемым промежуточным
соединением является О-замещенный гидроксиламин [222]. Однако эта реак-
ция, по-видимому, тесно связана с рассмотренными выше и может протекать
с циклизацией промежуточного амидоксима. При гидролизе З-уреидо-5-фенил-
c0h5conhc( --ncn)nh2 haNO1!' 'H2NCONHC^NOI-I -__________
Inhs L nhcocbii5 .
H2NCONHC-N
^ССцНд
oz
H2NC-N
> NZ 4CCBH5
1,2,4-оксадиазола образуется З-амино-5-фенил- 1,2,4-оксадиазол (т. пл. 164°)
[221, 222].
1,2.4-ОКСЛДИ АЗОЛЫ
391
Оксим диациламида, возможно, является промежуточным продуктом
в синтезе диамино-1,2,4-оксадиазола из дицианамида натрия и гидроксил-
амина [230].
NaN(CN)2 ^2», H2NC .NOH _________.. H2NC-N^
NHCN N CNH2
Окисление 1,2,4-оксадиазолинов. При конденсации ароматических (по
не алифатических) амидоксимов с ацетальдегидом [211, 217, 231—2331,
пропионовым альдегидом [234], изомасляным альдегидом [234], фенилацет-
альдегидом 1234] и салициловым альдегидом 1234] получены соответствую-
щие дигидро-1,2,4-оксадиазолы, а,0-Дихлорэтиловый эфир может заменить
ацетальдегид [233]. Полученные соединения окисляются перманганатом
A.rC(=NOH)NH2
N CHR N CR
в 3,5-дизамещенные 1,2,4-оксадиазолы. Сообщается, что при окислении про-
дукта конденсации бензамидоксима с салициловым альдегидом 1234], а также
при нагревании оксима бензоилсалициламидоксима [235] (пример приме-
нения метода Фалька, стр. 383) образуется один итотже оксадиазол с т. пл. 128°.
С6Н6С—NH
\ Ю]
N СНС6Н4ОН--------;
3 (или 5)-Фенил- |80,
5 (или 3)-(о-окси- <-----
фенил)-1,2,4-
оксадиазол
NOCOC6H5
,^\-CNH2
ML он
Если это наблюдение не будет опровергнуто дальнейшими исследованиями,
то, следовательно, в одной из приведенных реакций, по-видимому в реакции
Фалька, происходит молекулярная перегруппировка. Поэтому необходима
строгая проверка строения оксадиазолов, полученных любыми методами.
Дегидратация с перегруппировкой глиоксимов. а-Бензилдиоксим в поли-
фосфорной кислоте при 25° превращается главным образом в дифенил-1,2,4-
оксадиазол с примесью небольшого количества М-фенил-М'-бензоилмочевипы,
а при 120° — только в оксадиазол 1236 |. Серная кислота, хлористый водород
в уксусной кислоте, пятиокись фосфора в бензоле, пятихлористый или пяти-
бромистый фосфор и хлорокись фосфора также превращают а-бензилдиоксим
в дифенил-1,2,4-оксадиазол [237, 238]. Тот же продукт был получен из у-беп-
зилдиоксима и пятихлористого фосфора 1239]. а- или 0-Диоксимы метилфенил-
дикетона дают с хлорокисью фосфора З-фенил-5-метил-1,2,4-оксадиазол [2381.
CbH5C-N
CeH5C( = NOH)C(=^NOH)C0H5-----> bL \с«Н5
Ххо'7
Однако 0-диоксим с пятихлористым фосфором образует бензонитрил, хло-
ристый бензоил и другие продукты [238]. Фепиламиноглиоксим в присутст-
вии хлорокиси фосфора превращается в З-фепил-5-амино-1,2,4-оксадиазол
1238, 240).
Присоединение окисей нитрилов к нитрилам. Самопроизвольная диме-
ризация окисей нитрилов в фуроксаны сопровождается образованием углерод-
392
ОКСАДИАЗОЛЫ
углеродной и азот-кислородной связей. Другой вид димеризации, ведущий
к образованию 1,2,4-оксадиазолов, требует возникновения связей углерод—
кислород и углерод — азот, причем процесс, по-видимому, катализируется
сильными щелочами или кислотами. Под действием иода в водном растворе
О
CeH6C —N
CGH5C ( —NOH) N(,).NH4O11 / ЧССДК
или НС1 b d
о
IV
cbh5c-n
CcH5CH_=NOH____________ \г> ^CCBHS-|-1V
Na.COs / \\ /
о -o'
карбоната натрия бензальдоксим образует две изомерные Ы3-окиси произ-
водных 3,5-дифенил-1,2,4-оксаДиазола [241], одна из которых (IV) получена
из аммонийной соли бензонитрозоловой кислоты в азотной или соляной кис-
лоте [242]. Деполимеризация 1,3,5-триокиси 2,4,6-трифенил-1,3,5-триазина
в 20%-ной соляной кислоте приводит к образованию соединения IV [243], кото-
рое получается также из хлорангидрида бензгидроксимовой кислоты при ком-
натной темпер$туре в закрытом сосуде [221 ], однако в последнем случае при
высокой температуре N-окиси не образуются. Из хлорангидрида о-хлорбензгид-
С0Н5
% А 7°
4N N' 2О%-Ш1Я
ПС,.’-1 И с.п —IV <- C0H5C(-NOH)C1
N
роксимовой кислоты получен 3,5-ди-(о-хлорфепил)-1,2,4-оксадиазол [2441.
Во всех случаях промежуточным продуктом может быть окись бензонитрила.
C1CGH4C —N
IlarpeH.iiiue V
C1CGH4C(--.NOI1)C1----------> N CCGII4C1
о
При реакции окиси бензонитрила с ароматическим нитрилом в эфире при
комнатной температуре образуется дифенилфуроксан и З-фенил-5-арил-1,2,4-
оксадиазол [245]. Электроноакцепторные заместители в кольце ароматических
нитрилов облегчают протекание реакции, тогда как электронодонорные заме-
стители и все opmo-заместители препятствуют реакции. Аналогичная реакция,
вероятно, происходит при превращении ароматических альдоксимов в 3,5-
ArCN -1 CoH^CNO —>
CgH5C-N
СбН3С СС6Н5
диарил- 1,2,4-оксадиазолы под действием алкилпитритов [246], мононадсерпой
кислоты [247], щелочного раствора феррицианида [248] или щелочного рас-
твора гипохлорита [249]. При этом первоначально может образоваться N-окись
1,2.4-ОКСАДИАЗОЛЫ
393
альдоксимангидрида, которая далее будет диссоциировать с образованием
нитрила, окиси нитрила и воды. N-Окись бензальдоксимангидрида получена
О
[О] 1
ArCH = NOH — > |ArCH NON CHAr — > ArCN\ ArCH NO2H
[ArCH = NO2H] ArCNO + H2O
ArC —N
|ArCN + ArCNOJ —> N CAr
при действии на бензальдоксим щелочного раствора гипохлорита [249] или
феррицианида [248], N2O4 [250], а такжеалкилнитритов [246]. При нагревании
в хлороформе или бензоле эта окись превращается в 3,5-дифенил-1,2,4-окса-
диазол [248].
В присутствии бензамидина или другого сильного основания хлорангид-
рид бензгидроксимовой кислоты превращается в дифенил-1,2,4-оксадиазол
(I) [251].
Основание
CeH5C(^NOH)Cl---------> I
Превращения циклов. Превращение фуразанов и фуроксанов в 1,2,4-
оксадиазолы уже рассматривалось. Другим примером является превращение
бензоиламинофуразана в щелочном растворе едкого кали при 40° в амид
5-фенил-1,2,4-оксадиазолкарбоновой-3 кислоты [252], который для идентифика-
ции превращают путем гидролиза и декарбоксилирования в бензоилцианамид
(C6HSCONHCN). Изомерная 3-фенил-1,2,4-оксадиазолкарбоновая-5 кислота
свн5сос—cnh2
N N
кон
H2NCOC-N
о
декарбоксилируется в устойчивый 3-фенил-1,2,4-оксадиазол 1252]. Другими
ароматическими гетероциклами, которые могут превращаться в 1,2,4-оксади-
азолы, являются имидазолы, изоксазолы, пирролы и пиримидины.
В спиртовом растворе соляной кислоты 2,5(или 4)-дифепил-4(или 5)-нитро-
зоимидазол образует З-бензоил-5-фенил-1,2,4-оксадиазол 12531. Изомерный
З-фенил-5-бензоил-1,2,4-оксадиазол может быть получен при действии гидро-
ксиламина на 2,5-дифенил-З-иитрозопиррол 1254]; промежуточным продуктом
с8н5с—cno
n __2!SL
С2П5ОН
HN
C6H5COC-N
CCUH,
О
может быть оксим З-бензоил-5-фенилизоксазола 12541.
/N0 С6Н£
Д. р J4 HgNOH ,
у ^6П5 [\|
Н5С,
ССОС0Н5
N
Н
394
ОКСАДИАЗОЛЫ
Из 3,6-диацетамидобензизоксазола в растворе едкого натра получен
3-(2-окси-4-ацетамидофенил)-5-метил-1,2,4-оксадиазол [255].
NHCOCH3
С
//\/ Ч
I I1 N
CH3CONH-^X /
О
NaOH
CH3CONH — 1 Ч он
,N = CCH3
< I
Ч —о
Ыз-Окись аденина в кипящем уксусном ангидриде быстро превращается
как в 4-формамидо-, так и в 4-ацетамидо-5-(5-метил-1,2,4-оксадиазол-3-ил)-
имидазол (V) [256]. Независимый синтез соединения V (R = СН3) состоит
во взаимодействии 4-аминоимидазол-5-карбоксамидоксима и уксусного
ангидрида.
nh2 н
0 I N N-C------C = CNHCOR
Ч z^\/ \ ,гн Г(П Ч / \
N И Чи <снзсо)2о н сс N HN N
II уСН--------\ / \ /
XN/Xn 0 С
м
V
R=H, СН3
H2NC( NOH)C = CNH2
(СН3СО)2О
Н
V (R-CH3)
Различные методы получения. Дифенил-1,2,4-оксадиазол получен:
1) из его 4-окиси отнятием кислорода пятихлористым фосфором [22Г] или цин-
ком в уксусной кислоте [242]; 2) из хлористого бензоила и нитрометана в пири-
дине [257]; 3) из метилового сложного эфира ацы-формы фенилнитрометана
в соляной кислоте [2581.
Под действием азотистой кислоты N-ацилпроизводные 2-амииоциклогск-
сандиона-1,3 превращаются в 3-(<о-карбоксибутирил)-5-замещенные 1,2,4-
оксадиазолы [259].
/\^° HNO> HOOC(CH2)3COC-N
NHCOR 911
Пиридооксадиазолон получен нагреванием N-окиси 2-этоксикарбонилами-
попиридина [2601 или при обработке N-окиси 2-аминопиридина фосгеном [2611,
Ч- NHCOOC2H5
I Ч
ч0
Описаны тетрагидро-1,2,4-оксадиазолы. Взаимодействием [3-фенилгидрок-
силамина с ацетальдегидом и бензальдегидом в растворе едкого натра получен
N-фенилнитрон коричного альдегида. При реакции последнего с фенилизоциа-
1,2,4-ОКСАДИАЗОЛЫ
395
натом образуется 2,4-дифенил-3-стирил-5-кетотетрагидро-1,2,4-оксадиазол
[262].
Продукт взаимодействия основания Шиффа, полученного из уретана и бен-
зофенона, реагирует с гидроксиламином в присутствии метилата натрия с обра-
зованием 3,3-дифенил-1,2,4-оксадиазолидинона-5 [263]. При действии гидро-
ксиламина на хлорангидрид 1,1-дифенилметиленкарбаминовой кислоты полу-
чен, 5,5-дифенил-1,2,4-оксазолидинон-3 [263].
NaOH C6H5NCO
C6H5NHOH + CH3CHO + C6H5CHO------> C6H5N(O) = CHCH=-CHC6Hs-------->
C6H5CH = CHCH-N —C6H5
-> C6H5— N XC = O
О
(СвНа)гС — NH
(C6H5)2C h2noh (CgHg^CNHCOOCaHj cHaONa / \
|l------> | ------> NH C = O
c2h5oocn nhoh \ /
О
O - C--NH
H2NOH / '
(C6H5)2C = NCOC1--------> HN C(C6H5)2
Ъ"
Реакции 1,2,4-оксадиазолов
В противоположность дифенилфуроксану 4-окись дифенил-1,2,4-оксадиа-
зола реагирует как основание, образуя устойчивый хлоргидрат [2211. Под
действием как кислот, так и оснований эта N-окись распадается на бензойную
кислоту и бензонитрил [221]. Обе N-окиси дифенил-1,2,4-оксадиазола пре-
вращаются в дифенил-1,2,4-оксадиазол при обработке цинком в уксусной
кислоте или пятихлористым фосфором [241 ]. Цинк в уксусной кислоте не дей-
ствует на дифенил-1,2,4-оксадиазол [242], но цинк в соляной кислоте превра-
щает его в бензонитрил [204 ]; при взаимодействии с иодистым водородом и крас-
ным фосфором образуются бензойная кислота и аммиак [237].
Соляная кислота реагирует с оксимом как З-бензоил-5-метил-, так
и З-метил-5-бензоил-1,2,4-оксадиазола с образованием фениламинофуразана
[2641 — вещества, которое получается также из фениламинофуроксана дей-
ствием олова в соляной кислоте [265]. Наблюдался также аналогичный пиро-
лиз фенилгидразона З-бензоил-5-фенил-1,2,4-оксадиазола с образованием
2,4-дифенил-5-бензамидо-1,2,3-триазола [216].
. Восстановление З-фенил-5-амино-1,2,4-оксадиазола (т. пл. 153—154°)
действием фосфора и йодистого водорода в бензамидин послужило доказатель-
CeH-5C( = NOH)C — N C6H5C-CNH2 CH3C-N
Ч на \ HCI
N ССН3-----------> N N <----------- N CC(--=NOH)CoH5
О
О о о
CBH5NHN —С(С6Н5)С —N C0H5CONHC-CCuIIs
Ч 190“ Ч \,
N сс6н.
О
N
С6Н,
396
ОКСАДИАЗОЛЫ
ством строения оксадиазола [240]. При гидролизе 3-фенил-5-амино-1,2,4-
оксадиазола как соляной кислотой, так и едким натром образуется фенил-
окси- 1,2,4-оксадиазол (т. пл. 202°) [2401, полученной также щелочным гид-
ролизом З-амино-5-фенил-1,2,4-оксадиазола [222]. Предполагают, что при
этом происходит еще не изученная перегруппировка [222].
СвЩС — Is!
H2NC —N
HO(C2N2O)C6H5
N CNH2
При омылении этилового эфира 3-фенил-1,2,4-оксадиазолкарбоновой-5
кислоты получается соответствующая устойчивая кислота [266]. Щелоч-
ное расщепление 1,3-бнс-(3-толил-1,2,4-оксадиазол-5-ил)ацетона приводит
к 3-толил-1,2,4-оксадиазол-5-уксусной кислоте и 3-толил-5-метил-1,2,4-окса-
диазолу (VI) [233]. Оксадиазолилуксусная кислота легко декарбоксилирует-
ся [223].
(1) NaOH
(2) Н|О
С7Н7С —N С7Н7С —N
г/ *ССН2СООН + iZ \сн3
О О
VI
Активная метильная группа в 3-толил-5-метил-1,2,4-оксадиазоле (VI)
проявляется при катализируемой основаниями конденсации со щавелевым
эфиром [223]. Полученный кетоэфир обычными методами превращают
в 3-толил-5-цианметил-1,2,4-оксадиазол (VII) — соединение, которое с более
(СООС2Н5)2
V------------
C2H5ONa
С7Н7С —N
// V
N ССН2СОСООС2Н5
(1) Н2О
(2) HoNOII
С7Н7С —N
pZ \ch2c=noh
(СН3СО)2О
С7Н7С —N
У ч.
N CCH2CN
соон
VII
высокими выходами можно получить из толиламидокси.ма и хлорангидрнда
циануксусной кислоты в этиловом эфире при комнатной температуре с после-
дующим удалением эфира и кипячением смеси с уксусной кислотой [223].
Нитрил (VII) получен с низкими выходами действием цианистого калия
на 3-тол ил-5-бромметил-1,2,4-оксадиазол [223],
Реакционноспособность метиленовой группы в 5-положении 1,2,4-окса-
диазольного кольца проявляется в реакции Джаппа — Клингемана между
3-(/г-толил)-5-ацетонил-1,2,4-оксадиазолом и диазотированным о-анизидином
в щелочной среде [267].
С7Н7С —N С7Н7С—N
V \ NaOH V
N ССН2СОСН3-| CH3OC6H4N2C1--------> N СССОСН3
ХоVII * * Х ХоХ
NH
I
С6Н4ОСН3
1,2,3-ОКСАДИЛЗОЛЫ
397
Физические свойства
, Для некоторых 1,2,4-оксадиазолов известны данные по поглощению
в ультрафиолетовой области [268], дипольным моментам 12691 и тсплотам
сгорания [270].
1,2,3-ОКСАДИАЗОЛЫ
Производные 1,2,3-оксадиазолов, за исключением сиднонов, рассматри-
ваются как изомерные а-диазокарбонильные соединения (диазоокисп) [271].
RC-CR'
N О
Ч /
N
RC — CR'
н д
n2 о
Андерсон и Рёдель [272], исследуя спектры поглощения 1,2- и 1,4-диазо-
фенолов, пришли к заключению, что n-диазофенол имеет хиноидную структуру
и что орто-изомер не является бициклом, а, по-видимому, тоже имеет хиноид-
ное строение, что подтверждается дипольными моментами [273]. Строение
неустойчивого о-диазофенола можно представить в виде резонансного гибрида,
по крайней мере трех структур [274].
N2
о
Сидноны, открытые в Сиднейском университете в 1935 г. [275], пред*
ставляют собой мономолекулярные ангидриды некоторых N-нитрозопроиз-
водных а-аминокислот. При получении N-фенилсиднона (I) из N-нитрозо-
N-фенилглицина в уксусном ангидриде первоначально может образоваться
C6H5NNO
СН2СООН
pCgHjNNO
I
СН2СОСОСН3
C6H5N = N -
Н2С о
CsHgNzOz
I
смешанный ангидрид [276]. Чистый смешанный ангидрид, полученный из хло-
ристого ацетила и калиевой соли аминокислоты, при комнатной температуре
медленно теряет элементы уксусной кислоты с образованием N-фенилсид-
нона (I) [276].
Для сиднонов предложены различные структурные формулы [277].
В ’настоящее время молекулам этих соединений приписывают бетаиновую
[278] и мезоионную [277] структуры с координационной ковалентной связью
между двумя атомами азота в кольце [279]. У атома азота может быть алкиль-
ный или арильный заместитель (R), но не водород, а у атома углерода —
алкильный или арильный заместитель или водород. Известны сидноны с кон-
денсированными кольцами [280].
+
RN=N
R,4
о-
RN--N
I
О~
rn-»-n
R'C О
о
398
ОКСАДИАЗОЛЫ
N-Фенилсиднон (I) кристаллизуется из кипящей воды, а в горячем раз-
бавленном растворе едкого натра он гидролизуется с образованием N-нитрозо-
N-фенилглицина; при нагревании с бензиламином дает соответствующий
N-бензиламид [281]. С-Бром-Ы-фенилсиднон под действием едкого натра
расщепляется с образованием анилина, нитрита натрия и оксалата натрия [2791.
При действии водных растворов кислот на сидноны получаются алкил-
или 'арилгидразины, карбоновые кислоты и двуокись углерода [275, 2821.
В бензоле с эквимолекулярными количествами воды и хлористого водорода
сиднон образует гидразид [2831, что подтверждает предлагаемый ниже меха-
низм реакции [277]. Метод превращения первичных алифатических и арома-
тических аминов в соответствующие гидразины зависит от гидролитического
RNNH2 н2о
COR1
-со2
RNHNH2 + r’cooh
расщепления сиднонового кольца [284, 285]. N-Фенилсиднон (I) можно про-
нитровать в холодной концентрированной серной кислоте [281]. Хлориро-
вание и бромирование соединения I приводит к замещению водорода лишь
в сидноновом кольце [281, 286].
н sn CgHjN—N
н2ьо4~ у \
HN°3 O2NC ® О
-10° \ /
I
О-
N-Фенилсиднон устойчив к каталитическому гидрированию, но при дей-
ствии водорода под окисью платины медленно превращается в аммонийную
соль N-фенилглицина [281], которая" образуется также под действием цинка
в разбавленной уксусной кислоте [286]. Изучалось также полярографическое
восстановление сиднонов [287].
При взаимодействии сиднонов с n-бензохиноном образуются ди- и три-
циклические хиноны; при этом выделяется двуокись углерода [288].
R' R'
О I О |
НС N н с
! I NR RN j I NR
|| N с II N
о I О
R'
Моно- и диарилсидноны поглощают в области 310—340 мц, а N-бензил-
сиднон — около 290 мц [281, 289]. Полоса поглощения в инфракрасных спек-
1.3,4-ОКСЛДИАЗОЛЫ
399
трах в области 1720—1770 смт1 отвечает карбонильной группе [285, 290],
однако инфракрасные спектры сиднонов подробно не изучены. Найденные
дипольные моменты [279] (5,0—6,70 D) с отрицательным полюсом, направлен-
ным к карбонильному кислороду, находятся в согласии с теоретически рас-
считанными [291], которые показывают, что плоское кольцо этого гетеро-
цикла несет положительный заряд порядка 0,7—0,9, уравновешиваемый ана-
логичным отрицательным зарядом на экзоциклическом атоме кислорода.
Ы-(3-Пиридил)сиднон обладает фототропными свойствами; его бесцветные
кристаллы синеют после кратковременной экспозиции на солнечном свету
и вновь становятся бесцветными после нагревания примерно до 80° [285].
При действии на а-(Ы-метил-Ы-нитрозоамино)нитрилы азотной кислоты
или хлористого водорода в эфире получаются соли сиднониминов [292]. При
нейтрализации солей сиднониминов образуются соответствующие а-(Ы-метил-
Ы-нитрозоамино)амиды [292].
RCHCN нх RC--------CNH2 ' он_ RCHCONH'j
CH3NNO * СН3<® О X- ; CH3NKO
Алкил-ф-оксатриазолы получаются из соответствующих семикарбазидов
и азотистой кислоты [293] реакцией, аналогичной получению сиднонов из
RNHNHCONH2 —» RNNHCONH2 —> RN2CO2
rtro'
N-замещенных глицинов. Их также можно изобразить в виде бетаиновых
и мезоионных структур с координационной ковалентной связью между двумя
атомами азота. •
1,3,4-ОКСАДИАЗОЛЫ
Методы получения
Дегидратация 1,2-диацилгидразинов [294]. Циклодегидратация дибен-
зоилгидразина при 250—300° приводит к 2,5-дифенил-1,3,4-оксадиазолу [295].
Аналогичным образом при пиролизе Ы-ацетамидо-Ы-фенилуретана (при пере-
гонке) происходит отщепление этанола и образуется 2-метил-4,5-дигидро-4-
фенил-5-кето-1,3,4-оксадиазол [296]. Диацилгидразиды, по-видимому,
являются промежуточными соединениями как при образовании этих продуктов
из бензоилгидразида [297], так и при взаимодействии Ы-фенил-Ы-аминоуре-
N —N
- н2о \ -n2h4
(C0H5CONH)2-----> с6н5с сс6н5 - C6H5CONHNH2
—H2U
о
CrH5NNHCOCH3 -С0Н50Н C6H5N —N
СООС2Н5 o = cf \сн3
CH3CONH2
-NHg, Н2О
CeH5NNH2
СООС2Н5
о
400
ОКСАДИЛЗОЛЫ
тана с ацетамидом. При нагревании ди ацил гидр азиды превращаются в 2,5-
диалкил-1,3,4-оксадиазолы [298]. Так, например, ацилсемикарбазиды цикли-
зуются в 1,3,4-оксадиазолы с отщеплением амина. В кипящем пиридине семи-
карбазид 4,4-диметил-1-изоникотиноилсемикарбазида образует 5-(4-пиридил)-
1,3,4-оксадиазолон-2(ЗН) [299].
HN —N
CONHNHCON(CH3)2 -<ch3)2NH^ 0 = с/ ^cc5ii4n
Образованию 1,3,4-оксадиазольного кольца способствуют следующие
дегидратирующие агенты: хлорсульфоновая кислота [300], хлористый суль-
фурил [300], пятиокись фосфора 1301], и-толуолсульфокислота [300], ее
хлорангидрид [300], хлористый тионил [3021, хлорокись фосфора [302, 303],
хлорид цинка [304], ' ангидриды органических кислот 1305], пятихлористый
фосфор [306] и серная кислота [306].
N — N
Р2О5 х
(C2H5)2CHCONHNHCOC6H5-------> С„НЙС CCH(CfiH5)2
(СНзСО)аО X \
(C2H5CONH)2-------------> с2н5с СС2Н5
C6H5CONHNHCO— / Д
POCI3
С6Н5С
СС4Н3О
(C3H7CONH)2
ZnCla
C10H7C
N —N
реи
(a-Clr)H7CONH)2------
Диацилгидразиды образуются, по-видимому, в качестве промежуточных
продуктов при превращении биса хлорбензилиденгидразинов в 2,5-дизаме-
щенные 1,3,4-оксадиазолы действием горячей воды или раствора азотнокис-
лого серебра [306, 308]. Аналогичное замыкание цикла протекает при образо-
Ak'N 3
[/i-BrCGH4C(Cl) — N 2 --------’•
О
1,3,4-ОКСАДИ АЗОЛЫ
401
вании 1,3,4-оксадиазолов из бис-а-аминобензилиденгидразинов и азотистой
кислоты [309] или из ациламидразонов и азотистой кислоты |309].
[C«H5C(NH2)=N12
hno2
N— N
C6H5C( = NH)NHNHCOCeH5
hno2
N-N
Гидразиды и фосген. Окси-1,3,4-оксадиазолы или 3-замещенные 1,3,4-окса-
диазолоны-2 получаются из соответствующих гидразидов и фосгена. Бензоил-
гидразид и фосген в хлороформе дают 2-фенил-5-окси-1,3,4-оксадиазол
с количественным выходом [310, 311]. Аналогичным образом получены 5-окси-
N-N
СОС12 Л \
CeH5CONHNH2------> СвНвС СОН
0^
1,3,4-оксадиазолы с ароматическими, гетероциклическими [312] и алкиль-
ными [313] заместителями в положении 2. При замене фосгена Тиофосгеном
получены оксадиазолтионы [314].
При взаимодействии замещенных гидразидов с фосгеном образуются
3-замещенные 1,3,4-оксадиазолоны. Из фенилгидразида фенилуксусной кис-
лоты получают 3-фенил-5-бензил-1,3,4-оксадиазолон-2 [315, 316]. Диацетил--
гидразин, фенилкарбазиновый и фенилтиокарбазиновый эфиры вступают
с фосгеном в аналогичные реакции [311, 317].
C6H5N —N
СОС12 / \
c0h5ch2conhnhc0h5-----> о=с ссн2с0н5
CHgCON — N
coci2 *4 / \
(CH3CONH)2-----> о = с ссн3
о^
CeH5N —N
coci2 / \
C6H5NHNHC( = O)SR-----> ОС CSR
^о^
Конденсированный оксадиазолон получен при обработке N-аминопири-
дона-2 фосгеном [318].
СОС12
Ортоэфиры и гидразиды арилкарбоновых кислот. Монозамещенные
1,3,4-оксадиазолы можно- получить конденсацией гидразида ароматической-
26 Заказ № 978
402
ОКСАДИАЗОЛЬ!
карбоновой кислоты с этилортоформиатом, взятым в избытке [318]. Из высших
ортоэфиров получены соответствующие 2-алкил-5-арил-1,3,4-оксадиазолы
N — N
C6H6CONHNH2+RC(OC2H5)3 —> С6Н6С CR + C2H5OH
R=H, СН3, С2Н5
[319], причем были выделены промежуточно образующиеся 1-ацил-2-эток-
симетиленгидразины [319]. При замыкании цикла в реакции между гидра-
CON2H3
|| нс(ос2н6ь C5H4NCONHN=CHOC2H5 200\
N
N —N
—> c5H4Ncf \:с5н4ы
"видом пиразолкарбоновой-3 (или 5) кислоты и этилформиатом участвует
гетероциклический азот [319].
Реакция Гофмана с амидами N-ацил-Р-арил-Р-аланинов. Гипобромит
щелочного металла превращает амид N-ацил-Р-фенил-Р-аланина в 5-фенил-
1,3,4-оксадиазолон-2(ЗН), который идентичен продукту реакции гипобромита
HN — N
CgH5CHCH2CONH2 NaOBr / \ NaOBr
I ---> О=с СС6Н5 ----------CeH5CONHCONH2
NHCOR \ /
О
I II
с бензоилмочевиной [320]; авторы [320] высказывают предположение о про-
межуточных продуктах реакции. Соединение II также было получено по реак-
ции Гофмана из соединения III [320].
I
“C6H5CHCH2NCO
NIHCOR
С6Н5СН-СН2
RCON ''nH
С6НЬСН—СН2
-» C6H5CONHCONH2 —> C6H5CONHNCO -> II
N-ХлормочевинЫ, полученные при действии хлора на бензоил- и изони-
котиноилмочевины, дают со щелочью соответствующие оксадиазолоны [324].
HN-N
он- / \
ArCONHCONHCl----> О=С САг
Окисление гидразонов. Щелочные растворы феррицианида калия или
изоамилнитрита способны превращать бензоилгидразон бензальдегида
1,3,4-ОКСАДИАЗОЛЫ
403
в 2,5-дифенил-1,3,4-оксадиазол [322]. Продукт, полученный при действии
на семикарбазон бензальдегида гипоиодита или гипобромита натрия был
идентифицирован как 2-амино-5-фенил-1,3,4-оксадиазол [323]. В каждой
из этих двух реакций первоначальное окисление или галогенирование, по-види-
мому, предпочтительнее идет по атому углерода, а не азота.
CeH5CH=NNHCOCeH5
N-N
CeH5CH = NNHCONH2 ->
cnh2
N-NH
HC(OC2H6)
—------->
( y— CONH2
Ацилтиосемикарбазиды. В присутствии окиси свинца или окиси ртути
некоторые ацилтиосемикарбазиды, отщепляя элементы сероводорода, цикли-
зуются в 1,3,4-оксадиазолы [324]. Аналогичное замыкание цикла наблю-
дается и в присутствии едкого кали [325]. Циклизация 1-карбэтокси-4-фенил-
N—N
(C6H5)2CHCONHNUCSNH2
P b$O4
------H2NC CCII(CcH5)2
hconhnhcsnh2
PbO
20%-ное
KOH
C6H5NHNHCONHNHCSNHCeH5----------
тиосемикарбазида, протекающая в концентрированной соляной кислоте,
приводит к 2-этокси-1,3,4-оксадиазолтиолу-5 [326].
N —N
C6H5NHCSNHNHCOOC2H5 ->
HSC СОС2Н5
S-Метил производное 1-бензоилтиомочевины при пиролизе теряет метил-
меркаптан и превращается в 2-амино-5-фенил-1,3,4-оксадиазол [327].
N —N
-CH3SH
CeH5CONHNHC(=NH)SCH3 -------i
CeH5C CNH2
26*
404
ОКСАДИАЗОЛЫ
Реакция гидразидов карбоновых кислот с сероуглеродом. В 1952 г. Хог-
гарт наблюдал образование 2-замещенных 1,3,4-оксадиазолтиолов-5 при
нагревании калиевых солей 3-ароилдитиокарбазатов [328]. Аналогичные
результаты получены при нагревании смеси соответствующих гидразидов,
CS2 , Нагревание
rcon2h3 —гrconhnhcs2k----------------->
или
N —NH
сероуглерода и спиртового раствора щелочи (без выделения промежуточной
соли) [329, 330]. В качестве заместителей в положение 2 могут быть введены
ароматические, гетероциклические или алкильные группы [329—333]. Дан-
ные инфракрасных спектров указывают на наличие тионового таутомера
оксадиазолтиолов [330]. Циклизация карбазиновых эфиров [329] и амидов
[332] под действием кислот ведет к образованию 1,3,4-тиадиазолов [333].
Дигидро-1,3,4-оксадиазолы. Обработка серебряной соли бензоилгидразона
бензальдегида хлористым ацетилом или бензоилом в инертном растворителе
дает 2,5-дифенил-4,5-дигидро-4-ацил-1,3,4-оксадиазол [334]. Аналогичный
продукт получен при реакции между иодом и ртутной солью бензофенон-
N —NCOR
RCOC1 \
C6H5CH = NNHCOC6H5--------> С6Н6С СНС6Н5
(серебряная соль) \ /
бензоилгидразинового комплекса или между
ряной солью этого же комплекса [335].
хлористым бензоилом и сереб-
(СЙН5)2СО С(1Н5СОЖН3
N —NCOCeH5
(ртутная соль)
(CeH5)2CO-C0H5CON2H3 geHsCOCi
(серебряная соль)
С(СвН5)2
Диазотированный о-хлоранилин, взаимодействуя с хлормалоновой кис-
лотой в щелочной среде, дает 2,3-дигидро-2-кето-3-(о-хлорфенил)-1,3,4-окса-
диазол [336].
N—NCeH4CI
CtCH(COOH)2 У \
o-C1C6H4N2OH ----------> НС с = о
При реакции формилфенилгидразина с анилом фосгена в хлороформе
образуется анил 2,3-дигидро-2-кето-3-фенил-1,3,4-оксадиазола [337].
C6H5NHNHCHO + C6H5N = CCI2 —» CeH5N = C
С6Н5—N—N
/ ч.
” ~ сн
При обработке изоникотиноилгидразона ацетона уксусным ангидридом
получен 2,2-диметил-3-ацетил-5-(4-пиридил)-1,3,4-оксадиазолин [305]. Пред-
О
1,3,4-ОКСАДИАЗОЛ Ы
405
полагают, что реакция протекает следующим образом:
Y-C5H4NCONHN=С(СН3)2
(СН8СО)2О
-------> rC6H4NCONHNC(OCOCH3)(CH3)2
I
СОСН3
у* C5H4NC(OH) = NNC(OCOCH3)(CH3)2~
COCH3
CH3CON —N
(CH3)2C
CC5H4N
Другие методы синтеза. Из фенилтетразола и д-нитробензоилхлорида
в пиридине получен 2-(д-нитрофенил)-5-фенил-1,3,4-оксадиазол [338].
C(iH;-,C N
N Nil
X /
N
o2nc6h4c сс6н5
N —N
2-Тио-3-фенил-5-метил-1,3,4-оксадиазолон-2 при действии окиси ртути
превращается в соответствующее кетопроизводное [316].
С6Н5 —N —N
/ \
s = c ссн3
HgO
c6h5-n-n
/ \
о = с ссн3
Реакции с разрывом кольца
Многие 1,3,4-оксадиазолы легко расщепляются щелочью с образованием
производных гидразина [321 ]. В более мягких условиях, применяя гидрат окиси
бария [323, 336], углекислый натрий [316], гидразин в метаноле [312], анилин
[312, 321], спиртовый раствор аммиака [313] или едкое кали [339], удается
выделить гидразиды. В более жестких условиях при действии этилата натрия
[316] или едкого кали [304, 316] образуется гидразин или его алкильное или
арильное производное.
Расщепление кислотами встречается реже. При продолжительной обра-
ботке концентрированной соляной кислотой диалкил-1,3,4-триазолов проис-
ходит расщепление гетероциклического кольца [298]; в разбавленной серной
кислоте 2-амино-5-фенил-1,3,4-оксадиазол расщепляется с образованием гид-
разина и бензойной кислоты [323]. Нитрование 2-метил-4-фенил-А2-1,3,4-
CeH5COOH-l- N2H4
H2SO4
-----
C6H5-N-N
/ \
О-С ссн3
Ва(ОН)2
------> C6H5CONHNHCONH2
CeH5N2H3
C2H5ONa
или кон
Na2CO3
----- C6H5NHNHCOCH3
406
ОКСАДИАЗОЛЫ
N-N
НОС С — C5H4N-y
N2H4
y-C5H4NCONHNHCONHNH2
О
HN-N
о=с ссн2снс6н13
NH3
NHCOCH3
О
/j-O2NC6H4-N-N
C6H13CHCH2CONHNHCON112
r!lHCOCH3
О^С ССНЯ
KO I г
n-O2NC6II4NHNHCOCH3
CHgC CCIIg
NaOH
---------> N2H4-|-CH3COOH
или кислота
о
оксадиазолона-5 протекает по
рецикла [339.1.
При термической изомеризации соединения II с образованием соедине-
ния IV в качестве промежуточного продукта образуется, по-видимому, веще-
ство III [330]. Дигидро-1,3,4-оксадиазолы с ацильной группой в положении 3
легко гидролизуются разбавленной кислотой [322, 334, 335].
бензольному кольцу без расщепления гете-
HN-N
CeH5 —N —N
о=с ссн
hno3
n-O2NCeH4 — N — N
o=c
CCH3
\ z \ Нагревание
/N-------------
N
Н
II
CONHNCS
NH
Hl
N-N
NH
SH
IV
s = c
О
О
N —N
о
о
1
о
Восстановительное расщепление кольца 1,3,4-оксадиазола приводит
к образованию амидов. Обработка 2-(|3-бензамидоэтил)-А2-1,3,4-оксадиазолин-
тиона-5 и 2-фенил-А2-1,3,4-оксадиазолинтиона-5 избытком никеля Ренея
дает соответственно Р-бензамидопропионамид и бензамид [328, 340]. При этом
HN-N
CeH5CH —NNHCOR
Разб. HCI
25°
2 суток
CeH5CH
CR
Нагревание
------- с6н5сно + n2h4
Раэб. НС1
О
R = CH3r CeH5
N — N — СОС6Н5
/ \ н3о+ •
С(С6Н5)2 > (CeH5)2CO + (CeH5CONH)2
CeH5C
О
i,3,4-ОКСАДИАЗОЛЫ
407
был выделен промежуточный продукт — 2-замещенный 1,3,4-оксадиазол [332,
340]. В аналогичной реакции из 2-метилмеркапто-5-фенил-1,3,4-оксадиаэола
образуется бензамид [340]. При восстановлении соединения I оловом в соля-
ной кислоте получено некоторое количество п-фенилендиамина [339].
HN-N
Ni-Ренея
N-N
Ni-Ренея
-------> rconh2
R = C6H5CONHCH2CH2, Свн5, y-C5H4N
n-H2NCeH4 —N — N
/ V
O = C CCH3
hH2NCeH4NH2
Реакции без разрыва -кольца
Обработка соединения V (R — y-C5H4N) иодистым метилом в щелочи дает
S-метильное производное, которое при окислении превращается в соответ-
ствующий сульфон и оксадиазолон [332]. Последний может быть также полу-
чен окислением соединения V (R = y:C5H4N) щелочным раствором перекиси
водорода [332]. Под действием окиси ртути оксадиазолинтионы также пре-
вращаются в соответствующие оксадиазолоны [316]. Оксадиазолон (VI) всту-
пает в реакцию с пятихлористым фосфором с образованием соответствующего
геж-дихлорида (VII) [341]. Цинк, в спиртовом растворе соляной кислоты или
N-N
СН31 \ КМпОд
V(R = Y-C5H4N) ——> CH3SC CC5H4N--------->
in a Oil /
O
N —N HN-N
cn3so2cf \:c5h4n+o=c/ ^cc3h4n
металлический натрий в спирте превращают дихлорид в соответствующий
СВН5 — N — N CeH5 —N —N ' CeH5-N-N
/ \ HgO / \ PC's / Ч.
S=C ' ССН3 —О-С ССН3 —С12С ССН3
\ / \ / 150°3 • \ /
ООО
VIII VI VII
оксадиазолин. При действии воды на дихлорид регенерируется соединение VI.
Аммиак дает соответствующий имин, а сернистый аммоний — тион (VI11) [341 ].
С6Н5 —N —N
HN^cf **ССН3
NH3
<-----VII
150°
Zn
нсГ*
C6H5-N-N
HiX £СН3
N-Бензоильное производное 1,3,4-оксадиазолона-5 (синтезированного
из гидразида муравьиной кислоты и фосгена) получается из незамещенного
408
ОКСАДИАЗОЛЫ
оксадиазолона [310], однако диметилкарбамилхлорид ацилирует 2-метиль-
ный гомолог по атому кислорода [342]. Алкилоксадиазолоны (IX) в реакции
Манниха алкилируются по атому азота [343].
Ацилирование [323, 324, 344] и нитрозирование [324] 2-амино-1,3,4-
оксадиазолов протекает нормально. При восстановлении 2-нитрозамино-5-
N-N
(CII3)2NCOOC ССН3 Т-— О = с CR
\\ / K--GH3 /
Ъ о
С6Н5СО —N — N
/ \
о=с сн
фенил-1,3,4-оксадиазола образуется производное гидразина, которое под
действием азотистой кислоты превращается в соответствующий азидоокса-
диазол [324]. Окисление 2-амино-5-фенил-1,3,4-оксадиазола гипохлоритом
HOCH2N —N
о=с/ *CR
СН2О
IX
сн2о
R'NH
r;nch2 — n—n
/ Ч
O=C CR
приводит к получению азопроизводного, которое может быть восстановлено
в соответствующее гидр азосоединение [324].
N—N
RC CNHSO2Ar
N-N
N-N
.4 ч
RC CNHCOCH3
H5C6C CNHNO
О
О
(C8H5N2O)N = N(C8H5N2O)
г (NH<)2Sx
(C8H5N2O)NHNH(CsH5N2O)
О
Zn, H2O
N-N
H5C6C
N2H3
hno2
N-N
H5CeC
CN3
О
О
При обработке 2,5-дитолил-1,3,4-оксадиазолов азотнокислым серебром
получаются твердые производные, которые взрывают [304].
Перекись (X) при обработке серной кислотой и далее водой превращается
в 2-фенил-5-бензоил-1,3,4-оксадиазол, который с помощью амальгамы цинка
1,3,4-ОКСАДИАЗОЛЫ
409
может быть восстановлен в миндальную и бензойную кислоты [345]. Одним
из методов получения перекиси (X) является реакция замены атома хлора
, N—N N—N
/ \ \ (D HiSO4 \
с6н5с СС(С6Н5)2О 72;-^х- с6н6с ссос6н5
\ О /2
X
в 2-дифенилхлорметил-5-фенил-1,3,4-оксадиазоле (XI, R = С1) на кислород.
Этот процесс, в котором, по-видимому, образуются свободные радикалы,
катализируется ртутью [345].
N —N
7 \ о2
С6Н5С CC(C6H5)2R —X
о
XI
Сообщается об аналогичной реакции соединения XI (R = Н). Окисление
соединения XI (R = Н) бихроматом дает карбинол (XI, R = ОН), а броми-
рование — бромид (XI, R = Вг), который в свою очередь действием щелочи
превращается в карбинол (XI, R = ОН), а действием этилата — в соответ-
ствующий этиловый эфир (XI, R = ОС2Н5) [301].
Физические свойства и применение
Для некоторых производных 1,3,4-оксадиазола имеются данные по тепло-
там сгорания [346], дипольным моментам [347], поглощению в ультрафиоле-
товой области спектра [348, 349], а также данные по поглощению в видимой
[349] и инфракрасной [330] области спектра и рентгеноструктурные данные
[350]. Исследование [330] полос поглощения для 2-метил-Д2-1,3,4-оксадиазо-
линтиона-5 в области ниже 8ц при различных концентрациях указывает
на наличие равновесия мономер — димер с константой диссоциации в хлоро-
форме, равной 0,088 при 27°. Димеру приписывается следующее строение [330]:
О
НзСсГ 'c=S-.H-N —N
II I I II
N—N-H-..S = C ССН3
"с/
Некоторые антрахиноновые кубовые красители являются производными
1,3,4-оксадиазола [351].
Фенилбифенилилоксадиазол применялся в сцинцилляционных счетчиках
при изучении распада В10 [352].
