/









































































































































































































































































































































































































































































































































































































































































































































































































































































































































































Author: Вейганд-Хильгетаг
Tags: химические реакции отдельные химические методы химия органическая химия эксперименты
Year: 1968




Text
ИЗДАТЕЛЬСТВО • ХИМИЯ
1968
WEYGAND-HILGETAG
ORGANISCH-CHEMISCHE
EXPERIMENTIERKUNST
3. NEUBEARBEITETE AUFLAGE
HERAUSGEGEBEN VON
PROF. DR. PHIL. HABIL. G. HI LG ETAG
ZUSAMMEN MIT
DR. A. MARTINI
INSTITUT FUR ORCANISCHE CHEMIE
DER OEUTSCHEN AKADEMIE DER WISSENSCHAFTEN ZU BERLIN
1964
JOHANN AMBROSIUS BARTH / VERLAG ' LEIPZIG
k sn
ВЕЙГАНД-ХИЛЬГЕТАГ
МЕТОДЫ ЭКСПЕРИМЕНТА
В ОРГАНИЧЕСКОЙ ХИМИИ
ПЕРЕВОД С ТРЕТЬЕГО НЕМЕЦКОГО ИЗДАНИЯ
Л. В. КОВАЛЕНКО И А. А. ЗАЛИКИНА
ПОД РЕДАКЦИЕЙ
ПРОФ. Н. Н. СУВОРОВА
196 8
ИЗДАТЕЛЬСТВО -ХИМИЯ • МОСКВА
УДК 542.9 : 547
В 26
Вейганд-Хильгетаг. МЕТОДЫ ЭКСПЕРИМЕНТА
В ОРГАНИЧЕСКОЙ ХИМИИ.
Книга посвящена методам синтеза органических
соединений различных классов. По охвату материала
она представляет собой сжатую, но достаточно
полную внциклопедию методов синтеза. Эта уни-
кальная в мировой литературе книга является
прекрасным справочником, с помощью которого можно
выбрать наиболее удобную реакцию для получения
нужного вещества.
Книга рассчитана на самый широкий круг хими-
ков-органиков — исследователей, заводских работни-
ков и учащихся.
Книга содержит §44 стр., б рис., 6135 библио-
графических ссылок.
2—5—3
ВЗ-20-68-4
СОДЕРЖАНИЕ
Предисловие редактора ........... 18
Введение .......................... 19
РЕАКЦИИ, ПРОТЕКАЮЩИЕ БЕЗ ИЗМЕНЕНИЯ УГЛЕРОДНОГО
СКЕЛЕТА
Образование связей углерод — водород
Образование С—Н-связей в результате реакций присоединения
I. Присоединение водорода по олефиновым С—С-связям и к аро-
матическим системам ..................................... 21
I. Химические восстановители ............................ 21
Восстановление неблагородными металлами.............. 22
(Электрохимическое восстановление ................... 28
Восстановление литийалюминийтидридом и аналогич-
ными соединениями ................................... 28
Прочие восстановители............................... .30
2. Каталитическое гидрирование .......................... 31
Катализаторы ........................................ 32
Аппаратура для гидрирования л методы гидрирования 40
Каталитическое гидрирование ..................... 44
7/. Присоединение водорода по С=С-связям................. 52
1. Химические методы . . . ............................ 52
2. Каталитическое гидрирование .......................... 54
III. Присоединение водорода по С--О-связям............... 55
1. Восстановление неблагородными металлами.............. 55
2. Реакция Меервсйла — Пояндорфа — Верлея............... 57
3. Восстановление алюмогидридом лития................... 61
4. Восстановление комплексными боргндридами........... 65
5. Каталитическое гидрирование ......................... 68
Образование С—Н-связи в результате реакции обмена
I. Обмен галогена на водород.........................
1. Обмен галогенов у простых С—С-связей..............
Прямой обмен .....................................
Обмен галогена с получением промежуточных соеди-
нений ............................................
2. Обмен галогена у олефиновых С=С-связей и в аромати-
ческих системах.......................................
3. Обмен галогена у других связей (в хлорапгидридах кис-
лот) .................................................
II. Обмен кислорода на водород........................
1. Обмен гидроксильной группы........................
2. Обмен карбонильного кислорода ....................
Прямой обмен .....................................
Обмен карбонильною кислорода с получением проме-
жуточных соединений ..............................
Обмой карбонильного кислорода с промежуточным по-
лучением меркапталей .............................
3. Обмен кислорода в карбоксильной или сложноэфирной
группе ...............................................
4. Восстановление амидов кислот и лактамов...........
III. Обмен азота на водород...........................
IV. Обмен серы на водород.............................
Образование связи углерод — галоген
Образование связи С— На! в результате реакции присоединения
I. Присоединение галогена и родина по кратным С—С-связям 90
1, Свойства и очистка галогенов и родана................. 90
2. Общие положения о присоединении галогенов по С=С-
связи.................................................... 92
СОДЕРЖАНИЕ
3. Присоединение галогенов к ненасыщенным углеводоро-
дам ....................................................
4. Присоединение галогенов к полиенам..................
5. Присоединение галогенов к стероидам.................
6. Присоединение галогенов к ненасыщенным спиртам и эфи-
рам ....................................................
7. Присоединение родана к ненасыщенным соединениям . .
8. Присоединение галогенов к ненасыщенным карбоновым
кислотам ...............................................
9. Присоединение галогенов по С=С-связп................
10. Присоединение галогенов к ароматическим соединениям
II. Присоединение галогеноводородов по С=С- и С^С-сеязя.ч
1. Получение и свойства галогеноводородов...............
2. Общие положения о присоединении галогеноводородов
ио С — С-связям ........................................
3. Присоединение галогеноводородов к ненасыщенным угле-
водородам ...................-..........................
4. Присоединение галогеноводородов к ненасыщенным спир-
там, эфирам, карбонильным соединениям и нитрилам . .
5. Присоединение галогеноводородов по С=С-свнзям . . .
III. Присоединение галогенов по С=С- и С=С-связям с одно-
временным образованием С—С-, С—О- и С—N-связей . . .
1. Присоединение галогенов по С—О и С=С-связям с одно-
временным образованием С—С-Овязи.....................,
Присоединение а-галогенэфиров ио С=С-связям ....
Реакция Меервейна (взаимодействие галогенидов диазо-
пия с соединениями, содержащими С--С- и С=С-связи
Присоединение галогепангидрпдов кислот по С=С-свяви
Присоединение алкплгалогепядов н аналогичных соеди-
нений по С=С-связи ................................
2. Присоединение галогенов по С=С-связи с одновременным
образованием С—О-связи .................................
Присоединение гипогалогспитпых кислот по С=С-связи
(получение галогенгидринов) .....................
Получение хлоргидринов при помощи хромилхлорида
Получение галогенгидринов по реакции с N-галогеи-
амидами ..........................................
Алкоксигалогенировапио ...........................
3. Присоединение галогенов по С—С- и С^С-связям с одно-
временным образованием С—N-связи........................
94
97
98
99
101
101
103
104
107
107
111
113
117
120
122
122
122
123
124
124
125
125
128
128
130
131
Образование связи С—Hal в результате реакции замещения
I. Замещение водорода галогеном или реданом........... 132
1. Замещение галогеном водорода в насыщенных алифатиче-
ских углеводородах ................................... 132
2. Замещение галогеном водорода в олефинах . ........ 135
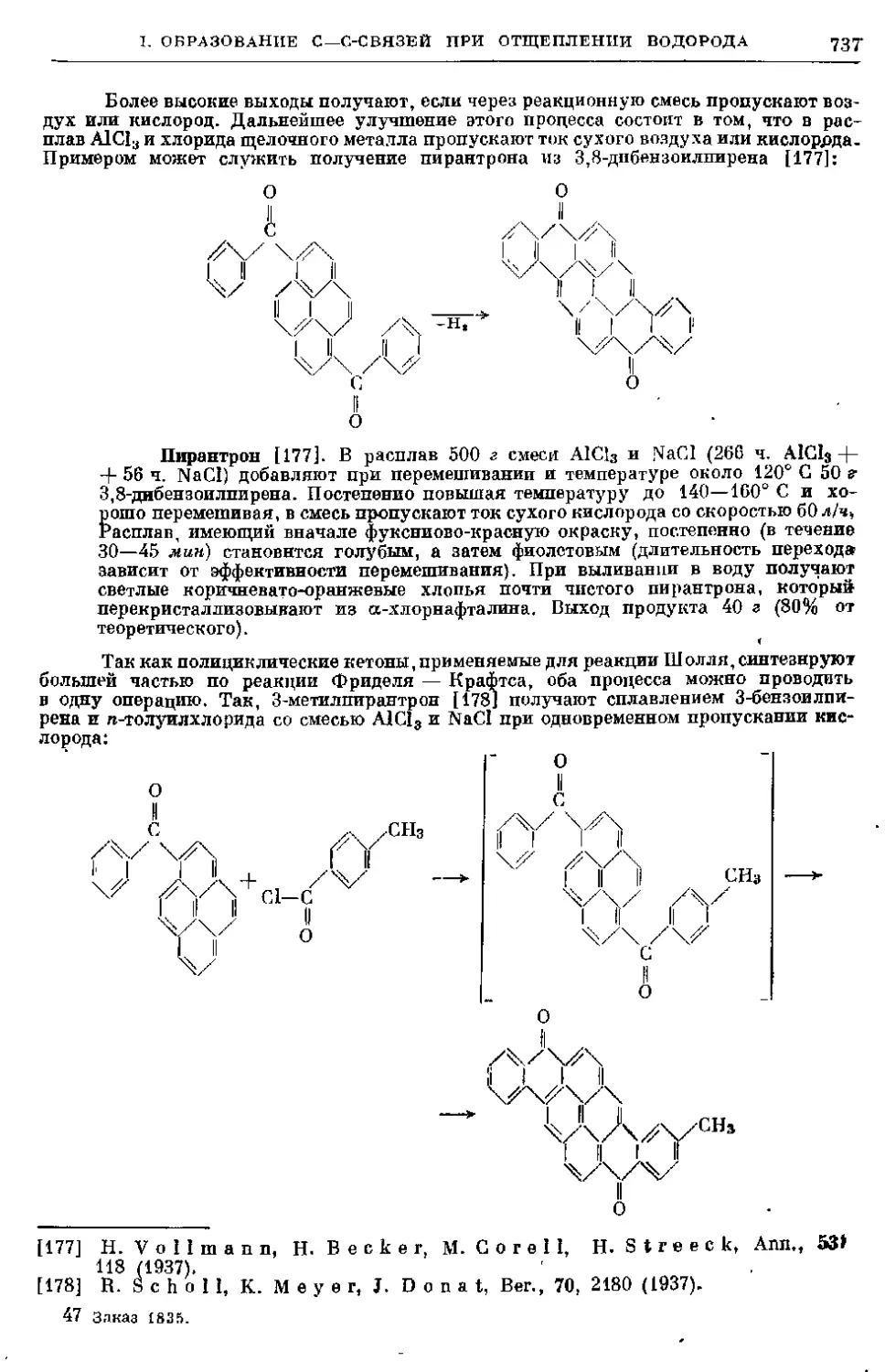
3. Общие положения о замещении галогеном водорода в аро-
матических соединениях .............................. 139
4. Замещение галогеном водорода в ядре ароматических угле-
водородов ...........‘................................ 142
5. Замещение галогеном водорода в боковой цепи ароматиче-
ских углеводородов ................................... 145
6. Замощение галогеном водорода в ацешленах.......... 148
7. Замещение галогеном водорода п фенолах, оксиарилалкап-
карбоновых кислотах, эфирах фенолов и ароматических
аминах ............................................... 150
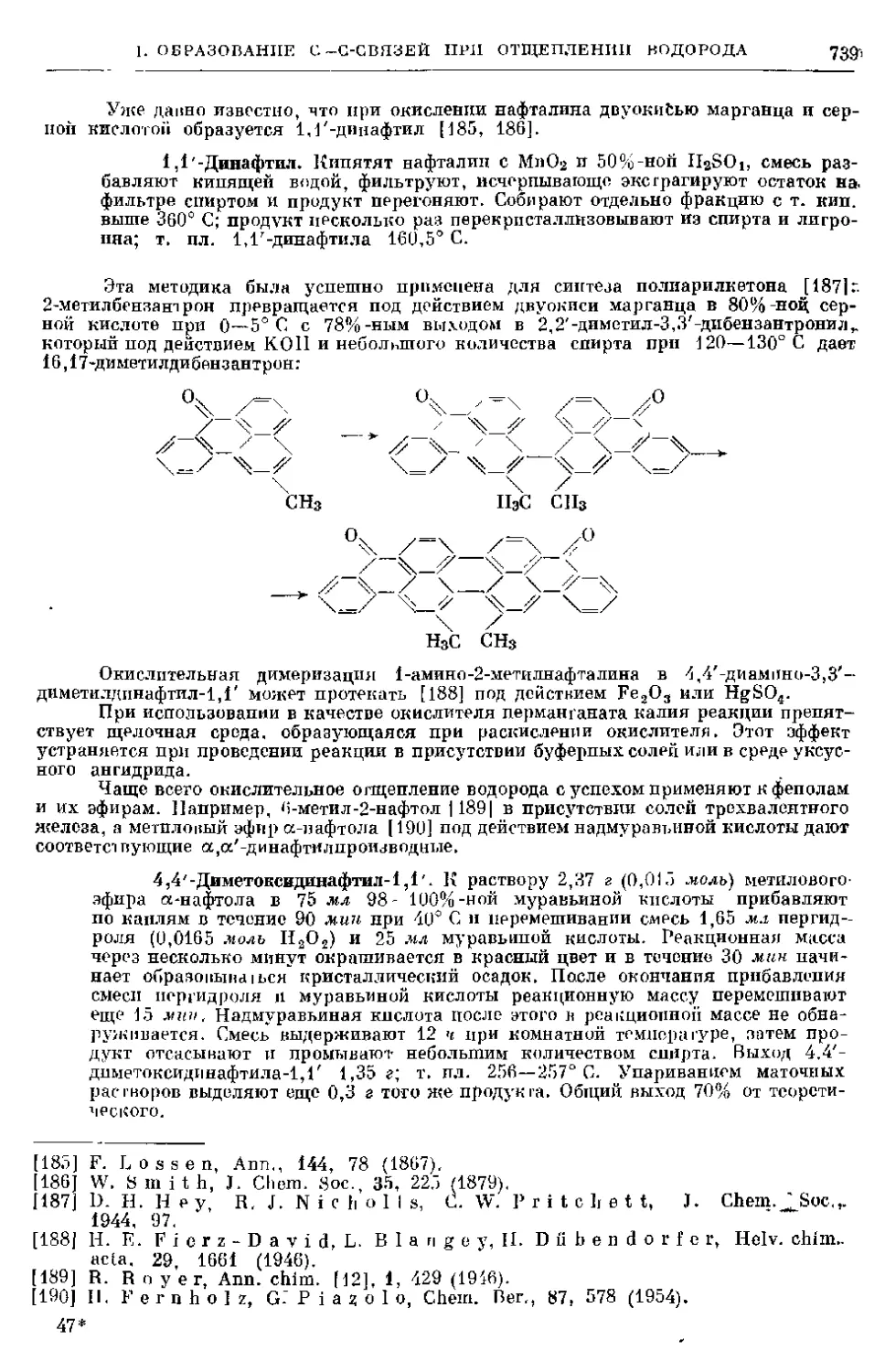
8. Замещение галогеном водорода в простых эфирах .... 159
9. Замещение галогеном водорода в алифатических моно-
карбодовых кислотах .................................. 161
10. Замещеипг галогеном водорода в алифатических дикар-
боновых кислотах п нитрилах.......................... 156
11. Замещение галогеном водорода в ароматических окси-
и амшюкарбоповых кислотах............................. 169
СОДЕРЖАНИЕ
12. Замещение галогеном водорода в ароматических интро-
соедипенпях ............................................ 173
13. Замещение галогеном водорода в альдегидах п кетонах 174
14. Замещение галогеном водорода в гетероциклических со-
единениях .............................................. 186
15. Непосредственное замещение водорода в ароматических
соединениях па SCN-rpynny............................... 190
II- Обмен галогена на другой галоген или на родан-группу . . 191
1. Обмен галогена па фтор.............................. 191
2. Обмен галогена на иод, бром или хлор................ 197
3. Обмен галогена на родан............................. 201
III. Замена гидроксильных, алкоксилъных и карбонильных
групп на галогены....................................... 202
1. Замела спиртового гидрокспла на галоген............. 202
Замена спиртового гидроксила на фтор............... 202
Замена спиртового гидроксида на хлор, бром и иод при
помощи галогеловодородов и галогенангидридов ки-
слот ............................................. 202
Замена спиртового гидроксила на галоген при помощи
трифенилфосфита и органических галоюпндои или
просто галогенов ................................. 21G
Замена спиртового гидроксила на галоген с промежу-
точным получением тозилатоп ...................... 218
2. Замена гидроксила на галоген в полуацеталях (синтез
а-галогенэфиров по Генрп)............................... 221
3. Замена ацилоксигруппы на галоген (получение галогеп-
замещенных углеводородов) .............................. 222
4. Замена С—О-связи связью С—Hal при расщеплении про-
стых эфиров н ацеталей.................................. 225
5. Замена гидроксила на галоген в фенолах и гетероцикли-
ческих соединениях ..................................... 229
6. Замена гидроксила в карбоксильной группе па галоген
(получение галогенангидридов карбоновых кислот) . . . 231
Замена ОН в карбоксильной группе па фтор............ 231
Замена ОН в карбоксильной группе на хлор, бром пли
иод при помощи неорганических галогенангидридов 231
Замена ОН в карбоксильной группе на хлор и бром при
помощи галогенангидридов органических кислот . . . 237
7. Замена карбонильного кислорода на хлор и бром .... 240
IV. Замена С—N-связи на связь С—Hal.................... 242
1. Замена С—N-связи на связь С—Hal в алифатических со-
единениях ............................................. 242
Замена С—N-связи па связь С —Па! в алифатических
аминах при помощи PCI5 и РВг5....................... 242
Замена С—N-связи на связь С— На! в алифатических
аминах при помощи бромциана......................... 243
Замена аминогруппы па галоген в алифатических со-
единениях при помощи нцтрозплгалогенилов .... 244
Получение галогенкетонов из диаялкетопов............ 245
2. Замена С—N-сцязи на связь С.—Hal в ароматических
и гетероциклических соединениях........................ 247
Замена аминогруппы па хлор и бром в ароматических
и гетероциклических соединениях (реакция Заидмей-
ера) ............................................... 247
Замена аминогруппы на под в ароматических соедине-
ниях ............................................... 250
Замена аминогруппы на бром в ароматических и гетеро-
циклических соединениях с промежуточным получе-
нием иербромидов диазонмя........................... 252
Замена аминогруппы па хлор и бром и ароматических
соединениях (реакция Швехтена)....................... 253
Замена аминогруппы на фтор в ароматических и гетеро-
циклических соединениях (реакция Шимапа) .... 253
Замена аминогруппы па группу SCN в ароматических
соединениях ........................................ 257
V. Замена С—S-связи на связь С—Hal..................... 257
VI. Замена С’^С-сеязи на связь С—Hal................... 259
Образование связей углерод — кислород
Образование С—О-связи в результате реакции присоединения
I. Присоединение кислорода по С=С-связи............ . 260
1. Молекулярный кислород................................ 260
2. Озон ............................................... 261
3. Получение алкиленоксидов из производных этилена 262
II. Присоединение воды по С—С-связи .......... 266
III. Присоединение воды по С^С-связи.................... 269
IV. Присоединение гидроксильных групп по С=С-сеязи . . . 272
1. Гидроксилирование при помощи органических надкислот 272
2. Гидроксилирование при помощи перманганата калия 274
3. Гидроксилирование при помощи четырехокиси осмия 275
4. Гидроксилирование при помощи комплекса иода с бензо-
атом серебра (реактив Прево) 276
5. 7^иг-Гидроксилирование по методу Вудворда............ 277
6. Гидроксилирование при помощи перекиси водорода и не-
органических катализаторов ........................... 277
V. Присоединение карбоновых кислот и спиртов по С—С-
и С=С-связям ........... ............................. 278
Образование С—О-связи в обменных реакциях с получением
гидроксил- и карбопилсодержащих соединений
I. Замещение водорода кислородом ..................... 284
1. Замещение водорода гидроксильной группой........... 285
2. Образование перекисей путем аутоокисления.......... 288
Моры предосторожности при обращении с перекисями 288
Образование гидроперекисей ....................... 288
Образование диалкилперекисей ..................... 293
3. Замещение водорода па карбонильный кислород .... 294
Получение альдегидов и кетонов.................... 294
Получение хинонов ................................ 298
4. Замещение водорода карбоксильным кислородом .... 299
5. Дегидрирование спиртов до карбонильных соединении 303
Получение альдегидов ............................. 303
Получение котонов ................................ 306
Окисление многоатомных спиртов.................... 309
6. Окисление спиртов и альдегидов до карбоновых кислот 310
7. Реакции Канниццаро и Тищенко....................... 312
II. Обмен галогена на кислород......................... 314
1. Обмен галогена на гидроксильную группу............. 314
2. Обмен галогена да карбонильный кислород............ 316
3. Обмен галогена на карбоксильный кислород........... 318
III. Обмен азота на кислород........................... 319
1. Обмен аминогруппы на гидроксильную группу......... 319
2. Обмен азота на карбонильный кислород............... 322
3. Обмен азота на карбоксильный кислород.............. 326
IV. Обмен сулъфогруппы на гидроксильную группу .... 328
Образование новых С—0-связей в результате обменных реакций
I. Получение простых эфиров .............. 329
1. Получение простых эфиров отщеплением воды от двух
молекул спирта ............................. 329
2. Получение простых эфиров алкилированием галогенсо-
держащими соединениями ..................... 333
3. Получение простых эфиров алкилированием диметил-
сульфатом .................................. 340
4. Получение простых эфиров алкилированием эфирами
и-толуолсульфокпслоты ............................... 341
5. Получение простых эфиров алкилированием диазомета-
ном ................................................. 342
11- Получение сложных зфиров......................... 343
1. Получение эфиров карбоновых Кислот из карбоновых ки-
слот и спиртов....................................... 343
Этерификация без катализатора.................. . 343
Этерификация в присутствии катализатора......... 345
2. Получение сложных эфиров алкилированием солеи карбо-
новых кислот......................................... 348
Алкилирование ацкилгалогенидами.................... 348
Алкилирование ди.метилсулг,фатом .................. 348
Алкилирование диазометаном......................... 349
3. Получение сложных эфиров ацилированием спиртов
п фенолов ..............'........................... 349
Ацилирование хлорапгидридамл кислот................ 349
Ацилирование ангидридами кислот.................... 351
4. Получение сложных эфиров переэтерификацией .... 352
III. Получение ацеталей ............................... 353
1. Получение ацеталей из карбонильных соединений и спир-
тов ................................................. 353
2. Получение ацеталей из карбонильных соединений и орто-
эфиров .............................................. 355
3. Получение ацеталей при помощи диметилсульфита . . . 356
4. Получение изопропилидеповых и бензилиденовых произ-
водных углеводов ....................................... 356
IV. Ангидриды карбоновых, кислот........................ 358
1. Получение ангидридов карбоновых кислот непосредствен-
ным отщеплением воды ................................... 358
2. Получение ангидридов карбоновых кислот по реакции
двойного обмена........................................’. 359
Разрыв связей углерод — кислород
I. Разрыв простой зфирной связи ............. 361
II. Омыление сложных эфиров карбоновых кислот . ... . 36.51
III. Омыление ацеталей, меркапталей, кеталей и ортоэфи-
ров ....................................... 369-
Образование связей углерод — азот
Образование С—N-связей в результате реакций присоединения
I. Присоединение азотсодержащих соединений по кратным
углерод-углеродным связям ...................... 372’
1. Присоединение аммиака и продуктов его замещения . . 372
2. Присоединение других азотсодержащих соединений . . . 373-
Присоединенпе диазоалканов и азидов ......... 373
Присоединение нитрилов ...................... 373
Присоединение окислов я оксихлоридов азота... 374
II. Присоединение азотсодержащих соединений по C=N-
и С~1Ч-связям .................... 375
1. Присоединение к производным цианистоводородной кис-
лоты ......................................... 375
2. Присоединение к цианатам п тиоцианатам........ 377
III. Присоединение азотсодержащих соединений по С—О-
связи ........................................... 378
IV. Присоединение азотсодержащих соединений по С=5'-
связи............................................ 379
Образование С—N-связи в результате реакции обмена
I. Замещение водорода азотом........................ 380
1. Нитрование ...................................... 380
Общие положения. Нитрующие йичпы.................
Нитрование алифатических соединении..............
Нитрование ароматических и гетероциклических со-
единений ........................................
2. Нитрозирование....................................
Общие положения .................................
Нитрозирование алифатических соединений..........
Нитрозирование ароматических соетнпсннй..........
3. Дзосочетапие .....................................
Общие положения .................................
Сочетание с соединениями, содержащими активные ме-
тиленовые группы ................................
Азосочетание с ароматическими компонентами.......
Окислительное азосочетанпе ......................
4. Прочие реакции ...................................
Введение аминогруппы.............................
Введение аммониевой группы.......................
Прямое введение дпазояиевой группы ..............
Реакция Эрлиха — Закса ..........................
II, Обмен галогена на азот............................
1. Взаимодействие галогениропзводнык с производными
аммиака ...............................................
Алкилирование ...................................
Арилирование ....................................
Ацилирование .................... ? ............
Прочие реакции ..................................
2. Взаимодействие галогснпропзводных с другими азотсо-
держащими соединениями ................................
Взаимодействие с нитритами.......................
Взаимодействие с азидами.........................
Взаимодействие с цианидами.......................
Взаимодействие с цианатами.......................
III. Обмен кислорода на азот..........................
1. Реакции азотсодержащих соединений с производными ки-
слот ................•.................................
Реакции азотсодержащих соединений со свободными ки-
слотами .........................................
Реакции азотсодержащих соединений с ангидридами
карбоновых кислот ...............................
Реакции азотсодержащих соединении с эфирами карбо-
ловых кислот ....................................
Реакции азотсодержащих соединении с амидами кислот
2. Реакции карбонильных соединений с азотсодержащими
соединениями ..........................................
Получение простых производных карбонильных соеди-
нений ...........................................
Получение аминов с функциональными группами в «-по-
ложении .........................................
Получение аминов восстановительным алкилированием
3. Реакции спиртов и фенолов с азотсодержащими соедине-
ниями ...............-.................................
Обмен спиртовых гидроксильных групп па аминогруппы
Реакции фенолов с аммиаком, аминами и гидразинами
4. Реакции простых эфиров и оксонпевых содей с азотсодер-
жащими соединениями ..................................
IV. Обмен других элементов на азот....................
1. Обмен азота па азот...............................
Перса.минироваиие (реакции обмена аминогрупп) . . .
Переампдированпе и аналогичные реакции ..........
Обмен азота в солях дпазонля и диазосоедипспиях . . .
2. Обмен углерода па азот............................
3. Обмен серы на азот . .............................
Обмен сульфогруппы па азот.......................
380
383
441
441
443
44?
445
450
454
408
470
Обмеп других серусодержашнх функциональных групп
на азот ......................................... 512
4. Обмен металлов па азот............................ 513
Изменение азотной функции в органических соединениях
I. Образование Л’—Н-связи ............................. 514
1. Образование N—Н-слнзп в результате реакции присоеди-
нения .............................................. 514
Получение гидразопроизводных из азосоедипений . . . 514
Получение гидразинов из диазоииевых солей........ 515
Получение аминов из нитрилов ................... 516
Получение вторичных аминов из оснований Шиффа . . 518
Получение замещенных гидразинов из гидразонов
и азинов.......................................... 519
2. Образование N —Н-связи н результате реакции обмена 520
Получение аминов восстановлением нитросоединений 520
Восстановление нитросоедцнений до гидроксиламинов 525
Получение аминов восстановлением оксимов и гидр-
оксиламцнов....................................... 525
Восстановление иитрозосоедицеиий до аминов .... 527
Восстановление нцтрозаминов до несимметричных ди-
замещенных гидразинов ............................. 528
Восстановление азос-осдинепия до амилов............ 529
Восстановление азинов, гидразонов и гидразинов . . . 530
Восстановление азидов............................... 530
Получение аминов расщеплением N-нлтрозамнпов , . . 531
II. Восстановление N-окисей............................ 531
III. Образование .V—О-связи ........................... 533
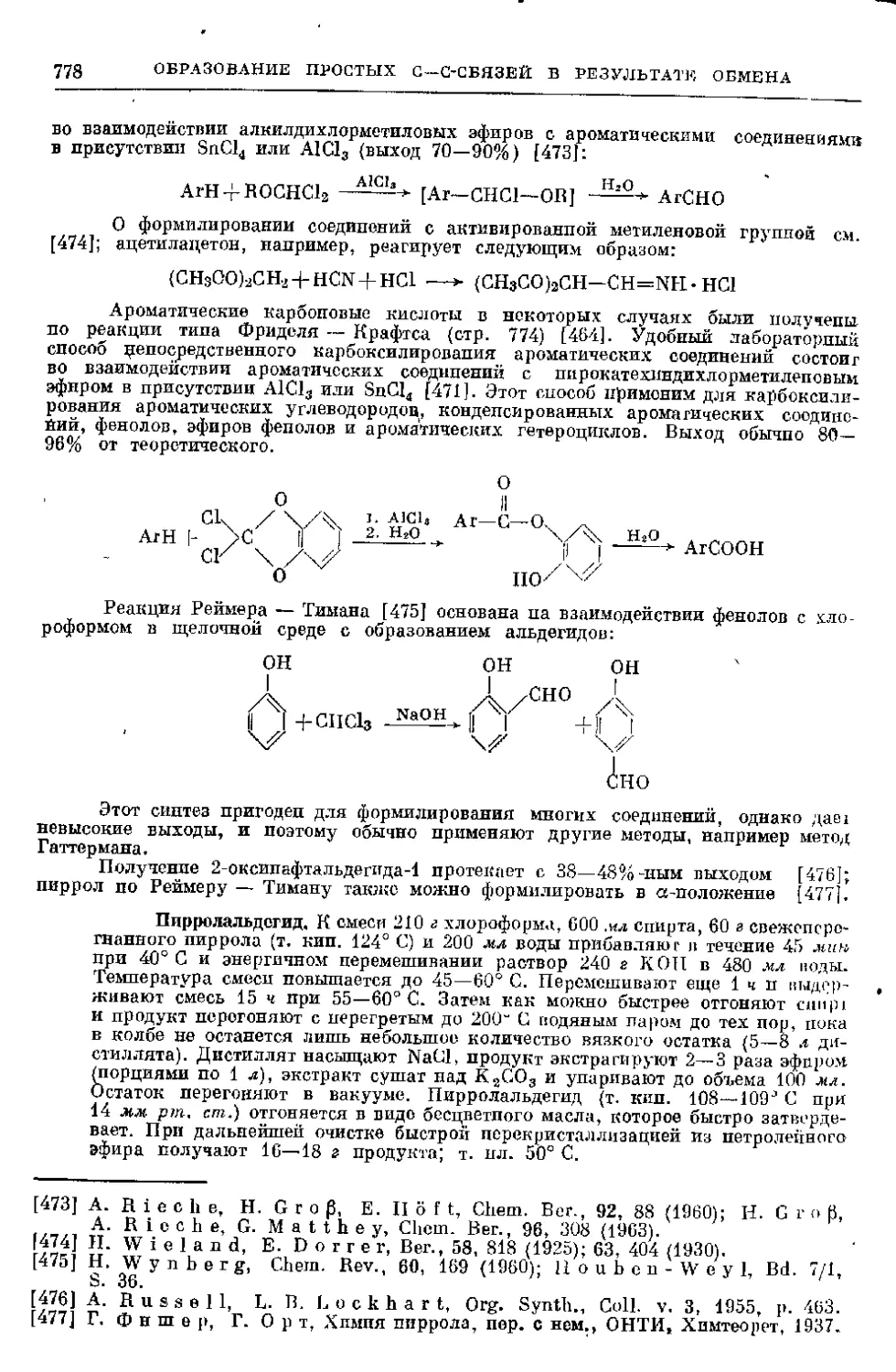
1. Образование N—О-связи в результате реакции присоеди-
нения .............................................. 533
Получение N-окисей ............................... 533
Получение аяокенлропзводных из азосоедпненпй . . , 534
Окисление нитрозо- л окепмной групп до нитрогруппы 534
2. Образование N — О-спязя в результате реакции обмена
(окисление аминов, гидрокспламинов и оксимов до нитро-
зосоедипенпй) ....................................... 535
IV. Образование N—N-связи в результате реакции обмена 536
1. Получение нптрозаминов ............................. 536
2. Получение нитраминов................................ 537
3. Диазотирование ароматических аминов................. 539
4. Получение азидов ................................... 541
5. Получение гидразинов ............................... 543
6. Получение дпазоамипосоединений..................... 544
7. Получение азосоедипений ............................ 545
8. Получение алифатических диазососдинений............. 546
V. Образование связи N—Hal ............................. 549
Образование связей углерод — сера
Образование С—s-связи в результате реакций присоединения
I. Присоединение соединений серы по кратным углерод-угле-
родным связям .................................... 550
1. Присоединение сероводорода и его производных .... 550
2. Присоединение других соединений серы.............. 554
II. Присоединение соединений серы по кратным углерод-азот-
ным связям . ................................. 556
III. Присоединение соединений серы по С—О-связи .... 558
Образование С—S-связи в результате реакций замещения
Д Обмен водорода на S-функцию......................... 559
1. Введение сульфогрупш.1 ........................... 559
Сульфирование алифатических соединений.......... 559
СОДЕРЖАНИЕ
Сульфирование ароматических соединений серной ки-
слотой п олеумом .................................. 561
Сульфирование серной кислотой и олеумом в присут-
ствии катализаторов ............................... 569
Сульфирование серным ангидридом и его аддуктами . . 570
Сульфирование ароматических соединений галоген-
сульфокислотами и другими сульфирутопщми аген-
тами ............................................. 572
2. Введение сульфохпоридиой группы.............. 573
3. Прямое введение сульфоновой и сульфоксидной групп 577
4. Прямое введение сульфиновой группы.. 578
5. Введение тионовой, тпоэфпрной и меркаптогрупп .... 579
II. Обмен галогена на 8-функцию........................ 580
1. Взаимодействие алкил- и арилгалогеыидов с солями и про-
изводными сероводорода ..... ......................... 580
Получение меркаптанов и дисульфидов................ 580
Получение простых тиоэфпров и солей сульфонил . . 583
Получение тиокетопов, меркапталей и меркаптолов . . 586
2. Взаимодействие ацнлгалогенидов с сероводородом и его
производными.......................................... 588
3. Взаимодействие алкилгалогенидов с сульфитами и сульфи-
повыми кислотами .................................... 589
4. Получение эфиров тиоциановой и изотиоциановой кислот 591
III. Обмен кислорода на S-функцию...................... 592
1. Взаимодействие спиртов с серусодержащими соединениями 592
Взаимодействие с сероводородом и его производными 592
Взаимодействие с бисульфитом натрия................ 592
2. Взаимодействие окисей алкенов с соединениями серы 593
3. Взаимодействие карбонильных соединений с соединениями
серы ................................................. 594
4. Взаимодействие производных карбоновых кислот с со-
единениями серы ...................................... 597
5. Взаимодействие алкил- и диалкилсульфатов с соедине-
ниями серы............................................ 598
IV. Замещение азота серой............................. 599
1. Замещение аминогруппы S-фуякцией............... -->99
2. Замещение иминогруппы S-функцией................ 602
3. Замещение третичного азота и аммониевой группы S-
функцией ............................................. 603
4. Замещение нитрогруппы S-функцией .................. 603
V. Замещение других функций........................... 604
1. Взаимодействие с реактивами Гриньяра................ 604
2. Взаимодействие с другими металлоорганическими со-
единениями ........................................... боб
Получение органических соединений серы путем превращения
имеющихся S-фуикций
I. Восстановление S-функций........................
II. Окисление S-фуикций ................
1. Окисление меркаптаной ..........................
2. Окисление дисульфидов ..........................
3. Окисление простых тиоэфиров.....................
4. Окислительное галогенирование органических соедине-
ний серы .........................................
Получение алкилсульфохлоридов .................
Получение арилсульфохлоридов...................
III. Расщепление органических соединений серы ......
1. Расщепление С—S-связи...........................
2. Расщепление S—Н-связи ..........................
3. Расщепление S—S-связи ..........................
4. Расщепление связи S—Ci .........................
Расщепление сульфохлоридов............. . . . .
Расщепление сульфинилхлоридов и сульфонилхлоридов
606
609
609
611
611
613
613
614
615
615
618
619
620
620
622
СОДЕРЖАНИЕ
13
5. Расщепление S—О-связи ............................. 623
6. Расщепление S—N-связи ............................. 624
IV. Получение производных тиоугольных кислот.......... 625
1. Тиоугольные кислоты .............................. 626
2. Эфиры хлорангидридов тиоугольных кислот........... 626
3. Эфиры тиоугольных кислот.......................... 627
Полуэфиры тиоугольных кислот...................... 627
Полные эфиры тиоугольных кислот................... 627
4. Производные тиокарбаминовой кислоты............... 628
Соли ............................................. 628
Тиокарбаминоилхлориды ............................. 629
Эфиры тиокарбаминовой кислоты (тиоуретаны) .... 629
5. Бис-(алкилксантогены) и тиурамдисульфиды........... 630
6. Изотиоцианаты (горчичные масла).................... 631
7. Соли изотиомочевины ............................... 631
Образование металл-углеродных связей
(металлоорганические соединения)
Способы получения металлоорганических соединений 634
I. Органические производные элементов первой группы . . . 636
1. Литийорганические соединения........................ 637
2. Натрийоргаяические соединения ...................... 646
II. Органические производные элементов второй группы . . 642
1. Магнийорганические соединения ..................... 643
2. Цинкорганическио соединения ....................... 647
3. Кадмийорганические соединения ..................... 649
4 Ртутьорганические соединения....................... 649
III. Органические производные элементов третьей группы 654
1. Борорганические соединения......................... 654
2. Алюминийорганические соединения.................... 656
IV. Органические производные элементов четвертой группы 659
1. Кремнийорганическне соединения .................... 659
2. Оловоорганические соединения....................... 662
3. Органические производные свинца ................... 664
V. Органические производные элементов пятой группы . . 665
1. Мьппьякорганическпе соединения .................... 665
2. Органические производные сурьмы.................... 668
3. Висмуторгапические соединения ..................... 670
Образование кратных углерод-углеродных связей
Этиленовые связи
Г. Изолированные этиленовые связи.................... 671
1. Отщепление воды от спиртов................... 671
2. Отщепление галогеповодородов.................. 676
3. Отщепление галогена ...................... 678
4. Расщеплепие азотсодержащих соединений............. 680
II. Сопряженные двойные связи......................... 682
1. Диены ............................................. 682
2. Полиены ........................................... 683
3. Ароматические и гетероциклические непредельные соеди-
нения ................................................ 683
III. Кумулированные двойные связи .................... 686
1. Аллены............................................ 686
2. Кетены ........................................... 687
Ацетиленовые связи
1. Дегидрогалогенирование дигалогеиидов или галоген-
олефинов.............................................. 689
2. Дегалогенирование................................. 692
3. Расщепление азотсодержащих соединений............ 693
14
СОДЕРЖАНИЕ
ОБРАЗОВАНИЕ НОВЫХ УГЛЕРОД-УГЛЕРОДНЫХ СВЯЗЕЙ
Образование новых С—С-связей
в результате реакции присоединения
I. Присоединение по С -С-связям ............ 696
1, Присоединение углеводородов по этиленовым связям . . 696
2, Полимеризация производных этилена................... 697
3. Диеновый синтез .................................... 700
4. Присоединение соединений с активированной С—Н-связью
(реакция Михаэля) .................................... 705
5. Присоединение формальдегида (реакция Принса) . . . 707
6. Присоединение органических гэлогеппроизводных . . . 708
7. Присоединение диазометана и синильной кислоты . . . 710
II. Присоединение по С=О-связи ......................... 712
1. Альдольная конденсация .............................. 712
2. Бензоиновая конденсация ............................. 715
3. Пинаконы.......................................... 715
4. Реакция Кольбе -- Шмитта ................ 717
5. Циангидриновые синтезы ......................... 717
6. Присоединение дназомстапа к карбонильной группе . . 719
7. Присоединение металлоорганических соединений к кар-
бонильной группе...................................... 720
Взаимодействие реактива Гриньяра с кетонами .... 721
Взаимодействие реактива Гриньяра со сложными эфи-
рами ............................................. 723
Взаимодействие реактива Гриньяра с альдегидами . . 725
Взаимодействие магнийорганических соединений
с окисью этилена и С.О2........................... 726
8. Синтезы с литийорганическими соединениями........... 727
III. Присоединение по кратным С—N-связям ....... 728
4. Присоединение магнийорганических соединений .... 728
2. Присоединение нитросоединепий ...................... 731
3. Циклизация нитрилов ............................... 732
4. Присоединение синильной кпелоты................... 733’
5. Присоединение кетонов к бензияиденанилину.......... 734
Образование простых С—С-связей в результате реакций обмена
I. Образование С—С-связей в результате отщепления водорода 735
1. Пи рохимическпе реакции ............................ 735
2. Каталитическое отщепление водорода.................. 736
3. Окислительное отщепление водорода ................... 738
4. Фотохимическая дегидродимеризация................... 741
II. Образование С—С-связей в результате отщепления гало-
гена в виде галогенида................................. И-
1. Отщепление галогена щелочными металлами............. 742
Реакции Вюрца и Фиттига............................ 742
Реакпдп ацетиленидов щелочных металлов с галогени-
дами ............................................ 745
2. Отщепление галогена другими металлами.............. 746
Отщепление галогена магнием........................ 746
Отщепление галогена серебром и медью............... 749
Отщепление галогена цинком......................... 750
3. Отщепление галогена алкоголятами, амидами и гидридами
щелочпых металлов и другими основаниями................ 753
Эфиры а-глмцидной кислоты.......................... 753
С-Алкилирование и С-ацилирование соединений с по-
движным атомом водорода............................ 754
4. Синтезы квтопоп при помощи металлоорганических со-
единений .............................................. 760
5. Синтезы нитрилов .................................. 763
III. Образование С — С-связей в результате отщепления гало-
гена в виде галогеноводорода .............. 765
СОДЕРЖАНИЕ
1. Синтез Фриделя — Крафтса ........................... 765
Ацилирование ....................................... 769
Алкилирование ...................................... 772
Особенности реакции Фриделя — Крафтса............. 774
2. Синтезы альдегидов и карбоновых кислот.............. 776
IV. Образование С—С-связей в результате отщепления
металла в виде его галогенида........................... 779
V, Отщепление воды с образованием новой С—С-связи . . . 779
1. Алкилирование ...................................... 779
2. Хлорметилирование ........................... 782
3. Оксимотилирование ........................... 783
4. А «метилирование (реакция Манпиха).................. 785
5. Сульфомстилированпе .......................... 786,
VI. Образование С—С-связи а результате отщепления спирта,
карбоновой кислоты и др................................. 786
1. Сложиоэфмрныс конденсации............................ 786
2. Отщепление карбоновых кислот......................... 792
VII. Образование С—С-связи в результате отщепления
азота, аминов и аналогичных соединений ................. 792
1. Отщепление элементарного азота..................... 792
2. Отщепление аминов ................................. 795.
Образование двойных С—С-связей в результате
реакций обмена
I. Отщепление водорода с образованием новых этиленовых
связей............................................ 799’
Ц. Отщепление галогена с образованием новых этиленовых свя-
зей ............................................... 800
1. Оыцепление галогена действием металлов......... 800
2. Отщепление галогена действием щелочей......... 800
III. Образование новых С=С-связей путем дегидратации 801
1. Синтез а,р-и р,у-ненасыщенных карбоновых кислот . . 801
Синтез Перкина ...................................... 801
Реакция Кневенагеля ................................. 803
Синтезы эфиров коричной кислоты ио Клайзену .... 805
Конденсация Штоббе.............................. 805
2. Синтез а, р-ненасыщенных альдегидов и кетонов .... 806
3. Прочие конденсации .................................. 808
4. Получение ненасыщенных нптросоединенпй............. 809
5. Получение ароматических систем..................... 810
IV. Отщепление азота с одновременным образованием новой
С=С-связи ........................................... 811
V. Отщепление серы с образованием новой С-=С-связи - . . 812-
VI. Отщепление фосфора с образованием новой С=С-свя.зи
(реакция Виттига)........................................ 812
РАЗРЫВ УГЛЕРОД-УГЛЕРОДНЫХ СВЯЗЕЙ
Термическое разложение
I. Отщепление двуокиси углерода......................
1. Декарбоксилирование по типу I.....................
2. Декарбоксилирование по типу II ...................
Кетоппое расщештепие р-кетокислот_...............
Декарбоксилирование малоновых кислот ............
Декарбоксилирование Р, у-ненасьпценных кислот . . .
Декарбоксилирование ароматических окси- и амино-
карбоновых кислот ...............................
II. Отщепление окиси углерода........................
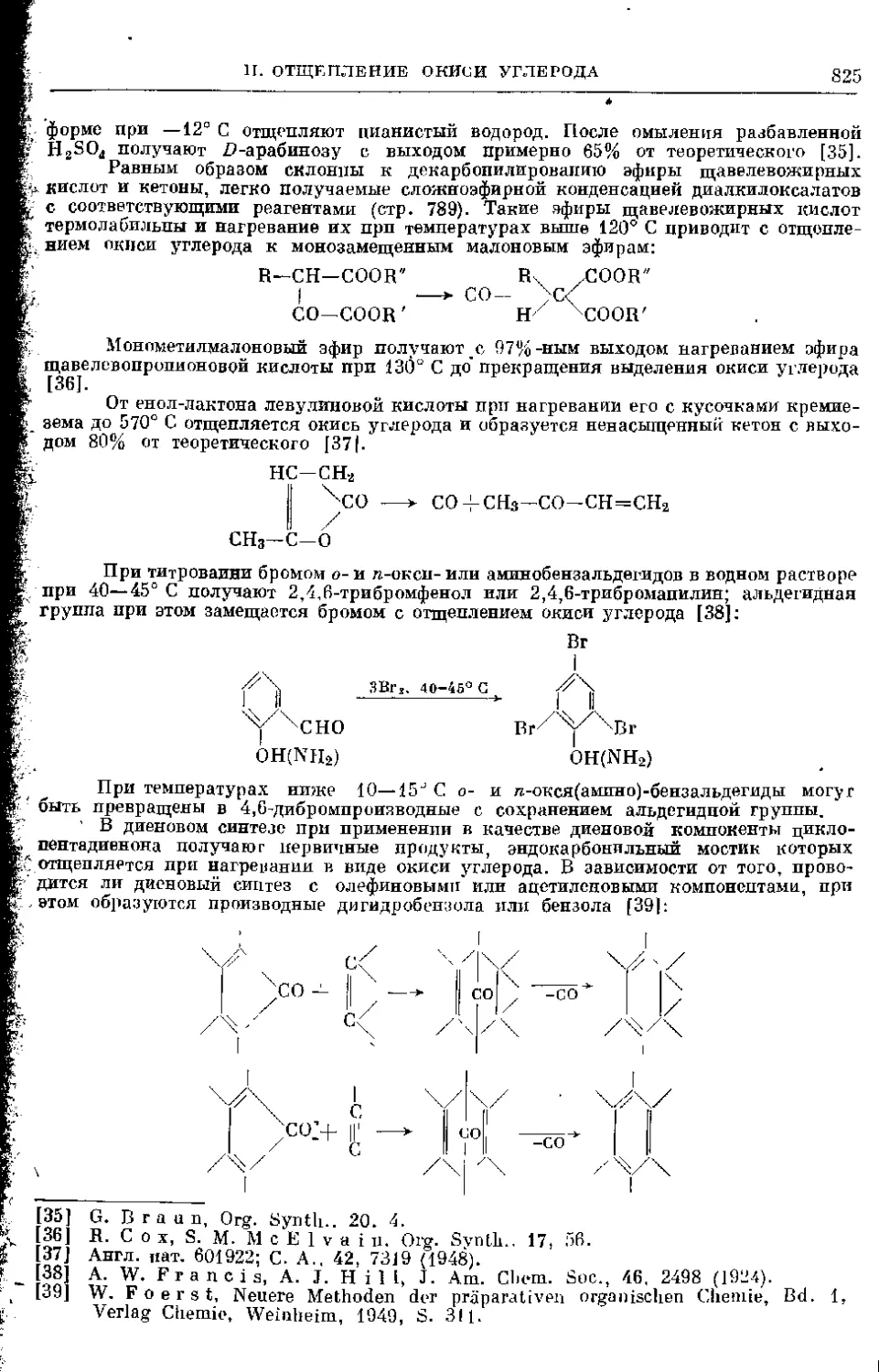
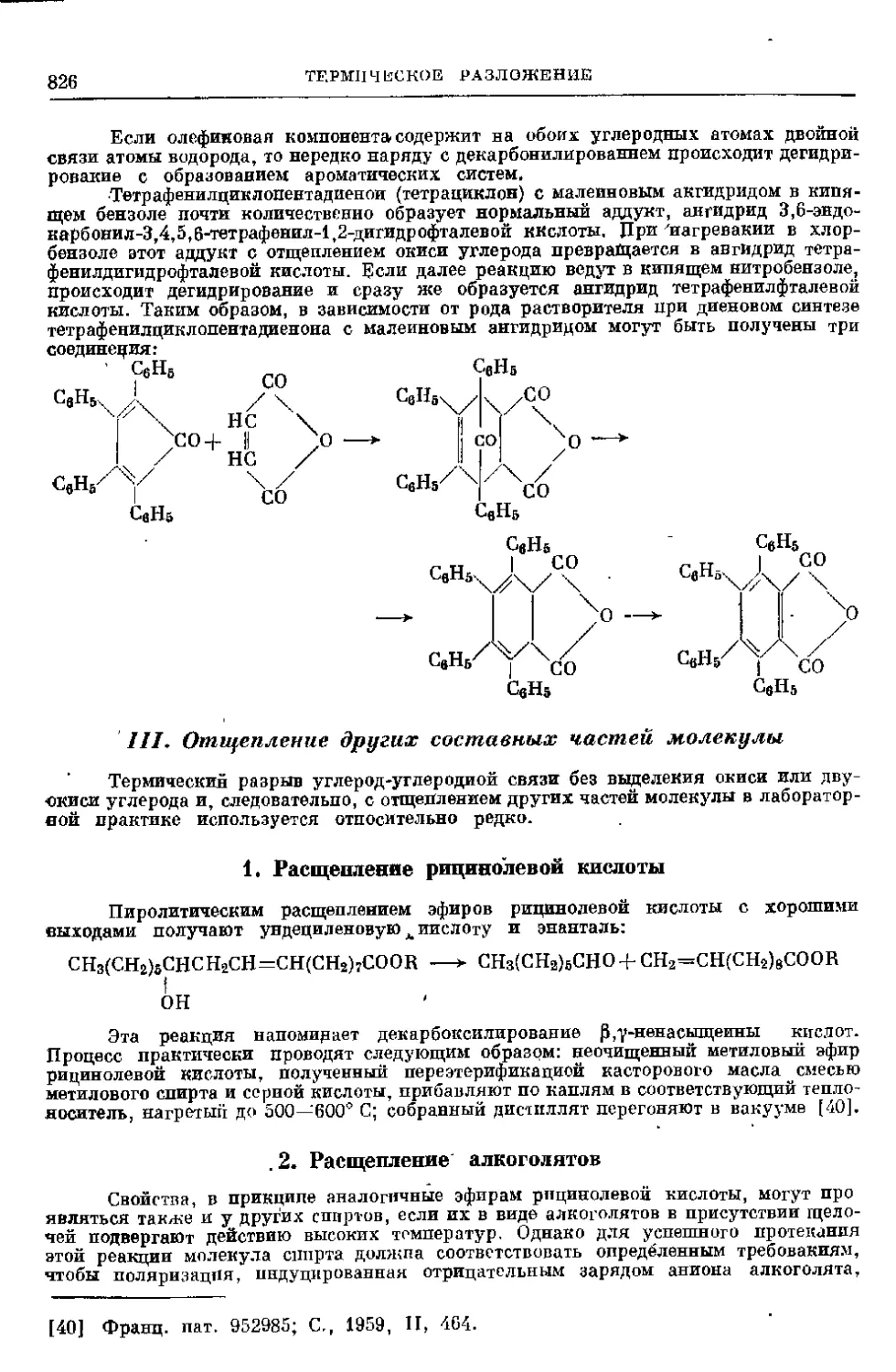
III. Отщепление других составных частей молекулы ....
1. Расщепление рицинолевой кислоты..................
2. Расщепление алкоголятов .........................
СОДЕРЖАНИЕ
3. Расщепление ацетона ................................ 828
4. Расщепление производных циклогексена................ 829
5. Превращение лумистерина в тахистерин................ 829
Окислительное расщепление С—С-связей
I. Окислительное расщепление углерод-углеродных связей в на-
сыщенных алифатических или алициклических углеводоро-
дах ................................................... 830
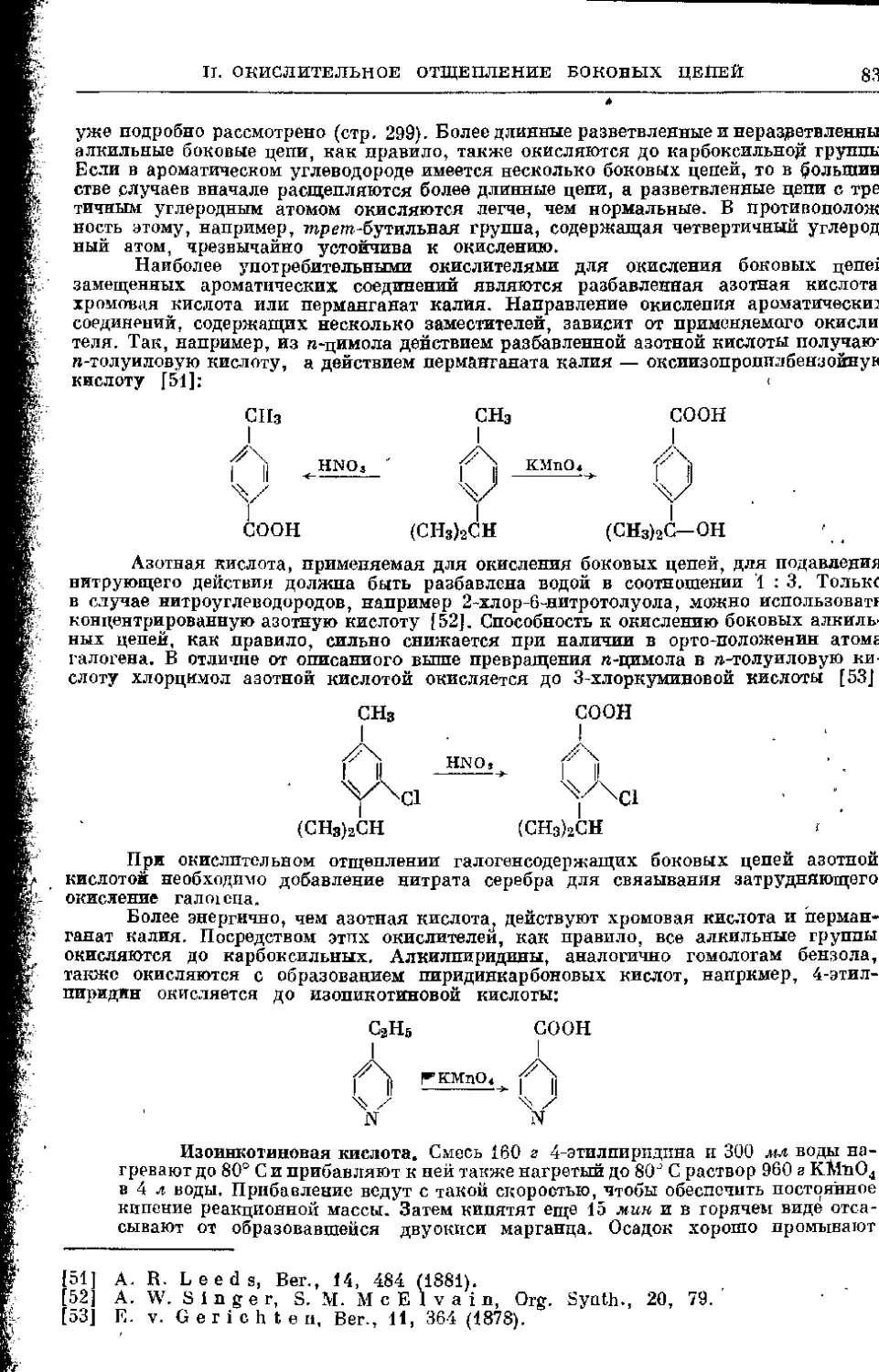
II. Окислительное отщепление боковых цепей, связанных
с ароматическим, или гетероциклическим ядром........... 830
1. Окислительное отщепление длинных боковых цепей аро-
матических и гетероциклических соединений............. 830
2. Расщепление гомологов фенола до фенолкарбоновых ки-
слот путем щелочного плавления........................ 832
III. Окислительное расщепление олефинов ........ 832
1. Окислительное расщепление ненасыщенных карбониль-
ных соединений посредством щелочного плавления . . . 833
2. Окислительное расщепление олефинов до альдегидов
обычными окислителями ................................. 833
3. Окислительное расщепление олефинов до кетопов и кар-
боновых кислот обычными окислителями................... 834
4. Окислительное расщепление олефинов озоном......... 835
5. Укорочение боковой цени по Виланду............... 836
IV. Окислительное расщепление С—С-связей в соединениях
с гетерофункцией в месте расщепления ......... 836
1. Окислительное расщепление вторичных спиртов и кето-
нов ................................................... 837
2. Онислительное расщепление метилкетонов............. 837
8. Окисление тетраацетатом свинца.................... 840
V. Окислительное расщепление кольцевых систем аромати-
ческих и гетероциклических соединений............... 841
Гидролитическое расщепление
ПЕРЕГРУППИРОВКИ УГЛЕРОДСОДЕРЖАЩИХ СОЕДИНЕНИИ
(ЗА ИСКЛЮЧЕНИЕМ
ПРОСТРАНСТВЕННЫХ ПЕРЕГРУППИРОВОК)
Перегруппировки с сохранением углеродного скелета
I. Перемещение кратных связей.......................
1. Перемещение двойной связи.......................
2. Аллильная перегруппировка ......................
3. Перемещение тройной связи.......................
II. Перегруппировки с изменением кислородной функции
1. Превращение а-окисей в карбонильные соединения . . .
2. Реакция ВильгеродТа ............................
3. Кето-енольная таутомерия .......................
4. Изомеризация кетоспиртов .......................
5. Перегруппировка Канниццаро .....................
6. Прочие перегруппировки .........................
III. Перегруппировки азотсодержащих соединений . . . .
1. Перегруппировка Амадори.........................
2. Перегруппировка Чепмена ........................
3. Перегруппировка окисей аминов ..................
4. Псевдооснования ................................
IV. Перегруппировки галогенсодержащих соединений ....
1. Перемещение галогена............................
2. Перегруппировки при реакциях галогенсодержащих со-
единений ...........................................
847 •
847
849
850
851
851
852
853
854
855
856
856
856
857
857
858
858
858
860
СОДЕРЖАНИЕ
17
2 Заказ J835,
Перегруппировки с построенном углеродного скелета
I. Перемещение от кислорода к углероду............
1. Перегруппировка Фриса..........................
2. Перегруппировка Клайзена ......................
3. Перегруппировка Бекера — Вснкатарамана ....
4. Перегруппировка бензиловых эфиров..............
II. Перемещение от азота к углероду...............
1. Бензидиновая перегруппировка ..................
2. Индольный синтез по Э. Фишеру . ...............
3. Перегруппировка Стивенса ......................
4. Перегруппировка Соммлс ........................
III. Перемещение от серы к углероду...............
IV. Перегруппировка Тиффено ......................
Перегруппировки с расщеплением углеродного скелета
862
862
863
864
865
865
865
866
867
868
868
869
I.-Перемещение от углеродами кислороду.................. 870
1. Перегруппировка Криге............................. 870
2. Перегруппировка гипохлоритов ..................... 871
II. Перемещение от углерода к азоту................... 871
1. Гофмановское расщепление амидов кислот............. 871
2. Расщепление по Курциусу............................ 872
3. Перегруппировка Бекмана............................ 874
4. Реакция Шмидта .................................... 875
Перегруппировки с перестройкой углеродного скелета
I. Перегруппировка Вагнера — Меервейпа и близкие к ней
реакции ...............................................' 8743
1. Перегруппировка Вагнера — Меервейна ................ 876
2. Пинаколиновая и ретропинаколиновая перегруппировки 878
3. Диенон-фенольная перегруппировка ................... 879
4. Изомеризация углеводородов ........................• 880
II. Прочие перегруппировки .............. 881
1. Реакция Якобсена ................................... 881
2. Перегруппировка типа бензиловой кислоты............. 881
3. Перегруппировка при окислении диалкилацетилеиов . . 882
Ш. Перегруппировки при разложении диазосоединений . . 882
1. Перегруппировка Вольфа — Шрётера .................. 882
2. Перегруппировки при взаимодействии карбонильных со-
единений с диазометаном .............................. 884 •
3. Перегруппировки при взаимодействии аминов с HNO2 885
Предметный указатель ......... 887
ПРЕДИСЛОВИЕ РЕДАКТОРА
В последние годы на нашем книжном' рынке появился ряд
прекрасных руководств по общим и теоретическим вопросам орга-
нической химии. В то же время синтетические аспекты этой науки
в широком плане освещены совершенно недостаточно в мировой
литературе, если не считать таких изданий, как выпускаемые отдель-
ными томами «Органические реакции» и «Успехи органической
химии», посвященные отдельным реакциям. Особое место занимает
руководство К. Вейганда «Методы эксперпмента в органической
химии». Оригинальная классификация органических реакций в син-
тетическом аспекте, тщательный отбор материала и прекрасно
подобранные в качестве иллюстраций прописи заслуженно принесли
этой книге мировую известность.
Этот справочник выдержал уже три издания. Мы предлагаем
читателю перевод третьего, последнего издания, которое было
выпущено в 1964 г. Пятнадцать лет, прошедшие со времени выхода
в свет второго издания, внесли столько существенных изменений
в материал, что потребовалась коренная переработка книги. Для
подготовки нового издания был привлечен большой коллектив авто-
ров-специалистов в разных областях органической химии, в основ-
ном сотрудников Института органической химии при Берлинской
Академии наук. Работу возглавил профессор Г. Хильгетаг, под
редакцией которого и вышел в свет этот справочник.
Советскому читателю знаком перевод второго издания этого
справочника, опубликованный в 1950 г. под редакцией профессора
В. Н. Белова. Книга быстро разошлась, И в 1952 г. вышло второе
ее издание, которое в настоящее время уже стало библиографиче-
ской редкостью.
При подготовке русского третьего издания кппгп мы сочли
целесообразным опустить I п III части киигп, в которых изложены
элементы лабораторной техники и физико-химического исследова-
ния в органической химии, так как эти вопросы более подробно
рассмотрены в ряде недавно выпущенных в свет монографий,
п включили только II часть, посвященную вопросам синтеза.
Мы воздержались от вмешательства в представленную авто-
рами библиографию и от составления специальных дополнений к
этому изданию, которые сильно увеличили бы объем кппгп и едва ли
при современном быстром развитии органической хпмпи полностью
отразили ее последние достижения. По сравнению с ранними изда-
ниями в книге возрос объем сведений о русских и советских работах.
Однако все же они отражены недостаточно, поэтому мы вынуждены
были дать некоторые примечания.
Следует выразить уверенность, что настоящее издание книги
Вейганда-Хпльгетага принесет пользу советским хпмикам-орга-
никам.
Н. Суворов
ВВЕДЕНИЕ
Целью препаративной работы в лаборатории органической
химии является получение определенных веществ заданной степени
чистоты из соответствующих исходных продуктов. Для создания
методически наиболее наглядной классификации большого числа
реакций, пригодных для препаративных целей, удобно исходить
из общего углеродного скелета, являющегося основой всех органи-
ческих соединений, и рассмотреть, какие изменения могут с ним
происходить в процессе реакций. Исходя из этого можно различить
четыре группы реакций:
1. Углеродный скелет целевого продукта не отличается от
скелета исходного вещества. Например: бензойная кислота из
толуола; ацетальдегид из этилового спирта.
2. Углеродный скелет нужного вещества строится из двух
или большего числа молекул за счет образования С—С-связей.
Например: ацетофенон из бензола и уксусного ангидрида (2 моле-
кулы); кетон Михлера из диметиланилпна и фосгена (3 молекулы);
каучук из изопрена (большое число молекул).
3. Углеродный скелет образуется в результате частичного
разрушения структуры .более богатой углеродом. Например: энан-
тол из касторового масла; фталевая кислота из нафталина.
4. Углеродный скелет образуется в результате перестройки
углеродного скелета исходного соединения. Например: пинаколин
из пинакона.
В процессе препаративных работ в одном синтезе могут
встречаться разные комбинации перечисленных основных превра-
щений, а именно:
1. Превращение молекул без изменения углеродного скелета.
2. Увеличение углеродного скелета.
3. Уменьшение углеродного скелета.
4. Перестройка углеродного скелета.
Особое положение занимают реакции, приводящие к образо-
ванию циклических структур. Однако поскольку протекающие при
этом реакции ничем не отличаются от других, при которых, как
и при каждой реакции вообще, разрываются и образуются новые
связи, то и они относятся к одному или одновременно к нескольким
из четырех основных типов реакций:
1. При образовании фталевого ангидрида из фталевой кис-
лоты, точно так^же как (при образовании пиррола из диальдегпда
янтарной кислоты и аммиака, в углеродном скелете ничего не
меняется.
2. Образование мезитилена из ацетона подобно образованию
окиси мезитила из ацетона.
3. Образование циклопентанона из адипината кальция пол-
ностью соответствует образованию ацетона из ацетата кальция.
4. Образование изоборнилхлорида из гидрохлорида камфена
протекает так же, как,-образование пинаколина из пинакона.
При установлении того, какие типичные химические про-
цессы охватываются такой систематизацией, были получены вполне
удовлетворительные результаты. В первую группу (превращение
без изменения углеродного скелета) попадают реакции гидрирова-
ния, хлорирования, нитрования, окисления, восстановления и др.,
которые обычно методически принято объединять. Ко второй группе
(увеличение углеродного скелета) относятся реакции конденсации
(например, образование конденсированных систем по Кемпфу),
и полимеризации (с образованием новых С—С-связей). Третья
2*
ВВЕДЕНИЕ
(уменьшение углеродного скелета) и четвертая группы (перегруппировки в угле-
родном скелете) введены впервые, но и они кажутся вполне логичными.
Правда, в результате такой систематики, в разделе «Альдегиды» ио приведены
методы получения бензальдегида: из толуола через хлористый бензилиден; из бензола
и смеси окиси углерода с хлористым водородом по Гаттерману — Коху или из
бромбензола (через магпийбромфенил) и эфира муравьиной кислоты по Гриньяру;
из стильбена при действии озона или из дифенилгликоля и тетраацетата свинца.
Однако все эти методы описаны в систематических учебниках и справочниках
по органической химии.
Преимуществом используемой в данной книге систематики является то, что
основные типы реакций рассматриваются вместе.
На вопрос, что является общим для всех реакций органической химии, может
быть дан лишь один ответ: все они сводятся к разрыву имеющихся и к образованию
новых связей.
Поэтому в пределах каждой из четырех главных групп в основу последователь-
ного расположения реакций следует положить не получение определенных групп,
а образование или расщепление связей. Большинство связей может образовываться
как в результате реакций присоединения, так и реакций обмена. По этому принципу
и построены основные разделы книги.
Построение очень большого первого осповною раздела этой части книги, «Реак-
ции, протекающие без изменения углеродного скелета», очень упростилось, после того
как оказалось, что в нем, должны рассматриваться только связи углерода с водородом,
галогенами, кислородом, азотом, серой, фосфором и металлами. Здесь же рассматри-
вается образование кратных С—С-связей в неизменном углеродном скелете. В подраз-
делах далее рассматриваются два типа реакций: присоединение ц обмен.
ОБРАЗОВАНИЕ СВЯЗЕЙ
УГЛЕРОД - ВОДОРОД*
ОБРАЗОВАНИЕ С—Н-СВЯЗЕИ
В РЕЗУЛЬТАТЕ РЕАКЦИИ ПРИСОЕДИНЕНИЯ
РЕАКЦИИ,
ПРОТЕКАЮЩИЕ
БЕЗ ИЗМЕНЕНИЯ
УГЛЕРОДНОГО
СКЕЛЕТА
Промышленное гидрирование ненасыщен-
ных С—С-связей проводится в настоящее время
преимущественно каталитическими методами.
Ввиду большой гибкости процессов каталитиче-
ского гидрирования они прочно вошли в препара-
тивную и аналитическую лабораторную практику.
Наряду с ними, а именно в химических исследо-
ваниях, прочно утвердились и другие методы вос-
становления, главным образом потому, что они
чрезвычайно удачно дополняют каталитические
методы и иногда даже превосходят их по изби-
рательности.
Например, при гидрировании ацетиленовой
связи в присутствии соответствующих катализа-
торов получаются почти исключительно ^ис-Ьти-
лсповые соединения, в то время как при восста- •
повлении комплексными гидридами металлов
или натрием в жидком аммиаке образуются со-
ответствующие трляс-соединения И]. Аналогич-
ные нвдения наблюдаются в случае присоедине-
ния водорода к ненасыщенным конденсирован-
ным циклическим системам [2]. Восстановление
С—О-групп в лаборатории проводят почти исклю-
чительно химическими восстановителями.
I. Присоединение водорода
по олефиновым С—С-связям
и к ароматическим системам
1. Химические восстановители
Важнейшими методами присоединения
водорода по этилеповой связи, к ароматическим
и к гетероциклическим системам являются ме-
тоды, использующие в качестве восстановителей
неблагородные металлы в соответствующих рас-
творителях. Все другие восстановители, как-то:
хлорид олова (II), модистый водород и даже
комплексные гидриды металлов — имеют гораздо
мопыпее значение для восстановления ненасы-
щенных С—С-связей.
* Материал обработали X. Бишофф, И. Г. Дит-
рих, Е. Хёфт, Д. Муравски. Подготови-
тельная работа по разделу проведена
Р. Филиппсоном.
[1] a) L. Grombie, Quart. Rev. (Chem.
Soc., London), 6, 128 (1952); b) K. N. Ca-
mpbell, L. T. Eby, J. Am. Chem.
Soc., 63, 216, 2683 (1941).
[2] W. S. Johnson, J. Ackermann,
J. г. Е a s t h а m, Н. A. de W a I t jr.,
J. Am. Chem. Soc., 78, 6302 (1956);
R. H. Jaeger, Tetrahedron, 2, 326
(1958).
22
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Раньше восстанавливающее действие систем с неблагородными метал-
лами приписывали так называемому водороду в момент выделения. В на-
стоящее время, главным образом из работ Вилыптеттсра [1], Хюккеля
[4] и Берча [5], стало известно, что необходимой предпосылкой для таких вос-
становлспий является легкость перехода электрона от металла к электрофильной
системе, за которым следует вторичная реакция — взаимодействие протона
с образовавшимся карбанионом. В зависимости от применяемых металлов,
растворителей и значений pH возникают восстановительные потенциалы
различной силы, что позволяет восстанавливать соединения с различным по силе
сродством к электрону.
Для объяснения восстановления изолированных ароматических .ядер
в системе щелочной металл — жидкий аммиак — спирт [5, 6] Хюккель в своей
более поздней работе [7] все же возвращается к старой гипотезе о «водороде
в момент выделения». Поскольку в такой системе нет металлической поверхности,
катализирующей ассоциацию атомов водорода (2Н —>Н2), то возможно, что
создающейся концентрации свободных атомов водорода достаточно для проте-
кания первой стадии реакции.
Восстановление неблагородными металлами
Восстановление натрием и спиртами. В то время как-
изолированные двойные связи и одноядерные ароматические соединения обычно не
изменяются под действием натрия в спирте, активированные двойные связи, как.
например, в стироле или в коричной кислоте, насыщаются достаточно легко. Поэтому
в соединениях типа Дг—СН—СН—(CIIj),,—СН—СН—R можно избирательно гидри-
ровать лишь сопряженные с ядром двойные связи |8]. Некоторые сопряженные диены
присоединяют два атома водорода и превращаются в олефины; конденсированные аро-
матические системы [9] тоже частично гидрируются. Так, например, из антрацена
и его замещенных образуются 9,10-дигпдропроизводные [9, 10], а из фенантрена
и аценафтена — тетрагидропроизводпые [9]. Из нафталина в этиловом спирте полу-
чается дпгидронафталин, в амиловом спирте — тетралин. Довольно легко гидрируются
азотсодержащие гетероциклы [11], еще легче восстанавливаются нафтолы и на'фтал-
ампны цо тетрагидропроизводных. Интересно, что при восстановлении р-пафтиламина
гидрируется замещенное ядро "[12], а при восстановлении а-нафтиламипа — преимуще-
ственно незамещенное. Из простых эфиров нафтолов можно с хорошими выходами полу-
чать циклические кетопы, проводя восстановление до стадии дигидропроизводного
и омыляя образующийся эфир енола [13].
Восстановление натрием и спиртами проводят аналогично восстановлению
сложных эфиров по Буво — Блану (стр. 83). Как правило, применяют этиловый
спирт, однако иногда если требуется более высокая температура предпочитают рабо-
тать с высшими спиртами. Обычно в результате реакции выделяется значительное
количество паров, с которыми нормальные холодильники но справляются. Поэтому
при проведении этой реакции выгодно использовать обратные холодильники с широким
просветом, которые пе захлебываются от большого количества конденсата. Многократ-
но предлагалось применять для этих целей металлические холодильники. Из прсдосто-
[3J R. Willstattcr, F.Seitz, Е. В и m m, Вег., 61, 871 (1928),
[4] W. Huckel, II. В г е t s с h n е i а е г, Ann,, 540, 157 (1939).
[5] A. J. Birch, Quart. ’Rev, (Chem. Soc., London), 4, 69 (1950).
[6] a) С. B. W с о s t e г, пат. CHIA 2182242; C., 1940, I, 3987; b) J. P. W i-
h an l, F, A. H a a k, Rec. trav. chim 67, 85 (1948).
[7] W. Huckel, B, G г a f, Г). Miinkner, Ann., 614, 47 (1958).
]8] B. L о e v, С. B. Da wsob, J. Am. Chem. Soc., 78, 1180 (1956).
[9] E. Bamberger, W. Lodter, Ber., 20, 3077 (1887),
И0] G. H. Daub, W. C. Doyle, J. Am. Chem. Soc., 74, 4449 (1952).
[11JD. Papa, E. Schwenk, E. Klingsborg, J. Am. Chem, Soc., 73,
253 (1951); O. Ncunhoeffer, H. Ulrich, Chem. Ber., 88, 1123 (1955).
[12] E. В. П. Waser, H. Mo e 11 e ring, Org. Synth., 9, 84 (1929).
£13] J. W. С о г n 1 о г t h, R. Robinson, J. Chem. Soc., 1949, 1855;
M. D. S offer, R. A. Stewart et al., J. Am. Chem. Soc., 72, 3704
(1950).
I- ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = С-СВ ЯЗЯМ
23
рожности не следует помещать реакционный сосуд в паровую баню или воронку Бабо;
наиболее безопасны песочные бани.
Пиперидин [14]. В круглодонную колбу с обратным холодильником
помещают 20 г пиридина в 150 г абсолютного спирта и нагревают на водяной
бане; затем прибавляют кусочками, не слишком медленно, 75 г натрия. Как
только течение реакции замедляется или начнет выделяться’алкоголят, приба-
вляют еще этилового спирта и заканчивают реакцию как можно скорее. Когда
весь натрий прореагирует, реакционную массу охлаждают, прибавляют равный
объем воды и перегоняют с водяным паром. Дистиллят нейтрализуют соляной
кислотой и упаривают досуха. В остатке получают с почти количественным выхо-
дом гидрохлорид пиперидина, который может быть перекристаллизован из спирта.
Восстановление амальгамой натрия. Восстановительное
действие амальгамы натрия аналогично действию натрия в спирте. Изолированные
двойные связи устойчивы и в этом случае, в то время как сопряженные связи реакцион-
носпособны. По данным Куна и Хоффера [15], водород всегда присоединяется к полие-
нам по концам сопряженной системы.
При использовании амальгамы натрия условия реакции более мягкие, чем при
гидрировании натрием в спирте. Поэтому амальгама натрия уступает по каталитиче-
ской активности при гидрировании циклических систем, но она зато более пригодна
для избирательного восстановления ненасыщенных карбоновых кислот, содержащих
фурановое [16] или тиофеновое [17] кольцо. Антрацен и его производные [18], акри-
дин [19] и резорцин превращаются в дигидропроизвоцные; терефталевая кислота
в зависимости от условий проведения реакции восстанавливается до дн- и тетрагидроте-
рефталевых кислот или до n-толупловой кислоты [3]. Для избирательного восстано-
вления С=С-связи в а,р-ненасыщспных нитрилах [20], кетонах [21], лактамах [22]
и карбоновых кислотах [23] также рекомендуют применять амальгаму натрия.
Способ получения амальгамы натрия см. [24]. Реакции с ней проводят в воде,
в абсолютном спирте [22], в щелочах [23] и в ледяной уксусной кислоте [21]. При этом
образующуюся гидроокись натрия рекомендуется нейтрализовать пропусканием дву-
окиси углерода. Для гидрирования значительных количеств ненасыщенных соедине-
ний этот способ вряд ли можно считать удобным. Если учесть необходимость регене-
рации ртути и неудобства работы с большими количествами ее, то следует отдать пред-
почтение каталитическим методам восстановления *.
Гидрокоричная кислота [25]. При нагревании растворяют 15 г коричной
кислоты в 75 мл 5%-ного раствора едкого натра, охлаждают и при интенсивном
[14
115
116
[17
[18
[19
[20
[21
122]
123]
124]
[25]
Удобным методом восстановления некоторых органических соединений амаль-
гамой патрия является так называемое «непрямое электровосстацовление»
1С. А. В о й т к е в и ч, авт. свид. 70300 (1947); Л. Н. Лаврищева,
П. М. П р ж и я л г о в с к а я, С. А. Войткевич, В. Н. Белов,
ЖОХ, 27, 1264 (1957)]. - Лрим. ред.
A. Ladenburg, Ann., 247, 51 (1888).
R. Kuhn, M. Hoffer. Вег., 65, 170 (1932).
A. S. Carter, J. Am. Chem. Soc., 50, 2303 (1928).
L. J. Owen, F. F. Nord, J. Org. Chem., 15, 988 (1950).
E. L. May, E. Mosettig, J. Am. Chem. Soc., 70, 688, 1077 (1948).
R. R. Burtner, J. W. Cusic, J. Am. Chem. Soc., 65, 1583 (194^).
S. Wi deg u i s t, Ark. Kemj, 26A, ^2 16, 10 (1948).
J. E. J. Dippy, R. H. Lewis, Bee. trav. chim., 56, 1000 (1937);
A. A. Challis, G. B. Clemo, J. Chem. Soc., 1947, 1692.
G. R. Clemo, L. K. Mishra, J. Chem. Soc., 1953, 192.
W. E. Bachmann, В. E. Holmen, J. Am. Chem. Soc., 73, 3660 (1951).
W. B. Brasen, C. R. Hauser, Org. Synth., 34, 56 (1954).
F. Henle, Anleitung fiir das organisch-chemische Praktiknm, Leipzig,
1927; L. Gattermann, H. Wieland, Die Praxis des organischen
Chemikers, Walter de Cruyter u. Co., Berlin, 1947.
24
ОБРАЗОВАНИЕ С—Н-СВЯЗЕИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
перемешивании постепенно вносят небольшими кусочками свежепригото-
вленную 2,5%-ную амальгаму натрия. Если после прибавления 300—350 г амаль-
гамы проба, подкисленная соляной кислотой и нейтрализованная содой, не
обесцвечивает раствор перманганата калия, водный слой отделяют декантацией
от ртути и фильтруют через складчатый фильтр. Фильтр промывают небольшим
количеством воды, фильтрат нейтрализуют соляной кислотой. При необходимости
фильтруют еще раз и прибавляют 15 мл концентрированной соляной кислоты.
Продукт реакции экстрагируют эфиром, вытяжку сушат над сульфатом натрия
и отгоняют эфир. Остается масло, кристаллизующееся при трении. Выход
кислоты составляет около 12 г (79% от теоретического). После перекристаллиза-
ции из достаточного количества теплой поды получают чищую гидрокоричную
кислоту; т. пл. 48,7° С.
Восстановление амальгамой алюминия. Ряд -нестойких
веществ можно с успехом восстанавливать амальгамой алюминия, так как при этом
легко удается поддерживать реакционную среду точно нейтральной. В фульвенах
и аналогично построенных производных индена и флуорена часто удастся восстанавли-
вать только одну из сопряженных двойных связей [26]. Этот метод особенно пригоден
для восстановления ненасыщенных сложных эфиров, склонных омыляться в щелочной
среде [27, 28].
Обычно восстановление амальгамой алюминия, полученной по способу Вислице-
нуса [27], ведут в течение 1—2 суток, прибавляя при необходимости небольшое коли-
чество воды.
Бепзилинден. К эфирному раствору 5 з беяаилидепинденз прибавляют
15 з свежеприготовленной амальгамы алюминия и добавляют время от времени
небольшие количества воды. Реакцию ведут до обесцвечивания реакционной
массы, на что требуется от 6 до 24 ч. Осадок отфильтровывают, тщательно промы-
вают на фильтре эфиром, сушат, упаривают эфир и перегоняют остаток. Полу-
ченный бепзилипден перегоняется при 13-ил pm. cm. в пределах 183—185Q С.
Аналогично восстанавливают бензальфлуорен, фурфуральфлуорен и дру-
гие производные [26].
Восстановление щелочными и щелочноземельным-и
металлами в жидком аммиак е. Физико-химические измерения говорят
о наличии свободных сольватированных электронов в растворах щелочных металлов
в жидком аммиаке, представляющих собой очень сильный нуклеофильный реагент.
Однако при гидрировании ненасыщенных соединений такие растворы почти не превос-
ходят по восстановительной способности описанные ранее системы.
За редким исключением [34] изолированные двойные связи.и одноядерные аро-
матические соединения не восстанавливаются. Зато сопряженные двойные связи [4, 29],
арилзамещенпые этилены [30] и полициклические ароматические соединения [4, 31]
с успехом частично гидрируются. В качестве побочпых реакций часто наблюдаются
димеризация иди глубокая полимеризация исходного пенасыщепного соединения.
Обычно применяют избыток натрия при температуре —70 или1—33° С (баня
с сухим льдом или при температуре кипения аммиака). Для улучшения растворимости
многих соединений в жидком аммиаке рекомендуется добавлять эфир, тетрагидрофуран,
бензол или его гомологи. Избыток щелочного металла затем удаляют прибавлением
аммонийных солей или спиртов; одновременно при этом протекает и протонирование
образовавшихся на первой стадии карбанионов.
[26] J. Thiele et al., Ann., 347, 249. 275, 290 (1906).
[27] H. W i s 1 i с e и п я, J. prakt. Chem., 54, 18 (1896).
[28] N. A. Mila я, D. E 1 e s, J. Am. Chem. Soc., 78, 5903 (1956).
f29j P. И. Левина, В. P. Скварчеико и др., Сборник статей по общей
химии, 1, 355 (1953).
[30] К. Ziegler, Н. Colonius, О. S с h fi f е г, Ann., 473, 36 (1929);
С. В. Wooster, J. F. Ryan, J. Am. Chem. Soc., 56, 1133 (1934);
II. Gilman, J. C. Bailie, J. Am. Chem. Soc., 65, 267 (1943).
[31] P. Lebeau. M.„ Picon, C. r., 173, 64 (1921); 158, 1514 (1914);
С. B. Wooster, F. B. Smith, J. Am. Chem. Soc., 53, 179 (1931).
I. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С«С-СВЯЗЯМ
25
Для восстановления С—С-связей в а,Р-ненасыщенных кетонах [2, 32] и про-
стых эфирах феполов и полициклических гидроароматических соединений [33] оправ-
дало себя применение растворов лития, который обладает большей реакционной
способностью.
Метод удалось значительно улучшить одновременным применением спиртов.
Эта найденная Вустером [6а] и разработанная главпым образом Берчем [35] методика
позволяет восстанавливать даже изолированные ароматические ядра. Бензол и его
гомологи восстанавливаются до п-дигидропроизводпых [7], изомеризующихся в при-
сутствии'амида натрия в жидком аммиаке [36] с образованием соответствующих
сопряженных диенов, которые, в свою очередь, могут быть восстановлены до произ-
водных циклогексепа. Очевидно, конденсированные ароматические соединения также
можно восстанавливать по этому способу: из нафталина, например, получается изо-
тетралин [37], из пирена — 3,8-дигидропирен [38], а сс-нафтол восстанавливается
до 5,8-дигидронафтола-1 с 97%-ным выходом [39]. Эта реакция подучила особенно
широкое применение при частичном гидрировании простых эфиров фенола [40], кото-
рые в отсутствие спирта обычно лишь дезалкилируются. В результате гидролиза Ьбра-
зующихся на первой стадии 3,6-дигидрировапп[,(х еиолоэфиров получаются ненасыщен-
ные циклические кетоны [35, 41].
Аналогичные продукты могут быть получены из соответствующих производных
анилина [42]. Восстановление ароматических карбоновых кислот идет также, в ядре
и почти исключительно [43] в положение 1,4. f
Этот метод с успехом видоизменялся различными авторами. Вустер [6а] и Берч
[35, 36] обычно прибавляли спиртовой раствор гидрируемого вещества к раствору
натрия в жидком аммиаке. Вибо и Хаак [6Ъ] добавляли натрий к раствору или суспен-
зии соединения в жидком аммиаке и спирте. Вилдс и Нельсон [44] рекомендуют при-
менять растворы лития, а«спирт вводить последним в реакционную среду. Какому из
этих методов следует отдать предпочтение —• решают в каждом конкретном случае. -
1,2-Диметилциклогексадиен-} ,4. Для его получения из о-ксилола Хюк-
кель и сотр. [37] рекомендуют применять метод Берча. Восстановление в жидком
аммиаке ведут в литровом цилиндрическом реакционном сосуде с внутрен-
ним диаметром 8 см. В верхней части этого сосуда имеются два горла с нормаль-
ными шлифами, одно для капельной воронки, другое для мешалки, две стеклян-
ные трубки с кранами для подачи и отвода аммиака, а также тубус для'Низко-
температурного термометра. Аммиак, высушенный над натронной известью
и едким кали, конденсируют в предварительно вакуумированном аппарате,
[32] F. Н. Howell, D. А/ Н. Taylor, J. Chem. Soc., 1958, 1248;
D. И. R. В а г I о n, D. A. J. I v с s, В. R. Thomas, J. Chem. Soc.,
1954, 903.
[33] A. Sandoval, G. II. Thomas ot al., J. Am. Chem. Soc., 77, 148
(1955); J. A. Barltrop, N. A. J. Rogers, J. Chem. Soc., 1958, 2566.
[34] N. A. Nelson, J. C. W ollensak, J. Am. Chem. Soc., 80, 6626 (1958).
[35] A. J. В i r c h et al., J. Chem. Soc., 1944, 430; 1946, 593; 1951, 1945.
[36] A. J. Birch, J. Chem. Soc., 1950, 1551.
[37] W. Huckel et al., Chem. Bor., 88, 338, 346 (1955).
[38] O. Neunhoeffer, H. Woggon, S. Dahnc, Ann., 612, 98 (1958).
[39] C. D. Gutsche, H. H. Peter, Org. Synth., 37, 80 (1957).
[40] R. G r-e w e, E. Nolte, R. II. R о t z о 1 1, Chem. Bcr., 89, 600 (1956).
[41] S. M. Mukhetji, N. K. Bha tt acharjy a, J. Am. Chem. Soc., 75,
4698 (1953); J. S. Mills, H. J; R i n g о 1 d, C. Djerassi, J. Am,
Chem. Soc., 80, 6118 (1958).
[42] G. Stork, W. N. White, J. Am. Chem. Soc., 78, 4604 (1956).
[43] M. E. Kuehne, B. F Lambert, J. Am. Chem. Soc., 81, 4278
(1959).
[44] A. L. Wilds, N. A. Nelson, J. Am. Chem. Soc., 75, 5360 (1953).
26
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
охлаждаемом смесью сухого льда и метилового спирта, и в жидком состоянии
освобождают от следов воды небольшими кусочками натрия. На талой установке
опыты можно проводить также в атмосфере азота.
К раствору 30 е натрия п 400 лл жидкого аммиака медленно по каплям при
температуре от —70 до —65° С прибавляют смесь 54 г о-ксилола и 51 г абсолют-
ного метилового спирта и перемешивают до исчезновения синей окраски. Затем
испаряют аммиак, добавляют к остатку воду и отделяют смесь углеводородов.
Дигидро-о-ксилол можно выделить через ди- или тетрабромид. Однако полезно
подвергнуть смесь'повторному восстановлению; в этом случае получают жела-
емый углеводород с выходом 91% от теоретического.
Твердый гексамип кальция, Ca(NH3)g, является восстановителем, аналогичным
по свойствам описанным растворам металлов в жидком аммиаке. Его можно применять
в ваде суспензии в эфире при комнатной температуре, т. е. без жидкого аммиака*.
При помощи этого^рсагепта удалось, папример, осуществить специфичное гидрирова-
ние незамещенного ядра в а-мотилнафталине [45].
Для более подробного ознакомления с реакциями восстановления металлами
в жидком аммиаке можно рекомендовать обзорные статьи [5, 46, 47].
Восстановление литием в органических аминах.
В сравнении с соответствующими растворами в аммиаке растворы лития в аминах
оказались хотя и более сильными, но менее избирательными восстановителями. Амины
лучше, чем аммиак, растворяют большинство органических соединений. Кроме того,.
более высокие температуры кипения аминов позволяют вести реакции при относительно
высоких температурах.’Поэтомупри восстановлении ароматических соединений облегча-
ется не только первая ступепь реакции (1,^присоединение), но и изомеризация первич-
ных продуктов реакции в сопряженные диены и тем самым дальнейшее восстановление
до гидроароматических соединении с одной двойной связью. И действительно, в сообще-
ниях об использовании этого метода редко упоминается о выделении дпгидропроиз-
водных ароматических углеводородов, в то время как из этилбензола с хорошим выхо-
дом получается 1-этилцпклогекссн 148]. Из сопряженных диенов успешно получают
стеричсски однородные олефины [49].
В некоторых случаях восстанавливаются даже изолированные двойные связи
[50, 51]; поэтому при гидрировании ароматических соединении в качестве побочных
продуктов чаше всего образуются производные циклогексана.
Восстановление проводят как при низких температурах, так и при температурах
кипения соответствующих амилов. В основном применяют метиламин и его ближай-
шие гомологи — этилендиамин [51] и алкиланилины [49]. Часто амины одновременно
являются донорами протонов; по другим данным в конце реакции следует прибавлять
соли аммония. Обзор этих реакций опубликовав Верней н Смитом [47].'
р-Оксиэтилциклогексен-1. Получают из р-феиилэтилового спирта [48],
Для этого в трехгорлую колбу емкостью 500 мл с мешалкой, обратным холодиль-
ником, охлаждаемым сухим льдом, и капельной воронкой помещают 14 г лити-
евой проволоки и 500 мл безводного этиламина. К этой смеси при перемешивании
в течение 8—10 ч прибавляют по каплям 48,8 г Р-фонилэтилового спирта. На-
блюдающаяся в начале опыта синяя окраска довольно быстро исчезает, и реак-
ционная масса окрашивается в различные тона. Все операции следует проводить
* Заслуга введения данного реагента в арсенал средств восстановления углеводоро-
дов принадлежит Б. А. Казанскому с сотр., которыйв 1937 г. начал проводить
исследования в этом направлении [Б. А. Казанский, Н. В. Смир-
нова, Пзв. АН СССР. Отд. матем. и естеств. наук, 1937, 547; Б. А. К а-
з анс кий, Н. Ф. Г л ушиев, ЖОХ, 8, 642 (1938); Б. А. Казан-
ский, И. В. Гостуискал, ДАН СССР, 76, 41)7 (1951)]. — Прим,
ред.
451 Н. В о е г, Р. М. Duiaker, Roc. trav. chim., 77, 346 (1958).
46] G. W. Watt, Chem. Rev., 46, 316 (1950).
47] A. J. Birch, H. Smith, Quart. Rev. (Chem. Soc., London), 12, 17 (1958).
48] R. A. BenkescT et al., J. Am. Chem. Soc., 77, 3230, 6042 (1955).
491 K.i Ziegler et al., Ann., 528, 101 (1937); 567, 1 (1950).
50] G. 0 h 1 о f I, II. Parnow, G. Schade, Chem. Her., 89, 1549 (1956);
E. J. Corey, E..W. Cantrall, J. Am, Chem. Soc., 81, 1745 (1959).
[51] L. Rcggel, R- A. F r i e d e 1, f. Wender, J. Org. Chem., 22, 891 (1957).
I. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = С-СВЯЗЯМ
27
в атмосфере азота. В конце реакции избыточный литий извлекают щипцами
и к раствору осторожно прибавляют хлорид аммония до обесцвечивания. После
этого отгоняют зтиламин и к остатку при охлаждении по каплям прибавляют
воду. Органический слой извлекают эфиром и сушат над сульфатом магния. Пере-
гоняя эфирпый раствор, выделяют Р-оксиэтилциклогексен-1 (т. кип. 74—75° С
при 2 мм pm. cm.); выход 67% от теоретического.
Восстановление цинком, оловом или алюминием.
Гидрирование этиленовых соединений цинком и кислотами имеет мало преимуществ
перед каталитическими методами. Все же этот метод по-прежнему применяется для
избирательного восстановления стероидов [52, 53], ненасыщенных дикетонов [53—55]
и производных этилена с лабильными функциональными группами [17, 56].
^Обычно реакцию с цинковой пылью проводят в ледяной уксусной кислоте или
в пиридине и ледяной уксусной кислоте [57]. Действенность этой комбинации, при-
менявшейся для мягкого восстановления полиенкарбояовых кислот и цианиновых
красителей, связана с наличием малых количеств воды или спирта. Восстановление
проводилось также цинком и минеральной кислотой в водной среде. При этом реко-
мендуется применять омедненную цинковую пыль. В последнее время для восстано-
вления солеи изохинолнния применяли олово и соляную кислоту [58].
Восстановление пиррола. Процесс довольно легко может быть оста-
новлен на стадии образования дигидропроизводного (пирролипа). Эндрюс
и Мак-Ильвэйя приводят следующую модификацию методики Кнорра и Рабе [59].
В колбе достаточных размеров охлаждают до 0° С (смесь льда с солью)
500 мл 20%-ной соляной кислоты, при интенсивном перемешивании добавляют
200 з цинковой пыли и затем медленно, в течение 1 ч, из капельной воровки
50 г пиррола. Если температура в начале Процесса превышает 10° С, то его ско-
рость настолько возрастает, что реакция уже не поддается управлению. После
того как весь пиррол введен, добавляют 300 мл концентрированной соляной кис-
лоты и перемешивают реакционную смесь еще 2 ч, поддерживая температуру
15—25° С. Затем прекращают охлаждение, а перемешивание’ продолжают
еще 4,5 ч при комнатной температуре. Непрореагировавший цинк отфильтровы-
вают и промывают небольшим количеством воды. К фильтрату прибавляют
щелочь до растворения выпадающего d начале осадка гидроокиси цинка, отго-
няют пирролин с водяным паром до исчезновения щелочной реакции по лакмусу
в дистилляте. Последний подкисляют соляной кислотой и выпаривают досуха
ла паровой бане. Смолоподобный остаток обрабатывают 40%-пым раствором
едкого натра, дважды экстрагируют эфиром, сушат под сульфатом натрия и пере-
гоняют. При этом получается 28,5 г пирролина; т. кип. 89—92° С; кристалличе-
ский гидрохлорид плавится при 182—163° С. Выход пирролина составляет,
таким образом, 56% от теоретического; в качестве побочных продуктов образу-
ются преимущественно вещества с более высокими температурами кипения.
Подобно Кекуле [80],
выделения» цапк и щелочь,
рекомендовавшему для получения «водорода в момент
в последнее время восстановление проводят сплавом
[52] L. F. F i е ь с г, S. Rajagopalan, Е. Wilson, М. Ti shier,
J. Am. Chem. Soc., 73, 4133 (1951).
[53] Е. М. Chamberlin, W. V. R и у 1 е et al., J. Am. Chem. Soc., 75, 3477
(1953).
[54] P. К а г г e г, С. H. Eugs ter, Helv. chim, acta, 32, 1934 (1949).
[55] H. B. Honbest, M. Smith, A. Thomas, J. Chem. Soc,, 1958,
3293.
[56] F. Berge 1, J. A. Stock, J. Chem. Soc., 1957, 4563; E. A. Fehnel,
M. Carmack, J. Am. Chem. Soc., 70, 1813 (1948).
[57] R. Kuhn, A. W interstein, Ber., 65, 1737 (1932).
[58] G. Wittig, H. Streib, Ann., 584, 1 (1953).
[59] L. H. A n d г e w s, S. M. M cE 1 vai n, J. Am, Chem. Soc., 51, 889
(1929); L. Knorr, P. Rabe, Ber., 34, 3491 (1901).
[60] A. Kekule, Ann. (Suppl.), 2, 85 (1862).
28
ОБРАЗОВАНИЕ С—Н-СВЯЗЕИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
никеля и алюминия (сплав Ренея) в водной щелочи, Переходящий при этом в раствор
алюминий восстанавливает нафталин и его замещенные [61], циклооктатетраен [621
и особенно 0-арилакриловые кислоты [63] с хорошими выходами.
Электрохимическое восстановление
Электрохимические методы гидрирования ненасыщенных С= С-связей более
чем общеизвестны. Так же, как при восстановлении сс,0-ценасыщенных нитросоедине-
ний до насыщенных аминов [64], присоединение водорода часто сочетается с восстано-
влением других функциональных групп. Из гетероциклических соединений во многих
случаях образуются частично или полностью насыщенные соединения. Например,
из 8-оксихинолина с 95%-пым выходом получается 1,2,3,4-тетрагидро-8-оксихииолин
[65]. Электрохимическое восстановление применял Аренс [66] еще в 1896 г. для син-
теза пиперидина из пиридина*.
Несмотря па хорошие выходы, достигаемые электрохимическими методами,
они не нашли широкого применения в лабораторной практике. Вероятно, это объяс-
няется не только отсутствием специальной аппаратуры, но и недостатком опыта в обла-
сти электрохимии. Для более широкого изучения литературы по этому вопросу может
служить обзор Сванна [67].
Восстановление литийалюминийгидридом
и аналогичными соединениями
Возможность применения литийалюминийгидрида и подобных веществ для
восстановления С=С-связей в значительной мере зависит от положения этой связи
в молекуле, от температуры реакции, а также от растворителя.
При обычном ведении процесса, то есть в кппящем эфире, водород присоеди-
няется лишь в особых случаях. Как изолированные, так и сопряженные двойные
связи и ароматические системы обычно устойчивы к действию этих восстановителей.
Исключением являются фульвспы [68], которые относительно легко переводятся
в алкилциклопоптадиены. Азотсодержащие гетероциклы гидрируются но С—N-связи
легче, чем по С—С-связп, поэтому пиридин и, в частности, хинолин превращаются
в 1,2-дигидропроизводные [69].
Даже этиленовые двойные связи в а,0-положении к полярным функциональным
группам не всегда восстанавливаются. Как правило, такая реакция протекает с груп-
пировками Аг— С=С—С= О (производные коричной кислоты [70, 71], халконы [72])
и Аг—С=С—N ((о-нитростиролы [73] и N-метилоксиндолы [74]). а,0-Ненасыщен-
* Электрохимическое восстановление хинолиновых оснований подробно изучалось
В. В. Левченко [ЖОХ, 11, 686 (1941); там же 17, 1656 (1947); 18, 1237 (1948)]. —
Прим. ред.
[61] D. Papa, Е, Schwenk, Н. В г е i g е г, J. Org. Chem., 14, 366 (1949).
[62] W. О. Jones, J. Chem. Soc., 1954, 1808.
[63] G. A. Page, D. S. T a r b e 1 1, Org. Synth., 34, 8 (1954); G. M. Badger,
J. Chem. Soc., 1948, 999.
[64] К. H. Slotta, R. К e t h ц r, .Ber., 71,. 62 (1938).
[65] S. Szmaragd, E. Brine r, Helv. chim. acta, 32, 1278 (1949).
[66] F. Ahrens, Z. Elektrochem. angew. physik. Chem., 2, 577 (1896).
[67] S. । S w a n n jr., в книге A. Weissberger, Technique of Organic Che-
mistry, v. Il, Intersci. Puhi., Inc., New York, 1956, p. 385.
[68] K. Hafner, Ann., 606, 79 (1957).
[69] F. Bohlmann, Chem. Ber., 85, 390 (1952).
[70] P. К a г r e r, P. В a n e r j e a, Helv. chim. acta, 32, 1692 (1949); A. R. К a-
t r i t z k y, J. Chem. Soc., 1955, 2586.
[71] F. A. H о c h s t e i n, W. G. Brown, J. Am. Chem. Soc., 70, 3484 (1948).
172] C. S. Rondes tvedt jr., J. Am. Chem. Soc., 73, 4509 (1951).
[73] J. M. Bobbitt, T. T. Chou, J. Org. Chem., 24, 1106 (1959); M. Erne,
F. Ramirez, Helv. chim. acta, 33, 912 (1950); J. F i n k e i n, J. Am.
Chem. Soc., 73 550 (1951).
[74] p. L. Julian, H. С. P r i n t y, J. Am. Chem. Soc., 71, 3206 (1949).
I. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = С-СВЯЗЯМ
29
ные циануксусные эфпры [75] и отдельные а,р-этнленкетоны [76] могут быть восстано-
влены такте с хорошими выходами.
При обычном проведении реакции эфирный раствор восстанавливаемого веще-
ства прибавляют к эфирному раствору гидрида; наряду с этиленовой группой в этом
случае восстанавливаются и другие группы. Но ири смешении компонентов реакции
в обратном порядке, т. е. при постепенном прибавлении ли^ийалюмипиигидрида к вос-
станавливаемому веществу, в большинстве случаев удаетсй избежать насыщения эти-
леновой связи. Например, из коричного альдегида по первой методике получается
гпдрокоричиый спирт, а по второй — коричный спирт, с хорошими выходами в обоих
случаях [71]. Низкая температура и отсутствие избытка гидрида способствуют вос-
становлению лишь функциональных групп. С другой сторопы, в некоторых случаях
удалось провести гидрирование только этиленоной связи, например при получении
1-фенил-2-нитропропана из 1-фенил-2-пптропрОпена [77] и дифспилуксусного альде-
гида из дифенилкетена [78].
При работе с высококипящими растворителями реакцию можно вести в значи-
тельно более жестких условиях п сократить время взаимодействия. Для этого пре-
имущественно используют гомологи эфира, тетрагидрофуран и диоксап. К примеру,
в ди-л-бутиловом эфире из пиридина получают небольшие количества пиперидина 179],
а аллиловый спирт можно, по крайней мере частично, перевести в и-иропиловыи
спирт [71). Для синтеза дигидроцроизводных акридина и аналогичных гетероциклов
рекомендуется применять литийалюминййгидрид в бензольно-эфирных смесях [69].
При действии гидрида диэтилалюминия в бензоле удалось восстановить хинолин до
ди- и тетрагидрохинолина [80]. Очень сильным, но ио всегда применимым восстанови-
телем является раствор литнналюмипийгндрида в моноэтиловом эфире диэтиленгли-
коля. Этот реагент термически устойчив до 200° С, и его преимуществом является
более мягкое протекание необходимого во всех случаях гидролиза. С ого помо^цью
удалось количественно восстановить даже аценафтилен до аценафтена [81].
При повышенных температурах или (если нужно) под давлением можно
и в отсутствие растворителя осуществить присоединение [82] гидридов алюминия
к этилену, монозамещенным этиленам RCH=CH2 и несимметричным дизамещенным
этиленам В.2С=СН2. В более жестких условиях эта реакция протекает и с цикло-
олефинами, но не с циклогексеном. Гидрид алюминия реагирует уже при 70° С;
лптпйалюминийгидрид лучше применять при температурах 110—120° С. Симметричные
дпзамощенныс этилопы RCH —CHR реагируют только в таких условиях, при которых
гидрид алюминия и лптийалюмипийгидрид начинают разлагаться. По в отдельных
случаях реакцию удалось провести и с термически более устойчивыми гидридами
дпалкилалюминия. При гидролизе образовавшихся в результате реакции алюминий-
органических соединений получаются насыщенные углеводороды. Значительные раз-
личия в способности веществ к присоединению позволяют проводить также селективное
восстановление, так, в частности, из 4-винилциклогексена можно легко получить
4-этилциклогексен.
Изопропилциклопентадиен [68]. Его получают легко осуществляемым
избирательным восстановлением. Раствор 53 г диметилфульвена в 200 мл
абсолютного эфира медленно добавляют по каплям к раствору 18 г литийалюми-
ннйгидрида в 200 мл эфира. Реакционная масса при этом нагревается, желтая
«краска фульвена исчезает и выпадает бесцветный осадок. По завершении реак-
ции, Примерно через 2 ч, продукт присоединения разлагают 100 мл метилового
спирта при охлаждении льдом и добавляют 2 я. раствор соляной нислоты до
образования прозрачного раствора. Эфирный слой отделяют, промывают раз-
бавленным раствором карбоната натрия и сушат над сульфатом натрия. Затем
испаряют эфир и перегоняют остаток в вакууме. Получается 38,4 *г •
[751 A. Dornow, G. Messwar b, Н. И. Frey, Chem. Ber., 83 , 445 (1950)'. -
[76] J. R. Catch, E. A. Evans, J. Chem. Soc., 1957, 2796.
[77] R. T. Gilsdorf, F. F. Nord, J. Am. Chem. Soc., 74, 1837 (1952).
[78] V. M. Micovic, M. M. Rogic, M. Lj. Mihailovic, Tetrahedron,
1, 340 (1957).
[79] P. de Mayo, W. Rigby, Nature (London), 166, 1075 (1950).
[80| W. P. Neumann, Ann., 618, 90 (1958).
[81] 1. Goodman, J. Chem. Soc., 1951, 2209.
[82] K. Ziegler, H. G. G e 11 о r t et al., Ann., 589, 91 (1954).
30
ОБРАЗОВАНИЕ С—И-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
(71% от теоретического) изопропилциклопентадиена; бесцветная жидкость,,
т. кип. 32—34° С (23 мм рт. ст.).
При работе с труднорастворимыми в эфире веществами восстановление удобно
проводить в аппарате Сокслета.
Триптамин [83]. В гильзе аппарата Сокслета экстрагируют 15 ;• 3-ю-
яитровинплнпдола в колбу, содержащую 15 г литийалюминнйгидрида и 500 мл
абсолютного эфира. К раствору медленно при охлаждении прибавляют 80 мл
воды и фильтруют. Эфирный раствор отделяют, сушат сульфатом натрия и насы-
щают хлористым водородом. Получают 12,7 г гидрохлорида триптамина, который
после перекристаллизации из смеси метилового спирта и этилацетата плавится
при 249—250° С (разл.).
Другие комплексные гидриды металлов редко употреблялись для восстановле-
ппя С=С-связей. Боргидрид патрия, который можно применять в водно-спиртовом-
растворе, по-видимому, в большинстве случаев не восстанавливает этиленовые связи.
Имеется лишь несколько сообщений об избирательном гидрировании циклим-
мопиевых солей [84] и избирательном восстановлении некоторых особенно актив-
ных этиленовых связей [70b, 85]. Для получения ряда нитропарафинов из нитро-
этиленов нашел применение очень мягкий восстановитель — тримстоксиборгпдрид
натрия [86].
Для подробного изучения таких реакций восстановления можно рекомендовать
монографию Гэйлорда, а также монографию Миновича и Михайловича [87].
Прочие восстановители
Часто употреблявшиеся ранее для восстановления ненасыщенных соединений
двухлористос олово, иодид фосфония, литионит натрия и подпетый водород в насто-
ящее время используются редко.
Так, например, при помощи хлорида олова (II) из амидов бензоилакриловых
кислот синтезировали некоторые амиды у-кетокарбоновых кислот. [88]. Дитионит
патрия применялся для насыщения этиленовой связи в бензилиденипдандионах [89]
и получения замещенных у-дикетопов4из 1,2-дибензоилэтиленов [90]. Трудно гидриру-
емый карбазол можно перевести в гексагидрокарбазол действием йодистого водорода
[91], а из 3-ниридилакриловой кислоты и йодистого водорода в присутствии фосфора
удалось получить 3-пиридплпропионовую кислоту [92].
В отдельных случаях для восстановления С—С-связей употреблялись и другие
реагенты, для восстановления . бензилидепхпнальдина и аналогичных соединений
применяли, например, п-тиокрезол [93]. Муравьиной кислотой восстановлен ряд
[83] Е. Н. Р.‘ Young, J. Chem. Soc., 1958, 3493.
[84] W. Awe, H. W i c h m a n n, R. В uer h о p, Chem. Ber., 90, 1997 (1957);
R. E. Lyle, R. E. Adel, G. G. Lyle, J. Org. Chem., 24, 342
(1954).
[85] J. Fajkos, Coll. . Czech. Chem. Comm., 23, 2155 (1958); M. M i у a n o,
M. Matsui, Chem. Ber., 91, 2044 (1958).
[86] H. Shechter, D. E. Ley, E. B. Roberson jr., J. Am. Chem. Soc.,
78, 4984 (1956).
[87] H. Г e й л p о д, Восстановление комплексными гидридами металлов, Издат-
инлит, 1959; В. М и ч о в и ч, М. Михайлович, Алюмогидрид лития
и его применение в Органической химии, Издатинлит, 1957.
[88] R. Е. Lutz, G. W. Scott, J. Org. Chem., 13, 284 (1948).
[89] Г. Я. В ан аг, Т. Т. Д у м и и с, ДАН СССР, 125, 549 (1959).
[90] R. Е. Lutz, S. М. King, J. Org. Chem., 17, 1519 (1952).
191] J. Schmidt, A. Sigwart, Ber., 45, 1779 (1912);
[92] P. С. Лифшиц и сотр., ЖОХ, 21, 1354, 1360 (1951).
[93] Н. Gilman, J. L. Towle, R. K. Ingham, J. Am. Chem. Soc., 76r
2920 (1954).
J. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С=С-СВЯЗЯМ
31
диалкнлепаминов [94] и циклиммопиевых солей [95] до соответствующих насыщенных
третичных аминов, а для восстановления сильно активированных этиленовых связей,
например в 1-циап-1,2-дифенйлэтилеие, рекомендуется бензиловый спирт в щелочной
среде [96].
Некоторые промежуточные продукты синтеза каротиноидов с кумулированными
двойными связями удалось с хорошими выходами перевести [97] н производные эти-
лена действием ацетата хрома (II).
Недавно описано применение диимина — восстановителя, часто образующегося
в качестве промежуточного продукта [98]. Получаемый различными путями диимин
HaN-NH3+[O]———
НООС—N= N-СООЦ ,асОг
ArSO3-NH-NHB _AraOiH
2NH2CI -)-2NaOH'
-2HSO ’
может гидрировать изолированные двойные и тройные связи.
\сн-сн/ + N2
Так, например, с его помощью удалось количественно перевести аллиловый
•спирт в пропанол. Диимпн отличается почти от всех других химических восстановите-
лей том, что под его действием, очевидно, происходит только ^ис-прпсоедипспие водо-
рода.
2. Каталитическое гидрирование
Очень удобным методом присоединения водорода но С= С-связям является
каталитическое гидрирование, протекающее в большинстве случаев легко и однозначно.
При повышенных температурах может проходить и обратная реакция — дегидриро-
вание, так что в таких случаях наряду с продуктами гидрирования в реакционной
массе могут содержаться и продукты дегидрирования в соответствии с положением
равновесия в данных условиях.
Гидрирование часто не удается осуществить из-за отравления катализатора.
Поэтому при гидрировании следует применять только чистыо-вещества. Это указание
относится не только к восстанавливаемому веществу, по и к реактивам и раствори-
телям, используемым для получения катализатора. Кроме того, очепь важно, чтобы
воздух в рабочем помещении также не содержал , примесей, отравляющих ка-
тализатор.
Водород из баллонов обычно можно применять без предварительной очистки.
Его пригодность лучше всего проверить в опыте по гидрированию известного вещества.
Загрязненный водород из баллонов и водород, получаемый н аппарате Киииа из цинка
и серной кислоты, следует очищать пропусканием через растворы перманганата калия
и нитрата серебра. Если кислород, содержащийся в водороде, полученном электроли-
тическим путем, мешает, его удаляют пропусканием газа через трубку для сожжения,
заполненную медью.
[94] Р. L. De Bonneville, J. Н. Macartney, J. Am. Chem. Soc. 72,
3073 (1950).
[95] A. H, Кост, Л. Г. Ю'дин, ЖОХ, 26, 1720 (1956).
[96] M. Avramoff, Y. Sprinzak, J. Am. Chem. Soc., 80, 493 (1958).
[97] W. Oroshnik, J. Am. Chem. Soc., 76, 5499 (1954).
198] E. J. Corey, W. L. Mock, D. J. Pasto, Tetrahedron Letters, 1961,
347; S. Hiinig, H. R. Muller, W. Thier, Tetrahedron
Letters, 1961, 353; E. E. van Tamelen et al., J. Am. Chem. Soc.,
83, 3725, 3729 (1961); E. Schmitz, R. О h m e, Angew. Chem., 73. 807
32
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Катализаторы.
Особое значение для успешного гидрирования имеют выбор и свойства подхо-
дящего катализатора. В качестве катализаторов наиболее пригодны благородные
металлы, никель, медь, кобальт, железо, а также смешанные, окисные и сульфидный
катализаторы. Мелко раздробленные металлические катализаторы готовят восстано-
влением производных этих металлов. Благородные металлы могут быть получены из
соответствующих соединений действием органических восстановителей, например форм-
альдегида или муравьиной кислоты, или восстановлением водородом при комнатной
температуре. Катализаторы, состоящие из неблагородных металлов, получают восста-
новлением при повышенных температурах в токе водорода соответствующих окп-
слов, гйдроокисеп, основных карбонатов, а также формиатов, ацетатов или
оксалатов.
Скелетные катализаторы получают по методу Ренея из сплава каталитически
активного и неактивного металла, растворяя неактивный металл большей частью
в щелочи, иногда в кислоте; в остатке получается катализатор*.
Катализаторы из благородных метал-лов отличаются
высокой активностью и пригодны для гидрирования уже при комнатной темпера-
туре. Это особенно важно в тех случаях, когда исходные вещества или продукты
реакции неустойчивы при повышенных температурах. Другим преимуществом пла-
тиновых и палладиевых катализаторов является их инертность по отношению к кис-
лотам, что позволяет вести гидрирование в кислых растворах, например в ледяной
уксусной кислоте. В качестве растворителей применяют, как и в других методах,
спирты, эфиры уксусной кислоты, простые эфиры и воду.
Элементарные платиновые контактные катализаторы. Платиновая
чернь [99]. Вилыптеттер и Вальдшмидт-Лейтц [99] ппи изготовлении плати-
новой черни несколько видоизменили известный способ Лоева [100]. Раствор
платинохлористоводородной кислоты (80 мл), содержащий около 20 « пла-
тины и подкисленный небольшим избытком соляной кислоты, смешивают со 150 мл
33%-иого раствора формальдегида и охлеждают до —10° С. К этой смеси при
хорошем перемешивании добавляют по каплям 420 г 50%-ного раствора едкого
кали, пе допуская повышения температз'ры выше 6° С. Затем'при интенсивном
перемешивании смесь нагревают 30 мин при 55—60° С. Выделившуюся платино- ,
вую чернь промывают, декантируя водой, до исчезновения щелочной реакции
и ионов хлора в промывных водах, переносят на воронку Бюхнера, следя за
тем, чтобы платина постоянно находилась под слоем воды, быстро отсасывают
л отжимают между листами фильтровальной бумаги. Катализатор помещают
в эксикатор и устанавливают глубокий вакуум. Через несколько дней продукт
превращается в сухую пыль. Эксикатор заполняют двуокисью углерода и в его
атмосфере храпят катализатор.
Если из платиновой черни в результате длительной выдержки в глубоком
вакууме не удаляются все газы, то через несколько дней из поглощенного водо-
рода и кислорода образуется вода [101].
Получение платиновой черни восстановлением муравьиной кислотой
описано Виландом (102].
* Повую интересную группу катализаторов гидрировании представляют бориды
никеля [К. Paul, Р. Buisson, N. Joseph, С. г., 232, 627 (1951))
и бориды металлов платиновой группы [Б. Д, Полковников,
А. А. Баландин, А. М. Т а б е р, ДАН СССР, 145, 809 (1962)]. Эти
катализаторы легко получаются взаимодействием сстветствующих хлоридов
с боргидридом натрия в водном растворе. Они очень активны и довольно устой-
чивы. Об их составе см. работу: Н. Н. Мальцев, 3. К. Стер л я д-
кин, В. И. Михеев, ДАН СССР, 160.’352 (1965). — Прим. ред.
[99] R, Willstatter, Е. Waldschmidt-Leitz, Вег., 54, 113
(1921).
(100J О. Loew, Вег., 23, 289 (1890).
[101] R. Willstatt tfr, М. Bommer, Вот., 51, 770 (1918).
/102] Н. Wieland, Ber., 45, 489 (1912),
t. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = С-СВЯЗЯМ
35
Часто применяют катализаторы на носителях *.
Палладий на животном угле [ЮЗ]. В сосуде для гидрирования к 2 а
животного угля прибавляют водный раствор 0,5 г дихлорида палладия. Послй-
восстановления водородом катализатор отсасывают, промывают водой, спиртом
и наконец эфиром. ,
Носителями катализаторов могут служить также асбест, пемза, кизельгур,
силикагель, кремневая кислота, активные угли, а также окисли, карбонаты и сульфа-
ты магния, кальция, бария, цинка, алюминия, железа, хрома и циркония. Метод полу-
чения палладиевого катализатора па сульфате бария приведен Мозинго [104].
Для особых целей, например для частичного гидрирования тройных связей
в двойные [105J или кумуленов в полиены [106], прпменяют'каталцзаторы, дезактиви-
рованные частичным отравлением. Такие катализаторы пе восстанавливают двойных
связей.
Дезактивированный палладий на карбонате кальция. Дезактивирован-
ный раствором ацетата свинца палладиевый катализатор готовят по Линдлару
[105]. В 400 мл дистиллированной воды замешивают 50 г чистого осажденного-
карбоната кальция, прибавляют 50 мл раствора хлорида палладия, содержащего-
5% палладия, перемешивают сначала 5 мин при комнатной температуре, затем
10 мин при 80° С. Горячую взвесь вносят в сосуд для гидрирования и встряхивают
в атмосфере водорода до прекращения поглощения водорода. После этого филь-
труют на воронке Бюхнера и тщательно промывают дистиллированной водой.
Осадок перемешивают с 500 мл дистиллированной воды. После получения суспен-
зии прибавляют раствор 5 г диацетата свинца в 100 мл дистиллированной воды;
перемешивают 10 мин при 20u С и затем еще 40 мин на кипящей водяной бане.
Катализатор фильтруют на воронке Бюхнера, тщательно промывают дистилли-
рованной водой и сушат в вакууме при 40—45° С.
Аналогичная дезактивация катализаторов наблюдалась также при действии
солей меди и висмута. Палладиевый катализатор на сульфате бария, дезактивирован-
ный хинолином и серой, применяют в реакции Розенмунда — Зайцева (стр. 74).
Платиновые или палладиевые катализаторы на асбесте (метод Зелинского-
и Борисова [107]). Чистый, промытый кислотой и прокаленный коротковолокяи-
стый асбест пропитывают концентрированным, слегка кислым раствором хлорида
платины или палладия, нагревая для более равномерной пропитки на водяной
бале. Затем волокна асбеста обрабатывают на холоду 35—40%-ным раствором
формальдегида, причем на 1 г платины берут 2,5—3 мл раствора формальдегида
(в случае палладия 4,5—5 мл). После охлаждения медленно добавляют полутора-
кратное по весу (в расчете на раствор формальдегида) количество 40—50%-иого
раствора едкого натра. Осаждение металла начинается уже на холоду, но-
для доведения реакции до конца нагревают на водяной байе. После этого при-
бавляют большое количество воды и промывают асбестовую массу от солей и-ще-
лочи. Для удаления следов щелочи черные асбестовые волокна вносят в сильно-
разбавленную уксусную кислоту. Обработку заканчивают промывкой массы
до нейтральной реакции, фильтрованием ^непродолжительной сушкой в сушиль-
ном шкафу. Полученный таким образом черный асбест является высокоактивным
катализатором гидрирования и дегидрирования.
* 0 получении платинированного угля, активированного хлоридами платины или
палладия, см. В. А. Казанский, А. Л. Либерман, А. Ф. Плат*
и др., ДАН СССР, 71, 477 (1950). — Прим. ред.
[103] Е. О t t, R. S с h г б t е г, Вег., 60, 633 (1927).
[104] В. М о z i n g о, ’ Org. Synth., 26, 1946, р. 77, 4
[105] Н. Lindlar, Helv. chim. acta, 35, 450 (1952).
[106] R. Kuhn, H. Fischer, Chem. Ber., 93, 2285 (i960).
[107] N. D. Zelinsky, P. В ori sol I, Ber., 57, 150 (1^24).
3 Заказ 1835.
:34
ОБРАЗОВАНИЕ G—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Зелинский и Турова-Поляк [108] описывают'специфические свойства осмиевого
катализатора. Гидрирование в присутствии осмиевого катализатора протекает обычно
при более низких температурах, чем в присутствии платиновых, палладиевых п никеле-
вых катализаторов. Осмий, нанесенный па асбсст, очень устойчив и способен работать
в течение нескольких месяцев без снижения активности. Недостатками контактных
осмиевых катализаторов является необходимость частой регенерации катализатора
в тех случаях, когда он применяется без носителя, и разложение гидрируемых веществ
в присутствии осмия при температурах выще 150° С.
Катализаторы На основе окислов и гидроокисей благородных металлов ныне
конкурируют с большим успехом с контактными катализаторами из элементов плати-
новой группы. Например, платиновая или палладиевая чернь практически вытеснены
более просто получаемыми окислами. Окисные или гидроокисные катализаторы вос-
.стаиавливают водородом в сосуде для гидрирования до металлов в очень мелкораз-
дробленном состоянии, которые и действуют как катализаторы. Продолжительность
восстановления колеблется от нескольких секунд до нескольких минут. Если восста-
новление ведут только в присутствии растворителя, то реакция обычно протекает
быстро, но если катализатор восстанавливают в смеси с гидрируемым веществом, то
длительность его восстановления зависит от природы восстанавливаемого вещества
Л может достигать 10—15 мин.
Катализатор на основе окиси платипы (катализатор Адамса) [109].
В фарфоровой чашке перемешивают 20 г нитрата натрия с раствором тетрахлорида
платины в 5 мл воды, содержащим 1 з платины. Смесь осторожно при перемеши-
вании стеклянной палочкой нагревают до удаления воды. Нагревание продол-
жают до расплавления массы (400 —500° С) и начала выделения бурых паров
окислов азота. После прекращения выделения окислов азота массу охлаждают
и растворяют в 50 мл воды, образовавшийся коричневый осадок промывают
сначала декантацией, а затем на фильтре до исчезновения реакции на нвтрат
в фильтрате. Однако получить продукт, совершенно свободный от щелочей,
трудно, и обычно в нем содержится около 2% щелочей, если сплавление про-
водилось при 400—500° С. При более высоких температурах сплавления содержа-
ние щелочей повышается. Коричневую окись платины сушат над концентри-
рованной серной кислотой или в вакууме. Это очень удобный в работе и исклю-
чительно эффективный катализатор.
По способу Брюса [110] для получения окиси платины исходят из хлорплати-
ната аммония. Аналогично готовят окись палладия по Шрайнеру и Адамсу [111].
Поскольку окись палладия легко пептизируется, промывать ее можно только 1 %-ними
растворами карбоната или нитрата натрия.
Как видно из приводимой ниже методики получения гидроокиси платины па
окиси магния, катализатор может образоваться также в результате гидролиза дихло-
рида платины под влиянием носителя — окиси магния [112].
Гидроокись платипы на окиси магния. В 500 ч. воды суспендируют
100 ч. окиси магния и суспензию обрабатывают раствором 2,9 ч. дихлорида пла-
тины в 400 ч. воды. Выделившаяся гидроокись двухвалентной платины осаж-
дается на окиси магния. После'промывки катализатор сушат при 100° С.
Катализаторы, на основе рутения [113] состоят из мелкодисперсного рутения
или из ого -окнелов и солей, которые могут быть нанесены па носители (древесный
[108] N. D. Zelinsky, М. В. Turowa-Pollak, Вег., 62, 2865 (1929).
[109] R. Adams, V. Voorhees, R. L. S hr iner Org. Synth., 8, 92
(1928).
[110] W. F. В г и c e, J. Am. Chem. Soc., 58, 687 (1936); ср. также Org. Synth.,
17, 98.
[Ill] R. L. S h г i n e r, R. Adams, J. Am. Chem. Soc., 46, 1685 (1924); 47,
1147 (1925).
[112] Герм. пат. 256500 (1911); Frdl., 11, 101.
1113] Пат. США 2494563 ^1947); С., 1951, 1, 122.
Т. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С=С-СВЯЗЯМ
35
уголь, сйликагель и др.). Наиболее целесообразно сплавлять металлический рутений
или его окисел с перекисью натрия и раствор полученной соли наносить на носитель.
Проработавшие какое-то время катализаторы могут быть в определенных
услввиях регенерированы воздухом или кислородом. Такую операцию полезно про-
водить даже во время гидрирования для того, чтобы' довести реакцию до конца или
повысить снизившуюся скорость поглощении водорода. Однако иногда при регенера-
ции может произойти полное дезактивирование катализатора. Если в процессе гидри-
рования катализатор спекается, что происходит обычно к'концу реакции, то его необ-
ходимо заменить свежим. Катализатор, который после встряхивания легко осе-
дает, но при взбалтывании быстро образует суспензию, считается еще годным для
работы.
Отработанный катализатор собирают и затем растворяют в царской водке.
После испарения избытка кислоты осаждают металл водным раствором аммиака.
Разработан [114] действенный, до трудоемкий метод переработки отработанных, т. е.
содержащих большое количество контактных ядов, платиновых катализаторов.
Коллоидные благородные металлы. Коллоидные катализаторы гидрирования
не имеют сейчас большого практического значения. Для их получения хлориды благо-
родных металлов восстанавливают, чаще всего гидразингидратом в слабощелочной
среде. Защитными коллоидами могут служить белковые вещества или их гидролизаты.
Данворт и Норд [115] получили коллоидный иридий с поливиниловым спиртом в каче-
стве защитного коллоида. Пааль [116] применял коллоиды платины и палладия с про-
тальбинатом или лизальбинатом патрия; Скита [117] использовал защитный колловд
гуммиарабик. Кельбер и Шварц [118] описывают устойчивый к действию кислот кон-
тантныи коллоидный катализатор с частично гидролизованным глутнвом в роли
защитного коллоида. Скита и сотр. [119] описывают получение коллоидных гидрооки-
сей платины и палладия.
Коллоидная платина [120]. В тридцатикратном количестве воды рас-
творяют 1 г лизальбината натрия и смешивают раствор с несколько большим
количеством едкого натра, чем необходимо для связывания всего хлора, содержа-
щегося в прибавляемой ллатинохлористоводо^пдной кислоте. Затем в щелочной
раствор вводят 2 г платинохлорисговодородион кислоты, растворенной в неболь-
шом количестве воды. К прозрачному темно-коричневому раствору добавляют
гидразипгидрат с небольшим избытком. Происходит выделение газа и жидкость-
окрашивается в темвый цвет. Восстановление быстро заканчивается. Затем чер-
ную жидкость, которая в тонком слое прозрачна и имеет черно-коричневый цвет,
подвергают диализу против воды. Таким способом после многократной смены-
внешней воды удается удалить из коллоидного раствора избыток едкого натра,
нспрореагиривавший тидразингйдрат и образовавшийся в реакции хлорид,
натрия. Профильтрованное содержимое диализатора осторожно концентрируют
на водяной бане и высушивают ввакуум-эксикаторе. Препарат представляет собой
черные, сильно блестящие, ломкие чешуйки, легко и полностью растворяющиеся
в воде с образованием черной непрозрачной жидкости.
Аналогично получают коллоидный палладий [120], Применяя водород.,
в качестве восстановителя.
Скелетные металлические катализаторы (металлы
Ренея). По методу, предложенному Ренеем, каталитически активный металл спла-
вляют с неактивным металлом и обрабатывают сплав реактивом, растворяющим неак-
тивпый металл. Вымываемыми неактивными компонентами могут быть алюминий,
кремний, магний и цинк. Из каталитически активных металлов находят применение
главным образом никель, кобальт, медь и железо.
114] Е. Wichers, J. Am. Chom. Soc., 43, 1268 (1921).
115] W. P. Dunworth, F. F. Nord, J. Am. Chem. Soc,, 72, 4197 (1950)^.
116] C. Pa al, C. Amberger, Ber., 37, 131 (1904); 35, 2197 (1902).
117] A. S k i t a, Ber., 57, 1977 (1924); 45, 3584 (1912).
118] C. Keiber, A, Schwarz, Вет., 45, 1946 (1912).
119] A. Skit a, W. A. Meyer, Ber., 45, 3584 (1912).
120] С. P a al, C. Amberger, Ber., 37, 126 (1904); 38, 1398 (1905).
3*
36
ОБРАЗОВАНИЕ С—Н-СВЯЗЕИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
По методу Ренея можно получать также катализаторы из благородных метал-
лов. Для этого сплавы алюминия с платиной (40%) или цинка С палладием (40%) раз-
лагают соляной кислотой [121].
Преимуществом металлов Ренея, и особенно пикеля Ренея, является то, что
они, подобно катализаторам на основе благородных металлов, активны в мягких
условиях. Не следует проводить гидрирование в присутствии никеля Ренея в среде
диоксана при температурах выше 210° С, так как в этих условиях диоксан реагирует
с водородом и никелем Ренея со взрывом. Прп работе с никелем Ренея температура
реакции снижается па 20—40° С по сравнению с температурой реакции в присутствии
других никелевых катализаторов. Однако при 100—150° С активность никеля Ренея
понижена. Таким образом, при повышенных температурах можно с равным успехом
.применять никелевые катализаторы, приготовленные по старым рецептурам (122].
Никель Ренея [123]. Расплавляют 400 г алюминия и нагревают его
до 1200° С. В расплав сразу вносят 300 а никеля в виде кубиков (такой никель
более пригоден для получения сплавов, чем компактный металл, подвергнутый
। механической обработке). В результате бурной реакции никель переходит в рас-
твор, температура которого повышается примерно до 1500° С. Сплав охлаждают,
разбивают и растирают в порошок. Небольшими порциями вносят 250 г сплава
в 100 мл приблизительно 25%-ного раствора едкого натра, охлаждаемого льдом.
Происходит рааложенве, сопровождающееся бурным выделением водорода (вспе-
нивание и разбрызгивание) и разогреванием. После внесенвя всего сплава темпе-
ратуру повышают до 90—1006 С и поддерживают ее до прекращения выделения
водорода. Реакционной массе дают отстояться, сливают жидкость и обрабатывают
массу дважды 1 л свежей щелочи. После тщательной декантации второй порции
щелочи осадок никеля промывают водой, перомептиняя и сливая воду с отстояв-
шегося осадка. Промывку ведут до нейтральной реакции промывных вод по
фенолфталеину. Таким же способом вытесняют воду спиртом. Катализатор хранят
d стеклянной посуде под слоем спирта.
Никель Ренея хранят в спирте цз-за его высокой пирофорности, связанной,
^вероятно, с наличием в катализаторе гидридов никеля [124].
Образование сплава протекает с значительным выделением тепла. Поэтому
вследствие повышения температуры часто удается получить и более высокоплавкие
сплавы с большим содержанием активного металла. В присутствии кислорода воздуха
в результате алюмотермической реакции может происходить дальнейшее повышение
температуры. Поэтому следует соблюдать осторожность! От
воздействия кислорода воздуха сплав можно защитить слоем соли или инерт-
ного газа.
В настоящее время выпускаются готовые сплавы.
После разложения алюминиевых сплавов в металле остается еще алюминий,
который, по-видимому, оказывает известное влияние па активность катализатора.
Если оставшийся алюминий удалить продолжительным воздействием щелочей, то
катализатор дезактивируется [125]. Сплавы, содержащие более 75% никеля, слабо
или совсем не разлагаются водной щелочью. Для их разложения применяют концен-
трированные растворы едкого натра пли сплавление с едким натром [126]. Для полу-
чения активного металла наиболее пригодны сплавы, в которых содержится 30—50%
активного металла.
Активность никеля Ренея можно повысить добавлением активаторов. В каче-
стве активаторов после разложения сплава или в процессе нромывки катализатора
[121] Пат. США 2384501 (1945); С. А., 39, 5422,(1945).
1122] Е. В. Max ted, R. А. Т i 11, J. Soc. Chem. Ind., 57, 197 (1938).
(1231 R. Paul, G. Hilly, Bull. Soc. chim. France [5], 3, 2330 (1936);
см. также В. Mozingo, Org. Synth. 21, 1941, p, 15; H. R, В i 1-
, 1 i с a, H. A d k i n s, Org. Synth. 29, 1949, p. 24; J. Am. Cliem. Soc.
70, 695 (1948).
(124J Пат. США 2461396 (1945); С., 1950, I, 2028.
[125] J. Aubry, BuH. Soc. chim. France [5], 5, 1333 (1938).
[126] Англ. пат. 282112 (1927); I. G. Farben. Ind.
I. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = С-СВЯЗЯМ
37
добавляют благородные металлы в виде их хлоридов, хлористоводородных кислот или
солей этих кислот [127].
Для повышения активности катализатора можно добавлять во время получения
сплава 3—10% хрома, молибдена или кобальта от массы никеля [128]. Гидрирование
в присутствии щелочей или органических оснований приводит к дальнейшему повыше-
нию активности.
Точно так же по методу Ренеи получают кобальтовые [129], железные [130]
и медные [130] катализаторы. Но область, их применения значительно уже области
применения никеля Ренея.
Для периодических гидрирований активный металл используют большей частью
в виде тонкого порошка. Для непрерывных процессов удобнее употреблять катализа-
тор в виде гранул, зерен илн кусков [131].
Металлические катализаторы. Никелевые катализаторы полу-
чают чаще всего в две стадии. Сначала готовят соответствующие соединения никеля,
например окислы, гидроокиси, основные карбонаты или ацетаты, формиаты или окса-
латы, иногда с добавками активаторов или носителей (так называемый эелепый кон-
такт), и затем подвергают их восстановлению.
Для приготовления ие содержащих серы катализаторов на нитрат никеля дей-
ствуют карбонатами, бикарбонатами или гидроокисями щелочных металлов. Образу-
ющиеся при осаждении соли щелочных металлов следует возможно полнее удалять
из продукта реакции. Затем гидроокись никеля- и основной карбонат никеля восста-
навливают, не переводя их в окись. Окись никеля чаще всего получают разложением
ацетата или нитрата никеля.
Формиат никеля разлагается при 200° С с образованием никеля. Для получения
очень мелкодисперсного пнвеля его формиат взвешивают в равном по весу количестве
парафинового масла и при перемешивании в токе двуокиси углерода или в токе водо-
рода нагревают до 260° С. Образовавшийся никель отфильтровывают, промывают
декантацией пстролейным эфиром и хранят под слоем последнего. При этом образуется
особо активный контактйый катализатор гидрирования под давлением.
Окись никеля можно восстанавливать [132] в трубке для сожжения в токе водо-
рода при 350° С. После того как масса полностью прореагировала, ее охлаждают,
продолжая пропускать водород. Затем .водород вытесняют током азота. Катализатор
вводят в гидрируемое вещество в атмосфере инертного газа. Свежсвосстановленныи
никель теряет на воздухе активность; поэтому если он не подлежит немедленному
использованию,,его хранит в инертной атмосфере, в вакууме, в метиловом или этило-
вом спирте, в бензоле.
Никелевый катализатор на кизельгуре [133]. В 80 мл дистиллированной
воды растворяют 58 а хексагидрата нитрата никеля и растирают с 50 г кизель-
гура (предварительно промытого соляной кислотой) в ступке в течение 30—
60 мин до гомогенизации смеси, которая приобретает ионсистенцию тяжелого
масла. Далее смесь медленно прибавляют н раствору 34 г моногидрата карбоната
аммония в 200 мл дистиллированной воды. Осадок отфильтровывают на воронке
Бюхнера, промывают двумя порциями воды по 100 мл каждая и сушат
при 110° С. Незадолго до употребления карбонат никеля восстанавливают 1 ч
в токе водорода при 450° С и скорости подачи водорода 10—15-мл/мин.
Катализаторы на носителях большей частью содержат 10—40% никеля. Нике-
.левые катализаторы обычно восстанавливают при 300—500° С; смешанные никелевые
[127] М. Deldpine, А. Н о г е a u, Bull. Soc. chim. France [5], 4, 31 (1937);
E. Lieber, J. Am. Chem. Soc., 72, 1190 (1950).
[128] R. Paul, Bull. Soc. chim. France [5], 13, 208 (1946).
[129] Пат. США 2166183 (1938); С., 1939, II, 3192; 628407; С. A., 44, 2675е (1950),
[130] L. Faucounau, Bull. Soc. chim. France [5], 4, 58 (1937).
[131] Пат. США 2094117; С., 1938, I, 4721; англ. пат. 628405; С. А., 44, 26758 (1950);
англ, пат. 628407; С. А., 44, 2675е (1950); англ. пат. 629405; С. А., 44, 44981 (1950).
[132] G. М. Schwab, L. Rudolph, Z. physik. Chem., Abt. В., 12, 427 (1931).
Д133] L. Covert, R. Connor, H. Adkins, J. Am. Chem. Soc., 54,
^651 (1932),
38
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
катализаторы, содержащие металлы более благородные, чем никель, можпо восстана-
вливать при более низких температурах. Так, сметанные медноникелевые катализа-
торы [134] восстанавливаются уже при 200—300° С.
Из катализаторов на носителях следует упомянуть никель на кизельгуре [135],
никель на пемзе [13о]. никель ла кизельгуре с окисью тория [137], никель на окистг
магния, бария или бериллия (138], никель на окиси алюминии [139] и никель на смеси
окислов цинка, бария и хрома [140]. Носителями никелевых катализаторов служат
также активный уголь, кремневая кислота, отбеливающая земля, каолин, пемза,
асбест, фуллерова земля, или же окислы, например, окись магния, окись алюминия
или боксит.
При мскрторых процессах гидрирования в гидрируемое вещество вводят «зеленый
контакт», причем сначала в большей или меньшой степени восстанавливается «зеленый
контакт» и лишь после этого начинается гидрирование вещества.
Аналогично никелю в качестве катализатора может использоваться также медь,
как в чистом виде, так и па посителяхжли в смеси с металлами всех групп периодиче-
ской системы элементов. Катализатор, содержащий медь, чаще всего в виде окиси,
гидрата окиси или основного карбоната, восстанавливается при 150—300° С. Медпый
катализатор на кизельгуре [141] получают так же, как никелевый катализатор на
кизельгуре.
Кобальтовые катализаторы получают восстановлением основного карбоната,
гидрата окиси плп окиси кобальта в токе водорода. Кобальтовый катализатор иа кизель-
гуре получен Хауком [142]. Из прочих кобальтовых катализаторов можно назвать-
кобальт на окиси бария и алюминия [143], кобальт на кизельгуре с окисью тория
[144] и кобальт на кизельгуре с окисью тория и магния [145].
Лангенбск [146] получал контактные катализаторы на основе смешанных
солей, восстанавливая последние в виде смешанных кристаллов в токе водорода.
Смешанные кристаллы удобны тем, что в них чрезвычайно тонко, вплоть до ионов,
распределены компоненты. Заряды и ионные радиусы катионов, а также химическая
природа анионов должпы быть идентичны. Для облегчения' разложения применяют
всегда оксалатные или формиатные ионы, обладающие восстановительными свойст-
вами. При разложении должпы образовываться не новые смешанные кристаллы, а два
разных вещества. Для этого один компонент разлагают не затрагивая другого или
разлагают оба компонента, но с образованием разных веществ. Так, например, при<
разложении смешанных кристаллов формиатов никеля и магния в токе водорода полу-
чают никелевый контактный катализатор на окиси магния. Исследованы системы сме-
шанных кристаллов оксалатов и формиатов кобальта и цинка, а также меди и кальция.
Контактные катализаторы на основе сметанных солей можпо приготовлять
также разложением комплексных солей типа А„[ВХт], где А — катиоп неблагород-
ного металла, а В — ион благородного металла в комплексном анионе [146]. В каче-
стве примера можно привести оксалатный комплекс Ы2 [Сц(С2О4)2]. Во всех случаях
образуются высокоактивные контактные катализаторы, обладающие максимальной
активностью при молярном соотношении компонентов 1 : 1, например оксалат никеля:
[134] G. Rienacker, R. Burman и, J. prakt. Chem., 2, 158, 10О
(1941).
[135] tlllmanns Enzyklopadie der Techn. Chemie, 2 AuIJ., Bd. 5, Berlin — Wien, 1932,
S. 171—172.
[136] BIOS Final Rep., 755, 14/15, I.* G. HSchst, герм. пат. 352439, по кнше-
Houben-Weyl, Bd. 4, T. 2, Stuttgart, 1955, S. 178.
[137] ГерМ. пат. 571898 (1930); С., 1933, I, 2899.
[138] Герм. пат. 307580 (1913); Frdl., 13, 174.
[139] CIOS Rep., 30, XXXII — 107, S. 118, I. G. Leuna, по книге Houbcn-
W e yl, Bd. 4, T. 2. Stuttgart, 1955, S. 180.
[140] Fiat Final Rep., 1313, I, 335/36.
[141] Герм. пат. 594481 (1930); С., 1934, II, 331.
[142] Пат. США 2166152 (1938); С., 1939, II, 3193.
[143] Герм. пат. 599103 (1932); Frdl., 21, 186; С., 1934, II, 4460; франц, пат. 761952;
С., 1934, II, 1022.
[144] F. Fischer, Н. Koch, Brennslofl-Cheni., 13, 62 (1932).
[145] CIOS Rep., 30, XXVII—69, 24/27, по книге Houben-Weyl, Bd. 4, Т. 2,
Stuttgart, 1955, S..185.
[146] W. Langenbeck, Angew. Chem., 68, 453 (1956).
I. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = С-СВЯЗЯМ
39
•океалат магния — 1:1. Активность их выше активности известных катализаторов
на основе того же металла.
Меднохромовые катализаторы (катализаторы Ад-
кинс в). Большое значение приобрели катализаторы на основе окисей меди и хрома
[147]. По Адкинсу, эти катализаторы получают не с переменным составом, а в виде
меднохромовои шпинели СиО • Сг2О3 (их часто называют меднохромитными катализа-
торами). В отличие от уже описанных катализаторов меднохромовый катализатор
представляет собой окисный катализатор, который не должен восстанавливаться
до металла даже в процессе гидрирования. Следует избегать восстановления черного
катализатора в красную неактивную форму. Окись хрома;зэщпщает окись меди от вос-
становления [148]; эту же функцию выполняют добавки окислов щелочноземельных
металлов [149].
В противоположность этой точке зрения Рабес и Шенк [150] полагают, что актив-
ными компонентами восстановленного катализатора являются металлическая медь
п активирующая окись хрома.
Вместо очень богатых хромом медных катализаторов часто применяют катализа-
торы с пониженным содержанием хрома. Однако такие катализаторы перед употре-
блением осторожно восстанавливают [151].
Адкинс с сотр. [152] получил высокоактивные меднохромовые катализаторы
разложением хромата меди и аммония при низкой температуре (100° С) и последующем
восстановлении в метиловом спирте в токе водорода при 100—200 ат.
Гидрирование в присутствии хромита меди протекает при повышенных давле
илях (от 40 до 200 ат) п температуре (100—280° С). Хромит меди по активности сравним
-с палладием, платиной или активным никелем.
Меднохромовые катализаторы можно получать путем тщательного механиче-
ского смешения окисей меди и хрома и термической обработкой таких смесей [153].
Лучшим лабораторным методом оказалось термическое разложение Аромата меди
-и аммонии.
Катализатор на основе окисей меди и хрома с окисью бария [153]. Вли-
вают 900 мл раствора, содержащего 260 г кристаллического нитрата меди
(Cu(NO8)2-3H2O) и 31 г нитрата бария, нагретого до 80° С, в 900 мл раствора,
который содержит 151'г бихромата аммония и 225 мл 28%-ного аммиака, при
•температуре 25° С. Образовавшийся осадок отфильтровывают на воронке Бюх-
нера, осадок на фильтре отжимают и отсасывают досуха. Продукт выдерживают
12 ч в сушильпом шкафу при 75—80° С и растирают. Его разделяют па три пор-
ции и подвергают каждую из пих термическому разложению в фарфоровой чашке
диаметром 15 сл, нагревая на голом пламени горелки. Разложение ведут при
постоянном перемешивании стальным шпателем, регулируя пламя так, чтобы
не происходило слишком бурного выделения газа. Разложение должно протекать
при возможно более'низкои температуре. Лучше всего нагревать одну сторону
чашки и усиленно перемешивать после начала распространения разложения
по всей массе. Во время разложения цвет порошка изменяется от оранжевого
через коричневый до черного. Когда масса приобретает однородный черйый
цвет и выделение газа ослабевает, порошку дают остыть. Все три порции соеди-
няют и обрабатывают 30 мин при перемешивании 600 мл 10%-пой уксусной кис-
лоты для растворения избыточной ,описи медп. Осадок отделяют на вороике
Бюхнера и промывают 6 раз водой порциями по 100 мл каждая. После высуши-
вания в течение 12 ч при 125° С катализатор измельчают. Выход составляет
170~г. Катализатор можно не восстанавливать перед употреблением.
[147] О. Schmidt, Вег., 64, 2051 (1931).
[148] Ch. Grundmann, в книге W. Foerst, Neuere Methoden der ргарага-
tiven organischen Chemie. 3 Aufl., Bd- 1. 1949, S. 118.
[149] R. -Connor, K. Folkers, H. Adkins, J. Am. Chem. Soc., 53, 2012
(1931).
[150] 1. Rahes, R. Schenk, Z. Eleklrochem., 52, 37 (1948).
1151] W. Normann, Z. Angew. Chem., 44, 714 (1931).
[152] H. Adkins, E. E. Bourgoyne, H. i. Schneider, J. Am. Chem.
Soc., 72, 2626 (1950).
.[153] R. Connor, K. Folkers, H. Adkins. J. Am. Chem. Soc., 54, 1138
(1932); H. Adkins., R. Connor, J. Am. Chem. Soc., 53, 1091 (1931).
40
ОБРАЗОВАНИЕ С —11-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Имеются и другие методы получения этого катализатора [154—155]. По данным
Ринера, при низких температурах разложения получаются более активные катализа-
торы, чем при высоких. Фрэйзер и Джексон [156] получали катализатор на основе'
окисей хрома и никеля состава 2NiO • Сг2О3, который опи, однако, восстанавливали'
перед употреблением. Зауер и Адкинс [157] описывают цинкхромитпый катализатор,
получение которого протекает аналогично получению меднохромитиого катализатора.
Разработан [158] способ приготовления катализатора на основе окиси цинка, хромита
кальция и серебра.
Сульфидные катализаторы. Сульфидные катализаторы в отли-
чие от чувствительных металлических катализаторов не отравляются серой. Поэтому
пх применяют в тех случаях, когда в гидрируемом веществе в виде примеси содержится
сера, для синтеза серусодержащпх органических соединений (например, тиолов) или
при деструктивном гидрировании серусодержащих органических соединений (на-
пример, гидрирование тиофена с отщеплением сероводорода). '
Катализаторы на основе сульфида железа илн кобальта [159]. Сульфид
железа, полученный осаждением из раствора соли железа раствором сульфида
аммония, обрабатывают в сухом виде сероводородом под давлением 10 am в те-
чение 3 ч и температуре 430° С. Сульфид кобальта обрабатывают сероводородом
12 ч при 200° С.
Для синтеза меркаптанов из альдегидов и сероводорода в условиях гидрирова-
ния прпгодея полисульфидный кобальтовый катализатор [160]. Его получают взаимо-
действием сплава кобальта и алюминия с сульфидом натрия.
Молибденсульфиднын катализатор [161]. К 36,5 в порошкообразного'
сплава молибдена с алюминием (50 : 50), суспендированного в 300 мл кипящей
воды, медленно прибавляют раствор 130 г серной кислоты в 100 мл воды. Суспен-
зию перемешивают 4 ч при 100° С. Продукт разложения промывают водой ДО1
нейтральной реакции и извлекают метиловым спиртом, после чего пропускают
сероводород до насыщения. Суспензию оставляют на две недели, затем снова
промывают катализатор метиловым спиртом. Катализатор хранят в метиловом,
спирте.
Аппаратура для гидрирования и методы гидрирования
Гидрирование со встряхиванием реакционной м а с-
с ы. Лабораторное гидрирование небольших количеств веществ проводят преимуще-
ственно при встряхивании реакционной массы и применяют катализаторы на основе-
благородных металлов или металлов Ренея. Форма реакционного сосуда не имеет реша-
ющего значения; это может быть грушевидная колба, утка или круглодонная колба.
Большое значение имеет быстрота встряхивания (интенсивность перемешивания).
Чаще всего прибор встряхивают в горизонтальном направлении, иногда в вертикаль-
1154] W. Lazier, Н. Arnold, пат. США 1964000; Org. Synth. 19, 1939, р. 31.
1155} Th. W. Л j е л е г, J. Am. Chem. Soc., 71, ИЗО (1949).
[156] J. C. W. Frazer, C. D. Jackson, J. Am. Chem. Soc., 58, 950'
(1936).
[157] J. Sauer, H. Adkins, J. Am. Chem. Soc., 59,1 (1937); H. II. Store h,.
J. phys. Chem., 32, 1744 (1928).
[158] Ch. Griindmann, Angew. Chem., 62, 558 (1950),
[159] Пат. США 2127383 (1936); С., 1939, I, 5110; франц, пат. 728913; С.,
1933, I, 545.
fl60] М. W. Farlow, W. A. L а г i е г, F. К. Sign algo, Ind. Eng. Chem.,
42, 2577 (1950). *
[161] Пат. США 2402613 (1939); С., 1947, 385.
J. ПРИСОЕДИНЕНИЕ ПОДОРОДА ПО С = С-СВЯЗПМ
41
ном [162]. Для работ при нормальном давлении в большинстве случаев достаточно
пользоваться установкой, изображенной па рис. 1.
Цилиндрический сосуд 6 (емкость между крайними делениями 1000 мл),
снабженный кранами 1 и 2, соединяют шлапгом с напорным резервуаром 7
для выравнивания давления в приборе. Цилиндр 6 может быть и большей емкости
и снабжен градуировкой, отвечающей поставленной задаче. Для градуировки
прибора в большинстве случаев достаточно заполнить его водой и отливать из
него по 50 мл иолы в мерный цилиндр. Дальнейшую градуировку по 10 мл можно
провести графически. Большей точности намерения для препаративных целей
не требуется. Перед началом опыта, поднимая резервуар 7, заполняют цилиндр 6
водой до горизонтальной части Т-образной трубки. Открыв краны 1—5, вытес-
_ ияют воздух йз прибора водородом. Последовательно закрывают краны 4—2,
опускают резервуар 7 и заполняют водородом цилиндр 6 при'незначительном
избыточном давлении до нижней отметки. После этого закрывают краны 1 и 5.
Для того чтобы установить опять
в системе атмосферное давление, /2 3 U 4
можно открыть краны 2 л 3 и вы- / / / /
ПуСТИТЬ ИзбЫТОК ВОДОрОДа, ОТКрЫВ т г-fe j?
на короткое время кран 4. Затем / ir**—
открывают кран 5, поднимают резер-
вуар 7 на 10—20 см выше уровня
воды в цилиндре и, встряхивая реак-
ционный сосуд#, начинают реакцию.
После того как запас водорода в ци-
линдре 6 использован, закрывают
-краны 2 и 3, прекращают встряхи-
вание, замечают уровень воды в ци-
линдре после установления атмо-
сферного давления с помощью со-
суда 7 и заполняют цилиндр 6 свежпм
водородом. Если для большего
хотят заполнить цилиндр 6
до начальной метки, то в этом слу-
чае кран 2 делают трехходовым
и после установления уровня воды на начальной отметке закрывают краны 1 и S
и выпускают избыток водорода через третий канал Крана 2, сообщающийся
с атмосферой. По другому варианту после закрывания крана 1 устанавли-
вают в цилиндре 6 с помощью резервуара 7 атмосферное давление и замечают
уровень воды. Записывают объем израсходованного водорода.
Рис. 1. Установка для'
гидрирования при
нормально^ давле-
нии;
1 —5 — краны; в — цилин-
прический сосуд; 7 —на-
порный резервуар; 8 —
реакционный сосуд.
Следует быть осторожным при введении в соприкосновение катализатора с водо-
родом. Некоторые окисные или гидроокисные катализаторы следует восстанавливать
под слоем защитной жидкости, так как они так сильно нагреваются при взаимодейст-
вии с сухим водородом, что при вытеснении воздуха из сосуда для гидрирования может
произойти взрыв гремучего газа. Из этих соображений контактный катализатор вос-
станавливают в том растворителе, и котором будет проводиться реакция, или покры-
вают его слоем эфира. Не встряхивая, вытесняют цз прибора воздух водородом, после
чего встряхивают до окончания восстановления. Затем водородом вытесняют эфир и,
не прерывая тока водорода, приливают в реакционный сосуд через воронку гидри-
руемый раствор.
При работе с окисными или гидроокисными катализаторами всегда образуется
некоторое количество поды. Еслп требуется добавить свежий катализатор или регене-
рировать кислородом старый, необходимо предварительно удалить водород вакууми-
рованием прибора. Перед введением водорода во избежание взрыва гремучего газа
нужно точно так же удалить кислород.
Для работ с большими загрузками при умеренном избыточном давлении (до
3 am} можно использовать аппарат, изображенный на рис. 2.'
1162] Е. В. Maxted, в справочнике Handbuch der Katalyse, Bd. 7, T. I, Springer-
Verlag, Wien, 1943, S. 654; E. B. Maxted, Trans. Farad. Soc., 13, 36
(1917).
42
ОБРАЗОВАНИЕ С —Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Аппарат рассчнтап на избыточное давление 6 ат п представляет собой-
стальной сосуд 4, емкостью 20 л, снабженный манометром 5 и тремя вентилями,
Гофера 1—3. Водород из баллона подастся по латунной трубке пли прочному
армированному резиновому шлангу.
При проведении первого опыта промывают предварительно водородом вос-
соединительные трубопроводы, а затем, закрыв вентиль 3, через вентили 1 и 2
Рис. 2. Аппарат для гид-
рирования при умеренном
давлении:
1—3 — вентили Гофера; 4 —
стальной сосуд; 5 — манометр.
откачивают масляным насосом воздух из сосуда и впускают водород, доводя
избыточное давление до 3 ат. Герметичность сосуда проверяют по манометру,
после чего открывают вентиль 3 п соединяют сосуд 4 с реакционным аппаратом.
В качестве реакторов применяют проверенные под давлением толстостенные
сосуды, которые закрыты специально закрепляемыми резиновыми пробками,
чтобы их не выбивало при повышенном давлении. Для
присоединения реактора пользуются толстостенной
высококачественной резиновой трубкой для работ при
повышенных давлениях.
Резиновые трубки и пробки кипятят в не-
скольких порциях 20%-ного раствора едкого натра
до прекращения окрашивания его в желтый цвет,
затем несколько раз тщательно промывают горячей'
дистиллированной водой.
После присоединения реакционного сосуда -за-
крывают вентиль!, открывают вентили 2 и 3 и отсасы-
вают воздух. Затем закрывают вентиль 2, открывают
вентили 1 и 3, отмечают давление и встряхивают реак-
тор в течение 6—8 ч. Если за это время показание-
манометра пе изменилось более чем па 0,02—0,03 ат,
аппаратуру можно считать готовой к работе. Прово-
дят восстановление эталонного вещества, записывают
изменение давления и определяют таким образом
перепад давлений, соответствующий поглощению -
1 моль водорода при данной температуре.
В качестве эталонного вещества используют
малеиновую кислоту; раствор 0,1 моль (11,6 г) кислоты
в 150 мл обычного спирта, к которому прибавляют
Если установка герметична и реактивы соответствуют*
0,1 г окиси платины. ___ „________ _.г_______ „ _________...
предъявляемым к ним требованиям, то от начала встряхивания до конца погло-
щения водорода проходит 20—30 мин.
Гидрирование при высоком давлении. Хотя большин-
ство процессов гидрирования можно вести при нормальном давлении в обычной аппа-
ратуре при встряхивании, иногда все же требуются более высокие давления. Для
гидрирования ароматических колец лучше применять давление 100—150 ат. Для вос-
становления сложных эфиров и амидов карбоновых кислот на меднохромовом катали-
заторе необходимы давления до 300 ат. В некоторых случаях, как, например, при гид-
рированиях с сульфидными катализаторами, могут потребоваться давления до 700—
1000 am. Все другие процессы гидрирования можно вести при давлениях от 10
до 100 ат.
Лабораторный автоклав для работ при высоких давлениях может быть заполнен
водородом из баллона с максимальным избыточным давлением 125 ат с доведением/
в нем давления до 100 ат при комнатной температуре. Это соответствует приблизи-
тельно избыточному давлению 175 ат при 250° С. Обычно давление водорода от 40*
До 200 ат создают подачей холодного газа. Для достижения более высоких давлений-
водорода пользуются компрессором. О протекании гидрирования можно судить по сни-
жению давления. С помощью графического изображения зависимости объема от дав-
ления и температуры [162а] можно на основании данных по давлению и температуре-
определять количество введенного или содержащегося в автоклаве водорода во время-
реакции.
Поскольку при повышенных давлениях водород лучше растворяется в гидрируе-
мых растворах или веществах, то уже не требуется столь интенсивное встряхивание^
[162а] Houben-Weyl, Mcthoden der organischen Chemie, Georg Thieme Verlag,.
Bd. 4, T. 2, Stuttgart, 1955, S. 260.
I. ПРИСОЙДИНЕНИЕ ВОДОРОДА ио с=с-связям
43
Рис. 3. Установка для восстановления катализа-
торов:
1 — электрическая печь; 2 ~ фарфоровая труба; 3 и 5 — про-
мывалки; 4 — приемник для конденсирующейся ноды.
как при нормальном давлении. Поэтому часто бывает достаточно проводить реакцию
в качающемся автоклаве, который проще в обращении и требует меньше ухода, чем
автоклав с мешалкой. 1
Конструкционный материал автоклава должен быть инертным и не должен
окалывать отрицательного влияния на ход реакции. Этого достигают футеровкой вну-
тренней части автоклава медью, серебром илн другими подходящими материалами
пли изготовляют автоклав из хромоникелевых сплавов.
Автоклав для гидрирования должен быть снабжен манометром, предохрани-
тельным вентилем и вентилем, через который во время опыта можно подавать допол-
нительное количество водорода. Температуру внутри автоклава измеряют при помощи
введенных в него или прочпо встроенных термоэлемента или защищенного трубкой
термометра.
Проводя исследования при повышенных давлениях следует руководствоваться
правилами техники безопасности, предусмотренными для работы в автоклавах опре-
деленных габаритов. Автоклавы должны находиться под постоянным наблюдением”
и для работы с пцми надо
атметь специальное разрешение.
После внесения гидри-
руемого вещества и катализа-
тора гидрирования автоклав
закрывают и промывают не-
сколько раз азотом, впуская
его под давлением и спуская
давление. Моткво удалять воз-
дух и при помощи вакуумного
насоса. Для испытания авто-
клава па герметичность его
оставляют на некоторое время
под давлением. После снятия
давления автоклав заполняют
водородом. Начальное давле-
ние выбирают таким, чтобы при нагревании аппарата оно пс превысило допустимого
предела. После окончания гидрирования автоклав охлаждают, а затем, открывая
вентиль, доводят давление до атмосферного. Только после этого можно открыть
автоклав. При разгрузке надо иметь в виду, что некоторые катализаторы, как,
например, никель Ренея, пирофорны. Поэтому даже при разгрузке они должны
постоянно находиться под слоем жидкости. Иногда для этой цели применяют вкла-
дыши из толстостенного стекла, но они не соответствуют полностью своему назначе-
нию, если вещество во время реакции испаряется.
Гидрирование в' контактной трубке. Газообразные' или
легко испаряющиеся вещества можно гидрировать в газовой фазе над никелевыми или
медными катализаторами на носителях. Для этого обычно используют проточную уста-
новку с вертикально расположенной контактной трубкой из тугоплавкого стекла, запол-
ненной гранулированным или волокнистым катализатором. Можпо применять трубки
с наружным олектрообогрево.ц. Внутри слоя катализатора должен находиться датчик
температуры. Защитная трубка для термопары, проходящая через весь слой катали-
затора, позволяет передвигать термоэлемент и замерять температуру по всей высоте
контактной трубки. При использовании нагревательных бань применимы U-образные
или подобные им контактные трубки, у которых впуск и выпуск газа нахо-
дятся вверху.
Катализаторы из окислов металлов тоже восстанавливают в трубках из туго-
плавкого стекла, кварца или фарфора, с внутренним диаметром 3—6 см, располагая
их под небольшим наклоном (рис. 3).
Температуру внутри трубки измеряют подвижной термопарой. Передняя зона
в трубке должна быть пустой, чтобы водород достигал катализатора при требуемой
температуре. Реакцию контролируют по изменению цвета катализатора или ио обра-
зованию воды. Если восстанавливаемый катализатор не должен соприкасаться с воз-
духом, то водород после охлаждения вытесняют чистым азотом или двуокисью
углерода, не содержащей серы. После этого катализатор можно сразу внести в вос-
станавливаемую жидкость или хранить в атмосфере инертного газа или под слоем
лгнертной жидкости.
ОБРАЗОВАНИЕ С—Н-СВЯЗЪ'Й В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Каталитическое гидрирование
Выбор катализатора. Несмотря па то что применимых ко всем слу-
чаям правил для выбора подходящих катализаторов гидрирования ненасыщенных со-
единений разных типов не существует, в некоторых случаях оправдывается применение
определенных типов катализаторов.
Для гидрирования газообразных и легколетучих олефинов в газовой фазе при-
меняют платину, палладий и никель. Жидкие олефины, ненасыщенные спирты, цикло-
олефины и их производные можно гидрировать при встряхивании с катализаторами
па основе благородных металлов (в спирте, ледяной уксусной кислоте или этилацс-
тате), с никелем Ренея или в автоклаве с никелевым катализатором. Ненасыщенные
кислоты можно гидрировать в виде их эфиров или нутриевых солей (низкомолекуляр-
ные кислоты) в водных пли водно-спиртовых растворах или свободными в расплаве
(кислоты от Се до С22) с помощью никелевых катализаторов. Для отой же цели при-
годны и катализаторы на основе благородных металлов.
_ Для гидрирования ненасыщенных боковых цепей ароматических соединений
можно применять никель Ренея или платипу в жидкой фазе, или медные катализаторы
в газовой фазе. Вследствие различных скоростей гидрирования олефиновых и аромати-
ческих двойных связей в большинстве случаев достигается избирательное насыщение
боковой цепи.
Для избирательного насыщения одной пз нескольких двойных связей можно
применять палладий или никель Ренея. Платина оказывает менее избирательное дей-
ствие, по иногда можно применять и ее. После поглощения стехиометрического коли-
чества водорода гидрирование прерывают. При гидрировании ароматических .колец
в присутствии металлов платиновой группы в реакционной среде ле должно содер-
жаться даже следов серы. Производные бензола, как, например, бензойная кислота
Л фенол, а также нафталин гидрируются легче, чем бензол. Но часто для этих соедине-
ний оправдывается гидрирование на никелевом катализаторе в автоклаве.
Гидрирование по олефиновым С=С-с вязям. Для гидриро-
вания по олефиновым двойным связям обычно не требуются особо активные катализа-
торы. По данным Шустера [163], скорость гидрирования уменьшается с увеличением
длины цепи. Гидрирование по двойным связям на концах цени идет быстрее, чем по
двойным связям в середине цепи. В свою очередь, такие соединения реагируют быстрее,
чем соединения с двойными связями, расположенными в местах разветвлений цепи
[164). Так, например, гидрирование по этиленовой двойной связи в трифенилэтилене
в присутствии катализаторов из благородных металлов протекает в десять раз медлен-
нее, чем гидрирование по двойной связи в стироле. Тетрафенилэтплен можно гидриро-
вать [165] только при 100° С и давлении 125 ат. Группировки R2C=CR2 и R2C=CHR
обычно не гидрируются в присутствии никеля Ренея, в то время как группировка
R2C-=CH2 еще может быть лрогидрировапа. Таким образом, этилен гидрируется легче,
если он менее замещен (правило Лебедева). Если в одной молекуле имеется несколько
групп, которые могут быть восстановлены каталитически возбужденным водородом,
то, как правило, гидрирование протекает в такой последовательности: гидрирование
по двойной связи и восстановление нитрогруппы происходит почти одновременно,
затем следуют восстановление карбонильной группы, циангруппы, гидроксильной-
группы, ароматической системы, связей гетероциклического ядра и очепь устойчивой
карбоксильной группы. ^ис-Соединения каталитически гидрируются легче, чем транс-
соединения. В одинаковых условиях малеиновая кислота гидрируется в три раза
быстрее фумаровой [166]. Из пары ^uc-коричпой и трянс-коричнои кпелот первая
гидрируется в два раза быстрее [166]. Это правило сформулировано Паалем и Шидевпт-
Цем [166|, однако, по данным Вейганда, Вернера и Ланцендорфа [167], существует
много исключении. Так, пгрдне-дибензоидэтилены гидрируются заметно быстрее, чем
изомерные им уис-формьк
[163] С. Schuster, Z. Elektrochem., 38, 614 (1932)<
[164] G. Vavon, D. Ivanoff, С., 177, 453 (1923).
1165] W. H. Zartman, H. Adkins, J. Am. Chem. Soc., 54, 1668
(1932).
[166] C. Pa al, H. Schledewitz, Ber., 60, 1221 (1927); 63, 766 (1930).
1167] C. W e у g a n d, A. Werner, W. Lanzendorf, J. prakt. Chem.,
151, 231 (1938). ‘
I. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С=С-СВЯЗЯМ
45
При каталитическом гидрировании наблюдается только уис-присоединение
водорода.
Избирательное гидрирование. При каталитическом гидриро-
вании бензола можно получить только один продукт реакции — циклогексан, так
как промежуточно образующиеся циклогексадиен и циклогексен выделить не удается
из-за того, что оба эти соединения гидрируются гораздо быстрее, чем ароматическая
система бензола. Гидрирование по неароматическим сопряженным двойным связям
протекает совершенно иначе. По данным Рихе, Гримма и Альбрехта [168], при гид-
рировании бутадиена-1.3 в присутствии палладиевого катализатора наблюдается
ступенчатое насыщение двойных связей. До тех пор пока не прогидрируется весь бута-
диен, образуются только бутилены, но не бутан. Лишь после этого бутилены гидри-
руются до бутана. В противоположность другим реакциям присоединения при катали-
тическом гидрировании бутадиена-1,3 протекает преимущественно 1,2-присоединение.
Частичное гидрирование кумуленов в присутствии катализатора Линдлара
[палладиевый катализатор на карбонате кальция, дезактивированный ацетатом'
свинца (стр, 33)} проходит избирательно с образованием цис-полиенов. После быст-
рого поглощения (п — 1)/2 моль водорода (п — число двойных связей) скорость гидриро-
вания становится практически равной пулю. По Куну и Фишеру [106], из тетрафенил-
бутатриена при поглощении 1 моль водорода образуется 1,1,4,4-тетрафенилбутадиен-1,Зг
R\ ZR R\ /R
>c=c=c-=c< —> >с=сн—сн=с<
Rz XR RZ 4R
1,1,4,4-Тетрафеннлбутадиен-1,3. К насыщенной водородом суспензии
800 мг катализатора Линдлара в 50 мл перегнанного над едким кали тетрагидро-
фурана прибавляют 500 мг тетрафеннлбутатриена и встряхивают в атмосфере
водорода при 21° С. Через 15 мин поглощается 35 мл (при нормальных условиях
31,5 мл) водорода, что соответствует поглощению 1 моль водорода. При
дальнейшем встряхивании в течение 1 ч поглощается только- 3 мл водорода.
После фильтрации отгоняют растворитель. Остаток кристаллизуют из цикло-
гексана и получают 420 з (84% от теоретического) продукта в виде бесцветных
.ромбов; т. пл. 201° С.
Тетрафенилгексапентаеп поглощает 2 моль водорода с образовапиом соответ-
ствующею цис-полиена;
R\ zR „ Rx zR
\з=с=с=с=с=с< >с=сн-сн=сн-сн=с<
Rz \R Rz \R
Бутадиен и пентадиен удалось -прогидрировать в паровой фазе до соответству-
ющих олефинов над никелем на хромите цинка [169].
Если сопряженные двойные связи вследствие различного замещения не равно-
значны, то замещенные двойные связи гидрпруются с большим трудом, чем незамещен-
ные [170]. В качестве примера можно привести гидрирование винилакриловой кислоты
в пентеновую кислоту:
СН2=СНСН=СНСООН —► сн3сн2сн=снсоон
Пентеновая кислота. В аппарате для гидрирования (см. рис. 2) раство-
ряют 29,5 г винилакриловой кислоты в 100 мл очищенного метилового спирта.
Добавляют 0,25 г катализатора Адамса и при встряхивании пропускают'
водород. После поглощения 1 моль водорода (0,602 г), на что требуется 20ли«,
гидрирование прекращают, катализатор отфильтровывают и продукт отделяют
перегонкой. Пентеновая нислота перегоняется при 93® С (15 мм рпг. ст.).
[168] A. Rieche, A. Grimm, Н. Albrecht, Brennstoff-Chem., 42. 177
(1961).
[169] G. N a t t a, R. R igainont i, P. Tono, Chimie Ind. (Milano), 29, 235
(1947).
[170] I, E, Musk a I, B. Knapp, Ber„ 64, 779 (1931).
46
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Аналогично протекает гидрирование цис-февнлбутадвена. в цис-фонялбутм-
леп [170]:
свп5-сн „ С6НБ~СН
II —и
СН2=СН-СН СНз-СН8-СН
Гидрирование изомерного mpasc-соедияения приводит к соответствующему
транс-фепилоутилену.
Получение цис- или тмнс-фенилбутилеаов. В аппарат Для гидриро-
вания помещают раствор 17,2 г соответствующего изомера фснилбутадиена
в 100 мл ледяной уксусной кислоты и добавляют 0,15 г катализатора Адамса.
После поглощения при встряхивании 1 моль водорода катализатор отфильтро-
вывают и продукт реакции перегоняют. ч«с-Фепилбутилеп отгоняется при 13 —
74,5° С (12 мм pm. cm.), транс-изомер — при 76—78° С (12 мм pm. cm.). Выход
поччи количественный.
При гидрировании цитраля
/О
(СН3)2С=СН—(СП3)2—С(СН3)=СН-С/
\ц
на катализаторе из окиси платины и платиновой черни в первую очередь гидрирова-
ние происходит по соседней с альдегидной группой С=С-связи, затем гидрируется
альдегидная группа и только после этого происходит гидрирование другой С=С-свлзи.
Но если применять этот катализатор с добавками сульфата железа (II) и ацетата цинка,
то сначала гидрируется альдегидная группа и после поглощения 1 моль водорода полу-
чается главным образом гераниол
(СНз)2С=СН—(CH8)s-C(CH3)=CH-CHaOH
После поглощения еще 1 моль водорода образуется цитронеллол
(СНз)3С=СН-(СН2)2-СН(СНз)-СНа-СНгОН
Дальнейшее гидрирование приводит к тетрагидрогераниолу [171].
Аналогичным образом из коричного альдегида в присутствии платиновой черни
с добавкой 0,01—0,02 ммоль хлорида железа (II) был получен коричный спирт [172].
При дальнейшем гидрировании образуется фенилпропиловый спирт. Но если добавить ;
к катализатору сульфат железа (II) и ацетат цинка, то после поглощения 1 моль водо-
рода гидрирование прекращается и получается коричный спирт, практически яс содер-
жащий альдегида. I
Аналогичное явление наблюдал Адамс с сотр. [173] при гидрировании а-фурил-
акролсина на платиновом катализаторе. В присутствии сульфата железа (II) и аце- ,=!
тата цинка при гидрировании сначала сохраняется двойная связь и получается ;
фурилаллиловый спирт, который затем поглощает еще 1 моль водорода и переходит
в фурпл-л-пропи.товый спирт. В присутствии только сульфата железа (II) восставав- I
ливастся главным образом фурановое кольцо и подучается тетрах’идрофурилйллиловый
спирт, который затем восстаналливается до тетрагидрофурил-я-пропилового спирта.
В присутствии ацетата ципка фурилаллиловыи спирт далее не гидрируется. j
При гидрировании ненасыщенных кислот обычно предпочтительно насыщается
двойная связь, Так, кротоновая кислота гидрируется в присутствии катализатора |
Адамса с образованием масляной кислоты [174J. Аналогично ундеценовая кислота |
гидрируется в присутствии окиси платины до ундекановой [175]. Сорбиновая кислота |
гиттрируртея в присутствии никеля Ренея или, лучше, палладия при комнатной тем-
пературе и нормальном давлении с образованием a-гексеновой кислоты, которая содер-
[171] R. Adams, В. S. Garvey, J. Am. Chem. Soc., 48, 477 (1926). |
[172] W, F. T u 1 e у, R. Adams, J . Am. Chem. Soc., 47, 3061 (1925). 1
[173] R. H. Bray, R. Adams, J. Am. Chem. Soc., 49, 2101 (1927). / ’I
[174] E. В. M a x t e d, V. Stone, J. Chem. Soc., 1934, 26.
[175] J. W. Kern, R. L. 8 hrine r,. R. Adams, J. Am. Chem. Soc., 47, A
1147 (1923). * 'Z
I ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С=С-СВЯЗЯМ
47
жит некоторое количество р-геисеиовой, сорбиновой и капроновой кислот [176].
Как и в случае випплакриловой кислоты (стр. 45), в молекуле сорбиновой кислоты
двойная связь, смежная с карбоксильной группой, гидрируется более медленно. Вой-
цик и Адкинс [177] восстанавливали в присутствии никеля Ренея (10—15% от веса
эфира) при комнатной температуре п 100—200 ат эфиры а-алкилиден-р-кетокислот,
которые они синтезировали пз альдегидов и этилового эфира ацетоуксусной кислоты'
или диэтилового эфира малоновой кислоты (отщепление воды), и получали эфиры
а-алкил-р-котокислот с выходами выше 90%; в качестве растворителя они использо-
вали эфир. Хорнинг с сотр. [178] применил палладий на угле для гидрирования эти-
леновой двойной связи в этиловом эфире а-ацетил-р-(2,3-диметоксифенил)-акриловой
кислоты, который получали из 2,3-диметоксибензальдегида н ацетоуксусного эфира
с отщеплением воды.
Если в качестве катализатора гидрирования использовать платину илн палла-
дий, то можно прогидрировать и сами кислоты. При употреблении никеля целесо-
образнее гидрировать эфиры. Аллен с сотр. [179] гидрировал соли некоторых
ненасыщенных кислот при высоких давлениях.
Янтарная кислота. В автоклав емкостью 325 мл помещают 100 мл вод-
ного раствора натриевой соли малеиновой кислоты, которая была получена рас-
творением 58 а малеиновой кислоты в 100 мл раствора едкого натра, содержащего'
на 5 а больше щелочи, чем это нужно для нейтрализации. Прибавляют 5—8 а
никеля Ренея [180], затем подают водород до начального давления 170 ат и гид-
рируют при 100° С, поддерживая эту температуру до окончания гидрирования.
Давление в процессе реакции начинает заметно падать. Примерно через 26 мин
падение давления прекращается, после чего отфильтровывают катализатор
(осторожно, катализатор пирофорен!). Реакционную массу нейтрализуют и уда-
ляют в вакууме воду. Янтарную кислоту экстрагируют абсолютным спиртом
и отгоняют спирт в вакууме. Выход янтарной кислоты 57 £ (98% от теоретиче-
ского). После перекристаллизации из воды кислота плавится при 184—185° С.
В описанных до сих пор процессах гидрирования ненасыщенных кислот или1
их эфиров карбоксильная группа не восстанавливалась. Зауеру и Адкинсу [157]
удалось осуществить гидрирование эфиров ненасыщенных кислот до ненасыщенных
спиртов па ципкхромптном катализаторе из окиси циика и окиси хрома. Ненасыщен-
ные спирты можно затем прогидрировать в обычных для соответствующих олефинов
условиях, например в присутствии платины или палладия в аппарате для встряхива-
ния, до насыщенных спиртов. При более высоких температурах в результате отщепле-
ния воды из этих спиртов могут образоваться соответствующие углеводороды.
Гидрирование ароматических соединений. Хотя для
гидрирования ароматических систем требуются более жесткие условия чем для гидри-
рования по олефиновым С=С-связям, трудности проведения таких процессов раньше'
переоценивали. Во всяком случае, ароматические системы гидрируются после всех дру-
гих ненасыщенных центров, за исключением гидроксильных и карбоксильных групп.
Применяя современные катализаторы, например окись платины, реакцию гидриро-
вания можно провести уже при комнатной температуре.
Циклогексан [181]. Гидрируют 15,6 г бензола в ледяной уксусной
кислоте при комнатной температуре и давлении водорода 2—3 ат в течение 2 ч<
над 0,2 г окиси платины. Выход циклогексана количественный. -
1176] Е. П. Farmer, L. A. Hughes, J. Chem. Soc., 1934, 1929.
[177] В. Wojcik, Н. Adkins, I. Аш. Chem. Soc., 56, 2424 (1934).
[178] E. C. Horning, J. Koo, M. S. Fish, G. N. Walker, Org. Synth..
31, 56 (1951).
[179] В. B. Allen, B. W. Wyatt П. R. Henze, J. Аш. Chem. Soc., 61,
843 (1939).
[180] W. Covert, H. Adkins, J. Am. Chem. Soc., 54, 4116 (1932).
1181] R. Adams, J. R. Marshall, J. Am. Chem. Soc., 50, 1970 (1928).
48
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Только совершенно не содержащие серы ароматические соединения гидриру-
ются в присутствии металлов платиновой группы при низких температурах. Виль-
штеттер и Хатт [182] количественно прогидрпровалп при комиатпоц температуре и
нормальном давлении 12 раз перекристаллизованный из уксусной кислоты нафталин
в декалпп. При гидрировании нафталина над никелевым катализатором при темпера-
туре около 200° С Шретер [163] получил при 40 am декалин, а при 15 am — тетралин.
Нафталин, так же как бензойная кислота, фенол и другие производные бензола,
гидрируется легче, чем бензол.
Тетралин. Используют нафталин, не содержащий сернистых примесей,
отравляющих катализатор. Успешным способом очистки оказалось многократное
сплавление нафталппа с натрием и последующая вакуумная перегонка. В авто-
клаве с мешалкой емкостью 4 л сплавляют 512 г нафталина с 15—20 а подходящего
соединения никеля (основной карбонат, окись), закрывают автоклав и подают
чистый водород до избыточного давления 12—15 am, проверяют герметичность
автоклава и при' перемешивании повышают температуру до 180—200° С. Давле-
ние сначала повышается, а затем падает (на 1 am за 45—60 сек). Когда давление
снизится до 5—8 am, вновь подают водород до тех пор, пока не поглотится не-
обходимое количество водорода, равное 178 л. Этот момепт распознают также
по уменьшению скорости поглощения водорода. К концу реакции прекращают
подачу водорода и продолжают процесс до почти полного поглощения водорода.
Опыт продолжается 1 ч или несколько больше. Образовавшийся тетралин можно
отогнать из автоклава в вакууме и, используя тот же катализатор, прогидриро-
вать следующую порцию нафталина. Шретер проводил до 25 гидрирований, не
открывая автоклава. Новые порции расплавленного нафталина впускали в авто-
клав, не изменяя разрежения.
9,10-Дигидрофенантрен [184]. Смешивают 40 г фенаитрепа со 100 мл
этилового спирта и гидрируют водородом при давлении 150—200 am и темпера-
туре 150° С в присутствии меднохромового катализатора. Реакция продолжается
около 3 ч. После гидрирования реакционную массу разгоняют на колонке Вид-
мера, собирая фракцию, отгоняющуюся при 176—178° С (20 мм pm. cm.). Выход
дпгидрофенантрена 87% от теоретического. После перекристаллизации из мети-
лового спирта получают дигидрофенантрен, плавящийся при 33,8—34,4° С.
Дальнейшее гидрирование дигидрофенантрепа протекает только при температу-
рах на 50—70° С выше температуры гидрирования фенантрена. Поэтому для
исчерпывающего гидрирования фенантрена лучше применять никелевые кон-
тактные катализаторы.
Примером гидрирования ароматического соединения в присутствии окиси
платины может служить получение гексагидро-п-амипобензойной кислоты [185].
Преимуществом этого процесса является возможность ведения его в водной суспензии
гидрируемого вещества без органических растворителей.
Гексагидро-л-аминобензойная кислота. В реакционном сосуде для гидри-
рования при встряхивании суспендируют 10 г дважды перекристаллизованной
из воды п-аминобензойной кислоты в 800 мл воды и прибавляют 0,5 а свежепри-
готовленной окиси платины. Гидрирование продолжается 14,5 ч; количество
поглощенного водорода несколько превышает расчетное. Коцец реакции лучше
всего распознается по осаждению катализатора в виде хлопьев. После оконча-
ния гидрирования катализатор отфильтровывают, фильтрат упаривают досуха
и перекристаллизовывают коричневый кристаллический осадок из небольшого
количества воды с добавлением активного угля. Для полноты осаждения кри-
сталлов добавляют метиловой спирт. Выход составляет 6—7 а (60—70% от тео-
[182] R Willstatter, D. Hatt, Вег., 45, 1471 (1912).
[1831 G. Schroeter, Ann., 426, 1 (1922); L. P а 11 г а у, С. г., 206, 1976 (1938);
М. Pier, Z. Elektrochem., 53, 297 (1949).
[1841 J. R. Durland H. Adkins, J. Am. Chem. Soc., 59 135 (1937);
A, Burger, E-Mosettig, J. Am. Chem. Soc., 58, 1857 (1936).
[185] Dr. R. Gabler, частное сообщение.
I. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С-С-СВ ЯЗЯМ
49
ретического). Бесцветные кристаллы гексагидро-л-аминобензойной кислоты
содержат 0,5 моль кристаллизационной воды и плавятся в запаянной трубке
при 275—280° С. По литературным данным температура плавления кислоты
может лежать в пределах от 260 до 330° С.
Можно исходить также из пптросоедпненпй и гидрировать их в водной среде
до соответствующих аминокислот в одну стадию.
Гексагидробензойная кислота [186]. Гидрируют смесь 50 г бензоата
Натрия в 75 лы воды в присутствии 7 а окиси никеля при начальном давлении
водорода 92 ат и температуре 300° С. Получается гексагидробензойная кислота
(т. кип. 235—238° С) с выходом 40% от теоретического и 9 г циклогексана.
При гидрировании ароматических оксикислот чаще всего происходит декарбок-
силирование. Соли могут быть только частично насыщены без декарбоксилирования.
Ипатьев и Разуваев [187] получили 40 г циклогексапола при гидрировании 65 г мети-
лового эфира салициловой кислоты в 65 г метилового спирта в присутствии
окиси никеля.
Гидрирование фенола также приводит к циклогексанолу. Гидрирование можно
проводить в газовой фазе [188] или под давлением в присутствии никелевого катали-
затора [189].’
Из резорцина под действием никеля Ренея или никелевого катализатора на
окиси кремния при 50Q С лод давлением в течение 10—12 ч получается дигидрорезор-
цип с выходом 85—95% от теоретического [190]. Греве, Нольте и Ротцолль [191].
использовали для гидрирования резорцина обработанный щелочами нииель Ренея.
Упрощенный способ получения дигидрорезорципа [192] заключается в гидрировании,
резорцина в присутствии свежеактивированного никеля Ренея при атмосферном давле-
нии п температуре 50Q С; реакция продолжалась 24 ч, выход дигидрорезорцииа соста-
вил 98% от теоретического.
При гидрировании 3,5-дпоксибензойпой кислоты в присутствии никеля Ренея
W-1 получается циклогександион-3.5-карбоповая кислота [193]. Гидрирование 3-этил-
фенола над никелем Ренея приводит к 3-этилцнклогексанолу [194].
р-Нафтол можно гидрировать избирательно, насыщая фенольное или бензоль-
ное кольцо. При ведении процесса с катализатором никелем Ренея W-4 при 65аС
в присутствии едкого натра в течение 1,5 ч происходит насыщение фенольного кольца,,
и с выходом &5% от теоретического получается 1,2,3,4-тетрагидронафтол-2. Без едкого
натра за 2 ч насыщается бензольное кольцо [195], и с выходом 87% от теоретического
получается 5,6,7,8-тетрагидронафтол-2. Гидрирование Р-нафтолов в присутствие хро-.
мита меди [19б] приводит к 1,2,3,4-тетрагидронафтолам. Хромит меди применим также
для гидрирования фенолов, крезолов или тимолов под давлением и при более высоких
температурах [197]. ' . .
Для гидрирования первичных ароматических аминов до первичных насыщенных
аминов рекомендуются окисные катализаторы [198], например окись кобальта или
[ 186]
1187]
[188
[189
[190
[191
[192
1193
1194]
[195]
[1961
1197]
[198]
W. Ipaticw, G. Rasuwajew, Ber., 59, 2028 (1926); 59, 306
(1926). Ч
W. Ipatiew, G. Rasuwajew, Ber., 59, 2031 (1926).
P. Sabatier, J. B. Senderens, C. r., 137. 1025 (1903).
A. Broche t, Bull. Soc. chim. France [4], 31, 1270 (1922).
R. B. Thompson, Org. Synth., 27, 21 (1947).
R. G г e w e, E. N о 1 t e, R. H. R о t z о 11, Chem. Ber., 89, 600 (1956).
H. S t e t t e r, VV. D ier ichs, Chem. Ber., 85, 61 (1952).
E. E. van Tamelen, G. T. H i 1 d a h 1, J. Am. Chem. Soc., 78, 4405
(1956).
С. V. Banks, D. T. Hocker, J. J. Richard, J. Org. Chem., 21,
547 (1956).
H. Adkins, G. К r s e k, J. Am. Chem. Soc., 70, 412 (1949).
B. G. McKusick, J. Am. Chem. Soc., 70, 2196 (1948).
W. R. В г о d e, R. W. van Dolan, Ind. Eng. Chem., 39, 1157 (1947).
Герм. пат. 528465 (1931); С., 1931, II, 1735.
4 Заказ 1835.
50
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
никеля. В этих условиях вторичные и третичные амины и продукты дезаминирования
не образуются. Вторичные и третичные ароматические амины можно гидрировать
с помощью никеля Ренея. При гидрировании первичных ненасыщенных аминов в при-
сутствии металлов платиновой группы с добавлением концентрированной соляной
кислоты получаются преимущественно первичные амины.
Циклогексиламин [199]. Получают гидрированием анилина в присут-
ствии катализатора коллоидной платины в смеси уксусной и соляной кислот.
Для приготовления катализатора прибавляют к смеси 30 -мл 10%-ного раствора
платинохлористоводородиой кислоты и 75 мл 2%-ного раствора гуммиарабика,
15 мл коллоидного раствора платины, который получают восстановлением кол-
лоидной гидроокиси двухвалентной платины (этот раствор является затравкой
и содержит 0,03 а платины в 1 мл). При встряхивании смеси в течение 0,5 ч
в водороде под давлением 1 ат образуется коллоидный раствор, содержащий
1,5 а платины.
К коллоидному раствору платины прибавляют раствор 8,37 е анилина
в 110 мл ледяной уксусной кислоты, затем 10 мл 36%-ной соляной кислоты.
Эвакуируют прибор для гидрирования и присоединяют его к источнику водорода.
Через 2,5 ч поглощается рассчитанное количестве водорода. Ббльшую часть
уксусной кислоты отгоняют в ванууме, остаток подщелачивают раствором едкого
натра, продукт гидрирования перегоняют с водяным паром, экстрагируют
эфиром, сушат, отгоняют эфир и перегоняют. Выход цикдогексиламина § г;
т. кип.-135° С.
В качестве катализатора гидрирования аминов в ядро рекомендована также
двуокись рутения [200].
Гидрирование гетероциклических соединений. Ката-
литическое гидрирование фурана можно проводить по методу Сабатье в присутствии
никеля при 170ь С; для гидрирования при 50° С пригоден никель Ренея [201J. Исполь-
зуя в качестве катализатора окись палладия, Старр и Хиксон [2о2] прогидрировали
без разбавителя фуран (120 г) при избыточном давлении 7 ат. Процесс длился 20 ч,
выход тетрагидрофурана 96%.
В молекуле фурфурола альдегидная группа гидрируется легче, чем кольцо.
В начестве катализаторов гидрирования фурфурола до фурфурилового спирта рекомен-
дованы медь на окиси хрома [203 J или медь на хромите бария [204]. При дальнейшем
гидрировании получается тетрагидрофурфуриловый спирт, который может быть полу-
чен непосредственно из фурфурола при комнатной температуре в присутствии платины
и хлорида железа (II). Волее жестние условия гидрирования приводят к разрыву
кольца и образованию пеитандиолов.
Гидрирование пиррола очень затруднено. Из катализаторов можно рекомен-
довать платину в ледяной уксусной кислоте [205], родий или палладий на асбесте [206],
окись платины [207].
Пирролидин [207]. В 60 мл ледяной уксусной кислоты растворяли
18 г дважды перегнанного над натрием пиррола н гидрировали в присутствии
[199] A. Skit a, W. В erend t, Вег., 52, 1519 (1919).
[200] L. С. Behr, J. Е. Kerby et al., .Т. Am. Chem. Soc., 68, 1296 (1946);.
A. E. Barkdoll, D. C. England et al., J. Am. Chem. Soc., 75, 1156
(1953).
[201] J. B. Cloke, C. Ayers, J. Am. Chem. Soc., 56, 2144 (1934).
[202] D. Starr, R. Hixon, J. Am. Chem. Soc., 56, 1595 (1934); Org. Synth.r
16, 78 (1934).
[203] H. D. Brown, R. M. Hixon, Ind. Eng. Chem., 41, 1382 (1949).
[204] R. Schr.6 ter, частное сообщение, см. Houben - W e у 1, Bd. 4, T. 2,
Stuttgart, 1955, S. 307; W. A. Lazier, H. R. Arnold, Org. Synth.,
19, 31 (1939).
[205] M. De Jong, J. P. Wibaut, Rec. trav. chim., 49, 2.37 (1930).
[206] N. D. Zelinsky, J. K. Jurjew, Ber., 64, 101 (1931).
[207) L. H. Andrews, S. M. M с E 1 v a i n, J. Am. Chem. Soc., 51, 889 (1929)..
I. ПРИСОЕДИНЕНИЕ ВОДОРОДА по с==с-связям
5
0,5 г окиси платины. Поглощение водорода протекало очень медленно и време
нами совсем прекращалось. Но встряхиванием реакционной массы с кислородов
удавалось активировать катализатор. Через 45 ч прибавляли 0,3 г свежей okhci
платины. За 96—100 ч поглотилось теоретическое количество водорода. Посл<
осаждения платины декантировали темноокрашенный}раствор, нейтрализовал!
уксусную кислоту концентрированным раствором едкого натра (во избежание
потерь легколетучего пирролидина эту операцию проводили с обратным холо
дильником), добавляли избыток едкого натра и отгоняли амин из щелочного рас
твора с водяным паром до исчезновения в дистилляте щелочной реакции п<
лакмусу. Дистиллят (около 1 л) подкисляли соляной кислотой и упаривали до
суха на водяной бане. Темноокрашенныи вязкий остаток обрабатывали 40%-ныь
раствором едкого натра и экстрагировали эфиром. После высушивания эфирног»
раствора сульфатом натрия в результате перегонки получилось 12 г (63% о-
теоретического) пирролидина; т. кип. 85—88° С.
Де Лонг и Вибо [205] гидрировали пиррол в ледяной уксусной кислрте
применяя платиновый катализатор из двуокиси платины (PtOa), при давлени!
водорода 1,8 ат и получили за 4 ч пирролидин с выходом около 80% от теоретн
ческого. Можно гидрировать [208] пиррол в спирте при избыточном давление
6 ат в присутствии двуокиси платины.
Как правило, замещенные у азота пирролы восстанавливаются гораздо легче
чем незамещенные. Так, N-фенилпиррол гладко восстанавливается до N-циклогедсия
пирролидина [208]. Для гидрирования 1-(К)-карбэтоксипиррола применяли никел]
Ренея [209].
Пиридиновое кольцо гидрируется аналогично бензольному, . но, например
в хинолине или в изохиполипе пиридиновое кольцо восстанавливается раньше беа
зольного. Для гидрирования этих соединении применяли платину [210], никель npi
высоком давлении [211] и осмий-цериевые катализаторы [2121. ‘ 2-Метилпириди!
удается прогидрировать в присутствии платины при 2—3 ат [213]., Производны»
пиридина гидрируются под давлением в присутствии никеля на силикагеле [214]
Под действием никеля Ренея при 200° С и давлении водорода 150—300 ат 2-метил
пиридин гидрируется за 35—40 мин до 2-мстилпиперидина с 90%-ным выходом [215]
Каталитическое гидрирование «связанным» в о до
родом. Киндлер и Пешке [216] использовали для гидрирования вместо молекуляр
ного водорода легко дегидрируемый тетралин. Им удалось таким образом достичь
некоторого избирательного действия; так, например, нитрил коричной кислоты почтх
количественно восстанавливался до нитрила гидрокоричной кислоты, т. е. груши
циана при этом не восстанавливалась. «-Хлоркоричная кислота гидрировалась д<
п-хлоргццрокоричной боз отщепления атома хлора. Таким образом, применение тетра,
линя в качестве растворителя при каталитическом гидрировании может оказать особо»
влияние на протекание реакции. 1
л-Хлоргидрокоричная кислота. Раствор 5 г «-хлоркоричной кислоть
в 180 мл тетралина нагревают 1,5 ч до кипения с 1 а палладиевой черни в колб»
Кьельдаля с вертикальным воздушным холодильником. Боковая цепь кнслоть
прн этом гидрируется полностью. Катализатор отфильтровывают, извлекаю-
из фильтрата раствором соды кислые компоненты, промывают содовую вытяжку
[208] L. С. С г a i g, R. М. Hixon, J. Am. Chem. Soc., 52, 804 (1930) .
[209] J. L. Rainey, H. Adkins, J. Am. Chem. Soc., 61, 1105 (1939).
[210] A. Skit a, W. A. Meyer, Ber., 45, 3593 (1912); Chem. Ztg., 38, 605 (1914>
Ber., 57, 1977 (1924); J. О v e r h о f f, J. P. W i b a u t, Rec. trav. chim.
50, 957 (1930).
[211] W. Ipatiew, Ber., 41, 991 (1908); J. v. Braun, A. P e t z о 1 d, J. Se
e m a n n, Ber., 55, 3779 (1922).
[212] W. S. Ssadikow, А. К. M i c h a i 1 о w, Ber., 61, 1800 (1928).
[213] A. Skit a, W- Brunner, Ber., 49, 1597 (1916).
[214] H. Adkins, H. I. Cramer, J. Am. Chem. Soc., 52, 4349 (1930).
1215] H. Adkins, L. F. Knick et al., J. Am. Chem. Soc., 56, 2427 (1934).
[216] K. Kindler, W. Peschke, Ann., 497, 193 (1932).
4*
52
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
эфиром, освобождая ее от растворенного тетралина, подкисляют соляной кислотой
и экстрагируют эфиром выделившуюся л-хлоргидрокоричнук> кислоту. После от-
гонки эфира получают 4,8 г чистой п-хлоргидрокоричной кислоты; т. пл. 123° С.
Применявшийся тетралин был очищен промыванием разбавленной соля-
ной кислотой, водой, осушкой над хлористым кальцием и перегонкой.
//. Присоединение водорода по С=С-связям
, Исчерпывающее гидрирование С=С-связи мало отличается от гидрирования
С=С-связи. Приходится лишь учитывать такие особенности, как, например, возмож-
ность образования взрывчатых ацетиленидов тяжелых металлов, характерное для аце-
тиленовых соединении, и большее потребление водорода вследствие большей ненасы-
щенности соединений. Обычно присоединение первой грамм-молекулы водорода про-
исходит легче, чем дальнейшее гидрирование до насыщенного соединении. Этим объяс-
няется то, что для гидрирования С=С-связи можно почти всегда применять методы, •
описанные в предыдущем разделе.
Однако препаративные методы перевода С=С-связи в С—С-связь несколько
отличаются от методов насыщения С=С-связей. Такое неполное восстановление
удается осуществить как с помощью химических реактивов, так и с помощью катали-
тически возбужденного водорода. В то время как по первому методу получаются чаще
всего устойчивые транс-этиленовые стереоизомеры, по второму методу образуются
преимущественно более богатые энергией метастабильные ^ис-изомеры.
1. Химические методы
Для частичного восстановления ацетилена и его производных нз обычных
химических восстановителей чаще всего применяют щелочные металлы в жидком
аммиаке и литнналюминнйгидрид или аналогичные соединения. В 1916 г. Траубе [217]
удалось провести частичное восстановление ацетилена хлоридом хрома (П); в некото-
рых других случаях такое восстановление осуществлялось также с помощью натрия
в спиртах, цинка в ледяной уксусной кислоте или омеднепного цинка (см. обзор [218]).
В то время как литийалюминийгидрид обычно ие восстанавливает изолирован-
ные тройные связи, подобные процессы удается провести с гидридом диалкилалюми-
ния [219] и с диалкилбораном [220J. Образующиеся при этом этилены имеют цис-
конфигурацию.
а,р-Ацетиленкарбоновые кислоты и их производные, как, например, прониоло-
вая кислота [221], ацетилендикарбоновая кислота [221 [ и фенилпропаргиловый альде-
гид [222], восстанавливаются и под действием литййалюмннннгидрида. При атом, 1
возможно, в первую очередь восстанавливается функциональная группа. В результате
образуются транс-аллиловые спирты. Как и следовало ожидать [223], изолированная
тройная связь в 1-карбэтоксигексадиине-1,5 устойчива к действию литийалюминнй- „
гидрида и в этом случае с хорошим выходом получается гептен-2-ин-6-ол-1:
НС=С.-СНа—СНГ- С—С -COOCaHs > HCsC-CH,—СН!-СН=СН-СН2ОН
Частичное восстановление ацетиленовой связи под действием литнналюмнний-
гидрида облегчается при наличии по соседству оксигрупп [224—228] или сопряжения
W. Т г a u b е, W. Passarg е. Вег., 49, 1692 (1916).
К. N. Camp bp II, В. К. Campbell, Chem. Rev., 31, 77 (1942).
G. Wilke, H. Millie г, Chem. Ber., 89, 444 (1956); A. E. G. Miller,
J. W. Biss, L. H. Schwartzman, J. Org. Chem., 24, 627 (1959).
H. C. Brown, G. Z we if el, J. Am. Chem. Soc., 81, 1512 (1959).
G. E. Benedict, R. R. Russell, 3. Am. Chem. Soc., 73 5444 (1951).
F. Wille, F. Knorr, Chem. Ber., 85, 841 (1952).
B, L. Shaw, M. C. Whiting, J. Chem. Soc., 1954, 3217.
C. A. G г о b, F. G a d i e D t, Helv. chim. acta, 40, 1145 (1957); E. F. Jenny,
J. Druey, Helv. chim. acta, 42, 401 (1959).
E.B. Bates, E. R. H. Jones, M. C. Whiting, J. Chem. Soc., 1954,1854.
W,, 0 ros hn i k, A. D. Mebane, J. Am. Chem. Soc., 76, 5719 (1954).
R. Ahmad, В. C. L. W e e d о n, J. Chem. Soc., 1953, 2125.
K. R. В h a r u ci а, В. C. L, W e e d о n, J. Chem. Soc., 1953, 1584.
11. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С-С-СВЯЗЯМ
53
с двойными связями [225]. Сопряженные олефиновые спирты можпо получать из про-
изводных ацетилена с сопряженной двойной связью и оксигруипой в соседнем поло-
жении [97, 228].
транс-Кротнловый спирт [228]. К 2 г литийалюминингидрида в 150 мл
абсолютного эфира при перемешивании в течение 0,5—1 ч прибавляют по кап-
лям раствор 3 г бутин-2-ола-1 в 25 мл абсолютного эфира. Смесь кипятят 3 ч
с’ обратным холодильником. После охлаждения избыточный гидрид разлагают
прибавлением* 14 мл этилацетата и затем 24 мл насыщенного раствора хлорида
аммония. После этого смесь фильтруют, промывают водой и сушат над сульфатом
натрия с небольшим количеством карбоната калия. Остаток после -отгоини
эфира перегоняют в вакууме. Получают 1,6 г лпранс-кротилового спирта;
т. кип. 122° С; ng 1,4278.
В большинстве случаев происходит 1,2-присоединение водорода к С=С-сВяаи.
Но в некоторых сопряженных ениновых системах происходит и 1,4-присоединение
[225, 228], причем интересно, что при этом образуются соединения с кумулированными
двойными связями, например;
/^/CseC-CI^CH-CH—СНз у^//СН=С-СН-СНа—СП-СНз
Другие примеры превращения тройных связей в двойные под действием литий-
алюминннгидрида можно найти в монографии Гэйлорда [87].
При действии натрием или литием в жидком аммиаке изолированные С=С-связи
гидрируются [1Ь, 46], что отличает их от аналогичных связей С=С. Из диалкилацети-
ленов [1Ь, 229, 230] можно таким образом с хорошими выходами получать транс-
этилены, не содержащие примесей предельных Соединений. Ацетилен [231] и моно-
алкилацетилены [1b, 230, 232] восстанавливаются только на одну треть? из остатка
образуются ацетилениды щелочных металлов, устойчивые к действию такого восста-
новителя.
Это свойство можно использовать в тех случаях, когда в соединениях с двумя
С=С-связями должна быть восстановлена только связь, находящаяся в середине цели.
Так [233], ундекадиин-1,7 • сначала переводят действием амида натрия в жидком
аммиаке в ацетиленид, а затем восстанавливают ацетиленовую группу в положении
7 и получают ундецен-7-ин-1.
Разложением ацетиленидов в реакционной массе солями аммония, лучше всего
сульфатом аммония, можно, однако, и из моноалкилацетиленов или диацетиленов полу-
чать олефины или соответственно днолефины с высокими выходами [230].
Гептадиен-1,6. (Аппаратурное оформление опыта и выделение конеч-
ного продукта см. на стр. 25.) К 1 л жидкого аммиака в трехлитровой колбе
прибавляют 295 г сульфата аммония и 101 г гсптадиина-1,6. Через боковой тубус
нрй перемешивания добавляют небольшими порциями натрин до появления
устойчивой сипей окраски (около 102 г). Обычным способом выделяют неочищен-
ный продукт с выходом 87%. После перегонки получают 64,4 а (61% от теорети-
ческого) гептад иена-1,6; т. кип. 90° С; ng 1,4142.
Парциальное восстановление по ацетиленовым связям щелочными металлами
в жидиом аммиаке применимо также к этинилкарбннолам [246] и ацетиленовым соеди-
нениям с кислотными [235] или гидроксильными группами [234] в молекуле.
Однако этот метод неприменим, когда в результате реакции образуются олефины,
которые взаимодействуют с натрием в жидком аммиаке. Например, из фенилацетилена
[229] В. В. Elsner, Р. F. М. Pau 1, J. Chem. Soc., 1953, 3156.
[230J A. L. Нейве, К. W. Greenlee, J. Am. Chem. Soc., 65 , 2020 (1943).
[231j H. Moissan, C. r., 136, 1217 (1903).
1232] P. Lebeau, M. Picon, C. r., 157, 137 (1913).
[233] N. A D obs on, R. A. R aphael, J. Cbem. Soc., 1955, 3558.
[234] F. Sondheimer, J. Chem. Soc., 1950, 877.
[235] D. R. How ton, R. H. Davia, J. Org. Chem., 16, 1405 (1951).
54
ОБРАЗОВАНИЕ С— Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
наряду с ацетиленидом получается только этилбензол [236]. Точно так же сопряжен-
ные диены, образующиеся из соответствующих диинов или енинов (стр. 24), восста-
навливаются дальше. Для этой цели удобно восстановление проводить омедненной
цинковой пылью [237]. При этом из дифенилдиацетилена в результате 1,4-присоеди-
нения на первой стадии получается дифенилбутатриен [238].
2. Каталитическое гидрирование
Для перевода ацетиленовых связей в этиленовые предпочтительно пользуются
каталитическими методами [218, 239], поскольку в таком случае процесс идет в мягких
условиях, что обеспечивает хорошие выходы. Преимущественно образующиеся при
этом чис-этилены обычно легко перегруппировываются в более устойчивые транс-
изомеры [97, 226, 240, 241].
Применение особо активных катализаторов не обязательно, так как гидриро-
вание по С=С-связи происходит довольно легко. Но катализаторы должны ускорять
главным образом избирательное гидрирование С^С-связи и не особенно затрагивать
С=С-связи.
Платиновая и палладиевая чернь из-за малой их избирательности для этой
цели не подходят. После поглощения рассчитанного количества водорода в продукте
реакции наряду с олефином содержатся значительные количества исходного веще-
ства и насыщенного соединения.
Применяя коллоидный палладий и, что несколько хуже, платиновые катализа-
торы по Адамсу (стр. 34), можно гидрировать С^С-связь с достаточной избиратель-
ностью [242]. После поглощения 1 .ио.гь водорода полученный продукт состоит глав-
ным образом из олефина, хотя при дальнейшем воздействии водорода реакция идет
до образования насыщенного соединения. Такими же избирательными свойствами
обладают катализаторы типа никеля Ренея [1Ь, 235, 243, 244].
В присутствии железа Ренея [245] и специально обработанных палладиевых
катализаторов, в отличие от предыдущих, скорость гидрирования заметно снижается
после поглощения 1 .ноль водорода. Но эти катализаторы гораздо менее активны, и про-
цесс приходится вести при высоких температурах и давлениях, а в таких условиях
может произойти изомеризация продуктов в транс-этилены. Поэтому в большинстве
методов используются катализаторы из палладия, осажденного на таких носителях,
как карбонат бария [246], сульфат бария [168, 247, 248], карбонат кальция [227,
234, 249] или окись алюминия [250], часто с добавками небольших количеств контакт-
ных ядов (пиридина [251], хинолина [105. 248. 252—254]). В новейшей литературе
[236] Р. Lebeau, М. Р i с о п, С. г., 157, 223 (1913); 175, 223 (1922).
[237] F. Straus, Ann., 342, 190 (1905); франц, пат. 237196; С., 1939. I, 3256; С. А.,
33, 6872 (1939).
[238] V. Grignard, Tcheoufaki, С. г., 188, 1531 (1929).
[239] G. S c,h i 11 е г, в книге Houben-Weyl, Methoden der organischen Chemie,
Bd. 4, T. 2, Stuttgart, Georg Thieme Verlag, 1955, S. 291.
[240] W. D. G e 1 m e r, I. A. S о 1 о m о n s, J. Am. Chem. Soc., 75, 3430 (1953)
[241] M. G. E 111 i n g e r, J. E. H о d g k i n s, J. Am. Chem. Soc., 77, 1831 (1955).
[242] P. Karrer, H. Rentschler, Helv. chim. acta, 27, 1297 (1944).
[243] S. В a n с e, H. J. Barber, A. M. W о о 1 m a n, J. Chem. Soc., 1943, 1.
[244] K. N. C a m p b e 11, M. J. O’C о n n о r, J. Am. Chem. Soc., 61, 2897 (1941);
M. Bourguel, Bull. Soc. chim. France [4], 45, 1067 (1929).
[245] R. P a u 1, G. H i 11 у, В u 11. Soc. chim. France [5], 6, 218 (1939); A. F. T h о m-
p s о n jr., S. B. W у a t t, J. Am. Chem. Soc., 62, 2555 (1940).
[246] G. F. Hennion, W. A. Schroeder et al., J. Org. Chem., 21, 1142 (1956).
[247] С. P aal et al., Ber., 42, 3930 (1909).
[248 D. J. C r a m, N. L. A 11 i n g e r, J. Am. Chem. Soc., 78, 2518 (1956).
[249 a) R. A. Raphael, F. Sondheimer, J. Chem. Soc., 1950, 3185; b) 1950,
120; c) 1950, 115; 1951, 2693.
[250] I. Marszak, M. Olomucki, Bull. Soc. chim. France, 1959, 182.
[251] H. Heusser, K. Eichenberger, P. A. Plattner, Helv. chim.
acta, 33, 370 (1950).
[252] H. Inhoffen, E. Bohlmannet al., Ann., 570, 54 (1950).
[253] W. Kimel et al., J. Org. Chem., 22, 1611 (1957); 23, 153 (1958).
[254] R. Riiegg, U. Glooret al., Helv. chim. acta, 42, 2616 (1959); J. W e i-
c h e t, L. В a h a, V. К v i t a, Coll. Czech. Chem. Comm., 25, 1914 (I960).
III. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = О-СВЯЗЯМ
приводятся в основном данные по применению описанного Линдларом [105] палладий-
свинцового катализатора на карбонате кальция [224, 241, 253—256] (его получение
см. стр. 33).
Избирательному гидрированию, по-видимому, лучше всего поддаются диалкил-
[1Ь, 168] и диарилацетилены [244], так как в этом случае дальнейшее гидрирование
образующихся олефинов протекает заметно медленнее. Например, из циклодецина
с 96%-ным выходом получается циклодецен [255]. Производные этилена, образующиеся
из монозамещенных ацетиленов [168, 257], этинилкарбинолов [105, 242, 246, 253,
254] и других алкинолов [97, 224, 227, 249а, 253], в большинстве случаев гидрируются
о неменьшей скоростью, чем исходные соединения. Тем не менее и в этом случае можно
с успехом приводить частичное восстановление с помощью избирательных катализато-
ров при работе с тщательной отмеренным количеством водорода.
При наличии других двойных связей, как изолированных [251], так и сопря-
женных [226, 249а], реакция тоже может быть направлена так, что преимущественно
будет восстанавливаться тройная связь. Из винилацетилена [168] над палладием
на сульфате бария можно, например, получать бутадиен-1,3 с выходом 69% от теоре-
тического. Поэтому каталитическое гидрирование широко применяется для синтезов
полиенов [97, 105, 226, 242, 252, 254]. Из соединений с двумя ацетиленовыми связями
получают соответственно производные диолефинов [248, 249Ь,с].
Сильно затрудняется или становится невозможным присоединение водорода
при наличии большого числа арильных заместителей, как, например, в тетрафенилбу-
тиндиоле или в ди-(трифенилметил)-ацетилене. Во всех других случаях, даже при
наличии в молекуле аминогрупп [224, 249а, 250], каталитическое гидрирование дает
хорошие результаты.
АцетиленДикарбоновые кислоты [235, 247, 249, 250] и их производные, напри-
мер амиды [241, 249Ь] и эфиры [240, 246, 248], в отличие от восстановления литий-
алюминийгидридом, могут быть переведены каталитическим гидрированием в произ-
водные этилена без изменения функциональной группы. Реакцию можно проводить
в таких мягких условиях, что даже легко гидрируемая нитрильная группа [243] или
чувствительная ацетальная группировка [149, 256] не подвергаются изменению; даже
из эфиров инолов этим способом удается получать эфиры енолов [251].
2-Метилбутен-3-ол-2 [253]. К раствору 336 г 2-метилбутин-3-ола-2
(4 моль) в равном объеме петролейного эфира прибавляют 16,8 г хинолина и 30 г
катализатора Линдлара. Смесь охлаждают до 10* С и встряхивают в атмосфере
водорода до поглощения 4 моль водорода. Через 3—5 ч поглощение водорода
заметно уменьшается. После отделения катализатора фильтрованием продукт
выделяют перегонкой на насадочной колонке. Получается 323 г (94% от теорети-
ческого) 2-мётилбутен-3-ола-2; т. кип. 97—98° С; rflfr 1,4141.
tjuc-Кротилфталимид [241]. В присутствии 1 г катализатора Линдлара
и 1 мл хинолина гидрируют суспензию 20 г К-бутин-2-илфталимида в 1100 мл
этилацетата при атмосферном давлении до начала уменьшения скорости погло-
щения водорода, на что требуется примерно 3,5 ч (поглощается около 1,03 моль
водорода). Раствор фильтруют и упаривают в вакууме. После перекристаллиза-
ции остатка из 50%-ного спирта получают 19,3 г (96% от теоретического)
N-ijuc-кротилфталимида; т. пл. 66—66,5° С.
Под действием двуокиси азота продукт перегруппировывается в транс-
изомер. Гидразинолизом из него можно получить ^uc-кротиламин с 67%-ным
выходом.
III, Присоединение водорода по С=0-связям
1. Восстановление неблагородными металлами
В большинстве случаев алифатические альдегиды довольно гладко восстана-
вливаются неблагородными металлами до первичных спиртов. Для этих целей можно
применять порошкообразный цинк или железо в ледяной уксусной кислоте; правда,
[255] V. Prelog, К. Schenker, Н. Н. Gunthard, Helv. chim. acta, 35, 1598
(1952).
[256] A. S. В a i 1 e у, V. G. К e n d a 11 et al., J. Chem. Soc., 1957, 3027.
[257] M. Bouguel, Bull. Soc. chim. France [4], 51, 253 (1932).
-56
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
тгри атом в результате вторичной реакции часто образуются ацетаты полученных
спиртов. Проведено исследование действия различных металлов в сочетании с уксус-
ной кислотой при восстановлении коричного альдегида [258J. Установлено, что при
применении железа замедляется побочная реакция образования гликоля и защищается
этиленовая связь. Другие исследователи [259] разработали подробную пропись полу-
чения «-гептилового спирта, используя результаты работы [258].
2-(а-Нафтил)-пропанол-1 [260]. Смесь 83,5 г 2-(а-пафтил)-пропионового
альдегида, 300 мл ледяной уксусной кислоты, 300 мл воды и 200 г порошка
железа нагревают 7 ч на паровой бане. Горячий раствор фильтруют, осадок на
фильтре промывают водой и эфиром. Фильтрат и промывные воды разбавляют
водой, и продукты восстановления экстрагируют эфиром. Остающееся после
отгонки эфира масло кипятят с раствором 15 г едкого натра в 30 мл воды и 50 лл
спирта для разложения образовавшегося ацетата. 2-(а-Нафтил)-пропанол-1
промывают, сушат его эфирныи раствор и перегоняют. Получают 66,3 г (78,5% от
теоретического) желтоватого вязкого масла; т. кип. 144—147° С (3 лслс рт, ст.).
Для восстановления альдегидов применяется часто амальгама алюминия.
Амальгаму натрия из-за щелочных свойств употреблять не рекомендуется.
Ароматические альдегиды более склонны к образованию гликолей при восста-
новлении, чем алифатические:
2АгСНО —> Лг-СН—СН-Аг
I I
ОН он
Можно подобрать такие условия проведения реакции, при которых будут полу-
чаться почти исключительно гидробензоины; в некоторых случаях они являются
единственным продуктом реакции [261]. Ароматические спирты удобнее получать
диспропорционированием по реакцпи Канниццаро (см. стр. 312), чем восстановлением.
Для восстановления котонов до вторичных спиртов обычно рекомендуется
применять щелочные восстановители, поскольку в кислой среде образуются преимуще-
ственно пинаконы. Часто применяют патрин в спирте или натрий во влажном эфире
или бензоле, амальгаму натрия, цинковую пыль в растворе едкого натра и аналогич-
ные восстановители. Гептанол-2 получается, например, с хорошим выходом при доба-
влении натрия к водно-спиртовому раствору метил-«-амилкетона [262]. Кетоны часто
восстанавливают натрием в водпо-эфирных суспензиях. Ненасыщенные кетоны при
этом частично гидрируются также по двойной связи.
З-Метилгептанол-2 [263]. Раствор 64 г З-метйлгептанона-2 в 300 мл
эфира приливают к суспензии 100 г бикарбоната натрия в 300 мл воды. К получен-
ной смеси при охлаждении добавляют небольшими порциями 40 г натрия, сильно
встряхивая реакционную массу перед добавлением каждой новой порции. 'Эфир-
ныи раствор отделяют, и после его осушки отгоняют растворитель. Остаток
перегоняют в вакууме. Выход З-метилгептанола-2 составляет 50 г (77% от теоре-
тического); т. кип. 172—173° С.
Фогель [264] встряхивал эфирный раствор спиро [3, 4]октанона-5 а насы-
щенным раствором карбоната калия и получил в результате прибавления метал-
[258] A. J. П i 1 I, Е. П. Naso n, J. Am. Chem. Soc., 46, 2236 (1924).
[259] Н. Т. С 1 а г k е, Е. Е. D г е g е г, Org. Synth., 6, 52 (1926).
[260] L F. F i e s e г, L. M. J о s h e 1, A. M. Seligman, J. Am. Chem.
Soc., 61, 2137 (1939).
[261] S. Dani low, Ber., 60, 2393 (1927).
[262] F. C. Whitmore, T. Otterbacher, Org. Synth., Coll. v. 2, 1943,
p. 317.
[263] 8. G. Powell, J. Am. Chem. Soc., 46, 2514 (1924); S. G. Powell.
F. Hagemann, J, Am. Chem. Soc., 66, 372 (1944).
[264] E. Vogel, Chent. Ber., 85, 25 (1952).
Ш. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = О-СЕЯЗЯМ
57
лического натрия соответствующий спирановый спирт — спиро [3, 4]октанол-4-
с выходом 79% от теоретического:
\Z\_I \/\-1
</z н/\ол
Восстановление кетонов амальгамой натрия более подробно исследовано Бах-
манном [265]. Флуоренон, бензофенон и его замещенные почти количественно восстана-
вливаются 2%-ной амальгамой натрия до соответствующих карбинолов. При этом
было доказано, что в качестве промежуточных продуктов образуются металлке-
тилы. Подробные данные о восстановлении амальгамой натрия ксантона см. [266]
и тиоксантона см. [267].
Взаимодействие спиртовых растворов кетонов с цинком и раствором едкого
натра или с цинком и концентрированным раствором аммиака тоже дает
хорошие результаты [268]. Лучшие выходы при меньшей продолжительности
реакции получают при восстановлении флуоренон-2-карбоновой кислоты цинком
и раствором едкого кали с добавкой небольшого количества сульфата меди [269].
При восстановлении флуоренон-1-карбоновон кислоты магнием в метиловом спирте
выход резко понижается [270], хотя Цехмейстер и Ром [271] восстанавливали по этой,
методике бензофенон и 1,3-дифенилацетоп и получали отличные выходы.
2. Реакция Меервейна — Понидорфа — Верлся
Одним из самых мягких способов восстановления, который в то же время оказы-
вает специфическое действие на группу С^О, является реакция Меервейна — Пон-
ндорфа — Берлея [272а, Ь]. Независимо друг от друга в 1925 г. Меервейн и Шмидт [273р
и Верлей [274] открыли, что альдегиды и некоторые кетоны восстанавливаются алко-
голятом алгомннпя в присутствии спирта до первичных спиртов. Почти одновременно-
этот метод восстановления открыл и Понндорф [275]. Он уже употреблял в некоторых
случаях вместо этилата алюминия изопропилат алюминия, преимущества которого
и возможность применения для восстановления кетонов обнаружили позже и другие-
исследователи [276, 277].
В-С-В'(П)+СНз-СИОН дЧ°сн<сн.).1.> Н_СН-В'(Н) + СНЭ-С-О
VI II
О . СНз ОН СНз
Взаимодействие альдегидов п кетонов с изонропилатом алюминия представляет
собой обратимую реакцию, которая постоянным удалением из среды одного из про-
дуктов реакции, например ацетона, может быть полностью смещена в одну сторону.
Вместо пзопропилата алюминия можно с успехом применять другие алюминиевые
[265] W. Е, Bachman n, J. Am. Chem. Soc., 55, 770 (1933).
[266] A. F. II oil с man, Org. Synth., 7, 88 (1927).
[267] П. F. Oehlschlaeger, I. R. Mac Gregor, J. Am. Chem. Soc., 72,.
5332 (1950).
[268] H. R. Frank, D. C. Tarbell, J. Am. Chem. Soc., 70, 1276 (1948).
[2C9] F. E. Ray, J. II. Weisbnrger, J. Org. Chem., 13, 655 (1948).
[270] J. Forrest, S. H, Tucker J. Chem. Soc., 1948, 1137.
[271] L. Zech meister, P. Rom, Ann., 468, 117 (1929).
[272]a) A. L. W i 1 d s, Organic Reactions, 2, 178 (1944); b) Th. В e г s i n, в книге-
W. Foerst, Neuere Methoden der praparativen organischen Chemie 3 Aufl.,
Bd. 1, Weinheim, Verlag Cheniie, 1949 S. 137.
[273] H. Meerwein, R. Schmidt, Ann., 444, 221 (1925).
[274] A. V e r 1 e y, Bull. Soc. chim. France [4], 537, 871 (1925); 41, 788 (1927).
[275] W. Ponndorf, Angew. Chem., 39, 138 (1926).
[276J W. G. Y о u n g W. H. Hartung, F. S. С г о s s I e у, J. Am. Chem. Soc.,
58, 100 (1936).
1277] IT. Lund, Ber., 70, 1520 (1937).
58
ОБРАЗОВАНИЕ С—Н-СВЯЗКИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
алкоголяты вторичных спиртов. Реакция, обратная этой, известна как окисление по
методу Оппенауера (стр. 304).
Изопропилат алюминия является пе только катализатором, но п восстановителем
(278]. Опыты с алкоголятами алюминия, полученными из спиртов с дейтерированными
атомами углерода [279], показали, что перенос водорода па карбонильную группу
происходит непосредственно от алкоголята без участия растворителя. Таким образом,
реакцию можно проводить с расплавленным изопропилатом алюминия и без изопро-
пилового спирта или в растворе толуола, или в растворах других углеводородов.
В отличие от других способов при таком способе восстановления не затрагива-
ются С=С-связи, находящиеся в а,p-положении к карбонильной группе. Кроме того,
сохраняются сложноэфирные и нитрогруппы и подвижный галоген. Преимуществом
этого метода являются также хорошие выходы спиртов.
Б качестве побочной реакции при восстановлении альдегидов возможна реак-
ция Тищенко (стр. 312); при восстановлении алифатических альдегидов она подав-
ляется введением избытка изопропилата алюминия. Кроме того, альдегид способен
вступать в реакцию альдольной конденсации сам с собой или с образовавшимся
ацетоном (стр. 712). Но эти побочные реакции встречаются редко и никоим образом не
снижают достоинств описанного способа восстановления.
Алкоголяты алюминия получают растворением в соответствующем спирте
алюминия, активированного с поверхности хлоридом ртути (II) или иодом.
Изопропилат алюминия [272а]. В круглодонную колбу емкостью 1000 мл,
загружают 300 мл безводного изопропилового спирта, 0,5 г хлорида ртути (II)
и 27 » (1 л*олъ) алюминиевой проволоки или фольги, которую тщательно очищают
наждачной бумагой и затем протирают. Колбу соединяют с обратным холодиль-
ником с хлоркальциевой трубкой и нагревают на водяной бане до кипения. После
начала кипения через холодильник приливают 2 мл четыреххлористого углерода.
Смесь окрашивается в коричневый цвет и через несколько минут начинается
бурное выделение водорода. Нагревание прекращают и регулируют скорость
реакции, охлаждая колбу ледяной водой. Когда реакция замедлится, нагревание
продолжают до тех пор, пока не растворится весь алюминий (6—12 ч). Горячий
раствор переливают в колбу для перегонки и после отгонки при небольшом раз-
режении изопропилового спирта отгоняют изопропилат алюминия (температура
масляной бани 180—190° С). Он отгоняется в виде бесцветной вязкой жидкости;
т. кип. 130—140° С (7 мм pm. cm.) и 140—150° С (12 мм, pm. ст.).
Изопропилат алюминия можно хранить в чистом виде й в виде раствора
в изопропиловом спирте в склянке с притертой запарафиненной пробкой. Во
многих случаях перегонка реактива не обязательна; полученный после растворе-
ния алюминия темноокрашенный раствор может быть непосредственно использо-
ван в реакции.
Подробное описание методов получения этилата и бутилата алюминия
см. [280, 281].
Некоторые альдегиды, кан, например, фурфурол [273], восстанавливаются
уже на холоду; но чаще приходится работать при повышенной температуре, что
и позволяет удалять обычно легколетучее карбонильное соединение перегонкой.
Кротиловый спирт (272а, 275]. К раствору изопропилата алюминия,
приготовленному обычным путем из 47 г (1,74 люль) алюминия и 500 мл изопропи-
лового спирта, приливают 210 г (3 моль) кротонового альдегида и 1 л безводного
изопропилового спирта. Реакцию проводят в круглодонной колбе емкостью
2 л, снабженной колонкой Вягре, которая соединена с нисходящим холодиль-
ником. Реакционную смесь нагревают на масляной бане (температура бани
110° С), причем образующийся ацетон медленно отгоняется при 6о—70° С.
[2781 L. Jack mann, J. Mills, Nature (London), 164, 789 (1949).
[279] ‘(Jgtjg) 1 i a m s, К. A. К r i e g e г, A. R. D a у, J. Am. Chem. Soc., 75,
[2801 W. C h a 1 m e r s’, Org. Synth., Coll. v. 2, 1943, p. 598.
[281] W. Wayne, H. Adkins, Org. Synth., 21, 8 (1941).
III. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = О-СВЯЗЯМ
5
После того как в дистилляте по реакции с 2,4-динитрофепилгидразином болыц
не обнаруживается ацетон (через 8—9 ч), оставшийся изопропиловый спир
отгоняют в вакууме. Остаток охлаждают до 40° С п гидролизуют, желательн
при охлаждении, прибавлением 900 мл холодного 6 н. раствора серной кислот]
(из 145 мл концентрированной серной кислоты л 790 мл воды). Маслянистый ело
отделяют, промывают водой и перегоняют при 80—70° С, постепенно понижа
давление с 275 до 65 мм рт. ст. Вторую порцию кротилового спирта получаю
при перегонке водной фазы н насыщении дистиллята карбонатом калия. Неочт
щенныи кротиловый спирт после осушки над карбонатом калия перегоняют с кс
ловкой. Выход 130 г (60% от теоретического); т. кии. 117—122° С.
При длительном процессе восстановления альдегида или когда в результат
реакции образуются неустойчивые па воздухе продукты, через реакционную масс
пропускают азот или водород. Обычно рекомендуется применять небольшой избыто
алкоголята алюминия; с недостаточным количеством алкоголята оптимальные выход
получаются только при восстановлении ароматических альдегидов.
Пиперониловый спирт [282]. В 500 мл абсолютного изопропиловох
спирта, полученного кипячением спирта в течение 4 ч с окисью кальция, перегот
кой, кипячением с изопропилатом алюминия и повторной перегонкой растворяй:
50 г пипероналя, добавляют 10 г изопропалата алюминия и нагревают на маелг
пой бане при 95° С в колбе с колонкой Бидмера п нисходящим холодильников
Через 4 ч, в течение которых при 77—81° С отгоняется 200 мл дистиллята, в дх
стилляте уже не удается обнаружить ацетона по реакции с раствором 2,4-дивд
трофепилгидразина в 1 н. растворе соляной кислоты. К остатку прибавляют воДч
доводят до слабокислой реакции соляной кислотой, продукт реакции экстраг5
руют эфиром, сушат и остаток после отгонки эфира перегоняют в вакуум;»
Выход пиперонилового спирта 45 г; т. кип. 151°С (13 мм рт. ст.). Продух
застывает с образованием кристаллов; т. пл. 51° С.
Подробное описание восстановления хлораля до трихлорэтилового спирта св
[280].
Восстановление кетопов до вторичных спиртов ио методу Меервейна — Пон]
дорфа — Берлея протекает аналогично восстановлению альдегидов, но скоросх
подобных реакций меньше. Метод Меервейна — Понндорфа — Верлея неприменв
для восстановления кетонов с сильно выраженной склонностью к енолнэации, напрх
мер, для восстановления эфиров Р-кетокислот, р-дикетонов, так как в этом случх
образуются еноляты алюминия, которые не восстанавливаются. Такие кетоны с болз
шим успехом восстанавливаются каталитическими методами или при помощи борги^
рида патрия или калия.
Для гидрирования низкокипящих кетонов можно пользоваться методикой
приведенной для восстановления кротонового альдегида (стр. 58).
Бензгидрол [272а]. Метод типичен для восстановления ароматически
кетопов. В круглодонной колбе емкостью 200 мл смешивают 18,2 г (0,1 мол
бензофенона с раствором 20 г (0,1 моль) иэопропилата алюминия в 100 мл сухох
изопропилового спирта. Колбу соединяют с коротким обратным холодильников
не подключая воду. Можпо применять также обратный холодильник с кипя
щим метиловым спиртом в рубашке или насадку для фракционирования ц
Хану [283]. Верхний конец обратного холодильника соединяют с небольшп
охлаждаемым водой нисходящим холодильником. Раствор нагревают на водянс
бане так, чтобы в одну минуту отгонялось 5—10 капель дистиллята (для труде
восстанавливающихся кетонов 1 — 4 капли в 1 мин). Через 30—60 мин в дисти;
ляте уже не обнаруживается ацетон. Если отгоняется более 50 или 60 мл из«
пропилового спирта, то в реакционную массу для сохранения объема добавляй;
20 мл сухого4изопропилового спирта. Через обратный холодильник пуская;
воду, нагревают реакционную смесь 5 мин до бурного кипения, затем опя1
[282] С. S с h о р 1, W. S а 1 z е г, Ann., 544, 14 (1940).
(283] A. Hahn, Вег., 43, 419 (1910).
60
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
отключают воду и испытывают первые капли погона на присутствие ацетона.
При положительной пробе на ацетон отгонку продолжают до его удаления.
После отрицательной пробы па ацетон большую часть изопропилового
спирта отгоняют при слегка пониженном давлении. По охлаждении остаток
гидролизуют разбавленной соляной кислотой (35 мл концентрированной соля-
ной кислоты в 175 мл воды), достигая полноты гидролиза энергичным встряхива-
нием суспензии. Кристаллический бензгидрол отфильтровывают, промывают
холодной разбавленной кислотой, затем водой и сушат. Выход неочищенного
продукта 18,4 г. Для очистки продукт растворяют в 50 мл горячего петролей-
ного эфира (т. кип. 60—70° С). Если получается мутный раствор, то добавляют
20 мл бензола и фильтруют горячий раствор. Бензгидрол кристаллизуется при
охлаждении в виде бесцветных игл. Выход 18,2 з (99% от теоретического);
т. пл. 67—69° С.
Легко восстанавливаются и циклические кетоны, например а-декалоп [277]
и тетралоны. 2,3-Дифенилтетралон-1 был восстановлен изопропилатом алюминия [284J
в соответствующее оксисоединение с выходом 93%:
О н ОН
। j, । —> । I ।
фурановое [273] и тиофеновое [285] кольца в условиях реакции Меервейна —
Понндорфа — Берлея не изменяются. Аналогично можно получить 4-метил-5-
(а-оксиутил)-тиазол с 71%-ным выходом из соответствующего ацетилтиазола [286]:
НзС—п---N HgC—й-----------N
II JI — II JI
CHsCo/\Z CHsCh/'Y
OH
Изопропилат алюминия восстанавливает н другие серуседержащие соединения, -
как, например, кетосульфоны и кетотиоэфиры. Таким способом [287] с хорошими
выходами были получены 1-(п-метилсульфонилфенил)-этанол н 1-(п-метилмеркапто-
фенил)-этанол:
СНзЗО2—^>—СОСНз —>- CH3S02—<^J4-CHCH3
(CH3S—) (CHgS—) ОН
Установлено [288], что удаление ацетона во многих случаях необязательно
и что при восстановлении бензофенона и бензосуберона в присутствии избытка изо-
пропилата алюминия получаются более чистые продукты.
Модификация этой реакции [289] заключается в том, что карбонильное соедине-
ние медленно вводят во время реакции, благодаря чему его концентрация всегда под-
держивается на низком уровне и уменьшается вероятность протекания побочных реак-
ций (стр. 58). Таким способом можно, например, осуществить восстановление бенз-
илидепацетона с 96%-ным выходом, в то время как при обычном методе он восстана-
вливается с трудом.
Как впоследствии было показано [290], скорость реакции Меервейна — Понн-
дорфа — Берлея повышается при работе со смесью изопропилата алюминия (65—
284
285
286
287
288
289
290
Н. М. Crawford, Н. В. Nelson, J. Am. Chem. Soc., 68, 134, (1946).
E. Campaigne, J. L. Diedrich, J. Am. Chem. Soc., 70, 391 (1948).
P. Baumgarten, A. D urn ow et al., Ber., 75, 142 (1942).
G. В. В a c n m a n n, C. L. Carlson, J. Am. Chem. Soc., 73, 2857 (1951).
W. L. T г u e 11, W. N. Moulton, J. Am. Chem. Soc., 73, 5913 (1951).
A. K. Macbeth, J. A. Mills, J. Chem. Soc., 1949, 2646.
G. Gal, G. Tokar, I. S i m о n у i, Magyar Kemiai Folyoirat [Ung. Z.
Chem.], 61, 286 (1955k C., 1957, 1150; G. G a 1, I. Si mon vi, G. Tokar,
Acta Chim. Hunger., 8, 163 (1955); C., 1957, 2436.
III. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = Ь-СВЯЗЯМ
61
80%) и хлоризопропилата алюминия (35—20%). Такой модифицированный способ
проведения синтеза полностью оправдал себя при получения бромгидрипов из а-бром-
кетоноп.
3. Восстановление алюмогндридом лития [87, 291]
Алюмогидрид лития, впервые полученный в 1947 г. Фанхолтом, Бондом и Шле-
зингером [292], оказался хорошим восстановителем и очень быстро получил широкое
применение в препаративной органической химии. Исследования Нистрома и Брауна
[293] показали, что ои очень быстро и гладко восстанавливает большое число функ-
циональных групп, причем в большинстве случаев кратные С—С-связи не изменяются.
Растворенный в эфире алюмогидрид лития готовят в лаборатории взаимодейст-
вием гидрида лития с галогенидом алюминия в эфире при 20° С:
4LiH + AlXs —> LiAlH4-)-3LiX
гдеХ—Cl, Вт.
Сначала для этой цели применяли хлорид алюминия, который затем заменили
бромидом, гораздо более удобным в работе [294}. В отсутствие веществ, с которыми
может реагировать алюмогидрид лития (на него влияет также воздух и двуокись
углерода), он не разлагается до 100° С. Выше 120° С разложение идет по следующей
схеме;
LIA1H* —> ЫН4-А14-1,5Н»
Алюмогидрид лития выпускается промышленностью в виде зернистой твердой
массы и хранится в запаянных жестяных банках. Поэтому его получение здесь но
описывается.
Растворение алюмогидрида лития в эфире часто протекает с большим трудом.
Перед растворением его растирают до тончайшего порошка в ступке в атмосфере азота
при полном отсутствии влаги, избегая местных перегревов, которые могут-привести
к сильным взрывам.
Удобнее всего растворять алюмогидрид лития в эфире в колбе с мешалкой
и обратным холодильником, защищенным трубкой с натронной известью. При
интенсивном перемешивании поддерживают слабое кипение раствора от несколь-
ких часов до двух суток, в зависимости от качества препарата. При растворении
технического продукта остается серый нерастворимый остаток. Поэтому при
использовании препарата для восстановления вводят 5—10%-пый избыток гид-
рида и проводят процесс в том же реакционном сосуде.
Для приготовления запаса раствора алюмогидрида лития через 1—2 дйя
раствор декантируют при помощи сифона, причем жидкость передавливают азо-
том. Раствор можно также фильтровать в атмосфере азота. Осадой на фильтре
и все остатки алюмогидрида лития разлагают, заливая его дибкеаном в приба-
вляя влажный диоксан или метиловый спирт. Только после разложения всего
активного гидрида аппаратуру промывают разбавленной соляной кислотой.
В некоторых случаях необязательно дожидаться полного> растворения алюмо-
гидрида лития в эфире; для восстановления можно непосредственно применять обра-
зующуюся суспензию*.
* Восстановление суспензией алюмогидрида лития является, как правило,
наиболее удобным и безопасным приемом. Во избежание взрывов следует
всегда проверять отсутствие перекисных соединений в употребляемых раство-
рителях. Само восстановление проводят в атмосфере сухого' азота. —
Прим., ред.
[291] w. G. Brown, Organic Reactions, VI, 469 (1951); U. Solms, Chimia, 5,
25 (1951).
[292] A. E. Figholt, А. С. В о n d, H. I. Schlesinger, J. Am. Chem. Soc.,
69, 1199 (1947).
[293] R. F. Nystrom, W. G. Brown, J. Am. Chem. Soc., 69, 1197, 2548 (1947);
70, 3738 (1948).
[294] E. Wiberg, M. S c h m i d t, Z. Naturforsch., 7b, 59 (1952).
62
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Наиболее удобно готовить раствор алюмогпдрида лития из выпускаемых про-
мышленностью ампул, в которых раздельно запаяны отмеренные количества гид-
рида лития н бромида алюминия. Ампулы вскрывают под слоем эфира и смешивают
полученные растворы. Таким образом избегают трудоемкого растворения твердого
алюмогпдрида лития и получают раствор с известным содержанием гидрида.
При восстановлении в качестве растворителя часто применяют дпэтиловып
эфир, тетрагидрофурап и реже дибутиловый эфир, диэтиловый эфир диэтиленгликоля
(диэтилкарбитол) и дибутиловый эфир диэтиленгликоля (дибутилкарбиюл) [295].
Хорошим растворителем является N-этилморфолин, но его трудно получить в чистом
виде [296]. Для приготовления растворов гидрида в других растворителях раствори-
тель прибавляют к эфирному раствору гидрида, затем отгоняют эфир. Все применя-
ющиеся растворители должны быть тщательно очищены [297].
При «нормальном» способе работы к раствору алюмогпдрида лития в соответст-
вующем растворителе прибавляют но каплям восстанавливаемое карбонильное соеди-
нение. Для гидрирования трудно восстанавливающихся веществ и соединений, которые
подвергают частичному восстановлению, применяют обратный порядок («инвертный
метод»), т. е. раствор или суспензию алюмогидрида лития прибавляют по каплямк рас-
твору карбонильного соединения.
Избыточное количество алюмогидрида лития можно разложить влажным эфи-
ром или осторожным прибавлением воды или раствора соды. Для разложения боль-
ших количеств алюмогидрида лития применяют вещества, разлагающие гидрид без
выделения водорода. Таким веществом является, например, этилацстат, поскольку
продукт его восстановления — спирт — обычно не мешает дальнейшему выделению
продукта реакции *.
Восстановление карбонильной группы алюмогидридом лития, протекающее
по общему уравнению
4Rx>C=O + LiAlH4 —»- [R ^>СНО—] AlLi -^>4R>CH-OH
R'Z |_R'/ J4 R'Z
выгодно отличается от восстановления по Мссрвсйну— Поппдорфу — Верлею меньшей
продолжительностью взаимодействия и отсутствием побочных реакций. Выходы при
этом не уступают таковым при восстановлении алкоголятамп алюминия.
Гептиловый спирт [293]. В трехгорлую колбу емкостью 2 л с мешалкой,
обратным холодильником, капельной воронкой и хлоркальциевой трубкой,
вносят раствор 19 г (0,5 .ноль) алюмогидрида лития в 600 мл эфира. При переме-
шивании добавляют 200 г (1,75 моль) и-гептаналя с такой скоростью, чтобы
поддерживать кипение реакционной массы. Через 10 мин при перемешивании
и сильном охлаждении осторожно вводят по каплям воду для разложения избытка
гидрида. Реакционную массу выливают затем в 200 мл ледяной воды и добавляют
1 л 10%-ной серной кислоты. После отделения эфирного слоя водный раствор
еще дважды экстрагируют порциями эфира по 100 мл. После высушивания эфир-
ных экстрактов и отгонки эфира содержапптйся в остатке /(-гептиловый спирт
ректифицируют с помощью колонки. Выход спирта 86%; т. кип. 175—175,5° С
(750 мм рт. ст.).
Аминоальдпгиды также могут быть восстановлены алюмогидридом лития.
Описано [298], например, восстановление 2,2-дпметил-З-мстиламинопрошгонового
* Удобным методом разложения является осторожное добавление воды с после-
дующим прибавлением строго определенного количества 15%-ного раствора
едкого натра, что обеспечивает получение хорошо фильтрующихся осадков
[V. М. М i с о v i с, М. Mi с h a i I о v i сУ J. Org. Chem., 18, 1196 (1953)].
В тех случаях, когда полученное J процессе восстановления вещество склонно
к образованию комплексов с соединениями алюминия, рекомендуется добавка
сегнетовой соли [II. И. С у в о р о в, В. С. Мурашова, ЖОХ, 30, 3112
(1960)]. —Прим. рсд.
[295] L. W. 'Г г е v о v, W. G. Brown, J. Ain. Chem. Soc., 71, 1675 (1949).
[296] F. A. Hochslein, J. Am. Chem. Soc., 71, 305 (1949).
[297] W. Bunge, в книге H ou ban - We vl, Methoden der organischen Chemie,
Bd. I, T. 2, 4 Aufl., 1959, S. 765.
[298] K. Hayes, G. Drake J. Org. Chem., 15, 873 (1950).
Ш. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = О-СВЯЗЯМ
f
альдегида избытком алюмогидрида лития, Соответствующий спирт получен с 72%-ны
выходом:
СНз СНз
CH3-NH-CH2-C—СНО L1AlH--> СНз—NH—СНа-С-СН20Н
I I
СНз СНз
По реакции Меервейпа — Нойндорфа — Верлен выход составляет лишь 27%
Реакция восстановления ароматических альдегидов протекает гладко. Феполалх
дегиды тоже могут быть восстановлены, хотя часто с плохими выходами. Примере»
гладко протекающей реакции может служить получение л-оксвбензилового спирт
[299] „о обычному методу. Попытки восстановить ванилин и вератровый альдеги
по увенчались успехом [300].
В случае очень реакционноспособных сопряженных систем иногда восстану
вливается и двойная связь. При восстановлении коричного альдегида в обычных угле
лпях образуется сразу гидрокоричный спирт. Если же восстановление проводить пр
—10° С, прибавляя раствор гидрида к раствору альдегида, то с хорошим выходе»
получается коричный спирт [301].
Если карбонильная группа в боковой цепи не сопряжена с ароматическим коле
цом, то двойные связи многократно сопряженной системы не восстанавливаются
Этот факт использовался при получении витамина Л из соответствующего альдегид
1302].
НзС СПз
\<^/СП=СН-С=СП-СН=СН-С=С11—сн2он
\/\сн3 СНз СИз
витамин А
Гидрирование алюмогидридом лития нашло широкое применение в химнн каро
тпноидов [87].
Этот метод может быть использован также для восстановления гетероцикличе
ских альдегидов. Например, 2-оксиметилпиррол получен [303] по «инвертному
методу из 2-пирролальдегида с 59%-ным выходом, в то время как пирролальдегидх
с большим числом заместителей восстанавливаются с трудом или совсем не восстапа.
вливаются [304].
Восстановление кетонов алюмогидридом лития проводится таким же способом
как и восстановление альдегидов; можно привести большое число примеров испольаова.
ния этого реагента для восстановления кетонов. Из галоидзамещенных кетонов полу-
чаются галоидгидрины. С хорошим выходом протекает восстановление 1,3-дихлораце
тона алюмогидридом лития; при восстановлении алкоголятами алюминия [305
1,3-дихлоргидрин получается лишь с выходом 20—25%. С хорошими выходами вое
стапавливаются также до соответствующих карбинолов сгерически затрудненны-
кетопы, как, наппимер, 1,1-дифенилацетон [306]. Из димезитилкетона получают димя
зитилкарбипол [307]; выход 93%.
[299] М. А. К а г i m, J. Indian Chem. Soc., 37, 244 (i960).
[300] Е. Larsson, Trans-Chaimers Univ. Techno]. Gothenburg, X» 94, 15 (1950)
C. A., 45, 1494 (1951).
[301] F. A. 11 ochstein, W. G. Brown, J. Am. Chem. Soc., 70, 3484 (1948)
[302] N. I,. Wen dler, C. R ose n b I u m, M. T is h] er, J. Am. Chem. Soc.
72, 234 (1950); J. F. A re ns, D. A. v. D or p, Rec. trav. chim., 68, 604 (1949>
[303] R. M. Silverstein, E. E. Ryskiewicz, S. W. Chaikin, J. Ata.
Cbem. Soc., 76, 4485 (1954).
[304] A. T re i bs, H. Scherer, Ann., 577, 139 (1952); A. Treibs, H. D err д
S c li e г e r, Ann., 589, 188 (1954).
[305] H, Schlenk, B. L a m p, J. Am. Cbem. Soc., 73, 5493 (1951).
[306] S. W i ns t e i п, В. К. M о rse et al., J. Am. Chem. Soc., 74, 1113 (1952).
[307] M. S. N c w m a n, N. C. Deno, J. Am. Chem. Soc., 73, 3644 (1951)
64
ОБРАЗОВАНИЕ С—Н-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Плохо растворимые в эфире пли трудно восстанавливающиеся вещества гид
рируют в аппарате Сокслета; правда, способ применим только для небольших загру-
зок *. При таком аппаратурном оформлении 1,8-дпбензонлнафталип восстанавливается
с 96%-иым выходом в соответствующий карбинол [308]:
\_/-co-CsHs L1AIH1 <^_^>-cikoh)-c,hs
)>-CO'-CsHs / СН(ОН)-C„HS
Кетопроизводные конденсированных циклических систем восстанавливаются
обычно с хорошими выходами. Из 9-ацетилантрацсна [309] соответствующий карби-
нол получается с 80%-ным выходом.
При восстановлении различных ароматических кетонов с аминогруппой в орто-
или пара-положении к кетогрупне в присутствии избытка алюмогидрида лития наряду
с соответствующими карбинолами образуются также и метиленовые соединения [310]
по следующей схеме:
о он
I* 1 ।
Чем болыпе избыточное количество алюмогидрида лития взято для реакции,
тем выше выход и-аминодифенилметана.
Аналогичная реакция протекала при восстановлении гетероциклических кето-
производных. Так, из 2-ацетплпиррола при обычном способе проведения реакции
образуется смесь небольших количеств пнрролметплкарбипола и 2-этилпиррола.
Прп обратном порядке введения реагентов [311], т. е. при добавлении алюмогидрида
лития к карбонильному соединению, получается толт.ко карбинол; выход 80%.
При восстановлении 2-ацетилтпофена алюмогпдридом лития образуется 1-(2-
тиенил)-этиловый спирт, в то время как прп каталитическом гидрировании восстана-
вливается и тиофеновое кольцо [312].
Асимметрический центр, находящийся рядом с карбонильной группой, при
восстановлении не изменяется. Так, из (—)-ментона получается смесь ментолов [(71%
( —)-ментола и 29% неоментола (г/uc-)], при окислении которых снова образуется
(—)-мептоп [313]:
I
I I
TJA1H4
Na^arjOj
Так как при восстановлении кетогруппы возможно возникает асимметрический
атом углерода или новые циклические чис-пгрямс-изомеры, то встает вопрос о стерео-
специфичности подобного метода восстановления. Хотя, в большинстве случаен обра-
зуется смесь обоих эпимеров или цис-транс-изомеров, известны примеры, когда один
из изомеров получается в большом количестве или даже с количественным выходом.
Если восстановлению подвергаются стерически незатрудненные кетоны, в основном
*
[308]
[309
[310
[311
[312
[313
Восстановление таких соединении может быть часто успешно проведено обыч-
ным способом при использовании в качестве растворителя тотрагидрофурана. —
Прим. ред.
R. L. Letsinger, Р. Т. Lansbury, J. Am. Chem. Soc., 81, 935 (19o9).
E. L. May, E. Mosettig, J. Am. Chem. Soc., 73, 1301 (1951)
L. H. Conover, D. S. Tarbell, J. Am. Chem. Soc., 72, 3586 (1950).
\V. Her z, Ch. F, Courtney, J. Am. Chem. Soc., 76, 576 (1954).
T. L. Cairns, В. C. Me К u s i c k, J. Org Chem., 15, 790 (1930).
D. S. N oyce, D. B. Denney, J. Am. Chem. Soc., 72, 5743 (1950).
III. ПРИСОЕДИПЕ'НИЕ водорода по с-=о-связям
65-
образуется более стабильный изомер, а при значптельпых стерических затруднениях
нодород присоединяется преимущественно с более доступной стороны молекулы.
Так, при восстановлении камфоры с 94%-ным выходом образуется смесь борнеол»
(10%) и изоборнеола (90%), т. е. преобладает цг/.с-пзомер [313].
Стереохимия восстановлений лптийалюминийгидридом, роль которой особенно
велика в области стероидов, ио может рассматриваться здесь более подробно; эти
вопросы освещены в обзорных статьях [87, 291].
Восстановление хпнонов протекает аналогично восстановлению кетопов. В то
время как из о-бензохинона с хорошим выходом образуется пирокатехин, из 1,2-наф-
тохпнона получается смесь 1,2-диоксииафталппа и транс-1,2-диокси-1,2-дигидронафта-
лина [3I4J. Из 1,4-пафтохинона наряду с 1,4-диокси-1.2.3,4-тетрагидронафталином
образуется также 4-окси-1-кето-1,2,3,4-тстрагмЛронафт«злпя [315].
4. Восстановление комплексными боргидридами
В последнее время наряду с алюмогидридом лития большое значение в качестве
новых восстановителей в препаративной органической химии приобрели различные
комплексные боргидриды [316, 317]. Среди них главное место занимают боргадриды
щелочных и щелочноземельных металлов, цинка и алюминия, триметокемборгидрид
натрия, а также диборан [317]. Восстановительные свойства слабее всего ныражеяы
у соединений чисто ионного характера, например у боргидридов щелочных металлов,
промежуточное положение занимают боргидриды щелочноземельных металлов, силь-
нее всего восстановительные свойства выражены у боргидрпда алюминия — соедине-
ния с ковалентными связями.
В настоящее время известно большое число методов получения комплексных
боргидридов. Боргидриды щелочных металлов и триметоксиборгидрнд натрия выпуска-
ются промышленностью; синтез других соединений описал в литературных обзорах
[87, 316-318].
Восстановление комплексными боргидридами во многих отношениях аналогично
восстановлению алюмогидридом лития, но имеет значительные преимущества. Ввиду
высокой стабильности боргидридов натрия и калия восстановление можно проводить
в водной и в спиртовой среде. Кроме того, при их употреблении почти не протекает
побочных реакции. Боргидриды щелочноземельных металлов, трпметоксиборгидрид
натрия п боргидрид лития применяются в эфирных растворах пли в растворе тетра-
гидрофурааа. Боргидрид натрия нерастворим в эфире вследствие незначительной склон-
ности иона натрия к сольватации, но он растворим в тетрагидрофуране, диоксанб
и диметиловом эфире диэтиленгликоля (диглиме). Пока еще редко применяемый при
восстановлениях диметилформамид является исключительно подходящим раствори-
телем для боргидрпда натрия. Восстанавливаемое вещество п боргидрид обычно
вводят в реакцию в виде растворов в различных растворителях *.
Для восстановления комплексными гидридами бора, как правило, нет необхо-
димости в исключения доступа влаги и кислорода воздуха. В отличие от алюмогид-
рида лития исходные растворы готовить не нужно, так как комплексные гидриды бора
выпускаются в виде хорошо растворимых мелкокристаллических веществ. В большин-
стве случаев восстанавливаемое соединение добавляют к гидриду, по применяют также
и обратный способ, при котором к веществу небольшими порциями прибавляется вос-
становитель. При восстановлении неустойчивых в щелочной среде соединений, напри-
мер альдегидов, следует пользоваться обычным способом смешения реагентов, так как
при этом действию щелочного раствора подвергается лишь незначительная часть исход-
ного материала. Обычно вводят избыток восстановителя, который после окончания
реакции разлагают небольшим количеством муравьиной кислоты, разбавленной
* Прекрасным растворителем для восстановлении боргидридом натрия является
влажный пиридин [Н. ТТ. Суворов, 3. Л. Ярославцева, ЖОХГ
29, 2889 (1959)]. — Прим. ред.
[314] J. Booth, Е. Boyland, Е. Е. Turner, J. Chem. Soc., 1950, 1188.
|315] Е. Boyland, D. Manson, J. Chem. Soc., 1951, 1837.
[316] B. Bormann, Angew. Chem., 68, (501 (1956).
[317] E. Schenker, Angew. Chem., 73, 81 (1961).
]318] E. Wiberg, Angew. Chem., 65, 16 (1953); F. G. A. Stone, Quart, Rev.
(Chem. Sec., London), 9, 174 (1955).
5 Заказ 1835.
бб
ОБРАЗОВАНИЕ С—Н-СВЛЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
минеральной кислоты пли ацетона [319}. Несмотря па то что с комплексными
боргидридами проще работать, чем с алюмогидридом лития, следует все же соблю-
дать некоторые меры предосторожности. Разложение избытка гидрида бора концен-
трированными кислотами может привести к бурной реакции с выделением взрывчатых
диборанов. Применение уксусной кислоты для разложения избытка гидрида, особенно
после восстановления алифатических альдегидов и кстопов, не рекомендуется, так
как при этом наряду с продуктами реакции могут легко образоваться эфиры
борной кислоты.
Тщательно изучалось [320] восстановление алифатических и ароматических
альдегидов боргидридом натрия. Обычно уже при комнатной температуре в результате
исключительно гладко протекающей реакции образуются с хорошими выходами соот-
ветствующие карбинолы:
4RCHO + NaBH4 —> NaB(0CH2R)4 —> 4RCH2OH
Боргидрид натрия является специфическим восстановителем карбонильных
групп и в большинстве случаев не восстанавливает двойных связей ненасыщенных
альдегидов. Точно так же питрогруппы, сложноэфирные, лактонпые, лактамные,
амидные и эпоксигруппы восстанавливаются лишь в исключительных случаях. Таким
образом, боргидрид натрия представляет собой ценность как избирательный восстано-
витель. Характерным примером является избирательное восстановление а-бромакро-
леина с гетероциклическим заместителем [321]:
Il I yr NaBH,, || I Вг
<)2n z ,/'' cii-c| 0,n/ ,/ cit=c
сно СН2ОН
В этой реакции бром, нитрогруппа и двойная связь остаются незатронутыми.
Благодаря хорошей растворимости в воде и метиловом спирте боргидрид натрия
часто находит применение для восстановления сахаров. При этом гликозидные связи
не затрагиваются. Дезоксисахара обладают большей реакционной способностью, чем
соответствуюптпе нормальные углеводы [322]. Часто при восстановлении углеводов
образуются комплексные соединения с борной кислотой, что затрудняет кристаллиза-
цию получаемых многоатомных спиртов. Такое осложнение можно преодолеть иосле-
дующим ацетилированием, при этом с хорошим выходом получают полностью ацетили-
рованные, хорошо кристаллизующиеся углеводы [323].
Восстановление ©-глюкозы, /7-маннозы, ©-галактозы, Ь-арабинозы,
D-ксилозы и ©-фруктозы. К раствору 1 г моносахарида в 20 мл воды при-
бавляют раствор 0,1—0,15 г боргидрида натрия в 10 мл воды. Реакционную
смесь, приобретающую слабощелочную реакцию, выдерживают при 20—25° С
до тех пор, пока подкисленная уксусной кислотой капля не даст отрицательную
реакцию с фелинговой жидкостью, или до постоянного значения удельного
вращения. Реакция заканчивается через 1 — 2 ч. Реакционную массу подкисляют
уксусной кислотой для разложения избытка боргидрида натрия и упаривают
в вакууме досуха. Сухой остаток встряхивают с 15 мл уксусного ангидрида,
содержащего 1 мл концентрированной серной кислоты, до растворения большей
части твердого продукта реакции и нагревают в течение 10 мин при 50—60° С.
После охлаждения 'реакционную массу выливают при перемешивании в 50 мл
воды со льдом. Хорошо кристаллизующийся ацетат отфильтровывают, промывают
водой, сушат и перекристаллизовывают обычным способом. Выходы составляют
от 75 до 87% от теоретического.
[319
[320
[321
[322
[323
D. J. С г a m, М. J. Н a t с h, J. Am. Chem. Soc., 75, 38 (1953).
S. W. С h a i k i n, W. G. Brown, J. Am. Chem. Soc., 71, 122 (1949).
G. Carrara, R. Ettorre et al., J. Am. Chem. Soc., 76, 4391 (1954).
J. B. L e e, Chem. Ind., 1959, 1455.
M. Abdel - Akher, J. K. Hamilton, F. Smith, J. Am, Chem. Soc.,
73, 4691 (1951).
III. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = О-СВЯЗЯМ
67'
Аналогично протекает селективное восстановление альдегидных групп сер-
дечных гликозидов и агликонов. Так были восстановлены [324] конваллотоксин, стро-
фантидип и аналогичные соединения.
Боргидрид калия действует совершенно так же, как боргндрид натрия, но для
восстановления альдегидов применяется реже. Более сильным восстанавливающим'
денствием обладает триметоьсиборгидрид натрия; боргидрид лития менее специфичен,
чем боргидрид натрия. Браун и Мид [325] использовали трпметоксиборгидрид натри»
для восстановления коричного альдегида.
Коричный спирт [317]. К суспензии 40 г триметоксиборгидрида натрия-
в 100 мл эфира прибавляют 21,7 г (0,165 моль) свежеперегнаиного коричного
альдегида; при этом реакционная масса разогревается до кипения. После нагре-
вания в течение 4 ч с обратным холодильником реакционный сосуд охлаждают
и прибавляют разбавленную серпую кислоту до прекращения выделения водо-
рода. Эфирный раствор сушат над сульфатом натрия и отгоняют растворитель.
При фракционированной разгонке получают 17,5 г (80% от теоретического)
коричного спирта; т. кип. 134—135° С (13 ,н.и pm. cm.).
Моноциаиборгидрид лития восстанавливает альдегиды при нагревании в диок-
сапе, например пиренальдегид (выход 85%), но не восстанавливает ароматических
и алифатических кетонов [326]. Из боргидридов щелочноземельных металлов следует-
упомянуть лишь трпметоксиборгидрид кальция, получаемый взаимодействием гид-
рида кальция с триметилборатом. Препаративное применение этого вещества для вос-
становления карбонильных соединений исследовано Хессе и Егером [327].
В сравнении с альдегидами большинство кетонов восстанавливается комплекс-
ными боргидридами довольно медленно. На основании различия в скоростях реакции
разработаны методы разделения альдегидов и кетонов [328]. Имеется ряд примеров
восстановления алифатических, алициклических, ароматических и гетероцикличе-
ских кетонов комплексными боргидридами, применение которых вместо алюмогидрида
лития особенно показано в тех случаях, когда в молекуле имеются одновременно дру-
гие функциональные группы, как, например, амидная, карбоксильная, эпоксидная,
сложпоэфирная, нитрильная, а также нитро- или меркаптогруппы.
5-Нитропентанол-2 [329]. Раствор 2,8 s (0,075 моль) боргидрида натрия'
в 50 мл воды, к которому добавлена капля Концентрированного раствора едкого
натра, в течение 1 ч при медленном перемешивании прибавляют по каплям
к смеси 19,7 г (0,15 моль) 5-ннтропевтанона-2 и 50 мл метилового спирта. Темпе-
ратуру смеси на уровне 20 — 25° С поддерживают охлаждением, а pH 3—4 регу-
лируют постоянным добавлением 3 п. раствора серной кислоты (контролируют-
pH-метром). Во время добавления боргидрида натрия выделяется водород и об-
разуется белое твердое вещество. После прибавления всего боргидрида смесь,
оставляют на 5 мил и разлагают избыток боргидрида натрия, добавляя i мл
концентрированной серной кислоты. После разбавления реакционной массы
водой до 350 мл и нейтрализации концентрированным раствором едкого натра
гомогенный раствор экстрагируют эфиром. Объединенные эфирные вытяжки
промывают трижды насыщенным раствором бисульфита натрия (порциями do-
150 мл в течение 5 мин) и насыщенным раствором хлорида натрия, затем филь-
труют через безводный сульфат натрия. После этого добавляют бензол и отго-
няют растворители. Вносят несколько кристаллов борной кислоты п перегоняют
в вакууме полученный 5-интропентакол-2, представляющий собой бесцветную-
жидкость; т. кип. 101 — 102,5° С (2 pm. cm.); выход 11,3 г (86,6% от теорети-
ческого).
[324] A. Hunger, Т Reichstein Chem. Вег., 85, 635 (1952); Helv. chim.
acta, 35, 1073 (1952).
[325 ] H. С. В г о w n, E. J. Me a d J. Am. Chem. Soc., 75, 6263 (1953).
[326] G. D ref a hl, E. Keil, J. prakl. Chem. [4], G, 80 (1958).
[327] G. Hesse, H. Jager, Chem. Ber., 92, 2022 [1959].
[328] J. P. С r i t c h 1 e y, J. Friend, T. Swain, Chem. a. Ind., 1958, 596.
[329] H. Shechter, 15. E. Ley, L. Zeldin, J. Am. Chem. Soc., 74,_
3G68 (1952).
5*
ku...
68
ОБРАЗОВАНИЕ С—Н-СНЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Типичным примером избирательного восстановления является взаимодействие
1-(2'-хлор-6'-нптробензил)-2,3-диоксопирролидина с боргидридом натрия, при котором
восстанавливается только одна кетогруииа [330]:
Cl CI
U\ LxA А/
w 4NOzO || AZ\NO2o j
о on
При восстановлении циклических кетонов триметоксиборгидрид натрия оказы-
вает более стереоспецифическое действие, чем боргидрид натрия. Восстановление 2-
п 4-метилциклогексавонов протекает с образованием тряке-изомеров [331] с выходами
соответственно 99 и 88%.
Алифатические, алициклические и ароматические дикетоиы обычно гладко
восстанавливаются до диолов эквивалентным количеством комплексных боргидридов.
С успехом проведено также восстановление хинонов боргидридом натрия или тримет-
оксиборгндридом патрия в различных растворителях.
5. Каталитическое гидрирование
Карбонильная группа в альдегидах и кетонах гидрируется в присутствии
катализаторов уже в мягких условиях. Выходы в большинстве случаев практически
количественные.
Карозерс и Адамс [332] исследовали каталитическое гидрирование альдегидов
в присутствии катализаторов на основе окиси платины и нашли, что добавка солей
железа ускоряет гидрирование.
ж-Метоксвбензиловый спирт [333]. В аппарат для гидрирования по
Адамсу (см. стр. 42) загружают 40 г ж-метоксибензальдсгида, 200 мл этилового
спирта, 0,2 г свежеприготовленной окиси платины по Адамсу и 2 мл 0,1 М рас-
твора сульфата железа. Реакционный сосуд начинают встряхивать при давлении
водорода 3 ат\ через 10 мии поглощается рассчитанное количество водорода
(0,294 моль). Катализатор может быть1.использован и для последующих гидриро-
ваний, однако скорость поглощения водорода уменьшается. После отгонки эти-
лового спирта при пониженном давлении остающийся ж-метоксибензиловый
спирт перегоняют в вакууме в токе азота; т. кип. 150° С (25 мм рт. ст.)', выход
количественный.
В то время как ацетон в присутствии окиси платины гидрируется с хорошим
выходом до изопропилового спирта, ряд кетонов, как, например, метилизопропилке-
тон, метил-н-амилкетоп и 2-метилгептанон-З, в этих условиях не восстанавлива-
ются [334].
Гидрирование карбонильных соединений в присутствии никеля Репея [335]
систематически исследовали Делепйн и Оро [127]. По данным этих авторов скорость
поглощения водорода различными соединениями колеблется в широких пределах.
При добавлении к карбонильным соединениям небольших количеств щелочи скорость
поглощепия водорода возрастает в два, три, а в некоторых случаях даже в десять раз.
При использовании никеля Ренея, активированного благородными металлами (пла-
тина, палладий), добавка щелочи Ьказывает еще более сильное действие. Хорошие
[330] Р. L. Bout hwick, S. Е. Cremer, J. Org. Chem., 24, 753 (1959).
[331] W. G. Doube в, G. J. Ponken, D. S. N о у c e, J. Am. Chem. Soc., 78,
2579 (1956).
[332] W. H. Carothers, R. Adams, J. Am. Chem. Soc., 4G, 1675 (1924).
[333] R. B. Woodward, J. Am. Chem. Soc., 62, 1480 (1940).
[334] p, C, Wh it e m ore, P. А. К r uege r, J. Am. Chem. 8oc., 55, 1531 (1933).
[335] R. S c h г 6 t e г, в. книге W. Foerst, Neuore Methoden dec praparaliven
organischen Chemie, 3 Aufl., Bd. 1, 1949, S. 98.
in. ПРИСОЕДИНЕНИЕ ВОДОРОДА ПО С = О-СВЯЗЯМ
69
результаты получены Делепином и Оро несомненно потому, что они работали со свеже-
приготовленным никелем Ренея, который получали непосредственно перед опытом
разложением эквивалентного количества сплава. При гидрировании альдегидов
и углеводов следует учитывать, что применение щелочей может вызвать побочные
реакции.
Оптимальное активирование никеля Ренея для восстановления карбопильной
группы достигается платинированием его в присутствии триэтиламина и последующей
обработкой щелочью [336].
Основания Манпиха типа R2NCH2C(CH3)8CHO гидрируются в виде гидрохло-
ридов в кислом растворе при pH 3—6 в присутствии никеля Реноя с большим успехом,
чем в присутствии благородных металлов. При этом с хорошими выходами образуются
соответствующие аминоспирты [337].
Особенно удачным катализатором является никель Ренея W-6, с которым
можно работать в стеклянной аппаратуре при низких температурах и низком давле-
нии. Имеется подробная пропись для получения такого катализатора [338]. Актив-
ность этого катализатора при восстановлении карбонильных соединений повышается
в присутствии триэтиламина [339]. С помощью никеля Ренея с добавкой триэтиламина
можпо нрогидрировать и эфиры кетокарбоновых кислот до эфиров оксикарбоновых
кислот. Так, из диэтилового эфира ацетондикарбоновой кислоты с почти количествен-
ным выходом получают диэтиловый эфир ji-оксиглутаровой кислоты [340].
Примепопие кобальтового катализатора для гидрирования кетонов с длинной
цепью при температурах около 200° С и давлении около 200 ат описано Авингером
с сотр. [342].
Каталитическое восстановление углеводов можпо проводить в присутствии
никеля Ренея. Однако из альдоз и кетоз соответствующие полиспирты удобнее полу-
чать простым кипячением в спирте с никелем Ренея [341].
Карбонильная группа альдегидов и кетонов гладко восстанавливается до спир-
товой группы при 125—150° С и давлении около 100 ат под действием меднохромовых
катализаторов.
По данным Адкинса и Коннора [153], ряд альдегидов и кетонов гидрировались
в этих условиях со 10и%-ным выходом. Так же гладко гидрируются кетослиргы, альде-
гидоспирты и эфиры кетокислот до соответствующих гликолей пли эфиров оксикислот.
Сели в соединениях имеются одновременно олефиновые двойные связи, то и они гидри-
руются в этих условиях.
Ароматические альдегиды и кетоны с гидроксильной группой в орто- иля пара-
положепии к карбонильной группе гидрируются уже при 110—130° С до углево-
дородов, [343].
Ряд арилметилкарбиполов был нолучеп гидрированием соответствующих кето-
пов при 120—150° С в присутствии медпохромового катализатора [344]. 3,4-Дихлор-
ацетофепон и 1-хлор-4-ацетилпафталин удалось прогидрировать с большим трудом,
так как галогенсодержащие соединения отравляют катализатор. По данным Ипатьева
п Хензеля [345], фенилалкилкетопы восстанавливаются при 115° С также в присутст-
вии катализатора на основе окиси меди и окиси алюминия без восстановления бензоль-
ного кбльца.
Изучалось восстановление метилциклоиропплкетона до метилциклопропмлкар-
бинола четырьмя различными методами [346]. При действии натрия и спирта
[336]
1337]
[338]
[339]
[340]
[341]
[342]
[343]
1344]
[345]
[346]
R. В. В 1 а и с е, D. Т. G i b s о n, J. Chem. Soc., 1954, 2487.
W. Wenner, J. Org. Chem., 15, 301, (1950).
H. R. В i I ] i с a, H. Adkins Org. Synth., 29, 24 (1949).
H. Adkins, H. R. В i I 1 i c a, J. Am. Chem. Soc., 70, 3118 (1948).
II. Stetter, O.-E. Bander, W. Neumann, Chem. Ber., 89, 1922
(1956).
J. V. Karabinos, A. T. Ballun, J. Am. Chem. Soc., 75, 4501 (1953).
F. Asi nger, If. E с k о 1 d t, Ber., 78, 579 (1943); F. As i n g er, Ber., 77,
73 (1944).
D. Nightingale, II. D. Bedford, J. Ore. Chem., 14, 1089
(1949). g
D. T. Mowry, M. R e n о 11, W. F. Huber, J. Am. Chem. Soc., 68» 1105
(1940).
V. N. I p a t i e f f, V. II a e u s e 1, J. Am. Chem. Soc., 84 , 520 (1942).
V. A. S 1 a b e у, P. H Wise, J. Ain. Chem. Soc., 71, 3252 (1949).
70
ОБРАЗОВАНИЕ С—Н-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
получались незначительные выходы карбинола; применение литийалюмпнийгидрида
наталкивается на трудности при больших загрузках; при каталитическом гидрировании
в присутствии никеля Ренея частично раскрывается циклопропановое кольцо; в при-
сутствии активировапного барием меднохромового катализатора при 100° С полу-
чается метилциклопропнлкарбинол с отличным выходом.
ОБРАЗОВАНИЕ С—Н-СВЯЗИ В РЕЗУЛЬТАТЕ РЕАКЦИИ ОБМЕНА
I. Обмен галогена на водород [547]
В ряду галогенов прямым обменом легче всего вамещается на водород иод:
F < С1 < Вг < I
Такая реакция протекает лучше с атомом галогена, связанным с третичным
атомом углерода, хуже — соответственно с вторичным и первичным. Кроме того,
обмен галогена облегчается при наличии по соседству электронодонорных заместите-
лей, ослабляющих связь углерода с галогеном.
Наиболее часто употребляемым восстановителем является смесь неблагород-
ного металла с кислотой (уксусной, соляной). Наряду с этим применяются и каталити-
ческие методы, как, например, гидрирование в присутствии никеля Ренея, палладия
ц окиси платины.
Не меньшее препаративное значение имеет восстановление через промежуточное
образование магнийорганических соединений. Иногда хорошие результаты получаются
при восстановлении галогенидов комплексными гидридами металлов, например литий-
алюминиигидридом и три-трет-бутоксилитийалюминийгидридом.
Существует ряд методов восстановления хлорангидридов кислот до соответ-
ствующих альдегидов; из них наиболее широко применяются методы Розенмунда —
Зайцева к Рейссерта.
1. Обмен галогенов у простых С—С-связей
Прямой обмен
Для прямого обмена галогена на водород можно использовать большинство
восстанавливающих систем из неблагородного металла и кислоты. Наиболее широко
применяется цинковая пыль в сочетании с соляной кислотой или со смесью уксусной
и соляной кислот. Обычно восстановление проводят при температуре кипения. Обмен
галогена в неустойчивых соединениях протекает в течение нескольких часов при ком-
натной температуре [348].
Ципковой пылью и уксусной кислотой можно восстанавливать с хорошими
выходами галогспомстилкетоны до метилкетонов без изменения кетогруппы [349].
Замену галогена в чувствительных к действию кислот соединениях можно проводить
и не применяя кислот, для чего такие соединения, как, например, галогеностероиды,
нагревают с цинковой пылью в еппрте [350]. Замещенные бенаилгалогениды восста-
навливаются до углеводородов цинковой пылью даже в 10%-ном растворе едкого
натра [351]. Для обмена галогена можно применять также системы: натрий в спирте
[352]; хлорид олова (II) в соляной кислоте [3531; железо в соляной кислоте [354];
[347]
Г-348]
[349]
[350]
[351]
[352]
[353]
[354]
R. Stroh, в кпиге II ouben-Weyl — Muller, Methoden der orga-
nischen Chemie, Bd. 5/4, 4 Aufl., 1960, S. 761.
W. B. Whalley, J. Chem. Soc., 1951, 3229.
Schnider, H. J. Bourquin, A. Grupner, Helv. chim. acta, 28,
510 (1945)
II. II. Inhof fen, G. Rolling, I. N ebel, Chem. Ber., 84, 361 (1951).
R. C. Fuson, J. J, Denton, J. W. Kneisery, J. Am. Chem. Soc., G3,
2652 (1941).
H. F. B. Hirschmann, M. A. Dans, J. Biol. Chem., 178, 751 (1949).
R. E. Lutz et al,. J. Am. Chem. Soc., 68, 1813 (1946k
K. Matsumura, J. Am. Chem. Soc., 52 , 4433 (1930).
Т, ОБМЕН ГАЛОГЕНА НА ВОДОРОД
71
иодид калия, кислоту и ацетон [355, 356]; амальгаму натрия; амальгаму алюми-
ния [357].
Наиболее распространенный способ замены галогенов будет проиллюстрирован
на примере получения н-гексадекана из цетплиодида.
н-Гексадекан [358]. В круглодонной колбе емкостью 2 л, снабженной
мешалкой и барботером, насыщают сухим хлористым водородом смесь из 915 мл
ледяной уксусной кислоты, 327 г цинковой пыли и 352 г цетплиодида. Затем при
перемешивании нагревают на паровой бане, пропуская через каждые 5 ч хлори-
стый водород. Через 25 ч реакционной массе дают остыть 1 отделяют всрхпий
слой, состоящий из гексадекана. Остаток переносят в 3 л воды, отфильтровывают
цинковую пыль, промывают ее 500 мл вады и 250 мл эфира, дважды экстрагируют
гексадекан из объединенных водных фильтратов эфиром порциями по 500 мл
каждая и растворяют в эфирных вытяжках отделенный рапее гсксадекан. Эфир-
ный раствор дважды промывают 20%-яым раствором едкого патра, расходуя
каждый раз по 250 мл щелочи. Отмывают водой щелочь, сушат над 150 г безвод-
ного сульфата натрия и после отгопки эфира фракционируют углеводород
в вакууме с помощью колонки. Получают 192 г ч-гексадекана (85% от теорети-
ческого); т. кип. 156—158° С (144 мм рт. ст.')', т. пл. 16—17° С.
Иногда обмен галогена на водород лучше всего протекает под действием безвод-
ного хлорида хрома (II). В случае некоторых бромкетостероидов восстановление
цинковой пылью в уксусной кислоте дает неудовлетворительные результаты. Между
тем при длительном взаимодействии галогенида с раствором хлорида хрома (II)
в ацетоне в атмосфере двуокиси углерода и 25—30“ С продукты дегалогенирования
получаются большей частью с хорошим выходом [359]. а-Бромкетоны тоже могут
быть восстановлены по этой методике [360].
Ценным восстановителем некоторых алифатических галогенидов и бепзилгало-
генидов является гидрид лития в присутствии каталитических количеств алюмо-
гидрида лития. Этот реактив применим не во всех случаях и иногда, особенно при
дегалогенировании випилгалогенидов, не приводит к желаемым результатам (361].
Из каталитических методов следует упомянуть применение никеля Ренея и бла-
городных металлов (палладия). При гидрировании замещеппых бепзилгалогенидов
по методу Вессели и Синвела в качестве катализатора применяют 20%-пый палладий
на сульфате бария в спирте в присутствии дпметиланилина [362]. Добавление основа-
ний необходимо для связывания образующейся кислоты, которая в противном случае
блокирует катализатор.
В„ связи с этим следует упомянуть побочную реакцию, наблюдающуюся при ката-
литическом гидрировании галогенидов. Буш и Вебер [363] нашли, что при использова-
нии в качестве источника водорода гидразина образуются продукты конденсации,
например дибензил из бензилхлорида.
Постадийный обмен галогена на водород в полигалогенидах имеет значение для
синтеза галогензамещенных углеводородов типа RCHX2. Синтезировать такие со-
единения удобно через альдегиды, если последние легко доступны. Однако получить
соединения RCHXa прямым галогенированием концевой метильной группы не всегда
удается из-за трудности фиксирования реакции па промежуточной стадии. Поскольку
трнгалогензамещенные соединения легко доступны, во многих случаях целесообразнее
355]
356
357
358
359
[360'
[361]
[362]
[363]
А. Н. В 1 a 11, Е. W. Т г i s t г а ш, J. Am. Chem. Soc., 74, 6273 (1952).
С. I. Bickel, R. Morris, J. Am. Chem. Soc., 73, 1786 (1951).
P. R n g g 1 i, M. Herzog, Helv. chim. acta, 29, 95 (1946),
P. A. L e v e n e, Org. Synth., Coll. v. 2, 320.
J. B. Conant, H. B. Cutter, J. Am. Chem. Soc. 48, 1016 (1926).
P. L. J uli an, W. Cole, A. Magnani, E. W. Meyer, J. Am. Chem.
Soc., 67, 1728 (1945).
H. Гейл род, Восстановление комплексными гидридами металлов, Издат-
иплиг, 1959; В. Мичович, М. Михайлович, Алюмогидрид лития
и его применение в органической химии, Издатинлнт, 1957.
F. Wessely, F. S i n w е 1, Mh. Chem., 81, 1055 (1950).
М. Busch, W. Weber, J. prakt. Chem., 146, 1 (1936).
ia
72
ОБРАЗОВАНИЕ С—Н-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
переводить их в дизамсщенные, обменивая лишь один галоген на водород. Одним
из лучших восстановителен для этой цели является арсенит натрия.
Дибромметан [364]. В трехгорлую колбу емкостью 2 л, снабженную
мешалкой, капельной воронкой и обратным холодильником, помещают 540 г
продажного 88%-ного бромоформа, содержащего спирт (rf26 = 2,59). В колбу
добавляют сначала 10 мл раствора 230 г трехокиси мышьяка и 440 г едкого натра
в 1,4 л воды (избыток щелочи). Затем умеренным нагреванием па паровой бапе
вызывают реакцию, добавляют примерно в течение 1 ч остальной раствор арсе-
нита с такой скоростью, чтобы реакционная масса умеренно кипела, и нагревают
еще 4 ч на паровой бане. Продукт реакции отгоняют с водяным паром, отделяют
пижпий слой, содержащий дибромметан, и экстрагируют водный слой 100 мл
эфира для извлечения суспендированных капель дибромметана. Этот экстракт
объединяют с отделенным ранее дибромметаном и сушат 10 г хлористого кальция.
При перегонке получают 290—300 г слегка желтоватого дибромметана, что соот-
ветствует 88—90%-пому выходу от теоретического; т. кип. 97—100° С.
Дииодметап можно получить также из йодоформа по методу Адамса
и Марвела [365].
Тригалогеизамещенные гетероциклические соединения восстанавливают в соот-
ветствующие со-дигалогеняамещенные [366] нагреванием со спиртом в 20%-ной серной
кислоте или с хлоридом олова (II):
спирт: HgSO,
Обмен галогена, с получением промежуточных соединений
При гидролизе магнийорганических соединений водой или гидроксилсодержа-
щими соединениями образуются углеводороды:
RMgX-[ R'OH -> RH + R'OMgX
Как известно, на этой реакции основан метод количественного определения
гидроксильных групп по Чугаеву — Церевитипову. Кроме того, эта реакция имеет
некоторое препаративное значение для получения углеводородов из соответствующих
галогенидов. Так, при действип разбавленной уксусной кислоты на реактив Гриньяра
из 2-иодметилдекалина получается с хорошим выходом 2-метилдекалин [367]. С хоро-
шим выходом получается также циклобутан при взаимодействии раствора циклобутил-
магнпйбромида в «-дибутиловом эфире с н-бутиловым спиртом в течение 30 мии при
50° С. Применение ди-«-бутилового эфира и я-бутилового спирта облегчает выделение
иизкокппящего циклобутана [368].
Эта реакция протекает не только с водой п спиртом, ио и с аммиаком, первнч-
пымп и вторичными аминами; например, по данным Губена, лучшим методом получения
насыщенных углеводородов из реактивов Гриньяра является разложение его хлоридом
аммония [369].
Подвижный хлор в гетероциклических соединениях может быть иногда замещен
на гпдразогруппу действием гидразина. При разложении образующегося при этом
[364] W. W. Hartman, Е. Е. Drcger, Org. Synth., 9, 56.
[365] R. Adams, C. Marvel, Org. Synth., 1, 57.
[366] L. K. Sharp, J. Pharm. a. Pharmacol.. 1, 395 (1949).
[367] P. И. Левина, С. Г. Куликов, ЖОХ, 16, 117 (1946).
]368J J. Cason, R. L. Way, j. Org. Chem., 14, 31 (1949).
]369] J. II on hen, ВЙГ., 38, ,3017 (1905).
I. ОБМЕН ГАЛОГЕНА НА ВОДОРОД
-3
производного водным раствором сульфата меди получают вещество, в котором исход-
ный атом галогена залгещен на водород 1370}:
NH—NH2
СпЗОч
с„п5о/^/''/ ceHsoz"^/'N
Альберт и Роджер [371] разработалп способ замены хлора, заключающийся
в получении «-толуолсульфогидразинов с последующим разложением их щелочами.
И, наконец, иод в алифатических соединениях можпо обменять на водород путем
взаимодействия алкилподида с тпомочевиной с последующей десульфуризацией тиуро-
ниевой соли встряхиванием с никелем Ренея [372].
2. Обмен галогена у олефиновых С—С-связей
и в ароматических системах
Обмен галогена на водород в галогеггорганическпх соединениях с галогеном
при двойной или тройной связи часто удается под действием амальгамы алюминия
[373], алюмогидрида лития [374], а также цинковой пыли в смеси пиридина и уксус-
ной кислоты ]375], причем кратные связи между атомами углерода не затраги-
ваются. Под действием амальгамы натрия в первую очередь чаще всего происходит
гидрирование по двойной связи, как, например, в випилгалогепидах.
Связанный с ароматическими системами галоген довольно легко обменивается
на водород только в том случае, если его реакционная способность повышена за счет
э.тектроноакцепторных заместителей в орто- пли пара-положении. Так, в 1-хлор-2,4-
динитронафталппе галоген обменивается на водород уже при нагревании до 150—
200° С с порошком меди в присутствии органической, например бензойной, кислоты
[376]. Обменять галоген на водород часто удается также под действием натрия в спирте
[377], ацетата меди (I) в пиридине пли окиси меди (I) и уксусного ангидрида 1378],
и особенно при каталитическом гидрировании. В качестве катализаторов можно при-
менять палладий на животном угле или на карбонатах щелочноземельных металлов,
а также окись платины (катализатор Адамса). Возможность гидрирования 2-хлорлепи-
дина в присутствии никеля Ренея в спиртовом растворе со щелочью для получения
лепидина показывает, что этот катализатор применим также для обмена галогена
на водород [379].
Лепидин. В 120 мл спирта растворяют 20 г 2-хлорлепидина и 6,3 г
едкого кали. Затем добавляют 3 г никеля Ренея и гидрируют при комнатной тем-
пературе и атмосферном давлении до поглощения 1 .моль водорода. Реакция за-
капчивается через 16 ч. Катализатор отфильтровывают и отгоняют спирт. Оста-
ток экстрагируют эфиром и промывают водой. После высушивания эфирного
экстракта растворитель отгоняют, а лепидин очищают перегонкой; т. кип.
126° С (40 мм рт. ст.). Выход 94% от теоретического.
[370 ] R. О. Clinton, С. М. Suter, J. Am. Chem. Soc., 69, 704 (1947).
[371 ] A. Albert, R. Roger, J. Chem. Soc., 1949, 1148.
[372 ] E. Наг d e g g e r, R. M. M on t a v on, Helv. chim. acta, 29, 1199 (1946)
[373 ] T. И. Темникова, С. А. Баскова, ЖОХ, 21, 1823 (1951).
374] L. F Hat ch, R. H. Perry, J, Am. Chem. Soc., 71, 3262 (1949).
375] A. Roedig, H. Niedenhriick, Chem. Ber., 90, 682 (1957).
376] E. V. Brown, J. Am Chem. Soc., 76, 3167 (1954).
377] A. Stepanow, Ber., 39, 4056 (1906).
378] W. G. H. E d w a r d s, R. G. St ewart, Chem. a. Ind., 1952, 472.
379] S. E. К r ahi er, A. Burger, J. Am. Chem. Soc., 63, 2367 (1941).
74
ОБРАЗОВАНИЕ С—Н-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
При использовании для гидрирования 2-хлорлепидина катализаторов
на основе благородных металлов [380] вместо никеля Ренея время реакции зна-
чительно сокращается и составляет лишь 1,5 — 2 ч.
3. Обмен галогена у других связей
(в хлорангидридах кислот)
Обмен галогена па водород в хлорангидридах кислот осуществляют по реакции
Розенмунда — Зайцева [381—383]. Избирательным гидрированием хлорангидридов
кислот в присутствии подходящего катализатора, чаще всего палладия (5%) на суль-
фате бария (95%), получают альдегиды. Восстановление проводят, пропуская ток
водорода через кипящий раствор хлораягидрида кислоты в ксилоле или в толуоле»
в котором суспендирован катализатор. При этом следует полностью исключить доступ
влаги и кислорода воздуха в реакционную массу, так как в противном случае образу-
ются ангидриды кислот.
Однако описанный метод применим лишь в том случае, если предотвращено
дальнейшее гидрирование образующегося альдегида до спирта или соответственно
до углеводорода. Это достигается введением описанного Розенмундом и Цетше [384]
контактного яда «хннолина-S», который может быть получен из хинолина и серы.
Позже Вейганд и Мендель [385] нашли, что хинолин-S, состав и строение которого'
неизвестны, можно заменить тиомочевиной. Другими контактными ядами являются
фенилгорчичное масло и 2-меркаптобепзтиазол. ,
Значительное влияние на выход альдегида оказывает температура реакции.
Поддерживая температуру на уровне, при котором началось выделение хлористого
водорода, получают альдегиды с самыми высокими выходами, тогда как при более высо-
ких температурах выход снижается вследствие образования побочных продуктов
реакции [386].
При таком способе восстановления не затрагиваются даже такие легко восста-
навливаемые группы, как кетогруппа и нитрогруппа и даже алифатически связанный
галоген. Правда, гидроксильные группы надо защищать ацетилированием.
Хинолия-S. Его получают кипячением в колбе с обратным холодильни-
ком в течение 6 ч 1 з серы с 6 г хинолина и разбавлением полученного продукта
70 мл обессеренного ксилола.
Получение катализатора — палладия на сульфате бария описано Мо-
зипго [387].
По другому методу хлорангидриды кислот переводят в альдегиды через соедине-
ния Рейссерта [388]. Рейссерт обнаружил, что при действии хлористого бензоила на
хинолин в присутствии водного раствора цианида калия протекает реакция, аналогич-
ная реакции Шоттена — Баумаппа, и с хорошим выходом образуется 1-бенэоил-
[380] F. W. Neumann, N. В. Sommer, С. Е. К as] о w,
R. L. S h г i n е г, Org. Synth., Coll. v. 3, 1955, p. 519.
{381 ] О. В а у e г, в книге Houben-Weyl — Mtiller, Methoden der orga-
nischen Chemie, Bd. 7, T. 1, 4 Aufl., Stuttgart, Georg Thieme Verlag, 1954,
S. 285.
[382] E. Mosettig, R. Mozingo, Org. Reactions, v. 4, New York, John Wiley
a. Sons, 1948, p. • 362.
[383] K. W. R о s e n m u n d, Ber., 51, 585 (1918); A. S a у t z e f f, J. prakt. Chem.
[2], 6, 130 (1872).
[384] K. W. Rosenmund, F. Zetsche, Bor., 54, 425 (1921); 56, 1481
(1923).
[385] C. We у g and, W. M e n z e 1, Ber., 76, 503 (1943).
[386] T. Boehm, G. Schumann, II. H. Hansen, Arch. d. Pharm., 271,
490 (1933).
[387] E. Schmidt, Ber., 52, 409 (1919); R. Mozingo, Org. Synth.,
Coll. v. 3, 1955, p. 685'.
[388] О. В а у e г, в книге Houhen-Weyl — Muller, Mcthoden der organi-
schen Chemie, Bd. 7, T. 1, 4 Aufl., Stuttgart, Georg Thieme Verlag, 1954,
S. 291.
II. ОБМЕН КИСЛОРОДА НА ВОДОРОД
2-циан-1,2-дигидрохинолнн, который гидролизуется кислотами, причем получается
бензальдегид и хинальдиновая кислота:
COCI —* (^Y^+CeH6-c/° +NH3
'V'VXcn Vv^cooh Чн
1
COCeHs
Соединение Рейссерта. Наиболее целесообразным способом его полу-
чения является взаимодействие 1 моль хлорангидрида кислоты в сухом бензоле
с i моль безводной сипильнои кислоты а 2 моль хинолина. Полученный аддукт
(выход 65—95%) омыляют кипячением с 5—Юн. раствором серной кислоты.
Расщепление обычно протекает с количественным выходом.
Зоин и Мюллер [389] действовали эфирным раствором тетрахлороловянной (II)
кислоты на имидхлориды и получили альдегиды ароматического ряда с хорошими
выходами. Для воссстановленпя имидхлоридов алифатических кислот этот метод
непригоден.
Из анилипов а,в-ненасыщенных -кислот через соответствующие имидхлориды
можно аналогичным образом получать а,р-ненасыщенные альдегиды [390]. Реакция
протекает через шиффово основание, которое далее гидролизуют до альдегида:
С1 Н
I I /Н
R—C=N—С8Н5 —► R— C=N—С6Н6 —► R—С< ~HI3N—С6Н6
^0
По более новым данным хлорянгидрипы кислот восстанавливают до альдегидов
также при —78е С три-торет-бутоксиалюмоглдридом лития в диметиловом эфире диэти-
ленгликоля (дитлим) [391].
При восстановлении алюмогидридом лития получается соответствующий спирт
с 60 — 100%-ным выходом. Из галоидангидрндов алифатических а, р-не насыщенных
кислот образуются преимущественно ненасыщенные спирты [392], хотя из хлорангид-
рида коричной кислоты получается гндрокоричный спирт. Несколько мягче, чем алюмо-
гидрид лития, реагирует этоксиалюмогидрид натрия, но даже с этим восстановителем
из хлорангидрида коричной кислоты не удается получить соответствующего альдегида;
при взаимодействии в тетрагидрофуране [393] в течение 4 ч при 65° С с сохранением
двойной связи образуется коричный спирт с 70%-ным выходом.
II . Обмен кислорода на додород
1. Обмен гидроксильной группы
Прямой обмен гидроксильной группы па водород имеет препаративное значение
лишь в некоторых особых, но довольно важных синтезах. Для восстановления первич-
ных и вторичных гидроксильных групп часто удобнее использовать косвенные способы.
В первом случае исходят из галогенидов, во втором, кроме того, используют и
ненасыщенные соединения, легко образующиеся из вторичных спиртов. Третичные
гидроксильные группы обычно обмениваются непосредственно на водород под
действием неблагородного металла со спиртом или кислотой, например натрия
та спирта, а также при помощи каталитического гидрирования.
Взаимодействие спиртов с иодистоводородной кислотой рассматривается здесь
как прямой обмен гидроксильной группы иа водород, потому что при таком способе
восстановления нельзя выделять галогенид в чистом виде из-за восстанавливающих
свойств иодистоводородной кислоты.
[389
[390
[391'
[392
[393
A. S о п п, Е. М ii 11 е г, Вег., 52, 1929 (1919).
J, V. Braun, W. Rudolph, Вег,, 67, 269, 1735 (1934).
Н. С. В г own, В. С. S u bb a R ао, J. Am. Chem. Soc., 80, 5377 (1958).
R. Nystroa, W. G. Brown, J. Am. Chem. Soc., 69, 1197 (1947).
G. Hesse, R. Schrodel, Ann., 607, 24 (1957).
76
ОБРАЗОВАНИЕ С—Н-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Учитывая высокую стоимость иода, обычно работают не с эквивалентными коли-
чествами иодистоводородной кислоты, а добавляют фосфор, который в присутствии
небольшого количества воды реагирует с выделяющимся в течение реакции иодом
с образованием иодистоводородной кислоты. Часто удается обойтись и смесью
фосфора с иодом, к которой иногда добавляют небольшие количества иодистоводо-
родной кислоты.
2-Этилбензойная кислота (получение из а-метилфталида по видоизме-
ненной методике Гпбе [394]. В литровой круглодонной колбе смешивают 390 мл
ледяной уксусной кислоты, 24 г красного фосфора и 8 г иода; эту смесь оставляют
па 20 мин при комнатной температуре, в течение которых со слабым разогрева-
нием проходит реакция между фосфором и иодом. Добавляют 8 мА воды, 2 мл
56,7%-ной иодистоводородной кислоты, 100 г а-метилфталида и кипятят 160 ч
с обратным холодильником. Еще не остывшую реакционную массу отфильтро-
вывают от фосфора и при перемешивании выливают в холодный раствор 35 г
бисульфита натрия в 1500 мл воды, причем исчезает коричневая окраска иода
и осаждается 2-этилбензойная кислота в виде бесцветных кристаллов. Послед-
нюю после охлаждения отфильтровывают, фильтрат упаривают и таким обра-
зом получают еще немного 2-этилбонзойной кислоты. Общий выход составляет
80 г. Из сконцентрированного фильтрата эфиром удается извлечь 10 г исходного
метилфтадида, и таким образом выход по прореагировавшему фталиду соста-
вляет 90% от теоретического. Для очистки продукт кристаллизуют из воды,
получая совершенно бесцветную 2-этилбензойную кислоту. -
Аналогично восстанавливают оксикарбоновые кислоты до карбоповых кислот.
Так, из бензиловой кислоты с выходом более 90% получают дифенилуксусную кислоту
[395]. Из лактонов полиоксикарбоновых кислот под действием йодистого водорода
часто образуются сначала лактоны монооксикарбоновых кислот жирного ряда. Из лак-
тона глюконовой кислоты получают лактон оксикапроновой кислоты и лишь при более
высоких температурах в запаянной ампуле получается капроновая кислота [396].
Из глюкозаминовой кислоты аналогично под действием иодистоводородной кислоты
получают аминокапроновую кислоту.
Прямой обмен гидроксильной группы на атом водорода можно осуществить
действием неблагородного металла в среде кислоты или спирта или при помощи ката-
литического гидрирования. Восстановлению способствует наличие в соседних положе-
ниях ненасыщенных связей, ароматических систем или карбонильных групп, а иногда
оно вообще протекает только при этом условии. Примером может служить не имеющее
большой препаративной ценности восстановление коричного спирта натрием в спирте
по Клагесу [397]. В реакционной массе содержится главным образом пропилбенэол
наряду с небольшим количеством проценилбензола С$Н5СН=С1ГСПЭ. Пропилбенэол
не может быть получен из фенилпропилового спирта СвН8СН2СН2СН2ОН, так как
последний совершенно не изменяется в условиях реакции. Наоборот, куминовый спирт
(СН3)2СНСвНаСН2ОН, гидроксильная группа которого находится рядом с ароматиче-
ской системой, восстанавливается до цимола уже при продолжительном нагревании
с одной лишь цинковой пылью. Восстановление ацетоинов, например бензоина, с хоро-
шим выходом до дезоксиацетоинов под действием цинка в' спиртовом растворе хлори-
стого водорода [398] свидетельствует о том, что спиртовая группа в а-оксикетонах
тоже сравнительно легко элиминируете^. Ацетоксигруппу этерифнцировапных уксус-
ной кислотой а^оксикетонов можио прямо заменить на водород, применяя длительное
кипячение с цинковой пылью в ледяной уксусной кислоте; соседняя кето-группа при
этом нс изменяется [399].
[394] G. Glebe, Вег., 29, 2533 (1896).
[395] С. S. Marvel, F. D. Hager, Е. С. Candle, Org. Synlb., 3, 45
(1923).
[396] Н. К i 11 a n i, S. Kleemann, Ber., 17, 1296 (1884).
[397] А. КI ages, Ber., 39, 2587 (1906).
[3981 H. St ob be, Ber., 35, 911 (1902).
[399] R.S. Roseafeld, T. F. Gallagher, J. Am. Chem. Soc., 77, 4367
(1955).
II. ОБМЕН КИСЛОРОДА НА ВОДОРОД
77
Гидроксильная группа, связанная с третичным атомом углерода, обменивается
значительно легче, чем связанная с вторичным или первичпым атомом. Так линалоол»
СНз
I
СН3-С=СН—сн2—сн2—с—сн-сн2
I I
СНз он
третичпая гидроксильная группа которого к тому же находится рядом с двойной
связью, переходит в линалоолен уже при нагревании с цинковой пылью в запаянной
ампуле [400]. Исходя из возможности протекания такой реакции, можно даже сде-
лать вывод о характере гидроксильной группы, т. е. отличить третичную гидроксиль-
ную группу от вторичной и первичной [401].
Чаще положительные результаты получаются при использовании сочетания
цинковой пыли с соляной или уксусной кислотой или же с раствором едкого натра.
Так, например, гидроксильная группа триарилкарбннолов легко обменивается на атом
водорода при кипячении с цинком и ледяной уксусной кислотой. По данным Кауф-
мана [402], метоксилированные трифенилкарбинолы можно восстановить до трифеиил-
метанов даже горячим спиртом в присутствии соляпой кислоты, но в этом случае
реакция, очевидно, идет через триарилхлорметан. Аналогично протекает и восстано-
вление фталеинов; так, по данным Байера [403], при кипячении 3,3-дифепилфталида
с цинковой пылью и раствором едкого натра образуется трифенилметаи-о-карбоновая
кислота:
С6Н6\ /С6Нь
/^/СН(СвНб)2
il I
\соон
Енольный гидроксил восстанавливается амальгамой натрия [404]. Этот же вос-
становитель в кислом растворе хорошо восстанавливает гидроксильную группу лакто-
нов. Так, из валеролактопа образуется валериановая кислота, а из капролактона —
капроновая. В отличие от этого лактоны полиоксикарбоновых кислот, особенно в ряду
углеводов, восстанавливаются амальгамой натрия в первую очередь по карбоксиль-
ной группе. По Берчу, винилкарбинолы и другие спирты можно восстанавливать-
натрием и спиртом в жидком аммиаке. 'Гак, из .D-сабннола образуется /нх-туйен:
СИ, СИ,
нс(сн3)г нс(сн,)
Бензиловый и фурфуриловый спирты восстанавливаются таким же способом соот-
ветственно до толуола и метилфурана [405].
Спирты можно восстанавливать до углеводородов также каталитическим гид-
рированием. 2,2,3-Триметилбутанол-1 гидрируется при 300° С за 18 ч в присутствии
катализатора на основе кобальта и окиси алюминия до триметилбутана (триптана)
с 56%-ным выходом. Эту реакцию не удалось провести под действием меднохромового-
400]
401
402
403
404
[405
F. W. S е m m I е г, Вег., 27, 2520 (1894).
F. W. S е m m 1 е г, Вег., 33, 776 (1900).
Н. Kauffman п, Вег., 38, 2702 (1905); 41, 4423 (1908).
A. v. В а е у е г, Вег., 12, 644 (1879).
D. V or] an der, О. Apelt, Вег., 37, 1134 (1904).
A. J. Birch, J. Chem. Soc.. 1945, 809.
78
ОБРАЗОВАНИЕ С—Н-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
катализатора 1406]. Алифатические спирты, кроме того, гидрируются до углеводоро-
дов в присутствии окиси ванадия и окиси алюминия [407 ]. Под действием никеля Ренея
из первичных спиртов с прямой цепью получаются предельные углеводороды, у кото-
рых на один атом углерода меньше, чем в исходном спирте [408].
Гемеллитол [409]. К раствору 61,5 а 2,3-диметилбензилового спирта
(т- пл. 65—65,5° С) в 70 .ил этилового спирта добавляют За меднохромового
катализатора, содержащего окись бария, и гидрируют эту смесь в автоклаве
с мешалкой при 220—235° С в течение 8,5 ч. Начальное давление водорода при
28° С составляет 100 ап, во время реакции оно достигает 190 ап. По окончании
гидрирования отделяют катализатор фильтрат разбавляют 600 мл воды и угле-
водород тщательно экстрагируют эфиром. После осушки эфирных растворов
сульфатом натрия отгоняют эфир и остаток перегоняют через короткую колонку
со стеклянной насадкой. Собирают фракцию 172—173’ С; выхед 50 г (92% от тео-
ретического).
Более подробно о применении медпохромовых катализаторов см. [410].
Никель Ренея расщепляет эфиры метансульфоновой [411] и бензойной кислот
'[412], окись платины — эфиры уксусной кислоты [413], а палладий на угле — эфиры
угольной кислоты [414].
Алюмогидрид лития не восстанавливает спирты. Гидроксильная группа обмени-
вается на водород лишь в некоторых исключительных случаях, например в замещен-
ных бензиловых спиртах с амино- или метокси-группой в орто- или пара-положении
к карбинольной группе [415].
Фенольный гидроксил удаляют перегонкой с цинковой пылью. Правда, этот
способ не имеет препаративного значения, но важен для доказательства строения
вещества. При перегонке фенантрола с ципковой пылью в токе водорода получается
с 72%-ным выходом фенаптрен [416].
2. Обмен карбонильного кислорода
Прямой обмен
Известны два основных способа обмена карбонильного кислорода на водород:
восстановление по Клемменсену и каталитическое гидрирование.
Наряду с пими для восстановления карбонильных групп в разных кетосоеди-
нениях можно использовать другие известные восстановители. Так, например, изоду-
рилфепклкетон в растворе ледяной уксусной кислоты можно частично восстановить
действием большого избытка иодистоводородной кислоты, перемешивая смесь в тече-
ние 2 ч при температуре кипения. Получается с хорошим выходом 2,4,5,6-тетраметил-
бензилфенилкетон [417]. Так же можно восстановить o-бромбензофенон смесью иоди-
'Стоводородной кислоты и красного фосфора до о-бромдифенилметана; выход 82%.
В этих случаях достигаются лучшие выходы, чем по методу Клемменсена [418].
[406] т. A. Ford, Н. W. J acohson, F. С. McGrew, J. Am. Chem. Soc., 70,
3793 (1948).
[407] V. 1. К о m are ws k у, C. F.'Pri се, J. R. Coley, J. Am. Chem. Soc.,
69, 238 (1947). -
[408] H.'AdkiB8. R. E. Burks, J. Am. Chem. Soc., 70, 4174 (1948).
[409 L. I. Smith, L. J. Spillane,!. Am. Chem. Soc., 62, 2639 (1940).
[410J W. Foerst, Weuere Methoden der praparativen organischen Chemie,
Bd. 1, 2 AufL, 1949, S. 117.
[411] A. S. D rei ding, A. J. Tomasewski, J. Am. Chem. Soc., 77, 411 (1955).
[412] M. W. Cronyn, J. Org. Chem., 14, 1013 (1946).
[413] T. A. G e i s s m a n, E. II i n reiner, J. Am. Chem. Soc., 72, 782 (1951).
4414} K. Kinder, K. Schrader, Ann 564, 49 (1949).
[415] L. H, Con о ver, D. S. T arh ell, J. Am. Chem. Soc., 72, 3586 (1950).
[416] A. Schonberg, F. L. Warren, J. Chem. Soc., 1939, 1838.
[417] R. C. F u s о n, P. E. H о c h, J. Am. Chem. Soc., 71, 1585 (1949).
;[418] С. К. В г a d s he r, F. A. Vingiello, J. Org. Chem., 13, 786 (1948).
II. ОБМЕН КИСЛОРОДА НА ВОДОРОД
79
Из 2-амипобензофенопа можно с очень хорошим выходом под действием избытка натрия
в абсолютном спирте получить 2-аминодифенилметан [419]. Имеются сообщения о про-
ведении аналогичных процессов восстановления под действием цинковой пыли
в кислом или щелочном растворе. Так, аллопрегпандпон-3,20 восстанавли-
вается £420} цинковой пылью и концентрированной соляной кислотой в спирте до
аллопрегнанона-20 с 72%-ным выходом. Описано также восстановление 1,2,5,6-ди-
бензантрахинон-4',8'-дисульфокислоты цинковой пылью в концентрированном
водноаммиачном растворе до соответствующего производного антрацена [421]; вы-
ход 95%.
Восстановление карбонильных соединений системой амальгамированный цинк—
концентрированная соляная кислота по Кломменсену применимо почти во всех слу-
чаях [422—425]. Амальгамированием цинка достигается перенапряжение водорода,
повышающее потенциал выделения водорода. Концентрация применяемой для вос-
становления соляной кислоты должпа лежать в пределах 20—40%; реакция проте-
кает при кипячении в среднем за 5—10 ч. Восстанавливая неустойчивые к действию
кислот карбонильные соединения, реакционную массу оставляют стоять на одпи пли
несколько суток при комнатной температуре. Иногда продукты реакции получаются
с весьма умеренными выходами. Выходы зависят как от химических свойств восстана-
вливаемого соединения, таи и в значительной степени от условий проведения реакции.
Необходимыми условиями для успешного протекания реакции являются некоторая
растворимость карбонильного соединения в воде и жидкое состояние вещества в усло-
виях реакции. Для увеличения растворимости можно применять добавки спирта,
уксусной кислоты или диоксана. Но при этом следует, однако, учитывать, что слишком
хорошая растворимость восстанавливаемого вещества облегчает образование побоч-
ных смолообразных продуктов, которые обволакивают металл и том самым инактиви-
руют ого. Такое явление устраняется добавками толуола.
По этому методу можно восстанавливать:
1. Алифатические и ароматические альдегиды.
2. Алифатичесиие, алициклические и жирно-ароматические кетоны. При вос-
становлении алифатических оксикетонов с первичными [426], вторичными [427] или
третичными [428] гидроксильными группами, находящимися не в a-положении к карбо-
нильной группе, гидроксильные группы ле затрагиваются — восстанавливаются
только карбонильные группы. При восстановлении дикетопов в некоторых случаях
возможен частичный обмен одной карбонильной группы, а при восстановлении
циклических 0-дикетонов иногда происходит превращение циклической систе-
мы [429].
3, Кетокислоты и их эфиры (а, р [430], у [431], 6 [432] и др.). Восстановление-
а-кетокислот [430] обычно приводит лишь к переводу группировки — СО—СООН
d -СН(ОН)-СООН.
4. а,р-Ненасыщенные карбонильные соединения. В этих соединениях [430]
группировка —СН=СН—СО— восстанавливается до —СН3—СН2 —СНг—, однако*
возможно восстановление или двойной связи, или карбонильной группы.
[419
[420
[421
[422
[423
[424]
425]
426
427
428
429
430
431
432
A. R. Т о d d et al., J. Chem. Soc., 1948, 292.
H. F. B. Hi rscbrn ann, M, A. D ans, J. Biol. Chem., 178, 751 (1949).
J. Cason, L. F. F i e s e r, J. Am. Chem. Soc., 62, 2681 (1941).
E. Clemmensen, Ber., 46, 1837 (1913); 47, 51, 681 (1914).
E. L. Martin, Org, Reactions, v. 1, New York, John Wiley a. Sons, 1942,.
p. 155.
O. Bayer, в книге Houben-Weyl — Muller Bd. 7, T, 1, 4 Aufl,^
1954, S. 491.