Биологические свойства
Описана антитуберкулезная активность , 2-(4-пиридил)-А2-1,3,4-окса-
диазолона-5 [321, 353]. Соответствующее 3-пиридильное соединение неак-
тивно [321].
HN — N
° = С/ Z
\ / 4---------
О
410
ОКСА ДИ АЗОЛЫ
ЛИТЕРАТУРА
1. V. Auwers, Meyer, Ber., 21, 784 (1888); Dodge, Ann., 264, 178 (1890).
2. Wolff, Ber., 28, 69 (1895).
3. В a n к s, A d a m s, J. Org. Chem., 21, 815 (1950).
4. Zincke, Ossenbeck, Ann., 307, 1 (1899); Ponzi o, Bertini, Gazz. chim.
ital., 61, 51 (1931).
5. В e h г, В r e n t, Org. Syntheses, 34, 40 (1954).
6. P о n z i o, Gazz. chim. ital., 62 , 854, 1025 (1932).
7. Durio, Sb urlato, Gazz. chim. ital,, 62, 1035 (1932).
8. Gunther, Ann., 252, 44 (1889).
9. M a 1 a v i у a, Dutt, Proc. Acad. Sci. United Provinces Agra Oudh, India, 4, 319
(1935) [C. A., 30, 1056 (1936)].
10. v. A u we r s, Me у er, Ber., 22, 705 (1889).
11. Malagnini, Gazz. chim. ital., 24, II, 1 (1894); В о e r i s, ibid,, 23, II, 165 (1893).
12. Русанов, Ber., 24, 3497 (1891).
13. Schmidt, Ber., 28, 1206 (1895).
14. P о n z i o, Gazz. chim. ital., 61, 704 (1931).
15. Boyer, Hernandez, неопубликованные данные.
16 Ingold, Shoppe e, J. Chem. Soc., 1928, 396.
17. Hammick, Edwards, Steiner, J. Chem. Soc., 1931, 3308.
18. Z i n с к e, S c h w a r z, Ann., 307, 28 (1899); Green, Rowe, J. Chem. Soc., 103,
897 (1913).
19. P о n z i o, Gazz. chim. ital., 36, II, 313 (1906).
20. Brom-me, Ber., 21, 391 (1888).
21. Nietzki, Knapp, Ber., 30, 1119 (1897).
22. Goldschmidt, Ber., 22, 3101 (1889).
23. v. Auwers, Meyer, Ber., 22, 1985 (1889); G о 1 d s c ii m i d t, Ber., 16, 2176
(1883); 17, 213, 801 (1884).
24. Goldschmidt, Schmid, Ber., 17, 2060 (1884).
25. H a n t z s c h, G 1 о v e r, Ber., 39, 4153 (1906); 40, 4344 (1907); S c h ni i d t, S о 1 !,
Ber., 40, 2454 (1907); 41, 3679 (1908); Schmidt, Mezger, Ber., 40, 4560 (1907).
26. Borsche, Weber, Ann., 489, 270 (1931).
27. Ильинский, Ber., 19, 340 (1886); Harden, Ann., 255, 148 (1889).
28. G r e e n, R о w e, J. Chem. Soc., Ill, 612 (1917).
29. C u s m a n о, T i b e r i o, Gazz. chim. ital., 81, 106 (1951).
30. Angelico, C u s m a n o, Gazz. chim. ital,, 66, 3 (1936).
31. A j e 1 1 о, C u s m a n o, Gazz. chim. ital., 68, 792 (1938); C u s m a n o, ibid., 78,
622 (1948).
32. P о n z i o, Gazz. chim. ital., 61, 138 (1931).
33. A j e 1 1 о, P e t г о n i c i, Gazz. chim. ital., 72, 333 (1942).
34. Wieland, Ki t a s a to, Utzino, Ann., 478, 43 (1930). .
35. Гетероциклические соединения, т. 5, ИЛ, Москва, 1959.
36. Q u i 1 i с о, F г е г i, Gazz. chim. ital., 60, 721 (1930).
37. Guha, Chakladar, Proc. 15th Indian Sci. Cong., 1928, 157 [C. A., 25, 2999
(1931)].
38. Dewar, J. Chem. Soc., 1944, 619.
39. Boyer, Toggweiler, J. Am. Cheni. Soc., 79, 895 (1957).
40. К о r e f f, Ber., 19, 176 (1886).
41. P о n z i o, Gazz. chim. ital., 36, II, 103 (1906).
42. Beckmann, Ber., 22, 1501, 1588 (1889); C i u s a, P a r i s i, Gazz. chim. ital.,
55,416 (1925).
43. В a n k s, R i c h a r d s, Taianta, 2, 235 (1958).
44. Meisenheimer, Lange, La m par ter, Ann., 444, 94 (1925).
45. P о n z i о, T о r r e s, Gazz. chim. ital., 59, 46 (1929); P о n z i o, ibid., 59, 713 (1929).
46. В о r s c h e, Ber., 40, 737 (1907).
47. Scholl, Ber., 21, 506 (1888); 23, 3490 (1890).
48. P о n z i o, Gazz. chim. ital., 66, 119 (1936); M i 1 о n e, ibid., 59, 267 (1929).
49. W i e 1 a n d, S e m p e r, Ann., 307, 49, 329 (1903).
50. Dorn о w, Fust, Jordan, Chem. Ber., 90, 2124 (1957).
51. Forster, J. Chem. Soc.. 83, 514 (1903).
52. W e r n e г, В u s s, 27, 2197 (1894); Wieland, Ber., 40, 1670 (1907).
53. P о n z i o, Gazz. chim. ital., 36, II, 288, 591 (1906).
54. Чугаев, Спиро, Ber., 41, 2219 (1908).
55. Q u i 1 i с о, P a n i z z i, Gazz. chim. ital., 72, 155 (1942).
56. M i n u n n i, C i u s a, Atti accad. Lincei, 14, II, 518 (1905).-
57. C i u s a, P a r i s i, Gazz. chim. ital., 53, 143 (1923); F r a n -z e n, Z i m m e г ni a n n,
J. prakt. chem., [2], 73, 255 (1907).
58. Boyer, A 1 u 1, J. Am. Chem. Soc., 81, 4237 (1959). В
ЛИТЕРАТУРА
411
59. Р о п z i о, Gazz. chim. ital., 39, I, 324 (1909).
60. В о u g a u 1 t, Bull. soc. chim. France, [4] 25, 384 (1919).
61. P о n z i o, Gazz. chim. ital., 33, I, 508 (1903).
62. Holleman, Rec. trav. chim., 6, 60 (1886).
63. Holleman, Ber., 20, 3359 (1887).
64. В о у e r, C h a n g, J. Am. Chem. Soc., 82, 2220 (1960).
65. Alexander, Kinter, McCollum, J. Am. Chem. Soc., 72, 801 (1950).
66. W i d m a n, Virgin, Ber., 42, 2798 (1909).
67. P о n z i o, Gazz. chim. ital., 62, 415, 424 (1932).
68. P о n z i o, Gazz. chim. ital., 56, 490 (1926); 62, 633 (1932); Ponzi o, Durio,
ibid., 61, 589 (1931).
69. Snyder, Boyer, J. Am. Chem. Soc., 77, 4233 (1955).
70. Wieland, H о c h t 1 e n, Ann., 505, 237 (1933).
71. Hayes, O’K e e f e, J. Org. Chem., 19, 1897 (1954).
72. S h i r 1 e y, G г о s s, D a n z i g, J. Org. Chem., 23, 1024 (1958).
73. Quil ico, Gazz. chim. ital., 61, 265 (1931); Q u i 1 i c o, S i m о n e t t a, ibid.,
77, 586 (1947).
74. Ungnade, Kissinger, J. Org. Chem., 24, 666 (1959).
75. Mills, Chem. News, 88, 228 (1903).
76. W i e 1 a n d et al., Ann., 444, 7 (1925).
77. Wieland, Ber., 42, 803 (1909).
78. Wieland, Bauer, Ber., 40, 1667 (1907).
79. Wieland, Ber., 42, 814 (1909).
80. Wieland, Ber., 42, 4207 (1909).
81. Werner, Skiba, Ber., 32, 1654 (1899).
82. Werner, Ber., 27, 2846 (1894).
83. В r a d у, P e a к i n, J. Chem. Soc., 1929, 2267.
84. S c h о 1 1, S c h о f e r, Ber., 34, 876 (1901).
85. Quilico, Parjzzi, Gazz. chim. ital., 72, 458 (1942).
86. Kissinger, Ungnade, J. Org. Chem., 23, 1340 (1958).
87. Hurd, Nilson, W i к h о 1 m, J. Am. Chem. Soc., 72, 4697 (1950).
88. Jovitschitsch, Ber., 28, 1213 (1895).
89. Kornblum, Weaver, J. Am. Chem. Soc., 80, 4333 (1958).
90. Carbone, Gazz. chim. ital., 62, 428 (1932).
91. Wieland, Rosenfeld, Ann., 484, 236 (1930).
92, Birkenbach, Sennewald, Ann., 489, 7 (1931).
93. S e h e r, Chem. Ber., 83, 400 (1950).
94. Muller, Metzger, Chem. Ber., 87, 1282 (1954).
95. Wieland. Bauer, Ber., 39, 1480 (1906).
96. Wieland, Ann., 329, 225, 239 (1903).
97. Levy, S c a i f e, J. Chem. Soc., 1946, 1100.
98. Angeli, Ber., 24, 3994 (1891).
99. Wieland, Ann., 329, 267 (1903).
100. Toennies, Ber., 13, 1846 (1880).
101. Wieland, Ann., 328, 206 (1903).
102. Wieland, Ann., 424, 107 (1921).
103. Wieland, Ber., 42, 4192 (1909); Sell, Biedermann, Ber., 5, 89 (1872);
Birckenbach, Sennewald, Ann., 489, 15 (1931); К e к u 1 e, Ann., 105,
279 (1858); D e P а о 1 i n i, Gazz. chim. ital., 60, 700 (1930); Birckenbach,
Sennewald, Ber., 65, 546 (1932).
104. Ponzi o, Cerrina, Gazz. chim. ital., 58, 34 (1928).
105. Титов, Русанов, ДАН СССР, 82, 65 (1952).
106. Е m mo ns, F r e e m a n, J. Org. Chem., 22, 456 (1957).
107. Zincke, Schwarz, Ann., 307, 28 (1899).
108. Noelting, Kohn, Chem. Ztg., 18, 1095 (1894).
109. Smith, Boyer, Org. Syntheses, 31, 14 (1951).
110. Fagley, Sutter, Oglukian, J. Am. Chem. Soc., 78, 5567 (1956).
111. Smith, Brown, J. Am. Chem. Soc., 73, 2435 (1951).
112. Boyer et al., J. Am. Chem. Soc., 75, 5298 (1953).
113. Boyer, Mil ler, J. Am. Chem. Soc., 81, 4671 (1959).
114. Mallory, Org. Syntheses, 37, 1 (1957).
115. G r e e n, R о w e, J. Chem. Soc., 101, 2452 (1912); 103, 897, 2023 (1913).
116. Gaughran, Picard, Kaufman, J. Am. Chem. Soc., 76, 2233 (1954).
117. Pausacker, Scroggie, J. Chem. Soc., 1954, 4499; Barlin, Pausacker,
Riggs, ibid., 1954, 3122.
118. В о у e r, M a m i к u n i a n, J. Org. Chem., 23, 1807 (1958).
119. Boyer, Toggweiler, Stoner, J. Am. Chem. Soc., 79, 1748 (1957).
120. Snyder, Boyer, J. Am. Chem. Soc., 77, 4233 (1955).
121. Богданов, Горелик, Хим. наука и пром., 3, 407 (Ю58).
412
ОКСАДИАЗОЛЫ
122. В о у е г, Е 1 1 z е у, J. Org. Chem., 24, 2038 (1959) и неопубликованная работа.
123. Kinney, J. Am. Chem. Soc., 51, 1592 (1929).
124. D г о s t, Ann., 307, 49 (1899).
125. Borsch e, Weber, Ann., 489, 270 (1931).
126. Wieland, Semper, Ann., 358, 36 (1907).
127. Po nzio, Gazz. chim. ital., 59, 713 (1929); Ponzi o, Carta-Satta, ibid.,
60, 150 (1930).
128. P о n z i o, Gazz. chim. ital., 61, 943 (1931).
129. Boyer, неопубликованные данные.
130. A n g e 1 i, Ber., 25, 1960 (1892).
131. P а о 1 i n i, A r m i t a n o, Gazz. chim. ital., 63, 917 (1933).
132. Holleman, Rec. trav. chim., 10, 211 (1890).
133. Ponzi o, Biglietti, Gazz. chim. ital., 63, 159 (1933).
134. P о n z i o, Gazz. chim. ital., 61, 943 (1931).
135. В i g i a v i, Gazz. chim. ital., 51, II, 324 (1922); Kinney, Harwood, J. Am
Chem. Soc., 49, 514 (1927).
136. Boyer, Buriks, Toggweiler, J. Am. Chem. Soc., 82, 2213 (1960).
137. Boyer, Schoen, J. Am. Chem. Soc., 78, 423 (1956).
138. Forster, Barker, J. Chem. Soc., 103, 1918 (1913).
139. F r i e s, N о 1 1, Ann., 389, 367 (1912).
140. Boyer et al., J. Am. Chem. Soc., 77, 5688 (1955).
141. Богданов, Королева, ЖОХ, 26, 259, 281 (1956).
142. D e P а о 1 i n i, Gazz. chim. ital., 57, 656 (1927).
143. V i a n e 1 1 o, Gazz. chim. ital., 62, 131 (1932).
144. Gabriel, Корре, Ber., 19, 1145 (1886).
145. Scholl, Brennei se n, Ber., 31, 642 (1898).
146. Cusmano, Giambrone, Gazz. chim. ital., 81, 499 (1951).
147. Schofield, Quart. Revs. (London), .4, 388 (1950).
148. A j e 1 1 o, Gazz. chim. ital., 67, 779 (1937).
149. D e P а о 1 i n i, Gazz. chim. ital., 57, 656 (1927).
150. P о n z i o, Gazz. chim. ital., 62, 415, 860 (1932).
151. Meisenheimer, Theilacker, Ann., 469, 128 (1928).
152. Ponzi o, Torres, Gazz. chim. ital., 59, 461 (1929).
153. P а о 1 i n i, Gazz. chim. ital., 63, 917 (1933).
154. P о n z i o, Gazz. chim. ital., 62, 127 (1932).
155. Quist, Acta Acad. Aboensis Math, et Phys., 5, № 2 (1928) (Chem. Zentr., 1929, 1,892).
156. R u g g e r i, Gazz. chim. ital., 55, 72 (1925) (Chem. Zentr., 1925, I, 2071).
157. Boeseken, Ross, Lennep, Rec. trav. chim., 31, 200 (1912).
158. Wieland, Semper, Gmelin, Ann., 367, 52 (1909).
159. Wieland, Frank, Kitasato, Ann.,' 475, 42, 1929; Wieland, К i t a-
s a t o, From, Ann., 475, 54 (1929).
160. P о n z i o, Gazz. chim. ital., 66, 819 (1936).
161. R u g g 1 i, В u c h ni e i e r, Helv. Chim. Acta, 28, 850 (1945).
162. A n g e 1 i, Gazz. chim. ital., 46, II, 300 (1916).
163. Lane, Williams, J. Chem. Soc., 1955. 1468.
164. В a k e г, О 1 1 i s, P о о 1 e, J. Chem. Soc., 1950, 1546.
165. Ponzi o, Rugger i, Gazz. chim. ital., 56, 733 (1926).
166. Bailey, Case, Tetrahedron, 3, 113 (1958).
167. Milon e, Borello, Gazz. chim. ital., 81, 368 (1951).
168. M i 1 о n e, Gazz. chim. ital., 61, 153 (1931).
169. Milon e, Borello, Gazz. chim. ital., 81, 677 (1951); M i 1 о n e, Borello,
N о c i 1 1 a, ibid., 81, 896 (1951).
170. Acly, French, J. Am. Chem. Soc., 49, 847 (1927).
171. В о у e r et al., J. Am. Chem. Soc., 77, 4238.(1955).
172. Boyer, Toggweiler, Stoner, J. Am. Chem. Soc., 79, 1748 (1957).
173. Tapp i, Gazz. chim. ital., 71, 111 (1941).
174. F a g 1 e у, частное сообщение.
175. Werner, Lehrbuch der Stereochemie, Gustav Fischer, Jena, 1904, p. 260.
176. Forster, F ierz, J. Chem. Soc., 103, 1918 (1913).
177. P о n z i o, Gazz. chim. ital., 66, 114, 119, 127, 134 (1936).
178. Borello, Colombo, Ann. chim. (Rome), 46, 1158 (1956); 47, 649 (1957)
179. E m m о n s, J. Am. Chem. Soc., 79, 5739 (1957).
180. P о n z i o, Gazz. chim. ital., 55, 698 (1925).
181. Yang, Johnson, J. Am. Chem. Soc., 54, 2066 (1932).
182. P о n z i o, Gazz. chim. ital., 57, 117 (1927).
183. G a s t a 1 d i, Gazz. chim. ital., 55, 201 (1925).
184. P о n z i o, Gazz. chim. ital., 53, 507 (1923).
185. Johnson, Menge, Am. Chem. J., 32, 362 (1904).
186. F a 1 c k, Ber., 18, 2467 (1885).
ЛИТЕРАТУРА
413
187. Adams, Kaiser, Peters, J. Org Chem., 18, 934 (1953).
488. M u s a n t e, Gazz. chim. ital., 68, 331 (1938).
189. F a 1 c k, Ber., 19, 1481 (1886).
190. M i 1 о n e, Gazz. chim. ital., 59, 266 (1929).
191. R о s s, К u n t z, J. Am. Chem. Soc., 74, 1297 (1952).
192. Pilot y, Steinbock, Ber., 35, 3101 (1902).
193. Virgin, Diss. Univ. Uppsala, (1914) [C. A., 14, 1319 (1920)].
194. Ponzi o, Bernardi, Gazz. chim. ital., 55, 70 (1925).
195. Behrend, Tryller, Ann., 283, 209 (1894).
196. Behrend, Steffens, Ann., 309, 241 (1899).
197. T u г e к, англ. пат. 298981 [С. A., 23, 3101 (1929)].
198. В о у e г, В u r i к s, J. Am. Chem. Soc., 82, 2216 (1960).
199. Forster, Fieri, J. Chem. Soc., 91, 1942 (1907).
200. Having a, Schors, Rec. trav. chim., 69, 457 (1950).
201. Rowe, Davies, J. Chem. Soc., 117, 1344 (1920).
202. Leandr i, Rebora, Ann. chim. (Rome), 46, 953 (1956).
203. Schulz, Ber., 18, 1081 (1885).
204. Tieman n, Kruger, Ber., 17, 1685 (1884).
205. Krummel, Ber., 28, 2231 (1895).
206. Stieglitz, Ber., 22, 3158 (1889).
207. Clarke, J. Chem. Soc., 1954, 4251.
208. С 1 e m m, Ber., 24, 833 (1891).
209. E к s t r a n d, Ber., 20, 226 (1887).
210. Richter, Ber., 22, 2456 (1889).
211. Miller, Ber., 22, 2793 (1889).
212. Oppenheimer, Ber., 22, 2445 (1889).
213. Knudsen, Ber., 18, 1071 (1885).
214. Gross, Ber., 18, 1076 (1885).
215. Wolff, Ber., 19, 1509 (1886).
216. Gramantieri, Gazz. chim. ital., 65, 102 (1935).
217. S c h u b a r t, Ber., 22, 2437 (1889).
218. Leandr i, Maioli, Ruzzier, Boll. sci. fac. chim. ind. Bologna, 15, № 3,
57 (1957) [C. A., 52, 7291 (1958)].
219. Buu-Hoi, Lecocq, Bull. soc. chim. France, 1946, 139.
220. Bergmann, Bendas, D’A v i 1 1 a, J. Org. Chem., 18, 64 (1953).
221. Wieland, Bauer, Ber., 40, 1667 (1907).
222. Adams, Kaiser^ Peters, J. Org. Chem., 18, 934 (1953).
223. Merckx, Bull. soc. chim. Beiges, 56, 339 (1947); 58, 58 (1949).
224. N о r d m a n n, Ber., 17, 2754 (1884).
225. M о d e e n, Ber., 27, 261 Ref. (1894).
226. Longo, Gazz. chim. ital., 61, 575 (1931).
227. Beckmann, Sandel, Ann., 296, 285 (1897). .
228. Yang, Johnson, J. Am. Chem. Soc., 54, 2066 (1932).
229. M u r r a y, D a i n s, J. Am. Chem. Soc., 56, 144 (1934).
230. Пат. США 2648669 [С. A., 47, 11258 (1953)].
231. T i e m а п п, Ber., 22, 2412 (1889).
232. Richter, Ber., 22, 2456 (1889).
233. Weise, Ber., 22, 2424 (1889).
234. Zimmer, Ber., 22, 3140 (1889).
235. S p i 1 к e r, Ber., 22, 2781 (1889).
236. Conley, Mikulski, J. Org. Chem., 24, 97 (1959).
237. Gunther, Ber., 21, 517 (1888).
238. Durio, Sb u r 1 a to, Gazz. chim. ital., 62, 1035 (1932).
239. Beckmann, Koster, Ann., 274, 21 (1893).
240. P о n z i o, Gazz. chim. ital., 62, 854 (1932).
241. В о u g a u 1 t, Robin, Compz. rend., 169, 341 (1919); Ciusa, Parisi, Gazz.
chim. ital., 53, 667 (1923).
242. Wieland, Bauer, Ber., 39, 1486 (1907).
243. Wieland, Ber., 42, 814 (1909)?
244. Werner, Bloch, Ber., 32, 1975 (1899).
245. Leandr i, Paollotti, Ann. chim. (Rome), 47, 376 (1957).
246. Franzen, Zimmermann, J. prakt. chem., [2] 73, 255 (1907).
247. Bamberger, Scheutz, Ber., 34, 2023 (1901).
248. Beckmann, Ber., 22, 1589 (1889).
249. Ponzi o, Busti, Gazz. chim. ital., 36, II, 339 (1906).
250. P о n z i o, Gazz. chim. ital., 36, II, 288 (1906).
251. M u s a n t e, Gazz. chim. ital., 68, 331 (1938).
252. P о n z i o, Gazz. chim. ital., 62, 415 (1932).
253. Cusmano, Ruccia, Gazz. chim. ital., 85, 1686 (1955).
414
ОКСАДИАЗОЛЫ
254. Ajello, Cusman о, Gazz. chim. ital., 70, 127 (1940).
255. Lindemann, Cissee, J. prakt. chem., [2] 122, 232 (1929).
256. Stevens, Smith, Brown, частное сообщение.
257. Terss, McEwen, J. Am. Chem. Soc., 76, 580 (1954).
258. Arndt, Rose, J. Chem. Soc., 1935, 1.
259. Stetter, Hoehme, Ber., 91, 1123 (1958).
260. Katritzki, J. Chem. Soc., 1956, 2063.
261. Boyer, Borger s, Wolford, J. Am. Chem. Soc., 79, 678 (1957).
262. U t z i n ger, Regenass, Helv. Chim. Acta, 37, 1892 (1954).
263. Safi r, Lopresti, J. Am. Chem. Soc., 80, 4921 (1958).
264. Ponzi o, Avogadro, Gazz. chim. ital., 53, 318 (1923).
265. V i a n e 1 1 o, Gazz. chim. ital., 58, 327 (1928).
266. Wurm, Ber., 22, 3130 (1889).
267. Merckx, Chim. & ind. (Paris), 63, 453 (1952).
268. M i 1 о n e, Gazz. chim. ital., 62, 154 (1932).
269. M i 1 о n e, Gazz. chim. ital., 65, 152 (1935).
270. Milon e, Allaven a, Gazz. chim. ital., 61, 75 (1931).
271. H u i s g e n, Angew. Chem., 67, 439 (1955) (обзор химии алифатических диазосоеди-
нений).
272. Anderson, Ro e d e 1, J. Am. Chem. Soc., 67, 955 (1945).
273. Anderson, Le Fevre, Nature, 162, 443 (1948).
274. Hodgson, Marsden, J. Soc. Dyers Colourists, 59, 271 (1943).
275. Earl, Mackney, J. Chem. Soc., 1946, 591.
276. В a k e г, О i 1 i s, P о о 1 e, J. Chem. Soc., 1950, 1542.
277. Baker, Ollis, Quart. Revs. (London), 11, 15 (1957).
278. Katritzky, Chem. & Ind. (London), 1955, 521.
279. Earl, Chem. & Ind. (London), 1953, 746 , 1284.
280. Hammick, Roe, Vo a den, Chem. & Ind. (London), 1954, 251; Kruger,
ibid., 1954, 465.
281 .Baker, Ollis, Poole, J. Chem. Soc., 1949, 307; 1950, 1542.
282. Eade, Earl, J. Chem. Soc., 1946, 591.
283. Kenner, Mackay, Nature, 160, 465 (1947).
284. Fugger, Tien, Hunsberger, J. Am. Chem. Soc., 77, 1843 (1955).
285. Tien, Hunsberger, J. Am. Chem. Soc., 77, 6604 (1955).
286. Earl, Rec. trav. chim., 75, 346, 1080 (1956).
287. Z u m a n, Z. physik. Chem. (Leipzig), 1958, 243.
288. H am mick, Voaden, Chem. & Ind. (London), 1956, 739.
289. Earl, Le Fevre, Wilson, J. Chem. Soc., 1949, S103.
290. Earle et al., J. Chem. Soc., 1951, 2207.
291. Hill, Sutton, Longuet-Higgins, J. chim phys., 49, 244 (1949); О r g e 1
et al., Trans. Faraday Soc., 47, 113 (1951).
292. Brookes, Wilson, J. Chem. Soc., 1957, 4409.
293. Boyer, Canter, J. Am Chem. Soc., 77, 1280 (1955); Boyer, Hernandez,
ibid., 78, 5124 (1956).
294. Stolle, J. prakt. chem., [2] 68, 130 (.1903) (обзор более старых работ).
295. Р е 1 1 i z z а г i, Atti reale accad. Lincei, [5], 8, I, 328 (1898) (Chem. Centr., 1899,
I, 1240).
296. Rupe, Gebhardt, Ber., 32, 15 (1899).
297. S i 1 b e r r a d, J. Chem. Soc., 77, 1185 (1900).
298. Stolle Schatzlein, J. prakt. chem., [2] 69, 503 (1904); Stolle, Dell-
schaf f, ibid., [2] 69, 506 (1904); пат. США 2512631 (С. A., 44, 9734 (1950)].
299. Yale et al., J. Am. Chem. Soc., 76, 2208 (1954).
300. Герм. пат. 825111 [С. A., 49, 630 (1955)].
301. A s p e 1 u n d, Acta Acad. Aboensis Math. et Phys., 5, No 1, 1 [C. A. 24, 4031 (1930);
Stolle, Foerster, J. prakt. chem., [2] 69, 382 (1904); Stolle, Hille,
ibid., [2] 69, 481, (1904); Stolle, Johannissien, ibid., [2] 69, 476 (1904);
Stolle, Thoma, ibid., [2] 73, 289 (1906).
302. Klingsberg, J. Am. Chem. Soc., 80, 5788 (1958); Stolle, В a m b a c h,
J. prakt. chem., [2] 74, 22 (1906); пат. США 2601179 [С. A., 47, 2500 (1953)].
303. Герм. пат. 806936 [С. А., 46, 7918 (1952)]; Hayes, Rogers, Ott, J. Am. Chem.
Soc., 77, 1850 (1955).
304. Stolle, Ber., 32, 797 (1899); Stolle, Stevens, J. prakt. chem., [2] 69, 379
(1904); Stolle, Zinsser, ibid., [2] 69, 491 (1904).
305. Labr io la, F e 1 i t t e, C. A., 39, 1405 (1945); Stolle, Gutmann, J. prakt.
chem., [2] 69, 497 .(1904); Yale et al., J. Am. Chem. Soc., 75, 1933 (1953).
306. A s p e 1 u n d, Acta Acad. Aboensis Math, et Phys., 6, No 4 (1932) [C. A., 28, 5457
(1934)]; Stolle, W e i n d e 1, J. prakt. Chem., [2] 74, 2 (1906).
307. Пат. США 2616891 [С. A'., 47, 3575 (1953)].
308. Beckmann, Gunther, Ann., 252, 44 (1889).
Литература
415
309. Pinner, Ann., 297, 221 (1897); 298, 16 (1897); Pinner, Caro, Ber., 27, 3288
(1894).
310. Dornow, Bruncken, Chem. Ber., 82, 121 (1949).
311. Lieser, N i s c h k, Chem. Ber., 82, 527 (1949).
312. Англ. пат. 729891 [С. A., 50, 6514 (1956); пат. США 2665279 [С. А., 49, 2521 (1955)].
313. Родионов, Зворыкина, Изв. АН СССР, ОХН, 1953, 70.
314. Freund, Вег., 24, 4178 (1891).
315. Rube, Labhardt, Ber., 33, 233 (1900).
316. Freund, Goldsmith, Ber., 21, 1240, 2456 (1888).
317. Busch, Stern, J. prakt. chem., [2] 60, 235 (1899).
318. H о e g e r 1 e, Helv. Chim. Acta, 41, 548 (1958).
319. Ainsworth, J. Am. Chem. Soc., 77, 1148 (1955).
320. Родионов, Киселева, Изв. АН СССР, ОХН, 1951, 57; 1953, 513.
321. Stempel, Zelauska s, Aeschli шапп, J. Org. Chem., 20, 412 (1955).
322. Stoll e, Munch, J. prakt. chem., [2] 70.393 (1904).
323. Valenti, Maggio, Ann. chim. (Rome), 42, 18 (1952).
324. D e, J. Indian Chem. Soc., [2] 7, 651 (1930); Stoll e, Fehrenbach, J. prakt.
chem., [2] 122, 289 (1929); Stoll e, Gaertner, ibid., [2] 132, 209 (1932); пат.
США 2832787 [С. A. 52, 14703 (1958)].
325. G u h a, Hye, J. Indian Chem. Soc., 7, 933 (1930).
326. M a j u m d a r, G u h a, J. Indian Chem. Soc., 10, 685 (1933).
327. H о g g a r t h, J. Chem. Soc., 1949, 1918; 1950, 612, 1579.
328. Hoggarth, J. Chem. Soc., 1952, 4811.
329. Y о ung, Woo d, J. Am. Chem. Soc., 77, 400 (1955).
330. Ainsworth, J. Am. Chem. Soc., 78, 4475 (1956).
331. Konig, Seifken, О f f e, Chem. Ber., 87, 825 (1954).
332. Yoshida, Assai, J. Pharm. Soc. Japan, 74, 946 (1954).
333. Baron, Wilson, J. Org. Chem., 23, 1021 (1958).
334. S t о 1 1 e, J. prakt. chem., [2] 68, 421 (1903).
335. Stolid, J. prakt. chem., [2] 70, 419 (1904).
336. Fu sco, Romani, Gazz. chim. ital., 76, 419 (1946); 78, 342 (1948).
337. Freund, Konig, Ber., 26, 2869 (1893).
338. Huisgen, Sauer, Sturm, Angew. Chem., 70, 272 (1958).
339. Freund, Haase, Ber., 26, 1315 (1893).
340. Ainsworth, J. Am. Chem. Soc., 78, 1636 (1956).
341. Freund, Kuh, Ber., 23, 2835 (1890).
342. Швейц, пат. 281963 [С. A., 49, 5532 (1955)].
343. В urckh a Iter, J. Am. Pharm. Assoc., 47, 799 (1958).
344. Brooks et al., J. Chem. Soc., 1950, 452.
345. Aspelund, Ber., 63, 1191, 1352 (1930).
346. Milon e, Allaven a, Gazz. chim. ital., 61, 75 (1931).
347. M i 1 о n e, Gazz. chim. ital., 65, 152 (1935).
348. Milone, Gazz. chim. ital., 62, 154 (1932).
349. Grammaticakis, Bull. soc. chim. France, 1953, 86.
350. Pfeiffer, Anal. Chem., 28, 206 (1956).
351. Пат. США 2511018 [С. A., 46, 270 (1952)].
352. Bollinger et al., Proc. Intern. Conf. Peaceful Uses Atomic Energy, Geneva,
1955 , 4 , 47 (1956) [C. A., 50, 12667 (1956)]; Hays, Rogers, Ott, J. Am. Chem.
Soc., 77, 1850 (1955).
353. Smith, Science, 119, 514 (1954).
Глава VII
ТИАДИАЗОЛЫ
В. Шерман
1,2,3-ТИАДИАЗОЛЫ
3
Номенклатура
В старой литературе 1,2,3-тиадиазолы именуются диазосульфидами,
тио-[аб 1-диазолами или 3-тио-1,2-диазолами. В настоящем обзоре номенкла-
тура и нумерация атомов заимствованы из справочника «The Ring Index».
Нумерация атомов 1,2,3-тиадиазола показана на формуле I. Устаревшая нуме-
рация, иногда еще встречающаяся в литературе, приведена на формулах
1а и 16.
Синтезы
Синтез из диазометана и фенилизотиоцианата. Первый метод получения
1,2,3-тиадиазолов описан Пехманом и Нолдом [1]. Действуя диазометаном
на фенилизотиоцианат, они получили продукт, которому приписали строение
5-анилино-1,2,3-тиадиазола (II). При этом считали, что конденсация диазо-
метана с фенилизотиоцианатом протекает по одному из двух вероятных меха-
S
CeH5NCS + CH2N2 -» C6H6HN-/ XN
---N
II
низмов с образованием четырех возможных продуктов в зависимости от того,
по какой связи идет присоединение: углерод — азот или углерод — сера.
Наличие триазольных структур исключалось вследствие устойчивости соедине-
ния по отношению к окиси ртути, что указывает на отсутствие тиокарбониль-
II
1,2,3-ТИЛДИЛЗОЛЫ
417
ной группы. Из двух тиадиазольных структур наиболее вероятно образование
структуры II. Повторное исследование 12] подтвердило наличие 1,2,3-
тиадиазольной структуры; этот вывод был сделан на основании изучения рас-
щепления соединения II и наблюдения, что метиленовая группа диазометана
предпочтительно атакует атом углерода карбонильной или тиокарбонильной
группы. При этом не было установлено, существует ли соединение в аминной
или иминной форме. Электронная интерпретация этой реакции приводит-
ся ниже.
Область применения реакции Пехмана, по-видимому, несколько огра1
ничена, так как оказалось, что метилизоцианат не реагирует с диазометаном
при комнатной температуре [2]. Другие алкилизоцианаты в этой реакции
не изучались.
Синтез из оксадиазолов и гидросульфида аммония. Второй метод син-
теза 1,2,3-тиадиазолов был открыт Вольфом 13] и состоял в действии гидро-
сульфида аммония на 1,2,3-оксадиазолы. Необходимый оксадиазол был полу-
чен диазотированием соответствующего а-аминокетона.
СН3СОСНСООС2Н5
nh2
HONO
О
Н3С— < N
11
C2HSOOC-___N
NH4SH
S
Н3С-Д \N
I'
СДООС "----N
Этот синтез позволил впервые получить незамещенный 1,2,3-тиадиазол
путем окисления 5-метил-1,2,3-тиадиазолкарбоновой-4 кислоты и последую-
щего ступенчатого декарбоксилирования.
Н3С-/
НООС —--N
КМпО4
S
S
НООС—/ XN
li
НООС —---N
НООС
Синтез из гидразонов и хлористого тионила. По третьему метрду, описан-
ному Хёрдом и Мори [4], 1,2,3-тиадиазолы получают исходя из ацил- или
бензолсульфонилгидразонов, которые обрабатывают хлористым тионилом.
S
СН3 NHCOOC2H5 SOC12 / 4N
I . I ----" II
НООС-С-----N НООС —----N
27 Заказ .№ 978
416
4иАДИАЗОЛЫ
Этим же путем карбэтоксигидразон ацетофенона был превращен в 4-фенил-
1,2,3-тиадиазол, бензолсульфонилгидразон дезоксибензоина — в 4,5-дифе-
нил-1,2,3-тиадиазол и т, д. Интересный тиадиазол получен при превращении
1-диокиси 4-карбэтоксигидразона тиохромана в 5-диокись 4Н-тиадиазол-
15,4-с1-бензотиопирана.
SOC12
----->
N = N
Представление о механизме этой реакции получено на основе выделения
промежуточного продукта (IV) в ходе превращения бензолсульфонилгидра-
зона дезоксибензоина (III) в 4,5-дифенил-1,2,3-тиадиазол. Обработка соедине-
ния III хлористым тионилом приводит к 12% соединения IV и 63% ожидаемого
дифенилтиадиазола. Нагревание соединения IV в кипящем метаноле дает
тиадиазол с выходом 55%. Кислотный или основной катализ повышает процент
превращения примерно до 90%. Катализируемая кислотами реакция’изобра-
жена на схеме. Аналогичный механизм предложен и для превращения, ката-
лизируемого основанием.
СвН5<^Н2 NHSO2CeH5
CjHjC—N
III
soci.
Я
CeH5C^^N-SOiCeH5
CeH5C-----NH
IV
H+
OH
I
CeH5Cj'+Xx'N—SO2C6H5
vNy—H__*
+ H+ + CjHjSO^H
Реакции и свойства
Многие 1,2,3-тиадиазолы окрашиваются при экспозиции на свету [4].
Природа этого явления не установлена. Сильные кислоты или основания
вызывают разложение 5-анилино-1,2,3-тиадиазола, который, однако, устойчив
в кипящей воде. Окисление 5-метил-1,2,3-тиадиазол карбоновой-4 кислоты
перманганатом в дикарбоновую кислоту [3] служит доказательством ста-
бильности этой циклической системы по отношению к окислителям.
Родоначальное соединение — 1,2,3-тиадиазол [3] — при комнатной тем-
пературе представляет желтую жидкость с т. кип. 157° (при атмосферном
t ,2,3-ТИЛДИАЗОЛЫ
419
давлении), растворимую в спирте, эфире и воде. Он устойчив к действию
кислот, но разлагается основаниями с выделением азота. 1,2,3-Тиадиазол —
слабое основание и образует чрезвычайно гигроскопичный хлоргидрат, кото-
рый разлагается водой на исходные компоненты. При нагревании с иодистым
метилом образуется моноиодметилат [3J.
Штаудингер и Зинварт 15] пытались синтезировать дифенилтиокетен
из 4,5-дифенил-1,2,3-тиадиазола. В обзоре, посвященном тиадиазолам,
Бамбдас [61 неправильно утверждает, что этот тиокетен якобы был получен.
В действительности же единственным органическим продуктом, полученным
Штаудингером и Зигвартом в этой реакции, был тетрафенилтиофен.
Нагревание
>200° ’
При облучении 4,5-дифенил-1,2,3-тиадиазола в бензоле ультрафиолето-
вым светом образуются два продукта: 2-бензгидрилиден-4,5-дифенилдитиол-
1,3 (IVa) и 2,3,5,6-тетрафенилдитиин-1,4 (IV6) [7]. При облучении раствора
4-фенил-1,2,3-тиадиазола получен 2-бензилиден-4-фенилдитиол-1,3 (IVb).
Авторы предполагают, что при этой реакции первоначально образуется проме-
жуточный бирадикал (IVr), который перегруппировывается соответственно
в дифенилтиокетен или фенилтиокетен. Тиокетен затем взаимодействует
с молекулой непрореагировавшего бирадикала, образуя дитиол, или же
димеризуется в дитиин (последний получается только в случае дифенилтио-
кетена). Дитиол (IVb) образуется также из 5-фенил-1,2,3-тиадиазола, но с низ-
ким выходом.
S
Н5С6-/ XN
hcU
Н5С6 N
т-г г _/$\ /С«Н5
5 6 II \=с<
н5с6Д8/ чс6н5
IIII
H5c6/4s/\c6H5
IVa
IV6
IVb
I
[С6Н5—С = СН —S • -> CeH5CH = C = S]
IVr
При нагревании тетрафенилдитиина (IV6) до температуры 180° происхо-
дит отщепление одного атома серы и образуется тетрафенилтиофен. Таким
образом, Штаудингер, по-видимому, все же получил дифенилтиокетен,
но в условиях реакции он превратился через дитиин в тиофен.
27*
420
ТиаДиазолЫ
1,2,3-Тиадиазолины
Единственным представителем этой группы является 1,1-диокись 2-фенил-
4-мстил-А3-1,2,3-тиадиазолипа, которая получена следующим образом:
SO3K
СН2 NHC(iHr,
i I
H3C-C=N
О о
'Х
s
/ 4N —СВН3
I
Н3С
Это соединение бронируется и нитруется в положение 5 [8].
1,2,3-Бензотиадиазолы и другие 1,2,3-тиадиазолы
с конденсированными ядрами
Введение и номенклатура. Литература, посвященная 1,2,3-тиадиазолам
с конденсированными ядрами, значительно обширнее, чем о самих 1,2,3-тиа-
диазолах. Однако, как отмечают Ходжсон и Доджсон [9] в отличном обзоре,
посвященном арилтиадиазолам, весьма удивительно то обстоятельство, что
несмотря на легкость образования этих соединений, они очень мало осве-
щены в литературе. Номенклатура, принятая для этой системы в «Chemical
Abstracts,— 1,2,3-бензотиадиазолы или просто бензотиадиазолы с нумера-
цией атомов, показанной в формуле V. Другая, встречающаяся в литературе
номенклатура,— бензо-1,2,3-тиадиазол, о-фенилендиазосульфиди изопиазтиол.
В более старой литературе принята нумерация, приведенная в формуле Va.
Синтез. Диазотирование о-аминотиолов и родственных соединений. Наи-
более широко исследованный и простейший метод получения 1,2,3-бензотиа-
диазолов — диазотирование о-аминотиофенолов с образованием так называе-
мых внутренних диазосульфидов. Этот метод впервые описан Бернтсеном
[10], а затем Якобсоном [11], которые в ряде последующих работ расширили
применение этой реакции [11—13]. Якобсон получил родоначальное соеди-
нение— 1,2,3-бензотиадиазол и далее [12, 13] распространил эту реакцию
на производные нафталина.
I,2.3-1 ИАДИАЗОЛЫ
421
Розе 1141 получил описанным выше методом 1,2,3-тиадиазопиримидин (VI)
из 2,5-диамино-4-метил-6-меркаптопиримидина.
Кроме того, в определенных условиях можно превратить S-замещенные
ZS------S\ S
\ Y I !] (1) HONO f'y ''N
! II I ! (2) Кипячение j; I J,
^NH2 H2N' 'V V—N
VII
тиофенолы в бензотиадиазолы. Так, например, Якобсон ЦП сделал порази-
тельное наблюдение, что при кипячении в воде диазотированного 2,2'-диамино-
дифенилдисульфида (VII) получается 1,2,3-бензотиадиазол. Ходжсон [151
позднее сообщил, что выход в этой реакции составляет только 50%. Буравой
и Тёрнер [161 подтвердили этот факт и установили, что даже в оптимальных
условиях при проведении диазотирования в разбавленной азотистой кислоте
выход бензотиадиазола никогда не превышал 60%. Эти авторы также пока-
зали, что в разбавленной азотистой кислоте диазотируется только одна амино-
группа с образованием монодиазониевой соли (VIII). Однако, если 2,2'-
диаминодифенилдисульфид нагревать с нитрозилсерной кислотой в кон-
центрированной серной кислоте, то образуется бпс-диазониевая соль (IX).
Последняя при реакции с водой на холоду в присутствии хлорида меди (I)
превращается в 1,2,3-бензотиадиазол (V) с выходом 84?».
| Конц. H5SO4
1ISO3ONO
VII Разб. HONO
VIII
(около 50%)
CuCl V (84%)
Было обнаружено, что диазотирование о-амппофенилбензилсульфида в
присутствии воды приводит к дебензилированию сульфида с образованием 1,2,3-
422
ТИАДИАЗОЛЫ
бензотиадиазола и бензилового спирта [17, 181.
HONO
+ свн5сн2он
Грин и Перкин [19] синтезировали интересный тиадиазол диазотирова-
нием n-фенилендиаминдитиосерной кислоты. n-Фенилендитиадиазол получен
при этом с количественным выходом.
h2nx^4/sso3h
Синтез из диазоокисей и пятисернистого фосфора. Бамбергер [20] разра-
ботал другой метод получения 1,2,3-нафтотиадиазолов путем обработки
диазоокисей пятисернистым фосфором, в результате чего кислород заме-
щается серой.
S-N
N = N
Синтез аминобензотиадиазолов реакцией Скраупа. Амино-1,2-3-бензо-
тиадиазолы вступают в реакцию Скраупа аналогично Р-нафтиламину. Фрис
и Рейтц [21] получили два тиадиазолохинолина методом Скраупа, однако
они заметили, что, когда положение 4 в 5-амино-1,2,3-бензотиаДиазоле занято
атомом хлора, циклизации в положении 6 не происходит. Авторы указывают,
nh2
Реакция
Скраупа
Реакция
Скраупа
1,2,3-ТИАДИАЗОЛЫ
423
что это справедливо также и для соединений ряда нафталина.
Различные синтезы. Первым бензотиадиазолом, описанным в литературе,
был 5-хлор-Г,2,3-бензотиадиазол. Он был синтезирован Бейльштейном и Кур-
батовым в 1879 г. [22] необычным способом — действием на 2-нитро-4-хлор-
тиофенол в спирте гидросульфидом аммония. Полученному продукту при-
писано строение X. При обработке этого соединения азотной кислотой
происходило энергичное окисление с образованием, как полагают, 5-хлор-1,2,3-
бензотиадиазола (XI). Сообщалось, что продукт реакции (XI) имеет т. пл. 103,5°.
НС1-СН3СООН
С12
HNO8
SnCl2
Cl
ccAAn
I В li
ноА/ N
CI
о-Феиилендиамин
N—-N
Cl
424
ТИАДИАЗОЛЫ
Позднее это соединение было получено Поллаком и сотрудниками 123], кото-
рые сообщили т. пл. 107°. Идентичность этих продуктов почти не вызывает
сомнений, однако реакция требует дальнейшего изучения, особенно в связи
с малой вероятностью тиадиазиридиновой структуры (X). Утверждение авто-
ров, что соединение X не обладает кислотными свойствами, ставит под сомне-
ние наличие в нем свободных сульфгидрильных групп.
Ряд интересных реакций, ведущих к образованию 1,2,3-тиадиазофеназина,
описан Фрисом и сотрудниками [24]. Это служит свидетельством устойчивости
бензотиадиазолов, так как они не поддаются Бездействию различных реаген-
тов, применяемых в этих реакциях.
Фрис получил также производное акридина из 5-амино-1,2,3-бензотиади-
азола через производное бензилидена; он сравнивает поведение в этой и дру-
гих реакциях 5-амино-1,2,3-бензотиадиазола с Р-нафтиламином, который
тем же путем образует аналогии! ый акридин [25].
свн3
СН
!1
Реакции и свойства. Нитрование 1,2,3-бензотиадиазола в концентриро-
ванной серной кислоте [21, 26]-дает два продукта: 4-нитро- и 7-нитро-1,2,3-
бепзотиадиазолы с преобладанием изомера с нитрогруппой в положении 4;
нитрование не идет при температуре ниже 100°. Это происходит, по-видимому,
HNO3
H2SO4
S
/V \N
I ILй
\/
NC>2
вследствие гетероциклического характера тиадиазола, благодаря чему воз-
можна протонизация гетероатомов, что дезактивирует бензольное ядро по
отношению к электрофильной атаке [9].
S
\N
I II !1
H,N_y И____N
S
I
N = NCBH3
В реакции диазосочетания 4-амино-1,2,3-бензотиадиазола с хлористым
фенилдиазонием диазогруппа присоединяется в пара-положение к амино-
группе [21]. Если в реакцию сочетания вступает 5-амино-1,2,3-бензотиадиа-
зол, то образуется 4-фенилазо-5-амино-1,2,3-бензотиадиазол 121, 24]. Пока-
зано также, что 4-хлор-5-амино-1,2;3-бедзотцадиазол не вступает в реакцию
1.2.Э-ТИАДИАЗОЛЫ
425
сочетания с хлористым фенилдиазонием. Все это напоминает поведение нафта-
лина, причем положения 4 и 7 в молекуле 1,2,3-бензотиадиазола сходны
с a-положениями в нафталине.
При диазотировании и последующем гидролизе 4-амино-1,2,3-бензотиа-
диазол превращается в соответствующее 4-оксипроизводное [211, которое
при бромировании обнаруживает, как и следовало ожидать, характерную
для фенолов орто- и пара-ориентацию.
Вг2_
СН8СООН
Вг
I S
Г |Г
ВгД^1—N
ОН
Окисление 4,7-диокси-1,2,3-бензотиадиазола хлоридом железа (III) ведет
к образованию п-хинона, однако 5,6-диокси-1,2,3-бензотиадиазол не удалось
окислить в о-хинон [211. Это вновь напоминает поведение соединений ряда
нафталина.
1,2,3-Бензотиадиазол весьма устойчив в самых различных условиях 1131.
Нагревание 1,2,3-бензотиадиазола в 20%-ном растворе едкого кали при
150° или в 27 %-ной серной кислоте при 200° не приводит к разрыву тиадиа-
зол ьного кольца и во всех случаях удалось выделить неизмененное исходное
соединение. Попытки окислить 1,2,3-бензотиадиазол перманганатом или фер-
рицианидом калия, хромовой кислотой или разбавленной азотной кислотой
оказались безуспешными; не было выделено ни сульфоксида, ни сульфона.
Сообщается [131, что 1,2,3-бензотиадиазол восстанавливается оловом в соля-
ной или железом в уксусной кислоте в о-аминотиофенил. Позднее Фрис 1241,
восстанавливая 5-нитро-1,2,3-бензотиадиазол в 5-амино-1,2,3-бензотиадиазол
' ?rSH
[H1 I
—' kJ'-NH2
\ /
оловом в соляной кислоте, не упомянул о разрыве тиадиазольного кольца.
Это можно объяснять стабилизующим влиянием аминогруппы, образующейся
в процессе восстановления. Подобная стабилизация гетероциклического
кольца электронодонорными группами хорошо известна в ряду птеридина [27 I.
1,2,3-Бензотцадиазолы при нагревании в интервале от 200 до 250° пре-
вращаются в соответствующие замещенные тиантрены с одновременным выде-
лением азота [131.
1,2,3-Бензотиадиазолы реагируют с иодистыми алкилами при повышен-
ной температуре с образованием главным образом сульфониевых солей [131.
Отмечается, что продукт присоединения нодистого метила к 1,2,3-бензотиа-
СН3
I
S S+ 1-
XN CH8I / V
II-----* I II
----N \ /------------N
426
ТИАДИАЗОЛЫ
диазолу обладает термохромными свойствами [281: окрашенный в желтый
цвет при комнатной температуре он становится коричневым при нагревании
до 100°. При охлаждении происходит обратный процесс. Известны другие
обратимые изменения цвета этого соединения, но природа этого явления непо-
нятна. Так, например, если соль растворить в воде, этаноле или хлороформе,
то растворы оказываются соответственно бесцветным, желтым и красным.
При длительном стоянии хлороформного раствора или при действии брома
происходит окрашивание в зеленый цвет, причем из раствора можно оса-
дить твердое вещество зеленого цвета, которое становится желтым при нагре-
вании. Подобные изменения цвета можно было бы, вероятно, объяснить
образованием соединения с открытым циклом, как, например, галогенида
алкилтиофенилдиазония. Такая соль, вероятно, была бы интенсивно окрашена
благодаря наличию резонанса, свойственного соединениям этого типа. Против
этого свидетельствует тот факт, что такая соль диазония, по всей вероятности,
разложилась бы с образованием о-иодалкилтиобензола 191. Однако в этом
случае соседняя алкилтиогруппа оказывает настолько сильное стабилизи-
рующее влияние на группу диазония, что предотвращает отщепление азота
и образование иодбензола. Предполагается, что окраска обусловена различ-
ными промежуточными соединениями между полностью раскрытым кольцом
XIII и циклической структурой XII. По мнению Ходжсона и Доджсона [9],
чем слабее связь S — N, тем интенсивнее окраска соединения. Такая струк-
тура может быть выражена формулой XIV, в которой связь S — N может
свободно изменяться по длине в зависимости от условий. 1,2,3-Бензоксадиа-
золы представляют собой окрашенные соединения, причем считают, что этот
тип резонанса ответствен за их окраску [291. Полагают, что у диазоокисей
связь О — N настолько слаба, что вклад циклической структуры незна-
чителен.
XIV
Предполагают, что, в то время как 1,2,3-оксадиазолы в основном суще-
ствуют в виде нециклической диазониевой формы, 1,2,3-тиадиазолы
представляют собой циклическую структуру весьма высокой прочности.
В пользу циклической структуры свидетельствует неспособность 5-амино-
1,2,3-бензотиадиазола претерпевать после диазотирования двойную реакцию
1,2,4-ТИАДИАЗОЛЫ
427
Зандмейера [26, 301. Единственными полученными при этом продуктами
являются 5-галоген-1,2,3-бензотиадИазолы. Не найдено ни одного дигалоген-
производного, в том числе и бис-Диазониевого соединения, образование
которого можно было ожидать. Дополнительным подтверждением циклического
строения служит сообщение о том, что в отличие от 1,2,3-бензоксадиазолов,
которые легко сочетаются с щелочным раствором 0-нафтола, 1,2,3-бензотиа-
диазолы в эту реакцию не вступают [30].
HONO
пр
+n*U-4
X-
Реакция
Зандмейера
Высокая устойчивость 1,2,3-бензотиадиазолов служит еще одним дока-
зательством циклической структуры, так как только при наличии значитель-
ной ароматической стабилизации можно ожидать, что диазосоединение этого
типа не будет разлагаться под действием многочисленных, упомянутых в нас-
тоящей главе реагентов. Предполагается [30], что 1,2,3-тиадиазольное кольцо
подобно тиофену стабилизировано благодаря наличию ароматического сек-
стета.
1,2,4-ТИАДИАЗОЛЫ
I 1а
Номенклатура
Употребляемая в настоящее время нумерация атомов 1,2,4-тиадиазолов
показана на формуле I. Более старая, не популярная теперь система была
предложена Рихтером (1а). В старой литературе встречаются также сле-
дующие названия для 1,2,3-тиадиазола: тио-labt 1-диазол, биазсульфол, азо-
сульфим и миазтиол.
Синтезы
Число препаративных методов получения 1,2,4-тиадиазолов намного пре-
восходит число методов синтеза 1,2,3-изомера. Синтезы замещенных 1,2,4-тиа-
диазолов и тиадиазолидинов описаны в самых ранних работах по органиче-
ской химии, хотя строение полученных соединений не было известно до конца
XIX столетия. Первое упоминание о продукте, который, как это было уста-
428
ТИАДИ АЗОЛЫ
новлено позднее, оказался 1,2,4-тиадиазолом, мы находим у Вёлера |31 I.
В 1821 г. он установил, что тиоциановая кислота разлагается с образованием
соединения C2H2N2S3. Позднее было найдено, что оно содержит тиадиазольное
ядро.
Синтез из амидоксимов. По-видимому, наиболее ранняя работа, пробу-
дившая интерес к 1,2.4-тиадиазолам, появилась в 1889 г. Это работа Шубар-
та (32|, который действовал на амидоксимы сероуглеродом в спиртовом рас-
творе едкого кали, причем полученному продукту была приписана структура
1,2,4-тиадиазола. Так, например, из бензамидоксима был получен 3-фенил-
5-меркапто-1,2,4-тиадиазол. Не известно, существует это соединение пре-
имущественно в енольной или в кетоформе.
S
N°H кон HS-.<
CeHsCNH, ' 2 ’ " nJ'________С„Н5
Вскоре после статьи Шубарта были опубликованы три работы, выпол-
ненные в лаборатории Тимана [33—35]. Показано, что при нагревании бенз-
амидоксима с избытком сероуглерода в отсутствие щелочи можно в конечном'
счете получить тот же продукт, что и при реакции с щелочным раствором
сероуглерода [34]. Предполагают, что продуктом реакции в нейтральной
среде является смесь (II), состоящая из бензамидсульфима и его N-дитио-
карбоната. Нагревание этой смеси до 100° привело к образованию тиадиазола.
NOH
II
H,NCCeH5
S NSH NSH
|! 1| I!
HSCNHCCeH5 • H2NCCeH5
II
Нагревание
NH
(H2NCC6H5)2- H2SC>3
Тима» [33] предоела£а*„, что. неизвестный гидросульфамип (тиогидро-
ксиламин) МОЖН»> получить гидролизом осу-
ществить не удалось, и в свободном виде вещество до сих пор остается неиз-
вестным.
Кох [35] синтезировал 5-анилино-3-фенил-1,2,4-тиадиазол (III) нагре-
ванием бензамидоксима в хлороформе с фенилизотиоцианатом. Несколькими
годами раньше Крюгер [36] показал, что бензамидоксим реагирует при ком-
натной температуре с фенилизотиоцианатом с образованием фепилтиомоче-
виппого производного бензамидоксима (IV), из которого, несомненно, может
быть получен тиадиазол. Вследствие этого схему реакции можно представить
следующим образом:
NOH
I
CgbljNCS-l H2NCCeH5 -
C.Ji.NliC S NOH На,.„с|1а11ие
; j । _____*_______
HN---------CC(iH5
IV
S
C„II5HN - /
N C„H5
III
Синтез из оксадиазолинов. Вскоре после того как Шубарт получил 5-мер-
кап то-3-фен ил-1,2,4-тиадиазол, Штиглиц 1371 показал, что 5-амино-3,5-дифе-
нйл-1,2,4-оксадиазолип можно превратить в меркаптофенилтиадиазол нагре-
1.2.4-ТИАДИАЗОЛЫ
429
ванием при 100° с сероуглеродом. Эта реакция, по-видимому, протекает через
промежуточное образование бензамидоксима, так как известно, что оксадиа-
золин разлагается при нагревании на бензонитрил и бензамидоксим.
II2N °
CeHs/; J
hn._
СвЩ
CS2
Нагревание
N-Ji CBH5
Синтез из тиоамидов. При окислении тиобензамида иодом Гофман [381
наблюдал, что 2 моля тиоамида, по-видимому, реагируют следующим образом:
2C7H7NS + 2I2 —» Ci4H10N2S + 4HI4-S.
Строение полученного продукта было описано спустя несколько лет Гофманом
и Габриэлем [39], которые показали, что при окислении образовался 3,5-дифе-
нил-1,2,4-тиадиазол. Персульфат аммония [40] и азотистая кислота [41 | также
могут служить окислителями. При использовании азотистой кислоты выход
дифенилтиадиазола составил 30%, а применение иода в спирте позволило
повысить выход до 77% [411.
S
II +
CBH5CNHo
Io
s
СвН5 ''n
N—U—ru
М5Г15
Шабрие и сотрудники [421 описали окисление тиобензамида и тионико-
тинамида иодом или метиловым эфиром М,М-дихлоркарбаминовой кислоты.
Сообщается, что таким путем были получены 2,5-дифенил- и 2,5-ди-(3-пири-
дил)-1,3,4-тиадиазолы. Это сообщение, без сомнения, ошибочно, поскольку
температура плавления дифенилтиадиазола, полученного Шабрие, очень
близка к температуре плавления аналогичного соединения, синтезированного
Гофманом [38]. По всей вероятности, полученные Шабрие соединения пред-
ставляют в действительности 3,5-дифенил- и 3,5-ди-(3-пиридил)-1,2,4-тиадиа-
золы.
||
CNHo
Ii
Два общих метода получения диарил-1,2,4-тиадиазолов из тиоамидов
описаны Ишикава [43, 44]. При обработке тиоамида хлористой серой в спир-
товом растворе едкого кали или хлористым тионилом, бензолсульфохлоридом
или хлористым сульфурилом в нейтральных условиях были получены 3,5-ди-
арил-1,2,4-тиадиазолы. Наилучшие выходы получены при использовании
хлористого тионила и бензолсульфохлорида.
При проведении этих реакций Ишикава выделил различные количества
побочного продукта, которому он приписал строение бензиминоизотиобенз-
амида (V) [44]. п- и л-Толилпроизводные этого соединения были получены
из соответствующего нитрила и тиоамида. Окисление толилиминоизотио-
толиламидов иодом дает дитолил-1,2,4-тиадиазолы. Этим способом была осуще-
ствлена конденсация- некоторых других арилнитрилов и тиоамидов, однако
только толилпроизводные получались с препаративными выходами, которые
430
ГИАДИАЗОЛБ1
в лучшем случае не превышали 23 °6.
SH NH
i II
С(!Нг,С N - - ССВ11.-,
V
S
Н3С - у,— (2NH2+ N - С — У— СН3 —>
SH NH
Z----х I II / X !2
-> Н3С—у— C = N — С- сн3
___ S
Синтез из тиоперимидовых кислот. Иной подход к получению 1,2,4-тиа-
диазрла через стадию образования необычного промежуточного продукта
описан Китамура 1451, который действовал на тиоамиды перекисью водорода
и получил соединения, описанные как «тиоперимидовые кислоты». Эти соеди-
нения имеют эмпирическую формулу RCH2NOS. Им приписывается структу-
ра VI. Фенилтиоперимидовая кислота устойчива в темноте; однако на свету
опа разлагается с образованием 3,5-дифенил-1,2,4-тиадиазола. При нагре-
вании До 100° фенилтиоперимидовая кислота переходит в бензонитрил и тиа-
диазол. Более важное свойство тиоперимидовых кислот заключается в их спо-
S
$ —* ° Нагревание Н5С6~|| N ; CBH5CN
свн5 —с—nh2 - iiX-СвН6
VI
собности конденсироваться с тиоамидами различных кислот с образованием
1,2,4-тиадиазолов с различными заместителями в положениях 3 и 5. В каче-
стве примера приведем получение 5-бензил-З-фенил-1,2,4-тиадиазола из бен-
зилтиоперимидовой кислоты и тиобензамида. Образование бензилтиоперими-
довой кислоты указывает на возможность синтеза этим путем 3,5-диалкил-
1,2,4-тиадиазолов, хотя этот вопрос, по-видимому, не освещен в литературе.
S
s -> о S свн5сн2 \
II V II II н
cbh5ch2cnh2 cbh5cnh2 N!--cbh5
Синтез из Ксантамида. В литературе имеются два сообщения о том, что
ксантамид может быть окислен в 3,5-диэтокси-1,2,4-тиадиазол с помощью
перекиси водорода [461 или хлорида меди (II) [47]. Это единственный метод
получения 3,5-диалкокси-1,2,4-тиадиазолов.
S
s |tl] С2Н-О-/
C2H-,OCNH2 N_J'__qc2h5
Синтез из амидинотиомочевин. В ряде работ [48—53 I Курцер показал, что
амидинотиомочевина и замещенные амидинотиомочевины могут быть окислены
i. 2,4-ТИАДИАЗОЛЫ
431
в амино- и замещенные амино-1,2,4-тиадиазолы. Если амидинотиомочевину,
полученную действием сероводорода на дициандиамид, обработать иодом или
перекисью водорода, то она окисляется в 3,5-диамино-1,2,4-тиадиазол [481
с выходом до 80% [491. Эта реакция сходна с окислением толилиминоизо-
тиотолиламида, описанном выше [44]. Курцер установил, что при окислении
S
S ‘уН [О| H2N ' \\ N
h,ncnhcnh2 n_1LNh2
иодом в спиртовом растворе поглощение иода прекращается после вступления
в реакцию одного эквивалента. Это позволило обнаружить, что иодгидрат
амидинотиомочевины не окисляется иодом. При проведении реакции в водном
растворе окисление иодом становится обратимым процессом, и только при
разведениях меньше 0,066 М реакция протекает с препаративными выходами.
Однако использование перекиси водорода в присутствии минеральных кислот
оказалось для препаративных целей вполне удовлетворительным.
Курцер применил эту реакцию для получения различных 3- и 5-заме-
щенных амино-1,2,4-тиадиазолов. Если полученные из гуанидинтиоцианата
и соответствующего алкилизотиоцианата [54] М-алкил-Й'-амидинотиомоче-
вины обрабатывать перекисью водорода в кислой среде, то с хорошим выходом
образуются 5-алкиламино-З-амино-1,2,4-тиадиазолы [49]. Этим способом были
получены 5-метиламино-, 5-этиламино- и 5-пропиламинопроизводные. Иод
в качестве окислителя для этой реакции, по-видимому, не применим вслед-
ствие неблагоприятных равновесных условий.
S
S NH ;,2О^ CH3HN^-/ \
ch3nhcnhcnh2 nJ____
При окислительной циклизации N-амидино-М'-фенилтиомочевины под дей-
ствием перекиси водорода или брома образуется З-амино-5-анилино-1,2,4-тиа-
диазол 1501. Этим путем могут быть также синтезированы 5-(п-толиламино)-
и 5-(п-бромфениламино)-3-амино-1,2,4-тиадиазол [50]. Замещенные амиди-
нотиомочевины могут быть получены из изотиоцианатов и гуанидинтиоцианата.
S
S NH [О] C«H5NH-pN
CeH5NHCNH(!NH2 ’ nJ1_Nh2
Этот способ был использован для синтеза 3,5-диариламино- и 3,5-диаралкил-
амино-1,2,4-тиадиазолов [50], как, например, 3,5-дианилино-, 5-анилино-З-
метиламино- и 3-анилино-5-метиламино-1,2,4-тиадиазолов.
S
S NH CeH5NH-/\
CeH5NHCNHCNHCH3 " f!}.JLNHCH3
В четвертом сообщении серии своих работ Курцер [51] рассматривает
окислительную циклизацию N-ароиламидинотиомочевин. При действии на эти
соединения перекиси водорода, содержащей один эквивалент хлористого водо-
рода, реакция протекает, как обычно, с образованием 5-амино-З-ароиламино-
1,2,4-тиадиазолов. Если, однако, проводить окисление перекисью водорода
в отсутствие минеральной кислоты, то оно протекает совершенно иначе и дает
с отличными выходами З-амино-5-арил-1,2,4-тиадиазолы. Ниже приводится
432
ТИЛДИ АЗОЛЫ
схема этих превращений на примере N-бензоиламидинотиомочевины. Образо-
вание З-амино-5-фенил-1,2,4-тиадиазола окончательно доказано путем срав-
нения продукта реакции с образцом, синтезированным при помощи другого
метода. Если при окислении этого соединения изменять количество мине-
ральной кислоты, то образуются смеси соединений VII и VIII, причем содер-
жание соединения VII увеличивается с повышением кислотности. При при-
менении брома в качестве окислителя оба продукта образуются в равных
количествах, что можно было ожидать вследствие выделения в процессе окис-
ления бромистого водорода.
S NH О н п
II II Н
H2NCNHCNHCCeH5 «+
| Н2°2
VIII
S
H2N~Q ? •
N ——NHCCoH5
VII
Механизм этой реакции точно не установлен, но Курцер предлагает два
варианта, из которых наиболее вероятный приводится ниже. Как будет пока-
зано, N-аренсульфонамидинотиомочевины циклизуются в тиадиазолы под
действием брома [53]. Попытки осуществить реакцию действием кислоты
или нейтрального раствора перекиси водорода оказались безуспешными —
основным продуктом реакции был аренс/льфонилгуанидин. Выделение заме-
щенного гуанидина подтверждает предположение об образовании ароилгуапи-
О NH S н п О NH Г S
Ч и I, Н2°2 II II I ч
‘I и |i ---------> II II 4- ll
ArCNHCNHCNH, ArCNHCNH, |_HOCNH2
S
Аг~Н n
N—LNHa
S NH
+ CO2 + NH3
дина при окислении ароиламидинотиомочевины. Трудно представить, чтобы
сульфонильная группа в процессе циклизации подвергалась тианированию,
S NH н n NH
Н Н —II
H2NCNHCNHSO2Ar H2NCNHSO2Ar
в то время как карбонильная группа ароилгуанидина может реагировать
подобным путем, образуя тиадиазол.
Под действием брома М-(п-толуолсульфонамидино)тиомочевина и N-(n-
толуолсульфонамидино)-М'-фенилтиомочевина циклизуются, образуя 5-ами-
S NH
II IJ Z---. Вг.,
RNHCNHCNHSO,— V~ СН3-----------"
S
RNH-/ \ _
n_IL_NHSq2 СНз
R — Н или С0Н5
1.2.4-ТИАДИАЗОЛЫ
433
но(или анилино)-3-(п-толуолсульфонамидо)-1,2,4-тиадиазол с почти коли-
чественным выходом [53]. Можно также провести циклизацию, но с более
низкими выходами, заменив бром аренсульфонилхлоридами в пиридине [53].
Амидинотиомочевина и N-арилзамещенные амидинотиомочевины могут этим
способом.одновременно ацилироваться и циклизоваться с образованием 5-ами-
но-3-аренсульфонамидо-1,2,4-тиадиазолов.
j| I ArSOgCl^
H2NCNHCNH2 Пиридин
NHSO2Ar
Синтез из тиобиуретов. Последовательно расширяя область применения
реакции окисления амидинотиомочевины и ее замещенных Курцер [52] пока-
зал, что полученные из изобиуретов 1-фенил-2-тио-4-изобиуреты и 1-фенил-2-
тиобиуреты могут быть окислены бромом или перекисью водорода в 3-алкокси
(или окси)-5-фениламино-1,2,4-тиадиазолы с отличными выходами. Полу-
S ОСН3 S О
CeH5NH(2NHC = NH —> CeH5NHCNHCNH2
|tO] [°]
S S
CeH5NH-/\ CeH5NH /
H II = II II
N—ОСНз N—-~OH
чены также метокси- и этоксипроизводные. Как будет показано ниже, алко-
кситиадиазолы можно также синтезировать из N-галогенмочевин [55].
Синтез из амидинов. Синтез, описанный Гёрделером [56], ведет к полу-
чению 5-амино-1,2,4-тиадиазола и З-замещенпых производных этого соеди-
нения. В основе этого метода лежит реакция между тиоцианатом калия
и N-галогенамидинами, например N-хлорформамидином:.
S
H2NC = NC1 + KSCN —» H2N— <
о
Опасности, связанной с применением N-хлорформамидина, можно избе-
жать, используя хлоргидрат формамидина, метилат натрия, бром и тиоцианат
калия в различных комбинациях. Этот метод, по-видимому, носит общий харак-
тер и используется для получения многих 5-амино-1,2,4-тиадиазолов, заме-
щенных алкильными или арильными группами в положении 3. При этом
выходы колеблются от 50 до 80%, что делает их ценными для практического
применения.
NH • S
CH3^NH2-HC14-2NaOCH34-Br24-KSCN —> H2N-j[ N
nJL-снз
Гёрделер [57] в патентной заявке описал способ получения 5-хлор-1,2,4-
тиадиазолов циклизацией амидинов или 5-алкилизотиомочевин под действием
трихлорметансульфонилхлорида. Этим методом из ацетамидина получен
5-хлор-3-метил-1,2,4-тиадиазол, а из 5-метилизотиомочевины — 5-хлор-З-ме-
28 Заказ №978
434
ТИАДИАЗОЛЫ
тилтио-1,2,4-тиадиазол; синтезированы также этилпроизводные.
NH S
ch3c!nh2+ci3csci С1“([ N
N-JL-СНз
SCH3 S
HN = C — NH24-C13CSC1 C1~l[ N
N-U—SCH3
Синтез из изомочевин. Аналогичным путем Гёрделер [551 успешно осу-
ществил синтезы 3-алкокси- или 3-арилокси-5-амино-1,2,4-тиадиазолов,
исходя из М-галоген-О-алкил(или арил)изомочевин. Эта реакция также,
по-видимому, носит общий характер и позволяет получить ожидаемый про-
дукт с выходом 40—90%. N-Галогенизомочевины ранее не были описаны.
Хлор- и бромпроизводные получены с высоким выходом при действии гипо-
галогенита натрия на О-замещенные изомочевины. N-Иодизомочевина
получена с низким выходом действием йодистого калия и гипохлорита натрия.
Уравнение реакции приведено ниже (R может быть метилом, этилом, бензо-
илом, фенилом или другим заместителем).
NH NC1
II II
ROCNH2-HC14-NaOCl ROCNH2 + NaC14-H2O
J, NaSCN
S
H2N~f N
nJ—or
Образование тиадиазола по описанному выше методу включает две ста-
дии: быстрое осаждение хлористого натрия и последующую медленную потерю
способности реакционной смеси окислять иодистый калий до иода. Доказа-
тельством того, что эта реакция имеет место в процессе получения N-тиоцианат-
алкоксиизомочевины служит выделение промежуточного продукта в опыте'
с метилизомочевиной. Показано, что прохождение циклизации промежуточ-
ного продукта зависит от pH и оптимальные выходы получались при pH
от 2 до 4. Циклизация не идет в растворах щелочей или сильных кислот.
NCI ? . NSCN ? S
CH3oi:NH2 ch3o<!nh2 h2n~
n_LOch3
Синтез из изотиомочевин. Реакция между алкил- или арилизотиомоче-
винами, гипогалогенитом и тиоцианатом натрия совершенно аналогична
описанным выше реакциям изомочевины. Этот метод также носит, по-види-
мому, общий характер и применялся для получения 3-метил-, 3-этил-, 3-лау-
рил- и 3-фенилмеркапто-5-амино-1,2,4-тиадиазолов и некоторых других 5-заме-
щенных производных [58].
NH S
RSCNH2 +NaSCN+ Вг —°2_> H2N~H N
N-U—sr
Синтез из гуанидинов. При попытке распространить описанные выше
реакции на М,М-дизамещенные гуанидины Гёрделер [59] обнаружил, что
1,2.4-ТИАДИАЗОЛЫ
435
хотя N.N-дизамещенные N'-хлоргуанидины получались с хорошими выхо-
дами, однако они не всегда циклизовались в тиадиазолы. Из N.N-дифенил-
и N.N-дибензилгуанидинов были получены N'-хлоргуанидины, которые были
выделены с хорошими выходами и затем превращены в 3-дифениламино-
и З-дибензиламино-5-амино-1,2,4-тиадиазолы. Реакцию удобно проводить,
NH N—С1
(CeH5)2NCNH2 —(CeH5)2NCNH2
| NaSCN
s
H2N-|
NJL~N(CeH5)2
получая бромгуанидин in situ в присутствии тиоцианата калия. Этим способом
тиадиазол получается сразу и с хорошим выходом.
NH S
R2N^NH2-HCl + Br + NaOCH3+ KSCN —>
nJL_NR2
В противоположность дифенил- и дибензилпроизводным Ы-хлор-Ы',Ы'-
диметилгуанидин и Ы-хлор-Ы,'Ы'-циклопентаметиленгуанидин были легко
получены обычным путем; однако хлорпроизводные превращались в тиадиа-
золы с очень низкими выходами. 3-Диметиламино-5-амино-1,2,4-тиадиазол полу-
чен другим способом из N.N-диметилгуанидина и родана с выходом 31% [59].
NH
(ch3)2n(!:nh2+(scn)2
S
H2N—,< N NH
Кт II H
N—11—N(CH3)2 + (CH3)2NCNH2- hscn
Выше описан осуществленный Курпером [51 ] синтез З-амино-5-фенил-
1,2,4-тиадиазола окислением N-бензоиламидинотиомочевины перекисью водо-
рода в нейтральном растворе. Гёрделер [60] получил это соединение более
прямым и однозначным методом синтеза из этилтиобензоата и гуанидина
с выходом 28%.
S NH S NH S
СвН5ЛоС2Н5 + H2N<!nh2 СвНвЬнЛыНг Свн5~||' N
* N-ll—Nh2
Синтез из тиоциановой кислоты. Надтиоциановая и изонадтиоциановая
кислоты. Производные надтиоциановой и изонадтиоциановой кислот пред-
ставляют наименее полно освещенную в литературе область химии 1,2,4-тиа-
диазолов. Эти соединения несколько подробнее рассмотрены Бамбасом [61]
в обзоре, посвященном тиадиазолам. Он считает, что этот вопрос довольно
запутан и полон противоречий. Лишь дальнейшие экспериментальные иссле-
дования дадут ясную картину, которая пока сильно усложнена различными
догадками. Кратко этот вопрос будет описан ниже, однако более подробные
сведения и ссылки на соответствующие работы можно найти в обзоре Бам-
баса.
В 1828 г. Вёлер [31] сообщил, что концентрированный раствор тиоциано-
вой кислоты разлагается с образованием соединения с эмпирической форму-
лой C2H2N2S3. В последующие годы это соединение называли надтиоциановой
кислотой, изонадтиоциановой кислотой и ксантановодородом. Вёлер обна-
3HSCN c2h2n2s3 + hcn
28*
436
ТИАДИАЗОЛЫ
ружил, что при действии на продукт конденсации тиоциановой кислоты вод-
ного раствора щелочи осаждается сера. Позднейшие исследователи пытались
объяснить эту реакцию либо как расщепление с образованием новой кислоты
с более низким содержанием серы [62], либо исходили из предположения,
что продуктами реакции являлись «дитиоциановая кислота» и сера [63].
Класон [64], изучая эту реакцию, пришел к выводу, что первоначально обра-
зующимися продуктами являются сера и «дитиоциановая кислота», но что
при стоянии большая часть серы вновь поглощается и образуется новая кис-
лота, изомерная продукту конденсации тиоциановой кислоты. С этого момента
началась путаница в номенклатуре этих соединений: продукт конденсации
тиоциановой кислоты стал известен под названием цадтиоциановой кислоты,
которой Глюц [65] приписал структуру IX. Класон назвал продукт кон-
денсации «ксантановодородом» (Xantanwasserstoff), а продукт, образовав-
шийся при обработке «ксантановодорода» щелочью,— нормальной надтио-
циановой кислотой. Таким образом, «ксантановодород» превратился в изо-
кислоту вследствие недоразумения. К счастью, после Класона номенклатура
стабилизировалась и установились наименования — надтиоциановая и изо-
надтиоциановая кислоты. Класон показал тесную связь между этими кис-
' лотами и их взаимопревращаемость при действии кислоты или основания.
Ниже приводятся схема реакций и номенклатура. Класон предложил струк-
туру IX для изокислоты и структуру для лабильной по отношению к кисло-
HSCN
I
Изонадтиоциановая кислота («ксантановодород»)
но' 11 н+
Надтиоциановая кислота
там надтиоциановой кислоты. Ганч и Вольвекамп [66] в более поздней работе
предложили для изонадтиоциановой кислоты дитиазолидиновую структуру
и высказали предположение, что надтиоциановая кислота может обладать
одной из таутомерных форм IX или X. Таким образом, в соответствии со схе-
мой, предложенной Ганчем и Вольвекампом, тиоциановая кислота претерпе,-
вает конденсацию с образованием 3-имино-1,2,4-дитиазолидинтиона-5 (изонад-
S S
s — / \ HS \
| NH П || N
hn_1=s n И SH
IX X
тиоциановой кислоты), который при действии основания перегруппировывается
в 3,5-димеркапто-1,2,4-тиадиазол (X) (надтиоциановую кислоту). Надтиоциа-
.новая кислота устойчива только в форме одной из ее металлических солей;
обработка кислотой вызывает перегруппировку в изонадтиоциановую кислоту.
Именно эта номенклатура и структурные формулы соответствуют принятым
в справочнике Бейлыптейна и, по-видимому, лучше всего отвечают свойствам
этих соединений. Однако бесспорное доказательство структуры отсутствует,
S s
S —/ \ но HS z
3HSCN S — Zt || N
HN___Unh но+ N_Lsh
XI X
и вполне возможно, что дитиазолидиновое строение изонадтиоциановой кислоты
не соответствует действительности.
1,2,4-ТИАДИАЗОЛЫ
437
Реакция изонадтиоциановой кислоты с едким кали детально изучена Ган-
чем и Вольвекампом. Флейшер [63] и Класон [64] прежде всего выделили
промежуточный продукт реакции изонадтиоциановой кислоты со щелочью,
который, как показали авторы, является двухосновной кислотой с формулой
C2H2N2S2. Он был выделен и идентифицирован как в форме свободной кис-
лоты, так и в виде некоторых солей металлов. Ход реакции показан ниже.
Класон предложил маловероятную структуру XII для «дитиоциановой кис-
C2H2N2S34-2KOH > K2C2N2S2 4" S-J- 2Н2О
лоты» и предположил, что этот промежуточный продукт реагирует с серой,
HS—й—N
II и
N-11 -SH
XII
образуя надаиоциановую кислоту. Ганч и Вольвекамп считали, что «дитио-
циановая кислота» может быть синтезирована действием сероуглерода на циан-
амид и что, по-видимому, она является N-циандитиокарбаминовой кислотой
или одной из ее солей (ХПа). Ганч и Вольвекамп утверждали, что, для того
чтобы образовалась соль N-циандитиокарбаминовой кислоты, ее предшествен-
ник — изонадтиоциановая кислота — должен иметь, скорее, строение 3-ими-
но-1,2,4-дитиазолидинтиона-5 (XI), чем 3,5-димеркапто-1,2,4-тиадиазола, как
предполагал Класон. Схема реакции приведена ниже.
Бамбас [61] в обзоре, посвященном тиадиазолам, указывает, что пред-
ложенное Класоном строение изонадтиоциановой кислоты в виде 1,2,4-тиа-
диазолидиндитиона-3,5 (IX) имеет несомненное преимущество. Бамбас утвер-
ждает, что многие аргументы, приведенные в пользу дитиазолидиновой струк-
туры, имеют силу также и для тиадиазолидиновой структуры. Он считает,
например, что тиадиазолидин мог бы также взаимодействовать с едким кали
с образованием калиевой соли N-цианднтиокарбаминовой кислоты и приводит
следующую приведенную ниже схему реакции. Бамбас высказывает далее
S S '
3HSCN _> s=fXs А°Д KS-pS
HN—_NH N—=NH
XI
j, KOH
KS s
XC=N —CsN4-S KS~C SK
KS7" Xlla N—C=N
предположение, что так как дитиазолидиновая структура для изонадтиоциа-
новой кислоты может быть неверна, то и другие соединения, изображенные
в виде дитиазолов, могут быть в действительности тиадиазолами. Так, напри-
мер, Фромм [67] проводил окисление фенилдитиобиурета иодом, перекисью
s, ks\
S==| '' уи К0Н» CNCN + S
HN-----===S /
KS
водорода и другими реагентами, получая «фенилтиурет», и описал э4от процесс
как нормальное окисление сульфгидрильных групп в дисульфид. Бамбас
предполагает, что окисление может приводить к образованию связи между
438
ТИАДИ АЗОЛЫ
атомами серы и азота, подобно тому как происходило при окислении тио-
бензамида в 3,5-дифенил-1,2,4-тиадиазол (см. работу Гофмана и Габриэля [39]).
SN SH S
H2NC=N-C = NCeH5 [21 HzN_|f
nJ=nc6h5
В этом случае продукт реакции был бы 5-анилино-З-меркапто-1,2,4-тиадиа-
золом. Как отмечает в своем обзоре по биуретам Курцер [68], согласно пра-
вилу Фромма [69], соединения типа R = С — S — S — C = R' исключительно
чувствительны к щелочам и аминам и разлагаются с образованием элемен-
тарной серы. Учитывая легкость, с которой «тиуреты» и изонадтиоцианаты
реагируют с основаниями, образуя серу и другие продукты, и сопоставляя
эти свойства с относительной устойчивостью замещенных 3,5-диамино-1,2,4.
SH NH S
CeH5NHC = N — (isH CeH5NH— N
N-LsH
тиадиазолов [49], можно считать, что до тех пор, пока не появятся новые
экспериментальные данные, наиболее приемлемыми для изонадтиоциановой
кислоты и тиуретов будут циклические дисульфидные структуры.
В реакции изонадтиоциановой кислоты со щелочью сера сначала оса-
ждается, а затем, если раствор оставить постоять некоторое время, медленно
вновь поглощается. Соль надтиациановой кислоты может быть затем выделена
из раствора [64, 66]. Эту последовательность можно представить как перво-
начальную атаку щелочи на изонадтиоциановую кислоту, в результате кото-
рой раскрывается кольцо и образуется N-циандитиокарбамат. Можно пред-
положить, что эта реакция будет протекать быстрее, чем последующая реакция
N-циандитиокарбамата с твердой элементарной серой. Реакция может быть
представлена следующей схемой:
1,2,4-Тиадиазол идины
Арилтиомочевины при окислении превращаются в тиадиазолидины,
обычно называемые «основаниями Гектора». Гектор [70] окислял фенилтио-
мочевину, дифенилтиомочевину, а-нафтилтиомочевину, а также толил- и кси-
лилтиомочевины различными окислителями и получил продукты, перво-
начально принятые им за 1,3,4-тиадиазолы. В более поздней работе [71],
в которой описаны результаты исследования расщепления оснований, Гектор
изменил свою точку зрения на строение продуктов окисления и пришел к выво-
ду, что они представляют собой 2,4-диарил-3,5-диимино-1,2,4-тиадиазолидины
(XIII). Гектор, в частности, обнаружил, что в результате кислотного гидро-
лиза или восстановления образуется не фенилгидразин, а дифенилгуанидин,
1,2,4-ТИАДИАЗОЛЫ
439
который мог получиться только из молекулы с атомами азота, находящимися
в 1,3 положениях.
Гофман и Габриэль [39] предложили другую структуру для оснований
Гектора, принимая их за 3,5-диариламино-1,2,4-тиадиазолы. Курцер [50]
опроверг это, синтезировав 3,5-дианилино-1,2,4-тиадиазол и показав его
неидентичность основанию Гектора, полученному из фенилтиомочевины.
Хаагер и Дот [72] синтезировали 2,4-дифенил-3,5-диимино-1,2,4-тиадиа-
золидин окислением фенилтимочевины азотистой кислотой. Одним из продку-
тов реакции был фенилизотиоцианат.
При действии перекиси водорода [706] или брома в спиртовом раство-
ре [73, 74] на М,М'-дифенилтиомочевину получают 2,4-дифенил-3,5-бис-(фенил-
имино)-1,2,4-тиади азолидин.
1 [°]> / \
CeHsNHCNHCeHs CeH5N=| N_CeH5
CgHs—N ! = NC0H5
Показано [75], что хлорметилсульфонилхлорид взаимодействует с N,N'-
дифенилмочевиной в кипящем хлороформе, образуя с вполне удовлетвори-
тельным выходом 2,4-дифенил-1,2,4-тиадиазолидинон-3.
S •
О > |Z 'n-CeH5
ClCHaSCl + CeHsNHCNHCeHs CeH5—N------==0
Реакции и свойства
Родоначальное соединение этого ряда—1,2,4-тиадиазол — получено
в 1956 г. [76]. Реакцией Зандмейера — Гаттермана 5-амино-1,2,4-тиадиазол
превращали в 5-бром-1,2,4-тиадиазол, который каталитически восстанавливали
в 1,2,4-тиадиазол. Незамещенное соединение представляет собой бесцветную
подвижную жидкость с т. кип. 121° и т. пл. —33° и запахом, напоминающим
S
Н2
N1
запах пиримидина. Представляет интерес, что температура кипения пирими-
дина 124°. Восстановление или окисление (30%-ной перекисью водорода)
вызывает разложение 1,2,4-тиадиазола. Наличие фенильных групп, как,
например, в 3,5-дифенил-1,2,4-тиадиазоле, заметно повышает устойчивость
к действию кислот [38].
440
ТИАДИАЗОЛЫ
Обработка 5-бром-1,2,4-тиадиазола горячей концентрированной серной
кислотой приводит к образованию 5-окси-1,2,4-тиадиазола с выходом 64% [761.
Метиламин замещает бром, при этом с высоким выходом образуется 5-метил-
амино-1,2,4-тиадиазол.