D. Ctaschewski, Angew, Chem., 71, 726 (1959).
E. Clemmensen, Ber., 47, 681 (1914).
R. E. Marker, E J. Lawson, J. Am. Chem. Soc., 61, 852 (1939).
R. E. Lutz, L. Smale, J. Org. Chem., 4, 220 (1939).
A. N. Dey, R. R. L instead J. Chem. Soc., 1935, 1063.
W. Steinkopf, A. Wolfram, Ann., 430, 113 (1923).
C. F. К о e 1 s св, J. Am. Chem. Soc., 55, 3885 (1933).
L. R u z i c k a, W. В uegger, C. F, Seidel, H. S chi nz. Helv. chin»
acta, 11, 496 (1928).
80
ОБРАЗОВАНИЕ С—Н-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Еслл восстановление карбонильного соединения но Клемменсену не приводит
к целп, то с большим успехом можно использовать метод Кпжнера (стр. 81).
«-Ундекан [433]. Кипятят 50 г метил-и-понилкетона (т. кип. 224—226° С)
и 300 г амальгамированного цинка в 330 мл смеси равных частей технической
соляной кислоты и воды в колбе с обратным холодильником в течение 24 ч,
непрерывно добавляя копцептрироваппую соляную кислоту для поддержания
бурного выделения водорода. После охлаждения отделяют маслянистый слой
от водного раствора, промывают водой и сушат. Прп перегонке получают 40 г
к ундекана (т. кип. 193—195u С), что соответствует выходу 88% от теорети-
ческого.
Точно такой же метод используется и для восстановления жирно-ароматических
кетонов.
н-Пропилбензол. В смеси равных частей технической соляной кислоты
п воды нагревают 25 г пропиофенона и 100 г амальгамированного цинка. Через
5 мин начинается бурная реакция; нагревание с добавлением время от времени
концентрированной соляной кислоты продолжают в течение 4 ч. Из реакционной
массы выделяют 20 г н-пропилбензола (фракция 155—160° С), что соответствует
90%-ному выходу от теоретического.
Мартин [434] предложил проводить реакцию Клемменсепа в присутствии
толуола. '
Арилмасляные кислоты. Для получения этих кислот из соответствующих
замещеппых бензоилпропионовых кислот встряхивают 100 г губчатого цинка
в течение 5 мин с 10 г хлорида ртути (II) и 5 мл концентрированной кислоты
в 150 мл воды. Водный слой сливают, к амальгамированному цинку приливают
75 мл воды, 175 мл копцептрировапнон соляной кислоты. 100 мл толуола и 50 г
восстанавливаемого карбонильного соединения. Для повышения раствори-
мости карбонильного соединения в водном слое иногда добавляют 3—5 мл ледя-
ной уксусной кислоты и интенсивно нагревают смесь с обратным холодильником
до кипения. Нагревание продолжают 24 ч, прибавляя через каждые 6 ч по 50 мл
концентрированной соляной кислоты. При этом оказалось, что восстановление
В-бензоилпропионовой кислоты, м- и п-В-толуилпропионовых кислот, 1- и 2-р-на-
фтоилпропионовых кислот, бензоилнафталипа, р-пафтилметилкетона и других
соединений протекает успешнее при добавке толуола. Для сравнения Мартин
проводил эти синтезы по старому и новому методам; выходы соответственно
составляли 72—78 и 90%.
Иногда по реакции Клеммепсепа не происходит однозначной замены кислорода
па водород. Например, при восстановлении бензофенона амальгамированными цин-
ковыми опилками и водным раствором хлористого водорода получается бензпипакоп
с 70%-ным выходом, а при применении спиртового раствора хлористого водорода
наряду с незначительным количеством ожидаемого дифенилметана образуются глав-
ным образом бензпииаколин и тетрафенилэтилен [435].
Возможно также каталитическое восстановление карбонильной группы до мети-
леновой. В качестве катализаторов применяют хромит меди и благородные металлы
(палладий на угле, палладий на сульфате бария, платину, окись платины). Так, арил-
кетоны восстанавливаются с хорошими выходами до алкилбензолов при 185—250° С
в присутствии медпохромового катализатора [436]. При 110—130° С о- и п-бензаль-
дегиды восстанавливаются до соответствующих крезолов, а о- и п-оксиарилкетоны гид-
рируются до оксиалкилбензолов.
Гидрирование карбонильного соединения проводят в стальном кача-
ющемся автоклаве с медным вкладышем вместимостью 500 мл. В него загружают
[433] Е. Clemmensen, Вег., 46, 1841 (1913).
1434] Е. L. М а г t i n, J. Am, Chem. Soc., 58, 1438 (1936).
[435] W. Steinkopf, A. Wolfram, Ann., 430, 113 (1923).
J436] D. Nighting-ale, J. Org. Chem., 14, 1089 (1949).
II. ОБМЕН КИСЛОРОДА НА БОДОРОД
81
0,25 моль кетона или альдегида и 4 г меднохромового катализатора [437 [. При
гидрировании оксиальдегидов во избежание образования смолистых продуктов
добавляют еще 100 мл безводного метилового спирта (ч. д. а.). Ароматические
кетоны восстанавливают при начальном давлении водорода 300—340 ат, окси-
альдегиды и оксикетопы можно гидрировать под давлением 220—240 ат. При
гидрировании в спиртовом растворе температуру поддерживают около 200—
250° С до прекращения изменения давления, а при ведении процесса без рас-
творителя для получения углеводорода достаточно температуры 180—195° С.
После окончания гидрирования реакционную массу вымывают метиловым спир-
том или бензолом, отделяют фильтрованием катализатор и обычным способом
выделяют продукт реакции.
2-Мстилфуран получают путем пропускания фурфурола в токе водорода при нор-
мальном давлении и 200—250°С над мелкораздроблепным хромитом меди на угле [438].
В мягких условиях восстановление удается провести в присутствии палладие-
вых катализаторов (3% палладия на сульфате бария или на угле). Растворителями
служат спирты, этилацетат и ледяная уксуспая кислота с небольшими количествами
серной или хлорной кислоты, ускоряющих гидрирование. Давление водорода колеб-
лется в пределах от нормального до 5 ат, температура — от комнатной до 90° С. Количе-
ство поглощаемого водорода не должно превышать 2 моль водорода на один карбониль-
ный кислород.
Восстановление карбонильной группы в альдегидах и кетонах до метильной
или метиленовой группы при помощи иомплексных гидридов металлов удается очень
редко. Ароматические альдегиды и кетоны с амино- или метокси-группами в орто-
или пара-положении к карбонильной группе восстанавливаются в очень жестких
условиях. При кипячении с обратным холодильником n-аминобензофенона в смеси
диэтилового и дибутилового эфиров в течение 1 ч с пятикратным избытком алюмо-
гидрида лития при температуре 80’ С наряду с n-амикобензгидролом (выход 15%) обра-
зуется также л-аминодпфенилметан [439] (выход 57%). В этих же условиях можно
восстанавливать производные анридопов [440] и ксантонов [441].
Карбонильная группа в гетероциклических соединениях восстанавливается
достаточно легко в ряду пиррола *, как это показали Трейбс и Дерра-Шеррер [442].
Обмен карбонильного кислорода с получением
промежуточных соединений
Метод Кижнера **. Исключительно удачным методом восстановления боль-
шинства карбонильных соединений является восстановление по методу Киж-
псра [443]. Метод состоит в получении гидразонов или семикарбазопов карбонильных
* О восстановлении комплексными гидридами металлов кетонов индольного
ряда см. Н. Н. Суворов, К. Б. Холодковская, М. Н. Преобра-
женская, Химия гетероциклических соединений, 1965, 265. — Прим. ред.
** В оригинале данная реакция неправильно называется реакцией Вольфа —
Кижнера. Между тем приоритет II. М. Кижнера бесспорен: 2 декабря 1910 г.
II. М. Кижнер сделал сообщение «О разложении алкилидеигидразииов в при-
сутствии едкого кали» [ЖРФХО, 42, 1669 (1910)]. Вольф опубликовал свою
Ваботу в 1911 г. [Вег., 44, 2760 (1911)], т. е. через год после первой публикации
'. М. Кижнера. Подробнее см. В. М. Родионов, Н. Г. Ярцева, Реак-
ции и методы исследования органических соединении, Госхимиадат, 1, 7
(1951). — Прим, ред,
[437] В. Connor, К. Folkers, II. Adkins, J. Am. Chem. Soc., 53,
2012 (1931).
[438] L. W. Burnette, I. В. J ofins, R. F. Holdren, R. M. Hixon, Ind.
Eng. Chem., 40 , 502 (1948).
[439] L. H. С о n о v e r, D. S. T a г b e 11, J. Am. Chem. Soc., 72 , 3586 (1950).
[440] G. M. Cadger, J. H. Sei dler, B. Thomson, J. Chem. Soc., 1951, 3207.
[441] A. Mustafa, M. К. H i 1 m y, J. Chem. Soc., 1952, 1343.
[442] A. Treibs, II. D e rra-S ch error, Ann., 589, 188 (1954).
[443] L. Wolff, Ann., 394, 86 (1912); H. Кижнер, ЖРФХО, 43, 582 (1911);
D. T о d d, Org. Reactions, v. 4, 1948, p. 378—422, V. Franzen, Reaktiona-
mechanismen, Heidelberg, 1958, 1 Folge, S. 140.
6 Заказ 183 5.
82
ОБРАЗОВАНИЕ С—Н-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
соединений и разложении их под действием едкого кали или алкбголята щелочного
металла:
zO
R-C< —> R—CH=N-NH2 —> R-CH3-J-N2
Присутствие едкого кали или алкотолята не всегда обязательно; гидразоны '
кетона Михлсра, флуоренона или бензофенона превращаются в соответствующие угле-
водороды уже в присутствии гидразингидрата [444]. Это же справедливо для некото-
рых кетонов, содержащих сульфоновые группы, например, для гидразона га-мстилсуль-
фонилацетофенона [445]. Разложение идет в отсутствие сильно основного катализа-
тора, очевидно, тогда, когда в образующейся метиленовой группе имеются подвижные
атомы водорода.
Протеканию реакции Кижнера иногда могут мешать две побочные реакции —
образование азина и восстановлений исходного карбонильного соединения до спирта.
Обе реакция идут в присутствии воды. Часть гидразона может расщепляться водой
на гидразин и карбонильное соединение, а затем гидразон и карбонильное соединение
реагируют с образованием азина. Наряду с этим образующееся при гидролизе гщ-ре-
зона и семикарбазона карбонильное соединение может восстанавливаться до спирта
содержащимися в реакционной среде алкоксильными анионами. Обе побочные реак-
ции удается подавить, предотвратив гидролиз гидразона нлн соответственно семикар-
базона. Этого можно достигнуть добавлением избыточного гидразина или удалением
освобождающейся при образовании гидразона воды.
При гидрировании гетероциклических или ароматических кетонов с активными
атомами галогена в молекуле реакция сопровождается замещением последних. Нитро-
соединения, стабилизующиеся в спиртовой среде в виде asfu-нитросоединеинй, не вос-
станавливаются. В некоторых условиях может произойти также расщепление простых
эфирных группировок.
По Вольфу гидразоны расщепляют действием алкоголята, по Кижнеру — при-
меняют твердое едкое кали.
Восстановление камфоры до камфана по методу Вольфа [446] проводят следу-
ющим образом.
Камфан. В 10 мл спирта растворяют 0,8 г натрия и 10 г хорошо высу-
шенного гидразона камфоры и нагревают в запаянной толстостенной ампуле
в течение 18 ч при 190° С. После прибавления 750 мл воды получают неочищен-
ный кристаллический камфан, который очищают перегонкой с паром; т. пл.
156—157° С; т. кип. 161° С (757 мм рт. ст.). Выход 7 г, или 84% от теорети-
ческого.
Приводим описание получения н-пропилциклобутана по методу Кижнера из
гидразона циклобутилэтилкетона [447].
я-Пропилциклобутан. Для получения гидразона на масляной бане на-
гревают в течение 3 ч при 110—130° С смесь 18 г кетона, 18 г 90%-ного гидр-
азингидрата и 50 мл абсолютного спирта. Затем отгоняют спирт и сушат остаток,
твердым едким кали. Гидразон сливают с едкого кали и разлагают нагреванием
в колбе Клайзена, снабженной капельной воронкой, при 120—140° С в присут-
ствии 2 г твердого едкого кали и двух небольших осколков глиняного черепка»
предварительно смоченных раствором соли платины и хлорида аммония и про-
каленных. Отогнанное вещество, обрабатывают разбавленной уксусной кисло-
той, выделяющийся углеводород промывают водой, сушат плавленым карбо-
натом калия и дважды перегоняют над металлическим натрием. Таким образом
получают 7 г углеводорода (44,5% от теоретического); т. кип. 99—lOO’С
(736 мм рт. ст.).
Без щелочей разложение по методу Кижнера протекает, например, при полу-
чении 3-метилпирена из пиреи-3-альдегида [448].
[444] Н. Staudinger, О. К up f е г, Вег., 44, 2197 (1911).
[445] Н. Klosterziel, Н. J. Backer, Rec. trav. chim., 71, 361 (1952).
[446] L. Wolff, Ann., 394, 96 (1912).
[447] N. D. Zelinsky, В. A. К asansky, Ber., 60, 1101 (1927).
[448] H. Bolmann, H. Decker, M. Corel!, H. Streeck, An>., 531,.
1 (1937).
1J. ОЬМВН КИСЛОРОД НА ВОДОРОД
8;
8-Метилпнрен, В стальной автоклаве емкостью 1 л в течение 8 ч нагре
вают при 200® С 20 г пирен-3-альдегида с 100 г гидразингвдрата (избыточно)
давление около 100 ат). Твердый неочищенный продукт промывают водой
перегоняют в вакууме и получают 17 е практически чистого З-метияггипенл
т. пл. 70—71® С.
Этот метод был значительно усовершенствован путем введения высококипящиз
растворителей и полного удаления реакционной воды, в результате чего отпала необ-
ходимость в высоком давлении [449].
у-(л-Фенокспфенил)-масляпая кислота [450]. Смесь 500 г (1,85 .моль]
0-(«-феноксибензоил)-пропионовой кислоты, 350 г едкого кали и 250 мл 85%-ногс
гидразингидрата в 2,5 л три- или диэтиленгликоля нагревают 1,5 ч с обратим^
холодильником. Образующуюся воду отгоняют с нисходящим холодильником
до тех пор, пока температура реакционной массы не достигнет 195° С. Затем
раствор нагревают еще 4 ч с обратным холодильником.
После охлаждения раствор разбавляют 2,5 л воды и медленно' вливают
в 1,5 л б н. раствора соляной кислоты. Вес отделенного высушенного продуйте
451 г (95% от теоретического выхода); т. пл. 64— 66® С. После перекристаяли-
вации из смеси лигроина и бензола получается чистая феноксифенилмасляная
кислота; т. пл. 71—72° С.
Разложение семикарбазона с образованием гетероциклического соединения
описано на примере восстановления 2-формилпиррола до 2-метйлпиррола [451].
Обмен карбонильного кислорода
с промежуточным получением меркапталей
Карбонильная группа в эфирах а- и 0-кетокарбоновых кислот легко реагирует
с меяилмеркаптаном в присутствии хлорида цинка и сульфата натрия с образованием
меркапталей. Меркаптали кипятят с Обратным холодильником в течение несиольких
часов с никелем Ренея в спирте [452]. В результате получаются эфиры карбоновых
кислот с выходом от 70 до 80%. В ряду углеводов из некоторых альдоз с достаточно
хорошими выходами получены так называемые 1-дезоксиполиолы кипячением в тече-
ние 2 ч меркапталей в спиртовом растворе с большим избытком никеля Ренея. Так,
например, восстанавливают .D-ксилозу до 1-дезокси-£>-ксилита [453].
' 3. Обмен кислорода в карбоксильной
или сложноэфирной группе
Восстановление карбонильной группы в карбоновых кислотах и их эфирах
до метиленовой переводит их в соответствующие спирты. Эту реакцию можно провести
тремя методами: а) по Буво — Блану; б) каталитическим гидрированием в присутствии
меднохромовото катализатора или никеля Реиея; в) комплексными гидридами металлов.
Открытое в 1903 г. Буво и Бланом [454] восстановление эфиров карбоновых
кислот натрием и спиртом проводится в довольно жестких условиях. Обычно для этого
раствор 1 моль эфира карбоновой кислоты в абсолютном спирте приливают по каплям
непосредственно к 6 г-атом натрия, нарезанного крупными кусками, и затем смесь
в течение нескольких часов нагревают с обратным холодильником до полного раство-
рения натрия, выделяя после добавления воды образующийся спирт.
[449] Huang-Minlon, J. Am. Chem. Soc., 71, 3301 (1949).
[450] Huang-Minlon, J. Am. Chem. Soc., 68, 2487 (1946).
[451] P. A. Cantor, R. Lancaster, C. A. Vander W erf, J. Org. Chem.,
21, 918 (1956).
[452] M.S. Newman, H.M. Walborsky.J.Am. СЬещ. Soc., 72, 4296 (1950).
[453] E. Z i s s i s, N. K. R i ch t m yer, J. Am. Chem. Soc., 75, 129 (1953).
[454] L. Bouveault, H. G. Blanc, Bull. Soc. chim. France [3], 29, 787; 31, 666
(1904); C. r., 136, 1676 (1903); 137 , 60 (1903).
6*
84
ОБРАЗОВАНИЕ С—Н-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Для успешного протекания реакции обязательно применение абсолютного*
спирта, поскольку в противном случае пропсходпт омыление с образованием значитель-
ных количеств кислоты. Обычно восстановление проводят в среде этилового cmipia,
но применяется также п н-бутнлолый спирт, в среде которого реакцию можно прово-
дить при более высокой температуре.
Вводя добавки, разрушающие образующийся алкоголят (двуокись углерода,
хлорид аммония), и соблюдая некоторые особые условия проведения реакции, можно
повысить выход конечного продукта [455].
Реакция протекает гладко с эфирами карбоновых кислот жирного ряда, но нс
с эфирами ароматических кислот, в которых карбоксильная группа связана непосред-
ственно с ядром. Соответствующие насыщенные спирты образуются при восстановле-
нии а,р-ненасыщопных кислот, но если двойная связь не сопряжена с карбоксиалкпль-
пой группой, то обычно она не гидрируется.
Разработан [456] улучшенный общий метод восстановления эфиров жирных
кислот натрием. По этому методу для восстановления рекомендуется применять вто-
рпчпыс спирты. Работают с небольшим избытком натрия и спирта (около 5%). Кроме
того, реакцию проводят в инертном растворителе, простом эфире или углеводороде,
например в ксилоле. Сложный эфир и применяемый для восстановления спирт раство-
ряют в достаточном количестве растворителя, для получения жидкой гомогенной реак-
ционной массы, и при перемешивании добавляют к мелкораздробленпому натрию в ки-
пящем растворителе. Выходы достигают 80—95%. Осооеппо удобен этот метод для
получения ненасыщенных спиртов из ненасыщенных эфиров.
Олеиловым спирт из этилового эфира олеиновой кислоты [457]. В кругло-
донную колбу емкостью 5 л, снабженную обратным холодильником с широком
просветом, вносят 200 г (0,65 люль) перегнанного этилового эфира олеиловой
кислоты и 1,5 л абсолютного спирта. Перед этпм спирт сушат лад кальцием
и перегоняют над т-рт-бутилатом алюминия сразу в реакционную колбу.
Через обратный холодильник постепенно вносят 80 г (3,5 г-атом) натрия,
поддерживая бурное протекание реакции. При этом колбу время от времени
встряхивают. После прекращения бурного выделения водорода добавляют еще
200 лл абсолютного спирта и нагревают па паровой бале до полного растворения
натрия. Затем прибавляют 500 мл воды и нагревают 1 ч с обратным холодиль-
ником для омыленпя непрорсагировавшего эфира. Неомыдяемую фракцию
извлекают эфиром, эфирный экстракт промывают 1%-пым раствором едкого
калп и затем водой до полного удаления щелочи (по фенолфталеину). После
осушки над сульфатом натрия отгоняют эфир и остаток разгоняют. Получают
84—89 г продукта (49—51% от теоретического); т. кип. 150—152° С
(1 мм рт. ст.).
Восстановление эфира карбоновой кислоты до спирта возможно также катали-
тическим гидрированием над меднохромовым окисным катализатором [458]. Для
проведения этого гидрирования требуются довольно жесткие условии: избыточное
давление водорода 200 — 300 ат и температура 200—250° С. В этих условиях эфиры
жирных кислот гидрируются до спиртов, обычно с выходами выше 90%; точно так же
эфиры дпкарбоновых кислот переходят в соответствующие диолы. Исключение соста-
вляет малоновый эфир и продукты его замещения; в этом случае ожидаемые 1,3-гли-
коли восстанавливаются дальше с образованием одноатомных спиртов [459]. То же
наблюдается и при восстановлении эфиров ароматических и гетероциклических карбо-
новых кислот, из которых получаются углеводороды. Эфиры бензойной кислоты с коли-
чественным выходом превращаются в толуол; из дпэтилового эфира фталевой кислот
с 84%-ным выходом образуется о-ксилол.
[455] L. В al f г а у, Р. A n g 1 а г е t, С. г., 224 , 404 (1947).
[456] V. L. Hansley, Ind. Eng. Chem., 39, 55 (1947).
[457] H. Adkins, R. H. Gillespie, Org. Synth., Coll, v. 3, 1955,
p. 671.
[458] Ch. Grund mann, в книге Neuere Methoden der praparativen organischen
Chemie, Bd. 1, Verlag Chemie, Weinheim Bergstr., 1943, S. 117; G. S c n i I 1 e r,
в книге Houben -Wey 1-Muller, Bd. 4, T. 1, 4 Aufl., 1955, S. 318.
[459] R. Connor, И. Adkins, J. Am. Chem. Soc., 54, 4678 (1932).
II. ОБМЕН КИСЛОРОДА НА ВОДОРОД
5
Гексаметнленгликоль из эфира адипиновой кислоты [458, 460]. В авт<
клав высокого давления, емкостью не менее 400 .мл, загружают 252 г (1,25 моа.
дпэтилового эфира адипиновой кислоты и 20 г меднохромового окпсного кат;
дпзатора [458, 461]. Автоклав закрывают, устанавливают в приспособлен!
для встрячппания и подают в него водород. Предельное давление зависит <
прочности автоклава, при этом следует помнить, что давление на .холоду в 1,8 ра:
меньше, чем давление при высокой температуре, которую нужно поддерживал
в начале реакции. При максимально допустимом рабочем давлении автоклат
около 350 ат начальное давление по должно превышать 140 ат\ в холодны
автоклав, рассчитанный на 650—700 ат, можно подавать водород, доводи давЛ'
нце до 200 ат.
После подачи водорода включают приспособление для встряхивания авт,
клана и как можно быстрее нагревают автоклав до 255° С. Уту температур
поддерживают до тех пор, пока давление не станет постоянным в течение 1
В зависимости от активности катализатора, начального давления и чисто!
исходного продукта гидрирование продолжается 6—12 ч. Если емкость авт
клава 2 л иди больше п начальное давление не очень велико, то во время реакцг
следует добавлять водород, чтобы давление не падало ниже 100 ат.
По окончании реакции автоклав охлаждают, снижают давление и пер,
носят содержимое автоклава в стакан емкостью 600 лм, ополаскивая автоклг
четырьмя порциями 96%-ного спирта (по 25 мл каждая). Катализатор отсасывай
на воронке Бюхнера или, что лучше, отделяют центрифугированием и промывай
четырьмя порциями 96%-ного спирта (по 25 .мл каждая). К фильтрату добаэ
ляют 50 мл 40%-пого раствора едкого натра и кипятят 2 ч с обратным холодил
пиком. ] [родукт реакции перекосят в перегонную колбу емкостью 1 л и отгоняй
спирт до температуры 95° С в парах. Горячий остаток переливают в жпдкостнь
экстрактор непрерывного действия, а перегонную колбу ополаскивают 50 л
воды. Продукт реакции экстрагируют эфиром плп бензолом, что в зависимое!
от эффективности экстрактора продолжается от 24 до 50 ч. После этого отгоияй
растворитель и после удаления спирта и воды гликоль перегоняют в вакууь
из колбы Длайзена емкостью 250 мл. Выход гексаметплеигликоля 125—132
(85—90%, от теоретического); т. кип. 143—144° С (4 мм рт. ст.)’, т. пл. 41—42° (
Другим способом восстановления эфиров карбоновых кислот является провод-
мое в мягких условиях восстановительное расщепление эфиров тиоловых кислот п<
действием никеля Ренея. При этом отщепляется меркаптан и образуются соответств;
ющие спирты [4621.
Каталитическому гидрированию подвергаются не только сложные эфир!
но и свободные кислоты. Так, наиример, из лауриновой кислоты [463] в присутствъ
катализатора окпеп меди при 300° С и давлении водорода 250 ат за 90 мин получай
лауриловый спирт; выход 94%.
Для прямого гидрирования карбоновых кислот до спиртов можно примепя:
катализаторы из окиси рутения или пз рутения на угле (10% рутения) [464]. Гидрир
ванне проводят при температуре около 150° С и давлении водорода 500—900 аг
Используя специальный катализатор типа никеля Ренея, можно при 50° С проводи:
। идрировапио эфиров сс-аминоцарбиновых кислот, причем образующийся амивоспи]
не подвергается рацемизации [465].
Неоценимое значение, особенно в химии природных соединений, имеет мет<
восстановления карбоновых кпелоч ц их производных при помощи комплекснь
[460] Л. W. Ingersoll, S, П. Babcock, Org. Synth., Coll. v.
p. 328.
[461] w. A. Laziej, H. R, Arnold, Org. Synth., Coll. v. 2, p. 142.
[462] O. Jeger, J. N oremberski, S. Szpilfogcl, V. Pregel, Hel
chim. acta, 29, 684 (1946).
[463] A. Guyer, A. Bieler, K. J а Ь e г g, Helv, chim. acta, 30, 39 (1947).
1464] J. E. Carnahan et al., J. Am. Chem. Soc,, 77, 3766 (1955).
1465] H. Adkins, H. H. Billica J. Am, Chem. Soc., 70, 695, 3118, 31:
(1948). -
86
ОБРАЗОВАНИЕ С—Н-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
гидридов металлов [466] и в первую очередь алюмогидрида лития [467, 468]. Свободные
кислоты, их эфиры и ангидриды легко восстанавливаются до соответствующих спиртов
[469]. Лактоны превращаются в диолы [470]. Реакция протекает довольно легко даже
при комнатной температуре. В качество растворителей применяются преимуще-
ствешю эфир, диоксал или тетрагидрофурап. Часто используют также метилаль, кото-
рый превосходит по своим качествам эфир (оп, например, лучше растворяет поликарбо-
новые кислоты п поликарбонильпые соединения [471]). Вместо алюмогидрида лития
можно применять также более дешевый алюмогидрид натрия *. В этом случае в каче-
стве растворителя применяют тетрагпдрофуран [472].
2-Метилбутапдиол-1,3 из а-метилацетоуксусного эфира [473]. В трех-
горлой колбе емкостью 500 мл с мешалкой, обратным холодильником в капель-
ной воронкой нагревают до кипения в течение 10 мин 4 г алюмогидрида лития
и 100 мл абсолютного эфира. Затем суспензию алюмогидрида лития охлаждают
во лъду л, продолжая охлаждение, медленно добавляют по каплям раствор
14,4 г а-метилацетоуксусного эфира в 50 мл абсолютного эфира. Перемешивают
смесь еще 1 ч при 0° С. и при хорошем охлаждении льдом осторожно до-
бавляют по каплям 20 мл воды. Отфильтровывают образующийся осадок
и восемь раз промывают его на воронке Бюхнера, перемешивая с небольшими
порциями эфира и опять отсасывая. После сушки объединенных эфирных филь-
тратов сульфатом натрия растворитель отгоняют и маслянистый остаток пере-
гоняют в вакууме. Получают бесцветное масло; т- ниц. 111—112° С (17 мм pm. cm.);
выход 6,2 г (60% от теоретического).
Наряду с алюмогидридом лития часто применяют в качестве восстановителей
боргидриды щелочных металлов, оказывающие более мягкое действие. Промежуточ-
ное положении между боргидридом натрия и активным алюмогидридом лития занимает
восстанавливающая система из боргидрида натрия и бромида лития в диглиме. С этим
реактивом можно проводить ряд процессов селективного восстановления. Так, из эти-
лового эфира и-хлорбеизойной кислоты при нагревании на водяной бане в течение 3 ч
н диглиме со смесью боргидрида натрия и бромида лития получают с 85—91%-ным
выходом п-хлорбензидовый спирт [474].
Боргидрид лития образует с диоксаном комплекс, более устойчивый к действию
воды, чем свободный гидрид [475].
4. Восстановление амидов кислот и лактамов
Амиды кислот и лактамы восстанавливаются аналогично кислотам и их эфирам.
Каталитическое гидрирование при помощи меднохромового окисного катализатора
проводят црп избыточном давлении 200—300 am и 250—265° С. Во избежание протека-
* Л. И. Захаркин и И. М. Хорлина показали, что восстановление эфиров карбо-
новых кислот дпизобутилалюминийгидридом является препаративным методом
получения соответствующих альдегидов (Язв. АП СССР, ОХИ, 316 (1963)]. —
Прим. ред.
[466] Н. Г е й л р о д, Восстановление комплексными гидридами металлов, Издат-
инлиг 1959; И. Нёгаапв, Angew. Chem., 68, 601 (1956); Е. Schenker,
Angew. Chem., 73, 81 (1961).
[467] В. M и ч о в п ч, М. Михайлович, Алюмогидрид лития и его приме-
нение в органической химии, Иэдатштлит, 1957.
[468] G. W. Brown, Org. Reactions, v. 6, New York, John Wiley Sons, 1951,
p. 469.
[469] R. F. N у s t ro m, W. G. Bro wn, J. Am. Chem. Soc., 69, 1197 (1947).
[470] R. F. N y’s t г о m, W. G. Brown, 3. Am. Chem. Soc., 70, 3739 (1948).
[471] E. Bernatek, Acta chem. Scand., 8, 874 (1954).
[472] A. E. F i n h о 1 t, E. C. J acobson, A. E. О g а г d, P. Thompson,
J. Am. Chem. Soc., 77, 4163 (1955).
[473] E. Buchta, H. Bayer, Ann., 73, 227 (1951).
[474] H. C. Brown, E. J, Mead, В. C. S ubba Rao, J. Am. Chem, Soc.,
77, 6209 (1955).
[475] R. Paul, N. J d s e p li, Bull Soc. chim. France, 1953, 758.
ПТ- ОБМЕН АЗОТА НА ВОДОРОД
87
ния побочных реакций следует проводить реакцию с максимально возможной ско-
ростью и прибавлять в качестве растворителя диоксан (около 350—400 ,чл на 1 моль
амида кислоты), который связывает выделяющуюся при восстановлении воду; беа
диоксана происходит омыление амида. В большинстве случаев получаются удовлетво-
рительные выходы аминов, но при гидрировании незамещенного амида продукт реакции
всегда состоит из смеси первичных и вторичных аминов, часто с преобладанием послед-
него. При восстановлении двузамещенных амидов кпелот возможность протекания
таких побочных реакций исключена и в качестве единственного продукта реакции
получается соответствующий третичный амин с хорошим выходом [476].
Исключительно гладко и с очень хорошими выходами проходит восстановление
амидов и лактамов алюмогидридом лития [467]. В таких условиях отсутствуют побоч-
ные реакции, протекающие при гидрировании на меднохромовых катализаторах.
Иногда используется также электрохимическое восстановление. Так, при элек-
тролизе 1-бутилпирролидопа-З при 60° С на кадмиевом катоде в 30%-ной серной кис-
лоте при плотности тока 0,053—0,073 а/см1 раскрывается кольцо и с 64%-ным выходом
образуется дибутиламин [477].
III. Обмен азота на водород
Прямой обмен аминогруппы на водород возможен только в определенных усло-
виях. Прежде всего стали известны процессы восстановительного расщепления третич-
ных аминов и четвертичных аммониевых соединений, причем легче всего расщепляются
аллил- или бензилзамещенные амины. В последнем случае бензильная группа отще-
пляется в виде толуола. Этим свойством бепзилзамещенных третичных аминов поль-
зуются для получения в чистом виде вторичных алифатических аминов из первичных.
Для проведения реакции первичный амин сначала конденсируют с бензальдегидом;
полученное основание Шиффа восстанавливают до бензилалкиламина, который алки-
лированием переводят в диалкилбензиламин. Затем каталитическим гидрированием
отщепляют бензильную группу от получившегося соединения [478]:
RNH2 + OHC-СвН5 —> RN=CH—СЙН5 —» RNH-СН2-СвНБ
R'\ Н\
—> >N-CH2CeH6 —> >NH+CH3C,H.
R / R /
Некоторое препаративное значение имеет восстановительное дезаминирование
третичных оснований^Манниха, которые получают конденсацией с формальдегидом
и вторичным алифатическим амином соединений с реакционноспособной С—Н-связью.
Таким образом удается заменить подвижный водород в соединении с подобной
С—Н-связью на метильную группу [479]:
COCH8 1 1 COCH3 COCH3 1 1
Qjj HCHO; NH(CH,)a > Ц/J -N11(CH,), *
COCH3 co co CH2CH2N(CH3)a CH2CH3
Восстановительное дезаминирование можно осуществить каталитическим гид-
рированием в присутствии никеля Ренея, палладия или хромита меди в качестве ката-
лизатора или действием амальгамы натрия или цинковой пыли в растворе едкого натра
в смеси метилового спирта и воды [480].
[476] В. W о j с i k, R. A d к i n s, J. Am. Chem. Soc., 56, 2419 (1934).
[477] N. J. Leonard, S. Swann jr., II. L. Dryden jr., J. Am. Chem.
Soc., 74 2871 (1952).
[478] J. S. Buck, R. Raltzly, J. Am. Chem. Soc., 63, 1964 (1941).
[479] B. Reichert, H. Roseman n, Arch. d. Pharm., 281, 189 (1943).
[480] F. M 6 11 e г, в книге Houhen - Weyl — Miiller, Methoden der or-
ganischen Chemie, Bd. 11, T. 2, 4 Aufl., 1958, S. 216.
88
ОБРАЗОВАНИЕ С—Н-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Обмен первичной аминогруппы, связанной с ароматическим ядром, на водород
происходит почти исключительно через солп дпазоппя [481], которые расщепляют
определенными восстановителя мп, например спиртами, гипофосфористой кислотой,
формальдегидом в щелочном растворе пли соединениями олова (II).
Обычно для проведения этой реакции свежеприготовленный раствор
соли дпазонця при 0° С прибавляют по каплям к охлаждаемому 50%-ному рас-
твору гипофосфористой кислоты при перемешивании. После окончания выделения
азота реакционную массу оставляют на несколько часов прп 0— 5е С и выделяют
продукт реакции.
Если в молекуле соединения, как, например, в п-ампнобенаиламине.
имеется ароматическая и алифатическая аминогруппы, то можно провести обмен
аминогруппы, связанной с ароматическим ядром, не затрагивая алифатическую
аминогруппу. Для этого ачпн растворяют в гипофосфористой кислоте и доба-
вляют по каплям водный раствор нитрита натрия, так как известно, что пер-
вичные алифатические амины при pH <3 не взаимодействуют с азотистой
кислотой 1482],
Егли в качестве восстановителя применяют спирт, то выходы продуктов дезами-
нирования часто бывают неудовлетворительными. В этом случае преобладает реакция
образования простых эфиров фенолов. Низшие спирты реагируют преимущественно
с образованием эфиров фенола, в то время как при употреблении высших спиртов
в основпом получаются углеводороды. Например, из хлорида беязолдиа.топия и мети-
лового спирта получается только анизол, с этиловым спиртом наряду с большим коли-
чеством фенегола образуется немнеио бензола, с бензиловым спиртом — и основном
бензол и лпшь незначительное количество бепзплфенилового эфира,
Кроме того, образованию углеводородов способствуют некоторые заместители
в ароматических аминах, например пптрогруппа и галогены. Хлорид трпбромбепзол-
дцазопия, в частности, взаимодействует даже с сильно разбавленным спиртом, причем
получается почти исключительно трибромбензол; трибромфенол содержится и реак-
ционной массе лишь в виде следов; аналогично из дпазосоодинений с нитрогруппой
в ядре с очень хорошими выходами образуются нптроуглеводороды.
Если для восстановления диазониевых солей используют суспензпю закиси
меди в спирте, то дезаминирование проходит исключительно быстро и продукты побоч-
ных реакций почти отсутствуют. По-видимому, этот метод является общим; особенно
хорошие выходы дают соединения с ярко выраженным» электрояоакцеиторными заме-
стителями, как, например, питроанплины и амцноаитрахнноны [483].
Обмен аминогруппы амидов кислот на водород приводит к соответствующим
альдегидам. По данным Вейганда [484], при восстановлении N-метпланилиДОв карбо-
новых кислот а.иомогпдридом лития подучаются хорошие выходы альдегидов. Таким
способом получено значительное число алифатических, ароматических и гетероцикли-
ческих моно- и днальдегидои. Другие функциональные группы в молекуле восстанавли-
ваемого соединения, а именно фенольные и спиртовые оксигрулпы, сульфгидрильные
группы, 1ноэфирпыс группы, 1а.югепы и гетероциклический азот, не мешают проте-
канию реакции.
Практически восстановление проводят следующим образом. Метиланилид
растворяют в сухом тетрагпдрофуране и к этому раствору при 0е С добавляют
небольшими порциями порошкообразный или растворенный в эфире алюмо-
гидрпд лития. На каждую карбметиланллидную группу берут 0,3—0,5 моль
алюмО| идрида лития. Реакция протекает со значительным выделением тепла,
которое отводят охлаждением смесью льда и соли. По истечении оптжмаль-
[481] N. Korn bin щ, Org. Reactions, v. 2, New York, John Wiley a. Sons,
1944, p. 262.
1482] N. Kornblum D (£ If fl and J. Am. Chem. Soc., 71, 2137
(1949).
[483] H. H. Hodgson H. S, T п г n e r, J. Chem. Soc., 1942, 748.
1484] F. Weygand G. Eberhardt et al., Angew. Chem., 65, 525
(1953).
TV ОБМЕН СЕРЫ НА НОДОРОД
пого времени взаимодействия реакционную массу ратинаюг разбавленное
тшелотой при охлаждении льдом. В зависимости от свойств альдегида его виде,
ляют экстракцией или перегонкой с паром.
Позже Рид п Кспипптейн [484а] нашли, что глдрогсиолптичсское расшепленп*
замещенных N'-ацплниразолов алюмогидридом лития является общим способов
получения альдегидов. Роакппя протекает при 0—20“ С D эфире с очень хороппгь
выходом *.
Амиды и гидразиды кислот можно восстанавливать и другими способами.
Так, Берч восстановил беппоилшпгорпдпи до бензальдегида натрием в жидком аммиак*
[485], а Вингфильд [486] получил альдегид никотиновой кислоты разложением гидра-
зида этой кислоты периодагом патрия в водном растворе аммиака. Возможно такж*
восстановительное отщепление азота в диазокетопах под действием хлорида олова (1Г
в смеси соляной кислоты, спирта и воды [487] или иодистонодородной кислотой [488]
Пре этом образуется метплкетоп.
IV. Обмен серы на водород
В такого з ппя превращениях важное место занимает обмен сулъфогруппы арома-
тических соединений на водород. Эта реакция элиминирования имеет большое препара-
тивное и промышленное значение. Сульфокислоты часто обладают различной устойчи-
востью к гидролитическому расщеплению. Этот факт может быть использован длг
очистки и разделения смесп углеводородов через сульфокислоты или их смссп, напри-
мер при разделении ксилолов. Возможность последующего отщепления сульфокис-
лотой группировки можно использовать в препаративных синтезах и для других
целей. Если хотят ввести заместитель (например, нитрогруппу) в молекулу аромати-
ческого соединения в такое положение, в которое это не осуществимо прямым замеще-
нием, то сначала замешают это положение (или положения) сульфогруппой, эате\
вводят заместитель и, наконец, отшеиляют сульфогрутшу при помощи гидролиза,
Так, с хорошим выходом получают 2,6-дпнптроанилин из хлорбензола через хлорбен-
лол-4-сульфокислоту, 2,6-Динитрохлорбензолсульфокислоту и 1-амипо-2,6-динитро-
бепзол-4-сульфокислоту [489]. Введение сульфогруппы в полпалкнлбеняолы чаете
способствует внутри- или даже мржмолекулярному перемещению алкильных групг
при нагревании до ЮО9 С полученной сульфокислоты с серной кислотой [490].
Практически для обмена сульфогруппы па водород применяют гидролиз в воде,
в разбавленной серной пли соляпой кислотах при температурах до 200° С
под давлением.
По данным имеющихся немногочисленных работ связь С—S в тпоэфирах [491]
меркапталях [492], эфирах тиоловых кислот [4931 и тионовых соединениях [494’
сравнительно легко поддается восстановительному расщеплению. Для этого обессери-
ваемое вещество нагревают в течение нескольких часов в спиртовом растворе с никелех * 8
* Л. И. Захаркин п И. М. Хорлина показали, что альдегиды могут быть полу-
чены восстановлением замещенных амидов |11зв. АН СССР, ОХИ, 214С
(1959)] и нитрилов [ДАТ! СССР, 116, 422 (1957)] дпизобутялалюмннийгпд
рттдом. — Прил<. ред.
[484а| W. R i е (I, F. J. Konigstein, Ann., 622, 37 (1959).
[485] A. J. В i г с h, J. С у ш е г m а п - С г a i g, М. 81 а у t е г, Austral. J. Chem.
8. 5)2 (1955).
[486] Н. N. W i и g f i e 1 d, W. H. Harlan, 17, R. Han mer, J. Am. Chem
•Soc.. 74, .5796 (1952).
[487] R. E. L u I z, J. Am. Chem. Soc., 68, 1813 (1946).
[488] M. L. Wolirom, R. L. Brown, J. Am. Chem. Soc., 65, 1516 (1843)
[489] H. P. Schult ?, Org, Synth.. 31, 1951. p. 45.
[490] L. I. Smith, Org, Reactions, v. 1, New York. John Wiley a. Sons, 1942
p. .370.
[191] G. S t о r k et al.. J. Am. Cliem. Soc.. 75, .384 (1953).
(4921 S. Archer et al.. J. Am. Chem. Soc,.. 76, 4915 (1954).
[493] A, V. McIntosh jr., R. ц. Levi n, E. M. Meinzer. J. Am. Chem
Soc., 70, 295.5 (1948).
[11'4] W. B. Whalfy et al., J. Am. Chem, Soc., 77, 745 (1955).
90
ОБРАЗОВАНИЕ СВЯЗИ С—Hat В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Ренея. Аналогичным способом можно элиминировать серу гетероциклического кольца,
например серу тиофена [495], с раскрытием кольца * **. Общим методом восстановитель-
ного расщепления тпоэфпров является также действие натрия в жидком аммиаке [496].
Арилиаотиониаиаты восстанавливаются алюмогидридом лития до вторичных метил-
аминов [497].
ОБРАЗОВАНИЕ СВЯЗИ УГЛЕРОД - ГАЛОГЕН ••
Органические галогепсодержащие соединения, применяющиеся в качестве
растворителей, исходных продуктов для получения полимеров, фармацевтических
препаратов, красителей, ядохимикатов, гербицидов и др., являются продуктами орга-
нического синтеза, широко используемыми в промышленности и научных последова-
ниях. Нс менее важна их роль п в качестве промежуточных продуктов. Через галоген-
содержащие соединения протекают многочисленные процессы образования связей
С—N, С—О п С—С, а также связей С=О, С^-С и С=С.
В данном разделе особое внимание уделено бромированию, наиболее часто при-
меняемому в лабораторной практике. Одновременно описано также большое число
лабораторных методов фторирования, хлорирования, иодирования, а также роданнро-
вания. Подразделение этих реакций проведено н основном по типам реакций и возмож-
ностям их осуществления применительно к различным классам соединений.
ОБРАЗОВАНИЕ СВЯЗИ С—На!
В РЕЗУЛЬТАТЕ РЕАКЦИИ ПРИСОЕДИНЕНИЯ
I. Присоединение галогена и родана
по кратным С—С-связям
1. Свойства и очистка галогенов в родана
Так как для присоединения галогенов (Х2) и родапа в большинстве случаев
Применяются свободные галогены — фтор, хлор, бром, иод и родан (SCN)2, то
следует сначала коротко остановиться па их свойствах и очистке.
Фтор — желто-зеленый газ с яапахом, напоминающим запах озона и хлора,
кипящий при —188° С. Получают чаще всего электролизом кислой соли KF-2HF,
затвердевающей при температуре около 100° С (таким образом электролиз можно
проводить лишь в обогреваемой паром ячейке). Катодом служит железо, анодом —
графит пли никель. Лабораторная аппаратура, применяемая для получения фтора,
• Восстановительное обессеривание тиофена и его производных подробно исследо-
вал Я. Л. Гольдфарб с сотр., что позволило применить способ для получения
углеводородов, аминокислот, макроциклов и др. [Я. Л. Гольдфарб,
И. С. Корсакова, ДАН СССР, 96, 283 (1954); Я. Л. Гольдфарб,
Б П Ф а б р и ч п ы й, там же, 100, 461 (1959); Я. Л. Гольдфарб,
С. 3. Т а й ц, Л. И. Беленький, ЖОХ, 29, 3564 (1959); ДАН СССР,
139, 1356 (1961)]. Процессы обессеривания органических соединений хорошо
описаны в книге: Ф. Челенджер, «Некоторые вопросы химии серусо-
держащих органических соединении», пер. с англ., Издатинлят, 1963,
стр. 111—136. — Прим. ред.
** Материал обработал X. Дорн.
[495] F. F. В licke, D. G. Sheets, J. Am. Chem. Soc., 70, 3766 (1948).
[496] F. E. Williams, E. Geb auer - Fiilncgg, J. Am. Chem. Soc., 5$,
352 (1931) ,
[497] W. П i e d, F. Muller, Chem, Ber., 85, 470 (1952).
I. ПРИСОЕДИНЕНИЕ Hal и SON ПО КРАТНЫМ С—С-СВЯЗЯМ
91
описана достаточно подробно (1, 2, За, 4а]. В лабораторной практике фтор всегда полу-
чают только в количествах, необходимых для непосредственного использования, нс
в литературе имеются указания о хранении фтора в стальных или никелевых балло-
нах и о способах их наполнения [Зс].
Все органические соединения, за исключением СС14, взаимодействуют с фтором
Из неорганических соединений реагируют с фтором даже такие вещества, как асбест
и вода, сравнительно более устойчивые к действию других галогенов. Реакция фтор!
с водой иногда протекает со взрывом. Металлы реагируют с фтором уже при нормально4
температуре, причем па поверхности некоторых из них образуются непроницаемые
пленки фторидов, которые предохраняют металл от дальнейшей коррозии. К ник
относятся монель-металл, никель, алюминий, магний, железо, сталь. Стекло устойчив!
<; действию сухого фтора, свободного от фтористого водорода.
Получение чистого .мора описано Брауером [4Ь]. Для очистки хлор пропускаю:
через две промывные склянки с концентрированной H2SO4, через трубку с СаО дл?
удаления хлористого водорода и лад Р3О; Для обезвоживания. Достаточно чисты!
хлор, почти не содержащий кислорода и окисей хлора, можно получать из гидрат*
двуокиси марганца и концентрированной соляной кислоты [4Ь].
Природный и искусственный пиролюзит непригодны для получения чистог<
хлора, так как в них содержатся карбонаты и сульфиды.
Чистый хлор [4Ь [ можно получать тремя способами:
1. По реакции бпхроиата калия с соляной кислотой при нагревании
В реакционную колбу, помещенную в воронку Бабо, вносят К2Сг2С>7, при
ливают из капельной воронки разбавленную TTCI [3 объема кислоты (d = 1,19
и 1 объем воды] и слабо нагревают по море надобности.
2. Из твердого КМпО4 п НС1 (d = 1,16), если требуется при охлаждения
Выделяющийся хлор всегда содержит довольно много кислорода.
3. В аппарате Кпппа из спрессованной в кубики хлорной извести и соля
ной кислоты.
Для дозировки удобно пользоваться жидким хлором (т. кип. —34° С; d — 1,557)
для чего его конденсируют в калиброванном сосуде, охлаждаемом смесью твердо!
углекислоты я ацетона.
В электролитическом хлоре из баллонов содержатся кислород, окпелы хлора,
азот, окись и двуокись углерода, хлористый водород и влага. Для очистки от этих
часто не мешающих при органических работах, примесей жидкий хлор испаряю'
и конденсируют в приемнике, охлаждаемом смесью эфира и углекислоты. Осушителе!
служит серная кислота.
Для препаративных целой вполне пригоден продажный бром (т. кип. —
58,8е С при 760 лл» рт. cm.; d%° — 3,119), который, если это необходимо, можн<
высушить встряхиванием с серной кислотой, в которой бром сравнительно мах
растворим. Получение чистого брома описано в литературе [4е, 5, 6, 7а]. Поел
бромирования избыточный бром удаляют SO2 или NaHSO.,. В качестве растворителе:
брома пригодны СС14, СНС13, СИаС12, CS2, пстро.тейный эфир, ледяная уксусна.
кислота и нитробензол. Первые три растворителя следует применять только пр-
низких температурах; при 0°С можно пользоваться также эфиром.
Иод (т. пл. 113,6° С) легко сублимируется, что используют для его очистки. Дл
устранения примесей хлора и брома можно при сублимации добавлять KI. Иод мал.
(1] L. М» Dennis, J. М. Vender, Е. G. Rochow, J. Am. Chem. Soc.
53, 3263 (1931).
[2] A. L. Henne, J. Am. Chem. Soc., 66, 96 (1938).
J3] M. II u d 1 i c k y, Chemie der organischen Fhiorverbindungen, Berlin, Deutsche
Verlag der Wi.ssenschaften, 1960: a) S. 28, b). S. 50; c) S. 152; d) S. 31; e) S. 2£
I) S. 47; g) S. 49; h) S. 296; i) S. 126; k) S. 76, 149; 1) S. 35; m) S. 45; n) S. 60:
o) S. 84; p) S. 167. (B 1961 г. вышел русский перевод.)
[4] G. Brauer, Handbuch der praparativen anorganischen Chemie, 2 Anfl., Bd. 3
Stuttgart, I960: a) S. 144; b) S. 249; c) S. 253; d) S. 254; e) S. 251; 1) S. 14E
g) S. 257; b) S. 258—59; i) S. 263; k) S. 264; 1) S. 191. (B 1956 г. вышел русски
перевод.)
[5] О. HSnigsclimid, Е. Zintl, Ann., 433, 201 (1923).
[6] О. H. S t r i e b e 1, H. S t г i e b e 1, Z. anorg. Chem., 194 , 294 (1930).
[7] A. R о e d i g, II о u b e n - W e у 1, В. 5. T. 4, 1960: a) S 14; b) S. 17; c) S. 54f
542; d) S. 484.
92
ОБРАЗОВАНИЕ СВЯЗИ С— На) В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
растворим в воле (0.28 г'л при 20'1 С), но хорошо растворяется и минеральных кислотах
и иодном растворе К1 (15,9 г/л в 0.12 и. растворе KI п 420 г/л в 1,9 и. растворе К]).
При компа । noli температуре он хорошо растворим в эфире, метиловом и этиловом спир-
тах, сероуглероде л бензоле, умеренно растворим в ледяной уксусной кислоте, хлоро-
форме, четыреххлорпстом углероде и петролейном эфире. Иод можно высушить в экси-
каторе пал CaCL. концентрированной II,SO4 пли РгО5; он не гигроскопичен. О получе-
нии чHcreifпюго иода см. |iej.
При работе с большими количествами иода его следует регенерировать из маточ-
ника, панримео окислением кислородом в присутствии окислив азота [4d. 8J. Удобный
п Hpoeioii способ регенерации мода из подпда см. 191.
К маточнику после получения п-лоданилила, содержащему приблизи-
тельно I моль Xal и примерно 0,8 моль NallCOs, добавляют 100 мл концентри-
рованной Il2SO4 п раствор 200 г Na2Cr2O7 в 200 мл воды. Выделившийся иод
трижды промывают декантацией водой, отсасывают п высушивают на часовом
стекле.
Родан (SCN)a применяется в основном точно так же, как и галогены [10а].
Это — жидкость [11], кристаллизующаяся при температуре от —3 до —2° С и легко
полимеризующаяся при комнатной температуре в красный аморфный продукт. В рас-
творе родам более стабилен, во все же полимеризуется при медленном взаимодействии
с соответствующим компонентом, особенно под действием света, тепла, влаги и кисло-
рода. Органические роданиды легко отделить от полимерного родана, так как он не-'
растворим в воде и органических растворителях.
Родап. Получение раствора. К приготовленной и охлажденной до 5—
10е С суспензии сухого роданида свинца Pb(SCN)2 в сухом эфире, мет ролёйном
эфире. СЙ2, С9Пе, СИСЬ, СС14 или в ледяной уксусной кислоте прибавляют
небольшой, против рассчптанного, избыток брома в виде 10%-пого раствора.
Бесцветный прозрачный раствор родана отделяют декантацией. Раствор следует
по возможности приготовлять непосредственно перед его использованием для
роданириванпя.
Нужный для работы роданид свинца получают из Pb(NOa)j п раствора
роданида натрпя в холодной воде ]10Ъ|.
Удобно применять родан «в момент выделения», получаемый, например, добавле-
нием брома к охлажденному раствору смеси роданпруемого соединения и роданида
щелочного металла в метиловом спирте, ацетоне, метилацетате или ледяной уксусной
кислоте [12, 13|. Родап в момент выделения образуется также при работе с роданидом
меди [14J:
2Cu(SCN)2 2CuSCN + (SCN)2-
Подвергаемое роданированию соединение обрабатывают в органическом раство-
рителе (метиловый спирт, этилацетат, ледяная уксусная кислота) или к кислой водной
среде черным роданидом меди, который также получается в ходе реакции из соли
меди (II) и роданида аммония или щелочного металла.
Родан расходуется в процессе реакции, и тем самым равновесие сдвигается
нираво, что заметно по обесцвечиванию раствора с образованием белого монородапида
меди CuSCN.
2. Общие положения о присоединении галогенов но С—С-связи
Присоединение талогепов к веществам с этиленовыми связями является очень
характерной реакцией, протекающей большей частью при низких температурах и само-
произвольно. Благодаря этой реакции, при которой из газообразных непредельных
соединений образуются маслообразные продукты реакции, непредельные соединения
[8J F. Аги d t, Вег., 52, 1131 (1919).
[9J К. Q. Brewster, Org. Synth., Coll. v. 2, 1943, p. 348.
(10] J. L. W ood, Org. Reactions, 3, 1946: a) S. 241; b) S. 25o; c) S. 257.
[И] E. MdcrMck, Arm., 419, 217 (1919).
[12] П. P. Kaufmann, Bor., 62, 390 (1929).
(13J If. P. Kaufmann, E. \V e b c r, Arch. d. Pharm., 267, 201 (1929).
[14] II. P. Kaul in a n n,* К. К u c h 1 c r, Ber., 67 , 944 (1934).
I. ПРИСОЕДИНЕНИЕ Hal и SCN ПО КРАТНЫМ С—С-СВЯЗЯМ
93
с этиленовыми связями были названы олефинами от французского слова olefiant —
маслообразующпй.
Присоединение фтора по двойной связи существенно отличается от присоедине-
ния других галогенов. Тепловой эффект этой реакции значительно превышает энергию
разрыва олефпповой С=С-связи, равной ~80 нкал/молъ. Реакции непосредствспного
присоединения фтора, а также замещения фтором столь экзотермичны, что большин-
ство органических соединений при этом сгорает иногда со взрывом, если не приняты
соответствующие меры предосторожности [15]. Присоединение фтора к олефинам не
имеет препаративного значения [ЗЬJ; в ароматических соединениях происходит одно-
временно замещение и присоединение. Из остальных галогенов легче всего присоеди-
няется к олефинам под, труднее всего — хлор.
Присоединение галогенов протекает но радикальному пли ионному механизмам.
Первый механизм преобладает при высоких температурах или при действии света.
Атом галогена, образовавшийся в стадии термического плп фотохимического
инициирования реакции или при введении инициатора, далее взаимодействует
по цепной реакции:
х . + Н3С=С112 —> ХСН-2—сн2.
ХСЦо—CHn’-J-Xg —>• XCII2—CHjX-j-X» и т. д.
Радикальное присоединение протекает без растворителей или в неполярных
растворителях, таких как пстролейвый эфир или четыреххлористый углерод (до 60u С).
Началом ионного галогенирования является для большинства реакций образо-
вание л-комплекса катиона Х+ с л-электронной парой С=С-снязи, который стабили-
зуется в результате присоединения аниона X" (присоединение галогена) или отщепле-
ния протона Н+ (замещение галогена).
Растворители с большой диэлектрической постоянной, например вода, ледяная
уксусная кислота, этиловый спирт, способствуют протеканию реакций по ионному
механизму. Но в их присутствии п-комплекс может присоединять молекулу раствори-
теля, после чего следует стабилизация комплекса с отщеплением Н* и образованием,
например, галогенгидрина или его эфира.
/А. Y V
>с=с< хсн2—ch2or+h+ ХСН2—СН2Х
НХ \н
замещение присоединение присоединение
Добавление аниона Х“ (например, хлорида или бромида патрия [16]) подавляет
образование галогенгидрина; связывание X", например нитратом серебра, способствует
его образованию. Для ионного галогенирования в индифферентных растворителях
требуется добавка иода, А1С13, FeCl3, HgCJ2 и других аналогичных веществ, способ-
ствующих поляризации молекулы галогена.
Некоторые олефины, например тетрафенилэтилен, полигалогенолефины,
а также малеиновый ангидрид трудно присоединяют или даже совсем ке присоединяют
1алогенов. Совершенно сухой этилен в темноте также по присоединяет брома. Более
подробно теория реакций присоединения галогенов и замещения галогенами описана
в специальной литературе [17, 18].
Присоединение брома по С=С-связям не обязательно начинается с электрофиль-
ного присоединения Вг+; возможен также механизм нуклеофильного присоединения.
115] A, L, Henn е, Org. Reactions, 2, 69 (1944).
116] Е. М. Т е г г у, L. Eichelberger, J. Am. Chem. Soc., 47, 1067 (1925),
[17] Е. М ю л л с р, Новые воззрения в органической химии,* пер. с нем., Издат-
инлит, 1960.
[18] Р. В. D. de la Mare, Quart. Rev., 3, 126 (1949).
94
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Так, например, обычное медленное присоединение брома в ледяной уксусной кислоте-
к малеиновой, фумаровой и коричной кислотам, стильбену, тетрахлорэтилену катали-
зируется бромидами лития или калия [19].
3. Присоединение галогенов к ненасыщенным углеводородам
С низкомолекулярными неразветвленными олефинами — этиленом, пропиленом,
и-бутиленом и н-амиленом хлор реагирует при комнатной температуре с образованней
продуктов присоединения (реакция 1)
CHS-CH=CH2 СНз-СНа-СН2С1 (1>
в то время как из разветвленных олефинов уже при комнатной температуре образуются
продукты замещения с сохранением двойной связи (реакция 2):
СП3-С=СН3 — -> СН2С1-С=СН2 + НС] (2)
СНз СНз
Неразветвленяые олефины можно хлорировать с замещением [20] только нр®
температурах порядка 400—500° С (реакция 3):
СЩ-СН=СН2 -^5* СИ2а-СН=СН2+НС1 (3>
В жидкой фазе протекает конкурирующее с реакцией (2) присоединение образу-
ющегося НС1 по С= С-связи; в газовой фазе эта реакция практически не идет. Выделя-
ющееся при присоединении хлора большое количество энергии (около 40 ккал/моль)-
может, если не пользоваться сильным охлаждением, индуцировать дальнейшее заме-
щение хлором первичных продуктов присоединения (ср. также [21]). Замещение хло-
ром можно подавить добавлением около 0,1—0,3 мол. % FeCla, способствующего при-
соединению хлора [22].
Присоединение хлора к этилену удается только при низких (—25° С) темпера-
турах [23]; при 20° С в результате присоединения и одновременного замещения обра-
зуется около 90% высших продуктов хлорирования. Пропуская хлор и этилен через-
охлаждаемую до —30° С насадочную колонку, получают дихлорэтан с 90%-ным выхо-
дом [24]. Реакция протекает еще легче при —70° С.
В качестве катализаторов, способствующих -присоединению хлора к этилену
с образованием дихлорэтана при более высоких температурах, используются хлориды
железа, меди, сурьмы [25], хлорид титана (растворитель — пеитахлорэтан) [26J
и хлорид кальция [27]. В присутствии железосодержащих порошкообразных бокситов
на пемзе (катализатор I) или крупнозернистого боксита (катализатор II) с понижением;
температуры реакции увеличивается содержание более высокохлорированных
продуктов [28].
' Например, из этилена с катализатором I при 12° С получается: дихлор-
этана 69%, трихлорэтана 24% и высших хлорэамещенных продуктов 7%; при
55—60° С соответственно 90%, 6% и 4%. С катализатором II при 55—65° С'
эти продукты получаются соответственно в количестве 94%, 3,5% и 2,5%; при
[19] R. A. Ogg, J. Am. Chem. Soc., 57, 2728 (1935).
[20] H. P. A. G г о 11, G. H e а г n e, Ind. Eng. Chem., 31, 1530 (1939).
[21] Ф. Азингер, Химия и технология моноолефинов, пер, с нем., Гостоптехиздат,.
1960.
[22] Е. G. Galitzenstein, С. Woolf, J. Soc. Chem. Ind., 69, 292 (1950k
[23] H. Bahr, H. Z i e 1 e r, Angew. Chem., 43, 233 (1930).
[24] A. Maier, герм. пат. 529524; С., 1931, II, 1921.
[25] Англ. пат. 147909; С., 1922, IV, 393.
[26] Франц, пат. 770943- С., 1935, I, 2255.
[27] Пат. США 2099231; G., 1938, I 3387.
[28] I. Garat, Вег., 76, 1115 (1943).
I. ПРИСОЕДИНЕНИЕ Hal и SCN ПО КРАТНЫМ С—С-СВЯЗЯМ
95
110—115° С из пропилена и хлора образуется 85—90% дихлорпропана; из смеси
а- и 0-бутилепов и хлора смесь 1,2- и 2,3-дихлорбутанов в количестве 70%.
Аппаратурное * оформление процесса подробно описано в специальной
литературе 128].
В качестве растворителей при реакциях присоединения хлора к олефинам
пригодны CClj, СНС13, СН2С1а. Жидкие олефины хлорируются большей частью бе»
растворителей.
Лри бромировании также необходимо отводить значительное количество тепла
реакции, иначе будет происходить замещение бромом в результате вторичных
и побочных реакций. Кроме того, образующийся НВт может присоединяться
по С=С-связям. Таким образом, при проведении реакций нужно применять охлажде-
ние и избегать избытка галогена.
Присоединение иода по С=С-связи с препаративпой точки зрения малоинтересно.
Иодирование проводят в эфире, этиловом спирте, CS2, СС14, водном растворе KI или
без ‘растворителя. Продукты присоединения иода неустойчивы, и большей частью уже
при комнатной температуре отщепляется иод. Поэтому в дальнейшем описывается
только присоединение хлора и брома.
Для присоединения хлора к ненасыщенным соединениям при сильном охлажде-
нии вводят непосредственно в вещество или в ого раствор рассчитанное по реакции ко-
личество хлора. Удовлетворительные результаты получаются даже, если ненасыщенное
соединение неполностью растворено. Можно также прибавлять по каплям раствор хлора
в соответствующем растворителе. Для хлорирования вполне пригоден сульфурил-
хлорид SO2C12, который употребляется без растворителя или с растворителями СС14»
СНС13, СН2С12.
Присоединение хлора к циклооктатетраену (29].
С1(Вг)
(Br)CL 1 /С1(Вг) /С1(Вг)
у у-у ch(Bn) у у sotd«(Brt); у\______/ ' '
(Вг) С1/Y~Xcl (Вг> \ci(Br)
С1(Вг)
К смеси 104 г (1 моль) циклооктатетраена и 208 г СН2С12 при 25° С и перемеши-
вания приливают 140 г (около 1 моль) SO2C12, поддерживая эту температуру
охлаждением и регулированием скорости прибавления сульфурилхлорида. В про-
цессе реакции выделяется сернистый ангидрид. После 1—2 ч перемешивания
при комнатной температуре и отгонки в вакууме СН3С12 коричневатое масло
дважды перегоняют при большом разрежении. Получается 120 г 7,8-дихлор-
бицикло [0,2,4]октадиена-2,4, в виде почти бесцветного масла; т. кнп.
60° С (0,1 мм рт. ст.) и 67—68° С (1,2 мм рт. ст.). Это вещество, как и соот-
ветствующий дибромид, димеризуется при стоянии.
При 0—5° С из циклооктатетраена и деойпого количества SO2C12 обра-
зуется тетрахлорбиццкло[0,2,4]октен; т. пл. 111—112° С. При пропусианиж
хлора в раствор циклооктатетраена в десятикратном количестве хлороформа,
при температуре от —30 до —25° С до полного его поглощения и дальнейшем
проведении реакции в течение нескольких часов при этой температуре и 8ч
при комнатной наряду с изомерным жидким гсксахлоридом получается гекса-
хлорбициклооктан; т. пл. 126—127° С.
Прибавлением 1 моль брома в двойном весовом количестве СН2С12 к 1 моле,
циклооктатетраена в двойном количестве СН2С12 при 0—5° С получают 7,8-ДЯ-
бромбицикло[0,2,4]октадиен-2,4; т. кип. 90—91° С (1 мм рт. ст.). Действие
2 моль брома в двойном весовом количестве СНаС12 на 1 моль циклооктатетраена
в десятикратном количестве этого растворителя прп 0—5° С приводит к обра-
зованию двух разных тетрабромидов, плавящихся при 147 — 148 и 94° С?
действие 3 моль брома в хлороформе при —20° С дает гексабромид^
т. пл. 150—151е С.
(29) W« Reppe et al., Ann., 560, 18, 50 (1948).
96
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
При проведении присоединения хлора с помощью SOaCl3 реакцию полезно
инициировать, добавляя сначала примерно 3% вводимого SO„Cls при осторожном на-
гревании, а затем вносить по каплям остальное его количество, применяя по мере
надобности слабое нагревание. Таким образом, например, из металлилхлорида полу-
чают 1.2,3-трихлор-2-метплпропап [30J.
Реакцию присоединения хлора под действием SO2Cl2 можно инициировать также
добавлением инициаторов радикальных реакций, так как этот процесс является ради-
кально-цепным.
К раствору 0,2 моль ненасыщенного соединения и 0,002 моль перекиси
бензоила в CCI4 пли СН2С1в по каплям прибавляют смесь примерно одинаковых
количеств SO2C12 (0.15—0,3 моль) и СС]4 с такой скоростью, чтобы смесь слабо
кипела, и после окончания прибавления кипятят примерно еще 1 ч. Например,
1,2-дихлорэтилеп не реагирует с избытком сульфурилхлорида даже при трех-
часовом кипячении; реакция начинается только при добавлении перекиси.
По этому способу синтезируют продукты присоединения хлора к цикло-
гексену, аллилхлориду, 1,2-дихлорэтилену с выходами соответственно 90, 80—90
и 85%; из стильбена [31J получается 45% дихлорида, плавящегося при 191—
193° С, и 33% дихлорида, плавящегося при 90—93° С.
Для присоединения брома к газообразным ненасыщенным углеводородам 'их
можно пропускать через бром при охлаждении. Так, например, получают в лабора-
тории 1,2-дибромэтан [32] и бутадиеитетрабромид [38]. Однако чаще всего раствор
брома прибавляют по каплям при перемешивании и охлаждении льдом к раствору
олефина в том же растворителе (стр. 91) до прекращения обесцвечивания брома.
Неустойчивые ненасыщенные соединения удобнее бронировать не бромом, а продуктами
присоединения брома к гидробромидам третичпых аминов или диоксану.
Галогснзамещенные олефины с галогеном у насыщенного атома углерода спо
собны довольно легко присоединять галогены. Например, присоединением брома
к аллплбромиду в четыреххлористом углероде при —5° С получают 1,2,3-трибромпро-
пан [39] с выходом 96—98%. Наличие галогена у пепасыщенпого атома углерода
затрудняет присоединение, галогенов, причем способность присоединять галогены
снижается [40] в ряду:
—СН=СН—; -СП=СНХ; СНХ«=СНХ; СНХ=СХ2; СХ2=СХ2
Присоединению галогена мепее всего препятствует фтор, сильнее всего — бром
В иодолефппах иод замещается другими галогенами уже на холоду. Например, из
1,1-дииодэти.чсна и брома (или хлора) в CCI4 получается 1,1,1.2-тетрабром или соответ-
ственно тетрахлорэтан [41]. Присоединение брома или хлора к полихлор(бром)оле-
фияам происходит легче всего фотохимически. В таких случаях жидкий или растворен-
ный в СС]4 олефин обрабатывают при температуре, не превышающей 25° С, и УФ-
[30] A. Mooradian, I. В. С 1 о k е, J. Am. Chem. Soc., 68, 785 (1946).
[31 ] М. S. К h а г a s с ii, Н. С. Brown, J. Am. Chem. Soc., 61, 3432 (1939).
[32] L. G a 11 е г т a n n, II. Wieland, Die Praxis des organischen Chemi-
kers, 38 Aufl., Berlin, VcrJag W. der Gruyter u. Co., 1958, S. 98,
[33a] K. W. R 0 s 0 11 m u n d, W. Ku hn be nn, Ber., 56, 1262 (1923); S. M. E.
Englert, S. M.’ Me Ё 1 vai n, J. Am. Chem. Soc., 51, 863 (1929).
33b] S. Nf E. Englert, S. M. M с E 1 v a in, J. Am. Chem. Soc., 51, 865 (1929).
33c| C. D jerassi, C. R. Scholz, J. Am. Chem. Soc., 70, 417 (1948).
33d] C. L. Arcus, H. E. Strauss, J. Chem. Soc., 1952, 2670.
34] C.L. Arcus, II. E. Strauss, J. Chem. Soc., 1952, 2669.
35] M. J. Frazer, W. Gerrard, J. Chem. Soc., 1955, 3628.
36] L. Farkas, O. Schachter, J. Am. Chem. Soc., 71, 2252 (1949).
37] А. В. Домбровский, ЖОХ., 24, 610 (1954).
38] J. Thiele, Ann., 308, 337 (1899).
[39] J. R. J oh ns on, W. L. M с E w 0 n, Org. Synthesen, F. Wieweg u. Sohn, Braun-
schweig, 1937, S. 522; Org. Synth., Coll. v. 1, 2 ed, 1956, p. 521.
[40] В. E. S w e d 1 u n d, P. W. Robertson, J. Chem. Soc., 1947, 630.
[41 ] II. P. Kaufmann, Ber., 55, 257 (1922).
I ПРИСОЕДИНЕНИЕ Hal И SCN ПО КРАТНЫМ С—С-СВЯЗЯМ
97-
облученип рассчитанным количеством брома (хлора). Присоединение хлора и брома
к фторатиленам протекает с очень хорошими выходами, особенно на солнечном свету
[Зс]. К тетрафторэтилену в автоклаве при 150° С с хорошим выходом присоединяется
иод [42].
Стильбен присоединяет бром относительно медленно. В случае ^ис-стильбена
в CSt на холоду в темноте происходит обычное для олефинов транс-присоединение
брома, причем получается 89% (± )-1,2-дибром-1,2-дифенилэтана (т. пл. 110—1110 С)
наряду с небольшим количеством ,«сзо-1,2-дибром-1,2-дифепплэтана (т. пл. 236° С).
Первый, между прочим, под действием брома пли иода в СС)4 в рассеянном дневном
свете перегруппировывается в „мезо-соединение 144]. .мезо-Соедипенис образуется в каче-
стве основного продукта при присоединении брома к транс-стильбену в сероуглероде
[45} или эфире [46]. При взаимодействии транс-стильбена с насыщенным раствором
хлора в безводном эфире под действием УФ-лучей или солнечного света получаются
одновременно (±)-и л«зо-1,2-дихлор-1,2-дифеиилэтаиы; т. пл. 93—94 и 191 —193° С
соответственно [44, 45].
1,1- Дифенлл- и 1,1,2-трифенилэтилейы очень легко реагируют с бромом [43].
Тетрафонилзтилен присоединяет хлор, но не присоединяет бром [47]. Стирол легко
присоединяет бром.
Стиролдибромид (1,2-дибром-1-фенилэтан). Его получают в достаточно-
чистом виде для дальнейшей переработки в фепилацетилен медленным приба-
влением по каплям при сильном перемешивании раствора 80 г брома в 100 мл
СС14 к 52 г стирола в 50 мл того же растворителя при температуре от —10 до-
—15° С. К концу реакции охлаждение ведут только льдом или водой, иначе
смесь преждевременно затвердевает. После непродолжительной выдержки
осадок отсасывают н высушивают на воздухе; выход 90—95% [48].
4. Присоединение галогенов к полиенам
При присоединении 1 моль брома (хлора) к полиенам обычно образуется смесь
дигалогензамещепных 1,2 (I) ,и 1,4 (II), причем содержание соединений I и II зависит
от растворителя и радикалов R' и R:
(Cl)BrCHR' CHR' (Cl)BrCHR'
| || Bff |
CHBr(Cl) CH CH
1 I 11
CH CH CH
n II 1
CHR CHR (C.l)BrCHR
I II
Исключительно 1,4-присоедипепие происходит, например, когда циклопентадиегь
обрабатывают бромом н петролейном эфире прп температуре от —30 до —40° С- Полу-
чается смесь нестойких при комнатной температуре цис-и транс-3.й-дибромциклопен-
тенов [49]. Кристаллический ^мс-3,5-дибромциклопептсн (т. пл. 45°С) прп перегонке
в вакууме (2 мм рт. ст.) частично перегруппировывается в продукт 1,2-прпсо-
едипспия — транс-3,4-дибромциклопентен [50]. Подобного рода перегруппировки
[42] R. .N. Haszeldine, К. Leedham, J. Chem. Soc., 1953, 1550. J
[43] J. Meisenheimer, Ann., 456, 141 (1927).
[44] R. E. В и c k 1 e s, W. E. Steinmetz, N. G. Wheeler, J. Am.
Chem. Soc., 72, 2497 (1950).
[43J P. Pfeiffer, Ber, 45 *817 (1912).
[46] L. I. Smit h, M. M. F a 1 k о p, Org. Synth., Coll. v. 3, 1955, p. 350.
[47] J, F. Norris, R. Thomas, В. M. Brown, Ber., 43, 2950 (1910).
[48] H. Fiesselmann, K. Sasso, Chem. Ber., 89, 1786 (1956).
[49] G. VV. Barber, J. Engljsh jr., J. Am. Chem. Soc., 73, 747 (1951).
[50] W. G, У о я n g, H. К. II a 11 ir S. Winstei n, J. Am. Chem. Soc., 78,
4338 (1956).
7 Заказ 1835.
•98
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
наблюдались также и для продуктов присоединения галогенов к бутадиену [51]; они
подробно описываются в приводимых примерах препаративных синтезов.
г ' 3,4-Дпхлорбутен-1 и 1,4-двхлорбутен-2 [52, 53[. В течение 45 мин при
перемешивании и охлаждении смесью сухого льда со спиртом (температура
реакции не должна превышать — 5° С) к раствору 180 г бутадиена в 350 мл сухого
СС14 добавляют раствор 150 г хлора в 750 мл сухого CCL. После отгонки раство-
рителя и ректификации остатка на колонке получают 35 г 3,4-дпхлорбутена-1;
т. кип. 123° С (766 мм рт. ст.), и 70 г 1,4-дихлорбутена-2; т. кип. 159° С
(766 мм рт. ст.). Последний имеет транс-конфигурацию [53] и представляет-
собой единственный продукт, образующийся при введении бутадиена и хлора
в объемном соотношении 3 ; 1 в СС14 или смесь СС14 и пиридина при охлаждении
смесью сухого льда и ацетона. Соответствующие грхс-соединения получают
из цис-бутеЕ-2-дв.ола-1,4 при 0° С с трихлорцдом фосфора в пиридине [53].
1,4-Дибромбутен-2 и 3,4-дибромбутеи-1. В течение 45 мин при —20° С
и перемешивании добавляют по каплям раствор 26 мл брома в 200 мл СС14 к рас-
твору 45 г бутадиена в 500 мл того же растворителя [52]. После удаления рас- *
творителя и перекристаллизации остатка из петролейного эфира получают 72 г
1,4-дибромбутела-2; т, пл. 54° С. Он имеет, как и соответствующее 1,4-дихлор-
соедипение, транс-конфигурацию [54]. Это соединение получают также при-
соединением при —30° С к бутадиену 1 моль брома в хлороформе [55]. 3,4-Ди-
бромбутен-1 в смеси с несколько меньшим количеством продукта 1,4-присоеди-
нсния (соответствекно 53,2 и 46,8%) получают добавлением в течение 2 ч при
—20° С 1 моль брома к раствору 4 моль бутадиена в 125 мл метилциклопен-
тана [56].
О присоединении галогенов к бутадиену см. работы [38, 51].
Чем больше С=С-связей в полиене, тем сильнее тенденция сохранить ненасыщен-
ное состояние. В этом случае все больше затрудняется насыщение всех двойных связей
галогеном и возрастает склонность к отщеплению галогеноводорода. Присоединение
хлора п брома к полиенам происходит с сохранением возможно более длинной системы
сопряженных связей, например при присоединении к фенилбутадиену — фенилви-
пилыюго сопряжения, к гексатриену — бутадиенового сопряжения [58]. Полиеновые
карболовые кислоты очень легко присоединяют бром по С=С-связи, наиболее удален-
ной от СООЙ-груины [57].
5. Присоединение галогенов к стероидам
Продукты присоединения брома к ненасыщенным стероидам играют роль про-
межуточных продуктов при синтезах в этом ряду. Присоединение брома проводят при
температуре ниже 20’ С в ледяной уксусной кислоте, эфире или хлороформе. В этих
условиях происходит обычное для С=С-связи траяе-нрисоединенпе. Первично образу-
ющиеся дибромиды транс-днаксиальной конформации могут перегруппировываться
в соединения транс-диэкваториальной конформации. Более подробные объяснения
и оригинальную литературу но этому вопросу см. [59]. Следует упомянуть, что для
[51] Е. Н. Farmer, 'С. D. Lawrence, J. F. Thorpe, J. Chem. Soc.,
1928, 730.
[52] L. N. Owen, J. Chem. Soc., 1949, 243. z
[53] K. Mislow, H. M. Hellmann, J. Am. Chem. Soc., 73, 244
(1951).
[54] К. M i s 1 о w, J. Am. Chem. Soc., 75, 2512 (1953).
[55] E. M. Shantz, J. Am. Chem. Soc., 68, 2557 (1946).
[56] J. C. Hill yer, С. H. Ice, пат. США 483049 (1949); С. A., 44, 652
(1950).
[57] О. Dann, Chem. Ber., 80, 427 (1947).
[58] P. B. D. de la M^re, E. D. H u g h e s, С. K. Ingold, J. Chem.
Soc., 1948, 21.
[59] Л. Флаер, M. Фпзер, Стероиды, нгр. с англ., изд. Мир, 1964.
I. ПРИСОЕДИНЕНИЕ Hal И SCN ПО КРАТНЫМ С—С-СВЯЗЯМ
99-
присоединения хлора к ненасыщенным стероидам, в том числе к холестерину, приме-
ним фепклиодидхлорид CjHsIClj в хлороформе [60].
6. Присоединение галогенов к ненасыщенным спиртам и эфирам
В разбавленных растворах при 0—25° С в большинстве случаев можно присоеди-
нить бром к ненасыщенным спиртам без одновременной замены ОН-группы.
2,3-Дибромпропанол [61]. На раствор аллилового спирта в десяти-
кратном объеме СС14 действуют рассчитанным по реакции количеством брома
в двойном объеме того же растворителя.
Реакцию присоединения брома к алкилвиниловым эфирам проводят большей-
частью в CCL или СНС13 при охлаждении от —5 до 0° С. Получение а,р-дибромэтил-
метялового эфира из винилмстилового эфира, равно как и сильно экзотермичное при-
соединение к эфиру хлора, см. [62]. Из винилэтилового эфира при охлаждении ниже
30° С (смесь льда с солью) получают а,р-дихлордиэтиловыи эфир [63]. Этот метод,
изученный на многочисленных примерах, широко применим [64]. Однако он имеет
препаративное значение только в том случае, если виниловые эфиры легко доступны
(например, из ацетилена спирта в присутствии едкого кали в автоклаве [65]).
а,р-Дигалоидэфиры представляют интерес в качестве промежуточных продуктов.
Они образуют, например, со спиртовым едким кали ацетали а-галоидальдегидов [66].
1,2-Дибромэтокскэтан. К раствору 1 моль винилэтилового эфира
в 4 моль СНС1э при —15° С и сильном перемешивании сначала медленно, затем
быстро прибавляют ио каплям 1 моль брома до прекращения обесцвечивания
последнего. Остаток после отгонки СНС1з разгоняют в вакууме (20 мм рт. ст.).
Получают 1,2-дибромэтоксиэтан; т. кип. 80° С при 20 мм рт. ст. Выход 88,5%
от теоретического.
Диэтилацеталь бромацетальдегида [67]. Раствор 1 моль 1,2-дибром-
этоксиэтана в 3,5 моль абсолютного СаН5ОН прибавляют при 0° С по каплям
и перемешивании к раствору 1,8 моль КОН в 8,5 моль абсолютного СаН6ОН.
После 5 мин кипячения на водяной бане реакционную массу выливают на лед,
отделяют слой бромацеталя, из водного слоя последний извлекают эфиром.
Объединенные вытяжки сушат над К2СОз. После отгонки эфира к этилового
спирта перегоняют диэтилацеталь бромацетальдегида на колонке в вакууме.
Выход 83% от теоретического; т. кип. 63° С (14 мм рт. ст.).
Присоединить иод к винилалкиловым эфирам невозможно, так как иод вызьь
вает полимеризацию эфиров.
О получении а-галоидальдегидов или кетонов из енолацетатов карбонильных
соединении см. стр. 177.
Реакцию присоединения брома к ненасыщенным спиртам и эфирам часто жела-
тельно проводить в особенно мягких условиях. С помощью пербромида пиридиний-
бромида можно избирательно присоединять бром по С=С-связи олефиковой боковой
[60] С. J. Berg, Е. S. Wallis, J. Biol. Chem., 182, 683 (1946).
[61] W. G. H. Edwards, R. Hodges, J. Chem. Soc., 1953, 3428.
[62] M. Ф. Шостаковскнй, П. В. Тюпаев, Синтезы органических со-
единений, Сборник 2, Изд. АН СССР, 1952.
[63] М. Ф. Шостаковскнй, Ф. П. Сиде лковская, Синтезы органи-
ческих соединений, Сборник 1, Изд. АН СССР, 1950.
[64] М. Ф. Шостаковскнй, ЖОХ, 13, 1 (1943); ДАН СССР, 41, 124 (1943);
ЖОХ, 14, 102 (1944); 17, 957 (1947).
165] А, Е, Фаворский, М. Ф. Шостаковскнй, Синтезы органических
соединений. Сборник 1, Госхимиздат, 1950.
[66] А. Е. Ф а в о р с к и й, М, Н. Щуиина, ЖОХ, 16, 385 (1945).
[67] Н. Bag ап г, Е. Brinckmann, Chem. Вег., 86, 1320 (1953).
7?
100
ОБРАЗОВАНИЕ СВЯЗИ С —Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
цепи фениловых эфиров, как, например, в боковой цепи сафрола, который бромп-
руется бромом также и в фенильное ядро. Этот реагент можно получать п непосред-
ственно в реакционной массе во время реакции, смешивая раствор бронируемого
вещества и соли пиридина в ледяной уксусной кислоте с рассчитанным количеством
брома.
Пербромид пиридинийбромида [33а] CeII5N • НБг • Вг2. Чистый пиридин
смешивают с избытком соляной кислоты и насыщают бромом. Выделяется темно-
коричпевоо масло, которое отделяют от водною слоя. При охлаждении смесью
снега с солью продукт вскоре кристаллизуется. Выпавшие кристаллы отсасы-
вают и дальнейшим охлаждением фильтрата проводят вторичную кристаллиза-
цию. Твердый пербромид пиридппийбромпда промывают СНС1з и перекристал-
лизовывают из этилового спирта (растворение ведут при 40—50° С).
По другому методу [ЗЗЬ] при температуре около 60° С к раствору 1 моль
гидробромида пиридина в 240 г ледяной уксусной кислоты добавляют при пере-
мешивании раствор 1 моль брома в 160 г ледяной уксусной кислоты. При мед-
ленном охлаждении из смеси кристаллизуются красные иглы или призмы пер-
бромида пиридинийбромида. Отфильтрованные кристаллы высушивают в ва-
куум-эксикаторе. Выход около 300 г, т. пл. 132—134° С.
Этот продукт получают также [33с, d], добавляя 1 моль брома к раствору
1 моль пиридина в 48%-ной бромистоводородпой кислоте (2 моль).
При присоединении к 1-фенилаллпловому спирту брома в сухом СНС13, СС14
и С8а получают исключительно 1,2,3-трибром-1-фенилпропан. С пербромидом диридп-
ницбромцда при 12—15° С, напротив преимущественно протекает реакция присоеди-
нения с образованием 2,3-дибром-1-фенилпропилового спирта [34]. Из аллилового
спирта так же, но без растворителя, получается 2,3-дибромпропиловыи спирт |35].
Аналогично действуют бромаддукты тетраметиламмонийбромида.
Бромадцукты тетраметиламмонийбромида [36]. Эвтектическая смесь двух
аддуктов (CII3)4NBr8 и (CH3)4NBt9 имеет состав (CH3)4NBr5,e; т. пл. 15,8° С.
J [ри содержании Вг511и Вге,2 выше 25° С эвтектическая смесь жидкая.
При охлаждении до 2'0—30° С к раствору 154 г (1 моль) тетраметилам-
монийбромида в 500 мл воды в течение 15 мин при перемешивании добавляют
400 г (2,5 моль) брома. Образовавшееся темно-красное масло (d = 2,33) отделяют
в делительной воронке. Выход 90% от теоретического; при повторном исполь-
зовании водного слоя в качестве растворителя выход около 100% от теорети-
ческого.
2,3-Дибромпропиловый спирт. При охлаждении льдом до 40° С к 58 г
(1 моль) аллилового спирта в течение 40 мин при перемешивании приливают
по каплям 222 г указанного выше бронирующего агента. Выпадающий при этом
тетраметиламмоннйбромид отсасывают и промывают эфиром. Практически его
можно количественно регенерировать. Промывной эфир соединяют с фильтра-
том и разгонкой получают 2,3-дпбромпропиловый спирт; выход 85% от теоре-
тического.
Для присоединения брома в «мягких» условиях применяют также дноксандибро-
мид (стр. 158):
СНг—СН2
О / ° ' БГа
СПг-СНг
Стиролдибромид [37]. Его получают постепенным добавлением при
охлаждении проточной водой 25 г диоксанднбромида к 10,4 г стпрола и после-
дующим разбавлением водой. Выход 100% от теоретического, т. е. замещение
полностью исключено.
2,5-Дибромгексен-З. При взаимодействии 25 г диоксапдибромида
п 11,8 мл гексадиена-2,4 образуется 86% 2,5-дибромгексена-З — продукта
1,4-присоедшгенпя.
Аналогично могут быть достигнуты хорошие выходы продуктов присоединения
брома к акролеину, акриловой кислоте, се нитрилу и эфирам, а также к винилацетату.
I. ПРИСОЕДИНЕНИЕ Hal И SCX ПО КРАТНЫМ С—С-СВЯЗЯМ
101
7. Присоединение родана к ненасыщенным соединениям
Родап (SCN)2 присоединяется при комнатной температуре с очень хорошими
пыходамп к большому числу ненасыщенных соединений: к этилену, аллиловому спирту,
олеиновой кислоте, стильбену, стиролу, апеголу и ацетилену [68]. Однако а,р-ненасы-
щенные карбоновые кислоты родап по присоединяют. При комнатной температуре
указанные реакции обычно протекают медленно (12 ч). К некоторым соединениям,
например к этилену, родан не присоединяется на свету. Указания па работы по при-
соединению родана к изопрену, бутадиену, терпенам и жпрам см. [69]. Реакцию при-
соединения родана пли иодродана ISCN к ненасыщенным соединениям можпо исполь-
зовать для определения подобных соединении [70, 71]. Преимущество методов с иодро-
даном по сравнению с методами, основанными на присоединении галогенов (12, С12,
Вг3. IC1), заключается в том, что невозможны реакции замещения.
Растворы родана с устойчивым титром. Самым подходящим растворителем
является безводная уксусная кислота с небольшим количеством уксусного ангидрида.
Чистый роданид свинца помещают в сухую скляпку, прокипяченную предварительно
с соляной кислотой, чтобы не выделялась щелочь. Добавляют затем растворитель —
1 объем уксусного ангидрида, 6 объемов уксусной кислоты, перегнанной лад 1% хро-
мового ангидрида, 3 объема безводного СС14 и встряхивают с бромом в темноте. После
фильтрования без .доступа влаги через высушенный фильтр получают бесцветный
раовор родана, устойчивый в течение нескольких недель.
Содержание родана определяют добавлением избытка 10%-ного водного рас-
твора KI и титрованием выделившегося иода Na2S2O3:
2KI + (SCN)2 —> 2KSCN-|-I2
Реакция присоединения (ЗСК)г К ненасыщенным кислотам используется в ана-
лизе жиров и высыхающих масел. В то время как иод присоединяется по всем
С—С-связям, родан присоединяется лшпь по некоторым из них. На основания разницы
между иодным (стр. 102) и родановым числами можно сделать вывод о составе смеси
жирных кислот, в которой наряду с другими кислотами содержатся олеиновая, линоле-
вая и линоленовая кислоты. Подробное описание этого метода см. [72а].
8. Присоединение галогенов к ненасыщенным карбоновым кислотам
Спорость присоединения галогенов к ненасыщенным кпелотам уменьшается
и последовательности хлор, бром, иод. К ненасыщенным карбоновым кислотам, а также
1. их эфирам и амидам в обычных растворителях бром присоединяется с хорошими
выходами уже при температурах от —5 до —25° С (к эфирам бром присоединяется
даже в отсутствие растворителя). Реакция протекает особенно быстро на свету.
а ,^-Диброммасляная кислота [73]. Ее получают по реакции присоеди-
нения брома к кротоновой кислоте в СС14 при 20° С. В конце реакции темпера-
туру повышают до 40® С. Выход кислоты 93% от теоретического.
Этиловый эфир а,р-дибромпропионовой кислоты [74]. Может быть полу-
чен с 98%-ным выходом прибавлением do каплям при перемешивания к этил-
акрплату рассчитанного количества брома при 5—10° С и последующей выдерж-
кой реакционной массы при комнатной температуре в течение 5 ч.
[68]
[69]
[7и]
[71]
[72]
173]
17'.]
Н. Р. Kaufmann. J. L i е р е, Arch. Pharm. Ber, dtscli. pharm. Ges.,
33, 139 (1923).
H. P. Kaufman 11, Neuere Melhodcn der praparativen organischen Chemie,
Bd. 1, Berlin, Verlag Chemie, 1944, S. 241.
H. P. Kaufmann, H. C rosse-Octringhaus, Ber., 70, 911 (1937).
H, P. Kaufmann, Stndien aui dem Fettgehiet, Berlin, Verlag Chemie, 1935,
H. P. К a u f ш a n n, Analyse der Fette nnd Fettproduktc, Bd. 1, Springer-VeT-
lag, 1958: ;<) S. 589—595; b) S. 570—584.
P. Seifert et al., Helv. chim. acta, 33, 725 (1950).
If. В r c t s cli n e i (I er et a!., Mh. Chem., 84, 1087 (1953).
102
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Р-Бром в этпловом эфире дибромпроппоиовой кислоты или а.р-дибромпропиови-
триле, получаемом в аналогичных условиях из акрилонитрила, легко замещается алк-
оксильным остатком.
Полученные таким образом а-бром-Р-алкоксиэфир или соответствующий нитрил
являются промежуточными продуктами для синтеза р-окси-а-аминокислот, как это-
описано для серина [75]. Реакция присоединения брома к кислотам с несколькими*
двойными связями, например к линолевой, линоленовой, и арахидоновой, служит для*'
количественного или полуколичестве много определения данных кислот в смеси жирных
кислот [70].
Свободный иод в спиртовом растворе почти не присоединяется или чрезвычайно"
медленно присоединяется к ненасыщенным жирным кислотам. В присутствии AgCl
присоединение иода ускоряется. На этом основан устаревший метод определения-
иодного числа [77, 78].
Иодное число. Величина принята для характеристики жиров и под-
разумевает число граммов галогена в пересчете на иод, которое в точно соблюда-
емых условиях присоединяется к 100 г жира. В большинстве методов, применя-
емых для определения кодного числа, раствор жнра или масла в СС14 или CHCla.
подвергают в течение '1—2 ч взаимодействию с титрованным раствором хлорида
иода 1С1 в смеси СС14 с ледяной уксусной кислотой [79], IBr в ледяной уксусной
кислоте [80] или NaBr и брома в метиловом спирте [81]; затем добавляют
10%-пый раствор KI и оттитровывают тиосульфатом свободный иод, выделив-
шийся из избыточного количества хлорида или бромида иода. Сопоставление"
стандартных и других методов определения иодного числа см. [72].
К ненасыщенным дикарбоновым кислотам бром присоединяется в горячих водных,
растворах.
а,а'-Дибромянтарная кислота [82]. Ее получают прибавлением по каплям
эквимолярного количества брома к кипящей смеси фумаровой кислоты с дву-
кратным количеством воды.
В обычных условиях, а также на свету шрдяс-коричная кислота в результат»,
транс-присоединения брома превращается в высокоплавкую [г. пл. около 200° CL.
(разл.)] а,р-дибромгидрокоричпую кислоту. К ^uc-коричной кислоте в CS2 бром при-
соединяется в темноте с образованием примерно 70% высокоплавкого и около 30%
низкоплавкого диастереомера [83, 84].
Подробное описание получения кумариндибромида из 1 моль кумарина и 1 молъ-
брома в СИС13 при комдатной температуре см. [85]. Кумаркндибромид превращается*
нод действием щелочей в соль кумариловой кислоты
кумарин
кон; c*H»OHtfip
\COOE
о
калиевая соль
кумариловой кислоты
[75]
[76]
[77]
78]
79
80
81
82
83
84
85
П.Вгосктапп, Н. М u s s о, СЬеш. Вег., 87, 590 (1954); J. L. Woodh
V. d и V i g n e a 11 d, J. Biol. Chem., 134 , 413 (1940).
G. Y. Sliinowara, J. В. В r 0 w n, J. Am, Chem. Soc., 60, 2734 (1938)..
B. v. II и b 1, Dinglers polytechn. J., 253, 281 (1884); J. Ephraim, Z-
angew. Chem., 8, 254 (1895).
J. J. A. W i i s, Z. angew. Chem., 11, 291 (1898).
J. J. A. Wijs, Ber., 31, 750 (1898).
J. H a n и s, Z. Unters. Nahrungs- u. Genupmittel, 4, 913 (1901).
H. P. Kaufmann, Z. Unters. Lebensmittel, 51, 3 (1926).
H. S. R hinesmith, Org. Synth., Coll. v. 2, 1943, p. 177.
C. Liebermann, Ber., 27, 2039 (1894).
E. Grovens^ein jr., D. E. Lee, J. Am. Chem. Soc., 75, 2640 (1953)»
R. C. F и s 0 n, I. W. Kneisley, E. W. Kaiser, Org. Synth., Coll-
у. 3., 1955, p. 209.
I. ПРИСОЕДИНЕНИЕ Hal И SCN ПО КРАТНЫМ С—С-СВЯЗЯМ
103
Акрилонитрил легко присоединяет бром с образованием а,Р-дибромпропиони-
трила, но хлор па него действует совершенно иначе. При пропускании хлора в акрило-
нитрил реакция пе идет. На прямом солнечном свету наряду с кипящей в широком
интервале жидкостью получается бе 1ая полимерная масса. При хлорировании акрило-
нитрила в пиридиновом растворе или в растворе другого гетероциклического основания
происходит гладкое присоединение хлора и получается а,р~дкхлориропиоянтрил
с практически количественным выходом.
а ,Р-Дихлорпропионитрил. В 55 г акрилонитрила к 14 г пиридина при
охлаждении водой пропускают хлор до достижения привеса в 71 г. Экстраги-
руют реакционную массу водой, высушивают над С,аС1а и разгонкой выделяют
а.р-дихлорнропионитрил; т. кип. 61° С (13 л.ч рт. ст.)-, выход 118 г, т. е. 95%
от теоретического. При дальнейшем хлорировании получают а,а,р-трихлор-
пропиопитрил. На солнечном свету наряду с последним образуется а,0,(3-три-
хлорпронионитрил.
а-Хлоракрилопитрил [86|. Его получают отщеплением хлористого водо-
рода ота,р-дихлорпропионптрила, действуя ацетатом натрия в метиловом спирте,
и перегонкой продукта реакции при атмосферном давлении с добавлением не-
большого количества гидрохинона для предотвращения полимеризации; т. кип.
87—89° С (736 мм рт. ст..)-
9. Присоединение галогенов по С=С-связн
Присоединение хлора к ацетилену имеет промышленное значение. При этом
получается смесь ди-и тетрахлорпроизводных. Лабораторного метода присоединения
к ацетилену 1 моль хлора не существует.
Фракционированием технического 1,2-дихлорвтилена получают ниэкоккпящую
транс-форму; т. кип. 47,85—47,87° С (769,5 мм рт. ст.).
При действии 18 г брома на 300 г транс-i ,2-дихлорэтилена в темноте в течение
24 ч получается равновесная смесь траке- и цис-форм, из которой цис-форма (т. кип.
59,6° С при 768 мм рт. ст.) может (эыть выделена перегонкой [87]. Для того чтобы
исключить окисление, перегонку такой смеси ведут [88) в токе СОг.
В работах, представляющих теоретический интерес, но мало применимых на
практике, исследовало фотохимическое присоединение хлора к ацетилену для выясне-
ния оптимальных условий получения дихлорэтилепа [89], а также механизм присоеди-
нения брома к ацетилену с образованием 1,2-дибромэтилена [90, 91] при 150 И
60—120°С.
Присоединением брома к насыщенному раствору ацетилена в абсолютном эти-
ловом спирте получают наряду с другими соединениями смесь цис- и транс-1,2-дибром-
зтиленов [94], 1,2-Дигалогенэтилены (Cl, Вт, I) образуются с очень хорошими выхо-
дами также при пропускании ацетилена при пониженном давлении (300 лек рт. ст.)
и 60—100° С в раствор солей меди (II) в 20%-ной соляной кислоте или 50%-ной бро-
мпгтоводородггой кислоте [98].
Апетилеп, предварительно очищенный содержащей нптрат меди концентриро-
ванной HNO3 и NaOH, пропускают в охлажденный льдом бром. Реакционную массу
[S6] Н. В г i n t z 1 n g е г, К« Pfanngtiel, Н. Koddebusch, Angew.
Chem., 60, 311 (1948).
[87] J. L. Jones, R. L. T а у 1 о г, J. Am. Chem. Soc.f 62, 3480 (1940).
[88] R. K. Wood, R. G. Dickinson, J. Am. Chem. Soc., 61, 3259
(1939).
[89] K. Pet e rg, L. Neumann Angew. Chem., 45, 261 (1932).
[90] J. E. Booker, G. K. Rollefson, J. Am. Chem. Soc., 56, 2288 (1934)..
[91 ] II. .1. S < h innacho r, Angew. Chem., 53, 504 (1940).
[92] P. VV. Robertson, W. E Dasent, R. M. Milburn, W. H. О I i-
v о r, J. Chem. Soc., 1950, 1628.
[93] В. П о in о I k a, F. S t, о I z, Ber., 18, 2284 (1885).
[941 A. Sahanojeff, Ann., 178, 113 (1875).
104
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
промывают раствором NaHSO3 и водой и сушат над СаС1г. Получается 1,1,2,2-тетра-
бромэтан [95—9?]. Выход 95% от теоретического, т, кин. 124 — 126° С (15 мм рт. ст.).
Присоединение пода по С=С-связя.м происходит часто легче, чем по С—С-связям;
например, пропуская при комнатной температуре ацетилен в раствор пода п абсолют-
ном этиловом спирте, легко получают [99, 100] транс-i.2-дшюдэтплсп; т. пл. 73° С.
Это вещество перегруппировывается [101] прп нагревании в запаянной толстостенной
трубке в жидкую i^uc-форму (т. кип. 185° С), а после 21 суток взаимодействия с мелко*
раздробленным натрием в абсолютном эфире превращается в 1,1-дпподэтплрн [100].
1,2- Дпподэтилен можно получить, пропуская ацетилен в 0,2 п. раствор
пода в водном K.I. Реакционную смесь оставляют стоять в течение двух дней
под небольшим избыточным давлением ацетилена; прп этом в реакцию вступает
96% иода и получается кристаллический транс-i ,2-дшгодэтплен [102J; выход
91% от теоретического.
Хотя ацетилены реагируют с хлором и бромом в общем медленнее, чем олефины,
при наличии электроноакцепторных групп, например карбоксильной группы, рядом
с С^С-связьго различие между скоростями реакций становится мспьшим. При наличии'
двух таких групп присоединение галогенов по С^С-связям происходит даже быстрее,
чем по двойным С=С-свяэям: к дпэтпловому эфиру ацетилепднкарбоновой кислоты
хлор присоединяется в уксусной кислоте при 25° С в 60 раз быстрее, чем к диэтиловому
эфиру фумаровой кислоты. Так, к а,р-непасыщенпым карбоновым кислотам' иод
в основном не присоединяется; ацетпленкарбоновые кислоты обладают большой реак-
ционной способностью. К пропаргиловой кислоте иод присоединяется в кипящем эфир-
ном растворе [93]. При медленном просасывании через раствор ацетилепднкарбоновой
кислоты в двойном количестве воды при охлаждении льдом в темноте насыщенного
парами брома воздуха получается в качестве основного продукта дпбромфумаровая-
кислота наряду с небольшим количеством диброммалеццовой кислоты [103, 104].
Дибром(дихлор)малеиповая кислота[106] образуется при окислении HNO3 (<1—1,52)
бром(хлор)мукоповой кислоты ПООС—СХ=СХ—СНО, которая может быть получена
из фурфурола п брома (хлора) [105].
К фенилацетилену в хлороформе при сильном охлаждении присоединяется
1 моль брома с образованием 78% а,р-дибромстирола [107]. Если реакцию проводят
при охлаждении льдом в избыточном количество брома (бром добавляют до появления
неисчсзающсго бурого окрашивания), то получают 1,1,2,2-тетрабромфенилэтан [108].
Выход 92% от теоретического; т. пл. 74—74,5° С. При взаимодействии серебряной сопи
фенилацетилена с иодом в растворе КД легко образуется трпиодстирол [109].
10. Присоединение галогенов к ароматическим соединениям
При взаимодействии галогенов с ароматическими соединениями реакция может-
в осповпом протекать в трех направлениях: присоединение, замещение в ядре и заме-
щение в боковой цепи. Последнее направление будет рассмотрено на стр. 141, 145 ел-
Замещение бенвола хлором (бромом) является ионной (электрофильной) реакцией [110],
A. Sabanejeff, Ann,, 216, 253 (1883).
Н. А п s с h ii t 7, Ann., 221, 133 (1883).
К. Е I b s, J. Newman, J. prakt. Cfioin. [2], 58, 245 (1898).
Англ. пат. 736375 (1953); С., 1956, 12684.
II. В i 1 t z, Ber.', 30, 1207 (1897).
H. P. К a u f m a n n, Ber., 55, 2o4 (1922).
H. P. Kanlmann, Ber., 55, 266 (1922).
A. C. Pappas, Acta Chem. Scand., 2, 292 (1948).
E. 0 t t, Ann., 392, 267 (1912).
R. E. L n t z, J. Am. Chem. Soc., 52, 3416 (1930).
H. Simonis, Ber., 32, 2084 (1899).
A. Salmony, H. Simonis, Ber. 38, 2583 2588 (1905).
J. V. Nef, Ann., 308, 273 (1899).
M. S. N e w man, H. E. Connor, J, Am. Chem. Soc., 72, 4003 (1950).
C. Liebermann, II. Sachse, Ber., 24, 4116 (1891).
С. C. Price, ,Chem. Rev., 29, 37 (1941).
1. ПРИСОЕДИНЕНИЕ Hal И SCN ПО КРАТНЫМ С—С-СВЯЗЯМ
105
в то время как фотохимическое или инициируемое перекисями присоединение галогенов
(хлора, брома) протекает ио радикалыю-цеппому механизму [111]. Замещение в боко-
вой цепи происходит также по радикальному механизму [112], причем высокая концен-
трация хлора способствует присоединению, а не замещению в боковой цепи [111].
Присоединение брома к чистому нафталину в СС14 при комнатной темпе-
ратуре значительно облегчается действием света и добавлением перекисей [ИЗ].
Такое же влияние оказывают зги два фактора на присоединение брома к чистому
фенантрену [114].
Присоединение фтора к ароматическим соединениям обычно протекает одно-
временно с реакцией замещения. При обработке фтором гоксахлорбснзола в СС1<
при 0° С были выделены два кристаллических продукта присоединения — тетрафтор-
гексахлорциклогексан и гексафторгексахлорциклогексап [115].
В результате присоединения 3 моль хлора к бензолу образуется 1,2,3,4,5,6-
гексахлорциклогексан. Выделены все восемь изомеров, возможных для более стабиль-
ной конформации циклогексанового кольца формы кресла [116, 117]. Один из них,
у-гиксахлорциклогексан, применяемый в качестве ядохимиката, является крупно-
тоннажным промышленным продуктом.
Сильноэкзотермичное присоединение хлора к бензолу, не подвергнутому специаль-
ной очистке, не требует подвода тепла и проводится чаще всего при УФ-облучении,-
в этом случае реакцию можно выполнять в присутствии водного раствора щелочи.
Катализаторы, такие, как железо, хлориды железа, сурьмы пли иод, благоприятствуют
электрофильному замещению, которое иод действием имеющихся в техническом бензоле
незначительных примесей протекает в качестве побочной реакции также при проведении
реакции присоединения.
Проведены исследования оптимальных условий, способствующих повы-
шению выхода у-изомера гексахлорциклогексана [118]. Установлено влияние
добавок веществ, участвующих вместе с хлором в цепной реакции, например
влияние хлористого метила, хлороформа, этилона и сероуглерода, а также
инициаторов радикальных реакций на слабом рассеянном свету При 20° С, таких,
как надбензойпая кислота, гидроперекись оксициклогексана и тетраизоамил-
олово. Эти инициаторы способствуют присоединению хлора даже в темноте,
незначительно повышая выход у-пзомера. Оба рода добавок увеличивают ско-
рость реакции. Добавка неорганических веществ не повышает выхода у-изо-
мера. Некоторые вещества, например иод или NalOs, тормозят присоединение
хлора, вследствие чего из хлорированной смеси удается выделить тетрахлор-
цицлогексен [119]. В темноте, при 20° С, фиксированном времени реакции и ско-
рости подачи хлора из многочисленных испытанных перекисей особенно сильно
ускоряют присоединение хлора к бензолу перекиси п,ге’-диметилбензоила и ди-
фенилацетпла и гидроперекись оксициклогексана. В полученном таким способом
[111] М. S. Kharasch, М. G. Bsrk man, J. Org. Chem., 6, 810 (1941).
[112] M. S. К h a r a s c h, P. C. White, F. B. Mayo, J. Org. Chem., 3,
33 (1938).
[113] F. R. Mayo, W. B. Hard y, J. Am. Chem. Soc., 74, 911 (1952).
[114] »M. S. Kharasch, P. C. White, F. R. Mayo, J. Org. Chem., 2,
574 (1938).
[115] L. A. Bigelow, J.H. Pearson, J. Am. Chem. Soc., 56, 2773 (1934).
1116] T. van der Linden, Ber., 45, 236 (1912).
[117] К. С. К a u e г, К. P. Du Vail, F. N. A 1 g и i s t, Ind. Eng. Chem.,
Ind. Ed., 39, 1335 (1947); C., 1948, I, 549; Ю. H. Безобразов, A. В.
Молчанов, К. А. Г a p, Гексахлоран, его свойства, получение и при-
менение, Госхимиздат, 1958.
[118] W. Langenbeck, G. Losse, Н. F u г s t, Chem. fTechn., 4, 247
(1952)
[119] II. Furst, J Lang, P.-P. Rammelt, Chem. Techn., 5, 359
(1953).
106
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
гексахлорциклогексане содержится несколько больше у-пэомсра, чем в продукте
присоединения хлора на свету [120J. В основном современные промышленные
способы получения гексахлорана, содержащего 12—17% у-изомера, основаны
на лабораторном методе Метыова [121].
Экспериментальные данные по разделению изомеров фракционной пере-
топкой и кристаллизацией см., в частности, [117, 122]. Обзор методов исполь-
зования изомеров, не обладающих инсектицидной активностью, но образующихся
в преобладающем количестве, см. [123].
При воздействии жидкого хлора на бензол в запаянной трубке на солнечном свету
[124| среди продуктов реакции найдены как различные продукты замещения, так и
продукты присоединения вплоть до полностью хлорированного додекахлорциклогек-
сана Cftdi g.
При обработке толуола избытком хлора выделены бензальхлорид, бензотрихло-
рид, хлорметилгексахлорциклогексан и дихлорметилгексахлорциклогексан [125].
В присутствии переносчиков галогенов последовательно или одновременно образуются
n-хлортолуол, о-хлортолуол, 2,3- и 2,4-дихлортолуолы, 2,3,4- и 2,4,5-трихлортолуолы
и, наконец, тетрахлор- и нентахлортолуол. .j
Галогенирование нафталина и бензольных углеводородов протекает различно. *
В то время как присоединение галогенов к бензольным углеводородам всегда приводит '<
к производным циклогексана, из нафталина без переносчиков галогенов легко полу-
чаются одновременно нафталиндихлорид Ci0H8CI2 и 1.2,3,4-нафталпнтетрахлорид 3
[126], а также моно-и дихлорнафталинтетрахлориды [127]. С переносчиками и при
нагревании образуются [128] а-хлорпафталин и перхлорнафталин С10С18.
Хлорирование антрацена протекает подобно хлорированию нафталина.
По Перкину [129], можно получить малоустойчивый 9,10-дихлорид, который под дей- .i
ствием избытка хлора переходит сначала в 9,10-дихлорантрацен [130] и затем в 9,10-
дихлорантрацен-1,2,3,4-тетрахлорид [131]. Перхлорирование провести не удается; .4
описан только октахлораптрацен, который, однако, пе был идентифицирован; при ,j
более глубоком хлорировании получаются гексахлорбензол и четыреххлористый угле-
Аналогично ведет себя фенантрен, дихлорид которого неизвестен, хотя дихлор- 1
фенантрентетрахлорид получен. При дальнейшем воздействии хлора образуется я
октахлорфенантрен; далее идет распад с образованием гексахлорбензола. 4
Так же как с хлором, чистый бензол не реагирует и с бромом в отсутствие ката- ,
лизаторов в темноте, фотохимическое присоединение брома и хлора является цепной
радикальной реакцией [133, 134). В реакциях замещения бром активнее хлора, но
скорость его присоединения к бензолу меньше [135]. Свет и добавка перекисей благо-
[120] W. Langenbeck, G. Losse, Н. Furst, Chem. Techn., 5 561 t
(1953).
[121] F.E.Matthewa, J. Chem. Soc., 1891, 166; 1892, 104; 1898, 243. ;
[122] K. Schwabe, H. Schmidt В. К iihnem ann, Angew. Chem.,
61, 482 (1949).
[123] H. Furst, H.-J. Dietz, Chem. Techn., 9, 657 (1957).
[124] T. van der Linden, Rec. trav. chim., 53, 45 (1934); 55, 282, 315 , 421,.
569 (1936); 57, 217 (1938).
[125] T. van aer Linden, Rec. trav. chim., 57,'1075 (1938). • e
[126] E. Fischer, Ber., 11, 735,1411 (1878). 4
[127] A. Faust, E. S a a m e, Ann., 160, 67 (1871); F. Schwarzer, Вег., з
10, 379 (1877). 9
[128] M. Berthelot, E. Jnngfleisch, Ann. Chim. [4], 15, 331 (1868); "
G. Ruoff, Ber., 9, 1486 (1876). J
[129] W. H. Perkin, Bull. Soc. chim. France [2], 27, 465 (1877). 1
[130] C. Graebe, C. Liebermann, Ann., Suppl., 7, 282 (1870). j
[131] F. Schwarzer, Ber., 10, 377 (1877); Hammerschlag, Ber., 19, |
1108 (1886). «
[132] G. Ruoff, Ber., 9, 1483 (1876). S
[133] E. Rabinowitsch, Z. physik. Chem., Abt. B., 19, 19o (1932). j
[134] D. L. Ha m mick, J. M. Hutson, G. T. Jenkins, J. Chenj, Soc., «
1938, 1959. 1
i
J
II. ПРИСОЕДИНЕНИЕ Н—На! ПО КРАТНЫМ С—С-СВЯЗЯМ
107
нриятствуют присоединению брома при низких температурах. При очень медленном
добавлении брома к бензолу, в присутствии 1%-ного раствора NaOH (раствор часто
меняют), при 0° С и на свету получены с малыми выходами два изомерных гексабром-
диклогексана 1136, 137].
Из конденсированных ароматических систем легче всего получаются такие про-
дукты присоединения брома, у которых пе все С=С-свяэи насыщены.
Присоединением брома к чистому нафталину в безводном СС14 при 0е С на свету
получают [138] 1,2,3,4-тетрабром-1,2,3,4-тетрагидронафталин (выход 30%), а в этом же
растворителе при комнатной температуре на свету и добавке перекиси (аскаридол)
[139] получается 12% этого вещества. К антрацену в сероуглероде при 0° С бром при-
соединяется с образованием 9,10-дибром-9,10-дигндроантрацена [129, 140]; из фенан-
трена в эфире [141] или сероуглероде [142] получается 9,10-дибром-9,10-дигидрофенан-
грен. Оба вещества при нагревании отщепляют бромистый водород и переходят
в 9-бромзамещенпыс.
II. Присоединение галогеноводородов по С- С- и С=С-связям
1. Получение и свойства галогеноводородов
Фтористий водород HF (т. пл. —83° С; т. кип. 19,4° С) получают из CaFa и кон-
центрированной H2SO4 в трубчатой вращающейся печи. Его можпо, например, абсор-
бировать водой и получить 70%-кую фтористоводородную (плавиковую) кислоту,
из которой при перегонке отгоняется практически сухой фтористый водород. Летучие
примеси (SiFt, SO2 и COS) удаляют повторной перегоикой [Зе]. Продажную 35%-ную
фтористоводородную кислоту можно очистить от ионов SOJ-, SiFj" и CI" перегонкой
над NaF с добавкой небольшого количества PbCOg-
Аппараты для работы с HF изготовляют из свинца, меди, серебра и пла-
тины; для плавиковой кислоты с концентрацией выше 58% пригодна также сталь.
Хранят плавиковую кислоту в сосудах из покрытого парафином стекла, поли-
этилена, целлулоида, политрифторхлорэтплена, политетрафторэтилена (тефлона).
Стекло быстро растворяется в водных растворах фтористого водорода.
Для смазки металлических шлифов и кранов этих аппаратов пригодны
фторированные углеводороды вязкой коясистенцпи. С наружной стороны шлифы
можно уплотнять пицсииом. При соединении металлической аппаратуры на
фланцах можпо использовать уплотняющие прокладки из свинца, меди или
мягкого железа. В качестве замазки пригодна смесь свинцового глета с глице-
рином.
Фтористый водород образует с водой азеотропную смесь (т. ккп. 112° С при
750 мм рт. ст.), содержащую 38,26% HF; температуры кипения водных растворов
фтористого водорода см. [143]. Из 48%-ного водного раствора фтористого водорода
двукратной, а из 70%-ного однократной перегонной можно получить практически
безводный фтористый водород. Для лабораторных целей перегонку лучше вести в сере-
бряной или платиновой аппаратуре.
Выпускаемый промышленностью фтористый водород содержит 96—99,95% HF
и поставляется в стальных баллонах; баллоны, содержащие 10—20 кг HF, наиболее
удобны для лабораторных работ.
[135] Т. van der Linden, Rec. trav. chim., 55, 291 (1936).
[136] F. E. Matthews, J. Chem. Soc., 1898, 243. ‘
[137] S. B. Hendricks, С. В i 1 i c k e, J. Am. Chem. Soc., 48, 3007 (1926).
[138] J. R. Sampey, J. M. Сох, А. В. К i n g, J. Am. Chem. Soc., 71, 3697
(1949).
1139] F. R. Mayo, W. B. Hard y, J. Am. Chem. Soc., 74, 916 (1952).
[140] E. de Barry Barnett, I. W. Cook, J. Chem. Soc., 1924, 1085.
141] R. Fittig, E. Ostermayer, Ann., 166, 363 (1873).
[142] M. Hay duck, Ann., 167, 180 (1873); H. Henstock, J. Chem. Soc.,
1921, 57.
£143] Ullmanns Enzyklopadie der technischen Chemie, Bd. 7, 3 Aufl., Munchen —
Berlin, 1956, S. 585.
(0g ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Фтористый водород выпускают из вентиля в виде жидкости или в виде
газа, для чего баллон подогревают теплой водой. При этом целесообразно про-
пускать фтористый водород через холодильник и собирать его в охлажденном
приемнике. Фтористым водород очень гигроскопичен. Однако безводный фто-
ристый водород можно переливать на воздухе без заметного поглощения воды
вследствие высокого давления пара испаряющегося IIF, препятствующего'
доступу влаги. Содержание малых количеств воды во фтористом водороде опре-
деляют измерением электропроводности [144|.
При работе с HF необходимо пользоваться защитными очками, резино-
выми перчатками и резиновым фартуком. Пораженные фтористым водородом
или фторидами места кожи как можно скорее обмывают 10%-ным раствором
(ТШ4)3СОз. Ожоги, вызванные фтористым водородом, рекомендуется также1
покрывать кашицей из магнезии с небольшим количеством глицерина.
В лаборатории наиболее чистый безводный HF приготовляют [4f, 145] нагрева-
нием до 500—600° С в медной аппаратуре бнфторида калия KIIF2, предварительно
высушенного прп 220° С.
Хлористый водород IIC1 (т. ил. — 114° С; т. кип. —85,0° С) получают для пре-
паративных целей из концентрированного водного раствора НС1 (dis -= 1,16, 31,5%
НС1; d16 = 1,19, 37,2% НС1). Он хорошо растворим в воде, этиловом спирте и эфире;
1 объем воды прп компатпой температуре растворяет около 450 объемов НС1. С водой
образует азеотропную смесь, содержащую 20,24% НС1 (т. кип. 110е С).
Наиболее удобно получать газообразный IIC1 прибавлением концентри-
рованной TIC1 (d — 1,19) в сосуд, наполненный концентрированной H2SO4,
через достигающую почти до дна сосуда капиллярную трубку, предварительно
заполненную концентрированной IIC1. Для уравновешивания давления газо-
отводную трубку соединяют с капельной воронкой. Применяют равные объемы
концентрированных НС1 и H2SO4. При этом из 200 мл копцоптрпровапной 11С1
получается 67,4 г газообразного НС1.
Для непрерывного получения больших количеств хлористого водорода
в трубку диаметром 5 см с отводом для хлористого водорода в верхней со части
и выпускным краном внизу, которая на высоту 25 см наполнена кварцем или
стеклянными бусами, одновременно приливают по каплям концентрированные-
соляную и серную кислоты [4g]. Выделяющийся хлористый водород пропускают
сначала через промывную склянку с копцептрироваппой H2SO4, затем через
склянку со стеклянной ватой. Для сушки применяют СаС12 и Н2ЗО4; пятпвкпсь
фосфора непригодна, так как она реагирует с хлористым водородом.
Хлористый водород пз баллонов не содержит влаги и заметных количеств
примесей.
Бромистый водород НВг (т. пл. —86,9° С; т. кип. —66,8е С) используется для
препаративных целей в виде газа или в виде водного раствора. Существует нсскольк
хороших методов получения безводного НВг.
а) II о л у ч е ц и е из водорода и брома |4Ь]. Аппарат со-
стоит пэ круглодонной колбы 4, капельной воронки, доходящей почти до дна
трубки Л для введения водорода и газоотводной трубки В. Колба, в которой
находится бром, опущена в водяную баню с температурой 25—35° С. Для пред-
охранения колбы от прямого света лучше всего покрасить ее в черный цвет,
оставив смотровое окошко. Трубка Л во время работы находится под поверх- -
ностью брома. Отбираемый из баллона водород перед поступлением в реакцион-
ную колбу проходит через промывную склянку, содержащую концентрирован-
ную II2SO4, и пустую предохранительную склянку. Для предварительной
очистки водород можно пропускать через трубку с нагретым платинированным.
[144] К. Fredenhagen, G. Cadenbach, Z. physfk. Chem., 146, 25i
(1930).
[145]- Дж. X. Симонс, Неорганические синтезы, Сборник 1, Издатинлпт,
’1951.
IT. ПРИСОЕДИНЕНИЕ II—Hal ПО КРАТНЫМ С—С-СВЯЗЯМ
109
асбестом и далее [146] через СаС12 п PaOs. Чтобы можно было уравнять
давление в системе, трубка В соединена трубкой В с капельной воронкой.
Трубка В соединена с обогреваемой горизонтальной трубной Г из туго-
плавкого стекла длиной 40—60 см, в которой находится платинированный
асбест или платинированный силикагель. От трубки Г отходит вертикальная
трубка Д. В ней находится красный фосфор, распределенный на стеклянных
бусах или кольцах Рапшга.
К трубке Г можпо также присоединить ловушку, охлаждаемую смесью
твердой углекислоты н эфира. Трубка Д соединена с промывной склянкой,
заполненной водой (для удаления фосфорных соединений), за которой следует
осушительная трубка с СаС12 * или СаВг2.
Перед началом реакции из установки водородом вытесняют воздух и на-
гревают трубку V до 330—380° С. Однако последний ее участок должен оста-
ваться холодным, во избежание термической диссоциации НВт. В колбу зали-
вают 50 мл брома п барботируют через него сильный ток водорода, чтобы обеспе-
чить избыток водорода. По мере расходования брома ого добавляют по каплям.
При слишком бурном течении реакции имеется опасность взрыва! О получении-
Н*Вг см. также |7Ь, 147, 148].
Для очистки бромистый водород концентрируют в ловушке, охлажда-
емой смесью твердой углекислоты и ацетона, и барботируют неочищенный про-
дукт через конденсат. Примесь брома можпо удалять с помощью красного фос-
фора, тонко распределенного на стеклянной вате или в копцентрироваппой
бромпстоводородяой кислоте; Р2О5 и концентрированная H2SO4 малопригодны
в качестве осушителей. Если предъявляются высокие требования к чистою
продукта, то для осушки лучше пользоваться [149] сухим СаВг2.
б) Получение из брома и тетралина.
С1о1112-|-4Вг2 1 * СщНвВга4НВг
Недостатком этого метода является использование только половины
вводимого брома. К высушенному над Na2SO4 и перегнанному тетралину (т. кип.
207° С), к которому добавлено немного железных опилок, по каплям приливают
бром. Реакционную колбу помещают в водяную баню, которую при замедлении,
реакции подогревают примерно до 40е С. Для удаления увлеченного брома
бромистый водород пропускают через обратный холодильник и промывную
склянку с перегнанным тетралипом (см. также [150]).
в) Получение из брома и красного фосфора. Не-
большие количества газообразного НВг можно также получить действием брома
на красный фосфор, суспендированный в насыщенном водном растворе бро-
мистого водорода. Описание лабораторной установки см. [151].
Растворимость НВг в воде при нормальном давлении следующая (в г/100 г-
воды); при О'* С — 22; прп 25е С — 193; при 100° С — 130. В насыщенном при 11° G
растворе НВг в ледяной уксусной кислоте содержится около 41% НВг.
Бромистый водород образует с водой азеотропную смесь, кипящую при постоян-
ной температуре (122,5° С при 740 мм рог. ст.; 126° С при 760 хх рт. ст.) и содер-
жащую 47,8% НВг. Эту величину можно повысить примерно до 65% НВг введением
бромистого водорода в воду при охлаждении смесью льда с солью.
* Рекомендуемый для осушки СаС12 лучите не применять, так как при этом
‘ получаемый бромистый водород может содержать примесь НС1. — Ярим. ред.
[146] С. Р. Smyth, С. S. Hitchcock, J. Am. Chem. Soc,, 55, 1831
(1933).
[147] H. Rh einbold l, Chemische Unterrichtsversucbo, 4 Aufl., Dresden —
Leipzig, Verlag Steinkopff, 1953, S. 107.
1148] J. R. Ruhoff, R. E. Burnett, E. E. Reid, Org. Synth., Coll,
v. 2, 1943, p. 338.
1149] G. P. Baxter, R. D. Warren, J. Am. Chem. Soc., 33,340 (1911).
[150] J. Houben, J. Boedlor, W. Fischer Ber., 69, 1772 (1936).
[151] E, Fahr, в книге Houben-Weyl, Melhoden der organischen Chemie,.
Bd. 5, T. 4, Stuttgart, 1960, S. 18.
no
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Водные растворы бромистого водорода.
а) В охлаждаемый водой раствор 120 г КВг в 200 лм воды медленно при-
ливают 90 лм концентрированной II2SO4, не допуская подъема температуры
выше 75° С. После охлаждения до комнатной температуры смесь отфильтровы-
вают от KHSO4 на стеклянном фильтре и фильтрат перегоняют.
б) В смесь 1200 г (377 мл) брома, 500 мл воды и 1500 г измельченного
льда пропускают сильный ток SO8 через барботер, погруженный в находящийся
внизу бром [152]. Ток SO2 следует так отрегулировать, чтобы не было про-
скоков. Вначале массу часто взбалтывают, для того чтобы инициировать реакцию
и предотвратить накопление больших количеств SO8, так как в противном случае
обычно образующийся в этих условиях сульфурилбромид SOaBr2 может бурно
реагировать с водой. Приблизительно через 2 ч смесь быстро желтеет, и реакцию
тотчас прекращают. Полученный продукт, содержащий 2 моль НВг на 1 моль
H8SO4, перегоняют до т. кип. 130° С и дистиллят ректифицируют. Выход 48%-ной
бромистоводородной кислоты 2400 г; т. кип. 125—126° С.
Йодистый водород HI (г. пл. — 50,7° С; т. кип. —35,4° С). В современных син-
тезах Ш применяется для присоединения по С=С-связям для обмена ОН-группы
на иод и в большинстве случаев выделяется непосредственно в реакционной смеси.
Безводный HI получают аналогично НВг из водорода и иода при 500° С над платини-
рованным асбестом [45]. В 1л воды при 10° С растворяется 12 моль HI.
Безводный иодистый водород. Применяя несложную аппаратуру, без-
водный HI можно получить следующим образом [153]. В круглодоииую колбу
емкостью 500 мл, соединенную через обратный холодильник с тремя охлажда-
емыми ловушками (первая до 0° С; вторая до —40° С; третья до —80° С), вливают
раствор 0,35 моль Р8(Х в 226 г (2 моль) 85%-ной НзРО4 и добавляют 1 моль Nal.
При нагревании колбы во второй и третьей ловушках собирается примерно
50% от теоретического количества йодистого водорода. По окончании выделе-
ния HI перекрывают соединение между первой и второй ловушками и пере-
гоняют продукт из второй ловушки в третью, доводя температуру во второй
ловушке до комнатной. При этом надо следить за тем, чтобы трубки не закупо-
ривались твердым HI. Из третьей ловушки HI можно испарять непосредственно
в реакционные растворы.
Водные растворы йодистого водорода. Их получают, пропуская H2S
в энергично перемешиваемую суспензию 120 г иода в 150 мл воды до-почти пол-
ного обесцвечивания иода. Отфильтрованный от выделившейся серы раствор
доводят до кипении для удаления H2S и затем перегоняют. Иодистоводородная
кислота, кипящая при постоянной температуре (126,5° С при 760 мм рт. ст.),
содержит 56,7% HI (d = 1,70) *. Водные растворы HI, окрашенные в коричне-
вый цвет выделяющимся иодом, обесцвечивают [4k] добавлением к горячей
кислоте по каплям в токе инертного газа 50%-ной Н3РО2.
Иодистоводородную кислоту следует хранить в коричневых запарафи-
ненных склянках, так как под действием кислорода воздуха и света она раз-
лагается. Для стабилизации добавляют красный фосфор в количестве 1 г на 1 л
кислоты.
В качестве растворителей в процессе присоединения HI при комнатной темпе-
ратуре употребляют СН8С18, СНС18 и GCl^; для присоединения НС1 и НВг применяют,
кроме того, бензол, толуол, ксилол, нитробензол, петролейный эфир, гексан, гептан
(бензин), нитрометан, ледяную уксусную иислоту, эфир и диоксан. Два последних
растворителя замедляют реакцию ионного присоединения.
* При перегонке иодистоводородной кислоты над красным фосфором образуется
фосфин, который с воздухом дает взрывчатую смесь. Во избежание взрывов
перегонку рекомендуется проводить в токе инертного газа. — Прим. ред.
[152] О. Kamm, С. S. Marvel, J. Am. Chem. Soc., 42, 304 (1920); Org.
Synth., Coll. v. 1, 1941, p. 29; v. 26, 2 Aufl., 1956.
1153] G. G. E c k e, N. С. С о 0 k, F. C. Whltm ore, Г. Am. Chem. Soc., 62,
1512 (1950).
If. ПРИСОЕДИНЕНИЕ H—Hal ПО КРАТНЫМ G-С-СВЯЗЯМ
lit
2. Общее положения о присоединении галогеноводородов по С=С-связям
Наиболее легко по С=С-связям присоединяется HI, труднее — НВг; присоеди-
нение НС1 без катализатора часто происходит значительно хуже. «Нормальное» при-
соединение галогеноводорода по С=С-связи начинается с присоединения протона Н*
к более отрицательному из двух ненасыщенных атомов углерода. Затем к образовав-
шемуся иону карбопия присоединяется аинон X-. Во многих случаях X" «нормально»
присоединяется к наименее «гидрогенизированному» атому углерода (правило Марков-
никова [154]).
.—СН2=СН—СН2Вг—.
НВг НВг
СНаВг-СН2-СПаВт СНзСНВт-СНйВг
«аномальное» «нормальное»
присоединение присоединение
Позднее будет показано, что «нормальное» присоединение в частных случаях
может происходить и с нарушением правила Марковникова. «Нормальное» электро-
фильное присоединение .ускоряется в присутствии полярных растворителей (например,
воды, уксусной кислоты [155]), сильных кислот, а также поляризующих катализаторов
(галогениды металлов). В темноте и в отсутствие кислорода чистый свежеприготовлен-
ный бромистый аллил за 10 суток количественно присоединяет НВг с образованием
«нормального» 1,2-дибромпропана. «Нормальному» присоединению бромкстого водо-
рода к бромистому аллилу значительно способствует добавка ZnBr2, а к хлоркстому
вппилу — FeBr2 или FeCl3 в ледяной уксусной кислоте.
В присутствии малых количеств кислорода или перекисей, добавляемых или
содержащихся в старых препаратах, за 30 мин из бромистого аллила образуется «ано-
мальный» 1,3-дибромпропан. Описаны многочисленные примеры таких реакций [156].
«Аномальное» присоединение является радикально-цепной реакцией, которая иниции-
руется специальными радикалообразующими веществами или фотохимически в жидкой
и газовой фазах [157]:
X • +Н2С=СНВ —> ХСН2-CHR (1)
ХСН2 — CHR-J-HX —► XCH2-CH2R + X • (2)
Эта реакция имеет препаративное значение только в случае присоединения бро-
мистого водорода. Для стадии 2 анергии активации хлористого и фтористого водородов
слишком высоки; в реакции с иодистым водородом образуются недостаточно активные
свободные радикалы—атомы иода [156, 158, 159]. Йодистый водород может восста-
навливать добавляемые перекиси, а образующийся при этом иод является катализа-
тором «нормального» присоединения.
«Аномальному» присоединению бромистого водорода к галогензамешеиным оле-
фннам, таким как бромистый аллил или хлористый винил, способствуют также мелко-
раздробленные металлы, в результате взаимодействия которых с бромистым водородом»
образуются атомы брома.
«Нормальное» присоединение НВг к стиролу, пропилену и изобутилену проте-
кает так быстро, что добавка железа не вызывает «аномального» присоединения. Введе-
ние некоторых антиокислителей, например катехина и тиофенола [160], полностью-
подавляет «аномальное» присоединение и к галогенированным олефинам.
*' [154] W. Markownikoff, Ann., 153, 256 (1870).
[155] М. S. Kharascb, J. A. Hinckley jr., M. M. Gladstone,.
J. Am. Chem. Soc., 56, 1643 (1934).
[156] F. R. Mayo, C. Walling, Chem. Rev., 27, 351 (1940).
[157] W. E. V a u g h a n, F. F. R u s t, Th. W. E vans, J. Org. Chem., 7,.
477 (1942).
[158] E. Muller, Angew. Chem., 64, 244 (1952).
ПР. R. May o, J. Am. Chem. Soc., 76, 5392 (1954).
M. S. Kharasch, W. R. Haefele, F. R. M a у 0, J. Am. Chen^
Soc., 62, 2074 (1940).
112
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
В двух следующих примерах, из которых первый, правда, является спорный,
показано, что «аномальное» присоединение НВг к олефинам катализируется не только
перекисями, азоизобутиронитрилом и подобными веществами. Так, при введении
сухого НВг (в условиях, исключающих доступ влаги) при нормальном давлении в пере-
мешиваемый раствор свободных от перекисей пентепа-1 или гептсна-1 в СС14 пли гек-
сане при температуре от —10 до —12° С или в ледяной уксусной кислоте при темпера-
туре от 0 до 5° С (1,5—3 ч) получаются исключительно 1-бромпситан или 1-бромгептан
[161]. При встряхивании этих олефинов в течение около двух месяцев при комнатной
температуре в темноте с 48% -ной водной бромистоводородной кислотой происходит при-
соединение с образованием исключительно 2-бромпеитана или 2-бромгептана. Про-
пилен, к которому в отсутствие растворителей НВг присоединяется с образованием
2-бромпропана [162], дает в w-пентане, несмотря на отсутствие перекисей и воздуха,
тоже продукт «аномального» присоединения — 1-бромпропан (эффект разбавле-
ния) [163].
Эффект разбавления в сочетании с добавкой перекиси используется для направле-
ния реакций в сторону «аномального» присоединения.
При отсутствии растворителей НВг очень быстро присоединяется к сти- i
ролу с образованием «нормального» а-фенилэтилбромида. В присутствии пере- '
кисп образуется только около 7% продукта «аномального» присоединения. j
Но если в раствор 1 моль стирола в 6 моль «-пентана с добавкой 0,02 моль пере-
киси лаурила в течение нескольких часов вводить НВг, то получается 80% i
р-фенилэтилбромида [164]. :
При фотохимическом «аномальном» присоединении НВг к олефинам также
используют эффект разбавления [165]. '?
Следует указать, что эффект разбавления оспаривался некоторыми исследова- •
телями. Так, в противовес литературным данным [161] пентен-1 присоединяет «апо- ;*
мально» ПВг с образованием 1-бромпентапа только в присутствии перекиси (аскари-1
дол). В уксусной кислоте, даже в присутствии аскаридола, предпочтительно происходит J
-«нормальное» присоединение и получается 2-бромпептап. В пентане также идет «нор- 3
малыше» присоединение НВг, однако в этом случае в качестве антиокислителя был 1
добавлен дифениламин [166]. J
Во всех случаях наличие галогенов в качестве заместителей при С=С-связях
снижает реакционую способность С=С-связей при электрофильном присоединении
и, следовательно, при присоединении галогсповодородов. Это действие ослабляется 1
в последовательности фтор, хлор, бром. Но даже в тех случаях, когда наличие галоген-
ных заместителей само по себе препятствует «нормальному» присоединению, последнее
становится возможным при работе в ледяной уксусной кислоте, при паличнп избытка 3|
HBj (работа под давлением в жидком НВг), при разбавлении и в присутствии некоторых (3
галогенидов металлов (ZnBr2, FeBra, FeCl3). Так, к бромистому винилу НВг присоеди-
няется при 100—200° С над стеклянной ватой с образованием 1,2-дибромэтана, а над а
асбестом, пропитанным бромидом ртути (II), — с образованием 80—90% 1,1-дибром.-
этана [167]. -J
Присоединение галогеноводородов к фторированным олефинам, а также к кар- -."а
бонильиым соединениям и нитрилам (стр. 117), происходящее не по правилу Марков- .я
никова, протекает фактически «нормально». В то время как в соединении я
СН3—СП=СНа средний атом углерода менее отрицателен, чем второй ненасыщенный -я
[161] М. L. Sherrill, К. Е. Mayer, G. F. Walter, J. Am. Chem. 3
Soc., 56, 926 (1934). 1
[162] M, S. Kharasch, M. С. M cNa b, F. R. M a у 0, J. Am. Chem. Soc., «
55, 2531 (1933). Э
[163] F. R. M а у о M. G. Savoy,!. Am. Chemi Soc., 69, 1348 (1947). i
[164] C. Walling, M.S. Kharasch, F. R. Mayo, J. Am. Chem.
Soc., 61, 2693 (1939). ”J
[165] Пат. США 2540126, 2540127; C.T 1952, 2580. ]
[166] M.S. Kharasch, J. A. Hinckley, M. M. Gladstone, J. j
Am. Chem. Soc., 56, 1642 (1934). !
[167] J. P. Wibaut; Z. Elcktrochem., 35, 602 (1929).
II. ПРИСОЕДИНЕНИЕ Н—Hal ПО КРАТНЫМ С—С-СВЯЗЯМ
ИЗ
атом, в соедяненииСР2—CH—CHjoh более отрицателен вследствие влияния группыСГ3.
Так, НС1 или НВг присоединяются к CF3—СН=СН, даже в присутствии А1С13 илж
А1Вг3 с образованием CF3—СН2—СН2С1 (Вг) [168]; под влиянием УФ-облучения вы-
ход повышается [169].
При присоединении галогеноводородов к а,0-ненасыщенпым карбоновым кис-
лотам и их эфирам галоген вступает в р-положение к карбоксильной или к карбалк-
оксильной группе. Так, присоединение HI к акриловой кислоте приводит к р-иодпро-
шгоновой кислоте [170], присоединение НВг или HI К этиловому эфиру кротоновой кис-
лоты — к соответствующим эфирам р-галогенмасляной кислоты [171 ]. В ненасыщенном
соединении с CF3- и карбалкоксильной группами преобладает влияние последней. Даже
в присутствии А1Вг3 или перекиси бензоила НВг присоединяется к этиловому эфиру
у,у,у-трифторкротоновой кислоты с образованием эфира р-бром-у,у,у-трифтормасля-
ной кислоты [172].
3. Присоединение галогеноводородов к ненасыщенным углеводородам
Присоединение HF к олефинам происходит очень легко, однако сопровождается
побочными реакциями полимеризации олефинов. Следы кислот и воды или температура
около 70° С вызывает отщепление HF от образовавшихся фтористых алкилов. Препара-
тивное значение имеет присоединение Н F к хлорзамещенным олефинам, которые тем
менее склонпы к полимеризации в его присутствии, чем больше атомов хлора связано
с ненасыщенными атомами углерода. Фтористый водород легко присоединяется к не-
симметричным галогензамещенпым олефинам, таким, как СН2=СНХ, RCH=CX8
или RCX—СН2, труднее к олефинам, имеющим атомы галогена у обоих ненасыщен-
ных атомов углерода. Реакция ускоряется в присутствии BF3, который образует
комплексное соединение и тем самым облегчает отрыв протона от молекулы HF;
однако одновременно ускоряются параллельно протекающие реакции обмена га-
логена на фтор и,смолообразование в результате полимеризации [173].
Фтористый водород присоединяется «нормально», т. е. водород направляется
к более отрицательному кз двух ненасыщенных атомов углерода. Следовательно,
{за исключением случая, когда рядом со связью С= С находится группа CFS (см; выше)],
фтор, согласно правилу Марковникова,1 обычно присоединяется к наименее гидрогени-
зированному атому углерода. Это относится также к фторированным олефинам. Есди
в последних кроме фтора имеются другие галогенные заместители, то при присоеди-
нении галогеиоводорода как по ионному, так и по радикальному механизму галоген
присоединяется к атому углерода, связанному с наибольшим числом атомов фтора
[174, 175]; например, присоединение бромистого водорода к CFS=CFC1 происходит
с образованием CBrF2—CHC1F.
Олефин добавляют (по возможности без катализатора BFS) к охлажден-
ному до температуры от —40 до —60° С жидкому HF, затем массу постепенно
нагревают и выдерживают 1—2 ч при 75—90° С. Реакционный сосуд может
быть выполнен из меди, монель-металла, нержавеющей стали, платины, магиня
или никеля. Реакцию с низкокипящими фторидами следует вести в автоклаве.
При работе с концентрированным водным раствором HF присоединении не
нроисходит.
1,2-Дихлор-2-фторпропан [31]. В охлажденный сухим льдом стальной
автоклав емкостью 2 л помещают 500 г 1,2-дихлорпропена-2 и 90 г безводного HF.
Автоклав закрывают навинчивающейся крышкой, снабженной вентилем и мано-
метром, и 18 ч нагревают при 50° С на водяной бане. Затем после охлаждения
, сухим льдом открывают вентиль, отвинчивают крышку, выливают реакционную
[168] A. L. Henne, S. К а у е, J. Am. Chem. Soc., 72 , 3369 (1950).
169] A. L. Henn е, М? N a g е г, J. Am. Chem. Soc., 73, 5528 (1951).
<70} J. Wislicenus, Ann., 166, 1 (1873).
171] W. Hemilian, Ann., 174, 324 (1874).
172] H. M. Walborsky, M. Schwarz, J. Am. Chem. Soc., 75, 3241 (1953),
173] A. L. H e n n e, R. C. Arnold, J. Am. Chem. Soc., 70, 758 (1948).
174 ] J. D. P а г k, M. L. S h а г r a h, J. R. Lacher, J. Am. Chem. Soc ,
71, 2339 (1949).
[175] R. N. H a & z e 1 d i n e, B. R. S t e e 1 e, J. Chem. Soc., 1954, 3747.
S Заказ 1835.
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
смесь па лед, нейтрализуют разбавленным раствором NaOH к перегонкой с водя-
ным паром отделяют органический слой. Высушенный над СаС12 неочищенный-
продукт (450 е) разделяют перегонкой па колонке и получают около 200 г 1.2-дн-
хлор-2-фгорпропана (т. ппп. 8.5—90° С) и приблизительно 200 г фракции, кипя-
щей при 90—94° С. Последнюю фракцию еще раз обрабатывают HF, как указано-
выше, и получают дополнительно 75 г 1,2-дихлор-2-фторпропаца.
О реакциях взаимодействия с HF см. также [176—178].
Ниже совместно рассматривается присоединение всех прочих галогеноводородов.
(ИХ). Олефины в газовой фазо очень медленно реагируют с IIC1, однако реакция про-
ходит легко [179J в присутствии безводного А1С13. К высшим нормальным олефинам-
галогеноводороды присоединяются легче, чем к газообразным низшим. Как правило,
галогеноводороды присоединяются труднее к концевым винильным группам, чем по-
двойным связям в середине цепи. В известных условиях это дает возможность разде-
лять изомеры. Например, к гексепу-2 (СН8СН2СН—СНСН3) хлористый водород при-
соединяется под действием дымящей соляной кислоты при комнатной температуре;
гексен-1 (СН3С1Т2СН2СН2СН —СН2) остается при этом неизменным [180]. Из двух изо-
меров RCBr~CHa и RCH=CHBr первый значительно легче присоединяет галогено-
водороды, чем второй, так что таким путем иногда удается разделять изомеры.
Этилен, реагирующий медленнее всех других олефинов с галогеноводородами, бы-
стро присоединяет в жидкой фазе (раствор в дихлорэтане.) НС1 в присутствии -A1C1Sj
даже при —80° С. В газовой фазе без катализатора этилен не реагирует с НС], НВг
в HJ, пропилен не реагирует с НС1 и НВг, а бутилен с НС1. Но в жидкой фазе присоеди-
нение происходит [181]. В газовой фазе в присутствии катализаторов этилени пропилен
уже при умеренных температурах[нрисоединяют ПС! и НВг с образованием хлористого*
(или бромистого) этилена и 2-хлор(бром)пропана [167]. Катализаторами реакции-
являются; BjCJg, BiBrs, пропитанный BiBr3 асбест, FeC]3, FeBr3, ZnCl2 и Д1С13.
Прп наиболее легко осуществимом жидкофазном присоединении галогеповодоро-
дов значительное влияние на реакцию оказывает растворитель. Например, пропилен
плохо присоединяет НВг в ледяной уксусной кислоте при комнатной температуре,,
а в гексапо, напротив, присоединение идет хорошо [182]. н-Бутилеп в ледяной уксус- '
пой кпелоте присоединяет НВг прп 5° С в толстостенной ампуле с образованием-
2-бромбутапа [183]. К пентену-1 НВг и III присоединяются без растворителя или в:
воде, и получается 2-бром(иод)пептап [184].
Наиболее легко НС1 присоединяете я к третичным олефвпам, например к изобу-
тилену и изоамилену. К изобутилену в гептане НС1 присоединяется при 0° С с образо-
ванием исключительно /ярт-бутллхлорида. В присутствии следов воды реакция уско-
ряется [185]. С добавкой А1С13 1-метилциклогексен полимеризуется, в то время как.
под действием сухого НС1 при 5° С в присутствии 5—10% SnCl^ получается 1-хлор-
1-метилциклогексан со 100%-ным выходом [186].
Хлорид алюминия непригоден также для получения циклогексилхлорида из-
пиклогексена и хлористого водорода. Эта реакция идет хорошо [187] в присутствии-
SnCl4 и TiCJ4.
[176] W. В о eke m ii 11 е г, Neuere Methoden der praparativen organischea-
Chemie, Bd. 1, Berlin, Verlag Chemie, 1944, S. 217—236.
[177] A. L. H e n n e, Org. Reactions, II, 67 (1944),
[178] J. H. Simons, Fluorine Chemistry, v. II. New York. Academic- '
Press Inc., 1954:-a) p. 213; b) p. 217; c) p. 340.
[179] J. P. Wibaut, J. J. D i e k ni a n n, A. J. Rutgers, Rec. trav
chim., 47, 477 (1928).
[180] Schorlemmer, Morgan, Ann., 177, 305 (1875).
[181J O. Maass, Can. J. Res., 3, 525 (1930); 5, 48 (1931).
[182] V. N. Ipatieff, H. Pines, R. C. WacklieUJ. Am. Chem Soc
56, 2398 (1934). *
[183] II. J. Lucas, R. T. D i 11 о n, W. G. Young J. Am Chem Soc
52, 1952 (1930).
[184] Ch. К. 1 n g о 1 d, E. Ramsden. J. Chem. Soc. 1931 2746
1185] F. R. Mayo, J. J. Katz, J. Am. Chem. Soc. 69 1339 (1947)
1186] M. Monsseron, G. Manon, C. r., 227, 533 (1948).
[187| M. Mousseron, G, Mano n, Bull. Soc. chim. France, 1949 392
П. ПРИСОЕДИНЕНИЕ Н—Hal ПО КРАТНЫМ С—С-СВЯЗЯМ
115
Для проведения реакции между сухим газообразным олефином и НВг смесь
газов пропускают через широкую нагретую до 100е С трубку, заполненную каталиэа-
торами— А1С1а, А1Вг3, BiCJ3, BiBra, ZnCl2 и HgCl2; применимы катализаторы, оса-
жденные на силикагеле или асбесте. Описание лабораторной аппаратуры см. (188].
В лабораторной практике чаще всего реакцию присоединения НВг проводят при 0—
15° С н ледяной уксусной кислоте или пропуская НВг при температуре около —20° С
в неразбавленный или растворенный в СНС13 олефин. Реакцию с легколетучимп олефи-
нами проводят в толстостенной ампуле в течение нескольких суток. Высококипящие
олефины можно обрабатывать п обычной аппаратуре; реакция идет примерно сутки.
Для того чтобы обеспечить «нормальное» присоединение НВг, необходимо
исключить доступ воздуха и убедиться в отсутствии перекисей в насыщенном соедине-
нии и в растворителе п в случае наличия удалить их соответствующим способом.
Можно также добавлять антиокислители, например дифениламин, тиофенол или
тиокрезол, которые устраняют влияние малого количества перекисей.
Такие растворители, как петролейный эфир, бензин, декалин, ароматические
углеводороды с боковой цепью (ксилол, кумол), тетралин и все эфиры большей частыр
содержат перекисные соединения.
Для удаления гидроперекисей растворители встряхивают с КОН или
КаОН, причем соли гидроперекисей осаждаются (охлаждение, защитные очки/)*.
Для перекисей тримерпых и полимерных альдегидов и кетонов рекомендуется
продолжительная обработка растворителя раствором FeSOt в 50%-ной H8SO4.
О дальнейших методах см. (189].
Положительное влияние полярных растворителей и галогенидов металлов на
•«нормальное» присоединение галогеноводородов к олефинам уже обсуждалось.
Для проведения «аномального» присоединения НВг добавляют перекиси или
применяют УФ-облучепис [190].
Легколетучио олефины загружают путем перегонки в соответственно
охлаждаемую толстостенную ампулу, в которой находится 0,04—0,15% пере-
киси и и которую таким же способом затем добавляют 1,2—1,5 моль предвари-
тельно ожиженного НВг. Ампулу запаивают и проводят реакцию при 0—20° С
в течение нескольких суток [191]. Трудполетучие олефины можно гпдроброми-
роватт» путем введения НВг в раствор олефина в Кипящем пентане, гексане или
гептане с добавкой перекисей иди путем УФ-облучения в кварцевой колбе.
Примером может служить способ гидробромировапия 1-бром(хлор)циклогек-
сепа-1, к которому при УФ-облучопми и комнатной температуре НВг присо-
единяется «аномально» с образованием цис-1,2-дибромцпклогексана [192], а в при-
сутствии FeCls — с образованием 1,1-дигалогенциклогексапа [193]. К аллил-
бензолу и его различным производным, замещением в ядро, сухой НВг присо-
единяется только в присутствии 3—8% перекиси дпацетила или бепзоила (охлаж-
дение смесью льда с солью/). Получается с хорошим выходом оэ-бромпропилбен-
зол [194].
* При этом возможны сильные взрывы; предпочтительнее обработка FeSO<. -
Прим. ред.
[188] .Т. Р. Wi b a u I, J. J. Diekmann, A. J. R и t g е г s, Rec. trav. chim.
‘ 47, 477 (1928).
[189] Hou hen- Werl, Methodcn der organischen Chemie, Bd. 8, Stuttgart
1952. S. 73.
[190] M. S. Kharasch, M. C. Me N
55, 2524 (1933).
[191] M. C. Kharasch, F. R. If a
2531 (1933).
[192] H. L. Goering P. I. Abel],
74, 3591 (1952).
[193] H, L. G oer i ng, L. L. S i m s J.
4194] R. Durand -Г) ran, Ann, Chim.
8*
b, F. R. Mayo, J. Am. Clhun. Soc.,
o, J. Am. Chem. Soc., 55, 2468,
B. F, А у с о c k, J. Am. Chem. Soc.,
Am. Chem. Soc., 79, 6270 (1957).
[13], 4, 43 (19591
116
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
В реакциях присоединения НС1 и НВг к бициклическим терпенам, особенно
в растворителях с высокой диэлектрической постоянной и при избытке галогеноводо-
рода, происходит перегруппировка первично образующихся галогенидов (перегруп-
пировка Вагнера — Меервейна). В качестве примера можно привести перегруппи-
ровку камфена в иэоборнилгалогенид:
камФен изоборнилхлорид (бромид)
Присоединение галогеноводородов к камфену [195]. Вводят 1 молы
сухого НС1 в раствор камфена в половинном объеме сухого эфира при охлажде-
нии смесью льда с солью и полном исключении влаги. С выходом 90% получают
камфенгидрохлорид.
Камфенгидробромид с выходом около 70% получают введением НВг
(85% от теоретического) в раствор камфена в петролейном эфире в тех же усло-
виях [196]. Полученные продукты загрязнены изоборнилхлоридом(бромидом).
Так как в изоборнилхлориде(бромиде) вторичный атом галогена значительно”
мепее подвижен, чем третичный атом галогена в камфенгидрохлориде(гидро-
бромиде), чистоту препаратов можпо определить титрованием спиртовым.
К ненасыщенным углеводородам с сопряженными С=С-связями (например,
к бутадиену) галогеноводороды могут присоединяться как в 1,4-, так ив 1,2-поло-
жения.*
СН3-СНВг-СН=-СН2 СН2=СН-СН=СПа ВгСН2—СН—СИ,—СН3
I И
Присоединение галогеноводородов и бутадиену [197]. При 30' С про-
пускают бутадиен в 66%-пый водный раствор НВг, отделяют собравшийся
сверху слой неочищенного продукта, промывают до нейтральной реакции, су-
шат над СаС12 и разгоняют. Получается кротилбромид (т. кип. 101—104° С),. -
который представляет собой смесь 80—85% 1-бромбутена-2 (II) и 15—2<i% .
З-бромбутена-1 (I). При комнатной температуре эта смесь участвует в реак-
циях так, как если бы она состояла только из 1-бромбутена-2, что например,.?
особенно важно при синтезе этилкротилбарбитуровой кислоты [198]. Аналогич-
ным образом при введении бутадиена в 57%-ныи водный раствор Ш при 2Od Q
получают кротилиодид; т. кип. 35° С при 12 мм рт. ст.
Присоединение галогеноводородов н циклопентадиену [199]. При тем-ц
вературе ниже 0° С (охлаждение твердой двуокисью углерода) в свежепригото-
вленный циклопеитаднен при перемешивания быстро вводят сухой НС1 (90% от *
теоретического), происходит 1,4-присоединение с образованием 3-хлорцикло- Л
пентена. Так как 3-хлорциклопентен быстро полимеризуется при комнатной
температуре с образованием черной смолы, аппаратуру после опыта следуетг.
тотчас же разобрать и вымыть! у
[195] II. Meerwein, К. van Emster, Ber., 53, 184 (1920); 55, 2500
(1922).
[196] Н. Meerwein, Ann., 453, 36 (1927).
[197] R. Voigt, I, prakt. Chem. [2], 151, 309 (1938).
[198] H. Goldhahn, Pharmazie, 8, 324 (1953).
[199] R. В. M о f f e t,t, Org. Syntn., 32, 1952, p. 41.
И. ПРИСОЕДИНЕНИЕ Н—На! ПО КРАТНЫМ С—С-СВЯЗЯМ
И
Присоединение галогеноводородов к циклогептатриену. В циклогепта
триене преимущественно насыщается только одна из трех сопряжении:
С=С-связей. С 40%-ным раствором НВг получают на холоду наряду с ди
бромидом 6-бромциклогептадиен-1,3 (выход 65%), который сыграл решающую
роль в первом синтезе тропина [200].
Для присоединения III, мало интересного с препаративной точки зрения
пропускают при 0—20° С газообразный олефин в не содержащий иода водный рэство?
HI с постоянной температурой кипения или в насыщенный раствор HI в ледяной уксус
ной кислоте. Можно также пропускать HI в олефин или его бензольный раствор
По другому методу, оказавшемуся пригодным также для иодирования спиртов и эфн
ров (см. стр. 210, 226), добавляют 0,5 моль олефина к смеси 2,14леоль 95%-ной Н8РО
и 1,5 дима KI (смесь готовят при охлаждении из 102 мл 85%-ной НаРО4> 47 а Р8О
л KI) и нагревают реакционную массу несколько часов с обратным холодиль
ником [201].
.cc-^-Ki-iijPOj —> ^>ch-ci+khspo4
Подробную пропись получения иодциклогексана из циклогексена см. [202]
Этот способ применим также для присоединения Hi к ненасыщенным кислотам [203]
4. Присоединение галогеноводородов к ненасыщенным спиртам, эфирам
карбонильным соединениям и нитрилам
Присоединение НС1 (НВг) к ненасыщенным спиртам без полной или частично!
замены ОН-группы на хлор (бром) возможно только в том случае, если ОН-группа от
делена от С=С-связи несколькими атомами углерода. Например, коричный спир*
реагирует при 0° С с газообразным НС1 или раствором НВг в ледяной уксусной кис
лоте с заменой ОН-группы на хлор или бром [204].
Для проведения «аномального» присоединения НВг к высшим ненасыщепныэ
спиртам нужно предварительно перевести спирты в сложные эфиры. К ундецеиолу
например, НВг присоединяется даже в присутствии перекисей с образованием
10-бромундеканола-1 — продукта «нормального» присоединения, а к упдецеиилацетат;
в присутствии перекисей он может присоединяться «аномально», но при добавлена
антиокислителей (водород и дифениламин) происходит «нормальное» присоедине
пие [205].
Из полученного в присутствии перекиси бензоила «аномально» гидробромиро
ванного ацетата бронированный спирт получают переэтерификацией с помощью мети
левого спирта и п-толуолсульфокислоты [206]. 11-Бромундеканол-1,13-бромтрндека
нол-1 и 15-бромпентадеканол-1 можно получать также, предварительно перевод^
соответствующий со-непасыщенпый ейирт в эфир борной кислоты.
Получение ненасыщенного спирта [207]. В раствор борного эфира вводя*
НВг в присутствии ди-трт-бутилперекиси при 0—2° С и по окончание
реакции после отгонки большей части растворителя (толуола) омыляют продук-
реакции добавлением воды и перемешиванием в течение 1 ч при 50° С.
[200] R. Wills tatter, Алл., 317, 87 (1901).
201] Н. S t о n е, Н. S h е с h t е г, J. Org. Chem., 15, 491 (1950).
202] Н. St one, П. S h е с h t е г, Org. Synth., 31, 1951, p. 66.
203] J. С о г s e, E. E. J ansen, J. Am. Chem. Soc., 77, 6632 (1955).
204] А. К 1 a g e s, K. Klenk, Ber., 39, 2552 (1906).
205] R. Ashton, J. C. Smith, J. Chem. Soc., 1934, 1308.
206] F. L. M, Pattison et al., J. Org. Chem., 21, 745 (1956).
207] M. G. J. В e e t s, W. M eer burg, Rec. trav. chim., 72, 414 (1953).
118
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Присоединение НС1 п НВг к фенил- или алкилвиниловым эфирам, приводящее
к образованию а-галогеналкиловых эфиров, следует проводить во избежание полиме-
ризации при низких температурах (от —5 до +5° С).
Имеется подробная методика получения а-хлорэтплбутилового эфира из винил-
бутилового эфира [208].
Присоединение галогеноводородов к другим ненасыщенным простым эфирам
также необходимо проводить при низких температурах, во избежание расщепления
эфира (стр. 225).
Р-Бромдигидроэвгенол. К 7г эвгенола, насыщенного НВг при 0° С, до-
бавляют при перемешивании п охлаждении льдом до 0° С насыщенный водный
раствор НВг и оставляют на 3—4 суток, изредка встряхивая. Получается 7,7 г
р-бромдпгидроэвгенола (7,7 г). Аналогичным образом синтезируют р-хлор-
дигидроэвгенол [209].
При электрофильном (первичная стадия присоединения Н+), а также нуклео-
фильном (первичная стадия присоединения X") присоединении галогеноводородов
к «.^-ненасыщенным альдегидам, кетонам, кислотам и нитрилам галоид вступает
в ^-положение:
R—СП=СН—С—R' R—СН—СН=С—R'
Реакция имеет препаративное значение для получения ^-галогенированных
кислот, эфиров п нитрилов.
В а,р ненасыщенные кетоны и альдегиды, которые можно растворить
в бензоле или CS2, при сильном охлаждении (ацетол 4- твердая углекислота)
осторожно пропускают НВг до начала ого проскока. Из окиси мезитила полу-
чают таким образом 2-бром-2-метилпептанон-4 [210].
При взаимодействии акролеина с НВг при —15° С в присутствии метилового
спирта бром в первую очередь присоединяется к 0-атому углерода. Конечным продук-
том, однако, является а,у-дибромэфир — 1,3-дибром-1-метоксппропан:
СН2=СН—СНО
I НВг Пг
j(cn,oH) уг
ВгСН2-СН2-СН(ОСНЙ)2 ВгСН2-СН2-СН(ОСНз)
| GlFjOH|
Это вещество после 12 ч пребывания п растворе метилового спирта превращается
в диметилацеталь З-бромпроппопопого альдегида [211].
Диэтилацеталь З-хлор-З-фенилпропионового альдегида. Если к насы-
щенному при 0° С раствору HCI в абсолютном этиловом спирте при температуре
от —5 до 0° С прилить по каплям 20 г коричного альдегида и выдержать смесь
при 0° С в течение 30 мин для завершения реакции, то получается 23 г диэтил-
ацеталя .З-хлор-З-фенилнропионового альдегида (1,1-диэтоксп-З-фенил-З-хлор-
пропан) [212].
[208] М. Ф. Ш ост а к ов с к и й, А. В. Богданова, Синтезы органических
соединений, Сборник 1, Изд. АН СССР, 1950.
[209] G. R-Clemo, J. И. Т и г п Ь л 11, J. Chem. Soc., 1945, 533.
[210] Н. Rupe, S. Kessler, Вег., 42, 4715 (1909),
[211] R. P i и e a и, J. Rech. Centre nat. Rech. Sei.. 1951, 292; C., 1954, 748.
[212] J. P. F о и r n e a u, S. Chant alou, Bull. Soc. chim. France, Mem.
[5], 12, 845 (1945)*.
И. ПРИСОЕДИНЕНИЕ Н—Hal ПО КРАТНЫМ С—С-СВЯЗЯМ
HS
В определенных условиях в результате реакции НС1 (НВг) с а, 0-ненасыщенными
кетопами помимо 0-галогенкетопов получаются окрашенные продукты присоедине-
ния [213].
Для присоединения НС1 (НВг) к ненасыщенным кислотам галогеноводород
вводят при охлаждении льдом в жидкую неразбавленную кислоту или в ее раствор
в ледяной уксусной кислоте пли эфире. Из кротоновой кислоты получают таким об-
разом 0-броммасляную кислоту [214].
Реакцию присоединения галогеноводородов к эфирам ненасыщенных карбоно-
вых кислот проводят при низких температурах (от —10 до 0° С) без раствори-
теля или в растворителях — эфире, петролейном эфире или ледяной уксусной
кислоте.
Метиловый эфир p-бромпропионовой кислоты. Получение этого эфира
из метилакрилата в сухом эфире см. [215].
Метиловый эфир 0-бромизомасляной кислоты [216]. Получают при дей-
ствии сухого НВг на метилметакрилат при температуре от —5 до —10° С в от-
сутствие растворителя.
Если прп охлаждении льдом в течение 20 мин в раствор 0-бензоилакриловой рис-
лоты в ледяной уксусной кислоте пропускают сильный ток НС1 (НВг) и затем выдер-
живают 3 ч при комнатной температуре для завершения реакции, то получают с 81%
ным выходом а-хлор-или с 90%-ным выходом а-бром-0-бенэоялпропиоповую кислоту
[217]. В этом случае 0-ориептирующее влияние кетогруппы преобладает над влиянием
карбоксильной группы.
Добавление Инициирующих образование свободных радикалов веществ пр®
реакции а,0-ненасыщенных карбоновых кислот или их эфиров с НВг не влияет н<
вступление брома в 0-положение. Однако возможно «аномальное» присоединение НВ;
к ю-ненасыщенным кислотам и их эфирам, у которых С= С-связи и карбоксильная
группа разделены группами СН2, с образованием со-бромкарбоновых кислот илт
эфиров.
К ундециленовой кислоте или ее этиловому эфиру в бензоле НВг при:
соединяется «нормально», если вводить его с водородом, добавлять в* качества
антиокислителя дифениламин или предварительно удалять загрязнения (альде
гиды], которые могут образовывать с воздухом перекиси. В присутствии пере
кисеи, которые специально вводят или которые самопроизвольно образуются
из примесей альдегидов в результате аутоокислспия кислородом воздуха, про
исходит «аномальное» присоединение НВг и получается 11-бромундеканова®
кислота или ее этиловый эфир [205, 218].
Для получения 11-бромундекановой кислоты в присутствии перекист
бензоила следует пользоваться ПВг, полученным из брома и тетралина. Не рено
мепдуется применять для этой цели свежеприготовленный НВг, синтезирован
пый из Вг2 Н2, содержащий примесь водорода [219].
К ы-ненасыщенным кислотам НгС = СН—(СНа)я—СООН, где п = 1—
присоединение НВг в гексане в присутствии перекисей происходит с образа
ванием а-бромнасыщенных кислот. В чистейшем гексане при добавке дифенил
амина и исключении доступа кислорода происходит «нормальное» присоединен!!,
бромистого водорода [220].
[213] D. Vorlander. Вег., 37, 1644, 3364 (1904); Ann., 341, I (1905).
[214] Н. Hunsdiecker, Вег., 75, 464 (1942).
[215] R. Mozingo, L. Л. Patterson, Org. Synth., Coll. v. 3, 1955
p. 576.
[216] H. L. Lochle, J. Am. Chem. Soc., 69, 15 (1947).
[217] E. Buclilo, S. Daunp r, Chem. Ber., 82, 63 (1949).
[218] R. Ash ton, J. C. Smit li, J. Chem. Soc., 1934, 435.
[219] R. G. Jones, J. Am. Chem. Soc., 69, 2352 (1947).
[220] A. Michael, П. S. M a s о n, J. Am. Chem. Soc., 65, 683 (1943).
120
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
К а,|3-пснасыщенным нитрилам сухой хлористый водород [221] и сухой бромистый
водород [222] присоединяются при комнатной температуре, причем галоген вступает
в ^-положение к нитрильной группе.
При взаимодействии в течение 3 ч акрилонитрила в сухом эфире с НС1
при комнатной температуре получают 3-хлорпропионитрил с выходом 80—85%
от теоретического; без растворителя [221 ] после 12 ч кипячения выход достигает
только 40%.
5. Присоединение галогеноводородов по С=С-связям
Алкины легко присоединяют HF уже без катализаторов, причем присоединение
ироисходит по правилу Марковникова:
R—С=С-R' —R—CF=CH—R' —R-CFa-CH2-R'
Ацетилен, в противоположность высшим алкипам, не реагирует с HF при нор-
мальном давлении,и температурах от —70 до 0° С.
После 72 ч взаимодействия ацетилена с HF при перемешивании в стальном авто-
клаве при комнатной температуре и давлении 8—12 ат около 15% ацетилена превра-
щается в смесь фтористого винила (35 мол. %) и 1,1-дифторэтана (65 мол. %) [223].
Фтористый винил. Его получают в газовой фазе при 83 — 109® С над ката-
лизатором HgCl2 — ВаС13, нанесенным на активный уголь. Из неочищенного
газа HF отделяют промывкой раствором едкого натра, а непрореагировавший
ацетилен отгоняют на низкотемпературной колонке в виде азеотропной смеси
с этаном, который специально для этой пели добавляют. Указания о пригото-
влении катализатора и о реакторе см. [224].
Реакция присоединения HF к гомологам ацетилена с образованием дифторалка-
нов описана как для маленьких [223], так и больших [225] (5—10 моль) загрузок.
В последнем случае следует избегать образования местных перегревов (осмоление!),
а также ведения процесса при слишком низких температурах, что может привести
к опасному накоплению еще не вступивших во взаимодействие компонентов реакции.
Пропин и бутин-1 пропускают в газообразной форме в жидкий, охлажденный льдом,
безводный HF (2,5 моль на 1 лсоль алкина); пентин и гексин приливают по каплям
при —50° С к фтористому водороду (реакции проводят в медной аппаратуре).
Для фторирования высококипящих алкинов употребляют раствор фтористого
водорода (5 моль) в эфире или кетоне (1 моль). Образующиеся из 2 моль фтористого
водорода и 1 моль растворителя оксониевые соединения хорошо растворяют алкины
и продукты реакции.
2,2-Дифтороктан [3g, 225]. В охлажденную льдом медную колбу ем-
костью 500 мл загружают 74 г эфира или 56 г ацетона и вводят через медную
трубку 100 г безводного HF. Затем к раствору возможно медленнее приливают
по каплям 1 моль октина-1 и нагревают до комнатной температуры. После 1 ч
выдержки смесь выливают иа лед’, отделяют органический слои и разгоняют его;
выход 2,2-дифтороктана 70—90%.
Реакция присоединения HCI к ацетилену представляет очень большой интерес
для промышленности, и данные по разработке этого процесса приведены в основном
в патентах. Присоединение НС] к алкииам происходит по правилу Марковникова.
Ацетилен реагирует с HCI в газовой фазе с заметной скоростью только в присутствии
[221] С. С. Р г i с е, J. Zomlefcr, J. Org. Chem., 14, 210 (1949).
[222] С. Moure u, H. L. В г о w n, Bull. Soc. chim. France [4], 27, 904 (1920).
[223] A. V, Grosse, С. B. L i n n, J. Am. Chem. Soc., 64, 2289 (1942).
[224] A. E. Newkirk J. Am. Chem. Soc., 68, 2467 (1946).
[225] A. L. Henne, E. P. P I ue d de m an, J. Am. Chem. Soc., 65, 587 (1943).
II. ПРИСОЕДИНЕНИЕ Н—Hal ПО КРАТНЫМ С—С-СВЯЗЯМ
121
катализаторов (галогенидов металлов). Скорости присоединения первой ц второй
молекулы НС1 зависят от применяемого катализатора.
iicscn -2^ нас^снс1 — > h3c-chci2
Очень эффективным катализатором присоединения HCI к ацетилену является
HgCl8, однако он совсем пе оказы вает влияния на присоединение ПС1 к хлористому
винилу. Эту реакцию ускоряет [226] ZnCl2. В интервале температур от 55 до 200° С
над катализаторами ZnCl2, HgC]2, BiCls и FeCI3 образуется только 1,1-дихлорэтан —
продукт нормального присоединения.
Хлористый винил. При пропускании ацетилена и НС1 над HgCla на сили-
кагеле со скоростями — для ацетилена 1,5 л/ч и для хлористого водорода 2,2 л/ч
обра<уются: 93% хлористого винила и менее 3% 1-1-дихлорэтана при 25° С
и 9-* и 1% этих веществ при 195° С.
Бромистый винил в газовой фазе реагирует с НВг легче, чем хлористый винил,
и гораздо быстрее, чем ацетилен. В противоположность хлористому водороду бромистый
водород в газовой фазе присоединяется в зависимости от применяемого катализатора:
по правилу Марковникова с образованием 1,1-дибромэтана или но, по правилу — с об-
разованием 1,2-дибромзтана. Например, в присутствии HgBr2 па асбесте при 100
я 150° С получается преимущественно 1,2-дибромэтан; так же действует FeBr3 на пемзе
при 200° С. На этом же катализаторе, но при 100° С образуется примерно 65% 1,2-
дибромэтапа и около 0,5% 1,1-дибромэтана. При использовании бЬВгдна асбесте при
100 и 150° С также получается смесь изомеров. Подробное описание аппаратуры, при-
меняемой для проведения реакций в газовой фазе, см. [227].
Бромистый винил [228, 229]. В небольших количествах бромистый винил
(т. кип. 17° С) получают лабораторным способом действием спиртового раствора
едкого кали или едкого натра на 1,2-дибромзтал. Дибромэтан синтезируют [228]
из ацетальдегида и РС13Вг2. В качестве ингибитора полимеризации к бромистому
винилу добавляют 0,1—0,2% гидрохинона.
Хлористый винил. Одни из промышленных способов получения хлори-
стого винила (т. кип. —13,9° С) заключается [230] в пропускании ацетилена
и НС1 при 140—200° С над активным углем, пропитанным HgCI2. Ингибиторов
полимеризации является фенол. Другие способы состоят в термическом дегидро-
хлорировании [230—232] 1,2-дихлорзтана в присутствии катализатора SnOy—
FeCla или под действием спиртового раствора щелочи. Ингибитором полиме-
ризации служит также фенол. л
Хлористый винил получают также в жидкой фазе, пропуская при 65—70° С
ацетилен при перемешивании через смесь 250 г СцС1.100 г NH4C1 и 30 мл 15%-иой
НС2. Аналогично бромистый винил получают, пропуская ацетилен при 60° С
через раствор СпВг в бромистоводородной кислоте [233].
Имеются также данные о методах присоединения НС1 к винилацетилену н era
алкильным производным [234, 235].
Хлоропрен [235]. При 0° С готовят смесь 45 г неочищенного винилаце-
тилена, 155 г концентрированной НС1, 20 г свежеосаждениого CuC], 9е NHtCl
0,5 г пирогаллола. После встряхивания при комнатной температуре смесь
226
227
228
229
230
231
232
233
234
235
J. Р. W i b a u t, J. van D а 1 f s е n, Rec. trav. chim., 51, 636 (1932).
J. P. W i b a u t, Rec. trav. chim., 50, 318 (1931).
J. P. W i b a u t, Rec. trav. chim., 50, 313 (1931).
F. R. Mayo, J. Am. Chem. Soc., 70, 1523 (1948).
Ф. Аз нигер, Введение в нэфтехимию, цер. с нем., Гостоптехиздат,
Москва, 1961.
F. К а 1 п е г, Koll.-Z., 113, 122 (1949).
J. С. G h о s Ъ, S. R. Das G u h a, Petroleum (London), 14, 261 (1951).
Н. Козлов, ЖПХ, Ю, 116 (1937); С., 1938, .1, 3326.
W. Н. С а г о t Ь е г в, D. D. С о f f m а п, J. Am, Chem. Soc., 54, 4071 (1932).
Н. J. В а с k е г. Th. А. Н. В I a s s, Rec. trav. chim., 61, 785 (1942).
122
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
разделяется на два слоя. Верхний коричневый слой промывают водой, сушат
над СаС12 с добавкой 1г пирогаллола и раз,сияют. Получают 12,5 г (20%)
2-хлорбутадиена-1,3 (т. кип. 59—61° С) и 12,5 г следующей фракции (т. кип.
61—75° С), содержащей ещо значительное количество хлоропрена.
2-Хлор-3-метплбутадиен-1,3. Получают аналогичным способом. После
очистки перегонкой с водяным паром выход составляет 75%; т. кип. 35° С.
Присоединение НВг к алкинам-1 и к олефинам можно направить так, что опО
будет происходить «нормально» с образованием 2-бромалкена-1 и 2,2-дибромалкана
пли «аномально» с образованием 1-бромалкена-1 и 1,2-дцбромалкана.
I—* ПСВг=СН« > BCBr..-CII;i
RC=CH —| «нормальное»
| присоединение
ИСЛТ=СПВг >ИСНВг-СН»Вг
«аномальное»
присоединение
Так, но реакции мотилацетнлена с НВг при —35° С образуется 2,2-ди-
бромпропан, а при добавке аскаридола—1,2-дибромпропан[236]. Из н-бутилацети-
,лена с НВг при 15° С с добавкой каталитических количеств гидрохинона и FeBrs
получается 2-бром-гексен-1; 1-бром-гексен-1 образуется при 0° С и добавке
перекисей [237].
Механизмы присоединения галогенов и галогеноводородов по С=С-связи
см. [238].
III. Присоединение галогенов по С~С- и С=С-связям
с одновременным образованием
С—С-, С—О- и С—N-связей
1. Присоединение галогенов по С=С- и С-'С-связям
с одновременным образованием С—С-связи
Присоединение и-галогенэфиров по С—С-связям
\ / । '
)С=С< -j-R'CII—X —> R'CH—С—-С—Х
7 4 I III
OR OR
а-Галоген- и а.р-дигалогспалкиловые эфиры могут в присутствии ZnCl2, В(С1,,
HgCl2 или А1С13 присоединяться по С=С-связям [239, 240].
Реакция, изученная ла многочисленных примерах [241—243], протекает при
низких температурах без растворителя или в CSZ. Она является общей и легко осуще-
ствима. Так, например, хлордиметиловый эфир легко присоединяется к изобутилену
[240], циклогексену [244], бутадиену {240, 241], винилацетилену [239], причем к двум
последним в 1,2- и 1,4-по.чожения.
[236]
[237]
239
240
241
242
243
244
М. S. Kharasch, J. G. McNab, J. Am. Chem. Soc., 57 2463
(1935).
C. A. Young, R. R. Vogt, J. A. Nieuwland, J. Am. Chem.
Soc., 58, 1807 (1936).
F. Bohlmann, Angew. Chem., 69, 82 (1957).
H, B. Dykstra, J. Am. Chem. Soc., 58, 1747 (1936).
F. Strauss, W. Thiel, Ann., 925. 151 (1936).
А. П. Пудовик, ЖОХ, 22, 773 (1952); 19, 241, 279 (1949).
R. Jung, Diplomarheit, Huinboldt-Universitat, Berlin, 1957.
E. H 6 f t, Dissertation, Humboldt-Universitat, Berlin, 1960.
C. D. N e n i t 7, e's с u, V. Przeme tzki, Ber., 69, 2706 (1936).
III. ПРИСОЕДИНЕНИЕ Hal С ОБРАЗОВАНИЕМ С—С-, С—О- Н С— N-СВЯЗЕЙ j£3
Реакция Меервейна * (взаимодействие галогенидов диазония
с соединениями, содержащими С —С- и С=С-связи)
Если в присутствии каталитических количеств СиС1а к водному раствору хло-
рида (бромида) Диазония, подкисленпому галогеноводородной кислотой или нейтрали-
зованному Са(ОН)2, MgO, NaHCO3, NasCO9 или ацетатом натрия, при 0—40° С при-
бавить ацетоновый раствор соединения с С=С- или CsC-связями, то, конкурируя
с реакцией Зандмейсра (см. стр. 247), происходит присоединение арила п галогена по
ненасыщенной связи. Ион Си* является одновременно катализатором реакций Меер-
вейпа и Зандмейсра. Вводимый в реакцию или образующийся из иона Си* за счет
взаимодействия с кислородом воздуха ион Си2* неизменно вновь восстанавливается
ацетоном в Си*-ион [245]:
2CuC]2+CHsCOCII3 2СиСЦ-С]СН2СОСН3
Следовательно, добавка ацетона благоприятствует также протеканию синтеза
по реакции Запдмейера. Считают [246], что реакции Зандмейера и Меервейна проте-
кают с промежуточным образованием арильных радикалов.
Реакция Меервейнд была изучена прежде всего для а,р-пснасыщенных карбо-
нильных соединений [247].
В а,0-ненаеыщенных карбонильных соединениях и нитрилах с алкильнъим
остатком в 0-положенли к В' галоген X вступает в а-положепие к В' (реакция 1):
ArN2X-|-RCH=CnR' —> ВСП—СНВ' (1)
Аг i
где В=Н, алкил; В'=СООН, СООСН3, CN, СНО.
Но если в ненасыщенных соединениях в 0-ноложеыип находится ароматический
остаток, то галоген вступает в 0-положение к В' (реакция 2):
AtN2X^-RbCH=CHR' —ь R"CH-CHR' (2:
где И'=как в реакции ((); R“—арил.
Имеются многочисленные примеры протекания реакции 1, п том числе присоедщ
пение хлоридов арплдиалонпя к эфирам и нитрилам акриловой и метакриловой кис
лот с образованием эфиров и нитрилов а-хлор-0-арилпропиоиовых кислот [248, 249}
Из а.р-ненасыщенных альдегидов получаются а-хлор(бром)-В-арилпропионовы<
альдегиды [250, 251]; бутадиен претерпевает 1,4-присоединенио [252—254]. В резуль
тате реакции фепилацетилена с нейтрализованным содой раствором хлорида фенил
диазония при 25—30° С с 46,5%-ным выходом образуется а-хлорсгильбен [255].
* О реакции Меервейна см. А. В. Домбровский, Реакции и методы исследова.
ния органических соединений (РиМИОС), Госхи»гпздат, 11, 285 (1982). —
Прим. ред.
[245] J. К. Kochi, J. Am. Chem. Soc., 77, 5090 (1955).
[246] J. К. Kochi, J. Am. Chem. Soc., 79, 2942 (1957).
[247] H. Mecrwefn, J. prakt. Chem., 152., 237 (1939).
[248] А. В. Д о м б p о в с к и й, А. П. Терентьев, А. М. Ю рке»ич
ЖОХ, 27 (89), 419 (1957).
[249] А. В. Домбровский, А. П. Терентьев, А. М. Юркевич
там же, 26 (88), 3214 (1956).
[250] А. В. Д о м б р о и с к и й, А. П. Т е р е н т ь е в, А. М. К) р к е в и ч, та»
же, 27 (89), 3047 (1957).
[251] S. Malinowski, S. Benbenek, Roczniki Chem. [Ann. Soc. chim.
Polonovum], 30, 1121 (1956); C., 1959, 15009.
[252] E. Muller. Angew. Chern., 61, 1/9 (1949).
[253] А. В. Д о м б p о в с к и й, ДАП СССР, 111, 827 (1956).
[254] А. В. Домбровский, А. П. Терентьев, ЖОХ, 27 (89), 2000 (1957^
[255] А. В. Д о м б р о в с к и й, ЖОХ, 27 (89), 3050 (1957),
124
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
а -Хлор-р-(п-нитрофенил)-пропионнтрил [252]. Растворяют при нагре-
вании 4,2 кг n-нитроанилина в смеси 9 л концентрированной HCI и 9 л воды,
охлаждают до 30° С и смешивают с 24 кг льда. Затем при сильном перемешивании
в течение нескольких минут вливают 7,3 л 30%-ного раствора NaNO2. Смесь
отсасывают и к полученному раствору соли диазоиия добавляют раствор 1,76 кг
акрилонитрила в 15 л ацетона. Затем вносят 600 г СиС1г; при 18° С через непро-
должительное время начинается выделение азота. Охлаждая льдом, поддержи-
вают температуру ниже 30° С. а-Хлор-p -(п-нитрофенил)-пропионитрил оса-
ждается и его перекристаллизовывают из метилового спирта; выход 5,3 кг;
т. пл. 110° С.
а -Хдор-р-фенилпропионовый альдегид [250]. К суспензии 10 г СиС12
н 4г СаО в растворе 22,5 г акролеина в 200 мл ацетона при 0—2° С приливают
по каплям нейтрализованный NaHCOs раствор 0,4 моль хлорида фенилдиазоиия
в 350 мл воды с такой скоростью, чтобы выделялось только 2—3 пузырька азота
в секунду. Через 2 ч реакционную массу экстрагируют эфиром, отгоняют эфир
и остаток фракционируют. Выход а-хлор-Р -фенилпропионового альдегида 52.8%
от теоретического; т. кип. 104—106° С (12 лл рт. ст.).
Присоединение галогенангидридов кислот по С—С-связи
Алкил-Р-хлорэтилкетоны [256] получают введением при 0—5° С этилена в при-
готовленную при 0® С смесь хлорангндрида алифатической кислоты и А1С13 *:
RCOC1 + CH2=CII2 ВСОСН2-СН2С1
р-Хлорэтилпропилкетон [257]. К 126 г н-бутирилхлорида при охлажде-
нии (лед с солью) и перемешивании добавляют в течение 80 мин 158 в иэмельчен-
’ •. ного непосредственно перед реакцией А1С13 и перемешивают еще 50 мин. Затем
при 20° С впускают сухой этилен до привеса в За г и перемешивают еще 30 мин
при той же температуре. Полученную массу смешивают со льдом, экстрагируют
GHCIg и перегоняют. Выход кетона 80%; т. кип. 67-^68° С (11 мм рт. ст.).
Присоединение алкилгалогенидов и аналогичных соединений по С=С-связи **
Имеется много вариантов реакции
RCH=CH2 + R'X —»- RCHX—ch2r'
протекающей под влиянием образующих радикалы веществ (перекиси ацетила и бен-
зоила, азосоединения), в которых R'X может представлять собой галоидзамещенные
углеводород^ [258] (СС14, СВг4) или галоидзамещенные эфиры карбоновых кислот
[R/CBrCOOR или СНВг(СООВ)2] [259]. Реакции протекает в атмосфере азота при
80—90° С; на 1 моль алкена расходуется около 0,05—0,1 моль перекиси.
В присутствии безводных А1С1а или FeCl3 олефины и хлоролефины могут при-
соединять хлоралканы со вторичным или третичным атомом хлора. Для проведения
* Реакция между олефиновыми углеводородами и хлорангидридами кислот в при-
сутствии безводного-А1С13 впервые описана И. Л. Кондаковым [Bull. Soc.
Chim.’ [3], 7, 576 (1892) J; см. также В. Н. Б е л о в, Т. А. Р у д о л ь ф и,
РиМИОС, 7, 255 (1958). — Прим. ред.
** Приспепипенве алисратическпх галогенпроиэлодных к олефинам известнокак реак-
ция Бутлерова — Эльтекова [Б. А. А р б у з о в, Реакции и методы исследо-
вания органических соединений (РиМИОС), Госхимиздат, 2, 7 (1952)]. — Прим,
ред.
[256] J. К е п пе г, F. S. S t a t h a m, Ber., 69,17 (1936).
[257] К. Bowden et al., J. Chem. Soc., 1946, 52.
[258] E. S. К h а г a s c h, E.V. Jensen, W. П. U г г у, J. Am. Chem. Soc.,
69, 1100 (1947).
[259] M. S. К h а г a s c h, P. S. S k e 11, P. F i s c h e r, J. Am. Chem. Soc., 70,
1055 (1948). *
HI. ПРИСОЕДИНЕНИЕ Hal С ОБРАЗОВАНИЕМ С—С-, С—О- Н С—N-СВЯЗЕЙ 125
присоединения таких хлор(бром)алканов к этилену последний пропускают при 0—
30° С в раствор, например, хлористого mpem-бутила в «-пентане в присутствии назван-
ных выше катализаторов [2бО].
2, Присоединение галогенов по С—С-связи
с одновременным образованием С—О-связи
Первой стадией этой реакции является присоединение Х+, причем из двух воз-
можных карбкатионов образуется наиболее энергетически выгодный [261].
СН3-СН=СН2 — -> СНз—бн-СН2 снз—си—сн2
[ I I
X ОН X
Так, НОВг и НОС1 присоединяются к пропилену [262] с образованием 1-бром-
<х.юр)прОпанола-2, а к «-бутилену [263] с образованием 1-бромбутанола-2. Источ-
ником катиона галогена являются гипогалогенитные кислоты НОХ (система Х2 -$
4- НаО) или N-галогенсоединения. Источником ОН" служит вода *, которая должна
присутствовать в реакции в достаточном количестве [264]. При проведении реакции
в спиртах образуются соответствующие эфиры галогенгидринов. В СС14 из серебряных
-солей карбоновых кислот и галогенов получаются [265—268] ацилгипогалогениты
RCOOX, которые при низких температурах присоединяются по С=С-связям в ваде
Х+ и RCOO".
Присоединение гипогалогенитных кислот НОХ в воде должно происходить
в нейтральной или слабокислой среде
Х2-[-Н2О ПОХ + Н+ + Х-
так как в присутствии кислот или хлоридов (бромидов) приведенное выше равновесие
сдвигается влево [16, 269], что благоприятствует образованию дигалогенидов. В щелоч-
ной среде получаются эпоксисоединения.
Присоединение гипогалогенитных кислот по С=С-связи
(получение галогенгидринов)
Реакция присоединения хлорноватистой кислоты к олефинам представляет
большой практический интерес. При пропускании этилена и хлора в воду (сгщгез
этиленхлоргндрина по Гомбергу [270]) с возрастанием концентрации НС1 увеличивается
'присоединение хлора. Эта нежелательная побочная реакция усиливается еще тем, что
олефины и хлор лучше растворяются в образующемся продукте присоединения, чем
в коде. Следовательно, опыт приходится прерывать и закапчивать при относительно
* Правильнее рассматривать в качестве нуклеофильного агента в данном случае не
ионы гидроксила, концентрация которых в кислой среде, естественно, вичтожиа,
а саму воду, присоединяющуюся за счет неподеленпой пары электронов кисло-
рода. — Прим. ред.
[2в0) L. Schmerling, J. Аш. Chem. Soc., 67, 1152 (1945); С. А., 44, 2006 (1950).
4261] Е. Мюллер, Новые воззрения в органической химии, пер. с нем., Издатпк-
лит, Москва, 1960.
[262] Е. Fourneau, J. Р и у а 1, Bull. Soc. chim. France [4], 31, 427 (1922).
[263] M. de M о n t m oil i n, P. M a t i 1 e, Helv. chim. acta, 7,. 108 (1924).
.[264] R. E. Buckles, J. E. M а и г er, J. Org. Chem., 18, 1586, (1953). -
265] M. В oc ke miillcr, F. W. Hoflmano, Ann., 519, 165 (1953).
266] M. 1. U s c h a k о w, W. O.Tchistow, Ber., 68, 824 (1935).
267] D. C. Abbott, C. L. Arcus, J. Chem. Soc., 1952, 1515.
268] W. G. H. E dwar ds, H. H о d g e s, J. Chem. Soc., 1954, 762.
,269] A. W. Francis, J. Аш. Chem. Soc., 47, 2340 (1925).
[270] M. Gomberg, J. Am. Chem, Soc., 41, 1414 (1919).
126
ОБРАЗОВАНИЕ СВЯЗИ С—Иа1 В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
малых концентрациях хлоргидрина [21Ь, 271, 272]. Так, при работе с этиленом выход,
хлоргидрина составляет 8—10%.
Этпленхлоргидрпн нельзя непосредственно выделить из реакционного
раствора, так как при перегонке отгоняется азеотропная смесь этиленхлор-
гидрпна с водой (т. кип. 97,8° С), содержащая 42,5% этиденхлоргидрина.
При добавлении к реакционной смеси известкового молока в щелочной среде-
образуотся окись этилена (т. кип. 10,7° С), которая легко отделяется от смеси.
Окись этилена вместе с эквивалентным количеством хлористого водорода можно-
затем пропустить в уже имеющийся отиленхлоргидрин и количественно вновь
превратить в этиленхлоргидрин (т. кип. 128,7° С прп 760 мм рт. ст.). О синтезе-
этиленбромгидрина см. [273].
С возрастанием молекулярного веса олефинов сильно увеличивается образова-
ние продуктов присоединения хлора. Однако хлоргидрины с очень хорошим выходом
можно получить, например, из иептена-2, гексепа-2, циклогексспа, бутадиена и пи-
непа. Для этого к водной эмульсии олефина, через которую пропускают под неболь-
шим избыточным давлением (0,35—3,5 ат) двуокись углерода, постепенно при охла-
ждении льдом и сильном перемешивании (температура 0 — 30° С) прибавляют"
концентрированный водный раствор гипохлорита (10—15%-ныи раствор хлорной
извести) [274].
1-Хлорбутен-3-ол-2 [275]. Смесь бутадиена и СО2 (6 : 1) вводят при
0—3° С в перемени ваемый 3,5%-ный раствор гипохлорита кальция (0,845 моль)..
Через 1,8 ч реакция заканчивается, что можно определить по отсутствию выделе-
ния иода из K.I, добавляемого к пробе. После отделения СаСО3 фильтрованием
на воронке Бюхнера реакционный раствор насыщают NaCl и экстрагируют
Продукт эфиром. Выход 52%; т. кип. 64,0 — 64,8° С (30 мм рт. ст.).
Дихлоргидрин аллилового эфира [276]. При перемешивании и охлажде-
нии до 3° С в смесь 0,25 моль аллилового эфира, 2,5 л воды и 102 г 35%-ного-
Са(ОС1)2 (0,25 моль) вводят СО2. Реакционный сосуд, охлаждаемый сухим льдом,
соединяют с обратным холодильником во избежание уноса эфира. Через 2 ч
после полного расходования гипохлорита кальция добавляют еще 51а Ca^ClK
и пропускают СО2 еще 2 ч при перемешивании. Отфильтрованный от СаСОд,
реакционный раствор насыщают NaC] и экстрагируют эфиром дихлоргидрив
аллилового эфира. Выход 94,4%; т. кип. 150—165° С (.и.и рт. ст.); после вто-
ричной перегонки т. кип. 138—139 °C (1 мм рт. ст.).
Этиленхлоргидрин. Существует аналогичный лабораторный способ полу-
нений этнленхлоргидрина [277]. В трехгорлую колбу емкостью 2 л загружают
через одно горло взвесь 250 г хлорной извести в 250 мл воды, в другое горло
вставляют достигающую дна трубку для ввода газов, в третье горло — отвод-
ную трубку, соединенную с промывной склянкой, в которой с помощью ртути
создают избыточное давление около 100 см вод. ст. Подводящая газы трубка-
соединена при помощи тройника с баллонами этилена и двуокиси углерода.
Двуокись углерода и этилен подают в соотношении 3 : 5 со скоростыЬ соответ-
ственно 15 и 25 л/'ч. Реакция заканчивается через 3 ч, что устанавливают по отри-
цательной иодкрахмальной реакции. Реакционную массу перегоняют с водяным
паром, дистиллят насыщают К2СО3, органический слой высушивают им же
и при перегонке в. интервале 125—129 °C получают 35 г (33%) этиленхлор-
гидрина.
[271]
1272]
273
274
275
276
277
W. G, D omask, К. А. К о b е, Ind. Eng. Chem., 46, 680 (1954).
Р. F е г г е г о L. R. F 1 a m m е, М. F о ц г е z, Ind. chinl. Beige,
133 (1954).
F. H. Me D owe II, J. Chem. Soc., 1926, 499.
В. T. В г о о к s, пат. США 2463850; С. А., 43, 4687 (1949).
В. С. К a d е s с h, J. Am. Chem. Soc., 68, 41 (1946).
J. R, R о a c b, S. E. Miller, J. Am. Chem. Soc., 71, 2667 (194
Метод разработан в лаборатории К. Вейганда, 1948.
Ш. ПРИСОЕДИНЕНИЕ Hal С ОБРАЗОВАНИЕМ С—С-, С—О- П с—1Й,
Наряду с этим очень простым методом существуют и другие способы при-
готовления растворов хлорноватистой кислоты, пригодных для проведения реак-
ции присоединения: введение хлора в охлаждаемую льдом взвесь свежеосажден-
ной окиси ртути [278] пли в раствор бикарбоната натрия в ледяной воде [279]
а также из хлорной извести н карбоната калия в водном растворе [280]. ’
Хлорноватистая кислота, 20%-ный раствору[281—282]. При сильном
перемешивании и охлаждении льдом с солью в воду пропускают хлор до ее на-
сыщения. Выпадающий кристаллогидрат хлора отфильтровывают па воронке
Бюхнера при охлаждении и смешивают с таким количеством окиси ртути (при-
мерно 3/4 от веса кристаллогидрата хлора), чтобы образующийся водный раствор
не имел запаха хлора. Раствор отфильтровывают и перегоняют в вакууме
(12 мм рт. ст.) при комнатной температуре. Дистиллят, содержащий до 20%
НОС1, хранят в темноте в замороженном состоянии. При —10° С он может со-
храняться в течение многих месяцев.
Для проведения реакции присоединения хлорноватистой кислоты смешивают
олефины при 10° С с 0,125 —0,5 М раствором этой кислоты. При работе с высшими
нормальными олефинами [282] выход хлоргидринов значительно повышается при до-
бавке пиридина и серной кислоты (начальное pH 6—6,5). Высшие хлоргидрипы можно
получать также взаимодействием алкилгипохлоритов с олефинами в водной среде.
Для этого пригоден, например, щрет-бутилгипохлорит. который получают обработкой
хлором третичного бутилового спирта в водной щелочи при 0—20° С.
пгретп-Бутилгипохлорит [283]. Получают хлорированием третичного
бутилового спирта в водной щелочи при 0—20° С. Это желтое масло; т. кип.
77—78° С (760 мм рт. ст.). Оно бурпо разлагается под действием интенсивного
света, перегрева или сопрпкосновения с резиной.
Для проведения реакции присоединения НОВг ненасыщенное соединение обычно
несколько часов перемешивают при комнатной температуре с бромной водой, после
чего избыток НОВг удаляют с помощью бисульфита натрия. Раствор насыщают
NaC] и извлекают бромгйДрпн из жидкой фазы эфиром или хлороформом. Вместо бром-
пой воды можно применять раствор гипобромитэ, приготовленный на холоду из брома
и карбоната щелочного металла [284]. У стирола склонность к образованию бромги-
дрииа так ярко выражена, что даже в горячей воде (60—90° С) под действием раствора
брома в присутствии КВг с отличным выходом образуется 2-бром-1-фенилэтиловый
спирт [264, 285].
Присоединение ОН и 1+ по С=С-связям происходит в насыщенном водой эфире,
к которому для удаления I" добавляют свежеприготовленную окись ртути в количестве
0,5 .чоль HgO на 1 моль олефина. Иод (1 .ноль) вводят частями при Охлаждении и пере-
мешивании и затем продолжают перемешивание до почти полного исчезновения окраски
иода [78, 286]. При проведении подобной реакции в абсолютном метиловом спирте по
С=С-связи присоединяются иод и метоксильная группа. Иодгидрины и их эфиры очень
неустойчивы. К а,0-ненасыщенпым карбоновым кислотам подповатистая кислота,
вероятно, пе присоединяется, а присоединяются иод и метоксильная группа
(см. стр. 130).
К С=С-спязям ненасыщенных карбоновых кислот HOCI и НОВг присоединяются
таким образом, что галогсп вступает в а-, а гидроксильная группа в p-положение к кар
боксильной группе.
[278
[279
[280
[281
'[282
[283]
[284]
1285]
(286]
W. Markownikoff, Ann., 336, 314 (1904).
A. Wohl, Н. Schweitzer, Вег., 40, 94 (1907).
Е. Bamberger, W. Lodter, Ann., 288, 81 (1895).
S. G ol dsch mi d t, Ber., 52, 758 (1919).
A. Guyer, A. Bieler, E. Pedrazzelli, Helv.
39, 423 (1956).
H. M. Teeter, E. W. Bell, Org. Synth, 32, 1952, p. 20.
E. Berner, C. N.Riiher, Ber., 54, 1954 (1921).
J. Read, W. G. Reid, J. Chain. Soc., 1928, 1487.
L. Brunel, C. r., 135, 1055 (1902).
chim.
acta,
iza
V£>J-AJUBAHMK СВЯЗИ C—Hal В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
а -Бром-р-оксимасляцая кислота [287]. Ес получают, пропуская к тече-
ние 5 ч насыщенный парами брома воздух в быстро перемешиваемый водный
раствор кротоновой кислоты (0,34 М).
а-Хлор-р-оксиизовалериановая кислота [288]. Получают приливанием
водного раствора хлорноватистой кислоты к суспензии 0,0-диметплакриловой
кислоты.
Получение хлоргидринов при помощи хромилхлорида
Направление присоединения НОС1 по С= С-связям, протекающее при взаимо-
действии с гипогалогенитами [289] или с N-галогенампдами по схеме:
RCH=CH4 —> RCH(OH)-CH2CL
можно изменить, пспользуя хромилхлорид СгО2С12. Так, при взаимодействии этого
реагента с пропиленом, «-бутиленом, пентеном-1 и гексеном-1 в СС14 или СНС13 при-
соединение происходит следующим образом [290]:
rch=cii2 —> RCHC1—СН2О11
То же относится и к соединениям ряда стероидов [291].
2-Хлорпропиловый спирт. В охлаждаемый льдом, быстро перемеши-
ваемый раствор 1 моль CrOjClg в 300 ял СС14 пропускают пропилен до исчезнове-
ния окраски хромилхлорида (23 ч). Выпавший в осадок твердый продукт гидро-
лизуют холодным раствором бисульфита натрия. Водный раствор насыщают
NaCl и экстрагируют продукт реакции эфиром. Эфирный раствор высушивают
над Na2SO4 и перегоняют; выход хлорпропилового спирта 43% от теоретиче-
ского.
Получение галогенгидринов по реакции с N-галогенамидами
В качестве источника Вт* при присоединении НОВг по С=С-связям с успехом
могут применяться N-бромсукципимид [292] или N-бромацетамид [293, 294].
Синтез и анализ N-бромацетамида см. [295], N-бромсукцинимида см. на стр. 137..
При комнатной температуре перемешивают в воде 1 леоль ненасыщенного»
соединения с 1 моль N-бромамида до полного растворения последнего и затем
экстрагируют образовавшийся бромгидрин эфиром или бензолом. Вливая неочи-
щенный продукт в раствор щелочи, его можно непосредственно перевести в эно-.
ксид, который при работе с колонкой будет непрерывно отгоняться. Так, на-
пример, получают 2,3-эпокси-2-метилбутан из 2-окси-3-бром-2-метилбутана
[296]. Добавка небольших количеств кислоты (серной или уксусной) ускоряет
реакцию.
При взаимодействии N-бромацетамида с меченным иС этиленом полу-
чается [297] с 90%-ным выходом Вт* 14СИ2—14СНг0Н.
287]
288]
289
290
291
292
293
294
295
296
С. О. Guss, Г.
S. W i n s t е i и,
Е. Schmidt,
Н. Е. С а г t е г, С. L. Z i г k 1 е, J. Biol. Chem., 178, 711 (1949)
Е. Ahderhalden, К. Heyns, Ber., 67, 537 (1934).
A. S'а у t z е f f," J. prakt. Chem. [2], 3, 88 (1871).
S. J. С r i s t 0 1, K. R. E i ha r, J. Am. Chem. Soc., 72, 4358 (1950).
ч т о , _ . _ _ N. L.Wendler, J. Am. Chem. Soc., 78, 3751 (1956).
R. Rosenthal, J. Am. Chem. Soc., 77, 2549 (1955).
=, J. Am. Chem. Soc., 61, 1576 (1939); 64, 2780 (1942).
, W. К n i 11 i n g, A. Aschorl, Ber., 59, 1279 (1926).
о, C. Gerold, Org. Synth., 31, 1951, p. 17.
n, L. L. Ingraham, I. Am. Chem. Soc., 74, 1163-
Am. Chem. Soc., 73».
(1952).
J297] D. E. Walz, M. Fields,!. A. Gihhs,
2968 (1951).
HI. ПРИСОЕДИНЕНИЕ Hal С ОБРАЗОВАНИЕМ С—С-, G—О- Н С—N-СВЯЗЕЙ 12S>
Реакция присоединения НОХ под действием N-галогенамидов представляет
ценность также прп синтезах стероидов. По 9(11)-С=С-связи гидроксил присоеди-
няется к CL3 (11₽), бром — к Сд (9а). В качестве растворителей применяют смеси аце-
тона. диоксана, пиридина или mpe/n-бутилового спирта с водой. В качестве кислоты
наиболее пригодной оказалась 0,46 н. НСЮ4, меньше всего способствующая образова-
нию эфиров галогенгидрина [298].
По реакции 21-ацетата Д*-9 ,и'-прегнадиендиол-17а,21-диона-3,20 (1>
с N-бромацетамидом получают с 80%-пым выходом ацетат 9а-бромгидрокор-
тизона П. Соединение II превращают в 9р,11р-окись III, из которой с хоро-
шими выходами могут быть получены [298] ацетаты соответствующих 9а-гало-
генгидрокортизонов IV действием [292—302] HI при —20° С или НС1 и HF'
при 0° С *.
Дальнейшие данные по присоединению гнпобромиой кислоты н стерои-
дам с помощью N-бромацетамида или N-бромсукцинимида см. [299—302].
Присоединение гипохлорцой кислоты по С=С-связям может происходить
также при взаимодействии с хлормочевиной [303].
лпранс-2-Хлорциклопентанол [304]. Монохлормочевину получают, про-
пуская хлор при температуре 0—15° С и энергичном перемешивании в водный
раствор мочевины, в котором содержится СаСО3 для связывания выделяющегося
НС1. К водному раствору хлормочевины при охлаждении добавляют уксусную
кислоту и соответствующий олефин. Из циклопентена наряду с 1,2-дихлор-
циклопентаном и ^uc-2-хлорциклопентанолом образуется 52—56% транс-2—
хлорциклопентанола.
* Указанная реакция имеет очень большое практическое значение для полу-
чения таких ценных лекарственных препаратов, кан дексаметазон,
триамсинолон, синалар н др. — Прим. ред.
[298] J. F г i с d, Е. F. S а Ъ о, J. Am. Chem. Soc., 79, 1136 (1957).
[299] L. В. Barkley et al., J. Am. Chem. Soc., 76, 5017 (1954).
[300] J. A. Hogg et al., J. Am. Chem. Soc., 77, 6401 (1955).
[301] R. H. Lena a rd, S. Bernstein, J. Am. Chem. Soc., 77, 6665-
(1955).
[302] H. L. S 1 a t es, N. L. Wendler, J. Am. Chem. Soc., 78 , 3749(1956).
[303] A. Detoeuf, Bull. Soc. chim. France [4], 31, 102, 171 (1922).
[304] H. B. Donahoe, C. . A. Vanderwerf, Org. Synth., 30, 1950,.
p. 24.
9 Заказ 1835.
130
ОБРАЗОВАНИЕ СВЯЗИ С—Hal Б РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
При взаимодействии олефина с N-бромацетамидом [294] или другими N-6poa>
амидами, например бензолсульфодибромампдом [305, 306], не в водной среде, а при
0_20° С в сухом метиловом или этиловом спирте получаются алкокснбромпды,
а в уксусной кислоте — ацплоксибромпды [294, 307].
Алкоксигалогенирование
Эфиры сс-бром-р-алкоксикарбоновых кислот являются важными промежуточ-
ными продуктами для синтеза оксиамипокислот, если соответствующие «^-ненасы-
щенные кислоты легко доступны.
Эфиры сс-бром-]3-метоксикарбоновых кислот получают с хорошими выходами
бромированием в хлороформе или этилацетате бромртутных соединений, легко полу-
чаемых из ацстоксиртутных производных и водного раствора КВг.
T> T
\c=CH—COOR CH*°H
R»Z
R
R"
ОСНа HgOCOCII3
I
-СН
I
COOR
ОСН3 HgBr ОСНз
R\ I । Bri R'\ I
>C----СП >C-CHBr-hHgBr.,
R»Z I R"Z |
COOR COOR
Из эфира р,р-диметилакриловой кислоты (R' и R" = СН3) таким способом
получают эфир а-бром-Р-метоксиизовалериановой кислоты [308], из эфира кротоновой
кислоты (R' = Н, R" = СН3) — эфир а-бром-р-метоксимасляной кислоты [288,
310] и из эфира акриловой кислоты (R' и R" = II) — эфир а-бром-р-метоксипропионо-
вой кислоты [309]. Эти вещества являются промежуточными продуктами в синтезах
Р,р-диметилсерина, 2),Л-треонина и 2),£-серина.
Иногда удобнее проводить реакцию метоксибром(иод)ртутных соединений
с иодом в кипящем метиловом спирте, смеси метилового спирта с водой или в СС14,
как, например, при превращении N-аллилфталимида в 2-метокси-З-фталимидпропил-
галогенид.
При присоединении ацетата ртути к N-аллилфталимиду в кипящем метиловом
спирте и последующем обмене с бромидом (иодидом) калия в кипящей смеси метиловый
спирт — вода образуется в осповном бромид или иодид 2-метокси-З-фталимидпропил-
ртути наряду с небольшим количеством изомеров. Этот продукт плохо превращается
при действии брома в соответствующий пропилбромид [311], но с иодом, напротив,
легко образует пропилиодид. *С N-аллилфталимидом реакция протекает достаточно
быстро [317].
Метоксибромирование легко протекает также при непосредственном взаимодей-
ствии с бромом. Например, пропуская охлажденный насыщенный парами брома ток
воздуха в раствор 0,р-диметнлакриловой кислоты в метиловом спирте, получают
а-бром-р-метоксивалериановую кислоту с 75%-ным выходом от теоретического [313].
Для связывания НВг добавляют окись магния. Бромистый водород можпо удалить
также добавлением нитрата серебра и карбоната кальция [314].
305] А. А. Пет ров, ЖОХ, 8 (70), 487 (1938).
306] Н. L. Holme s, К. М. Man n, J. Am. Chem. Soc., 69 2001 (1947).
307] М. В. Л и х о ш е р с т о в, А. А. П е т р о в, ЖОХ, 9 (71), 2012 (1939).
308] W. S с h г a u t h, Н. Geller, Ber., 55, 2788 (1922).
309 Н. Е. Carter, Н. D. West, Org. Synth., Coll. v. 3, 1955, p. 774.
3101 H. E. Carter, H. D. W e s t, Org. Synth., Coll. v. 3, 1955, p. 813»
311] D. E.Peareoi, M. V. S i g a 1 jr., R. II. К г и g, J. Org. Chem., 15. 1048
(1950).
[3121 B. R. Baker et al., J. Org. Chem., 17, 74 (1952).
[313] K. Rufenacht, Helv. chim. acta, 35, 764 (1952).
[314] K. Meinel, Ann., 516, 231 (1935).
HI. ПРИСОЕДИНЕНИЕ Hal С ОБРАЗОВАНИЕМ С—С-, С—О- И С—^-СВЯЗЕЙ £3.
Если в присутствии свежеосажденпого карбопата кальция при комнатно!
температуре подействовать спиртами на легко получаемые а-под-0-хлоркарбоновьв
кислоты (готовят действием 1,25 .ноль ТС1 и СС14 на 1 .ноль а.р-ненасыщеппой кислоты)
то наряду с а,0-пепасыщеппыми кислотами, количество которых зависит от применен
ного спирта, образуются а-иод-р-алкоксикарбоновые кислоты. Такие кислоты такж<
получают добавлением к суспензии 7—15 г СаСО3 в растворе 5—10 г ненасыщенно!
кислоты в метиловом («-пропиловом, изопропиловом) спирте хлорида иода (1,25 мол)
ла 1 моль кислоты); после 20 ч перемешивания реакционную массу фильтруют
При алкоксигалогенпровапии коричной кислоты по этому методу продукты реакцит
выпадают после выливания фильтрата в воду; выход 88 (62 и 22)% от теоретического
Реакция протекает через ROI — алкилгипоиодит [315].
3. Присоединение галогенов по С=С- и С^С-связям
с одновременным образованием С—N-связи *
Реакция присоединения нитрозилхлорида NOCI к соединениям с С=С- j
С=С-связями
(СП3)2С—СНСНз (СНз)2С-СНСН3 —> (СН3)2С-СС11з
II I II
Cl NO Cl NOH
1 И
изучена на многочисленных примерах [316а]. в частности, она приобрела препаратив-
ное значение в химии терпенов [317, 318].
Из олефинов аллильной структуры RCH2CH=CH2 иитрозохлориды обра-
зуются труднее, чем из алкенов типа пропилена RCH—'CHCIIS и олефинов с концевой
двойной связью, например CeHsCII=CH2 или RCH2C(CH3)—СН2. Из «^-ненасыщен-
ных кислот и пнтровипильпых соединений нитрозохлориды не образуются, но ид
можно получить из ненасыщенных кислот, у которых С=С-связь и карбоксильнар
группа разделены несколькими СН2-группами. К олефинам с заместителями, снижа-
ющими реакционную способность С—С-связи к нуклеофильным реагентам и повыша-
ющими реакционную способность к электрофильным реагентам, присоединение нитро-
зилхлорида происходит особенно хорошо. Па первой стадии присоединяется NO+,
па второй стадии С1~, причем хлор присоединяется к наименее гидрогенизированному
атому углерода [319].
В большинстве случаев нитрозохлориды получают в виде бесцветных кристал-
лических веществ, являющихся димерами соединения I. Окрашенные в синий цве*]
мономеры образуются при плавлении или при нагревании растворов. Одпано прь
этом, чаще всего одновременно, происходит перегруппировка в а-хлороксииы (II),
сопровождающаяся пслезновением синей окраски. Необходимым условием перегруп-
пировки является наличие в соединении I атома водорода у углерода, связанногс
с NO-i'pyiuioil. Если такового нет, то получаются стабильные синие мопомеры, напри-
мер тетраметилэтиленнитрозохлорид [320].
Выходы продуктов реакции присоединения нитрозилхлорида в сильной мере
зависят от условий реакции и прежде всего от температуры и растворителя. Если
олефин обладает достаточной реакционной способностью, то кристаллические про-
дукты легко получаются при пропускании газообразного NOC1 при 0° С в жадное
ненасыщенное соединение [Ь16а ] или в раствор олефина в ледяной уксусной кислоте
при 5—10° С. В качестве растворителей пригодны также СС14, СПС13, СН2С1а, нитро-
метан и нитроэтан; в качестве катализаторов [316а, 321] — Ni, FeCl3, CuCl.
* См. также стр. 373, 374.
[315] Е. L. J а с к я о П, L. Р а s i u t, J. Am. Chem. Soc., 50, 2249 (1928).
[316] T,. J. Beckham, W. A. F е s s 1 е г, М. А. К i s е, Chem. Rev., 48 (1951)z
a) p. 369—378; b) p. 324.
[317] O. Wallach, Ann. 245, 241 (1888); 252, 106 (1889); 253, 249 (1889); 258
319 (1890); 345, 127 (1906); 360, 26—81 (1908); Ber., 28, 1308 (1895).
[318] A. Bayer, Ber., 28, 1586 (1695); 29, 1078 (1896).
|319] N. T h 0 r n e, .]. Chem. Soc., 1956, 4271.
|320] J. T h i c 1 e, Ber., 27, 454 (1894).
[321] P. J. Gaylor, Petrol. Proc., 2, 870 (1947).
132
ОБРАЗОВАНИЕ СВЯЗИ С — Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Нитрозилхлорид (т. кип. —5,8° С) получают |316Ь] пропусканием смеси NO
и С12 (2 : 1) над активированной окисью алюминия при 40—50° С.
Для препаративных целей нитрозилхлорид преимущественно получают непо-
средственно в реакционной массе. Для этого к смеси ненасыщенного соединения
с амил- или этилнитритом при —15° С добавляют концентрированную соляную
кислоту или раствор НС1 в этиловом спирте или к раствору ненасыщенного соединення
в спиртовом растворе НС1 приливают по каплям концентрированный раствор NaNOs.
Нитрозохлорид 2-метилбутена-2 [322, 323J. При температуре ниже 0° С
(охлаждение льдом с солью) в перемешиваемую смесь 30 г 2-метилбутена-2
и 50 мл амилнитрита приливают по наплям в течение 1,5 ч 50 мл охлажденной
льдом дымящей НС1. Смесь тотчас же окрашивается в синий цвет и постепенно
затвердевает в белую кристаллическую кашицу. Через 30 мин охлажденную
смесь отсасывают и промывают холодным этиловым спиртом. Выход 29 г;
т. пл. 76° С.
Для получения а-хлороксима растворяют 5 г полученного продукта
в 50 мл этилового спирта и нагревают до кяпекяя, после чего раствор кипит
несколько минут даже без дальнейшего нагревания. При этом синяя окраска
переходит в желтую. После удаления спирта получается оксим 2-хлор 2-метил-
бутанона-3. Выход 4,5 г; т. пл. 50° С.
Разработан более интересный метод получения а-хлороксимов [324], заключа-
ющийся в восстановлении нитроолефинов эфирным раствором SnCla в эфирном
растворе НС1 при охлаждении смесью льда с солью.
Нитрозилхлорид применяют также для определения С—С-связей в жирах,
жирных кислотах и ненасыщенных углеводородах (ср. обзор [325]).
Нитрилхлорид NO^Cl также присоединяется по С=С-и С = С-связям, причем
из галогензамещенных этиленов, стирола, стильбена, коричной кислоты, циклогексена
и феиилацетилена образуются а-хлорннтросоединения, из этилена — дихлорэтан,
.а из кетена — с незначительным выходом легко разлагающийся нитроацетилхлорид.
JJpu работе с нитрилхлоридом легко могут происходить взрывы (подробности см. [326]).
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ РЕАКЦИИ ЗАМЕЩЕНИЯ
I. Замещение водорода галогеном или роданом
1. Замещение галогеном водорода
в насыщенных алифатических углеводородах
Реакция замещения водорода на фтор сильно экзотермична:
I I
—С—H-f-Fa —> —С—F4-HF4-IO2 ккал
I I
Поэтому последовательное замещение атомов водорода на фтор связано с большими .
трудностями (при замещении на хлор выделяется 22,9 ккал! .ноль, а при замещении
на бром — 6,2 ккал/моль}. Большей частью фтором замещается сразу несколько '
атомов водорода н расщепляется С—С-связь, в результате чего основным продуктом -й
реакции является CF4. Моиофторзамещенные чаще получают путем обмена других
галогенов на фтор (стр. 191). Промышленное значение имеет прямое получение поли-
фтор- и полностью замещенных перфторсоединенин [178с]. Особый интерес пред-
ставляет электрохимическое фторирование органических соединений в безводной
[322
[323
[324 ... _
[325 H. P.
[326 W. S
J. Schmidt, Вег., 35, 3730 (1902).
N. Thorne, J. Chem. Soc., 1956, 2588. ‘
A. D orn о w, Н. D. J or d a n, А. М u 11 е г, Chem. Вег., 94, 67 (1961).
Н. Р, Kaufmann, Р, flover, Fette и. Seifen, 47, 103 (1940).
Steinkopf^ М. Kuhnel, Ber., 75, 1323 (1942).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
133
•фтористоводородной кислоте. Процесс ведут в стальных или никелевых электролизе-
рах ври 20° С на никелевых анодах и никелевых или железных катодах при плотности
тока 0,02 а/см2 и напряжении 5—6 в. При фторировании углеводородов для повыше-
ния проводимости добавляют KF или LiF. Образующийся в реакции HF непрерывно
возвращается в цикл *.
В то время как в лаборатории хлорированные алканы в чисто препаративных
целях редко получают замещением водорода хлором, хлорирование алканов в про-
мышленном масштабе имеет очень большое значение. Выделяющееся в течение реакции
большое количество тепла может быть отведено из газовой фазы введением избыточ-
ного количества алкана иди путем разбавления инертным газом, а из жидкой фазы
с помощью охлажденного растворителя. В технике используются фотохимические,
каталитические и термические Процессы. Во всех процессах наряду с монохлорзаме-
щепнымп получаются ди- и полпхлорзамещенпые.
Жидкие, хлорированные, а также ожиженные (при нормальной температуре
газообразные) и расплавленные (т. пл. до 70° С) парафины хлорируют, пропуская
в парафин хлор при перемешивании и УФ-облучении, пысокоплавкие алканы и поли-
этилен хлорируют в СС14. Подробности аппаратурного оформления фотохимического
хлорирования изобутана до хлористого изобутила и mpe/n-бутилхлорида, хлористого
метила до хлористого метилена, хлористого метилеиа до хлороформа, а также хлори-
рования zi-додекана см. [327а].
При жидкофазном гомогенном каталитическом хлорировании к жидким или
растворенным в СС14 парафинам добавляют хлориды иода, фосфора, серы, сурьмы,
железа, олова (патентную литературу см. [327Ь) или вещества, инициирующие ради-
калы, например тетраэтилсвинец [328]. Олефины, которые могут присутствовать
в парафинах в виде примесей, также оказывают каталитическое действие [329, 330].
Гомогеппое каталитическое хлорирование осуществимо и в газовой фазе в при-
сутствии паров калия, натрия или SbCl5. Гетерогенное каталитическое хлорирование
происходит в темноте при повышенных температурах в газовой фазе над такими кон-
тактами, как активный уголь, кизельгур, пемза, глина, каолин, силикагель или
боксит, которые могут быть пропитаны солями металлов, особенно солями меди [327с].
Оно протекает по криптоиопному механизму в поэтому на процесс не влияет, напри-
мер, присутствие кислорода, являющегося ингибитором (обрыв цепи) радикальных
процессов фотохимического и термического хлорирования. При этом даже добавляют
воздух для того, чтобы так же, как в процессе Дикона, перевести выделяющийся
хлористый водород в хлор.
По оправдавшему себя в промышленности варианту подаваемая в иониче-
ский реактор газовая смесь поддерживает взвешенный контакт (активный уголь)
в постоянном движения («кипящий слой»). Преимущество состоит в том, что
процесс не нарушается из-за отложения угля и смолистых продуктов на поверх-
ности катализатора, как это происходит при чисто термическом или каталити-
ческом хлорировании с неподвижным контактом [327dJ.
Термическое хлорирование (400—500° С) в отсутствие катализаторов и света
является цепной реакцией, инициируемой термической диссоциацией хлора [331,].
Сведения о лабораторной аппаратуре для этого способа хлорирования, особенно
важного для низкомолекулярных парафинов, см. [327е, 332]. По атому способу пара-
фины можно хлорировать сначала до монохлорзамещенных, а затем до более высоко-
замещенных. Правда, получать СИС13 и СС14 прямым взаимодействием метана с необ-
ходимым для их образования количеством хлора невозможно, так как в этих пропор-
циях компоненты реагируют со взрывом и с выделением углерода.
* Электрохимическое фторирование см. И.Л.Кнунянц, Г. А. С ок о л ъ ск ля,
Реакции и методы исследования органических соединений (РиМИОС), Госхимиз-
цат, 6, 343 (1957). — Прим. ред.
[327) F. A s i n ge г, Chemie und Techoologie der paralfin-Kohlenwasserstoffe, Berlin*
Akademie-Verlag, 1956; a) S. 157; b) S. 164; c) S. 169; d) S. 191; e) S. 177;
f) S. 184; g) S. 611. (Имеется русский перевод).
J628] W. E. Vaughan, F. F.Bast, J- Org. Chem., 5, 467 (1940),
Г329] R, M. De anesly, J. Am. Chem. Soc., 56, 2502 (1934).
1330] H. P. A. G г 0 11, G. Hearne, Ind. End. Chem., Ind. Ed., 31, 1239 (1939).
1331] W. E. V a u g h a n, F. F. Rust, J. Org. Chem., 5, 449 (1940).
1332] H. B. Hass, Ind. Eng. Chem., 27, 1192 (1935).
134
ОБРАЗОВАНИЕ СВЯЗИ С—На! В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Лее же СС14 можно получать в одну стадию. Для чего метан при температуре-
около 450° С пропускают через многократно изогнутую трубку, в которую подают-
хлор через сопла, расположенные на таких расстояниях друг от друга, чтобы реакция-
завершалась за время прохождения метана от одного сопла к соседнему [327f, 333J.
Количество изомеров, образующихся при молохлорпрованип парафинов,
определяется относительными скоростями реакции замещения различно связанных,
атомов водорода. Соотношение скоростей [327g] замещения атомов водорода п группах
СИ3—, — СН2—, >СН— при 300° С равно 1 : 3,25:4.43. Катализаторы и облучение-
не изменяют этого соотношения. Из этого следует, что с ростом длины цепи количе-
ство образующихся 1-хлорзамещепных все больше снижается. Так, из н-пропана-.
образуется 48% 1-хлорпропзводного, а из н-додекана только 8,5%.
Вместо хлора в присутствии веществ, образующих радикалы, источником,
СЬ может служить также SO2CJ3.
Из SO2C12 без радикалообразующих веществ выделяется только молекулярный,
хлор по схеме:
SO2CI2 -t * SO2 + CI2
Свободнорадикальное хлорирование хлором (фотохимическое, термическое пли
с добавкой веществ, образующих радикалы) протекает по цепному механизму:
Cb-J-RII —> R.4-HCI
R’ + Cl2 —> RCl-j-Cl»
Хлорирование SO2C12 в присутствии инициаторов — перекисей пли азосоедц-
непий может протекать и таким путем [334-—336J:
R«4-SO2C12 —► RC1H-SO2C1.
SO2Cb —> SO2-|-Cb
В присутствии перекисей Ллорировапио с помощью SO2C12 протекает даже-
в темноте [337|. Хлорирование смеси 1 моль SO2CI2, 1— 3 моль углеводорода и
0,02 моль перекиси бензоила заканчивается в большинстве случаев за 1—2 ч. Прп этом
вторичный атом водорода замещается хлором легче, чем первичный. Наличие гало-
генных заместителей затрудняет дальнейшее галогенирование и притом особенно
сильно у атома углерода, уже имеющего галоген. Ио мере удаления от этого
атома влияние галогенного заместителя ослабляется. Наличие двух илн трех атомов
хлора у одного и того же атома углерода препятствует дальнейшему свободнорадикаль-
ному хлорированию SO2CI2 таких соединений, как, например, хлороформ, тетра-
хлорэтая или хлористый бензилиден. В качестве растворителей для свободнорадикаль-
ного хлорирования парафинов и боковой цепи ароматических углеводородов с по-
мощью SO2C12 (см. стр. 146) пригодны, в зависимости от требуемой температуры
реакции, СП2С12 (40° С), СНС13 (61° С), СС14 (77° С), бензол (80° С), хлорбензол
(132° С) и о-цпхлорбензол (180° С).
Бромирование алканов представляет меньший интерес. Водород
метильной группы замещается бромом с наибольшим трудом, водород метинной группы
наиболее легко, так что, например, из 2,3-диметилбутана и брома на свету при комнат-
ной температуре легко получают 2,3-дйбром-2,3-дпметилбутдн [338]. О цепном меха-
низме термически или фотохимически инициируемого свободнорадикального броми-
рования см. [339].
[333] Е. Т. McBee, Н. В. Hass, Ind. Eng. Chem.. 34, 296 (1942).
[334] H. C. Brown, М. S. Kharasch, 'Г. Й. С n а о, I. Am. Chem. Soc.,.
62, 3435 (1940).
335] H.-J, Sc’hu m a c he r, J. Stauff, Angew. Chem., 55, 341 (1942).
336] M. C. Ford, W. A. Waters, J. Chem. Soc., 1951, 1851.
337] M. S. К harasch, H. C.Brow n, J. Am. Chem. Soc., 61, 2142 (1939).
338] A. V. G г 0 s s e, V. N. I p a t i 0 f f, J. Org. Chem., 8, 440 (1943).
339] M. S. Kharasch, W. s.Zimml, W. Nudenberg, J. Org. Chem.,,.
20, 1430 (1955). ‘
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА JJaJ ПЛИ SCN
С вопросом о ra ioronnpoBauuii алифатических углеводородов можно познако-
миться также в ряде более старых работ [340—344].
2. Замещение галогеном водорода в олефинах
Замещение водорода галогенами в насыщеипых углеводородах, как уже было
отмечено, не имеет столь большого препаративного значения, как замещение водорода
галогенами в углеводородах с С=С-связями.
Галогензамешенные олефины с галогеном у атома углерода, связанного
двойной связью, получают в препаративных синтезах и в промышленности отщепле-
нием галогена Х2 или галогеноводорода НХ от полигалогенироваппых углеводородов.
Для отщепления галогена применяют термические методы или взаимодей-
ствие с металлами (Fe, Mg, Zn, Al).
Например, 1,2-дихлорэтилен получают в промышленности отщеплением
хлора от 1,1,2,2-тетрахлорэтана с помощью Железных опилок и водяного пара
[345], дифтор-1,2-дихлор-зтален — из 1.2-дифтортетрахлорэтана с помощью
цинковой пыли в этиловом спирте [346, 347].
Отщепление галогеноводорода с образованием галогеноолсфинов проте-
кает по правилу Зайцева [343], по которому протон предпочтительно отщепляется
от менее гидрированного атома углерода. В препаративной химии и особенно
в промышленности значительную роль играют термическое и водно-щелочное
отщепление галогеноводородов [349]. Отщепление галогеноводорода поддействием
твердого едкого натра или едкого кали на примере получения 2,3-дибромпропи-
лена-1 из 1,2,3-трибромпропана см. [35OJ; возможно добавление индифферентных
растворителей (петролейный эфир, высококипящие алканы) |351]. В качестве
примера реакции отщепления галогеноводорода под действием водно-спиртового
NaOH можно указать на получение а-хлорстирола из 1,2-дихлор-1-фенилэтана;
выход около 90% от теоретического [352]. ₽ -Галогенстлролы получают с одно-
временным декарбоксилированием путем отщепления галогеноводорода от
а, р -днталогенгидрокоричиых кислот с помощью ацетатов щелочных металлов
в кипящем этиловом спирте или с помощью раствора соды [353]. В качестве
реагентов для отщепления галогеноводорода применяют также третичные амины;
пиридин, коллидип, хинолин, диалкиланилины или триэтиламин. Бромстероидьг
можно дегидробромировать в очень мягких условиях [354—355], для чего их
раствор в пиридине, содержащем 5—10% AgNO3, оставляют стоять при комнат-
ной температуре в течение 30—48 ч.
A. I). II е г z f с 1 de г, Вег., 26, 2432 (1893); 27, 489 (1894).
A. Michael, Вег., 34, 4037 (1901).
Н. Kronsteia, Вег., 54, 1 (1921).
Р. Wertyporoch, Вег., 66, 732,(1933); Ann., 493, 153 (1932).
F. Asinger, Ber., 75, 66 (1942).
[340
[341
[342
[343
[344
[345] Ullmanns Enzyldopadie der technischen Chemie, Bd. 5, 3 [Aufl., Miin-
chen — Berlin, 1954, S. 428.
[346] H. 8. Booth et al., J. Am. Chem. Soc., 55, 2231 (1933).
[347] J. C. Sauer, Org. Synth., 36, 1956, p. 19.
[348] A. Saytzeff, Ann., 179, 296 (1875).
,.[349] R. Stroh, в книге И о u b e п - W о у 1, Methoden der organisheH
Chemie, Bd. 5, T. 4, Stuttgart, I960, S. 731 — 742.
[350] R. Lespioau, M. Bourguel, Org. Synth Coll. v. 1, 1956,
p. 209.
[351] H. Baganz et al., Angew. Chem., 66, 307 (1954); Chem. Ber., 87,
1622 (1954); 89, 1560 (1956).
[352] W. S. E ш e r s о n, E. P. Agnew, J. Am. Chem. Soc., 67, 520 (1945).
[353] F. Straus, Ber., 42, 2878 (1909); H. В i 1 t z, Ann., 296, 266 (1897).
[354] E. Dane, Y. Wang \V. Schulte, Hoppe-Seyler’s Z. physiol.
Chem., 245, 80 (1936).
.[355] A. Bntenandt, G. Schramm, П. К и d s z и s, Ann., 531.
205 (1937).
136
ОБРАЗОВАНИЕ СВЯЗИ С —Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Методы непосредственного галогенирования олефинов в положение, соседние
с двойной связью, в основном разработаны за последние 20 лет. Аллилхлорирование
хлором уже упоминалось на стр. 94. Особенно важным для препаративных целей
является аллилбромирование (реакция Воля — Циглера). Этот способ был впервые-
описан Волей [356], который подвергал взаимодействию олефины с N-бромацетамп-
дом в эфире или ацетоне.
Способ был значительно улучшен Циглером [357]. проводившим реакцию*
с N-бромсукцинимидом:
ПгС-СО , г НгС-СО
I \ \ I I I \ \ I I
'NBr+\С=С-СН—> /NH— \С=С— СВг
П2С-СО 1 нас-со 1
N-Бромсукцинимид относительно стабилен, однако N-галогенамиды и имиды
карбоновых кислот, в том числе 1,3-дихлор-5,5-диметилгпдантоин, N-хлорсукцинимид
и N-бромсукципимид, термически лабильны и при нагревании могут разлагаться
со взрывом [358].
Аллилбромирование N-бромсукцинимидом является цепной радикальной реак-
цией, протекающей на поверхности кристаллов имида. При помощи бромсукципимида,
растворенного в тетрахлорэтане или нитрометане, бром присоединяется по С=С-связям
[365]. Поэтому применяют растворители, в которых N-бромсукцинимид и по возможно-
сти также и образующийся сукцинимид плохо растворимы. Предпочтительно употреб-
ляют сухой СС14. При работе с компонентами, обладающими значительной реакционной
способностью, применимы также циклогексан и бензол. Чем сильнее поляризована '
связь N—галоген в N-галогенированных соединениях, то есть чем сильнее положи-
тельно заряжен бром, тем легче происходит замещение атомов водорода по двойной
связи. Пространственное строение N-бромсукцинимида и почти неполярная связь.
N—Вг делают его особенно пригодным для свободнорадикального аллилбромп-
рования.
N-Бромпроиэводные могут действовать как окислители. Например, вто-
ричные спирты окисляются N-бромацетамидом и N-бромсукцинимидом в кетопы;
это свойство используется при синтезах стероидов [359]. При взаимодействии
первичных спиртов с N-бромсукцинимидом через альдегиды и полуацетали обра-
зуются эфиры, из меркаптанов — дисульфиды [365]. Качественное обнаружение-
и определение не вступившего в реакцию N-бромсукцинимида производят по-
реакции с подкисленным раствором KI, из которого при этом выделяется иод.
Описано хлорирование аллильных групп в ССЦ при помощи некоторых N-хлор-
ациланилидов с галогенными заместителями в ядре. Этот способ, однако, значительно-
хуже общепринятого метода аллилбромирования при помощи N-бромсукцинимида
[357]. N-Хлорсукцинимид применяют для аллилхлорирования только в некоторых,
специальных случаях, например для получения 5-хлорметилурацила или 6-хлор-
метил-2-метилтиоурацила из 5-метилурацила или 2,6-диметилтиоурацила в кипящем’
СНС18 с добавкой 15 мол. % перекиси бензоила [360].
Реакцию с N-бромсукцинимидом можно активировать термически, УФ-облу-
чепием [361, 362], добавкой инициаторов радикалов (перекись бензоила, динитрнл-
а,а'-азоизомасляной кислоты, диметиловый эфир а,а'-азоизомасляной кислотьр
[363, 364], или же путем увеличения поверхности кристаллов N-бромсукцинимида
нанесением его на нейтральную двуокись кремния [365].
[356] A. Wohl, Вег., 52, 51 (1919); 54, 476 (1921).
[357] К. Z i е g 1 е г et al., Ann., 551, 80 (1942).
[358] R. H. Martin, Nature (London), 168, 32 (1951).
[359] L. F. Fieser, S. R a j ago pal an, J. Am. Chem. Soc., 72, 5530 .
(1950).
[360] R. A. West, H. W. Barrett, J. Am. Chem. Soc., 76, 3146 (1945).
[361] M. S. Kharasch, R. M aloe, N. C. Y a n g, J. Org. Chem., 22, 1443
(1957).
[362] J. C.Martin, P. D. В а г 11 e 11, J. Am. Chem. Soc., 79, 2533 (1957)..
[363J H. Schmid, P. К a г г e r, Helv. chim. acta, 29, 573, 1965 (1946).
[364] M. C. F о г d, W. A. W a t e г s, J. Chem. Soc., 1952, 2240.
[365 J L. Horner, E. ,H. Winkelman n, Angew. Chem., 71, 349 (1959).
I. ЗАМЕЩЕНИЕ ВОДОРОДА ИЛ Hal ИЛИ SCN
137
При аллилбромировании с помощью N-бромсукцинимида необходимо учиты-
вать протекание побочных реакций
;С —С—CHBrR
ВгС—t—CHR
)
которые можно затормозить ведением реакции при низких температурах ц активирова-
нном, но полностью подавить их не удается.
Аллильной перегруппировке (ср. стр. 849) первичных продуктов бромирования
способствует стремление к сопряжению с соседней кратной связью, например, если
R является >С=С<Г, —С=С—, —C=N, >С=О или СвН6.
Это особенно характерно для олефинов с кокцевой С—С-связью или диолефинов
с изолированными С=С-связями. После аллильной перегруппировки может происхо-
дить вторичное аллилбромирование, а также отщепление НВг, бели оно приводит
к дальнейшему сопряжению или ароматизации. Отщепившийся НВг вытесняет из
N-бромсукцинимида бром, который может присоединяться по. С=С-связям. Много-
численные литературные данные по этому вопросу см. [365].
Для аллилбромирования моно-, ди- и алициклических олефинов действительны
следующие закономерности [357, 365]. Без активаторов метиленовые группы брони-
руются легче, чем метильные, а последние немного легче, чем метиновые. Олефины
с прямой и разветвленной цепью могут бронироваться под действием N-бромсукцин-
пмида с замещением только одного атома водорода в каждом аллильном положения.
В циклических олефинах возможно двукратное бромирование по каждому аллиль-
ному положению, однако в результате аллильных перегруппировок и отщепления НВг
происходит дальнейшее превращение таких продуктов. Например, циклогексан
превращается в м- и п-дибромбепзолы [366]. Если на одну С=С-связь приходятся
четыре аллильные группы, то возможно, как, например, в случае тетраметилэтилена,
четырехкратное аллилбромирование.
Вторичное бромирование многоядерпых циклических олефинов всегда происхо-
дят в то же кольцо, а именно по второму, еще свободному аллильному положению.
Если это положение занято, бромируются аллильные положения других циклов.
Бромирование веществ, растворяющих N-бромсукцинимид, затрудняется при
наличии заместителей, стерически блокирующих аллильную группу, а также в тех
случаях, когда угол между С=С-связыо и аллильной группировкой сильно отличается
от 120° или в случае поляризации аллильного положения нитро-, циан-, сульфогруп-
пами или С—С-связью.
Аллилбромированию с помощью N-бромсукцинимида хорошо поддаются не
только олефины, но и а,0-непасыщенные кетоны, эфиры карбоновых кислот, нитрилы
п лаптопы. При аллилбромировании сопряженных диенов, бромировании боновой
цени ароматических и гетероциклических соединений (см. стр. 186), а также при
замещении третичного атома водорода у С=С-связи бромом с помощью N-бромсукцин-
пмпда необходимо добавлять инициаторы радикалов.
N-Броисукцинимид [365] (ср. также [357]). В охлаждаемой смесью
льда с водой колбе для сульфирования (или подобной колбе с широким горлом)
с механической мешалкой растворяют 50 а сукцинимида (т. нл. 125—126° С)
в растворе 20 г NaOH в 100 мл воды. (Сукцинимид, регенерированный из
N-бромсукцинимида, предварительно перек ристаллизовывают из двойного количе-
ства этилового спирта с активным углем.) В колбу добавляют 100 е измельчен-
ного льда и при энергичном перемешивании в один прием приливают 27 мл
, брома, перемешивают 10 мин и быстро отсасывают. Бром необходимо возможно
быстрее суспендировать в смеси, так как N-бромсукцинимид тотчас же выпадает
в виде плотной кристаллической кашицы.
Рекомендуется [367] также прибавлять бром в виде раствора в равном
количестве СС14.
Полученный N-бромсукцинимид один-три раза замешивают в нашицу
ледяной водой и быстро отсасывают так, чтобы последняя промывная вода не
содержала брома. Далее N-бромсукцинимид высушивают в эксикаторе сначала
|3lifi] R. А. В arnes, J. Am. Chem. Soc., 70, 145 (1948).
't367] E. Campaign e, B. F. T u I I a r, Org. Synth., 33, 1953, p. 97.
138
РАЗОВЛНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
над NaOH, а затем над Р2О5; выход около 70 г, что составляет 98% от теоретиче-
ского; т. пл. 174—175Q С, содержание активного брома 44,9%.
N-бромсукцинимид можно перекристаллизовать почти без разложения,
для чего его растворяют в десятикратном количестве воды при 75—80° С, быстро
отфильтровывают и охлаждают льдом (т. пл. 176—177° С); из нитрометана он
кристаллизуется без разложения. Перекристаллизованный N-бромсукцинимид.
менее активен, чем неочищенный промытый продукт.
Адлилбромирование с помощью N-бромсукцинимида. Обычно к раствору
безводного жирно-ароматического олефина в безводном СС14 добавляют сухой
N-бромсукцинимид и активатор и, поместив в баню и изредка встряхивая, нагре-
вают примерно до 80° С (колба должна быть снабжена эффективным обратным
холодильником с осушительной трубкой). Вещества с большой реакционной
способностью приливают к суспензии N-бромсукцинимида в СС14 по каплям при
комнатной температуре и перемешивании; с легко летучими олефинами можно
работать при более низких температурах, используя УФ-облучепие. Применять
только фотоактивированио, однако, но очень целесообразно, так как реакция
протекает медленно. Олефины обычно можно сушить перегонкой над натрием;
технический СС14 кипятят в течение суток с обратным холодильником над пяти-
окисью фосфора и перегоняют на колонке. Соотношение олефин : СС14 соответ-
ствует интервалу от 1 : 10 до 1 : 20; соотношение N-бромсукцинимид : СС14
равно 1 ' 4. При аллплбромировании веществ с малой реакционной способно-
стью применяют меньшие количества растворителя; жидкости можно брониро-
вать и без растворителей.
Очень реакционноспособные соединения реагируют с N-бромсукцинимидои
в отсутствие катализатора; в остальных случаях добавляют инициаторы радикалов.
В качестве инициаторов (активаторов) применяют часто дипитрил ajx'-азоиэомасляной
кислоты (1 ч. на 100—1000 ч. N-бромсукцннимида) и перекись бензоила (1 ч. и больше
на 100 ч. бромсукцинимида).
а ,а '-Азоизомасляная кислота. Получают взаимодействием бромной воды
с динитрилом а,а'-гидразоизомасляной кислоты, который синтезируют из аце-
тона, K.CN и гидразинсульфата в горячей воде [368, 369].
Температура разложения перекиси бензоила выше, чем температура разложе-
ния азоизомасляной кислоты, и примерно на 30° С выше температуры кипения СС14.
Реакции с N-бромсукципимидом в присутствии перекиси бензоила трудно
поддаются управлению, и поэтому ее рекомендуется применять при работе
с малореакционноспособпыми веществами, главным образом с жирно-ароматическими
соединениями. Активатор можно смешивать с N-бромсукцинимидом (не растирать!)
или добавлять растворенным в небольшом количестве хлороформа. Большие коли-
чества перекиси бензоила нельзя нагревать с хлороформом!
Особеппо действенны окислительно-восстановительные катализаторы [365].
К реакционной массе добавляют на холоду несколько капель примерно 60%-ного
раствора отрсот-бутилгидроперекиси в метилфталате и затем при 70’ С 1—2 капли
концентрированного хлороформного раствора лаурината кобальта (II) или меди (II).
Реакция начинается тотчас и протекает спокойно.
При достижении 80° С начинается бромирование, сопровождающееся
выделением тепла. Охлаждение реакционной массы нежелательно, но на случай
бурного развития реакции нужно иметь паютове воду со льдом для периодиче-
ского охлаждения. Если начало процесса задерживается, смесь нагревают в те-
чение нескольких минут при 80’ С или добавляют еще активатор. В большинстве
случаев реакция закапчивается за несколько минут (при загрузках до несколь-
ких сот граммов). Если реакция продолжается при 80° С более 1 ч, то это ука-
зывает, особеппо прп работе с олефинами, на протекание побочных реакций.
Окончание реакции определяют по исчезновению тяжелого осадка N-бромсук-
цинимида па дне колбы и образованию слоя более легкого сукцинимида на по-
верхности жидкости. Если нужно, разбавляют концентрированную массу не-
[368] J. Thiele, К. Heuser, Ann., 290, 30 (1896).
[369] A. W, Dox, J. Аш. Chem. Soc., 47, 1473 (1925).
I. ЗАМЕЩЕНИЕ ВОДОРОДА HA Hal ИЛИ SCN
139
большим количеством СС14 и прерывают нагревание. При получении многократно
бронированных соединений их отсасывают в горячем виде, в остальных слу-
чаях — после предварительного охлаждения. Сукцинимид промывают СС14,
который удаляют из фильтрата в вакууме; остаток перекристаллизовывают или
фракционируют в вакууме.
Следует указать еще две обзорные работы, посвященные реакциям с N-бром-
«сукцинимидом [365, 370]*.
3. Общие положения о замещении галогеном водорода
в ароматических соединениях
Прямое замещение водорода галогеном в ароматических соединениях (гало-
тенировапие в ядро) является препаративно важной реакцией. Конкуренция реакций
присоединения и замещения галогеном в ароматических соединениях рассмотрена
на стр. 104. Фтор целесообразнее вводить в ядро и в боковую цепь путем замены
С—N-связи (стр. 253) или другого галогена (стр. 191) на фтор. Поэтому ниже будут рас-
сматриваться только хлорирование, бромирование и иодирование ароматических сое-
динений. Так же как нитрование и сульфирование, галогенирование в ядро является
реакцией электрофильного замещения. Реакция ускоряется под влиянием растворите-
лей с высокой диэлектрической проницаемостью (на пример, нитробензола, ледяной
уксусной кислоты) и действующих поляризующе катализаторов (железные оиилки,
галогениды железа, алюминия, цинка, сурьмы, а также иод). В этих условиях жирно-
ароматические соединения, у которых галогенируется также и боковая цепь, можно
галогенировать при низких температурах (10—20° С) преимущественно в ядро.
Легкость реакции галогенирования в ядро ароматических и гетероциклических
соединении, обладающих свойствами ароматических, в большой мере зависит от
имеющихся в молекуле заместителей. Если заместителями являются галогены
я группы —СООН, —SOsH п —N02, то, как правило, галогенирование надо вести
в присутствии катализатора. Наличие, например, групп —ОН, —О-алкил и —NHa
настолько благоприятствует галогенированию в ядро, что реакция протекает и без
катализатора.
В замещеппых бензолах заместители влияют на скорость и направление гало-
генирования в ядро. Заместители первого рода, такие, как —Вг; —CI; —F; —СН3;
—NHC0CH3; —ОСН3; —ОН; —NHS и—N(ClI3)2, расположенные в ряд по возраста-
ющей (но сравнению с водородом) скорости хлорирования и бромирования, являются
орто- и преимущественно пара-ориецтантамп. Заместители второго рода: —СНО,
—СОВ, — СООС2Н5, — С001Г, —NO2, — SO3H, CF3 ориентируют галоген в мета-поло-
жение и снижают скорость галогенирования [371]. Фтор-, хлор- и бромбензолы бро-
мируются и газовой фазе при 400—450ь С преимущественно в мета-, а не в орто-
•и пара-положения. В присутствииFeCl3 температура, при которой происходит инверсия
направления замещения, выше, а при УФ-облученпи ниже, из чего можно сделать
вывод о свободнорадикальном механизме мета-бромирования галогенбензо-
лов [372].
Для галогенирования ароматического -соединения с заместителями первого
рода в мета-, а с заместителями второго рода в орто- и лара-положения, следует при-
менять в общем случае косвспные способы.
1,3,5-Трибромбензол [373]. Его получают из 2,4,6-триброманилина,
, который диазотируют и кипятят с этиловым спиртом. 2,4,6-Триброманилин легко >
* См. также И. В. М а ч и и с к а я, В. Л. Б а р х а ш, Реакции и методы иссле-
дования органических соединений (РиМИОС), Госхимиздат, 9, 287 (1959).—
Нри.ъ. ред.
1370] С. JJjerassi, Chem. Bev., 43, 271—317 (1948).
1371] P.W. Robertson, P.B.D.de la Маге, B. E.Swedlund, J.
Chem. Soc., 1953, 782.
[372] J. P.Wibautet al., Bee. 1rav. chim., 56, 815 (1937); 69, 1031 (1950).
(373] G. H. Colom an, W. T. T a 1 b о t, Org. Synth,, Coll. v. 2, 1943,
p. 592.
140
ОБРАЗОВАНИЕ СВЯЗИ С—На] В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
синтезируют, пропуская воздух, насыщенный парами брома, через водный рас-
твор гидрохлорида анилина.
2,4,6-Трибромбензойная кислота [374]. Ее получают аналогичным нуте»
из 3-амино-2,4,6-трибромбензойной кислоты, в которой диазотированием в при-
сутствии гипофосфористой кислоты обменивают аминогруппу на водород.
Роль катализаторов при упомянутом выше электрофильном галогенирована»;
ядра галогенами заключается в образовании катиона Х+:
Br2 + FeBr3 Br+(FeBr4)'
Другой метод получения галогеикатионов Х+ описан в работах Биркеабаха^
Губо, Уотерса [375—377]. При взаимодействии эквимолекулярных количеств иода,
(брома) с ацетатом ртути или ацетатом, трихлорацетатом, перхлоратом, сульфатом^
или трифторацетатом серебра в отсутствие воды в реакционной смеси в качестве^
промежуточных продуктов образуются ацилгипогалогениты:
AgC1044-I3 —> Agl-f-IClOi
RH-f-IClOi —> RI-f-HClOi
CF3COOAg + I2 —► AgI-f-CF3C00I
RH + CF3COOI —> RI + CFgCOOH
По этому способу можно бронировать и иодировать незамещенные ароматику
ческие соединения (стр. 144) и соединения с заместителями, ориентирующими в орто-
и пара-положения (стр. 156) и, прежде всего, в мета-положение (стр. 173). Описан»; >
также некоторые аналогичные методы хлорирования [378]. Эти методы применим»)
в безводной и в водной средах. Часто работают и без растворителей.
Перхлорат серебра применяют в сухом виде или в эфире или бензоле с добав-4
кой CaO, MgO или СаСО3 для связывания образующейся хлорной кислоты. Пример*^
такого галогенирования приведен на стр. 145. ’Й
Для ацетата серебра в качестве растворителя пригодна ледяная уксусная м
кислота, для трифторацетата серебра — СС14 и до 150° С — нитробензол. Как конку-^
рирующий процесс может протекать реакция Бородина * — Хунсдикера [ 379] (стр. 819); :*
при совместном нагревании иода, трифторацетата серебра и нитробензола в отсутствие---з
способного иодироваться вещества образуется трифториодметан: о;
CF3COOAg + I2 —>- CF3l4-AgI-f-COa &
Получение трифторацетата серебра из окиси серебра и трифторуксусной
кислоты в водной среде и очистка сырого продукта (остатиа после выпаривания.^
воды) экстракцией эфиром см. [380]. ..0
Трифторацетат серебра обладает преимуществами перед остальными солямОД;
[381а]. Он лучше всего растворим в органических растворителях (например, в бензоле^
и эфире); выделяющаяся трифторуксуцная кислота (т. кип. 71,5fl С) легко отгоняется;^
не образуется опасная хлорная кислота, как при работе с перхлоратом серебра. При- >
мер использования трифторацетата см. на стр. 156. v
374
375
376
377
379
380]
381]
Эта реакция открыта А. П. Бородиным еще в 1861 г. [Ann. Chem. u. Pharm.,..
119, 123 (1961)].—Прим. ped.
M. M. R obison, В. L. Robison, Org. Synth., 36, 1956, p_. 94.
L. Birckenbach, J. Gou beau, Ber., 65, 395 (1932); 66, 1280 (1933)..,
D. H. Derbyshire, W. A. W at ers, J. Chem. Soc., 1950, 573, 3694—
I. R. L. Barker, W. A. Waters, J. Chem. Soc., 1952, 151.
J. H. Gorvin, J. Chem. Soc., 1953, 1237.
С. V. W 11 s 0 n, Org. Reactions, 9, 332 (1957).
D. E. J a nss e n, С. V. Wil s on, Org. Synth., 36, 1956, p. 46.
R. N. Haszelaine, A.- G. S h a r p e, J. Chem. Soc., 1952, 993 (a), 1000 (b)-
1. замещение водорода на Hai или son
14Г
Для галогенирования соединений, содержащих СООН-группы (например,,
бензойная [382] или жирно-ароматические кислоты |383]), их сухие соли серебра
(0,05 моль) в сухом СС14 обрабатывают бромом или иодом.
В водных растворах галогенов содержится чрезвычайно мало катионов X*..
Для того чтобы сдвинуть вправо равновесную реакцию
Н2О-ЬВгг НОВг-|-Н+Ч-Вг-
добавляют соли серебра или ртути (II), или КВгО3, связывающие Вг_:
5Н+4-5Вг_4-НВгОз—> ЗВга-}-ЗН2О
При одновременном добавлении минеральных кислот (пригодна и 75%-ная1
уксусная иислота [384]) бромноватистая кислота становится сильным бронирующим
агентом:
Н++НОВг [НаОВг]+
Те же рассуждения относятся и к иодированию (примеры см. на стр. 144
и 171).
В то время как бромирование и иодирование ядра по методу Биркеибаха —
Губо — Уотерса происходят в примерно одинаковых условиях, почти все остальные-
способы иодирования ядра ароматических соединений отличаются от обычных методов-
хлорирования и бромирования, так как реакция иодирования иодом обратима:
RH-f-I2 RI-f-HI
При этом образующийся иодистыи водород целесообразно удалять следующими’'
способами;
а) путем солеобразования (добавлением гидроокисей, карбонатов, бикарбона-
тов щелочных металлов, буры J385], окиси ртути, ацетата ртути (II), аммиака, алифа-
тических аминов);
б) с помощью окислителей (НаО2, Na2S2O8, Н1О3 или MeIO3, HNOS, Н28О4,_
или олеума), которые вновь приводят к выделению иода из HI.
По окончании реакции избыточный иод удаляют пропусканием S02
или встряхиванием с растворами гидросульфита или тиосульфата натрия; иногда^
также применяют раствор K.I или ртуть. Кроме воды в качестве растворителей^
применяют ледяную уксусную иислоту, спирты, бензоя, реже эфир и СС14.
Довольно широко испольэуется-иодирование в присутствии Na2S2O8, Н108
или HN03; реагенты, перечисленные в пункте а, как и перекись водорода,,
рименимы тольио для иодирования таких ароматических или гетероцикличе--
ских соединений, строение которых благоприятствует электрофильному замеще--
нию; многочисленные примеры см. на стр. 144, 152, 171сл., 187.
Для свободнорадикального хлорирования и бромирования боковой цепи, еслвг
растворители вообще применяются, используют неполярные растворители CS2, СС14г
менее пригоден бензол.
Уже незначительные количества серы в CS2 или СС14 заметно замедляют
бромирование боковой цепи толуола [386] иа свету и при 57° С. В иесиольио
раз обработанном встряхиванием с ртутью и перегнанном CS8 бромирование боко-
вой цепи происходит значительно быстрее. Хлороформ является не очень хоро-
шим растворителем при бромировании, так как при 57’ С ои уже через несколько
минут вступает в реакцию с бромом. Однако хлороформ (75 лл), десять раз про-
мытый водой (по 50 мл), высушенный и перегнанный, устойчив к действию-,
брома [387].
[382] К. Birnbaum, Н. Reinherz, Вег., 15, 456 (1882).
[383] D. Papa, Е. Schwenk, Е. Klingsberg, J. Аш. Chem. Soc., 72^.
2623 (1950).
[384] S. J. Branch, В. J ones, J. Chem. Soc., 1954, 2317.
[385] A. Classen, W. Lob, Ber., 8, 1605 (1895).
[386] J. B. Sampley, E. M. H icks, J. Am. Chem. Soc., 62, 3252 (1940).
[387] J. R. Sa mey, F. S. Fa wee tt, B. A, Morehead,' J. Am. Chem-.
Soc., 62, 1839 (1940).
142
ОБРАЗОВАНИЕ СВЯЗИ С —Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Галогенированию боковой цепи способствует нагревание при солнечном свете,
УФ-облученпе или освещение сильной нематовой лампой (200—.300 вт), а также
добавление инициаторов радикалов. Прибавление 0,03 моль перекисного соединения
(аскаридол, перекись бензоила) на 1 моль брома вызывает при 25° С в темноте уже
через 25 мин бромирование толуола на 85%, причем образуется бензилбромид [388];
выход 98%. Большие концентрации НС1 или НВг в реакционной смеси оказывают
неблагоприятное действие на реакцию.
Галогенирование боковой цепи метилированных пли более высокоалкилирован-
иых ароматических соединений но а-углероду протекает аналогично аллилгалогени-
рованию олефинов (стр. 94 и 136). Поэтому понятно, что N-бромсукцинимид может
применяться также для бромирования боковой цепи. Но и то время как действуя
хлором или бромом на метилароматические соединения, можно заместить галогеном
все три атома водорода в боковой цепи, с помощью N-бромсукципнмида удастся ввести
в боковую цепь только два атома брома. Для замещения одного а-атома водорода
бромом применяют не более 1 моль N-бромсукцинимида на каждую метильную группу,
для замещения двух а-атомов — 2 моль реагента. В большинстве случаев в качестве
активатора добавляют перекись бензоила; реакция продолжается 5—60 мин (стр. 138).
Таким способом получают, например из толуола бензилбромид [363, 389],
из 1- и 2-метилпафталина — соответствующие ороммстилсоединенпя [390, 391 ]; дальней-
шие примеры см. [365].
В тех случаях, когда условия благоприятствуют галогенированию в ядро,
бромирование ядра происходит также под действием N-бромсукцинимпда. Аромати-
ческие углеводороды с конденсированными ядрами, например иафталин, антрацен
и фенантрен [392], а также такие феноловые эфиры, как 1- и 2-метоксинафталин,
вератрол, диметиловыс эфиры резорцина и гидрохинона |392], триметиловыи эфир
пирогаллола [393], бромируются в ядро под действием N-бромсукципимпда без ката-
лизатора. Из пирокатехина и 2 моль N-бромсукцинимида образуется 4,5-днбромпиро-
катсхин, из резорцина и 3 моль N-бромсукципимида — 2,4,6-трибромрезорцин [394,395],
из антраниловой кислоты, а также из 2- или 4-окснбспзойпой кислоты и 2 моль
N-бромсукцинимида — 4,5-дибром- или 3,5-Дибромпроизводиыс [394—396]. Бензол
и толуол бромируются N-бромсукцинимидом в ядро только в присутствии эквимоле-
кулярных количеств A1CI3, ZnCl2, FeCl3 или JI2SO4.
4. Замещение галогеном водорода в ядре ароматических углеводородов
Для галогенирования в ядро чаще всего применяют хлор, который, в зависи-
мости от обстоятельств, можно разбавлять двуокисью углерода. Хлорбензол получают
с обычными переносчиками галогена ([2, Л1С13, FeCl3). Несмотря на то что при хлори-
ровании ппф|алина получается главным образом 1-хлорпафталпп, его трудно отделить
от образующегося в качестве побочного продукта 2-хлорнафталина, п поэтому 1-хлор-
нафталин предпочитают получать по Зандмейеру из 1-аминонафталипа (а-нафтил-
амина). Чистые орто- и пара-галогентолуолы также лучше готовить из соответствующих
толуидинов.
Вместо хлора можно применять также SO2C12 (стр. 150) или хлор в момент
выделения, который для препаративных целей получают из хлористого водорода
и перекиси водорода (стр. 157), а в промышленности — из хлористого водорода на
контакте, содержащем соли меди. Под действием хлорамина Т (/i-CH3C5H4SO2NC13)
в растворе хлористого водорода в ледяной уксусной кислоте 2-метилмезобснзаптроп
хлорируется в ядро с образованием З-хлор-2-метилмезобензантрона [397]. При кипя-
[388] М. S. Kharasch, J. Am. Chem. Soc., 59, 1405 (1937).
[389] II. Schmid, Helv. chim. acta, 9, 1144 (1946).
[390] Ng. Ph. В u u - H о i, J. Lecocq, J. Chem. Soc., 1946, 830.
[391 j N. B.Chapma n, J. F. A. William s, J, Chem. Soc., 1952, 5044.
[392] Ng. Ph. Buu-Uoi, Ann., 556, 1 (1944).
[393] Ng. Ph. Buu-Hoi, Rec. trav. chim., 73, 197 (1954).
|394] W. I.Bailey, J. В e I 1 o, J. Org. Chem., 20, 525 (1955).
[395] M. F. Abdel -Wahab, M. Z. В araka t, Mh. Chem., 88, 692 (1957).
[396] M. Z. В a r a k a t, M. F.Abdel-Wabab, J. Am. Chem. Soc., 75, 5731
(1953); 77, 1670 (1955).
1397] D. H. II e у, II. I. N icholls, C. W.Pritchett, J. Chem. Soc., 1944, 97.
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
143
чении бензтриазола в течение Зч с обратным холодильником в царской водке полу-
чается 4,5,6,7-тетрахлорбензтриазол с 87%-ным выходом от теоретического [398].
Обычно монобромирование проводят, прибавляя по каплям 0,8—1 моль
брома при перемешивании и охлаждении льдом к 1 моль углеводорода с добавкой
переносчиков галогена и выдерживая смесь 12 ч. Затем для завершения реакции
мржно некоторое время выдерживать реакционную массу при 50° С или сразу же
промыть ее водой и разбавленной щелочью. Если возможно, продукт очищают пере-
гонкой с водяным паром. Бромирование бензола бромом в присутствии железных
опилок подробно описано Гаттерманом [399]. Перед началом бромирования полезно
вводить в углеводород сухой бромистый водород [400].
В качестве еще одного примера галогенирования следует указать на бро-
мирование о-ксилола до 4-бром-о-ксилола бромом в присутствии иода или желез-
ных опилок и иода [401].
Некоторые ароматические углеводороды легко бронируются в ядро в индиф-
ферентных растворителях (CCL или CS2) при нагревании без катализатора при
добавлении 10%-ного избытка брома. Это относится, например, к меэитилопу [402],
нафталину [403], антрацену (образуется 9-бром- [404] или 9,10-дибромантрацея
[4051) и фенантрену [406] (образуется 9-бромфенантрен). Мезитилен, который может*
также бронироваться в боковую цепь, бронируется в ядро в СС14 при 10—15° С.
Продукт бромирования боковой цепи, получающийся наряду с примерно 80% бром-
мезитилена, удаляют кипячением в течение 1 ч неочищенного продукта бромирования
с раствором этилата натрия в этиловом спирте.
Хлормезитилен и другие монохлорполиалкилбензолы тоже получают хлориро-
ванием в СИС13 при 0’ С без переносчиков [407].
4,4'-Днбромдифенил. Получается с выходом 75—77% даже при выдер-
живании в течение 8 ч тонкоизмельченпого дифенила (0,1 моль) и броня
(0,24 моль) в вакуум-оксикаторе, кран которого немного приоткрыт, для того
чтобы НВг мог улетучиваться [408].
Если при охлаждении льдом к 7 мл брома и 0,-2 г железа в порошке добавить
1 мл алкилбепзола в течение 1 ч и перемешивать еще 1 ч, то получается полибром-
соединение, в котором вторичный и третичный алкилы замещены бромом, а первичный
алкил не затронут [409].
В качестве призера бромирования ароматического углеводорода по способу
Уотерса приведено получение бромбензола.
Бромбепзол [376]. В эмульсию, приготовленную из 0,25 моль бензола,
1 л 2 н. HN03 и 0,125 моль (20 г) брома, при комнатной температуре в течение
90 мин приливают по каплям при перемешивании раствор 0,125 моль AgNO,
в 100 мл воды; выход бромбензола 80% от теоретического.
[398] R. Н. W i 1 е у, К. Н. Н и s s u п g, J. М о 11 a t, J. Am. Cbem. Soc., 77,
5105 (1955).
[399] L. Ga t te r m ann, Die Praxis des organischen Chemikers, 37 Aufl., Berlin
1956, S. 95.
1400] Пат. США 2659760 (1950/53); С., 1955, 5897.
[401] W. A. W i s a n s k y, S. A n s Ь a c h e r, Org. Synth., Coll. v. 3
1955 p 138
1402] L? J.Smit’h, F. II. M c D о u g a 11, J. Am. Chem. Soc., 51, 3002 (1929>
L. I. Smith, Org. Synth., Coll. v. 2, 1943, p. 95.
I403J F. F. В 1 i c k e, J. Am. Chem. Soc., 49, 2846 (1927); H. T. Clarke, M. R.
Brothen, Org. Synth., 10, 14 (1930); Coll. v. 1, 121 (1941).
[404] E. do В a г г у Barnett, J. W. Cook, J. Chem. Soc., 1924, 1086.
[405] 1. M. П о i 1 fi г о и, J. 8. Hoaton, Org. Synth., 3, 41 (1923).
1406] C. A. Dornf eld, J. E. Callen, G. H. Cole ma n, Org. Synth., Coll
v. 3, 1955, p. 134.
[407] L. I. S m i t n. C. L. M о у 1 e, .1. Am. Chem. Soc.. 58, 7 (1936).
[408] R. E. Buckles, N. G. Wheeler, Org. Synlh., 31, 29 (1951).
[409] G. F. 11 enn i он, J. G. Anderson, J. Am. Chem. Soc., 68, 424 (1946^
144
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Аддукт диоксана с бромом (см. стр. 158) так?Ке пригоден для введения брома
и ядро [410].
4-Бромтолуол. Смешивают 9,5 г толуола с диоисандибромидом (из 15,1 а
брома) и нагревают до 35—40° С; смесь быстро обесцвечивается. После разба-
вления 10%-ным раствором NaOH получают 16,8 г 4-бромтолуола. Аналогично
монобромируются нафталин в положение 1, антрацен в положение 9 и флуорен
в положение 2.
Бромид иода 1Вг в ледяной уксусной кислоте, в СНС13 и даже в нитробензоле
[411 ] является мягким бронирующим агентом и рекомендуется для получения 4-бром-
1-нафтола, а-бромнафталина [412] и 4-бром-1-метоксинафталина [413]. Бромид иода
приготовляют из эквимолекулярных количеств иода и брома в используемых для
реакции растворителях (ледяная уксусная кислота, СНС13).
Для прямого введения иода в ядро ароматических углеводородов из упомянутых
на стр. 141 методов обычно могут быть использованы только те, в которых реакция
проходят особенно активно. Так, в наиболее пригодном для бензола и алкилбензолов
сильно экзотермичном процессе иодирования [414] при помощи иода и HNO5 (d — 1,5)’
в качестве побочных продуктов получаются фенолы и нитросоединения. Фенолы
удаляют встряхиванием неочищенного продукта реакции с 10%-ным раствором NaOH,'
нитросоединения — восстановлением железными опилками и НС1; иодароматичеоки®
соединения отгоняют с водяным паром из подкисленного раствора (индикатор конго}.
В перемешиваемую, нагретую на водяной баие до 50° С смесь 1,5 люль
иода и 5,1 моль бензола по иаплям вводят в течение примерно 1 ч 275’жл HNOj
(d — 1,5). Выделяются значительные количества окислов азота, и смесь постер
пенно разогревается до кипения. Подробные данные о получении иодбензола*
см. [415] и 2-иодтиофена см. [416].
Можно использовать не только систему иод — азотная кислота, по и проводить-'
иодирование вещества в смеси ледяной уксусной кислоты, концентрированной H2SO4r
1 моль иода и NaNO2 [417]. При этом особое значение имеет качество нитрита натрия.'
Рекомендуется спекшийся нитрит измельчать в ступке так, чтобы получались кусин*
объемом не более 1 см2. Вместо азотной кислоты употрсбимы также нитрозилсернай*
кислота и нитрующая смесь [418].
Персульфат натрия Na2S2O8 также может применяться в качестве окислителя^
Hi, образующегося при прямом иодировании алкил- и арилбензолов [419J:
2RCeH6-f-l2 + Na2S2O8 —► 2RCeHJ4-2NaHSO4
Растворителем служит ледяная уксусная кислота, к которой для растворений^
Na2S2O8 добавляют 10—20 объемп. % воды; иод и Na2S2O8 вводят в стехиометриче*-
-скпх количествах [420а]. "
[410] А. П. Т о р е и т ь е в, Л. И. Б е л е н ь к п й, Л. А. Я н о в с к а я, ЖО X
24, 1265 (1954).
[411] F. W. В е n n е t t, A. G. S h а г р е, J. Chem. Soc., I960, 1383. ч
[412] W. Militzer, J. Am. Chenf. Soc., 60, 256 (1938).
[413] E. C. S p a e t h, . T. A. Geipman, T. L. J scobs, J. Org. Chem., 11, -
399 (1946). J
1 [414] R. L. D a t t a, N. R. Cha tterjee, J. Am. Chem. Soc., 39, 437 (1917);“*
41, 292 (1919 . -
[415] F. B. Dains, R. Q. Brewster, Org. Synth., 9, 46 (1929); Collzi/
v. 1, 323 (1947). .
‘[416] H. Y. Lew, C. R. Noll er, Org. Synth., 30, 53 (1950).
[417] ?J. T. P 1 a t i, W. H. Strain, S. L. W а г г о n, J. Am. Chem. Soc., 65,
4 274 (1943). ' .?
[418] P. S. Varma et al., J. Indian Chem. Soc., 12, 343 (1935); 14, 156 (1937);
А. Л. I{ о в и к о в, ЖОХ, 24, 655 (1954).
[419] К. Е I b я, A. J а г о s 1 a w z е w, J. prakl. Chem. [2], 88, 92 (1913).
Д420] Н. О. W i г t h, * О. Konigstein, W. Kern, Ann., 634, 84(a), 87(b),
96(c), 100(d) (1960).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
145
4-Иоддифенил [420с]. Раствор 15,4 г дифенила в 100 мл ледяной уксус"
пой кислоты при 80° С разбавляют до помутнения водой (около 25 мл) в доба-
вляют 12,7 г иода, 12,5 г Na2S2O8 и 8 мл СС14. Последний служит для смывания
из холодильника возгоняющегося иода. Смесь интенсивно перемешивают до
исчезновения окраски иода (приблизительно Зч). Добавляют 300 мл воды,
отсасывают выделившийся в осадок продукт и высушивают. Образовавшийся
4-иоддифенил (т. иип. 118—125° С при 0,05 мм рт. ст.) перегоняют вместе
с непрореагировавшим дифенилом. После перекристаллизации из метилового-
спирта получают 17,4 г (62% от теоретического) 4-иоддифенила; т. пл. 112° С.
Довольно широко используются способы иодирования иодом и йодноватой
кпслотой или иодатами ]420Ь](см. также стр. 156). Как растворитель пригодна ледя-
ная уксусная кислота, которая до 80° С пе реагирует с йодноватой кислотой.
При 100® С она разлагается с выделением двуокиси углерода. Для повышения раство-
римости можно применять нитробензол, который иодируется только при 120* С.
Чтобы растворить нодноватую кислоту, необходимо добавлять около 10 объеми. %
поды. На 2 моль иода берут 1.1—1,15 моль йодноватой кислоты, т. е. 10—15%-яый
избыток. Иодируемое ароматическое соединение вводят с небольшим избытком:
5КСвН6-Ь2124-ШОз —*• 2RCeH4l + 3H2O
Иодирование системой 12—Н1О3 пли с добавкой Na2S2O8 является реакцией,
катализируемой кислотами. Поэтому в большинстве случаев к смеси добавляют
3 объема концентрированной H2SO4. Реакция акзотермична п при 80—85° С закан-
чивается через 2—3 ч.
Иодбензол [420d]. Смесь 25 г беплола, 80 мл ледяной уксусной кислоты,
15 мл 50%-ной H2SO4, 24,4 е иода и 9,5 <? ШОа нагревают 1 ч при перемешивании
до 90° С, затем перемешивают при 95° С до полного израсходования иода (2 ч).
Выделенный водой продукт реакции растворяют в 50 мл СИС13, обрабатывают
раствором NaHSO3, высушивают и отгоняют бензол и СНС13. Неочищенный
продукт (47,5 г) разбавляют 50 мл петролейного эфира и пропускают для очи-
стки через колонку с окисью алюминия, которую предварительно обрабатывают
аммиаком, высушенным над КОН, до прекращения первоначального разогре-
вания. Перегонкой элюата в вакууме получают иодбензол; выход 83% от теоре-
тического.
1-Иод-4-хлорбензол (выход 53%), 1-иод-4-бромбензол (выход 65%)
и л-дииодбензол (выход 56%) получают аналогично.
Способ иодирования по Бнркенбаху — Губо — Уотерсу описан в следующих
примерах:
Иодбензол. В бьщтро перемешиваемую смесь 0,3 моль бензола, 0,1 моль
Ag2SO4, 200 мл концентрированной НС1 и 200 мл воды в течение 2 ч порциями
добавляют 0,22 моль тонкоизмельченного (1) иода, перемешивают еще 1 ч, раз-
бавляют 600 мл воды и декантируют от осадка Agl и непрореагировавшего иода.
После обработки бисульфитом экстрагируют эфиром продукт реакции из декан-
тированного раствора и твердой фазы. Экстракт перегоняют в вакууме и полу-
чают иодбензол; выход 78% от теоретического [377J.
По другому способу [375] суспендируют 22 г AgClO.и 10 г СаСО3 в 100 мл
бензола и при охлаждении смешивают в течение 15 мин с 25 г иода. Затем смесь
отфильтровывают, трижды промывают водой, сушат над СаС1г и перегоняют;
выход иодбеизола 80% от теоретического.
I-Йоднафталин. Иодируют нафталин, как описано выше, но в ионцептри-
рованном (50%-ном) эфирном растворе; выход продукта 85% от теоретического.
Эфярн$пз раствор иода неустойчив при длительном контакте с перхлоратом,
однако вследствие быстрого протекания реакции разложение не проявляется.
5. Замещение галогеном водорода в боковой цепи
ароматических углеводородов
Хлорирование боковой цепи горячего толуола хлором на свету с образованием
хлористого бензила п бензилидена (бензальхлорида) описано Гаттерманом [421].
[421] L. Ga Hermann, Die Praxis des organlschen Chemikers, 37 Aufl., W. de
Cruyter u. Co., Berlin, 1956, S. 92, 184.
10 Заказ 1835,
146
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
При получении бензальхлорида прямым хлорированием толуола одновременно обра-
зуется трихлорбензол, с трудом отделяемый от бензальхлорида вследствие близости
их температур кипения (205 и 214° С). Одпако при гидролизе бензальхлорида до
бензальдегида трихлорбензол превращается в легко отделимую бензойную кислоту.
Для хлорирования боковой цепи требуется добавка перекисей. Не содержатп-й
перекисных соединений толуол не реагирует в темноте с SO2C12 даже при кипячении
в течение 6 ч.
К смеси хлорируемого соединения с SO2C12 добавляют 0,001—0,005 моль пере-
киси бензоила или лаурила на 1 моль SOaCl2, нагревают с мощным обратным холодиль-
ником до слабого кипения и кипятят (15—30 мин) до прекращения выделения HCI
и S02, что указывает на конец реакции (при работе с кумолом кипятят 90 мин). Про-
дукты хлорирования выделяют разгонкой реакционной смеси. Из 1 моль толуола
и 1 моль SO2C12 (или менее) с очень хорошим выходом получается хлористый бензил,
с 2 моль или больше SO2C12 — бензальхлорид. Бензотрихлорид не образуется.
Так же как при свободнорадикальном бромировании боковой цепи при хлори-
ровании боковой цепи под действием SOaCla и перекиси галоген вступает преимуще-
ственно в «-положение к ядру: из этилбензола главным образом образуется а-хлор-
этилбензол, из кумола — 90% а- и 10% р-хлоризопропплбензола. При хлорировании
mpem-бутилбензола хлор вступает в p-положение. При действии сульфурилхлорида
и перекиси на о- и и-нитротолуолы соответствующие хлористые бензилы не образу-
ются [422].
Вместо перекисей можно применять также а.сс'-азоизобутиронитрил, где
R = CN, или диметиловый эфир а,а'-азоизомасляиой кислоты, где R = СООСНд.
В растворе при 80—120° С они гладко разлагаются с образованием азота и тре-
тичных ароматических радикалов:
R R R
II I •
(CH3)2C-N=N-C(CH3)2 —-> 2(СНз)2С. -[-N2
Эти вещества не действуют на такие растворители, как бензол, толуол и СС14.
При добавлении 0,3—0,5 мол. % инициатора к кипящей смеси толуола или
трифепилметада с S02CI2 получают хлористый бензил (выход 70% от теоретического)
или тряфепилхлорметан (выход 77% от теоретического) [336].
Хлористый бензил [336]. В нагреваемую с обратным холодильником
на водяной бане смесь 0,8 моль сухого толуола и 0,4 моль SO2CIs добавляют 0,2 а
динитрила а, а'-азоизомасляной кислоты. Через несколько секунд начинается
бурная реакция с выделением HCI и SO2; реакция заканчивается через 15 мин.
Реакционную массу дважды перегоняют и получают наряду с избыточным толу-
олом хлористый бензил с выходом 70% от теоретического.
Трифенилхлорметап [422]. Смесь 8,4 г трифенилметана, 13,5 г SO2CIa.
и 0,1 г перекиси лаурила кипятят 30 мин с обратным холодильником. После-
отгонки избыточного SO2Cl2 полученный трифеннлхлорметан перекристалли-
зовывают дважды из бензина (т. кип. 90—100’ С). Выход 5,7 г; т. пл. 111 —112° С.
При хлорировании боковой цепи бромтолуолов под действием SO2C12 и пере-
киси бензоила протекает побочная реакция замещения брома в ядре на хлор [423].
Обычный способ получения замещенных бензилбромидов заключается в сле-
дующем. В соответствующий замещеппый толуол при перемешивании и нагревании
до 120—160° С на масляной баке или в его кипящий раствор в СС14 при УФ-облучении
или освещении лампой в 100—600 впг приливают по каплям 1,1 моль брома или
раствор брома в СС14 в течение 1,5—2 ч. При работе с бромом трубка капельной
воронки должна быть погружена в жидкость.
Примером бромирования бромом при 145—150° С является получение 4-нитрс~
бензилбромнда из 4-нитротолуола [424].
[422] М. S. К h а г a s с h, Н. С. Brown, J. Am. Chem. Soc., 61, 2144, 2148 (1939)..
[423] G. L. Goerner, R. C. Name tz, ,T. Am. Chem. Soc., 73, 2940 (1951).
[424[ G. П. Cole m a n, fi. E. Honpywel I, Org. Synth., Coll. v. 2, 1943»
p. 443. ,
1. ЗАМЕЩЕНИЕ НОДОРОДЛ НА Hal ПЛИ SCN
4-Нитробепзилбромид. К 4-нИгротолуолу в течение 2 ч при 145—150® 1
по каплям прибавляют бром; реакцию ведут еще 10 .чин а затем выливают масс
в горячий бепзин, из которого после обработки активным углем выкристаллизс
нынают 4-нитробензилбромид. Кренке, опубликовавший прописи для получали
замещенных бензилгалогенидов, в том числе .ч-нитро-, .ч-хлор-п-иод-, 2,6-д!
хлор-[425] и п-хлорбензилбромидов [426], а также краткий литературный обзо
методов получения таких соединений [427], усовершенствовал прпведеннуз
в «Органических синтезах» |428] пропись получения 4-нитробензплбромиде
По его способу необходимо па каждые 80 г ге-иитротолуола добавлять 4 г ртут
и 1 мл воды. Реакционную массу выливают и воду и растворяют лерастворенны
остаток в 240 мл горячего этилового спирта. При 0° С выкристаллизовываете
80% i-нитробензилбромида, в то время как по первоначальному способу выхо
составлял 55—66% от теоретического.
Друтпм примером бромирования может служить получение а,а'-диброк
о-ксплола [428|.
Хлористый или бромистым бензилы могут быть также легко получены замено
хлором (бромом) гидроксильной группы путем хлормстилировапия [429] пли броь
метилирования [7(1].
Ароматические соединения с галогенированной боковой цепью чувствительн
к влаге и действуют раздражающе на слизистые оболочки. Эти соединения термическ
пс очень устойчивы. Их термостойкость тем меньше, чем менее ароматично ядре
Па легкой подвижности брома в бромзамещенпом бензиле основан простой спосо
определения брома в боковой цепи [387].
Навеску сухого бромида кипятят 3 ч с большим избытком ацетата натри:
в абсолютном этиловом спирте, подкисляют 6 н. HNO3. добавляют отмеренньд
избыток стандартного раствора AgNO3 и оттитровывают его остаток растворе*
Бензальхлориды и бромиды представляют интерес как промежуточные пр<
дукты для синтеза альдегидов. Их получают аналогично моногалогснсоединениям -
действием на бензил только 2 моль галогена.
Подробно описано: приготовление неочищенного 4-хлорбензальхлориде
из которого при действии ионцентрированной H2SO4 получают п-хлорбензалз
дегид [430]; неочищенного 4-бромбензальбромида, который по реакции с водны
карбонатом кальция переводят в п-бромбензальдегид [431]; чистого а,а,а',а
тетрабром-п-ксилола — исходного продукта для синтеза терефталевого альд<
гида [432J; а,а,а',а'-тетрабром-о-ксилола—исходного продукта для получ<
ния о-фталевого альдегида [433].
Бромирование ацетилированного .ч-крезола в СС14 при облучении лампе»
в 500 вт дает 75% л-ацетоксибензальбромида, который по реакции с формиата
натрия в спирте превращается в л-оксибензальдегид; выход 70% от теоретич«
ского [434].
F, Krohnke, Н. SchmeiB, W. Gottstein, Chem. Bet., 8*
135 (1951).
F. Rrooke, Chem. Ber., 83, 43 (1950).
F. Krohnke, I. Vogt, Chem. Ber., 85, 373 (1952).
E. F. M. Stephenson, Org. Synth., 34, 100 (1954).
R. Adams, Org. Reactions, 1, 63 (1947).
W. L. McEwen, Org. Synth., Coll. v. 2, 1943, p. 133.
G. II. С и I e m a n, G. Ё. Honeywell, Org. Synth., Coll, v, 3
1943, p. 89.
J. M. Snell, A. Weiasberger, Org. Synth., Coll. v. 3, 1955, p. 788»
J. С. В i 11, D. S. T а г b e 11, Org. Synth., 34, 82 (1954).’
E. L. Eliel, K. W. Nelso n, J. Chem. Soc., 1955, 16^8.
10*
148
ОБРАЗОВАНИЕ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
6. Замещение галогеном водорода в ацетиленах
Замещение водорода у С=С-связи галогеном может происходить непосред-
ственно или через С = С-металлоорганпческпе соединения, из которых затем, без про-
межуточного их выделения, можно получать галогенацетилены.
Методы хлорирования, бромирования и иодирования ацетиленов в щелочных
растворах гипогалогенптов [435] применимы для галогенирования алкинов, алкино-
лов и ацетиленмоиокарбоновых кислот, хотя бы с одним атомом водорода у С^С-связи;
RC=CH + NaOX —► RC=CX + NaOH
Наиболее быстро протекает взаимодействие с гипоподптом, наиболее медленно
с гипохлоритом.
При взаимодействии свободных гипогалогенитных кислот НОХ с алкинами .
замещения не происходит, а образуются а,а'-дигалогопкарбонильпые соединения:
RC=CH-|-2HOX —> [RC(0H)2-CHX2l —> RC-CHX2
Газообразные алкппы вводят при 0° С в щелочной (до 23% в пересчете на КОП)
раствор гипогалогенита. Жидкие, предварительно растворенные в петролейном эфире,
и твердые алкины встряхивают или перемешивают при комнатной температуре
с щелочным раствором гипогалогенита, иногда с добавкой эмульгатора.
Растворы гипогалогенптов [435]. В раствор 180 г КОН в 800 мл воды .
при 0° С и интенсивном перемешивании вливают по каплям 25 мл брома. Полу-
чается раствор примерно 0,55 н. по КОВги 2 н. по КОН.
В водный 12,5%-ный раствор КОН при 0° С пропускают хлор до мгновен-
ного обесцвечивания лакмусовой бумаги и затем разбавляют равным объемом -
25%-ного раствора КОН. Получается примерно 0,4—0,7 и. раствор по КОС1
и 2,2—1,7 н. по КОН.
Щелочной раствор гипоиодита довольно неустойчив. Лучше всего его
готовить непосредственно в реакционной смеси из KI и раствора NaOCl (стр. 149).
Гипогалогенлтный способ может служить также для получения дихлорацети- <
ленов. В качестве исходного продукта наиболее пригоден ацетилен из карбида каль-
ция. Ознакомиться с методом рекомендуется в работах [435, 436]. Дихлорацетилен )
(т. кип. 29° С при 743 мм рт. ст.) ядовит и при нагревании, а также при соприкосно-
вении с кислородом (даже с растворенным в воде воздухом) легко взрывает. Дибром- ‘к
ацетилен тоже очень взрывоопасен. С увеличением числа атомов углерода и молеку- «
лярного веса галогена в молекуле галогенацетиленоп стабильность соединений .2
возрастает, но все они термически нестойки и требуют осторожного обращения. v
Описан простой опыт [436], демонстрирующий склонность дихлораце-
тилена и взрыву, который также показывает, что совместное хранение хлорной
извести, карбида кальция и щелочей в присутствии влаги опасно.
В высокий химический стакан емкостью 250 мл, содержащий холодный
раствор щелочи, приготовленный из 20 г КОН и 50 г мелкорастолченного льда, -
добавляют 15 з хлорной извести. Смесь размешивают до жидкой кашицы. Стаиан
помещают в баню со льдом и вносят в смесь в течение 20—30 мин 3—5 кусочное
технического карбида иальция величиной с горошину. Затем вынимают стакан
из бани и, после того как смесь вследствие гидролиза карбида постепенно
нагреется, наблюдают при перемешивании самовозгорание выделяющейся пены, >
а также легкий треск, происходящий от хорошо заметных в темноте отдельных * 1
взрывов (тяга!).
Дихлорацетилен удобнее получать отщеплением НС1 от трихлорэтилена [436,
437]. Эсрирные растворы дихлорацетилена, который с эфиром образует молекулярное ’
[435] F. S t г a u s, L. К о 11 е k, W. Н е у п, Вег., 63, 1868 (1930).
[436] R. Riemschneider, К. Brendel, Ann., 640, 8, 14 (1961).
[437] Е. О t t, W. О t t e m е у е г, К. Р а с к е n d о г f f, Вег., 63, 1941 (1930);
Е. Ott, К. Р а с к е n d о г f Г. Вег.. 64, 1324 (1931).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
149
соединение, не подверженное аутоокислению (1 : 1), лучше всего получать при про-
пускании смеси паров трихлорэтилена гг эфира над нагретым до 125е С КОН в атмо-
сфере азота; подробности аппаратурного оформления см. [436].
1-Бромгексин-1 [438]. К щелочному раствору гипобромита (стр. 148)
прибавляют 45 г гексина-1 и энергично перемешивают при комнатной тамперу,
туре в течение 18 ч. Органическую фазу извлекают петролейным эфиром и высу-
шивают над Na2SO4. вакуумной перегонкой получают 1-бромгекслн-1. Выход
72% от теоретического, т. кип. 40—41° С при 15 мм рт. ст.
Дииодацетилен. При охлаждении льдом и перемешивании пропускают
ацетилен в раствор 20 г К1 в 100 мл 1 и. раствора Na ОН и одновременно прили-
вают по каплям около 150 мл 1 н. раствора NaOCl до почти полного исчезновения
первоначальной желтой окраски смеси. Выпавший белый осадок дииодацетилена
промывают водой, хорошо отжимают и сушат 24 ч над Р2О5 в эксикаторе прп
нормальном давлении (так как он летуч, то его нельзя сушить в вакууме). Выход
96,5% от теоретического; т. пл. 82° С. Ацетилен для синтеза получают, приливая
по каплям воду к 600 г карбида кальция. Из такого ацетилена образуется чи-
стый белый продукт, а из чистого ацетилена, отбираемого из баллона, наряду
с дииодацетиленом образуется желтый тетраиодэтилен. Раствор гипохлорита
готовят [489], пропуская 12 г хлора в 200 мл охлажденного льдом 2 н. рас-
твора КОН.
Считают, что в темноте при комнатной температуре дииодацетилен может
храниться, не разлагаясь, в течение несколько месяцев [439]; однако имеется
сообщение о несчастном случае [440], когда сухой продукт (2 кг!) разложился -
со взрывом.
По более старой, подробной прописи дииодацетилен получают из ацетилена
и иода в жидком аммиаке [441].
Вместо гипоиодита щелочного металла для получения иодацетиленов можно
применять также ацилгипоиодит в форме комплекса Симонини [(C8HiCOO)3Ag]I.
Реакцию бензоата серебра и иода с алкином проводят [442] в бензоле или СС14;
R'COOAg+I2-|-RC=CH —R'GOOH-J-RC^C-l -
Алкин-1-олы-З (этинилкарбинолы) с хорошими выходами хлорируются и бро-
нируются по С=С-связи водно-щелочными растворами гипогалогенитов [443, 444],
в то время как для их иодирования применяется реакция, протекающая с промежу-
точным образованием —С=С-металлооргапических соединений [443].
Такой способ имеет также известные преимущества при галогенировании
высших алкилацетиленов. Сначала по реакции алкина с этилмагкийбромидом в сухой
эфире или с амидом натрия или калия в жидком аммиаке готовят соответствующий
металлоорганические соединения, из которых далее получают талотеналкивц. Так,
из мягниибромалкина (без его выделения) при действии брома в сухом эфире при
—32° С получаются с хорошим выходом 1-бромалкияы:
CH3(CH2)4C-CMgBr —CH3(CH2)flC=CBr4-MgBr2
Из калиевого пропзводпого в сухом эфире получают при действии хлора
и —70° С 1-хлоралкииы [445]:
СЦз(СН2)вС^СК4-С12 —> СНз(СН2)вС=С-С1 + КС1
[438] К. Е. Schulte, М. Goes, Arch. d. Pharm., 290/62, 124 (1957).
[439] V. Franzen, Chem. Ber., 87, 1148 (1954).
[44oJ H. 3 aeschke, Chem. Ber., 88, 101 (1955).
[441] Th. H. Vaughn, A. Nieuwland, J Am. Chem. Soc., 54, 78Z-
(1932).
[442] C. Prevost, С. r., 200, 942 (1935); 204, 989 (1937).
[443] A. В a v 1 e y, J. Org. Chem., 19, 570 (1954); 20, 109 (1955). •
[444] J. С о 1 о n g e, L. Cumet, Bull. Soc. chim. France, 1947, 841.
[445] P. A. Me Cusker. R. R. Vogt, J. Am. Chem. Soc., 59, 1307 (1937).
150
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Из натриевых производных с иодом в жидком аммиаке образуются 1-иодалкнны
[446]. Иодалкины могут быть синтезированы и из магнийбромацетилена и вода при
0° G в растворе абсолютного эфира [447].
7. Замещение галогеном водорода в фенолах,
окспарилалканпарбоновых кислотах, эфирах фенолов
и ароматических аминах *
Получение моно- и дигалогенпроизводных фенолов и ароматических аминов
представляет особый интерес для препаративных целей. Нежелательное в этом случав
замещение хлором и бромом, иногда даже во всех активируемых заместителями
положениях, происходит часто уже при проведении реакции в мягких условиях.
Монохлорирование (бромирование) фенола хлором (бромом) идет на холоду преиму-
щественно в 4-положение, а при 150—180° С — в 2-положение.
4-Бромфенол. Бромирование фенола в CS3 при охлаждении смесью льда
и соли приводит к получению около 80% 4-бромфекола. Предварительно отго-
няется фракция, содержащая трудноразделяемую смесь 2- и 4-бромфенолов.
Выход 4-бромфенола можно еще повысить применением 20%-ного избытка фе-
нола и приливанием по каплям при 0° С раствора брома в дихлорэтилене [448,
449].
4-Хлорфенол [450]. При обычной температуре смешивают эквимоле-
кулярные количества фенола и SO2C12 и после окончания бурною выделения
SO2 и HG1 нагревают ка водяной бане до прекращения выделения газов. Затем
реакционную массу промывают раствором соды, сушат над СаС12 и разгоняют;
выход почти количественный; т. кип. 216—218° G; т. пл. 37° С.
Для получения орто-галогенированных фенолов можно предварительно ввести
сульфогруппы в положения, не подлежащие замещению хлором или бромом. Соответ-
ствующие оксисульфокислоты хлорируют или бронируют без их выделения и затем
гидролизуют реакционную смесь перегонкой с водяным паром в присутствии H2SO4.
2-Вромфенол [451]. Его получают с выходом около 40% бромированием водно-
щелочного раствора о- н п-фенолдисульфокислот.
Лучшие результаты дает галогенирование неочищенных оксисульфокислот
в смеси нитробензола с H2SO4. Таким способом получают 2,6-дихлорфеяол или
2,6-дйбромфенол с выходом около 70%, 2-хлорфенол с выходом 72% и 2-бромфенол
[452] с выходом 46,5%.
Можно также галогенировать соответствующие фенолкарбоновые кислоты
с последующим декарбоксилированием полученных продуктов.
2,6-Дихлорфенод [453]. Этиловый эфир 4-оксибензойной кислоты нагре-
вают 1,5 ч на паровой бане с 2,2 моль SO2C12 и получают с 85%-ным выходом
этиловый эфир 3,5-дихлор-4-оксибензойной кислоты, который омыляют затем.'
446'
447'
448
449
450
[451]
[452]
[453]
См. также А. П. Т е р е н т ь е в, Л. Л. Я н о в с к а я, Реакции и методы иссле-
дования органических соединений (РиМИОС), Госхимиздат, 6, 7 (1957).—
Прим. ред.
Th. Н. Vaughn, J. A.Nicuwland, J. Am. Chem. Soc., 55, 2152 (1933).
V. Grignard, H. Perrichon, Ann. Chim. [10], 5, 5 (1926).
R. Adams, G. S. Marvel, Org. Synth., 1, 39 (1921).
H. E. P о d a 1 1, W. A. Foster, J. Org. Chem., 23, 280 (1958).
L. Vanino, Handbuch der praparativen Chemie, Bd. II, F. Enke, Stuttgart,
1937, S. 430.
R. C. Huston, M. M. Ballard, Org. Synth., Coil. v. 2, 1943, p. 97.
R. G. H u s t о и. A. li.Neeley, J. Am. Chem. Soc., 57, 2176 (1935).
D. S. Tarbell, J. W. Wilso n, P. E. Pant a, Org. Synth., 29,
35 (1949); Coll/v. 3, 268 (1955).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА На! ИЛИ SCN
метанольным раствором КОНвЗ,5-лихлор-4-окспбензопную кислоту. Эту кислоту
(1,2 моль) медленно нагревают с 575 г диметиланнлина до 190—200° С и выдер-
живают при такой температуре до окончания выделения СО2 (около 2 ч). 2 6-
Дпхлорфенол получают с выходом 80—90% от теоретического. ’
Для избирательного бромирования фенолов и феноловых эфиров могут быть
использованы л некоторые комплексные соединения брома.
С дпоксандибромидом без растворителя или в растворе эфира или диоксана
можно получать монобромзамещепные фенола, многоатомных фенолов, феноловых
эфиров и салициловой кислоты [454].
4-Бромфенол. К 9,4 г фенола при охлаждении водой постепенно доба-
вляют 25 г диоксандибромида (получение см. на стр. 158), выливают смесь в воду
п экстрагируют эфиром. Получают 4-бромфепол с выходом 88% от теоретиче-
ского; т. кип. 115—117° С при 11 —12 мм рт. ст.-, т. пл. 64—65° С. Без охла-
ждения образуются значительные количества 2-бромфенола!
Многоатомные фсполы, папример пирокатехин, пирогаллол, а также эфиры
фенолов можно мояобромировать гидробромидом дибромпда пиридипия (пербромидом
пиридинийбромида) при комнатной или более низкой температуре [33, 455]. Приме-
няют готовый реактив (получение см. на стр. 100) или прибавляют по каплям раствор
брома известной концентрации в ледяной уксусной кислоте к смеси бронируемого
вещества с гидрохлоридом пиридина (получают пропусканием сухого НС1 в эфирный
раствор пиридина) или к смеси вещества с сернокислой солью хинолина и ледяной
уксусной кислотой.
Многоатомные фенолы легко бронируются бромом с образованием ди- и полн-
бромпроизводных. Из пирокатехина с 2 .коль брома в ледяной уксусной кислоте полу-
чают 4,5-дибромпирокатехпн, который легко бронируется в хлороформе до 3,4,5-три-
бромпирокатехина [456]. Аналогично этому резорцин сначала бронируется в пара-
положение к двум гидроксильным группам с образованием 2,4-дибрОмрезорцина,
затем в общее орто-положение до 2,4,6-трибромрезорцина [457J. 4-Бромрезорцин
получают [458] бромировапием 2,4-диоксибснзойной кислоты [459] 1 моль брома
в ледяной уксусной кислоте при 30—35° С и последующим декарбоксилированием
образующейся 2,4-диокси-5-бромбензойной кислоты кипячением с водой в течение
24 ч. 2,4-Диоксибензойную кислоту готовят из резорцина.
2.4,6-Трибромфенол легко синтезировать добавленном к водному раствору
Фенола бромной поды до прекращения обесцвечивания реакционной массы. При избытке
рома образуется трибромфенолбромяд (получение см. [460]), который легко отдает
катион брома и поэтому действует как бронирующий агент. Он вытесняет иод из
иодидов [481], так что при титровании фенола бромом (или раствором бромид-бромата),
KI и NagS-jOg расходуется шесть атомов брома на одну молекулу фенола н образуется
трибромфенол.
При бромировании о- и и-оксибензойных, а также о- и n-аминобеизойных или__
бензолсульфокислот в воде уже при обычной температуре карбоксильные нлп сульфо-
группы замещаются бромом с выделением СО2 или H2SO4. При 40—45° С в о- и п-окси-,
а также в о- и п-амипобензальдегпдах альдегидная группа тоже замещается бромом
с отщеплением окиси углерода. Конечными продуктами являются 2,4,6-трибромфенол
или 2,4.6-триброманилин [462]. Поэтому можно, например, с помощью 0,1 и. раствора
бромид-бромата количественно определять п-оксибензойную и сульфаниловую кис-
лоты.
[454]
[455]
[456
457
458
459
460
461
Л. А, Яновская, А. П. Т е р е н т ь е в, Л. И. Б е л е н ь к и й, ЖОХ,
22 (84), 1594 (1952); С. А., 47, 8032b (1953).
К. Freudenberg., Н. Fikentscher, М. Harder, Ann., 441
178 (1925).
М. Kohn, J. Am. Chem. Soc., 73, 480 (1951).
M. Kohn, G. Loff, Mh. Chem., 45, 592 (1924).
R. B. S an din, R. A. Me К e e, Org. Synth., Coll. v. 2, 1943, p. 100.
M. Nierenstein, D. A. Clihbens, Org. Synth., Coll. v. 2,1943, p. 557.
M. К о h n, S. Sussmann, Mh. Chem., 46, 578 (1925)..
Ph. Fresenius, Angew. Chem., 64, 473 (1952).
A. W. Francis. A. J. Hill. J. Am. Chem. Soc., 46, 2498 (1924).
ОБРАЗОВАНИЕ СВЯЗИ С—На! Б РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Для иодирования фенолов и оксифенилалкановых карбоновых кислот применим
ряд мягких методов замещения водорода иодом в ароматическом ядре, перечисленных J
в способе а) (стр. 141, а также стр. 187), и, кроме того, иодирование хлоридом иода л
в аммиачной или щелочной средах (стр. 170). д
При иодировании иодом в присутствии щелочей или аминов применяют коротко- »
обрядный иод или раствор иода в водном растворе NaI(Nals) или К1(К13), в метиловом
спирте или иногда в эфире. Недостатком этого способа является накопление в маточ-
ных растворах большого количества иодида, из которого при больших загрузках реге-
нерируют иод (стр. 92). Однако в лаборатории этот метод часто применяется
и дает хорошие выходы. В щелочных растворах иодом были проиодированы много- :
численные перазветвлеиные и разветвленные <г>-(4-оксифенил)-жирные кислоты [463]-
3,5-Динод-4-оксифенилуксусная кислота. К раствору 0,1 моль 4-окси-
фенилуксусной кислоты в 800 мл 0,5 н. КОН при комнатной температуре и пе- $
ремешивании прибавляют по каплям раствор 50,8 г иода и 50,8 з К] в 250 мл J
воды. После отсасывания осадка реакционную массу охлаждают до 0—5° С
и при перемешивании медленно пропускают в фильтрат SO2 до pH 2—3. Выпав- (
ший неочищенный продукт переосаждают из раствора NaHCO3 и кристалли-
зуют из водного этилового-спирта. Выход 3,5-дииод-4-оксифсннлуксусной кис- -и
лоты 92% от теоретического; т. пл. 216(5—217,5° С. !
При иодировании иодом фенолов со свободными орто- и пара-положениями •
в водно-щелочной среде наряду с желаемыми иодпроиэводными могут образоваться
красно-коричневые полимерные побочные продукты, папример «красный Лаутемана» .
из фенола [464—465] и аристол из тимола [466]. Эти нежелательные побочные реакции • i
можно свести к минимуму ведением процесса в метанольном растворе щелочи [467] <-’1
и в концентрированных водных или метанольных растворах аммиака [468]. Медлен- •
ным добавлением концентрированного раствора иода в K.I к раствору фенола в кон- '
центрированном NH4OH получают 2,4,6-трииодфенол с почти количественным выхо-
дом [469].
При работе по описанному способу в виде промежуточного продукта образуется 1
чорпый взрывчатый иодид азота Nl3, поэтому способ следует применять для иодиро '
нания только легко иодируемых фенолов (ср. [467]), которые быстро связывают иодид- 7
азота по мере его образования. Реакцию ведут при комнатной температуре и быстром--
перемешивании, причем раствор иода (в водном К1 или метиловом спирте) медленно е]
приливают по каплям по мере его расходования. Во избежание возможного образо-
вания избыточного иодида азота к концу реакции добавляют концентрированный-.;
раствор NaHSO3 и перемешивают.
Вместо аммиака можно применять водные или метанольные растворы первич-4*-
ных или вторичных алифатических аминов, не образующих взрывчатых иодпроизвод-- >
ных [470, 471]. Этот рекомендуемый для препаративных целей метод полностью
оправдал себя при получении тироксина из 3,5-дииодтиронина в 20%-ном водном./
этиламнне (К13), метилового эфира тироксина [471] из метилового эфира 3,5-дииодти-
ронина (12) в растворе бутиламина в метиловом спирте (1 : 2), 3,5-диподтирозина (Nalg)
в 20%-ном водном этиламине [472] и 'при иодировании (К13) ряда ароматических;
аминов и фополов с применением 0,5 моль 60%-ного водного раствора этилендиамина;
па 1 .коль иодируемого соединения [473].
[463]
464
465
466
467
468
469
470
[471]-
[472]
[473]
D. Papa, Е. Schwenk, ,Е. Klingsberg, 3. Am, Chem. Soc., 72,
2623 (1950); D. Papa, H. Breiger, E. Schwenk, V. P e t e r s о nt
J. Am. Chem. Socs, 72, 4907 (1950); M. G. Pratt, J. O. Hoppe, S. Ar-
cher, J. Org. Chem., 13, 578 (1948).
W. H. Hunter, G. H. Woollet, J. Am. Chem. Soc., 43, 135 (1921).
G. H. W о о 1 1 e 11 et al., J. Am. Chem. Soc., 59, 861 (1937).
G. H. W о о 11 e t t, J. Am. Chem. Soc., 43, 553 (1921).
H. Cassebaum, 3. prakt. Chem. [4], 13, 141 (1961).
J. C. Colbert et al., 3. Am. Chem. Soc., 66, 122 (1944).
R. L. Cat la, N. Prosad, J. Am. Chem. Soc., 89, 441 (1917).
С. V. Borde ianu, Ann. sci. Univ. Jassy, 20, 131 (1935); C. A., 30, 1760.
(1936).
3. С. С 1 a у t p n, В. A. H e m s, 3. Chem. Soc., 1950, 842.
3. H. Barnes, В. A. H e m s et al., J. Chem. Soc., 1950, 2829. .
К. T. P о t t s, 3'. Chem. Soc., 1953, 3711.
ЗАМЕЩЕНИЕ ВОДОРОДА НА На! или SCN
153
Р-(3-5 Дипод-4-оксифенил)-лропмоновая кислота [472]. Раствор 50 г
иода и 48 г К] в 150 мл воды постепенно добавляют при перемешивании к рас-
твору 16,2а В-(4-оксифенил)-пропионовой кислоты в 200 мл 20%-ного водного
метиламина. После 10 мин перемешивания смесь подкисляют (по конго) медлен-
ным добавлением 2 н. раствора ПС1. Выпавший белый неочищенный продукт
промывают водой и перекристаллизовывают из этилового спирта; выход 90%
от теоретического; т. пл. 166—168’С.
Нежелательного образования (особенно при больших загрузках) значитель-
ного количества иодида в маточном растворе можно избежать, применяя 1GI вместо
иода. Преимуществом 1С1 является также его растворимость в йодных растворах NaCl
u КС1; о получении 1С1 и его растворов см. стр. 170. Очень легк.0 кодируемые фенолы
.h-RC6H4OH (R=NH2 или ОН) при обычной температуре уже гладко образуют
трпподзамещенные, взаимодействуя с солянокислым раствором хлорида иода [467,
474]. Иодирование фенола 1С1 в ледяной уксусной кислоте не рекомендуется, так как
образующийся 2,4,6-трииодфенол содержит много примесей. Иодирование с помощью
[С1 в уксусной и соляной кислоте будет подробней рассмотрено на стр. 171.
Метод иодирования IC1 в щелочной или аммиачпой среде оказался пригодным
для получения 2,4.6-трииодфенола, а также ряда 2,4,6-трииодирбваииых «-замещен-
ных фенолов *-R-C.H4OH (R=OCH4, NHGOCH3, NHGOCsH7, CH3, Cl, I, COC,H,;
COOH, N02, OCH2COOH, OCHaGOOCHs) [467].
При иодировании IC1 в аммиачной среде, так же как при иодировании иодом,
в качестве промежуточного продукта образуется иодид азота Nla. Несмотря на
что до сих пор не поступало сообщении о взрывах образующегося при иодированйй
иодида азота, все же необходимо принимать меры предосторожности (защитий'0
экраны, очки, добавление фенолов, легко взаимодействующих с иодидом азота [467].
Совершенно безопасным и равноценным является применение 1С1 'в метанольных
растворах метиламина или NaOH.
Ниже приведены общие способы получения трииодпроизводных фенола и «-заме-
щенных фенолов действием £01 в присутствии NH3, NaOH или метиламина [467]),
Трииодзамещениые фенолы
а) Растворяют 0,1 моль фенола в 600 мл 10—15%-ного раствора NHg.
При перемешивании в течение 20 мин приливают по каплям 155 мл 2М
раствора NaICl2 (получение см. стр. 171), перемешивают 10 m и добавляют
10—20 мл концентрированного раствора NaHSO3, чтобы разложить избыточ-
ный Nl3, который может образоваться в этих условиях. Выпадающий в осадок
иодированный фенол отсасывают, суспендируют в воде и подкисляют НС1.
Если продукт реакции остается в растворе, то по окончании реакции иодирован-
ный фенол выделяют НС1 или H2S04; выход трииодзамещениых 73—96,5% для
фенолов, у которых В. = OCHS, NHCOC3H7, NHCOCHa, GHS, Н, I, COOH,
OCH2COOH, OCH2COOCH3. При получении иодзамещениой л-окснбензойиой
кислоты (R = GOOH) для иодирования требуется больше времени, чем указы-
валось выше. В то время иак первые две молекулы 1С1 реагируют еще относи-
тельно быстро, для введения третьего атома иода необходимо 30 мин.
б) Растворяют 0,1 моль фенола в 500 мл 5—10%-ного раствора NH*. в ме-
тиловом спирте. Вводят при перемешивании в течение 20 мин 155 мл 2 М рас-
твора NaICla, перемешивают еще 10««к, разбавляют 500 мл воды, избыток
иодирующего агента разлагают 10—20 мл концентрированного раствора NaHSOg
и выделяют продукт реакции подкислением НС1 или H2SO4. За потреблением
1С1 удобно следить по исчезновению темной окраски, обусловленной Nig.
Выход трниодзамещенных продуктов 80—95% при R — NHCOGaH7, NHCOCHg,
Н, *NO2. При иодировании «-нитрофенола (R — NO2) приливают по каплям
2 М раствор NaIC]a порциями по 50 мл в течение 10 мин, после чего перемеши-
вают смесь до исчезновения окраски, обусловленной N13, а именно: около 15 мин
после введения первой порции NalGla, около 30 мин после введения второй п
около 60 мин после введения третьей порции.
в) Реакцию проводят, как описано в пункте б), по вместо 5—10%-ного
раствора аммиака в метиловом спирте применяют раствор метиламина в метило-
вом спирте; выход трийодзамещениого 80—95% при R =NHCOGgH7, NHCOCHg, Н.
[474] Н. С а н 3 е b а и m, Pharmazie, 15, 313 (1960).
154
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
г) Растворяют 0,1 моль фенола и 18 г NaOH (в случае фенолмонокарбоно-
вых кислот 22 г NaOH) в 500 мл метилового спирта. При перемешивании в тече-
ние 20 мин вводят 155 мл 2 М раствора NalCl2, отсасывают выпавший в осадок
NaCl и промывают 55 мл метилового спирта. Фильтрат разлагают 10—20 лл
концентрированного раствора NaHSO3, разбавляют 500 мл воды и выделяют
продукт реакции подкислением концентрированной НС1: выход трииодзамещен-
ного фенола 75—95% при R = NHCOC3H7, NHCOCH3, Н, COCeHfi, COOH,
NO2. Чтобы легко получить трииодзамещеииый л-нитрофенол (R = NO2),
требуется двойное количество 2М раствора NalClj.
Ароматические соединения с заместителями, облегчающими электрофильное
замещение, можно иодировать в ядро с помощью иода и Н2О2. Для получения относи-
тельно чистого 2,4,6-трииодфенола с хорошим выходом в раствор фенола и иода в эти-
ловом (55° С) или в метиловом (45° С) спирте медлеппо приливают но каплям при
энергичном перемешивании 3,5—4 моль 30%-ной перекиси водорода [475]. Недостат-
ком этого способа является возможность бурно протекающей реакции разложения
перекиси. Опасность взрыва может быть сильно уменьшена добавлением небольшого
количества двуокиси марганца [467]. В присутствии следов других тяжелых металлов
возможно образование значительных количеств (30—40%) тетраиодзамещен-
ных фенолов.
Способ, в котором применяется 12—Н2Оа, пригоден также для иодирования
некоторых гетероциклических соединений (ср. стр. 187). Например, из 8-оксихиао-
лина образуется 5,7-дииод-8-оксихинолип с выходом 89,4% от теоретического [476]
Медленно приливая по каплям при перемешивании 30%-ную Н2Оа к анилину в мети-
ловом спирте при 45° С, получают 2,4-дииоданилин при соотношении иод : ани-
лин = 3 : 1 и 2,4,6-трииоданилии [477] при соотношении реагентов 4:1: Феноловые
эфиры можно иодировать иодом в присутствии Н2О2 только при добавлении концен-
трированных минеральных кислот (серной, фосфорной или .азотной) [478—480].
В кислой среде скорость иодирования значительно возрастает и в большинстве случаев
реакция протекает бурно.
4-Иодаиизол [478]. К смеси 5,93 г анизола, 6,97 г порошкообразного
иода, 30 Л4л 95%-ного этилового спирта и 1,0 мл H2SO4 {d — 1,83) в течение
20 мин маленькими порциями приливают 10,0 мл 30%-ной Н2О2. В промежутках
между добавлениями перекиси смесь слабо подогревают и энергично переме-
шивают. После введения примерно 5 мл раствора Н2О2 происходит бурная реак-
ция, которой дают утихнуть перед дальнейшим добавлением перекиси. После
охлаждения и разбавления водой выделяется темное масло, которое извлекают
эфиром. Экстракт промывают раствором Na2S2O3 и водой, сушат над СаС]2
и отгоняют эфир. Остаток кристаллизуют в холодильнике и получают 11,2 г
продукта; т. пл. 49° С. После перекристаллизации ив 90%-ного этилового спирта
получается 10,18 г (80% от теоретического); т. пл. 50° С.