S S S
но-/ \ Вг-/• \ CH3NH / \
II II II ' II II
N и N---U N ~
Диазотированный 2-амино-1,2,4-тиадназол сочетается с замещенными бен-
золами, образуя азокрасители [77].
При действии йодистого метила на 5-амино-1,2,4-тиадиазол [78] полу-
чается метилированный в ядре продукт — 5-имино-4-метил-1,2,4-тиадиазолин.
S S S'
CH3I HN-/ АггО \
П21Ч—N —> П1М—| N-------N
М нД-Н м
сн31
SCH3
ch3nh(!;=nh
Обработка иодистым метилом в более жестких условиях приводит к образо-
ванию N-метилметилизотиомочевины. Однако 5-амино-З-фенил-1,2,4-тиадизол
метилируется сначала по атому азота в положении 4, а затем по иминному
азоту без разрыва кольца. Обработка 5-имино-4-метил-1,2,4-тиадиазолина
окисью серебра вызывает перегруппировку в 5-метиламино-1,2,4-тиадиазол.
5-Амино-1,2,4-тиадиазолы представляют собой слабые основания, обра-
зующие соли с минеральными кислотами, которые при растворении в воде
гидролизуются с образованием свободных оснований. Последние легко аци-
лируются, образуя 5-ациламино-1,2,4-тиадиазолы [56].
При восстановлении 3,5-диамино-1,2,4-тиадиазола цинком в соляной кис-
лоте он превращается в амидинотиомочевину, из которой это соединение
может быть получено [48]. В этом отношении диаминотиадиазол отличается
от других 1,2,4-тиадиазолов, которые теряют при восстановлении серу и пре-
. S S NH
н м /• \ [Н] II II
м2^— h N ----------- H2NCNHCNH2
^NH2 IO1
вращаются в замещенные амидины или гуанидины. Так, например, восста-
новление 3,5-дифенил-1,2,4-тиадиазола приводит к образованию бензилбенз-
амидина [43].
S NH
г н / \ [Н] ||
свм5-н N ------------, ceH5CNHCH2CeH5
II II
N---СвЫ5
При обработке 3,5-диамино-1,2,4-тиадиазола одним эквивалентом хлори-
стого сульфурила образуется продукт монозамещения, который считают
3-сульфонамидопроизводным [49]. Получить 5-амино-З-ароиламино-1,2,4-тиа-
диазолы можно только окислительной циклизацией N-ароиламидинотио-
мочевин [51 ]. Действие хлористых ароилов на 3,5-диамино-1,2,4-тиадиазолы
всегда дает ди- и тризамещенные продукты. При взаимодействии 3,5-диамино-
1,2,4-ТИАДИАЗОЛЫ
441
1,2,4-тиадиазола с двумя эквивалентами хлористого бензоила образуется
3,5-дибензамидо-1,2,4-тиадиазол [49]. При использовании избытка хлори-
стого бензоила получается трибензоилтиадиазол невыясненной структуры,
однако тетраацилпроизводное не образуется.
При ацилировании З-амино-5-метиламино-1,2,4-тиадиазола хлористым
бензоилом или аренсульфонилхлоридом вступает одна ацильная группа
(в положение 3), если в реакции участвует один эквивалент, и три ацильные
группы — при большом избытке реагента [49]. Полученным продуктам при-
писывается строение замещенных тиадиазолидинов.
Реакции 3-амино-5-ариламино-1,2,4-тиадиазолов с ацилирующими аген-
тами аналогичны реакциям соответствующих 5-алкиламинопроизводных [50].
Однако 3,5-дианилино-1,2,4-тиадиазол ведет себя подобно «основаниям Гек-
тора», давая только моноацетильные и бензоильные производные независимо
S
„ H*N-|fXN
H3C-^__>-SO2CI ! || ---
N---— NHSO2->_____СН3
S ==
I || 2СвН5СОС1
N—— NH2
S
c6h5cohn-/\
N——NHCOC6H5
от условий. Строение этих моноацилпроизводных точно не известно. Пред-
ставляет интерес тот факт, что n-толуолсульфохлорид легко давал дисуль-
фонилпроизводное, принимаемое за 2,4-дизамещенный тиадиазолидин.
S S
CH3NH—Z ceH5coci CH3N = |// СОС6Н5
Д LNH2 * C6HSCO—N----------------! = NCOC6Ha
Как уже упоминалось, «основания Гектора» ацилируются только по одной
из двух иминных групп [70а]. Вследствие большей реакционной способности
3-аминогруппы в 3,5-диамино-1,2,4-тиадиазоле вполне вероятно, что ацилиро-
ванные «основания Гектора» представляют собой 3-ацилиминосоединения [49].
S S
HN”j N—С6Н5 (сн3со)2о t!N- N—С6На
С6На—N -=NH C8H5-'n—! = NCOCH3
При нагревании 3,5-диимино-2,4-дифенил-1,2,4-тиадиазолидина с кон-
центрированной соляной кислотой одна из иминных групп отщепляется
и образуется продукт, которому приписано строение 2,4-дифенил-З-имино-
1,2,4-тиадиазолидинона-5 [79]. При восстановлении этого соединения цинком
в уксусной кислоте получается К,М'-дифенилгуанидин. При попытке гидро-
лизовать оставшуюся иминогруппу в более жестких условиях происходит
S S
HN=|Z N-С6На ’ н+ °=|/ 'n—С6На
C6Ha-N — = NH CeH5—N---- = NH
лишь разрыв кольца и образуется также дифенил гуанидин.
442
ТИАДЙАЗОЛЫ
При нагревании 3,5-диимино-2,4-дифенил-1,2,4-тиадиазолидина со спир-
товым раствором аммиака при 150° получено вещество с эмпирической фор-
мулой C14H12N4S [79]. В справочнике Бейлылтейна этому продукту пред-
положительно приписывается строение 1,4-дифенил-5-имино-1,2,4-триазоли-
динтиона-3. Курцер [80], однако, показал на основании независимого синтеза
из N-фенил-М'-фениламидинотиомочевины, что это соединение является 3,5-ди-
анилино-1,2,4-тиадиазолом [50].
А
СЛН5ЫН—< N
N---И—NHc6h5
А
HN=< N-C6H5
C6H5—N---1=NH
Фромм и Биттерих [74] изучали расщепление 2,4-дифенил-3,5-бис-(фенил-
имино)-1,2,4-тиадиазолидина в различных условиях. Они установили, что
обработка бромом в спирте или крепкой соляной кислотой приводит к обра-
зованию трифенилгуанидина. При нагревании тиадиазолидина с уксусной
кислотой получается 2-анилинобензотиазол, а с анилином — 2-(трифенил-
гуанидино)бензотиазол. Эти интересные реакции приведены на схеме.
S
НС1 C6H5N=.Z \ _СбНз СНзСООН
H5C6-N—1=nqh5
nc6h5
II
c6h5nhcnhc6h5
ceH5NH2
| || V- NHCeH5
ф vy
N NC6H5
/V x n
I X_N-C-NHC6H5
S C6H5
1,2,5-ТИАДИАЗОЛЫ
la
Введение и номенклатура
Одноядерные 1,2,5-тиадиазолы были синтезированы только в перцод
написания настоящего обзора. Однако бензо- и конденсированные гетеро-
циклические производные, а также некоторые 1,2,5-тиадиазолидины были
известны ранее. Первые попытки получить родоначальный гетероцикл отно-
сятся к 1897 г. [81], когда в надежде получить 1,2,5-тиадиазол безуспешно
1,2,5-ТИАДИАЗОЛЫ
443
пытались провести реакцию этилендиамина с сернистым ангидридом. Удиви-
тельно, что простой метод окисления бензопроизводного, обычно применяе-
мый для получения многих других гетероциклов, не был известен до 1958 г.
Паттерсон предложил номенклатуру и нумерацию этой гетероцикличе-
ской системы, изображенную формулой I. Рихтер приводит другую нумерацию,
соответствующую формуле 1а. Известны другие наименования: тио- [aat 1-ди-
азол и пиазтиол. Последнее применяется для бензопроизводного этой системы.
Синтез и реакции
Последним из одноядерных тиадиазолов, который предстоит рассмотреть,
является 1,2,5-тиадиазол. Окисление 2,1,3-бензотиадиазола озоном [82] или
4-нитро-2,1,3-бензотиадиазола перманганатом калия приводит к 1,2,5-тиа-
диазолдикарбоновой-3,4 кислоте. Ступенчатое декарбоксилирование [83] поз-
воляет получить родоначальное соединение— 1,2,5-тиадиазол.
Из моно- и дикарбоновых кислот приготовлены различные производные:
сложные эфиры, хлорангидриды, амиды и гиразиды. Попытка получить 4-ами-
но-1,2,5-тиадиазол реакцией Курциуса оказалась безуспешной ввиду нестой-
кости 4-карбэтоксиамино-1,2,5-тиадиазола к щелочному гидролизу и инерт-
ности к кислотному гидролизу.
1,2,5-Тиадиазол [83] — бесцветная жидкость с т. кип. 94°, т. пл.—50,1°
и запахом, напоминающим запах пиразина. Он растворим в большинстве орга-
нических растворителей и при растворении в воде образует нейтральный
раствор. Подобно замещенным 1,2,5-тиадиазолам, родоначальный цикл рас-
крывается при восстановлении цинком в соляной кислоте с образованием
в качестве одного из продуктов реакции — сероводорода. Окисление 1,2,5-тиа-
диазола надуксусной кислотой также приводит к расщеплению кольца. Бром
не вступает в реакцию с 1,2,5-тиадиазолом в четыреххлористом углероде
в присутствии железных опилок или на солнечном свету.
1,2,5-Тиадиазолидин
Некоторые представители ряда 1,2,5-тиадиазолидина синтезированы [84]
из N-замещенных этилендиаминов и двухлористой серы. Хлористый суль-
фурил дает 1,1-диокись, а хлористый тионил — 1-окись. Этим способом были
444
ТИАДИАЗОЛЫ
получены: 2,5-дидодецил-1,2,5-тиадиазолидин, 1,1-диокись 2,5-диметил-1,2,5-
тиадиазолидина и 1-окись 2,5-дибензил-3-метил-1,2,5-тиадиазолидина. Сооб-
щается также, что при действии гипохлорита натрия и затем сульфида натрия
на дибензоил- или дибензолсульфонилэтилендиамин можно получить 2,5-дибен-
зоил- или 2,5-дибензолсульфонил-1,2,5-тиадиазолидин [85]. Попытка полу-
чить этим методом 2,5-бис-(п-толуолсульфонил)-1,2,5-тиадиазолидин оказалась
безуспешной [83]. Эти авторы [83] отмечают, что данные, приведенные в рабо-
те [85], основаны лишь на результатах элементарного анализа содержания
азота и что в случае бензолсульфонилтиадиазола у исходного соединения и про-
дукта реакции температуры плавления очень близки (168 и 163—164°). О тем-
пературе плавления смеси ничего не сообщается.
S
Ci2H25NHCH2CH2NHC12H26 ----ч- Ci2H25—N N—С12Н25
so2ci2 /\
CH3NHCH2CH2NHCH3-------> СН3—N N—СН3
О
t
S
SOC12 / \
C6H5CH2NHCH2CHNHCH2C6H5-------> С6Н5СН2—N N —СН2С6Н5
ЧСН3
2,1,3-Бензотнадиазолы и родственные конденсированные циклические системы
I
Введение и номенклатура. Большинство литературных данных по'1,2,5-
тиадиазолам относится к бензо-, нафто- и другим конденсированным системам,
в состав которых входит этот гетероцикл. Нумерация, предложенная Паттер-
соном, показана на формуле I. Наиболее принятые названия 2,1,3-дибензо-
тиадиазол и бензизотиадиазол (1,2,3-бензотиадиазол обычно называют просто
бензотиадиазолом). В большом числе работ для этой системы приводится
название пиазтиол, которое, к несчастью, было применено к неизвестному
родоначальному соединению, самому 1,2,5-тиадиазолу. Нумерация, как пока-
зано в формуле 1а, приведена в справочнике Рихтера, но широко не приме-
няется. В справочнике Бейльштейна эти соединения именуются 3,4-бензо-
1,2,5-тиадиазолами.
Синтез. Гинсберг [86], синтезировавший некоторые 2,1,3-бензоселен-
диазолы, попытался получить аналогичные соединения, содержащие фосфор,
бор, теллур и серу. Опыты оказались успешными лишь в отношении серы
и являлись одним из первых синтезов 2,1,3-бензотиадиазола. Этот сийтез
Гинсберг проводил либо в автоклаве, в котором о-фенилендиамин нагревался
1,2,5-ТИАДИАЗОЛЫ
445
с сернистой кислотой или с концентрированным раствором бисульфита натрия,
либо пропуская в кипящий о-фенилендиамин сухой сернистый газ. Метод
с использованием автоклава преподчтительнее, так как дает лучшие выходы,
а при применении SO2 происходит образование смолистых побочных продук-
тов. Таким же путем Гинсберг синтезировал нафто-[1,2] [1,2,5]-тиадиазол
из 1,2-диаминонафталина [87]. Другие производные, как, например, 5-метил-
и 5-ЭТОКСИ-2,1,3-бензотиадиазол, были также синтезированы этим методом.
^/XNH2
+ 2NaHSO3 —» | | S+ Na2SO3 + 5H2O
Михаэлис [88] получил 2,1,3-бензотиадиазол из о-фенилендиамина и хло-
ристого тионила или тиониланилина. В более поздней работе [89] показано,
что метод Михаэлиса носит общий характер и дает высокие выходы.
ИЛИ
C6H5N=S=O
soci3
Интересный аналог пурина или птеридина был получен с выходом 73%
из 1,3-диметил-5,6-диаминоурацила и хлористого тионила [90].
Сообщается, что при действии гидроксиламина на изонитрозотиокамфору
образуются а-камфорхинондиоксим, (3-камфорхинондиоксим и борнилен-
1,2,5-тиадиазол [91]. По-видимому, сероводород, выделяющийся в процессе
СН3 СН3
Ov N nh2 Ck N N
V\/ 2 snr. v \
। II I I s
/\/\ \/\ /
CH3 II NH2 || N
О о
реакции, реагирует с диоксимом с образованием тиадиазола. Сообщается
также, что сероводород превращает диоксим (3-камфорхинона в борнилен-d/-
1,2,5-тиадиазол. Диметилглиоксим и диоксим циклогександиона-1,2 не всту-
пают в этих условиях в реакцию с сероводородом [83].
2,1,3-Бензотиадиазолин
Единственным описанным синтезом 2,1,3-бензотиадиазолина является
получение 2,3-диацетил-2,1,3-бензотиадиазолина из Ы,Ы'-диацетил-о-фени-
лендиамина [85]. Диацетил-о-фенилендиамин превращается в ди-Ы-хлор-
446
ТИАДИАЗОЛЫ
амид, который циклизуется под действием сульфида натрия в бензотиадиа-
золин.
NHCOCH3
NHCOCH3
СОСН3
li-Cl
У\/
k/\
N —CI
<Loch3
NaaS
I
COCH3
Реакции и свойства. Вопрос о структуре 2,1,3-бензотиадиазола в течение
многих лет являлся спорным и только недавно он стал получать удовлетво-
рительное решение. Были предложены следующие структуры: 1,'П и III. Резуль-
таты исследования дифракции электронов [92] подтверждают: правомерность
структур I и II; при этом предполагается, что вклад этих двух резонансных
III
форм приблизительно одинаков. Строение этого гетероцикла может изобра-
жаться мезоионной структурой (формула IV или V) [93]. Это, по-видимому,
не является существенным для понимания строения 2,1,3-бензотиадиазола,
так как его дипольный момент (1,73 D) [94] ниже, чем можно было ожидать.
Некоторые мезоионные соединения, например сидноны, имеют дипольный
момент выше 5,0 [95]. Тем не менее при рассмотрении ионных реакций 2,1,3-бен-
Li
б" I
N N
/\/ \ /Х/ \
I S + C6H5Li -> II S
\/\ / \/\ /
N N
6+ |
СеНэ
CeHjLi |
y\/NL12 ^\/NLi2
(C6H5)2S +||| <- | ||
^/xNLi SC6H5
СбЩ с6н5
h2o |
nr
^/\nhc6h5
1,2.5-ТИАДИАЗОЛЫ
447
зотиадиазолов мезоионные структуры, вероятно, имеют преобладающее зна-
чение. Так, например, показано [96], что при нуклеофильной атаке 2,1,3-бен-
зотиадиазола нуклеофильный реагент присоединяется к атому азота гетеро-
цикла. При действии фениллития на 2,1,3-бензотиадиазол образуется N-фенил-
о-фенилендиамин и дифенилсульфид. Ход реакции, которая представляет
собой удобный препаративный метод получения N-замещенных о-фенилен-
диаминов, показан на схеме на стр. 446.
Исследования Эфроса и Левита [97, 98] показали, что электрофильные
агенты атакуют бензольное кольцо 2,1,3-бензотиадиазолов в положениях 4 и 7.
Это, по-видимому, следствие общей электроноакцепторной природы тиадиазоль-
ного кольца, которая проявляется в электронной недостаточности в положе-
ниях 5 и 6 бензотиадиазола. При нитровании 2,1,3-бензотиадиазола образуется
4-нитропроизводное [97, 98], а не 5-нитро-2,1,3-бензотиадиазол, как сообща-
лось ранее [99]. Сульфирование тоже ведет к образованию 4-замещенных
производных [98]. Выходы в этих реакциях высокие. Восстановление 4-нит-
ро-2,1,3-бензотиадиазола цинком в соляной кислоте дает с 67%-ным выходом
1,2,3-триаминобензол [97] и представляет собой препаративный метод его
получения.
nh2
Zn NH2
нЕГ> U~nh2
В присутствии железа в качестве катализатора 2,1,3-бензотиадиазол хло-
рируется в положения 4 и 7. Монохлорпроизводные при этом не были выделены.
Реакция в отсутствие катализатора не проходила [98].
Среди производных 2,1,3-бензотиадиазола, о которых сообщает Гинсберг,
имеется полученный из соответствующего диаминотолуола 5-метил-2,1,3-бен-
зотиадиазол [86]. Это соединение при обработке одним эквивалентом брома
в уксусной кислоте дает монобромпроизводное. О том, что произошло бро-
мирование бензольного ядра, свидетельствует устойчивость соединения к азот-
нокислому серебру или основанию. Получено также мононитропроизводное
5-метил-2,1,3-бензотиадиазола.
448
ТИАДИАЗОЛЫ
4- и 5-Нитро-2,1,3-бензотиадиазолы были синтезированы из соответ-
ствующих диаминонитробензолов и хлористого тионила. [97—100]. Восста-
новление нитробензотиадиазолов цинком в уксусной кислоте приводит к обра-
зованию аминобензотиадиазолов. Аминосоединения представляют собой сла-
бые основания, по своему поведению сходные с нормальными ароматическими
аминами. Например, 4-хлор- и 5-хлор-2,1,3-бензотиадиазолы могут быть
получены из аминобензотиадиазолов по реакции Зандмейера. Амино-2,1,3-
бензотиадиазолы сочетаются с азосоединениями, образуя интенсивно окра-
шенные азокрасители.
2,1,3-Бензотиадиазолы по своим физическим свойствам (температурам
кипения, запаху и т. п.) сходны с хиноксалинами [86]. Они являются сла-
быми основаниями и образуют с сильными минеральными кислотами соли,
неустойчивые по отношению к воде. 2,1,3-Бензотиадиазол устойчив по отно-
шению к спиртовому раствору хлорида ртути (II) (при 250°) и хромовой кис-
лоте, однако перманганат медленно окисляет его с образованием продуктов
неизвестного состава [86]. Как уже указывалось, олово или цинк в соляной
кислоте восстанавливают 2,1,3-бензотиадиазолы в о-аминосоединения и серо-
водород.
1,3,4-ТИАДИАЗОЛЫ
й ;й
1 / 1а
Введение и номенклатура
1,3,4- Тиадиазолам и его дигидропроизводным посвящена большая часть
литературы по тиадиазолам, и с ними проведено больше исследований, чем
со всеми другими изомерами этого ряда. Производные этой циклической
системы нашли применение в качестве лекарственных препаратов, ингибиторов
окисления, цианиновых красителей и комплексообразователей с металлами.
Нумерация атомов в этом изомере отвечает общепринятой (I). Другая
нумерация предложена Рихтером (1а). Правильное наименование— 1,3,4-
тиадиазол. Среди других встречающихся в литературе названий — тиобиазол,
тио-[&&1]-диазол и 1,3,4-тиодиазол.
Синтезы
Синтез из тиосемикарбазидов. Метод I. Многие синтезы 1,3,4-тиадиазолов
исходят из. тиосеми карбазида или замещенных тиосемикарбазидов. Фройнд
и Мейнеке [101] показали, что сам тиосеми карбазид при взаимодействии с хло-
ристым ацетилом циклизуется непосредственно в 2-амино-5-метил-1,3,4-тиа-
диазол. Это простой и, по-видимому, наиболее общий способ получения 2-амино-
5-замещенных 1,3,4-тиадиазолов. В приведенном примере R может быть
метилом [101], норгиднокарпилом [102], бензилом [103], циклопропилом
’[104] и др. Единственным в своем роде является соединение, полученное из
s
О
R(ici + h2ncnhnh2
S
S 0 0
II II II
2H2NNHCNH2 + C1C(CH2)8CC1
s s
HgN —ij7 \— (CH2)g —/ — NH2
n-n Й-n
1,3,4-ТИАДИАЗОЛЫ
449
хлорангидрида себациновой кислоты [105]. Во многих случаях предпринима-
лись попытки, не всегда удачные, превратить эти аминотиадиазолы в сульф-
аниламидные производные.
Пульвермахер [106] впервые показал, что хлористый ацетил может вызы-
вать циклизацию алкил- или арилзамещенных тиосемикарбазидов. Напри-
мер, из хлористого ацетила и 4-метилтиосемикарбазида был синтезирован 5-ме-
тил-2-метиламино-1,3,4-тиадиазол. Аналогичный продукт получен из хлори-
стого ацетила и 4-аллилтиосемикарбазида.
S
S сн80С1 СН3— / NHCH3
H2NNHCNHCH3
Синтез из тиосемикарбазидов. Метод II. Когда Пульвермахер [106]
попытался ввести фенильную группу в ядро тиадиазола с помощью хлористого
бензоила, он натолкнулся на неожиданную трудность. Обработка 4-аллилтио-
семикарбазида хлористым бензоилом приводила к 4-аллил-1-бензоилтиосеми-
карбазиду и только при нагревании бензоилпроизводного с хлористым аце-
тилом происходила циклизация с образованием 2-аллиламино-5-фенил-1,3,4-
S OS
II С6Н5СОС1 II II СН3СОС1
H2NNHCNHCH2CH = CH2-------> CeH5CNHNHCNHCH2CH = CH2---------->
. S
? С6н5— nhch2ch = ch2
N-N
тиадиазола. Наблюдалось также, что при взаимодействии хлористого ацетила
с 1-формил-4-метилтиосемикарбазидом получался 2-метиламино-1,3,4-тиа-
диазол.
Ацилгалогениды в качестве агентов циклизации тиосемикарбазидов при-
менялись Марквальдом и Боттом [107]; они указывают, Что, в то время как
1-бензоил-4-фенилтиосемикарбазид удалось превратить при действии хлори-
стого ацетила в 2-амилино-5-фенил-1,3,4-тиадиазол (II), обработка хлористым
S
О S CHgCoci / \ NHCH3
II II --------------3
HCNHNHCNHCH3 N—N
бензоилом приводила к образованию 3,4-дифенил-5-меркапто-4,1,2-триазо-
ла (III). Хоггарт [ 108 ] показал, что выводы Марквальда и Ботта являются отно-
сительно правильными, так как при более тщательном изучении обнаружено,
что дегидратация 1-бензоил-4-фенилтиосемикарбазида при действии хлористого
ацетила или хлористого бензоила дает как тиадиазол, так и триазол — факт,
установленный ранее немецкими авторами. В каждом случае преобладаю-
щим изомером был тот, о котором сообщили Марквальд и Ботт.
S N
CeH5-/ \-NHC6H6 HS-^ \ '
N-N c6H5-ri—LceH5
II III
CH3COC1
I----------------> 11(44%), III (5%)
О I s
|| II CeH6COCl
CeH5CNHNHCNHCeH5----------> II (20%), III (32%)
I----------------> 11(40%), III (20%)
29 Заказ № 978
450
ТИАДИАЗОЛЫ
Синтез из тиосемикарбазидов. Метод III. Хоггарт [1081 получил ряд
2-амиио-5-арил-1,3,4-тиадиазолов,. используя в качестве дегидратирующего
агента фосфорную кислоту; для этих реакций можно также применять серную
кислоту. Хоггарт отмечает, что хотя ацилгалогениды являются столь же
эффективными агентами циклизации 1-бензоилтиосемикарбазидов, как фос-
форная или серная кислота, однако они нередко частично ацилируют амино-
группу тиадиазолов, давая смесь продуктов. Из всех тиосемикарбазидов,
для циклизации которых применяли фосфорную кислоту, только 1-бензоил-
4-фенилтиосемикарбазид дает смесь тиадиазольного и триазольного производ-
ных, как это имеет место при использовании в качестве циклизующего агента
ацилгалогенида. Примером мягкой циклизации с высоким выходом при дей-
ствии фосфорной кислоты является образование 2-бензамидо-5-фенил-1,3,4-
тиадиазола из 1,4-дибензоилтиосемикарбазида. Показано также, что 1-ацил-
тиосемикарбазиды циклизуются под действием бен'золсульфокислоты, напри-
мер при получении 2:амино-5-амил-1,3,4-тиадиазола [109].
О S O „ _ S 9
II II II Н3РО4 / V II
CeH5CNHNHCNHCC6H5 * свН5—ц NHCCeH5
N—N
Хоггарт установил, что при дегидратации 1-бензоилтиосемикарбазидов
в щелочной среде, например в присутствии этилата натрия, получаются только
замещенные меркаптотриазолы.
Муравьиную кислоту также можно применять для циклодегидратации
ацилтиосемикарбазидов. Например, 1-формил- или 1-бензоилтиосемикарба-
зид могут быть с помощью муравьиной кислоты легко превращены соответствен-
но в 2-формамино-1,3,4-тиадиазол или 2-формамино-5-фенил-1,3,4-тиадиазол
OS S
HCNHNH^NHa (1) нсоон> / NH2
(2) Н+ N N
OS s
CeH5CNHNHCNH2 -(1) hco°^ CeH5—/ nh2
(2)H+ || 11
N—N
[ПО]. Формильная группа легко может быть удалена, в результате чего обра-
зуется свободное аминосоединение. При действии уксусного ангидрида на
1-ацетилтиосемикарбазид получен 2-ацетиламидо-5-метил-1,3,4-тиадиазол.
Синтез из тиосемикарбазидов. Метод IV. Пульвермахер [106] йаблюдал,
что муравьиная кислота может действовать подобно галогенангидридам жирных
кислот, осуществляя ацилирование и циклизацию в одну стадию. Так, он
установил, что при нагревании 4-фенилтиосемикарбазида с муравьиной кисло-
той образуется 2-анилино-1,3,4-тиадиазол. Очевидно, здесь сначала происхо-
дит формилирование, а затем циклодегидратация, как это имело место в преды-
дущем методе. Однако вследствие простоты этот метод заслуживает примене-
ния специально для синтеза тиадиазолов.
S S
H2NNH(3NHCeH5 НС°°Д V~ NHCeHs
N—N
Синтез из тиосемикарбазидов. Метод V. Удобный препаративный метод
получения 2-амино-5-меркапто-1,3,4-тиадиазолов разработал Гуа [111]. При
1,3,4-ТИАДИАЗОЛЫ
451
действии на тиосемикарбазид сероуглерода и едкого кали образуется калиевая
соль тиосемикарбазиддитиокарбоновой-4 кислоты (IV). При нагревании соеди-
нения IV до 140° происходит циклизация с образованием соли 2-амино-5-
меркапто-1,3,4-тиадиазола. При добавлении иода к раствору соединения IV
S S S S
h2nnh^nh2+cs2 —°Д kscnhnhcnh2 ks— ц 4j—NH2
N-h
IV
112
s s
H2N-/ \ S-S-Z nh2
. II I ll
N-N N — N
образуется дисульфид. Эта реакция является общей: из 4-замещенных тио-
семикарбазидов образуются 2-замещенные амино-5-меркапто-1,3,4-тиадиазолы,
а из 1-замещенных и 1,4-дизамещенных тиосемикарбазидов — тиадиазоли-
дины, которые будут рассмотрены ниже. Так, например, 4-фенилтиосемикарба-
зид дает 2-анилино-5-меркапто-1,3,4-тиадиазол. Промежуточное соединение
типа IV было выделено только в случае самого тиосемикарбазида.
S S
H2NNHCNHC6H5 + CS2 А°Д HS—J J" NHCeH5
Гуа [111] показал также, что в некоторых случаях сероуглерод в ней-
тральной среде реагирует непосредственно с тиосемикарбазидами, образуя
аминомеркаптотиадиазолы. По-видимому, положение 1 в тиосемикарбазиде не
должно быть замещено, поскольку 1-фенилтиосемикарбазид не реагирует
с сероуглеродом даже при 190°, а 4-фенилтиосемикарбазид гладко вступает
в реакцию при 150°. При взаимодействии сероуглерода с тйосемикарбазидом
в нейтральной среде образуется небольшое количество 2,5-димеркапто-1,3,4-
тиадиазола, по-видимому, в результате замены аминогруппы при действии
образующегося в ходе реакции Сероводорода.
S S S
H2NNHcJnH2 + CS2 ——> HS— ''j-NHa _j_ HS—^^-SH
N-N N—Is
При проведении реакции в диметилформамиде при 80° 2-амино-5-меркапто-
1,3,4-тиадиазол был получен с выходом выше 90% [112].
Синтез из тиосемикарбазидов. Метод VI. Из калиевой соли дитиоизони-
котиновой кислоты и тиосемикарбазида получен тиоизоникотиноилтиосеми-
карбазид, который при кратковременном нагревании при 260р [113] 'йфевра-
щался в 2-амино-5-(4-пиридил)-1,3,4-тиадиазол с умеренным выходом. %
s s s s Ч
N^ ^SK+H2NNK(i!NH2 -> N^________________CNHNHcJnH2 22_греваи22
NC/-ij/ ¥nh'
N—N
29*
452
ТИАДИАЗОЛЫ
Синтез из тиосемикарбазидов. Метод VII. В другом методе получения
1,3,4-тиадиазолов с применением тиосемикарбазида используется ортому-
равьиный эфир. Эйнсуорс [114], основываясь на более ранних сообщениях
о реакции ортомуравьиного эфира с тиокарбогидразидом, при которой обра-
зуются 1,2,4-триазолы, провел реакцию с тиосемикарбазидом. Вместо триазола
получился 1,3,4-тиадиазол. Первоначальным продуктом реакции в случае
избытка эфира, по-видимому, является М,М'-бпс-(1,3,4-тиадиазол-2-ил)форм-
амидин (V). Под действием кислоты он легко превращается в 2-амино-1,3,4-
тиадиазол, и, взаимодействуя с ортомуравьиным эфиром, может снова превра-
щаться в замещенный формамидин. В присутствии одного эквивалента орто-
эфира единственным продуктом, который удалось выделить, был аминотиадиа-
S
---------H2NNHCNH2 + HC(OC2H5)3
Избыток
НС(ОС2Н5)3
100°
100°
н+
----------->
НС(ОС2Н5)з
зол. Реагируя при более высоких температурах с избытком ортомуравьиного
эфира тиосемикарбазид дает эфир VI. При действии на соединение VI одного
эквивалента 2-амино-1,3,4-тиадиазола образуется бпс-тиадиазолоформами-
дин (V).
s s
II HC(OC2H5)3 if fl— N = CHOC2H5
h2ncnhnh2 ------—|| ||
N-N
VI
При замене ортомуравьиного эфира ортоуксусным образуется замещенный
тиосемикарбазон, которому приписывается строение VII. Это соединение при
180° теряет молекулу спирта, причем образуется смесь равных количеств
2-амино-5-метил-1,3,4-тиадиазола и 5-меркапто-3-метил-1,2,4-триазола [114].
Главным продуктом реакции тиосемикарбазида с ортопропионовым эфиром
является 2-амино-5-этил-1,3,4-тиадиазол. Последняя реакция протекает при
температуре кипящей водяной бани.
S СН3 S
(C2H5O)3CCH3+H2NNHCNH2 -> C2H5OC = NNH(^NH2
VII
180°
s
сн3-/ >-nh2
N — b
S zS4
II CgH3—1| ||—NH<
(C2H5O)3CCH2CH3+ h2nnhcnh2 -> || |
N—N
1,3,4-ТИАДИАЗОЛЫ
453
При кратковременном нагревании тиосемикарбазида с одним эквивалентом
ортомуравьиного эфира образуется тиосемикарбазон этилформиата (VIII)
[114, 115], который, весьма вероятно, является промежуточным продуктом при
образовании аминотиадиазола. Это подтверждается превращением соединения
VII под действием уксусного ангидрида в 2-ацетиламино-1,3,4-тиадиазол [115].
Интересно отметить, что при действии на соединение VIII теплого разбавлен-
ного раствора карбоната натрия образуется 3-меркапто-1,2,4-триазол [115].
S S S
II |1 (СН3СО)2О NHCOCH3
HC(OC2H5)3+H2NNHCNH2 —> C2H5OCH = NNHCNH2------------->
VIII н Я
Синтез из тиосемикарбазидов. Метод VIII. Попытка синтезировать 1-(а-
метилтиобензилиден)тиосемикарбазид (IX) для изучения его противотубер-
кулезной активности оказалась безуспешной, так как это соединение в ней-
тральной или кислой среде, по-видимому, самопроизвольно циклизовалось
в 2-амино-5-фенил-1,3,4-тиадиазол [116]. При действии на иодид 4-(а-метил-
тиобензилиден)морфолиния (X) тиосемикарбазида в пиридине или другом
S SCH3 j_
свн5<^ —Д ceH5J:=N\ \)
X
I s
Н+ II
I h2nnhcnh2
S г SCH3 S
С6Н3 —.( NH2 -ch3sh I II
<-------LCeH5C = NNHCNH2J
N—N
IX
основании реакция протекает в ином направлении с образованием 1-(а-морфо-
линобензилиден)тиосемикарбазида (XI). Обработка соединения XI разбав-
ленными кислотами приводит к 2-амино-5-фенил-1,3,4-тиадиазолу, а кипящим
спиртовым раствором щелочи — к 3-меркапто-5-фенил-1,2,4-триазолу [116],
подобно тому как это имеет место в случае 1-бензоилтиосемикарбазида [117].
X + H2NNHCNH2 --> CeH5C= NNHCNH2 :--> || ||
1 XI N-N
Синтез из тиосемикарбазидов. Метод IX. При взаимодействии тиосеми-
карбазида с сероуглеродом и иодистым метилом в спиртовом растворе щело-
чи образуется промежуточное соединение, которое, по-видимому, имеет строе-
ние S-метилтиосемикарбазиддитиокарбонового эфира [118]. При нагревании
его в растворителях или с разбавленной кислотой происходит циклизация
с образованием 5-амино-2-метилтио-1,3,4-тиадиазола.
S
SCH3 S
,, кон I || н+
H2NNH(jNH2 + cs2+ CH3I--> HN = CNHNHCSCH3-------
S
H2N —/ >— SCH3
N —N
454
ТИАДЙАЗОЛЫ
Синтез из тиосемикарбазонов. Метод X. В 1901 г. Янг и Эйр [ 119] сообщили,
что бензальтиосемикарбазон может при действии хлорида железа (III) под-
вергаться окислительной циклизации, превращаясь в 2-амино-5-фенил-1,3,4-
тиадиазол. Показано, что 4-фенил- и 4-метилбензальтиосемикарбазоны анало-
гичным путем образуют 2-анилино- и 2-метиламино-5-фенил-1,3,4-тиадиазолы
соответственно. Этим путем Дэ и Рой-Чоудхари [120] синтезировали большое
число 5-замещенных 2-амино-1,3,4-тиадиазолов.
c6h5ch=nnhcnh2
FeCl3
s
с6н5_/ \-nh2
N-N
S
S
FeCl3 .
C6H5CH= NNH<3NHCH3
s
C6H5-/ NHCH3
Il II
N-N
Хольмберг [121] установил, что ряд тиосемикарбазонов альдоз может
быть превращен методом Янга и Эйра в производные тиадиазола. Например,
из тиосемикарбазона L-арабинозы с небольшим выходом был получен 2-амино-
5-(Ь-арабо)тетраоксибутил-1,3,4-тиадиазол (XII). Некоторые тиосемикар-
базоны кетонов способны вступать в реакцию Янга и Эйра только в том случае,
если группа, находящаяся в a-положении по отношению к карбонильной,
способна отщепляться. Например, тиосемикарбазон фруктозы окисляется
хлоридом железа (III) в D-изомер соединения XII с Потерей одного атома угле-
рода. Этот тип расщепления также имеет место при взаимодействии с хлори-
дом железа (III) тиосемикарбазонов а-оксиацетофенона, бензоина или фенил-
глиоксиловой кислоты. Каждый из этих тиосемикарбазонов образует один и тот
же конечный продукт — 2-амино-5-фенил-1,3,4-тиадиазол.
При попытках окислить тиосемикарбазоны перекисью водорода вместо
ожидаемого тиадиазола (стр. 455) [120] получены 1,2,4-триазолы. Показано,
S
II
ch=nnhcnh5
неон
HOCH Fecig
HOCH
CH20H
i-
носн
CHjOB.
II
c=nnhcnh2
HOCH
неон.
неон.
ch2qh
HOCH
CH2OH
XII
однако, что феррицианид кальция действует подобно хлориду железа (III)
[122]. С помощью этого реагента из соответствующего тиосемикарбазона был
получен 2-(п-аминобензолсульфониламино)-5-метил-1,3,4-тиадиазол.
1,г,4-ТИАДИАЗОЛЫ
455
S
c6h5ch==nnhcnh2 —
C6H5C=NNHCNHj—-
СН2ОН
s
C6H5—c=nnh^nh2 —
СА-снон
s
CeH5C=NNHGNH2--
соон
FeCl3
s
II H2O2
CeH5CH = NNHCNH2---->
S
II /,-4. HO-
CH3CH = NNHCNHSO,— f NHCOCH3------------->
d J X-----/ 3 Ca3[Fe(CN)e]2
— NHSO2 —NH2
Дэ и Рой-Чоудхари [1201 окисляли тиосемикарбазон а’цетона перекисью
водорода и сообщили о получении 2,5-бпс-(ацетонгидразоно)-1,3,4-тиадиазола.
При этом авторы основывались на ранней работе Гектора (см. стр. 438 о 1,2,4-
тиадиазолидинах), который первоначально установил, что при окислительной
конденсации двух молекул тиоамида образуется 1,3,4-тиадиазол. В более
поздней работе Гектор показал, что продуктами этих реакций в действитель-
ности были 1,2,4-тиадагаэолидины. Как отмечает Бамба [123] в своем обзоре,
посвященном тиадиазолам, Дэ и Рой-Чоудхари, по всей вероятности, полу-
чили 1,2,4-тиадвазолидин, а не описанный ими 1,3,4-тиадиазол.
Синтез из тиокарбазидов. Описанные выше реакции тиосемикарбазидов
близко сходны с превращением тиокарбазида и тиокарбазонов в гидразино-
производные 1,3,4-тиадиазола. Так, например, при нагревании 1-фенилтио-
карбазида с муравьиной кислотой образуется 2-фенилгидразино-1,3,4-тиадиа-
456
ТИАДИАЗОЛЫ
зол [124]. Действие муравьиной кислоты напоминает аналогичную реакцию
между муравьиной кислотой и тиосемикарбазидами [106].
S $
|| нсоон / NHNHC6H5
h2nnhcnhnhc6h5------> |
N—N
Другой реакцией, сходной с окислением тиосемикарбазонов [119], являет-
ся окисление 1-фенилбензальтиокарбогидразона в 2-фенил-5-фенилгидразино-
1,3,4-тиадиазол [125]. Эта реакция распространена и на другие тиокарбогид-
разоны, так что она носит общий характер.
S $
II FeCig c6h5nhnh-Z \-с6н5
C6H5CH = NNHCNHNHC6H5---->
N—N
В качестве третьего примера, относящегося к замещенным тиокарбазидам,
приведем образование 2-меркапто-5-фенилгидразино-1,3,4-тиадиазола из ксан-
тогената калия и фенилтиокарбазида [124].
S S $
II II CbHjNHNH-/ SH
C6H5NHNHCNHNH2 + C2H5OCSK —>
N—N
Синтез из дитиокарбазатов. Метод I. Другим методом получения 1,3,4-тиа-
диазолов являются реакции с промежуточным образованием замещенных
дитиокарбазиновых (дитиогидразинугольных) кислот и их эфиров. К этой
группе относится реакция образования 2,5-ди меркапто-1,3,4-тиадиазола в ре-
зультате взаимодействия сероуглерода и гидразина в основной среде [117, 126 ].
S
нп_ HS-/ SH
H2NNH2+2CS2 -ДД. I
Li
Синтез из дитиокарбазатов. Метод II. Эфиры 3-ацилдитиокарбазиновой
кислоты при обработке кислотами циклизуются с образованием замещенных
тиадиазолов [127, 128]. Реакция эта, по-видимому, носит общий характер. При
этом применялись как бензиловые, так и метиловые эфиры 3-ацилдитиокарба-
зиновой кислоты. Выходы составляли приблизительно 80%. Ацильные группы
могут содержать арильные или алкильные радикалы. В двух случаях было
О S
Н II
ch3cnhnhcsch2c6h5
S
н-ь СН3—'''jj SCH2C6H5
N— N
II (CF3CO)2O CF3 —/ SCH2C6H5
H2NNHCSCH2C6H5---------->
N—N
показано, что выделение ацилдитиокарбазата из смеси не обязательно. При
нагревании бензилового эфира дитиокарбазиновой кислоты с муравьиной
кислотой или трифторуксусным ангидридом образуются соответственно
2-бензилмеркапто-1,3,4-тиадиазол и 2-бензилмеркапто-5-трифторметил-1,3,4-
тиадиазол. Механизм, предложенный для катализируемой кислотой реакции,
приведен ниже.
1,3,4-ТИАДИАЗОЛЫ
457
Il п
c6h5cnhnhcsch3
При нагревании эфиров 3-ацилдитиокарбазиновой кислоты в щелочной
среде образуются 1,3,4-оксадиазолы. Так, например, бензиловый эфир 3-бен-
зоилдитиокарбазиновой кислоты превращается в 2-меркапто-5-фенил-1,3,4-
оксадиазол.
Синтез из дитиокарбазатов. Метод III. Этиловый эфир дитиокарбазиновой
кислоты может быть получен взаимодействием диэтилтритиокарбоната с гидра-
зином [129]. В результате реакции эквимолекулярных количеств этилового
эфира дитиокарбазиновой кислоты и диэтилтритиокарбоната в щелочной
среде образуются 2,5-диэтилмеркапто-1,3,4-тиадиазол и 2-этилмеркапто-5-
меркапто-1,3,4-тиадиазол, причем первый получается с более высоким выходом.
Предположено, что неустойчивое промежуточное соединение, которое может
быть выделено из продуктов реакции, является 1,2-бш>этиЛдитиокарбогид'ра-
зидом. При стоянии в течение нескольких дней диэтилмеркаптосоеди-
нение превращается с высоким выходом в 2-меркапто-5-этилмеркапто-1,3,4-
тиадиазол. Этиловый эфир дитиокарбазиновой кислоты реагирует также с
с этилксантогенуксусной кислотой, что приводит к получению 1-этилксанто-
s s
HO- II II
—> [ c2h5scnhnhcsc2h5
C2H5S
S S
II II
C2H5SCNHNH2 + c2h5scsc2h5
ген-2-этилдитиокарбогидразида [129]. При его нагревании выделяется серо-
водород и образуется 2-этокси-5-этилмеркапто-1,3,4-тиадиазол.
s s s s Haroe.
II II IL- II ванне
c2h5scnhnh24-c2h5ocsch2cooh—>c2h5sgNhnhcoc2h5--------->
s
C2H5S-/ \-OC2H5
N —N
При взаимодействии этилового эфира дитиокарбазиновой кислоты с карбок-
симетилдитиобензоатом предполагаемое промежуточное соединение — этил-
458
ТИАДИАЗОЛЫ
тиобензоилдитиокарбазат немедленно циклизуется с образованием 2-меркап-
то-5-фенил- и 2-этилмеркапто-5-фенил-1,3,4-тиадиазолов, причем первый полу-
чается с более высоким выходом [129].
S S $
II II с6н5—/ \—SC2H5
C2H5SCNHNH2 + C6H5CSCH2COOH —> II
N-N
s
C6H5
N-N
Синтез из дитиокарбазатов. Метод IV. Эфиры дитиокарбазиновой кислоты
могут быть превращены в эфиры тиосемикарбазидодитиокарбазиновой кисло-
ты действием тиоциановой кислоты, алкил изотиоцианатов или арилизотиоциа-
натов. Производные тиосемикарбазидов могут быть затем циклизованы в 1,3,4-
тиадиазолы действием кислот средней силы или при нагревании [118]. При об-
работке метилового эфира дитиокарбазиновой кислоты тиоцианатом калия
в соляной кислоте нет необходимости выделять промежуточно образующийся
тиосемикарбазидодитиокарбоновый эфир, так как он непосредственно цикли-
зуется
в 2-метилмеркапто-5-меркапто-1,3,4-тиадиазол.
S
|| HSCN
h2nnhcsch3------>
“ 6 НС1
S S
II II
h2ncnhnhcsch3
HS-|/ SCH
N—N
з
Показано, что в условиях, аналогичных условиях циклизации тиосеми-
карбазидодитиокарбоксилатов калия [118], арил- и алкилизотиоцианаты кон-
денсируются с метиловым эфиром дитиокарбазиновой кислоты. При этом
образуется промежуточное соединение (XIII), которое при благоприятных
условиях сразу же циклизуется в 2-замещенный амино-5-метилмеркапто-
1,3,4-тиадиазол [130]. Например, фенилизотиоцианат при кипячении с метило-
вым эфиром дитиокарбазиновой кислоты в разбавленной соляной кислоте
образует 2-анилино-5-метилмеркапто-1,3,4-тиадиазол. Эта реакция проводи-
лась также с некоторыми арилизотиоцианатами и аллилизотиоцианатом. Про-
ll Г Н Н 1 CH3S -/ nhc6h5
C6H5NCS + H2NNHCSCH3 -* Lc6h5nhcnhnhcsch3J —>
N—N
XIII XIV
межуточное соединение (XIII) может быть выделено, если реакция проводится
в отсутствие кислоты. При взаимодействии соединения XIII с уксусным ангид-
ридом образуется ацетильное производное соединения XIV, а при его нагрева-
нии с концентрированной соляной кислотой происходит отщепление одного
эквивалента анилина и получается 2-меркапто-5-метилмеркапто-1,3,4-тиадиазол.
Нагревание соединения XIII с 2н. раствором едкого натра приводит к отщеп-
лению метилмеркаптана и образованию 2-анилино-5-меркапто-1,3,4-тиадиазола.
При нагревании соединения XIII в отсутствие реагентов при 140° получается
соединение XIV [118].
НС1
XIII
NaOH
/ S
CH3S ii ii SN
II || +C6H5NH2
N —N
/ S
HS -< \- nhc6h5
II || +CH3SH
N— N
1,3,4-ТИАДИАЗОЛЫ
459
Синтез из дитиокарбазатов. Метод V. Этиловый эфир дитиокарбазиновой
кислоты вступает в реакцию с альдозами с образованием соответствующих
производных дитиокарбогидразона. Для проведения окислительной цикли-
зации двух из этих дитиокарбогидразонов в тиадиазолы был использован
S
II
ch=nnhcsc2h5
I
неон
I
HOCH FeCl3
HOCH
I
СН2ОН
НСОН N---N
HOCH
I
HOCH
I
CH2OH
хлорид железа (III) [129]. Таким путем получены 2-(Ь-арабо)тетраоксибутил-
5-этилмеркапто-1,3,4-тиадиазол и 2-(О-галакто)пентаоксиамил-5-этилмеркапто-
1,3,4-тиадиазол. Другие производные сахаров этилдитиокарбазата не дали
при окислении замещенных тиадиазолов.
s s s
II но- II FeCl3 CH3S— / \
CH3SCNHNH2 + CH2O—> ch3scnhn = ch2---------> ||
N — N
Метиловые и бензиловые эфиры дитиокарбазиновой кислоты реагируют
также с формальдегидом в щелочной среде, образуя дитиокарбазоны метил-
и бензилформальдегида [131]. Окисление хлоридом железа (III) дает 2-метил-
меркапто-1,3,4-тиадизол и 2-бензилмеркапто-1,3,4-тиадиазол. Последний был
также получен из бензилового эфира дитиокарбазиновой кислоты и муравьи-
ной кислоты [128].
Синтез из тиоацилгидразинов. Тиоацилгидразины могут служить исход-
ными веществами для получения 1,3,4-тиадиазолов. При цагревании тиобен-
зоилгидразина с ортомуравьиным эфиром образуется 2-фенил 1,3,4-тиадизол
[132]. При замене ортомуравьиного эфира ортоуксусным получается 2-метил-
5-фенил-1,3,4-тиадиазол [133].
S , S.
II и ц—СвНз
CeH5CNHNH2+HC(OC2H5)3-> || ||
N —N
При действии сероуглерода в щелочной среде тиоизоникотиноилгидразин
превращается в 2-меркапто-5-(4-пиридил)-1,3,4-тиадиазол [113].
$ ____ s
\ II но- N\ \\ 'ir-SH
ЬГ ^-CNHNH2 + CS2-------> \=/ || ||
N — N
Под действием муравьиной кислоты тиобензгидразид Гладко превращается
в 2-фенил-1,3,4-тиадиазол [121], который с более низким выходом получают
при действии хлорида железа (III) или трибромида пиридиния на тиобензгидра-
зон формальдегида.
S / s\ s
|| нсоон CeH5—/ >, [О] ||
CeH5CNHNH2-------> I || <—CeH5CNHN = CH2
N — N
Тиобензгидразон ацетальдегида при окислении персульфатом калия или
хлоридом железа (III) дает 2-метил-5:фенил- 1,3,4-тиадиазол. Тиобензгидра-
460
ТИАДИАЗОЛЫ
зон бензальдегида под действием перекиси превращается в 2,5-дифенил-1,3,4-
тиадиазол. Другие тиобензгидразоны альдегидов ведут себя аналогично.
При взаимодействии тиобензгидразида с ксантогенуксусной кислотой
получен 2-этокси-5-фенил-1,3,4-тиадиазол [134]. Тот же тиадиазол может быть
синтезирован из ксантогенгидразида и тиобензоилтиогликолевой кислоты [ 135 ].
Возможно, что обе эти реакции протекают с образованием одного и того же
невыделенного промежуточного соединения — М-тиобензоил-М'-ксантоген-
гидразида.
S S
II II
C2H5OCSCH2COOH + h2nnhcc6h5
s s
II II
C2H5OCNHNHCC6H5
, s.
-»C2H5O-/ C6H5
N — N
Л
I
I
s s
II II
C2H5OCNHNH2 + C6H5CSCH2COOH
Холмберг описал превращение тиобензгидразида в 2,5-дифенил-1,3,4-
тиадиазол, протекающее с небольшим выходом при нагревании в бензоле [136].
Эта реакция может быть осуществлена путем нагревания гидразида до темпе-
ратуры плавления, либо в разбавленном этаноле, либо в разбавленной кислоте
или основании [ 134 ], либо, лучше всего, окислением перекисью водорода [ 136 ].
II С6н5 / \-с6н5
cgh5cnhnh2-- II ||
N — N
Попытка получить тиоизоникотиноилгидразин действием гидразина на тиоизо- ’
никотинамид окончилась неудачей. Вместо него был выделен с умеренным
выходом 2,5-ди-(4-пиридил)-1,3,4-тиадиазол (XV) [137 ]. Этот дипиридилтиадиа-
зол образуется также в качестве побочного продукта при получении изонико-
тинамидразона из тиоизоникотинамида и гидразина в более мягких условиях,
чем указывалось выше [138]. Обе эти реакции, возможно, протекают через
S 7 S.
I h2nnh2 N
— CNH2------->
z 130°
N
N
N — N
XV
стадию промежуточного образования М,М'-дитиоизоникотиноилгидразина
(XVI), который самопроизвольно циклизуется в тиадиазол. Предполагается,
что получение 2,5-ди-(4-пиридил)- 1,3,4-тиадиазола из калиевой соли дитио-
изоникотиновой кислоты также включает промежуточное образование N,N'-
дитиоизоникотиноилгидразина [113].
S
CSK
H2NNH2
S S
л--Ч 11 /' ч.
ф—CNHNHC—XN
XVI
XV
Синтез изацилгидразинов. Штолле получил 2,5-дифенилтиадиазол различ-
ными методами, весьма сходными с описанными выше реакциями. Он пока-
1.3.4-ТИАДИАЗОЛЫ
461
зал, что бензоилгидразин [1391 или М,М'-дибензоилгидразин [140] реагирует
с пятисернистым фосфором с образованием 2,5-дифенил-1,3,4-тиадиазола.
В реакции с М,М'-дибензоилгидразином промежуточным продуктом является
М,М'-дитиобензоилгидразин. В условиях, при которых Холмберг синтезиро-
вал 2,5-дифенил-1,3,4-тиадиазол из тиобензоилгидразина [1361, по-видимому,
можно получить неустойчивый М,М'-дитиобензоилгидразин посредством транс-
тиоамидирования. Аналогичные превращения, по всей вероятности, происхо-
дят при реакции бензоилгидразина с пятисернистым фосфором.
О О
II II P2S5
c6h5cnhnhcc6h5—>
|| II
CeH5CNHNHCCeHs
-H2s
/ s\
CeHj —j| Ц’— CeH5
N —N
S S
II
CeH5CNHNH2
jp2S5
О
II
CeH5CNHNH2
Штолле и Кинд [1391 синтезировали дифенил-1,3,4-тиадиазол из бен-
зальбензгидразида или бензальазина при действии на них пятисернистого
фосфора. Дихлорбензальазин, полученный из М,М'-дибензоилгидразина и
пятихлористого фосфора, также давал с пятисернистым фосфором 2,5-дифе-
нил-1,3,4-тиадиазол [141].
Реакция М,М'-диацилгидразинов с пятисернистым фосфором использована
Штолле и его сотрудниками для получения большого числа 2,5-диарил- и диал-
килзамещенных 1,3,4-тиадиазолов [142, 1431. Например, N.N-диацетил-
S4 •
0 0 w г__/ \____сн.
II II 3 || || 3
CH3CNHNHCCH3 II II
гидразин дает 2,5-диметил-1,3,4-тиадиазол и т. д. Штолле и Кинд синтезиро-
вали несколько дитиадиазолов из диацилпроизводных оксалилдигидразина.
Таким путем были получены 5,5'-диметил- и 5,5'-дифенил-2,2'-ди-1,3,4-
О 00 О
II Illi II
CHgCNHNHCCNHNHCCHg
P2S5
тиадиазолы. Несимметрично замещенные 1,3,4-тиадиазолы также можно
получить методом Штолле. Так, при действии пятисернистого фосфора N-дифе-
нилацетил-М'-бензоилгидразин превращается в 2-бензгидрил-5-фенил-1,3,4-
тиадиазол [1441, а N-ацетил-М'-бензоилгидразин — в 2-метил-5-фенил-1,3,4-
О О
CgHsx II II
/CHCNHNHCCeH5
свн/
P2S5
тиадиазол [145]. Аналогично из N-изоникотиноил-М'-ацетилгидразина с не-
большим выходом получается 2-метил-5-(4-пиридил)-1,3,4-тиадиазол [137].
462
ТИАДИАЗОЛЫ
О О
/—\ ч 'I1
CNHNHCCH,
\=/ 3
p2s6
Синтез из тиоамидов. Шабрие с сотрудниками [146] ошибочно указали,
что тиобензамид и тионикотинамид окисляются в 2,5-дифенил и 2,5-ди-(3-
пиридил)-1,3,4-тиадиазол под действием метилового эфира М,М'-дихлоркарба-
миновой кислоты или иода [146]. Как указано в разделе, посвященном 1,2,4-
тиадиазолам (стр. 429), окисление тиоамидов приводит к 3,5-дизамещенным
1,2,4-тиадиазолам. Гофман и Габриэль [147] описали окисление тиобензамида
иодом с образованием 3,5-дифенил-1,2,4-тиадиазола, (с т. пл. 90°), который
по своим свойствам очень близок к продукту, полученному Шабрие (т. пл.
94,5°). Сообщается, что 2,5-дифенил-1,3,4-тиадиазол имеет т. пл. 141° [139].
Отсюда следует, что вещество, описанное Шабрие как 2,5-ди-(3-пиридил)-
1,3,4-тиадиазол в действительности является 3,5-ди-(З-пиридил)-1,2,4-тиадиа-
золом.
Синтез из дитиомочевин. Метод I. Дитиомочевину и замещенные дитио-
мочевины превращали в 1,3,4-тиадиазолы несколькими методами. Сама дитио-
мочевина при обработке 3%-ной перекисью водорода циклизуется в 2,5-ди-
амино-1,3,4-тиадиазол [148].Этим путем 1,6-дифенилдитиомочевинаокисляется
в 2,5-дианилино-1,3,4-тиадиазол, а 1-фенилдитиомочевина — в 2-амино-
- /Sx
I? И.О. H!N1 Г№2
H2NCNHNHCNH2 —Л llj
5-анилино-1,3,4-тиадиазол [149]. Подобным образом несимметрично заме-
щенные тиадиазолы получают окислением соответствующим образом замещен-
ных дитиомочевин [150].
S S [0] c6h5nh /Sy nh2
c6h5nhcnhnhcnh2 I II
N — N
Синтез из дитиомочевин. Методы II и III. При действии уксусного ангид-
рида на дитиомочевину образуется диацетильное производное 2,5-диамино-
1,3,4-тиадиазола [151]. Ацетильные группы легко удаляются гидролизом
с образованием родоначального тиадиазола. 1,6-Дизамещенные дитиомочевины
аналогичным образом превращали в диацетилдизамещенные аминопроизводные.
По-видимому, дитиомочевины могут также циклизоваться в присутствии
щелочи. Указывается, что Ганилино-6-фенилдитиомочевина при действии
20%-ной щелочи дает 2-анилино- и 2-меркапто-5-(0-фенилгидразино)-1,3,4-
тиадиазол [124]. Та же дитиомочевина под действием хлорида железа (III) пре-
вращается в 2-анилино-5-фенилазо-1,3,4-тиадиазол.
s s 2 0%-ное^
C6H5NHNHCNHNHCNHCeH5 К°н
S S
CeH5NHNH-| SH CeH8NHNH-|/ NHCeH5
N-N N — N
Синтез из дитиомочевин. Метод IV. Обработка дитиомочевины и заме-
щенных дитиомочевин соляной кислотой, фосгеном и другими реагентами
дает продукты, которые ошибочно описаны в старой литературе как триазолы.
1,3,4-ТИАДИАЗОЛЫ
463
То, что эти соединения являются в действительности тиадиазолами, было
впервые показано Бушем, [152, 153] и позднее Гуа [154]. Фройнд с сотруд-
никами [155] обрабатывал 1,6-дифенилдитиомочевину соляной кислотой и
C6HS С6Н5
HS~lf V54 + Н8~^Ч|[~МНС®Н=
s & hci N~N N—N
II If JK xvn XVIII
C4 HgNHCNJHMMcWHCeH?, Г
ийх HC\
hs 'Sj|—nhc6h5 c6h5n.h—NH.C6HS
N—N + N—N
MX XX
получил два соединения, которым он приписал триазольные структуры XVII
и XVIII. Фройнд [156] также установил, что при действии на дифенилдитио-
мочевину фосгена образуется лишь одно вещество, которому было припи-
сано строение XVIII [156]. Буш и Шмидт [152], показали, что при нагревании
дифенилдитиомочевины до температуры ниже 200° получаются два вещества,
идентичные тем, которые описаны Фройндом [155, 156]. Буш приписал этим
продуктам изомерные тиадиазольные структуры (XIX) и (XX) на следующем
основании: 1) соединение XX не обессеривается при обработке окисью ртути
и, следовательно, не содержит серы, способной существовать в форме тиокарбо-
нильной группы; 2) соединение XIX в щелочном растворе в присутствии
алкилгалогенидов образует моноалкилпроизводные, которые были устойчивы,
по отношению к окислителям (не образовывали дисульфидов) и которые имели
свободную группу N — Н, способную образовывать нитрозаминопроизводное.
Дитиомочевина также реагирует с фосгеном и с соляной кислотой подобно
замещенным дитиомочевинам. Полученным продуктам Фройнд [155, 156]
первоначально приписал также триазольную структуру; однако позднее Буш
иЛотц [153], атакжеГуа [154] показали, что они в действительности являются
тиадиазолами. Один из продуктов взаимодействия дитиомочевины с соляной
s s HCI HS-/S\-NH2 + h2n-/S\ nh2
H2NCNHNHCNH2 Нагревание |l I || II
2 л N — N N — N
XXI
н
/N\
HSq| |TSH
N —N
XXII
кислотой был назван дитиоуразолом и ему было приписано строение XXII.
Это соединение изомерно тиадиазолу XXI. Гуа [154] изучил это соединение
и представил доказательство невозможности существования двух тиольных
или тиокарбонильных функциональных групп, а также с несомненностью
установил тиадиазольную природу этого гетероцикла.
Модификации тиадиазолов, отличающиеся по температурам плавления.
Гуа [111] первый наблюдал, что 2-амино-5-меркапто-1,3,4-тиадиазол (XXI)
способен существовать в формах с различными температурами плавления.
464
ТИАДИАЗОЛЫ
Соединение, полученное Фройндом и Имгартом [155], плавится при 245°.
Однако Гуа установил, что при стоянии его в течение 3 месяцев температура
плавления этого соединения стала 234°. Гуа также показал, что нагревание
до 150° с концентрированной соляной кислотой дает соединение с т. пл. 224°.
Все эти модификации, согласно анализу, являются изомерами и обнаруживают
идентичные химические свойства. Так, например, с иодом они образуют один
и тот же дисульфид, с иодистыми алкилами — одни и те же моноалкилпроиз-
водные и т. д. Джанниа и Гуа [157] пытались объяснить различия между
изомерными соединениями, но, по-видимому, приуспели только в загроможде-
нии химической литературы. При рассмотрении этой проблемы они сначала
предположили, а затем отклонили возможность того, что модификации с раз-
ными температурами плавления являются кето-енольными таутомерами 2-
амино-5-меркапто-1,3,4-тиадиазола. Предложенные структуры приведены ниже.
Последние две были отвергнуты, так как в этих соединениях ни одним из мето-
дов не удалось обнаружить присутствия аминогрупп. Первые две структуры,
по мнению авторов, исключаются вследствие того, что такие таутомеры не
могут существовать как индивидуальные соединения. Между тем в той же
статье Джанниа и Гуа указывают, что дитиомочевина существует в виде двух
модификаций с разными температурами плавления (223 и 203°), которые опи-
саны как таутомеры. Показано, что эти две дитиомочевины при обработке
кипящей соляной кислотой дают один и тот же тиадиазол с т. пл. 245°. Будучи
не в состоянии выявить какое-либо химическое различие, Джанниа и Гуа
показали, что соединение ст. пл. 245° имеет более коротковолновый максимум
поглощения в ультрафиолетовой области спектра, чем соединение с т. пл. 234°.
На основании данных ультрафиолетовых спектров сделано предположение,
что более легкоплавкое соединение имеет строение 2-имино-5-меркапто-1,3,4-
тиадиазола (XXIII), а более высокоплавкая форма — мостиковую структуру
XXIV. Последняя противоречит правилу Бредта [158],'согласно которому
HSYfН (т.пл.2340)
N---NH
HSvS
| НИ (т.пл. 245°)
HN--N
XXIII
XXIV
в соединениях, имеющих кольцо с внутренним мостиком, атом углерода в месте
соединения двух колец не может иметь двойной связи. Исключения из правила
Бредта встречаются только тогда, когда кольца достаточно велики. Это пра-
вило в основном применимо к малым циклам, о которых здесь идет речь. Если бы
такое соединение действительно существовало, то оно, несомненно, должно
было бы быть очень неустойчивым, в то время как соединение, которому Джан-
ниа и Гуа приписали эмдо-иминную структуру, получено в результате обра-
ботки дитиомочевины кипящей концентрированной соляной кислотой. При-
рода модификаций с разными температурами плавления этого меркаптоамино-
тиадиазола пока еще не может быть объяснена, но с полной уверенностью
можно исключить такие структуры, как XXIV. Дальнейшее рассмотрение
этого вопроса приведено на стр. 473.
Синтез из монотиодимочевин. Известно, что монотиодимочевины цикли-
зуются аналогично дитиомочевинам, однако при этом образуются аминоокситиа-
диазолы вместо соответствующих меркаптопроизводных. Первые исследова-
,3,4-ТИЛДИАЗОЛЫ
4 65
тели полагали, что при циклизации монотиодимочевин подобно дитиомочеви-
нам образуются триазолы. Фройнд и Шандер Г159] нагревали монотиодимоче-
вину с концентрированной соляной кислотой и получили соединение, которое
ими было принято за оксимеркаптотрназол. Арндт и его ученики 11601 синте-
зировали триазол, используя щелочь вместо кислоты в качестве агента цикли-
зации, и показали, что полученное вещество отличается от соединения, синте-
зированного Фройндом, который, вероятно, получил 2-амино-5-окси-1,3,4-
тиадиазол. В связи с этим снова возникают осложнения. Соединение, сиптези-
О S S
I il ll(-l jin_/ \____Nil.,
II.,NCNHNHCNH, --------- j| I1 N“2 (т. пл. 177 )
N —N
рованное Фройндом, плавится при 177’. Однако если монотнодимочевипа
циклизуется в присутствии уксусного ангидрида [1511, то образуется моноаце-
тилпроизводное (т. пл. 295°), которое при гидролизе соляной кислотой и после-
дующей нейтрализации хлоргидрата (т. пл. 108') дает соединение, изомерное
полученному Фройндом тиадиазолу, но с т. пл. 240°. Оба изомера при нагре-
вании с уксусным ангидридом образуют одно и то же моноацетильное произ-
водство с т. пл. 295”. Джанниа и Гуа 11571 объясняют различие температур
плавления наличием соединения с мостиковой структурой XXV, однако это
объяснение неудовлетворительно, так как противоречит правилу Бредта.
,s^nh2
H'O'I (т. пл. 240°)
N--NH
XXV
Имеются указания, что эта изомерия не ограничивается незамещенными
производными. Так, например, 2-анилино-5-окси-1,3,4-тиадиазол также суще-
ствует в виде двух форм (т. пл. 206 и 246°). Обработка 1-фенил-2-тиодимочевины
уксусным ангидридом и гидролиз образующегося при этом моно- или диаце-
тата дает 2-анилино-5-окси-1,3,4-тиадиазол с т. пл. 206° [161]. Сообщается [162]
также, что изомер этого соединения с т. пл. 246° получается в качестве одного
из нескольких продуктов реакции между 4-фенилтиосемикарбазидом и моче-
О s s'
1[ || (I )(<’.![цСОЦО т тп / \ МНГ II
I1.>NCNIINHCNHC(!II5 - --- 11ОП| 1“N ° 5 (т. п.ч. 206 )
(2)HCI N—N
виной при повышенных температурах. Соединение с т. пл. 246 получено
также из 6-анилино-1-фенил-2-тиодимочевины действием соляной кислоты 11631.
Взаимосвязь между производными анилипоокситиадиазола с т. пл. 206 и 246"
не. была так тщательно изучена, как в случае незамещенного аминоокситиадиа-
зола, так как не удалось получить производных для этих двух форм.
S О
II II -2NH.,
C(iHf,NHCNHNH2-| IbNCNIL-----------1
S
н°п/ \_nhci;h5(t пл 2|б)
OS N — N
II Ii HCI j
c6h5nhnhcnhnhcnhc6h5 -1
—СбН51Мг11\П2
Обработка 6-фенил-2-тиодимочевины уксусным ангидридом приводит
к образованию моно- и диацетильных производных 2-амино-5-окси-1,3,4-
тиадиазола [161 ]. Из этого и других примеров следует, что заместитель в поло-
30 Заказ № 978
466
ТИАДИАЗОЛЫ
жении 6 всегда отщепляется от молекулы в процессе циклизации, ведущей
к образованию тиадиазолов.
О S
|| |1 (1)(СН3СО)2О
C6H5NHCNHNHCNH9---------------
3 “ (2)Гидролиз
S
НО-
I! h
N —N
1,3,4-Тиадиазолины и тиадиазолидины
Синтез из замещенных дитиокарбазатов. Различные 1,3,4-тиадиазолидины
можно получить, по-видимому, с помощью общей реакции между альдегидами
или кетонами и 2-замещенными дитиокарбазиновыми кислотами. Буш [164]
показал, что калиевая соль 2-фенилдитиокарбазиновой кислоты и ее метиловый
эфир легко вступают в реакцию с альдегидами, образуя ДМ,3,4-тиадиазолииы.
11а примере получения 5-метил-5-этил- и 5,5-диметил-2-меркапто-4-фенил-
S
CeH5NHNHCSK / CH.,CI JO
H/S
СНз-И 'p- SK
CeH5-N-N
s
II
C(,l Ir.NHNHCSCH.) : CIJ..0 —>
cBH5
s
/ sch::
N — N
S О
II И
CeH5NHNHCSK I CH:iCC2H5 —>
А2-1,3,4-тиадиазолина Буш и Штерн [165] установили, что с кетонами эта
реакция протекает так же, как и с альдегидами.
Буш и его ученики [166, 167] синтезировали также ряд4-арил- или4-алкил-
2-меркапто-А2-1,3,4-тиадиазолинтионов-5 реакцией соответствующих замещен-
ных калийдитиокарбазатов с сероуглеродом. При замене ’сероуглерода фос-
геном образуется А2-тиадиазолинон-5 [166].
S S S
II CSa \ / S к
CH3NHNHCSK------> | IF к
СН3 —N — N
S OS
II As/ \_ g[Z
CeH6NHNHCSK + СОС12 -» | ||
С6Н5 —N — N
Калиевая соль 2-фенилдитиокарбазиновой кислоты в зависимости от
условий реакции может взаимодействовать с фенилизотиоцианатом по двум
путям [168]. Сплавление при 115° дает тиадиазолин, тогда как при комнат-
ной температуре в щелочном растворе образуется изомерный трпазол.
S
II 115»
CeH5NHNHCSK +CgHjNCS-------->
CeH5N s
sk
— N — N
При взаимодействии хлористого бензоила с калиевой солью 2-фенилди-
тиокарбазиновой кислоты Буш [169] получил два вещества, из которых одно,
как указывается, представляет собой 1,2-дибензоилфенилгидразин, а дру-
гое — окисленный тиадиазолин, которому была приписана мостиковая струк-
тура XXVIa [170], отвергнутая в настоящее время, так как она противоречит
правилу Бредта [158]. Это же соединение получено в результате термического
1.3.4-ТИАДИАЗОЛЫ
расщепления 2,2'-дитио-быс-(4,5-дифенил-А5г-1,3,4-тиадиазолина). Природа его
более детально рассматривается в разделе, посвященном реакциям и свой-
ствам 1,3,4-тиадиазолов (стр. 464). Опубликованные данные в большинстве
? 8
С6Н5СС1 + c6h5nhnhcsk
С6Н5—N--N
XXVIa
CeHjNNHCOQHs
COCe^
СеН.г—N---N
XXVI
случаев позволяют приписать этому соединению мезоиоиную структуру, одна
из резонансных форм которого представлена формулой XXVI. Соединения
такого типа, не содержащие заместителей в положении 5, могут быть получены
реакцией хлоргидрата этилового формиминоэфира 1171]. или натрийдитио-
формиата |172] с солью арилдитиокарбазиновой.кислоты.
Синтез из тиоацилгидразинов. При обработке замещенных гидразинов
дитиокарбоновыми кислотами можно получить большое число тиогидразидов.
Установлено, что эти тиогидразиды легко реагируют с альдегидами и кето-
нами в присутствии соляной или уксусной кислоты с образованием А2-1,3,4-
тиадиазолннов 1173, 174]. Так, 1-тиоацетил-2-фенилгпдразин при взаимодей-
ствии с ацетальдегидом образует 2,5-диметил-4-фенил-А2-1,3,4-тиадиазолин,
а с ацетоном — 2,5,5-триметнл-4-фенил-А2-1,3,4-тиадиазолин. Найдено, что
эта реакция протекает также и с альдозами. Получены производные глюкозы,
галактозы, маннозы, арабинозы и других сахаров, однако с фруктозой продукт
реакции не был получен 1175, 176]. Обнаружено, что производные сахаров
СН3
Ч s ‘ S
I / S СН3ССН3 \
С11;(- СИ, Н 4 СН3-/ \-СИ:!
С0Н5—N — N C6H5NHNHCCH3-----------> ceH5—N—N
показывают более сильное вращение плоскости поляризации,
чем исходные сахара 1176].
"А
|| С!1,ОИ(С!ЮН)4— И CGIIr,
CI HOI 1(С1 К)11)/,СН О I CjjHr.NHNHCCoIl.-, — " г н J, J!
Синтез из а-хлоргидразонов. Бюлов и Зайдель [1771 показали, что этило-
вый эфир (2,4-дихлорфенилгидразоно)хлоруксусной кислоты взаимодействует
с ксантогенатом калия с образованием 2-карбэтокси-4-(2,4-дихлорфенил)-
А2-1,3,4,-тиадиазолинтиона-5 в качестве основного продукта. Фуско |178]
расширил сферу этой реакции путем использования а-хлорбензальфенилгид-
разона и разнообразных реагентов для получения некоторых гетероцикличе-
ских систем, в том числе и представителей ряда тиадиазола. При нагревании
С! Cl S
z---1 I Н
Cl-V ^-NHN^CCOOCaHs+KSCOCaHs
S S
V \ СООС,НГ1
—> С1 у— Л — N
CI
30*
468
тилдилзолы
с тиомочевиной в растворе это хлорпроизводное дало 2,4-дифенил-5-имино-
А2-1,3,4-тиадиазолин. N-Замещенные тиомочевины образуют ожидаемые тиа-
диазолины, а тиосемикарбазид реагирует обычным путем, давая производное
гидразона. Ксантогенат натрия реагирует так, как показано Бюловом и Зай-
делем (см. выше), образуя 2,4-дифенил-А2-1,3,4-тиадиазолинтион-5.
s
HN
S
ibNCNH»
ci I
Y и С0Н5
c6h5-n — 1Q
CelI5NHN — С —СвН5
s
H,NN S
H2NCNHNH.2
- Свн5
Синтез из замещенных тиосемикарбазидов. Известно, что замещенные
тиосемикарбазиды реагируют с фосгеном, тиофосгеном и другими реагентами
с образованием тиадиазолинов. Например, 1,4-дифенилтиосемикарбазид и тио-
фосген образуют 2-анилино-4-фенил-А2-1,3,4-тиадиазолинтион-5 [179]. Фосген
реагирует с дифенилтиосемикарбазоном с образованием соответствующего
тиадиазолинона [180]. Тиосемикарбазид, замещенный в положениях 2,4
и 1,2,4, может также циклизоваться при взаимодействии с фосгеном 1181 L
В последнем случае продуктом реакции является тиадиазолидин.
S
II CSC12
CoH5NHCNHNHC6H5-------
S S
Y Vnhc8h5
CgH5 — N — N
Показано, что различные 1-, 4--и 1,4-замещенные тиосемикарбазиды при
действии сероуглерода в щелочи циклизуются в соответствующие тиадиазо-
лины 11111. Сероуглерод в нейтральной среде легко вступает в реакцию, с
4-фенилтиосемикарбазидом, давая 2-анилино-5-меркапто-1,3,4-тиадиазол, одна-
ко с 1-фенилтиосемикарбазидом тиадиазолин не образуется. Фенилизотиоцианат
QJl.NHCNNH, -S0-2.
I
свн5
s NCeH5
ho-/ 'Y
N - N-- C,;H 5
s
II
C(1H5NHCNNHCrH5
CH3
COC[.2
0 s nc0h5
4/
I I
C6H5 -N —N—CeH5
при взаимодействии с 2-метил-4-фенилтиосемикарбазидом дает тиадиазолип.
Эта реакция, вероятно, протекает через промежуточное образование замещен-
S
S S
R'NHNHCNHR Y |j~NHR
КОН R'_ N —N
ной дитиомочевины. Тот же тиадиазолин образуется при сплавлении хлоргид-
рата замещенного тиосеми карбазида. Эта реакция протекает с выделением
метилгидразина в виде его хлоргидрата [179, 182].
1. .3.1 - Т И А Д И Л 3 О Л Ы 4 6g
При действии альдегидов в спиртовом растворе соляной кислоты на
S
II
C,;H:.\CS -- -------------- -- -----!
СИ;, -j.
C(iH;,N S
Il
CH, - N —N
2CeH5NHCNNH,-HCl —----------------------------------------
CH3 •
1,4-дифепилтиосемикарбазпд образуется 2-апилпио-4-феш1л-5-арил-или 2-апи-
лино-4-фенил-5-алкил-А2-1,3,4-тпадпаз()ЛШ1ы с почти количественным выхо-
дом [183].
S Н S
CG1I5NHCNHNHC6H5-| RCI1 О—> [' / г
! 1|
C0H5-N— N
N!1COH;,
Синтез из замещенного семикарбазида. Этиловый эфир а-(4-семикарба-
зпдо)пропионовой кислоты может быть превращен действием фенилизотиоциа-
пата в тиадиазолин [184]. Полагают, что промежуточно образующийся тиоги-
дантоип претерпевает раскрытие кольца,
обработке гидроокисью бария.
О СН3
C6H5NCS-|-H.,NCNHNHCH
I
COOC.J I
г
S NC0H-,
110 IT
N —N -CI1COOH ------
CH3
а затем снова циклизуется при
СФФ,
N
S=-C с=о
I
jhncnhn—сн
II '
О С113
i Ва(ОН)2
С«Н5
ICN S NH
i X .
ОС с соон
I I /
HN N - СН
СН3
Имидазо-[2, l-^J-тиадиазолы
Конденсированные 1,3,4-тиадиазолы могут существовать лишь в виде
производных тиадиазолина или тиадиазолидина. Пример такой системы опи-
сан в работе Бана и сотрудников, посвященной имидазо-[2,\-Ь 1-тиадиазолам.
При обработке 2-амино-1,3,4-тиадиазола а-галогенкетоном образуется 3-заме-
щенный 2-иминотиадиазолин. При кипячении в воде эти тиадиазолины легко
470
ТИАДИАЗОЛЫ
циклизуются в имидазотиадиазолы. Конденсация 2-амино-1,3,4-тиадиазола
с «-бромацетоном с последующей циклизацией приводит к 6-метилимидазо-
C1L,
. S , N
II Г Vй
N N - ;
СООСчНг,
5 О
а. irNI^l CI |,ССНСС)ОС211
। N N Ci
12,1-61-тиадиазолу 1185]. В результате реакции этилового эфира 1-хлорацето-
уксусной кислоты с 2-амино-5-метил-1,3,4-тиадиазолом получается 5-карб-
этокси-2,6-диметилимидазо-[2,1-6]-тиадиазол. Синтезирован также ряд других
производных, включая 6-фенил- и 6-(п-нитрофенил)-имидазо-12,1-61-тиадиа-
золы [1861.
Реакции и свойства
Незамещенный 1,3,4-тиадиазол синтезирован в 1956 г. 2-Бром-1,3,4-
тиадиазол получен реакцией Гаттермана из 2-амино-1,3,4-тиадиазола. Восста-
новление 2-бром-1,3,4-тиадиазола дает родоначальное соединение— 1,3,4-
тиадиазол |187|. 1,3,4-Тиадиазол — бесцветное достаточно устойчивое соеди-
нение с т. пл. 42', не дающее максимума в ультрафиолетовом спектре поглоще-
ния в области выше 220 лгр. Цинк в соляной кислоте или 30%-пая перекись
водорода разлагают это соединение,.хотя 1,3,4-тиадиазол более устойчив к пере-
киси, чем 1,2,4-изомер. Водный раствор щелочи также разлагает 1,3,4-тиадиа’
зол, однако он вполне устойчив по отношению к минеральным кислотам.
Интересно отметить сходство температур кипения при атмосферном давлении
1,3,4-тиадиазола (204 ) и пиридазина (208%.
Амипо-1,3,4-тиадиазолы вступают в реакции, характерные для аромати-
ческих аминов. Как показано выше, можно продиазотировать и заместить ами-
ногруппу. Диазотированный амин может также вступать в реакцию сочета-
ния с образованием азокрасителей. При диазотировании 2,5-диамино-1,3,4-
тиадиазола в концентрированной соляной кислоте образуется только моно-
диазониевая соль. Сочетание с фенолом дает 2-(п-оксифенилазо)-5-амино-1,3,4-
тиадиазол [188]. Диазотирование в концентрированной бромистоводородпой
кислоте приводит к 2,5-дибром-1,3,4-тиадиазолу. Как 2,5-дихлор-, так и 2,5-ди-
s ч
II2N — / %—NH2 (1) 1IONO
II </фснол Нм / . х X 7 > <)Н
N N || || /
|HONO, 1113г N-N
I
S
B'i irBr
N — N
1.3,4-ТИАДИАЗОЛЬ!
471
бромтиадиазол инертны по отношению к горячему спиртовому раствору азотно-
кислого серебра.
Аминогруппы 2,5-диамино-1,3,4-тиадиазола ацилируются нормальным
образом с образованием диацилпроизводных, однако конденсация с бензаль-
дегидом, фенилизотиоцианатом и цианом происходит лишь по одной из амино-
групп 11481.
Сообщается, что 2-амино-1,3,4-тиадиазол при нитровании дымящей азот-
ной кислотой 11891 дает 2-амшю-5-нитро-1,3,4-тиадиазол, который может быть
продиазотировап в концентрированной серной кислоте и введен в реакцию
сочетания с образованием азокрасителя.
/ s
HoN -1 N =СНС6Н5
11 1! 65
ii i*
N — N
Метнлт-иадиазолы взаимодействуют с бензальдегидом с образованием
стирилпроизводных, что указывает на электроноакцепторпый характер кольца
тиадиазола. Проведена конденсация 2-метил-5-фенил-1,3,4-тиадиазола с аро-
матическими альдегидами и п-нитрозодиметиланилином [145]. Реакцию бен-
зальдегида с 2,5-диметил-1,3,4-тиадиозолом можно провести в направлении
образования моно- или дибензилиденпроизводных |190]. Дальнейшим под-
тверждением электронооттягивающего характера ядра 1,3,4-тиадиазола слу-
жит сообщение о том, что нитрование 2-феиил-1,3,4-тиадиаэола смесью азотной
и серной кислот дает 50% мета-, 33% пара- и 1796 орто-нитрофепил-1,3,4-
тиадиазолов [191].
Попытка восстановления 1,3,4-тиадиазолов в 2,3-дигидронроизводныс
оказалась безуспешной 1192]. Были также предприняты попытки восста-
новить 5-фенил-2-(п-ацетиламннофенил)- и 5-метил-2-амино- 1,3,4-тиадиазолы
гидросульфитом натрия, цинком в спиртовом растворе щелочи и 5%-ной амаль-
гамой натрия в спирте. Лишь фенил- и п-ацетиламинофенилтиадиазолы всту-
CuHj-!7 г- СН =N - ( \-N(CH3)2
I ! ------------------" ! II
N — N N-N
-CH---CHC6Hr,
11 6 ’ C6H5CHO
: I
N - N
,SX
CeH5CH = CH—/
il
N —N
CH = CHC6H6
пали is реакцию, причем только при действии амальгамы натрия. Продукты
восстановления представляли собой тиосеми карбазоны бензальдегида и
n-ацетиламинобензальдегида. Предполагается, что реакция протекает через
т
ТИАДИАЗОЛЫ
стадию образования 2,3-дигидросоединений, однако вследствие неустойчи-
вости они немедленно разлагались до семикарбазонов. Существует предполо-
жение, что 2,3-дигидропроизводное может быть промежуточным продуктом
в процессе окисления тиосемикарбазонов альдегидов в тиадиазолы по методу
Янга и Эйре [1191.
/s\
С«НЬ - -и и
il !
N - - N
NH.,
“ Na(llg)r
s И 1 S
,./v , ' II
jj I N|[j ----------•CJICll ХХНСМЬ
N- Nil
Большинство меркапто-1,3,4-тиадиазолов вступает в обычные реакции
арилмеркаптанов, такие, как окисление в дисульфиды и сульфоновые кислоты,
алкилирование и т. д. Имеется, однако, несколько реакций, которые, по-види-
мому, более или менее специфичны для этого изомера тиадиазола. Окисление
2,5-димеркапто-1,3,4-тиадиазола иодом приводит к образованию дисульфида,
а при применении хлорида железа (III) образуется полисульфид [1931. Если
дитиол обработать уксусным ангидридом [194] или нагревать в течение
нескольких дней вводном или спиртовом растворе 1127], то образуется
сульфид.
^хНагревание или
\|(СН3СО)2О
У некоторых тиадиазолпидпсульфпдов при обработке аминами или аммиа-
ком происходит раскрытие кольца. При действии на дисульфид 2-меркапто-
5-метнл-4-фенил-А2-1,3,4-тиадиазолина аммиака образуется 1-фенилтиосеми-
карбазид и родоначальный меркаптан (XXVII). При замене аммиака анилином
получен 1,4-дифенилтиосемикарбазид и соединение XXVII [195]. Взаимодей-
ствие аминов или аммиака с дисульфидом 2-меркапто-5-фенил-А2-1,3,4-тиа-
c6h6nhnhcnh2 +
С6Н5—-N N
XXVP
диазолинтиона-5 протекает иным путем — с образованием смеси сульфенамида
(XXVIII) и свободного меркаптана (XXIX) [196]. При действии алкиламинов
получены алкилсульфенамиды, однако в реакциях с ариламинами, такими,
как анилин, был выделен не промежуточный N-арилсульфенамид, а продукт
перегруппировки, который, как было показано, является арилтиадиазолин-
сульфидом. Такая перегруппировка N-арилсульфенамидов в сульфиды встре-
чается также в карбоциклических системах [197].
1,3,4-ТИАДИАЗОЛЫ
473
Интересный ряд производных 1,3,4-тиадиазолина
который изучал эти соединения в течение нескольких
был описан Бушем,
лет. Буш 1164, 1981
XXVIII
установил, что при нагревании замещенных б«с-(-А2-1,3,4-тиадиазолин)дисуль-
фидов получаются два продукта: родоначальный меркаптан и соединение,
содержащее на два атома водорода меньше, чем исходное соединение. Буш
сначала предположил, что второе соединение имеет строение XXX. После
того как он выяснил, что это соединение не обессеривается под действием окиси
ртути (т. е. не имеет тиокарбонильной функциональной группы), он приписал
XXXI
XXXII
XXXIII
этому веществу строение XXXI 1170]. Шёнберг [199] обратил внимание на
тот факт, что такая структура, как XXXI, находится в противоречии с правилом
Бредта [158], и если все же она способна существовать, то соединения такого
типа должны быть крайне неустойчивыми. На самом деле соединения Буша
обладают высокой термической и химической устойчивостью. Шёнберг пред-
ложил представлять эти соединения в виде гибрида резонансных структур
типа XXXII и XXXIII, а также других, вытекающих из теории резонанса.