Аналогично получают 1-иод-2-метокси(этокси)нафталин.
3,5-Дииодсалициловая кислота [479]. К энергично перемешиваемой
смеси 4,0 г салициловой кислоты, 7,4 г порошкообразного иода, 2,0 мл H2SO4
(d = 1,83) и 20 мл 95%-ного этилового спирта при 60° С порциями по 0,5 мл
в течение 10 мин прибавляют 4,5 мл 30%-нои Н2О2. После введения 3 мл Н2О2 -
наступает бурная реакция, температура повышается до 85° С и иод полностью
растворяется. По .окончании бурной реакции добавляют оставшуюся перекись, f
продолжают процесс еще 5 мин при 60—65° С, охлаждают и добавляют 80 мл
воды. Неочищенный продукт промывают водой, трижды 4 мл уксусной кислоты
и вновь водой, растворяют в 15 мл кипящего ацетона и выделяют кристалличе-
скую 3,5-дииодсалициловую кислоту медленным добавлением 80 мл воды при
перемешивании.
475] J. Е. М arsh, J. Chem. Soc., 1927, 3164.
476] Т. N og га di, Chem. Ber., 85, 104 (1952)
477] II. D о гл, Chem. Ber., 90, 464 (1957).
4781 T„ Jurd, Austrpl. J. Sci. Res., 2A, 595 (1949).
479] L. Jur d, Austral. J. Sci. Res., ЗА, 587 (1950).
[480] L. J u rd, J. Am. Chem. Soc., 77, 5747 (1955).
Г. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SGN
153
Иногда при иодировании в ароматическое ядро бывает целесообразно перево-
дить С—Н-связь сначала в С—металл-связь и затем замещать металл иодом. Из метал-
лов большей частью применяется ртуть, за исключением особых случаев, в которых
используется литий [481].
CeH6OH + (CH8COO)2Hg ———o-CH3COOHgCeIl4OH
—LHaGOUH
o-CH3COOHgCeH4OH ——o-ClHg—CeH4—OH + CHsCOONa .
2o-ClHg—С,Н,- ОН — -► 2'i-l С, И,—OH + HgCl2 + Hgl,
Введение ацетоксимеркур- или хлормеркургруппы в ароматические соедине-
ния или в гетероциклические соединения, обладающие ароматическими свой-
ствами, легко осуществимо, но и при этом в большинстве случаев образуется
грудноразделимая смесь изомеров.
о-Иодфенол [483]. о-Хлормеркурифенол (получение см. [482]) в хлоро-
форме 2 ч перемешивают с иодом, отсасывают смесь HgCl2 и Hgl2. отгоняют
большую часть хлороформа и остаток встряхивают с раствором KI для удаления
растворенного Hgl2. Перегонкой в вакууме получают o-иодфенол. Выход 63%
от теоретического; т. кип. 130° С (18 мм рт. cm.). -л >
2,4-Дииодфенол [485]. К энергично перемешиваемой суспензии 2Q ?
2,4-диацетоксимеркурифенола [484] в 10%-ном растворе KI добавляют раствор
33 г иода и 33 г KI в 300 мл. воды. Получается 2,4-дииодфенол с хорошим вы-
ходом.
Хлормеркурисоединекия настолько устойчивы, что перед обменом группы
— llgCl на галоген их можно подвергнуть значительным превращениям. Так, напри-
мер. получаемое из толуола [486] или из п-толуолсульфокислоты [487] соединение
n-CeH4IlgClCH8 может быть окислено щелочным раствором перманганата калия
в n-хлормеркурибензойиую кислоту [488], которая по реакции с иодом превращается
в 4-иодбензойную кислоту [489]; ряд примеров приведен на стр. 189.
Эфиры фенолов можно хлорировать в ядро хлором, более удобно дозируемым
SO2C12 в ледяной уксусной кислоте, а также при помощи РС1б.
Дихлор- и трихлорфеноксиуксусные кислоты. Из феноксиуксусной кис-
лоты с 2 моль хлора в ледяной уксусной кислоте образуется 2,4-дихлор-, а прщ
избытке хлора 2,4,6-трихлорфепоксиуксусная кислота. При медленном нагре-
вании феноксиуксусяои кислоты с избытком 8О2С12 от 0 до 75° С и ввдержива-
лпи при этой температуре до окончания выделения газа получается 2,4-дихлор-
фрцокспуксусная кислота с выходом 74% от теоретического. При взаимодей-
ствии с SO2CI2 в кипящем СС14 хлорирование не происходит [490].
[481] Н. Gilman et al., J. Am. Chem. Soc., 67, 350, 1479 (1945); 3. Org. Chem,»
19, 560 (1954).
[482] F. C. \V h i t in о г e, E. R. Hanson Org. Synth., 4, 13 '(1925).
[483] F. C. Whitmore, E. R. Hanson, Org. Synth., 4, 37 (1925).
[484] J. C. Cains, 3. H. Wadi a, J. Indian Chem. Soc., 6, 613 (1929);
C., 1930, I, 46.
[485] S, Vawzonek, S. C. Wang, J. Org. Chem,, 16, 1271 (1951).
I486] O. Di mroth. Ber., 32 761 (1899)- W. Steinkopi, Ann., 413, 329
(1917).
[487] W. Peter s, Ber., 38, 2569 (1905); F. C. W h 14 m о г e, F. H. Hamil-
ton, N. Thurman, Org. Synth., 3, 99 (1923).
[488] F. C. W h i t m о re, G. E. W oo d w ar d, J. Am. Chem. Soc., 48, 534
(1926); Org. Synth., 7, 18 (1927).
[489] F. C. Whitmore, G. E. Woodward, Org. Synth., 7, 58 (1927).
[490] M. Fiei ds, S. R о t hchi Id M. A. Lealfei, 3. Am. Chem. Soc.,
74, 2435 (1952).
156
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Для бромироважя в ядро спрдипаний с окси-, алкокси- или амидогруппами
в мягких условиях рекомендуется пропускать насыщенный парами брома воздух,
азот пли двуокись углерода через водный раствор или суспензию ароматического
соединения. Такое бромирование может быть осуществлено и в отсутствие влаги,
если до и после реакционного сосуда присоединить хлоркальциевые трубки.
4-Броманнаол [492]. В охлажденный раствор 200 г анизола в 750 мл
ледяной уксусной кислоты при перемешивании пропускают воздух, насыщенный
парами брома. Реакцию ведут в трехгорлой колбе, снабженной газоподводящей
трубкой, опущенной в раствор, н отводной трубкой, соединенной с водоструй-
ным насосом. К газоподводящей трубке присоединяют двухгорлую колбу, в ко-
торой находится 310 г брома с погруженным в него широким капилляром. Полу-
ченный в результате реакции бледно-желтый раствор вливают в 4 л воды, бром-
анизол отделяют и перегоняют в вакууме; выход около -83% от теоретического;
т. кип. 120° С (12 мм рт. ст.).
Аналогично получают 4-бромвератрол (выход 80% от теоретиче-
ского) [493].
4-Бромаиизол образуется из анизола также по реакции с РВг6; 4-хлор-
анизол [491] получают аналогично по реакции с РС15.
Моно- и дибромзамещениые эфиры фенолов можно получать при действии на
растворы эфира в сухом бензоле или СС14 N-бромсукцинимидом (1 — 2 моль), который
вносят в раствор в несколько приемов при перемешивании и охлаждении водой
(ср. стр. 138, 142).
Метод Биркенбаха —Губо — Уотерса (стр. 140) пригоден также для введения
брома н пода в ядро феноловых эфиров.
4-Иоданизол. Нагревают 20 мл анизола с 4,40 г трифторацетата серебра
до 100° С и затем охлаждают до комнатной температуры. После добавления 5,10 а .
порошкообразного иода тотчас выпадает Agl. Реакционную массу нагревают
15 мин при 160° С, охлаждают, фильтруют и перегоняют. Выход 75% от теоре-
тического; т. кип. 235—237° С.
4-Броманизол. Получают аналогично по реакции с бромом при 120° С.
Выход 76% от теоретического, т. кип. 110—115° С (23 мм рт. ст..}.
Способ пригоден также для получения из анилина 4-иодапилина (выход
51%) и 4-броманилина (выход 62%).
4-Иод-1,2-диметоксибепзол [380, 494]. Раствор 1 моль иода в CHClg
приливают по каплям при комнатной температуре и перемешивании к суспен-
зии 1 моль сухого трифторацетата серебра в 1 моль сухого вератрола и ведут •:
реакцию в течение 3 ч. Отсасывают осадок Agl, промывают CHClg и перегрняют л
фильтрат. Получается 4-иод-1,2-диметоксибензол; выход 85—91% от теорети- >
ческого.
4-Иоданизол удобнее получать иодированием анизола IC1 в ледяной уксусной
кислоте [495], хотя выход продукта при этом ниже.
Прииером иодирования эфиров фенолов системой 12—Н1О8 является получе- ;
ние 4-иоданнзола п 2,4-дииоданнзола f420d].
4-Иоданизол. При энергичном перемешивании смесь 12 г анизола, 45 мл rs
ледяной уксусной кислоты, 20 мл воды, 0,7 мл концентрированной H2SO4, 15лл к
СС14, 10,2 г иода и 3,9 г йодноватой кислоты нагревают при 40° С до полного J
израсходования иода (3 ч). Осажденный водой продукт иодирования растворяют А
в СНС1з, раствор высушивают и пропускают через колонку с окисью алюминия, J
[491] W.Authcnrieth, Р. Miihlinghaus, Вег., 39, 4098 (1906),
[492] К. П. Slotta, Н. Heller, Вег., 83, 3043 (1930).
[493] W. М. W h al е у, С. White, 3. Org. Chem., 18, 184 (1953),
[494] D. Е. J a n ss en, J. v an Al len, С. V. Wilson, J. Org. Chem.,
20, 1328 (1955).
[495] H. R. Frank. P. E. Fanta, D.S. Tarbel, J. Am. Chem. Soc., 70,
2317 (1948). *
Т. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
15‘
которая предварительно обработана NHa, высушенным над КОН до нрекраще
ния первоначального разогревания. После отгонки растворителя получаю
4чгоданпзол после двогЧиой перекристаллизации из метилового сппрта. Выхо;
73% от теоретического; т. пл. 52° С.
2,4-Дииоданизол. Реакцию ведут, каи описано выше, во берут половинно
количество анияола (6 г) и воды (10 мл) и проводят реакцию 2 ч при 40° С, 2
при 60° С и наконец, при 75° С. Выход 2,4-дииоданизола 72% от теоретичесного
т. пл. 67° С.
Давпо известный метод иодирования подом в присутствии IlgO описан н;
стр. 187. Для связывания образующегося Н1 вместо HgO может применяться такж
ацетат ртути (11) в ледяной уксусной кислоте. Так, например, из 3,3'-диметоксида
бензила получают б.б'-дииодпроизводпое с выходом 93% от теоретического [496]
Получение феноксиметилхлоридов описано на стр. 258. Солеобразовани,
(среда — минеральная кислота), N-ацилирование и N-алкилирование снижают спо
собность ароматических аминов вступать в реакцию с галогенами. Взаимодействие
с галогенами подвергают водные растворы, гндрогалогеяндов аминов или применяю
в качестве растворителя ледяную уксусную кислоту, и которой для связывании обра
зующегося галогеноводорода добавляют ацетат натрия. Так же как при бромирова
нии фенолов, особенно большой интерес представляет получение моно- и дибромпро
взводных аминов.
4-Броманмлин [497]. Можно получить, приливая по каплям при охла
ждении льдом немного менее 1 моль брома в ледяной уксусной кислоте к рас
твору 1 моль анилина в том же растворителе.
Хлорирование (бромирование) обоих орто-положений анилина достигается
с помощью искусственного приема, аналогичного применяемому для фейола, а имени,
замещением пара-положения легко отщепляющейся группой. В качестве примеры
одновременно демонстрирующего метод галогенирования с помощью хлористого (бро
мистого) водорода и перекиси водорода (более старые данные см. таиже [498, 499]
можно привести получение 2,6-днхлор- или 2,6-днброманилина:
NH2 nh2
I х I x
0 + 2HX ; 2HBO3 —0Y h.°;h.so^
I I
SO2NII2 SOBNI1B
r.ie X =С1 или Br«
2,6-Дихлор(днбром)анилин [500]. К раствору 1 лоль сульфаниламид:
в приблизительно 6 моль НС1 (концентрированная соляная кислота, разбавлен
ная в отношении 1 : 1) или в примерно 2,3 моль бромистоводородной кислоте
(40%-ная ПВг, разбавленная в отношении 1 : 8,5) при быстром перемешивани!
и температуре 45° С (или для диброманнлина соответственно 70—75° С) при
бавляют 2 моль 30%-ной Н8Оа. Экзотермическую реакцию проводят в теченн-
ЗОЛшм при температуре не выше 60° С (соответственно 85—90е С). В обоих слу
чаях осаждается §,5-дигалогенсульфапиламид, из которого нагреванием с 70%
ной H2SO4 и перегонкой с водяным паром получают 2,6-дигалогенанилины
[496] 1, w. С о г n f о г t h, R. R о b i n s о n, J. Chem. Soc., 1942, 684.
[497) W. Fuchs, Mh. Chem., 36, 113 (1915).
[498| A. Leulier, Bull. Soc. chim. France [4], 35, 1325 (1924).
[499] A. Zinke, Ber., 58, 330 (1925).
1500] M. K. Seikel, Org. Synth., 24, 47 (1944); Coll. v. 3, 262 (1955).
158
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Для мягкого бромирования ароматических аминов пригоден диоксандибромид
(оранжево-желтое вещество, т. пл. 64° С), легко получаемое смещением эквимоле-
кулярных количеств диоксана и брома и охлаждением разогревшейся реакционной
массы ледяной водой (501]. При использовании раствора диоксандибромида в диоксане
достаточно добавить к диоксану при перемешивании и охлаждении желаемое количе-
ство брома.
Диоксаядибромид можно получать также и следующими способами.
Диоксаядибромид
а) К 500 а диоксана прп перемешивании быстро прибавляют 990 г брома.
Горячий раствор вливают в 2 л ледяной воды, отсасывают оранжевый осадок
и высушивают его на глиняной пластинке; выход 94% от теоретического; т. пл.
60° С. Диоксандибромид умеренно растворим в дпоксане, СНС13, 1,2-дихлор-
этане, СС14, слабо — в эфире, с которым постепенно образует двухфазную си-
стему. Он малорастворим в воде и гидролизуется при комнатной температуре
через несколько дней, быстрее — при нагревании[453]. В качестве растворителей
употребляют диоксан или эфир.
б) При энергичном перемешивании добавляют 320 г высушенного брома
к 160 г перегнанного над натрием диоксана [502]. Разогревшийся до 60° С
раствор выливают в 2 л охлажденного до —20й С петролейного эфира (т. кип.
ниже 40° С), отсасывают желтый осадок и, как можно быстрее (чтобы исклю-
чить потери из-за значительной летучести диоксандибромида), в вакууме осво-
бождают осадок от оставшегося растворителя; т. пл. 60—61° С; может храниться
при 0° С *.
Амины бронируются диоксандибромидом в присутствии рассчитанного коли-
чества концентрированной щелочи; при охлаждении и перемешивании в диоксиновый
раствор амина прибавляют диоксаядибромид.
4-Бромаяилин [501]. К 9,3 г анилина в 30 мл диоксана и растворе
едкого кали (5,6 а КОН в 20 мл воды) приливают по каплям при энергичном
перемешивании и5?Св течение 2 ч 16 г брома в 160 мл диоксана. Органический
слой промывают 15 мл 40%-ного раствора КОН, отгоняют в вакууме раствори-
тель и перекристаллизовывают осадок пз разбавленного этилового спирта;
выход броманилипа 68% от теоретического.
Для иодирования ядра ароматических аминов обычно применяют иод в присут-
ствии основных акцепторов HI (а также перекиси водорода) или IC1 в соляной или
ледяной уксусной кислоте.
2, 6-Дииод-4-нитроанилин [503]. К раствору 1 моль 4-нитроанилина
в 370 мл горячей ледяной уксусной кислоты при перемешивании в течение 2 ч
приливают по каплям раствор 2 моль IC1 в 100 мл ледяной уксусной кислоты
(экзотермическая реакция!) и затем перемешивают еще 2 ч на кипящей водяной
бане. Более подробное описание см. [504].
* Удобным способом Получения диоксандибромнда является следующий.
К охлажденной льдом смеси 68 мл диоксана и 74 мл гептана приливают
охлажденный раствор смеси 141,6 г брома и 144 мл гептана, перемеши-
вают 5 мин, охлаждая стакан с реакционной массой в бане со льдом.
Выпавший оранжевый осадок отфильтровывают, промывают два раза 20 мл
гептана и на воронке подсушивают 5—6 мин током воздуха. Получают
150 г диоксандибромида, который тотчас же используют для реакции
[(Н. Н. Суворов и сотр., ЖОХ, 31, 3715 (1961)]. — Прим. ред.
[501] G. М. Kosolapoff J. Am. Chem. Soc., 75, 3596 (1953).
[502] J. 1). В i 11 i m ori a, N. F. Mad agan, J. Chem. Soc., 1954,
[503] A. Michael, L. M. Notion, Ber., 11, 113 (1878).
[504] R. B. S a u di n. W. V. Drake, F, Lege r, Org. Synth., Coll. v. 2,
1943, p. 196.
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА На! ИЛИ SCN
159
4-Иоданилин [505, 506]. Раствор 1 жоль IC1, приготовленный из 127 а
иода и 150 мл ледяной уксусной кислоты (стр. 170), приливают по каплям при
перемешивании к раствору 1 моль ацетанилида в 150 мл ледяной уксусной кис-
лоты (экзотермическая реакция!) и затем перемешивают еще несколько часов.
Выпадает 4-иодацетанилид. Через 12 ч добавляют 2 л воды и сырой продукт,
промытый водой и разбавленным раствором NaOH, перекристаллизовывают
из этилового спирта. Выход 90%; т. пл. 184° С. Омылением концентрированной
IIC1 получают 4-иоданилпн.
Получение 4-иоданилина взаимодействием анилина (1,2 холь) с порошко-
образным иодом (1 моль) в 1 л воды в присутствии 1,8 моль NaHCO8 см. [507].
Соответствующее иодирование o-толуидина в пара-положение к аминогруппе
осуществляется в эфирно-водной среде [508] в присутствии СаСО3.
8. Замещение галогеном водорода в простых эфирах
Эфиры, в которых атомы водорода замещены галогеном, можно получать непо-
средственным действием хлора или брома на соответствующий эфир. Также важны
способы получения галогензамещеииых из альдегидов, полуацеталей и ацеталей
(стр. 221. 228)» в которых, следовательно, по крайней мере формально, одна С—О-связь
заменяется связью С—Hal.
Методы непосредственной замены водорода галогеном имеют препаративное
значение только для получения хлорэфиров. О хлорировании диэтицового эфира,
исследованием которого занимались длительное время^ см. [509, 510]. При комнатной
температуре хлор вступает в диэтиловый эфир в такой последовательности: а-; а В-;
а,Р,₽,р-, что, вероятно, происходит в результате чередования отщепления НС1
и присоединения хлора. Хлорирование при температуре от —25 до —30° С при облу-
чении в значительной степени предотвращает отщепление НС1 и позволяет получить
из сухого диэтилового эфира а,а'-дихлордиэтиловый эфир с выходом 57% от теоре-
тического [511].
При работе по этому способу с меньшим количеством хлора в качестве основ-
ного продукта образуется а-хлорэтилэтиловый эфир, который фракционной перегон-
кой зпачительпо легче отделить от побочных продуктов, чем эфир, полученный по
синтезу Генри (стр. 221). Аналогично при хлорировании тетрагидрофурана при тем-
пературах от —30 до —40° Си УФ-облучении образуются 2-хлор- и 2,5-дихлортетра-
гидрофураны [512], а при хлорировании диоксана при температурах от —5 до —10’ С
в СС14—2,5-дихлордиоксан [513]. Из дииаопропилового эфира при низких темпера-
турах образуется смесь не установленного состава [514].
а ,р-Дихлорэтилэтиловыи эфир из диэтилового эфира [515]. В начале
реакции хлор необходимо пропускать в эфир медленно при охлаждении ледякой
водой, так как накопление его в реакционной массе легко может привести к бур-
ному взаимодействию, сопровождающемуся воспламенением смеси. В ходе реак-
ции смесь насыщается НС1 и опасность воспламенения снижается. Однако при
недостаточно интенсивном перемешивании из-за пересыщения смеси НС1 может
произойти выброс массы вследствие мгновенного резкого выделения НС1.
A. Michael, L. М. Norton, Вег., И, 108 (1878).
F. D. Chat t away, А. В. Constable,!. Chem. Soc., 1914, 124.
R. Q. Brewster, Org. Synth., Coll. v. 2, 1943, p. 347.
A. A. G о 1 d b о r g, J. Chem. Soc., 1942, 713.
A. L i e b e n, Ann., 146, 180 (1868).
O. Jacobsen, Ber., 4, 215 (1871).
G. E. Hall, F. M. Ubeitinl, J. Org. Chem., 15, 715 (1950).
H. Grog, Angew. Chem., 72, 268 (1960).
L. A. Bryan, W. M. Smedley, R. K. Summerbell, J. Am. Chem.
Soc., 72 , 2206 (1950).
G. E. П a 11, I. S i г e 1, J. Am. Chem. Soc., 74, 836 (1952).
E. A. Wildman, H. G г a y, J. Am. Chem. Soc., 41, 1122 (1919).
160
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Хлорированием 800г диэтилового эфира сначала при охлаждении льдом (35ч до
плотности реакционной массы 0,785 г/сл3) и затем при постепенном повышении
температуры до комнатной (еще 47 ч до плотности 0,96 г/см?) получают а.£-ди-
хлорэтиловый эфир; выход 24,2% от теоретического.
При хлорировании диметилового эфира, в котором нет р -углерода, замещение
происходит преимущественно в последовательности а,а'-[516]; а,а,а'-[517, 518};
а,а,а,а' ,а'-;а,а,а,а' ,а'',а'-.
Для получения полихлордиметиловых эфиров любой степени хлорирования
в жидкий хлордиметиловый эфир одновременно вводят диметиловый эфир и хлор при
охлаждении водой (внутренний холодильник) и интенсивном УФ-облучении [519].
Во избежание взрыва реакция должна проводиться в атмосфере азота до тех пор,
пока не начнется интенсивное выделение HCL
сс.а-Дихлордиметиловый эфир получают хлорированием димегилового эфира.
Быстрый лабораторный способ получения малых количеств а,а-дихлорметилалкило-
вых эфиров, основанный на превращении иарбоннльной группы в группу > CCL,
заключается во взаимодействии алкиловых эфиров муравьиной кислоты с РС1в
(стр. 241).
а,а- и а,а'-дихлорднметиловые эфиры. Хлорирование в газовой фазе моно-
хлорциметилового эфира хлором, разбавленным азотом, проводят при УФ-облу-
чении и 90—95° С в насадочной колонке (подробное описание см. [520]), состо-
ящей из трубки для возврата жидкости и расположенного сбоку от нее более
широкого реактора. Хлорируемая в реакторе газовая смесь конденсируется
в обратном холодильнике, установленном таким образом, что конденсат стекает
в колонку через трубку для возврата жидкости, минуя реактор, а более н-иэко-
кипящий (60° С) моиохлордиметиловый й>ир постоянно возвращается в реак-
ционный объем. В перегонной колбе собираются а,а'-(т. кип. 101—103° С) и
а.а-дихлордиметиловый (т. кип. 84—86,5° С) эфиры, а также более высокохлори-
рованные продукты.
При периодическом процессе хлорирование ведут до тех пор, пока темпе-
ратура выходящих из колонки паров не достигнет 64° С. После фракционной
перегонки продуктов хлорирования из 599 г монохлордиметилового эфира полу-
чают 225 г а,а- и 459 г а,а'-дихлордиметилового эфира. Пропускание выделя-
ющегося НС1 в смесь метилового спирта и формалина [521] дает дополнительное
количество мопохлордпметилового эфира.
Хлорирование диоксана ведет, в зависимости от условий реакции, к получению
Многочисленных продуктов установленного строения (см.’обзор [522]). 2,5-Дибром-
дигидрофуран удается получить 1,4-присоединеннем брома к фурану [523].
Бромирование диалкиловых эфиров приводит к бромальдегидам и ацеталям
[524,525]. а,Р-Дибромалкиловые эфиры получают из сс-хлоралкиловых эфиров
и брома:
CH3CHC1OC2H8 + Br2 —> СН2ВгСНВгОС.2П5 + НС1
Образующийся хлористый водород уДаляют в конце реакции в вакууме в токе дву-
окиси углерода.
516] F. М. L i t t е г s с h е i d, Ann., 330, 112 (1904).
517] A. de S ona y, Ber., 27R, 337 (1894).
518] H. S. Booth, P. E. Burchfield, J. Am. Chem. Soc., 57, 2070 (1935),
519] O. Neu n ho eff er, G. Schmidt, Chem. Techn., 10, 103 (1958).
520] H. Grofi, Chem. Techn., 10, 659 (1958).
521] C. S. Marvel, P. K. Porter, Org. Synth., 9, 58 (1929).
[522] H. Stumpf, Chemie und Anwendungen des 1,4-Dioxans, Verlag Chemie
Weinheim, 1956.
[523] C.-H. Schmidt, Angew, Chem., 68, 175 (1956).
[524] C. Mauguio,, С. r., 147, 747 (1908).
[525] К. К г a t z 1, K. Schubert, Mh. Chem., 81, 988 (1950).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
161
а ,р-Дибромэтилэтиловый эфир [526]. В смесь 100 г паральдегида и
100 г этилового спирта, охлаждаемую смесью льда с солью, осторожно про-
пускают сухой НС1; при этом образуется два слоя. Верхний слой высутпивлют
над СаС12 и разгоняют, собирая фракцию в интервале 93—96° С; выход 69%.
К полученному таким образом а-хлорэтплэтиловому эфиру при охлаждении льдом
постепенно (каждый раз после прибавленной порции ждут, когда произойдет
обесцвечивание) приливают по каплям теоретическое количество брома; при
этом выделяется НС1. Неочищенный продукт перегоняют в вакууме, собирая
фракцию в интервале 92—95° С (17 мм рт. ст.), выход а,р-днбромэтилэти~
левого эфира 88—91% от теоретического.
1,5-Диэтокси-1,2,4,5-тетрабромпентан [527]. Тетраэтилацеталь глута-
рового диальдегида обрабатывают РС1б при 30—40и С и перемешивании и бро-
мируют образовавшийся 1,5-дпэтокси-1,5-дихлорпегггаи при 0® С.
а-Галогенэфиры легко отщепляют галогеноводород даже при относительно низ-
ких температурах. Они чувствительны к нуклеофильным реагентам, следовательно
и к воде. В этих эфирах галоген значительно более подвижен, чем в алкилгалогенидах,
к обменивается легче, чем алкоксильная группа. Обзор способов получения а-гаяо-
генэфнров. см. [528].
9. Замещение галогеном водорода в алифатических монокарбоновых
кислотах
При хлорировании неразветвленных алифатических карбоновых кислот пред-
почтительное замещение в то или иное положение зависит от того, преобладает ли ион-
ный или свободнорадикальный механизм хлорирования [529, 530]. Ионное (электро-
фильное) замещение хлором происходит в сс-положение к карбоксильной группе.
Ему благоприятствуют добавка поляризующих катализаторов (FeCl8; иод) и исклю-
чение доступа света. а-Хлорнрованаё облегчается также индуктивным действием
групп СООН, COOR (в эфирах) или С0С1 (в хлорангидридах кислот). Хлорангидрид-
ная группа оказывает значительно более индуктивное действие, чем сложноэфирная
или карбоксильная группы. Поэтому хлорирование хлорангидридов кислот Протекает
с особенно хорошими выходами, а хлорирование эфиров жирных кислот с незначитель-
ными выходами.
Уже давно известно получение а-хлормасляной кислоты хлорированием
ее хлорангидрида ппи температуре кипения в присутствии иода с последующим
омылением водой [531].
В систематической работе Гриляссотр. [532] исследовано хлорирование жириИх
кислот со средней длиной цепи в присутствии катализаторов, оказывающих поляри-
зующее действие или промежуточно образующих хлорангидриды (иод, сера, фосфор»
SOCL, РС1#); лучше всего действует смесь иода и PC1S. Хлорирование ведут, применяя
эквимолекулярные количества хлора и кислоты, скорость подачи хлора 25—50 л/м
при температуре — для масляной кислоты 100—160° С, а для н-капроновой и высших
кислот — не выше 100—120° С. Поскольку при хлорировании высших жирных кислох
повышается вероятность протекания побочных реакций, необходимо поддерживать
возможно более высокую концентрацию образующегося па промежуточной стадии
[526] (I. S w all en, С. Е. В о ord, J. Am. Chem. Soc., 52, 654 (1930).
[527] P. Baudart, Bull. Soc. chim. France, 11, 336 (1944).
1528] T,. Summers, Chem. Rev., 55, 301—342 (1955).
[529] M. S. Kharaech, H. C. Brown, J. Am. Chem. Soc., 62, 925 (1940).
[530] В. E i g t о г t, Chemismus und Konstitution, Bd. I, F. Enke^ Stuttgart, 1948,
S. 321
[531] W. W. Markownikoff, Ann., 153, 241 (1869).
[532] W. Griehl, W.-J. Schulze, H. Furst, Chem. Ber., 91, 1165 (1958),
И Заказ 1835-
162
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
хлорангидрида кислоты в возможно более низкую концентрацию пропускаемого
хлора. Установлено, что УФ-облучение гораздо сильнее благоприятствует протека-
нию побочных реакций, чем видимый свет или ИК-облучение. Несмотря на то что
облучение способствует свободнорадикальному хлорированию, ведущему к образо-
ванию наряду с а-хлорзамещенными жирными кислотами также жирных кислот,
замещенных хлором у остальных атомов углерода (см. киже), в двух следующих
примерах кислоты и хлорангидриды хлорируются при добавочном воздействии види-
мого света. При проведении реакции в темноте выходы, по-видимому, снижаются [532].
Однако если несколько продлить время протекания реакции, то, вероятно, можно
отказаться от освещения.
а-Хлормасляная кислота 1532]. Смесь 1 моль масляной кислоты,
0,005 моль иода, 0,038 моль фосфора и 0,006 моль РС1а обрабатывают при пере-
мешивании, 100° С и освещении лампой в 200 вт 1 моль хлора (скорость подачи
хлора 50 л/ч). Затем небольшие количества образовавшегося хлорангидрида
омыляют в течение 30 мин 10 мл воды. Простой перегонкой выделяют а-хлорма-
сляную кислоту; выход 71% от теоретического, считая на прореагировавшее
вещество. Дальнейшим тонким фракционированием выделяют чистую а-хлор-
масляную кислоту; т. кип. 104,2° С при 20 мм рт. ст. (колонна заполнена
насадкой на высоту 1 ле и соответствует 18 теоретическим тарелкам).
Хлорангидриды кислот до Св хлорируются с хорошими выходами; слишком
высокая концентрация хлора снижает выход [532].
Получение хлорангндридов а-хлоркарбоновых кислот. На бане нагре-
вают 1 моль хлорангидрида кислоты до 100° С и при перемешивании со скоростью
25 л/ч вводят в него 1,1 моль хлора, все время освещая реакционный раствор
лампой в 200 от. По окончании реакции продукты хлорирования разделяют раз-
гонкой. а-Хлорбутирилхлорид (т. кип. 130—131° С) получают с выходом 92% от
теоретического, считая на хлорангидрид, а хлорангидрид а-хлориаовалериановой
кислоты — с выходом 89% ; т. кип. 155—156° С.
Рекомендуется добавка поляризующих катализаторов, например иода.
Так, пропуская хлор в кипящий хлорангидрид (30 г) с добавкой 0.5 г нода до
достижения теоретического привеса [533] получают а-хлорпропиопил- и -бу-
тирилхлориды.
В темноте без катализаторов SOgClg при 70—90° С слабо хлорирует али-
фатические кислоты и их хлорангидриды. Однако в присутствии переносчиков
галогена получаются а-хлорзамещенные соединения.
При кипячении d течение 12 ч с обратным холодильником 0,5 моль про-
лиоиилхлорида с 0,3 моль SOaCl2 и 0,5 г иода получается 22,0 г чистого а-хлор-
пропиоиилхлорида, не загрязненного (1-изомером [529].
е-Беизоиламино-а-хлоркапроновая кислота [534]. Растворяют 35,3 г
(0,15 моль) е-бензонламииокапроновой кислоты и 0,5 г тонкоизмельчодиого
иода в 70 мл SOaCla и нагревают смесь на водяной бане при 60—65° С до пре-
кращения выделения газа (60—90 мин). В течение следующих 60—90 мин водя-
ную баню медленно доводят до кипения, затем отгоняют избыточный SOaCl2 и
остающуюся неочищенную е-бензоиламипо-а-хлопкапроновую кислоту промы-
вают водой; выход 96—97,5% от теоретического. Полученный продукт без даль-
нейшей очистки используют для получения лизина.
При применении к нерааветвленным гомологам хорошо изученного способа
хлорирования уксусной кислоты хлором в присутствии красного фосфора на солнеч-
ном свету (синтез монохлоруксусяой кислоты см. [535]) получается смесь хлорзаме-
щенных [536]. В разветвленных кислотах гладко замещается лишь третичный а-атои
[533] В. Wolffcnstein, J. Rolle, Вег., 41, 735 (1908).
[534 ] A. G а 1 a t, J. Am. Chem. Soc., 69, 86 (1947).
[535] L. Gattermann, Die Praxis des organischcn Chemikers, 37 Aufl., W. de
Gruyter u. Co., Berlin, 1956, S. 109.
[536] H. H. Guest, Ch. M, Goddard jr., J. Am. Chem. Soc., 66, 2074 (1944).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НЛ II.il ИЛИ SCN
163
водорода. Пз изомасляной кислоты получают а-хлоризомасляную кислоту, из изо-
валериановой [(СН3)2СНСН2СООП] — р-хлорпроизяодиое.
Свободнорадикальной реакции замещения хлора способствует отсутствие
поляризующих катализаторов, освещение прямым солнечным светом или УФ-облуче-
пие, а при работе с SO3CIs добавка перекисей. При хлорировании жирных кислот и их
хлорангидридов в таких' условиях получается незначительное количество а-хлор-
замещенных и много 0 - и у-хлорзамещенных [537]. При фотохлорированин бутирил-
хлорида в ССЦ (солнечный свет, 0° С) образуется около 20% а-, 60% 0 - и 20% у-хлор-
бутирилхлорпда [538].
Жирные кислоты и их хлораягидриды при помощи сульфурилхлорида в при-
сутствии перекисей почти иоличественио превращаются в хлорпроизводные. Ледяная
уксусная кислота является исключением; не более 50% кислоты превращается в хлор-
уксусную кислоту; ацетилхлорид не Хлорируется сульфурилхлоридом и в присут-
ствии перекисей лаурила и бензоила. Ввиду бурного протекания катализируемой
перекисями реакции хлорирования жирных кислот сульфурилхлоридом рекомендуется
добавка разбавителей (CCL, хлорбензол). Хлорангидриды кислот реагируют гораздо
менее бурно и поэтому добавки разбавителей не требуется. ч
На следующем примере показано, в каком соотношении образуются изо-
меры при хлорировании некоторых жирных кислот SO2Cla в присутствии пере-
кисей [529]. Смесь 0,8 моль жирной кислоты, 0,4 моль SO2CI2, 0,4 моль СС14
и 0,002 моль перекиси бензоила умеренно нагревают с обратным холодильником
в темноте до окончания выделения SO2 и НС1 (60—90 мин). Затем нагревание
ведут в течение еще 4 ч с 1,6 моль тионилхлорпда и получают смесь хлорангид-
ридов, которую фракционируют после отгонки избыточного тионилхлорпда
п четыреххлористого углерода. Состав смеси: 45% а- и 55% р-хлорпропионил-
хлорида, 15% а- и 85% р-хлоризобутирплхлорида и 10% а-, 45% |3~ и 45%
у-хлор-и-бутирилхлорида.
«-Бромирование идет чище, чем «-хлорирование. При а-хлорироварии жирных
кислот и их хлорангидридов трудно избежать образования побочных продуктов, при
бромировании получаются только а-бромпроизводные. В изовалериановой или изо-
капроновой кислотах в условиях ионной реакции бромом замещается а-атом водо-
рода, а не третичные р- или у-атомы [539]. а-Бромалкановые кислоты и их эфиры
можно синтезировать непосредственно или через стадию образования галогенангид-
ридов кислот.
Электрофильное замещение бромом а-СН2-группы карбонильных соединений
протекает тем легче, чем меньше мезомерная стабилизация карбонила. Трудность
замещения увеличивается в ряду; кетон, хлорангидрид кислоты, аигидркд, сложный
эфир, карбоновая кислота, амид кислоты [540]. Эту закономерность можно, в част-
ности, продемонстрировать на примере бромирования левулиновой (у-кетовалериано-
вой) кислоты. В присутствии красного фосфора эта кислота бронируется 2 моль
брома не в «-положение к карбоксильной группе, а по обоим атомам углерода, сосед-
ним с карбонильной группой, с образованием 3,5-дибромлевулнновой кислоты [541].
Но Геллю — Фольгарду — Зелинскому [542—544] безводную кислоту подвер-
гают взаимодействию с красным фосфором и бромом. Красный фосфор предварительно
промывают водой от следов кислот и высушивают при 100° С; бром обезвоживают
встряхиванием с концентрированной H2SO4. Можно применять 1 моль красного фос-
фора, ио также и каталитические его количества (примерно 3%). Вместо красного
фосфора можно вводить каталитические количества PC]S. Для получения свободных
а-бромзамещепных жирных кислот 1 моль низшей или средней жирной нис-
лоты подвергают взаимодействию с 1,1 моль брома (10%-ный избыток) и отгоняют
N. О. V. Sonntag, Chem. Rev., 52, 359 (1953).
A. Michael, W. W. Garner, Ber., 34, 4046 (1901).
K. Fischer, Ber., 36, 2988 (1903).
I n £ ° 1 d, c. W. S h о p p e c, J. F. Thorpe, J. Chem. Soc., 1926,
Ch. D. ГГ u r d, J. R. F e г г а г о, I. Org. Chem., 16 1640 (1951).
С. H e 11, Ber., 14, 891 (1881).
J. V оД h а г d, Ann., 242, 141 (1887)
N. Belinsky, Ber., 20, 2026 (1887).
11*
164
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
соответствующую сс-бромкислоту из реакционной смеси в вакууме, создаваемом водо-
струйным насосом. При бромировании высших жирных кислот применяют немного
более 2 моль брома и разлагают водой неочищенный бромангидрид а-бромкислоты,
разбавленный CCI4.
Этиловый эфир а-бромфенилуксусной кислоты [545]. Растертую в ступке
смесь 275г фснилуксусной кислоты и 15г красного фосфоравысушивают несколько
суток в эксикаторе надСаС18. Затем к этой смеси в колбе емкостью2л при встрлхи- \
вании прибавляют 200 г брома, высушенного концентрированной H2SO4. Реакци-
онную массу нагревают, сначала осторожно, а затем на кипящей водяной бане
п добавляют к ней еще 320 г брома. Затем нагревают еще 2 ч, охлаждают, раз- .<
бавляют 200 мл абсолютного этилового спирта, оставляют на ночь и кипятят • J
еще 1 ч с обратным холодильником. После выливания в воду высаливают этило- ?
вый эфир сс-бромфенилуксусной кислоты и извлекают СНС13; выход 75% 3
от теоретического. 1
В качество примеров бромирования средних или высших жирных кислот '^
см. синтезы а-бромизовалериановой [546], а-бромизокапроновой [547], а-бром-н-кап- 3
роновой [548] и а-бромдодекаяовой [549] кислот. >]
а -Бромизовалериановая кислота [546]. Смесь 8,6 моль изовалериановой
кислоты, обезвоженной азеотропной перегонкой с бензолом, 1500 г брома, вы- 1
сушенного встряхиванием с 1 л концентрированной H2S04, и 15 мл РС13 нагре- то
вают при 70—80° С до исчезновения в обратном холодильнике темно-красной я
окраски паров брома (10—20 ч). Затем вносят еще 25 мл брома, нагревают до 1
исчезновения бурых паров, медленно повышают температуру масляной бани
до 100—105° С и выдерживают массу 1,5—2 ч при такой температуре. После J
этого разгоняют в вакууме и фракцию с интервалом кипения 110—125° С (15 мм -Я
рт. ст.) используют для получения О t-валина; выход фракции 87,5—88,6% . «я
а -Бромизокаироновая кислота [547]. Получается из 4,3 моль кислоты, J»
4,65 моль брома и 10 мл РСЦ; выход 63—66% от теоретического. Фракция 125— ;
131° С (12 мм рт. ст.) служит для синтеза Г) Г-лейциня
а-Бромдодекановая кислота [549]. Для получения этой кислоты в при- ’
сутствии PC1S вполне достаточно 2 моль брома на 1 моль кислоты. J
Можно обойтись и меньшим количеством брома, прежде всего для бромирования:
кислот с длинной цепью, если эти кислоты действием тионилхлорида SOC12 сначала?
перевести в хлорангидриды и затем бронировать [550]. Небольшая добавка иода'
при этом ускоряет реакцию, но зачастую необходимости в ней нет. Хлорангидрид^
а-бромзамещенных кислот можно перевести в кислоты действием воды, а в соответ-i
ствующие эфиры — действием спиртов. По этому способу легко, например, полу-5
чается этиловый эфир а-бромпрониоибвой кислоты [551].
Метиловые эфиры неразветвленных жирных (С4_12; С14; С]в и С]8)а-бром^
кислот [552]. Смесь 1 моль жирной кислоты и 150 г SOC12 нагревают при 80—|
100е С до окончания выделения газов (3—4 ч). Затем добавляют несколько кри^
сталлов иода и приливают по каплям 1,2 моль брома в течение 4 ч при работе'
с низшими кислотами и 1,4 моль брома в течение 10 ч при работе с высшими кис-'
лотами, следя за тем, чтобы в верхней части холодильника не было заметно брома^
[545]
Г546
547
548
549
550
551
552
В. М. Родионов, Н. Н. С у в о р о в, К. С. Михайлов, Син<
тезы органических соединений, Сборник 2, Изд. АН СССР, 1952.
С. s. м а г V е I, Org. Synth., 20, 106 (1940); Coll. v. 3, 848 (1955).
C. S. Marvel, Org. Synth., 21, 74 (1941); Coll. v. 3, 523 (1955).
H. T. Clarke, E. R. Taylor, Org. Synth., Coll. v. 1941, p. 115.
C. F. A 11 e n, M. 3. К a 1 m, Oig. Synth., 37, 29 (1957).
Ch. K. Ingold, 3. Chem. Soc., 119, 316 (1921).
A. I. Vogel, J. Chem. Soc., 1948, 648. ?
H. Reinheclcel, Chem. Ber., 93, 2227 (1960). 4
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
165
и нагревают еще 2—4 ч на кипящей водяной бане. Реакционную смесь выливают
в 125 мл кипящего метилового спирта, причем высшие кислоты образуют два
слоя. Бромэфир выделяют, выливая реакционную массу в холодную воду,
затем растворяют его в этиловом эфире и последовательно промывают водой,
растворами NaHSOs, Na2CO3 и водой (при работе с высшими кислотами соду
не применяют во избежание образования эмульсий). После высушивания над
Na»SO4 отгоняют эфир и остаток фракционируют в вакууме, создаваемом прц
работе с кислотами (до С18 водоструйным, а с более высокомолекулярными —
масляным насосом). Чистоту эфира оценивают по коэффициенту преломления.
Ниже, на примере синтеза метилового эфира а-бром-н-масляной кислоты пока-
зано, что при работе с небольшим избытком спирта этот способ еще более упрощается.
Метиловый эфир а-бром-н-масляной кислоты [553]. К 112 г техниче-
ского SOC12 приливают в течение 75 мин при обычной температуре и перемеши-
вании 70,4 г «-масляной кислоты. Затем смесь нагревают 30 мин на водяной баие
и отгоняют избыток SOC12. В заключение при 80—90° С прибавляют 135 а брома
в течение 1 ч при перемешивании и нагревают реакционную массу 5 ч на кипя-
щей водяной бане. Затем ее охлаждают и, продолжая охлаждение, приливают
по иаплям 46 мл метилового спирта, после чего кипятят 2 ч с обратным холодиль-
ником. В делительной воронке отделяют ненужный незначительный верхний
слой. Перегонку ведут сначала при атмосферном давлении и отбрасывают не-
большую фракцию, отгоняющуюся при 100° С, далее перегонку ведут в вакууме
и собирают фракцию, кипящую в интервале 70—75° С (при 15 .нж рт. ст.); выход
эфира 75—86% от теоретического.
Синтез а-бром-н-масляной нислоты омылением горячей водой неочищен-
ного бромангидрида, образовавшегося по реакции масляной кислоты с красным
фосфором и бромом, см. [554].
Этиловый эфир бромуксусной кислоты. Получают бромированием
17,5 моль кипящей ледяной уксусной кислоты, к которой добавлено 200 жл ук-
сусного ангидрида и 1 мл пиридина, 7,03 жаль брома. Получается бромуксус-
ная кислота, которую этерифицируют смесью этилового спирта с бензолом при
непрерывном удалении воды [555]. Более старые методы бромирования красный
фосфором и большим избытком брома см. [556, 557].
Эфиры карбоновых кислот не бронируются бромом. Одиако можно бронировать
смесь эфиров с хлорангидридами кислот в молярном отношении от 1 ; 1 до 5 : 1, при-
чем достигаются такие же выходы, как при бромировании 2—6 моль хлор-
ангидрида [558]. При этом происходит переэтерификация промежуточного а-бром-
хлорангидрида кислоты эфиром кислоты с образованием эфира а-бромированной
жирной кислоты и хлорангидрида:
RCHGOC1 + RCH2COOCH3 1=7 RCHCOOCHs-J-RCHaCOCl
Из полуэфиров хлорангидридов дикарбоновых кислот, ROOC—(СНг)„—СОС1,
в^иоторых имеются как эфирная, так и хлораягидридная группы, можно без особых
искусственных приемов, но с умеренными выходами получать неочищенные полу-
эфиры моно-а-бромхлораяпадридов дикарбоновых кислот ROOC (CH2)«-i CHBrCOCJ
(см. бромирование в тионилхлориде, стр. 167). При применении 1 моль брома
образуются еще и монобромировапные диэфиры и диэфиры а,а'-дибромдикарбоновых
кислот или их хлорангидриды. Таким образом, в результате превращения образуется
смесь диэфиров а-моно^ром- и а,а'-дибромдикарбоновых кислот.
[553] II. Cassebaum, Arch. d. Pharm., 292/64, 569 , 570 (1959).
[554] Е. Kischer, A. Mouneyrat, Вег., 33, 2387 (1900).
[555] S. Natelson, S. Gottfried, Org. Synth 23, 37 (1943); Coll. v. 3,
. 381 (1955).
7556] K. A u w e г s, R. Bernhard i, Ber., 24, 2216 (1891).
[557] L. Vanino, Handbuch der nraparativen Chemie, Bd.‘ II, F. Enke.
, Stuttgart, 1937, S. 108.
1558] II. Reinheckel, Chem. Ber., 93, 2222 (1960).
166
ОБРАЗОВАНИЕ СВЯЗИ G—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Получение Р-галогепкарбоновых кислот или их эфиров путем присоеди-
нения галогеноводородов к а,р-ненасышепны.м карбоновым кислотам рассмо-
трено на стр. 119-
Большое значение для препаративных целей имеют также у-бром-а,р-нена-
сыщенные карбоновые кислоты, которые получают бромированием N-бромсукцин-
имидом (стр. 136) при кипении. В качестве растворителя в большинстве случаев при-
меняют СС14, иногда не содержащий тиофена бензол, инициатором радикалов служит
перекись бензоила. По этому способу получают, например, метиловый (этиловый) эфир
у-бромкротоновой кислоты [357, 559] и этиловые эфиры следующих кислот: 4;бромпен-
тен-2-овой [560), 4-бром-4-метилпентен-2-овой [363] (СН3)’2СВгСН=СНС00С2Н5 и
со-бромсорбиновой [561] ВгСН2СН = СНСНт=СПСООС,2Н6.
Бутиролактон легко бромируется при сильном охлаждении в присутствии
красного фосфора [562] или триоромида фосфора с образованием бромангидрида
а.у-диброммасляной кислоты, который осторожным прибавлением более 1 моль воды
при 80s С переводят к кислоту. При отщеплении бромистого водорода от кислоты
опа превращается в а-бромбутпролактон — важный промежуточный продукт в син-
тезе метионина [562].
Склонность алифатических кислот к а-бромированню так сильно выражена,
что при взаимодействии кислоты с бромом в толстостенной ампуле в присутствии
переносчиков галогена (сера, фосфор, иод) образуются а.а-дибромкислоты. Так,
из ледяной уксусной кислоты и брома с добавкой серы при 150° С получают дибром- •
уксусную кислоту [564].
Трихлор- или трибромуксусные кислоты получают окислением хлораля или
бромаля. а,а-Дихлорзамещениые алифатические кислоты образуются при взаимо-
действии а-кетокислот с пентахлоридом фосфора.
10. Замещение галогеном водорода
в алифатических дикарбоновых кислотах и нитрилах
Для галогенирования дикарбоновых кислот применяют в основном те же методы, .
что и для галогенирования монокарбоновых кислот. Такими являются предпочтитель-
ное а-бромирование и а-хлорирование в условиях ионной реакции, понижение доли
а-хлорпроизводпых при радикальном хлорировании и обычно применяемое галогеии- <
рование галогенангидридов кислот как таковых илн с промежуточным их образова-
нием. Течение реакции осложняется тем, что во многих случаях, например при полу-
чении а.сс'-дигалогепдикарбоновых кислотиих эфиров, образуются два диастереомера, ;
лезо-форма и рацемическая форма.
<х,а'-Дихлорадипиновая кислота [565]. При 80® С в темноте в дихлор-
ангидрид адипиновой кислоты вносят 5 г FeCl3 или соответственно 2 г иода на
1 моль дихлорангидрпда и пропускают хлор до поглощения 2 моль хлора на
1 моль исходного дихлорангидрпда. После этерификации продукта реакции ,
метиловым спиртом получают 55% (при употреблении FeCl8) и 69% (при употреб- 1
ленпи иода) дпметилового эфцра дихлорадипиновой кислоты; т. кип. 122—
128® С (0,6 мм рт. ст.). После охлаждения дистиллята и внесения затравки
выкристаллизовывается 43% общего количества эфира а,а'-дихлорадипиновой
[559] J. English jr., J. D. Gregory, J. Am, Chem. Soc., 69, 2123 (1947);
D. T о tl d, S. Teich, J. Am. Chem. Soc., 75, 1895 (1953); R. D. Schuetz,
W. П. Houff, J. Am. Chem. Soc., 77, 1839 (1955).
[560] W. Herz, J. Am. Chem. Soc., 78, 1488 (1956).
[561] H, E, Ungnade, T. К. H о p k i n s, J. Am. Chem. Soc., 73, 3091 (1951).
[562] H, P 1 i f и i n g e r, Chem. Ber 83, 267 (1950).
[563] K, Folkers et al., J. Am. Chem. Soc., 77, 5142 (1955).
[564] A. Uicetu, E. Scoffono, Gazz. chim. ital., 81, 133 (1951); C., 1951,
II, 2325; P. (renvresse, Bull. Soc. chim. France [3], 7, 365 (1892),
[565] W. Treibs, H.'Walther, Chem. Ber., 88, 396 (1955).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ПЛИ SCN
167
кислоты, который омыляют кипячением с концентрированной НС1 до полного
растворения (1 ч). Полученную кислоту можно разделить перекристаллиза-
цией из воды на плохорастворимую .мезо-а,а'-дихлорадипиновую кислоту (т. пл.
186й С) и легкорастворимую рацемическую а,а'-дихлорадипиповую кислоту
(т. пл. 145° С).
Рф'-Дихлорадипиновая кислота. Вез применения поляризующих ката-
лизаторов и при УФ-облучении в аналогичных условиях [получают дпметило-
вый эфир дихлорадипиновой кислоты (61%; т. кип. 108—112° С при 0,6 мм рт.
ст.), который омыляют в кислоту. В противоположность обеим а,а'-дихлоради-
пиновым кислотам полученное вещество состоит па 94% из растворимой в бен-
золе р,Р'-дихлорадипиповой кислоты (т. пл. 126° С), которая является также
главным продуктом хлорирования сульфурилхлоридом в темноте в присутствии
4 г перекиси оензоила на 1 .ноль дихлорангидрида [56,5].
О хлорировании глутаровой кислоты см. [566].
Хлорирование нитрилов RCII2CN в условиях ионной и даже свободнорадикаль-
ной реакций (УФ-облучсвие) дает преимущественно а,а-дихлорпитрилы. Это справед-
ливо также и для динитрилов дикарбоновых кислот. Йе
Пропуская хлор при 70—75° С в динитрилы глутаровой [567], адипиновой,
пробковой и себациновой [568] кислот, их переводят в динитрилы а,а,а' а'-тетрахлор-
дикарбоновых кислот, которые омыляют кипящей концснтрироваивой НС1, и полу-
чают соответствующие тетрахлордикарбояовые кислоты.
Очень легко галогенируются малоновая кислота, малоновый эфир и их алкил-
и аралкилпроизводные. Хлормалоновую кислоту готовят омылением хлормалонового
эфира, который образуется при пропускании хлора При 70° С в малоновый эфир [569].
О получении а-хлор-я-масляной кислоты через этилмалоновый эфир см. [570].
Под действием 1 моль брома суспендированная в эфире малоновая кислота
превращается в растворимую в эфире броммалоновую кислоту [5б9];''под действием
2 моль брома в муравьиной кислоте получается, особенно быстро на свету, дибром-
малоновая кислота [571]. Приливая по каплям бром в сухие эфирные растворы
алкил- или аралкилзамещеииых малоновых кислот с такой скоростью, чтобы реак-
ционная масса непрерывно кипела, с очень хорошим выходом образуются замещен-
пые броммалоновые кислоты, из которых декарбоксилированием получают большое
число а-бром(арил)алифатических кислот:
RCH(COOH)a —* RCBr(COOH)2 —> RCIIBrCOOH + CO2
Этот способ подробно описан на примере сс-бром-р-фенилпропионовой кис-
лоты [573] — важного продукта для синтеза £>,£-фепилаланина [572].
Высшие дикарбоновые кислоты бронируют по способу Гелля — Фольгарда —
Зелинского как таковые или в виде дихлорангидридов, получаемых с помощью SOC12,
Такими способами готовят, например, а,«'-дибромяптарную [542, 574), -глу-
таровую [575], -адипиновую [576] и пимелнновую [577] кислоты. Для синтеза ди-
эфиров неочищенные дихлорангидриды а.а'-дибромдикарбоновых кислот подвергают
W. Treibs, К. Mi chaolis, Chem. Ber., 88, 402 (1955).
W. Treibs, J. Herrmann, G. Zimmermann, Chem. Ber.. 93,
2198 (I960).
W. Treibs, Q. Zimmermann, Chem. Bor., 90, 1146 (1957).
M. Conrad, H. Reinbach, Ber., 35, 1816 (1902).
A. M. Cloves, Ann.. 319, 357 (1901).
R. Willstattar, Ber., 35, 1375 (1902).
E. Fischer, Ber., 37, 3063 (1904).
C. S. Marvel, Org. Synth., Coll. v. 3, 1955, p. 705.
C. Hell, J. Gorodetzky, Ber., 21, 1731 (1888).
J. M. van der Zanden, Rec. trav. chim., 53, 483 (1934).
.1. M. v a n der Zanden, Rec. trav. chim., 63, 113 (1944).
E. A. Fchnel, G. C. Oppenlandcr, J. Am. Chem. Soc., 75, 4660
(1953). » г г , ,
168
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
взаимодействию с соответствующими спиртами. Типичным примером является синтез
диэтилового эфира а,а'-дибромадипиновой кислоты с промежуточным получением
дихлорангидрида 1578}.
Диметпловые эфиры а,а'-дибромадипиновой [579—581] или а,а'-дибром-
себациновой кислоты синтезируют через бронированные дихлорангидриды.
Наиболее легко получаются их л«зо-формы (т. пл. 77 и 52° С соответственно)»
которые выкристаллизовываются из разогнанной в вакууме смеси изомеров.
Рацемический жидкий диметиловый эфир а.а'-дибромадипиновой кислоты пере-
группировывается в лмо-форму частично уже при перегонке, а полностью при
обработке раствором метилата натрия [580].
Так как наличие хлорангидридной группы СОС1 гораздо больше благоприят-
ствует а-бромированцю, чем наличие эфириой группы COOR, следовало ожидать, что
полуэфиры монохлорангидридов дикарбоновых кислот предпочтительно будут бро-
нироваться 1 моль брома в «-положение и COCl-группе, что даст возможность легко
получить диэфиры а-бромдикарбоновых кислот. Однако практически выход их очень,
мал. Так, диэтиловый эфир а-бромглутаровой кислоты получается с 58%-ным выхо-
дом [582], диметиловые эфиры а-бромадипицовой кислоты—с 42%-ным выхот
дом [583], а-бромпробковой кислоты — с 18,5%-ным выходом и а-бромсебациповой
кислоты — с 11,5%-ным выходом. Причина плохих выходов приведена на стр. 165.
Значительно лучшие выходы достигаются бромированием эфиров монохлорангидри-
дов в SOC12 в качестве растворителя. При бромировании моноэфиров дикарбоновой
кислоты нет необходимости выделять моноэфир хлорангидрида кислоты.
Дмэфмры а-бромдикарбоновых кислот. Примерно 2 ч кипятят в колбе-
с обратным холодильником 1 моль моноэфира дикарбоновой кислоты с 300 мд
SOCI2; затем в течение 2—3 ч к реакционной массе приливают по каплям при
слабом кипевии 1,05 моль брома, оставляют на ночь и потом осторожно выливают
в 500 мл этилового спирта. Можно сначала отогнать в вакууме избыток SOC12 и
затем цеочищениый эфир «-бромхлорангидрида обработать 200 мл этилового-
спирта. После 2—3 ч стояния при комнатной температуре смесь выливают в воду,
неочищенный бромэфир извлекают тремя порциями эфира (по 150 мл каждая)»
эфирный раствор последовательно промывают водой, растворами NaHSO-, и
Na2CO3, вновь водой и высушивают над Na2SOj. После отгонки эфира продукт-
реакции перегоняют па колонке высотой 15—30 см с хорошим водоструйным или
масляным вакуум-насосом. Выходы диэтиловых эфиров а-бромглутаровои»-
а-бромадипиновой и а-бромсебациновой кислот около 90% от теоретиче--
ского [584].
Этот способ пригоден также для получения эфиров а-броммонокарбоновых -
кислот, папример этиловых эфиров а-бромциклогексил- и а-бромфенилуксус-
иых кислот.
Диэтиловые эфиры а-хлордикарбояовых кислот [584]. Такие эфиры-
с хорошим выходом получают по описанному выше способу. Вместо 1 моль брома-
добавляют 450 мл SO2CI2 и перед обработкой этиловым спиртом неочищенного
мопоэфпра хлорангидрида а-хлордикарбоновой кислоты удаляют в вакууме-
а,а;а',а'-Тетрабромдикарбоновые кислоты получаются при взаимодействии ди-
карбоновых или а,а'-дибромдикарбоновых кислот с бромом в присутствии малых коли*
[578] Р. С. G и Ь a, D. К. Sanka r-а n Org. Synth., Goll. v. 3, 1955, p. 623.
[579] H. Stephen, Ch. Weizmann, J. Chem. Soc., 103, 271 (1913).
[580] A. Bern ton, H. I n g, W. Perkin, J, Chem. Soc.. 126, 1500 (1924).
[581] W. Trcibs, G. Leichsenrjng, Chem. Ber., 84, 54 (1951)»
W. Treibs H. Reinheckel, Chem. Ber., 89 , 51 (1956).
[582] P. Karr er, F. Kohrer, Helv. chim. acta, 27, 142 (1944).
[583] H. Reinheckel, Chem. Ber., 93. 2554 (1960).
[584] E. Schwenk, D. Papa, J. Am. Chem. Soc,, 70, 3626 (1948).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
169
честв иода в запаянной трубке при 120—245° С. По другому способу [385] омыляют
тстрабромдинитрилы NC— СВг.,(СН2')„СВг2—CN. Тетрабромдинитрилы (кроме тех,
у которых п равно 0 или 1) синтезируют пропусканием паров брома в токе азота
в нагретый до 80—125° С динитрил соответствующей кислоты с добавкой иода
(0,3 г на 100 з адиподинитрила) до окончания поглощения брома (8 ч); выход
около 40%. Хлорированием при атмосферном давлении получают только тетра-
хлординитрилы кислот с малой и средней длиной цепи; бромированием в итих
условиях могут быть получены и тетрабромдинитрилы высших кислот, например
тапсиевой (п = 14) кислоты [586].
При бромировании ацетонитрила в присутствии красного фосфора при 50—
60° С образуется 2,4,б-три-(дибромметил)-!,3,5-триазин [587].
а-Бромиитрилы типа R(CH2)nCHBrCN малоустойчивы. Их окраска изменяется
при стоянии при комнатной температуре, они перегоняются с разложением даже
в вакууме при 30е С. Неочищенные а-бромнитрилы получают [588], медленно приба-
вляя по каплям 1 моль брома при 70° С к 1 моль нитрила и примерно 1 г РВг3.
При действии на а-бромнитрилы в течение 3 мин концентрированной HsSO4
получаются устойчивые амиды а-бромкислот. а-Хлор- и а-бромнитрилы типа
R2CBr(CI)CN устойчивый могут перегоняться в вакууме, создаваемом водоструйтпли
насосом. Они получаются с хорошими выходами [589] по реакции нитрилов с PCL
или РВг8.
И. Замещение галогеном водорода
в ароматических окси- и аминокарбоновых кислотах
В свободные орто- и пара-положения к гидроксильной или аминогруппе арома-
тических окси- и аминокарбоновых кислот (а также соответствующих сульфокислот
и эфиров) можно в мягких условиях вводить хлор, бром или иод. В воде под действием
избытка брома замещается карбоксильная 1-рунпа (см. стр. 151). Легче всего полу-
чаются дигалогенпроиаводные о- или n-оксибензойных кислот с атомами галогева
в о-, п- или о-.о-положениях к оксигруппе. Из салициловой кислоты в ледяной уксус-
ной кислоте с небольшим избытком против 2 моль хлора, брома [590] или хлорида
иода [591] образуются 3,5-дигалогенсалициловыс кислоты, из 4-оксибензойной кис-
лоты— 3,5-дигалоген-4-оксибснзойная кислота.
О хлорировании этилового эфира 4-оксибензойной кислоты SOjCIs см.
стр. 150; 3,5-дибром-4-оксибензойная кислота легко получается при добавлении
раствора 2 моль брома в ледяной уксусной кислоте к раствору 4-оксибензойной
кислоты в том же растворителе и последующем нагревании на паровой бане.
Антраниловая кислота бромнруется бромом в ледяной уксусной кислоте с обра-
зованием 3,5-дибромпроизводного [592]. Еще лучше ]593] реакция протекает при
пропускании в солянокислый раствор антраниловой кислоты тока воздуха, насыщен-
ного парами брома (2 моль). Йз раствора в галогеноводородной кислоте 3,5-дибром-
2-аминобензойная нислота выделяется наряду с незначительным количеством три-
броманнлина; выход 92% от теоретического. При введении в солянокислый раствор
антраниловой кислоты при температуре не выше 30° G около 2 моль.хлора аналогично
получается 3,5-дихлор-2-аминобензойная кислота; подробную пропись см. [594[.
[585] W. Treibs, J. Herrmann, W. Gerhardt, Chem. Ber., 91, 290 (1958).
f586f W. Treibs, J. Herrmann, G. Zimmermann, Chem. Ber., 93,
2198 (I960). ‘
[587] E. Ghigi, Gazz. chim. ital., 71, 641 (1941).
[588] G. L. Stevens, W. Holland, J. Org. Chem., 18, 1112 (1953).
[589] C. L. Stevens, T. H. Coff ield, J. Am. Chem. Soc., 73, 103 (1951).
[590] R. B. Earle, H. L. Jackson, J. Am. Chem. Soc., 28, 109 (1906).
[591 [ G. H. WooIIett, W. W. Johnson, Org. Synth., Coll. v. 2, i943,
p. 343.
[592] A. S. Wheeler, W. M. Oates, J. Am. Chem. Soc., 32, 7?1 (1910).
[593 [ F. Ullman n, E. Kopetsch n'i, Ber., 44, 426 (1911).
[594] E. R. A t k i n s о n, D. M. Murphy, J. E. Lufkin, Org, Synth., 31,
96 (1951).
170
ОВРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Из галогенированных окси- и аминокарбоновых кислот большой интерес пред-
ставляют иодзамещенные- являющиеся промежуточными продуктами синтеза рентгено-
контрастных веществ. Особенно пригодными для их получения оказались методы
иодирования с применением IC1, которые удобнее рассмотреть именно в этом разделе
(ср. также стр. 153).
Хлорид иода. Пользуясь часто рекомендуемым методом получения ICI, i
заключающимся в пропускании тока хлора через порошкообразный иод до
рассчитанного привеса- и последующей перегонке, нельзя получить чистый про-
дукт. Для получения чистого препарата следует руководствоваться приводимой ‘
ниже методикой [595]. {
Во взвешеппую колбу емкостью 1 л со шлифами, охлаждаемую смесью
сухого льда и ацетона, конденсируют 300 мл хлора (d = 1,557). Газообразный .
хлор предварительно пропускают через пустую промывную склянку, склянку j
с концентрированной Щзба и склянку со стеклянной ватой. Затем в колбу
добавляют 6,5 моль иода (по весу), после чего содержимое колбы затвердевает. ;
Колбу постепенно вынимают из охлаждающей бани (тяга!), причем улетучи- 4
кается избыточный хлор, взвешивают и в случае надобности добавляют недоста-' ~
ющее для образования IC1 небольшое количество иода. Неочищенный жидкий .?!
ICI выдерживают в закрытой пришлифованной пробкой колбе не менее 24 ч ;
при температуре на несколько градусов выше обычной комнатной *. Уже доста-
точно чистый продукт может быть очищен кристаллизацией; жидкий ICI (т, пл.'~-:
27,2° С) охлаждают и, после того как закристаллизуется около 80% всей массы,
сливают жидкий остаток.
Для препаративного иодирования вполне пригоден полученный такнм
образом неочищенный IC1. Содержание в нем примеси избыточного иода (в ра- ;
боте не мешает) определяют титрованием раствором KIO3 (50 г/л^ навески ICI <
(1—2 .ил), которую добавляют к смеси 25 мл концентрированной HCI, 25 мл
воды и 5 мл хлороформа. Титрование ведут до исчезновения фиолетовой окраски
хлороформного слоя; 1 мл К1О3 соответствует 0,1188 г иода [596]. /
Перехлорировапие (или недостаток иода) определяют титрованием на-
вески ICI раствором К1 (50 г/л) до появления окраски иода в хлороформном .)
слое:
ci2+ki=kci + ici ?
ICI + KI = KCI + 1г J
1 мл Ki соответствует 0,0766 г недостающего иода. Рассчитанное недо-
стающее количество иода добавляют тс неочищенному IC1 или его свежеприго-
товленным растворам в соляной или в ледяной уксусной кислоте.
Хлорид иода, не содержащий иода и 1С13, анализируют, добавляя водный
раствор KI и титруя 0,1 н. раствором NaaSsOs; 1 мл 0,1 н. Na2S2O3 соответствует
0,00812 г 1С1.
Хлорид иода проще получать в виде раствора н применять его не выделяя.
Для этого существуют различные способы.
а) В темноте при 15° С пропускают рассчитанное количество хлора (при
малых загрузках ожиженный хлор отбирают из калиброванного сосуда) в иод,
находящимся под слоем ледяной уксусной кислоты. Во время пропускания ле-
дяную уксусную кислоту, находящуюся над более тяжелым иодом, перемеши-
вают. К концу реакции иод почти полностью переходит в раствор [553, 597].
б) Растворяют 110,7 г KI и 71,3 г К1О3 в 833 мл 6 н. раствора HCI и раз-,
бавляют водой до 1 л. Получается солянокислый 1 М раствор К1С1а. При не-
обходимости солянокислый раствор хлорида иода можно извлечь эфиром
и эфирный раствор высушить над хлористым кальцием [598].
* Хлорид иода хорошо хранить в темноте в склянках с притертой пробкой
с добавкой 5 вес. % концентрированной HCI, вследствие чего он стано-
вится жидким и удобным для употребления. —Прим. ред.
[595] J. Cornog, R. А. К а г g е s, J. Am. Chem. Soc., 54, 1882 (1932). ‘
[5961 V. H. Wallingford, H. G. D acker, M. Kr uty, J. Am. Chem. ;
Soc., 74, 4367 (1952).
[597] F. D. C h a t t a w а у, A. B. Constable, J. Chem. Soc., 1914. 125.
[598] K. Cleu,’W. J a g e m a n n, J. prakt. Chem. [2], 145, 260 (1936).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИДИ SOM
171
в) Эфирный раствор IC1 получают также из 1 моль IIgCl2 и 2 моль иода.
Такой раствор используется для присоединения иода кС = С-скязям жиров (иод-
ное число по Гюблю, стр. 102} п олефинов (5991-
Хлорид иода является иодирующим агентом только в условиях, благоприят-
ствующих его диссоциации иа I* и С]‘, что, например, происходит в уксусной кислоте
и нитробензоле. В большинстве случаев иодирование проводят в ледяной уксусной
кислоте. Пары хлорида иода хлорируют твердую салициловую кислоту [411].
Ароматические амины, аминокарбоновые кислоты и амипосульфокпслоты [508]
можно иодировать хлоридом иода в солянокислом растворе.
Хлорид иода разлагается в водных растворах [600]:
5IClj + 3H3O 2I2-I-IO3+10CI- + 6H*
Разложение можно устранить, применяя достаточно концентрированную
соляную кислоту или используя для иодирования водные растворы К1С12 или NalQ.,
содержащие избыточный KCI или NaCl, что тоже сдвигает равновесие влево [601].
Водные растворы К )С12 или NaICl2 можно получать из ранее приготовленного
IC1 или, что проще, непосредственным приготовлением 2 М раствора К1С12. Ннжр
приведено несколько таких способов.
а) Из хлорида иода [602]. Добавляют 1 моль IC1 к раствору 120 г К.С1
в 350 мл воды и разбавляют водой до 500 .ил.-Если в растворе недостаточно хлора,
отфильтровывают нерастворенный иод через стеклянный фильтр или стеклянную
вату, если имеется избыток хлора — добавляют рассчитанное количество KI.
б) Непосредственное получение раствора.
КЮ3+2К1 + 6НС1 —► ЗК1С12 + ЗНгО
В энергично перемешиваемую смесь 71 г К1О3, 40 a КС1, 5 мл концентрированной
НС1 и 80 мл поды одновременно приливают по каплям раствор 111 г KI в 100 .чд.
воды и 170 мл концентрированной HCI (d — 1,19) с такой скоростью, чтобы
ле происходило выделения хлора. Затем доводят водой объем смеси до 500 мл.
в) Непосредственное получение раствора NaICl2. Исходят из молярных
количеств хлора и иода. Суспендируют 520 г порошкообразного иода в растворе
400 г NaC] в i,2 л воды, вводят при комнатной температуре и перемешивании
142 г хлора (2—3 ч) и перемешивают еще 1 ч. Отсасывают избыточный иод, про-
мывают водой, раствор разбавляют до 2 л. На титрование 1 мл раствора после
обработки KI должно расходоваться 40 мл 0,1 н. раствора NaaS2O3. .
Нейтральные растворы К1С12 или NaIC]B особенно пригодны для иодированна-
фенолов в присутствии аммиака, аминов или щелочей (стр. 153). С таким же успехов
их можно применять для иодирования в слабой солянокислой среде (пример см. н^
стр. 172)'.
3-Амино-2,4,6-трииодбензойная кислота [596]. В раствор 68,5 е
(0,5 моль) 3-аминобепзойной кислоты в 2 л воды, содержащий 50 мл концентри-
рованной HCI (d = 119), медленно при перемешивании приливают смесь 290 ?
1С1 и 290 мл концентрированной HCI. Реакционную массу постепенно иагревакиг
до 80—85° С и перемешивают еще 3 ч при этой .температуре. Выпавшую неочи-
щенную 3-амин»-2,4,6-трииодбепзойную кислоту (около 92%) очищают дву-
кратной перекристаллизацией ее натриевой соли иа примерно пятикратногс
(по весу) количества воды с добавкой небольшого количества NaHSO3. Полу-
чается бесцветный продукт; т. пл. 196,5—197,5“ С (разл.); выход 70% от теоре-
тического.
[599] S. W i п s t е i п, Е, Grunwald, J. Am. Chem. Soc., 70, 836 (1948).
[600] C. J. Klemme,]. H. Hunter, J. Org. Chem., 5, 232 (1940).
[601] H. W. Cremer, D. R. Duncan, J. Chem. Soc., 1932, 2031,
[602] J. O. Hoppe et al., J. Am. Chem. Soc., 78, 3214 (1956).
172
ОБРАЗОВАНИЕ С—На! В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
2-Амино-3,5-динодбензойная кислота. Получают аналогичным способом,
применяя 185 г 1С1 и 0,5 лоль 2-аминобензойиой кислоты. Таким же способом -
можно синтезировать 4-амино-3,5-даиодбензойную кислоту.
2>Амвно-5-иодбензойная кислота [603}. Получают с хорошим выходом '
взаимодействием 1 .коль антраниловой кислоты с 1 моль IC1 при 18—22° С в раз- ,
бавленной HCl. •
В 3,5-диаминобензойную кислоту с двумя- способствующими электрофильному
аамещению.аминогруппами при помощи NaICI2 или К1С12 в солянокислой среде можно га
внести три атома иода. Между тем иодированием водной суспензии З-ацетамидо-5- J
аминобснзойяой кислоты при помощи 2 М раствора К1С12 удается получить только J
2,6-дииодпроизводное. При этом реакционная среда становится солянокислой. Если же fj
для нейтрализации карбоксильной группы и образующейся соляной кислоты добавить £
нужное или даже избыточное количество щелочи (около 0,5 моль NaOH на 1 холь..]
СООН), то иодом замещается и третий атом водорода в ядре. Следовательно, в ней- *
тральной или слабощелочной средах, в которых свободные амино- и карбоксильная, ;
1 руппы являются аннонами, электрофильное замещение иодом происходит легче,?;?
чем в кислой среде [602].
3,5-Диамино-2,4,6-трииодбензойная кислота [602]. К полученному ката-41
литическим гидрированием 1 моль 3,5-дннитробензойной кислоты раствору ?
3,5-диаминобензойной кислоты в 12 л разбавленной НС1 (содержащей 168 мл
концентрированной HCI, d — 1,19) добавляют при перемешивании и комнатной ч
температуре 1,6 л 2 М раствора К1С12 и перемешивают еще 30 мин. Выпавший .•
коричневый неочищенный продукт промывают водой и высушивают на воздухе; -
выход 475 г. Кислоту очищают через аммониевую соль.
4
Получение трииодзамещенной 3-оксибензойпой кислоты с помощью NalCl» !
описано на стр. 153.
До сих пор рассматривалось галогенирование ароматических окси- и амино- ?
карбоновых кислот в орто- или пара-положения к окси- или аминогруппе, т. е. в поло- *
ження, для которых облегчено электрофильное замещение. При замещении болен
двух атомов водорода в 2- и 4-оксибензойных кислотах требуется также замещением!
мета-положения по отношению к оксигруппе, и поэтому галогенирование возможное
только в более жестких условиях, например в олеуме. Проведение реакции в олеуме, "
содержащем 60—70% SO8, позволяет вводить в 3,5-дибромсалициловую кислоту еще
два атома брома, в 3,5-дихлорсалициловую кислоту — еще один атом хлора илн одия -з
атом хлора и одип атом брома [604] п в 3,5-дибром-4-оксибензойную ккслоту — еще,1
два атома брома [605]. <
2,3,5,6-Тетрабром-4-оксибензойная кислота. В колбе для сульфирова- <
ния, снабженной механической мешалкой на шлифе (смазывается концентриро---
валной H2SO4), обратным холодильником н капельной воронкой, трубка которой^
немного погружена в олеум, в течение 30 мин энергично перемешивают смесьг
150 г 3,5-дибром-4-оксибензойной кислоты и 750 мл 70%-ного олеума на водяной-?
бане с температурой 28—29° С. Затем при 29° С пятью порциями (6; 6; 6; 5 ’
и 5 мл) через каждые 10 мин в течение 45 мин приливают по каплям высушенный •;
концентрированной II2SO4 бром и перемешивают еще около 5 ч при 29° С. i
Смесь оставляют па ночь и затем при перемешивании стеклянной палочкой мед-?’
леппо выливают на лед. Выпавшую в осадок неочищенную 2,3,5,6-тетрабром-4- .;
оксибензойную кислоту переосаждают из аммиака; выход 76% от теоретического; t
т. пл. 222° С. После перекристаллизации из разбавленного этилового спирта
т. пл. кислоты 228° С.
При действии избытком брома на эту полигалогенировапиую оксибен- т<
зойную кислоту в растворе 30%-ной уксусной кислоты (60° С) происходит за-“
мещение карбоксильной группы на бром и отщепление СО2. i
[603] V. Н. Wallingford, Р. A. Krueger, Org. Synth., Coll. v. 2.
1943, p. 349. >
[604] L. H. F a r i n h о I t, A. P. Stuart, D. Twiss, J Am. Chem. Soc., 62. :
1237 (1940). Л ;
[605] H. P f a n z, H.*D о г n, Arch. d. Pharm., 61, 657 (1956).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
173
Дальнейшее введение атомов иода в 3,5-дигалоген-2 (или 4)-оксибенаойные
кислоты иодированием в олеуме не удается. Однако, галогенируя ангидрид о-сульфо-
бензойпой кислоты в 60%-ном олеуме, можно получить ангидриды 3,4,5,6-тетрахдор-о-
сульфобензойных кислот и из них соответствующие кислоты [606] (то же относится
и к тетрабром- и тетраиодзамещенным). Этот способ впервые применил Ювалта [607]
для бромирования фталевого ангидрида в 50%-ном олеуме и получил наряду с дру-
гими продуктами ангидрид тетрабромфталевой кислоты [607, 608].
Подробное описание синтеза ангидрида тетраиодфталевой кислоты при
взаимодействии 1 моль фталевого ангидрида с 2,12 моль иода в 600 мл 60%-ного
олеума при 65° С и далее в течение 2 ч при 170° С см. [609].
Способ Ювалта применим также для полибромирования гетероциклических
соединений. Так, из 3,5-Дибром-4-пиридона по реакции с бромом в 70%-ном олеуме
при 80° С получается с 85%-ным выходом 2,3.5,6-теграбромпиридон. Из этого веще-
ства взаимодействием с РВг6 в запаянной трубке при 180° С с хорошим выходом
получают пентабромпиридин [605].
Описанные до сих пор способы синтеза галогенированных ароматических кар-
боновых кислот относились к кислотам, которые содержат группы, облегчающие
электрофильное замещение. Если таковых не имеется, то, не отдавая предпочтения
косвенным методам (реакции Зандмейера, окислению галогенированных альдегидов или
толуолов), можно по методу Биркенбаха — Губо — Уотерса (стр. 140) довольно изящно
вводить бром или нод в мета-положение к карбоксильной группе. Бензойную кислоту,
которая взаимодействует с бромом только в зацаянной трубке, а с бромом и желез-
ными опилками [610] только дри 170—260° С, можно быстро галогенировать ионами
liljiorcHOB в смеси уксусной и азотиой кислот в присутствии AgNO3 или в смеси уксус-
ной и серной кислот в присутствии КВгО3, как описано в следующих примерах [376J.
З-Бромбензойная кислота. В раствор 0,1 моль бензойной 'кислоты и
2,1 мл брома в смеси 250 мл ледяной уксусной кислоты и 150 мл концентриро-
ванной HCI при 25° С (охлаждение!) в течение 15 мин при перемешивании при-
ливают по каплям раствор 4 г КВгО3 в 50 мл воды. Во время последующего
перемешивания (1 ч) начинает выпадать 3-бромбензойпая кислота. Затем в те-
чение 30 мин приливают по каплям еще 4 г КВгО3 в 50 мл воды; выход 3-бром-
бензойной кислоты 11 г.
З-Иодбензойная кислота. В энергично перемешиваемую смесь 0,1 моль
бензойной кислоты, 0,05 моль Ag2SO4 (AgjSO4 растворим в 90%-ной H2SO4),
240 мл концентрированной H2SO4 и 30 мл воды, нагреваемую на кипящей водя-
ной бане, порциями вносят 0,11 моль тонко измельченного (!) иода. Во время,
последующих 80 мин перемешивания и нагревания непрерывно выпадает Agl.
После охлаждения смесь разбавляют 2 л воды, удаляют избыточный иод бисуль-
фитом и экстрагируют эфиром 3-иодбензойную кислоту из раствора и из выпав-
шего продукта. Полученную после отгонки эфира неочищенную 3-иодбензойную
кислоту перекристаллизовывают из водного ацетона; выход 75% от теоретиче-
ского; т. пл. 185—186° С.
12. Замещение галогеном водорода в вроматическнх ннтросоединеннях
Получение 3-бромнитробензола по Уотерсу [376]. К 0,1 моль нитро-
бензола и 0,1 моль брома в 90 мл концентрированной HaSO4 и 10 мл воды
прибавляют 0,1* моль Ag2SO4, встряхивают в течение 16 ч при комнатной
температуре. После отделения AgBr фильтрат-разбавляют водой и экстрагируют
3-бромнптробензол эфиром; выход 70% от теоретического, т. пл. 55—56° С.
[606] О. Т w i s s, L. Н. F а г i n h о I t, J. Am. Chem. Soc., 58, 1561 (1936).
[S07[ N. Ju wait а, герм. пат. 50177 (1889); FrdL, 2, 94 (1887—1890).
[608] D. S. P r a t t, С. O. Y о u n g, J. Am. Chem. Soc., 40, 1416 (1918).
[609] C. F. H. A11 e n, П. W. J. Cress man, H. B. Johnson, Org.
Synth., 27, 78 (1947); Coll. v. 3, 796 (1955).
[610] H.L. Wheeler, B. W. McFarland, Am. Chem. J., 19, 364 (1897).
|74 ОБРАЗОВАНИЕ СВЯЗИ С--Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ-
2-Бром-4-нитрокумол [611]. Его можно синтезировать либо аналогично
синтезу 3-бромнитробензойной кислоты, либо классическим методом бромирова-
ния нитробензола бромом в присутствии порошкообразного железа [612].
13. Замещение галогеном иодорода в альдегидах и кетонах '*
При прямом хлорировании и бромировании альдегидов замещение галогеном J
может происходить как по атому углерода альдегидной группы, так и по соседнему 1
атому углерода. Последняя реакция представляет наибольший интерес. й
Хлорированные в a-положение ацетальдегиды являются важными промежу-ч
точными продуктами. Хлорацетальдегид (т. кип. 85—85,5° С при 748 мм рт.
может быть получен хлорированием ацетальдегида хлором при 18—20° С в соляноин
кислоте [613] или безводного ацетальдегида [614] при температуре ниже 10° С. В по-1
следнем случае во избежание воспламенения, возможного при действии хлора на пароле
образный ацетальдегид, смесь необходимо, особенно вначале, сильно охлаждать.*!
Реакцию начинают с малым количеством ацетальдегида, и остальное количество^
охлажденного сухим льдом ацетальдегида прибавляют в несколько приемов. Скорость;
подачи хлора (до — 1,37) регулируют по отводу реакционного тепла.
i
Хлорируя ацетальдегид сухим хлором при температуре 18—20° С, поддеру
живаемой охлаждением, получают продукт состава 2С1СН2СНО • СН;)СНО • НС!]'
из которого перегонкой выделяют хлорацетальдегид; выход 60%. Если хлорид
рова'хь ацетальдегид при очепь медленном повышении температуры до 80°
то в зависимости от количества введенногд хлора получается дпхлор- и трпхлор-i
ацетальдегид [615[.
Безводный хлорацетальдегпд в лабораторном масштабе получают с хоро-'
шим выходом из этиленкарбоната по следующему способу [616, 617]: ;
I а
HjC-O. нс-ох I
| с=о -JJ7T* I /С=о —gg-- сн2-сно
Н2С—<У Н2С—0х
Пропускают хлор через 3,44 моль свежеперегнанного этиленкарбоната
при 63—70° С и УФ-облучении до увеличения массы на 119 г (24 ч). Фракцио»
ной перегонкой в вакууме, создаваемом водоструйным насосом, получают смес|
из 5,2% 1,2-дихлорэтиленкарбопата (т. кип. 78—79° С при 19—20 м.ч рт. emty
и 69% монохлорэтиленкарбоната (т. кип. 106—107° С при 10—11 мм рт. cm.)i
К 19 г монохлорэтиленкарбоната добавляют каплю триэтиламина н ме®
лепно нагревают в перегонном аппарате до 200° С. Образующийся при отщепли
пии двуокиси углерода хлорацетальдегид собирается в приемнике в виде зел#
иоватои прозрачной жидкости. Реакция продолжается около 1 ч. Продукт
очищают перЬгонкой на маленькой колонке; выход 9 г; т. кип. 84—86° С.
[611] М. Crawford. F. Н. С Stewart, J. Chem. Soc., 1952, 4445. .Л
[612] J. R. Johnson, C. G. G a u e г k e. Org. Synth., 8, 46 (1928). 'J
[613] H. Guinot, I. T a b u t e a u, C. r., 231, 234 (1950). .
[614] J. Soli, пат. ФРГ 8445 95; Ullmans Enzyklopadje der technischen Cherniy
Bd. 5, 3 Aufl., Miinchen — Berlin, 1954, S. 319.
[615] M. H. Щукина, ЖОХ, 18, 1653 (1948); С. A., 43, 2575 (1949).
[616] H. Grot}, Dipiomarbeit F. Polly, Humboldt-Universitat, Berlin, 1901J
[617] M.S. Newman, R. W. Adder, J. Am. Cbem. Soc., 75, 1264 (1953);
77, 3791 (1955).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
175
В промышленности хлорацетальдегид получают непрерывным способом. В хло-
рируемую смесь пропускают хлор и одновременно вводят ацетальдегид ниже уровня
жидкости. УФ-облучение ускоряет хлорирование, продукт реакции получается в виде
полимера,
а,|}-Дихлорэтилэтиловый эфир (см. стр. 159) реагирует в определенных
условиях, например при синтезе 2-аминотиазо.та из а,р-дихлорэтилэтилового
.эфира и тиомочевипы в воде, так жо, как смесь хлорацетальдегида, этилового
спирта и хлористого водорода.
Дихлорацетальдегид [618] (т. кип. 88° С при 760 лл рт. ст.) получают
двухстадийным хлорированием (1-я стадия: 10—14° С, 1,6—1,7 моль хлора
на 1 моль ацетальдегида; 2-я Стадия: 28—32° С, 1,9—2,1 моль хлора на 1 моль
ацетальдегида) смеси ацетальдегида (или паральдегида) с водой в молярном
отношении 1 -. 3,5—4,5.
Трихлорацетальдегид (хлораль), кипящий при 97,8° С, получают хлори-
рованием этилового спирта при облучении и перемешивании. В настоящее время
применяют пе абсолютный этиловый спирт, рекомендованный Либихом [619]».
л 70—80%-пый спирт и непосредственно получают хлоральгидрат
СНзСН2ОН+4С12 + Н2О —> СС1зСП(ОН)2-|-5НС1
Облучение снижает время реакции (72 ч) в 3—4 раза [620].
Так как пары этилового спирта могут реагировать с хлором со взрывом,
прп этой реакции хлор и водный спирт вводят под поверхность хлорируемой
реакционной массы, которую непрерывно отводят через слив. Реакцию прово-
дят, например, ступенчато, в двух последовательно соединенных сосудах, по-
степенно повышая температуру, и заканчивают при плотности d2o — 1,44.
Прямой перегонкой отделяют хлоральгидрат (т. кип. 97,5° С; т. пл. 53° С), из
которого перегонкой с концентрированной серной кислотой (в соотношении
1 : 1) выделяют хлораль, Содержащий хлористый водород. Хлористый водород
удаляют повторной перегонкой с небольшим количеством карбоната кальция.
Хлораль является промежуточным продуктом в синтезе дихлордифенилтри-
хлорэтапа (ДДТ):
2C,HtCl+Ca3CHO Н,30‘; S°* > (С„Н4С1)2СНСС13 + Н2О
Хлораль может быть получен также хлорированием водного ацетальдегида
в растворе уже готового хлораля [621].
При исчерпывающем хлорировании диметилацеталя при температуре не выше
6Q° С в качестве главных продуктов реакции получаются формальдегид, а-мовохлор-
диэтиловый эфир, триоксиметилен и хлористый водород. Исчерпывающее хлорирова-
ние диэтплацеталя (60—70° С) приводит к образованию продукта, после обработки
которого концентрированной H2SOd получается с хорошим выходом хлораль [622].
Описание восстановления хлораля этилатом алюминия до трихлорэти-
лового спирта по Меервейну [623], равно как и взаимодействие с СаСОэ с обра-
зованием дихлоруксусной кислоты, см. [G25], более подробно об этом см. [624].
[618] F. С. Potts апгл. пат. 644914; С. А., 45, 5176 (1951).
[619[ J. V. Т. i е b i g, Ann., 1, 189 (1832).
[620] О. W.C ass, пат. США 2443183, 2478152; С. А., 42, 7318 (1948); С. А.,
44, 1G7 (1950).
[621] IT. Petri, Е. М a h I е г, пат. ФРГ 874303 (1948).
[622] J. S. R е i с h е г t, J. II. В a i ] с у J. A. N 1 с u w 1 а п <3, .Т. Am. Chem.
rt.n Soc.. 45. 1554 (1923).
623| П. Meerwpin. Ann., 444, 221 (1925).
W. Chalmprs, Org. Synth.. Coll. v. 2, 1943, p, 598.
[62.,] c. Cope, J. R. Clark, R. Connor, Org. Svnth., Coll. v. 2, 1943,
p. 181.
176
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Уже давно известная реакция образования галогенангидрпдов кислот из аро-^
магических альдегидов и галогенов {626] может быть использована в препаративных^
целях, если соответствующий альдегид более доступен, чем карбоновая кислота».5
Так, пропуская хлор в течение около 15 ч в 1 моль энергично перемешиваемого рав-'а
плавленного 2-хлорбензальдегида при 140—160° С, получают 2-хлорбензоилхлорид;'й
выход 80.%. Более подробные данные см. [627]. J
а-Бромальдегиды можно получать прямым воздействием 1 .моль брома па альде-я
гид в СНС13 или CS2 при облучении и низкой температуре (—15° С). Прп этом мотива
(но не обязательно) связывать НВг при помощи СаСО3. Таким способом были полу~Я
чены, например, а.а'-дибромсукциндиальдегид [628], а-бромвалеральдегид [62ч
и а-бромфсиилацетальдегпд [630]. а-Бромальдегнды лучше всего хранить в вида
ацеталей, для чего к бронированной реакционной смеси добавляют спирт. Так. в част
ности, получают ацетали а-бром-«-валеральдегида [631], а-бромизовалеральдо
гида [632] и а,а'~дибромадипиндиальдегида [633]. При бромировании ацеталей 2
брома образуются а,а-дибромацетали [634]. Хлорированием (бромированием) в хжй
ристом метилене при — 20’С получают 70% а-хлор- и 50% а-бромфенил-ацетальд^
гида, а также а-оромгептаналь, выход 67% от теоретического [635]. -
При помощи диоксандибромида [636] можно получить ряд а-бромзамещенныЗ|
алифатических альдегидов с выходами 50—90% и а-бромфенилацетальдегид с вых«Й
дом 40%. ;
Бромацетальдегид СН2ВгСНО. К 8,8 г ацетаЛьдегица^50 мл эфира npi
5—10° с прибавляют раствор 50 г диоксаядибромида в эфире. После исчезновц
ния окраски массу промывают водой, раствором соды'и вновь водой. Получаете*
7,5 г бромацетальдегида; т. кип. 104—106° С. •’
Из паральдегида также можпо получить а-бромальдегид. Паральдегид спачал
бромпруют, а затем ацеталируют добавкой спирта [637]. Диэтилацеталь бромацетал>
дягиля получают с высоким выходом (85%) бромированием паральдегида при —10%
н облучении в присутствии перекисей [638] и последующей обработкой реакционно
смеси абсолютным этиловым спиртом. ' ’д
Особенно простым способом является бромирование паральдегида (3 м<>4
брома на 1 моль паральдегида) в спиртах при энергичном перемешивании и охлажде
нии водой, в результате чего, в зависимости от применяемого спирта, образую^)
диэтилацеталь (выход 72,5% от теоретического), ди-н-пропилацеталь [639] (вых<
65%), ди-н-бутилацеталь (выход 65%), диизопронилацеталь [640] (выход 22%) брО1
ацетальдегида. %
Ацеталь брома'цетальдегпда и бромаль. В течение примерно 5 ч к сиирМ
вому раствору паральдегида прибавляют по каплям бром, нейтрализуют броМЙ
стый водород добавкой взвеси Na2CO3, высушивают верхний слой KaCQ
и фракционируют после отгонки низкокипящей фракции на колонке в вакуум
создаваемом водоструйным насосом. -?
626
627
628
629
630
631
632
633
634
635
636
[637]
[638]
[639]
[640]
L. Wohler, J. Liebig, Ann., 3, 262 (1832).
И. Т. Clarke, Е. R. Taylor, Org. Synth., 9, 34 (1929). *
С.. II a r r i e s, II. Krtttzfeld, Ber., 39, 3671 (1906).
H. Erlenmeyer, J. P. .J u n g, Helv. chim. acta/ 32, 37 (1949).
H. Erlenmeyer, Helv. chim. acta, 30 , 2058 (1947). .
R. Kuhn C". G г и n d m a n n, Ber., 70, 1898 (1937). j;
F. G. Fischer, K. L d w e n b e г g, Ann., 494, 272 (1932); Ber., 64, 31 (193Я
F. G. Fischer, K. LSwenberg, Ber., 66, 667 (1933).
S. M. Me El vain, L. R. Morris, J. Am. Chem. Soc., 74, 2659 (1952
J .-J. Riehl С. r., 245, 1321 (1957). £
Л, А. Яновская, А. П. Терентьев, ЖОХ, 22(84), 1598 (19«?^
C. A., 47, 9258e (1953).
R. D w о г z a k et al., Mh. Chem., 46, 253 (1925); 48, 252 (1927); 50, 467 (192S]
52, 142 (1929); 53/54, 588 (1929).
G. D а г z e n s, M. Meyer, С. r., 236, 292 (1953). Ji
H. В a g a n z, K.-H. Dossow, W. Hohmann, Chem. Ber., 86,
(1953).
H. В a g a n z* С. V i t z, Chem. Ber., 86, 398 (1953).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
177
По другому способу [641] добавляют 0,5 моль паральдегида к снеси
4,5 моль брома и 1,5 г серы. После 2 ч нагревания при 60—80е С получается
бромаль.
Ацетали бромируются бромом в хлороформе в присутствии карбоната каль-
ция [642]. В качестве акцептора бромистого водорода можно применять также пири-
дин [643].
Диэтилацеталь а-бром«р-этокснпропионового альдегида [644]. К пере-
мешиваемой смеси 1 .моль диэтилацеталя p-зтоксипропионового альдегида
и1 моль пиридина по каплям прибавляют 1 моль брома и оставляют стоять при
60—65° С до исчезновения красной окраски (2—4 ч). Получается неочищенный
диэтилацеталь а-бром-р-этоксипропионового альдегида, выход 83%. В этом
случае в качестве промежуточного продукта образуется мягкий бронирующий
агент — пербромид пиридиндибромида.
Диэтилацетали ацетальдегида, пропионового альдегида, изо- и «-бутираль-
дегида бромируются также вносимым в несколько приемов N-бромсукцинимидом при
25—40° С на солнечном свету или при облучении лампой 60 вт с образованием а-бром-
ацеталей [645]. Так как реакция экзотермическая, ее проводят при Охлаждении.
По этому способу получаются более чистые продукты, чем по описанным выше мето-
дам, но с такими же выходами.
Довольно широко применяется синтез а-галогенальдегидов с присоединением
галогена по С-связям ♦.
При кипячении альдегида в колбе с обратным холодильником с уксусным
ангидридом и ацетатом калия в течение 1 ч альдегид переходит в енолацетат. К послед-
нему (1 .ноль) в СС14 при 0—10° С присоединяется бром (1 моль). Реакционную смесь
оставляют стоять 2—3 суток с 99,5—100%-ным метиловым спиртом и получают а-бром-
диметилацеталь, из которого перегонкой с концентрированной HCI выделяют а-бром-
альдегид:
Вг Вт
ПСНаСНО (сн.с0).°» RCH=CHOCOCHS ——•» RCHCHOCOCHs гн,оти
—► RCHCH(OCHs)a -.—RCHCHO
I I
Вг Вг
По этому способу из ацетальдегида, изобутиральдегида и неразветвленных
алифатических альдегидов С7—CJS получается 65—86% бромпроизводных, из феиил-
ацетальдегпда [646]—только 25%. Синтез а-бромгептальдегида таким способом
см. [647].
Диэтилацеталь подацетальдегида. Хлорид иода в концентрированной
HCI (d = 1,19) прибавляют к раствору винилацетата в СС1< при 5° С. В течение
, 20 мин образуется а-хлор-р-иодэтилацетат с почти теоретическим выходом.
Если к раствору последнего в СС14 добавить абсолютный этиловый спирт и СаС1в
[641
642
643
£44
645
646
647
В указанном процессе можно исходить из простых виниловых эфиров (М. Ф. Ш о-
стаковский, Ю. Б. Каган, Ф. П. Си д е л ь к о в с к а я, ЖОХ, 17е
957 (1947)]. — Прим. ред.
F. A. Long, J. W. Howard, Org. Synth., Coll. v. 2, 1943, p. 87.
E. Fischer, K. Landsteiner, Ber., 25, 2549 (1892).
S. M. M с E 1 v a i n, P. M. Walters,!. Am. Chem. Soc., 64, 1963 (1942).
W. T. Simpson, J. Am. Chem. Soc., 71, 754 (1949).
E. N. Marvell, M. J. Joncich, J. Am. Chem. Soc., 73, 973 (1951).
P. Z. Bedoukian, J. Am. Chem. Soc., 66, 651, 1325 (1944); 79, 891 (1957).
P. Z. Bedoukian, Org. Synth., 29, 14 (1949).
12 Заказ 1835.
178
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
и оставить смесь на трое суток при 10—15° С, то получается диэтилацеталь иод-
ацстальдегида с выходом около 80% от теоретического [648].
Для получения свободных а-бромальдегидов [649] бромацеталь кратковременно
(1 мин) нагревают в смеси дпоксана, воды и концентрированной НС1 (10 : 5 : 1),
быстро охлаждают, разбавляют водой и экстрагируют продукт реакции эфиром.
О синтезе а-галогенацеталей через а,р-дигалогенэфиры см. также стр. 99.
Галогенированные в ядро ароматические альдегиды и кетоны редко получают
галогенированием альдегидов или кетонов. Их синтезируют из галогенированных
исходных веществ различными методами или, исходя из нитроальдегидов, используют '
реакцию Зандмейсра. Ниже описан каталитический способ синтеза 3-бром(3-хлор)бенз-
альдегида и 3-галогенацетофенона прямым галогенированием, основанный на при-?
менопии больших количеств безводного А1С13 (swamping catalyst effect). ;
В колбе для сульфирования при перемешивании и исключении доступа. '
влаги при 25—100°- С к расплавленному комплексу 1 моль карбонильного соеди-
нения и 2,5 моль свежевозогнанного А1С13 прибавляют около 1,2 моль брома-I
или около 1,25 моль хлора (хлор сжижают в ловушке, охлаждаемой смесью су- j
хого льда и ацетона) под поверхность расплава. Подробное описание способа t
см. [650].
Окси-, алкокси- и аминоальдегиды подобно фенолам и ароматическим ампиам
галогенируются в мягких условиях. О возможности отщепления альдегидной группы
уже упоминалось (стр. 151). О конкурентном галогенировании в ядро и в боковую
цепь соответствующе замещенных жирно-ароматических кетонов говорится на стр. 183. -
Из всех карбонильных соединений кетоны наиболее легко галогенируются Й
в сс-положение к карбонильной группе. Реакции галогенирования карбонильных J
соединений предшествует сильно ускоряемая кислотами мономолскулярная перегруп-1
ппровка в активную енольную форму [651—653]. Поэтому пока в реакционной массе-?
нет кислоты реакция начинается с трудом. При работе с большими загрузками нельзя 3
вводить большие количества галогена в начале реакции, так как в таком случае после
индукционного периода (образование галогеноводорода в медленно протекающем ч
сначала процессе) может начаться бурная экзотермическая реакция. При бромирова-,,
пии наиболее целесообразно предварительно нагреть несколько миллилитров кетонан
с бромом и эту смесь добавить к основному количеству бронируемого вещества («за-4-
травка»). J
Прямым хлорированием ацетона получают монохлор-, 1,1-дихлор-;1,1,1,3-тетра-
хлор-, пеитахлор- и гексахлорацетон. Следовательно, хлор имеет тенденцию замещать1 л
все водороды при одном атоме углерода. То же происходит и с бромом, что будет пока- :
зано на примере дальнейшего бромирования 1,3-дибромацетопа. . 5
При мопохлорировании и монобромировании асимметричных кетонов с атомами j
водорода у обоих соседних с карбопильпой группой атомов углерода необходимо-;
учитывать возможность образования смесей изомеров. Так как при катализируемом]
кислотами галогенировании кетонов определяющей скорость реакции стадией является
енолизация, то предпочтительное направление енолизации асимметричных насыщен-*-'
ных ациклических и циклических кетонов определиет предпочтительное положение
замещении. В обоих случаях, за некоторыми исключениями, замещение происходит
по модифицированному правилу Зайцева: галоген предпочтительно присоединяется
к а-атому углерода, соседнему с атомом углерода, к которому присоединено наиболь-3
шее число атомов водорода [654]. Если карбонильная группа находится между ме<
[648] S. Akiyoshi, К. Okuno, J, Am, Chem. Soc., 74, 5759 (1952).
[649] E. R. Alexander, J. Am. Ghem. Soc., 70, 2592 (1948). ,»
[650] D. E. P e а г s 0 n, H. W. Pope, W. W. H а г g г о v e, W. E, S t a m-'
per, J. Org. Ghem., 23, 1412 (1958).
[651] A. Lapworth, J. Ghem. Soc., 1904, 30: 1908, 2188, >
[652] F. Krohnke, Ber., 69, 921 (1936). -
[653] R. Hu i sge n, F. J a k 0 b, Ann., 590, 40 (1955); О. H. Wheeler.- ]
J. L. M a t e 0 >, J. Org. Chem., 22, 605 (1957).
[654] H. M. E. Cardwell, A. E. H. Kilner, J. Chem. Soc., 1951, 2430.
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА На! ИЛИ SCN
179
тильпой и любой алкильной группами, то водород <х-атома углерода алкильяои группы
замещается галогеном легче, чем водород метильной группы. Далее, например, 2-ме-
тилциклогексапон предпочтительно галогенируется в положение 2; 3-метилцикло-
гексанон — в положение 6 (звездочкой отмечено положение предпочтительного заме-
щения):
СН8 СНз
О соотношении изомеров, образующихся при монобромироваиии диалкилкето-
пов с разветвленными и неразветвленными радикалами, см. также [655].
Здесь пет возможности более подробно остановиться на довольно сложных
явлениях, происходящих при бромировании 3-кетостероидов. В то время как,
например, монобромирование холестапопа следует указанному правилу, про-
изводные копростанона [656], в частности метиловый эфир З-кето-12-а-ацет-
онсихолаповой кислоты, монобромируются (против правила) в положение 4.
Многочисленные литературные данные по галогенированию стероидов см. [657].
Бромирование карбонильных соединении является равновесной реакцией:
RCOCHR'R" + Br2 RCOCBrR'R"-FHBr (а
Если не допускать сильного нарастания концентрации бромистого водорода
или большого падения концентрации брома, то при бромировании большинства кето-
нов равновесие полностью сдвигается вправо [658]. Хлоркетоны не восстанавлива-
ются хлористым водородом. а-Бромкарбопильные соединения легче восстанавливаются
бромистым водородом, если R' является электропоакцепторным заместителем, напри-
мер карбоксильной группой или бромом.
а-Бромацетоуксуспый эфир, который может быть получен только при условии
удаления бромистого водорода, перегруппировывается в растворе бромистого водо-
рода в ледяной уксусной кислоте в у-бромацетоуксусный эфир с промежуточным обра-
зованием свободного брома.
Если дать прореагировать 1 моль метилкетопа в ледяной уксусной кислоте,
CS2, СС14 или других растворителях в запаянной трубке с 1 моль брома при 20—
40° С и немедленно после исчезновения бромной окраски выделить продукты реакция,
то в зависимости от использованного растворителя получают разные количества
а-моно- и а,а-дибромкетонов. При оставлении реакционной массы еще на 30—60 мин
при 20° С Для установления равновесия (б)[см. реакцию (а)}, независимо от раство-
рителя, например при бромировании ацетофенона, 80% брома расходуется на обра-
зование моно- и 20% па образование дибромацетофенона ]652].
НВг
2RCOCH2Br RCOCII3+ RCOCHBr2 (б)
Следовательно, с помощью 1 моль брома нельзя из метилкетопа получить только
монобромсоединение. Для того чтобы уменьшить количество образующегося дибром-
производного, необходимо применять избыток кетона, который должен быть тем
больше, чем больше сдвинуто вправо равновесие (б).
Бромирование ацетона в ледяной уксусной кислоте (запаянной трубке
приводит через промежуточное образование 1,1-дибромадетона к 1,3-ди-
[655] A. A. Sacks, J. G. Aston, J. Am. Chem. Soc., 73, 3902 (1951).
[656] H. H. Inh of fen, G. К 6 11 i n g, G. Koch, Chem. Ber., 84,
363 (1951).
[657] Л.^ф и з e p, M. Ф и 3 e p, Стероиды, пер. с англ., Москва, Изд. Мир,
[658] F. Krohnke. II. T i m m 1 e r, Ber., 69, 614 (1936).
12*
180
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
бромацетону. Удаляя бромистый водород, можно увеличить содержание 1,1-'^
дибромацетона [659].
2,2-Дибром-З-кетостероиды перегруппировываются в 2,4-дибром-З-ке- 3
тостероиды [660]. J
Кетоны можно хлорировать хлором при охлаждении льдом или простой водой.-',?*
Подробное описание такого метода получения 2-хлорциклогексанона см. [661]. Виде- J
ляюпщися хлористый водород способствует образованию продуктов конденсации»?!
и поэтому его удаляют из сферы реакции. •-«
Монохлорацетон [662]. В колбу, снабженную обратным холодильником,.;
капельной воронкой, газоподводящей трубкой и опущенным до дна термометром^
(или в соответствующую колбу для сульфирования с механической мешалкой),^
загружают 500 г ацетона и 125 г измельченного в порошок мрамора. Охлаждают^
проточной водой, пропускают в массу умеренный ток хлора Е при этом медленн^
добавляют по каплям 315 мл воды. Температура докжна быть не выше 30° и це£'
ниже 10° С. После растворения почти всего мрамора температуру повышают
до 40° G и поддерживают ее в течение нескольких часов до окончания выдала^
нкя двуокиси углерода. Следят за тем, чтобы в смеси все время оставался мра~;
мор; в случае надобности его добавляют в небольшом количестве. По окончании?
реакции отделяют в делктельной воронке образовавшийся верхний слой и под*
вергают его фракционной перегонке. Из 4200 г ацетона получают 1100 г чистого'
монохлорацетона (т. кип. 118—120° С); последняя фракция содержит дихлор*?
ацетон. °
Мрамор препятствует образованию окиси мезитила под действием хло*
ристого водорода. Добавляемая вода служит для растворения СаС1а, но ее Л
вводят заранее, так как в ее присутствии хлор не сразу вступает в реакции^
При накоплении хлора возможны внезапные и непредвиденные реакции. Поэтом#
если во время хлорирования появляется обусловленная хлором отчетлива)
окраска, прекращают добавление воды, пока приблизительно при 25° С раствор
вновь не обесцветится. Так как по окончании реакции органический слой Шк
ходится над высококонцентрированным раствором СаС12, его вряд ли ПУНАМИ
сушить перед разгонкой. Перемешивание массы во время пропускания тов|
хлора сокращает время реакции и улучшает выход.
В иачестве хлорирующего агента можно использовать также сульфурилхлФ
рид SOaCl2. - '
Монохлорацетон [663], не содержащий 1,1-дихлорацетона. Продуй
получают взаимодействием в течение 5,5 ч без доступа влаги 2,37 моль SOjCb
с 40 моль ацетона при охлаждении. '£
2-Хлор-2-метилциклогенсавон [664]. При комнатной температуре (охлй|
ждение водой!) в течение 1 ч раствор 1,1 моль SO2GI2 в сухом СС14 прклквают ей
каплям при перемешивании к раствору 1 .моль 2-метилциклогексанона в том ЯМ
растворителе, перемешивают 2 ч при комнатной температуре и промывают pearf
ционную массу водой, раствором NaHCOs н насыщенным раствором Nadt
После сушки и перегонки получают 2-хлор-2-метилциклогексанон; выхо))
83—85% от теоретического. 4
3-Метил-3-хлорбутаиоиг2 [665]. Приливают по каплям 1 моль метИЖ
бутаноиа к 0,96 моль охлажденного ньдом SOsCla. По окончании реакции yfllli
ляют при нагревании SOe и HG1 н перегоняют продукт реакции; т. кип. 117,2° G
выход 84% от теоретического,
[659] F. Krohnke, 0. Liideritz, Ghem. Вег., 83, 60 (1950).
[660] Н. Н. Inh often, G. Ziihlsdorff, Вег., 76, 233 (1943). •
[661] M. S. Newman, M. D. Farbmann, H. Hjpsher, Org. Synth., 2U|
22 (1945); GoU. v. 3, 188 (1955 .
[662] Fritsch, Ann., 279, 313 (1894). ?
ПЕ. R. Buch m an, H. S argent, J. Am. Chem. Soc., 67, 401 (1945). , *
E. W. Warn h off, D.G. Martin, W. S. Johnson, Org.
Synth., 37, 8 j(1957). ’
[665] P. Delbaere, Bull. Soc. chim. Belgique, 51, 1 (1942); C., 1942, II, 763.
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
181
В качестве растворителей при бромировании кетонов применяют СС14, CSa
СН2С12. СНС18 и эфир, причем последние три пригодны для работы при низких тем-
пературах. Основное количество образующегося при бромировании НВт можно уда-
лять различными способами. Можно добавлять СаСО8 или NaHCOa к растворам ке-
тона в СН2С12, СНС18 или СС14- Однако если при атом не добавить к реакционной массе
предварительно нагретую пробу («затравку»), возможен длительный индукпиптпты^
период и последующее бурное выделение двуокиси углерода.
Алифатические кетоны удобно бронировать по методу Норриса [666], по кото-
рому к смеси раствора кетона, воды и КС1Оа при 35—40® С по каплям прибавляют
бром;
eHCHsCOCH,H' +ЗВГ, + КС1О3 eRCHBrCOCH2R' + KCl + 3H2O
Из метилэтилиетона по этому способу получается смесь 2-бромбутанона-З
(основной продукт) и 1-бромбутанона-2 (см. правило замещения на стр. 178).
Смесь можно разделить перегонкой в вакууме [667] на эффективной колоиие
при флегмовом числе 6 и разрежении 150 » рт. ст. Способ пригоден также
для синтеза монобромацетона.
Очень простым способом является бромирование по Фаворскому [668]. В иетон,
большей частью при охлаждении ледяной водой, приливают по каплям 1 моль брома,
отдувая НВг азотом или С02; можно также при О’ С через меток пропускать ток азота,
насыщенный парами брома. И в этом случае рекомендуется инициировать реакции;
«затравкой» или добавлением двух капель концентрированной бромистоводородиоё
кислоты (НВг в воде или уксусной кислоте).
Для бромирования кетостероидов в ледяной уксусной кислоте раствором брома
в том же растворителе рекомендуется [669] добавлять в раствор небольшое количестве
НВг и несколько крунниок А1С13.
Иногда бромистый водород удаляют только по окончании реакции, как, напри-
мер, при получении бромацетона.
Бромацетан [670]. При 70—80° С к смеси 6,8 моль (500 мл) ацетона,
372 лсд ледяной уксусной кислоты и 1,6 л sopfii при перемешивании добавляют пс
каплям 7,3 моль брома, регулируя скорость добавления по обесцвечиванию реак-
ционной массы (1—2 ч). По окончании реакции смесь разбавляют 0,8 л воды
охлаждают до 10? С и нейтрализуют твердым Na2C0s. Бромированный продую
отделяют, сушат над СаС12 и фракпиоякруют; выход 43—44% от теоретического;
т. кип. 40—42° С (13 мм рт. ст.).
Бромацетон можно, кроме того, получать так же, как хлорацетОЕ
(стр. 180), для чего к 4 ч. ацетока и 1 ч. мрамора при 28—31° С постепенно доба-
вляют 2,5 ч. воды, одновременно просасывая через массу воздух, насыщенны^
парами брома [671].
Бромиетоны раздражают кожу и слизистые оболочки (wcsal). Неперегнан-
ные в вакууме препараты загрязнены и вскоре окрашиваются. Для их стабили-
зации рекомендуется добавлять небольшое количество MgO-
Для получения в качестве основного продукта реакции более высокобромироваи-
лого ацетона следует увеличить количество вводимого брома.
[666] J. F. Norris, Ind. Eng. Chem., 11, 828 (1919).
[667] J. R. C a t c h, D. F. E 1 I i о 11, D. H. H e у, E. R. H. J on es
J. Chem. Soc., 1948, 272.
[668] A. Fa worst i, J. prakt. Chem. [2], 88, 641 (1913).
669 P. A. Plettner et al., Helv. chim. acta, 30, 385 (1947).
‘670] P- A. L e v e n e, Org. Synth., Coll. v. 2, 1943, p. 88.
[671] R. Scholl, G. Matthaiopoulos, Ber., 29, 1555 (1896).
182
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
1,3-Дибромацетон. К смеси 500 мл ацетона, 1,6 л воды и 375 мл ледяной
уксусной кислоты при 70° С медленно прибавляют по каплям 650 мл брома
(1,8 моль на 1 люлъ ацетона). П_ри этом получают 60 г монобромацетона, 20 г
1,1-длбромацетона, 50 г высокооромированных продуктов и 473 г 1,3-дибром-
ацетопа; т. кип. 97 —98° С (21—22 мм рт. ст.).
1,1,3-Трибромацетон [672]. В нагретый до 80 — 90° С раствор 210 г
1,3-днбромацстона в 500 мл ледяной уксусной кислоты в течение 1 ч без даль-
нейшего подвода тепла прибавляют по каплям 150 г брома в 500 мл ледяной
уксусной кислоты. После отгонки в вакууме.кислоты и первой фракции полу-
чают 180 г 1,1,3-трибромацетона (т. кип. 114—116° С при 14 мм рт. ст.)
и 21 г 1,1,1,3-тетрабромацетона (т. кип. 132—133° С при 13 мм рт. ст.).
Для синтеза бромкстонов вместо брома мощно применять комплексные соедине-
ния брома. При бромировании очень малых количеств веществ, например кетостеро-
идов, пригоден кристаллический, и поэтому легко дозируемый, пербромид пирндиний-
бромида (стр. 100). Для проведения реакции эквимолекулярные количества этого
комплекса и кетона нагревают до 40—60° С в ледяной уксусной кислоте или этиловом
спирте [33с].
Под действием раствора комплекса диоксандибромида в эфире на ацетон тоже
получается бромацетон с выходом 83% от теоретического [636]. Пригодность данного
метода для синтеза малоустойчивых циклических бромкетонов оспаривается [673].
При бромировании ацетилкарбинолов в эфире [502] способ себя оправдал *. Комплекс -
брома с эфиром — темно-красное масло, получаемое прибавлением сухого эфира •;
к охлажденному льдом брому, также рекомендуется для мягкого бромирования цикли-
ческих кетонов в сухом эфире [674].
При использовании N-бромсукцинимида (стр. 137) устраняются побочные
реакции, протекающие с бромом или бромистым водородом. С помощью N-бромсукцин-
имида получали, например, 2-бромциклогексанон [675] и 2-бромциклогептанон [676]
В течение примерно 30 мин нагревают с обратным холодильником в СС1<
1 моль кетона и 1 моль N-бромсукцнпимида с добавкой пезпачительпого коли-
чества инициаторов радикалов (0,1 г динитрила азоизомасляной кислоты или
около 2 г перекиси бенвоила). После охлаждения отсасывают раствор бромке-
тона от сукцинимида.
При действии N-бромсукцинимидом на а,р-ненасыщенные кетоны бром всту-
пает в аллильное положение. Так, тестостеронацетат [677] бронируется в положение 6.
тестостероиацетэт
ОСОСНэ
* Хорошие результаты получены при бромировании кетостероидов в укрупненном
масштабе диоксандибромидом в метаполе [II. II. Суворов и сотр., ЖОХ,
31, 3715 (1961)1. — Прим. ред.
[672] F. Weygand, V. Schmied-K owarzik, Chem. Ber., 82,
335 (1949).
[673] И. В. Малинская, А. С. Подберезина, ЖОХ, 28 (90), 1501
(1958).
[674] N. R. Е а н t о n, S. J. N е 1 s о н, J. Am. Chem. Soc., 75, 640 (1953).
[675] Н. S с h т j d, Р. К а г г е г, Helv. chim. acta, 29 , 579 (1946).
[676] Е. J. Core y/j. Am. Chem. Soc., 75, 2303 (1953).
[677] C. Meysfre, A. Wettstein, Experientia (Basel), 2, 408 (1946).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
18:
С примерами бромирования N-бромсукцииимидом можно ознакомиться в обзор
ной работе [678], исключения см. [679].
Получение а-бром- и а-иодкетонов через енолацетаты будет рассмотрено н;
стр. 184.
Жирно-ароматические кетоны без заместителей, способствующих электрофиль
ному замещению, легко бронируются обычными мягкими методами в а-положени<
к карбонильной группе. Реакцию ведут большей частью в ледяной уксусной кислот©
CH2CI3, CHCI8, СС.14 или в эфире при комнатной или более низкой температуре с при
мененнем рассчитанного количества брома. Можно также вести реакцию в хлорист©!
метилене или бензоле с добавкой соды. «Затравка» (см. выше), УФ-облучение и до
бавка каталитических количеств AlBr3 (A1CIS) (около 1,2 г на 1 моль кетона) ускоряю1
реакцию.
Из ацетофенона в сухом эфире с А1С13 и 1 моль брома при 0° С получаю-
а»-бромацетофенон (фенацилбромид); выход 88—96% от теоретического
т: пл. 51° С. Бромистый водород отдувают воздухом (подробности см. [680])
При действии на ацетофенон 2 моль брома в ледяной уксусной кислоте таким яс,
образом легко получают ш.о-дибромацетофенон [681].
п-Бромфенацилбромид [682]. В раствор 4-бромацетофенока в двойное
количестве ледяной уксусной кислоты при 20° С прибавляют по каплям 1 мол
брома; выделяется кристаллический n-бромфенацилбромид, ВгС6Н4СОСН4В
(т. пл. 108—109° С). При действии 2 моль брома на 4-бромацетофепоп [683
так же легко образуется п-а,а-трибромацетофенон, п-ВгСвН4СОСНВга
При добавлении щелочного раствора w-бромацетофенона или п-бромфенацил
бромида к нейтрализованной содовым раствором карбоновой кислоте получаете,
хорошо кристаллизующийся из этилового спирта эфир со-оксиацетофенона; эта реан
ции пригодна для идентификации жирных кислот.
RCOONa + BrCH2COC6H6 —► RCOOCHaCOCeHs + NaBr
Жирно-ароматические кетоны с заместителями в ядре, способствующими галс
генированию ядра, уже в очень мягких условиях можно избирательно бромироваг
в ядро или в боковую цепь. При этом учитывают, что перед а-галогенироваиием пре
исходит катализируемый кислотами переход кетона в активную форму. Устраня
енолизацию и связывая НВг добавкой ацетата натрия или калия, удается, например
бронировать 4-метоксиацотофенон в ледяной уксусной кислоте при 20° С до З-бром-4
метоксиацетофенона с 95%-яым выходом от теоретического [684] и 4-оксиацетофено
ДО 3,5-дибром-4-оксиацетофенона [685]. Бромированию боковой цепи благоприяэ
ствует применение «затравки». В холодной ледяной уксусной кислоте избирательа:
бронируется боковая цепь 2- и 4-оксиацетофенонов, а также 2- и 4-оксипропиофеис
нов; в водкой уксусной кислоте происходит бромирование в ядро [686]. Для больше
надежности избирательного бромирования боковой цепи оксигруппы блокирую
ацилированием или бензоилированием к только после дальнейших превращена:
а-бромкетонов освобождают оксигруппы гидролизом или каталитическим гидре
рованием.
[678] С. Djerassi, Chem. Rev., 43, 282 (1948).
[679] Ng. P., Buu-Hoi, Experientia (Basel), 2, 310 (1946); P. Karrej
A. R. Naik, Helv. chim. acta, 36, 1527 (1953).
[680] R. M. Cowper, L. H. Davidson, Org. Synth., Coll. v. 2, 194<
p. 480.
[681] F. Krohnke, Chem. Ber., 83, 56 (1950).
[682] W. D. Langley, Org. Synth., 9 , 20 (1929).
[683] J. J. Klingenberg, Org. Synth., 35, 11 (1955).
684] F. Krohnke, К. E 11 e g a s t, Chem. Ber., 86, 1562 (1953).
[685] K. W. Rosenmund, K. Pfroepffer, Chem. Ber., 90, 1922 (1957’
[686] Ng. Ph. В u u - H о i, Ng. D. X u о n g, D. L a v i t, J. Chem. Soc., 195«
1034; 1955, 18.
184
ОБРАЗОВАНИЕ СВЯЗИ С —Hal Б РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Наконец, а-бромкетоны аналогично а-бромальдегидам можно получать такжу.Я|
через енолацетаты [687]. К енолацетатам в СС14 при охлаждении льдом присоединяю» •л
точно рассчитанное количество брома, после чего бронированный раствор обрабаты* а
вают абсолютным метиловым спиртом и оставляют на двое суток. " "
ОСОСНз ОСОСНз
КСН2СОСНз —> КСН=ССНз — -> КСНВгСВгСНз СНзОН->
—> RClIBrCOCHa+НВг + СНзСООСНЙ
Эту реакцию можно использовать в том случае, если нужные енолацетаты легко1?
доступны. Из кетонов RR'CHCOCH3 могут образовываться два изомерных енолаце~,
тата I и II.
ОСОСНз 0 ОСОСНз 3
I II I
R'RCHC=CHa<— R'RCHCCHs—> R'RC=CCH3 4
II
Возможность образования изомеров I и II зависит от R и R' и от условий подуц?
чения енолов. •
Енолацетагы синтезируют с умеренными выходами по реакции с уксусная^
ангидридом в присутствии п-тилуолсульфокислоты [687] или, что лучше, пропуску
нием кетена при 60—80° С в соответствующий кетон с добавкой 0,5% сульфоуксусно!^
кислоты [688, 689]. Изящным способом получения енолацетатов является «перёЙ
этерификация» кетонов [690] изопропенилацетатом НаС=С(ОСОСН3)СН3, который?
получают при 55° С из ацетона и кетена [688J.
Изопропенилацетатным методом готовят, например, из метилэтилкетона иве-*
мер II с 96%-ным выходом, а из метилизобутилкетона изомер I с 92%-ным выходом/,
Из 20-кетостероидов по реакции с уксусным ангидридом и п-толуолсульфокисл^
той [691] образуется енолацетат II, а с изопропенилацетатом [692] — енолацетат
Для введения в 20-кетостероид с С=С-связями в ядре А или в ядре В атома;
галогена в положение 21 енолацетаты подвергают взаимодействию с галогенирующими
агентами, не затрагивающими С=С-связь. В качестве такого агента предложен N-иод^
сукцинимид [693].
COCOCH,
CH3COO
Ацетат 21-иод-Д6-прегненол-ЗР-она-20. Нагревают 45 мин 2,0 г Д>Н;'
ацетата Д6>го-прегнандиендиола-ЗР,20 с 1,18 г N-иодсукцинимида н 2 мл очн-j
щенного диоксана при 85° С. Охлажденный темно-красный раствор обрабат»^
вают сначала раствором KI, затем раствором Na2SaOg и большим количества®
воды. Выпявттгий неочищенный продукт (2,1 г; т. пл. 115—123° С) перекристалЛИ^
зовывают из смеси метилового спирта и воды (9 : 1); получают 1,31 г ацетат^
21-иод-Д6-прегненол-ЗР-она-20; т. пл. 135—137° С. * В.
[687
[688
[689
[690
[691
[692
[693
Р. Z. Bedoukian, J. Am. Chem. Soc., 67, 1430 (1945). - . 3
В. H. Gwynn, Е. F. D о g е г i n g, J. Am. Chem. Soc., 64, 2216 (1942)t
F. G. Y 0 u ng et al., J. Am. Chem. Soc., 72, 3635 (1950).
H. J. H age meyer, D. C. Hull, Ind. Eng. Chem., 41, 2920 (1949).
T. F. Gallagher et al., J. Am. Chem. Soc., 70, 1837 (1948).
T. F. Gallagher et al., J. Am. Chem. Soc., 74, 2810 (1952).
C. D j e r a s s i, С. T. Lenk, J. Am. Chem. Soc., 75, 3493 (1953).
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
185
N-иодсукцинимид [693]. К раствору 392 г сукцинимида в 12 л кипящей
воды прибавляют 510 г с-вежеосяжданной окиси серебра, фильтруют и оставляют
кристаллизоваться серебряную соль сукцинимида, которую затем отсасывают
и промывают холодной водой; выход 450 г. При 5—10° С 49,5 г тонко измельчен-
ной соли прибавляют в несколько приемов при перемешивании к раствору
50,8 г иода в 300 мл ацетона. После исчезновения окраски (30 мин) отсасывают
Agl. От фильтрата отгоняют ацетон в вакууме при комнатной температуре и не'
очищенный продукт промывают эфиром; выход 43 з; т. пл. 189—191° С. Его
растворяют в возможно меньшем количестве горячего диоксана и осаждают CClt.
Получаются бесцветные иглы; т. пл. 200—201° С. Для иодирования пригоден
продукт, плавящийся не ниже 197° С.
Если в енолацетате нет дополнительных С=С-связей, иодкетоны можно полу-
чать наряду с ацетилхлоридом также по реакции с хлоридом иода в хлороформе [693].
Так как 21-иод-20-кетостероиды являются важными промежуточными продук-
тами для синтеза 21-окси-20-кетостероидов, например дегидрокортикостерона * и кор-
тизона [694], а также деэоксикортикостерона (6957, следует указать, что такие
а-иодкетоны можно также получать по очень интересной реакции с иодом**:
RCOCH3 .(.сооснр,; c,H,ON a^ rcoch^CONb .
СООСаНй
--». RCOCHIC^0 CH,ONa; СШОН^ RCOCH21
'СООС2Н5
21-Иод-Д5-прегненол-Зр-он-20 [696]. К суспензии 0,1 моль натриевой
сели этилового эфира 21-оксалил-Д6-прегненол-Зр-она-20 в 500 мл абсолютного
метилового спирта в течение 15 мин при —15° С прибавляют при перемешивании
раствор 25,4 г иода в 600 мл метилового спирта. Через 30 мин после прибавле-
ния иода медленно добавляют раствор метилата натрия (136 ллметиловогс
спирта 4- 2,5 г натрия) и выдерживают 1 ч. Затем в течение 1 ч в несколько
приемов приливают 2 л воды, причем выпадает 21-иод-Д5-прегненол-30-он-2О.
Оставляют на ночь, отсасывают и промывают 30%-ным метиловым спиртом.
Слегка влажный неочищенный продукт подвергают дальнейшей переработке:
иод замещают на СН3СОО-группу.
а,а,а-Тригалогвнмегилкетоньг расщепляются щелочами на тригалогеиметакь
и карбоновые кислоты. Эта галоформная реакция [697] происходит также при непо-
средственном воздействии щелочных растворов гипогалогенитов на метилкетояы
Она имеет препаративное значение для получения карбоновых кислот из метилке-
тонов.
RCOCX8 + NaOH —> RCOONa + CHXg
RCOCH3 + 3X2 + 4NaOH —> RCOONa + CHX3+3NaX + 3H2O
* О синтезе 11-дегидрокортикостерона см. О. К. Н и к п ф о р о в а, Н. Н. С у в о
ров, ЖОХ, 29, 2428 (1959). — Прим. ред.
** Помимо этих «косвенных» методов получения 21-иод-20-кетостероидов существует
метод прямого иодирования 20-кетопрегнановых производных в положение 21
Он состоит в действии избытка иода иа кетон в присутствии окиск кальцик
в среде тетрагидрофуран — метаиол или метилендихлорид—метанол. В Д4-3-ке-
тоне предельная группировка кольце А и положения 2, 9 и 12 (в случа»
11-кетостерокдов) при этом не затрагиваются [Н. Ringold, G. Stork
J. Am. Chem., Soc., 80, 250 (1958); О. К. Никифорова, H. H. С у в о
ров, ЖОХ, 80, 238 (i960); Н. Н. Суворов, 0. К. Никифорова
М. В. Соколова, Мед. пром. СССР, № 12, 9 (I960)]. — Прим. ред.
694] L. н. S а г е t1 et al., J. Am. Chem, 80c., 74 , 4974 (1952). .
[695] H. RJu s c h 1 g, Angew. Chem., A 60, 247 (1948).
[696] H. R u s c h i g, Chem. Ber., 88 , 882 (1955).
[697] R. C. Fuson, B. A. Bull, Chem. Rev., 15, 275 (1934).
186
ОБРАЗОВАНИЕ СВЯЗИ С—Иа1 В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Аналогичную реакцию с иодом используют для идентификации веществ типа
RCOCII3 (где Н=Н, алкил, арил) илп соединений, окисляемых гипоподитом до
RCOCU3, например этилового спирта (йодоформная проба Либена).
Йодоформная проба. Растворяют 0,1 г или 0,1 мл вещества в 5 мл
диоксана, прибавляют 1 мл 10%-ного раствора NaOH и по каплям раствор К13
(10 г иода, 20 г KI и 80 мл воды) до появления устойчивого темного окрашива-
ния, после чего нагревают не более 2 мин при 60s С. Если раствор обесцвечи-
вается, прибавляют по каплям еще немного раствора К13 и кратковременно
нагревают. Затем смесь обесцвечивают несколькими каплями 10%-ного раствора
NaOH и разбавляют водой. При известных условиях йодоформ выделяется в виде
желтых кристаллов (т. пл. 119° С) из метилового спирта [698].
Йодоформ можно получать также, действуя 4 М раствором NalCL па водно-
щелочной раствор ацетопа [699].
14. Замещение галогеном водорода в гетероциклических соединениях
О-Гетероциклические соединения (фуран, бенз- и дибепзфуран) очень легко
бронируются преимущественно бромом в CS2, СНС13, СС14 или ледяной уксусной
кислоте. Хроман и бенздиоксан, являющиеся циклическими эфирами фенолов, бро-
нируются в бензольное ядро. Хлорирование и бромирование изохромана и пере-
группировка 1-галогенпроизводного в 2-х.юр- или 2-бромэтилбензальдегид описаны
на стр. 859.
Тиофен легко бронируется в положения 2 и 5. Под действием N-бромсукцни-
имида получается 2-оромтиофен с довольно низким выходом [700], лучший выход
достигается по реакции с бромом.
2-Бромтиофен [701]. К охлаждаемой в бане со льдом смеси из 1 моль
тиофена и 100 мл СС14 в течение 4 ч при перемешивании прибавляют по каплям
1,1 моль брома в 300 мл СС14. Остаток после отгонки растворителя нагревают
в течение 4 ч иа паровой бане при периодическом перемешивании с 15 г измель-
ченного в порошок NaOH. Затем раствор отделяют декантацией от едкого натра,
который дополнительно промывают СС14. Ректификацией на колонке получают
2-бромтиофен (выход 55% от теоретического, т. кип. 153—154° С) и 29,2 г 2,5-ди-
бромтиофена (т. кип. 95—98° С при 16 мм рт. ст.).
2,5-Дибромтиофен [702]. К смеси 297 г тиофена с равным объемом бен-
зола прибавляют по каплям 950 г брома с такой скоростью, чтобы не выделялись
пары брома. Когда выделение НВг замедляется, добавляют 700 мл C2HgOH
и 250 г NaOH и кипятят смесь 16 ч с обратным холодильником. После разбавле-
ния водой отделяют органический слой и перегоняют на колонке: получают
152 а 2-бромтиофена (т. кип. 158—162° С) и 425 в 2,5-дибромтиофена (т. кип.
200—210° С).
Применяя большие количества брома, легко получить 2,3,5-трибром-
тиофен [703] и тетрабромтиофен [704].
2-Хлортиофен лучше получать [706] взаимодействием тиофена в эфире
с SO2C1« в присутствии небольшого количества А1С13. При использовании N-хлор-
ацетамида [705] получаются худшие .результаты.
[698] R. С. Fuson, С. W. Т и I I о с k, J. Am. Chem. Soc., 56, 1638 (1934).
[699] А. И. Григорович, Н. Г. Симчаев, Мед. пром., 12, 27 (1958)‘
С., 1959, 8840.
[700] Ng. Ph. В u u - Н о i, Ann., 556, 9 (1944).
[701] F. F. В I i с к е, J. H. В u г с к h a I t e г, J. Am. Chem. Soc., 64»
478 (1942).
702] K. Folkers et al., J. Am. Chem. Soc., 67, 2094 (1945).
703] C. Troyanowski, BuU. Soc. chim. France, 1955, 424.
704] W. Steinkopf, H. Jacob, H. P e n z, Ann., 512, 136 (1934).
705} W. Steinkopf, A. Otto, Ann., 424, 68 (1921).
706] А. Тб Ы, O. EJberhard, Ber., 26, 2947 (1893).
1. ЗЛЧЫЦ1ШИ& ьидигидл ua nai пни
2-Бромпроизводпые фурана и тиофена могут быть с хорошим выходом пол
чспы бромированием фурана или тиофена при охлаждении льдом диоксаидиброми
и диоксапе или эфире [410].
Из упомянутых в пункте а) на стр. 141 методов иодирования для гетероцикл
ческпх соединении применяется, в частности, уже давно известный способ иодирован
иодом в присутствии желтой HgO. Смешивают 1 моль иодируемого вещества или е
раствора в спирте, ледяной уксусной кислоте, бензоле или лигроине (т. кия. 100
120° С) с 1 моль иода и 0,6 — 1 моль HgO при встряхивании или энергичном перемет
нянин. Иод и HgO добавляют попеременно маленькими порциями. В болыпинст
случаев уже при обычной температуре образуется красная Hgl2. Подробную рабоч}
пропись получения 2-иодтиофена этим способом см. [707]. 2-Иодтиофен [416] моя?
быть легко получен также иодированием тиофена иодом в присутствии копцентри^
ванной HNO3.
Метилтиофепы (2- и 3-) легко бронируются в ядро. При действии N-бромсу
цинимида в СС14 образуется смесь бронированных в ядро и в боковую цепь мети
тиофепов. В некоторых случаях целесообразно не разделять их перегонкой, а неп
средствеино использовать в дальнейших реакциях, например для синтеза 2-(пипер
динметил)-тиофена по реакции с пиперидином или З-тиофенальдегида по реакщ
с гексаметилентетрамином [708].
Подробное описание бромирования 3-метллтиофопа в боковую цепь под де
ствием N-бромсукцинимида в сухом бензоле в присутствии перекиси бензоила см. [70S
Катализируемое кислотами разложение 3-бромметилтиофена, которое иногда мояс
происходить со взрывом, замедляется добавкой СаСО3 или третичных аминов.
2-Хлорметилтиофсн. Его лучше всего получать хлорметилировани,
тиофена [710]. В смесь 525 мл концентрированной НС1, 450 мл 40%-ного форм
липа и 465 мл тиофена при перемешивании пропускают до насыщения энерга
ный ток EICI, поддерживая температуру в пределах 0—10° С. Затем реакционна
массу выливают и 2 л воды, отделяют выделяющееся масло, а продукт из водно
слоя несколько раз экстрагируют эфиром, соединяют масло с эфирным экстра
том, промывают эфирный раствор несколько раз водой и сушат над СаСО3. Пос.
отгонки эфира 2-хлорметилтиофен перегоняют, выход 47% от теоретическог
т. кип. 78—82° С при 18 мм рт. ст. 2-Хлорметилтиофен нельзя хранить в плотж
закрытой склянке, так как он может самопроизвольно и бурно разлагаться!
В пирроле и имидазоле атомы водорода всех СН-грунн очень легко замещаютс
хлором, бромом или иодом. Получение 2,3,4.5-тетраподпиррола является примере
галогенирования с помощью галогеповодорода и перекиси водорода (см. такя
стр. 157).
2,3,4,5-Тетраиодпиррол [711]. Растворяют 6,7 г свежеперегнанкох
пиррола и 66 8 KI при 30° С в 400 мл 25%-ного этилового спирта и прибавляй;
28 мл ледяной уксусной кислоты, 10 г ацетата натрия и 38 мл 10%-нон Н2С
в 50 мл воды. Раствор разогревается и окрашивается в красно-коричневый цве*
Температура пе должна превышать 45° С; эту температуру поддерживают еи
20 жил. Через 1ч смесь разбавляют двойным количеством воды. Серо-бурь;
осадок перекристаллизовывают из этилового спирта, содержащего активны:
уголь.
Получаются белые иглы; выход 80% от теоретического; т. раз.
162-164° С.
Электрофильное замещение галогенами водорода в пиридине, пиримидин:
хинолине и подобных веществах затруднено, в то время как нуклеофильное замещ<
ние па амино- или оксигруппы (в а- и у-цоложение к азоту) или синтезы с замыкание
[707] W. М inni s, Org. Synth., Coll. v. 2, 1943, p. 357.
[708] T. W. Campbell, W. W. Kaeding, J, Am. Chem. Soc., 73, 4018(1951
[709] E. Cam p aigne, B. F. Tullar, Org. Synth., 33, 96 (1953).
[710] F. F. В 1 i c k e, F. Leonard, J. Am. Chem. Soc., 68, 1934 (1946).
[711] A. Treibs, H. G. Kolm, Ann., 614, 187 (1958).
188
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
кольца амнпо- иля оксизамсщенных N-гетероциклов происходят довольно легко»
Поэтому в лаборатории, если это возможно, пользуются методами замещения амино-
или оксигруппы на галоген, как, например, при получении 2- п 4-бромпиридина
(стр. 252, 230) и 2-хлорпиримидина (стр. 248, 229).
Электрофильное замещение водорода галогеном в пиридине происходит в по-
ложения 3 и 5. Бромированием расплавленного гидрохлорида пиридина в металличе-
ской бане при 212—215° С с добавкой 10 г HgCla па 100 г пиридина получают в каче-
стве основного продукта 3-бром- или 3,5-дибромпиридин, что зависит от количества
брома, взятого для бромирования [712, 713]. Повышение температуры галогениро-
вания пиридина приводит к изменению типа замещения (ср. аналогичное явление
при бромировании галогенбензолов па стр. 139).
2-Бромпиридин [713]. В колонке с электрообогревом, высотой 40 ем
и диаметром 3,5 см, заполненной высокоплавкой насадкой из стеклянных трубок
длиной 5 мм и диаметром 6 жл, нагретой до 500° С, подвергают взаимодействию
в паровой фазе 6 моль пиридина с 9 моль брома. Бром приливают по каплям
через стеклянную спираль длиной 12 см, расположенную в колонке над насад-
кой на расстоянии 2 см от нее, а пиридин приливают одновременно через спи-
ральную насадку, присоединенную к верху колонки (длина 60 см), также нагре-
тую до 500° С. Продолжительность реакции около 5 ч. Скорость подачи реагентов
должна быть такой, чтобы в црисоедивеиной к колонке двухгорлой колбе (ем-'
кость 1 л), снабженной воздушным холодильником (выпуск НВг!), не появлялись
пары брома. Неочищенный продукт подщелачивают раствором NaOH и пере-
гоняют с водяным паром, собирая Зл /хистпллнта. • Фракционной перегонкой
извлеченной бензолом смеси бромидов получают 46% 2-бромпиридина (т. кип.
90—92° С при 25 мм рт. ст.) и 17% 2,6-дибромпиридина (т. пл. 118—119° С) v
Газофазное хлорирование пиридина на пемзе в присутствии азота приводит.
уже при 270° С к a-замещению. Получают 2-хлорпиридин с выходом 31% от теорети-
ческого; при более высоких температурах (до 400° С) в качестве основного продукта
образуется 2,6-дихлорпиридин [714]. Хлорирование расплавлепноуо гидрохлорида
пиридина при 165—175° С ведет главным образом к получению 3,5-дихлорпири-
дина [713, 714]. 4-Хлорпиридин лучше всего готовить непосредственно из гидро-,
хлорида 4-К-пиридилпиридиинйхлорида [715, 716].
- cl_ -
Гидрохлорид 4-К-пиридилпиридипийхлорида. При охлаждеики льдоод;
до 8—12° С и перемешивании к 600 г SOCI2 прибавляют по каплям 200 г пири-:;..
дина, высушенного над едким иали, и перемешивают еще несколько часов, давая
температуре смеси медленно повыситься до 20° С. После трех суток* стояния ири
20° С избыточный SOCI2 (и прочие низкокипящие продукты) отгоняют в вакууме, х
а остаток выдерживают 1 ч на кипящей водяной бане- Охлажденную до 0е С. :,
серо-бурую кристаллическую массу обрабатывают (охлаждение льдом!) 200 мл
абсолютного этилового спирта, охлажденного льдом, отсасывают и дважды
промывают холодным абсолютным этиловым спиртом (порциями по 200 мл).,
Высушенный в вакууме неочищенный продукт растворяют в смеси 100 мл воды
и 300 мл 2 и. НС1, кипятят 5 мцн с активным углем, доводят объем дд 180 мл :
в вакууме, создаваемом водоструйным насосом, смешивают с 400 мл этилового
спирта и охлаждают до 0° С. Выпавший светло-желтый гидрохлорид 4-N-nHpB«<-
дилпиридинийхлорида промывают абсолютным этиловым спиртом и высушивают
в вакууме; выход 167 г; т. пл. 158—160° С.
4-Хлорпиридин. Смешивают 120 г гидрохлорида 4-пиридилпиридВний«
хлорида со 110 г PCI* и сплавляют на масляной бане при 130—140° С. Быстро* ‘
затвердевающий плав вновь разжижают, повышая в течение 10 мин температуру i
[712
[713
[714
[715
[716
Н. Maier-Bode, Вег., 69, 1535 (1936).
S. М. М с Е I v a i и, М. A. G о е s в, J. Am. Chem. Soc., 65, 2231 (1943).
J. Р. W i b a u t, J. R. Nicolai, Rec. trav. chim., 58, 709 (1939).
E. Koenings, H. Greiner, Ber., 64, 1052 (1931).
K. Bowden, P. N. Green, J. Chem. Soc., 1954, 1796.
I. ЗАМЕЩЕНИЕ ВОДОРОДА НА Hal ИЛИ SCN
18!
до 150° С. После охлаждения реакционную массу смешивают с водой, слаб<
подщелачивают раствором Na2CO3 и продукт тщательно экстрагируют эфиром
Экстракт промывают водой, сушат над Na2SO4, отгоняют с колонкой эфир и оста
ток перегоняют в вакууме. После добавления к дистилляту воды отделившееся
тяжелое масло растворяют в эфире, сушат и перегоняют. Получают 30 г 4-хдор
пиридина, выход 50% от теоретического; т. кип. 53—55° С (20 лл* рт. стЛ,
Аналогичным образом (РВг5, 130° С) может быть получен 4-бром
пиридин |717].
Значительно легче происходит замещение водорода галогеном в амино- и окси
замещенных N-гетероциклических соединениях. При этом применяют методы, аиа
логичные методам галогенирования ароматических аминов и фенолов (стр. 150)
При бромировании 4-оксипиридииа до 3,5-дибром-4-оксипиридина даж<
в очень мягких условиях следует учитывать протекание побочной реакции
окислительной деструкции [605]. 3,5-Динод-4-оксипиридин получают [718
взаимодействием 4-пиридона с иодом в водном растворе, который поочередн<
подщелачивают раствором NaOH и подкисляют НС1.
Совершенно так же получают 2-амино-5-иодпиридин [719].
Натриевую соль 2-оксипиридииа переводят иодом в водном раствора
К2С03 в 3,5-дииод-2-оксипиридин [719J.
Обсуждавшийся па стр. 155 способ замещения водорода иодом через ртуть,
органические соединения оказался очень подходящим для получения 2-амино-5-
иодпиримидина [720].
Для этого водную суспензию ацетоксимеркурисоединения, образовав
шегося в течение нескольких минут из амина в ацетата ртути, быстро обрабаты
вают раствором иода в горичем дноксане при 70’ С. Из реакционной массь
водным раствором K.I осаждают Hgl2; выход 2-амипо-5-иодпиримидина 70% от.
теоретического.
2-Хлормеркури-3-метилгиофен превращается в водном растворе иодг
и иодида калия с 92%-ным выходом в 2-иод-3-метилтиофен [721], а легко доступ
ный 2,5-ди-(хлормеркури)-3-метилтиофен [722] с 70%-ным выходом — в 2,5-дп
иод-3-метилтиофен [721]. Аналогичным способом моно- или дихлормериурж
производные 2,5-диметилфурана превращаются в 3-иод- или 3,4-дииод-2,5-
диметилфураны [723].
Существуют два способа прямого галогенирования боковой цепи алиил-If*
гетероциклических соединений:
а) галогенирование в ледяной уксусной кислоте в присутствии ацетата натрия
или калия [725, 726], протекающее по механизму, подобному механизму бромирова-
ния кетонов [724];
]717] D. Jerchel, Н. Fischer, К. Thomas, Chem. Вег., 89, 2928
1718) Я.к. г, A, S. Briggs, J. Soc. Chem. Ind., 62, 189 (1943).
[719] W. T. С a 1 d w e 11, F. T. T ys on, L. L a u er, J. Am. Chem. Soc.
66, 1481 (1944).
[720] R. G. Shepherd, С. E. Fellows, J. Am. Chem. Soc., 70, 157 (1948)
[721] W. Steinkopf, \v. Hanska, Ann., 532, 241 (1937).
[722] W. Steinkopf. A. Killimgstad, Ann,, 532, 292 (1937).
[723] Ch. D. Hurd, k. Wilkinson, J. Am. Chem. Soc., 70, 741 (1948)
1724} B.R. Brown, D. L. Hammick, В. H. Thewlis, D. J. Wat
bridge, J. Chem. Soc., 1953, 1369.
725] W. Koenigs, Ber., 31, 2364 (1898).
[726[ B. R. В г о w n, D, L. H a m m i ck, В. H. T h e w I i s, J. Chem. Soc.
1951, 1145.
190
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
б) бромирование с помощью N-бромсукцивимида.
При бромировании N-гетероциклов с активными СП3-груцпами по первому
способу обычно образуются только трихлор- или трибромметилпроизводные. Для
бромирования 3-метилппридина, 3- и 4-метплхпнолцна и 1-, 3- и 4-метилизохинолина
этот способ неприменим. 2- и 4-Метилпиридицы таким путем хорошо хлориро-
вать [726, 727], но не бронировать.
4-Трихлорметилпиридип. В смесь 21 мл 4-метилииридина, 20 мл уксус-
ного ангидрида, 60 г ацетата натрия и 250 мл уксусной кислоты при 80° С про-
пускают 50 г сухого хлора (3 моль на 1 моль пиколина). Реакционную массу
выливают на лед, подщелачивают аммиаком и продукт экстрагируют эфиром. •
Двукратной вакуумной перегонкой экстрагированного эфиром масла получают
15,4 г 4-трихлорметилпиридина, т. кип. 105—107° С (18 мм рт. ст.). Это ве- '
щество разлагается в течение нескольких дней; в водной уксусной кислоте ,
с AgNO3 он превращается в изоникотиновую кислоту, в ацетон-солянокислой 71
среде с оловом — в 4-дихлорметилпиридин.
По второму способу можно, в частности, получить 4-бромметилхинолип (лепи-
дилбромид) [728], который не может быть приготовлен галогенированием в ледяной
уксусной кислоте.
4-Бромметилхииоаин (лепидилбромид). К перемешиваемой суспензии ’
0,08 моль N-бромсукцинимида в 50 мл СС14 прибавляют при 60° С 0,1 моль лепи- 1
дина. Смесь кипятят 30 мин с обратным холодильником и в горячем виде филь-
труют с отсасыванием. Из фильтрата в виде белого кристаллического продукта
выпадает лепидилбромид (10 г), который для очистки от примеси сукцинимида
тщательно промывают водой и сушат в вакууме; т. пл. 88—91° С. Продукт необ- J‘
ходимо тотчас же подвергнуть дальнейшим превращениям, так как он разла- ч
гается в течение нескольких часов. 4
Литературу о дальнейших реакциях N-гетероциклических соединений ?
С N-бромсукцинимидом см. [729].
Броммотилпропзводные N-гетсроциклических соединении можно синтезиро-
вать также окислением металпроизводных N-гетероцпклическпх соединений окисью- |
селена, восстановлением получающихся альдегидов формальдегидом и
обменом гидроксильной группы образующегося спирта на бром при
в бензоле [726].
15. Непосредственное замещение водорода в ароматичесиих
на SCN-группу *
'-’я
Первичные, вторичные и третичные ароматические амины, а также фенолы $
со свободными орто-и (или) пара-положениями легко роданируются родапом (SCN)a.
последующим
помощи PBr5
соединениях я
Группа SCN вступает в пара-положение к ОН- или NRa-rpynnc, а если пара-
положение занято, то в орто-положение [И, 12]. В последнем случае легко происхо- я
* О замещении водорода на родан-группу’см. II. Н. Мельников, ТТ. Д. Суха- 3
рева, Реакции и методы исследования органических соединений (РиМИОС), ч!
8 , 9 (1959). — Прим. ред. 3
[727] Р, D yson, D. L. Н a m m i с k, J. Chem. Soc., 1939, 781. а
[728] К. N. Campbell, J. F. Acker ma п, В. К. Campbell, J. Am. 1
Cbem. Soc., 71, 2906 (1949). 1
[729] L. Horner, В. H. W i n к e 1 m a n n, Angew. Chem., 71, 361 (1959). a
II. ОБМЕН Hal НА ДРУГОЙ Hal ИЛИ SCN
191
дит замыкание кольца [14, 730]. Так, из «-толуидина образуется 2-амино-6-метил-
бензтиазол, из этилового эфира «-аминобензойпой кислоты — 2-амано-6-карбэгокси~
бензтиазол.
Бензол и нафталип пе роданируются роданом, а высшие конденсированные
ароматические соединения—антрацен, бензпирен и другие роданируются {731].
Роданируемое вещество можно вносить в раствор родана (получение раствора см. на
стр. 92), но проще работать с родапом в момент выделения. В реакционной смеси
родан образуется из роданида щелочного металла при действии брома (хлора) или
из Cu(SCN)g. В качестве растворителей для работы с хлором или бромом пригодны
уксусная кислота или метиловый спирт и метилацетат, насыщенные NaBrили NaCl.
Тимолроданид (3-окси-6-родан-1-метил-4-изопропилбензол). Растворяют
15 г тимола и 35 г роданида натрия (NaSCN • 2НгО) в 100 мл метилового спирта,
насыщенного NaBr, и при комнатной температуре приливают по каплям при
перемешивании раствор 20 г брома в 30 мл метилового спирта, насыщенного
NaBr. После 30 мин стояния к реакционной массе добавляют шести-, восьми-
кратное количество воды; тимолроданид отделяется в виде масла и выкристал-
лизовывается после 2 ч охлаждения льдом. Продукт высушивают на глиняной
пластиние и перекристаллизовывают из лигроина с активным углем; выход
95% от теоретического; т. пл. 105° С.
Аналогичным способом получают в метиловом спирте при 5° С 4-родан-
анилин с 97%-ным выходом [12].
Подробное описание родаиирования с помощью NH4SCN и брома в ледяной
уксусной кислоте на примере получения 4-родандиметиланилина см. [732].
4-Родапаннлип [14]. Растирают 11 г NH4SCN и 12 г CuSO4. Образо-
вавшуюся черную вязкую кашицу прибавляют при 50° С к перемешиваемому
раствору 2,3 а анилина в 50 мл 40%-иой H2SO4 и, повысив через 10 мин темпе-
ратуру до 75° С, ведут реакцию до перехода черного Cu(SCN)2 в бесцветны
CuSCN. После фильтрации продукт экстрагируют из остатка горячей водой.
Из объединенного фильтрата после нейтрализации Na2COa получают 2,9 в 4-ро-
дананилина, который перекристаллизовывают из воды; т. пл. 57° С.
Реакцию можно также вести в ледяной уксусной кислоте, этилацетат^
или метиловом спирте с Cu(SCN)2, предварительно приготовленным осаждением
из раствора CuSO4 эквивалентным количеством NaSCN, отсасыванием и промыв-
кой этиловым спиртом или эфиром.
Родан можно получать электролизом солянокислого водно-спиртовото раствора
роданида щелочного металла или роданида аммония в присутствии роданируемогс
вещества [10с]. Этот способ не имеет препаративных преимуществ перед описанными
выше способами. О введении SCN-группы в ароматические соединения через диазо-
соединення см. стр. 257.
II. Обмен галогена на другой галоген или на родан-группу
1. Обмен галогена на фтор
Реакция обмена одного галогена на другой имеет особенно большое значение
для получения алифатических фтор- и иодпроизводных. Обзорные работы по этомд
вопросу см. [3, 176, 733, 734]. При обмене на фтор наблюдается следующий порядод
[730
[731
[732
[733
[734]
R. Q. Brewster, Р. В. D a i n s, J. Am. Chem. Soc., 58, 1364 (1936).
J. L. Wood, L- F. Fieser, J. Am, Chem. Soc., 63, 2323 (1941).
R. Q. Brewster, W. Scbroeber, Org. Syntn., Coll. v. 2, 1943, p. 574:
A. L. Henne, Org. Reactions, 2, 49 (1944).
G. Schiemann, Die organischen Fluorverbindungen und ihre Bedeutunj
fur die Technik, Darmstadt, 1951; J. H. Simons, Fluorine Chemistry, v. Г
Academic Press Inc., New York, 1950; Ch. S I e s s e r, St. R. Schram, Pre
parations, Properties and Technology of Fluorine and Organic Fluorine Compounds
McGraw-Hill, New York, Toronto, London, 1951; E. F о г c h e, в энциклопедии
Ullmanns Enzyklopadie der technischen Chemie, Bd. 7, 3 Aufl., Munchen —
Berlin, 1956, S. 605—641.
192
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
бром замещается легче, чем хлор, иод — легче, чем бром. Наиболее легко обмени-
вается галоген в галогенированных карбоновых и сульфокислотах в а-положенив
к карбонильной или карбоксильной группе и в a-положении к С=С-связи (в аллиль-
ном положении), труднее в насыщенных галогеналкнлах RCX2R' и RCXS. Перевод
групп СНХ или СН2Х в CHF или CH2F осуществляется с помощью галогенидов метал-
лов. В винилгалогенидах обмен галогена на фтор чаще всего невозможен, в аромати-
ческих соединениях обмен возможен только в том случае, если в орто- или пара-поло-
жении к галогену имеется ннтрогруппа. Наличие атома фтора у соседнего атома угле-
рода затрудняет обмен галогенов.
В промышленности предпочитают проводить обмен с помощью безводного HF
(стр. 107); прн малых лабораторных загрузках хорошие результаты дают проводи-
мые в стеклянной аппаратуре реакции с фторидами металлов, SbFs пли смешанный
фторгалогенидом сурьмы SbFsX2. Последний получают в основном непосредственна
в реакционной массе из 1 моль Sbr3 и 1 моль хлора или брома. В большинстве случаен
приходится вести реакцию без доступа влаги. Обычно обмен атома брома на фтор»
снижает температуру кипения вещества примерно на 70° С, а обмен атома хлора —’
на 40е С. Поэтому органический фторид, образующийся в ходе обменной реакции,
можно отогнать на колонке.
В отсутствие катализаторов HF вступает в реакцию только с очень реакционно-
способными галогенидами, т. р. с галоидангидридами кислот или аллилгалогенидами.,
Так, в беизотрихлориде все атомы хлора легко замещаются фтором при 0—140° С
(735, 737--739]. .
Бензотрифторид [735]. В медную колбу, снабженную доходящей до дна1
медной трубкой для подвода таза, газоотводной трубкой и медной мешалкой^
помещают 500 г бензотрихлорида и при 0° С и перемешивании медленно про-,
пускают в него 200 г HF (72 ч). Из отводной трубки сначала выходит только^
HCI, затем во все возрастающем количестве HF и уносимый бензотрифторцц,
которые конденсируют в медном приемнике, "охлаждаемом смесью поваренной'
соли со льдом. Продукт реакции, оставшийся в колбе, соединяют с конденсатом
из приемника, нагревают до комнатной температуры и связывают еще содер-
жащийся в нем фтористый водород встряхиванием с NaF. Затем смесь фильтруют,
н перегонкой фильтрата получают 300 г бензотрифторида; т. кип. 102,3* С. Тяже»,
лую фракцию, состоящую только из частично фторированного бензотрнхлорида^
можно использовать опять в реакции. i
О получении бензотрифторида с помощью В Fs с примерно одинаковым"
выходом см. [736].
Соединения RCX2R' и ЙСХ8 реагируют с HF в присутствии SbF3 или SbCl8J
Действующие как катализаторы смешанные галогениды сурьмы (V) непрерывно'
регенерируются фтористым водородом, причем образующийся SbCI3 реагирует с фто-"
ристым водородом и превращается с выделением хлористого водорода в SbF3. 'j
Дифтордихлорметан. CCI2F2 (т. кип. — 30° С). Промышленный способ)
получения состоит в введении HF (т. кип. 19,4° С) и СС14 (т. кип. 76° С) в реак-’
циониый сосуд, в котором находятся SbF3 и небольшое количество пентагало-.'i
генида сурьмы. Через колонку непрерывно отгоняют хлористый водород и ди-’
фтордихлорметан, в то время как HF, CCI8F (т. кип. 25° С) и СС14 остаются^
в зоне реакции [740]. Лабораторный способ с применением SbCI6 и HF под давлетс
нием см. [ЗЬ, 741]; с использованием ” л ”
см. (733].
SbF3 и 0,1 моль SbCla на 1 моль SbF,
[735] J. Н. Simons, L. J. Lewis, J. Аш. Chem. Soc., 60 , 492 (1938). -i
[736] E. Pouterman, A. G iradet, Helv. chim. acta, 30, 107 (1947).
[737] K. Wiechert, Angew. Chem., 56, 333 (1943). )
[738] J. H. В г 0 w n, C. W. Suckl ing, W. B. W h a 11 e y, J. Cbem. Soc.»
1949, 95.
[739] F. S m i t h et al., J. Appl. Chem., 2, 97, 127 (1952).
[740] A. L. Henne, пат. США 2007208, 1930129, 1833847; С. A., 29, 545»
(1935); 28, 179 (1934); 26, 1047 (1932). ,
[741] L. J a r k 0 v*s k у, M. H u d 1 i c k y, Chem. Listy, 51, 625 (1957).
II. ОБМЕН Hal На ДРУГОЙ Hal ИЛИ SCX
193'.
Способ получения фторидов по реакции Свартса уже давно известен н сохранил
свое значение до настоящего времени. Соответствующий галогенид подвергают взаимо-
действию с SbF3 (0,35 моль SbFs на 1 моль подлежащего обмену галогена). Реакцион-
ную способность не особенно активного SbF« повышают добавкой 2—5% SbCl6 илн
примерно i% хлора, или 5% брома [733, 742]. Обмен галогенов происходит посте-
пенно и ему благоприятствует увеличение добавки SbCl8, хлора или брома. Особенно-
активное действие оиазывает комплекс SbF9CI«>2HF, получаемый [7-43] нагреванием
до 150° С в автоклаве 1 моль SbCl5 с 5 моль HF.
Обмен галогена в соединениях с группировками типа —СНХ—при действии HF
так же неосуществим, как и в случае SbF3 или SbF3X2.
Трифторид сурьмы [41, 744]. Получают растворением Sb2O3 в избытке-
водного раствора HF, выпариванием досуха и перегонкой в медной аппаратуре;
т. кип. 376° С.
Примером работы со смешанными фторгалогекидами сурьмы (V) является получение-
этилового эфира трифторуксусной кислоты и трифторуксусной кислоты череа
1,3,5-трис-(трифторметил)-сижж-триазин [3i, 745].
СС13 CF8
I I
N N N N
|| | SbF3; SbCt,^ || । . HCl:C!HtQH> 3CFaCOOG2H5
CI3C'/Xn'X'CCI3 РзС/\ГЧРв
Трифторуксусная кислота. Трихлорацетонитрил насыщают сухим НВг
в присутствии около 0,01 моль А1Вг3 или А1С13, причем он циклизуется в 1,3,5-
трис-(трихлорметил)-еил«-триазин. Возрастающими порциями вводят 500 а
этого вещества в течение 4 ч при перемешивании в горячую смесь 800 г SbFa
и 105 a SbCl3, в которую предварительно при 150° С пропускают 100 а хлора.
Реакцию ведут в аппарате, снабженном колонкой с обратным холодильником.
Реакционную массу нагревают до слабого кипения и через 30 мин [31] или через
2 ч [745] после окончания прибавления галогенидов сурьмы прй флегмовом
числе 10 начинают отбирать фракцию при 98—105° С (43 г) и вторую фракцию
при 105—125е С (153 г). Эту фракцию еще раз фторируют смесью 500 г 8bFa
и 105 г SbClg, насыщенной при 150е С хлором. Отобранную при последующем
фракционировании высококипящую фракцию вновь фторируют. В совокупности
получают около 160 г (выход 49% от теоретического) трис-трифторметилтри-
азина; т. кип. 98—100° С. По Нортону [745], выход должен составлять 90%
от теоретического.
кипячением этого продукта с водно-спиртовым раствором НС1 получаю!
этиловый эфир трифторуксусной кислоты, который сначала находится в виде
азеотропной смеси с водой; т. кип. 52,5—56° С. Азеотропную смесь можно обез-
водить концентрированной H2SO4 или омылить раствором NaOH в трифтор-
ацетат натрия. Эту тонконемельченную соль добавляют при охлаждении и кон-
центрированной H2SO4 и отгоняют затем безводную трифторуксусную кислоту;
т. кип. 72—74е С.
Трифторунсусная кислота образует с водой азеотропную смесь (т. нип,
105,5° С) с содержанием 79,4% CF3COOH. Она может быть также получеив
в лаборатории окислением ж-аминобензотрифторида (из бензотрифторида нитро-
ванием и восстановлением) бихроматом калия [746].
742
743
744
745
746
F. Swans, Bull. cl. sci., Acad. roy. Belgique [3], 24, 474 (1892).
W. B. Whalley,?. Soc. Chem. Ind., 66, 427 (1947).
O, Ruff, Die Chemie des Fluors, Springer-Verlag, Berlin, 1920.
T. R. Norton, J. Am. Chem. Soc., 72, 3527 (1950).
F. Swarts, Bull. cl. sci., Acad. roy. Belgique, 8, 343 (1922); C., 1923, I» 86,
13 Заказ 1835.
194
ОБРАЗОВАНИЕ СВЯЗИ С—Hal Б ₽ЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Смешением 1 г трифторуксусной кислоты с 0,87 г Р2О5 и последующей перегонкой
получают ангидрид трифторуксусной кислоты [747] (т. кип, 39° С), легко образующий
с карбоновыми кислотами или ангидридами смешанные ангидриды, которые расще-
пляются на катион — ацил и анион — трифторацетат:
,0 /О
(CF3CO)2O-l-RC<f —> CF3C<f I
\)Н XO-|CR
Этим объясняется то, что добавка ангидрида трифторуксусной кислоты дает
•возможность успешно проводить этерификацию трудно этерифицируемых кислот [748]
и спиртов [749].
При взаимодействии образовавшихся в реакционной массе смешанных ангид-
ридов с олефинами получают а, ^-ненасыщенные кетоны
X)C^CH-+CF3C/ —^C=C-CR
/ \q-cr / ||
II о
о
а при взаимодействии с алкинами—1,3-дикетоны [750]. Сведения о применении
трифторуксусной кислоты см. [751].
Трифторуксуспая кислота получается также окислением 2,3-дихлоргексафтор»
бутена-2 перманганатом калия [752]:
СС12=СС1СС1=СС12 —> СС13СС1=СС1СС13 —>
—► CF3CC1=CC1CF3 - КМ°0,: K°g CFaCOOH
В промышленности трифторуксусную кислоту производят электролизом 4% -ного
раствора уксусного ангидрида в безводном HF и абсорбцией выделяющегося фторан-
гидрида трифторуксусной кислоты [753].
Способ обмена галогенов на фтор с помощью фторидов металлов (AgF, Hgl1.
HgFX, HgF2, KF) применим и в тех случаях, когда описанные до сих пор методы
непригодны, главным образом при обмене галогенов в алкилгалогенидах.
Фторид серебра AgF получают взаимодействием карбоната [744] или окиси [754]
серебра с водным фтористым водородом. Раствор выпаривают и затем быстро высуши-
вают остаток. Фторид серебра трудно получить безводным; кроме того, на 1 моль
обмениваемого галогена (X) приходится вводить 2 моль AgF, так как выделяющийся
галогенид серебра образует с фторидом двойную соль [755]. В настоящее время AgF
применяется сравнительно редко; он вполне заменим KF. Однако преимуществом AgF
является возможность ведения реакции в мягких условиях.
, 21-Фторстероиды могут быть получены добавлением небольшого избытка
50%-ного водного раствора AgF при 30—40° С к раствору 21-иодстероида во
[747
[748
[749
[750
[751
[752
[753
[754
1755]
3. М. Т с d d е г et а]., 3. Chem. Soc., 1949, 2976.
R. Rood jr., J. Am. Chem. Soc., 77, 3403 (1955).
A. H. Ahlbrecht, D. W. Codding, J. Am. Chem. Soc., 75, 984 (1953),
А. Г<. H e n n e, J. M. Tedder, J. Chem. Soc., 1953, 3628.
J. M. Tedder, Chem. Rev., 55, 787 (1955).
A. L. H e n n e, P. Trott, J. Am. Chem. Soc., 69, 1820 (1947).
E. А. К a u c k, A. R. D iesslin, Ind. Eng. Chem., 43, 2332 (1951).
H. J. Emeleua, H. G. H e a I, J. Chem. Soc., 1946, 1126.
F. Swarts, Bui], cl. sei,, Acad. roy. Belgique [5], 7, 438 (1921); C., 1921,
II. ОБМЕН Hal НА ДРУГОЙ На! ИЛИ SCX
1S
влажном ацетонитриле [756]. Реакцию можно вести также без растворителе
в сухом фтористом водороде, углеводородах или эфире *.
Для обмена галогена и водорода в галогензамещенных углеводородах на фтс
применим [758] также AgF2 и CoF3. Фторное серебро получают пропусканием фтог
при 150—200° С через галогенид серебра AgX; CoF3 готовят [3m, 757] из СоС1а и CoJ
или взаимодействием СоаО3 с фтором при 250—300° С. Оба вещества пригодны такн
для фторирования углеводородов, тан как в этом случае на 1 моль образующегос
фтористого водорода выделяется примерно вдвое меньше тепла, чем при фторирован®
чистым фтором [Зп, 759, 760].
Фторирование с помощью AgFa или CoF3 проводится ступенчато с повг
шением температуры в соединенных последовательно четырехгранных peaj
циоиных трубках, в которых тонким слоем распределен фторид металла. В этс
Же аппаратуре предварительно готовят фториды металлов, а после проведена
фторирования регенерируют их пропусканием тока фтора.
Фторид ртути HgF является более активным агентом, чем AgF, при получена
алкилфторидов из алкилиодидов или алкилбромидов. Добавка иода благоприятствуй
протеканию реакции обмена. В реакциях с алкилхлоридами HgF мало активен, дл
фторирования соединений типа RCX2CX2R' он непригоден, так как при атом происх<
дит отщепление аалогена с образованием галогенированных олефинов и соле
ртути (II).
Более действенными агентами, чем HgF, являются HgClF и UglF, которь
получают добавкой к HgF эквивалентных количеств галогена [761].
Особенно активным агентом для обмена галогенов на фтор является HgF
оба атома фтора которого обмениваются на хлор, бром или иод. С иодидами Hgl
реагирует так бурно, что иодид приходится разбавлять углеводородами, фторирова»
ными углеводородами, СНС13 или СН8С12; эфир и кетоны непригодны в качестве ра<
творителей. С помощью HgF2 возможен обмен на фтор до трех атомов брома и до дву
атомов хлора у одного атома углерода [762]. Эта соль в чистом виде гораздо мен<
доступна, чем AgF, HgF й KF.
Фторид ртути (II). Его получают [744] взаимодействием HgF с хлоро
и последующим удалением сублимацией образующегося HgCJ2. По другое
способу [762] пропускают ток фтора над HgCl2< распределенным тонким слое
в медной трубне. Проще получать HgF2 из HgO и HF [763, 764] или HgCI2 и Н
[765] непосредственно в реакционной смеси, однако в этом случае процесс иео<
ходимо вести при низких температурах и в металлическом аппарате.
* О получении фторкетонов стероидного ряда при помощи перхлората фтор
см. 10. А. Т и т о в, И. Г. Р е ш е т о в а, А. А. А х р с м, РиМИОС
15, 7 (1966). — Прим. ред.
[756 ] Р. Т a n и Ь a user, R. J. Р г a 11, Е. V. J ensen, J, Am. Cheft
Soc., 78, 2658 (1956).
[757 ] II. F. Priest, Inorg. Synth., 3, 175 (1950); R. D. Fowler et al., In<
Eng. Chem., 39, 343 (1947); E. T. McBee, Ind. Eng. Chem., 39, ЗЮ (1947
758] E. T. M с В e e et al., Ind. Eng. Chem., 39, 310 (1947).
759] E. T. McBee, L. D. Bechtol, Ind. Eng. Chem., 39, 380 (1947).
760] R. D. Fowler et al., Ind. Eng. Chem., 39, 292 (1947).
761] A. L. Henna, M. W. RenoU, J. Am. Chem. Soc., 60, Ю60 (1938).
762] A. L. Henna, T, M i d g 1 e у jr., J. Am. Chem. Soc.,- 58, 884 (19o6).
763] A. L. H e n n e, J. Am. Chem. Soc., 60, 1569 (1938).
764] A. L. H e n n e, J. V. F 1 a n a g a n, J. Am. Chem. Soc., 65, 236
(1943).
[765 ] F. E. R а у, С. E. Albertson, J. Am. Chem. Soc., 70, 1954 (1948).
13*
196
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
При охлаждении сухим льдом в стальном автоклаве, в котором находится-)
1 моль HgO, конденсируют 10—12 моль HF и туда же добавляют охлажденное»
сухим льдом галогенированное соединение (2 моль замещенного галогена ня»)
1 моль HgO). Автоклав закрывают и несколько часов встряхивают, давая тед-J
пературе повыситься до комнатной.
Для приготовления фторорганических соединений без применения фтора, фто^
ристого водорода или металлической аппаратуры следует попытаться провести заме^
щенис тозилоксигруппы, n-CHsCgH4SO2O (стр. 218) или галогена фтором с помощь*^
KF. Фторид калия, кристаллизующийся с двумя и четырьмя молекулами воды, слё»;
дует, за редкими исключениями, употреблять в абсолютно сухом виде. Л
Для получения сухого KF его сушат 24 ч при 125° С, после чего очень';
тонко измельчают и сушат еще 24 ч в вакуум-сушильном шкафу при 150°
Большие количества фторида обезвоживают азеотропной сушкой с бензолом*
После отгонки азеотропной смеси бензол — вода добавляют более высококидя;
щий растворитель (см. ниже) и отгоняют оставшийся бензол. '
Из сульфохлоридов с помощью KF легко получают фторангидриды сульфокиФа
лот (766, 767]. Фторангидриды карбоновых кислот, отгоняющиеся в процессе реакцййь
через колонку, готовят нагреванием хлорангидридов с KF, а формил- и ацетилфтф--;
риды (768, 769] — взаимодействием кислот с бензоилхлоридом и KF. В качеств^
разбавителей можно применять ксилол или ацетамид. При 100—150° С фторид кал?№
реагирует также с эфирами, нитрилами и^дмидами а-хлор- и а-бромзамещенных алй?
фатических кислот.
Этиловый эфир фторуксусной кислоты [30, 770]. В трехгорлой колбё'
емкостью 2 л, снабженной герметичной мешалкой и колонкой с охлаждаемый?
водой дефлегматором, на масляной бане нагревают до 110° С смесь 8 моль ацог?-!
амида и 5,9 моль этилового эфира хлоруксусной кислоты. При интенсивном^
перемешивании к этой смеси в несколько приемов прибавляют 8,2 моль тонкой;
измельченного сухого KF и затем повышают температуру баин до 140° С. При*,
мерно через 30 мин, (при флегмовом числе 10) начинают отгонять неочищенный^
продукт; при 76—115,5° С отгоняется 245 г, а при 115,5—117,5° С ещ<
210 г. Повторной перегонкой обеих фракций получают этиловый эфир фтор*§
уксусной кислоты с выходом 52% от теоретического; т. кип. 117—118° С.
Превращение алкилгалогенидов RCH2X в алкилфториды с помощью KF про»4’
it в этиленгликоле или, лучше, в диэтиленгликоле без доступа влаги при интепН
ИСХОДИТ с diruieuiл-пниле или, лучше, в диэтилемглилиле иеа доступа вл гни при nurw-i
сивном перемешивании. Необходимо вводить не менее 2 моль безводного KF на 1 моль*
подлежащего обмену галогена. Таким способом (2,5 моль KF, этиленгликоль;)
150—185° С) из .со.со'-дихлоралкалов получают ы.ы'-дифторалканы (771], из алкил-ч
галогенидов (этиленгликоль, 180° С) — алкилфториды [772] и из со-хлор(бром)гЙ
спиртов или эфиров <о-хлор(бром)алифатических кислот (диэтиленгликоль, 120-*- •<
130е С) — соответствующие офторорганические соединения [773]. Описание этого
способа на примере получения н-гексилфторида из «-генсилбромида см. [774]. .j”
(766
(767
(768
J769
(770
[771
(772
(773]
(774]
W. Т. Truce, F. D. Н oerger, ?. Am. Chem. Soc., 76, 3230 (1954).
W. Davies,?. H. Dick,?. Chem. Soc., 1931, 2104; 1932, 483.
A. N. N e s m e j a n о w, E. I. Kahn, Ber., 67 , 370 (1934),
А. И. Машенцев, ЖПХ, 20, 854 (1947); C. A., 42, 5418 (1948).
E. D. Bergmann, I. Blank, ?. Chem. Soc.. 1953, 3786.
F. W. Hoffmann, ?. Org. Chem., 14, 105 (1949).
H. Kitano, K. F u k u i, J. Chem. Soc. ?apan, ind. Chem. Sect., 58,
(1955)- C. A., 50, 3995 (1956).
F. L. M. Pattison et al., ?. Org. Chem., 21, 739 (1956).
A. I. V о g e I, ?. L e i c e s t e r, W. A. T. M acey, Org. Synth., 36,
(1956).
352 ;
II. ОБМЕН Hal НА ДРУГОЙ Hal ИЛИ SCN
197
2-Фторэтвловый спирт из этиленхлоргидрипн [775]. В перемешиваемую
смесь 350 s сухого тоикоизмельченного KF, 320 г этиленгликоля и 130 г ди-
этиленгликоля при 170—180° С в течение 3 ч приливают по каплям 322 а этилен-
хлоргидрина, причем 2-фторэтиловый спирт (т. кип. 97—104° С) отгоняется
в процессе реакции через колонку высотой 30 см. Для полноты выделения спирта
через аппаратуру к концу отгонки просасывают медленный ток воздуха. Чтобы
удалить следы HF из неочищенного продукта (152,5 г), к нему добавляют 10г
NaF и оставляют на двое суток, после чего еще раз перегоняют; выход 109 г
(42,5% от теоретического).
2-Фторэтиловый спирт получают также из этиленхлоргидрнна и KF
в глицерине [776] при 50—60° С или в автоклаве без растворителя [777] при
130—135’ С.
Для обмена галогенов на фтор в ароматических соединениях KF может приме-
няться только в том случае, если галоген особенно подвижен вследствие наличия нитро-
группы в орто- и (или) пара-положении. Скорость реакции н количество побочных
продуктов в большой мере зависят от природы растворителя. Гликоли непригодны
для этой цели, в нитробензоле реакцию приходится вести при 190—205° С, в диметил-
формамиде или диметилсульфоксиде [778] обменная реакция с 2- или 4-нитро- и 2,4-дн-
нитрохлорбензолами протекает уже при температуре около 100° С.
2,4-Динитрофторбеизол. Подвергают взаимодействию 25,5 г 2,4-дииитро-
хлорбензола в 25,5 г диметилформамида с 14,5 г сухого KF (30 мин при 140—
150° С или 13 ч при 95—100° С), реакционную массу разбавляют и образовав-
шееся масло хорошо промывают водой, сушат и перегоняют в вакууме. Выход
2,4-динитрофторбензола 77% от теоретического; т. кип. 122—124°С (1лл* рт. ст.)
и 156—158° С (12 мм рт. ст.).
По другому способу [779] смесь 2,4-динитрохлорбензола и сухого K.F
нагревают в течение 7 ч при 180—200° С. После охлаждения извлекают продукт
из реакционной смеси горячим сухим бензолом, бензол отгоняют, остаток пере-
гоняют в вакууме и перекристаллизовывают из метилового спирта; выход 92%
от теоретического; т. пл. 27° С.
О получении 2,4-динитрофторбензола нитрованием фторбензола
см. [Зр, 780].
Значитеньно реже, чем KF, применяются LiF, NaF и NH4F. Но иногда хоро-
шие выходы достигаются при использовании KHFa, например при получении фтор-
ангидридов карбоновых кислот (С2—С8) нагреванием кислот в течение 1 ч при 100° С
с сухим KHFa и бензоилхлоридом [781].
2. Обмен ганогена на нод, бром или хлор
Галогены легче всего обмениваются на иод при взаимодействии органического
галогенида с неорганическим иодидом, преимущественно с Nal, реже с KI и Cals.
RX+Г Rl+X-
где X==C1, Вг.
Скорость равновесной реакции сильно зависит от растворителя. Особенно она
велика в ацетоне, который, кроме того, обладает тем преимуществом, что в нем очень
[775] F. W. Н о f f m a n n, J. Am. Chem. Soc., 70, 2596 (1948).
[776] G. О I a h, A. P a v 1 a t h, Acta chim. Acad. Sci. Hung., 3, 199 (1953).
[777] В. C. S aun dera, G. J. S t a cey, I. G. E. W i 1 d i n g, J. Chem,
Soc., 1949, 773.
778] G. C. F i n g e г, C. W. К ruse, J. Am. Chem. Soc., 78, 6034 (1956).
779] H. H. В о p о ж ц о в, Г. Г. Якобсон, ЖОХ, 27 (89)» 1672 (1957).
[780] Н. Zahn, A. W ii г z, Angew. Chem., 63, 147 (1951).
[781] G. О 1 a h, S. К u h n, S. В е k е. Chem. Ber., 89, 862 (1956).
198 ОБРАЗОВАНИЕ СВЯЗИ С —Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ Ц
хорошо растворим Nal (1,29 молъ/л) и очень плохо NaBr и NaCl (5,5-10-6 моль/л).
В сухом ацетоне Nal растворяется при комнатной температуре примерно в 20 раз а
лучше, чем KI. Особой реакционной способностью обладают а-галогенкарбонильные я
соединения, аллил-и бензилгалогениды, причем бром обменивается легче хлора; пер- ч
вичные алкилгалогениды реагируют легче, чем вторичные и третичные [782, 783], Я
В ароматических соединениях галоген может обмениваться на иод лишь в том -I
случае, если связь углерод — галоген ослаблена наличием электронопрптягивающих -
заместителей, например нитрогруппой в орто- и (или) пара-положении. 1-Иод—
2,4-динитробензол получают с 70%-ным выходом кипячением (15 мин) 1 моль*
2,4-динитрохлорбензола с 5 ммь K.J в диметилформамиде [784], Разработан также>
простой способ обмена галогена на иод в других ароматических соединениях (стр. 200).^,
Хотя в основном взаимодействие галогенорганических соединений с Nal.^
протекает очень гладко, приходится считаться, в известных условиях, с побоч-Л
ними реакциями. Так, HI, возможно образующийся при реакции очень подвиж- ?
ного атома иода, может оказывать восстанавливающее действие:
RI-j-HI —* RH+I2 ?
Поэтому рекомендуется при переработке продуктов реакции связывать./
иод не бисульфитом, а тиосульфатом или разбавленным раствором NaOH.
При действии иодидов на 1,2-дигалогениды могут образовываться олефины!
RCHX—CHXR' —RCH=CHR'+2X-+I2
Иногда эта реакция желательна, например, для введения 0= С-связей в сте-/'
роиды [785]. 1
Условия взаимодействия органических галогенидов подбирают в соответствии•
с реакционной способностью галогенидов. На 1 моль галогенида вводят 1—1,25 моль-.
сухого Nal (при взаимодействии ценных и малореакционноспособных галогенидов^
больше) и, как правило, примерно шестикратное по отношению к иодиду количество^
сухого ацетона. J
Смесь перемешивают при комнатной температуре или при температуре]
кипения до прекращения осаждения NaBr или NaCl. После их отсасывания,
из фильтрата удаляют растворитель, остаток смешивают с эфиром и связывают'
иод обработкой раствором тиосульфата. Л<
Работая с галогенорганическими соединениями типа RX, обладающими малой:;
реакционной способностью, можно использовать более высококипящие растворители^’
например метилэтилкетон [786, 787] или ацетонилацетон. При действии на RX амй<
нами или фенолами иногда может оказаться полезным перевести на промежуточной^
стадии обладающий малой реакционной способностью RX в RI. Для этого в реакциоя-,
ную смесь вводят небольшие количества Nal или K.I. J
Обмен галогенов по реакции с Nal или Kj может быть также осуществлен в воде;
или метиловом или этиловом спиртах. /
Иодуксусная кислота[788]. Нагревают в течение 2 ч при 50° С 1 моль хлоруксус-i
ной кислоты с 1 моль KI в концентрированном водном растворе. Смесь обесцвечиваю^
сернистой кислотой, извлекают продукт реакции эфиром, упаривают высушенный^
над СаС1? эфирный раствор и перекристаллизовывают иодуксусную кислоту из неболЬ-;
пгою количества воды или из петролейного эфира; т. пл. кислоты 82° С.
[782] Н. Finkelstein, Вег., 43, 1528 (1910).
[783] J. В. Conant. R. Е. Hussey, J. Am, Chem. Soc., 47, 476 (1925).
[784] J. F. В u n n e t t, R. M. Conner,!. Org. Chem,, 23, 305 (1958); Org. Synth,,.
40, 34 (1960).
[785] H. H. Inhoffen, Ber., 69, 1134 (1936).
[786] A. H. Ford-Moore, Org. Synth., 30, 10 (1950).
[787] E. Klingsberg, J. Am. Chem. Soc. 72, 1031 (1950).
[788] E. Abderhaiden, M, Guggenheim, Ber., 41, 2853 (1908).
11. ОБМЕН Hal НА ДРУГОЙ Hal ИЛИ SCN
199
Примерами использования метилового или этилового спиртов могут служить
получение 2-и одгептана [789] из 2-6 ромгептана (Nal, 6 ч кипячения)
или а-иоддигидрохаульмугровой кислоты [790] из соответствующего бромсоединевпя
(KI, 12 ч кипячения).
Обмен хлора или иода на бром имеет гораздо меньшее препаративное значение,
чем рассмотренные до сих пор реакции обмена галогенов. В припципе он протекает
аналогично рассмотренным ранее, но в менее мягких условиях; иод подвижнее хлора.
Галоген, находящийся рядом со связями С=-=С или С=С, можно обменять на бром
с помощью LiBr в кипящем метиловом спирте или ацетоне, NaBr в кипящем метило-
вом или этиловом спирте или с помощью СаВг2. Особенно активен безводный А2Вг3,
с участием которого удается заменить хлор бромом даже в СС14.
Трихлорбромметан [791]. Продукт Представляет интерес как раство-
ритель при фосфорилировании. Получают его в простом приборе, выдерживая
в течение трех суток при комнатной температуре 350 мл СС14 с 260 г А1Вг8. Это
вещество можно также получить парофазпым бромированием хлороформа [792]
при 420—450° С.
Тетрабромметан [793]. К смеси 130 г СС14 и 300 г этилбромцда при тем-
пературе от —10 до О’ С в течение 30 мин прибавляют приготовленный при
охлаждении раствор 300 г А1Вг3 в 400 г этилбромида. Смесь выдерживают 30 мин,
В качестве основного продукта реакции образуется тетрабромметап.
Бромид алюминия [794]. Его получают с отличным выходом, вводя
по каплям 250 г брома под насадку из смеси алюминиевых стружек и колец
Рапгига. В качестве катализатора в начале реакции вводят немного А]Вг3. Реак-
цию проводят в колбе емкостью 1 л, которую заполняют на 4/6 ее объема. Уже
во время двухчасового приливания брома за счет того, что реакция очень экзо-
термичпа, отгоняется А1Вг3. По окончании добавления брома А1Вг3 отгоняют,
нагревая колбу на воздушной бане; т. кип. А]Вг3 255° С; т. пл. 94,9° С.
Хлор- и броморганические соединения можно получать также действием хлора
или брома на органические галогениды. Так, например, под действием хлора или
брома в петроленном эфире на 2-иодоктан угяе при —76° С образуется с хорошим
выходом 2-хлор- или 2-бромоктан [795]. Такого рода превращения не имеют препара-
тивного значения, однако с ними приходится сталкиваться в основном в качестве
нежелательных побочных реакций. Например, хлорирование боковой цепи о-, м-
и n-бромтолуолов приводит к образованию смеси галогенированных бензил-
галогонидов, замещенных хлором и бромом как в ядре, так и в боковой цепи [796].
Если в бромбензол в течение 1 ч пропускают хлор при 90° С и освещении лампой
в 200 вт, то образуется 91% хлорбензола. Бром может обмениваться |797] также
на 82Вг. в присутствии FeClg или А]С1з бромбензол хлорируется до о- и п-хлор-
бромбензола [798]. Ароматически связанный бром может гладко обмениваться на хлор
при действии CuClc небольшой добавкой СиС1„. Реакция особенно хорошо проходит
в а-пиколине и в диметллформамиде [799]. Ароматически связанный иод вообще
не обменивается на хлор или бром.
1789] М. Schirm, Н. Besendorf, Arch. d. Pharm., Вег., 280, 64 (1942).
[790] Ng. Pg. В и и -Hoi, Р. Cagniant, Вег., 75, 1181 /1942).
[791] Н. G. V е г р е г, G. К. R о 11 е f s о n, J. Am. Chem. Soc., 56, 1456
(1934).
[792] Л. И. 3 а х а р к и н, Синтезы органических соединений, Сборник 2,
изд. АП СССР, 1952.
[793] Н. S. N u 11 i n g, P. S. Petrie, пат. США 2120675; С., 1938,
II, 2840.
[794] V. Koutnik, J. Benes, Chem. Prum., 8 (33), 187 (1958); C., 1958,
12911.
[795] F. M. В e г i n g e r, H. S. Schultz, J. Am. Chem. Soc., 77, 5533 (1955).
[796J F. Asinger. Mh. Chem., 64, 153 (1934).
1797] B. Miller, Ch. Walling, J. Am. Chem. Soc,, 79, 4189 (1957).
[798] M. A. F. Holleman, T. v a n der Linden, Rec. trav. chim., 30,
r 253 (1911).
[799] W. В. H ar dy, R. B. Fortenb augh, J. Am. Chem. Soc., 80, 1716
(1958).
200
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Кроме неорганических галогенидов и свободных галогенов в реакциях гало-
генного обмена могут использоваться алкнлгалогенпды R'X:
AlCh
RC1H R'X RX + R'C1
>дный А1С13 (0,02—0,03 моль) катализирует установление равно-
1Й реакции. Если применяют примерно 5%-ный избыток R'X (R'aCH^
то при 30—40° С и непрерывной отгонке легколетучих метил- или этил-*!
Безводный
песня в этой r___,__ ____ _r _____... , , __
или C2H5), то при 30—40° С и непрерывной отгонке легколетучих метил- или этил-*
хлорида равновесие реакции сдвигается вправо. Таким способом, в частности, иолу*-,
чают с очень хорошими выходами тетраиодметан из CCI4 и метилиодида [800], иод<м
форм из хлороформа и этилиодида [801] или метилиодида [800] и бромоформ из хлоро-,
форма и этилбромида [800].
Катализируемое AIC1S взаимодействие двух многократно галогенированных)
алканов приводит к смеси хлорбромалканов:
AlCh
СНВг3 + СнС13 СНС12Вг + СПС1Вг2
Aids |
Сн2ВгСП2Вг + СН2С1СН2С1 2СН2С1СН2Вг
Подробности об этих синтезах см. [802, 803]. Введение галогена в органически^
галогенид вместо относительно прочно связанного с ним другого галогена может быт^
осуществлено с помощью промежуточного получения металлоорганических соедине-
ний, чаще всего соединений Гриньяра:
2RX + 2M? —»- 2RMgX
2RI + MgX3 + MgI2
Эта реакция имеет особенно большое значение для получения иодированны^
ароматических и гетероциклических соединений из хлор- и бромпроизводных.
Наиболее простой способ проведения синтеза состоит в том [804], чт<4
к приготовленному из 1 моль арилбромида и магния эфирному раствору реактиву
Гриньяра приливают по каплям раствор 0,65 моль иода в абсолютном эфирв^
т. е. значительно меньше расчетного количества. Реакцию считают закончетаои
после обесцвечивания иода. Реаициоииую массу смешивают с ледяной водой
и разбавленной НС1; высушивают и обрабатывают эфирный раствор, содержат
кроме арилиодида еще и арилбромид и продукт восстановления — арома
веский углеводород (АгН).
Магнийорганические соединения могут реагировать нс только с элементарн
галогенами, но и с органическими галогенидами:
RMgX + R'I —> RI + R'MgX
например с бром- или иодциаиом [805] или иодацетоннтрнлом [806].
Соответствующие реакции с дииодацетиленом протекают очень быстро уЖ^
при 0° С, причем алкил-и арилиодиды получаются с 75—95%-ными выходами от тес^
ретического [439]: •’
2RMgX+C=C
XMgC=CMgX-)-2RI
J. В. Н I n k a m р, J. Am. Chem. Soc., 67, 1642 (1945).
e r, J. Chem. Soc., 1904, 1082.
[800] H. S о.г о 0 s,
[801] J. W. Walk.., .. ... ...
[802] M. S. К harasc h, В. M. К u d e г n a, W. U г г y, J. Org. Chem., 1>й
895 (1948).
[803] G. Calingaert et al., J. Am. Chem. Soc., 62, 1545 (1940). '
[804] R. L. Datta, H. K. Mitter, J. Am. Chem. Soc., 41, 287 (1919).
1805] V. Grioard et al., Ann. Chim. [9], 4, 28 (1915); [10], 5, 5 (1936). ’
[806] J. v. Braun, -H. Deutsch, A. Schmatloch, Ber., 45, 1246 (1912).'
II. ОБМЕН На] НА ДРУГОЙ Hal ИЛИ SGN
201
В данной реакции равновесие почти полностью сдвинуто вправо вследствие
плохой растворимости гриньяровского соединения ацетилена в эфире (комплекс
Иоцича^. Выделение продукта не вызывает трудностей, так как при гидролизе разба-
вленной H2SO4 ацетилен удаляется в виде газа. Недостатком этого способа является
взрывоопасность сухого дииодацетилена [440] (получение см. на стр. 149).
Иодбензол [439]. К эфирному раствору гриньяровского соединения
из 0,2 моль хлор- Или бромбензола приливают по каплям при охлаждении льдом
раствор 0,1 моль дииодацетилена в 50 мл эфира (помутнение). После 15 жим
выдерживания при комнатной температуре отделяют декантацией осевший
на дно сиропообразный мутный слой или непосредственно всю массу разлагают
разбавленной H2SO4. Из высушенного эфирного раствора выделяют иодбензол;
т. кип. 188° С; выход 95 или 97% от теоретического.
Аналогичным образом из приготовленного взаимодействием индола с этил-
магнийбромидом эфирного раствора гриньяровского соединения получают
3-иодиндол; выход 83% от теоретического.
3. Обмен галогена на родан
Классическим и общепринятым методом получения алифатических роданидов
(эфиров тиоциановой кислоты) является взаимодействие галогенидов с роданидами
металлов:
Алкилроданнд. В кипящую смесь 1,5 моль тонкоиамельченного KSCN
к 340 мл этилового спирта (около 1{3 роданида растворяется) при перемешивании
приливают по каплям 1 моль алкилбромида и кипятят еще 2 ч. Затем отгоняют
часть растворителя, реакционную массу разбавляют водой, экстрагируют алкнл-
роданид эфиром, сушат эфирный раствор иад CaCis или Ka2SO4 и выделяют
алкилроданнд перегонкой в вакууме. Выходы роданидов от амил- до тридецил-
роданида 80—90% от теоретического [807].
Можно раствор галогенида [например, С1(СНа)яОН] в этиловом спирте
кипятить несколько часов с концентрированным водным раствором KSCN н
после разбавления водой экстрагировать продукт хлороформом [808]. В качества
реакционной среды можно использовать также диоксаи. Производные родан-
гндринов, RO(CH,)„SCN, применяются в качестве иисеитицидов [809].
Реакцию с NaSCN, также как и другие реакции обмена галогенов, можно
вести в ацетоне. Для получения олеплроданида [810] 54 а бромида подвергают
взаимодействию с 75 г NaSCN • 2HSO в 500 мл ацетона при 80Q С в автоклаве (20 ч).
Низшие алкилроданиды очень просто получают постепенным добавлением
(встряхивание и охлаждение по мере надобности) 1 моль диалкилсульфата к рас-
твору 1 жолъ KSCN в 50 мл воды [811].
Чем’подвижнее замещаемый галоген в органическом галогениде, тем легче
образуются эфиры изотиоциановой кислоты (горчичные масла):
, Изотиоцианаты кипят при значительно более низкой температуре и более ста-
бильны, чем алкилроданиды; их можно получать нагреванием алкилроданидов. До-
бавка ZnCl2 ускоряет перегруппировку [812]. Часто уже сразу получается смесь
[807] Р. Allen Jr., J. Am, Chem. Soc., 57, 198 (1935).
[808] H. D. Vogelsang, T. Wagner-Jauregg, R. Rebling,
Ann., 569, 190 (1950).
aT. Wagner-Jauregg, Ann., 561, 94 (1949).
T. Warner - J auregg et al., J. prakt. Chem., 155, 216 (1940).
[811] P. Walden, Ber., 40, 3215 (1907).
[812] E. Schmidt et a]., Ann., 568, 193 (1950).
202
ОБРАЗОВАНИИ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
алкплроданлда и изотиоцианата. При проведении реакции с KSCN или при обра-
ботке реакционной массы при высоких температурах (120—130° С) из галогенидов
с легкоподвижным галогеном непосредственно получаются изотиоцианаты.
Таким способом из хлорметилметилового или хлорметилэтилового эфира
и KSCN в петролейном эфире получают метокси- или этоксиметялизотиоциа-
нат [813], из а-ацетобромглюкозы и AgSCN в толуоле— 2,3,4,6-тетраацетил-
а-Р-тлюкозо-1-иаотиоцианат [814].
III. Замена гидроксильных, алкоксилъных
и карбонильных групп на галогены
1. Замена спиртового гидроксила иа галоген
Замена спиртового гидроксила на фтор
Как правило, непосредственное замещение гидроксильной группы на фтор •
с помощью HF не удается осуществить. Все способы получения алкилфторидов иа •
спиртов основаны на превращениях их эфиров. Спирт превращают в алкилхлорид, ’
бромид, иодид или тозилат и затем обменивают галоген (стр. 191) или тозилокспгруппу ;
(стр. 218) на фтор.
В присутствии малых количеств борфторида алкиловые и циклоалкиловые
эфиры фтормуравьиной кислоты разлагаются уже при температуре около 50е С с обра-
зованием алкилфторида [815].
FCOOR — BF + CO, ;
Эфиры фтормуравьиной кислоты получают из эфиров хлормуравыгной кислоты
обменом галоида под действием фторида таллия [816] или, если нет дорогого T1F,
из карбоннлбромфторида, OCFBr и спирта, причем образуется исключительно фтор-
муравьиный эфир.
C1C00RH-T1F —► FCOOR *—• OCFBr-]-ROH
Подробные данные, в том числе и по получению карбоннлбромфторида из BrF».J
и СО, см. [817].
Замена спиртового гидроксила на хлор, бром и иод 5
при помощи галогеноводородов и галогенангидридов кислот .
Замена спиртовой ОН - группы на галоген при помо щИ- ,
галогеноводород а. Самым простым способом замены спиртовой гидроксиль-.
ной группы на С1, Вг или I является реакция с галогеноводородом:
ROH+HX RX4-h2O J
Легкость прохождения реакции увеличивается в последовательности НС1,
НВг, HI. Третичные спирты реагируют с галогеноводородом легче, чем вторичные# '2
последние легче, чем первичные [818]. Реакционная способность спиртов по отношению
к галогеповодородам возрастает, таким образом, в последовательности, обратной их j
реакционной способности по отношению к карбоновым кислотам; в этом случае легче всего j
этерифицируются первичные спирты [818, 819]. Чем чище спирт, тем труднее он взанмо- *
[813] Е. Schmidt, W. Striewsky, Вег„ 73, 286 (1940).
[814] F. Р. van de К am р, F. Micheel, Chem. Ber., 89, 133 (1956).
[815] S. N a k a n i s k i, T. C. Myers, E. V. J ensen, J. Am. Chem, Soc.,
77, 5033 (1955).
[816] S. N a k a n i s k i, T. C. Myers, E. V. Jensen, J. Am. Chem. Soc., 77,
3099 (1955).
[817] G. A. Olah, S. J. Kuhn, J. Org. Chem., 21, 1319 (1956).
[818] G. M. Bennett, F. M. Reynolds, J. Chem. Soc., 1935, 131,
[819] R. B. Bradbury, J. Am. Chem. Soc., 74, 2709 (1952).
II. ЗАМЕНА ГИДРОКСИЛЫ!., АЛКОКСПЛЬН. И КАРБОНИЛЬН» ГРУПП НА Па] 203
действует (20° С, 30—40 ат) с сухим хлористым водородом [820]. Чем легче образуется
алкилгалогенид, тем больше его склонность к отщеплению талогеноводорода и, сле-
довательно, первичные н-алкилгалогениды наиболее стабильны. Однако и первичные
алкилгалогсниды с разветвленной цепью легко отщепляют галогеноводород при на-
гревании.
Так как взаимодействие спиртов с галогеноводородом проходит не коли-
чественно, почти всегда получается смесь галогенида со спиртом, которая пе
может быть полностью разделена фракционной перегонкой из-за того, что спирты
часто образуют азеотропные смеси с соответствующими галогенидами. Спирты
отделяют от продукта реакции встряхиванием с концентрированной серной
или, лучше, с концентрированной соляной кислотой.
Первой стадией взаимодействия спиртов с галогеноводородом является образо-
вание оксониевого комплекса R — ОН2, поэтому электроноакцепторные заместители
(галогены, NO2, CN, COOAlk) затрудняют реакцию обмена. Их влияние тем слабее, чем
более удален заместитель от ОН-группы. Так, например, в 1,2-гликолях одна гидр-
оксильная группа легко обменивается на бром, тогда как обмен второй затруднен.
Галогеноводород, тионилгалогенид и галогениды фосфора вызывают вальде-
новское обращение оптически активных спиртов с ОII-группой у асимметрического
атома углерода. Так как в зависимости от условий реакции обращение конфигурации
происходит более или менее полно, часто, смотря по обстоятельствам, получают гало-
гениды с различными значениями угла вращения. О стерическом направлении обмена
ОН-группы на бром в стероидах см. [821].
Обмен на галоген гидроксильной группы в сильноразвствленных первичных
спиртах и во вторичных спиртах с третичным атомом водорода в а-ноложении под
действием галогеноводорода может легко вызвать перегруппировку углеродной цедн.
В этом случае образуются преимущественно третичные галогениды; например, из
неопентилового спирта в качестве главного продукта реакции получается вгревг-амил-
бромид:
СПз Вг
СНз~С-СН2ОН СНз-С-СН2—СПз
I I
СНз СНз
В исследованиях обмена ОН-группы на галоген в спиртах типа
RR'CHCH2OH (R = R'=C2H5) установлено [822], что при действии концентрирован-
ной IIC1 в присутствии ZnCI2 (стр. 205) за счет перегруппировок углеродного скелета
образуется по меньшей мере семь изомерных алкилхлоридов. Между тем при прове-
дении замещения с SOC12 в пиридине (стр. 214) реакция проходит без перегруппи-
ровки и получается 82% (С2Н5)2СНСН2С1. При действии концентрированной НС1
и ZqC12 па нормальные спирты происходит промежуточное образование олефинов
и присоединение к ним ВСЯ, вследствие чего получается смесь хлорзамещенных.
Например, при обработке я-амилового спирта концентрированной НС1 и ZnCl2 обра-
зуется 57% н-амилхлорида и 10% смеси 2- и 3-хлорпентанов. Взаимодействие с тио-
нилхлоридом в пиридине приводит к 80% ч-амилхлорида [823]. При действии НВг
и РВг3 на вторичные спирты нельзя получить только нормальные вторичные алкил-
бромиды, если возможно образование изомерных вторичных алкилбромидов. В то
время как взаимодействие пентанола-1 с НВг или РВг3 дает 1-бромпентан, из пен-
танолов-2 и - 3 в тех же условиях получается смесь 2- и 3-бромпеитанов [824].
1820] Н. Schlubach, Е. Elsner, Н. Knoop, Angew. Chem., 47, 131 (1934).
[821] L. F. F i e s e r, Experlentia (Basel), 6, 312 (1950).
[822] F. C. Whitmore, F. А. К a r n a t z, J. Am. Chem. Soc., 60, 2533 (1938).
[823] F. C. Whi t more, F. A, Karnatz A. II. P о p k i n, J. Am. Chem.
Soc., 60, 2540 (1938).
[824] H. Pines A. Rudin, V. N. Ipatieff, J. Am. Chem. Soc., 74, 4063
(1952).
204
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Обмен вторичной ОН-группы на галоген через тозилат происходит без перегруппиро—j
вок [824—826] (см. также стр. 218). *
Изобутилгалогениды и другие производные изобутана легко перегруппиро-J
вываются в mpem-бутилпроиэводные. По реакции с РВг, (стр. 212) из изобутанола;!
с хорошим выходом образуется изобутнлбромид и менее 1% трет-бутилбромида [827]
Для удаления mpem-бутилбромида 1 кг изобутилбромида перемешивают^
5 ч с 300 мл дистиллированной воды, промывают органический слой еще ра$
дистиллированной водой, сушат над К2СО3 и перегоняют на иолоша
при 135 мм рт. ст. (т. кип. 41,8—42,5° С). Холодная дистиллированная вода
гидролизует только mpem-бутилбромид. Титрованием водного слоя по Фоль>
гарду, можно определить содержание трет-бутилбромида [828]. До некоторой
степени устойчивые к холодной воде вторичные гало!-ениды в отличие от нерав
ветвленных первичных галогенидов отщепляют галогеноводород при встряхи
вании с раствором карбоната щелочного металла, что также может быть исполь^
зовано для ориентировочного их определения.
При замещении ОН-группы на галоген в аллильных производных I необходима
учитывать возможность аллильной перегруппировки с образованием соединения П;
ОН
j— RCHCH=CHR' —Г
RCHCH=CHR'
RCH=CHCHR'
X
X
in
Преобладает ли образование соединений I] или Ш, зависит от реакционно*
среды, а также от радикалов R и И'. Равным образом возможна аллильная перетру»»
пировка аллилгалогенида III в соединение II, и, наконец, протекающие иногда вм&
имиые перегруппировки при дальнейшем взаимодействии усложняют идентификаций
конечных продуктов реакцик. Аллилхлориды стабильнее соответствующих бромидом
Первичные и вторичные аллилхлориды мало склонны к изомеризации при обычны®
операциях очистки. Аллилгалогеннды обладают значительно большей реанционня
способностью, чем аллилгалогеннды; особенно активны и нестабильны иодйды.
иейшие сведения о механизмах реакций и многочисленные литературные даннЫ
см. [829]. Здесь следует только указать, что при соблюдении определенных услов®
удается достичь полной аллильной перегруппировки или полного ее исключений
Так, например, применяя SOC12 в разбавленном эфирном растворе, можно провеет!
приведенные ниже реакции обмена со 100%-ной аллильной перегруппировкой [83(ШЯ
СНзСН=СНСН2ОН —► СНзСНС1СН«=СН2 ’
СНдСН(ОН)СН=СН2 —> СН8СН=СНСН2С1 -•
СвН5Сн=СнСН2ОН —> СвН6СНС1СН=СН8
Особенно странной кажется последняя реакция, поскольку обычно в реакция^
обмена сохраняется наиболее длинная цепь сопряжения [831, 832]. Если в аллильных
спиртах общей формулы RCH(OH)CH=CHR' заместители R или R'и R имеют соответ^
825]
826
827
828
829
[830
[831]
[832]
Н. Phillips, J. Chem. Soc., 1925, 2552. *
A. J. H. House a, J. Kenyon, H. Phillips, J. Chem. Soc., 1929, 1700»
F. C. Whitmore, A. R. Lux, J. Am. Chem. Soc., 54, 3448 (1932).
A. M i c h a e 1, H. LeupOl d, Ann., 379, 263 (1911). %
R. H. D e W о 1 f e, W. G. Young, Chem. Rev., 56, 801 (1956).
F. F. C age ri o, G. E. Dennis, R. H. DeWolfe, W. G. Young»
J. Am. Chem. Soc., 77, 4182 (1955).
A. Klages, К. К 1 e n k, Ber., 39, 2552 (1906). J
L. V1 a i s e n, E. T i e t z e, Ber., 58, 279 (1925).
JIJ. ЗАМЕНА ГИДРОКСИЛЫ!,, АЛКОКСИЛЬН. И КАРБОНИЛЬП. ГРУПП НА Hal 205
ственно расположенные С—С- или С==С-связи, то происходит полная аллильная пере-
группировка и образуется соединение с самой длинной цепью сопряжения [833—835]
Исходя из кротилового спирта или метилвинилкарбииола получают-
смесь 1-бромбутеиа-2 (т. кип. 107° С) ,и З-бромбутена-1 (т. кип. 86,5° С):
СН9СН=СНСН2ОН НВ* СНзСН=СНСнаВг (86%)
ИЛИ РВг, а
СНзСН(ОН)СН=СН8 СНзСНВгСН=СН2 (14%)
Оба бромида медленно перегруппировываются при комнатной темпера-
туре (при 100° С быстро) с образованием равновесной смеси из 86% первого
и 14% второго бромида. Медленным фракционированием этой смеск при нор-
мальном давлении получают З-бромбутен-1, кипящий при более низкой тем-
пературе, который непрерывно образуется по равновесной реакции [836].
При получении иодидов из спиртов кроме уже упомянутых перегруппировок
могут проходить и другие побочные реакции. Чем подвижнее иод в органическом
иодиде, тем легче происходит восстановление по схеме
RI-J-HI—> RH-f-Is
Этой побочной реакции можно избежать переводом спирта в хлорид, бромид
пли тозилат перед обменом галогена или тозилгруппы на иод.
Получаемые из гликолей 1,2-дииодиды тотчас же отщепляют иод с образованием
ненасыщенных соединений, которые могут вступать в дальнейшие реакции. Таким
способом из глицерина и Ш при избытке глицерина получают аллилиодяд, а при
избытке HI— изопропилиодид; механизм реакции см. [819].
Г]Ь(ОН)СЯ(ОН)СН2(ОН) —СН21СН1СН2ОН-^* СН2=СНСН21~—*
—► CHaCHICHjI -zj* СНзСН-СНа —CHjCHIClb
Относительно обмена спиртовой группы на хлор с помощью НС] установлено
что первичные насыщенные спирты плохо реагируют при нагревании в открытом сосуде
с концентрированной НС1 без катализатора. Между тем нэ бензилового, коричногг
и аллилового спиртов таким способом с хорошими выходами получают соответству
ющие хлориды [8о7]- Эти спирты реагируют с концентрированной НС1 уже при ком
натной температуре [837, 838]. Во всех случаях [839—840] применима реакция с коц
центрированной HCI и ZnCl2.
способ. Растворяют 2 лмъ ZnCl2 в 2 моль концентриро
ванной НС], прибавляют 1 моль спирта и кипятят с обратным холодильнике!
3—4 и (при добавлении 5 моль спирта кипятят 9 ч). После охлаждения отделяю*
верхний слой, галогенид кипятят в течение 30 мин с обратным холодильником
добавив равный объем концентрированной H2SO4, отгоняют хлорид, промываю*
его водой, сушат над СаС1а и перегоняют.
При получении нестойких хлоридов, например циклопентилхлорид;
[841], нагревают 60 мин на паровой бане, промывают отделенный верхний ело!
[833 ] A. R о е d i g, H.-J. Becker, Chem. Ber., 89, 1726 (1956).
[834 ] Ch. Prevost, F. Bi d on, Bull. Soc. chim. France, 1955, 1408.
[835 ] I. M. H e i 1 b г о n et al., J. Chem. Soc., 1945, 77.
[836 ] W. G. Young et al., J. Am. Chem. Soc., 57, 2013 (1935); 58, 10
(1936); 59, 2051 (1937); 60, 847 (1938).
837] J. F. Norris, J. Am. Chem. Soc., 38, 1071 (1916).
838] R. McCullough, F. Cortes e, J. Am. Chem. Soc., 51, 225 (1929).
839] J. F. Norris, H. B. Taylor, J. Am. Chem. Soc., 46, 753 (1924).
840] /. F. N о г г i s, H. В. T а у 1 о r, Org. Synth., 5, 27 (1925).
841] M. T. Rogers,/. T. Roberts,/. Am. Chem. Soc., 68, 843 (1946).
206
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
водой, раствором NaHCOs, водой и насыщенным раствором СаС12, сушат над
СаС12 и перегоняют. Низкокипящие алкилхлориды лучше отгопять из реак-
ционной массы в процессе продолжающейся около 1 ч реакции через служащий
ректификационной колонной вертикальный холодильник; при этом подачу
охлаждающей воды регулируют так, чтобы температура, показываемая термо-
метром наверху холодильника, не превышала температуры кипения алкил-
хлорнда болео чем па 2° С. Таким способом готовят н-пропилхлорвд (выход
70—72%) и «-бутилхлорид (выход 76—77% от теоретического) [842].
Вместо ZnCK применяют также СаС12, например для получения цикло-
гексплхлорида [843J. О синтезе циклогексилхлорпда насыщением раствора
цикло1ексанола в 1,5-кратном количестве концентрированной HCI (d = 1,19)
хлористым водородом и 2 ч кипячением с обратным холодильником см. [844].
В многоатомных спиртах гидроксильные группы обменивают на хлор чаще всего
кипячением с концентрированной НС1 или пропусканием тока сухого НС1. Особенно
большой интерес представляют способы, при которых в спирте остаются свободные
ОН-группы.
Пропилснхлоргидрин CH2CICH2CH2OH. В 25—30 мл нагретого до 150—
170° С пронандиола-1,3 СН2ОНСП2СНаОН пропускают очень быстрый ток
НС1, причем происходит отгонка смеси пропиленхлоргидрииа, воды, НС1 и ие-
ирореагировавшего пропандиола. По мере расходования пропандиола-1,3 его
непрерывно приливают по каплям в реакционную массу (2—3 мл/мин); подроб-
пости см. [845].
Еще удобнее пользоваться следующим способом [846]. Встряхивают
250 г пропандиола-1,3 с 450 г S2C12, причем смесь разогревается. Происходит
бурное выделение SO2 и выпадает сера. Реакция протекает в течение 1 ч без
подвода тепла, после чего массу нагревают 6 ч с обратным холодильником на
водяной баие и затем 30 мин на открытом пламени. Когда прекратится выделе-
ние SO2, смесь охлаждают, продукт реакции извлекают эфиром, выпавшую серу
промывают эфиром, из объединенного эфирного раствора встряхиванием с рас-
твором соды удаляют SO2 и высушивают продукт над Na2SO4. После отгонки
эфира смесь фракционируют и фракцию 140—180° С еще раз перегоняют. Полу-
чают 160 г (60% от теоретического) пропилепхлоргидрнна; т. кип. 160—164° С
(или 60—64° С при 10мм рт. ст.).
ю-Хлорспирты, С1(СН2)„ОН. Спирты, у которых п = 5-?-10 и 18, полу-
чают из а,и-диолов следующим способом [773]. Смесь диола и концентриро-
ванной ПС1 (около 5 моль НС] на 1 моль ОН), к которой добавлена вода в коли- .
честве приблизительно 11а от начального объема кислоты, нагревают с толуолом
или петролейпым эфиром (т. кип. 100—120° С). При этом образовавшиеся о-хлор-
спирт и небольшое количество а,ю-дихлоралкана непрерывно экстрагируются.
Реакцию проводят в аппарате для экстракции жидкости, например по Кучеру —
Штойделю, или в простом специальном аппарате [847]. Изменяя объем соляной
кислоты, добиваются возможно меньшего объема фазы экстрагента, находя-
щейся над кислой водной фазой реакционной смеси, и возможно более быстрого
непрерывного вывода образующегося to-хлорспирта из зоны реакции.
По такому же принципу получают со-бромспирты. Например, при нагре-
вании в течение 18 ч при 80° С 1 моль гександиола-1,6 с 1,2 моль 48%-ной НВг
(экстрагент — петролейный эфир, т. кип. 90—100° С) образуется 6-бромгекса-
пол-1 с выходом около 80% от теоретического [848]. •
[842] А. М. W h а 1 е у, J. Е. Copenhaver, J. Am. Client. Soc., 60,
2497 (1938).
[843] A. 1. V 0 g e 1, J. Chem. Soc., 1948, 1811.
[844] H. A. Mayes, E. E. Turner, J. Chem. Soc., 1929 , 502.
[845] C. S. Marvel, H. O. Cal very, Org. Synth., 8, 112 (1928); Coll,
v. 1, 5G3 (1941).
[846] C. G. Derick, D. M. В i s s e 1, J. Am. Chem. Soc., 38, 2481 (1916).
]847] K. N. C a m p b e 11, A. II. Sommers, Org. Synth., Coll. v. 3,
1955, p. 446.
[848] E. F. D egering, L. G. Boatright, J. Am. Chem. Soc., 72,
5138 (1940).
г
ш. ЗАМЕНА ГИДРОКСИЛЫ!., АЛКОКСИЛЬН. II КАРБ0НИЛЫ1. ГРУПП НА Hal 207
При добавлении в качестве катализатора карбоновых кислот в глицерине изби-
рательно обмениваются на хлор одна или обе первичные гидроксильные группы.
З-Хлорпропандиол-1,2 (а -монохлоргидрнн глицерина) [849]. Его легко
получают, пропуская НС1 в смесь 500 а 90%-ного глицерина и 10 г ледяной
уксусной кислоты при 105—110° С до привеса в 190 з.
По Смиту [850] этот же спирт готовят пропусканием ПС1 в тщательно
обезвоженный глицерин с добавкой 2% янтарной кислоты без подвода тепла
в течение 8 ч. За счет экзотермичности процесса температура массы возрастает
до 60—70° С.
1,3-Дихлорпропанол-2 (а,у-дихлоргидрин глицерина) получают анало-
гичным способом, но в приведенное выше количество глицерина пропускают
НС1 до привеса в 440г; о выделении продукта см. [851].
Особенно легко обменивается третичная гидроксильная группа.
ь трет-Бутилхлорид [852]. Для его получения встряхивают в делитель-
* ной воронке 1 моль трет-бутилового спирта или его азеотропной смеси с водой
» (около 88% спирта) с 3 моль концентрированной HCI. Слоям дают в течение
15 мин разделиться, промывают хлорид водой и 5%-ным раствором NaHCOs,
; сушат лад СаС12 и перегоняют; т. кип, 50—52° С.
Обмен спиртового гидроксила на бром с помощью НВг происходит при взаимо-
действии спирта с 48%-ной НВг, с сухим НВг или с водной НВг в присутствии концен-
трированной H2SO4. Последний способ наиболее удобен, но его недостатком является
склонность полученных алкилбромидов отщеплять НВт при хранении. Получающиеся-
олефины дают с бромистым водородом полимерные продукты, которые мо1*ут окраши-
вать бромид [838].
Простейшим методом осуществления реакции является очень медленная?
(1 — 2 капли дистиллята в секунду) перегонка смеси 1 моль спирта с 4 моль 48%-ной?
НВг и выделение из дистиллята неочищенного продукта. Галогенирование спиртов от Са
л выше по этому способу дает плохие выходы. Низкокипящие алкилбромиды извле-
кают смесью воды со льдом.
Гидробромиды р-бромэтиламинов. BrCH3CH2NR2 • НВг (R = алкил или H)v
Их получают действием 48%-ной НВг на соответствующий аминоэтиловый
спирт. Охлажденный льдом аминоспирт приливают по каплям к 3 моль охла-
жденной льдом 48%-нон НВт. Затем колбу соединяют с колонкой и вроми от вре-
мени отгоняют значительные объемы дистиллята. В промежутке между отгон-
ками колонка работает как обратный холодильник. Подробное описание этогсь
довольно простого способа, дающего очень хорошие выходы, см. [853].
Р -Оксипроннонитрил (получают из зтиленхлоргидрина) непосредственна
переводится кипящей 40%-ной НВг в р-бромпроппоповую кислоту [854].
Лучшим методом получения высших алкилбромидов (выше Q) является взаимо"
действие сухого НВг со спиртами [855—857].
849
850
851
852
853
854
J. В. Conant, О. R. Quayle, Org. Synth., 2, 33 (1922).
L. Smith, Z. physik. Chem., 94, 701 (1920).
J. B. Conant, 0. R. Quayle, Org. Synth., 2, 29 (1922).
J. F. Norris, A. W. Olmsted, Org. Synth., 8, 50 (1928).
F. С о r t e s e, Org. Synth., Coll. v. 2, 1943, p. 91.
В. C. Kendall, В. M c Ken zie, Org. Synth., Coll. v. 1, 1956».
p. 131; W. Voigtiander, Chem. Techn., 12, 1343 (I960).
3. R. Ruhoff R. E. Burnett, E. E. Reid, J. Am. Chem. Soc., 56*.
2784 (1934).
L. I. Snith et al., 3. Org. Chem., 4. 336 (1939).
[855]
[856] „. _______________ _в. ...................... _
[857]] A. I. Voge], J. Chem. Soc., 1943, 636.
.•208
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Высшие алкилбромиды. В 1 моль спирта при 100° С через опущенную,.!
до дна колбы трубку, которая может иметь на конце расширение с больший,а
числом отверстии, или через пористый распылитель пропускают 1,5 моль сухого?!
НВг (1 моль расходуется на реакцию обмена, около 0,5 моль на насыщение^!
образующейся воды). Для улавливания отходящего НВг обратный холодильник Я
соединяют со взвешенной приемной колбой с головкой от промывной склянким
длинная трубка которой погружена в воду. Конец реакции определяют по разо*ы
греванию воды и явному привесу промывной склянки за счет выделяющегосяйи
из реакционной массы непрореагировавшего НВг. Во время пропускания
НВг температуру реакционной массы поддерживают в пределах 100—120°
Так как реакция экзотермична, внешний подогрев необходим только в К|яи[4я
реакции. Неочищенный бромид охлаждают и отделяют от образовавшейся брО*
мистоводородной кислоты. Подробное описание этого способа на примере полу*
чения додецилбромида см. [858]. Выходы додецилбромида около 90%, цикл»
гексилбромида около 70% от теоретического.
а,й)-Дибромалканы Вг(СН2)яВг. В гликоль сначала пропускают сильней
ток НВг при 100° С до насыщения и затем его продолжают медленно пропуская)
еще 6 ч при 135° С. После охлаждения реакционной смеси отделяют нижнюй
водную фазу. Для лучшего разделения слоев реакционную массу можно раз1
бавить бензолом, петролейным эфиром или эфиром. Неочищенный дибром^
промывают равным объемом теплой воды, затем маленькими порциями 10%-ной
раствора Na2CO3 до полного удаления НВг, вновь теплой водой и фракцнм
нируют в вакууме. Выходы а,со-дибромалканов с различным п следующие (в
3 .............................. 75 I859I 1
8.........•..................... 75 [880] J
6. 9 и 10 ...................... 90 1859]
Таким же способом получают 0-фенилэтилбромид; т. кип. 99° (
(15 ммрт. ст.)-, выход 92% От теоретического [861]. \
При взаимодействии гликолей НО(СН2)ЯОН с сухим НВт при темпере?
турах 80—200° С происходит внутри- и межмолекулярное отщепление вода
и в качестве побочных продуктов получаются дибромдиалкиловые эфир^
Вг(СН2)яО(СН2)„Вг,. а когда стерические факторы благоприятствуют образе
ванию циклических эфиров, то и эти эфиры [862]. Из циклических эфиров над
большее значение имеет тетрагидрофурак, получаемый из бутайдиола-1,4; ф
обратной реанции см. стр. 225. д
Реакция спиртов с 48%-ной НВг и концентрированной H2SO4 находит широкой
применение [863]. Однако добавка H2SO4 способствует образованию эфиров и ол»
финов, особенно из вторичных и третичных спиртов. Протекание этих нежелательный
побочных реакций можно уменьшить, видоизменив общепринятый способ получ^
ния н-алкилбромидов. Ниже приведены примеры.
«-Алкилбромиды [857, 864, 865]. К смеси 2 моль 48%-ной НВг и 1 моЛЦ
концентрированной H2SO4 прибавляют при перемешивании 1—1,6 моль спирта».
Затем при перемешивании в несколько приемов добавляют • ещё 0,8—1,6 л*а*|
концентрированной H2SO4 и кипятят, лучше при пяраммипийнии, 3—4 ч с обрат-*
ным холодильником. "
[858]
[859
I860
[861
[862
1863
1864
1865]
Е. Е. R е i d, J. R. R u п о f f, R. E. В u г n e 11, Org. Synth., Соц.-
v. 3, 1943, p. 246.
W. L. McEwen, Org. Synth., Coll. v. 3, 1955, p. 227; 20, 24 (1940).
R. Adams, N. К ornblum, J. Am. Chem. Soc., 63, 199 (1941).
F. C. Whi t more et al., J. Am. Chem. Soc., 67, 2059 (1945). ' 4
A. MiilleT, W. Vane, Chem. Ber., 77—79, 669 (1944—1946). '
О. К a m m, C. S. Marvel, J. Am. Chem. Soc., 42, 299 (1920).
О. К a m m, C. S. Marvel, Org. Synth., 1, 1—13 (1921); Coll v. 2,1
29 (1941):-26 (1956). j
F. C. Whitmore, L. H. Sutherland J. N. Cosby, J. Am,'.
Chem. Soc., 64, 1361 (1942). Л
in. ЗАМЕНА ГИДРОКСИЛЫ! , АЛНОНСПЛЬН- И КАРБОНИЛЫ!. ГРУПП НА На! 20
Эгилброинд (т. кпп. 39п С). Отгоняется л.? реакционной массы за счет
слабого ее разогревапия во время приливания по каплям H2SO4 (1,5 моль).
Отгопку ведут через эффективный нисходящий холодильник, форштосс которого
опущен в ледяпую воду в охлаждаемом йрпемпике. Аналогично получают аллил-
бромид; т. кип. 69—72° С. Кипячение с обратным холодильником вызвало бы
большие потери.
Алкилбромиды до Св, кипящие прп не очень высокой температуре, не-
посредственно отгоняют из реакционной смеси до прекращения отгопки нерас-
творимых в воде веществ (около 1 ч). Загрязненный спиртом, эфиром и другими
примесями бромид отделяют от водного слоя дистиллята. Реакционную Maccj
можно также перегонять с водяным паром; этот способ оправдал себя, например
при выделении н-бутил- и к-децилбромидов [865].
Реакционную смесь, содержащую высшие бромиды, фильтруют мере;
стеклянную вату или кизельгур, разбавляют водой и отделяют неочищенные
бромид в делительной воронке.
Очистка алкилбромидов. Неочищенные бромиды встряхивают с кон-
центрированной H2SO4 (объем кислоты равен 1/1 объема бромидов), которая
извлекает примеси. Из спиртов частично образуются алкилсерные кислоты
которые плохо растворимы в концентрированной H2SO4, но зато хорошо рас-
творяются в водно-аммиачном метиловом спирте. Низшие (до С9) алкилбромидь
после отделения H2SO4 промывают водой и раствором Na2COs. Алкилбромидь
выше С,о после обработки концентрированной H2SO4 п отделения от нее смеши
вают с равным объемом 50%-ного метилового спирта и затем при встряхиваниь
добавляют водный аммиак до появления красного окрашивания фенолфталеина
Отделенный бромид промывают еще раз равным объемом 50%-ного метиловогс
спирта, вновь отделяют и сушат над СаС]2. При очистке бромидов Св—С9 таких
способом происходят потери. В этом случае применяют возможно меньшее коли
чсство спирта, но достаточное для разрушения иногда образующейся эмульсих
из алкилбромида и аммиачной промывной воды. Для того чтобы предотвратит!
образование эмульсии, рекомендуется [857] добавлять СаС]2. Целесообразна»
применять следующую обработку. Неочищенный бромид дважды взбалтываю-
в делительной воронко с равным объемом концентрированной HCI (d -=-1,19)
причем извлекаются спирты и эфиры, а затем промываю г его водой, растворох
NaHCO3, вновь водой и сушат над СаС12. Температуры кипепия и другие физи-
ческие копстапты большого числа алкилхлоридов, -бромидов и -иодпдов см. [857]
Спирты, от которых легко отщепляется вода, бронируют в более мягких уело
виях 48%-ной НВг и концентрированной H2SO4.
mpem-Бутилбромнд [866]. При 20° С к охлажденной смеси 3,2 мол^
48%-ной НВг и 100 мл концентрированной II2SO4 при перемешивании при:
ливают по каплям 1,6 моль третичного бутилового снирта, через 30 мин отделяю-
неочищенный бромид, дважды промывают его водой и сушат СаС12 п небольших
количеством СаО. Наряду с небольшим количеством изобутилена получаю-
85% трт-бутилбромида.
В то время как из пропандиола-1,3 по стандартному способу с сорнол
кислотой (см. выше) получается около 90% 1,3-дибромпропапа [864], из бутаа
диола-1.4 образуется большое количество буро-черной смолы.
1,4-Дибромбутап (т. кип. 78° С при \0ммрт. ст.}. Его готовят с выхе*
дом 83% от теоретического по следующему видоизмененному способу [867]
К охлажденной льдом смеси 138 г 48%-ной ПВг и 117 г концентрированно^
H2SO4 приливают по каплям 27 г бутандиола-1,4. Смесь выдерживают 24
при комнатной температуре, нагревают 3 ч на паровой бане и после охлажденпа
отделяют неочищенный дибромид в делительной воропке.
Вместо готовой 48%-пой ПВг можно применять НВг, полученную из бромид,
щелочного металла и I12SO4 или из брома и SO2. Первый способ пригоден для получени •
[866] 1). Bryce - S m i t li, К. E. Howlett, J. Chem. Soc., 1951, 1141.
[867] C. D. N e n i t z e s c u, J. Necsoiu, J. Am. Chem. Soc., 72, 348
(,950).
14 Закав 1835
210
ОБРАЗОВАНИЕ СВЯЗИ С -Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
алкилбромидов только до С4 и 1,3-дибромпропана [8641. Он описан на примере
получения л-бутилбромида [864] и этилбромпда [868]. При больших загрузках можн
непосредственно применять смесь бромистого водорода с серной кислотой, пригото
вленную J864J из брома и SO2 (стр. 110).
?1-Бутилбромид. В смесь 1300 s измельченного льда и 1200 г брома П|
охлаждении льдом пропускают SO2 до исчезновения красной окраски броьр
Получается продукт, соответствующий смеси 15 моль 48%-ной НВг и 7,5 х®
}T2SO4. К полученной смеси прибавляют сначала 12 моль н-бутилового спнрп
затем при перемешивании в несколько приемов еще 600 г концентрирование
H2SO4. После Зч кипячения массы с обратным холодильником меняют хоЗ
дильнпк на нисходящий п в течение 1 ч отгопяют готовый продукт. Обработт
неочищенного н-бутплбромида см. выше. Выход 95% от теоретического; т. шб
101—104= С. Ч
Ниже рассмотрен обмен спиртового гидроксила на иод с помощью иодИст®
водорода [857, 869].
Универсальный способ. Алкилиодиды получают медленной перегощу
(1—2 капли в секунду) 0,5 моль спирта с 1,5—2 моль HI, кипящей при постой
'ной температуре (d = 1,7). Сначала отгоняют половину реакционной ма®
(отгонку соединений Ci— Св ведут 4—6 ч, а соединений Cs—Са ведут 7—frii
выделяют из дистиллята неочищенный иодид, водный слой возвращают в peck
ционную среду и продолжают отгонку еще некоторое времн. Неочищенный иод
промывают равным объемом концентрированной HCI, водой, 5%-ным растворе
Na2CO3, вновь водой, сушат над СаС12 и перегоняют. i
Под действием тепла или света от алкилиодпдов отщепляется под, которй
может быть удален встряхиванием с порошкообразным серебром. • *.
Бензиловые спирты почти количественно реагируют с иодистоводородной
лотой (d = 1,7) за короткое время уже при комнатной температуре [870].
Аллилиодид [838]. К 1 объему аллилового спирта прибавляют 6 о(
емов И( (d = 1,7) и оставляют на пять суток при комнатной температуре. (
деляют неочищенный иодид, промывают его разбавленным раствором NaOl
и водой и после высушивания над СаС12 перегоняют в токе азота. Выход 7^
80% от теоретического; т. кип. 101° С.
а, ш-Дииодалканы, 1(СНа)я1. Иодалканы с п = 8 или 12 получают narj
ванном в течение 6 ч с обратным холодильником при перемешивании соотш
ствующего диола со 100%-ным избытком HI (d = 1,94). После охлаждена
тт разбавления водой выделившееся тяжелое масло экстрагируют эфиром, эфц
ие,тй раствор промывают водой, растворами NaHCOg и NallSOg и после высуну
вания над Na2SO4 перегоняют в вакууме [871]. Выход около 80% от теоре®
веского.
Можно предварительно получать HI в реакционной смеси из KI и 95%-Щ
ТТ3РО4. По этому способу готовят поднды из первичных, вторичных, третнчнр
и многоатомных спиртов с очень хорошими выходами [201]: .
ROH+KI + HsPO4 —> RI+КН2РО4 + н2о
Смешивают 135 г 85%-пой Н3РО4 с 65 г P20g, охлаждают и прибавляИ
2 моль тонкоизмельченного KI и затем 1 .моль спирта. Смесь нагревают с обр®
иым холодильником при перемешивании в течение 3—5 ч при 100—120° С. По<$
охлаждения иодид извлекают эфиром и эфирный раствор промывают раствор®
Na2S2O3 и водой. ,
{868] L. Gattermann, Я. Wieland, Die Praxis des organischrn Cbemikeri
35 Aufl., W. de Gruyter u. Co., Berlin, 1953. S. 86.
[869] ,T. Г. N о г r i s, Am. Chem. J., 38.. «39 (1907).
Г8701 G. IT. D a u b, Д. N. C a s 11 e, J. Org. Chem., 19, 157.3 (1954).
[871] E. P. Taylor, ,T. Chem. Soc., 1952, 144.
Ш. ЗАМЕНА ГИДРОКСИЛЬН., АЛКОКСИЛЪН. II КАРБОНИЛЬН. ГРУПП НА Hal 211
С деталями этого способа рекомендуется ознакомиться на примерах по-
дробных прописей для получения 1,6-дииодгексана [872] п м~октадецил-
подпда J873J.
Замена спиртовой ОН-группы на галоген при
помощи га л о г ей ан гид р ид ов. Помимо галогеноводородов для замеще-
ния гидроксила на галогены можно использовать также некоторые галогеиангидриды.
Для обмена ОН-группы на бром пли иод наиболее пригодны галогениды фосфора РВг8
и Р1а, для обмена на хлор — SOC12. По реакции с РВг3 образуется меньше олефинов,
чем ио реакции с НВт пли системой НВг — H2SO4, п поэтому он особенно пригоден
для обменных реакций с вторичными и третичными спиртами. Реагент РВг, приме-
няют как таковой или получают в реакционной смеси из брома и красного фосфора;
Р13 почти всегда образуется в реакционной среде из иода и красного фосфора, к кото-
рому для активации можно добавлять белый фосфор. Реакция не протекает прямо по
суммарному уравнению
ЗВОНЧ-РХз —► 3RX4-HPO(OH)8
Большей частью сначала галогены в РХ3 постепенно замещаются эфирными группами
и образующийся эфир фосфористой кислоты расщепляется выделяющимся при этом
галогеноводородом:
ЗКОН + РХз —> Р(ОВ)з + ЗНХ—>3RX + HPO(OH)2
Подробнее о механизме обмена гидроксила на галоген с помощью РХ3 см. [874].
Связывание галогеповодорода при проведении реакции в эфире или в присут-
ствии пиридина затрудняет расщепление алкиловых эфиров фосфористой кислоты
галогеноводородом. 5та реакция облегчается избыточным количеством спирта, рас-
творяющего галогеноводород, нагреванием после завершения первой стадии процесса,
а также более длительным временем взаимодействия. В то же время связывание боль-
шей части галогеноводорода эфиром или пиридином часто желательно при галогени-
ровании неустойчивых в кислых средах спиртов.
Взаимодействие спиртов с ЗОС12 или SOBr2 протекает с промежуточным обра-
зованием эфиров галогенангидрида сернистой кислоты (галогснсульфинатов):
ROH + SOXj —» HX-f-ROS(O)X
ROS(O)X —Н-' BX + SO2
Подробнее о механизме этой реакции см. [875].
Препаративное значение имеет синтез хлоридов с помощью SOC12 с добавкой
пиридина [876] илп других третичных аминов [877—879]. Этот способ не вызывает
изомеризации и с успехом применяется для галогенирования оксисоединений, чув-
ствительных к ЦС1. Таким путем из алкокси- [878] или алкоксиарилалка-
яолов [879], из тетрагидрофурфурилового спирта [880], а также из алкинолов
[881, 882] получают соответствующие хлориды с хорошими выходами.
[872]
[873]
[874]
1875]
[876
[877
[878
[879
4 880
[881]
[882]
Н. Stone, Н. S h с с h t е г, Org. Synth., 31, 31 (1951),
G. W. Wood, J. Chem. Soc., 1953, 3327.
W. Gerrard et al J. Chem. Soc., 1940, 1464; 1944, 85; 1945, 106; 848;
1946, 741; 1949, 2309; 1952, 914; 1953, 1920, 2069; 1955, 277.
A. S ta b 1 e r, E. S c h j r m, Ber., 44, 319 (1911); E. D. H u g h e s, С. K. In-
gold et al., J. Chem. Soc., 1937, 1196; E. S. Lewis, Ch. W. Boozer,
J. Am. Chem. Soc., 74, 308 (1952).
G. D a r z e n s, C. r., 152, 1314 (1911).
A. Cohen, J. Chem. Soc., 1935, 429.
D. E. Ames, R. E. Bowman. J. Chem. Soc., 1950, 406.
H. Rapoport, J. E. Campion, J. Am, Chem. Soc., 73, 2239 (1951).
L. A. Brooks, П. R, Snyder, Org. Synth., 25, 84 (1945); Coll. v. 3, 69&
(1955). /
M. S. N e w m an, J. H. W о t i ?, J. Am. Chem. Soc., 71, 1292 (1949).
M. G. E t 11 i nge r, J. E. H о d g k i n s, J. Am. Chem. Soc,, 77, 1835 (1955);
K.-E. S c h и I t e, M. Goes, Arch. d. Pharm., 290/62, 124 (1957).
14*
ОБРАЗОЯАНПЕ СВЯЗИ С —Hui В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Асимметричный дихлордиметиловый эфир, который можно рассматривать как
дихлорангидрид метилового эфира ортомуравышой кислоты НС(ОСП3)С1», реагирует
со спиртами неоднозначно. Поэтому он непригоден для получения алкплхлоридов |883].
Общий метод замещения спиртового гидроксила на бром с помощью РВг3 описан
на следующем примере.
Изобутилбромид (885] (СНз^СНСНаВг. При перемешивании и охлажде-
нии смесью льда с поваренной солью, исключая доступ влаги, к 7 моль охла-
жденного до —1UJ С сухого изобутнлоного спирта приливают по каплям 2,56 моль
(244 мл) свежеперегианпого РВг8 с такой скоростью, чтобы температура пе под-
нималась выше 0е С (примерно 4 ч). Затем прекращают охлаждение и перемеши-
вают смесь до тех пор, пока ее температура по повышается до комнатной, после
чего оставляют на ночь. Реакционную колбу соединяют с колонкой высотой
30 см и отгоняют неочищенный бромид, т. кип. около 50° С (200 мм рт. ст.).
Дистиллят охлаждают до 0° С, трижды промывают концентрированной II2SO4r
охлажденной до 6° С (по 50 мл), п встряхивают с 25 г сухого К3СО3 до исчез-
новения запаха НВг. Затем перегоняют на колонке высотой 1 м при атмосферном
давлеппп (т. кип. 91—933 С) пли п вакууме на колонке с флегмовым числом 5 : 1
(т. кип. 41—433 С при 135 мм рт. ст.); выход 55—60% от теоретического.
Аналогично получают «/nop-бутилбромид СН3СНВгС2Нб, выход 80%;
м-ироиилбромид, выход 95%; изопропилбромид, выход 68% от теоретического,
и цнклопентилбромид [841].
Трибромид фосфора (т. кип. 171 —173° С). Его синтезируют, добавляя
бром к перемешиваемой суспензии красного фосфора в СС14. Для перегонки
РВг3 применяют эффективную ректификационную колонну [885, 886].
Условия проведения обменной реакции спиртов с РВг3 и выделения продуктов
реакции можно широко варьировать.
Спирт можпо разбавлять бензолом, петролейным эфиром, эфиром или СС14.
При бромпровапии пенасыщеппых спиртов и спиртов с эфирными связями, которые
могут вступать в побочные реакции с образующимся НВг, добавляют пиридин, но
обычно в этом пет надобности.
Тстрагидрофурфурилбромид [887]. Для его получения приливают
по каплям 1 моль тетрагидрофурфурилового спирта и 5 г сухого пириднпа при
—5° С (охлаждение льдом е поваренной солью) к смеси 0,36 .ноль РВг3, 15 г
сухого пиридина и 50 мл сухого бензола и выдерживают 24—48 ч при комнатной
температуре.
р-Этоксиэтилбромид [888]. Получают без добавления пириднпа из Р-эт-
оксиэтилового спирта и трибромпда фосфора при постепенном повышении темпе-
ратуры до слабого кипения. Наряду с этим бромидом образуется значительное
количество этилбромида.
Пропаргилбромид [889]. К охлажденной до 0° С смеси 224 г безводного
пропаргилового спирта и 48 г безводного пиридина при перемешивании при-
ливают по каплям 378 г РВг3, смешанного с 2 мл пиридина. Затем кипятят 1 ч
на водяной бане, отгоняют пропаргилбромид в приемнике над сухим К3СОа
п через некоторый промежуток времени перегоняют еще раз; т. кип. 84° С; выход
59% от теоретического.
При взаимодействии оптически активных спиртов с РВг3 обычно происходит
вальденовское обращение. Для получения возможно более чистых оптически актив- '
[883] A. Rieche, Н. G г о р, Chem. Вег., 92, 83 (1952).
[884] .1. Colonge, G. Р о i 1 a n е, Bull. Soc. chim. France, Мёт. [5j, 1955, 499.
[885] G. R. N о 1 Io r, R. Dinsmore, Org. Synth., Coll. v. 2, 1943, p. 358.
[886j J. ?. Gay, R. N. Maxson, Inorg. Synth., 2, 147 (1946).
[887] L. H. Smit h, Org. Synth., 23, 88 (1943); Coll. v. 3, 793 (1955).
[888] G. C. Harrison, H. Diehl, Org. Synth., 23, 32 (1943); Coll. v. 3,
370 (1955). 4
[889] K.-E. Schultt M, Goes, Arch. d. Pharm., 290/62, 124 (1957).
Ш. ЗАМЕНА ГИДРОИСПЛЬН., АЛКОНСИЛЬН. И КАРБОННЛЬН. ГРУПП НА Hal 213
ных бромидов необходим постоянный избыток спирта. Например, [890—891] при
низкой температуре (вначале охлаждение сухим льдом) к еппрту медленно приливают
по каплям 0,6 моль от эквимолекулярного количества РВг3.
Обмен вторичных и третичных ОН-групп на бром с помощью РВг3 рекомен-
дуется вести в очень мягких условиях. Для получения 2-метИл-2,4-дибромпентана
п 3-метил-2,4-дибромгексана РВг3 приливают по каплям при температуре не выше
—15° С к соответствующему спирту, поднимают температуру массы и течение двух
суток до 20° С и выдерживают массу еще 1—3 суток при этой температуре. Выход 90%
от теоретического [892].
В других случаях для доведения реакции до конца приходится применять
нагревание. При этом, а также при оггонке неочищенного бромида из реакционной
смеси необходимо повышать температуру медленно во избежание образования фосфи-
нов, способных самовоспламеняться. При получении пентаэрлтритбромида С(СН2Вг)4
приходится нагревать смесь 20 ч па масляной бане при 170—180° С для доведения
реакции до конца [893].
В отличие от общей прописи для выделения продукта реакции можно вылить
реакциоппую массу в ледяную воду, отделить неочищенный продукт или извлечь
его эфиром пли петролейным эфиром, промыть водой и 5%-ным раствором Na2COs
и высушить над СаС12. Можно добавить воду к реакционной смеси и отогнать неочи-
щенный бромид, например циклопентилбромид [894], с водяным даром.
Можно также РВг3 предварительно получать в реакционной смеси из красного
фосфора и брома. При этом необходимо следить за тем, чтобы бром не соприкасался
с сухим красным фосфором. Сначала хорошо смешивают спирт с 1 моль красного
фосфора и затем при сильном перемешивании приливают по каплям около 2 моль
брома с такой скоростью, чтобы температура смеси достигала 80—100° С. Этот способ
описан на примере получения 1,3-дибромпропанола-2 из глицерина [895].
В то время как при обмене гидроксила на бром по реакции с РВг3 большей
частью применяют предварительно приготовленный трибромид, при обмене на иод Р13
обычно получают в реакционной смеси из фосфора и пода.
В общепринятом методе обмена спиртовой ОП-группы па иод по реакции с фос-
фором и иодом исходят из следующего соотношения: на 3 моль сухого спирта берут
1,5 моль иода м 1 — 1,2 моль фосфора. Экзотермическая реакция обычно гладко прохо-
дит с красным фосфором, освобожденным ос кислотных примесей повторным кипяче-
нием с водой и сушкой при 100° С. Добавка около 50% желтого фосфора ускоряет
реакцию, однако желтый фосфор нс должен входить в непосредственное соприкосно-
вение с иодом. При добавке желтого фосфора следует пользоваться описанной ниже
специальной аппаратурой и следить за тем, чтобы прп обработке реакционной массы
остаток желтого фосфора всегда находился под слоем реакционной массы или воды,
т. е. не соприкасался с воздухом.
Сначала хорошо смешивают спирт с красным фосфором и затем при пере-
мешивании маленькими порциями добавляют тонкоизмельченпый иод, при необ-
ходимости время от времена охлаждая смесь. Затем выдерживают смесь не-
сколько часов при комнатной температуре для завершения реакции н в заклю-
чение кипятят 2 ч с обратным холодильником. Примером может служить полу-
чение этилиодида [896]. Легколетучие иодиды отгоняют непосредственно из
реакционной массы. Реакционную массу с высококипящими иодидами выливают
в ледяную воду, отфильтровывают от непрореагировавшего фосфора, экстра-
гируют подид эфиром, экстракт встряхивают с раствором Na2SsO8, 5%-ным
раствором Na2CO3 и водой, сушат над СаС12 и после отгонки эфира перегоняют
в вакууме.
890
891
892
893
894
895
896
Н. Branns, Rec. trav. chim., 65, 799 (1946).
A. Streitwieser jr. J. Am. Chem. Soc., 75, 5014 (1953).
J. D. Bartleson et al., J. Am. Chem. Soc., 68, 2516 (1946).
H. B. Schurink, Org. Synth., Coll. v. 2, 1943, p. 476.
C. R. Noller, R. Adams, J. Am. Chem. Soc., 48, 1084 (1926).
G. Braun, Org. Synth., Coll. v. 2, 1943, p. 308.
L. Gattermann, Die Praxis des organischen Chemikers, 3-5 Aufl.
W. de Gniytor u. Co., Berlin, 1953, S. 88.
214
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
В то время как обменную реакцию с вторичными и третичными спиртами ведут
около 8 ч при 0—5° С и затем выдерживают смесь при комнатной температуре, для
получения высших первичных иодидов требуется многочасовое последующее нагре-
вание при температурах выше 100° С, как это показано на подробно описанном при-
мере синтеза 1-иодгексадекана из цетилового спирта [897].
При работе с добавкой желтого фосфора и больших загрузках иод вводят в смесь
фосфора и спирта по мере его расходования в виде раствора в соответствующем спирте. 1
Реакционную колбу соединяют с сосудом, в котором находится иод, в дан- .
ном случае с капельной воронкой, снабженной трехходовым краном. Воронка
связана с колбой припаянной к ос верху не слишком узкой нисходящей стек-
лянной трубкой и, кроме того, еще с обратным холодильником. В колбу вносят •'
смесь спирта с фосфором и нагревают. Поднимающиеся по трубке пары спирта ••
конденсируются в обратном холодильнике и постепенно растворяют находя- .
щийся в мернике иод. Для того чтобы трехходовой кран не засорялся, под слоем >
иода имеется слой стеклянной ваты. Скорость реакции можно регулировать
степенью открывания кранЗ. По этому рекомендованному Фогелем [898. 899]
способу можно успешно вести процесс также с одним только красным фосфором.
После израсходования всего иода закрывают крап и перегоняют лсгколстучпе ’
иодиды через стеклянную трубку в мерник. Волее старые данные об аналогичных \
установках см. [900, 901].
С помощью SOC12 почти все спиртовые пщроксильные группы, a idKAe ]
ОН-группы оксикарбоновых кислот обмениваются на хлор. Этот метод представляет
особую ценность, когда обмен спиртовой ОН-группы при действии НС1 сопровождается .]
нежелательными побочными реакциями (изомеризацией, присоединением НС1, рас-
щеплением простых эфиров). В таких случаях можно использовать многочисленные
варианты способа Дарзана [876], по которому к смеси 2 моль сянрта и 3 моль пиридина
при охлаждении льдом или смесью льда с солью приливают по каплям 3 моль SOCI2.,j
Обычно при галогенировании алкпнолов и алкоксиалкадолов применяют на 1 моль’Д
спирта 1 —1,1 моль пиридина и 1,1 моль SOC12, а при галогенировании алканолов, 4
склонных к изомеризации в присутствии хлористого водорода, 1,3—1,4 моль SOCI2. .я
Для получения н-алкилхлоридов иногда рекомендуют вводить еще больший избыток J
SOCl2 (2 моль SOCl2 па 1 моль ОН), а для получения высших алкилхлоридов, напри- з
мер ундецил- и додецилхлорида [857], без добавки пиридина — 4 моль SOC]2. Спирты 1
можно разбавлять бензолом [902], хлороформом [879] или эфиром [881, 882]; при J
галогенировании алкоксиалканолов можно вместо пиридина применить диметилани-
лип [877—879] или хинолин [878]. Применять можно только очищенный (стр. 215) J
или свежеперегнанный SOCl2. М
1-Хлорпентан [823]. В смесь 5 моль н-амилового спирта и 5 моль с\ хою
пиридина при —10® С приливают по каплям 6,5 моль SOCl2. Реакционную массу а
в течение б ч подогревают до 104° С и выдерживают 90 мин при этой темпера- ' а
туре. Примерно при 73° С начинается выделение SO2. Охлажденный поочищен- <2
ный продукт промывают разбавленной НС1, сушат К2СО3 и перегоняют; т. кип. J
106—106,5° С (741 мм рт. ст.); выход 80% от теоретического. -1
1-Хлоргептин-2 [881]. В раствор 1,1 моль SOCI2 в 100 мл сухого эфира>Л
приливают по каплям при перемешивании раствор 1 моль алкипола п 1 моль Я
сухого пиридина в 100 мл сухого эфира, причем смесь закипает за счет тепла. |
экзотермичной реакции. По завершении реакции отгоняют около 150 мл эфира, -л
повышая температуру смеси до 80° С, затем добавляют еще 13 г SOCI2 и кипятят . j
при перемешивании в течение 1 ч. После этого к массе приливают 500 мл холод-..-3
[897]
[898
[899
[900
[901
[902
J. R. В у е г s, J. В. D i с к е у, Org. Synth., Coll.
v. 2, 1943, р. 322.
A. I. Vogel, англ. пат. 565452 (1944); С. А., 40, 5066 (1946).
A. I. Vogel, J. Chem. Soc., 1948, loll.
R. Adams, V. Voorheis, J. Am. Chem. Soc., 41, 797 (1919).
H. S. King, Org. Synth., Coll. v. 2, 1943, p. 399.
C, Barkovsky, Ann. Chim., 19, 487 (1944).
1П. ЗАМЕНА ГИДРОКСИЛЬИ., АЛКОКСИЛЬН. П КАРБОННЛЬН. ГРУПП НА Hal 21'
ной воды, отделяют эфирный раствор, промывают ого раствором NaaCO3 и водой
сушат над CaCls п быстро отгоняют хлоргептин в вакууме; т. кип. 73° С
(24 .ил рт. ст.); выход 77% ог теоретического.
Аналогично получают 1-хлоргептпн-З с выходом 72% от теоретического
2-Этокеиэтилхлорид 1878]. К охлажденной до —20° С смеси 1 лол»
сухого основания и 1 .ноль 2-этоксиэтилового спирта при перемешивании при
ливают по каплям 1,1 моль SOCls так, чтобы температура не повышалась выш*
0® С. Затем дают температуре подняться до комнатной и в заключение смес!
выдерживают 2 ч при 100° С. Охлажденный продукт обрабатывают разбавлен-
ной НС1, этокеиэтилхлорид экстрагируют эфиром, экстракт промывают раз
бавленным раствором NaOH и водой и сушат над NaaSO3; выход 61% (с диметид
анилином), 80% (с пиридином), 84% (с хинолином).
а-Хлорфенилуксусная кислота [903]. Растворяют 0,75 моль этиловог<
эфира миндальной кислоты (из миндальной кислоты п этилового спирта в прж
сутствии НС1) в 0,82 моль 8001%. После 16 ч выдерживания при комнатной тем
пературе раствор нагревают 30 мин на паровой бане и выливают смесь в ледануж
воду. Полученный этиловый эфир а-хлорфенилуксусной кислоты (выход 81 —
85% от теоретического) извлекают эфиром и после отгонки эфира омыляют кипя
пением в смеси ледяной уксусной кислоты с концентрированной НС1.
Метиловый эфир а-хлорфеннлуксусной кислоты [904J. Получают при
бавлением в несколько приемов 11,5 г РОС13 к смеси 12,5 г месилового эфире
миндальной кислоты и 12 г пиридина при охлаждении смесх,к> льда с соль»
с последующей выдержкой в течение 1 ч в охлаждающей смеси и 12 ч при ком
пэтной температуре. Выход ниже, чем в предыдущем примере.
Дезилхлорид, CeIIbCHClCOCfiIIb. Подробно оппсанпый синтез из 0,17 мол/
бензоина, 0,63 моль пиридина и 0,63 моль SOCla см. [905]-
Очистка тионилхлорпда. По часто применяемому методу кипятят S0C1.
примерно с 10% льняного масла и перегоняют с колонкой *. Для удаления SO,
и HaSO4 рекомендуют также перегонку с хинолином [906]; не следует применят]
для этой цели диметиланплин [907]. Можно, например, перегнать 50 г S0C1.
сначала с 10 мл хинолина, а затем с 20 мл льняного масла [908]. Хорошо заре
комендовал себя следующий простой способ [909]. В течение 4,5 ч кипятя1:
с обратным холодильником смесь 900 мл неочищенного SOCla с 25 г серног<
цвета и затем быстро перегоняют через колонку. Получается окрашенный про-
дукт (94%) и в остатке остается монохлорид серы (т. кип. 135,6° С). Этот продукт
вповь перегоняют па колонке, отбирая сначала в течение 12 ч первую оирашеж-
ную фракцию (в основном 8СЦ; т. кип. 69° С), после чего приблизительно за 2 «
перегоняется почти бесцветный SOCla; выход 88%; т. кип. 78,8° С.
Обмел ОН-группы на хлор с помощью SOC1% в аминоалнаполах, которые еле
дует разбавлять хлороформом или бензолом, также происходит с хорошим выходом
Аминоалканолы можно непосредственно подвергать взаимодействию с SOClJt н<
лучпге исходить из их гидрохлоридов.
Гидрохлорид ^1М-бис-(|8-хлорэтил)-вми1]а [910]. Его получают, напри:
мер. по реакции 2 моль неочищенного гидрохлорида диэтаноламина с 5,33 мол г
SOCI2 (388 мл) и 400 мл бензола при температуре около 30° С.
Труднодоступные спирты можно переводить в соответствующие бромиды такж«
по реакции с SOBr2 ® присутствии пиридина. Правда, сам SOBr2 не очень легко полу-
чае гея.
903
904
905
906
907
908
909
[910
Вместо льняного масла можно успешно использовать медицинское персика
вое пли абрикосовое масло. — Прим. ред.
Е. L. Е I i е I, М. Т. Fisk, Th. Prosser, Org. Synth., 36, 3 (1956).
Th. Wagner-Jauregg, Helv. chim. acta, 12, 63 (1929).
A. M. Ward, Org. Synth., Coll. v. 2, 1943, p. 159.
H. Meyer, K. Schlegel, Mh. Chem., 34, 569 (1913).
E. Best horn, Ber., 42, 2697 (1909).
E, L. M art in, L. F. F i e s e r, Org. Synth., Coll. v. 2, 1943, p. 569.
T>. L, Cottle, J. Am. Chem. Soc., 68, 1380 (1946).
K. Ward jr., J. Am. Chem. Soc., 57, 914 (1935).
216
ОБРАЗОВАНИЕ СВЯЗИ С —Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Тпонилбромид [911]. Получают взаимодействием сухого НВг с SOC12
при 0° С. По другому способу 1912] к КВг добавляют жидкий St)2. Температура
кипения SOBts равна 60° С (40 .ч.м рт. ст.} и 45° С (22 лм рт. ст.}. 'Гпони.т-
бромид при комнатной температуре разлагается медленно, при 100° С — быстрее:
48ОВг2 —> 2SO2 + S2Br2 + 3Br2
Для получения бромалканов приливают по каплям при температуре
от —10® С до 0° С п перемешивании 1,3 моль 8ОВгг к смеси 1 моль соответству-
ющего спирта и 1,1 моль пиридина; затем доводят смесь до комнатной темпера-
туры и дополнительно еще немного нагревают до прекращения выделения SO2.
Таким способом получают, например, 2-, 3- и 4-бром-н-октаиы [913]; выходы
около 40% от теоретического
Замена спиртового гидроксила на галоген при помощи трифенилфосфита ,
и органических галогенидов или просто галогенов 1
По предложенному Ландауером и Райдопом [914] способу алкилгалогекиды ?,
получают нагреванием эквимолекулярных количеств трифенилфосфита, спирта и со- ~
едипепия R'X, где Х=С1, Вт пли I; R'может быть аралкилом, алкилом, Н, X, Li,
Na или NH4:
(СбН5О)3Р4-ROH+ П'Х —> RX + Н'Р(ОСеН5)г + CeHROII (1) |
В том случае, когда Н'=аралкпл или алкил, соединение R'X должно быть
более реакционноспособным, чем соедпнение RX, если только RX нельзя непрерывно ]
выводить из зоны реакции. В трифепилфосфитном методе на первой стадии взаимо- '
действия образуется квазифосфопиепое соединение А, которое затем переэтерифи-
цируется
OR ?
I т
(CeHsO)3PR'X 4- ROH—> (C6H50)2PR'X-FCeH5OII (2)
л |
Далее аналогично второй стадии перегруппировки Арбузова (реакция 3) |
(RO)jP + B'X -1 сташ1я^ (но)зРК'Х ” g^S-. (RO)Jk-+ RX (3) -j
происходит образование желаемых а.ткилгалогепидов (реакция 4)
?
(CeH6O)2(RO)PR.’X —> (C6H&O)2PR' + RX (4) •.
Этот способ особенно пригоден для получения алкилбромидов и алкплиодидов из спир" '
тов, не способных к реакции ввиду стерических затруднений, и из ненасыщенных -
[911] X. Хибберт, Дж. С. 11 у л л м е н, Неорганические синтезы, Сбор- 4
ник I, 1939, стр. 113; Руководство по неорганической препаративной •
химии, под редакцией Г. Браусра, пер. с нем., Издатинлит, 1956. ?
[912] M.J. Frazer, W. Gerrard, Chem. a. Ind., 1954, 280; Angew. <?
Chem., 67, 42 (1955). t
[913] F. A s i n g e r, G, Geiseler, M. Hoppe, Chem. Ber., 90, 117
{Wol}.
[914j S. R. Landafter, H. N. R у don, J. Chem. Soc., 1953, 2224.
Ill. ЗАМЕНА ГИДРОКСИЛЫ!., АЛКОНСИЛЬН, II КАРБОНИЛЬН. ГРУПП НА НМ 217
спиртов. В качестве RrX для препаративных целей наибольший интерес представляют
бензилхлорид и хлор и особенно бензилбромид, метилиодид, бром и мод; соединение,
в котором R'= Н, можно применять лишь в том случае, если R'X=IICI. При проведе-
нии реакции обмела с термически неустойчивыми спиртами следует сначала получить
квазпфосфониевое соединение (СвН5О)эРВ'Х и затем подвергнуть его взаимодействию
со спиртом. Если вместо алкилгалогенида в реакции участвуют галогены хлор, бром
или иод, то при галогенировании ненасыщенных спиртов надо также предварительно
получать неочищенное соединение (СвН5О)яРХ2. Но обычно все три компонента
могут непосредственно реагировать по реакции 1.
Метилтрифепоксифосфоиийиодид [914]. В течение 36 ч кипятят с обрат-
ным холодильником 31 г трифенилфосфита с 21 г метилиодида (без доступа
влаги!). При этом температура кппения смеси повышается с 70 до 115° С. После
охлаждения смеси и обработки абсолютным эфиром выпадают желтовато-корич-
невые иглы метилтрифеноксифосфониниодида. Их повторно промывают абсо-
лютным эфиром и высушивают над Р2О5; выход 42 г (94% от теоретического).
Для последующей стадии, которую следует вести без доступа влагп, лучше всего
использовать сохраняемый под абсолютным эфиром неочищенный продукт.
Его можпо очистить быстрым растворением в сухом ацетоне и осаждением абсо-
лютным эфиром; т. пл. 146° С.
Получение алкилиодидов при помощи метилтрифеиоксифосфоннйводи-
да [914]. К 1 моль неочищенного метилтрифеноксифосфонийиодида осторожна
прибавляют 1 .ноль соответствующего спирта. При работе с реакционноспособ-
ными спиртами необходимо охлаждение, с нереакционноспособными — слабое
нагревание. В зависимости от свойств алкилиодидов применяют один из следу-
ющих способов их выделения: а) алкилиодид, который кипит при более низкой
температуре, чем фенол, непосредственно отгоняют в вакууме; б) алкилиодид
отгоняют вместе с фенолом в вакууме, дистиллят разбавляют эфиром и фенол
извлекают разбавденпым раствором NaOH, охлажденным льдом; в) реакцион-
ную массу разбавляют эфиром, фенол вымывают разбавленным раствором NaOH
и охлажденной льдом водой, эфирный раствор сушат л перегоняют.
Пеопентилиодид, (СН3)3ССН21 (прямой способ получения [914, 915]
по реакции 1). На бане нагревают в течение 24 ч 1 моль трифенилфосфита
с 1,4 моль метилиодида и 1 моль пеопентиловою спирта. По мере расходования
метилиодида температура смеси возрастает с 75 до 130е С. Неочищенный иодид
п фепол совместно отгоняют в вакууме и из дистиллята удаляют фенол промыв-
кой охлажденным льдом и разбавленным раствором NaOH и водой. Полученный,
продукт содержит около 6% гпре/п-амилиодида [915]. Для его удаления неочи-
щенный неопентилиодид встряхивают 5ч с трехкратным объемом воды, органи-
ческий слой отделяют и встряхивают еще 1 ч с однократным объемом 0,1 и. вод-
ного раствора AgNO3, затем промывают водой, сушат и фракционируют; выход
неопентилноднда 53—57% от теоретического; т. кип. 71° С (100 мм рт. ст.).
Многочисленные другие примеры см. [914, 916].
Получение трифсноксифосфонийгалогенида и прямая обменная реак-
ция (СвН56)3РХ2 со спиртами [917]. К 1 моль трифенилфосфита, полученного-
нагрсванием 1 моль РС13 с 3 моль фенола до прекращения выделения ЦС1 и после-
дующей перегонкой в вакууме [918J (т. кип. 209—210° С при 1 мм рт. ст.),
прибавляют при Тщательном исключении доступа влаги, перемешивании и:
охлаждении 1 моль галогена. К приготовленному таким образом неочищенному
(С8Н6б)3РХ2 при комнатной температуре, если необходимо при охлаждении»
при перемешивании приливают по каплям 1 моль спирта. В большинстве случаев
синтезируемый а.ткилгалогепид выделяют непосредственно разгонкой в ваку-
уме. Выход насыщенных алкилиодидов 60—98%, аллилбромида — 75% от тео-
ретического.
Прямое взаимодействие трифенилфосфита, спирта н галогена [917].
При 0° С и смеси эквимолекулярных количеств трифенилфосфита и спирта
[915] N. К о г n b I u m, I). С, I f f 1 а n d, J. Am. Chem. Soc., 77, 665&
(1955).
[916] A. Campbell, Н, N. Rydon, J. Chem. Soc., 1953, 3004.
[917] D. G. С о е, D. R. Lan dauer, H. N. Яу don, J. Chem. Soc.».
1954, 2281.
[918] H. B. Gottlieb, J. Am. Chem. Soc., 54, 749 (1932).
218
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
постепенно при перемешивании прибавляют 1 моль галогена. Смесь некоторое
нремя выдерживают при комнатной температуре и выделяют алкилгалогенид,
например перегонкой при пониженном давлении. Выход алкилгалогенидов
62—92% от теоретического. Для получения алкилгалогенидов из ненасыщенных
спиртов этот способ не применим.
Замена спиртового гидроксила на галоген
с промежуточным получением тозилатов
Несмотря на ю что в ряде рассмотренных до сих пор способов обмена спирто-
вого гидроксила на галоген спирты превращались на промежуточной стадии в суль-
финаты или производные эфиров фосфористой кислоты, которые затем подвергали
алкилирующему расщеплению, обычно не было необходимости выделять эти проме-
жуточные продукты. В тозилатном методе, напротив, сначала из спирта получают
эфир толуолсульфокислоты (тозилат), который и подвергают взаимодействию с гало-
генидом щелочного или щелочноземельного металла:
n-CH3CeH4SO2CI + HOB , n-CHAH4SO2OR
n-GH3C6H4SO2OR + MeX -------га-СНзС6Н48О2ОМе-| RX
Эта реакция имеет особенно большое значение для получения алкилфторцдов,
иоддезоксисахаров и для обмена ОН-группы на галоген в соединениях, способных
к перегруппировкам углеродного скелета (перегруппировке Вагпера — Меервейна)
пли к аллильным перегруппировкам. ч
Получение тозилатов [919]. В охлажденный до —5° С раствор 10 г
спирта в 100 мл сухого пиридина прибавляют тозилхлорид n-CH8CeH4SO2CI
в количестве 1,1 моль на 1 моль спирта. Встряхивают смесь, не вынимая из ледя-
ной бани, до полного растворения всего тозилхлорида, выдерживают еще 2 ч
при 0° С и затем при температуре не выше 5° С приливают по каплям воду (три
раза по 1 мл и затем 2 и 5 мл), добавляя каждую порпию в течение 5 мин. Потом
приливают еще 100 мл воды. Если тозилат выкристаллизовывается, его отсасы-
вают, в противном случае тозилат экстрагируют эфиром или СПС19. Экстракт
промывают охлажденным льдом разбавленным раствором П-ЯО4, водой, рас-
твором NaHC03 и сушат над Na2SO4. После удаления растворителя неочищен-
ный тозилат часто непосредственно используют в обменной реакции.
Тозилаты низших спиртов, быстро реагирующие с пиридином уже при 0° С,
получают при более низких температурах [920]. '
Описанный ниже способ перевода тозилатов в алкилфториды пригоден для 5
получения первичных, вторичных и галогензаМещениых алннлфторидов [920]. '
Алкилфториды [920]. При перемешивании и в большинстве случаев
при пониженном давлении (20—500 мм рт. ст.) нагревают в диэтилепгликоле ’
(т. кип. 245° С) 1 моль тозилата с 5 моль KF. В соответствующей охлаждаемой г-
ловушке улавливают алкилфторид, улетучивающийся главным образом j тече-
ние первых 2 ч. Применяют равные весовые количества и KF, и диэтиленгли- (-
коля. Время реакции 3—9 ч, температура 70—240° С. Из циклогексилтозилата
» этих ^условиях образуется циклогексен; фенилтозилат не реагирует с KF.
Подобным же образом можно получать алкилфториды через эфиры бензол- [921] j
и метансульфокислот [922J. <
Метод обмена спиртовой ОН-группы на бром через тозилаты доле-
.сообразно применять тогда, когда другие способы приводят к изомеризации продуктов '
[919] R. S. Tipson, J. Org. Chem., 9, 238 (1944).
[920] W. F. Edgell, L. Parts, J. Am. Chem. Soc., 77, 4899 (1955). '•>
[921] В. В. Разумовский, A. E, Фриденберг, ЖОХ, 19, 22 (1949).
,[922] F. L. M. P a 11 i s о n, I. E. M i 11 i n g t о n, Canad. J. Chem., 34, 757 s 1
(1956).
Ш. ЗАМЕНА ГИДРОКСИЛЬН.. АЛКОКСИЛЬН. И КАРБОННЛЬН. ГРУПП НА Hal 21'
реакции. Обмен вторичной спиртовой ОН-группы на галоген через тозила1
не сопровождается перегруппировками углеродного скелета [825]. Только этю
способом можно, папример, получить чистые 2- и 3-бромпентаны [824]. Из 1-нС-алли
лоиого спирта при переходе через тозилат получается только 1-14С-аллилбромид [923]
т. е.. следовательно, реакция идет без аллильной перегруппировки.
В то время как из а,Р-ацетиленовых спиртов по реакции с SOCL и пиридин»)
пли е РВг3 в сухом эфире получают соответствующие галогениды с хорошими выхо
дамп [881]. р,у- и у,о-ацетпленовые спирты дают соответствующие хлориды, бромид:
и иодиды с хорошими выходами только через тозилаты при взаимодействии с LiC
н 2-этокспэтиловом спирте, с сухим СаВг2 в 2-(этоксиэтокси)-этиловом спирте ид]
с Nal в ацетоне. Первые две реакции протекают экзотермично; получаемые продукт:
непрерывно отгоняют при несколько пониженном давлении [924].
Для обмена тозилоксигруппы на хлор или бром можно применять также СаС1
или Lidl в ацетоне [925] или LiBr в ацетоне. При действии безводного СаВг2 на дито
знлат бутин-2-диола-1,4 в абсолютном этиловом спирте получают 1,4-дибромбутин-2
выход 90% от теоретического [926]; безводный СаВга получают действием 2НВг н,
СаС03 с последующей сушкой при 600° С.
Пентаэритритбромид С(СН2Вг)4 [927]. В течение 12 ч при перемешивани]
нагревают при 140—150 °C смесь полученного из 1 моль пентаэритрита неочи
щенного бензолсульфоната с 5,8 моль NaBr в 1 л диэтиленгликоля. Выход пев
•гаэритритбромида, перекристаллизованного из ацетона, 67—68%. Аналогична
взаимодействует тозйлат пентаэритрита в диэтиленгликоле с NaBr (20%-ны1
избыток) [928].
Замена спиртовых ОН-групп на иод через тозилаты происходит аналогично замен
галогенов, чаще всего под действием Nal в ацетоне, причем по реакционной способ
ностн по отношению к Nat тозилаты близки к хлоридам [929]. Как правило, тозилат®
первичных спиртов реагируют легче, чем тозилаты вторичных.
Эта обменная реакция часто применяется для установления структуры угле
водов (правило Олдхема — Резерфорда) [930]. Из наблюдения, что под действием Na
в ацетоне (2 ч при 100° С в запаянной трубке) в полностью тозилированном ме
тил-р-Р-глюкозиде обмен происходит только у Сд, а в углеводах с тозильными груд
пами у С2, С3, С4 или С6 обмена не происходит, был сделан вывод, что в этих условия:
на иод обменивается только первичная ОН-групна, а вторичная тозилировапна:
гидроксильная группа не обменивается. Однако имеются многочисленные исключенИ;
ил этого правила. В некоторых 1,6-дитозилированных многоатомных спиртах ряд,
углеводов обе первичные тозилированные ОН-группы обмениваются на иод [931
только при увеличении обычного времени реакции до 5 ч, а в других — за 2 ч пр:
100° С обменивается на иод только одна из двух первичных тозилированных труп:
[932, 933]. Правда, в последнем случае с помощью уксусного ангидрида можно с ха
рошим выходом провести обмен обеих первичных тозилированных оксигрупп иа иод
[923]
[924]
[925
[926
[927
[928
[929]
[930]
[931]
'[932]
[933]
К. F. Nystrom, I. С. L е a k, I. Am. Chem. Soc., 75 , 3039 (1953).
G. Eglinton, M. C. Whi ting, J. Chem. Soc., 1950, 3650.
P. L. Julian, A. M a g n a n i, J. Am. Chem. Soc., 71, 3209 (1949).
W. J. G e n s 1 e г, A. P. Ma ha de van, J. Am. Chem. Soc., 78, 168 (1956)
H. L. Herzog, Org. Synth., 31, 82 (1951).
E. R. В ц c h m a n, D. H. D о u t s c h, G. 1. Fujimoto, J. Ann
Chem. Soc., 75, 6228 (1953).
R. S. Tipson, M. A. С 1 a p p, L. H. С г e t c h e r, I. Org. Chem., 1Й
133 (1947).
J. W. О I d h a m, J. K. Rutferford, J. Am. Chem. Soc., 54, 38
(1932).
L. W а г g h a, T. Puskas, Ber., 76, 861 (1943).
A. T. N ess, R. M. Ha nn, C. S. Hu d s on, J. Am. Chem. Soc., 66, 190
(1944).
R. M. Hann, J. K. W о 1 f e, C. S. H u d s о n, J. Am. Chem. Soc., 6S
1900 (1944).
220
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
1,6-Дпдсзокси-1,6-дииод-2,4;3.5-диметилен-0-сорбит. Смесь 4,0 г
1,6-дитозил-2,4;3,5-диметилен- ZJ-сорбита. 3,4 г Nal и 40 мл уксусного ангидрида
кипятят 2 ч с обратным холодильником п затем избыток (СНзСО)2О разлагают
иодой. Постепенно выкристаллизовывается дниодид (выход 91%); после пере-
кристаллизации из этиловою спирта т. пл. 151 —152° С.
С другой стороны, при действии Nal в ацетоне при 100° С может обмениваться
на иод вторичная тозилированная гидроксильная группа многоатомных спиртов
производных углеводов [934] и других спиртов, например диэтилкарбинола [929],
изопропилового спирта и 1,3-бутандиола [935]; в последних двух случаях достаточно
применить концентрированный водный раствор KI.
Реакционная способность тозилатов в значительной степени зависит от стери-
чсских факторов: монотоэилат транс-циклогександиола-1,2 реагирует с КТ и LiCl
значительно быстрее, чем t/uc-соединение [936]. Особенно вяло реагируют тозилирован-
ные гидроксильные группы, находящиеся непосредственно у многократно замещен-
ного кольца; более подробные указания и критическое обсуждение имеющихся до
настоящего времени данных см. [937].
Соседние тозилировапные оксигруппы, особенно концевые, не обмениваются
на иод. Образуются ненасыщенные соединения и выделяется иод:
II I I
—С—С—+ 2NaI —> —С=С—+2TsONa+12
। ।
TsO ,OTs
Многочисленные литературные данные о наблюдении этой реакции и о ее меха- J
низме см. [938]. - ’*
О получении иодстероидов через тозилаты и мезилаты CH3SO2OR см. [939, 940].
Перевод тозилатов в иодиды. В склянке для работы под давлением или
в толстостенной запаянной трубке нагревают 2—5 ч при 100° С 1 моль тозилата •:
с 1,2 моль высушенного Nal в сухом ацетоне. Реакцию тозилатов с низкой реак-
ционной способностью можно проводить также в метилэтилкетоне, ацетонил-
ацетоне или диэтилепгликоле; тозилаты низших первичных спиртов реагируют <
уже при комнатной температуре. За ходом реакции следят по выпадению натрие- J
вой соли «-толуолсульфокислоты, которую по окончании реакции отсасывают А
и промывают ацетоном. После отгонки растворителя добавляют воду, иодид ;
извлекают эфиром и промывают эфирную вытяжку раствором Na2SaO8 и водой, j
Низшие алкилиодиды, которые во время реакции непрерывно отгоняются через
колонну, можно получать также взаимодействием тозилатов с концентрированным =
водным раствором KI (0,2 моль KI в 10 мл воды); тозилат фенола не реагирует с Nal 3]
в ацетоне.
В то время как неопептилиодид можно получать [941] по реакции с Nal через "а
тозилат, неопентилбромид (выход 86%) и неопентилхлорид (выход 60%) рекомендуется
получать через триэтилнеопентоксисилан [942]. 'ч
хинолин; •
(СНз)зС-CH2OH + ClSi(C2H5)8—j
РВг8;
—> (ClI,)3C_CH2-OSi(C2Hs)8 (Cl)BrSi(C!H5),+ (CH3)3C-CH2Br(Cl) 1
[934 ] R, С.- H о c k e t t et al., J. Ant. Chem. Soc., 68, 927 (1946), 1
[933 ] F. Drabowtal, D. К 1 a m a n n, Mh. Chem., 82, 970 (1951). ’
936] M. F. Clarke, L. N. Owen, J. Chem. Soc., 1949, 315. j
937] N. K. Matheson, S. J. Angy a], J. Chem. Soc., 1952, 1 £33. *
938] A. B. Foster, W. G. Overcnd, J. Chem. Soc., 1951, 3452. ‘
939] P. Borrevang, Acta Chem. Scand., 9, 587 (1955). ]
940] P. Tannhauser, R. J. Pratt, E. V. Jensen, .1. Am. Chem. Soc., •,
78, 2658 (1956). ?
[941 ] F. G. Bordwcll, В. M. Pitt, M. Knell, J. Am. Chein. Soc., 73 5004 "1
(1951). 1
[942 ] L. H, Sommer, H. D. Blankma n, P. C. Miller, J. Am. Chem. ’
Soc., 76. 803 (1$>4). t-
J1I. ЗАМЕНА ГЛДРОКСПЛЬП., АЛКОКСИЛЬН. II КАРБОННЛЬН. ГРУПП HA Hal 221
2. Замена гидроксила на галоген в полуацеталях
(синтез а-галогенэфиров по Генри)
Для получения а-галогеналкилалкиловых эфиров (ср. стр. 159) чаще всего
используют синтез Генри, по которому н эквимолекулярную смесь альдегида и спирта
при охлаждении пропускают сухой НС1:
B'CHO + ROH+HCI R'GHCIOR4-n2O
Формально этот синтез можно рассматривать как обмен ОН-группы па галоген
в полуацеталях.
Основными побочными продуктами, образующимися прежде всего при
недостаточном охлаждении, являются плохо отделяющиеся ацетали и в некото-
рых случаях темная смола — продукт отщепления НС1 и полимеризации. Чтобы
избежать перемешивания цвух образующихся жидких фаз, приводящего к обра-
зованию темноокрашенных продуктов, НС1 пропускают в реакционную массу
медленно. Если плотности обеих фаз почти одинаковы, их смешение и обра-
зование эмульсии можно все же предотвратить добавлением связывающих воду
солей.
2-Фенил-1-хлорэтилэтиловый эфир С8Н6СН2СНС1ОС2Пб [943]. В охла-
жденную льдом смесь 32,5 г фенилацетальдегида, 12,5 г этилового спирта и 30 е
безводного Na2S0d при перемешивании быстро пропускают 11,5 г сухого НС1.
Жидкий продукт декантируют, добавляют к нему 16 г СаС12 и для удаления НС1
выдерживают 45 мин при 20 мм рт. ст.; выход неочищенного продукта 94%
от теоретического.
Взаимодействием 30 г параформальдегида со смесью 1 моль спирта и 1 -чоль
НВг при охлаждении получают соответствующие сс-бромметилалкииовые эфиры;
добавление 1 моль СаС12 повышает выход продукта [944].
По синтезу Гепри можно, в частности, получить монохлордиметмловый эфир
из формалина [945, 521], иараформальдеглда [946] или триоксана 1947]; а-хлор-
этилэтиловый эфир из паральдегида [948, 526], а также а-бром- [944, 949] и а-иодал-
киловые эфиры [945].
При галогенировании ненасыщенных альдегидов одновременно происходив
присоединение галогеноводорода; например, из акролеина, Метилового спирта и HCJ
получается а,р-дихлорпропилметиловый эфир [950, 95[].
Взаимодействие алифатических альдегидов с галогеноводородом в описанных
выше условиях в отсутствие спиртов приводит к образованию бис-(а-галогепалкило-
вых) эфиров:
2RCIIO + 2HC1 —► (ВСНС1)2О+Н2О
Таким способом получают бис-а-хлорэтиловый эфир [952, 953] и бис-а-гало-
генмотиловые эфиры [954].
[943 ] С. D. Н u г d, Н. L. W ₽ h г m е i s t е г, J. Am. Chem. Soc., 71, 4007
(1949).
[944 ] Н. W. Lucien. G. Т. Mason. J. Am, Chem. Soc., 71, 258 (1949).
1945] L. Henry, Ber., 26 R 933 (1893); Bull. Acad. roy. Belg. [3], 25, 439.
1946] E. Wedekind, Ber., 36, 1384 (1903).
[947] J. F. Walker, A. F. Chadwick, Ind. Eng. Chem., 39, 974 (1947).
948] I), fiaulhie r, Ann. Chim. [8], 16, 289 (1909); C. r., 143, 831 (1906).
949] G. M. Blair, H. R. Henze, J. Am. Churn. Soc., 54, 399 (1932).
,950] W. Duliere, Bull. Soc. chim. France [4], 33, 1647 (1923).
951] R. V о e t, Bull. Soc. chim. France [4], 41, 1308 (1927).
952] A. Gcuther, R. G a r t m e 11, Ann., 112, 1 (1859).
953] A. Geather H. La at sch, Ann., 218, 13 (1883).
951] W. Tisrhl<cl<cjib), !3, 2286 (188.3); 20 Л, 70! (1887).
222
ОБРАЗОВАНИЕ СВЯЗИ G —Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Бнс-хлорметиловый эфир можно получать также, приливая по каплям в те-
чение 5,5 ч при температуре ниже 10° С (охлаждение!) 452 мл хлорсульфоновой
кислоты (6,9 .коль) к смеси 168 мл концентрированной НС1 и 240 г параформаль-
дегида (около 8 моль СН2О) [955].
Из параформальдсгида, концентрированной серной кислоты и бромида натрия
получают бис-бромметнловый эфир [956].
Для получения бис-а.р-дибромэтилового эфира на 70 г паральдегида дей-
ствуют 320 г брома при температуре от —5 до —10° С, затем медленно нагревают сна-
чала до 15, а через 15 мин до 20° С, выливают реакционную массу на лед и нейтрали-
зуют ее содой [957]. В. этом случае необходимый для реакции бромистый водород
образуется во время реакции.
3. Замена ацнлоксигруппы на галоген
(получение галогензамещенных углеводородов)
Замещение ацилированных гидроксильных групп в полуацеталях, спиртах
или алифатических оксикарбоновых кислотах на 1алоген можно осуществить в основ-
ном в мягких условиях. Эта реакция имеет особенно большое значение в химии угле-
водов, а именно для получения очень реакционноспособных ацетогалогеноз, приме-
няемых для синтеза многочисленных О- и N-гликозидов. Гликозидные а цп в ок си гру ттп м
замещаются очень легко, в то время как остальные ацилоксигруппы не затрагиваются.
В большинстве способов образуется более стабильная а-галогеноза, независимо от
того, исходят ли из а- или ^-углевода [реакция (а)].
Оас Н Оэс Н
н СН,Оас Н ' СНрас
Ацетохлоруглеводы (ацетилгликозллхлориды) получают [958] взаимодействием аци-
лированных углеводов — с ацетилхлоридом и ZnQ2, с РС15 и А1С13 [959] или
только с Л1С1з [960], с TiCl4 в спиртовом безводном хлороформе [961] и с а.а-дихлор-
метилметиловым эфиром [962].
Особенно простым способом обмена, протекающего по уравнению (б), является
действие сухого НС1 на раствор или суспензию ацетата углевода.
Ацетохлор-р-Р-глюкоза [963]. В смесь 20 г пента-О-ацетил-В-глюкоэы
и 250 мл абсолютного эфира при 5° С в течение 40 мин пропускают при переме-
шивании 50 г НС1; при этом основное количество углевода переходит в раствор.
Смесь оставляют в закрытом сосуде на льду на двое суток. Получившийся про-
зрачный раствор упаривают в вакууме, причем выпадает 65% неочищенного.
[955] S. R. Вис, Org, Synth., 36, 1 (1956).
[956] Н. Stephen. W. F. Short, G. Cladding, J. Chem. Soc., 1920, 5It.
[957] H. Hibbert, S. Z. Perry K. A. Taylor, J. Am. Chem. Soc., 51,
1553 (1929).
1958] D. H. Brauns, J. Am. Chem. Soc., 46, 1484 (1924); 47, 1280 (1925).
[959] F. v. Ari t, Mh. Chem., 22, 144 (1901); Z. П. Skraup, R. Кге m а и в,
Mh. Chem., 22, 375 (1901); E. M. M о n t g о m e г у, C. S. Hudson, J. Am
Chem. Soc., 64, 247 (1942).
KW. К о ry t n i k, J. A. M i 11 s, J. Chem.. Soc., 1959, 636.
E. Pacsu, Ber., 61, 1508 (1928).
[962] H. Grop, I. Farkas, Chem. Ber., 93, 95 (I960).
[963] J. J. Fox, I, Goodma n, J. Am. Chem. Soc., 73, 3257 (1951).
Ш. ЗАМЕНА ГИДРОКСИЛЫ!., АЛКОКСИЛЬН. И КАРБОИПЛЬН. ГРУПП HA Hal 223
продукта; после перекристаллизации из сухого эфира т. пл. 98—99° С; [а]^{ —
12° С. Ацетохлор-p-©- глюкозу можно получить также, пропуская сухой
НС1 в смесь 100 мл РС13 и 10 г пента-О-ацетил-р-©-глюкоаы при '90—100° С
(5ч). Аналогично получают 0-ацетохлоргалактозу [964].
В условиях, указанных в приведенном примере, в противоположность другим
методам [958, 959, 961, 962] не происходит превращения в a-форму. Раньше нестабиль-
ную ацетохлор-р-©-глюкозу получали из ацетобром-а-©-глюкозы галогенным обме-
ном со свежеосаждеппым AgX или воздействием жидкого ПС1 на пента-О-ацетил-а-О-
глюкозу [965].
Бензохлор-а-©-глюкоза образуется [966] с 77%-ным выходом от теоретического
при взаимодействии в течение 4 ч пентабензоил-р-©-глюкозы с 1 моль TiCI4 в СНС13
при 60° С.
Синтез ацетобром-а-©-глюкозы через пента-О-ацетил-Р-©-глюкозу (которую по-
лучают при действии уксусного ангидрида и ацетата натрия на ©-глюкозу) давно
известен [967, 968]; более простой метод ее получения ем. 1969].
Ацетобром-а -©-глюкоза. Ацетилируют 0,33 моль моногидрата ©-глю-
козы 280 мл уксусного апгидрида с тремя каплями концентрированной HjSO4.
Затем отгоняют 200 мл смеси уксусного ангидрида и уксусной кислоты,
прибавляют 60 мл свежего уксусного ангидрида и при охлаждении льдом про-
пускают 140—160 г НВг. Смесь оставляют на ночь, а затем при температуре
бани не выше 60° С при пониженном давлении и под конец в вакууме, создавае-
мом масляпым насосом (ниже 5 мм рт. ст.), отгоняют НВг, уксусный ангидрид
и уксусную кислоту. Остаток перекристаллизовывают из диизопропиловог’о
эфира.
Обычно для получения бромпроизводных ацетаты углеводов подвергает в те-
чение 12—24 ч при 0°.С взаимодействию с насыщенной НВг ледяной уксусной кисло-
той, добавляют ледяную воду и экстрагируют'продукт реакции СНС]8. По другому,
широко применяемому способу ацетаты углеводов получают взаимодействием'угле-
водов с уксусным ангидридом и 70%-ной НС104 (0,5 объсмн. %) при 30—40°'С; не-
обходимый для обменной реакции НВг получают, добавляя к полученной реакцион-
ной массе при охлаждении красный фосфор, бром и воду (90% от расчетного количе-
ства). Обработку ледяной водой н экстракцию СНС13 проводят, как обычно [970/971].
Реакционную массу можно непосредственно перерабатывать далее, например для по-
лучения три-О-ацетил-©-глюкаля [972]. Ниже приведен метод получения бромпроиз-
нодпых, исходя из ©-глюкозы или из пента-О~ацетил-6-©-глгокозы; он может бьиъ ре-
комендован в тех случаях, когда отсутствует сухой НВг.
Ацетобром-а-©-глюкоза [973]. При охлаждении к суспензии 30 г крас-
ного фосфора в .300 мл ледяной уксусной кислоты приливают по каплям 180 г
брома п затем фильтруют массу через стеклянную вату или стеклянный фильтр.
[964] .1. d е Pascual Teresa. F. G а г г i d о Е я р i п о s a, An, Real
soc. csp. fis. quim., Ser. B., 52, 347 (1956); C., 1958, 8643.
[965] H. H. 8 c h 1 n b a c h, P. Stadler, I. Wolf, Ber., 61, 287 (1928).
1966] R. K. N e ss, H. G. Fie tcher, C. S. II n d s о n, J. Am. Chem. Soc.,
72, 2200 (1950); 73, 296 (1951).
1967] E. Fischer, Ber., 49, 584 (1916).
[968] L. Gattermenn, Die Praxis des organischen Chemikers, 35 Aufl., W. de
Gruyter u. Co., Berlin, 1953, S. 340.
[969] С. E. Redemann, C. Niemann Org, Synth., 22, 1 (1942); Coll. v. 3,
11 (1955).
[970] M. В a rcza i-M a rt os, F. Korosy. Nature (London), 165, 369 (1950).
[9V1] В. Helferich et al., Chem. Ber., 86, 873 (1953); 87, 1489 (1954).
[972] В. H e I f e r i c h, E. N. Mnl cahy, H. Z i e g I e r, Chem. Ber., 87, 234
(1954).
[973] P. G. S chenr рг, F. Smith, .1. Am. Chem. Soc., 76. 3224 (1954).
224
ОБРАЗОВАНИЕ СВЯЗИ
Hal И РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
К 100 мл полученного раствора прп встряхивании прибавляют 10 а глюкозы,
поддерживая температуру на уровне 40° С пли ниже, и в заключение выдержи-
вают смесь 2 ч при комнатной температуре. Затем разбавляют 100 мл СНС13
и массу выливают при перемешивании в 200 мл смеси льда с водой. Отделяют
хлороформный слой, водный слой встряхивают с. СПС13. Объединенные хлоро-
формные растворы промывают водой, раствором NaHCO3 и после высушивания
пад СаС1„ отгоняют СНС13 в вакууме. Остаток растворяют п возможно меньшем
количестве эфира, из которого при сильном охлаждении выкристаллизовы-
вается ацетобром-D-i люкоза; т. пл. 87—89° С; выход 66% от теоретического.
По другому способу все количесгпо описанного выше реакционного раствора
и 216 г пента-О-ацетил-{1-1>-г.тюко.1ы выдерживают 2 ч при комнатной темпера-
туре, разбавляют 300 мл СНС13, выливают на смесь льда с водой (800 мл). обра-
батывают, как указано выше, и получают ацетобром-2)-глюкозу с выходом 84?(>
от теоретического.
Ацетилированные аминосахара в основном взаимодействуют с галогеноводоро-
дом точно так же. как и обычные ацилированные углеводы, одпако образующиеся
1-галогенаминоуглеводы (ацилгликозамипилгалогениды) легко перегруппировываются.
1-а-Вром-М-ацетил-3,4,6-три-О-ацетил-Р-глюкозамии *, получаемый из ненга-О-аце-
тил-а-О-или пента-0-ацетил-р-Л-глюкозамина по реакции с НВг в ледяной уксусной
кислоте, очень легко переходит в присутствии воды в гидробромид 1,3,4,6-тетра-б-аце-
тИл-а-Л-i люкозамина [974J. N-Ацетил-3,4,6-грп-О-ацетил-а-Л-глюкозаминилхлорид,
образующийся с хорошим выходом из N-ацегИлглюкозамина и ацетнлхлорида, более
стабилен, однако перегруппировывается, подобно ацетобромуглеподам (ацетилгликозил-
бромидам), особенно в присутствии кислот [975]. Напротив, N-ацети,т-3,4,6-три-О-аце-
тял-а-2?-глюкозаминилфторид. получаемый как обычные ацстофторсахара (ацетнлглико-
зилфториды) [976], из певта-О-ацстил-о-или пента-0-ацотил-Р-2)-глюкозамина и жид-
кого HF, является довольно стабильным соединением. Этот аминосахар может быть даже
деэацетилирован ио Земплепу [677].
Лцстоиодсахара (анетилглюкознлиодиды) [978]. образующиеся под действием
раствора III в ледяной уксусной кислоте, могут быть получены и из бромсоединений
по реакции обмена с иодидом патрия в ацетоне прн комнатной температуре [979).
Обмен ацилированных гидроксильных групп на галоген используется не только
в химии углеводов, по и для синтеза дибромидов из диолов со смежными гидроксиль-
ными группами через их ацетаты [980, 981J.
При насыщении сухим НВг ^-бутиролактона, являющегося внутримолеку-
лярно ацилированной окепкарбоновой кислотой, образуется у-броммасляная кислота,
которую многочасовым кипячением с 3 моль метилового спирта, 300 мл СН2С12 и 3 мл
концентрированной H2SO4 переводят в метиловый эфир у-броммаедяной кислоты
с выходом 85—90% от теоретического ]983] (этерификация по Клинтону — Ласков-
скому [982]). Очень простой способ получения соответствующего этилового эфира
заключается в кипячении в течение 4 ч 0,5 моль у-бутпролактона с раствором 1 моль-
НВг в 500 мл абсолютною этилового спирта [984].
* Далее для соединений этого типа будет приводиться более точное название:
1^-ацетил-3,4,6-три-0-ацетил-а-0-1люкозаминилгалогепид. — Прим. ред.
[974] F. М i с h е е I, Н. Petersen, Chem. Вег., 92, 302 (1959).
[97,5] F, М i с h е е 1, F.-P. vau d е Kamp, Н. Ре ! ersen, Chem. Вег.»
90, 524 (1957). .
[976] D. II. Brauns. .1. An. Chem. Soc., 45, 833 (1923).
[977] F. Mic h о e 1, II. \V u 1 11, Chem, Ber., 89, 1,526 (1956).
[978] E. Fischer, Fl. Fischer, Ber., 43, 2535 (1910).
[979] В, He I lerich, R. Gootz, Ber., 62, 2791 (1929).
[980] II. J. I. u cas, M. J. Sc hla 11 e r, R. C. J о n e s, J. Am. Chem. Soc.»
03, 22 (1941).
[981] W. G. Young, Z, Jasaitis, L. Levaua s, J. Am. Chem. Soc.. 59
403 (1937).
[982] R. O. Clinton, S, C. Laskowski, J, Am. Chem, Soc., 70, 3135 (1948).
[983] J. F, Tinker J. Org. Chem., 16, 1418 (1951).
[984J W. A. Hcckho w, D. S. Tafbel 1, I. Am. Chew. Soc., 74, 4961 (1952).
Ш ЗАМНИ \ ГЛДРОПСПЛЬН,, АЛКОЬСЛЛЬН. И КАРБОНИЛЬН. ГРУПП НА Hal 225.
Вместо рассматривавшейся до сих пор сложноэфнрцой группировки ROC(=O)R'
для обмена ацил оксигруппы на галогеп можно использовать также диалкилсульфатьь
ROSO2(OR). При атом наряду с органическим галогенидом образуется соответству-
ющая кислота IIOSO2(OR).
Этот способ имеет практическое значение для синтеза метилиодида, который
получают непрерывной от гонкой при введении по каплям 630 г диметилсульфата
в перемешиваемую смесь 800 г KI, 60 г СаСО3 и 430 мл воды при 60—65° G.
Метилиодид кипит при 41—43° С; более подробные указания см. [985, 986].
4. Замена С—0-связи связью С—Hal
при расщеплении простых эфиров и ацеталей
Расщепление простых эфиров с помощью галогеноводородов, хлорангидридон.
кислот, Р0Х3 и РХ8, а также А1Х3 является препаративно важным способом для полу-
чения не только гидроксилсодсржащихсоединенпй, но и галогенсодержащих соединений
из циклических эфиров и арилалкиловых эфиров, причем пэ последних образуются
алкилгалогеннды. Из галогеноводородов наиболее активным является III, менее
активен НВг, еще менее активен HCI. Теоретические сведения и многочисленные ли-
тературные данные о расщеплении эфиров см. [987]. Методы получения ш-галогенал-
канолов или а,й-дИ1алогеналканов расщеплением циклических эфиров описаны на
следующих примерах:
Br(CHs)4Br ____
I | ( СП.СОС!^ CHsCOOfCIhJjCl
С1(СН2)4С1 <32-1
4-Хлор-н-бутплацетат и 4-хлорбутанол-1. В течение 90 мин кипятят
с обратным холодильником без доступа влаги 65 г ацетилхлорида, 50 г тетра-
гндрофурана (т. кип. 66° С) и 0,01 г ZnCl3. Перегонкой реакционной массы по-
лучают 4-хлорбутилацетат (т. кии. 78—79° С при 15 мм- рт. ст., 90—91° С
при 20 мм. рт. ст.); выход 76% от теоретического. Без ZnCl2. с большим его-
количеством или с А1С13 выходы значительно меньше [988]. Переэтерификацией
метиловым спиртом в присутствии небольших количеств концентрированной
НС1 или п-толуолсульфокислоты получают 4-хлорбутанол-1 (т. кип. 64—65° С
при 3 мм рт. ст.); выход 80% от теоретического. При этом реакцию ведут при
температуре не выше 40° С, а к концу реакции не выше 50° С, так как иначе-
вповь образуется тетрагидрофуран.
Аналогичным способом из тетрагидропирана получают 5-х л ор-«-амил~
ацетат [989, 990] (выход 85% от теоретического); из 2,5-диметилтетра—
гидрофурана - 1-метил-4-х до р-н-амила цетат [988] (выход 77%).
При добавке 2,2 г ZnCl2 па 1 моль циклического эфира, по-видимому, дости-
гаются еще более высокие выходы [991].
4-Хлорбутанол-1 получают еще проще, пропуская НС1 в кипящий
тетрагидрофуран до повышения температуры кипения реакционной массы
до 103,5—105,5е С (около 5 ч). Реакционную массу перегоняют в вакуумц-
[985]
[986
[987
[988
Т989
[990
[991]
L. Gattermann, Die Praxis des organischen Chemikers, 35 Aufl.,
W. de Fruytor u. Co., Berlin, 1953, S. 88.
W. W. Hartman, Org. Synth., Coll. v. 2, 1943, p. 404.
R. L. Burwell jr., Chem. Rev., 54, 615 (1954).
J. B. Cloke, F. J. Pilgrim, J. Am. Chem. Soc., 61, 2667 (1939).
M. E. Synerholm, J. Am. Chem. Soc., 69, 2581 (1947).
D. E. Ames, R. E. Bowman, R. G. Mason, J. Chem. Soc.,.
1950, 176.
F. L. M. Pa 11 ison of al., J. Org. Chem., 21, 745 (1956).
15 Зака-, 1835.
226
ОБРАЗОВАНИЕ СВЯЗИ С —Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
(ли возможности при 15 ммрт. ст.), причем сначала улетучивается большое ко-
личество НС1; выход 4-хлорбутапола-1 54—57% от теоретического [992]. Если при
проведении этой реакции маленькими порциями добавляют безводный ZnCl2
и общей сложности 50% от общего веса, то получают 1,4-дихлорбутан; выход
59% от теоретического [993].
4,4'-Дпхлордибутиловый эфир. Из 1 моль тетрагидрофурана, 0,33 моль
РОС13, 10 мл концентрированной H2SO4 в результате экзотермической реакции
при 60 — 100° С получают эфир с выходом 70% от теоретического [994].
1,4-Дибромбутан. Для получения этого а, ы-дибромалкана пропускают
НВг в кипящий тетрагидрофуран [995], пока температура кипения
смеси не повысится до 150° С, или добавляют 1 моль тетрагидрсфурана к холод-
ной смеси 6 моль 50%-ной бромистоводородной кислоты и 50 мл концентриро-
ванной H2SO4, кипятят 3 ч с обратным холодильником и выделяют продукт
реакции перегонкой с водяным паром [996]. Особенно прост следующий метод
[997]. К раствору 500 г NaBr в 600 мл воды при интенсивном перемешивании
прибавляют 2 моль тетрагидрофурана и 750.ил H2SO4 с такой скоростью, чтобы
температура поддерживалась на уровне 70—72° С. После 8 ч нагревания на
> паровой бане раствор декантируют от солей, отделяют органический слой, а вод-
ный слой и соли промывают 500 мл бензола. Органический слой соединяют
с бензолом, промывают раствором Na2CO3 и NaHSO3, сушат над СаС12 и пере-
гоняют; т. кип. 194—196° С и 81° С при 15 мм рт. ст.\ выход 86% “ от тео-
ретического.
1 -Хлор-4-бромбутап. Это соединение (т. кип. 80—82° С при 30 мм рт. ст.)
лучпГе всего получать обменной реакцией между РВг3и неочищенным со-хлор-н-
бутиловым спиртом [998] [С1(СН2)4ОН] или^между красным фосфором, бромом
и этим спиртом [997]. Спирт готовят пропусканием НСЛ в тетрагидрофуран.
1-Хлор-4-бромбутан можно синтезировать также прямым способом,
для чего к смеси 1,2 моль NaBr, 1,3 моль NaCl, 400 мл воды и 1 моль тетрагидро-
фурана при перемешивании Приливают по каплям 400 мл концентрированной
H2SO4 с такой скоростью, чтобы температура не поднималась выше 70° С. При
этом наряду с 10% 1,4-дибромбутана образуется 44% 1-хлор-4гбрЬмбутаиа;
переработку реакционной массы см. [997].
1,5-Дибромпентан. Получают кипячением в течение 3 ч смеси 1,5 моль
48%-ной НВг, 74 г концентрированной H2SO4 и 0,25 моль тетрагидропирана;
подробные указания см. [999].
Расщепление простых эфиров HI используется обычно для освобождения фе-
нольных ОН-групп или для аналитических целей (определение метоксильных групп
по Цейзелю). Для получения иодидов эта реакция мало пригодна, так как при тре-
бующихся высоких температурах и длительности реакции от органичееннх иодидов от-
щепляется HI. Кроме того, HI действует как восстановитель. Можно избежать по-
бочных реакций, если вести процесс с KI и 95%-ной Н3РО4 (ср. также стр. 117, 210).
ROR + 2KI + 2H3PO4 —> 2В1 + 2К11гРО4Н-Н2О
Этот способ вполне оправдал себя при расщеплении алифатических, циклических
и арвлалифатических эфиров [201].
[992] D. S I a-r г, R. М. Hixon, J. Am. Chem. Soc., 56, 1596 (1934)- Org.
Synth., Coll. v. 2, 1943, p. 571.
[993] Sh. Fried, R. D. Kleene, J. Am. Chem. Soc., 63, 2691 (1941).
[994] K. Alexander, L. E. Schniepp, J. Am, Chem. Soc., 70, 1840
(1948).
[995] Sh. Fried, R. D, Kleene, J. Am. Chem. Soc., 62, 3258 (1940).
[996] Ch, L. Wilson, J. Chem. Soc., 1945, 48.
[997] А. К a I 11 s z у n e г, J. Org. Chem., 22, 834 (1957).
[998] M. S. Newman, J. H. Wotiz, J. Am. Chem. Soc., 71, 1294 (1949);
D. A. Shirley, Organic Intermediates, J. Wiley a. Sons, New York,
1951.
[999] D. W. Andrus, Org. Synth., 23, 67 (1943); Coll. v. 3, 692 (1955).
III. ЗАМЕНА ГПДРОКСИЛЬН., АЛКОКСИЛЬН. II КАРБОНИЛЬН. ГРУПП НА Hal 227
[ ,4-Дииодбутан [1000], В трохгорлой колбе емкостью 1 л к 65 г Р2О5
при медленном перемешивании прибавляют 135 мл (2 моль) 85%-ной HSPO4.
Дают смеси охладиться до комнатной температуры, прибавляют 2 жоль КГ
и, наконец, 0,5 жаль тетрагидрофурапа. Массу нагревают с обратным холодиль-
ником 3 ч при перемешивании и после охлаждения до комнатной температуры
смешивают со 150 мл воды и 250 мл эфира. Эфирный слой промывают раствором
Na2S20s и насыщенным раствором NaCI и сушат с помощью Na2SO4. После от-
гонки эфира и перегонки остатка в вакууме получают 1,4-дииодбутан; выход
92—96% от теоретического; т. кип. 108—110° С (10 дл рт. ст.).
Аналогичным способом из ди-н-бутилового эфира получают н - б у т и л-
и о д^и д (выход 81%), а из диизопропилового эфира — изопропилиодид
(выход 90% от теоретического).
а,<о-Дигалогеналканы с длинной углеродной цепью получают по Брауну [1001].
из соответствующих эфиров фенолов. Эфиры фенолов легко синтезировать по способу
В горца из (о-галогеналкилариловых эфиров:
Na;
2Х(СНа)„Х 2ArON*» 2Х(СНа),ОАг -5®™» ЛгО(СН-г)г,ОЛг
а,(о-Дифсноксиалканы можно синтезировать также-следующим способом [1003],.
ArO(CH2)nX-f-NaC=CH —> АгО(СН2)„С=СН
N8NH»;
Лк)(С||:)„С=СИ + АгО(СП2)„Х —
— АЮ(СНг),С=С(СН,)„ОЛг — * АгО(СН2)„„2ОАг
Для расщепления эфиры кипятят 24—48 ч в колбе с обратным холодильником
или в запаянной трубке при 100—180° С с 48%-вой НВг или с трехкратным теоре-
тическим количеством 56,7%-ной кипящей при постоянной температуре HI.
ArOfCHjJs.OAr НЮ Н1)* Х(СН2)2„Х + 2ЛгОН
где Х=Вг или I.
С увеличением длины цепи эфира снижается его способность к расщеплению..
Нуклеофильность эфиров фенолов, играющая решающую роль па первой стадии
расщепления эфира, заключающейся в присоединении протона к эфирному кислороду,
ослаблена мезомерией. Следовательно, необходимо предотвратить возможность мезо- -
мерии, например, гидрированием дифениловых эфиров [1002], применением соединений
с Аг — и-СНоОСдН4 (гваякол вместо фенола), или использованием Эфиров, уиоторых
Аг_ лг-СН3ОС8Н4 (монометиловый эфир гидрохинона вместо фенола) [1003, 1004].
Так, если исходят из гваякола, то до п не более 40 получают очены.
хорошие выходы а,(о-дииодидов.
Если расщепление проводят кипячением в колбе с обратным холодиль--
пиком, сообщающимся с атмосферой, то после кипячения в течение нескольких,
часов смесь фильтруют с отсасыванием и отфильтрованный продукт еще раз на-
гревают с новой порцией HI. Неочищенный иоднд промывают водой, высушивают-
и экстрагируют в аппарате Сокслета петролейиым эфиром для очистки отгуми--
ноподобных продуктов разложения гидрохинона.
11000] И. Stone, Н. Schechter, Org. Synth., 30 , 33 (1950).
11001] J. V. Braun, Ber., 42, 4541 (1909).
[1002] I. v. В r a u n et al., Ber., 70, 973, 1598 (1937).
[1003] A. W. Nineham, J. Chem. Soc., 1953, 2601,
[1004] K. Ziegler, H. Weber, H. G. Gellert, Ber., 75, 1715 (1942)..'
228
ОБРАЗОВАНИЕ СВЯЗИ С —Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Особенно легко расщепляются эпокспсоединснля, вступающие уже при охла-
ждении льдом в экзотермическую реакцию с 48%-ной НВг или концентрированной
Н1 с образованием галогенгидринов. В щелочной среде легко вновь получаются
эпоксисоединения.
Особый интерес при расщеплении эпоксидов представляют стерические соотно-
шения, связанные с вальденовскпм обращением:
В\ R'
Вг-
R\/R?
С—ОН
I
Вг—С
R" /\r"'
R\ ,.R'
В г—С
I
С —ОН
R" -ZX'RW
Если R' — R"'= Н (i/uc-эпокспд) и R = R ", то образуются трео-галоген-
гидрнн (±-смесь); при R = Д’" Н (тракс-эпокспд) и R' = Н” получается зрилгро-
галогенуидрин. Эти соотношения изучены на примере 2,3-эпоксибутапа [1005]. Если
К =# R" или R' R", то одновременно появляется структурная изомерия, как это
было показано для 2,3-эпоксипентана [1006].
Препаративное расщепление эпоксмсоединений осуществляется очень
просто. При перемешивании и охлаждении льдом к 48%-пой НВг или 55%-ной
Ml постепенно прибавляют эпоксисоединение или его раствор в петролейном
эфире или СНС15. По окончании реакции массу осторожно нейтрализуют карбо-
патом щелочного металла и извлекают галогенгидрин эфиром. Иодгидрины на-
столько нестабильны, что, как правило, не выдерживают перегонит. В каче-
стве примера приведено получение 2-бромэтилового спирта [1007] пропусканием
3 моль окиси этилена при охлаждении смесью льда с поваренной солью в 4,25 моль
по меньшей мере 46%-ной НВг; выход спирта 87—92% or теоретического.
Присоединяя HF к эпоксисоединениям, получагот фторснирты *, например,
из окиси этилена и HF в эфире образуется фторэтпловый спирт [1008].
Взаимодействие ацеталей или полуацеталей с галогенангидрпдами ьислот (РС13,
РС%, РВг3, PBrs, ClSOsH, SOCI2, СП3СОС], CgHjCOCl) является общим методом
получения а-галогенэфиров:
R'CII(OR")OR R'CHCIOR-^-POClg-l R"C1
(ср. также стр. 159, 221).
Так синтезируют, в частности, ct-хлорэтиловый эфир [1009] из диэтилацеталя
ацетальдегида и РС1Э, а-хлорбензиловый эфир [1010] из диметилацеталя бензальде-
гида, ацетилхлорида и тионилхлорида. аналогично готовят бромалкиловые эфиры [1011].
а~Бромллкиловые эфиры получают также пропусканием при 20° С НВг в а-хлор-
алкиловые эфиры; а-иодалкиловые эфиры образуются по реакции хлоралкиловых
эфиров с Nal и ацетоне.
* Метод разработан II. Л. Кнунянцем [ДАТ! СССР, 55, 287 (1947)]. — Прим. ред.
[1005] S. W j n s I е I n Л. .1. Lucas, J. Am, Chem. Soc., 61, 1576 (1939).
[101)6] li. J. Lucas, M. J. Schlatte r, R. C. June $, J. Am. Chem. Soc.,
63, 22 (1941).
[1007] F. K. Thayer, C. 8. Marvel, G. S. Hicrs, Org. Synth., 6,
12 (1926).
[1008] О. В. Ки л д и nt e в а, И. II. Петро в, ЖОХ, 19, 95 (1950).
[1009] A. Gcuther, A. Bachmann, Ann., 218, 39 (1883).
[1010] F. Straus, H, Heinze, Ann., 493, 191 (1932).
[1011] A. К J i p ц 1, K’ Haas, Рог, 42, 2581 (1900).
III. ЗАМЕНА ГИДРОКСИЛЬН., АЛКОКСПЛЬН. И КАРБОПИЛЬН. ГРУПП НА Hal 229
5. Замена гидроксила на галоген в фенолах
и гетероциклических соединениях
Для замещения ОН-группы па галоген в фенолах или, что чаще, в гетероцикли-
ческих соединениях иногда применяют РХ6 и РОХ3, где X — С1 или Вг.