В таких структурах находят отражение как правило Бредта, так и химические
свойства этих соединений. Например, известно [200], что при кипячении соеди-
нения XXX (R = R' = С6Н5) в течение нескольких минут со спиртовым
раствором йодистого метила образуется иодметилат. Шёнберг приписывает
иодметилату строение XXXIV. На основании изложенных представлений
о строении так называемых э«до-тиотиадиазолов Шёнберг объяснил наличие
большого числа этих соединений, которые существуют в виде двух форм с раз-
личными температурами (см. об этих соединениях в разделе, посвященном
синтезу 1,3,4-тиадиазолов, стр. 464). Буш [200] изобразил одно из этих соеди-
нений в виде XXXV. Это вещество существует в двух формах с т. пл. 236 и 255°.
474
ТИАДИАЗОЛЫ
Согласно концепции Шёнберга, это соединение может существовать в виде двух
взаимопревращающихся изомерных форм (XXXVI и XXXVII), каждая из
СН2С6Н,
c,'№lrscHs сл~А
С6Н5_N— N C6H5-A-L{
Г
XXXIV XXXV
сн2с6н5
которых представляет собой гибрид нескольких резонансных структур. Можно
ожидать, что в рядах триазола и тиадиазола также будут обнаружены соеди-
нения с различными температурами плавления.
Бейкер [172] предположил, что представления Шёнберга правильны,
но неполны, поскольку не представлены все возможные резонансные струк-
туры, в том числе XXXII и XXXIII. С целью более удовлетворительного изо-
бражения подобных соединений Бейкер предложил применять мезоионную
формулу с использованием символа ±, как показано в формуле XXXVIII. Таким
XXXVIII
о'бразом, более адекватным графическим изображением является циклическая
структура с одинарными связями и с распределенным зарядом, показанным
символом +. Бейкер предложил для этого соединения название ф-(4-арил)-
2,4-дигидро-2-тио-1 -тиа-3,4-диазол.
Как уже указывалось в разделе, посвященном синтезу тиадиазолинов,
наиболее удобным методом получения мезоионных тиадиазолов является
S
кон II
---СНдСООН + c6h5nhnhcsk
S
NH3 II
—>- c6h5nhnhcnh2
реакция хлорангидрида кислоты с калиевой солью арилдитиокарбазино-
вой кислоты. На эту реакцию имеется много ссылок в статьях Бейкера [1721
по мезоионным соединениям.
При щелочном гидролизе мезоионные соединения расщепляются на исход-
ную кислоту и соль дитиокарбазиновой кислоты [164]. В некоторых случаях
амины действуют аналогичным образом. 4,5-Дифенилзамещенное соединение
при обработке аммиаком превращается в 1-фенилтиосемикарбазид [200]
Бензиламин вызывает образование 1-фенил-4-бензилтиосемикарбазида наряду
с другим соединением, которому приписано строение XXXIX, но, по-видимому,
ЛИТЕРАТУРА
475
S
C6HSCH,NH, II
c6h5nhnhcnhch2c6h5
xxxix
имеющего строение XL или XLI. При действии анилина и других ароматиче-
ских аминов образуются только циклические структуры, но не тиосемикар-
базид.
ЛИТЕРАТУРА
1. Pechmann, N о 1 d, Вег., 29, 2588 (1896).
2. Sheehan, I z z о, J. Am. Chem. Soc., 71, 4059 (1949).
3. Wolff et al., Ann., 325, 129 (1902); 333, I (1904).
4. Hur d, Mor i, J. Am. Chem. Soc., 77, 5359 (1955).
5. Staudinger, Siegwart, Ber., 49, 1918 (1916).
6 В a m b a s, The Chemistry of Heterocyclic Compounds, Interscience Publishers, New
York, 1952, p. 5. '
7. Kirmse, Horner, Ann., 614, 4 (1958).
8. Mazak, Suszko, Roczniki Chem., 9, 431 (1929) |C. A., 23, 4187 (1929)1
9. Hodgson, Dodgson, J. Soc. Dyers Colourists, 64, 65 (1948).
10. Bernthsen, Ann., 251, 1 (1889).
11. .1 acobson, Ber., 21, 3104 (1888).
12. J а с о b s о n, N e y, Ber., 22, 904 (1889).
13. J а с о b so n et al., Ann., 277, 209 (1893).
14. Rose, J. Chem. Soc., 1952, 3448.
15. H о d g s о n, J. Soc. Dyers Colourists, 40, 330 (1924).
16. В u r a w о у, T u r n e r, J. Chem. Soc., 1950, 469.
17. Burawoy et al., J. Chem. Soc., 1954, 82, 90.
18. S p e c k I i n, M e у b e c k, Bull. soc. chim. France, 18, 621 (1951).
19. G r e e n, P e r k i n, J. Chem. Soc., 83, 1201 (1903).
20. В a m b e r g e r et al., J. prakt. Chem., [2] 105, 251, 266 (1923).
21. Fries, Reitz, Ann., 527, 38 (1937).
22. Бельштейн, Курбатов, Ann., 197, 81 (1879).
23. P о 1 1 a k et al., Monatsh., 49, 221 (1918).
24. Fries et al., Ann., 454, 172 (1927).
25. Haase, Ber., 36, 588 (1903).
26. Hodgson, Dodgson, J. Chem. Soc., 1948, 1006.
27. Albert, Quart. Revs., 6, 197 (1952).
28. H a n t z s c h, Ber., 42, 68 (1909).
29. Hodgson, Marsden, J. Soc. Dyers Colourists, 59, 271 (1943).
30. Hodgson, Dodgson, J. Chem. Soc., 1948, 870.
31. Wohler, Gilbert’s Ann. Physik., 69, 273 (1821).
32. S c h u b a r t, Ber., 22, 2441 (1889).
33. T i e m a n n, Ber., 24, 369 (1891).
34. С г a у e n, Ber., 24, 385 (1891).
35. Кос h, Ber., 24, 394 (1891).
36. К r ii g e r, Ber., 18, 1053 (1885).
37. S t i e g 1 i t z, Ber., 22, 3148 (1889).
38. Hofmann, Ber., 2, 645 (1869).
39. Hofmann, Gabriel, Ber., 25, 1578 (1892).
40. Walther, J. prakt. Chem., [2] 69, 44 (1904).
476
ТИАДИАЗОЛЫ
41 Cronyn, Nakagawa, J. Am. Chem. Soc., 74, 3693 (1952).
42 Ch a br i er et al., Bull. soc. chim. France, [5] 16, 237 (1949).
43. Ishikawa, Sci. Papers Inst. Phys. Chem. Research (Tokyo), 3, 147 (1925) [C. A.,
19, 3087 (1925); Chem. Zentr., 96 [2], 2206 (1925)1.
44. Ishikawa, Sci. Papers Inst. Phys. Chem. Research (Tokyo), 7, 237 (1927) 1C. A.,
22, 1581 (1928); Chem. Zentr., 99 [1], 1763 (1928)].
45. Kitamura, J. Pharm. Soc. Japan, 58, 246 (1938) [C. A., 33, 1726 (1939); Chem.
Zentr., 110 [I], 4606 (1939)].
46. Holmberg, Svensk Kern. Tidskr., 41, 249 (1929) [C. A., 24, 2111 (1930)].
47. D u b s к у, T r t i 1 e k, Chem. obzor., 8, 1 (1933) [C. A., 27, 2137 (1933)].
48. Kurzer, J. Chem. Soc., 1955, 1.
49. Kurzer, J. Chem. Soc., 1955, 2288.
50. Kurzer, J. Chem. Soc., 1956, 2345.
51. Kurzer, J. Chem. Soc., 1956, 4524.
52. Kurzer, Chem. & Ind. (London), 1956, 1482.
53. Kurzer, J. Chem. Soc., 1957, 2999.
54. Slot ta et al., Ber., 63, 208 (1930).
55. Goerdeler, Bechlars, Chem. Ber., 88, 843 (1955).
56. G о e r d e 1 e r et al., Chem. Ber., 87, 57 (1954).
57. Goerdeler, герм. пат. заявка, G 14382 (September, 1956).
58. Goerdeler, Chem. Ber., 89, 2742 (1956).
59. G о e r d e 1 e r, W i 1 1 i g, Chem. Ber., 88, 1071 (1955).
60. G о e r d e 1 e r, F i n с к e, Chem. Ber., 89, 1033 (1956).
61. В a m b a s, The Chemistry of Heterocyclic Compounds, Vol. 4, Interscience Publis-
hers, New York, 1952.
62. Volckel, Ann., 43, 74 (1842).
63. Fleischer, Ann., 179, 204 (1875).
64. KI a son, J. prakt. Chem., [2] 38, 366 (1888).
65. Glutz, Ber., 3, 343 (1870).
66. Hantzsch, Wolvekamp, Ann., 331, 265 (1904).
67. Fromm, Ann., 275, 20 (1893).
68. Kurzer, Chem. Revs., 56, 154 (1956).
69. Fromm, Ann., 348, 144 (1906).
70. Hector, (a) Ber., 22, 1176 (1899); (6) Ber., 23, 357 (1890).
71. Hector, Ber., 25, Referate 799 (1892).
72. Haager, Doht, Monatsh., 27, 277 (1906).
73. H u g e r s h о f f, Ber., 36, 3121 <1903).
74. F г о m m, В i t t e r i c h, Ann., 394, 284 (1912).
75. Brintzinger, Schmahl, Chem. Ber., 87, 314 (1954)
76. G о e r d e 1 e r et al., Chem. Ber., 89, 1534 (1956).
77. Герм. пат. 927944 [С. A., 49, 12842 (1955)].
78. G о e r d e 1 e r et al., Chem. Ber., 87, 68 (1954).
79. Dos t, Ber., 39, 863 (1906).
80. Kurzer, Chem. & Ind. (London), 1956, 526.
81. Michaelis, G r a e n t z, Ber., 30, 1011 (1897).
82. Лесин, X а лецкий, ЖОХ, 28, 2126 (1958).
83. Weinstock, Synthesis and Properties of 1,2,5-Thiadiazole and Its Derivatives,
Ph. D. thesis to Indiana University (1958), University Microfilms, Ann Ardor, Michi-
gan, Library of Congress Card No. Mic. 58-2947, Carmack, Weinstock, and Shew, paper
presented before the 136th American Chemical Society Meeting (1959).
84. Пат. США 2624729 [С. A., 47, 11256 (1953)].
85. P e у г о n, Bull. soc. chim. France, 1949, 381.
86. H i n s b e r g, Ber., 22, 2895 (1889).
87. H i n s b e r g, Ber., 23, 1393 (1890).
88. Michaelis, Ann., 274, 262 (1893).
89. Халецкийи др., ДАН СССР, 106, 88 (1956) |С. А., 50, 13885 (1956)].
90. В 1 i с k е, G о d t, J. Am. Chem. Soc., 76, 2798 (1954).
91. Sen, J. Indian Chem. Soc., 15, 537 (1938).
92. L uzzait i, Acta Cryst., 4, 193 (1951).
93. В a к e r et al., J. Chem. Soc., 1950, 1542.
94. H i 1 1, S u t t о n, J. chim. phys., 46, 244 (1949).
95. Hill, Sutton, J. Chem. Soc., 1.949, 746.
96. L a n e, W i 1 1 i a m s, J. Chem. Soc., 1955, 1468.
97. Эфрос, Левит, ЖОХ, 23, 1629 (1953).
98. Эфрос, Левит, ЖОХ, 25, 183 (1955).
99. Халецкий, Песни, ЖОХ, 20, 1981 (1950).
100. Халецкий, Песни, ЖОХ, 24, 131 (1954).
101. Freund, Me i пес k е, Ber., 29, 2511 (1896).
102. Arnold, Ber., 75, 87 (1942).
ЛИТЕРАТУРА
477
103. Пат. США 2524729 (1950) [С. А., 45, 2020 (1951)].
104. Пат. США 2623877 [С. А., 47, 9356 (1953)].
105. Y а 1 е, J. Am. Chem. Soc., 75, 675 (1953).
106. Pulvermacher, Ber., 27, 613’ (1894).
107. Marckwald, Bott, Ber., 29, 2914 (1896).
108. Hoggarth, J. Chem. Soc., 1949, 1163.
109. Brooks et al., J. Chem. Soc., 1950, 452.
110. Ba n, J. Pharm. Soc. Japan., 74, 695 (1954) ]C. A. 48, 10741 (1954)].
HI. G u h a, J. Am. Chem. Soc., 44, 1510 (1922).
112. Англ. пат. 726045 (1955) [С. A., 50, 5037 (1956)].
ИЗ. К о n i g et al., Ber., 87, 825 (1954).
114. Ainsworth,!. Am. Chem. Soc., 78, 1973 (1956).
115. К a n а о к a, J. Pharm. Soc. Japan, 75, 1149 (1955) [C. A., 50, 5647 (1956)].
116. P e a k, S t a n s f i 1 d, J. Chem. Soc., 1952, 4067.
117. В u s c h, Ber., 27, 2507 (1894).
118. В u s c h, В i e h 1 e r, J. prakt. Chem., [2] 93, 339 (1916).
119. Young, Eyre, J. Chem. Soc., 79, 54 (1901).
120. De, Roy-Choudhurv, J. Indian Chem. Soc., 5, 269 (1928) |C. A., 22, 4123
(1928)].
121. Holmberg, Arkiv Kemi, 9, 65 (1955).
122. Пат. США 2447702 (1948) [С. A., 42, 8823 (1948)1.
123. В a m b a s, The Chemistry of Heterocyclic Compounds, Vol. 4, Interscience Pitbli-
shers 1952.
124. Guha, Roy-Choudhury, J. Indian Chem. Soc., 5, 163 (1928) [C. A., 23,
1397 (1929)].
125. Guha, Roy-Choudhurv, J. Indian Chem. Soc., 5, 149 (1928) [C. A., 23,
139 (1929)].
126. Losanitch, J. Chem. Soc., 121, 2542 (1922).
127. Fuji! et al., J. Pharm. Soc. Japan, 74, 1056 (1954) [C. A., 49, 11592 (1955)].
128. Young, Wood, J. Am. Chem. Soc., 77, 400 (1955).
129. Sandstrom, Arkiv Kemi, 4, 297 (1952).
130. G u h a, G u h a, Quart. J. Indian Chem. Soc., 4, 161 (1927) [C. A., 21, 3199 (1927)].
131. Sandstrom, Arkiv Kemi, 9, 255 (1956).
132. Ainsworth, J. Am. Chem. Soc., 77, 1148 (1955).
133. Пат. США 2733245 (1956) [С. A., 50, 12115 (1956)].
134. H a 1 m b e r g, Arkiv Kemi, Mineral. Geol., 25A, № 18 (1947) [C. A., 42, 5918 (1948)].
135. W a n g e 1, Arkiv Kemi, 1, 431 (1950).
136. Holmberg, Arkiv Kemi, Mineral. Geol., 17A, № 23 (1944) [C. A., 39, 4065 (1945)].
137. McMillan et al., J. Am. Pharm. Assoc., 42, 457 (1953).
138. Van der Burg, Rec. trav. chim., 74, 257 (1955).
139. Stolle, К i n d, J. prakt. Chem., [2] 70, 423 (1904).
140. S t о 1 1 e, J. prakt. Chem., [2] 69, 145 (1904).
141. S t о 1 1 e, T h о m a, J. prakt. Chem., [2] 73, 288 (1906).
142. Stolle, Ber., 32, 797 (1899).
143. Stole et al., J. prakt. Chem., [2] 69, 145, 366, 382, 474, 506 (1904).
144. A s p e 1 u n d, Acta Acad. Aboensis Math, et Phys., 5, №1,1 (1929) [Chem. Zentr.,
100 [1], 2414 (1929)].
145. О h t a et al., J. Pharm. Soc. Japan, 73, 1127 (1953) [C A., 48, 12091 (1954)].
146. C h a b r i e r et al., Bull. soc. chim. France, [5], 16, 237 (1949).
147. Hofmann, Gabriel, Ber., 25, 1586 (1892).
148. Fromm et al., Ann., 433, 1 (1923).
149. Fromm, J о к 1, Monatsh., 44, 297 (1923).
150. Patel, Ch a к r a v a r t i,.J. Indian Inst. Sci., A13, 85 (1930) [C. A., 24, 4770
(1930)].
151. Guha, J. Am. Chem. Soc., 45, 1036 (1923).
152. Busch, Schmidt, Ber., 46, 2240 (1913).
153. Busch, Lotz, J. prakt. Chem., [2] 90, 257 (1914).
154. Guha, J. Am. Chem. Soc., 44, 1502 (1922).
155. Freund, Imgart, Ber., 28, 946 (1895).
156. Freund, Wi'schewiansky, Ber., 26, 2877 (1893).
157. J a n n i a h, Guha, J. Am. Chem. Soc., 52, 4860 (1930).
158. В r e d t, Ann., 437, 1 (1924).
159. Freund, Schander, Ber., 29, 2506 (1896).
160. A r n d t et al., Ber., 55, 341 (1922).
161. Guh a, Chakrabort y, J. Indian Chem. Soc., 6, 99 (1929) [C. A., 23, 2974 (1929)].
162. G u h a, S e n, J. Indian Chem. Soc., 4, 43 (1927) [C. A., 21, 2900 (1927)].
163. G u h a, H у e, J. Indian Chem. Soc., 7, 933 (1930) [C. A., 25, 3633 (1931)].
164. Busch, Ber., 28, 2635 (1895).
165. Busch, Stern, J. prakt. Chem., [2] 60, 233 (1899).
478
ТИАДИАЗОЛЫ •
166. Busch, Ber., 27, 2507 (1894).
167. Busch et al., J. prakt. Chem., [2] 60, 51, 206, 212 (1899).
168. Busch, W о 1 p e r t, Ber., 34, 304 (1901).
169. Busch, J. prakt. Chem., [2] 60, 216 (1899).
170. Busch, J. prakt. Chem., [2] 67, 201 (1903).
171. Busch, Schneider, J. prakt. Chem., [2] 67, 246 (1903).
172. Baker et al., J. Chem. Soc., 1951, 289; 1950, 1542; 1949, 307.
173. W u V t s, L а с о u r t, Bull. sci. acad. roy. Belg., 19, 543 (1933) |C. A., 27, 5075
(1933)].
174. Wuyts, Wachsmuth, J. pharm. chim., 22, 289 (1935).
175. Wuyts, Compt. rend., 196, 1678 (1933).
176. W uyts, Verstraeten, Bull. sci. acad. rov. Belg., 20, 168 (1934); 21, 415 (1935)
[C. A., 28, .3407 (1934); 29, 5112 (1935)].
177. Bulow, Seidel, Ber., 57, 357 (1924).
178. Fusco, Musante, Gazz. chim. ital., 68, 147 (1938) [C. A., 32, 7456 (1938)];
ibid., 68, 665 (1938) [C. A., 33, 2518 (1939)]; Fusco, Rend. ast. limbardo sci., 71,
425 (1938) [C. A., 34, 3267 (1940)].
179. Busch, Holzmann, Ber., 34, 320 (1901).
180. Freund, Kuh, Ber., 23, 2821 (1890).
181. Busch, Limpach, Ber., 44, 560 (1911).
182. Marckwald, Sedlaczek, Ber., 29, 2920 (1896).
183. В i s c h, R i d d e r, Ber., 30, 849 (1897).
184. В a i 1 y, J. Am. Chem. Soc., 26, 1006 (1904).
185. В a n, J. Pharm. Soc. Japan, 74, 658 (1954) [C. A., 48, 10740 (1954)].
186. Matsukawa, Ban, J. Pharm. Soc. Japan, 72, 610 (1952) ]C. A., 47, 6409 (1953)]
187. Goerdeler, Ohm, Tegtmeyer, Chem. Ber., 89, 1534 (1956).
188. Hoile, Teherenbach, J. prakt. Chem., |2] 122, 289 (1929).
189. Пат. США 2708671 (1955) [С. A., 49, 15252 (1955)].
190. О h t a, К i m о t o, J. Pharm. Soc. Japan, 76, 10 (1956).
191. О h t a et al., J. Pharm. Soc. Japan, 73, 701 (1953) [C. A., 48, 7005 (1954)].
192. Медовщикова, Постовски й, ЖОХ, 24, 1989 (1954).
193. В u s с h, Z i е g е 1 е, J. prakt. Chem., [2] 60, 40 (1899).
194. Guha, Janniah, J. Indian Inst. Sci., 21A, 60 (1938) [C. A., 33, 599 (1939)].
195. Busch, Munker, J. prakt. Chem., [2] 60, 212 (1899).
196. Busch, Ber., 29, 2127 (1896).
197. Moore, Johnson, J. Am. Chem. Soc., 58, 1091 (1936).
198. Busch, J. prakt. Chem., [2] 60, 25 (1899).
199. Schonberg, J. Chem. Soc., 1938, 824.
200. В u s c h et al., J. prakt. Chem., |2| 67, 216 (1903)
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азакрин 167
Азаринин 7, 8, 9, 10
DL, синтез 10
Азациклопропен 311 321
2-Азидо-1,2,4-триазол 347
3,3'-Азо-сц.и.и-триазол, соли 348
5-Алкиламино-3-амино-1,2,4-тиадиазолы 431
2-Алкил-5-арил-1,3,5-оксадиазолы 402
Алкиларилфуроксаны 370, 374, 375
1-Алкилбензотриазолы 321, 322, 323
2-Алкилбеизотриазолы 313, 322, 323
1-Алкил-Р-карболины 216
4-Ал кил-2-меркапто-А2-1,3,4-тиадиазол и н-
тионы-5 466
3-Алкил-5-меркапто-1,2,4-триазолы 325
Алкилиафтиридины 177
Алкилоксадиазолоны 408
Алкил-ф-оксатриазолы 399
З-Алкил-4-оксикумарины 62
3-Алкил-1,2,4-триазолтиолы-5 345
1-Алкилтриазолы 314
2-Алкил-виц-триазолы 322
4-Алкил-1,2,3-триазолы 298, 324
1 -Алкил-4-феиил-5-амино-1,2,3-триазолы
299
З-Алкокси-5-амино-1,2,4-тиадиазолы 434
З-Алкокси-5-фенил амино-1,2,4-тиадиазолы
433
2-Аллиламино-5-фенил-1.3,4-тиадиазол 449
7-Аллилоксикумарииы 34
Аллобергаптен 17, 18
Аллобергаптол 17
Алловиснагии 40, 41
Алловиснагон 41
Аллодигидротоксикароловая кислота 136
Аллоимператорин 22
Аллоксантоксилетии 71, 76, 79
Аллопейцении 70. 71
монометиловый эфир 70
Аллоротеиои 125, 126
Аллороттлерин НО, 113
О-пентаметиловый эфир 1 11
Альстонилин 259, 260
синтез 261, 262
строение 260
Альстэнилинол 261
Альстонин 259, 260
Амидразоны 330
Амизол, см. 3-Амино-1,2,4-триазол
3-Амино-5-алкил-1,2,4-триазолы 327, 332,
341
2-Амино-5-амил-1,3,4-тиадиазол 450
2-Амино-5-анилиио-1,3,4-тиадиазол 462
3-Амино-5-ан ил ин о-1,2,4-тиадиазол 431
2-Амино-5-(L-ар або)тетраоксибутил-1,3,4-
тиадиазол 454
5-Амино-З-аренсульфонамидо-1,2,4-тиади-
азолы 433
2-Амино-5-арил-1,3,4-тиадиазолы 450
З-Амино-5-арил-1,2,4-тиадиазолы 431
5-Амино-3-ароиламино-1,2,4-тиадиазолы
431, 440
Аминоацилфуразаны 374
2-(п-Аминобензолсульфониламино)-5-
метил-1,3,4-тиадиазол 454
4-Амино-1,2,3-бензотиадиазол 424, 425
5-Амино-1,2,3-бензотиадиазол 423, 424, 425,
426
Амиио-1,2,3-бензотиадиазолы 422
Амино-2,1,3-бензотиадиазолы 448
4-Аминобензотриазол 317
5-Аминобензотриазол 317
Аминогарман 195
4-Амииогарман 209 и сл.
8-Аминогарман 209
4-Аминогуанозол 332, 336
4-Амино-3,5-дигидразииоН ,2,4-триазол
334
2-Амино-5,7-диметил-1,8-иафтиридин 177
5-Амино-3,5-дифеиилоксадиазолин 428
1 - Амино-4,5-дифенил -1,2,3-триазол 318
3-Амино-5-карбокси-1,2,4-триазол 344, 345
2-Аминолепидин 162
2-Амиио-5-меркапто-1,3,4-тиадиазол 450,
451, 463, 464
3-Амино-5-меркапто-1,2,4-триазол 328
3-Амиио-5-метиламиио-1,2,4-тиадиазол 44 1
11-Амино-5-метил-5,11-дигидрохиндолин
240
8-Амино-2-метил-Р-карболин, см. 8-Амино-
гарман
2-Амино-5-метил-1,3,4-тиадиазол 448, 452,
470
5-Амино-2-метилтио-1,3,4-тиадиазол 453
З-Амино-5-метил-1,2,4-триазол 346, 349
3-Амино-4-метилфуразан 373
7-Амино-5-метилхиндолиний, хлорид 239
11-Амино-5-метилхиидолиний. соли 239
и сл.
11-Амино-11-метокси-5-метил-5,11-дигидро-
хиндолин 240
2-Амино-1,5-иафтиридин 180
2-Амиио-1,7-нафтириДин 180
2-Аминоиафтиридины 178
2-Амино-5-нитро-1,3,4-тиадиазол 471
2-Амиио-1,3,4-оксадиазолон 408
480
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
5-Амино-5-окситиадназол 327
2-Амино-5-окси-1,3,4-тиадиазол 465
З-Амино-5-окси-1,2,4-триазол 336
2-Амино-5-(4-пиридил)-1,3,4-тиадиазол 451
3-Амино-5-пропил -1,2,4-триазол 346
5-Аминотетразол 329
2-Амино-1,2,4-тиадиазол 440
2-Амино-1,3,4-тиадиазол 452, 454, 469, 470,
471
4-Амино-1,2,5-тиадиазол 443
5-Амино-1,2,4-тиадиазол 433, 439, 440
Амииотиадиазолы 328
Амино-1,2,4-тиадиазолы 431
Амино-1,3,4-тиадиазолы 470
5-Амино-1,2,4-тиадиазолы 440
4-Аминотноуразол 327
5-Амино-З-(п-толуолсульфонамидо)-1,2,4-
тиадиазол 432
1-Амино-1,2,3-триазол 318
3-Амино-1,2,4-триазол 327, 344, 346, 348,
350
4-Аминотриазол 330, 336, 347
5-Амииотриазол 344
4-Амин о-1,2,4-триазол ди карбоновая-3,5
кислота 336
З-Амино-1,2,4-триазолкарбоновая-5 кисло-
та 336, 347
Аминотриазолкарбоновые кислоты 325
Амино-си.и.м-триазолы 348
4-Аминоуразол 337
9-Амино-л«-фенантролин 282
9-Амино-п-фенантролин 282
10-Амино-л«-фенантролин 282
2-Амино-5-фенил-1,3,4-оксадиазол 403, 405,
408
З-Амино-5-фенил-1,2,4-оксадиазол 380, 389,
396
2-Амино-5-фенил-1,3,4-тиадиазол 453, 454
3-Амино-5-фенил-1,2,4-тиадиазол 432, 435
5-Амино-З-фенил-1,2,4-тиадиазол 440
З-Амино-5-фенил-1,2,4-триазол 349
2-Аминофенилтриазол карбоновая кислота
310
7-Аминохиндолин 237
11-Аминохиндолин 237, 239
9-Аминохиннндолин 222
2-Амино-3-хлор-5,7-диметил-1,8-нафтирп-
дин 177
2-Амино-5-этил-1,3,4-тиадиазол 452
З-(Р-Аминоэтил)-! ,2,4-триазол 350
11-Амино-11-этокси-5-метил-5,11-дигидро-
хиндолин 240
Ангелицин 13, 16, 27 и сл.
Аигидробиакангелицин 20, 21
Ангидроизоэводиамин 248
Ангидрониевые основания 211 и сл.
Ангидронодакенетин 23
Ангидрофульвиновая кислота 85, 94, 95
3-(п-Анизил)-6-фенилтрйазол- [с.]-изоксазол
307
2-Анилинобензотиазол 442
2-Аиилино-5-меркапто-1,3,4-тиадиазол 451,
458, 468
З-Анилино-5-метиламиио-1,2,4-тиадиазол
431
5-Анилиио-3-метиламино-1,2,4-тиадиазол
431
2-Анилино-5-окси-1,3,4-тиадиазол 465
5-Анилино-1,2,3-тиадиазол 416, 417, 418
2-Ан и лино-1,3,4-тиадиазолтион-5 346
5-Ани л ино-3-(п-толуолсул ьфонамидо)-
1,2,4-тнадпазол 433
4-Анилино-1,2,3-триазол 311, 318
2-Анилино-5-фенилазо-1,3,4-тиадиазол 462
4-Анилино-4-фенил-5-алкил-А2-1,3,4-
тиадиазолины 4-69
2-Анилино-4-фенил-5-арил-А2-1,3,4-
тиадиазолины 469
2-Анилино-5-(Р-фенилгидразиио)-1,3,4-тиа-
диазол 462
3-Анилино-5-фенил-1.2,4-оксадиазол 390
2-Анилино-5-фенил-1,3.4-тиадиазол 449.
454
5-Аиилино-3-фенил-1,2,4-тиадиазол 428
2-Анилино-4-фенил-Д2-1,3,4-тиадназолннтп-
он-5 468
Антранилилхиндолин 235
Апогармин 223
Апоксантилетин 80
Апоксантокснлетин 18, 77. 78
Апоротиорамин 46
Апотоксикарол 72
Арабин 191, 192, 195
2-(Ь-Арабо)тетраоксибутил-5-этилмеркап-
то-1,3,4-тиадиазол 459
Арбутовая кислота 129 и сл.
2-Арил-5-аминобензотриазол 318
4-Арил-3-амино-5-меркапто-1,2,4-триазолы
328
2-Арнл-4-арилазо-5-ампнобензотриазол 318
Арилациламинофуразаны 358
1-Арилбензотриазолы 321
4-(4-Арил)-2,4-дигидро-2-тио- 1-тиа-3,4-дп-
азол 473, 474
1-Арил-4-карбэтокси-5-окси-1,2,3-трназолы
301
4-Арил-2-мерка пто-А2-1,3.4-тиадиазолинти-
он-5 466
Арилметилфуроксаны 380
р-(3-Арил-1,2,4-оксадиазол-5-ил) пропионо-
вая кислота 389
3-Арилокси-5-амино-1,2.4-тиадиазолы -434
2-Арил-1,2,3-триазолы 316
3-Ароил-5-фенил-1,2,4-оксадиазол 388
Атамантин 29,30
Аураптен 30,31 и сл.
Аураптенгидрат 31
Аураптеновая кислота 30,31
4-Ацетамид о-5-(5-метил-1,2,4-окса дна зол-3-
ил) имидазол 394
10-Ацетамино-п-фенантролин 275
2-Ацетиламино-5-метил-1,3,4-тиа диазол
450
2-Ацетиламино-1,3,4-тиадиазол 453
3-(п-Ацетиламинофенил)-5-бензилтио-1,2,4-
триазол 382
Ацетилгармалин 200-, 206
1-Ацетнл-1,5-дигидро-5-фенил-1,2,3-триазо
ло-[с!]-триазол 321
1-Ацетил-5,6-диметилбензотриазол 316
О-Ацетилкараньиновая кислота 44
6-Ацетил-4-карбэтокси-5-окси кумарин 3!
3-Ацетил-2-карбэтоксихромон 90
З-Ацетилкеллин 36
1-Ацетил-4-метил бензотриазол 315
1-Ацетил-5-метилбензотриазол 315
4-Ацетил-5-метил-1,2,3-триазол 307
8-Ацетил-4-метилумбелл иферон 32
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
481
1-Ацетил-5-метоксибензотриазол 315, 317
1-Ацетил-6-метоксибензотри азол 315
2-Ацетил-5-метоксибензотриазол 315
5-Ацетил-6-окси-4,7-диметоксикумаран 38
3-Ацетил-4-окси-6,7-диметоксикумарин 88,
89 и сл.
8-Ацетил-7-окси-2,3-диметоксихромон 45
6-Ацетил-5-окси-4-метил кумарин 58
6-Ацетил-7-окси-4-метил кумар ин 58
5-Ацетил-4-окси-З-мети л кумарон 12
8-Ацетил-7-окси-2-метилхромой 66
8-Ацетил-7-окси-5-метокси-2,2-диметил хро-
май 71
Ацетилротенолоны 119
6-Ацетил-8-этил-5-окси-4-метил кумарин 59
6-Ацетил-8-этилумбеллиферон 53
Ацетоацетилкумарины 88
3-Ацетоацетил-4-окси-6,7-диметокси кума-
рин 88
4-Ацетоксикумарон 44
2,5-бис-(Ацетонгидразоно)-1,3,4-тиади-
азол 455
5-Ацетонил-1,2,4-оксадиазол 389
Ацетоноксипеуцеданиновая кислота 25
5-Ацил амино-1,2,4-тиадиазолы 440
Ацил аминотриазолы 308
1-Ацилбензотриазолы 302
6-Ацил-4-мети л-5-(карбэтоксиметокси) ку-
марины 34
З-Ацил-4-оксикумарины 89, 90
6-Ацил-5-окси-4-метил кумарины 34
Банистерин 191
1-Бензамидо-4-метил-1,2,3-триазол ЗСЗ
1-Бензамидо-5-метил-1,2,3-триазол 303
3,5-Бензамидо-1,2,4-тиадиазол 441
1-Бензамидо-1,2,3-триазол 303
2-Бензамидо-5-фенил-1,3,4-тиадиазол 450
З-Бензамидо-4-фенилфуразан 360
2-(Р-Бензамидоэтил)-Д2-1,3,4-оксадиазо-
линтион-5 406
2-Бензгидрил-5-фенил-1,3,4-тиадиазол 461
З-Бензиламино-5-амиио-1,2,4-тиадиазол
435
3-Бензил-4-амино-5-оксиизоксазол 375
1-Бензилбензотриазол 321
2-Бензил-4,5-дигидро-Р-карболин 201
Бензилиденгарман 208
3-Бензилмеркапто-5-арил-1,2,4-триазолы
347
2-Бензилмеркапто-1,3,4-тиадиазол 456, 459
2-Бензилмеркапто-5-трифторметил-1,3,4-
тиадиазол 456
З-Бензил-5-метил-1,2,4-оксадиазол 388
1-Брнзил-5-метил-1,2,3-триазол 310
6-Бензилокси-2-изопропенилкумарон 75
1-Бензил-3-оксиметил-1,2,4-триазол 339
1-Бензил-5-оксиметил-1,2,4-триазол 339
6-Бензил окси-4-метоксикумаронкарбоно-
вая-2 кислота, этиловый эфир 18
N-Бензилсиднон 398
2-Бензил-2,3,4,5-тетрагидро-Р-карболин 204
1-Бензил-1,2,3-триазол 313, 315, 322
1-Бензил-3-фенил-5-окси-1,2,4-триазол 338
5-Бензил-З-фенил-1,2,4-тиадиазол 430
4,5-Бензо-Р-изокарболин 224
3-Бензоил-5-амино-1,2,4-оксадиазол 375
Бензоиламинофуразан 393
31 Зак № 978
Бензоиламинофуроксан 367
1-Бензоилбензотриазол 315, 316
3-Бензоил-4,5-дифенилизоксазол 373
3-Бензоил-5-метил-1,2,4-оксадиазол 395
3-Бензоил-5-фенил гидразин-1,2,4-оксади-
азол 375
З-Бензоил-5-фенилизоксазол 393
3-Бензоил-5-фенил-1,2,4-оксадиазол 360,
393
бис-Бензои лформальдоксиминофуроксан
363
Бензоин 11
2,3-Бензо-у-карболин 227, 229
2,3-Бензо-б-карболин 217, 232 и сл.
3,4-Бензо-а-карболии, см. Хининдолин
3,4-Бензо-б-карболин 217, 233
4,5-Бензо-а-карболин 217
4,5-Бензо-Р-карболин 217, 222—224, 225
6,7-Бензо-а-карболин 217 219
Бензокарболины 217 и сл.
2,3-Бензо-у-карболины 227—233
1-Бензолсульфонил-5-бромнафто-11,2|-три-
азол 316
1-Бензол сульфон ил-5-метил бензотри азол
316
1 -Бензол сул ьфони л-6-метил бензотри азол
316
2,3-Бензо-1,5-нафтиридин 165
Бензо-1,6-нафтиридин 171
2,1,3-Бензоселендиазолы 444
1,2,3-Бензотиадиазол 420, 421, 424, 425
2,1,3-Бензотиадиазол 444, 445, 446, 447
2,1,3-Бензотиадиазолины 445
1,2,3- Бензотиадиазолы 420 и сл.
номенклатура 420
реакции 424—427
свойства 424—427
синтез 420—424
2,1,3-Бензотиадиазолы 444
номенклатура 444
реакции 447 и сл.
свойства 448
синтез 444 и сл.
структура 446
а-Бензот.риазоилмасляная кислота 314
Бензотриазол 302, 312, 315, 316, 323, 324
кислотность 322
комплексы 322
1-окись 305
основность 322
реакции 313, 317 и сл.
свойства 323, 324
синтез 304, 305 ,,
1-Бензотриазол, производные 314 '
Бензотриазолкарбоновая-1 кислота, мети-
ловый эфир 310
Бензотриазолкарбоновая-4 кислота 302
Бензотриазолы
кислотность 321 и сл.
номенклатура 297
основность 322
реакции 308 и сл., 314 и сл.
свойства 323 и сл.
— биохимические 325
соли и комплексы 322
Бензотриазо-4,7-хиион 323
Бензофуразан 359, 360, 373, 375, 376, 377,.
378
5,6-Бенз.охинолин 269
482
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Бергамоттин 17
Бергаптен 17, 18, 20, 21
Берга.птенхинои 19, 20, 21
Бергаптин, см. Бергамоттин
Бергаптол 16, 17, 20, 23, 24
гераниловый эфир 17
у,у-диметилаллиловый эфир 23
метиловый эфир 17, 19
Бергаптолхиион 25
Бергаптохииои 23
Биакангеликол 21
Биакангелицин 20, 21
Биакангелициидиацетат 21
Биакангелицнновая кислота 20, 21
Бифлавонилы 69, 70, 71
Борнилен-г//-1,2,5-тнадиазол 445
5-Бромбензотриазол 317
7-Бромгарман 210
3-Бром-4-окси-1,5-нафтиридин 177
2-Бром-1,3,4-тиадиазол 470
5-Бром-1,2,4-тиадиазол 439, 440
3-Бром-1,2,4-триазол 344, 345
9-Бром-о-фенаитролин 279
9-Бром-п-фенантролин 280
10-Бром-л-фенантролин 280
10-Бром-п-фенантролин, моноиодметилат
275
5-(п-Бромфеииламиио)-3-амино-1,2,4-тиади-
азол 431
3-(п-Бромфеиил)-5-метил-1,2,4-оксадиазол
388
С-Бром-Ы-фенилсиднон 398
7-Бромхиидолин 237
7-Бромхиндолиикарбоновая-11 кислота 237
Веделолактон 50
Визамминол 42, 43
Виснаганы 82
Висиагии 35, 38 и сл., 41, 42
Виснагон 39, 41, 42, 45
Виснадин 82
Вогонин 41
2-(D-Галакто) пентаоксиамил-5-этил меркап-
то-1,3,4-тиадиазол 459
Галловая кислота 53—56
Галлофлавин 54
5-Галоген-1,2,3-бензотиадиазолы 427
З-Галогеи-1,2,4-триазолы 344
' Гальфордии 13, 29
Гаматин 44
Гармалан 196, 198, 200, 201
Гармалин 191, 192, 195, 197, 203
реакции 198 и сл.
синтез 193, 199, 200 и сл.
структура 199, 200
Гармалиний катион 200
Гармалол 192
Гарман 192, 197
бромметилат 215
свойства 196
синтез 195, 196, 197
4-Гарманилкарбаминовая кислота, бензи-
ловый эфир 210
Гармановая кислота 193
Гармин 191, 192, 193, 195, 196, 197, 198, 203
Гармол 195
О-алкиловые эфиры 197 и сл.
Гексагндроаллоимператорин 22
Гексагидроизоимператорин 22
Гексагидроимператорин 21
5,6,9,10,11 а, 12-Гексагидроиидоло- 13,2-feJ-
хинолизинон-11-(8Н) 247
Гексагидрокарболин 191
Гексагидроороселон 30
Гексагндроосайин 97, 102, 103
1 - Гексадецил-3- карбэтоксиметилбензотри-
азолий, бромистый 313
2,4,6,2',4',6'-Гексаокси-5,5'-диацетил-3,3'-
диметилдифенилметан 110
Гексаоксидифенилметан 110, 112
1 - Гексил-4,7-дикето-4,7-дигидро-5,6-диок-
сибензотриазол 320
1-Гексил-4,5-диформил-1,2,3-триазол 319
Гемипиновая кислота 57
Гинкгетин 69, 70
тетраметиловый эфир 70
Гомоптерокарпин 47, 49, 50
Гомофусциигидрат 65
Грамин 247
Гуаиазил, см. 4-Аминогуанозил
Гуаназол, см. 3,5-Диамино-1,2,4-триазол
Гуаиазолы 349 и сл.
Гуаназопиразолои 342
Гуаназотетразин 342
Дегвелин 82, 117, 118, 138
оптическая активность 130, 133, 137
Дегидровизамминол 43
Дегидрогомоптерокарпии 49
Дегидродегвелин 138
Дегидро-Р-дигидроротенон 126
Дегидроизоротенон 124, 126, 130
Дегидрокетоиобирин 253 и сл.
Дегидроротеион 119, 120, 126, 128, 130, 131
Дегидросуматрол 133
Дегидроэллаговая кислота 56
Дегидроэллиптон 139
Дезерпидин 256
Дезоксиапоксантилетин 80
Дезоксиапоксантоксилетин 77
Дезоксидигидроореоселои 23
Декарбориссовая кислота 120, 121
Деметилмангостин 115
Деметоксигармин 193
Дерризовая кислота 116, 119, 120, 126
Дерритол 126, 127, 129
метиловый эфир 126 и сл., 141
Дерровая кислота 120, 121, 134, 135, 139
этиловый эфир 128
1,5- Диазаантрацеи 267, 271
1,8- Диазааитрацен 270
1,5- Диазафенантрен, см. л-Фенантролин
1,9- Диазафеиантрен 286
2,7-Диазафеиаитрен 284 и сл.
2,10-Диазафенантрен 285 и сл.
1,8-Диазафлуоренои-9 272
1,15-Диазафлуоренон-9 272
4,5-Диазафлуоренон-9 272
М,М'-Диалкилбензотриазолий, солн 325
2,5-Диалкил-1,3,4-оксадиазолы 400
3,5-Диалкил-1,2,4-тиадиазолы 430
Диалкил-1,3,4-триазолы 405
1,4-Диалкил-1,2,3-триазолы 321
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
483
Диалкилфуроксаны 361, 374
3,5-Диалкокси-1,2,4-тиадиазолы 430
1,2-Диамино-4-азидо-5-иитробензол 371
4,5-Диаминобензофуразан 369
2,7-Диамнно-4-метил-1,8-нафтирндин 178
Диамино-1,2,4-оксадиазол 391
2,5-Диами>но-1,3,4-тиадиазол 462, 470,
471
3,5-Диамино-1,2,4-тиадиазол 431, 440, 441
3,5-Диамино-1,2,4-триазол 334, 342, 348,
350
3,5-Диамино-4-фенил-1,2,4-триазол 344
2,4-бис-(Диаминофенил)-1,2,3-триазол 309
2,5-Дианилино-1,3,4-тиадиазол 462
3,5-Дианилино-1,2,4-тиадиазол 431, 439,
441, 442
Диаиилииофуразан 361
3,5-Ди а рал килами ио-1,2,4-тиадиазолы
431
3,5-Диариламино-1,2,4-тиадиазолы 431
2,4-Диарил-3,5-диимиио-1,2,4-тиадиазоли -
дииы 438
3,5-Диарил-4-(о-карбоксиарил)три азолы
337
3,5-Диарил-1,2,4-оксадиазолы 392
Диарилсидноны 398
3,5-Диарил-1,2,4-тиадиазолы 429
Диарилфуроксаиы 363, 374
Диароилфуроксаиы 375
2,3-Диацетил-2,1,3-бензотиадиазолин 445
1,2-Диацетил-1,2,4-триазолои-5 350
3,4-Диацетилфуроксан 364, 386
Диацилфуразаиы 372, 374, 384 и сл.