Выше 100° С равновесие РВг5 РВг3 Вг2 так сильно сдвинуто вправо,
что при употреблении РВг5 следует в качестве побочных реакций ожидать бро-
мирования и, возможно, дегидрирования.
Этот способ имеет главным образом препаративное значение для синтеза хлор-
замещенных N-гетероциклических соединений. Обычно гидрохлориды соответству-
ющих окси-М-гетероциклических соединений несколько часов нагревают с обратным
холодильником с избытком РОС13, затем в течение нескольких часов отгоняют непро-
реагировавшую РОС13, реакционную массу разлагают льдом, подщелачивают и экстра-
гируют хлорзамещенное гетероциклическое соединение эфиром. Так получают 2-хлор-
пирпмидины [1012], 2-хлор-4-метилпиримидин [1013] и 2-хлор-4,6-диметилпирими-
дины [1014], 4,б-дихлор-2-мсгилпиримидин 11015] и 2-хлор-4-метилхинолин [1016].
Процесс можно вести также с добавкой третичных аминов. 2,6,8-Трихлорпурин
образуется из 2,6-дихлор-8-оксипурина по реакции с РОС13 и диметиланилином [1017],
3-хлориндазол [1018] — из 0,2 моль сухого индазолопа, 0,2 моль сухого пиридина
и 0,3 моль РОС1Я.
Вместо РОС13 можно применять РС]5 или их смесь [1014, 1019]. 1,4-Дихлорфтал-
азин [1020] получают, например, нагреванием в течение 4 ч при 150° С 32 г фталил-
гидразида со 100 г РС15.
3,8-Дихлорпиридазип [1021, 1022]. В течение 5 ч кипятят с обратным
холодильником 25 г 3,6-диоксипиридазина (из дигидрохлорида гидразина и
малеинового ангидрида [1021]) с 300 мл РОС13. Поело отгонки в вакууме избытка
РОС]3 охлажденный остаток выливают па лед и смесь слабо подщелачивают по
лакмусу концентрированным раствором NH3. Выпавший продукт сушат в ва-
кууме, а из полученного фильтрата экстрагируют 3,6-дихлорпиридазпн хлоро-
формом. Из высушенного экстракта выделяют дополнительные количества неочи-
щенного продукта; общий выход 87% от теоретического. Дихлорпиридазин
очищают вакуумной перегонкой (т. кип. 89—91° С при 1,2 мм рт. ст.) или пере-
кристаллизацией из гексана (т. пл. 69—70° С).
2-Хлорпиримидин [1014]. Смесь 5,3 г гидрохлорпда 2-оксипиримидина,
15 г РС15 и 4 мл РОС13 кипятят в течсияе 45 мин при 140—150° С. После отгонки
части РОС]3 остаток выливают на лед, подщелачивают NaOH, добавляют NaCl
и извлекают эфиром 2-хлорпиримидин. Поело отгонки эфира продукт, перекри-
сталлизованный ил петролейного эфира, плавится при 65° С; выход 93% от тео-
ретического. '
[1012] J. W. Copenhaver R. F. Kleinschmidt, апгл. пат. 663302
(1951); С. А., 46, 10212 (1952).
[1013] Т. Matsukawa, В. О h t a J. I’harm. Soc. Japan, 69, 489 (1949); С. А.,
44. 3456 (1950).
[1014] Т. М a tsutawa, В. О b t a, J. Pharm. Soc. Japan, 69, 491 (1949); С. А.,
44, 3456 (1950).
[1015] I. В a d d i I е у, В. L у t h g о е D. М с N е i 1, A. R. Т о d d, J. Chem.
Soc., 1943, 383.
[Ю16] S. E. К rah] er, A. Burger, J. Am. Chem. Soc., 63, 2367 (1941).
[1017] J. D a v о 1 1, B. L у t h g о e, A. R. Todd, J. Chem. Soc., 1946, 833.
[1018] E. F. M. Stephenson, Org. Synth., 29, 54 (1949).
[1019] J. P. Wibaut, F. W. В г о e k ni a n, Rec. trav. c^im., 58, 885 (1939).
1.1020] II. D. K. Drew, H. H. Hatt, J. Chem. Soc., 1937, 16.
[1021] И. II, Mizzoni, 1’. E. Snoerri, J. Am. Chem. Soc,, 73, 1874
(1951).
[1022] E. A. Steck, R. P. Brun dage, L. T, Fletcher, J. Am.
Chem. Sue., 76, 3226 (1954).
ОБРАЗОВАНИЕ СВЯЗИ С —Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Особенно легко обменивается на хлор ОН-группа в пикриновой кислоте, кото-
рая, однако, реагирует с РОС13 только в форме пиридинпикрата.
Пнкрилхлорид [1023]. К горячему раствору 22,9 г лпкрвновой кислоты
в 200 мл 95%-пого этилового спирта прибавляют 7,9 г пиридина. При охлажде-
нии выделяется 30,1 г (98%) пиридинпикрата; т. пл. 166° С. Смесь 1 моль сухого
пиридинпикрата, 0,67 моль РОС13 и 250 мл бензола кипятят 15 мин с обратным
холодильником. Бензольный слой и выделившееся масло разделяют и каждый
в отдельности промывают горячей водой. После отгонки бензола из бензольного
слоя получают 73%, а из масла 25% пикрилхлорида (т. пл. 79—81° С).
Из 4-оксипиридипа под действием смеси РС1& и РОС1а получается 4-хлорпиридин
с выходом 75% от теоретического; с РБг6 при 110° С образуется только 47% 4-бромпири-
дина [1019]; побочным продуктом является 3,4,5-трибромпиридин.
Б общем способе замены ОН-группы на бром в гетероциклических соеди-
нениях применяют РВг5 или РОБг3.
Смешивают 1 -иолъ оксисоединения с 1,05 моль РБгй и, исключая доступ
влаги, нагревают 8—8 ч при 100° С. Затем реакционную массу разлагают льдом, •
подщелачивают NaOH или NaHCO3 и выделяют продукт реакции перегонкой /
с водяным паром или экстракцией подходящим растворителем. В качестве при-
мера см. синтез З-бром-6-метилпиридазина [1024].
Нагревают 1 моль оксисоединения несколько часов при 120—150° С -
с 1 моль или большим количеством РОВг3, иногда с добавкой толуола или кси- ,
лола, и после охлаждения разлагают льдом. В качестве примера можно озна-
комиться с получением 2,4,6-трибромпиримидина из барбитуровой кислоты
[1025] или 3,6-дибромпиридазина Ill из гидразида малеиновой кислоты I .
(3,6-диоксипиридазина) [1022]:
ill
В некоторых случаях РОБг3 может действовать, как дегидрирующий агент. )
Так, из 3,6-диоксигексагидропиридазина и РОБг3 при 70—80° С то$ке получают^
3,6-дибромпиридазин, причем добавка брома повышает выход [1026]. ,
Пригодный для препаративных целей]РВг6 готовят, приливая по каплям^
1 моль РВга при интенсивном перемешиванииt охлаждении и защите от влаги ]
к раствору 1 моль брома примерно в десятикратном по объему количестве Петра-j
леиного эфира (т. кип. 30—60° С) [1027]. .
[1023] R. Во усг, Е. V. Spencer, G. F. W г I g h t, Canad. J. Research,?;
24 В, 200 (1946); С. A., 41, 108 (1947).
[1024] C, Grund ma nn, Chem. Ber., 81, 7 (1948).
[1025] D. R. V. Golding, A. E. Senear, J. Org. Chem., 12, 293 (1947). ’
[1026] C. Pedrali, A. Mantegani, J. Org. Chem., 23, 778 (1958).
[1027] С. E. Kaslofr, M. M. Marsh, J. Org. Chem., 12, 456 (1947).
III. ЗАМЕНА ГИДРОКСИЛЫ!., АЛКОКСПЛЬН. И КАРБОНИЛЬН. ГРУПП НА Hal 031
Бромокись фосфора РОВг3 (т. пл, 56 С, т. кип. 193° С) получают следу-
ющими способами |1028, 1029]:
ЗРВг5 + Р2О5 5РОВг3
РВг5+НСООН —* РОВг3 —2НВг-^СО
Для обмена ОН-гр" ипы фенолов или гетероциклических соединений на хлор,
бром или иод может быть и: яменсн также трсфенилфосфитный метод (стр. 216). По-
дробности см. [1030J.
(ArO)8PXi + Ar'uU —* Аг'Х (АгО)дРО 4-НХ
6, Замена гидроксила в карбоксильной группе на галоген
(получение галогенангидрндов карбоновых кислот)
Замена ОН в карбоксильной группе на фтор
Фторангидриды карбоновых кислот не имеют большого препаративного зна-
чения, так как проводимые с ними реакции могут быть осуществлены проще с анало-
гично действующими, но более доступными хлорапгидридами. Фторангидриды кар-
боновых кислот очень устойчивы, даже к гидролизу. Формилфторвд устойчив при ком-
натной температуре в течение нескольких часов, после чего происходит разложение
на HF и СО. О получениифторангидридов из хлорангидридов кислот и KF упомина-
лось на стр. 192. При постепенном нагревании ангидрида масляной кислоты с KHF,
до 180° С из реакционной массы непрерывно отгоняется бутирилфторид; т. кип. 67° С
(767 мм рт. ст.)-, аналогично из ангидрида бензойной кислоты при 190—240° С полу-
чается бензоилфторид; т. кип. 155—156° С, Вместо KHF2 можно применять также KF;
выходы достигают 80—90% от теоретического [1031].
При работе по этому способу приходится предварительно готовить ангидриды
кислот. По другому способу [1032] соответствующие фторангидриды можно получать
нагреванием низших жирных кислот Сх—Сд, а также моно-и трихлоруксусных кислот
с бензоилфторидом, при непрерывной отгонке продуктов реакции из равновесной реак-
ционной смеси (ср. стр. 237)
RCOOH-|-CeH5COF
RCOF-|-CeH6COOH
Эту реакцию можно комбинировать с реакцией обмени галогена под действием
KF или КНР2, нагревая смесь карбоновой кислоты, бензоилфторида и фторида к$-
лия [768] или сухого бифторида калия [781].
Замена ОН в карбоксильной группе на хлор, бром или иод
, при помощи неорганических галогенангидридов
Существует несколько вполне пригодных методов получения остальных галогенан-
гидридов карбоновых кислот. Так как наиболее часто употребляются хлорангидриды
карбоновых кислот, то основное внимание будет уделено способам их получения. Как
правило, ОП-группу карбоксильной группы обменивают на хлор с помощью неорга-
нических хлорангидридов. В большинстве случаев не весь хлор, вводимый в форме
[1028] Л. S. Booth, С. G. Secgmiller, Inorg. Synth., 2, 151 (1946).
[1029J С. Grundmann, Chem, Ber., 81, 7 (1948).
[1030] D. G. Coe, H. N. R у d о n, B. L. T о n g e, J. Chem. Soc., 1957, 323.
[1031] А. И. M а ше нце в, ЖОХ, 15 , 915 (1945).
[1032] А. И. Машенцев, ЖОХ. 16, 203 (1946).
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
неорганического или органического хлорангидрида кислоты, используется в реакции
обмена, часть его выделяется в виде НС1 в результате вторичных реакций.
ВСООН + РС15—> RCoCl + POCla-l-HCl (1)
3RCOOH 2РС13 —> 3RCOC1 + Р2О3 + ЗНС1 (2)
RC.00II-J-S0CI3 —> RCOC1 + SO2 + HC1 (3)
2ВСООМа + РООз—> 2RC0Cl-|-NaPO3+NaCI (4>
Из неорганически л хлорангидридов для этой реакции главным образом при-
меняют РС]а, РС13 и SOCI2: хлорокись фосфора РОС]3 реагирует только с солями кар-
боновых кислот (реакция 4). Под действием SiCI4 при 50° С можно из изомасляной
кислоты получить соответствующий хлорангидрид с 85%-ным выходом от теоре-
тического [1033].
Пентахлорид фосфора является энергичным хлорирующим агентом. Он приме-
ним во всех случаях, когда температура кипепия образующегося хлорангидрида
ниже или выше температуры кипения выделяющейся хлорокиси фосфора (т. кип.
105,4° Си 45° С при 85 мм рт. ст.). Так как в большинстве случаев реакция (1) сильно
экзотермичпа. то иногда применяют разбавление, напрпмер, бензолом, ПАтрпляйным
эфиром, CHClg или CS2.
Хлорангидриды карбоновых кислот (по реакции с РС15). Смесь 1 моль
карбоновой кислоты и 1,05 моль РС1Д в сухом бензоле нагревают 3—4 ч при
80° С до прекращения выделения IIC1 и затем бензол и РОС]3 отгоняют в ва-
кууме. Остаток РОС13 можно удалить добавлением сухого толуола и повторной
отгонкой. Многие хлорангидриды плохо растворяются в петролейном эфире,
в то время как РОС13 хорошо растворима; поэтому, применяя разбавление пет-
ролейншм эфиром, можно разделить смесь неочищенного хлорангидрида с хлор-
окисью фосфора /1034]. Мри проведении реакции карбоновой кислоты с PClfr
в петролойпом эфире образующийся хлорангидрид выкристаллизовывается из
реакционной массы.
Польшей частью просто смешивают при охлаждении льдом 1 моль кислоты
с 1—1,1 моль РС1}, иногда слегка нагревают па водяной бане для инициирования
реакции (начало выделения НС1) и продолжают нагревать, исключая доступ
влаги, до окончания реакции (около 30 мин) или ведут реакцию в течение не-
скольких часов при комнатной температуре. Если хлорангидрид кипит при более-
высокой температуре, чем Р0С13, последнюю отюпяют чаще всего при понижен-
ном давлении, а хлорангидрид очищают перегонкой в вакууме [(см., например,
сиптез м-нитробензонлхлорпда [1035] и хлорангидрида итаконовой кислоты
[1036] CH2=-C(COCI)CII8COCI)J. Если хлорангидрид кипит при более низкой
температуре, чем Р0С13, то его отгоняют из смеси на колонке. Так получают
оксалилхлорид /1037].
В некоторых случаях в качестве растворителя для проведения реакции
с РС15 пригодна также РОС13. Этим пользуются, например, при получении
беташтхлорида из гидрохлорида бетаипа [1038].
Выход хлорангидридов зависит от качества примененного РС1}. Рекомен-
дуется удалять возможные примеси РС13 и РОС13 патреванием РС15 в вакууме-
11039] или, что лучше, специально готовить PCIS в лаборатории. Ниже приведен
метод получения РС]5. Сухой хлор пропускают над поверхностью жидкого-
РС13 примерно до 50%-ного привеса. Прп этом массу периодически переме-
[1033] И. II. Назаров, И. Л. К о т л я р е в с к а я, ЖОХ, 20, 1441 (1950)..
[1034] Н. Staudinger, Вег., 4J, 356 (1908).
[1035] R. Adams, R.L. Jenkins, Ore. Synth,, 3 75 (1923).
[1036] H. Feuer, S. M. Pier, Org. Synth., 33, 41 (1953).
[1037] O. Wallach, Ber., 8, 300 (1875).
[1038] P. A. P 1 u t t n e г, M. Geiger, Helv. chim. acta, 28, 1362 (1945).
[1039] E. () t t. Org. Sylith., Coll, v. 2, 1943. p. 528.
in. ЗАМЕНА ГИДРОКСПЛЬН., АЛКОКСИЛЬН. И КАРБОППЛЬН. ГРУПП ИА Hal 233
NH.
пшвают, чтобы подводящая газ трубка не забивалась РС15. К концу реакции
РС18 затвердевает [1035].
Из аминокарбоновых кислот или их гидрохлоридов по реакции с РС15 в ацетил-
хлориде[1040] пли, лучше, в СС14, в котором РС15 растворяется [1041], получают гидро-
хлориды хлорангидридов амипокарбоновых кислот. Эти вещества очень чувствительны
к влаге и поэтому реакцию проводят в герметичной аппаратуре. Продукт реакции
отсасывают через погружной пористый стеклянный фильтр, промывают подходящим
сухим растворителем (петролейный эфир, СС14) и отсасывают досуха.
Гидрохлорид хлорангидрида аминоуксусной кислоты (гидрохлорид гли-
цилхлорида [1041]. К суспензии 5 г глицина в 200 мл С11С]3 прибавляют 15 а
РС]5. Плотно закрытый реакционный сосуд энергично встряхивают в течение
10 ч при комнатной температуре. Затем продукт реакции отсасывают без доступа
влаги через пористый стеклянный фильтр и трижды промывают сухим СС14,
трижды сухим петролейным эфиром и один раз сухим эфиром. Аналогичным спо-
собом получают гидрохлорид алапилхлорнда; выход 90% от теоретического,
после перекристаллизации из ацетилхлорида выход 75—80%.
Чистые (для анализа) гидрохлориды хлорангидридов аминокарбоповых кислот
можно непосредственно получать пропусканием тщательно осушенного НС1 (колонна
с СаС12, две промывные склянки, одна с концентрированной H2SO4, другая с Р2О5
на стеклянной вате) в прозрачный раствор N-карбоксиангидрида аминокарбоновой
кислоты в диоксане до начала кристаллизации [1042].
R—СН—СО
К—СН—СООН C0Clt. mcaH I \0 нак диоксан > R-CH-COCl^^
JJN—СО NHa' I1C1
Раствор N-карбоксиангпдрида готовят, пропуская сухой, промытый концентри-
рованной H2SO4 фосген в энергично перемешиваемую суспензию аминокарбоновой
кислоты в диоксано при 40—50° С до полного пли почти полного растворения кислоты;
избыток фосгена отдувают сухим воздухом и отфильтровывают продукт реакции без
доступа влаги (ср. стр. 239).
На примере этой реакции видно, что иногда целесообразнее сначала получить
из кислоты ангидрид, а затем хлорангидрид. Как правило, ангидриды карбоновых
кислот довольно устойчивы к действию неорганических хлорангидридов; при действии
неорганических хлорангидридов, например SOC]2, в обычных условиях па соответ-
ствующую дикарбоновую кислоту — малеиновую, янтарную или фталевую, предпочти-
тельно образуются циклические ангидриды [1043]. Однако из фталевого ангидрида’
I по реакции с РС]6 при повышенных температурах с очень хорошим выходом полу-
чается еил<л<-а-фталоилхлорид П.
Реакция протекает, вероятно, через асилл-о-фталоилхлорид 111, который
при перегонке при атмосферпом давлении перегруппировывается в 2-фталоил-
хлорид 11. С другой стороны, под действием А1С18 соединение 11 перегруппиро-
вывается [1039] в соединение Ш (реакции 1).
При высоких температурах можно перевести циклические ангпдридгл в дихлор-
ангидриды дикарбоновых кислот также при помощи SOCI2 с добавкой ZnCl2
(реакция 2):
СО
,СОС1
А1С13
1. РС1$
2 SOG)t—ZnClg
со
',0
со
[Ю40] Е. Fischer, Вег., 38, 606, 2914 (1905); 39 , 544 (1906).
[1041] S. Levine, J. Am. Chem. Soc., 76, 1382 (1954).
[1042] M. Brenner, I. Photaki, Helv. chim. acta, 39, 1525 (1956).
[1043] JJ. Meyer, Mh. Chem., 22, 437 (1901).
234
ОБРАЗОВАНИЕ СВЯЗИ С —Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
силм<-о-Фталоилхлорид [1039] (из фталевого ангидрида и РС1Й). В кругло-
донной колбе емкостью 500 мл, снабженной обратным холодильником с хлор-
кальциевой трубкой, нагревают в течение 12ч при 150° С на масляной бане 1 моль
сублимированного фталевого ангидрида с 1,06 моль чистого РС15. Затем обрат-
ный холодильник заменяют на нисходящий и постепенно повышают температуру
бапи^до 250° С, причем отгоняется большая часть образовавшейся РОС13. Оста-
ющийся в колбе неочищенный о-фталоилхлорид перегоняют в вакууме; т, кип.
131 — 133° С (9—10 лл рт. ст.); выход 92% от теоретического. Препарат содер-
я?ит небольшое количество фталевого ангидрида. '
та.ч.ч-о-фталоилхлорид [1044] (из фталевого ангидрида и SOCIj.). В трех-
горлой колбе с обратным холодильником нагревают до 220° С при перемешивании
300 г фталевого ангидрида с 1,5 а безводного ZnCl2. Затем в течение 10 ч прили-
вают по каплям 241 з SOC12 с таким расчетом, чтобы температура не опускалась
ниже 220° С, и в заключенно перегоняют реакционную смесь при сильно пони-
женном давлении. При 119 —122° С (4,5 мм рт. ст.) отбирают 380 г дистиллята.
Из него при охлаждении выкристаллизовывается 22 г фталевого ангидрида,
который отфильтровывают. Фильтрат представляет собой фталоилхлорид 94%-
ной чистоты (определение по хлору), и следовательно, достаточно чистый для
разнообразного использования.
Дихлорангидрид янтарной кислоты [1045]. Получают аналогичным спо-
собом взаимодействием 20 г янтарного ангидрида, 1 г безводного ZnCl2 и 34 а
SOC12 при 120° С в течение 10'/; выход дихлорангидрпда 74% от теоретиче-
ского.
Для получения легколетучих хлорангидридов используется реакция ыавным '
образом с РС]8 (т. кип. 74,5е С). Обычно дают прореагировать 1,1 моль карбоновой
кислоты с 0,5 моль РС13 при комнатной температуре или при слабом нагревании до
прекращения выделения НС1 и затем непосредственно отгоняют хлорангидрид (см.,
например [1046], получение ацетилхлорида из уксусной кислоты и РС18). Можно также
по окончании реакции извлечь хлорангидрид из реакционной массы растворителем
(бензол, петролейный эфир, CS2) и непосредственно в растворе подвергнуть дальней-
шим превращениям.
Хлорангидрид фенилуксусной кислоты [1047]. В течение 1 ч нагревают
на паровой бане 0,5 моль фенилуксусной кислоты с 0,25 моль РС13. К еще теплой
смеси прибавляют 400 мл сухого бензола и отделяют декантацией бензольный
раствор хлорангидрида фенилуксусной кислоты от выделившейся фосфористой
кислоты. Если добавить этот раствор к 0,56 моль безводного А1С18, то по реакции
Фриделя — Крафтса образуется дезоксибензоин с выходом 82% от теоретиче-
ского.
Для получения хлорангидридов карбоновых кислот при помощи неорганических
хлорангидридов чаще всего применяют тионилхлорид [1048] (т. кип. 78,8° С). Его
преимущество заключается в том, что в качестве побочных продуктов образуются
только газообразные вещества (реакция 3, стр. 232) и что избыточный SOC12 может
быть легко отогнан из реакционной массы. Указания по очнетце SOC12 приведены
на стр. 215. На ход реакции с SOCla могут влиять [1049] такие примеси в техническом
SOC12, как SO3 и H2SO4.
[1044] L. P. К у г i d е s, J. Am. Chem. Soc., 59, 206 (1937).
[1045] P. R u g g ] i, A. Maeder, Helv. chim. acta, 26, 1476 (1943).
[1046] L. Gat termann, H. Wieland, Die Praxis des organischen Chemikers, ч
35 Aufl., Berlin, 1953, S. 111.
[1047] C. F. H. Allen, W. E. Barker, Org. Synth., Coll. v. 2, 1943,.
p. 156. B
[1048] H. Meyer, Mh. Chem., 22, 109, 415 (1901).
[1049] H. Meyer, R. Turnau, Ber., 42, 1163 (1909).
III. ЗАМЕНА ГИДРОКСИЛЬН., АЛКОКСИЛЬН. И КАРЕОИИЛЬН. ГРУПП НА Hal 235
При перегонке хлорангидрида фенилпропиоловой кислоты (т. кип.
105—107° С при 11 мм рт. ст.), полученного по реакции с техническим SOCI2,
дважды произошло разложение со взрывом. При применении для
синтеза SOCI2, очищенного от труднолетучих примесей однократной ректифика*
цией, перегонка неочищенных хлорангидридов фенилпропиоловой и коричной
кислот проходила без осложнений [1050]. Таким образом, чистый SOCi2 можно
использовать и для получения хлорангидридов ненасыщенных карбоновых
кислот.
Б отличие от РС1Б кипящий SOC12 не затрагивает карбонильные группировки
— СНО, —СО или —COOR в кето-, альдегиде- или карбалкоксикарбоновых кислотах.
Алкоксильные группы довольно устойчивы к действию РС]Б и SOCI2, в то время как
спиртовые, а также фенольные гидроксильные группы реагируют с этими хлорангид-
ридами [1051]. Лучше всего перед превращением карбоксильной группы оксикарбоно-
вых кислот в хлорангидридную защищать гидроксил ацетилированием, как, например,
прп синтезе хлорангидрида ацетилминдальной кислоты [1052].
При наличии определенного рода заместителей в молекуле оксикарбоно-
вой кислоты удается в мягких условиях провести прямую реакцию обмена с тио-
лилхлоридом. По реакции с тионилхлоридом при 50° С из 2,3,5,6-тетрабром-4-
оксибензойной кислоты почти количественно получается хлорангидрид, в то
время как при более высоких температурах происходит конденсация с образова-
нием высокомолекулярных продуктов [605].
Пиридин катализирует взаимодействие карбоновых кислот с тионилхлоридом.*
Добавление 1 моль пиридина на 1 моль тионилхлорида позволяет получать нестойкие
хлорангидриды в мягких условиях [1053—1055]. Добавка диметплформамида также
благоприятствует образованию хлорангидридов под действием тионилхлорида [1056].
Так как эта реакция протекает через хлорангидрид амида муравьиной кислоты, ее
описание приводится на стр. 240.
Тионилхлорид реагирует также с солями щелочных металлов карбоновых
кислот.
Хлорангидриды карбоновых кислот (получение с помощью SOC]2). При-
мерно к 1,2 моль SOC12 по каплям приливают при перемешивании 1 моль кар-'
боновой кпслоты, при необходимости охлаждая реакционную колбу проточной
водой. Можно применять больший избыток тионилхлорида, по это не приводит
к существенному увеличению выхода. К твердым кислотам приливают или при-
бавляют по каплям SOC12; для разбавления реакционной массы можно добавлять
бензол. Необходимо пользоваться эффективным обратным холодильником.
Реакционную смесь нагревают на водяной бане (50—80° С) до прекращения выде-
ления SO2 и НС], затем отгоняют избыток SOC12, чаще всего в вакууме, созда-
ваемом водоструйным насосом, и фракционируют неочищенный хлорангидрид
(см., например, синтезы бензоилхлорида [1057], н-бутирилхлорида [1058]
Применение пиридина является частным случаем катионного катализа, открытого
М. Я. Крафтом и сотр. [ДАН СССР, 88, 725 (1952); 109, 312 (1956)]. —
Прим. ред.
К. v. Auwers, Е. R i s s е, Вег., 64, 2220 (1931).
Н. М е у е г, Mh. Chem., 22, 429 (1901).
К. Thayer, Org. Synth., 4, 1 (1925).
P. Carre, D. Libermann, C. r., 199, 1422 (1934).
W. R. Brown, J. Chem. Soc.,1945, 601.
J. P. E. Human, J. A. M ills, Nature, 158, 877 (1946).
II. H. Bosshard, R. M о г у, M. Schmid, 11. Z о I I i n g e r, Helv.
chim. acta, 42, 1653 (1959).
L. Gattermann, H. Wieland, Die Praxis des organischen
Chemikers, 35 Aufl., Berlin, 1953, S. 112.
В. H e 1 f e г 1 c h, W. Schaefer, Org. Synth., 9, 32 (1929).
236
ОБРАЗОВАНИЕ СВЯЗИ С- Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
и изобутирплхлорида [1059]. Выходы хлорангидридов в большинстве случаев
достигают примерно 90% от теоретического.
Во многих случаях неочищенные хлорангидриды карбоновых кислот ис-
пользуют дальше без дополнительной очистки. Так, например, д и х л о р ан-
гидрид адипиновой кислоты получают нагреванием смеси 1 моль
адипиновой кислоты и 3 моль SOCI2 при 50—60° С до прекращения выделения
IlCI и 80g (4 ч). и после отгонки в вакууме избытка SOCIg неочищенный дихлоран-
гидрпд используют для синтеза 1,4-дибензоилбутана [1060].
Для получения хлорангидрида у-ф енил масляной кис-
лоты осторожно нагревают 0,2.коль у-фенилмасляной кислоты с 0,27 моль
чистого SOC]«, ведут реакцию 30 мин без подвода тепла, выдерживают 10 мин
па паровой бане п отгоняют непрореагировавший тионплхлорлд в вакууме,
создаваемом водоструйным касосом; продукт реакции извлекают сероуглеродом
н применяют в неочищенном виде для синтеза а-тетралона по реакции замыкания
кольца в присутствии хлорида алюминия [908].
Моноэфиры неполных хлорангидридов дикарбоновых кислот [1061].
Хлорангидрид [J-к арбо метокси п ропиополой кислоты
получают нагреванием в течение 1 ч при 40° С 2 моль мопометнлового эфира
янтарной кислоты с 2,4 моль SOCI2, оставляют на ночь при комнатной темпера-
туре, нагревают еще 2 ч при 40° С, отгоняют в вакууме избыток SOC12 и пере-
гоняют полученный [J-карбометоксипропионилхлорид (т. кип. 92—93° С при 18 мм
рт. ст.); выход 90—93% от теоретического. При сильном нагревании отщеп-
ляется метилхлорид и образуется ангидрид янтарной кислоты.
Аналогичным способом получают моноофиры неполных хлорангидридов
глутаровой, адипиновой и себациновой кислот, которые как можно быстрее
перегоняют при 4 мм рт. ст. или в более глубоком вакууме.
Мопоэтпловый эфир неполного хлорангидрида
щавелевой кислоты получают из калиевой соли моноатялового эфира
щавелевой кислоты. К 50 г соли, залитой слоем абсолютного эфира, при охла-
ждении льдом приливают по каплям 80 г SOC]2. Затем прекращают охлаждать,
выжидают, пока реакционная смесь пе нагреется до комнатной температуры, и
нагревают 15 ч с обратным холодильником на водяной бане. Выделившийся
KCI отфильтровывают, промывают осадок абсолютным эфиром и фракциони-
руют фильтрат. Отгоняющаяся при 125—138° С фракция состоит в основном из
эфира полухлорангидрида и может без дополнительной очистки применяться
в синтезах по Фриделю — Крафтсу. Выходы достигают 70% от теоретического.
Повторной обработкой предгона получают дополнительное количество эфира
оксалилхлорида.
Хлорангидриды карбоновых ннслот (получение с помощью SOCIg и пири-
дина) [1053—1055]. Карбоновую кислоту растворяют в десятикратном объеме
сухого растворителя (преимущественно эфир, а также бензол, СНС13, СС14)
И 1 моль сухого пиридина. Приливают по каплям при перемешивании при 0° С
точно 1 моль чистого SOC12, при этом выпадает гидрохлорид пиридина (за исклю-
чением случая, когда реакцию проводят в СПС13). Затем смесь выдерживают
1 ч при 12—20° С. Нестойкие хлорангидриды тотчас же подвергают дальнейшему^
взаимодействию, например, со смесью спирта (или вилла) и 1 моль пиридина/
Бромапгидриды карбоновых кислот имеют гораздо меньшее препаративное зна-
чение, чем хлорангидриды. Их можно получать из карбоновых кислот и РВг3(илн
красного фосфора и брома), а также из'хлорангидридов и сухого НВг.
Ацетилбромид. К 150 г ледяной уксусной кислоты прибавляют 10 г
Красного фосфора, постепенно приливают по каплям 400 г брома, после чего
кипятят смесь 1 ч с обратным холодильником и отгоняют образовавшийся аце-
тилбромид, кипящий при 76° С. Его получают также пропусканием при комнат-
[1059] R. Е. Kent, S, М. McElvain, Org. Synth., Coll. v. 3, 1955,
p. 490.
[1060] R. C. Fuson, J. T. Walker, Org. Synth., Coll. v. 2, 1943,
p. 169.
[Ю61] J. Cason, Org. Synth., Coll. v. 3, 1955, p. 170.
Ш. ЗАМЕНА ГИДРОКСИЛЬН., АЛКОКСИЛЬН. И КАРБОНИЛЬН ГРУПП НА На! 237
ной температуре 3 моль сухого НВг в ацетилхлорид с последующей перегонкой
готового продукта [1062].
Иодангидриды карбоновых кислот получают ио реакции ангидридов кислот
с красным фосфором и иодом (пли Р13), или по реакции хлорангидридов карбоновых
кислот с сухим иодистым водородом 11062] или с иодидом кальция [1063].
Замена ОН в карбоксильной группе на хлор и бром
при помощи галогенангидридов органических кислот
Для обмена гидроксила карбоксильной группы на галоген могут применяться'
органические галогенангидриды, преимущественно хлорангидриды кислот, причем
при непрерывной отгонке образующегося хлорангидрида равновесие сдвигается вправо
(реакция 1):
RCOOH-FR'COCl RCOCI-rR'COOH (1)
Если же отгонку не производить, го вслед за первичной обменной реакцией
протекают вторичные реакции с отщеплением НС] (реакции 2—6):
^\/сос1
RCOOH + I || ~Znci'
СО
/0
СО
(2)'
RCOOH-I-C1COCOC1 RCOCl-f-C0-(-CO2 (3)-
RCOOH-J-C1C0C1 ——RCOCH-CO2 (4)
RCOOII4-CfiH&CCl3 -^j-> RCOC]4-CeH6COCl (5>
RCOOH--C12CHOCH3 _hL-> RCOCI+НСООСНз (6)
Реакция 1 используется для синтеза легколетучих хлорангидридов карбоно*
вых кислот из 1 моль соответствующей кислоты и 12—2 моль бензоилхлоридя [1064,-
1065].
Хлорангидрид акриловой кислоты [1066]. Смесь 3 моль акриловой ки-
’ слоты, 6 моль бонзоилхлорида и 0,5 г гидрохинона перегоняют с умеренной ско-
ростью через колонку высотой 25 см. Фракцию, которая кипит до 85° С (основное
количество кипит при 60—70° С), собирают в охлаждаемый льдом приемник,
содержащий 0,5 г гидрохинона. Дистиллят (215—225 г) еще раз перегоняют
через такую же колонку, отбирая фракцию, кипящую и отгоняющуюся в интер-
вале 72—74° С (7*40 мм рт. ст.)\ выход 185—195 г. При употреблении неорга-
нических хлорангидридов получаются худшие выходы. Кипячением остатка от
перегонки в течение 2 ч с водой и неболыпим количеством серной кислоты реге-
нерируют бензойную кислоту с выходом 90%.
[1062] Н. Staudinger, Е. Anthes, Вег., 46, 1431 (1913).
[Ю63] II. Spindler, Ana., 231, 272 (1885).
[1064 II. С. Brown, J. Am. Chem. Soc., бб, 1325 (1938).
Г1065] J. Forrest, D. A. L i d d e 1, S. H. T n c k e r. J. Chem. Soc., 1946, 454,
[1066] G. II. S t e m p e 1 jr., R, P. Cross, R. P. Mariella, J. Am'.
Chem. Soc., 72, 2299 (1950).
238
ОБРАЗОВАНИЕ СВЯЗИ С —Hat В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Таким же способом синтезируют хлорангидриды кротоновой п трнхлор-
уксуспой кислот [1064].
Перегонкой смеси 1-14С-ацетата калия, бензойной кислоты и бензоплбро-
мида получают 1-14С-ацетилбромид с 75—90%-ным выходом от теоретического
[1067).
Приливая по каплям 1 моль карбоновой кислоты при 130—140е С к 1,1 моль
o-фталоилхлорида, получают по реакции 2 соответствующий хлорангидрид. Если он
кипит при температуре ниже 130—140° С, то его можно непрерывно отгонять через
колонку; выход 90%. С помощью o-фталоилхлорида при добавке небольшого коли-
чества ZnCl2 ангидриды кислот также переводятся в хлорангидриды;
СП—СО СОГ1 ч со
и \ ^\/ u СН-СОС1 ^\/ \
\)-н II —> II +i II ;о
аос^сн
Дихлорангидрид фумаровой кислоты [1068]. В колбу, снабженную почти
доходящим до дна термометром, колонкой высотой 30 см с нагревающей ру-
башкой и нисходящим холодильником, вносят 98 г малеинового ангидрида, -
230 г примерно 94%-ного о-фталоилхлорида, 2 г безводного ZnCl2 и нагревают
реакционную смесь в течение 2 ч на масляной бане при внутренней температуре •'
нс выше 130—135° С. Затем смесь охлаждают до 90—95° С и как можно быстрее
отгоняют в вакууме неочищенный дихлорангидрид фумаровой кислоты (т. кип.
60—85° С при 13—14 мм рт. ст.). Затем его еще pas медленно фракционируют ;
(т. кип. 62—64° С при 13 мм рт. ст.)', выход 82—95% от теоретического. Про-
дукт лучше хранить в запаянном сосуде.
По реакции с оксалилхлоридом (реакция 3), действующим более энергично, ;
чем о-фталоилхлорид, хлорангидриды получают из нестойких карбоновых кислот •
[1069] или их натриевых солей [1070] в бензоле уже при комнатной пли более низкой
температуре. Из ряда нитропроизводных бензойной кислоты образуются только л
смешанные ангидриды из 2 моль ароматической кислоты и 1 моль щавелевой кислоты •
[1071]. Если из карбоновой кислоты с основными группами в молекуле хотят по- 1]
лучить свободный хлорангидрид, а не его гидрохлорид, соль щелочного металла ',
карбоновой кислоты подвергают взаимодействию с оксалилхлоридом. ,
Хлорангидрид никотиновой кислоты [1072]. К суспензии 16,1 г высу- [
шейной, тонкоиэмельченной калиевой соли никотиновой кислоты в 75 мл сухого '
бензола при перемешивании и охлаждении в бане со льдом приливают по кап- •
лям 12,5 г оксалилхлорида в 25 мл сухого бензола. После 12—20 мин перемеши-
вания удаляют из бани нерастаявший лед, нагревают баню до комнатной темпе-
ратуры, затем в течение 30 мин смесь доводят до кипения и кипятят 30 мик. После -'
охлаждения отсасывают КС] без доступа влаги и после отгонки растворителя .
перегоняют фильтрат в вакууме, создаваемом водоструйным насосом; выход
85% от теоретического и более. Из натриевой соли хлорангидрид получается '^
с более низким выходом.
[1067
[1068
[1069
[1070
[1071
[1072
Н. S. Anker, J. Biol. Chem., 176, 1333 (1948).
L. P. Ky rides, Org. Synth., 20, 51 (1940); Coll. v. 3, 422 (1955).
J. Heer, K. Miescher, Helv. chim. acta, 29, 1071, 1895 (1946).
A. L. Wilds, С. H. Shunk, J. Am. Chem. Soc., 70, 2427 (1948).
R. Ada ms, L. H. Ulich, J, Am. Chem. Soc. 42. 599 (1920k
H. N. Wingfield [T W. R. Harlan, H. R. H a n m e r, J.
Chem. Soc., 75, 4C64 (1953).
Am.
ЗАМЕНА ГПДРОКСПДЬН., АЛКОКСИЛЬН. П КАРБОНИЛЫ! ГРУПП НА Hal 2.3$)
Аналогичным образом с помощью оксалплбромида из карбоновых кислот
о.иг, лучше, их натриевых солей получают бромангидриды [1071].
Оксалилбромид. Его получают, пропуская ПВг (8 моль НВг на 1 моль
оксалпдхлорпда) в 100 г очищенного оксалилхлорида (около 12 ч), и после 6 ч
выдержки выделяют продукт реакции фракционной перегонкой; т. кин.
102—103° С (720 мм рт. ст); выход 85% от теоретического. Окраску брома-
устраняют встряхиванием со ртутью [1062].
Хлорангидриды жирных кпслот могут быть получены также взаимодействиевг
фосгена с жирными кислотами (реакция 4). Тонкое распределение газа и быстрая'
подача С0С12 в реакционный сосуд, представляющий собой обогреваемый в серноки-
слотной бане цилиндр с впаянной в него пористой стеклянной пластинкой, повышает
выход. Лучше вести реакцию л интервале 140—160° С; выходы составляют 70—90%-
от теоретического [1073].
Бензотрихлорид и а.а-дихлорметилметиловый эфир (получение см. на стр. 160,
241), которые могут рассматриваться как хлорангидриды ортобензойной кислоты или-
мо.чометилового эфира ортомуравьиной кислоты, соответственно, реагируют с карбоно-
выми кислотами по реакции 5 или 6 (стр. 237). чаще всего в присутствии незначительных
количеств ZnC]2. Апгидриды кислот реагируют с этими веществами также с образова-
нием хлорангидридов, но без выделения НС1. Так как при работе с бензотрихлоридом
наряду с целевым хлорангидридом RCOC] неизменно образуется бензоилхлорид, реак-
цию .5 можно использовать только в том случае, когда температура кипения получа-
емого хлорангидрида достаточно сильно отличается от температуры кипения бензоил-
хлорида. По реакции с бензотрихлоридом получают, например, фторацетилхлорид
из фторуксусной кислоты [1074] или о-фталоилхлорид из фталевого акгидркда [1044].
Б то время как реакции с бензотрихлоридом обычно протекают при температурах
выше 100° С, а,а-дихлорметилметиловый эфир реагирует с карбоновыми кислотами
уже па холоду, Для достижения хороших выходов к реакционной смеси добавляют
0.01 -0,1 мол. % ZnCl2 и кратковременно нагревают при 70—100° С. Если исходят
из натриевых солей карбоновых кислот, приходится нагревать 2 ч при 90—100° С.
Хлорангидриды карбоновых кислот (получение из карбоновых кислот
и а, (х-дихлорметилметилового эфира С12СНОСН3) [1075]. Смешивают 1 моль кар-
боновой кислоты с 0,01 моль ZnCl2 и быстро приливают по каплям при пере-
мешивании 1,25-ноль а.а-дихлорметилметилглэого эфира. После того как прекра-
тится выделение НС1, массу нагревают около 1 ч при 70—100° С. При этом
отгопяются метиловый эфир муравьиной кислоты (т. кип. 30—32° С) и воз-
Shjjh избыток с^а-дихлорметилмотилового эфира (т. кип. 84—86,5° С);. •
ток подвергают фракционной перегонке,
Из тозил- и фталоиламинокарбоновых, кислот кратковременным нагреванием'
до’80—85° С с избытком а,а-дихлорметилметийового эфира с хорошим выходом полу-
чают соответствующие хлорангидриды. В отличие от них хлорангидриды, первона-
чально образующиеся из а-карббепзоксиаминокарбоновых кислот и тионилхлорида
пли избытка ClgCHOCHg. отщепляют бензилхлорид и гладко переходят в ангидриды
1\-карбокспаминокислот [1076], являющиеся важными промежуточными Продуктами
в синтезе пептидов;
R—СН-СООН
I
hn-coocu2c6h5
СЬСНОСПа
SOCh
R—СН—СО
| ^О + СвН5СН2С1
HN-CO
[1073] J. Р г a t, A. Etienne. Bull, Soc. chim. France [5], 11, 30 (1944).
[1074] E. Gryszkiewicz-Trochi mowski, A. Sporzy nski,
J. Wnuk, Rec. trav. chim., 66, 419 (1947).
[1075] A. Rieche, H. G г о p, Chem. Ber., 92 , 83 (1959),
[107&] К. P о d u s k a, J. G г о p, Chem. Ber., 94 , 527 (1964).
240
ОБРАЗОПАНИЕ СВЯЗИ С —Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Аналогично а.а-дихлорметилметилоному эфиру реагирует [1077) а,а,а'-три-
хлордиметпловый эфир С12СПОСН2С1.
Очень реакционноспособным агентом для получения хлоргшгидрпдов является
диметиламидохлорид муравьипой кислоты C12CHN(CH3)2, который подобно <х,а-
дихлорметилметиловому эфиру может рассматриваться как производное ортомуравьи-
ной кислоты. Этому соединению Цоллингер приписал солеобразпую структуру [1056]:
[(CH3)2N=CHC.l <- -> (CH3)2N-CIIC1] Cl-
Он получается при обменном взаимодействии диметилформамида с SOC12, РС13, РС16,
оксалилхлоридом, а также с С0С12 и реагирует с карбоновыми и сульфокислотами
с образованием хлорангидридов:
RCOOH + [(CHs)2N = CHC]]C]- “ici^ RCOC1 + (CH3)2NCHO (7)
Вместо Cl в катионе А может стоять также группа 0S0C1 или соответствующий
остаток других известных хлорирующих веществ. Для активирования реакции получе-
ния хлорангидридов при помощи Ы0С12 достаточно уже каталитических количеств
диметилформамида (1/20 моль на 1 моль карбоновой кислоты), так как, согласно урав-
нению 7, диметилформамид непрерывно рсгенсрируе i ся. Вводя добавку диметилформ-
амида, можно получать хлорангидриды из карбоновых кислот, плохо реагирующих
с одним SOC12.
п-НитробенЗоилхлорид [1056]. Его получают, например, нагреванием
в течение 30яин до 90—95° С 50,1 а л-нитробензойпой кислоты, 160 мл хлор-
бензола, 2,4 мл диметилформамида и 22,4 мл тиопилхлорида; выход около 90%
от теоретического.
По реакции с SOC12 в присутствия диметилформамида получаются также суль-
'фохлориды из сульфокислот пли их натриевых солей.
Из имид хлоридов, образующихся при взаимодействии амидов карбоновых.
кислот с РС16 в бензоле или хлороформе [1078— 1080], и карбоновых кислот в нейтраль-
яой среде получают хлорангидриды [1078]:
RCC1=NR' + R“COOH —> RCONHR'+R”COC1
Имидхлорид и карбоновую кислоту кипятят 30 мин в бензоле или эфире и вы- ,
деляют хлорангидрид, например, перегонкой.
7. Замена карбонильного кислорода на хлор и бром
Получение альдегид- и кетопдихлоридов методически не отличается от полу-
чения хлорангидридов кислот. Примером такой реакции является синтез бепзальхло-
рида из бензальдегида. Получить этот хлорид в чистом виде прямым хлорированием «
толуола очень трудно. >4
Бензальхлорид. В запаянной трубке нагревают 2 ч при 130—140° С -
1 молъ бензальдегида с 1,5 моль оксалилхлорида и получают бензальхлорид
с почти теоретическим выходом [1081]. По другому способу [1082] к бонзаль-
дегиду постепенно при охлаждении прибавляют SOC12. По окончании выделения .
SO2 встряхивают смесь с водой, растворяют отделившееся масло в эфире, эфир- '
1077] A. R i е с h с, И. G г о В, Е. Н о f t, Angew. Chem., 70, 602 (1958).
1078 F. Cramer, K. Baer. Chem. Ber., 93, 1232 (1960).
1079] O. Wallach, Ann., 184, 1 (1877); 214, 193 (1882).
1080] J. v. Braun, F. Josies, W.iHnch, Ann., 453, 125 (1927).
1081] H. S t a u d i n«g e r, Ber., 42 3976 (1909).
1082) F. L oth, A. Michael is, Ber., 27, 2548 (1894).
Ill ЗАМЕНА ГИДРОКСИЛЬН., АЛКОКСИДЫ!, И КАРБОННЛЬН. ГРУПП ПА Па] 241
ный раствор встряхивают с раствором бисульфита для удаления непрореагиро-
вавшего бензальдегида, фильтруют и отгоняют эфир. Перегонкой остатка полу-
чают чистый бепаальхлорид с хорошим выходом.
Часто для перевода >С— О в ]>СС12 применяют РС18. Для этого алифатический
альдегид пли кетон приливаю! при 0° С к взятому с небольшим избытком тоякоиз-
мельченноиу PCI.,:
RCOCH2R' 4- РС15 —► RCCla — ch2r'+poci3
Побочная реакция протекает с выделением ПС] п образованием монохлор-
этплепов:
RCOCH2R'+PC15 —»- RCC1-CHR' + HC14-POC13
Эта реакция становится главной прп использовании в качестве исходных
веществ арилметилкетонов.
Основной и побочный продукты реакции являются исходными для синтеза
ацетиленов.
Для получения бромзамещенных РВгБ применим только при бромировании
очень реакционноспособных соединений. В ацетальдегиде возможен обмен карбониль-
ного кислорода на бром с помощью РС13Вг2, который образуется в реакционной колбе
при взаимодействии брома с РС13 при охлаждении |228]. Из бензальдегида бензаль-
бромггд получается уже поп действием бромистого водорода [1084].
Карбонил карбалкоксильпой группировки обычно не взаимодействует с РС15.
Известны отдельные случаи, когда такое взаимодействие имеет препаративное
значение. Из алкиловых эфиров муравьиной кислоты по реакции с РС1Ь с хорошим
выходом образуются а.а-дихлорметилалкплолые эфиры [1085, 1086], которые отделяют
от хлорокиси фосфора фракционной перетопкой.
а ,а-Дихлорметилалкиловые эфиры. К 1 моль алкялформиата при 0° С
прибавляют в несколько приемов при перемешивании 0,9 моль РС16. После уда-
ления ледяной бани продолжают перемешивать смесь при комнатной темпера-
туре, причем смесь разодевается и РС15 полностью растворяется. Смесь разде-
ляют фракционной перегонкой. \
IICOOR + PC15 —> CI2CHOR+POC(3
Моноэтиловый эфир неполного хлорангидрида щавелевой кислоты
(ср. стр. 236) можно подушить через дихлорэтоксиуксусный эфир, который при
перегонке при нормальном давлении иногда самопроизвольно, лучше в присут-
ствии небольшого количества металлической платины, отщепляет этилхлорпд
[1087].
СООС2Н5
I
СООС2Н5
СООС2Н5
I
СС12
I
ос3н5
РС1»
(10 ч; 13i>°C)'>
СООС;Н5
I ' + С2Н5С]
СОС1
По данным Берта и Баррэ [1088], применение тщательно очищенного
РС]8 повышает выход эфира до 70—80% от теоретического. Однако следует пред-
почесть обменную реакцию с промежуточным образованием моноэтилового эфира,)
так как при работе по описанному выше способу ио неизвестным причинам сильно
колеблются выходы.
[1083] Tli. L. Jacobs, Org. Reactions, v. V, J, Wiley a. Sons, New York, 1949
p- 20.
1084] D. Vorlander, Ann., 341, 22 (1905).
1085J 11. F i s c li e r, A. Schwarz, Ann., 512, 240 (1934).
1086] A. Rieche II. G г о В, E. 11 0 f t, Ber., 93, 93 (I960).
1U87J K. Scholl, W. Eger er, Ann., 397, 326 (1913).
[1088] L. Bert, R. Barre, Bull. Soc. chim. France [4], 37, 1041 (1925);
41, 47, 1165 (1927).
16 Заказ 1835.
242
UhHA3UBA±LHE СВЯЗИ С—На! В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
IV. Замена С—N-связи на связь С—Hal
1. Замена С—N-связи на связь С—Hal
в алифатических соединениях
Замена С—N-связи на связь С—Hal J
в алифатических аминах при помощи РС1Ь и РВгъ ]
Нмидхлориды разлагаются термически с образованием нитрилов и алкилхло-1
радов [1089]. Па этом основана реакция Брауна [1090], по которой алкиламнны^
фенилалкиламины, феноксиалкиламины, а,й)-диамипы или циклические вторичные^
амины сначала бензоилируют, затем бензоильные производные превращают в имицга-ад
логениды
R-NH;
R-NH-COC6H5 _РОХз.
R—N—СХ—CgHa
или амидгалогепиды:
RX-|-C6HbGNj
'СН; QH.COC1
^>nh -—------
ч:н2
:N-COCfiib
-см, ..-сире
СНг -СГЦХ
Расщепление имид- или амадгалогенидов обычно происходит при вакуумио!
перегонке реакционной смеси. В сочетании с синтезом Кольбе (замена галогена на
циан) можно по реакции Брауна удлинить цепь алкилгалогенида на один атом угле-
рода; в сочетании с расщеплением амидов карбоновых кислот по Гофману можно пере-
вести карбоновые кислоты Са в алкилгалогепиды C„_j (ср. стр. 259).
Реакция Брауна [1091]. Сначала в присутствии раствора NaOH бензо*
илируют 1 моль амина при помощи 1 моль бензоилхлорида. Затем 1 моль высу»
шейного бензоиламина осторожно без доступа влаги нагревают с 1 моль PCI®
или РВг6 до образования гомогенного расплава или раствора. Можно также сшй
чала получать РВгв в реакционной смеси, для чего к 1 моль бензоиламина пр^
перемешивании и охлаждении прибавляют около 1,02моль РБг3 и затем 1,01 мол^
брома [1092—1094]. Вместо РВг5 для приготовления бромидов можно такж«
использовать РС13Вг2, который получают предварительно медленным вводе»
нием 1 моль брома в РС]8 и встряхиванием при охлаждении, после чего вводя!
бензоилированный амип. “
Для выделения продукта из реакционной массы известно несколько раз-
личных способов. Хлорокись фосфора (т. кип. 105,4® С) в большинстве случае!
отгоняют из реакционной массы при нормальном давлении. При получении орга*
нических хлоридов с низкой температурой кипения применяют ректификацией^
ную колонку, но так как реакцией Брауна пользуются преимущественно для
получения высококипящих галогенидов, то к ректификации прибегают редкой
Бромокись фосфора (т. кип. 193° С) почти всегда перегоняется в вакууме, созН
даваемом водоструйным насосом совместно с бензонитрилом (т. кип. 191eCj
и органическим бромидом (температура вне колбы 70—120° С). При получении
очень высококипящих бромидов сначала отгоняют основное количество РОВт^г
[1089] Н. v. Pechraann, Вег., 33, 611 (1900).
[1090] 5. V. Braun et а]., Вег., 37, 2915, 3210 (1904); 38, 2336 (1905); 39, 20Я
(1906); 43, 2837 (1910); 55, 3526 (1922).
[1091] J.v. Braun, W. Sobecki, Вег., 44, 1464, 2867 (1911). '
[1092] N. J. L eonard, Z. W. Wicks, J. Am, Chem. Soc., 68, 2402
(1946).
[1093] J. A. Arvin, R. Adams, J. Am. Chem. Soc., 50, 1983 (1928).
[1094] N. J. Leonard, E. W. Nommensen, J. Am. Chem. Soc., 71Й
2809 (1949). " ?
[1095] J.v. Braun. G. Irmisch, Ber., 65, 880 (1932).
IV. ЗАМЕНА С—N-СВЯЗИ НА С-Ва1
243
в вакууме, создаваемом водоструйным насосом, и затем разлагают имндбромид
ври перегонке в вакууме, создаваемом масляным насосом [1096].
Из дистиллята РОХ3 удаляют обработкой ледяной водой. Бензонитрил
обычно ие удается отделить от органического галогенида перегонкой. Его можно
омылить до бензойной кислоты многочасовым кипячением с концентрированной
НС1 пли 40—48%-ной НВг. После отгонки бромида с водяным паром или извле-
чения его эфиром бензойная кислота полностью удаляется промывкой раствором
соды. Для отделения бензонитрила от органического бромида можно также
обработать высушенную смесь абсолютным этиловым спиртом (1 моль спирта
на 1 моль исходного бензоиламина) и пропустить в смесь сухой НС]. Через 2—5
суток добавляют эфир и отфильтровывают нерастворимый в эфире гидрохлорид
бензиминоэтилового эфира. Эфирный раствор органического галогенида далее
промывают в делительной воронке водой и выделяют галогенид перегонкой.
В качестве примеров перевода циклических вторичных аминов в а,со-
дибромалканы следует указать на синтез 1,5-дибромггентана из N-бензоилпнпе-
ридпна [1097] и 1,5-дпбром-З-метилпентана из 1-бензоил-4-метилпипериднна
[1092].
Замена С—N-связи на связь С—Hal
в алифатических аминах при помощи бромциана
Расщепление по Брауну [1098, 1099] циклических третичных аминов при по-
мощи BrCN имеет меньшее препаративное значение, чем реакция бензоиламиноя
с РЕп. и используется главным образом при выяснении структуры алкалоидов. При
действии BrCN на ациклические третичные амины
И ч А .
>N—R + BrCN —> RBr-f- \N-CN
R / R"Z
отщепляется алкилбромид предпочтительно с наиболее короткой алкильной цепью;
если R= аллил или бензил, алкилбромид легко отщепляется, если R = СвН6, арвл-
бромид не отщепляется. В качестве побочных продуктов образуются бромиды четвер-
тичных аммониевых оснований по реакции алкилбро.мида с исходным третичным ами-
ном. поэтому надо следить, чтобы BrCN всегда был в избытке. Вследствие того что
BrCN очень ядовит и легколетуч (т. кип. 62° С), необходимо работать обязательно под
тягой п с малыми загрузками.
Подробное описание синтеза бромциана из NaCN и Вг2 см. [1100].
Циклические третичные амины реагируют с UrCN с сохранением кольца
(реакция 1, дезалкилирование) или с расщеплением его (реакция 2):
CH2 cn2
(CHs)?NR (CH^)„ N-CN + RBr (1)
^CHa CH2
| BrCN
ХН2Вг /СН2Вг
(Йн3),, Н^Ц1£^Лгная)(СНг)д (2) .
^СЩ—N(R)—CN \сн2—NH(R) • НВг
[1096] W. John, II. Pint, Hoppe-Seyler’s Z. physiol. Chem,, 273,' 225
(1942).
[1097] J. V. Braun, Org. Synth., 9, 70 (1929).
[1098] J. v. Braun, Ber., 33, 1438 (1900); 40, 3914 (1907); 42, 2219 (1,909).
[1099] H. A. Hageman, Org. Reactions, v. VII, J. Wiley a. Sons. New York,
1953, p. 198.
[1100] W. W. Hart man, E. E. D reger, Org. Synth., Coll, v.- 2, 1943, p. 150.
16*
ОБРАЗОВАНИЕ СВЯЗИ С—На] В РЕЗУЛЬТАТЕ 3 СМЕЩЕНИЯ
-------------------------------------------------------------------------------_ j
Из Ьн-бутплнпрроллдпна п BrCN п бензеле по реакция (2) с количественным ?
выходом образуете» м-бутил-д-бромбутилципнамлд [1101]. Прп взаимодействии BrCN '
с I-этилппрролпдпном [1102J на 94% проходит реакция (2). с 1-этилпиперидином *
на 66% реакция (/). Из плпсридпнацетопптрила [1103] (п — 3, R = CH2CN) под ден- *
ствием BrCN по реакции (1) получается с хорошим выходом бромацетона грид [1104]. ,5
Прп В -- бензил идти- и шестичлепиые циклы не расщепляются [1105].
Общий способ проведения реакции (ср. [1099]). Раствор амина в сухом !
эфире, хлороформе или бензоле постепенно приливают к раствору BrCN (ишяд!)„ 1
Реакция экзотермична. Можно также нагревать смесь амина с BrCN (1—1 1 моль "
BrCN на 1 моль амина) на паровой бане. В качестве побочных продуктов выпа--4
дающих в большинстве случаев при работе в растворителях, помимо бромидов j
четвертичных аммониевых соединений (см. выше) могут получаться также гидро» J
бромиды третичных аминов. Необходимый для этого НВт выделяется и резуль- ?
тате образования олефинов. При проведении реакции без растворителя получае-’*
мый продукт извлекают из реакционной смеси эфиром. Нспрореагирсвавший ямин>
и его соль удаляют встряхиванием в делительной воронке эфирного раствора-
N-замещепного ©-бромалкилцианамида с разбавленными водными кислотами- '
N-Замещенные со-бромцианамнды можно выделять перегонкой в вакууме
или применять в неочиптенпом виде для дальнейших превращений. Например,1'
бром можно заменить на группу N^R)2, где R = алкил [1101], или отщепить-1
циангруппу кипячением с 48%-пой НВг. Примером такой реакции служит ;
получение гпдробромида №(©-бром-н-амил)-анилипа [1106] в результате 40»'
кипячения Br(CH,)3CII2N(CeH6)CN, приготовленного из К-фенилпилеридина-
и бромцнана, с 48%-ной "НВт.
В других случаях образующийся при реакции дезалкилирования замещен-
ный цианамид может являться целевым продуктом реакции, как это показано
на примере синтеза метил-а-нафтилцианамида из диметил-а-нафтиламина и бром-
циана [1107].
Замена аминогруппы на галоген в алифатических соединениях ;
при помощи нитрозилгалогенидов
Обменная реакция алифатических аминов [1108] или амипокарбоновых кислог^
[1109] с нитрозилгалотенидами приводит к образованию галогенидов 1
RNH2-]-NOX —<RX-rN2^H2O
а не солей диазония, как в аналогичной реакции с ароматическими аминами.
Нитрозилхлорид NOC1 или питрозилбромид NOBr получают в реакционной;
смеси, медленно прибавляя NaNOj при 0° С к раствору аминокарбововой кислот»?
в HCI или соответственно НВг, или в 2,5 н. H2SO4 с добавкой бромида калия [1110]:: ч
HX4-HNO2 NOX4-H3O
Если к охлажденному раствору аминокислоты или ее эфира в 10—20 %-ной
НВт приливают по каплям бром и затем при 0° С пропускают окись азота, то в качеств»’
промежуточного продукта образуется нитрозилбромид [1111]: • • '
2NO-i-Bra 2NOBr
[1101
1102
1103
1104
1105
1106
1107
[1108
[1409
[1110
[1111
R. С. Е 1 d е г f i е 1 d, П. А. Н age m a n, J. Org. Clienj., 14, 626 (1949)».:
J. v. Braun, Ber., 44, 1252 (1911). A
E. Knoevenagc], Ber., 37, 4082 (1904). [
J. v. Braun, Ber., 41, 2113 (1908).
J. v. Braun, Ber., 49, 2629 (1916); 50, 45 (1917). :
II. J. N i t z s c h k e, G, Faerber, Chem. Ber., 87, 1637 (1954). 3
W. J. Cressman, Org. Synth., Coll. v. 3, 1955, p. 608.
В. Солонина, ЖРФХО, 30, 431 (1898).
P. Walden, Bor., 28, 2766 (1895); 29, 133 (1896).
K. Pfister ct al. J. Am. Chem. Soc., 71, 1096 (1949).
E. F i sc h c r.et a]., Ber., 39, 2929 (1906); 40, 489, 1054 (1907); 41, 890 (1908). •]
JV. злмиил С—Л-СВЯЗЛ НЛ С -II,И
2
В общем случае при взаимодействии а-амипокарбоновых кислот с шпрозллгал
гепидом не происходит обращения конфигурации у a-атолш углерода Щ12], хо
может меняться направленно вращения; например, из правовращающего L-аланп]
oopaiyeicH левовращающая Z-хлор- или Л-бромцропИОноиая кислот. По реакщ
с пнтрознлгалогенпдаил из оптически активных а-амппокпслот можно получать опт
чески активные а-галогс’пзамещепные жирные киситы.
D- пли L-хлорпропиоповая (пли бромпропионовая) кислота [НК
В раствор 0,2 моль оптически чистого D- или £-алалмпа в 250 мл 6 п. ПС1 (и,
НВг) вносят при энергичном псремеингвании и О—5е С. к течение 2,5 ч свея
paciepibril NaNO2 (0,32 моль) и перемешивают еще 4 ч. После экстрагирован'
эфиром, сушки над СаС12 и фракционной порегокни выход Г)- наш £-хлоррропг
новей кислоты (т. кип. 77° С при 10 мм. рт. ст.) достигает 32% от теоретическо!
а выход £7-пли £-бромпропиоиоиоп кислоты 60—65%; т. кип. 78й С (4 .м.ч рт.т
Получение галогенкетонов из диазокетонов
Из диазоалклнов [1114}, эфиров диазокарбоиовы.х кислот [1115] и диазокетод
под действием галогеноводородов или щлогенов получаются моп<>- или дигалогена
каны, эфиры моно- или дигалогенкарбоновых кислот, а также мопо- илн дигалоге
кетоны.
RCllN2-}-HX —> RCH2X4-N2
где HX-=HF, HCI, НВг.
где Х-=С1, Вг, I.
RCI1N2 + Xa > КСНХ2 + ^2
Реакция чаще всего протекает при комнатной пли более низкой томперату
и имеет препаративное значение только в синтезе галогенкетонов, образующих
из диазокеюнов без примеси изомеров, обычных в прямом галогенирован
(ср. стр. 178).
Для синтеза ыоногалогепкетонов можно исходить из чистых диазокетон<
для получения которых в эфирный или бензольный раствор 3 моль диазометала и
0—5° С и перемешивании медленно приливают по каплям эфирный или бензолыг
раствор или суспензию 1 моль хлорангидрида карбоновой кислоты, оставляют см«
па 1—2 ч при 20—25° С, после чего отгоняют растворитель в вакууме и nepeKj
стал лизовывают диазокетоп [1116]:
RCOCI4-CH2N2 —> BCOCHN2+HCI
В качестве побочных продуктов при получении диазокетонов, особенно ц
недостаточном избытке диазометана, возможно образование а-хлоркетонов. Час
эфирный реакционный раствор, состоящий из .хлорангидрида и диазометана, Hej
средствснно подвергают взаимодействию с галогеноводородом [1117, 1118].
Если при этом применяется, например, бромистый водород, то образующий
а-бромкетон может быть загрязнен а-хлоркетоном. Для получения чистого брома
[1114
[1115
[1116
[1117
[1118
[1112] К. Freudenberg, W. Kuhn, 1. liumann, Вег., 6.3, 2382 (19&
W. A. Cowdrey, Е. D. Hughes, С. К. Ingold. J. Chem. Soc., 19;
1243; A. Neuberger, Adv. Protein Chem.. 4, 297 (1948),
[1113] S.-C. J. Fu, S. M. Birnbaum, .1. P. G r e e n s t e i n, J. A
Chem. Soc., 76, 6054 (1954). 4
Л. v. Pechinann, Bor., 27, 1889 (1894).
T. Curtins, J. prakt. Chem. [2], 38 , 430 (1888).
W. E. Bachman 11, W. 8. Struve, Org. Reactions, I, 48 (1942).
R. E. L u t z, J. W. Wilson, J. Org. Chem,. 12, 769 (1947).
J. W. Wilson R. E. Lutz, R. H. J о r d a 11, J. Org. Chem., 12, «
(1947).
246
ОБРАЗОВАНИЕ СВЯЗИ С—На) В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
тона можно к бромангидрпду кислоты добавить диазометан и в полученный эфирный • •
реакционный раствор при 0° С пропустить сухой бромистый водород [1119].
Фторкетопы, например со-фторацетофенон [1120], получают из диазокетонов
взаимодействием с безводным HF в эфире, хлоркетоны — взаимодействием с сухим
11С1 в эфире или хлористом метилене [1121], с концентрированным раствором НС1
и диоксане [1122] или с водно-ацетоновой соляной кислотой [1123]. Для получения
бромкстонов применяют сухой или 42—48%-ныи НВг в эфпре, диоксане или петро-
лейном эфире. Ледяная уксусная кислота, рекомендованная также как растворитель
[1124, 1125], оказалась мало пригодной, так как при ее взаимодействии с диазокетонами
[11261 могут получаться ацетилированные кетолы RCOCH2OCOCH3. Йодистый водород
реагирует с диазокетонами с образованием метилкетонов и иода [1122]. Реакция
с галогеноводородами протекает в большинстве случаев при 0—20° С.
3-Бром-1,1-дифенилпропанон [1117, 1127]. К охлажденному льдом
'эфирному раствору диазометана (из 20 г ннтрозометилмочевины) при перемеши-
вании приливают по каплям раствор 0,057 моль дифенилацотилхлорида в 100 мл
сухого эфира. Реакционную смесь выдерживают 30 мин при 0° С, затем 1 ч при
комнатной температуре, отгоняют избыток диазометана и эфир и растворяют
остаток в 100 мл сухого эфира. К этому раствору приливают по каплям при пере-
мешивании смесь 14 мл 48%-ной НВт и 14 мл эфира; смесь можно охлаждать
льдом. После прекращения выделения азота раствор упаривают; выкристалли-
зовавшийся бромкетон перекристаллизовывают из метилового или изопропило-
вого спирта; т. пл. 66—67° С.
1,12-Дибромдодекандион-2,11 [1118]. Раствор 0,243 моль дихлорангид-
рида себациновой кислоты в 75 мл сухого эфира приливают по каплям в течение
Зч при 5—10° С к 1,43 моль дназометапа в 2 л эфира. Выпадающий твердый
диазокетон не выделяют, а оставляют па ночь при комнатной температуре.
После этого ио каплям медленно приливают при. 18° С смесь 235 в 42%-нои
НВг и 200 мл эфира. Через 8—10 ч (конец выделения азота) смесь охлаждают
що 0° С и отсасцвают дибромдикетон; выход 87% от теоретического; т. пл.
-97—98° С; после повторной перекристаллизации из этилового спирта т. пл.
102-103° С.
Примеры применения этой реакции к производным углеводов см. [1128].
а,а-Дихлор- или дибромкетоны [1129] получают, например, действием хлора
или брома на растворы диазокетонов в CCI4; сс,а-дииодкетоны — из дназокетонов
и иода в хлороформе [ИЗО, 11.31].
J. Chem. Soc.. 1948, 278.
и сотр., Изв. АН СССР, ОХП. 1956, 377; С. А., 50,
Mill?, А. С. Соре, J. Am. Chem. Soc., 72, 3477
[1119]
[1120]
[1121]
1
J. R. С a t с Ь et al
И. Л. Кнунянц
5454 (1956).‘
R. В. Turner, .1.
(1950).
[1122] Н. В. W a g п е г, J. М. Тот е, J. Am. Chem. Soc., 72, 3477 (1950).
[1123] Р. Ruggli, К. Knecht, Helv. chim, acta, 27, 1108 (1944).
[1124] К. Balonov ic, D. С e г a r, L. F i 1 i p о v i c, J. Org. Chem., 18, 868
(1)53).
(1125] H. Schlenk B. G. L a in p, B. W. De II a a s, J. Am. Chem. Soc., 74,
2552 (1952),
' 11120] J. F. Lane, R. L. Feller, J. Am. Chem. Soc., 73, 4231 (1951).
£1127] W. G. Dan ben, C. F. H i s k с у, M. A. M u h s, J. Am. Chem.
Soc., 74, 2083 (1952).
.11128] M. L. Wolfrom R. L. Brown, J. Am. Chem. Soc., 65, 1516
(1943).
,[1129] A. Roedi g Ц. L u n k, Chem. Ber., 87, 975 (1954).
[ИЗО] L. Wolff, Ann.w 325, 143 (1902); 394, 40 (1912).
£1131] F. Arndt, В. E i s t e r t, W. P а г t a 1 e, Ber., 60, 1369 (1927).
JV. ЗАМЕНА C— N-СВЛЗП HA C—Hel
24
2. Замена C—N-связм на связь С—Jlal
в ароматических и гетероциклических соединениях
Обмеп ароматической аминогруппы па галоген и родан через диазосоединени
является очень важным препаративным методом для введения фтора, хлора, броме
иода и родана в определенные положения ядра или в положения, в которых прямо
замещение водорода галогеном затруднено или даже невозможно (например, метг
галогенирование ядра при наличии заместителей, ориентирующих в орто- и пара-п<
ложевия; получение галогеяальдегидовд Методически различают три возможноегг
обменную реакцию растворов солей диазония с галогеноводородпыми кислотами в npi-
сутствии моногалогепндов меди (реакция 3апдмейера[1132]) пли без катализатора (пр<
имущественно замена — Х2Х-группы па иод); расщепление диазонийпербромидо!
термическое расщепление соединений диазопия (реакция Шве.хтена и Шнмана).
Замена аминогруппы на хлор и бром
в ароматических и гетероциклических соединениях
(реакция Зандмейера)
Реакция Зандмейера протекает, вероятно, через радикалоподобные промеж^
точные стадии [1133, 1134] или через свободные арилрадикалы [1135].
[АгХ2]+С1_ц-СиС1 ArN2CuCI2 (1
. ArN2CuCl2—*- Аг. 4-CnCla + Nz , (J
__ Ar.+CuCl3—> ArCl-^CuCl C
Подробнее о первой стадии реакции см. [1134, 1135]. Решающую роль игра*
ион меди как редокс-катализатор. С повышением концентрации попов галогена сиз
ждется скорость реакции Зандмейера, так как, например [1134], из действующего кг
катализатор Cud и аниона хлора образуется комплекс (CuC]s)2-. Однако с увблич
ни ем концентрации ионов галогена еще сильное затормаживаются побочные реакце
4 и 5, так что несмотря па замедление скорости реакции выход арилгалогенида во
растает [1134].
В водной среде арильные радикалы могут реагировать не по реакции
а возможна димеризация с образованием диарилов
2[ArN2]*C]- + 2CuCl —> Ar—Ar + 2N34-2CuCl2 (
или из попов диазония могут получаться азосоедпнения:
2[ArN2]+Cl-4-2CuCl —> Ar—N=N—Ar4-N3+2CuCl2 (
В этих побочных реакциях расходуется хлорид меди (1].
Разбавление водой и снижение концентрации ионов хлора способству
протеканию побочных реакций. Концентрацию ионов хлора можно снизив
например, прибавляя, в отличие от обычного способа проведения реакции, со®
нокислый раствор CuCl к раствору соли дпазокия.
Концентрация СпС] снижается также при образовании комплекса с CuCIs
НС1. Присутствие СиС]2 так сильно тормозит реакции 4 и 5, что можно обойтись оче:
малым количеством СпС] (0,01 моль СиС] на 1 моль диазоеоединения вместо обычз
применяемых 0,1—0,2 моль) [1134]. Каталитическое действие только СиС12 объ/з
няется [1136] промежуточным восстановлением части СиС12.
Следовательно, для того чтобы достигнуть высоких выходов арилгалогенид
при взаимодействии солей диазония с хлористым водородом в присутствии СиС
надо подавить побочные реакции 4 и 5 добавкой комплексообразователей (HQ
[1132J Т. Sandmeyer, Вег., 17, 1633, 2650 (1884).
[1133] Е. Р f е i 1, О. V е 11 е л, Ann., 562, 163 (1949); 565, 183 (1940).
[1134] Е. Р f е i 1, Angew. Chem., 65, 155 (1953).
[1135] J. К. Kochi, J. Ara. Chem. Soc., 79, 2942 (1957).
[1136] H. Hodgson, J. Chem. Soc., 194ч, 18; 1946, 745.
248
ОБРАЗОВАНИЕ СВЯЗИ С -Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
СиСк), однако при этом одновременно будет снижаться скорость основной реакции.
Ее можно повысить проведением реакции при более высокой температуре, по в этом
случае будет усиливаться реакция образования фенолов:
[ArN-2]*Cl- + II2O —► ArOH-J-Ns + IICl
В отсутствие галогенида меди из хлоридов и бромидов диазопия по этой реак-
ции в основном образуются фенолы.
В некоторых случаях диазотированные амины можпо переводить в хлорсоедп-
нения с помощью одного хлористого водорода; например, диазотированием 2-амино-
ппрпмидипа в концентрированной ПС] при температуре от —15° С до —10е С и пере-
мешиванием солянокислого раствора соли диазония в течение 1 ч (от —10J С до —5° С)
получают около 27% 2-хлоринрпмидипа; подробные указания см. [1137]. ।
Вместо CuCl катализатором может служить металлическая медь в порошке I
[1138, 1139]. Рассмотрение механизма реакции Запдмейера см. и обзорах | J140, 1141]. i
'Гак как нптросрудпы в орто- и пара-положеппи к диаэогрунпе относительно
легко вступают в реакции нуклеофильного замещения, необходимо считаться с воз-
можностью протекания побочной реакции замещения таких групп ионами хлора или?
брома Ц142].
Методика проведения реакции Запдмейера показана ниже на ряде примеров.
.м-Хлорбензальдегпд [1143]. В стакане емкостью 3 л растворяют 2 м<,ль
SnCl.j • 211,0 в 600 мл концентрированной НС1, охлаждают до 5° С и при спли-
ном перемешивании и охлаждении льдом сразу смешивают с 0,66 моль .и-нитро-
бензальдегида (температура повышается примерно до 100° С). Полученный крас-
ный раствор л-ампнооензальдсгида охлаждают до 2е С смесью льда с повареппой
солью и в образовавшуюся кашицу при 0—5° С в течение 90 мин при энергичном
перемешиваний па капельной воронки, трубка которой погружена в реакциоцнхю
массу, приливают раствор нитрита натрия (0,67 моль NaNO2 в 150 мл виды)
до положительной иод-крахмальпой пробы па свободную HNOS (должно оста-
ваться около 5 .ил нитритного раствора).
Раствор хлорида диазония прибавляют при встряхивании к нагретому
до 75° С раствору CuCl, приливают затем 840 мл концентрированной НС1, оста-
плятот на ночь ц отгоняют продукт реакции с водяным паром. .ч-Хлорбензаль-
дегнд, отгоняющийся в первых 1,5 л дистиллята, экстрагируют эфиром (2 пор-
ции по 150 мл): пых-ол 75—79% ог теоретическою; т. кип. 84—86° С при
8 мм рт. ст.: 107—109° С При 26 мм рт. ст.
.и-Бромбензальдегид [1144] получаю! аналогичным способом, проводя
восстановление с помощью SnBr2 — НВг и обрабатывая раствор бромида диазо-
ния горячей суспензией ОпВг и 48%-ной НВг.
Приготовление раствора СиС]. В колбе емкостью 5 л растворяют 0.75 моль
CuSO4 • 5Н2О п 161 г NaCl в 600 мл воды и приливают раствор 41 г Na2S2O3
и 27 г NaOH в 300 мл воды [1143].
Солянокислый 5 М раствор CuCl для синтеза п- и о-хлортолуолов из
п- и а-толуидипов [1145. 1146] готовят следующим образом. К раствору 5 мол.
CuSO4 • 2Н2О и 5.6 моль NaCl и 4л горячей воды в точение 5—10 мин приливают
раствор 265 г NaIIS03 и 175 г NaOH и 2 л воды. Смесь охлаждают и выделив-
шийся осадок промывают декантацией. Хлорид меди (I) получается в виде белою
порошка, темнеющего при соприкосновении с воздухом. Неочищенный хлорид
меди (I) растворяют в 2 «г 28%-нои соляной кислоты (d — 1,14).
[1137]
I. С. К. о g а ц. R. М i п i п, С. С. О ve г berge г, Org. Synth., 35, 34
(1955).
L. Galtermann, Вег., 23, 1218 (1890); 25, 1086 (1892).
W. A. Waters, J. Chem. Soc., 1942, 266.
W. A. Cowdrey, D. S. Davies, Quart. Rev., 6, 358—379 (1952).
H. H. Hodgson, Chem. Rev., 40, 251 (1947).
R. llnisgon, Angew. Chem., 67, 439 (1955).
J. S. В и c k, W. S. Ide, Org. Synth., Coll. v. 3, 1943, p. 130.
F. T. Tyson, Org. Synth., Col).’ v. 2. 1943, p. 132.
G. S. M a r v e J. S. M.'M с E 1 v a i n, Org. Synth., 3, 33 (1923).
G Deller, Angew. Chem., 23, 392 (1910).
IV ЗАМЕЛА С-хХ-СПЯЗП ПА с — Па]
24'
Получение CuCl из СиС12 и Си, Нагревают до обесцвечивания смесь 250
GuSO4 • 5П..О. 120 г NuCI, 500".u.i поды, 1000 г концентрировапной HCI и 130 ,
Ou. После разбавления концентрированной IIC1 до 2036 г н растворе содержите}
oi.ci.io 10% CuCl; после отделения декантацией от осадка ого можно хранит!
бе 1 доступа воздуха. Для получения кристаллического CuCl к декантированному
пли отфильтрованному раствору прибавляют поду и оллаждаюг. К частичнс
окислившемуся прп хранении препарату прибавляют pacinop .М14С1 в НС
и нагревают с, медью илн Na,SOs до обесцвечивания.
Вместо CuCl для синтеза названных и других .хлор-и бромароматическиэ
соединений применяется также металлическая медь в порошке. Ее получаю1:
по способу Гаттермана {14381 добавлением в перемешиваемый насыщенны^
на холоду водный раствор CuSO4 просеянной через тонкое сито цинковой
пыли. Полученную смесь бледно-голубого цвета па>ревают до 80° С, тонко-
распредслснпый порошок меди отделяют дскантацпеп от^раствора ZnSO4, не-
сколько раз промывают водой, отмывают or цт.пка разбавленной НС1 и затек
водой до отсутствия следов кислоты. Очень реакционноспособную «медь Гаттер-
мана» тотчас же использу ют пли храпят в виде влажпой пасты.
Подробное описание синтеза о-бромтолуола из диазотированного о-толуидинЕ
в присутствии порошкообразной меди в качестве катализатора см. [1147].
о-Хлорбромбепзол J1118], К охлажденной до 0° С смеси 1 моль о-хлор-
анплпна и 31)0 мл (2,5 моль) 48%-ной НВг при перемешивании и’охлаждении
быстро прибавляют раствор 1 моль ХаХ02 в 125 зм воды. Добавлением Маленьких
кусочков льда поддерживают температуру смеси ниже 10° С. Диазотирование
ведут прп отчетливой кислой реакции среды по кош о, последние Милли-
литры раствора NaISO2 добавляют медленно (до посинения иод-крахма.тьной
бумаги).
В колбе, снабженной нисходящим холодильником, капельной воронкой
и вначале перекрытой пароподводящей трубкой, нагревают до кипения 0,55 моль
GuBr и 80 мл 48%-пой ПВг и к полученной смеси приливают в несколько при-
емов в течение Зи мин холодный раствор соли диазония. При атом из реакцион-
ной смеси, непрерывно нагреваемой до кипения, отгоняется большая часть
о-хлорбромбепзола. После прибавления всего раствора соли диазония пускаюг
сильный ток водяного пара и дополнительно отбирают дистиллят до сум-
марного объема I — 1,5 л. От дистиллята отделяют органический слой и обра-
батывают его концентрированной П2ЦО4до тех пор, пока кислота станет слабо
окрашиваться (примерно 4 раза по 10 мл H2SO4). Затем промывают водой, 5%-ныэд
раствором NaOH и вновь водой, суптат над 3 г СаС12 и перегоняют; выход 89—
95% от теоретического; т. кин. 199—201° С (742 мм рт. ст.).
Аналогично получают лг-хлорбромбензол. 1,3-дибромбензол и о-бромани-
зол с выходами около 90% от теоретического. При получении о-броманизола
обработка серной кислотой не нужна.
о-Бромнитробензол [1149]. Синтезируют таким же способом. о-Нитро-
.пш лип в виде суспензии медленно диазотируют в 400 мл 48%-ной ТТВг (при тем-
пературе ниже 10° С). Вместо 80 мл в атом синтезе добавляют 120 мл 48%-ной
НВг. Перегоняющееся с водяным паром масло затвердевает при охлаждении
(т. пл. 41-42.5° С).
Получение CuBr [1148]. К перемешиваемому раствору 2,4 моль
CuSO,, • 5Н2О п 3,4 моль NaBr и 2 л теплой поды в течеппе 10 мин прибавляют
1.2 моль x%’a2SO.(; иногда до исчезновения синею окрашивания приходится до-
бавлять немного больше сульфита. Смесь охлаждают, быстро отсасывают СиВг
и сушат в течение ночи на воздухе; выход 320 г (93% от теоретического).
Раствор CuBr в ПВг. нужный для синтеза о-броманизола (из 4 моль ди-
азотировапяо!о р-аипзпдина), готовят [1150] смешением раствора 260 г CuSO4-
•5П2О и 625 г NaBr в 500 мл воды со 120 г концентрированной H3SO4. Полу-
ченную смесь кипятят 4 ч с 80 г меди в виде порошка или опилок. Аналогично
готовят раствор CuBr для синтеза п бромтолуола пз п-толуи;шна, но воды
|1147] 1.. A. Bigelow, Org. Svntli.. 9, 22(1929): Coll., v. 1, <35 (1956).
f 1148| .1. b. Hartwell, Org. Synth., 24, 22 (1944); Coll., 3, 185 (1955).
111491 В, В. С a r 1 i n, G. W. L a rs on, j. Ara. Chem. Soc., 79, 939 (1957).
11150] E. И и rdegger, D. Rcdlich, A. Gal, Helv. chim. acta, 28,
631 (1945).
250
ОБРАЗОВАНИЕ СВЯЗИ C-Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
применяют в восемь раз больше [1151 ]. В обоих случаях приготовленный в раз-
бавленной II2SO4 раствор сульфата диазония приливают ио каплям в кипящий
раствор СиВг, одновременно отгоняя продукт разложения с водяным паром.
При получении арплгалогенидов из очень слабоосновпых ароматических
аминов указанные амины приходится диазотировать в концентрированных кислотах,
например в концентрированной H2SO4. Однако последняя образует с NaNO2 нитрозил-
серную кислоту, и поэтому для выделения свободной азотистой кислоты во время диазо-
тирования необходимо добавлять ледяяую уксусную [1152, 1153) или фосфорную
кислоту 11154, 1155]. Можно также диазотировать амины водным раствором NaNO2
в смеси Н3РО4 (84%-пой) и IINO3 (d = 1,4) прп температуре от —8 до —12’ С. Разло-
жение ведут концентрированной НС1 или 48%-ной НВг в присутствии гаттер^ановской
меди или СиВг на холоду. Так получают 2-хлор- и соответственно 2-бром-4гметокси-
юензтиазол [1156], этиловый эфир 2-бромтиазол-4-карбоновой кислоты [1157] к 2-бром-
тиазол [1158, 1159]. j
n-Дихлорбензол (из ге-фениленднамина) [1160]. К раствору 3,6$! г гидро-
хлорида л-фенилендиамина в 70 мд НзРО4 (d = 1,7) при 5° С (охлаждение смесью
льда с поваренной солью) при энергичном перемешивании приливаю? по каплям
раствор 3 г NaNO2 в 30 мл концентрированной H2SO4. Через 30 мин “прибавляют
2 з топкоизмельченной мочевины. Раствор диазоеоединения прп сильном пере-
мешивании приливают по каплям к охлажденному до 0г С раствору CuCI-,i при-
готовленному кипячением с обратным холодильником 10,8 г CuSO4-5HsO,
5,4 з NaCl, 45 мл воды, 150 мл HG1 (d = 1,19) и 30 г металлической меди, п-Ди-
хлорбензол выделяют перегонкой с водяным паром; выход около 70% от теоре-
тического; т. пл. 53° С.
п-Дииодбензол, В раствор диазоеоединения (см. выше) добавляют не-
сколько кусочков льда и приливают смесь к концентрированному раствору 8 г
К.]. Смесь оставляют на ночь, разбавляют водой и промывают выпавший в осадок
л-дииодбепзол водой и раствором Na2S20g; выход почти количественный.
Галогенированные ароматические соединения получают [1161] через проме-
жуточно образующиеся диазоеоединения взаимодействием нитрозосоединения с солью
гидроксиламина в присутствии CuCI или СиВг и концентрированной НС1 или соответ-
ственно НВг:
Ar-N=O4-NllaOH'HX —[ArNa]+X- —+ ArX-]-N2
Этот метод пригоден для получения n-хлор- н п-бромфенолов.
Замена аминогруппы на иод в ароматических соединениях
На примере получения я-дииодбензола (см. выше) показано, что при помощи
йодистого водорода очень легко можно заменить диазогруппу на иод без добавления
соли меди (I). Холодный раствор иодида щелочного металла (1—2 моль иодида на -
[1151] L. A. Bigelow, Org. Synth., 5, 21 (1925).
[ 1152] Н. Л. Hodgson, J. Walker, J. Chem. Soc., 1933, 1620; 1935, 530. ’
(1153] А. K. Stepan, C. S. Hamilton, 1. Am. Chem. Soc., 71, 2438 (1949).
(1154] H. A. J. Schoutissen, J. Am. Chem. Soc., 55, 4531 (1933).
(1155] J.. H. Welsh, .1. Am. Chem. Soc., 63, 3276 (1941).
(1156] H. Erlenmeyer, H. U e b e г w a s s e r, Helv. chim, acta, 25, 515
(1942).
[1157] II. Erlenmeyer, Ch. J. Morel, Helv. chim. acta, 25, 1073 (1942).
(1158] K. Ganapatiii, A. Venkataraman, Proc. Indian Acad. Scl., '
22A; 362 (1945).
(1159] G. К 1 e i n В. P r i j s, Helv. tfhim. acta, 37, 2064 (1954).
£1160] H. A. J. Schoutissen, J. Am. Chem. Soc., 55, 4537 (1933).
(1161] H. II. Hodgson, W. H. H. Norris, J. Chem. Soc., Suppl. Issue,
1949, 181.
IV. ЗАМЕНА С—N-СВЯЗИ НА С—Hal
1 моль диазотированного амина) приливают по каплям п соляпо- или серною
раствор диазосоединения, к которому для удаления остатка азотистой кислоты
варительпо добавляют немного мочевины или, наоборот, кислый раствор диазосО(
ния приливают к холодному раствору иодида. Выделение азота закапчивается
многочасового стояния и, если необходимо, заключительного нагревания на во,
бапе. В качестве побочных продуктов могут образоваться фенолы, для удаления'
рых добавляют щелочь перед выделением арилиодида (отфильтровывают, перег<
с водяным паром или экстрагируют эфиром, хлороформом, бензолом или толу<
При этой реакции диарилы и а'зосоединсния не образуются, так как ее мех,
отличается от механизма реакции Запдмейера [1141]. Добавка иода иногда пов]
выход арилиодида [1162, 1163].
I2+ I —>• 18
[ArNgJ^-J-lg’
—> Arl + N2 + I3
В качестве примера обычного способа проведения реакции следует, у»
на синтез иодбензола из анилина [1164].
Дпазотироваппые амилы, неустойчивые в кислой среде, подвергаю’
имодействию с кислым раствором иодида в присутствии несмешивающ
с водой растворителя (например, СНС1з), который непрерывно экстраг
•бразующийся органический иодид [1163, 1165].
При взаимодействии водных растворов K.I с растворами солей д.
ния, полученными из слабоосновных аминов в концентрированной j
яри помощи нитрозплсерной и 85?-#-ной НзРО4 иодзамещенпы!? соединения
дуются с хорошими выходами. Примерами такой реакции являются полу
3,4,5-трпиод-1-нитробензола из 2,6-дииод-4-нитроанилина [1166] и аналог]
синтезы 3-иод-, 3,4-дииод-, 2,5-Дииод н 2,3,5-трииод-1-нитробензолов [:
Аминокарбоновые кислоты можно диазотировать «непосредств«
а) в разбавленной серной кислоте; б) в концентрированной серной ки<
в) «косвенным способом», для чего раствор смеси щелочных солей амнион
новых кислот с нитритом натрия вливают в концентрированную сод
кислоту.
Так, например, по способу (а) получают 2- и 3-иод-4-мотилбенз«
кислоты [1168], по способу (б) — 2,3,5-трииодбепзойпую кислоту [1169,
и по способу (в) — 6-иод-2-нитробензойную кислоту [1171], используя
ветствующим образом приготовленные холодные растворы солей днаа
В случае 3-метилбензойных кислот, имеющих в. одном из орто-полр>з
к СООН-груипе аминогруппу, нри взаимодействии разбавленных расг;
соответствующих сульфатов диазопия с иодидом калия образуются .т,
оксикислоты [1172].
[1162] А. Ваеует, Вег., 38, 2760 (1905).
[И63| В. A. Hems et al., J. Chem. Soc., 1949, 3432.
[1164] H. J. Lucas, E. R. Kennedy, Org. Synth., Coll. v. 2, 1943, p
[1165] K. Freudenberg, M. Reichert, Chem, Ber., 87, 1837 (1954).
[1166] R. B. Sandin, T. L. Cairns, Org. Synth., Coll. v. 2,
p. 604.
[1167] П. C a s s e b a u m, Arch. d. Pharm., 292/64, 571 (1959).
[1168] E. Kloeppel, Ber., 26, 1733 (1893).
[1169] C. J. Klemme, J. H. Hunte r, J. Org. Chem., 5 , 509 (1940).
[1170] S. C. J. Olivier, W. P. Combe, Rec. trav. chim., 69 , 25 (1950);.
C. A., 44, 5330 (1950).
[1171] B. R. Baker et al., J. Org. Chem., 17, 164 (1952).
[1172] ]-]. Abbes, Ber., 26, 2955 (1893).
2'>2
ОБРАЗОВ \HI1E С1Ш.Ш С-If if U РЕЗУ.ЧЬТ \ТЕ ЗАМЕЩЕНИЯ
Иногда рекомендуется добавлять медь. Так, добавляя 1,2 .«ель KI и 1 г
медной бронзы к разбавленному сернокислотному раствору 1 з(оль диазоти-
рованного п-аминофепола получают п подфенол; выход 69—72% от теорети-
ческого |1173}.
о-Иоднитробепзол [1162]. Заливают 100 г о-нитроанилина 1 л воды
и прибавляют 600.ua концентрированной H2SO4. Полученный прозрачный рас-
твор охлаждают до 5“ С и диазотируют соответствующим количеством NaNO2,
не допуская подъема температуры выше 10° С. Раствор сульфата диазонпя в не-
сколько приемов приливают к раствору 200 г KI и 200 г иода в 200 .чл воды.
После прекращения выделения азота смесь некоторое время нагревают на водя-
ной бане и встряхивают еще теплую смесь в делительной ворош\с с раство-
ром Na2SOs. При охлаждении количественно выпадает кристаллический
о-цодннгробоизол; т. нл. 54° С. Аналогично получают м- и н-поднитробензолы.
Замена аминогруппы на бром '
в ароматических и гетероциклических соединениях ;
с промежуточным получением пербромидов диазония
При взаимодействии бромидов диазония с бромом в бромистоводородной кислоте
образуются плохо растворимые в воде пербромиды диазония, которые в присутствии
воды или при нагревании в этиловом спирте или в ледяной уксусной кислоте разла-
гаются по реакции (стр. 251):
[RN-.f Вт’ —> RDr-J-W24-Br4
Реакция имеет препаративное значение для получения бромзамещенных гетероцик-
лических соединений из ампногетероцпклов (способ Крэйга).
2-Бромпцрндин [1174, 1175]. В 790 мл (7 моль) 48%-пой НВг (Крэйг
применял 63%-лую кислоту) в течение 10 мин при 10—20° С (охлаждение льдом
с солью) вносят 150 г (1,59 моль) 2-имппопиридипа. Затем прп 0° С (и ниже) при-
ливают по каплям при сильном перемешивании 120 мл брома (30 мин), причем
реакционная масса густеет вследствие осаждения оранжево-желтого пербромида.
Далее прибавляют по каплям еще 120 мл брома (15 мин), т. е. всего 4,7 моль,
и затем п течение 2 ч раствор 4 моль NaNO2 в 400 мл воды, поддерживая темпе-
ратуру 0и С пли ниже. К копцу добавления раствора NaN02 начинается
выделение азота. После 30 мин выдержки н смеси прибавляют раствор 15 моль
NaOII в 600 мл воды с такой скоростью, чтобы температура не превышала 20—
25° С. Продукт экстрагируют 4 раза эфиром порциями по 250 мл, экстракт сушат
100 г КОП и перегоняют. Выход 86—92% от теоретического; т. кпп. 74—75° С
(13 мм рт. ст.)- Большое значение имеет хорошее перемешивание (требуется
хорошая мешалка, способная перемешивать загустевающую по краям реак-
ционную массу).
Получить 2-иодпиридин по этому способу не удалось.
Ио способу Крэйга из соответствующих ампносоедпнений аналогично
[1175] получены 2-бром-З- и 2-бром-З-мстилпиридип [1176] (выходы 87 и 80%
Ог теоретического), 2-бром-б-мстплпирядии [1177] (выход 79%, устойчив только
е виде гидрохлорида), 4-бромпиридин [1178] (выход 85—95%), 2-бро.м-З-карб-
метоксипиразин [1179] (выход -43%), 1-бром-5,0,7,8-тетрагиДрОИЗОХинолии
[1173] F. В. Dai ns, F. Eberly, Org. Synth., Coll. v. 2, 1943, p. 355
[1174] L. C. Craig, J. Am. Chem. Soc., 56, 231 (1934).
[1175] C. F. H. Allo n, J. R. T h i г t 1 e, Org. Synth., 26, 16 (1946); Coll
v. 3, 136 (1955).
[1176] II, L. Bradlow, C. A. Vanderwerl, J. Org. Chem., 14, 512
(1949).
[1177] B. Ada ms,S. Miyano, J. Am. Chem. Soc., 76, 3170 (1954).
] 1178] A. Murray III, W. II. L a n g 11 a m, J. Am. Chem. Soc., 74, 6289
(1952).
11179] К. С. E 11 i n g s о n, R L. Henry, J. Am. Churn. Soc., 71, 2798
(1949).
IV. ЗАМЕНА С -N-СВЯЗП НА С-Н-Л
253
[1180] (выход 77,5?^) п 2-бром-4-\-лорбензтиазол [1181] (выход 72%). Условия
синтеза иногда варьируют. Например, при получении 2-бром-4-хлорбензтцазола
осле добавления раствора NaNO3 смесь выдерживают в охлаждающей бане
при перемешивании в течение 2 ч вместо 30 мин.
Замена аминогруппы, на хлор и бром в ароматических соединениях
(реакция Шеехтена *)
Аминодпфеннлы, амлмонафталпны и ампнофенантрены переводятся по способу
Запдмейера или по способу, видоизмененному Гаттермапом (стр. 247), в соответству-
ющие хлор- пли бромарилы, но зачастую с очень плохими выходами, в то время как
реакция Швехтепа, являющаяся развитием реакции Шимапа (термическое разложе-
ние фторборатов диазония), дает хорошие результаты.
При добавлении водных растворов равных весовых количеств HgC]3
и КС] или HgBr2, а также IIg(NO3)3, и КВг к растворам диазотированных аминов
до уравнениям 1 и 2 образуются комплексные соли, выпадающие в осадок.
[ArNsI+X- •>НеХ'» [ArN,]tHgXJ —► ЛгХ + Na+HgX, (1,
2[ArN.]+X-[АгК2Ц+ HgXV—► ЗАгХ+гМг + НвХг (2)
> д< \=С1 или Вг.
Соли высушивают промывкой ацетоном, метиловым спиртом пли эфиром
пли просто на воздухе п после смешения с двойным весовым количеством КС]
it J\H4CI пли КВт разлагают нагреванием. Так, по реакции 1 получают 2,2-
и 4,4-дихлор-, а также 2,2-дпбромдифенилы; выход около 80% от теоретиче-
ского[1182], а по реакции 2—2-бромнафталип; выход 60% от теоретического
[1182, 1183].
Разложение можно проводить осторожным нагреванием соли в кругло-
донной колбе с воздушным холодильником, в широкой трубке из йенского стекла
или в выпарной чашке. Последний способ применяют для получения 1-, 2- и
3-хлор- или 1-, 2- и 3-бромфенантренов из соответствующих амипосоединений
11184]. Амины диазотируют при 0° С, для чего их раствор в пиридине приливают
по каплям к раствору пптрозилсерпой кислоты, приготовленному из NaNO3
и смеси концентрированной HaSOi с водой в отношении 2 : 1.
Замена аминогруппы на фтор
в ароматических и гетероциклических соединениях
(реакция ПТимана)
Чаще всею для введения фтора в ядро ароматических соединений применяется
термическое разложение фторборатов диазония (реакция Шимана) [1185—1187].
Фторбораты диазония довольно устойчивы и нечувствительны к ударам. Фторбораты,
* Образование иодбепзола при разложении фепилдиазонийтрииодмеркуроата
открытов1929 г. А. Н. Несмеяновым [Z. anorg. allg. chem., 178, 300 (1929), цит
по книге А, Н. Несмеянов, Избранные труды, т. II, 19.59, стр. 361]
поэтому название «реакция Швехтена» едва ли уместно. — Прим. ред.
[ 1180] К, Ore we, А. М on do n, Е. Nolte, Ann., 564, 180 (1949).
IH81J R. С. Е 1 d е г 1' i е 1 d, F. W. S ho г t, J. Org. Chem., 18, 1099 (1953).
[1182] 1-1.-W. Schwecliten, Ber., 65, 1005 (1932).
[1183| M. s. Newman, P. H. W i s e, J. Am. Chem. Soc., 63, 2847 (1941).
[1184] W. E. Bachmann, Ch. H. Hoatner, J. Am. Chem, Soc., 58
2194 (1936).
[1185] G. В a I 7, G. S c h i e m a n n, Ber., 60, 1186 (1927).
[1186] G. Schicmann, J. prakt. Chem., 140, 97 (1^34)
[1187] Д. к о e, Org. Reactions, v. V, J. Wiley a. Sons, New York, 19'49, p. 193—228.
254
ОБРАЗОВАНИЕ СВЯЗИ С— На! В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
выпадающие в осадок в процессе реакции, получают двумя способами: взаимодействием |
борфторводородной кислоты пли ее натриевой соли NaBF4 с полученными обычны» ij
способом солянокислыми растворами диазосоединений . $
ArNHo • ИХ4-HNOa-ЬBFJ —> 2НаО + X" + [ ArN2]+ BF 4 .j
[ArNa]+BFj —► ArF4-Na + BF8 J
и диазотированием растворенных в HBF* аминов водным раствором NaNO2. Объе»Я
реакционной массы, содержащей ион Вг4", должен быть возможно меньшим, чтоби(д
концентрация прогонов была менее 1 молъ/л, так как многие^торбораты дназоння&а
плохо растворимы в холодной воде, а в сильнокислой'среде их .растворимость вое«я
растает. Фторбораты арилдпазония с фенольными и карбоксильнымигруппами хорошей
растворимы в воде. Фторированные в ядро ароматические кислоты молено получата^
по реакции Шимана через их эфиры (ср. стр. 255). Диазоеоединения, синтезированные^
по второму способу, сразу выпадают в осадок в виде фторборатов, что сводит к минж^Я
муму побочные реакции, например образование фенолов. Однако недостатками спо~Я
соба являются: загустевание реакционной массы и работа в среде, содержащей ив-(я
быток агрессивной HBF4.
Фторбораты диазония, полученные любым из обоих способов, выдерживают
менее 30 мин при температуре не выше 0° С, быстро отсасывают, промывают холодны»^
растворами HBF4 или NaBF4, спиртом и эфиром и тщательно высушивают под тягоцЯ
на фильтровальной бумаге, положенной на проволочную сетку. ?Л
Если разложению подвергают влажные фторбораты диазония, то выходы галоЭДЯ
генндов низкие. При термическом разложении фторборатов, начало которого определи
ляют по выделению белых паров трифторида бора, выходы галогенидов тем вышщйИ
чем ниже температура разложения. Разложение ведут в стеклянной колбе, заполнениоюИ
примерно наполовину фторборатом диазония, или в широкой трубке из иснскогЗД
стекла, присоединенной к приемнику с обратным холодильником. При разложений™
легколетучих фторборатов верхний конец холодильника соединяют с еще одним охла^Я
ждаемым приемником. Трифторид бора можно поглощать в приемнике водой, паствоДИ
ром щелочи или суспензией NaF в воде, причем образуется NaBF4 (следует помниткИ
о том, что кристаллы могут закупорить трубку). Трубку с сухой солью нагпеваюЙМ
снаружи у верхней границы заполнения, после чего разложение в большинстве слиЯ
чаев продолжается самопроизвольно. Соединения, содержащие иитрогруппы, частмВ
разлагаются очень бурно. Поэтому их смешивают с трех-пйтикратпым весовым колзЙЯ
чеством песка, BaSO4 или NaF и разложение ведут порциями по 5—25 г. '
Фторбораты диазония, ив которых при сухом термическом разложении ариД«Н
фториды получаются с плохими выходами, можно разлагать отдельными порциямжЯ
в безводном углеводороде, нагретом на несколько градусов выше температуры разлодМ
жения фторбората. Так получают, например, л-фтор- или о-фторпропиофеноп в кэдИ
пящем толуоле или гептане J1188J.
Помимо реакции Шимана (термическое разложение сухих фторборатов диазониадИ
существуют более новые способы обмена аминогруппы ароматических соединенияЯ|
на фтор, несколько напоминающие реакцию Зандмейера или ее видоизменение ПяМ
Гаттерману.
Из фторборатов диазония можно получать арилфториды* даже при комяатао!^В
температуре [i 189} перемешиванием ацетоновых, водно-ацетоновых или водных раствояИ
ров фторборатов с гаттермановской медью или CuCl. Этот способ, который в основндшИ
характеризуется хорошими выходами, с успехом применяется для разложения ра<&Я
творимых в воде фторборатов диазония или таких фторборатов термическое разлояфЗИ
ние которых (нитрозамещенные) протекает очень бурно, вследствие чего их можна^Н
разлагать только маленькими порциями. аЯ
* В ряде случаев ароматические фторзамещенныо могут быть с успехом получен^
при прямом диазотировании арнламина и разложении фтористого анилдпааонн^
в безводном HF [R. Ferm, С. Van dor Werf, J. Am. Chem. Soc.,
4809 (1950)J. — Прим. ped. j
[1188] B. L. Z e n i t г, W. II. Hartung, J. Org. Chem., 11, 449 (1946). ’?
[1189] E. D. Ber gjn a n n, S. Berko vic, R. I k a n, J. Am. Chem. SocJ
78, 6037 (1956). J
IV. ЗАМЕНА C-N-СВЯЗИ НА С—Hal
255
Фторбензол [1185, 1190]. В аппарате емкостью 40 л или толстостенном
стеклянном стакане (300 X 300 лж) смешивают 1350 мл воды с 1650 мл НС1
{<1 — 1,19) и при энергичном перемешивании охлаждают смесью льда с поварен-
ной солью. Затем в полученный раствор вносят при 5° С и перемешйЬании третью
часть от 2075 е гидрохлорида анилина и при температуре не выше 7° С медленно
приливают раствор 1200 г NaNO2 в 1500 мл воды.
В процессе введения нитрита 'продолжают вносить отдельными порциями
гидрохлорид анилина, вследствие чего на дне сосуда всегда имеется некоторое
количество непрореагировавшего гидрохлорида. Ко времени введения половины
раствора нитрита в реакционную массу должен быть внесен весь гидрохлорид
анилина. Для полного диазотирования используется почти все количество рас-
твора нитрита (проба с иод-крахмальной бумажкой). Одновременно в покрытом
воском стеклянном, медном пли свинцовом сосуде готовят раствор HBF4, для
чего в несколько приемов при перемешивании и температуре не выше 25е С
(охлаждение ледяной водой) вносят 1000 г НзВОз (16,2 лоль) и 2150 г 60%-ной
HF (65лоль). Приготовленный раствор охлаждают и приливают в охлажден-
ный ниже 0ф С раствор соли диазония. Раствор вносят возможно быстрее, но так,
чтобы температура не поднималась выше 10° С. Для энергичного перемешива-
ния густой массы применяют медленно вращающуюся металлическую или дере-
вянную мешалку, покрытую кислотоупорной краской пли гумированиую. Через
20—30 мин после добавления всей HBF4 выпавший осадок отфильтровывают
на нутч-фильтре и промывают 800 мл ледяной воды, равным количеством мети-
лового или этилового спирта и 900 мл эфира, каждый раз возможно полнее
отсасывая массу. Фторборат расстилают тонким слоем и сушат па воздухе в тече-
ние почи. Затем его разлагают в колбе емкостью 12 л или двух колбах емкостью
по 5 л. Приемниками служат три соединенные между собой широкими трубками
колбы Эрленмейера, присоединенные к реакционной колбе с помощью холодиль-
ника большого диаметра, погруженные в охлаждающую смесь. Отходящие газы
отводят в тягу или пропускают предварительно через ледяную воду пли раствор
соды. Для того чтобы вызвать разложение фторбората диазония, Йолбу нагре-
вают снаружи открытым пламенем у верхней границы наполнения и затем под-
держивают спокойное течение реакции, по мере необходимости охлаждая или
подогревая массу до тех пор, пока не прекратится выделение паров BFs даже
при сильном нагревании колбы.
Собравшийся в приемнике дистиллят отделяют от выкристаллизовав-
шегося фенола и несколько раз промывают 10%-ным раствором NaOH (до почти
бесцветных промывных вод) и водой. Так как плотность фторбензола 1,025,
ю последние порции промывной щелочи необходимо очень тщательно отделять,
ибо, в противном случае, при дальнейшей промывке водой образуется разба-
вленная щелочь почти такой же плотности, которую очень трудно отделять
от фторбензола. Промытый продукт сушат СаС12 н перегоняют на колонке. Выход
780—870 г (50—56% от теоретического); т. кип. 84—85° С.
Описание способа получения малых количеств фторбензола с примене-
нием NaBF4 и разложением в трубке из йенского стекла (выход 64% от теоре-
тического) см. [1191J.
Для приготовления раствора фторбората натрия [1192], содержащего
примерно 5,38 моль NaBF4, к смеси 338 г НзВОз и 1338 мл концентрированной
НС1 прибавляют при перемешивании и охлаждении 900 г NaF. После 2 ч вы-
держки раствор фильтруют.
п-Фторбензойная кислота [1193]. В колбе емкостью 5 л при нагревании
на паровой бане и перемешивании обрабатывают 165 г этилового эфира п-амино-
бепэоиной кислоты (1 моль) 204 мл соляной кислоты (d — 1,19) и 300 мл воды.
Образуется бесцветная паста гидрохлорида. Ее охлаждают до 0° С смесью
льда с поваренной солью и медленно прибавляют к пой при перемешивании
и температуре не выше 7° С раствор 72,6 г NaNO2 (1 моль 95%-ного NaNOa)
[1190] В. Т. Flood, Org. Synth., Coll. v. 2, 1943, p. 295.
[1191] M. Г у д л и ц к и й, Химия органических соединений фтора, Госхим-
издат, 1961, стр. 103.
[1192] A. Roe. Org. Reactions’. 5, 203 (1949).
[1193] G. Schiemann, W. W inkelm tiller, Org. Synth., Coll. v. 2,
1943, p. 299.
256
ОБРАЗОВАНИЕ СВЯЗИ C-Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
п небольшом количестве воды до сохранения слабополощитрльной под-
ьрахмальной реакции смеси в течение 10 мин.
Одпонременно готовят раствор I1BF4. В покрытом изнутри парафином
с гек.гяниом стакане или свинцовом сосуде при охлаждении льдом (температура
(!* должна превышать 25° С во избежание расплавления воска) растворяют 68 г
11зВО3 (J ,1 молъ) в 133? 60%-ной HF пли применяют продажную 40%-ную
НВ1‘4 (200 г). Охлажденный льдом раствор довольно быстро приливают при
перемешивании к раствору соли диазония; температура не должна превыигать
10° С. Затем перемешивают еще 20—30 .чин if отфильтровывают па пугч-фильтре
выпадающий в виде густой пасты фторборат диазония- Его промывают после-
довательно 300 мл холодной воды, 300 мл метилового спирта и 200 мл эфира,
отсасывая каждый раз как можно тщательнее, и, наконец, сушат в вакуум-
эксикаторе над концентрированной 1I2SO4. Выход фторбората дпазоппя
[n-CaII5OOCC6Ii4N2IBF4 198—205 г, что составляет 75—78% от теорети-
ческого; т. разл. 93— 94° С-
Термическае разложение фторбората удается провести без потерь вслед-
ствие осмолеппя только в том случае, если оп тщательно высушен. Его вносят
порциями но 85 г в колбу емкостью 2 л, связанную широкой изогнутой стеклян-
ной трубкой с приемником емкостью 1 л. Нескопдспспровавшисся пары В Fa
пропускают в воду, а непоглотившпйся газ отводят в хорошо действующую тягу.
Колбу сначала нагревают сверху коптящим пламенем горелки до начала выде-
ления белых паров В Fa, поело чего разложение фторбората протекает самопро-
извольно. Прп затухании реакции прибегают к периодическому нагреванию.
Реакцию считают законченной, когда под действием сильлого нагревания вся
твердая масса переходит в жидкость. Основное количество эфира п-фторбензой-
цой кислоты (т. кип. 105—106° С при 25 мм рт. ст.) остается в реакционном
сосуде, меньшая часть уносится в приемник, откуда его извлекают эфиром,
фильтруют и отгоняют эфир.
Дли омыления карбэтоксильной группы эфира п-фторбопзойпой кислоты
эфир, не подвергая очистке, нагревают в колбе с обратным холодильником с рас-
твором 56 г КОН в 80 мл 95%-ного этилового еппрта п 120 мл воды. Еще горя-
чую смесь фильтруют п кислоту осаждают подкислением концентрированной
НС1 до кислой реакции по бумаге конго. После охлаждения выпавшую п-фтор-
бензойную кислоту отсасывают и сушат. Для очистки ее растворяют в горячем
пастворе 40 г К2СОз в 400 мл воды, раствор обесцвечивают углем, фильтруют
в горячем нпде и осаждают продукт ПО. Выход 38—40 г (63—69% от теорети-
ческого, считая на этиловый эфир n-аминобензойной кислоты); т. пл. 186° С.
п-Фторфенол [1189]. К 200 мл 56%-ной HBF4, охлажденной смесъкг
льда с повареппой солью, при —6° С одновременно при перемешивании в не-
сколько приемов прибавляют растворы 10,9 г п-аминофенода в 50 мл 56%-ной
HBF4 и 6,9 г КаК'О2 в 50 мл воды с таким расчетом, чтобы в реакционной массе
всегда был избыток нитрита. К полученному прозрачному раствору добавляют
5 г CuCl н2ч перемешивают при 80—90° С, После охлаждения льдом получают
n-фторфенол; выход 71% от теоретического; т. пл. 46° С.
Таким же способом с выходом 87% от теоретического получают п-фтор-
фенилуксусную кислоту.
В отличие от довольно устойчивых фторборатов диазонпц ароматических соеди
пений фторбораты диазония 2- и -З-аминопиридинов весьма нестабильны [1194].
2-Фторйиридии. Получают диазотированием раствора 2-амплойиридипа
в 40%-пой НВГ4 при 10u С (нитрит добавляют в несколько приемов), причем
образующийся фторборат тотчас разлагается с выделением тепла; выход 34%
от теоретического.
З-Фторпиридин. Фторбораг З-пиридннднязонпя, смоченный вфнром,
устойчив некоторое время прп 10е С. но бурно разлагается прп испарение
растворителя. Он выпадает в осадок при диазотировании 3-аминопцридина
и смеси 40%-ной HBF4 и 95%-ного этилового спирта (1 : 2) при 0° С к добавле-
нии и конце реакции эфира. Фторборяг медленно разлагают в виде суспензию
в пысококипящем летролепном эфире при 15—20° С; выход 50% от теорети-
ческого.
[1194] A. Roe, G. F. Hawkins, J. Am. Chem. Soc., 69. 2443 (1947).
ЗАМЕНА С—S-СВЯЗП НА С—Н.П
257
Замена аминогруппы на группу SCN
в ароматических соединениях
Для введения группы SCN в ароматические соединения значительно чаще поль-
зуются реакцией Зандмейера или ее видоизменением по Гаттерману, чем прямым
замещением водорода на родан (стр. 190). К полученному обычным способом раствору
соли дпазония прибавляют водный раствор роданида щелочного металла и CuSCN
или, наоборот, раствор соли диазония постепенно приливают к водному раствору
K3[Cu(SCN)4J. Можно применять также порошок металлической меди [1195—1197].
Реакция протекает с выделением азота часто уже при комнатпой температуре пли при
умеренном нагревании.
[,ед,сх-+8См- C"SCN <"'C“U a.scn+n2+x-
Наряду с арилроданидами (арилтиоцианатамп) могут образовываться ароматы
ческие горчичные масла (арилизотиоцианаты) [1198].
5-Родансалициловая кислота [1199]. Растворяют 7,65 г 5-ампносалици
ловой кислоты в 600 мл воды и 14,5 мл концентрированной НС1 и диазотирую1:
J,8 г NiiNO2, как обычно. Серо-голубую соль диазония быстро отжимают и ио
степенно вносят при 65° С в раствор 6,1 г CuSCN, 30 г KSCN и 17 мл воды. П<
мере выделения азота раствор окрашивается в светло-коричневый цвет. По охла
жденип в осадок выпадает 5-родансалициловая кислота, которую для очистк!
иереосаждаю? НС1 из натриевой соли, получендой из разбавленного раствор}
Na2COj, и перекристаллизовывают из бензола; выход 73% от теоретического
т. пл. 167 —168° С.
V. Замена С—S-связи на связь С—Hal
В известных условиях сульфогруппы ароматических соединений довольно легк
обмениваются на хлор и бром. Реакция обмена протекает особенно легко, если в орте
пли пара-положении имеются окси- или аминогруппы [1200]. При проведении эти,
реакций следует учитывать возможность замещения атомон водорода галогепом. Так
например, при действии бромид-броматного рас, пора в солянокислой среде при комнач
ной температуре на сульфаниловую кислоту получается с количественным выходе»]
2,4,6-триброманилин [462] (ср. стр. 151). Если в молекуле ароматической сульфокис
лоты имеются заместители, препятствующие электрофильному замещению галогенам:
(NO2 или еще одна группа SO3H), то эту реакцию можно использовать для препарг
тивных целей, например для синтеза 2-нитро-4,6-дибромфенола или 2-бром-6-питрофс
пол-4-сульфокислоты [1201] из 2-нитрофенол-4,6-дис.ульфокислоты и ПВг при 0—15s ।
или хлорированных нитронафталинов [1202] из нитронафталпнсульфокислот и рас
твора NaClOs в IIC1 при 90—95° С.
1- и 2-хлор, а также 1,5-, 1,6-, 1,7- и М-ддхлораитрахицоны получают взаимс
действием соответствующих сульфокислот с NaC103 или хлором в НС1 при 100° С
с бромом и небольшим количеством воды под давлением при 190—2£0° С образуйте
соответственно бромантрахиноны [1203—1205].
[1196
[1197
[1198
[1199
[1200
[1201
[1202
[1203]
[1204]
[1205]
L. Gattermann, W. Hausknccht, Ber., 23, 738 (1890).
G. Thurnaucr, Ber., 23, 769 (1890).
A. Hantzsch, B. Hirsch, Ber., 29, 947 (1896).
J. W. Dienske, Rec. trav. chim., 50, 407 (1931).
H. P. Kaufmann, E. Rofbacli, Ber., 58, 1559 (1925).
R. L. D a 11 a, J. Cli. Bhoumik, J. Am, Chem. Soc., 43, 303 (1921).
E. SakeJlarios, Ber., 55 , 2846 (1922).
P. Friedlander, S. К a r a moss in i s, O. Schenk, Ber., 55,
(1922)
Frdl’, 9, 673 (1908-1910),
H. E. Fierz-D a vi d, А. К г e b s о r, W. And era и, JJeJv. chjr
acta, 10, 197 (1927).
A. A. Goldberg, J. Chem. Soc., 1932, 77.
17 Заказ 1835.
258
ОБРАЗОВАНИЕ СВЯЗИ С—Hal В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
В 1,2,3,4,5,6,7,8-октагидроантрацен-Э-сульфокислоте, в которой исключена
возможность дополнительного галогенирования ядра, сульфогрулпа легко обмени-
вается [1206] на бром под действием бромнд-бромата или иодид-иодата в 1 п. растворе
НС1 при 35е С. Сульфохлоридная группа в арилсульфохлоридах также может обмени-
ваться на хлор, например при нагревании (около 220° С) с РС16 в запаянной трубке или
в трпхлорбензоле или с SOC12 [1204, 1207].
Алифатические и ароматические сульфохлориды способны термически расще-
пляться с образованием аякил- или арилхлоридов и SO8:
RSO2Cl —> RC1 + SO2
Однако обычно при этом протекают» и побочные процессы, которые зачастую
не позволяют использовать ^.реакцию для препаративного получения арплхлорпдов;
подробно об этой реакции см. [1208].
Расщепление алифатических сульфохлоридов происходит с хорошими выходами.
В качестве примера можно привести расщепление додекансульфохлорида в кипящем
ксилоле [1209] с образованием додецилхлорида и SO2; довольно хорошо протекает
также расщепление бензилсульфохлорида [1210].
Особенно легко происходит отщепление SOg от арилоксиметансульфохлоридов.
В то время как при действии РС15 на фенилметансульфокислоту (CeH5CH2SO5H) бен-
зилхлорид [1211] образуется при 100° С, а при действии РВт5 на ео натриевую соль
бекзилбромид [1212] образуется при 90° С, хлорметил- или бромметилариловые эфиры
[1213] получают с 80—90%-ным выходом простым растиранием в ступке 1 моль сухой
натриевой соли арилоксиметансульфокислоты с 2 моль РС15 или РВг5.
ArOCH2SO3Na + PC15 —> ArOCH2C14-SO2 + NaCl + POCl3
Возможность обмена связи С—S на связь С—Hal используют также для полу-
чения хлорированных гетероциклических соединений. Пропуская хлор при охлажде-
нии в водную суспензию этилмеркаптопиримидина, как правило, получают сульфон
(реакция 1); в некоторых случаях наряду с хлорзамещенным гетероциклическим соеди-
нением образуется этансульфохлорид (реакция 2), доля которого заметно возрастает
при незначительном повышении температуры.
Примером этого является получение 2-хлор- к 2,4-дихлор-5-карбэтоксипири-
мидина [1214, 1215].
ух /СООС2Н5
N |
Л А
СзНг.З/ V ХС1
уу ZCOOC2H:,
СЬ; 0-20° с |
C2H5SO2//Xrf4''cl
ух уСООС2Н5
С!»;40—50°С NT , „ т.
- II j
ci/yXci
Меркаптосруппа также может быть обменена на хлор.
1206] G. S с h г о е t е г. Вег., 60, 2042 (1927).
1207] Е. Nolting, Вег., 8, 1091 (1875).
1208) A. R i е с h е, Е. Naumann, J. prakt. Chom. [4], 9, 1Q8 (1959),
1209] F. As in ger, Ber., 77—79, 193 (1944—1946).
1210] H. Limpricht, H. v. Pochmann, Ber., 6, 534 (1873).
1211] T. B. J 0 h n s 0 n, J. A. Abmler, J. Am. Chem. Soc., 36, 381 (1914).
[1212] W. H. Hunter, Б. E. Sorenson, J. Am. Chem. Soc., 54, 33&5 (1932).
[1213] Ц. J. Barber et al., J. Appl. Chem., 3, 266 (1953).
[1214] J. M. Sprague, T. B. Johnson, J. Am. Chem. Soc., 57, 2252 (1935).
[1215] E. Ballard, T.,B. J ohnson, J. Am. Chem. Soc., 64, 797 (1942).
VI. ЗАМЕНА G—С-СВЯЗИ НА С—Hili
25'
2-Хлорбензтиазол [1216]. В колбе емкостью 3 а с мешалкой п воздузд
ним холодильником к 1 моль 2-меркаптобензтиазола прибавляют маленьким]
порциями 1,1 .ноль SaCl2. Когда экзотермическая реакция ослабевает, смес
осторожно подогревают до прекращения новообразования, затем кипятят 90 ми,
с обратным холодильником, оставляют па ночь и перегоняют черный продукт
отбирая фракцию 150—170° С (55—65 мм рт. ст.). Эту фракцию промываю1
10%-ным раствором Na2C03 и водой, сушат над Na2SO4 и еще раз перегоняют
Выход 72% от теоретического; т.‘ кип. 158—162° С (50 мм рт. ст.).
Реакция перевода тиоэфиров в органические галогензамещенные соединени;
имеет очень малое препаративное значение. Указания до расщеплению соеднпенщ
В — S—R в присутствии хлора, брома, бромциана или метилиодида с заменой С—S
связи связью С—Hal см. [1217]. При действии 1 моль брома на тиоацетали в безводно;
среде (эфир) при комнатной температуре образуются бромполумеркаптали (а-бромтис
эфиры) и сульфенилбромиды. Из этилтиогликозпдов аналогично получаются гликозил
бромиды;
По этой реакции из 2,3,4,6-тетраапотнл-₽-этилтио-£’-глюкозида и броме
перегнанного над H2SO4 в абсолютном эфире при 20° С, получают а-ацетобро^
глюкозу, а из 2,3,4,6-тетраацетил-а-этилтио-Г)-глюкозида — р-ацетобромглк
козу, которую нельзя синтезировать другими методами [1218].
Дихлорангидриды дикарбоповых кислот можно получать с очень хорошим
выходами из циклических тиоангидридов карбоновых кислот (если последние легк
доступны):
СО
^/с°а
^/\coci
+ SC15
Пропуская хлор при 245° С через расплав тиоангидрида фталевой кислот
до прекращения образования SC12, почти со 100%-ным выходом получают о-фтг
лоплхлорид [1220]. Тиоангидрид легко синтезировать [1219] пз фталевое
ангидрида и NaS.
VI. Замена С—С-связи на связь С—Hal
Расщепление С—С-связи с одновременным образованием галоидпроизводньа
рассматривается в других разделах, поэтому здесь приводятся только ссылки на эт
разделы. На стр. 151 описано самопроизвольное отщепление СО2 или СО при бромир*
вании ароматических о- к n-окси- и амипокарбоновых кислот. Галоформная реакци
обсуждается в разделе о расщеплении С—С-связи (стр. 888, 845), а также в наст«
ящем разделе па стр. 185- Реакции сухих серебряных солей карбоновых кислот с гал«
генами в молярном соотношении 1 ; 1 (реакция Бородина — Хунсдикера), представ
ляющие препаративный интерес для получения алнилгалогенидов и эфиров со-гал.
генкарбоновых кислот, описаны в разделе о расщеплении С—С-связей (стр. §1&
[1216] W. S с о 11, G. W. Wa 11, J. Org, Chem., 2, 149 (1937).
[1217] D. S. Tarbell, D. P. Harnish, Chem. Bev., 49, 1—45 (1951).
[1218] F. Weygand, H. Z i e щ a n n, II. J. Bostmann; Chem. Bee
91, 2534 (1958).
[1219] A. R e i s s e г t, II. Hoile, Ber., 44, 3029 (1911).
[1220] E, О 11, A, Langenohl, W. Zerwcck, Ber., 70, 2360 (1937).
17*
obFAjujiAFiiiE с. о-связи в результате присоединения
260
ОБРАЗОВАНИЕ СВЯЗЕЙ
УГЛЕРОД — КИСЛОРОД *
ОБРАЗОВАНИЕ С—О-СВЯЗИ В РЕЗУЛЬТАТЕ РЕАКЦИИ ПРИСОЕДИНЕНИЯ
I. Присоединение кислорода по С = С-связи
1. Молекулярный кислород
Молекулярный кислород присоединяется по двойным связям таких олефинов,
как стирол, 1,1-дифенилэтилен, диметил- и диэтилкетен, винилацетат, циклопента- '•
диен или циклогсксадиен с образованием полимерных перекисных соединений. Про- •
текающая при этом реакция является реакцией сополимеризации олефина с кисло» '
родом [1, 2].
^>C=C<f —С—С—00 /с”с\ —С—С—00—С—с—
z х I I till
.1 II.
—> —С—С—00—С—С—00
till
Сополимеры с кислородом большей частью чрезвычайно взрывчаты. При обра-
щении с ними необходимо принимать специальные меры предосторожности, описание
которых приведено в разделе «Образование перекисей путем аутоокисления» (стр. 288).
Перекись дмфенилэтмлена. По методу Штаудингера [3] пропускают
кислород в 10 а 1,1-дифенилэтплспа, помещенного в маленькую реакционную
трубку с внутренним холодильником, которую облучают лампой в 500 свечей.
Через трое суток извлекают петролейным эфиром 0,5—1 г бесцветной аморфной
перекиси дифенилэтилеиа. После промывки эфиром получается препарат, пла-
вящийся при 131—132е С (разд,).
В отличие от вышеприведенной реакции образования полимерных перекисей
из олефинов и кислорода «фотосснсибилизированное аутоокисление» диенов приводит
к образованию циклических мономерных перекисей:
Реакция протекает аналогично диеновому синтезу с кислородом в качестве
диенофила [4]. Такие же перекиси могут быть получены из аценов, соответствующих
стсринов и низкомолекулярных диенов (например, циклогексадиена) [5]. Для ини-
циирования реакции необходимо облучение, иногда даже добавление сенсибилиза-
торов.
* Материал обработали М. Шульц и Г. Зеебот.
[ 1} W. Kern, Н. W i 11 е г s I п и, Angew. Chem., 67, 573 (1955); R. С г j е g е е,
в книге llouben-Wcyl, Melhoden der organischen Chemie, Bd. 8, 4 Aufl.,
Stuttgart, 1952, S. 1.
[2] A. A. M i 11 e r, F. R. Mayo, J, Am. Chem. Soc., 78, 1017 (1956),
[3J П. Standinger Ber., 58, 1075 (1925).
14] К, A 1 d er, M. S ch u ma с пег, в книге Fortschritte der Chemie organischer
Naturstolfe, 13d. X, Springer-Verlag, Wien, 1958, S. 96ff.
[5] А. Шенберг, Препаративная органическая фотохимия, пор. с нем., Издат-
инлцт, 1963.
i. присоединение кислорода по с^с-спнзи
261
Ау юокпсленио аценов особенно подробно научено Дюфрессом и сотр. [6].
Я растворах (например, в CS2) на свету, без добавления сенсибилизаторов ацены
быстро превращают» в соответствующие перекиси. Так, например, образуется пере-
кись рубрена
Перекись стероидного ряда впервые получена Виндаусом и Брункеном из
эргостерина. Новейшие исследования показали, что перекись эргостерина имеет
структуру внугрициклической перекиси [8)
Фотосепспбплизпрованное аутоокпсление приобрело особенно большое значе-
ние после опубликования работ Шенка с сотр. [9], благодаря которым этот способ
получил широкое распространение. Шенк и Цщлер [10] сначала синтезировали
фотосенсибнлпзпрованным аутоокнсленпсм а-терпипена встречающуюся в природе
перекись — аскаридол:
Перекись эргостерина [7]. К нагретому до 60° С раствору 4 г эргосте-
рина и 1200.нл 95%-пого спирта прибавляют 6 мг эозина, и смесь освещают
в большом химическом стакане следующим образом: в сосуд, содержащий рас-
твор, погружают более узкий стакан, снабженный приспособлением для подвода
и отвода воды, в котором горкт газонаполненная лампа в 200 вт. Через раствор
эргостерина медленно пропускают струю кислорода до тех пор (примерно через
3 ч), пока не прореагирует весь эргнетррин (в пробе со спиртовым раствором
дигитопина не должен выпадать осадок). Затем большую часть спирта отгоняют
в вакууме, отделяют выпавшие кристаллы перекиси и перекристаллизовывают
их из спирта или ацетона до получения вещества с постоянной т. пл. 178° С;
выход 70% от теоретического.
2, Озон
При расщеплении олефипов озоном первым продуктом взаимодействия озона
с пенасыщепным соединением является неустойчивый «первичный озонид», существо-
вание которого было недавно экспериментально доказано Григе и Шредером [11]
[6] Ch. Dutraiss е, Bull. Soc. chim. France [5], 6, 422 (1939); Ch. D u f г a i s s e,
1-7) ' ° I I n z, Bull. Soc. chim. France |5], 5, 1073 (1938).
MJ A. WiHaus, J, Brunke n, Ann., 460, 225 (1928).
181 W D „ _______ T? 1 < _T’ nr Л , ' - _nt,.... ». nr,
[9] G. O. Schenk, Angew. Chem., 64, 12 (1952); 69, 579 (1957).
ni °- Schenk, K. Ziegler, Naturwiss., 32, 157 (1944).
U'J H. Grieger, G. Schroder, Chem. Bor., 93, 689 (1960).
иьРАЗОВАНИЕ С—О-СВЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
(на примере озонирования /пранс-ди-трет-бугилэтилена при —75° С. Первич]
озонид экзотермически распадается на альдегид пли кетон и перекисный биполяр]
ион. Выделяемый озонид образуется лишь в результате повторно! о соединения об
«сколков молекулы:
Получающиеся озониды, следовательно, уже не имеют, как это впервые пока
Штаудингер 112], углеродного скелета исходного ненасыщенного соединения;
подтвердил также Рихе, получив озонпды отщеплением воды от диоксидиал:
перекисей [13].
Озониды и их расщепление описаны в разделе «Разрыв углерод-углерод
связей» (стр. 835).
3. Получение алкиленоксидов из производных этилена
Окисление ненасыщенных соединений органическими надкислотами в
водных растворителях, таких, как хлороформ, эфир, диоксап, бензол, ацетон, пр
кает с образованием этиленоксидов по уравнению: *
^С=С<^4- RCOOOH
>С---с/ <- RCOOH
o’
Особенно большое значение для эпоксидирования приобрели надкисл1
надбепзойная, монопадфталевая, надуксусная и трифторнадуксуспая.
Ввиду неустойчивости образующихся алкиленоксидов они получаются с X
шими выходами только при проведении реакции при возможно более низких т<
ратурах [14] и в отсутствие сильных кислот [15].
При использовании надбензойной и монопадфталевой кислот необхр
употреблять инертные растворители; в отсутствие сильных минеральных кщ
часто применяющихся в качестве катализаторов при синтезе надкислот, и при т!
ратуре реакции ниже 30° С эпоксидирование надуксусной кислотой можно провс
в уксуснокислом растворе.
При окислении надуксусной, трифторпадуксусной и особенно надмуравь
кислотами образующиеся вначале эпоксиды легко превращаются в ироизво;
а-гликолей.
Скорость взаимодействия олефинов с надкислотами зависит ог числа и ,
заместителей у ненасыщенных атомов углерода [16]. Алкильные и особенно ая.
сильные группы у двойной связи повышают скорость реакции. Очень быстро о
ляется тстраметилэтилен; эпоксидирование випилэтилового эфира [17] протер
в течение нескольких мипут с выделением большого количества тепла. Фенил?
группы обычно действуют как алкильные. Непредельные соединения, в кото
двойная связь сопряжена с карбоксильной, карбалкоксильпой или карбонит
группами, как, например., коричная, малеиновая, фумаровая, кротоновая кнел
или их эфиры, а также «.^-ненасыщенные кетоны, с трудом переводятся надкислот
в эпоксиды или вовсе не эпоксидируются.
* Эта реакция открыта и подробно изучена Н. А. Прилежаевым (II. А. П р и.
ж а е в, Органические перекиси, их применение для окисления пспредель]
соединений, 1912). — Прим. ред.
[12] Н. Staudinger, Вег., 58, 1088 (1925).
[13] A. Rieche, Й. Meister, Вег., 65, 1274 (1932).
[14] Т. W. F j ц d 1 е у, D. Swern, J. Т. Scanlan, J. Am. Chem. Soc.p
412 (1945), ' :
[15] D. Sworn, G. N. В i 11 e n, T. XV. Findley, J. T. Scanlan, J. i
Chem. Soc., 67, 1786 (1945).
[16] D. Swern, J. Am. Chem. Soc., 69, 1692 (1947).
[17] M. Borgmann, A. Mickeley, Ber., 54, 2150 (19211.
I. ПРИСоЕДИНЕИПГ КИСЛОРОДУ ПО С=ОСВЯЗП
263
Это в рапной мере относится п к соединениям с несколькими С=С-связями
(изопрен, гераниол, цитраль и др.). Обзор методов эпоксидирования надбензойными
кислотами см. 118].
Для синтеза алкиленоксидов в первую очередь применяют надбензойную
кислоту, котору ю можпо удобно получать расщеплением перекиси бензоила с помощью
алкоголята натрия [19].
Надбензойная кислота. Растворяют 10 г перекиси бензоила * в 20 лм
юлуола и раствор охлаждают при перемешивании до —5° С. Затем в течение
j мин прибавляют охлажденный до —2е С раствор 2 г натрия в 50 мл 96%-ного
' этилового спирта. Тотчас же выпадает осадок надбензоата патрня. К смеси
прибавляют 200 мл ледяной воды, в которой надбвнзоат растворяется. Отделен-
ный в делительной воронке от толуола водный раствор надбензоата натрия
дважды промывают эфидом для удаления остатка толуола и этилового эфира
бензойной кислоты. В очищенный водный раствор прибавляют смесь 5 г концен-
трированной H2SO4 с 69 г ледяной воды. Выделившуюся под действием Нг304
свободную надкислоту извлекают дважды 60 мл хлороформа. Обе полученные
хлороформные фракции объединяют и сушат Ma2SO4. Этот хлороформный рас-
твор можно использовать для окисления. Для получения кристаллической над-
бензойной кислоты растворитель выпаривают, просасывая через раствор сухой
воздух при 30 мм рт. ст. и 25—30° С. Содержание активного кислорода опре-
деляют йодометрическим титрованием.
Алкпленоксиды из олефинов и надкислот, в частности надбензойной кис-
лоты в большинстве случаев получают следующим способом,
Олефин растворяют в хлороформе и после охлаждения смешивают с холод-
ным раствором надкислоты в хлороформе, взятой с небольшим избытком. Реак-
ционную смесь выдерживают при 0° С, олефины с малой реакционной способ-
ностью — при комнатной температуре. Ход реакции контролируют титрованием
небольших проб реакционной массы.
По окончании реакции непрореагировавшую надкислоту и образовав-
шуюся карбоновую кислоту тщательно экстрагируют раствором NaOH и затем
из хлороформного слоя выделяют полученный продукт.
Ввиду опасности работы с органическими перекисями обязательно соблюдать
меры предосторожности, описанные в разделе «Образование перекисей путем ауто-
окисления» (стр. 288). При окислении неизвестных олефинов необходимо сначала
изучить их поведение на малых'пробах, прежде чем подвергать превращению большие
загрузки. К переработке реакционной массы можно приступать только после разло-
жения пли экстрагирования избытка надкислоты щелочью (опасность взрыва!).
Окись стирола [22]. К раствору 42 г (0,30 моль) падбензойной кислоты
и 590 С1Ю1з прибавляют 30 г (0,29 моль) стирола. Раствор выдерживают 24 ч
ори 0е С. несколько раз встряхивая в первые часы опыта. Окончание окисления
у стана нл и мают титрованием аликвотной части хлороформного раствора. Затем
неоднократным встряхиванием с 10%-пЫМ раствором щелочи удаляют образо-
вавшуюся бензойную кислоту и небольшой избыток непрореагировавшей йад-
бензойпой кислоты, органический слой промывают от щелочи водой и сушат
над NagSOj. Фракционной перегонкой получают 24—26 г окиси стирола в виде
бесцветной жидкости; выход 70—75% от теоретического; т. кип. 101° С
(4l) ,и.н рт- ст.).
* Перекристаллизацию перекиси бензоила ни в коем случае нельзя проводить
при пагренапип. Наилучшим способом является способ Нозаки и Барт-
летта [20], но которому перекись бензоила растворяют при комнатной
температуре в хлороформе л осаждают холодным метиловым спиртом.
Она выпадает в виде хорошо оформленных кристаллов. Перекристалли-
зация из горячего хлороформа опасна! [21].
18] Г). Sworn, Org. Reactions, 7, 378 (1953).
19] . i/Д i) «. j. Mattner, W. Busse, Angew. Chem., 65, 57 fl953).
2QJ K. Nozaki P. D. Bartlett; J. Am. Chem. Soc., 68, 1686 (1946).
21J A. R i e c h e, M, S c h u 1 z, Chem. Techn., 11, 264 (1959). '
22J П. Hibhort, P. Burt, Org. Synth., 8, 1Q2 (1928).
ч
264 ОБРАЗОВАНИЕ С-О-СВЯЗИ В РЕЗУЛЬТАТЕ ЛГПСОЕ Д1 [НЕН НЯ
Бензальдегид легко реагирует с молекулярным кислородом с образованием1^
надбензойной кислоты (см. стр. 288). Эту склонность бгл.щ.иьдегпда к аутоокислению^
используют для эпоксидирования [23], при котором можно обойтись без выделения*
чистой надбеизойной кислоты. Для образования эпоксидов с хорошим выходом доста-Ё
точно обработать кислородом или воздухом смесь олефина с бензальдегидом. Однако7
из алифатических альдегидов эпоксиды получаются анпп. с истачмтелыидмп ны.хо-'!
дами [24]. [
Мононцдфталевую кислоту' можно использовать для получения эпокспсоедине-^
ний так же, как п надбензойную. Окисление олефинов с ее помощью происходит^
в общем, с хорошими выходами и почти в таких же условиях, как и при работе с над«Ф
бензойной кислотой. Однако применение мононадфтдлевой кислоты имеет некоторые-'
преимущества вследствие того, что ее растворы более стабильны, чем растворы над—
бензойной кислоты. Поэтому падфталовая кислота особенно пригодна для эпоксиди^
рования медленно реагирующих олефинов. Кроме того, прп проведении реакций?
в хлороформном растворе образующаяся нерастворимая п хлороформе фталевая]
кислота легко отделяется от продукта реакции.
Монопадфталевая кислота. Ес получают преимущественно из фталевого^
ангидрида и щелочного раствора перекиси водорода по методу Беме [25], моди-г
фмцироваиному Пайном [26]. В снабженную мешалкой и термометром и охла^
ждаемую льдом с солью колбу емкостью 1 л вливают раствор 62 е (0.5 .«оль^
Na2COs в 250 мл воды. Раствор охлаждают до 5° С и и один ирном добавляю^
70 г (0,6 моль) 30%-пой 112Ог. После охлаждения до О1' С раствор смешиваний
с 75 г (0,75 моль) тонкоизмельчеппого просеянного фталевого ангидрида и при?
температуре от —5 до 0° С в течение 30 мин быстро перемешивают до иочти^
полного растворения. Раствор переносят в делительную воронку емкостьжм
2 л, добавляют 350 мл эфира, встряхивают с холодным раствором 30 мл кон-^
центрированной H2SO4 в 150 мл воды и в заключение еще трижды экстрагирую^
эфиром, порциями по 150.мл каждая. Объединенные эфирные вытяжки промы-»
лают 200 мл 40%-ного раствора (NTTd)2SO4 и оставляют для сушки па ночь в хо-;
лодильиике над 50 г безводного MgSO4. Содержание моцонадфтялевой кислоты?
определяют титрованием; выход 78% от теоретического, считая па фталевый^
ангидрид,
О получении кристаллической мопопадфта.'генон кислоты см. [27]. ;
Эпоксидирование с помощью мононадфталевой кислоты * приобрело наибольшее^
значение в химии природных соединений [48, 28). р
Наряду с упомянутыми надкислотами имеет значение также надуксусная'?
кислота вследствие удобства ее получения. Надуксусная кислота почти всегда при-,
меняется в уксуснокислом растворе, так как в чистом виде или кппцептрпрованнаяА
опа чрезвычайно взрывоопасна. Уксуснокислые растворы надуксусной кислоты7
получают смешением уксусной кпелогы или ацетаигидрида с 25—90%-пой Н2О2^;
иногда в присутствии небольшого количества концентрированной H2SOd в качестве-^
* Эпоксидирование енолацегатов надбеизойной и иадфталевой кислотами с после-у
дующим омыленном является важным методом введения 17n-i ндроксильлоХ;
группы в 20-кетопрегпаны при синтезе кортизона (I. Krilchevsky.;
et al., J. Am. Ghem. Soc., 74, 483 (1952); 11. H. Суворов, 3. Я. Яро-у
славцева, ЖОХ, 29, 2889 -(1959). — Прим. ped. j
[23] D. S wern, T. W. ' F i n d] e y, J. Scanlan, J. Ащ, Chem. Soc., 66h 4
1925 (1944). J
[24] D. Sw₽rn, T. W. Findley, J. Ain. Chem. Soc., 72, 4315 (1950); i
T. W. Findley, D. Swern, пат. США 2567930; С. A., 46, 3560 (1952).v-J
,25] H. Bohme, Org. Synth., 20, 70 (1940).
[26) G. B. Payne, J. Org. Chem. 24, 1354 (1959).
[27] H. Bdhme, Org. Synth., Goll. v. 3, 1955, p. 619. J
[28] A. Bowers, E. D e n о t, H. L'rqui za, L. M. Sanchoz-Hidalg or‘
Sleroidcpoxyde; Tetrahedron, 8, 116 (1960); P. Кагтег, в книге Fortschrjttfr ;
der Chemie organischer Naturstofle, Bd. V, Springer-Vcrlag, Wien, 1948, S. 1. '
I. ПРИСОЕДИНЕНИЕ КИСЛОРОДА ПО С.—С-СВЯЗИ
2G5
катализатора (защитный экран, опасность взрыва)). В зависимости от концентрации
применяемой перекиси водорода получают 10—40%-пые растворы надкислоты.
Работы с растворами надуксусной кислоты надо проводить по возможности
быстро [14] п при 20—25° С. Сильные минеральные кислоты (H„SOJ должны отсут-
ствовать в реакционной смеси, поэтому часто рекомендуется добавка ацетата патрия
в качестве буферного вещества.
1,2-Эпоксидодекап [29]. В точение 24 ч при 20—25° С перемешивают
смесь 50,5 г (0,3 моль) додсцева-1 п 408 г примерно 0,9Af раствора надуксусной
кислоты (0,36 люль). При этом расходуется 80—85% теоретического количества
надуксусной кислоты. По истечении указанного времени перемешивания смесь
выливают в холодную воду п экстрагируют продукт реакции эфиром. Эфирную
втяжку промывают водой и затем растворитель отгоняют. Фракционной пере-
гонкой получают 29 г (52% от теоретического) 1,2-эпоксидодекала; г. кшт. 97—
98е С (3,5 .ч.ч рт. ст.); 1Л356.
По даппым Эммонса и Пагано [30], эффективным эпоксидирующим агентом
является трифторнадуксусная кислота- Ее получают из трифторацетангидряда и пере-
киси водорода в Хлористом метилене. Взаимодействие с олефинами происходит в том
же растворителе в присутствии Na2COs, NaHCO3 или Na2HPO4. В присутствии
•последних эпоксидироваться могут и олефины с электропоакцепторными заместите-
лями, такие, как эфир метакриловой кислоты (выход 84%)
7» №.НРО.; СИ,
СЩ^ССООСН, с1,»с00°н.> Н2С----------сСООСНз
\ /
о
и этиловый эфир кротоновой кислоты (выход 73%).
Метиловый эфир а-метилглицидной кислоты [30]. В течепие 20 мин
к кипящей смеси 20 г (0,2 моль) метилового эфира метакриловой кислоты, 200 мл
хдорпстого метилена и 113 г (0,8 моль) Na2IIPO4 прибавляют при энергичном
перемешивании раствор трифторнадуксуспой кислоты, получеппый из 7,0 мл
(0,25 моль) 90%-нОЙ НаО2, 42,3 мл (0,3 моль) трифторацетангидрида и 50 мл
хлористого метилена. Когда экзотермическая реакция затихнет, смесь нагре-
вают еще 30 мин, затем выливают в воду и продукт реакции экстрагируют хло-
ристым метиленом. После перегонки получают 19,4 г метилового эфира а-метил~
глицидной кислоты, что составляет 84% от теоретического; г. кип. 62—65° С
(32 ,цх рт. ст.).
Как было установлено [31], аф-пепасьпценпые кетоны реагируют с перекисью
водорода, в щелочных растворах с образованием эпоксикетонов.
Окись бензальпцетофенона. Раствор 2,08 г (0,01 моль) бензальацето-
фенона в 25 мл метилового спирта смешивают с 3 мл 0,012 моль) 15%-ной
ТТ2О2 и 2,5 мл 2 н. раствора NaOlI. При смешении массу охлаждают так, чтобы
температура ее не поднималась выше 30е С. Через короткое время раствор быстро
затвердевает в кристаллическую кашицу. После отсасывапия п промывки не-
большим количеством метилового спирта получают 1,8 г бесцветного кристал-
лического вещества; из маточника при смешении с водой выделяют еще 0,2 г
продукта. Выход 89% от теоретического; т. пл. 90° С.
Опубликовали и другие методы эпоксидирования щелочным раствором перекиси
водорода, например для получения изофороноксида (2,3-эпокси-3,5,5-триметилцикло-
гексанона) [32] и мезитилоксидэпоксида [33].
[29] D. Swern G. N. Е i 11 е n, J. Т. Scanlan, J. Am. Chem. Soc.,
Joni 68, 1504 (1946).
130] W. D. E m m о n s, A. S. Pagan o, J. Am. Chem. Soc., 77, 89 (1955).
lol E. Weitz, A. Scheffer, Her., 54, 2327 (1921).
Hol Wasson, H. O. House, Org. Synth., 37, 58 (1957).
133J О. В. P a у n. e, J. Org. Chem., 23, 310 (1958).
266
ОБРАЗОВАНИЕ С —О-СВЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Щелочимо растворы перекиси иодорода, очевидно, специфически реагируй
по а,0-двойным связям * ненасыщенных кетовов {34].
Возможно также окисление пеписыщеиных кетонов, например окиси мезитил
метплвинилкетопа, метилизопропепилкетопа, лалкона, циклогексенона-3, до соотв<
ствующпх эподоисоедпненпй гидроперекисью mpem-бутила в присутствии трилси
Г (тримртилбенл1ламмонпя) [35].
Некоторые а,0-ненасыщенпые нитрилы с разветвлением цепи у а-атома углеро
при окислении перекисью водорода в присутствии Na,_,CO3 и водного ацетона образу
эпокснамиды; например, из нитрила а-фенипкоричноц кислоты получается ам
а,0-дифенилглицидной кислоты [36]. {
Из тетраарилэтилепон при помощи СгОя в ледяной уксусной кислоте бН
получены соответствующие диоксиды [37]; КМпО4 в ледяной уксусной кисзд
окисляет различные стероиды в 5,6-положении до эпоксидов [38]. 4,
II, Присоединение воды по С = С-связи .1
Гидратация олефинов до спиртов происходит при каталитическом влип
кислот (чаще всего применяют II»SO4), причем промежуточно образующиеся эф,
минеральных кислот легко гидролизую।ся в кислой среде: л
R—СП—СП—ИЧ-НгО —> B-CH2-CII-R .л
он
Присоединение воды к олефинам в присутствии серной кислоты проте!
по иравилу Марковникова. причем независимо от положения двойной связи вс]
образуются вторичные или третичные спирты. Первичный этиловый спирт получу
только из этилена. Добавка к реакционной массе перекиси бензоила не влияев
реакцию гидратации, и, следовательно, перекисный эффект, подобный проявляй
муся при присоединении к олефинам бромистого водорода, в данном случае не И]
места [39].
Скорость гидратации отдельных олефинов различна и зависит от струят
олефина. Это проявляется в том. что некоторые олефины гидратируются уже в j
сутствии разбавленной серной кислоты, а другие только в присутствии концентр
ванной. В гомологическом ряду тенденция присоединять воду возрастает от этиа
к гексену, вновь снижаясь у олефинов с более длинной цепью. Отношение стругу
ных изомеров к присоединению воды также различно. Например |40], способ^
бутена-1 присоединять воду вдвое больше, чем бутена-2. ЭлектроноакцептЭД
заместители у двойной связи снижают реакционную способность вещества (напрЖ
в случае коричной кислоты и дихлорэтилена), алкильные группы, напротив, 4
шают ее. Во всех случаях при применении серной кислоты необходимо вести реай
при возможно более низкой температуре. Концентрированная сорная кислота 41
способствует полимеризации олефинов, которую ипогда можно избежать, исполу
вместо серной кислоты органические кислоты, например 5—10%-ный водный раса
щавелевой кислоты [41].
[34]
35
36
37
38
39
[40]
[41]
При эпоксидировании ацетата Д5,16 -прегнадиопол-30-опа-2О перекисью^
дорода в щелочной среде затрагивается только сопряженная двойная
[Р. J u 1 1 a n et al., J. Am. Chem. Soc., 72, 5145 (19o0)]. — Прим, ped.-'-’
S. Wins teia, R. B. 11 о n d e r s о и, в книге R. С. Е 1 d е г f 1 е’
Heterocyclic Compounds, John Wiley a. Sons. New York — London, 1950< Й
N, C: Y ang, R. A. Finnigan, J. Am. Chem. Soc., 80, 5845 (1958). -1
1. V. Murray, I. B. Cloke, J. Am. Chem. Soc., 56, 2749 (1934). J
W. Bockemiiller, ,B. J a n 0 e n, Ann., 542. 166 (1939). •'£
M. E h re n s t e i n, M. T. D ccke r, J. Org. Chem,, 5, 5u4 (194Q)r
В. T. Brooks, J. Am. Chem. Soc., 56, 1998 (19.34). .'.g
A. M i c h a e 1, R. F. Brunel, Am. Chem. J 41, 118 (1909).
J. G у г, в книге Houben-Weyl, Methoden der organischen Chem]
Bd. Ill, 2 Aufl., Leipzig, 1923, S.77. ' 5
II. ПРИСОЕДИНЕНИЕ ВОДЫ ПО С=С-СВЯЗИ
267
трепг-Амиловый спирт из триметнлэтнлена (р-амилепа) [42].
/СН3 /СПз ХПз
СПзСП=С< —> СНЧСП2С< —> СН9СН,С<
ЧШз IХСП3 рСПз
owi дн
Смесь 500 г льда и 500 г концентрированной I12SO4 охлаждают до. 0°С.
В полученный раствор при энергичном перемепгиванпи вносят 500 мл р-амнлена,
перемешивают еще несколько часов и затем отделяют водный слой от пепро-
реагировавшего амилена. Водный слой выливают на 2 кг льда и осторожно
нейтрализуют концентрированным раствором 720 г NaOII. Затем перегоняют
реакционную смесь до прекращения выделения амилового спирта из пробы
конденсата под действием К2СОз- Из дистиллята с помощью карбоната калия
выделяют спирт п сушат его пад К2СОз- Перегонкой получают чистый mpem-ами-
ловый спирт; т. кип. 1Q3U С; выход около 90% от теоретического.
Промышленное значение гидратации олефинов освещено в монографии Азин-
гера [43] и работе Сека [44].
^^-Ненасыщенные кислоты легко присоединяют воду с образованйем соответ-
ствующих оксикислот. Так, из раствора 3-нитроакриловой кислоты в 70 %-пой муравь-
иной кислоте в результате Зч нагревания при 85—100° С получают 2-окси-З-нвтро-
пропиововую кислоту с 83%-пым выходом от теоретического [45].
СН=СН—СООН СН2-СН-СООП
I I I
no2 no2 он
Р-Оксчцропяоповую (гидракрпловую) кислоту готовят из акриловой иислогы
прп нагревании ее с раствором едкого натра [46]. К фумаровой и малеиновой кислотам
вода присоединяется без катализаторов при 150—200° С. В присутствии щелочи
гидратация может проходить [47] при длительном нагревании уже при 100° С. Гидра-
тация малеиновой кислоты возможна также в присутствии кислот (СО2, H2SO4)
и тяжелых металлов (Hg, Cd, Zn, Си) при повышенных температурах [44].
0-Окснмасляная кислота может быть получена гидратацией кротоновой кислоты
с водной серпой кислоте [48].
К 2-ме'1ИЛГоптон-2-ону-6 вода присоединяется в присутствии 35%-пой H2SOi
при комнатной температуре с образованием 2-метилгептанол-2-она-6:
СНзк пго СНзх
)С~ СН-(СП2)2-СО-СНз —~>С-(СТ12)з-С0-С11з
CEl3Z СН/ |
ОН
2-Метплгептанол-2-он-6 [49]. Смесь 2-метцлгептен-2-опа-6 и 35%-кой
112SО4 встряхивают в течение 9 ч прп комнатной температуре в токе азота. Оста-
вляют па ночь в холодильнике, разбавляют ледяной водой и при температуре
не выше 5° С нейтрализуют водным раствором NaOH. После исчерпывающей
экстра!,-дни эфиром выделяют перегонкой третичный спирт. Выход 85% от теоре-
тического; т. кип. 100—102° С (7 мм рт. ст.}.
[42] R. Adams, О. kamm, С. S. М а г v е 1, J. Am. Chem. Soc., 40,
1955 (1918).
. 143] ф. А з и п г е р, Химия и технология мопоолефшюв, пер. с нем., Гостоптех-
издат, Москва, 1960.
>44] R. s с к а, в книге G. М. Schwab, Handbuch der Katalyse, Bd. 7/2, Sptinger-
. Vcrlag, Wien, 1943, S. 37.
145] H. S bech ler, F. Conrad, A. L. D a u 1 t о n, R. B. Kaplan, J. Ain.
r/ • ^'hem. Soc., 74, 3052 (1952).
Н6 И. Erlenmeyer. Ann., 191, 281 (1878).
47 F. L о у d 1, Ann., 192, 80 (1878); II. J. v a u’t Hoff, Ber., 18, 2713 (1885).
4« D. Pressman, H. J. Lucas, J. Am. Chem. Soc., 61, 2276 (1939).
I4JJ j. Meinwald, J. Am. Chem. Soc., 77, 1617 (1955).
268
ОБРАЗОВАНИЕ С—О-СВЯЗИ Б РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Металлиловый спирт СН2=С(СП3)—СП2ОЦ гидратируется смесью 25%-но»
Н2ЗО4 и изобутиральдегида с образованием циклического апеталя изобутиленгликоля^
Гидролизуя ацеталь разбавленной минеральной кислотой, получают изобутилен-*
гликоль с 94%-ным выходом от теоретически о [50]. j
Циглер с сотр. [51] разработали способ превращения олефинов с конце-i
выил — СН2-груниами в первичные спирты, т. е. способ гидратации не по правилу^
Марковникова *
\с=СП2 \еН-СП2О11
По этому способу соответствующий олефин сначала подвергают взаимодей-
ствшо с алюмининоргапнческпм соединением, в результате чего образуется первичны!
алкилалюминий. Последующее окисление воздухом млн кислородом приводит к обра
зованию алкоголята алюминия, гидролизом которого затем получают первичны!
спирт: ;
.СН-CllaOal
где al = VgAl.
Гидратроповый спирт из а -метилстирола. Смешивают 125 ?. (0,63 .моль
триизобутилалюминин с 354 г (3 .моль) а-метплстирола и перегоняют. После
отделения 19 г головной фракции собирают 200 г гидратропоиого спирта; т;
кип. 107,5—108° С (15 мм рт. ст.); выход 78% от теоретического, считая а
алюминий.
Еще один метод превращения олефинов в спирты, протекающего против
правила Марковникова, описан Брауном и Сабба Гао [52, 53]. Метод заключаете
во взаимодействии простых олефинов, таких, как этилен, пентен-1, пентен-2, цикл!
гексен или стирол, с боргидридом натрия в присутствии А1С13 при 25° С. При эта
образуются триалкил^ораны с выходами до 90% от теоретического. Последние лег»
окисляются щелочным раствором перекиси водорода в соответствующие эфиры борнд
кислоты, которые могут гидролизоваться до спиртов. Выделение промежуточны
продуктов на отдельных стадиях необязательно. Для подобного рода реакций мож|
использовать [54] также аддукт пиридина с бораном CeH6N -ВН3.Этот метод гидратаця
нашел применение для получения многих первичных спиртов, в том числе спирте
стероидного ряда [55]. ‘ ‘
2,2-Дмфенилэтиловый спирт [52].
(С6Н6)2С=СП2 —> [(CeH5)2CH-CHj3B —> [(СвН5)2СН-СН2-О]3В —-> !
—-> 3(СвН5)2СН-СН2о|
* Цпглеровский процесс едва ли можно называть п строгом смысле слова «гидр^
тацией (т. е. присоединением воды к двойной связи) против правила Маркович
кова». — Прим. ред.
[30] G. II е а г п е, М. Т a m е 1 е, W. Con verse, Ind. Ёпд. Chora.. 33. 8б
(1941).
[511 К. Ziegler, F. Krupp, К. Zosel, Ann., 629, 241 (1960).
|52] II. C, Brown., В. C. Snbba Rao, J. Am. Chem. Soc., 78, 5694 (1956)
[53] H. C. Brown, В. C. Subba R a o, J. Org. Chem., 22, 1136, 1137 (1957)
[54] M. F. Hawthorne, J. Org. Chem., 23, 1788 (1958).
[55] 8. Wolf e, M. Nussi m, Y. Mazur, F. Sondhc i mer, J Org. Chemi
24, 134 (1959k R. P a p p o, J. Am. Chem. S c., 81J HG
Ilf. ПРИСОЕДИНЕНИЕ РОДЫ ПО С С-СВЯЗИ
26!
При исрсмсшивапии к раствору NaBH4 и А]С1з в диметпловом эфир,
дпэтпленгликоля (диглим) медленно прибавляют 1,1-дифенплэтилен. Смес]
выдерживают 3 ч при комнатной температуре в токе азота, затем нагревают 1
на водяной бане и в заключение при температуре ниже 40° С в вакууме отгоняю'
около половины растворителя. Оставшийся растворитель вымывают разбавлен
ной НС1 и водой, остаток смешивают с раствором NaOH у этиловом спирте и при
лпвают по каплям 20%-вый избыток 30%-ной ПгО2 с такой скоростью, чтобг
смесь слабо кипела (обратный холодильник). После этого продукт реакци]
экстрагируют эфиром и перегонкой выделяют 86,4 г (87% от теоретического
2,2-дпфенилэтнлового спирта; т. кип. 192 —194е С (20 ,-u.w рт. ст.).
Исследования циклических олефинов показали, что гидратация по двойней
связи с промежуточным присоединением борана происходит как цис-присоединени.
по двойном связи; из 1-метилциклопснтепа с 85%-ным выходом от тсоретическог,
образуется транс-2-метилциклопентанол [56].
III. Присоединение воды по С=С-связи
Присоединение воды к ацетиленам * лучше всего осуществлять в кислой сред
в присутствии ртутных соединений. Из ацетилена образуется ацетальдегид [57]
НС=СН + Н2О —> СНзСНО
При присоединении воды к ацетиленовым соединениям с концевыми тройным!
связями образуются только кетоны:
О
К-С=СН -2^ Н-С-СНа
Исключение составляет трифторметилацетилен [58].
Гидратация моно- и дизамещенных ацетиленов может в некоторых случая;
происходить под действием одних только кислот, особенно серной кислоты. п-Брои
фенилпропиоловая кислота в концентрированной серной кислоте количественна
превращается в n-бромбензоилуксуспую кислоту [59]:
О
CSC-COOH Н1О L СЩ-сооп
Л ! —’ л 11
ц/v
п,п'-Диметилтолан [60] и фепилацетилен [61] гидратируются в ледяной
уксусной кислоте, содержащей соответствующие количества разбавленной cepnoj
кислоты.
Большее значенпе, чем гидратация алкинов в присутствии кислот, имеет при:
соединение воды к этим соединениям под влиянием солей ртути. Обычно для этог«
Применяют растворы HgSO4 в серной кислоте или (CH3COO)2IIg в уксусной кислоте
Если образующийся продукт растворим в воде, гидратацию можно проводить в при:
сутствии водных растворов катализаторов; при гидратации нерастворимых в вод.
* Эта реакция открыта М. Г. Кучеровым [ЖРХО, 13, 533, 542 (1881)]. — Прим. ре&
[56] Н. С. Brown, G. Zweifel, J. Ain. Chem. Soc., 81, 247 (1959).
[57] Gattermann -Wieland, Die Praxis des organischen Chemikers, WaL
ter de Gruyter a. Co., Berlin, 1953, S. 183.
[58] R. N. II a s z с 1 d i n e, K. Lee d ham, J. Chem. Soc., 1952, 3483;
R. N. II a s l с 1 d i n e, J. Chem. Soc., 1952, 3490.
59] M. Reimer, E. Tobin, J. Am. Chem. Soc., 63, 2490 (1941).
60] W. P. Buttenberg, Ann., 279, 335 (1894).
[61] J. U. Ncf, Ann., 308, 294 (1899).
270
ОБРАЗОВАНИЕ С-О-СБЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
ацетиленов рекомендуется вводить растворители, например 70%-пый метиловый
спирт, 70%-ный ацетон или уксусную кислоту [62].
Гексанон-2 из гевсина-1. В трехгорлой колбе емкостью 500 мл, снабжен-
ной мешалкой, капельной воронкой и обратным холодильником, нагревают
до 60° 0 смесь 1 г HgSO4, 1 г концентрированной H3SO. и 150 г 70%-ного мети-
лового спирта (или 150 г 70%-ного ацетона, или 50 г 60%-пой уксусной кислоты)
и в течение 1 ч приливают к ней по каплям прп перемешивании 41,0 г (0,5 моль)
гексина-1. Перемешивают 3 ч прп указанной температуре, отгоняют после охла-
ждения метиловый спирт (пли ацетон) и из остатка твердым NaCl высаливают
гексанон-2. После отделения, промывки и нейтрализации его сушат над СаС18
и перегоняют; т. кип. 120° С; выход 78,8% от теоретического. При использовании
уксусной кислоты ее сначала нейтрализуют холодным раствором Na2COg и затем
выделяют спирт оппсапным способом.
Особенно эффективным катализатором гидратации алкинов является смесь
окиси ртути, эфирата трифторида бора и трифторуксусной кислоты [63]; применяют
также смеси окиси ртути и трифторида бора [64], окиси ртути и серной кислоты [65].
При гидратировании неустойчивых в кислой среде ацетиленов присоединение воды
проводят в нейтральных условиях с помощью ртутьацетамида или ртуть-п-толуол-
сульфамида [66]. Для гидратации пригодны также кислые ионообменные смолы,
насыщенные ионами ртути [67].
Присоединение воды к дизамещенным ацетиленам может происходить по двум
направлениям;
0 °
|< н.о Н.О II
RCH2CR' rc=CR' ---------------* RCCII2R
II I
что зависит от заместителей R и R'. Кисйород присоединяется к наиболее удаленному
от электроноакцепторной группы атому углерода ацетиленового соединения. Если
нет существенной разницы в полярности заместителей R и R', образуется смесь кето-
нон (I) и (II). Так, гидратация гептнпа-2 в метиловом спирте по методу Томаса с сотр.
приводит к двум кетонам — гептанопу-2 и гептанону-3, почти в равных количествах
[62, 68].
К а,Р-ацетилепкарбоновым кислотам вода присоединяется с образованием
соответствующих р-кетокислот:
О
нс=ссоон ^2- r<5ch2cooh
По Мурё и Делапжу [69], реакцию проводят кипячением кислоты со спиртовым
раствором КОН; при кипячении с водными растворами щелочей одновременно проис-
ходит декарбоксилирование и образование метилкетопов.
[62] R. J. Thomas, К. N. С a m ф b е 11, G. F. II е n n 1 о n, J. Am. Chem.
Soc., 60, 718 (1938).
[63] J. С. II а га ] е t Н. В. Henbest, Е. R. Н. Jones, J. Chem. Soc., 1951# :
2652. ;
,[64] N. A, Dobson, R. A. Raphael, J. Chem. Soc., 1955, 3558.
[65] G. W. Stacy, R. A. Miculec, Org. Synth., 35, 1 (1955).
[66] M. W. Gol d berg, R. Aeschbacher, Helv. chim. acta, 22, 1185 (1939);
M. W. Goldberg, R Aeschbacher, E. Hardegger, Helv. chim. '
acta, 26, 680 (1943).
[67] M. S. New man, J. Am. Chem. Soc., 75, 4740 (1953); J. D. Billimoria,
N. F. Maclagan, 3. Chem. Soc., 1954, 3257.
[68] G. F. II 0 n n i 0 n, C. J. Pillar, J. Am. Chem. Soc., 72, 5317 (1950).
[69] Ch. Mourou, R. Delange, C. r., 136, 753 (1903); C., 1903, I, 1018.
27
Муро и Лпзенек [70J переводили а.р-ацетпленнптрилы или амиды ацетиленовы
кислот в соответствующие кетонитрилы или кетоамиды кипячением их сппртовы
растворов с аминами.
Ацегпленкетоны присоединяют воду п дают р-дикстоны:
R—СО—C=CR’ _?.».?-» R—СО—СН3—СО—R’
Иоф [71] синтезировал дибензоилыетан. действуя концентрированной II2SO4 н
бенэонлфенплацетилен; по атому методу могут быть получены многие р-дикетон!
[72]. 1'снзоплацетплен не изменяется в концентрированной H2SO4 при 20° С; npi
более высоких температурах наступает разложение; под действием разбавленно:
H2SO4 и сульфата ртути [73] при 80—100° С образуется 1-фенилпропапдион-1,2
C6H6-CO-CsCH Cfl|s--c<> -СО- СПз
1-Фепилпропандион-1,2. К раствору 6,5 г бензоиладетилена в 100 м.
этилового спирта прибавляют 50 мл 2 н. раствора H2SO4, содержащей 0,25
HgSO4. Смесь нагревают 6 ч на водяной бане при перемешивании, постепенн<
добавляя еще 50 .ил 2 н. раствора H2SO4, содержащего IIgSO4. Экстрагирование!
эфиром и перегонкой выделяют 4,1 г 1-фенИдпропандиона-1,2 в виде желтог,
масла; т. кип. 88° С (17 лл pm.. cm.)] njj 1,5350.
К алкоксиацетиленам вода присоединяется при комнатной температуре в при
сугствии НС1 и (CH3COO)2Hg с образованием сложных эфиров. Из бутилфенокса
ацетилена (4,96 г) получается 2,8 г фенилового эфира капроновой кислоты [74]:
С4Н9—С=С—OCgHg * С4Н9—СН2—СО—OCgHg
При гидратации ацетиленовых спиртов образуются оксикетоны:
R\ HiO R\
)С-С=СН \с—С —СНз
R/ | R/ । и
ОН но о
При действии сульфата ртути в избытке разбавленной серной кислоты нс
2-метилбутин-3-ол-2 с хорошим выходом получается [75] 2-метилбутанол-2-он-3,
(СН3)2С(ОН)С=СН (С118)2С(ОН)СОСН3
Это вещество образуется [76] также при кипячении в НС1 белого нерастворимогс
осадка, полученного при добавлении HgCla к водному раствору З-метилбутинола-3
[70] Ch. Moureu, J. Lazennec, С. г.. 143, 553 (1906); 144, 491 (1907); 144
804 (1907); С., 1907, II, 37; Bull. Soc. chim. France [4], I, 1062 (1907); [3], 35,
1179 (1906).
[71] J. U. Ncf, Ann., 308, 277 (1899).
[72] M. Yvo n, C. r., 180, 748 (1925); Ch. Moureu, R. Delange, Bull. Soc.
chim. France, 25, 306 (1901); Ann. Chimie, 2, 277 (1914); R. C. Fuson,
G. E. Ц 11 у о t, J. L. Hickson, J. Am. Chem. Soc., 61, 410 (1939)*
R. A. If о г t 0 n, A. H a 5 s a n, T. C, Calloway, J. Chum. Soc., 1934 j
899.
[73] K. Bowden, E. A, Braude, E. R. H. J 0 n e s, J. Chem. Soc., 1946, 945.
[74] T. L. Jacobs, R. Cramer, F. T. W e i 6, J. Am. Chem. Soc., 62, 1842
(1940).
[75] H. Scheibler, A. Fischer, Bet., 55, 2903 (1922).
•[76] К. II e P, H. Munderloli, Ber., 51, 377 (1918).
[77] E. M. McMahon, J. N, Roper jr. et al., J. Am. Chem. Soc., 70, 2971
(1948).
272 ОБРАЗОВАНИЕ С-О-СВЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
По реакции с ацетатом ртути в ледяной уксусной кислоте при 75—95° С из 2-метил-
бутпп-З-ол-2 получают ацетилированный оксикетоп с выходом 98,5% от теорети-
ческого [77].
1-Э|Пнплцпклогексанол гидратируется [67] до 1-ацетплциклогсксапола ионо-
обменной смолой, обработанной 1%-ным раствором HgSO4; аналогичным способом
можно синтезировать оксиацетон из пропаргилового спирта [78].
Оксиацетон. Раствор 10,4 г (0,2 .«ель) пропаргилового спирта в 100 мл
поды перемешивают при 23е С с 2 г ионообменной смолы дауэкс 50, которую
предварительно обрабатывают раствором HgSO4 в разбавленной H2SO4, тща-
тельно промывают п высушивают. Температуру в течение 13 мин повышают
до 55° С. Далее массу перемешивают 25 мин при 50г С и затем при 40° С. Через
1 ч 40 мин отфильтровывают смолу и промывают ее 20 мл воды. Отбирают алик-
вотную часть раствора (12 мл) и смешивают с фенилгндразппом. Получают
2,85 г (87% от теоретического) феппдгидразона окснацетона; т. пл. 99—101° С.
При нагревании с 85%-ной муравьиной кислотой третичные ацетиленовые
спирты отщепляют воду и превращаются в а,Р-нонасыщеппые кегопы [79, 80].
Описана [81] каталитическая гидратация этинилкарбинолов стероидного ряда
н ледяной уксусной кислоте при помощи окиси ртути н трифторида бора в присут-
ствии незначительных количеств ангидрида уксусной кислоты. Наряду с присоеди-
нением воды одновременно происходит перегруппировка с расширением кольца, как,
например, в случае Д8-17-этиниландростендиола-3,17. Мопдон [82] осуществил
в чрезвычайно мягких условиях присоединение воды к третичному ацетилепгликолю'
при помощи охлажденного льдом раствора HgSO4 в 85%-пой муравьиной кислоте;
выход количественный*.
IV. Присоединение гидроксильных групп по С = С-связи
1. Гидроксилирование при помощи органических надкислот
При действии органических надкислот на олефины образуются эпоксиды
(C'lp. 262); при более высоких температурах, более продолжительном взаимодействии
и в присутствии сильных кпелот получаются а-гликоли. Первично образующиеся
эпоксиды в этих условиях нестабильны и с переходом через оксиэфиры гидролизуются
до а-гликолей [18];
* Фундаментальную работу по изучению гидратации дивпнилацетиленов
см. Й. Н. Назароп И сотр., Усп. хим., 20, 71 (1951). — Прим. ред.у
[78] М. S. Newman, J. Am. Chem. Soc., 75-, 4740 (1953).
[79] II. Rupe, W. Messnc r, E. Kamh] i, Helv. chim. acta, 11, 449; C., 1928
I, 2811; F. G. F i sc he г, K. Lo we u berg, Ann., 475, 203 (1929);
J. I). Chanle y, J. Am. Chem. Soc., 70, 246 (1948).
[80] G. F. Henni on, R. B. Davis, D. E. Malone у, J. Am. Chem. Soc.
71, 2813 (1949).
81] L. Ruzicka, H. F. M e d a h 1, Nature, 142, 399 (1938); C., 1939, I, 1996,
,[82] A. M undo n, Ann., 585, 43 (1954),
IV. ПРИСОЕДИНЕНИЕ ГИДРОКСИЛЬНЫХ ГРУПП по с=с-связи
27:
Гидролиз эпоксидов происходит по механизму транс-размыкания кольца
например [83], из цнкло!ексеноксида образуется траис-циклогсксандиол-1,2.
Для окисления олефинов чаще всего применяют надуксусную и надмуравьинув
кислоты. Ввиду того что эти кислоты в чистом виде легко взрывают, применяют и:
разбавленные растворы.
Разработан метод [15], ио которому олефин подвергают взаимодействию с почт!
стехиометрическим количеством 25—30%-ной Н2О2 в ледяной уксусной кислоте
содержащей небольшие количества концентрированной серной кислоты. Этот мето;
позволяет получить в течение нескольких часов а-гликоли с хорошими выходам!
при температуре около 40° С.
транс-Циклогександиол-1^2 [84]. К 28,9 г циклогексена при энергич-
ном перемешивании и начальном охлаждении медленно приливают смесь 43,9 ;
ледяной уксусной кислоты, 43,9 г 30%-ной Н2О2 и 4,5 з концентрированно^
H,SO4. Скорость приливания регулируют так, чтобы температура оставаласг
в пределах 20—25° С. Затем еще мутную смесь нагревают 2—Зч при 45° С дс
полного осветления раствора, нейтрализуют раствором NaOH и исчерпывающа
экстрагируют продукт реакции эфиром. Остающийся после отгонкн раствори-
теля кристаллический осадок транс-циклогексапдиола-1,2 (31,2 г; 75% от тео-
ретического) перекристаллизовывают из этилацстата; выход 22 г (53% от теоре-
тического); т. пл. 103—104° С.
Особенно удобным гидроксилирующим агентом является надмуравьиная кислота,
которую получают смешением небольшого избытка 30%-ной Н2О2 с 98—100%-ной
муравьиной кислотой (чистая надмуравьиная кислота сильно взрывает!). Окисление
олефинов происходит при температуре около 40° С и вскоре заканчивается; потен-
циально образующиеся оксиэфиры омыляются до а-гликолей разбавленной щелочью
или просто нагреванием с водой.
транс-Циклогександиол-1,2 [85]. К смеси 105 г 98—100%-ной муравьи-
ной кислоты и 13 ? (0,115 .ноль) 30%-пой Н2О2 прибавляют 8,0 г (0,097 моль)
циклогексена. После непродолжительного встряхивания начинается энергичное
разогревание, и при 65—70° С суспензия становится гомогенной. Ее нагревают
на водяной бане при 65—70° С еще 2 ч, и отгоняют в вакууме избыток муравьиной
кислоты. Остаток нагревают 45 мин на водяной бане с 20%-ным раствором
NaOH и после охлаждения нейтрализуют HCI. Воду отгоняют в вакууме, остаток
перегоняют. Отогпавшаяся при 128—132° С (15 мм рт. ст-) фракция вскоре
кристаллизуется. Перекристаллизацией из ацетона получают 7,9 г транс-цпкло-
гексапдиола-1,2 (70% от теоретического); т. пл. 102—103° С.
По данным Гринспена [86], гидроксилирование олефинов можно, проводить
при помощи смеси уксусной кислоты и 90%-ной Н2О2, причем получаются очень
высокие выходы. Весьма активна также смесь концентрированной муравьиной кислоты
с 90%-ной Н2О2. Эта смесь особенно пригодна для гидроксилирования «^-ненасыщен-
ных кислот ]87].
Трифторнадуксусная кислота [88] также является активным окислителем
олефинов до а-гликолей (о получении кислоты см. стр. 265). Реакция протекает с обра-
зованием окситрифторацетатов, которые при действии метилового спирта превра-
щаются в а-гликоли. Очень хорошие выходы гликолей получают при добавлении
к реакционной смеси трифторацетата триэтпламмония.
[83] S. W instein, J. Am. Chem. Soc., 64, 2792 (1942).
[84] H. Meerweia et al., герм. пат. 574838; С., 1933, I, 4038.
[85] A. R о e Ь u с k, II, Adkins, Org. Synth., 28, 35 (1948).
[86] F. P. Greenspan, J. Am. Chem. Soc., 68, 907 (1946); Ind. Eng. Chem., 39,
847 (1947).
[87] J. English jr., J. D. Gregory, J. Am. Chtem. Soc., 69, 2120 (1947).
[88] W. D, E mmons, A. S. P a g a n o, j, P. Free man, J. Am. Chem.
Soc., 76, 3472 (1954).
18 Заказ 1835.
274 ОБРАЗОВАНИЕ С-О-СВЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Пентапдиол-2,3. К охлажденной суспензии 6,0 мл (0,22 моль) 90%-ной А
Н3Ог в 50 мл хлористого метилена прибавляют при перемешивании 37,2 мд ’)
(0,264 моль) ангидрида трифторуксусной кислоты. Полученный раствор пере- J
мешивают 10 мин на холоду и затем переносят в капельную воронку, снабжен- Т
ную приспособлением для выравнивания давления. В течение 30 мин смесь >
приливают по каплям к раствору 14,0 г (0,2 моль) пентена-2 и 21,4 г (0,1 .моль} <
трифторацетата триэтиламмония в ЙО мл хлористого метилена, причем начи- i
нается сильное кипение смеси. После внесении всего раствора смесь перемеши-Ч
вают еще 15 мин при комнатной температуре и затем в вакууме отгоняют летучие 1
компоненты. Перегонкой бесцветного остатка при 38—60° С (2 мм рт. ст.) *
получают 50,5 г эфира трифторуксусной кислоты. Этот эфир кипятят 2 « с 300 мл; й
3%-ного раствора НС1 в метиловом спирте, отгоняют в вакууме растворитель^
и перегонкой получают 15,1 г бесцветного пентандпола-2,3 (74% от теоретиче-; Я
ского); т. кип. 58—59° С (0,5 мм рт. ст.)', 1,4412.
2. Гидроксилирование при помощи
перманганата калия *
Олефины легко окисляются разбавленным раствором КМпО4 до »/ис-1,2-глико%>
лей [89]. Эта реакция использовалась для гидроксилирования многих ненасыщенных^
соединений различных классов [90].
Выбор условий взаимодействия имеет большое значение, так как в некоторых^
случаях может проходить дальнейшее окисление образовавшихся гликолей. Напрн-^
мер [91], при быстром добавлении КМпО4 к нейтральному раствору олефина обра^
зуются главным образом ацилоины RCH(OII)—COR'. Гидроксилирование проводят^
чаще всего па холоду разбавленными нейтральными или щелочными растворамвИ
КМпО4; растворителями служат вода, ацетон, спирты, метилциклогсксан или смесьЦ
спирта с водой. При проведении реакций в нейтральной среде [92] часто добавляют^
MgSO4.
Окисление олефинов в щелочном растворе (pH > 12) в смешанном растворителе ^
(mpem-бутиловый спирт — вода) протекает легче, чем в нейтральной среде. Гликоли'^
получаются более чистыми и с высокими выходами [93, 94]. %
В качестве примера этого, в принципе весьма простого способа, приводится;,
получение аритро-9,10-диоксистеарпновой кислоты из олеиновой кислоты [93]. %
эритро-9,10-Диовснстеариновая кислота. Смесь 1,0 г (0,0035
олеиновой кислоты, 1,0 г (0,025 лоль) NaOH растворяют при нагревании в 100 лйб
воды и после охлаждения разбавляют 800 мл ледяной воды и 100 г ралдробле®*}
ного льда. К холодной смеси (5° С) при сильном перемешивании, как можнв*(
быстрее, приливают раствор 0,80 г (0,005 моль) КМпО4 в 80 мл воды. Чере&<
5 мин смесь обесцвечивают SO2, прибавляют 30 мл концентрированной HCL*!
п в течение 1 ч охлаждают ледяной водой. Отфильтровывают осадок и послед
сушки в сушильном пистолете при комнатной температуре получают 1,05%^
(94% от теоретического) неочищенной «ритро-9,10-диоксистеаршювой кислоты.'^
Ее очищают перекристаллизацией кз 95%-ного этилового спирта. Выход чистого2
продукта 0,90 г (81% от теоретического,!; т. пл. 130- 131° С.
* Окисление по Вагнеру. — Прим. ред. Я
[89] G. Wagner, Вег., 21, 1230, 3343, 3347 (1888); 23, 2307 (1890); 27, 1636 (1894)^
J. Boeseken, Rec. trav. chim., 47, 683, 839 (1928). 4
[90] Стероиды: В. A. Windaus, Вег., 40, 257 (1907); R'. Tull, R. Е. Jone s,.^
S. A. R о b ins on,M. T i s h 1 e r, J. Arn^ Chem. Soc., 77, 196 (1955); M. F i e%;
s e r, A. Quilico, A. Nicion et al., J. Am. Chem. Soc., 75, 4066 (1953^-1
Сахара: R. A. Raphael, С. M. Roxburgh, J. Chem. Soc., 1955, 3405^S
ipc-Днметилциклогексан: H. Meerwein, Ann., 542, 123 (1939). г
[91] G. King, J, Chem. Soc., 1936, 1788.
[92] F. Straus, A. Rohrbacher, Ber., 54, 69 (1921).
[931 К. B. W i b e eg, K. A, S a e ge b a r t h, J. Am. Chem. Soc,, 79, 2822 (1957)."C
[94] A. Lap wortu, E. N. Mothram, J. Chem. Soc., 1925, 1628.
IV. ПРИСОЕДИНЕНИЕ ГИДРОКСИЛЬНЫХ ГРУПП по с=с-связи
2;
При работе с 1,10 а (0,007 моль) КМпО4 в 110 мл воды и обесцвечивав^
реакционной массы через 1 мин получают 1,10 г неочищенного продукта (98<
от теоретического) и соответственно 0,88 г (79% от теоретического) чистог
продукта.
Олефины можно окислять также щелочным раствором КМпО. до уис-а-глик<
лей [96].
Ненасыщенные альдегиды гидроксилируют преимущественно в виде ацетале!
При окислении ацеталя акролеина КМпО4 в водном растворе получают ацеталь глг
церннового альдегида [95].
3. Гидроксилирование при помощи
четырсхокиси осмия
Водные растворы хлората калия [97] или, лучше, растворы хлората серебр
[98] в присутствии небольшого количества OsO4 стереоспецифически окисляют ол«
флны до yuc-a-гликолей (метод К. Гофмана). Этот метод пригоден главным o6paaoj
для окисления водорастворимых олефинов [99, 100].
Промежуточными продуктами окисления олефинов по Гофману являютс,
выделенные Криге [101] эфиры осмиевой кислоты
\с-о, ,О
f I
' )>С-о/ %
При кипячении с водными растворами хлоратов эфиры расщепляются до гликс
лей, причем регенерируется OsO4, которая может вновь присоединяться к молекул
олефина..,О механизме этой реакции см. [99, 100].
Препаративно наиболее целесообразно проводить омыление эфиров осмиево]
кислоты водным раствором Na2SOs, при этом осмий выпадает в виде труднораствори
мото комплекса с сульфитом.
Работы Криге с сотр. [101, 102] показали, что гидроксилирование при помощи
OsO4 применимо также для водонерастворимых олефинов. Эфиры осмиевой кислотх
образуют с 2 моль пиридина стойкие и хорошо кристаллизующиеся аддукты. Эп
аддукты могут быть также непосредственно получены из олефина, четырехокиси осмил
ц пиридина в соответствующем растворителе (хлороформ, этилацетат, бензол
СС14). Они легко и с хорошими выходами омыляются в tyuc-a-гликоли. Гидролиз про
водят методом восстановительного расщепления, кипячением' с водно-спиртовыь
раствором Na8CO3 или с формальдегидом в слабых щелочах [103] или переэтерифа
нацией под действием маннита в разбавленных щелочах [102].
__ Для проведения переэтерификации маннитом измельченный твердый пириди
новый комплекс встряхивают с водным щелочным раствором маннита (из расчета
2 моль КОН и 2 моль маннита на один атом осмия) до полного растворения или сначала
растворяют пиридиновый комплекс в хлористом метилене (или хлороформе) и ветра
хивают с щелочным раствором маннита до полного обесцвечивания раствора
[9й[ Е. J. Wi t zc mann et al., Org. Synth., Coll. v. 2, 1943, p. 307.
[96] W. Rigby, J. Chem. Soc., 1956, 2452.
[97] К. A. Jlof man n, Ber., 45, 3329 (1912); K. A. Hofmann, O. Ehrhart
O. Schneider, Ber., 46, 1657 (1913).
[98] G. Braun, J. Am. Chem. Soc., 51, 288 (1929).
[99] R. Criegee, в книге G. M. Schwab, Handbuch der Katalyse, Bd. 7/1
Springer-Verlag, Wien, 1943, S. 597.
[100 J N, A. M ilas, в книге The Chemistry of Petroleum Hydrocarbons, v. 2, Rein-
, hold Publishing Corporation, New York, 1955, p. 419.
'[101] R. Criegee, Ann., 522, 75 (1936).
[102] R. Criegee, B. Marchand, H. W a n n о w i u s, Ann., 550, 99 (1942)
[103] E. li. H. J о nes, R. J. Meekins, J. Chem. Soc., 1940, 456; H. Reich
M. Sa ttai, T. Reichstein, Helv. chim. acta, 23, 170 (1940).
18*
276
ОБРАЗОВАНИИ С-О-СВЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
В большинстве случаев диолы получаются с превосходными выходами. Так как
процесс можно вести без нагревания, побочных реакций почти не наблюдается-
Этот метод имеет особое значение в химии стероидов (104].
Изучение кинетики этой реакции проводилось Баджером [105], который
осуществил окнеление OsO4 многих конденсированных ароматических соединении.
^«с-Цнклогептандиол-1,2 [102]. В 100 мл абсолютного эфира при 0° С
вносят 1,92 г циклогептспа, 5,10 г ОзО4 и 3,5 мл пиридина. Смесь почти тотчас
же затвердевает -в коричневую кристаллическую кашицу. Через 30 мин кри-
сталлы отсасывают и промывают их эфиром. Получают светло-коричневый кри-
сталлический порошок пиридинового комплекса; выход 10,01 г (98,5% от тео-
ретического). Его перекристаллизовывают из смеси хлористого метилена с петро-
лейньгм эфиром.
Встряхивают 9,5 г-полученного аддукта с раствором 10 г маннита в 100 мл
водного раствора КОН. Аддукт быстро растворяется. Затем в течение 12 ч диол
экстрагируют хлористым метиленом, отгоняют из экстракта растворитель и оста-
ток перегоняют в высоком вакууме. При температуре бани 90—100° С и раз-
режении О.1 мм рт. ст. перегоняется почти теоретическое количество чистого
г{ис-циКлОгептандиола-1,2, который затвердевает; т. пл. 48° С.
4. Гидроксилирование при помощи комплекса иода
с бензоатом серебра (реактив Прево)
Прево с сотр. [106—111] показал, что олефины стереоспецифически взаимо-
действуют с раствором комплекса [(CeH5COO)2Ag]I с образованием транс-i,2-дибен-
зоатов, из которых легко получаются соответствующие трднс-1,2-гликоли. Комплекс
готовят взаимодействием бензоата серебра с иодом в бензоле.
Краткий обзор этой реакции см. [112].
Гексадекандиол-1,2 [ИЗ]. К суспензии 26,5 г бензоата серебра в 150 лл ’
бензола при встряхивании прибавляют раствор 10,6 г иода в 100 мл бензола.
Затем к смеси медленно приливают при встряхивании 10,5 г теисадсцона-1 в 50 мл
бензола и кипятят 1 чс обратным холодильником. После охлаждения фильтруют
и отгоняют растворитель. Остающийся дибензоат гексадекандиола-1,2 омыляют
в течение 3 ч кипячением с 12 г КОН в 75 мл этилового спирта и 25 мл воды...
Гидролизат вливают в 500 мл горячей воды и после охлаждения отсасывают
выпавший в осадок продукт, который перекристаллизовывают сначала из метц-
лового спирта, затем из лигроина и вновь из метилового спирта. Выход 4 г (33%
от теоретического); т. пл. 73,1—73,6° С.
[104] A, Butenandt, Z. Naturforsch., 1, 222 (1946); J. W. С о о k, J. J а с k,
J. D. Loudon et al., J. Chem. Soc., 1951, 1397; L. H, Sarett, J. Am.
Chem. Soc., 71, 1169 (1949); ft. B. Woodward, F. Sondheimer et al.,
J. Am. Chem. Soc., 74, 4223 (1952); R. H trsch m ann et al., J. Am. Chem.
Soc., 76, 4013 (1954); W. S. A 11 e n, S. Bernstein,!. Am. Chem. Soc., 78,
1909 (1956); К. В i e m a nn, G. Biichi, В. H. Walker, J. Am. Chem.
Soc., 79, 5558 (1957).
[105]- G. M. Badger, J, Chem. Soc., 1949, 456; 1950, 1809; G. M. Badger,
K. R. Lynn J. Chem. Soc., 1950, 1726.
106] C. Prevost, С. r., 196, 1129 (1933); 197, 166 (1933).
107] C. Prevost, Losson, C. r., 198, 659 (1934).
108] С. P r ё v о s t, B. L и t z, C. r., 198, 2264 (1934).
109] C. Prevost, C. r., 200, 408, 942 (1935).
НО] C. Prevost, J. Wiemann, C. r., 204, 700 (1937).
Ill] C. Prdvost, C. r., 204, 989 (1937).
112] N. A. Milas, в книге The Chemistry of Petroleum Hydrocarbons, v. 2, Rein-
hold Publishing Corporation, New York, 1955, p. 431.
[ИЗ] C. Niemann, G. D. Wagner, J. Org. Chem., 7, 227 (1942).
IV. ПРИСОЕДИНЕНИЕ ГИДРОКСИЛЬНЫХ ГРУПП по с=с-связи
27'
5. г/мс-Гидроксилироваиие по методу Вудворда
Вудворд и Брутчер мл. [114] предложили новый метод синтеза цис-сс-гликолеЙ
по которому олефины подвергают взаимодействию с иодом и ацетатом серебра в уксус
ной кислоте. Первая стадпя реакции приводит к образованию уис-оксиацетатов
гидролизом которых получают цис-а-гликоли. Этот использованный сначала в хини,
стероидов [115] метод получил затем широкое распространение для цис-гидроксилв
роваппя [116—118],
цис- Гидроксилированое по Вудворду базируется на работах Випштейна с сотр
о влиянии заместителей в органической химии (см. обзорную статью [119]).
г^ис-Цпклогександиол-!,2 [118]. В трехгорлой колбе, снабженной обрад
ним холодильником, термометром и мешалкой, к суспензии 16 г (0,096 ляэлб
ацетата серебра в 150 мл ледяной уксусной кислоты прибавляют 3,42
(0,042 моль) свежеперегнанного циклогексена (т. кии. 83—85° С). Затем в течени
30 мин при комнатной температуре и сильном перемешивании в реакционную
смесь впосят 11,7 г (0,046 моль) порошкообразного иода, смешивают с 6,67
воды п при перемешивании нагревают 3 ч при 90—95° С. После охлаждена]
фильтруют, осадок на фильтре промывают горячим бензолом и этилацетатом
фильтраты объединяют и отгоняют растворитель в вакууме водоструйного на
coca. Остающееся желтое вязкое масло растворяют в метиловом спирте, и рас
твор фильтруют. Фильтрат нейтрализуют небольшим количеством спиртового
раствора КОН и затем 1,5 ч кипятят с обратным холодильником, добавив раство]
3,5 г КОН в 20 мл метилового спирта. После отгонки в вакууме метиловог<
спирта остаток растворяют в 500 мл горячего эфира, фильтруют и отгоняют эфир
Остается 3,92 г (81% от теоретического) неочищенного продукта. Перекристад
лизацией из СС14 получают 3,2 г (66% от теоретического) чистого цис-цикло-
гександиола-1,2; т. пл. 97—98° С.
' 6. Гидроксилирование при помощи перекиси водорода
и неорганических катализаторов
Прямое присоединение перекиси водорода по двойным связям олефинов соответ-
ственно уравнению
клстзатор \с-с/
z /| Iх
но он
в большинстве случаев протекает [120] в органических растворителях в присутствии
каталитических количеств окислов металлов: V8O5, W03, МоО3, СгО3, SeO2. Для этой
цели пригодны такие окисльт металлов, которые легко дают с перекисью водорода
соответствующие неорганические надкислоты [121 — 122]. Подробные исследования
о возможностях использования указанных выше катализаторов проведены Магденои
и Юпгом [123].
[И4]
[115]
116]
117
118
119
120
121
[122]
[123]
R. В. Woodward, F. V. Brulcher jr., J. Am. Chem. Soc., 80, 209
(1958).
W. S. К n о w I e s, Q, E. Thompson,!. Am. Chem. Soc., 79, 3212 (1957);
L. B. Barkley, W. S. Knowles, H. Raffelson, Q. E. Thomp-
son, J. Am. Chem. Soc., 78, 4111 (1956).
D. Ginsburg, J. Am. Chem. Soc., 75, 5746 (1953).
F. D. Gunstone, L. J. Morris, J. Chem. Soc., 1957, 487.
F. V. Brutcher jr., G, Evans, J. Org. Chem., 23, 618 (1958).
W. L w о w s k i, Angew. Chem., 70, 490 (1958).
N. A. Milas, J. Am. Chem. Soc., 59, 2342 (1937).
W. Treibs, Ber., 72, 1194 (1939).
W. Treibs, G. Franke, G. Le ichsen ring, H. R 6 d er, Chem.
Ber., 86, 616 (1953).
M. Mugdan, D. P. Y о u n g, J. Chem. Soc., 1949, 2988.
278
ОБРАЗОВАНИЕ С-О-СВЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Гидроксилирование олефинов при помощи Н2О2 в присутствии OsO4 приводит
к ц ас-гликолям. Реакция протекает через промежуточное образование циклических
эфиров осмиевой кислоты, при гидролизе которых получаются yuc-гликолп. Другие
катализаторы способствуют транс-гидроксилированию, происходящему с промежу-
точным образованием эпоксисоединений [124].
Окисление олефинов Н2О2 в присутствии каталитических количеств ОзО4
может проводиться в абсолютном третичном бутиловом спирте (реагент Майласа) [125],
в эфире [126], в водном метиловом спирте [127] или в ацетоне [128]. Вместо перекиси
водорода может также применяться трет-бутилгидроперекись [129].
Гидроксилирование полиолефинов при помощи реагента Майласа может проис-
ходить так, что реагируют только некоторые двойные связи. Например, из цикло-
пентадиена образуется смесь 1,2- и 1,4-дициклопентендиолов [125, 131]. Этот метод
применяется, в частности, для селективного окисления 0-каротина в альдегид вита-
мина А [130].
Окисление при помощи VaOe и Н2О2 ведут в безводном третичном бутиловом
спирте, в разбавленном метиловом спирте или в ацетоновом растворе. В этом случае
окисление происходит яеспецифически, так как наряду с гидроксилированием про-
текают другие реакции окисления, особенно по аллильным положениям [121, 122].
Магден и Юнг [123] рекомендуют WO8 в качестве катализатора гидроксилиро-
вания перекисью водорода. При этом наиболее целесообразно проводить реакцию
в водном или уксуснокислом растворе при 50—70° С. Окисление происходит стерео-
специфически с образованием тракс-а-гликолей. Этот способ применяется в промыш-
ленности; выходы, как правило, хорошие. Окислы молибдена или селена [132] равным
образом являются пригодными катализаторами гидроксилирования, однако они.’
почти не дают преимуществ по сравнению с окислами осмия и вольфрама.
У. Присоединение карбоновых кислот и спиртов по С~С-
и С=С-связям
Препаративное значение этой реакции, приводящей к образоиапию простых
или сложных эфиров, кеталей и ангидридов кислот, можно пояснить на нескольких-
примерах.
Органические карбоновые, кислоты присоединяются по углеродной двойной
связи тем легче, чем эта связь более активирована соседними замещающими группами.!
и чем больше кислотность карбоновой кислоты. Особенно легко образуются эфиры*
третичных спиртов
(CH3)2C=CH2+RCOOH (CH3)3C0C0R
Реакция обратима и катализируется сильными кислотами. Хорошими катали-
заторами являются 95%-ная H2SO4, органические сульфокислоты и хлорид цинка*
[124] G. В. Р а у и е, С. W. Smith, J. Org. Chem., 22, 1682 (1957).
[125] N. A. M i I a s, S. Sussmaa, J. Am. Chem. Soc., 58, 1302 (1936); 59, 2345'
(1937); N. A. Milas, S. Sussman, II. S. Mason, J. Am. Cnem. Soc.,A
61, 1844 (1939); N. A. Milas, L, C, Malnn e y, Is Am. Chem. Soc., 62,4
1841 (1940); R. C. Hockett, A. C. Sapp. S. R. Millmann, J. Am^J
Chem. Soc., 63, 2051 (1941); C. D. H u г d, С. D. К e 1 s o, J. Am. Chem. Soc.f«
70, 1484 (1948). t
[126] R. Crie gee, Ann., 522, 75 (1936). *
[127] N. Claus on-Kaas, J. Fakstrop, Acta Chem. Scand., 1, 216 (1947). j
[128] J. W. С о о k, R. Sc ho ent a I, J. Chem. Soc., 1950, 47.
[129] T. L. Gresham, T. R. Steadman, J. Am. Chem. Soc., 71, 737 (1949).J
J130J N. L. Wendler, C. Rosenblum, M. Tishler, J. Am., Chem.\
Soc., 72, 234 (1950): G. C. L. Goss, W. D. McFarlane, Science, 106,-^
375 (1947).
[131] L. N. Owen P. N Smith, J. Chem. Soc., 1952, 4035.
[132] P. Seguin, C r., 216, 667 (1943).
V. ПРИСОЕДИНЕНИЕ КАРБОНОВЫХ КИСЛОТ И СПИРТОВ ПО С=С-И С-С-СВЯЗЯМ 2'
|133| пли трифторид бора J134—136]. Однако при применении более 10% BF3 пров
мшит преимущественно полимеризация. Для достижения хороших выходов сложнь
эфиров необходимо проводить реакцию при возможно более низкой температур
яуэе/п-Бутиловый эфир о-бензоилбенаойной кислоты [137]. Раств<
10,0 г о-бепзоилбензойпой кислоты в 70 мл эфира, содержащий 1 мл концентр
рованяой H2SO4, смешивают со 100 г жидкого изобутилена в склянке емкость
500 мл для работ под давлением. Закрытую склянку оставляют на ночь при КО]
латной температуре. После этого реакционный сосуд охлаждают смесью jn»i
<• поваренной солью, открывают и его содержимое вливают в холодный раствс
40 <- NaOH в 500 мл ледяной воды. Отделяют органическую фазу ц сушат не
К.СОз. После отгонки эфира получают 8,81 г (70% от теоретического) почд
чистого продукта; т. пл. 66—70° С.
При присоединении органических кислот к терпенам, особенно к камфен
и пинену, большей частью наряду с реакцией присоединения происходят перегрупщ
ровки.
Изоборнилацетат [138]. Смесь 100 г камфопа, 250 г ледяной уксусно
кислоты и 10 г 50%-пой H2S0^ пагревают в течение нескольких часов при 50—
60’’ С. При этом верхний слои постепенно исчезает и образуется прозрачны
красноватый гомогенный раствор, -из которого после охлаждения высаживаю
водой изоборпнпацетат. После промывки и сушки отгонкой выделяют франци]
с т. кип, 107° С (13 .млг рт. ст.). Пзоборнеол получают из неочищенного нас
борнилацетата.
Протекающее аналогичным путем превращение пинена в борнеол, которо
можно осуществить через борнеолбензоат [139] или оксалат [140], представляв
промышленный интерес.
Е то время как приводящее к образованию сложных эфиров ирисоединени
карбоновых кислот идет в кислой среде, присоединение спиртов по двойным связям
олефинов катализируется также и основаниями.
Метод присоединения спиртов представляет интерес для получения просты:
эфиров, особенно таких, в которых имеются и другие функциональные группы. Оченл
легко образуются третичные алкиловые эфиры [141] из изобутилена и изоамилена.
Реакция катализируется HCI, H2SO4, кислыми солями и А1С13. Первичные спирте,
присоединяются легче, чем вторичные; третичные спирты почти не присоединяются
Присоединение спиртов к олефинам [134] протекает также в присутствии катализа,
тора BF3.
Реакция фенолов с олефинами может протекать с образованием’простых эфиром
или приводить к алкилированию ядра. Катализаторами являются минеральные иислоте»
и BF3. При низких температурах получаются простые эфиры, при повышенных —
идет перегруппировка, приводящая к С-алкилпрованию [142, 143].
Реакция присоединения спирта к камфену происходит с перегруппировкой
приводящей к соответствующему изоборниловому эфиру. Меканием реакции выяс-
нен Меервейпом [144].
[133] (I, Meerwein, Ann., 455, 227 (1927); J. Kondakoff, Вег., 26, Ref,
1012 (1893); 25, Ref. 864 (1892); J. prakt. Chem. [2], 48, 467 (1893).
1134] jl. Meerwein, Ber., 66, 411 (1933); J. prakt. Chem. [2], 141, 126 (1934).
[135] T. B. Dorris, F. J. S о w a, J, A. Nieuwland, J. Am. Chem. Soc.,
56, 2689 (1934).
[136] Й. L. Wunderly, F. J. Sowa, J. Am. Chem. Soc., 59, 1010 (1937).
[137] W, S. J obnson, A. L. McCloskey, D. A. Dunnigan, J. Am,
Chem. Soc., 72, 514 (1950).
] 138] J. Bertram H. Walbaum, J. prakt. Chem. [2], 49, 1 (1894).
139] G. Boucbarda t, J. L a f о n t, С. r.. 125, 111 (1897).
140] Герм. пат. 134553 (1902).
141] T. W. E v a ns, K. R. Edlund, Ind. Eng. Chem., 28, 1186 (1936).
142j j. B. N ie derl, S. N a tels on, J. Am. Chem. Soc., 53, 272, 1928 (1931).
143] F. J. Sowa, H. D. Hinton, J. A. Nieuwland, J. Am. Chem. Soc.,
54, 2019, 3694 (1932).
[144] H. Meerwein, L. Gcrad, Ann., 435, 174 (1924).
280
ОБРАЗОВАНИЕ С-О-СВЯЗП В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Взаимодействием диметиловых эфиров малеиновой и фумаровой кислот с метила-
том натрия получен диметиловый эфир метоксиянтарной кислоты [145]. Более легко
в том же направлении реагируют алкилиденмалоповые эфиры общей формулы
Н2С=С(СООВ)2; Либерману [146J иэ бензальмалоновою эфира и этилата натрия
удалось сначала выделить натрийсодержащее соединение.
Реберг с сотр. [147] установили, что в безводной среде в присутствии слабых
щелочей первичные н вторичные (но не третичные) алифатические спирты легко при-
соединяются по двойным связям эфиров акриловой кислоты, причем с хорошими
выходами получаются эфиры р-алкоксипропионовой кислоты. Фенолы присоединяются
подобным образом.
Алифатические а,Р-ненасыщенные альдегиды реагируют с низшими спир-
тами с обрааовапием ацеталей, Р-алкоксиацеталей и р-алкоксиальдегидов. При-
соединение катализируется ионами водорода и ОН-ионами. Присоединение высших
спиртов происходит труднее. Из акролеина [148], например, при нагревании со спир-
тами в присутствии HCI пли толуолсульфокислоты образуется диацеталь р-алкокси-
пропионового альдегида наряду с p-алкоксипропионовым альдегидом и ацеталем
пропионового альдегида. Применение избыточного количества спирта повышает выход
диацеталя p-алкоксипропионового альдегида.
р-Метоксипропионовый альдегид из акролеина и метилового спирта [149],
Смесь 600 г 93,5%-ного акролеина (10 молъ), 1300 г метилового спирта (40 м,оль)
и 3 г концентрированной H2SO4 кипятят в колбе с обратным холодильником
в течение 6 ч. В начале кипячения температура смеси достигает 60° С, а после
5 ч кипячения достигает 75—76° С. Для нейтрализации и полного удаления
ионов SOJ~ к смеси при перемешивании прибавляют 6 г порошкообразной без-
водной Ва(ОН)2 или эквивалентное количество гидратированной Ва(ОН)а
и 4 ч интенсивно перемешивают при комнатной температуре. Затем прибавляют
1,8 г порошкообразной безводной щавелевой кислоты и еще 2 ч перемешивают.
Титрованием проверяют содержание свободной щавелевой кислоты, которое
должно равняться 0,04%. Омыление промежуточно образующегося диметил-
ацеталя р-метокеппропионового альдегида заканчивается за 2 ч. Реакционную
массу фильтруют п разгоняют на колонке. Получается ]3-метоксппропионовый
альдегид (т. кии. 121° С при 760 мм рт. ст.}, образующий с водой азеотропную
смесь (т. кип. 92е С при 760 мм рт. ст.).
Под влиянием оснований (алкоголятов, щелочей, третичных аминов) из крото-
нового альдегида и метилового спирта на холоду образуется В-метоксибутиральде-
гид [150].
₽-Метоксибутиральдегид [151]. Смесь 25 г кротонового альдегида и 50 мл
абсолютного метилового спирта охлаждают до —10° С и после нейтрализации
в большинстве случаев кислой реакционной массы несколькими каплями рас-
твора метилата калия прибавляют еще 1 мл 10% кого раствора метилата калия.
Температура повышается до 40° С и замах кротонового альдегида исчезает.
По охлаждении массу слегка подкисляют уксусной кислотой, извлекают про-
дукт реакции большим количеством эфира и эфирный раствор сушат над K2COg-
[14.5] Th. Р и г d i е, .1.’ Chem. Soc., 47, 8o9 (1885); Th. Purdie, W. Marshall,
J. Chem. Soc., 59, 469 (1891); Beilstein, Bd. 3, S. 437.
[146] C. Liebermann, Ber., 26, 1876 (1893); L. Claisen, L. Crismer,
Ann., 218, 143 (1883).
[147] С, E. R eh berg, M. B. D iron, С. H. Fish er, J. Am. Chem. Soc.,
68, 544 (1946); 69, 2970 (1947); С. E. Rehberg, M. B. Dixon, J. Am.
Chem. Soc., 72, 2205 (1950)- R. H. H a 11, E. S. S t e r n, J. Chem. Soc., 1Я49.
2035.
[148] L. Claisen, Ber., 31, 1014 (1898).
[149] H. Schulz, H. Wagner, Angew. Chem., 62, 115 (1950).
[150] Герм. пат. 554949; С., 1932, 11, 2107.
[151] H. Meerwein, в книге Houhen-Wey I, Methoden der orgams-
chen Chemie, Bd. 3, 2 Anti., Leipzig, 1923, S. 127.
V. ПРИСОЕДИНЕНИЕ КАРБОНОВЫХ КИСЛОТ И СПИРТОВ по с-с- и с-с-связям
2
р-.Метокснбутиральдегид, остающимся после отгонки эфира, перегоняют п
атмосферном давлении; т. кип. 128° С (760 .н.п рт. ст.)\ выход 50% от теорез
ческого.
В присутствии щелочных катализаторов акролеин и кротоновый альдег
ле! ко осмоляются.
Спирты присоединяются по особенно реакционноспособным двойным связ;
шшнлкетонов с образованием р-алкоксикетонов, причем реакция катализирует
п щелочами, и кислотами. Добавление ингибиторов (гидрохинон и др.) ограничив^
побочные реакции. Примером такой реакции является синтез 1-метоксибутанона-З
метилвинилкетона:
СН3СОСН=СН2+СН3ОН —> СНзСОСН2СН2ОСНз
1 -Метоксибутаион-З. Получение в присутствии мег
лата натрия [152]. К смеси 368 г (11,5 моль) безводного метилово
спирта и 4,5 г метилата натрия при 10—15° С в течение 2 ч прибавляют при nej
мешивании 231 г (3,-Злкмъ) метилвинилкетона. Охлаждая в ледяной бане, сме
перемешивают еще 3 ч л затем нейтрализуют раствором НС1 в метиловом сайр
(pH 7). Раствор оставляют на почь в холодильнике (5° С) и затем перегоняв
Выход 246 г, что составляет 73% от теоретического; т. кип. 137 — 140° С (и-
760 мм рт. ст.), 64—66° С (при 50 мм рт. ст.)-, 1,4050.
Получение в присутствии окиси ртути и тр
фторида бора [153]. В литровую трехгорлую колбу вносят 1—2 г кре
ной окиси ртути, 1 мл эфирата трифторида бора и 2 мл безводного метилово
спирта. Массу нагревают, добавляют еще 26 г метплового спирта и при пер
мешивании приливают по каплям 65 г (1,08 моль) метилвинилкетона и 4£
(1,05 моль) метилового спирта. Далее кипятят 1 ч с обратным холодильник»
па водяной бане и оставляют па ночь. После нейтрализации безводным КлС<
продукт выделяют перегонкой. Выход 61% от теоретического; т. кип. 137
138° С (745 мм рт. ст.).
Дюфресс и Жеральд [154] изучили присоединение спиртов к ненасыщеннь
кетонам, содержащим галоген при двойных связях. Так, к а-бромбензальацетофешц
в присутствии небольшого количества алкоголята в абсолютном спиртовом раство'
па холоду присоединяются метиловый и этиловый спирты, нормальные и разветвив
ные пропиловые и бутиловые спирты:
C6HbCOCBr=CHCeH6+ROH °Н~ СвН6СОСНВгСНСвН6
I
OR
При более высоких температурах в щелочной среде от этих продуктов отщв‘
ляется бромистый водород и образуются эфиры енолов следующего состава:
CeH5COCH=C(OR)R
Р-Алкоксипропионитрилы получаются в результате присоединения к акрил
нитрилу первичных и вторичных спиртов под влиянием основных катализаторов[155
р-н-Бутоксипропионитрил [156]. В колбе с обратным холодильнинс*
смешивают 148 з (2 моль) я-бутилового спирта с 2 г 40%-ного водного раствор
гидроокиси триметилбензиламмония (тритон Б). К смеси прибавляют 10&
[152] R. С. Elderfield, В. М. Pitt, I. Wempen, J. Am. Cher
Soc., 72, 1334 (1950).
[153] D. P. Kilian G. F. Henn ion, J. A. Nieuwland, J. At
Chem. Soc., 58, 892 (1936).
. [154] Ch. D 11 f г a i s s e, P. Gerald, C. r., 173, 985 (1921); 174, 1631 (192S
Bull. Soc. chim, France [4], 31, 1292 (1922).
[155] C. F. К о e I s c h, J. Am. Chem. Soc., 65, 437 (1943); R. V. C Ii г i s t i a ,
jr., R. M. Hixon, J. Am. Chem. Soc., 70, 1333 (1948).
[156] W. P. Utermohlen jr., J. Am. Chem. Soc., 67, 1505 (1945).
282
ОБРАЗОВАНИЕ С-О-СВЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
(2 миль) акрилонитрила с такой скоростью, чтобы температура не поднималась
выше 45° С. По окончании добавления акрилонитрила смесь перемешивают
еще 1 ч, подкисляют уксусной кислотой и перегоняют в вакууме; т. кип. 98° С
(20 ,ч.1! рт. ст.); выход 219 г (86% от теоретического).
Присоединение карбоновых кислот к кетенам приводит к образованию анги-
дридов кислот:
О 0 0
(I II II
R—с—ОН+О=С=СН2 —*• R—С—О— С— СНз
Так, из кетена и ледяной уксусной кислоты получается уксусный ангидрид. С другими
карбоновыми кислотами по этой реакции образуются смешанные ангидриды [157—
159], которые, однако, легко диспропордионируются в симметричные ангидрйды
кислот. Этот метод дает возможность получать с хорошими выходами ангидриды,
которые с трудом могут быть синтезированы иными способами.
Получение ангидрида н--капроновой кислоты из капроновой кислоты и кетена
см. [160].
Ацилирование жидких карбоновых кислот можно проводить без растворителей;
твердые карбоновые кислоты растворяют в эфире [157], ацетоне [161] или бензоле
[162]. Температуры, при которых проводят реакции, различны: наряду с охлажде-
нием льдом [160] указаны температуры от 20 (см. [161]) до 90° С (см. [162]). Обзорную
статью «Кетен в препаративной органической химии» см. [163].
Низшие алифатические спирты гладко ацетилируются кетеном при комнатной
температуре:
О
II
ROH+O=C=CH2 —> RO-С-СНз
Взаимодействие с высшими спиртами и третичным бутиловым спиртом удается
провести в присутствии кислотных катализаторов [164], иногда применимы также
и основания [159, 165].
Наличие других функциональных групп в молекуле одноатомных спиртов
в общем не препятствует ацетилированию; многоатомные спирты [166] плохо ацети-
лируются даже в присутствии катализаторов. Очевидно, накопление гидроксильных
групп в одной молекуле снижает способность спирта реагировать с кетеном.
С фенолами кетены способны реагировать только в присутствии катализаторов.
В присутствии серной кислоты, n-толуолсульфокислоты или других кислотных ката--
лизаторов выходы продуктов почти всегда высокие [164, 167, 168]. Из многоатом-
ных фенолов с кетенами реагируют только гидрохинон [169] и резорцин [168]..
Реакция кетена с флороглюцином протекает медленно и с плохими выходами даже
в присутствии серной кислоты.
Ch. D. Hur d, И. F. D u 11, 1. Am. Chem. Soc,, 54, 3427 (1932).
A. R. Emery, V. Gold, J, Chem. Soc,, 1950, 1443,
A. B. Boese jr;. Ind, Eng. Chem., 32, 16 (1940).
J. W. Williams, J, A. Kry nf tzky. Org, Synth,, 21, 13 (1941).
A. H. Gleason, пат. США 2178752; С., 1940, I, 1566,
G. D e Witt Graves, пат. США 2135709; С., 1939,. I, 796.
G. Quadbeek, Angew. Chem., 6&, 361 (1956).
Ch. D. Hur d, A. S. R о e, J. Am. Chem. Soc., 61, 3355 (1939).
F. С h i c k, N. T. M. Wilsmore, J. Chem. Soc., 93, 946 (1908); 97; 1978
(1910). .
Ch. 1). Hurd, S M. Can t or, A. S, R oe, J. Am. Chem. Soc., 61, 426
(1939).
G. De Witt Graves, пат. США 2007968; С., 1935, II, 3299.
R. N о d,zu, ,T. I s о s h i m a, Cr, 1955, 8358.
F. O. Rice, J. Greenberg et al., J. Am. Chem. Soc., 56, 1760 (1934).
V. ПРИСОЕДИНЕНИЕ КАРБОЛОВЫХ КИСЛОТ И СПИРТОВ ПО С = С-II С С-СВЯЗЯМ 28
' третп-Бутилацетат [164], Нагревают до 60° С mpcm-бутиловый сппр
(74 р), к которому добавлена концентрированная II2SO4 (0,5 .мл), и в течени
1,5 н пропускают кетен. Слегка окрашенный в коричневый цвет продукт реакцп
промывают 10 мл 2 н. раствора NaOH и затем 20 мл воды. Оба промывных вол
пых раствора экстрагируют эфиром и объединяют обе эфирные вытяжки с не
очищенным продуктом. Получсппый раствор сушат над К2СОз и перегоняю
при атмосферном давлении. После отгонки небольшой первой фракции пере
гоняется mpem-бутилацетат; т. кип. 94—96° С (750 мм рт. ст.)-, выход 83 г
С ацетиленом спирты реагируют под давлением в присутствии щелочных катали
заторов с образованием виниловых эфиров* (винилирование) [170]:
CH=CH+HOR —> CH2=CH-OR
В присутствии кислотных катализаторов, таких, как BFS и смесь HgO с BFg
к ацетилену присоединяются две молекулы спирта и получаются ацетали [171—174]
CH=CH+2HOR —-> СНз-CH(OR)s
Лиюратурный обзор см. [175],
Днизоамилацеталь ацетальдегида. В 200 г изоамилового спирта, с’одер
жащего 10 г 63%-ного раствора BFs в метиловом спирте и 1 г HgO, пропускаю1
29,5 г ацетилена. По завершении реакцинТмассу промывают небольшим коли
чеством воды и соды и экстрагируют продукт реакции эфиром. Эфирный раство]
сушат над К2СОз и перегоняют.
При получении неустойчивых в кислой среде ацеталей, особенно кеталей
перед смешением с водой необходимо тщательно нейтрализовать реакционную
массу безводной содой.
Точно так же, как с одноатомными спиртами, ацетилен реагирует с многоатои
ными спиртами, а-оксикислотами и их производными [171].
Из замещенных ацетиленов образуются кетали [176, 177]. Взаимодействие»
бутилацетилена с метиловым спиртом получают с 70%-иым выходом диметилацетал!
н -бутилметилкетона:
С4Н9С=СН+2СН3ОН С4Н8С(ОСНз)2СНз
В качестве катализатора можно добавлять эфират трифторида бора и окись ртути
В присутствии BF3 и HgO реакция протекает гладко только с метиловым спир
том; другие одноатомные спирты реагируют по неустановленному механизму с образо-
ванием полимерных продуктов. Однако добавление к катализатору небольшого ноли
чсства трихлоруксусной кислоты позволяет проводить реакцию также и с высшими
спиртами [178].
К ацетиленам, тройная связь которых находится в сопряжении с олефиновой
двойной связью, в присутствии окиси ртути и борфторидалкоголята присоединяются
* Эта реакция отбыта А. Е. Фаворским‘[ЖРХО, 20,518 (1888)], — Прим, ред,
[170] W, R е р р е, Chemie und Tectinik der Acotylen-Druck-Roaktionen, Verlag
Chemie, Weinheim/Bergstrape, 1952.
[171] J. A. Nieuwland, R. R. Vogt, W, L, F о о h e y, J. Am, Chem, Soc..
52, 1018 (1930),
[172] H. J). II i n t о n, J. A. Nieuwland, J. Am. Chem. Soc,, 52 , 2892 (1930).
[173] H. Bowlus, J. A. N ieuwlan d, J, Am. Chem. Soc., 53 , 3835 (1931).
[174] L. A. O’L e а г у, H. H. W e n z k e, J. Am. Chem. Soc., 55, 2117 (1933).
[175] D. Kastner, в книге W. Foerst, Neuere Methoden der praparativeo
organischen Chemie, 2 Aufl,, Verlag Chemie, 1944, S. 419.
[176] G. F. HenBion, D. B. Killian et al., J. Am. Chem. Soc,, 56, 113Q
(1934).
[177] D. B. Killian, G. F. Henni on, J. A. Nieuwland, J. Am. Chem,
. ,, Soc., 56, 1384 (1934).
[178] D. B. Killian, 6. F. H e n n i о n, J. A. Nieuwland, J. Am: Chem.
Soc., 58, 80 (1936).
284
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
три молекулы спирта; например, из винилацетилена с метиловым спиртом получается
2,2,4-триметокснбутан [179, 180]:
СН.=СН-С^СН -+эсн-о1!-х СНзО—CH.CII.-С(0СН8)г-СНз
2,2,4-Триметоксибутан. К катализатору, состоящему из 15 г красной
TTgO, 4,5 мл эфирата трпфторпда бора, 2 г трихлоруксусной кислоты и 10 мл
метилового спирта прибавляют 480 г (15 моль) метилового спирта. Смесь пере-
носят в трехгор.тую колбу емкостью 3 л, снабженную эффективным холодиль-
ником, и прп охлаждении водопроводной водой и постоянном перемешивании
в течение 3 ч пропускают 312 г (6 моль) винилацетилена. Затем температуру
водяной бани повышают до 50° С и еще 1 ч непрерывно перемешивают. После
этого прибавляют 10 г безводного К2СОз л перемешивают еще 15 мин. Жидкую
фазу отделяют п перегоняют. Получают 487 г 2,2,4-триметоксибутана (65%
от теоретического и расчете на метпловый спирт); т. кип. 63—65° С
(25 мм рт. ст.); пг£ 1,4082.
К ацетиленам, тройная связь которых находится в сопряжении с карбоксильной
или фенильной группой *, первичные спирты присоединяются в присутствии апкого-
лята натрия [181 —184]. Так из фенилацетилена при 135е С получают В-алкоксисти-
рол 1182]; .
CeHiC^CH+ROlT сбн5сн=снок
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
С ПОЛУЧЕНИЕМ ГИДРОКСИЛ- И КАРБОНИЛСОДЕРЖАЩИХ СОЕДИНЕНИЙ
Z. Замещение водорода кислородом
Замещение водорода .кислородом в органических соединениях может осуще-
ствлю ься под действием различных окислителей. Однако во избежание окислительного
разрушения молекулы необходимо тщательно соблюдать условия проведения реакции.
Из наиболее употребительных окислителей мало специфичны перманганат
калия, окись хрома (VI) и азотная кислота. Тетраацетат свинца и SeO2, напротив,
обладают специфическим действием. В качестве окислителей применяются также
перекись водорода и ее производные, четырехокись азота, двуокись марганца, четырех-
окись рутения [185] и феррицианид калия [186].
В промышленности для окисления предпочтительно используют кислород
(чаще всего воздух) в присутствии катализатора. Реакции аутоокислеиия, осуществля-
емые без катализатора под действием кислорода воздуха при нормальных условиях,
приводят, как правило, к образованию перекисных соединений.
Замена водорода на кислород возможна ив некоторых реакциях конденсации,
когда образование связи между двумя атомами протекает с обменом степенями
окисления. В этих целях окислителями служат главным образом нитрозосоедипения
О реакции А, Е, Фаворского - (для винилацетилена) и се синтетических
возможностях см. обширные исследования И. Н. Назарова и его школы
(И. П. Назар о’в, Избранные труды, Изд. All СССР, 1961). — Прим. ред.
D. В. Killian, G. F. Hennion, J. A. N ieuwland, J. Am. Chem.
Soc., 56, 1786 (1934).
D. B. Killian, G. F. Hennion, J. A. N ieuwlantl, J, Am. Chem.
Soc., 58, 892 (1936).
J. U. Nef, Ann., 308, 270 (1899).
Ch. M о u reu, С. r., 138, 286 (1904); Bull. Soc. chim. France [3], 31,527(1904).
К. А и wer s, Ber. 44, 3514 (1911).
K. Auwers, B. Ottens, Ber., 58, 2067 (1925).
L. M, Berko wit z, P. N. R у 1 a n d e r, J. Am. Chem. Soc., 80, 6682
(1958). ’ ’
B. S. Thyagajajan, Chem. Rev., 58, 439 (1958).
I. ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
25
м.111 производные азотистой кислоты. Они дают с активными метнленоЬымп группам
шиффовы основания или оксимы, которые гидролитически расщепляются е образ<
ванием оксосоединений.
Дегидрирование гидроксилсодержащих. соединений также может рассматрз
кагься как реакция окисления. Окисление углерода в этом случае происходит пут<
перевода ординарной связи С—О в двойную связь С=О (например, при .дегидрир
вании спиртов или альдегидгидратов).
1. Замещение водорода гидроксильной группой
Окисление углеводородов до первичных и вторичных спиртов практичесв
применяется в лабораторной практике лишь в отдельных случаях. Толуол и его пром
водные можно переводить в соответствующие бензиловые спирты с помощью, наприме'
МпО2, РЬО2, тетраацетата свинца и кислоты Каро. Наиболее целесообразно применя;
тстраацетат свинца в ледяной уксусной кислоте, так как в. этих условие
образующиеся спирты этерифицируются, что защищает их от дальнейшего окислена
Получение бензилацетата из толуола [187]; В течение 4 ч кийятят'12О
тетраацетата свинца с 40 г толуола в 200 ,нл ледяной уксусной Кислоты,’ tfp&;
варигельно перегнанной над КМпО^. Затем отгоняют большую часть 'уксуснс
кислоты, остаток разбавляют водой, продукт извлекают эфиром, вытяжку встр.
хивают с раствором бикарбоната, калия и сушат. Разгонкой в вакууме пол;
чают 7,5 2 неочищенного бензплацетата..
Для получении тетраацегат? свинца 200 г чистого свинцового сурих
вносят При 80° С при перемешивании турбинной мешалкой очень маленькие
порциями в 1000 мл ледяной уксусной кислоты, следя за тем, чтобы не измен,
дась температура. Из профильтрованного, в случае надобности, раствора п{
охлаждении выкристаллизовывается в виде игл бесцветный тетраацетат свинц,
его отфильтровывают, промывают небольшим количеством ледяной уясуснс
кислоты и сушат в эксикаторе. О получении больших количеств см. [18.
189, 189а]. ' ' '
В алифатическом ряду СН8-группы, находящиеся рядом с атомами углерод
связанными с кислородом, во многих случаях окисляются т&трйацетатом свинца д
СН8ОН-грушг. Из ацетона получаются как моноокси-, так it диоксия^етон в вид
их ацетатов, из уксусного ангидрида — диацетат ангидрида гликолевой КИелотх
из ацетофенона — ацетат бензоилкарбинола [190]. ' 11
Образование вторичных спиртов происходит при помощи тех же окислителе]
Например, по реакции дпфенилметана с тетраацетатом свинца получается ацетл
дифенилкарбинола; окисление дифенилметана идет несколько Легче, чей окисленв
толуола. Из малонового эфира получается ацетоксималоновый эфир, из ацетоуксу
ного эфира—а-ацетоксиацотоуксусйый эфир,' соединения, которые "Очень труда
синтезировать другими Путями. 1 " ' ''
В качестве дальнейшего примера можно указать на превращение ацеНафтеь
в аценафтенол По реакции с двуокисью свинца в ледяной уксусной кислоте 119
и по реакции с суриком в лёдяпой уксусной кислоте 1'192].
Новые исследования по окислению тетраацетатом свипца опубликованы Криа
[193]. Кроме того, следует упомянуть обзорную статью по окислению тетраацетатс
свинца и иодной кислотой [194].
В кетонах соседняя с карбонильной СН2-груипа не реагирует с кислотой Кар.
напротив, по аналоцщ с биологическим окислением жирных кислот в организм
окисляются 0-атомы углерода. Например, из метилпропилкетона образуете
[187] О. Dimrotb, R. Schweizer, Ber., 56, 1384 (1923).
H88J R. Criegee, Ann., 481, 283 (1930).
1189J 11. Mendel, Hee. trav. chim., 59, 720 (1940).
1190] O. D imrdth, R. Schweizer, Ber., 66, 1375 (1923).
[191] R. Marquis, C. r., 182, 1227 (1926).
[192] J. Casow, Org. Synth 21, 1 (1941).
[193] R. Criegee, Angew. Chem., 70, 173 (1958).
[194] R. Cr iege e Neuere Methoden der praparativen organischen Chemie, 3 AufJ
Verlag Chemie, 1949, S. 21.
286
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
СНаСОСП2СН(ОН)СН3. Исключением из этого правила является диэтилкетон, из
которого при окислении получается С2НБСОСН(ОН)СН8.
Специфическим окислителем соседних с двойными связями метиленовых групп
до спиртовых групп является SeO2. Но этот метод получения ненасыщенных спиртов,
по-видимому, может с успехом применяться только для окисления олефинов более
чем с пятью атомами углерода в цепи. Например [195], из З-мстилпонтена-2 получается
3-мстилпентен-2-ол-4:
СНз-СН=С-СН2-СН3—J *СНз—СН—С—СН—СНз
I II '
СНя Н3С ОН
Подробно изучена реакция алициклических олефинов'с Se02. Циклопентен <
окисляется при нагревании с SeO2 в уксусном ангидриде (ацилирующее окисление) ’’
с образованием моноацетокси- наряду с небольшим количеством диапетоксицикло-
пентена. При проведении реакции в запаянной трубке при 90—100° С, наоборот, '
получается в основном диацетоксисоединение и небольшое количество моноацетокси-
соединения [196]. Следовательно, в зависимости от условий проведения реакции 1
в большей или меньшей степени окисляется также и вторая, соседняя с двойной связью, :Ь‘
метиленовая группа. •;
Ацетокснциклопептеиы. Раствор 50 а циклопентена в 50 мл уксусного
ангидрида нагревают на водяной бане с 50 г SeOa до прекращения кипения смеси !
(6—8 ч). Затем нагревание на водяной бане продолжают еще 3 ч. Красно-корнч-
невую массу перегоняют в сильно охлажденный приемник, сначала при атмо-
сферном давлении, а затем при разрежений (12 мм рт, ст.). В отгоняющейся
при нормальном давлении и температуре бани 80—130° С фракции наряду с ук-
сусной кислотой и уксусным ангидридом содержится моноацетат. Продукт реак-
ции очищают повторной перегонкой (т. кип. 154—155° С при 760 мм рт. ст.).
Фракция, перегоняющаяся при 120—130° С (15 мм рт. ст.) состоит в основном
из диацстата, выход которого после повторной перегонки составляет 4—5 г.
д
В жирных кислотах с разветвленной цепью водород у третичного атома угле-
рода замещается гидроксилом под действием щелочного раствора КМпО4. Например,
из изомасляной кислоты получается а-оксинзомасляная кислота, а из изокапроновой Й
кислоты иэокапролактон 1197]. ]
Третичные спирты можно иногда синтезировать из соответствующих углеводо- . у
?одов также с помощью перекиси водорода и ее производных. Например, 10-метил- t
О-оксиантрон может быть получен действием щелочного раствора перекиси водорода Д
на 10-метилантрон [198]. J
Производные трифенилметана переводят в трифенилкарбинолы с помощью £
различных окислителей. Сам трифенилметан окисляется HN03 или СгО3 в ледяной *
уксусной кислоте в трифенилкарбинол, а тетраацетатом свинца -г- в ацетат трифенил-
карбинола. Наибольшее значение реакция имеет в производстве красителей трнфе- '<
нилметанового ряда из лейкоосноваяий; для этой цели применяются самые различные
окислители — мышьяковая кислота, нитробензол, азотистая или иитрозилсериая Л
инслоты.
Выделенные лейкоосноваиня лучше всего окислять в солянокислом растворе . Л
МпО2 или РЪО2 до солей красителей и затем выделять из ких щелочью карбинолы: л
н он
СП- СНз СНзх I ,СНз ;
>N—с—Ч—N/ —> >N—V- с—{ %—N< ’
сц/ I \сн3 СН/ Х=/ I \снч \
б) о
[195] A. Gnillmocat, С. г., 201, 904 (f935).
]196] Е. D а и е, 3. 8 с h m i t t, C. Rautenslrauch, Ann., 532, 29 (1937).
[197] R. M e у e r, Ann., 219, 240 (1883),
[198] H. Hey m-а n n, Trowridge, J. Am. Chem. Soc., 72, 85 (1950),
I. ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
28-
Растворяют 3,3 г лейкоосновапия малахитового зеленого в 40 мл горячо;
1 и. IIC1, разбавляют 300 мл воды ц к раствору после охлаждения (при сильно!
встряхивании) в течение 5 мин прибавляют взвесь 2,4 г РЬОг в 20 мл воды
встряхивают еще 5 мин при нагревании на водяной бане и к прозрачному рас
тлору прибавляют 4 г 7.цС1а. Продукт реакции высаливают насыщенным рас
твором NaCI, отсасывают, растворяют в горячей воде и вновь высаливают. Выхо,
двойной соли красителя с ZnC|a равен 3 г.
По Виллигеру и Конечному [199], к разбавленному водному раствор-
соли красителя при размешивании постепенно по каплям приливают снльц'
разбавленный раствор NaOH или NaaCOs, отфильтровывают выпавшее карбц
мольное основание и после перекристаллизации из лигроина получают его в’вид
кристаллических корок с т. пл. 120—122° С, а из эфира в виде кубиков с т. пл
109—110° С. Очевидно, это связано с диморфизмом, который Эмиль и Отто Фише]
наблюдали также и для лейкооснований [200].
. В ряду ароматических соединений классическим примером прямого введение
фенольного гидроксила при помощи окисления является получение ализарина и<
p-оксиантрахинона. Препаративное значение имеют и другие синтезы; так по Чита
бабину, хинолин превращается при щелочном плавлении в а-оксихинолин (карбости
рил) [201].
Более универсальным способом является окисление фенолов до дифеноло]
ио Элбсу [202] с помощью 1 моль надсерной кислоты в щелочной среде. При этць
вторая ОН-группа вступает в пара-положение. Пара-замещенные одноатомные феноль
окисляются 3 моль надсерной кислотой с образованием орто-замещенных продукте^
окисления с хорошими выходами.
• Получение 2,5-диокси-6-метоксиацетофенона из 2-окси-6-метокснацето
фенона [203]. Растворяют 16 г 2-окси-6-метоксиацетофенОна в 200 мл 10%-ног<
раствора NaOH и охлаждают до 15° С. Затем к полученному раствору при 15е <
в течение 4 ч приливают по каплям раствор 26 е K2SgOe в 500 мл воды, выдер
живают реакционную массу 24 ч при комнатной температуре и подкисляю*
по конго концентрированной НС1. При этом выпадает не вступивший
в реакцию исходный продукт. Далее добавляют еще 150 мл концентрированно!
НС1, нагревают 1 ч на паровой бане, горячий раствор кипятят с животным углеь
и фильтруют. При охлаждении раствора до 0° С выделяется 3,5 г 2,5-диокса-
6-метоксиацетофенопа; светло-желтые призмы; т. пл. 90е С (из метиловог<
спирта). Из маточника извлекают эфиром еще 1,5 г продукта.
Из о-нитрофенола таким же способом получают 2-нитрогидрохинон; выход
30% от теоретического [204].
Цротокатеховую кислоту получают окислением гг-окслбензойной кислоты
выход 50% от теоретического [205].
Берман и Питт [206] применили метод окисления по Элбсу для соединений
пиридинового ряда и полумили, например, 2,5-диоксипиридин из 2-оксипиридин®
с выходом 42% от теоретического. Обзорная статья по методу окисления персульфа-
тами по Элбсу см. [207, 208].
V. V i I i а е г, Е. Kopelschni, Вег., 45, 2916 (1912),
Е. Fischer, 0. Fischer, Вег., 12, 798 (1879).
А. Е. Tschitschihahin, Вег., 56, 1883 (1923).
К. Rihs, J. prakt, Chem., 48, 179 (1893).
К. Wallenfels, Вег., 75, 790 (1942).
М. J. As tie, St. P. Stephenson, J. Am. Chem. Soc., 65, 240
(1943).
К. В a h u Rao, N. V. Suhha Rao, C., 1957, Ю179.
E. J. Behrmann, В. M. P i 11, J. Am. -Chem. Soc., 80, 3717 (1958).
R. G. R. В а с о n, Sci. Proc. Roy. Dublin Soc., 27, 177 (1956).
S. M. Setlina, Chem. Rev., 49, 91 (1951).
288
ОБРАЗОВАНИЕ С—О-СВЯЗП В ОБМЕННЫХ РЕАКЦИЯХ
2. Образование перекисей путем аутоокисления
Многие органические вещества самых различных классов могут подвергаться
аутоокислению, т. е. изменяться под действием кислорода воздуха [209], При этом
часто образуются перекиси, т. е. соединения, содержащие группировку —00—.
По обменной реакции водорода с кислородом получаются только алкилгидро-
перекиси ROOH, надкислоты RCOOOH и диалкилперекиси ROOR. Другие типы
перекисей рассмотрены в разделе «Образование С—С-связи в результате реакции
присоединения».
Меры предосторожности при обращении с перекисями
Обычно взрывы перекисей вызываются нагреванием, ударом, толчком пли
катализаторами. Но следует также учитывать возможность самопроизвольного взрыва
даже таких перекисей, которые считаются безопасными. К самым важным мерам,
предосторожности относится ношение защитных очков. Ряд органических перекисей
(перекись mpem-бутила, гидроперекись изопропилбензола, перекиси циклогексанона,
метилэтилкетона и диацетила) вызывают тяжелые травмы при попадании в глаза
[210]. При такого рода несчастных случаях необходимо тотчас же промыть глаза
водой или лучше 2%-ным раствором NaHCO3. Применение маслянистых веществ
полностью исключается.
Не допускается получение, переработка и хранение больших количеств пере-
кисей. Хорошую защиту обеспечивают применение защитных экранов из небьющегося
стекла или передвижных защитных решеток из мелкоячеистой сетки. Для защиты
рук рекомендуется ношение прочных кожаных перчаток. Сосуды с перекисями даже
в лаборатории следует переносить в маленьких корзинах из мелкоячеистой сетки.
Перекиси должны храниться в холодильнике, сосуды с перекисями нельзя оставлять
без контроля вне холодильника. Использование толстостенных сосудов с притертыми
пробками опасно! Взрывчатые перекиси нельзя отжимать на пористой тарелке метал-
лическим или стеклянным шпателем, их следует сушить на фильтровальной бумаге
и пользоваться при этом роговым шпателем.
Жидкие перекиси можно перегонять только в вакууме при температуре водяной
бани не выше 70—80° С. Остатки от таких перетопок необходимо тотчас же осторожно
разбавлять растворителем и выливать.
Исполнители, проводящие микроанализы и ИК-спектроскопические исследо-
вания, должны быть предупреждены о взрывоопасности соединения. Температуру
плавления сильновзрывчатых перекисей нельзя определять в аппарате с серной кисло-
той: для этого пригоден металлический блок или микроаппарат для определения
температур плавления.
Образование гидроперекисей
При аутоокислении соединений с активированными CI-I-связями (олефины,
простые эфиры, спирты, альдегиды, кетоны, парафины) сначала всегда образуются
гидроперекиси ROOH по цепной радикальной реакции [211]:
r.+O2 —► ROO. ROOH-f-R« и т. д.
Перед проведением реакции окисления исходные продукты следует подвергнуть
очистке, главным образом для удаления ингибиторов аутоокисления. Крнсталличе- =
ские органические соединения растворяют в бензоле, хлороформе, этилацетате, серо-
углероде иля хлорбензоле, жидкие вещества можно подвергать аутоокислению
непосредственно или в растворителе. Обработку кислородом лучше всего проводить 1
[209] R. С г i е g е е, в книге Н о и Ь е n - W е у I, Methoden der organischen Che-
mie, Bd. 8, Stuttgart, 1952, S. 1; Fortschritte chem. Forschung, Bd. 1, Springer-
Verlag, Berlin, 1949/50, S. 508.
|210] H. J. Kiichle, ZbI. Arbeitsmed., Arboitsschntz, 8, 25 (1958).
[211] A. Riechc Angew. Chem., 50, 520 (1937); Die Bedeutung der organischen
Peroxyde fiir die chemische Wissenschaft und Technik, F. Enke, Stuttgart, 1936,
Angew. Chem., 5Ц 707 (1938).
I. ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
28!
в аппарате для встряхивания, исполь <уя кислород из баллона или пропуская тонко
распределенный ток кислорода пли воздуха. В последнем случае потери летучи?
продуктов можно предотвратить, соединив реактор с колонкой Рашита. Очень легкс
самоокисляющиеся тяжелолетучие вещества окисляются воздухом до гидроперекисе!
пли диалкилперекисей уже в открытых кристаллизаторах.
УФ-Облучение ускоряет поглощение кислорода, поэтому в большинстве олу
чаев без пего нельзя обойтись. Добавление каталитических количеств солеи тяжелы?
металлов, а также других перекисей сокращает индукционный период и тем самыь
ускоряет реакцию аутоокислепия. Однако следы тяжелых металлов вызывают такж<
усиленное разложение образующихся гидроперекисей, что затрудняет их выделение,
*1аЩс всего целесообразно прекращать реакцию окисления при содержании в реакци
опной массе 5—10% перекиси.
Аутоокисление углеводородов [212, 213]. Насыщенные
углеводороды довольно трудно реагируют с кислородом. Для их аутоокисления тре-
буется температура порядка 100—ИО5 С и почти всегда применение катализаторов,
При статическом воздействии кислорода на неразветвленныс парафины образуется
смесь возможных гидроперекисей, так как по реакционной способности по отношение
к кислороду различные СН-группы мало отличаются. Предпочтительно окисляется
водород" у третичного атома углерода. Так, по Криге [214], из декалина образуется
9-гидроперекись дскалина-
ООН
Добавление НВг (индуцированное бромистым водородом аутоокисление) может
значительно ускорить газофазное аутоокисление изопарафинов, например изобутан®
[215]. При помощи этого видоизмененного способа аутоокислсния, при котором
прибавленный НВг служит источником свободных радикалов, удалось осуществить
промышленное производство гидроперекиси и перекиси трет-бутила.
Для получения индивидуальных гидроперекисей парафинов более целесооб-
разно применять не прямое аутоокисление, а иные методы; аутоокисление соответству-
ющих соединений Гриньяра [216] или пергидролиз спиртов [217], диалкилсульфатоь
[218] или метансульфонатов [219].
Если в а-положении к ароматической системе имеется СН2- или СН-группа»
то кислород взаимодействует с пен предпочтительно с образованием «замещенной
бепзилгидроперекиси» [212], как, например, в тетралние, гидриндене, флуорене,
кумоле, n-ксилоле и этилбензоле. Температура аутоокисления этих соединений нижа
температуры аутоокисленип парафинов.
[212] Н. Hock, Н. Kropf, Angew. Chem., 69, 313 (1957).
[213] W. Langenbeck, W. Pritzkow, Chem. Techn., 2, 116 (1950); 4, 39t
(1952); Fette u. Seifen, Abstrichmittel, 55, 435, 506 (1953); W. Pritzkow,
K. A. Muller, Ann., 597, 167 (1955).
[214] R. Criegee, Ber., 77, 22 (1944).
[215] E. R. Bell, F. fl. Dickey et al., Ind. Eng. Chem., 41, 2597 (1949);
В. В a r n e t t, E. R. Bell et a]., Ind. Eng. Chem., 41, 2612 (1949); пат. С1ЙА.
2395523, пат. США 2403771.
[216] C. W а 11 j n g, S. A. Buckler, J. Am. Chem. Soc., 77, 6032 (1955).
[217] N. A. Milas, A, Harris, J. Am. Chem. Soc., 60, 2434 (1938); N. A. M i-
las, D. Surgenor, J. Am. Chem. Soc., 68, 205, 643 (1946); N. A. Milas».
L. H. Perry, J. Am. Chem. Soc., 68, 1038 (1946); R. Criegee, H. Diet-
rich, Ann., “560, 135 (1948).
[218] A. v. Bayer, V. V i 11 i g e r, Ber., 27, 1510 (1894); 34, 738 (1904); A. R i-
cche, F. Hitz, Ber., 62, 2458 (1929); E. J. Harris, Proc. Roy. Soc.»
Ser. A., 173, 132 (1939); W. Egger sgliiss, Monographie zur Angew. Che-
mie und Chemie-Ing.-Techn., As 61, Verlag Chemie. 1951.
[219] H. R. Williams, II. S. Mosher, f. Am. Chem. Ssc., 76, 2984, 2987»
3495 (1954).
19 Заказ 1835.
290 ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
Для выделения образующихся гидроперекисей можно пользоваться отгонкой .1
в вакууме (иногда в глубоком вакууме) растворителя и избытка исходного продукта. 1
Остаток от перегонки представляет собой гидроперекись в наиболее обогащенной '3
форме. Кристаллизация гидроперекиси происходит немедленно пли после длительного .3
стояния в холодильнике. Гидроперекиси с низкими температурами кипения чаще
всего выделяются в чистом виде уже при перегонке в глубоком вакууме. Гидропере- 1
кисп, обладающие кислотными свойствами, экстрагируют холодным раствором NaOH. Л
из реакционной массы, разбавленной смесью эфира с петролейным эфиром. Щелочную :
вытяжку осторожно нейтрализуют и затем гидроперекись извлекают эфиром.
Получение гидроперекиси тетралина аутоокнслением тетралина [220]. •’
Н ООН
В 1л технического тетралина при температуре около 75а С в течение *
50—60 ч пропускают энергичный ток воздуха при хорошем ого распылении;
через некоторое время жидкость желтеет. Потери вещества снижаются nptc\-
использованни эффективного обратного холодильника. После указанного'Вре*^
мени на водяной бане (75° С) при разрежении 1 — 2 мм рт. ст. отгоняют нв1’’^
вступивший в реакцию тетралип, что составляет примерно 4/5 общего объема^
смеси. Остаток частично закристаллизовывается при длительном стоянии, бы-м
стрее при охлаждении до 0° С. Перекись отсасывают и осторожно сушат. Выхода
неочищенного продукта составляет^—17% от массы окисленной смеси; т. пяЛ
около 52° С. Для очистки препарат несколько раз перекристаллизовываю^
из большого количества петролейного эфира. Получается бесцветная и почти
лишенная запаха гидроперекись в виде игл; т. пл. 56° С.
Аутоокисление олефинов. Олефины реагируют с кислородом^
в «-положении к двойной связи. При этом могут образовываться не только гидрой
перекиси с неизмененным положением двойной связи, но и изомерные гидроперекиси
с перемещенной двойной связью. Как было установлено [221], такие продукты подурю
чаются в результате мезомерии образующихся сначала сцР-иенасыщеиных радикалов^
{«-метиленовый механизм): $
I -I I . '<
—СНа-С=С< —► —СН—С=С< <----------> — СП=С-С<
|°-
I I
-СН-С=С< ~СП=-С-С<
I I 1
ООН ООН •.
Этот механизм объясняет получение двух гидроперекисей при аутооки с ленив.:;
1,2-диметилциклогексева [221] и четырех гидроперекисей при аутоокислении метилО-'j
вого эфира олеиновой кислоты [222]. Д
Циклические олефины, например циклогексен [223], взаимодействуют с кнсло-::/
родом легче, чем алифатические олефины.
[220] II. Hock, W. S use mi hl, Вег., 66, 61 (1933).
[221] E. H. F a г m e г, D. A. Sutton, J. Chem. Soc., 1946, 10; E. H. Farm»
H. P. Koch, D. A. Sutton, J. Chem. Soc., 1943, 541; E. H. Farme
Trans. Farad. Soc., 38, 340 (1942); 38. 348 (1942); 42, 228 (1946).
[222] J. R о [$, A. J. Gebhart, J. F. Gcrecht, J. Am. Chem. Soc., 71, 282h?
(1949). £
[223] R. Cr i e g с e, H. P i 1 z, H. F 1 у g a r e, Ber., 72, 1799 (1939). / *
I. ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
291
«Фотосенсибилизированное аутоокисление» соединений с изолированными
двойными связями подробно изучалось Шенком с сотр. [224]. Так называемая реакция
«косвенного заместительного присоединения в аллильное положение» приводит также
к шдроперекпсям, которые только в особых случаях идентичны гидроперекисям,
образующимся при несенсибилизированном аутоокислении.
В этой реакции молекула кислорода присоединяется к первому атому углерода
двойной связи, после чего один атом водорода у Cs (аллильное положение) переходит
к кислороду с одновременным перемещением двойной связи в положение С2—С8:
О?; сенсибилизация 3
। 1 Г4 zi । ।
Н Н II ноо н н
Подробное описание реакции см. в обзорной статье Шенка [225].
Аутоокисление простых эфиров имеет особенно важное значение, так как обра-
зующиеся при этом перекиси часто являются причиной сильных взрывов. Подробнее
об этом см. в работе Рихе [226].
Как установили Рихе и Мейстер [227], гидроперекись диэтилового эфира
превращается только при нагревании в сильно взрывающую полимерную перекись
этллидена:
л, нагревание |
С->116-О-СН2—СНз — + С2Нб—О—СН—СНз-----------------* [-СН-ОО-
I
ООН
Аналогично полимеризуется также гидроперекись диизопропилового эфира [228].
Эфиры, содержащие перекиси, особенно диэтиловый эфир, тетрагидрофуран,
диизопропиловый эфир и диоксан, представляют большую опасность в лабораторной
практике. При выпаривании растворителей могут происходить сильные взрывы.
Перекисные соединения часто содержатся не только в упомянутых эфирах, но и в дру-
гих растворителях. Таковы, например, бензин, петролейный эфир, декалин, ксилол,
кумол и тетралин.
За небольшим исключением присутствие перекисных соединений можно уста-
новить нагреванием с подкисленным раствором K.I (подкисление уксусной нли в иеио-
торых случаях концентрированной соляной кислотой).
Гидроперекиси удается удалить [229] из растворителей встряхиванием с твер-
дым КОН и последующей перегонкой над КОН (содержащий много перекиси тетра-
гвдрофуран может при этом бурно реагировать!). Малые количества перекиси удаляют
погружением натриевой проволоки в растворитель.
Под действием таних восстановителей, как бисульфит, медноцинковый сплав
[230| пли алюмогидрид лития, гидроперекиси превращаются в спирты. Перманганат
калия [231] или двуокись свинца [232] равным образом разрушают перекиси. Длитель- .
ное действие раствора FeSO4 в 50%-ной H2SO4 разрушает также я полимерные пере-
киси альдегидов и кетонов. Петролейный эфир и бензин удобно освобождать от пере-
кисей фильтрованием через колонку, заполненную А12О3.
G. О. Schenck, Fiat-Berichte, Naturforschung und Medizin in Deutschland,
1939—1946; Prap. organ. Chem., 1948, S. 167ff; G.' 0. Schenck, H. E g-
gert, W. De nk, Ann., 584, 177 (1953).
G. (). Schenk, Angew. Chem., 69, 579 (1957).
A. Rieche Angew. Chem., 70, 251 (1958).
A. Rieche, R.Meister, Ber., 64, 2335 (1931).
A. Rieche, K. Koch, Ber., 75, Ю16 (1942).
W. Lepper, Chem. Ztg., 66, 314 (1942).
H. Fierz-D a vid, Cmmia, 1, 246 (1948).
A. H a r m s e, Pharmac. weekbl., 78, 1085 (1941); C., 1942, I, 382.
F. R. Fislicr, R. A. Baxter, Mines Mag., 1940. 447; C., 1941, I, 445.
19*
292
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
Широко применяемый в качестве растворителя тетрагидрофурап образует
при аутоокислении гидроперекись тстрагидрофурана [233]:
V \оои
31.0 была первая гидроперекись, которую удалось выделить из окисленных
эфиров. В последнее время выделены кристаллические гидроперекиси простых эфнрод,
полученные аутоокислением циклических бензиловых эфиров, например окислением
пзохромапа [234] и фта'Ьана [235].
Аутоокиеление 1,1-диметнлфталана до гидроперекиси 1,1-дпметилфта-
лана [235].
Н8С СНз
Н3С СНз
;о —L-
:о
сп2
сн
ООН
При комнатной температуре и УФ-облучонии 100 г 1,1-диметилфталана '
встряхивают с кислородом. При содержании 39% перекиси реакцию аутоокис- '•
Ленин обрывают. Затем массу растворяют в равном объеме эфира и прибавляют ’
к этому раствору равное количество петролейного эфира. Раствор встряхивают ,,
сначала с 25 мл 5%-ного раствора NaIICO3 и затем с 10 мл воды. После эт<но '
экстрагируют гидроперекись сначала 30 мл и затем три раза по 25 мл охлажден- ';
него льдом 2 н. раствора NaOH. Объединяют слегка окрашенные в желтый цвет/
щелочные вытяжки и для удаления растворимых в эфире веществ нстряхиваЪг.-
с 10 мл холодного эфира. В сильно охлажденный щелочной раствор при вепре-*
рывном перемешивании по каплям приливают 2 н. раствор H2SO4 до образовав
пия устойчивой эмульсии (pH ~ 9), при этом гидроперекись 1,1-диметилфталана ’
выделяется в виде масла. Затем перекись исчерпывающе экстрагируют эфиром,^,
эфирный экстракт поочередно встряхивают с 2 н, раствором NaHCO3 и водой^
и сушат в течение ночи над Na2SO4. После отгонки эфира остается вязкая масса,«
которая при растирании с холодным петролейным эфиром затвердевает в кри-j
стал.чическую кашицу. Ее перекристаллизовывают из смеси эфира с петролей-^
ным эфиром; т. пл. 63—64° С. *
Ацетали также очень склоппы к аутоокислепию. Из кристаллических гидро-)"
перекисей ацеталей удалось выделить гликольацетали бензальдегида и eto произвол- ^
ных [236]. <
Аутоокисление кетонов протекает с промежуточным образованием большей ;
частью нестойких гидроперекисей кетонов и приводит к альдегидам и кислотам [237]: .
[237]: Я
R—СО—CH2R —г-+ R—СО—СНООН—R
RCHO-I-RCOOH
•Ота реакция имеет большое значение при окислении парафинов.
Аутоокисление альдегидов приводит к образованию надкислот [238]:
RCHO+O2 —> RCOOOH
1233]
I234J
1235]
1236]
1237]
1238]
R. Criegee, R. Tanne n herger, Dissertation Taimonherger, Karlsruhe,
1946; A. Robertson, Nature (London), 162, 153 (1948).
A. R i e c h e, E. Schmitz, Chem. Ber., 90, 1082 (1957).
A. Rieche, M. Schulz, Ann., 653, 32 (1962).
A. Rieche, E. Schmitz, E. Beyer, Chem. Ber., 91, 1935 (1958).
A, Rieche, Wiss. Ann. dtsch. Akad. Wiss. (Berlin), 2, 721 (1952).
G. Wittig, Ann., 558, 201 (1947).
I. ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
293
Например, аутоокислением бензальдегида можно получить надбензойную
кислоту [239]; в присутствии ацетангидрида получается перекись ацетилбензоила
[240].
Образование диалкилперекисей
В некоторых случаях диалкилперекиси ROok получаются в качестве главных
пли побочных продуктов яри аутоокислении органических соединений. Они почти
всегда образуются из алкилгидроперекисей в результате вторичных реакций. Непосред-
ственное соединение относительно долгоживущих радикалов с кислородом
R.-|-O3 -► ROO.
ROO«+R. —> ROOR
имеет второстепенное значение и происходит только в особых случаях, например
при получении перекиси трифенилметила из радикала трифонилметила [241], при
индуцированном бромом аутоокислении аллилбромида до перекиси а.а'-дибромизо-
пропила [242] и при газофазном аутоокислении изопарафинов [215] в присутствии
бромистого водорода.
В болыпинстве случаев диалкилперекиси образуются из первично получаемых
алкилгидроиерекисей, как, например, При аутоокислении изохромана [234], фталана
1235], гликольацеталя [243] или тетрагидроацепафтена [244]. Образование диалкил-
перекисей из алкилгидроиерекисей может происходить под влиянием минеральных
кислот. При этой «ионной димеризации» [234, 235] одновременно получается перекись
водорода:
Н+
2ROOH ——- ROOR+H2O4
В качестве примера ионной димеризации приводится получение перекиси 1,1-ди-
метилфталана из алкилгидроперекиси 1,1-диметилфталана.
В качестве примера реакции ионной димеризации гидроперекиси ниже описан
синтез перекиси 1,1-диметилфталана из соответствующей гидроперекиси [235]:
Н3С СНз Н3С СПз НзС СН3
Перекись 1,1-диметилфталапа. Растворяют 500 мг гидроперекиси 1,1-ди-
метилфталана в 10 мл холодного 2 н. раствора NaOH и прибавляют к раствору
5 мл метилового спирта, а затем 20 мл 2 н. H2SO4; при этом выделяется масло,
кристаллизующееся в холодильнике. Выход 390 мг (86% от теоретического).;
После перекристаллизации перекиси из смеси бензола с петролейным эфиром
т. пл. перекиси 114—Иб0 С.
Вторая возможность образования диалкилперекисей заключается в радикаль-
ной реакции под действием тяжелых металлов в качестве катализаторов [245, 246].
[239] W. Р. Jorissen, Р. A. A. van der Beck, Rec. trav. chim., 45, 245
(1926); P. A. A. van der Beck Rec. trav. chim., 47, 286 (1928).
[240] J. U. N e f, Ann., 298, 280 (1897)- см. A. Baeyer, V. Villiger, Ber.,
r 33, 1569 (1900).
[241] K. Ziegler, Angew. Chem., 61, 168 (1949). ,
[242] w. В ockc miiller, L. P f e u f f e r, Ann., 537, 178 (1939).
[243] R, Criegee, M. Lederer, Diplomarbeit Lederer Karlsruhe, 1950.
[244] W. Treibs, J. ThSrmer, Chem. Ber., 90, 94 (1957). - .
[245| VV. Treibs, G. Pellmann, Chem. Ber,, 87, 1201 (1954). '
[246] M. s. К harash, A. F о n o, J. Org. Chem., 23, 324 (1958).
291 ОБРАЗОВАНИЕ С — 0-СВЯЗП В ОБМЕННЫХ РЕАКЦИЯХ
R качестве примера реакции, протекающей по радикальному механизму, при-
водится получение перекиси циклопентенилкумила из гидроперекиси кумола и цикло-
нентен.с
V
(СН3)2С—ООН
Перекись циклопентенилкумила (245). В трехгорлой колбе емкостью^
250 .w.i, снабженной газоподводящей трубкой, к смеси 20 г гидроперекиси кумол#;
п 70 г циклопентепа в токе азота приливают по каплям 2 мл 18%-ного бепзоль-'
ного раствора стеарата кобальта (II), что соответствует 30 мг кобальта, при этом»
температура массы повышается до 40° С. После 5 ч термостатирования в атмо--
сфере азота при 45° С остаток перегоняют в токе азота при 2 мм рт. ст. Полу-Й
чают две основных фракции: диметилфенилкарбинол (т. кип. 63—68° С) й uepft^
кись циклопентенилкумила (т. кип. 112—113° С; 1,5196; rf2® 1.0195).
3. Замещение водорода на карбонильный кислород )
Получение альдегидов и кетонов }
Для олислеиня метильных и метиленовых групп в карбонильные лучше ие-1
пользовать мягко действующие окислители — соединения марганца (IV), хромалхло^
рид, нитро- или нитрозосоединения и SeOg. Лишь в отдельных случаях, например!
при получении бензофенона из дифенилметана, ввиду исключительной устойчивости;
продукта окисления применимы энергичные окислители. Тетраацетат свинца пригоде#
только при образовании альдегидов, устойчивых к дальнейшему окислению.
а-Мегиленовые группы кетонов особенно легко окисляются с промежуточны»^
образованием соответствующих изонитрозокетонов по схеме: Л)
RCO—CH2R —5------ RCO—CR —> RCO—COR
NOH -
Так, взаимодействием метилэтилкетона с изоамилнитритом и омыление*^
образовавшегося монооксима дикетона (без его выделения) серной кислотой получаю^
диацетил [247].
При переводе малонового эфира в эфир мезоксалевой кислоты с помощь^
нитрозных газов также нет необходимости выделять промежуточно образующим^
ПрОДуКТЫ [248].
Общее применение имеет метод Закса, по которому активированные метили*
новые группы конденсируют с «-нитрозодиметиланилином и полученные шиффодй!
основания расщепляют разбавленными минеральными кислотами: • ’!
Этот метод был использован для превращения 4-замещекного тетрафеннлциклй*’
нентадиена в соответствующий тетрафенилциклопентадиенон [249].
[247] О. Diels, Е. Stephan, Вег., 40, 4337 (1907).
[248] A. W. Dox, Org. Synth,, 4, 27 (1925).
[249] L. Mohr et ak, J. Am. Chem. Soc., 77, 989 (1955).
!
I. ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
295
Тетрафепилциклопентадиенон. Диеп растворяют в смеси бензола с мети-
ловым спиртом (3 : 1 или 3:4), смешивают примерно с трехкратным молярным
количеством п-нитрозодиметиланилина и нагревают до кипения. Затем приба-
вляют несколько капель 10% -ного раствора метилата натрия в метиловом спирте
и выдерживают 5—10 мин при кипении. Полученный раствор смешивают с кон-
пептрированной НС1, кипятят 5—10 мин и экстрагируют тетрафенилциклопен-
тадиеяон бензолом.
Шиффово основание можно также получать в спиртовом растворе в при-
сутствии каталитических количеств соды или пиперидина. В большинстве
случаев темноокрашенные шиффовы основания выпадают в осадок, и их гидро-
лизуют кислотой обычным путем.
Реакционноспособные метильные группы также взаимодействуют с п-нитрозоди-
метиланилином, причем в конечном итоге получаются альдегиды [250]. Так, из 1,3-ди-
метил-4,6-динитробензола получают 4,6-динитронзофталевый альдегид. Для подобных
реакций оказался очень пригодным этилиитрит.
Превосходным окислителем реакционноспособных метильных и метиленовых
групп является предложенная Райли двуокись селена. Особенно легко реагируют
с этим окислителем метилкетоны и их шиффовы основания. Легко реагируют также
и метиленовые группы, соседние с карбонильной группой. Поэтому закие вещества,
как глиоксаль и фенилглиоксаль в настоящее время легко готовят окислением соответ-
ствующих соединении при помощи SeO2. Упомянутый выше эфир мезоксалевой кис-
лоты может быть получен по этому способу с хорошим выходом. Существенным для
процесса окисления является состояние окиси селена *; в качестве растворителей
для проведения реакций рекомендуются диоксан, ледяная уксусная кислота, этиловый
и бутиловый спирты, толуол и вода. Например, из циклогептавона соответствующий
а-дикетон получается с 90%-ным выходом £251]. Фенилглиоксаль может быть синте-
шропан как из фенилацетальдегида, так и из ацетофенона.
Фенилглиоксаль из ацетофенона (252]. В смесь 500 -ил дпоксапа a 20 мл
воды при 60° С и перемешивании вносят 112 г SeO2 (1 л«мь). После полного ее
растворения прибавляют в один прием 120 г (1 -ноль) ацетофенона и нагревают
около 3 ч в колбе с обратным холодильником при энергичном перемешивании.
После охлаждения отфильтровывают выпавший селен и осторожно отгоняют
на водяной бане диоксан, лучше при пониженном давлении. Дальнейшую очи-
стку фенилглиоксаля, который очень легко полимеризуется, производят фрак-
ционной перегонкой (т. кип. 96° С при 22 мм рт. ст.) или перекристаллизацией
его гидрата из Этилового эфира; выход 70% от теоретического. Гидрат получают
также перекристаллизацией из небольшого количества воды.
На примере синтеза 2-нафтальдегида из 2-метилнафталина [253] очевидно,
{ что метильные группы ароматических соединений тоже реагируют с селенистой кисло-
той **. В толуидинах, аминогруппы которых защищены фталоильными, и в крезолах,
гидроксильные группы которых защищены бешолсульфонпльпыми группами, метиль-
ные группы при окислении SeO2 в зависимости от соотношения реагентов превращаются
S альдегидные или карбоксильные [254]. Таким образом Баджер при помощи SeO2
у в водной среде и температуре 230—240® С перевел флуорен в флуоренон [255].
Рекомендуется применять свсжевозогнанный продукт. — Прим. ред.
Эта реакция осуществлена также и в гетероциклическом ряду; см., на-
пример, В. М/ Родионов, М. А. Беркенгейм, ЖОХ, 14, 330
(1944). — Прим. ред.
Р. Ruggli, Р. Hindermann, Helv. chim. acta, 20, 279 (1937).
R. W. Harr et al., J. Org, Chem., 14, 836 (1949).
Houben -Weyl, Methoden der organischen Chemie, Bd. 7/1, 4 Aufl., S. 149;
ср. H. L. Riley et al., J. Chem. Soc., 1932, 1875.
В. M. Родионов, Синтезы органических соединений, Сборник 2, Изд.
АН СССР, 1952.
G. Zemple n, L. Kisfaludy, Chem. Вег., 93, 1125 (1960).
G. М. Badger,!. Chem. Soc., 1941, 535.
296
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
Обработка эфира малеиновой кислоты селенистой кислотой приводит к образование
эфира дикетоянтарной кислоты [256]:
О О
II II >
НС—С—OR О=-С—С—OR 1
II I 3
НС—С—OR О—С—С—OR J
II II ’ j
О о
Обзор работ по окислению двуокисью селена см. [257]. При получения SeOs нз!
селена следует соблюдать необходимые при работе с взрывоопасными веществами"1
предосторожности. 7
Вейганд с сотр. [258] показал, что при окислении КМпО4 можно легко подцер>*
живать нейтральную реакционную среду добавленном некоторых солей, оказывающих
буферное действие, например солей магния. Образующуюся по реакции щелочь можнси
связывать также добавкой ангидрида уксусной кислоты. В нейтральной среде устой-5
чивы также такие неводные растворители, как, например, ацетон, что позволяет про-,
водить окисление нерастворимых в воде соединений в гомогенной системе. .<
Ниже приведены примеры окисления КМпО4 в нейтральной среде п-этилаце-
тофснона и тетралина.
л-Дпацетилбензо.т. Раствор 30 г Mg(NО3)2 • 6Н3О в 250 мл воды приливаю^
к раствору 15 г этилацетофенона. Затем смесь нагревают до 50° С и при интенсив-
ном перемешивании маленькими порциями вносят 16 з порошкообразного*
КМпО4; при этом температура смеси возрастает до 70° С. Окисление оканчиваете^
через 2 ч. Реакционной массе дают остыть, отфильтровывают образовавшуюся
двуокись марганца, промывают осадок эфиром и извлекают продукт реакции
из водного фильтрата эфиром. Объединенные эфирные вытяжки сушат над СаС12»!
фильтруют и отгоняют эфир; при этом в осадок выпадает п-диацетилбензол^
выход 13,8 з (84% от теоретического); т. пл. 114° С.
а-Тетралон. В трехгорлой колбе с мешалкой и обратным холодильню
ком растворяют 38 з тетралина в 500 мл ацетона. Раствор нагревают до кипени^
и в него при перемешивании постепенно вносят через третье горло порошко
образную, хорошо перемешанную смесь 66 г КМпО4 и 49 г Ca(NOs)2 • 4Н2СК
Весь перманганат расходуется за 10 ч, после чего массу фильтруют и при
мальном давлении отгоняют ацетоп. Остающееся масло фракционируют в ваку-
уме. После отгонки при 95—99° С (19 мм рт. ст.) 20.5 г непрореагировавшеН
тетралина при 134—137° С (19 мм рт. ст.) отгоняется 14.5 з а-тетралона, окра-
шенного в слабо-желтый цвет, который можно очистить через семикарбазой
Выход 65% от теоретического в расчете на прореагировавший тетралин. >
Способ окисления циклоолефинов и циклоалифатических соединепий в органи-
ческих растворителях в кетоны был разработан Трейбсом с сотр. [259]. Окислителе®
в этом методе служит перекись водорода, катализатором — надванадиевая инслота..
При окислении циклоолефинов кислород взаимодействует с атомом углерода, нахо?
дящимся по соседству с двойной связью (при окислении тетралина реагирует атоЯ
углерода, находящийся в a-положении к ароматическому ядру). В качестве раствор®
теля во всех случаях применяют ацетон. Конец реакции определяют но переход]
окраски из желто-красной в красно-коричневую илк зеленую. Этим способом, например^
с хорошими выходами синтезированы циклогексенон из циклогексена и а-тетрало|1
из тетралипа.
.Метильные группы, связанные с ароматическим ядром, можно превращать
и альдегидные группы с помощью смеси пиролюзита (МпО2-Н2О) и серной кислоты
умеренной концентрации [260]. Этот способ особенно рекомендуется в тех случаях»
[256] S. A s h i n, L. de V. М о u 1 d s, Н. L. R 11 е у, J. Chem. Soc., 1935, 902;
[257] G. Stein, Neuere Mcthodon der praparativon organischen Chemie, Bd.
3 Aufl., Verlag Chemie, 1949, S. 1; N. Rabj ohn, Org. Reactions, 5 , 331 (1949)j;
G. R. Waitkins, C. W. Clark, Chem. Rev., 36, 235 (1945). 1
[258] W. Winkler, Chem. Ber., 81, 256 (1948). *
[259] W. Treibs et a]., Angew. Chem., 52, 698 (1939); Chem. Ber., 86, 616 (1953).
[260] A. Boner, Chem. Ztg., 24, 446 (1900) (Korrespondenz). *
I ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
297
когда окисляемое вещество растворимо в 90—95%-ной H2SO4. Если возможность
сульфирования исключается, можно применять также олеум или смесь серной и ук-
сусной кислот. Таким путем можно перевести в альдегиды почти все производные то-
луола, например из толуол-2-сульфокислоты и толуол-2,4-дисульфокислоты этим
способом получаются соответствующие альдегиды с выходом свыше 80% от теорети-
ческого [261].
Вместо МпО2 окислителем производных толуола может служить смесь СгО3
и уксусного ангидрида иди серной кислоты. Образующиеся при этом альдегиды вы-
деляют в йвде устойчивых к окислению диацплатов альдегидов.
Тот факт, что о-нитротолуол в кислой среде Не взаимодействует с МпО2, но
окисляется СгО3 в о-нитробензальдегид, показывает, что этот способ является более
общим, чем приведенный выше метод окисления МпО2. В качестве примера окисления
при помощи СгО3 приведено превращение о-нитротолуола в о-нитробензальдегид [262].
Диацетат о-нитробензальдегида. Растворяют 50 г о-нитротолуола
в смеси 570 мл ледяпой уксусной кислоты и 565 мл уксусного ангидрида и осто-
рожно приливают 85 мл концентрированной HaSO4. К реакционной массе при
перемешивании и температуре 5 — 10° С в течение 2 ч маленькими порциями
прибавляют 100 г СгО3 и перемешивают еще 3 ч. Затем смесь выливают на мелко-
раздробленный лед, разбавляют водой до 6 л и перемешивают до тех пор, пока
не затвердеет выделившееся масло. Твердый продукт отсасывают, промывают
водой и еще раз 2%-ным раствором NafCOs при перемешивании. После сушки
продукт обрабатывают 150 мл петроленного эфира (60—70Q С) при кипении
и после охлаждении отсасывают. Выход 21—22 г (23—24% от теоретического);
т. пл. 87—88° С. Экстрагируя кислый маточник хлороформом, выделяют еще
часть продукта. Хлороформный раствор промывают раствором NaHCO3 и сушат
над Na2SO4. После отпаривания растворителя получают коричневый сироп,
который кристаллизуется после обработки гептаном и охлаждения до 0° С.
о-Нитробензальдегид. Смесь 51,6 г дяацетата о-нитробензальдегида,
272 мл концентрированной ПС), 450 мл воды и 80 мл спирта нагревают 1 ч
в колбе с обратным холодильником при перемешивании. Если после охлаждения
до 0° С альдегид выделяется в виде масла, добавляют примерно равный объем
воды и оставляют на длительное время на холоду. Неочищенный продукт отса-
сывают, промывают водой и очищают быстрой перегонкой с водяным паром.
Выход 23,7 г (74% от теоретического); т. пл. 44—45° С.
Для получения бромированных альдегидов из соответствующих замещенных
толуолов [263] лучше применять СгО3, чем SeOa или МпО2.
Окисление углеводородов хромовой кислотой может быть использовано также
для синтеза кетонов. Так, при действии на аценафтен Na2Cr2O7 в кипящей ледяной
уксусной кислоте получают аценафтенхинон[264]. Этим же способом, применив смесь
ледяной уксусной кислоты с бензолом после 40—48 ч окисления при 0° С, Физер
[265] осуществил превращение холестерина в М-холестендион-3,6 с 39—40%-ным
выходом от теоретического. Механизм окисления соединениями хрома и марганца
изучен Уотерсом [266]; кинетика и механизм окисления КМпО4 описаны Ледбери
и Каллисом [267].
Метод превращения метильных групп в альдегидные с помощью хромнлхло-
рида по Этарду может представлять ценность для перевода ксилолов, мезитиленов
и других соединений с несколькими Метильными группами в моноальдегиды. Так,
из соответствующих ксилолов с удовлетворительными выходами были получены три
изомерных толуиловых альдегида [268].
1261] Frill., 7, Ю8 (1902-1S04). '
[262] S. М. Tsang ct al., Org. Synth., 24, 75 (1944); A. F. Waltonet al.,
J. Am. Chem. Soc,, 67, 11501 (1945).
[263] Б. M. Б о г о с л о в с к и й и др., ЖОХ, 24, Ю43 (1954); ср. Angew. Ghem.,
67, 237 (1955).
[264] С. S. Maxwell, С. F. IT. Allen. Org. Synth 24, 1 (1944),
[265] L. F. Ficser, Org. Synth., 35, 36 (1955).
266] w. A. Waters, Quart. Rev., 12, 277 (1958).
[267] J. W. La dhury, C. F. C u 11 i s, Chem. Rev., 58, 403 (1958).
[268] H. D. L a w, F. M. P e г k i n. i. Chem. Soc., 1907, 258.
298
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ PW АКЦИЯХ
Ряд ароматических соединений с алкильной группой окисляются в жидкой*
фазе до кечонов воздухом в присутствии СгО8: i
АгСЩСНз АтСОСНз 1
Пропуская воздух в течение 40 ч при 130° С в суспензию 1% СгОа и 4% СаСО,
в л-диэтилбензоле, получают м-этилацетофенон с выходом 50% от теоретического
[269]. Таким же образом из эфиров ароматических карбоновых кислот, ацетофеноног
и галогенбснзолов, содержащих алкильные группы, синтезируют соответствующем
котоэфиры, дикетоны или галогенкетоны [270]. j
Получение хинонов *
Во многих случаях осуществимо прямое превращение ароматических углеводе^
родов в хиноны. По одному из патентных описаний [271], n-бепзохинон синтезирую
с 50—55%-ным выходом от теоретического нагреванием бензола в течение 5 мин пта
56е С с влажной двуокисью свинца в 100%-ной серной кислоте. Для выделении хиною
массу выливают на лед и продукт экстрагируют эфиром. Электролитическим
нием бензола можно синтезировать хипон с выходом 65% от теоретического [272],
Гребе и Либерманн получили антрахинон окислением антрацена смесью' хр©
мовой и уксусной кислот [273]. Лучшие результаты достигаются [274] при применена!
в качестве катализаторов КаСЮ, и V2O5. Однако антрахинон можно получить с хд£
рошим выходом и окислением ЙМпО4.
Антрахинон. По методу Вейганда растворяют при нагревании 2.5 --I
антрацена в 125 мл уксусного ангидрида и при 80—90° С постепенно, осторожЩ
прибавляют 10 г тонкоизмельченного КМпО4. Окисление заканчивается чер«
3—4 ч. Массу охлаждают льдом и фильтруют через пористый стеклянный фильтр
на фильтре остаются антрахинон и ацетат калия, а коллоидный МпО2 проходи
через фильтр. Осадок на фильтре промывают уксусным ангидридом и 'извлекаЮз
ацетат калия водой. В остатке получают 2,2 г антрахинона, что соответствуй
выходу 78% от теоретического.
Антрахинон, иногда с количественным выходом, готовят сплавлением антрй
цена с такими окислителями, как нитраты, перманганаты и хлораты [275]. _>
Фенантрен под действием хромовой смеси дает 9,10-фенантренхинон с выходе!
80% от теоретического [276]. Окислением 2,3-диметилнафталина смесью Сг03 и унсув
ной кислоты легко получают 2,3-диметил-1,4-нафтохинон [277]. О каталитической
окислении нафталина до 1,4-пафтохинона см. статью [278]. §
Одноатомные фенолы и нафтолы превращаются в хипоны при их взаимодед
ствип с нитрозодисульфонатом калия [279]. При незамещенном пара-положении peart
ция с 2 моль нитрозодисульфоната обычно приводит к синтезу n-хинонов; из фенозй
образуется 1,4-бензохинон, а из а-нафтола — 1,4-нафтохинон. 5-Оксицафтохинон-1^
[269] D. Т. Mowry, J. Am. Chem. Soc., 67, 1050 (1945).
[270] W. S. Emersonet al., J. Дт. Chem. Soc., 68, 674, 1666 (1946); 69 190?
(1947); 70, 1180 (1948). j:
[271] Герм. пат. 533128; С., 1931, Ц, 3042.
[272] A. Seyewetz, G. Miodon, Bull. Soc. chim. France [4], 33, 449 (1923}X;
[273] C. G r a e b e, C. Liebermann,. Ann. der Chemie und Pharmazie, 7, 28$
(1870).
[274] H. W. U n d e rw о о d jr., W. L. W a 1 s h, Org. Synth., Coll. v. 2, 1943Z
p. 554. 3
[275] Герм. пат. 568784, Erf. J. S i о 1 i s c h; C., 1933, II, 280. <
[276] R. P. L i n s t e a d, P. Levine, J. Am. Chem. Soc., 64, 2023 (1942). M
[277] L. I. S m i t h, I. M. Webste r, J. Am. Chem. Soc., 59, 664
[278] H. E. Fierz-D avid, L. В 1 a n g e y, W. v. К r a n n
Helv. chim. acta, 30, 247 (1947).
[279] H.-J. Ten ber et al., Chem. Ber., 85 , 95 (1952); 86, 1036 (1953); 87, 1236.1
1251 (1954); 88, 802 (1955).
I. ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
299
может быть получен как пз 1,5-, так и из 1,8-диоксинафталина. Фенолы с замещенным
пара- и свободным орто-положением окисляются до o-хинонов. Так, пз 3,4-диметил-
фенола получается 4,5-диметилбензохинон-1,2, из п-крезола—4-метилбензохинон-1,2.
Выходы хинонов лежат в пределах 70—80%; реакция протекает в исключительно бла-
гоприятных условиях: температурный интервал 5—25“ С, растворитель — вода;
продолжительность реакции колеблется в отдельных случаях от 10 лшя До 3—4 ч.
Нптрозодисульфонат калия легко синтезировать. В сухой виде он устойчив в течение
нескольких месяцев [279].
4. Замещение водорода карбоксильным кислородом
Метильная группа в ароматических или гетероциклических соединениях окис-
ляется до карбоксильной по общей схеме:
ВСНз —> RCOOH
В качестве окислителей применяются главным образом перманганат калия
и хромовая кислота. При наличии в одном ядре нескольких метильных групп по мень-
шей мере одна из них окисляется концентрированной азотной кислотой, а остальные
часто остаются неизмененными.
В общем виде окислительная способность реагентов снижается в последова-
тельности:
KMnOi^-HsCrOa^HNOs разбавл.
Если возможность нитрования исключена, для окисления используют концен-
трированную nNO3; применяя дымящую HNO3, получают из гексаметилбензола
с 35%-ным выходом меллитовую кислоту [280].
Окисление хромовой кислотой ведут в концентрированной уксусной кислоте
пли в сернокислых растворах любой концентрации. Наиболее часто используемая
хромовая смесь состоит из 40 ч. К2СгО4 или КагСгО4. 55 ч. концентрированной H2SO4
и двойного объема воды.
При проведении окисления КМпО4 в нейтральной среде образуются гидроксиль-
ные ионы
2КМп04 + НаО —* 2МпО2 + 2КОН4-ЗО
которые лучше всего нейтрализовать добавлением MgSO4. В кислых растворах
используется также окислительная способность МпО2:
2KMn04 + 3HaS04 —» K2SO4-[-2MnSO*+5O+3HaO
Получение бензойной кислоты из толуола описано в многочисленных патентах
[281]. Легность превращения трех изомерных ксилолов в толуиловые кислоты раз-
лична: о- и п-ксилолы окисляются в толуиловые кислоты уже при кипячении с кон-
центрированной HNOS (rf=i,4), разбавленной в отношении! ; 2—1 : 4; о-толуяловая
кислота [282] образуется при этом с выходом 53—55% от теоретического; ж-толуило-
вая кислота [283] получается при окислении более концентрированной HNO3 (2 : 3).
При наличии различных боковых цепей иногда удается одним окислителем окислить
сначала более длинную цепь, другим — более короткую. Так, из n-цимола при дей-
ствии разбавленной HNO3 образуется толилметилкетон, из которого далее образуется
п-н»луиловая кислота:
(СНз)2СНСвН4СН3
u СИзСОСвЩСНз —► НООССвНаСНз
По реакции с щелочным раствором КМпО4 получается а-оксипропилбензойная
кислота:
(CHj)2CHC„H1CHj KMnQ‘-°H, (CHj)2CH(OH)CeH4COOH
[280] J. Р. Wibau t et al., Rec. trav. chim., 60, 743 (1941); M. C h a i g n e ,a u,
С. r., 233, 657 (1951).
[281] Герм. пат. 360528; FrdL, 14, 439; герм. пат. 377990; FrdL, 14, 441; герм,
пат. 261775; FrdL, 11, 207; герм. пат. 311051; FrdL, 13, 271.
[282] IT. Е. Zang, R. T. R a p a 1 a, Org. Synth., 27, 84 (1947).
[283] A. Reuter, Ber., 17, 2028 (18841.
300
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
Общие закономерности, па основании которых можно было бы предсказать .
течение такого процесса окисления, не установлены.
Окисление изомерных ксилолов до фталевых кислот также протекает различно,
.к- и л-кси.чолы реагируют как с хромовой кислотой, так и с перманганатом калия.
о-Ксплол разрушается хромовой кислотой, в то время как по реакции с перманганатом .
калия в любых условиях тотчас же образуется о-фталевая кислота, а о-толуилован J
кислота в реакционной массе не обнаруживается [284]. При нагревании п-ксилола
с 5—30%-ной IINO3 при 150—200° С под давлением 10—70 ат в результате гладко
протекающей реакции получается терефталевая кислота [285]. Толуиловые кислоты, 3»
полученные окислением ксилолов HNO3, при обработке щелочным раствором КМпО4 -3
также легко превращаются в соответствующие фталевые кислоты [286]. . 1
В производных бензола, содержащих наряду с метильными группами и другие !
заместители, окисление метильных групп происходит иногда легче, иногда труднее, *
чем в незамещенных другими группами метилбенволах. Хлор- и бромзамещенные
толуолы, так же, как ароматические нитроуглеводороды, в общем гладко превра-j
щаются в карбоновые кислоты. Крезолы плохо окисляются указанными окислителями'/
в оксибензойные кислоты. Напротив, эфиры крезолов легко окисляются КМпО4 до';
алкоксибензонных кислот. . <
Некоторые гомологи нафталина исключительно устойчивы к обычным окисли-*
толям. Так, например, Вайсбергеру и Круберу не удалось окислить 1.6-диметилнафтагс
лин ни хромовой кислотой, ни КМпО4. Они получили 1,6-нафталиндикарбоцовуЮ
кислоту с умеренным выходом лишь при 48-часовой обработке 1,6-диметилнафталинв»
феррицианидом калия [287]. ’
1,6-Нафталиндикарбоновая кислота. Смесь 4 г 1,6-ди.метилнафталина j
с раствором 250 г K3Fc(CN)e и 43 г КОП в 1 л воды нагревают при 60° С в
ние 24 ч и перемешивании. Затем добавляют еще 80 г K3Fe(CN)6 в 14 г КОН'
и перемешивают еще 24 ч при 60° С. После этого непрореагировавший диметил-
нафталин отгоняют с водяным паром, отфильтровывают остаток, подкисляют-:
и извлекают продукт эфиром. Ия эфирной вытяжки получают 0,5 г дикарбоновой
кислоты.
Интересно, что одна из метильных групп 1,6-диметилнафталина сравни-*
тельно легко окисляется уже разбавленной азотной кислотой [288].
Так же как гомологи бензола, гомологи пиридина, хинолина, тиофена и других-
гетероциклических соединений окисляются до карбоновых кислот с сохранением*
гетероциклического цикла. В ряду пиррола окисление прямым путем осуществить*
не удается, оно достигается через промежуточные продукты [289]. В ряду хинолина
иногда также бывает целесообразно использовать обходпые пути. Так, Рабе с сотр^
осуществил полный синтез гидрохинина не прямым окислением 4-метил-6-метоксИ-:
хинолина *
СН3
N
а превратил его в продукт конденсации с бензальдегидом, из которого получил карбо-
новую кислоту [290]. '
В пиридипе а- и у-алкильные группы окисляются легче, чем группы, находя-
щиеся в (5-положении. При окислении а- и у-пиколинов КМцО4 легко получаются пв-
[284 A. Claus, Е. Г 1 о ~ L, -
[285 Франц, пат. 961030; С. 1950, Л. 26
[286 W. Welt h, Вег., 7 1058 (1874).
[287 ° -• ° - 1 - - - - - 1 - -
[288
[289
[290
A. Claus, Е. Pieszek, Вег., 19, 3083 (1886).
Франц. пат. 961030; С., 1950, Л. 2614.
В. W е i р g с г Ь е’г, О. К ruber, Вег., 52, 352 (1919).
Герм. пат. 301079; С., 1917, П; 713; Frdl., 13. 210.
Ср. Н. Fisch^T et al., Ann., 461, 244 (1928).
P. Rabe et al., Ber., 64, 2493 (1931).
ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
301
колиновая и изоникотиновая кислоты [291]. Для окисления у-пиколина до изонико-
типовой кислоты вполне пригодна также двуокись марганца, образующаяся
при окислении КМпО4 некоторых органических соединений [292]. Хорошие выходы
достигаются при низких температурах и непродолжительном времени реакции.
Окислитель следует вводить маленькими порциями.
о-Хлорбензойная кислота [293]. В колбе емкостью 12 л с обратным
холодильником при перемешивании медленно нагревают до кипения 200 г
o-хлортолуола с 7 л воды и 600 е КМцО4 (чтобы реакция пе протекала очень
бурно, начинать нагревание надо осторожно). Через 3—4 ч после исчезновения
перманганатной окраски отгоняют с водяным паром через нисходящий холо-
дильник некоторое количество непрореагировавшего толуола (25—30 г), еще
горячую реакционную смесь фильтруют, осадок МпО3 дважды промывают пор-
циями по 500 мл горячей воды и в вакууме упаривают фильтрат до объема 3,5 л.
Затем, в случае необходимости, обесцвечивают фильтрат углем, отделяют уголь
и при непрерывном встряхивании подкисляют 250 мл HCI (d = 1,19). После
охлаждения выпавшую хлорбензойную кислоту отфильтровывают, промывают
водой и сушат; выход кислоты 163—167 г (74—78% от теоретического); т. пл.
137—138° С. Для очистки ее перекристаллизовывают из 600 мл толуола. Выход
после первой перекристаллизации 135—140 г; т. пл. 139—140° С.
п-Нитробензойная кислота [294]. Смешивают 680 г Na2Cr2O7 с 1500 лм
воды и 230 г п-нитроюлуола и приливают к смеси 1700 мл концентрированной
HoS04. При этом «-нитротолуол плавится и вскоре начинается энергичная реак-
ция; поэтому вторую половину серной кислоты необходимо добавлять осторожно
в несколько приемов. Затем смесь нагревают в течение получаса до кипеиня,
охлаждают, разбавляют 2 л воды и фильтруют выделившийся неочищенный
продукт через фильтровальную ткань. Осадок на фильтре промывают 1 л воды,
еще раз нагревают его на водяной бане с 1000 мл 5%-ной H2SO4 и вновь филь-
труют. Осадок растворяют в 5%-ном растворе NaOH, фильтруют в избыток
разбавленной серной кислоты и получают 230—240 г слегка желтоватого неочи-
щенного продукта, что соответствует выходу 82—86% от теоретического. Для
очистки продукт перекристаллизовывают из бензола; т. пл. 238° С.
Окисление боковых цепей галогенсодержащих гомолоюв' бензола азотиой
кислотой протекает с трудом, но облегчается добавлением нитрата серебра для свя-
зывания галогена. Галоген в ядре также нередко затрудняет окисление; он часто
защищает от окисления находящуюся в орто-положенин алкильную группу. Так,
окисление п-хлор-м-ксилола и п-дибром-п-ксилола хромовой кислотой протекает только
до ,хлор-л4-юлуиловой или дибром-п-толуиловой кислоты [295]:
^\-/СНЗ СО //\/С°0Н
I II I II
ci/y а/у
СНз СН3
Дябром-и-толуиловая кислота превращается в дибромтерефталевую кислоту
только под действием щелочного раствора перманганата [296]:
I II
щс/^/Хвг
Вг, /ч ZCOOH
I II
КМпОд
Br\z\/COOH
। I
ноос/^^вг
[291] R. L. Malan.P. М. Dea n, J. Am. Chem. Soc., 69, 1797 (1947); A. W. S i n-
ger, 8. M. McElvain, Org. Synth., Coll. v. 3, 1955, p. 740.
1292J A. M. T у a t j i, Chem. Ber., 92, 2677 (1959).
J293J O. Emmerli ng, Ber., 8, 880 (1875); H. T. Clarke, E. R. Taylor,
Org. Synth., Coll. v. 2, 1943, p. 135.
1294 J О. К a m m, A. O. Matthews, Org, Synth., 2, 53 (1922).
[295] 0. Jacobsen, Ber., 18, 1761 (1885).
I296J B. Schultz, Ber., 18, 1862 (1885).
302
ОБРАЗОВАНИЯ С— О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
Наличие в ядре нитрогрупп тоже затрудняет окисление боковой цепи, так чт<Й
нитробенэойные кислоты удается получать из нитротолуолов только при помощи пер-й
манганата или хромовой смеси. При этом смесь К2СгаО7 с HaSO4 оказывает еще бол&й
энергичное окисляющее действие, чем хромовая кислота в ледяной уксусной кислоте»!
Например, тринитротолуол окисляется хромовой смесью в тринитробензойную кир»Я
лоту, но не окисляется хромовой кислотой в ледяной уксусной кислоте. АналогичийЭ
протекает окисление сульфокислот. Например, о-толуолсульфокислота в виде амид»-
получаемого действием аммиака на ее хлорид, окисляется щелочным раствором КМд<^
при 60° G в сахарин:
СО
/СООП
-СНз
мп
'so2
t
При одновременном наличии в ядре нитро- ц сульфогрупп для окислений
боковых цепей иногда рекомендуется применять персульфат [297). 4
Окисление боковых цепей в присутствии таких легко окисляемых заместителей
как аминогруппа, осуществимо, естественно, только при защите последних. Аминам
группу можно ацетилировать, после чого, например, из n-ацетотолуиднца при д«дН
ствии КМпО4 получается ч-ацетиламинобензойная кислота [298]. АнтраниловщЯ
кислота образуется соответственно из о-ацетотолуидина. При этом реакционную масрЦЯ
нейтрализуют добавлением MgSO4. Я
В промышленности при получении бензойной кислоты из толуола, чтобы сзцаеД
номить окислитель, поступают иначе. Толуол хлорируют при нагревании до бензиздем
бензаль- и бензотрихлорида и затем омыляют в присутствии окислителя или окислязовЯ
толуол смесью перманганата и гипохлорита, что приводит к таким же результатами™
Концевая метильная группа жирно-ароматических метилкетонов относительно гладким
окисляется перманганатом до карбоксильной группы. Хотя эфиры образуюпщхаЙИ
при этом арилглиоксиловых кислот могут быть получены и другим способом, нюяЯ
на одном примере показано применение этого метода. Клаус и Нейкранц подробном
изучили условия реакции [299].
Фенилглиоксиловая кислота [299]. При перемешивании к 12 г ацеимИ
фенона в 200 мл воды в 5—6 приемов прибавляют нагретый до 70° G раствщЯ
32 г КМпО4 и 12 г КОН в 200 мл воды, каждый раз ожидая обеспвечиваймМ
реакционной массы. Полное окисление продолжается около 30 мин. После филыИ
троваяия, подкисления, извлечения эфиром и т. д. получают более 7 г фенйдаИ
глиоксиловой кислоты. Из n-толилметнлкетона выход п-толялглноксиловойМ
кислоты выше 70% от теоретического.
Стоит упомянуть также о получении диметилмалоновой кислоты из триметадчИ
уксусной кислоты [300] нагреванием ее с щелочным раствором КМиО4. ;'^М
В известных случаях окисление боковых цепей ароматических углеводород&им
проводят с помощью окислительного щелочного плавления, нагревая исходное ЩиИ
щсство до 200—300° С с 3—4-кратным количеством NaOH или КОП в присутствие
небольшого количества воды. Такой способ можно рекомендовать для окисления гОмтаМ
логов фенола, так как при этом можно исходить из свободных фенолов. На нримемяВ
превращения аеилл-.и-ксиленола в о-окси-л-толуиловую кислоту видно, что предДййИ
чтителъпо окисляется соседняя с гидроксилом алкильная группа [301];
НзС\^\/СНз H’CW\/COOH 'I
1 Ji —> । j "Я
[297] Герм. пат. 80165; Frdl., 4, 146.
[298[ I. Me isenhei me г et al., Ann,, 423, 86 (1921).
[299] A. Claus, W. Neukran z, J. prakt. Chem., 44, 77 (1891).
[300] P. D. Bartlett, G. L. Fraser, R. B. Woodward, J. Am. Chei
Soc., 63, 498 (1941)..
[301] O. Jacobsen. Ber., 11, 376, 570 (1878).
I. ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
303
5. Дегидрирование спиртов до карбонильных соединений
Получение альдегидов
Вследствие доступности первичных алифатических спиртов метод их де-
гидрирования до соответствующих альдегидов имеет особенно большое препаратив-
ное значение. Наиболее важным способом дегидрирования является высокотемпера-
турное каталитическое отщепление водорода с применением или без применения акцеп-
торов водорода. Наряду с этим для получения карбонильных соединений еще до сих
пор широко используются классические окислители: хромовая кислота, двуокись
марганца я азотиая кислота.
Дегидрирование первичных спиртов при помощи хромовой кислоты проводят
при 0—100° С. Выходы альдегидов составляют 40—60%. Побочными продуктами
являются главным образом карбоновые кислоты и сложные эфиры. Наиболее подхо-
дящей оказалась окислительная смесь, которую приготовляют из 1 моль K3CrgO7,
2.5 моль HsSO4 и 300 г водй (смесь Бекмана), и к этой смеси по каплям приливают
спирт. Для того чтобы защитить альдегид от дальнейшего окисления, через реакцион-
ную массу пропускают азот или СОг и регулируют охлаждение так, чтобы спирт кон-
денсировался. проходя через небольшой обратный холодильник. Выделяющийся
альдегид лучше всего улавливать в хорошо охлаждаемых ловушках. Процесс ведут
с избытком хромовой кислоты; реакция протекает по уравнению:
3RCH2OH + 2CrO3 + 3H2SO4 —> ЗКСНО + Сг2(5О4)з + 6112О
Изомасляньш альдегид [302]. В колбу, снабженную капельной ворон-
кой и нисходящим холодильником, вливают 100 г изобугнлового спирта и 750 мл
воды, нагревают смесь до 70—80° С и, пропуская ток СО2, приливают по каплям
Раствор 135 г КгСг2О7 в 675 мл воды и 50 г концентрированной H2SO4. Альдегид
выделяют из дистиллята в виде бисульфитного соединения раствором бисульфита
и снова получают в свободном состоянии перегонкой с избытком раствора NagCOj;
т. кпп. чистого альдегида 61° С.
Получение пропионового и пропаргилового альдегидов из соответству-
ющих спиртов см. [303].
Пропаргиловый альдегид (раздражает слизистые оболочки глаз и носа!)
можно также получать окислением пропаргилового спирта активной двуокисью
марганца [304].
Интересный вариант окисления спиртов хромовой кислотой до альдегидов
разработали Оппенаузр я Оберраух [305]. При помощи трет-бутилхромата
в присутствии раствора тпрелг-бутилового спирта в органическом растворителе
можно дегидрировать спирты до альдегидов с выходом 90% от теоретического.
Описание способа см. [30&].
Замещенные бензальдегиды получают с отличными выходами окислением бен-
зиловых спиртов димером двуоинси азота (N2O4), который легко доступен и исполь-
зуется и виде раствора в хлороформе или в четыреххлористом углероде [307, 308].
Еще один метод получения альдегидов заключается в действии N-бромсукции-
пмида на первичные спирты (см. также стр. 309). Реакция протекает гладко при крат-
ковременном кипячении компонентов в органическом растворителе. Этим способом
из 4,5-диметоксифталилового спирта был получен 4,5-диметоксифталальдегид [309].
1302] A. L i р р, Ann., 205, 2 (1880); ср. также A. Pfeiffer, Вег., 5. 699 (1872).
[3i>3] С. I). Н и г d, R. N. Meiner t, Org. Synth., Coll. v. 2, 1943, p. 541;
1. C. Sauer, Org. Synth,, 36, 66 (1956).
[304] Uouben-Weyl, Bd. 7/1, 4 Aufl., 1954, S. 177.
1305] R. V. О p p e n a u e г, H. Oherrauch, C. A., 44, 3871 (1950).
[306] llouben -Weyl, Bd. 7/1, 4 Aufl., 1954, S. 173.
1307] В. O. Field, J. Grundy, J. Chem. Soc., 1955, 1110.
[3n8J J. R. Park, J. R. Partington, J. Chem. Soc., 1924, 74.
[3i>9] j. Blair, W. R. Logan, G. T. Newbold, J. Chem. Soc., 1956, 2443.
301
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
Во многих случаях при действии концентрированной азотной кислоты на про-
изводные бензилового спирта примерно с 80%-ным выходом образуются соответству-
ющие бензальдегиды [310].
В качестве дегидрирующего средства, особенно для окисления растворимых :
в воде оксибен.зиловых спиртов, часто выгодно применять л-нитробензолсульфокис- *
лоту [311].
Для дегидрирования ценных спиртов часто бывает целесообразно использовать
реакцию Меервейна — Поиндорфа — Берлея — Оппенауэра (см. также стр 308).
При действии альдегидов или кетонов на алкоголяты первичных спиртов протекают j
следующие равновесные реакции: |
ZO /V J
RCII2OMe+R'C<f RC< 4-R'ClhOMe 1
хн хн
-О \
RCH2OMe + R'CR' RC<f -j-R'CHR"
Il ХН I
О ОМе ;
Этот метод оказался особенно пригодным для получения альдегидов терпено-j;
вого ряда. Для смещения равновесия реакции в направлении образования требуемого^
карбонильного соединения (Оппенауэр) можно увеличить концентрацию акцептора--,
водорода или подобрать акцептор с такой температурой кипения, чтобы можно было
отогнать образующийся альдегид.
При дегидрировании а.р-ненасыщснных спиртов, реагирующих уже при отно-4
сительно низких температурах, концентрация акцептора водорода должна быть вы-.;
сокой. Для связывания водорода при получении алифатических альдегидов наиболее ।
пригодным оказался коричный альдегид, а при получении алициклических альдеги-1
дов — анисовый альдегид. Окисляемый спирт переводят сначала в алкоголят взаимо-'^
действием с рассчитанным количеством изопропилата алюминия, а затем подвергают
окислительно-восстановительному взаимодействию с 120—200%-ным против теоре-4
тического избытком высококипящего альдегида *.
О^бзор работ, посвященных этому способу, составлен Джерасси [312], с кри-4
тической оценкой реакции можно познакомиться в статье Лаухенауэра и Шпнца [313]. -
а -Циклоцитраль из а -циклогераниола [313]. В трехгорлой колбе, снаб-^
женной капилляром, капельной воронкой и колонкой высотой 10 с.и нагревают.!
(12 ля рт. ст.) 3,75 г а-циклогераниола с 1,66 г изопропилата алюминия (тео-^
ретическое количество) при температуре баин 70—100° С до тех пор, пока при-^
мерно через 45 мин не закончится превращение. Затем в один прием прибавляют?
5,1 г анисового альдегида (155% от теоретического количества) и смесь нагре*^
вают при 12 мм рт. ст. таким образом, чтобы можно было медленно отгонять ’,
альдегид. Полученную фракцию очищают повторной перегонкой. Выход 66%--^
от теоретического; т. иип. 75° С (12 мм рт. ст.).
Каталитическое дегидрирование спиртов лучше всего протекает в паровой фазе,
так как равновесие реакции 1
RCH2OH RCHO + H2
сдвигается вправо главным образом при повышенных температурах. Для дегидриро-1
вапия ненасыщенных 'спиртов требуются акцепторы водорода, в противном случае.
* Окисление аф-ненасыщенных спиртов в соответствующие альдегиды можно;
осуществить с хорошими результатами по модифицированному методу Кренке/
[Р. К а г г е г, А. Е р р г е ch I, Helv. chim. acta, 24, 1039 (1941)]. —Прим.'
[310] В. Helferich, R. Streeck, E. GUnther, J. prakt. Cheni. [2], 151,J
251 (1938). >
[311] F. Hanns, J. prakt. Cheni. [2], 158, 254 (1941). i
[312] C. Djerassi, Org. Reactions, 6, 207 (1951). j
[313] A. Louchenauer, H, Schinz, Helv. chim. acta, 32, 1265 (1949); ’
ср. И о u b e n , W о у 1, Bd. 7/1, 4 AufL, 1954, S. 188. /
I. ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
305
может произойти гидрирование образующегося альдегида. Чтобы избежать этого, во
время дегидрирования пропускают воздух. Прп дегидрировании насыщенных спиртов
нет необходимости в применении акцептора. Процесс дегидрирования ненасыщенных
и высококипящих спиртов часто полезно вести прп пониженном давлении. Реакция
протекает при 300—400° С. Насыщенные алифатические спирты до Св дегидрируются
по этому способу с выходами выше 80% or теоретического. Лучшими катализаторами
являются соединения меди, серебра п пипка. Приготовление катализаторов см. [314|.
Рис. 4. Лабораторная уста-
новка для каталитического
де! идрировзния спиртов:
1 — колба; 2 — трубка с катали-
затором; 3 — нагреватель; 4 —
насадка для термометра; 5--
фракционная колонка; 6 — хо-
лодильник; 7--приемник.
На рис. 4 изображена схема лабораторной установки для каталитического
дегидрирования спиртов. Она особенно удобна для получения альдегидов из
высококипящих спиртов п для работы в вакууме. Прибор имеет колбу, из кото-
рой пары спирта попадают в нагретую до нужной температуры трубку с катали-
затором. Температуру в реакционном объеме контролируют по термометру.
Насадка для термометра соединена с фракционной колодкой для отгонки обра-
зующегося альдегида, который, пройдя холодильник, собирается в приемнике.
Пепрореагировавший спирт стекает обратно в колбу. Выделяющийся водород
улетучивается. Размеры отдельных частей установки подбираются в соответ-
ствии^ требованиями. При высоте трубки для катализатора в 100 см диаметр ее
должен равняться 25—30 мм.
Для приготовления, например, медного катализатора в трубку для ката-
лизатора помещают свернутую в рулон медную сетку, на которую наносят пасту
из окиси меди с добавкой 1% окиси серебра. Катализатор сначала сушат в токе
водорода, медленно поякиляя температуру, и затем медленно восстанавливают
при 300° С. В общей сложности этот процесс занимает 8—10 ч; затем катализа-
тор охлаждают без доступа воздуха.
При получении альдегидов с т. кип. до 200° С можно дегидрирование
вести при атмосферном давлении, если же синтезируют более высококипящие
(314J И on hen - WeyJ, Bd. 7/1, 4 Aufl., 1954, S. 162.
20 Заказ 1835.
306
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
альдегиды, то работают при пониженном давлении. Температура в реакционн
пространстве должна несколько превышать температуру кипения получаемс
альдегида.
Из гераниола при атмосферном давлении получаются а- к p-цитрали, из ф©
нялэтилового спирта — фенилацеталъдегид; коричный альдегид [315] образуете!
с хорошим выходом из коричного спирта на серебряном катализаторе при 200“ С i
разрежении 20 мм рт. ст. Описан лабораторный метод окисления тетрагидрофуя
фурилового спирта воздухом на серебряной вате [316].
Ниже на примере дегидрирования /(-нонилового спирта описан общий сносе)
дегидрирования спиртов.- 1
«-Нониловый альдегид [317]. Успехи синтеза в значительной MajJ
зависят от тщательности подготовки катализатора. К водному раствору чисто»
Cu(NO8)_, приливают рассчитанное количество раствора чистого NaOH и вьшм
шую Си(ОН)2 промывают декантацией слегка подогретой водой до полной не
тральностп. Избыток воды отсасывают на нутч-фильтре, не допуская слншкс
сильного высыхания осадка гидроокиси меди. Ее можно хранить в виде пася
в закрытой склянке. Пасту Си(ОН)г наносят тонким слоем ка кусочки медне
сетки размером 10 X 5 см, которые скручивают в рулоны длиной 10 см и аагр
жают в медную трубу высотой 80 см, вмещающую 30 таких рулонов. Ра?м
ячеек медиой сетки не должен быть очень мал, так как это снижает активное
катализатора. Сначала всю систему продувают водородом, который для промы
ки и сушки проходит две промывные склянки с КМпО4 и H2SO. соответствен^
Трубку обогревают при помощи электронагревателя до 300° С, причем гидр
окись меди восстанавливается в губчатую металлическую медь. Восстановлен]
продолжается 8 ч. Если к дегидрированию приступают не сразу, то катализату
охлаждают в токе водорода. При оставлении на ночь через аппарат пропуска!
медленный ток водорода. Перед началом проведения реакции трубку нагрева!
в течение 1/2 ч до 300® С и продувают водородом. Затем в колбу наливают и-й
ниловый спирт, сокращают подачу водорода, эвакуируют до 3—5 мм рт.
устанавливают в контактном объеме температуру 240° С и доводят спирт
кипения. Вакуумный приемник приспособлен для улавливания различи
фракций; если при слишком большой скорости перегонки временно перегоняет]
непрореагировавший нониловый спирт, что заметно по повышению температу)
в верхней части колонии, эту фракцию собирают отдельно. По окончании дегя
рироваиня установку заполняют водородом и дают ей охладиться без досту,
воздуха, чтобы сохранить активность катализатора. Выход нонилового алвд
гида 90% от теоретического; т. кип. 78э С при 3 мм рт. ст.
Высшие многоатомные спирты удается дегидрировать лишь частично; спиря
производные углеводов не выдерживают реакции дегидрирования (разрушаются).-
Высококипящие спирты иногда выгоднее дегидрировать в жидкой фазе в
сутствин акцепторов водорода, например меди н нитробензола или .и-динитробензоЛ
Таким способом получают о-хлорбензальдегид из о-хлорбензилового спирта при 20$
210° С, выход 86% от теоретического [318].
Получение кетонов у
Дегидрирование вторичных спиртов до кетонов происходит аналогично тольв
что описанному методу синтеза альдегидов; применяются те же окислители и та же те?
ника работы. Реакции протекают обычно более гладко, так как образующийся кетОЙ
более устойчивы к действию избытка окислителя, чем альдегиды. '
[315] С. М о и г с п, G. М i g п о и а с, С. г., 171, 652 (1920).
[316] I. G. М. Bremner et al., J. Chem. Soc., 1949, Suppl. 25.
[317] A. Lewi d so h n, Perfum. a. Essent. Oil. Rec., 15, 13 (1924).
[318] F. Zetsche* P. Zala, Helv. chim. acta, 9, 288 (1926).
I. ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
307
Окисление хромовой смесью ведут в водной пли уксуснокислой среде при 20—
40° С, реже при более высоких температурах (50—80° С). При дегидрировании трудио-
растворимых спиртов применяют интенсивное перемешивание. Выходы кетонов дости-
гают 60—80% от теоретического.
Ментон [319]. К нагретому до 30° С раствору 60 г (1 моль) К2Сг2От
в 50 г концентрированной HsSO4 и 300 мл воды прибавляют 45 г кристалличе-
ского ментола. При перемешивании смеси происходит окисление спирта, сопро-
вождающееся разогреванием. Образуется черно-бурая масса, которая разжи-
жается в масло при температуре выше 53° С. Если в течение 30 мин температура
реакционной массы не достигнет 55° С, массу подогревают, если температура
подымается выше 55° С — смесь охлаждают. После охлаждения масляный слой
растворяют в эфире, промывают эфирный раствор 5%-ным раствором NaaCOs,
сушат и перегоняют в вакууме; т. кип ментона 98—100° С (18 мм рт. ст.)-
выход 83—85% от теоретического.
Пентип-З-он-2 удивительно легко получается окислением при 5—10° С пентин-3-
о.та-2 хромовой кислотой в ацетоне [320].
Вместо К2Сг2О7 можно применять КМпО4, но это обычно не дает никаких пре-
имуществ. Дн-тр^яг-бутилкарбинол может быть окислен концентрированной HNO3 до
гексаметилацетона [321].
Алкиларилкетоны синтезируют с хорошими выходами из соответствующих
спиртов по Груиди при помощи четырехокиси азота [322] (ср. стр. 303). Для этого
спирт растворяют на холоду в сухом хлороформе и прибавляют избыток N3O4. После
продолжительного взаимодействия при комнатной температуре массу промывают
раствором соды и водой, удаляют растворитель и окислы азота и очищают кетон
перегонкой.
Раньше бензоины окисляли до днкетоиов азотной кислотой, однако дегидриро-
вание протекает лучше под действием соединений меди. Например, при помощи суль-
фата меди в пиридине бензоин переводится в бензил с 86%-ным выходом [323]. Холе-
стенон получают по Дильсу и Аодергальдену по следующему способу [324].
Холестенон. Нагревают 100 а холестерина до 300° С, в несколько приемов
вносят 20 г пе слишком тоикоизмельчепной оклей меди и 15 мин нагревают при
температуре бани 300—310° С. Затем еще не совсем остывшую смесь растворяют
в 1л горячего метилового спирта, фильтруют и при 0° С при перомешивавин
вносят затравку — кристалл холестенона. После прекращения кристаллиза-
ции метанолу дают улетучиться в вакууме над серной кислотой н остается холе-
стенон с общим выходом 60 а.
Очень рекомендуется метод окисления в присутствии каталитических количеств
ацетата меди, который непрерывно регенерируется в присутствии нитрата аммония.
Нитрат аммония превращается при этом в нитрит, который медленно разлагается
на воду и азот [325]. Этим способом были подучены дпкетоны из различных
замещенных бензоинов, а из 2-оксициклодеканона — циклодекандион-1,2 [326].
Установлено, что бензоины превращаются в бензилы при обработке их свежеосайс-
деяной двуокисью марганца [327]. В этой же работе имеются данные о дей-
ствии МпО4 на легко окисляемые органические соединения в воде и органических
растворителях.
[319] Е. Beckmann, Ann., 250, 325 (1889).
[320] Е. A. Braude et al., J. Chem. Soc., 1949, 612.
[321] P. D. Bartlett, A. Schneider, J. Am. Chem. Soc., 67, 143 (1945),
[322] J. Grundy, J. Chem. Soc., 1957, 5087.
[ЗЙЗ] H. T. Clarke, E. E. Dre ger, Org. Synth., 6, 6 (1926).
1324] O. Diels et at, Ann., 459, 21 (1927).
[325] M. W e i p, M. Appel, J. Am. Chem. Soc., 70, 3666 (1948).
[326] A. T. Blomquist, A. Goldstein, Org. Synth., 36, 77 (1956).
[327] M. Z. Barakat et al., J. Chem. Soc., 1956, 4685.
20»
308
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
Спирты с двойными и тройными связями можпо мягко окислить в кетоны или
альдегиды при комнатной температуре встряхиванием с десятикратным избытком
синтетического гидрата двуокиси марганца Мп02-Н20 в инертных растворителях:
бензине, хлороформе, бензоле или ацетоне [328J. Реакция рекомендуется для окисле-
ния гидроксильных групп чувствительных соединений. Таким способом получают,
например, 1-фенилпропин-2-он-1 из 1-фенилпропин-2-ола-1 с 67,6%-ным выходом
[329]. Обобщенные данные о применимости этого способа, условиях реакции и спо-
собах приготовления активной МпО2 приведены в работе Эванса [330].
В некоторых случаях вторичные спирты можно переводить в кетосоединения
с помощью тетраацетата свинца. Так, например, из эфиров а-оксикарбоновых кислот
образуются эфиры а-кетокарбоновых кпелот [331], в то время как у-кетокислоты могут
быть легко получены окислением у-лактонов бромом в присутствии гидроокиси маг--‘
ния [332]. '
В ряду стероидов особенно зарекомендовал себя мягкий метод получения кето- ;
нов дегидрированием вторичных спиртов по реакции Оппенауэра при каталитическом-!
действии алкоголятов металлов (стр. 304).
R4 R\ R\ R\
>CHOMc< >C=O >C=O + }CHOMc
R'Z R^Z RlZ R^Z
Рассмотрению этого метода посвящены обзорные статьи [312, 333]. ’
Для того чтобы сдвинуть равновесие реакции в сторону образования карбонильч^
него соединения, добавляют большой (40—80-кратпый) избыток акцептора водорода i
В качестве таковых оказались пригодными ацетон, этилметилкетон и циклогексанон^
Эти вещества вместе с образующимися из них продуктами конденсации легко удаляются^
из реакционной смеси перегонкой с водяным паром после завершения реакции. Дляз
получения кетонов, которые могут отгоняться из реакционной смеси при температурах^
ниже 100° С, в качестве акцептора водорода с успехом применяется бензил. Иногда^!
в качестве дегидрирующего средства рекомендуется хинон.
Продолжительность реакции и концентрации реагентов выбирают в соответ-
ствии с характером исходных компонентов. Чаще всего реакцию проводят при темпер
ратуре около 60° С. Образование продуктов конденсации значительно снижается прЯ
работе в инертпых растворителях — бензоле, диоксане и толуоле. Из алкоголям»
металлов наряду с изопропилатом алюминия оказался пригодным изобутилат алюми^
ния; как правило, его вводят в реакционную массу в количестве 0,5 моль в расчете!
на исходный спирт. По методу Оппенауэра окисляются также азот- и галогенсодер-
жащие вещества. В качестве примера приведено дегидрирование холестерина в холе*
Холестенон. Смешивают раствор 10 г холестерина в 120 г горячего аце-
тона с раствором 12 г кристаллического mpem-бутилата алюминия в 300
бензола и кипятят 10 ч в колбе с обратным холодильником. Для Отделения алю-
миния реакционный раствор основательно встряхивают с разбавленной H2SO4^
бензольный слой отделяют, промывают, сушат и выпаривают бензол. Выход
8,9 г; т. пл. 79—80q С.
Мягкое, не затрагивающее структуры дегидрирование вторичных спиртов^
до кетонов может быть также осуществлено с помощью N-бромацетамида. Этот cnocof
[328] J. Attenburrow et al., J. Chem. Soc., 1952, 1094.
329] I. Iwai et al., C.. 1959, 7093.
330] R. M. Evans, Quart. Rev., 13, 61 (1959). ’
331] V. Prelog, H. L. Meier, Helv. chim. acta, 36, 322 (1953). *
332] R. R. Russel. C. A. Vandewerf, J. Am. Chem. Soc., 69, 12 (1947)^
333] Th. В e г s i n, Ncuere Methoden der praparativen organischen Chemie, 2 Aufl.*
Verlag Chemie, Weinheim, 1944, S. 137. ’
[334] Th. R e r s i n, Neuere Methoden der praparativen organischen Chemie, 3 Aufl.?;
S. 147; cp. R.’V. Oppenauer, Org. Synth., 21, 18 (1941). >
I. ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
309
особенно охотно применяют в ряду стероидов [335]. Аналогично для окисления первич-
ных и вторичных спиртов * очень пригоден N-бромсукцинимид [336].
Следует отметить также исследования Хейнса и Блазеевича [337]. По их данным
при каталитическом окислении первичных спиртов кислородом на платиновом кон-
такте в зависимости от условий реакции получаются альдегиды или карбоновые кис-
лоты. Вторичные спирты превращаются в кетоны. Такой способ рекомендуется для
получения альдегидов с длиной цепью из соответствующих спиртов.
Хорошие результаты дает каталитическое дегидрирование вторичных спиртов
в паровой фазе на катализаторах, подобных применяемым для дегидрирования первич-
ных спиртов. В этом случае реакция протекает еще легче, так как из кетонов образуется
меньше побочных продуктов. Хард и сотр. [338] получили из циклогексанола
с 60%-ным выходом циклогексанон при помощи меднохромового катализатора Ад-
кинса [338]. Возможно также дегидрирование вторичных спиртов в жидкой фазе.
Б качестве катализатора наряду с другими можно использовать никель Ренея. Окис-
ление целесообразно проводить в присутствии акцептора водорода, например цикло-
гексанона. Для проведения реакции кратковременно нагревают смесь спирта, раство-
рителя, катализатора и акцептора водорода [339].
Для окисления фенолов до хинонов можно применять различные окислители. На-
пример, гидрохинон окисляется в n-хинон хромовой смесью [340] или NaC103 в при-
сутствии пятиокиси ванадия [341] с выходом выше 90% от теоретического. Замещен-
ные гидрохиноны окисляются до хинонов при помощи солей трехвалентного железа
[342] или окиси серебра Ag2O (см. [343]). Реакцию можно проводить также в органи-
ческой среде, например в спирте или уксусной кислоте. Для окисления замещенных
л-амин®фенолов до га-хинонов предпочтительно применяют соли железа (111); выходы
выше 60% от теоретического.
Дегидрирование пирокатехина с помощью окиси серебра приводит к образова-
нию и-.хинопа [344]. О получении амино-о-хинопов см. [345]. Дегидрирование гидро-
хинонов можпо также с успехом проводить с помощью тстраацетата свинца [346].
Обзор возможных методов получения бензохинонов окислением см. [347].
Окисление многоатомных спиртов
По предложенному Фентоном и Джексоном [348] методу а-гликоли переводят
в а-окспальдегиды действием 3—6%-ной Н2О2 и солей железа (II) при комнатной тем-
пературе. Выходы умеренные. Простые первичные и вторичные спирты в этих условиях
не окисляются.
Для окисления первичных и вторичных спиртов в слабощелочной среде до
карбонильных соединений весьма пригоден при наличии других групп»
чувствительных к окислению, комплекс хромового ангидрида и пиридина
СгО3 • 2CjH6N [G. Р о о s et all., J. Am. Chein. Soc., 75, 422 (1953)]. — Прим. ped.
M. D. S о f f e r, M. A. J e vuik, J. Am. Chem. Soc., 77, lOOo (1955);
E. P. Oliveto et al., J. Am. Chem, Soc., 78, 1414 (1956).
L. Horner, E. H. Winkelman n, Angew. Chem., 71, 354 (1959).
К. He у ns, L. Blazejewici, Tetrahedron, 9, 67 (I960).
Ch. 1). Hurd, H. G r e e n g a r d, A. S. R о e, J. Am. Chem. Soc., 61, 3359
(1939).
E. С. К 1 ei de rer, E, С. К о rn f el d, J. Org. Chem,, 13 , 455 (1948);
ср. англ. пат. 767093; J. appl. Chem., 8, 615 (1958).
E. В. V 1 i e t, Org. Svnth., 2, 85 (1922).
H. W. Underwood, W.L. Walsh. Org. Synth., Coll. v. 2, 1943»
p. 553.
t. 1. Smit h, P. F. Wile у, J. Am. Chem. Soc., 68, 889 (1946).
J. M. Bruce, F. K. Sutcliffe, J. Chem. Soc., 1956, 3820.
R. Wiilstatter. A. Pfannenstiel, Ber., 37, 4744 (1904).
L. Horner, H. Lang, Chem. Ber., 89, 2768 (1956).
R. Criegee, Angew. Chem., 70, 173 (1958).
J. Cason, Org. Reactions, 4, 305 (1948).
JI. J. H. Fenton, H. Jackson, J. Chem. Soc., 75, 1 (1899).
310
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
Манноза из маннита. В небольшом количестве воды растворяют 50 e''j
маннита, добавляют водный раствор 12,5 г FeSO4-7IIsO и 150 лл 5—6%-НОЙ'З
Н2О2. После затухания реакции массу обрабатывают избытком свежеосажденного
В&СО3,фильтруют, фильтрат сильно упаривают при 50°Си давлении 30 мм рт.
л остаток доводят в вакууме до сиропообразной консистенции. К сиропу доба-я
вляют десятикратный объем абсолютного спирта, фильтруют и смешивают с из-Ц
битном эфира. Выпадающие при атом бесцветные хлопья расплываются, образуя,^
светло-желтый сироп, который частично кристаллизуется при растирании со$
спиртом и эфиром. Для идентификации маннозы из нее получают фенилги.^
дразон.
Литературный обзор по этому методу см. [349]. «
Описан способ получения маннозы окислением маннита азотной кислотой [350]<
6. Окисление спиртов и альдегидов до карбоновых кислот
Для превращения спиртов и альдегидов в карбоновые кислоты можно практик
чески использовать все общепринятые окислители. При окислении спиртов кислотными!1
агентами в качестве побочных продуктов образуются эфиры, поэтому иногда окислений
предпочитают вести щелочным раствором КМпО4. Реакция идет при перемешивании'
при комнатной температуре и последующем охлаждении; выходы не бывают коли-
чественными. В качестве примера следует указать иа синтез пропионовой кислотй
ил пропилового спирта [851].
10-Фтордеканол в ледяной уксусной кислоте переводится при помощи СгО||
с 93%-ным выходом в соответствующую фторкарбоновую кислоту [352]. J
Получение карбоновых кислот из первичных спиртов в присутствии катализ^р
торов дегидрирования при 120—150° С описано Любомпловым с сотр. [353].
%
Пирослизевая нислота (видоизмененный способ Фольгарда) [354]. Рао*
творяют 19 г фурфурола в 50 мл пиридина и раствор охлаждают в смеси льдщ
с поваренной солью. Затем к раствору при энергичном перемешивании прилив
вают по каплям раствор 21 г КМпО4 в смеси 280 мл пиридина и 120 мл воды*
так, чтобы температура в реакторе не превышала —3° С. После того как вес!
окислитель прибавлен, перемешивают еще полчаса, отфильтровывают выпавшей
МпО2 и промывают осадок небольшим количеством пиридина. От фильтрата
отгоняют в вакууме растворитель, сухой остаток подкисляют HCI и извлекаю!
продукт реакции эфиром. После отгонки эфира остается 19 а пирослиэевой кисй
лоты в виде желтоватых кристаллов (86% от теоретического).
Фурфурол можно окислять также кислородом в присутствии окиског^
Медно-ссребряного (Cu2O/Ag;O) катализатора [355]. Реакцию ведут в щелочной
среде, получается фуран-2-карбововая кислота с выходом 86—90% от теоретик
ческого. j
Для превращения альдегидов в карбоновые кислоты в большинстве случае^
используют перекись водорода [356]; равным образом для окисления с успехом пр®»
меняются и надкислоты [357]. ’ !?’
-------------- • I
[349] В. Criegee, Handbuch der Katalyse, 7, I. Halfte, Springer-VerlariiS
Wien, 1943, S. 590. Л
[350] E. Fischer, J. llirschberger, Ber., 22, 365 (1889). .
1351] H. Fournier, C. r.,144, 333 (1907).
[352] К L. M. Pa ttison I B. S t о t h e г s, R. G. W о о d f о r d, J. Am,-
Chem. Soc., 78, 2256 (1956). J
[353] В. И. Л ю б о м и л о в, А. И. Куценко, P. Л. Абрамова, ЖОХ,
(89), 2054 (1957); С., 1959, 15317.
'1(354] J. Volhard, Ann., 261, 380 (1891). 4
J355J R. J. II a r r i s s о n, M. Moyle, Org. Synth., 36, 36 (1956).
]356J A. Dobrowsky, Mh. Chem., 86, 325 (1955). 5
1357] A. v. Wacek» A. v. Bezard, Ber., 74. 857 (1941).
I ЗАМЕЩЕНИЕ ВОДОРОДА КИСЛОРОДОМ
311
Наиболее рекомендуемым методом является превращение альдегидов в карбоно-
вые кислоты с помощью окиси серебра. Окись серебра легко доступна, не взаимодей-
ствует с другими окисляемыми группами и при ее применении получаются очень чи-
стые кислоты. Азингер приготовил таким способом энантовую кислоту из энантола
и пальмитиновую кислоту из гексадеканаля с выходом 97,5% от теоретического [358].
О получении ненасыщенных кислот из ненасыщенных альдегидов при помощи Нв0г
в присутствии SeO2 сообщили Смит и Холм [359]. Проводя окисление 0-хлорпропио-
паля HNO3, удалось получить p-хлорпропионовую кислоту с 81%-ным выходом
[360]. Окисление альдегидов до кислот в газовой фазе на платниово-силикагелевом
катализаторе Исследовано Фостером и Кэйсом [361].
В химии углеводов используются специальные методы окисления альдегидных
групп, различные для аналитических и для препаративных целей. Для превращения
альдоз в альдоновые кислоты с успехом применяется электрохимический метод, впер-
вые предложенный Исбеллом и Франтом [362].
Позднее Килиани [363] модифицировал метод Исбелла; он оказался вполне
пригодным для окисления не только гексоз, но и пентоз.
7)-Манноновая кислота из Л-маннозы по Килиани [364]. Растворяют
100 г Л-маннозы в 2 л воды, смешивают с 17,7 i СаВг2 и 55 г СаСО3 и при пере-
мешивании подвергают электролизу в подходящем цилиндре с угольными элек-
тродами (диаметр электродов 12 мм, расстояние между ними около 8 ем, сила
тока 0,5 —0,7 а). Через 50—55 ч реакционная масса почти полностью теряет вос-
становительные свойства. Ее фильтруют, возможно полнее упаривают, дают
затвердеть и измельченную аморфною массу сушат. Получение кристалличе-
ского манггоповокисяого кальция описано в оригинальной работе.
Для превращения манноновой кислоты в лактон 1 ч. кальциевой соли:
растворяют на водяной бане в 3 ч. воды, медленно смешивают с избытком щаве-
левой кислоты, отсасывают выпавший оксалат кальция, концентрируют и упа-
ривают раствор до исходного веса кальциевой соли и оставляют кристал-
лизоваться.
Для превращения альдоз в альдоновые кислоты можно применять также окис-
ление бромом в водной среде [365]. Препаративное значение приобрело окисление
углеводов и их производных кислородом в присутствии катализаторов, содержащих
платину [366]. Таким способом можно получать и альдоновые кислоты из альдоз.
Пентозы при этом окисляются значительно быстрее. При 22° С они окисляются уже
в течение 45 мин, в то время как для окисления Л-глюкозы требуется 5 ч. На основа-
нии наблюдения, что в присутствии платины кислородом предпочтительно окисляются
первичпые, а не вторичные спиртовые группы, разработан удобный синтез уроновых
кислот. Для этого необходимо блокировать карбонильную группу у С-1 любым
пригодным способом, например переводом альдозы в гликозид [367]. Об окислении
концевых первичных спиртовых групп в углеводах с помощью N2O4 см. [368].
а-Кетокарбинолы легко окисляются. Единственный существующий пригодный
способ нх окисления использовался еще Броером и Цинке [369]. По этому способу
па спирты действуют соединениями меди (II) в водной среде. Например, оксипировино-
1 радную кислоту получают обработкой диоксиацетона избытком ацетата меди (Ц) с вы-
ходом 87% от теоретического [370].
[358] F. Asinger, Вег., 75. 658 (1942).
[359 С. W. Smith, R. Т. Holm, J. Org. Chem., 22, 746 (1957).
[360] С. Moureu, R. Ch au d, Org. Synth., 8, 54 (1928).
[361] П. 1). Foster, D. B. Keyes, Ind. Eng. Chem., 29, 1254 (1937).
[362] II. S. 1 a b e I 1, II. L. F r u s h, Bureau of Standards J. of Res., 6, 1145 (1931);
C., 1931, II, 3331.
1363] KKiliani, Ber., 66, 117 (1933).
[364] K. Bernhauer, K. Irrgang, Biocliem. Z., 249, 216 (1932).
[365] W. G. Overend, M. Stacey, L. F. Wiggins, J. Chem. Soc., 19'i9_
1358.
1366] K. Heyns et al., Ann., 558, 187, 192 (1947).
367] K. lleyns, H. Paulsen, Angew. Chem., 69, 603 (1957).
368| K. Maurer, G. D r e f a h I, Ber., 80, 94 (1947).
369] А. В г c u e r, Th. Z i n k e, Ber., 13, 636 (1880); Ann., 216, 313 (1883).
37U[ W. E. Evans jr. et al., J. Am. Chem. Soc., 60, 1628 (1-938).
312
ОБРАЗОВАНИЕ С —ОЧ.ИИЗК И ОБМЕННЫХ РЕАКЦИЯХ
7. Реакции Канниццаро п Тищенко *
Альдегиды, в которых сс-атом углерода не связан с атомом водорода, превраща-
ются в соответствующие спирты и карбоновые кислоты под влиянием водных пли спир-
товых растворов щелочей (реакция Каннпццаро):
2K-CI1O —> RCOO- ТНСП-.ОН
Обычно в эту реакцию, названную по имели ее первооткрывателя реакцией
Канниццаро, вступают альдегиды ароматического п гетероциклического рядов, хотя
известны и исключения пз этого правила. Подобные окислительно-восстановительные
реакции играют значительную роль в биологических процессах, происходящих в клет-
ках под влиянием ферментов. В лаборатории реакция Канниццаро используется для
получения кислот, а также спиртов, причем выходы каждого компонента обычно со-
ставляют 50%
Пирослизевая кислота и фурфуриловый спирт [371]. В сосуд емкостью 4 л
вносят 1000 г технического фурфурола, охлаждают при перемешивании до
5—8° С и из делительной воронки приливают раствор 275 г NaOH в 350 мл воды
так, чтобы температура смеси не поднималась выше 20° С. После этого переме-
шивают еще 1 ч и добавляют количество воды, нужное для растворения выпав-
ших кристаллов натриевой соли нирослизевой кислоты. Затем в аппарате для '
экстракции жидкостей экстрагируют из раствора фурфуриловый спирт 1500—
2000 мл эфира. Основные количества фурфурилового спирта можно также
удалить встряхиванием с эфиром в делительной воронке.
Оставшуюся после экстрагирования эфиром щелочную жидкость подкис-
ляют по индикатору конго 40%-ной H2SOj (примерпо 400 мл). При охлаждении
выпадает в осадок темноокрашепная смесь нирослизевой кислоты и бисульфата
натрия; смесь отсасывают, растворяют в 2300 лм кипящей воды, добавляют
60 г животного угля и выдерживают при кипячении около 45 мин, после отделе-
ния угля фильтрат охлаждают до 16° С (не ниже). Выход пирослизевон кислоты,
достаточно чистой для проведения многих реакций, составляет 60—63% от тео-
ретического.
Для очистки малые количества кислоты возгоняют, желательно в вакууме.-
(2 мм рт. ст.); большие количества перегоняют при пониженном давления;
т. пл. 133—134° С.
Фурфуриловый спирт получают разгонкой эфирной вытяжки. Сначала .
при нормальном давлении собирают фракцию, отгоняющуюся при температуре.:
кипящей жидкости 95° С, а затем фракционируют остаток в вакууме. Выделяю!
310—325 г спирта (т. кип. 75—77° С при 15 мм рт. ст.), что соответствует вы-
ходу 61—63% от теоретического. Для хранения фурфуриловый спирт стабили-
зируют добавкой 1% мочевины.
Нитробепзонные и галогенбензойные кислоты получены из соответствующих ..
альдегидов с выходами 84—90% от теоретического[372]. При сплавлении л-оксибенз- ..
альдегида при 190° С с КОН он почти количественно превращается в лыоксибензой- ...
ную кислоту, так как промежуточно образующийся бензиловый сцирт реагирует
с КОН по уравнению:
HOv - КОк
иьон+гкои —> гн2о
При взаимодействии двух различных альдегидов с концентрированными щело-
чами происходит перекрестная реакция Канниццаро. Этот метод особеппо рекомен-*
дуется для получения первичных спиртов. В качестве второго компонента реакции -
применяется избыток формальдегида, при этом формальдегид преимущественно окис-1
ляется до кислоты, а высший альдегид восстанавливав icn до спирта; J
RCHO+CH2O -SC. RCII.OII + HCOO- i
[371] W. C. Wilson Org. Synth., 6, 44 (1926),
[372] G. Lock, Ber., 63. 835 (1930).
I. ЗАМЕЩЕНИЕ ВОДОРОД \ ЪЛСЛОГОДОМ
Способ не требует особой аппаратуры н специальных реагентов, спирты полу,
чаются Gbicipo п с отличными выходами. Дэвидсон п Вейсс [373] приводят общеприме-
внмый и хорошо воспроизводимый способ превращения л-толилальдегида в п-толил.
карбинол. О перекрестной реакции Канниццаро * моно-, ди- и тризамещенных бенз-
альдегидов с формальдегидом см. также (374).
Перекрестная реакция Канниццаро с формальдегидом имеет препаративно^
значение для альдегидов с водородом у а-агома углерода. Эгог атом водорода заме-
щается па окенметильную группу, а альдегидная группа восстанавливается избытком
формальдегида в спиртовую. Так, из альдегидов с двумя а-атомамп водорода полу-
чают триметплолалкаиы [375]:
RCH2c/° +2Пс/° -он~^ RC(CHoOH)2c/°
чи хн хн
/О /О п
RC(CI12OH)2C^ +HC<f RC(('.HoOH)3+HCOO-
4 н хн
Пентаэритрит С(СН3ОН)4 получают этим способом из ацетальдегида [376].
Реакция Канниццаро применима почти ко всем без исключения двузамещенным
бензальдегидам. Но если оба орто-положения к альдегидной группе замещены элек-
троноакцепторными заместителями, как, например, в 2,6-динитробензальдегиде или
в 2,6-дигалогенбензальдегидах, то происходит отщепление карбонильной группы.
Наилучптие выходы при проведении реакции Канниццаро достигаются
с 50%-ными растворами КОН или NaOH. При проведении реакции с нитро-
бензальдегидами необходимо применять только 15—35%-ные щелочи. В случае
плохорастворимых альдегидов пользуются 25%-ным спиртовым раствором
КОН. В водных средах требуется сильное встряхивание. Перекрестная реакция
Капнлццаро проводится в метиловом спирте. Если во время реакции выделяется
тепло, массу слегка охлаждают. Для медленно протекающих реакций рекомен-
дуется слабый подогрев. Часто благоприятной температурой реакции является
Обзор работ по реакции Канниццаро составлен Гейссманом [377]. Исследова-
ния механизма реакции опубликованы Каратом и Снайдером [378].
Под действием алкоголятов происходит димеризация альдегидов. Это положение»
по наблюдениям Тищенко [379], можно препаративно использовать для получения
сложных эфиров, в спиртовом и кислотном компонентах которых содержится одина-
ковое число атомов углерода. Реакция протекает под каталитическим действием алко-
голятов алюмвпия или магния по общей схеме:
2RCIIO —► RCOOCII2R
Эфиры такого типа помечаются в природе (например, спермацет, пальмитило-
вый эфир пальмитиновой кислоты). Этот способ представляет ценность в том случае»
когда применяемый альдегид (например, фурфурол) более доступен, чем соответству-
ющие спирт и кислота. Для реакции требуются очень чистые исходные продукты.
Она применима также к альдегидам, содержащим водород у а-атома углерода.
* Перекрестная реакция Канниццаро была впервые препаративно разработана
В. М. Родионовым и А. М. Федоровой [ЖОХ, 7, 947 (1937)]. — Прим. ред.
[373] D. Davidson, М. W е i 0, Org. Synth., Coll. v. 2, 590 (1943).
[374] G. S. Misra, S. B. Sri vastav a, J. Indian Chem. Soc., 32, 201 (1955);
C., 1957, 6118; J. prakt. Chem.. [4], 6, 170 (1958).
[375] M. G. Dupont, R. D u 1 о u, A. Duplessis-Kergomard, Bull.
Soc. chim. France, 1949, 314.
[376] H. T. Clarke, R. Philips, Org. Synth., 4, 53 (1925).
[377[ T. A. Ge i 0m an, Org. Reactions, 2, 94 (1944).
[378[ M. S. Kharasch, fl. Snyder J. Org. Chem., 14, 819 (1949).
[379] W. T i s c h t s c h e н k о, C., 1906, II, 1309, 1552.
314
ОБРАЗОВАНИЕ С—0-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
Как установил Клайзен [380], прп работе с ароматическими альдегидами в кай
честве катализатора можно с успехом использовать алкоголят натрия. Таким способом
в присутствии бензилата натрия с выходом 90—93% от теоретического получают бета
зплбензоат из бензальдегида [381]. Между тем, при проведении реакции с алифатиче^
сними альдегидами в присутствии алкоголята натрия происходит альдольная
денсация.
Подробные исследования по изучению оптимальных условий реакции проводи
Чайлд и Адкинс [382]. В отличие от реакции Канниццаро, для проведения которой
требуется по меньшей мере 1 моль щелочи, в этой реакции достаточно каталитически
количеств алкоголята. Из алифатических альдегидов с 2—8 атомами углерода в таки
условиях получаются соответствующие эфиры свыходом 69—100% от теоретическом
Проведение димеризации альдегидов до эфиров в присутствии алкоголята алюмину
приводит в большинстве случаев к образованию около 30% побочных продуктов
По-видимому, лучшими катализаторами являются алкоголяты цинка или галогенов
держащие алкоголяты алюминия. При их помощи удается даже из ацетальдегид
получить этилацетат [383]. В присутствии алкоголята магния или соединена
Mg[Al(OC2H5)4]2 может быть осуществлена перекрестная реакция Тищенко [384f
при этом из промежуточно образовавшегося альдоля по реакции его с исходным ал&
дегидом образуется эфир 1,3-диола: *'
R
О
НСП2-СН(ОИ)-СН-СН2-0-С-СНгП
11. Обмен галогена на кислород
Алифатически связанный галмеи легко обменивается на кислород с образова-
нием спиртов, альдегидов, кетонов или карбоновых кислот. Легкость обмена снижает^
в ряду: R —I >• R—Вг> R—С1 или в ряду: третичный > вторичный > первичны]
алкядгалогениды. Обмен фтора на кислород до сих пор играет незначительную рол
в препаративной органической химии. Для получения фенолов из арилгалогенинш
требуются жесткие условия ведения процесса, что применяется преимущественно в npdi
мышленности.
1. Обмен галогена на гидроксильную группу
Гидролиз алкилгалогенидов на практике проводят кипячением их с водньШЯ
щелочами, с суспензиями карбонатов щелочноземельных металлов или с суспензия
окиси свинца (I1J. Иногда используют обходный путь: галогенпроизводное превр^
щают обработкой ацетатом щелочного металла в соответствующий эфир, которм
затем омыляют. Этот способ применяют главным образом для получения гликонеа
так как гидролиз щелочами 1,2-дигалогенсоединений в большинстве случаев предо
ходит с плохими выходами вследствие образования побочных продуктов. , J
З-Хлор-циклопентен-1 превращается под действием охлажденного льдом рас*
твора NaHCO3 в циклопентен-1-ол-З [385]. Двухчасовой обработкой 1,3-дихлорбз^
тена-2 4,2%-ным раствором соды получают 3-хлорбутен-2-ол-1 с выходом 63% от теоре-
тического; галоген у ненасыщенного атома углерода не омыляется [386]. Для успей**
ного проведения гидролиза алкилгалогевидов можно также использовать нитрат с-Й»
ребра [387]. В некоторых специальных случаях омыление алкилгалогенпдов проводят
1380]
[381]
[382]
[383]
[384]
[365]
[386J
f.387J
L. Claisen, Вег., 20, 646 (1887).
Но tib о n-’w e'yl,8/nii 558е (1952)7.......
W. С. Child, Н. Adkins, J. Am. Chem. Soc., 47, 798 (1925).
H. Henecka, в книге Houben - Weyl, 8/1П, 558 (1952).
F. J. V i ] 1 a n i, F. F. N о г d, J. Am. Chem. Soc., 69, 2605 (1947).
K. Alder, F. H. Flock, Chem. Ber., 89, 1735 (1956).
L. F. H a t c h, V. C h i о 1 a, J. Am. Chem. Soc., 73/361 (1951).
H. Gil man. C. G. Branne n, R. K. Ingna m, J. Am. Chem.
78, 1690 (1956).
О. К a m m, F. W. Kam m, Org. Synth., 2 , 5 (1922); H. Henecka, в книг^
Soc.
и ОБМЕН ГАЛОГЕНА НА КИСЛОРОД
л кжлой среде. Так, например, n-нитротрифенилметилхлорид превращают в п-нитрс
трпфепилкарбинол [388] в ледяной уксусной кислоте с добавлением НС1О4. 4-Хлор
карбостирил [389] получают кипячением в течение 2 ч 2,4-дихлорхииолина в диоксан
с Ви. НС1. Мак-Клоски и Колеман [390] описали превращение ацетобромглюкоз;
в 2,3,4.6-тетраацетат 0-/>-глюкозы при помощи Ag2CO3. Особенно легко омыляютс.
бензнлгалогеииды. Для получения бензилового спирта бензилхлорид кипятят 2
с трехкратным количеством свежеосажденной окиси свинца и десятикратным коли
чеством воды [391]. Мелье [392] кипятил в течение нескольких часов эквимолекуляр
ные количества бензилхлорида и К2СО8 до исчезновения масла на поверхности реакцв
онпой смеси. Выход бензилового спирта не указан.
Гидролиз а-галогенкарбонильных соединений следует вести очень осторожно
так как сс-оксикарбонильные соединения чувствительны к щелочам *. Для получена;
бензилгликолевого альдегида [393] гидролиз проводили в водной суспензии ВаСОа
Большое значение имеет получение а-оксикарбоновых кислот. Хороший лаборатории]
способ получения гликолевой кислоты приведен Витцеманом [394]. Он отличаете;
от ранее опубликованного способа [395] тем, что для омыления вместо карбоната наль
ция применяют ВаСО3.
Гликолевая кислота. Растворяют 50 г монохлоруксусной кислощ
в 500 мл воды и медленно прибавляют 120 г ВаСО8. Смесь нагревают около 30
в колбе с обратным холодильником прп перемешивании до полного превращена;
выделения СО2. Далее отфильтровывают избыток ВаСО8 и из горячего фильтрат,
осаждают перешедший в раствор барий прибавлением 52,1 г 95%-ной HaS04
После выдерживания смеси в течение некоторого времени на горячен бане отса
сывают осадок и многократно промывают его водой. Фильтрат и промывные вода
смешивают с небольшими порциями H2SO4 до тех пор, цока в растворе оста
путся лишь следы ионов бария. Снова фильтруют и упаривают фильтрат в вану
уме, причем выпадает еще немного BaSO4. Остающийся енроп, сильно пахнущи]
соляной кислотой, нагревают 2 ч на паровой бане для удаления основного вола
чества НС], после чего остается 40 г остатка. При потирании стеклянной палоч
кой или при внесении затравки (кристаллическая гликолевая кислота) начи
нается кристаллизация; получается 16,5 г гликолевой кислоты, что соответ
ствует выходу 41 % от теоретического. Выход кристаллической кислоты в значи
тельной мере зависит от степени упаривания раствора; следует избегать чрезмер
ного упаривания. Если гликолевая кислота остается в аморфном состоянии, ©
можно еще раз перевести в бариевую соль и повторить вышеописанные операции
Общий выход гликолевой кислоты составляет 88,7%, ио половина этого количе
ства может быть выделена только в виде кальциевой соли.
Для синтеза а-оксимасляной кислоты этот способ непригоден. Ее полу
чают следующим образом.
а-Оксимасляная кислота [396]. Кипятят в течение 5—6 ч 100 а а-броь®
масляпой кислоты с эквимолекулярным количеством К2СО8 (83 г) и 500 •».
воды. Затем раствор упаривают, остаток смешивают с рассчитанным количест
вом НС], отсасывают вязкий раствор от выпавшей смеси КВт и КС1 и продук
исчерпывающе экстрагируют из фильтрата эфиром в аппарате для экстракци]
жидкостей. После сушки над Na2SO4 и отгонки эфира остаток пере
гоняют в вакууме и после многократного фракционирования получают а-окси
масляную кислоту; т. кип. 140° С (12 мм рт. ст.)-, т. пл. 42° С; выход не указаа
Методику см. I. К г i t с h е v s k у ct al., J. Am. Chem. Soc., 74, 483 (1952)
В общем случае целесообразно сначала заместить галоген на ацетоксигрупп;
для образования ацетоксикетонов и затем осторожно их гидролизовать. —Лрил*
ред.
Р. D. Bartlett, J. D. Cotman jr., J. Am. Chem. Soc., 72, 3098 (1950)
R. J. Rowlett jr., R. E. Lutz, J. Am. Chem. Soc., 68, 1290 (1946).
С. M. Me Cl oske v, G. H. С о 1 e m a n, Org. Synth., Coll. v. 3, 43
(1955).
C. L a u t h, E. G г i m a u x, Ann., 143, 81 (1867).
J. Meunier, Bull. Soc. chim. France [2], 38, 159 (1882).
S. Danilo w, E. Venus-Danilowa, Bor., 63, 2769 (1930).
E. J. W i t z e m a n n, J. Am. Chem. Soc., 39, 109 (1917).
A. Holzer, Ber., 16, 2955 (1883).
C. Bischoff. P. Walden, Ann.. 279, 102 (1894).
31G
ОБРАЗОВАНИЕ С О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
В качестве побочной реакции при омылении алифатических галогенидов может
происходить отщепление галогеноводорода, которое часто удается в значительной
мере предотвратить применением свежеосаждепной окиси серебра, реагирующей уже
па холоду. Однако можпо более надежно избежать образования олефинов омылением
галогенидов через соответствующие ацетоксипроизводпые.
Если гидролиз алкплгалогенидов затруднен или происходит отщепление гало-
геповодорода, применим способ, предложенный Буво 1397]: обработка кислородом
гриньяровских соединений галогенидов в эфирных растворах с последующим разложе-
нием водой. Этот способ рекомендуется также И для ароматических соединений. Обычно
при этой реакции плохие выходы часто могут быть улучшены добавкой алифатических
металлоорганических соединений [398[.
Находящийся в ароматическом ядре галоген обменивается непосредственно
на гидроксильную группу с трудом. Гидролиз галогензамещенных бензолов проводится
под давлением в присутствии щелочей или катализаторов [399]. Превращение хлор-
бензола в фенол описано в многочисленных патентах [400]. Из арилбромидов
соответствующие фенолы можно получать по реакции с разбавленными щелочами
в автоклаве в присутствии медных катализаторов [401]. Легче протекает гидролиз арил-
галогенидов, у которых имеются электроноакцептордыс заместители н орто- или пара-
положении к галогену.
2. Обмен галогена на карбонильный кислород
Превращение есл-дпгалогенидов в альдегиды или кетоны достигается с помощью
кислотных или щелочных агентов. Часто омыление гладко протекает и в водной среде
в присутствии FeCIs. Для гидролиза чисто алифатических дигалогенцроизводных
требуются более жесткие условия, чем для гидролиза бензальгалогевидов. В бепзаль-
хлориде, получаемом хлорированием толуола, всегда содержатся примеси бензил-
хлорида и бензотрихлорида. В присутствии борной кислоты при 130—160’ С эта смесь
с хорошим выходом превращается в бензальдегид [402]. Метод применяется в про-
мышленности. n-Хлорбепзальхлорид омыляется при комнатной температуре при
взаимодействии с концентрированной серной кислотой [403] в течение 5 ч. При гидро-
лизе 1,1,2-трифтор-2-хлор-3-фенилдиклобу'гена 92%-ной H2SO4 и 100° С образуется
фенилциклобутендион с выходом 75% от теоретического [404]. Для получения
?1-метил-пиридона-2 (выход 80% от теоретического) смесь Х-метил-2-иодпиридинийио-
дида и 6 п. НС! нагревают 24 ч с обратным холодильником и затем нейтрализуют твер-
дой содой[405]. n-Бромбензальбромид гидролизуется при нагревании в течение 15 ч
с водной суспензией СаСОч. Образующийся в-бромбеизальцегид выделяют перегонкой
с водяным паром [40GJ. о-Фторбензальдегид [407] получают омылением о-фторбевзаль-
бромида К2СО3. Виттиг и Видал [408] осуществили синтез флуоренона из 9.9-дибром-
флуорена действием ацетата натрия в 60%-ной уксусной кислоте. В известных случаях
переход от ге-к-дигалогепидов к карбонильным соединениям рекомендуется проводить
обменный взаимодействием с эквимолекулярным количеством безводной щавелевой
кислоты при 110—130° С, причем все побочные продукты улетучиваются из реакцион-
ной массы. Например, при нагревании 2-(дихлорметил)-бензимидазоЛа с порошкообраз-
[397] М. L. Bouveault Bull. Soc. chim. France [3], 29, 1053 (1903).
[398] C. D. Hurd, K. L. Kreuz, J. Am. Chem. Soc., 72, 5543 (1950).
[399J Герм. пат. 539176, Frdl., 18,‘ 805; 588649, Frdl., 20, 421; W. Mathes,
Angew. Chem., .52, 591 (1939).
[400] G. S. Sanshaw, Chem. Age, 45, 61 (1941); E. Hausmann, Erdol u.
Kohle, 7, 496 (1954); C., 1955, 1603.
[401] I. Lowenthal, J. M. Pepper, J. Am. Chem. Soc., 72, 3292 (1950).
[402] 1. J, Makarof i-Semliansky, S. t’rokin, J. prakt, Chem.,
[N. F.I, 147, 317 (1937).
[403] W. L. McEwen, Org. Synth., Coll. v. 2, 133 (1943).
[404] E. J. S mu tny, I. D. Roberts, J. Am. Chem. Soc., 77, 3420 (1955).
[405] H. L. В r a d 1 о w, С. A. V a n d e r w о r f, J. Org. Chem., 16, 1151 (1951).
[406] G. H. Cole ni an, G. E. Heneywell, Org. Synth. Coll., v. 2, 89 (1943).
(4.(3 7 j В. В. Коршак, Г. С. К о л е с и и к о в, Синтезы органических соеди-
нений. Сборник 2, Изд. АН СССР, 1952.
[108] G. W i 11 Г g, J?, Vida 1, Chem. Ber., 81, 370 (1948).
II. ОБМЕН ГАЛОГЕНА ПА КИСЛОРОД
317
пои щавелевой кислотой образуется 2-альдегидобензимидазол с выходом 80% от теоре-
тического [4091. о-Фта.та.тьдегид и пафталнн-2,3-днальдегпд можно получить нагрева-
пнем в течение 40 ч а,а,а',а'-тетрабром-о-ксилола и соответственно 2,3-ди-(дибром~
метил)-нафта.тина с водно-спиртовым раствором оксалата калия [410].
Для синтеза альдегидов или кетонов не всегда обязательно применять г₽л-дц-
1алогениды. Имеются способы, по которым карбонильные соединения легко, иногда
с отличными выходами, получаются также из моногалогенидов. Например, при при-
бавления З-хлорциилолентена-1 при энергичном перемешивании и 0° С к водному
раствору Na2Cr2O? образуется хромовый комплекс цИклопентен-1-она-З, который
разлагают до кетона, осторожно приливая к нему по каплям прп охлаждений 50%-ную
H„SO4.‘ выход 60—68% от теоретического [385], ЦиклопеНтапдион-1,2 может быть
получен добавлением по каплям при сильном перемешивании и 100° С водного рас-
твора FeCI3 к водному раствору 2-хлорциклопентанона; выход 80% от теоретиче-
ского [411].
Болео широко применим Метод Соммле [412], По этому методу из галогепметил-
арильных соединений в спирте или хлороформе по реакции с гексаметилентетрамином
образуются четвертичные аммониевые соли, которые при кипячении с водой или раз-
бавленными кислотами разлагаются с образованием альдегидов, формальдегида,
метиламина и аммиака. При ведении всего процесса в разбавленной уксусной кислоте
(1 : 1) выделение промежуточных продуктов в болыпипстве случаев пе является необ-
ходимым.
о-Толуиловый альдегид. Смешивают 60 г и -бром-о-ксилола с 250 мл
спирта, прибавляют 48 г гексаметилентетрамина (небольшой избыток) и 50 мл
воды; при встряхивании смесь разогревается и уротропин переходит в раствор.
Через 5 мин запах о-ксилолбромида исчезает; добавляют еще 200 мл воды, кипя-
тит 2 ч с обратным холодильником и перегоняют с водяным паром. Когда после
отгонки около 1,5 л дистиллята все еще мутный конденсат больше не имев!
запаха альдегида, из него экстрагируют продукт реакции эфиром, промывают
экстракт водой, сушат и фракционируют. Получается 27 г о-толуилового альде-
гида (70% от теоретического); т. кип. 86—88° С (19 мм рт. ст.).
1-Нафтальдегид [413]. Смесь 106 г 1-хлормегплнафталина, 106 г гекса-
метилентетрамина, 250 мл ледяной уксусной кислоты и 250 мл воды кипятя!
в колбе с обратным холодильником в течение 2 ч. Через 15 мин смесь гомогени-
зируется, затем начинается отделение масла. По окончании кипячения приба-
вляют 200 мл концентрированной НС1 и нагревают еще 15 мин. После охлажде-
ния продукт экстрагируют эфиром, эфирный слой несколько раз промыва»!
водой, раствором Ха2Сб3, вновь водой и сушат NaeCO3. После отгонки эфире
альдегид перегоняют при 105—107° С (0,2 мм рт. ст.) или при 160—162° С
(18 рт. ст.). Альдегид затвердевает при 0—2,5° С. Выход 70—77 г (75—8296
от теоретического).
1-Хлорметилнафта.гин и 1-нафтальдегид раздражающе действуют нс
кожу и глаза!
Следует указать на синтез 2-тиофенальдегида пз соответствующего хлорметиль-
ною соединения [414] и стильбен-4-альдегида из 4-метилстильбепа через бромметид-
стильбен [415]. При помощи реакции Соммле можно также получать м- и л-дяальдв-
гиды; о-диальдегиды по этой реакции не образуются.
Алифатические альдегиды редко удается получать таким путем, так как oat
готчас же вступают в дальнейшие взаимодействия. Однако иногда это оказываете.»
Н. Baganz, Angew. Chem., 68, 151 (1956).
А. С. Со ре, S. W. Fenton,]. Am. Chem. Soc., 73, 1672 (1951); W. Rie4
11. Bodem, Chem. Ber., 89, 711 (1956).
11. H. Inhoffen, IT. К r a m e r, Chem. Ber., 87, 493 (1954).
W. 8o mmelct, C. r., 157, 852 (1913).
S. J. Angy al, J. R. Tetaz, J. G.k Wilson, Org. Synth., 30
67 (1950).
К. B. Wiberg, Org. Synth., 29, 87 (1949).
G. Dre fa hl, W. II art г о dt, J. prakt. Chem. [4], 4, 127 (1956).
318
ОБРАЗОВАНИИ С—0-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
возможным. Выделенное четвертичное соединение алкилгалогенпда и гексаметилед
тетрамина медленно вносят в 50%-ную уксусную кислоту и тотчас же отгоняют с И
дяным паром образующийся альдегид. J
Обзорную работу по реакции Соммле см. [416],
Нренке установил, что соединения с особенно реакционноспособным галог^
ном — типа АгСН=СНСЙгС1, АгСбСНаС1, а также и АгСНаС1, образуют пиридина^
вые соли, превращающиеся при взаимодействии с n-нитрозодиметиланилином в нв
троны, на которых при кислотном гидролизе кислотами гладко образуются альдегида^
ArCHjCOCHjCl -И™™, [АгСНгСОСНг-NCsHslCl- -!
| ONC.ll.X(CHa), J
ArCHgCOCIIO Н — ArCH2COCH=N—CsH4N(CIl3)2 ' 'Я
Все эти реакции можно проводить в мягких условиях при низких температура
поэтому способ особенно полезен для получения а,В-ненасыщенных альдегидов и а
мещенных глиоксалей. Обзор реакций составлен прение [417]; там же приведя
экспериментальные данные. Рид н Бендер [418] впервые синтезировали гетероцикл
ческий о-диальдегид (тионафтендиальдегид-2,3) по способу Кренке-
Еще один общий метод превращения замещенных бензилгалогенидов в соотвс
ствующие альдегиды основан на действии натриевых солей алифатических нитропар
финов на бензилгалогониды. Особенно пригодны для этой цели натриевые соли 2-нитр
пропана и нитроциклогенсана. Они образуют с бензалгалогенидами неустойчив!
эфироподобные соединения, разлагающиеся на оксим и альдегид: '
(CH3)2C=NOCH2C6H5 —► (CH3)2C = NOHH-C6H5CHO
Например, во такому методу с выходом около 70% от теоретического удалец
получить из о-ксилилмонобромида о-толуиловый альдегид [419]. i*
3. Обмен галогена на карбоксильный кислород
Обмен галогенов в тригалогенидах на карбоксильный кислород происходи
аналогично омылению моно- и гвм-дигалогенидов. Относительно этой реакции 1
имеется новых данных. Обычно применяют известковое молоко при 50° С, спиртов®
щелочи, иислоты или ведут процесс в водной среде с FeCl3. Способ не имеет болышй
препаративного значения, так нак соответствующие исходные продукты часто трудв
доступны. В некоторых специальных случаях (хлораль; 1,1,1-тригалогенацетон) ti
дролиз происходит с расщеплением С—С-связи. t
Описан интересный синтез а-алноксиизомасляной кислоты [420] конденсации
ацетона с хлороформом в последующим, щелочным щдролиаом в присутствии спирт
продукта конденсации до кислоты: *'
(CHsJiC-CCl, (CHS)2C-COOH '
ОН OR
Гидролиз, вероятно, протекает через стадию образования эпоксисоединени!
(СНз)зС---СС12 3
''o' -i
[416] S. .T. A ng j al, Org. Reactions, 8, 197 (1954).
(4171 F. Krohnke, Angew. Chem., 65, 613 (1953).
f418] W. R i e d, H. Bender, Chem. Ber,, 89, 1574 (1956). '
[419] IL B. Hass, M. L. Bender, Org, Synth., 30, 99 (1950); [G. Vtzinge№
Ann.. 556, 50 (1943). — Прим. ped.].
[420] Ch. W v j z то a n n, M Sulzbacher, E. Bergmann, J. Am. Chenh'
Soc., 70, 1153 41948).
Ш. ОБМЕН АЗОТА НА КИСЛОРОД
319
При действии щелочей на 1,1-ди-(п-хлорфенил)-2,2,2-трихлорэтан (ДДТ) в за-
висимости от условий получают ди-п-хлорфенилуксусную кислоту или 1,1-ди-(и-хлор-
фе1Шл)-2,2-дихлорэтилен [421].
п-Бромбензойвая кислота. Получают гидролизом п-бромбензотрихлорцдц
нагреванием его в течение 5 ч с 85%-ной HsS04 при 30—50° С. Кислоту виде»
ляют, выливая реакционную массу на размельченный лед; выход 95% от тео>
ретического.
Бензойная кислота. Бензотрифторид омыляют нагреванием со 100%-ной
H2SO4 до исчезновения слоя бензотрифторида; выход бензойной кислоты 94%
от теоретического [422].
Карбоновые кислоты редко получают из их галогенангидридов. Для гидролиза
галогенангидридов их приливают по каплям при интенсивном перемешивании в ледя-
ную воду или охлажденный льдом раствор Na8CO8. Тольно в исключительных случаях
приходится применять для разложения спиртовый КОП. Ароматические галоген-
атидриды более устойчивы, чем алифатические.
III. Обмен азота на кислород
1. Обмен аминогруппы на гидроксильную группу
В алифатическом ряду аминогруппа обменивается на гидроксильную под
действием азотистой кислоты:
rch2nh8h-hno2 —► денной+N2 4-Н2о
Реакция не всегда протекает по приведенной схеме, а при дезаминировании:
/<-а.ткиламинов наряду с первичными н вторичными спиртами образуются олефины,
сложные эфиры, алкилгалогениды (при ведении процесса в соляной кислоте) и простые
эфиры (при ведении процесса в спиртах); основными продуктами реакции являются
первичные спирты (30—50%) и олефины (около 25—35%). Образования сложных
эфиров можно избежать проведением реакции в растворах фосфорной иля хлорной
кислот. Как правило, амин растворяют в рассчитанном количестве 10—15%-ной уксус-
ной кислоты, приливают по каплям в реакционную смесь эквивалентное количества
насыщенного раствора NaNO, и затем в течение нескольких часов нагревают при
80—90е С.
Для превращения Z-аспарагина в Z-яблочную кислоту Вальден [423] растворял
аминокислоту в разбавленной азотной кислоте и раствор обрабатывал окислами азота
на горячей водяной бане при частом встряхивании до полного прекращения выделения
азота. Переход а-ампнокарбоновых кислот в а-оксикарбоновые кислоты под действием
азотистой кислоты идет без изменения конфигурации асимметрического центра
молекулы.
Из циклических аминов под действием HNO2 образуются вторичные спирты
и циклоолефины *. Дезаминирование алкилциклогэксиламинов обычно происходит
с сохранением конформации. При экваториальной аминогруппе образуется цикло-
гексанол с экваториальной оксигруппой; прп аксиальной аминогруппе образуется
небольшое количество экваториального оксисоединения, а в качестве основного
продукта реакции — циклогексен [424].
В то время как 1,3- и 1,4-аминоспирты переводятся при помощи H'NO8 в основ-
ном в двухатомные спирты, 1,2-аминоспирты реагируют с одновременной перегруппи-
ровкой и чаще всего образуются кетоны:
n .,C<Olli- Clt..MI3 R'COCH2R
R ’/
• Этот процесс приводит также к сужению цикла (перегруппировка Н. Я. Демья-
нова), см. Н. Я. Д е м ь я н о в, Сборник избранных трудов, Изд. АН СССР,
1936, стр. 235—482. — Прим. ред.
[421] О. Grummi tt, A. Buck, Н. Egan, Org. Synth., 26, 21 (1946).
[422] G. M. Lefave, J. Am. Chem. Soc., 71, 4148 (1949).
|423J P. Walden, Ber., 28, 2771 (1895).
[4241 A. K. Bose, Experfentia, 9, 256 (1953).
320
ОБРАЗОВАНИЕ С- О-СВЯЗП В ОБМЕННЫХ РЕАКЦИЯХ
Подробное и наглядное представление об отношении аминов к азотистой
кислоте см. [425].
13 большинстве случаев алифатические амины менее доступны, чем соответству-
ющие оксисоединения. Поэтому превращение первичных алифатических аминов в пер-
вичные спирты имеет гораздо меньшее препаративное значение, чем превращение
первичных ароматических аминов в соответствующие фенолы.
Существует несколько путей для перевода первичных ароматических аминов
в фенолы. В некоторых случаях возможен обмен аминогруппы на оксигруппу под
действием разбавленных кислот или щелочей. Этот способ используется главным обра-
зом в промышленных процессах и не всегда пригоден в лабораторной практике. 1-Ами-
нонафталин-3,5-, 3,6- и 3,7-дисульфокислоты можно количественно перевести в соот-
ветствующие 1-оксинафталиндпсульфокислоты [426] нагреванием кислых солеи в воде
до 180° С. Кипячением 2-амиио-5-нитроанизола в течение 30 ч с водным раствором
NaOH получают 4-нитрогваякол с выходом 86% от теоретического [427].
Общеприменимым способом превращения ароматических аминов в фенолы илн
нафтолы является нагревание водных растворов ароматических диазосоединеяий
(стр. 539).
Во избежание протекания побочных реакций при разложении галогенидов
или нитратов диазония диазотирование нельзя вести в азотнокислых растворах или
растворах галогеноводородных кислот. Приходится работать с сернокислыми рас-
творами, что вследствие плохой растворимости сернокислых солей ароматических
аминов вносит некоторые усложнения.
[ArNj]+HSOJ ArOH + HaSOj + Na
Метод поясняется на нескольких примерах.
^-Нитрофенол из м-нитроанилина [428]. Охлажденной смесью 450 мл
воды и 330 мл концентрированной H3SO4 заливают при перемешивании 210 е
тонкоизмельченного ле-нитроанилина и смешивают с 800 г льда. После того как
образуется гомогенная смесь, из капельной воронки приливают в. течение 8—
10 мин раствор 105 г NaN02 в 250 мл воды до положительной иод-крахмальной
реакции, поддерживая температуру 0—5® С. Затем перемешивают 5—10 жин,
дают осесть сульфату л-нитрофенилдиазония, сливают отстоявшуюся жидкость
и для очистки соли диазония, в случае необходимости, промывают ее деканта-
цией ледяной водой. К фильтрованию прибегают только при наличии примесей
в виде крупных частиц.
Тем временем в колбе емкостью 5 л доводят до кипения смесь 750 мл
воды с 1 л концентрированной H2SO4 и к кипящему раствору из капель-
ной воронки добавляют слитую с сульфата диазония жидкость так, чтобы смесь .
в колбе энергично кипела (около 50 мин). Затем маленькими порпиямн приба-
вляют твердый сульфат диазония, избегая потерь вследствие выбросов пены.
После добавления всей соли кипятят еще несколько минус, выливают содержи-
мое колбы в горячем состоянии в большой химический стакан, охлаждаемый
проточной водой, и дают смеси охладиться при энергичном перемешивании.
В этих условиях выделяющийся в виде темного масла л-нитрофенол затверде-
вает в Мелкодисперсном виде. После охлаждения фенол отфильтровывают, воз-
можно тщательнее отсасывают осадок и промывают 450 мл ледяной воды. Неочи-
щенный продукт сушат на воздухе, выход составляет 170—180 з (81—86% от тео-
ретического). Для очистки продукт перегоняют в вакууме (т. кип. 160—165° С
црн 12 мм рт. .ст.) или перекристаллизовывают из бепзола.
Способ получения оксипроизводных из диазосоединений применим и для других,
относительно устойчивых нитродиазосоединеиий. Если соединения диазоиня легко-'
разлагаются, то их не следует выделять в твердом состоянии.
[425] IT. F г е у t a g, F. Moller, G. Pieper, H- Soli, в книге Н о u b е n -
W е у 1, Methoden der organischen Chemie, 4 ЛиП., Bd. 11/2, 1958, S. 133.
[426] FrdL, 4, 518; A. Rieche, H. Seeboth, Ann., 638, 102 (i960).
[427] N. L. D г a k e, H. С. II a r r i s, С. B. J age r, J. Am. Chem. Soc., 70,
170 (1948).
[428] К. II. F, Ma^ske, Org. Synth., 8, 80 (1928).
III. ОБМЕН АЗОТА НА КИСЛОРОД
3
м-Хлорфенол [429]. Раствор сульфата диазония, полученный обычиь
способом из .и-хлораннлина, медленно приливают по каплям к нагретой j
1-40° С смеси концентрированной H2S04 и воды (2 : 1), после чего продукт реа
кип перегоняют с водяным паром, экстрагируют эфиром, сушат над Na2S<
и перегоняют в вакууме; т. кип. 214е С; выход 67% от теоретического.
Общеприменимый путь синтеза фенолов оппсан на примере превращения 3-бро,
4-а.минотолуола в 2-бром-п-крезол [430].
2-Бром-га-крезол. В раствор амина в серной кислоте при перемешивай!
и температуре 0—5е С к течение 15 мин вводят по каплям под поверхность жи
кости иодный раствор NaNO2, перемешивают еще 5 мин и последовательно см
шпвают с водой, мочевиной и льдом. Полученную массу в несколько приему
добавляют к нагретому до 130—135° С раствору 150 г Na2SO4, 200 г II2SO4
100 мл воды; выход 80—92% от теоретического.
Аналогичное превращение м-ампнобензальдегида в лг-оксибензальдег®
ВМ. [431].
Описан [432] способ перевода галогенидов диазония в фенолы в присутствй
фосфорной кислоты. При обработке фторборатов диазония ледяной уксусной кислоте
образуются ацетилоксисоединения, которые дезацетилирутот обычным путем [433
Относительно устойчивы трифторацетаты диазония, из которых нагреванием равны
образом можно получать фенолы с Хорошими выходами [434].
Аминогруппы в псевдоароматических гетероциклических соединениях тон
были заменены во многих случаях на гидроксильные группы через соответствующе
диазоеоединения. Таким образом, в частности из гуанина, был получен ксантин [435
В большинстве случаев аминогруппу в а- и ^-нафтиламинах можно замени^
оксш руппой действием бисульфита натрия (реакция Бухерера; см. также стр. 495
XaHSO*
C10H7NH2 + H2O : CioII7OH + NH3
Нафтиламин обрабатывают 4—8-кратным избытком NaHSOg в течение 6—30
(продолжительность реакции различна для разных аминов) при 90—150° С в водно
среде; при этом образуются .м-тетралонсульфокислоты. Сульфокислоты устойчив
в кислой и нейтральной среде, но под действием щелочей разлагаются на сульфи
и нафтол [438]. Выходы почти количественные. Таким путем можно перевести такж$
5-аминохинолин в 5-окснхинолин [436]. Если в мета-положении к аминогруппе нахе
дится сульфогруппа, реакция не идет. Обзор по реакции Бухерера составлен Дрейко
[437]. Новые исследования реакции Бухерера проведены Рихе и Зееботом [438]. Автор-
выяснили механизм реакции и приводят богатую библиографию *.
Омыление амидов карбоновых кислот происходит подобно омылению эфиро
при кипячении со щелочами или сильными кислотами. Например, нагревание в течена
30 мин бепзоилглицил-2>£-лейцинамнда с 1 н. раствором NaOH при 100° С получаю
бен.юилглицил-Z)L-лейцин с выходом 68% от теоретического [439]. Для омылена
Вопросам механизма реакции Бухерера посвящены классические исследовали.
Н. II. Ворожцова ст. (И. Н. Ворожцов, Основы синтеза промежуточны,
продуктов и красителей, Госхимиздат, 1955, стр. 411—416). — Прим, рес
А. Б. Holleman, I. J. R i n k e s, Rec. trav. chim., 30, 81 (1911).
II. E. Ungnade, E. F. Orwell, Org. Synth., 23, 11 (1943).
R. N. I eke et al., Org. Synth., 29, 63 (1949).
H. Brockman n, A. D orlars, Chem. Ber., 85, 1180 (1952).
L. E. Smith, H. L. II a 11 e r, J. Am. Chem. Soc., 61, 143 (1939).
M. R. Pettit, M. Stacey, J. C. Ta flow, J. Chem. Soc., 1953, 3081
F. Korte, II. Barkemeyer, Chem. Ber., 89, 2404 (1956).
E. B. Hartshorn, S. L. Baird jr., J. Am. Chem. Soc., 68, 1562 (1946^
N. L. Drake, Org. Reactions, 1, 105 (1942).
A. R jpclie, H. 8 e e b о t h, Ann., 638, 57; 66 (I960).
Th. Wieland, H. Fritz, Chem. Ber., 86, 1198 (1953).
322
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
нерастворимых амидов рекомендуется употреблять смесь соляной р ледяной уксус-
ной кислот.
Применяя для трудногидролизуемых амидов обработку азотистой кислотой
по Буво, их можно перевести с хорошими выходами в карбоновые кислоты [440].
Трифенилуксусная кислота [441]. При слабом нагревании растворяют
0,2 г тонкомзмельченного трифенилацетиламида в 2 г концентрированной H2SO4,
охлаждают раствор льдом и к нему очень медленно приливают охлажденный
льдом раствор 0,2 г NaNO2 в 1 мл воды. Затем постепенно нагревают на водяной
бане. При 60—70° С начинается энергичное выделение азота, которое заканчи-
вается при 80—90° С; смесь нагревают еще несколько минут (но не больше)
на кипящей водяной бане. После охлаждения трифенилуксусную кислоту оса-
ждают ледяной водой и перекристаллизовывают из ледяной уксусной кислоты;
т. разл. 255° С-
Об омылении амидов см. также [442, 443].
Если азотистая кислота при этом может оказать нитрозирующее действие,
то этот метод не может быть использован.
Для превращения амидов в карбоновые кислоты рекомендуется также при-
менять трехокись азота N2O3. Например, для получения трибутилуксусной кислоты
пропускают в охлажденный льдом раствор трибутилацетамида в ледяной уксусной
кислоте N^Oj, оставляют смесь на ночь и затем 2 ч нагревают на паровой бане; выход
80% от теоретического [444]. Описано превращение амида глюкозокарбоновой кислоты
под действием жидкой N„O3 в соответствующую карбоновую кислоту [445].
2, Обмен азота на карбонильный кислород
Обмен азота на карбонильный кислород имеет очень небольшое препаративное
значение. Используемые при атом методы не всегда применимы.
Джонсон и Дегеринг [446] получали альдегиды и кетоны из первичных и вто-
ричных алифатических нитросоедипенпй, для чего проводили гидролиз натриевых
солей их ациформ.
, При действии сухого НС1 на нитрилы в абсолютном эфире или в смеси эфира
с хлороформом образуются имидхлориды, продукты восстановления которых гидро-
литически расщепляются SnCl2 с образованием альдегидов. Способ, по-вндимому,
может с успехом применяться только для получения ароматических ини гетероцикли- ;
ческих альдегидов; наибольшее значение для реакции [447] имеет активность упо-
требляемого для восстановления SnCl2. Этим путем синтезированы р-нафтальдегид ’
(выход 95% от теоретического) [447] и 4-метилтиазол-5-альдегид (выход 40% от тео- '
ретического) [448]. ’
Об аналогичных превращениях нитрплов в альдегиды сообщает Хенле [449],
который использует вместо SnCl2 амид натрия п ведет реакцию в присутствии фенил- ".
гидразина. При этом образуются фенилгидразоны, расщепляемые обычным путем д
в альдегиды. '
Аналогичным способом был получен о-толилальдегид [450]: по реакции с РС16
анилид о-толуиловой кислоты превращали в пмпдохлорид, который затем растворяли 'i
[440] М. L. Bouvcaiilt, Bull. Soc. chim. Franco [3], 9, 368 (1893). ?
[441] G. Heyl V, Meyer, Ber., 28, 2783 (1895). *
[442] J. J. Sudborough, Ber., 28, Ref. 917 (1895).
[443] V. Meyer, W.Molz, Ber., 30, 1279 (1897).
[444] N. Sperber, D, Papa, E. Schwenk, J. Am. Chem. Soc., 70, 3093 /1
(1948). |
445] R. Grewe, G. R ockstroh, Chem. Ber., 86, 546 (1953). |
446] K. J о h п s о n, E. F. D egering, J, Org. Chem., 8, 10 (1943). Vs
447] W. Williams, Org. Synth., 23. 63 (1943).
448] C. R. H ari л gt on, R. C. G. Moggri dge, J. Chem. Soc., 1939, 445. ]
449] F. Henle, Ber., 35, 3039 (1902); 38, 1362 (1905).
450] W. Williams С. H. Witten, J. А. К г у n i t s k y, Org. Synth,, 1
26, 97 (1946). ' 1
1П. ОБМЕН АЗОТА НА КИСЛОРОД
в эфирном растворе SnCls, насыщенном сухим НС1:
/СИз Z\ZCHs
| h PC). t Y fl snch: нс)
X- XcO.NHCeHs ^C—NCeH5
I
Cl
^\zCHa z\zCH*
—> ] Y HCEH.o _ f \( _|_ceHjNB
XZ\c=NCeH6 ^/XCHO
[
H
При последующей перегонке с водяным паром образовавшийся аяил расп
плялся на альдегид и анилин. Таким же способом был получен fi-ионилнденацетаа
дегид [451], причем SnCl5 был заменен эфирной суспензией СгС12.
Общий метод превращения нитрилов в альдегиды заключается в действии натри
этоксиалюминийгидрида на нитрил. Этим способом после 1 — 2 ч взаимодейста
3-цианпиридина с натрийэтоксналюминийгидридом в тетрагидрофуране был получ
лиридин-з-альдегид; выход 83% от теоретического [452]. Еще лучшие результат
получаются при употреблении литийэтоксиалюминийгидрида; процесс ведут в эф®
ном растворе при 0° С. Алифатические альдегиды образуются с выходами 70—80*
ароматические — с выходами 80—90% от теоретического [453].
Уместно указать и на получение альдегидов из N-метиланилидов иарбояов]
кислот [454]. Последние переводятся алюмогидридом лития в альдегиды часто с xoj
шпми выходами. Восстановление * лучше всего проходит в тетрагидрофуране, в 6oj
шинстве случаев при 0® С.
Еще один простой метод получения альдегидов состоит в том, что каталитих
ское гидрировянио нитрилов останавливают на промежуточной стадии образовав:
альдимина, для чего наиболее пригоден семикарбазид. По этому способу удало<
в частности, получить альдегиды из производных бензилцианида с хорошими выз
дами [455]. Образующийся семикарбазоп фенилацетальдегида расщепляют раствор,
формальдегида.
По одной из модификаций реакции Соммле (см. стр. 317) первичные н вторичн:
бензиламины превращают в альдегиды при помощи формальдегида и гексаметиле
тетрамииа. Например, Греймор и Девис получили этим путем бензальдегид из гид{
хлорида бензиламина [456]. Гидрохлорид 2-аминометилфлуорена при таком же СЕ
собе дает флуорен-2-альдегид с выходом 70% от теоретического [457]. О вэаимодейств:
третичных аминов с гексаметилентетрамином см. работы [458, 459].
Другим путем, позволяющим осуществлять превращение первичных алифа*а
ческих аминов в альдегиды, по-видимому, является воздействие на амин кисиоро
воздуха в присутствии порошкообразной меди [460]; реакция еще мало изучена.
Превращение алифатических нитросоединений в кетоны известно как реакщ
Нефа. Оно протекает с промежуточным образованием ациформы нитросоединенд
* Особенно хорошие результаты получаются прп использовании этиленимяд
карбоновых кислот [II. Brown, A. Tsuka m о t о, J. Am. Chem. Soc., 5
2016, 4549 (1961)]. — Прим. ред.
[451] R. Kuhn, С. Morris, Ber., 70, 857 (1937).
[452] G. Hesse, R. Scbrodel, Angew. Cnem., 68, 438 (1956).
[453] H. C. Brown, C. J. Shoaf, C..P. Garg, Tetrahedron Letters, №
9 (1959).
[454] F. W e у g a n d et a]., Angew. Chem., 65, 525 (1953).
[455] H. 1’lieninger, G. Werst, Angew. Chem., 67, 156 (1955).
[456] J. Greymore, D. R. Davies, J. Chem. Soc., 1945, 293.
[457] S. J. Angya], G. B. Barlin, P. C. Wailes, J.'Chem. Soc., 195
3512.
[458] H. R. Snyder, S. S wa m i и a t Iran, H. J. Sims, J. Am. Chem. So»
, 74, 5110 (1952).
[459] M. M. Robinson, B. L. Robinson,!. Am. Chem. Soc., 77, 457 (1951
[460] С. И. Шуннина, ЖОХ, 7, 983 (1937); С., 1937, 11, 3152.
324
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
Например, для получения З-ацетплцпклогексанона [461] растворяют 3-(1-нптроэтил)-
циклогексанон в разбавленном растворе NaOH, прибавляют при 0° С при перемеши-
вании 30%-ную II2SO4 и медленно нагревают сначала до комнатной температуры, а
затем до 55u С.
Карбонильные соединения редко получают из оксимов и гидразонов прямым
путем. Однако эти производные представляют интерес для очистки и выделения аль-
дегидов и кетонов.
Легче всего расщепляются производные кетонов, немного труднее — производ-
ные ароматических альдегидов; расщепление производных алифатических альдегидов
происходит в наиболее жестких условиях. Оксимы расщепляются легче, чем гидра-
зоны. Гидролиз простых оксимов и гидразонов протекает гладко, в то время как при
расщеплении изонитрозокетонов иногда возникают затруднения. Гидролиз ведут
в соляной или разбавленной серной кислотах и для повышения растворимости иногда
добавляют спирт. Для расщепления легко разлагающихся карбонильных производных
с успехом применяются щавелевая, фталевая или уксусная кислоты. Общеприменимый
метод предложен Тиманном [462]. Он заключается в том, что оксимы пли семикарба-
зоны летучих с паром альдегидов перегоняют в токе водяного пара с фталевым анги-
дридом: выходы почти количественные.
В качестве примера кислотного гидролиза следует указать па превращение
мопооксима фенилметплдикетона при помощи водной H2SO4 в дикетон [463]. п-Хи-
нонокепмы с хорошими выходами гидролизуются [464] в водном растворе СиС12. 2,4-Ди-
нитрофенилгидразоны очень гладко расщепляются в присутствии СиСО3 80%-ноц
или более концентрированной муравьиной кислотой [465] или раствором левулиновой
кислоты (9 объемов) в 1 н. соляной кислоте (1 объем) [466], иногда в присутствии орга-
нического растворителя для повышения растворимости. Последний метод оказался
также вполне пригодным для гидролиза оксимов. Для расщепления соединений Жи-
рара рекомендуется [467] применять 0,5—2 н. НС1. Расщепление происходит при
комнатной температуре в течение 12—16 ч.
При нагревании фенилгидразона бепзоина в ледяной уксусной кислоте в тече-
ние 2 ч получают бензил с выходом 77% от теоретического [468]. Образующийся при
атом фенилгидразин оказывает дегидрирующее действие на вторичную оксигруппу.
Оксимы очень мягко окислительно расщепляются также азотистой кислотой или изо-
амилнитритом па холоду. Этот способ особенно рекомендуется для выделения не-
устойчивых альдегидов. Таким путем был получен янтарный диальдегид [469].
Янтарный диальдегид. Суспендируют 30 г диоксима янтарного диальде-
гида в 40 мл диоксана, суспензию охлаждают льдом и к ней в течение примерно ।
40 мин прибавляют 45—50 мл этилнитрита с такой скоростью, чтобы темпера-
тура все время оставалась в пределах 5—15® С, до тех лор, пока от добавления
новых порций нитрита не перестанет повышаться температура. Смесь оставляют
на почь и затем фракционируют в токе СО2 при разрежении 13 мм рт. ст.
Сначала отгоняется диоксан, а при 67° С перегоняется 13—14 г мономерного
альдегида, что соответствует выходу около 60% от теоретического. Мономерный
диальдегид можно довольно долго хранить без доступа влаги; постепенно он
превращается в стекловидный полимер, из которого при перегонке вновь выде-
ляется мономер.
Глутаровый дпальдегпд получают из диоксима [470] с выходом 85 — 95%
от теоретического, дейеюуя па диоксим NaNO2 в солянокислой среде при тем-
пературе от —10 до — 5Г' С. •
461
462
463
464
465
466
467
468]
469]
470]
Л. McCoubrey, J. Chem. Soc., 1951, 2933.
F. Tiemann, Ber., 33, 3721 (1900).
W. W. Hartmann, L. J. Roll., Org. Synth., 23, 1 (1943).
W. T. Su me r f or d, D. N. Dalton, J. Am. Chem. Soc., 66, 1330 (1944).
R. Robinson, Nature, 173, 541 (1954).
С. II. De P u у, B. VV. Ponder, J. Am. Client. Soc., 81, 4629 (1959).
E. Lederer, G. Nachmias, Bull. Soc. chim. France, 1949, 400.
W. Theilacker, P. Tros ter Ann., 572, 144 (1951).
C. Mannich, H. Budde, Arch. <1. 1’harm., 270, 283 (1932).
A. C. Cope et* al., J. Am. Chem. Soc., 73, 3416 (1951).
F
III. omn АЗОТА НА КИСЛОРОД
325
Этот метод [471] применим также для обработки ссмикарбазонов.
Для получения чувствительных альдегидов, особенно в ряду углеводов, с успе-
хом используется особый вид «переэтерификации», прп которой желаемое карбониль-
ное соединение вытесняется из его производного другим карбонильным соеди-
нением:
RCH=NR' + R’CHO R'CH=NR'-kRCHO
В качестве «вытесняющих компонентов» применяют бензальдегид, 2,4-дипитро-
бензальдегид, формальдегид и пировиноградную кислоту. Формальдегид вызывает
нежелательные последующие реакции. В химии углеводов применяют только бенз-
альдегид. Примером реакции разложения бензальдегидом может служить способ,
описанный Фишером [472].
Гдлагептоза. Растворяют 10 г фснилгидразона галагептозы в 400 мл
горячей воды и встряхивают с 5 мл чистою бензальдегида. После почти подлоге
выделения маслянистого продукта реакции и осветления жидкости в ней рас-
творяют при нагревании еще 6 г фенилгидразона галагептаноэы и встряхивают
с 2,5 мл бензальдегида. Эту операцию повторяют еще раз. Затем всю смесь нагре-
вают 15 мин, дают ей охладиться, отфильтровывают выделившийся фенилгидра-
зон бензальдегида и извлекают из фильтрата эфиром непрореагировавшие
бензальдегид и бензойную кислоту. Обесцвечивают водный раствор животным
углем, фильтруют и полностью отгоняют в вакууме растворитель. Галагептоза
остается в виде бесцветного сиропа.
Аналогичным способом была получена 3,5-монометилен-7?-ксилоза
[473].
Озазоны также расщепляются этим способом. По Э. Фишеру и Арм-
стронгу [474], растворяют 20 г фенилмальтозазона в 1600 мл кипящей воды
и энерючно перемешивают с 16 ч. чистого бензальдегида. Если бензальдегид
достаточно тонко распыляется, реакция продолжается около 20 мин. Пс
охлаждении смеси отфильтровывается фенилгидразон бензальдегида, обра-
зующийся почти в теоретическом количестве, и поступают далее, как описана
выше.
Для получения карбонильных соединений из соответствующих ссмикарбазонон
в ряду стероидов обменное взаимодействие проводят предпочтительно с пировиноград-
ной кислотой [475].
Расщепление шиффовых основапий в карбонильные соединения и амины про-
исходит аналогично гидролизу оксимов и гидразонов. Протекание реакции зависит
от основности амина и реакционной способности альдегида, па которые разлагается
азометин. Гидролиз можно проводить в относительно мягких условиях, например
при кратковременном нагревании с разбавленными минеральными кислотами, или
ингида в разбавленных спиртовых или уксуснокислых средах. Продукты кон-
денсации аминов с альдозами разлагаются ужо в присутствии следов кислот. Шиф-
фовы основания обычно не разлагаются щелочами. Так же как в оксимах и гидра-
зонах, альдегидный остаток шиффового основания можно заместить остатком
другого
[476].
альдегида.
Для этой цели предпочтительно применяется формальдегид
И71] Е. Р. 011 ю1 о el al., J. Am. СЬеш. Soc., 78, 1736 (1956).
[472] E. Fischer, Ann., 288, 145 (1895).
[473] O. Th. Schmidt, G. N ieswand t, Chern. Ber., 82, 5 (1949).
[474] E. Fischer, E. F. Armstrong, Ber., 35, 3142 (1902).
[475] 1). T a u 1), R. D. П о f f s о m ш e r, N. L. Wen dler, J. An
Soc., 79, 454 (1957).
[476] R. Adams, G. H. С о 1 e ш a n, Org. Synth., 2, 17 (1922).
Chem.
326
ОБРАЗОВАНИЕ С—О-СВЯЗИ В ОБМЕННЫХ РЕАКЦИЯХ
3. Обмен азота на карбоксильный кислород J
Получение карбоновых кислот из обычно легко доступных нитрилов имеете
большое значение для соединений алифатического, ароматического и гетероцикла
лического рядов. Омыление нитрилов проводится в жестких условиях, например дей^Я
ствиеы сильных кислот или щелочей. Применять ли омыление кислотами или щелочами,
нужно решать в каждом отдельном случае. Предпочтение следует отдавать гидролпк1
кислотами, особенно концентрированной ПС1. Для лучшего растворения нитрила оод
ление часто проводят в присутствии уксусной кислоты. С той же целью щелочной
омыление иногда ведут в присутствии спирта или пиридина, иногда под давление^
Первичные цианиды омыляются легче, чем вторичные и третичные, а алифатические &
легче, чем ароматические. Последние остатки нитрила или образующегося в качеств
промежуточного продукта амида не всегда легко отделяются от реакционной масс*
В некоторых случаях омыление ведут только до стадии амида кислоты и завершай
гидролиз другим способом, например взаимодействием с азотистой кислотой [477]
Для перевода алифатических динитрилов в дикарбоновые кислоты целесообразв
применять концентрированную HCL Так поступают при получении глутаровой кЩ
лоты из динитрила глутаровой кислоты [478]. Ароматические нитрилы, напрнмф
o-толунитрил, омыляют 75%-ной H2SO4.
о-Толуиловая кислота [479]. В колбу с мешалкой и обратным xojKJ
дильником заливают 3 кг 75%-ной H2SO4 (d = 1,67), нагревают до 150° С и п&
перемешивании в течение 2 ч прибавляют 1 кг о-толунитрила. Перейешивап
массу еще 2 ч при 150—160° С, затем повышают температуру до 190° С и продо^
жают перемешивание еще 1 ч. Полученную кислоту высаживают водой, nepj
водят в натриевую соль (нерастворимый амид толуиловой нислоты отфильтрф
вывают), подкисляют и перекристаллизовывают из бензола. Выход от 930 Д
1030 г (80—89% от теоретического); т. пл. 102—103е С. .
Трудноомыляемые нитрилы, например о-дизамещеиные бензонитрилы, былв
с успехом переведены в карбоновые кислоты 100%-ной фосфорной кислотой [48QI
в то время как полифосфорная кислота омыляет простые ароматические и алиф&^Й
ческие нитрилы (110° С; 1 ч) с хорошими выходами только до амидов кислот [48!Й
Фенилуксусная нислота. Фонилацстонитрил омыляют до фенилуксусясй
кислоты [482] разбавленной H2SO4. А
На 700 г бензилцианида берут 840 мл концентрированной H8SO4 и 1150 4
воды. Реакция заканчивается после 3 ч нагревания и перемешивания. Выхсб
кислоты 630 г, что составляет 77,5% от теоретического. 4
Для получения необладающей запахом фенилуксусной кислоты (ДЙ
парфюмерии) лучше применять способ Манна [483], по которому омылейи
проводят не серной кислотой, а щелочью. Бензилцианид нагревают в вода?
спиртовом растворе КОН до полного прекращения выделения аммиака, отгоняй
спирт, из охлажденного водного раствора экстрагируют эфиром непрореаги^
вавшие бензилцианид и фенидацетамид, водный слои подкисляют и обоабатЫЯ
вают далее, как обычно. . 3
Перед перекристаллизацией фенилуксусную кислоту лучше очищай
перегонкой в вакууме; т. пл. 76,5° С.
Алифатические нитрилы ле1че всего омыляются щелочами. А;
[477
[478
[479
[480
[481
[482
[483
S. 8 а г е 1, М. 8. Newman, J. Am. Chem. Soc., 78, 5418 (1956),
С. S. Marvel, W. F. Tuley, Org. Synth,, 5, 69 (1925).
Я, T. Clarke E. R. Taylor, Org. Synth., Coll. v. 2, 588 (1943).
G. Berger S. C. J. Olivier, Rec. trav. chim., 46, 600 (1927).
H. R. S n у der, С. T. Elstoi). J. Am. Chem. Soc., 76, 3039 (1954).
R. Adams, A. F. Thai, Org. Synth., 2, 63 (1922).
W. M a n n, Ber., 14, 1645 (1881).
Ш. ОБМЕН АЗОТА НА КИСЛОРОД
32’
Маргариновая кислота из цетилцианида [484]. В колбе с обратным холо
дольником кипятят 30 г цетилцианида (нитрил н-гексадецнлкарбоновой кислоты
с 60 мл спирта. 15 мл воды и 10 г КОН в течение 31 ч до полного прекращен»!
выделения аммиака. Остающуюся после отгонки спирта соль разлагают НС1
выделившуюся маргариновую кислоту хорошо промывают водой и сушат npi
умеренном нагревании. После двукратной перекристаллизации из иетролей
ного эфира она плавится при 61,2° С; выход 78,5% от теоретического.
Синтез малоновой кислоты щелочным омылением циануксусной кислоты см
[485]. Превращение ацилцианидов в а-кетокарбоновые кислоты происходит при много
суточном их выдерживании в холодной концентрированной соляной кислоте [486J
Циангидрины переводятся концентрированной НС1 в а-оксикарбоновые кислоты
При щелочном омылении большая часть циангидрина расщепляется до исходны:
продуктов. Синтез миндальной кислоты из нитрила см. [487].
Аминонитрилы, полученные действием NH8 и HCN или NH4C1 и biaCN на аль
дегиды или из NH8 и циангидринов, лучше всего омыляются концентрированной НС1
При этом аминокислоты получаются в виде гидрохлоридов, которые переводят в сво
бодные кислоты с помощью PbO, NH3 или пиридина. Критический обзор реакци!
гидролиза аминонитрилов см. [488]. Синтез DL-аланина см. [489, 490]. Полученне2>£-а
амиио-а-фенилпропионовой кислоты из ацетофенона, NaCN и NFIaCl через соответ
ствующий аминоцианид см. [491]. Для получения ^-аланина прибавляют Р-амино
пропионитрил при 90—95° С в течение 40 мин к раствору Ва(0Н)2 и выдерживаю-
еще 40 мин при этой температуре; выход 85—90% от теоретического [492].
Как уже упоминалось выше, омыление нитрилов до карбоновых кислот в неко
торых случаях удобнее проводить косвенным путем. Пфейффер и Маттой [493] уста
повили, что при пропускании хлористого водорода в горячий раствор цианстнльбеи:
в метиловом спирте циапгруппа превращается в карбоксиметильную. Нитрильпаз
группа может быть переведена в сложноэфирную в один прием кипячением нитрил;
в спирте в присутствии хлористого водорода или серной кислоты. При введении НС
в качестве промежуточных продуктов образуются гидрохлориды имидоэфиров [494]
иадролизующиеся водой до эфиров карбоновых кислот и NII4C1. Дренфал с сотр. полу
чил гидрохлорид 4-амино-4'-карбметоксистильбена кипячением в течение 2 ч 4-аце
гпламино-4'-цианстильбена в насыщенном ИС1 метиловом спирте [495]. По даиньп
Шпигеля [496], на 1 моль нитрила следует вводить до 10 моль спирта и 1 моль H2SOf
результаты, получающиеся при нагревании на водяной бане, не всегда удовлетвори
тельны. Теоретические выходы получают при работе в запаянных трубках.
Описай [497} простой метод превращения нитрилов в амиды кислот действие]
на нитрил 3—30%-ной H2SO4 в щелочной среде при температуре около 50° С. Эти
способом получен амид никотиновой кислоты из p-цианпиридина, трудно омыляемог
другими способами [498]. Алифатические диннгрилы от дивитрила янтарной кисиота
и до динитрила пробковой кислоты, ароматические динитрнлы, динитрилы терефтале
[484] К. L. Shriner, J. И. Fulton, D. Burks jr., J. Am. Chem
Soc., 55, 1496 (1933).
’48.3] N. Weiner, Org. Synth., Coll. v. 2, .376 (1943).
[486] T. S. Oakwood, C. A. W e i sg e r b c r, Org. Synth., 24, 14, 16 (1944)
[487] В. B. Corson et al., Org. Synth., 6, 58 (1926); Gattermann -Wie
land. Die Praxis des organischen Cheniikers, 35 Aufl., 195.3, S. 198.
[488] W. Cocker, A. Lapworth, J. Chem. Soc., 1931, 1391.
|489] Gattermann-W island, Die Praxis des organischen Chemikers
35 Aufl., 1953, S. 200.
1490] E. C. Kendall, B. F. McKenzie, Org. Synth., 9, 4 (1929).
[491] R. E. Steiger, Org. Synth., 24, 9 (1944).
[492] j. H. Ford, Org. Synth., 27, 1 (1947).
[493] P. Pfeiffer, К. M a t t о n, Ber., 44, 1115 (1911).
[494] A. Pinner, Die Iminoather und ihre Derivate, Verlag Robert Oppenheim
Berlin, 1892.
[495] G. D rel a hl, H. S e e b о t h, W. Degen, J. prakt. Chem. [4], 4, Ю
, (1956).
[496] L. Spiegel, Ber., 51, 296 (1918).
[497] C. R. Noll er, Org. Synth., Coll. v. 2, 586 (1943).
[498] A. Georg, P. Bachmann, Helv. chim. acta, 26, 358 (1943).
328
ОБРАЗОВАНИЕ С—О-СНЯЗИ И ОБМЕННЫХ РЕАКЦИЯХ
вой и изофталевой кислот, а также 2,6-днцланпирид»1н омыляются при 60—95е С. до
амидов (о-цианкарбоцовых кислое при ио.м<лцп сильноосновных ионообменных смол
па основе полистирола (499].
IV. Обмен сульфогруппы на гидроксильную группу
Обмен сульфогруппы на гидроксильную является важнейшим методом синтеза
фенолов. Применяемый для этого метод щелочного плавления сульфокислот не всегда
проходит гладко по схеме:
HSO3K+KOH — > K2SO^ROH
Известно, что наряду с нормальной реакцией обмена часто происходит окисле-
ние, классическим примером которого является превращение антрахинон-₽-сульфо-
кислоты в ализарин.
Несмотря на подобие в действии щелочей КОН и NaOH они по-разному реаги-
руюг с сульфокислотами. Так, при действии КОН бензолсульфонат калия почти коли- ;
чественно дает фенол, а при действии NaOH — лишь частично. Однако при превраЩе-
нии бензолтрисульфокислоты во флороглюцин [500] поведение оснований как раз
обратное: по реакции с КОН удается заместить только две сульфогруппы, а по реакции
с NaOH, хотя и с плохим выходом, замещается и третья сульфогруппа.
При соответствующем подборе температурного режима и необходимой концен-
трации щелочи часто удается с помощью КОН и NaOH провести последовательный
обмен нескольких сульфогрупп. Так, в зависимости от концентрации щелочи, из л-бен- v
золдисульфокислоты получают л-оксибензолсульфокислоту (501] пли резорцин [5o2Jv
Реакцию проводят следующим образом: в плавильном тигле из серебра, никеля, меди •
или железа к расплавленной смеси КОН или NaOH с небольшим количеством воды •
при 200—300° С в несколько приемов добавляют сульфонат. Подробно разработан- * 1
ный синтез р-нафтола приведен в книге Гаттормана [503]. О получении n-крезола см.
[504]. 3-0ксипиридин получен из пиридин-3-сульфокислоты с выходом около 80% •:
от теоретического [505].
1,8-Диоксинафталин ]50б]. В серебряном тпгле сплавляют 60 г КОН ;
с 20 мл воды и 14 г ангидрида 1-нафтол-8-сульфокислоты (нафтосультон), плотно
закрывают тигель и нагревают при 200—300°С и перемешивании еще 15—2о мин.• v
Потемневшую реакционную массу охлаждают, разлагают 300 мл 13%-нои HCI,
прибавляют такое количество концентрированной НС1, чтобы полностью пре-
кратилось бурное выделение газа, и нагревают, добавляя воду (около 1 л), >
причем все переходит в раствор, за исключением небольшого количества смолы. .
При охлаждении фильтрата выкристаллизовывается 6 г неочищенного 1,8-диокси-
нафталина; из маточника извлекают эфиром дополнительно еще 2 г продукта. >-
После перекристаллизации из воды, в которой растворепо некоторое количество
SO2, получается продукт с т. пл. 140° С.
Метод обмена меркаптогруппы на гидроксильную заключается во взаимодей-
ствии меркаптанов с хлоруксусной кислотой, в результате чего образуется простой _
тиоэфир. Тиоэфиры иногда гидролизуются в оксисоединения и тиогликолсвые кислоты.?
уже горячей водой, а иногда только под действием концентрированной соляной
кислоты. ,
п.'11С|С||2соон _ЦС|* BSCH2COOH ROH-HSCH.C.00H
* 3
Этим путем может быть получен 2-окси-4-аминопиримидип (цитозин) из 2-мер-
капто-4-аминопиримидина [507]. О
[499
[500
(501
(502
[503.
[504]
[505]
(506J
1507]
С. Bert he г, Спеш. Вег., 92, 2616 (1959).
L. Barth, J. Schreder, Вег., 12, 422 (1879).
F. Willson, К. Н. Meyer, Вег., 47, 3162 (1914). 2
Н. Е. F i е г z - D a v 1 dt G. Stamm, Helv. chim. acta, 25, 368 (1942).
Ga t tor m a nn - W i e] an d, Die Praxis des organischen Chemikers, 35 '
Aufl., 1953, S. 208.
W. W. Hartmann, Org. Synth., 3. 37 (1923).
H. Furst, II. J. Dietz, J. prakl. Chem. [4], 4, 154 (1956). )
H. Erdmanfi, Ann., 247, 356 (1888),
G. II. Hi tchings, P, B. Russ < I, J. Chem. Soc., 1949, 2454.
] ПОЛУЧЕНИЕ ПРОСТЫХ ЭФИРОВ
ОБРАЗОВАНИЕ НОВЫХ С—О-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ
I. Получение простых эфиров
1. Получение простых эфиров отщеплением воды
от двух молекул спирта
Простые эфиры можно получать каталитически отщеплением воды от двуз
молекул спирта:
r—он-; НО—R —> R—О-В+Н2О
Эфиры образуются таким путем легче всего из третичпых спиртов, труднее
из первичных. Реакцию почти всегда проводят в присутствии катализаторов дегидра-
тации. При этерификации в жидкой фазе применяют кислотные катализаторы, преиму
щественно концентрированные минеральные кислоты, а также органические сульфо-
кислоты, кислые соли и галогенсодержащие соединения, например BF3, ZnCl2, А1С1а
При этерификации в газовой фазе пары спиртов пропускают над твердыми катализа
горами (Л12О3, Ti02. обезвоженные квасцы и т. п.) [508].
Некоторые спирты, например трифепилкарбииол и даже бензгидрол, этерифи:
цируются уже при перекристаллизации из метилового или этилового спирта в при:
сутствии следов кислот, оставшихся после их сиптеза. Фторфенол превращается в соог
ветствующий эфир уже при кипячении в колбе с обратным холодильником. Эфирь
низших алифатических спиртов обычно получают нагреванием смеси спирта с cepHOj
кислотой. В этом методе необходимо тщательно соблюдать установленный режим
реакции, особенно температуру, так как очень легко образуются олефины.
Диэтиловый эфир [509]. Нагревают при 140° С этпловый спирт (2 вес. ч.
с концентрированной H2SO4 (9 вес. ч.) Температура реакции может быть пони,
жена добавлением 9—10% безводного сульфата алюминия [510]
В этих жестких условиях эфиры с хорошими выходами получают только и-
сиирсов с длиной цепи Ct—С3; спирты с большим числом атомов углерода дают глав
ным образом олефины [511—513]. Одпако по модифицированному способу, при кото
ром поддерживалась точно установленная температура реакции и непрерывно отгоня
лось рассчитанное количество воды, Фогелю [511] удалось приготовить эфиры и И:
высших спиртов (до С8).
Несимметричные алифатические эфиры получаются с хорошими выходами до
гидратацией соответствующих спиртов только в том случае, если вторым компонентов
реакции является третичный спирт [514]. В качестве конденсирующего агента чащ»
всего применяют серную кислоту.
'прет-Бутилэтиловый эфир [515]. К кипящей смеси 2 моль этиловог,
спирта с 2.5-юэатным количеством 15%-ной H2SO4 постепенно прибавляю-
1 моль mpem-рутилового спирта и непрерывно отгоняют азеотропную смем
эфира с водой; выход 95% от теоретического.
mpem-Бутилизопропиловый эфир (514]. При получении этого спирте
замена 15%-ной H2SO4 водным раствором l\aIISO4 значительно увеличивав*
выход.
[508] F. К 1 a g е s, в книге G. М. Schwab, Handhuch der Katalyse, Bd. 7/2
Springer-Verlag, Wien, 1943, S. 292.
1-509) A. Williamson, Ann. Chinlie [3], 40, 98 (1854).
[510] J. B. Senderens, С. r., 151, 392 (1910).
[511] A. I. Vogel, J. Chem. Soc., 1948, 616.
[512] L. M. Norton, С. O. Prescott, Am. Chora. J., 6, 245 (1884).
[•’>13] G, Schroeter, W. Sondag, Ber. 41, 1921 (1908).
[514 J. F. Norris, G. W. Rigby, J. Am. Chem. Soc., 54, 2088 (1932).
[515] I. N. H n 1 t in a n, A. W. Davis, I]. T. Clarke, J. Am. Chem
Soc., 43, 366 (1921).
330 ОБРАЗОВАНИЕ НОВЫХ С—0-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ
Трнфенилизоамиловый эфир [516]. Трифенилкарбннол растворяют'*
в 100%-ной HoSO4 (ил 96%-ной H2SO4 и S03) и прп —10° С раствор смешивают
с изоамиловым спиртом. Образовавшуюся смесь выливают в ледяную воду-si
и выделяют эфир. Выход 88% от теоретического.
Бензгидрил-^-хлорэтиловый эфир [517]. Его синтезируют аналогичным 5
способом, нагревая на водяной бане смесь этиленхлоргвдрпна, концснтриро--]
ванной H2SO4 и бензгидрола в бензоле. °J
При применении концентрированной серной кислоты часто протекают нежела-
тельные побочные реакции. Поэтому для этерификации неустойчивых соединений
рекомендуется употреблять в качестве конденсирующих агентов органические сульфо*?
кислоты, например п-толуолсульфокислоту [518]. Так, циклические эфиры енолов’
[519, 520] можно получать кипячением раствора исходного вещества в бензоле с не-*'
большим количеством п-толуолсульфокислоты прп непрерывном удалении образу-'
ющейся воды с помощью водоотделителя.
Моноэтиловыи эфир дигидрорезорцнна [520]. Смесь 215 г дигидрорезор*)
цииа, 300 мл 96%-ного этилового спирта, 1500 мл бензола и 7 г п-толуолсульфЬ-'
кислоты нагревают в аппарате с водоотделителем до окончания реакции. Выхой)
епблового эфира 90% от теоретического. Л
Из соответствующих спиртов и л-толуолсульфокислоты удалось синтезировал^
также симметричные эфиры с длинной цепью [521]. После 4 ч кипячения в колбе с об*1
ратным холодильником смеси тиодигликоля, изоамллового спирта и л-толуолсульф<$
кислоты получают тиодигликоль-бис-изоамиловып эфир с выходом 83% от теоре^
тического [522]: *
ХН2СН2ОН /Ct^CHoOCjHu-uao •
gz цзо-CtHiiOH gz -ч
^СЩСЩОН ^CHgCHgOCgHii-uae •>
Этерификация под влиянием концентрированной НС1 рассматривается па пр»*
мере синтеза А2’2 -дициклопентснилового эфира из циклопентеп-1-ола-З [523]: ?$
ип о 'S
Л2,2 -Дициклопентеииловый эфир [523]. Растворяют 10 мл концентр»
рованной НС1 в 420 г циклопентен-1-ола-З. Через несколько часов отстаивает»
водный слой, который отделяют, и маслянистый продукт реакции перегоняй^
в вакууме над NaHCO3. Небольшая первая фракция состоит из воды и непреврА
щениого циклопентанола (т. кип. 48° С при 11 мм рт. ст.). Основная фракцй)
перегоняется при 81° С (11 мм рт. ст.) в виде бесцветной жидкости. ВыхсЙ
почти количественный. ?
При нагревании^ смеси и-бутплового спирта, концентрированной НС1 и N-W
тилол-М'-фенилмочевины до 50° С получают соответствующий н-' ' "" ~i,_
(выход 86% от теоретического) [524]*
C6H5NH-CO-NHCH2OH + C4H9OH —» CeHjNH-CO-NHCHoOCiHe + HsO .7
i-бутиловый эфй|
516
517
518
519
520
521
[522]
[523]
[524]
Н. A. Smith, R. J. Smith, i. Am. Chem. Soc., 70, 2400 (1948).
S. S и g a s a w a, K. Fujiwara, Org. Synth., 33, И (1953). &
F. Krafft, Ber., 26, 2829 (1893).
R. L. Frank, И. К. II a 1 1 jr., J. Am. Chem. Soc., 72, 1645 (1950).
B. Crewe, E. Nolte, B. IT. R otzoll, Chem. Ber., 89, 600 (1956).
R. Perron. C. Panuot, C. r., 228, 584 (1949); Bull. Soc. chim. France.
1949, W. л t'
F. R ichter F. В. Augustine et al., J. Am. Chem. Soc., 74, 4070
(1952).
K. Adler, F.'H, Flock, Chem. Ber., 89. 1732 (1956).
S. Zi geuncr, K. Voglar, R. Pitter, Mh. Chem., 85, 1196 (1954).
I. ПОЛУЧЕНИЕ ПРОСТЫХ ЭФИРОВ
331
Легко этерифицируются бензиловые спирты [525]. Например, салигенин [526]
превращается в o-оксибевзилметиловый эфир при нагревании с метиловым спиртом
я запаянной трубке при 150° С:
Г || 4-СМзОН | Y +Н2О
Ч>:/\сн2ОП '^'/\сн2осн3
При нагревании фталилового спирта в присутствии незначительных количеств
г-толуолсульфокпслоты получается циклический простой эфир — фталан (выход
около 60% от теоретического [527]):
^VCIl!0H ^VC\
I ) — [ ¥ о+н2о
^/\СН2ОН СН?
Это соединение легко осмоляется в присутствии кислот, и поэтому его необхо-
димо тотчас же после образования удалять из реакционной смеси отгонкой в токе
азота. Производные фталилового спирта так же, как и сам фталиловый спирт, легко
отщепляют воду под действием кислот.
Для синтеза производных фурана из 1,4-гликолеи в качестве дегидратиру-
ющего средства особенно пригодна фосфорная кислота. Тетрагидрофуран получают
из бутандиола-1,4 в промышленном масштабе.
НгС—СН8
НО он о
Химия фурана и нирана подробно описана Джонсом п Тейлором [528].
Синтез оксетанов [529] (производные триметиленоксида) осуществляется про-
стым способом, для чего раствор соответствующего диола в концентрированной серной
кислоте вносят в нагретый концентрированный раствор щелочи и отгоняют образу-
ющийся при этом циклический эфир.
Получение смешанных жирно-ароматических эфиров совместным отщеплением
воды от спиртов и фенолов в общем не имеет большого значения. Однако с помощью
соляной кислоты флороглюцин можно превратить в ди- или триалкиловый эфир [530].
Некоторые нафтолы, оксиантрацены и оксифенантрены [531], например а-нафтол,
^-нафтол, 1,5-диоксиантрацен, а-антрол и 0-антрол [533], также можно этерифици-
ровать спиртами в присутствии соляной или серной кислоты.
а-Нафтилметиловый эфир [532]. На масляной бане в течение 4 ч нагре-
вают прн 125° С в колбе с обратным холодильником 25 г а-нафтола, 25 г абсолют-
ного метилового спирта и 10 г концентрированной H2SO4. Реакцию рекоменду-
ется вести под небольшим давлением. Реакционную массу выливают в воду, мас-
лянистый слой промывают разбавленной щелочью для удаления небольшого
[525] J. Meisenheimer, Вег., 41, 1421 (1908).
[526] J. de Jonge, В. IJ. Bibo, Rec. trav. chim., 74, 1448 (1955).
[527] A. J. Weinheimer, S. W. Kantor, C. R. Hauser, J. Org, Chem.,
18, 801 (1953).
1528] D. G. Jones, A. W. C. Taylor, Quart. Rev., 4, 195 (1950).
[529] L. F. Schmoyer, L. C. Case, Nature (London), 183, 389 (1959).
1530] W. Will, K. Albrecht, Ber., 17, 2107 (1884)- W. Will, Ber., 21, 603
(1888); J. llerzig, H. Kaserer, Mh. Chem., 21, 875 (1900); K. Freu-
denberg, Ber., 53, 1425 (1920).
[531] F. R. Japp, A. Findlay, J. Chem. Soc., 71, 1122 (1897); L. Gatter-
m a n n, Ann., 244, 72 (1888).
15321 O. N. Witt, F. Schneider, Ber., 34, 3173 (1901).
[533] Br. Lampe, Ber., 42, 1416 (1909); ср. C. Liebermann, A Hagen,
Ber., 15, 1427 (1882); 11. Dienel. Ber., 38. 2864 (1905).
332 ОБРАЗОВАНИЕ НОВЫХ С—О-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ
количества пепрореагировавшего нафтола и затем перегоняют (т. кип. 258е С).
Выход почти количественный. Еще легче этерифицируется р-нафтол.
Прп взаимодействии глюкозы с метиловым спиртом и незначительным коли-
чеством, сухого НС1 (в большинстве случаев 0,25% пли меньше) при нагревании в за-
паянной трубке или при кипячении с обратным холодильником образуется почти ис-
ключительно а-метплглюкозид, наряду с небольшим количеством ' В-мегилглюко-
зида [534, 535].
а -Метил-Д-глюкозид [534]. В 200 г (251 мл) не содержащего воды и аце-
тона метилового спирта пропускают НС1 до привеса в 5 г и разбавляют 1800 мл
метилового спирта. К полученному 0,25%-ному раствору хлористого водорода,’
в мегпловом спирте прибавляют 500 г тонкоизмельченной, сухой а-Д-глюкозы i
и кипятят 72 ч в колбе с обратным холодильником (обратный холодильник во'^
избежание доступа влаги воздуха должен быть снабжен трубкой с натронной^
известью), причем через 15 мин вся глюкоза переходит в раствор. Затем раствор '
охлаждают, вызывают кристаллизацию потиранием палочкой о стенки или^
затравкой а-метил-Ь-тлюкозида и после 12 ч выдержки при 0° С отфильтровы-d
вают выпавший частично продукт реакции и промывают его двумя порциями -^
холодного метилового спирта по 100 мл каждая; выход 85—120 г; т. нл. 165° С. J
Маточник кипятят еще 72 ч и концентрируют раствор. В сумме получаетсЯ'-я
260—266 г продукта, что составляет 48,5—49,5% от теоретического; для допоя*‘!
пительной очистки метилглюкозид можно перекристаллизовать из 5 ч. метило-з
вого спирта, причем температура плавления глюкозида существенно пе меняется.-!
По более простому способу 150 мл метилового спирта насыщают HCI,;!
как указано выше, до привеса 12 г. доводят объем метиловым спиртом до 500 мл,й
прибавляют 200 г глюкозы и кипятят в течение 18 ч в колбе с обратным холодиль-^
пиком. После первой кристаллизации получают 95 в, а после отгонки из маточ--^
инка (240 мл спирта) еще 20 г а-метнлглюкозида.
В маточниках, остающихся после описанных опытов, содержатся незна-<
читальные количества р-мстилглюкозпда.
Способ получения а-мотилгалактозида. несколько отличающийся от описанный;
и применимый для получения других углеводов, описали Михель и Литмапн [536],"
а-Метилгалактозид. В колбе с обратным холодильником кипятят неко- J
торое время 25,8 г а-2?-галактозы с восьмикратным количеством метилового 4
спирта и в еще горячий раствор при -постепенном охлаждении до 15° С пропу-^
скают сухой НС1 (до концентрации НС1 13,6%). Раствор темнеет и после выдер.-ч
живания в течение ночи теряет восстановительные свойства. Затем часть НС1;
отгоняют в вакууме, остаток удаляют с помощью РЬС.О3. Фильтрат при стоянии
в вакууме полностью затвердевает в кристаллическую массу; выход 20,4 г4
(73,9% от теоретического). а-МогилгалактОзид перекристаллизовывают из этил- -,
ацетата, воды или 50%-него ацетона. ‘
В качестве примера этерификации на твердых катализаторах в газовой фазе^
описывается получение фталана из фталилового спирта [537]. -
Фталан. Расплавляют 276 г фталилового спирта и дегидратируют, про--
пуская его пары при 300° С через нагретую колонку, заполненную активиро-;
ванной окисью алюминия. При этом образуется 240 г красно-коричневого масла л
и 36 мл воды. Перегонкой с водяным паром в присутствии 10%-ной водной .,
щелочи получают 214 г фталана, что составляет 89% от теоретического.
[534] В. Hejferich, W. Schafer, Org. Synth., 6, 64 (1926).
[535] F. S c h mi t h, J. W. V a n Clewe, J. Аш. Chem. Soc., 77, 3159 (1955). 4
[536] F. M i c h e e 1, O. Li t tmann, Ann., 466. 124 (1928). i
[537] J. En tel, С,- H. Ru of, H. C. Howard, J. Am. Chem. Soc., 74, 441
(1952).
I. ПОЛУЧЕНИЕ ПРОСТИХ ЭФИРОВ
зз:
2. Получение простых эфиров алкилированием
галогенсодержащими соединениями
Классический метод получения эфиров по Вильямсону заключается во взаимо
действии алкилгалогенидов с алкоголятами или фенолятами щелочпьтх металлов:
RONa-KR'X —> R—О—R'-'-NaX
Этим простым и удобным методом можпо получать несимметричные и симметрии
ные эфиры.
В качестве галогенсодержащих соединений применяют по возможности алкил
подиды, реагирующие легче, чем соответствующие алкилбромиды; алкилхлорищ
в большинстве случаев обладают меньшей реакционной способностью. Однако в бев
аилхлориде [538, 539]. фурфурилхлориде [540], аллилхлоридах [541—544] и эпи
хлоргидрине [545] галоген легко замещается алкоксильной группой. Особенно актин
ные галогенсодержащие соединения, такие, как дифенил- и трифенилгалогенметаш
[546], о- п и-нитробензилхлорид [547], реагируют со спиртами с образованием эфиро
уже при стоянии или при кратковременном нагревании.
При взаимодействии алкоголятов натрия с вторичными или третичными алкил
галогенидами в результате отщепления галогеноводорода наряду с эфирами легк
образуются олефины. Этот побочный процесс можно ограничить подбором подходящи
компонентов реакции: хорошие результаты достаются при введении вторичны
н чретпчных компонентов в виде алкгц олятов, а также при использовании алкс
голшов магния [548].
Обычный способ получения алкоголятов заключается по взаимодейства
щелочного металла с соответствующим спиртом. При использовании окспсоедъ
нений сложного строения, например частично замещенных углеводов, прнм<
няются растворители: абсолютный эфир [549], диоксан [55О’| или жидкий ак<
миак [551]. Спирты, трудно реагирующие с. натрием, часто с успехом переводя
в алкоголяты взаимодействием с калием или с жидким сплавом натрия и кала
(1:2) [552]. По реакции с амидом натрия алкоголяты можно получать ка,
в жидком аммиаке [554}, так л в органических растворителях [553].
[552]
[55.3]
[554]
W. J. М о п а с е 11 i, G. F. Hennion, J. Am. Chem. Soc., 63, 1722 (1941’
W. S. E merson, J. W. Heyd et al., J. Am. Chem. Soc., 69, 1905 (194?‘
W. R. Kirner, J. Am. Chem. Soc., 50, 1955 (1928).
M. Conrad, C. Bruckner, Z. pfiysik. Chem., 4, 631 (1889).
W. T. Olson, H. F. Hi psher et al., J. Am. Chem. Soc., 69, 2451 (1947
M. Tamel e, C. J. О 11, Ind. Eng. Chem , 33, 115 (1941).
Cp. D. S. Tar bell, Org. Reactions, 2, 22, 26 (1944).
M. J. В 6 e s e k e n, Rec. trav. chim. 34, 102 (1915).
M. M. C. Friedel, J. M. Crafts, Ann. Chimie [6], 1, 502 (1884).
Герм. пат. 136680.
О. Diels, P. Blumberg, Ber., 44. 2847 (1911)
A. S. Meyer, T. Reichstein, Helv. chim. acta, 29, 152 (194B
K. Freundenberg, R. M. Hixon, Ber., 56, 2119 (1923).
K. Freudenberg, K. Friedrich, 1. Bumann, Ann., 49«
57 (1932).
K. Freudenberg, H. Boppel, Ber.. 71, 2505 (1938); В. C. Hej
dricks, R. E. Rundle, J. Am. Chem. Soc.. 60, 2563 (193&
M L. Wolirom, W. W. Binkley et al., J. Am. Chem. Sog
73, 3553 (1951).
A. Bae yer, Ber., 26, 826, 2560 (1893).
J, B. Wright, H. G. К о 11 о f f. J. II. Hunter, J. Am. Chen
Soc., 70. 3098 ’(1948); R. P a n 1, S. T c h e 1 i t c h e f f, Bull. Soc. chin
France, Mem. [5], 14, 341 (1947).
C. L. Stevens, A. J. \V e i n h e i m e г, J. Ain. Chem. Soc., 8«
4072 (1958).
334 ОБРАЗОВАНИЕ НОВЫХ С—0-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ
Алкоголяты часто образуются уже при добавлении спиртового раствора [
едкЪго кали [555] пли суспензии гидроокиси [556] или карбоната [557] щелоч- §
ного металла к соответствующему спирту.
Для синтеза эфиров нагревают суспензию алкоголята в соответствующем гало- •]
генсодержащем соединении или раствор алкоголята п галогенида в растворителе, -4
например в том же. спирте. J
Получение пзобутилэтилового эфирапо Вильямсону [558]. Растворяют J
натрия в чистом изобутиловом спирте (разогревание) и избыточный спирт отго- -Ц
нягот при нагревании до 180° С в токе водорода. Образовавшийся изобутилат
натрия быстро растирают в порошок и нагревают при 75° С в запаянной трубкеч
с этилиодидом до тех пор, пока весь этилиодид не прореагирует (около 7 суток).А
При вскрытии трубки улетучивается небольшое количество изобутилена. Полу-;
ленный изобутилэтиловый эфир отгоняют из трубки, промывают водой, (Лшат * 1
п перегоняют над натрием; т. кип. 79 —80° С; выход хороший.
Без специального выделения алкоголятов можно получать 2-алкоксиэтиловыв ч
спирты [559] из этиленгликоля и алкилгалогенидов, симметричные диалкоксипро-,5
ппловые спирты [560] — из симметричного дпхлоргидрина глицерина, соответ*:::
ствующие оксиэфиры — из триметиленгликоля [561]. Диэфиры гликолей типа!)
ROCH2CH,OR' удобно синтезировать, добавляя алкилгалогениды к раствору натрия*
в избытке моноалкилового эфира этиленгликоля [562]. ;
Диметиловый эфир этиленгликоля [562]. В колбе емкостью 3 л. снабжен- *
нон мешалкой и обратным холодильником, к 1830 г (24 моль) монометллового
эфира этиленгликоля добавляют при перемешивании в несколько приемок 138 а '
(о,0 моль) натрия. После растворения всего натрия в раствор медленно пропу-*,
екают хлористый метил, поддерживая температуру 60° С. Через 3 ч массу охла-ч!
ждают и отделяют жидкую фазу от выпавшего NaCl. Отгоняется 422 г диметило*-»
вого эфира этиленгликоля (78% от теоретического в расчете иа натрий), т. кнп<5
83.5—84,0° С п2<>1,3813.
Галогепэфиры типа R0(CH2)«X получают, добавляя спиртовые растворы алко^
голята патрия к раствору соответствующего дигалогенида в безводном эфире. Напри^х
мер, из 1,6-дибромгексана и теоретического количества метилата натрия в метиловому
спирте образуется 1-метокси-6-бромгексан с выходом 80% от теоретического [563]Л]
а,p-Дихлоральдегиды реагируют при температурах ниже 15° С с разбавленным^
растворами алкоголятов натрия, давая хорошие выходы соответствующих диалк-ц
оксиальдегидов [564]. Из этилового эфира 0-бромпропионовой кислоты и избытка^
натрия в абсолютном спирте образуется этиловый эфир 0-этоксипропионовой кислоты^
с выходом 88% от теоретического [565]. Синтез алкоксикислот, например 1-метоксИ*^
уксусной кислоты, описан Леффлером и Калкинсом [566]. Нагреванием диэтилацеталй Я
1
[355
[556
[557
[558
[559
[560
[561]
[562]
[563]
[564]
[565]
J566]
I. U. N о f, Ann., 318, 7 (1901).
Пат. США 1459177; С., 1925, II, 1224.
L. С 1 a i s е п, О. Е j s 1 е Ь, "Aon., 401, 29 (1913).
W. L i р р е г t,, Ann., 276, 160 (1893).
F. С. Cooper, М. W. Partridge, J. Chem. Soc., 1950, 462. . "й
H. R. Henze, B. G. R о g e г s, J. Am. Chem. Soc., 61, 433 (1939): 62. 175$J
(1940). 5
L. I. «Smith, J. A. S p r n n g, J. Am. Chem. Soc., 65, 1276 (1943h-A
G. M. Bennett, A. L. H о c k, J. Chem. Soc., 1927, 472, 4
.1. V. C a p i n j о 1 a, J. Am. Chem. Soc., 67, 161.5 (1945). 4
P. Ba u dart, Bull. Soc. chim. France [5], 11, 336 (1944); N. L. D r a k
et al., J. Am. Chem. Soc., 68, 1537 (1946).
J- Lichtenberger, M. Naftali, Bull. Soc. chim. France [5], 4, 325»д
(1937). •
M. Swaminatha n, J. Indian Chem. Soc., 23, 388 (1946). J
M. T. Leffler, A. E. Calkins, Org. Synth., 23, 52 (1913); R. C. F u-)
son, B.-(J. Woje ilc. Org. Synth., Coll. v. 2. 1943, p 26l).
1- ПОЛУЧЕНИЕ ПРОСТЫХ ЭФИРОВ
33!
бромацетальдегида н алкоголята натрия в течение 24 ч на водяной бане получаю
диэтилацеталь этоксиацетальдегида [567]. По этому методу можно синтезировал
и алифатические аминоэфиры.
(5,Р'Р"-Триэтокснтриэтиламин. N(CH2CH2OR)3 [568]. Раствор 6,24
(0,11 моль) КОН в 25 мл 95%-ного этилового спирта и 4.42 г (0,0183 моль) гидре
хлорида трихлортриэтпламина кипятят 3 ч в колбе с обратным холодильников
После отделения выпавшего в осадок КС1 спиртовой фильтрат упаривают пр
10 мм рт. ст. и остаток растворяют в воде. Из раствора Извлекают продух
реакции эфиром и сушат над MgSO4. Отгонкой при 134—137° С (12 мм рт. ст
получают 2.80 г триэтоксиэтиламина (66% От теоретического); т. пл. гидрохлс
рида триэтокситриэтиламина 193—195° С.
Синтезы эфиров по Вильямсону осуществимы также л с гетероциклическим
соединениями. Нагреванием в запаянной трубке в течение 24ч при 150° С 2-xnoj
4-аминохинолина с раствором натрия в абсолютном н-бутиловом спирте получаю
2-«-бутокси-4-аминохинолин с выходом 88% от теоретического [569]. 2-Хлорбен;
тиазол переводится при 0е* С раствором эквивалентного количества натрия в этилово
еппрге в 2-эгоксЕбензтиазол [570]. Из этилового эфира 2-хлорхииоксалин-З-карбонс
ной кислоты этим путем получают этиловый эфир 2-этоксихпноксалин-З-карбоновр
кислоты [571].
В 2,4,6-трибромпиримидине возможен частичный обмен брома на алкоксильньз
группы. При действии на него раствором двух эквивалентов натрия в абсолютно
этиловом спирте я применяя в качестве растворители абсолютный бензол, при комнат
ной температуре образуется 2,4-диэтокси-б-бромпиримндин [572]; выход 89% от теор»
шческого. При взаимодействии 4-хлор-2-амниоДз-метоксипиримидниа с раствор»
натрия в этиловом спирте и ксилоле, от которого отогнан избыточный спирт, поел
2 ч нагревания в колбе с обратным холодильником образуется 2-амино-4-этокси-(
мсюксипиримидин [573]; с большим количеством этилового спирта 6-метоксигрупп
также обменивается на этоксильную и образуется 2-амино-4,6-диэтоксипиримидин.
По указанному способу удается также алкилирование фенолов с образование
эфиров фенолов. Примером может служить описанное Вейгандом и Габлером [57<
получение n-иитрофенолового эфира. Взаимодействие происходит гладко в высок<
кипящем кетоне, используемом в качестве растворителя (например, в циклопеатаноне
Аналогичным способом удается также этерификация n-оксибензальдегнда вы»
пиши спиртами.
п-Нитрофенил-м-гептиловый эфир. При энергичном перемешиванд
кипятят в колбе с обратным холодильником 20 г ц-нитрофенолята калия, 25
я-гептилбромида и 60 г циклопентанона. Уже через 30 мин выделяется плотиц
кашица КВт, реакция заканчивается через 2,5 ч. После охлаждения смесь ра;
бавляют эфиром, встряхивают с раствором NaOH н выделяют продукт обычный
способами, выход 89% от теоретического. Основное количество добавленное
цпклопентанона регенерируют.
н-Октиловый эфир ?!-оксибензальдегида. Раствор 6,1 г (0,05 лол»
n-оксибензальдегида и 4 г КОН в 50 мл циклогексапола смешивают с раствор»
14.4 г (20%-ный избыток) н-октилиодида в 50 мл циклогексанола. Затем раство
при энергичном перемешивании нагревают 4 ч на масляной бане при 120—130° С
причем в осадок выпадает KI. После охлаждения продукт экстрагируют эфиров
1567] Е. Spath, Mh. Chem., 36, 5 (1915).
1568] Н. Н. Richmond, G. F. Wright, J. Am. Chem. Soc., 67, 22«
(1945).
[569] .1. В ii c h i, H. Hurni, R. Lieberherr, Helv. chim. acta, 32, 18C
(1949).
l.>701 T(. G i 1 m а п, К. E. Lentz, J, A. Bee 1, J. Am. Chem. Soc., 74, 105
(1952).
[571 ] A. If. G о w e n I о c k, G. T. Newbold, F. S. Spring, J. Chem. So»
1945, 622.
[•"►721 B. W. Langley J. Am. Chem. Soc., 78, 2136 (1956).
[•>73] F. L. Rose, G. A. P. T n e y, ,T. Chem. Soc., 1946, 81.
[574] C. Weygand, R. Gabler, J. prakt. Chem., 155, 334 (1940).
33В НОВЫХ С—0-СВЯЗЕЙ в РЕЗУЛЬТАТЕ обменных реакций
вытяжку промывают водой и разбавленной НС1, сушат над Na2SO4 п после от-
гонки эфира фракционируют в вакууме. При 162—163° С (4 мм рт. ст.) пере-
гоняется 7,6 г смешанного эфира (65% от теоретического).
Для получения алкилариловых эфиров можно использовать также метод Клай-
зепа [557]. По этому способу фенол кипятят на водяной бапе с алкилбромидом в ацето- ,
новом растворе в присутствии К2СО3. Вместо алкилбромидов можно применять также
алкилхлориды в смеси с KI; ацетон заменим метилэтилкетоном [575, 576J. Если кетоны
в указанных условиях реагируют с фенолами (например, в случае фенолальдегидов),- ...
в качестве растворителей используют спирты [557, 577]. Обзор синтезов аллил-.-’
ариловых эфиров см. [544]. 3
Получение аллилфенилового эфира по Клайзену [578]. Смесь 188 г
фенола, 242 г аллилбромида, 280 г тонкоизмельченного К2СО3 и 300 г ацетона .
кипятят в колбе с обратным холодильником на водяной бане в течение 8 ч. при-
чем выпадает осадок КВг. После охлаждения добавляют воду и извлекают <1
продукт реакции .эфиром. Эфирный раствор встряхивают с 10%-ным раствором j
NaOH и сушат над К2СО3. Остающийся после отгонки эфира остаток перего- л
пяют в вакууме; т- кип. 85° С (19 мм рт. ст.); выход 230 г (86% от теоретиче-• Ч
ского).
<
Чисто ароматические эфиры типа дифенилового эфира могут быть получены Ч
пз галогенароматических соединений и фенолятов [579—583]. Добавка небольшого ч
количества порошка меди или солей меди к реакционной смеси способствует повыше- у;
пию реакционной способности галогена в арилгалогснидах.
Дифениловый эфир. На масляной бане в течение 2—2,5 ч при 210— д
230° С нагревают в колбе с обратным холодильником смесь 15,7 г бромбензола, 4
11,8 г фенола, 6,2 г КОН и около 0,1 г медной бронзы (природная медь С). Затем.’*
из реакционной массы отгоняют с водяным паром сначала нопрореагпровавший ’
бромбензол, а потом дифениловый эфир. Выход 12,7 г (90% от теоретического); л
т. пл. 28° С; т. кип. 257° С.
Из галогенароматических соединений с нитрбгруппой в орто- и пара-положе-д
ниях к галогену эфиры получают при сравнительно низких температурах [584—587].
2,4-Динитрофторбензол со спиртами в присутствии триэтиламипа дает алкид-2,4-дН- :
нитрофениловые эфиры [588]. Реакция может быть использована для идентификации"!
спиртов.
2,4-Динптрофенил-и-пропнловый эфир [588]. К раствору 0,5 г динптрофтор- 1
бензола в 0.2 г н-пропилового спирта приливают 3 капли триэтиламина. Через >
12 ч смесь подкисляют избытком 1 н. раствора НС] и выделившееся масло триж- 1
ды экстрагируют эфиром (порциями по 20 мл). Эфирный слой промывают 2 и. ,
раствором NaHC03 и сушат. Остаток после отгонки эфира мерекристаллизовы-
вают пр водного метилового спирта; выход 0,4 г; т. ил. 29° С. Динитрофтор- |
бензол раздражает кожу!
________________ i
I
[575J
[576J
577
578
579
580
581
582
583
[584
[585
[586
[587
1588
L. I. S m 1 t h, П. Н. Н о е h n, A. G. W h i t п е у, J. Am. Chem. Soc., >
62, 1863 (1940). ’ i
F. Mauthner, J. prakt. Chem. [2], 148, 95 (1937). л
В. Loe v, C. R, Da w son, J. Am. Chem. Soc., 78, 6095 (1956). ’
L. Claisen, Ann., 418, 97 (1919). *
F. Ull mann, S. Sponagel, Ber., 38, 2211 (1905). -J
R. Q. Brewster, Th. Grocning, Org. Synth.. Coll. v. 2, 1943, p. 445.,.,
H. E. U ngn a do, E. F. Orwoll, Org. Synth,, 26, 50 (1946).
H. S t e 11 e r, G. Duvo, Chem. Ber., 87, 1699 (1954). %
W. M. Whaley, L. S t a r k e r, M. Mea dow, J. Org. Chem., 18, 833’3
(1953).
L. C. Raiford, J. C. Colbert, J. Am. Chem. Soc., 48, 2652 (1926).
F. Ullmann, Ber.. 29, 1880 (1896).
C. Willgerodt, Ber., 12, 762 (1879).
R. W. Bost.'F. Nicholson, J. Am. Chem. Soc., 57, 2368 (1935).
W. B. Whalley, J. Chem. Soc., 1950, 2241.
4
Т. ПОЛУЧЕНИЕ ПРОСТЫХ ЭФИРОВ
331
Для алкилирования алкплгалогенидами наряду с алкоголятамп щелочныя
металлов применяют также соответствующие соединения серебра, особенно в случае
силанокислых фенолов. Однако в некоторых случаях взаимодействие с солями се-
ребра приводит к образованию продуктов, совершенно отличных от получаемых п<
реакциям с соответствующими солями щелочных металлов. Например, в присутствие
окиси серебра происходит О-метилирование флороглюцина, а в присутствии произ-
водных щелочных металлов — С-метилпрованис (589). О получении пикриловы?
эфиров из пикратов серебра см. 1590].
Синтез серебряных солей часто связан с препаративными трудностями. Однако
пользуясь методом Пурди и Ирвина [591], можно избежать специального получение
серебряных солей, для’ чего соответствующий спирт подвергают взаимодействии
с алкилгалогенидом в присутствии окиси серебра. Этот метод особенно важен длх
метилирования углеводов.
Этерификация гидроксильных групп в углеводах имеет большое значение длг
определения структуры и для синтеза углеводов. Так как в молекуле углеводов содер
жатся не только спиртовые оксигруппы, но и гликозидные группы, чисто формальн<
следовало бы рассматривать образование собственно эфиров и гликозидов в разные
разделах. Однако методически более целесообразно сделать это одновременно. (Спите;
гликозида из свободного углевода н спирта рассмотрен в предыдущем разделе).
В ' большинстве синтезов гликозидов исходят из а-ацетогалогепоз, кото
рг.те под действием спиртов или фенолов в присутствии акцепторов кислоты пре
терпевают вальденовскос обращение и превращаются в р-ацотнлгликозиды. В качеств»
акцепторов кислот служат окись и карбонат серебра, окись ртути, окись цинка и хино
.тин [592—594]. Образующуюся при взаимодействия с карбонатом серебра вод?
целесообразно связывать добавкой сульфатов натрия или кальция или гидрид;
кальция.
Тетра-О-ацетил-^-метилглюкозид |595]. Растворяют 8,6 г ацетобром
глюкозы в 126 мл абсолютного метилового спирта и при комнатной температур
встряхивают с 8,5 г сухого порошкообразного Ag2CO3. Происходящее вначал
бурное выделение СО2 примерно через 1 ч прекращается, после чего в закрыто]
сосуде на аппарате для встряхивания продолжают реакцию еще 6 ч. Нераство
римую часть отфильтровывают, осадок хорошо промывают эфиром, фильтра,
смешивают с водой и небольшим количеством чистого ВаСОа, вновь фильтрую
н растворителям дают испариться в вакуум-эксикаторе над H2S04. Сухой оста
ток встряхивают с водой и повторно с эфиром. Объединенные эфирные вытяжкз
встряхивают с раствором Na2CO3 и водой и сушат над Na2SO4. После отгонк;
растворителя остается хорошо кристаллизующийся остаток, который перекрм
сталлизовывают из метилового спирта, а затем из лигроина; выход глюкозид
3,75 г; т. пл. 104—105° С.
Под действием хлорида титана (IV) [596] или трнфторида бора [597] ацетат:
р-гликозида перегруппировываются в трудно доступные сс-глниозвды.
[589] J. Her zig, S. Zeisel, Mh. Chem., 10, 144 (1889); 11, 291 (1890); J. Hej
z i g, Вг. E г t h a 1, Mh. Chem., 32, 491 (1911); ср. E. Ot t, E. N aue t
Ber., 55, 924 (1922).
[590] G. E. Phil brook, D. J. Massey, J. Am. Chem. Soc., 73, 3454 (1951'
[591] Th. Pur'dje, I. С. I rvine, J. Chem. Soc., 75, 157, 485 (1899); 83, IQ
(1903); 79, 957 (1901).
[592] G. Zemple n, в книге Fortschritte der Chemie organischer Naturstoffe, Bd. ]
Springer-Verlag, Wien, 1938, S. 1.
[593] F. м i c h e e I, Chemie der Zucker und Polysaccharide, 2 Aufl., Akadem. Vea
lagsgesellschaft Geest u. Portig K. G., Leipzig, 1956, S. 75.
]594) F. Klagfts, в книге G. M. Schwab, Handbuch der Katalyse, Bd. 7/S
Springer-Verlag, Wien, 1943, S. 310.
[595] W. Koenigs, E. Knorr, Ber., 34, 965 (1901).
15961 E. Pascu, J. Am. Chem. Soc., 52, 2563, 2568 (1930).
[597] B. Lindberg, Acta Chem. Scand., 2 , 426 , 534 (1948);
22 Заказ 18.35.
338 ОБРАЗОВАНИЕ НОВЫХ С —О-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ
Наряду с описанным способом Кеппгса п Кнорра удобным методом является
получение гликозидов по Земплсну [592, 598] из сс-ацетогалогсноз п спиртов или фено- .
лов в бензольном растворе в присутствии ацетата ртути. В зависимости от количества -
используемых спиртов и катализатора по этому методу можно получать а- или
р-ГЛИКОЗИДЫ.
Тетра-О-ацетнл-р-бензилглюкозпд [599], Растворяют 10 з ацетобром-
глюкозы в 60 ли бензола и, добавив 3,8 г ацетата ртути и 10,4 г бензилового
спирта, кипятят 2 ч в колбе с обратным холодильником. Затем массу промывают, а
встряхивая с водой, отгоняют в вакууме бензол и остающийся сироп растворяют
в небольшом количестве метилового спирта. Прд размешивании с водой обычно . Д
сразу наступает кристаллизация. Неочищенный продукт перекристаллизовывают J
из эфира. Выход 60—80% от теоретического. ,j
По Гсльфериху [600], фепплглинозпды образуются при сплавлении фенолов/3
с ацетатами углеводов в присутствии кислотных катализаторов.
Обзорные работы по синтезу гликозидов см. [592—594].
Алкилирование спиртовых гидроксильных групп углеводов протекает значи- А
тельно трудней, чем образование гликозидов. Особенно большое значение приобрели *1
метиловые [601], этиловые, бензиловые [602] и триэтиловые [603] эфиры углеводов. *4
Важнейшим методом метилирования углеводов является взаимодействие углеводов
с метилподидом п окисью или карбонатом серебра [591]. Для того чтобы избежать
окисления восстанавливающих групп углеводов их защищают, лучше всего пере-
водом в гликозиды. А
Тетра-О-метил-а -метилглюиозид [604]. В смесь 60 я метплглюкозида .
(1 моль), 200 мл метилового спирта и 900 е метилиодида постепенно вносят 680 г
(9,5 леоль) окиси серебра. Сначала реакционный сосуд охлаждают, а к концу J
реакции некоторое время подогревают на водяной бане. Продукт реакции экс- «
драгируют эфиром, вытяжку фильтруют и перегоняют в вакууме; выход 62,5 г; *
т. кип. 148—150° С (13 мм рт. cm.). v
О-Алкилированпе углеводов часто проходит неполно; в большинстве случаев
для достижения нужного содержания метоксильных групп приходится несколько pas *
дополнительно метилировать. Проведепо [605] метилирование углеводов метилиодидом
и окисью серебра в диметилформамиде; при этом отпала необходимость в дополни-
тельном метилировании, требующемся в других методах.
Исчерпывающее метилирование тростникового сахара. Растворяют я
10,2 г сахарозы в 120 мл теплого диметилформамида [т. кип. 43—44° С пр» «а
12 мм рт. ст.) и при 20° С смешивают с 45 мл йодистого метила. Затем при силь- з
ном перемешивании в течение около 15 мин вносят 45 г Ag»O. После добавле- z,5|
ния около 20 г Ag»O температура смеси становится рациой примерно 30’ С, л
при дальнейшем добавлении окисла температуру поддерживагОт не выше 30° С, j
при необходимости охлаждая массу. Ко1Да примерно через 40 мин после я
[598] G, Zemplcn et al., Ber., 63, 368, 2720 (1930); 64, 744, 1545, 1852 (1931).
J599] F. К 1 a g e s, R. Niemann, Ann., 529, 201 (1937).
[600] B. Helf erich, E, S c i mi t z - H il 1 e b rech t, Ber., 66, 378 (1933).
[601] E. J. Bourne, St. Peat, в книге Advances in Carbohydrate Chemistry,
v. 5, Academic Press, Inc. Publishers, New York, 1950, p. 145.
[602] Ch. M. McCI oskey, в книге Advances in Carbohydrate Chemistry, v. 12,
Academic Press, Inc. Publishers, New York, 1957, p. 137.
[603] В. H e 1 f e г i c h, в книге Advances in Carbohydrate Chemistry, v. 3, Acade-
mic Press, Inc. Publishers, New York, 1948, p. 79.
[604] W. N. Па wo Mb, J. Chem. Soc., 107, 13 (1915).
f 605 J R. Kuhn, H. Trischmann, T. L 6 w, Angew. Chem., 67, 32 (1955);
R. КвЬ л, Л. }[. Baer, Л. Seeligcr, Ann., 6И, 236 (1958).
I. ПОЛУЧЕНИЕ ПРОСТЫХ ЭФИРОВ
33'
добавления всей описи серебра температура начнет самопроизвольно снижаться
реакционный сосуд (rmipoKorop.’ian склянка с притертой пробкой) затериваю
и механически встряхивают в течение 12 ч. Выпавший осадок центрифугирую
и промывают 50 мл дпметилформамида и 50 мл хлороформа. Объединенный рас
твор смешивают с 500 мл воды и 5 г KCN и извлекают продукт реакции хлоро
формой в 4—5 приемов порциями по [00 мл. Объединенную хлороформную вы
тяжку 3—1 раза промывают водой порциями по [00 мл и сушат над Na2SOe
Получается 11,5 г октамешлеахарозы; т. кип. 140—145° С (0.001 л/.u рт. ст.)
«22 1,1559.
Окись серебра можпо заменить на ВаО, который не оказывает окисляющег
действия. При помощи смесей SrO и Sr(OH)2 или ВаО и Ва(ОН)5 в диметилформамид
как в растворителе метилирование идет в особенно мягких условиях [606].
По еще одному из известных методов эфиры углеводов получают из натриевы.
производных углеводов и алкилгалогеппдов. Для синтеза алкоголятов углевод ИЛ1
соответствующее производное обрабатывают натрием в жидком аммиаке, эфире ил,
диоксине.
3-0-Бснзил-1,2;5,6-ди-0-пзопропилидсп-а-ТАглюкоза [607]. В колб
емкостью 250 мл, снабженной осушительной трубкой, растворяют 45 г (0,17 моль
диизопропилиденглютсозы в 500 мл абсолютного эфира и прибавляют 9
(0,39 моль) натриевой проволоки. Оставляют смесь на 16 ч, после чего удаляю
непрореатпровавшип патрпй. Затем, одновременно отгоняя эфир, приливаю
по каплям 22 мл (0,18 моль) белзгглбромида, Температуру смеси постепенв:
повышают до 70° С и поддерживают в течение 5 ч. Остаток растворяют ц 300 м
лигроипа (т. кип. 60—70е С) и раствор промывают в 5 приемов порциями вод:
по 200 мл каждая. Растворитель отгоняют и вакууме, а остаток перегоняю
в глубоком вакууме (т. кип, 146—149° С при 0 05 мм p»t. ст)-, выход 38—44
(63- 73% от теоретического).
Наконец, в химии углеводов имеют известное препаративное значение их три
фенилметиловые эфиры. Трифенилметилхлорид реагирует предпочтительно с перви\
ними оксигрупиами, поэтому при реакции его с обычными углеводами происходи
алкилирование только оксигруппы в положении 6. Так как тритильный остаток па
действием разбавленных кислот'вновь легко отщепляется (стр. 362), со таким цуте
можно получать производные углеводов с одной свободной оксигруипой в положении f
Углевод сначала подвергают взаимодействию с трифснилмстилхлоридом, затем ацете
лируют остальные оксигруппы и в заключение отщепляют тритильный ост?
ток [603, 608].
6-О-Тритил-а-/9-глюкоза [609]. К охлажденному раствору 36 г безвод
ной глюкозы в 180 мл абсолютного пиридина прибавляют 58 г трифенилметио
хлорида и оставляют смесь при комнатной температуре на 1—2 суток. Зате
отфильтровывают выпавптлй аддук г хлористого водорода с трпфенилкарбиноло
и при 0° С к фильтрату по каплям добавляют воду до начала появления мути
Смесь оставляют на 30 мин. во льду, выливают ее в 1,75 л ледяной воды, н<
сколько раз перемешивают образовавшийся желтоватый сироп с водой, декантэ
руют воду и остаток растворяют в 200 мл метилового спирта. При стояла
выделяется сначала небольшое количество трифепилкарбинола (2 г), пос»
чего при 0° С выкристаллизовывается тритилглюкоза. Ее сушат на лорнете
тарелке и получают 30 г неочищенного продукта (36% от теоретического»
О-О-'Грнтил-а-Р-глюкозу перекристаллизовывают из абсолютного спирт:
и получают кристаллосольват с двумя молекулами спирта; т. пл. 57—58Q С.
[606J S. 3. Kuhn Pt al., Angew. Cliem., 72, 808 (1960).
]6O7J См. ссылку [602]. стр. 146.
{608J В. H e I f e r i c h, J Bockpr, Ann., 440, 1 (1924); В. H e 1 f e г 1 c ;
Angew. Chem., 41, 871 (1928).
(609J B. Helferich, M о о g, A. lunger, Ber., 58, 675 (1925).
22*
340 ОБРАЗОВАНИЕ НОВЫХ С—О-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ Ц
Тритиловые эфиры играют известную роль также прп синтезе смешанных гли-У|
церидов. Глицерин реагирует с одной или двумя молекулами трифепплхлорметана»^
Тритиловый эфир глицерина [610]. Растворяют 8 г глицерина и 12
трифенилхлорметана в 30 мл пиридина, оставляют на 16 ч при комнатной темпе-’!
ратуре, выливают в воду и, не обращая внимания на небольшое количествам
выкристаллизовавшегося гидрохлорида пиридина, извлекают выделившееся'
масло эфиром. Эфирную вытяжку последовательно промывают HCI, водой?
и раствором NaHCOs, сушат и растворитель отгоняют. Прп хранения во льдз^
маслянистый остаток через некоторое время затвердевает. После перекристаллвл
зации последовательно из спирта, бензола и этилапетата получают 3 г чистом^
тритилового эфира глицерина; т. пл. 92—94° С. ?
Для получения дитритилового эфира глицерина (предположительна^
1,3-эфира) 1 г глицерина нагревают 30 мин на водяной бане с 20 мл пиридиш
п 7 г трифенилхлорметана. Обработку реакционной массы ведут, как указа»
выше; т. пл. эфира 170—171° С.
3. Получение простых эфиров алкилированием диметилсульфатом ;
Для алкилирования спиртов и фенолов вместо алкилгалогенидов можно при
менять дпалкилсульфаты. В синтезе метиловых эфиров особое значение имеет диметил
сульфат [611]. ч
Этот метод находит преимущественное применение для алкилирования фенолов
по Ульману (612—614]. Реакция проходит в две стадии:
ROII + (CH3)2SO4—> ROCH3+CH3HSO4 I
ROH + CH3HSO4 —> ROCH34-H.>SO4 ч;
Первая стадия легко протекает уже при слабом нагревании. Часто удается
использовать и вторую алкильную группу алкилирующего агента для этёрификаци
еще одной молекулы оксисоединения, однако для этого требуется более высокая те®
пература. Обычно для проведения метилирования диметилсульфат медленно приливаю
по каплям к Щелочному раствору оксисоединепия. У
Анизол [615]. В трехгорлой колбе емкостью 5 л, снабженной мешалке^
обратным холодильником и капельной воронкой, растворяют 235 г фенол
(2,5 моль) и 100 г NaOH (2,5 моль) в 1 л воды и раствор охлаждают при перЙ
мешивании до температуры несколько ниже 10° С. Затем к нему в течение 1*1
при перемешивании приливают из капельной воронки 315 г диметилсульфатй
прекращают охлаждение, 30 мин нагревают на водяной бане, прибавляЩ
вторую порцию (в данном случае в течение 15 лин) такого же раствора фенол
(235 г фенола и 100 г NaOH в 1 л воды) и кипятят 15 ч на открытом пламени
Образовавшиеся слои разделяют; продукт из водного слоя экстрагируют 200
бензола и после’сушки и двукратной перегонки получают 388—405 г аиизоД)
(72—75% от теоретического); т. кип. 153 —154° С (1& мм рт, ст,), ;
Для алкилирования неустойчивых в щелочной среде соединений, например noaji
фенолов, рекомендуется обрабатывать соответствующие ацетилпроизводные диметЯЛ
сульфатом и раствором NaOH (иногда в присутствии водного ацетона); при этом одн®
610] В. Н е 11 о г х с h, Р. Е. S р е i d е 1, W. Т о е 1 d t е, Вег., 56, 769 (192$
611] V, Сеге he z, Bull. Soc. chim. France, 43, 762 (1928). ’ ,
612] F. Ullman n, P. Wenner, Ber., 33, 2476 (1900); Ann., 327, 114 (1903h
613] D. M. Musser, H. Adkins, J. Am. Chem. Soc., 60, 667 (1938). 4
614] W. И. P e г k i*n jr., C. W e i z m a n n, J. Chem. Soc., 89, 1649 (1906). 4
615] G, S. TIiers. F. D. Hager, Org. Synth., Colt, у. 1, 1941, p. 58. w
I. ПОЛУЧЕНИЕ ПРОСТЫХ ЭФИРОВ
34
временно происходит омыление и метилирование [616J. В некоторых случаях [617
раствор NaOH целесообразно заменять растворами NasCOa или NaHCO3.
Для метилирования неустойчивых в щелочах веществ можно также осторожн
приливать по каплям раствор щелочи к смеси метилируемого соединения и диметил
сульфата с такой скоростью, чтобы щелочь быстро расходовалась [618]. По друтом-
способу к водпому раствору неустойчивых в щелочах алкилируемых соединений из дву,
капельных воронок при энергичном перемешивании приливают одновременно диметил
сульфат и 30%-ный раствор NaOH так, чтобы реакционная среда оставалась Практи
чески центральной.
Пента-0-метил-Р-2>’Глюкоза [619]. Растворяют 37 г а-2>-глюкозы в 10 м.
воды, смешивают примерно с 30 г (22 мл) диметилсульфата к погружение]
в теплую воду кагревают до 35° С. Затем медленно по каплям при энергично]
перемешивании добавляют 38 мл раствора 109 я NaOH в 190 мл воды. Темпе
ратуру повышают сначала до 40° С, а затем постепенно в течение 3 ч до 60° С
поддерживая ее далее на этом уровне. Одновременно, продолжая кепрерывд
перемешивать, из двух капельных воронок примерно эквивалентными порциям]
медленно приливают остаток щелочи н диметилсульфат, всего 143 г (109 мл)
В заключение температуру повышают до 100е С, перемешивают еще 30 ми,
л охлаждают. Продукт экстрагируют хлороформом, сушат и фракционируют
После отгонки хлороформа почти вся масса перегоняется при 108—110° <
(0,23 лм* рт. ст.). Продукт затвердевает сразу При внесении затравки пента
О-метпл-р-2>-глюкозы; т. пл. 39—41° С; [а]р — 13,3°.
Исчерпывающее метилирование сахарозы диметилсульфатом в эфирно]
растворе достигается по методу Бредерека и сотр. [620].
4. Получение простых эфиров
алкилированием эфирами п-толуолсульфокислоты *
Эфиры ароматических сульфокислот подобно диметилсульфату являются силъ
пыми алкилирующими агентами. Для алкилирования фенолов эфирами арнлеульфа
кислот могут применяться как водные растворы фенолятов, так и их растворы в абсо
лютных спиртах [621]. Часто при взаимодействии сухого фенолята щелочного металл,
в ацетоновом растворе кли по Клайэену [557] смеси фенола, ацетона и безводного
К2С03 с эфиром сульфокислоты реакция алкилирования протекает гладко. По Дра
ховцалю и Кламанву [622], фенолы алкилируются эфирами п-толуолсульфоиислоп
в водном растворе NaOH, причем эфиры n-толуолсульфокислоты н первичных одна
и многоатомных спиртовТполучаются с хорошими выходами. Эфиры сульфокисло
и вторичных или алициклических спиртов реагируют с фенолами хуже.
• Вопросам алкилирования фенольного гидроксила алкил-га-толуолсульфонатамт
посвящены классические исследования Б. М. Родионова. Особенно эффективен
способ алкилирования аминофенолов четвертичными аммониевыми соедщ
нениями на их основе [В. М. Родионов, Избр. труды, АН СССР, 19511
стр. 26—71]. — Прим. ред.
[616] К. Freudenberg, Ann., 433, 230 (1923); W. N. H a w о г t h, H. Ma
chemer, J. Chem. Soc., 1932, 2270.
[617] Герм. пат. 122851, ср. H. Meyer, Mh. Chem., 24, 837 (1903); G. Wolf
J. Am. Chem. Soc., 75, 2673 (1953); G. W. G г a y, J. В. H a r 11 e у. A. I h
b о tson, J. Chem. Soc., 1955, 2686; F. Faltis, G. Wagner, E. Adler»
Ber., 77, 686 (1944); см. также H. S hapiro, K, A. Smith,!. Chem. Soc.
1946, 143.
[618] H. D ecke г, О. К о c h, Ber., 40, 4794 (1907); К. Freudenberg, Ber.
53, 1424 (1920).
[619] W. N. Haworth, G. C. L e i t c h, J. Chem. Soc., 113, 194(1918).
[62oj H. Bredereck, G. Hagelloch, E. Hambach, Chem. Ber., 87, 3J
(621| d i, Ber., 53, 1839 (1920).
• [622] F. Drahowzal, D. Klamann, Mh. Chem., 82, 588 (1951).
1
342 ОБРАЗОВАНИЕ НОВЫХ С —О-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ
Фенетол (феннлэтиловый эфир) [622], Растворяют 19 г (0,2 моль)
фенола в 65 мл 3 н. раствора NaOH (0,2 моль NaOH), смешивают с 40 г этил-п-
толуолсульфоната и при перемешивании нагревают в колбе с обратным холо-
дильником на водяной бане 1 ч. Затем добавляют еще 10 мл б п. раствора NaOH
и нагревают ЗО-иин. После охлаждения отделяют органический верхний слой,
растворяют в эфире, раствор промывают разбавленным раствором NaOH, водой
и сушат. После отгонки эфира фенетол перегоняют в вакууме; выход 19,2 а •
(78,7% от теоретического); 1,5075.
Выделение чистого алкилсульфоната требуется не во всех случаях; соответству-
ющие эфиры можно получить и из п-толуолсульфохлорида [623, 624], спирта и фенола. •:
Однако при этом необходимо, чтобы после взаимодействия сульфохлорида со спиртом J
в реакционной смеси перед добавлением фенола была установлена требуемая для;-/
оптимального алкилирования концентрация щелочи.
Неролин (р-нафтилэтиловый эфир) [623]. При 15° С смешивают 47,5 a- J
(0,25 моль) n-толуолсульфохлорида и 20 г (0,45 моль) этилового спирта с 40 мл
25%-ного раствора NaOH и 3 ч перемешивают при этой температуре. Затем у
к полученной массе добавляют 29 е (0,2 мОль) g-пафтола, 8,0 г (0,2 моль) NaOH 'j
и 10 мл воды; таким образом достигается примерно 4 и. концентрация щелочи.
Эту смесь нагревают 30 мин на кипящей водяной бане, смешивают с 10 мл 'Я
6 н. раствора NaOH и снова нагревают 30 мин. После охлаждения отсасывают
образовавшуюся кристаллическую массу, промывают разбавленной щелочью^
и водой и высушивают. Выход неролина 33,0 г (96% от теоретического в расчета »
на Р-нафтол); т. пл. 36° С.
Алифатические спирты тоже можно алкилировать до соответствующих эфиров.з
при помощи эфиров л-толуолсульфокислоты. Для Этого к безводному спирту доба- J
вляют необходимое для образования алкоголята количество натрия илп в избытке а
спирта растворяют рассчитанное количество КОН и раствор вводят в реакцию с эфи-'д
ром rt-толуолсульфокислоты. Выходы по обоим методам равны 70—80% от теорети-а|
ческого. При алкилировании менее доступных высших алифатических спиртов алко-Ж
голяты натрия получают в присутствии бензола и полученную реакционную смесь»
подвергают взаимодействию с сульфонатом при температуре кипения. ' '
Этил-н-бутиловый эфир [623]. В 50 мл абсолютного этилового спиртаШ
растворяют 5 е натрия (0,22 моль) и постепенно добавляют 46 г м-бутил-л-толуол*У|
сульфоната (0,2 моль). Реакцнопвую смесь медленно подогревают прд переезд
мешивании, причем начинается довольно бурное взаимодействие. Когда прекря---®
щается экзотермическая реакция, массу кинятят еще 30 мин в колбе с обратий»^
холодильпиком, охлаждают, добавляют 10%-ный раствор NaCl и встряхивают.^
Выделившийся после этого эфир отделяют, несколько раз промывают раство-^З
ром NaCl, сушат над Na2SO4 к перегоняют. Выход 15,8 г (77,5% от теоретиче-.я
ского); т. кип. 90—91® С. • «
5. Получение простых эфиров алкилированием диазометаном 1
Взаимодействие диазометапа с фенолами происходит в мягких условиях в ии- •,
дифферентной среде (эфир, спирт или хлороформ) и может быть использовано главным^
образом для количественного метилирования неустойчивых и труднодоступных
фенолов:
С6Н5ОН—CH2N2 —> CeH60CH3+N2 j
Диазометан — газообразное вещество, чрезвычайно ядовитое, поэтому.)
работу следует вести в хорошо действующем вытяжном шкафу. В неразбавленному
состоянии* диазометан сильно езривает]
♦ Взрывоопасен диазометап и в эфирном растворе, см. Синтезы органических-*
препаратов, сб. 12, пер. с англ., Изд. «Мир», 1964, стр. 24. Прим. ред. 2
[623] F. Drahowzal, D. Klamann, Mh. Chem., 82, 594 (1951). ,
[624] J. C. Clayton, G. F. H. Green, B. A. Hems, J. Chem. Soc., 1951.
2467; P. S c h’o rigin, J. Makaroff-Semljanski, Ber., 65, 1293 1
[1932).
II. ПОЛУЧЕНИЕ СЛОЖНЫХ ЭФИРОВ
343
Тян лак этерификация алифатических спиртов диазометапом обычно протекает
медленно, то этерификацию карбоновых кислот или фенолов диазометаном можно
вести в спиртовых растворах. Однако, по данным Меервейна и Гинца [625], метилиро-
вание спиртов диазометаном значительно облегчается при добавлении к реакционной
смеси комцлексообразователей, повышающих кислотность, таких, как ZnCla, MgCle,
FcCl3 м.тп алкоголяты алюминия. Точно так же метилирование становится возможным
при наличии в молекуле спирта заместителей, повышающих подвижность водорода.
Метил-н-бутиловый эфир [625]. В 5%-ный раствор бутилата алюминия
в «-бутиловом спирте пропускают 13 г диазометана. Выход эфира 19,3 а (83% от
теоретического); т. кип. 70—71° С.
При использовании в качестве растворителя гексана или гептана вместо
эфира в некоторых случаях удается провести метилирование алифатических
спиртов диазомеганом и без катализатора [626].
II. Получение сложных эфиров
1. Получение эфиров карбоновых кислот из карбоновых кислот
и спиртов
Спирты реагируют с кислотами с выделением воды и образованием слож-
ных эфиров [627]:
ВСоОН-]-Й'ОИ ЛСООВ'4-Н20
Эта реакция обратима; по закону действия масс выход сложного эфира повы-
шается с увеличением концентрации одною из реагирующих веществ. Такой же эффект
дает удаление образующейся воды или эфира из сферы реакции. Обычно реакция эте-
рификации практически осуществима только при повышенной температуре, особенно
в присутствии кислотного катализатора.
Скорость образования сложных эфиров зависит от строения кислот и спиртов.
Первичные спирты этерпфицируются с большей скоростью, чем вторичные иии тре-
тичные; та же последовательность наблюдается и в реакционной способности первич-
ных, вторичных и третичных карбоновых кислот. Этерификация третичных спиртоа
обычным способом в присутствии кислотного катализатора затруднена, так как прж
этом легко образуются олефины. В ароматическом ряду стеричсскке факторы могут*
вызвать уменьшение скорости этерификации; в частности, орто-замещенные карбоно-
вые кислоты этерифпцируются с трудом.
Этерификация без катализатора
Поскольку каждая карбоновая кислота, соответственно се силе, должнщ
являться катализатором собственной этерификации, образование сложного эфирщ
возможно уже при простом нагревании кислоты н спирта. Сильные карбоновые кис-
лоты, например муравьиная, щавелевая или пировиноградная, образуют соответству-
ющие сложные эфиры при обычном нагревании со спиртом. Этот метод исполь-
зуется в тех случаях, когда применение минеральных кислот в качестве катализаторов
исключается в связи с Неустойчивостью этерифицируемых карбоновых кислот в сНльно-
кислой среде.
Выход сложного эфира может быть значительно увеличен непрерывным удале-
нием воды из сферы реакции. Если все компоненты реакции кипят при более высокой
температуре, чем вода, то воду можно просто отогнать. Примерами таких реакций
являются синтез триглицеридов из глицерина и труднолетучих высших жирных кис-
лот и синтез амилового эфира пировиноградной кислоты. По Симону [628], этот эфи^
1625] Н. Meerwein, G. Hinz, Ann. 484, 1 (1930).
(626) Н. Meerwein, Th. В era in, W. Burneleit, Ber,, 62, 1006 (1929)
[627] H. Henecka, в книге Hou ben - Weyl, Methoaen der organischen Che-
mie, Bd. 8, 4 Aufl., Georg Thieme Verlag, Stuttgart, 1952, S. 516.
(628) L. Simon, Ber., 26, Ref. 769 (1893).
344 ОБРАЗОВАНИЕ НОВЫХ С—О-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИ1
получается с 99%-ным выходом при нагревании эквимолярных количеств пировино-;
градной кислоты и амилового спирта в вакууме, создаваемом водоструйным насо*
сом. После установления равновесия, что достигается через несколько часов, мед:
ленно отгоняют образующуюся воду.
Бензиловый эфир фенилуксусной иислоты. Нагревают до 150—160°
смесь 1 моль фенилуксусной кислоты и 2 моль бензилового спирта в круглодонно
колбе емкостью 500 мл с нисходящим холодильником. При этом отгоняет
смесь бензилового спирта н воды. От дистиллята отделяют верхний воде
слой, а бензиловый спирт возвращают в колбу. Через 1 —1,5 суток ре
заканчивается, причем за это время отгоняется 1 моль воды, образующ
прн этерификации. Реакционную массу разгоняют в вакууме и получают бензВЁ
ловый эфир фенилуксусной кислоты с выходом 95% от тсоретическог
т. иип. 270° С (166 мм риг. ст.).
Непрерывное удаление реакционной воды азеотропной перегонкой после д
вления соответствующих компонентов, например бензола, толуола, ксилола или гал|
генсодержащих алифатических растворителей («азеотропная этерификация»), пр
водит к быстрой этерификации, большей частью с высокими выходами. Течение реа
ции можно непосредственно контролировать по количеству выделяющейся воды. П
меняемые для этого приборы — водоотделители, общеизвестны [629].
Тильпапе [630] рекомендует пропускать отгоняющуюся азеотропную см
через гильзу экстрактора, заполненную карбидом кальция, и удалять таким обра
реакционную воду из органической фазы перед возвращением ее в реакцию.-
При этерификации труднолетучей кислоты низкокипящим спиртом мои
непрерывно отгонять водный спирт из кипящей реакционной смеси и непрер
возвращать последний после сушки над прокаленным карбонатом калия в реак
Диэтиловый эфир щавелевой иислоты [631]. В колбу помещают с
180 г безводной щавелевой кислоты и 500 мл этилового спирта и отгон
водный спирт через короткую колонку во вторую колбу, в которой нахо
200 а прокаленного К2СО^ под слоем 250 мл спирта. Вторую колбу нагрева
до слабого кипения, чтойд пары обезвоженного спирта по наклонной тр
поступали в колбу с реакционной массой. После 5 ч нагревания по
диэтиловый эфир щавелевой кислоты с выходом 80—90% от теоретическо
Для этерификации нестойких спиртов и кислот оправдали себя методы,
которых выделяющаяся вода связывается химически уже во время реакции, напр
добавленным молярным количеством карбодиимида [632].
Эфиры муравьиной кислоты образуются при нагревании этой кислоты с
ветствующим спиртом [633]. Этнлформиат, например, получают медленной nepej
кой смеси 35 мл этилового спирта, 20 мл муравьиной иислоты и 50 мл воды. Про
отгоняется при 54° С; выход 83,4% от теоретического.
Моноэтиловый эфир винной кислоты гладко образуется при перегонке вини
кислоты с абсолютным этиловым спиртом; продукт реакции кристаллиз
после добавления воды [634].
Моноэтиловый эфир щавелевой кислоты [635}. Медленно нагрев
до 135° С 1 ч. обезвоженной Щавелевой кислоты с 1 ч. абсолютного этилов*
спирта. После охлаждения отфильтровывают избыточную щавелевую кисл
и упаривают фильтрат при пониженном давлении на масллиой бане прн т
ратуре не выше 140° С. Процесс этерификации контролируют по разлож
[629]
630
631
632
633
634
635
Gat ter m an n - W i el a n d, Die Praxis des organischen Chemikers,.
33 Aufl., Berlin, 1948, S. 177.
E. Th i e I ej> a p e, Ber., 66, 1454 (1933).
J. Kenyon, Org. Synth., Coll. v. 1 1933, p. 258,
Примеры см. ссылну [627], стр. 521.
F. В о d г о u х, С. г., 160 , 204 (1915); М. П. Р а 1 о m а а, С., 19f3, 11, 1
Guerin, Ann., 22, 252 (1837).
R. Anschtitz, Вег., 16, 2413 (1883).
11. ПОЛУЧЕНИЕ сложных ЭФИРОВ
34
растворенной свободной щавелевой кислоты, определяемому по повышения
давления. По окончании разложения давление вновь понижается. В ааключмт,
реакционную смесь перегоняют при разрежении, создаваемом водоструйнщ
насосом. Фракционированием получают чистый моноэтиловый эфир щавелевО]
кислоты; т. кип. 117° С (15 мм рт. ст.).
Особенно легно протекает этерификация глицерина высшими жирными кисло
тами. Реакция идет без всяких добавок, однако значительно ускоряется в пр®
сутствии цинковой пыли.
Три-н-тридецилглицерид (получение по Феркаде, Ли н Меербургу [638
в модификации Вейганда). Смесь За н-тридекановой кислоты, 1,1 а перегнав
ного безводного глицерила и щепотку цинковой пыли постепенно нагреваю
от 130 до 200° С (температура бани) в медленном токе СО, при разрежена
150 мм рт. ст. в течение 7 ч. Затем понижают давление до 120 мм рт. ст,
а температуру доводят до 240° С. Еще теплое содержимое колбы растворят
в эфире, отфильтровывают цинковую пыль и цинковую соль и, не охлаждая
нейтрализуют спиртовым раствором КОН по фенолфталеину. Далее обесцвеч®
вают жидкость животным углем и снова фильтруют. Приливают двойной объаз
этилового спирта и, охлаждая льдом, оставляют для кристаллизации на иоч®
Выпавший триглицерид отсасывают и тщательно промывают холодным спиртом
После сушки т. пл. 43,0’ С.
Этерификация в присутствии катализатора
Б качестве катализаторов этерификации применяют преимущественно HaSO4
НС1, НдРО4, арилсульфоновые кислоты, кислые соли или кислые катионообменньа
смолы. Минеральные кислоты действуют не только каталитически, но и-как водоотн®
мающие средства, поэтому часто применяются в больших, превышающих каталитв
ческие, количествах, обычно 5—10% от массы карбоновой кислоты.
Метод этерификации в присутствии HsSO4 состоит в нагревании смеси нарбс
новой кислоты с четырех-, пяткиратиым количеством спирта в течение 5—7 ч. Д<
Этерификации пирнднн- или хинолинкарбоновых кислот [637] требуется значителЬд
большее количество минеральной кислоты, чем для этерификации обычных карбонбвыг
кислот. Однако очень высокая ионцентрация серной кислоты часто привода
к осложнениям-.
Соляную кислоту (чаще всего газообразный НС1) пропускают в спиртовой ра<
твор карбоновой кислоты до его насыщения. Этот метод позволяет проводить этер®
фикацию нестойких соединений в мягких условиях при комнатной температуре
а иногда даже при охлаждении. Однако обычно нагревание с обратным холодильника
необходимо л в случае применения соляной кислоты; постоянная концентрация НС
при этом поддерживается дополнительным медленным введением газообразного ИС;
Этиловый эфир бензойной кислоты [638]. К 50 г бензойной кислот:
и 100 г абсолютного этилового спирта приливают 10 г концентрированной Н,8С
и смесь нагревают 3 ч с обратным холодильником. После этого бдльшую час*
спирта отгоняют на водяной бане. К остатку приливают пятикратное ноличест®
воды и нейтрализуют твердой содой. Выделившееся масло извлекают эфнрох
эфирный раствор сушат К2СО8 и после отгонки эфира остаток перегоняю^
Выход этилового эфира бензойной кислоты 55 г (90% от теоретического;
т. кип. 95° С (17 мм рт. ст.).
При использовании в качестве катализатора хлористого водород
(100 г абсолютного этилового спирта, содержащего 3% НС1) смесь нагреваю
2 ч и выделяют эфир бензойной кислоты аналогичным способом. Выход 76% с
теоретического.
1636] Р. Е. Verka de, J. van der Lee, W. Meerburg, Rec. trax
chim., 51, 851 (1932).
1637] R. Camps, Arch. d. Pharm., 240 , 346 (1902).
[638] E. Fischer, A. Speier, Ber., 28, 3252 (1895).
346 ОБРАЗОВАНИЕ НОВЫХ С—О-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ
Азеотропный метод этерификации с кислотным катализатором позволяет зна-
чительно сократить количество добавляемых минеральных кислот. Их часто можно '
заменять алкил- или арилсульфокислотами, вследствие чего уменьшаются побочны^ •
процессы. Азеотропной этерификацией переводят в эфиры первичные и вторичные
спирты; третичные спирты обычно ие этерифицируются и таким способом.
Метиловые эфиры карбоновых кислот не могут быть получены обычным методом '
азеотропной этерификации. По методу Клинтона и Лесковского [639] для получения'
метилового эфира карбоновую кислоту нагревают с метиловым спиртом 6—15 ч в при-
сутствии HjjSOg в качестве катализатора и хлористого метилена или дихлорэтана,
не удаляя реакционную воду. После окончания реакции отделяют органический слой, .
содержащий сложный эфир, промывают его раствором NaHCO3 и выделяют продукт '
реакции.
Разработан другой метод получения метиловых эфиров, по которому для свя- ?
зывания воды, выделяющейся при этерификации, используют диметилацеталь j
ацетона [640]:
лсоон+снзон — - псооснз ;-и,о !
(СНзО)аС(СНз)а-ЬНаО —> 2СНзОН + (СНз)гС-О '
Алифатические эфиры аминокислот получают нагреванием аминокислоты со •
спиртом и избытком хлористого водорода.
Гидрохлорид эфира глицина [641]. Смесь глицина со спиртом насы-,,,
щают НС1 и кипятят 4 ч в колбе с обратным холодильником. По охлаждении ,
гидрохлорид эфира выпадает в кристаллическом виде; выход может быть повы- .'1
шен выделением эфира из маточного раствора. J
Гидрохлорид n-аминофенилуксусной кислоты [642] уже при перекристаллнза- 1
ции из спирта переходит в гидрохлорид соответствующего этилового эфира.
По разработанному Мейером [643] методу этерификации карбоновую кислоту X
растворяют в избытке концентрированной серной кислоты (иногда при нагревании)^
и к раствору осторожно приливают небольшой избыток соответствующего спирта, л
После окончания часто бурной реакции (если масса не разогрелась, ее подогревают) «1
реакционную массу выливают на твердый карбонат натрия. Прн этом выделяется слож- а
ный эфир. Метод находит применение при этерификации стеричеснн затрудненных кис-
лот, карбоновых кислот класса азотсодержащих гетероциклов и при получении эфиров J
ацетокдикарбоновой кислоты из лимонной кислоты и спиртов [644]. -й
Наряду с уже упомянутыми выше катализаторами этерификации для этой
цели часто с успехом применяют безводные соли [645], например сульфаты меди, же-
леза, никеля, цинка, кобальта к марганца, а также пиросульфат калия. Оксикислоты I
(молочная, а-оксимасляная и оксивалериановая) гладко этерифицируются в присут- J
ствии обезвоженного сульфата меди [646]. Я
Исключительно эффективными катализаторами этерификации оказались хлор- 3
ангидриды кислот, например тконнлхлорид, ацетилхлорид, стеароилхлорид и эфиры J
клормуравьиной кислоты [647]. Эти катализаторы следует предпочесть минеральным «|
кислотам, особенно при этерификации веществ, чувствительных к нагреванию. В при- *1
сутствии небольших количеств хлорсульфоновой кислоты (0,01—0,05 моль) с хорошими $8
[639] R. О. С 1 i n t о и, S. С. L a s к о w s к i, J. Am. Chem. Soc., 70, 3135 (1948). |
[640] N. B. L ore t t e, J. H. Brown jr. J. Org. Chem., 24, 261 (1959). fl
[641] Th. Curtins, F. Goehel, J. prakt. Chem. [2], 37, 159 (1886). J
[642] II. S alkowski, Ber., 28, 1917 (1895); B. F I dr sc he j m, J. prakt. 3
Chem. [2], 68, 347 (1903). 1
[643] H. Meyer, Mh. Chem., 25, 1202 (190-1). J
[644] G. S c Ii г о c t e r, Ber., 49, 2710 (1916). j
J645] A. В ogn Jawlensky, J. Narbutt, Bor., 38, 3344 (1905). '3
[646] E. CI e m ш e nsen A. H. С. H e i t ma n, J, Am. Chem. Soc., 42, 319 -3
(1909). , I
[647] K. Freudenberg, W. J akob, Bor., 74, 1001 (1941). я
*4
J
II. ПОЛУЧЕНИЕ СЛОЖНЫХ ЭФИРОВ
34
выходами получаются эфиры даже трудно этерифицпруемых кислот, например эфи
2-февилх11иолив-4-карбоновоп кислоты [648].
Кроме того, в качестве катализаторов этерификации оправдали себя диметил
сульфит [<j49], трифторид бора [650—652], ароматические сульфохлориды [653
и хлорокись фосфора [654].
Этерификации в присутствии хлорокиси фосфора. Растворяют (или cj
спендпруют) 0,1 моль карбоновой кислоты в трех-пятикрагиом количеств
спирта. При перемешивании и охлаждении в течение 20—30 мин прибавляв:
по каплям 3—5% РОС1Э (в расчете на карбоновую кислоту), поддержива
температуру пе выше 30—40° С.
Можно также прибавлять РОС13 в больших количествах при встряхив<
пни; и этом случае реакционную массу нагревают 2—3 ч в колбе с обратны
холодильником или оставляют на 1—3 суток при комнатной температуре.
Для синтеза сложных эфиров, которые не могут быть получены обычным
методами этерификации, Штааб [655] рекомендует применять К,М'-карбонилдиими,
азол. Этим методом получают эфиры из кислот и спиртов, разлагающихся в снльн>
кислой среде.
Фенпловыи эфпр бензойной иислоты. К смеси 2,44 з бензойной кислот
и 30 мл сухого тетрагидрофурана при комнатной температуре прибавляв
3,24 г карбонилдиимидазола. По окончании выделения СО2 добавляют 1,88
фенола и нагревают еще 1 ч с обратным холодильником. После испарения тетр
гидрофурана и промывки водой остается почти чистый фениловый эфир бензо
ной кислоты; т. пл. 70°; выход 85% от теоретического.
Для получения сложных эфиров третичных спиртов раствор кислоты и спир
обрабатывают п-толуолсульфохлоридом [653]. Эта реакция протекает через ангидрид:
которые немедленно реагируют с соответствующими спиртами *.
Эфиры третичных спиртов [653]. Карбоновую кислоту растворях
в 20—50 ч пиридина, причем в некоторых случаях выпадает соль. К это»
раствору добавляют 2 моль бензол- или «-толуолсульфохлорида. После охлаж/
ния до 0и С приливают 1 моль спирта или фенола, смесь выдерживают 1 ч п
охлаждении льдом, затем выливают в трех-четырехкратный объем воды. Пол
чаются сложные эфиры с превосходными выходами.
По этому методу могут быть этерлфицированы даже третичные ацетиленкарС
нолы [656], которые из-за их малой реакционной способности в обычных условиях
реагируют.
Аммониевые соли карбоновых кислот реагируют со спиртами по следующее
уравнению:
RCOONHi + R'OH RCOOR'+NII3+H2O
На этой реакции основан метод этерификацпВ [657], особенно пригодный ц
* Третичные спирты и фенолы удобно ацилировать действием хлорангидрида в П]
сутствшг магния [A. Spasov, Вег., 75, 779 (1942)].— Прим., ред.
[648] J. Erdos, Aogew. Chem., 63, 329 (1951).
[649] J. С. S h е е h a n, R. С. O’N ell, J. Am. Chem. Soc., 72 , 4614 (1950).
[650] H. D. Hi n t on, J. A, N i e u wl an d, J. Am. Chem. Soc., 54, 2017 (19c
[651] T. В D orris, F. J. S owa, J. A. N ieuwland, J. Am. Chem. Soc.,
2689 (1934).
[652] F. J. Sowa, J. A. N i eu wl a n d, J, Am. Chem. Soc., 58, 271 (193<
T. B. Dorris, F. J. Sowa, J. Am. Chem. Soc 60, 358 (1938).
[653] J. H. Brewster, C. J. C i о t t i jr., J. Am. Chem. Soc., 77, 6214 (19?
[654] J. К 1 о s a, Arch, d, Pharm., 289, 125 (1956).
[655] H. A. Staab, Angew. Chem., 71, 194 (1959).
[656] J. К I о s a, Angew. Chem., 69, 135 (1957).
[657] E. M. Pilaehione, E. J. Costello, С. H. F 1 s h e J. Am. Ch.
Soc,, 73, 5265 (1951).
348 ОБРАЗОВАНИЕ НОВЫХ G—О-СВЯЗЕЙ Б РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ
исходных веществ, чувствительных к кислотам. По видоизмененному методу можно |
применять не безводные аммонийные соли, а нагревать карбонат аммония, карбоновую
кислоту и спирт в присутствии воды [658]. .
2. Получение сложных эфиров алкилированием солей карбоновых кислот-1
Алкилирование алкилгалогенидами ;
В некоторых случаях, особенно при стерических препятствиях (этерификацищ^
о,о-дизамещенных ароматических карбоновых кислот или третичных карбоновых)
кислот), рекомендуется получать сложные эфиры алкилированием солей карбоновых^
кислот по схеме:
RCOOMe-f-R'X —>- RCOOR'-f-MeX '•<
Этот метод имеет значение и в тех случаях, когда в реакцию могут вступать-
несколько гидроксильных групп или различные карбоксильные группы, в то время'
как желательно получить моноацильные соединения. . 7
Так, моноацилировэнные производные гликоля удобно получать из хлоргидрине
по схеме:
RCOONa + ClCH2—СН2ОП —* RCOOCH2—CH30H + NaCl *
Для проведения алкилирования соль карбоновой кислоты нагревнЮП
с галогенидом (при необходимости в запаянной трубке или в автоклаве) и выдей
ляют продукт одним из подходящих способов.
Для этерификации неустойчивых и малодоступных карбоновых кислот приме-,
няют чаще всего трудпорастворимые, легко получаемые серебряные соли, однако можяо>ч
использовать также свинцовые соли и соли щелочных металлов.
По наблюдениям Брюля [659], натриевые соли зачастую не реагируют гладкШ
с алкилгалогенидами; в этом случае взаимодействию благоприятствует добавление;
небольших количеств метилового спирта. Для успешного проведения реакции необЗД^
дпмо соль тонко измельчить. После получения свипцовых или серебряных солей ИЙ
промывают водой, спиртом и эфиром и затем сушат в вакуум-эксикаторе; следует избе-
гать сушки При нагревании.
Из галогенидов наиболее энергично реагируют иодиды, которые рекомендуете^
применять также из-ва их относительно высоких температур кипения. При этерифика-
ции некоторых соединений оправдало себя применение растворителей илн разбавит^
лей, например, нсилола, бензола, толуола, лигроина, хлороформа, эфира или ацетон!?»*
Можно обходиться и без выделения серебряных солей, если смесь кислоты с аЛ^
килгалогенидом в разбавителе подвергать взаимодействию с окисью серебра [660^
Алкилирование д.иметилсулъфатом
Для алнилирования солей карбоновых кислот применяются также диалкилсулн^
фаты; наибольшее значение имеет днметилсульфат. -<
Реакция протекает в мягких условиях по уравнению: ,.5
RCOONa-HCH3)2SO4 —> RCOOCH3 + CH3OSO3Na j
Полное использование диметидсульфата может быть достигнуто нагреванием-
в толстостенной запаянной трубке:
2RCO'ONa + (CH3)iSO4 - > 2ВСООСНз + ^а38О4 r-j|
Диметилсулъфат — сильный яд\ Поэтому с этим реагентом необходимо,
всегда работать под тягой и промывать использованную для реакций посуд$
раствором аммиака в метиловом спирте. '
Реакцию можно проводить в водном растворе при комнатной температуре ИЛ
при слабом нагревании [661]. Для этого карбоновую кислоту растворяют в рассчитан^
[658] R. R. Da Haro, Rev. Fac. Ci. quim., Univ. пас. La Plata, 25, 31 (1952)$
C„ 1957, 7891.
[659] J. W. Briihl, Ber., 35, 3627 (1902).
[660] C. Lieberm a«i n, S. Lindenbau., Ber.,
[661] A. Werner, W. Sey bold, Ber., 37, 3658
42, 1397 (1909).
(1904).
fl. ПОЛУЧЕНИЕ СЛОЖНЫХ ЭФИРОВ
349
ном количестве щелочи и при перемешивании по каплям прибавляют алкилирующий
агент. Дополнительное метилирование при более высокой температуре (60° С) в боль-
шинстве случаев гарантирует полное превращение. Избыток диметнлсульфата удаляю1]
нагреванием с раствором соды па водяной бане. Если метилирование в водном раствор»
невозможно, нагревают сухую калиевую соль карбоновой кислоты с диметилсудьфа-
том (662].
В органических растворителях свободные карбоновые кислоты удобно алкилц.
руются диметилсульфатом следующим способом [663]: ацетоновый раствор нарбойо-
вой кислоты, содержащий молярное количество диметнлсульфата, при перемешиваний
по каплям приливают к кипящей смеси ацетона с молярным ноличеством тонкоизмель-
ченного карбоната налия. Затем смесь кипятят при перемешивании в течение несколь-
ких часов.
Алкилирование диазометаном
Во всех случаях, когда другие методы по каким-либо причинам, например из-зе
неустойчивости молекулы кислоты, оказываются непригодными или при этерификации
малых количеств труднодоступных карбоновых кислот, можно проводить алкилирова-
ние диазоалканами. Реакция протекает в индифферентной среде, спирте или эфира,
без существенных побочных реакций и удобна в исполнении (см. [664]). Она исполь-
зуется предпочтительно для получения метиловых эфиров при помощи диазометана:
RCOOH + CHaNa —> RCOOCH3 + N2
Следует помнить, что диаэометан чрезвычайно ядовитый газ, работз
с которым необходимо проводить под хорошо действующей тягой. Неразбавлен-
ный диазометан сильно взрывает, поэтому при этерификации большого коли-
чества няслоты диазометаном требуется принимать меры предосторожпоспг
Лучше всего пользоваться эфирным раствором дназометана *. Растворы неустой-
чивы и постепенно обесцвечиваются с выделением азота. Растворы диазометана можн<
сушить при помощи КОН, но при атом всегда происходят потери. Спиртовые растворы
диазометана еще более неустойчивы, чем эфирные.
Этерификацию карбоновых кислот ведут в эфирных растворах при комнатной
температуре. Трудиорастворимые в эфире кислоты растворяют предварнтельнс
в индифферентном по отношению к диааометану растворителе и затем этот pacTBOj
постепенно вводят в эфирный раствор диазометана до превращения выделения азота.
Иногда смесь умеренно нагревают; небольшая добавка метилового спирта ускоряет
метилирование [665]. Если после прекращения выделения азота сохраняется желта»
окраска диазометана, то можно считать, что этерификация пфошла полностью; в про-
тивном случае проводят добавочное алкилирование новой порцией раствора диазо-
соединения. Дальнейшая обработка реакционных растворов зависит от природы обра-
зовавшегося эфира. Обычно раствор фильтруют, чтобы отделить возможно обра.-
золавшиеся полиэтиленовые соединения, и отгоняют растворитель.
3. Получение сложных эфиров ацилированием спиртов н фенолов
Ацилирование хлор ангидридами кислот
Взаимодействие хлорангидридов карбоновых кислот со спиртами иди фенолам»
тоже приводит к образованию эфиров карбоновых кислот
RCOCl + R'OH —> RCOOR'4-HCl
Реакция имеет особое значение для идецтификацпн спиртов и фенолов по и;
ацильпым производным. Кроме того, по этому способу легко получаются сложны
* См. примечание на стр. 342-
[662] С. Graebe, Ann., 340, 246 (1005).
[66.3/ Си. ссылку [627], стр. 542.
[664] В. Е i s t е г t, в книге W. Foerst, Neuere Methoden der praparativen orga
nischen Chemie, 2 Aufl., Verlag Chemie G. in. b. II., Berlin, 1944, S. 359ff.
[663] В. E i в t e т t, F. А г a d t, L. Loa we, A. A v c a, Chem. Ber., 84, IS-
(1951). J ’
350 ОБРАЗОВАНИИ НОВЫХ С О-СВЯЗПП в РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ
эфиры фенолов, являющиеся, например. пслодпымп продуктами для перегруппировки "
(Бриса. Хлорангпдридный метод применяется к том случае, когда эфиры не могут быть
получены другим методом, а также для этерификации третичных спиртов. В наиболее
простых случаях ацилирования оксисоединений с целью их идентификации [666}
оксисоедипение встряхивают с бензоилхлоридом или замещенным бензоилхлоридом
в присутствии 10%-ного раствора NaOH до полного израсходования хлорангидрида'
(реакция Шоттена— Бауманна [666)). Для возможно более полного бензоилирования j
рекомендуется применять 20 ч. раствора NaOH (20%-ныл раствор) на 6 ч. беизоилхло-'.
рида [667]. По данным Скраупа [668], для достижения почти количественного выхода ’
следует вводить 7 моль NaOH (10—30%-ный раствор) и 5 моль хлорангидрида на каж- '
дую гидроксильную группу. При ацилировании соединений, неустойчивых в щелочной
среде, можно применять разбавленные щелочи или растворы карбоната или бикарбо-
ната патрия. Реакция с алифатическими спиртами в большинстве случаев протекает: j
па холоду, с фенолами — лучше прп слабом нагревании. L
В методе Шоттена — Бауманна может применяться также ацо1илхлорид.4
Однако из-за высокой склонности ацетплхлорида к разложению лучше пользоваться ^
другими методамп ацилирования (реакция с уксусным ангидридом).
Усовершенствованием метода Шоттена — Бауманна является метод Эйнгорна^
[669], в котором вместо щелочи применяется пиридин. -ч
Растворяют ацилируемое вещество в пяти-десятикратном количестве^
сухого пиридина, охлаждают и постепенно прибавляют хлорангидрид; в неко-л
торых случаях рекомендуется добавлять хлороформ. .После многочасового?
стояния (6—10 ч) реакционную смесь вливают по каплям в холодную разбавлен-
ную серную кислоту или в воду.
В некоторых случаях этерификацию хлорапгидрпдамн удается провести и в от-$
сутствие щелочи или пиридина.
зй
Дифениловый эфир фталевой кислоты [670]. Смесь эквивалентных.^
количеств фталоилхлорпда и фенола оставляют на несколько часов, промываю!?
продукт реакции спиртом и затем перекристаллизовывают из спирта. Выходу
почти количественный; т. пл. 74—76° С. ™
']
Добавление магниевых опилок при получении фениловых эфиров способствуете
легкому и полному ацилированию [671]. ™
Вместо хлорангидридов можно применять смесь фенола, карбоновой кислоты |[
и переносчика галогена (Р0С18, РС13, РС15. SOC12) [672]. Этот способ имеет особенней
большое значение, когда хлорангидриды трудно доступны. «
Фениловый эфир гиппуровой кислоты [673]. Смешивают 10 г tohko-j
измельченной гиппуровой кислоты с 7 г фенола и при температуре водяной^
бани постепенно прибавляют 6—8 г РОС13. Смесь нагревают до полного раство-%
рения, выливают в ледяную воду, нейтрализуют раствором соды, сливают^
водный слой с мазеобразной массы и растирают с водой до полной кристалла*'^
эации; т. пл. 104° С (из этилового спирта). %|
[666] Е. Baumann, Вег., 19, 3218 (1886). |
[667] A. Panormow, Вег., 24, Ref. 971 (1891). -3
[668] Zd. Н. Skraup, Mh. Chem., 10, 390 (1891). |
[669] A. Einhorn, F. Н о 11 a n d t, Ann., 301,' 95 (1898); A. D е n i n g e
Ber., 28, 1322 (1895); F. U 1 1 m a n n, G. N a d a i, Ber., 41, 1870 (1908). JS
[670] F. I. В 1 i с к e, O. J. W e i n к a u f f, J. Am. Chem. Soc., 54, 331 J
(1932).
[671] A. Spassow, Ber., 75, 779 (1942); Org. Synth., 20, 21 (1940); E. H. M a n,J
F. W. Swarn c r, Ch. R House r. J. Am. Chem. Soc.. 73, 902 (1951). J
[672] M. Ncncki, J. prakt. Chem, [2], 25, 282 (1882); R. Seifert. J. prakt.?
Chem. [2], 3k 467 (1885). I
[673] F. Woi₽, Bor., 26, 1700 (1893). <|
п. получение СЛОЖНЫХ. ЭФИРОВ
Ацилирование фенолов удается проводить также в присутствии малых колпчес
серной кислоты [674] инн хлорида олова (IV) [671, 675, 676}.
Сложные эфиры а-нафтола [674]. а-Пафто.т в количестве 50—100
обрабатывают в колбе с обратным холодильником 10%-ным, против таорет
ческого, избытком соответствующего хлорангидрида. Если реакция протека
слишком вяло, в качестве катализатора добавляют одну каплю HaSO4. Пос.
2 ч стояния при комнатной температуре реакционную смесь нагревают 1 ч j
водяной бане и выливают в ледяную воду; продукт извлекают эфиром, про№
вают раствором бикарбоната и перегоняют в вакууме. Выход 90—95% от теор,
тического.
Ацилирование ангидридами кислот
Ацилирование спиртов и фенолов ангидридами кислот часто происходит прощ
чем ацилирование хлорапгидридами:
(RCO)2O + R'OH —> RCOOR'H-RCOOH
Добавка незначительных количеств концентрированной серной кислоты [677-
679], хлорной кислоты [680, 695], хлорида цинка [681—685], ацетилхлорида [63'
687], ацетатов щелочных металлов [688—692] или третичных оснований [693] значз
тельно увеличивает скорость реакции.
Эта реанция имеет особенно большое значение в химии углеводов (ci
обзор [694]).
Пента-О-ацетил-а-D-глюкоза [695]. Осторожно смешивают 1 л внгч
дрида уксусной кислоты с 0,5—1 мл 70%-ной НС104. В смесь при переметив,
нии и охлаждении вносят 250 г глюкозы, оставляют на 2 ч при комнатной тема
ратуре и образовавшийся раствор медленно выливают при перемешивай®
в 4 л ледяной воды. При этом выпадает пента-О-ацетил-а-Д-глюкоа,
Для разложения избыточного ангидрида уксусной нислоты массу оставляй
на 10—12 ч и затем отфильтровывают. Полученный продукт суспендируй
в 5 л воды, отсасывают и сушат на воздухе на фильтровальной бумаге. Выхс
390 г (70% от теоретического); т. пл. 109—110° С (из этилового спирта).
674]
675
676
677
678
[679
[680
[681]
[682
[683
[684
[685
[686
[687}
[688[
[689]
[690]
[691]
[692]
[693]
[694]
[695]
R. W. Stoughton, J. Am. Chem. Soc., 57, 203 (1935).
H. Huber, К, Brunner, Mh. Chem., 56, 325 (1930).
Герм. пат. 463518; H. Reihlen, С., 1929, I, 2235.
W. W. Prichard, Org. Syntb , 28. 68 (1948).
A. P. N. Franchimont, Ber., 14, 1920 (1881).
R. Burns, D. T. I ones, P. D. Ritchie,!. Chem. Soc., 1935, 403.
D. Kruger, W. Roman, Ber., 69, 1830 (1936).
E. Er wig, W. Koenigs, Ber., 22, 1464, 2207 (1889).
A. P. N. Franchimont, Ber., 12, 2059 (1879).
C. S. Hudson, J. K. Dale, J. Am. Chem. Soc., 37, 1264 (1915).
R. H. Baker, F. G. Bordwell, Org. Synth., 24, 18 (1944).
W. H. Perkin jr., J. L. Simonsen, J. Chem. Soc., 1905, 858.
F. A d i c kes, J. prakt. Chem., 161 275 (1943).
G. R. Clemo, S. B. Graham. J. Chem. Soc., 1930, 215.
C. Liebermann, Ber., 11, 1619 (1678).
И. Meyer, Mh. Chem., 34, 651 (1913).
W. S. E m e r s о n et al., J. Am. Chem. Soc., 68, 1665 (1946).
Ga 11 e rm a nn - W i c ] a n d, Die Praxis des organischen Chemikers»
33 Aufl., Berlin, 1948, S. 358.
T. J. Otterbacher, Org. Synth., Coll. v. 1, 285 (1941).
C. Liebermann, Ann., 331, 362 (1904).
F. M i c h о e 1, Chemie der Zucker und Polysaccharide, 2 Anfl., Akadem. V<=s
lagsgesellscliaft Geest n. Portig K. G Leipzig, 1956, S. 102.
S. D. Nicholas, F. S mi t li, Nature (London), 161, 349 (1948).
1
352 ОБРАЗОВАНИЕ НОВЫХ С—О-СВЯЗЕП П РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ
Ангидрид диацетилвинной кислоты [696]. К 10О г тонкоизмельченной
винной кислоты приливают смесь 120 мл уксусного ангидрида и 3 мл концентри- л
рованной H2SO4. Реакционная масса сильно разогревается и винная кислота*'
переходит в раствор. После непродолжительного кипячения раствор охлаждают,‘ ।
отфильтровывают кристаллизующийся в почти теоретическом количестве ангид- 'j
рид диацетилвинной кислоты п промывают его бензолом. Неочищенный продукт
может непосредственно использоваться для получения недокиси углерода. .
Для дальнейшей очистки ангидрид перекристаллизовывают из бензола; j
т. пл. 1359 С. I
Циклические ангидриды дикарбоновых кислот расщепляются спиртами до кис--*
лых эфиров [697—702]: *
RCH—СО RCH—СООП
I I '
RCH—СО RCH—COOR' ;
Этот метод используют для выделения спиртов из смесей при помощи фталевого-
ангидрида [698, 702]. $
4. Получение сложных эфиров переэтерификацией
g
При нагревании сложных эфиров со спиртами протекает реакция двойного
обмена, именуемая «переэтерификацией». Кислоты [703—706] или основания [707-я-.
712] оказывают на реакцию каталитическое действие:
RCOOR' R"OH RCOOR" + R'OII '•
Для смещения равновесия в желаемом направлении применяют большой избы*
ток спирта. . :
Алкоголиз сложных эфиров карбоновых кислот под влиянием щелочных ката*
лизаторов имеет особенно большое препаративное значение для синтеза эфиров тер#
мически нестойких карбоновых кислот с длинной углеродной ' цепью (иапрнмер#
0-кетокарбоповых[713—719] и эфиров спиртов, неустойчивых в кислых средах[715«.
[696]
[697]
[698]
[699]
[700]
[701]
[702]
[703]
[704]
[705]
[706]
[707]
[708]
[709]
[710]
[711]
[712]
[713
[714
[715
[716
|717
[718
[719
A. Wohl, С.' О е s t е г 1 i п, Вог., 34, 1139 (1901).
J. Cason, Org. Synth., 25, 19 (1945).
Е. L. Е 1 i е 1, A. W. В u г g s t a h 1 е г, J. Am. Chem. Soc., 71, 2252 (1945
R. Adams, J. M. Wilkinson jr., J. Am. Chem. Soc., 65, 2207 (194?
J. W. Bruhl, Ber., 26, 284, 1097 (1893).
E. Blaise, A. Koehler, C. r., 148, 489 (1909).
K. Freudenberg. K. Friedrich, I. Bumann, Ann., 494, 56 (193!
J. C. S a u e г, В. E. H a i n, P. W. В o’u t we 11, Org. Synth., 20, 67, 69 (194(
С. E. R о h b e r g, С. H. Fisher, 3. Am. Chem. Soc., 66, 1203 (194*
С. E. Re h beTg, Org. Synlh., 26, 18 (1946).
E. Fischer, M. Bergmann, Ber,, 52, 833 (1919).
R. В a 1 t z 1 y, J. S. Buck, J. Am. Chem. Soe., 63, 2022 (1941).
E. E. R c i d, Ind. Eng. Chem., 29, 1344 (1937).
J. Purdie, Ber., 20, 1555 (1887).
M. Reimer, H. R. Downes, J. Am. Chem. Soc., 43 , 945 (
R. Kremann, Mh. Chem., 26, 783 (1905); 29, 23 (1908); M. P f - - - ...
Chem., 31, 301'(1010).
E. Fischer, Ber., 53, 1637 (1920).
G. Ze mpUn, A. Kunz, Ber., 56, 1705 (1923); G. Zemplen, Ber., S
1258 (1926); G. Zemplen, A. Gerecs, I. Hadacsy, Ber, 69, 18
. eters, Ber., 20, 3323 (1887).
Conrad, W. Epstein, Ber., 20, 3057 (1887).
Cohn, Mh. Chem., 21, 200 (1900).
Lapworth, A. C, O. Hann, J. Chem. Soc., 81, 1499 (1902).
Rape, E. Lenzinger, Ann., 395, 87 (1913).
Kagj, Ann., 420, 33 (19201-
... R, В a d e r. L. O, Cum miuif s, II. A. V о g e 1, J’Am. Chem. StK
73, 4195 (1951).
(1936).
Th. Р <
М. ”
Р.
И.
Н.
(1921).
> 1 а п и 1, МЬ
1П. ПОЛУЧЕНИЕ АЦЕТАЛЕЙ
353'
718], которые нельзя получать обычными методами этерификации. В качестве катализа-
торов таких реакций оказались пригодными алкоголят натрия ]708], едкий натр ]709,
710] и карбонат калия [711].
Трудно синтезируемый другими способами эфир антраниловой кислоты и гера-
ниола можно получить следующим образом.
Эфир антраниловой кислоты и гераниола. При 100—120° С нагревают
1 моль метилового эфира антраниловой кислоты с 1 моль гераниола в присут-
ствии 0,05 моль алкоголята натрия, алюминия или магния. При этом медленно
отгоняется метиловый спирт. В вакууме взаимодействие несколько ускоряется.
После того как прекратится отгонка, промывают остаток для удаления неболь-
шого количества непрореагировавшего метиловою эфира 10%-ной H2SO4, не-
разрушающей эфира гераниола, и после обработки получают золотисто-желтое-
масло, кипящее при 188° С (4 мм рт. ст.).
Таким образом удается провести обменное замещение первичных спиртов дру-
гими первичными или вторичными спиртами с более высокой температурой кипения.
Однако для получения эфиров третичных спиртов способ не пригоден. Эфиры третич-
ных спиртов получают взаимной переэтерификацией двух различных эфиров карбоно--
вых кислот [720], например эфира муравьиной и какой-нибудь другой кислоты:
НСООС(СНз)з + ЙСООСН3 ^2. КСООС(СНз)з + НСООСНз
Сначала получают эфир муравьиной кислоты и того третичного спирта,
которым желательно этерифицировать данную карбоновую кислоту, для чего
в течение нескольких суток выдерживают смесь третичного спирта с избытком
смешанного ангидрида муравьиной и уксусной кислот. Затем выделенный
обычным способом эфир смешивают в молярном соотношении с метиловым;
эфиром другой кислоты, прибавляют 0,05 моль свободного третичного
спирта и 0,05 моль натрия и нагревают до 100—120° С. При этом медленно
отгоняется наиболее низконипящий компонент равновесной смеси — метил-
формиат. Реакция иногда протекает довольно бурно и обычно заканчивается;
через 3—4 ч. После нейтрализации свободной щелочи разбавленной уксусной
кислотой обработку остатка проводят обычным образом.
Переэтерификация может быть осуществлена также действием свободных карбо-
новых кислот на эфиры:
RCOOH —R'COOR' RCOOR"+ R'COOH
Эта реакция широко употребляется для получения эфиров неустойчивых спир-
тов, например для синтеза эфиров различных карбоновых кислот и винилового спирта
из винилацетата [721].
Алкоголиз лактонов происходит с раскрытием кольца аналогично расщеплению:
сложных эфиров до соответствующих эфнров оксикарбоповых кислот [722—724].
III. Получение ацеталей [725, 726]
1. Получение ацеталей из карбонильных соединений и спиртов
Образование ацеталей из карбонильных соединений и спиртов в присутствие
кислотных катализаторов проходит гладко только в том случае, если устанавлива-
ющеося равновесие сильно сдвинуто вправо или сдвигается в сторону образованна
ацеталя удалением выделяющейся по реакции воды.
ROH
Альдегид-!-Спирт < * Полуацеталь Ацеталь-l-Вода
]72о] А. V о г 1 е у, Bull. Soc. chim. France (4], 41. 803 (1927).
1721] u. Swern, E. F. J ordaii jr., Org. Synth.., 30, 106 (1950).
f722f A. Baeyer, V. V i 1 1 i g e r, Ber., 33, 862 (1900).
[723] E. R. Mejncke, S. M. M с E 1 v a i n, J. Am. Chem. Soc., 57, 1444 (1935)
[72'jJ 'p. Gresham, J. E. Jansen et a]., J. Am. Chem. Soo., 70, 100-
(1948).
23 Заказ 1835.
354
ОБРАЗОВАНИЕ НОВЫХ С—О-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕННЫХ PEAK]
Эта реакция сильно зависит от строения реагирующего карбонильного соедине-
ния и от строения спирта. Первичные спирты реагируют лучше, чем вторичные н тре-
тичные. Трудность взаимодействия карбонильных соединений возрастает в последо-
вательности: формальдегид, алифатические альдегиды, а,р-ненасыщепные альдегиды,
ароматические альдегиды, кетоны. Циклические кетали очень легко образуются нз аце-
тона л 1,2- и 1,3-гликолрй.
Адамс и Адкинс [727] подробно изучили эффективные катализаторы ацеталнэа-
цин. Хорошие результаты были получены при применении хлорида кальция £728],
хлорида железа (III) [727], а также хлорида цинка, гидрохлорида пиридина и бисуль-
фата натрия [725].
Формальдегид, три изомерных нитробензальдегида, изо- и тарефтальдиальдегиды
можно превратить в ацетали при помощи диметилсульфата и щелочи. Из о-фтальдиаль-
дегида в этих условиях образуется 1,3-диметоксифталан [729].
Получение днэтилацеталя ацетальдегида с применением в качестве катализато-
ров СаС12 и ПС1 (выход 65% от теоретического) см. [730],
Метилаль [731]. К тонкоизмельченному параформальдегиду приливают
2,5-кратное количество 1%-ного раствора НС1 в метиловом спирте и нагревают,
при 100° С в течение 12—15 ч, причем весь параформальдегид растворяется
и запах формальдегида почтщисчезает. Раствор нейтрализуют раствором NaOH:
и фракционируют на аффективной колонке. Выход метилаля 80% от теорети-
ческого; т. кип. 41—42° С.
По другому способу метилаль'получают из 35—40%-ного формалина
прн действии 2,5-кратного количества 2%-ного раствора НС1 в метиловом
спирте. Для связывания воды к раствору добавляют равное количество CaCls,
причем смесь разогревается. Уже через 15 мин метилаль выделяется в видн;
масла. Через 15 ч его перегоняют и получают чистый метилаль с выходом 75% ах,
теоретического.
Особенно пригодными для получения ацеталей оказались способы, в которых
реакционная вода непрерывно удаляется из реакционной массы азеотропной пере-
гонкой [732]. В этом случае требуется относительно небольшой избыток спирту
(20—50%) и устраняются снижающие выход побочные реакции (осмоление, обра-’»
зование простых эфиров и олефинов), так как количество катализатора можно ав&-,
чнтельно уменьшить. Обычно бывает достаточно 0,1% толуолсульфокислоты; пригодны
также кислые ионообменники [733].
Ацетали ннэкоинпящих спиртов или карбонильных соединений, например ди-J
метил-и диэтилацетали ацетальдегида или диалкилкетали ацетона, такими методамИ?(
получать нельзяАиз-за трудности удаления реакционной воды отгонкой.
[725] О. В а у е г, в книге Houben-Weyl, Methoden der organischen Chemie,
Bd. 7/1, 4 Aufl., Stuttgart, 1954, S. 417.
[726] F. К 1 a g e s, в книге G. M. Schwab, Ilandbuch der Katalyse, 7/2, Spna-.
ger-Verlag, Wien, 1943, S. 305.
[727] E. W. Adams, H A d k i n s, J. Am. Chem. Soc., 44, 2749 (1922); 47, 1358,J
1368 (1925); 50, 178 (1928); W. H Hartung, H. Adkins, J. Am. Chem.
Soc., 49, 2517 (1927). , '1
[728] R. D. Haworth A. Lapworth, J. Chem. Soc., 1922, 80; Org. Synth.
1, 1 (1932).
[729] E. Schmitz, Chem. Ber., 91, 410 (1958). <
[730] II. Adkins, В. H. Nissen, Org. Synth., 3, 1 (1923).
[731] E. Fischer, G. Grebe, Ber., 30, 3054 (1897).
1732] E. J. Salmi, Ber., 71, 1803 (1938). „
,[733] Пат. США 2566559 (1947)- A. A. Dolnick, M. Potash, P. Mastagli, Z.4
Z a f i r i a rt i s, С. г., 23Й, 616 (1953): P. Mastagli, Z. Z a f i r i a d i s,
G. L a g г а n’g e, C. r., 237, 187 (1953).
III. ПОЛУЧЕНИЕ АЦЕТАЛЕЙ
2. Получение ацеталей из карбонильных соединений и ортоэфиров
Общий метод получения ацеталей или кеталей заключается во взаимоденствь
карбонильных соединений с ортоэфирами муравьиной кислоты или другими ортоэфз
рами (например, с метиловым и этиловым эфирами ортокремиевой кислоты) под вли;
нисм небольших количеств сильных кислот по схеме [734]:
R2CO + HC(OR')3 —* R2C(OR')2tHCOOR'
В качестве катализаторов применяются свободные кислоты, различные неорг.
ппческпе соли, например безводный FeCl3 или NH4C1.
Синтез осуществляется просто. Растворяют карбонильное соединена
в соответствующем спирте (в некоторых случаях 2 моль), прибавляют ортоэфй
(около 1.2 моль) и затем катализатор о ио окончании реакции выделяют ацетал
Если катализатор FeCl3, то реакция протекает очень быстро и бурно, в ирису
сгвии же NH4C1 — в большинстве случаев несколько медленнее. Учитыва
чувствительность ацеталей к действию кислот, их выделяют из щелочной нл
нейтральной среды.
Диэтилацеталь бензальдегида [734]. Смешивают 37,5 г бензальдегид
с, 57 г ортоэтилового эфира муравьиной кислоты и 49 г этилового спирта, прз
бавляют 0,15 мл концентрированной НС1, причем температура смеси быстр
повышается до 48° С, переносят на водяную баню и быстро нагревают до кии
нпя. Затем массу быстро охлаждают, слабо подщелачивают несколькими каплям
спиртового раствора КОН и перегоняют. После отгонки спирта и эфира муравы
пой кислоты при 217—223° С перегоняется диацоталь бензальдегида; вых<
63 г (99% от теоретического).
При применении вместо соляной кислоты тонкоизмельченного NH4(
(0,75г) смесь кипятят Юлим в колбе с обратным холодильником и получая
ацеталь тоже с почти количественным выходом. В качестве катализатора може
применять KHSO4 (0,3 г) или FeCl3.
Из альдегидов с реакционноспособными двойными связями (акролеин, кротон,
вый альдегид) ацетали могут быть получены действием этилового эфира ортомуравы
ной кислоты в присутствии нитрата аммония.
Диэтилацеталь акролеина [735]. К смеси 22 г стабилизованного акролеин
и 72 г этилового эфира ортомуравьиной кислоты прибавляют горячий растве
1,5г NH4NO3 в 15.0 абсолютного спирта; смесь кипятят 8—Юлин в кол?
с обратным холодильником, фильтруют и смешивают темио-коричневыи раствс
с, двукратным по объему количеством эфира. После этого раствор встряхиван
с разбавленным раствором NH4OH до прекращения выделения аморфноа
осадка, промывают водой и сушат над К2СО3. При перегонке в интервале 120'
125° С перегоняется 37,5 г диэтилацеталя акролеина (73% от теоретического
Образование кеталей из кетонов протекает точно так же, как и из альдегида;
Выходы обычно несколько ниже, однако большей частью достигают 80% от теоретз
ческого.
Ацетали получают с помощью эфиров ортокремиевой кислоты следующим обр;
зом [736]: смешивают 1 моль альдегида или кетона с 2—3 моль соответствующего бе;
водного спирта и 1,1 моль эфира ортокремиевой кислоты и добавляют 10 капель н<
сыщенного при комнатной температуре спиртового раствора IIC1 или пропускав
несколько пузырьков сухого НСГ. Затем смесь выдерживают л течение несколько
суток при комнатной температуре или некоторое время кипятят с обратным холодила
НИКОМ.
Диметилкеталь бензофенона непосредственно выкристаллизовывается с
реакционной массы с хорошим выходом, в то время как при получении многа
других ацеталей реакционную массу подвергают фракционной перегони,
[734] L. Claisen, Вег., 29. 1005 (1896); 40, 3903 (1907); 47, 3171 (1914).
[735] II. О. L. F i s с 11 е г, Е. В а е г, Helv. chim. acta, 18, 516 (1935).
1736] В. 11 с 1 f е г i с h, J. U a u s, е n, Ber., 57, 795 (1924).
ОБРАЗОВАНИЕ НОВЫХ С—О-ОВЯЗЕП В РЕЗУЛЬТАТЕ ОБМЕННЫХ
иногда в вакууме. Правда, выделенные таким способом ацегалн содержат п
меси соединений кремния, для разложения которых дистиллят кипятят с небо
шим количеством спирта и КОНЗили подвергают еще неразогианную см
следующей обработке. Массу выливают (если необходимо, при охлажден
в примерно 30%-ный раствор КОН (500 мл щелочи на 1 моль ортоэфщ
встряхивают около 10 мин при комнатной температуре для омыления или (j
работе с высшими спиртами) кипятят некоторое время в колбе с обратным хо
дильником. Спиртовой слой, освобожденный от соединений кремния, отделя:
сушат К2СО3 и разгоняют; выход 70—90% от теоретического.
3. Получение ацеталей прн помощи диметилсульфита
Для получения ацеталей диметилсульфит был впервые применен Фоссом [737
Реакция’протекает почти в таких же условиях, как и с ортомуравьиными эфирами^ в
в присутствии каталитических количеств хлористого водорода. Этот способ непригоДе
для получения ацеталей из алифатических кетонов.
Диметилкеталь циклогексанона. Смесь 49 г циклогексанона, 6о г ди»
тилсульфита, 60 мл метилового спирта и 1 мл 11,2%-ного раствора НС1 в мел
ловом спирте кипятят в колбе с обратным холодильником на водяной бащ
Через несколько минут начинается бурное выделение SO2. Массу нагрева^
3 ч, дают ей охладиться, подщелачивают несколькими каплями метилата натри
и встряхивают для разложения не вступившего в реакцию диметидсульфлт
с 200 мл 40%-ного раствора КОН. Затем разбавляют водой, извлекают продуй
эфиром и сушат К2СО8. Разгонкой получают 57 г неталя (выход 79% от теоретз
ческого); т. кип. 63—65° С (22,5 мм рт. ст.).
С таким же успехом могут применяться гомологи диметилсульфита. Ацетал
-высших спиртов получаются прн взаимодействии альдегида с диметилсульфитом в пра
сутствии соответствующего спирта. i
Ди-к-бутнлацеталь бензальдегида. Нагревают на масляной бане 10,5.
бензальдегида с 12 г диметилсульфита, 27 г н-бутилового спирта и нескольким
каплями 20%-ного раствора-ЙС1 в метиловом спирте. Через несколько мину
начинается выделение SO2. В течение 2 ч температуру бани повышают до 150-
160° С; при этом отгоняется 20 мл смеси метилового спирта с небольшим нолт
чеством бутилового спирта (температура 65—70° С в верху колонны). Остато
разгоняют в вакууме н получают 18,5 г ди-к-бутилацеталя бензальдегйд
(80% от теоретического); т. кип. 149—150е С.
Этим же методом удается получать ацетали высших спиртов, например ацет;
(цетилового спирта; таким способом можно превращать также углеводы в глн
зиды.
4. Получение изопропилиденовых и бензилиденовых производных
•углеводов
Образование простых гликозидов уже было описано (стр. 337). Здесь короче
будут рассмотрены ацеталеподобные изопропилиденовые и бензилиденовые пронзвОД
ныв углеводов. Обзор методов получения кеталей иа тетрнтов, пентитов и гексито!
см. [738].
Изопропилиденовые производные углеводов (ацетониды) [739] получав
обработкой углеводов или соответствующих их производных избытком ацетон
[737] W. V о р, Ann., 485, 283 (1931).
[738] S. A, Barker, Е. J. Bourne, Advances in Carbohydrate Chemistry, v.
Acad. Press, Inc. Publishers, New York, 1952, p. 137. • ,
£739] F. Micheel,» Chemie der Zucker und Polysaccharide, 2 Anil., Akadem. V
lagsgesellschaft Geest u. Portig K. G., Leipzig, 1956, S.J86.
Ш. ПОЛУЧЕНИЕ АЦЕТАЛЕЙ
357
в присутствен катализаторов — кислот [740}, хлорида цинка [741] или безводного
сульфата медн [742]. Кроме того, полезно добавлять вещества, связывающие воду,
напрнмер сульфат натрия.
Из D-глюкозы и ацетона гладко образуется 1,2;5,6-диизопропилиден-Д-глю-
коза, которая„является производным фуранозы, а не пнранозы.
1,2;5,6-Диизопропшшдем-В-глюкоза (диацетонидглюкоза) [743].
Суспендируют 65 в топкоизмельчепной и просеянной глюкоды в 1800 мл чистого
ацетона и механически встряхивают с 55 мл концентрированной HsSO4 (через
4—5 ч остается нерастворенным только около 5 в глюкозы). Осадок отфильтро-
вывают, добавляют к фильтрату избыток безводного NaaCO3 до тех пор, пока
темный раствор не станет светло-желтым, еще раа фильтруют раствор и выпари-
вают в ванууме досуха. Затем обрабатывают небольшим количеством
эфира, причем моноизопропилиденглюкоза выпадает в осадок, оставляют на
ночь в холодильниие для полного выделения мононзопропипиденглюкозы,
отделяют ее фильтрованием и к фильтрату постепенно прибавляют петролейный
эфир. Выделившуюся плотную кристаллическую кашицу диизопропнлиден-
глюкозы отсасывают, промывают петролейный эфиром и сушат; выход 45—50 з;
т. пл. 102° С (по другим данным, НО— 111е С); fcs]^ 19°.
Моноизопроцилиденглюноза, образующаяся в приведенном методе в качестве
побочного продукта, может быть получена по способу Фишера [744] из глюкозы и насы-
щенного НС1 ацетона при более кратковременном взаимодействии.
По данным Фишера [745], из D-маннита при действии ацетона образуются три-
изопроннлнденовые производные (триацетониды). Изучению строения этих соединений
посвящена работа [746].
Триацетонидманнит. Встряхивают 1 ч. тонкоизмельченного маннита
с 10 ч. сухого ацетона, содержащего 1% НС1. Через несколько часов весь-маи-
нит растворяется, еще через 12 ч кислоту нейтрализуют порошкообразным РЬСО3,
раствор фильтруют и выпаривают на водяной бане. Остающееся желтоватое
масло прн охлаждении затвердевает. Для очистки его переосаждают водой из
спирта; после сушки над H2SO4 т. пл. 68—80° С; [a]D -4-12,5°.
Циклические ацетали получают конденсацией альдегидов с полиоксисоедииениямн
ряда углеводов методами, аналогичными описанным для получения нзопропилидено-
вых производных. Реакция между D-глюкозой и бензальдегидом в присутствии димера,
пятиокиси фосфора [747] или хлорида цинка [748] приводит н получению 4,6-бензили-
ден-D-глюкозы.
4,6-Бензилиден^-глюкоза [748]. В течение 24 ч 130 г сухого виноград-
ного сахара встряхивают на качалке со 100 г тонкоизмельченного ZnCls и 300 мл
свежеперегнанного бензальдегида. К вязкой массе прибавляют 400 мл ледяной
воды, причем бензилиденглюкоза сразу выкристаллизовывается в виде длинных
[740]
[741
[742
[743
[744
[745
[746]
[747]
[748]
Е. F х s с h е г, Ch. П u n d, Вег., 49 , 93 (1916); Р. A. Levene, G. М. Me-
yer, J. Biol. Chem., 48, 233 (1921)- 57, 319 (1923); К. Freudenberg,
W. Durr, H.v. Hochstetler, Ber. 61, 1738 (1928); D. J. Bell,
J. Chem. Soc., 1935, 1874; II. van Gru ne n b erg, C. Bred t, W. Freu-
denberg, J. Am. Chem. Soc., 60, 1507 (1938).
H. O. L. Fischer, C. Taube, Ber., 60, 485 (1927).
II. О hie, Biochem. Z., 131, 611 (1922).
K. Freudenberg, K. Smeykal, Ber., 59, 107 (1926).
E. Fischer, Ber., 28, 2496 (1895).
E. Fischer. Ber., 28, 1168 (J89.’>).
L. F. Wi gg ins, J. Chem. Soc., 1946, 13.
W. A. van Ekenstcin. J. J. Blanksma, Rec. trav, chim., 25, 153 (1906).
L. Zervas, Ber., 64, 2293 (1931).
358 ОБРАЗОВАНИЕ НОВЫХ С—О-СВЯЗЕП В РЕЗУЛЬТАТЕ ОБМЕННЫХ РЕАКЦИЙ
игл. После промывки холодной водой и петролейным эфиром остается 60________
"0 г неочищенного продукта; т. пл. 130° С (плавится нерезко). При повторной
перекристаллизации из аммиачной воды температура плавления повышается
до 188° С и получается чистая 4,6-бензилидсн-21-глюкоза в количестве 4% От
исходной глюкозы.
Синтез 1,3-бензилиденглицерина см. [749].
71'. Ангидриды карбоновых кислот
Ангидриды карбоновых кислот [750] лучше всего получаются по классическому;
методу — взаимодействием хлорангидридов кислот с солями кислот. Этот способ можеР
быть использован также для синтеза смешанных ангидридов. При добавлении пиридина^
к реакционной смеси вместо солей можно с успехом применять свободные кислоты^
Можно также получать ангидриды непосредственным термическим отщеплением воднй
от карбоновых кислот, причем некоторые алифатические и ароматические дикарбонов^
кислоты образуют циклические ангидриды. л
' I
1. Получение ангидридов карбоновых кислит *
непосредственным отщеплением воды
Легче всего дегидратируются ароматические о-дикарбоновые кислоты. Особое
положение занимают алифатические дикарбоновые кислоты из четырех и пяти атомов
углерода; в этом случае отщеплению воды способствует образование пяти-и шести*
членных циклов (см. правило Блана, стр. 818). ю,т'-Дикарбоновые кислоты с длинно!
цепью при высоких температурах легко дают полимерные ангидриды.
От глутаровой кислоты вода отщепляется уже при медленном нагревании д(
230—280° С и образуется ангидрид; т. пл. 56—57* С. Малеиновый ангидрид [751
можно получить с 90%-ным выходом при отгонке реакционной воды в виде азеотропна
смеси с тетрахлорэтаном. Фумаровая кислота перегруппировывается при нагревали
в малеиновый ангидрид, Адипиновая кислота лишь после многочасового кипячеНШ
с уксусным ангидридом образует ангидрид (полимерный), от которого затем отщеЦ;
ляется СО2 и образуется циклопептанон. |
Образованию ангидридов дикарбоновых кислот способствует нагревание в пря£
сутствпи ацетилхлорида [752—758], хлорокиси фосфора -[752—753], тиопилхлори$
[764] или ангидрида уксусной кислоты [759—763]. .
___
[749] Н. S. Hill М. S Whalen, Н. Н i b Ь е г I, J. Am. Chem. Soc.'J
50, 2239 (1928). J
[750] JI. Ilonecka, в книге Houben-Weyl, Methodea der organischeai
Cbemio, Bd. 8, 4 Aufl , Stuttgart, 1952, S. 746.
|751] A. Mason. J. Chem. Soc., 1930, 701. Jj
[752] L. F. Fieser E. L. Martin, Org. Synth., Coll. v. 2, 1943, p. 560.
[753] J. B. Conn, ’G. B. Kis’tiakowsky et al., J. Am. Chem. Soc., 64j
1749 (1942). • 4
[754] Ch. C. Price, A. J. Tomisek, J. Am. Chem. Soc., 65. 440 (1943). v
[755] B. R. Baker, J. Am. Chem. Soc., 65, 1577 (1943). '
[756] L. F. Fieser, W. H. Daudt, J. Am. Chem. Soc., 63, 784 (1941). J
[757] E. С. К о r n f e 1 d, R. G. Jones, T. V. Park e, J. Am. Chem. Soc., 7fj
158 (1949).
[758] W. E, В a c h m a n n, S Ku?hner, A. C. Stevenson, J. Am. Chem,
Soc., 64, 977 (1942). 1 J
I759J O. Grum mitt, R. E g a n, А. В u c k, Org. Synth., 29, 49 (1949). ’<
[760] E.C. Horning, A. F. Fi nel 1 i. J. Am. Chem. Soc., 71, 3205 (1949).*;
J761] R. C. R о b e r t s, T. B. J о h n s о n, J. Am. Chem. Soc., 47, 1399 (1925&
[762] J. C. Roberts, B. S li a w, J. Chem. Soc., 1950, 2844. 2
[7631 В. II. N i c o*l e t, J. A. Bende r, Org. Synth., Coll. v. 1, 1941, p.
[764] L, McMaste r, F. F A h in a n n, J. Am. Chem. Soc., 50, 146 (1928). 5
F
IV. АНГИДРИДЫ КАРБОНОВЫХ КИСЛОТ
Ангидрид янтарной кислоты (765J. Нагревают 100 г янтарной кислоту
с 65 г РОС1я до прекращения выделения HCI п реакционную массу Перегоняют
Получают 80 г ашпдрида янтарной кислоты (95% от теоретического); т. кип
261° С; т. пл. 120° С.
Ангидрид глутаровой кислоты [766]. Глутаровую кислоту осторожи<
нагревают до 40° С с двойным количеством ацетилхлорида и после прекращен^
выделения НС1 перегоняют в вакууме; т. кип. 150° С (10 мм рт. ст.у, т. пл. 56—
57° С (из эфира).
Многочасовым нагреванием глутаровой кислоты с двойным или тройные
количеством SOCI2 получают ангидрид с выходом 78% от теоретического [764]
Ангидрид адипиновой кислоты [767]. Адипиновую кислоту нагреваю:
6—7 ч на водяной бане с десятикратным количеством ацетилхлорида.
2. Получение ангидридов карбоновых кислот
по реакции двойного обмена
Для получения ангидридов кислот из хлорангидридов и щелочных солей карбо
новых кислот исходные вещества смешивают и отгокяют образовавшийся ангидрид
Уксусный ангидрид [768]. К 8 г порошкообразного безводного ацетате
натрия при охлаждении по каплям прибавляют 5 г ацетилхлорида, смесь уме
ренно нагревают и отгоняют образовавшийся ангидрид. Для удаления остатко>
ацетилхлорида ангидрид перегоняют над ацетатом натрия. Выход ангидрида
5—6 г (80—90% от теоретического); т. кип.„136,5° С.
Можно избежать приготовления чистых хлорангидридов кислот, используя
реакции, в которых они образуются в качестве промежуточных продуктов. Например,
нагревать натриевые соли карбоновых нислот с переносчиками галогена — гОС]а,
РС16 или SOC12.
Чтобы обойтись без получения солей карбоновых кислот, можно ацилировать
свободные кислоты хлорангидридом.
Ангидрид масляной кислоты [769]. Эквимолекулярные количества масля-
кой кислоты и се хлорангидрида нагревают до кипения сначала 1 ч ка водяной
и затем 7 ч на масляной бане. Перегонкой реакционной смеси получают ангидрид
масляной нислоты; т. кип. 198—199° С (765 мм рт. ст.).
Третичные основания, особенно пиридни, образуют с хлорангидридами кисло»
продукты присоединения, которые уже при низких температурах являются эффектив-
ными ацилирующими средствами. При добавлении пиридина к хлорангидриду и по-
следующем разбавленки небольшим количеством воды получаются ангидриды кисло»
с хорошими выходами [770].
Ангидрид бензойной кислоты. В 40 мл сухого диоксана растворяют 14 а
(0,1 моль) свежеперегнаниого бензоилхлорида и при охлаждении ледяной
водой смешивают с 40 мл сухого пиридниа. Не прекращая охлаждения, добав-
ляют 1 мл воды, лучше разбавленной небольшим количеством дибкеана, остав-
ляют при охлаждении льдом на 10 мин. и выливают в смесь 75 мл концентриро-
ванной НС], 75 г льда и 75 мл воды. Отсасывают выпавший кристаллически!»
ангидрид, промывают ледяной водой, сушат в вакууме и перекристаллизовывают1
из смеси эфир — петролейный эфир. Выход 11 г (97% от теоретического);
т. пл. 42—43-° С.
[765
[766
[767
[768
[769
[770
J. Volhard, Ann., 242, 150 (1887).
D. М о 1, Rec. trav. chim., 26, 381 (1907).
G. L. Voerman, Rcc. trav. chim., 23, 269 (1904).
Ch. Gerhardt, Ann. Chimie [3], 37, 313 (1853).
E. Linnemann, Ann., 161, 179 (1872).
H. Adkins, Q. E. Thompson, J. Am. Chem. Soc., 71, 2242 (1949).
3Q0 ОБРАЗОВАНИЕ НОВЫХ С—О-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕННЫХ
Аналогично реагирует смесь хлорангидрида и пиридина со свободными карбо-
новыми кислотами. J
Важным и общеприменимым косвенным методом получения ангидридов кнсда^
является взаимодействие свободных карбоновых нислот с уксусным ангидридом [77Ця
Для этого соответствующую кислоту нипятят в колбе с обратным холодильнике»
около 2 ч с пяти- или шестикратным количеством уксусного ангидрида. Затем
образовавшуюся уксусную кислоту и избыток уксусного ангидрида отгоняют, лучяйв
в вакууме, и остаток осторожно нейтрализуют холодным 2—3%-ным раствор»
Na2CO3 или тотчас же перерабатывают. /Д
Ангидрид бензойной кислоты [772]. В течение 6 ч кипятят в кодвв
с обратным холодильником смесь 48,8 г бензойной кислоты, 41 г уксусном
ангидрида и 200 з сухого бензола н затем фракционируют реакционную ма&9
После отгонки бензола и уксусной кислоты получают ангидрид бензоЙмИ
кислоты. Выход 36,8 г (81% от теоретического); т. кип. 347—348° С; т. пл. 42ЭДИ
Уксусный ангидрид реагирует с хлорангидридами кислот, при этом нар*Я
с более высокомолекулярным ангидридом образуется ацетнлхлорид [773]; карбоной»
кислоты тоже реагируют с ацетялхлоридом с образованием ангидридов кислот]774Я
По реакции с хлорангидридами можно подучать и смешанные ангидриды.
при синтезе ангидридов из карбоновых кислот и ангидрида уксусной кислоты в наяВ
стве промежуточных продуктов получаются смешанные ангидриды, которые.в некотИ
рых случаях могут быть выделены; таков, например, смешанный ангидрид муравьияЯ
и уксусной кислот [775]. Этот ангидрид может заменить неустойчивый ангидрид мушЯ
вьиной кислоты; он переносит в процессе реакции только реакционноспособный ф|Я
мильный остаток и употребляется для получения многочисленных эфиров муравьишЦ
кислоты, которые в свою очередь путем переэтерификации превращаются в труяЯ
доступные иными методами сложные эфиры третичных спиртов (стр. 353)- ПкЯ
Смешанный ангидрид муравьиной и уксусной кислот [775]. При охлааЙя|
кки льдом смешивают 138 г безводной муравьиной кислоты с 408 з уксусном
ангидрида и оставляют на некоторое время. Затем нагревают смесь на водямИ
бане до 50° С и снова охлаждают. После этого перегоняют в вакууме с иорд
кой колонкой. При 18 мм рт. ст. собирают отдельно фракции, кипящие дД
27—31° С, 31—32° С и выше 32" С. Смешанный ангидрид муравьиной и уксус^уИ
кислот отгоняется в средней фракции; т. кип. 29° С (17 мм рт. ст.). СредяМ
. фракцию обрабатывают петролейным эфиром, лучше всего растворяюдДИ
уксусную кислоту, довольно хорошо уксусный ангидрид н очень плохо расмйИ
ряющим смешанный ангидрид. В нерастворимой в петролекиом эфире чям|
содержится около 70% смешанного ангидрида. Ее еще раз фракционируя®
в вакууме; выход 80,3% от теоретического.
По другому способу [776] ангидриды кислот с хорошими выходами получм
нагреванием в течение 6—20 ч при-100—150° С серебряных солей карбоновых кищй
с С8г. Ангидриды получаются в очень чистом виде. В качестве побочного продукта Й
разуется только свободная карбоновая кислота.
[771'
[772j
[773
[774
[775
[776
W. A u t е n г i е t b, G. T b о m a e, Ber., 57, 430 (1924). Ш
A. Kaufmann, A. Luterbacher, Ber., 42, 3484 (1909).
F. Zetzsche, Ch. Fliitsch et al., Helv. chim. acta, 9, 181 (1926).‘Ж
M. H. Fournier Bull. Soc. chim. France [4], 5 , 920 (1909).
A. Behal, Ann. Chim. Phys. [7], 2, 417 (1900); C. r., 128, 1460 (1899). 4®?
D. Bryce-Smith, Proc. Chem. Soc., 1957, 2o. '•J
]. РАЗРЫВ ПРОСТОЙ ЭФИРНОЙ связи
36
РАЗРЫВ СВЯЗЕЙ УГЛЕРОД - КИСЛОРОД *
Разрыв С—О-связи может происходить в следующих классах соединений
в ангидридах карбоновых иислот, ортоэфирах, ацеталях, полуацеталях, эфирах карбо
новых кислот, простых эфирах.
Разрыв происходит тем легче, чем больше гетероатомов находится у соседней
с кислородом углеродного атома С—О—С-группы. Следовательно, устойчивости
С—О-свяэи указанных типов соединений возрастает в приведенном выше порядке
В качестве специфических особенностей следует отметить, что эфирная евнэ]
в лактонах и лактидах разрывается легче, чем в нормальных сложных эфирах, и чт<
стойкость иислородсодержащих гетероциклических ядер тем меньше, чем болыне'атомо]
кислорода в них содержится. Так, например, паральдегид расщепляется легче, чеь
диоксан.
I, Разрыв простой эфирной связи
Расщепление эфирной связи в большинстве случаев протекает в довольно же
стких условиях, часто по невыясненному механизму, и используемые для расщепление
реагенты не всегда применяются с одинаковым успехом. Расщепление эфиров прово
дится под действием кислотных агентов, таких, как галогениды алюминия А1Ха
H2SO4, ангидриды и хлорангидриды кислот и галогеноводороды, а также под действие*
оснований, металлоорганических соединений и щелочных металлов. В некоторых слу
чаях связь С—О—С может быть разорвана также гидрированием.
Из галогеноводородов наиболее пригоден HI. При применении эквивалентные
количеств реагентов получаются нодалкан и спирт, причем обычно иэ более высоко
молекулярной группировки образуется спирт, а из низкомолекулярной — иодалках
[1]. Так, эфиры, содержащие СН3-группу и более высокомолекулярный остаток, гладк<
и довольно быстро расщепляются с образованием йодистого метила и спирта. На этол
методе основано определение метоксильных групп по Цейзелю: пробу кипятят в колб<
с обратным холодильником с концентрированной водной Ш и определяют улетучива-
ющийся иодистый метил.
При действии избытка HI на алифатические эфиры следует ожидать образовали*
подалканов из обеих половин молекулы. Из алкилариловых эфиров, напротив, наряду
с иодалканами всегда образуются фенолы:
C8H5OR + H1 —> CeH6OH + RI
Расщепление арплметиловых эфиров часто используют для получения фенолов
в большинстве случаев выходы фенолов достигают 72—93% от теоретического. Дл*
этого эфир кипятят в колбе с обратным холодильником с водными растворами HI иад
НВг, часто в смеси с ледакой уксусной кислотой.
Пирокатехин [2J. Кипятят гваякол в течение 6—7 ч с 48%-ной НВг.
Непрореагировавший гваякол, отгоняющийся при этом с водяным паром
отделяют в отстойнике и возвращают в реактор.
.и-Оксифенилуксусная кислота [3]. Получают кипячением л-метокси-
фенилуксусной кислоты в течение 1,5 ч с HI; выход 72% от теоретического [3]
п,и’-Диоксидифениловый эфир. В течение 4 ч кипятят 24 г п,а'-димето-
ксидифенилового эфира с осторожно приготовленной смесью 60 мл 48%-ной НВз
и 30 мл уксусного ангидрида и затем выливают в 300 мл теплой воды. При охла-
ждении в виде бесцветных пластинок кристаллизуется 19,7 г (93% от теорети-
ческого) диокендифенилового эфира; т. пл. 160—161° С. При дезалкилирование
при помощи А1С18 образуется большое количество снопообразных примесей-.
О методах расщепления эфиров фенолов см. обзор [4].
’ Материал обработали X. Зеебот и М. Шульц.
[1] R. D. S i I v а, Вег., 8, 1352 (1875); W. S i р р е г t, Ann., 276, 196 (1893).
[2] Н. Т. С 1 а г k е, Е. R. Т а у 1 о г, Org. Synth.; 3, 28 (1923).
3J Е. С. К о г n f о I d, J. Am. Chem. Soc., 70, 1375 (1948).
[4] A. Liittringhaus, F. V. Saaf, Angew. Chem., 51, 915 (1938).
362
РАЗРЫВ СВЯЗЕЙ УГЛЕРОД-КИСЛОРОД
Для расщепления эфиров применяют водную иодистоводородную кислоту,
иногда в смеси с ледяной уксусной кислотой, или насыщают эфир при 0° С и в запаянное
трубке нагревают иа водяной бане. Из-за сильного восстановительного действия Н]
его применение сильно ограничено. Для того чтобы исключить это восстановительное
действие, можно с успехом применять смесь иодида щелочного металла с фосфорной
кислотой. При взаимодействии эфиров с избытком этой смеси получаются иодалкаиы
[5]. Эфиры фенолов не реагируют в этих условиях.
Вместо HI почти всегда можно употреблять’НВг, при этом реакция идет не-
сколько медленнее. Эфиры со значительной реакционной способностью-можно разла-
гать также соляной кислотой.
Эфп ры трифенилкарбинола нестойки уже к действию разбавленных минераль-
ных кислот: они расщепляются под их действием в ацетоновом или спиртовом рас-
творе при комнатой температуре, а в 80%-ной уксусной кислоте — при нагревание
с образованием трифенилкарбинола п спирта [6J.
Эфиры третичных алифатических спиртов под действием HCI гладко превра-
щаются в третичные галогениды [7].
При нагревании 1,4-диметокси-2,6-дибензоксибензола в течение 1 ч при 65® С
с соляной кислотой подвергаются гидролизу обо бензильные группы, а метоксильные
группы не затрагиваются [8]. При обработке в течение 10 ч бензилметилового эфира
концентрированной соляной кислотой при 90® С и энергичном перемешиванки с одно-
временным пропусканием НС1 происходит почти количественное расщепление на хло-
ристый бензил и хлористый метил [9].
Имеющиеся в молекуле активные группы оказывают решающее влияние на рас-
щепление эфиров. Виниловые эфпры, простые эфиры 1,3-дикетонов и эфиры 1,3-дикето-
кислот так же чувствительны к действию кислот, как н гликозиды. Эфиры 1,2-ке*
.гослиртов, как, например, эфиры циклогексанол-1-она-2, тоже очень легко расщепля-
ются [10].
Циклические эфиры довольно гладко расщепляются галогеноводородами.
На этом основан изящный способ получения 1,4-дигалогеналканов из тстрагидрофуран®
и его производных. Наибольшей реакционной способностью в этой реакции обладает
HI, менее эффективен НВг и еще менее HCI. Многочасовым пропусканием HI, НВг
или HCI в присутствии ZnCI2 в горячий тетрагидрофуран получают 1,4-дигалогенидм
с выходами 59—75% от теоретического [И]. Введение НС] в тетрагидрофуран беа
добавкк ZnCl2 приводит к 4-хлорбутанолу-1 с 65%-иым выходом [12]. Аналогичные-
реакции осуществимы также и с тетрагидропиранами.
Клеменс установил, что при сплавлении эфиров с гидрохлоридом анилина про-,
исходит эфирное растепление [13]. Этот метод особенно пригоден для таких эфиров,
при расщеплении которых образуются легколетучне алкилгалогениды; пропускание
сухого НС1 облегчает протекание реакции 114]. Вместо гидрохлорида анилина молото
использовать гидрохлорид или гидробромид пиридина при температуре около 200е С.-
Ряд фениловых эфиров с хорошими выходами разлагается серной кисло-*
той [15]. $
В присутствии кислот Льюиса, например ZnCI2, А1С1„ TiC]#, SnCl4, FeCIs,.
SbCl5, а также BF8, хлорангидриды и ангидриды кислот переводят простые эфиры*,
в сложные эфиры и галогеналкилы. Эта реакция применяется в основном только для,
алкиловых эфиров, так как ариловые эфиры при этом большей частью вступают в реак-'
дню Фриделя — Крафтса. Меервейи и сотр. [16] провели глубокие исследования:
[5] II. S t о n е, II. S h е с h t е г, J. Org. Chem., 15, 491 (1950).
[6] В. II е ] f е г i с h, Advances in Carbohyd. Chem.. 3, 81 (1948). ‘
[7] J. L. E. Erickson, W. H. Ashton, J. Am. Chem. Soc., 63. 1769 (1941)^
[8] W. Baker, B. Nodzu, P. Robinson, J. Chem. Soc., 1929, 74. 4
[9] А, Я ieche, II. S e e b о t b, Chem. Techn., 10, 614 (1961).
[10] M. Bergmann, M. Gierth, Ann., 448, 65, 69 (1926). v
Ill) S. Fr i e d,R. D. К J e e л e, J.Am.Chem. Soc., 63, 2691 (1941);D. S. Tar*,
bell, C. Weaver J. Am. Chem. Soc., 63, 2939 (1941).
[121 D. Starr, R. M. Hixon. Org. Synth., Coll. v. 2, 1943, p. 571.
[13] A. Klomenc, Ber., 49, 1371 (1916).
f 14 E. A. Zof in, K. A. Tschch ikowadse, C., 1933, II, 3118.
[15] O. Diols, F.Bunil, Ber., 38, 1491 (1905); G. K. Hughes, F. L i о u sr,
C., 1938, j, 4033. !
[16] H. Meerwein el al., J. prakt. Chem., [N. F.J, 134, 51 (1932); 147, 257 (1937).
I. РАЗРЫВ ПРОСТОИ ЭФИРНОЙ связи
363
механизма реакции и опубликовали данные о выходах для ряда кислот Льюиса. Луч-
шими катализаторами, по-видимому, являются ZqCI2 и SnCl,. Так, в присутствии
0,1 моль SnCl4 (в расчете на 1 моль бензоилхлорида) из этилового эфира и беггзоил-
хлорида в течение 4 ч при 110° G получается с 80%-ным выходом хлорэтан и с 90%-ным
выходом этиловый эфир бензойной кислоты [17]. При действии на тетрагидрофурав
хлорангидридов в присутствии ZnCl2 образуются 4-хлорбутиловые эфиры[18]. Броман-
гидриды и особенно иодангидриды расщепляют эфиры даже без катализаторов [19J.
Галогениды алюминия имеют препаративное значение, особенно для отщепления
метоксильных и этоксильных групп от ароматических эфиров. Диариловые эфиры
таким путем не расщепляются. Расщепляющее действие А1С13 иа эфиры было открыто
на примере расщепления анизола [20], при котором образуется кристаллическое двой-
ное соединение, распадающееся при нагревании на галогеналкил и хлорфенолят алю-
миния.
При нагревании 10 г анизола с 15 г Л1С18 в течение 3 ч при 120° С выде-
ляется метилхлорид. Остающийся фенолят разлагают водой, подкисляют HCI
экстрагируют фенол эфиром и выделяют его обычным способом. ’
Жирно-ароматические эфиры легче расщепляются А1Вг3; бромид алюминия
растворим в таких индифферентных растворителях, как бензол, сероуглерод и лигроин.
По Пфейфферу и Хааку [21], реакция протекает по следующей схеме:
ROCH3 + AlBr3 —> ROCH3-AlBr3 —»- ROAIBr2 + CH3Br
ROAIBr2-FH2O —> НОН4-А1ВгаОН
Если в соединения имеется карбонильная группа, то в реакцию встуиает вторая
молекула бромида алюминия [21]. В простых случаях для этого достаточно [22] нагре-
вать в колбе с обратным холодильником раствор вещества в бензоле с требуемым коли-
чеством А1Вг3. (Бензол не должен содержать тиофена!)
в-Оксибензофенон. Нагревают в колбе с обратным холодильником в тече-
ние 4 ч 0,82 г 2-метоксибензофепоиа с 3,1 г А1Вгэ и 27 мл бензола. Смеси даюх
охладиться, добавляют к ней избыток HCI, разделяют слои, экстрагирую^
продукт из водного слоя эфиром и извлекают фенол из объединенного эфирно-
бензольного раствора 2 н. раствором NaOH. Щелочную вытяжку выливаю-!
в соляную кислоту, вновь экстрагируют эфиром и после выпаривания эфира
получают 0,74г маслянистого о-оксибензофенопа (96% от теоретического);
т. пл. 39° С.
в-Оксибеизойная кислота. Растворяют 0,79 г анисовой кислоты, высу-
шенной над Р2О6, в 35 мл горячего бензола, добавляют раствор 4.9 е А1Вга
в 30 мл бензола и кипятят 4,5 ч в колбе с обратным холодильником. Посл^
охлаждения смесь разлагают концентрированной НС], продукт экстрагирую-!
эфиром, извлекают из эфирной вытяжки оксикислоту прк помощи NaOH,
повторно подкисляют щелочную вытяжку, снова экстрагируют кислоту эфиром
и после выпаривании получают 0,37 г п-оксибеизойной кислоты.
Аналогичным образом были дезалкилированы папаверин и пиперонал.
Эфирное расщепление может происходить также под действием кодида магния
[23], трихлорида бора |24], тс!рахлорида кремния [25] и литийалюминийгидрида
[17]
[18]
[19]
[20
[21
[22
/23
[24
[25
Л. М. Сморгонский, ЖОХ, 17, 416 (1947); С. А., 42, 858 (1948).
М. Е. Sy пег holm, Org. Synth., 29, 30 (1949).
Е. L. G u a t и s, P. G. Stevens, J. Am. Chem. Soc., 55, 378 (1933);
N. O.V. Sonntag, Chem. Rev., 52, 353 (1953).
C. Hartmann L. Gattermann, Ber., 25, 3531 (1892).
P. Pfeiffer, E. Haack, Ann., 460, 156 (1928).
P. Pfeiffer, W. Loewe, J. prakt. Chem. [N. F.J, 147, 293 (1936).
V. Grignard, J. Hitz, Bull. Soc. chim. France [5], 3, 1181 (1936).*
W. Gerrard, M. F. Lappert, Chem. Rev., 58, 1091 (1958).
R. Schwarz, W. Kuchen, Chem. Ber., 89, 169 (1956).
361
РАЗРЫВ СВЯЗЕЙ УГЛЕРОД-КИСЛОРОД
CeH6OMgX-]-RR'
[26]. До настоящего времени эти реагенты мало применяются на практике для эфирного
расщепления. i
Некоторые аралкиловые эфиры, которые не поддаются кислотному расщепленн»
или расщепление которых приводит к нежелательным продуктам, разлагают щелоча!»
или алкоголятами при температурах выше 150° С, лучше в присутствии этиловой»
спирта или более высококипящих спиртов. Например, при нагревании в течение 3
иератрола с равными частями КОН и спирта он частично гидролизуется с образованиям'
гваякола [27]. 2-Метоксифениловый эфир после 10 ч кипячения в диэтиленглико
расщепляется при помощи NaOH в 2-оксифениловый эфир; выход 91% от теор
ческого [28].
Как уже упоминалось, легкость расщепления эфиров фенолов соответствуй
легкости их образования; например, эфиры нитрофенолов омыляются уже при
репном нагревании со спиртовым раствором КОН. Эфиры полинитрофенолов час
расщепляются под действием аммиака, первичных или вторичных аминов [2
Эфиры гидрохинонов равным образом легко разлагаются разбавленным холо
раствором едкого натра [30].
Для расщепления С—О—С-связи можно использовать также металлоорг
окне соединения *. Соединения Гриньяра могут реагировать с Простыми эфирами
следующему уравнению:
CeH5OR + R'MgX
Эту реакцию иногда используют для расщепления аралкиловых, и особеВД
бензиловых эфиров. Обычно она протекает при температурах порядка 150—200^
[31]. Между тем, расщепление В.у-непасыщеиных эфиров фенолов происходи уже ШЙ
55° С [4].
Другие металлоорганические соединения оказывают более энергичное действ^
чем соединения Гриньяра. Например, этиловый, изопропиловый или шреш-бутилов^
эфиры холестерина разлагаются под влиянием амилиатрпя при 10° С до холестерч^
пентана и олефина [32] согласно уравнению:
Хол — OC2Hs-|-H-C5HiiNa —> Хол—ONa-{.И-С5Н12-1-С2Н4
В качестве побочного продукта получается холестадиен, образующийся в резуЙ
тате следующей реакции:
Хол—OR+н~СБНцНа —> Холестадиен+п-С5П1г4-НО№ У
Я
Если в алкильном остатке R, как, например, в метиловом и бензиловом эфир®
холестерина, отсутствует р-атом водорода, то образуется исключительно холестадц,
Эфирному растеплению иатрийалкилами посвящены работы Летзингера с ОСЧ
[33]; об эфирном расщеплении фениллитием см. [4].
Для расщепления эфиров фенолов могут применяться также металличеСЦ
натрий или калий. Так, например, дифениловый эфир, один из наиболее устойчив
эфиров, разлагается жидким калий-натриевым сплавом при комнатной темперай
[34]. Вебер и Сова, действуя натрием в жидком аммиаке на 4,4’-дизамещеннйй дифея
г»
[26]
[27
28
29
30
31
32
[33]
[34]
Вопросам расщепления простых эфиров под действием натрия и натрийалнилЙ
посвящены классические исследования П. П. Шорыгина [П. П. Шорыг.И;|
Избранные труды, Изд. АН СССР, 1950, стр. 197—257].—Прим. ред. $8
Р. к а г г е г, О. Riittner, Helv. chim. acta, 33, 812 (1950); V. T w e e d ff
M. Cuscuri da, Angew. Chem., 66, 646 (1954).
L. Bouveault, Bull. Soc. chim, France [3], 19, 75 (1898). .j
П. E. Ungnade, К. T. Zilch, J. Org. Chem., 15, 1109 (1950). 4
R. J. W. L e Fevre et al., J. Chem. Soc., 1927, 1168. , n
U. N e f, J. prakt. Chem., 42, 168 (1890). '
E. S p S t h, Mh. Chem., 35, 319 (1914). J
D. H. G on 1 d, К. H. Sch a af, W. L. R u i g h,, J. Am. Chem. Soc., Я
1263 (1951).
R. L. L e t s i n g e r et al., J. Am. Chem. Soc., 70, 3342 (1948); 73, 5708 (193Й
75, 2649 (1958)
E. Muller, W. Bunge, Ber., 69, 2171 (1936).
И. ОМЫЛЕНИЕ СЛОЖНЫХ ЭФИРОВ КАРВОНОВЫХ кислот
365
ловый эфир, расщепили его на производные бензола и фенола [35]. Диаллиловой эфир
разлагается натриевой пылью при 35° С с образованием аллилнатрпя и аллилата на-
трия. Эта реакция является удобным методом получения аллилнатрия [36]. При кипя»
чеппп анизола, фенетола, фенилбензилового эфира и дифенилового эфира с калием
пли ширнем в пиридине без побочных продуктов образуется фенол с выходом 90%
от теоретического [37].
Во всех рассмотренных до сих пор способах расщепления эфирной связи тре-
буки ел агрессивные реагенты. Фрейдояберг с сотр. установил, что для расщепления
бензиловых эфиров можпо использовать также каталитическое восстановление водоро-
дом [38]. По этому способу бензилалкиловые эфиры в присутствии платиновых метал-
лов пли никеля Ренея очень мягко превращаются в толуол и спирт. Беизилфениловый
эфир лучше всего подвергать восстановительному расщеплению при помощи паллади-
рованного животного угля.
З-Галлоилглюкоза [39]. При 25° С 10 г 3-трибенэилгаллоилглюкоэы-
в 250 мл метилового спирта гидрируют в присутствии 2 г 2%-ного палладиро-
вапного животного угля. Гидрирование заканчивают через 3,5 ч, после погло-
щения 1260 мл (теоретически 1240 мл) водорода. Раствор отфильтровывают
от катализатора, промывают осадок этиловым спиртом и выпаривают в вакууме.
Бесцветное, вспененное вещество растворяют в небольшом количестве води»
и упаривают в эксикаторе. Остается бесцветная, хрупкая масса; выход 5,4 а
(98% от теоретического).
В препаративной работе можно, пользуясь этим методом, защитить оксигруппы'
бензилированием к затем вновь регенерировать их в мягких условиях.
Обзор работ по расщеплению бензиловых и циклических бензиловых.-
эфиров гидрированием см. [40]. О расщеплении фурановых колец см. [41J.
Расщепление эфиров полно и подробно описано л работе [427, в которой'
освещена также теория вопроса, а в заключение приведены некоторые данные-
о расщеплении тиоэфиров; об их расщеплении см. также [43].
II. Омыление сложных эфиров карбоновых кислот
Гидролиз эфиров карбоновых кислот с образованием карбоновых кислот и спир-
тов проходит в щелочной пли кислой среде:
RCOOR*+H2O RCOOH4-ITOH
В то время как для кислотпого гидролиза требуются только каталитические
количества кислоты, для щелочного омыления необходимы молярные количества
щелочей, так как щелочи действуют не только каталитически, но и образуют с карбо-
новыми кислотами соли, которые выводятся из равновесной реакции [44, 45].
[45]
F. G. Weber, F. J. Sowa, J. Am. Chem. Soc., 60, 94 (1938).
П. L. Le taincer, J. G. T г a у n h a in, J. Am. Chem. Soc., 70, 3342
(1948).
V. Prey, Ber., 76, 156 (1943).
K. Fte adenberg, W. Dfin, H. v. Ilochslc t ter, Ber., 61, 1739
(1928).
O. Th. Schmidt, A. Schach, Ann., 571, 35 (1951).
W. H. н а г t u ng, R. S i m о n of f, Org. Reactions, 7, 263 (1953).
D. G. Jones, A. W. C. Taylor, Quart. Rev., 4, 199 (1950).
R. L. Burwell jr., Chem. Rev., 54, 615 (1954).
D. S. T а г b e 11, D. P. Harnish, Chem. Rev., 49, 1 (1951).
Подробное описание реакции см. Н. Henecka, в книге Houben-Weyl,
Metnoden der organischen. Chemie, Bd. 8 4 Aufl Georg Thieme Verlag, Stutt-
gart, 1952, S. 418.
F. К 1 age s, в книге G. M. Schwab, Handbuch der Katalyse, Bd. 7/2, Sprin-
ger-Verlag, Wieri, 1943, S. 325.
РАЗРЫВ СВЯЗКИ УГЛЕРОД—КИСЛОРОД
С повышением температуры скорость омыления сложных эфпров увеличивается^
Протеканию реакции способствует также непрерывная отговка образующегося спирта.
Скорость кислотного гидролиза зависит от концентрации ионов водорода: эта реакция
протекает обычно значительно медленнее, чем щелочное омыление.
Скорость омыления сложных эфиров как в Щелочной, так и в кислой среде за-
висит от строения остатков карбоновой кислоты [46] и спирта [47]. Стерические факторы'
оказывают большое влияние на способность сложных эфиров к омылению. Расщепление:"
затрудняется при наличии разветвления углеродной цени у соседнего с карбоксиль-;
ной группой атома углерода. Такое же действие оказывает разветвление цепи спирто-;
вого компонента. Скорость омыления сложных эфиров зависит также от кислотност#!
отерифидпрованиой кислоты. Чем сильнее выражены кислотные свойства кислоты,4
тем быстрее происходит расщепление щелочами образуемых ею эфиров [48]. Эфирьг
первичных спиртов омыляются щелочами легче, чем эфиры вторичных, а последние —|
легче, чем эфиры третичных спиртов. Напротив, в кислой среде особенно легко про^
исходит расщепление эфиров третичных спиртов. Чисто алифатические эфиры реаГиЙ
руют лучше, чем ароматические. Установлено, что чем легче образуются эфирнщ
тем легче протекает их гидролиз.
Омыление щелочами. В качестве опыляющего реагента в бо-ттглтлпин
стве случаев применяют водные или спиртовые (метиловый или этиловый спирты^
диэтиленгликоль) растворы NaOH или КОН. Более мягко действуют растворы гидро^
окисей бария или кальция. Если в результате’гидролиза образуются неустойчивей
к действию сильных щелочей компоненты, то часто удобнее проводить омыление pang
творами Na»COs, NaHCO3 или метилата натрия [49]. Для омыления особенно устойЗ
чивых эфиров, например эфиров о,<?'-диаамещенных ароматических карбоновых кислот
приходится иногда прибегать к щелочному плавлению.
Концентрация применяемых щелочей и температура реакции зависят от entf
собности сложных эфпров к гидролизу и от устойчивости образующихся продуктов
гидролиза. ‘
Неразветвлецпые алкиловые эфиры низших алифатических карбоновых кисло
обычно омыляют кипячением в течение 0,5—2 ч с 25%-ным раствором NaOH. При гидро
лизе нерастворимых эфиров для гомогенизации среды рекомендуется применять спир
товые щелочи. Для омыления высококппящих эфиров удобнее пользоваться раствО
рами КОН в диэтиленгликоле [50]. Ио окончании реакции избыток щелочи удаляй
пропусканием СО2.
Некоторые эфиры расщепляются уже при обработке водой. К ним, например
относятся метиловый или этиловый эфиры муравьиной кислоты [45], полуэфиры глЦ
коля [51], этерифицироваиные по карбоксильной группе оксикарбоновые кислоты [52
эфиры ацилгликолевой кислоты [53] и некоторые сложные эфиры алкалоидов групц
трепана [54].
К легко гидролизуемым эфирам относятся также эфиры простых а- и |3-амия1
кислот. Эти эфиры часто можно расщепить кипячением с водой |55]; но обычно рей
мендуется применять для гидролиза водный раствор гидроокиси бария [56, 57]. Эя
реагент имеет особые препаративные преимущества: многие бариевые соли карбой* 1
вых кислот плохо растворимы и поэтому могут быть отделены; избыточный Ва(ОН)
удаляют послб реакции обработкой СО2 или H2SO4. 4
[46]
[47]
[48]
[49]
[50]
[51
52
. 53
54
55
56
457
К. Kindler Ann., 450, 1 (1926); 452, 90 (1927); 464, 278 (1928): С. N. Н 11
shelwood et al., J. Chem. Soc., 1936, 1337; 1935, 1482; 1938, 1801;'G. D <
vies, D. P. - E v a n s, J. Chem. Soc., 1940, 339. 2s
II. Olsson et al., Z. physik. Chem., 102, 26 (1922); 118, 99, 107 (1925); 1»
243 (1927).
H, Olsson, Z. physik. Chem., 133, 233 (1928).
L. Claisen, Вет., 38, 703 (1905). J
С. E. Redemann H. J. Lucas, Ind. Eng. Chem., Anal. Ed., 9, 52
(1937).
E. Erlenmeyer, Ann, 192, 249 (1878). 5
L. Schreiner, Ann., 197, 7, 12, 15 (1879).
M. Senff, Ann., 208, 274 (1881). -
S. P. Findley, J. Am. Chem. Soc., 76, 2855 (1954). <
E. Fischer, Ber. 34. 445 (1901).
F. Weygarfd, Ber., 74, 257 (1941). )
T. Kobayashi, Ann., 536, 158 (1938).
2
II. ОМЫЛЕНИЕ СЛОЖНЫХ ЭФИРОВ КАРБОНОВЫХ КИСЛОТ
367
[3-Аланин [56]. Из 30 г эфира циануксусной кислоты каталитическим
гидрированием в среде ледяной уксусной и серной кислот в присутствии PtOa
получают этиловый эфир р-аланина. Отделяют катализатор возможно полнее
удаляют уксусную кислоту в вакууме и выливают остаток в 300 мл воды. При-
бавляют 100 г тонкоизмельченной Ва(ОН)2 и кипятят 3 ч. После охлаждения
осаждают барий точно рассчитанным количеством разбавленной II2SO4. Осадок
BaSO4 отделяют центрифугированием, а раствор сильно концентрируют в ва-
кууме. р-Аланин быстро выкристаллизовывается; выход 17 г (72% от теоретиче-
ского); после переосаждения его эфиром из раствора в водном спирте т. пл.
Эфиры сильных карбоновых кислот, например циануксусной [58], омыляются
2и%-ным водным NaOH уже па холоду; аналогично эфиры дикарбоновых кислот (мало-
новой [59], а-метилпимелиповой [60]) превращаются в соответствующие эфирокислоты.
Полное омыление эфиров дикарбоновых кислот происходит лгппь при длитель-
ном кппячепип со сниртовым раствором КОН. При щелочном омылении эфиров ненасы-
щенных карбоновых кислот может произойти миграция двойной связи [61]. Напротив
|идролиз этих эфиров HCI приводит к получению карбоновых кислот с неизмененным
положением двойной связи.
Эфиры а-кетокислот лучите всего омыляются водными щелочами пли спиртовым
pac-i вором К2СО8; спиртовые растворы едких щелочей часто вызывают разложе-
ние [62].
Фенилглиоксиловаи кислота [63]. Суспензию 140 г метилового эфира
фенилглиоксиловой кислоты в 700 мл 2 и. раствора NaOH встряхивают до раство-
рения большей части вещества. Омыление доводят до конца нагреванием в течение
30 мин на водяной бане. После охлаждения удаляют нерастворившийся остаток
экстрагированием эфиром. Водный слой подкисляют 350 мл 5 н. НС], экстраги-
руют вновь эфиром и выделяют кислоту обычным способом. Выход 115,5 г (90%
от теоретическою); т. кип. 105° С (0,1 мм рт. ст.)\ т. ил. 64,5—65,5°'С (пере-
кристаллизация из СС14).
Для омыления при комиатной температуре эфиров р-котокислот, особенно эфиров
высших кетокислот, целесообразно применять смесь ледяной уксусной и соляной кис-
лот [64].
Лактоны легко переводятся водными растворами оснований в соответствующие
оксикарбоновые кислоты.
Омыление ацетатов углеводов щелочами не представляет труда в том случае,
если восстанавливающиеся группы сахаров защищены устойчивыми к щелочам замести-
телями. Соответствующие гликозиды или ацетоинды могут быть дезацегилированы
действием спиртовых щелочей, раствора гидроокиси бария, жидкого аммиака [65]
или спиртовою раствора аммиака [66].
[58]
[59]
[60]
[61]
[62]
[63]
164]
[65]
[66]
II. Не п е с к а, в книге Houben-Weyl, Methodeii der organischen Che-
mie, Bd. 8, 4 Aufl., S. 422.
D. S. Breslow, E. Bau mgarten, C. R. Ha user, J. Am. Chem.,
Soc., 66, 1286 (1944).
L. F. Fieser, M. T. Leffler et al., J. Am. Chem. Soc., 70, 3206 (1948).
B. R. Baker, M. V. Querry, A. F. К a d i s h, J. Org. Chem., 13, 123
(1948). ’ *
L. Bon veaul t, Bull. Soc. chim. France [3], 15, 1017 (1896); F, F. В 1 i c k e,
M. U. T s a o, J. Am. Chem. Soc., 66, 1645 (1944); F. F. В 1 i c k e, R, F. F e 1 d-
kamp, J. Am. Chem. Soc., 66. 1087 (1944).
E- Baer, M. К a t e s, J. Am. Chem. Soc., 67, 1482 (1945).
M. A. Mitz, A. E. Axelrod, K. Hofmann, Am. Chem. Soc., 72
1231 (I960).
E- F ischer, H. S traup, Ber., 45, 2467 (1912).
E. Fischer, M. Rcigmann, Ber., 50, 1047 (1917).
РАЗРЫВ СВЯЗЕЙ УГЛЕРОД—КИСЛОРОД
Омыление ацетатов углеводов лучше всего происходит по методу Земплена [бя
в присутствии каталитических количеств метилата натрия в метиловом спирте, а
Получение маннита из гекса-О-ацетилманнита (68]. Заливают 8з
гекса-О-ацетилманпита 20 мл абсолютного метилового спирта, нагревают ЭД
водяной бане и добавляют 2 мл 0,1 н. раствора метилата натрия. Через 3 л»
начинается выделение маннита. Реакционную смесь выпаривают досуха ИяЦ
пониженном давлении, растворяют остаток в 5 мл воды и добавляют к раствви
при нагревании 25 мл абсолютного спирта. При охлаждении выход кристаллиЯ
ского маннита 2,7 г (80,5% от теоретического); т. пл. 165° С. /’Я
Омыление ацетатов может также произойти при многодневной выдержке см|
при комнатной температуре или даже при 0° С. Реакция проходит до конца и в томляВ
чае, когда дезацетилируемое вещество плохо или почти нерастворимо в используем
растворителе. Ацетилцеллюлозу полностью омыляют нагреванием с раствором метиЭДД
натрия в метиловом спирте [67J; пользуются обычпо 0,1—0,01 н. растворами мятил^Д
натрия. дЙЯ
Трифторуксусные эфиры углеводов расщепляются уже под действием воды.^И
метилового спирта (69J. ;:'ЙЯ
Омыление кислотами. Омыление сложных эфиров кислотами аяН
бораторной практике используется реже. Оно имеет значение для омыления НеустйИ
чивых в щелочной среде соединений или тогда, когда омыление щелочами не достигИ
цели, как, например, в случае эфиров тетраметилянтарной кислоты (70]. Для проэдЯ
ния гидролиза эфир кипятят с водными минеральными кислотами, при омылении вэдВ
нерастворимых соединений добавляют ацетон, диоксан или ледяную уксусную кисщДВ
Скорость гидролиза сложных эфиров увеличивается с повышением темпера^Ц
и концентрации ионов водорода. В качестве кислот, катанизирующих гидролиз, мН
всего применяют рабавленные серную или соляную кислоту; пригодна также п-тойявИ
сульфокислота. ^9
Под действием концептрированпых НС1 и НВг наряду со свободными карбмВ
выми кислотами образуются соответствующие алкилгалогениды, которые легко уанВ
ются из реакционной массы отгонкой[71]. Этот метод иногда применяют для полута^В
карбоновых кислот. даЯ
Неустойчивые карбоновые кислоты можно получать из их эфиров каталиэдЩ
емой кислотами реакцией переэтерификации. Например, акриловую кислоту мо^В
получить из ее метилового эфира переэтерификацией муравьиной кислотой в прП^Н
ствии серной кислоты (72].
Кислотное расщепление эфиров широко используется в промышленностям^
омыления жиров. Для этого применяют катализаторы, так называемые «рапид-расЭДнИ
тели» (реактив Твитчелла). Природа и действие этих катализаторов подробно иэуя^В
Шлутиусом [73].
Расщепление эфиров гидрированием*. Сложные ая^В
бензилового спирта расщепляются на палладиевом катализаторе (Pd/H2) с обра^В
ни ем соответствующих карбоновых кислот и толуола (74]. Этот способ имеет др^оВВ
тивное значение, например для расщепления ацилированных эфиров малоновоЖИ^В
[67]
[68]
[69]
(70
[71
(72
(73
(74
Наряду с бензильной защитой карбоксильной группы, удаляемой каталитиЧета
гидрированием, большое значение имеет тиретп-бутильная защита. ПрИ Ж
лизе таких эфиров получаются карбоновые кислоты п изобутилён [Т.
et al., J. Am. Chem. Soc., 85, 488 (1963)]. — Прим. ped. -Jg
G. Zemplen et al., Ber., 56, 1705 (1923); 59, 1258 (1926); 69, 1827 $1
G. ZempUn, E. Pacsu, Ber., 62, 1613 (1929). ga
E. J. Bourne, С. E. M. T a 11 о w, J. С. T a 11 о w, J. Chem. Soc.,‘ И
1367. |
K. v. Auwers, Ann., 292, 180 (1896).
W. Theil acker, G. Wend tl and, Ann., 570, 50 (1950). -• 5
С. E. Rehberg, Org. Synth., 29, 5 (1949). J
E. Schlu t ins, J. prakt. Chem., 142, 49 (1935). J
K. W. R osen mun d, F. Zetzsche, F. Heise, Ber., 54, 2038
Резюме в статье W. H. Hartung, R. Simonoff, Org. Reactions, 7>|
(1953). ' 4
Ш ОМЫЛЕНИЕ АЦЕТАЛЕЙ, МЕРКАПТАЛЕЙ, КЕТАЛЕЙ И ОРТОЭФИРОВ
зе
лоты R —СО—СП'(СООС2Н5)2, так как под действием щелочей ацильные группы
щеиляюгся быстрее, чем происходит омыление. Однако при гидрировании соответ
ствующпх бензиловых эфиров на палладированном угле можно в мягких условия
отщепить бензильные группы; этот путь иногда выбирают для получения кетонов (75'
IV
I /СООСНгСбНб
К -СО—с<
\:ооснгсвн.',
R’
pd.'ir. I /СООП
------* R—со-С< —> В-СО-СН2К'
хсоон
Расщепление сложных эфиров гидрированием нашло важное применение дл
получения пептидов по «карбобснзоксиметоду» Бергманпа [76]. По этому метод
аминокислоты сначала подвергают взаимодействию с бензиловым эфиром хлор
угольной кислоты и после сочетания с другими аминокислотами бензильнь!
остаток отщепляют от эфира бепзилкарбаминовой кислоты в виде толуола. Образу
гощиеся при этом производные карбаминовой кислоты тотчас же распадаются на СО
и пептид:
Ccl]5-CH3-O-CO-NlI-Cn-CO—NH-CH-COOH Pd/Ht^
—> H2N—сп-со—NH —С Н—СО ОН 4- с6н8—сп3 4- СО
III. Омыление ацеталей, меркапталей, кеталей и ортоэфиров
Образование ацеталей, кеталей и меркапталей является наиболее удобны!
способом стабильного блокирования карбонильной группы. Защищенная ацетализа
цией карбонильная группа очень легко регенерируется после проведения изменение
в молекуле.
Омыление ацеталей имеет большое значение также потбму, что ненасыщенны,
альдегиды иногда получают через их ацетали. Так, при синтезе альдегидов по Гринь»
яру альдегиды часто образуются в виде ацеталей. Обычно ацетали гладко гидролизу
ются разбавленными минеральными кислотами, но устойчивы к действию щелочей
Расщепление, как правило, проводят 0,01—3 и. H2SO4, применяют и разбавленную
НО, NaHSO3 и органические кислоты, например уксусную, щавелевую или виннукь
Нерастворимые в воде ацетали разлагают в присутствии органических растворителей
таких, как спирт, ледяная уксусная кислота пли ацетон.
Фепилпропаргиловый альдегид [77]. Его диэтилацеталь нагреваю-
30 мин прп периодическом встряхивании па паровой бане с примерно 12%-ноз
H2SO4; выход альдегида 70—81% от теоретического.
Глицериновый альдегид [78]. Диэтилацеталь глицеринового альдегид:
расщепляют встряхиванием в течение 7 суток с 0,1 н. II2SO4. Выход альдегид.
80% от теоретического.
Кетали в большинстве случаев моментально расщепляются сильно разбд.
вленными кислотами. Относительно устойчивы циклические ацетали и кетали, а такие,
ацетали, образованные пз альдегидов с основными группами. Последние при гидролиз,
могут вступать в альдольную конденсацию или образовать шиффовы основания
Поэтому их омыляют концентрированными кислотами.
Ниже приведен способ омылепия чувствительных ацеталей, например диацетал»
p-метилкротоиового альдегида J 79].
[7о] R. Е. Bowman, J. Chem. Soc., 1950, 325.
J76J M. Bergmann, L. Zervas, Ber., 65, 1192, 1201 (1932).
77] C. F. H. Allen.C. О, E dens jr., Org. Synth., Coll. v. 3, 1955, p. 731.
[78| E. J. Witzemann et al., Org. Synth., Coll. v. 2, 1943, p. ,305.
[79( F. G. Fischer, K. Lfisenberg, Ann., 494, 272 (1932).
24 Заказ 1 835.
370
РАЗРЫВ СВЯЗЕЙ УГЛЕРОД - КИСЛОРОД
p-Метилкротоновый альдегид. Встряхивают 53 г диацеталя с Ю лЙ
насыщенного на холоду водпого раствора впппой кислоты; смесь при этом сильи
охлаждают и гомогенизируют. Затем при встряхивании добавляют 200 мл пас»?
щеипого на холоду раствора СаС12, в результате чего в виде масла выделяется
р-метплкротоновый альдегид. к
По другому способу [80] чувствительные ацетали расщепляют обрабс
ацетолом в присутствии бензолсульфокислоты.
Ступенчатый гидролиз триацетовида маннита по Ирвину и ПаттепеынИ
[81]. Растворяют 30 г триацетонида маппита в 450 мл спирта, содержащего 3?
воды и 0,1% НС1. Раствор перемешивают 2,5 ч в термостате при 4° С, заг
осаждают хлориды карбонатом серебра и встряхивают фильтрат с животш
углем. При упаривании фильтрата в вакууме сначала выпадает неизменен#
трпацетонид маннита; его отфильтровывают и выпаривают фильтрат досуз
Прп повторном растворении в холодном ацетоне свободный маппит пе рас®
ряется, а моно-и диацетонид маппита переходят в раствор. При отгонке ацеМ
в вакууме получают смесь моно-и диацетонидов в количестве 50% от в8ят<
триацетонида. Для разделения их кипятят с большим избытком бензола. П
охлаждении выпадает кристаллический моноацетонид маппита; пэ фильтре
удаляют растворитель, а остающуюся сиропообразную массу перегоняют п
172° С и разрежении 11 льм рт. ст. Д
После многих недель стояния в холодильнике диацетонид выкристащ
зовывяется в форме игл; т, пл. 37—39° С. Синтезированный продукт представай
собой, 3,4;5,6-диацетонид маннита, так называемый диацетонид маппита.
мерный ему симметричный 1,2;5,6-диацетонпд маппита получают частичН!
ацстонированием маппита [82].
Продолжая гидролиз диацетонида маннита, Ирвин и Паттерсон получи
3,4-моноацетонид маннита [81, 83]; изомерный 1,2-моноацетонид маииита ®
получен через эфир борной кислоты [84]. '[
Гидролиз 1,2-изопропилиден-3,5,6-трибензпл-а-глюкозы до 3,5,6-м
беизил-а-глюкозы происходит в течение 4 ч в смеси 96%-ного спирта н 3/i
HsSO4 при 809 С; выход 73% от теоретического [85].
Для омылепия 4,6-бензилиден-2-деаокси-1-галактонитрила его нагрев^
с 20%-ной уксусной кислотой и, медленно приливая по каплям 10%-ную ум$
ную кислоту, отгоняют смесь уксусной кислоты и бензальдегида [86]. РеаКД
заканчивается через 30 мин.
Частичный гидролиз бис-диметилацеталя ацетоуксусного альдегида до дииея
ацеталя ацетоуксусного альдегида можно осуществить, нагревая бис-диметилацет#
20—30 мин при 50—60° С с эквивалентным количеством воды; выход 91% от теории
ского [87].
Избирательное расщепление, ацетальиой связи в сложных эфирах ацетай
может быть проведено с помощью уксусной кислоты [88]. ' '
[80]
[81]
[82]
83]
84
85
86
87
88
Н. Sehin z, G. Schappi, Helv. chim. acta, 30, 1483 (1947). «
I. C. Irvine, В. M. Patterson, J. Chem. Soc., 105, 907 (1914).
E. Fischer, Cb. Bund, Ber., 49, 41 (1916); L. v. V a r g h a,
66, 1397 (1933).
A. Mtiller, Ber., 65, 1056 (1932).
L. v. Vargha, Ber., 66, 1396 (1933). >
F. Weygand, O. Trauth, Chem. Ber., 85, 60 (1952). j
R. G r e w e, H. Pocbaly, Chem. Ber., 87, 52 (1954). *
W. Frankest al., Chem. Ber., 86, 793 (1953). '
R. Gre we, E. Nolte, Ann., 575, tl (1952). V-
ПТ. ОМЫЛЕНИЕ АЦЕТАЛЕЙ, МЕРКАПТАЛЕЙ, КЕТАЛЕЙ И ОРТОЭФИРОВ 3'
При гидрировании в присутствии металлов платиновой группы ацетали бензал
дсгида расщепляются с образованием толуола и исходного спирта. Этот способ яме
значение для получения моноэфиров полиоксисоединений, например р-моиоэфир<
глицерина [80]:
ОН
I
СН2-СН-СН2
I I
О—СН—о
I
СвНб
ОСОСНз
сн3-сн-сна зНд
о—СН—о
1
СвНб
ОСОСНз
!
сн2—СН—СП-2
1 | + С6НбСНз
он он
Бензаль-а-метилглюкозид был восстановительно расщеплен водородом в пр:
сутствии платиновой черни с образованием толуола и а-метилглюкозида (9oj.
Аналогично мягким и в некоторых случаях рекомендуемым методом расщепл
ния ацеталей и кеталей является расщепление при помощи ионообменных смол [91
Еще один метод расщепления ацеталей заключается в их взаимодействии с анга,
рядами кислот, например с ангидридом уксусной или бензойной кислоты, в прису
ствии ионов водорода [92]. Из 1 моль ацеталя и 1 моль ангидрида прн этом обр
зуются альдегид и сложный эфир. При кислотном гидролизе ацеталей в прясутстви?
например, гидроксиламина, гидразина или аминов можно непосредственно получа’
оксимы, гидразоны или шиффовы основания.
Меркаптали более устойчивы к гидролизу, чем ацетали. Их расщепление можа
проводить с помощью HgCl2 и СаСО8 или CdCO3 или, что еще лучше, при помои
HgO. Этот метод приобрел наибольшее значение в химии углеводов [93].
5-Дезокси-Р-арабнноаа [94]. В течение 8 ч при 40° С перемешивай
0,01 моль меркапталя 5-дезокси-Р-арабинозы с 60 мл ацетона, 6 мл воды, 6
желтой HgO и 6 г HgCl2. Затем осадок отсасывают, промывают водой, выпаривав
фильтрат при 40° С в вакууме в присутствии 1 г HgO, продукт из остатка трижд
экстрагируют теплой водой (порциями по 30 мл). Из объединенного экстрак:
осаждают Н2$ ртуть в виде сульфида ртути и отфильтровывают его. Раство-
из которого удаляют соляную кислоту при помощи ионообменной смолы (в’
фатит), упаривают до сиропообразного состояния и сушат в вакууме; вых<
дезоксиарабинозы 0,9 г (67% от теоретического).
Уреидостпльбенальдегид. Растворяют диэтилмеркапталь 4-уреидостил
бспальдегида-4' в смеси ацетона с водой, смешивают с HgCl2 и HgO, перемет:
вают 1 ч при комнатной температуре и отфильтровывают ртутное соединена:
После добавления воды выкристаллизовывается уреидостпльбенальдегид; выхс
89% от теоретического [95].
Обзорную работу по расщеплению меркапталей и меркаптолов см. [96].
[89] М. Bergm ann, N. М. Carter, Z. physiol. Chem., 191; 211 (1930).
[90] К. Freudenberg, Н. Toepfler, С. Anderson, Ber., 61, 175
(1928).
[91] W. L. G 1 e n, G. S. M e у e г s, G. A. Grant, J. Chem. Soc., 1951, 257<
С. E. Ballou, H. O. L. Fischer, J. Am. Chem. Soc., 76, 1660 (195(3
[92] A. Wohl, H. Roth, Ber., 40, 217 (1907).
[93] E. Sovkin, T. Д ei chstein, Helv. chim. acta, 28, 940 (1945); H. Z i a
n e r et al., Chem. Ber., 91, 1; 48; 427; 1006; 1657 (1958); 92, 1618; 2893; 315
(1959).
[94] H. Zinner, K. Wessely, H. Kristen, Chem. Ber., 92, 162
(1959).
[95] G. I) ref ahi, H. Seeboth, W. Degen, J. prakt. Chem. [4], -
102 (1956).
196] D. S. Tarbell, D. P. Harnish, Chem. Rev., 49, 67 (1950).
372
ОБРАЗОВАНИЕ с—n-связей в результате присоединения
Ортоэфпры могут рассматриваться как кетали эфиров карбоновых кислот.
Они гидролизуются аналогично кеталям. Для препаративных целен расщепление орто-
эфпрон нс пмеет значения, ибо оно приводит к обычным карбоновым кислотам, которые
все без исключения могут быть получены другими, более удобными путями.
Ангидриды карбоновых кислот гладко гидролизуются под действием воды, что
описано в учебниках органической химии.
ОБРАЗОВАНИЕ СВЯЗЕЙ УГЛЕРОД - АЗОТ *
ОБРАЗОВАНИЕ C-N-СВЯЗЕЙ В РЕЗУЛЬТАТЕ РЕАКЦИЙ ПРИСОЕДИНЕНИЯ
Т. Присоединение азотсодержащих соединений по кратным
углерод-углеродным связям
1. Присоединение аммиака н продуктов его замещения
Присоединение аммиака и аминов к простым олефинам происходит только
в присутствии катализаторов в жестких условиях. Например, N-этиланилин можно
получить из анилина и этилена при 300° С и 200 ат в присутствии натрия [1 ]; выход
80—90%. Повышенное давление и катализаторы необходимы также для присоединение
амипоп по С=С-связям (2—4]. По эта реакция обычно пе останавливается на стадии^
простого винилирования [2, 3] или в результате ее вместо ожидаемых виниламиноБ,
получаются изомерные им основания Шиффа [4]. Возможно также винилирование тре--;
тичных аминов. Так, при 60—70° С, давлении 20 ат и отношении ацетилен : азот =
= 2 : 1 с хорошими выходами получается нейрин (2, 5J:
(CH3)3N + CH=CH-MI2O ОН-
Значительно легче реагируют олефины, двойная связь которых активирован^
другими соседними кратными связями, например а,р-ненасыщоииые карбоновые
кислоты, кетоны [6], нитрилы [7] и аналогичные соединения. В простейшем случай
достаточно уже активирования соседней двойной связью в диене или бензольным ядроМ£
Например, анилин взаимодействует со стиролом уже при нагревании в колбе с обратный
холодильником в присутствии небольших количеств патрия с образованием N-фенилй
р-фенилэтиламина с выходом 70% от теоретического [8J. р-Дминокислоты получаю^
нагреванием а,р-ненасыщенных карбоновых кислот при температуре около 100вС«
с аммиаком в запаянной ампуле [9[. Соответствующие эфиры р-амипомасляпой кислотЬК
[10] были получены с выходом до 85% в результате 14 суток взаимодействия прЯ?
комнатной температуре эфиров кротоновой кислоты с первичными или вторичным^
аминами в спиртовом растворе. Присоединение морфолина к производным акриловой^
кислоты в уксусной кислоте протекает настолько гладко, что реакция пригодна ДЛЯ;
количественного определения этих ненасыщенных соединений [И]. Примером скли#-
[II
[2]
[3]
[4]
16]
[7]
[8]
[9]
[10]
[11]
Материал обработали Г. Лемап и X. Тейхман. 3
И. Stroh, J. Е bersberger, et al., Angew. Chem., 69, 130 (1957).
W. Reppe et al., Ann., 601, 128 (1956). 1
C. W. Kruse R. F. К leinschmi <11, J. Am. Chem. Soc., 83, 216 (1961)<ь
C. W. Kruse, R. F. Kleinschmidt, J. Am. Chem. Soc., 83, 213 (1961ЭД
О приемах работы с ацетиленом и мерах предосторожности см. W. R е р р е fit;
al., Ann., 596, 10 (1955).
N. II. С гр in we 11, Chem. Rev., 38, 83—137 (1946).
F. C. W h i t m от e, H. S. Mo sh s r et al., J. Am. Chem. Soc., 66 , 725 (1944)fc
R. Wegler, G. Pieper, Chem, Ber., 83, 1 (1950). ^’5
R. Engel, Bull. Soc. chim. France [2], 50, 102 (1888).
D. W. Adamson, 1. Chem. Soc., 1950, 886. *
F. E. Critchfield, G. L. Funk, J. B. Johnson, Anal. Chem., 23«
76 (1956).
Г. ПРИСОЕДИНЕНИЕ АЗОТСОДЕРЖАЩИХ СОЕДИНЕНИИ ПО КРАТНЫМ СВЯЗЯМ 3'
ности а.р-неписыщеыпых кетонов к присоединению может служить легкость синте;
диацетонампиа пз окиси мезитила при перемешивании с водным аммиаком в течей]
нескольких часов [ 12]; так же легко присоединяется гид рокси л амин, причем образует<
диацетопгидроксиламин (13].
Амиды кислот получаются в результате присоединения аммиака или амине
к кегенам; так, из дикетена с высокими выходами легко приготовить амиды ацетоуксу
вой кислоты [14, 15].
N-Галогенамины легче присоединяются даже по переакционноспособпым двО]
ным связям, чем сами амины, причем одновременно в a-положение вводится атом гал
гена. Аналогично реагируют N-галогенсульфамиды, тогда как почти все N-галогенир
ванные амиды карбоновых кислот реагируют совершенно по-другому.
2. Присоединение других азотсодержащих соединений
Присоединение диазоалканов и азидов
Исключительно легко присоединяются диазоалканы и азиды, особенно по акп
внрованным двойным связям. Иногда продукты присоединения, образовавшиеся m
первой стадии реакции, отщепляют азот уже иа холоду, по чаще при нагревании с oi
разованием С—С-связей. Тем пе мепее во многих случаях такие реакции присоедиа,
ния являются наиболее изящным путем синтеза азотсодержащих гетероциклически
соединений.
Например, прп внесении метилового эфира коричпой кислоты в охлаждаемь
эфирный раствор диазомегана получается практически с количественным выходе
метиловый эфир 4-фепилпиразолип-З-карбоновой кислоты [16]:
С6Н=,СН=СНСООСН3 —- СвН6СН-СНСООСНз
I I
Н2С у
Из бензальацетофенона таким способом с почти количественным выходом обр.
зуется 4-фенил-З-бепзоилпиразолип [17]. По этому принципу из диазомстана или диаа.
уксусного эфира можно синтезировать большое число производных пиразолин,
Аналогично из алкинов получаются производные пиразола; папример, реакци
пропаргилового альдегида с эфирным раствором диазометапа дает пиразолальдегид-
с выходом 84%, с диазоуксусным эфиром: — 86% этилового эфира пиразолальдегид-;
карбоповой-4 кислоты [18]. Сам пиразол легко образуется из диазометана и ацетт
лена [19].
Таким же способом получают триазолы, присоединяя по С=С-или С=<
связям азотистоводородную кислоту или азиды вместо диазопроизводных [18].
Присоединение нитрилов
Важным в препаративном отношении методом получения алкилировад
ных амидов с вторичными или третичными алкильпыми группами при атоме aaoq
является присоединение синильной кислоты или нитрилов к олефинам в присутстви
П2]
ИЗ
[14
[15
[16
[17
[18
[19
Р. В. И а е s е 1 о г, Org. Synth., 6, 28 (1926).
С. Harries, R. Gley, Вег., 31, 1808 (1898).
С. Е. К a s 1 о w, D. J. Cook, J. Am. Chem. Soc., 67, 1969 (1945).
J. W. Williams, J. А. Krynitzky, Org. Synth., 21, 4 (1941).
H. v. Pechmann, E. Burkard, Ber. 33 3594 (1900).
L. 1. Smith, W. B. Pings, J. Org. Chem., 2, 23 (1938).
R. Hilt tel, Ber., 74, 1680 (1941).
H. v. Pechmann, Ber., 31, 2950 (1898); W. Hiickel, J. Da tow-
E. Simmcrsbach, Z. physik. Chem. Abt. A, 186f йп9 (1940).
374
ОБРАЗОВАНИЕ С—N-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
А
концентрированной серной кислоты или других сильных кислот (реакция Графа—
Риттера) [20—26]:
rc=n+r;c_cb; (r-c=n-cr;-chr;i н-cn-nh-cr;-chr; J
Эта реакция протекает с хорошими выходами при комнатной температуре илтем
при умеренном нагревании в ледяной уксусной кислоте иди в дибутиловом эфире.
Смесь 10,4 г стирола и 6,1 г ацетонитрила вносят в раствор 17 г бепзолсуаьЯ
фоновой кислоты в 50 мл ледяной уксусной кислоты и оставляют па 15 ч, Затейте
ее выливают на 200 г раздробленного льда и нейтрализуют аммиаком; выделяете!
вязкое масло, которое экстрагируют тремя порциями эфира (по 100 мл каждая
Объединенные вытяжки сушат над К2СОз и после отгонки эфира перегоняя
(т. кип. 175—180° С при 7 мм рт. ст.). После перекристаллизации из петроле:
ного эфира выход N-a-фенилэтилацетамида [20] 7 г (43% от теоретического)
t. ПЛ. 72—73° C. " ’ ‘ ‘
"A
Формамиды, образующиеся по реакций олефинов с синильной кислотой, особен!
легко омыляются до аминов, этот синтез является изящным способом получения обычй
труднодоступных третичных алкиламинов [21]. Реакция широко применима и мояя
использоваться также для получения соединений других классов, например N-ацилай!
нокислот [23], изохинолинов [24] или оксазолинов [25].
С равным успехом вместо олефинов можно исходить из соответствующих втори1
ных или третичных спиртов.
Присоединение окислов и оксихлоридов азота *
Димер двуокиси азбта присоединяется по олефиновым двойным связям с обрай
ванием динитросоединеиий и нитронитритов; последние неустойчивы и легко окисл$
ются до нптронитратов или гидролизуются до иитроспиртов [27]. Несмотря па хоропЦ
выходы, часто получаемые по этой реакции, она не имеет большого значения в препай
тивной химии, так как при ее проведении необходимо точно соблюдать установлений
режим и применять очень чистые реактивы; кроме того, в результате реакции всвг|
образуются смеси веществ. Указанное относится также к присоединению нитрон^
хлорида.
Присоединение нитрозилхлорида по двойной связи тоже протекает неоднознач!
[28]. Препаративное применение эта реакция может, по-видимому, найти для присоеД
нения нитрозилхлорида к эфирам ненасыщенных спиртов; так, например из ацевЛ
коричного спирта был получен 1-фенил-1-хлор~2-оксимино-3-ацетоксипропан [29ji$
СвН6СН=СНСН2ОСОСН8 СвН5СНССН2ОСОСН3 л
I II
Cl NOH
[20]
[21]
[22]
[23]
(24]
(25]
[26]
[27]
[28]
[29]
Изучение присоединения окислов азота по двойной связи было начато Н. Я. Дей
яновым в 1893 г., работы которого в данной области могут быть названы клйСЛ
ческими (Н. Я. Демьянов, Избранные труды, Изд. АН СССР, 1936)/^
Прим. ред. ’ Л
J. J. Ritter, Р. Р. М i n i е г i, J. Am. Chem. Soc., 70, Ю45 (1948).
J. J. R i t1 е г, J. К а 1 i s h, J. Am. Chem. Soc., 70, 4048 (1948), .'il
F. R. Benson, J. J. Ritter, J. Am. Chem. Soc., 71, 4128 (1949)..
. R i 11 er, J. Am. Chem. Soc., 71/4130 (1949). '’$
И u rp h y, J. Am. Chem. Soc., 74, 763 (1952); H. Wob
R.
weber, R. Ы i 11 m a n n, Angew. Chem., 72, 1001 (1960). ?
R. M. Lusskin, J, J. Ritter, J. Am. Chem. Soc., 72, 5577 (1950). -S?
II. Plant, J. J. Ritter, J. Am. Chem. Soc., 73, 4076 (1951); Ch. L. P tyl
r i s, R. M. Christenson, J. Org. Chem., 25, 331 (i960). д,
N. L e v у et al., J. Chem. Soc., 1946, 1093, 1096, 1100; 1948, 52; 1949, 2M
L. J. Beckham, W. A. Fessler, M. A. Kise, Chem. Rev., 48, 34
369 (1951). r ' !
R. P f 1 e g e r, F. Landauer, Ann., 610, 115 (1957).
II. ПРИСОЕДИНЕНИЕ АЗОТСОДЕРЖАЩИХ СОЕДИНЕНИЙ ПО C—N- Н С=Ы-СВЯЗЯМ
Из тетраалкилэтиленов в результате присоединения иитрозилхлорида, восстц
вления ZnCl2 и замыкания кольца под действием щелочи получают 2,2,3,3-тет
алкнлэтиленимины с очень высоким выходом [30].
II. Присоединение азотсодержащих соединений по C—N
и С=1У-сеязям
1. Присоединение к производным цианистоводородной кислоты '
Реакционную способность тройной C^N-связи в синильной кислоте или;
производных можно использовать для образования С—N-связей. В простейшем слу»
при взаимодействии с аминами образуются амидины, для чего необходимо усилить по.
ризацию С—N-связи. Так, Шорт с сотр. [31J получил с хорошими выходами амидд
при нагревании нитрилов с соответствующими аминами в присутствии А1С13. Неза:
щенные амидины получаются с отличными выходами при проведении реакции в ни
ком аммиаке под давлением в присутствии солей аммопия [31а]. Иногда для синт<
амидинов используют также присоединение амидов щелочных металлов к нитрил
[32J; но чаще всего эта реакция протекает недостаточно гладко. Гораздо более глад
и с отличными выходами получают амидины при взаимодействии ароматичесн
нитрилов с магниевыми производными вторичных аминов, которые одновременна об]
зуются в реакционной среде из этилмагнийбромида и амина [33, 34].
Амидины. К эфирному раствору этилмагнийбромида (50%-ный избы*]
в расчете на нитрил) прибавляют с небольшим избытком вторичный амин; noq
окончания выделения этана смесь нагревают в колбе с обратным холодилы
ком около 20 мин, после чего добавляют раствор нитрила в эфире; после 2—3
кипячения с обратным холодильником реакционную массу разлагают льд<
В некоторых случаях этот путь дает лучшие результаты, чем синтез амиди®
по Пиннеру.
Значительно легче, чем амины, присоединяются к нитрилам гидроксилами;
с образованием амидоксимов [34а].
R—С—NH3
II
NOH s
Бензамидоксим [35]. В течение нескольких часов нагревают воде
спиртовой раствор эквивалентных количеств гидрохлорида гидроксилами®
бензонитрила и K2COs при 60—80° С.
Взаимодействие изоннтрилов с первичными аминами приводит к N,N'-flH3a&.
щенным формамидинам [36].
Присоединение производных аммиака к цианамиду приводит к образован®
гуанидинов. Соли незамещенною гуанидина получают сплавлением дициандиамж
с солями аммония, например с нитратом аммония [37]. Большей частью амины глад;
взаимодействуют с цианамидом только в виде гидрохлоридов; реакция протекает П]
кипячении в колбе с обратным холодильником в абсолютном спирте [38]. Такой мет*
[30] G. L. С 1 о s s, St. J. В г о j з, J Am. Chem. Soc., 82, 6068 (I960).
[31J P. О x 1 e у, M. W. P art ri dge, W. F. S h о г t, J. Chem., Soc., 19<
1110.
[31a] F. C. Schaefer, A. P. Krapcho, J, Org. Chem., 27, 1255 (1962).
132] G. N ewbery, W. Webs ter, J. Chem. Soc., 1947, 738; K. Z i e g 1 e
H. Ohlinger, Ann., 495, 99 (1932).
[331 E. L о г z, R. В a 11 z 1 y, J. Am. Chem. Soc , 70, 1904 (1948).
[34] E. L о r z, R. В a 1 t z 1 y, J. Am. Chem. Soc., 71, 3992 (1949); 73, 93 (1951).
[34a] Резюме в статье F. E 1 о у, R. L enears, Chem. Rev., 62, 155 (1962).
[35] F. T i e m a n n, Ber., 17, 128 (1884).
[36] T. L. D a v i s, W. E. Y e 11 a n d, J. Am. Chem. Soc., 59, 1998 (1937).
[37] T. J,. Davis, Org. Synth., 7, 46 (1927).
[38] A. Kampf, Ber., 37, 1681 (1904).
376
ОБРАЗОВАНИЕ С—N-СВЯЗЕ,. В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
неприменим к слабоосновным аминам, например к нитроапплинам. Но и в этом случае
выходы гуанидинов практически количественные, если сплавлять амин с избытком
цианамида и обрабатывать затем избытком коцентрированной соляной кислоты [39].
В результате присоединения гидразина к цианамиду с высокими выходами полу-
чается аминогуанидин; простой способ получения аминогуанидина из динатриевой
соли цианамида и сульфата гидразина описали Фаитл и Зильберман [40].
Гуанидины синтезируют также из изомерных цианамидам карбодиииилов.
которые исключительно легко реагируют с аммиаком или аминами. Так, например,
при смешении дифенмлка^эбодиимида и анилина в результате экзотермической реакции'
образуется симметричный трифенилгуапидин [41]. Соответственное присоединение фе-<
нилгидраэпна к дифенилкарбодиимиду, приводящее к трифениламипогуапидину,
является первой стадией синтеза нитрона [42]. j
Азидный ион присоединяется по тройной связи нитрилов с образованием тетра-J
зольного кольца. Эта реакция обычно протекает очепь гладко, но большей частью в те-^
чение длительного времени [43] или при нагревании в толстостенной запаянной-^
трубке [44]. )
5-(га-Метоксифенил)-тетразол [43], В колбу с обратным холодильником^
загружают ,33 г (0,25 моль) п м •токсибензонптрила, 22 г (0,33 моль) азида иа<*
трия, 20 г (0,33 моль) ледяной уксусной кислоты и 100 мл «-бутилового сплрта-J
и нагревают смесь в течение 4 ч. После этого добавляют еще 5 г азида натрия, Ч
10 г ледяной уксусной кислоты и 10 мл бутилового спирта и нагревают еща’У
2 суток. Затем добавляют 300 мл воды и упаривают в вакууме до объема 100 мл.-3
Образовавшуюся суспензию подщелачивают 10%-ным раствором NaOH, филь-J
труют п экстрагируют примеси двумя порциями бензола, по 50 мл каждая.^
Водный щелочной раствор подкисляют разбавленной НС1 и выделившийся оса--^
док после промывки холодной водой дважды перекристаллизовывают пз 20%-иого^
изопропилового спирта: выход 81%; т. пл. 23S—239° С. э
Применяя в качестве растворителя диметцлформамид [45], время реакциям
можно сократить примерно до 24 ч. -я
Особенно легко реагирует дициандиамид с азидами, образуя 5-.тмипотетразод'&
[44]. Точно так же получают тетразол [46] с выходом около 80% нагреванием спиртов
вого раствора азотистоводородной кислоты с безводной синильной кислотой в течение:
2—3 суток при 100° С.
И, наконец, следует упомянуть реакции димеризации, тримеризации и полиме-
ризации производных синильной кислоты. Так, хлорциан очень легко тримеризуетсйК
в присутствии кислотных катализаторов с образованием цианурхлорида [47]. По^
действием щелочей гладко протекает димеризация цианамида до дициандиамида,
[48]; тримеризация до меламина осуществляется нагреванием дициандиамида с аммиак
ком под давлением [49]. '
Нитрилы тоже тримеризуются, причем в зависимости от природы нитрила полу<
чаются триазииы или аминопиримидины.
Например, при нагревании ацетонитюила в течение 5 ч с эквимолекулярным коли- /.
чеством сухого метилата калия при 140° С образуется 2,4-диметил-б-аминопиримидиЯч
(кианметин) с 70% -пым выходом. Из дихлорацетонитрила в присутствии А1С13 и хлори- >
[39] F. Arndt, Вег., 46, 3522 (1913); F. Arndt, В. Rosenau Вег., 50,
1260 (1917).
[40] Р- F a n 11 Н. g j ] h е г ш a n n, Ann,, 467, 279 (192S). .
[41] W. Wei th, Ber., 7, 10 (1874). J
[42] M. Busch, Ber., 38, 858 (1905).
[43] R. M. II e г b s t, К. В Wilson, J. Org. Chem,, 22 1142 (1957).
[44] J. S. Mi hina, R. M. Herbst, J. Org. Chem., 15, 1082 (1950).
[45] W. G. Finnegan, R. A. Henry, R. Lofq uist, J. Am. Chem*-;
Soc., 80, 3908 (1958).
[46] O. D imroth, G. Fester, Ber., 43, 2219 (1910). 1
[47] O. Diels, Ber., 32, 692 (1899).
[48] L. А. Г i n c k.* Inorg. Synth., 3, 43 (1950).
[49] Швейц, пат. 189406 (1935); С., 1937, II, .3233.
II. ПРИСОЕДИНЕНИЕ АЗОТСОДЕРЖАЩИХ СОЕДИНЕНИЙ ПО C = N- И С=.\-СВЯЗЯМ •
с того водорода при комнатной температурь получается 2,4,6-трис-(дихлорметв;
1,3,5-трпазин с 85%-ным выходом [51]. Бензонитрил тримеризуется в присутств
кислотных катализаторов даже при стоянии в течении 1—2 суток его раствора в хл<
сульфоновой кислоте при 0° С и дает симме»ричный 2,4,6-трифенилтриазин (квас
пин) [52]. При действии на бензонитрил суспензии натрия в качестве главного продув
с выходом до 85% образуется 2,2,4,6-тетрафенпл-2,3-дягидро-силл-триазин [5
Несимметрично замещенные 1,3,5-триазины получают сополимеризацией разл]
ных нитрилов [51] или в результате реакций обмена [54] между хлорангидрида
кислот и нитрилами [55]\
2. Присоединение к цианатам и тиоцианатам
Весьма многосторонний метод получения с высокими выходами производи
мочевины основан па принципе синтеза Вёлера, В эту реакцию вступают, с одной с
роны, цианаты щелочных металлов, эфиры изоциановой кислоты и горчичные Macj
с другой стороны, — аммиак, амины, амиды, гидразины и аналогичные соединен!
Прп использовании цианата щелочного металла реакцию проводят обычно в вода
растворе и применяют амины в виде солей; можно также вести реакцию со свободны
аминами в разбавленной уксусной кислоте; синтез проводится чаще всего при kqmhj
ной или при умеренно повышенных температурах. Ряд примеров получения моноар!
мочевин по этому методу см, [56].
Изоцианаты реагируют с первичными аминами в индифферентном растворите
при комнатной температуре с выделением тепла, причем образуются соответствуют
смешанные мочевины с практически количественными выходами. Несколько медленн
реагируют вторичные амины, плохо — вторичные ароматические амины. Растворили
в воде амины часто вступают в реакцию даже в виде водных растворов, посколь;
изоцианаты взаимодействуют с аминами легче, чем с водой.
Таким путем можно количественно переводить а-аминокислоты в соотве
ствующие уреидокисдоты, для чего встряхивают шелочпой раствор аминокисло*
с а-нафтилизоцианатом. После отделения нерастворимой динафтилмочевиа
(продукт конденсация избыточного изоцианата) уреидокислоту осаждают по
кислением [57],
Аналогично кипячением водных растворов гидрохлорида амина и роданид
щелочного металла [58] или амина и горчичного масла в спирте [59] получают тиомоч
вины; ароматические горчичные масла реагируют обычно уже па холоду. Так же глада
взаимодействуют водные растворы аммиака с горчичными маслами с образование
моноалкилтиомочевин [60].
Для получения тиосемикарбазида пользуются следующей хорошо зарекоменд
вавшей себя методикой [61].
Тноеемикарбазид. В раствор 28 мл 100%-ного гидразингидрата в 80 л
поды при перемешивании вносят небольшими порциями 72 г гидразинсульфаг.
устанавливают pH 3—4 и при 40° С постепенно добавляют 120 г NH4NCS. Пер
мешивают еще J0 мин и прибавляют Й10 мл метилового спирта. Продолжат
A. R. R о н z 1 о, W. В. Cook, Org. Synth-. 24, 6 (1944).
Ch. Gru n dm a n n, G. Weiss c, S. S e i d o, Ann., 577, 77 (1952).
A. H. Cook, D. G. Jones, J. Chem. Soc., 1941, 278.
J. J. Ritter, R. D. Anderson, J. Org. Chem., 24, 208 (1959).
H. Meerwein et al., Chem. Ber., 89, 217 (1956).
F. К г a f f t et al., Ber., 22, 803 (1889); 23, 2382 (1890); 25, 2263 (1892).
F. К u r z e r, Org. Synth., 31, 8 (1951).
C. N e u berg, A. M a n a s s e, Ber., 38, 2359 (1905).
F. К u г z e r, Org. Synth., 31, 21 (1951).
Th. О 11 e r b a ch er, F. C. Wh i t m ore, J. Am. Chem. Soc., 51, 190
(1929).
M. L. M о о r e, F. S. Crossley, Org. Synth., 21, 83 (1941); E. Schmidj
W. Striewsky et al., Ann., 568 192 (1950).
Пат. ФРГ 1067426; C-, i960. 3377.
378
ОБРАЗОВАНИЕ С—N-СВЯЗВЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
перемешивание в течение 30 мин и отфильтровывают выпавший (NHd)aSO4.i
К фильтрату добавляют от 2 до 3 мл ацетона и кипятят 24 ч с обратным холодиль-;:
инком. Через 12 ч добавляют еще 1 мл ацетона для восполнения потерь. Уже при
кипячении начинается выделение кристаллического тиосемикарбазида. Его*
отфильтровывают па воронке Бюхнера и промывают метиловым спиртом. Вы-5
ход 90—95 г из расчета на загруженный гидразин (89—94% от теоретического)^
т. пл. 180—181° С. . <
Для получения ацилированных мочевин из амидов кислот и изоцианатов тр$м
буются более жесткие условия, но реакция протекает, как правило, с хорошими ВнИ
ходами.
К-Бензоил-К'-фенилмочевина [62]. Медленно нагревают смесь 12,1 >
бензамида и 11,9 в фенилизоцианата до 115° С. При этой температуре начин»*
ется бурная реакция. Когда температура поднимается до 165° С, нагревание
прекращают. Быстро застывающее при охлаждении содержимое колбы кристая^
лизуют из диоксана; выход 19,8 е (82% от теоретического); т. пл. 203—204° бй
Особенно легко, часто уже нахолоду, реагируют с изоцианатами и горчичным:
маслами [63] сульфамиды в водно-щелочном растворе [64], причем образуются выделя
ющиеся при подкислении сульфонилмочевины. ,
Гидразины взаимодействуют с солями и эфирами циановой кислоты так же лег»
и в таких же условиях, что и амины. Сам гидразин подвергается обычно двов
ному замещению. Алкилгидразины реагируют обычно по а-, арилгидразины — D
р-атому азота. .4
Эфиры изоциановой кислоты аналогично эфирам цианистоводородной кислой
могут подвергаться ди- и тримеризации. В присутствии триалкилфосфиновых катализе
торов образуются димерные уретдионы [65, 66], а в присутствии ацетата калия [К
или алкоголята натрия [67] — гримерные эфиры изоциануровой кислоты.
III. Присоединение азотсодержащих соединений по С=0-связц «
Реакции присоединения азотсодержащих соединений к карбонильным соедини
ниям протекают обычно с отщеплением воды и образованием двойной C=N-cb®BI
Поскольку при этом в конечном счете происходит обмен кислорода на азот, такие ре£?
ции рассматриваются в данном разделе (стр. 470 сл.). 4
Только в сравнительно немногих случаях первичные продукты присоединен^
настолько стабильны, что обычное отщепление воды не происходит. Это характеру
например, для таких
Например, хлораль присоединяет ряд---------- ------ „-----
соедннения [68]: Я
альдегидов, которые образуют также стабильные гидраты. у’* 1’
'раль присоединяет ряд аминов и дает довольно устойчивы.
/Н
СС13-С< 4-H2NR
^0
СС13—СН(ОН)—NHR
Аналогичные «O.N-полуацетали» можно синтезировать также из бензальдегид
[69] и формальдегида [70], но они нестабильны. На основании химических своЙСП
[62
63
64
65
66
67
68
69
70
Р. F. Wiley, J. Am. Chem. Soc., 71, 1310 (1949).
F. Kurzer, Chem. Rev., 50, 7 (1952).
S. Petersen, Chem. Ber., 83, 551 (1950).
L. C. R a i f о r d H. B. F г e у e r m u t h, J. Org. Chem., 8, 230 (1943).
A. W. Hofmann, Ber., 18, 764 (1885). U
A. Michael, Ber., 38, 22 (1903). у
L. Rugheim.er, Ber, 39, 1653 (1906). A
A. D о r n 0 w, H. T h i e s, Ann., 581, 224 (1953). у
H. Hellmann, G. О p i t z, Cnem. Ber., 89, 81 (1956).
IV. ПРИСОЕДИНЕНИЕ АЗОТСОДЕРЖАЩИХ СОЕДИНЕНИЙ ПО C = S-CBH3M
з-
оксазолидинов, получаемых из 1,2-аминоспиртов и карбонильных соединений, t
можно рассматривать как циклические O.N-ацетали [71].
Несколько более устойчивы соответствующие продукты присоединения амид
кислот к альдегидам. N-Оксиметилфталпмид, имеющий значение для препаративнт
[72] и аналитических целей [73], получают с выходом выше 90% уже при непродолж
тельном кипячении в колбе с обратным холодильником фталимида с водным раствор<
формальдегида [73, 74]. Ряд других N-оксиметилкарбопамидов Эйнхорн [75] синт
зпровал нагреванием амидов кислот и формалина в иодной среде в ирису
ствии щелочных катализаторов конденсации, например К2СО3, NaOH или тр
этиламина.
IV. Присоединение азотсодержащих соединений по C—S-сеязи
Препаративный интерес представляет лишь присоединение аммиака и 4;
производных к сероуглероду с образованием дитиокарбаматов, которые находи
применение в качестве промежуточных продуктов в самых разнообразных синтеза
Аммиак, алифатические и алициклические [76] амины реагируют с CS8 с ад
чптельным выделением тепла, образуя соответствующие аммониевые соли диад
карбаминовых кислот:
2RNII2 + CS2 -> [RNHs[+[RNn—CSS]"
B случае алифатических диаминов образование соли может протекать за сч<
второй аминогруппы; получающиеся при этом бетаины конденсируются при нагревакс
с соляной кислотой и переходят в алкилентиомочевины с хорошими выходами [77, 78
NH
H,N—(СЩ),—NHi + CS, —. H8N4'-(CH2)„-NHCsS-^h3* <С.Н2)„
Хотя ароматические амины, являющиеся слабыми основаниями, тоже присд
единяются к CS2, они не дают солен с полученными дитиокарбаминовыми кислотамд
неустойчивые свободные кислоты немедленно распадаются на сероводород и горчи’
ное масло, к которому присоединяется в конце концов избыточный амии и образуют^
тиомочевины [79, 80]. Небольшие количества серы действуют каталитически на ац
реакцию [81].
Ароматические амины гладко реагируют с сероуглеродом с образованием дитнч
карбаматов при молекулярном соотношении ароматического амина и сероуглерод
1 : 1 и в присутствии сильного основания (NaOH или третичный амин) для связыван£|
кислоты.
Описан [82] метод, по которому с высокими выходами из ряда алифатические
ароматических и гетероциклических аминов в хлороформе в присутствии триэтиламиа
получаются дитиокарбаматы с высокими выходами.
171]
172]
173]
[74]
[75]
[76]
[77]
:78
'79
80
8Г
82;
Е. D. Bergmann, Chem. Rev., 53, 309 (1953).
И. Hellmann, Angew. Chem., 69, 463 (1957); H. Hellman n,G. О p i t 3
a-Aminoalkylierung, Verlag Chemie G. m. Ь. H., 1960.
M. B. Winstead, H. W. Heine, J. Am. Chem. Soc., 77, 1913 (1955).
S. R. Buc, J. Am. Chem. Soc., 69, 254 (1947).
A. Einhorn Ann., 343, 207 (1905); Герм. пат. 157355; FrdL, 7, 616 (1902-
1904).
A. S k i t a, H. R o,l 1 e s, Ber., 53, 1246 (1920).
C. F. H. Allen, С. O. Edens, J. Van Allan, Org. Synth., Coll. v. 5
1955, p. 394; T. B. Johnson; С. О. E d e n s, J. Am. Chem. Soc., 64, 270
(1942).
G. E. P. S m i t h jr., G. A 11 i g e г et al., J. Org. Chem.., 14, 935 (1949}
L. C. R a i f о г d, G. M. McNulty, J. Am. Chem. Soc., 56, 680 (1934).
S. H ii n i g, H. Lehmann, G. Grimmer, Ann., 579 , 77 (1953).
A. Hugershoff, Ber., 32, 2245 (1899).
С. M. В u e s s, J. Am. Chem. Soc., 77, 6613 (1955).
380
ОБРАЗОВАНИЕ С—N-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
Аналогично реагируют и гидразины. Так, из фенилгпдраэипа в эфире получают
соль Р-фенилдитиокарбазиновой кислоты (выход почти количественный), которая
служит исходным продуктом для синтеза дитизона [83].
Литературный обзор по реакциям аммиака и аминов с сероокисыо углерода
см. [84].
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ РЕАКЦИИ ОБМЕНА
J. Замещение водорода азотом
1. Нитрование
Общие положения. Нитрующие агенты [84а]
Замещение водорода нитрогруппой является одной из важнейших реакций
в органической химии, особенно в ароматическом ряду. Для проведения этой реакции
применялась как сама азотная кислота в самых разных концентрациях, смесях
и растворителях, так и большое число других реагентов, но все они имеют меньшее
значение, чем азотная кислота, особенно в виде нитрующей смеси.
В некоторых случаях находит применение разбавленная азотная кислота,.,
например при нитровании легко нитрующихся фенолов или парафиновых углеводо-
родов. Концентрированная азотная кислота и нитрующие смеси в большинстве слу-
чаев'совершенно непригодны для нитрования фенолов, так как при этом образуется
большое количество побочпых продуктов. Разбавленная азотная кислота является,
слабым’нитрующим агентом и довольно сильным окислителем, что объясняется йена-'
пенным присутствием азотистой кислоты, которая вообще затрудняет нитрование, но
оказывает сильное каталитическое влияние на питрованне фенолов и ароматических
ав^инов [85]. Однако разбавленная азотная кислота имеет ограниченное применение,
так как ео нитрующее действие проявляется лишь при повышенных температурах,',
когда уже превалируют побочные реакции окисления.
Окислительные свойства разбавленной азотной кислоты можно использовать
для одновременного введения в ароматическое ядро одной гидроксильной группы
и нескольких нитрогрупп в присутствии солей ртути в качестве катализатора. ЭтО(
открытое Вольффенштейпом [86] «оксинитрование» позволяет, например, в одну
операцию получать из бензола [87] пикриновую кислоту с выходом 50%, а из бензой-,
ной кислоты [88], правда с мепыпим выходом, — 2,4,6-тринитро-З-оксибензойную '
кислотуА(о механизме реакции см. [89]). т
2,4 -Динитрофенол [90]. В колбе емкостью 1,5 л сметалкой, обратным,
холодильником и капельной воронкой растворяют 60 г HgO в 541 мл 70%-ноЗй
HNO3 и рзбавляют водой до 750 мл. После прибавления 0,1 г NaNOs приливают
по каплям 50 г бензола при 50° С в течение 200 мин. Перемешивают еще 160 мин-
при 50° С и дают смеси остыть; через 12 ч 2,4-динитрофенол отфильтровывают И
[83] J. Н. Billmann, Е. S. Cleland, Org. Synth., Coll. v. 3, 1955, p. 360.
[84) R. J. Ferm, Chem. Rev., 57, 630 (1957).
[84a) T. Urbanski, Chemie una Technologie der Explosivstoffe, Bd. 1, VEb
Deutscher Verlag fur Gmndstoffindustrie, Leipzig, 1961, S. 18—73.
[85] К. К. И п г о и ь д. Механизм реакций и строение органических соединении,
пер. с англ., Издатинлят, 1959.
[86] R. Wolffenstein, О. Boters, Вег., 46, 586 (1913). т
[87] Е. Е. Aris toff et al., Ind. Eng. Chem., 40, 1281 (1948).
88] R. Wolffenstein, W. Paar, Ber., 46, 589 (1913).
[89] F. H. Westhcimer E. Segel, R. Schram m, J. Am. Chem. SOC.
69, 773 (1947).
[90] W. E. Bachman J. M. Chamerda et al., J. Org. Chem., 13,
390 (1948).
I. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
и промывают разбавленной азотной кислотой и водой; выход 87 г; т. пл. 11
ЦЗа С. Дополнительным извлечением динптрофелола из фильтрата можно
вести суммарный выход до 68% от теоретического.
Концентрированная азотная кислота оказывает нитрующее действие при бо
alum температурах и менее склонна вызывать нежелательное окисление, но так j
опа постоянно разбавляется образующейся при питровании водой, для получе]
удовлетворительных результатов приходится вводить очень большой избыток азот]
кислоты, что сильно ограничивает применение этого способа.
Во избежание этих осложнений уже с давних пор нитрование проводят в ср
концентрированной серной кислоты. Образующаяся при этом нитрующая си
главным образом употребляется для нитрования ароматических соединений. В заве
мости от реакционной способности отдельных классов нитруемых соединений при
няю1 нитрующие смеси, получаемые смешиванием IINO3 самой различной кояцент
ции с концентрированной HaSO4 или с олеумом. Установлено, что чем труд
нитруется соединение, тем меньше воды должно содержаться в нитрующей см«
Так, в промышленности используют ряд стандартных нитрующих смесей, из котор
например, смесь для мононитровапия толуола имеет следующий состав (в вес. 1
35% HNO3, 53% H2SO4 и 12% воды, тогда как для введения второй нитрогрух
работают с безводной смесью, состоящей из 33% HNO3 и 67% HaSO4. Для mohqhhx
ванпя количество нитрующей смеси подбирают с таким расчетом, чтобы в ней содер:
лось стехиометрическое количество азотпой кислоты, а для динитрования более цс
сообразно брать избыток азотной кислоты. Очень большое значение имеет 6hctj
отвод тепла реакции, то есть непрерывное энергичное перемешивание. Посколз
все реакции нитрования являются экзотермическими реакциями, в том месте, к
по каплям подают реагирующее вещество, может произойти значительный мест!
перегрев, который при запоздавшем перемешивании или при недостаточной иптепс
ности его может привести к взрыву.
Серная кислота в нитрующей смеси пе только связывает воду, а, что гора
важнее, непрерывно поддерживает достаточную концентрацию нитронильных катио
NO£. которые, собственно, п являются нитрующим агентом [91, 92]. Этим обусловь
и значительное подавление окислительных побочных реакций. Кроме того, прей
ществами серной кислоты являются превосходная растворяющая способность, высо
температура кипения и большая теплоемкость.
С другой стороны, конечно, применение нитрующей смеси недопустимо, &
нитруемое вещество взаимодействует с серной кислотой, например омыляется :
сульфируется. В таких случаях в качестве растворителей используют ледяную ук«
ную кислоту, хлороформ, четыреххлористый углерод, хлористый метилен, бен?
эфир и даже такие реакционноспособные растворители, как спирт и ацетон. Ангид;
уксусной кислоты хорошо связывает воду н в смеси с азотной кислотой оказы^
почти такое же действие, как нитрующая смесь, но легко сам нитруется и поэг
может применяться только при низких температурах*. Интересно, что как в а
случае [93], так и при нитровании в ледяной уксусной кислоте [94] или в рассмач
васмом далее ацетилнитрате. соотношение орто- и пара-замещенных ароматичесы
ядра может сильно отличаться от их соотношения при нитровании нитрующей смее
В качестве растворителя при нитровании бензонитрила с успехом употребляли анх
рид трифторуксусной кислоты, в то время как в этих же условиях в нитрующей см
почти половина этого нитрила омылялась [95, 96].
Делались попытки заменить серную кислоту в нитрующей смеси друга
минеральными кислотами. Так, для нитрования алкиловых эфиров малоновой е
лоты успешно применялась смесь безводной азотной кислоты и полифосфор
* Нитрование в уксусном ангидриде надо проводить с большой осторожное,
из-за возможности сильных взрывов. — Прим. ред.
[91] Ссылка [85], стр. 269.
192] R. .1. Gillespie, D. С. Millen, Quart. Rev. (Chem. Soc., London), 2,
(1948).
193] 0. N. Witt, A. L’tor man n, Ber.. 39, 3901 (1906).
[94] C. Schwalbe, Ber., 35, 3301 (1902).
[95] E. J. Bourne, M. Stacey et al., J. Chem. Soc., 1952, 1695.
[96] J. j. Blanksma, E. M. Petri, Rec. trav. chim., 66, 353 (1947).
382
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
кислоты [97]. Смесь фтористого водорода с азотной кислотой обладает сильными
нитрующими свойствами [98]. При ее помощи пз бензола прп температуре около 0е С
образуется нитробензол с выходом 83%.
В некоторых случаях смесь нитрата щелочного металла и концентрированной
серной кислоты оказывается более эффективной, чем нитрующая смесь.