Диацилфуроксаны 372, 384 н сл.
2,5-Дибензил-3-метил-1,2,5-тиадиазоли-
дин 444
2,5-Дибензоил-1,2,5-тиадиазолидин 444
Дибензоилфуразан 370
Дибензоилфуроксан 363, 369, 370, 375,
378, 384
2,5-Дибензолсульфонил-1,2,5-тиадиазоли-
дин 444
4,5-Дибром-1,2,3-бензотриазол 316
2,5-Дибром-1,3,4-тиадиазол 470
4,5-Дибром-1,2,3-трназол 316
Дибромфуроксан 373, 375
Дигалогеифуроксаны 366, 367
2,7-Дигидразино-4-метил-1,8-нафти ридин
178
Дигидроаллоксантоксилетин 79
Дигидроаураптеновая кислота 31
Дигидро-2,3-бензо-у-карболин 227
3,4-Дигидробиакангелицин 20
Дигидрогарман, см. Гармалан
Дигидрогармин, см. Гармалин
Дигидрогомоптерокарпин 47, 48, 49
L-Днгндрогомоптерокарпон 48
Дигидродегвелин 131, 138
Дигидродегидроаллотоксикарол 136
Дигидродегидродегвелин 138
Дигидродегидроизоротенон 125, 126
Днгидродегидроротенон 126, 144
Дигидродегидротоксикарол 134, 135
а- 137
Р- 137
Дигидродегидротоксикароловая кислота
137
Дигидродегидроэллиптон 139
Дигндродерризовая кислота 126, 144
Дигидроизоаллороттлерин НО, 111, 113,
114
пентаметиловый эфир 114
тетраметиловый эфир 114
Дигидроизодерризовая кислота 126
Дигидроизодерритол 126
Дигидроизоосайетин 104, 105
Дигндроизоосайин 102, 103
моноацетат 102
монометиловый эфир 103, 105
Дигидроизопомиферин 102, 103
диацетат 102
диметиловый эфир 104, 105
Дигидроизопомиферитин, эфиры 104
Дигндроизоротеиол 126
Дигидроизоротенон 125, 126, 130
Дигидроизотубовая кислота 122 и сл.
Дигидроинофиллолид 84
2,3-Дигидро-3-кето-3-фенил-1,3,4-оксади-
азол, анил 404
2,3-Дигидро-2-кето-3-(о-хлорфенил)-1,3,4-
оксадиазол 404
Дигидрокоринантеин 243, 244, 247
Днгидроксантилетин 80, 81
Дигидроксантоксилетин 77, 78, 79
Дигидрометилгомофусцин 65
Дигидромувангетин 81
Ди гидром ундулетон, метиловый эфир 106
Дигидро-1,8-нафтиридин 173
Дигидроновобиоцин 64
Дигидроновобиоциновая кислота 64
Дигидроноргарман 248
Днгидро-1,2,4-оксадиазол 391
Днгидро-1,3,4-оксадиазолы 404, 406
Дигидроореоселон 26
Дигидроореоселоновая кислота 26
Дигидроороселон 30
Дигидроосайин 97, 102
Дигндропейценин 71
Дигидропомиферин 97, 100, 102
4',5'-Дигидропсорален 16
Дигидроптерокарпин 47 и сл., 49
Дигидроптерокарпон 48
Дигидроротеноиы 118, 125, 126, 128, 144
Дигидросамидин 82
Дигидросезелнн 81, 82
1,2-Дигндро-1,2,3,4-тетразии 336
Дигидротетразины 306
Дигидротоксикарол 134
Дигидротоксикароловая кислота 134
метиловый эфир 135, 136
Дигидротубаиновая кислота, метиловый
эфи р 106
Дигидротубовая кислота 122, 123, 125
Р- 123, 125
9,10-Днгидро-п-фенантролин 273, 274
Дигидрофусцин 64
Дигидроэллиптовая кислота 139
Днгидроэллиптол 139
Дигидроэллиптон 139
2,5-Дидодецил-1,2,5-тиадиазолидин 444
3,5-Диимино-2,4-днфенил-1,2,4-тиадиазо-
лидин 441, 442
4,5-Дикарбокси-1,2,3-трназол-1 -илуксусная
кислота 310
Дикарбэтоксифуроксан 365
2,5-Димеркапто-1,3,4-тиадиазол 451, 456
3,5-Димеркапто-1,2,4-тиадиазол, см. Над-
тиоциановая кислота
31*
4'84
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
3-Диметиламино-5-амино-1,2,4-тиадиазол
435
2,4-Диметил-7-амиио-1,8-нафтиридин 160
О-Диметилаигидрофульвиновая кислота
94, 95
2,4-Диметил-7>-ацетамиио-1,8-нафтиридин,
1-иодметилат 177
2,2-Диметил-3-ацетил-5-(4-пиридил)-1,3,4-
оксадиазолии 404
2,3-Диметил-4,5-беизо-|3-изокарболии 225
4,5-Диметил-2,3-беизо-у-изокарболии 230
1,2- Диметил-4,5-беизо-|3-карболии 224, 225
1,5- Диметил-2,3-беизо-у-карболин 229 и сл.,
231
1,6- Диметилбеизотриазол 313, 314
5,6-Диметилбеизотриазол 314, 315
Диметилгармии хлористый 216
2,6-Диметил-4,8-диокси-л1-фенантролин 279
2,7-Димети л-4,5-диокси-л-фенантролин 279
Диметилдипиридилы, комплексы с метал-
лами 291, 292
5,5'-Диметил-2,2'-ди-1,3,4-тиадиазолы 461
11,12-Диметил-11Н-индоло- [2,3-Ь]-5-хино-
лизиний, иодид 232
1,3-Диметил-2-кето-2,3-дигидро-|3-карбо-
лин 207
5,5-Диметил-2-меркапто-4-фенил-А2-1,3,4-
тиадиазолии 466
1,2-Диметил-8-метокси-|3-карболин 21 7
2,4-Диметил-7-метокси-1,8-и афти ридин,
1-иодэтилат 177
1,4-Диметил-З-метокситриазолон 340
1,3-Диметил-5-иитробеизотриазолий, йоди-
стый 313
2,4-Диметил-6-окси-1,5-нафтиридин 166
4,4'-Диметилпсоролен 32
2,3-Диметил-6,7,8,9-тетрагидро-4,'5-бензо-
8-изокарболии 226
2,3-Диметил-2,3,4,5-тетрагидро-|3-карбо-
лин 204
1,8-Диметил-1,2,7,8-тетрагидро-п-феиант-
ролиндион-2,7 276, 281
2,5-Диметил-1,3,4-тиадиазол 461, 471
2,5-Диметил-1,2,5-тиадиазолидии 444
3,5-Диметил-1,2,4-триазол 340, 343, 348,
349
4,5-Диметил-1,2,3-триазол 303, 316, 318, 323
3,5-Диметил-1 -(1,2,4-триазол-3-ил)-1,2,4-
триазол 349
1,3-Диметил-2,4,5-три кето-2,3,4,5-тетра-
гидро-|3-карболии 207
4,8-Диметилумбеллиферои 52
1,2-Ди.метилуразол 340
1,10-Диметил-о-феиаитролин 270
2,4-Диметил-п-фенаитролии 273
Диметил-о-феиаитролииы 270
1,3-Диметил-9-феиил-2,10-диазафеиаитреи
286
2,5-Диметил-4-фенил-А2-1,3,4-тиадиазолин
467
1,2-Диметил-4-феиилуразол 340
С-Диметилфлороглюцин 107
Диметилфуразаи 369, 370, 378
Диметилфуроксаи 361, 371
О-Диметилфусцииовая кислота 64, 65
2,2-Диметилхромен 72
2,2-Диметилхромеиоиовое ядро 71—76
О-Диметилцитромицетин 93, 94 и сл.
О-Диметилцитромицин 86, 87, 90, 92, 93
О-Диметилцитромицинилхлорид 90
О-ДиметилцитромицииоЛ 86, 87, 90, 91, 92,
93
метиловый эфир 94
О-Диметилцитромициион 57, 86, 87, 88, 90,
92, 93
метиловый эфир 93, 95
5,6-Диметоксиаигелицин, см. Пимпииел-
лин
5,6-Диметокси-2-ацетилкумаранои-3 86
2-(3,4-Диметоксибензил)-2,3,4,5-тетрагидро-
В-карболинкарбоиовая-2 кислота 204
5,7-Диметокси-2,2-диметилхроман 78
4,6-Диметокси-2-изопропил-К-кумараиои
74
4,6-Диметокси- 7 -метилкумароикарбоновая-2
кислота, этиловый эфир 18
2,4-Ди метокси-6-метоксиацетофен ои 40
2,4-Диметокси-1,8-нафтиридин 182
5,8-Диметоксипсорален, см. Изопимпинел-
лин
2,3-Диметоксихининдолии 220
8,9-Диметоксихиииидолии 220
1,2-Динитрозо-4-азидо-5-нитробензол 371
о-Динитрозоароматнческие соединения
реакции 369—378
свойства 378
синтез 367—369
структура 379—388
1,2-Динитрозоацеиафтеи 372
о-Динитрозобеизол 368, 371, 372, 374, 376,
377, 379, 386
1,2-Дииитрозо-3,6-диаиилино-4,5-динитро-
беизол 377
1,2-Динитрозо-3,6-дибромбензол 369
1,2-Дииитрозо-3,5-динитробензол 377, 378
1,2-Дииитрозо-4,5-дииитробензол 377, 378,
386
1,2-Динитрозо-3,5-Дииитро-4-метилбеизол
377
о,о'-Динитрозодифенил' 384
цисЛ ,4-Динитрозо-1,4-дихлорциклогексаи
384
2,3-Динитрозо-5-метилпиридии 371
1,2-Динитрозоиафталии 387
2,3-Динитрозо-1,4-иафтохинои 387
1,2-Динитрозо-4-иитробеизол 374
1,2-Дин и трозо-3-окси-4,6-ди нитробензол
387
1,3-Динитро-4,5-дииитрозобеизол 371, 374
3,5-Дииитро-1,2-динитрозобеизол 377
1,2-Динитрозо-4-хлор-5-метоксибензол 368
4,6-Динитро-7-оксибёизофуразаи 373
1 (2,4-Динитрофеиил)-4-карбометокси-5-
окситриазол 315
2,4-Диокси-6,7-беизо-1,8-нафтиридии 169
4,7-Диокси-1,2,3-беизотиадиазол 425
5,6-Диокси-1,2,3-беизотиадиЭзол 425
5,7-Диокси-2,2-диметилхроман 70, 76, 108
5,7-Диокси-2,2-диметилхроманои-4 73
1,4-Диокси-2,3-дииитрозонафталин 387
спл1Л1-3,3’-Диоксн-5,5'-днтриазоил 328, 348
2,4-Диокси-2,6-дифеиил-л1-феиантролии-
дикарбоиовая-3,7 кислота, диэтиловый
эфир 267
4,5-Диокси-2,7-дифеиил-п-фенаитролииди-
карбоиовая-3,5 кислота, диэтиловый эфир
268
4,6-Диокси-2-изопропилкумараи 76
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
485
1,4-Диокси-3-карбометокси-2,7-нафтиридин
175
2,4-Диокси-З-карбометокси-! ,8-нафти ридип
169,- 180
5,8-Диокси-7-карбометокси-1,6-нафти ридин
172, 174
4,7-Диокси-8-метилкумарин 63
2,7-Диокси-4-метил-1,8-нафтиридин 178
2,7-Диокси-5-метил->,8-нафтиридин 177
5,7-Диокси-2-метилхроман 104
7,8-Диокси-6-метоксикумарин, см. Фрак-
сетин
2,4-Диокси-1,5-нафтиридин 169
2,4-Диокси-1,8-нафтиридин 169
5,8-Диокси-1,6-нафтиридин 174, 179
6,8-Диокси-1,7-нафтиридин 173
Диоксиороселон 30
1,8-Диокси-о-феиантролин 279
4,5-Диокси-п-феиаитролин 279
1,8-Диокси-о-фенаитролинди карбоновая-
2,7 Кислота 279
4,8-Диокси-Л1-фенантролиндикарбоиовая-
3,7 кислота 283
5,7-Диокси-8-(В-фени л пропионил)-2,2-диме*
тилхроман 108
3,4-Диоксихиндолин 243
10,11-Диоксихиндолин 235
2,2'-Дипиридил, комплексы с Металлами
287, 288, 289, 290, 293
2,3'-Дипиридил 271
3,3'-Дипиридил 271
2,3' -Ди пиридилди карбоновая-3,2' кислота
271
3,3'-Дипиридилди карбоновая-3,3' кислота
271
2,5-Ди-(4-пиридил)-1,3,4-тиадиазол 460
3,5-Ди(-3-пиридил)-1,2,4-тиадиазол 429
3,5-Ди-(3-пиридил)-1,3,4-тиадиазол 462
Дистемонантин 57
3,5-Ди-(п-сульфамидофенил)-4-амино-1,2,4-
триазол 331
Дитиоуразол 328
3,5-Ди-(о-толил)-1,2,4-оксадиазол 388
2,5-Дитолил-1,3,4-оксадиазол 408
Дитолил-1,2,4-тиадиазолы 429
Ди-(1,2,3-триазолы) 298
3-Дифениламино-5-амино-1,2,4-тиади азол
435
1,4-Дифенил-5-амино-1,2,3-триазол 318
3,5-Дифенил-4-амино-1,2,4-триазол 331
3,4-Дифени л-5-анилино-1,2,4-триазол
329
2,4-Дифенил-5-бензамидо-1,2,3-триазол
395
3,5-Дифенил-4-бензил-1,2,4-триазол 338
1,4-Дифенил-2,3-бензо-6-карболин 233
6,7-Дифенил-2,5-диазафенантрен 284
6,7-Дифенил-2,5-диа за фенантролин кар-
боновая-8 кислота 284
2,5-Дифенил-4,5-дигидро-4-ацил-1,3,4-
оксадиазол 404
3,6-Дифенил-1,2-дигидро-1,2,4,5-тетра-.
зин 331
3,6-Дифенил-7,8-дигидро-8-тиа-1,2,5-три-
азаиндолизин 347
2,4-Дифенил-3,5-диимино-1,2,4-тиадиазо-
лидин 479
3,6-Дифенил-4,5-динитрозопиридазин 369
Дифенилдифуразан 385
32 Заказ № 978
2,4-Дифенил-З-и мино-1,2,4-тиади азол и -
динон-5 441
2,4-Дифенил-5-и ми но-А2-1,3,4-тиади азол и и
468
3,4-Дифепил-5-меркапто-4,1,2-триазол 449
1,3-Дифенил-5-метил-1,2,4-триазол 332
1,5-Дифенил-3-метил-1,2,4-триазол 335
1,5-Дифенил-3-метокси-1,2,4-триазол 348
Дифенил-1,2,4-оксадиазол 391, 393, 394
N-окиси 395
2,5-Дифенил-1,3,4-оксадиазол 399, 403
3,5-Дифенил-1,2,4-оксадназол 365, 379, 388,
390, 392, 393
3,3-Дифенил-1,2,4-оксадиазолидинон-5
395
5,5-Дифенил-1,2,4-оксадиазолидинон-З
395
3,5-Дифенил-4-окси-1,2,4-триазол 331
2,4-Дифенил-3-стирил-5-кетотетрагидро-
1,2,4-оксадиазол 395
2,5-Дифенил-1,3,4-тиадиазол 460, 461,462
3,5-Дифенил-1,2,4-тиадиазол 429, 430, 439,
440
4,5-Дифенил-1,2,3-тиадиазол 418, 419
2,4-Дифенил-1,2,4-тиадиазолидинон-З 438
1,5-Дифенил-1,2,4-триазол 334, 349
2,4-Дифенилтриазол 310
3,5-Дифенил-1,2,4-триазол 327, 331, 335,
340
Дифенилуразин 336
2,6-Дифенил-л1-фенантролИн 284
2,7-Дифенил-п-фенантролиндикарбоновая-
4.5 кислота 284
2,3-Дифенил-о-фенантролинкарбоновая-1
кислота 283
2,6-Дифенил-л1*фенантролинкарбоНовая-4
кислота 284
2,4-Дифен и л-3,5-бис-(фен ил имино)-1,2,4-тиа-
диазолидин 439, 442
Дифенилфуразан 37! \ 372, 373, 374
Дифенилфуроксан 361, 363, 364, 365, 366,
.367, 369, 370, 371, 374, 375, 380
2-Дифенилхлорметил-5-фепил*1,3,4-окса-
диазол 409
1,4-Дифенил-5-хлор-1,2,3-триазол 318
1,5-Дифенил-3-хлор-1,2,4-триазол 348, 349
4,5-Диформилтриазолы 298
3,5-Дифурил-1,2,4-триазол 329
2,2'-Дихинолил, комплексы с металлами
292, 293
Дихлорбензо-(3-карболин 226
2,7-Дихлор-4,5-бензо-(3-карболин 226
5-(а,Р-Дихлорвинил)триазол-4-и л глиокси-
ловая кислота 317
2,7-Дихлор-4-метил-1,8-нафтиридин 180
2,6-Дихлор-1,5-нафтиридин 176, 179, 182
2,4-Дихлор-1,8-нафтиридин 180, 181, 182
2,5-Дихлор-1,3,4-тиадиазол 470
2,7-Дихлор-л-фенантролин 277, 281
3,5-Ди-(о-хлорфеиил)-1,2,4-оксадиазол 392
4,5-Дициан-1,2,3-триазол 304, 322
1 -(4-Диэтиламино-1 -метилбутиламино)-10-
метокси-о-фенантролин 282
4-(4- Д иэти ламино- 1-метилбу тиламино)-2-ме-
тил-л-фенантролин 283
1,8-бис-(4-Диэтиламино-1 -метилбутилами-
но)-о-фенантролин 282
2-(4-Ди эти л амино-1 -мети л бути л амино)-/г-
фенантролин 283
486
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
4-(4-Диэти ламино-1 -мети лбути ламино)-п-
фенантролин 283
4-(3-Диэти ламино-2 оксипропиламино)-4-
окси-л-фенантролин 283
4,8-бис-(3-Диэтиламино-2-оксипропиламп-
но)-л1-фенантролин 283
1,10-бнс-(З-Диэтиламинопропиламино)-п
фенантролин 283
4-(3-Диэтиламинопропиламино)-9-хлор-п-фе-
нантролин 283
2-(Р-Диэтиламиноэтиламино)-4,5-бензо-р-
карболин 283
2-(|3- Диэтил амино) эти л ами но-1,4-диме-
тил-|3-карболиний, дисалицилат 211
2-(|3-Диэтиламиноэтиламино)-1 -мети л-4,5-
оензо-|3-карболин 226
4-(2-Ди эти л аминоэтил ами но) -2-мети л-п-фе-
нантролин 283
6-(2-Диэти ламиноэти лами но)-8-метил-л«-фе-
нантролин 283
8-(2-Диэти ламиноэти ламино)-б-метил-л-фе-
нантролин 282
3-(2-Ди эти л аминоэтил амин о)-о-фенантролин
282
9-(8-Диэтиламиноэтиламино)-п-фенантролин
282
2,5-Диэтилмеркапто-1,3,4-тиадиазол 475
4,4-Ди эти л-2,3,4,5,10,11,12,13-октагидро-
2-окси-1 -мети л-2,3-бензо-у-карболиний,
иодид 231
3,5-Диэтокси-1,2,4-тиадиазол 430
1 -Додецил-З-бензилтриазолий, бромистый
325
Дракрвая кислота 146
Дракокармин 145
Драконол 146, 147
Дракорубанол 145
Дракорубин 50, 116 и сл., 145, 146, 147
Изоазаринин 9
Изоаллороттлерин 110, 111, 113
Изоаураптен 31, 32
Изобергаптен 19
Изовиснагин 40
Изовиснагон 40
Изогаллофлавины 54
Изогальфордин 13, 29
Изогиикгетин 69, 70
Изодеметилмангостин 115
Изодерризовая кислота 124, 126
Изодерризовые кислоты 126
Изодерритол 141
Изодигидроореоселоп 26
Изодигидроротенон 131
Изоимператорин 23
рИзокарболин 214
Изокеллин 38
Изонадтиоциановая кислота 435, 436—438
Изооксипеуцедании 24
Изоосайетин 103, 104
Изоосайин 101, 102, 103
Изопейцении 70
Изопимпииеллин 19, 20, 21, 23, 29, 81
Изопомиферин 102, 103, 104
Изопомиферитин 104
Изопропилкумараиоп 73
Р)зопсорален, см. Апгелицип
Изорезерпин 258, 259
Изоротенон 124, 126, 130, 131, 132
Изороттлерин, см. Изоаллороттлерин
Изосемазин 7, 9, 10
1,2,3-Изотриазолы 312
Изотубанол 123, 124
Изотубовая кислота 122, 123, 124, 127
Изофлаваноны 49, 115
Изохининдолин 221
Изоцианиловая кислота <376
Изоэводиамин 248, 249
З-Имино-1,2,4-дитиазол ид шт ion-5 436
5-Имино-4-метил-1,2,4-тиадиазолин 440
Иминотиоуразол 326
Императорин 21, 22
Индолодиазин 201, 202
Индоло-[3,2-Ь]-хинолизин 247
11Н-Индоло-[2,3-Ь]-хинолизины 231 и сл.
Инофиллолид 83, 84
Иобирин 252, 253, 259
З-Иод-1,2,4-триазол 344, 345
Йохимбин 191, 248, 251, 253—256
Кайафлавон 70
Калофилловая кислота 83, 84
Калофиллолид 83 и сл.
Калофиллолиды 83
Камала 106, 107
Каранииновая кислота 139
Караниол 28
Караньин 28, 35, 43—45
Караньиновая кислота 43, 44, 45
Караньол 43, 44, 45
1 -Карбалкоксибензотриазолы 310
1 -Карбамоил-3,5-диметил-1,2,4-триазол
343
11 -Карбокси-5-метилхиндолиний хлорид
240
З-Карбокси-1,2,4-триазол 344
5-Карбокси-1,2,4-триазол 343
2-(п-Карбоксифенил)-1,2,3хриазол 310
2-(п-Карбоксифенил)-1,2,3-триазолди кар-
боновая-4,5 кислота 310
2-(2-Карбоксиэтил)-2,3,4,5-тетрагидро-2-кар-
болинкарбоновая-2 кислота 206
а-Карболин 185, 187, 189, 211
Р-Карболин 185, 193, 194, 195, 208, 209
у-Карболин 185, 187 и сл., 189, 214
6-Карболин 185, 188
Карболиния катион 212, 213
Р-Карболинкарбоновая-2 кислота 208
Р-Карболинкарбоновая-4 кислота 209
Карболины 185 и сл.
ангидрониевые основания 211 п сл.
номенклатура 186
синтез 321
структура 212 и сл.
<1-Карболины 185
ангидрониевые основания 211—216
синтез 187—190
Р-Карболины
ангидрониевые основания 214 и сл.
синтез 192—194
у-Карболины 185
ангидрониевые основания 211, 213
и сл.
свойства 190 и сл.
синтез 187—190
6-Карболины 185, 188, 189, 192, 199
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
487
4-Карбометокси-5-окситриазол 315
4-Карбэтоксиамино-1,2,5-тиадиазол 443
4-Карбэтокси-5-анилиио-1,2,3-триазол 311
5-Карбэтокси-2,6-диметилимидазо- [2,1-6]-
тиадиазол 470
2-Карбэтокси-4-(2,4-дихлорфенил)-А2-1,3,4-
тиадиазолинтион-5 467
7-Карбэтоксиметоксиумбеллиферон-8-альде-
гид 28
3-Карбэтокси-4-окси-7-ами но-1,8-нафтири-
дин 158
4-Карбэтокси-5-оксибензотриазол 310
3-Карбэтокси-4-окси-1,5-нафтиридин 164
3-Карбэтокси-4-окси-1,8-нафтиридины 158
Келлии 35, 36—38, 41, 42
Келлииои 36, 37, 38, 41
Келлол 35, 38, 42
2-Кето-2,3-дигидро-4,5-бензо-р-карболин
192, 201—203, 225, 226
2-Кето-2,3-дигидро-р-карболинкарбоно-
вая-4 кислота 193, 209
Кетоиобирин 252, 253, 254
2-Кето-1-метил-2,3-дигидро-р-карболин
193, 211, 225
2-Кето-3-метил-2,3-дигидро-р-карболин 194
2 -Кето-2,3,4,5-тетра гидро-6-карболин 194,
207, 251
2-Кето-7-этокси-1,2,3,4,10,11 -гексагидро-
карболин 208
2-Кето-7-этокси-11 -метил-2,3,4,5-тетрагид-
ро-р-псевдокарболин 208
Коринантеидин 243
Корииаитеии 243
Криптолепин 219, 239, 240, 242
Ксантилетин 71, 76, 79 и сл., 81, 82
Ксантоксил S 7
Ксантоксилетин 71, 76—79
Ксаитотоксилетин 81, 82
Ксантотоксин 19, 20, 81
Ксантотокси хинон 17
Ксаитотоксол 20, 21 и сл.
Кубебин, альдегид 8
Кумэстрол 50
Лапахеиол 72
3-Лаурилмеркапто-5-амиио-1,2,4-тиадиазол
434
71отурии 192, 195
Луваигетии 81 и сл.
Малаккол 117, 118, 140
Маигостин 115
Мармелозин.см. Императорин
3-Меркапто-4-амино-5-гидразино-1,2,4-три-
азол 326
3-Меркапто-4-амиио-1,2,4-триазол 326, 338,
345
2-Меркапто-5-метилмеркапто-1,3,4-тиа-
диазол 458
5-Меркапто-3-метил-1,2,4-триазол 452
2-Меркапто-5-метил-4-фенил-А2-1,3,4-тиади-
азолин 472
2-Меркапто-5-(4-пиридил)-1,3,4-тиадиазол 459
Меркапто-1,3,4-тиадиазолы 472
3-Меркапто-1,2,4-триазол 347, 453
Меркаптотриазоламины 326
Меркапто-1,2,4-т.риазолы 328, 348
2-Меркапто-5-фенилгидразино-1,3,4-тиади-
азол 456
2-Меркапто-5-(Р-фенилгидразино)-1,3,4-тиа-
диазол 462
2-Меркапто-5-фенил-1,3,4-оксадиазол 457
2-Меркапто-5-фенил-1,3,4-тиадиазол 458
2-Меркапто-5-фенил-А2-1,3,4-тиадиазолин-
тион-5 472
3-Меркапто-5-феиил-1,2,4-триазол 453
2-Меркапто-5-этилмеркапто-1,3,4-тиадиазол
457
Метазоновая кислота 171
7-Метил-6-азаиидол, см. Апогармин
5-Метиламиио-3-амиио-1,2,4-тиадиазол 431
1-Метил-5-аминопиридон-2 166
2-Метиламиио-1,3,4-тиадиазол 449
5-Метиламино-1,2,4-тиадиазол 440
3-Метил-5-амино-1,2,4-триазол 339, 350
2-Метиламино-5-фенил-1,3,4-тиадиаз0л 454
Метиламинофуразан 360
Метиламинофуроксан 371
1 -Метил-5-ф-аминоэтил)-1,2,4-триазол 350
Метил-п-анизилфуразан 372
Метил-п-анизилфуроксан 370
1-Метил-5-анилино-1,2,3-триазол 311, 318
Метиларилфуроксаны 378
2-Метил-7-ацетоксихромои 66
1-Метил-2-бензилбензотриазолий, сульфо-
метилат 315
Метилбензилфуроксан 375
4-Метил-2,3-бензо-у-изокарболин 229
3-Метил-5-бензоил-1,2,4-оксадиазол 395
Метилбензоилфуразан 374
1-Метил-4,5-бензо-Р-карболин 224
2-Метил-4,5-беизо-р-карболин 224
4-Метил-2,3-беизо-6-карболин 232
5-Метил-2,3-бензо-у-карболии 229
5-Метил-2,1,3-беизотиадиазол 445, 447
1-Метилбензотриазол 310, 315, 317, 319,
322, 323 /-
2-Метилбензотриазол 310, 313, 317, 322
2-Метил-4,5,6,7-беизотриазол 317
5-Метилбеизотриазол 308, 315, 317
2-Метил-6-бром-1,8-иафтиридии 174
Метилгармалин 198, 199, 203
Метилгарман 21 К
Метилгармин 203, 217
Метилгомофусцин 65
О-Метилдезоксиапоксантоксилетин 77
6-Метил-2,5-диазафенаитрен 284
3-Метил-4,5-Диамиио-1,2,4-триазол 332, 344
2-Метил-4,5-дибром-1,2,3-триазол 316
О-Метилдигидроаллоксантоксилетииовая
кислота 79
а-Метил-4',5'-дигидроаигелицин 34
О-Метилдигидродегидротокси карол 135,
136
2-Метил-4,5-дигидро-0-карболии 196
О-Метилдигидроксаитилетииовая кислота
77, 80
1-Метил-1,2-дигидро-п-феиантролии 275
1-Метил-1,4-дигидро-п-феиаитролии 275
1-Метил-1,2-дигидро-л(-фенаитролиион-2
281
1-Метил-1,2-дигидро-п-феиаитролииои-2
276
2-Метил-4,5-дигндро-4-феиил-5-кето-1,3,4-
оксадиазол 399
Метил-О-диметилцитромицетии 86
32.4.
488
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Метил-О-диметилцитромицетинон 57
Метил-(3,4-диметоксифенил)фу роксан 374
2-Метил-6,7-диметоксихромонкарбоновая
кислота 90
5-Метилдиоксихиндолин 243
1 -Метил-3,5-дифенил-1,2,4-триазол 332
10-Метил-2,6-дифенил-.и-фенантролинди-
карбоновая-4,8 кислота 283
О-Метилдракоиол 146, 147
О-Метилдракорубанол 146, 147
2-(3', 4'-Метилен диоксибензил)-2,3,4,5-тет-
рагидро-Р-карболин 204
2,3-Метилендиокси-5-метилизохининдолин
221 •
2,3-Метилендиокси-6-метилхининдолин 221
2-Метилеидиоксихиииндолии 220
2,3-Метилеидиоксихиндолин 242, 243
3,3-Метилен-б«с-(4-оксикумарины) 61 и сл.
2-Метил-а-изокарболин 211, 212, 213
4-Метил-у-изокарболин 213, 214
6-Метилимидазо- [2,1-6]-тиадиазол 470
Метил-З-О-карбамил-4-О-метилновобиоза
63
1-Метил-а-карболин 189, 211, 216
1-Метил-В-карболии 194
2-Метил-р-карболин, см. Гарман
Метилкарболииий катион 212
5-Метил-11-карбометоксихиндолиний, суль-
фометилат 240
3-Метил-2-кето-2,3-дигидро-Р-карболин 208
О-Метилксаитоксилетиновая кислота 77,
80
3-Метилмеркапто-5-амиио-1,2,4-тиадиазол
434
2-Метилмеркапто-5-меркапто-1,3,4-тиадиа-
зол 458
2-Метилмеркапто-1,3,4-тиадиазол 459
2-Метилмеркапто-5-фенил-1,3,4-оксадна-
зол 407 .
5-Метил-2-метиламино-1,3,4-тиадиазол 449
5-Метил-5-метил-2-меркапто-4-фенил-Д2-
1,3,4-тиадиазолии 466
Метил-п-метоксифенилфуроксан 377
4-Метил-1,8-нафтиридии 180
З-Метил-5-нитрамино-1,2,4-триазол 297,
348, 350
5-Метил-3-нитрамино-1,2,4-триазол 350
1-Метил-5-иитробензотриазол 313
1-Метил-6-иитробеизотриазол 313
1-Метнл-7-иитробеизотриазол 317
9-Метил- 10-иитро-о-фенаитролин 282
Метилнитрофуроксаи 366, 371
2-Метил-4-окси-6-алкокси-1,5-нафтиридин
164
2-Метил-4-окси-7-амино-1,8-нафтиридин
160
2-Метил-4-окси-5,7-бензо-1,5-нафтиридин
164
6-Метил-8-окси-2,5-диазафенантрен 284
3-Метил-4-окси-6,7-диметоксикумарин 91
2-Метил-6-окси-1,5-иафтиридин 166
Метилокси-о(л1, п)-фенантролины 279
2-Метил-5-(4-пиридил)-1,3,4-тиадиазол 461
О-Метилтетрагидроал л оксантоксилети но-
вая кислота 79
2-Метил,-6,7,8,9-тетр аги дро-4,5-бензо-|3-кар-
бол ин 226
Метилтетрагидрогарман, см. 2,3-Диметил-
2,3,4,5-тетрагидро-Р-карболин 204.
N-Метилтетрагидрогармин 203
3-Метил-2,3,4,5-тетраги дрога рмин 198
2-Метил-2,3,4,5-тетрагидро-|3-карболин, см.
Тетрагидрогарман
2-Метил-2,3,4,5-тетр а гидро-у-к ар бол ин 190
2-Метил-2,3,4,5-тетрагидро-р-карболин-
карбоновая-2 кислота 200 и сл., 205
2-Метил-2,3,4,5-тётрагидро-|3-карболнн-
карбоновая-4 кислота 196
О-Мети л тетр аги дроке анти лети нова я кис-
лота 77, 80
Метилтетрагидроноргармин 199
2-Метилтетрагидроосайнн, димети левый
эфир 99
1 -Метил-1,2,3,4-тетрагидро-л1(о, ^-фенан-
тролин 275
Метил тетраокси-4,5-бензо-Р-карболин 223
5-Метил-1,2,3-тиадиазолкарбоновая-4 кис-
лота 417, 418
1-Метил-1,2,3-триазол 313, 314, 316
2-Метил-1,2,3-трназол 314, 316, 322
1-Метил-1,2,4-триазол 330, 335
З-Метил-1,2,4-триазол 344
4-Метил-1,2,3-триазол 316
1 -Метил-1,2,3-триазол-4-альдегид 319
З-Метил-1,2,4-триазол-5-нлдиазоний хло-
рид 344
1-Метил-1,2,3-трназолкарбоновая-4 кис-
лота 322
3-Мети л-2,4,5-тр и кето-2; 3,4,5-тетраги дро-
Р-карболин 208
1 - Мети л-4,5,6-три хлорбензотри азол 317
4-Метилумбеллиферон 32
4-Метилуразол 340
1-Метил-о-фенанрролин 283
2-Метил-л1-фенантролин 283
2-Метил-п-фенантролин 283
З-Метил-п-фенантролин 273
9-Метил-о-фенаитролин 2.83, 290, 291
Ю-Метил-.и-фенантролпн 283
6-Метил-ж-фенантроли н карбонов а я-8 кис-
лота 283
2-Метил-п-фенантролин карбоновая-4 кис-
лота 283
N-Метилфенантролон 280, 281
1 -Метил-З-фенил-5-амино-1,2,4-триазол 326
4-Метил-1 -фенил-2,3-бензо-6-карболин 233
2-Метил-4-фенил-1,8-нафтирпдин 161
2-Метил-4-фенил-А3-1,3,4-оксадиазолон-4
406
3-Метил-5-фенил-1,2,4-оксадиазол 390
1-Метил-3-фенил-5-окси-3,5-дигидро-1,2,4-
триазол 344
2-Метил-5-фенцл-1,3,4-тиадиазол 459, 461,
471
З-Метил-5-фенил-1,2,4-триазол 340
Метилфенилфуразан 370, 374
МетилфенилфурокСан. 362
С-Метилфлороацетофенон 108, НО
С-Метилфлороглюцин 107, 108, 109
Метилфуразанкарбоновая кислота 369
4'-Метил-5,6,2',3'-фур аноку мар пн 32
Метилхиндоланол 248
11-Метилхининдолии 220
5-Метилхиндолиний, хлорид 239
1-Метил-2-хлор-|3-карболин 210, 211
2-Метил-6-хлор -1,5-нафтнридпн 166
1 -Метил-4-хлор-1,2,3-триазол 316
1-Метил-5-хлор-1,2,3-триазол 318
предметный указатель
489
2-Метилхромон 58
З-Метилхромон-З-глиоксалевая кислота 90
2-Метилхромоны 37, 58
8-Метилхромоны 60
Метилэтилкарболиний, соли 212
Метод
Дёбнера 162, 168
Дёбнера — Миллера 162
Кнорра 156, 159, 164, 165, 168
Комба 160 и сл., 164, 165, 168
Конрада — Лимпаха 156, 159, 164,
165, 168
Костанецкого 53, 59
Ниментовского 169—171
Померанца — Фритча 163
Пфитцингера 169
Скраупа 162, 165, 168
Фридлендера 169, 171
Чичибабина 161 и сл.
6-Метоксиангелицин, см. Сфондии
8-Метокси-3-ацетил-2-кето-2,3,4,5-тетра-
гидро-Р-карболин 206
5-Метоксибензотриазол 315
5-Метоксибензофуразан 361
5-Метокси-8-(у,у-диметил ал л ил окси) псора-
лен 25
5-Метокси-8-изоамилпсорален 25
4'-Метокси-5'-изопропилпсорален, см. Пеу-
цеданин
8-Метокси-Р-карболин, см. Норгармнн
8-Метокси-Р-карбол ин карбоновая-2 кис-
лота, см. Норгарминкарбоновая кислота
Метокси-Р-карболины 197
8-Метокси-2-кето-3-метил-2,3,4,5-тетрагид-
ро-Р-карболин 206
8-Мето кси-2-кето-2,3,4,5-тетр аги дроф-кар -
болин 207
7-Метоксикума рин-8-ацетальдегид 31
7-Метоксикумарин-8-уксусная кислота 31
15-Метокси-5-метил-2,3-бензо-у-карболин
229
7-Метокси-2-метил-Р-карболин 197
9-Метокси-2-метиЛ-р-карболин 197
8-Метокси-З-мети л-2-кето-2,3-дигидроф-
карболин 198
7-Метокси-8-метилкумарин 31
З-Метокси-4-метилтриазолон 340
2-Метокси-11-метилхининдолии 220
8-Метокси-1,6-нафтиридин 180
4-Метокси-6-нитробензотриазол 325
5-Метокси-8-нитропсорален 21
2-(3-Метокси-4-оксибензил)-2,3,4,5-тетра-
гидроф-карбол ин карбоновая-2 кислота
204
5-Метокси-8-оксипсорален 20, 21
6-Метоксипиперониловая кислота 105
8-Метоксипсорален,. см. Ксантотоксип
7-Метокси-2,8,2',3'-тетраметил-5',4' ,5,6-
фуранохромон 35
8-Метоксиумбеллиферон 81
З-Метоксиуразол 340
9-Метокси-п-фенантролин 272
а-Метоксифенацтролины, иодметилаты 276
7-Метоксихроманкарбоновая-З кислота
47
7-Метокси-А3-хроменкарбоновая-3 кисло-
та 47
Монаскорубрин 45, 46
Моиоарилсидноны 398
Моноацилфуразаны 374
О-Монометилуснетол 45
Мононитробергаптен 17
Монотиоуразол 329
Мундулетон 106
Мундулоксиновая кислота 106
Мундулон 105 и сл.
Мунетон 141
Мутаротенон 131
Надтиоциановая кислота 435, 436
4-(1-Нафтилсульфонил)карбоксамидо-5-
окси-1,2,3-триазол 312
Нафтиридилнафтиридиний, соли 177 и сл.
1,5-Нафтиридин 156, 165, 180, 181, 182
1 ф-Нафгиридин 156
1,8- Нафтиридин 156, 157, 181 и сл.
1,7- Нафтиридин-2-альдоксим 172
Нафтиридинкарбоновые кислоты 158
Нафтиридины
N-окиси 175, 176
реакции 175—182
1,4- Нафтиридины 175
1,5- Нафтиридины
реакции 175, 176, 177, 178, 179, 180,
181, 182
синтез 163—175
1,6- Нафтиридины 170 ’
реакции 178, 179.
синтез 167—175
1,7- Нафтиридины 163, 164, 166
реакции 180
синтез 171, 172, 174
1,8- Нафтиридины
иодметилаты 175
реакции 175, 177, 178, 180
свойства 158
синтез 155—162, 169—171
1,2,3-Нафтотиадиазолы 422
Нафтофуразан 374
Неторовая кислота 128
3-Нитрамино-5-метил-1,2,4-триазол 297,
348
2-Нитрамино-1,2,4-триазол 329
3-Нитрамино-1,2,4-триазол 348, 350
1-(п-Нитробензаль)аминотриазол 302
ацп-Нитробензилметилфуразан 376
4-Нитро-1,2,3-беизотиадиазол 424
5-Нитро-1,2,3-бензотиадиазол 424 .
7-Нитро-1,2,3-бензотиадиазол 425
4-Нитро-2,1,3-бензотиадиазол 443, 447,
448
5-Нитро-2,1,3-бензотиадиазол. 448
4-Нитробензотриазол 317, 323
5-Нитробензотриазол 313, 317, 323
4-Нитробензофуразан 369
7-Нитрогармалин 210
7-Нитрогарман 210
7-Нитро-1,5-диметилбензотриазол 313
1,2-Нитро-4,5-динитрозобензол 371
2-Нитро-9,10-динитрозофенантрен 369, 387
2-Нитрозамино-5-фенил-1,3,4-оксадиазол
408
10-Нитрозохиндолин 243 . :>
7-Нитро-6-карболинкарбоновая-3 кислота
189, 243
5-Нитроксантотоксин 19
Нитроловые кислоты- 363, 365
490
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
4-Нитро-6-метилбензотриазол 313
7-Нитро-2,3-метилендиоксихиндолин 243
9-Нитро-5-метилизохининдолии 222
7-Нитро-5-метилхиндолиний, хлорид 239
4-Нитро-5-метоксибеизотри азол 318
Нитрон 332, 348
3-Нитро-1,2-нитрозобензол 377
З-Нитро-4-окси-1,5-нафтиридин 171, 177
9-Нитро-л1-фенаитролин 282
9-Нитро-о-фенантролии 282, 290, 291
10-Нитро-л<-фенантролин 282
6-(п-Нитрофеиил)имидазо- [2,1-Ь]-тиади-
азол 470
Нитрофенил-1,3,4-тиадиазолы 471
2-Нитрофенил-1,2,3-триазол карбоновая
кислота 310
2-(п- Нитрофеиил)-1,2,3-триазол карбоно-
вая-4 кислота 310
2-(п-Нитрофеиил)-5-фенил-1,3,4-оксади-
азол 405
Нитроферроии 291
7-Нитрохиндолин 237
11-Нитрохиндолии 237
,7-Нитрохиидолиикарбоновая-11 кислота
237
9-Нитрохиииидолии 222
Новобиоцин 62, 63, 64
Новобиоциновая кислота 63 и сл.
Нодакенетии 23 и сл., 82
Нодакенин 23
Норгарман, см. р-Карболин
Норгармии 208
Норгарминкарбоиовая кислота 208
1,2,3-Оксадиазол 357
1,2,4-Оксадиазол 357, 358
1,3,4-Оксадиазол 357
1,3,4-Оксадиазолои-5 407
Оксадиазолоны 407
1,3,4-Оксадиазолоны-2 401
1,3,4-Оксадиазолтиолы-5 404
1,2,3-Оксадиазолы 397—399
1,2,4-Оксадиазолы
реакции 395 и сл.
свойства 378, 397
синтезы 388—394
бир-1,2,4-Оксадиазолы 390
1,2,5-Оксадиазолы 357—378
1,3,4-Оксадиазолы
применение 409
реакции 405—409
свойства физические 409
синтезы 399—405
1,3,5-Оксадиазолы 378
4-ОксамиДО-1-оксамолил-1,2,4-триазоли-
диидиои-3,5 338
1-Окси-3-алкил-2,7-нафтиридины 173
З-Окси-б-алкил-сильи-триазолы 325
7-Окси-8-аллилкумарииы 34
о-Оксйальдегидокумарины 15
4-Окси-7-амино-1,8-нафтиридин 158
З-Окси-5-амино-1,2,4-триазол 349
4-Окси-5-арилазо-1,2,3-триазолы 317
1-Окси-3-арил-2,7-нафтириднны 173
3-Окси-5-арил-си.ил1-триазолы 325
3-(2-Окси-4-ацетамидофенил)-5-метил-1,2,4-
оксадиазол 393
7-Окси-8-ацилхромоиы 45
3,3'-(о-Оксибензилиден)-бис-(4-оксикума-
рнн) 62
2-(л1-Оксибензил)-2,3,4,5-тетрагидро-Р-кар-
болии 256
2-(з1-Оксибензил)-2,3,4,5-тет'ра1идро-Р-
карболинкарбоновая-2 кислота 205
бис-Оксибензилфуроксан 375
2-Окси-4,5-бензо-|3-карболин 225
5-Окси-2,3-бензо-у-карболин 220
4-Окси-1,2,3-бензотиадиазол 425
1-Оксибензотриазол 314
4-Оксибензотриазол 317
5-Оксибеизотриазол 310, 317, 318
5-Оксибензотриазол карбоновая-4 кислота
318
6-Оксибензофуран 15
1-Оксн-4,9-диазафенантренкарбоновая-9
кислота 286
7-Окси-3,4-дигидрокумарин 26
5-Окси-4,7-диметилкумарин 59
7-Оксн-2,2-диметилхроман 74, 75, 80
7-Окси-2,2-диметилхромаион 74
6-Окси-4,7-диметоксикумаран 37
4-Окси-6,7-диметоксикумарин 53, 88, 92
7-Окси-6,7-диметокси-3-метилкумарнн 86
3-Окси-6,7-диметокси-2-метилхромон 86
7-Окси-8-изовалерил-4-метнл кумарин 120
6-Окси-2-изопропилкумаран 75
6-Окси-2-изопропилкумаранон 75
5-Окси-6-карбоксибензотриазол 310
3-Окси-5-карбокси-1 ,'2Л-триазол 344
1 -Окси-5-карбэтоксиметил-6-нитробензо-
триазол 305
о-Оксикетокумарииы 15
6-Оксикумаран 16
4-Оксикумарин 62
7-Оксикумарин 33
7-Оксикумарины 34
4-Оксикумарои 43 и сл.
З-Окси-5-меркапто-1,2,4-триазол 326, 327
2-Окси-4-метил-7-амино-1,8-нафтиридин
159
1-Оксиметилбензотриазол 314, 315, 318
1-Окси-3-метил-4,9-диазафенантрен 286
3-Окси-1-метил-4,9-диазафеиантреи 286
1 -Оксиметил-5-оксибеизоТриазол 315
4-Оксиметил-5-оксибензотриазол 315
2-Оксиметил-2,3,4,5-тетрагидро-|3-карбо-
лии 200
5-Оксн-5-метил-1,2,8-триазаиидолизин
345
5-Окси-7-метнл<-1,2,8-триазаиндолизин
342
5-Оксн-7-метил-1,3,8-триазаиндолизин
342
З-Оксиметил-1,2,4-триазол 339, 346
2-Окси-5-метилтриазол карбоновая-4 кис-
лота 304
5-Окси-7-метокси-2,2-диметил-7-изовале-
рилхромаи 105
5-Оксн-7-метокси-2,2-димети л хроман 135,
136
6-Окси-4-метоксикумаран 41
6-Окси-4-мето кси кумарон карбоновая-2 кис-
лота 18
7-Окси-3-метокси-2-метил хромой 61
5-Окси-7-метокси-4-фенилкумарин 83
7-Окси-З-метоксифлавои 60
а-Оксиминонитроловая кислота 366
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
491
2-Окси-1,5-нафтиридин 165, 177
2-Окси-1,7-нафтиридин 172
2-Окси-1,8-нафтиридин 159
4-Окси-1,5-нафтиридин 164, 165, 166, 169,
170, 171, 177, 178
4-Окси-1,7-нафтиридин 164
4-Окси-1,8-нафтиридин 158, 159, 170
8-Окси-1,6-нафтиридин 180
8-Окси-1,7-нафтиридин 165
Оксинафтиридины 181
1-Окси-6-нитробензотриазол 305, 308
4-Окси-6-нитробензотриазол 325
2-Окси-3-нитро-1,5-нафтиридин 177
3-Окси-5-нитро-1,2,4-триазол 339
5-Окси-1,3,4-оксадиазол 401
5-Окси-1,2,4-оксадиазол-З-илуретан 390
Окси-1,3,4-оксадиазолы 401
Оксипеуцеданин 22, 24, 25
Оксипеуцеданииовая кислота 25
5-Оксипсоралеи, см. Бергаптол
7-Окси-5,6-тетраметилен-1,3,8 (или 1,2,8)-
триазаиндолизин 342
2-Окси-4,5-тетраметилен-1,2,3-триазол 304
5-Окси-1,2,4-тиадиазол 440
4-Окси-1,2,3-триазол 301, 317, 322
5-Окси-1,2,3-триазол 312, 322
З-Окси-1,2,4-триазол 339
3-Окси-сил(Л1-триазол 340
1-Окси-1,2,3-триазолдикарбоновая-4,5 кис-
лота 308
Окси-1,2,4-триазолы 318, 348
2-Окси-л« (или п)-фенантролин 281
4-Окси-п-фенантролин 277 и сл.
6-Окси-л(-фенантролин 280
10-Окси-л(-фенантролин 270, 277, 282
Оксифенантролины 277 и сл.
2-(п-Оксифенилазо)-5-амиио-1,3,4-тиади-
азол 470
3-Окси-5-феииламино-1,2,4-тиадиазол 433
З-Окси-5-фенил-1,2,4-оксадиазол 382, 383
3-Окси-5-фенил-1,2,4-триазол 348, 349
7-Окси-6-формил-2,2-диметилхроман 80
7-Окси-8-формил-5-метокси-2,2-диметил -
хромаи 79
7-Окси-8-формилхромоны 60
Оксифуранокумарины 14
11-Оксихининдолин 220
7-Окси-8-хлорацетилкумарин 33
15-Оксиэллиптон, см. Малаккол
8-(2-Оксиэт ил аминоэтил амино)-4-окси-л1-
фенантролин 283
Октагидроаллороттлерон 112, 113
Октагидро-1,8-нафтиридин 175
1,2,3,4,5,6,12,12а-Октагидро-12-окси-
11,12-диметил-11 Н-иидоло-[2,3-&]-хино-
лизин 232
Октагидророттлерон 108, 109, 113
1,2,3,4,5,6,7,8-Октагидро-п-фенантролин
273
5,5 а, 6,7,8,9,9а, Ю-Окта гидро-«-фенантро-
лин 273, 274
Ореоселон 25, 26 и сл., 30
Ороселон 29, 30
Орцин 11
Осайетин 97, 99
Осайин 85, 95, 96, 97, 101. 11)2, 103, 105
Остенол 31
Остол 31, 32
Острутол 24
Пахиррхизон 118, 140 и сл.
Пахиррхицин 50
Пейценин 70
Пелтогинол 57, 58
1,3,3,5,5-Пентаметил-2,3,4,5-тстрагидро-у-
карболин 191
Пергидророттлерин 114
Пергидророттлерон 114
Перициаииловая кислота 360
Пеуцеданин 25
Пимпинеллин 23, 28 и сл.
Пиниатин 44
Пинорезинол 7,9, 10
Пиперонилметилфуроксан 377
1,4-Пиранокумарины 61 и сл.
2-(4-Пиридил)-Д2-1,3,4-оксадиазолон-5 409
М-(3-Пиридил)сиднон 399
Пиридиноксифенантролины 279
Пиридонафтиридин 160
Пиридооксадиазолон 394
Пиримидонафтиридины 160
Поли-4-амино-1,2,4-триазолы 333
Помиферин 85, 95, 96 и сл., 100 и сл.,
102, 103, 104, 105
Помиферитин 100, 101, 104
5-Пропил амино-З-амино-1,2,4-тиадиазол
431
З-Пропил-4-метилфуроксан 367
Р-Псевдокарболин 208
Псорален 13, 15 и сл.
Птерокарпйн 47 и сл., 49, 50
Резерпин 191, 248, 255 и сл., 258 и ел.
Ресцинамин 256
Риссовая , кислота 120, 121
Ротенол 126, 128
Ротенолоны 124 и сл.
Ротенон 115, 117, 118 и сл., 120, 122, 124,
125 и сл., 128, 130, 131, 132, 142 — 144
Ротенонон 128, 129
Ротеноновая, кислота, см. [5-Дигидророте-
нон
Ротенононовая кислота 129
Ротеноны 130, 131, 132, 142—144
Ротиорин 45, 46
Роттлерин 82, 85, 106, 107, ПО, 111, 113
Роттлерон 108
Роттлероновая перегруппировка 114
Рубропунктатин 45, 46
Рутекарпин 191, 207, 248, 249
Самидин 82
Сезамин 7, 8, 9, 10
Сезамолии 7, 10
Сезелин 71, 80, 81
Семпервирин 246, 259, 260
Серпентин 259
Сиднонимины 399
Сидноны 397, 398
Склеротиорин 45, 46
Скополитин-8-альдегид 29
Сотетсуфлавон 70
3-Стирил-5-фенил-1,2,4-оксадпазол 388
Суматрол 117, 118, 133, 134
Сфондилин 29
Сфондин 29
Сциадопитизин 69, 70
492
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Телепатии 191
Тетрабромтетраметиленфуразан 359, 378
Тетрабромтетраметиленфуроксан 369, 378
Тетрагидроаллороттлерин III, 112, 113, 114
Тетрагидроальстонилинол 261
2,7,8,9-Тетрагидро-2,3-бензо-у-карболин
229
Тетрагидробензо-1,7-нафтиридин 171
Тетрагидрогармалин 197
Тетрагидрогарман 196, 204
2,3,4,5-Тетрагидрогарман 215
Тетрагидрогарманкарбоновая кислота 196
Тетрагидрогармин 198, 203
Тетрагидродегидромаллакол 140
Тетрагидродегидроротенон 130
Тетрагидродегидросуматрол 133
Тетрагидродерризовая кислота 119, 120,
122, 126
Тетрагидродерритол 127
1,2,3,4-Тетрагидро-11,12-диметил-11 Н-ин-
доло-[2,3-&1-5-хинолизиний, иодид 232
1,2,3,4-Тетрагидро- 12Н-индоло- [2,3-а]-хи-
нолизиний, бромид 244
Тетрагидроиобйрин 251, 259
Тетрагидро-р-карболин 203 и сл., 205
2,3,4,5-Тетрагидро-р-карболин 193, 195
Тетрагндро-у-карболин 190, 191
2,3,4,5-Тетрагидро-у-карболии 191
6,7,8,9-Тетрагидро-у-карболин 191
Тетрагидрокарболиний, иодид 190
Тетрагидро-р-карболинкарбоновая-2 кис-
лота 193, 204
2,3,4,5-Тетрагидро-р-карболинкарбоновая-
4 кислота 193, 195, 209
Тетрагндролувангетин 81
Тетрагидромалакколовая кислота 140
Тетрагпдронодакенетин 23
Тетрагидрэ-1,2.4-оксадиазолы 394
Тетрагидроороселон 3)
Тетрагидрэосайетин 98, 99, 100
Тетрагидроосайетиновая кислота 101
Тетрагидроосайетинон 100, 101
Тетрагидроосайин 97, 98, 99,-102, 103, 105
Тетрагидропомиферин 97, 100, 102, 103, 105
Тетрагидропомиферитин 100, 101
Тетрагидропомиферитон 101
Тетрагидроротенол 128
Тетрагидроротенон 118, 119, 120, 125, 126,
128, 130
Тетрагидроротенонон 128
Тетрагидророттлерин 107, 108, 109, 110,
112, 113, 114
Тетрагидророттлерон 109
Тетрагидросуматрол 133
Тетрагидросуматроловая кислота 133
Тетрагидротубанол 120, 123, 124, 127
Тетрагидротубовая кислота 123
1,2,3,4-Тетрагидро-п-фенантролин 276
5,6,7,8-Тетрагидро-п-фенантролин 278
1,2,3,4-Тетрагидро-п-фенатролинон 272,
273
Тетрагидрофуранокумарины 14
Тетрагидроэллиптовая кислота 140
Тетрагидроэллиптон 140
Тетразины 337
Тетра-Р-карболин 205
О-Тетраметилгинкгетин 69
Тетраметиленфуроксан 361, 371
Тетраметилизогаллофлавин 54
1,3,4,5-Тетраметил-! ,2,4-трпазолип, иодид
340
1,2,3.4-Тетранитрозо-5-нитробензол 378,
386
1,3,5,6-Тетраокси.кеантон ,105
2,2',2",2"'-Тетрапиридил 291, 293
2,3,5,6-Тетрафенил-2,3-дигидро-вт{-тет-
разин 306
Тетрафенилнафтодифуран 1 I
Тетрафенилтетразин 338
4,5,6, 7-Тетрахлорбензотриазол 314, 317,
322
Тефровая кислота 138
Тефрозин 117, 118, 133, 137
1.2,3- Тиадназоп 418 и сл.
1,2,4- Тиадиазол 439, 443
1,3,4-Тиадиазол 470
1,2.5- Тнадиазолдикарбоновая-3.4 кислота
443
1,2,5- Тиадиазолидин 443 и сл.
1,2,4- Тиадиазолидины 438, 439
1,2,5- Тиадиазолидины 442
Д2-Тиадиазолинон-5 466
1,2,3-Тиадиазолины 420
1,3,4- Тиадиазолины 466—469
А2-1?374-Тиадиазолины 466, 467
Тиадиазолохинолины 422
1,2,3- Тиадиазолы
с конденсированными ядрами 420—426
номенклатура 416
реакции 418
свойства 418
синтезы 416—418
1,2,4- Тиадиазолы
номенклатура 427
синтезы 427—439
1,2,5- Тиадиазолы 442
номенклатура 443
реакции 443
синтез 443
1,3,4- Тиадиазолы
модификации 463 и сл.
номенклатура 448
реакции 470
синтезы 448—462
1,2,3- Тиадиазопиримидин 421
1,2,3-Т иадиазофеназин 424
Тиантрены 425
Тиоперимидовые кислоты 430
5-Тио-1,3,4-трифенил -1,2,4-триазолон 345
Тиоуразол, см. З-Окси-5-меркапто-1,2,4-
триазол
2-Тио-3-фенил-5-метил -1,3,4-оксадиазолон-
2 405
Токсикаровая кислота 128, 134
Токсикарол 72, 82, 117, 130, 133, 134—137
5-(п-Толиламино)-3-амино-1,2,4-тиадиазол
431
3-(я-Толил)-5-ацетонил-1,2,4-оксадиазол
396
З-Толил-5-бромметил-1,2,4-оксадиазол 396
З-Толил-5-метил-1,2,4-оксадиазол 396
1,3-бис-(3-Толил-1,2,4-оксадиазол-5-ил)аце-
тон 396
3-Толил-1,2,4-оксадиазол-5-уксусная кис-
лота 396
5-Толил-5-цианметил-1,2,4-оксадиазол 396
бис-Триазен 299
Триазоилацетамид 320
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
493
Триазоилацетонитрил 319
1,2,3-Триазол 314, 315, 316, 323
1,2,4-Триазол 335, 340, 343, 344, 348, 350
номенклатура 296
синтезы 325—337
Триазолазид 299
1,2,3-Триазол-4-альдегид 322
бис-Триазолбензолы 305
бис-Триазолбензохиноиы 305
Триазолдикарбоновая-4,5 кислота 308
1,2,4-Триазол-З-илкарбинол 346
1,2,3-Триазол-1-илуксусная кислота
310
Р-(1,2,4-Триазол-1-ил)этанол 346
Триазолииы 299, 300
Триазолия соли 313
1,2,3- Триазолкарбоновая кислота 321
Триазолкарбоновые кислоты 323
Триазолотриазолы 338
Триазолоциннолон 302
1,2,4- Триазолсульфоновая кислота 347
сн.м.м-Триазолтиолы 347
Триазолтриазен 299
Триазолфеназины 320
в«1{-Триазолы(1,2,3-Триазолы)
кислотность кольца 320
номенклатура 296
основность кольца 322
реакции 312—320
синтезы 297—310
термическая изомеризация 311 п сл.
сн.ч.М'-Триазолы (1,2,4-Триазолы)
биохимические свойства 350 и сл.
кислотность кольца 347 и сл.
комплексы 349
основность кольца 348
применение 350
реакции боковой цепи 346 и сл.
— замещения 339—346
синтезы 337, 338, 454
1,2,4-Триалкил-у-карболиний, иодид 189
2,4,5-Триаминонитробензол 371
3,5,7-Триамино-1,2,6,8-тетразаин дол из ин
342
3,4,6-Триацетоксикумаран 17
4,5,М-Трибром-1,2,3-триазол 316
2,4,5-Три кето-2,3,4,5-тетрагидро-[}-карбо-
лины 207
1,5,6-Триметилбензотриазол 314
2,5,6-Триметилбензотриазол 314
Три-О-метилведелолактон 50
Триметилизогаллат 54, 55
О-Триметилпелтогинол 57, 58
О-Триметилпелтогинон 57, 58
1,3,5-Триметил-1,2,4-триазол 340
3,4,5-Триметил-1,2,4-триазол 340
1,2,4-Триметилуразол 340
Триметил-о-фенантролины 270
2,5,5-Триметил-4-фенил-Д2-1,3,4-тиадназо-
лин 467
Триметилфлороглюцин 107
2-(3,4,5-Триметоксибензил)-2,3,4,5-тетра-
гидро-р-карболинкарбоновая-2 кислота
204
2,2',2"-Трипиридил 291
Три(триазол)бензол 306
2-(Трифенилгуанидино)бензотриазол 442
1,3,4-Трифенил-1,5-дигидро-5-тиокето-
1,2,4-триазол 336
3,6,7-Трифен и л-1,2,5,8-тетр аза индол изи н
347
1,3,5-Трифеннл-1,2,4-триазол 329, 349
2,4,5-Трифенил-1,2,3-триазол 306
3,4,5-Трифенил-1,2,4-триазол 330, 331
Р-Тубаиновая кислота 105
Тубанол 123
Тубовая кислота 122, 123, 125, 127,
129
Умбеллиферон 27, 28, 51, 81
Умбеллиферои-8-альдегид 28. 30, 52, 82
Умбеллиферонкарбоновая-6 кислота 26
Уразол 340
3-Уреидо-4-бензоил-5-феннл-1,2,4-триазол
343
З-Уреидо-5-метнл-1,2,4-триазол 334
З-Урендо-5-фенил-1,2,4-оксадиазол 390
З-Уреидо-5-фенил-1,2,4-триазол 334, 338
Фарагол 10
Феллодтерин 25
Фенантрен-9,10-фуразан 372
Фенаитридин 321
.к-Фенантрол ин
метилаты 274. 275
N-окиси 276, 277, 280
производные 277—282
реакции 272
свойства 271
синтез 270, 271
о-Фенантролии
комплексы 269, 287—290
метилаты 274, 275
N-окиси 277
производные 277—281
реакции 270, 272
синтез 269
н-Фенантролин
метилаты 274, 275, 276
N-окиси 276, 277, 280
производные 272, 273, 277—282
реакции 272, 273 и сл.
свойства 272
синтез 268
л1-Фенантролин-2-альдегид 283
л1-Фенантролин-8-альдегид 283
п-Фенантролии-4-альдегид 283
м (или п)-Фенантролинкарбоновая-2 кис
лота 283
о-Фенантролинкарбоновая-9 кислота 283
.«-Фенантролинкарбоновая-10 кислота 283
м (о или п)-Фенантролин-9,10-хинон 272
Фенантролины 266
классификация 266
номенклатура 267
производные 273
реакционная способность 272
синтез 267—269
устойчивость 272
9,10-Фенантролохинон 272
Фенацилфенилфуразан 374
4-Фенилазо-5-амино-1,2,3-беизотиадиазол
424
З-Фенил-5-алкил-1,2,4-оксадиазолы 389
Фенилалкоксифуроксаны 375
494
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1-Феиил-3-амиио-5-анилиио- 1,2,4-триазол
329
2-Феиил-4-амиио-5-ацетамидо-1,2,3-три-
азол 318
3-Феиил-4-амнно-5-меркапто-1,2,4-трназрл
331, 347
З-Феиил-5-амиио-1,2,4-оксадиазол 395, 396
2-Феиил-4-амиио-5-оксибензотриазол 309
3-Феиил-4-амиио-5-оксиизоксазол 375
2-Феиил-4-амиио-5-окси-1,2,3-триазол 318
N-Феииламииотриазол, см. Дифенилура-
зии
1-Феиил-5-амиио-1,2,3-триазол 299, 318
3-Феиил-4-амиио-1,2,4-триазол 345
1-Фенил-5-амино-1,2,3-триазол карбоно-
вая-4 кислота 310
Феииламинофуразан 360, 395
Феииламииофуроксаи 370
Феиил-п-аиизилфуроксаи 362
1-Фенил-3-аиилиио-1,2,4-триазол 337, 350
3-Феиил-5-арил-1,2,4-оксадиазол 392
2-Фенил-4-ацетил амино-1,2,3-триазол 321
З-Феиил-5-беизоил-1,2,4-оксадиазол 393
2-Феиил-5-бензоил-1,3,4-оксадиазол 408
3-Феиил-5-беизоил-1,3,4-оксадиазолои-2
401
1-Феиил-4-бензоилоксимиио-1,2,3-триазо- ,
лон-5 324
2-Феиил-4-беизол азо-5-метил-1,2,3-три-
азол 308
1-Феиилбеизотриазол 323
2-Феиилбеизотриазол 306, 323
4-Феиилбензотриазол 306
1-Фенилбеизотриазол-4,7-хиион 324
2-Фенилбеизотриазол-4,7-хиион 324
Фенилбифеиилилоксадиазол 409
2-Феиилгидразиио-1,3,4-тиадиазол 455
1-Феиилгуаиазол 343, 350
З-Фенил-4-дезилфуразан 373
10-Феиил-4,9-диазафеиаитреи 287
2-Феиил-4,5-диамиио-1,2,3-триазол 318
2-Фенил-4,9-дигидро-4,9-дикето-5,8-диок-
синафто-[2,3]-триазол 320
2-Фенил-4,5-дигидро-Р-карболии 201
1-Феиил-2,3-дигидро-2-метил-3-кето-1,2,4-
триазол 349
1-Фенил-4,5-дикарбэтокси-1,2,3-триазол
320
1-Фенил-3,5-димеркапто- 1,2,4-триазол 348
1-Феиил-3,5-диметил-1,2,4-триазол 335,
349, 350
2-Фенил-4,5-диметил-1,2,3-триазол 302
п-Фенилеидитиадиазол 422
5-Фенилимидазо-[2,1-Ь]-тиадиазол 470
1-Феиил-4-карбалкокси-5-метил-1,2,3-три-
азол 299
1-Феиилкарбамилбензотриазол 315
1-Фенил-5-карбокси-1,2,4-триазол 343
2-Феиил-4-(о-карбоксифеиил)-1,2,3-три-
азолкарбоиовая-5 кислота 310
1 - Феиил-4-карбэтокси-5-ами ио-1,2,3-три-
азол 311
1-Фенил-2-карбэтокснацетилгуаиазол 343
1-Фенил-4-(а-карбэтоксиацетил)-1,2,3-три-
азол 320
3-Феиил-5-меркапто-1,2,4-тиадиазол 428
3-Фенил-5-меркапто-1,2,4-триазол 341
2-Фенил-4-метил-5-амино-6-беизол азобеи-
зотриазол 315
1-Фенил-5-метиламино-1,2,3-триазол 311
2-Фенил-4-метил-5-амиио-1,2,3-триазол 318
2-Фенил-4-метил-5-ацетоиил-1,2,3-триазол
307
2-Фенил-4-метнл-5-бензоил-1,2,3-триазол
307
2-Феиил-6-метилбеизотриазол 306 .
1-Фенил-З-метилгуаиазол 350
2-Феиил-4-метил-5,6-диамииобеизотрпазол
315
1-Феиил-3-метил-4-карбамоил-5-кето-4,5-
дигидро-1,2,4-триазол 343
1-Феиил-2,4-метил-5-кето-3,5-дигидро-1,2,4-
триазол 344
2-Феиил-4-метил-1,8-иафтиридии 161
1-Феиил-3-метил-5-окси-1,2,4-триазол 334,
336
1-Феиил-4-метил-5-окси-1,2,3-триазол 323
1 -Феиил-З-метил-5-стирил-1,2,4-триазол
344
2-Фенил-4-метил-А3-1,2,3-тиадиазол ин 420
3-Феиил-5-метилтио-1,2,4-триазол 328
I-Феиил-3-метил-.| ,2,4-триазол 327. 344
1 -Феиил-5-метил-1,2,3-триазол 310
2-Фенил-4-метил-1,2,3-триазол 316
1 -Фенил-5-метил-1,2,3-триазол карбоно-
вая-4 кислота 320
1-Фенил-2-метилуразол 340, 341
Фенилметилфуроксаи 370, 371, 375
1-Феиил-3-метокси-4,5-дигидро-4-метил-5-
кето-1,2,4-трназол 340
4-Феиил-5-(п-метоксифеиил)амино-1,2,3-
триазол 321
2-Феиил-1,7-иафтиридин 171
2-Феиилиафтотриазол 322
Феиилиитрозоловая кислота 366
Феиилиитроловая кислота 365
4-Феиил-5-(п-иитрофеиил)амино-1,2,3-три-
азол 321
Феиилиитрофуроксаи’367, 370, 375
З-Феиил-1,2,4-оксадиазол '393
2-Феиил-Д2-1,3,4-оксадиазолиитиои-5 406
З-Феиил-1,2,4-оксадиазолкарбоиовая-6 кис-
лота 393, 396
5-Фенил-1,2,4-оксадиазолкарбоновая-З кис-
лота 393
5-Фенил-1,3,4-оксадиазолон-2(ЗН) 402
1 -Фен ил-З-окси-5-алкил-симм-три азолы
325
1-Феиил-5-окси-4-иитрозо-1,2,3-триазол 317
Феиилокси-1,2,4-оксадиазол 381, 396
2-Феиил-5-окси-1,3,4-оксадиазол 401
1-Фенил-3-окси-1,2,4-триазол 349
1-Фенил-5-окси-1,2,3-триазол 317
2-Феиил-4-окси-1,2,3-триазол 318
3-Феиил-5-окси-| ,2,4-триазол 344
1-Фенил-5-окси-1,2,3-триазолкарбоновая-4
кислота 317
Феиилоксифуразаи 383
Феиилоксифуроксан 375
2-(1-Феиил-1-прото-1,2,3-триазол-3-ил)-1 -
хлорсульфоиилэтанхлорид 300
2-( 1-Феиил-1-прото-1,2,3-тр и азол-3-ил)
этаисульфоиат 300
N-Фенилсидиои 397, 398
2-Фен ил-2,3,4,5-тетрагидро-Р-карбол ин 204
1-Фенилтетразолкарбоиовая-5 кислота 324
2-Фенилтетразолкарбоновая-5 кислота 324
1-Фенил-4,5-тетраметилен-1,2,3-триазол 300
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
495
2-Феиил-7,8,15,16-тетраметокси-4,5-беизо-
f-карболин 223
2-Фенил-1,3,4-тиадиазол 459, 471
4-Фенил-1,2,3-тиадиазол 418, 419
5-Фенил-1,2,3-тиадиазол 419
Фенилтиоперимидовая кислота 430
3-Фенил-5-(л-толил)-1,2,4-триазол 329
1-Фенил-1,2,3-триазол 297, 298, 300, 323
2-Фенил-1,2,3-триазол 303, .323
4-Фенил-1,2,3-триазол 301, 321, ,323
1-Фенил-1,2,4-триазол 335, 343
З-Фенил-1,2,4-триазол 334, 344
4-Феннл-1,2,4-триазол 336
1-Фенил-1,2,3-триазол-4-альдегид 319, 322
2-Фенил-1,2,3-триазол-4-альдегид 316, 319
1-Фенилтриазол ди карбоновая-4,5 кислота
297
(I -Фенил-1,2,3-триазол-4-ил)изонитрозо-
ацетанилид 308
2-Фенил-1,2,3-триазол-4-карбннол 319
I-Фенил-1,2,3-триазолкарбоновая-4 кисло-
та 309
2-Фенил-1,2,3-триазолкарбоновая-4 кисло-
та 320
N-Феиилтриазолотриазол 318
4-Фенил-1,2,4-триазолдитиол-3,5 346
2-Фенил-1,2,3-триазол-4-уксусная кислота
319
1-Фенилуразол 340, 341
4-Фенилуразол 326, 330, 340
2-Фенил-л-фенантролинкарбоновая-4 кис-
лота 267
6-Фен ил-м-фенантрол ин карбоновая-8 кис-
лота 267
Фенил-о-феиантролииы 270
I -Фенил-4-фенилазо-5-окситриазол 324
2-Феиил-5-фенилгидразин-1,3,4-тиадиазол
456
Фенил(фенилглиоксил)фуразан 385
3-Фенил-5-фенил-1,2,4-оксадиазол 391
Фенилфуразан 374
Фенилфуроксан 362, 367, 374, 375, 381,
384
3-Фенил-5-хлор-1,2,4-оксадиазол 382
1-Фенил-5-хлор-1,2,3-триазол 318
1 -Феиил-5-хлор-1,2,3-триазолкарбоиовая-4
кислота 301
Фенилхлорфуроксан 375
1-Фенил-4-циаи-1,2,3-триазол 308
Фенилциаифуразан 371
Фенилцианфуроксан 370
1 -Фенил-3-этил-4-метил-5-окси-1,2,4-три-
азол 341
1-Феннл-3-этил-5-метил-1,2,4-триазол 335
I -Фенил-3-этил-5-окси-1,2,4-триазол 340
Феррин 289
Ферроии 270, 289
Фнкусии, см. Псорален
Флаванолы 69
Флаваноны 115
Флавогалловая кислота 55
Флавогаллол 55, 56
Флавоны 68
4-Формамидо-5-(5-метил-1,2,4-оксадназол-
3-ил)имидазол 394
2-Формамино-1,3,4-тиадиазол 450
2-Формамино-5-фенил-1,3,4-тиадиазол 450
3-Формил -5,7-диметокси-2,2-диметил хро-
май 79
Фуразаны
6-Ф ормил-5-мето кси-7-карбэто кси метокс и-
кумарин 18
3-Формил-4-окси-6,7-диметоксикумарин 91
5-Формил-4-оксикумарои 28
7-Формил-6-окси-4-метоксикумарон 18
8-Формил-7-окси-3-метоксифлавоны 61
1-Формил-4-формил амино-1,2,4-триазол и-
диндион-3,5 338
Фраксетин 29
Фульвииовая кислота 85 , 94 , 95
бис-Фуразанбензолы 369
Фуразаидикарбоновая кислота 369
Фуразаидинитроловая кислота 360
N-окиси, см. Фуроксаны
реакции 369—378
синтез 357—361
физические свойства 378
Фураиобензофураны 10 и сл.
синтез 10—13
структура 11
Фуранокумарины 13 и сл.
изомеры 13 и сл.
реакции 14—15
синтез 15 н сл., 23 и сл.
2',3',7,8-Фуранофлавон 45
'Фураиофлавоиы 38
Фуранофураны 7 и сл.
Фуранохромоны 32
изомеры 35
реакции 35 и сл.
синтез 36
Фуроксандикарбоиовая кислота 376
. Фуроксанкарбоновая кислота 369
Фуроксаны
инфракрасные спектры 379
монозамещенные 381—388
реакции 369—378
синтез 361—367
структура 379
Фусцин 62, 64, 65
Фусциновая кислота 64
Хиндолин (3,4-Беизо-Р-карболии) 217,233,
235, 241, 242
иодметнлат 237 и сл.
синтез 234, 236
структура 233 и сл.
Хиндолинкарбоновая-11 кислота 222. 233.
234, 235, 236 и сл., 240
Хининдолин 217, 219—220, 222
Хииокифлавин 70
4-Хлор-5-амино-1,2,3-беизотиадиазол 424
2-Хлор-4,5-бензо-р-карболин 225
4-Хлор-2,1,3-бензотиадиазол 448
5-Хлор-1,2,3-бензотиадиазол 423 и сл.
5-Хлор-2,1,3-бензотиадиазол 448
5-Хлорбеизотриазол 322
8-Хлор-6,7-бензохинолин 269
2 - Хлор-Р-карболинкарбоновая-4 кислота
195
7-Хлор-2-кето-2,3-дигидро-4,5-бензо-[}-кар-
болин 226
5'-Хлормеркурметилфуранокумарии 34
2-Хлор-1 -метил-4,5-бензо-Р-карболин 226
I - Хлорметилбеизотриазол 314, 318
5-Хлор-З-метил-1,2,4-тиадиазол 433
5- X лор-3-метилтио-1,2,4-тиа диазол 433
496
ПРЕДМЕТНЫЙ-УКАЗАТЕЛЬ
2-Хлор-1,5-нафтиридин 176, 18Г
4-Хлор-1,5-нафтиридин 179
г-Хлор-З'-нитро-! ,5-нафтиридин 181
5-Хлор-8-окси-1,6-нафтиридин 179
2-(п-Хлорсульфонилфенил)-1,2,3-триазол
316
5-Хлор-1,2,4-тиадиазол 433
З-Хлор-1,2,4-трназол 344, 345, 348
5-Хлор-1,2,4-триазол 348
1-Хлор-п-фенантролин 280
2-Хлор-л-фенаитролин 276, 277, 280, 281
2-Хлор-п-фенантролин 277, 281
4-Xлор-.и-фенантролин 277
6-Х лор-л<-фенантролин 280
9-Хлор-о-фенантролин 279, 291
10-Хлор-л!-фенантролин 271, 280
10-Хлор-п-фенантролин 275
3-Хлор-5-фенил-1,2,4-оксадиазол 382, 383
Хромано-у-пнроны 70
Хромон-8-альдегиды 60
Хромоно-у-пироны 66, 67 и сл., 69
Цатон 30
Цианнловая кислота 376
4-Циан-5-метил-1,2,3-триазол 301
З-Цианметил-4-фенил-1,2,4-триазол 336 ,
3-(3-Цианпропил)-2,3,4,5-тетрагидро-Р-кар-
болинкарбоновая кислота 247
5-Цнаитетразол 300
4-Циан-1,2,3-триазол 300
Цианфуразан 360
I 1-Цнанхиндолин 237
Циклоновобиоцнновая кислота 64
Цитромицетин 64, 85, 86, 93
Цитромнцин 86, 92, 93, 95
Эйпарин 82
Эллаговая кислота 54
Эллиптол 141
Эллиптон 117, 118, 138 и сл., 139
Эпипинорезинол 9, 10
Эпиротенон 131
Эпиэвдесмин 10
Эрознин 50
5-Этнламино-З-ампно-1,2,4-тиадиазол 431
2-Этил-4,5-бензо-Р-карболин 224
О-Этилвиснагон 39, 41
О-Этилдезоксиапоксантоксилетин 78
о-Этилкеллинон 36
З-Этилмеркапто-5-амнно-1,2,4-тиадиазол
435
2-Этнлмеркапто-5-меркапто-1,3,4-тиади-
азол 457
2-Этилмеркапто-5-фенпл-1,3,4-тиадиазол
458
4-Этил-10,11,12,13 тетрагидро-1-метил-2,3-
бензо-у-карболиний, иодид 231
1-Этил-1,2,3-триазол 310
1 -Этил-1,2,3-триазолкарбоновая-5 кислота
310
О-Этилфлороглюцин 39
2-Этил-4-формилфлороглюцин 39
4'-Этнл-5,6,2',3'-фуранокумарин 32
5-Этокси-2,1,3-бензотиадиазол 445
З-Этокснметил-1,2,4-триазол 346
5-Этокси-5-метокси-8-мети л кумар ин 78
5-Это кси-7-метокс и-6-метил кумарин 78
2-Этокси-1,3,4-оксадиазолтион-5 403
2-Этокси-5-фенил-1,3,4-тиадиазол 460
2-Этокси-5-этилмеркапто-1,3,4-тиадиазол
457
6-Этокси-5-этил-4-метокснбензофуран 39
Чатрейзнн 56
Эвдесмин 7, 8, 9, 10
Эвдонамин 191, 207, 248, 251
Ягеин 191
Якарепбин 85. 105
С Ямаицин 85, 105
СОДЕРЖАНИЕ
Предисловие Эльдерфильда к русскому изданию................................... 5
Глава I
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ДВА ГЕТЕРОАТОМА КИ.Л Г< ДА
В РАЗЛИЧНЫХ ЦИКЛАХ
У . Б. Валли
Введение.......................................................... . 7
Фуранофураны. Бисфурапондные лигпаиы................................... 7
Фуранокумарины....................................................... 13
Фуранохромоны ........................................................ 35
Кумаранохроманы .................................... 47
Кумаро-а- и кумаро-у-пироны........................................... 51
Хромоно-у-пироны .................................... 66
Хромено-а-пироны ................................... 71
Хромено-у-пироны .................................... 84
Хроманохроманоны, хромеиохромоиы и хромонохроманы ................... 115
Литература............................................................148
Глава II
НАФТИРИДИНЫ
И. Вейс, Ч. Гаузер
Введение........................................................; . . 155
Синтезы ............................................................. 155
Синтезы типа А.................................................... 155
Синтезы типа Б . . . .................................. 169
Разнообразные синтезы............................................. 175
Реакции.............................................................. 175
Литература........................................................... 182
Глава III
КАРБОЛИНЫ
В. Кермак, Дж. Мак-Дейл
Введение............................................................. 185
Простые карболины.................................................... 187
Введение.......................................................... 187
а-, у- и 6-К.арболины............................................. 187
Р-Карболины....................................................... 191
Ангидрониевые основания, получаемые из карболинов................. 211
N-Алкилпроизводные Р-карболина.................................... 216
498 • содержАнйе
Бензокарболины ............................................................ 217
Введение............................................................... 217
6,7-Бензо-а-карболин .................................................. 219
Хининдолин ............................................................ 219
4,5-Бензо-р-карболин .................................................. 222
2,3-Бензо-у-карболин................................................... 227
Хиндолии[3,4-бензо-6-карболин]......................................... 233
Индоло-[2,3-а]-хинолизнны ................................................. 243
Индоло-[3,2-Ь]-хинолизин................................................... 247
Пентацнклические p-карболиновые алкалоиды.................................. 248
Эводиамин и рутекарпин................................................. 248
Йохимбин и родственные алкалоиды....................................... 251
Четвертичные пентацнклические p-карболиновые алкалоиды................. 259
Литература................................................................. 262
Глава IV
ФЕНАТРОЛИНЫ
В. Кермак, Д. Мак-Кейл
Введение.............................................................. 266
Общие методы синтеза.................................................. 267
о-, м- и п-Фенантролины............................................... 269
Устойчивость и реакционная способность фенантролинов.................. 272
Метилаты. Продукты восстановления и окисления......................... 274
N-Окиси фенантролина.................................................. 276
Окси- и галогенпроизводные фенантролина............................... 277
Нитро- и аминофенантролины............................................ 282
Фенантролииальдегиды ................................................. 283
Фенантролиикарбоновые кислоты и сложные эфиры......................... 283
Различные диазафенантрены............................................. 284
Комплексы 2,2'-дипиридила и о-фенантролнна с металлами................ 287
Литература..,......................................................... 294
Глава V
МОНОЦИКЛИЧЕСКИЕ ТРИАЗОЛЫдИ БЕНЗОТРИАЗОЛЫ
Дж. Бойер
Введение.................................................................. 296
виц-Триазолы.............................................................. 297
Методы получения..................................................... 297
Реакции замещения.................................................... 312
Реакции боковых цепей у виц-триазола................................. 318
Выделение азота из виц-триазольного кольца........................... 321
Кислотность виц-триазольного кольца.................................. 321
Основность виц-триазольного кольца................................... 322
Соли и комплексы виц-триазольного кольца............................. 322
Водородные связи в виц-триазолах..................................... 322
Спектры поглощения виц-триазолов..................................... 323
Различные свойства................................................... 323
Биохимические свойства виц-триазолов................................. 324
силл-Триазелы............................................................. 325
Методы получения..................................................... 325
Реакции замещения в симм-триазольном кольце.......................... 339
Реакции боковой цепи............................:.................... 346
Кислотность производных симм-триазола................................ 347
Основность производных симм-триазола................................. 348
Соли и комплексы си.и.и-три азолов................................ 349
Спектры поглощения сизея-триазолов................................... 349
Различные свойства и применение...................................... 350
Биохимические свойства............................................... 350
Литература............................................................... 351
СОДЕРЖАНИЕ 495
Глава VI
ОК.САДИАЗОЛЫ
Дж. Бойер
Введение................................................................... 357
1,2,5-Оксадиазолы ........................................................ 357
Получение фуразанов.................................................... 357
Получение фуроксанов................................................... 361
о-Дипитрозоароматические производные................................... 367
Реакции фуразанов, фуроксанов и о-диннтрозоароматических производных. 369
Физические свойства.................................................... 378
Проблема строения фуроксана............................................ 379
1,2,4-Оксадиазелы ......................................................... 388
Методы получения....................................................... 388
Реакции 1,2,4-оксадиазолов............................................. 395
Физические свойства.................................................... 397
1,2,3-Оксадиазолы ......................................................... 397
1,3,4-Оксадиазолы ......................................................... 399
Методы получения ...................................................... 399
Реакции с разрывом кольца.............................................. 405
Реакции без разрыва кольца . . . 407
Физические свойства и применение....................................... 409
Биологические свойства ................................................ 409
Литература ................................................................ 410
Глава VII
ТИАДИАЗОЛЫ
Б. Шерман
1,2,3-Тиадиазолы............................................................ 416
Номенклатура ........................................................... 416
Синтезы ................................................................ 416
Реакции и свойства...................................................... 418
1,2,3-Тиадиазолины...................................................... 420
1,2,3-Бензотиадиазолы и другие 1,2,3-тиадиазолы с конденсированными ядрами 420
1,2,4-Тиадиазолы............................................................ 427
Номенклатура ........................................................... 427
Синтезы ................................................................ 427
1,2,4-Тиадиазолидииы.................................................... 438
Реакции и свойства...................................................... 439
1,2,5-Тиадиазолы........................................................... 442
Введение и номенклатура................................................. 442
Синтез и реакции....................................................... 443
1,2,5-Тиадиазолидин .................................................... 443
2,1,3-Беизотиадиазолы и родственные конденсированные циклические системы 444
2,1,3-Бензотиадиазолин.................................................. 445
1,3,4-Тиадиазолы............................................................ 448
Введение и номенклатура................................................. 448
Синтезы ................................................................ 448
1,3,4-Тиадиазолины и тиадиазолидины..................................... 466
Имидазо-[2,1-6]-тиадиазолы ............................................. 469
Реакции и свойства.................................................... 470
Литература.................................................................. 475
Предметный указатель.................................................... . 479
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЙ
Том 7
Редактор Г. Б. Шкляева
Технический редактор Е. С. Потапенкова
Корректор Л Д. Кучерова
Сдано в производство 18/1II 1965 г.
Подписано к печати 14/V1H 1965 г.
Бумага 70Х 1 081/16=] 5,60 бум. л.
, 43,75 ггеч. л.
Уч.-изд. л. 40,76. Изд. № 3/2520
Цена 3 р. 05 к. Зак. 978
ИЗДАТЕЛЬСТВО «МИР»
Москва, 1-й Рижский пер., 2
Московская типография № 16
Главполиграфпрома Государственного
комитета Совета Министров СССР по печати
Москва, Трехпрудный пер., д. 9