Так. удалось провести нитрование практически нереагпрующего с азот-Й
ной кислотой или нитрующей смесью пиридина с выходом 15—204ц при добавле*^
пип 1'ебольшпми порциями нитрата калия в нагретый до 330’ С (!) раствор ппрй^
дина в 18%-ном олеуме [99]. Из столь же индифферентного к нитрования^
б-нитрохинолина удалось получить динитропроизводное нагреванием его в течей
нпе 10 час с концентрированной H2SO4 и рассчитанным количеством KNtJjj
при 130—140° С в запаянной трубке [100].
Смеси нитрита натрия’с серной или соляной кислотой тоже могут иногда оказвй
вать нитрующее действие наряду с ожидаемым нитрозированием и вызывать далек1!
идущее нитрование бензольного ядра, что имеет значение для получения некоторь^
диалкилнитроанилинов (стр. 388, ср. 397 сл.). <
Такие смешанные ангидриды, азотной кислоты [101], как ацетил- и бензоил^
нитраты, являются отличными нитрующими агентами для ароматических соединений^
при работе с ними часто количественно получают мононитропроизводные и, что удивв^
тельно, замещение происходит преимущественно в орто-доложение. К сожалений»^
эти легкодоступные соединения не совсем безопасны. При работе с ними, особещ'"*™
с ацетилнитратом, происходили несчастные случаи в результате самопроизвольнь
взрывов не ТОЛЬКО При нагревании [102], но и на холоду [103].
Для нитрования ароматических соединений иногда применяют эфиры азотн
кислоты [104], главным образом этилнитрат, в присутствии концентрировали ________
серной кислоты [105] или хлорида алюминия [106, 107]. Большее значение имев!
их применение для нитрования соединений с активной метиленовой группой (см. ниже#
в присутствии щелочных катализаторов, протекающее по типу сложноэфирной кан«
денсации.
Нитрование ароматических соединений тетранатрометаном ведут в щелочной
среде, например в растворе пиридина [108, 109]. По этому способу из диметил^
и диэтил-п-толуидица, а также из n-крезолас высоким выходом получаются мононитрф
производные [110]. Реакция протекает гладко прн охлаждении льдом, тогда как дрИ
нагревании на водяной бане третичные основания дезалкилируются и переходя^
в N-нитросоединения ] 111]. В таких условиях тетранптрометаномв ацетоновом раствору
можно нитровать в ₽-положение даже ароматические пропенильные производный
как, например, о-анетол, изосафрол и эфиры изоэвгенола, с сохранением двойной!
[97]
[98]
[99]
[ЮО]
101
102
103
104
[105]
106]
107]
108]
109J
J. Р. Kispersky, К. К I a g е г, J. Am. Chem. Soc., 77, 5433 (1955). ’
J. Н. S i m о n s H. J. Passino, S. Archer, J. Am. Chem. Soc., 68^’
608 (1941).
F. Friedl, Ber., 45, 428 (1912); A. К i г p a 1, E. Reiter, Ber., 58,,
699 (1925); H. J. Den Hertog jr., J. О v e г h о f f, Rec. trav.^
chim., 49, 552 (1930). *
A. Kaufmann, II. Decker, Ber., 39, 3648 (1906). /
V. G о 1 d, E. D. H u g h e s, С. K. Ingold, J. Chem. Soc., 1950, 2467.
’A. Pictet, E. К hot insky, Ber., 40, 1163 (1907).
W. К 6 n i g, Angew. Chem., 67, 157 (1955). k
R. Boschan, R. T. Merrow, R. W. van D о 1 a h, Rev., 55, 49fr
(1955). '
H. Raudnitz, Ber., 60, 738 (1927).
H. Willstaedt, G. Scheiber, Ber., 67, 471 (1934).
Б. В. T p о н о в, и др., ЖРФХО, 62, 2267 (1930): С., 1931, II, 421.
Литературный Обзор: К. F. Hager, Ind. Eng. Chem., 41, 2168 (1949).
Синтез см. Р. Liang, Org. Synth., Coll. v. 3. 1955, p. 803.
I, ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
3
связи и ароматического кольца; изомерные аллильные соединения при этом не peai
руют [110, 112].
В последнее время разработан еще один способ нитрования в щелочной сред
Он основан на применении нитрата ацетонциангидрина, легко получаемого нз циа
гидрина ацетона и дымящей азотной кислоты. Смесь пригодна для нитровав
соединений с активной метиленовой группой, как, например, арилацетонитрилов
эфиров малоновой и ацетоуксусной кислот [113].
В качестве нитрующего агента испытывали также ангидрид азотной кислот
Как и комплекс этого ангидрида с трехфтористым бором [114], он оказался сильщ
нитрующим агентом [115]. исего помощью при 0° С можно перевести бензол с кол
явственным выходом в нитробензол [116].
Димер двуокиси азота (N2O4) обладает меньшей реакционной способность
чем ангидрид азотной кислоты N2O5. Чистый N3O4 почти не взаимодействует с бен;
лом. но переводит даже пиридин в 3-интропиридин с выходом около 10% от теоретю
ского [117].
Смеси димера двуокиси азота с углеводородами взрывоопасны!
В сочетании с серной кислотой [118] или с хлоридом алюминия [119, 120] Na
проявляет очень сильное нитрующее действие; в данном случае это объяскяется бод
легким образованием ионов нитрония, которые также, несомненно, возникают и
употреблении твердого комплекса N2O4-BF31 обладающего сильным нитруиищ
действием [121]. ’
Хлористый нитронил, который в чистом виде действует на ароматическ
соединения скорее как хлорирующий агент [122—123], в присутствии кислотно
катализаторов — HF, BF3 или А1С13, способствующих образованию ионов нитрону
становится сильным нитрующим агентом [124]. Фтористый нитронил проявлю
нитрующее действие и в отсутствие катализаторов, но выходы нитросоединения оче
низкие [125].
Тетрафторборат нитронила [NO2]+ [BFJ”, полученный из фтористого нит|
нила и BF3 или из N2OS, HF и BF3, оказался пригодным для нитрования аромати*
ских соединений с' высокими выходами, иногда даже при охлаждении смесью, сн^
с поваренной солью {126—128].
Нитрование алифатических соединений [128а]
Замещение водорода питрогруппой в алифатических углеводородах им<
второстепенное значение в лабораторной практике. То что эта реакция вообще мо»
протекать с удовлетворительными выходами, было впервые показано Коновалов!
[ИО]
[111]
[112]
Ц13]
[114]
[115]
[И6]
[117]
[118]
[119]
[120]
[121]
[122]
[123
[124
[125
[126
[127
[128
[128а]
Е. Schmidt, Н. F i s с h е г, Вег., 53, 1529 (1920).
Е. S с h m i d t, Н. F i s с h е г. Вег., 53, 1537 (1920).
Е. Schmidt, R. Schumacher et al., Ber., 55, 1751 (1922).
W. D. Emmons, J. P. Freeman, J. Am. Chem. Soc., 77, 4391 (195.
G. B. Bachman, J. L. Dever, J. Am. Chem. Soc., 80 , 5871 (1958).
V. G о 1 d, E. D. Hughes, К. С. Г n g о 1 d, G. II. Williams,!. Che
Soc., 1950, 2452.
L. B. Haines, II. Adkins, J. Am. Chem. Soc., 47, 1419 (1925).
P. S chorigin, A. T о p tschie w, Ber., 69, 1874 (1936).
L. A. Pin-ck, J. Am. Chem. Soc., 49, 2536 (1927).
A. Schaarschmidt, Ber., 57, 2065 (1924).
A. Schaarschmidt, H. Balzerkiewicz, J. Gante, Ber., £
499 (1925).
G. B. Bachman, С. M. Vogt, J. Am. Chem. Soc., 80, 2987 (1958).
F. P. G i n t z, D. R. Goddard, M. J. С о 11 i s, J. Chem. Soc., 195
445.
M. J. Collis, D. R. Goddard, J. Chem. Soc., 1958, 1952.
С. С. P r i с e, C. A. S e а г s, J. Am. Chem. Soc., 75, 3276 (1953).
G. Hetherington, P. L. Robinson, J. Chem. Soc., 1954, 3512.
G. Olah, S. Kuhn, Chem. a. Ind., 1956 98.
G. Olah, S. Kuhn, A. M 1 i n k 6, J. Chem. Soc., 1956, 4257.
G. A. OI a h, S. J. К u h n, J. Am. Chem. Soc., 80 , 6541 (1958).
Резюме в статье: N. Kornblum, Org. Reactions, 12, 101 (1962).
3S4
ОБРАЗОВАНИЕ С— ?Ч-СВЛЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Г
[129]. проводившим эту реакцию с 13%-ной НХОЯ в запаянной трубке при 130—->
150е С.
Однако методы прямого нитрования парафиновых углеводородов, представая?
ющис практический интерес, появились лишь в течение трех последних десятилетий^
Так, Хасс [130] разработал применяемый в промышленности метод мононитрования Ч
низших алифатических углеводородов в газовой фазе с -~70%-ной Hi\O3 при темпе-.'
ратуре выше 400° С. Но этот метод мало пригоден для лабораторных целей, так на*4
наряду с прямым нитрованием всегда протекает расщепление С—С-связей и в резуль-.^
тате реакции обычно получается смесь изомерных нитропроизводных нитруемого!
углеводорода и продуктов его расщепления. Попытки распространить область примене^-т
ния метода -Хасса на алифатические простые эфиры, спирты, кетоны и карбоновые^
кислоты привели к весьма неудовлетворительным результатам [131].
Высшие парафиновые углеводороды (с температурой кипения около 200’ (О
и выше) моясно подвергать мононитрованнго по методу Грундмана [132] в достаточно^
простой лабораторной аппаратуре при нормальном давлении в жидкой фазе. Для этог<йа
в нагретый до 140—200° С углеводород через трубку с пористой стеклянной пластин«-2
кой пропускают перегретую HNO3 (d — 1,4). В этом случае всегда получается стати-J
стически вероятная смесь изомеров [133—135].
Мононитро-ч-додекаы. В нагретый до 180 — 190° С н-додекан пропускают-^
в течение 3 ч азотную кислоту. Выход мононптрододекана 40% от теоретическоио.2
Из реакционной массы выделяют также 43% непрорсагированшего исходного»
додекана и незначительные количества ди- и полипитроироизводных. Примри
пение избытка IINOs и увеличение продолжительности взаимодействия приводит1
к увеличению доли дипитроироизводпых [132]. .s
Для парафиновых углеводородов с числом углеродных атомов от 7 до 12, кото^
рые не нитруются ни по методу Хасса, пи по методу Грундмана, Гейзелер [136] раз*(
работал способ нитрования двуокисью азота под давлением. Реакция протекает по£
общему уравнению;
2HH + 3NO. ---► 2RNO2 + NO + H2O !,
Нитрование проводят в толстостенной стальной трубке при 150—200° С и давл**"'
нии около 40 ат. Этот метод может представлять практический интерес лишь для
непрерывного процесса нитрования в полупромышленном масштабе*. 4
Нитрогруппы можно вводить в соединения с активными метиленовыми группами^
при помощи упоминавшейся выше щелочной конденсации с алкилнитратами.
Так, например, прибавляя смесь бензилцианида с метил- или этилнитратО^
к охлаждаемому раствору алкоголята, получают с 80%-ным выходом фенил-ачи^
питроацетопитрил, который омылением и декарбоксилированием превращают в фенилу
нитрометан [137, 138]. Другие примеры см. [139]- С таким же выходом по аналогичной^
реакции получают фенилнитрометан из эфира фенилуксуспой кислоты [140]. По этому'
методу Виланд [141] превратил циклогексанон в калиевую соль 2-ауи-нитроциклоге»^
[129]
ИЗО]
[131
132
133
134
135
136
137
138
139
140
[141
Нитрование окислами азота подробно изучено П. П. Шорыгппьгм и А. В. Топ-%
чиевым, см., папример, ЖОХ, 5, 549 (1935). — Прим. ред.
М. К on о wal о w, Вег., 26, Ref. 878 (1893).
Ф. А з и п г е р, Химия и технология парафиновых углеводов, Гостоптехиздат,.
1959.
II. В. Hass, D. Е. Hudgin, J. Am. Chem. Soc., 76, 2692 (1954). !,
Ch. Grundmann, Die Chemie [Angew. Chem.], 56, 159 (1943).
F. Asinger, Ber., 77, 73 (1944). i
Ch. Grundmann, Ber., 77, 82 (1944).
F. Asinger, K. Halcour, Chem. Ber., 94, 83 (1961).
G. Geigeler, Angew. Chem., 67, 270 (1955).
W. Wislicenus, A. Endres, Ber., 35, 1755 (1902).
A. P. Black, F. H. В a h e r s, Org. Synth., Coll. v. 2, 1943, p. 512.
W. Wislicenus, H. Wren, Ber., 38, 502 (1905).
W. Wislicenus, R. Gru Izner, Ber., 42, 1930 (1909).
II. W i о 1 a n d, P, Garbsch, J. J. Cha van, Ann., 461, 295 (1928); ,
см. также [142].
X ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
з;
санопа с выходом около 40%, а циклопептапоп почти с количественным выходом •
в 2,5-ди-(калий-аци-нитро)-циклопентанон.
Особенно оправдало себя применение амилнитрата в сухом тетрагидрофура!
в присутствии трети-бутилата калия в качестве катализатора конденсации [ 142, 143
например, по такому способу из динитрила адипиновой кислоты при —50° С получех
а,а'-дннитропроизводное с 93%-пым выходом, которое омылением и декарбоксилир,
ванпем можно перевести в 1,4-дпнитробутан [1431.
Соединения с активными метиленовыми i руппами, например эфиры малонов<
в ацетоуксусной кислоты или арилацетонитрилы, можно нитровать при помоц
цитрата ацетонциангидрина [ИЗ].
Для этого раствор такого метиленового соединения в тетрагндрофурано сначаз
переводят действием гидрида натрия в ^натриевую соль и затем после добавлен!
1—2-кратного количества нитрата ацетонциангидрина нагревают 2 ч в колбе с обра
ным холодильником. Из замещенных эфиров ацетоуксусной или малоновой кисл»
по этому методу с хорошими выходами получают эфиры а-нитрокарбоновых кисло
При аккуратной работе можно проводить и прямое нитрование малоновоз
эфира; так. Висблат и Литтл [144], действуя концентрированной HNO3 (d = 1,,
при 15—20° С, получали нитромалоповый эфир с выходом выше 90%; ср. также [9
Нитрование ароматических
и гетероциклических соединений
Как известно, легкость введения ннтрогрушты в ароматическое ядро в энач]
тельной мере зависит от имеющихся заместителей. При переходе ог бензола к в]
метильным гомологам легкость замещения заметно возрастает. В то время как дл
нитрования бензола необходимо применять нитрующую смесь и работать при hobj
шейной температуре (около 50° С), ксилолы нитруются уже концентрированной HNC
при комнатной температуре. Мононитрование дурола азотной кислотой неосуществим»
так как реакция чаще всего идет дальше с образованием динитропроизводног<
При нитровании полиалкилбепзолов часто наблюдалась миграция алкильных груш
или их обмен на нитрогруппу [146]. Высшие гомологи, такие, как этилбензол и кумоа
тоже нитруются с хорошими выходами нитрующей смесью в ядро. При мононитровани
алкилбензолов обычно получают смесь всех трех возможных изомеров, причем в смес
всегда содержится меньше всего мета-изомера. С возрастанием длины боковой цеп
за счет уменьшения количества орто-изомера увеличивается доля пара-изомер
и в меньшей мере доля мета-изомера [147].
Нитрование боковой цепи, причем исключительно в a-положение, происходи
при нагревании углеводорода с разбавленной HNO3 в запаянной трубке. При нитр<
вании ненасыщенных или более длинных разветвленных боковых цепей проявляете
также окислительное действие HN03.
Для введения нескольких питрогрупп требуются гораздо более жесткие усл<
вия, чем для мололитрования, т. е. более концентрированная кислота и более высока
температура. И в этом случае алкильные заместители облегчают замещение. Taj
например, тринитротолуол можно сравнительно просто получить прямым тринитрч
ванием углеводорода, тогда как для синтеза тринитробензола по этому методу потр»
бовались бы исключительно жесткие условия; его проще получать косвенным путе\
например окислением тринитротолуола хромовой кислотой с последующим декарс
оксидированием [148).
Прямым нитрованием в бензольное ядро нельзя ввести более трех нитрогрупл
в образующемся при этом соединении должно содержаться не менее двух нитрогруч
[142 ] II. F е и е г J. W. S h е р h е г d, Ch. Savides, J. Am. Chem. Soc., 7<
r 4364 (1956).
[143J H. F e н e r, Ch. Savides, J. Am. Chem. Soc., 81, 5826 (1959).
144] D. I. Wcishlat, D. A. Lyttle, J. Am. Chem. Soc., 71, 3079 (1949).
145] N. К orn b lu m, J. H. Eicher, J. Am. Chem. Soc., 78, 1494 (1956).
146] D. V. Nightingale, Chem. Rev., 40, 117 (1947).
147] K. L e R о i Nelson, H. С. Bro wn, J. Am. Chem. Soc., 73, 5605 (1951 '
148] II. T. Clarke, W. W. Hartman, Org. Synth., 2, 93 (1922).
25 Заказ 1835.
386
ОБРАЗОВАНИЕ С—К-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
в орто-положении друг к другу, а такие соединения обладают очень большой реайй
ционной способностью. Оба изомерных тетранптробензола получают окислению»
тринптрозамещенных азотсодержащих производных бензола [149]. Обычно при ди-®
тринитрованиях, согласно известному правилу замещения, нитрогруппы вступавж
п мета-положение друг к другу. В то же время прн достаточной активации другими!
заместителями, оказывающими более сильное влияние, чем нитрогруппы, плотен
возможно образование и орто-динитропроизводных. Так, м-нитротолуол, образуя
ющийся при мононитровании толуола с выходом около 4%, при дальнейшей
нитровании дает смесь 2,3-, 3,4- и 3,6-динитротолуолоя. '
В таких же условиях, как бензол, нитруются и конденсированные аромяМЙИ
ческие системы; однако общего правила о месте вступления нитрогруппы в эти соадД
нения вывести не удается. В присутствии концентрированной H2SO4 при комнатаД
или несколько повышенной температуре нафталин нитруется азотной кислотой [1я8
151 ] или двуокисью азота [ 118], давая 1-нитронафталин с выходом около 90%. 2-Н1М
ронафталин, образующийся в очень незначительных количествах [151] (около 4.5ЙЯ
при прямом нитровании нафталина, получают из диазотированного р-нафтиламииЯ
по модифицированной реакции Зандмейера [152]. Дальнейшее нитрование 1-нитЧ^И
производного нитрующей смесью приводит к образованию смеси 1,5- и 1,8-динитр®И
нафталинов, причем содержание 1,8-изомера повышается с понижением температуря
реакции [153]. Болев высокоплавкое 1,5-производное плохо растворяется в пирпдиЯВ
и, следовательно, может быть отделено от 1,8-изомера. ‘чД
В то же время при нитровании тетралина нитрующей смесью при 0° С, пиЯ
котором замещение происходит исключительно в бензоидном кольце, образуюяИ
приблизительно равные количества 5- и 6-нитротетралина с общим выходом 80—90,тИ
От теоретического [154]. При разделении смеси нитротетралинов перегонкой
взрывы [ 154аJ! В более жестких условиях наряду с 5,7-динитротетралином неожиданЙИ
получается также 5,6-динитротетралин [154]. ,
Отношение антрацена к нитрующим агентам в значительной мере определяемо
повышенной реакционной способностью 9,10-положений. В зависимости от УСЛовдЙИ
проведения реакции образуются продукты окисления, как, например, антрахиияйЯ
и соответственно продукты его нитрования или же через первичные продукты ппиаИ
единения — 9-интро- или 9,10-динитроантрацен. Метод получения 9-нитроантраЦетИ
см. [155]. 'ам
Флуорен легко нитруется [156] в положение 2 концентрированной HNbffl
в ледяной уксусной кислоте при 50—65° С, тогда как под действием этилиитршИ
в присутствии этилата калия реагирует метиленовая группа и в результате экзотерф^Д
ческой реакции калиевая соль яци-формы 9-нитрофлуорена образуется с примемяИ
70%-ным выходом [157].
Хлорбензол нитруется нитрующей смесью в течение 4 ч при температуре яймИ
комнаткой, образуя с почти количественным выходом смесь, состоящую на 8/3 из
на Vg из о-нитрохлорбензола. Дальнейшее нитрование приводит к смеси, препмуЛмЯ
ственно из 2,4-динитрохлорбензола наряду с. небольшим количеством 2,6-динитРЙН
хлорбензола. Фторбензол нитруется почти исключительно в пара-положение. тЯ|
2,4-Динитрофторбензол. К охлаждаемой нитрующей смеси, состояЯмД
из 120 з HNOg {d = 1,52) и 280 г H2SO4 [d — 1,84), прибавляют по кпП1МИИ1
40 г фторбензола и нагревают 2 ч на водяной бане. Выход 87% от теоретичеик^йИ
[149] W. В о г s с h е, Вег., 56, 1939 (1923); W. В о rsc he, Е. Р е s к е, Вег.< «Я
815 (1926). ' ’
[150] J. М. Berkebile, А. Н. Fries, J. Chem. Educat., 25, 617 (1948). ?
151] Н. Е. Pierz-David, В. Sponagel, Helv. chim. acta, 26, 98 (1943$
152] H. H. Hodgson, E. Marsden, J. Chem. Soc., 1944, 22. $
1531 Ch. Gassmann, Ber., 29, 1243, 1521 (1896). 1
154] G. Schroeter, Ann., 426, 17 (1922).
154a] E. Hecker, R. Lattrell, E. Meyer, Chem. Ber., 95, 985 (1962). $
155] Ch. E. В г a u n, C. D. Cook, Ch. Merrit jr., J. E. Rousseau, Orft
Synth., 31, 77 (1951). 14
[156] W. E. Kuhn, Org. Synth., Coll. v. 2, 1943, p. 447.
[157] W. Wislicbnus, M. Waldmiiller, Ber., 41, 3334 (1908).
[158] H. Zahn, A. Wiirz, Angew. Chem., 63, 147 (1951). -I
I. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
387
Это важное для химии пептидов соединение может быть также получено
обменом галогенов в более доступном 2,4-динитрохлорбепзоле [159, 160].
Обмен галогена является также наиболее удобным методом для получения 2,4-
длпптролодбензола [161].
Как уже упоминалось, фенолы легко нитруются [162]. Ввиду высокой чувстви-
тельности фенолов к действию окислителей нитрование HNO3 приходится вести в под-
ходящих 'растворителях (ледяная уксусная кислота или хлороформ) и при охлажде-
нии. Лучшие выходы получаются под действием N2O4 в смеси петролейного эфира
н бензола [163, 164]. В результате реакции всегда образуется смесь переменных коли-
чество о- и n-питрофепола, из которой летучее о-производное легко выделяется пере-
гонкой с водяным паром. Однако доля га-нитрофенола повышается с увеличением
диэлектрической проницаемости применяемого растворителя [163]. Один п-нитрсфенол
можно получить нитрованием фепола, этерифицировапного взаимодействием с бензол-
или толуолсульфохлоридом, с последующим омылением продукта нитрования.
Наиболее целесообразно, особенно для введения нескольких нитрогрупп,
исходить из фенолсульфоновых кислот и обменивать в них сульфогруппы на нитро-
группы (стр. 512). Можно также обменивать хлор в хлорнитробензолах на оксигруппу
кипячением с водной щелочью.
Ароматические амины так же легко нитруются, нак и фенолы, ио первичные
и вторичные амины очень склонны вступать в нежелательные побочные реакции
окисления. Поэтому рекомендуется защищать аминогруппы ацилированием или
взаимодействием с альдегидами с образованием оснований Шиффа.
Прибавление большого избытка концентрированной серной кислоты уже обеспе-
чивает достаточную защиту аминогруппы вследствие образования аммонийной соли у
правда, при этом вводимая нйтрогруппа направляется главным образом в мета-поло—
жепие. В качестве N-ацилпроизводных чаще всего используют ацетильные производные,
по применялись и бензоильные, n-толуолсульфопильные, оксалильные, карбэтоксиль-
ные (из эфира хлоругольной кислоты) и фталильные производные.
Нитрование анилина в виде ацетильного производного нитрующей смесью»
приводит главным образом к образованию n-нитроацетанилида наряду с неболыпик
количеством орто-изомера. Соотношение изомеров сильно сдвигается в сторону орто-
производного при нитровании в отсутствие серной кислоты в смеси уксусного ангид-
рида и ледяной уксусной кислоты.
Нитроанилины [93]. К раствору 45 г ацетанилида в 34 г уксусного
ангидрида и 22 г ледяной уксусной кислоты прибавляют приготовленную при
нагревании и охлажденную смесь 23 г HNO3 (d ~ 1,5), 1 г мочевины и 23 г ледя-
ной уксуспой кислоты. Смесь оставляют на 24 ч, осаждение продукта затем завер-
шают добавлением 360 г льда и отделяют осадок на воронке Бюхнера. Выхоц
неочищенного вещества 52 г (87% от теоретического). Неочищенный продукт"
растирают при 0° С со смесью 50%-ного раствора КОН (1 объемн. ч.),
воды (4 объемн. ч.) и спирта (1 объемн. ч.), при этом растворяется только-
орто-производное. После отделения на воронке Бюхнера-и промывки водой
получают пара-производное в чистом виде, которое при нагревании с разбав-
ленным раствором NaOH омыляется до n-нитроанилина. При стоянии филь-
трата при комнатной температуре в течение суток заканчивается омыление
о-производпого и о-нитроапилии осаждается в виде больших кристаллов.
Соотношение орто- и пара-производного составляет примерно 3 : 1.
Чистый о-нитроанилин можно также получать, прибегая к блокированию,
пара-положения. В качестве исходного соединения берут сульфаниловую кис-
лоту, которую ацетилируют, обрабатывают на холоду нитрующей смесью а
нагреванием с концентрированной НС1 отщепляют сульфогруппу.
[159] Н. Н. Ворожцов, Г. Г. Якобсон, ЖОХ, 27 (89), 1672 (1957).
[160] G. С. Finger, С. W. Kruse, J. Am. Chem. Soc., 78, 6034 (1956).
[161] I. F. Bunnett, R. M. Connie r, J. Org. Chem., 23, 305 (1958); Org.
Synth., 40, 34 (I960).
[162] C. A. Bunton, E. D. Hughes, С. K. Ingold et al., J. Chem. Soc,,
1950, 2628.
[163] J. Podkowa, A. Tarnawski, Mh. Chem., 90, 179 (1959).
[164] H. Wieland, Ber., 54, 1776 (1921).
25*
388
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
По другому методу в качестве исходного продукта используют оксани-
лпд, получаемый из анилипа и щавелевой кислоты прп 200° С. Его сульфируют
концентрированной H2SO4 при нагревании па водяной бапс, после охлаждения
до 50° С сульфопродукт нитруют нитрующей смесью, состоящей из равных
частей IINO3 (d = 1,44) и концентрпроваппой HaSO4 и последовательно отщеп-
ляют оксалильную и сульфогруппы длительным нагреванием реакционной
массы, предварительно разбавленной водой.
При нитровании бензальанилина [165] или фталанила образуется почти исклю-
чительно n-нитроанилип. Последний метод [166] особенно удобен и рекомендуется
для всех ароматических амилов, которые вступают в реакции конденсации с фталевый
ангидридом. ’
п-Нитрпянилин. При умеренном нагревании растворяют 2,23 ч. Г^-феттд^
фталимида (фталанила) в 14 ч. концентрированной H2SO4 и охлаждают Дф
0° С, причем фталанил опять частично выпадает из раствора; затем прибавляю^
по каплям 2,55 ч. смеси HNOg и моногидрата ссриой кислоты, содержащей 25%]
HNOg, поддерживая температуру ниже 3° С, после чего смесь выливают на лед^
отделяют выпадающий нитрофталанил и промывают его водой. Для переамийи*^
ровапия высушенный продукт нагревают в сосуде, выдерживающем повышенно^
давление, в течение 1 ч при 170—180° С с 1,1 ч. анилина. Из образующейся реак^
ционной массы, содержащей фталанил, n-нитроанилин и анилин, последний отдвя
ляют перегонкой с водяным паром, и n-нитроанилин извлекают пз остатки
горячей водой. Остающийся фталапил может быть использован для следующего
штровапия [166].
Примером мета-нитрования в сильпокислом растворе в условиях образования 8M>i:
монневой соли служит получение зс-нлтродиметилапилина [167]. I
м-Нитродиметиланилип. К приготовленному при охлаждении раствору^
363 г димотиланилипа в 1270 мл концентрированной H2SO4 при 5° С и перемеши
вании медленно приливают по каплям смесь 200 мл концентрированной HgSO
и 200 мл IINO3 (d ~ 1,42). Продолжают перемешивание при 5—10° С в течет
1 ч, выливают смесь в 6 л воды и нейтрализуют ер концентрированным йоднд
раствором аммиака до полного осаждения пара-производного, что замечаете
по изменению цвета осадка от желтого (пара-изомер) до оранжевого (меИ$>
изомер). После отделепия пара-прои.зводного продолжают добавление аммиак
до pH 3; выход перекристаллизованного из 400 мл спирта л-питродиметилаИ||
лина составляет 280—316 г (56—63% от теоретического); т. пл. 59—60°ЧС.
Если нитрование ведут в менее кислом растворе, то образуются главным обрй
зом пара- и орто-производные, из которых первое особенно легко нитруется дая4
до 2,4-динитродиметиланилина [168].
Введение более двух нитрогрупп в диметиланилии протекает уже неоднозиачд
и в результате деметилирования и N-нитрования приводит в конце концов к N-srerW
2,4,0-тринитрофенилнитрамину, который известей как взрывчатое вещество ГГО
названием «тетрил».
Необычно протекает и реакция некоторых пара-замещенных третичных ароьй
тических аминов с азотистой кислотой. Так, при обработке 1Ч,К-диметил-п-толуидин
[169, 170], Г^М-диметил-п-анизидпна [171] и п-диметиламиноацетанилида [172
[165] Герм. пат. 72173; FrdL, 3, 48 (1890—1894). *
[166] Герм. пат. 141893; FrdL, 7, 64 (1902—1904); ср. герм. пат. 148874; FrdL, 7, б
(1902-1904). ;
[167] Н. М. Fitch, Org. Synth., Coll, v. 3, 1955, p. 658. 1
[168] J. G 1 a z e г, E. D. Hughes, С. K. Ingold, et al., J. Chem. Soc., 1950®
2657. 1
[169] G. P. Crowley, G. J. G. Milton et al., J. Chem. Soc., 1940, 1286.'i
[170] II. II. II 0 dgson, Л. Kershaw,]. Chem. Soc., 1930, 277. я
[171] H. H. Ilod’gson J. H. Crook, J. Chem. Soc., 1932, 1812. |
[172] H. H. Hodgson; J. H. Crook, J. Chem., Soc., 1932, 2976. |
1. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
3{
нитритом щелочного металла и соляной или серной кислотой на холоду с высоким^
иногда количественными, выходами получаются соответствующие З-нитро-4-димети.
аминопроизводные; в то время как при действии азотной кислоты образуются 2-нитр
производные:
N08
Следует указать, что ароматические аминонитроироизводные обычт
удобнее получать нагреванием галогеннитропроизводных с аммиаком или ам;
нами, например с о- или п-нитродиметиланилином [173], или частичным восстань
влением полинитропроизводных (ср. стр. 429 и сл., а также стр. 522).
Альдегиды нитруются при охлаждении концентрированной IINO3 или нитрз
ющей смесью с образованием преимущественно л-нитропроизводных. Так, напримв]
при действии на бензальдегид нитрующей смесью ирн 5—10° С получается л-нитр,
бензальдегид с выходом около 80% наряду с небольшим количеством орто-изомех
[174]. Практически свободный от изомеров л-нитробензальдегид получают действие
нитрующей смеси на гидробепзамид [175].
лсПитробензальдегид. При 10—12° С впосят небольшими порциям
350 г размолотого в порошок гидробензамида в 1650 г моногидрата серной кц
лоты. После полного растворения раствор охлаждают до 0° С и в течение 15
прибавляют к нему 930 г нитрующей смеси (30% HNOs и 70% моногидрата се]
пой кислоты) так, чтобы температура реакционной смеси была ниже 15° <
Затем в течение 10 —12 ч повышают температуру до 15—18° С и смесь вылив«
ют в такое количество льда (около 1500 ч), чтобы установилась температур
45° С. Через несколько часов остывший раствор фильтруют на воропке Бв>;
нера и отмывают .ч-питробензальдегид от кислот. Выход л-пптробензальдегих
450 з; т. пл. 58° С. В виде примеси препарат содержит 0,5% орто-изомера.
Действуя азотной кислотой на ароматические альдегиды при низких темпер!
турах, удалось получить продукты присоединения, так называемые нитраты альдеп
дов. которые, будучи внесены в серную кислоту, переходят в нитрозамещеиные в ядх
альдегиды [176].
Если исходят из диацетата бензальдегида, то количество орто- и пара-изомерс
увеличивается [177]; однако для приготовления о- и n-ниТробепзальдегидов следу<
отдать предпочтение косвенным путям синтеза, например окислению соответствующе)
нитротолуолов [178, 179] или нитрокорпчных кислот [177]. О получении о-нитр»
коричного альдегида см. [180].
Введение нескольких нитрогрупп в ароматические альдегиды прямым нитров^
пнем удается лишь в редких случаях, например при нитровании аписового альдегид
[181]. Обычно и тут приходится прибегать к косвенным методам.
[173]
[174]
[175]
[176]
[177
[178
[179
[180
[181]
Т. W. С a m р h е 1 1, J. Am. Chem. Soc., 71, 740 (1949).
В. N. I с k е, С. Е. Redemann, В. В. W f s е g а г v е г, G. А. А 1 1 е
Org. Synth., 29, 72 (1949).
О. Ba yer, в книге Houbeo - W е v 1, Methoden der organischen Chemi.
Bd. 7/1, 4 Aufl., Stuttgart, 1954, S. 407.
G. Reddelien, J. prakt. Chem. [2], 91, 213 (1915); Angew. Chem., 35, 5g
(1922)
W. Davey, J. R. Gwilt, J. Chem. Soc., 1950, 204, 3348.
S. M. T s a n g, E. H. Wood, J. R. Johnson, Org. Synth., 24, 75 (1944
S. V. Lieberman, R. Connor, Org. Synth., Coll. v. 2, 1943, p. 44
R. E. Buckles, M. P. Bellis, Org. Synth., 33, 60 (1953).
E. Wiirnor, Ber., 29, 157 (1896).
390
БРАЗОВАНИЕ С—N-СВЯЗП В РЕЗУЛЬТАТЕ ОБМЕНА
При этом чаще всего используют в качестве исходных продуктов питротолуолье
и переводят их в аниды питробензальдегидов, которые затем легко разлагаются кон-
центрированной НС1 на альдегид и амин. Толуолы взаимодействием с анилином ц
окислением КМпО4 переводят в анилы через бензил галогениды [182] или использую»!
впервые описанную Заксом [183] конденсацию нитротолуолов с п-нитрозодиметнл-
анидином [184—186а]. Продукт конденсации 2,4,6--тринитротолуола и п-нитрозодиаад^
тиланилина взрывоопасен [185] (см. также стр. 412). 1
При нитровании ацетофенона препаративное значение имеет только мета-нитро^
вапие. Как и при нитровании бензальдегида, этой реакпди способствует большой
избыток концентрированной или, лучше, дымящей H2SO4 и как можно более низке®
температура реакции [187]. Таким путем получают л-нитроацетофенон с 90%-иыЗ
выходом [188, 189]. Нитрование одной HNOS и повышение температуры, напротив
увеличивает выход орто-изомера [190], но синтезировать его таким путем неудобий
О получении оксимов о- и и-нитроацетофенона прямым окепмирование
о-или ге-нптроэтилбензола [191] см. стр. 395, а их получение из о- и п-нигробв}
зоилхлордда и малонового эфира см. [192].
В качестве примера нитрования нитрила приводится синтез 3-нитробензря
нитрила [96]. '
З-Нитробензонитрил. В 25 мл дымящей HNO& растворяют 5 е бен8<й
нитрила п нагревают 30 мин до 60° С, затем выливают в воду и продукт реакцй|
перекристаллизовывают из кипящей воды; бесцветные иглы; выход 6 г: т. пж
116° С. ' ,’j
При нитровании карбоновых кислот нитрогруппа становится почти исключу
тельно в мета-положение, но иногда бывает довольно трудно выделить свободные f
примеси других изомеров продукты. Поэтому для получения чистой л-нитробензойвд
кислоты более целесообразно исходить из метилового эфира бензойной кислоты; Hfl
действием нитрующей смеси при хорошем охлаждении он переходит с выходом окой
80% в чистое л-нитропропзводное [193], из которого после омыления образуете
с 90%-ным выходом чистая .м-нитробензойная кислота [194]. Понятно, что введен®
второй нитрогруппы связано с значительно большими трудностями. Так, из бензоина!
кислоты при действии нитрующей смеси, для приготовления которой применяла^
дымящая HNO3, получают в довольно жестких условиях 3,5-дипитробензониУ1
кислоту с 55%-ным выходом [195]. Нитробензойные кислоты с нитрогруппой в орт$
или пара-положепии готовйт иосвенным способом, например окислением соответств^
ющих нптротолуолов хромовой кислотой. *
[182]
[1831
[184]
[185]
[186]
К. V. Auwers, Е. Fense, Вег., 58, 1369 (1925). 1
F. Sachs, R. Kempf, Вег., 35, 1224 (1902).
A. Lowy, В. В. Wescott, J. Am. Chem. Soc., 42, 849 (1920).
St. S e с а г e a n u, Ber., 64, 834, 837 (1931). , J
uuj A. Lowy, E. H. Balz, J. Am. Chem. Soc., 43, 341 (1921).
[186a] G. M. Bennett, E. V. Bell, Org. Synth., Coll. v. 2, 1943, p. 223; Пр8
веденную методику для 2,4-динитробензальдегида ср. с критическими зам®
чаниями I. Tana^sescu, V. Farcasan, С., 1958, 9477. Л;
J. W. Baker, W. G. Moffitt, J. Chem. Soc., 1931, 314. 3
[187]
[188
[189
[190
[191
[192
[193]
[194]
[195]
H. Rupe, A. Braun, K. v. Zembruski, Ber., 34, 3522 (1901). '
V. G. Morgan, H. B. Watson, J. Soc. Chem. Ind., 55, 29T (1936).
C. Engler, Ber., 18, 2238 (1885).
А. П. Ford-Moore, H. N. Ry don, J. Chem. Soc., 1946, 679.
C. A. Reynolds, Ch. R. Hauser, Org. Synth., 30, 70 (1950)
II. G. Walker, Ch. R. Hauser, J. Am. Chem. Soc., 68, 1386 (1946).
О. К a mm, J. B. Segur, Org. Synth., 3, 71 (1923). 7
О. К a m m, J. B. Segur, Org. Synth., 3, 73 (1923). .
R. Q. Brewster, B. Williams, R. Phillips, Org. Synth.,
Coll. v. 3, 337 (1955). 1
В.
R.
I. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
3'
Нитросалициловая кислота [196]. В минимальном количестве ледян<
уксусной кислота растворяют 1 моль салициловой кислоты (или ее эфира
при комнатной температуре быстро добавляют i моль HNOg (d = 1,42), расгв
репный в девятикратном объеме ледяной уксусной кислоты, и пагревают до и
явления темной окраски. После охлаждения продукт реакции осаждают водо
Выход питросалициловой кислоты (или ес эфира) около 50% от теоретическог
Образуются исключительно 5-нитронроизводные.
Пз 1-оксинафтойной-2 кислоты и ее эфиров получаются 4-нптропроизво
ные; в этом случае выходы выше.
При нитровании фталевой кислоты и ее ангидрида образуется смесь 3- и 4-нитр
фталевых кислот [197]. Плохо растворимая в воде 3-нитрофталевая кислота ион?
быть легко выделена из смеси изомеров; выход около 30%. 4-Нитрофталевую кисло-
удобнее получать нитрованием фталимида [198] смесью дымящих азотной и серн*
кислот при охлаждении с последующим омылением [199]; общий выход около 50%
теоретического.
Совершенно различно относятся к действию нитрующих агентов гетероцикл
ческпе соединения [200]. В то время как пиридин исключительно устойчив к действе
f(NO3, пиррол претерпевает при нитровании далеко идущие изменения, для предотвр
щения которых реакцию приходится проводить при возможно более низких темпер
турах. Все же интровапие пиррола удалось провести при помощи HNO3 и уксуса*
ангидриде при температуре от —10 до —15° С; при этом был выделен 2-нитропирр<
с выходом 21%, при дальнейшем нитровании которого с достаточно высоким выход*
получилась смесь, состоящая на 80% из 2,4- и на 20% из 2,5-динитропиррола [201
Наличие заместителей, особенно метильных групп, значительно облегчает нитрон
ние [202]. О питрованни замещенных пирролов см. [202—204].
З-Нитропиррол. Получают декарбоксилированием 4-нитропиррол-2-ка
боновой кислоты. Этиловый эфир этой кислоты легко синтезировать пут,
конденсации натриевой соли нитромалонового диальдегпда с эфиром глицп
[205], выход 75%.
Труднодоступны также нитрофураны. 2-Нитрофуран удалось получить
фурана [206] или фуран-2,5-дикарбоновой кислоты [2О7( в уксусном ангидриде а-
действии HNO8 (d = 1,51); причем из дикарбоновой кислоты выход составил лих
около 5%. Наличие метильных заместителей в фуране тоже значительно облегча
реакцию нитрования. В аналогичных условиях из 10 г 2-мстилфурана получе:
3—3,5 г 2-метил-5-нитрофурана [208]. Имеющий большое значение в промышленное
5-нитрофурфурол получают прямым нитрованием альдегида или его диацетата с 40
45%-ным выходом [209].
[196] Н. С. В а г а п у, М. Р i a n k a, J. Chem. Soc., 1946, 965.
1197] Р. J. С u 1 h a n е, G. Е. Woodward, Org. Synth., 7, 70 (1927); С. М. М
з е г. Th. G о m р г, J. Org. Chem., 15, 583 (1950).
1198] Е. Н. Huntress, R. L. S h г i п е г, Org. Эта th., Coll/' v. 2, 1943, p. 4?
[199] E. H. Huntress, E. L. $h loss jr., P. Ehrlich, Org. Synth., Cc>
v. 2, 1943, p. 457.
[200] K. Schofield, Quart. Rev. (Chem. Soc., London), 4, 382 (1950).
[201] [. J. Rinkes, Rec. trav. chim., 53, 1167 (1934).
202] H. Fischer, W. Z e r w e c k, Be?., 55, 1949 (1922).
2t)3] I. J. Rinkes, Rec. trav. chim., 56, 1142 (1937).
204] H. J. Anderson, Canad. J. Chem., 35, 2} (1957); 1958, 4?76.
2051 W. J. Hale, W. V. Hoy t, J. Am. Chem. Soc., 37 , 2538 (1915).
206] В. T. Freure, J. R. Johnson, J. Am. Chem. Soc., 53, 1142 (1931).
207] I. j. Rinkes, Rec. trav. chim., 49, 1169 (1930).
208] I. J. Rinkes, Rec. trav. chim., 49, 1118 (1930).
209] H. Gilmann, G. F. Wright, J. Am. Chem. Soc., 52, 2550, 4165 (19S
R. Behnisch, в энциклопедии «Ullmans Encyklopadie der Techoiscti
Chernies, Bd. V, 3 Aufl., 1954, S. 239.
392
ОБРАЗОВАНИЕ С-К-СВЯШ В РЕЗУЛЬТАТЕ (П.МЕНХ
Диацетат 5-нитрофурфурола [209]. Прп 0° С смешивают 143 г уксусного
ангидрида и 4.3,7 г дымящей H1NO3 (d 1,5). Приливают по каплям к этой смеся
и точение 30—40 мин раствор 49,5 г днацстата фурфурола в 51 г уксусного ангид-
рида с такой скоростью, чтобы температура по превышала —5Q С, и перрметшт.
ВИЮТ 3 ч. Выливают смесь на 1 кг льда и к ней при перемещпианпп прибавляют
40%-ный раствор NaOH до полного выделения маслянистого продукта. После
отделения водного слоя осторожно, небольшими порциями добавляют ранний
объем пиридина. При этом происходит замыкание кольца, так как промежуточ-
ный продукт реакции не имеет циклического строения. Реакционную массу
затем некоторое время нагревают, разбавляют двух-трехкратным объемом ледя-
ной воды и через 30 мин отделяют образовавшийся осадок диацетата 5-нитрофур»'
фурола, который промывают разбавленной уксусной кислотой и водой до пол^
кого удалении пцрвдииа; выход 24,5 г (40% от теоретического); т. пл. 85Q С?
после перекристаллизации из спирта 92,5° С.
Тиофен нитруется относительно легко. В уксусном ангидриде под действием
нитрующей смеси пз дымящей HN05 (d 1,51) и ледяной уксусной кислоты, обрй
зуется 2-нитротИофен с 70—80%-пым выходом [210, 211]. З-Нитротиофеп получаю^
с общим выходом около 20% из тиофена через тиофен-2-сульфонилхлорпд, нитровй4
пием его до 4-нитропроизводного и последующим отщеплением сульфонильной групш^
[211]; о синтезе динитропроизводных тиофена см. [211].
Пиридин нитруется только в очень жестких условиях и образуется смета
2- и З-нптрониридина с плохими выходами [99]. Все три мононитропиридипа легщ
получать окислением соответствующих аминопиридинов кислотой Каро [212—214],
Гораздо болео гладко, хотя большей частью тоже в очень жестких условиях, протекав;
нитрование, если п пиридиновом ядре имеются заместители, как, например, гидроксил
[215], аминогруппа [216] или метильная группа [217]. Так, действием KNO3 и дымж
щей H2SO4 на 2,4,6-триметилнирвдин было получено с 90%-ным выходом 3-нитро
производное, а пз 2,6-диметилпиридина аналогичным способом с 66%-ным выходом--*
3-нитро-2,б-диметилпиридин [217]. 4-Питропиридин удобно получать через N-OKHCi,
пиридипа [218—219а]. •’
4-Нитропиридин. Па кипящей водяной бане в течение 24 ч нагреваии
26 г пиридина, 50 мл пергидрола и 100 мл ледяной уксусной кислоты. Отгоняй»
в вакууме уксусную кислоту, остаток при охлаждении льдом растворяют в 70 де
концентрированной H2SUd, добавляют смесь 110 мл концентрированной HNO
(с/ — 1,52) и 70 мл концентрированной H2SO4 и 2,5 ч нагревают на масляной бан<
upti температуре 130° С (в реакционной массе). После этого пропускают ток NO;
постепенно повышая температуру до 200е С. Когда выделение коричневых napd
NOj ослабевает, реакционной массе дают остыть в токе NO, выливают ее в Л<У
и, добавляя Na2COa, устанавливают pH 6. Отфильтрованный осадок растворяя»
в хлороформе ц из слегка щелочного фильтрата трижды экстрагируют продуМ
реакции хлороформом. Хлороформные растворы объединяют, промывают водой»
обесцвечивают активным углем и суптат над Na2SO4. После отгонки
хлороформа в вакууме остаток растворяют в иетролейном эфире, раствор натр®-,
вают до кипепия и оставляют для кристаллизации при охлаждении льдом.'
Выход 4-питрониридина, кристаллизующегося в виде белых чешуек, 26,4 я
(71% от теоретического); т. ял. 50° С.
^.-Нитропиридип раздражает кожу (вызывает образование пузырей) I
[210]
211]
212
213
214
215
216
217
218
219
219а]
V. S. Babasinian, Org. Synth., Coll. v. 2, 1943, р. 466.
Л. Н. Blatt, S. Bach, L. W. К г е s с h, J. Org. Chem., 22, 1693
Л. К i г р а 1, W. Bohm, Вег., 64, 767 (1931).
О. V. S с h i с k h, А. В inz, A. Schulz, Вег., 69, 2593 (1936).
А. К i г р а 1, W. Bohm, Вет., 65, 680 (1932).
Е. Koenigs, К. Fretcr, Вет., 57, 1187 (1924).
М. A. Phillips, I. Chem. Soc., 1941, 9.
Е. Р 1 a z е к, Вет., 72, 577 (1939).
Е. О с h i a i, J. Org. Chem., 18, 534 (1953).
К. Thomas? D. Jerchel, Angew- Chem., 70, 719 (1958).
F. Krbhnke, H. Schafer, Chem. Ber., 95, 1098 (1962).
(1957)^
I. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
3
2. Нитрозпрование
Общие положения
Из-за относительно низкой электрофильности катиона нптрозония Электр
фнльпое замещение водорода нптрозильной группой возможно только в соединение
с достаточно подвижными атомами водорода (соединения с активными метиленовые
группами, фенолы и третичные ароматическиеамины). С-Питрозопропзводные вооба
малоустойчивые соединения. Более устойчивы соединения, нитрозогруппа которъ
связана с третичным атомом углерода или с вторичным атомом, связанным одновр
мепно с стабилизующими группами, например с NOa, COOK или галогеном. Мног)
нитрозосоединения в виде яркоокрашенных мономеров существуют только в растворг
или в газообразном состоянии. В твердом состоянии они переходят в бесцветна
димеры, для которых предложена структура азодиокисей и из которых иногда mojkj
выделить цис-тран-с-изомеры [220].
Первичные и вторичные нптрозопроизводныс необратимо стабилизуются сбыте
уже в процессе получения, превращаясь в изомерные оксимы. Такое превращен!
третичных алифатических нптрозосоединсний сопровождается одновременным ра.
щеплением соседних С—С-связей в результате гидролиза или декарбоксилирована;
Более устойчивы ароматические нитрозосоедипепия; если они и превращают!
в оксимы, то обычно реакция обратима в рамках истинного таутомерного равновесн.
Так, например, пара таутомеров п-питрозофенол— монооксим n-бензохинона мож,
быть подучена как нитрозировапием фенола, так и оксимировапием п-бензохиноц
В качестве нитрозирующих средств обычно применяют азотистую кисло:
(NaNO2 it НС1) или ее эфиры, в первую очередь этил- и амилнитрит. При работе с эф>
рами можно применять как кислые (ПС1), так и основные (алкоголяты) катализатор!
Преимуществом сочетания этилпитрита с хлористым водородом является во
мощность удаления всех побочных продуктов в вакууме при комнатной температур
В особых случаях в качестве нитрозирующих средств, большей частью с таким
успехом, применялись нитрозилсерная кислота, нитрозилхлорид и окислы азот
Растворителями служат чаще всего вода, спирт, ледяная уксусная кислота или эфи-
Реакция протекает обйчно достаточно гладко уже при 0° С; менее реакционноспосо'
ные соединения нитрозируются прп умеренном нагревании до 50—60° С.
Нитроаирование алифатических соединений
Получение оксимов. В алифатическом ряду реакция нитрозирования служх
главным образом для получения оксимов [221], которые всегда образуются вместо истиз
ных нитрозосоединений, если только выбором особых условий не противодействуй
перегруппировке. Как правило, эта реакция протекает тем легче, чем сильнее актива
ровап замещаемый водород соседними электропоакцепторпьгми группами.
При нитрозировании кетонов непосредственно образуются а-оксиминокетоа
[222]; пз а-хлоркетопов — хлорангидриды гидроксамовых кислот [223]. Моноокси
диацетила [224] получают примерно с 60%-ным выходом прн пропускании этилнитри-;
в метилэтилкетон при 40—55° С в присутствии небольшого количества концентрир
ванной НС1. При нитрозировании гомологичных несимметричных кетонов возможе
замещение как в длинной, так и в короткой части углеводородной цепи. При избыта
нитрозирующего реагента в катализируемой кислотами реакции возможно замещена
обоих a-положений, как, например, в тропиноне [225] или циклогексаноне 1226, 22Z
[220] В. G. G о wen lock, W. L ii t t к о, Quart. Rev. (Chem. Soc., London
12, 321 (1958).
Резюме в статье: О. Touster, Org. Reactions, 7, 327 (1953).
“22] E. Karg, Arch. d. Pharm., 282/54, 49 (1944).
223 G. Hesse, G. Krehbiel, Chem. Ber., 88, 130 (1955).
““/1 S e ш о n, V. R. Damerell, Org. Synth., Coll. v. 2, f943, p. 20.
225] R. W i И s t a 11 e r, Ber., 30, 2698 (1897).
*26] A. Treibs, A. Kuhn, Chem. Ber., 90, 1691 (1957).
227] A. F. Ferris, G.S. Johnson et al., J. Org. Chem., 25, 492 (1960
D. С. В a t о s k y, N. S. Moon, J. Org. Chem., 24, 1694 (1959).
394
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Если вместо кетонов исходят из соответствующих 0-кетокислот, нитрозировш
ние протекает, конечно, легче, так как в этом случае метиленовая группа активир'ц*
вана сильнее; проведение реакции с NaNO2 и разбавленной кислотой в водной ср£ш
приводит к одновременному декарбоксилированию и образованию тех же а-оксимииЗн
кетонов, по с лучшими выходами. Из а-этилацетоуксусной кислоты Дильс [228$
получил таким путем З-оксиминопентанон-2 с 94%-пым выходом. Ацетондикарбоно,^
кислота может реагировать как по с„_ 2 ~ ' ,ТГ______ , _г._
пминоацетоуксусной кислоты [229], так и но двум метиленовым группам с образование
(ис-изонитроаоацетона [230. 231]. ' '
ианон-2 с 94%-пым выходом. Ацетондикарбонов,
одной метиленовой группе с образованием
Бис-изоннтрозоацетон [230]. Раствор 150 г неочищенной ацетондший
боновой кислоты в 275 мл воды хорошо охлаждают в смеси льда с попарен^
солью и прибавляют к нему при перемешивании раствор 100 г NaNO2B20Q"
поды с такой скоростью, чтобы температура не поднималась выше 0° С. Зм?
охлаждают до —5°С и немедленно отсасывают выпадающий осадок, промыЙ,
его малыми порциями ледяной воды. Еще некоторое количество продукта по,
чают добавлением к фильтрату 200 мл холодпой 6 н. HNOs. После четырехкр!
ной промывки небольшими количествами ледяной воды бесцветный проду
сушат в вакуум-эксикаторе над НгЗО4; выход 59 г (51 % от теоретического
т. разд, 133° С. ‘
Эфиры и амиды р-кетокислот с незамещенной метиленовой группой могут бы
нитрозированы до эфиров или амидов а-оксимино-р-иетокислот. Например, a-cfli
иминоацетоуксусяый эфир [232] образуется с почти количественным выходом „ЗЯ
приливании по каплям водного раствора NaNO2 к охлаждаемой смеси ацетоуксусй,
эфира и 18%-ной H2SO4 (ср. также [233]). Ацилгомологи ацетоуксусного эфира рек
руют также обычно гладко.
Изопитрозобензоилуксусный эфир [234]. К раствору 50 г бензоилукв'
ного эфира в 125 г ледяной уксусной кислоты, охлажденному до 5—8° С, М
ленно приливают по каплям раствор 18,5 г NaNOs в небольшом количестве во;
причем выкристаллизовывается часть образовавшегося оксима. Остаток Q
ждают ледяной водой из раствора, постоявшего 2—3 ч при комнатной температу
После перекристаллизации соединения из горячего спирта оно выпадает в В,
больших пластинок. Выход чистого оксима 52 г; т. пл. 121 ° С.
<
Диэтиловый эфир ацетондикарбоновой кислоты нитрозируется тольне >
одной метиленовой группе; при попытке провести динитроэирование под действа!
амилнитрита Пехман [235] получил 3,5-дикарбэтоисн-4-оксниэоксазол ’
ОН
I ' S
ROOC— С—С^С—COOR
[228] о. Diels, G. Plant, Ber., 38, 1917 (1905).
[229] J. В. Neilands, A. Neuberger, J. J. Scott, J. Am. Chem. S<X
82, 214 (1960). 1 4
[230] T. A. Geissman, M. J. S c h 1 a t t e r, J. D. Web b, J. Org. Cheni
H, 736 (1946).
[231] O. Achmatowicz, M. Leplawy, Roczniki Chem. [Ann. Soc. chU
Polonorum], 32, 1375 (1958); C., 1960, 10545. I
[232] M. Z. J 0 v i t schi t sch, Ber., 28, 2683 (1895); 35, 151 (1002). 1 .
[233] H. Adkins, E. W. Reeve,!. Am. Chem. Soc., 60, 1328 (1938); N. F. Ag
bertson, B. F. Tullar et al., J. Am. Chem. Soc., 70, 1150 (1948). й
[234] L. W о 1 f f, A. A. H a 11, Ber., 36, 3612 (1903). ,
[235] H. v. Pechmann, Бег., 24, 857 (1891).
I. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
39
Нптрозирование а-замещенных эфиров 0-кетокислот протекает с одновремев
ным отщеплением ацильной группы;
4 R R R
I I I
СНзСО—СН—СООВ —► СНдСО—С—СООВ —► С—COOR
I II
NO NOH
Таким путем из а-алкилацетоуксусных эфиров получают эфиры а-оксимияо
алкилуксусных кислоту имеющие препаративное значение для синтеза а-аминокисло
[236} л а-кетокислот [237]. Особенно хорошие выходы оксиминовых соединенн}
достигаются при проведении нптрозирования алкилнитритами в присутствии алкого
лита [2381 или, лучше, в 85%-пой серной кислоте [236, 239].
а-Оксиминокапроновая кислота [239]. В химическом стакане емкосты
400 мл к 30 г 30%-ной HaSO4, охлаждаемой смесью льда с поваренной солью
при перемешивании медленно прибавляют 18,6 г (0,1 моль) этилового эфир;
н-бутилацетоуксусной кислоты, следя за тем, чтобы температура не поднималас.
выше —5° С. Затем при температуре около 0° С туда же прибавляют по капля)
11 г (0,1 моль 5%-ный избыток) бутилнитрита с такой скоростью, чтобы на.
блюдаемое легкое вскипание не переходило в заметное выделение окислов азота
При прибавлении к реакцонной смеси маленьких кусочков льда начинается ввд
деление оксиминоэфпра в виде творожистого белого осадка. Затем приливам)'
холодную воду и продукт экстрагируют эфиром. Эфирный раствор встряхиваю'
с охлажденным 10%-ным раствором NaOH, в котором оксймипопроизводно<
растворяется с красным окрашиванием. Щелочной раствор нагревают 15 ми,
на водяной бане, охлаждают и подкисляют. Выделившуюся а-оксимипокапроно
вую кислоту отфильтровывают и дополнительно экстрагируют ее эфиром и
фильтрата. После перекристаллизации из бензина выход кислоты 86% от теоре
тического; т. пл. 136“ С (разл.).
Образование эфиров tx-оксиминокислот из эфиров циклических р-кетокисло
неминуемо связано с раскрытием цикла. При действии этилнитрита и этилата натри,
на эфир циклопентан-2-карбоновой кислоты получают эфир а-оксиминоадипиново;
кислоты [240]. Но если в условиях реакции омыллется эфирная группа, то нитрозе
соединение стабилизуется с декарбоксилированием и раскрытия кольца не ироне
ходит. Например, из эфира циклогексанон-2-карбоновой кислоты получен моноокснэ
циклогексапдиона-1,2 [241], который с трудом можно синтезировать из циклогекса
нона [227].
В качестве исходных веществ для синтеза а-оксиминоэфпров особенно удобн:
использовать алкилмалоновые эфиры. Хорошие выходы оксимипоэфиров достигают^
при нитрозировании эфиров этилиитритом и эквимолекулярным количеством этилат?
натрия [238] или эфирным раствором NaNO2 и хлористым водородом [239].
Циануксусиые эфиры нитрозируются с образованием оксиминоциануксусни.
эфиров [242]. Замещенные циануксусиые эфиры одновременно омыляются и декарбс
ксилируются, превращаясь в а-оксиминонитрилы, в то время как свободные нислотз
в эту реакцию не вступают [243].
Алкильные группы, находящиеся в орто- и пяра-положениях н ннтрогруппа.:
ароматических соединений, также нигрозируюгся в а-поло;кенно. Так, нигрозирове
нием соответствующих нитроэтилбенэолов mpem-бутилнитритом в присутствн:
трят-бутилата натрия удалось получить о- и в-питроацетофеионоксим с выходах
К. Е. Hamlin jr., W. Н. Hartung, J. Biol. Chem., 145, 349 (1942
R. Fischer, Th. Wieland, Chem. Ber., 93, 1387 (1960).
J. C. Shivers, Ch. R. Hauser, J. Am. Chem. Soc., 69, 1264 (1947).
R. H. В a rry, W. H. Hartung, J. Org. Chem., 12, 460 (1947).
W. Dieckmann, Ber., 33, 579 (1900).
T. A. G e i s s m a n, M. J. Schlatter, J. Org. Chem.,' 11, 771 (1946).
M. Conrad, A. Schulze, Ber., 42, 735 (1909).
Th. K. Walker, J. Chem. Soc., 125, 1622 (1924).
396
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
соответственно 73,5 и 67% от теоретического [191] (о приготовлении трет-бутил-'^
нитрита см. [244]).
Нитрозировать углеводороды (за исключением цпклопентадиена [245] и флуо-
рена [246]) описанными выше методами нс удается, но этого можно достигнуть фото-;
химическим путем — действием нитрозилхлорида или окислов азота в присутствии
хлора. Так, при освещении солнечным светом раствора нитрозилхлорида в толуоле^
в течение 2 ч при комнатной или несколько пониженной температуре был получен i
Р-беиэалъдоксим с практически количественным выходом (из расчета на пптрозил—]
хлорид) [247]. Подобным же образом синтезировали оксим циклогексанона с выходи**
до 71'/6 (ио нитрозилхлориду), пропуская в циклогексан нитрозилхлорид на свету!
прп низких температурах (от 0 до —30° С) [248]. Недостатком этого метода являете*!
то, что работу надо проводить при сильном разбавлении. J!
Для фотонитрозировапия насыщенных углеводородов можно вместо нитрозилжв
хлорида с успехом использовать смесь окиси азота и хлора. Из циклогексана в зависжаЦ
мости от условий и соотношения NO : С12 в качестве основного продукта получается»
1-хлор-1 -нитрозоциклогексан, димер нитрозоциклогсксана илп циклогексанопоксим^
Подробности об этой интересной реакции см. [249].
Получение истинных нитрозосоединенпй. До настоящего времени активные*
метиленовые соединения редко переводились в истинные цитрозосоединепия или ИХ
димеры. Во избежание немедленной перегруппировки в оксимы необходимо уян
в процессе получения нптроэосоединений удалять даже следы щелочи и, ио воэмояр
ности, отказаться от применения растворителей, особенно содержащих гидроксильные
группы. д
Бейер синтезировал некоторые димерные нитрозосоединеикя терпенового ряд|
действием этил- ц амилнитрита на терпенкетоны в присутствии небольших количеств
ацетилхлорида или в лигроиновом растворе действием амилнитрита и нескольких
капель концентрированной соляной кислоты [250]. Аналогичный метод был исполь*
зован [251] при получении димерного 2-питрозо-2-карбэто'ксициклопснтанопа и ertt
гомологов. •
Димерный 2-нитрозо-2-карбэтоксициклопентанон [251].
ROOC ° J COOR
При охлаждении льдом смешивают Юг этилового эфира циклопентанон-2г
карбоновой кислоты и 6—7 г (рассчитанное количество 5 г) этилнптрита и доба-
вляют несколько капель ацетилхлорида. Через 10—15 ч осадок отделяют4 П8
воронке Бюхнера и промывают- эфиром. Выход бесцветного бис-нитрозопроизвод^
ного 60—80% от теоретического. . >i
Виланд и Блох [252] получали бис-нитрозосоедипепия, пропуская пптрозн»!
газы [из As2O3 и HNO3 (й 1,38)1 в охлаждаемые эфирные растворы 1,3-дикетонож
Таким способом получены димерные нитрозопроизводные дибензоилметана, п-анизоия|
бензоилметана и бензоилацетона. • 3
Несколько иначе реагируют с питрозпыми газами а-замещенные ацетоуксусный
эфиры с хотя бы одним атомом водорода в a-положении. При пропускании нитрозны!
[244]
[245
246
247
248
249
250
251
[252]
С. S. Сое, Th. F. Doumani, J. Am. Chem. Soc., 70, 1516 (1948). ‘
J. Thiele, Ber., 33, 666 (1900). ’
C. F. К о e 1 s c h, J. Org. Chem., 26, 1291 (1961). ]
E. V. L у n n, H. L. Arkley, J. Am. Cnem., Soc., 45, 1045 (1923).
M. A, Naylor, A. W. Anderson, J. Org. Chem., 18, 115 (1953).
E. Mfiller, H. Metzger et al., Angew. Chem., 71, 229 (1959).
A. Baey er, Ber., 28, 639 1586 (1895); 29, 1078 (1896). <’
W. Dieckmann, Ber., 33, 590, 594 (1900); W. D ie c k ma n n, A. Gr«?
eneveld, Ber., 33, 604 (1900). I
H. Wieland, S. Bloch, Ber., 37, 1524 (1904). s
I. ЗАМЕЩЕНИИ ВОДОРОДА ДЗОТОМ
3?
газов и такие эфиры без растворителей одновременно с введением нитрозогрупг
рпдро.нпичсскн отщепляется ацетильный остаток [253]. Из а-мстил-, а-этил- и а-бутц
ацетоуксусного эфира этим способом получены синие мономерные а-нитрозопроизво
вис пропионовой, масляной и капроновой кислот:
СЦаСО—CR—COOR °0С!-*- СНзСО—CR—COOR —»- HCR—COOR
I 111
Н NO NO
где R ——СНЯ; — Cells; —CIbCOOR; —CaII9; — CII(COCH3)COOR.
Примеры такой реакции см. [254, 255].
В аналогичных условиях эфиры а-нитрокислот жирного ряда не образуй
inn розосоедипений, подобных псевдонитролам; происходит отщеплепие питрогрупц
с образованием эфиров нитрозокислот жирного ряда [255] (ср. [145, 256]). При нитр
зированпп а-метилбензоилуксусного эфира можно избежать гидролиза и подучи?
а-нптрозо-а-метилбензоплуксусный эфир [257].
Кроме упоминавшегося выше метода фотонитрозирования [247 — 249], друг]
способы получения нитрозоуглеводородов прямым нитрозпрованием нензвестн]
Такие соединения получают окислением других азотсодержащих производнь
(стр. 535), фотохимическим илн термическим разложением алкилнитритов ,[258
пли окислительным расщеплением оксазиранов [259].
Питрозирование ароматических соединений
Питрозирование фенолов п третичных аминов. Самые стабильные иитрозосоед;
нения относятся к ароматическому ряду. Легче всего протекает С-нитрозированх
фенолов и третичных аминов, не имеющих заместителей в пара-положении. Произво,
ные бензола питрозируются почти исключительно [260] в пара-положение к окси- ил
аминогруппе. В ряду нафталина реакция протекает иногда неоднозначно. В то врех-
как из р-нафтола почти количественно получается а-нитрозо-р-пафтол [261], х
а-пафтола образуется смесь примерно равных количеств 2- и 4-нитрозонафтола-1 [262
В качестве нитрозирующего агента чаще всего применяют азотистую нислот;
Нитрозируемые фенолы обычно растворяют в щелочи, приливают водный растве
NaNO2 и выделяют азотистую кислоту, приливая по каплям разбавленную минерал;
ную кислоту. Третичные амины растворяют в избытке минеральной кислоты и npj
бавляют по каплям рассчитанное но реакции количество раствора NaNOa. Температур
реакции при этом не должна превышать 10° С.
Нитрозофенол (классический способ) [263]. Растворяют в 1500 мл вод
60 г фенола, 27 г NaOH и 54 г NaNO2 и прп охлаждении до 7—8° С к этому ра.<
твору прибавляют постепенно смесь 150 г ковцентрированнойН2304и400л£л водх
Через 2 ч выделившийся нитрозофенол отфильтровывают, промывают холодне
водой и растворяют его в эфире. Раствор встряхивают при комнатной температур
с активным углем, который отделяют из фильтрата после испарения эфир^
Получают п питрозофенол в виде желтоватых игл; выход 80% От теоретического
[253] J. Schmidt, К. Th. Widmann, Вег., 42, 497, 1886 (1909).
1254] J. Schmidt, Л. Н a i d, Ann., 377, 23 (1910).
[255] J. Schmidt, H. Dielerle, Ann., 377, 30 (1910).
[256] N. К orn Ы u m, R. K. Black wood, D. D. Mooberry, J. Am.
Chem. Soc., 78, 1501 (1956).
[257] J. Schmidt, E. Aeckerle, Ann., 398, 251 (1913).
[258] B. G. Gowenlock et al., J. Chem. Soc., 1955, 4190; 1956, 1670; 196l
2222. 40
[259] W. D. E m m о n s, J. Am. Chem. Soc., 79, 6522 (1957).
[260] St. Veibel, Ber., 63, 1577 (1930).
261] C. S. Marvel, P. K. Porter, Org. Synth., 2, 61 (1922).
[262] R. H e n г i q u e s, M. I 1 i n s k i, Ber., 18, 704 (1885).
[263] J. L. Bridge, Ann., 277, 85 (1893).
398
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
т. пл. 125° С (разл.). Для очпсткп можно провести перекристаллизацию из
зола или толуола.
Иногда удобнее проводить реакцию в спиртовом растворе, например
получения нптрозотимола
СН3
Ал°
ж//
СН(СН8)2
Нитрозотимол [264]. К раствору 100 г тимола в смеси 500 мл 95%-ш
спирта и 500 мл концентрированной соляной кислоты при 0° С медленно при
вляют небольшими порциями 72 г NaNO2, осаждают неочищенный прод
большим количеством воды и перекристаллизовывают из бензола; выход 87
т. пл. 160—164° С. J
Из многоатомных фенолов можно получать динитрозосоединения. 4-Нитрс
резорцип был получен в виде калиевой соли приливанием по каплям амилнитр
к охлажденному раствору резорцина и КОН в абсолютном спирте; в то же врй
резорцин в кислом растворе легко динитрозируется азотистой кислотой в 2,4-пола
ния [265, 266]. Следует отметить, что при синтезе тринитрозофлороглюцина произоЕ
взрыв [266а|. •
Заместители в орто- или мета-положении не мешают реакции нитрозировая
Так, из о- и jn-крезола получают 5- или 6-нитрозопроизводные [267]. Фенолы-, з$
щенные в пара-положение, пе реагируют в этих условиях или вступают в иные реакЦ
Например, n-крезол нитруется азотистой кислотой в орто-положение к гидроксилу
группе [267]. .
Нитрозофенолы часто синтезируют также из п-нитрозодиалкиланилинов гид
литическим отщеплением аминогруппы. Для проведения этой реакции, использу®
главным образом в синтезе вторичных аминов, п-нитрозодиалкиланилины кише
с водными растворами щелочей [268] или действуют на них растворами бисул»
та [269]. ,
Нитрозо- и оксигрунпы можно одновременно вводить в орто-положение Д]
к другу по реакции Баудиша [270]. При этом в результате окисления гидроксилаьА
или восстановлепия азотистой кислоты образуются радикалы NOH, которые в а
сутствии окислителя и солей меди в качестве катализатора атакуют ароматичен
ядро. Выходы целевых продуктов по этой реакции обычно не очень высоки, но nui
чение о-нитрозофеполов другими способами чаще всего также сопряжено с значите
ными трудностями. Кронхейм J271], используя реакцию Баудиша, синтезирЫ
более 50 моно- и дизамещеиных о-нитрозофенолов.
[264] Е. Kremers, N. Wakeman, R. М. Hixon, Org. Synth. J
92 (1926). Y
265] F. H о nr i ch, Ber., 35, 4191 (f902).
266] W. R. Orndorff, M. L. Nichols, J. Am. Chem. Soc., 45, 1536 (192
266a] H.-E. Freund, Angew. Chem., 73, 433 (1961).
267] II. II. H о d g s о n E. А. С. С г о u c h, J. Chem. Soc., 1943, 221. '$
268] А. В a e у e г, H. С a r o, Ber., 7, 963 (1874). , J
269] Герм- пат. 74628- Frdl., 3, 957 (1890—1894); J. v. Braun, K. Heid«
E. Muller, Ber., 51, 737 (1918); R. M u n c h, G. T. T h a n n h a u¥i
D. L. С о 111 e, J Am Chem. Soc., 68, 1297 (1946); W, R. Boon, J. ChM
1947, 307. Ji
[270] O. Baudisch, Naturwiss., 27, 768 (1939); Science [New York], 92, 336 (194
C., 1941, I 2100; O. Baudisch, Sch. H. Smith, Naturwiss., 27, "
(1939); O. Baudisch, J. Am. Chem. Soc., 63, 622 (1941); О. В a u d 1 8 d
Arch. Biochenl., 5, 401 (1944); C. A., 40, 5323 (1946). Л'
[271] G. Cronhoim, J. Org. Chem., 12, 1, 7, 20 (1947). • Я
I. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
Из третичных ароматических аминов удается нитрозировать диметиланилв
лилия- и дипропиланилин; дибутил- и днизоамиланилин нитрозировании) не п<
даются [272].
Хороший метод нитрозирования диметиланилина см. [273, 274].
Значительное влияние на выход нитрозопроизводных третичных ами^
оказывает количество нитрита, которое должно быть равно теоретическому д
лишь незначительно превышать его. Кроме того, выбирают такую концентрат
минеральной кислоты, чтобы растворимость образующегося нитрозопроизводна
в реакционной массе была минимальной, в противном случае могут протека
нежелательные побочные реакции, например отщепление N-алкильных гру
с последующим N-иитрозировапием или нитрованием в ядро [274, 275]. Hhtj
ванне в ядро происходит также при попытке нитрозировать п-замещенвг
амины [169—172, 276] (ср. также стр. 388). Иногда я-нитрозпрование затрз
нпется заместителями в орто-положении к атому азота. Известны также mhoi
численные другие исключения [277, 278].
Нитрозирование вторичных аминов. С-Нитрозопроизводные вторичных аро&з
тических аминов могут быть получены при помощи так называемой перегруппировз
Фишера — ^еппа [279] действием галогеноводорода на соответствующие N-интра
амины. В этом случае происходит отщепление N-нитроаогрупны в виде нитрозилгал
генида, который затем нитрозирует вторичный амин в ядро {280]. Обычно превращев:
достигается при стоянии вещества в спиртовом растворе хлористого [279] или бром
стого [281] водорода при комнатной температуре; при этом из раствора через иекох
рое время выделяются желтые или оранжевые кристаллы гидрогалогенидов С-нитр
зопроизводных. В качестве катализаторов перегруппировки иногда применяет!
также концентрированная водная соляная кислота [277, 282] или раствор хлориста
водорода в ледяной уксусной кислоте [283].
Эта реакция протекает не только с простыми И-нитрозо-Жалкиланилниа»
[279, 284], но и с замещенными производными анилниа со свободным пара-полож
лшем,. такими, как К-нитрозо-К-этил-о-толуидин[279|, N-нитрозо-ж-хлор-и N-нитроз
л-бром-И-метиланилин [286] или N-HnTpoao-N-Метилантраниловая кислота [277, ,287
С-Нитрозопроизводное этой кислоты может быть омылено до нитрозосалицилов*
кислоты [288]. О перегруппировке производных а- и В-нафтиламина см. [281, 23
285, 289].
4-Нитроао-К-этил-а-нафтиламин с выходом 73% от теоретического получ*
также прямым нитрозированием вторичного амина [290] (см. ниже).
[272[
[273]
[274]
[275]
[2761
[277]
[278]
Р. К а г г е г, Вег., 48, 1398 (1915).
G. М. Bennet t, Е. V. Bel], Org. Synth., Coll. v. 2, 1943, p. 223.
П. H. Hodgson, D. E. Nicholson, J. Chem. Soc., f941, 470.
W. G. Macmillan, Th. H. Reade, J. Chem. Soc., 1929, 2863.
H. H. Hodgson,!. H. Crook, J. Chem. Soc., 1936, 1500.
J. H о u b e n, Ber., 42, 3188 (1909).
J. v. Braun, Z. Arkuszewski, Z. Kohler, Ber., 5f, 2?
(1918).
O. Fischer, E. Hepp, Ber., 19, 2991 (1886).
P. W. Neber, Hl Rauscher, Ann., 550, 182 (1942).
O. Fischer, E. Hepp, Ber., 20, 1247 (1887).
J. Houben, Ber., 46, 3984 (1913).
J. II ou h en, W. Brassert, Ber., 40, 4740 (1907).
L. Wacker, Ann., 243, 290 (1888).
E. Kock, Ann., 243, 307 (1888).
M. Ikuta, Ann., 243, 272 (1888).
J. Honben, E. Kellner, Ber., 42, 2757 (1909); J. Hou he»
G. Schreiber, Ber., 53, 2352 (1920).
J. Hou ben, E. Kellner, Ber., 42, 2757 (1909); J. Houbet
G. Schreiber, Ber., 53, 2352 (1920).
O. F i s c h e г, E. H e p p, Ber., 20, 2471 (1887).
L. Blangey, Helv. chim.^acta, 211 1579 (1938).
400
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Нптрозпровапис первичных аминов. ТЗланги [290] удалось осуществить прямое
С-нптрозирование первичных аминов добавлением амина к охлаждаемому льдом
раствору Na.NOo в концентрированной H2SO4 без доступа влаги. В качестве продуктов
реакции, осагкдаемых ледяной водой после израсходования всей азотистой кислоты,
всегда образовывались п-нитрозамины. Так были получены 4-питрозопроизводные.
й-нафтиламина и его 2-,6-,7-и 8-моносульфокислот, а в бензольном ряду —нитрозо:
производные .и-анизидина, -м-толуидина и аналогичных соединений. От нафтионовой.:
кислоты отщепляется сульфогруппа и получается тот же продукт, что и из а-пафтял--
амина. О синтезе С-нитро.зопроизводпых первичных аминов из нитрозофеиолов ей-'
стр. 494. 4
3. Азосочетание $
Общие положения,
'Й
Под азосочетанием [291] понимают все реакции катионов диазония с анноной
идпыми реагентами, при которых происходит разряд ионов и получаются нейтральный
молекулы:
[Аг-N—NJ4-X- —► Ar—N—N—X '
Строго говоря, к этому типу реакций относятся также перегруппировки содей
диазопия с образованием диазотатов, например образование диазоциапндов из цианм
дов диазония [292}. В зависимости от элемента, с которым сочетается терминальна^
(копцевой) атом азота, образуя новую ковалентную связь, речь может идти и С-, О
N- или S-азопроизводных. Здесь рассматривается только получение С-азопроизводнвд
В качестве диазокомпонепты применяются почти исключительно соли диазоиж
Поскольку из-за неустойчивости этих соединений проведение реакции в жестк!
условиях (например, при повышенных температурах) большей частью исключаете
С-азосочетапие из-за относительно низкой электрофильности диазониевой групп
удается осуществить лишь с особенно реакционноспособными компонентами, наприлв
используя соединения с активными метилеповыми группами, фенолы или арома®
ческие амины.
Значительное влияние на протекание реакции сочетания оказывает таил
природа диазокомпонепты. Диазонпевые соли с элекгроноакцепторными заместп/Я
лями легче вступают в реакцию сочетания и при более низких значениях pH, ж
соли с электронодонорными заместителями. Поданным Рааба [293], способность диад
тированных анилинов к азосочетанию снижается в ряду: полипитроанилины, нптр
хлорапилины, х.торапилины, апилинсульфокпслоты, анилин, апизидипы, амИИ
фенолы. Чем сильнее активирована диазокомпопента элсктроноакцепторпыми .замесу
телями, тем меньше могут быть активированы азокомпононты в реакции сочетав®
Так, например, диазотированный 2,4,6-трипптроапилин может вступать в реакци
сочетания даже с мезитиленом, образуя ярко-красный 2,4,6-тринпгробензолазОЫ
зитилен [294, 295]. В тетразотиропанных диаминах диазогруппы чаще всего обладав
разной реакционной способностью, чго позволяет проводить ступенчатое сочетание,!
с различными аэокомпонентамп. _ у
[291] ’Монографии: К. Holzach, Die aroniatischen Diazovcrbindungen, Ferdlntf
Enke, Stuttgart, 1947; К. H. S a u n d e г a, The Aromatic Diazo-CompOUO)
and their Technical Applications, 2 ed., Edward Arnold a. Co., London, 194
II. Zollinger, Chemie der Azofarhstoffe, Birkhauser Verlag, Basel — Stuf
gart, 1958 (имеется русский перевод).
[292] D. Anderson, R. L W. Le Fevre, J. S a v a g e, J. Chem.
1947, 445; N. Sheppard, G. В. В. M. Sutherland, J. Chem. S«
1947, 453; D. Anderson, M.E, Bedwell, R. J. W. Le РИГ
J. Chem. Soc., 1947, 457.
[293] H. Raabe энциклопедии «Ullnianns Encyklopadie der Tochnischen CheniM
Bd. 4, 3 Aufl., Munchen — Berlin, 1953, S. 76ff. •;
[294] К. H. Me y*e г, H. Toclitermann, Ber., 54, 2283 (1921).
[295] L. F. Fieser, W. P. Campbell, J. Am. Chem. Soc., 60, 1142 (1938
I. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
4
К соединениям, которые могут служить компонентами при азосочетании, отн
сятся главным образом те же соединения, которые вступают в реакцию прямого нитр
зирования. Эта аналогия проходит еще дальше: подобно тому как алифатическз
нитрозопроизводные легко перегруппировываются в оксимы, продукты сочетал]
с алифатическими компонентами легко претерпевают аналогичную перегруппиров]
в гидразоны [296, 297]. Поэтому при сочетании с алифатическими азокомпоиентаь
редко получаются истинные азопроизводные. Даже чисто ароматические азопроиаво
вые иногда перегруппировываются в гидразоны; однако в этом случае, вероятж
имеет место истинное таутомерное равновесие. Так, давно известно [298], что пр
дукт сочетания диазобензола с а-нафтолом идентичен монофенилгидразону 1,4-нафт
хинона. Изучая спектры поглощения, Кун и Бэр [299] показали, что преимуществу
ное содержание в соединении азо- или гидразонной формы зависит от растворитед.
Но такая таутомерия наблюдается нё во всех случаях: для о-, л- и п-оксиазобензозд
даже в растворах обнаружена только азо-форма [299].
Если принять для истинных азосоединений формулу производных диимиз
R— N=N—R, то можно ожидать наличия стереоизомерных форм; денствительн
часто описывается превращение образующихся обычно >праме-изомеров в уис-форьг
например под действием УФ-облучения [300—304].
Сочетание с соединениями, содержащими активные метиленовые * групп
Сочетание соединений с активными метиленовыми группами (I) <; молярных
количествами солей арилдиазония обычно протекает гладко [296, 297], причем обр
зуются соответствующие арилгидразоны (II); иногда в качестве побочного продукт
получаются формазаны (III):
хч Хч . Хх- ,N=N-R
>СН2—> >CH-N=N-R —► >C=N-NH-R —- Х-С<
У/ у/ у/ ''N—NH-R
I II Ш
Реакционная способность активной метиленовой группы в значительной степей
определяется природой активирующих соседних групп X и У, оказывающих сумма -
ное влияние. По данным Хюнига [305], встречающиеся обычно активирующие заместп
толи располагаются в следующий ряд по убывающему активирующему действие
NOs>CIIO>COCH3>CN>COOCsH6>CONH2>COOII>SO2CH3>CeH6
Из этого можно, например, сделать вывод, что ацетоуксусный зфир сочетаете
легче, чем малоновый, а малоновый, в свою очередь, легче, чем малоновая кисло»
или фснилуксусная кислота. Чем сильнее активирована метиленовая группа, т^
ниже может быть значение pH реакционной среды. Обычно реакцию сочетания пр
водят в водном или водно-спиртовом растворе при 0—10° С. Для понижения кисло
ности в большинстве случаев достаточно прибавить большое количество ацетата натри;
Слишком высокое значение pH, а также избыток диазосоединения способствует двоз
ному сочетанию с образованием формазанов (стр. 403).
[296
297
298
299
300
301
[302
См. также «Оргапическпо реакции», сб. 10, Издатинлит, 1963. — Прим, ред,
3. М. Parmenter, Org. Reactions, 10, 1 (1959).
R. R. Phillips, Org. Reactions, 10, 143 (1959).
Th. Zincke, H. Bindewaid, Ber., 17, 3026 (1884).
R. Kuhn, F. Bar, Ann., 516, 143,(1935).
G. S. Hartley, Nature (London), 140, 281 (1937); J. Chem. Soc., 1938, 63
A. H. Cook, J. Chem. Soc., 1938, 876.
L Ze chmei ster, O. Frehden, P. Fischer Jorgensen, NjrtAa
wiss., 26, 495 (1938).
[303] A. H. С о о k, D. G. J ones, J. Chem. Soc., 1939, 1309; A. H. С о о 1<
л D. G. J ones, J, В. P о 1 у a, J. C hem. Soc., 1939, 1315.
[304] H. H. Hodgson,! I. Chem. Soc., 1948, 1097; W. R. В г о d e, J. H. G
u 1 d, G. M. Wyman, I. Am. Chem. Soc., 74, 4641 (1951).
[305] S. Hu nig, O. Boes, Ann., 579 28 (1953).
26 Заказ 1835.
402
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Фенилгпдразон диметилового эфира мезоксалевой кислоты [306]. Прили-
пают раствор соли диазония, приготовленный из 12 г анилина, 36 г 30%-ной.
HCI, 150 г льда и воды и 10 г 98%-ного NaNO2, к охлажденному раствору 20 а
дпметилового эфира малоновой кислоты в 100 г этилового спирта. После этого
медленно до помутпепия добавляют концентрированный водный раствор ацетата"
натрия и оставляют раствор па 24 ч в холодильнике. Гидразон выделяется в кри«
ггаллпческом виде; т. пл. 62® С (перекристаллизация из спирта). .
Соединения с активными метиленовыми группами, претерпевающие при нитрсй
зировании из-за превращения в Оксимы далеко идущие структурные изменения^-!
например декарбоксилирование или отщепление ацила, подвергаются тем же превра4
щениям при азосочетаниях. Так, а-монозамещенные эфиры р-кетокислот, как и прж!
нитрозировании, отщепляют ацильную группу и превращаются в производные а-кето*ч
кислот* (реакция Джэппа — Клингемана [297, 307]): j
R—СО—CR*—COOR’ —> R—СО—CR'—COOR’ — > CR'—COOK"
। । и |
H N—N—R N-NH-R A
Например, сочетанием эфира циклопентанон-2-карбоновой-1 кислоты (I) с пйй
ридом бензолдиазопия и последующей обработкой щелочью получают фенилгидраащЙ
а-кетоадипиновой кислоты (III) с выходом 73% от теоретического [308, 309]. Из этого
же эфира при сочетании с солями диазония, полученными из и-питроанилина£|
3,4,5-трииодапилина и 2,6-дииод-4-аминоапизола, удалось получить в виде промер
жуточных неустойчивых кристаллических продуктов азопроизводпые (II), которМ^
все же переходили в гидразоны при нагревании или обработке разбавленными^
щелочами [310]:
COOR COOR СООП
Н2С—СН Н2С—C-N=NR' Н2С—C=N-NHR'
I I —>• I I —> I
Н2С С-0 н2с с=о п2с соон • .
сн2 сн2 сн2 J
I II III •'1
Свободные 0-кетокислоты при сочетании большей частью декарбоксилируются
Например, при сочетании циклопентанон-2-карбоновой-1 кислоты с хлоридоЦ
бензолдиазония получают [311] монофеинлгидразон циклопентандиона-1,2, а ЛВ:
ацетоуксусной кислоты — 1-фепилгидразои метилглиоксаля с выходом 80% от теорв»
тпческого [312]. Ч
В отдельных случаях удалось также провести сочетание алифатических угле?,
водородов с диазопиевыми солями. Так, Мейер [313] получил кристаллические аэ<Й
производные из бутадиена и его гомологов. j
[306]
[307
[308
[309
[310
[311
[312
[313
Эта реакция с успехом была использована В. В. Феофилактопым для синтева,
разнообразных а-аминокислот восстановлением указанных гидразонов цинком]
в соляной кислоте [(ДАН, 24, 755 (1939); ЖОХ, 17, 993 (1947)]. — Прим. редД
Сочетание солей арилдиазония с 2-кетопиперидин-З-карбоновой кислотой с цосле-~
дующей ипдолизацией по реакции Э. Фишера позволило Абрамовичу и Шапиро^
разработать общий синтез триптаминов (J. Chem. Soc., 1956, 4589). — Прим.
С. Bulow, A. Ganghoder, Вег., 37, 4169 (1904). г
F. R. Japp, F. К 1 i n g е m а п п. Вег., 20, 2942 (1887).
L. Kalb, F. Schweizer, G. S с h i m р f, Ber., 59, 1858 (1926), ‘
R. P. Linstead, A. B.-L. Wang, J. Chem. Soc., 1937, 807.
L. Kalb, D. F. Schweizer, H. Zellner, Ber., 59, 1860 (1926). ; з
W. Dieckmann, Ann., 317, 63 (1901).
G. A. R с у no]ils, J. A. Van Allan, Org. Synth., 32, 84 (1952).
К. H. Meyer, Ber., 52, 1468 (1919). >
J
Т. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
Формазаны, Особое место среди продуктов сочетания солей диазо
с соединениями, содержащими активные метиленовые группы, занимают формаз
[296, 314], которые обычно готовят из гидразонов альдегидов. Соответственно эц
способу получения их обычно рассматривают как соединения с одной азо- и од
гидразонной группой у одного и того же атома углерода (соединение I или П).
скольку все же все попытки получить пары изомеров, аналогичные (I) и (П), с раза
ними арильными радикалами на концевых атомах азота закончились неудачей [3
их теперь болео справедливо оценивают [316] как резонаценые гибриды (III):
Аг’—N=N. Ar'—NH—
>CR 'ЧСК
Аг—NH—Ж Ат—N=N/
I II
-Аг'-N=N. Ar'-N—
H ^.CR <-------> H ^CR
_ Ar-N-N^ Ar-?J=N/
III
Поэтому независимо от порядка сочетания двух различных групп ArN2 с i
тральным атомом углерода получаются идентичные продукты. Уже Пехман [Э
в качестве продукта сочетания фенилгидразона бензальдегида с хлоридом п-тозз
диазония получил тот же формазан, какой получается из n-толилгидразона бензе
дегида и хлорида бензолдиазония (ср. [317]).
Со старой номенклатурой формазанов можно ознакомиться в работах [£
315. 318, 319], вопросы стереохимии см. [320, 321].
Сочетание гидразонов с солями диазония проводится чаще всего в слабощел:
ной среде при температурах от 0 до 10° С. Лучше всего добавлять уже часта
нейтрализованный ацетатомяатрияраствор солидиазония к спиртовому раствору гв
азопа, который также содержит ацетат натрия. Вместо ацетата натрия можно прв
нпть пиририп [322].
3-Этил-1,5-дифенилформазан [320]. Растворяют в 25 мл метилов
спирта 3,2 г фенилгидразона пропионового альдегида и добавляют 7 г крисд
лнческого ацетата натрия в 35 .ил метилового спирта. После охлаждения до <
проводят сочетание с раствором соли диазония, полученной пз анилина (2 га
липа -J-9 мл концентрированной НС1 + 2 мл воды и 2 а 80%-ного раств
NaN02 -j- 3 мл воды). Формазан, выделяющийся Сначала в виде красного мае
Кристаллизуется через 14 ч.
Реакция сочетания обычно проходит с хорошими выходами, но иногда выхя
неожиданно низки. Выходы снижаются, если в фенилгидразонах альдегидов имею
электроноакцепторные заместители в мета- или пара-положении или если использую
ароматические альдегиды с заместителями в тех же положениях ядра [3 •
В случае применения фенилгидразопов с окси- иди аминогруппами в ядре, а тад
нафтилгидразонов [323] вместо взаимодействия диазокомпоненты с центральным «
мом углерода может произойти ее сочетание с ароматическим ядром. По данв
Псхмана [315], гидразоны типа R2N — N=CHR, т. е. не имеющие водор
У атома азота в гидразонной группе, не способны [к образованию формазанов. Исх.
из этого, Буш и Пфейффер [324] предположили, что па первой стадии синтеза формам
314
315
316
317
318
319
320
321
322
323
[324
26*
Обзорная статья: A. W. Nincham, Cheni. Rev,, 55, 355 (1955).
Н. v. Р echma nn, Вог., 27, 1679 (1894).
L. Hunter, С. В. Roberts, J. Chem. Soc., 1941, 820, 823.
R. Kuhn, D. Jerchel, Ber., 74, 941 (1941).
H. v. P e c Ii ш a n n. Ber., 25, 3175 (1892).
E. Bamberger, E. Wheelright, Ber., 25, 3201 (1892).
I. Hausser, D. Jerchel, R. Kuhn, Chem. Ber., 82, 515 (1949).
R. Kuhn, H. M. Weitz. Chem. Ber., 86, 1199 (1953 -
J. N. Ashley, В. M. D a v i s et al., J. Chem. Soc., 1953, 3881.
I. Hausser, D. Jerchel, R. Kuhn, Chem. Ber., 84, 651 (1951).
M. Busch, H. Pfeifer, Ber., 59, 1162 (1926).
404
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
в результате замещения водорода в гидразонной группе происходит образовали
тетразена. Установлено, что значение pH влияет иа направление реакции. Так, и
фенилгидразона бензальдегида и хлорида бензолдиазония при pH 9 получаете
формазан, при pH 4—8 — тетразен, а при pH 3 — п-бензолазофенилгидразон бене
альдегида [325}.
Иногда нет необходимости предварительно специально получать гидразоны
'так как последние сами могут образоваться в результате реакции сочетания. Много
численные соединения с активными метиленовыми группами могут реагировав
с 2 моль соли диазония, давая сразу формазаны. Например, этим путем при обработк
пировиноградной кислоты раствором КОН и хлорида бензолдиазония может быт
получен 3-оксалил-1,5-дифенилформазан (формазилглиоксалевая кислота) с выха
дом до 94% от теоретического [326]. Часто при таких реакциях происходит отще
нление ацильных групп (СН3С0— или —СООН) [318, 319, 327]. Так, ацетоуксусни
эфир почти количественно переходит в 3-карбэтокси-1,5-дифенилформазан [319, 327]
а этилмалоновая кислота — в З-этил-1,5-дифенилформазан [328].
В некоторых случаях, например при получении формаза®
СвН5—NH—N=CH—N=N— СвН8 из малоновой кислоты [328], удобнее вводил
1 моль диазосоединения, так как иначе легко протекает тройное сочетание с ов
разованием бензолазоформазанов.
В сильнощелочных растворах формазаны с подвижными заместителями, кан
например,— СООН, —СОСН9, —C0C00H, могут сочетаться еще с третьей диазоком
понентой в положение 3, ,отщепляя эти заместители и образуя так называем^
3-бензолазоформазаны. 3-Бензолазо-1,5-дифенилформазан
,N—NH—Аг
Аг- N—N— C<f
\N=N—At
в частности, получают в качестве конечного продукта при исчерпывающем соча
тании с диазосоединением ацетона [319[, ацетальдегида [319, 326, 329], пировина
градаой кислоты [319, 326], ацетоуксусной кислоты [319, 327], малоновой кислот®
[318], ацетондикарбоновой кислоты [318] и др.
Азосочетание с ароматическими компонентами J
Гораздо большее значение имеет азосочетаяие с ароматическими, а такяй
с гетероциклическими компонентами, прежде всего для получения красителей [330.],
Кроме того, талое сочетание имеет препаративное значение для синтеза первичны^
аминов, которые могут быть получены из азосоединений восстановительным расщеплю
нием (стр. 529).
Реакция азосочетания сильно зависит от pH среды. Легко протекающие азооф
четания проводят при более низких pH, чем сочетания,’ протекающие с трудов
Для фенолов область сочетания лежит в пределах pH 5—9, для аминов—приблизительна
3,5—7; на область оптимальных значений pH оказывают влияние и заместители
диазо- и азокомпоиентов. О том, что диазосоединения с электроноакцепторныцй
заместителями, к которым следует отнести и тетразосоединеиия, сочетаются легчй
при более низких значениях pH, упоминалось уже ранее.. В азокомпоненте (амигг^
фенол) электроноакцепторные заместители (—NO3>— СООН, — Cl, — SOaH) понижай^
способность к сочетанию, электронодонорные заместители (СН3, —ОН, — NH2) повьй
тают ее. Чем менее активна азокомпонента, тем обычно уже область pH, в кев
торой реакция протекает успешно.
[325] Н. Hauptmann, A. Cid de Mello Perisse, Chem. Ber.,
1081 (1956). *
[326] E, В a m h er ger, J. M u 1 1 e r, J. prakt. Chem. [2], 64, 199 (1901).
[327] E. Bamberger, E. W. Wheelwright, J. prakt. Chem. [2], 65$
123 (1902).
[328] Th. K. Walker, J. Chem. Soc., 124, 2775 (1923).
[329] E. В a m h e r g e r, J. Muller, Ber., 27, 147 (1894).
[330] Г. Э. Фнрц - Давид, Л. Бланжей, Основные процессы производств*
органических красок, пер. с нем., Издатинлпт, 1957.
I. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
Фенолы. В ряду бензола азогруппа вступает почти исключительно в наг
положение к оксигруппе. Незначительное количество образующегося орто-пров
водного иногда удаетси отделить перегонкой с водяным паром [331]. Если в пара-t
ложении есть заместитель, то сочетание проходит в орто-положение, как, вапримс
в случае п-хлорфенола [332] или п-крезола [333]; но если и в орто-положениях иь
ются заместители, то сочетание ие происходит или оно протенает с Отщеплени
блокирующего заместителя, как, например, прн сочетании с n-окснбензойной нислот
[334]; в таком случае может произойти также и О-азосочетание, например с 2,4
триметилфенолом [335]. Достаточно активные азокомпоненты могут сочетать
с избытком диазопроизводного дважды (в 2,4-положения) или даже трижды (в 2,4
положения). Трехкратное сочетание в одно ядро, например образованна 2,4,6-пгр;
(бензолазо)-фенола 1336] или 2,4,6-трио(бензолазо)-резорцина [337], идет обыч
с плохими выходами.
сс-Нафтол сочетается легче, чем ^-изомер и преимущественно в положение
иногда менее активные диазониевые соли вступают сначала в положение 2. Для разд
ления 2-и 4-азоизомеров смесь обрабатывают разбавленными щелочами, растворяющие
преимущественно 4-азопроизводиое. р-Нафтол и его производные сочетают
исключительно в a-положение, в некоторых случаях даже с вытеснением занести^
лей, таких, как —СООН [338], ио не — СН8 [339]. Сульфокислоты а-нафТола в зав
симости от местонахождения сульфогруппы сочетаются в положения 2 или 4. Орто-
пара-днфенолы, аминофенолы-'И диамины легко окисляются дназономпонентой до и
ионов. Простые эфиры фенолов сочетаются с бблыпим трудом и обычно толк-,
с особенно реакционноспособными диазокомполентами; в качестве растворителя
этого с успехом применяют ледяную уксусную кислоту [340].
Для проведения сочетания растворимые в воде фенолы и фенолсульф
кислоты растворяют в растворе NaaCOa, труднорастворнмые — в эквивалентне
количестве раствора NaOH с добавкой NasCO». Соду добавляют в таком колич
стве, чтобы ее было достаточно для связывания всей кислоты в растворе диаз
ниевого соединения и превращения в бинарбонат. Затем медленно и при хороша
перемешивании пркливают кислый раствор диазониевого соединения к щело
ному раствору азокомпоиенты. Если сочетание протекает быстро, то реакци
можно вести без особых предосторожностей при комнатной температуре. IIj
работе с медленно сочетающимися реагентами лучше, во избежание разложен®
диазосоединения, охлаждать смесь льдом и защищать реакционную массу <
прямого воздействия яркого света. Реакция считается законченной, кргд
нанесенная на фильтровальную бумагу проба не образует нового красите®
с водным раствором особо активной азокомпоненты, например R-соли или |
нафтолята натрия. Прн проведении реакции с диазосоединенинми, которые уж
в содовом растворе легко перегруппировываются в дназотаты (например, .
и п-нитродиазобанзол), вместо соды лучше употреблять ацетат натрия, СаСС
или пирндни.
Амины. Прн взаимодействии первичных или вторичных ароматических ам®
нов с солями диазония наряду с сочетанием в ароматическое ядро или даже вмес®
[331] Е. Bamberger, Вег., 33, 3188 (1900).
[332] М. Krause, Вег., 32, 124 (1899).
[333] Е. Nolting, О. Kohn, Вег., 17, 351 (1884).
[334] St. V. Kostanecki, J. D, Zibell, Ber., 24, 1695 (1891).
[335] 0. Dimroth, H. Leichtlin, O. Friedemann, Ber., 50, 153
(1917).
[336] E. Grandmougin, H. Freimann, Ber., 40, 2662 (1907); J. praki
Chem. [2], 78, 384 (1908).
[337] W. R. Orndorff, B. J. Ray, Ber., 40, 3211 (1907).
1338] R. N i e t z k i, A. L. G u i t e г m a n, Ber., 20, 1274 (1887).
[339] K. Fries, E. H u h n e r, Ber., 39, 435 (1906).
[340] К. H. Meyer, S. Lenhard t, Ann., 398, 74 (1913); К. H. Meje »
A. Irscjiick, H. Schlosser, Ber., 47, f741 (1914); K. v. Auwer?
F. Michaelis, Ber., 47, 1275 (1914); K. v. Auwers, E. Borsch?
Ber., 48, 1716 (1915); J. F. Bunnet, d. В. H о e y, J. Am. Chem. Soc., 8£
3142 (1958).
406
ОБРАЗОВАНИЕ С -Х-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
такого сочетания может протекать N-азосочетание с образованием триазенов [341]. Но
с повышением кислотности среды эта реакция затрудняется. Соответственно этому
из активных диазокомпонент, и особенно реакционноспособных аминов, сочетаю-
щихся уже в кислой среде, получаются сразу л-аминоазопроизводные [342]. Тенденция
к образованию триазепов снижается в 'следующей последовательности; анилин,
o-толуидии, л-толуидин, крезидин, 2,5-диметоксианилин [293]. В обычных условиях
n-ксилидин, крезидин, 2,5-диметоксианилин, а также а- и 0-нафтиламин сонета-'
клея почти всегда с образованием аминоазосоединений. Даже при проведении реак-
ции с анилином можно в соответствующих условиях, например в растворе муравьиной
кислоты, почти полностью подавить образование триазена [342].
Уже образовавшийся триазен в большинстве случаев превращается^
в аминоазопроизводное при умеренном нагревании с разбавленными кислотами
Более гладко протекает превращение при нагревании триазена с применявшимся
для сочетания амином и небольшим количеством гидрохлорида этого амина
[343]. Катализируемое кислотами превращение триазенов протекает с промежу-
точным повторным образованием соли диазония и амина [344, 345]. Если пере?
группировку триазена ведут в присутствии другого амина, легче вступающею
в реакцию сочетания, образующаяся диазокомпонента предпочтительно взаимо-
действует с последним: например, из азоаминобензола и диметилаинлина поду-'
чается п-диметиламиноазобензол [343].
Образование триазена можно полностью предотвратить, есни вместо свободных;
аминов применять их N-метил-о-сульфокислоты, которые легко получаются из
амина и бисульфитного соединения формальдегида [346]
ArNH2-f-HCHO-f-NaHSO3 —>ArNH-CH2—SO3Na-f-H2O
и после сочетания омылять сульфометильный остаток [347].
В то же время ацилирование амина в большинстве случаев препятствует соче-
танию; правда, некоторые амины являются исключением, например N-ацетия*
и N-бензоил-а-нафтиламин [348], N-тозил-а- и N-тозил-р-нафтиламин [349].
Первичные амины сочетаются медленное, чем соответствующие фенолы. N-За-
местители значительно увеличивают способность амина к сочетанию. Так, напрнмы»^
скорость сочетания ’диметилаинлина и дифениламина заметно больше скорйстМ
сочетания анилина. Описан ряд примеров сочетании с N-диалкиланилинами [350, 351]/
Лара-замещенные производные анилида могут сочетаться также в орто-положения
к аминогруппе, хотя склонность к такого рода реакции н© очень велика. Например
димстил-п-толуидин и n-бромдиметиланилин не реагируют с диазотировйнцож
сульфаниловой кислотой, тогда как п-диметиламинобензойная кислота вступаю
в реакцию с отщеплением СО2, образуя п-диметиламиноазобензол-тГ-сульфокислрТж
(гелиаитин) [352]. сс-Нафгиламин [353] сочетается обычно в положение 4, но реакция!
может проходить и в положение 2. 0-Нафтиламин сочетается исключитеиьтс
в а-положение, причем, как и в ^-нафтоле, заместители в a-положении могут вытеснять^
[341] О структуре и таутомерии ароматических триазенов см.: Т. W. CamphelK
В. F. D а у, Chem. Rev., 48, 299 (1951). J
[342] К. Н. Meyer, Ber., 54, 2265 (i921).
[343] E. Rosenhauer, H. Unger, Ber., 61, 392 (1928).
[344] H. V. Kidd, J. Org. Chem., 2, 198 (1938). $
1345] 0. Wallach, Ann., 235, 233 (1886). '.I
[346] A. Eibner, Ann., 316, 89 (1901); H. Bucherer, A. Schwalbe, BeSS
39, 2796 (1906). 1
[347] Герм. лат. }3}860; FrdL, 6, 872 (1900—1902). Я
[348] W. К 6 n i g, E. Kohler, Ber., 54, 981 (1921).! J
[349] O. N. Witt, G. Schmitt, Ber., 27, 2370 (1894); W. Konig, E. K3W
1 e r, Ber., 55, 2139 (1922). 1
[350] К. II о 1 z a c h, A. Simon, Ber., 75, 166 (1942). -Ц
[351] E. Philippi, G. U lmer-Р leak, Ber., 74, 1529 (1941). 3
[352] W. Scharrtin, Kaljanov, Ber., 41, 2056 (1908). Я
[353] H.S. Turner, J. Chem. Soc., 1949, 2282. ‘!4
I. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
ся азогруппой, О месте вступления азогруппы в ариламипосульфокислоты, име
щие важное техническое значение, см. [293].
Нерастворимые в воде амины растворяют в разбавленных кислотах ц
необходимости также в метиловом или этиловом спирте, ацетоне или даже в п
ридпне. В реакцию вводят не амипосульфокислоты, а их натриевые соли. Ес
имеется опасность нежелательного многократного сочетания амина, мож
в виде исключения прибавлять диазониевый раствор к раствору амина; в обц
пых случаях применяется обратный порядок. Если реакция протекает спит»,
медленно, минеральную кислоту нейтрализуют ацетатомпатрия или постепдпт
прибавлением соды, причем нейтральная среда должна достигаться лишь к ко
цу реакции. При сочетании тетразосоединений (например, из бензидина) с обр
зованием смешанных бис-азосоединений следует избегать проведения реакт
в условиях, благоприятствующих образованию симметричного бис-аэосоедив
ния. Для этого реакцию всегда проводят сначала с менее реакционноспособна
азокомпонентой, затем с более реакционноспособной и ведут сначала проце.
при возможно минимальных значениях pH. Если в соединении одновремен]
имеются амино-и оксигруппа, то место сочетания с диазокомпонентой в кислс
растворе определяется аминогруппой, а в щелочном растворе — оксигруппо.
Поэтому, папример, при сочетании с имеющими промышленное значение амин:
нафтолсульфокяслотами требуется тщательный контроль значений pH. В так®
случаях сначала проводят сочетание в кислых по индикатору конго раствора;
а после вступления.в реакцию первого моля раствора соли диазония прибавили
> соду для установления щелочной среды, а затем — вторую половину раствор
соли диазония или другую диазокомпоненту.
Способ выделения продукта азосочетания в значительной мере завись
от природы азосоединеиия. Кислые красители (сульфокислоты) часто высал]
вают из растворов в виде натриевых солей добавлением большого количеств
поваренной соли. Основные красители можно высаживать в виде гидрохлориде
добавлением соляной кислоты и поваренной соли. Оксиаэокрасители. обычн
малорастворимы и выпадают непосредственно в процессе реакции.
Подробные прописи синтезов азокрасителей, имеющих промышление
значение, см. [330].
Окислительное азосочетание
Азосоединения можно получать также окислительным азосочетанием. Это
метод применяется главным образом для получения азосоединений гетероциклическое
ряда, ибо вследствие неустойчивости соответствующих диазониевых соединений он:
не могут быть получены прямым сочетанием [354, 355]. Под так называемым окис
лительным азосочетанием понимают окислительное взаимодействие гидразоне»
сфенолами, ароматическими аминами или соединениями, содержащими активные мети
леновые группы, с образованием продуктов, которые могут рассматриваться формальн
как азосоединеиия и как азины. Ниже приведен пример такой реакции:
[354] Обзорная статья: S. Н u n i g et al Angew. Chem., 70, 215 (1958); 74, 81S
„ (1962).
1355] S, H u n i g, H. Balli, H. N 6 t her, II. Geiger, Ann., 628, 75 (1959)..
408 ОБРАЗОВАНИЕ С—N-СБЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Применяемый для реакции гидразон должен иметь строение амидразона >'
I j
(i>N—C=N—NH2) или его винильного аналога ' *
(>N-(CH=CH)„— C=N-NHa)
В качестве растворителя можно применять воду в смеси с метиловмм спиш
том, монометиловый эфир гликоля или диметилформамид, окислителем служив
чаще всего KaFe(CN)e, но можно употреблять и NaaCrO4, Ц8Оа, соли Си (II), Fe (lira
или Ag. Точно так же, как и при прямом азосочетании, и в этом случае решатот^д
влияние оказывает значение pH. Сочетание с фенолами проводят в слабощелочной
среде, амины сочетаются в разбавленных минеральных кислотах или с ацетаппвЗ
буфером.
Примером такого способа проведения синтеза является сочетание гщ
азона Н-метилбензтиазолона-2 с фенолами или с соединениями, содержащщ
активные метиленовые группы [356]. В смеси 50 мл воды, 50мл метилового спил
и 10 мл 25%-ного водного раствора NH4OH растворяют 22 ммоль KsFe(CN
Одновременно готовят раствор 5 ммоль гидразона N-метилбензтиазолош
и 5 ммоль азокомпоненты в 70 жл метилового спирта и 30 мл воды. К этому да
твору в течение 2—5 мин при температуре 25—30° С, поддерживаемой охлащ
нием, и перемешивании приливают раствор KsFe(CN)e. Сразу образуется крае
тель. Через 15 мин добавляют 250 мл воды, отфильтровывают выпавший крас
тель, промывают водой и сушат в вакууме над силикагелем. ~ ,
Из Р'нафтола, например, получают таким образом 1,4 г (88% от теораг
веского) неочищенного (3-метилбензтиазолил)-2-азиио-1-(2-кето-1,2-дигидр
нафталина); т. пл. 237° С (после перекристаллизации из хлорбензола — тоню
красные иглы; т. пл. 243—244° С).
4. Прочие реакции ь
Введение аминогруппы |
Прямое замещение водорода аминогруппой удается только в отдельных ос
бых случаях, но тем не менее представляет значительный препаративный интер<
В качестве аминирующих агентов применяются амиды щелочных металл»
гидроксиламин и N-галогенамины.
Уже Закс [357] кашел, что определенные производные нафталина (яафтиламвд
или нафтолы) при плавлении с амидом натрия аминируются в незамещенное коль!}
предпочтительно в положение 5. Эта реакция имеет большее значение для по^Е
чения амивосоедниений некоторых азотсодержащих гетероциклов [358, 359].
Наиболее известным примером является получение 2-аминопириднва |
способу Чичибабина [360]. Пиридин нагревают с амидом натрия в толуоле Ш
ксилоле при температуре от 120 до 150° С. Особенно удобен в качестве реакций
иой среды диметиланилин [361]. При более высоких температурах (150—180®
удается вводить еще одну аминогруппу и получать 2,6-диамииопиридин (вы$
82—90% от теоретического). и даже 2,4,6-триаминопиридин [361]. Схорони
выходами аминируются также гомологи пиридина, например а-пиколин [3
362], и производные хниолина [358]. Но выходы резко снижаются, если вЦ$С
амида натрия применяют натриевые производные первичных или вторично
аминов [363]. Обязательным условием гладкого протекания реакции являет
[356] S. Hu nig, К. Н. Fritsch, Ann., 609, 143 (1957). *
[357] F. Sachs, Вег., 39, 3006 (1906). '
[358] Резюме в статье: М. Т. Leffler, Org. Reactions, 1, 91 (1942).
[359] С. L. D е a s у J. Org. Chem., 10, 141 (1945).
[360] А. Ч ич нб а б ин, О. Зейде, ЖРФХО, 46, 1216 (1914)7 С., 191
I, 1064. ’
[361[ Герм. пат. 663891; FrdL, 25, 357 (1942); С., 1938, II, 2843. <
[362] Е. D. Р а г к е г, W. Shiv е, J. Am. Chem. Soc., 69, 63 (1947). Л1
[363] К. К о v й с s, Т. V a i d a, Chem. a. Ind., 1959, 259;
I. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
использование чистого амида натрия. Наиболее удобные способы получек
амида натрия см. [358, 364, 365J. О мерах предосторожности при работе с амид
натрия см. [364, 366J. Для аминирования производных хинолина и изохинолд
при комнатной температуре употребляли с большим успехом амид бария и
калия в жидком аммиаке [367[.
Вместо амида натрия можно применять натриевые соли гндразина или иесм
метричного диметилгидразина; пиридин нагревают с такой солью в течение 8 ч в &
водном гидразине. Кауфман и Шенек [368] получили а-гидразинпиридины с выхода;
около 40% от теоретического.
Ароматические нитропроизводные, которые можно сравнить по реакционк
способности с производными пиридина, тоже могут иногда подвергаться прямо;
аминированию. Из 1-нитронафталина и пинеридида натрия (амид натрия и пипарида
удалось получить 4-пиперидил-1-нитронафталин [369]. Нитробензол образует с nj
пзводными щелочных металлов и карбазола [37О[ или дифениламина [371] соответ»
вующие п-аминосоединения: М-(л-интрофенил)-карбазол (выход 70% от теоретическоа
и n-нитротрифениламин (выход 45% от теоретического). Реакцию нитробензола с пих
ридидом лития см. [372]. __,
Прямое аминирование ароматических нитросоединений часто удается такз
путем достаточно гладко протекающей конденсации с гидроксиламином в щелочк<
растворе, причем обычно аминогруппа вступает в орто-или пара-положение к нитр
группе. Наличие даже одной нитрогруппы в производных нафталина способству
достаточной их активации; в бензольном ряду эта реакция протекает только с динитю
производными. Из 2-нитронафталина получен 2-иитроиафтипамин-1 с выходом 80
от теоретического [373], из 1-иитронафталина — соответственно 4-иитроиафтиламкв
с выводом 60% от теоретического [374]. При аминировании производных хинола;
аминогруппа также вступает в ядро, замещенное нитрогруппой.
5-Амино-6-нитрохинолин [375]. Растворяют при нагревании 5 а 6-ннтр
хинолина и 6 г гидрохлорида гидроксиламина в 90 мл 96 %-ного спирта. Для пол
чения как можно более мелкого осадка труднорастворимого нитрохинолина ра
твор охлаждают при интенсивном встряхивании. Затем при комнатной теме
ратуре добавляют сразу 30 мл 20%-ного раствора КОН в метиловом спирч
Выделяется КС1, а 6-нитрохинолии после непродолжительного встряхиванд
переходпт в раствор, окрашивая его в желтый цвет; температура при этом
несколько минут повышается на 20° С. В скором времени начинается выделеьс
[364] Н. II е n е с к а, в книге Houben-Weyl, Methoden der organised
Chemie, Bd. 8, 4 Aufl., Stuttgart, 1952, S. 571—572; cp.: G. S p i &
berger, там же, Bd. 11/1, 1957. S. 74.
[365] F. W. Bergstrom, Org. Synth., Coll. v. 3, 1955, n. 778.
[366] F. W. Bergstrom, W. C. Fern el i us, Cnem. Rev., 12, *
(1933); 20, 413] (1937).
[367] F.' W. Bergstrom, J. Am, Chem. Soc., 56, 1748 (1934); Ann., 5t
..... 34 (1935); J. Org.iChem., 2, 411 (1938); 3, 233, 424 (1939).
[368] Th. Kauflmann, W. Schoeneck, Angew. Chem., 71,- 285 (195?
Th. Kauffman n, J. Hansen, Ch. Rose I, W. Schoeneck, Ant
" 656, 103 (1962).
[369] W. Bradley, R. Robinson, J. Chem. Soc., 1932, 1254»
[370] G. de Montmollin, M. de-Montmoll in, Helv, cbim. acta,
f 94 (1923).
[371] F. W. Bergstrom, I. M. G r a n a г a, V. Erickson, J. Org. Chert
, 7, 89 (1942).
1372] R. H u i s g e п, H. Rist, Ann., 594, 159 (1955).
[373] J. Meisenheimer, E. Patzig Ber., 39, 2533 (1906).
[374] Ch. C. Price, S.-T. V о о n g, Org. Synth., Coll. v. 3. 1955,. p. 664.
[375] R. Huisgen, Ann., 559, 142 (1948).
-410
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
аминонитрохинолипа, причем раствор превращается в кашицу. После Потряхи-
вания с 250 мл теплой воды, фильтрования и сушки получают 5,2 г (94% от тео-
ретического) шелковистых желтых игл; т. ил. 272° С (из большого количества
спирта). Желтый гидрохлорид плохо растворим в воде.
Проведено [376] прямое аминирование ароматических соединений, не содержа-
щих нитрогрупп, гидроксиламин-о-сульфокислотой в присутствии А1С13. Однако вслед-
ствие неоднозначного протекания этой реакции такой метод может представлять инте-
рес лишь в особых случаях. Так, из толуола с выходом около 50% от теоретического
получают смесь, состоящую главным образом из о- и п-толуидинов [377].
Методом внутримолекулярного аминирования является циклизация N-галоген-
производвых вторичных алифатических аминов (реакция Гофмана — Леффлера) [377а]
СН8-СН2
R-CH СН8
\
NBr
I
СНз
СПе-СНа
н-(!н сн2
N
I
СНз
Действуя концентрированной H2SO4 на N-бромметилбутиламик с последующей
обработкой реакционной массы раствором щелочи, Леффлер [378] получил N-метил-
пирролидии, а из N-бромпроизводиого 4-метиламиногептана по аналогичной реакции—
№метил-2-пропилпирролидин [379].
N-Хлорзамещенные амины более пригодны для синтеза пирролидинов, чем
соответствующие N-бромпроизводные. Таким путем замещенные пирролидины были
получены с выходом до 75% от теоретического. Метод нашел применение также для
получения бициклических третичных аминов [381—383] и N-дибромпроизводиых
первичных аминов [384]. В результате двойного замыкания кольца из Й-дибром-4-
аминогентана синтезирован пирролизидии с выходом 35% от теоретического [385}.
И в этом, как и в других случаях [363. 386, 387], лучшие выходы достигались ш>И
УФ-облучении реакционной массы.
В последнее время удалось также синтезировать циклические амипы из N-мопо-
хлорпроизводных первичных аминов [387а]. Из бутиламина с выходом 70% от теоре-
тического получен пирролидин, из к-амиламина с выходом 80% от теоретического —
2-метилнирролидин, а из 1-амино-н-гептана или 4-амино-к-гептана с выходами соот-'
ветственво 50 и 70% от теоретического -г- З-я-пропллпирролидии [387а]. Механизм
этой реакции см. [387].
[378]
[379
[380
[381
[382
[383
[384]
[385]
[386}
[387]
[387а]
[376] R. N. Keller, Р. A. S. Smith, J. Am. Chem. Soc., 66, 1122 (1944); 68ь
899 (1946),
[377] P. К ov a c i c, R. P. Beune t, J. Am. Chem. Soc., 83, 221 (1961); P. К o-_
v a c i с, 3. L- Foote, J. Am. Chem. Soc., 83, 743 (1961).
[377a] Обзорная статья: M. E. Wolff, Chem. Rev., 63, 55 (1963).
K. Loffier, C. Frey tag, Ber., 42, 3427 (1909).
K. Loffler, Ber., 43, 2035 (1910).
G. H. Coleman, G. E. Goheen, J. Am. Chem. Soc., 60, 730 (1938). '
G. Menschikoff, Ber., 69, 1802 (1936).
St. Wa wzonek, P. J. Thelen, J. Am. Chem. Soc., 72, 2118 (1950).
St. W a w г о n e k, M. F. Nelson jr., P. J. Thelen, J. Am. Chem.^
Soc., 73, 2606 (1951).
E. Sch.mitz, Angew. Chem., 73, 23 (1961). f
E. Schmitz, D. Murawski, Chem. Ber., 93, 754 (1960). 5
W. R. II с г t I e г, E. J. Corey, J. Org. Chem., 24, 572 (1959). J’
St. W a w z о n c k, T. P. Culbertson, J. Am. Chem. Soc., 81, 3367'
(1959).
E. Schmitz, D. Murawski, Z. Naturforschung, 17b, 127 (1962).
I. ЗАМЕЩЕНИЕ ВОДОРОДА АЗОТОМ
Следует упомянуть также о методе образования С—N-связей, сопровождающе
ся циклизацией. Так, при действии на оксимы реактивами Гриньяра получают про®
водные этпленимина (388, 389J:
CeH6-С—СН(СН3)а —(С6Н6)аС-------------С(СНа)а
II \ /
NOH NH
Введение аммониевой группы
Примером реакции, при которой происходит замещение атома водорода аммовд
вой группой, является образование пиридип-4-пиридиниевых солей из пириди:
и тиоиилхлорида. Поскольку эти соли часто служат исходными продуктами для пол
чения других труднодоступных 4-замещенных производных пиридина [219], зде
приводится получение гидрохлорида М-пиридил-4-пиридинийхлорида [219].
ЬГ-Пиридил-4-ниридинийхлорид. В трехгорлой колбе емкостью 1 л, снабже
ной капельной воронкой, мешалкой с вращающимся шлифом и обратна
холодильником с хлоркальциевой трубкой, к 300 мл сухого пиридина nj
интенсивном перемешивании прибавляют по каплям 900 г SOCla (техничеекз
продукт!). Охлаждая реакционпый сосуд снаружи проточной водой и регулирт
скорость приливания ЗОС12, легко удается поддерживать температуру реакцт
около 20° С. После добавления всего SOC]g смесь оставляют на три дня щ
комнатной температуре. Избыточный SOC18 удаляется при вакуумной перегони
при которой температуру водяной бани постепенно повышают до 100° С и подце
живают на этом уровне еще 2 ч после полной отгонки всего SOC18. Остающий<
в колбе твердый продукт превращается после кипячения с 200 мл абсолютна:
метилового спирта в однородную кристаллическую кашицу, которую пос.
охлаждения до 0е С отфильтровывают на воронке Бюхнера. Промывают кро
сталлы небольшим количеством спирта и сушат при 110® С; выход 260 а (60% <
теоретического); т. пл. 145—148® С. Для дальнейшей очистки продукт раств
ряют при нагревании в небольшом количестве 2 н. НС1, фильтруют и многократ»
обрабатывают фильтрат животным углем. После упаривания в вакууме и доб
вления спирта выделяются почти белые кристаллы, которые после охлаждена
отделяют, промывают спиртом и сушат. После перекристаллизации из метилово}
спирта т. пл. продукта 151® С. (Механизм этой реакции см. [219].)
Прямое введение диазониевой группы
Ароматические соединения во многих случаях могут быть сразу переведем
в диазониевые соли действием азотистой кислоты или ее производных [390—395
ArH + 2HNO24-HX —► [Аг-N*]X-+2HaO+2O
Реакция протекает с промежуточным образованием нитрозопроизводиыд
Сульфо- и карбоновые кислоты, нитропроивводные и аналогичные соединения с трудо
вступают в такую реакцию. Из фенолов (но не из нафтолов, реагирующих с Ьбравов<
нием только иитрозопроизводных) и из третичных аминов чаете получают соли
азоиия с хорошими выходами; например, из’Ы.Ы-диэтиланилииа [390] п-диазопроизвод
ние получаются с выходом 70% от теоретического, из фенола [393] с выходом 96%
из о- и л-крезола [393] — с выходом 98%, из хлорфенола — с выходом 95% и
фенол-о-сульфокислоты [395] с выходом 75—85%.
[388 ] К. N. Campbell, В. К. Campbell, J. F. McKenna, Е. Р. Che
put, J. Org. Chem., 8, 103 (1943); К. N. Campbell, L. G. Hesa
I. I. Schaffner, J. Org. Chem., 9, 184 (1944).
[389 ] H. M. К i s s m a n, D, S. T a r b e 11, J. Williams, J. Am. Chert
Soc., 75, 2959 (1953).
390] J. M. Tedder, J. Chem. Soc., 1957, 4003,
391] J. M. Tedder, G. T h e a k e r, J. Chem. Soc., 1957, 4008.
392] J. M. Tedder, J. Am. Chem. Soc., 79, 6090 (1957).
393] J. M. Tedder, G. Thcaker, J. Chem. Soc., 1958, 2573..
394] J. M. Tedder, B. Webster, J. Chem. Soc., 1960, 3270. • .
395] W. R od ionow, W. Matwee w, Ber., 57, 1711 (1934).
412
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Прямое диазотирование фенолов имеет препаративное значение. Для поду-,
чения растворов фенолдиазопиевых солей по методу Теддера [393] реакцию прсн^
водят в сильно разбавленных водных растворах в присутствии ацетона при 0е Ц
(с менее реакционноспособными фенолами, такими, как хлор- и нитрофенолу
реакция идет при 20’С), добавляют 14-кратный избыток NaNO8 и 4-кратны^
избыток НС1, применяемой в виде 2 н. НС1, и оставляют смесь на 20 ч в темнота.
Реакция Эрлиха — Закса
1
Ароматические иитрозосоединения реагируют с многочисленными соединениями, i
содержащими активные метиленовые группы, с образованием оснований ШиффЫ
(реакция Эрлиха — Закса [396]), из которых в результате гидролиза получаются соо»
ветствующие карбонильные и аминопроизводные: |
R\ R\ R'\ I
r2CH2 + 0N-R' —> r^>C=N-R’ —> r^>C-O + H2N-R' _ J
В качестве метиленсодержащей компоненты могут служить 0-дикарбонильщш
соединения, производные фенил-и циануксусного эфира, нитротолуолы, иодметилаийа
а- и у-пиколина и другие соединения [396—401] (ср. также стр. 390). я
Чаще всего реакцию проводят в присутствии таких щелочных натализатороцЦ
как гидроокиси щелочных металлов, сода, пиридин или алкоголят.
П'Диэтиламнноанил 2,4-ДИнитробеизальдегида [183]. Растворяют в 500 лйи
этилового спирта 91 г 2,4-динитротолуола и 90 г л-нитрозодиэтилаиилина, ирД
бавляют 150 з кристаллической соды и кипятят смесь в течение 5 ч в колбе с
ратным холодильником. После охлаждения отфильтровывают осадок на воронш
Бюхнера и тщательно промывают его кипящей водой. Выход 188 г (88% от
ретического); т. пл. около 173° С (разл.).
Иногда реакция протекает так бурно, что исходные вещества приходит^д
вводить в реакционную массу постепенно, небольшими порциими или применяя®
незначительные количества катализатора. В тех случаях, когда реакция в воддр%|
спиртовой среде протекает недостаточно быстро, ее удобнее проводить в раствоцв
абсолютного спирта в присутствии алкоголята [287]. -зЯ
Получение азометинов протекает с промежуточным ’образованием первичным
продуктов присоединения. В некоторых случаях эти промежуточные продукты моздн
дегидрироваться иитрозосоединением до нитрона, причем одновременно получаюпн
азоксисоедииения [400, 401]: -
R\cHNR' + 2R'NO -ЖГ» R^C-NR' + R'-N=NR”
R'/ I н‘° И'/ I I
он Об й
Образование нитрона при Избытке метиленового соединения в болыпинбЯ
Случаев подавляется. Если при антивной метиленовой группе находится атом галогёв
то нитрон получается даже в отсутствие избытка нитрозосоединення, поскольку Щ
этом возможна стабилизация первичного продукта присоединения вследствие отщепЯ
[396 ] Р. Ehrlich, F. Sachs, Ber., 32, 2341 (1899).
£397] F. Sachs, Ber., 33, 959 (1900); 34, 494 (1901); F. S a c h s, H. В a r s c h a I4
Ber., 34, 3047 (1901); 35, 1437 (1902); F. S a c h s, E. В г у, Ber., 34, 118 (190i
F. Sachs, A. Rohmer Ber,, 35, 3307 (1902); F. Sachs, P. Beck’
re sen, Ber., 36, 1132 (1903). '4
[398] A< Kaufmann, L. G. Vale tt e, Ber., 45, 1736 (1912); 46, 49 (1913).
[399] S. Skr а ирЛ. Bohm, Ber., 59, 1007 (1926).
[400] F. К г 6 h n k e, Ber., 71, 2583 (1928).
[401] F. Krohnke, H. Leister, I. Vogt, Ber., 90, 2792 (1957).
II. ОБМЕН ГАЛОГЕНА НА АЗОТ
4
пия галогеноводорода, как, например, при реакции n-нитробензилхлорида с нитрод
бензолом [400]:
O2N-Cl,TI4-CIJ2Cl + ON—В —> O2N-СеП4-СНС1-N—R
I
ОН
O2N-CeH4-CH=N-R4-H(
О
II. Обмен галогена на азот
1. Взаимодействие галогенпронзводных с производными аммиака
Алкилирование
В качестве алкилирующих агентов в первую очередь применяются алкилгалс
гониды, из которых, как известно, наибольшей реакционной способностью обладаю
иодиды. Первичные алкилгалогениды реагируют легче, чем вторичные; трагичны
галогениды легко отщепляют галогеноводород под действием аминов и переходя
в олефины. Винилгалогениды более склоним к полимеризации, тогда как аллилгалс
гениды реагируют обычным образом. Реакция а-алкилированиых аллилгалогенидо
с аминами почти всегда сопровождается аллильной перегруппировкой. Полярно
растворители повышают скорость реакции алкилирования; чаще всего применяются
вода и этиловый спирт; из-аа более высокой температуры кипения употребляют такая;
бензиловый спирт и простые эфиры гликоля. Ацетонитрил и нитрометан примекотгат
ся как растворители при кватериизации иодистым метилом [402].
Взаимодействие аммиака, первичных и вторичных аминов с алкилгалогенидама
практически всегда приводит к образованию смеси продуктов высших степенен алкилж
рования. Поэтому при взаимодействии с избытком галогенида можно иногда за однч
операцию получить четвертичную соль в присутствии основания, исключающего воа
можность образования гцдрогалогенида. Однако для синтеза четвертичных солей
лучше проводить алкилирование третичного амина.
Для получения первичного амина с удовлетворительным выходом кеобходимх
вводить достаточно большой избыток аммиака (по крайней мере десятикратный).
Такой избыток применяется в предложенном Брауном [403] методе алкилировании
в жидком аммиаке. Понятно, что образующаяся при всех условиях смесь изомеров
разделяется на компоненты тем легче, чем больше алкильный остаток примененногс
галогенида, так как в этом случае различие в температурах кипения свободных осно-
ваний больше. Поэтому этот метод неприменим для получения первых членов гомоло-
гического ряда моноалкиламинов; он неприменим также для получения аминов с очей®
длинными цепями, вследствие того, что соответствующие галогениды ие растворяются
в жидком аммиаке [404].
При проведении синтеза в запаянной трубке со спиртовым раствором аммиа-
ка получается главным образом вторичный амин [405]. н-Вутиламин можно синтезиро-
вать по довольно трудоемиому способу из бутилбромида и спиртового раствора аммиака
при комнатной температуре и нормальном давлении с выходом 47% от теоретиче-
ского [406].
Аналогичным способом синтезировал ряд замещенных в ядре первичных 6-феннл—
етиламинов с выходом 70—80%. Для этого раствор 15 в соответствующего р-фенил-
этилбромида в 500 мл спирта, насыщенный аммиаком, выдерживали при 5° С в течение
5—8 суток [406].
402] F. G. Mann, J. Watson, 3. Org. Chem., 13, 502 (1948).
403] J. v. Braun, Ber., 70, 979 (1937).
404] J. v. Braun, R. Klar, Ber., 73, 1417 (1940).
405] O. Westphal, D. Jerchel, Ber., 73, 1002 (1940).
406]J?. C. W h i t m о г e, D. P. L a n g I о i s, J. Am. Chem. Soc., 54, 3441 (1932'
414
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Аналогично замещенные в ядре 2-амино-1-фенилпропаны могут быть получены
взаимодействием соответствующих хлоридов со спиртовым раствором аммиака под.
давлением (408]. В качестве примера синтеза с аммпаком выбрак синтез фенамина
(бензедрина).
Фенамин [408]. В 450 мл насыщенного спиртового раствора аммиаиа
(125 г/л) растворяют 25 г 2-хлор-1-фепилпроиана и раствор нагревают при 160° С
в качающемся автоклаве в течение 9 ч. После охлаждения и фильтрования спирт
отгоняют, остаток подщелачивают 6 а. раствором NaOH и амин экстрагируют
бензолом 4 —5 раз порциями по 20 мл. Бензольный раствор 3—4 раза встряхивают-
с 6 н. раствором НС1, извлекая гидрохлорид бензедрина. Раствор гидрохлорида
подщелачивают NaOH и многократно экстрагируют фенамин бензолом. Посла-,
сушки над К 2СО3 и перегонки получают 11,1 г (51% от теоретического) фенамина;,
т. кип. 80—82° С (11 мм рт. ст.).
В основном те же трудности встречаются и при взаимодействии дигалогензаме~>
щенных с аммиаком. При использовании а,<в-дигалогензамещенных углеводородов;
с числом углеродных атомов С4—Св появляется новое осложнение, заключающееся
в том, что эти соединения легко реагируют с аммиаком, давая циклические соедине-
ния; чаще всего образующийся при этом азотсодержащие гетероциклы быстро реаги-'
руют далее с образованием четвертичных спиранов [403].
Наибольшее значение имеет получение первичных алифатических аминов nov
методу Габриэля [409], при котором образование вторичных и третичных аминов"?
исключено. Исходным следиирпием является фталимид калия, который подвергают,
взаимодействию с алкилгалогенидом; при этом образуется N-алкилфталимид, который
подвергают гидролизу *.
Метод Габриэля применяется в самых различных вариантах, в эту реакцию-;
вступают также дигалоидзамещенные соединения, которые могут реагировать с 1 или;
2 моль фталимида калия; реакция протекает гладко почти со всеми алпфатическимЙ
галогенидами, даже с галогенидами очень сложной структуры. В случае малой реай«
циоииой способности галоидного алкила лучшие результаты получаются при испольЬ’’
зовании таких растворителей, как ацетон или, еще лучше, формамид, диметилфорМ^
амид или ацетамид [410]. Si-
Для расщепления замещенных фталимидов их кипятят с 20%-ным растворой)
НС1 или нагревают в запаянной трубке при более высокой температуре; применяется^
также омыление по методу Познера [411] сначала раствором КОН, причем образу-^
ются фталаминовые кислоты, окончательный гидролиз которых протекает в присутст-i
вии соляной кислоты. Более удобным методом расщепления является непродолжнтель-Ч
ное нагревание замещенного фталимида со стехиометрическим количеством гидразин-3
гидрата в спирте и последующее омыление образующегося на этой стадии фталазонн]
соляной кислотой [412]. Карбонильные группы, содержащиеся в N-алиильноМ^
остатке фталимида, ацетализирутот перед расщеплением гидразином [413]. Для рас-^
щеплеиия фталимидокарбоновых кислот молоко применять также фенилгидразин [414]. <
Ввиду устойчивости фталимидных произбодных их можно использовать во 'мно-^
гих реакциях. В частности, реакципииоспособные группы в N-алкильном остатке могуч|
* В методе Габриэля вместо галогеппроизводных с успехом могут быть испольэО^
ваны эфиры n-толуолсульфокислоты [Е. Sakellarios, Helv. chim. acta, 20J
' 1675 (1946)]. — Прим. ред.
[407] J. Н. Speer, A. J. Hill J. Org. Chem., 2, 139 (1938). 3
[408] Т. М. Patrick jr., Е. Т. McBee, Н. В. Hass, J. Am. Chem. SoM
[409]
[410]
[411]
[412]
[413]
[414]
68, 1009 (1946). ” ’I
S. Gabriel, Вег., 20, 2224 (1887).
J. С. S h е е h а п, W А. В о I h о f е г, J. Am. Chem. Soc., 72, 2786 (1950)ie
Th. Posner, Ber., 26, 1856 (1893).
H. R. Ing, R. П. F. Maoske, J. Chem. Soc., 1926, 2348.
К. В a 1 e ii o,v ic, N. Bregant et al., J. Org. Chem., 18, 297 (1953).
I. Schumann, R. A. Boisonnas, Nature (London), 169, 154 (1952)fJ
Helv. chim. acta, 35, 2235 (1952). Я
1
II. ОБМЕН ГАЛОГЕНА НА АЗОТ
4
подвергаться многочисленным превращениям до омыления имидной группировю
Например, при взаимодействии фталимида калия с хлораигпдридами галогеизамеща
вых кислот в реакцию вступает не хлор карбоксильной группы, а галоген в алифатч
ческой цепи и получаются хлорангидриды фталимидокарбоновых кислот, из которь
под действием натриймалонового эфира или других енолятов щелочных металле
образуются fj-дпкетоны или ^-котокарбоновые кислоты и в конечном итоге аминокет<
вы. Взаимодействием хлорангидридов фталимидокарбоновых кислот с ароматически^
соединениями по реакции Фриделя — Крафтса получают жирно-ароматическне амин<
кетоны. Аминоспирты можно синтезировать также по методу Габриэля. И, наиоке]
взаимодействием оромалкилфталпмидов с гидросульфидом калия можно заменят
бром на сульфгидрильную группу; в этом случае при омылении получаются аминомй)
каптаны [415, 416].
фталимид калия [417]. К раствору 20 г фталимида в 400 мл абсолют
него этилового спирта приливают раствор 7,6 г КОН в 30 мл 75%-ного спирта
быстро охлаждают и отфильтровывают выделившийся фталимид калия. Фильтра
нагревают, добавляют ещв 20 г фталимида и 7,6 г КОН в 30 мл 75%-ного спирта
снова охлаждают и фильтруют. Фталимид калия размешивают с ацетоном
фильтруют и при умеренном нагревании сушат на воздухе; выход 40—45 в
Для препаративных целей удобно также применять в качестве растворителе
пиридин [418].
0-Вромэтилфталимид [415, 419]. Смесь 100 г фталимида калия н 300
(3 моль) этиленбромида кипятят в колбе с обратным холодильником около 7 ,
до превращения реакционной массы в сироп. Перегонкой с водяным паром отде
ляют непрореагировавший этиленбромид; оставшаяся вязкая масса кристалла
зуется при охлаждении под слоем воды. Воду сливают, остаток растворяю»
в 150 мл кипящего 96%-ного спирта, фильтруют, охлаждают, после завершенна
кристаллизации осадок отфильтровывают и сушат на воздухе. Получаете*
около 120 а неочищенного продукта, который извлекают примерно 240 мл кипя-
щего CSs; при этом в остатке получают около 8 г этилендвфталимида. Из профиль,
трованиого сероуглеродного раствора выкристаллизовывается бромэтилфтал
имид. Маточные растворы упаривают и получают 100 г (73% от теоретического
0-бромэтилфталимида [415]; т. пл. 79—80° С (после перекристаллизации и?
разбавленного спирта т. пл. 82—83° С [419].
р-Бромэтиламин [420]. Нагревают 20 г p-бромэтилфталимида с 50—
60 мл концентрированной НВг (d = 1,49) до 180—200е С в запаянной трубку
в течение 2 ч. После охлаждения добавляют холодную воду, отфильтровывают
нерастворившуюся фталевую кислоту и упаривают фильтрат на водяной бан^
досуха. Затвердевающий прн охлаждении темноокрашеннмй остаток перекри-
сталлизовывают из 15—20 мл абсолютного спирта. При охлаждении образуются
крупные кристаллы гидробромида бромэтиламина и наряду с ними выделяется
тонкий порошок, который отделяют от маточного раствора декантацией и про-
мывают холодным абсолютным спиртом. Вещество перекристаллизовывают еще
раз из абсолютного спирта; т. пл. 155—160° С. Действуя концентрированным
раствором КОН на соль бромистоводородной кислоты, получают неустойчивое
свободное основание.
Кадаверни (1.5-диаминопентан) [421]. В перегонной колбе нагревают
эквимолярные количества бензоилпиперидина и PClg; дистиллят встряхивают
с холодной водой для разложения образующейся POCI3. Полученную смесь»
бензонитрила и 1,5-дихлорпентаиа сушат падСаС12.добавляют избыток фталимида.
[415] S. Gabriel, Вег., 22, 1137’(1889).
[416] S. Gabriel, Вег., 24, 1110 (1891).
[417] W. J. Hale, Е. С. В г i 11 о n, J. Am. Chem. Soc., 41, 841 (1919).
[418] J. Reitmann, в книге Houben-Weyl, Methoden der organischen
Chemie, Bd. 8, 4 Aufl., Stuttgart, 1952, S. 658.
[419] P. L. Salzberg, J. V. Supnfewski, Org. Synth., 7, 8 (1927).
[420] S. Gabriel, Ber., 21, 566 (1888).
[421] j. v. Braun, Ber., 37, 3583 (1904).
416
ОБРАЗОВАНИЕ С—N-СВЯЗЯ В РЕЗУЛЬТАТЕ ОБМЕНА
калия (2,5 моль на 1 моль бензоилпиперидина) и быстро нагревают при перемени
вании в открытой колбе до 190—200° С. Масса плавится, окрашиваясь в кормчий
вый цвет, и превращается в вязкий сироп. Указанную температуру поддерживаю
в течение 1—2 ч. Реакционную смесь охлаждают, вепрореагировавпшй фталимя
калия извлекают кипящей водой и с водяным паром отгоняют бепягюттрн
и репрореагировавший 1,5-дихлорпентан, При промывании остатка после
гонки сначала 50%-ным, а затем абсолютным спиртом он постепенно преви
щается в светлый порошок, который в завершение кипятят с достаточным колиёЙ
ством спирта. После охлаждения осадок отфильтровывают, промывают небод|
шим количеством спирта, растворяют в горячем хлороформе и осаждают спиртм
Неочищенный продукт перекристаллизовывают из смеси спирта с хлороформ^
и сушат прп 100° С. Получают 1,5-дифталимидпентан, выход 60—70% от теорем
ческого, т.пл. 186° С. Этот продукт нагревают при 200°С в течение 2ч в яяпаята|
трубке с трехкратным количеством концентрированной НС], после охлаждеМЙ
отфильтровывают выделившуюся фталевую кислоту, упаривают фильтрат Й
водяной бане досуха, скова растворяют в воде и снова упаривают, продоляЩ
эту операцию до полного удаления избытка соляной кислоты. Остаток промыв»
спиртом и эфиром и получают с почти количественным выходом чистый дигидр}
хлорид 1,5-диаминопентана; т. пл. 255° С. &
В некоторых случаях можно отказаться от предварительного получения фтм
имида калия и непосредственно проводить взаимодействие фталимида с алкилгалани
видом в присутствии KSCO3. Приведенный ниже пример иллюстрирует этот снося
синтеза и расщепление замещенного фталимида гидразином. 3
Бензиламин [412]. Тщательно приготовленную смесь 300 г фтяятгмд
и 150 з сухого,КаСО3 нагревают 3 ч с 300 з (20%-иый избыток) бензилхлорйй
на масляной бане в колбе с обратным холодильником. После этого отгоняй
избыточный бензилхлорнд с водяным паром, фильтруют и промывают твердаи
бенаилфталимид водой; выход 360—375 з; т. пл. 116’ С (после перекристаллН
ции из ледяной уксусной кислоты).
Для получения бензиламина нагревают суспензию тонкоиэмельчеияя
бензилфталимида с эквимолекулярным количеством гидразиигидрата в (япймй
Образовавшийся желатинообразный осадок разлагают при нагревании избыСЯ
ним количеством соляной иислоты; выделяющийся при этом фталилгидраяй
отфильтровывают и промывают водой. Фильтраты объединяют, отгоняют СПЙ|1
отфильтровывают незначительное количество выпавшего при охлаждении .фЯ
лилгндразида, добавляют щелочь и экстрагируют бензиламин эфиром. ЭфиряЯ
раствор сушат КОН и фракционируют. Выход 90—95% от теоретическая
т. кип. 185—187° С. л
На методе Габриэля основан разработанный Серенсеном [422] синтез а-амйй
кислот, промежуточным продуктом в котором является фталимидомалоновый. эяН
[422, 423]. 13
Фталимидомалоновый эфир. На масляной бане при 100—120° С наги
вают до начала реакции 210 з броммалонового эфира с 165 з фталимида к&»|
после чего реакции протекает самопроизвольно. Затем реакционную мм|
охлаждают, промывают холодной водой для удаления КВг и непрореагиройЦ
шего фталимида калия. Высушенный остаток перекристаллизовывают сиаЧМ
из бензола, а затем из абсолютного спирта. Выход фталимидомалопового эф>Я
70—80% от теоретического; т. пл. 74° С. ’Л
В молекулу фталимидомалопового эфира через его натриевое производное момД
ввести алифатический радикал. Последующее омыление приводит через различи!
промежуточные продукты к а-аминокарбоновым кислотам. Хорошую пропись для овЯ
[422] S. Р. L. Sorensen, С. г. Trav. Lab. Carlsberg, 6, 1 (1903): С., 1903, II,
(4231 А. Е. Os ter berg, Org. Synth., 7, 78 (1927).
II. ОБМЕН ГАЛОГЕНА НА АЗОТ
чения аспарагиновой кислоты см., например, [424]. В качестве других примеров мо>
привести синтезы лантионила и этионина [425], а также кинуренина [426].
Натриевое производное фталимидомалонового эфира. В круглодоп;
колбе емкостью 500 мл, снабженной обратным холодильником с хлоркальцие
трубкой, растворяют 4,6 г натрия в 80—100 мл свежеперегианного над натр
спирта. К этому раствору при 60—70° С прибавляют 62—63 г (несколько бодз
ряссчитяиного количества) абсолютно сухого фталимидомалонового эфв
который при встряхивании реакционной массы растворяется, придавая расти
желтую окраску. При точном соблюдении температурного режима вскоре Ht
пается выделение кристаллического желтого натриевого производного; непрер
ным встряхиванием и соответствующим охлаждением массы достигается под
выделение достаточно мелкокристаллического продукта. После этого, полное
исключив возможность увлажнения реакционной массы, отгоняют в ваку
спирт, повышая температуру бани к концу отгонки до 140° С. Последние сл
спирта удаляют продувкой сухим воздухом, очищенным от углекислоты, и е
кукрованием. - \
По данным Серенсена [427], в зависимости от условий проведения с
теза натриевое производное фталимидомалонового эфира выделяется в виде сту
или кристаллов различного размера. Студнеобразный продукт не может б
отделен от избытка спирта без разложения, а крупнокристаллический прод
обладает малой реакционной способностью.
Аналогично получают и-аминокислоты, для чего на первой стадии провс
взаимодействие фталимида калия с а.сэ-дигалогенидом, а на второй стадии обменив
второй атом галогена по реакции с натриевым производным малонового эфира,
такому способу Фишер [428] получил ориитип. Исходя из дигалогензамещенд
кислот, можно одновременно вводить две аминогруппы; при этом в качестве раствс
теля особенно пригоден днметилформамнд. Шихан и Болхофер [410] получила
cz.d-дибромадипииовой кислоты соответствующее а.б-дифталимидиое произвол
с выходом до 90%, а из него омылением раствором НВг в ледяной уксусной иислог<
а,б-диамииоадипиновую кислоту с выходом 91% от теоретического.
Другой способ синтеза первичных алифатических аминов разработан Дел<
ном [429]. Алкилируя уротропин, получают соответствующую соль мопочетвертичЕ
основания, которую расщепляют спиртовым раствором НС1. Поскольку многие хл<
ды’и бромиды реагируют с уротропином очень медленно, можно во время алкил*
вания получать соответствующие иодиды, добавляя эквимолекулярные количе<
Nal.
Первичные алифатические амины (получение по упрощенному
ТОДУ) [430]. Растворяют 1 моль уротропина в 8—10-кратном весовом кол:
стве горячего 95%-ного спирта, медленно добавляют 1 моль Nal и затем 1 л
алкилхлорида или алкилбромида. После окончания выделения осадка реакц*
ную массу насыщают газообразным НС1, затем осадок вновь растворяется и be
дает NH4C1, который отфильтровывают на воронке Бюхнера. От прдзрачц
фильтрата отгоняют спирт, к остатку добавляют избыточное количество щец
и отгоняют выделившийся амин. Для полного выделения четвертичной с
Г424] М. S. Dunn, В. W. Smart, Org. Synth., 30, 7 (1950).
[425] R. Kuhn, G. Quadbeck, Ber., 76, 527. 529 (1943).
[426] A. Butenandt, W. Weidel et al., Hoppe-Seyler’s Z. physiol. Ch.«
279, 27 (1943).
[427] S. P. L. Sorensen, Hoppe-Seyler’s Z. physiol. Chem., 44, 454 (1905).
[428] E. Fischer, Ber., 34, 454; cp. 2900 (1901).
[429] M. D e lepine, C. r., 120, 501 (1895); 124, 292 (1897); Bull. Soc. chim. £8
ce [3], 17, 290 (1897); [4],31, 108 (1922).
[430] A. G a 1 a t, G. Elion, J. Am. Chem. 8oc., 61, 3585' (1939).
27 Заказ 1835.
ОБРАЗОВАНИЕ С—М-СБЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
уротропина по этому методу часто требуется довольно длительное время (от не-
скольких часов до многих недель); выходы высокие.
По уротропиновому методу легко получаются а-аминокетоны.
w-Аминоадетофснон [431]. Сначала из эквимолекулярных количеств
w-хлорацетофепопа и уротропина в хлороформе получают четвертичную соль.
При комнатной температуре выделение осадка заканчивается через 12 ч; выход
около 60% от теоретического; т. пл. 145’ С.
К 30 г тонкоизмельченной четвертичной соли приливают холодную смесь
из 30 мл 38 %-ной НС1 и 240 мл метилового спирта. Реакционную массу оставляют
на 3 суток, встряхивая .ее время от времени. Затем отфильтровывают выпавший
NH4C1 и фильтрат упаривают в вакууме досуха. Остаток растворяют при нагре-
вании в 4 мл воды; при охлаждении выкристаллизовывается 9—10 г гидрохлорида,
(о-аминоацетофенона; т. пл. 186—187° С. Еще 2—3 г продукта можно получить
из маточного раствора.
Этот способ нашел применение также для синтеза аминокислот [432, 4331,
«-(аминоалкил)-бепзойных кислот [434] и аминов гетероциклического ряда [435, 43б1.„.
Так, из р-бромпропионовой кислоты был получен р-аланин [433] с выходом 85^>.
В литературе описано сравнительно мало удовлетворительных методов для
• синтеза вторичных и третичных аминов моноалкилированием первичных и соответ-
ственно вторичных аминов действием алкилгалогенидов, а также для аналогичных ,
реакций с аммиаком. Основные осложнения при проведении таких синтезов уже '
упоминались.
N-Алкил- и К,.К-диалкил-0-фенетиламины образуются с хорошими и даже
отличными выходами [407] при нагревании в течение 6—8 ч (100° С) в запаянной
трубке 1 моль Р-фенетилбромида с 5 моль первичного или 3 моль вторичного амина.
Получение 0-днэтиламиноэтанола из этиленхлоргидрина и диэтиламина с выхо-
дом около 70% от теоретического см. [437].
Диэтилизопропиламин [438]. Алкилирование диэтиламина изопропнл-
бромидом не удается осуществить без растворителей и при нормальном давлении,
но в глицерине или гликоле эта реакция протекает с удивительно высокими выхо-
дами. Смесь 123 г изопропилбромида, 94,9 г диэтиламина и 50 г глицерина на-
гревают до слабого кипения в колбе с обратным холодильником в течение 72 *.
Затем ее подщелачивают, сушат выделившиеся амины над КОН, фракционной t
перегонкой выделяют диэтнлнзопропиламин. Выход 67 г (60% от теоретического); »
т. кип. 108q С.
Как правило, несколько более однородно протекает алкилирование первичных
ароматических аминов. Так, из хлористого бензила и анилина (4 моль) в присутствии j
1,25 моль NaHCO8 можно получить N-бензиланилин [439] с выходом до 95%; при мень- |
шем избытке анилина и в этом случае преимущественно образуются третичные >
основания. >?
Во многих случаях реакцию.можно проводить без конденсирующих агентов.
Хикииботтом [440] получал моноалкиланилины кипяченном в течение многих часов г
[431] С. Mannich, F. L. Hahn, Ber., 44, 1542 (1911).
[432J A. Baniel, М. Frankel, I. Friedrich, rA. Katchalsky, J
J. Org. Chem., 13, 791 (1948). - j
[433] N. L. Wendler, J. Am. Chem. Soc., 71, 375 (1949).
[434] F. F В 1 i c k e, W. M. L i 1 i e n f e 1 d, J. Am. Chem. Soc., 65, 2281 (1943), 1
[435] F. F. В 1 i c k e, J. H. В u rc k h a 1 ter, J. Am. Chem. Soc., 64, 477 (1942).
[436] H. К e s k i n. C. D. Mason, F. F. Nord, J. Org. Chem. 16, 1333 (1951). ,3
[437] W. W. Hartman, Org. Synth., Coll. v. 2, 183 (1943). ?
[438] S. Caspe, J. Am. Chem. Soc., 54, 4457 (1932). • ?
[439] F. G. Willso’n, T. S. Wheeler, Org. Synth., 8, 38 (1928).
. (440] W. J. H i c k i ii b о t t о m, J. Chem. Soc., 1930, 992. *
п. обмен галогена па азот
4'
пропел-, изопропил- и бутилбромида с 2,5—4-кратным избытком анилина; вых<
75%. Для отделения избытка анилина из образующейся смеси оснований он дримею
иодный раствор ZnCl2; в этих условиях только апилин дает нерастворимый устойчив®
и воде комплекс, который после продолжительного отстаивания отфильтровываю
Выделение вторичных аминов из образующихся смесей аминов возможно пут»
перевода их в нитрозамины.
Моноэтиланилин [441]. В колбе с обратным холодильником нагрева!
до слабого кипения 100 г анилида и 130 г этилбромида в течение 1—2 ч. Поч-
полностью затвердевающую при охлаждении реакционную массу растворя®
в воде и кипячением удаляют небольшое количество избыточного этилбромид
После этого добавляют 300 мл 20%-ного раствора NaOH, а выделяющуюся мд
лянистую смесь аминов отделяют и из водного слоя экстрагируют эфиром et
некоторое количество оснований. Эфирные вытяжки объединяют с основнь
количеством оснований и отгоняют эфир на водяной бане. Остаток растворят
в смеси 200 мл концентрированной НС1 и 1000 мл воды, хорошо охлаждас
льдом и добавляют 60 г NaN02. Выделяющийся в виде темного масла этилфенис
нитрозамин извлекают эфиром. Непрореатировавший анилин остается в раствс»-
в виде хлорида фенилдиазония. Для окончательной очистки эфирную вытяну
можно промыть разбавленным раствором NaOH. Эфир отгоняют и нервное,
маслянистый остаток в раствор 350 г SnCl2 в 400 мл концентрированной НС
охлаждая при необходимости водой. Затем раствор подщелачивают NaO]
отгоняют продукт реакции с водяным паром и извлекают его эфиром. Эфирн]
вытяжки сушат над КОН. При перегонке получают 40—50 г моноэтиланилин
т. кип. 205° С. Чистый моноэтиланилин не дает цветной реакции с раствор»
хлорной извести.
Более новый способ моноалкилирования л-толуидина с высоким выходом алки
амина основан на том же принципе [442]. О получении чистых аралкиламинов, j
содержащих третичных продуктов, см. также стр. 420, 462.
Первичные алифатические [443] или ароматические [444—446] амины мог
вступать в реакцию с а,<1)-дигалогензамещеиными углеводородами с замыканием колыд
при этом образуются N-алкилированные или N-арилироваиные гетероциклическ.
соединения. Для подавления образования а,<в-диаминосоединений рекомендует
проводить циклизацию с эквимолекулярными количествами амина и дигалогени;
в присутствии соды [446, 447].
^(о-Толил)-пиперидин [446]. Смесь 10,7 г (0,1 моль) о-толуидив
23 г (0,1 моль) 1,5-дибромпентаиа и 10,6 г (0,1 моль) безводной соды в 30 мл ч
луола нагревают в колбе с обратным холоднльпиком в течение 24 ч при перемена
вании. Толуольный раствор декантируют, остаток растворяют в воде, к раство
добавляют NaOH до сильнощелочной реакции и продукт экстрагируют тол^
лом. Объединенные толуольные растворы перегоняют. Повторное фракциоа
роваине в вакууме дает 10,3 а (59% от теоретического) М-(<?-толил)-пиперидие
т. кип. 65—66° С (0,6 мм рт. ст.).
Осложнения, вызываемые при алкилировании аминов образованием гидрогатз
гснидов, часто можно легко устранить добавлением к реакционной массе стеричес;
затрудненного (т. е. неалкилируемого) амина для связывания кислоты. Особен;
удобно применять для этой цели триизопропаноламин.
[441] JI. Erdmann, Anleitung zur Darstellung organischer Praparaj
Ferdinand Enke, Stuttgart, 1894 , 8 . 428.
[442] J. S. В u c k, C. W. Ferry, Org. Synth., Coll. v. '2, 290 (1943).
[443] R. C. Elderfield, H. A. H a g e m a n, J. Org. Chem., 14, 605 (194’
[444] J. v. Braun, Ber., 37, 3210 (1904).
[445] M. S c h о 1 t z, E. W a s s e г m a n n, Ber., 40, 852 (1907).
[446] A. JI. Sommers, S. E. A a 1 a n d, J. Am. Chem. Soc., 75, 5280 (195
[447] J. Sauer, R, Huisgen, A. Hauser, Chem. Ber., 91, 1461 (1958).
120
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
N.N-Дибутиланилин [448]. В колбе с обратным холодильником, тща-
тельно защищенной от доступа влаги, нагревают при перемешивании 9,3 г
(0,1 моль) анилина, 30,14 г (0,22 моль) н-бутилбромида и 57,3 а (0,3 лоль) трииао-
пропаноламина до начала умеренного кипения бутилбромида (100—110° С).
Через 2 ч кипение бутилбромцца прекращается даже при постепенном повышении
температуры до 180° С. Реакционную массу перемешивают еще 2 ч при этой’
температуре, затем при интенсивном перемешивании добавляют двукратный
объем воды. Суспензию переносят в делительную воронку. Через 2 ч сырой про--
дуит выделяется в виде масла. Незначительное дополнительное количеством
продукта можно извлечь разбавлением водного слоя, перемешиванием с 17 *.]
(0,3 моль) КОН и экстрагированием эфиром. Промытый водой и высушенный
КОН и К2СО3 неочищенный дибутиланилин перегоняют, используя колонку-
со спиральной насадкой. Получают 18,5 г дибутилаинлина; т. кип. 151* Qi
(15 мм рт. ст.). Кубовый остаток содержит еще 0,9 г продукта. Общин выходу;
95% от теоретического. 4
Вторичные амины в чистом виде получают алкилированием и последующим!
омылением ацилированных первичных аминов. Этот способ аналогичен синтезу Габ-'З
ризля, ио он не так универсален. Вследствие более кислого характера амидного водо-Л
рода во избежание О-алкилирования и в этом случае приходится применять производ-1
ные щелочных металлов. В более старых методах для получения амидных производных?
щелочного металла пользовались металлическим натрием. fl
N-Метил ацетанилид [449]. В колбу емкостью 2 л загружают последо-^
вательно 400 а сухого бензола, 11,5 г свежеприготовленной натриевой проволоки,
67,5 г ацетанилида и нагревают смесь до растворения ацетанилида. В спором i
времени при интенсивном выделении водорода начинается образование белой"
зернистой натриевой соли, которое заканчивается после 2 ч иипячения с обрат-Н
ным холодильником. К реакционной массе сразу добавляют 80 е метилиодида 5
и нагревают еще 1 ч; охлажденную смесь встряхивают с 100 мл воды. Охлажден- $
ный бензольный раствор сушат над Na2SO4 и упаривают (не досуха), при этом.й
кристаллизуется N-метилацетаннлид (60 а после промывки 20 мл бензола)] g
из маточного раствора и бензола, применявшегося для промывки, получают й
еще 13,3 а продукта. Общий выход N-метилацетанилнда 98,3% от^еоретичесиогоря
после перекристаллизации нз лигроина т. пл. 100е С (испр.).
’4
Аналогичным методом получают N-метил-п-метокснацетанилид с выходом 96%’?
ют теоретического. j
Для металлирования р-ацетнафталида требуются более высокие температуры, j
N-Метил-р-нафтил амин [450]. Ацетнафталид кипятят с. рассчитанным ’
количеством натрия в толуоле. Не выделяя натриевую соль, добавляют экви- ‘
молярное количество метилиодида; при этом происходит выпадение Nal, который
отфильтровывают. Толуольный раствор кипятят с разбавленной НС1, водный 4
слой для удаления толуола встряхивают с эфиром и подщелачивают. Выделив-/
шееся масло перегоняют в ваиууме, выход 55% от теоретического; т. кип. 165— •;
170° С (12 мм рт. ст.),
Были сделаны попытии заменить в этой реакции натрий на гидрид натрия [451)» *
но оин не увенчались особым успехом. Впоследствии было установлено [452], что полу-
J448
[449 xuiviepape, uer., DO, \_iuoo/.
[450 R. p s c h о г ru w. Karo, Ber., 39 , 3140 (1906).
[451 W e P ~ ~ - т л /--l.’™ »r. 1ЛПП ИПШ
[452
S. Hu nig, М. Kiessei, Chem. Вег., 91, 380 (1958).
Е. Thielepape, Вег., 68, 751 (1935).
W. 8. FonesS J. Org? Chem77iV1699*(1949)7 ''
I. J. P ас h t er, M. C. Kloetzel, J. Am. Chem. Soc., 74, 1321 (1952).
П. ОБМЕН ГАЛОГЕНА НА АЗОТ
4
чение амидов щелочных металлов совсем ие обязательно в тех случаях, когда в ка*
стве растворителя применяют ацетон и реакцию проводят в присутствии избытка КО
При этом хорошие выходы достигаются даже с амидами кислот, не реагирующие
с гидридом натрия.
N-Метил-п-ннтроацетанилид. В колбе с обратным холодильником р<
творяют 5 г n-интроацетанилида в 100 мл ацетона и прибавляют 6 а тонкоиамел
ченного NaOH. Смесь нагревают до умеренного кипения и прибавляют раств
6 г метилиодида в 15 мл ацетона. Через 1 мин осадок отфильтровывают, фильтр
упаривают, разбавляют водой н охлаждают. Выход продукта 4,36 г (81%
теоретического для чистого продукта); т. пл. 153—154° С. Еще 0,67 8 продук
получают из маточного раствора.
Еще Пикте и Крепье [453] алкилировали формаинлид в присутствии спиртово
раствора КОН и омылением (непродолжительное нагревание со спиртовым раствор*
КОН или концентрированной НС1) получали моиоалкиланилины с радикалами
метильного до амильного.
Легче и более гладко чем амиды карбоновых кислот, реагируют более нислз
N-алкилсульфоиамиды [454]. Это различие ясно видно из приведенного ниже др
мера, где метилирование одновременно сульфоинлироваиного и ацетилирование»
зтилеидиамниа происходит исилючительио по сульфонамидному атому азота.
Монометилэтилендиамин [455]. Сначала получают моноацетилэтиле
диамин, который образуется при многодневном стоянии 528 е етилацетата с 155(
70%-ного водного раствора этилендиамина. После гомогенизации смеси провод
двукратную франциоиную перегонку; выход продукта 365 г (60% от теоретик
ского; т. кип. 125—130° С (5 мм рт. ст.).
К раствору 306 в моноацетилэтилендиамина в равном количестве вод
при перемешивании медленно прибавляют по каплям одновременно 530 г бензо
сульфохлорида и 1200 е 10%-ного водного раствора NaOH. Оставляют смесь i
несколько часов, слегка подкисляют минеральной кислотой и отфильтровывай
выпавший амид (№бензолсульфо-№-апетилэтиленднамин).
Для перекристаллизации сульфамида его растворяют в разбавленном этп
ловом спирте и фильтруют горячий раствор для отделения незначительна
примеси дисульфосоединения. Выделение маслянистого продукта предотвращай
применением большого количества растворителя; выход 500 а (70% от теорет:
ческого); т. пл. 103° С (испр.).
Для метилирования 121 а 1\7-бензолсульфо-1\7'-ацетилатилеидиами^
растворяют в эквивалентном количестве кипящего спиртового раствор
КОН (35 а 85%-пого КОН в 200 мл спирта) и к полученному раствору в течен]
15 мин приливают по каплям 142 г метилиодида и нагревают 2 ч. После охлаящ
ння отфильтровывают выделившийся KI (около 60 г) и отгоняют с паром метилм.
дид и этиловый спирт. Остающийся метилированный амид омыляют кипячение
в течение 12 ч с 500 мл концентрированной HCI в колбе с обратным холодИльнэ
ком, добавляя периодически немного свежей концентрированной НС1. Гидрола
зат упаривают в вакууме почти досуха. После добавления твердого NaOH *
концентрированного водного раствора отгоняют полученный амиш К ;фтилля]
прибавляют твердый NaOH и сушат выделившийся амин сначала над свежщ
NaOH, а затем над металлическим натрием при кипячении с обратным холодил]
ником. После перегонки с иолоикой получают 28 г (80% от теоретического
монометилэтилендиамина; т. иип. 115—116° С.
Для расщепления сульфамидов [456] обычно требуются еще более жестка
условия, чем для расщепления амидов карбоновых кислот. Такое расщепление дост*
гается многочасовым нагреванием их с концентрированной НС1 в колбе с обратнее
[453] A. Pictet, Р. Стёр ie их, Вег., 21, 1106 (1888).
[454] О. Н i п а Ь е г g, Ann., 265, 178 (1891).
[455] S. R. A s p i n a 11, J. Am. Chem. Soc., 63, 852 (1941). ’
[456] S. Searles, S. N ukina, Chem. Rev., 59, 1077 (1959).
422
ОБРАЗОВАНИЕ С—N-СБЯЗИ Б РЕЗУЛЬТАТЕ ОБМЕНА
холодильником, лучше в запаянной трубке при 150° С, или продолжительным нагре-
ванием с концентрированной H2SO4. Толуолсульфамиды, по-видимому, довольно
гладко расщепляются хлорсульфоновой кислотой 1457] при нагревании в течение
2—3 ч при 130—150° С. В отдельных случаях проводилось также восстановительное
расщепление толуолсульфамидов натрием и кипящим бутиловым спиртом или цинком
и соляной кислотой [458]. Ниже нриводится пример восстановительного расщепления,
очень удобного в работе.
Метилаллиламин [459]. К 43,2 г мелкоизмельченного КОН в небольшом^
количестве спирта прибавляют 143 г метиламида n-толуолсульфокислоты и затем
85 г аллилхлорида; смесь при этом разогревается до кипения (обратный холо-
дильник!). После растворения большей части щелочи и замедления реакции
смесь нагревают на паровой бане в течение 6 ч. Затем отгоняют спирт и аллид-
хлорид, образовавшееся масло декантируют с выделившейся соли и перегоняют
в вакууме. Выход М-метил-К-аллил-п-толуолсульфамида 155 г (89% от теоретн--
ческого); т. кип. 190—193° С (12 мм рт. ст.); 1,5340.
К кипящему раствору 45 г М-метил-К-аллил-п-толуолсульфамида в 500 мл>
бутилового спирта мепяртлто прибавляют 46 а натрия в виде небольших кусочков
и нагревают 1 ч в колбе с обратным холодильником. После охлаждения добавляют'
воду и отгоняют амин в приемник с соляной кислотой; перегонку прекращают,',
когда начинает перегоняться нейтральный дистиллят. Содержимое приемник»';
упаривают в вакууме досуха, обрабатывают концентрированным растворов
NaOH и отгоняют метилаллиламин; выход 6,7 г (48% от теоретического); т. кип.
65° С; «а» 1,4065.
Другим препаративным способом перевода первичных аминов во вторичные
является кватеринзация оснований Шиффа, образующихся в качестве промежуточных
продуктов. Четвертичные соли, получающиеся почти с количественным выходом при;
продолжительном нагревании первичных аминов с низшими алкилгалогенндам»1!
в запаянной трубке, легко гидролизуются до вторичных аминов. Для получения освоца-г
ний Шиффа обычно применяют бензальдегид. Таким способом с отличными выходам»;
синтезировали ряд р-фенилпропилалкиламинов [460], Упоминавшийся ранее метилаль?
лиламин был получен, исходя из аллиламина [461], через апил с выходом 71%.
Метилнониламин. Единственным пригодным методом оказался следуй
ющий [462]. Нагревают 9 г нониламина (т. кип. 85° С при 13 мм рт. ст.) и 7
бензальдегида на водяной бане 1 ч, образующийся аинл перегоняют в вакуум®|
(т. кип. 179° С при 14 мм рт. ст.). Полученный анил (13,6 а) запаивают в трубК$|
с 8,4 е йодистого метила, через 1 ч помещают в водяную баню и нагревают в те-с
чепие 3 ч. К образовавшейся в виде темно-красного вязкого масла четвертичной]
соли приливают 40 мл 90%-ногО этилового спирта и нагревают 1 ч на водяной^
бане. После этого отгоняют спирт, остаток подкисляют и экстрагируют эфпроИ?
бензальдегид. Основание выделяют из кислого раствора щелочью и перегоняю»^
выход продукта 6,6 г; т. кип. 95° С (14 мм рт. ст.).
Вторичные амины были получены алкилированием цианамида с последующим^
омылением. Преимуществом метода является дешевизна и доступность исходного^
цианамида кальция. -,>1
[457] W. Marckwald, A. v. Droste-Hiilshoff, Вег., 31, 3261 (1898X1
[458] D. К lamann, G. Hofbauer, Chem. Вег., 86, 1246 (1953). I
[459] A. W. West on, A. W. ’Ruddy, С. М. S u t е г, J. Am. Chem. Soc.*i
65, 674 (1943). 1
[460] Е. Н. Woodruff J. Р. Lam bo оу, W. E. Bust, J. Am. Chem Я
Soc., 62, 922 «(1940).
(461J A. L. Morrison, H. Rinderknecbt, J. Chem. Soc., 1950, 1478. a
[462] II. King, T. S. Work, J. Chem. Soc., 1942, 401. 1
1
II. ОБМЕН ГАЛОГЕНА НА АЗОТ
Днбензпламин [463]. В 100 мл 70%-ного спирта суспендируют 14,(
тонкоизмельченного технического цианамида кальция, содержащего 50% осн с
лого вещества, прибавляют 25,3 г бензилхлорида и нагревают 12 ч в колбе с с
ратным холодильником. После этого осадок отфильтровывают, промывают спи
том и спиртовой фильтрат насыщают НС1. Полученный раствор нагрева:
в автоклаве при 140° С в течение 4—5 ч. При этом дибензиламин выделяет
в виде гидрохлорида, кристаллизующегося в форме крупных листочков; вых:
13 г (55% от теоретического).
Аналогичным образом был получен [464] диаллилцианамид с выход,
51 — 56%. Он гидролизуется разбавленной H2SO4 до диаллиламина; выход око
85% от теоретического.
Кватеринзация третичных аминов под действием алкилгалогенидов в больше
стве случаев протекает без осложнений, если реакция не затруднена стерически]
факторами и основные свойства амина достаточны. Из эквимолекулярных количес
третичного амина и алкилталогенида, лучше всего в метиловом спирте или в ацетои
четвертичные соли чаще всего получают уже на холоду или при кипячении с обратив
холодильником; выходы хорошие или даже количественные. Длд их выделения ра
твор упаривают и добавляют эфир.
Получение простых четвертичных аммониевых солен с разными эамесг
телями описано и в более новых работах [465, 466]. Здесь же следует упомяну
о широких возможностях применения в препаративных целях N-алкилированнь
пиридинов [467].
Более сильными алкилирующими средствами, чем простые алкилгалогенид]
являются галогенкетоны и галогенальдегиды, а также галогензамещенные проста
эфиры и карбоновые кислоты.
Алкилирование а-галогензамещенными простыми эфирами имеет препарата
ное значение лишь в некоторых особых случаях и часто сопровождается побочный,
реакциями, например отщеплением галогеноводорода (см. обзорные статьи [468]
При проведении реакции с галогензамещениыми кетонами и альдегидами сл
дует иметь d виду, что эти соединения могут реагировать с аммиаком и первичные
-аминами и по карбонильной группе. Обычно такие реакции исключают ацетализаци*
карбонильной группы.
Диэтилацеталь аминоацетальдегида [469]. Раствор хлорацеталя в мет:
ловом спирте и 18 моль безводного аммиака нагревают в аппарате для рабох
под давлением в течение 10 ч при 140° С; выход 74% от теоретического.
В аналогичных условиях получены также ацетали а-амино- и а-мети.
аминопропионового альдегида [470] с выходами соответственно 55 и 40%
[463]
[464]
[465]
[466]
[467]
[468]
[469]
'[470]
W. Traub е, A. Engelhardt. Вег., 44, 3149 (1911).
Е. В. V 1 1 е t, Org. Synth., 5, 43, 45 (1925).
М. Finkelstein, R. С. Peterson, S. D. Ross, J. Am. Chen
Soc., 81, 2361 (1959).
E. Grovenstein jr., E. P. Blanchard jr. et al., J. A'm. Chen
Soc., 81, 4842 (1959).
F. Krohnke, Angew. Chem., 65, 605 (1953); 74, 811 (1962); 75, 18-
317 (1963).
L. Summers, Chem. Rev., 55, 326 (1955); H. В a g a n z, Angew. Chem.
71, 366 (1959).
R. B. Woodward, W. E. Doering, Org. Synth., Coll. v. 2
50 (1955).
J. R. J о h ii s о n, A. A. Larsen et al., J. Am. Chem.' Soc., 69, 236
(1947).
424
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Ароматические амины целесообразнее применять в виде их N-метялличяг.ип^
производных.
При алкилировании вторичных и третичных аминов галогензамещениыми пото-
пами или альдегидами защита карбонильной группы в большинстве случаев ие обяза-
тельна.
а-Днметиламиноизомасляный альдегид [471]. К 300 г холодной
35%-ного водного раствора диметиламина приливают по каплям 40 г д-бромизр.
масляного альдегида, поддерживая температуру не выше 10° С. Через сутиИ
продукт экстрагируют эфиром, сушат над MgSO4 и перегоняют с колонной;
выход 9,9 г (32% от теоретического); т. кип. 126—129° С.
Диметиламиноацетон [472]. К раствору 45 г диметиламина в 1 л сухогО
эфира в течение 15 мин при 2—10° С добавляют при прррмаптиряпии раствор
46,3 г хлорацетона в 100 мл сухого эфира. Перемешивают еще 7 ч при комиатнся
температуре и оставляют на 3 суток. После этого отфильтровывают выдадпД
шинся гидрохлорид диметиламина, промывают его эфиром и сушат фильтруй
вместе с эфиром от промывки над Мд8О4. После отгонки эфира остаток перегм
няют в вакууме; выход 37,5 г (74% от теоретического);!, кип. 35—36еИ
(25 лл рт. ст.)-, п2^ 1,4128. |
Аналогичным способом (в некоторых случаях при нагревании) с хороши™
выходами получен ряд аминокетонов [473—475].
В качестве примера алкилирования аминов галогепзяметенным нитрилом
веден синтез y-N-пнперкдвнбутиронитрила [476].
y-N-Пинеридинбутиронитрил. В колбе емкостью 5 л, снабженной мешада
кой, обратным холодильником и капельной воронкой, нагревают до кипений
600 г (7 моль) пиперидина в смеси с 500 мл сухого бензола и 100 мл хлороформу
и к кипящей смеси прибавляют в течение 30 мин 310 г (3 моль) у-хлорбутнрон*
трила. После 5 ч кипячения смесь оставляют на ночь, отфильтровывают выдели^
шился гидрохлорид пиперидина н промывают его эфиром. Объединенные фиЯ|
траты перегоняют на колонне до температуры 108° С в парах. Дополинтельа
выделившийся гидрохлорид пиперидина отфильтровывают от охлажденной
остатка и промывают эфиром; общее количество гидрохлорида пиперидина с
ставляет 350 в (96% от теоретического). Фильтрат и промывной эфир перегоняв
и получают 393 в y-N-пиперидинбутнронитрила (86,2% от теоретического^
т. кип. 129—131° С (25 мм рт. ст.)', 1,4653.
Аналогично из у-бромбутиронитрила и диэтиламина получают у-диэтн
аминобутиронитрил; выход 86% от теоретичесиого; т. инп. 101—103е
(21 мм рт. ст.)-, 1,4351. |
Кларк и Мошер [477] нашли," что добавление 0,05 мол. % Nal облегчай
ведение реакции, и получили таким образом из у-бромбутиронитрила 96,7?
у-диэтиламинобутиронитрила. £
Без применения растворителей обычно выходы хуже [478]. Тем не мей!
из Е-бромкапронитрила был синтезирован е-диэтиламиноиапронитрнл [47i
с выходом 90% от теоретического; т. кип. 102—102,5° С (4 мм рт. ст.). '
[471]
[472]
[473]
[474]
[475]
[476]
[477]
[478]
Е. R. Alexander,!. Am. Chem. Soc., 70 , 2592 (1948).
H. E. Z a u g g, В. W. Horrom, J. Am. Chem. Soc., 72, 3004 (1950).
J. W. Magee, H. R. Henze, J. Am. Chem. Soc., 60, 2148 (1938.
H. R. Henze, Ch. B. Holder, J. Am. Chem. Soc., 63, 1943 (1941).
R. C. Elderfield, Ch. Ressler, J. Am. Chem. Soc., 72,
(1950).
F. C. Whit more, H. S. Mosher et al., J. Am. Chem. Soc., 66,
(1944).
D. E. Clark, H. S. Mosher, J. Am. Chem. Soc., 72, 1026 (1950).
W. P. U term ohlen jr., C. S. Hamilton, J. Am. Chem.
63, 156 (1941)’.
[479] S. Breslow, Ch, H. Hauser, J. Am. Chem. Soc., 67 , 686 (1945).
II. ОБМЕН ГАЛОГЕНА НА АЗОТ
4:
Во многих случаях аминокислоты можно синтезировать взаимодействием гая
гензамещениых жирных кислот с аммиаком. Часто тание синтезы проводились св
обычно высоким избытком 'аммиака. Однако Фишер [480] получил хорошие выход
всего лишь с пятикратным иоличеством аммиака при непродолжительном нагревай®
в запаянной трубке.
Аланилглицин [480]. К-(а-Бромпр'опионил)-глицин нагревают в запаяз
ной трубке с пятикратным количеством 25%-ного водного аммиака в течет
20 мин при 100° С. Реакционную массу упаривают, добавляют спирт и повтори»
упаривание для полного удаления воды. При обработке остатка горячим абсолкк
ным спиртом (60 мл на 5 г исходного вещества) весь выпавший NH4Br через в
которое время растворяется. Остающийся дипептид растворяют в минимально
количестве горячего 50%-ного спирта, фильтруют и к полученному раствор
прибавляют абсолютный спирт до появления мути. При длительном столиц
аланилглицин-кристаллизуется в виде коротких игл; выход 87% от теоретик
ского; т. пл. 228° С (разл.).
При аммонолизе простых галогензамещенных жирных кдслот выходы обычв
ниже; здесь, очевидно, следует применять большой избыток /аммиака. Установив®
[481], что добавка (NH4)2COS повышает ;выход первичных аминокислот. Ниже прив,
дены прописи получения а-аминокислот с числом углеродных атомов от 2 до 6.
а-Аминомасляная кислота [481]. 450 г карбоната аммония (8 лм>л
аммиака) нагревают с 140 мл воды до 55° С, перемешивают и после^охлаждеий
до 40° С прибавляют 410 мл водного раствора аммиака (6 лсоль аммиака.
При этой температуре медленно, в течение 30 мин добавляют 167 г а-броммаслл
ной кислоты. Реакционную массу нагревают 24 ч при 40—50° С, отгоняют в
водяной бане NH» нСО2 н затем упаривают раствор в чашке до начала выделена
аминокислоты. После охлаждения раствор фильтруют и осадок дважды промь
вают на фильтре небольшим количеством метилового спирта. После упариванй
фильтрата до объема 125 мл и прибавления 250 мл метилового спирта получавс
вторую порцию продукта. Общий выход чистой аминокислоты 59—62 е (57-~
60% от теоретического). . '
О синтезе аминокислот из галогензамещенных жирных кислот и жиднох
аммиака см. [482].
N-Алиилзамещенные аминокислоты получают точно так же — в результат
взаимодействия галогензамещенных жирных кислот с алкиламинами. Так, например
при стоянии в течение двух дней смеси 5 в Б-а-бромпроиионовой(иислоты с 15 ле
33%-иого водного раствора метиламина получается L-N-метилаланин с выходом 71Э
от теоретического [483].
Для синтеза N-алкилированных аминокислот применяется также описание
выше алкилирование сульфамидов [483, 484]. Фишер получал N-метиллейцин из лейще
на метилированием его N-толуолсульфонильного производного метилнодидом [484
Выделение свободных аминокислот часто затрудняется присутствием ябразс
вавшихся в результате реакции или добавленных аммонийных солей; легче вядотях:
эфиры аминокислот *.
Этиловый эфир а-метилдминомаеляной кислоты [485]. Смесь 56 а а-брок
масляной кислоты н 1,5 л 36%-ного водного раствора метиламина перемешиваю»
[480
1481
[482
[483
[484
[485
Для выделения свободных аминокислот из нх гидрохлоридов удобно пользе
ваться окисью этилена [В. М. Родионов, Н. Г. Ярцева, Из®
АН СССР, ОХН, 108 (1950)]. - Прим. ред.
Е. Fischer, W. Axhausen, Ann., 340, 130 (1905).
N. D. Cheronis, К. H. Spitzmeuller, J. Org. Chem., 6, 349, (1941'
H. H. S i s 1 e r, N. D. Cheronis, J. Org. Chem., 6, 467 (1941).
E. Fischer, L. v. Mechel, Ber., 49, 1355 (1916).
E. Fischer, W. Lipschitz, Ber., 48. 860 (1915).
N. J. Leonard, W. V. R u у 1 e, J. Am. Chem. Soc., 71, 3094 (1949).
426
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
при комнатной температуре в течение четырех суток. После этого в вакууме,
создаваемом водоструйным насосом, отгоняют избыток водного метиламина,
причем регенерируется около 1,34 л 29%-ного раствора метиламина. К остатку
прибавляют 39 е КОН в виде 50%-ного водного раствора и 200 мл 95%-ного
спирта и упаривают в вакууме досуха. Для полного удаления метиламина
п воды к остатку еще раз добавляют 150 мл спирта и упаривают в вакууме. Эту
операцию повторяют еще два раза. Образовавшийся сиропообразный, частично
закристаллизовавшийся продукт размешивают с 500 мл абсолютного спирта,
пока весь сироп не растворится, а кристаллы останутся. Полученный раствор
насыщают сухим газообразным НС1, оставляют на ночь, затем кипятят 1,5 ч
в колбе с обратным холодильником и отгоняют спирт в вакууме досуха. К остатку
добавляют 300 мл эфира и охлаждают смесь в бане со льдом, затем медленно,
при хорошем перемешивания добавляют холодный раствор 50 г КОН в 50 мл
воды, следя за тем, чтобы температура не поднималась выше 10° С. После этого
колбу закрывают и энергично встряхивают. Желтоватый эфирный слой Отделяют
и повторно экстрагируют аминоэфир сначала 200 мл эфира, затем трижды пор-
циями эфира по 100 мл каждая. Объединенные эфирные вытяжки сушат над
MgSO4. В результате пепегонки получают 30,4 е (63% от теоретического) амино*-
эфира; т. кип. 64—65,5^С (20 мм рт. ст.); nty 1,4174.
Несколько меньшей реакционной способностью отличается галоген в жирных-
кислотах, находящийся не в a-положении. Для получения метилового эфира у-диэтил-
аминомасляной кислоты у-хлорппоизводное переводили действием Nal в соответ-
ствующее у-иодпроизводное [486].
В галогенированных лактонах обмен галогена на аминогруппу можно проводить,
без одновременного раскрытия кольца [487, 488].
Особенно легко протекает, конечно, кватернизация под действием галогенза--,
мещеиных производных карбоновых кислот. В качестве примера приводится получе-
ние хлорида триметилбетанпгидразида (реактив Жирара Т) [489]
[(СИз)8Й—снв—co-nhnh2]ci-
Хлорид триметилбетаингидразида. Растворяют 984 г этилового эфира;
хлоруксусной кислоты и 280 г сильно охлажденного безводного триметиламина
в 2 л абсолютного спирта. На следующий депь к раствору прибавляют еще 200 в
триметиламина и через 24 ч при хорошем перемешивании в один прием приливают
400 $ гидразингидрата. После прекращения сильно экзотермичной реакции
помещают реакционную массу в холодильник. Выпавшее вещество выделяют^
защищая его от влаги воздуха. Из маточного раствора при упаривании получаю^
вторую фракцию. Выход продукта около 90% от теоретического; т. пл. 192** <3£
(разл.).
При алкилировании гидразинов моно- и диалкилгидразины, как правило, алкж-л
лируются даже легче, чем сам гидразип. Прн этом, так же, как и при алкилирования^
аммиака, лишь в отдельных случаях удается получить моно-* и диалкилгидразины,,
с удовлетворительными выходами [490]. Проще всего получаются К,К,К-триалкилгт
гидразиниевые соли [491, 492]. Относительно легко алкилируется гидразингидрата
в щелочной среде действием хлоргидринов, давая гидразиноспирты [493]. С
1(1Л:
Мопоаралкилгидразины удобно получать по методу А. И Коста, Р. С. Сагитул-j
липа и Сунь К)й-шань [ЖОХ, 30, 3280 (i960)]. — Прим. ред. 1
F. F. В 1 i с k е, W. В. Wright jr., М. F. Z i е n t у, J. Am. Chem».!
Soc., 63, 2488 (1941). . !
J. E. Li vak, E. C. Britton et al., J. Am. Chem. Soc., 67, 2218 (1945),*
W. Keil, Hoppe-Seyler’s Z. physiol. Chem., 171, 242 (1927). 1
A. Girard, G. Sandulesco, Helv. chim. acta, 19, 1095 (1936)? A. G Hl
raid, Org. Synth., Coll. v. 2, 85^(1955). J
ver., <**, (ин/. -i
G. M. Omietanski, B. Rudner, Chem. Rev., 5ТЛ:
[486]
[487]
[488]
[489]
[490] О. WestplTai, Вег., 74, 759 (1941).
[491] Н. И. S i з ] е г, С - - - • ------’
1021 (1957). ,
[492] С. Harries, Т. Haga, Вег., 31, 56 (1898).
[493] G. G е v е г, J. Am. Chem. Soc., 76, 1283 (1954).
J
II. OSMEH ]\1ЛОГЕЯА НА АЗОТ
После металлирования натрием в жидком аммиаке фенилгидраэин лег
и с хорошими выходами взаимодействует с алкилгалогеиндами, образуя N-алкил-
фенилгидразины [494]. При алкилировании гидразина М,М'-диалкилгидразины вооб]
не образуются, но они относительно гладко получаются При алкилировании N.N'-д
ацилгидразинов и последующем омылении [495—498]. Об алкилировании моноацг
гидразинов см. [499].
Более гладко реагируют с гидразином ct-галогенкарбоновые кислоты.
Этиловый эфир а-гидразинуксусной кислоты [500]. К 500 г (5 мо4
водного 32%-ного раствора гидразина прибавляют в несколько приемов 94,£
(1 моль) хлоруксусной кислоты. Через 48 ч добавляют 85 а (около 2 моль) NaC
и упаривают реакционную смесь в вакууме, причем вновь освобождается око
4 моль гидразина. К сухому остатку прибавляют 650 мл 30%-ного спнртово
раствора НС1 и осторожно нагревают смесь до кипеиня. После этого охлажда;
смесь и насыщают газообразным НС1 (приблизительно в течение 2 ч), добавля]
500 мл абсолютного спирта, нагревают до кипения и фильтруют горячий раствс
Прн охлаждении кристаллизуется гидрохлорид гидразиноэфира в количест
103 г (66,5% от теоретического); т. пл. 150—153е С.
В заключение следует упомянуть, что алкилирование гидрокснламина алкила
дидами протекает также до Й,М-диалкилпроизводных, которые, особенно их визит
гомологи, в большинстве случаев очень легко реагируют далее с образованием окис
триалкиламинов [501].
Арилирование
Ароматически связанный галоген с трудом вступает в реакции *. Если атоь
галогена не активированы сильными электроноакцепторными заместителями в орт:
или пара-положеини, то они располагаются по возрастающей подвижности в такой >
ряд, как алкилгалогеинды. В препаративной работе практически применяются лит]
хлор- или бромзамещенные ароматические соединения, взаимодействие которых с а.
миаком или аминами протекает только в жестких условиях, и то лишь в ирису
ствни катализаторов — медного порошка или солей меди. Особенно хорошим катал:
затором является медь Ренея из сплава Деварда [502]. Каталитическое действие ме;
в этой реакции, известной под названием реакции Ульмана, может быть усилено сл
дами иода. Для связывания кислоты обычно применяют сухой К2СО3, в качестве ра
творитсля удобнее всего применять нитробензол. В таких условиях удается да>з
провести арилирование дифениламина.
Трифениламин [503]. В колбу емкостью 2 л, которая снабжещ^ффе
тивной мешалкой и обратным воздушным холодильником длиной окол^ю5 с-
[494]
495
496
497
498
499
500
[501
[502J
[503]
Имеются в виду реакции нуклеофильного замещения. — Прим. рвд.<
L. F. A u d г i е t h, J. R. W e i s i g e г, H. E. Carter, J. Org. Chettj
6, 417 (1941).
C. D. Harries, Ber., 27, 2276 (1894).
T. Fo] praers, Rec. trav. chim., 34, 34 (1915).
H. H. Hatt, Org. Synth., Coll. v. 2, 208 (1943).
H. C. R a m s p e г g e r, J. Am. Chem. Soc., 51, 918 (1929).
R. L. Hinman, M. C. Flores, J. Org. Chem., 24, 660 (1959).
A. Carmi, G. P о 1 1 a k, H. Yellii, J. Org. Chem., 25, 44 (I960).
W. R. Dunstan, E. Goulding, J. Chem. Soc., 75, 792 (1899),
K. Bauer, Chem. Ber., 83, 10 (1950).
F. D. Hager, Org. Synth., 8, 116 (1928).
428
ОБРАЗОВАНИЕ G—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
соединенным с нисходящим холодильником Либиха, помещают 176 а мелко*
измельченного дифениламина, 204 г иодбензола, 1 л ие содержащего кислой
нитробензола, 138 е мелкоразмолотого безводного КаСО8 и 5 а медного катализа*
тора (можно применять обыкновенную бронзу). При сильном перемешиваичЙ
реакционную смесь нагревают так, чтобы нитробензол конденсировался в верхи
ней части воздушного холодильника. Небольшая часть его при этом попадав
в нисходящий холодильник. Этот дистиллят время от времени сушат над Na8SC§
и возвращают в реакцию. Реакция заканчивается^ерез 12—24 ч, когда прекрда
щается выделение воды. Нитробензол и избыток дифениламина отгоняют м
колбы емкостью 5 л с паром. Собирают дистиллят (около 8—10 л), заканчивая
перегонку, иогда 1 л дистиллята содержит менее 5 г нерастворимых в воде м|
ществ. Из остатка при охлаждении кристаллизуется неочищенный трифенвин
амин, который дважды промывают 400 мл воды, Неочищенный продукт экстрЯйй
руюх 1 л бензола и затем еще треия порциями бензола (по 75 мл каждая), Энм
тракты отделяют от основного количества воды и перегоняют до тех пор, поиа 1М
начнет отгоняться прозрачный бензол. -Затем к остатку приливают сухой 6ЙВ
зол до объема 1,2 л, фильтруют, охлаждают и пропускают в него гавообрЯ
ный НС1. Через несколько часов отфильтровывают выпавший гидрохлррйд
дифениламина и промывают его бензолом. После отгонки бензола от объеднздя
иых фильтратов отгоняют в вакууме неочищенный трифениламин, польвущИ
короткой колонкой и достаточно широким воздушным холодильником. Перм|
фракция состоит из небольшого количества бензола и сильноокрашеннцх ЦВМ
месей, затем перегоняется трифениламин; выход от 220—235 г; т. кип. 195—205уЯ
(10—12 мм рт. ст); т. пл. 120—124° С. Продукт перекристаллизовывают Я
700 мл этилацетата; общий выход после выделения продукта из маточных рае
творов 200—210 в (82—85% от теоретического); т. пл. 126° С. ЧЦ
Аналогичным способом из карбазола получают N-фенилкарбазол с выхЦ
дом 88% от теоретического [503]. ‘ $8
Если исходным продуктом является не дифениламин, а N-февилантранилм
кислота, то с хорошим выходом получается трифениламин-о-карбоновая кислота [50
Сама N-фенилантраниловая кислота может быть также приготовлена по реакции Уз
мана из о-хлорбензойной иислоты с выходом около 90% от теоретического [502,
Часто вместо свободных аминов с большим успехом применяются их N-ацетя
производные.
•м-Нитродифениламин [506]. Растворяют 12 а .м-нитроацетанилида и 21
бромбенэола в нитробензоле и прибавляют 5 а К2СО3. Добавляя иеболы»
количества KI и медиого порошка, смесь скачала нагревают 10 ч при 18у*
затем в течение 8 ч медленно повышают температуру до 210° С. Содержимое "“Я
подвергают перегоняв с водяным паром и получают в остатке коричневое мае!
которое при охлаждении сильно густеет. Водный слой отделяют, маслообраяй
растворяют в этиловом спирте, фильтруют и для омыления иипятят в течей
3 ч с 20—25 мл концентрированной HG1. При добавлении воды л-нитродифеЙ
амин выделяется в виде коричневого масла, которое в скором времени нрней
лизуется. Выход 11,3 г (80% от теоретического). После перекристаллизация г
этилового спирта получают'кирпично-красиые кристаллы; т. пл. 112° С.
Значительно легче протекает взаимодействие неактивированиых галогенза^
щенных ароматических гпрдиярпий с металлированными аминами. В простейшем Й
чае достаточно проводить взаимодействие в присутствии амида щелочного мета®!
N-Фецилпиперидин [507]. В колбе с обратным холодильником нагреве!
7,8 г амида натрия в 30 мл сухого пиперидина в течение 15 мин, затем через обр|
[504] I. Goldberg, М. Nimerovsky, Вег., 40, 2448 (1907).
[505] С. F. Н. Allen, G. Н W. McKee. Org, Synth., СоП. v. 2, 15 (1943). J
[506] I. Goldberg, Вег., 40, 4541 (1907). ь
[507[ J. F. В u n n e t, T. К. В г о t h e г t о n, J. Org. Chem., 22, 832 (1957).
II. ОБМЕН ГАЛОГЕНА НА АЗОТ
ный холодильник прибавляют по каплям 15,7 г бромбензола; при этом выделяет!
аммиак и начинается бурная реакция, которая затихает после окончания доб
вления бромбензола. В заключение смесь нагревают 2 ч до кипения, охлаясдй]
и осторожно прибавляют 25 мл воды и 25 мл бензола. Бензольный слой отдела!
и промывают тремя порциями 10%-ной соляной яислоты (по 25 мл каждая
Объединенные солянокислые экстракты сильно подщелачивают коицеНтрировц
ным раствором КОН; при этом выделяется масло, которое извлекают 15 j
эфира. Водный слой встряхивают с тем же количеством эфира, эфирные вытяжд
объединяют и после отгонки эфира перегоняют остаток. Получают 16 а (991
от теоретического) N-фенилпиперидина; т. кип. 93—97° G (3—4 мм рт. ст,
1,5593.
N-Цнклогексиланилин [508[. Растворяют 12 а металлического нале
в 100 лм жидкого аммиака и приливают к этому раствору 100 мл cyxoj
циклогексиламина. После испарения аммиака прибавляют 47 е сухого 6poj
бензола, причем смесь разогревается и окрашивается в коричневый цвет. Е
нагревают при 120—130° С и перемешивании еще 3 ч, затем к темноокрашенис
смеси добавляют 100 мл воды и выделившийся огранический слой промывац
150 мл воды. Амии из объединенных водных вытяжек экстрагируют 100 *
эфира. Эфирный экстракт присоединяют к органическому слою и сушат на
Na2SOj. Растворитель отгоняют при нормальном давлении. Остаток перегоняй;
в вакууме; выход циклогексиланнлина 22 а (42% от теоретического); т. них
142—147° G (9 мм рт. ст.). Следующая фракция, отгоняющаяся при 198—205е
(9 мм рт. ст.), вероятно, представляет собой циклогексилдифениламин.
Тем не меиее применение этой реакции арилирования к замещенным и конденсб
рованным галогенэамещенным ароматическим соединениям очень сильно ограниченс
поскольку в большинстве случаев в качестве промежуточного продукта получаете,
арин [509], что обычно приводит к перегруппировкам. Если подобная перегрушю
ровка протекает количественно, то она иногда может иметь препаративное значение
.м-Анизидин (получение из о-хлоранизола) [510]. К раствору 0,85 молл
амида натрия в 800 лм жидкого аммиака в течение 30 мин прибавляют по каплях
60 г о-хлоракпзола, смесь перемешивают еще 30 мин и разлагают избытоэ
амида натрия при помощи NH4CL Растворитель испаряют, из остатка вкстрап»
руют эфиром амин и из эфирного раствора осаждают его в виде гидрохлорида.
Выход 30 а (57,7% от теоретического).
Реакционная способность ароматически связанного галогена значительно новы?
шается при наличии нитрогрупп в орто- или пара-положении; меньшее влияние ог^азк
вают карбоксильные, сульфо- и циангруппы. В активированных таним образом арома.
тических галогензамещенных подвижность атомов галогена имеет обратный порядок:
так атом фтора обладает максимальной подвижностью. В препаративном отидрениЕ
этот факт используется, например, для определения при помощи 2,4-дннитроф’^убен-
зола N-концевых аминокислот в пептидах [511]. Легкость протекания реакции видке
из следующего примера.
2,4-Динитро-ГН0-оксиэтил)-анилин [512]. Встряхивают 0,5 г йпвах-
амина и 1 a NaHCOg в 10 мл воды с раствором 0,8 г 2,4-динитрофторбензолщ
в 20 мл этилового спирта при комиатной температуре в течение 3 ч. Для удаление
[508] R. A. S е i b е г t, F. W. В е г g з t г о m, J. Org. Chem., 10, 544 (1945).
[509] R. Н u i s g е n, J. Sauer, Angew. Ghem., 72, 91 (I960).
[510] H. Gilman, S. Avakian, J. Am. Ghem. Soc., 67, 349 (1945).
[511] F. Sanger, Biochem. J., 39, 507 (1945); A. L. L e v y, D. C h u n g, J. Auk
Chem. Soc., 77, 2899 (1955).
[512] w. Graft mann, H. H б г m a n n, H. Endres, Chem. Ber., 86^.
1477 (1953).
430
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
избытка динитрофторбензола прибавляют 0,2 г гликоля, встряхивают еще 2 <
отгоняют в вакууме спирт и добавляют к остатку воду. Затем продукт экстрам
руют эфиром, эфирный экстракт упаривают и остаток перекристаллизовываю
из этилового спирта; получают 1,62 г (86,4% от теоретического) красно-желты
игл; т. пл. 92° С.
Бройнигер и Шпангенберг [513] описали большое число 2,4-динитрофенильны
производных амипов, полученных таким способом.
Очевидно, что реянципттяя способность галогена повышается с увеличение)
числа нитрогрупп в орто- или пара-положениях к нему. Так, например, реакци
с n-нитрохлорбензолом протекают еще в довольно жестких условиях. <
4-Нитродиэтиланилин [514]. Нагревают 100 г (1,4 моль) диэтиламшз
с 97,8 а (0,6 моль) 4-нитрохлорбензола в качающемся автоклаве при 175е С в Т|
чение 8 ч. Продукт реакции выливают в 1 л воды, отфильтровывают осадок, прв
мывают его водой, растворяют при 60° G в 200 мл 20%-ной НС1, фильтру^
через стеклянный фильтр и кз фильтрата осаждают аммиаком 114 г (94% от
ретического) неляпптрнпог» 4-нитродиэтиланилнна с т. пл. 71—73° С. После nepj
кристаллизации из этилового спирта получают 85 г продукта с т. пл. 76° С.
При проведении реакции с гидрохлоридом амина можно избежать применена
избыточного давления. .л
4-Нитродиметиланн.тин [173]. В колбе емкостью 500 мл к 42 г п-бром
китробепзола, 300 мл пиридина и 50 г NaHCOs прибавляют раствор 30 г гидр&
хлорида диметиламина в 10 мл теплой воды и нагревают 10 ч смесь до кипений
Затем отфильтровывают выделившийся NaBr и промывают его 200 мл ацетом
Фильтрат объединяют с промывным ацетоном, нагревают до кипения и прим
вляют к смеси воду до появления мути. При охлаждении выделяются иглм
которые перекристаллизовывают из метилового спирта; т. пл. 164° С. Вторя
фракция кристаллов может быть выделена после упаривания маточиого раствОв
до 1/3 исходного объема. Общий выход 32,4—33,6 а (94—97% от теоретической^
Аналогичным способом из o-хлорнитробензола получают о-питродиметнж
анилин; выход 85%; т. кип. 149° С (20 мм рт. ст.). -43
Пропись получения 2,4-динитроанилина из динитрохлорбензола см. [51.
Для проведения этой реакции при нормальном давлении динитрохлорбензол cnj
вляют с ацетатом аммония при 170° С и над полученным расплавом в течение 6 ч nj
пускают аммиак. По другому методу дииитрохлорбензол сплавляют с мочевиной [5Й
Пикрнлхлорид соответствует по реакционной способности алкилгалогенида
Линке [517] провел реакции большого чксла ароматических аминов с пикрилхлор
дом, оставляя их взаимодействовать в течение 15 ч в спиртовом растворе при 7’’
и получал соответствующие производные всегда с практически количественным выход®
Реакция пикрилхлорида с анилином в этилацетате в присутствии NaHCO3 была испйй
зована даже для количественного определения анилина [518].
Гидразин, подобно аммиаку и аминам, тоже легко реагирует с активирований
галогснзамещенными ароматическими соединениями. 2,4-Динитрофенилгидразин [51
часто применяемый в качестве реактива на карбонильную группу, получают в во®
спиртовом растворе из 2,4-динитрохлорбензола и гидразина с выходом выше 80
Таким же образом можно арилировать гидразоны, что представляет интерес в ч
случаях, когда 2,4-динитрофенилгидразоны образуются непосредственно из кето®
с большим трудом или вовсе не образуются. В качестве примера, который можно И * 2
С. L. В еЬт, J. Е. К i гЪ у et al., J. Am. Chem. Soc., 68, 1296 (1946). ,,
F. B. Wells, C. F. H. A lie n, Org. Synth., Coll. v. 2, 221 (1943).
515
516
517
518
519
513] Н. В rauniger, К. Spangenberg, Pharmazie, 12, 335 (1957). J
514 С. L. В е h т, J. Е. К i г Ъ у et al., J. Am. Chem. Soc., 68, 1296
2 „..-.К - n ..
Англ. пат. 263552; С., 1927, I, 2013.
В. Linke. Вег., 56, 848 (1923).
В. L i n k е, Н. P re issecker, J. Stadler, Ber., 65, 1280, 1282 (193
C. F. II. Allen, Org. Synth., Coll. v. 2, 228 (1943).
II. ОБМЕН ГАЛОГЕНА НА АЗОТ
4<
прострапить и на другие соединения, описывается перевод гидразона ацетона в ег
2,4-дипитрофенилгидразоп [520].
2,4-Динитрофенилгидразон ацетона. Растворяют 1,5 г (0,02 моль) гидр
азона ацетона в 10 мл 95%-ного спирта и прибавляют к раствору 4,0 г (0,019 мол\
2,4-динитрохлорбензола в 30 мл 95%-ного этилового спирта. Смесь нагреванэ
1 ч до кипения, охлаждают и фильтруют. 'Остаток промывают небольшим коле
чеством теплого спирта для удаления непрореагировавшего динитрохлорбензол
и сушат. После однократной перекристаллизации из 95%-ного этилового спирт
получают 2,4-динитрофенилгидразон ацетона; т. пл. 123° С.
Ацилирование
Среди производных кислот (стр. 445—463), применяемых для ацилирования азот
содержащих соединений, важную роль играют галогенангидриды кислот.
Из галогенангйдридов кислот практическое применение находят лишь соответ
ствующие хлорангидриды. Часто бывает достаточно вводить неочищенный хлоранги
дрид [521], получаемый из кислоты и РС13, или SOC1,. Для ацилирования аммиак^
проще всего растереть хлорангидрид кислоты с (NH^)2CO3, нагреть эту смесь на води
ной бане для завершения реакции и отмыть от солеи аммония холодной водой. Реко
мепдуется также проводить взаимодействие хлорангидридов кислот с ацетатом аммо
ния при комнатной температуре в ацетоне [521а]. По другому способу хлорангидри,]
медленно приливают по каплям к избытку концентрированного водного раствора
аммиака при хорошем охлаждении и интенсивном перемешивании. Затем смесь ещ<
некоторое время перемешивают и упаривают в вакууме досуха. Ввиду растворимости
амидов низших карбоновых кислот в воде их следует отделять от NH4G1 при помоще
органических растворителей. Для этого можно применять, например, абсолютный, эти:
ловый спирт; для выделения изобутирамида очень удобно употреблять кипящий этил
ацетат [522], из которого прн охлаждении изобутирамид кристаллизуется с выходок
около 80%. Особенно удобно проводить синтез с водным раствором аммиака, ио, по
йятно, лишь в тех случаях, когда образующийся амид нерастворим в воде и epaaj
выпадает в осадок, как, например, а-фенилбутирамид [523]и р-нафтилацетамид [524]
При проведении синтеза в органическом растворителе можно избежать разделе*
ния амида и NH4G1 экстрагированием растворителем. Чаще всего в качестве раствори-
телей применяют бензол, петролейный эфир и хлороформ; в эфире часто получаются
более низкие выходы. При проведении синтеза не следует вводить газообразный ам-
миак в раствор хлорангидрида кислоты, поскольку при этом иногда могут образо-
ваться значительные количества диацилированиых производных; поэтому хлорангид-
рид к раствору аммиака рекомендуется приливать по каплям. При использовании
органических растворителей можно после окончания реакции отделить NH.C1 филь-
трованием или при получении нерастворимых в воде амидов отмыть NH4C1 водой.
Метод ацилирования в неводных растворителях, который обычно дает высокие вы-
ходы и чистые продукты, демонстрируется па приведенном ниже примере; таким же
способом был получен ряд других амидов всегда с хорошими или даже отличныма
выходами.
Бутирамид [525]. В трехгорлой колбе емкостью 500 мл с обратны»®
холодильником, защищенным От влаги воздуха, капельной воронкой и газо-
проводящей трубкой, нагревают на кипящей водяной бане 250 мл сухого бензола.
Сухой аммиак пропускают в бензол со скоростью около 250 мл!мин. Одновременна
в течение 1 ч приливают по каплям раствор 21,3 г хлорангидрида масляной кис-
лоты в 50 мл сухого бензола, пропускают аммиак еще 30 мин, отфильтровывают-
15201 L. 1. Braddock, М. L. Willard, J. Org. Chem., 18, 313 (1953).
[521) О. A s с h а и, Вег., 31, 2344 (1898).
521а] V. a. Finan, G. A. Fothergill, J. Chem. Soc., 1962 , 2824.
[522] В. Е. Kent, S. И. McElllia, Org. Synth., Coll. v. 3, 490 (1955).
1523] s. M. M с E 1 v a i n, C. L. Stevens, 1. Am. Chem. Soc., 69, 2663 (1947).
'524| M. s. Newman, J. Org. Chem., 9. 518 (1944).
1525] G. e. Phil brook, J. Org. Chem., 19, 623 (1954).
432
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
образовавшийся NH4C1 и дважды промывают остаток 25 мл сухого бензола.
Фильтрат и промывной бензол упаривают до объема 75 мл и к кипящему раствору
осторожно добавляют петролениый эфир (т. кип. 30—60° С) до начала кристалл
лизации амида. Еще раз нагревают до исчезновения мути и быстро охлаждают.
Выделившийся бутирамид отфильтровывают на воронке Бюхнера и дважды
перекристаллизовывают таким же образом из бензола и петролейного афера
и сушат на воздухе. Выход 13,7 а (78,7% от теоретического); белые кристаллы
без запаха; т. пл. 115—116° С.
Ацилирование первичных и вторичных аминов может в принципе проводить^
.аналогично ацилированию аммиака; при этом одна молекула амина расходуется на
связывание соляной нислоты и теряется. *
И в этом случае в зависимости от растворимости аминов реакцию можно весЩ
как в водной, так и в безводной среде. Так, например, для получения Ы-метиламид<й
низших жирных кислот [528] соответствующие хлорангидриды приливали но каплет
к взятому с избытком водному раствору метиламина при температуре от —10 до —20° Q]
Для получения многих диэтиламидов преимущественно ароматических кислот опращ
дала себя следующая общая методика [527]. J
Диэтиламиды. К раствору 0,2 моль сухого диэтиламина в 100 мл сухога
эфира соответствующей кислоты медленно приливают по каплям 0,1 моль хлор!
ангидрида при хорошем охлаждении н интенсивном перемешивании. Поел*
этого смесь перемешивают еще 1 ч при комнатной температуре и в заключен^
добавляют холодную воду до полного растворения гидрохлорида аминя. Орг®
ническнн слой отделяют и промывают последовательно порциями по 100 яш
5%-ной НС1, водой, 5%-ным раствором NaOH, опять водой и насыщенным рш
твором NaCl. После сушки, фильтрования и отгонки эфира продукт перегоняв
в вакууме. Выходы обычно высокие. 'й
Вместо эфира можно применять также другие неводные растворятся®
как, например, бензол или хлорзамещенные углеводороды; хлорангидрид и аМЯЯ
рекомендуется растворять в одном и том же растворителе. ‘
При ацилировании труднодоступных аминов выделяющийся галогеноводорвй
•связывают не самим амином, а более дешевым основанием. При проведении синим
в органическом растворителе можно применять для этой цели сухие карбонаты щещя
яых металлов.
2-Беиэоиламино-н-бутанол [528]. Смесь 500 мл бензола, 25 г сухвЯ
Na8CO3 и 18,3 г 2-амино-«-бутанола охлаждают до 10° С. Затем к этой смеси ml
перемешивании прибавляют по каплям раствор 28,1 г бензоилхлорида в 1002Я
бензола с такой’скоростью, чтобы температура не превышала 10° С. ПримеиД
через 45 мин прибавляют еще 25 г Na2CO3 и опять перемешивают 4 ч при КЙЯ
и затем 4 ч при комнатной температуре. В заключение реакционную Мй4я|
кипятят в колбе ?с обратным холодильником до исчезновения запаха беНзЛМ
хлорида. Неорганический осадок отфильтровывают и промывают дважды/100ет
Snnero бензола. Объддттченныя бензольные растворы упаривают до обмЯ
мл н охлаждают для кристаллизации. Вторую фракцию кристаллов получи
после упаривания маточного раствора до объема 150 мл. Общий выход пР°Д?зд|
34—35 г (90% от теоретического); т. пл. после промывки бензолом 97—9ЭТЯ
после перенристаллизации нз толуола 98—99° С.
Почти количественное ацилирование аминов в отсутствие агентов, связ^Д
тощих кислоты, достигается длительным кипячением их гидрохлоридов с хйорМ
гидридом нислоты в инертном растворителе до прекращения выделения галогеновой
рода [529—531]. дя
[526] G. F. D А 1 е 1 i о, Е. Е. В е i d. J. Am. Chem. Soc., 59, 109 (1937).
[527] E. T. McCabe. W. F. Barthel et al., J. Org. Chem., 19, 493 (1954),
[528] J. H. В illman, E. E. Parker,]. Am. Chem. Soc., 66, 538 (1944).
[529] H. Franken, Ber., 42, 2465 (1909),
(530JH. Suida F. Drahowzal, Ber., 75, 991 (1942).
(5311 E. Ronwln, J. Org. Chem., 18, 127, 1546 (1953). *
II. ОБМЕН ГАЛОГЕНА НА АЗОТ
Бензанилид [529]. В колбе с обратным холодильником в течение 8
кипятят смесь 9 г гидрохлорида анилина, 9 г бензоилхлорида и 50 мл бензола. П
истечении этого времени выделение HCI закапчивается. Реакционную масс
•хлаждают, выпавший амид промывают бензолом и водой и перекрнсталлизовг
вают из спирта. Выход почти количественный; т. пл. 162° G.
Этот метод применим для ацилирования как ароматических [529], так и алиф;
•гических яминод [530], и при ацилировании больших количеств алифатических амии*
удобнее, чем описываемый ниже метод Шоттена — Бауманна [530]. Таким я
способом можно ацилировать аминокислоты [529, 531], представляющие собой ва;
тренние сопи.
При ацилировании по известному методу Шоттена — Бауманна [532] реакци
проводят в водной суспензии с гидроокисями щелочных металлов, которые связывай
выделяющиеся кислоты. Поэтому метод особенно пригоден для ацилирования труда
гидролизуемыми ароматическими хлорангидридами. Большей частью бывает дост
точпо встряхивать в течение некоторого времени амин, суспендированный в раство]
едкого натра, с хлорапгидридом при комнатной температуре; иногда для завершена
реакции и гидролиза непрореагировавшего хлорангидрида реакционную массу
заключение непродолжительное время нагревают и образовавшийся амид отфильтр
вывагот или экстрагируют органическими растворителями. Поскольку реакционн,-
масса должна оставаться слабощелочной до конца реакции, а хлорангидрид все >з
в некоторой степени гидролизуется, щелочь и хлорангидрид обычно вводят с 25%-нь
избытком.
При ацилировании алифатическими или другими очень реакционноспособных
хлорангидрпдами рекомендуется применять охлаждение. В этих условиях тоже бол
шей частью достигаются высокие выходы.
N-Метилизобугирамид [533]. Осторожно смешивают раствор 320
(8 моль) NaOTI в 1 л воды с раствором 278 г (4 моль) гидрохлорида метилами^
в 690 мл воды и охлаждают. К смеси при перемешивании прибавляют по капля
424 а (4 моль) хлорангидрида изомасляной кислоты с такой скоростью, чтоС
температура не превышала 10° С. После этою доводят температуру до коми^
ной, отделяют слой амида, а из водной фазы три раза экстрагируют амид эфире
порциями по 200 мл. Слой амида и эфирные растворы объединяют, сушат и пер
гоняют. Выход амида 303 г (75% от теоретического); т. кип. 120—121° С (27
рт. ст.); 1,4350.
Аналогично получают N-этильпое производное [533] (т. пл. 65—67’
и N-метилмстакриламид [534].
Реакция Шоттена — Бауманна часто применяется для ацилирования амил
кислот, особенно для их бензоилирования или введения карбобензоксигруппы.
Универсальный способ бензоилирования аминокислот по Шопену — Баумана
и обзор ранней литературы приводит Штайгер [535]. Отдельные прописк дляйбензо
дарования глицина [536] и е-амияоканроновой кислоты см. [537]. "
Ниже приведен пример синтеза карбобензоксиаминокислот, имеющих болып<
значение для получения пептидов; N-ацильный остаток этих кислот легко подвергает,
гидрогенолитическому отщеплению [538].
С. Schotten, Вег., 17, 2544 (1884); 23. 3430 (1890); Е. Baumann, Беа
19, 3218 (1886); L. v. Udransky, Е. Baumann, Ber., 21, 2744 (1885
О. Hinsberg, L. v. U J г a n s к у, Ann., 254, 252 (1899).
S. М. М с Е 1 v a i n, С. L. Stevens, J. Am. Chem. Soc., 69, 2&
(1947).
J. Ileyboer, A. J. Staverman, Rec. trav. chim., 69, 787 (1950).
R. Steiger, J. Org. Chem., 9, 396 (1944).
A. W. Ingersoll, S. H. Babcock, Org. Svnth., Coll. v. 2, 328 (194;
J. С. E c k, C. S. Marvel, Org. Synth., Coll. v. 2, 76 (4943).
M. Bergmann, L. Z e г v a a, Ber., 65, 1192 (1932).
Заказ 183&.
434
4 ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
N-Карбобензоксиглицин [539]. К охлаждаемому льдом раствору 7,5
глицина в 50 мл 2 н. раствора NaOH при перемешивании в течение 20—25 ми;
одновременно прибавляют по каплям 17 г бензилового эфира хлормуравьино,
кислоты и 25 мл 4 н. раствора NaOH. После этого смесь перемешивают ещ
10 мин, примеси экстрагируют эфиром и подкисляют водный слой по конг
концентрированной НС1. Осадок отфильтровывают, промывают небольшим коли
чеством воды и после сушки на воздухе перекристаллизовывают из хлороформа
Выход 18—19 г; т. пл. 120° С.
п-Нитро-[540] и п-бромкарбобензоксиаминокислоты [541] обычно легче кри
сталлизуготся, чем незамещенные карбобензоксиаминокислоты, но и они легко подвер
гаются гидрогенолитическому расщеплению. Известны также защитные группы амил
ных функций, сообщающие веществу окраску, имеющие особенно большое значёни
при хроматографической очистке аминокислот, и пептидов, см. [542].
Для связывания образующегося по реакции Шоттена — Бауманна галоген?
водорода вместо едкого натра или кали применяют гидроокиси магния [538], кальцп
[543], бария [544], бикарбонат натрия [545] и ацетат натрия. Во многих случал
отличные результаты получены при использовании пиридина, который иногда можв
одновременно служить также растворителем (см., например, [546]).
Метиловый эфир п-(п'-нитробензоиламино)-бензойной кислоты [547]
К раствору 2.6 г метилового эфира ге-аминобензойной кислоты в 20 мл cylor
хлороформа, не содержащего метанола, прибавляют раствор 2,9 г п-нптробензои£
хлорида в 10 мл хлороформа и затем 5 мл абсолютного пиридина. Смесь рад?
гревается и начинается выделение светло-желтого вещества. Через 15 ч (й®
20° С) осадок отфильтровывают. Выход 4,6 г (98% от теоретического), посд
перекристаллизации из этилацетата т. пл. 244° С. ,2
Диэтиловый эфир бензомламиномалоновой иислоты [548]. К смеси 5$?
диэтилового эфира аминомалоновой кислоты, 50 мл пиридина и 250 мл воды ни
ливают по каплям при температуре не вьппе 50° С и перемешивании 48.5 г бензои
хлорида. Смесь охлаждают до комнатной температуры, отфильтровывают вЫ2Ш
тий осадок, сушат его в вакууме и растворяют в минимальном количеств
бензола. Из полученного раствора осаждают продукт медленным добавлеВте
шестикратного объема петролейного эфира; выход 92,4 г (90% от теоретическом!
т. пл. 73—74° С. Ц
При ацилировании в пиридине по методу Эйнгорна в некоторых случаях моя
отказаться от применения готовых хлорангидридов, так как они могут получювЯ
в процессе синтеза. Для этого кислоту совместно с амином растворяют в пиридв
добавляют тионилхлорид (SOC12) и оставляют на некоторое время при охлажден
после чего смесь выливают в воду и промывают выпавший амид последовательно
бавленной кислотой разбавленной щелочью и водой.
. • ;
[539] Н. Е. С а г, t е г, R. L. F г а п k, Н. W. I о h в s t о n, Org. М
Coll. v. 3, 167 1955).
[540] F. Н. Carpenter, D. Т. G i s h, J. Am. Chem. Soc., 74, 3818 (19f
D. T. Gish, F. H. Carpenter, J. Am. Chem. Soc., 75, 950 (1953).;
[541 ] D. M. Channing P. B. Turner, G. T. Young, Nature (Lon<*
167, 487 (1951). ’ „
[542] R. Schwyzer, P. Sieber, K. Zatskd, Helv. chim. acta, 41, 491 (18
[543] J. Biehringer, A. Busch, Ber., 36, 137 (1903).
[5441 A. E t a г d, A. V i 1 а, С. r., 135, 698 (1902).
[545[ E. Fischer, Ber., 32, 2454 (1899).
[546] R. Walther, St.- Wlodko wski, J. prakt. Chem. [2], 59, 266 i(18
(547] R. Kuhn E. F Moller, G. Wendt, II. В einert, Ber.,-*
717 (1942). - ‘ \
[548] С. E, Redemann, M. S. Dun n, J. Biol. Chem., 130, 345 (1939)* ;
II. ОБМЕЛ ГАЛОГЕНА НА АЗОТ
Иногда вместо пиридина можно использовать и другие третичные амины, и
пример триэтиламин [549] или диэтиланилин [550].
Следует указать также на возможность образования кетенов из хлорангидрид
алифатических кислот и третичных аминов [551].
Высокая реакционная способность галогена, связанного с карбонильной гру
пой, позволяет почти всегда применять для ацилирования по описанным выше снос
бам даже такие галогенангидриды, в молекуле которых имеются и другие функцв
нальные группы. Полное исключение нежелательных побочных реакций достигает
в этом случае выбором достаточно низкой температуры проведения реакции. Так, и
взаимодействии а-бромацетилбромида [552] и а-ёромпропионилбромида [553], а так?
дихлорацетилхлорида [554] с разными аминами в этиленхлориде при температуре
— 10 до —20° С были получены с высокими выходами соответствующие амиды, прич
кислоты связывались избытком амина.
Монохлорангидриды эфиров дикарбоновых кислот могут быть переведены в с
ответствующие моноамиды эфиров дикарбоновых кислот с сохранением сложноэфи
яой группы. Так, например, хлорангидрид монометилового эфира себациновой кв
лоты реагирует с водным аммиаком с образованием эфирамида с 95%-ным выходу,
[555]. Для получения уретанов часто пользуются реакциями эфиров хлормуравьинг
кислоты с аммиаком [539, 556], первичными [557] и вторичными аминами [55?
Во многих случаях можно проводить амидирование дихлорангидридов как
одной, так и по двум функциям. Хлорангидриды карбаминовой кислоты, образу
щиеся из фосгена и аммиака или аминов в строго определенных условиях [559], пр
менялись для синтеза амидов ароматических карбоновых кислот по реакции Фр
деля — Крафтса и для получения несимметрично замещенных мочевин.
Гораздо проще идет двустороннее амидирование фосгена в симметричные мо*
вины. Особый интерес приобрело взаимодействие имидазола с фосгеном, приводят
к образованию N.N'-карбонилдиимидазола [560], обладающего значительной реакцис
ной способностью вследствие подвижности амидных связей.
Ввиду легкости обмена имидазольных остатков на другие группы это соедие
цие может быть широко использовано для синтеза в исключительно мягких условд,
-других производных ’угольной кислоты, эфиров, амидов и гидразидов карбоиова
кислот, пептидов, эфиров фосфорных кислот и аналогичных соединений [560а]. Г]
скольку карбонилдиимпдазол гидролизуется уже влагой воздуха, его получеа
должно проводиться при тщательном исключении доступа влаги.
М,1Ч'-Карбонилдиимидазол [560Ь]. В приборе на шлифах растворяют 1(
имидазола в 120 мл безводного тетрагидрофурана. К этому раствору из граду
рованной пипетки для кислот прибавляют 2,50 мл фосгена, конденсированно
в ловушке, охлаждаемой смесью ацетона с сухим льдом. Тщательно исключу
доступ влаги, отсасывают выпавший гидрохлорид имидазола па воронке с nopt
тым стеклянным фильтром и упаривают в вакууме тетрагидрофурановый раста
досуха. Выход М.М'-капбонилциимидазола 90—95% от теоретическо*
т. ил. 116—118° С.
Вместо тетрагидрофурапа иногда применялись и другие инертные раста
рители, например бепзол [561].
II. Suter, И. Zut ter, Н. W i d 1 е г, Ann., 576, 223 (1952).
Р. R u g g I i, J. Rohner, Helv. cbim. acta, 25, 1533 (1942).
W. E. Hanford, J. C. Sauer, Org. Reactions, 3, 124 (1946).
W. E. Weaver, W. M. Whaley, J. Am. Chem. Soc., 69, 515 (1947) .
W. E. Weave r, W. M. Whale y, J. Am. Chem. Soc., 69, 1144 (194“
A. D. Swensen, W. E. Weaver, J. Am. Chem. Soc., 70, 4060 (194?
W. S. Bishop, Org. Synth., Coll. v. 3, 613 (1955).
W. M. Kraft, R. M. Herbst, J. Org. Chem., 10, 483 (1945).
W. W. II art man, M. R. Bre then, Org. Synth., Coll. v. 2, 278 (194Z
C. S. Marvel, W. A. No yes.-J. Am. Chem. Soc., 42, 2276 (1920).
H. П о p f f, H. О h 1 i n g e r, Angew. Chem., 61, 183 (1949).
H. A. Staab, Ann., 609, 75 (1957).
Обзорная статья: В. A. S t a a b, Angew. Chem., 74, 407 (1962).
II. A. Staab, K. W e n d e 1, Chem. Ber., 93/ 2902 (1960).
B. Paul, G. W. Anderson, J. Am. Chem. Soc., 82, 4596 (I960).
28*
436
ОБРАЗОВАНИЕ G—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Для получения тиокарбонилдиимидазола к раствору имидазола в хлоро-
форме прибавляют по каплям бензольный раствор тиофосгена [561а].
Дихлорапгидрид малоновой кислоты и дизамещенные аналоги его взаимодейст-
вуют с сильноосновными аминами с образованием преимущественно диамндов соответ-
ствующих малоновых кислот. Замыкание кольца в циклические имиды удается лишь
При взаимодействии дихлорангидридов дизамещевпых малоновых кислот со слабым»
аминами ароматического или гетероциклического ряда; аналогично реагируют
N.N-дизамешснные гидразины с образованием 3,3-дизамещеиных 1-аминоацетидиндио-
нов-2,4 [562].
СО
R\
R^
R'
R"
Так же гладко реагируют с первичными аминами галогениды синильной кис-
лоты. Для получения цианамидов ату реакцию рекомендуется проводить с бромциа-
ном. Во избежание образования гуаиндинов следует применять интенсивное охлаждение.
Бензилцианамид [563]. К раствору 4,3 г бензиламина при охлажденив-
в бане со льдом и перемешивании медленно прибавляют по каплям раствор--
2,1 г бромциана в 14 мл сухого эфира. Гидробромид бензиламина отфильтровы-
вают и упаривают фильтрат в вакууме досуха. Получают 2,6 г (95% от теорети-
ческого) неочищенного продукта; т. пл. 42—44° С.
Дизамещенные цианамиды часто выгоднее получать не взаимодействием вто-
ричных аминов с бромцианом, а подвергая третичные амины так называемому расще-
плению по методу Брауна [564]. При этом обычно в виде алкилбромида отщепляете^
самый короткий алкильный остаток.
N-Метил-Д[-гт.-пйфтилп1ианямид [565]. В вытяжном шкафу в течение 16
нагревают на паровой бане до кипения 171 г .Ч.М-диметил-а-нафтиламина-
и 125 г бромциана и выливают продукт в 2,5 л сухого эфира; при этом выпадает’
•садок образующегося в качестве побочного продукта бромида триметил-а-;
нафтиламмония (выход 10 г, т. пл. 160° С). Осадок отфильтровывают, фильтра?-,
иромывают четыре раза примерно 15%-ной НС1, порциями по 800 мл каждая,.,
и пять раз порциями воды по 500 мл. Эфирный раствор сушат 30—35 г безводного*
CaSO4 и после отгонки эфира перегоняют остаток в вакууме; выход продукт»’
115—122 г (около 65% от теоретического). Н-Метил-М-а-нафтилцианамид пред*"
ставляет собой слегка желтоватое масло с т. кип. 171° С (1 мм рт. ст.) или 185--*
187°С(Злл рт. ст.). .
Менсе гладко, чем с аммиаком, первичными и вторичными аминами, протекав
взаимодействие хлорангидридов кислот с гидрокскламином, гидразином, третичныг
амилами и амидами кислот.
[561а] Н. A. Staab, G. W а 1 t h е г, Ann., 657, 98 (1962).
[562] А. Е b no t her, Е. J ucker et al., Helv. chim. acta, 42, 918 (1959).
[563] H. K. N a g y, A. J. T о m s о n, J. P. Horwitz, j. Am. CheJ
Soc., 82, 1609 (1960).
[564] H. А. П a g*e m a и. Org Reactions, 7. 198 (1953).
1565] H. W. J. Cressman. Org. Synth., Coll, v, 3, 608 (1955).
II ОБМЕН ГАЛОГЕНА ПА АЗОТ
4
Лишь в редких случаях гидроксамовые кислоты готовят взаимодействием гид
оксиламина с хлорангидридами кислот [566, 567]; гидроксиламин более целесообраз]
ацилировать эфирами карбоновых кислот.
Гидразиды карбоновых кислот тоже проще синтезировать гидразниолнзом ело:
вых эфиров, поскольку из хлорангидридов кислот часто таким образом (по-видимом
особенно легко в водной среде) получаются ТЧ.Х'-диацильные производные^ [56?
Так, по реакции Шоттена — Бауманна [497] с выходом 70% от теорет
' ческого получают М,М'-дибензоилгидразин, имеющий большое значение
1 синтеза 1Ч,Ы'-диалкилгидразинов; в то же время карбобензоксигидразв
& применяемый иногда для синтеза пептидов, получают взаимодействием эфи;
f хлормуравьиной кислоты с избытком гидразина в хлороформе [569]. Между те
, в водном растворе образуется в основном N.N'-дикарбобензоксигидразин [56?
Третичные алифатические амины не ацилируются простыми хлорангидрида^
* карбоновых кислот. Если все же реакция и происходит, то от хлорангидридов отпз
'ё нляется хлористый водород и образуются кетены [551], которые легко подвергают!
дальнейшим превращениям. Этот метод вряд ли имеет какое-либо препаративное зв
•L чение для получения кетенов, хотя в некоторых случаях взаимодействие протеку
Г с хорошими выходами, например при синтезе дифенилкетена [570]. Иногда галогенд
в ангидриды кислот оказывают на третичные амины дезалкилирующее действие [57]
Ацилирование третичных алифатических аминов под действием хлорангидрид,
Г ' кислот с образованием солей триалкилациламмония удалось осуществить лишь ие т<
давно [572] в присутствии SbCl6; при этом действительным ацилирующим агент<
г следует считать гексахлорантимоиат карбоксония:
h HCOCl + SbCls —- |RCO)+[SbCl,]- -ЛЕ», [BCO-NB;i+|SbCl,r
Такие триалкилациламмониевые соли являются более сильными ацилирующие
агентами, чем сами хлорангидриды кислот.
В отличие от третичных алифатических аминов пиридин N-ацилируется дал
в отсутствие комплексообразующих кислот Льюиса [573, 574]. (Обзор, посвящении
солям N-ацилпиридиния, см. [467].)
Вследствие пониженной основности амидов кислот их ацилирование, естес
венно, протекает с большим трудом, но значительно облегчается в присутствии пирз
дина [546, 575, 576]. Возможно, что причиной такого сильного ускорения реакцк
является промежуточное образование упоминавшихся выше солей Ы-ациллиридин®
[577]. Так, дибензамид получается из бензоилхлорида и бензамида в среде пиридин
с почти количественным выходом даже при охлаждении, тогда как без пиридина реа:
ция вообще не идет [578]. Томсону [575] удалось получить триацильные произвсь
ные с высокими выходами действием ароматических галогенангидридов на алифатич
ские и ароматические амиды в присутствии пиридина даже при температурах от —f
до —70° С.
Ацилирование амидов по другим методам, в которых не применяется придал
обычно проводят в гораздо более жестких условиях, чем ацилирование аминот. Таз
r
L
[566] М. A. S t о 1 b е г g, W. A. Mosher, Th. Wagner-Jauregg, J. Arj
Chem. Soc., 79, 2615 (1957).
[567] A. W. Scott, W. О К e а г s e, J. Org. Chem., 5, 598 (1940).
[568] C. Naegel i, G. Stefanovitsch, Helv. chim. acta, 11,'621 (1923
569] H. Boshage n, J. Ullrich, Chem. Ber., 92, 1478 (1959).
570] H. Staudingcr, Ber., 44, 1619 (1911).
571| O. Hess, Ber., 18, 685 (1885).
572] F. Klages, E. Z a n g o, Ann., 607, 35 (1957).
573] V. Prey, Ber., 75, 537 (1942).
574] II. Adkins, Qu. Ё. Thompson, T. Am. Chem. Soc.. 71, 2242^(1949).
[575] Qu. E. Thompson, J. Am. Chem. Soc., 73, 5841 (1951).
[576] N. О. V. S о и n t a g, Chem Rev., 52, 308 (1953).
1577] W. Hiickel, Ann., 540, 283 (1939).
[578] A. W. Tit her ley, J. Chem. Soc., 85, 1673 (1904).
438
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
например, для моно- и диацилирования мочевины галогенангидридами жирных кислот
смесь нужно продолжительное время нагревать [579, 580].
н-Бутирилмочевина [579]. К смеси 10 г мочевины и 25 мл бензола при-
бавляют две капли концентрированной НаЗОл и нагревают до кипения, приба-
вляют к этой смеси 15 г м-бутирилхлорида в течение 15 мин при перемешива-
нии и нагревают еще 4 ч. При охлаждении выделяется ацилированная мочевина,
которую отфильтровывают и промывают раствором NaHCOs и водой; выход
75—85% от теоретического; т. пл. 173—174° С.
В аналогичных условиях моноацилированные мочевины могут подвер-
гаться дальнейшему ацилированию по второму атому азота' [579, 580].
Во многих случаях ацилирования амидов можно отказаться от нагревания,
если для синтеза применять предварительно металлированные амиды. Например,
Дильс [581], пользуясь этим методом, провел диацилирование этилового эфира карб-
аминовой кислоты (уретан) 2 жаль эфира хлормуравьиной кислоты и получил этиловый
эфир дикарбэтоксикарбаминовой кислоты (так называемый триэтиловый эфир азот-
трикарбоновой кислоты);
HjNCOOH +2С1СООН + 2Na N(COOJI)3-'<NaCl — н2
Для идентификации амидов кислот иногда используют их реакцию с симметрич-
ным фталилхлоридом с образованием N-ацилфталимидов [582]:
^\/С0С1 /\/С\
' I I +H3NCOR ZHcT- II I N-COR
\coci X';7\o
Амид нагревают с рассчитанным количеством фталилхлорида без растворителя
или в толуоле до прекращения выделения хлористого водорода [582]. В некоторых
случаях при синтезе N-ацилфталимидов этот путь более удобен, чем ацилирование
фталимида [583]. Например, Х-(п-толуолсульфонил)-фталимид удалось получить
только таким способом [582].
Ацилирование амидов карболовых кислот хлорангидридами карбоновых кислот
нередко сопровождается нежелательными побочными реакциями. Так, например, при
взаимодействии хлорангидридов с анилидами может произойти обмен ацильных групп
[584] или образование амидина [585]:
RCOCl + R'CO-NHCeH5 —“ R'COCl--RCO-NIlCeH5
Об ацилировании цианамида галогенангидридами см. [586].
Прочие реакции
Получение нзонианатов из фосгена и амииопроизводных [587, 588]. В то время
как реакция фосгена с избыточным количеством свободного амина приводит к образо-
[579]
[580]
[581]
582
583
584
585
586
[587]
[588]
R. W. Stoughton, J. Org. Chem., 2, 514 (1938). \
R. W. S t о u g h t о n, H. L. Dickison, 0. G. Fitzhugh, J. Am.
Clicm. Soc., 61, 408 (1939).
O. Diels, Ber., 86, 736 (1903); С. H. F. Allen, Д. Bell, Org. Synth.;
Coll. v. 3, 415 (1955).
Th. W. Evans, W. M. D e h n, J. Am. Chem. Soc., 51, 3651 (1929).
Ch. D. Hurd, M. F. Dull, J. Am. Chem. Soc., 54, 2432 (1932).
A. Pictet, Ber., 23, ЗОН (1890).
C. A. Friedmann, O. G. В a c k e h e r g, J. Chem. Soc., 1938, 469.
O. D i с 1 s, A. W a g n e r, Her., 45, 874 (1912); A. F. Crowther,
F. H, S. Curd, F. L. Rose, J. Chem. Soc., 1948, 586.
Обзорная ста<ья: W. Sielken, Ann., 562, 75 (1949).
Обзорная статья: J H Saunders, R. J. Slocomb e, Chem. Rev., 43,
203 (1948).
II ОБМЕН ГАЛОГЕНА ПА АЗОТ
ж
ванию симметричных мочевин, действие избытка фосгена на гидрохлориды первичных
аминов дает возможность получать изоцианаты. Реакция протекает при повышенной
температуре и, по-видимому, следующим образом. Сначала отщепляется НС1 и обра-
зуется хлорангидрид карбаминовой кислоты, который при высокой температуре рас-
падается на изоцианат и НС1:
RNH2 • HCI4-COCI2 _анС1- RNH—СОС1 - RNCO
Этот способ исключительно удобен в лабораторной практике и непригоден толь-
ко при фосгенировании первых членов гомологического ряда аминов. Обычно синтез
проводится в высококяпящем' инертном растворителе (толуол, ксилол, хлорбензол^
хлорнафталин!, температура кипения которого должна как можно сильнее отличаться
от температуры кипения получаемого изоцианата. Для облегчения взаимодействия
соль амина хорошо измельчают или, еще лучше, ее осаждают пропусканием газо-
образного НС1 в раствор амина в используемом для фосгенирования растворителе.
Смесь нагревают до 130—150е-С при интенсивном перемешивании (в некоторых слу-
чаях и до более высокой температуры), и пропускают фосген в суспензию (или раствор}
или, лучше, над суспензией до прекращения выделения НС1.
Фенилизоцианат [588а|. Раствор 93 г анилина в 1 л толуола при 70° С
. насыщают сухим газообразным НС1. Затем при температуре кипения пропу-
скают СОС12 до образования прозрачного раствора. Лри разгонке с эффектив-
ной колонкой получают 99 г (83% от теоретического) фенилизоцианата;
т. кип. 158—168° С.
Гексаметилендиизоцианат [587]. В 1 л дихлорбензола суспендируют
190 г тщательно размолотого дигидрохлорида гексаметилендиамина и фосгени-
руют смесь при 190—195° С. Через 18 ч образуется прозрачный раствор. После
отгоняй растворителя продукт перегоняют с колонкой Видмера и получают
160 г (95% от теоретического) гексаметилендиизоцианата; т. иип. 132° С
(15 мм рт. ст,}.
Синтез можно проводить и со свободными первичными аминами. Для этого
сначала па холоду проводят реакцию амина с избытком фосгена; обычно амин (чаще
всего раствор амина) при охлаждении прибавляют к раствору избытка фосгена.
Вместо симметричной мочевины в таких условиях образуется смесь гидрохлорида
амина и хлорида карбаминовой кислоты, которую фосгенируют далее при нагревании,
как указано выше. Этим способом был получен дифенилметан-4,4'-диизоцианат [587].
Другой вариант используется для фосгенирования сильноосновных алифатиче-
ских аминов, особенно для фосгенирования диаминов, гидрохлориды которых не взаи-
модействуют с фосгеном. В раствор диамина в подходящем для фосгенирования раство-
рителе пропускают сначала сухую СО2, причем образуется карбаминовокислая соль:
H2N-R-NH2 + CO4 —> Нз^-R—NH-COO-
которая затем под действием фосгена на холоду превращается в хлорид карбаминовой
кислоты, а при нагревании — в изоцианат [587]:
H3N-R_NH-COO- -zgJT- |H3N-H-NH-COC1]C1-OCN-H-NC^ '
Таким способом, например, получен циклогексил-1,4-диизоцианат [587].
В отдельных случаях исходными продуктами для получения изоцианатов слу-
жили также мочевины. ’ •
Фенилизоцианат [587]. При 150° С в течение 4—5 ч проводят реакцию
смеси 1,7 кг дифенилмочевины, 4 кг 1-хлориафталина и 800 г фосгена. Затем
продувают через смесь в течение 30 мин воздух для удаления задержавшегося
НС1. Чистый фенилизоцианат выделяют фракционной перегонкой; выход 1,6 кв;
т. кип. 55—57° С (16 мм рт. ст.). Остаток после перегонки можно использовать
в следующем синтезе фенилизоцианата.
Простейшие алифатические изоцианаты часто с успехом получают термическим
разложением некоторых мочевин или- уретанов.
[588а] D. V. N. Hardy, J. Chem. Soc., 1934, 2011.
440
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Метилизоцианат (587]. По реакции дифениламина с фосгеном получают
хлорангидрид дифенилкарбаминовой кислоты, который реагирует с метилями-
ном с образованием симметричной 1Ч-дифенил-1М'-мвтидмочевины. При пагрева-.
нии 226 г этой мочевины до 240—290° С получается 57 г метилизоцианата*
т. кпп. 38—40° С. Остаток после перегонки представляет собой дифенилами^,-
Особенно легко — уже при перегонке — расщепляются мочевины, получаю-
щиеся из эквимолекулярных количеств первичных аминов и 1^,№-карбонилдиимид-.
азола
r-_nh-co-n;
притом образуется изоцианат и имидазол [589].
Циклогексилизоцианат (589]. К раствору 10,06 г N,N '-карбоиилдиимид-
азола в тетрагидрофуране при перемешивании медленно прибавляют по кап-<
лям 6,15 г циклогексиламина. После отгонки растворителя в вакууме продукт
собирают в интервале 80—96° С (45 мм рт. ст., температура бани 170—185° С)}:
выход 6,41 г (83% от теоретического).
Аналогично термическим разложением тиомочевин получают изотиоцианаты;,
и в этом случае особенно хорошие результаты дает имидазолидпый способ с исполь-
зованием в качестве исходного соединения Й'.К'-тиокарбонилдиимидазола (589а].
а-Нафтилизотиоцианат [590]. В колбе с обратным холодильником нагре-
вают 16,16 г а-нафтилтиомочевины и 180 мл хлорбензола в течение 8 ч при 150° С.
Затем хлорбензол отгоняют сначала при остаточном давлении 20 лм* рт. ст.,
затем при 1 мм рт. ст. и экстрагируют а-нафтилизоцианат из остатка четырьмя
порциями гексана, по 30 мл каждая; при этом перастворенной остается смесь,,
состоящая нз 0,65 г а-нафтилтиомочевины и такого же количества ди-tz-нафтил--
тиомочевины. Из объединенных гексановых растворов после удаления раствори-
теля получают 12,75 г (86% от теоретического) а-нафтилизотиоцианата; иглы
с т. пл. 58-58,5° С.
Получение изонитрилов н формамидов пз хлороформа и аминов. Первичные
амины реагируют с хлороформом в присутствии гидроокиси щелочного металла с обра-
зованием изонитрилов *. Эта реакция, часто применяемая для идентификации первич-
ных аминов, иногда используется благодаря хорошим выходам в препаративном мас-
штабе. Реакция экзотермическая и обычно протекает уже при смешении реагентов.
Употребляя бепзол в качестве растворителя, Малатеста [591] получил из некоторых
алифатических и жирно-ароматических аминов (бутил-, изоампл-, амил-, бензиламин)
изонитрилы с выходами S0—85% от теоретического.
Получаемый ив анилина феяилизонитрил лишь с трудом поддается очистке от
непрореагировавшего анилина. Препараты, содержащие анилин, болыпен частью не
окрашены; чистый фенилизонитрил, напротив, легко полимеризуется с образованием
днавила индиго, окрашенного в синий цвет [592]. j
Феинлизоннтрил (5921. В трехгорлой колбе емкостью Зле мешалкой,;
на вращающемся шлифе и двумя холодильниками Фридриха при перемешивании-
нагревают на асбестовой сотке до начала спонтанной реакции 200 мл (2,2 моль)
* При синтезе изонитрилов не следует забывать об пх высокой токсичности (хоро-
шая тяга, перчатки!). — Прим, ред-
(5891 II. A. S I а а Ь, W. Benz Angew. Chem., 73, 66 (1961); Ann., 648, 72 (1961).
[589a] H. A. Staab, G. Walther, Anu., 657, 104 (1962).
[590] J. N. Baxter et al., J. Chem. Soc., 1956, 659.
[591] L. Malalesti, Gazz. chim. ital., 77, 238 (1947); C. A., 42, 869 (1948).
[592] Ch. Grundmann, Chem. Ber., 91, 1360 (1958).
II. ОБМЕН ГАЛОГЕНА НА АЗОТ
441
анилина, 360 мл (4,5 моль) хлороформа, 320 а (8 моль) NaOH в виде чешуек
и 10 мл абсолютного этилового спирта. Охлаждая время от времени смесь в бане
со льдом, регулируют скорость этой очень бурной реакции, поддерживая ее
в границах, определяемых мощностью обратных холодильников. Происходящее
ипогда вспенивание можно устранить только дальнейшим сниженном скорости
реакции. По завершении спонтанной реакции смесь нагревают еще 1 ч при пере-
мешивании до слабого кипения, следя за тем, чтобы непрерывно густеющее со-
держимое колбы не пристало к стенкам колбы. Общая продолжительность реак-
ции от 1,5 до 3 ч. После охлаждения смеси образовавшийся осадок отделяют на
воронке Бюхнера в вытяжном шкафу и промывают 250 мл эфира. От темно-
коричневого фильтрата отделяют небольшое количество водной фазы и отгоняют
растворители на водяной бане при температуре не выше 50° С. Остаток после
отгонки растворителей немедленно подвергают дальнейшей перегонке в вакууме
с эффективной колонкой. При 57—62° С (14 мм рт. ст.) перегоняется фенилизо-
питрил (73—75 г), затем при 62—73° С (14 мм рт. ст.) отгоняется главным
образом апилин (89 г), который может быть использован в следующем синтеае.
При повторной псрегопке с колонкой из первой фракции получают 71 г чистого
фенилизонитрила, т. кип. 53,5—54° С (13 мм рт. ст.), который в скором вре-
мени окрашивается в глубокий синий цвет, что говорит об отсутствии примеси
анилина. Выход по прореагировавшему анилину 55% от теоретического.
Об изонитрплах см. также стр. 445. Кроме того, следует упомяпуть способ их
получения из N-замещенных формамидов, основанный на отщеплении воды [593].
Вторичные амины реагируют с хлороформом в тех же условиях. Правда, в этом
случае образуются не изонитрилы, а N-дизамещеиные формамиды [594, 595].
N.N-Дибхтилформамид [594]. К 129,2 г днбутиламина и 160 мл хлоро-
форма прибавляют по каплям раствор 160 г NaOH в 1л метилового спирта (экзо-
термическая реакция, см. предыдущий синтез) и кипятят в колбе с обратным хо-
лодильником в течение 24 ч. Охлажденный раствор фильтруют. При перегонке
его в вакууме наряду с 19,9 г (15%) непрореагировавшего днбутиламина полу-
чают 102 г (65% от теоретического) Х,1М-дибутилформамида; т. кип. 63е С
' (0,1 мм рт. ст.)-, 1,4419.
2. Взаимодействие галогенпроизводных
с другими азотсодержащими соединениями
Взаимодействие с нитритами
Алкилгалогениды могут реагировать с неорганическими нитритами с образо-
ванием нитросоединений [128а]. По старым прописям для этих целей применяют искли>
чительно нйтрит серебра и ведут реакцию при повышенной температуре (80—110° С).
Однако вследствие протекания многочисленных побочных реакций удовлетворитель-
ные выходы (около 50% от теоретического) получались практически лишь прн рафе
с простейшими первичными иодидами (примерно до СО. Применение бромидов давЖо
хорошие результаты только в редких случаях [596]. Все же проведением реакции
в гораздо более мягких условиях удалось сделать этот метод достаточно экономичным.
Синтез проводят при возможно более низкой температуре в среде эфира. Из нормаль-
ных алкилиодидов и даже бромидов до Се и AgNO, получаются первичные литроал-
каны с выходами около 80% от теоретического [5971. В эту реакцию вступают также
галогениды изостроения, но только в тех случаях, когда разветвление находится ые
[593] I. U g i, R. М е у г, Chem. Вег., 93, 239 (1960); 1. U g i, W. В е t z U, F е t-
zer, K. Offerman n, Chem. Bor., 94, 2814 (1961).
1594] M. B. Frankel. H. Feuer, J. Bank, Tetrahedron Letters, Л» 7 5 (1959)*
C. A., 53, 21640 (1959); Angew. Chem., 71. 682 (1959).
595] M. Saunders, R. W. Murray, Tetrahedron, 6, 88 (1959).
1596] A. I. Vogel, J. Chem. Soc., 1948, 1847.
1597] N. Kornblum, B. Taub, IT. E, Ungnade, J. Am. Chenu Soc.. 76,
3209 (1954).
442
ОБРАЗОВАНИЕ С—N-СВЯЗП В РЕЗУЛЬТАТЕ ОБМЕНА
ближе, чем в ^-положении к атому галогена [597]. К вторичным и третичным галогени*
дам метод практически неприменим [598]. Приведенная ниже пропись синтеза 1-нйтро^
октана может быть использована для получения низших первичных нитросоединений
из подидов или бромидов и нитрита серебра.
1-Нитрооктан [597, 599]. В трехгорлой колбе, снабженной капельно!
воронкой, мешалкой и обратным холодильником, охлаждают смесь 100 «
<0,65 моль) AgNO2 и 150 мл сухого эфира до 0° С и прибавляют по каплям 120Л
(0,5 .моль) октилиодида в течение 2 ч при перемешивании в темноте. Смесь пере
мешивают еще 24 ч при 0° С и еще 36 ч или дольше при комнатной температ;
до отрицательной пробы Бейльштейна в жидкой фазе. Соли серебра отфнльт
выдают и промывают большим количеством эфира, фильтрат и промывной эс|
объединяют и отгоняют эфир с колонкой при нормальном давлении. Оста:
разгоняют в вакууме. Сначала отгоняется октилннтрит (выход 8,4 г; т. К1
51° С при 5 мм рт. ст.; 1,4124—1,4125), а после промежуточной фракг
(т. кип. 56—71° С при 3 мм рт. ст.) отгоняется 1-нитрооктан; выход 64,9
(83% от теоретического); т. кип. 71,5—72° С при 3 мм рт. ст.; 1,4321—1,432
Способ получения нитрита серебра см. [5991.
Аналогично из а-фталимидо-о-галогеналканов могут быть получены а-амшт
(й-нитроалканы, например 1-амино-5-нитропентан [600].
Раныпе для синтеза нитроалканов нитриты щелочных металлов вместо нитри:
серебра применялись редко. Примером такого синтеза может служить получение нитр
алканов по методу Кольбе, при котором в водном растворе при температуре кипени
подвергают взаимодействию соли а-галосензамещенных жирных кислот и нитрв
щелочного металла; от образующихся при этом а-питрозамощенных жирных кисло
в условиях реакции отщепляется СО2 и полученные питроалканы отгоняют с водяньй
паром. Практически синтез Кольбе применим только для получения нптрометайа а
хлоруксусной кислоты [601] (выход 38% от теоретического) и нитроэтана из a-tipoi
пропионовой кислоты [602] (выход около 50% от теоретического). С высшими галоп»
замещенными жирными кислотами синтез Кольбе почти не идет [604, 603].
Между тем при взаимодействии эфиров a-бромзамещонных жирных кисл<
и NaNO2 в водно-спиртовом растворе в течение нескольких дней прн комнатной темл<
ратуре с хорошими выходами получаются а-оксмминоэфиры (реакция Леперка [145}
Впоследствии Корнблюм [604] показал, что нитриты щелочных металлов, о©
бенно NaNO2, можно использовать вместо AgNO, для получения нитросоединеш
если применять в качестве растворителя диметил'формампд. Одним из преимуще.
этого метода является также то, что в реакцию легко вступают не только первичн]
но и вторичные алкилиодиды и алкилбромиды. И в том, и в другом случае выходы
ставляют около 60% от теоретического. Для проведения синтеза большей частью дос
точно перемешивать галогенид с избыточным количеством NaNO2 в диметилформамг
при комнатной температуре; для первичных иодидов продолжительность реаки
составляет оуоло 2,5 ч, для первичных бромидов — около 6 ч. При проведении ре»
ции с вторичными галогенидами для повышения растворимости NaNO4 в диметилфох
амиде и для сокращения продолжительности реакции добавляют мочевипу [60
Рекомендуется также добавлять флороглюцин для разложения образующихся в на’
стве побочных продуктов алкилнитритов, которые, взаимодействуя с нитросоедВД
ниями, могут значительно уменьшить их выход [604, 145, 256]. Значительное cokjH
щение продолжительности реакции достигается применением в качестве растворите?!
диметилсульфоксида. В этом случае синтез проводят без добавки мочевины или фло]Й
глюцина [604, 605]. у
(598]
[599
[600
(601
(602
(603
[604
[605]
N. Komblum, R. A. Smiley, et al., J. Аш. Chem. Soc,', 77, 5528
(1955).
N. Korn blum, H. E. Ungnade, Org. Synth., 38, 75 (1958).
V. P г e 1 о g et al., Helv. chim. acta, 43, 901 (I960). ...
F. C. W hi tin ore, M. G. Whitmore, Org. Synth., 3, 83 (1923).
V. Auger, Bull. Soc. chim. France [3], 23, 333 (1900).
W. Treibs, H. Reinlieckel, Chem. Ber., 87, 341- (1954).
N. Korn blum H. O. Larson et al., J. Am. Chem. Soc., 78, 1497-
(1956).
N. Kornblum, J. W. Powers, I. Org. Chem., 22, 455 (1957).
П. ОБМЕН ГАЛОГЕНА НА АЗОТ
445
4-Нитрогептан [604]. При перемешивании приливают 67,8 г (0,3 моль)
4-иодгептана к смеси 36 г (0,52 моль) NaNO2. 40 г (0.67 моль) мочевины и 600 мл)
диметилформамида. Умеренно охлаждая водой реакционную колбу, температуру
смеси поддерживают около 20—25° С. Перемешивание продолжают еще в тече-
ние 5,5 ч, затем реакционную массу выливают в 1,5 л воды со льдом, покрытой
слоем из 100 мл петролейного эфира (т. кип. 35° С). Водную фазу отделяют в
продукт экстрагируют четыре раза петролейным эфиром (порциями по 100 мл).
Объединенные экстракты промывают водным 10%-ным раствором NaaSOs,
четырьмя порциями воды (по 75 мл каждая) и сушат над MgSO4. После отгонки
петролейного эфира при комнатной температуре остаток перегоняют с колонной
в вакууме. Сначала отгоняется 10,9 г (25% от теоретического) 4-гептилнитрита
(т. кип. 44° С при 18 мм рт. ст.\ 1,4032), а затем 26,4 г (60% от теоретического)
4-нитрогептана (т.--йип. 59° С при 8 мм рт. ст.\ 1,4219).
Аналогичным способом из соответствующих а,а)-дибромалканов получают
а,са-динитроалканы, например 1,6-динитрогексан (606].
При проведении реакции Корнблюма с эфирами галогензамещенных карбоновых
кислот в диметилформамиде или в диметилсульфоксиде следует обязательно добавлять
флороглюцин, так как в противном случае в качестве основных продуктов реакции
получаются а-оксиминоэфиры (607] (ср. [145, 256]).
Подробную пропись получения этилового эфира а-нитромасляной кис-
лоты (выход 75% от теоретического), которую можно использовать и для синтеза
других эфиров галогензамещенных кислот, см. [608]. Эфир нитроуксуснойкис-
лоты нельзя получить таким способом [609]. Его получают [610] взаимодействием
эфира иодуксусной кислоты с AgNO2 в течение нескольких дней при комнатной
температуре. Для этой же цели рекомендуется [611] окисление изонитрозоацето-
уксусного эфира смесью К2Сг2О7 и H2SO4.
Взаимодействие .с азидами
В алкил- и ацилгалогенидах под действием азида натрия возможен обмен
атома галогена на азидный остаток, причем получаются алкилазиды.
Меры предосторожности. Самопроизвольные взрывы органических ази-
дов уже часто приводили к тяжелым несчастным случаям. Литературным данным
о предположительной устойчивости отдельных азидов, особенно низкомолеку-
лярных, не следует доверять (см., например [612]). Все работы с азидами надо
проводить с исключйтельной тщательностью и с соблюдением правил техники
безопасности, обычных при работе с перекисями и списанных на стр. 288- Кроме
того, следует напомнить, что азотистоводородная кислота ядовита.
Алкилазиды получают обычно по реакции алкилиодидов или алкилбромиЖв
с избытком азида натрия в соответствующем растворителе при нагревании [613].
(606] J. К. Stille, Е. D. V е s s е 1, J. Org. Chem., 25, 478 (I960).' .
[607] N. Korn Hum, R. K, Blackwood, J. W. Powers, J. Am. Chem.
Soc., 79, 2507 (1957).
[608] N. Kornblum, R. K. Blackwood, Org. Synth., 37, 44 (1957).
[609] N, Kornblum W. M. W e a v e r, J. Am. Chem. Soc., 80, 4335
(1958).
[610] N. Kornblum, M. E. Chalmers, R. Daniels, J. Am. Chem.
Soc., 77, 6654 (1955).
(611] .В. M. Родионов и сотр., Синтезы органических соединений,
Сборник 2, Изд. АН СССР, 1952.
[612] Ch. Grundmann, К. Haldenwanger, Angew. Chem.? 62, 410
(1950).
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Для получения алкилазидов Ся — С1П рекомендуется применять в качеств^
растворителя простые эфиры гликоля [614].
Амилазид [614]. В трехгорлой колбе емкостью 1 л с обратным холодиль
ником, термометром и мешалкой к смеси 27 8 активированного азида натрия [615]
450 мл моноэтилового эфира диэтиленглпколя (карбитол) п 75 мл воды при пере
мсшивапии прибавляют сразу 60 г чистого амилиодида. В течение несколькиз
минут все компоненты растворяются. Продолжая перемешивание, повышаю!
температуру медленно н течение 3 ч до 95° С и выдерживают раствор еще 20 ’
при этой температуре. После охлаждения реакционную массу делят на две частт
и выливают каждую из них в 600 мл ледяной воды. Водные слои отделяют л
дважды экстрагируют из пих амилазид 200 мл эфира. Эфирные растворы объеди
няют с полученными ранее органическими слоями и отгоняют эфир. Остатоа
(42 г) перегоняют в вакууме; выход амилазида 27.8 з (81,4% от теоретического};
т. кип. 77—78’ С (122 мм рт. ст.); 1,4266.
Примеры синтеза алкилазидов с использованием иных растворителей
см. [616-618].
Азиды карбоновых кислот получают обычно из гидразидов, по в принципе
можно получать и непосредственно из хлорангидридов карбоновых кислот и азида
натрия. Обычно азиды используются лишь как промежуточные продукты при получе-
нии изоцианатов, уретанов, мочевин, аминов л подобных соединений и часто подвер-
гаются дальнейшим превращениям без выделения в чистом виде.
Синтез азидов можно проводить как в инертных органических растворителях,
так и в водно-органической среде, особенно в случае ароматических хлорангидридов»
Если получаемый азид не нужно далее разлагать до изоцианата, то реакцию проводят
при охлаждении. <
лс-Нитробензоилазид [619]. К раствору 50 г ленитробензоилхлорида
в ацетоне приливают охлажденный концентрированный водный раствор 20 а
азида натрия при перемешивании и охлаждении. Через некоторое время из охла-
жденной смеси выделяется кристаллический азид. Его очищают переосаждением
из ацетона водой; выход 53 г; т. пл. 68° С.
При проведении синтеза в инертных органических растворителях скорость взаимо-'
действия становится достаточной лишь при повышенной температуре (60—80° С) воде^*
ствие плохой растворимости азида натрия. Для ускорения взаимодействия азид кат*
рия перед началом реакции активируют [615]. Однако в таких условиях от азидОЦ
отщепляется азот и они превращаются в изоцианаты. Таким способом, например?:
синтезировали метил-, «-гексил-, фенилизоцианаты и др. [620]. Пропись синтеза ундё^
цилизоцианата с промежуточным получением азида карбоновой кислоты в водной
ацетоне и его разложением до изоцианата в бензоле см. [621]. Полученные изоцианаты
часто тоже не выделяют, а подвергают дальнейшим превращениям до уретанов, аминов-
и других соединении. . , ;
[613] J. II. Boyer, F. С. С a n t е г, Chem. Rev., 54, 1 (1934). ><
[614] Eu. Lieber, Т. S. Chao, С. N. R. Rao, J. Org. Chem., 22, 238 (1957),?
[615] Р. A. S. Smith, Org. Reactions, 3, 382 (1946); ср. J. Nelles, Bet?*
65, 1345 (1932). у
[616] J. H. Boyer, J. Hamer, J. Am. Chem. Soc., 77, 951 (1955);*]
J. H. Boyer, F. С. C a n t e r, J. Hamer, R. K. Putney,;
J. Am. Chem. Soc., 78, 325 (1956).
[617] Th. Wieland, II. J. Hennig, Chem. Ber., 93, 1236 (1960).
[618] L. Horner, A. G г о B, Ann., 591, 117 (1955).
[619] C. Naegel i, A. Tyabji, Helv. chim. acta, 16, 349 (1933).
[620] G. Schroetei* Ber., 42, 3356 (1909). , 1
[621] C. F. H. A 11 e n, А. В e 1 1, Org. Synth., Coll. v. 3, 846 (1955).
Ш. ОБМЕН КИСЛОРОДА НА АЗОТ
445
Взаимодействие с цианидами
В то время как алкилирование или ацилирование легко диссоциирующих циа-
нидов металлов, например цианидов щелочных металлов, протекает почти исключи-
тельно с образованием С—С-связи, причем получаются нитрилы [622] или соответст-
венно ацилцианиды [623], недпссоциирующие цианиды могут алкилироваться и во
азоту.
Изонитрилы (получение по методу Готье). Цианид серебра нагревают
на паровой бане в течение нескольких часов с 0,5—1 моль алкилиодида. Синтез
из низкокипящих алкилиодидов проводят под давлением, при этом реакционная
масса не должна окрашиваться в коричневый цвет. Изоиитрил связывается в ком-
плекс цианидом ерребра, который после отгонки избытка алкилиодида разлагают
концентрированным раствором KCN; выделяют изонитрил перегонкой.
Подробную пропись получения этилизопитрила см. [624].
Нитрилы могут подвергаться N-алкилированию и N-ацилированию в инертных
растворителях в присутствии некоторых комплексообразующих хлоридов металлов,
например SbCls, FeCl3 и др., причем получаются соли нитрилия [625]. При щелоч-
ном гидролизе этих солей с хорошими выходами получаются N-алкилированные или
N-ацилированные амиды кислот [625].
Взаимодействие с цианатами
Эфиры изоциановой кислоты очень редко получали алкилированием алкилгало-
генидами солей циановой кислоты [626] (ср- стр. 438, 466). Несколько большее значение
имеет реакция ацилгалогекидов с цианатом серебра.
Вследствие высокой чувствительности ацилизоцианатов к влаге с ними
следует работать при тщательном исключении доступа влаги. При проведении
синтеза в среде абсолютного эфира при комнатной температуре или слабом на-
гревании [627, 628] реакция обычно заканчивается через 1 — 3 ч. Для получения
бензонлизоцианата цианат серебра и бензоилхлорид нагревают до кипения
в четыреххлористом углероде в течение 6 ч [629]. По старым прописям
для получения ацетилизоцианата [630] процесс ведут также без растворителя
при охлаждении.
111. Обмен кислорода на азот
1. Реакции азотсодержащих соединений
с производными кислот
Реакции азотсодержащих соединений со свободными кислотами •
Аммиак н первичные и вторичные амины могут ацилироваться карбоновыми
кислотами при нагревании. Отщепление воды протекает в условиях равновесной реак-
ции, по константе равновесия которой образование заметных количеств амида следует
[622]
[623]
[624]
[625]
[626
[627
[626
[629
[630
D. Т. Mowry, Chem. Rev., 42, 189 (1948).
J. Т h е s i n g, D. Witzel, A. Brehm, Angew. Chem., 68, 425 (1956).
H. L. J ackson, В. С. M c Kus ic k, Org. Synth., 35, 62 (1955).
H. Meerwein, P. L a a s c h, R. Mersch, J. S p i 1 1 e, Chem. Ber.,
89, 209 (1956).
J. J. D о n 1 e a v у, J. English Jr., J. Am. Chem. Soc., 62, 218 (1940).
A. J. Hill, W. M. Degnan, J. Am. Chem. Soc., 62, 1595 (1940).
Th. Lieser, K. Macura, Ann., 548, 243 (1941).
C. L. A r c u s, B. 8. Prydal, J. Chem. Soc., 1954, 4018.
О. C. Billeter, Ber., 36, 3213 (1903).
446
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
ожидать лишь при температурах выше 100° С (чаще всего при 180—200° С). Поэтому"
обычно одну из компонент реакции применяют в избытке (кислоту или амин) и постов
явно удаляют образующуюся воду из реакционной массы. Только при формилирова-1
нии можно применять водную кислоту. Выходы обычно высокие.
Для получения незамещенных амидов чаще всего достаточно сухой перегонки^
аммониевой соли кислоты; добавка NH4HCOa позволяет проводить реакцию при болед
низкой температуре. Хорошие выходы ацетамида достигаются при введении значащ
тельного избытка ледяной уксусной кислоты [631]. Амиды высших жирных кислой
(С8 — С1в) синтезируют пропусканием аммиака в течение 10—14 ч в нагретые до 190—Я
210° С кислоты [632, 633]. Удобным методом получения незамещенных амидов я*|
ляется также сплавление карбоновых кислот с мочевиной или тиомочевиной [634}||
Амид энантовой кислоты [635]. Смесь 1 моль мочевины с 0,5 лммш
энантовой кислоты нагревают при 170—180° С в течение 4 ч. Выход около 704^3
80% от теоретического. я
Вместо мочевины применялись также амиды других легко летучих кислота!
так, например, для амидирования олеиновой кислоты использовался формамид [63од|
N-Замещенные амиды карбоновых кислот получают аналогичным способом^
так, для синтеза И,!У-диметиламидов простых жирных кислот в нагретую кислот®!
пропускают днметиламин [633, 637]. Для синтеза бензанилида эквимолекулярным
количества анилина и бензойной кислоты нагревают до 180—190° С; выход 84% от тейД
ретического [638]. Упоминавшийся выше метод с использованием мочевины примешиЯ
и для получения N-замещенных амидов, если исходными соединениями являются
соответственно замещенные мочевины [639] или тиомочевины [639а]. ' 'Я
Анилиды карбоновых кислот [639]. Тщательно переметанные эквимолДвЯ
кулярньте количества карбоновой кислоты и симметричной дифенилмочевинфя
медленно нагревают на масляной бане в колбе Гизлера. Смесь начинает пенитывЙЗ
через 20—30 мин при температуре бани около 200—205° С. Еще через 0,5 4И
при температуре бани около 210—220° С вспенивание прекращается и реакцйжЗ
заканчивается- Реакционную массу охлаждают, растворяют в эфире, для удале&МЯ
образовавшегося анилина два раза встряхивают с небольшим избытком $%-нога|
раствора НС1 и промывают водой. После отгонки высушенного над CaCls эфирйге
остается неочищенный анилид. При получении плохо растворимых в эфире аИм
лидов образующийся анилин можно отделить промывкой эфиром. Выход анилдаЯ
дов около 90% от теоретического. чД
' Из янтарной кислоты по этому способу готовят N-фепилсукцинимид. 'Я
Отщепление воды от аммонийных солей может проводиться также в высоким
кипящих инертных растворителих, с непрерывной отгонкой образующейся вфдД
в виде азеотропной смеси. Для этой цели применялись нитробензол и ксилол. НмВ
сравнительно легко протекающем процессе формилирования, например при формплфИ
ровании N-метиланилина, реакция идет уже в кипящем толуоле [640]. ,
[631] G. Н. Coleman, А. М. Alvarado, Org. Synth., 3, 3 (1923). ^1
[632] A. W. Ralston, C. W. Eoerr, W. О. P о о 1, J. Org. Chem., 8, .4Я
(1943).
[633] J. A. M i t c h ё 1 1, E. E. R e i d, J. Am. Chem. Soc., 53, 1879 (1931). Айя
[634] E. C her b u 1 i e г, F. L a n do It, Helv. chim. acta, 29, 1315, 1438 (1948|
A.-U. Rahman, M. A. Medrano, О. P. Mittal, Rec. trav. chim., 1®
188 (1960).
[635] J. L. Gut hrie, N. Ra bjohn, Org. Synth., 37, 50 (1957). Д
[636] E. T. R о e, J. T. Scanlan, D. Swern, J. Am. Chem. Soc., 71,
(1940).
[637] J. R. Ruhoff, E. E. Reid, J. Am. Chem. Soc., 59, 401 (1937). ;£|
[636] C. N. Webb, Org. Synth., 7, 6 (1927). Ж
[639] A. R a h m a n, M. O. Farooq, Chem. Rer., 86, 945 (1953). • “Л
[639a] A.-U. Rahman, M. A. Medrano, В. B. J eanneret, J. Ог£<й9
Chem., 27, 3315 (1962). "$|
[640] L. F. F iese'r, J. E. Jones, Org. Synth., Coll. v. 3, 1955, p. 590.
Ш. ОБМЕН КИСЛОРОДА НА АЗОТ
447
Предпринималось немало попыток смягчить при помощи определенных добавок
довольно жесткие условия реакции ацилирования и сделать этот метод приемлемым
и для более чувствительных веществ. Так, при ацилировании ароматических аминов
рекомендуется добавлять небольшие количества концентрированной H2SO4 или А1С18.
Для связывания выделяющейся воды в некоторых случаях вводили Р2О6, в других —
оправдало себя добавление к аммонийной соли соответствующего кислоте ангидрида.
Правда, действие ангидрида не ограничивается связыванием воды; являясь более
сильным ацилирующим средством, он сам, несомненно, в первую очередь будет всту-
пать в реакцию ацилирования.
N-я-Пропнлацетамид [641]. В колбе емкостью 250 мл с обратным холо-
дильником^ 25 г н-пропиламина медленно прибавляют 30 г уксусной кислоты
{бурная реакция). После добавления 50 мл уксусного ангидрида нагревают смесь
0,5 ч до кипения и отгоняют уксусную кислоту с колонкой. Когда собирается
70 мл дистиллята, перегонку продолжают уже без колонки. Вслед за небольшой
промежуточной фракцией получают 34—35 г (около 90% от теоретического)
N-н-пропилацетамида; т. кип. 208° С (при нормальном давлении) или 100° С
(16 лл рт. ст.).
По чаще всего каталитические добавки значительно изменяют химизм реакции
вследствие образования с амином или кислотой особо реакционноспособ-
ного промежуточного соединения, которое часто может быть даже выделено.
Так, взаимодействие аминов с карбоновыми кислотами в присутствии РС13
происходит в гораздо более мягких условиях; эта реакция протекает не через проме-
жуточно образующийся хлорангидрид кислоты, как предполагалось ранее, а через
«фосфазопроизводное», образующееся на первой стадии из амина и РС13:
5RNH2 + PCI3—» RN=P-NHR + 3RNH2-HC1
Это соединение далее реагирует с карбоновой кислотой и даст амид [642]:
RN=P—NHR + 2RCOOH _HpoZ 2R—СО—NjlR
Обе стадии реакции можно проводить раздельно [642, 643], но обычно фосфор-
азосоединение не выделяют. В последнее время таким способом часто синтезировали
пептиды из эфиров аминокислот и аминокислот с защищенными аминогруппами, см.
J643—647]. R качестве растворителей применяли бензол, диоксан и пиридин.
..--Этиловый эфир карбобензокси-£-аланилглицина [646]. К 10,3 г эфира
глицина в 150 мл сухого пиридина при охлаждении и перемешивании приливают
по каплям 4,36 мл чистейшего РС13 в 25 мл пиридина. После непродолжительного
стояния при комнатной температуре прибавляют сразу 22,5 г карбобензоксн-
L-аланина и нагревают смесь'3 ч на водяной бане при перемешивании. После
охлаждения выделившуюся полимерную фосфористую кислоту желательно
отфильтровать, так как последующая обработка реакционной массы при этом
часто значительно упрощается. Далее растворитель отгоняют в вакууме яри 40—
50° С, остаток переносят в смесь этилацетата и разбавленной 1Ж1 так,
чтобы реакция водного слоя была кислой по конго, t Органический слой
тщательно отделяют, промывают раствором КНСО^ (для удаления непрореаги-
ровавшей или взятой в небольшом избытке карбобензоксиаминокислоты) и
водой, а затем сушат над Na2SO4. После отгонки растворителя получают эфир
4641J К. Н е у n s, W. v. Bebenburg, Chem. Вег., 86, 278 (1953).
[642] II. W. G г i m ш е 1, Л. Guenther, J. F. М о г g a n, J. Am. Chem.
Soc., 68, 539 (1946).
[643] St. Goldschmidt, H. Lautensch lager, Ann., 580, 68 (1953).
1644] St. Go 1 dsch mi d t, Ch. J u t z, Chem. Ber., 86, 1116 (1953); 89, 518 (1956);
* St. Goldschmidt, G. Rosculet, Chem. Ber., 93, 2387 (1960).
1645] O. Sus, Ann., 572, 96 (1951).
[646] W. GraBmann, E. Wiinsch, Chem. Ber., 91, 449 (1958).
[647] W. Grapmann, E. Wunsch, A. Riedel, Chem. ber., 91, 455 (1958).
448
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕН \
карбобензоксимептида в виде длинных белоснежных игл. Выход 26,8 г (81% от
теоретического). После перекристаллизации из смеси зтилацстата и петролсй-
.пого эфира т. пл. 100° С.
Для получения анилидов карбоновых кислот Р0С13 применяется реже, чем
РС13. Описание проведения этой реакции си. [648]. При получении цианацетилмоче-
вины по методу, аналогичному синтезу мочевой кислоты по Траубе, также пользуются
хлорокисью фосфора.
Цианацетилмочевина [649]. К смеси 6 моль мочевины и 2 моль циан-
уксусной кислоты при перемешивании медленно прибавляют 1 моль Р0С13.
За счет теплоты реакции масса разжижается и затем снова застывает. Сильного
разогревания реакционной смеси следует избегать, так как это вызывает ее потем-
нение. При обработке водой цианацетилмочевина остается в осадке; т. пл. 209—
212° С (разд.).
Ацилирование аминов карбоновыми кислотами протекает с отличными выходами
уже при комнатной температуре, если проводить реакцию в инертном растворителе,
например в тетрагидрофуране, D присутствии расчетного количества карбонилдиимид-
азола. [560]. Реакция протекает с промежуточным образованием пмидазолида кис-
лоты
RCOOH + I J || J| ГП|
XN-CokN N NH
I
RCO
квтгрыж вод действием амина переамидирустся^ [650, 650а]:
N---й N----г,
J-j-HNRa —* RCO—NRj-|-|| ||
N HN
I
RCO
Синтез следует проводить с сухими реагентами и в отсутствие влаги. Расчетное
количество карбонилдиимидазола (см. стр. 435) прибавляют к раствору карбоновой
кислоты в сухом тетрагидрофуране и после окончания выделения СО2 добавляют
расчетное количество амина. Примерно через 1 ч при 20е С реакция заканчивается.
Выход почти количественный [650].
Ввиду легкости образования амида в этом случае описанный метод пригоден
также для образования пептидных связей [561].
Так же как N.xN'-карбонилдиимидазол, в этой реакции применялись и N,N'-tho-
пилдиимидазол [651] п 14,№-тиокарбонилдиимидазол [651а].
Образование амида происходит уже при комнатной температуре, если к смеси
карбоновой кислоты и ямипя прибавлен какой-либо карбодиимид, например N,N'-flH-
циклогексилкарбодиимпд [652—656]:
RCOOH-|-NH2R’ + CeHi1N=C=NCeHH —> RCONHR'4-CeHuNH-CO-NHC6Hn
648] J. К I о s a, Arch. d. Pharm., 286/58, 253 (1953).
649] W. Traube, Ber., 33, 3043 (1900). t
650] H. A. Staab, Angew. Cbcm.. 71, 164 (1959).
650a] H. A. Staab, M. Luking, F. H. Darr, Chem. Ber., 95, 1275 (1962).
651] H. A. Staab, K. Wendel, Angew. Chem., 73, 26 (1961).
651a] H. A. Staab, G. Walther, Ann., 657, 98 (1962).
652] J. C. Sheehan, G. P. II e s s, J. Am. Chem. Soc., 77, 1067 (1955).
[653 ] II. G. Khorana, Chem. a. Ind., 1955, 1087.
[654 ] J. C. S h e e h a n, J. J. H 1 a v k a, J. Org. Chem., 21, 439 (1956).
[655 ] J. C. Sheehan, M. Goodman, G. P. Hess, J. Am. Chem. Soc., 78*
1367 (1956). *
[656 ] B. Helferich, H. Boshage n, Chem. Ber., 92, 2813 (1959).
ITI. ОБМЕН КИСЛОРОДА НА АЗОТ
*4?
Часто эту реакцию можно проводить даже в водпой среде. Образующаяся в качестве
побочного продукта ЬГ.Н'-дициклогексилмочевипа плохо растворима в органических
растворителях и может быть легко отделена.
Этиловый эфир фталиллейцилвалина [656]. К 1,82 г (10 ммоль) гидро-
хлорида этилового эфира 2),£-валина в 20 мл сухого тетрагидрофурана при
0° С прибавляют 1,38 мл триэтиламина. Через 1 ч отфильтровывают гидрохлорид
триэтиламина и хорошо промывают его тетрагидрофураном. В фильтрате раство-
ряют 2,57 г (10 ммоль) фталил-£-лейцина и после охлаждения до —8° С доба-
вляют 2,26 г (11 ммоль) дициклогексилкарбодиимида. Раствор оставляют на 2 ч
при —5° СдгЗатем па почь прп комнатной температуре. Отфильтровывают выкри-
сталлизовавшуюся Н,Н'-дициклогексилмочевнну, фильтрат упаривают в ва-
кууме. полученный сироп растворяют в 25 мл этилацетата. Раствор промывают
25 мл 2 п. НС] и два раза по 25 мл воды. Упариванием в вакууме высушенного
пад Na2SO4 и профильтрованного раствора получают 3,5 г (90% от теоретиче-
ского) продукта в виде смолы, застывающей в стеклообразную массу. Эта массц
легко растворима в спиртах, хлороформе, этилацетате, пиридине и диметилформ-
амиде, бензоле и толуоле и нерастворима d эфире, петролсйном эфире и воде.
Аналогично при взаимодействии с гидроксиламинами, например с О-бензил-
гидроксиламином [657], по карбодиимидному методу получают гидроксамовые кислоты
с хорошими выходами.
Здесь не рассматриваются многочисленные другие варианты ацилирования
аминов карбоновыми кислотами, например в присутствии ацетиленовых эфиров [658J
или тетраэтилпирофосфита [659] и др.; обзорные работы По синтезу пептидов методой
Виланда и другими методами см. [660, 661].
Реакция аминов с карбоновыми кислотами может применяться также для
синтеза гетероциклических систем. Алифатические 1,2-диамины реагируют с карбоно-
выми кислотами и их производными и дают имидазолины [662]. При кипячении «-фени-
лендиамина с карбоновыми кислотами в течение 0,5 ч образуются 2-алкилбензимид—
азолы [663[:
| | +НООСВ
Из p-бромэтиламина и угольной кислоты (NaHCO3) образуется [664] оксазолидон-2:
/ВГ /°\
112^ НО. Н2С \
। + \с=о —> ] С=О
Нгс НОХ Н2С /
4
[6э7] Р. A. Platt пег ct al., Helv. chim. acta, 40, 1545 (1957).
6э8| J. F. Arens, Rec. trav. chim., 74, 769 (1955).
[659] J. В 1 о d i n ge r, G, W. Anderson, J. Am. Chem. Soc., 74, 5514 (1952);'
G. W. Anderson, J. Biodinger et al., J. Am. Chem. Soc., 74, 5304
(1952).
[660] Th. W i e 1 a n d et al., Angew. Chem., 63, 7 (1951): 66, 507 (1954): 69, 362
(1957); 71, 417 (1959); 75, 539 (1963).
(661] w. Grap m a n n, E. Wunsch, Fortschr. Chem. org. Naturstoffe (Wien),
, 13, 444 (1956).
- [662] R. j. Ferm, J. L. R ie bsomer, Chem. Rev., 54, 593 (1954).
[663] w. O. Poo], П. J. Harwood, A. W. Ralston, J. Am. Chem. Soc.,
t 59, 178 (1937).
[664] S. Gabriel, G. Eschenbach, Ber., 30, 2494 (1897).
i 29 Заказ 1835.
L
150
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Если все упоминавшиеся ранее реакции аминов с карбоновыми кислотами''
приводили к образованию амидов, то при взаимодействии с муравьиной кислотой/
можно непосредственно получать ?1,1Ч'-диарилформамидины. Для этого смешивают’
без доступа влаги равные весовые количества формиата натрия и гидрохлорида амине!
с пятикратным весовым количеством первичного ароматического амина; при темпера-3
туре кипения реакция заканчивается через 10—15 мин. Реакция пригодна для обнару^
жения муравьиной кислоты в смеси с другими одноосновными жирными кислотами [6б5];|
Реакции азотсодержащих соединений
с ангидридами карбоновых кислот
Ангидриды являются более сильными ацилирующими средствами, чем самвд
кислоты. При взаимодействии ангидридов с аммиаком, первичными и вторичным»
аминами амиды кислот образуются легко и с очень хорошими выходами. Правда^
соответственно уравнению реакции ,з
zR' - /R' 1
(RCO)2OJ-HN< —> RCO-N< +RCOOH - J
\R' xR' j
для ацилирования используется лишь половина кислотной компоненты, что ножное
отнести к недостаткам метода. Это имеет значение при ацилировании труднодоступЛ
ными ангидридами кислот. Поэтому из ангидридов жирных кислот применяется!
главным образом дешевый и легко доступный уксусный ангидрид, хотя при исполь-Э
зовании его гомологов выходы амидов кислот так же высоки. Преимуществом употре^
бления для реакции ангидридов карбоновых кислот является возможность проведения
ацилирования в более мягких условиях. В отличие от условий ацилирования галоген,^
ангидридами кислот выделяющуюся при реакции кислоту обычно не обязательна
связывать избытком амина или другим оспованием. ?
Условия проведения реакции можно варьировать в широких пределах. Обычно
взаимодействие протекает уже при смешении амина с ангидридом, сопровождающемся
выделением тепла. Но чаще реакцию завершают нагреванием смеси на паровой баш
в течение 1—2 ч или при температуре кипения. При проведении реакции с жидким!
компонентами не всегда обязательно применять растворители.
N-Бензилацетамид [641]. К 21 г бензиламина приливают по капля!
20 г уксусного ангидрида и кратковременно нагревают смесь до кипения. Образо
давшуюся уксусную кислоту отгоняют с колонкой. Отогнав 9 мл дистиллята*
перегонку продолжают в вакууме без колонки, конденсируя пары в воздушно^
холодильнике. После отбора небольшого предгона перегоняется N-бензилацеТ
амид; выход 95% от теоретического; т. кип. 191Q С (23 ммрт. ст.); т. пл. 61е С
Все же в большинстве случаев реакцию лучше проводить в среде какого-либ<
растворителя. Таким растворителем при ацетилировании может служить избыто!
уксусного ангидрида или ледяная уксусная кислота. Во всех случаях можно прим^
нять инертные растворители, например эфир, бензин,'бензол или толуол; реже исполь;
зуют диметилформамид и пиридин. Спирт пригоден только при низких температурах,
п-Нитроацетанилид [666]. К горячему концентрированному раствор!
13,8 г л-нитроанилина в бензоле прибавляют 10,5 г уксусного ангидрида. PeaiC
ционная масса вскипает и в скором времени практически количественно выкри-
сталлизовывается n-нитроацетанилид; т. ил. 215—216° С.
/
Для формулирования можно применять легко доступный смешанный аНгидрЩ
муравьиной и уксусной кислот [667].
[665] W. В. Whalley,!. Chem. Soc., 1948, 1014.
[666] A. Kaufmann, Вег., 42, 3480 (1909).
[667] С. W. Huffman, J. Org. Chem., 23, 727 (1958).
III. ОБМЕН КИСЛОРОДА НА АЗОТ
451
Ароматические ангидриды реагируют обычно медленнее, чем алифатические.
Так, для бензоилирования п-питроанилина [666] его кипятили с ангидридом бензой-
ной кислоты в бензоле в течение 1,5 ч.
Поскольку гидролиз ангидридов карбоновых кислот на холоду протекает мед-
ленно, во многих случаях реакцию можно проводить и. в водной среде.
N-Ацетилглиции [668]. В 300 мл воды растворяют 75 г глицина и при
перемешивании в один прием вливают 215 г 95%-ного уксусного ангидрида;
смесь разогревается. Перемешивание продолжают в течение 15—20 мин. Начина-
ющая кристаллизация завершается при охлаждении. На следующий день от-
фильтровывают выпавший осадок, промывают его холодной водой и сушат при
100—110° С; выход продукта 75—85 г; т. пл. 207—208° С.
Фильтрат и промывные воды упаривают досуха в вакууме при 50—60° С;
перекристаллизацией остатка из 75 мл кипящей воды получают еще 20—30 в
продукта. Из маточного раствора можно выделить еще 4—6 г. Общий выход
104—108 г (89—92% от теоретического).
Даже ароматические амины можно ацилировать в водной среде. Например,
для ацетилирования л-анизидина основание растворяли в разбавленной уксусной
кислоте [669, 670]. Часто эта реакция проводилась не с аминами, а с их солями с не-
органическими кислотами в водном растворе [671]; выделявшуюся при этом минераль-
ную кислоту связывали ацетатом натрия [672].
Диацилирование аминов или ацилирование амидов кислот протекает, конечно,
в более жестких условиях [673, 674]. Так, например, для получения диацетильного
производного 2,6-дибром-л-толуидина [675] основание нагревали с двойным количе-
ством уксусного ангидрида в течение 2 ч при 150—160° С, а синтез N-пропионилфтал-
имида [676] из фталимида и ангидрида пропионовой кислоты проводится при темпера-
туре кипения в течение 12 ч.
Дипропионамид [674] (CHjCHjCOJzNH. Эквимолекулярные количества
пропионамида и ангидрида пропионовой кислоты нагревают в течение 1 ч при
100° С с небольшим количество»! концентрированной H2SO4; выход 74% от теоре-
тического.
Даже ацилируемые с трудом амины, такие, как* карбазол или дифениламин,
ацилируются большей частью в более мягких условиях в присутствии кислотных
катализаторов, например концентрированной H2SO4, НС1О4, А1С13 и FeClj.
Так, из. дифениламина и уксусного ангидрида с добавкой около 0,4% НС1О4
при 80—90° С уже через 6—8 мин почти количественно получается N-ацетильное про-
изводное [677]. Из карбазола под действием уксусного ангидрида и одной капли
30%-ной НС1О4 при комнатной температуре через 3—5 мин получается ацетильное
производное, с выходом 95% от теоретического [677].
Циклические ангидриды дикарбоновых кислот легко реагируют с аммиаком,
первичными и вторичными аминами с раскрытием кольца, образуя моноамиды дикар-
боновых кислот. Так, при смешении растворов фталевого ангидрида и анилина в хлоро-
форме в результате экзотермической реакции образуется моноанилид фталеирй кис-
лоты [678]. При яагревании этих моноамидов при более высокой темпера^ре или
668] Н. М. Herbst, D. Shemin, Org, Synth., Coll. v. 2, 1943, p. 11.
669] P. E. Fanta, D. S. T a r b e 1 1, Org. Synth., Coll. v. 3, 1955,’ p. 661.
670] H. P 1 i e n i n g e r, Ch. E. Castro, Chem. Ber., 87, 1760 (1954).
671] J. Pinnow, Ber., 33, 417 (1900).
672] С. H. Roeder, A. R. Day, J. Org. Chem., 6, 25 (1941).
67.3 ] J. B. Polya, T. M. Spotswood, Rec. trav. chim., 67, 927 (1948).
674] D. Davidson, H. Sk о vron ek, J. Am. Chem. Soc., 80, 376 (1958).
675] F. U 1 f f e г s, A. V. Janson, Ber., 27, 93 (1894).
67£] Ch. D. Hur d, M. F. Du] 1, J. Am. Chem. Soc., 54, 2432 (1932).
[677] А. А. Берлин, ЖОХ, 14, 438 (1944).
[678] M. L. Sherrill. F. L. Schaeffer, E. P. S h о у e r, J. Am. Chem.
Soc., 50, 474 (1928).
29*
452
ОБРАЗОВАНИЕ C-N-СВЯЗН В РЕЗУЛЬТАТЕ ОБМЕНА
в присутствии водоотнимающих средств эти моноамиды снова претерпевают цикли
цию, т. е. они переходят в циклические имиды кислот. При ведении реакции с сам,
начала в достаточно жестких условиях сразу получаются циклические имиды. Нан]
мер, N-фенилсукцинимид образуется уже при кипячении ангидрида янтарной кислс
и анилина в водном растворе [679J; сукцинимидоуксусная кислота образуется
ангидрида янтарной кислоты и глицина при 170—180Q С за 2 ч в вакууме [680]. Ана.
гичная реакция фталевого ангидрида с аминокислотами, протекающая при нагре
нии в течение 15 мин при 180—185° С, может применяться для идентификации амт
кислот [681]. Все же для препаративного получения фталоиламинокислот [68:
синтез лучше проводить в диоксаие [655] при 105° С. Особенно легко и количествен
протекает реакция фталевого ангидрида с аминами в ледяной уксусной кислоте [68
Имид малеиновой кислоты (III) легко и в мягких условиях образуется Из
ангидрида и мочевины, причем промежуточные продукты (1) и (II) могут быть вы,
лены в чистом виде [683]:
НС—СО—NHCONHo НС-СО НС-СО
| —> | ^NCONH3 —| \ш
НС—СООН НС-СО НС-СО
I II III
Аналогично получают фталимид; добавление NHSNO3 к расплавленной мочевин!
приводит к образованию циклического бис-амидина фталевой кислоты (дииминофтаж
имида) с хорошим выходом [684]. »
Для получения|1Ч-алкилфталимидов можно нагревать с фталевым ангидридом
не амины,’ а соответствующие алкилмочевины [685].
N-mpem-Бутилфталимид [686]. При температуре около 220° С в тече-
ние 15 мин нагревают ?првт-бутилмочевину с несколько более, чем
2.н<?л& ангидрида фталевой кислоты; выход 75% от теоретического.
Для расщепления уретанов [685], плохо расщепляющихся гид]
рекомендуется аналогичная 'реакция с ангидридом фталевой кислоты:
СО
/ \ NH-R
I II О + |
/ COOR
СО
со
NR + CO2 + R'OH
Образование циклической системы при взаимодействии циклических ангидри-
дов кислот с гидразином происходит легче, чем при взаимодействии с аминами, однако
в этом случае большое значение имеют строение ангидрида и способ проведения реанх
ции. Ангидрид янтарной кислоты [687] дает только лилейные гидразиды; из фталевого;
[679] G. Koller, Вег., 37, 1598 (1904).
[680] J. Scheiber, Н. Reckleben, Вет., 46, 2412 (1913). -
[681] J. Н. Billman, W. F. Harting, J. Am. Chem. Soc., 70, 1473 (1948fe
[681a] Литературный обзор: F. Weygand, J. Kaelicke, Chem. Ber., 95^
1031 (1962). 1
[682] G. Wanag, A. Veinbergs, Ber., 75, 1558 (1942).
[683] P. O. Tawney, R. H. Snyder et al., J. Org. Chem., 25, 56 (1960).
(6841 F. Baumann, B. Bfenert et al., Angew. Chem., 68, 137 (1956).
[685] R. H. F. M a n a k e, J. Am. Chem. Soc., 51, 1202 (1929). ;
[686] L. I. S m i t h, О. H. Emerson, Org. Synth., Coll. v. 3, 1935, p. 151. ,s
[687] II. Feuer, G.,B. Bachman, E.H. White, J. Am. Chem. Soc., 73^
4716 (1951). ' i
Ш, ОБМЕН КИСЛОРОДА Т1Д АЗОТ
453
ангидрида [688] п замещенных фталевых ангидридов ]689] в различных условиях
поручаются разные циклические гидразиды [690]; из малеинового ангидрида [691]
в зависимости от соотношения исходных продуктов образуются как циклические, так
и линейные гидразиды.
Циклический гидразид малеиновой кислоты [691]. В трехгорлой колбе
емкостью 2 л растворяют при перемешивании 161,7 г ангидрида малеиновой иис-
лоты в 1 л кипящей ледяной уксусной кислоты. Нагревание прекращают и в те-
чение 10 МШ1 прибавляют 75 г 100%-ного гидразингидрата; при этом образуется
частично растворимый желтый осадок. Затем нагревают еще 30 мин, и после охла-
ждения отфильтровывают осадок, многократно промывают небольшими порциями
спирта и эфира и сушат на воздухе. Получается 100 г бесцветного продукта.
Вторую фракцию выделяют из фильтрата после отгонки 800 мл растворителя,
добавления 200 мл воды и охлаждения; дальнейшее разбавление фильтрата
400 мл воды и упаривание до 100 мл дает третью фракцию. Общий выход 144 е
(83% от теоретического); т, пл. выше 300° С (разл.).
Основания Шиффа, имеющие строение R —СН2—CH —NR', исключительно
легко реагируют с уксусным ангидридом даже на холоду с образованием ацетилиро-
ванных енаминов:
иен,—ch=nr' rch-ch-nZ
-ЦНгСООП \COCH3
Выделяющуюся уксусную кислоту необходимо связывать; особенно пригоден
для этой цели триэтиламин.
Н-Пропил-Н-виыилацетамид [692]. В трехгорлой колбе емкостью 1л -.
с мешалкой, капельной воронкой и термометром ксмеснббг N-пропилацетальд-
имина и 78 е триэтиламина в 350 мл бензола при охлаждении до 5—8° С прили-
вают по каплям 79 г уксусного ангидрида. После прибавления всего ангидрида
реакционной массе дают нагреться до комнатной температуры. При пере-
гонке в вакууме получают 59 г К’-пропил-ТЧ-винилацетамида; т. кип. 77—78° С
(14 мм рт. ст.). Из фракции, отогнавшейся при 40—77° С (14 ммрт. ст.), после
встряхивания с раствором 10 г NaOH в 40 мл воды и повторной перегонки по-
лучают еще 8 г продукта. Общий выход 68% от теоретического; 1,4725.
При нагревании а-аминокислот с ангидридами карбоновых кислот * в пири-
дине происходит.декарбоксилирование и образование N-ацилироваппых а-аминокето-
нов (реакция Данина — Веста) [693—695]:
R-CH-NH, Н—CH-NH- COR-
। (К GO)gO [
COOII COR'
* Об аномальном поведении N-метилтриптофана при реакции с уксусным ангидри-
дом см. А. Н. Кост, Т. В. К о р о н е л л и, Р. С. Р а г и т у л л и к^Хим.
прир. соед., 3, 205 (1966). — Прим. ped. w
(688] И. D. К. Drew, Н. И. Hatt, J. Chem. Soc., 1937. 16.
[689] Н. D. К. D г е w, F. Н. Р е arm an, J. Chem. Soc., 1937, 26.
[690] Обзорная статья о циклических фтальгидразидах и других соединениях,
W. R. Vaughan, Chem. Rev., 43, 447 (1948).
[691] Н. Feuer, Е. Н. White, J. Е. Wyman, J. Am. Chem. Soc., 80, 3790
f (1958).
1692] П. Brecdorveld, Rec. trav. chim., 79, 401 (1960).
(693] I]. D. Daki n, R. Wes t, J. Biol. Chem., 78, 91, 757 (1928); P. A.Levene,
R. E. Steiger.J. BioL Chem., 74,^689 (1927); 79, 95 (1928); M. Proste-
n i k, N. Geroncevic, J. Pl use e с, M. S a t e v a, Naturwiss., 47, 496
[694] R. j|. Wiley, J. Org. Chem., 12, 43 (1947); F. E. Lehmann et al.,
, -Helv. chim. acta, 33, 1217 (1950).
(695] G. H. C 1 e 1 a n d, C. Niemann,!. Am. Chem. Soc., 71, 841 (1949),
454
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
2-Ацетамвдо-1-фенилбутанон-3 [365]. Смесь 12,4 г фенилаланв
39,3 г пиридина и 65,2 г уксусного ангидрида нагревают на паровой бане в т<
ние 5 ч. Затем перегонкой с водяным паром удаляют пиридин, остаток обраба
вают избытком водного раствора NaHCO3 и продукт экстрагируют шеет
порциями эфира, по 100 мл каждая. Эфирные растворы сушат, отгоняют ad
и воскообразный остаток, окрашенный в оранжево-желтый цвет, дважды пеа
кристаллизовывают из ксилола. Выход кетопа 12,1 г (79% от теоретического
бесцветные иглы; т. пл. 98—99° С (испр.). ' /
Реакции азотсодержащих соединений с эфирами карбоновых кислот^
При взаимодействии аммиака и его производных со сложными эфирами еле,
различать два принципиально разных типа реакций; в обоих случаях реакция П{
кает с обменом кислорода на азот:
SNi/^kC-^R
AccP-R~\n==
I II
J
В то время как эфиры карбоновых кислот, за редкими исключениями, обусло!
ными характером заместителей [696], ацилируют по атому азота (1), многие эфиры к
ральных кислот, а также эфиры арилсульфоновых кислот являются сильными длк
рующими средствами (II).
Ацилирование азотсодержащих соединений эфирами карбоновых кислот. Д
ствие эфиров карбоновых кислот па аммиак, первичные и вторичные амииы привод
обычно уже в довольно мягких условиях, к образованию амидов карбоновых кисл
Аналогичные реакции эфиров карбоновых кислот с гидразином и гидроксиламии
являются важнейшими методами получения простых гидразидов карбоновых ниа
и гидроксамовых кислот.
Реакционная способность эфиров карбоновых кислот по отношению к пронзв<
ным аммиака в гораздо большей степени зависит от строения ацилирующего среде®
чем реакционная способность других производных карбоновых кислот. Так, иаи]
мер, диэтиловые эфиры малоновой и алкилмалоновых кислот легко реагируют с аМ»
ком, превращаясь в соответствующие амиды, а диэтиловый эфир диэтилмалояо
кислоты в обычных условиях в эту реакцию не вступает [697]. Большое значение ил
также природа амина; например, метиловый эфир молочной кислоты гладко реагир
с диметиламином, образуя диметиламид, и с большим трудом взаимодействует С I
тил- или дибутиламином [698]. Возможно, что такое различие в реакционной слое
ности частично объясняется стерическими факторами. Реакции аминолиза эфиров
щелочного гидролиза симбатны. Метиловые эфиры легче подвергаются аминолг*
чем этиловые [699, 700]. Эфиры первичных спиртов расщепляются легче эфиров i
ричных спиртов [699]. Первичные амины реагируют с большей скоростью, чем амм
[701], вторичные в большинстве случаев реагируют значительно медленнее амин
[701, 702]. Реакция катализуется небольшими количествами воды [703, 704], бо
[696] F. D. Chattaway, J. Chem. Soc., 1936, 355; ср. Н. Irving, J. Chi
Soc., 1936, 797; А. С. Pierce, М. М. Joullie, J. Org. Chem., 27, З1
(1962).
[697] Е. Fischer, A. D i 1 t"h е у, Вег., 35, &44 (1902).
[698] W. Р. Ratchford, С. Н. Fisher, J. Org. Chem., 15, 317 (1950)..
[699] М. Gordon, J. G. М G 11 е г, A. R. Day, J. Am, Chem. Soc., 70, i
(1948).
[700] P. R. Russel, J. Am. Chem. Soc., 72, 1853 (1950).
[701] R. Baltzly, 1. M. Berger, A. A. Rothstein, J. Am. Chem. S
72, 4140 (1950).
[702] E. M с C. Arnett, J. G. Miller, A. R. Day, J. Am. Chem. Soc.,
5393 (1951).
[703] M. Gordon, J. G, Miller, A. R- Day, J. Am. Chem. Soc., 71, 1
(1949).
[704] P. К Glasoe^L, D. S с о 11, L. F. A u d г i e t h, J. Am. Chem.- Soc.,
2965 (1941).
FFF. ОБМЕН КИСЛОРОДА НА АЗОТ
455
эффективным катализатором является алкоголят [700, 701, 705]. Особенно выгодно
применять алкоголяты при ацилировании ароматических аминов [706].
Для получения незамещенных амидов кислот часто бывает достаточно встряхи-
вать или перемешивать сложный эфир с концентрированным водным раствором аммиа-
ка. Продолжительность реакций для разных эфиров весьма различна. Из этилового
эфира циануксусной кислоты на холоду при действии концентрированного водного
раствора аммиака образуется в течение 1 ч амид с выходом 88% от теоретического
[707]. Так же быстро реагирует метиловый эфир фторуксусной кислоты (осторожно,
сильный яд[)Д
Фторацетамид [708]. К 20 г метилового эфира фторуксусной кислоты
при охлаждении ледяной водой прибавляют избыток (20 мл} водного раствора
аммиака (d = 0,88). Через 1 ч выделившийся осадок (15 г) отфильтровывают
и сушат. Получается практически чистый продукт. Упариванием маточного рас-
твора в вакууме выделяют еще некоторое количество амида. Выход количествен-
ный. После перекристаллизации из хлороформа или смеси хлороформа с бензи-
ном получают фторацетамид с т. пл. 108° С.
Для амилолиза эфира муравьиной кислоты (709J его приходится 7 ч перемеши-
вать с концентрированным аммиаком при 25—30° С. Еще медленнее реагирует эфир
метакриловой кислоты [710]. 1
Метакриламид. В течение семи суток встряхивают 76 г метилового эфира
метакриловой кислоты с 390 мл аммиака (d = 0,885). Затем гомогенный раствор
небольшими порциями упаривают в вакууме при остаточном давлении 19 мм
рт. ст. на водяной бане (78° С). Неочищенный продукт сушат над СаС12 до по-
стоянного веса (65 г), измельчают белую массу и растворяют ее при кипячении
с обратным холодильником в 580 мл хлористого метилена. Раствор фильтруют
на воронке для горячего фильтрования и продукт из остатка еще раз экстраги-
руют 60 мл хлористого метилена. При охлаждении в бане со льдом кристалли-
зуется мономерный метакриламид; еще две фракции могут быть получены из маточ-
ных растворов. Общий выход амида 48 г' (75% от теоретического); т. пл. 95—
109° С. После повторной перекристаллизации получают чистый амид в виде
тонких пластинок; т. пл. 110—111° С.
Вместо водного аммиака в некоторых случаях применяли жидкий аммиак [711,
712]. Очень хорошие выходы получаются при проведении реакции с раствором ам-
миака в этиловом или, еще лучше, в метиловом спирте.
Ацетамид [701]. В течение 24. ч встряхивают 0,5 моль метилового эфира
уксусной кислоты с 8 .моль аммиака в метиловом спирте при комнатной тем-
пературе и выделяют ацетамид вакуумной перегонкой; выход 90% от теоре-
тического.
Диамид этилмалоновой кислоты [700]. В течение 96 ч при комнатной
температуре встряхивают смесь 10 г соответствующего эфира в 50 мл метилового
спирта, 100 мл насыщенного при 0° С раствора аммиака в метиловом спирте
и 0,1 г натрия; выход диамида 91% от теоретического. В отсутствие натриялыход
снижается. w
.Взаимодействие эфиров с аминами, если оба компонента жидкие, можно прово-
дить и без растворителей. Так, па метилового эфира молочной кислоты и амина,
[705] В. L. Betts, L. Р. Hammett, J. Am. Chem. Soc., 59, 1568 (1937).
[706] Ch. C. Price, В. H. Velzen, J. Org. Chem., 12, 386 (1947).
1707] В. B. Corson, R. W. Scott, С. E. V о s e, Org. Synth., 9, 36 (1929).
[708] F. J. Buckle, R. Heap, В. C. Saunders, J. Chem. Soc., 1949,
. 912.
[709] D. T. Mowry, J. M. Battler, Org. Synth., 30, 46 (1950).
[710] C. L. Arcus, J. Chem. Soc., 1949, 2732.
[711] J. К Jeinberg, 1. F. Audrieth, Org. Synth., Coll. v. 3, 1938, p. 516.
[712] L. F. Audrieth, J. Kleinsberp, J. Org. Chem., 3, 312 (1938).
456
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
взятого с 10%-пым избытком, при комнатной температуре получен ряд моно- и диалкил—3
амидов, в среднем с хорошими выходами [713|. Л
Н-/<-Гексиллактамид [713]. К 208 г (2 .моль) метилового эфира молоч-4
ной кислоты прибавляют 222 г (2,2 моль) н-гексиламнна, встряхивают смесь ж?
оставляют се на 15 суток при комнатной температуре. При перегонке с кололи'
кой Вигре при нормальном давлении сначала отгоняют метиловый спирт (55 е^».
затем в вакууме при остаточном давлении 1 ммрт. ст. — За промежуточной;-'
фракции (т. кип. 28—120° С). В качестве основной фракции (т. кип. 120—128° Ф
при 0,3 мм рт. ст.) получают 341 г (98% от теоретического) N-н-гексиллант-'
амида; после трехкратной перекристаллизации из смеси эфира и петролейного*
эфира т. пл. продукта 41—42° С.
Если термическая стабильность и летучесть компонентов позволяют проводит^'
реакцию при повышенных температурах, то продолжительность реакции значительно^
сокращается. N-Формильные производные анилина, Р-пафтиламина, 1-фенилэтиламина?
и ментпламина были получены [714] практически с количественным выходом пр»*!
нагревании в течение 1 ч при 100—110° С. 'J
N-м-Децилпикотинамид (715]. В колбе, снабженной колонкой и холо-,'
дильником, нагревают при 214—250° С в течение 190 мин 61,8 г этилового эфира*/
никотиновой кислоты и 70,8 г н-децнламина. При этом отгоняется 10,6 г этилового*
спирта. Остаток растворяют в 500 мл хлороформа и осветляют раствор кипяче-''
нием с углем. Из профильтрованного теплого раствора осаждают амид метро***
лейпым эфиром. После охлаждения и фильтрования получают 90,8 г (82,7% от-'
теоретического) N-w-децилпикотинаМИда; т. пл. 69,8—70,4° С. Путем многократ-
ных перекристаллизаций из смеси хлороформа и петролейного эфира получают-;
амид с т. пл. 72,2—72,6° С.
При получении амидов из эфиров кстокарбоновых кислот необходимо реакция^
вести при нагревании, так как иначе в результате взаимодействия кетогруппы с амн-,
ном образуются енамины [716] (ср. стр. 473, 480).
Анилид бензоплуксусяой кислоты. К кипящему раствору этилового эфир»:
бензоилуксусной кислоты в ксилоле при 145—150° С прибавляют по каплям-
анилин и одновременно отгоняют образующийся спирт [717, 718]. Аналогичным'.
способом в кипящем толуоле получают апилид ацетоуксусной кислоты с выходом^ ’
85% от теоретического [719].
Эфиры карбоновых кислот или амины с малой реакционной способностью легко$
вступают в реакцию не в виде свободных аминов, а в виде их производных с щелочными ;
или щелочноземельными металлами. В ароматическом ряду такие производные легко»
образуются из амина и металла в инертных растворителях и могут применяться дли*'|
дальнейших превращений без выделения их в чистом виде [720].
[713] W. Р. Ratchford, J. Org. Cliem., 15, 326 (1950); W. P. Ratchfor d.J
C. 11. F i s h e r, J. Org. Chem., 15, 317 (1950). *
[714] J. P. E. Human, J. A. Mills, J. Chem. Soc., 1948, 1457. ii
[715] С. O. Badgett, J. Л. P r ovost jr, et al., J. Am. Chem. Soc., 67.-1
1135 (1945). ?
[716] L. Limpaeh, Bor., 64, 970 (1931). £
[717] C. J. Kibler, A. Weissberger, Org. Synth., Coll. v. 3, 1955,-;
p. 108.
[718] С. H. F. Allen, W. J. Ilumphlett, Org. Synth., 37, 2 (1957). ‘ i
[719] См. ссылку [330].
[720] E. S. Stern, Chem. a. Ind., 1956, 2.77.
Ш. ОБМЕН КИСЛОРОДА. НА АЗОТ
Диапмлнд диэтилмалоновой кислоты. Образуется при кипячении диэти-
лового эфира диэтилмалоновой кислоты в течение 30 мии с 2 моль апилида натрия
в толуоле; выход дианилпда хороший [720].
Аналогичным способом в кипящем толуоле получены соответствующие
анилиды из этилового эфира М,К-диэтилглицина и натриевых производных ани-
лина и из N-ятиланилина и 2,6-днметиланилина; выходы 65—90% от теоретиче-
ских [720].
Вместо производных аминов и щелочных металлов мояшо применять и соответ-
ствующие амвпомагннйгалогепиды R^N — MgX, полученные из первичных или вторич-
ных аминов (реакция Бодру) [721];
R\
)N-MgX
R'"/
OMgX п О
I ZR Hto II /R
H—C-----N\r'" -R'on: -MgcuHjx"* R~C~N\r'"
OR'
Таким способом Вейганд [722] синтезировал ряд N-метиланилидов карбоновых
кислот.
Куп и Моррис [723] получали из эфира р-ионилиденуксусиой кислоты соответ- ’
ствующий о-то.туидид с выходом 80% от теоретического. Этим методом были синтези-
рованы также ?1,1\т-дифеиилацетамид (выход 85% от теоретического), И.М-дифенил-
бензамид (88% от теоретического), анилид капроновой кислоты (87% от теоретического)
и анилид лауриновой кислоты (80% от теоретического). Ниже приведена общая мето-
дика [721].
Раствор 0,1 моль амина в 30 мл эфира медленно прибавляют по каплям
к 0,1 моль метилмагнийиодида в 50 мл сухого эфира. После окончания .бурной
реакции добавляют раствор 0,05 моль сложного эфира в 20 мл эфира и нагревают
2 ч на водяной бане. Продукт реакции разлагают осторожным добавлением 50 мл
воды,-a._0.ee основные соли магния растворяют в 2 н. НС1. Эфирный слой отде-
ляют и сушат, эфир и непрореагировавптий амин отгоняют, из остатка при пере-
кристаллизации получают анилид.
Аналогичным способом ^-аминокислоты были переведены в N-метилани-
лиды, карбазолиды и индолиды [724].
Скорость взаимодействия эфиров карбоновых кислот и аминов во многих слу-
чаях повышается также с применением в качестве ацилирующих средств таких слож-
ных эфиров, спиртовые остатки которых легко поддаются обмену. Швейц^ [725]
рекомендует использовать для этой цели цпанметиловЫе эфиры R — СО—О —Си|—CN.
Цианметиловые эфиры аминокислот с защищенными аминогруппами легко реагируют
с эфирами аминокислот уже при комнатной температуре и поэтому с успехом приме-
няются при синтезе пептидов. Для трифторацетилирования аминокислот и пептидов
рекомендуется фениловый эфир трифторуксусной кислоты [726].
Обзорная работа Вилапда и др. [660, 661] дает хорошее представление о других
способах синтеза пептидов при помощи активированных таким образом сложных
эфиров.
L. Bassett, С. R. Thomas, J. Chem. Soc., 1954, 1188.
W о у g a n d et al., Angew. Chem., 65, 530 (1953).
Kuhn, C. J. O. R. Morris, Ber., 70, 853 (1937).
В i r k li о f e r, E. Frankus, Chem. Ber., 94, 216 (1961).
S c h w у z e г et al., Helv. cliim. acta, 38, 69. 80, 83 (1955).
Weygand, A. Ropsch, Chem. Ber., 92. 20Й5 (1959).
458
ОБРАЗОВАНИЕ G-N-СВЯЭИ В РЕЗУЛЬТАТЕ ОБМЕНА
Активированный эфир уксусной кислоты — изопропевилацетат, получаем
из кетена и ацетона так же легко, как и сам кетен, реагирует с аминами уже при к<
натной температуре, причем выделяется большое количество тепла [721]. С его i
моптьто может быть получен даже такой труднодоступный амид, как N-ацетилимидаЗ!
образующийся из имидазола с выходом 94% от теоретического [728].
N-Ацетилморфолин [727]. Помещают 100 г изопропенилацетата в кол
для перегонки и медленно прибавляют к нему по каплям 87 г морфолина. Ц
этом начинается бурная реакция и образующийся ацетон непрерывно отгоняв!
Перегонкой в вакууме продукта реакции получают N-ацетилморфолии; вы
80% от теоретического; т. кип. 89° С (1 лсж рт. ст.}\ 1,4337.
Лактоны обычно являются такими же ацилирующими средствами для айв
и аммиака, как и линейные эфиры карбоновых кислот.
' N-Метиламид й-оксиметилбензойной кислоты [729]. Растворяют в 1-
29%-ного водного раствора метиламина при нагревании 175 г фталида (I). J
охлаждении льдом N-метиламид о-оксвметилбензойной кислоты (II) выдел»
в виде бесцветных кристаллов; выход 159 г (74% от теоретическс
т. пл. 122—123° С.
CO-NH-СНз
АГ
'V'XchjOH
II
Амины с низкой реакционной способностью могут и в этом случае пр
нятьсн в виде N-металлпроизводных [730].
Если у атома азота образующихся амидов оксикарбоновых кислот имеется х
бы один атом водорода, то они переходят при нагревании в лактамы. Так, уггомт
шийся выше N-метиламид о-окскметилбензойиой кислоты (И) при 160° С цю
зуется в соответствующий лактам (III). При такой циклизации наряду с отщеплет
воды происходит и выделение метиламина с образованием исходного фталида
поэтому реакцию следует проводить под давлением [729].
Однако в некоторых случаях лактоны реагируют и как алкилирующие сред
и дают аминокислоты. Так, при кипячении 4-бутиролактона с фталимидом калия в
метилформамиде образуется 4-фталимидомасляная кислота [731]; в тех же услоа
из фталида (I) получается о-(фталимидометил)-бензойная кислота [732]. Осо&
сильно алкилирующие свойства выражены у р-лактонов, но направление peai
с 0-лактонами в значительной мере зависит от условий взаимодействия [733].
добавлении пропиолактона к раствору диметиламина в эфире при 0° С образу
диметиламид гидракридовой кислоты (IV) с выходом 95% от теоретического, т<
как при обратном порядке смешения реагентов, т. е. при пропускании диметилаь
[727
[728
[729
[730
[731
[732]
[733]
Н. J. Hagemeyer jr., D. С. Hull, Ind. Eng. Chem., 41, 2920 (18
J. H. Boyer, J. Am. Chem. Soc., 74, 6274 (1952).
W. Theilacker, K. Kalends, Ann., 584, 87 (1953).
W. Reppe et al.. Ann., 5ft6, 175 (1955).
G. Talbot, R. G a u d г y, L. Berlinguet, Canad. J. Chem., 36,
(1958).
J. В or ns tc in, P. E. Drummond, S. F. Bedell Олу. Syn
38, 81 (1958). '
T. L. G r e s h a m, J. E. Janson et al., J. Am. Chem. Soc., 73, 3168 (13
095-?’ Huid- Я НаУао, J. Am. Chem. Soc., 74, 5889 (1952); 76, c
Ш. ОВМЕН КИСЛОРОДА НА АЗОТ
459
в эфирный раствор лактона при 0° С, может быть получен И.М-диметил-р-аланин (V)
с выходом 84% от теоретического £733):
СН2~СН2 Н2С-СН2 СН2—СН2—СООН
I | *— I I —► I
ОН СО—N(CHah О-СО N(CHs)2
IV V
Из реакций эфиров карбоновых кислот с амидами наибольшее значение имеет
циклизация, протекающая при взаимодействии мочевины с диэфирами дикарбоновых
кислот. В соответствии с условиями эфирной конденсации синтез проводится со стехио-
метрическими количествами алкоголята.
Значительное влияние на направление реакции оказывает также растворитель.
Барбитуровая кислота [734[. К раствору 48 г натрия в 400 мл абсолют-
ного метилового спирта при перемешивании прибавляют 320 г малонового
эфира и кипятят в течение 10 мин в колбе с обратным холодильником. При до-
бавлении горячего раствора 120 г мочевины в 300 мл абсолютного метилового
спирта реакционная масса превращается в густую кашицу, которую для завер-
шения конденсации кипятят еще 5 ч с обратным холодильником. После этого
реакционную массу выливают в 3 л воды и подкисляют 200 мл концентрированной
НС1. Сначала реакционная масса растворяется, а затем в виде красивых
кристаллов осаждается барбитуровая кислота. После перекристаллизации
из воды и двухдневной сушки при 100° С выход составляет 210 г', т. пл.
выше 260° С (разл.).
Проведение синтеза в более жестких условиях с применением в качестве
растворителя этилового спирта см. [735].
Так же гладко протекает аналогичная реакция конденсации мочевины
с эфиром апуксусной кислоты по Траубо [736].
Следует заметить, что для получения производных барбитуровой кислоты
можно применять и обратный путь, а именно: конденсировать диамиды малоно-
вой или диалкилмалоповых кислот с дпэтилкарбонатом. Реакцию проводят
• в жидком аммиаке в присутствии NaOH или амида натрия в качестве катализато-
ров конденсации. По-видпмому, в большинстве случаев такой метод не имеет
никаких преимуществ перед обычным ]737].
Гидразий реагирует с эфирами карбоновых кислот обычно легче, чем амины.
Простейшие эфиры жирных кислот чаще всего превращаются в гидразиды кислот уже
при комнатной температуре в результате экзотермической реакции; однако лучше
прибегать к непродолжительному нагреванию на водяной бане. Побочная реакция
образования ^№-диацилированных гидразинов, часто наблюдаемая при использова-
нии хлорангидридов и ангидридов кислот, в этом случае встречается очень редко.
Приведенные далее примеры характеризуют данный метод.
Гидразид гг-нитрофенилуксусиой кислоты [738]. На паровой бане в тече-
ние 30 мин нагревают 34 г метилового эфира гг-нитрофенилуксусной кислоты
и 50 г 42%-ного гидразингидрата. При этом сначала реакционная масса окраши-
вается в красный цвет, который постепенно исчезает, и к концу реакции продукт
застывает в твердую желтую массу. Гидразид промывают водой и кристаллизуют
из абсолютного спирта. Выход 33 г (97% от теоретического); иглы; т. пл. 167° С.
Дигидразид адипиновой кислоты [739]. К кипящей смеси 120 мл 85%-ного
водного гидразингидрата (2 моль) и 25 мл абсолютного этилового спирта
[734] W. Р f 1 с i d с г е г, Е. Nubel, Ann., 631, 168 (1960).
[735] J. В. Dickey, A. R. Gray, Org. Synth., Coll. v. 2, 1943, p._ 60.
[736] W. R. Sherman, E. C. Taylor jr., Org. Synth., 37, 15 (1957).
[737] K. Shi mo, S. Wakamatsu, J. Org. Chem., 24, 19 (1959).
[738) R. L. Shriner, J. M. Cross, J. Am. Chem. Soc., 60, 2338 (1938).
[739] p. A S. Sipith, Org. Synth., 36, 69 (1956).
460
ОБРАЗОВАНИЕ G-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
в течение 1—2 ч по каплям прибавляют 101 г (0,5 моль) дпэтилового эфира
адипиновой кислоты. Кристаллизующийся при охлаждении гидразид про
мывают спиртом, эфиром и сушат. Выход около 90% от теоретического
т. пл. 174-181° С. -3
п-Этоксибензгидразид [740]. Но менее 8 ч кипятят смесь 1 моль этиловой
эфира п-этоксибснзойной кислоты и 1,25 моль гидразингидрата в виде 20%-ной
водного раствора. При охлаждении выделяется гидразид в виде бесцветных итд;
выход 95% от теоретического. Перекристаллизацией их из разбавленнОп
этилового спирта получают бесцветные прямоугольные пластинки; т. пл. 126-i
Так же как при аминолизе, эфиры карбоновых кислот с разветвлением в a-nojtci
жении реагируют с гидразином медленнее, чем неразветвлепные сложные эфиры. Пр1
должительность реакции сокращается при использовании в качестве растворителе
спиртов с более высокой температурой кипения, например бутилового спирта, или пр
взаимодействии с безводным гидразином.
Взаимодействие эфиров карбоновых кислот с алкилвамещенными гидразинай
может привести к образованию смесей изомерных гидразидов Ас—NH—NH1
и Ac—NR—NH2, которые не всегда удается разделить (см., например, [741]). |
Гидроксиламин ацилируется эфирами карбоновых кислот с образованием гидр<
ксамовых кислот. При смешении реагентов в спиртовом растворе реакция часто npoft
кает уже при комнатной температуре в течение нескольких часов. Рекомендуете!
вести реакцию в присутствии эквивалентного количества алкоголята. В этом случй
гидроксамовые кислоты получаются в виде солей, из которых легко выделить свобод
ные кислоты. ;
Малоидигидроксамовая кислота [742, 743]. К 80 а (0,5 .моль) диэтиловаб
эфира малоновой кислоты приливают 1 л раствора свободного гидроксиламиЯ
в абсолютном спирте (1 моль), полученного из гидрохлорида гидроксИиамнЯ
и рассчитанного количества алкоголята в этиловом спирте, и добавляют раствЭ
этилата, приготовленного из 25 г (1,1 моль) натрия в 500 мл абсолютного спирту
Начинающееся выделение динатриевой соли малондигидроксамовой кислоты 31
канчивается через несколько часов; выход 78 г. £
Для получения свободной кислоты прибавляют 17 г соли к рассчитанной
количеству (50 мл) 2,8 н. спиртового раствора HCI и оставляют на 2 ч при периС
дическом встряхивании. Выделившийся NaCl отфильтровывают, спирт отгоняй
D вакууме (прн 25° С) и получают неочищенную малондигидроксамовую кислоту
т. пл. 144—147° С; чистая кислота плавится при 155° С.
Па том же принципе основан метод получения бензгидроксамовой кислой
1744]; примеры получения различных, замещенных в ядре бензгидроксам^вй
кислот см. [566]. ’ ;
Ацилирование азотсодержащих соединений ортоафярами. Образование айв
динов из ортоэфиров (главным образов из ортоэфиров муравьиной кислоты) и аммиак
или ароматических аминов было известно уже почти сто лет тому назад (ср. обзорну!
работу [745]). В качестве примера ацилирования аммиака приведен современна
метод, по которому ацетамидин получается в виде ацетата [746]. ’
[740] Р. Р. Т. Sab, K.-S. Chang, Ber., 69, 2763 (1936).
[741] R. L. Hinman, D. Fulton, J. Am. Chem. Soc., 80, 1895 (1958). ;
[742] Ch. D. Hurd, D. G. Botteron, I. Org. Chem., 11, 207 (1946).
[743] A. llantzsch, Ber., 27, 799 (1894). %
[744] C. R. Hauser, W. B. Renfrow jr., Org. Synth., Coll. v. 2
1943, p. 67. . '
[745] H. W. Post, The Chemistry of the Aliphatic Orthoesters, Reinhold PublishlUl
Corp., New York, 1943, p. 84If.
[746] E. С. T a у 1 о r, W. A. Eh rh art, J. Am. Chem. Soc., 82, 3138 (1960)
III. ОБМЕН КИСЛОРОДА НА АЗОТ
461
Ацетат ацетамидина. В колбе с обратным холодильником нагревают
в течение 45 мин смесь 32,4 г этилового эфира ортоуксусной кислоты и 15,4 г
ацетата аммония, пропуская одновременно ток сухого аммиака. Затем реакцион-
ную массу перегоняют при температуре бани 155—160° С до тех пор, пока тем-
пература перегоняющихся паров ве понизится до 75° С. При этом примерно-
за 25 мин отгоняется 18,5 г дистиллята. Остаток охлаждают до комнатной тем-
пературы4^ фильтруют. После промывки этиловым спиртом и сушки получают-
16,5 г ацетата ацетамиднна; т. пл. 189—191° С. Из фильтрата может быть выде-
лено еще некоторое количество продукта. Общий выход 19,75 г (84% от теорети-
ческого).
Аналогичным способом в среде ледяной уксусной кислоты получен ацетат-
формамидина с таким же высоким выходом [746].
Много неясностей встречалось при изучении механизма реакции и приводы
конечных продуктов взаимодействия ортоэфиров с ароматическими аминами. Сейчас
можно с уверенностью сказать, что на первой стадии реакции образуются имидоэфиры,
которые в случае эфира ортомуравьиной кислоты [746а] с избытком амина переходят-
в амидины [747]:
/OR vNHAr
ПС(ОН)3Д±*Ь> нс< HC<
XNAr -NAr
Приведенная ниже методика может применяться также и для получения других
ароматических формамидинов.
М^'-Бис-(о-хлорфенил)-формамидин [748]. В колбу емкостью 500 мл
с присоединенной к ней колонкой с насадкой (длина около 40 см) с наружным
•богревом помещают 104 г (0,7 моль) этилового эфира ортомуравьиной кислоты
и 179 е (1,4 моль) а-хлоранилина. Колбу нагревают в течение 1,5 ч на бане с теы-
ературой ИВ’Х, поддерживая в колонне температуру 90—100” С. После этого
температуру бани повышают до 180® Сив течение 30 мин отгоняют этиловый
спирт. Содержимое колбы нужно быстро вылить в фарфоровую чашку, так как
родукт реакции при охлаждении быстро затвердевает.
Выход неочищенного продукта практически количественный; т. пл. 137—
141° С. После перекристаллизации из сухого бензола получают 150 г (81% от
теоретического) 14,№-бис-(о-хлорфенил)-формамидина в виде бесцветных призм;
т. пл. 139—141® С.
Однако если взаимодействие проводят с эквивалентными количествами реаги-
рующих веществ или с избытком ортоафира и в присутствии каталитических колиМств
кислоты, то реакция останавливается на первой стадии и с хорошим выходом полутени
А -арилимидоэфиры.
Этиловый эфир N-фсннлимидомуравьином кислоты [749, 750]. В течение
30 мин нагревают с обратным холодильником 1 моль анилина, 1,5 моль этилового
эфира ортомуравьиной кислоты и 10 капель H2SO4. Затем отгоняют 117 мл
этилового спирта, нейтрализуют остаток 3,5 г mpem-бутнлата натрия и перего-
няют в вакууме. После отгонки избыточного ортоэфира получают 121,6 г (82% от
теоретического) имидоэфира; т. кип. 117—119° С (40 мм рт. ст.).
[746а] R. Н. De Wolfe, J. Org. Chem., 27, 490 (1962).
747] R. M. R о b e г t s, R. H. De Wolf e, J. Am. Chem. Soc., 76, 2411 (1954).
174s] R. M. Roberts, J. Org. Chem., 14, 277 (1949).
749] R. M. Roberts, J. Am. Chem. Soc., 7i, 3848 (1949).
I7o/j] R. M. Roberts. P. J. Vogt, Org. Synth., 35, 65 (1955).
462
ОБРАЗОВАНИЕ C-N-CBH3H В РЕЗУЛЬТАТЕ ОБМЕНА
Поскольку при высоких температурах в присутствии кислотных катализато]
N-арилформимидоэфиры перегруппировываются с образованием изомерных N-али]
форманилидов
ZOR II /Ai
НС< —> HC-N<
^NAr XR
последние можно в соответствующих условиях непосредственно получать взаимо;
вием амина с ортоэфиром [751, 752].
N-Этилформанилид [751]. В течение 30 мин нагревают, применяя обр|
ный холодильник, 18,6 з (0,2 моль) анилина, 44,5 г (0,3 моль) этилового эфи
ортомуравьиной кислоты и 0,78 г концентрированной H2SO4. Затем кол
соединяют с насадочной колонкой (длина около 30 см), температуру бани нов
тают до 175° С и поддерживают ее еще в течение 30 мин. При этом отгоняй
21,8 г этилового спирта. После охлаждения реакционную массу перегони
в вакууме (40 мм рт. ст.). Сначала отгоняется 9,1 г избыточного ортоэфк
(т. кип. 65—67° С), затем несколько капель имидоэфира (т. кип. 118—120°I
и, наконец, 22,4 г (75% от теоретического) N-этилформанилида; т. кип. 150
154° С (40 мм рт. ст.).
При нагревании с разбавленной НС[ формильный остаток легко отщепляете
что позволяет применять эту реакцию для получения чистых N-мЬноалкилакиЛ
нов [751, 752].
Ацилирование азотсодержащих соединений имндоэфирами. Рассматривавшие!
вт,пне синтезы амидинов из ортоэфиров протекают, как уже упоминалось, через ст
дию образования имидоэфиров (обзорные работы но амидинам и имидоэфирн|
см. [753—755]). Соответствующим образом могут, конечно, реагировать с аммиаком Л
аминами сами имидоэфиры, получаемые обычно из нитрилов в виде гидрохлорид!
[754]. При проведении реакции с первичными аминами следует учитывать, что П]
повышенных температурах может обмениваться не только эфирная группа, но и ай
ный остаток имидоэфира:
/OR'
R-C^ R—С:
*NH
NHR' W4
К 1'П,
zNIIIV
^NR"
Попятно, что с вторичными аминами такой обмен невозможен. Ароматичесщ
амины, обладающие слабоосновпыми свойствами, реагируют обычно с имидоэфира»
плохо и неоднозначно. Во всех случаях реакцию проводят без доступа влаги.
Для получения гидрохлоридов незамещенных амидинов рекомендуется след
ющий простой общий метод [756]. -
-Гидрохлориды амидинов. Приблизительно 10%-ный раствор (или сусш
зия) 0,1 моль гидрохлорида имидоэфира, в абсолютном спирте, содержаЩ
0,13 моль аммиака, встряхивают в закрытом сосуде при комнатной томперятЭ
в течение двух-трех суток. Выделившийся NH4C1 отфильтровывают, фильтр
разбавляют эфиром и отделяют гидрохлорид амидина, который перекрист^
лизовывают из смеси мотилэтилкетона и этилового спирта.
Получение гидрохлорида ацетамидина из ацетонитрила см. [757]. ’
[751] R. М. R о b е г t s. Р. J, V о g t, J. Am. Chem. Soc., 78, 4778 (1956). 'i
[752 R. M. Hoberts, P. J. Vogt, Org. Synth., 38, 29 (1958).
[753] R. L. Shriner, F. W. Neumann, Chem. Rev., 35, 351 (1944).
[754] P inner, Die Tmidoathei’ und ihre Derivate, Verlag Oppenheim,
[755] R. Roger, D. *G. Neilson, Chem. Rev., 61, 179 (1961).
[756] C. D j e r a s s i, C. R. Scholz, J. Am. Chem. Soc., 69, 1688 (1947). J
[757] A. W. Do x, Org. Synth., 8, 1 (1928).
F
III. ОБМЕН КИСЛОРОДА НА АЗОТ
463
N-p-Диэтиламиноэтиламидины [758]. Растворяют 1 моль гидрохлорида
имидоэфира в восьми-десятикратном весовом количестве абсолютного этилового
спирта, прибавляют 1,05 моль 0-диэтиламиноэтиламина и перемешивают в тече-
ние 8 ч при 40—45° С. Спирт отгоняют в вакууме, остаток растворяют в воде
и подкисляют HCI по индикатору копго. После непродолжительного нагревания
раствор охлаждают и отделяют фильтрованием или экстрагированием эфиром
примеси, не обладающие основными свойствами. После этого раствор упаривают
в вакууме^ри 40° С досуха; последние следы воды отгоняют в виде азеотропной
смеси с бензолом и этиловым спиртом. Выход продукта 75—80% от теоретиче-
ского.
Аналогичное взаимодействие с этилендиамином [756] приводит к образованию диза-
мещенных имидазолинов, которые следует рассматривать как циклические амидины.
Так же легко под действием аминов [759] или аминокислот [760] переходят
циклические имидоэфиры (О-алкиловые эфиры лактимов) в циклические амидины!
О-Алкилизомочевины реагируют как простые имидоэфиры; из О-метилизомоче-
вины и орнитина образуется аргинин с выходом 94,5% от теоретического [761].
Наконец, следует упомянуть, что вакуумная перегонка свободных форм-
и ацетимидобензиловых эфиров приводит к отщеплению бензилового спирта и циклизу-
ющей тримеризации и является, таким образом, простым методом получения симмет-
ричного триазина и его 2,4,6-триметилпроизводного [762].
Гидразин реагирует с гидрохлоридами имидоэфиров аналогично аминам и в тех.
же условиях с образованием амидразонов [754, 763, 764].
Алкилирование азотсодержащих соединений эфирами серной кислоты. Из эфи-
ров серной кислоты для алкилирования производных аммиака применяются в основном
только диметил- и диэтилсульфаты. В достаточно мягких условиях на аминную ком-
поненту переносится всегда только одна алкильная группа эфира. Полное использова-
ние диалкилсульфатов удается обычно лишь при температурах 160—200° С.
Алкилирование аммиака и первичных аминов до первичных и, соответственно,
вторичных аминов чаще всего протекает с плохими выходами, так как одновременно'
образуются и все возможные продукты высшей степени алкилирования. Наилучшие
результаты получаются-при алкилировании первичных ароматических аминов до тре-
тичных. В этом случае образование четвертичных аммониевых солей меньше сказы-
вается на выходе, так как при кипячении с алкоголятом или при перегонке с паром из
щелочной среды такие четвертичные соли снова разлагаются до третичных аминов. '
Хюниг [765] рекомендует следующую пропись для метилирования первичных,
ароматических аминов до третичных.
К взвеси 3—41* *оль NaHCO3 в воде прибавляют ароматический амин (1 моль)-
и в один прием диметилсульфат (2,5—3,5 моль). При перемешивании начинается реак-
ция, сопровождающаяся выделением СО2. Хорошие выходы алкиламниов получаются,
если основность амина достаточно высока, чтобы реакция метилирования могла проте-
кать при температурах ниже 35° С. При более высокой температуре диметилсульфат
заметно гидролизуется. Кроме того, амин и днметилсульфат должны хоть немного
растворяться друг в друге, что иногда достигается добавлением иесмешивающегося
с водой растворителя. '
М,Х-Днметил-0-нафтиламип [765]. В трехгорлой колбе, снабженной'
мешалкой, термометром и счетчиком пузырьков выделяющегося газа, к 14,5 в’
0-нафтиламина, 34aNaIICOs и 50 мл воды прибавляют 34 мл диметилсульфата.
При 10° С начинается интенсивное выделение газа, прекращающееся через
45жмм. Нагревая до 55—60° С, омыляют избыток диметнлсульфата. Реакционную-
[758] F. Н. S. Curd, С. G. R a i s о n, J. Chem. Soc., 1947, 160.
[759] R. Е. Benson, Т. L. С a i г n s, J. Am. Chem. Soc., 70, 2115 (1948).
[760] S. Petersen, E. Tietz e, Ann., 623, 166 (1959).
[761] F. Turba, K. Schuster, Hoppe-Seyler’s Z. physiol. Chem., 283, 27 (1948).
[762] F. Cramer, K. Pawelzik, J. Kupper, Angew. Chem., 68, 649 (1956);.
* ср. также F. C. Schaefer, G. A. Peters, J. Org. Chem., 26, 2778 (1961)..
[763[ D. Jerchel, H. Fischer, Ann., 574, 96 (1951).
[764] A. Pinner, Ann., 297, 221 (1897); 298, 1 (1898).
[765] S. II u n i g, Chem. Ber., 85, 1056 (1952).
464
ОБРАЗОВАНИЕ G-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
массу отделяют от остатков NaHCO3, сильно подщелачивают NaOH и кипя-
тят 1 ч в колбе с обратным холодильником. Образовавшееся масло экстрагируют
хлороформом. Остаток после отгонки хлороформа перегоняют при 160—161® G
(12 мм рт. ст.) в виде бесцветного масла; выход 16,8 г (98% от теоретического)^
Присутствие бикарбоната иеобходпмо для выделения нз образующейся на пер^
вой стадии алкилсернокислой соли свободного основания для дальнейшего его алкилн^
рования. Преимуществом бикарбоната в сравнении с более сильными основаниях^
является возможность избирательного алкилирования аминофенолов по аминогруппу
при устанавливающихся в реакционной среде значениях pH [765|.
По старым прописям для связывания образующейся алкилсерной кислотИ
часто использовали водные растворы NaOH. Щелочную среду поддерживали поперек
менным добавлением диметилсульфата и раствора щелочи. В этом случае выходы в зна-
чительной мере зависели от точного соблюдения условий и от чистоты диметилсульфаТа
о-Бромдиметилаинлин [766). В трехгорлой-колбе емкостью 1 л к 100
о-броманилина и 100 мл воды прибавляют 3 екв (219 г) диметилсульфата тренд
равными порциями. Первую порцию добавляют к смеси прп перемешиваний
оставляют ее до гомогенизации и тщательно нейтрализуют 25%-пым растворов
КОН при охлаждении в бане со льдом; так же добавляют вторую порцию, S
при последующей нейтрализации рекомендуется вводить небольшой избытф
щелочи; затем при перемешивании прибавляют остальной димотилсульфат
оставляют смесь на 1 ч, амин экстрагируют из щелочного раствора эфиром
промывают экстракт водой и сушат над К2СО3. При перегонке получаю,
81.5 г (70% от теоретического) "о-бромдиметиланилина; т. кип. 100—101° (
(12 мм рт. cm.). f
Аналогично получают л-бромдпметиланилпп; выход 54% от теоретиче*
ского; т. кип. 118—119° С (8 мм рт. ст.). ?
В некоторых случаях можпо алкилировать амины и простым нагревание!
с диметилсульфатом. Так, из 1 моль n-анизидина и 2,2 молъ дпэтилсульфата нагревЙ
днем в течение 4 ч при 120—130° С получили К,Г\-диэтил-я-анизидин с выходом 61$
от теоретического; аналогичным способом после 6 ч нагревания смеси получил)
К,К-диэтил-«-толуидин (выход 91 % от теоретического); а после 8 ч нагревания —
К^-диэтил-.«“фенетидин (выход 52% [767]).
Важным способом синтеза этиленимина и его производных является циилиаа*
ция"'сернокислых эфиров этаноламинов при нагревании с щелочами [768—770]:
CH2-OS03 Н2С. ’
| + NaOH^ | \NH Ч
CH2-NH3 Н2(У ;
Чистые первичные амины могут быть получены алкилированием уротропин!
диалкилсульфатами и последующим расщеплением четвертичных солей [771].
При алкилировании цианамида образуются диалкилциапамиды, из которьи
омылением выделяют вторичные амины. Например, из цианамида кальция и диметил;
сульфата получили диметнламин с выходом 70—80% от теоретического [772). Однаш
чаще всего этот метод не имеет никаких преимуществ перед методом с применение!
алкилгалогенидов. • '
При алкилировании диалкилсульфатами амидов карбоновых кислот и лактама
следует учитывать, что свободные амиды легко претерпевают О-алкилирование с обра;
[766] FI. Gilman,!. Banner, J, Am. Chem. Soc., 62, 344 (1940). i
[767] H. A. Fahim, A. M. F 1 e i f e 1, J- Chem. Soc., 1951, 2761.
[768] С. H. F. А И e n, F. W. Spangler, E. B. Webste r, Org.^S^nthi
30, 38 (1950).
[769] K. N. C a m p bell, А. П. Sommers, В, K. Campbell, Org. Syntb«
27, 12 (1947). ,
[770] R. С. E I d о г f i e I d, H. A. Hageman, J. Org. Chem., 14, 605 (19491
[771] F. L. H a h n, ’ll. Waite r, Ber., 54, 1531 (1921).
[772] W. Traube, A. Engelhardt, Ber., 44, 3149 (1911). ?
Ill, ОБМЕН КИСЛОРОДА НА АЗОТ
'465
зопанпем имидоэфиров. Так, например, из капролактама и диметплсулъфата в зави-
симости от условий реакции получают О- цчи N-метплпролзводноо [ 759 J. Для синтеза
из амидов кислот исключительно N-аяки (производных рекомендуется применять
в качестве исходных соединений N-иропзводиые амидов с щелочными металлами и про-
водить алкилирование в инертном растворителе.
Так, например, для получения N-метнл-п-анцзиднна N-ацетильное про*
изводное сначала цеталлнруют амидом натрия в кипящем толуоле, затем при
100° С добавляют диметилсульфат п омыляют продукт реакции в щелочной среде
[670].
Большее значение имеет алкилирование амидов сульфокислот диметил- или
длэгилсульфатом. Алкилирование анилидов арилсульфокислот проводят следующим
образом.
Растворяют в 0,042 моль 10%-ного водного раствора NaOH 0,04 моль
анилида арилсульфокислоты и при перемешивании в течение 30 мин при 25° С
приливают по каплям 0,042 моль диалкилсульфата. Выделение N-алкиланипида
арилсульфокислоты начинается через несколько минут поело начала добавления
диалкилсульфата. Перемешивание продолжают еще 1 ч при 40° С, отфильтровы-
вают осадок и перекристаллизовывают его из 95%-ного спирта [773].
Взаимодействие гидразина с диалкилсульфатами протекает неоднозначно. Более
гладко алкилируются ацильные производные гидразина. Так, например, для полу-
чения М,Л'-диметилгидразняа в водно-щелочной среде метилируют К,К'-дибензоил-
гидразин диметилсульфатом и омыляют продукт алкилирования [497].
Алкилирование азотсодержащих соединений эфирами фосфорной кислоты. .
До настоящего времени случаи алкилирования азотсодержащих соединений эфирами
фосфорной кнелоты/^едко описывались в литературе. Общин способ алкилирования
ароматических аминов приводит Биллман [774]. Условия реакции подобраны так,
что эфиры фосфорной кислоты полностью дезалкилируются.
Алкилирование ароматических аминов. В колбе емкостью 500 мл с обрат-
ным холодильником нагревают 2 ч смесь 0,3 моль анилина и 0,2 моль соответ-
ствующего триалкилфосфата. В большинстве случаев реаиция сначала протекает
с значительным выделением тепла и температура смеси сильно повышается.
После прекращения бурного кипения холодильник с водяным охлаждением ме-
няют на воздушный. К охлажденной до 50° С смеси приливают раствор 25 г
NaOH в 100 мл воды, нагревают ее 1 ч до кипения и охлаждают в химическом
стакане до комнатной температуры. Верхний маслянистый слой амина отделяют
от выделившегося фосфата натрия, который промывают эфиром. Амин и эфирный
раствор объединяют к сушат над Na8SO4. Остаток после отгонки эфира оставляют
иа ночь с равным объемом уксусного ангидрида, смесь встряхивают с 20 мл
концентрированной НС1 и 30 мл воды до полного растворения амина. Првмесь
амида из раствора удаляют двукратной экстракцией эфиром, порциями по 30 мл
каждая, затем амин выделяют 25%-ным раствором NaOH и извлекают его эфи-
ром. После сушки над NazSO4 раствор перегоняют. С соответствующими триал-
килфосфатами были получены, например, следующие вещества: днметилаяилин
(выход 68%); диэтиланилнн (78%), ди-н-пропиланилин и ди-ч-бутилаиилик
(79%).
Аналогичным способом подвергали дналкилированию а- и Р-нафтиламины [774],
а также ряд замещенных в ядре анилинов [775]. Из 2-аминофлуорена и триэтилфос-
фата при 205° С с почти количественным выходом получен 2-диэтиламинофлуорен
[773] М. N е е m а п, A. Modiano, J. Org. Chem., 21, 667 (1956).
[774] J. Н. Billman, A. R a d i k е, В. W. Mundy, J. Am. Chem. Soc., 64,
2977 (1942).
[775] D. G. Thomas J. H. Billman, Ch. E. Davis,!. Am. Chem. Soc., 68,
895 (1946).
30 заказ 1835
466
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
f776], в несколько более мягких условиях из морфолина — N-этилморфолин с выходом
70% от теоретического [777].
При алкилировании цианатов' щелочпых металлов до алкилизоциапатов ис-
пользование триалкилфосфатов [778] дает лучшие результаты, чем использование
эфиров серной кислоты и сульфокислот [7791-
Вместо третичных эфиров иногда в качестве алкилирующих средств применя-
лись также диалкилхлорфосфаты. По этому методу из анилина при 240—250° С полу-
чали моноалкильпые производные с выходом около 95% от теоретического [780].;
Алкилирование азотсодержащих соединений эфирами арилсульфокислот
Широкое применение в качестве алкилирующих средств нашли эфиры арилсульфокис-»
лот; особенно эффективными алкилирующими агентами являются эфиры толуолсульг*
фокислоты и первичных спиртов. При взаимодействии p-хлорэтилового эфира толуод-
сульфокислоты с фталимидом калия па азотную функцию обменивается не галоген#
а остаток сульфокислоты [781]. •'
Как и при алкилировании эфирами серной и фосфорной кислот, в этом случае
следует исходить из свободных аминов, так как при алкилировании, например, гкдрв>
хлоридов аминов замещается почти исключительно анион хлора и основным продуц;
том реакции является хлористый алкил.
При нагревании 0,05 моль и-октилового эфира п-толуолсульфокислоп
С 0.1 моль гидрохлорида диэтидамина в 35 мл хлороформа за 6 ч получают н
октилхлорид с выходом 91% от теоретического [782].
Продукты реакции, образующиеся из свободных аминов и эфиров сульфокислот
также, как при использовании диалкилсульфатов. почти всегда представляют собо#
смесь аминов всех возможных степеней алкилирования. Однако можно подобрать таки
условия реакции, которые благоприятствуют образованию определенных продух
тов алкилирования. При необходимости реакцию проводят в растворителе, обычи!
в спиртах пли ацетоне.
Моно-н-пропиланилип [783]. Смесь 18,8 г (0,2 моль) анилина и 24 '
(0,1 моль) «-пропилового эфира n-толуолсульфокислоты нагревают на масляис
бане при 110° С в точение 5 ч. При охлаждении реакционная масса застывав
вследствие кристаллизации n-толуолсульфоната анилина, который растворив
в 70 мл воды. Выделившееся красно-коричневое масло извлекают эфиром, эфиг
иый раствор промывают водой и сушат над Na2SO4. При перегонке получав
12 г (87% от теоретического) мопо-и-пропиланилина в виде слегка желтовато!
масла; т. кип. 95—105° С (10 мм рт. ст.).
Ди-/1-пропиланнлиы |783]. Смесь 9,4 г (0,1 моль) анилина, 48
(0,2 лодь) «-пропилового эфира п-толуолсульфокислоты п 12,8 г (0,2 мол\
КОН нагревают на водяной бане. Реакция начинается сразу, сопровождая
бурным вспениванием реакционной массы.
* Фундаментальные работы в этой области принадлежат В. М. Родионову [Ви
Soc. Chim. [4], 39, 322 (1926); [4]. 45, 121 (1929)]. - Прим. ред.
[776] Т. L. Fletcher, М. Е. Taylor, A. W. Dahl, J. Org. Chem., 2
1021 (1955).
[777] W. Н, С. R и е gge berg J. С h е г n а с k, J. Am. Chem. Soc., 70, 18'
(1948).
[778] Th. I. Bieber, J. Am. Chem. Soc., 74, 4700 (1952).
[779] К. H. Slotta, L. Lorenz, Ber., 58, 1320 (1925); К. H. Slott
R. Tschesche, Ber., 60, 295 (1927).
[780] W. Gerrard, G. J. leacocke, Chem. a. Ind., 1954, 1538.
[781] E. J. Sakellarios, Helv. chim. acta, 29, 1675 (1946).
[782] D. К I a m a n n, Ann., 583, 63 (1953).
[783] К. II. Slotta, W. Franke, Ber., 63. 678 (1930).
Ill ОБМЕН КИСЛОРОДА НА. АЗОТ
467
Для завершения процесса массу нагревают еще 2 ч на -масляной бане
до 110° С. Кристаллизующийся при охлаждении и-толуолсульфопат калия рас-
1 воряют в воде, выделяющееся красное масло после очистки п сушки перегоняют
с небольшим количеством уксусного ангидрида. Выход 12,6 е (70% от теоретиче-
ского), светло-желтое масло; т. кип. 127° С (10 мм рт. ст.).
При взаимодействии вторичных аминов с бис-сульфоповымп эфирами гликолей
получают 1Ч,Гч,Г<',]\'-тетразаме1ценные диамины [784]:
KSO.O- (СН2)„-0SO2R --->
R\ -R'
—> >N—(CIb)„—N<" 4-2RSOaOH-NHR'R"
R"Z \R"
N,N,N'.N'-TeTpa3aMenjeHHbic диамины [784]. Бис-сульфоновый эфир
гликоля и соответствующий вторичный амин (20а«в) в безводной среде нагревают
до кипения в течение 20 ч при перемешивании. Затем отгоняют непрореагировав-
шпй вторичный амин и добавляют к раствору избыток 40%-ноп» раствора NaOH.
Маслянистый слои отделяют от водной фазы, экстрагирую! па водного слоя
продукт эфиром п сушат объединенные органические фазы над К2СО3. Остаток
после отгонки эфира перегоняют в вакууме.
По этому методу получены, например. N,N,N'. N'-тетрациклогексилэти^ен-
диамин (выход 86% от теоретического; т. пл. 102—104° С); N,N,N',N'-TeTpasTHn-
эчилендиамин (55% , т. кип. 89° С при 31,5 мм рт. ст.); N,N,N',N'-reTpaH3onpo-
пилтриметилендиамин (86%; т. кип. 83°С при 10—11 мм рт. cm.); N,N,N',N'-re-
тра-н-бутилтстраметилендиамин (78%; т. кип. 107—108° С при 1 мм рт. ст.);
^^М'.М'-тетрафецилтрпметИлепдиамин (65%; т. кип. 118—121е С при
0,3 .«л рт. ст.)?' р,р'-дпэгиламинодиэтидовый эфир (13?-6; т, кип. 69—70° С
при 1 мм рт. ст.).
N-Алкилированные гетероциклические соединения получаются из первичных
аминов и бис-сульфоновых эфиров гликолей за одну операцию [785]. Так, пз бис-
бснзолсульфоната 1,5-пептандиола и циклогексиламина получается N-циклогексил-
пиперидин; выход 81% от теоретического; т. кип. 234° С. Из эфира диэтиленгликоля
(1) и бензиламина образуется N-бонзилморфолия (И); выход 90% от теоретичесного;
т. кип. 79° С (2 мм рт. ст.):
/
Н2С
Аг8О2О-Н2С
Н
О о
С На
(Jn,-OSO2Ar —> Ц2С СН2
\/
Н N
/ 1
R
I
R
I п
где R=CeHeCH2; Ar = CsHs
К кипящему раствору 0,5 моль бис-арилсульфоната гликоля в 500 мл
сухого диоксана прибавляют по каплям в течение 2 ч 1,5 моль первичного амина.
Затем добавляют водный раствор 60 г (1,5 моль) NaOH и упаривают раствор
до половины объема. Остаток охлаждают, перемешивают с эфиром, эфирную
вытяжку фильтруют и фракционируют фильтрат.
[784] Th. М. L a a k s о, D. D. Reynolds, J. Am. Chem. Soc., 73, 3518 (1951).
[785] D. D, Reynolds, W. O. Kenyon, J. Am. Chem. Soc., 72, 1597 (1950),
30*
468
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Фениловые эфиры арилсульфокислот обычно непригодны для арилированше
как показал Ульман, исключение составляют лишь некоторые нитрофениловв|
эфиры [786, 787]. о
Так, при пропускании аммиака в кипящий бензольный раствор эфира п-толуол
сульфокислоты и 2,4-дпнитро-1-нафтола с количественным выходом образуется 2,Я
динитро-1-нафтиламин; аналогично с анилином получают 1-фениламино-2,4-динитр<
нафталин с выходом 98% от теоретического [787].
В некоторых случаях первичные амины, например аллиламин [771], получа®
алкилированием уротропина и гидролизом образующихся четвертичных уротропинн(
вых солей.
Важный метод синтеза чистых первичных или вторичных аминов основан $
алкилировании амидов сульфокислот с последующим расщеплением продуктов али^
лпрования.
При моноалкилировании амидов арилсульфокислот со свободной аминогруппа!
реакцию проводят в присутствии соды; в растворе КОН л^гко образуются диалкилр
рованные продукты [788].
N-Монометил-п-толуолсульфамид [788]. К горячему насыщенному ра<
твору 21,2 г (0,2 моль) безводного Ка2С0з в 25 мл воды прибавляют 19 г (0,11
n-толуолсульфамида и нагревают в колбе с обратным холодильником до полной
растворения. Затем прибавляют 18,6 s (0,1 моль) метилового эфира п-толуо^
сульфокислоты, кипятят еще 2 ч при перемешивании, охлаждают, добавляй
воду и фильтруют. Из фильтрата при подкислении выделяют 1,4анепрореагир<
вавшего п-толуолсульфамида (7,3% от загрузки). Для получения чистого монй
алкильного производного продукт реакции кипятят с избыточным количество!
раствора NaOH, отфильтровывают нерастворившийся диметилсульфамид (2 Ц
10% от теоретического; т. пл. 86,5° С) и подкисляют фильтрат HCI (1 : 1)
Выделяющийся N-монометил-п-толуолсульфамид достаточно чист; выход 13-<
(70,3% от теоретического); т. пл. 77° С. i
Так же получены N-моноэтил- и N-моно-н-бутил-п-толуолсульфамида
с выходами соответственно 86 и 65% от теоретического. ;
Диалкилирование амидов сульфокислот и дальнейшее алкилирование N-алкиЛ]
и N-арилсульфамидов [788] проводится в растворе КОН (ср. J773]).
N-Этил-га-толуолсульфанилид [788]. В 108 мл 10%-ного раствора NaOl
(0,3 .ноль) растворяют 74 г (0,3 моль) и-толуолсульфанилнда, прибавляют 60 4
(0,3 моль) этилового эфира и-толуолсульфокислоты и смесь нагревают в нолб
с обратным холодильником в течение 2 ч при 100° С при перемешивании. Поа#
окончания реакции смесь охлаждают при перемешивании, отфильтровывав
закристаллизовавшийся этилсульфанилид, промывают его водой и суша^
выход 78,8 г (94,8% от теоретического); т. пл. 88° С. Из фильтрата прк подкисл^
нии НС1 можно выделить 3,2 е (4,3% от загрузки) непрореагировавшего п-толуо$
сульфаиилида. ' ,
Этот метод может быть распространен на получение других ^М-дизамещениЫ^
амидов сульфокислот. Если исходят из незамещенных амидов, то реакцию проводя
с удвоенным количеством NaOH и эфира. Так же гладко протекает алкилировани
при помощи эфиров высших спиртов [773, 778]. ‘ j
Получение амидов арилсульфохлоридов и их алкилирование можно проводив
в одну операцию [788]. '
Омыление амидов сульфокислот рассматривается на стр. 421 и 624.
Реакции азотсодержащих соединений с амидами кислот <
Обменом карбонильного кислорода амидов кислот на атом азота первичный
или втори’гных амипов можно во многих случаях достаточно легко получить амидин^
Реакция протекает в присутствии галогенангидридов неорганических кислот, иапр®
[786] F. UJImann, G. N a d a L Вег., 41, 1870 (1908). ' %
[787] F. иПтапв, W. Bruck, Вег., 41, 3932, 3939 (1908).
[788] D. К 1 а ш а п*п G. Hofbauer, F. Drahowzal, Mh. Chem., 83, 870
(1952); ср. D. К lama nn, H. Bertsch, Chem. Ber., 89, 2007 (1956).
III. ОБМЕН КИСЛОРОДА НА АЗОТ
469
мер РС1Э, РОС13. PC1R, SOC12 и др. (иногда добавляют также алкилирующие средства).
Каталитическое действие галогенангидридов объясняется промежуточным образова-
нием импдохлоридов, которые в некоторых случаях можно выделить в чистом вида.
По этому методу получают главным образом формамидины. Ниже приведен метод
синтеза амидинов в присутствии PClg в среде сухого хлороформа.
М,М'-Дифенилформамидин [789]. К охлаждаемому раствору 4,8 а
(0,023 моль) PClg в 20 мл хлороформа небольшими порциями медленно приба-
вляют 2.4 г (0,02 моль) форманилида. После окончания реакции к смеси доба-
вляют раствор 1,9 мл (0,02 моль) свежеперегнанного анилина в хлороформе;
выпадает осадок. Реакционную массу нагревают 2,5 ч в колбе с обратным холо-
дильником, охлаждают в бане со льдом, фильтруют, промывают холодным аце-
тоном и сушат в вакууме. Из образовавшейся соли свободное основание форм-
амидина получают обработкой спиртовым раствором этилата натрия. Неочищен-
ный продукт, кристаллизующийся в бане со льдом, отфильтровывают, промывают
холодным спиртом и после сушки экстрагируют ацетоном. Полученный ацето-
новый раствор выливают в воду, выпадающий амидин еще раз переосаждают из
ацетона водой; выход 2,6 г (66% от теоретического); т. пл. 140—141° С.
Бредерек с сотр. [790] проводили реакцию с РОС13 в бензоле и получили таким
способом много разных амидинов с хорошими выходами.
М,М-Диметил-М'-фенилформамидин. К 27,4 г диметилфопмамида в 50 мл
абсолютного бензола в защищенной от влаги аппаратуре при 20—25° С прили-
вают по каплям при перемешивании 23,1 г РОС13 в 50 мл абсолютного бензола
и оставляют реакционную массу при комнатной температуре на ночь. На следу-
ющий день при ^5* С медленно по каплям прибавляют 11,6 г анилина в 40 .ил
абсолютного бензола, удерживая выделившееся масло путем интенсивного пере-
мешивания во взвешенном состоянии. Перемешивание продолжают 3—4 ч,
верхний бензольный слой сливают, остаток дважды промывают декантацией
по 50 мл бензола.
Для получения свободного основания продукт реакции растворяют в ледя-
ной воде, добавляют равный объем бензола и сильно подщелачивают 2 н. раство-
ром NaOH. Продукт выделяют из смеси встряхиванием с бензолом, отделяют
бензол и еще два раза экстрагируют продукт из водной фазы бензолом. Бензоль-
ные растворы объединяют, сушат над NaaCO8 и перегоняют; выход 15,6 е (85% от
теоретического); т. кип. 119—122° С (10 мм рт. ст.)\ 1,5955.
Аналогичным методом из диметилацетамида получен К,К-диметил-К'-
фепилацетамидин; выход 58% от теоретического.
Иначе протекает образование формамидинов в присутствии алкилирующих
средств; в этом случае на первой стадии реакции образуется формимидоэфир, кокорый
немедленно вступает в реакцию с избытком формамида, превращаясь в аиидин.чретод
исключительно простой.
Метилсульфат формамидина [791]. В защищенном от влаги приборе
нагревают при 50—60° С в течение 3 ч 126 г диметилсульфата и 90 г формамида.
После отгонки в вакууме образовавшегося метилформиата получают ейроп,
кристаллизующийся при трении стеклянной палочкой о стенки сосуда. Через
Сутки продукт реакции растирают с сухим тетрагидрофураном, отфильтровывают
в промывают сухим тетрагидрофураном и абсолютным эфиром. После сушки
в вакууме получают 117,5 г продукта (65% от теоретического); т. пл. 100—105° С
(после'кристаллизации из метилового спирта т. пл. 105° С).
[78Ъ] Н. G. Mandel, A. J. Hill, J. Am. Chem. Soc., 76, 3978 (1954).
[790] H. Bredereck, R. G о ш p p e г, К. Klemm, H. R e m pf er, Chem.
Ber., 92, 837 (1959).
[791] H. В re d о rec k, R. Go m p pe r et al., Chem. Bor., 92, 329 (1959).
ОБРАЗОВАНИЕ С- N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
2. Реакции карбонильных соединений
с азотсодержащими соединениями
Получение простых производных карбонильных соединений
Первичной реакцией при взаимодействии аммиака и его производных с карбо-
нильной группой является реакция присоединения с образованием так называемых
O.N-полуацеталей (стр. 379). Однако эти соединения но могут быть выделены в чистом
виде. Обычно они стабилизуются, причем отщепляется вода и между углеродом и азо-
том получается двойная связь (ср. стр. 379, 480, гак что формально реакция может рас-
сматриваться как обмен карбонильного кислорода на = N—R-группу. Конечные про-
дукты реакции представляют собой производные карбонильных соединений, имеющие
большое значение в синтезах и в аналитической химии. Такой способ образования
С— N-связи представляет особый интерес для препаративных целей, когда в молекуле
карбонильного соединения имеются другие функциональные группы или соответ-
ственно ориентированные двойные или тройные связи, так как при этом с одновремен-
ным образованием новых С—С-связей образуются гетероциклические системы. '•
Получение иминов, оснований Шиффа [792]. Мономерные имины К — CH=NHr
образования которых можно было бы ожидать при взаимодействии аммиака с альдеги-
дами, неустойчивы: некоторые из них полимеризуются (например, 2.4,6-триметилгек-
сагидро-1,3.5-трпазин из ацетальдегида), другие конденсируются с карбонильным
соединением (гидробензамид из бензальдегида) или претерпевают одновременно кон*
денсацию и полимеризацию (уротропин из формальдегида ,[793]).
Кетимины R2C=NH обычно тоже не могут быть получены из кетонов и аммиака.'
Одним из немногих исключений является флуоренопимин, который был получен про-
пусканием аммиака в флуоренон при 165° С с выходом 66% от теоретического [794]..
Недавно было показано, что прямое превращение кетонов в их имипы может прово-:
диться при помощи фосфинпминов. ।
Дифенилкетимин [795]. Раствор 5,85 г трифенилфосфинимина и 3,4 s.
бензофенона в 200 мл бензола нагревают 2 ч до слабого кипения и отгоняют бея-1
зол. В остатке получается масло, которое перегоняют в вакууме при 108° С'
(1 мм. рт. ст.). Выход дифенилкетнмина 3,24 г. При пропусканий! газообразному
НС1 в бензольный раствор кетимина выпадает его ^гидрохлорид, сублимируй
ющийся при 230—250° С.
О получении кетиминов из нитрилов и магнийорганических соединений см>
[796], а по методу Хеша см. [797]. I
Значительно большее значение, чем незамещенные имипы, имеют их Х-алниль-4
ные и N-арильные производные (основания Шиффа, азометины, анилы), получаемые*
пз первичных аминов и альдегидов или кетонов. >]
Для синтеза чисто алифатических альдиминов RCH=NR реакцию рекомендует-!
ся [798] проводить при 0° С без растворителя, прибавляя альдегид к амину и связывая!
образующуюся воду твердым КОН. Приведенный ниже пример можно распространить.?
па получение других иминов. " >
«
[792] Обзор реакций альдегидов с аммиаком и аминами см. М. М. S prunff Chenyl
Rev., 26, 297 (1940). ПИ
[793] Н. Н. Richmond, G. S. М у е г s, G. F. W г i g h t, J. Am. Chem. Soc..^|
70, 3659 (1948). j
[794] G. II. H а г r i s, B. R. H а г г i m a n, K. W. Whee ler, J. Am. Chernas
Soc., 68, 846 (1946). ' 7Я
[795] R. Appel, A. Hauss, Chem. Ber., 93, 405 (1960). .ifl
[796] P. L. Pickard, 9. J. Vaughan, J. Am. Chrnn. Soc., 72, 876, 5017 (1950)51
P. L. Pickard, T. L. Tolbert, J. Org. Chem., 26, 4886 (1961) 1
[797] P. E. S p oerr i, A. S. D u В о i s, Org. Reactions, 5, 387 (1949). ч
[798] К. N. Campbell, A. H. Sommers, В. K. Camp bell, J, Am Chem.:«
Soc., 66, 82 (1944). 'Л
Ill ОБМЕН КИСЛОРОДА НА АЗОТ
N-Пропилбутиральцимин [798]. В трехгорлой колбе емкостью 250 мл
снабженной мешалкой, обратным холодильником и капельной воронкой, к 23,6 ,
(0,4 моль) н-пропиламина прибавляют по каплям при охлаждении льдом в тече
ние 2 ч 28,4 г (0,4 моль) масляного альдегида. Смесь перемешивают еще 15 мин
добавляют твердый КОП и оставляют до четкого разделения смеси на два слоз
(около 10 .иип). Органический слой ос1авляют на ночь в холодильнике над мелко
раздробленным КОП и перегоняют над свежим КОН. Основное количество про
дукта отгоняется при 120—124° С; выход 70% от теоретического; ла° 1,4149
Следует отметить, что основания Шиффа этого типа, как и незамещенные у атом<
азота имины, довольно неустойчивы. У ких тоже явно выражена склонность к триме
рпзацми с образованием производных гексагидротриазина. Кроме того, при высоко!
температуре чисто алифатические основания Шиффа легко вступают в реакцию, анало
гнчнуто альдольной конденсации. Так, при нагревании «-бутилиден-н-бутиламина (I
в колбе с обратным холодильником (140—150° С) в течение 3 ч образуется основани.
Шиффа (11); выход 65% от теоретического [7Й9]:
2C3H;CII-NC4H9 —> Г СаН5—СН—CH = NC4Ha1 —>
1 [ С4НЭКН—СНС3Н7 ]
—CjIIj-C—CH=-NC4He + C4H9NH2
II
СН—С3Н7
II
Реакцию между альдегидом и амином можно проводить также в таких условиях
при которых в основном образуется не простейшее основание Шиффа, а продукт ег
дальнейшей конденсации [800].
а.р-Ненасыщенпые альдегиды реагируют с аминами в большинстве случав!
с одновременным присоединением по С=О- и С=_С-связям. Для сохранения двойной
С—С-связи имин получают из альдегида и N-алкилкетпмипа [801, 802[:
В\ Н\
R-CHO+ >C=NRW —► R—СН—NR"'-|- >С^О
R»/ Rff/
Б отличие от чисто алифатических оснований Шиффа продукты конденсаца
первичных, особенно ароматических, аминов с ароматическими альдегидами являйте,
достаточно устойчивыми соединениями. Ввиду их ярко выраженной способности к крВ
сталлизации они могут служить для идентификации аминов и альдегидов. Обыча
реалия протекает с хорошими выходами уже при смешении реагирующих Лцестгз
ппогда при непродолжительном нагревания (например, в спиртовом растворе™Част*
склонность к конденсации столь велика, что реакцию можно проводить в водной срСд^
Бензальметиламин [803]. К 106 г бензальдегида в течение 20 мин прг?
бавляют 150 г 33%-ного водного раствора метиламина. Реакция протекает с вы-
делением значительного количества тепла. Для завершения реакции смесь осте
вляют на 12 ч при койнатной температуре, затем насыщают раствор поваренно
[799] W. S. Emerson, S. М. Hess, F. С. U h 1 е, J. Am. Chem. Soc., 63, 8?
(1941).
[800] А. М. Paquin, Chem. Вег.. 82, 316 (1949).
[801] С. W. S т i t h, D. G. N о г I о n, S. A. В a 11 а г <3. J. Am. Chem. Зое
75, 3316 (1953).
[802] Пат. США 2513996; С. А., 44, 8361 (1950).
[803 j N. II. С г о тп w е 1 1, R. D. В a bson, Ch. Е. Harris, .J. Am. Chert
Soc., 65, 312 (1943).
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
солью и продукт экстрагируют эфиром. Высушенный эфирный раствор перего-
няют на эффективной колонке; выход 83 г (70% от теоретического); бесцветная
жидкость; т. кип. 183—185° С.
Бензальанилин [804]. В колбе емкостью 500 мл к 106 г бензальдегида
при хорошем перемешивании прибавляют 93 г анилина. Реакция начинается
через песколько секунд п сопровождается разогреванием и выделением воды.
Через 15 мин смесь выливают в химический стакан емкостью 600 мл, содержащий
165 мл 95%-ного спирта. Кристаллизация начинается через несколько минут.
Раствор охлаждают льдом, осадок отфильтровывают и сушат на воздухе; выход
152—158 г (84—87% от теоретического); т. пл. 52“ С. При упаривании маточного
раствора в вакууме получают еще 10 г бензальанилина. Рекомендуется применять
свеженерегианные бензальдегид и анилин.
Бензальанилнн является полиморфным соединением и не имеет постоян-
ной температуры плавления; у разных препаратов т. пл. колеблется от 42 до 56° С.
В отличие от альдегидов кетоны, особенно ароматические, обычно с трудом всту-
пают в реакцию с аминами. Для получения соответствующих кетиминов реакцию при-
ходится проводить при повышенной температуре и в присутствии катализаторов кон-
денсации, которыми, по данным Редделина [805], являются ZnCI2 и комплексные
соединения его с аминами.
Бензофепонанил [805]. К смеси 10 з бензофенона и 10 г анитина при
160“ С в течение 0,5 ч прибавляют 1 — 2 а свежерастерюю безводного ZnCxa.
После охлаждения желтую массу кипятят с небольшим количеством бензола
или эфира. Из полученного раствора анил выпадает в виде желтых кристаллов;
выход 12 г (85% от теоретического); т. пл. 117° С.
Аналогичным образом проводилось взаимодействие 2-метилцпклогокса-
нона с анилином и циклогексанона с анилином и о-толуидином, с применением
толуола в качестве растворителя [806].
Вместо ZqCI2 применяли также РОС13. Этот способ рекомендуется в тех слу-
чаях, когда аддукты ZnCl2 с аминами нерастворимы и не оказывают достаточного
каталитического действия [807]. Особенно эффективным катализатором конденсация
флуоренона-9 с ароматическими аминами является эфират трифторида бора [808].'
Простой метод получения чисто алифатических кетиминов приводит Нор-
тон [809].
Амин и кетон нагревают, удаляя образующуюся реакционную воду в виде
азеотропа с бензолом, толуолом, ксилолом или избытком кетона.
Растворимые в воде амины и кетоны, которые не вступают в реакцию в та-
ких условиях, оставляют на 24 ч при комнатной температуре в присутствии кон-,
центрированной IIC1 (3 г копц. НС1 на каждые 5 моль амина и кетопа), связывают*
образующуюся в результате реакции воду твердым NaOH и перегоняют органик
ческий слой. Этими двумя способами синтезировано около 40 алифатических1
кетиминов с очень хорошими выходами [809]. . а
Ацетопанил не может быть синтезирован прямым взаимодействием аце-р
тона с анилином; при этом получается окись мезитила, реагирующая дальше
с анилином с образованием 2,2,4-триметилдигидрохиполина [810]. Ацетонанилг'
[804] L. А. В i g е 1 о w, Н. Е a t п о u g h, Org. Synth., 8, 22 (1928). $
[805] G. Reddelien, Ber., 43, 2476 (1910); 42, 4759 (1909); J. H. BillmaDJ
К. M. T a i, J. Org. Chem., 23, 5.35 (1958).
[806] C. Ilan sc h, F. Gschwend, J. В am es berger, J. Аш. Chenin
Soc., 74, 4554 (1952). J
[807] A. W. Weston, R. J. M i c h a e 1 i s jr., J. Am. Chem. Soc. 73, 1381’
<1951).
[808] M. E. Taylor, T. L. F 1 e t c h e r, J. Org. Chem., 26, 940 (1961). ?
[809] D. G. N о г t о n, V. E. H а и r y, et al., J. Org. Chem.. 19 1054 (1954). ••
[810] G. R e d d e 1 i*e n, A. Thurm, Ber., 65, 1511 (1932); I. W. Elliot jr.,;
P. Yates, J. Org. Chem., 26, 1287 (1961).
F
III. ОБМЕН КИСЛОРОДА HA AJOT
легко получается при конденсации кеталей ацетона с анилином [811]. Однако
этот способ применим не во всех случаях, так как многие ацетали и кетали про-
являют необычную устойчивость к действию аммиака или аминов (см. [469
470]).
Взаимодействие эфиров 0-кетокарббновых кислот с аммиаком или первичными
и вторичными .аминами протекает с выделением тепла чаще всего уже при смешении
реагентов. При этом получаются не соответствующие иминокислоты, а таутомерные
ненасыщенные аминокислоты. Так, из эфиров р-кетокислот образуются производные
эфира р-аминоакриловой кислоты:
R-CO-CH2-COOR R-C=CH-COOR
nii2
Этиловый эфир p-аминокротоновой кислоты [812]. В 97,5 г ацетоуксус-
ного эфира при перемешивании пропускают в течение 5 ч сильный ток сухого ам-
миака. В течение первого часа температура повышается до 40°С; далее, применяя
охлаждение (баня с холодной водой), ее поддерживают в пределах 35—40° С.
Затем добавляют 100 мл эфира и отделяют образовавшуюся воду. При вакуумной
перегонке с колонкой Видмера отгоняется 88,2 г (90% от теоретического) этило-
вого эфира p-аминокротоновой кислоты; т. кип. 91—93° С (8—9 мм рт. ст.);
1,4980.
Аналогичным способом получены эфиры р-метиламяно- и р-диметпламино-
кротоновой кислоты.
Ароматические амины часто реагируют только при повышенной температуре
или в присутствии кислотных катализаторов (например, гидрохлорида амина) [813];
конденсацию ацетоуксусного эфира с анилином проводят в кипящем бензоле в присут-
ствии небольшого количества ледяной уксусной кислоты [814]. Все же нагревание
до слишком высокой температуры не рекомендуется, так как при этом в реакцию
вступает эфирная группа и образуется амид [716, 815]. (ср. стр. 456).
Следует также упомянуть, что под действием аммиака и первичных аминов
1,4-дикетоны циклизуются с образованием производных пиррола (реакция Пааля —
Кнорра [816]).
В настоящей книге не рассматриваются и многочисленные синтезы гетероци-
клических соединений, при которых наряду с новыми С—С-связями между углеродом
и азотом образуется двойная связь из карбонильной и аминной функции. Ири Ж)м
только в редчайших случаях удается выделить стадию образования C=N-cbh3« в само-
стоятельную реакцию. Новым примером такой реакции является необычно гладко
протекающий сиптез производных пиридина по Вольману [817].
На первой стадии реакции происходит присоединение активированных снамп-
пов, например эфира p-аминокротоновой кислоты, к ацетиленам, содержащим в а-по-
ложении карбонильпую группу, а образующееся аминокарбопильное соединение (1)
[811] L. Claise'n, Вег., 29, 2931 (1896).
[812] 8. A. G1 ick man, А. С. С о ре, J. Am. Chem. Soc., 67, 1017 (1945).
[813] 8. С о f f е у, J. К. Т h от s о n, F. J. W i 1 s о л. J. Chem. Soc., 1936, 856.
[814] G. A. Reynolds, Ch. R. Ilaiisc r, Org. Syulh., Coll. v. 3, 1955, p. 374.
[8154 L. Knorr, Ber., 25, 775 (1892).
[816] L. Lederer, С. P a a 1, Ber., 18, 2591 (1885); L. Knorr, Ber., 18, 299
(1885).
[817] F. Bohlmann, D. Ralilz, Chem. Ber., 90, 2265 (1957).
474
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
переходит при нагревании в производное пиридина — алкиловый эфир 2,6-диметил-
яикотиновой кислоты (II):
CH CH \ СИ \
C lie—COOR ПС C—COOR НС C—COOR
1 + II -> 1 II -> 1 II
l-С С- СПз СПз-С С—СНз CH3—C C-C[J3
II /
o h2n 0 nh2 N
I II
Этиловый эфир 2,6-диметилникотиновой кислоты [817]. К раствору
12,9 г этилового эфира p-аминокротоновой кислоты в 30 мл спирта прибавляют
6,8 г бутин-1-она-З. По завершении реакции смесь нагревают на водяной бан^
още 15 мин, а затем охлаждают. После перекристаллизации из метилового
спирта получают аддукт (I) в виде бесцветных призм; выход 90% от теоретиче
ского; т. пл. 135° С.
При нагревании до 120—140° С аддукта (I) в вакууме протекает циклизд
ция с образованием этилового эфира 2,6-диметилпикотпновой кислоты; выхб;
90% от теоретического; т. кип. 60° С (0,1 мм рт. ст.).
Получение гидразонов, семикарбазонов и аналогичных соединений. Гидразин,
и особенно несимметричные продукты его замещения, образуют с альдегидами и кете*
нами исключительно ценные в препаративной химии и в анализе производные.
Гидразоны — производные назамещенного гидразина, часто являются неустой-
чивыми соединениями. Они представляют малый интерес для препаративных цели!
и применяются, например, в реакции Кижнера [818] или в синтезе гетероциклически!
соединений. Получение таких гидразонов часто усложняется тем, что в гидразине оба
NH2-группы имеют тенденцию вступать в реакцию, причем образуются азины. Во избёг
жание этого синтез гидразона чаще всего проводят с избытком гидразина. Катализа-
торами конденсации служат триэтпламин [821], ВаО или NaOH в спиртовом раствор
Вместо гидразипгидрата можно применять и сульфат гидразина; серную КН-
лоту при этом связывают ацетатом натрия и осаждают образовавшийся сульфат натрв
спиртом. Конденсация протекает при комнатной температуре или при умеренном наРр
вании. Продолжительность взаимодействия кетонов несколько больше, чем дл
альдегидов.
Гидразон ацетона [819, 520]. В 15 г гидразипгидрата вносят несколы
кусочков ВаО и приливают 15 г ацетона, причем реакционная масса сильно раз'
гревастся. Через несколько дней реакционную массу фильтруют и перегоняю1
выход продукта около 31% от теоретического; т. кип. 124—125° С. При хран
пии гидразон медленно разлагается с выделением аммиака и азота.
Способ получения гидразона бензила см. [820]; о синтезе гидразона и азив
бензофенона см. [821].
Алифатические кетазины получаются при взаимодействии стохпометрическя
количеств реагентов.
Диметилкетазин [822]. К смеси 1 моль гидразинсульфата и 2 .«ty
ацетона небольшими порциями при встряхивании прибавляют несколько больП
2 люль NaOH в виде 20%-ного водного раствора. Образовавшийся кетазин извл
кают эфиром, раствор сушат и перегоняют; т. кип. 131° С.
[818] D. Т о d d, Org. Reactions, 4, 378 (1948).
[819] Th. C u r t i u s, L. P f 1 u g, J. prakt. Chem. [2], 44, 53,5 (1891).
[820] С. II. F. A 1 1 e n, Org. Synth., Coll. v. 2, 1943, p. 497.
[821] D. П. R. Barton, R.E. O’Brien, S. Sternhell, J. Chem. Soe
1962, 470.
[822] Th. C u г t i u s, *E. Zinkeisen, J. prakt. Chem. [2], 58, 315 (1898
Th. C u rt i u s, K. Th u n, J. prakt. Chem. [2], 44, 161 (1891).
Ill ОБМЕН КИСЛОРОДА НА АЗОТ
475
Этот способ непригоден для синтеза алифатических альдазпнов.
Подробное описание получения бензальазина см. (823].
В отличие от обычных реакций карбонильных соединений из формальдегида
и гидразина образуется продукт более высокой степени конденсации — «тстраформаль-
трпс-азпн» [824]
нгГ'\/\н
I I I
UN N Nil
при термической деполимеризации которого образуется мономерный формальдазип
[825]. Точно так же не удается выделить простой фенилгпдразон формальдегида;
в уксуснокислом растворе образуется преимущественно симметричный 1,4-дифенил-
гексагидротетразин, однако выделены и другие продукты конденсации [826].
Гораздо большее значение, чем простые гидразоны, имеют феиилгидразоны и
их 4-нитро- и 2,4-динитропроизводные. Они часто служат для выделения и идентифи-
кации карбонильных соединений.
Феиилгидразоны лучше всего получаются в разбавленном уксуснокислом рас-
творе; применение более концентрированной уксусной кислоты может привести к обра-
зованию ацетилфенилгидразина (т. пл. 130—131° С, испр.). Если рядом с карбонильной
группой имеется гидроксильная группа, возможно образование озазона. Во избежа-
ние этого синтез следует проводить в отсутствие кислоты в среде, близкой к ней-
тральной. Обычно конденсация протекает уже на холоду, но практически почти всегда
реакционную массу нагревают некоторое время на водиной бане. Чаще всего компо-
ненты вступают в реакцию и без растворителя. а,р-Нснасыщенные альдегиды и кетоны
могут реагировать с фепилгидразином с замыканием кольца и образованием производ-
ных пиразола.
Фенилгпдразон ацетофенона (827]. Эквимолярные количества ацето-
фенона (40 г) и фенилгидразина‘(36 г) нагревают 1 ч на паровой бане. Горячий
продукт реакции растворяют в 80 мл этилового спирта. При охлаждении льдом
и трении стеклянной палочкой о стенки сосуда фенилгпдразон кристаллизуется.
Его промывают 25 мл этилового спирта; выход 54—57 г; еще 4—10 г фенилгидр-
азопа получают при упаривании маточного раствора и промывного спирта- Фенил-
гидразон сушат в вакуум-эксикаторе над СаС12; общий выход 61—64 г (87—91%
от теоретического); т. пл. 105—106° С.
Карбонильное соединение может быть выделено из фснилгидразона нагрева-
нием с соляной кислотой, а во многих случаях и нагреванием с бенз- или форм-
альдегидом.
2,4-Динптрофенилгидразоны обычно хорошо кристаллизуются и обладают чет-
кой температурой плавления. Правда, они склонны к образованию смешанных и<и-
сталлов. что может привести к ошибкам при идентификации карбонильных соединеЯй.
Для выделения и идентификации простых кетонов особенно пригоден 2,4-динитрофе- •
нилгидразин. Применение его для получения производных а-оксикетонов не рекомен-
дуется [828].
2.4-Динитрофенилгидра.зин конденсируют с карбонильными соединениями
в присутствии минеральных кислот (11С1, II2SO4, НГ1РО4) [828—830]. В некоторых
случаях серцуго кислоту очень трудно удалить из продукта реакции [830]. В присут-
ствии уксусной кислоты 2,4-динитрофенилгидразин может еще и ацетилироваться [829],
[823] Н. II. Hatt, Org. Synth., Coll. v. 2, 1943, p. 393.
К. A. Hofmann, D. Storm, Ber., 45. 1725 (1912).
[82aj N. P. Neureiter, J. Am. Chem. Soc., 81, 2910 (1959).
826] E. S c h m i t z, R. Oh m e, Ann., 635 82 (1960).
1827] *R. L. Shriner, W. C. Ashley, E. W e 1 c h, Org. Synth,, Coll. v. 3,
1955, p. 726.
1828] Ch. F. I-I, Allen, J. Am. Chem.-Soc., 52, 2935 (1930).
[849] II. II. Strain, J. Am. Chem. Soc., 57, 738 (1935).
476
образование с—n-связи в результате обмена
что иногда приводит к недоразумениям. Образовавшийся 2,4-дпнитрофепплгидразов^
иногда бывает трудно отделять от нспрореагировавшего 2,4-дппитрофенилгв»
дразина, поэтому реакцию следует проводить с избытком карбонильного спедтлте- й
ния |830].
Для идентификации пользуются раствором 1 а 2,4-дииитрофепплгидразина
в 3 мл концентрированной H2SO4 и 10 мл спирта. При умеренном нагревании реактив <
взаимодействует со спиртовым раствором карбонильного соединения [829, 831]. С ним
конденсируются даже такие карбонильные группы, которые не вступают в реакцию
с другими реагентами [831].
Джонсон [832] рекомендует применять фосфорнокислый раствор дпнитрофеннлт’4
гидразина: 50 г 2,4-динитрофепилгидразииа нагревают на паровой бане с 600
85%-ной Н8РО4 и добавляют к получеппому раствору 395 мл 95%-ного спирта. Й
Шани [833] проводит реакцию спиртового раствора карбонильного соединения!
с раствором 2,4-динитрофенилгидразина в диметиловом эфире диэтпленгликодя
(«диглим»), прибавляя в_ качестве катализатора несколько капель концентрировав-:^
ной НС1. ' /й
2,4-Динитрофенилгидразоны стерически затрудненных кетонов, которые лишь')
с большим трудом или вообще не получаются прямым путем (например, из дибутил-,J?
диамил-, динонилкетона), большей частью легко получаются взаимодействием неэа>$
мещениых гидразонов с 2,4-дииитрохлорбензолом [520] (стр. 430).
Для выделения и идентификации карбонильных соединений наряду с фенил-'
гидразином и его нитропроизводными использовали также много других арилгидра*?
зинов со свободной ИН2-группой. Однако чаще всего их применение не дает никаких^
преимуществ по сравнению с применением 2,4-динитрофенплгидразппа. Правда,
известный интерес представляют некоторые производные гидразина, являющиеся/:
специфическими реактивами на определенные группы карбонильных соединений»ш
й
Влиянию заместителей на реакцию замещенных фепилгидразинов с угле-^
водами и о селективности их действия на смеси углеводов посвящена работа Штр<1-,1
[834]. Например, так называемый дифенилметандиметилдиглдразин й
СН3 СН3
H2N— N—СН2—N—NH2 j
часто применяется для разделения смесей углеводов, так как он значительной
легче конденсируется с альдозами, чем с кетозами [835]. Несимметричный фенилу
метилгидразин также может быть использован для идентификации альдо»,?
и кетоз: с альдозами он образует только моногидразопы, тогда как при действия?
его на кетозы легко получаются соответствующие озазоны [836]. п-Карбокся-^
фенилгидразин дает производные карбонильных соединений, которые можнОй
определять титрованием [837]. Меньший интерес представляют гидразоны,,;
получаемые при помощи алифатических гидразинов, папример из метил-?
и NjN-диметилгидразина ,[838]. Я
[830]
[831]
[832
[833
[834
[835
[836
[837
[838]
C. F. H. Allen, J. H. Richmond, J. Org. Chem., 2, 222 (1938).
О. В а у e г, в книге II ou ben -Wey 1, Methoden der organischen Chemie,
Bd. 7/1, 4 Aufl., Stuttgart, 1934, S. 465.
G. D. Johnson,!. Am. Chem. Soc.. 73, 5888 (1951); 75, 2720 (1953).
П. J. Shine, J. Org. Chem., 24, 252 (1959).
H. H. S t г о h, Chem. Ber., 91, 2645, 2657 (1958); 90, 352 (1957).
J. v. Braun, Ber., 41, 2169, 2604 (1908); 43, 1495 (1910).
C. Neuberg, Ber., 35, 959 (1902).
St. Veibel, Acta Chem. Scand., 1, 54 (1947);
H. W. Schmidt, Acta Chem. Scand., 2, 545 (1948).
R. H. Wiley, St. C. S 1 а у ш a k e г, H. Kraus,
22, 204 (1957); fi. H Wiley, G. Irick, J. Org.
(1959).
St.
J. Org, Chem.,'
Chem., 24. 1925)
Ill ОБМЕН КИСЛОРОДА H\ АЗОТ
477
Гидразиды кислот конденсируются с карбонильными соединениями так же,
как алкил- и арилгидразины.
Значительный пптерес для аналитических целей представляют семккарбазопы,
которые хорошо кристаллизуются, имеют четкие температуры плавления п поэтому
особенно пригодны для идентификации низших алифатических альдегидов.
Семикарбазоны можно получать в водной среде, добавляя карбонильное соеди-
ненно (в случае необходимости в спиртовом растворе) к водному раствору гидрохло-
рида семикарбазида в присутствии ацетата щелочного металла [839].
Для расщепления семикарбазонов их нагревают с разбавленной минеральной
кислотой или вытесняют карбонильное соединение бензальдегидом или 2,4-дивитро-
бензальдегидом.
Тиосемпкарбазоны также являются большей частью трудпорастворимыми и
хорошо кристаллизующимися соединениями. Их получают при действии свободного
нюсемикарбазида в спиртовом растворе на карбонильное соединение.
Тиосемикарбазон л-нитробензальдегвда [840]. Смешивают теплые рас-
творы 30,2 з (0,2 моль) n-нитробензальдегида в 300 мл 95%-ного этилового
спирта и 18,2 з (0,2 моль) тиосемикарбазида в 300 мл воды. Сразу же начинается
кристаллизация тиосемикарбазона. После охлаждения его отфильтровывают;
выход 38 г (85% от теоретического); т. пл. 232—233° С (разл.).
Основания Шиффа, арилгидразоны, семикарбазоны и оксимы при нагревании
их с тиосемикарбазидом в спиртовом растворе также почти всегда переходят в тио-
сс'микарбазоны. Это очень устойчивые соединения и в отличие от других производных
карбонильных соединений обычно не расщепляются с выделением карбонильного сое-
динения (за исключением тносемикарбазона ацетона, стр. 377).
Особенно быстро выделяются альдегиды и кетоны, далее из очепь разбавленных
растворов при действии питроаминогуанидияа [841 — 843]
,NH
h2n-nh-€C
XNH-NO3
Реакцию проводят в водной или водно-спиртовой среде в присутствии неболь-
шого количества уксусной кислоты [841]. Для выделения углеводов нитроампногуани-
дин непригоден [841].
Очень легко подвергаются обратному расщеплению гидролизующиеся в мягких
условиях продукты конденсации карбонильных соединений с 5-(а-фенплэтил)-семи-
оксамгидразидом [844]
СНз
С6Нб—CH—NH-CO—CO-NH-NHa
Такие производные приходится получать в абсолютно безводной среде. Сиф
компонентов в сухом бензоле нагревают в течение нескольких минут в присутствии
следов иода |844].
При помощи оптически активных форм этого гидразида можно разделять
на оптические антиподы рацематы карбонильных соединении [844]. Для этой же
цели применялись следующие соединения: Р-гартрамидгидразид [845]
[839] N. Zelinsky, Вег., 30, 1541 (1897).
[840] J. Bernstein, II. L. Y а 1 е et al., J. Am. Chem. Soc., 73, 906 (1951).
[841] W. F. Whitmore, A. J. R e v u k a s, G. B. L. Smit h, J. Am. Chem.
Soc., 57, 706 (1935).
i842> G. B. L. Smith, E. P. Shoub, J. Am. Chem. Soc., 59, 2077 (1937).
[843] R. A. Henry, G. B. L. Smit h, J. Am. Chem. Soc., 74, 278 (1952).
[844] N. J. Leonard, J. 11. Boyer, J. Org. Chem., 15, 42 (1950).
[ 845] F. N er de 1, E. Henkel, Chem. Ber., 85, 1138 (1952).
478
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
H2NNH—СО—СН(ОН) — СН(ОП)— СО — NH2, и £-6-ментплсемикарбазид [846],
H2NNH — СО—NH — С10Н19. При расщеплении антиподов камфоры был с успе-
хом использован Z-мснтиловый эфир карбазиновой кислоты [8471
NH.,NII—СО—ОС10Т118. ,
Растворимые в воде гидразоны альдегидов и кетонов получают из толуолсуль*
фоната гидразида N-мстилникотиновой кислоты
N CH3CeH4S0?
при кипячении смеси реагентов в абсолютном спирте в течение 15 мин. Обрязуюттгиас^
продукты конденсации обладают четкими температурами плавления и легко подвер-^
гаются очистке. Они вновь легко расщепляются кислотами или переводятся в 2,4-дини-^
трофенилгидразоны. .
Такие же растворимые в воде гидразоны получают из реактивов Жирара
СН,-со -NH-NH.ji
peaKi ив Жирара р
[(CfT3)3N -CH2-CO-NII-MI2]C1-
Особенно хорошие результаты дают эт гидразиды при разделении смесей природны:
соединений [849, 850J.
Обычно конденсация проводится в среде водного метилового спирта в npi
сутавии ацетата натрия [489].
В тех случаях, когда уксусная кислота почему-либо мешает или в
присутствии усложняются операции по выделению копенного продукта, реакцв
проводят в нейтральной среде, пользуясь катионообмеппыми смолами тш
амберлит IRC 50 (пли пермутит В, вофатит СР 300), которые после проведен!
конденсации отделяют фильтрованием [851].
Дальнейшая обработка реакционной массы производится обычно без выделу
пил гидразонов. Полученный раствор встряхивают с соответствующим нерастворимы!
в воде растворителем, например эфиром; при этом гидразоны остаются в водной фазсм
а сопровождающие их органические вещества переходят в ор1анический слой. Карб»
нильные соединения легко регенерируются из водного раствора при помощи мягкой
гидролитического расщепления гидразонов в присутствии кислот- При соответствую-
щем подборе условий реакции, особенно-темиературы и pH, можно провести достаток
но четкое разделение различных карбонильных- соединений как па стадии конденса]
цни, так и при расщеплении образовавшихся гидразонов. Неустойчивые в кислой сред]
карбонильные соединения (например, цитраль) рекомендуется выделять обменяй!
реакцией с избыточным количеством формальдегида [851]. ;
[846] А. В. Crawford, F. J. W i 1 s о n, J. Chem. Soc., 1934. 1122.
[847] R. В. Woodward, T. В. К oh man, G. С. Ы а г г i 5, J. Am. Chei
hoc.. 63, 120 (1941).
[848] C. F. H. A 1 1 e n, J. W. Gates jr., J. Org. Chem., 6, 596 (1941).
[848a] Обзорная статья: О. [I. Wheeler. Chem. Rev., 62, 205 (1962).
[849] A. St. Pfa u, P. A. P la tine r, Helv. chim. acta, 22, 64u (1939).
[850] T. Reichsteinf Helv. chim. acta, 19, 1107 (1936).
[851] C. L, T p j t e 1 b a u in, J. Org. Chem., 23, 646 (1958).
III. ОБМЕН КИСЛОРОДА НА АЗОТ
479
Получение оксимов. Для превращения карбонильных соединений в оксимы
реакцию проводят обычно в водно-спиртовой среде с избытком гидрохлорида гидроксил-
амина, нейтрализуя свободную кислоту ацетатом патрия или содой [852, 853]. Опти-
мальное значение pH ~ 4,7. Альдегиды и простые кетоны реагируют обычно при ком-
патпой температуре. (О получении оксимов нитрозированием соединений с активной
метиленовой группой см. на стр. 393.)
Энанталъдоксим [854]. К смеси 348 г (5 .воль) гидрохлорида гидроксил-
, амина, GOG мл воды и 4G0 г (4 моль) энаптола прп перемептивадии прибавляют
по каплям раствор 265 г (2,5 моль) Na2CO3 в 500 мл воды, не допуская подъема
температуры выше 45° С. После окончания прибавления карбоната смесь еще
1 ч перемешивают при комнатной температуре. Маслянистый слой отделяют,
промывают двумя порциями воды (по 100 мл) и перегоняют в вакууме. При тем-'
псратуре бани 140—147° С отгоняется 420—480 ? оксима, т. кип. 103—107° С
(6 мм рт. ст.). Перегнанный оксим постепенно кристаллизуется, т. пл. 44—
46° С; после перекристаллизации из 60%-пого спирта т. пл. 53—55° С.
По данным Виланда [852], часто применяемый для оксимированпя выспшх
потопов (например, камфоры [855], ацетофенона [856], пропиофенона [856] и бензо-
фенона [857]) метод, по которому реакция проводится при нагревании в растворах едких
щелочей, не имеет никаких преимуществ перед обычным методом, использующим
ацетатный буфер. Например, из 7-витро-а-тетралона .и гидрохлорида гидроксиламина
в водно-спиртовой среде в присутствии ацетата патрия оксим образуется с выходом
98,5% от теоретического [858].
Исключительно сильным оксимирующим реактивом является смесь гидроксил-
амина с пиридином [859]. Кетоны с объемистыми углеводородными радикалами, кото-
рые не реагируют в обычных условиях с гидроксиламином, гладко оксимируются
этим реактивом при нагревании, при необходимости в спиртовой среде [860, 861],
Пиридиновый метод применялся даже для количественного определения карбониль-
ных соединений [862].
Диарилкетоксимы [86f ]. Раствор 0,01 моль кетона и 0,011 моль соляно-
кислого гидроксиламина в смеси пиридина и абсолютного спирта (в большинстве
случаев достаточно 4—5 мл каждого из растворителей) нагревают 2 ч на паровой
бане, отгоняют в вакууме растворители и промывают водой обычно быстро кри-
сталлизующийся остаток.
Для оксимированпя некоторых стерически затрудненных кетопов, например
о-толиларилкетонов, требуется очень длительное нагревание (около 40 «)•
Оксимы углеводов, используемые при расщеплении углеводов по методу Воля,
могут быть также с успехом получены из гидрохлорида гидроксиламппа в присутствии
ппридина [863].
При взаимодействии альдегида со смесью солянокислого гидроксиламина и
пиридила в присутствии уксусного ангидрида одновременно происходит отщеплений
поды и образуется нитрил [863]. w
[852J W. Н й с к е 1, М. Sachs. Ann., 498, 176 (1932).
[853] F. Nerdel, I. Н u I d s с li i n s к у. Chem. Вег., 86, 1005 (1953).
[854] E. W. Bousquet, Org. Synth., Coll. v. 2, 1943, p. 313.
[855] K. v. A и w e г s, Ber., 22, 604 (1889).
[856] K. N. Ca m p be]], В. K, Ca mp bel 1, E, P. Ch apul, J. Org. Chem.,
8, 99 (1943).
[857] A. Lachmaii Org. Synth., Coll. v. 2, 4943, p. 70.
1858] II. J. Shine, J. Org. Chem., 23, 318 (1958).
[859] J. Meisenheimer, E. M a h ] c r, Ann., 508, 191 (1934).
[860] W. E. Bachmann Ch. H. Boatner, J. Am. Chem. Soc., 58, 2097
(4 936).
[861 ] W. E. Bachmann M. X. Bart о n, J. Org. Chem., 3, 300 (1939),
[862] W, M, D, Bryant, D. M. Smith. J. Am. Chem. Soc., 37, 57 (1935).
[863] С. H. Trabert, Arch. d. Pharm.. 294/66, 246 (1961).
480
ОБРАЗОВАНИЕ G-N-СВЯЗИ В^РГЗУЛЬТАТЕ ОБМЕНА
2-Нитробензонитрил [863]. В колбе емкостью 100 мл растворяют в 30 и,
пиридина 5,0 з 2-нитробензальдегида (т. пл. 42.5° С) и 4.0 г гидрохлорида гидр*
ксиламина. К полученному раствору небольшими порциями прибавляют 13 д
уксусного ангидрида, причем температура раствора повышается до 80° С. Peai
цпопную массу нагревают 1 ч на кипящей водяной бане, охлаждают и выливая)
на 500 г льда. Когда лед расплавится, кристаллический осадок отфильтровываем
промывают водой и перекристаллизовывают из этилового спирта.
4,1 г; игольчатые кристаллы; т. пл. 110° С.
Выход нитршб
Вместо относительно дорогого гидроксиламина для синтеза оксимов моче
применять более дешевый гидроксиламин-М^М-дисульфокат натрия [864) (легко в
лучаемый из NaNO,, NaHSO3n S02), причем выделение этого соединения из реакцно
ной массы не обязательно. Подробное описание такого способа получения оксим
на примере ацетоноксима см. [864] и циклогексапоноксима см. [865]. В некотор
случаях применялась также натриевая соль гпдроксиламин-М-моносульфокисл*
Из-за низких температур плавления оксимы мало пригодны для идентификаг
карбонильных соединений. Они часто образуются в виде медленно кристаллизующи:
масел. Оксимы низкомолекулярных карбонильных соединений часто удобнее очищ;
при помощи вакуумиой перегонкк.
Многие оксимы дают стереоизомеры [867, 868|. Из ароматических карбонн.
ных соединений обычно образуются син-, или а-оксимы, у которых гидроксильная гр;
па и альдегидный водород (соответственно в случае кетонов более короткий радик:
находятся в цис-положении, тогда как в алифатическом ряду чаще образуются б*'
реакционноспособные анти-, или p-изомеры, с транс-конфигурацией.
R—С-Н R-C-H
I ’I
N—ОН НО—N
син- или я-оксим анти- илн З-оксим
Нередко в результате реакции образуются оба изомерных продукта; та
смеси, плавящиеся в широком интервале температур, разделяют фракционной крис'
лизацней или переводят при помощи парциальной изомеризации в один из стерео!
меров [868].
Получение аминов с функциональными группами
в а-положении
Взаимодействие аминов с карбонильными соединениями может протекать
только с образованием двойной связи между углеродом и азотом, как это paccMai
валось выше. В некоторых случаях возможна также стабилизация образующихся
первой стадии реакции O.N-полуацеталей в результате замены гидроксильной rpyi
на второй атом азота (I).
>С = О_н^^
R'z
R\ zOTT
/С,
R’Z ХХ
hnr; R znR2
"Hl° * R'/^X
где (T) X = NR"; (П) X-CNJ (III) X«SO3H.
Аналщичный тип реакции наблюдается при взаимодействии карбоннльны
соединений с синильной кислотой (II) нлн бисульфитом (III) в присутствии амииО!
[864] W. L. Semon, Org. Synth., 3, 6f (1923).
[865] J. C. Eck, C. S. Marvel, Org. Synth.. Coll. v. 2, 1943, p. 76.
[866] W. L. Semon, V. R. Damerell, J. Am. Chem. Soc., 46, 1290 (191
[867] K. ,Fr e и d e n berg, Stereocheftlie, Leipzig — Wien, 1933, S. 963ff, ,»
[868] G. Wittig, Stereochemie, Akademische Verlagsgesellschaft m. b. II., Leij>
zig, 1930, S. 182IL
'fl
Ill ОБМЕН КИСЛОРОДА НА АЗОТ
481
в этом случае первая стадия реакции представляет собой присоединение по С = О-
спязи с образованием аддукта (циангидрина или бисульфитного соединения), соответ-
ствующего О.N-полуацеталю, гидроксильная группа которого обменивается на
атом азота.
Получение N.N-ацеталей и аналогичных соединении. O.N-Полуацетали, обра-
зующиеся в результате присоединения вторичных аминов к карбонильным соедине-
ниям (стр. 378), не могут стабилизоваться в результате отщепления воды и образова-
ния оснований Шиффа, как это возможно при присоединении первичных аминов.
Вместо этого происходит обмен гидроксильной группы на второй аминный остаток
и получаются N.N-ацетали [869, 8701-
К',К-Ацетали, или «аминали» [871 ], являются в сравнении с О, N-полуацеталями
довольно устойчивыми соединениями, и образование их протекает без осложнений даже*
при проведении синтеза с недостаточным количеством амина. Если взаимодействие-
протекает слишком медленно, то получающуюся в реакции воду связывают К2СО3;.
под действием кислот М,М-ацетали тотчас гидролизуются.
Диморфолинофеинлметан [870].
0/-4N-CH—
К 21,2 г (0,2 моль) бензальдегида при охлаждении прибавляют 17,4 г (0,2 моль)*
морфолина. Раствор мутнеет и через 1 ч начинает выпадать белый осадок. Реак-
ционную массу оставляют на ночь, затем фильтруют. Фильтрат представляет
собой непрореагировавший бензальдегид (примерно 10 мл). Выход сухого не-
очищенного продукта 20,4 г (77% от теоретического). После перекристаллизации1
из 95%-ного спирта получают бесцветные кристаллы; т. пл. 101 —101,5° С.
Аминали с подвижным атомом водорода в р-положеннн к азоту легко отщепляют-
1 моль амина при нагревании и переходят в енамины [872—874]:
R-CH2-Cn(NRi)2 Zhnr; R-CH=CH-NR;
На этой реакции основан простой метод получения енаминов из карбонильных
соединений и вторичных аминов, причем синтез можно проводить без выделения’
^,Х-ацеталей.
1-Морфолиноциклопентен-1 [875].
В круглодовной колбе емкостью 500 мл. снабженной обратным холодильником»
и ловушкой Дина — Старка, нагревают до кипения 42 г (0,5 моль) циклопента^|
нона и 65,4 а (0,75 моль) морфолина в 200 мл бензола в течение 5 ч. Начинающееся
сразу выделение воды заканчивается через 4—5 ч. После отгонки бензола реак-
ционную массу фракционируют в вакууме; выход 67,3 г (88% от теоретического);
т. кип. 105 — 109° С (13 Мм рт. ст.).
Па получении енаминов из морфолина и циклопентапона [875] пли циклогек-
санона [876] основан разработанный Хюннигом изящный метод удлинения’
[869] В. л. Henry, W. М. Dehn, J. Am. Chem. Soc., 71, 2271 (1949).
[870] M. Z i e f, J, Ph. Mason, J. Org. Chem., 8, 1 (1943).
871] H. Bohme, Angew. Chem.. 68, 224 (1956).
1872] C. Mfi n n j oh, H. Da v j dsen, Ber., 69, 2106 (1936).
[873] E. Bcnzing, Angew. Chem., 71, 521 (1959).
[874] G. О p i t z, H. Hellmann, II. W. Schubert, Ann., 623, 112 (1959)..
[875] S. H ii n i g, W. Lendle, Cliem. Ber., 93, 909 (1960).
[876] S. H ii n i g, E. Benzing, E. L ii c k e, Chem. Ber., 90, 2833 (1957).
31 Заказ 1839.
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
углеродной цепи алифатических карбоновых кислот па 5 или 10 и соответственно1^
6 или 12 атомов углерода [877]. Jj
а,Р-Ненасьпценные альдегиды могут присоединять амин одновременно по С=О«1
и С—С-связи: .Я
/О
rch2cii=chc/ -F3NHR2 —> rch2chch2ch(nr;)24-ЩО
ХН I
NRj
Получающиеся из этих аддуктов З-амнно-1-енамины легко отщепляют вторуй
аминогруппу с образованием сопряженных С^С-связсй [878|:
R-CH2-CH-CH=NR' —> r—сн=сн—сп—сн—nr;
I
nr;
При взаимодействии дивторичных диаминов с карбонильными соединениям^
получаются циклические аминали, конечно, лишь в тех случаях, когда аминогруппу
находятся в 1,2-, 1,3-или 1,4-положениях. Так, например, 1,3-диамины легко кондей
сируются с альдегидами и кетонами и дают производные гексагидропиримидина [879а
880], а дивторичные этилендиамины образуют производные имидазолпдинов [880*
1,3-Диарилимидазолидины[881]. К раствору N.N'-flnapnnBTHneHflHaMHEfll
в метиловом спирте или этилацетате прибавляют избыточное количество 309Й
ного раствора формалина и подкисляют реакционную массу уксусной кислотой^
Конденсация 1\г,№-дифенилэтилсндиамина с альдегидами протекает настолько
гладко, что ее можно применять для выделения и идентификации алифатическга
и ароматических альдегидов [882]: >3
СН2—Nil—Аг
I 4-ОНС—R
СН2—NH—Аг
Аг
СН2—
| ^-сн-в
сн3-ц/
чАг
Реакцию рекомендуется проводить в растворе в метиловом спирте в присутствИ
одной капли уксусной кислоты, иногда при непродолжительном нагревании. Обрй
зующиеся 2-замещенные 1,3-дифенилимидазолидины хорошо кристаллизуются и нйй
вятся в узком интервале температур [882].
Гетероциклические соединения со структурным элементом М,М-ацеталей могу!
быть получены и по реакции карбонильных соединений с производными гидразинй
[877] S. Hunig et al., Chem. Ber., 90, 2833 (1957); 91, 129 (1958); 92, 652 (193$
93, 909, 913 (1960).
[878] C. Mannich, K. Handb e, K. Rot h, Ber., 69, 2112 (1936); W. L aj
genbcck, O. Godde, L. Weschky, R. Schaller, Ber., 75, 2<
(1942); H. Fi nch, E, A. P e t e г s о n, S. A. В a 1 1 a г d, J. Am. CheE
Soc., 74, 2016 (1952). .
[879] E. Bergmann, D. Her ma r, E. Zimkin, J. Org. Chem., 13, 3|
(1948). ;
[880] J. L. R i e li so m er, G. It. Morey, J. Oil-. Chem., 15, 245 (1950).
[881] L. Jaenicke, E Erode Ann., 624, 134 (1959).
[882] I-l.-W. W anzhck, W. Locliel, Chem. Ber., 86, 1463 (1953).
Ifl ОБМЕН КИСЛОРОДА НА АЗОТ
483.
Тик, из тетрагпдрофталазипа (I) и формальдегида с количественным выходом обра-
зуется 1,2,4,5-бис-(о-ксилилен)-гексагидро-™.ил-1етравин (II) [883]:
Зга реакция не идет с дысптими альдегидами н может поэтому служить характер-
ной реакцией на формальдегид (чувствительность реакции 10-5 г!мл).
В качестве реактива применяют 0,1%-ный водный раствор гидрохлорида тотра-
гидрофталазина. Значение pH испытуемого раствора должно лежать в пределах 4,5—6
(ацетатный буфер). Соединение (II) выпадает в течение 5 мин', из воды — белые призмы,
из горячего хлорбензола — листочки; т. пл. 265—270° С (разл.).
Реакции формальдегида с гидразином [824] и фенилгмдразипом [826], при кото-
рых происходят подобные циклизации, уже упоминались ранее (стр. 475).
ITj первичных аминов аминали получались лишь в редких случаях. В таких
реакциях явно преобладает тенденция к образованию оснований Шиффа, лишь в ред-
ких случаях сопровождаемая побочной реакцией образования аминалей. Правда,
основным продуктом реакции бензальдегида с n-нитроанилином является соответст-
вующий аминаль [884].
Несколько проще получаются N-ацилироваппые аминали из альдегидов и ами-
дов кислот, не содержащих заместителей при азоте [885]:
0 zNHCOR'
пе/ +2HsNCOR'--------► П-СИ
Хп \nhcoh'
Известны также смешанные М,М-ацстали из амидов кислот и аминов [73, 886,
887]. N-Лриламинометнлфталимиды образуются с высокими выходами при кипячении
в течение 30 мин спиртового раствора смеси фталимида, формальдегида и первичных
ароматических аминов:
Они могут применяться для идентификации аминов [73], так как представляют
собой кристаллические вещества с четкими температурами плавления. Аналогично
реагируют вторичные ароматические и алифатические амины [886].
Получение а-аминонитрилов, гидантоинов в аналогичных соединений. Гидре*
сильные группы в аддуктах карбонильных соединений с бисульфитом и синильнои
кислотой исключительно легко обмениваются на аминогруппы.
Большое число а-ампносульфокислот [888] получено из аддуктов с бисульфитом
(а-оксисульфокислот) и аммиака или аминов при комиатной температуре или при уме-
ренном нагревании. Кроме того, эти соединения являются важными промежуточными
[883] Е. Schmitz R. О h m е, Monatsber. dtsch. Akad. VViss. (Berlin), 1, 366
(i 939).
[884] A. 11 a n t z sc h. 0. S c li w a h, Ber.,,34, 833 (1901).
[885] W. A. Noyes D. B. Forman, J. Am. Chem. Soc., 55, 3493 (1933);
A. E. Martell R. M. Herhst, J. Org. Chem., 6, 878 (1941); T. R. L e-
wis jr., F. R. Butler, A. E. Martell, J. Org. Chem., 10, 145 (194o).
[886] *H. W. Heine M. B. Winstead, R. P. Blair, J. Am. Chem. Soc., 78,
672 (1956).
[887] H. Zinner II. II e r b i g, II. W i g e r t, Chem. Ber., 89, 2131 (1956).
[888] L. Neelakentan, W. H. Hartung, J. Org. Chem., 24, 1943 (1959).
31*
484
образование с—n-связи в результате обмена
продуктами прп синтезе аминокислот по Штреккеру (ср. стр. 717 сл.) в варианте Кневев
геля — Бухерера). По атому методу бисульфитное соединение альдегида подверга]
сначала взаимодействию с аммиаком или аминами; при действии цианида щелочив
металла или синильной кислоты на образующуюся при этом а-аминосульфокисло
получается а-аминонитрил, который далее омыляют:
/Н
R-c<0
/н
R—С—SO3Na
ЧОН
,Н
В-С-ЗОз^а
XNH2
,Н
R-C-CN
\nh3
R—СН—СООН
I
NHa
Разработан [889] общий метод получения а-аминонитрилов по этой схс
ори необходимости натриевые соли а-амипосульфокислот можно выделить в чис
виде.
Подробную пропись получения диэтиламиноацетонитрила из бисуль
ного соединения формальдегида, дизтиламина и цианида натрия см. [!
Аналогично перевод циангидринов в а-аминоиитрилы является одной из ст?
синтеза Штреккера в варианте Тимана и тоже проводится при комнатной темпера!
«ли при умеренном нагревании *.
Нитрил а-амшюизомасляной кислоты [891, 892]. Сухой аммиак пр<
скают в свежеприготовленный ацетонциангидрин, не допуская подъема
пеРатуры выше 65° С. Реакционную массу некоторое время выдерживают
60° С и извлекают эфиром образовавшийся а-аминонитрил. Полученный раот
перед перегонкой тщательно (!) сушат над СаС18, так как продукт реакции от
легко разлагается. Перегонкой в вакууме получают аминонитрил с выхо,
64% от теоретического; т. кип. 48—50° С (11 мм рт. ст.). Лучшие выходы^
85% от теоретического, получают при добавлении водоотнимающих сре
(Na8SO4) уже при пропускании аммиака.
Разработано получение N-замещенных нитрилов а-амикоизомасл
кислоты из ацетонциангидринов и аминов при комнатной темпера
[893J.
Описаны различные примеры использования синтеза Штреккера — Тш
с другими компонентами; с а-аминодизтилуксусной [894], а-аминофенилуг
ной [895J и а-амино-а-фенилпропионовой кислотой [896].
* Важным вариантом метода Штреккера является одностадийный синтез а-ам
кислот, имеющий большое практическое значение [Н. Д. Зе линек
Г. Л. С т.а д н и й о в, ЖРФХО 58, 722 (1906)]. — Прим. ред.
[889] D. В. Luten jr., J. Org. Chem., 3, 594 (1939).
[890] С. H. F. A 1 1 e n, J. A. V a n A 1 1 a n, Org. Synth,, Col], v. 3, 1
p. 275.
[891] If. T. Buch ere r W Steiner, J. prakt. Chem. [2], 140,
(1934).
[892] II. T. Clarke, II. J. Bean, Org. Synth., Coll. v. 2, 1943, p. 29.
[893] L. .1. Exner, L. S. Luskin, P. L. De Benne vjlle, J.
Chem. Soc., 75, 4841 (1953).
[894] R. E. Steiger, Org. Synth., Coll. v. 3, 1955, p. 66.
[895] R. E. Steiger, Org. Synth., Coll. v. 3, 1955, p. 84.
[896] R. E. Steiger, Org. Synth., Coll. v. 3, 1955, p. 88.
III. ОБМЕН КИСЛОРОДА НА АЗОТ
485
При взаимодействии циангидринов не с аммиаком, а с (№Н4)гСО3, обычно с хо-
рошими выходами образуются 5-замещепвые гидантоины (синтез Бухерера — Хенце)
1891);
Нх ,CN
,><
R'z ХОН
R4 /CN
СО— NH3
NH
HxJ-NH
R'/ -\H—CO
R /CO—NH
>4 I
XNH-CO
Эта реакция протекает очень гладко и может быть использована для идентифи-
кации карбонильных соединений [897).
5,5-Диметнлгидантоия [891]. Нагревают 0,1 моль ацетонциангидрина,
0,2 леоль (NH4)aCO3 и 80 мл воды до 30—40° С. Энергичная реакция, заканчива-
ющаяся через 30 мин, сопровождается гомогенизацией реакционной массы.
При упаривании в вакууме получают гидантоин с почти количественным выходом;
после перекристаллизации из лигроина или этилового спирта т. пл. 174—175° С.
Для синтеза подобных соединений можно исходить также пе из циангидрина,
а непосредственно из цианида и карбоната аммония.
Часто при омылении гидантоинов получают а-аминокислоты с лущпнми выхо-
дами, чем нз соответствующих а-аминонитрилов. Из аминокислот, полученных через
циангидрины и гидантоины, можно назвать метнодин [898], лизин [899, 900), аланин.
[901] и фенилаланин [901].
Получение аминов восстановительным алкилированием
Взаимодействие аммиака и амииов с альдегидами и кетонами в присутствии
соответствующего восстановителя является важным методом получения аминов. В ка-
честве восстановителей применяются главным образом каталитически возбужденный
водород [902], муравьиная кислота и ее производные (реакция Лейкарта — Валлаха,
обзор [903]). Если карбонильной компонентой является формальдегид, то в некоторых
случаях он одновременно действует и как восстановитель.
Подробно описаны способы получения из NH4C1 и формальдегида метиламина
(выход 50% от теоретического) [904] и триметиламина (выход 89% от теоретичес-
кого) [905].
Восстановительное алкилирование в принципе не отличается от гидрирования
оснований Шиффа, но оно более удобно, так как и получение и восстановление про-
водятся в одну стадию.
Каталитически возбужденный водород в качестве восстановителя. Восстанови»
тельное алкилирование каталитически возбужденным водородом обычно проводятЯ|
в автоклаве. В качестве катализаторов гидрирования чаще всего применяют никель
Ренея (реже кобальт Ренея) и платиновые металлы. Давление водорода и температура
[897] Н. R. Н е n z е, R. J. S р е е г, J. Am. Chem. Soc., 64, 522 (1942).
[898] Е. Pierson, М. G i е 1 1 а, М. Т 1 з h 1 е г, J. Am. Chem. Soc., 70, 1450
(1948); D. О. Holland, J. Н. Nay] er, J. Chem. Soc., 1952, 3403.
899} o. A. R ogers, R. D. E mm ick et al., J. Am. Chem. Soc., 71, 1837 (1949).
900] R. Gaudry, Canad. J. Res., Sect. B., 26, 387 (1948).
901] R. Gaudry, Canad. J. Res., Sect. B, 26, 773 (1948).
902] w. S. Emerson, Org. Reactions, 4, 174 (1948).
903] M. L. M о о г e, Org. Reactions, 5, 301 (1949).
9041 C. S. Marvel, R. L. Jenkins, Org. Synth., 3, 67 (1923).
905) R. A da ms, C. 8. Marvel, Org. Synth., 1, 79 (1921); R. Adams,
В. K. Brown, Org. Synth., 1, 75 (1921).
48«
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
реакции и значительной мере-.определяются реакционной способностью карбонил
него соединении и активностью катализатора, поэтому давать какие-либо рекоменд
цнп для выбора условий реакции не представляется возможным. При работе с никел
вымп катализаторами давление водорода составляет от 20 до 150 ат л рабочая темпер
гура от/10 до 150е С. Однако известно немало случаев, когда гидрирование достаток
гладко протекало у?вд при комнатной температуре и незначительном давлеш
(1—3 ат}.
Наибольшее значение имеет восстановительное алкилирование для получен!
первичных аминов из альдегидов и кетонов. Обычно такой синтез проводится в спирт
вом растворе с избытком аммиака в присутствии никеля Ренея.
Формальдегид мало пригоден для алкилирования аммиака, так как реакцв
протекает в основном с образованием уротропина. Низшие алифатические альдегид
рекомендуется прибавлять к реакционной массе постепенно, во избежание альдольна
конденсаций. С высшими алифатическими и ароматическими альдегидами- реакци
большей частью протекает гладко и не требует применения особых мер пре;
осторожности.
н-Гептиламин (906). В автоклав с мешалкой загружают 113 г (1 мол
энанталя, 150 мл метилового спирта, 10 г никеля Ренея и 50 мл безводного амш
ака. Смесь гидрируют при 90° Си давлении водорода 100 ат. Примерно чер<
30 мин поглощение водорода прекращается. Катализатор отфильтровывай
и отгоняют аммиак и метиловый спирт через колонку высотой 20 см до тех по]
пока температура бани не достигнет 100° С. Азеотропной перегонкой с бензоло
удаляют воду. После удаления бепзола при 50° С (14 мм рт. ст.) отгоняете
н-гептнламин; выход 93,5 г (81% от теоретического).
Бенаиламитт [907]. К раствору 51 г (3 моль) аммиака в 300 ,ил холо;
ного этилового спирта прибавляют 3 моль бензальдегида и 10 г никеля Рене/
Гидрирование начинается при 40° С и начальном давлении водорода 90 ат
заканчивается через 30 мин при 70е С. После фильтрования фильтрат перег<
няют, выход бензиламина 287 г (89,4% от теоретического); т. кип. 70—80е i
(8 мм рт. ст.); выход дибепзиламина 21,7 г (7,1%); т. кип. 140 — 150е 1
(7 мм рт. ст.).
Мстилалиилкетоны реагируют с аммиаком так же, как альдегиды, с хорошим
выходами, например, из гептанона-2 получается 2-аминогептан (выход 75 — 80% от тес
ретического) [908]. Высптис кетоньг, особенно кетоны, разветвленные в а-по.то?к6яй
к кетогруппе, даже при избытке аммиака легко восстанавливаются до вторичнИ
спиртов. Добавление NH4C1 или уксусной кислоты (1% от количества когона) повй
шает выход амина [909]. Нортон [809] получил первичные амины сочень хорошим^
выходами при гидрировании смеси молярных количеств кетона и безводного аммиаЮ
(без растворителя) в присутствии никеля Ренея при 120—150е С и 35—70 am. j
Изопропиламин [809]. В качающемся автоклаве гидрируют 696л
(12 моль) ацетона и 204 г (12 моль) жидкого аммиака над 50 г никеля Ренея др
140° С и давлении водорода 52 ат. Прн перегонке смеси получают 545 г (77л
от теоретического) пзопропиламина; т. кип. 31,5—32,5° С; 1,377.
В аналогичных условиях из метилэтилкетона получен 2-аминобут81
с выходом до 90% от теоретического; из метиламилкетона — 2-аминогепта1
с выходом 90% и даже из дийзобутилкетона [809] при 150° С и 70 ат — соответ4
ствующий амин с выходом 48%.
Реже проводилось восстановительное алкилирование аммиака при более низки]
давлениях водорода. По данным .Хаскельберга (910), некоторые кетоны, напримв]
[906] F. М б ] 1 е г, в книге Н о u b о n - W е у 1, Methoden der organised
Chemie, Bd. 11/1, 4 Aufl., Stuttgart, 1957, S. 605.
[907] Ch. F. Winau s, J. Am. Chem. Soc., 61, 3t>66 (1939).
[908] E. П о h г m a n п, H. A. S h о n 1 e, j • Am. Chem. Soc., 66, 1516 (1944
[909] H. Wilms, в книге H ou ben -We yl, Methoden der organischen Chemi
Bd. 11/1, 4 Aufl. Stuttgart, 1957, S. 611.
[910] L. Ilaskelberg, J Am. Chem. Soc., 70, 2811 (1948).
III. ОБМЕН КИСЛОРОДА НА АЗОТ
487
фенилацетоп и 1-диэтПламмно1Гептапон-4, гидрируются с хорошими выходами (85%
от теоретического) до первичных аминов в спиртовом аммиаке при комнатной темпера-
туре и нормальном давлении водорода в присутствии никеля Ренея. На платиновых
катализаторах при низких давлениях получаются, по-видимому, преимущественно
вторичные амины [911]. Однако, поданным Александера и Майзегэйдса [912], первич-
ные амины можно получать с удовлетворительными выходами и на платиновых
катализаторах при низких давлениях водорода, проводя восстановление в присутствии
несколько более чем молярного количества NH#C1.
Восстанавлииают водородом взвесь 0,2 г окиси платипы в 10 мл воды, доба-
вляют 0,3 .моль кетона, 20 г (0,37 моль) NH4C1, 225 мл насыщенного раствора
аммиака в метиловом спирте и 25 мл водного раствора аммиака и гидрируют при
давлении 1 — 3 ап. По этому методу из соответствующих кетонов, в частности,
получены 4-амино-2-метил-пептан (выход 57—65% от теоретического), 4-амино-
гептан (40—60%), 3-амино-2,4-димстилпентан (55%) и а-фепилэтпламин (69%)
[912].
Восстановительное алкилирование не является таким общим методом для полу-
чения вторичных аминов, как для первичных. Взаимодействие аммиака с 2 моль кар-
бонильного соединения в алифатическом ряду приводят обычно к образованию смеси
моно-, ди- и триалкиламинов. Но из бепзальдегида, а также из о-хлор- и о-метилбензаль-
дегида соответствующие дибензиламины получаются с выходом 80—90% от теорети-
ческого [907].
Дибензиламин [907]. Его получают так же, как монобепзиламин
(стр. 486), с той лишь разницей, что реакцию проводят с 3 моль бензальдегида
и 1,5 моль аммиака; выход дибензиламина 81%, выход монобензиламииа 12%
от теоретического.
Более гладко, чем диалкилирование аммиака, протекает моноалкилировакие
первичных аминов. Только формальдегид легко реагирует дальше с образованием тре-
тичных аминов. С другими алифатическими альдегидами выходы вторичных аминов
получаются достаточно йдсокимп, но во избежание альдольной конденсации альдегид
рекомендуется добавлять к реакционной массе постепенно.
, Первичные ароматические амины подвергаются восстановительному алкили-
рованию алифатическими альдегидами с удовлетворительными выходами в спир-
товом растворе на никелевых или платиновых катализаторах уже при давлении
водорода 3 аж в присутствии ацетата натрия [913].
Хорошие выходы вторичных аминов достигаются при действии ароматических
альдегидов на первичные алифатические и ароматические амины, как при низких,
так и при высоких давлениях водорода [914].
N-Бензил-м-толупдин [915]. Смесь 1 моль бензальдегида и 1 моль .и-т^у-
идина в 200 мл эфира гидрируют в присутствии 8—10 г никеля Ренея при комнат-
ной температуре и давлении водорода около 70 ат. Выход продукта 89—94%
от теоретического; т. кип. 153—157° С (4 мм рт. ст.).
pH] A. S к i t a, F. Keil, Ber., 81, 1452 (1928).
[912] Е. R, Alexander, A. L. M i s e g a d e s, J. Am. Chem. Soc-, 70, 1315
r (1W
[913] W. S. Emerson, P. Jl. Walters, J. Am. Chem. Soc., 60, 2023
(1938); W. S. Emerson, W. D. Bobb, J. Am. Chem. Soc., 61,3145
r (1939).
[914] D. M. Balcom, C. R. Nollcr, Org. Synth., 30, 59 (1950); K. Boden-
d о r f, H. Raaf. Arzneimittol-Forsch., 5, 695 (1955).
1915] C.H.F. Allen, J. Van A 11 an, Org. Synth., Coll. v. 3. 1955,
p. 827.
488
ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
N-Метилбензиламин [&16]. Сначала готовят катализатор гидрирование*
смеси 0,5 г PdCl2 и 5 г активного угля в разбавленном солянокислом растворе
Многократным декантированием с метиловым спиртом удаляют воду, добавляю-]
к катализатору 45 г бензальдегида, раствор 31 г метиламина в метиловом спирт(
п гидрируют смесь водородом при атмосферном давлении. Поглощение водороде
протекает быстро, примерно 1 л в течение 5 мин. Выход продукта 44 г; т. кип,
78° С (14 мм рт. ст.).
При восстановительном алкилировании ароматических аминов нередко перед
гидрированием удаляют образовавшуюся воду азеотропной перегонкой или гидрируют
предварительно полученное основание Шиффа (стр. 518). Такая модификация синтеза
особенно выгодна при восстановительном алкилировании кетонами (809, 917], которые
легко восстанавливаются до вторичных спиртов. Но и в этом случае введение
каталитических количеств NH4C1 или уксусной кислоты подавляет образование
спирта (ср. стр. 486).
Исключительно легко протекает восстановительное алкилирование этаноламина
альдегидами и кетонами при давлении водорода 2 ат в присутствии платинового ката-
лизатора [918(.
Третичные амины обычно получаются методом восстановительного алкилирова-
ния с неудовлетворительными выходами. Некоторое значение имеет лишь метилиро-
вание вторичных и диметилирование первичных аминов формальдегидом. Боумен [919]
описывает синтез ряда алифатических диметиламинокислот гидрированием смеси амино-
кислот с формальдегидом в водной среде при нормальном давлении в присутствий
палладия на активном угле. Лучшие результаты получаются обычно при постепенном
добавлении альдегида к реакционной массе. При метилировании ароматических
аминов приходится принимать специальные меры, чтобы предотвратить ноиденсацию
формальдегида с ароматическим ядром (920(.
Для восстановительного алкилирования часто применяют не только первичны^
амины, но и такие азотсодержащие соединения, которые при восстановлении превра-
щаются в первичные ямины, как, например, нитрозо- и нитросоединения, нитрилы н
другие производные. Так, из нитробензола в одну операцию можно получать N-алкил-,
и N.N-диалкиланилины (921, 922]. На стр. 519 приведен синтез алкилариламинов и»;
ароматических нитрлслединеттий. ‘
Циклические вторичные амины можно получать гидрированием таких карбо-*
нильных соединений, в которых на соответствующем расстоянии от карбонильной?
группы имеется другая функциональная группа, восстанавливающаяся'до первично!^
аминогруппы [923, 924]. Например, при гидрировании p-бензоилпропионитрила в среде]
метилового спирта в присутствии никеля Ренея водородом при нормальном давлений;
происходит циклизация и образуется 2-фенилпирролидин с выходом 77,5% от теорет'
тического [923].
В некоторых случаях проводилось также восстановительное алкилирование^
гидразина. Например, из ацетона и гидразина в кислой среде в присутствии платив
нового катализатора и давлении водорода 2 ат с практически количественным выходома
получается 1Ч,]Ч'-диизопропилгидразин (925]. 1
[916]
[917]
918
919
920
921
922
923
924
(925]
В. W е g 1 е г, W. Frank, Chem. Вег., 69, 2071 (1936). 4
R. Е. L ц t z et al., J. Org. Chem., 12, 760 (1947); J. S. В u c k, J. Am. Chem.
Soc., 53, 2192 (1931).
А. С. С о p e, E. M. Tlancoc k, J. Am. Chem. Soc., 64, 1503 (1942). ]
R. E. Bowman, IT. H. Strou d', J. Chem. Soc., 1950, 1342.
D. E. Pearson, J. D. Bruton, J. Am. Chem. Soc., 73, 864 (1951). '
W. S. Emerson, H. W. Mobrma n, J. Am. Chem. Soc.. 62, 69 (1940M
W. S. Emerson, C. A. U г a n e c k, J. Am. Chem. Soc., 63, 749 (1941). '
E. B. Knot t, J. Chem. Soc., 1948, 186. ',1
H. H e n e c k a, Chem. Ber., 82, 104 (1949). j
H. L. L о c h t e, J. R. В a i I e y, W. A. Noyes, J. Am. Chem. Soc.,-’
43, 2597 (1921)4 H. L. L о c h t e, W. A. No yes, J. В. В alley, J. Аш^
Chem. Soc., 44, 2556 (1922).
III. ОБМЕН КИСЛОРОДА НА АЗОТ
489
Реакция Лейкарта — Валлаха *. Реакция Лейкарта — Валлаха представляет
собой восстановительное алкилирование аммиака или аминов карбонильными соедине-
ниями в присутствии муравьиной кислоты или ее производных, играющих роль вос-
становителей [923]:
R, , R’ R\ ZR"
V,O + nN< Ч-НСООН —► уСН — N< Ч-СО2 + Н2О
В'/ \R"' R'/ \R"
Наплучшие результаты дает взаимодействие аминов с формальдегидом и выс-
шими кетонами, температура кипения которых выше 120® С. Первичные и вторичные
амины получаются в виде N-формильных производных, ив которых свободные основа-
ния выделяют гидролизом. Реакция Лейкарта — Валлаха применяется более ограни-
ченно, чем описанное ранее каталитическое восстановительное алкилирование. Однако
использование в качестве восстановителя муравьиной кислоты позволяет применять
при алкилировании такие компоненты, в которых имеются легко гидрирующиеся груп-
пы, как, например, двойные связи, нитро- и нитрильпые группы. Эти группы, восста-
навливающиеся каталитически возбужденным водородом, не изменяются в условиях
реакции Лейкарта — Валлаха.
Первичные и вторичные амины метилируются формальдегидом до третичных
иминов с хорошими выходами. На каждую вводимую метильную группу берут 1 —
1,25 моль формальдегида и 2—4 моль муравьиной кислоты.
Метилирование первичных и вторичных алифатических аминов до тре-
тичных [926]. К смеси 5 моль 90%-ной муравьиной кислоты и 2,2 моль 35%-ного
раствора формальдегида при охлаждении прибавляют 1 моль первичного амина.
Для алкилирования вторичных аминов достаточно половины указанного коли-
чества муравьиной кислоты и формальдегида, но избыток рсагонтой не мешает.
Смесь нагревают на паровой бане. Сначала реакция протекает с бурным выделе-
нием СО2, затем выделение СО» прекращается и нагревание реакционной массы
продолжают еще в течение 2—4 ч; общее время нагревания 8—12 ч. После доба-
вления более 1 моль НС1 отгоняют формальдегид и муравьиную кислоту. Бес-
цветный остаток растворяют в воде, разлагают гидрохлорид амина 25%-ным
раствором NaOH и отгоняют амин с вОдяным паром. Дистиллят насыщают
сухим КОН, отделяют масляный слой и сушат над КОН, после чего амин пере-
гоняют над натрием. По атому методу были получены следующие амины с выхо-
дами более 80% от теоретического: диметил-ч-бутиламин, т. кип. 94® С; диметил-
бензиламин, т.. кип. 176—180° С; N-метилпиперидин, т. кип. 106° С.
Метилированные гидразины нс могут быть получены иа гидразингидрата таким
способом [927].
Поскольку формальдегид может реагировать с ароматическими аминами не
только по атому азота, но и по атомам углерода ароматического ядра, амины типашн-
лпна могут метилироваться по этому способу только в том случае, когда замест^^ли
в 2-, 4- и 6-положепиях исключают возможность подобной конденсации. По методу,
опробованному Борковски и Вагнером [928], с хорошими выходами метилируются
и амины с одним или с двумя заместителями в орто- или пара-положении.
На паровой бане нагревают 2,5 моль параформа и не мепее 3 моль 98%-
ной муравьиной кислоты. К этой смеси при интенсивном перемешивании в тече-
ние 10—15 мим, прибавляют 1 моль первичного ароматического амина и продол-
жают нагревание еще 5 мин. Если получаемый метилированный амин склонен
[926]
[927]
[928]
L
В изучении этой реакции деятельное участие принимал Н.М.Кижнер [ЖРХО.31,
872, 1033 (1899); 32, 381 (1900)]. В литературе бна известна как реакция Лей-
карта. — Прим. ред.
П. Т. С 1 а г k е, Н. В. G i 11 е s р i е, S. Z. Wei sshaus, J. Am.
Chem. Soc.,, 55, 4571 (1933).
F. К 1 a g e s, G. No ber, F. Kircher, M. Bock, Ann., 547, 1 (1941).
W. L. Dorbowski, E.C. Wagner, J. Org. Chem., 17, 1128 (1952).
490
ОБРАЗОВАНИЕ С-N- СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
к реакциям конденсации по ядру, то исходный амин прибавляют в два раза бь
и реакционную массу не нагревают. Полученную смесь выливают в охлаждс
раствор NaOH (1,3 экв в расчете на муравьиную кислоту) и перегоняют с вод
паром. Полученное масло извлекают эфиром, сушат над К2СОа и выделяют
зовавшийся амин перегонкой.
Высшие алифатические альдегиды использовались сравнительно редко в
стве алкилирующих средств. В достаточно мягких условиях из вторичных алиф;
ских аминов и алифатических альдегидов могут быть получены третичные а]
Так, с выходом 60% от теоретического из диметиламина получены [929]: димети
пиламин, диметилизопропиламин, диметилгептиламин и др.
Циклические вторичные амины, такие, как морфолип [929] и пипераз!
[930], также подвергались N-алкилированию при помощи алифатическв
альдегидов. Для получения бензиламина из бензальдегида требуются бол,
жесткие условия (3 ч нагревания при 190° С); выход 60% от теоретического. 3
мощенные в ядре первичные бензиламины (за исключением n-метильного ирон
водного) получаются лишь с плохими выходами [931]. Бензилирование бензал
дегидом таких вторичных аминов, как пиперазин [9301 или пиперидин [932
протекает исключительно гладко.
Низшие алифатические кетоны мало пригодны для алкилирования по метох
Лейкарта — Валлаха. Но превращение ацетофенона и других высококипящих кет
нов в первичные амины с выходом 70—80% от теоретического легко осуществить п[
действии смеси формиата аммония и формамида (реактив Ингерсолла) [933], получа
мой нагреванием формиата аммония или смеси карбоната и карбамата аммония с м!
равьиной кислотой до 165° С.
Алкилирование кетонами [933—935]. В трехгорлой колбе емкостью 3 .
снабженной термометром, доходящим до дна колбы, широким нисходящим хоЛ(
дильником и капельной воронкой, к 215 г продажной смеси карбопата/кар*
амата аммония (количество, соответствующее 4 моль аммиака) прибавляют н-
степенно 215—230 г (4,1 моль) 85—90%-ной муравьиной кислоты. Смесь ост
рожно нагревают, постепенно повышая температуру до 165° С, при этом отг<
няется вода. К горячей смеси прибавляют 1 моль кетона и нагревают далее И
небольшом пламени горелки. Через холодильник отгоняются вода, двуок®
углерода, аммиак и некоторое количество кетона, который можно периодичевв
возвращать в реакционную колбу. Реакцию с легколетучими кетонами проводи
в колбе с обратным холодильником и ловушкой для воды. Когда реакционна
масса нагреется до 175—185° С, выделение воды практически прекращаете!
нагревание при этой температуре продолжают еще 3—4 ч. При алкилирована
стерически затрудненными кетонами, например камфорой или фенхолом, пр<
должительность реакции увеличивается до 10 ч. Реакция заканчивается, когд
в холодильнике практически прекращается осаждение (NH4)2CO3. После охЛ4
жденпя реакционную массу размешивают с двойным объемом воды. Нераствор!
[929] Р. L. DeBenne ville, J. Н. Macarthney, J. Am. Chem. SOC-
72, 3073 (1950).
[930] W. T. F о г s e e jr., С. B. Pollard,/. Am. Chem. Soc., 57, 178
(1935). J
[931] K. G. Lewis, J. Chem. Soc., 1950, 2249.
[932] E. S t a p 1 e, E. C Wagner, J. Org. Chem., 14. 559 (1949). ''
[933] A. W. Ingersoll, J. H Brown et al., J. Am. Chem. Soc., 58, 180
(1936). ;;
[934] F. Nerdel, Н» Liobig, Chem. Ber., 87, 221 (1954). •
[935] A. W. I n g e г s о 1 I, H. 1). De Witt, J. Am. Chem. Soc., 73, 33?
Ш. ОБМЕН КИСЛОРОДА НА АЗОТ
491
мыя в воде неочищенный продукт кипятят с 100 мл концентрированной НС1
в течение 30—50 мии и из полученного раствора нопрореагировавший кетон
или продукты его конденсации удаляют многократной экстракцией бензолом.
Легко осмоляющиеся кетоны можпо также отгонять с водяным паром перед кис-
лотным гидролизом. Из солянокислого раствора выделяют амин едкой щелочью
и дальнейшую обработку ведут обычным способом.
Для перевода кетонов в первичные амины были предложены многочисленные
модификации реакции Лейкарта — Валлаха. детали которых здесь пе рассма-
триваются. Опубликованы сведения о наиболее целесообразных способах проведения
этой реакции применительно к отдельным кетонам |936—938] и о катализаторах реак-
ции [938-910].
Алифатические [941] и жирно-ароматические кетоны [937] с длинными алкиль-
ными цепями превращали в первичные амины, например, в следующих условиях.
а-Амипододсцнлбензол [903, 937]. В трохгорлой колбе емкостью 500 мл,
снабженной капельной воронкой, термометром и нисходящим холодильником,
медленно нагревают до 160° С смесь 105 г (1,72 .моль) 28%-ного водного раствора
аммиака и 88 г (1,72 моль) 90%-ной муравьиной кислоты; при этом отгоняется
вода. Затем к смеси в один прием добавляют 89,5 г (0,344 моль) лаурофсяона и
нагревают в течение 24 ч при 160—170° С. Отгоняющийся кетон время от времени
возвращают в реакционную колбу. Образовавшийся N-формилироваппый амин
омыляют в реакционной массе кипячением с 120 мл концентрированной НС1
в течение 8 ч. Реакционную массу оставляют на 12 ч при комнатной температуре,
добавляют 200мл воды, измельчают кристаллы выпавшего гидрохлорида амина,
отфильтровывают и промывают холодной водой. После перекристаллизации
из горячей воды получают 76 г (78% от теоретического) гидрохлорида а-амино-
додецилбепзола; т- пл. 115—116° С (из этилового спирта). Свободный амин
кипит при 170—172° С (1 — 2 мм рт. ст.); 1,4903.
Аналогично получают из аминов и кетопов вторичные [916, 942—944] и третич-
ные [932, 940, 944—946] амины. Вместо аммиака (пли формиата аммония) или форм-
амида в этом случае применяют муравьинокислые соли аминов и соответственно N-ал-
кил- и N.N-диалкилформамиды. Так, при взаимодействии циклогексанона и муравьиной
кислоты с метилформамидом получают N-метилциклогексиламин, с диметилформами-
дом — М,К-диметилциклогексиламин, а с формамидом и муравьиной кислотой —
нсзамещенйый циклогексиламин [944].
N-Циклогексилпиперидин [945]. При умеренном охлаждении водопро-
водной водой к 23 г (0,5 моль) 98%-ной муравьиной кислоты быстро прибавляют
43 г (0,5 моль) пиперидина. К еще горячей смеси добавляют 25 г (0,25 моль)
циклогексанона и кипятят в колбе с обратным холодильником в течение 5,5 ч
(для равномерного кипения обязательно добавить кипятильники!). Охлажденный
раствор выливают в большой избыток воды, подкисляют 50 мл концентрирован-
ной НС1 и тризкды экстрагируют амин бензолом. Для гидролиза N-формилп»
перидина водный раствор оставляют на 6 ч, затем добавляют концентрированный
водный раствор 30 г NaOH и отгоняют с водяным паром 400 мл дистиллята.
Маслянистый слой амина отделяют от дистиллята, основание из водного слоя
С. В. Pollard, D. С. Young jr., J. Org. Chem., 16, 661 (1951).
F. S. Crossley, M. L. Moore, J. Org. Chem., 9, 529 (1944).
V. J. Webers, W. F. В r u c e, J. Am. Chem. Soc., 70, 1422 (1948)..
A. H. Кост А. П. Терентьев, Г. А. Швехгеймер, Известия
АН СССР, ОХН, 1951, 150; С., 1954, 1221.
J. F. Bunnet, J. L. Marks, J. Am. Chem. Soc., 71, 1587 (1949).
B. L. M u г r, Ch. T. L e s t e r, J. Am. Chem. Soc., 77, 1684 (1955).
A. Novell!, J. Am. Chem. Soc., 61, 520 (1939).
H. Suter, II. Zutter, Ann., 576, 220 (1952).
M. Mousseroii, R. Jacquier, R. Zagdoun, Bull. Soc. chim.
France, 1952, 197; C. A., 47, 5905 (1953).
P. A. S. Smith. A, J - Macdonald,!. Am. Chem. Soc., 72, 1037 (1950).
J. F. В u ь n e t, J. L. Marks, II. M о о, J. Am. Chem. Soc., 75, 985 (1953).
492
ОБРАЗОВАНИЕ С—N-СВНЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
экстрагируют петролейный эфиром, полученный экстракт объединяют с
ной порцией амина, сушат и перегоняют в вакууме. Выход М-циклогексилпице]
дина 25,4 г (61 % от теоретического); т. кип. 114—118° С (25 мм рт. ст.); ,
1,4662.
При взаимодействии ацилоинов с избытком формамида образуются с хорош
выходом 4,5-дизамещенные имидазолы [946а], а не ожидаемые аминоспирты:
- R—СО СН R > R——R
он «Н
сн
Подобно ацилоинам реагируют и а-галогея- и а-аминокстопы [946а].
4,5-Ди-н-пропилимидазол [947]. Умеренно нагревают до кипения (17JJ
190° С; воздушный холодильник) в течение 3 ч 20 г н-бутироина и 40 мл фри
амида. Вакуумной разгонкой продукта реакции получают 17 г (81% от теорш
ческого) желтоватого масла; т. кип. 168° С (10 мм рт. ст.); т. пл. 66° С. Гии
хлорид 4,5-ди-я-пропилимидазола плавится (из малонового эфира) при 163®
3. Реакции спиртов и фенолов
с азотсодержащими соединениями
Обмен спиртовых гидроксильных групп на аминогруппы -
Обмен на аминогруппы неактивированных гидроксильных групп, связав
с алифатическим атомом углерода, может происходить только в присутствии кат
заторов и в исключительно жестких условиях.
При использовании таких катализаторов дегидратации, как окись алюмк
каолин, боксит, фосфат алюминия или соответствующим образом подобранные смм
ные контактные катализаторы, реакция протекает при температурах порядка 2
500° С и давлениях до нескольких сот атмосфер. Для проведения такой реакции тр
ются специальная аппаратура н катализаторы. Поэтому метод применим только в !
мышленности, тем более, что в таких жестких условиях возможно аминирование л
простейших соединений и получающиеся амины в лаборатории проще синтезира
другими способами.
Алкилирование аммиака, а также первичных и вторичных аминов иервичн
п вторичными/спиртами в присутствии катализаторов гидрирования, например 1
тины, палладия, никеля или хромита меди, протекает в несколько более мягких
виях, при температурах порядка 100—250° С и давлениях до 25 ат (реже от 16(j
150 ат). Температура реакции в значительной мере определяется каталиватором. Д
ствие катализаторов иа основе благородных металлов проявляется уже при темпа
турах, начиная со 100° С, аминирование спиртов в присутствии никелевых каталй
тор(Ов проводится обычно при 150—220° С, а при использовании катализаторов на'$
нове хромита меди, рабочие температуры достигают 190—250° С. СинтеЖгрн OTt
нередко проводится в атмосфере водорода.
При алкилировании аммиака обычно образуются смеси первичных, вторив
и третичных аминов; из циклогексанола при 150° С в атмосфере водорода при давЛ'
20 ати в присутствии восстановленного никеля довольно гладко получается цг
гексиламин с выходом 91% от теоретического [948]. Многочисленные способы 4
[946а] Обзорная статья Н. Bredereck, В. G о m р р е г et al., Ar
Chem., 71, 753 (1959).
[947] II. Bredere k, G. T h e i 1 i g, Chem. Ber., 86, 88 (1953).
[918] A. Guyot, M. Fournier, Bull. Soc. chim. France [4], 47, 203 (1
C., 1930, I, 2730.
Ill, ОБМЕН КИСЛОРОДА НА АЗОТ
493
.пирования различных аминов спиртами, преимущественно над никелевыми контактами
и при повышенных давлениях, описаны Адкинсом [949—952].
Соединения с несколькими гидроксильными группами, расположенными в мо-
лекуле у разных атомов углерода, могут реагировать с образованием циклических ами-
нов [951—952]. Так, из бутандиола-1,4 и бензиламина прп 250е С в присутствии хро-
мита меди в автоклаве получают 1-бензилпирролидин [951] с выходом 76%, а из гексан-
дпола-1,5 и я-амиламина в аналогичных условиях 1-амил-2-метилпиперидин с выхо-
дом 75% от теоретического [952].
Особый интерес для лабораторной практики представляют реакции, протека-
ющие при нормальном давлении. Поэтому здесь будут в основном рассмотрены реакции
высококипящих спиртов с аминами. Однако известны и случаи алкилирования низ-
шими спиртами (за исключением метилового), которые дают высокие выходы продуктов,
алкилирования при атмосферном давлении. Так, например, возможно моноалкилкро-
вание анилина и бензидина алифатическими спиртами С2—Св, а также бензиловым
спиртом при кипячении смеси исходных соединений в присутствии никеля Ренея [953].
N-Пропиланилин [953]. Смесь 25 мл анилина, 15 г хорошо промытого
пропиловым спиртом никеля Ренея и 100 мл пропилового спирта нагревают
в колбе с обратным холодильником при перемешивании в течение 16 ч. После-
охлаждения катализатор отфильтровывают н несколько раз промывают спиртом.
Фильтрат и промывной спирт объединяют и перегоняют в вакууме через колонку
Вигре высотой 25 см. Выход амина 30,3 г (82% от теоретического); т. кип. 98,5—
100е С (И мм рт. cm.)-, ri$ 1,5406.
Мопоалкиланилниы с. выходами того же порядка получаются при проведе-
нии реакции с другими неразвегвленнымн спиртами, но с изобутиловым н изо-
амиловым спиртом выход соответствующих моноалииланилинов составляет соот-
ветственно только 41 и 49% от теоретического [953].
N-Этнл- ^-нафтиламин [954]. Смесь 14,3 ^-нафтиламина, 100 мл 95%-ного-
этилового спирта и 40 г влажного никеля Ренея нагревают 4 ч в колбе с обратным
холодильником и после фильтрования фракционируют в вакууме. Выход N-
этил-В-пафтиламина 14 г (82% от теоретического); т. кип. 162° С (12 мм рт. ст.);
п™ 1,6402.
Минеральные кислоты (H2SO4, НС1) и их соли [ZnCl2, Zn(NH3)2Cl2, CuCl2^
FeCl3 и др.*] тоже оказывают каталитическое действие на реакции спиртов с аминами.
В данном случае особый интерес для лабораторной практики представляет алкилиро-
вание ароматических аминов, протекающее при 180—250° С под давлением.
Диметиляпилии [955, 956]. В автоклаве с чугунным вкладышем нагре-
вают смесь 93 г анилина, 105 г чистого метанола и 9,4 г 94%-ной H2SO4 в течение-
6 ч до 215° С. После охлаждения смеси к ней добавляют 25 г 30%-ного раствора
NaOH и снова нагревают 5 ч при 170° С для разложения частично образовавшем
гося четвертичного основания до третичного амина. Реакционную массу neperd(|
няют с водяным паром, высаливают из дистиллята амик, отделяют его й дели-
тельной воронке и перегоняют через колонку. Выход почти чистого диметил-
анилина 117 з (96% от теоретического); т. кип. 192° С.
949] С. F. Winans, II. A d k i n s, J. Am. Chem. Soc., 54, 306 (1932).
950] E. J. Scb woegkr, H. Adkins, J. Am. Chem. Soc., 61, 3499 (1939).
951] J. H. Paden, H. Adkins, J. Am. Chem. Soc., 58, 2487 (1936).
952] R. m. II i 1 I, II. Adkin s, J. Am. Chem. Soc., 60, 1033 (1938).
9o3J R, G. В i с e, E. J. К о h n, J. Am. Chem. Soc., 77, 4052 (1955); Org. Synth.,
36, 21 (1956).
[9.?4] C. Ainsworth, J. Am. Chem, Soc., 78, 1635 (1956).
[955] См. ссылку [330], S. 129.
[9,)6] R. N. Shreve G. N. Vriens, D. A. Vogel Ind. Eng. Chem.,
42, 791 (1950).
494
ОВР ХЗОВ ЧИПЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Диэтиланплин. Его получают с xopotnniM выходом аналогичным спс
бои из шдрохлорида анилина и этилового спирта при нагревании в течение
до 180—200° С. Образовавшийся наряду с пйм в небольшом количестве мо
этиланплин отделяют п-толуолсульфохлоридом. В этом случае не рекомендув
применять в качестве катализатора серную кислоту, так как она вызывает дег
ратанию этилового спирта до этилена J955].
Кневонагсль [9571 употреблял в качестве катализатора под. Нагревая 18,€
анилина, 36,8 я этилового спирта и 0,5 г иода в течение 10 ч при 220—230е С, он пол
чпл чистый диэтиланплин с выходом 87% от теоретического. В этой же работе прив
дится большое число других примеров.
Реже проводилось взаимодействие аминов со спиртами в присутствии щелочит
катализаторов конденсации (NaOH, MgO, алкоголяты). На ряде примеров установлю
|958], что особенно хорошие результаты таким способом достигаются при монобеиз
лпрованип первичных ароматических и гетероциклических аминов.
N-Бепзил-а-нафтнламин [958]. В колбе, снабженной обратным ход
дильником и ловушкой для воды, нагревают до кипения 35,6 г а-нафтиламин
40 г бензилового спирта и 3 г сухого КОН. Через 15 мин температура в napj
достигает 267° С и реакционную массу начинают охлаждать. При добавлен!
равного объема воды амин выделяется в кристаллическом виде; выход 95
от теоретического.
Способ получения 2-бепзилампнопиридина (выход 99% от теорстическо:
из 2-аминоппридина и бензилового спирта см. [959].
В
аминогруппу, когда гидроксильная группа активирована соседней электроноакцепто
пой группой. Так, Бодендорф I960] уже па холоду получил 1-(у-кетобутил)-п'ипер
дпн из З-кетобутанола-1 и пиперидина, а 1-диметиламинобутаноп-З из 3-котобутанола
и диметиламипа.
Реакции енолизованных эфиров кетокарбоновых кислот с аммиаком и амина^
рассматривались па стр. 473; па стр. 480 сл. описаны аналогичные превращен)
а-аминоспиртов, циангидринов и а-оксисульфокислот.
гораздо более мягких условиях удается обмен гидроксильной группы,]
v Реакции фенолов с аммиаком, аминами и гидразинами
Обмен связанной с ароматическим ядром оксигруппы на аминогруппу с до<
точной для препаративных целей скоростью протекает обычно только в присутст
кислотных катализаторов и притом в довольно жестких условиях. Исключением
ляются некоторые нитрозофенолы и полиоксипроизводные, которые вступают в р<
цию обмена уже в относительно мягких условиях и иногда без катализатора.
Так, цапример, ппи нагревании n-нитрозофепола с солямшаммония 1
паровой бане в течение 30 мин образуется га-нитрозоанилни с выЯодом 50% 1
теоретического [961, 962], а из а-нитрозо-[5-нафтОла и водного раствора мети
амина при 35° С получается сс-нитрозо-р-метиламинонафталип [963] с выход!
[957] Е. Knoevenagel, J. prakt. Chem. [2], 89, 1 (1914). >
1958] Y. Sprinzak, J. Am. Chem. Soc., 78, 3207 (1956).
I959J Y. Sprinzak, Org. Synth., 38, 3 (1958).
[960J К. Badendorf G. v’oralewski, Arch. d. Pharm., 271/43, 113 (1933
[961] O. F i s c he r, E. H e p p, Ber., 20. 2475 (1887); 21, 684 (1888). ’ 4
[962] J. Wjllenz, J. Chem. Soc., 1955, 2049.
1963] E. W. MalmJjere, C. S. II a m i 11 о n, J. Am. Chem. Soc., 7
2415 (1948). j
1П. ОБМЕН КИСЛОРОДА НА АЗОТ
495-
90%, Легкость такого обмена, по-видимому, обьясняется таутомерией этих
фенолов с соответствующими кетонами:
ОН О NJI NH2
NO NOH NOII NO
Флороглюцин, который под действием гидроксиламина может превра-
щаться даже в триоксим [§64], реагирует со смесью аммиака и NH4C1 уже при
комнатной температуре, переходя с почти количественным выходом л 5-амино-
резорцин [965].
Наличие нитрогруппы тоже повышает реакционную способность феполов, но
тем не менее замена оксигрупп на аминогруппы протекает при 150—200° С, и поэтому
реакцию приходится проводить в автоклаве [966]. Для получения первичных аминов
пз ди- и тринитрозамещенных фенолов и нафтолов при нормальном давлении их спла-
ц.тягот с мочевиной [967, 968]. Таким путем был получен пикрамид [968] с выходом
88% от теоретического (т. пл. 188° С), для чего пикриновую кислоту и 3 .ноль моче-
вины нагревали в течение 36 ч при 173* С.
Важнейшим способом получения ароматических аминов из соответствующих
фенолов является реакция Бухерера, несмотря на ограниченные возможности ее приме-
нения [969]. Под реакцией Бухерера понимают превращение некоторых феполов
и амины в присутствии сернистой кислоты и обратный процесс, а также катализуемое-
бисульфитом переаминирование ароматических амилов (ср. стр. 502).
Механизм этой интересной реакции в течение днительного времени оставался
спорным и выяснен лишь в последнее времи благодаря работам Рихе и Зееботд [970] *.
Конечный результат взаимодействия является следствием ряда равновесных реакций,,
показанных па примере а-нафтола и соответственно а-нафтиламина:
* См, примечание на стр. 321.
964] А. В а е у е г, Вег., 19, 159 (1886).
965] J. Pollak, Mh. Chem., 14, 401 (1893).
[966J V. Merz, С, Bi s, Ber., 19, 1749 (1886); O. N. Wit t, Ber., 19, 2032 (1886);
A. Barr, Ber., 21, 1541 (1888); E. Diepolder, Bor,, 42, 2916 (1909).
967] о. Кущ, J. prakt. Chem. [2], 75, 323 (1907).
[968] E. Y. Spencer, G. F. Wrigh t, Canad. J. Res., Sect. B, 24, 204 (1946):
C. A., 41, 723 (1947).
[369] N. L. D ra k e, Org. Reactions, 1, 105 (1942).
1970) A. flieche, II. Seeboth, Ann., 638, 43—110 (i960).
496 ОБРАЗОВАНИЕ С—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Решающее значение в этой реакции имеет положение равновесия И
гавпсящсе не только от концентрация реагирующих веществ, но и в значител!
мерс от направления дальнейшего превращения имина (III), который может отщегг
бисульфит пли подвергаться гидролитическому расщеплению [971]. *
Реакция Бухерера применима главным образом для производных нафта;
[972—976]. Иза- и р-нафтола гладко получаются соответствующие первичные ама
Заместители в положении 5, 6, 7 или 8 мало влияют на скорость реакции. а-Нафт
с сульфогруппами в положении 2 или 3, а также 0-нафтолы с заместителями, особ(
с карбоксильной группой, в положении 3, в реакцию Бухерера обычно не вступ,
Исключительной реакционной способностью обладает оксигруппа в положении 1 в 1
фтол-4-сульфокислотах; даже при наличии других гидроксильных групп эта оксягр
обменивается в первую очередь. В ряду бензола в реакцию Бухерера вступакЛ I
ным образом резорцин и его гомологи [977, 978]. В хинолинах и изохинолинах <
нивяются только оксигруппы, находящиеся в бензоидном кольце [979].
Для перевода нафтолов в первичные амины нафтол, сульфит аммопия и ам.
fмолярные соотношения АтОП : ЗО2 '• около 1 : (1,5—2) : (5—8)] нагре
в автоклаве при 130—160° С; продолжительность реакции 6—30 ч.
^-Нафтиламин [980]. В автоклаве с мешалкой в течение 8 ч вагр<
при 150° С 144 г (1 лоль), р-нафтола с раствором сульфита аммония, получе
пропусканием 100 г SO2 в 400 жл раствора аммиака (d — 0,90) при охлажд
Давление в автоклаве достигает 5—7 ат. Затвердевшую при охлаждении
ционную массу размешивают с небольшим количеством воды, отфильтровы
промывают водой и растворяют при нагревании в 1,5 л воды с 150 мл конце
рованной НС1. Мутный от непрореагировавшего нафтола и незначительных _
чести вторичного амина раствор кипятят с 10 г животного угля и фильтр
Из отфильтрованного в горячем состоянии раствора осаждают р-нафтилг
добавляя при перемешивании 20%-ный раствор NaOH до щелочной реаКф
фенолфталеину. После охлаждения амин отфильтровывают, промывают и с
в вакууме при 30—50° С. Получается 130—140 г красноватого нсочищеЕ
продукта с т. пл. 109—1110 С. Для дальнейшей очистки вещества его перегО
в вакууме в колбе с саблевидной трубкой на металлической бане с темпера!
210° С. Выход чистого амина 125—135 г; бесцветные кристаллы, т. пл. 11
Аналогичным способом синтезируют применяемую в промышленг
2-нафтиламин-1-сульфокислоту из 2-нафтол-1-сульфокислоты [981].
Вторичные амины можно получать из фенолов и из соответствующих перви
•аминов. р-Нафтиламины реагируют с аминами легче, чем ^-нафтолы, которые в
очередь более реакционноспособны, чем а-пафтолы. Обмен гидроксильной групп
остаток сильного основания можно проводить так же, как при синтезе первичных
нов, в присутствии сульфита алкиламмония и алкиламина [молярные соотнош
[971]
[972]
[973]
[978
[979]
1980]
1981 ]
Н. Seeboth, Monatsber. dtsch. Akad. Wiss. [Berlin], 3, 43 (1961).
H. Th. Bucherer, J. prakt. Chem. [2], 69, 49 (1904); 70, 345 (1904); Z.
ben- u. Textjlchemie, 2, 193 (1903); C., 1903, П, 42. ' ?
H. Th. Bucherer, A, Stohmann, J. prakt. Chem. [2], 71, 433 (i
Z. Farben-u. Textilchemie, 3, 57 (1904); C., 1904, I, 1012.
H. Th. Bucherer, F. Sey de, J. prakt. Chem. [2J, 75, 249 (1907).
H. Th. Bucherer, M. Schmidt, J. prakt. Chem. [2], 79, 369 (1
H. Th. Bucherer, A. Uhlmann, J. prakt. Chem. [2], 80, 201 (1
H. Th. Bucherer, E. Hoffmann, J. prakt. Chem. [2], 121, 113 (1
W. H. Hartung L. J. M innick, H. F. Koehl er, J. Am. C
Soc., 63, 507 (1941).
N. N. Woroshtzow, J. M. Kogan, Ber., 65, 142 (1932); R. A.
b i n s о n, J. Am. Chem. Soc., 69, 1944 (1947); R. Й. F. M a n s k e, M. К U-i
J. Am. Chem. Soc., 72, 4907 (1950).
R, S chr 6 t er, в книге H ou ben - We у I, Methoden der organii
Chemie, Bd. 11/1, 4 Aufl., Stuttgart, 1957, S. 150.
См. ссылку [330].
III. ОБМЕН КИСЛОРОДА НА АЗОТ
497
ЛгОН : SO2 : RNH. около 1:2: (2—3)|. Если скорость реакции не имеет существен-
ного значения, то реакцию проводят путем кипячения фенола с 2—3 моль амина и би-
сульфита натрия при добавлении к реакционной Массе сульфита аммония (всего 4—
12 моль S03). Ароматические амины можно вводить в небольшом избытке, но такая
реакция протекает гладко лишь с ^-производными нафталина. О синтезе N-арилиро-
ванных Р пафтиламипов см. |970|, а также стр. 503.
Симметричные диариламины, например ди-(р-нафтил)-амин [982], могут быть
получены также взаимодействием первичных ариламинов с бисульфитом.
В принципе по реакции Бухерера можно получать и арилгидразииы [983—986]
взаимодействием соответствующих фенолов с гидразингидратом и SO2. Так, из 2,3-ди-
оксинафталина получают 2,3-нафтилендигндразин с выходом 57% от теоретического
[983, 984], а из 2,7-диоксинафталниа — 2-гидразино-7-оксинафталии [985J с выходом
82%. Все же большого значения эти реакции не имеют.
При взаимодействии арилгидразинов с нафтолами в присутствии сернистой
кислоты соответствующие гидразопроизводные не образуются. Из а-нафтола, фенил-
гидразниа и NaHSO3 сначала образуется фенилгидразон а-тетралон-3-сульфокислоты,
который при действии кислот (по аналогии с синтезом ипдола по Фишеру) циклизуется
п получается 1,2-бензкарбазол; выход 85% от теоретического. Подобным способом
из Р-пафтола получается 3,4-бензкарбазол [970].
Амины из фенолов, гидроксильные группы которые не активированы соответ-
ствующими заместителями (ср. стр. 494) и не обмениваются на аминогруппы по реак-
ции Бухерера, получают при повышенных температурах (180—300° С) и поэтому в боль-
шинстве случаев под давлением. В качестве катализаторов часто с успехом применяют
ZnClj, Zn(NH3)2Cl2 (получение см. [987]), реже — СаС12, NaHSO3, FeCl3 и сульфани-
ловую кислоту. Очень хорошие выходы при арилированяи ароматических аминов
нафтолами получал Кневепагель [957], проводивший реакцию в присутствии катали-
тических количеств иода. Часто рекомендуется вести реакцию не со свободными ами-
нами, а с их гидрохлоридами. Лабораторные методики по аминированию с участием
простых компонентов реакции приводятся главным образом в старой литературе
[967, 988].
2-Амино-З-нафтойная кислота [989]. 2-Окси-З-нафтопнуто кислоту, вод-
ный аммиак и ZnCl2 нагревают в автоклаве 36 ч; выход кислоты 70% от теорети-
ческого. Эта кислота не может быть синтезирована по методу Бухерера.
2-Фенил-7-аминохииолян [990]. В запаянной трубке в течение 3 ч нагре-
вают 6,6 г 2-фенил-7-окснхинолииа и 25 г аммиаката хлорида кальция при 250° С,
а зат^м еще 7 ч при 280—290° С; выход продукта 80% от теоретического.
Недавно Хекер [991] предложил простой метод перевода п-алкилфенолов
в л-алкил анилины. Этот метод, использовавшийся для получения аминозамещенных
эстрогенов, может иногда применяться и для синтеза других соединений. Алкилфенол
окисляют тетраацетатом свинца до n-хинолацетата, при взаимодействии которого с се-
микарбазидом или 2,4-динитрофенилгидразином образуется азосоединение; вос-
становительным расщеплением этого аэосоединсния получают соответствующий
п-алкилапилин.
Герм. пат. 114974; FrdL, 6, 198 (1900—1902).
Н. Franzen, Вег., 38, 266 (1905).
IT. Franzen, J. prakt. Chem. [2], 76, 205 (1907).
Н. Franzen, W. Deihel, J. prakt. Chem. [2J, 78, 143 (1908).
H. Franzen, Th. E i c h 1 e r, J. prakt. Chem. [2], 78, 157 (1908).
V. Merz, P. Muller, Ber., 19, 2901 (1886).
V. Мегг, P. Muller, Ber., 20, 544 (1887); R. Lloyd, Ber., 20, 1254
(1887); K. Buch, Ber., 17, 2634 (1884).
С. II. F. Allen, A. Bell, Org. Synth., Coll. v. 3, 1955, p. 78.
W. Borsch e, M. Wagner - К о e m mi c h, Ann., 544, 292 (1940).
E. Hecker, Chem. Ber., 92, 3198 (1959); E. Hecker, E. Walk, Chem.
Ber., 93, 2928 (I960).
32 Заказ 1835.
498
ОБРАЗОВАНИЕ С-К-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
4. Реакции простых эфиров и оксониевых
солей с азотсодержащими соединениями
Замена алкокспльной группы в простых ациклических алифатических эфи
на аминогруппу представляет незначительный препаративный интерес. В не оч
жестких условиях удовлетворительные результаты получаются лишь в тех случа
когда эфирная связь в достаточной мере активирована находящимися в ^-положа
карбонильными, карбоксильными или нитрильными группами. Некоторые р-метот
кетоны (например, I) переводятся в 0-аминокетоны [992] уже при температурах ш
100° С. Еще лете протекает расщепление эфирной связи в активированных еполоэ
рах типа этоксиметиленацетоуксусного эфира (II), этоксиметплепмалоноиого эф]
(III) *, а также этоксиметиленацетилацетона (IV).
R—СО—СП-
I
Н'-СН-ОСНз
CH3C0-C—COOR
II
СП-ОС2Н5
и
ROOC—С—COOR
II
СП-ОС2Н6
III
СНзСО-С-СОСНз
СН — ОС3Ц5
IV
Под действием аммиака и первичных алифатических аминов они часто уже npi
комнатной температуре с очень хорошими выходами превращаются в соотвда
ствующие аминометиленовые производные [993], используемые для многочисленны!
синтезов азотсодержащих гетероциклических соединений, например 4-оксихинолнши
[995—999].
Этиловый эфир аминометиленщавелсвоуксусной кислоты [1000]. К раС;
твору 61 г (0,25 моль) этоксиметиленщавелевоуксусного эфира в 250 мл сухоте
эфира при охлаждении смесью льда с поваренной солью приливают в один прием
26 в 18%-ного раствора аммиака в абсолютном спирте (0,27 моль аммиака);
Реакционную массу встряхивают и оставляют на 20 ч в закрытом сосуде, затея
желатинообразный осадок отфильтровывают и тщательно промывают сухвд
эфиром. Из объединенных фильтратов при упаривании в вакууме получают
сироп, который быстро кристаллизуется. Выход продукта 45—49 г (84—91% <И
теоретического); т. пл. 68—09е С (из смеси эфира и петролейного эфира). J
Нс всегда нужпо исходить из выделенных в чистом виде алкоксиметиленовыей
соединений (например, II, II], IV и др.); их получение из соединений с активной метя*
леновой группой (ацето- или циануксусного эфира и т. д.) и эфира ортомуравьиной
кислоты и взаимодействие с амипом можно проводить в одну операцию [999]. '
* Способ получения-из малонового эфира, ортомуравьиного эфира и уксусного»
ангидрида описан в работах [994], [995]. С
[992]' И. II. Назаров, С. А. Б а р т а н ь я н, ЖОХ, 22, 1668, 1794 (1952)^
С. А., 47, 9968, 9969 (1953).
[993] L. Claisen, Ann., 297, 1 (1897). -.(i
]994] W. Е. Parham, L. J. Reed, Org. Synth., Coll. v. 3, 1955, p. 395. _'j
[995] G. F. Duffin, J. D. Kendall, J. Chem. Soc., 1948, 893.
[996] Ch. C. Price. R. M. Roberts, Org. Synth., Coll. v. 3, 1955, p. 272. 4g
[997] Ch. C. Price, N. J. Leonard, H. F. II e г Ъ r a n d s о n, J. Am. ChettUi
Soc., 68, 1251 (1946). ’
[998] Ch. C. Price, R. M. Roberts, J. Am. Chem. Soc., 68. 1204 (1946). .4
[999] H. R. Snyder, R. E. J ones, I. Am. Chem. Soc., 68, 1253 (1946).
[1000] R. G. Jones,! Am. Chem. Soc., 73, 3684 (1951). •
III. ОБМЕН КИСЛОРОДА НА АЗОТ
499
Реакции простых эфиров с ароматическими аминами [995—999, 1001, 1002]
и амидами кислот [993, 1001, 1003], например с мочевинами, протекают с хорошими
выходами лишь при 100—150ч С.
При взаимодействии алкоксиметиленовых соединений с гидроксиламинами,
гидразинами или бифункциональными амцнопроизводными одновременно происходят
обмен алкоксильной группы па азотпые функции и циклизация и образуются разные
гетероциклические соединения [993, 1003—1006].
Кроме алкоксиметиленовых производных с активной метиленовой группой
расщеплению под действием аминов подвергаются также многие другие енолоэфиры,
как, папример, эфиры р-алкоксиакриловой кислоты [1007], у-пироны [1008, 1009]
и_ФУРфУРОл [1010].
1,2^6-Триметилпирндон-4 [1009]. Поддерживая температуру ниже 40° С,
к 150 мл 40%-пого водного раствора метиламина при перемешивании в течение
1 ч прибавляют раствор 70 г 2,6-диметил1(ироца-4 в 200 мл воды. Реакционную
массу оставляют на 1 — 2 ч, охлаждают до 0° С, осадок отфильтровывают и пере-
кристаллизовывают из горячей воды. Выход продукта 88% от теоретического;
т- пл. 245—246° С.
1-(4'-Ннтрофенил)-пиррол-2-альдегид [1010]. В колбе с обратным холо-
дильником кипятят в течение 2—3 ч фурфурол с п-нитроанилипом в среде водного
метилового спирта в присутствии кислоты. Выход продукта практически коли-
чественный.
В ароматическом ряду расщепление эфирной связи аммиаком и его производ-
ными используется для препаративных целей главным образом в реакциях с простыми
эфирами некоторых ннтрофенолов. Так, например, эфиры ди- и тринитрофенолов рас-
щепляются аммиаком, первичными аминами, гидразингидратом или гидроксиламином
при температурах от 50 до 200° С, в зависимости от активирующего влияния замести-
теля. Диариловые эфиры реагируют более гладко, нежели алкариловые эфиры. При
расщеплении алкариловых эфиров атом азота всегда замещает алкоксильную группу,
а в диариловых эфирах замещается наименее нитрованная или непитрованная
ароксильная группа.
, Так, Борте [1011] получил 2,4-динитроапилин при действии аммиака
на 2,4-диннтродифениловый эфир, а при действии анилина на это же соединение
был получен 2,4-динитродифениламин. 2,4-Дннитрофенил-р-нафтиловый эфир
при кипячепии с анилином дает 2,4-дипитродифениламин с выходом 97%; т. пл.
156-157° С [1012].
[1001] R. II. Baker, А. Н. Schlesinger, J. Ат. Chem. Soc., 68, 2009
(1946).
[1002] R. Н. Baker, J. G. Van Oot et al., J. Am. Chem. Soc.,71, 3060
(1949).
[1003] C. W. Whitehead, J. Am. Chem. Soc., 74, 4267 (1952). ,
[1004] P. S chmid t, К. E ichen berger, W. VV i 1 h e 1 m, Angew. Chem.,
73, 15 (1961).
[1005] A. Dornow et al., Chem. Ber., 91 1830 (1958); 93, 1103 (1960).
[1006] A. Kreutzberger, Ch. Grunamann, J. Org. Chem., 26, 1121
(1961).
[1007] P. t. De Benneville, J. H. Macarthney, J. Am. Chem.
Soc., 72, 3725 (1950).
[1008] J. W. A rm it, T. J. Nolan, J. Chem. Soc., 1931, 3023.
Il009] K. N. Campbell, J. F. Ackerman, В. K. Campbell, J. Org.
Chem., 15, 337 (1950).
11010] JI. А. Яновская, Синтезы органических соединений, Сборник 1, 1Гзд.
АН СССР, 1950.
11011] W. Borsch с, Вег., 56, 1488 (1923).
И012] Y. О g a t а, М. О k а п о, J. Am. Chem. Soc., 71, 3211 (1949).
32*
500
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ\0БМЕНА
Аналогично протекают реакции эфиров нитрофенолов с гидразином [1011
я гпдроксиламином [1013]. При нагревании 2,4-дннитроапизола с 90%-ным гидряя^
гидратом на водяной бане в течение 0,5 ч с почти количественным выходом образует»
2,4-динитрофенилгидразин [1011].
В ряду производных пиридина и хинолина расщепление эфирной связи предел
вляет интерес в основном для введения аминогруппы в положение 4. Так, 6-нитрол
аминохинолин [1014] был получен с выходом 84% от теоретического при нагревав
в течение 25 мин соответствующего 4-феноксипроизводного с избытком ацетата ам
ния при 170—180° С.
Активации эфирЕОй связи нитрогруппами в производных пиридина не требует
если реакция проводится не с аминами, а нх гидрохлоридами. Йерхель и Якоб [10
получали N-замещеиные 4-аминопиридины с хорошими выходами из 4-пириди^
ниловых эфиров и ряда гидрохлоридов амилов.
4-Анилинопиридин [1015]. Получается из 4-фепоксипиридина и из
точного количества гидрохлорида анилина при нагревании смеси при 18(
в течение 3 ч; выход 89% от теоретического.
Насыщенные циклические простые эфиры с пяти- или шестичленными кольц
реагируют с аммиаком или аминами с заменой эфирного кислорода на NH- или I
группу. Однако эта реакция может представить интерес для лабораторной практ
лишь в исключительных случаях, так как она протекает в чрезвычайно жест:
условиях *.
Так, для перевода тетрагидропирана в N-фенилпиперидин под действием а
лина требуется температура порядка 330—340° С и наличие катализатора на оса
окиси алюминия [1016]. В аналогичных условиях из тетрагидрофурана можно пс
чить пирролидины [1017].
Циклические эфиры с меньшим числом членов в кольце реагируют при б<
низких температурах. При этом получаются только линейные соединения (ам
спирты). Для перевода триметиленоксида в N-эамещенные 3-оксипропиламины ре
рующие вещества длительно нагревают в запаянной трубке при 150° С в присутс:
небольшого количества воды [1018].
Этилеяоксиды реагируют с аммиаком и более основными аминами боль'
частью уже при комнатной температуре; реакция протекает с выделением тепла и у
ряется в присутствии спиртов или воды. Следует напомнить, что при работе с oxi
этилена возможны взрывы/
Из окиси этилена и аммиака в зависимости от взятого соотношения реагщ
образуется смесь различных количеств моно-, ди- и триэтаноламина, которые мо;
разделить перегонкой [1019]. Более однозначно протекает взаимодействие описи 1
лена с первичными и особенно с вторичными аминами.
р-Диэтвламиноэталол [1020]. В смесь 880 г (12 моль) диэтиламииг
440 з метилового спирта при перемешивании пропускают 220 г (5 моль) OKI
* Реакция замены кислорода на NH-грунпу в циклических эфирах, а тяц
фуранах открыта и- всесторонне изучена Ю. К. Юрьевым и его шиой
[Ю. К. Ю р ь е в, Ученые записки МГУ, вып. 79 (1945)]. — Прим. ред. -
[1013] W. В о г sc h о, Вег., 56, 1494 1939 (1923); W. Borsch е, Е. FesK
Вег., 59, 683 (1926).
[1014] J. С. Е. Simpson, Р. Н. Wright, J. Chem. Soc., 1948, 1707.
[1015] D. Jerchel, L. Jakob, Chem. Ber., 91, 1266 (1958).
[1016] A. N. Bourns H. W. E mble ton, M. К. H a n s u 1 d, Org. Syrii
34, 79 (1954).
[1017] W. R e p p e et al., Ann., 596, 143ff (1955).
[ 1018j S. Searles, V. P. Gregory, J. Am. Chem. Soc., 76, 2789 (1954).
[1019] L. Knorr, Ber., 30, 909, 915, 918 (1897).
[1020] F. Moller, в жпиге Houben-Weyl, Methoden der organise!
Chemie, Bd. 11/1, 4 Aufi., Stuttgart, 1957, S. 312.
Ill ОБМЕН КИСЛОРОДА НА АЗОТ
501
этилена, скорость подачи которой регулируют, чтобы температура не поднима-
лась выше 60° С. По окончании реакции при нормальном давлении отгоняют
метиловый спирт и непрореагировавший диэтиламин; диэтиламиноэтанол пере-
гоняют d вакууме; выход 550 г (94% от теоретического); т. кип. 54—55® С
(12 мм рт. ст.).
Описан общий метод получения других Р-диалкиламипоэтанолов [1021].
В реакцию с окисью этилена вступают и третичные амины; так, из триметил-
амина и окиси этилена при комнатной температуре получают холин [1022].
Первичные ароматические амины [1023] реагируют с окисью этилена чаще всего
уже при комнатной температуре или при умеренном нагревании. Слабоосновные
амины вступают в реакцию при температурах 100—140° С (автоклав).
Реакционная способность замещенных этиленоксидов снижается с увеличением
числа заместителей. В несимметричных этиленоксидах аминогруппа присоединяется
к наиболее гидрогенизированному атому углерода.
Гидроксияамин реагирует с окисью этилена уже при комнатной или даже при
более низкой температуре с образованием М,М-бис-(р-оксиэтил)-гидрокснламина;
с увеличением продолжительности взаимодействия образуется трис-(р-оксиэтил)-амин-
оксид [1024].
С хорошими выходами протекает взаимодействие эпоксидов с гидразингидра-
том до гидразиноспнртов [493] и с азидом натрия до азидоспиртов [1025].
Двуокись азота реагирует с окисью этилена в среде' хлороформа (0—20° С),
образуя с почти количественным выходом 2-яитроэтилнитрат, который при 35—45° С
омыляется 10%-ным раствором соды до 2-нитроэтилового спирта [1026]. Нитроэтило-
вый спирт может быть получен с выходом 85% от теоретического также прямым дей-
ствием окиси этилена и СО3 на водный раствор NaNO2 при 25—30° С [1027].
Важным методом получения производных пиридина является взаимодействие
пирилиевых солей [1028] с аммиаком, первичными аминами, гидроксиламином или
гидразинами:
где R = алкил, арил; R' = алкил, арил, ацил, — NHQHe, —NHC0NH3, —ОН.
Действуя аммиаком на легкодоступные пирилиевые соли, можно получать боль-
шое число пиридинов, синтез которых другими методами часто связан с значительными
затруднениями. К таким производным пиридина относятся 2,4,6-триалкил- или
[1021] Е. Rohrmann, Н. A. S h о n 1 е, I. Am. Cliem. Soc., 66, 1641
, (1944).
11022] К. Н. Meyer, Н. Н о р f f, Вег., 54, 2279 (1921).
[1023] Ю. К. Ю р ь е в, и сотр., Известия АН СССР, ОХН, 1951, 317; С.. 1952, 203.
[1024] L. W. Jones, G. R. Burns, J. Am. Chem. Soc., 47, 2966 (1925).
[1025] C. A. Va nder Wert, R. Y. H e i s 1 e r, W. E, McE wen, J. Am.
Chem. Soc., 76, 1231 (1954); W. E. M с E wen, W. E. С о n r a d, С. A. V a n-
derWerf, J. Am. Chem. Soc., 74, 1168 (1952).
1026] G. Darzens, С. r., 229, 1148 (1949); C. A., 44, 4412 (1950).
1027] Яп. пат. 6910; С. A., 48, 1412 (1954).
[1028] Обэор работ по получению и реакциям пирилиевых солей см. К'. D i m-
г о t h, Angew. Chem., 72, 331 (1960).
502
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
2.4.6-триарилзамсщенные пиридины. По старым прописям реакцию ведут в водной среде
Димрот[1028] рекомендует суспендировать соли пирилня в абсолютном третичном бути
ловом спирте и пропускать в суспензию сухой газообразный аммиак. Смесь разогре
вается и соль переходит в раствор; при охлаждении выделяется аммониевая coju
неорганической кислоты, анион которой входил в состав пирилиевой соли (например
NH1CIO4, NII4BF4); производное пиридина осаждают пз раствора добавлением воды.
2,4,6-Трифснилпнридин [1028]. В 100 мл абсолютного третичного бутц-
лового спирта суспендируют 10 г тотрафторбората 2,4,6-трифенилпирилия
и смесь нагревают до кипения; затем пропускают через нее сильпый ток сухоте
аммиака. Соль пирилпя переходит в раствор. Через 30 мии отфильтровывают
выпавший NH4BF4 и добавляют к фильтрату небольшое количество воды дс
появления мути. В холодильнике через некоторое время выпадает кристалличе-
ский продукт в виде листочков, выход 8 г (90% от теоретического); т. пл. 138° С,
При действии первичных алифатических и ароматических аминов на 2,4,6-три-
замещеппые соли пиридин в результате аналогичной реакции образуются соот-
ветствующие N-алкил- или N-арилпиридиниевые соли, обычно с превосходными
выходами [1028].
Если заместители в положении 2 и 6 не оказывают слишком больших стерических
затруднений, то при действии гидроксиламина образуются пиридин-Х-оксиды [1029],
при действии семикарбазида — N-уреидопирндииы [1029], а при действии фенилгид-р*
азина — представляющие интерес [1030] N-ариламинопиридины [1030, 1031], которые
другим способом вообще не могут быть получены.
За редким исключением соли хромилия и ксантнлия не переводятся в соответ^
ствующис производные хинолина или акридина. i
IV. Обмен других элементов на азот
1. Обмен азота на азот
В этом разделе рассматриваются представляющие наибольший интерес реакций
переамидировапия л пореаминирования, а также различные возможности образования
С—N-связи при помощи диазониевых солей и диазосоединений. Обмен нитрогрупв
па аминогруппы возможен практически только в о- и n-динитропроизводных и вряд Ш?
имеет какое-либо значение. Получение а-нитрозо- и а-оксиминоэфиров из а-нитроэфи-
ров и нитрита натрия [145, 255, 256] уже рассматривалось па стр. 397 и 442- J
Переаминирование (реакции обмена аминогрупп)
Под реакциями персаминирования или обмена аминогрупп понимают такое
взаимодействие аммиака с аминами или двух аминов между собой, при которых фор*
малыш аминогруппа одного амина обменивается на аминогруппу другого; некоторая
примеры обмена азотной компоненты в основаниях Шиффа, гидразонах и аналогичны^
соединениях упоминались в разделе, посвященном их получению (стр. 470—480), ЯЯ*
пример: - '
r-nh2+r'—nr;—>- r-nr;+r'-nh2
При нагревании первичных ароматических аминов в присутствии кислот (НС1£
П3РО4, п-толуолсульфокислога) в некоторых случаях гладко протекает конденсация
с образованием диариламинов. Например, дифениламин можно получать нагревание^
в течение 20 ч при 230е С и давлении 6 ат смеси анилипа с его гидрохлоридом [1032j^
[1029] А. Т. В а 1 а Ъ а п, С. D. Nenitzcscn, Ann., 625, 74
Е. Schmitz, Chem. Вег., 91, 1488 (1958).
[1030] К. Dimroth, G. Arnoldy et al., Ann., 604, 221 (1957).
[1031] W. Schneider et al., Ber., 54, 2285 (1921); 61, 2445 (1928); 74, 1252
(1941); Ann., 438, Ц5, 147 (1924).
[1032] См. ссылку [330].
IV, ОБМЕН ДРУГИХ ЭЛЕМЕНТОВ НА АЗОТ
503
N-Фснил-а-нафтиламич [1O33J. (Для получения этого амина реакция
Бухерера мало применима.) В колбе с термометром п длинным воздушным
холодильником кипятят 143 а а-нафтпламина, 175 г анилина и 3 е сульфаниловой
кислоты в телепне 40—48 ч. При этом выделяется аммиак, а температура кипения
смесп повышается со 195° С до приблизительно 215° С. Но окончании реакции
продукт разгоняют в вакууме (12 лл рт. ст.) при 70° С. Сначала отгоняется
80—85 г анилина, затем с толчками и вспениванием перегоняется промежуточная
фракция (8—10 г), в которой уже содержится некоторое количество N-фенил-
а-иафтиламипа. Основная фракция (190—195 г) перегоняется при 212 — 216° С,
остаток переходит при 225° С. Общий выход около 200 г (91 % от теоретического).
Бесцветные иглы с т. пл. 62° С (из небольшого количества этилового спирта).
Хорошие выходы N-арил-а-нафтиламинов были получены также в присутствии
каталитических количеств пода, например при переаминировании а-нафтиламина
анилином, толуидинами, м- и л-хлоранилинами, о- и «-анизидинами [957], 2,3- и 3,4-
диметиланилинами [1034]. При нагревании р-иафтиламипа в течение 4 ч при 230°С
в присутствии 0.5—1% иода с почти количественным выходом образуется ди-(р-наф-
тил)-амин, а из n-амипофенола в аналогичных условиях получается 4,4-диоксндифе-
ниламин с выходом 70% от теоретического [957].
При нагревании 2.2'-диаминодифенила и его производных с НаРО4 они почти
количественно превращаются в карбазолы [1035].
Карбазол [1035]. В открытом сосуде при хорошем перемешивании нагре-
вают при 200—210° С 50 г 2,2'-диаминодифенила и 278 г Н3РО4 (<7 = 1,85).
Уже через 15 мин начинает выделяться карбазол. Через 5 ч отфильтровывают
выделившийся карбазол из горячей реакционной массы и промывают водой до
нейтральной реакции; выход 45 з (количественный); т. пл. 238° С.
В алифатическом ряду в результате такой же циклизации получают пирролидины
и пиперидины [1036, 1036а].
Обмел аминогруппы возможен и в 0-нафтиламипе, и его производных; эти соеди-
нения можно также обратимо превращать в N-аляил- и N-арил-Р-нафтиламины
в условиях реакции Бухерера (ср. стр. 497).
, 8-Окси-2-фенилам»нонафталин-6-сульфокислота («фепил-у-кислота»)
[1037]. Кипятят в течение 24 ч 239 г чистой 8-окси-2-аминонафталип-6-суль-
фокислоты (у-кислота), 750 г раствора NaHgO3 (содержащего 25% SO2), 750 мл
воды и 200 г анилина. Затем подщелачивают реакционную массу концентриро-
ванным раствором Na5CO3 и отгоняют избыток анилина с водяным паром. Прн
подкислении раствора IIC1 выпадает фенил-у-кислота почти в чистом вКде;
выход 75—80% от теоретического.
Третичные и четвертичные основания Манниха часто легко и в мягких условиях
вступают в реакцию обмена аминогруппы, если аминный компонент применяется в из-
бытке, а выделяющийся вторичный или третичный амин достаточно летуч (ср. обзоры^
[72, 1038]. Таким образом можно получать и первичные амины, обменивая аминныи
остаток основания Манниха на фталимидную группу [1039] и подвергая продукт реак-
ции омылению.
[1O33J См. ссылку [330].
[1034] N. Р. Bun-Hoi, J. Chem. Soc., 1949, 670.
[1035] И. Leditschke, Chem. Вег., 86, 522 (1953).
[1036] A. La denburg, Ber., 18, 3100 (1885); 20, 442 (1887); W. Keil, Hoppe-
Seyler’s Z. physiol. Chem., 171, 242 (1927).
[1036a] K. Kindler, D, Ma It h ies, Chem. Ber., 95, 1992 (1962).
[1037] См. ссылку [330].
[1038] H. Hellmann, Angew. Chem., 65, 473 (1953).
[1039] R. O. Atkinson, J. Chem. Soc., 1954, 1329; A. В u t e n a n d. t, U. Ren-
ner, Z. Naturforsch, 8B, 454 (1953); пат. ФРГ 933339; С., 1956, 1738.
504
ОБРАЗОВАНИЕ G-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Обмен аминогруппы в основаниях Манниха может протекать также под д
ствием сульфонамидов [1040], гидроксиламинов [1041] и фенилгндразина [1Г*
Основания Манниха, полученные из кетонов, при взаимодействии с гидразинами
чала превращаются в гидразоны, которые затем циклизуются с обменом аминогр)
и превращаются в пиразолины [1043]:
R—СО—СН2 ArNTTNH, , R—С—СН2
I 'I 1 HN н •
СН2-NRo n ch2-nr; *
NH
L
R-C---CH2
li I
N CH2
V
L
Четвертичные пиридиниевые соли [467] с электроноакцепторными группир
ками при атоме азота, например 2,4-динитрофенил' или пиридпл-Д-ттгтрипттиист
соли, могут реагировать с аминами, являющимися нуклеофильными агентами,
двум направлениям: четвертичное ядро пиридиния вытесняется введенным в реанг
амином (реакция 2) или оно расщепляется с образованием производного глутаноноэ
альдегида (расщепление Цинке) [1044], причем атом азота пиридина отходит в Е
первичной аминогруппы с заместителем, находившимся у этого атома в соли пнриди!
(реакция 2). Производное глутаконового диальдегида в свою очередь может ся<
циклизоваться с образованием гетероциклической системы, так что конечным резу.
татом этой реакции является обмен заместителей при атоме азота [1044, 1045]:
О ^&F-0+hnhr-
N N
I
RCI-
сн=сн-сн2
| | + rnh2
СП СН
NR' NR'
‘—О’
N
I
П'СГ*
[1040] J. J. Lie аг i, G. D о u g h е г t у, J. Am. Chem. Soc., 76, 4039 (19^
J. J. L i с а г i, L. W. H а г t z e 1, G. Dougherty, F. R. Benatf
J. Am. Chem. Soc., 77, 5386 (1955).
[1041] J. Th esing, Chem. Ber., 87, 507 (1954); J. T h e s i n g, A. Mull*
G. Michel, Chem. Ber., 88, 1027 (1955).
[1042] J. T h e s i n g, C.-H. W i 1 1 e г s i n n, Chem. Ber., 89, 1195 (1956).
[1043] M. S t a m p e r, B, F, Aye ock, J. Am. Chem. Soc., 76, 2788 (195*
H.B. Nisbet, J. Chem. Soc., 1945, 126.
[1044] T. Zincke, Ann., 330, 361 (1903); 333, 296 (1904); T. Zincke, W. Wu
ker, Ann., 341, 865 (1905).
[1045] H. Lettre, W. Haede, E. Ruhbaum, Ann., 579, 123 (1953).
IV. ОБМЕН ДРУГИХ. ЭЛЕМЕНТОВ НА АЗОТ 5Q5
Протекание реакции по схеме 1 или 2 зависит От строения пиридиниевой соли
и условий проведения реакции (ср. обзоры [219, 467]).
При нагревании М-(2,4-динитрофенил)-пиридинийхлорида с гидрохлори-
дом анилина при 200° С через некоторое время образуется 2,4-диннтродифенил-
амин [1046] (реакция Z); известны другие примеры згой реакции [1047].
Реакция, протекающая по схеме I, представляет значительный препаративный
интерес для аминирования солей пнридил-4-ниридиния, так как позволяет вводить
аминогруппы в положение 4 пиридина.
Так получают 4-аминопиридин [1048], для чего хлорид М-пирндил-4-пиридиния
нагревают с водным аммиаком в течепие 8 ч при 150° С или пропускают в течение 3 ч
аммиак в раствор этой соли в феноле при 180—190° С (выход 80% от теоретического)
[1049]. Аналогичным способом из этой же соли в расплавленном феноле при пропуска-
нии диметиламнна синтезируют 4-диметиламинопиридин [1050]. При действии арома-
тических аминов в этих условиях легко протекает расщепление четвертичного пириди-
ниевого кольца; поэтому для получения 4-ариламипопиридннов реакцию рекомендуется
проводить с гидрохлоридами ароматических амипов [1015].
4-Фениламинопиридин [219]. Смесь 2,3 г гидрохлорида М-пиридил-4-
пиридиннйхлорида и 2,6 г гидрохлорида анилина нагревают при 180° С в тече-
ние 90 мин. После охлаждения смесь растворяют в воде и при нагревании обра-
батывают животным углем. Полученный раствор сильно подщелачивают
раствором NaOH и нагревают до кипения. При охлаждении выделяются крис-
таллы 4-феннламинопиридина; выход количественный. После перекристаллиза-
ции из водного метилового спирта т. пл. 172° С.
Расщепление кольца, подобное расщеплению по реакции Цинке, в некоторых
случаях для получения альдегидов проводилось и с другими азотсодержащими гетеро-
циклическими соединениями. Так, например, в щелочной среде в присутствии гидр-
оксиламина можно в результате расщепления пиррола получить диоксим янтарного
диальдегида [1051]. Аналогичным способом иа дигидропириднна, который готовят
восстановлением пиридина натрием в метиловом спирте, синтезируют диоксим глута-
рового днальдегида [1052].
Цинлическая система этиленимина расщепляется аммиаком или аминами с го-
раздо большим трудом, чем кольцо окиси этилена; в качестве катализаторов при этой
реакции применяют хлорид аммония [1053] и хлорид алюминия [1054].
Переамидирование и аналогичные реакции
Группа NH2 амидов карбоновых кислот может обмениваться на остатки других
аминов, гидразинов и аналогичных соединений. Особенно легко эта реакция протекает
в отсутствие заместителей у амидного атома азота. Так, при нагревании аминов с форм-
амидом в вакууме часто уже при температурах до 70° С достаточно гладко получаются
формильные производные аминов |1О55]. Амиды высших карбоновых кислот подвер-
гаются аналогичным превращениям при температурах около 150—200° С [1056].
[1046] F. К г о h n k е, Angew. Chem., 65, 605, 624 (1953).
[1047] А, Ф. Вомпе, Н. Ф. Т у р к ц и н а, ДАН СССР, 64, 341 (1949);
С. А., 43, 4671 (1949).
[1048] Е. Koenigs, Н. Greiner, Вег., 64, 1049 (1931); J. Р. W i Ь a u t,
S. Herzberg, J. Schlatmann, Rec. trav. chim., 73, 140 (1954).
[1049] A. Albert, J. Chem. Soc., 1951, 1376.
[1050 D. Jerchel, H, Fischer, K. Thomas, Chem. Ber., 89. 2921 (1956).
[1051 ] R. Willstatter, W. Heubner, Ber,, 40, 3871 (1907); L. С. К e a g 1 e,
W. H. H a r t и n g, J. Am. Chem. Soc., 68. 1608 (1946).
[1052] А. С. С о p e, H. L. D г у d e n jr, et al., J. Am. Chem. Soc., 73, 3416 (1951). *
[1053] L. B. Clapp, J. Am, Chem. Soc., 70, 184 (1948).
[1054] G. H. Coleman, J. H. Callen, J. Am. Chem. Soc., 68, 2006 (1946).
[1055] B. Lachowicz, Mh. Chem., 9, 695 (1888); герм. пат. 449112; Frdl., 15,
234 (1925—1927).
[1056] J. II. M c G r e g о r, F. W e г d, J. Soc. Chem. Ind., 66, 344 (1947).
506
ОБРАЗОВАНИЕ C-N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
Переамидированио протекает легче при сплавлении амида карбоновой кислоты с гидро-
хлоридом амина [641, 1057]. Диациламиды, включая циклические имиды дикарбоновых
кислот, папример сукцинимид или фталимид [1058], обладают болыпей реакционной
способностью, чем простые амиды.
Интересные реакции переамидирования осуществимы с исключительно реак-
ционноспособными N-ацилимидазолами [560а]. Легко получаемый из фосгена и имид-
азола карбонилдиимадазол (стр, 435) реагирует с аминами иногда уже при комнатной
температуре с образованием соответствующих мочевин [560]. Например, из анилина
в среде тетрагидрофурана при комнатной температуре за 30 мин образуется дифенил-,
мочевина с выходом 91% от теоретического [560]. Имидазолиды карбоновых кислот,
легко получаемые в среде тетрагидрофурана из карбонилдипмидазола и карбоновые
кислот (см. уравнение реакции па стр. 448), легко переходят в другие амиды при взаимо-
действии с аминами при комнатной температуре, причем реакцию проводят в одну
стадию без выделения имидазолидов [650, 650а]. Таким способом, например, былв-
получены анилиды н диэтиламиды n-алкилзамещенных бензойных кислот с выход*»»:
90—95% от теоретического [650]. Этот же метод с успехом применялся для синтеза,
пептидов [561]. Имидазол- или триазол-1^-карбалкоксипроизводные, образующиеся'
из имидазола пли триазола с эфирами хлоругольной кислоты, легко превращаются
в эфиры карбаминовой кислоты (уретаны) под действием аминов [1059]. Имидазолиды,
и трпазолиды ароматических кислот обладают меньшой реакционной способностью,':
чем соответствующие производные алифатических кислот. Так, для получения диятт-
лида достаточно нагревать диимидазолид адипиновой кислоты с анилином в течения
15 мин при 120° С, тогда как аналогичная реакция диимидазолида терефталевой кис-.'
лоты с анилином [1060] протекает за 3 ч при 150° С.
Реакции переамидирования часто применяются для перевода мочевины в N-'
и М,1Т-замещонные мочевины и в соответствующие гидразиды. Мочевипу можно сплав-)
лять с амином [1061] или некоторое время нагревать компоненты в ледяной уксусной'
кислоте [1062]. В зависимости от температуры реакции и молярпого соотношения ре-’
агентов при этом получают моно- и симметрично дизамещенные мочевины, но нередко,
реакция протекает с образованием смеси моно- и дизамещенных продуктов. Гидрёд.
хлориды аминов могут реагировать также с кипящими водными растворами мочевины.)
Известным примером является образование метилмочевины из мочевины при нагре-,
вании ее в течение 3 ч с раствором гидрохлорида метиламина в процессе получения иит-
розометилмочевипы [1063]. Синтез фенил-и М,№-дифенилмочевины при действии вод-
ного раствора гидрохлорида анилина на мочевину см. [1064].
При использования в качестве исходного соединения нитромочевицы почт
всегда получаются амиды с очень хорошими выходами [1065—1068]. В этом случа
на новый амидный остаток обменивается исключительно нитроамидпая группа, ш
этому такой метод особенпо пригоден для синтеза монозамещенных мочевин и иногй
даег лучшие результаты, чем обычный метод их получения через изоцианаты [1066]
Этилмочевина [1069]. В сосуде емкостью 2 л растворяют 44 з атиламиа
в смеси 200 мл воды и 100 з льда, прибавляют 104 з интромочевины и пагревавт
на водяной бане до начала интенсивного выделения азота. Во избежание выброс!
массы сосуд охлаждают ледяной водой, а после прекращения выделения азот<?
Ann., 609, 83 (1957).
Chem. Вег., 90, 1326 (1957).
H. W. Underwood jr.,
47. 2437 (1914).
[1057] A. G a 1 a t, G. К 1 i s о n, J. Am. Chem. Soc., 65, lo66 (1943); D. Kia
m a n n, Mh. Chem., 84, 925 (1953).
[1058] F. S. Spring, J. C, Woods, J. Chem. Soc., 1945, 625.
[1059] H. A. Staab, ' - -,‘л ™
[1060] H. A. Staab,
[1061] T. L. Davis,
(1922).
[1062] A. S о n n, Ber., ... .
p д r n l. L о e w o, S. A v a n, Ber., 73, 606 (1940).
T. L. Davis, К. C. Blanchard, Org. Synth., 3, 95 (1923).
T. L. Davis, К. C. Blanchard, J. Am. Chem. Soc., 51, 1790 (1924
J. S. buck, C. W. Ferry, J. Am. Chem. Soc., 58, 854 (1936).
R. W. Charlton, A. R. Day, J. Org. Chem., 1, 552 (1936).
J. K. S h i 1 1 i n,g ton et al., J. Ain. Chem. Soc., 80, 6551 (1958).
E. Biilmann. А. К 1 i t, Ber., 63, 2205 (1930).
Am. Chem. Soc., 44, 25S
1063]
1064
1065
1066
1067
1088
10691
IV.' ОБМЕН ДРУГИХ. ЭЛЕМЕНТОВ ПА АЗОТ
507
для завершения реакции смесь нагревают до кипения. Этилмочевина кристал-
лизуется при упаривании раствора в вакууме. Затем массу охлаждают до 0° С,
отфильтровывают этилмочевину и промывают ледяной водой; выход 55 г; т. пл.
90—92° С. Из маточного раствора и промывных вод при упаривании выделяют
дополнительно 15,7 г этилмочевины; т. пл. 89—92° С. Еще 7—8 г этилмочевины
получают из остатка после кристаллизации при растворении его в абсолютном
спирте и осаждении эфиром. Общий выход продукта 78 г (90% от теоретического).
Описано также взаимодействие с амидами питрогуапидина [1070].
Гидразиды карбоновых кислот могут быть получены действием гидразингидрата
и.in замещенных шдразинов на соответствующие амиды. В зависимости от условий
проведения реакции, молярного соотношения реагентов и уже имеющихся в молекуле
гидразина заместителей реакция приводит к образованию моно- или диацилироваппых
гидразинов.
М,К'-Диформилгидразин [1071]. Раствор 25 мл (0.5 моль) гидразин-
гидрата в 45 г (1 моль) формамида нагревают 2 ч на паровой бане. Пепрореагиро-
вавшие исходные соедипения отгоняют в вакууме и добавляют к остатку 100 мл
этилового спирта. Получают Г4,№-диформилгидразиц; выход 80% от теорети-
ческого; т. пл. 160° С
Бензгидразид [1072]. Бензамид нагревают в трехкратном количестве
воды с эквивалентным количеством гидразингидрата до прекращения выделения
аммиака и охлаждают- Выкристаллизовавшийся бензгидразид перекристаллизо-
вывают из горячей воды. Образование гидразида из амида протекает более гладко
и быстро, чем из этилового эфира бензойной кислоты.
Интересной областью применения описанной реакции является взаимодействие
пептидов с гидразином (8—10 ч при 100° С). При этом все аминокислоты, за исключе-
нием С-концевых, превращаются в гидразиды. Так как С-концевые аминокислоты
легко отделяются от гидразидов кислот, то при помощи данного метода можно опре-
делять последовательность аминокислот в пептиде (см., например, [1073]).
При нагревании в запаянной трубке мочевины с молярпым количеством гидр-
азмнаидрата при 100° С в течение 3 ч образуется семпкарбаэпд [1074]; т. ил. 96° С.
4-Фенилсемикарбазид [1075] CO — NHNII2 был получен нагреванием в те-
чение 12 ч па паровой бане фенилмочевины (0,5 .моль) с 42%-ным гидразингидратом
(1 моль).
Синтез 4-замещенных семикарбазидов можно осуществить также из семпкарб-
азонов, обменивая при нагревании с высококипящпмя аминами свободную аминогруппу
на замещеппую [1076].
В исключительно мягких условиях протекает синтез гидразидов карбоновых
кислот по Штаабу [650а, 1077] через имидазолиды. Для этого соответствующий гидр-
азин прибавляют к эквимолярной смеет карбоновой кислоты и карбонилдиимцдазола
в сухом тетрагидрофурано (реакцию контролируют по выделению СОг). Для синтеза
незамещенных гидразидов можно применять гидразингидрат, так как гидразинолиз
имидазолидов протекает быстрее, чем их гидролиз [650а. 1077].
Но для того чтобы перевести гидразид карбоновой кислоты в амид, часто реко-
мендуется сначала получить азид из гидразида и азотистой кислоты и затем провести
реакцию с амином; азидный остаток обменивается легче, чем гидразидная группа.
[1070] Л. F. McKay, Chem. Rev., 51, 301 (1952).
1071 ] С. A i ns wort h, И, G. Jones,!. Am. Chem. Soc., 77, 621 (1955).
1072] Th. Curtins, G, Struve, J. prakt. Chem. [2], 50, 296 (1894).
1073] G. В г а и n i t z e r, Chem. Ber., 88, 2025 (1955).
1074) Th. Curtius, K. Heidenreich, Bor., 27, 55 (1894).
1075] A. S. Wheeler, Org. Synth., 6, 74 (1926).
1076] W. Porsche, Врг., 38, 831 (1905).
[1077] IF. A. S t a a b, Angew. Chem., 71, 385 (1959).
508
ОБРАЗОВАНИЕ C-N-СВЯЗИ Б РЕЗУЛЬТАТЕ ОБМЕНА
Обмен азидного азота иа амидный имеет особенно важное значение в химии пептид?»
|660, 661J.
Интересный обмен азотных функций происходит в симметричном трнаэине (полу*
чение см. [762, 1078}). При действии первичных аминов каждая метиловая групп*
триазина конденсируется с двумя молекулами первичного амина с образование
М,№-дизамещенного формамидина, в то время как три атома азота гетеродиклическ(
системы выделяются в виде аммиака. ,
СН
N
НС
N
I +6NH2R
СН
zNR
3HCtf 4-3NH3
\NHR
Реакция протекает с превосходными выходами в среде инертных безводных ра<
творителен (бензол, тетрагидрофуран, диоксан) илн без растворителей. Низшие а
фатические амины реагируют при комнатной температуре, высшие — при умерен:
нагревании (обзорную статью см. [1079а]).
Применяя в этой реакции диамины, аминофеполы, аминотиофенолы и аналог
ные соединения с соответственно расположенными функциональными группами, моя
осуществить замыкание кольца в гетероциклическое соединение, при этом обе Функ
опальные группы амина связываются метин свой группой триазина, а в качестве .
бочного продукта практически образуется только аммиак [1079]. Симметричн
триазнн реагирует с гидразингидратом и дает дигидразон М^'-диформилгидраа:
HaNN = CH—NH—NH—CH=NNH2, при помощи которого можно также проводить
тересные синтезы гетероциклических соединений [1080]; замещенные гидразины л
фенилгидразина реагируют аналогично первичным аминам с образованием фор
завов [1080].
Обмен азота е солях диазония и диазосоединениях
Ароматические аминогруппы могут обмениваться на другие азотсодержав
остатки с промежуточным образованием диазониевых солей.
Основное значение реакции заключается в возможности переводить амя
в нитросоединенин. Многие иитросоедннения, которые не могут быть получены прям
нитрованием, легко синтезируются по этому методу с удовлетворительными или да;
отличными выходами из нитритов и нейтральных или слегка щелочных растворов сой
диазония. Успех реакции в значительной степени зависит от выбора подходяще
переносчика нитрогруппы [1081]. По данным Ходжсона и Марсдена [152], лег
всего в нитросоединения переводятся гексаннтритокобальтиаты диазония.
Диазотируют 0,1 моль амина 7 г NaNO2 в возможно меньшем по объей
соляно- нли сернокислом растворе. Раствор соли диазония нейтрализуют СаСО
фильтруют и прибавляют к фильтрату при перемешивании 45 г тринатрийгекс
нитритокобальтиата; при этом большей частью выделяется трис-диааониев
соль в твердом виде. Для перевода в нитросоединенне 10 г тонконзмельчеиН
* соли диазония вносят при комнатной температуре небольшими порциями и;
хорошем перемешивании в раствор 10 г NaNOa и 10 г CuSO4 в 60 мл воды, в В
тором суспендировано 4 г Сн2О. После окончания выделения азота образовав
[1078] Ch. Grun d mana, A. Kreut z berger, J. Am. Chem. Soc,. 76. 504
(1954).
[1079] Ch. Grundmann, A. Kreut z berger, J. Am. Chem. Soc., 77, 65Й
(1 15'); J. Polymer Sci., 38, 425 (1959). >
[1079a] ( h. Grundmann, Angew. Chem., 75, 393 (1963). '
[1080] CD. Grundmann, A. Kreutzberger, J. Am. Chem. Soc., 79, 2831
(1957); cp. Ch. G r*u n d m a n n, R. Rat z, J. Org. Chem. 21, 1037 (1956).
[1081] A. Hantzsch, J. W. В lagd en, Ber., 33, 2544 (1900).
IV. СЕМЕН ДРУГИХ ЭЛЕМЕНТОВ ПА АЗОТ
509
шееся нитросоединение экстрагируют подходящим растворителем (например,
хлороформом) или отгоняют с паром из щелочного раствора [152].
По этому методу получены [152J: а-нитронафталин (выход 68% от теоре-
тического), Р-нитронафталин (60%), п-питротолуол (69%), п-нитроаниаол (68%)
и о-нитротолуол (61%).
Аналогичным способом, но с худшими выходами получен ряд динитронаф-
талинов |1082].
В качестве исходных соединений могут применяться также твердые сульфаты
диазония (получение см. [1083]). Их отмывают от свободной нислоты спиртом или эфи-
ром и вносят в виде водного раствора или в твердом виде в водный раствор NaNOa,
в котором суспендирован сульфит меди (1) и (II) [1084].
Для получения сульфитов меди к водному раствору сульфата меди приба-
вляют равное весовое количество водного раствора NaaSO3 н промывают осадок
[1084, 1085].
Такой метод особенно пригоден для синтеза динитронафталинов. Подробную
пропись получения 1,4-динитронафталнна из 4-нитро-1-нафтиламина, которую можно
использовать также и для синтеза аналогичных соединений, см. [1085]. Вместо суль-
фатов диазония в этой реакции можно применять также гексанитритокрбальтиа-
гы [1084].
Во всех случаях, когда амин диазотируется в водном растворе н образующаяся
соль диазония устойчива в нейтральном растворе, проведение реакции можно еще
более упростить [1086, 1087], как видно из следующего примера.
о-Дилитробензол [1087]. Растворяют 10 г o-нитроанилипа в горячей
смеси 12 мл HaSO4 (d = 1,84) и 30 мл воды н выливают полученной раствор при
интенсивном перемешивании в 50 мл ледяной воды. После добавления 20 а раз-
дробленного льда к полученной смеси очень быстро (1) и при хорошем перемеши-
вании. прибавляют раствор 8 г NaNOa в 13 мл воды. Перемешивают 5 мин и полу-
ченный раствор соли диазония приливают отдельными порциями, но достаточно
быстро, через широкую стеклянную трубку к хорошо перемешиваемому раствору
t 100 г NaNOa и 45 г NaHCO3 в 1 л воды при 60е С, к которому добавлен силиконо-
вый пеногаситель (трубка должна быть опущена в жидкость). Через 5 мин после
того, как добавлено все количество смеси, осадок отфильтровывают и промывают
2 н. раствором НС1 и большим количеством воды. Выход практически чистого
о-дннитробепзола 11,8 е (97% от теоретического); т. пл. 116—118° С. Совершенно
чистый о-динигробензол получают пропусканием бензольного раствора неочищен-
ного продукта через колонку с окисью алюминия; выход 90% от теоретического;
т. пл. 118° С.
п-Динитробензол [1087]. Его получают таким же способом, но в этом
случае достаточно половинного количества HaSO4 и NaHCO3. -
При взаимодействии других диааониевых солей приходится добавлять
катализатор [1086]; достаточно добавить к горячему раствору NaNOa несколько
граммов CuSO4 и СпаО.
В принципе диазогруппа может быть замещена не только на нитрогруппу,
но и на другие азотсодержащие заместители. Так, например, при взаимодействии
[1082] Н. Н. II о d g s о n, Е. R. Ward,!. Chem. Soc., 1947, 127.
[1083] II. II. Hodgson, A. P. Mahadevan, J. Cnem. Soc., 1947, 325.
[1084] H. H. Hodgson, A. P. Mahadevan, E. R. Ward, J. Chem, Soc.,
1947, 1392.
[1085] H. H. H о dgson, A. P. M a lia de van, E. R. Ward, Org. Synth.,
Coll. v. 3, 1955, p. 341.
[1086] H. H. Hodgson, F. Heyworth, E. R. Ward, J. Chem. Soc., 1948,
1512.
[1087] E. R. Ward, C. D. Johnson, J. G. Hawkins, J. Chem» Soc., 1960,
894.
510
ОБРАЗОВАНИЕ С—N-СВЯЗП В РЕЗУЛЬТАТЕ ОБМЕНА
с цпанатами щелочных металлов получаются арилизоцианаты [ 1088], а с азидом натрия i
или с азотистоводородной кислотой образуются арплазиды [1089, 613]. Другие превра- .3
щения см. [291].
Кроме того, следует упомянуть, что алифатические и ароматические амины ал-
килируются под действием диазометаяа [1090] в присутствии BF3 или под действием -
диазокетонов [1091] в присутствии порошкообразной меди, однако препаративное ’
значение таких реакций невелико. ?!
Из ароматических изоцианатов и диазометана можно получать Р-лактамы [1092]
G6H5-N-CO j
Cen6-NCO | | |
H2c—CII2 J
2. Обмен углерода на азот t
Образование С—N-связи с разрывом С—С-связи наблюдается в некоторых,1
случаях в качестве нежелательной побочной реакции при нитровании (обзор работ о та- -
ких аномальных процессах см. [146, 1093]), нитрозировании и азосочетании. Некото-
рые примеры подобных реакций рассматривались в соответствующих разделах. Такой
обмен представляет препаративный интерес лишь в тех случаях, когда он становится
основной реакцией, как, например, в некоторых случаях оксимирования азотистой .
кислотой (стр. 394) или в реакции Джэппа — Клищемана (стр. 402).
Нитрование n-цимола дымящей НГчО3 приводит к образованию 2,4-ди>ти-»3«
тротолуола [1094]. 3,5-Динитро-1-метилпиридон-2 [1095] образуется с хорошим J
выходом при кипячении 6~кето-1-метил-1,6-дигидропикотиновой кислоты^
с HNO3 (d --= 1,52). 4-Окси-3,5-дибромбензойпая кислота в водно-спиртовом^а
растворе количественно превращается в 4-нитрозо-2,б-днбромфенол [1093] а
под действием NaNO5. Оксиметильныо группы в фенолах обмениваются на нитро-
группы под действием азотистой кислоты [1096] и на азогруппу под действием J
солей диазония [1097]. «|
Изящным методом ацилирования достаточно силънаосновных аминов являете»’;
их взаимодействие с ацилцианидами [623]:
R—СО—CNH-R'NH2
R—СО-NHR' + HCN
Преимуществом этого метода [1098, 1099] в сравнении с обычным ацилированиеМ;!
поШоттену— Бауманпу или по Энцхорну является возможность ацилировать амины^З
неустойчивые в водной, кислой или щелочной среде. Я
Реакция проводится при комнатной температуре в инертных растворителях, 4*з
причем связывание выделяющейся синильной кислоты нс обязательно. В этих уело- 1
_________________ 4
[1088
[1089
1091
[1092
L. Ga t t ermann, Л. Ca n h ler, Ber., 23, 1225 (1890); 25, 1086 (1892). 1
11WO O. Noelting,.O. Michel Rer., 26, 86 (1893). J
[1090 E. Mu 1 ] e г, II. Huber-Emden, W. Rnndel, Ann., 623, 34 (1959). J
[1091 P. Yates, J. Am. Chem Soc., 74, 5376 (1957). Д
[1092 J. C. Sheehan, P. I. Izzo, J. Am. Chem. Soc., 70, 1985 (1948); 71, 4059 Я
(1949). Д
[1093] R. A. Henry, J. Org. Chem., 23, 648 (1958). 1
[1094] J. A If th an, Ber., 53, 78 (1920). 1
[1095] A. II. В e г r i e, G. T. Newbold, F. S. Spring, J. Chem. Soc., J
1951, 2590. Я
[1096] E. Ziegler, K. Gartler, Mh. Chem., 80, 634 (1949). 4
[1097] E. Marder, I. W. Rud erman J. Am. Chem. Soc., 73 5475, (1951); »
E. Ziegler, G. Zigeunei, Mh. Chem., 79, 42, 89, 358 (1948). -J
[1098] A. Dornow, H. Theidol, Angew. Chem., 66, 605 (1954). 4
[1099] A. Dornow H. Theidd, Chem. Ber., 88, 1267 (1955). j
J
V. ОБМЕН ДРУГИХ ЭЛЕМЕНТОВ НА АЗОТ
511
пнях аминосппрты только N-ацилируются даже в присутствии избытка реактива;
лить при температурах выше 100° С, например в кипящем диметилформамиде, в реак-
цию вступает и гидроксильная группа.
N-Бензовл-Л-эфедрин [1099], К раствору 1 г (6 ммоль) L-эфедрина
в приблизительно 100 мл эфира постепенно прибавляют 0,9 г (7 ммоль) бензоил-
цианида в 50 мл эфира. Через несколько часов из умеренно упаренного рас-
твора начинается кристаллизация чистого ^бензоил-^-эфедрина; выход 1,2 а
(71,5% от теоретического); т. пл. 110° С.
Аналогичным способом, исходя из дихлорацетилцианида, который может быть
получен [1100] в реакционной массе из хлораля и NaCN, синтезировали хлорамфени-
кол (левомицетип) [1099].
Малая прочность С—С-связи в хлорале, легко расщепляемой сильноосновными
аминами, с успехом используется для N-формилирования, тем более что реакция про-
текает обычно в мягких условиях с очень высокими выходами [1101 —1103].
Для этого к раствору амина в хлороформе в отсутствие доступа влаги по
каплям прибавляют эквимолярное количество хлораля, смесь перемешивают
несколько часов при комнатной температуре и затем нагревают 30 мин на паро-
вой бане [1102].
При нагревании днэтилового эфира ацетондикарбоновой кислоты с трехкрат-
ным количеством амина происходит двукратное расщепление С—С-связи. Исходя
из замещеппых р-фенилэтиламинов таким способом получали соответствующие моче-
вины [1104].
3. Обмен серы на азот
Обмен сулъфогруппы на азот
Ввиду доступности сульфокислот ароматического ряда обмен их сульфогруппы
на другие функциональные группы часто является самым простым методом введения
заместителей в ароматическое ядро. Этот путь имеет особенно большое значение при по-
лучении аминов антрахинонового рядя. Так, 1- и 2-амяноантрэхивоны и 1,5-, 1,8-
м 2,6-диаминоантрахннопы получают нагреванием соответствующих сульфокислот с ам-
миаком под давлением; при этом очепь важно удалять отщепляющийся сульфит из
среды вследствие равновесного характера процесса, что достигается осаждением (на-
пример, хлоридом бария) нли окислением до сульфата (мышьяковой кислотой илн
л-нитробензолсульфонатом натрия).
1-Аминоантрахннон [1105]. Смесь 60 е антрахиноп-1-сульфоната калия,
120 а 24%-ного раствора аммиака и 21 г л-нитробензолсульфоната патрия нагре-
вают в течепие 4 ч до 170—175° С в автоклаве о мешалкой и поддерживают эту
температуру в течение 12 ч (давление около 25 ат). После охлаждения реакцион-
ную массу тщательно фильтруют через воролку Бюхнера и осадок промывают
небольшим количеством горячей воды. Осадок снимают с фильтра и кипятят
с 300 лл воды, подкисленной НС1. Вновь отфильтровывают осадок, промывают
горячей водой и сушат при 90° С. Выход технического продукта около 39 г
(95% от теоретического); т. пл. 238° С. После перекристаллизации из ксилола
получают чистый 1-аминоаптрахннон, выход 75% от теоретического; т. пл. 241е С.
[1100] Англ. пат. 712745; С., 1955, 3736.
[1101] G. В. L. Smith, М. Silver, Е. S. Becker, J. Am. Cbem. Soc., 70,
4254 (1948).
[1102] F. F. Blieko, Chi-Junj L u, J. Am, Chem. Soc., 74, 3933 (1952).
[1103] F. F. Blicke, Chi-JungLu J. Am. Chem. Soc., 77, 29 (1955).
[1104] M. I. Moyer, W. E. McEwen, J. Am. Chem. Soc., 73, 3075 (1951).
[1105] См. ссылку [330].
512
ОБРАЗОВАНИЕ С—N-СВЯЗН В РЕЗУЛЬТАТЕ ОБМЕНА
Точно так яке можно синтезировать 2-аминоантрахинон [1105}. В соответ- >
ствующей прописи получения 1-метиламиноантрахинона [1106] в качестве^
окислителя используют NaC103.
1,3-Диаминонафталин. Нагревают i-нафтиламин-3-сульфокислоту ч
с аммиаком или аминами под давлением при 160—180° С (сульфогрунпы, находя^
щиеся в другом кольце, в этих условиях не обменнваются). Одновременный обмеТ"
сульфо- и оксигруппы происходит при проведении реакции с 1-нафтол-З-сульфо
кислотой (1107}.
При сплавлении солей сульфокислот с амидом натрия реакция протекает п]
нормальном давлении, но с худшими выходами [357]. В то же время в синтезе N-ара
пиперидинов из ароматических сульфокислот проведение реакции в кипящем пипер
дине с амидом натрия полностью себя оправдало [1108].
Алкил- и арнлсульфоннльные остатки обмениваются на амины так же, к<
сульфогруппы. Подобное расщепление сульфонов протекает прп взаимодействии с вт
ричными циклическими аминами в присутствии амида натрия, часто с высокими вых
дами третичных аминов [1108—1110].
Известен также обмен сульфогрунпы на дназогруппу. Иногда при сочетай
о- нли п-амипобензолсульфокислот и о-, м- и n-оксибензолсульфокислот с соля)
диазония такое превращение протекает исключительно легко [342].
Особое значение имеет замена сульфогруппы на нитршруппу [1111], Иног)
удается очень изящно предотвратить окисление фенолов, происходящее при их пряма
нитровании, для чего сначала фенолы сульфируют, а затем вытесняют сульфогрутп
остатком азотиой кислоты [1112}- Многие нитропроиэводные, представляющие техн
ческий интерес, получают в промышленном масштабе именно таким образом. Извесп
лабораторный способ синтеза пикриновой кислоты, который можно распространи
на получение 2,4-динитро-1-нафтол-7-сульфокислоты (нафтоловый желтый S) и нити
крезолов [1113,1114]. В некоторых случаях фенол-, крезол- и ксиленолсульфокислот
с успехом переводились питрозными газами в соответствующие ннтрофенолы [1115
Обмен других серусодержащих функциональных групп на азот.
Часто используемый метод получения гуанидинов основан на взаимодейстЩ
S-алкилизотиурониевых солей с аммиаком или аминами [1116—1118]. .‘£
м_с/гаЬ 52Ь,
4SR
/NH2
'NHR'
[1106] С. V. Wilson, J. В. Dickey, С. Н. F. Allen, Org. Syntii.
29, 66 (1949). Г
[1107] Герм. пат. 89061, 90905, 90906, 94075; Frdl., 4, 598—600 (1894—1897).
[1108] Т. К. Brotherton, J. F. Bunnett, Chem. a. Ind., 1957, 80; J. Ад
Chem. Soc., 78, 155 (1956); J.’F. Bunnett, T. K. BrothertoJj
S. M. Williamson,' Org. Synth., 40, 74 (1960).
[1109] W. Bradley, J. Chem. Soc., 1938, 458.
[1110] H.-J. Nitzschke, H. В u d k a, Chem. Ber., 88, 264 (1955).
[1111] Ch. M. Suter, The Organic Chemistry of Sulfur, John Wiley a. Sons, Nfll
York, 1944, p. 406ff.
[1112] R. King, J. Chem. Soc., 119, 2105 (1921).
[1113] См. ссылку [330], стр. 144. н
[1114] L. Gattermann, If. Wieland, Die Praxis des organischen Chert
kers, 35 Aufl., Berlin, 1953, S. 172.
[1115] R. L. D a 11 a, P. S. Varma, J. Am. Chem. Soc., 41, 2039 (1919).
[1116] С. E. Braun, J. Am. Chem. Soc., 55, 1280 (1933.
[1117] K. King, S. M. T о n k i n, J. Chem. Soc., 1946, 1063.
[1118] A. F. McKay, W. G. Hatton, R. O. Braun, J. Am. Chem. Soft
78, 6144 (1956).
IV ОБМЕН ДРУГИХ ЭЛЕМЕНТОВ НА АЗОТ
515.
При использовании тиурониевых солей с незамещенными атомами азота реак-
ция протекает обычно уже па холоду.
Гуанвдоуксусная кислота (гликоциамин) [1119]. При охлаждении льдом
к 92,5 г бромида S-этилизотиурония приливают 252 мл 2 н. раствора NaOH
п затем быстро добавляют горячий (80” С) раствор 41 s гликоколя в 90 мл воды.
Корда температура реакционной массы достигает 25°С, прекращают охлаждение.,
Кристаллизация продукта начинается примерно через 30 мин, после чего в реак-
ционную массу добавляют 100 мл эфира и оставляют ее на ночь. На следующий
день смесь охлаждаю? в течение 2ч в бане со льдом, сливают эфир и отфильтро-
вывают продукт. После промывки его ледяной водой (2 порции по 20 мл), этило-
вым спиртом (2 порции по 150 мл) и эфиром (2 порции по 150 мл) и сушки на воз-
духе получают 47—53 г (80—90^о от теоретического) гуанидоуксусиой кислоты;
т. пл. 280-284° С.
В случае N-замещепных тиурониевых солей необходимо нагревание. Таким,
же образом были получены и нитрогуанидины из N-нитропроизводных тиурони-
свых солей |1120]. Особенно гладко протекает взаимодействие тиурониевых солей с
гпдразингидратом с образованием аминогуапидинов [1121]. Вместо изотиурониевых
солей можно применять также тиомочевины, проводя их взаимодействие с аммиаком
или аминами в присутствии обессеривающих средств, например окиси свинца
(ср. [1122, 1123]).
Расщепление эгилеысульфидов аминами можно использовать для получения
р-аминоэтантиолов [1123а]. В то время как взаимодействие с алифатическими аминами
протекает при кипячении в бензоле в нолбе с обратным холодильником [1123а], для
проведения реакции с ароматическими аминами применяются жесткие условия (нагре-
вание под давлением до 100Q С и выше) [1124].
4. Обмен металлов на азот
Из большого числа металлоорганических соединений препаративное зпачение-
для получения С—N-связей имеют главным образом реактивы Гриньяра. Алкил- и
арилмагаийгалотениды подвергаются многочисленным превращениям под действием
азотсодержащих соединений [1125], однако определенный интерес представляют лишь,
немногие из них.
Так, папример, из магнийоргацических соединений и хлорамина можно получать,
первичные алифатические и ароматические амины:
2RMgCl I-NH2CI —> RNHMgCl + MgCh+RII
[it,о
BNHs + MgfOHJCl
ГИ19] Е. Brand, F, С, Braid, Org. Synth., Coll. v. 3, 1955, p. 440.
И 120] L. F i s h h e i n, J. A. Callaghan, J. Am. Cham. Soc., 76, 1877 (1954).
[1121] G. W. Kirsten, G. B. L. Smith, 1. Am. Chem. Soo., 58, 800 (193S);
R. A. Henry, G.B.L. Smith, J. Am. Chem. Soc., 73, 1858 (1951);
W. G. Finnegan, R. A. Henry, E. Lieber, J. Org. Chem.. 18, 779-
(1953); E. S. Scott. L. F. Audrieth, J. Org. Chem., 19, 1231 (1954).
[1122] S. Petersen, в книге 1] ouben-Wey], Methoden der organischen
Chemie, Bd. 8, 4 Aufl., Stuttgart, 1952, S. 183.
[1123] J. Macholdt-Erdniss. Chem. Ber., 91, 1992 (1958).
[1123a] Литературный обзор: R. J. Wineman et al., J. Org. Chem., 27, 4222
(1962).
[1124J П. R. Snyder, J. M. Stewart, I. B. Z j e g 1 e r, J. Am. Chem. Soc.,
69, 2672 (1947); герм. пат. 631016; С., 1936, 11, 1615; англ. пат. 445805; С., 1986,
II, 3946.
[1125] М. S. К h а г а е с Ь, О. Reinmuth, Grignard Reactions of Nomnetallic.
Substance?, Prentice-Hall. Inc., New York, 1954, p. 1199Н.
33 Заказ 1635.
514 ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Взаимодействие протекает в эфирном растворе, лучшие результаты получаются
при использовании алкилмагнийхлоридов; побочным продуктом реакции является
соответствующий алкилхлорид.“Этст метод может представлять интерес главным
образом при синтезе первичных аминов, аминогруппа которых связана с вторичным
нли третичным атомом углерода. Примеры применения этой реакции, а также метод
получения хлорамина приведены в обзорной статье [1126].
Вместо неустойчивого хлорамина можно применять более удобный в обращении
О-метилгидроксиламин *.
Третичный бутиламин [1127]. Раствор 141 г О-метилгидронсиламииа
в 300 мл сухого эфира постепенно прибавляют при интенсивном перемешивании
и температуре от —10 до —15° С к реактиву Гриньяра, приготовленному из
609 г mpem-бутилхлорида в 3 л эфира. После добавления всего О-метилгидр-
оксиламина смесь выдерживают при —10° С еще в течение 30 мин, а затем 2 ч
пагревают в колбе с обратным холодильником. Раствор, охлажденный до 0° С,
разлагают Зл 5 н. раствора НС1. Водный слой упаривают в вакууме до мипималт.-.
ного объема. Затем его сильно подщелачивают 50%-ным раствором КОН и отго-
няют неочищенный амин, собирая фракцию в интервале 35—70° С. Полученный
амин сушат над КОН и перегоняют еще раз; выход 153 г (70% от теоретического
из расчета на О-метилгидрокспламин); т. кип. 40—45° С. После многократной
перегонки получают чистый ямин с т. кип. 44,4° С; п°£ 1,3789.
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ *♦ 1
Я
I. Образование N-П-связи Л
Образование N—Н-связи может происходить в результате реакции присоеди-
нения или замещения. Все применяемые для этого методы основаны на реакции вос-
становления.
1. Образование N—Н-связн в результате реакции л
присоединения
Получение гидразопроизводных из азосоединений
(R—N=N—R —> R-NI1-NH-R)
Азосоединения легко восстанавливаются до Ь’,К'-дизамещенных гидразинов^
называемых также гпдразосоединекнями. Простейшим ароматическим представителе^
этого ряда является гидразобензол, получаемый преимущественно восстановление^
нитробензола цинковой пылью и щелочами [1] или электрохимическим восстановлю^
нием [2] через стадию образования азобензола***. Для прямого восстановления азобеЯУЗ
зола до гидразобензола могут применяться, в частности, такие восстановители, как
цинковая пыль и спиртовой раствор КОН, амальгама алюминия [3], взвесь железе
* Этот метод предложен Н. И. Шевердиной и К. А. Кочешковым [ЖОХ, 8, 1825Й
(1938)], показавшими также, что О-метилгидроксиламин может быть замене®
более доступным О-бензилпроизводным [Изв. АН СССР, ОХН, 75 (1941)].
Прим. ред. эд
** Материал обработал Р. Оме.
*** Основным промежуточным продуктом при этом являются азоксисоединения, кол®й
чеотво ааосоединений сравнительно невелико. Процесс детально изучал В. О. Лж
кашевич [Усп. хим., 17, 892 (1948)]. —,Прим. ред.
[11261 W, Theilacker, Е. Wegner, Angew. Спет., 72, 127 (1960).
[1127] R. Brown, W. Е. J о п е э, J. Chem. Soc., 1946, 781. 3
[1] L. Gattermann, Н. Wieland, Die Praxis deg organischen СЬепйкеМй
Walter de Gruyter u. Co., Berlin, 1953, S. 162.
[2] E. Mil Iler, Elektrochem. Praktikum, Dresden u. Leipzig, 1913, S. 186.
[3] H. Wislioenus, J. prakt. Chem. [2], 54, 65.
I ОБРАЗОВАНИЕ N—Н-СВЯЗИ
515'
в щелочном растворе [4], а также электрохимическое восстаповлспие суспензии азо-
бензола в щелочной среде |5]. Гидразосоединения образуются с хорошими выходами
и при разложении водой продуктов присоединения этилмагнийбромида к азосоедине-
ниям [С].
Прп проведении реакции с избытком восстановителя происходит расщепление
N — N-связи и образуются две молекулы первичного амина. Такое восстановительное
расщепление азосоединеннй облегчается наличием в ядре электроноакцепторных
заместителей. Азосоедипения удивительно устойчивы к действию алюмогидрида лития
и только длительное взаимодействие с этим восстановителем приводит к образованию'
дшндропропзводных с плохими выходами. Между тем азогруппа, входящая и состав
ароматического гетероцикла, легко восстанавливается алюмогидридом лития [7].
Процесс получения гидразобензола и других гидразосоедннений необходимо
нести непрерывно и без доступа воздуха, так как эти соединения легко окисляются кис-
лородом воздуха до исходных азопроизводных.
Описано частичное восстановление бис-азосоединений цинковой пылью в пи-
ридине в присутствии небольшого количества ледяной уксусной кислоты. Неустой-
чивые азосоедипония мягко восстанавливаются цинковой пылью в аммиаке в атмосфере
азота [8]. Нитроазосоединопия, суспендированные в растворе аммиака пли щелочи,
восстанавливаются сероводородом с сохранением нитрогруппы.
Алифатические азосоединения восстанавливаются в соответствующие N,N'-flnaa-
вилгидразины с хорошими выходами. Тиле [9] описывает восстановление азомотана.
до симметричного димотилгидразина цинковой пылью в 8%-ном растворе NaOH при
0" С. Восстановление можно проводить также амальгамой натрия.
Получение гидразинов из диазониевыт солей
(R—N2X —> RNH-NH2)
Арилдиазониевые соли обычно восстанавливают до арилгидразипов сульфитами
щелочных металлов по методу Фшпсра или действием SnCl2 по методу Мейера. При-
действии сульфита на арилдиазониевые соли в нейтральной или щелочной среде сна-
чала образуются диазосульфонаты (R—N— N — SO;jNa), которые далее восстанавли-
ваются цинковой пылью и уксусной кислотой или сернистой кислотой. Прп этом они
переходят в соли арилгидразннсульфокислот, сульфогруппа которых отщепляется при
нагревании с разбавленной H2SO4.
Ведение процесса по старым прописям часто приводит к неудачам. В по-
следнее время предложен ряд модификаций этих методов, которые можно
использовать при восстановлении аналогичных соединений. Так, рекомендуется
после подкисления реакционной массы, обработанной раствором .Na2SOs, нагре-
вать се в течение нескольких часов [9а].
Дли восстаповлопия солей ари.тдиазония в кислой среде часто применяется
В результате реакции, как правило, получаются комплексные соли олова
с гидрохлоридами арилгидразипов, которые легко разлагаются щелочами. Этот метод
проще сульфитною и особенно пригоден для лабораторных синтезов; восстановление
протекает при низкой температуре (около 0°С). Метод оказался непригодным для
восстановления длазонпевых солей антрахинонового ряда, хлорида п-нитробензол-
диазония и хлорида о-бензолтетраэоппя. Пример восстановления хлорида бензолди-
азоиия SnCl2 до фенилгидразина приводит В. Мейер [10].
[4] Герм. пат. 138496 (19.1.1903). Chem. Fabr. vorm. Weiler-ter Meer, дополнение
к герм. пат. 181116.
[5] Е. Darmstadter, герм. пат. 196979 (С., 1908, I, 1505; 1907, I, 1649).
[6[ И. Franzen, W. D о j b е 1, Вет., 38, 2716 (1905).
[7| F. Bolilmanu, Chem. Вег.. 85, 390 (1952).
[8] Р. R и g g 1 i, К. Н б I z J е, Helv. chim. acta, 26, 814, 1190 (1943).
[9] J. Thiele, Ber., 42, 2578 (1909).
[9a] Org. Synth., СоП. v. 1, 1941, p. 432.
[10] V. Meyer, Bor., 16, 2976 (1883).
33*
516
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Гидрохлорид фенилгидразина (получение по методу Бриптцннгера [Ц
К 9,3 г анилина прибавляют 40 мл НС1, разбавленной в отношении 1 : 1, и
азотируют при охлаждении 70 мл 10%-ного раствора NaNOa). ПрозрачЕ
раствор соли диазония медленно (в течение 15—20 мин) приливают к 1 х
щепного раствора SO2, охлаждая реакционную массу водой и продолжая п
пускать SO2. После непродолжительного ^стояния (5—10 мин) добавля
50 мл концентрированной НС] и упаривают смесь до объема около 250 .
При этом начинается выделение почти бесцветного гидрохлорида фенилги,
азнна, который после охлаждения получают с выходом 13,5 а (94% от теб
тического).
п-Нятрофенилгигтяяии, Растворяют 20 а n-ннтроанилина в 42 мл к<
центрированной НС] и40 мл воды, выливают в 100 г измельченного льда и п
охлаждении и перемешивании диазотируют реакционную массу раствор
12 в NaN08 в 12 мл воды. Оставленный на 3 м во льду раствор фильтру
и при интенсивном перемешивании в течение 15 мин приливают к охлажд
пому до 0° С раствору 45 г безводного NaaSOa и 4 г NaOH в 100 мл воды. Че
5 мин к темно-красному раствору приливают 140 мл концентрированной Т
и оставляют на 12 ч. Выделившиеся кристаллы га-нитрофенилгидряяинсул*.
ната отсасывают и нагревают на водяпой бане в течение 20 мин с 40 мл кош
трированной НС]. После охлаждения отфильтровывают смесь NaCl и гидро!
рида ге-нитрофенилгидразина, растворяют последний в спирте, фильтр;
и упаривают спиртовой раствор; выход продукта 9—12 г.
Для синтеза арилгидразинов из солей арилдиазония рекомендуются и дру
методы восстановления.
В последнее время осуществлено электрохимическое восстановление хлор1
фенилдиазония [12J. При этом гидрохлорид фенилгидразина получается с кош
ственным выходом.
В качестве общего метода синтеза арилгидразинов рекомендуется восстаио]
ние солей арилдиазония трифенилфосфином [13]. Хорошие выходы получаются таз
при использовании SO2 в качестве восстановителя [14].
Фенилгидразин и его гомологи являются сильными ядами крови, кото
могут привести к хроническим отравлениям организма. У некоторых лг
фенилгидразин вызывает па коже сыпь н сильный зуд.
Получение аминов из нитрилов
(R—C=N-»R —СНа—NH2)
Все большее значение для получения первичных амипов приобретает во<
новленле нитрилов. Несмотря на сильно ненасыщенный характер нитрильной гру
восстановление ее до аминогруппы иногда затруднительно. Подходящими мето;
восстановления являются каталитическое гидрирование и восстановление Li)
или натрием в каком-либо спирте.
Каталитическое гидрирование. При каталитическом гидрировании нитрил
образуются ие только первичные амины. В иачестве побочных продуктов образую"
также вторичные и третичные амины. Ввиду большого значения, которое приоб;
промышленный синтез амниов, каталитическое восстановление нитрилов подвертя
детальному исследованию. Замечательный обзор применяемых в настоящее вр(
методов см. [14а]. Для получения главным образом первичных аминов рекомендую
следующие методы каталитического гидрирования.
[11 ] Н. Brintzinger, Die Chemie, 66, 233 (1943).
[12] Р. Ruetschie, G. Trumpler, Helv. chim. acta, 36, 1649 (1953).
[13] L. Horner, H. Hofmann Angew. Chem., 68, 484 (1956).
[14] K. Pfannstie], J. Janecke, Ber., 75, 1096 (1942).
[14a] II о u b e n - W e y1, Methoden der organischen Chemie, Bd. 11/1, 4 Al
1957, S. 454-571.
[. ОБРАЗОВАНИЕ N—Н-СВЯЗИ
517
Связывание образовавшегося амина в виде соли
[15]. Реакцию ведут в спиртовом растворе в присутствии 3 моль кислоты на моль
нитрила (HC},H3SO4, НС1О4) в качающемся автоклаве под давлением с применением
палладиевого катализатора. Выходы по этому методу достигают 90% от теоретического.
Связывание выделяющегося амина в виде амида [16]:
Нитрилы гидрируют в присутствии ангидридов кислот или сложных эфиров. При
этом образуются соответствующие амиды с хорошими выходами. В качестве катали-
заторов применяют контакты на основе благородных металлов или обезвоженные ме-
таллы Ренея. Гидрирование протекает при избыточном давлении —3 ат и нормальной
температуре в присутствии платинового катализатора или при 100 ат н температуре
около 100° С в присутствии никеля Ренея.
Гидрирование в присутствии аммиака [17]. Метод нашел
широкое применение. Очевидно, в результате проявления закона действия масс в этом
случае предотвращается отщеплеине аммиака. Гидрирование можно вести как прн
атмосферном давлении и комнатной температуре, так и при повышенном давлении и ком-
натной или повышенной температуре. Восстановление при атмосферном давлении ведут
всегда в присутствии растворителя и с большим количеством катализатора (кобаль-
товые катализаторы или катализаторы Ренея). При гидрировании под давлением
аммиак может частично или полностью играть роль растворителя. Необходимое коли-
чество аммиака колеблется от 1,2 моль (для восстановления ароматических нитриль-
ных групп) до 5 моль на 1 моль нитрила (для восстановления низших динитрилов).
Восстановление нитрильной группы в присутствии аммиака при нормальных
условиях под действием никеля Ренея на примере получения бензиламин-4-карбоновой
кислоты из 4-циапбензойной кислоты описано Альбертом [18]; гидрирование под
давлением в присутствии аммиака см. [19].
Фенилэтиламин. В автоклав емкостью 2 л загружают 1 кг перегнанного
над никелем Ренея бензалцианида и столовую ложку никеля Ревея и закрывают
его. Затем подают 180 мл жидкого аммиака н водород до давления 140 ат
и встряхивают около 1 ч при 120—130° С. После охлаждения продукт гидри-
рования отфильтровывают от катализатора и перегоняют в вакууме. Выход
83—87% от теоретического; т. кип. 90—93° С (15 мм рт. ст.).
Восстановление алюмогидридом лития. Под действием LiAlH* нитрилы пере-
ходят в первичные амины с выходами от 32 до 98% от теоретического. В отличие
от продуктов каталитического гидрирования по этому способу вторичные и третичные
амины не образуются, вследствие чего метод особенно пригоден для лабораторного
синтеза первичных аминов. Для восстановления 1 моль нитрила достаточно 0,5 моль
1лД1Н4, однако введение избытка восстановителя повышает выходы (как правило,
работают с молярным отношением компонентов 1 : 1). Амундсен [20] пзучал оптималь-
ные условия восстановления нитрилов LiAlH4. Общий метод описан им на примере
гидрирования нитрила каприловой кислоты.
Октиламип. К охлаждаемому в бане со льдом раствору 3,8 г (0,12 моль)
LiAlH4 в 200 мл абсолютного эфира по каплям прибавляют раствор 12,5 г
(0,10 лоль) нитрила каприловой кислоты в 20 xit эфира. По окончании восста-
новления при интенсивном перемешивании, продолжая охлаждение реакцион-
ной массы, понемногу последовательно прибавляют 4 мл воды, 3 мл 20%-ного
раствора NaOH и 14 мл воды. Эфирный раствор сливают с белого осадка неор-
ганических веществ, промывают осадон небольшим количеством эфира, эфирные
растворы объединяют, отгоняют эфир, а затем в вакууме — октиламин.
Выход амина 11,5—11,9 г (89—92% от теоретического); т. кип. 53° С (6 м рт. ст.).
[15] W. Н. Hartung, J. Am. Chem. Soc., 50, 3370 (1928).
[16 J W. H. Carothers et al., J. Am. Chem. Soc., 49, 2912 (1927).
117J Англ, пат. 282083 (8.11.1928); герм. пат. 541229 (19.1.1932); G. Hahn,
О. Schales, Ber., 67, 1491 (1934).
[18] A. Albert, D. Magrat h, J. Chem. Soc., 1944, 678.
[19J Org. Syalh., Coll. v. 3, p. 720.
I*0] L. H. Amundsen et al., J. Am. Chem. Soc., 73, 243 (1951).
518
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Восстановление натрием. Нитрильные группы, связанные с первичными али-
фатическими радикалами, восстанавливаются натрием в спирте. Метод непригоден для
восстановления нитрильпых групп, связанных с ароматическим ядром или с вторич-
ным атомом, имеющим в а-положенип бензольное ядро, [CeH3—CH(CN)—RJ. В этом
случае происходит отщепление нитрильной группы:
RCN4-H2 —* RH-f-HCN
При восстановлении в качестве растворителя применяется обычно
спирт- Натрий вводят в избытке из расчета 7 г-атом натрия на одну нитрильную группу*
введение большего количества натрия приводит к дальнейшему повышению выхода.’
Для полного растворения 1 г-атом натрия достаточно несколько более 3 моль бутиле^
вого спирта. Восстановление рекомендуется проводить прп механическом переменны'
вании. Ниже приведена методика такого восстановления [21], применимая во многих
случаях.
В двухгорлой колбе емкостью 5 л с широким холодильником (диаме-
2,5 см, длина 100 см) нагревают до кипения раствор 1 жоль нитрила или окси!
в 2300 мл я-бутилового спирта. К кипящему раствору прибавляют 161 г натр)
(порциями по 10—20 г через каждые 10—20 лепи). После замедления реакп?
продолжают нагревать с обратным холодильником до практически полно:
растворения всего натрия. К охладившейся реакционной массе прибавляй^.
1,5 л воды и отгоняют бутиловый спирт и амии, проверяя дистиллят на содер^
жание амина; при необходимости добавляют еще некоторое количество вода]
и продолжают отгопку до полного удаления амина. Дистиллят подкисляю?
до слабокислой. реакции и упаривают до объема 300 мл. К концентрату добаА
вляют еще 500 мл воды и перегоняют до полного удаления бутилового спирта;
Концентрированный раствор гидрохлорида амина, свободный от бутилового
спирта, пересыщают щелочью, амин отделяют (при необходимости его извяФ
кают эфиром) и сушат сначала твердой едкой щелочью, затем натрием. Пери-
гонкой получают амин в чистом виде без предгона и остатка.
При употреблении для восстановления натрия *, содержащего 0,1-^
0,01% калия, восстановление нс идет [49].
Получение вторичных аминов из оснований Шиффа
(R-CH=N-R —> R-CIb-NH-R)
Гидрирование оснований Шиффа протекает обычно легко и с удовлетворителен
пыми выходами. Восстановлению поддаются основания Шиффа алифатических и арО;
матическпх альдегидов с алифатическими и ароматическими первичными аминами.
Восстановление проводится как каталитическими методами (над никелем Ренея, катв:
лизатором Адамса, палладием па угле), так и в присутствии LiAlH4, особенно в набору
торных условиях. Во многих случаях гппффовы основания не выделяют в чистом виде,
а непосредственно гидрируют смесь амина и карбонильного соединения. Если пример
няемые альдегиды неустойчивы или склонны к альдольной конденсации, карбонильное
соединение постепенно добавляют к смеси первичного амина, водорода и катализатора
гидрирования. В особых случаях в реакционной массе получают и используемые *
реакции амины. Вместо аминов иногда берут соединения, легко восстанавливающиеся ДО'
перцичпых аминов (нитро-и нитрозопромзводные, оксимы, нитрилы), см. такЖ^
стр. 485, 488. ч
Методы каталитического восстановления демонстрируются на приведения^
ниже примерах.
N-Метилбензиламин [22]. При комнатной температуре под избыточн
давлением 3 алг встряхивают 83 г перегнанного бензальметиламина и 7 г ник(
Ренея в 100 мл абсолютного спирта. Восстановление практически закая*
* Сплав натрия с 2% калия равподснеп чистому натрию [А. Н. К о о 1)
А. П. Т о р е н т ь е в, ЖОХ, 17, 105 (1947)1- — Прим. ред.
[21] С. М- S u t е г, Е.* W. М о f f е t, J. Am. Chem. Soc., 56, 487 (1934).
[22] N. H. Cromwell et al., J. Am. Chem. Soc., 65, 312 (1943).
I. ОБРАЗОВАНИЕ N—П-С.ВЯЗИ
519
вается через 2 ч. Продукт реакции выделяют перегонкой с эффективной ко-
лонкой; выход 60 г метилбензиламина; т. кип, 184—186° С (760 мм рт. ст.).
N-Метпланилии [23]. К смеси 640 г 29,8%-ного раствора водного форм-
альдегида и 6 г триэтиламина в течение 15 мин при перемешивании прили-
вают 558 г анилина, поддерживая охлаждением температуру на уровне 20—
25° С. После прибавления всего анилина перемешивание продолжают еще
10 мин, после чего отделяют органический слой и смешивают его с 500 мл
метилового спирта и 46 г триэтиламина. Эту смесь гидрируют в присутствии
катализатора никель на кизельгуре, при температуре 60—85° С и избыточном
давлении водорода 10 am. Поглощение водорода закапчивается через 1,5 ч.
Катализатор отфильтровывают, к фильтрату добавляют 20 мл 30%-ного
раствора NaOH и перегоняют в вакууме (120 мм рт. ст.). Сначала отгоняется
440 мл метилового спирта, содержащего 46 мл триэтиламина. Перед дальней-
шей перегонкой остаток переносят в делительную воронку и отделяют 230 мл
воды. Получается 575 а продукта, содержащего 90% N-метиланилина, 4,9%
анилина и 5,1% М^-диметиланилина.
Алкллэрнламипы ]24]. В 150 мл спирта растворяют 0,1 моль аромати-
ческого нптросоединения, 0,3 моль альдегида и 2,0 г ацетата натрия. После
добавления 3 — 6 г никеля Ренея смесь гидрируют при начальном избыточном
давлении водорода 3 ат. Через 12—24 ч поглощение водорода заканчивается.
Катализатор отфильтровывают, фильтрат подкисляют НС] и растворитель
отгоняют на паровой бале. Остаток подщелачивают раствором NaOH, экстраги-
руют эфиром амин и выделяют его перегонкой.
По этому методу получены следующие вторичные амины: N-этиланилин
(выход 57—63% от теоретического), N-бутиланилин (94—96% от теоретиче-
ского), N-бутил-п-толуидин [88% от теоретического).
С хорошими выходами получаются вторичные амины при восстановле-
нии азометинов LiAlH4. Описано восстановление бензальанилина до N-бензиланили-
на [25]. Проведение восстановления полностью соответствует описанному выше вос-
становлению нитрилов до первичных аминов под действием ЫА1На.*
Получение замещенных гидразинов из гидразонов и азинов
(R—NH— N=CH-R —> R-NII-NH-CH2-R)
(R—CH=N—N=CH—R —> R—СП2—NH—NH—СН2—R)
Восстановление гидразонов и азинов приводит к образованию №,Н'-аамещенпых
гидразинов. Для этого применяется каталитическое гидрирование’(палладиевые или
платиновые контакты), восстановление натрием и спиртом или восстановление алюмо-
гидридом лития, которому следует отдать предпочтение при проведении реакции
в лабораторных условиях (ср. [666], а также стр, 525). При каталитическом
гидрировании не рекомендуется применять никелевые контактные катализаторы, таи
как в их присутствии возможно расщепление N—N-связи.
* Прекрасными восстановителями оснований Шиффа во вторичные амины япляют-
ся боргидрид натрия [J. В i 1 1 m a n, Л. D i е s i ng, J. Org. Chem., 22, 1068
(1957)[ и диметиламинобораи [J. Billman, W. McDowell, J. Org.
Chem., 26, 1437 (1961)]. — Прим. ped.
[23] Швейц, пат. 310827 (1952), Ciba (Basel), 15.11.1955.
]24] W, 3. Emerson, H. W, Mohrmann, J. Am. Chem. Soc., 62,
69 (1940).
[251 R. F. N у strom, W. G. Brown, J. Am. Chem. Soc., 70', 3738 (1948).
520
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
2. Образование N—Н-связя в результате реакции обмена
Получение аминов восстановлением нйтросоединений
(R-NO2 —► R-NH2)
Восстановление нитрогруппы до аминогрушйл — одна из важнейших титла
реакций органической химии — происходит во всех нитросоединениях без исключи
с хорошими выходами *. Химизм реакции в каждом отдельном случае определи,
связью нитрогруппы в молекуле соединения, применяемым восстановителем и сред
в которой проводится восстановление. На первой стадии восстановления всегда об
зуется нитрозогруппа независимо от строения нитросоединения в условий реакц
Нитрозогруппа, образующаяся из нитрогруппы, связанной с первичным или йтор
ным атомом углерода, реагирует обычно далее в изоформе. Она восстанавливав'
подобно оксимной группе через гидроксиламино- или иминогруппу до аминогруш
Промежуточно образующаяся нзонитрозогруппа придает известную неустойчиво
реакционной массе в кислых гидролитических средах. Поэтому лишь немногие 8
становители, применяемые для восстановления ароматических нитросоединещ
могут быть использованы для получения алифатических аминов из соответствую^
нитросоединений. Если образующаяся нитрозогруппа связана с третичным атом
углерода, то она либо переходит в аминогруппу с промежуточным образованием ги*
оксиламина, либо амии получается в результате конденсации нитрозогруппы с обра
вавшимся гидроксиламином и промежуточным переходом через азокси- и гидра
производные:
О
&
R-NO + HO-NH-R —> R-N = N-R —►
—> R—N=N-R —> R-NII-NH-R —> 2RNH2
По первой схеме восстановление протекает преимущественно в кислой ср<
тогда как щелочная среда благоприятствует протеканию реакции с промежуточг
образованием азоксипроизводных. Нитрофенолы и нитроапилины непосредстве
восстанавливаются даже в щелочной среде. При соответствующем подборе уело:
все промежуточные продукты обоих направлений реакции могут быть выделены в
стом виде. Само собой понятно, что эти промежуточные продукты восстанавливая
до аминов, даже если они получены другими способами. Однако препаративное L
ченне имеют только азосоединеиия, получаемые по реакции азосочетания.
Восстановление нитрогрупп может осуществляться различными способа;
Важнейшими из них являются: каталитическое гидрирование на Pt, Pd, Си, ник
Ренея (в промышленности применяются также специальные смесевые катализатор
восстановление железом, оловом, SnCls, Na2SaO4, Fe(OH)a, H2S и его соля
гидразином, сульфитами, цинком, алюминием; электрохимическое восстановлен
Если нитрогруппа является единственной функциональной группой в молеку
то выбор восстановителя определяется лишь способом выделения образующе/ч
амила. Реакция почти всегда проводится при температурах не выше 100° С; кои
трации реагирующих веществ подбирают с таким расчетом, чтобы сильноэкзотерми^
реакция восстановления не протекала слишком бурно.
При наличии в восстанавливаемом нитросоединении других функциональ
групп, с трудом поддающихся гидролизу или восстановлению в обычных уело?
(например, алкоксильные, сульфокил’ьные, карбоксильные, сульфоновые, амид
в сульфамидные группы), сохраняется достаточно широкий выбор восстановите
Если в молекуле нитросоединения имеются сульфамидная, сульфо- или амИЬ
группы, реакцию ведут в кислой или щелочной среде, что определяется свойств
конечных продуктов реакции. Металлическое олово или SnCl2 могут вызывать рал
пление алкоксильных групп и декарбоксилирование, поэтому их следует приме
с известной осторожностью. При наличии в молекуле реакционноспособных г]
следует учитывать следующие факторы: устойчивость заместителя к кислотному
щелочному гидролизу, к действию восстановителя, возможность других превращу
под действием восстановителя. Может оказаться необходимым проводить реая
в условиях, отличных от общепринятых. В таких случаях рекомендуется обраща
к специальной литературе, так как в этой книге приводятся лишь выборочные о(
методы восстановления нитросоединений.
* Реакция открыта Ц. Н. Зининым в 1842 г. — Прим. ред.
I. ОБРАЗОВАНИЕ M—Н-СВЯЗИ
521
Непрерывное каталитическое восстановление нитросоединений в жидкой или
газовой фазе применяется главным образом в промышленности. Для препаративных
целей используется преимущественно периодический способ гидрирования. Восста-
новление проводят При нормальном или повышенном давлении; если есть возможность
выбора, то предпочитают вести гидрирование при повышенном давлении (до 50 ат).
В промышленности чаще всего используются комбинированные катализаторы;
для лабораторных целой применяют Pd, Pt, Ni, Си. Восстановителем служит обычно
молекулярный водород, в особых случаях применяется химически связанный водород,
например водород из гидразина, муравьиной кислоты или ненасыщенных гидроароыа-
тических соединений (циклогексен). В качестве растворителя рекомендуется применять
соединения, достаточно хорошо растворяющие воду, образующуюся в результате
реакции (низшие спирты и моноэфиры гликолей, тетрагидрофурап, диоксан, пиридин,
диметилформамид, ледяная уксусная кислота). Для растворимых солей (например,
солей нитрокарбоновых кислот) рекомендуется вести процесс в воде.
Реакция каталитического гидрирования протекает с выделением большого
количества тепла; при недостаточном отводе тепла реакция может протекать со взры-
вом [26]. Течение процесса регулируют подбором соответствующих концентраций,
парциального давления водорода и количества катализатора. Особое внимание следует
уделять отводу тепла — охлаждению. Наиболее гладко протекает гидрирование
при нормальном давлении. Чаще всего реакцию проводят при разбавлениях
от 1:1 до 1 : 20.
5-Аминохинолин. Суспензию 34,8 г 5-нитрохинолина в 200 мл абсолют-
ного спирта встряхивают в атмосфере водорода в присутствии 0,4 г катализа-
тора Адамса (окись платины.). Реакция, сопровождающаяся заметным повы-
шением температуры, закапчивается приблизительно через 2 ч. Раствор освет-
ляют углом, растворитель удаляют в вакууме и перегоняют остаток (т. кип.
180—181° С при 7 мм рт. ст.); выход технического продукта 27 г (Й5% от
теоретического); т. пл. 100—107° С. После перекристаллизации из эфира полу-
чают 23,5 г 5-аминохиполина (выход 82% от теоретического); т. пл. 108—110° С.
Аналогичным способом можно получать 8-амннохинолин с выходом
96% от теоретического; т. кип. 140,5—141,5° С (7 мм рт. ст.); т. пл. 64—
65° С.
’ З-Хлораяилин [27]. При комнатной температуре под избыточным давле-
нием водорода 2—3 ат гидрируют 30 г 3-нитрохлорбензола, растворенного
в 100 мл абсолютного спирта, в присутствии 2,5 г цикеля Ренея. Из профиль-
трованной реакционной массы выделяют продукт перегонкой; выход 3-хлор-
анилина 24 г (98,5% от теоретического); т. кип. 113° С (18 мм рт. ст.); п-то-
луолсульфонильное (тозильное) производное после кристаллизации из спирта
плавится при 134° С.
4-Хлоранилин [28]. В качающийся автоклав емиостью 900 мл загружают
смесь 300 г технического 4-нигрохлорбензола, 400 мл спирта и 15 г нилеля
Ренея, нагнетают водород, доводя давление до 100 ат, и включают перемеши-
вание и обогрев. При 50° С достигается достаточно большая скорость поглоще-
ния водорода, и нагревание реакционной массы прекращают. Когда давление
упадет до 20 ат, добавляют водород из баллона. Через 23 мин при температуре
но выше 100° С поглощается теоретическое количество водорода. Реакционную
массу охлаждают, отфильтровывают катализатор и перегоняют фильтрат.
Получается 236 г (97% от теоретического) 4-хлоранилина; т. пл. 70—71’ С.
Катализатор можно использовать несколько раз.
Аналогичным образом получают 2-хлоранилин (выход 96,5% от теорети-
ческого) и 2,5-дихлоранилин (выход 97% от теоретического).
Метод восстановления ароматических ннтросоединений до аминов при помощи
гидразингидрата в присутствии никеля Ренея описал Бэлкомом и Ферстом (29, 49].
При нормальных условиях восстановление ароматических интрогрупп гидразниом
протекает очень медленно, а в более жестких условиях восстанавливаются не только
[26] Н. A d к 1 п a, Org. Synth., 22, 10 (1942).
127] N. Lofgren, Acta Chern. Scand., 9,-1079 (1955).
1281 G. F. Winans. J. Am. Chem. Soc., 61, 3564 (1939).
[29] D. Ba'lcom, A. Furst, J. Аш. Chem. Soc., 75, 4334 (1953).
я
i-1
522 ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
нитрогруппы, но и другие функциональные группы нитросоединения. В то же время прн |
восстановлении нитросоединепий в присутствии небольших количеств никеля Ренея д
при комнатной температуре получаются амины с выходом 80—99% от теоретического, ‘и
причем другие функциональные группы не затрагиваются. Реакцию проводят в спир- а
товом растворе с 2—3 моль 100%-ного гидразпнгидрата, я
Восстановление железом в кислом растворе применяется главным образом в про-
мытпленности (расход соляной кислоты очень мал)*. Образующийся FeCl3 реагирует J|
с избытком металла и дает FeCl„, который играет в дальнейшем роль восстановителя. .'Ц
В результате реакции осаждается Fe(OH)a и получается свободное основание в смеси^Я
с небольшим количеством гидрохлорида амила. На выделспис амина пз такой смеси
расходуется незначительное количество щелочи, что сильно удешевляет процесс<-Я
Сильно уменьшается также образование хлорзамещенных продуктов.
Общий метод восстановления нитросоединений 130]. На водяной бане^
кипятят 1 моль нитрососдинепия с 10 мл концентрированной НС1 в 500
спирта. К этой смеси в четыре приема примерно через каждые 5 мин прибавляют^
170 г железных опилок в расчете па каждую нитрогруппу. Для предотвращения^
спекания железных опилок поддерживают интенсивное кипение массы. ПосщИ$
добавления всего железа кипятят еще 2 ч. Метод выделения зависит от свойств^
получаемого амина, ’я
Летучие с паром амины перегоняют с водяным паром после добавлений
к смеси 10 г Na2CO3, причем перегоняющийся вначале спирт собирают отдельно
от мутного дистиллята, содержащего амин. Можно также разложить реакциои-^!
ную массу рассчитанным количеством щелочи, профильтровать горячий растворяя
и промыть железный шлам спиртом. От полученного фильтрата отгоняют оснодЧй
ное количество спирта и осаждают из остатка гидрохлорид амила концентрировав
ванной НС1. Если образующаяся соль легко растворима, то после отгонкС^
основного количества этиловою еппрта остаток пасыщают газообразным НСО
и осаждают соль эфиром.
Восстановление оловом, так же как и SnCl2, дающее в некоторых случаях даад
лучшие результаты, нашло широкое применение в препаративных работах, несмотря
на некоторые недостатки этого метода. Но в последнее время оба эти восстановителе
все больше вытесняются современными каталитическими методами. у
При проведении реакции с этими восстановителями иногда наблюдаются неж®*
лательные превращения, например, отщепление галогенов, сульфо- и карбоксильнМ$
групп, находящихся в орто- или пара-положении к питрогрупне, образование галр*
гензамещенных целевых аминов, гидролитическое превращение аминогруппы в оксМ
группу, которые отчасти обусловлены необходимостью вести процесс в сильнокислоЖ
среде [31].
При восстановлении оловом или SnCI2, как правило, образуются комплексны»
соединения олова и гидрохлорида амина, из которых едкой щелочью или сероводород
дом выделяют свободные амины. '
Восстановление ж-питробензальдегида [32]. Растворяют 450 г (2 молИ
SnCl2-2H2O в 600 мл концентрированной HCI. К охлажденному до 5° С рЛ
твору постепенно прибавляют 100 г (0,60 моль) ж-нитробензальдег'нда,
держивая температуру ниже 30’ С. ж-Нитробензальдегид растворяется и o&pl
зуется красный раствор, из которого при охлаждении выделяется пастообразна
двойная соль амина и SnCl2, которую отделяют фильтрованием через порист?#
стеклянный фильтр.
Из способов восстановления нитросоединений производными сероводо]
в щелочной среде часто используют спиртовые растворы сульфида аммония. При I
* См. В. О. Лукашевич, Усп. хим., 17, 692 (1948). — Прим. ред.
[30] R. W. West, J. Chem. Soc., 127, 494 (1925). -4
[31] J. J. Blanksma, Rec. trav. chim,, 23, Й10 (1904); 24, 320 (1905); L. J a c J
son, S. Calve»t, J. Am. Chem. Soc., 18, 466 (1896). ''
[32] Org. Synth., Coll. v. 3, p. 453 . z
I. ОБРАЗОВАНИЕ X-Н-СВЯЗИ
523
пускании сероводорода в спиртовой раствор аммиака и нитросоединспия питрогруппы
восстанавливаются гладко и полностью уже при умеренных температурах (например,
восстановление п-нитрофенилуксусной кислоты 136]); все же в некоторых случаях
реакцию приходится проводить при повышенных давлениях. Вилльштеттер [33] на-
блюдал, что на холоду восстановление часто останавливается на стадии образования
глдроксиламйна.
В качестве восстановителей применяются также сульфид и гидросульфид на-
трия. Если в орто-положении к нигрогруппе имеется вторая нитрогруппа пли гало-
ген, то реакция может протекать с заменой орто-заместителя и с образованием серусо-
держащих продуктов. В особых случаях может применяться дисульфид натрия, Na2S2,
реагирующий следующим образом:
RNO2 + Na2S2 + H2O —> RNH2-)-Na2S2O3
Так как в результате реакции сера не образуется, то тиосульфат легко отделить
от амина.
Восстановление производными сероводорода позволяет проводить частичное
восстановление полинитросоединений.
В некоторых случаях при использовании смеси NaHCOg с Na3S получаются
лучшие выходы аминов, так как образующийся при этом гидросульфид натрия служит
также восстановителем. Водный метиловый спирт является лучшим растворителем,
чем водный этиловый спирт [34]. Плохо растворяющиеся в спирте нигросоедпиения
можно восстанавливать сероводородом в пиридине [35].
Анилин [37]. В реакционном сосуде с мешалкой и обратным холодиль-
ником при кипячении и перемешиваннн растворяют 32 г серы в растворе 78 з
Na2S в 360 мл воды (или 240 г Na2S .9Н2О в 200 мл воды). К раствору при-
ливают 123 г нитробензола. После кипячения при перемешивании в течение
около 12 ч реакция заканчивается (водный раствор осветляется). Реакционной
массе дают некоторое время отстояться, отделяют анилин от водного слоя, высу-
шивают и очищают перегонкой.
-и-Нитрпапилии [38]. К раствору 10 г 1,3-динитробензола в 74 мл мети-
‘лового спирта прибавляют раствор 5,5 г (по расчету 5,0 г) NaliS в 150 мл
врдного метилового спирта и нагревают в колбе с обратным холодильником
в течение 25 мин. Затем отгоняют 150 мл метилового спирта и выливают остаток
в 500 лиг ледяной воды; выход лс-нитроанилина 8,3 а (около 90% от теорети-
ческого).
Восстановление дитионитом патрия по схеме:
BAO24-3Na2S2O4-f-4H2O —> RNH2 + 6-NaHSO3
проводят в нейтральной или слабощелочной среде; эта реакция протекает обычно
гладко, но возможны и побочные реакции (образование сульфаминовых кислот или
аминосульфокислот). Благоприятствующая протеканию реакции слабощелочная среда
устанавливается уже с помощью третичных ароматических или гетероциклических
оснований. Нитросоедниения с кислотными группами вводят в реакцию в впде солей
щелочных металлов. Растворителями могут служить спирт, ацетон, вода или формамид.
Восстановитель можн) вводить в избытке (120% от теоретического).
З-Амино-4-окситолуол [39]. Раствор 10 г З-пнтро-4-окситолуола в 200 мл
воды и 50 мл концентрированного раствора аммиака медленно прибавляют
133] R. Willstatter, Вег., 41, 1936 (1908).
134] И. II. Hodgson, Е. R. Ward, J. Chem. Soc., 1945, 663, 794.
135] II. Н. Hodgson, Н. S. T u г n e r, J. Chem. Soc., 1943, 318.
[36] Org. Synth., 3, 11 (1923).
[37] Герм. пат. 144809 (1902); J. Kunz, Frdl., 7, 57.
138] II. H. Hodgson, E. R. Ward, .1. Chem. Soc., 1949. 1316.
[39] H. M. Woodburn, C. F. Stuntz, J. Am. Chem. Soc., 72, 1361
(1950).
524
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
к раствору 75 г Na2S2O4 в 300 мл воды. Через 5 мин начинается выделяй^
белых хлопьевидных кристаллов и красная окраска раствора переходит в светлой
желтую. Из реакционной массы, не фильтруя ее, экстрагируют эфиром
эфирные бЫтяжкп сушат, обрабатывают углем и отгоняют эфир. Получаете
6,8 а (81% от теоретического) желто-коричневых кристаллов; т. пл. 134—135° С
Попытки получить это соединение с помощью других восстановителей, ВД
видимому, не увенчались успехом. При помощи Na?S«O1 можно также чяг.тиа^
восстановить 2,4-динитрофенол в 2-амнно-4-нитрофенол; выход хороший [42J
Реже применяется восстановление нитросоединений гидроокисью железа (Г
хотя в некоторых особых случаях этот метод имеет значительные преимуществ
Fe(OH)2 используется для восстановления таких нитросоединений, в которых napi
с нитрогруппой содержатся другие легко восстанавливающиеся или гидролизующи
кислотами группы. Так как реакция протекает в щелочной среде, ее можно исполь
вать для восстановления растворимых в щелочах нитросоедннений.
Гидроокись железа (II) может применяться также для восстановления нит
замещенных углеводородов. Аллен [40] получал из нитробензола анилин с количеств
ным выходом. Несмотря на щелочную среду реакции бимолекулярные промежуточ»
продукты образуются редко. Количественные соотношения реагирующих веще
вытекают из уравнения:
RNO2 + 6Fe(OH)2 + 4H2O —> RNH2 + 6Fe(OH)9
Общий метод синтеза описан Якобсом н Гейдельбергером [41].
В последнее время возрастает значение восстановления алифатических нит
соединений до аминов. Однако ввиду большей реакционной способности алифатичее
питрогрупп выбор условий реакции более ограничен, чем при восстановлении аром
ческих иитросоединений. В щелочной среде могут протекать реакции конденсаг
а в кислой среде возможно гидролитическое отщепление первичных иитрогрупп.
Промежуточными продуктами восстановления являются оксимы [43], из н
рых, вероятно, образуются вторичные амины. В иислой среде оксимы могут гидр
зоваться до карбонильных соединений; альдоксимы могут претерпевать перетру
ровку Бекманиа.
Для восстановления алифатических нитросоединений с успехом примени,
каталитическое гидрирование над никелем Ренея [44], над окисью платины [
восстановление железом в ледяной уксусной или соляной иислоте [46], алюмогидрс
лития [47] и натрийэтоксиалюмогидридом [48|.
Восстановление нитромочевни приводит к образованию семикарбазидов:
/NHR /NHR
ОС< —> ОС<
^NHNOa xNHNH2
В иачестве восстановителя может применяться гидразингидрат [49, 50]. IIJ
водное нитромочевины приливают по каплям к кипящему раствору гидразингид]
в спирте и нагревают еще 1 ч на водяной бане. Получаются семикарбаэиды с выхс
около 60% от теоретического. Сульфат семикарбазида образуется при электро*!
песком восстановлении нитромочевины свыходом 69% от теоретического [51].
[40
41
[42
43
44
45
46
47
48
49
50
[51
Н. С. А 1 1 е п. J, phys. Chem., 16, 131 (1912). г
W. А. Jacobs, М. Heidelberger, J. Am. Chem. Soc., 39, 1435(191
Org. Synth., Coll. v. 3, p, 82.
C. Grundmann, Angew. Chem., 62, 558 (1950). /
К. I. Johnson, E. F. Degering, J. Am. Chem. Soc., 61, 3194 (19J
D. C. Iff land, F. A. Cassis, J. Am. Chem. Soc., 74, 6284 (1952). J
Ср. с ссылкой [44].
Ср. с ссылкой [25]; A. D о г п о w, F. Boberg, Ann., 578, 94 (1952).
G. Hesse, R. S c li r 6 d e 1, Ann., 607, 24 (1957).
T. L. Fletcher, jd. I. Namkun g, J- Org. Chem., 23, 680 (1958). ’
L. C. McVeigh, f, D. Rose, J. Chem. Soc., 1945, 713.
Org. Synth., 5, 93 (1925).
I. ОБРАЗОВАНИЕ X—Н-СВЯЗИ
525
Восстановление нитросоединений до гидроксиламинов
(RNO2 —*• RNHOH)
В соответствующих условиях восстановление нитросоедпненнй останавливается
на стадии образования N-замещениых гидроксиламинов. Обязательной предпосылкой
для выделения гидроксиламинов является проведение восстановления в нейтральной
или близкой к нейтральной среде. В качестве восстановителей применяют в первую
очередь цинковую пыль в растворе хлорида аммония [52J, амальгаму алюминия [53],
сульфид аммония [54], а также используют каталитическое гидрирование на катализа-
торе Адамса в среде этилацетата [55].
Феиилгидроксиламин [56]. В 8 л воды растворяют 250 г NH4CI и приба-
вляют 500 г нитробензола. Затем при интенсивном перемешивании в течение
15—20 мин прибавляют 620 г 88%-ной цинковой пыли. Реакция протекает
с разогреванием смеси до 60—65° С. После добавления цинковой пыли пере-
мешивают еще 15 мин, до начала понижения температуры. Горячий раствор
фильтруют и промывают осадой Zn(OH)2 1 л горячей воды. Фильтрат насы-
щают 3 ка NaCl и охлаждают до 0° С. Феиилгидроксиламин кристаллизуется
в виде длинных светло-желтых нгл. В осадке содержится некоторое количество
NaCl. Выход фенилгидроксиламина 275—300 г (62—67% от теоретического).
Получение метилгидрокенламина из нитрометана описал Бекман [57].
Получение аминов восстановлением оксимов и гидроксиламинов
(R=NOH —*- RNHa R-NHOH —> RNM2)
Оисиминотруппа =NOH превращается в аминогруппу с промежуточным
образованием гидроксиламинов или иминов. Восстановлением оксимов можно:
1. Получать амины из соответствующих карбонильных соединений.
2, Вводить аминогруппы в активные метиленовые группы (через получаемые при
нитрозировании изонитрозопроизводные).
3. Получать амины из продуктов присоединения окислов азота (или нитрозил-
хлорцда) к олефинам.
Для восстановления окенминогруппы можно применять большое число восстано-
вителей'[58]: амальгаму натрия в кислом растворе [59], натрий в присутствии^пирто»
[60, 61], каталитически возбужденный водород (никелевые или медные катализаторы),
цинк в кислом или щелочном растворе [62], олово или SnCl2 в солянокислом растворе
[63], дитионит натрия [64], алюмогидрид лития [66а, 66в], амальгаму алюминия [65],
а также электрохимическое восстановление [67}.
[52] Е. Bamberger, Вег., 27, 1350 (1894).
[53] V. Migridichian, Organic Synthesis, v. 1, New York, 1957, p. 490.
[54] O. N eunhoef f er, H. G. Liebich, Ber., 71, 2248 (1938).
[55] L. F. Fieser, E. В. H e r s h b e г g, J. Am. Chem. Soc., 62, 1643 (1940).
[56] C. S. Marvel, О. К a m m, J. Am. Chem. Soc., 41, 279 (1919).
[57] E. Beckmann, Ann., 365, 204 (1909).
[58] Houben-Weyl, Methoden der organischen Chemie, Bd. 11/1, 4 Aufl.,
495—502 (1957).4
[59] H. Goldschmidt, Ber., 19, 3232 (1896); 20, 492 (1897).
[60] F. G. M a n n, I. W. G. Porter, J. Chem. Soc., 1944, 456.
[61 Org. Synth,, Coll. v. 2, 1950, p. 318.
[62] N. F. A ] bertson, J. Am. Chem. Soc., 70, Ц50 (1948); L. Claisen,
r Ann., 274, 90 (1893).
[63] E. Gio vannini, P. Portman n, Helv. chim. acta, 31, 1381 (1948),
64] l. J. Smith, M. Schubert, J. Am. Chem. Soc. 70, 2656 (1948).
[65] V. Cerchez, C. Dumitresco-Colesiu, Bull. Soc. chim. France [5],
1, 852 (1934).
166a] в. Миновнч, M. Михайлович, Алюмогидрид лития н его применение
в органической химии, пер. с англ., Издатпнлнг, 1957.
[666] Н. Г е й л р о д, Восстановление комплексными гидридами металлов, пер.
с англ., Издатиплит, 1959.
167] 1. Tafel, Е. Pfeffermann, Вег., 85, 1514 (1902); S. Kaplansky,
там же, 60, 1842 (1927); 36, 219 (1903).
526
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Метод восстановления амальгамой натрия описан ниже на примере восстановле-
ния 5-хлорфурфуральдоксима до 5-хлорфурфурилахгина [68].
5-Х.торфурфуриламин. Раствор 7,25 г 5-хлорфурфуральдоксима в 100 мл
95%-ного спирта перемешивают 1 ч с 237 а 2,5%-ной амальгамы натрия, одно-
временно медленно приливая в общей сложности 15,5 г ледяной уксусл “
кислоты для поддержания постоянной слабокислой реакции. Раствор слива,
с ртути, разбавляют водой и встряхивают с эфиром для удаления непрореагщ
павшего альдоксима. Кислый водный слой .подщелачивают и извлекают ам
эфиром. Эфирный раствор амина сушат над Na2SO4. отгоняют эфир, а остат
перегоняют в вакууме; выход 5-хлорфурфуриламииа 5 г (76% от теоретик
ского); т. кип. 50—55° С (25 мм рт. ст.).
По этому методу Гольдшмидт [59] восстанавливал оксимы бензальдегид,
бензофенона, камфоры, бензоина и изомасляного альдегида. Грэнахер рекоменду
проводить восстановление оксима фенилпировиноградной кислоты до фенилалани
амальгамой натрия в присутствии молочпой кислоты (выход 80—90% от теорети
ского). Оксимы восстанавливаются амальгамой лития до первичных аминов, нитриа
цые группы остаются незатронутыми |70].
Восстановление оксимов натрием в спирте проводится так же, как восстанок
ние нитрилов (стр. 516). В качестве стандартной методики может служить прош
получения н-гоитиламина [71]. Аналогично синтезируют 2-ашшооктан [60]. В отли*
от общепринятого метода Прелог [72] очень быстро прибавляет натрий к кипяще
спиртовому раствору оксима и получает из оксимов многочленных циклических снст
соответствующие амины с выходами до 90% от теоретического.
Каталитическое восстановление оксимов проводят в тех же условиях, что
каталитическое восстановление нитрилов (стр. 516). При использовании в качес?
катализатора пикеля Репся альдоксимы претерпевают перегруппировку Бекш
[73, 74[. Ацетилирующее восстановление оксимов можно^проводить d присутсп
пикеля Ренея в уксусном ангидриде *; так, например, из изонитрозомалонового эфЯ
получают ацезиламиномалоновый эфир с выходами выше 80% от теоретического [7
Каталитическое восстановление изонитрозоацетоуксусного эфира в уксусном ангидрг
в присутствии катализатора палладия на угле приводит к образованию а-ацетилаэ
ноацетоуксусного эфира с количественным выходом [62]. С количественным выход,
гидрирование протекает и в более сложных случаях в присутствии платииированно
никеля Ренея в содовом растворе [77].
При гидрировании оксима пентанона в 95%-пом этиловом сиирте на ник
Ренея при комнатной температуре при избыточном давлении водорода 3 ат
поглощения расчетного количества водорода Иффлэнд [78] получил 2-аминопеН'
с выходом 85% от теоретического. Гораздо более жесткие условия требуются, нап
мер, для восстановления оксима 2-этилциклогексанона (130° С, 83 ат, нэсыщенЕ
аммиаком спирт, никель Ренея) [79]. На примере получения н-амиламина. н-гепт
амила, бензиламина, изоиропиламина, циклопснти.тамина и других соедние]
Удобным способом проведения такого восстановления является обработка ок
мов цинковой пылью в среде муравьиной кислоты или смеси уксусный ангидрид
уксусная кислота [К. Shaw, Ch. N о 1 an, J, Org. Chem., 22, 1668 (1957)].
Прим. ped.
F. A. Hochstein, G. F. W r i g h t, J. Am. Chem. Soc., 71, 2257 (194'
C. Granacher, Helv. chim. acta, 5, 610 (1922); 6, 458 (1923).
T. Kametani, J. Nomura, J. Pharrn. Soc. Japan, 74, 1037 (1954).
Org. Synth., Coll. v. 2, 1950, p. 318.
V. Prolog, Helv. chim. acta, 28, 178/576 (1945).
R. Paul, Bull. Soc. chim. France [5], 4, 1115 (1937).
A. G. Cald well, E. R. H. Jones, J. Chem. Soc., 1946, 599.
M. V i g n a u, Bull. Soc. chim. France, 1952, 638 (Documentation).
E. Larson, Svensk kem. Tidskr., 61, 242 (1949).
Delepine, Horeu Bull. Soc. chim. France [5], 4, 3 (1937).
D. С. I f f 1 a n d,-T oh'-Fu - Yen, J. Am. Chem. Soc., 76, 4180 (1954). .
F. E. К i n g et al., J. Chem. Soc., 1945, 277.
I. ОБРАЗОВАНИЕ N—Н-СВЯЗП
527
показано, что восстановление па никелевых контактах во многих случаях протекает
с высокими выходами [83].
Восстановление 'оксимов оловом идет ч спиртовой сильнокислой среде. К рас-
твору прибавляют избыток олова, кипятят до образования прозрачного раствора и осаж-
дают олово сероводородом. Поело упаривания спиртового раствора получают гидро-
хлориды аминов. По этому методу получено много соединений, в том числе 2-амино-
пеятаиок-З, аминоацетон [80]. дламиноацетон [81].
Для восстановления оксимов применяется также LiAlH4, правда только в тех
случаях, когда в соединении имеются другие лабильные группы, требующие проведе-
ния восстановления в мягких условиях иин при необходимости одновременного вос-
становления карбоксильной группы. Прп восстановлении кегокелмов наблюдалась
перегруппировка Бекмапа.
Общая методика [76]. В 400 мл абсолютного эфира суспендируют 15 е
80%-ного ГЛА1Н4 (0,25 моль) и к полученной смеси при интенсивном перемеши-
вании приливают по каплям раствор 0,2 моль оксима в 100—200 мл абсолютного
эфира. После прибавления всего оксима реакционную массу нагревают на водяной
бане в течение 3—4 ч при интенсивном перемешивании, затем охлаждают,
добавляют к пей воду и разбавленную H2SO4 до явно выраженной кислой реак-
ции в водном слое. Эфир отделяют, водный слой несколько раз встряхивают
с эфиром для удаления нейтральных веществ, при охлаждении прибавляют
к пему раствор NaOH и в случае выпадения осадка воду. Амин из мутноватог»
щелочного раствора экстрагируют эфиром, эфирные вытяжки сушат и выде-
ляют амии перегонкой. Выходы 50—70% от теоретического.
Реже и главным образом лишь в особых случаях применялось восстановление-
дитионитом нагрия (Na2S2O4), амальгамой алюминия, а также электрохимическое
восстановление оксимов; поэтому для более подробного изучения этих методов сле-
дует обратиться к оригинальной литературе. Восстановление Na2S2O4 проводится
в щелочном или аммиачном растворе [82]. Амальгама алюминия применяется в без-
водных средах; расчетное количество воды прибавляют по каплям при охлаждении
льдом [65].
Для восстановления ненасыщенных оксимов с сохранением двойных связей
лучше всего применять амальгаму натрия при умеренно повышенных температурах.
Лучшим методом получения соответствующих аминов из диоксимов хинонов является
восстановление Na2S в иипящем спирте [83]. При помощи (NH4)2S можно восстанавли-
вать нитрсн-руппу, сохраняя оксиминогруппы [84]. Аминокарбоновые кислоты полу-
чают через оксимы соответствующих котокарбоновых. кислот, при этом в первую очередь
применяют каталитическое гидрирование.
Восстановление t-идроксиламинной группы проводят в принципе теми же сред-
ствами, что и восстановление оксиминогруппы. Ароматические гидроксиламины,
которые могут перегруппировываться в кислой среде в аминофрнолы, рекомендуется
восстанавливать амальгамой алюминия [85].
Восстановление нитрозосоединений до аминов
' (F! -N=O —> TV-.H.J
Условия, в которых протекает восстановление питрозогруппы (образующейся
на первой стадии восстановления нитросоединений) до аминов, конечно, мало отли-
чаются от условий восстановления нитрогрупп.
Первичные и вторичные питрозосоеднненвя алифатического ряда встречаются
обычно в димерной форме и при нагревании или иод действием света могут переходить
в и.зонитрозосоедииеиия [86], Поэтому практически восстановление алифатических
801 S. Gabriel, G. Plnkus, Вег., 26, 2197 (1893).
cl] К. К. Koessler, М. Т. Hanke, J. Am. Chem. Soc., 40, 1716 (1918).
82] L. J. Smith, W. M. Schubert, J. Am. Chem. Soc., 70, 2656 (1948).
83] P. D. Bartlett et al., J, Am. Chem. Soc,, 72, 1003 (1950).
84] W. R. Boon J. Chem. Soc., 1949, 230.
85] II. W i s 1 i с c n u s, Ber., 29, 495 (1896).
186] E. Muller, II. Metzger, Ber., 88, 165 (1955); 88, 1891 (1955).
528 ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
нитрозосоединений проводится в таких же условиях, как и описанное ранее boccti
вленио оксимов.
В ароматическом ряду практическое значение имеет лишь восстановление ле
получаемых нитрозопроизводных фенолов [87] и третичных аминов. Они восста
вливаются в таких ясе условиях, как аналогичные нитросоединения. Восстановив
может проводиться как в кислой, так и в щелочной среде. В кислой среде прииен)
обычно каталитическое гидрирование (палладий или никель Ренея); в щелочной ср
с хорошими выходами протекает восстановление дитионитом натрия [88]. В отделы
случаях в качестве восстановителей применяются также гидросульфид аммония [f
железо, олово [90], фенилгидразин [91] и иодистый водород [92].
Восстановление а-нитрозо-0 -пафтола дитионитом см. [87]. Пропись
становления n-нитрозодиметиланилина до п-аминодиметиланилина ZnCl- в
лянокислом растворе приведена в практикуме Гаттермана — Виланда [93]
При восстановлении n-нитрозодиметиланилина- LiAlH4 с 80%-1
выходом получается 4,4'-бис-(диметиламино)-азобензол [666].
Восстановление нитрозаминов до несимметричных
дизамещенных гидразинов
Восстановлением нитрозаминов можно получать несимметричные дизамеще
тидразпны. В принципе гидразины, замещенные простейшими группами, пол;
аналогичным способом через производные нитрозоалкилмочевин. Их восстанавл»
обычно цинком в уксуснокислом растворе; действуя минеральными кислотами, м
отщепить нитрозогруппу. Впервые этот метод описан Фишером [94].
Несимметричный дифенилгидразин. В 250 мл спирта растворяют
дифенипнитрозамина, вводят 75 г цинковой пыли и при хорошем охлаж;
и постоянном встряхивании постепенно прибавляют ледяную уксусную кц
до прекращения выделения тепла и до тех пор, пока отфильтрованная ;
реакционной массы не перестанет окрашиваться в сине-зеленый цвет от
яления концентрированной НС1. Горячий раствор отфильтровывают от г
вой пыли, фильтрат упаривают до 1/4 его объема, разбавляют равным об
воды и при охлаждении и перемешивании приливают большой избыток дьп
НС.1. При охлаждении выделяется смесь гидрохлоридов дифснилгид|
и дифениламина в виде синих игл. Неочищенный продукт перекриста;
вывают из горячей сильно разбавленной НС1, отфильтровывая образую!
в результате гидролиза маслянистый дифениламин. При добавлении н .
трату дымящей НС1 снова выделяется гидрохлорид дифепилгидразина.
необходимости эту операцию повторяют несколько раз. В заключение nj
перекристаллизовывают из спирта и получают гидрохлорид днфендлгид]
в виде тонких бесцветных игл. Свободное основание выделяют действием
точного раствора NaOH и извлекают эфиром в виде слегка желтоватого »
Аналогичным способом синтезируют несимметричный диметилгидразмя
и метилфенилгидразин [96]. Этилгидразнн получают по Фишеру через N-нит
этилмочевину [97]. Нитрозамины восстанавливаются с хорошим выходом до С0<
ствующих гидразинов также под действием ЫА1Н4.
[87]
[88]
[89]
[90]
[91]
[92]
[93
[94
(95
[96
[97
А. Е. Albert, J. Am. Chem. Soc., 76, 4985 (1954).
Org. Synth., Coll. v. 2, 1943, p. 38.
W. A. J а с о b s, M. Ileidelberger, J. Am. Chem. Soc., 39, 2188 (:
J. Lindner, Mh. Chem., 42, 421 (1921).
E. Ф. Э ф p о с, A. E. По рай-Кошиц, ЖОХ, 17, 1801 (1947).
I. H. В о у e r, W. S с h о е n, J. Am. Chem. Soc., 78, 423 (1956).
Ссылка [1], стр. 273.
F. Fischer, Ann., 190, 175 (1878).
Org. 8ynth., Coll. v. 2, p. 211.
Org. Synth., Coif. v. 2, 418.
E. Fischer, Ann., 199, 287 (1879).
Г. ОБРАЗОВАНИЕ N—Н-СВЯЗИ
529
Восстановление азосоединений до аминов
Восстановление азосоединений до аминов с расщеплением связи N—N имеет
практическое значение, так как многочисленные азосоединения легко получают даже
в промышленном масштабе сочетанием диазониовых солей с соответствующими реак-
ционноспособными соединениями. Таким способом можно синтезировать большое
число первичных я милов без примеси изомеров, что не всегда достижимо другими
методами. В качестве восстановителей в основном применяют такие же вещества, как
и при восстановлении нитрогруппы до аминогрупп. Ввиду высокой химической стой-
кости азогруппы исходные азосоединения можно подвергать многообразным превра-
щениям, которые трудно осуществить с аминосоединениями. Таким образом, с исполь-
зованием азогруппы в качестве защитной группировки открывается возможность
для получения различных замещенных первичных аминов. Кроме того, восстанови-
тельное расщепление азогруппы имеет большое значение для установления строения
азокрасителей.
Однако при восстановлении азосоединений в кислом растворе при недостаточ-
ной скорости дальнейшего восстановления промежуточно образовавшихся гидразо-
производных могут протекать бензидиновые или семидиновые перегруппировки.
В таких случаях предпочитают вести восстановление в щелочной среде.
Чаще всего можно непосредственно восстанавливать растворы или пасты,
образующиеся при азосочетании; только при каталитическом гидрирования следует
выделить и очистить полученное азопроизводное.
Для получения аминов из азосоединений применяются преимущественно сле-
дующие восстановители: дитионит натрия 198], цинк [99], хлорид олова (II) [100],
иодистоводородная кислота [101] и активированный водород.
Восстановление дитионитом натрия протекает обычно с высокими выходами,
, я этому методу отдают предпочтение перед всеми остальными вследствие простоты
его проведения. В нагретый до 40—60° С нейтральный или щелочной водный или спир-
товой раствор 1 моль азосоединения вносят 2 моль Na2S2O4 и кратковременно нагре-
вают до обесцвечивания раствора 198].
2-Амвяо-4-метоксифенол [102]. В 1 л воды растворяют 100 г NaOH
, « и 100 г 2-бензодазо-4-метоксифеиола, раствор нагревают на водяной бане и при
перемешивании постепенно прибавляют Na2S2O4 до обесцвечивания раствора.
Образующийся анилин извлекают эфиром. При нейтрализации оставшегося
щелочного водного раствора выпадает аминофенол. Его быстро отфильтровы-
- вают, сушат и сразу переводят в гидрохлорид раствором хлористого водорода'
в эфире. Выход продукта 57,5 г; т. пл. 205—212° С (разл.). Еще 7,8 г продукта
с более низкой температурой плавлении получают экстрагированием эфиром
. из водного маточного раствора. Общин выход 2-амино-4-метоксифенола 85% от
‘ теоретического.
При каталитическом восстановлении азосоединений до аминов могут приме-
ту вяться любые эффективные катализаторы гидрирования. Применение давления не
требуется, так как увеличение скорости гидрирования достигается повышением тем-
? пературы реакции. Расщепление азобензола с образованием анилина протекает при
& нормальном давлении и 180—200° С. При использовании катализаторов, в присутствии
; которых гидрируются и ароматические системы, можно в одну операцию получать
* гндроароматические амины. Восстановление азосоедннсний в присутствии никеля
; Ренея подробно изучил Уайтмор [103]. Он проводил гидрирование при нормальных
| условиях и неизменно получал амины с высокими выходами. В качестве растворителей
* можно применять воду, спирт и диоксан, среда реакции должна быть нейтральной
£ или слабощелочной.
Г 198) Е. G randmougin, Вег., 39 , 2494 , 2561 (1906); J. prakl. Chem. [21, 76,
£. 124 (1907); И. Fischer, Hoppe-Seyler’g Z. physio]. Chem., 60, 69 (1909).
I 199] O. N. Witt, Ber. 19, 1719 (1896); T. Zinke, Ann., 278, 173 (1894).
k 1100] O. N. Witt, Frdl., 4, 67 (1894-1897); C. Liebermann, Ann., 211,
I. r 36 (1882).
f UOf] R. Meyer, Ber., 53, 1265 (1920).
E 1102] W. 1. C 1 о s e et al., J. Am. Chem. Soc., 71, 1265 (1949).
E 1103] W. F. Whitmore, A. J. Revukas, J. Am. Chem. Soc., 59, 1509 (1937);
L 62, 1687 (1940).
t 34 Заказ 1835.
530
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИЙ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
п-Фенилендиамнн [104]. Раствор 0,85 г n-аминоазооензола в 100
95%-ного спирта встряхивают с 0,1 г PtOa в атмосфере водорода при избыточном^
давлении 3 ат. Через 20 мин поглощение водорода заканчивается. Из реак^
ционной массы выделяют 5,12 г «-фенилендиамина и 2,92 г анилина. 3
Восстановление азинов, гидразонов и гидразинов *
(R=N-N=R —»- 2RNHa) '
Азины и гидразоны при восстановлении в мягких условиях переходят в соответ*^
ствующие дизамещенные гидразины, а в более жестких условиях протекает восстанд^
вительное расщепление N—N-связи и образуются амины. Таким образом, наряду?
с восстановлением оксимов появился еще один, правда редко используемый, мэдвв|
перевода карбонильных соединений в амины. В качестве восстановителя Тафель [10&S
рекомендует применять амальгаму натрия в кислой среде, а с помощью цинка в кисл<яи
среде можно переводить арилгидразоны а-кетокарбоновых кислот в соответствуя?
ющие аминокислоты *. Азины и гидразоны хорошо восстанавливаются и сплавом НИН
келя с алюминием. Общую методику такого восстановления см. [106]. а®
Восстановление азидов
(RN3 ------> RNH2) tyJ
При действии восстановителей на азиды происходит отщепление элементарного®
азота и образуются амины; механизм этой реакции еще не совсем выяснен. Такой ЙИни]
соб получения аминов применяется сравнительно редко, например для синтеза первщнН
ных алифатических аминов, так как соответствующие азиды являются легкодост™я
ними соединениями. В качестве восстановителей применяются преимущественно,<Я
же реагепты, что и при восстановлении питросоединений. Азиды получают из алкиздЦя
логенидов и азида патрия (стр. 443 сл.) или из окиси этилена и азотистоводородн^И
кислоты [109]. %йя|
Арилазиды тоже можно переводить в амины. Так, прн каталитическом j
ровапии [НО] фенилазида образуется анилин с 88%-ным выходом. Во всех случали
применимо каталитическое восстановление азидов на платиновых или палладневячИ
катализаторах [НО]. При каталитическом восстановлении тетраапетилглюкозилаз|шИ
на пикелевом контакте был получен тетраацетплглюкозамин с выходом 95% от теоиИа
тического [111]. При восстановлении LiAlII4 выходы аминов преимущественно хоаДИ
шие. Так, из 0-фенилптилазида с 89%-лым выходом получается 6-фенплэтиламнн [Н31я[
Используя реакцию с трифенилфосфпном, Хорнер и Гофман [113] переводили ази^Ш
с фосфинимипы:
n3P + .N,R -► RsP-N-H + N2
которые под действием II Вг в ледяной уксусной кислоте омыляются с образоваИИЙд
амина it окиси трифенилфосфина.
* Метод В. В. Феофилактова. — Прим. ред. ‘ЛЦ
Г104[ L. II. A n d г е w s, A. L о w у J. Am. Chem. Soc., 56, 1411 (1934). , Jji
[105] J. T a f e 1, Ber., 19, 1924 (1886); 20, 398 (1887); 22, 1854 (1889). JS
[106] v. Rudokirooff, Gazz. chim. ital., 81, 725 (1951); ссылка [58], стр. 5Я
[107] Ссылка [666]. г. Лж
[108] Там же.
[109] С. A. van der Werf et al., J. Am. Chem. Soc., 76, 1231 (1954).
[110] A. В e r t h о, J. Maier, Ann., 498, 50 (1932).
[111] В. H e 1 f e г i c li, A. M i t г о w s k y, Chem. Ber., 85, 1 (1952).
[112] [. H. Boyer, J*. Am. Chem. Soc., 73, 5865 (1959).
[113] L, Horner, H. Hofmann, Angew. Chem., 68, 485 (1956).
II. ВОССТАНОВЛЕНИЕ N-ОКИСЕЙ
531
Получение аминов расщеплением N-нитрозаминов
Вторичные амины можпо идентифицировать по реакции образования нитроз-
амипов. Для обратного выделения аминов из N-нитрозопроизводных применяются
различные методы, например восстановительное расщепление нитрозаминов соляной
кислотой в присутствии FeCl2:
R2N—NO-f-HClFeCI2 —>- R2NH+NO + FeCl3
Утот метод применим также для количественного определения нитрозаминов
[114], так как количество образующейся окиси азота легко определить волюметрически.
Аналогично протекает восстановительное расщепление при помощи CuCl и соляной
кислоты.
К нитрозамипу добавляют избыток охлажденного раствора CuCl в соляной кис-
лоте (d = 1,17); тотчас же с выделением окиси азота начинается реакция, заверша-
ющаяся при нагревании. Таким способом из нитрозопроизводных получают вторичные
амины с хорошими выходами.
В то время как мягко действующие восстановители (амальгама натрия, цинко-
вая пыль в соляной кислоте) переводят питрозамипы в несимметричные дизамещенные
гидразины, при восстановлении Zn в H2SO4 или Zn в НС1 происходит восстановитель-
ное расщепление цитрозамина, которое приводит к образований» исходного вторичного
амина [115]. Гладко протекающее расщепление нитрозаминов наблюдается также под
действием разбавленной H2SO4 в присутствии мочевины [116].
З-Питро-Х-метиланилин. При добавлении 20 г метил-(З-нитрофенил)-
нитрозамипа к нагретому до 50° С раствору 15 а мочевины d 75 мл воды и 75 мл
H2SO4 сразу начинается выделение газов (N2, COS и NO). Реакционную массу
нагревают при 100° С еще 15 мин. После охлаждения вторичный амин осаж-
дают аммиаком и перекристаллизовывают из лигроина; выход продукта 15,5 г;
т. пл. 67,5° С.
Расщепление нитрозаминов при простом кипячении с НС1 протекает с образова-
ние^ вторичного амина и азотистой кислоты;
ll.lv— NO R.NH + HNOa
II. Восстановление N-окисей
В этом разделс|рассматриваются изменения азотпых функций.]не имеющих N—Н-
связей, происходящие при их восстановлении, например при восстановлении окисей
аминов и аэоксипроизводных.
С тех пор, как стало известно, что электрофильные заместители вводятся в N-
окиси азотсодержащих гетероциклов легче, чем в исходные N-гетероциклы, химия
N-оксидов приобрела большое значение. Ряд обзорных работ дает примерное предста-
вление о том, как много исследований проведено в этой области [117—119]. Присоеди-
нение и обратное отщепление кислорода часто используется при изучении азотсодер-
жащих природных веществ.
В то время как для восстановления алифатических и арилалифатических N-оки-
сей в соответствующие третичные амилы можно применять и мягкодействующие вос-
становители, восстановление N-оксидной группировки в ароматических гетероциклах
Протекает обычно лишь под действием сильных восстановителей. Хорошие результаты
получены при восстановлении N-окисей пиридинов водородом в присутствия никелк
Ренея в среде уксусного ангидрида и ледяпой уксусной кислоты; реакция при этом
протекает быстро и пиридиновое кольцо не гидрируется. Гидрирование в присутствии
платиновых или палладиевых катализаторов обычно тоже приходится вести в кислой
[114] К. Lehmstedt, Вет., 60, 1910 (1927).
1115] Е. Fischer, Ann., 190, 152 (1878).
[116] W. G. Macmillan, Th. Reade, J. Chem. Soc., 1929, 585.
[117] E. Ochiai, J. Org. Chem., 18, 534 (1953).
<[118] A. R. Katritzky, Quart. Rev., 10, 395 (1956).
.[119J K. Thomas, D. Jerchel, Angew. Chem., 70, 731 (1958).
34*
532
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
среде, во, например, N-окиси 3-метилпиридина и 4-метилпиридина легко восстававши
ваются в присутствии PtOj и в среде метилового спирта (119]. N-Оксиды восстававши
ваются до третичных аминов также при действии железа в уксуснокислом раство}
[120]. В жидком аммиаке восстановление происходит как под действием натрия, та
и под действием элементарной серы. Возможно также термическое отщепление^ка,
порода. N-Окись пиридина расщеплялась при 140—210° С в присутствии цинковс
пыли или медного порошка на пиридин п другое основание не усталовлепно
строения [121]. При наличии в молекуле N-окиси других заместителей, поддающих
восстановлению, например нитрогруплы, удается отщепить кислород при помов
РС1з в хлороформе; так, из N-окиси 4-нитропиридина получают 4-нитропиридин с в]
ходом 70—80% от теоретического [117].
Окиси аминов реагируют с алкилгалогенидами с образованием О-алкильны
производных, которые расщепляются щелочами на третичный амин и альдегид^
l + CeHaCH2Br —> || j
'n ч'+/
f ГВг’
0 O-CH,C,Hs
J + C6H&CHO
N
Эту реакцию можно использовать для элиминирования N-оксидного кислорода
4-Метилпиридин [119]. В 60 мл ледяной уксусной кислоты и 10 м
уксусного ангидрйда растворяют 20 г N-окиси 4-метилпиридияа и после доба
вления 1 г никеля Ренея при интенсивном встряхивании гидрируют при коц
натной температуре. После окончания поглощения водорода основное коли
чество катализатора легко отделяют фильтрованием. К фильтрату прибавляю1
около 20 мл концентрированной НС1 и упаривают его в вакууме, создаваемая
водоструйным насосом, при температуре 80° С до сиропообразной консистенции
При добавлении разбавленного раствора NaOH выпадает Ni(OH)2. Через нека
торов время отгоняют свободное основание с водяным паром, выделяют eri
из дистиллята NaOH и в течение нескольких суток сушат над твердым NaOH
Последние следы основания извлекают из водной фазы эфиром. Эфипные эк®
ранты и основание объединяют и перегоняют; получается 39,2 е (84% от теорй
тического) чистого 4-метилпирндина; т. кип. 144—145° С (760 мм рт. ст.^
При восстановлении окисей аминов образуются третичные амины очень вн*
сокой чистоты. Описано получение чистых метилпиридинов яз смесей метилпиридиио
восстановлением их N-окисей [142].
С иеменьшнм успехом применялись для восстановления N-окисей алюмогидрн
лития [129], азотистая кислота [122], олово и хлорид олова (II) [123], цинк в кисЛ0|
среде [124], дитиоиит натрия (125] и сернистый ангидрид (126].
Азоксисоединения переводятся в азо- или гидразопроизводные сравннтелья
мягкими восстановителями. Восстановление азокенбензола цйнком в щелочном рай
творе до гидразобензола описано в практикуме Гаттермана и Виланда [127]. АзокеЦ
соединения переводятся ЫА1Н4 в азосоединения большей частью с количественны!
выходом [128]. 1
[120] I. G. Murray, С. В. Hauser, J. Org. Chem., 19, 2008 (1954).
[121] М. К a t a d a, J. Pharm. Soc. Japan, 67 , 53 (1947).
(122] A. P i c t e t, M. Mattison, Ber., 38, 2782 (1905).
[123] G. T. Newbold, F. S. Spring, J. Chem. Soc., 1948, 1864.
[124] P. Friedlander, H. Osterman n, Ber., 14, 1916 (1881).
[125] D. L. Vivian, J. Am. Chem. Soc., 73, 457 (1951).
[126] H. Z. Lecber, W. D. H a г d y, J. Am. Chem. Soc., 70, 3789 (1948)-
27] Ссылка (11, с-вр. 168.
28] Ссылка [666].
29] Там же.
Ш. ОБРАЗОВАНИЕ N—0-СВЯЗИ
533
III. Образование N—О-связи
N—О-Связь образуется как в результате реакции присоединения, тан и в ре-
зультате реакции замещения. В любом случае это превращение является окислитель-
ным процессом.
1. Образование N—О-связи в результате реакции присоединения
Получение N-окисей
(RSN —> R3N—>0)
>' Химия N-окисей начала практически разрабатываться лишь в последние двад-
£ цать лет. Как уже отмечалось в предыдущем разделе, значение N-окисей, особенно в
; ряду азотсодержащих гетероциклов, непрерывно возрастает. Важнейшими способами
г получения таких соединений является окисление третичных амнков перекисью водо-
к рода, надкислотами или озоном.
Л Окисление переписью водорода. Для получения N-окисей
применяются обычно 30%-ные растворы Н2О2. Количество вводимого окислителя,
- а также температура реакции зависят от реакционной способности окисляемого амина.
Г Во избежание возможного взрыва избытка перекиси водорода по завершении реакции
s разложение М2О2 рекомендуется проводить с помощью обрезка платиновой жести
г ИЗО].
Y Гидрат окиси триметиламнна [131 ]. При комнатной температуре смеши-
г вают 100 мл 33%-ного водного раствора триметиламина с 600 мл 3%-ного
. раствора чистой Н2О3. Если через 24 ч еще ощущается запах триметиламина,
добавляют дополнительно 100—200 мл Н2О2. После того как в реакционной
массе уже не обнаруживается триметиламнн, раствор упаривают в вакууме
г и перекристаллизовывают остаток из этилового спирта и эфира. Гидрат окиси
ь , триметиламина выпадает в виде игл; выход до 95% от теоретического;
т. пл. 96° С.
с
~ Безводную окись триметиламина получают обезвоживанием гидрата при 120—-
г 150° С в вакууме (12 мм рт. ст.). В промышленности гидраты окиси триалкиламинов
к получают при низкой температуре (—40° С) [132].
fe: ' Окисление надкислотами. Применяя в качестве окислителя
- надбензойную кислоту. Мейзенгеймер [133] получил N-окись метилэтиланилина, ко-
торую не удается синтезировать окислением амина водной перекисью водорода. Этим
к, методом можно получать и другие N-окиси диалкиланилинов [134]. При синтезе
£ ряда N-окисей в качестве окислителей наряду с надбензойной кислотой применяли
| также надуксусную* 1 н мононадфталевую кислоты [135]. Для получения окиси л-бром-
Б дпметиланилина применялась кислота Каро [136].
Окисление триалкиламинов озоном [137] проводят в хлоро-
& форме при —80° С. По завершении окисления в реакционную массу пропускают газо-
g образный НС1 и выделяют гидрохлорид N-окиси; выходы до 90% от теоретического.
| О получении и свойствах окисей аминов ряда пиридина см. [119]. Окись цнри-
к дчна получена Мейэенгеймером [138] еще в 1926 г. действием надбензойной кнслоты
Г на пиридин. В качестве окислителей пригодны и другие органические надкислоты.
Г Образование окисей аминов из пиридинов удается только в том случае, если в качестве
Ъ ИЗО] А. С. Соре, Р. Н. Towle, J. Am. Chem. Soc., 71, 3426 (1949).
E !131| J. M eisenheimer, K. Bradin g, Ann., 397, 286 (1913).
[ [132] Пат. ФРГ 937058 (1952); BASF, Erl. E. Rotter, E. H a a r e r, C., 1956»
| 11563.
r f49° J. Meisenheimer, Ber., 52, 1667 (1919).
J. Meisenheimer, Ann., 449, 188 (1926).
M. A. Stahmann, M. Bergmann, J. Org. Chem., If, 586 (1946).
L. W. Jones, E. B. Hart sh о r n, J. Am. Chem. Soc., 46, 1840 (1924)»
W. Strecker, M. В a’l t e s, Ber., 56, 2693 (1921).
J. Meisenheimer, Ber., 59, 1848 (1926).
534
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
растворителя применяется карбоновая кислота. Так, окислением соответствующих
пирицинкарбоновых кислот 30%-ной Н..О2 в ледяной уксусной кислоте получений
N-окиси изоникотиновой [139] и нигсотиновой [140] кислот. Установлено [117], что"'
< смесь пергидрола с ледяной уксусной кислотой является реагентом, пригодным дли \
.получения любых N-окисей пиридинового ряда. .;
N-Окись пиридина. Раствор пиридина в ледяной уксусной кислоте нагре- |
вают с избытком 30%-ной НВО2 в течение нескольких часов при 70—80’С,
упаривают реакционную массу в вакууме, подщелачивают сс и извлекают ^
продукт реакции хлороформом. N-Окись пиридина очищают перегонкой 1
в вакууме. d
По этому способу аминоокиси гомологов пиридина и многих другнх-м
его производных получают обычно с очень высокими выходами [117]. 4
В настоящее время известно большое число N-окисей пиридинового ряда, изЗ
«вторых большое препаративное значение имеют в первую очередь окиси самого пири--3
дина и его простейших гомологов. Семиполярный характер N—0-связи придает этим |
-соединениям свойства солей; они хорошо растворимы в воде, плохо растворяются |
« бензоле и эфире. Подобно четвертичным солям пиридиния N-окиси пиридина очень 4
гигроскопичны. Температуры кипения и плавления окисей всегда выше, чем у qoor-.i
.вегствующих аминов Аминоокиси имеют также основной характер и образуют соли.^
.Для их идентификации можно применять пикраты и пикролонаты [141]. .
Получение азоксипроизводных из азосоединений
(R-N=N—R —> R—N=N-R)
Азосоединения гладко и почти количественно окисляются до азоксисоединений пе-^
зрекисыо водорода в ледяной уксусной кислоте. Реакцию проводят при комнатной темч|
пературе, но в некоторых случаях требуется продолжительное нагревание на водяно#|
бане 1142]. Этим же методом получают л полиазоксисосдинения [143]. Однако, как!
.правило, для окисления применяют органические надкислоты; в мягких условиях^
реакция протекает почти количественно. Обзор полученных таким путем азоксисоеди-J
нений приводит Сверн [144]. Подробно изучено окисление азосоедипений падбензойнойд
•кислотой [145]. Фенилгидразон бензальдегида тоже окисляется надфталевой кислотоО
до бензилазоксибензола [146]: Ч
О
Т
CjHs-CH-N-NH-Cjn.^i CjH6-CH2-N=N-C,H[ — С,Н6-СН2—N=N-CtH- '
Окисление нитрозо- и оксимной групп до нитрогруппы.
Окисление нитрозоцроизводНых до нитросоединений имеет особенно большое*
значение в тех- случаях, когда введение нлтрозогр.уппы (окислением аминогруппы)^
-осуществляется легче, чем прямое нитрование (так, например, аминопиридины и хи-у
'нолины, а также некоторые о-нитроапилины являются сравнительно доступными соедИ^Я
нениями; окисление их до нитрозосоедидений и последующее дальнейшее окислении
^приводит к нужным нитропроизводным). , J
.1139]
.[140
.1141.
-1142.
U43J
(144J
,[145]
Я46]
Е. G h i g i, Ber., 75, 1318 (1942).
С. R. Clemo, Н. Koenig, J. Chem. Soc., 1949, 231.
A. B. Ka tri tzky, J. Chem. Soc., 1956 , 2404.
J). J erchel, W. Melloh, Ann., 613, 144 (1958).
P. Ruggli, G. Bartuschl, Helv. chim. acta, 27, 1371 (1944).
J-\ Swern, Chem. Rev., 45, 38 (1949).
G. M. Badger cl al., J. Chem. Soc., 1953, 2143—2158.
В. M. Lyne h, К. H. Pausacker, J. Chem. Soc., 1953, 2517.
Ш. ОБРАЗОВАНИЕ N—О-СВЯЗИ
53&
Для окисления о-нитрозонитросоединсний рекомендуют применять смесь- пер-
гидрола и азотной кислоты [147].
4,5-Динитро-1,2-диметилбензол. К раствору 10 г 4-нитрозо-5-питро-1,2-
димотилбензола в 300 мл ледяной уксусной кислоты прибавляют смесь 150 ад-
ледяной уксусной кислоты и 150 мл 33%-ной Н2О2, а затем 10 мл HNO3 (d =
— 1,40). При нагревании на паровой бано темно-зеленый раствор в течение не-
скольких минут становится оранжевым. Встряхиванием пробы реакционной массы.'
с хлороформом проверяют наличие исходного нитрозосоединения (зеленое-
окрашивание) и после окончания реакции осаждают динитроксилол 600 мл.
воды; выход 9 в (83% от теоретического).
Аминопиридипы окисляются через нитрозопиридины до соответствующих нитро-
ппридииов смесью пергидрола и серной кислоты за одну операцию [148].
Описано [149] окисление нитрозогруппы смесью азотной кислоты с водой при>
40— 50® С. На примере 2-нитрозо-5-нитротолуола [150] описано одновременное окис-
ление бихроматом калия в сорнокислом растворе нитрозогруппы до нитрогруппы-
н метильной группы до карбоксильной, При нагревании гептафторнитрозопропана
с кислородом при 70° С в запаянной трубке образуется соответствующее нитросо
едннение [151]. Можно рекомендовать обзорную ра&оту по С-питрозосоединеникм.
[152].
Оксимная группа тоже может окисляться до нитрогруппы. Так, при окислении/
диатилового эфира изонитрозомалоновой кислоты МпО2 в ледяной уксусной кислоте-
1153] (24 ч при 30° С) образуется диэтиловый эфир нитромалоновой кислоты с 88%-ным.'
выходом *.
2. Образование N—0-связи в результате реакции обмена (окисление*
аминои, гидроксиламинов и оксимов до нитрозосоединений)
Амины окисляются до нитрозосоединений с промежуточным образованием гидро-
ксил аминов:
RNH2 —> RNHOII —> RNO
В качестве окислителей могут применяться кислота Каро [154], бихромат и сер—
пая кислота [155], хлорид желоза (III) [156], надбензойная и надуксусная кислотье-
[157]. Окисление арилгидроксиламинов до пптрозосоединений достигается обычно
действием бихромата и серной кислоты. Ароматические нитрозосоединения можна
получать также через диоксймы n-хинонов, применяя для их окисления феррицианид
калия [158]. Бамбергер и Зелигман детально изучили окисление алифатических аминов.
[ 159]; они показали, что строение конечных продуктов окисления зависит от характера»
замещения у аминированного атома углерода. Если аминогруппа связана с третичным-
атомом углерода, то при ее окислении образуются нитрозосоединения; при окислении.
Окисление изопитрозоацетоуксусного эфира хромовой кислотой дает с хорошим
выходом нитроуксусный эфир [В. М. Родионов, И. В. Мачинская»,
В. М. Беликов, ЖОХ, 18, 917 (1948]. — Прим. ред.
R. Kuhn, W. Klaveren, Ber., 71, 779 (1938).
Л. К 1 г р а 1, W. Bohm, Вег., 65, 680 (1932).
С. F. К о е 1 s с h, J. Am. Chem. Soc., 66, 2019 (1944).
Org. Synth., Coll. v. 3, 1955, p. 334.
D. A. Barr, N. R. H a s z e 1 d i n e, J. Chem. Soc., 1956 , 3416.
В. C. Gowenlock, Quart. Rev,, 12, 321 (1958).
L. Canomica, Gazz. chim. ital., 77, 92 (1947).
Org. Synth., Coll. v. 2, 1942, p. 334, cp. [1], стр. 159.
Ссылка fl], стр. 158.
R. E. Lotz, M. Lytton, J. Org. Chem., 2, 73(1937); Barrow, Thor-
neycroft, J. Chem. Soc., 1939, 773.
J. D’A n s, A. К n e i p, Ber., 48, 1144 (1915).
Francesconi, Brescianl, Gazz. chim. ital., 34, 11 (1904).
E. Bamberger, R. Seligman n, Ber., 86, 685 701 (1903).
536
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
аминогрупп, связанных с первичным или с вторичным атомом углерода, образуются!
соответствующие оксимы. Так, например, при окислении кислотой Каро (HaSO5) трет-%
бутиламина образуется третичный нитрозобутан, а при окислении изопропиламина — $
оксим ацетона £162]. Оксимы были получены при каталитическом окислении первичных'!
аминов. Алифатические, арилалифатические и алициклические моно- и диамины окне-]
ляются до соответствующих оксимов Н2Оа в присутствии катализаторов — раствори-1
мых в воде солей вольфрамовой, молибденовой и урановой кислот. i
2-Нитрозо-5-нитротолуол [154]. К 50 г (0,33 .моль) 5-нитро-2-амино-|
толуола прибавляют охлажденный льдом раствор 200 жл концентрированной!
H2SO4 в 50 мл воды. Полученную смесь перемешивают при комнатной темпера-1
туре. Одновременно готовят кислоту Каро, для чего к 175 мл охлажденной!
льдом H2SO4 (d = 1,84) при перемешивании добавляют 300 г K2SoOe; затем.|
к этой смеси прибавляют 800 г льда и 300 мл воды и хорошо перемешиваются
Полученный таким образом раствор кислоты Каро при перемешивании прилив!
вают к суспензии нитроаминотолуола. Перемешивание продолжают еще 2 чя
при 40° С, затем в один прием добавляют еще 100 г порошкообразного K.S.O^g
смесь перемешивают снова 2 ч при 40° С и разбавляют водой до объема 4 ал
Осадок отфильтровывают, промывают водой и очищают полученный 2-нитрозоЗ
5-нитротолуол перегонкой с водяным паром; выход 30—39 г.
-/
Получение нитрозобензола окисленном фенилгидроксиламина бихромата!
в серной кислоте описано в практикуме Гаттермана и Виланда [155]. j
IV. Образование N — N-связи в результате реакции обмена ;
1. Получение нитрозаминов
N-Нитро^опроизводные вторичных аминов могут применяться для идсатифика
ции вторичных аминов; кроме того, они являются исходными соединениями в синте^
несимметричных дизамещенных гидразинов. Их получают действием азотистой кн<
лоты на алифатические и ароматические вторичные амины. Для этого к раствору амиц
в кислой водной среде прибавляют небольшой избыток NaNO2. Часто реакцию прв
водят в солянокислом, реже в уксуснокислом растворе. Ароматические питрозамиВ
обычно синтезируют при нагревании [160]. N-Нитрозоглицины можно получа?
в водной суспензии без добавления минеральной кислоты [161]. Амины, плохо раств
римые в водных кислотах, интроаируют в ледяной уксусной кислоте [ 163] илн в спирт
Ннтрозокапролактам был получен действием интрозных газов на раствор калролакга»
в ледяной уксусной кислоте [164]. Большинство питрозамивой являются нейтрадь
ними соединениями, плохо растворимыми в воде. Диметилпитрозамин обладает слаЙ
основными свойствами и поэтому может быть осажден из эфирного раствора в виД
гидрохлорида. Нитроэамины обычно перегоняются с водяным паром или в вакууму
правда иногда при этом наблюдалось их разложение со взрывом [165]. у
Описаны синтезы многих, имеющих практический интерес N-нитрозопроизвр
ных, иапримеп интрозодиметиламипа [166], N-нитрозометиланилина 1167], нитрозои
тилмочевины [168] и'нитрозометилуретана [169]. Два последних соединения являю#
промежуточными продуктами при получении диазометана.
160
161
162
163
164
165
166
167
168
[169
Org. Synth., Coll. v. 2, 1950, р. 211.
W. Baker et al., J. Chem. Soc., 1949, 307.
K. Kaier, С. В e r t h e r, Chem. Ber., '93, 132 (i960).
E. J. Forbes, J. Chem. Soc., 1956, 513.
R. Huisgen, R. R e i n e r s h о f.e r, Ann., 575, 174 (1952).
Org. Synth., Coll. v. 3, 1955, p. 244.
Org. Synth., Coll. v. 2, 1943, p. 211.
Org. Synth., Coll. v. 2, 1943, p. 460.
Org. Synth., Coll. v. 2, 1943, p. 461.
Org. Synth., Coll. v. 2, 1943, p. 464.
ОБРАЗОВАНИЕ N—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
537
2. Получение нитраминов
(RR'NH —► RR'N-NO2)
Первичные алифатические нитрамины (RNH—NO,) получают: 1) нитрованием1
N-алкиламидов; 2) алкилированием нитрамидов; 3) расщеплением N .N-днхлораминов-
азотной кислотой [294]; 4) нитрованием аминов алкил- или ацилнитратами (179].
При получении нитраминов нитрованием амидов переводят сначала амин в со-
ответствующий амид, нитруют амид безводной или дымящей HNOg или смесью HNO»
и II2SO| и гидролизуют полученный ннтрамид до нитрамина в щелочной среде.
Метилнитрамин (N-HHTpo-N-метил-п-толуолсульфамид) [170]. К 130 ж*
70%-ной HNO3 при 5° С н перемешивании прибавляют в несколько приемов
23 г N-метил-п-толуолсульфамида. После полного растворения сульфамида
температуру понижают до 0° С н, продолжая перемешивание, добавляют ещ»
130 мл 98%-ной HNO3. При той же температуре смесь перемешивают еще
в течение 10 мин и выливают ее в воду со льдом. Нитросульфамид отфильтро-
вывают и перекристаллизовывают из спирта; выход продукта 21 г (73% от тео-
ретического); т. пл. 57° С.
Для расщепления ГС-нитро-М-алкил-п-толуолсульфамидов пиперидином
[171] прибавляют к раствору 10 г нитрамида в 35 мл ацетонитрила при 5—10е С
и перемешивании 10 г пиперидина. Реакция протекает с заметным выделением
тепла. Для завершения реакции смесь кипятят 30 мин в колбе с обратным
холодильником н выливают ее в 100 мл 10%-ного раствора NaOH. Выпавший
N-тозилпипериднн отфильтровывают, фильтрат осторожно подкисляют и экстра-
гируют продукт реакции эфиром. После сушки эфирный раствор перегоняют
в вакууме. Выходы нитраминов 81—96% от теоретического.
Нитрамины часто получают также алкилированием нитрамндов, образующихся
при нитровании амидов арилсульфокислот или уретанов [17Щ.
Общая методика [170]. К эквимолярному количеству нитроурегава»
' растворенному в минимальном объеме эфира, прибавляют 0,1—0,5 М бензоль-
ный раствор дназоуглеводорода. Смесь оставляют на ночь, затем нагревают
30 мин до 40° С, охлаждают и пропускают в полученный раствор аммиак^
Аммонийную соль нитрамина отфильтровывают, промывают бензолом, раство-
ряют в воде и осаждают свободный нитрамин осторожным добавлением раз-
бавленной НС1.
При обработке К,Х-дихлораминов азотной кислотой образуются N-x.iop-N-цитр-
амины, от которых отщепляется хлор и с удовлетворительными выходами образуются
соответствующие нитрамины [172]. Реакция протекает по следующей схеме:
/С1 zN02
R—N< —► R—N< —► R-NH—NO3
V.l
Реакцию проводят в среде уксусного ангидрида с 99%-ной HN03, при этой
кислоту и дихлорамин одновременно приливают по каплям к растворителю с таким
расчетом, чтобы в реакционной массе постоянно имелся 5%-ный избыток HNOg. Реак1»-
ционную массу упаривают до Vs объема, выливают в холодную воду и добавляют 100 мл
раствора NaOH для перевода нитрамина в раствор в виде натриевой солн; маслянисты»
побочные продукты извлекают эфиром. После этого водный слой подкисляют уксусяоЬ
кислотой, экстрагируют свободный ннтрамин эфиром и перегоняют в вакууме.
Вторичные алифатические нитрамины можно получать нитрованием вторичныщ
аминов и окислением нитрозаминов.
[170] М. I. Gillibrand, А. Н. Lambertso п,г J. Chem. Soc., 1949U
1883.
[171] W. D. Emmons, I. P. Freeman, J. Am. Chem. Soc.. 77, 6061 (1955).
[172] G. N. R. Smart, G. F. Wright, J. Am. Chem. Soc., 70, 3141. (194^)к.
538
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕННИХ
Лишь слабоосновные вторичные амины переводятся при действии HNO3 в нитр-
амины. Более сильные основания, как, например, диметиламин, нитруются HNOS
01 уксусном ангидриде только в присутствии хлористого водорода или ацилхлоридов
[175]. Для нитрования вторичных аминов можно применять также ацилнитраты [173].
Окисление нитрозаминов до нитраминов представляет большой интерес для
получения этого класса соединений, так как исходные питрозамины являются легко
доступными соединениями. Отличным окислителем оказалась трифторнадуксусная
кислота [176]. Окисление ведут безводной трифторнадуксусной кислотой в иетилея-
хлориде или 90%-ной Н2О2 в трифторуксусной кислоте. Выходы составляют около 70%
от теоретического.
Ароматические нитрамины получают нитрованием аминов или окислением
диазотатов.
При нитровании ароматических аминов часто получаются пе нитрамины, а С-яи-
тропроизводные, образующиеся в результате нитрования в ядро- Нитрампны получаются,
•как правило, лишь в тех случаях, когда в исходном амино в орто- или пара-положе-
няи имеются электроноакцепторпые заместители. Для нитрования используют дымя-
щую иля 100%-ную азотную кислоту как таковую или в смеси с ангидридом уксусной
кислоты [177]. Не применяют для нитрования смеси азотной и серной кислот, так как
в присутствии серной кислоты образующиеся нитрамины перегруппировываются в ни-
троариламины.
Для получения некоторых нитраминов используют также третичные ямины.
Так. например, тетрил (2,4,6-тринитрофенилметилнитрамин) получают нитрованием
димстиланплина, причем одна метильная группа подвергается окислительному отще-
плению [178]. Для нитрования с успехом применялись бензои.тнитрат [179] и азотный
ангидрид [180].
Окисление диазотатов облегчает получение нитраминов из аминов, по имеющих
электроноакцепторных заместителей в ароматическом ядре. Превращение аминов
в нитрамины может происходить в процессе одной операции. Амин диазотируют, рас-
твор соли диазония приливают к раствору едкой щелочи и добавляют окислитель?
.K3Fe(CN)e, NaClO, CaOCL [181], Н2О2 [182] или КМпО4 [183].
По этому методу Бухнер [184] получил феяилнитрамин.
Вторичные нитрамины являются нейтральными соединениями, первичные об-
разуют с основаниями соли, избыток щелочи вызывает часто разложение нитрамина
на карбонильное соединение, азот и воду [185]. Вторичные нитрамины разлагаются
водными щелочами па азотистую кислоту, альдегид и первичный амин. До настоящего-
времени пе выяснен механизм разложения первичных алифатических нитраминов под
действием кислот, которое протекает с выделением закиси азота NSO. Таким образом,
нитрамины не гидролизуются кислотами подобно нитрозаминам с образованием ами-
нов, по гак же, как и нитрозамины, превращаются восстановителями в соответству-
ющие гидразины. Первичные и вторичные арилнитрамипы легко перегруппировываются^
-в кислом растворе с образованием нитроариламинов. Большинство нитраминов раз-v
латаются со взрывом при нагревании, однако пизтпие вторичные алифатические нитр-
амины перегоняются в вакууме.
Нитрамины ароматических гетероциклов до недавнего времени были йена-;
вестны; лишь в последнее время получены некоторые N-нитропиразолы [186].
[173]
[174]
1175]
[176]
1177]
<173]
1179
W. D. Emmons, I. Р. Freeman, J. Am. Chem. Soc., 70, 4387 (1955).
W. I. Chute et al., Canad.-J. Res., 26 B, 114 (1948).
T. Connor, G..N. R. Smart, G. F. Wright, Canad. J. Res., 26 Bi
294 (1948).
W. D- Emmons, A. F. Ferris, J. Am. Chera. Soc., 75, 4623 (1953); 76j
3468 (1954).
A. E. Bradfield, I. К. P. Orton, J. Chem. Soc., 1929, 915.
С. E. С 1 a r k s 0 n, I. G. Holden, T. Malkin, J. Chem. Soc., 1950,
1556.
<180
<181
[182
<183
<184
<185
£186]
F. E. Francis, J. Chem. Soc., 89, 1 (1906).
E. Hol f, Ann., 311, 91 (1900).
T. Zincke, A. Kuchenbccker, Ann., 330, 1 (1904).
E. Bamberger, O. Baudisch, Ber., 42, 3568 (1909).
E. Bamberger K. Landsteiner, Ber., 26, 482 (1893).
M. Bnchue r, <Rer., 35, 266 (1902).
I. В а г г о L et al., J. Chem. Soc., 1951, 1282.
R. H ii tie 1, F. В u chele, Chem. Ber., 88, 1586 (1955).
IV. ОБРАЗОВАНИЕ N—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
53?г
Нитрамины находят применение главным образом в качестве взрывчатых
веществ (гексоген, тетрил, нитрогуанидин).
Изящным методом получения диазоалканов является взаимодействие концен-
трированных водных растворов щелочей с N-anKHn-N-HHTpoao-N'-HHTporyaHHflHHaMB
1187]. Обзорную работу по нитраминам см. [188], по нитрогуанидинам — см. [189].
3. Дназотировавве ароматических аминов
Соли первичных ароматических аминов реагируют с азотистойТкислотой с обра-
зованием солей диазония:
ArNlI2 + HCl + HNO2 —> ArNJCl--|-2H2O
Все амины, растворимые л водных кислотах, можно диазотировать по так назы-
ваемому «прямому методу». К холодному раствору ариламинов в водной минеральной
кислоте приливают по каплям холодный раствор NaNO2- Оптимальной температурой
диазотирования простых ариламинов является 0—2° С, л-галогепариламинов &—
5° С, бензидинов, пафтиламинов, о- и n-галогенаминов около 10—12° С. В отдельные
случаях работа ведется и при более высоких температурах: например, п-нитроаниля»
диазотируют при 34° С, а о-аиизидин при 40° С. Конец реакции определяют по- врв-
сутствию избытка HNO2, что устанавливается при помощи иодкрахмальной бумаги,.
От применения амидосульфоновой кислоты для разложения избыточной HN02 в на-
стоящее время отказались, так как это соединение реагирует с некоторыми реакцион-
носпособными диазосоединениями [190].
Во избежание сочетания диазониевой соли с непрореагировавшим амином
с образованием дназоаминопроизводного применяют избыток минеральной кислот»
(3 моль). Плохо растворимые соли аминов диазотируют в мелкокристаллическом
состоянии [191] или, лучше, по методу Клауса в безводной кислоте (серпая кислота).
При этом считают, что действующим агентом является нитрозилсерная кислота. Ме-
тод Клауса применяется главным образом для диазотирования слабоосновных аминов:
с электроноакцепторными заместителями, например для диазотирования 4-нитро-1-наф—
тпламина [192], пикрамида, 4-интро-2,6-дииодапилина [193]. Амины, не реагирующие-
с концентрированной HNOg (d = 1,48), рекомендуется диазотировать по методу Вктта
1194]. Для этого амины (например, динитроаинлины) растворяют в концентрированной
HNOg, добавляют метабисульфит калия, который восстанавливает часть азотной кис-
лоты до азотистой кислоты
K2S2OiH-2HNO3 —> K2S2O7-J-2HNO2
а азотистая кислота реагирует с амином с образованием соответствующего диано*-
сосдинения.
Ариламины, содержащие кислотную группировку (сульфокислотную или карбо-
ксильную), диазотируют по так называемому «косвенному методу». Их растворяют в вод-
ном растворе щелочи, добавляют рассчитанное по реакции количество раствора NaN02
и приливают эту смесь по каппям к избытку кислоты. При этом выделяется дназосоеди-
непие, обычно в твердом виде. Такой метод с успехом применяется для диазотирования»
амицобензойной кислоты, анилин- и нафтилаиннсульфоинслот; так же можно диазоти-
ровать и слабоосновпые амины, как, например, нитроанилины, о-нитро-п-фенетидин^.
аминоазобепзол и др. (ср. [198]).
Твердые соли диазония * получают по следующему методу [195].
[187
188
189
190
191
192
193
194
195
Внимание! Твердые соли диазония в сухом виде взрывают! — Прим. реа~.
A. F. М с К а у, J. Am. Chem. Soc,, 70, 1974 (1948).
А. Н. Lambertson. Quart. Rev., 5, 75 (1951).
A. F. McKay, Chem. Rev., 51, 301 (1952).
H. W. Grimmel, J. F. Morgan, J. Am. Chem. Soc., 70, 1750 (1948)4-
Org. Synth., Coll. v. 1, 1941, p. 113.
A. Claus, С. В e у s e n, Ann., 266, 224 (1891); Org. Synth., 28, 52 (1955K
C. Niemann, С. E. Redemann, J. Am. Chem. Soc., 63, 1550 (1941K
O. N. Witt, Ber., 42, ^953 (1909).
A. Hantzsch, E. Jochem, Ber., 34, 3337 (1901).
540
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Солянокислую соль амина растворяют или суспендируют в трехкратном
количестве ледяной уксусной кислоты и при ю° С и интенсивном перемешива-
нии добавляют рассчитанное количество амилинтрита; к полученному раствору
осторожно добавляют эфир, осаждая соль диазония. Этот метод был впослед-
ствии значительно улучшен [196].
Твердый хлорид феттилдиазония (196]. Суспендируют 5 г гидрохлорида
анилина в смеси 30 мл ледяной уксусной кислоты и 30 мл сухого очищенного
от перекисей диоксана. Полученную суспензию охлаждают в смеси льда с пова-
ренной солью и пропускают в нее этилнитрит до полного растворения соли
амина (избыток этилптттритл определяют по иодкрахмальной бумаге). По дости-
жении комнатной температуры к смеси сразу добавляют 150 мл сухого диоксана,
после чего выпадает соль феннлдиавопия в виде тонких* белых кристаллов.
Продукт реакции отфильтровывают и дважды промывают 25 мл диоксана.
Хлорид р-нафтилдназония (получение раствора прямым методом) [197].
В смесь 450 мл концентрированной НС1 и 500 мл воды вносят 143 г (1 моль)
Р-нафтиламина. Взвесь гидрохлорида охлаждают до 5° С добавленном 500 а
льда; затем при интенсивном перемешивании добавляют около 69 г твердого,
NaNOj до обнаружения незначительного избытка азотистой кислоты (по иод-’
крахмальной бумаге). Во времи реакции постепенно добавляют около 600 а
льда, поддерживая температуру не выше 5° С. По завершении реакции раствор,,
фильтруют.
Сульфат сульфапплдпазония (косвенный метод диазотирования) [198].
Растворяют 33 г (0,1 моль) сульфанилата натрия и 7 г NaNO2 в 120 мл воды
и охлаждают раствор льдом. Затем этот раствор медленно приливают-по каплям'
при перемешивании к охлаждаемому льдом раствору 17 мл конггентрирлиянной1
H2SO4 в ’100 мл воды. Сразу выпадает твердая соль диазония. j
Ниже приведен способ диазотирования слабоосновных аминов в концен--
трировапной H2SO4 (метод Клауса).
Общая методика [199]. Раствор основания в горячей ледяной уксусной-
кислоте (кислоту добавляют из расчета 12 мл кислоты па 1 г амина) быстро?
охлаждают до комнатной температуры и постепенно добавляют при перемеши-ц
вании к раствору NaNO8 в концентрированной H8S04 [17 мл H2SO4 (d 1,84);’
на 1 г NaNOg]. Этот раствор готовят следующим образом: мелко размолотый’
NaNOg добавляют при интенсивном перемешивании к кислоте, смесь нагревают^
до 70° С и после полного растворения охлаждают до комнатной температуры,|
(Применяют 10%-яый избыток NaNO2 в расчете па амин.) у
Непрерывный метод получения диазосоединений описан Хупфером [203J.A
Кинетика и механизм реакции диазотирования тщательно исследованы [200]
Дяазониевые соли неустойчивы как в твердом состоянии, так и в растворах. С твердыми^
солями диазония следует обращаться крайне осторожно, используя их лишь в не-®
больших количествах, так как при нагревании или толчке, а иногда и без всякого^
заметного повода, эти соединения разлагаются с очень сильным взрывом [201]- Аниуй
ониая компонента влияет на устойчивость соли. Хлориды и сульфаты, как правило^
более безопасны, чем нитраты и перхлораты. Менее опасны растворы дпазоняевЫХ|
солей, но и они постепенно разлагаются (часто уже при температурах выше 5е С)й|
поэтому нх следует всегда готовить непосредственно перед употреблением. Па свету!
разложение солей значительно ускоряется. В последнее время усовершенствования
методы стабилизации диаэониевых солей. Так, например, стабилизация достигается®
получением соответствующих солей с 1,5-нафталиндисульфокислотой или 2-нафтол-1-^
сульфокислотой [202], двойных солей с хлоридом ртути (Л) [197] или взаимодействием
[196 ] W. Smith, С. Е. Waring, J. Am. Ghem. Soc., 64, 469 (1942). J
[197 ] Org. Synth., Goll. v. 2, 1943, p. 432. |
[198 ] К. H. Saunders, The Aromatic Diazo-Compounds and Their ТесЬпЙЗ
cal Applications, 2 ed., E. Arnold a. Co., London, 1949, p. 10. - S
199] Там же, стр. 13. 3
200] H. Schmidt, Ghem. Ztg., 78, 565 (1954). |
201] E. Bamberger, Ber., 28, 538 (1895). '•
202] Organic Reactions, v. II, John Wiley and Sons, Inc., New York, 1944, p. 285»,-
203] H. H u p f e r, Angew. Ghem., 70, 244 (1958).
IV. ОБРАЗОВАНИЕ N—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
541
2 моль соли диазония с 1 моль пиперазина [204]. Часто применяются также фторбо-
раты диазония [205].
Возможно также тетразотирование диаминосоединений [206]. Некоторые за-
местители, находящиеся в орто-положении к новообразующейся диазогруппе, могут
реагировать с ней, образуя циклическую систему, иногда с выделением азота; так,
например, при диазотировании о-фенилевдиамина получается 1,2,3-бензтриазол с выхо-
дом 51% от теоретического [207]. Если орто-заместителем является активированная
метильная группа, то образуется индазол [208]. Получение, свойства, реакции и стро-
ение ароматических диазосоединений подробно описаны в монографии Саундерса [198].
Диазотирование протеинов * описано Рорлихом [209].
4. Получение азидов
Азиды можно получать следующими методами:
1) действием пербромидов диазония па аммиак [210]
ArN2Br3 + NIl3 —► ArN3-{-3HBr
[выходы около 55% от теоретического);
2) взаимодействием гидразинов с азотистой кислотой [211]:
ArNH-NHs + IIONO —> ArN3+2H2O
(выходы 65 — 68% от теоретического);
3) из дпазосоединепий и гидразингидрата [212]:
ArN2X + NHa-NH2 —> HX-|-ArN2—Nil—NII2 —> AcN3 + NH3
(выходы около 70% от теоретического);
4) из арплдиазониевых солей и О-алкилгидроксиламипов [213]:
ArN2Cl+ RONH2 —> ArNa+ROH + HCI
.(выходы хорошие);
5) из диазонпевых солей и сульфамидов [214]:
ArN2C]r+Ar'SOaNH2 —> ArN3+ Ar'SO2Hт HCI
(выходы 60—80% от теоретического);
6) из диазониевых солеи и азотистоводородной кислоты [215]:
ArN2X + HN3 —► ArN3-rN2-IIX
(выходы очень хорошие);
7) окислением фснилсемикарбазида [216]:
CcH5Nii-NH-CONH8 —► CeHgN=N—СО—NH2 —► CeH5N=N—NH2 —> CeH5N3
(выход 53% от теоретического);
* Речь идет о модифицировании остатков тирозина в белках, по-видимому,
через нитрозопроизнодные. —Прим. ред.
[204] Р. I. Drumm, W. F. О’С о n п о г, I. Reilly, Sci. Proc. Roy. Dublin.
Soc., 22, 223—227 (1940); C., 1943, II, 521.
[205] Org. Synth., Coll. v. 2, 1943, p. 188, 225; A. Wayne R u d d у, E. B. S t а гч
key, W. H. H orting, J. Am. Chem. Soc., 64, 828 (1942).
[206] Ссылка [198], стр. 21.
{207] Org. Synth., 20, 16 (1940).
[208] Org. Synth., 20, 73 (1940).
[209] M. Rohrlich, Chem. Ztg., 80, 847 (1956).
[210] K. Clnsius, 11. Hurzeler, Helv. chim. acta, 37, 383 (1954).
[211] Org. Synth., Coll. v. 3, 1955, p. 710.
[212] c. K. Banks, О. M. Gruhzit, J. Am. Chem. Soc., 70, 1268 (1948).
[213] А. В. В 0 e s e et al., J. Am. Chem. Soc., 53, 3520 (1931).
[214] A. Key, P. K. Dutt, J. Chem. Soc., 1921, 2088; 1928, 2035; H. Brets-
chneider, H. Rager, Mh. Chem., 81, 970 (1950).
[215] E. Noelting, O. Michel. Ber., 26, 86 (1893).
1216] A. Darapsky, Ber., 40, 3033 (1907).
542
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
8) из арилдиазовиевых солей и галогенаминов [217] (выходы 50 — 80% от тооре
тического).
Феинлазид (из пербромида диазония и аммиака) [210]. К 15 мл 21%-ног<
раствора аммиака приливают 10 мл эфира и при охлаждении льдом и хорошее
перемешивании небольшими порциями добавляют 4,05 г пербромида диазония
Аммиак и пербромид моментально реагируют на границе раздела водного и эфир
ного слоев, и образующийся азид растворяется в эфире. Эфир отгоняют в вакуу-
ме. а реакционную массу, состоящую из двух фаз, перегоняют с водяным паром
Дистиллят, содержащий аммиак, подкисляют разбавленной H2SO4, экстраги-
руют азид эфиром и перегоняют в высоком вакууме, собирая азид в приемнике
охлаждаемом сухим льдом. Выход 0,775 г (55% от теоретического).
Пербромид фенилдиазония готовят по способу, приведенному в практи-
куме Гаттермана и Виланда [218].
Фенилазид (из фенилгидразина и азотистой кислоты) [211]. К смеси
300 мл воды и 58,5 мл концентрированной НС1 прн охлаждении смесью льда
и соли и перемешивании в течение 10 мин приливают по каплям 33,5 г чистей-
шего фенилгидразина. Продолжая перемешивание, добавляют при 0° С 100 лм
эфира и медленно приливают по каплям свежеприготовленный раствор 25 а
NaNO2 в 30 мл воды с такой скоростью, чтобы температура не поднималась
выше 5° С (20—30 мин). С водяным паром отгоняют 400 мл жидкости, отде-
ляют эфирный слой, а водный слой встряхивают с 25 мл эфшра. Объединенные
эфирные вытяжки сушат над 10 г СаС1а и при пониженном давлении отгоняют
эфир на водяной бане (26—30° С). Затем понижают давление до 5 мм рт. ст.
и осторожно отгоняют фенилазид на водяной бане, температура которой около
65° С; выход продукта 24—25 г (65—68% от теоретического); т. кип. 49—50° С
(5 .«at рт. ст.).
Арилазиды (из диазониевых солей и и-толуолсульфамида). Общая мето-
дика [214]. Растворяют 1 л1?ль ароматического моноамина (0,5 моль диамина)
в 2,5 моль концептрировяттиой НС1 и диазотируют полученный раствор или
суспензию гидрохлорида амина рассчитанным количеством NaNO2. Раствор
соли 'диазония смешивают с раствором 1 моль п-толуолсульфамида и 5 моль
NaOH при охлаждении до —5° С и оставляют на 1—2 суток. Для выделения
продукта реакции доводят pH раствора до 8, продукт экстрагируют эфиром
(твердые азнды сначала отфильтровывают и затем экстрагируют их из маточных
. растворов). Перегонку ведут так, как описано выше.
Арилазиды чрезвычайно сильно взрывают * при нагревании!
Ацилазиды получают действием азотистой кислоты на гидразиды кислот:
RCONHNIb Н™!- RCON3
Реакцию проводят аналогично диазотированию первичных аминов.
п-Нитрофенилацетазид [219]. Растворяют 12 г соответствующее» гидр*
азида в 2 г воды, к которой добавлено 8 мл концентрированной НС1. При 20° С
и перемешивании добавляют 5 г NaNO2. Выделившийся азид отфильтровывают;,
выход 83,5% от теоретического.
Как правило, ацилазиды получают из галогенангидридов кислот и азида натрия.7
Для этого раствор ацилгалогенида в ацетоне, эфире или диоксане прибавляют к вод-,;
ному раствору азида патрия при температурах 0—25и С. Реакция протекает с очень*
хорошими выходами [220].
♦ Осторожность требуется п при работе с описываемыми ниже ацилазидами. —)
Прим. ред.
[217] Герм. пат. 456857, S. Skraup, К. St einruck (16.2.1928); FrdL, 16,<
452 (1931).
[218] Ссылка [1], стр. 250.
[219] R. L. Shin*o, J. М. Cross, J. Am. Chem. Soc., 60, 2339 (1938).
[220] E. W. Barrel, J. Am. Chem. Soc.. 63, 3434 (1941).
IV. ОБРАЗОВАНИЕ N—N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
54
I
5. Получение гидразинов
Наряду с уже известными методами получения монозамещениых или несимед
гричыых дизамещенных ароматических и алифатических гидразинов восстановление
солей диазония и ннтро.заминов, препаративное значение имеет и перенесенный в
алкил- н ариламины синтез гидразинов из амина и N-галогенамнна по методу Рашиг»
В последнее время особенно подробно исследовалось получение алкилгидразинов [221
Широкое применение может найти, например, такая методика.
Алкилгидразины. К холодному раствору 0,04 моль хлорамина, NHjC]
в 250 мл воды, полученному из аммиака и NaClO, прибавляют 0,25 г желатин
и соответствующий амин (молярное отношение хлорамина к алккламину 1 : 8
В течение 1 ч температуру реакционной массы повышают до комнатной, зате
смесь 10—30 мин нагревают на водяной бане. Образующийся алкилгидрази
можно выделить двумя путями:
избыток амипа и часть воды отгоняют и подкислепием остатка выделяю
гидрохлорид или сульфат алкилгидразина;
реакционную массу подкисляют уксусной кислотой, добавляют бензалх
догвд, экстрагируют эфиром, к эфирному раствору добавляют щавелевую ки<
лоту (дигидрат) и отгоняют бензальдегид с водяным паром. При упаривантх
остатка получают оксалат алкилгидразина.
Выходы алкилгидразинов составляют 40—60% от теоретического.
Таким же методом из вторичных аминов можно получать несимметричные дв
замещенные/гидразипы [222]. Третичные амины реагируют с хлораминами с образова
нием 1.1,1-триалкилгидразописвых солей [223].
Арилгидразины можно синтезировать из арпламинов и галогенамнпов [224]
но бодее удобно синтезировать эти соединения восстановлением диазониевых солей
В другом очень хорошем методе получения алкилгидразинов [53. 225] приме
Нили вместо хлорамина гидроксиламин-0-сулъфокислоту (NH2 —О—SO3H) и получал-
Продукт с выходом около 50% от теоретического. Этот метод был использован дл,
синтеза мпогих арплгидразпнов. Ниже приведена типичная пропись проведени.
•синтеза.
п-Амилгидразин [225]. Смешивают 180 мл воды. 47,5 г (0,55 .ноль
«-амиламина и 9,4 г КОН (образуются два слоя). Эгу смесь нагревают в колб
с обратным холодильником при перемешивании и в течение 30 мин приливаю
к ней по каплям раствор 9,9 г (0,083 моль) гидроксиламин-О-суЛьфокислот*
в 50 мл холодной воды. Реакционную массу охлаждают и подкисляют 70 м.,
ледяной уксусной кислоты; при атмосферном давлении отгоняют от смссн 50 м..
жидкости. После охлаждения отфильтровывают неорганические соли, фильтра,
нагревают до 50° С и перемешивают в течение 10 мин с 9 г бензальдегида
Образующуюся эмульсию охлаждают и трижды встряхивают с 50 мл эфир^
Объединенные эфирпые вытяжки смешивают с раствором 20 г дигндрата щаве
левой кислоты в 100 мл воды и после добавления небольшого количества актиа
ного угля перегоняют с водяным паром до тех пор, пока пе прекратится отгонк;
бензальдегида. Остаток фильтруют и охлаждают; при этом выделяется белы!
осадок оксалата и-амилгидразина, который отфильтровывают. Фильтрат упара
вают до объема 20 мл и получают таким путем еще некоторое количество окса
лата. Оксалат перекристаллизовывают из 150 мл этилового спирта. Выход
продукта 5 г (31% от теоретического).
[221] L. F. A u d г i е t li, L. Н. Diamond,!. Am. Chem. Soc., 76, 4869 (1954^
77, 3131 (1955); G. M. О m i et a nski, A. D. Kel m ers et al., J. Am.
Chem. Soc., 78, 3874 (1956); E. S c h m i t z, D. Habisch, Chem. Вег., 9Ь
680 (1962): E. Schmitz, R. Ohm e, Angew. Chem., 73, 220 (1961).
[222] R. A. Rowe, L. F. Audrieth, J. Am. Chem. Soc., 78, 563 (1956),
1223] g. M. Omietanski, H. H. S i s 1 e r, J. Am. Chem. Soc., 78, 1211 (1956>
[224] франц, пат. 735020; О. T. К ref ft, 2, 1, 1932.
[225] G. Gever, К. Hayes, J. Org. Ghem., 14, 813 (1949).
544
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Гидроксвламин-О-сульфокнслота [295, 226]. К 13 г сульфата гидроксщ
амина очень медленно прибавляют 30 мл хлорсульфоновой кислоты, приче{
сульфат гидроксиламина почти полностью растворяется. При нагревании смев
до 100° С выпадает гидроксиламин-О-сульфокислота. Смесь нагревают ыд
5 мин, затем реакционную массу охлаждают в эксикаторе и-осторожно вно^я
ее в абсолютный серный зфнр, охлажденный льдом. Смесь тщательно растирая
до образования тонкой взвеси, отфильтровывают на воронке Бюхнера суш!
в эксикаторе над Р2О5. Выход 95% от теоретического. *
При окислении днарнламинов легко образуются N—N-связи тетраарнлгидраяй
нов. В качестве окислителей могут применяться двуокись свинца в эфире [22м
КМпСЬв ацетоне [228] или, лучше всего, Ag,0 в смеси пиридина и эфира [229].
В практикуме Гаттермана и Виланда [230] приводится метод получения тет>||
фенилгндразина окислением дифениламина.
6. Получение диазоамииосоединешш
Диазониевые соли реагируют с первичными и вторичными ароматический
аминами с образованием диазоаминосоединений:
R'N2X+NH2R R'“N=N-NHR + ПХ .
Часто эти соединения образуются как нежелательные побочные продукты nJ
проведении диазотирования в недостаточно кислой среде. У желтых, хорошо кристЯ
лизующихся диазоаминосоединений основные свойства слабо выражены; оин образу!
с кислотами соли, но могут образовывать соли и с металлами, обменивая атом водород
связанный с азотом. у;
Диазоаминосоединения получают: . J
1) сочетанием диазониевых солей с первичными и вторичными аминами в в]
ферном растворе [231, 234]; ’
2) действием нитрозных газов (алкилнитритов) на спиртовой раствор coJH
первичных аминов;
3) действием нитрозаминов на первичные амины; м
4) смешанные диазоаминосоединения образуются при действии реактв#в
Гриньяра на арилазиды [232].
При действии диазониевых солей на диазоаминосоединения в спиртовом расгвЙД
в присутствии метилата натрия образуются бис-диазоаминосоединения [233]: .з
Ar-N=N
>N-Ar
Ar-N=N/ лЦ
Диазоаминосоединения разлагаются минеральными кислотами на исход
компоненты: диазониевую соль и амин. В слабокислой среде они могут перетрут
ровываться в аминоазопроизводные [235].
[226]
227
228
229
230
231
232
233
234
235
F. Sommer О. F. Schulz М. Nassau, Z. anorg. allg. fiheM
147, 142 (1925). ' •
H. Wieland, Вег., 45, 2602 (1912). • i
Н. Wieland, Вег., 40, 4270 (1907). 1
Н. Wieland, Вег.,-53, 1322 (1920). 4Я
Ссылка [1], стр. 308.
О. Dim? о th, Вег., 38, 2328 (1905). „ая
О. Dimroth, Вег., 36, 909 (1903); 38, 670 (1905).
Н. v. Pechm-ann, Вег., 27, 703 (1894). -У
Org. Synth., Coll. v. 3, 1955, р. 163. ':.а
Р. Ruggli, Courtin, Helv. chim. acta, 15, 90 (1932). -.’д
IV. ОБРАЗОВАНИЕ N—N-СВЯЗН В РЕЗУЛЬТАТЕ ОБМЕНА
545
7. Получение азосоединений
Азосоединения, R — N = N—R, можно получать следующими способами:
1) по реакции азосочетания (стр. 400);
2) восстановлением нитро-, нитрозо- н азоксисоединений;
3) конденсацией нитрозосоединенин с аминами;
4) окислением гидразинов и аминов;
5) из N-арилгидроксиламинов и тиониларнламииов [252];
6) перегруппировкой диазоаминосоедииеинй; ,
7) разложением диааониевых солей в присутствии солей одновалентной меди;:
8) диспропорционированием гидразосоединений;
9) сочетанием дназониевых солей с алифатическими диазосоединениями. По-
следним способом можно получить аралифатические азосоединення; эта реакция проте-
кает с отщеплением азота алифатической диазогруппы.
Восстановление нитро-, нитрозо-и азоксясоедияе-
ний (способ 2). При соответствующем подборе восстановителя восстановление нитро-
соединений можно остановить на стадии образования азопроизводных. Так, например,
при восстановлении нитробензола цинковой пылью в щелочной среде получают азо-
бензол с выходом 86% от теоретического [236]. Хорошие выходы азосоединений до-
стигнуты также с алюмогидридом лития [237]. Галогензамещеиные нитросоединения
превращают в азосоединения по реакции с гидразином в спиртовом растворе в присут-
ствии палладиевого катализатора [241]. Нитросоединеиня восстанавливаются до азо-
производных также хлоридом олова (II) [240], амальгамой натрия [240], сульфидами
щелочных металлов в сильнощелочной среде, электрохимическим методом [242],
натрийэтоксиалюминийгидридом [259], а в некоторых случаях даже метилани-
лином [243] и фенилгидразнном [244]. Азоксибензол превращается в азобензол нрн
перегонке с железными стружками [245]. Азоксисоединения, образующиеся в ка-
честве промежуточных продуктов при восстановлении нитросоединений, становятся
основными продуктами при использовании в качестве восстановителя метилата натрия
[238] или арсенита натрия [239], выходы оноло 85% от теоретического:
. 4RNO24-3CH3ONa —> 2R—N=N— R + 3HC0ONa-f-3H2O
Азобензол [236]. Раствор 250 г нитробензола в 2,5 л метилового спирта
смешивают с раствором 325 з NaOH в 750 дистиллированной воды; затем7
добавляют 288 г цинковой пыли и при перемешивании нагревают 10 ч с обрат-
ным холодильником. Смесь фильтруют в горячем состоянии и отфильтрованный
цинкат натрия промывают небольшим количеством горячего метилового спирта.
Отгоняют метиловый спирт, охлаждают и отфильтровывают выкристаллизо-
вавшийся .азобензол. Следы солей цинка извлекают из продукта нагреванием
его до 70° С с 500 мл 2%-ной НС1; выход 160 з (86% от теоретического); т. пл.
66-67,5° С.
Конденсация ариламинов с арилнитрозопроизвод-
и ы м и (способ 3). Проведение реакции в ледяной уксусной кислоте или в пиридине'
приводит к образованию азо- или полиазосоединения, часто с хорошими выходами
(около 80% от теоретического). Реакция, как правило, протекает уже прн непродол-
жительном нагревании исходных веществ [246].
Org. Synth., 22, 28 (1942).
К. F. Nystrom, W. G. Brown, j. Am. Chem. Soc., 70, 3738 (1948).
H. S. Fry, I. L. Cameron, J. Am. Chem. Soc., 49, 864 (1927).
Org. Synth., Coll. v. 2, 57 (1943).
O. N. Witt, Ber., 18, 2912 (1885).
M. Busch, K. Schulz, Ber., 62, 1458 (1929).
Ссылка 2, стр. 186.
E. Bamberger, M. Vuk, Ber., 35, 713 (1902).
C. Mills, J. Chem. Soc., 67, 925 (1892).
Schmidt, Schulz, Ann., 207, 329 (1881).
P. Ruggli, C. Petitjean, Helv. chim. acta, 21, 711 (1938).
35 Заказ 183Ь.
•546 ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Окисление гидразинов (способ 4). По этому способу легко и просто '
образуются ароматические, алифатические и смешанные азосоединения. В присутствии
щелочей ароматические гидрааосоединения дегидрируются до азосоединений уже под.
действием кислорода воздуха [247]. В качестве окислителей можно использовать
также гипобромиты [247], окись ртути н перманганат калия. Для дегидрирования
1У,М'-диметилгидразина применялся нейтральный раствор хромата калия [248] или
хлорид меди (it) в ацетатном буфере [249]. В работе [249] сообщается о взрыве, про- '
исшедшем при употреблении азометана, СН3— N=N—СН3. Для дегидрирования эфи-
ров гидразодикарбоновых кислот применялась концентрированная азотная кислота
[254]. Симметричные азосоединения можно получать также окислением первичных
ароматических аминов. В качестве окислителей применяли надкислоты [250],
феррицианид калия [251], гипохлорит натрия [252|, но при этом всегда полу-
чалось значительное количество смол («анилиповый черный»).
II з N-a рил гидрокси л амипов и т и они л а р и л амин ов
(способ 5) с очень хорошими выходами (около 90% от теоретического) образуются азосо-
единения (метод Михаэлиса) [253]
ЗАг— NHOIl + Ar'N=SO —► Ar—N=N—Аг’+Аг— NHSOgH+ArNIk+HgO
При разложении диазониевых солей (способ 7) с электроне- •
акцепторными заместителями в ядре в присутствии солой меди (I) отщепляется азот I
и образуются азосоединения [255]. *
Диспропорционированием гидразобензола (способ 8)
при повышенной температуре получают анилип и азобензол [247].
Азосоединения являются сравнительно устойчивыми обществами. Многие из них
перегоняются без разложения. Они не изменяются под действием кислот « щелочей, ’
могут непосредственно бронироваться, нитроваться и сульфироваться. Азосоедн-
нениц существуют в цис- и mpa'tc-формах. Более стабильная транс-форма переходит
в ifuc-форму при облучении коротковолновыми лучами [256]. Для разделения цис- н
транс-форм используют метод хроматографии на окиси алюминия [257, 258],
8. Получение алифатических диазосоединепий
В отличие от арнламинов алкиламины не диазотируются. Поэтому для получения
алифатических диазосоединений наряду с некоторыми специальными методами большей
частью используют косвенный путь через питрозоацплалкиламины, от которых в ще-
лочной среде отщепляется ацильная группировка [260]. Алифатические диазосоедине-
пия можно получать:
1) из нитрозоалкилуретанов [261]:
СНз—N—СООС3Нд-]-2КОН —> CHsNs 4“ К2СО3 4- С2Н5ОН Н8О
I
NO
[247]
248]
249]
250]
251]
252]
253]
[254
[255
[256
[257
[258
[259
[260
[261
Ссылка \ стр. 163.
J. Thiele, Вес, 42, 2578 (1909). ,
F. Р. Jahn, J. Am. Chem. Soc., 59, 1761 (1937)'. i
N. Prileschaycw, Ber.. 42, 4815 (1909).
E. Bamberger, F. M eimberg. Ber., 26, 497 (1893). 4?
E. Bamberger, Ber., 31, 1523 (1898). , -
A. M ichael is, К. P e t on, Ber., 31, 984 (1898); H. E. Fierz-David,’
IIclv. chim. acta, 34, 846 (1951). *
O. Diels, D. Fritsche, Ber., 44, 3018 (1911).
В. M. Bogoslowski, 7. S. Kazakova, C. A., 46, 2003 (1952). J
R. Huisgen, Angew. Chem., 65, 40 (1953).
Ссылка г, стр. 164. J
A. H. Cook, J. Chem. Soc.. 1938, 976; 1939, 1309.
G. Hesse, R. Schrodel, Ann., 607, 24 (1957).
R. Huisgen, I. R ein ertsh of er, Ann., 575, 174 (1952).
H. v. Pcchmann, Ber., 27, 1888 (1894).
IV. ОБРАЗОВАНИЕ N -N-СВЯЗИ В РЕЗУЛЬТАТЕ ОБМЕНА
547
2) из нитрозоалкилмочевин [262]:
СНз-N—C-NH2 + 2KOH —> CH2N2 + K0CN + 2H2O
I II
NO О
3) из М-метил-М-нитрозо-п-толуолсульфамида [264]:
CH3C6H4S02N-CH3 + C2H50- —> CH2N2^CH3CeH4SO2C2H5+nO-
i
NO
4) из 1У-алкил-ЬГ-нитрозо-Х'-нитрогуанидинОв [265]:
CH3N-C-NHNO2 + KOH —> CH2N2+K[NCNN02]+H20
I 1!
ON NH
5) из нитрозометиламиноизобутилметилкетона [263]:
СНз СНз
СНз-N—с-СН2СОСН3 СНгМ2-гС=СНСОСНз + Н3О
I I I
ON СНз СНз
6) окислением гидразонов альдегидов и кетонов [266]:
R2C=N-NH2 -----RiCNs+HeO + Hg
7) действием спиртового раствора КОП на хлороформ и гидразин [270]:
CHCI3-I-H2N-NH24-3KOII —> CH2N2 + 3KCI4-3H2O
Химия дназоалканов, представляющая большой практический и теоретический
интерес, хорошо освещена в обзорных работах [267—269].
Все алифатические диазоеоединения ядовиты и окрашены в цвета от желтого
до пурпурно-красного. Низшие диазоалканы газообразны при комнатной температуре
и исключительно взрывоопасны; высшие представляют собой жидкости или твердые
вещества и гораздо более устойчивы. На основании сравнительных исследований
скорости разложения диазолканов кислотами, алифатические диазоеоединения
[262] Е. A. Werner, J. Chem. Soc., 115, 1093 (1919); Org. Synth., 15, 3 (1935);
F. Arndt, 1. Amende, Angew. Chem., 43, 444 (1930); F. Arndt,
H. Scholz, Angew, Chem., 46, 47 (1933).
[263] Org. Synth., Coll. v. 3, 244.
[264] Rec. trav. chim,, 73 , 229 (1954); Org. Synth., 34, 96 (1954).
[265] A, F, McKay, Chem, Rev., 51, 331 (1952); A. F. M с К a y, J. Am. Chem.
Soc., 70, 1974 (1948); J. Am. Chem. Soc., 71, 1968 (1949).
[266] Th. C ur t i us et al., Ber., 44, 2197 (1911); 49, 1897 (1916); 49, 19231(1916);
49,1928 (1916); 49,1951 (1916); Org. Synth., Coll. v. 3, p. 351; W. Schroeder,
L. Katz, J. Org. Chem., 19, 718 (1954).
[267] В. E is tert, Angew. Chem., 54, 99, 124, 308 (1941); 55, 118 (1942); 61, 185
(1949).
[268] Org. Reactions, 8, 364 (1954).
[269] R. Hnisgen, Angew. Chem., 67, 439 (1955).
[270] H, Staudinger, O. Kupfer, Ber., 45, 501 (1912).
35*
548
ИЗМЕНЕНИЕ АЗОТНОЙ ФУНКЦИИ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
расположены в следующий ряд по уменьшающейся основности, т. е. ио снижению
реакционной способности по отношению к кислотам [271]: -м
II СПз 1 c6n5 1 CeH4x
СНгХ*»- CNo > cn2 > CN.-> > 1 3N2 >
1 ' 1 ceHZ
CH3 ceH6 CgHg
ceHa H H COC9H6 COOR
> cn2 > 1 • CN2 > . CN3 > CN2 > j CN2
COCeII5 COOR COCgHs COC6H5 COOR
Диазодикарбонильные
,, . .„ г__________соединения, расположенные в конце этого ряда, очеизд
устойчивы к действию кислот. Диазометан количественно разлагается слабыми кис-д
лотами по уравнению: ' j
CeH5COOH+CH»N2 —> CeHsCOOCHs + N-i ’J
Эта реакция может применяться для количественного определения содержания*
диазометана в его растворах [272]. Кислоты могут вызывать разложение диазоалканов,1
не вступая в реакцию, т. е. оказывают чисто каталитическое действие. Это всегда на-й
блюдаетс.я в тех случаях, когда промежуточно образующийся ион карбонил соеди^
няется не с анионом кислоты, а с растворителем, содержащим гидроксильную группу^
Поскольку вода является более нуклеофильным агентом, чем ионы сульфата или пер^я
хлораза, разложение диазоуксусного эфира водной хлорной кислотой протекает^
например, полностью по каталитической схеме:
НС1О4-|-НгО —> H30+ + C10j I
H3O+ + :N=N~CH-COOR 7^ Н2О+ :N=N-CH2-COOR ——
•—► H2O+CH2-COOR —> Н26—СН2—COOR НО—СН2—COOR-f-H2O 'Д
Циклические изомеры диазоалифатических соединений содержат ненасыщенной
кольцо из трех членов с С—N- и N — N-связями; они бесцветны и устойчивы к дедам
-ствию кислот [276а]. . Я
Диазометан [272]. К 100 мл эфира, налитого поверх 30 мл сильно охлЙй|
ждепного 40%-ного раствора’КОН, небольшими порциями прибавляют 10ав|
нитрозометилмочевипы. Поддерживая температуру 5° С, встряхивают смёайа
10 мин. После этого сливают темно-желтый эфирный раствор и сушат его 3»яЯ
пад твердым КОН *. Раствор диазометапа можно хранить некоторое времИ
в холодном затемненном месте в сосуде, закрытом пробкой с капилляром; диазм|
метан медленно разлагается с выделением азота (ср. метод его получения [272аШ|
Днфенилдиазометан [273J. В сосуде для работы под давлением в те«ед|
яие 75 мин встряхивают смесь 13 г (0,066 моль) ^гидразона бензофенона, 15*»
обезвоженного Nd2SO4, 200 мл эфира, 35 г желтой (нлн красной) HgO и 5 JHgi
(при использовании красной HgO 10 мл) насыщенного раствора КОН в этилДД
вом спирте. Реакционную массу фильтруют и отгоняют растворитель в вакуумдД
при комнатной температуре. Полученное темно-красное масло растворгЕЯМН
в петролейном эфире (т. кип. 30 — 60° С), раствор фильтруют и отгоняют раствйИ
* Не следует употреблять шлифы! Для сушки рекомендуется гранулировав
ный КОН (Синтезы органических препаратов, сб. 12, пер. с англ., 1964
стр. 25). — Прим. ред. , _ , '
[271] Н. Staudinger, A. Ganle, Вег., 49, 1897 (1916).
[272] Ссылка х, стр. .615.
1272а] Org. Synth., Coll. v. 3, 244.
,[273] I. В. Miller, J. Org. Chem., 24, 561 (1959).
V. ОБРАЗОВАНИЕ СВЯЗИ N—Hal
549
ритель в вакууме при комнатной температуре. Полученное масло охлаждают
сухим льдом н дают ему постепенно нагреться до комнатной температуры;
при этом получаются темно-красные кристаллы; выход 89% от теоретического;
т. пл. 29—32° С (ср. также ]273а]).
Этиловый эфир диаэоуксусной кислоты [274]. При работе с этим ядови-
тым и взрывоопасным эфиром необходимо соблюдать исключительную осто-
рожность! В четырехгорлой колбе емкостью 2 л, снабженной мешалкой, капель-
ной воронкой, термометром и газоподводящей трубкой, к раствору 140 г (1 моль)
-гидрохлорида этилового эфира гликоля в 250 мл воды прибавляют 600 лл
.могиленхлорида и смесь охлаждают до —5° С. К полученной смеси в атмосфере
азота при перемешивании добавляют охлажденный раствор 83 г (1,2 моль)
NaNO2B250«rf воды, понижают температуру до —9° Сив течение 3 мин при-
ливают по каплям 95 г 5%-ной H2SO4. Температура реакционной массы при
этом не должна подниматься выше -|-10 С (температура в охлаждающей бане
— 23° С). Реакция закапчивается через 10 мин. Реакционную массу переносят
в предварительно охлажденную делительную воронку емкостью 2 л, отделяют
воду и тщательно перемешивают золотисто-желтый органический слой с 1 л
холодного 5%-него раствора NaHCO». Продукт из водного слоя экстрагируют
75 мл метиленхлорида. Экстракт объединяют с основным продуктом, интен-
сивно встряхивают с раствором NaHCO3 и сушат над 15 г обезвоженного
XazSO4. Основное количество растворителя отгоняют через колонку при
остаточном давлении 350 мм рт. ст., остаток отгоняют при 20 мм рт. ст.',
температура бани при этом нс должна превышать 35° С. Получается 90—100 г
(79—88% от теоретического) желтого масла; п$ = 1,4б2. Продукт достаточно
чист для большинства синтезов, но может быть дополнительно перегнан
с водяным паром в вакууме [275].
Разработан [276] безопасный метод получения больших количеств этого
эфира из эфира Т^-интрозо-М-ацетилглнцина,
V. Образование связи N—Hal
• Органические N-галогспамины открыты в 1850 г. Вюрцсм [277]. Для получения
соединений этого класса Вюрц пропускал хлор или соответственно бром в жидкий '
алифатический амин. При таком способе работы, пригодном только для получения
дигалогенаминов, две трети амина связывались в виде соли и терялись:
3RNH2+2CI2 —>• 2RNH2-1IC1 + RNC12
Последующие исследователи устранили этот недостаток, применив соли хлор-
новатистой кислоты. Как правило, реакцию проводят, действуя на амины гипохлори-
тами щелочных металлов:
RNH2 + 2NaOCl —RNClj-|-2NaOH
При получении бромпроизводных употребляют раствор брома в щелочи. По-
скольку выходы и устойчивость образующихся моно- или дигалогепированных аминов
зависят от pH среды [278], их рекомендуется синтезировать по методу Аллигера [279],
который хлорировал первичные амины до мопогалогенамннов при температуре ниже
10° С и pH меньше 8,5. Дигалогеналкиламины и диалкилгалогенампны наиболее устой-
чивы в среде, близкой к нейтральной; поэтому их получают обычно в буферных рас-
творах (бикарбонат [280], эквивалентные количества ацетата натрия и уксусной ннс-
лоты [281]). Разложение образующихся галогенаминов часто предотвращают, удаляя
[273a] Org. Synth., 24, 53 (1944).
[274] Org. Synth., 36, 25 (1956).
1275] Org. Synth., 24, 53 (1944).
[276] H. Reimlinger, Angew. Chem., 72, 33(1960); Chem. Ber., 93, 2162(1960).
[276a] E. Schmitz, R. Ohme, Chem. Ber., 94, 2166 (1961); 95, 795 (1962).
1277] A. Wurtz, Ann. Chimie ]3J, 30, 443 (1850).
[278] I. W e i 1 I. C. Morris,!. Am. Chem. Soc., 71, 3123 (1949).
[279] G. A 11 i g e г, пат. США 2459759 (18.1.1949); G. E. P. Smith jr., G. A 1-
liger et al., I. Org. Chem., 14 , 935 (1949).
[280] L. K. Jackson et al., J. Am. Chem. Soc., 69, 1539 (1947). .
[281] F. D. Chat t away, I. Chem. Soc., 87 , 381 (1905).
550
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
их из реакционной среды с помощью органических растворителей (эфир [282], лиг-
роин [283], хлороформ [284, 285] или четыреххлористый углерод).
При получении галогснаминов применяются также органические переносчики
галогенов. Так, например описано получение N-бромаминов при помощи N-бромамн-
дов [286], дихлормочевины [287] и ЬГ-хлорэтилкарбамата [288]. Для М-аминохлори-
рования в органических растворителях с успехом используются эфиры хлорноватистой
кислоты с этиловым [289] и трет-бутиловым [290] спиртами, а для N-хлорирования
очень неустойчивых и труднодоступных аминов стероидного ряда было предложено
применять N-хлорсукцинимид [291].
Как правило, N-галогснамины являются тяжелыми жидкостями с отвратитель-
ным запахом. Некоторые из них довольно устойчивы, другие, как сообщают, бурно
разлагаются, ипогда даже со взрывом. N-Галогенариламины [292] можно сохранять
в течение нескольких иней только в растворах при температурах около —70® С. Моио-
галогенамины тоже легко разлагаются, причем образуются дихлорамины и амины:
2RNHC1 ---> RNCI2 + RNH2 /
Такое диспропорционирование легче протекает в присутствии разбавдеппмт
кислот, тогда как концентрированная соляная кислота окисляется всеми хлораминами,
до хлора. Для разложения галогенаминов применялись и другие восстановители Н1г
NaaSO3 и H2S. В последнее время появились две обзорные работы по синтезам на основе
N-галогенаминов [293— 297].
ОБРАЗОВАНИЕ СВЯЗЕЙ УГЛЕРОД-СЕРА *
ОБРАЗОВАНИЕ C-S-СВЯЗИ В РЕЗУЛЬТАТЕ
РЕАКЦИИ ПРИСОЕДИНЕНИЯ
Z. Присоединение соединений серы по кратным углерод-углеродным
связям
1. Присоединение сероводорода и его производных
Присоединение сероводорода и меркаптанов по С = С-связям в отсутствие инн*
циаторов происходит предпочтительно по правилу Марковникова:
Ч /Н I I /
>С=--С< +П28 —> >С—С<
Г \r- R/ 4V
Материал обработал Д. Мартин.
Н. Кижпер, ЖРФХО, 31, 872 (1899).
Org. Synth., Coll. v. 3, 1955, *р. 159.
G. N. R. Smart, G. F. Wright, J. Am. Chem. Soc., 70, 3141 (1948).
J. S t i e g 1 i t z, I. V 0 s b u r g h, Ber., 46, 2151 (1913).
T. Seliwanow, Ber., 26 , 423 (1893).
R. L. Datta, J. Chem. Soc., 101, 166 (1912).
R. L. D a t t a, S. D. Gupta, J. Am. Chem. Soc., 36, 386 (1914).
St. Goldsch m id t et al., Ber., 58, 572 (1925).
H. Zimmer, L. F. Audrieth, J. Am. Chem. Soc., 76, 3856 (1954).
II. Rusch ig et al., пат. ФРГ 896803 (16.11.1953).
St. Goldschmidt, Ber., 46, 2728 (1913); 55, 2450 (1922).
D. J. S t a n n 0 n i s, J. Org. Chem., 22, 475 (1957).
W. D. Emmons,!. P. F r e e m a n, Angew. Chem., 67, 349 (1955).
R. G 0 s 1, A. Meuwsen, Ghem. Ber., 92, 2526 (1959).
W. The Slacker, E. Wegner, Angew. Chem., 72, 127 (1960).
E. Schmitz, Angew. Chem., 73, 23. (1961).
I. ИРИСОЕДППЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПО КРАТНЫМ С-С-СВЯЗЯМ
551
Однако под действием Уф-облучения или в присутствии перекисей присоедине-
ние происходит против правила Марковникова [1]. В лабораторной практике присо-
единение сероводорода имеет препаративное значение только в том случае, если воз-
можно проведение реакции в жидкой фазе. При этом используются главным образом
основные катализаторы. Так, из кротоновой кислоты п сероводорода в присутствии
диэтиламипа при 70° С была получена р-мсркаптомасляная кислота [2], а из окиси
мезитила и сероводорода в присутствии триэтиламина — метил-р-меркаптоизобутил-
кетон с выходом 80% от теоретического [3].
На первой стадии реакции происходит присоединение сероводорода к ненасы-
щепнол?у соединению, затем, особенно при избытке олефина, образуются тиоэфиры [4].
Р,Р'-Тиодипропионовая кислота [5]. Насыщенный сероводородом рас-
твор 40 ч. NaOH в 100 ч. воды смешивают с раствором 94 ч. акрилата натрия
в 130 ч. воды и после добавки 0,1 ч. гидрохинона нагревают в течение 20 ч.
при 100° С. После подкисления реакционной смеси получается р,р'-тиодипро-
пионовая кислота.
Меркаптаны и тиофенолы присоединяются по С = С-связям легче, чем серово-
дород, причем реакционная способность этих соединений по отношению к двойной
связи снижается в ряду тиофенолы ’> первичные меркаптаны вторичные мер-
каптаны ’> третичные меркаптаны [6].
Так как S —Н-связь мало поляризована и гетеролиз се на протон и меркаптид-
ной затруднен 17]. присоединение к олефинам SH-групп происходит преимущественно
по радикальному механизму в присутствии перекиси бензоила [8] или при УФ-облуче-
нии [9]. К сильцополярным двойным связям в а,0-нспасьгщенпых карбопильных со-
единениях, нитрилах или карбоновых кислотах и т. д. меркаптаны и тиофеполы могут
присоединяться под влиянием основных катализаторов и по ионному механизму, при-,
чем С—S-связь всегда образуется у карбонисвого иона, находящегося в р-положении:
R—S—H-рR'-CH- CH—С—R'
R'-CH-CH2-C-R''
О
Таким образом из бензилиденацетона и тиофепола был получен 0-фенилмерканто-
бензилацстон [10], а из метилмеркаптана и метмлвинилкетона — 4-метилмеркапто-
бутанон-2 [11].
4-Метилмеркаптобутанон-2. К 5,6 г метилвинилкетона, охлажденного
в смеси льда с солью, прибавляют 9 г метилмеркаптана и 0,5 г сухого К.2СОэ.
По окончании реакции выдерживают еще 15 мы-н при комнатной температуре,
затем К2СОд отфильтровывают на воронке Бюхнера и фильтрат перегоняют.
Выход 4-метилмеркаптобутанона-2 7,3 г (77% от теоретического); т. кип. 72—
74° С (18 .хм рт. ст.).
Присоединение меркаптанов к а,р-пенасыщепнкгм альдегидам лучше всего про-
водить в присутствии триэтиламина [12]. Однако из акролеина и метилмеркаптана
[1
[2
(3
[4
(5
16]
W. Е. Vaughan, F. F. Rust, J. Org. Chem., 7, 472 (1942).
R. Dahlbom, Acta Chem. Scand., 5, 697 (1951).
Z. F б I d i, J. Kolionitsch, J. Chem. Soc., 1948, 1683.
V. N. Ipatieff, B. S. Friedmann, J. Am. Chem. Soc., 61, 71 (1939).
Франц, пат. 845793 (1938); С., 1940, I, 1571.
M.S. К h а г a s c h, W. Nudenberg, G. J. M a n t e 11, J. Org. phem.,
16, 524 (1951); C. S. Marvel, R. R. Chambers, J. Am. Chem. Soc.,
70, 993 (1948).
[7] И. Л. К н у н я н ц, А. В. Фоки н, Усп. химии, 19, 545 (1950).
[8] С. Т. L е s t е г, G. F, R о d g е г s, Е. Е. R е i d, J. Am. Chem. Soc., 66, 1674
(1944).
19] B. Wei bull. Ark. Komi, 23A, № 18 (1947).
[10] S. Ruhemann, J. Chem. Soc., 87, 17 (1905).
[11] H. Bohme, P. H e 1 1 e r,f Chem. Ber., 86, 443 (1953).
112] J. R. Catch, A. H. С о о k et al., J. Chem. Soc., 1947, 1609.
552
ОБРАЗОВАНИЕ С—S-СВЯЗН В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
в присутствии ацетата меди был получен 0-метнлмеркаптопропионовый альдегид с вы—|
ходом 94% от теоретического [13]. '•*
Меркаптаны и тиофеиолы могут присоединяться также к а,0-неиасьпценнымй]
карбоновым кислотам и их эфирам [14]. Например, из коричной кислоты и тиофеиол^
синтезировали 0-февилмеркаптогидрокоричную кислоту [15].
р-Фенилмеркаптогндрокоричная кислота. Смесь 10 г коричной кислоты^
8 г тиофенола и 10 г насыщенного раствора НВг в ледяной уксусной кислоту
нагревают в течение 9 ч при 100° С в склянке для работы под давлением. ЗатШ
реакционную смесь разбавляют большим количеством воды и отгоняют непро?
реагировавший тиофенол с водяным паром. Выделившееся масло отделяю®!
кристаллизуют при охлаждении с внесением затравки и затем перекристаллИ*
зовывают из лигроина. Выход продукта составляет около 75% от теоретичен
ского; т. пл. 85—86° С. -’А
Присоединение меркаптанов к ненасыщенным дикарбоновым кислотам [16л
и меркаптокарбоиовых кислот к акриловой кислоте [17] удается осуществить уже
в водно-щелочной среде. ’л
Присоединение меркаптанов и тиофенолов к «^-ненасыщенным интрнлаж
(цианалкилироваиие) легко происходит в присутствии следов щелочей иногда уяи
на холоду [18]. Из p-алкилакрилонитрила и тиофенола получают 0-алкил-р-феишы
меркаптопропионитрилы [19]; из 1-цианциклогексена-1 и меркаптанов — 1-циа»И
2-алкилмеркаптоциклогексаны [20].
Меркаптаны могут реагировать также с винилхлоридом [21], винилацетатам
[22], а-нитроолефинами [23] и азлактонами [24]. При взаимодействии с азлактонайИ
происходит одновременное раскрытие кольца и образуются N-ацильные производим
тиоэфиров аминокислот: -а
Свп6—сн=с—C=:O + 2RSH
О
CeH5-СН-СН-COSR
1 I
S NH
С— Свн5
СО-СвН5
Хиноны [25, 26] реагируют с меркаптанами с образованием алкилмеркапт®
гидрохинонов, которые можно дегидрировать до алкилмеркаптохинонов действием »
бытка хинона или других ч
окислителей.
Ann. Soc. chim. Polonorum, 33, 217 (1959); С., 1961. 321
H. Brzozowski, 71U1L. out.. М111Ш. •- u„x, uu, , 1 fvi , <
C. D. H u r d, L. L. Gershbein, I. Am. Chem. Soc., 69, 2328 (1947).
F. Arndt, Ber., 56, 1269 (1923).
E. J. Morgan, E. Friedmann, Biochem. I., 32, 733 (1938).
A. Schoberl, A. Wagn e'r, Chem. Ber., 80, 379 (1947).
O. Bayer, Angew. Chem., 61, 229 (1949).
R. M. Ross, J. Am. Chem. Soc., 71, 3458 (1949).
[13
[14
[15
[16
[17
[18
[19] ................--- х-----------------
[20] R. М. R о s s, F. W. R a t h s, J. Am. Chem. Soc., 73, 129 (1951).
[21] W. H. C. R ueggeberg, W. A. Cook, E. E. R e i d, J. Org. uue®
13, 110 (1948).
[J2 ] W. H. C. R u e g g e Ь e г g J. C h e г m a c k et al., I. Am. Chem. Soc., ?
2292 (1948).
[23] R. L. Heath, A. Lambert, J. Chem. Soc., 1947, 1477. >1
[24] R. Ruhemann, J. Chem. Soc., 87, 461 (1905); В. И. N icolet, J.
Chem., 95 , 389 (1932); R. N e h e г, M. Spillmann et at, Helv. ehH
acta, 29, 1874 (1946); A. Butenand t, H. Jatzkewitz, P. FoueK
Hoppe-Seyler’s Z. physiol. Chem., 282, 268 (1947). Л-
[25] O. Dimroth/L. Kraft, K. Aichinger, Ann., 545, 124 (1940).
[26] M. Schubert, J. Am. Chem. Soc., 69, 712 (1947).
E. E. Reid,
N
R
1
1 ПРИСОЕДИНЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПО КРАТНЫМ С—С-СВЯЗЯМ
553
2-Метилмеркапто-1,4-нафтохинон [27]. Растворяют 16,5 г нафтохинона
в 1000 мл абсолютного спирта при 40° С. Раствор после охлаждения до комнат-
ной температуры фильтруют, затем охлаждают до 15° С и при перемешивании
сразу прибавляют 10 г холодного метилмеркаптана. Реакционную смесь выдер-
живают 30 мин при комнатной температуре, посяе чего добавляют 15 мл 70%-
ного раствора FeCl3 (считая на FeCl3-6H2O) и через 15 мин еще 22 мл того
же раствора. Полученную суспензию охлаждают до 7° С, фильтруют и осадок
промывают на фильтре петролейным эфиром. Выход продукта (желтые кри-
сталлы) 11,9 г (56% от теоретического); после перекристаллизации из спирта
т. пл. 186,5—187е С.
а-Меркаптокетоны сходны по свойствам с меркаптанами. Хотя в а-меркапто-
кетонах S —Н-группа поляризована сильнее из-за присутствия соседней карбонильной
группы, их присоединение происходит легче по радикальному механизму. Присоеди-
нение 2-меркаптопентанона-З [28, 29] к симметричным и несимметричным замещенным
олефинам в присутствии катализаторов, способствующих как ионному, так и радикаль-
-ному присоединению, приводит к а-кетосульфидам:
^С-С^Н-СНз—CII-C-C2H5 —> СНз-СН-С-СгНб
z \ II I II
HS О ЙО
\ I/
рсн-с^
З-Оксолеятил-2-циклогексилсульфид. Смесь 0,5 моль 2-меркаптопента-
нона-З, 0,5 моль свежеперегнанного циклогексена и 400 мг перевиси бензоида
нагревают в колбе с обратным холодильником в течение 8 ч с последующей
фракционной перегонкой в вакууме. Выход продукта 44% от теоретического;
т. кип. 89° С (2 мм рт. ст.).
В качестве олефиновых компонент для реакции с 2-меркаптопентаноном-З и
другими меркаптанами были использованы стирол, триметилэтилен, 1-метилцикло-
гексен~1, а также олефины, содержащие функциональные группы [30],—винилхлорид,
1-хлорциклогексен-1, а-хлоркротоновый альдегид, аллиламин, аллиловый и коричный
•спирты, аллилбромид и аллилхлорид.
Очень легко и с хорошими выходами к олефинам присоединяются сульфеиил-
хлориды с образованием 0-галогентиоэфиров:
RSC1 + CH2=CH2 —> RS—CH2CH2CI
0-Хлорзтплфенилсульфид [31]. В раствор 15 г фенилсульфеинлхлорида
в 80 мл СС14 без доступа влаги пропускают этилен до обесцвечивания раствора.
После перегонки в вакууме получают 14,6 г чистого 0-хлорзтилфенилсульфида;
т. кип. 122° С (13 мм рт. ст.).
Если С-^С-связь блокирована группами с сильным эффектом сопряжения, то
присоединение затрудняется или не идет совсем. Обзор всех областей применения этой
реакции см. [32].
При взаимодействии тиокарбоновых кислот с ненасыщенными соединениями
получаются эфиры тиолкарбоновых кислот, которые могут быть омылены до меркапто-
[27] J. Е. Little, Т. J. S р г о s t о n, М. W. F о о t е, J. Am. Chem.
Soc., 71, 1124 (1949).
(28] К. Riihlmann, D. Gramer et al., J. prakt. Chem. [4], 10 , 316 (1960).
.29] F. Asinger, M. Thiel, E. Pallas, Ann., 602, 37 (1957).
[30] K. Ruhlmann, U. Schrapler, D. Gramei, J. prakt. Chem. [4],
10, 325 (1960).
31] H. Lecher, Ber., 58, 414 (1925).
132] R. A, T urner, R. Connor, J, Am. Chem. Soc., 69, 1009 (1947).
554
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
карбоновых кислот [33]. Реакция с тиоуксусной кислотой всегда происходит так^З
что ацетилмеркапто-остаток присоединяется к р-углеродцому атому (по отношению, д
к карбоксильной группе): , ’
ВСН-СНСООП-гСНзСОЗП КСПСП2СООН 7
I
SCOCHg
p-Меркаптовалерианоная кислота [34]. К 25 г пропилидепуксуснон
кислоты прибавляют 25 г тиоуксусной кислоты. Па следующий депь вакуумной
перегонкой при 133—134° С и 2 мм рт. ст. получают 39,5 г кристаллического-
вещества; т. пл. 43—45° С. 35 г полученного соединения смешивают с раствором^
25 г NaOH в 200 мл воды, смесь на следующий день подкисляют раствором, -
содержащим 60 г концентрированной H2SO4 в 60 мл воды. р-Меркаптовалериа
новую кислоту из реакционной смеси извлекают эфиром; эфирный раствор
высушивают над СаС1г и перегоняют. Выход р-меркаптовалериановой кислоты-f
22,5 0; т. кип. 108—110° С (4 мм рт. ст.}.
Тиокарбоновые кислоты реагируют, в основном, со всеми описанными олефи-,
новыми системами и в условиях радикального присоединения [35]. 'i
Совершенно однозначно в стерическом отношении протекает присоединение^
тиоуксусной кислоты к цис- и тране-2-хлорбутсну-2 с образованием 90% трео- и 10%^
аритро-2-ацетилмеркапто-3-хлорбутанов [36].
Тиоловые соединения присоединяются также и к ацетиленам. При этом обычно|
получается смесь цис- и трдас-винилсульфидов [37]. '•(
2. Присоединение других соединений серы
Двуокись серы реагирует с олефинами в присутствии катализаторов, иниции- j
рующих образование радикалов, с образованием полисульфонов [38]. В определенных
условиях при взаимодействии бутадиена, 2,3-диметйлбутадиена, изопрена и гекса*-
диена [39] с двуокисью серы получаются циклические сульфоны: .
r_C=C-R
S°2 - 1
Присоединение к олефинам серного ангидрида и олеума дает р-оксисульфокне-
лоты через промежуточные карбилсульфаты [40]. Взаимодействие циклогексена с сер-
ной кислотой в ледяной уксусной кислоте приводит к ifис-циклогексанол-2-сульфо-1
кислоте (продукт присоединения), а также к циь-логексен-1-сульфокислоте (продуит.:
замещения). Этилендихлорид [42] и этилендибромид [43] тоже реагируют с серной кисло-
[33] В. Holmberg, Е. Schjanberg, Ark. Kemi, 14 А, № 7 (1940); С., 1941, .
П, 2553; Е. SchjSnberg, Svensk kem. Tidskr., 53, 282 (1941); C., 1942,.
I, 604.
[34] E. S c h j a n b e fg, Ber., 74, 1751 (1941). -j
[35] R. Brown, W. E. Jones, A. R. Pinder, J. Chem. Soc., 1951, 2123.
[36] N. P. Neurei ter.F. G. В ordwell, J. Am. Chem. Soc., 82, 5354 (I960).!
[37] E. P. Kohler, H. Potter, J. Am. Chem. Soc., 57,1316 (1935); L. N.O w е-Ил
M. U. S. Sul ta n ba w a, J. Chem. Soc., 1949, 3109.
[38] H. Standings r, B. Ritzent haler, Ber., 68, 455 (1935).
[39] Герм. пат. 236386 (1910); Frdl., 10, 1039 (1910—1912); H. J. Backey
J. S trat ing, С. M. H. К ool, Rec. trav. chim., 58, 778 (1939). J
[40] A. Michael, N. Weiner, J. Am. Cbem. Soc., 56, 294 (1936); С. M. S 1W
t e г, P. В. E vans, j. M. К i e f e r, J. Am. Chem. Soc., 60, 538 (1938). 7
[41] R. Sperling, J. Chem. Soc., 1949, 1938. 3
[42] Герм. пат. 362744 (1921); Frdl., 14, 223 (1921—1925). *
[43] C. S. Rondestvedtir J C. W у g a n t, J. Am. Chem. Soc., 76 , 509*
(1954). '
Г. ПРИСОЕДИНЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПО КРАТНЫМ С-С-СВЯЗЯМ
555
той. При расщеплении карбнлсульфатного кольца образуются хлор- и бромацеталь-
дегидсульфокислоты:
СНС1
/ \
„со С1НС О н о
ClCH-CHCl I [ -SiiU С1СП-СИО-:-С1$ОзП
0-2^ SO2 i
\ / 503и
о
С ацетиленом серный ангидрид реагирует с образованием ацетальдегиддисуль-
фокислоты 144).
Бисульфит патрия присоединяется к олефинам под давлением; в присутствии
перекисей процесс протекает с достаточной скоростью и при атмосферном давлении
[45]. Легкость присоединения зависит от условий реакции и применяемого растворителя.
О применении этой реакции в зависимости от вида олефина и условий взаимодействия
см. обзор [46], К а-нитроолефинам бисульфит присоединяется в водном растворе при
0е С с образованием 1-нИтро-2-сульфонатов.
2-Нитроэтансульфокислота [47]. К раствору 39,4 г NaIISO3 в 70 мл
воды при —5—0° С по каплям прибавляют 27,5 г нитроэтилена при сильном
перемешивании. Затем смесь перемешивают 2 ч при этой температуре и 4 ч
при 20° С, после чего раствор выпаривают при 40° С и пониженном давлении
и из остатка экстрагируют абсолютным этиловым спиртом натриевую соль
2-нитроэтансульфокислоты; выход 75% от теоретического. Соль перекристал-
лизовывают из спирта.
Бисульфит натрия или калия реагирует с аллиловым спиртом с образованием
соответствующих солей З-оксипропансульфокислоты [48]. Бисульфит присоединяется
также и к ненасыщенным жирным кислотам [49], акрилонитрилу [21], олефиндикар-
боновым кислотам и их эфирам [50].
Суяьфиновые кислоты уже без катализаторов гладко присоединяются по крат-
ным углерод-углеродным связям с образованием сульфонов, если эти связи активи-
рованы наличием соседней карбонильной или нитрилыюй групп.
р-фспилсульфомасляная кислота [51]. Растворяют 7 г кротоновой кис-
лоты в 200 мл 0,4 н. раствора фепилсульфиновой кислоты. После самопро-
извольного испарения растворителя кристаллы высушивают на воздухе. Выход
р-фенилсульфомасляной кислоты 14 г; после перекристаллизации из 15 мл
воды т. пл. 102,5— 103.5е С.
С6Пв8О2Н + СПзСН=-СИСООН —СИ3СНСНгСООН
I
йОгС6Пй
Сульфарильпый ocTdioK всегда присоединяется и р-положение к карбок-
сильной группе, причем взаимодействие с тряяс-олефинкарбоновыми кислотами
происходит значительно медленнее, чем с дас-кмелотами.
144] Цат. США 2552421 (1951); С. А.. 46, 2565 (1952).
[45] Пат. США 2504411 (1950); С. А., 44, 5897 (1950).
[46] М. s. Kharasch, Е. М. М а у, F. К. М а у о, J. Org. Chem., 3,175 (1939).
[47] R. L. Heath, П. A. Piggott, J. Chem. Sue., 1947, 1481.
[481 J. П. Helberger, Ann., 588, 71 (1954).
[49] R. T. E. Schenck, J. D a n i h e 1 к у, J. Org. Chem., 16, 1683 (1953).
[50| Цат. CHIA 2028091 (1933); C.. 1936 I, 3019.
[51] E. S c h j а и Ь e r g, Врг., 76. 287 (1943).
556
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
«.^-Ненасыщенные кетоны [52] реагируют с сульфоновыми кислотами с обрат
зованием у-кетосульфонов. Из хинонов и арилсульфиновых кислот в результате 1,4»;
присоединения образуются арилсульфонилгидрохинопы. Если в молекуле хинон»,
свободно орто-или пара-положение и отсутствуют группы ОН и NH2, эту реакцию1*
можно использовать для непосредственного обнаружения хиноидной структуры [53];
К хинонам может присоединяться также тиосульфат натрия; восстановление?
образующихся при этом S-арилтиосульфатов приводит к о-меркаптогидрохинонам^
Меркаптогидрохинои [54, 55]. Растворяют 43,2 ч. хпнопа в 150 ч. ледя^
ной уксусной кислоты. Теплый раствор (40—50° С) добавляют к раствору 150 ч*
NaaS2Og в 200 ч. воды, так чтобы температура смеси не поднималась выше 10’С;
После кратковременного перемешивания прозрачный и почти бесцветный раствс
насыщают КС1; при этом выпадает соль гидрохинонмонотиосульфокислоп
Раствор 2,6 г этой соли в 10 мл воды смешивают с 20 мл концентрированна
НС1 я затем постепенно добавляют 5 г цинковой пыли. Температуру реакцией
ной смеси поддерживают в пределах 40—50° С до окончания выделения газ
После экстракция эфиром получают меркаптогидрохинон в виде длинных нг.
т. пл. 118° С.
ОН
Na8S3O3; сн,соон
Пго. Zn+HCt
------------->
он
к/и
он
II. Присоединение соединений серы по кратным углерод-азотные
связям
Эфиры тиогликолевой кислоты присоединяются по двойным С—N-связям ШИ(_
фовых оснований; при этом сначала возникаете—S-связь, а затем происходит замЫКв
нпе кольца с образованием 2,3-дизамещенных тиазолидонов-4 [56П
RCH=NR'-i-CH2-COOR" —► RCH-NH-R' —> RCH—NR'
I ',11
SH S COOR S CO
CH2 CH2
Описано присоединение меркаптанов к беизальантраниловой кислоте [57$
При присоединении меркаптокетонов к альдаминам (по Азингеру) образуйте"
Д4-тиазолины [58].
Д4-Тиазолины. В колбе с насадкой Дина — Старка кипятят 1 мо.
альдимина и 1 моль а-меркаитодиэтилкетона в 300 мл бензола д© полно:
[52] Н. Gilman, L. F. Cason, J. Am. Chem. Soc., 72, 3469 (1950); D. V 0
lander, A. Friedberg, Ber., 56, 1144 (1923).
[53] О. H i n s b e r g, Ber., 36, 107 (1903); J. W a 1 k e r, J. Chem. Soc., 1945, 62
[54] W. A 1 с a 1 a y, Helv. chim. acta, 30, 578 (1947).
[55] Герм. пат. 175070 (1905); Frdl., 8, 140 (1905—1907).
[56] H. D. Troutman, L. M. Long, J. Am. Chem. Soc., 70, 3436 (1:
A. R. Surrey, 4ч Am. Chem. Soc., 71, 3354 (1949); 74, 3450 (1952).
[57] G. W. Stacy, R. J. Morath, J. Am. Chem. Soc., 74 , 3885 (1952).
[58] K. R ii h 1 m a n n, M. Thiel, F. A s i n g e r, Ann., 622, 94 (1959).
II ПРИСОЕДИНЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПО КРАТНЫМ C-N-СВЯЗЯМ 557
прекращения отделения воды в водоотделителе. Затем отгоняют растворитель
и остаток перегоняют в вакууме или перекристаллизовывают.
C2H6—CO N—R "C2H&-CO NH—R “I C2HS— C N—R
СНз—СН + СН—R' —► СНз-СН 1 1 CH—R | —> CH3-C CH—R
1 \ \ Z
SII s s
Выход T.кип.
R R' % °C
CH3 C8H5 85 138-139 (3 мм pm. cm.)
(т. НЛ. 58 °C)
CfiH5 CeH5 70 197—198 (5 мм pm. cm.)
Циклогексил CgHg 29 140 (5 мм pm. cm.)
C4H9 C3H7 70,5 120 (4 мм pm. cm.)
CeH5 C3H7 72 155 (Ь мм pm. cm.)
Большое препаративное значение имеет реакция органических соединений
серы с нитрилами, приводящая к амидам тиокислот. Эта реакция особенно легко про-
текает с ароматическими нитрилами или в тех случаях, когда на нитрильную группу
влияют системы с двойной связью. Алифатические нитрилы реагируют с H2S только
в присутствии основных катализаторов [59]. Обычно в качестве катализаторов исполь-
зуют гидросульфиды аммония и щелочных металлов, диметиламин, триэтиламин и
другие сильные основания. Реакцию необходимо проводить в безводной среде и при
возможно более высокой концентрации H2S.
Тионамиды [60]. Нитрил растворяют в равном весовом количестве пири-
дина и добавляют 1 моль триэтиламина. Через полученный раствор пропускают
сухой сероводород в течение 1 — 2 ч. Тнонамид выделяют выливанием реакцион-
ной смеси в воду.
Если в нитриле содержатся нитрогрупны, то при присоединении сероводорода
одновременно происходит их восстановление до аминогрупп. В ct-оксинитрилах ОН-
группы предварительно защищают ацилированием [61]. О получении и химических
свойствах Чиоиамидов см. обзор ]62].
В безводной среде нитрилы [63] и синильная кислота [64] в присутствии хлори-
стого водорода присоединяют меркаптаны с образованием гидрохлоридов ^имидо-
тиоэфиров:
>NH • HCI
RC=N + HSR' + HC1 —> RCf
'SR'
Гидрохлорид бензимидотиофеинлового эфира [65]. В охлажденную льдом
смесь равных частей бензонитрила и тиофенола пропускают хлористый водород
и затвердевший продукт реакции тщательно перемешивают с эфиром. Образу-
ющийся с почти величественным выходом гидрохлорид представляет собой
белый кристаллический порошок; т. пл. 178° С.
Присоединение меркаптоуксусной кислоты к нитрилам, протекающее очень
легко, может быть непосредственно использовано для идентификации нитрилов [66].
159]
[60]
[61
[62
[63
[64
[65
[66]
К. Kindler, Ann., 431, 187 (1923).
А. Е. S. F а 1 г f u 11, J. L. Lowe, D. A. Peak, J. Chem. Soc., 1952,
742.
A. Albert, Ber., 48,-470 (1915).
R. N, H urd, G. D ela Mater, Chem. Rev., 61, 45 (1961).
E. Schmidt, Ber., 47, 2545 (1914).
J. H ouben, R. Zivadinovitsch, Ber., 69 , 2352 (1936).
W. Autenrieth, A. Bruning, Ber., 36, 3464 (1903). •
F. E. Condo, E. T. H i n k e 1 et al., J. Am. Chem. Soc., 59, 230 (1937)..
558
ОБРАЗОВАНИИ С-S-СВЯЗИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Насыщение раствора нитрила и меркаптоуксусной кислоты сухим НС1 проводят в абсо-
лютном эфире при 0а С. Выход продуктов присоединения составляет 80—90% от тео-
ретического.
III. Присоединение соединений серы по С=0=связи
При взаимодействии альдегидов и кетонов с меркаптанами на первой стадии';
образуются полумеркаптали, которые, однако, удается выделить без затруднений,.'
только в том случае, если соседний с карбонильной группой углеродный атом несет^
сильно электроотрицательный заместитель. Уже давно известны полумеркаптали^
хлораля |67], изатина [68], фенантреяхинона [69] и пировиноградной кислоты [68]^
Полумеркапталь хлоралд [70]. К охлажденному до —10° С эфирном^
раствору 8 г метилмеркаптана добавляют 24,5 г хлораля. После отгонки эфира’’
полученный продукт перекристаллизовывают из петроленного эфира. Выход;
продукта 26,6 г (82% от теоретического); т. пл. 56° С. ф
Из трихлорацетона, напротив, при взаимодействии с метилмеркаптаном
и хлористым водородом сразу же образуется меркаптол (полный меркаптале
кетона). ц
Препаративное значение полумеркапталей заключается в возможности их пре*
.вращения в а-галогентиоэфиры. причем последние могут быть получены в одну ста-?’
дию, например при одновременном взаимодействии формальдегида, меркаптана^
и хлористого водорода:
СН2О— HSR —>
г п
I
H-C-SR
ОН -
I
С1
Хлорметилэтилсульфид [71]. В охлаждаемую смесью льда с солью суспен-^
зию 15 г полиоксиметилена в 31 а этилмеркаптана пропускают хлористый:
водород до полного растворения. К раствору добавляют СаС12, закрываю^
пробкой с капилляром и оставляют на 24 ч. После этого отделяют верхний^
слой, удаляют оставшийся хлористый водород током сухого воздуха и пере*
гоняют, предохраняя от влаги воздуха. Выход хлорметилэтилсульфида 60—70 i
(60% от теоретического); т. кип. 128—131° С.
а-Галогентиоэфиры можно также получать из ацетальдегида и бензальдегида^
К С —О-связям большинства альдегидов и кетонов, карбонильная группа КОТО*
рых не слишком сильно стерически затруднена заместителями у а-углеродного атом^
(например, ароматические кетоны), в результате экзотермической реакции легко при*
соединяется бисульфит с образованием бисульфитных соединений — а-окси-а-сульфО*
кислот. Последние под действием кислот или щелочей легко расщепляются на исходу
пые продукты. 1')
R. В\ /ОН
>С- О 4- NaHSO3 —> >С<
R'/ R'/ xSOsNa
Бисульфитные соединения обычно трудно растворимы в избытке раствора
сульфита и в спирте и поэтому могут быть легко выделены.
[67] С. A. Martins, Р. М cndelsoh
[68] Е. Baurnann, Вег., 18, 883 (1885).
[69] A. Schonberg, О. Schutz, G.
J70]
171]
., 3, 443 (187О)|
„, ,. ________ -, .Вег., 60, 234«И
(1927). .°’ *
Н. Bohme, Н. D. L о h m е у е г, J. W i с к о р, Ami., 587, 51 (1954).
И. Bohme, Вег., 69, 1610 (1936). "»
F
I. ОБМЕН ВОДОРОДА НА S-ФУНКЦИЮ
559
Бисульфитные производные получают, как правило, простым встряхиванием
карбонильного соединения или его раствора с концентрированным раствором NaHSOs.
Для увеличения растворимости карбонильного соединения к раствору рекомендуется
добавлять небольшие количества спирта и мыла в качестве эмульгатора.
При получении бисульфитных производных ненасыщенных карбонильных сое-
динений возникают осложнения, так как бисульфит может присоединяться и по двой-
ным С=С-связям (стр. 555). В результате этой реакции образуются различные про-
дукты присоединения, состав которых зависит от относительной реакционной способ-
ности С=С- и соответственно С — 0-связи. Одновременное присоединение по двойной
С=С-связи можно предотвратить, если применять свежеприготовленный (из 1 моль
чистого кристаллического NasSOo и 1 моль уксусной кислоты) бисульфит и проводить-
реакцию в нейтральной среде (буфер), избегая длительного взаимодействия и повыше-
ния температуры. Таким образом, папример, были получены «нормальные» бисульфит-
ные производные коричного альдегида, цитронеллаля и цитраля [72].
При присоединении к альдегидам сульфиновых кислот образуются а-оксисуль-
фопы:
ОН
.0 1
R-C< -]-R'SO2H —> R-C-SO2R'
ХН I
II
а-Оксисульфоны, ла исключением низших чпето алифатических членов, легко
кристаллизуются, однако эти вещества неустойчивы, скорость разложения зависит
от природы компонентов.
Разработан способ получения а-окспметилсульфонов [73].
Несколько граммов алкилсульфината магния нагревают с 35%-ным
формалином до 40—50° С и при сильном перемешивании по каплям добавляют
немного 12 н. H3SO4. Через 0,5—2ч отделяют образовавшийся алкилоксиметил-
сульфоп от формалина. Изоамил- и л-гексилоксиметилсульфоны представляют
собой жидкости, гептил- и высшие оксиметилсульфоны — твердые вещества.
Из высших алифатических альдегидов а-оксисульфоны получают кипя-
чением в токе азота эфирного раствора сульфиновой кислоты с избытком альде-
гида в течение 15 мин — 1ч. При работе с ароматическими альдегидами упо-
требление азота излишне, так как образующиеся при этом аддукты значительно-
стабильнее, чем аддукты, полученные из алифатических альдегидов.
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ
РЕАКЦИИ ЗАМЕЩЕНИЯ
/. Обмен водорода на S-функцию
1. Введение сульфогруппы
Сульфирование алифатических соединений
В алифатическом ряду введение сульфогруппы путем замещения водорода не
имеет такого значения, как в ароматическом. При непосредственном сульфировании
алканов серной кислотой, олеумом или трехокисью серы протекают главным образом
побочные реакции (изомеризация, дегидрирование, окисление, образование сульфо-
нов), и этот метод до сих пор не нашел практического применения.
Однозначное взаимодействие с серной кислотой с образованием сульфокислот
наблюдается у алифатических карбонильных соединений, кислот и тому подобных,
соединений.
[72] F. Ticmann, Вег., 31, 3297 (1898).
[73] Н. Brederock, Е. Bader, Chem. Вег., 87, 129 (1954);
560
ОБРАЗОВАНИЕ С —S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Камфорсульфокислота [74). При обработке 100 г (±)-камфоры 135
уксусного ангидрида и 98 г концентрированной H2SO4 в течение четырех сутс
образуется оптически неактивная камфорсульфокислота. После перекристаз
лнзации из ледяной уксусной кислоты выход продукта 40% от теоретического
При сульфировании жирных кислот H2SO4 выходы а-сульфокарбоновых ки
лот обычно невысоки. Но при сульфировании в присутствии пиридина сульфокисло-
получаются с практически количественными выходами [75].
Для сульфирования алифатических соединений наряду с хлорсульфонов
кислотой применяются аддукты SO3 с диоксаном и пиридином. При помощи этих аддз
тов *, которые следует рассматривать как бетаииоподобные ониевые соли, очень лег
осуществляется перенос трехокисн серы к сульфируемому соединению:
H->so3+rh —> rso3h+<^2^
с/ X6->SO3+RH —> RSO3H + C/
что дает возможность проводить сульфирование в индифферентных растворителях П
очень мягких условиях **. Особенно гладко в этих условиях протекает взаимодейств
альдегидов и кетонов с образованием а-кето- или альдегид-а-сульфокислот [«71
Аддукт серного ангидрида с диоксаном [77]. Из стеклянного' сосу
с 60%-ной дымящей H2S04 перегоняют S03 в охлаждаемую холодной вод
взвешенную колбу, содержащую сухой дихлорэтан. К полученной смеси г
сильном перемешивании добавляют эквивалентное количество прокипяченН!
над натрием и перегнанного диоксаиа; температура смеси не должна и
вышать 5Q С.
Ацетофенон-и-сульфокислота [76]. К 0,35 моль аддукта SO3 с диой
иом в течение 1 ч прибавляют 0,35 моль (42 г) ацетофенона так, чтобы темпе
тура смеси не поднималась выше 35° С. Реакционную смесь переметив
еще 2 ч, а затем разлагают, выливая ее в 300 мл воды. После отделе
органического слоя водную фазу нейтрализуют холодным раствором Na
и упаривают, продувая ток воздуха. Из твердого остатка продукт реаи
экстрагируют кипящим 60%-иым водным спиртом. После охлаждения на э
раита кристаллизуется 54 г (70% от теоретического) натриевой соли ацетофез
©-сульфокислоты.
Аналогичным образом получают натриевые соли 2-ацетотиофеи
ацетомезитилен-к»-, р-ацетонафталин-к»-, пииаколон-а-, пропиофенон-а- и :
бутирофеноя-а-сульфоквслот. Такие альдегиды, как фенилацетальдегид, :
бутиральдегид и гептальдегид, этим способом также можно превра*
в а-сульфокислоты.
При взаимодействии фенилалкилкарбоновых кислот с аддуктом серного аи
рида с диоксаном образуются а-сульфвкислоты [78], в то время как прямое сульф)
ванне H2SO4 приводит к замещению в бензольном ядре.
Промышленное значение имеет сульфирование действием SO 2 и кисло
(сульфоокисление). Реакция протекает по свободнорадикальному механизму 1
* Строение аддуктов описано в учебниках теоретической органической хи
см. также G. С i 1 е n t о, Chem. Rev., 60, 147 (1960).
** См. работы А. П. Терентьева и его школы. — Прим. ред.
[74] В. Rewald, Вег., 42, 3136 (1909); Р. L i р р, Н. К пар р, Вег.,
915 (1940).
[75] Пат. США 2268443 (1942): С. А., 36, 2564 (1942).
[76] W. Е. Truce, С. С. Alfie г i, J, Am. Chem. Soc., 72, 2740 (1950).
[77] F. G. В о г d w el 1, C. S. Rondestvedt jr. j. Am. Chem. I
70, 2432 (1948).
178] W. E. Truce, С. E. Olson. J. Am. Chem. Soc., 75, 1651 (1953).
I. ОБМЕН ВОДОРОДА НА 8-ФУНКЦИЮ
561
Сульфирование ароматических соединений серной кислотой и олеумом
Сульфирование, как одна из основных реакций химии ароматических соедине-
ний, имеет очень большое практическое значение. Под сульфированием понимают
введение в молекулу ЗО3Н-группы, осуществляемое в простейшем случае действием
ПЛЗО4 [80):
R-H + H0—SO3H R-S08H + Hs0
Продукты реакции — свободная сульфокислота или ее соли большей частью
растворимы в воде. Ароматические сульфокислоты являются исключительно важными
промежуточными продуктами для получения фенолов, в первую очередь фенолов наф-
(алппового ряда.
Свободные сульфокислоты в большинстве случаев не имеют характеристических
температур плавления; все они значительно более сильные кислоты, чем карбоновые.
Для успешного сульфирования отдельных соединений решающее значение
имеют сульфирующий агент, растворитель и условия проведения реакции — концен-
трация кислоты, температура и продолжительность процесса.
Применение различных сульфирующих агентов описано ниже; в этом разделе
кратко рассмотрено влияние условий проведения реакции на ход сульфирования. ,
Для сульфирования используют обычную концентрированную серную кислоту
(68%-ная), моногидрат (100%-ная H2SO4) и дымящую серную кислоту (олеум), содер-
жащую 5,7% 8О3. При сульфировании образуется вода, что может привести к разба- -
влению реакционной массы и преждевременному прекращению реакции. Поэтому
необходимо либо брать соответствующий избыток серной кислоты, либо восстанавли-
вать первоначальную концентрацию добавлением олеума (при слишком сильном разо-
гревании охлаждают!), или азеотропной отгонкой [81] удалять образующуюся воду.
Описано [81, 82) получение n-толуолсульфокислоты с использованием азеотропного
обезвоживания реакционной смеси. При получении ж-крезолсульфокислоты удаление
воды осуществляли вакуумной перегонкой [83].
Под влиянием воды, выделяющейся в процессе реакции, сульфогруппа может
'также вступать в другие положения. Кроме того, равновесие между углеводородом
и сульфокислотой может настолько сильно сдвинуться в сторону углеводорода, что
будет протекать катализируемое кислотой отщепление сульфогрупп. С другой стороны,
при высоких концентрациях олеума необходимо учитывать возможность вступления
в молекулу нескольких сульфогрупп.
В зависимости от реакционной способности исходных соединений применяемые
при сульфировании температуры колеблются в широких пределах, от 0° С до примерно
200° С, причем с повышением температуры скорость реакции увеличивается, ио воз-
растает опасность изомеризации продуктов или их дальнейшего замещения.
О продолжительности реакции нельзя сказать ничего определенного; некоторые
соединения (например, тиофен) сульфируются в течение нескольких минут, в то
время как для исчерпывающего сульфирования других соединений требуется
несколько суток.
При сульфировании неустойчивых соединений, осмоляющихся под действием
серной кислоты, рекомендуется проводить реакцию с растворами, причем растворитель
выбирают в зависимости от растворимости исходного продукта или сульфокислоты.
В качестве растворителей предложены ацетонитрил, ледяная уксусная кислота, уисус^-
ный ангидрид, жидкий сернистый ангидрид, петролейный эфир, четыреххлористый
углерод и др. Так, например, 1-нафтол-2-сульфокислоту получают из а-нафтола дей-
ствием H2SO4 в ледяной уксусной кислоте [84].
Влияние катализаторов на реакцию сульфирования сказывается в большей
степени, чем на реакцию нитрования. Важнейшими катализаторами являются
(79) R. Graf, Ann., 578, 80 (1952); L. Orthner, Angew. Chem., 62, 302 (1950).
[80] О механизме реакции см. С. С. Price, Chem. Rev., 29, 37 (1941); С., 1942,
I, 984.
[81] Н. Meyer, Ann., 433, 327 (1923).
[82] L. G a Hermann, H. Wieland, Die Praxis des organischen Chemikers,
r 35 Aufl., Berlin, 1953, S. 170.
< [83] Герм. пат. 281054 (1914);-Frdl., 12, 160 (1914—1916).
[84] M. Conrad, W. Fischer, Ann., 273, 107 (1893). 4
. 36 Заказ 1835.
L
562
ОБРАЗОВАНИЕ G— S-СВЯЗТТ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
бисульфаты щелочных металлов прп повышенных температурах, пятиокись ванадия [85],
иод [86] (применение спорно, см. [150]). трифторлд бора [87] и фтористоводородная
кислота, действие которой, вероятно, основано на образовании фторсульфоновой кис-
лоты в качестве активного промежуточного соединения. Иногда в присутствии солей
ртути сульфогруппу удастся вводить против правил замощения. Так, при действШ
па антрахинон только H:S04 обрадуется почти исключительно ₽-антрахинонмоносуль-
фокислота, а прп добавлении небольшого количества ртути — практически только
а-моносульфокислота (для этого достаточно уже 0,5% ртути [88]). Правда, как пока--
залп систематические исследования [89]. случаи такого замещения чрезвычайно редкий
При сульфировании чрезвычайно большое значение имеет способ выделения^
сульфокислот из реакционной среды. Отделение избытка H2SO4 от образовавшейся’
сульфокислоты легче всего происходит при сульфировании аминов. Аминосульфо-t
кислоты большей частью гак плохо растворимы в воде, и особенно в разбавленной-;
II2SO4. что практически полностью выпадают при разбавлении реакционной массй,
после чего их остается только отфильтровать и промыть. Свободные ароматичееншй»
сульфокислоты в водных минеральных кислотах (например, в H2SO4, НС1) растворимой
хуже, чем в воде. Поэтому их часто можно выделить простым разбавлением реакцион^З
ной массы менее концентрированной H2SO4 или медленным добавлением воды илн’;
льда. Таким путем, например, отделяют n-толуолсульфокислоту. плохо растворимую]
в 66—71%-ной H,SO4. от орто-изомера, выпадающего из 45—55%-иой H2SO4 [90].
Свободные сульфокислоты можно выделить, внося сульфомассу в органические
растворители, смешивающиеся с серной кислотой (ацетоп, спирт) [91], или непрерывно
экстрагируя сульфокислоты бензолом или толуолом. .а
Выделение сульфокислот в вице солей осуществляется обычно двумя способами^
По одному способу растворенную в воде сульфомассу нейтрализуют гатпеной известья?^:
отфильтровывают выпавший CaSO4 и обрабатывают фильтрат, содержащий кальциевую
соль сульфокислоты, Na2COa. причем образуется натриевая соль сульфокислоты^
Выпадающий СаСО3 отфильтровывают, фильтрат упаривают до начала кристаллина^
ции или. если необходимо, досуха. Этот так называемый известковый метод примени»!
практически для всех сульфокислот, кроме тех, которые образуют трудно растворимы^
кальциевые 'соли (например, при получении 1.4-аминонафтол*2-сульфокислоты)А"
Однако он неудобен из-за использования больших количеств жидкости, которую нео&ч
ходимо пыпаривать. Можно попытаться выделять сульфонаты щелочных металлов д(Н
бавленнем спирта. По другому способу разбавленную водой сульфомассу смешивав^
с неорганическими солями щелочных металлов [чаще всего с NaCl или IVajSO*, режф
с KCI пли (NH4)2SO4], высаливая соответствующую соль сульфокислоты. Одиаио «А
не всегда применим, так как не все сульфокислоты высаливаются достаточно полней
Кроме того, выделяемые этим методом соли сульфокислот, как правило, загрязнены
неорганическими солями, что, однако, обычно не мешает их дальнейшей переработке*
Для получения солей сульфокислот,’ содержащих очень незначительное количеств?
Heopi анически.х солей пли свободных от этих примесей, их промывают сначала наем*
щенным, затем разбавленным раствором NaCl и далее, если сульфонат не слишком
легко растворим, ледяной водой. Можно также попытаться очистить сульфонат пере?
кристаллизацией из органических растворителей, например водных метилового ИЛ*
этилового спиртов.
Для выделения свободных сульфокислот из их солей обычно твердые натрием
соли этих кислот смешивают с концентрированной НС1; образующийся NaCl отфял
тровывают на пористом стеклянном фильтре и фильтрат упаривают в вакууме. С»
бодные сульфокислоты получаются в виде сиропа или в виде расплывающейся криста;
лической массы. Дальнейшая очистка сульфокислот от незначительных количесч
со.трй и других примесей осуществляется растворением в спирте. Свободные сульф'
[85] Герм. пат. 214156 (1909): Frdl., 9, 670 (1908—1910).
,86] Е. Kn ое venagel, J. prakt. Chem. [2], 89, 1 (1914).
j87J R. J. Thomas, W. F. A n z t I о 11 i, G. F. П en л t on, Ind. Eng. Chem
32, 408 (1940); G. F. Hennion, С. J. Schm idle, J. Am. Chem. Soc
65, 2468 (1943).
[88] M. Il jinsky. Ber., 36, 419" (1903).
[89] K. Holdermann, Ber., 39,1250 (1906); O. D imrolh,W. V. Schmai
del, Ber., 40, 2411 (1907).
[90] Герм. пат. 137935 (1901); Frdl., 7, 53 (1902-1904). ,
] 91 ] G. V. S 1) i г о 1 k a r. I. S. U p p a I, V. Ven ka t a rama n, J. India*
Chem. Soc., 17, 443 (1940); C., 1941, I, 2103. j
1. ОБМЕН ВОДОРОДА НА S-ФУНКЦИЮ
563
кислоты удается надежно выделить из бариевых или свинцовых солей серной кислотой
it.iH сероводородом.
При взаимодействии с органическими основаниями арилсульфокислоты, как
правило, образуют хорошо кристаллизующиеся соли с четкой температурой плавле-
ния. Эти соли можно использовать для идентификации сульфокислот [92]. Описан,
например, способ разделения различных нафталинсульфокислот через их соли с арил-
аминамп [93|. Бензидиновые и дианизидиновые соли в большинстве случаев трудно
растворимы и могут быть использованы для разделения и количественного определения
сульфокислот [94].
Толуидиновые соли арилмоносульфокислот [95]. «Эквимолярные количе-
ства арилсульфоната калия и гидрохлорида п-то.туидина растворяют при 60°С
в возможно малом количестве воды и быстро охлаждают при хорошем переме-
шивании. п-Толулдинопую соль отфильтровывают, промывают небольшим
количеством ледяной воды и перекристаллизовывают из воды, добавляя, если
необходимо, активированный уголь. Затем перекристаллизовывают еще раз
из воды и, наконец, из водного этилового спирта.
S-Бензил- и Я-а-нафтилметилизотпуронийхлориды тоже образуют с солями
сульфокислот кристаллические соли, пригодные для идентификации сульфокислот
и часто для их разделения (96):
RSO3Na-MR'CH3-S-C(NH2)2]+C1- —> [R'CH2-S-C(NH2)2]+(RSO3)'^NaCl
Для очистки простейших представителей арилсульфокислот можно применять
высоковакуумную перегонку [97]; однако обычно чистые препараты получают через
сульфохлориды или сульфамиды.
Сульфирование и влияние заместителей в бензольном ряду. В бензольном ряду
,скорость сульфирования и положение, в которое вступает сульфогруппа, зависят от
имеющихся в ядре заместителей. Сульфирование облегчается при наличии в ядре-фе-
ппльных оксигрупп; так же, но несколько менее эффективно, действуют алкокси-,
.амино- и ациламнногруппы; наиболее слабое влияние оказывают алкильные группы.
Напротив, наличие галогенов, карбоксильных и карбонильных групп, уже имеющихся
сульфогрупп, и особенно нитрогрупп, затрудняет сульфирование. Так, динитропро-
изводные обычно уже не сульфируются.
Сульфирование почти всегда протекает согласно правилам замещения [98].
Заместители первого рода при низких температурах направляют, сульфогруппу пре-
имущественно в орто-положение, при повышенных — в пара-положение; в мета-поло-
жение сульфогруппа вступает при наличии заместителей второго рода.
Бензолсульфокислота [99] (простой способ получения см. [82]). В сталь-
ном, фарфоровом или эмалированном сосуде, снабженном мешалкой и обратным
холодильником, и течение примерно 30 мин осторожно смешивают 200 г бен-
зола с 450 г 10%-ного олеума, причем температура не должна подниматься
[92J R. Wendiand, J. Rode, R. Meintzer, J. Am. Chem. Soc., 75, 3606
(1953).
[93] Й. B. Forster, С. M. Key worth, J. Soc. Chem. Ind., 46, T 25 (1927);
C., 1927, I, 1676.
[94] H, E rd ma пл, Ber., 32, 3186 (1899); В. Ф. Б о p о д к и н, Т. В. М а л к о в а,
ЖАХ, 3, 166 (1948); А. А. Спрысков, Б. И. Караваев, ЖОХ, 22,
1620 (1952).
[95] A. D. Barton, L. Young, J. Am. Chem. Soc., 65, 294 (1943).
[96] E. Chambers, G. W. Watt, J. Org. Chem., 6, 376 (1941); W. A. Bon-
ner, J. Am. Chem. Soc., 70, 3508 (1948).
[97] F. К r a f f t, W. W i 1 k c. Ber., 33, 3207 (1900); J. V. В r a u n, K. Wei-
b и a c h, Ber., 63, 2639 (1930).
[96] K. Lane r, J. prakt. Chem. [2], 143, 130 (1935).
199] Г. E. Фирц-Давид, Л. Б л а и ж e й, Основные процессы произ-
водства органических красок, пер. с нем., Издатинлит., 1957.
564
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
выше 75° С. Полученную смесь нагревают до 110° С (при более высокой темпа!
Йатуре довольно легко образуются дисульфокислоты) и выдерживают 1,5___2 яы
осле того как прореагирует весь бензол, реакционную массу выливают в
воды, нейтрализуют при перемешивании кипящую смесь примерно 450 г пород
шкообразното СаСОа и отфильтровывают выпавший CaSO4 на большом нутш
фильтре. Осадок промывают водой (около 1,5 л) в перешедшую в раствор иялдй!
циевую соль сульфокислоты нагреванием с примерно 110 g Na2COs (до щелочвдМ
реакции по фенолфталеину) переводят в натриевую соль. Выделившийся
этом СаСОя отфильтровывают в горячем состоянии и упаривают фильтрат ~Я1
открытом пламени до начала выпадения кристаллов бензолсульфоииллемИ
натрия. При охлаждении образуется паста из соли и воды, содержащая окамВ
15% твердой фазы; она состоит (в расчете на сухое вещество) примерно 1Й
90% бензолсульфоната. 7% Na2SO4 и Na2CO3 и небольшого количества кдпммД
евых солей. Эту пасту можно непосредственно перерабатывать в фенол (щелДИ
ное плавление) или выпариванием досуха выделять бензолсульфонат патпиЙЯ
Если требуется возможно более полное отсутствие неорганических солей, отйЯ
сывают кашицу и высушивают осадок, что, правда, сопряжено со значитэдЯ
ными потерями вещества. '-$М
Азеотропной осушкой с избытком бензола можно получить моногйдтйВ|
бедзолсульфокислоты (т. пл. 45—46° С) [81].
При дальнейшем сульфировании бензолмоносульфокислоты образуется бензозг-яЯ
дисульфокислота [100].
Бензол-.н-дисульфокислота. К 78 г (1 леоль) бензола при хорошем пердИ
мешиваиии в течевке 2 ч приливают по каплям 250 г 20%-вого олеума
чтобы температура смеси не поднималась выше 40—50s С. Затем таким же сшвЯ
собой в течение еще 2 ч добавляют 200 г 66%-ного олеума, причем температэдИ
повышается до 75° С. Полученную смесь нагревают еще 1 ч при 90° С, выш|В
вают в 2 л воды и в горячем состоянии при перемешивании нейтрализуют гпЙ|И
мерно 400 г СаСО3. После отсасывания и промывания осадка фильтрат обраэдЯ
тывают, как описано выше, примерно 100 г Na2CO3 и упаривают. ОстатиЯ
после упаривания сушат при 130—140° С. Выход продукта около 250 г (90яЯ
от теоретического).
Толуол сульфируется легче, чем бензол, давая смесь о- в и-толуолсульфоквей^И
[90], причем при нагревании образуется главным образом пара-соединение [1Щ^В
(стр. 562). Описан способ получения толуол-2,4-дисульфокислоты [102]. Высшие гст^И
логи бензола, вероятно вследствие стерических затруднений, сульфируются тоЛМ^Н
в пара-положение [103]. Из трех изомерных ксилолов легче всего сульфируется
лол; различную реакционную способность ксилолов в реакции сульфирования можМИ
использовать для их разделения методом фракционного сульфирования [104].
При наличии в бензольном ядре атома галогена сульфогруппа практичМяЦИ
направляется исключительно в пара-положение [81, 105].
Описано получение n-бром- и п-иодбензолсульфокислот [106].
Хлорбензолдисульфокислота. Ее целесообразно получать из л-хлорбеБМн^Н
сульфохлорида медленным нагреванием с 4 ч. моногидрата серной кксй^^^В
при 160—180° С до окончания выделения HCI. После выливания реакционаяМ
смеси в воду продукт выделяют «известковым» методом, как описано въбм^И
[100] Г. Е. Ф и р ц -Д ав ид, Л. Б л а и ж е й, см.9». pfJ
(Ю1[ Герм. пат. 57391 (1890); Frdl., 3, 905 (1890—1894); Е. Е. G i 1 b ег t, Е. Р.
nes, Ind. Eng. Chem., 43, 2030 (1951). -Ла
1102J Г. E. Ф и p ц - Д а в п д, Л. Б л а н ж e й, см.99, •
(103) М. Senkowski, Вег., 23, 2417 (1890); R. L. Frank, R. E. В e rtft
O. L. Shotwell, J. Am. Chem. Soc., 71, 3891 (1949). Лй
[104] T. S, Patterson, A. McMillan, R. S. S о m e r v i 11 e, J. С“ЧЯ
Soc., 125, 2488 (1924). ЛаЗ
[105] G. F. Lis k, Ind. Eng. Chem., 41, 1025 (1949); герм. пат. 116759; Frdl., 5;J|
(1897-1900); герм. пат. 260563 (1912); Frdl., 11, 141 (1912-1914). :-Л8
[106] R.R. Baxter. F. D. Chattawav, J. Chem. Soc., 107, 1815 (19±Я|
I. ОБМЕН ВОДОРОДА НА S-ФУНКЦИЮ
565
Фенол сульфируется исключительно легко; при низких температурах образуется
орто-производное, при повышенных (около 100° С) — пара-соединение наряду При-
мерно с 10% фенилового эфира сульфокислоты.
Фенол-о-сульфокислота (107). Расплавляют 200 г фенола, к расплаву
добавляют при хорошем перемешивании 100 г H2SO4 (моногидрата) и охлаж-
дают; полученная смесь уже не так легко затвердевает. Прп температуре ниже
20° С и перемешивании добавляют еще 200 г моногидрата, продолжают пере-
мешивать 6—8 ч, поддерживая температуру ниже 20° С, и выливают реакцион-
ную смесь в 1,5 л воды. Основное количество избыточной H2S04 нейтрализуют
сначала PhCO34 (около 500 г), затем порциями осторожно добавляют ВаСО3
до перехода синей окраски бумажки конго в красную, с тем, чтобы сульфоки-
слота оставалась в растворе в виде кислой бариевой соли. Необходимо избегать
длительного кипячения с избытком карбоната, так как при этом выпадает
трудно растворимый нейтральный сульфонат бария. Смесь отфильтровывают
от PbSO4 и BaSO4 и хорошо промывают осадок, фильтрат еще раз отфильтро-
вывают от вновь выпадающего осадка и выпаривают, пока удельный вес маточ-
ного раствора, находящегося над выпавшими кристаллами, станет равным
1,18—1,20 (измеряется на холоду). Необходимо следить за тем, чтобы наряду
с грубыми, срастающимися в корку кристаллами о-сульфоната не выпадали
мелкие волокнистые иглы изомерного продукта, которые в случае их образова-
ния следует растворить, добавляя холодную воду. Выделенный таким способам
. неочищенный о-сульфонат отфильтровывают 'и промывают холодной водой
до удельного веса промывной воды 1,08—1,09. Выход неочищенного фенол-
о-сульфоната бария около 25% от теоретического.
Фенол-п-сульфоиислота [107]. Реакционная масса, полученная по опи-
санному выше способу, состоит приблизительно из 40% о-сульфокислоты
и 60% «-сульфокислоты. После выделения неочищенного о-сульфоната маточ-
ный раствор содержит около 80% n-сульфоната, который после добавления
MgSO4 выкристаллизовывается в виде магниевой соли.
Если необходимо получить только л-сульфокислоту, то 200"г фенола
нри хорошем перемешивании нагревают 6—8 ч при 90—100° С с 200 г 95%-ной
H2SO4, охлаждают до 70—75° С, смешивают с 50 мл воды, для того, чтобы
предотвратить затвердевание смеси, и охлаждают до комнатной температуры^
Из реакционной смеси, содержащей более 80% n-сульфокислоты, можно непо-
средственно выделить свободную n-толуолсульфокислоту (см. оригинальную
работу).
о-Крезол действием концентрированной H2SO4 превращают в 2-метнлфенол
4-сульфокислоту [108], а гваякол — в соответствующую 2-метоксифенол-4-сульфо-_
кислоту [108].
При наличии в молекуле аминогруппы сульфогруппа вступает в орто- н пара-
положения. Сульфирование олеумом с повышенным содержанием серного ангидрнда
дает преимущественно м-сульфоиислоты. При низких температурах из анилина воз-
никает главным образом аннлин-о-сульфокислота, при повышенных температурах —
в основном сульфаниловая кислота [109]. Сульфаниловую иислоту получают с хоро-
шим выходом взаимодействием анилина с концентрированной H2SO4 в течение 4—5 ч
при 180—190° С [110].
Назревание бисульфатов аминов (процесс «запекания») приводит к аминосульфо-
кислотам. Этот простой способ применим для получения сульфокислот из ароматических
аминов, например толуидинов, ксилидинов, хлоранилинов, а-нафтиламина, бензи-
дина и третичных аминов. Преимуществом метода в сравнении с обычным сульфирова-
нием серной кислотой является то, что при этом не образуются более высокосульфиро-
ванные продукты, так как берется точно рассчитанное количество серной кнслоты.
Как правило, при таком превращении ие образуются изомеры, поскольку сульфогруппа
[107] J. Obermiller, Вег., 40, 3637 (1967).
[108] М. Е. Hultquistet al., J. Am. Chem. Soc., 73, 2558 (1951).
109] E. R. Alexander, J. Am. Chem. Soc., 68, 969 (1946); 69, 1599 (1947).
[1,10] L. Ga t t ermann, H. Wieland, см.82, стр. 172; . Г. E: Фирц-
Давид, Л. Б ланж ей, см.99.
566
ОБРАЗОВАНИЕ С —S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
почти всегда однозначно вступает в пара-положение или, если оно замещено, в орто-'
положение к аминогруппе, но никогда не вступает в мета-положение.
СН3 СН3 СНз
H.SO* «запекание" —1 II * iz^i
учи, учи, “O3SZV4CH3
АН, +NH3HSOJ +NH3
другие методы сульфирования
Для процесса «запекания» практически важно наличие возможно большей по*:'
верхности бисульфата амина, что достигается распределением его тонким слоем на листе-?
жести, растиранием корки, применением вакуума и тому- подобными мерами. Паи-"
более благоприятными температурами процесса являются 170—220° С; однако в каж- <
дом отдельном случае температуры различны и должны устанавливаться путем проб-’
ных опытов. Процесс лучше вести в вакууме (111), так как при этом сульфирований
проходит быстрее, более гладио и устраняется опасность обугливания. Применяемые'
для «запекания» бисульфаты должны быть возможно более чистыми, поскольку содёрУ
жащанся иногда в соли серная кислота может способствовать иному направлении^
сульфирования. Сульфируемое соединение лучше всего растворить в ледяной уксусной ?
кислоте, вылить раствор в рассчитанное количество концентрированной серной кно*-^
лоты п удалить уксусную кислоту в вакууме; остаток количественно coctoIhkj
из бисульфата. J
О сульфировании ароматических аминов по методу «запекания» см. обзор [1121* 1
Этим методом были получены следующие сульфокислоты: 2,4-д(1метил-1-аМИНОУ
бензол-6-сульфокислота (113), 2-амино-1-метилбензол-5-сульфокислота [114) и$
1-аминобензол-4-сульфо-2-карбоновая кислота [115]. Перегруппировку бисульфатов^* 1
можно осуществить также нагреванием их в высококипящем индифферентном раствор
рителе [116]. . л
Менее одпозначно, чем методом «запекания», протекает прямое сульфирование?’
аминов серной кислотой или олеумом. Таким образом из N-этиланилнна получена’,
17-этилаиилин-4-сульфокислота [91], из диметиланилина — N.N'-диметиланилин-З^
и -4-сульфокислоты [117]; из этилбензиланилина — этил-(л-сульфобензил-аниУ
лин |118): , -
(у-к-сн2уу O-N“CHa~\=/ -?
СЛ ’ C2Hs \ Л
[ill] Г. E. Ф и p ц - Д а в и д, Л. Б л а н ж e й, см.8».
(112) W. Huber, Helv. chim. acta, 15, 1372 (1932); A. F. H oil ema n, C., 1933^;
I, 2809. -f
[113] A. Junghahn, Ber., 35, 3750 (1902).
[114J C. F. H. Allen, J. A. Van Allan, Org. Synth., 27, 88 (1947). J
[1151 Пат. США 2353351 (1944); С. A., 38, 5845 (1944). 0,
(116) Герм. пат. 549136 (1931); Frdl., 19, 705 (1934). ;
[117] Е. А. Шилов, 4. Н. Куракин, ЖОХ, 18, 2092 (1948): С. А., 43, 3803?
(1949). -i
[118] L. В I a n g е у, Н. Е. F t с т z, G. Stamm. Helv. chim. acta, 25, 416*1'
(1942); Г. E. Ф и p ц - Д а в и л, Л. Б л а и ж e й, см."; стр. 131; герм. .
пат. 697/7 (1891); Frdl., 3, 39 (1890—1894). '
I ОБМЕН ВОДОРОДА НА S-ФУНКЦИЮ
567
Этил-(.и-сульфобензил)-аннлин. К 150 г H2SO4 (моногидрат) при пере-
мешивании в течение 15 осторожно приливают по каплям 150 а атилбен-
зиланилина, поддерживая температуру ниже бО^С. К полученной массе прили-
вают по каплям 150 г 60%-ного олеума и нагревают при 60° С около 3 ч.
Конец реакции определяют по отсутствию помутнения при добавлении
разбавленного раствора соды к небольшой пробе реакционной смеси, смешанной
с водой.
Затем реакционную массу выливают в 1 л воды, через 12 ч отфильтро-
вывают выделившуюся сульфокислоту и промывают ее водой. Выход продукта
72% от теоретического. Маточпый раствор нейтрализуют известью, растворен-
ную кислоту переводят в натриевую соль добавлением сульфата натрия и фильт-
рат упаривают до объема 400 мл. После подкисления часто уже при трении
стеклянной палочкой и внесении затравки получают вторую фракцию кислоты
(около 5%). Добавлением NaCl из маточного раствора можно выделить и натрие-
вую соль n-кислоты (около 15% от теоретического).
Ацилированные амины могут сульфироваться в пара-положение с сохранением,
отЕцеплепием или изменением ацильного остатка. Так, например, из N-метилацет-
анилида получают М-метиланилии-4-сульфокислоту {119].
о- п тг-Аминофенолы сульфируются труднее, чем фенол; сульфогруппа при этом
вступает в орто- или пара-положение к оксигруппо. Из o-аминофенола образуется
исключительно 2-аминофенол-4-сульфокислота [120], из л<-аминофеиола — 4-амино-
2-оксибензолсульфокислота [121], а из л-аминофенола — 5-амино-2-оксибензолсуль-
фокислота [122].
Нитробензолсульфокислоты обычно получают нитрованием ароматических
сульфокислот. Обратный путь применяют только в тех случаях, когда сульфирование
протекает более однозначно, чем нитрование. Сульфирование нитросоединепий следует
вести очень осторожно, так как при этом возможно их разложение со взрывом. Приме-
рами сульфирования нитросоединений являются синтезы л-иитробензолсульфокислоты
из нитробензола [123] и 5-нитро-2-метнлбензолсульфокислоты из «-нитро-
толуола -[124].
Карбоксильная и формильная группы ориентируют вступление сульфогрунпы
в мета-положение. Так, из бензойной кислоты получают 3-с,ульфобензойную кислоту
[125], из бензальдегида — бензальдегид~3,5-дисульфокислоту [126].
Сульфирование и влияние заместителей в нафталиновом, антраценовом и фе-
нантреновом рядах. При сульфировании соединений нафталинового ряда уже при
вступлении первого заместителя возможно образование двух изомеров, аир. При суль-
фировании этих соединений обычно получают смесь изомеров, состав которой зависит
от температуры реакции; при проведении реакции на холоду преобладает а-изомер,
при 120—130° С образуется [^сульфокислота. При нагревании а-сульфокислоты с сер-
ной кислотой она может изомеризоваться в р-сульфокислоту. На направление заме-
щения прп сульфировании оказывают влияние уже имеющиеся заместители, правда
не в такой степени, как в бензольном ряду. Заместители первого рода облегчают обыч-
пое электрофильное замещение, направляя сульфогруппу в орто- и пара-положения.
Заместители второго рода, напротив, затрудняют замещение, при этом менее затруд-
ненным оказывается мета-положение; одпако легче происходит сульфирование неза-
мещенного ядра. Согласно правилу Армстронга и Винна, при прямом сульфировании
две сульфогрунпы никогда пе вступают в орто-, пара- или пери-положения друг
к другу.
[119] J. II а 1 b е rk а п п, Вег., 54, 1836 (1921); G. A. Smyth, Вег., 7, 1241
(1874).
[120] J. Post, Ann., 205, 51 (1880).
11211 A. L. М ill er, Н. S. Mosher et al., J. Am. Chem. Soc., 7j, 3559 (1949);
герм. пат. 70788 (1892); Frdl., 3, 59 (1890—1894).
[122] R. Bauer, Ber., 42, 2107 (1909).
[123) Г. E. Фирц-Давид Л. Бланжей, cm.99.
И24] Г. E. Ф и p ц - Д а в и д, Л. Б л а н ж е й, см.вв.
[125] Н. О Hermann, Ann.,' 280, 5 (1894).
[126] Г. Е. Фирц-Давид, Л. Бланжей, см.98.
508
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Нафталин-а-сульфокислота [127]. В 260 г II2SO4 (моногидрат), охла*^
жденпой до 0° С, при перемешивании быстро вносят 128 в тонкоизмельченного'!
нафталина. Для того чтобы предотвратить внезапное выделение сульфокислот®
из пересыщенного раствора, после окончания добавления нафталина кристал-1
лизацию вызывают'внесением небольшого количества ct-сульфокислоты (полу-.<1
ченной нагреванием на водяной бане пробы нафталина с серной кислотой И&
последующим охлаждением). Если часть нафталина не вступила в реакций^
сульфирования (непрореагировавший нафталин обнаруживается по обрааовай]
нию мути при разбавлении небольшой пробы реакционной смеси водой), массга
необходимо нагревать некоторое время при 60° С. За
Нафталнн-р-сульфокислота [128,_ 129]. В колбе, снабженной тормомеО
трои, капельной воронкой и мешалкой, расплавляют 100 г нафталина и npjda
160° С в течение примерно 15 мин приливают по каплям 160 г концентрирован*!
ной HsSO4. Смесь перемешивают еще 5 мин при 160° С, выливают в 120 мм
холодной воды, через 24 ч отфильтровывают выпавшие кристаллы и отжимают^
на пористой тарелке. Для очистки полученный продукт порциями по 10 «и
растворяют в 5 мл воды, фильтруют в горячем состоянии, смешивают горячими
раствор (температура выше 70° С) примерно с 2 мл концентрированной Н(ЗЯ
и охлаждают. Выход сульфокислоты в виде тригидрата 160 г. Кислоту сушам
на порйстой тарелке или в эксикаторе над NaOH. Д
При более высокой температуре и более длительном воздействии олеума на на$Й!
талин получают ди- и трисульфокислоты. О получении нафталин-1,5- и нафталина
1,6-дисульфокислот см. [130], нафталин-2,7-дисульфокислоты, см. [131], о получен™
яэфталин-1,3,5- и нафталин-1,3,6-трнсульфокислот см. [99]. ;J|
В ряду нафтолов прямому сульфированию обычно подвергают только Р-нафтоДи
а-нафтолсульфокислоту чаще всего получают другими путями (например, щелочные
плавлением нафталинди- и нафталинтрисульфокислот или по реакции Бухерера Иж
соответствующих 1-нафгиламинсульфокислот). Однако в отдельных случаях проводя®
прямое сульфирование. Так, например, при взаимодействии а-нафтола с серной кши
лотом [132] в течение 2 ч при 50° С или с моногидратом [133] в течение 36—48 ч пр®
20° С, если необходимо с последующей однодневной обработкой прп 40° С. получаю®
1-нафтол-2,4-дисульфокислоту в виде кислой натриевой соли с выходом 95% от тега
ретического. -
Место вступления сульфогруппы в молекулу Р-нафтола в большой мере зависла
от температуры реакции. При низких температурах получается 2-нафтол-1-сульф*ВД
кислота [134J; с повышением температуры или в присутствии избытка H8S04 из эгаМ
кислоты в результате миграции сульфогруппы образуется 2-нафтол-8-сульфокислОЙЙ
(кроцеиновая кислота) и, наконец, при 110° С — почти исключительно 2-нафтоЛ^М
сульфокислота (кислота Шеффера). Дальнейшее сульфирование избытком На9ЦЯ
приводит к р-нафтолди- и р-нафтолтрисульфокислотам [135], используемым дая синя
теза красителей. "я
[134]
[135]
Г. Е. Ф н р ц - Д а в н д, Л. Б ланжей, см. [99].
L. Gattermann, Н. Wieland, см. [82], стр. 171.
О. N. Witt, Вег., 48, 751 (1915).
Герм. пат. 45229 (1887); Frdl., 2, 244 (1887-1890).
Герм. пат. 48053 (1888); Frdl., 2, 243 (1887-1890).
М. Conrad, W. Fischer, Ann., 273, 105 (1893). j
Houben - Weyl, Methoden der organischen Chemie, Bd. IX, 4 Aufl., Geo®
Thieme Verlag, Stuttgart, 1955, S. 485. Я
Герм. пат. 74688 (1893); Frdl., 3, 440 (1890—1894); R. N ietzki, Ber., 1»
.305 (1882).
Г. E. Фирц-Давид, Л. Бланжей, см.99; Frdl., 4, 623—643 (189M
1897); К. В епкатараман. Химия синтетических нрасителей, пв«
с англ., т. 1, Госхимиздат, 1956; Ullmanns Encyklopadie der tecnnischen Chemjg
Bd. 12. 3. Aufl., Urban u. Schwarzenberg, Munchen — Berlin, 1960, S. 58®
609; Ч. Сьютер, Химия органических соединений серы, пер. с англ., ч.
Издатинлит, 1951. . *
I. ОБМЕН ВОДОРОДА НА S-ФУНКЦВЮ
569
При сульфировании ct-нафтиламина всегда образуется смесь сульфонислот, при-
чем сульфогруппа вступает в положение 4, 5 или 6. Однако обычно 1-нафтиламин-
сульфокислоты получают не сульфированием а-нафтпламина., а другими путями, на-
пример восстановлением яитронафталинсудьфокислот.
1-Нафтиламин-4-сульфокислота [136] (скитез методом «запекания» [111]).
Получение яислого сульфата. В трехгорлой колбе, снабженной
капельной воронкой, мешалкой и нисходящим холодильником, нагревают
до 120—125° С 73,5 г 70%-ной H2SO4 и при энергичном перемешивании в тече-
ние 30 мин приливают по каплям нагретый до 50° С раствор 75 г а-нафтиламина
в 15 г бензола, непрерывно отгоняя бензол. Твердый остаток сушат 18 ч
при 120° С.
Получение сульфокислоты. Нагревают 75 г тонконзмель-
ченного сульфата до 180° С при 10—15 мм рт. ст. в течение 8 ч. После охла-
ждения полученный светло-серый осадок растворяют примерно в 500 мл воды,
добавляя 20 г безводного Na2COs. Раствор доводят до кипения, фильтруют,
экстрагируют бензолом непрореагировавпптй нафтиламин, еще раз нагревают
до кипения. К этому раствору добавляют НС1 до начала помутнения и далее
активированный уголь и отфильтровывают в горячем состоянии. Охлажденный
раствор подкисляют НС], отфильтровывают выделившуюся нафтионовую ки-
слоту и сушат ее при 100° С. Выход кислоты 60—65 г (85—95% от теоретиче-
ского).
Более однозначно протекает замещение при сульфировании ^-нафтила мина.
При соблюдении соответствующих условий (температура и концентрация серного ан-
гидрида в сульфирующей смесн) можно получать 2-нафтилампн-5- и 2-нафтилаиин-8-
сульфокислоты или 2-нафтиламин-6- и 2-нафтиламин-7-сульфокислоты. С увеличением
концентрации SO3 в сульфирующей смеси из моносульфокислот образуются ^-нафтил-
аминди- и в-нафтиламиятрисульфокислогы [135].
2-Нафтиламип-1-сульфокислоту (кислота Тобиаса) можно получить с выходом
90% от теоретического взаимодействием ^-нафтнламина с SO3 в тетрахлорэтане [137].
При сульфировании антрацена наряду с Р-сульфояпслотой образуются дисуль-
фокпслоты [138]. Более подробно изучено сульфирование антрахинона [139]
и фенантрена [140]. При сульфировании фенантрена серной кислотой получают фенан-
треп-2- и фенантрен-З-моносульфокислоты, которые можпо разделить [141].
Сульфирование серной кислотой и олеумом в присутствии катализатора
В присутствии солей металлов [например, Na2SO4 и (NH4)aSO4] агрессив-
ное действие серной • кислоты вследствие эффекта разбавления ослабляется.
Описано [142]. например, сульфирование пирена в присутствии NaaSO4. 1-Амиио-
аптрахинон-2-сульфокислоту также целесообразно получать [143] в присутствии
NaaSO4.
Наиболее давно известно каталитическое влйянпе, оказываемое на реакцию
сульфирования ртутью и ртутными солями, которые не только увеличивают скорость
реакции, ио и определяют направление замещения вследствие промежуточного
образования металлоорганических соединений [139]. Так, без катализатора нз
[136]
[137]
[138]
[139]
[140]
[141]
[142]
[143J
R. Н. С. Nevile, A. W i n t h е г, Вег., 13, 1948 (1880).
Пат. США 1969189 (1932); С., 1935, I, 960.
Герм. пат. 77311 (1893); Frdl., 4, 271 (1894—1897).
К. Lauer, J. prakt. Chem. [2], 135, 173 (1932); к. La uer, J. prakt. Chem.
[2], 138, 81 (1933).
L. F. Fieser, J, Am. Chem. Soc., 51, 2460, 2471 (1929); cp. F. L. С о h e uj
U. Cormier, J. Am. Chem. Soc., 52, 4363 (1930).
L. F. Fieser, Org. Synth., Coll. v. 2, 1955, p. 482.
E. T i e t z e, O. Bayer, Ann., 640, 189 (1939).
Герм. пат. 484997 (1927); Frdl., 16, 1248 (1931).
570
Ol>r VfOU.kHIIF C-S-СВЯ-ЗИ Б РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
антрахинона u олеума образуется антрамшои-2-сульфокислота Illi], в присутствии'1
же солен ртутц — антрахинон-1-сульфокислота [144].
Антрахинон-1-еульфокислота. В 200 мл 20%-ного олеума вносят прк^
перемешивании хорошо растертую смесь 208 г чистого сухого антрахинону/
с 4 г отмученной IlgO или HgSC>4 и перемешивают при 50° С до полного раство-
рения. Полученный раствор нагревают в течение 1 ч до 130—135° С, в течение/
последующих 2 ч приливают по каплям 50 г 60%-ного олеума, после чего пере-
мешивают еще 1 ч при 135° С. После охлаждения смесь выливают и 2 л ледяной/
воды, пагревают до кипения и отфильтровывают от пепрореагировавшего антра^
хинона (около 60 г). Фильтрат вновь доводят до кипения и прп перемешиваншГ?
добавляют к нему нагретый до той же температуры раствор 80 г КС! в небодь^
шом количестве воды. Полученный раствор охлаждают до 60° С и выдерживаю»/
прн этой температуре 6 ч. Выпавший осадок отсасывают при 60° С иа пред-г
варнтельно нагретом нутч-фильтре, промывают насыщенным раствором KCtJ
и небольшим количеством воды. Выход калиевой соли анграхинон-1-сульфо-*
кислоты 190 г (58% от теоретического). w
При сульфировании фталевого ангидрида действием SO3 образуется 4-сульфо*
фталевый ангидрид, а в присутствии HgSO4 — 3,5-дисульфофталевый ангидрид [145]^
Ртуть и ртутные соли катализируют сульфирование 4-метилпиридина до 4-метплпириЧ
дин-3-сульфокислоты [146] и пиридина до пиридин-З-сульфокислоты [147]. Примени?
ние катализаторов в этих случаях способствует значительному повышению выходЙ
продуктов. Хинолин в присутствии 0,2—3% ртути можно превратить в хинолин-5-'И
сульфокислоту (148] без одновременного образования хинолиц-8-сульфокислоты.?
Катализаторами реакции сульфирования являются также фтористый водород^
трифторид бора [87] (например, при сульфировании бензола, толуола, нафталина^
нафтиламина, нафтола, фенола, кароазола и дифенила) и борная кислота [149] (нацря/*
мер, при получении хишшарин-6-сульфокислоты). Иод не оказывает влияния на реак<
дню сульфирования [150]. ,
Сульфирование серным ангидридом и его аддуктами
Преимущество прямого сульфирования действием серного ангидрида [151Н
по сравнению с сульфированием серной кислотой заключается в том, что в процессе^
реакции не образуется вода и тем самым однозначно определяется направление замеще-^
ния [152]. В большинстве случаев реакция протекает очень бурно, часто даже с обут *
гливанием, поэтому целесообразно проводить ее при низких температурах или в прн- -
сутствии растворителей. Сульфирование можно осуществлять пропусканием газооб’я
разного серного ангидрида в жидкое сульфируемое вещество или в его раствор. ' .«
Метиловый эфир сульфосалициловой кислоты (153]. Серный ангидрВД.л
пропускают в метиловый эфир салициловой кислоты до затвердевания реакцион-
ной массы. Температуру при этом медленно повышают с 25 до 107° С. Л
В качестве растворителя при сульфировании серным ангидридом особенно .
пригоден жидкий сернистый ангидрид [154]. ,
144]
145]
146]
147]
148]
149]
150]
151]
[152]
[153]
[154]
Г. Е, Фирц-Д. авид, Л. Б л а н ж е й, см.9».
Н. Waldmann, Е. Schwenk, Ann., 487, 287 (1931). •
J. L. W ebb, A. H. Cr o-w in, 3. Am. Chem. Soc., 66, 1456 (1944).
S. M. M с E 1 I va in, M. A. G о e s e, J. Am. Chem. Soc., 65, 2233 (1943)»
Пат. США 26 89850 (1952)' С., 1956, 6811.
Герм. пат. 492000 (1928); Frdl., 16, 1247 (1931).
V. A u g е г, М. V а г у, С. г., 173, 239 (1921); С., 1922, I 492.
С. М. Suter, A. W. Weston, Org. Reactions, 3, 141; G. F. L i s к, Inik.
Eng. Chem., 40, 1671 (1948); 41, 1923 (1949); 42, 1746 (1940). <
K. Lauer, J. prakt. Chum. [2], 143, 127 (1935).
Пат. США 2527880 (1950); С. A., 45, 1164 (1951). J
L. Leiserson R. W В Ost R. LeBaron, Ind. Eng. Chem., 40, 505
(1948); W. H. C* R u e g g e b e r g, T. W. S a u I s, S. L. N о r w о о de
J. Org. Chem., 20, 455 (1955).
I. ОБМЕН ВОДОРОДА НА S-ФУНКЦИЮ
571
Сильного окислительного действия серного ангидрида можно избежать, исполь-
зуя уже упоминавшиеся аддукты серного ангидрида (стр. 560), сульфирующая актив-
ность которых зависит от их стабильности [151]. Димер сорного ангидрида или про-
дукты присоединения серного ангидрида к минеральным кислотам (например, H2SaO7)
являются более сильными сульфирующими агентами, чем стабильные аддукты с диок-
сапом [77, 155] и третичными аминами, с помощью которых можно вести сульфирование
в очень мягких условиях [156]. Неустойчивые в кислой среде соединения сульфи-
руются без разложения аддуктом серного ангидрида с пиридином.
Аддукт серного ангидрида с пиридином (пиридинсульфотрноксид) [157].
При перемешивании и хорошем охлаждении к раствору 1 моль S03 в трехкрат-
ном количестве СС]4 прибавляют 1 моль пиридина. Образующийся аддукт
отсасывают, промывают для удаления незначительных количеств пиридинсуль-
фата небольшим количеством ледяной воды и высушивают. Выход продукта
около 90% от теоретического.
Вместо 8О3 можно использовать хлорсульфоновую кислоту (1 моль).
При помощи пирпдинсульфотриоксида особенно легко сульфируются гетеро-
циклические соединения; из пиррола получают пиррол-2-сульфокислоту [158], из
2-ацетилпиррола — соответствующую 4-сульфокислоту {159J.
Фуран-2-сульфокиелота [160]. Чистый фуран (т. кип. 31,2—31,5° С),
полученный из технического фурана разложением его аддукта с малеиновым
ангидридом при 140—150° С, нагревают 8—10 ч при 100° С в толстостенной
трубке с пиридинсульфитриоксидом. Реакционную массу обрабатывают в тече-
ние 30—40 мин водной пастой ВаСО3 и фильтруют в горячем состоянии. Фильт-
рат упаривают и осаждают сульфонат бария этиловым спиртом. Выходы про-
дукта зависят от количества пиридинсульфотриоксида. Так, из 1,9 г фурана
получают следующие выходы сульфокислоты:
Пиридинсульфотрноксид выход сульфокислоты
г %
4.4 30
8.8 56
13.4 90
Я-Бенаилизотиурониевая соль фурап-2-сульфо/сислоты плавится при
205° С.
2-Ацетил-5-фурансульфонат бария. Аналогичным образом 2,0 г 2-ацетил-
фурана и 5,8 г пиридинсульфотриоксида нагревают в течение 10 ч при 140° С
в 10 мл дихлорэтана и затем смесь обрабатывают ВаСО3. При этом наряду
с 0,8 г исходного продукта получают 2,5 г (82,5% от теоретического) 2-ацетил-
5-фурансульфоната бария [161], который перекристаллизовывают из разбавлен-
ного спирта.
Из индола и 3-мстилиндола образуются 2-сульфокислоты [162].
1155] С. Kl. S u t е г, Р. В. Evan s, J. М. Kiefer, J. Am. Chem, Soc., 60. 538
(1938).
[156] P. Baumgarten, Ber., 59, 1976 (1926).
157] Герм. пат. 514821 (1926); Frdl., 16, 534 (1931).
1158] А. П. Теревтье», JI. А. Японская, В. Г. Я in у н с к и й, ЖОХ
20, 510 (1950); С. А., 44, 7828 (1950); ср. С. А., 43, 7015 (1949).
[159] А. II. Т е р еить ев, Л. А. Яновская, ЖОУ, 19, 2118 (1949); С. А., 44.
3973 (1950).
1160] А. П. Терентьев, Л. А. Каз if цы на, ЖОХ, 18, 723 (1948);
С. А., 43, 214 (1949).
[161] A. IL Терентьев, J1. А. Казицяна, Л. М. Туровская.
, . ЖОХ, 20, 185 (1950); С. А., 44, 5862 (1950).
[162] A. Ii. Терентьев, С. К. Голубева, Л. В. Цымб.ал, ЖОХ,
19, 781 (1949); С. А., 44, 1095 (1950).
572 ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Некоторые ароматические соединения, например анилин, фенол и нафталии,-
также можно сульфировать при 170° С пиридинсульфотриоксидом [156]. >
Сульфирование ароматических соединений галогенсулъфокислотами ,<
и другими сульфирующими агентами
При сульфировании ароматических соединений хлорсульфоновой кислотой
в большей степени, чем при сульфировании другими реагентами, происходят последу*
ющие реакции, в частности образование сульфохлоридов и, возможно, сульфонов!
Так, например, в промышленности действием избытка хлорсульфоновой кислоты ш
толуол получают о- и n-толуолсульфохлориды. Однако при определенных условий
(соотношение реагентов, температура, применение растворителей) хлорсульфоноваи
кислота может с успехом использоваться и для введения сульфогруппы.
Сульфирование хлорсульфоновой кислотой целесообразно проводить в инерф
ных растворителях, папример в сероуглероде, четыреххлористом углероде, хлоро^
форме, хлорбензоле, дихлорбензоле и нитробензоле.
Преимуществом сульфирования хлорсульфоновой кислотой по сравнению с суль
фировакием серной кислотой является возможность проведения "Ьроцесса в более мят
ких условиях, что приводит к получению более чистых и однородных продуктов
Заместители, изменяющиеся под действием H^SO* (например, нитрильная группа)
пе затрагиваются при сульфировании хлорсульфоновой кислотой.
Этим методом были получены 2-нафтолсульфокислота-1 [163], хризенсульфо
кислота*-6 [164] и л-крезолсульфокислота-б [165]. Особенно легко таким способе!
сульфируются ароматические амины.
1-Амино-3-трифторметилбензол-4*сульфокислота (166]. Растворяют 161 *
1-амино-З-трифторметилоензола в 1 л тщательно высушенного о-дихлорбен-
зола, прибавляют при пяре.мелнипянии 120 г свежеперегнаиной хлорсульфрно*
вой кислоты и медленно нагревают до 180° С; при этом происходит энергичней)
выделение хлористого водорода (4—5 ч). Смесь охлаждают, отсасывают осадоМг
промывают его эфиром. Сухой порошок растворяют при нагревании в 600'Jkf
2 н. раствора Na2C0g и 1 л воды, экстрагируют эфиром непросульфированн^
основание и подкисляют НС] (по конго); сульфокислота выпадает в осадой;
Аналогичным образом сульфируют различные замещенные аминотрифто^»
метилбензолы. J
9,9 -Спиробифлуорен-2,2'-дисуЛьфокислота [167].
К кипящему раствору 12,8 г 9,9'-спиробифлуорена в 80 мл хлороформ®
в течение 75 мин, прибавляют раствор 5,4 мл перегнанной хлорсульфоново^
кислоты в 20 мл хлороформа. После Зч нагревания смесь охлаждают, отфнльт*
Бовывают продукт, промывают его хлороформом и сушат в вакууме над КОН*
ыход продукта 17,9 г; т. пл. 225° С.
[163] Г. Е. Фирц-Давид, Л. Б л а н ж е й, см.9».
[164] М. S. Newman, J. A. Cathcart, J. Org. Chem., 5, 621 (1940).
[165] R. D. Haworth, A. Lapworth, J. Chem. Soc., 125, 1303 (1924).
[166] ГерМ. пат. 629257 (1934); Frdl., 22, 287 (1939).
[167] J. H. Weisbdrger, E. K. Weisburger, F. E. Ray, J. Anu
Cbem. Soc., 72, 4253 (1950).
I, ОБМЕН ВОДОРОДА НА S-ФУНКЦИЮ
573
Аналогичным образом ароматические углеводороды можно сульфировать фтор-
сульфоновой кислотой, если необходимо — под давлением [168]. Для сульфирования
терпенов в качестве сульфирующих агелтов иногда применяется пнросульфурпл-
хлорид [169], для сульфирования третичных ароматических аминов — тионил-
хлорид [170].
В фенольные соединения сульфогруппы можно непосредственно вводить еще
одним способом — взаимодействием их с сульфитом или бисульфитом натрия (сульфит-
ная реакция).
Из резорцина получается натриевая соль 1,3-диоксициклогексантрисульфокис-
лоты-1,3,5, пз которой под действием щелочи отщепляются 2 моль NaHSOs и 1 моль
Н2О и образуется фенол-ж-сульфокислота [171].
Фенол-ж-сульфокислый натрий [172]. В течение 22 ч нагревают 100 а
резорцина с 1 л раствора бисульфита натрия (d = 1,32), затем осаждают суль-
фит- и сульфат-ионы ацетатом бария, фильтруют и фильтрат упаривают до
объема 500 мл. После обработки полученного раствора 1 л 85%-ного этилового
спирта получают 242,8 г тринатриевой соли 1,3-диокси-1,3,5-цпклогексаятри-
сульфокислоты. К 100 г этой соли в 100 мл воды прибавляют 40 мл 50%-ного
раствора NaOH и оставляют на ночь. Затем смесь нейтрализуют НгЗО4, до-
бавляют 400 мл 85%-ного этилового спирта, фильтруют, упаривают и остаток
экстрагируют горячим 99%-ным этиловым спиртом. Из экстракта высаживают
натриевую соль фенол-ж-сульфокислоты 500 мл эфира. Выход продукта 23,55 в;
т. пл. 314° С после переиристаллизацин из этилового спирта.
Этим методом получены также 1,4-диоксиаи грахинон- 2-сульфокислота из хи-
инзарина [173] и 1,2,4-триоксиантрахинон-З-сульфокислота из пурпурина [174]
В присутствии окислителей таким же способом можно сульфировать амины,
полиамины и аминофеполы, склонные и образованию хинопов [175]. Например, из
а-нафтиламина получена 1-амино-2-оксинафталинсульфокислота-4 [176], а из п-фени-
лен^иамяна — моносульфокислота [177].
При сульфировании питрозосоединений бисульфитом одновременно происходит
их восстановление и образуются аминосульфокислоты [178]. При взаимодействии
ароматических нмтросоединеняй с бисульфитом одновременно сульфируется ядро
и возникают ариламин-№сульфокислоты, которые после отщепления N-сульфогрупны
также превращаются в аминосульфокислоты. Эта реакция известна в литературе как
реакция Пириа, см. обзор [179]. , ,
2. Введение сульфохлорнднон группы
В алифатическом ряду замещение водорода на сульфохлоридную группу осуще-
ствляют главным образом но способу Рида [180] одновременным воздействием на
[168]
[169]
[170]
[171]
172
173
174
175
176
[177]
[178]
[179]
[180]
J. Н. Simons, Н. J. Passino, S. Archer, J. Am. Chem. Soc., 63,
608 (1941).
Пат. США 2220678 (1937); С., 1941, I, 2750.
4- Michaelis, E. Godchaux, Ber., 23, 553 (1890).
W. M. Lauer, С. M. Langkammerer, J. Am. Chem. Soc., 56, 1628
(1934).
В. H. Уфимцев, ЖПХ, 20, 1199 (1947); C. A., 43, 2595 (1949).
Герм. пат. 287867 (1914); Frdl., 12, 436 (1914—1916).
Герм. пат. 288474 (1914); Frdl., 12, 437 (1914—1916).
ср. A. Seyewetz, C., 1936, I, 2923.
E. В a m a n n, K. Schriever, G. Muller, Arch. d. Pharm., 287, 570
(1954).
Герм. пат. 64908 (1892); Frdl., 8, 40 (1890—1894).
J. M. Kogan, Ber., 64, 2150 (1931); Г. E. Ф ирц -Давид, Л. Б л а н-
ж е й, см.99.
W. Н. Hunter, М. М. Sprung, J. Am. Chem. Soc., 53, 1432—1447
(1931).
Пат. США 20460090 (1933); С., 1937, I, 720; пат. США 2174110 (1934); G., 1940,
I, 311; пат. США 2174492 (1938); С., 1940, I, 465; пат. США 2263812 (1938);
С., 1945, 11, 296; пат. США 2370421 (1945); С. А., 39, 3420 (1945).
574
ОБРАЗОВАНИЕ G—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
алифатические соединения двуокиси серы и хлора (су.чьфохлорированпе) [181] по сум- •
парному уравнению:
HH + SOs + CI2 —> HSO2CI4-HCI
Реакция, механизм которой был неверно истолкован первооткрывателем, прово-
дится при УФ-облучении пли ₽ присутствии катализаторов при 20—25° С и протекает '
но свободнорадикальному цепному механизму [1816]:
С12 —— > Cl • +С1 • образование свободных радикалов
RH+C1- —*• R-+HCI |
R- +SO3 —> RS02- г рост цепи
ЯЗО, • + Я2 —. RSO-jCl + CI • J J
Cl.-j-сЬ —»-Cl2 обрыв цепи
r. +cl- —*• rci ;
Прп сульфохлорврованим одновременно происходит обычное хлорированиег
углеродной цени:
RH4C12 —> RCI + HC1 !
Однако при УФ-облучепии эта конкурирующая реакция неожиданно почти сов--г.-
сем отстукает на задний план и, такам образом, ход реакции почти избирательно на- j
правляется в сторону сульфохлорирования [182]. Изменяя температуру и соотноте-.-®
ние сернистого ангидрида и хлора, также можно подавить реакцию хлорирования,-
Оптимальные условия сулъфохлорированпя: УФ-облучение (X = 3000—3600 А)Л-
температура 20—25° С (начальная температура реакции 35—40° С) и соотношение
хлора и сернистого ангидрида 1 : 1,3. Прп пропускании хлора и сернистого ангидрида. ‘
в находящуюся в темноте смесь совершенно чистых парафиновых углеводородов реак-’-*
ция практически не идет. Однако в присутствии органических перекисей [183] и дру-^:
гих радикалобразующих катализаторов [184[ реакция протекает даже в темноте.:
Наиболее пригодными катализаторами оказались перекись ацетона, обработанный:;
кислородом скипидар, перекиси «ipem-бутила [185] и ацетилпиклогекспдсульфо- '
нила [186]. ;
Наиболее гладко и однозначно в указанных выше условиях происходит сульфо-
хлорирование «-парафинов и циклопарафинов простого строения. Разветвленные пара-
финовые углеводороды и алкилированные ликлопарафины менее прпгодны, так как
третичные атомы водорода не замещаются на SO»CI-rpyuny.
Вопреки данным патентной литературы 1180] ароматические соединения этим
путем заметно не сульфохлорируются [181, 182]; например, толуол только хлорп-
руется, этилбензол и «-пропилбенэол сульфохлорируются только в боковую цепь
на 8 и 20% соответственно.
Сульфохлорпрованпе кислородсодержащих соединений (спиртов [187], альде-
гидов, кислот, сложных эфиров [180], эфиров, ацеталей и др.) происходит почти всегда
[181] Обзорные статьи по с>льфохлорирс»валию: a) J. Н. Н е I Ь е г g е г, Angew.
Chem., 55, 172 -(1942): h) Н. J, Schumacher. J. Stauff,’ Angew. *
Chem., 55, 341 (1942); с) Ф. А з и н г е р, Химия и технология парафиновых л
углеводородов, пер. с нем., ГостояГехиэдат, 1959.
[182] Н. К г о е р е 1 i n. W. О р 1 t z, W. Freis s. Erdol u. Kohle,2, 498 (1949);
Angew. Chem., 64, 273 (1952); ср. также C., 1950, 11, 487. -J
[183] G. F. Lisk, Ind. Eng. Chem., 40. 1671 (1948). j
[184] Герм. пат. 765790 (1953); С, 1954 188: англ. пат. 628014 (1949); С.Л., 44, 2556 1
(1950); пат. ФРГ. 833808, 839351 (1949): С.. 1953, 465; пат. С1П А 2503253 (1947);
С., 1952, 4244; К. Ziegler, Brennstoffchcm., 30, 181 (1949). •
[185] Пат. США 2536008 (19о0)- С. А., 45, 4262 (1951); пат. США 2542578 (1951); J
С. А., 45. 8027 (1951).
[186] Пат. ФРГ. 841147 (1952); С. А. 47 4897 (1953); R. G г a f, Ann., 578, 50 (1952).
[187] Франц, па-i. 8701^ (1941)-С. 1942, I Г, 478; англ. пат. 538407 (1941); С. А.. 36, i,
1335 (1942).
I. ОБМЕН ВОДОРОДА НА S-ФУНКЦНЮ
575
неудовлетворительно но сравнению с сульфохлорприванием парафиновых углеводо-
родов. В большинстве случаев в молекулу преимущественно вступает хлор без при-
соединения серы.
Из 1-хлорбутана получают 1,2-, 1,3- и 1,4-хлирбутапсульфохлориды [188], гид-
ролиз которых дает соответствующие хлорсульфокис.юты. 1-Хлорбутансульфокпс-
.ioif.i-З и -4 при нагревании отщепляют хлористый водород и превращаются в суль-
ьшм (внутренние ангидриды оксисульфокислот )[189].
Обычно после сульфохлорирования в реакционной массе содержатся следующие
продукты [181в|: моносульфохлориды; ди- и полисульфохлориды; хлормоносульфо-
хлориды; хлорди- и полисульфохлориды; алкилхлориды п непрореагировавший ис-
ходный продукт (неомыляемые нейтральные масла).
При сульфохлорированни низкомолекулярных парафиновых углеводородов
отдельные продукты реакции могут быть разделены лереюнкой. Оказалось также, что
моносульфохлориды можно довольно четко отделить от иолисульфохлоридов [190],
если к их смеси добавить не менее пятикратного количества пентана и охладить
до —30° G.
Показано [191, 193—195], что строение мокосульфохлоридов может быть опре-
делено по правилу Хасса [192], согласно которому замещение вторичного атома водо-
рода происходит примерно в 3,25 раза быстрее, чем первичного. Соответственно этому
при сульфохлорированни пропана образуется 50% иропап-1- и 50% пропан-2-сульфо-
хлорида [193], из бутана [194] — 33% бутап-1- и 07% бутап-2-сульфохлорида, а при
сульфохлорировании н-додекана [195] после удаления из смеси нейтрального масла
и ди- и полисульфохлоридов получается смесь, состоящая из 8,5% додекан-1-и по
18,3% додекан-2-, додекан-3-, додекан-4-, додекан-5-и додекан-6-сульфохлоридов.
Обычно в лабораторных условиях жидкие углеводороды сульфохлорируют
в вертикальной кварцевой трубке (в крайнем случае, в стеклянной трубке) диаметром
около 60 мм, снабженной внутренним термометром и охлаждающим змеевиком. При
сульфохлорированип газообразных углеводородов Трубку заполняют СС14; углеводо-
род и сернистый ангидрид с хлором вводят через пористые распылители. Источником
сЪета служит кварцевая лампа, устанавливаемая па расстоянии 25 см.
Сульфохлорирование пропана [193, 196]. Прн 15 мм рт. ст. перегоняют 1000 г
уже освобожденной от растворителя смеси, полученной пропусканием при освещении
кварщчюй лампой 2,5 объеми. ч. пропана, 1,1 объемн. ч. SO2 п 1 объемп. ч. хлора
в СС14 до достижения в реакционной массе примерно 20%-ного содержания продукта
реакции. При 70—150° С перегоняется 85% загрузки (основная часть перегоняется
при 70—90° С). Дистиллят состоит в основном из пропанмоносульфохлорида, неболь-
шого количества хлорпропан- й дихлорпропанмопосульфохлорпдов, остаток — из
дисульфохлорцда. Последующей перегонкой дистиллята на колонке Рашига длиной
50 см (флегмовое число 1 : 5) получают 88% смеем изомерных ироп&нмоносульфо-
хлоридов: т. ьип. 72—79° С (15 жж рт. cm.}', d2Q — 1,268; 1,452. Остаток от пере-
гонки состоит из монохлор- и дихлорпропансульфохлоридов.
Как уже указывалось, сульфохлоридную группу нельзя ввести в ароматические
углеводороды до методу Рида, однако сульфохлориды ароматических соединений
удается получать взаимодействием ароматических соединений с хлорангидридсм
серной кислоты [197]. По реакции с хлорсульфоновой кислотой сульфохлориды обра-
зуются при наличии достаточно большого избытка хлорсульфоновой кислоты высокой
чистоты. В качестве побочных продуктов могут образоваться сульфоны, которые
[188] Англ. пат. 516214 (1938); С., 1940, II, 1076.
[189] J. 11. II е1 b er? er, G. Na леске,' П. М. Fischer, Ann., 562, 23
(1949).
[190] Герм. пат. 711821 (1939); С., 1942, 1, 1448; герм. пат. 734562 (1941); С. 1943,
11. 1426.
[191] ср. F. Asin ger. Вег., 75, 668 (1942).
[192] if. В. Hass,E. Т. McBee Р. Weber, Ind. Eng. Chem., 27, 1190 (1935);
28 333 (1936)- H. B. Has s, E. T. McB e e, L. F. 11 a t c h, Ind. Eng.
Chem., 29, 1335 (1937).
[193] F. As i n ger, W. S c h m i d t, F. Ebeneder. Ber., 75, 34 (1942).
[194] F. As i nger, F. Ebeneder, E. Bock. Ber., 75, 42 (1942); F. A s i n-
ger, F, Ebeneder, Ber., 75, 344 (1942).
[195] F. Asinger, Ber., 77, 191 (1944).
[196] Франц, пат. 842508 (1938); С., 1939, II, 2712.
576
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
можно/подвергнуть дальнейшему сульфохлорированию [198]. Из бензола таким спо£
собом получают бензолсульфохлорид с выходом 75—77% от теоретического [199]^
п-ХлорбензЬлсульфохлорид [200]. К 60 г охлажденной до —15° С хлор*
сульфоновой кислоты прибавляют по каплям в течение 2 ч 20 г хлорбензол*^
Смесь периодически встряхивают в течение 2 ч при температуре от —5 д&
—10° С и затем выдерживают 12 ч при комнатной температуре. После этого,
ее выливают на лед, отделяют масло и перегоняют его в вакууме при 144’ С
(15 мм рт. ст). Выход сульфохлорида 22 г; т. пл. 53° С. Из остатка пефе
кристаллизацией из ледяной уксусной нислоты получают 5 г чистого п,я*5
'дихлордифенилсульфона.
Сульфохлориды различных замещенных ароматических соединений можно MH
лучать, добавляя по каплям при 0е С пятикратное количество хлорсульфоновой кислота
к раствору сульфируемого соединения в пятикратном количестве хлороформа и поем
дующей выдержкой (20 ли«) при комнатной температуре. Для выделения продук?
реакции реакционную массу разлагают льдом, промывают хлороформенный слой вода
и отгоняют растворитель. Остаток перекристаллизовывают из низкокипящего сухог
петролейного эфира или из хлороформа. Продукт высушивают в вакууме над конпед
трированной H2SO«. *
По этому способу синтезированы, например, следующие бензол- п нафталпнсульф<Й
хлориды [201]: д
Бензолсульфохлорпды
4-Фтор- ...................
4-Хлор- ...................
4-Бром- ...................
3,4-Дпхлор- ...............
2,5-Дпбром- ...............
З-Метил-4-фтор- ................
2-Метлл-4-хлор- ................
Нафталпнсуль’фохлориды
4-Хлор- ..................
7-Хлор- ..................
4-Бром- ..................
7~Бром- ..................
Т. пл., °C
35— 36 | из бензола
53.5 J нлп эфира
75.4
18—19
71 (после 1 ч
нагревания)
Жидкость
52—53 (после
10 мин прп 50° С)
92—93
124—126
81-83
142-143
Описано также получение околбензол- [202] и оксинафта.тнеульфохлй*
рядов [203].
Для получения ампноарилсульфохлорпдов целесообразно предварительно запр^
тить аминогруппу ацилированием [204—206). Сульфохлориды ароматических карбо®
[197] Е. Е. Gilbert, Е. Р. Jones, Ind. Eng. Chem.. 51,- 1151 (1959), 50, 141СГ
(1958); 49, 1553 (1957) (ежегодные обзорные сообщения). 1
[198] J. Pollak, М. 11 е i m b е rg-Кг а и Е. Katschcr, О. Lustig,
Chem., 55, 363 (1930).
[199] Н. Т. Clarke, G. S. Babcock, T. E.'Murray, Org. Synth, Coll. v.
78 (1937).
[200] F. U 11 m a n n, J. К о г s e I t, Ber., 40, 641 (1907). i
[201] E. H. Huntress F. H. Carlen. J. Am. Chem. Soc., 62, 511 (1940k’j
[202] J. Pollak, E. G e b a и er- Fill n egg, Mh. Chem., 47, 537 (1927); C.V
1928, II, 1081. "
[203] J. Pollak, E. G e ba иег - Fii In egg, E. Blumen s t ock-Hal?
w а Г d, Mh. Chem., 49, 187 (1928); C., 1928, 11. 1772; J. P a 11 a k, E, В 1
m e ns t ock-H a I w a rd, Mh. Chem), 49, 203 (1928); C., 1928, 11, 1779.J
[204| G. F. Lisk, Ind. Eng. Ghem., 42, 1751 (1950).
[205] Герм. пат. 573193 (1931); Frdl., 19, 699 (1934).
[206] R. N. Johnson, S. Smiles. J. Chem. Soc., 123, 2384 (1923).
I. ОБМЕН ВОДОРОДА НА S-ФУНКЦИЮ
577
новых кислот можно получать из хлорангидридов карбоновых кислот при помощи суль-
фирующих средств, не образующих воды при сульфировании [207].
З-Карбоксибензолсульфохлорид. В 100 г бензоилхлорида при 110е С
пропускают ЯО3 до привеса 71,3 г и затем смесь выдерживают 2 ч при 130° С.
После этого реакционную массу растворяют в хлороформе, раствор промывают
ледяной водой, отгоняют из него хлороформ. Остаток перекристаллизовывают
из толуола; т. пл. 3-карбоксибензолсульфохлорида 133° С.
Чтобы устранить побочные реакции и избежать многократного сульфохлориро-
вания часто рекомендуется проводить описанные выше синтезы в инертных раствори-
телях — СНС13, СС14 и др. Побочные реакции могут происходить также под влиянием
образующейся при реакции воды, которую можно связать добавлением избытка хлор-
сульфоновой кислоты или, лучше, добавленном олеума, пятиокиси фосфора или тиопил-
хлорида.
Для введения сульфохлоридной группы в ароматические соединения могут
быть использованы также пиросульфурилхлорид [208] и в присутствии конденсиру-
ющих средств, например хлорида алюминия [209], сульфурилхлорид.
3. Прямое введение сульфоновой н сульфоксидной групп
Прямое введение сульфоновой группы в ароматические соединения происходит
в качестве побочной реакции в процессе сульфирования' ароматических соединений
серной кислотой. При соответствующих изменениях условий реакции, например повы-
шении температуры сульфирования [210] или в присутствии водоотнимающих средств
[211], удается в достаточной мере увеличить выход сульфонов. Особо следует отметить,
общеприменимый метод получения простых и сметанных сульфонов [212] по Мейеру-
181], когда из равновесной смеси непрерывно удаляют образующуюся в реакции воду.
Этим способом были, например, синтезированы: дифенилсульфон, ди-п-толилсульфон,.
.и-диксилилсульфон, ди-п-хлорфекилсульфон, дитетралилсульфон, фенилтолилсуль-
фон, п-толил-л-ксилилсульфон, фенил-п-хлорфенилсульфон, р-нафтилтолилсульфон
и тетралнлтолилсульфон.
Ароматические сульфоксиды получают препаративно главным образом двумя
методами. Первый метод основан на том, что ароматические соединения могут взаимо-
действовать с тионилхлоридом в условиях реакции Фриделя — Крафтса; из толуола
при этом образуется ди-n- толилсульфоксид [213], а из бензола — дифенилсульфоксид
[214, 215].
Дифенилсульфокеид. К смеси 50 г бензола и 16 г SOCl2 прибавляют
порциями при охлаждении ледяной водой 30 s тонкоизмельченного А1С13.
Затем добавляют еще 20 г бензола, смесь нагревают 30 мин на водяной бане
и после охлаждения выливают в воду. Бензольный слой отделяют, промывают,,
йушат и отгоняют растворитель. Выход днфенилсульфоксида 21 г; после пере-
кристаллизации из петролейного эфира т. пл. 70—71° С.
207] Франц, пат. 872771 (1941); С., 1942, 11, 2219.
208] W. Steinkopf, К. Buchheim, Вет., 54, 2963 (1921).
209] Н. С. Brown, Ind. Eng. Chem., 36, 788 (1944).
210] Г,. E. Hinkel, G. H. R. Summers, J. Chem. Soc., 1949, 2854.
211] R. Otto, Ber., 19, 2421 (1886); G. M a c h e k, H? Haas, J. prakt. Chem. [2] r
, 160, 41 (1942); A. M i c h a e 1, A. Adair, Ber., 10, 583 (1877).
[212 ] H. Drews, S. Meyerson, E. K. Fields, Angew. Chem., 72, 493
[213 ] &19p°a’r k e r, Ber., 23, 1844 (1890).
[214 ] A. Schonberg, Ber., 56, 2275 (1923).
T215j R. L. S h r i n e r, H. C. Struck, W. J. J о г i s 0 n, J. Am. Chem. Soc.t
52, 2065 (1930).
37 Заказ 1835.
578
ОБРАЗОВАНИЕ О -S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Второй метод получения ароматических сульфоксидов заключается во взаимо-
действии ароматических аминов с бепзолсульфпповой кислотой [216, 217]:
\ /-8О«Н ч Н-<^ SO—NH.J :
4. Прямое введение сульфмновой группы *4-
В присутствии катализаторов реакции Фриделя — Крафтса углеводороды pea-J
гпруют с двуокисью серы с образованием сульфиновых кислот: ’Л
RH + SO2 HSO2H J
В случае алифатических углеводородов реакцию лучше проводит!, под давлей
нием и при повышенных температурах [218], в то время как из алкенов сульфиновы»;
кислоты образуются уже при атмосферном давлении [219].
Ароматические углеводороды и арилгалогениды могут быть превращены в суль*
фпновые кислоты со средним выходом 80% от теоретического. Реакцию необходим#
проводить при низких температурах, инициируя ее пропусканием сухого НС1, по анаж
логии с синтезом альдегидов по Гаттермапу. Образующиеся тетрахлоралюмппийсуль-^
финаты разлагают не кислотами, а сначала щелочами и лишь затем полученные таким.
образом соли щелочных металлов обрабатывают кислотой [220]. Из- эфиров фенола#
сульфиновые кислоты образуются уже на холоду; однако реакция легко протекавТ-
дальше до сульфоксидов и сульфониевых соединений ]221 ]: *
во-^2/-н+8оа —> во-so,h ;
2ВО-^-H + SO2 —> (R0-\ /~),S0
ЗВО-^^-н-bSOa [(h°-<_>-)3s]+C1-
Беизолсульфиновая кислота [220]. В суспензию 200 г А1С13 в 400 мЛ
бензола пропускают при охлаждении льдом 5 г сухого НС1 и затем 126 г сухого
SO8- Смесь выдерживают 12 ч при комнатной температуре, выливают наледа
затем смешивают с раствором 260 г NaOH в 1 л воды и нагревают с обратными
холодильником на водяной бане до полного разложения тетрахлоралюминищ-
А12О3 и, насыщая фильтрат СО2, удаляют остаток солей алюминия. Отфильтр<Д
ванный раствор упаривают и обрабатывают его концентрированной НС1. Выход^
чистой бензолсулъфиновой кислоты 170 г (80% от теоретического, считая на.1
хлорид алюминия); т. пл. 83° С. - f* 1
Этим же способом можно получить n-толуолсульфиновую кислоту с вы-2
ходом 94%; т. пл. 84° С. d
пц оиднхиш иаас ди уоа.ютсшщ icipa.uivpet411UaUUim^v;.
сульфината. Избыток бензола отгоняют с водяным паром, отфильтровывают*
А12О3 и, насыщая фильтрат СО», удаляют остаток солей алюминия. Отфильтро^’
При сульфинированпи труднолетучих углеводородов и производных углеводоро-^
дов целесообразно разбавлять реакционную массу сероуглеродом. Л
n-Ксилолсульфиновая кислота [220]. Прп взаимодействии 10 г n-ксилола; г
40 г сероуглерода и 12,6 г А1С13 при 0° С с хлористым водородом и сернистым.^
ангидридом после двухчасовой выдержки получают 12,5 г ге-ксилолсульфиново4-
кислоты; т. пл. 84—85° С.
Этим же способом синтезированы мезитилен-, исевдокумол-, п-хлор^*
бензол-, л-бромбснзол- и нафталин-а-сульфиновые кислоты.
О. Hins berg, Вес., 36, 113 (1903).
Р. W. В. Harrison, J. К е п у о п, И. Р h 11 I i р я, J.
1926, 2079.
Англ. пат. 321843 (1928); С., 1930, 1, 1366.
Герм. пат. 727205 (1938); С., 1943, I, 580.
Е. KDOcvciagol, J. Kenner, Ber., 41, 3315 (1908).
С. Е. С о 1 Ъ у, С. S. М с L о u g li 1 i п, Ber., 20, 195 (1887).
[216]
[217]
[218]
[219]
[220]
1221]
Chem. Soc Л
I. ОБМЕН ВОДОРОДА НА S-фУНКЦИЮ
579'
б. Введение тионовой, тиоэфирной и меркаптогрупп
Для получения тиокетопов замещением водорода серой можно иногда применять,
обработку производных дифенилметапа серой [222] или сульфидом натрия [223].
В присутствии катализаторов реакции Фриделя — Крафтса ароматические
атомы водорода замещаются серой с образованием простых тиоэфиров; этот метод,
например, был использован для синтеза дифенилсульфида [224], фенотиазпна [225[’
и его производных [226].
Тпоэфпры можно также получать, применяя вместо серы ее галогениды:
SCla Ь2СвН6 ——+ (C6H5)2S.A1C13 [-2НС1
S2Cl2-2CeH6 А1СЬ> (C.6H5)2S-A1CI3+2HC1 ^S
Дифенилсульфид [227]. В круглодонную колбу емкостью 5 л, снабжен-
ную мешалкой, капельной воронкой и обратным холодильником с отводной
трубкой для образующегося HCI, помещают 980 мл сухого бензола и 464 г
безводного А1С13. К смеси после охлаждения в бане со льдом в теченке-
1 ч прибавляют при 10° С раствор 405,1 г S2Ci2 в 450 мл бензола. Взаимо-
действие наступает внезапно с выделением хлористого водорода и осаждением1
желтого вязкого алюминиевого комплекса. Массу перемешивают еще в течение
2 ч при комнатной температуре и затем около 1 ч при 30° С до окончания выде-
ления хлористого водорода. После этого выливают реакционную смесь на 1 кг-
льда, отделяют органическую фазу и отгоняют бензол на паровой бане. Охлаж-
денный до 0J С остаток отфильтровывают от серы на воронке Бюхнера, смеши-
вают с 500 мл метилового спирта и перемешивают 3 ч при 0° С. Затем вновь
фильтруют и фракционируют перегонкой, причем при 155—170е С (18 мм рт.ст.у
перегоняется 470—490 г желтого масла. Для получения бесцветного продукта
* дистиллят нагревают при перемешивании н течение 1 ч с 70 г цинковой пыли
и 200 г 40%-ного раствора NaOH. Отделяют органический слой, промывают
дважды по 500 мл воды, сушат над Na2SO4 и перегоняют. Выход бесцветного-
дифенилсульфида 450—464 г (81 — 83% от теоретического); т. кип. 162—163° С
(18 мм рт. ст.).
Фенолы взаимодействуют с SCI2 в четыреххлористом углероде при —10J С.
с образованием тиоэфиров, выпадающих в виде бесцветных пли желтых осадков,,
которые перекристаллизовывают из бензола [2281.
Описано взаимодействие диалкилапилннои с хлоридами серы, приводящее-
к образованию соответствующих тиоэфиров и дисульфидов [229]. Из анилина п серы
в присутствии РЬО получается 4,4'-диаминодифенилсульфпд [230].
4.4'-Диамннодвфенилсульфид. При перемешивании 93 г свежеперегнан-
ного анилипа и 32 г серы нагревают на масляной бане до 135—145° С; в течение-
5^5 ч прн згой температуре добавляют порциями 160 г РЬО и перемешивают-
еще 30 мин. Темное масло смешивают после охлаждения с 30 мл 20%-ного-
раствора NaOH и продукт реакции трижды экстрагируют кипящим спиртом.
После отгопки спирта избыток анилина удаляют перегонкой с водяным паром.
[222] A. Е. Tschilscbibabin, I. L. К n u n j a n z, Вег., 62, 3051 (1929).
[223] Герм. пат. 287994 (1913); Frdl., 12, 207 (1914—1916).
[224] W. D i 1 t h е у, L. N е и h а и я et al., J. prakt. Chem. [2]. 124, 108 (1930).
|225] Герм. пат. 222879 (1909); Frdl., 10, 144 (1910—1912).
[226| Герм. пат. 224348 (1909); Frdl., 10, 144 (1910—1912).
[227] W. W. Hartmann, L. A. Smith, J. B. Dickey, Org. Synth.,.
Coll. v. 2. 1955, p. 242.
[228] F. D un n i n g, B. D и n n i n g jr.. \V. F.. Drake, J. Am, Chem. Soc.,.
53, 3466 (1931).
[229] E. Hoizmann, Ber., 20, 1636 ((.887); E. Holzmann, Ber., 21, 2056-
(1888).
[230] ftl. J.. AJ о о г e, T. В. Johnson, J. Am. C.hem. Soc., 57, 1287 (1935).
37*
580
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Полученный продукт действием разбавленной НС1 переводят в гидрохлорид^
Гидрохлорид растворяют в воде, кипятят и высаживают основание щелочью^
Можно произвести еще очистку через сульфат. В заключение продукт пера»
кристаллизовывают из 35%-ного спирта; т. пл. 107—108° С. ;
Взаимодействие гидрохлоридов первичных ароматических аминов с избытков
S2C12 (реакция Герца) [231] и последующий щелочной гидролиз дают тиазтионий^
хлориды соответствующих о-аминотиофенолов [232]. Если в амине свободно napaji
положение, то образуются 2-меркапто-4-хлоранилины. 3
Описано получение тиоэфиров взаимодействием Сульфеиилхлоридов с соедини
.пнями, содержащими подвижный атом водорода [254, 255]. _
II. Обмен галогена на S-функцию •,
1. Взаимодействие алкил- и арилгалогеиидов с солями >
и производными сероводорода j
Получение меркаптанов и дисульфидов 3
Наиболее употребительным методом получения тиолов является взаимоде^
•ствпе гидросульфидов щелочных металлов с алкилгалогенидами и достаточно актиз
ними арилгалогенидами. При этом вследствие установления характерного для гидр»
•сульфидов равновесия .
SNaSH ^=i Na.jS - HiS ' Я
возможно образование тиоэфиров. Этой реакции можно избежать, если пропуская
н реакционную массу IIaS или вести реакцию ирк температуре не выше 50—60° I
пли в автоклаве, в возможно более безводной среде. В таких условиях получен ря
низших алкаптиолов (до нонантиола) с выходами 36—74% от теоретического [233;
В случае арилгалогеиидов такие превращения удаются только ирк налИЧЙ
в них очень реакционноспособного галогена. Так, например, нагревая под давление]
о-галогенбензойные кислоты с гидросульфидом, щелочного металла в присутсТЫЙ
порошка меди, их можно перевести в тиосалициловую кислоту [234]. При миогочаы
вом нагревании n-нитрохлорбензола с избытком NaaS в водном растворе одиовремеи!
происходит восстановление питрогруппы и образование п-аминотиофенола [236
Очень гладко и уже в водном растворе происходит образование тиогликолевой кислот^
Тиогликолевая кислота [236]. К примерно 15%-пому раствору KS
(2 моль) при перемешивании постепенно добавляют раствор 100 г ХЯО
уксусной кислоты в 500 мл воды, нагревают 15 мин на водяной бане, смет
вают с концентрированным раствором ВаС12 и затем с 25%-ным раствором-М
миака (около 1,25 моль). Отделяют выпавшую соль (соскабливание н встрял
ванне), отсасывают жидкость, обрабатывают гликолят трехкратным коЛИЧ
ством 12%-ной НС1, извлекают образовавшуюся тиогликолевуто кислоту эфйрс
и очищают ее перегонкой; т. кип. готового продукта 107—108° С (16 .ил рт. ст.
Получение меркаптанов и тиофенолов с помощью тиокарбоновых кислот -ocyriJ
ствляют взаимодействием соответствующих алкилгалогенидов с тиокарбоновыми кц
лотами, преимущественно тиоуксусной или тиобенаойной; образовавшиеся эфир
тиолкарбоновых кислот гидролизуют в кислой или щелочной среде: -
СвП6СНСОСН8 - +С?квг8К > СвНбСНСОСНз —— > СвНбСНСОСНз ?
4 вг scocHg sh
[231] Герм. пат. 367346 (1914); Frdl.,” 14 , 918 (1921—1925); герм. пат. 360690 (1911
Frdl., 14, 908 (1921 — 1925). ' 1 1
[232] J. М. F. Lea per, J. Am. Chem. Soc., 53, 1891 (1931). *
[233] L. M. E 11 i s jr., E. E. R e i d, J. Am. Chem. Soc., 54, 1674 (1932).
[234] Герм. пат. 189200 (1906); Frdl., 8, 474 (1905—1907). >
[235] H. Gilman,^}. C. Gainer, J. Am. Chem. Soc., 71, 1747 (1949). /
{236] P. KlasOB, T. Carlson, Ber., 39 , 732 (1906). 3
II. ОБМЕН ГАЛОГЕНА НА S-ФУНКЦИЮ
581
Преимуществом этого п описанных шике способов является получение меркаптанов
без прпмеси тпоэфпров [237].
а-Ацетилмеркапто-а-фенилацетон. -К раствору 29 г тиоуксусной кислоты
в 200 мл спирта, предварительно точно нейтрализованному возможно более
концентрированным раствором КОН, по каплям прибавляют 70 а а-бром-а-
фснилацетона при температуре не выше 50° С. При этом выпадает КВг. После
охлаждения массу смешивают с водой, извлекают выделившееся масло эфиром,
сушат его над Na2SO4 и разгоняют в вакууме. Фракцию, отгоняющуюся при
140—160° С (12 мм рт. ст.), еще раз перегоняют на колонке Видмера. Выход
продукта 36 г (50% от теоретического); т. кип. 157—158° С (12 мм рт. ст.).
а-Меркапто-а-фенилацетон. При 20° С 14,5 г полученного ацетильного
соединения энергично встряхивают со 180 мл 5%-ного раствора NaOH до почти
полного растворения (примерно 45 мин). Затем раствор дважды быстро промы-
вают эфиром, подкислением осаждают меркаптан и извлекают его эфиром.
Полученный раствор сушат над Na2SO4 и отгоняют эфир. Остающееся масло
медленно кристаллизуется. После перекристаллизации из системы спирт — вода
получают 10—11 г (80% от теоретического) а-меркапто-а-фенилацетона в виде
бледно-желтых кристаллов; т. пл. 108—110° С.
Образование С—S-связи в результате обмена галогена во многих случаях может
происходить при взаимодействии соответствующих алкил- и арилгалогенидов с произ-
водными тиоугольпой кислоты, тиокарбаматами, тиомочевиной, дитпокарбаматами
п ксаитогенатами. Ценность этих методов заключается в том, что промежуточные со-
единения легко гидролизуются до меркаптанов (о ксантогенатном методе см. стр. 599).-
Так, в алифатическом ряду дитиолы [238] и ненасыщенные меркаптаны [239] с успехом
получают по дитиоуретановому методу Брауна:
RX + NH4SCNH2 —> RSCNH2 + NH4X
RSCNH2 --------> RSH-t-IISCN
II
s
Для синтеза тиолов используют тиомочевину, поскольку она легко взаимодей-
ствует с алкилгалогенидами |240]. смесями спиртов с НВг [241], галогенидами соот-
ветствующих ароматических [242] и гетероциклических [243] соединений с образова-
нием солей S-алкил- или S-арилизотиурония. Щелочной гидролиз этих солей обычно
приводит к тиолам с хорошими выходами:
Об алкировании днметплсульфатом и арилированиц сарями диазония см. [244],
[237] А. V. W.a с е к, К. К г a t z 1, A. v. В ё z а г d, Вег., 75, 1352 (1942).
[238] J. v. Braun, Вег., 42, 4568 (1909).
U39] J. у. Braun, Т. Plate, Вег., 67 , 281 (1934); J. v. Braun, R. М ur-
rnr jahn, Ber., 59, 1208 (1926).
[240] G. G. Urquhart, J. W. Gates jr., R. Connor, Org. Synth, 21, 36
r< t (1941); A. J. S p e z i a I e, Org. Synth., 30, 35 (1950).
12411 R, l. F r a nk, P. V. Smith, J, Am. Chem. Soc., 68 , 2103 (1946).
242] С. P r i c e, G. W. Stacy, J. Am. Chem. Soc., 68 , 498 (1946).
[243] M. A. Phillips, H. Cnapiro, J. Chem. Soc., 1942 , 584.
1244] F. Arndt, Ber., 54, 2236 (1921); M. Busch, K. Schuli, J. prakt. Chem.
[2], 150, 173, 180 (1938).
582
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
«-Додецилмеркаптан [240]. Смесь 125 г (0,5 моль) долецплбромидд~ \
38 г (0.5 моль) тномочевины и 250 мл 95%-ного этилового спирта в течение*.
3 « нагревают на паровой бане. Затем смешивают с раствором 30 ? (0,75
NaOH в 300 мл воды и кипятят смесь 2 ч. Частично выделившийся меркаптак*
отделяют от водного слоя, подкисляют водный слой разбавленной II2SO4 (7 Мл.
концентрированной H2SO4 в 50 мл воды) и экстрагируют меркаптан 75
бензола. Экстракт объединяют с неочищенным меркаптаном в после двукратно®
промывки водой (порции по 200 мл) сушат над 20 г безводного Na2SO4 и фра»-7'
ционирутот. Выход н-додецилмеркаптана 80—84 г (79—83% от теоретического)!
т. кип. 165—169° С (39 мм рт. ст.).
Для получения тиолов пригоден также тиосульфат натрия, при взаимодействии!
которого с алкилгалогенидами образуются S-алкилтиосульфаты (соли Бунте); нослед4:
one даже без предварительного выделения обычно в условиях восстановления (для
подавления образования дисульфидов) переходят в меркаптаны:
CH2COONa + Na2S-O3 —*• CII2COONa —► CH3COONa ji
I II
CI S-SO3Na SH
Тиогликолевая кислота [245]. Монохлоруксусную кислоту нойтряля*,
дуют 3 н. раствором NaOH, смешивают с эквивалентным количеством NaaS^Oj]
и нагревают 1 ч на водяной бане. Окончание реакции определяют по отсутствию^
помутнения (выделение серы) при смешении пробы реакционной массы с кислой
той. Полученную реакционную массу гидролизуют 2 ч 25%-ной НС1. Продуй?]
выделяют экстракцией эфиром и последующей вакуумной перегонкой. Выхадй
тиогликолевой кислоты 95% от теоретического. л!
S-Алкилтиосульфаты окисляются элементарным подом до дисульфидов с отлич-*]
ними выходами, вероятно, без промежуточного образования меркаптанов [246]:
2R-S + 2H2O + I2 —> R-SS-R+2NaHSO4-[ 2Ш
Л
SO3Na .JH
Ддя получения дисульфидов этим методом выделения промежуточно образу-
ющихся алкилтиосульфатов не требуется. Например, из метилового эфира бромуксус--1^И
нон кислоты получают диметиловый эфир длтполнгликоленой кислоты [246], из бе®->®^И
зилхлорида — дибензплдпеульфид с выходом 99% от теоретического [246]. >
Ди-н-бутилдисульфид [247]. При нагревании раетворяют 38,1 г бутил-
бромида и 74,4 г Na2Sa03 в 450 мл 50%-пого спир,а. К раствору небольшими
порциями прибавляют иод до появления устойчивой окраски. Продукт из вод-.-я^Н
ного слоя экстрагируют эфиром, отгоняют эфир и остаток соединяют с предав" .ЗИ
рительно отделенным от реакционной массы маслянистым слоем. Объединенный7^^И
продукт промывают раствором NaHSOs, сушат над Na2SO4 и перегоняют в ва-
кууме. Выход ди-н-бутилдисульфида 57% от теоретического; т. кип. 90—100°
(3 мм рт. ст.).
Дисульфиды образуются с хорошими выходами также при взаимодействии алкил-Й^И
и арилгалогеппдов с Na2S2. По Блаиксма [248], который синтезировал этим методом^^И
[245] A. Wagner, Dissertation Hannover, 1951; ср. С.. 1953, 4954; ср. Н о Ur^
ben-Weyl, Methoden der organischen Chemie, Bd. JX, Georg-Thiem®»
Verlag, Stuttgart, 1955 S. 10.
[246[ T. S. Price, D. F. Twiss. J. Chem. Soc., 95, 1489 (1909). '3
[247] H. E. Westlake jr. G. Dougherty, J. Am. Chem. Soc., 64й1
149 (1942). , Я
[248] J. J. В 1 a n k s m a, Rec. trav. chim., 20. 132 (1901). I
11. ОБМЕН ГАЛОГЕНА НА S-ФУЛКЦШО
583 '
большое число алифатических и ароматических дисульфидов, галогенид нагренают
л спиртовом растворе с эквивалентным количеством дисульфида натрия. Таким обра-
ти, в частности, получены о.о'-динитродифенилдисульфид (выход №% от теорети-
ческого ) [249], дпсульфиддималоповая кислота [250] и дисульфиддифенилдигликолсвая
ьпслота [251].
Несимметричные дисульфиды впервые были получены взаимодействием сульфе-
пилхлоридов с меркаптанами [252]. Реакцию проводили без доступа влаги в абсолют-
ных растворителях (эфир, четыреххлористый углерод), иногда с добавлением медной
бронзы в качестве катализатора [253].
Этилфенилднсульфид [254]. К раствору 11 г тиофенола в 50 мл эфира
при перемешивании и 0° С по каплям прибавляют 9,6 г этилсульфенилхлорица.
Затем смесь слегка нагревают на водяной бане, отгопяют растворитель и пере-
гоняют в вакууме; т. кип. этилфенилдисульфида 126° С (15 мм рт. ст.).
При взаимодействии хлорметплсульфенилхлорпда с меркаптанами или тпофено-
лами образуются соединения, которые наряду с дисульфидной группой содержат тио-
эфирную группу [255]:
CH2SC1-2RSH —> CHo-SS-R
I I
CI • SR
Получение простых тиоэфиров и солей сульфонил
Тиоэфиры по аналогии с меркаптапами и дисульфидами можно получать взаи-
модействием алкил- и некоторых арилгалогенидов с NaaS. Реакцию обычно проводят
в спиртовом, водно-спиртовом или в водном растворах, используя, как правило, сте-
хиометрические количества исходных продуктов.
Низшие диалкилсульфнды [256]. В трехгорлой колбе, снабжеппой обрат-
ным холодильником, капельной воронкой и мешалкой, растворяют 1,5 моль
Na2S • 9Н2О в 250 мл воды и при сильном перемешивании медленно добавляют
2 моль алкилгалогепида. По завершении сильно экзотермичной реакции массу
нагревают еще 3 ч с обратным холодильником и затем отгоняют тиоэфир с водя-
ным паром. Продукт реакции промывают водой, 10%-ным раствором NaOH,
вновь водой, сушат над СаС12 и перегоняют. Этим способом получены: диметил-
(т. кип. 37,3° С), диэтил-(т. кип. 92° С) [257], диизопропил- (т. кип. 119,8° С)
и ди-н-пропилсульфиды (т. кип. 142,8° С) [258].
Аналогичным образом мо:кно получить тиоэфиры из аралкил- и арилгалогепи-_
дов, имеющих достаточно реакционноспособный атом галогена.
Бис-(р-нафтилметил)-сульфид [259]. К 36 г p-хлорметилнафталина при-
бавляют раствор 25 г Na2S-9H2O в 15 мл воды и 230 мл абсолютного этнло-
М. Т. Bogert, A. Stull, Org. Synth., Coll. v. 1, 1937, p. 213.
A. Schoberl, H. E ck, Ann., 522, 107 (1936).
A. Schoberl, E. Berninger, F. Ilarrcn, Ber., 67, 1548 (1934).
H. Lecher, Her., 53, 577 (1920); H. L e c h e r, Ber., 53, 591 (1920).
S. Magnusson, J. E. Christian, G. L. Jenkins, J. Am. Pharm.
Assoc. Sci. Ed., 36, 257 (1947): C. A., 42, 877 (1948).
H. Brintzinger, M. Langheck, Chem. Ber., 86, 557 (1953).
H. Brintzinger et al., Chem. Ber., 87, 300, 314, 320, 325,(19.54).
D. T. McAlla n, T. V> С п I 1 н m et aL. J. Am. Chem. Soc., 73,
3627 (1951).
H. В 6 h ше, H. J. Gran, Ann., 577, 68 (1952).
R W. Bost, M. W. Conn. Org. Synth., Coll. v. 2, 1955, p. 547.
C. G. О verberg e r, S. P. L i g t h e 1 m, A. E. Swi re, J. Am.
Chem. Soc., 72, 2856 (1950).
584
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
вого спирта и нагревают 8 ч с обратным холодильником. После охлажден
массу выливают в воду, отсасывают осадок и перекристаллизовывают его,
этилового спирта. Выход продукта 26,5 в (84,4% от теоретического): т в
123—124° С. »
Бис-(п-хлорбензил)-сульфид [259] получают аналогично из п-хлорбеиз
хлорида с выходом 85,7% от теоретического.
n-Нитро- и динитрохлорбеизолы реагируют с Na2S с образованием тиоэфир
при проведении реакции с о-нитрохлорбензолом во избежание восстановления нит
группы раствор NaaS необходимо прибавлять по каплям [260]. В случае ж-нит
хлорбензола из-за незначительной реакционной способности атома галогена прозе
дит преимущественное восстановление нитрогруппы. ,
2,4,2',4'-Тетранитродифенилсульфид [261]. В 1000 мл этилового сш
растворяют 100 в 2,4-динитрохлорбензола. К раствору небольшими порпи
прибавляют сплав 65,5 г Na2S • 9Н2О и 9 г серы. Происходит бурная реак!
которую доводят до конца кипячением на песчаной бане в течение 3 ч. П<
охлаждения продукт реакции отсасывают, промывают спиртом и затем теп
водой до полного удаления NaCl. Выход продукта 86 г; т. пл. 280° С (раз
Сметанные тиоэфиры получают взаимодействием алкил- или арилгалогенв
с меркаптанами [262] или, что более целесообразно вследствие большей скорости pi
ции, с меркаптидами:
RSNa-f-R'Cl —* R-S-R' + NaCl
Реакция лучше всего протекает с алкил- или арилбромидами, так как соотв
ствутощие хлориды обычно слитком мало реакционноспособны. При использовав
очень реакционноспособных галогенсодержащих соединений, особенно иодидов, щ
является влияние нежелательной побочной реакции окисления — восстановлен]
приводящей к образованию дисульфидов [263]:
2RSH + R'! —* R—SS—R4-R'H-f-HI J
Карбоксиметилбутилсульфид [264]. К раствору 120 г NaOH в 600 л
воды прибавляют 360 г неочищенного бутилмеркаптана и при перемешиван^
и охлаждении вносят нейтрализованный содой концентрированный раствО
285 г хлоруксуснон кислоты. Через несколько часов полученный растай
нагревают и примеси отгоняют с водяным паром. Затем подкисляют остМЬ
разбавленной H,S04 и фракционируют выделившийся в виде масла карбоксЬ^
метилбутилсульфвд перегонкой в вакууме. Выход продукта 245 г; т. кип. 140^
144° С (Ю—15 мм рт. ст.). \
Таким же способом из тиосалициловой н монохлоруксусной иисдг’
получают о-карбоксифенилтиогликолевую кислоту с выходом 80% от теореп
ческого [265].
Особенно гладко тяоэфиры образуются при взаимодействии алкилгалогенид»
с 2,4-дииитротиофенолом. Эта реакция может быть использована для пдентпфинацг
органических галогенсодержащих соединений [266].Ниже приводится общая методй»
получения тиоэфиров взаимодействием 2,4-динитротмофенола с алкилбромидам
и алкилиодидами.
[260]
[281
[262
[263
. [264
[265
[266]
R. Nietzki, П. Both of, Вег., 29, 2774 (1896); F. Kehrman
Е. Bauer, Ber., 29, 2362 (1896).
J. Терр em a, L. В. S e Ь г e 11, J. Am. Chem. Soc., 49, 1755 (1927).
W. N e k г a's sow, N. N. M elnikow, Ber., 62, 2091 (4929).
N. Hellstrom, Z. physik. Chem., Abt. A, 163, 33 (1933).
Y. Uyeda, E. E. Reid, J. Am. Chem. Soc., 42, 2385 (1920).
Г. E. фярц -Давид, Л. Б л а п ж e й, см. [99].
R. W. В о s t, Р, К. Starnea, Е. L. Woo d, J. Ain. Chem. Soc., 7.T
1968 (1951).
II. ОБМЕН ГАЛОГЕНА НА S-ФУИКЦИЮ
585
Типафирм, К 5 г (0,025 моль) 2,4-динитрОтпофенола в 50 мл бутилкар-
битола (монобутиловый эфир диэтиленгликоля) прибавляют 5 мл 28%-ного
раствора КОН и затем 0,025 моль галогенида. В некоторых случаях, если
реакция не начинается сразу, смесь нагревают 10—30 мин при 70° С или, что
иногда более удобно, оставляют массу на длительное время при комнатной
температуре. После окончания реакции охлажденную массу разбавляют ледя-
ной водой, отсасывают выпавший продукт и перекристаллизовывают его (в каче-
стве растворителей пригодны этиловый и н-бутиловый спирты, диоксан, бензол
и ацетон).
Реакцию удается провести и с алкилхлоридамц, если в реакционную
смесь кроме указанных выше реагентов вносят 3 г KI.
В условиях этой реакции из ацетобромглюкозы образуются S-глюкозиды (267].
Так, взаимодействием 2-, 3- или 4-тиол-1-оксибспзолов с а-ацетобромглюкозой в водно-
ацетоиовом растворе с добавкой КОН получены соответствующие тстраацетил-|}-
D- [2(3 или 4)-оксифенил]-тиоглюкозиды с хорошими выходами [268].
Арилгалогениды, содержащие менее реакционноспособные атомы галогена,
с меркаптанами также образуют тиоэфиры, если проводить реакцию при 225—230° С,
используя меркаптиды тяжелых металлов (Pb, Zn, Hg). Этим способом синтезированы
а,Р-динафтилсульфид [269], фенил-а- и фенил-0-нафтилсульфиды [270], и о-, м- и п-
толил-а-(и -Р)-нафтилсульфиды [271]. В тех случаях, когда меркаптиды тяжелых ме-
таллов проявляют слишком большую устойчивость и при 240° С еще не вступают
во взаимодействие с арилбромидами, арилсульфиды удается получать по общеприме-
нпмой реакции арилиодидов с меркаптидом натрия в присутствии медного катализа-
тора [272].
Днфенилсульфнд [272]. К раствору 1,2 г металлического натрия в 7 мл
спирта прибавляют 7 г тиофенола, отгоняют спирт, добавляют затем 0,2 г
медной бронзы и 12.9 а иодбензола и смесь нагревают в течение 2,5 ч на масля-
ной бане при 235—240° С. После охлаждения массу растворяют в небольшом
количестве спирта, подкисляют разбавленной Н28О4 и перегоняют с водяным
паром в присутствии цинковой пыли. При этом отгоняется непрореагировавший
иодбензол и восстанавливается до тиофенола небольшое количество дифенил-
дисульфида, образовавшегося в качестве побочного продукта. После охлаждения
раствор фильтруют, цинковый шлам тщательно промывают эфиром, сушат
эфирный раствор 'над СаС12 и фракционируют. Выход дифеннлеульфида 6,1 г;
т. кип. 295° С.
Таким методом получены различные замещенные дифенилсульфиды, напри-
мер метил-, метокси- и нитродифенилсульфиды.
Описано получение 6-алкилтпопуринов взаимодействием 6-меркаптопурина
с алкилгалогенидами или 6-хлорпурина с меркаптидами натрия [273]:
RSNa
<-----
CI
I N
Дальнейшее взаимодействие тпоэфиров с алкилгалогенидами приводит к обра-
зованию сульфониевых солей
Г К
\S + R"X —> )S-R'
К'/ Lr,z
Ср. Т. Sabalitschka, Arch. d. Pharm., 267, 675 (1929).
G. Wagner, H. Kiihmstedt, Arch. d. Pharm., 294, 117 (1961).
F. Krafft, Ber., 23, 2364 (1890).
F. Krafft, E. Bourgeois, Ber., 23, 3045 (1890). „
E. Bourgeois, Ber., 24, 2264 (1891).
F. Mauthner, Ber., 39 , 3594 (1906).
T. P. J о h n s t. о n, L. В. H olum, J. A. Montgomery, J. Am. Chem.
Soc., 80, 6265 (1958).
586
ОБРАЗОВАНИЕ С,—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
воде и нерастворимы в эфире. Дан простых алифатических,
ляется общей, в то время как ароматические тиоэфирыь
которые легко растворимы в ..
соединений эта реакция является____, х .. . г__________...... w
реагируют различно; например, тпокрезолметиловый эфир не метилируется метил-':
нодидом, но может метилироваться дпметнлсульфатом [274]. В качестве растворителей'?
обычно применяют спирт, ацетон, бензол и нитрометан. Достаточно реакционность?
собпыс компоненты реагируют при комнатной температуре. Обрдсовдпис
пых солей часто можно значительно ускорить добавлением эквивалентных количеств*
солей металлов [275]:
температуре. Образование сульфоние-i
добавлением эквивалентных иоличйстж^
R
>S 1 R'Cl
Rz
l'R\ г
>S- R [РеС14]-
Lrz J
В случае очень чувствительных к влаге сульфониевых солей, например бутил-J
октил-, лаурил- п цетилдпметллсульфонпйподидон [27(5], процесс рекомендуете»!
вести в токе сухого азота. 'у
спиртом. Продукт отфильтровывают и перекристаллизовывают из водиог<$
•та. Выход 10,4 г (75% от теоретического); т. пл. 150° С (разл.). '
Пиметилйенягтил^лкАпттйбгюмии F2781. Ряствотиютт О » 1л.(1тт11й„<»гл. ’
Метионинметилсульфонийиодид [277]. В течение 6 ч нагревают 7,45 г*
(0,05 моль) метионииа при 45J С с 7,8 г (0.055 моль) метилиодида в 40 мл воды. ]
Раствор упаривают в вакууме до сиропообразного состояния и растирают с горя<?
спирта. Выход 10?4 г (75^(. о/ теоретического); . ..
Диметилфенацилсульфонийбромид [278]. Растворяют 9 г са-бромацето-1
фенона в 30 г смеси 1 объемн. ч. воды и 20 объемн. ч. ацетона и к этому раствору^
прибавляют 3 г димстилсульфида. После примерно получасового стояния?
выделяется сульфопиевая соль в виде длинных, бесцветных игл. Выход про-,
дукта 10,2 г (85% от теоретического); т. пл. 145° С. •
В более жестких условиях дисульфиды [279—281 ] также переходят в соли суль-'
фония; и в этом случае взаимодействие происходит легче в присутствии металлический-
солей [282, 283]. Я
Получение тиокетонов, меркапталей и меркаптолов
Прп взаимодействия серусодержащих соединений (например, гидросульфвда^
натрия, тиоуксусной кислоты) с геминальными дихлорпроизводпыми дпарилкетонов ,!
и гетероциклических кетонов атомы галогена обмениваются на серу и образуются^
тпокртоны [284]. Таким путем из бензофоионхлорпда и спиртового раствора гидро-;
[274]
[275[
К, v. Auwers, F. Arndt, Вес., 42, 2713 (1909): ср. F. Kebrmann,'
A. Duttenho Ге г, Вег., 38, 4197 (1905). J
К. A, Hofmann, К. О t t, Ber., 40, 4930 (1907); R. R. Renslia w,„
D. A. S e a I e, J. Am. Chem. Soc., 55 , 4951 (1933).
,iIU|R. Kuhn, O. D a n n, Ber., 73, 1092 (1940).
[277] R. O. A t k i.n в о n, F. Poppelsdorf, J. Chem. Soc., 1951, 1378.
[278] H. В 6 li m e, W. Krause, Chem. Ber., 82, 426 (1949). %
[279| S. H. Davies, Ber., 24, 3548 (1891).
/ [280| W. Sieinkopf, S. M ii II e r, Ber., 56. 1926 (1923).
[281] M. L. Selker, Ind. Eng. Chem., 40, 1467 (1948).
[282] D. P. H i I d i t c h, S. Smiles. J. Chem. Soc., 91. 1394 (1907). %
[283] P. C. Ray, N.'Adhikary, J. Indian Chem. Soc.. 7. 297 (1930). д
[284] L. Gattermann. H. Schulze, Ber., 29, 2944 (1896).
И. ОБМЕН ГАЛОГЕНА НА S-ФУПКЦПЮ
587
сульфида натрия получен тиобензофепон с выходом 16% от теоретического
[285]:
(C8H5)3CCl2 + 2NaSII —> (CeH5)3C=S + 2NaCi+n2S
По общепрпменимой реакции ароматических кетохлорпдов с тиоуксусной
кислотой можно легко и с хорошими выходами получать многочисленные производные
тпобензофепонового ряда [2861
Аг Аг С1
\q-_q рс1> илп (COCl)^ Х'С/ CHaCOSH >
Аг/ ArZ 'ЧС1
Г Агч а 1
>С<
[агх 4shj
Аг
—> >C=S '
Аг^
например, тпоксантиоп:
Тпоксантиоп. В течение 20 « кипятят на водяной бапе без доступа
влаги 5.7 г тиоксантона [287] с 10 г оксалилхлорпда и затем высушивают
при 100° С в вакууме досуха. Остаток растворяют в 75 мл сухого бензола.
К раствору в токе СО2 небольшими порциями прибавляют 10 г тиоуксусной
кислоты. Полученную'смесь нагревают на кипящей водяной бане до прекраще-
ния выделения НС.1 (около 5 ч). выпаривают досуха в вакууме и остаток пере-
кристаллизовывают сначала из бензина (т. кип. 80—90° С) и затем из ксилола.
Полученный продукт представляет собой черные иглы с зеленоватым оттенком;
т. пл. 168° С. В
В завершение раздела о получении тпокегопов следует еще упомянуть взаимо-
действие ароматических соединений с тиофосгеном по реакции Фриделя — Крафтса
[288. 289].
[fa замещенных галогенпиридвниевых и хинальдипиевых солей действием гидро-
сульфидов получаются тиопиридоны [2901 и тиохинальдопы [291J:
S
N СНз
I- СНз
СНз
[285] И. Standi и ge г, F. Freudenberger, Org. Synth., Coll. v. 2, 1955,
p. 573.
286] A. Schonberg, O. Schutz, S. Nickel, Ber., 61, 1375 (1928).
287] E. G. Davis, S. Smiles,4 J. Chem. Soc., 97, 1296 (1910).
288| Герм., пат. 37730 (1886); Frdl., 1, 94 (1877-1887).
289] L. Gattermann, Ber., 28, 2869 (1895).
290] A. Michaelis, A. H 6 1 k e n, Ann, 331, 245 (1904).
291] E. Champaigne, R. E. Cline, С. E. К a s 1 о w, J. Org. .Chem., 15,
600 (1950).
588
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
При взаимодействии геминальных дигалогенсодержащих соединений с мер]
тидами в условиях, исключающих отщепление галогеново^орода, обычно гладко о!
зуются меркапталя [292] или меркаптолы [293]:
R-, ,С1 /SR'
)Cf +2R'SII —> >с<
нх Чс1 hz 4r'
меркапталь
Ry ySIT
Н'/ \SR‘ ,
меркаптол
R. ,С1
>С< 4-2R"SH
R'/ \ci
Смешанные меркаптали с выходами до 90% получают пз а-галогентиОэфир
и меркаптанов 1294].
Тиоэтилтиобензилметан. При смешении 11 з хлорметилэтилсулыф]
с 12,4 з бензилмеркаптана происходит очень бурная реакция с выделен]
хлористого водорода. Реакционную смесь необходимо охлаждать водой. Ма
оставляют на ночь и фракционируют перегонкой в вакууме. Выход проду
60% от теоретического: т. кип. 105° С (19 мм рт. ст.).
Бис-(тиобензил)-метан. Его получают из 3,5 г хлорметилбензилсу.
фида и За бензилмеркаптана. Выход продукта 4,8 г (90% от теоретического); т.;
55° С (после перекристаллизации из изопропилового спирта).
2. Взаимодействие ацилгалогенидов с сероводородом
и его производными
Обмен галогена на серу в ацилгалогеяидах приводит к образованию тио]
боновых кислот или их производных. Например, важнейшим способом’получе^.
тиокарбоновых нислот является ацилирование сероводорода. Реакцию провод
в безводном пиридине, и она, по-видимому, имеет общий характер [295|. Для ДОС"
ження высоких выходов необходимо достаточно интенсивное охлаждение, сильв
ток сероводорода п энергичное перемешивание во избежание местного скоплея
избытка хлорангидрида. Таким способом были получены простые тиокпслоты вши
до тиононановой кислоты [295].
Тиоуксусная [296] и тиобензойная [296, 297] кислоты. В колбе емкой»
1 л, снабженной мешалкой и обратным холодильником, насыщают 200^
сухого пиридина сероводородом, промытым раствором иода и высушеннь
СаС12, п при температуре ниже 15° С при охлаждении и перемешивании в теч
ние 2—3 ч прибавляют 40 г ацетилхлорида (или при 5° С 40 г бензоилхлорида
Затем смесь подкисляют рассчитанным количеством 5 н. H2SO4, продуктизвл
кают эфиром, сушат над Na2SO4 и фракционируют в токе азота. Выход,!®
кислоты 60—65% от теоретического; т. кип. тиоуксусной кислоты 87,5—88*
т. кип. тйобензойиой кислоты 85--870 С (10 мм рт. ст.).
[292] Е. Fromm, Ann., 253, 155 (1889); R. Otto, К. Miihle, Ber., 28, 111
(1895). •
[293] A. Schonberg, O. Schutz, Ann., 454, 47 (1927); A. Schonbei!
O. Schutz, V. Bruckner, J. Peter, Ber., 62, 2550 (1929).
[294] H. Bohme, H. Fischer, R. Frank, Ann., 563, 54 (1949).
[295] A. F г e d g a, H. Bauer, Ark. Kemi, 2, 113 (1905): C., 1952, 989.
[296] S. Sunn er, T. Nilson, Svensk кеш. Tidskr., 54, 163 (1942); 6
1943, I, 829. •
[297] P. Noble jr., D. S. T a r b e 1 1, Org. Synth., 32, 101 (1952).
II. ОБМЕН ГАЛОГЕНА НА S-ФУНКЦИЮ
589'
Ацилированием меркаптанов получают эфиры тиолкарбоновых кислот. Эту
реакцию, часто очень гладко протекающую и в отсутствие оснований [298J, проводят
обычно по методу Шоттена — Бауманна [299, 300] или в присутствии пиридина [301].
Гидрохлорид S-бензоилцистеамина [302]. Нагревают 2 г гидрохлорида
цистеамина с 3 мл бензоилхлорида до начала выделения хлористого водорода
(155—170° С). Далее реакцию проводят без дополнительного подогрева. За-
твердевшую смесь растирают с сухим эфиром, промывают несколько раз те»
же растворителем и перекристаллизовывают из сухого днметилформамида,
смешанного с несколькими каплями концентрированной НС1. Из маточника,
можно извлечь эфиром вторую фракцию. Выход продукта 2,7 г (70% от теорети-
ческого); т. пл. 167° С (разл.). ।
1,5-Бие-(бензонлмеркапто)’пентан. 1,5-Димеркаптопентан встряхивают
с бензоилхлоридом и избытком NaOH. При этом образуется 1,5-бис-(бензоил-
меркапто)-пентан с почти количественным выходом; т. пл. продукта 45° С после-
перекристаллизации из этилового спирта ]299].
3. Взаимодействие адки.тгалогенидов с сульфитами
и сульфиновыми кислотами
Обмен галогенов в алкилгалогенидах па серу при взаимодействии с сульфитами-
щелочных металлов для получения алифатических сульфокислот впервые осуществлен!
Штреккером [303].
RX + NaaSOs —> RSO8Na + NaX
Этансульфокнслота. Нагревают 20 г зтилиодяда с раствором 20 г пристал- -
лического (NHa)2SO3 в 40 мл воды в течение 6 ч [304], затем смесь охлаждают.
При этом образуется аммониевая соль этансульфокислоты. Для получения-
бариевой соли смесь разбавляют водой и нагревают с РЬО до окончания выделе-
ния аммиака. Фильтрат освобождают от свинца обработкой сероводородом.
После вовой фильтрации к раствору добавляют избыток ВаСО3, фильтруют
и выпаривают досуха.
Особенно хорошие выходы получаются в случае н-алкилгалогенидов, в то
время как атомы галогенов у вторичного атома углерода обмениваются только на 25%,
а третичные галогексодержащие соединения в этих условиях превращаются в олефины.
При взаимодействии дибромэтана с концентрированным водным раствором
NasSO3 образуется эгаидисульфокислота J305], при недостатке сульфита — натриевая-
соль бромэтансульфокислоты [306]. Общая методика получения алкан-а,ш-дисульфо-
натов приведена нище [307].
R. В. В a k er, Е. Е. R е i d, J. Am. Chem. Soc., 51, 1567 (1929).
W. Autenrieth, A. Seyer. Ber., 41, 4253 (1908).
С. E. D a 1 g 1 i e s h, F. G. Й a и n, J. Chem. Soc., 1947, 559.
P. N. R у ] a n d e r, D. S. T a r b e 1 I, J. Am. Chem. Soc., 72, 3021 (1950).
T. Wieland, E. Bokelman n, Ann., 576, 20 (1952).
A. Strecker, Ann., 148, 90 (1868).
W. Hemilia n, Ann., 168, 145 (1873); cp. F. C. Wagner,.
E. E. R e i d, J. Am. Chem., Soc., 53, 3409 (1931).
В. C. Saunders, J. Chem. Soc., 1950, 686.
C. G. О v e i b e r g e r, D. E. Baldwin, H. P. Gregor, J. Am. Chem..
Soc., 72, 4864 (1950). '
G. С. H. S tone, J. Am. Chem. Soc., 58, 488 (1936); ср. В. H e 1t e г i c h
H. G r ii n e r t, Ber., 74, 1531 (1941).
590
ОБРАЗОВАНИЕ G—S-C.RH.3II В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Бутандисульфокислота (по Вейганду). В двугорлую колбу емкостью 1 д,
снабженную мешалкой п обратным холодильником, помещают 100 г дибром*
бутана (0,46 моль), 150 г безводного Na,SO3 (2,5 • 0,46 моль) и 400 мл водц:
и при энергичном перемешивании нагревают 20 ч па водяной бане. Затем смесь
разбавляют до объема 1 л и еще теплый раствор встряхивают с животным углем
до исчезновения запаха лука. Раствор оставляют на ночь, затем отфильтровы-.
вают уголь й пропускают в раствор хлористый водород. После начала выпадения'
NaCl колбу охлаждают ледяной водой и продолжают пропускать хлористый-
нодород до полного прекращения выпадения NaCl. Затем раствор фильтруют^
упаривают фильтрат до 300 мл и вновь пропускают хлористый водород при*
охлаждении льдом. При этом выпадает дигидрат бутандисульфокислоты. Дигидч
рат сушат в вакууме над твердым NaOH, после чего он имеет т. пл. 86—1
93® С. Дигидрат обрабатывают уксусным ангидридом и удаляют образовав*
шуюся уксусную кислоту. Выход безводной кислоты 92 г (92% от теоретик
чсского); т. пл. 136е С.
Аралкилбромпды [308, 309] реагируют с сульфитом натрия легче, чем аралкил^
хлориды; при проведении реакции с бензилхлоридом оказалось полезным добавляй»
в реакционную смесь NaOH. 5-j
Бензилсульфокислота [310]. В течение 1,5 ч кипятят с обратным холб*
дильпиком 250 г кристаллического тонкоизмельченного Na2SO3 с 125 г беий
зилхлорида и 200 мл 10%-ного NaOH. Горячий раствор тотчас же перегоняя^
с водяным паром для удаления избытка бензилхлорида и бензилового спирта^
затем упаривают. Выход натриевой соли бензилсульфокислоты 97% от теоретв»
четкого. Соль кристаллизуется из спирта с одной молекулой воды, но легк&
выветривается па воздухе. л
Так же с образованием соответствующих сульфокислот реагируют с сульфите»
«атрия галогенированные карбоновые кислоты и их производные [311], галогенир<$
ванные спирты [312], галогенированные кетопы [313] и галогенированные эфиры)
[314].
Ио реакции Штреккера из арилгалогенидов с подвижным атомом галогейа,
и сульфита натрия также образуются сульфокислоты. Так, при нагревании о-хлор-т'
•бензальдегида с Na2SO3 под давлением в течение 8 ч с очень хорошим выходом полу-'
чают бензальдегид-о-сульфоиислоту [315]. 5-Нитробензальдегид-2-сульфокислота обрат*
зуется уже при кипячении 5-нитро-2-хлорбензальдегида со спиртовым растворов!
сульфита натрия [316]. В нитроарилгалогенидах может происходить частичной
восстановление нитрогруппы до аминогруппы (стр. 573): полигалогепированныв
соединения в этих условиях дают полисульфокислоты [317]. , Д
Реакция образования сульфонов [318] при взаимодействий алкилгалогенидов
•с сульфинатами щелочных металлов уже давно известна:
RSO2Na4-R'X —► RSO2R' + NaX 'j
Эту реакцию обычно проводят в водной или спиртовой среде на кипящей
водяной бане. Пригодными для синтеза галогенидами являются первичные и вторийч
яые алкилгалогеняды, а также бензил- и арилгалогениды, если в последних атовий
галогена активированы нитрогруппами, находящимися в орто- или пара-полоЖенИи.;
С алифатическими сульфинатами выходы обычно меньше, чем с арилсульфинатами»;
[308]
[309]
|310|
|311
[313
314
315
316
317|
! 318]
Ч. С ь ю т е р, Химия органических соединений серы, пер. с англ., ч. 2, Издать
инлит, 1951.
С. A. Bunton, Е. А. Н а I с v i, J. Chem. Soc., 1952 , 4541. А
Е. F г о m га, J. d'e S e i x a s Palma, Ber., 39, 3312 (1906). .'*
Франц, пат. 788748 (1935): С., 1936, I, 1713. «
j. M. S t e w а г t, H. p. С о r d t s, J. Am. Chem. Soc., 74 , 5884 (1952). -4
G. D. Parkes, S. G. Tinsley, J. Chem. Soc., 1934, 1861. J
Пат. США 2394834 (1946); С. A., 40, 2658 (1946). 'Я
Герм. пат. 88952 (1896)- Frdl., 4, 133 (1894—1897).
Герм. пат. 165613 (1904)- Frdl., 8, 158 (1905—190/). J
Герм. пат. 98321 * (1897);'Frdl., 5, 207 (1897—1900). '
В. О t1 о. Вег., 13, 1272 (1880); R. Otto, Ann., 283, 181 (1894). ii
И ОБМЕН ГАЛОГЕНА ПА S-ФУНКЦПЮ
591
Метил- к этилбеизилсульфокы [319]. Водный раствор бенэплсульфипата
натрия кипятят с обратным холодильником в течение нескольких часов с неболь-
шим избытком метил- пли этплподида в присутствии NaOH. Продукт реакции
извлекают эфиром л перекристаллизовывают из воды. Метилбензилсульфон
плавится при 127° С, этилбепзилсульфон— при 84''С.
Фенилбеизилсульфон [320]. Нагревают 178 г бепзолсульфпната натрия
п 127 г бензилхлорида в 500 мл абсолютного спирта в течение 7—8 ч. После
выливания горячего реакционного раствора в 1000 мл ледяной воды продукт
реакции отсасывают, высушивают и перекристаллизовывают из спирта. Выход
продукта 120 г (52% от теоретического); i. пл. 146 —146.5° С.
Реакция с аридгалогенидами протекает в более жестких условиях: при более-
высокой температуре или под давлением.
п-Ацетиламино-н'-иитродифенилсульфон. п-Ацетиламинобеязолсульфп-
новокислый натрий и н-питрохлорбензол нагревают 3,5 ч при 142° С в этилен-
гликоле и метилкарбитоле. При этом образуется п-ацетиламино-п'-нитроди-
фепилсульфон (выход 51%), из которого восстановлением молено получить.
п .п' -диампнодифенилсульфоп [321].
о- и н-Питродифенплсульфоны [322]. В течение 3 ч прп 160° С в запаян-
ной трубке пагревают 4 г бепзолсульфиновой кислоты. 4,5 г е-хлорнптробен-
яола п 2.5 г ацетата натрия (или соответственно 5 г бензолсульфиновой кислоты,
5.5 г п-длорнптробензола и 3,5 г ацетата натрия) с необходимым для растворе-
ния количеством спирта (около 10 ai.i). После охлаждепия продукт реакции
отсасывают и промывают водой. Выход орто-соедлнения 4,5 г. иара-соедияе-
пия — 5,5 г. После перекристаллизации па спирта с добавкой активирован-
ного утля т. пл. соответственно 147,5 и 143° С.
Этим методом получено большое число сульфонов (см. обзор [308]).
Получение эфиров' тиоциановой и изотиоциаиовой кислот
При взаимодействии алкилгалогенидон с роданидами щелочных металлов;
происходит замощение водорода с образованием эфиров тиоциановой кислоты (орга-
нических роданидов). Однако заранее нельзя предвидеть, образуются ли по этому
методу эфиры тиоциановой кислоты пли изомерные им горчичные масла. Поскольку
горчичные масла являются, по-видпмому, более стабильными соединениями, роданиды-
можно перевести нагреванием в горчичные масла.
В препаративно важных методах получения эфиров тиоциановой кислоты-
исходят также из диродана, который ведет себя в реакциях как псевдогалоген и по-
этому способен присоединяться по олефиповым двойным связям и замещать водород
в ароматических соединениях. О получении органических роданидов (стр. 101, 190)
сч. обзоры [323. 324].
Эфиры тиоциановой кислоты, кроме того, могут быть получены взаимодействием»
арил- [325] и алкилсульфенхлоридов [326], а также перхлорметилморкаптана [327}
с цианидом калия:
RSC14-K.CN —► RSCN4-KC1
[319] Е. Fromm, J. de Seixas Palma, Врг., 39. 3308 (1906).
[320] R. L. Shriner. H. C. Strac k, W. J. J о r i s о n, J. Am. Chcm_
, Soc., 52. 2067 (1930).
[321 ] W. Ferry, J. S. В u c k, R. В a I t z 1 y. Org. Synth., 22, 31 (1912).
[322] F. II liman n, G. Pasdermadjian, Ber., 34, 1150 (1901).
[3231 H. P. Kaufmann. Angew. Chem., 54. 195 (1941): H. P. Kaufmann,
Neuere Me^hoden der praparativen organischen Cnerniv, Verlag Chemie, Berlin,
1944, S. 237.
[324] J. I,. Wood, Org. Reactions, 3, 240 (1949).
[325| T. Zincke, К. E Ismay er, Ber., 51, 751 (1918).
[32Н] H. В r i nt zinger, M. Langheck, Chem. Ber., 86. 560 (1953).
[327] Пат. США 2650240 (1953); С. A., 48, 8819 (1954).
-592
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
III. Обмен кислорода на 8-функцию
1. Взаимодействие спиртов с серусодержащими соединениями
Взаимодействие с сероводородом и его производными
Обмен кислорода в спиртовых гидроксильных группах на серу с образовав
меркаптанов часто удается осуществить с хорошими выходами пропусканием а
паров спирта с сероводородом при 350—380° С над окисью тория. Описаны оптим
ные условия получения простых алкплтиолов (от метил- до изоамилмеркапта:
[328]. Такое замещение возможно и в более мягких условиях [329]. Например,
взаимодействии N-оксиметилпиперидина, легко получаемого из пиперидина и форм
дегида, с сероводородом в результате экзотермической реакции возникает coot
ствующий тиол.
При взаимодействии с меркаптанами спирты с отщеплением воды образ
тиоэфиры. Например, из бензоина и тиогликолевой кислоты получают дезплтиог
колевую кислоту [330]:
С6Н5-СН-ОН НС1 С6Н5-СП-8СНгСООН
СИ с о +HSOW0H ^5* I
CGtl5—С=О Cfj-Hb—с=о
Дезплтиогликолевая кислота. Нагревают на водяной бане 5 г бензо:
с 6,5 г тиогликолевой кислоты в течение 30 мин, одновременно пропус
сухой хлористый водород. Полученную массу обрабатывают холодным рас
ром соды и фильтруют в разбавленную соляную кислоту. Продукт реак
через некоторое время выкристаллизовывается из раствора. Его перекрис
лизовывают из четыреххлористого углерода или водного метилового спи:
Т. пл. 105° С.
Этим же методом из трифеиилкарбпноиа и п-тиокреэола получен я-тоа
трифенилметилсульфид [331].
В смеси с кислотами первичные спирты могут служить алкилирующими
•ствами; при взаимодействии спиртов с тиоэфирами- отщепляется вода и образу
соли сульфония:
Нч Г R\ 1*
>S-f-R"OH-bH+ X- —► >S-R" Х- + Н2О
R'/ |_R'Z J
Перхлорат диметилфенилсульфония [332]. Кипятят 2 г метилфеннлеу
фида с 3 мл 70%-ной НС1О4 и 6 мл метилового спирта. Затем отгоняют избы1
метилового спирта и выливают остаток в воду; при этом выпадает в ocaj
соль сульфония. После перекристаллизации из воды т. пл. продукта 160®
Взаимодействие с бисульфитом натрия,
Обмен спиртовой оксигруппы под действием бисульфита натрия в некого]
«случаях происходит с образованием солей сульфокислот:
R3COH + NaHSO3 RsCSO8Na
[328] R. L. Kramer, E. E. Reid, J. Am. Chem. Soc., 43, 880 (1921).
[329] A. Hinz, L. U. Pence, J. Am. Chem. Soc.. 61, 3134 (1939).
[330] O. Behaghel, E. Schneider, Ber., 68, 1588 (1935).
’[331 ] M. P. В a 1 f e, J. Kenyon, С. E. Searle, J. Chem. Soc., 11
3309. *
1332] O. Hinsberg, Ber., 69, 492 (1936).
Ш. ОБМЕН КИСЛОРОДА НА S-ФУНКЦИЮ
593
Так, вз три-п-толилкарбинола получают натриевую соль три-п-толилметан-
сульфокислоты [333] и из трифенилкарбинола — натриевую соль трифенилметаи-
сульфокислоты [334].
2. Взаимодействие окисей алкенов е соединениями серы
При взаимодействии окисей алкенов с серусодержащими соединениями, напри-
мер тиоцианатами, тиосульфатом натрия или тиомочевиной, происходит обмен кисло-
рода иа серу и образование соответствующих алкнленсульфпдоа [335, 336):
R-CH—CH-R'^KSCN —► R—СН—СН—R'--KOCN
S V
Реакция часто протекает очень бурно, так что приходится применять интенсив-
ное охлаждение.
Цнклогексиленсульфид [336, 337). В течение 1,5 ч при 60° С 5 г окиси
циклогексена в 20 мл метилового спирта обрабатывают 5 г тиомочевины. Затем
выливают реакционную смесь в 150 мл воды. Продукт реакции извлекают
хлороформом и фракционируют перегонкой в вакууме. Выход циклогексилен-
сульфида 58% от теоретического; т. кип. 67—68° С (16 мм рт. ст).
При взаимодействии окисей алкенов с сероводородом происходит расщепление
кольца с образованием Р-оксимеркаптанов; таким способом при обработке глициди-
лового спирта раствором Ва(ОН)», насыщенным сероводородом, получают а-монотно-
глицерин с 68%-ным выходом [338], а прн пропускании окиси этилена с небольшим
избытком влажного сероводорода через нагретую до 150—160° С глиняную трубку —
монотиоэтиленгликоль [339, 340].
Расщепление окисей алкенов, которое может происходить также под действием
меркаптанов, приводит к получению простых p-окситиоэфиров. Для этой реакции
были использованы окись этилена, окись циклогексена, эпихлоргидрин, а в качестве
тиоловых компонентов — этил-, пропил-, фенил- и бензилмеркаптаны в присутствии
небольшого количества активированного угля как катализатора [340].
Этил-(Р-окси-у-хлорнропнл)-сульфид. В течение 4 ч нагревают при 50° С
46 г эпихлоргидрина и 31 г этилмеркаптаяа. Выход продукта 90% от теорети-
ческого; т. кип. 114—115® С (16 мм рт. ст.}.
Фенил-[Р-оксиэтил)-сульфид. В закрытом сосуде в течение 24 ч выдержи-
вают смесь Иг тиофенола, 4,4 г окиси этилена и небольшого количества актив
внрованного угля. Выход продукта 12 г; т. кип. 119—120° С (4 мм рт. ст.)Л
Расщепление эпоксидов происходит с очень хорошими выходами под-действием
меркаптидов калия в спиртовом растворе в отсутствие катализатора [341].
[333] A. Mothwurf, Вег., 37, 3158 (1904).
[334] А. В а еу er, V. V i] 1 ig er, Ber., 35, 3016 (1902).
[335] франц, пат. 797621 (1934); С., 1936, II, 1062.
[336] С. С. J. Culvenor, W. Davies, К. Н. Pausacker, J. Chem. Soc.,
1946, 1050..,
[337] Е. Е. v a nj Та mel en, Org. Synth., 32, 39 (1952).
[338] L. S m i 11£, В. S j 6 b e г g, Ber., 69, 678 (1936).
[339] Франц, пат- 769216 (1933); С., 1935, I, 1770.
[340] C. D, Nenitzescu, N. Scarlatescu, Ber., 68, 587 (1935).
(341 f С. C. j. С и J v e n о r, W. Davies, N. S. Heath, J. Chem. Soc., 1949,
278.
38 Заказ 1835.
594
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Важный для препаративных целей метод, основанный на обмене кислорс
на серу, заключается во взаимодействии окисей алкенов с бисульфитом натрия [34
в результате чего образуются натриевые соли Р-оксисульфокислот [343]:
Н2С--CH2 + NaHSO3 —► CII2OH-CH2SO3Na
V
Натриевая соль 2-оксипропан-1-сульфокислоты [312]. Смесь 95
(0,5 моль) метабисульфита натрия и 67 г (1,16 моль) окиси пропилена перец
шивают 8 ч в 200 мл воды при компатной температуре и затем выпаривщ
досуха. После перекристаллизации из 90%-ного спирта получают 138 г (85,2
от теоретического) натриевой соли 2-оксипропан-1-сульфокислоты; после wj
ричной перекристаллизации из 95%-ного спирта т. пл. продукта 225—227*
3. Взаимодействие карбонильных соединений
с соединениями серы
Обмен атома кислорода в карбонильных соединениях на сору с образовали
меркаптанов может происходить под действием сероводорода или серы в присутств
водорода и не отравляемых серой катализаторов гидрирования [344].
„ „ катализатор „ „
R—С—R' + H2S+H2 ------=Гн7б—* R—СН—R'
О SH
Реакция протекает под давлением около 140 ат при 150—200° С в орисутстк
кобальт- иля никельполисульфидньгх катализаторов. Выход продукта в среда
70% от теоретического.
В отсутствие водорода, но под давлением, которое в зависимости от характе;
карбонильного соединения колеблется в пределах 35—85 ат, альдегиды и кетот
реагируют с сероводородом с образованием гем -дитиолов [345]:
R-C-R' + 2H2S —R-C-R'
|| "н’° /\
О HS SH
При действии Нг3 на карбонильные соединения в присутствии соляной ннсл<
или других конденсирующих средств образуются тиоальдегиды и тиокетоны, ’
является наиболее общепринятым методом получения этих соединений. Однако у тц
альдегидов и тиокетонов в еще более сильной степени, чем у их кислородных аналоге
проявляется стремление к полимеризации, предпочтительно с образованием цикл
ческих тримеров:
R-C-R'
|н,5
R-C-R'-----
HO SH
слабокислая
среда
сильнокислая
среда
Линейный полимер
с пеболыпим содер-
жанием кислорода [346]
[342] Герм. пат. 569148 (1931); С., 1933, I, 3004.
[343] R. Ten Eych Schenck, S. Kaizerman, J. Am. Chem. Soc., 71
1636 (1953).
[344] M. W. Farlow, W. A. Lazier, F. JG S igna i go, Ind. Eng. Chem.
42, 2547 (1950). *
[345] T. L. Cairns, G. L. Evans, et al., 3. Am. Chem. Soc., 74, 3982 (1952)
III. ОБМЕН КИСЛОРОДА НА ^ФУНКЦИЮ
595
Эти тримеры, являющиеся производными 1,3,5-тиациклогексана, обычно назы-
вают e-тритианами. Тиоформальдегид и симметричные тиокетоны образуют тримеры
только одной формы, остальные соединения (кроме соединений с пространственно
блокирующими заместителями) могут существовать в двух стереоизомерных формах
(а и р). Для а (^ис)-формы характерно расположение всех трех заместителей R или
R' по одну сторону плоскости кольца, в то время как у 0 (тпронс)-формы только два
заместителя R или R' расположены по одну сторону плоскости кольца. Под влиянием
катализаторов г^ис-соединеиии могут переходить в более устойчивые и высокоплавкие
0-формы [347—349].
Тиоальдегиды приобрели препаративное значение, так как при действии на
них восстановителей отщепляется сера (в виде сероводорода) и образуются олефины
и полимеры [350]:
2RC< —> RCH«CHR + 2S
^S
Методику получения тритиоформальдегида см. [351].
а-Тритиоацетальдегид [352]. В смесь равных частей воды, ацеталь-
дегида и концентрированной соляной кислоты пропускают сероводород. Выде-
ляющееся при этом масло постепенно затвердевает; через 24 ч продукт отфиль-
тровывают и многократно перекристаллизовывают из ацетона; т. пл. 101° С.
0-Тритиоацетальдегид [352]. В смесь 1 ч. ацетальдегида и 3 ч. насы-
щенного хлористым водородом спирта пропускают сероводород. Продукт пере-
кристаллизовывают из ацетона; т. пл. 125—126° С.
0-Тритиобензальдегид [352]. В смесь бензальдегида и спиртового рас-
твора хлористого водорода пропускают сероводород. Образовавшийся осадок
кипятят с бензолом; при охлаждении смеси 0-тритиобензальдегид сооса-
ждается с одной молекулой кристаллизационного бензола, который улетучивается
при температуре около 140° С; т. пл. ₽-трИтиобензальдегида 225° С.
Получение а-тритпобензальдегида см. [353].
Моногалогенбензальдегиды превращают в соответствующие тримерные
тиоальдегиды действием газообразных НС1 и H2S в абсолютном этилацетате [354]
Тиокетоны являются окрашенными соединениями, но все они, особенно диалкил'
и алкарилтиокетоны, очень склонны к образованию бесцветных трцмерных тритианов.
Так, тпоацетон обычно существует только в виде тримера и лишь при нагревании
Превращается в мономер (фиолетовые пары) [355]. Однако при взаимодействии ацето-
фенона с сероводородом [355] наряду с тримером возникает мономерный тиокетон
в виде разлагающегося синего масла, которое получается также при пиролизе тримера.
Простые мономерные тиокетоны до сих пор мало изучены из-за присущего им непе-
реносимого запаха [356].
Тритиоциклопентанон и тритиоциклогексанон [357]. В раствор цикло-
пентанона или циклогексанона в пятикратном количестве абсолютного этило-
вого спирта пропускают сначала хлористый водород, а затем насыщают серово-
[346]
S. W. Lebodew, М. Platonow, Вег., 69, 762 ' (1926); Е. Muller.
G. Schiller, J. prakt. Chem. [2], 116, 175 (1927); J. H. Wood,
R. W. Bos t, J. Am. Chem- Soc., 59, 1011 (1937).
0. Hassel, H. V i e r v о 1 1, Acta Chem. Scand., 1, 149 (1947).
A. Schdnberg, M. Z. Barakat, J. Chem. Soc., 1947, 693.
A. Schonberg, J. Chem. Soc.. 1948, 891.
R. Kuhn, Angew. Chem., 50, 703 (1937).
R. W. В 6 s t, E. W. Constable, Org. Synth., 16, 81 (1936).
E. Baumann, E. Fromm, Ber., 22, 2600 (1889).
H. Klinger. Ber., 9, 1895 (1876); 10, 1877 (1877).
J. A. Stanfield, L. B. Reynolds jr., J. Am. Chem. Soc.,
74, 2878 (1952).
E. В a u m a n и, E. F г o m m, Ber., 28, 895 (1895)i
E. Baumann, B. Fromm, Ber., 22, 2592 (1889).
E. Fromm, Ber., 60, 2090 (1927).
38*
596
ОБРАЗОВАНИЕ C-S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
дородом. Через некоторое время из раствора выделяются кристаллы, ко
рые отсасывают н промывают разбавленным спиртом. После перекристаллнзав
из этилового спирта т. пл. тритиоциклопентанона 99Q С, т. пл. тритиогеи
нона после переосаждения на хлороформа этиловым спиртом 101 —102’
Мономеры, а также тиокамфора получены Сеном [358].
Ароматические и гетероциклические тиокетоны не склонны к полимериза:
и, как правило, хорошо кристаллизуются. Для получения тиокетонов ароматнчес
кетоны обрабатывают сероводородом в присутствии хлористого водорода (напри*
при синтезе тиобензофенона [359] и тиофлуоренона [360] или пентасульфидом фосф
(например, при получении диметилтиохромона [361] и 4-тиофлавона [362]).
4-Тио-у-инрон [363]. у-Пирон кипятят 30 мин с двойным колнчесл
чистого желтого пентасульфида фосфора. Отфильтрованный раствор и бензо
ный экстракт осадка на фильтре выпаривают. После перекристаллизации оста'
из иизкокипящего петролейного эфира получают желтые иглы; т. пл. 49е
При взаимодействии меркаптанов с альдегидами н кетонами происхо;
отщепление воды и образуются геминальные диалкилмеркаптосоединения: меркапт!
(производные альдегидов, R' = Н) и меркаптолы (производные кетонов):
R4 R4 /SR’
\C=O + 2R”'SH ——>C<
n./ -H.0 Д./ \SH.
меркаптол
Обычно альдегиды более реакционноспособны, чем кетоны. Из тиолов бенз!
меркаптан конденсируется легче, чем этнл-, амил- и фенилмеркаптаны [364].
Меркаптали. Общий способ получения [365]. В смесь 2 моль меркапт»
и 1 лоль альдегида пропускают сухой НС1. Реакция протекает быстрее ц
нагревании и удалении воды. Меркаптали перекристаллизовывают из эфи,
петролейного эфира или бензола. Ди-л-бромфенилмеркапталь бензальдег!
после перекристаллизации из эфира имеет т. пл. 79—80° С; дифенилмерк
таль коричного альдегида, перекристаллизованный из петролейного эфи
— т. пл. 80—81° С; дифенилмеркапталь пиперонала после перекристаллиэав
из ацетона — у. пл. 48° С.
Общую методику получения дибутилмеркаптолов и дпбутилмеркаптаЛ
см. [366].
Описано также получение меркапталей углеводов [367]. Эта реакция примени!
ко всем альдозам и алифатическим тиоспиртам, но не идет с тиофенолами а таю
с фруктозой и сорбозой [368], поэтому таким путем можно, например, ’дт-тдели
глюкозу из смеси фруктозы и сорбозы.
D. С. Sen, J. Indian Chem. Soc., 13, 268 (1936); 12, 647 (1935).
H. Stand Inger, H. Freudenberger, Ber., 61, 1576 (1928).
E. C a m p a i g n e, W. B. R e i d jr., J. Am. Chem. Soc., 68, 769 (1946).
H. Simonis, S. Rosenberg, Ber., 47, 1232 (1914).
W. Baker, J. В. Harborne, W. D. Ollis, J. Chem. Soc., 1952, 1302
F. Arndt, E. Scholz, P. Nachtwey, Ber., 57, 1903 (1924)’.
T. Posner, Ber.. 34, 2643 (1901).
E. Baumann, Ber., 18, 883 (1885).
T. C. Wb itner jr., E. E. Reid, J. Am. Chem. Soc., 43, 639 (1921).
E. Fischer, B*er., 27, 673 (1894).
H. Zinner, Chem. Ber., 83, 275 (1950); 84, 780 (1950); 86, 495 (1953).
JU. ОБМЕН КИСЛОРОДА НА S-ФУНКЦИЮ
597
Диэтилмеркапталь глюкозы [367]. В склянке с притертой пробкой
прп встряхивании растворяют при комнатной температуре 70 г глюкозы в 70 г
дымящей соляной кислоты (d — 1,19). Раствор охлаждают льдом и смешивают
(в четыре приема) с 40 г этилмеркаптана, энергично встряхивая. Через корот-
кое время большая часть меркаптана растворяется и смесь разогревается.
Через 4 ч отсасывают плотную кристаллическую кашицу, промывают ее неболь-
шим количеством холодного этилового спирта и хорошо отжимают. Выход
неочищениого продукта 59 г. После перекристаллизации из четырехкратного
количества этилового спирта, двукратной перекристаллизации из воды и еще
одной из спирта получают чистый для анализа препарат; т. пл. 127—128° С.
Для идентификации и выделения карбонильных соединений особенно пригодны
меркаптали, получаемые на основе п-нитробензилмеркаптана [369] п меркаптокарбо-
новых кислот [370].
При взаимодействии кетонов с меркаптанами в тех же условиях, что и с альде-
гидами, образуются меркаптолы, хотя и с большим трудом [365]; при этом в качестве
конденсирующего агента обычно рекомендуется добавлять хлористый цинк [371].
4. Взаимодействие производных карбоновых кислот
с соединениями серы
Ацилирование сероводорода (ср. стр. 588) приводит к тиокарбоновым кислотам.
Так, например, при пропускании сероводорода в уксусный ангидрид образуется тио-
лксусная кислота с 74%-ным выходом [372, 373]. Этот метод применим также и для
других ангидридов [374].
При ацилировании меркаптанов ангидридами кислот получаются эфиры тио-
карбоновых кислот. Для синтеза низших эфиров используют метод Шоттепа — Бау-
манна [375, 376].
Эфиры тиокарбоповых кислот. Общий способ получения. Смесь
40 г NaOH, 75 мл воды и 1,2 моль меркаптана выливают на 500 а мелкораз-
дроблениого льда и при энергичном перемешиваний быстро добавляют 1,25 моль
уксусного ангидрида. Перемешивают еще 5 мил, после чего образовавшийся
эфир отделяют, промывают, сушат и перегоняют.
Для получения высших алифатических эфиров (от С4 до Cg) тиоуксус-
ной кислоты [376] смесь 1 моль меркаптана, 1,2 моль уксусного ангидрида н 1/5 от
его массы безводного ацетата натрия кипятят 1 ч с обратным холодильником
па масляной бане, температуру которой повышают от 130° С (начальная тем-
пература) до 150° С. Реакция обычно протекает так бурно, что колбу периоди-
чески необходимо вынимать из бани. По окончании реакции охлажденную смесь
выливают в воду и выделяют эфир, как описано выше.
Этим способом получен ряд эфиров тиоуксусной кислоты — от метилового
до «-октилового с выходами в среднем 90% от теоретического. Тиофенол ацилируют
смешанными ангидридами кислот, содержащими желаемый ацильный остаток и оста-
ток моноэтилового эфира угольной кислоты [377].
Из эфиров ортотиокарбоновых кислот наиболее доступны эфиры орютиому-
равьиной кислоты, которые получают взаимодействием муравьиной кислоты с меркап-
танами [378].
[369] A. Schaeffer. Л. Murua, Вег., 40, 2007 (1907).
[370] J. J. R i t t е г, М. J. Lover, J. Am. Chem. Soc., 74, 5576 (1952).
1371] J. Bongartz, Ber., 19, 1931 (1886).
1372] E. К. Ellingboe. Org. Synth., 31, 105 (1951).
1373] B. Sioberg, Svensk kern. Tidskr., 63, 90 (1951); C. A., 47, 8640 (1953).
1374 Пат. США 2587580 (1952); С. А., 46, 10192 (1952).
1375 О. Hinsberg, Вет., 39, 2433 (1906).
[376 F. W. Wenzel jr., Е. Е. Reid, J. Am. Chem. Soc., 59, 1089 (1937).
[377 T. Wieland, W. Schafer, E. В okelmann, Ann., 573, 99 (1951).
[378 J. Ilonben, К. M. L. Schultze, Ber., 44, 3235 (1911); J. H ouh en,
Ber. 45, 2942 (1912); ср. также H, Grop, A. Riechc, Chem. Ber., 94, 543
(1961)--
598
ОБРАЗОВАНИЕ C—S СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Еще одним препаративно важным методом обмена кислорода в карбонильных
соединениях на серу является взаимодействие амидов карбоновых кислот с сульфи-
дами фосфора, приводящее к образованию тиоамидов [379]:
5RCNR2-^P2S6 —> 5RCNR2^P2O5 •
II II '.(i
О s
Эта реакция, обычно проводимая в кипящем бензоле или толуоле, довольна
широко применима. Она протекает со вторичными и третичными амидами [380] зна-
чительно более гладко, чем с первичными, которые легко дегидратируются образуй
ющейся пятиокисью фосфора до нитрилов. J
Тиоамиды [381]. Амиды кислот обрабатывают смесью PaS5 и K2S сначала
при комнатной температуре, затем при 70—80° С. Этим способом получены
в частности, тиоацетамид, тиобензамид, фенилтиоацетамид, тиоамид гидро
коричной кислоты и их N-метилпроизводные. ч
Для синтеза тиобензанилида [382] в качестве растворителя лучше исполь*
зовать пиридин.
О методах обмена кислорода на серу в амидах кислот см. обзор [3831.1
5. Взаимодействие алкил- и диалкилсульфатов
с соединениями серы л;
Алкилирование соединений серы алкил- и диалкилсульфатами имеет меныпй
значение, чем алкилирование алкилгалогенидами. Так, например, получение меркан*
танов [384, 385] перегонкой эфиров серной кислоты с гидросульфидом калия в настой
ящее время почти не применяется в лабораторной практике. 1 7.
Простые тиоэфиры, особенно низшие, можно получать алкилированием сулй
фидов щелочных металлов как алкилсульфатами [386, 387], так и диалкилсульфа^
та.ми [388-390].
Диэтилсульфид [391]. Растворяют 120 г Na2S-9H2O в 120 мл воды^
раствор нагревают до кипения в колбе для перегонки емкостью 750 мл, слаб*
жени ой эффективным нисходящим холодильником и помещенным в лед прием*
ником. Затем в раствор приливают по каплям 155 г диэтилсульфата с такс®
скоростью, чтобы реакция протекала бее подвода тепла извне. Дистиллят наст/*
щают NaCl, отделяют сульфид в делительной воронке н после высушивания
над СаС12 фракционируют. Выход диэтилсульфида 35 г (78% от теоретического)?'
т. кип. 90-92° С. «
При алкилировании простых тиоэфиров могут получаться сульфоиийалнвл-;
сульфаты. Применение диалкилсульфатов иногда необходимо, так как, например/-,
диарилсульфиды [392] и метиловый эфир и-тиокрезола не образуют сульфонпевыхг
солей с алкилгалогенидами.
A. W. Hof шапп, Вег., П, 340 (1878).
A. Reiss ert, Ber., 37, 3708 (1904).
К. Kindler, Ann., 431, 209 (1923).
E. Klingsberg, D. Papa, J. Am. Chem. Soc., 73, 4989 (1951).
R. N. Burd, G. DeLaMater, Chem. Rev., 61, 47 (1961).
J. Obermeyer, Ber., 20, 2918 (1887).
Vesely, Ber., 47, 1516 (1914).
Ber., 20, 3407 (1887).
[379]
[380
[381
|382
[383
[384 . . . _ ,
[385] E. Votocek, V.
386] Г. Y.
387 H. H epworth, J. Chem. Soc., 119, 1254 (1921).
388 H. Bohme, W. Krause Chem. Ber., 82, 426 (1949).
389
390
391
1392]
11. и и и ш е, vv. лгаиэе, vuuiu. uei., ui, \iinvj.
Н. L. Gray, G. О. Gutekunst, J. Am. Chem. Soc., 42, 856 (1920).
W. Strecker. R. S p i t a 1 e r, Ber., 59, 1763 (1926).
H. Bohme, H.*J. Gran, Ann., 577, 72 (1952).
F. Kehrmann, A. Duttenhofer, Ber., 38. 4197 (1905).
rv. ЗАМЕЩЕНИЕ АЗОТА СЕРОЙ
59$
Диметил-я-толилеульфоинёметилсульфат [393]. В течение 15—20 мин
па водяной бане нагревают эквивалентные количества метилового эфира тио-
крезола и диметилсульфата. Сначала получается вязкое масло, которое при охла-
ждении затвердевает в кристаллическую массу; т. пл. продукта 97° С.
Метилирование диметилсульфатом используют для получения метиловых
эфиров из тиокарбоновых кислот [394] и алифатических сульфокислот из сульфитов.
Метаисульфонат бария [395]. В 600 .ил 26%-ного раствора (NH^gSOg
при 10° С медленно вносят 126 г чистого диметилсульфрта. После растворения
диметилсульфата к раствору добавляют пасту из 75 а кристаллического (NHJaSOg.
с 75 мл воды п смесь нагревают несколько часов на водяной бане. Затем
массу разбавляют в горячем виде до трехкратного объема, разлагают небольшой
избыток (NH^SOg серпов кислотой, добавляют рассчитанное количество-
Ва(ОН)2 и выпаривают аммиак. Избыток Ва(ОН)2 осторожно удаляют серной
кислотой, затем нейтрализуют массу ВаСО3 и фильтруют. Фильтрат выпари-
вают и получают в остатке метаисульфонат бария.
IV. Замещение азота серой
1. Замещение аминогруппы S-фуннцией
Замещение аминогруппы с образованием С—S-связи проводится через легко-
получаемые ия ароматических аминов соли диазония. Как и при аналогичных реакциях
алкилирования, для того, чтобы в большей или меньшей степени исключить образова-
ние простых тиоэфиров, необходимо применять в качестве акцепторов арила только
такие гидросульфиды щелочных металлов, в которых водород формально замешен
легко отщепляемым остатком, главным образом ацилом. Так, для получения тиофе-
колов преимущественно используются ксантогенаты щелочных металлов. Образу-
ющиеся вначале ксантогенаты диазопия с отщеплением азота легко превращаются
в арилксантогеиаты, из которых щелочным гидролизом получают тиофенолы [396,
{ s-с-оон6 он-~ > /^>-SH
Xe=Z II
S
Для предотвращения взрывов [398] реакцию рекомендуется проводить очень
осторожно при перемешивании, лучше всего путем добавления холодного раствора
соли диазония к нагретому до 60—70° С раствору ксантогената. На 1 моль диазосоеди-
нения целесообразно брать 1,4 моль ксантогената в десятикратном количестве воды
[393] К. Auwers, F. Arndt, Вег., 42, 2713 (1909).
[394] L. S. Р г a t t, Е. Е. R е i d, J. Am. Chem. Soc., 37, 1934 (1915).
[395] C. Wey gaud (неопубликованная работа).
[396| R. Lcuckart. J. prakt. Chem. [2], 41, 187 (1890).
[397] D. S. Tarbell, D. K. Fukushima, Org. Synth., 27 , 81 (1947).
[398] К. H о 1 z a c h, Die aromatischen Diazoverbindungen, F. Елке, Stuttgart,
1947, S. 216.
[399] К. H. Saunders, The Aromatic Diazo-Compounds and -their Technical
Applications, E. Arnold a. Co., London, 1949, p. 326.
600
ОБРАЗОВАНИЕ G—S-СВЯЗП В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Тногидрохинон [400]. Растворяют 110 г (1 моль) п-аминофенол
в избытке 10%-ной НС1 (2,25 лоль), охлаждают ниже 15° С и при 15—20*.
медленно добавляют 70 г (1 моль) NaNO2. Полученный раствор соли диазони
при хорошем перемешивании и 70—75° С медленно вводят в раствор 224
(1,4 моль) этилксантогената калия в 650 мл воды. Затем смесь нагревают еш
30 мин при 90° С, добавляют 160 г (4 моль) NaOH (для разложения ксантом
ната) и кипятят полученную массу в течение нескольких часов. Далее эту смев
сильно подкисляют избытком 50%-ной H2SO4 и нагревают в присутствии цин^
и бензола для восстановления образовавшегося дисульфида. Тиогидрохиив
выделяется в виде масла. Его промывают разбавленной водой и перегоняв
в вакууме. Выход тпогидрохинона 61 г; т. кип. 133—137° С (11 мм рт. спЦ
При взаимодействии солей диазония с Na2S2 также выделяется азот и глада
образуются дисульфиды:
1
2[ArNa]+ Cl--t-Na.2Ss —► Ar-SS-Ar + 2NaCl + 2N2
2,2'*Бне-(Р-карбокеивиннлфенил)-дисульфид [401]. Зто соединение пол^
чают, добавляя о-диазокоричную кислоту в горячий концентрированный рй
створ избытка Na2S2 с последующим подкислением. Для очистки продуй
растворяют в холодном растворе соды, фильтруют, подкисляют и перекриста^
лизовывают из этилового спирта; т. пл. 221° С. ,2-
Этим же методом синтезирована дитиосалициловая кислота (о,о'-дикаЙ
боксидифенилдисульфид) [402].
Ароматические простые тиоэфиры часто получают взаимодействием соле}
диазония с меркаптидами щелочных металлов. Во избежание взрывов реакцию лущф
всего проводить в приведенных выше условиях [403, 404]. Этим способом были синти
зиропаны а- и Р-нафтилтиогликолевые кислоты [405], Р-тноантрахиноил-1-антраХЙ
нон-2-карбоновая кислота (1,2'-диантрахиноилсульфид-2-карбоновая кислота) [408|
п-этоксифенилбутилсульфид [400] и в присутствии порошка меди, катализирующее
разложение диазосульфида, — дифенилсульфид [407].
Большое препаративное значение имеет распространенный метод замены диаз^
группы остатком сульфиновой кислоты действием SO2 в присутствии порошка мей
или Сп2О, действующих как восстановители [408].
Сульфиновые кислоты. Общий метод синтеза [409]. Полученный из амина
сернокислый раствор соли диазония, если необходимо, подкисляют избытрйй
H2SO4 и при хорошем охлаждении насыщают SO2 до поглощения не мещМ
15 г SO2 на каждые 100 мл раствора. Раствор должен оставаться полиостйЖ
прозрачным (помутнение наступает в случае недостаточного подкисления или
охлаждения). При продолжающемся охлаждении, перемешивании и пропускай
нии SO2 в реакционную массу вносят порошок меди [410, 411] до прекращений
выделения азота. Сульфиновую кислоту извлекают из раствора и осадка эфиром;
[400] Е. Mill е.г, R. В. Read, J. Am. Chem. Soc., 55, 1224 (1933). J
[401] C. Chmelewsky, P. Friedlander, Ber., 4(j, 1903 (1913).
402] C. F. II. Allen, D. D. McKay, Org. Synth., Coll. v. 2, 19553
p. 580.
[403] J. H. Ziegler, Ber., 23, 2469 (1890). • .«
[404] F. Mayer, Ber., 42, 3046 (1909).
[405] Герм. цат. 194040 (1905); Frdl., 8, 1370 (1905-1907).
[406] Герм. пат. 460087 (1926); Frdl., 16, 1250 (1931); ср. герм. пат. 475688 (1927$
Frdl., 16, 1360 (1931). - л
407] G. Е. Hilbert, Т. В. Johnson, J. Am. Chem. Soc., 51, 1526 (1929fc
408] Герм. пат. 100702 (1897); Frdl., 5. 44 (1897-1900).
409] L. Gattermann, Ber., 32, 1136 (1899). •
410] L. Gattermann, Ber., 23, 1219 (1890).
411] F. Ullmann, Ber., 29, 1S78 (1896).
IV. ЗАМЕЩЕНИЕ АЗОТА СЕРОЙ
эфирный раствор встряхивают с раствором соды и подкисляют водный экстр,
в зависимости от растворимости сульфиновой кислоты в воде ее отсасыв
или извлекают эфиром. Этим способом с выходами в среднем 90% от теор,
ческого были получены бензол-, толуол-, ксилол-, галогенбензол-, метоксид
.зол, этоксибензол-, карбоксибензол- и нафталинсульфиновые кислоты.
Таким методом синтезированы также различные галогенбензолсуль
новые кислоты [412, 413]-
При получении ароматических сульфиновых кислот в качестве восстановит
вместо меди с успехом можно применять соли Fe2+.
Так, например, из диазотированного 2,4-динитроанилина и двуокиси е
в присутствии FeSO4 с выходом 74,1% от теоретического образуется 2,4-дниитрсм
золсульфиновая кислота [414].
О методах получения сульфиновых кислот из солей диазония см. обзор [4
При реакции дифенилдиазометана с двуокисью серы выделяется азот и о?
зуется циклический сульфон — тетрафенилэтиленсульфон [416]:
2(CeH6)2CN2^-SO2 —> (СбН5)2С----C(CeH5)2 + 2N2
\ /
SO2
Показано [419], что разработанный Меервейном и сотр. [417] метод синд
п-хлорбензолсульфохлорида [418] из хлористого n-хлорфенилдиазония в жид
SO 2 в присутствии CuCl является общим методом получения ароматических суль
хлоридов:
[ArN2]+Cl’ + SO2 — -> ArSO2Cl + N2
Вместо жидкого SO2 целесообразно применять его насыщенный раствор (и
мерно 30%-ный) в ледяной уксусной кислоте, а вместо твердого хлорида диазони»:
возможно более концентрированный его раствор в соляной кислоте. В качестве ката
затора наиболее пригоден СиС12*2Н2О, восстанавливающийся в ходе реакции с об
зованием мелкодисперсного и частично растворяющегося CuCl, который и являе
катализатором. Только хлориды диазония с сильными электроноакценторными за
стителями, например динитробеизолдиазонийхлорид, реагируют с SO2 даже в от<
ствие катализатора. Температура реакции должна поддерживаться таким образ
чтобы происходило энергичное выделение азота и реакция протекала достатсг
быстро; в этом случае побочные реакции подавляются и увеличивается выход £j
дукта. Значительное ускорение реакции в некоторых случая^ может быть достигв:
введением в реакционную массу несмешивающихся с водой растворителей, имеюп
небольшую диэлектрическую постоянную (например, СС14. СвП9), или увеличен]
концентрации ионов хлора введением MgCI2. Этим способом с выходами в среда
70-80% от теоретического получены бензол-, толуол-, нафталин-, галогеибенз<
нитробензол-, метоксибензол- и карбоксибензолсульфохлориды.
n-Бромбензолсульфохлорид. Приготовленный из 43 г (0,25 л«мь) п-бр.
анилина, 85 мл 36%-ной НС1 и 19 г (0,275 моль) NaNO2 в 30 мл воды раст;
n-бромбензолдиазонийхлорида, содержащий 9.5% свободной НС1, прилива
к смеси 200 мл 30%-ного раствора SO, в ледяной уксусной кислоте и концепт*
рованного водного раствора Юг СиС12 • 2НгО. Происходит бурное выделе]
азота, температура смеси повышается с 12 до 35° С. Дальнейшее повыше]
[412] М. Е. Hank е, J. Am. Chem. Soc., 45, 1325 (1923).
[413] W. A. Silvester, W. P. Wynne. J. Chem. Soc , 1936, 691.
[414] H. M eerwein, G. Dittmar et al., Chem. Ber., 90 , 859 (1957).
[415] W. E. Truce, A. M. Murphy, Chem. Rev., 48, 86 (1951).
416] H. Staudinger, F. Pfenni л ger, Ber., 49, 1941 (1916).-
[417] H. M eerwein, E. В ii c h n e r, K. van E ms ter, J. prakt. Ch^
[2], 152, 251 (1939).
[418] Герм. пат. 859461 (1942); С., 1953, 7932.
[419] H. Meerwein, G. Dittmar et al., Chem. Ber., 90, 841 (1957)
602
ОБРАЗОВАНИЕ С—S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
температуры предотвращают охлаждением ледяной водой, Через 3—4 мин
реакция заканчивается. Реакционную массу смешивают с трехкратным коли-
чеством ледяной воды, через 15 мин продукт отсасывают, промывают и сушат.
Выход гс-бромбензолсульфохлорида 58,6 г (92% от теоретического); т. пл.
76,3—76,5° С (из бензола); т. кил. 153° С (15 мм рт. ст.}.
При взаимодействии диазобензол- и диазонафталинсульфокислот с раствором
двуокиси серы в ледяной уксусной кислоте в присутствия CuCl вместо сульфохлоридов
образуются дисульфокислоты, что обусловлено неустойчивостью моносульфохлоридов
дисульфокислот в водных растворах [419]. Реакция протекает гладко и с хорошими
выходами. Так, например, из 1-диазо-2-оксянафталяп-4-сульфокислоты иолучают
2-оксинафталин-1,4-дисульфокислоту [420J.
Бензол-1,4-дисульфокислота. Раствор 18,4 е (0,1 ммь) диэзосульф-
аниловой кислоты в 100 мл концентрированной НС! приливают к суспензии
2 г CuCl в 150 мл раствора SO2 в ледяной уксусной кислоте. Температура
смеси при этом повышается с 18 до 23° С, дополнительным нагреванием темпе-
ратуру поднимают до 42° С. Выделение азота (2,3 л при нормальных условиях)
заканчивается через 4 ч. Прозрачную реакционную смесь выпаривают в вакууме
досуха и добавляют насыщенный раствор NaCl; образуется каганца бензол-1,4-
дисуцьфопата натрия. После отсасывания его несколько раз промывают метило-
вым спиртом, перекристаллизовывают из насыщенного раствора NaCl и вновь
тщательно промывают метиловым спиртом для удаления NaCl. Выход продукта
21,5 г (76,3% от теоретического).
Аналогичным способом были получены бснзол-1,2~, бензол-1,3-, нафта-
лин-1,4- и нафталин-1,6-дисульфокислоты, а также п-бензолсульфоарсоиоваф
кислота.
2. Замещение имииогруппы S-фуикцией
При взаимодействии этилеииминов с сероводородом или меркаптанами проис-
ходит разрыв С—N-связи и образуются 6-меркаптоалкиламины или 6-аьшноэтил-
сульфиды [421]:
Н2С--CH2 + HSH(RSH) —> CII2-CH2
NH - (R)HS NHs
Таким методом был получен цистеамин [422].
S-Метил-и S-этилцистеамнны [423]. К охлажденному до —15° С раствору
метил- кли этилмеркаптана в метиловом спирте постепенно добавляют этнлея-
имин и выдерживают смесь при комнатной температуре.
N-Ацетилцистеамип [424]. При 5 — 10° С к раствору 250 г тиоуксусной
кислоты в 1700 мл абсолютного метилового спирта по каплям добавляют
142 г этиленимина и затем кипятят с обратным холодильником в течение 30 мин.
Продукт выделяют фракционированием в вакууме, причем вначале перего-
няется небольшое количество S-ацетилцистеамина, который кристаллизуется
в приемнике. Далее перегоняется N-ацетильное производное в виде бесцветного
масла. Выход продукта 175 г (45% от теоретического); т. кип. 138—140° G
(7 мм рт. ст.}.
[420] Франц, пат. 826402 (1937); С., 1938, II. 185. J
[421] Н. Bestian, Ann., 566, 210 (1949). '
[422] Е. J. М i 1 ] я, №. Т. Bogerl, J. Am. Chem. Soc., 62, 1173 (1940).
[423] T. Wieland, E. F. Moller, G. Dieckelmann, Chem. Ber-r
85, 1041 (1952).
[424] R. Kuhn, G. Q u a d b e c k, Chem. Ber., 84, 844 (1951). <;
IV. ЗАМЕЩЕНИЕ АЗОТА СЕРОЙ
Взаимодействие имидоэфиров с сероводородом является препаративно важнъ
методом получения эфиров тионкарбоновых кислот [425]:
ZNH
R—rH5S—► R-C<f +NH3
x0R’ XOR-
3. Замещение третичного азота и аммониевой группы S-фуикцией
Замещение третичного атома азота серой в некоторых шиффовых основания
приводит к образованию тиокетонов. Этим методом был получен тиобензофенон [42f
Тиобензофенон [426]. В аппарате для гидрирования (в утке) растворят
7 г анида бензофенона в 50 мл бензола, затем обрабатывают его сухим хлор
стым водородом, причем выпадает желтая солянокислая соль. Хлористый вод
род вытесняют сероводородом и встряхивают в течение шести суток под давд
иием аппарата Киппа. Образующийся темно-синий раствор отфильтровыва!
от солянокислого анилина и пепревращенного гидрохлорида шиффова основ
ння бензофенона, упаривают в токе СО2 бензол и остаток фракционируя;
перегонкой в вакууме. При 176—178° С (18 мм рт. ст.) в виде темно-сине
масла перегоняется 3 г тиобензофенона, затвердевающего при охлажден]
и плавящегося от тепла рук.
4-Тиофлавон [427]. В раствор 0,5 г флавонбензилимина в 15 лм эт
левого спирта в течение 30 мин пропускают сероводород, через 2 ч продул
отсасывают, осадок промывают водой и сушат. Выход 4-тиофлавона 0,277
(72% от теоретического). После перекристаллизации из петролейного эфи]
(т. кип. 60—80° С) т. пл. 88° С. Реакция протекает по следующей схеме:
NCH2CeH5 S
H,S
---->
Действием сульфида натрия на четвертичные аммониевые соли можно получи*
простые тиоэфиры; наряду с ними образуются соответствующие' третичные амин:
Однако расщеплепие происходит гладко только в том случае, есяи в четвертичн,
соли наряду с арильной имеется бензильная или аллильная группа, так как имен]
эти группы алкилируют сульфид натрия [428].
4. Замещение нптрогруппы S-функцией
В производных о-динитробензола одна иитрогруппа достаточно реакциона
способна и под действием сульфида, меркаптида или сульфита натрия может бьг
замещена на меркапто-, алкилмеркапто- или сульфогруппу. Одиако в препаративнъ
целях этот метод не нашел широкого применения.
При взаимодействии o-динитробензола с Na2S образуется <?,о'-динитродифена
сульфид [429]; из тринитроанизола — димстокситстранитродифенилсульфид [43С
[425] Y. Sakurada, Mem. Coll. Eng. Kvoto Imp. Univ., A 10, 67, 79 (192g
C., 1927, I, 1300, 1301.
[426] G. R eddelicn, H. D a n i 1 о f, Her., 54, 3x41 (1921).
[427] W. Baker, J. В. II а г b о г n e, W. D. Ollis, J. Chem. Soc., 19S
1299.
[428] H. R. S n у d e r, J. C. Speck, J. Am. Chem. Soc., 61, 668, 2895 (1939).
[429] C. A. L о b г у <be В r u у n, J. J. Blanksma, Rec. trav. chim., 20, 1-
(1901).
[430] j. J. Blanksma, Rec. trav. chim., 23, 114 (1904).
604
ОБРАЗОВАНИЕ С S-СВЯЗИ В РЕЗУЛЬТАТЕ ЗАМЕЩЕНИЯ
Из о-динитробензола и тиогликолевой кислоты получена о-нитрофенилтногликолевая>
кислота 1431].
Особенно легко протекает замещение нитрогруппы сульфогруппой в случач
нитроантрахинонов. Реакцию проводят многочасовым кипячением нитросоединений
с водным раствором сульфита натрия или калия [432, 433].
V. Замещение других функций J
1. Взаимодействие с реактивами Гриньяра
Высокая реакционная способность соединений Гриньяра широко использует^
для получения сероорганических соединений. Действие элементарной серы на грин^
яровские соединения из-за возможных побочных реакций (4341 является менее ваяй
ным, чем остальные, методом синтеза меркаптанов, однако реакция магнийорганич^
ских соединений с сульфепилхлоридами может широко использоваться для получений
простых тиоэфиров. и
RMgBr + CISR' —*• К—S—R' [ MgClBr (
Так, реакция фенил- и толилсульфенхлоридов с фенилмагнийбромидом приво]
дит к дифенил- и фенил-п-толилсульфидам [435]. )
Фенилбензилсульфид [255]. К раствору фенилмагнийбромида, получав
ному из 2,8 г магниевых стружек и 17 г бромбензола в 50 мл абсолютно:^
эфира, при перемешивании и охлаждении добавляют 5,85 г хлормегялсульфеинж
хлорида в 10 мл абсолютного эфира. После нагревания с обратным холодили
ником в течение 30 мин смесь охлаждают и выливают на лед. Добавляют 10 дм
концентрированной НС1, отделяют эфирный слой, сушат его над СаС1, и фрай|
ционируют перегонкой в вакууме. Первая фракция отгоняется при 60—90о’Я
(14 лел рт. ст.). Выход фенилбензилсульфида 2,4 г (55% от теоретического)^
т. кин. 145—155° С (14 мм рт. ст.). •;
При действии на соединения Гриньяра двуокиси серы [436] или сульфурил
хлорида [437] образуются сульфиновые кислоты. Выходы при этом редко превышай
€0% от теоретического. Побочные реакции могут быть подавлены проведением пр<
цесса при температурах ниже 0°С; количество SO2 не должно превышать необхоДв
мого для реакции.
2RMgBr 2HSO2MgBr , . (HSO2)aMg
Ниже приведен общий способ получения сульфиновых кислот. >3
Сульфиновые кислоты [436, 438]. В эфирный раствор алкилмагнийгаде
геиида при хорошем охлаждении пропускают SO2 до прекращения выпадет^
осадка. После удаления эфира белый кристаллический остаток сульфина'Г)
магния можно перекристаллизовать из воды. Этим способом получены магниевы
соли этан-, пропан-, и-бутап-, изопентан-, циклогексан- и бензолсульфниовМ
кислот, а при температуре от —35 до —40° С магниевая соль додекаи-1-сульф^
новой кислоты [439].
[431] Р. Friedlander, Вег., 39, 1065 (1906).
[432] Герм. пат. 164292 (1903); Frdl., 8, 231 (1905—1907). "
[433] Пат. США 2499003 (1950); С. А., 44, 7883 (1950).
[434] М. S. Kh ага sob, О. Reinmuth, Grignard Reactions of Nonmetall®
Substances, Prentice-Hall, Inc., New York, 1954, p. 1274. £
[435] H. Lecher, F. H olschn ei d er et al., Ber., 58, 414 (1925). '
[436] J. v. Braun, K. WeiBbach, Ber., 63, 2838 (1939). •
[437] B. Oddo, Gazz. chim. ital., 35, П, 136 (1905); C., 1905, 1Г, 763; ср. C., IMS
I, 1145. , *
|438] A. Rosenheim, L. Singer, Ber., 37, 2152 (1904).
[439] C. S. Marvel, R. S. Johnson, J. Org. Chem., 13, 824 (1948). -
V. ЗАМЕЩЕНИЕ ДРУГИХ ФУНКЦИЙ
60
Для выделения свободных сульфиновых кислот магниевую соль суспет
дируют в небольшом количестве воды и при очень хорошем охлаждении обрвб>
тывают концентрированной НС1 или H2SO4. Выделившееся масло экстрагируй
эфиром; из эфирного слоя извлекают кислоту раствором соды, вновь осаждай:
кислотой, еще раз экстрагируют эфиром и из экстракта удаляют эфир токо
воздуха при низкой температуре.
Другие примеры получения сульфиновых кислот см. [415, 440, 441
В некоторых случаях при взаимодействии арилмагнийбромидов с сульфоксв
дами удается получить сульфониевые соли [442]:
(C6H5)3SO+ArMgBr —> [(CeH5)2SAr]+[OMgBr]-
—> [ (CeIi6)2SАг] +Вг- 4- MgBra 4- НаО
Для синтеза тиокарбоновых кислот [443] может быть использована реакцк.
соединений Гриньяра с сероокисью углерода, при этом наряду с карбинолами обра
зуются тиокислоты с примерно 70%-ным выходом:
RMgX I-COS —► RCSMgX RCSH-I Mg(OII)X
Этим путем получены тиопропионовая, тиобензойная, тио-n- и тио-о-толуило
вые кислоты.
При взаимодействии сероуглерода с магнийорганическими соединениям!
образуются дитиокарбоновые кислоты [444]. Реакция имеет общий характер, н>
в ароматическом ряду дает лучшие выходы. Кроме того, следует упомянуть о получе
нки эфиров тионкарбоновых кислот взаимодействием соединений Гриньйра с эфирам]
хлорангидрида тионугольной кислоты [445]
RMgX + ClCOR' —> RCOR'4-MgXCl
и и
а также о получении тиоамидов к [446—448] действием соединений Гриньяра на
изотиоцианаты:
RN=C=S4-R'MgX —> RN=C— SMgX —► RN—С—SH —» RNH—C=S
Эта реакция протекает с выходами в среднем 90%, если применяют 50%-ны4
избыток гриньяровского соединения.
N-Цнклогексилтиобензамнд [447]. К раствору 0,08 моль феннлмагний-
бромида в 200 мл эфира, охлажденному до 5s С, при перемешивании добавляю1:
10,89’г (0,077 моль) циклогексилизотноцианата в 50 мл эфира. Затем реакционную
[440] Р. Allen jr., J. Org. Chem., 7, 23 (1942).
[441] J. Houben, H. Doescher, Ben, 39, 3503 (1906).
[442] B. S. W i 1 d 1, S. W. T а у 1 о г, H. A. P о t ra t z, J. Am. Chem. Soc.
73, 1965 (1951).
[443] F. Weieert, Ber., 36, 1007 (1903).
[444] J. Houben et al., Ben, 35, 3695 (1902); 39, 3503 (1906); 40, 1303, 172
(1907).
[445] M. D e 1 e p i n e, C. n, 153, 279 (1911); C., 1911, II, 1213; M. Delepine
Bull. Soc. Chim. France [4], 9, 904 (1911); C., 1911, II, 1584.
[446] D. F. W о г r a 1 1, J. Am. Chem. Soc., 47, 2974 (1925).
[447] G. A 11 i n g e r, G. E. P. Smith jr., E. L. С а г г, H. P. Stevens
J. Org. Chem., 14, 962 (1949).
[448] R. N, Hurd, G. DeLaMater, Chem. Rev., 61, 54 (1961).
606
ПОЛУЧЕНИЕ СОЕДИНЕНИЯ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ S-ФУНКЦИН
смесь перемешивают еще 1 ч при 5° С, постепенно нагревают до комнатной
температуры и выливают на лед. Массу подкисляют, эфирный слой отделяют,
сушат над Na^SOi н отгоняют эфир. Выход продукта 15 г (88% от теоретиче-
ского); после нескольких перекристаллизаций из бензина т. пл. 90—92° С.
2. Взаимодействие с другими металлоорганическими
соединениями
Получение тиоамидов из изотиоцианатов путем присоединения по С= S-связям
характерно не только для соединений Гриньяра, но и для других металлоорганиче-
ских соединений [449, 450J. Наиболее подробно описаны реакции изотиоцианатов
с фенилиатрием п фениллитпем [451], натриевыми солями ацетоуксусного эфира
[452], ацетилацетопа [453] и малонового эфира [454]. Реакция протекает гладко
я может быть использована для идентрфикации органических соединений щелочных
металлов [455]. Для получения сульфниовых кислот из двуокиси серы и металло-
органических соединений наиболее пригодны литий- [456] и натрийорганические
соединения [457]. В заключение следует отметить, что тиофенол образуется при взаи-
модействии фениллития с элементарной серой [458].
ПОЛУЧЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ
ИМЕЮЩИХСЯ S-ФУНКЦИЙ
К этим методам относятся восстановление, окисление и расщепление органи-
ческих соединений серы, Получение тиоугольной нислоты и ее производных рассматри-
вается отдельно в конце этого раздела, о перегруппировках органических соединений
серы см. стр. 868-
Z. Восстановление S-функций
Восстановление, которому могут быть подвергнуты практически псе органи-
ческие соединения серы, в зависимости от условий реакции приводит к образованию
меркаптанов или дисульфидов. Препаративное значение имеют преимущественно
восстановление ароматических сульфохлоридов и восстановительное расщепление
дисульфидов.
Ароматические сульфохлориды под действием различных восстановителей
в большинстве случаев с хорошими выходами превращаются в тиофенолы. Наиболее
простым методом является восстановление цинковой пылью в соляной или серной
кислоте.
Тиофепол [459]. При 0° С к смеси 400 ч. концентрированной H2SO4,
1200 ч. воды и 200 ч. цинковой пыли прд перемешивании приливают 100 ч.
бензолсульфохлорида. Смесь несколько часов выдерживают на холоду, затем
кипятят до исчезновения мути и перегоняют с водяным паром. Тиофенол из
дистиллята экстрагируют эфиром, экстракт сушат и ректифицируют. Выход
продукта 80—85% от теоретического; т. кип. 169,5° С,
[449] W. S с h 1 е n k, Е. Bergmann, Ann., 463, 1 (1928).
[450] W. S с h 1 е и к, Н. Н i 1 Те m a n n, I. R о ell о f f, Ann., 487, 135 (1931).
[451] И. Gilman, F. Breuer, J. Am. Cliem. Soc., 55, 1262 (1933).
[452] D. E. Worral, J. Аш. Chem. Soc., 40, 415 (1918).
[453] D. E. Worral, J. Аш. Chem. Soc., 42, 1055 (1020).
[454] D. E. Worral, J. Am. Chem. Soc., 50, 1456 (1928).
[455] A. A. M orton, A. R. Olson, J. W. Blattenberger, J. Am. Chem.
Soc., 63, 314 (1941).
[456] W. E. Truce, J. F. Lyons, J. Am. Chem. Soc., 73, 126 (1951).
[457] W. Schlenk, R. Ochs, Ber., 49, 613 (1916).
[458] H. C 1 1 ni а ц, L. Fullharl, J. Am. Chem. Soc., 71, 1480 (1949).
[459] E. Bourgeois, Ber., 28, 2319 (1895); Rec. trav. chim., 18, 432 (1899);
R. Adams, C. S. Marvel, Org. Synth., Coll. v. 1, 1937, p. 505.
I. ВОССТАНОВЛЕНИЕ S-ФУНКЦИИ
607
В качестве восстановителей используют также олово (для получения ч-хлор-
гнофенола [460], дихлорид олова (для синтеза дифенил-4,4'-дитиола [461]), порошок
железа (для получения о- и и-тиокрезолов [462]) и литийалюмннийгидрид (для полу-
чения »-бутплмеркаптана [463])
Сульфохлориды могут быть восстановлены нагреванием в течение 1 ч с полу-
торакратным весовым количеством амальгамы алюминия [464] в спирто-эфирной
смеси с последующим постепенным разбавлением смеси водой [465]. Таким спосо-
бом получены 2,5-днхлортиофенол, 0-тионафтол и я-ацетиламинотиофенол с выходами
50—70%. В антрахиноновом ряду для восстановления сульфохлоридов оказались
пригодными сульфиты щелочных металлов [466].
При соответствующих условиях восстановление сульфохлоридов можно оста-
новить па стадии сульфиновых кислот. Несмотря на то, что всегда возможно дальней-
шее восстановление сульфиновых кислот до меркаптанов и дисульфидов, эта реакция
широко используется для получения сульфиновых кислот [467]. В качестве восстано-
вителей применяют цинк в нейтральной, водно-щелочной или спиртовой среде [468—
470], амальгаму натрия в абсолютном эфире или бензоле [471—473], магний в эфире
[474] и железо или цинк в ледяной уксусной кислоте [475].
Тиофен-2-сульфиновая кислота [476, 477]. Тиофенсульфохлорид раство-
ряют в нескольких объемах спирта и постепенно при охлаждении смешивают
с цинковой пылью. После исчезновения запаха сульфохлорида отсасывают
кашицеобразную реакционную массу и осадок тщательно промывают водой
для удаления ZnCL. Остаток, состоящий из цинковой соли тиофенсульфиновой
кислоты и цинковой пыли, суспендируют в воде и добавлением раствора соды
переводят цинковую соль в легко растворимую в воде натриевую соль. Раствор
фильтруют, фильтрат упаривают ва водяной бане до небольшого объема, под-
кисляют НС1 и экстрагируют эфиром сульфиновую кислоту. После отгонки
эфира в виде масла остается свободная сульфиновая кислота. Она затверде-
вает при выдерживании в вакууме над H2SO# в иглы; т. пл. 67е С.
Ароматические сульфохлориды, за исключением сульфохлоридов, неустойчи-
вых в щелочной среде, почти всегда удается восстановить действием сульфита натрия
в нейтральной водной или спиртовой среде или, лучше всего, в водном растворе би-
карбоната щелочного металла:
BSO2Cl-bNa2S03 + 2Na0H —> BSO2Na + NaC14-Na2SO4-l-H2O
Сульфиновые кислоты. Общий способ получения [478]. Встряхивают
смесь 20 г сульфохлорида, 50 г Na2SO3-9H2O и 100 г раздробленного льда
до растворения сульфохлорида (около 3 ч). Во избежание выделения SO2
Н. J. В а с k е г, J. Kramer, Rec. trav. chim., 53, 1101 (1934).
C. S. Marvel, P. D. Caesar, J. Am. Chem. Soc., 73, 1097 (1951).
E. Profit, Cbem. Techn., 5, 239 (1953).
C. S. Marvel, P. D- Caesar, J. Am. Chem. Soc., 72, 1033 (1950).
H. W i s 1 i с e n и s, J. prakt. Chem. [2], 54, 18 (1896).
E. Gebauer-Fiilnegg, J. Аш. Chem. Soc., 49, 1386 (1927).
Герм. пат. 292457 (1914); С., 1916, II, 42; Frdl , 13, 394 (1916—1921).
W. E. Truce, A. M. Murphy, Chem. Rev., 48, 69 (1951).
M. Bazlen, Ber., 60, 1470 (1927).
C. Pauly, Ber., 9, 1595 (1876).
F. C. Whitmore, F. H. Hamilton, Org. Synth., Coll. v. 1, 1937,
p. 493.
R. Otto, Ann., 143, 207 (1867).
R- Otto, O. v. Gruber, Ann., 142, 93 (1867).
S. Gabriel, A. Deutsch, Ber., 13, 388 (1880).
II. Gilman, R. E. Fothergill, J. Am, Ciicm. Soc., 50 , 804 (1926).
W. Kalle, Ann., 119, 160 (1861).
L. Weitz, Ber., 17, 800 (1884).
A. Biedermann, Ber., 19, 1616 (4886).
S. Krishna, H. Sing, J. Am. Chem. Soc., 50, 794 (1928).
608 ПОЛУЧЕНИЕ СОЕДИНЕНИИ СГЛ’Ы ПУТЕМ ПРЕВРАЩЕНИЯ 8-ФУККЦИИ
периодически проверяют щелочность среды и добавляют разбавленный раствор
NaOH. Смесь постоянно охлаждают добавлением льда. После полноте
растворения смесь фильтруют и сульфиновую кислоту медленно осаждаем
концентрированной НС1. Выход кислоты 80—90% от теоретического.
Этим способом получены бензол-, толуол-, галогенбензол- и карбокси
бензолсульфиновые кислоты.
Кроме сульфита натрия для получения сульфиновых кислот в качестве восста
повителей используют дихлорид олова f479j, сульфиды [480, 481] и арсеинты [482
щелочных металлов, тиофеноляты [483] и металлоорганические соединения циню
[484], свинца [485] и магния [486].
Сульфиновые кислоты могут быть восстановлены до меркаптанов и дисульфи
дов [487—489] преимущественно действием цинка в кислой среде.
Общий способ восстановления сульфиновых кислот [490]. Сульфино
вую кислоту растворяют в воде, добавляют четырех- или пятикратное количе
ство цинковой пыли и при пропускании водяного пара постепенно смешиваю!
с небольшим избытком 30%-ной H2SO4. Отогнанный с водяным паром меркапта!
экстрагируют эфиром, сушат над Na2SO4 и ректифицируют.
Этим способом получены алкил-, алкокси- и карбокситиофенолы.
Если сульфиновые кислоты восстанавливают бромистым водородом в уксусной
кислоте, то образуются дисульфиды [488, 491—493]:
2RSO2H-J-бНВг —► R—SS-R + 4H2O + 3Br2
Меркаптаны могут быть получены также восстановительным расщеплением
простых тиоэфиров натрием в жидком аммиаке [494]; очень широко применяется
также восстановление дисульфидов. Например, для синтеза тиосалициловой инслоты
рекомендуется использовать цинк в ледяной уксусной кислоте [495], для п-бром-
тиофенола — цинк в соляной кислоте [496], для низших алкантиолов — металли?
ческий натрий в эфире [497], для цистеина — натрий в жидком аммиаке [498], для
бутил-, амил-, фенил-, бензил- и додецилмеркаптанов — литийалюминийгидрвд
в эфире или тетрагидрофуране [499] и, наконец, для различных алкил- и арилмеркащ
танов — сульфид калия [500].
Герм. пат. 224019 (1908); С., 1910, II, 513.
Герм. пат. 263340 (1912); С., 1913, I, 829.
A. Gutmann, Вег., 42 480 (1909).
R. Otto, Вег., 24, 713 (1891).
W. Kalle, Ann., 119, 158, 161 (1861).
R. Schiller, R. Otto, Ber., 9, 163C ""
H. Burton, W. A. Davy, J. -------- ---
K. Fries, W. Vogt, Ann., 381, 331 (1911).
A. ReiBert, Ber., 55, 858 (1922).
W. B. Price s. Smiles, J. Chem. Soc., 1928, 2372.
9, 1636, 1637 (1876).
J. Chem. Soc., 1948, 529.
[479] M. Cl a as z, Ann., 380, 314 (1911).
480 -- — —nx. «ua
481
482
483
484
485
486
487
488]
|489] ..v, m. . -
490] L. Gattermann, Ber., 32, 1136 (1899).
491] K. Fries, G. Schflrmann, Ber., 47, 1195 (1914). ]
492] K. Fries, E. Engelbert z, Ann., 407, 217 (1915). !
493] K. Fries, H. Koch, H. Stuken brock, Ann., 468, 177 (1929). ]
[494] F. E. Williams, E. Gebauer-Fulnegg, J. Am. Chem. Soc., 53^
353 (1931) -
[495] C. F. H. A 1 1 e n D. D. M с К a y, Org. Synth., Coll. v. 2, 1955, p. 580. .
[496] E. Bourgeois A. Abraham, Rec. trav. chim., 30, 407 (1911).
[497] C. G. Moses E. E. R e i d, J. Am. Chem. Soc., 48, 776 (1926). *
[498] V. du V i g n e a u d, L. E. A udri e th, H. S. Loring, J. Am. Chem,
Soc., 52, 4500 (1930). . *
[499] R. C, Arnold, A. P. L i e n, R. M. A 1 m, J. Am. Chem. Soc., 72, 73V
(1950) * J
[500] R. О 11 o. A. R 6 ss in g, Ber., 19, 3129 (1886).
II. ОКИСЛЕНИЕ S-ФУНКЦИЙ
609
Монотиоэтиленгликоль [301]. Растворяют диэтокспдйсульфид в сппрте
и смешивают с избытком слегка разбавленной H.,SO4. В эту смесь постепенно
вносят цинковую пыль и оставляют па ночь при температуре около 40° С. После
фильтрования кислоту нейтрализуют содой, ановь фильтруют и фракциони-
руют полученный монотиоэтиленгликоль перегонкой в вакууме; т. кип. про-
дукта 61° С (18 мм рт. ст}.
н-Бутилмеркаптан (натриевая соль) [502]. К 100 мл сухого ксилола
добавляют 1,25 г металлического натрия и 5 г ди-н -бути л дисульфида. После
12 ч нагревания на паровой бане смесь фильтруют и промывают эфиром.
о-Нитротиофенол [503]. Нагревают до кипения 30 г топкоизмельчеиного
ди-(о-нитрофенил)-дису.1ьфида в 100 мл спирта и постепенно смешивают с вод-
ным раствором гидросульфида натрия (полученного из 2,2—2,5 в NaOH и HaS)
н 8 г NaOH. Дисульфид при этом быстро растворяется с образованием корич-
невого раствора. Раствор разбавляют водой и осаждают о-нитротиофенол соля-
ной кислотой.
Гидрохлорид цветения [504]. В солянокислый раствор цистпна вносят
оловянную фольгу, причем вначале олово растворяется без выделения водо-
рода. Когда начнется отчетливое выделение газа, смесь разбавляют, осаждают
олово сероводородом и упаривают смесь досуха.
ZZ. Окисление S-функций
1. Окисление меркаптанов
Дисульфиды, находящиеся с тиолами в окислительно-восстановительном
равновесии
зН v
II--SS-В <17=-- 2BSH
очень легко могут быть получены дегидрированием меркаптанов. Выбор подходящего
окислителя зависит от легкости окисления, которая возрастает в ряду от третичных
через вторичные до первичных меркаптанов и далее до тиофенолов [505]. В более
жестких условиях окисление может идти дальше с образованием сульфокислот.
В качество окислителей используют кислород воздуха в водно-аммиачной среде
(о,о'-диацетилдифенилдисульфид [506, 507]), перекись водорода (о,о'-дициннамилами-
дофенилдисульфид [508]), иод (дистирилдпсульфиддлкарбоыовая кислота [509]), гипо-
галогениды (низшие алкандисульфиды [510]), ГеС18 [1,Г-ди-(карбэтоксинафтол)-
4,4’-дисульфид [511]] и K5f Fe(CN5)] [ди-(о-цианбензил)-дисульфид [512]].
Ди-(я-цпанбензил)-дисульфвд [513]. В 100 -ил 95%-ного этилового
спирта растворяют 5 е п-ццанбензилмеркаптана, смешивают с 25 мл концен-
трированного NHS и в течение четырех суток подвергают раствор действию
Е. Fromm, Н. Jorg, Вег., 58, 305 (1925).
R. Е. Stutz. R. L. S h г 1 п е г, J. Am. Chem. Soc., 55, 1244 (1933).
К. Brand, Ber., 42, 3465 (1909).
E. Baumann, Hoppe-Seyler’s Z. physiol. Chem., 8, 300 (1884).
M. S. К h a r a s c h, W. N u d e n h e г g, G. J. Mantell, J. Org. Chem.,
16, 524 (1951).
Герм. пат. 198509 (1907); С., 1908, I, 2118.
Герм. пат. 565967 (1930); С., 1933, 1, 1855.
W. Н. М i 11 s J. В. W h i t m о г t h, J. Chem. Soc., 1927, 2747.
A. Schober’l. H. Eek, Ann., 522, 97 (1936).
D. T McAllan, 1 V. Cullum et al., J. Am. Chem. Soc., 73, 3627
(1951).
T. Zincke, J. Ruppersberg, Ber., 48, 120 (1915).
A. W. Bay, S. Gabriel, Ber., 23, 2478 (1890).
С. В а г k e n Ь u s, E. B. Friedman, R. K. F 1 e g e, J. Am. Chem.
Soc., 49, 2552 (1927).
39 Эака8 1835.
ГГ0.П5ЧЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ S-ФУПКЦИИ
кислорода воздуха. Выделившиеся светло-желтые кристаллы перекристалли-
зовывают из 95% -пого этилового спирта. Выход продукта 3,5 а (70,4% от теоре-
тического); т. пл. 148° С.
Ди-(о- и л-метоксифенил)- и ди-(этоксифенил)-дисульфиды [490]. Алко-
кситиофенолы растворяют в небольшом избытке 10%-него раствора NaOH
и добавляют к полученному раствору теоретическое количество тонкоизмель-
ченного иода. Дисульфид выпадает в кристаллической форме. Добавляют
небольшое количество сернистой кислоты, отсасывают осадок и перекристалли-
зовывают из этилового спирта.
Ди-(бензтиазолил)-дисульфид [514, 515]. Раствор 2-меркаптобензтиаэола
в рассчитанном количестве разбавленного раствора NaOH смешивают с теоре-
тическим количеством водного раствора K3[Fc(CN)e]. Выпавший дисульфид
отсасывают, промывают водой и перекристаллизовывают из бензола; т- пл.
продукта 186° С.
Из бифункциональных дитполов соответствующего строения можно получать
циклические дисульфиды. В качестве окислителя для синтеза 1,2-дитиагидриндана
и 2,3-дптиатетралина [516] применяли FeCl3. При окислении цис- и транс-1,2-дя-
(меркаптометил)-циклогексанов образуются цис- и транс-2,3-дитиадекалииы, име-
ющие наиболее стерически благоприятную конфигурацию двойного кресла:
FcC],
2.3 • дитиадекпллн
цис- и транс-2,3-Дитиадекалины [517]. Раствор 1 г цис-(плп. трине)
1,2-ди-(меркаптометил)-циклогексана в 50 мл смеси метилового спирта и ледя-
ной уксусной кислоты (1 ; 1) при перемешивании медленно приливают к раствору
2 г FeCl3 в 12 мл метилового спирта и 5 мл ледяной уксусной кислоты и через
некоторое время разбавляют трехкратным объемом воды. При этом выделя-
ется молочного цвета масло, которое затем становится прозрачным и'затверде-
вает в холодильнике с образованием бледно-желтых кристаллов. Перекристал-
лизацию проводят из метилового спирта. Выход г/ис-дитиадекалпна 71% от
теоретического; т. пл. 41,5° С; выход транс-дитиадекалина 77,5% от теорети-
ческого; т. пл. 56,5—57° С.
При действии более энергичных окислителей меркаптапы можно окислить
до сульфокислот. Этот .метод широко применим и часто используется для синтеза
некоторых сульфокислот. Для получения алифатических сульфокислот и у-пириднн-
сульфокислоты в качестве окислителей оказались пригодными азотная кислота [518,
519] и перманганат калия [520]. Бевзтиазол-2-сульфокислоту с отличным выходом
[514] Р. Jakobson, Вег., 21, 2626 (1888).
[515] Герм. пат. 613300 (1933); Frdl., 22, 1, 301 (1939).
516] Л. Liittringhaus, К. П a g е 1 е, Angew. Chem., 67, 304 (1955).
517] Л. Liittringhaus, A. Brechlin, Chem. Вег., 92, 2271 (1959).
518| D. L. Vivian, Е. Е. Reid, J. Am. Chem. Soc.., 57, 2559 (1935).
519| К. Koenigs,/}. К i n n e, Ber., 54, 1357 (1921).
[520] B. F 1 a s c h e n t r a g e r, G. Waiinscbaff, Ber., 67, 1121 (1934).
II. ОКИСЛЕНИЕ S-ФУНКЦПЙ
611
можно получить взаимодействием 2-мсркаптобензтиазола и щелочного раствора пере-
писи водорода или гипохлорита [521].
Бензтиазол-2-сульфокислота. К раствору 8,4 ч. меркаптобеизтиаэола
в примерно 150 ч. разбавленного раствора NaOH (40 г NaOH в 1 л воды) доба-
вляют 6 ч. перекиси водорода в концентрированном водном растворе. Образо-
вавшуюся сульфокислоту высаливают NaCl.
Окисление меркаптидов свинца [522] и бария [518] обычно дает более высокие
выходы сульфокислот.
2. Окисление дисульфидов
Сульфокислоты могут быть получепы также окислением дисульфидов [52,3—526]:
2R-S—S—R^5O24-2H2O -> 4RSO3II
Окисление- дисульфидов в лабораторной практике широко используется для
препаративного синтеза эфиров тиосульфиновых кислот и эфиров тиосульфокислот.
эфир тиосуль-
фшювой кислоты
K-S-S-R в—SOj-S-Л
эфир тиосульфо-
кислоты
Окисление дисульфидов до эфиров тиосульфиновых кислот лучше всего проис-
ходит при действии эквивалентных количеств надбензойной или надуксусной кислот
в хлороформе [527], в то время кай эфиры тиосульфокислот образуются при окислении
дисульфидов небольшим избытком перекиси водорода в уксусной кислоте [528, 529].
3. Окисление простых тиоэфиров
В зависимости от выбранного окислителя окисление простых тиоэфиров при-
водит к сульфоксидам или сульфонам,-
r2s -> r2so R2SO2
Сульфоксиды удается получить, используя различные окислители. Так, напри- •
меР, дифенилсульфоксид [530], дибензилсульфокспд [528], л,п'-ди-(ацетиламинофо-
пц.л)-сульфоксид [528] и диэтилсульфоксид [531] получают окислением соответству-
ющего тиоэфира рассчитанным количеством перекиси водорода в ледяной уксусной
кислоте или ацетоне.
J18 (1949); C. A., 44, 1528 (1950).
ell, A. N. Say in, J. Am. Chem. Soc., 73, 5127 (1951).
W. Wenk, Ber., 45, 1373 (1912).
J. H. Bailey, C. J. Ca vallito. J. Am. Chem. Soc.,
[521] Герм. пат. 615132 (1933); Frdl.. 22, 1, 296 (1939).
[522] J. W. McBain, R. С. Williams, J. Am. Chem. Soc., 55, 2250 (1933).
[523] W. А. РгоеН, С. E. A da ms В. H. Schoemaker, Ind. Ene. Chem.,
40, 1129 (1948).
[524] Пат. США 2489:
1525] W. T. С a 1 d w
1526] F. Fichter,
[527] L. D. Small,
69, 1710 (1947).
[528] О. H insberg, Ber.. 41, 2836, 4294 (1908); J. С у m er ma n, J. B. W il-•
1 i s, J. Chem. Soc., 1951, 1332.
[529] G. Lpandri, A. T u n d o, Ann. Chimica (Roma), 44, 63 (1954): C. A., 49,
4563 (1955).
[530] О. H insberg, Ber., 43, 289 (1910).
[531] R. Pummcrer, Ber., 43, 1407 (1910). *
39*
(’|2 ПОЛУЧЕНИЕ СОЕДИНЕНИИ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ S-ФУНКЦИИ
Бензилфенилсульфоксид [532]. Отфильтрованный раствор 52 г бензпл-
фенилсульфнда в 250 мл ацетона смешивают с 40 г 30%-ной Н2О2, сильно
встряхивают и оставляют на 72 ч. После отгонки ацетона остаток тотчас же
при слабом охлаждении кристаллизуется. Его перекристаллизовывают из
60%-ного этилового спирта. Выход продукта 40 г; т. пл. 122—123° С.
Дициклопентилсульфоксид [533]. Раствор 3,4 г дициклопентилсульфида Л
в 30 мл ледяной уксусной кислоты смешивают с 2,5 г пергидрола и нагревают-,
на водяной бане в течение 2 ч. После этого в вакууме отгоняют уксусную кис-
лоту и кристаллический остаток перекристаллизовывают из метилового спирта; j
т. пл. продукта 71,5° С. 1
1
В качестве окислителей используют также надкислоты [534, 535] (в зквивалеит-1
ных количествах), хромовую кислоту в ледяной уксусной кислоте [536] и азотную л
кислоту [537]. I
При окислении сульфидов в более жестких условиях образуются сульфоны, |
которые могут быть также получены окислением сульфоксидов. В качестве окислите-а
лей наряду с перекисью водорода применяют перманганат калия, хромовую н азотную 1
кислоты. Условия реакции в сильной степени зависят от строения исходного сульфида 1
и выбора окислителя. Применение перекиси водорода имеет то преимущество, что Ц
в этом случае не образуется неорганических побочных продуктов, вследствие чего1!
значительно упрощается выделение сульфонов. ;
Дифенилсульфон 1530]. Раствор дифенилсульфида в ледяной уксусной-
кислоте смешивают с избытком (около 2,5 моль) перекиси водорода. После-
многосуточного стояния при комнатной температуре начинается выделение
бесцветных кристаллов, количество которых увеличивается при разбавлении ;
раствора водой; т. пл. дифенилсульфона 124° С,
Аналогичным образом получены п-нитрофенйлметилсульфон [538], различные ’
алкилтолилсульфоны и р-хлорэтилтолилсульфон [539]. •
Сульфиды могут быть окислены до сульфонов также при помощи надкис-’!
лот [534, 540[.
Дихлорметилметилсульфен. К 1,93 г дихлорметилметилсульфида доба-1
вияют раствор 6,3 г мононадфталевой кислоты в 200 мл эфира и выдерживают*
в течение многих суток. Затем отгоняют эфир, днхлорметилметилсульфон эк-’*
страгируют хлороформом. Заключительную перекристаллизацию продукта про-
водят из системы хлороформ — петролейный эфир или бензол — циклогексан.;
Выход дихлорметилметилсульфояа 2,02 ? (84% от теоретического); т. пл.5
71-72,5° С.
Окисление сульфидов перманганатом калия чаще всего проводят в растворе/,
уксусной или серной кислоты. При этом для выделения сульфона необходимо отде-?
лить массу от образующегося Мп(ОН)4. В случае труднорастворимых сульфонов эт$й
операцию осуществляют восстановлением Мп(ОН)4 необходимым количеством раствора^
бисульфита натрия или двуокисью серы. Для выделения легкорастворимых в реакцион^-
ной смеси сульфонов отфильтровывают гидроокись марганца и либо добавляют водул
[532]
533]
534
535
536
537
538
[539
[540
R. L. Shriner, Н. С. Struck, W. J. J о г i s о n, J. Am. Chem4
Soc., 52, 2066 (1930). J
S. Lo е ven ich, Н. U t я с h et al., Ber., 62, 3092 (1929).
H. Bohme, Ber., 70, 379 (1937).
L. N. Lewin, J. Tschulkoff, J. prakt. Chem. [2], 128, 171 (1930). .'•/
F. Mayer, Ber., 43, 584 (1910). -i
L. Gattormann, Ann., 393, 140 (1912). <;
T. Zincke, Ann., 400, 16 (1913). 1
H. G i 1 m a n,’N. J. В e a b e r, I. Am. Chem. Soc., 47, 1449 (1925). :•[
H. Richtzenhain, B. Alfredson, Chem. Ber., 86, 142 (1953). i
II. ОКИСЛЕНИЕ S-ФУНКЦИЙ
613
либо после подкисления реакционной массы экстрагируют продукт эфиром. Таким
образом был получен этиловый эфир а,а-ди-(этилсульфонил)-пропионовой кислоты:
SOgCaHe
I
СНз—С—СООС2Н6
I
SO2C2H5
Этиловый эфир а,а-ди-(этилсупьфонил)-проиноновой кислоты [541].
Диэтилмеркаптол пировиноградной кислоты встряхивают с избытком холод-
ного раствора КМпО<, периодически добавляя разбавленную H2SOj. Затем
раствор обесцвечивают пропусканием SO8, сульфон экстрагируют эфиром.
Если выделенное вещество не затвердевает, необходимо повторить окисление.
После перекристаллизации из 50%-ного этилового спирта т. пл. дисульфона
60—62° С.
Описана общая методика получения 2,4-динитрофенилалкилсульфоаов дей-
ствием перманганата калия в ледяной уксусной кислоте 1542].
n-Нитрофенилалкил- и ге-нитрофениларилсульфоны могут быть получены
окислением соответствующих сульфидов хромовой кислотой в ледяной уксусной
кислоте с выходом в среднем 80% от теоретического [543].
Сульфоны. Общий способ получения. Нагревают до кипения раствор 10 г
сульфида в 100 мл 80%-ной уксусной кислоты н добавляют насыщенный вод-
ный раствор хромовой кислоты до оранжевой окраски раствора (избыток окис-
лителя). Кипятят еще 15 мин, добавляют равный объем воды, • охлаждают
и сульфои перекристаллизовывают из 50%-ной уксусной кислоты.
Применение азотной кислоты в качестве окислителя ограничено главным обра-
зом из-за возможного нитрования ароматических сульфидов [544].
О получении сульфонов окислением сульфидов см. [545].
4. Окислительное галогенирование органических соединений серы
Получение олкилсулъфохлоридов
Различные органические соединения двухвалентной серы: сульфиды, дисуль-
фиды, ксантогенаты и пзотиурониевые соли действием хлора могут быть превращены
в сульфохлориды. Из циклических сульфидов получают галогеналкансульфохлориды,
например из пропиленсульфида — 1-хлорпропан-2-сульфохлорид [546].
1 Высшие а-хлорированиые сульфохлориды [547] с хорошими выходами можно
получить взаимодействием альдегидов с сероводородом и последующей обработкой,
продукта реакции хлором в присутствии воды.
Об окислительном хлорировании диалкил- и днаралкилсульфидов см. [548].
1541] Т. Posner, Вег., 32, 2801 (1899).
[342] В. W. В о s t, J. 0. Т 11 г n е г, R. D. N о г t о п, 3. Am. Chem. Soc., 54,
1986 (1932).'
[543] W. R. Waldron, E. E. Reid, J. Am. Chem. Soc., 45, 2399 (1923).
[544] D. W. G о h e e п, C. F. Bennet,!. Org. Chem., 26, 1331 (1961).
1545] 4. C ьютер, Химия органических соединений <<еры, пер. с англ., ч. Зш
Издатинлит, 1951.
[546] J. М. Stewart, Н. Р. С о г d t s, J. Am. Chem. Soc., 74, 5880K(1952).
[547] Пат. ФРГ 836492 (1944); С., 1952, 5486.
'548] S. W. Lee, G. Dougherty, J. Org. Chem., 5, 81 (1'940).
J
Г. 14
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ Б-ФУНКЦИП
Реакцию сульфидов (или дисульфидов) с хлором проводят в дву- или
пятикратном объеме уксусной кислоты, содержащей необходимое количества
воды в качестве донора кислорода. Хлор пропускают до тех пор,пока реакцион-
ная масса не перестанет обесцвечивать раствор КМпО^. Ил дп-н-бутилсульфида
с выходом 80% получают н-бутансульфохлорид: ил дибензплсульфида — толу-
ол-а-сульфохлорид.
Этим методом получены также этансульфохлорпд и н-пента«сульфохлорид.
Алкилтиоцианаты [549] и алкилтиосульфаты [5501 при взаимодействии с хло-
ром также превращаются в алифатические сульфохлорпды.
Этансульфохлорид 1549]. Суспендируют 20 г этилроданида в 200 мл
воды и при энергичном перемешивании пропускают хлор со скоростью 2—3 л/ч. '
Температура реакции 0—5° С. Когда реакционная масса примет устойчивую
зелено-желтую окраску (избыток хлора), подачу хлора прекращают и для уда-
ления его через массу медленно просасывают воздух. Выделившееся масло-,
извлекают эфиром, промывают вытяжку растворами NaHSO3 и NaHCO3, сушат ;
над СаС12, отгоняют эфир и остаток перегоняют в вакууме. Выход этансульфо-Т
хлорида 70% от теоретического; т. кин. 71 —72° С (20 мм рт'. ст.).
При действии хлора или брома на водные растворы алкилизотиурониевых
солей образуются сульфохлориды или сульфобромиды [550, 551]. Этот способ особенна"
удобен тем, что производные изотиомочевины очень легко получить реакцией алкил-
1алогенидов с тиомочевяной (стр. 631).
Получение арилсулъфохлоридов
Описанный выше способ получения сульфохлоридов можег быть распространен
и на ароматические соединения серы. Так, при пропускании хлора в присутствии
воды в раствор дисульфидов в уксусной кислоте образуются сульфохлориды;
Аг—S—S—Аг + 5С12 + 4Н2О -> 2ArSO2Cl + 8HCI
З-Нитротолуол-4-сульфохлорид [552]. К раствору 2 г 3,3-дп'-(нитро-
толил)-4.4'-дисульфида в 10 мл ледяной уксусной кислоты добавляют 2 мл ’
воды и без охлаждения пропускают хлор до насыщения. Жидкость при этом
разогревается и после охлаждения в виде бесцветных кристаллов выпадает
часть сульфохлорида; остальную часть сульфохлорида получают выпариванием
раствора. Для очистки продукт перекристаллизовывают из смеси бензол —
бензин; т. пл. продукта 98—99° С.
Обработкой дисульфида бромом получают сульфобромид [553] с т. пл.
Хлорирование можно проводить в среде НС1 в присутствий HNO8, как,
например, при получении о-нитробензолсульфохлорида 1554]. Тиофенолы [555]
и гетероциклические меркаптаны [556] также могут быть хлорированием превращены
в сульфохлориды.
п-Толуолсульфохлорид. n-Тиокрезол, растворенный в пятикратном коли-
честве ледяной уксусной кислоты, насыщают хлором. После непродолжптель-
[549] Т. В. Johnson, I. В. Douglas я, J. Am, Chem. Soc., 61, 2548 (1939).
[550] С. Z 1 о g 1 е г, J. М, Sprague, J. Org. Chem., 16, 621 (1951).
[551] Т. В. Johnson, J. М. Sprague, J. Am. Chem. Soc., 58, 1348 (1936);
59, 1837 (1937); 59, 2439 (1937); 61, 176 (1939).
[552] T. Zincke, H. Rose, Ann., 406, 134 (1914).
[553] T. Zincke, II. Rose, Ann., 406, 108 (1914).
[554] E. Wertheim, Qrg. Synth. Coll. v. 2, 1955, p. 471.
[555] T. Zincke, W. Frohneberg, Ber., 42, 2728 (1909); 43, 840 (1910).
[556] R. O. R о b 1 i n jr., J. W. G 1 a p p, J. Am. Chem. Soc., 72, 4890 (1950).
III. РАСЩЕПЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИИ СЕРЫ
615
ного выдерживания и отгонки растворителя получают п-толуолсульфохлорид
с т. пл. 69° С.
л-Толуолсульфобромид синтезируют из 3 г п-тиокрезола, 15 г ледяной
уксусной кислоты в 6 г брома. После перекристаллизации из небольшого коли-
чества бензина т. пл. продукта 93—94° С.
III. Расщепление органических соединений серы
1. Расщепление С—S-связи [557]
Расщепление С—S-связи иногда используется для получения тноэфиров из
меркаптанов:
2RSH -> RSR-l-HgS
Реакция, например в случае 0-меркаптоантрахиноиа [558], происходит уже
при простом нагревании, однако идет гораздо лучше в присутствии акцепторов серо-
водорода. В качестве катализатора применяется смешанный контакт из сульфидов
кадмия, цинка и окиси алюминия [559].
Этиленсульфиды в присутствии ВР3 и уксусной кислоты могут расщепляться
спиртами с образованием р-алкоксимеркаптанов; меркаптаны в этих условиях дают
соответствующие моиоэфиры дитиогликоля [560].
Общий способ проведения реакции. К смеси 0,11 молъ меркаптана и
2 капель эфирата бортрифторида в течение 45 мин при перемешивании и нагре-
вании на паровой бане по каплям добавляют 0,05 моль сульфида. Затем нагре-
вают еще 2 ч, экстрагируют продукт эфиром, экстракт промывают водой и насы-
щенным раствором NaCl, сушат и перегоняют в вакууме.
Этим способом из циклогексенсульфида и н-алкилмеркаптанов получают
a лкил-2-меркаптоциилогексил сульфиды
/\ /\ ZSH
[си f Y
I I I
\/ \/ \SR
температуры кипения которых приведены ниже:
R т. киц., °G
С4Н9...............109—111 (2,5 мм рт. ст.)
С5Нц ............ 123—126 (3,5 мм рт. ст.)
С8Н1з ........... 130—133 (3 мм рт. ст.)
С7Щ5 ............141 — 144 (3 л.н рт. ст.)
Для расщепления С—S-связи в циклических сульфидах были использованы
также карбоновые кислоты [561], хлорангидриды кислот [562] н амины [563].
Малая термическая стойкость меркаптолов делает препаративно возможным
их пиролитическое, расщепление до тиокетонов.
[557] D. S. Т а г Ь е 11, D. Р. Н а гп i я h, Chem’ Rev., 49, 1 (1951).
[5о8[ Герм. пат. 254561 (1912); Frdl., 11, 606 (1912—1914).
1559] P. J. w ieze vi c h,L. B. Turner, P. K. Frohlich, Ind. Eng. Chem.,
25, 295 (1933).
[560] H. R. Snyder, J. M. Stewart, J. B. Ziegler, J. Am. Chem. Soc.,
69, 2675 (1947).
[561] С. C. J. Culvenor, W. Davies, N. S. Heath, J. Chem. Soc., 1949
282.
1562] W. Davies, W. E. S a v i g 0, J. Chem. Soc., 1950, 317.
1563] H. R. Snyder, J. M. Stewart J. B. Ziegler, J- Am. Chem. Soc.,
69, 2672 (1947). »
(;ц; ПОЛУЧЕНИЕ СОЕДИНЕНИИ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ S-ФУНКЦИИ
Ксантион [565]. Нагревают 4 г ксантон,чибснзплмеркаитола [564] в ва-
кууме прн 205° С в течение 15 мин. Остаток экстрагируют 10 мл горячего
бензола и охлаждают. Выход ксантиона 0,5 а; т. пл. 156я С после перекристал»
лизации из бензола.
Реакция протекает по схеме:
Для расщепления особенно пригодны термически неустойчивые бензилмеркан-
тилы ароматических кетонов, из которых наряду с тиокетоном и бензилмеркаптаномЬ,
образуется ряд продуктов дальнейшего разложения [565]. Чисто ароматическая^
меркаптолы типа АггС(5Аг')„ при нагревании дают соответствующие тиокетоны /й’
дисульфиды, наряду с другими продуктами разложения [565, 566]. •
Путем расщепления С—S-связи в эфирах тиоциановых кислот действие»*
щелочей получают дисульфиды [567—569]. Однако эта реакция не имеет большого*:
препаративного значения.
Ароматические тиокетоны при взаимодействии с гриньяровскими соедд-^
нениями [570] пли магнийиодидом в присутствии магния [571], а также чере»'-
дпазосоединения [572, 573] могут быть превращены в тетраарилзамещенные этилен-*
сульфиды:
2
Агх
)C=S ------
Аг/
ЙАГ'МеТ At.
~Ar--g1^ \с----с/ -i-Ar'-Ar' + Mgl2 + MgS
Агх \ / чАг
S '
„ Аг\ /Аг
-Mg )С---------С< +MgS
Arz \ / хАг
Этим методом были получены тетра-п-анизил-, тетрафенпл-, дифенплдиани-*
зил- и п.и'-тетраметилдиаминодифенилднфенйлэтиленсульфиды.
Прп щелочном расщеплении сульфонов, лучше всего действием алкоголятов^
образуются сульфиповые кислоты. Так, например, бепзолсульфиновую кислоту
получают из фенил-р-феннлзтилсульфона с выходом 72% от теоретического [574{
CeH6SO2CH2CH2CeH5 + KOlI -+ С6Н58О2КН-СН2==СНСвН6 + Н2О
а из 2,4-дцнитродифенилсульфоиа — соответствующую кислоту с выходом 79%
теоретического [575].
A. Schonberg, О. Schutz, Ann., 454, 52 (1927).
A. Schonberg, О. Schutz, Ber., 62, 2322 (1929). ;
A. Schonberg, O. Schutz, V. Bruckner, J. Peter, Ber., 62."
2550 (1929).
S. Gabriel, W. E. Lauer, Ber., 23, 87 (189Q).
H. P. Kaufmann, R. RoBbach, Ber., 58, 1556 (1925).
K. v. Auwers, C. Schumann, Ber., 34, 4267 (1001).
A. Schonberg, Ann., 454, 37 (1927).
A. Schonberg, O. Schutz, Ber., 60, 2351 (1927). ),
П. Staudinger, J. Siegwart, Helv. chim. acta, 3, 833 (1920).
II. Staudinger, J. Siegwart, Helv. chim. acta, 3, 840 (1920).
G. W. Fenton, €. K. Ingold, J. Chem. Soc., 1930, 705.
N. Kharasch, R. Swidler, J. Org. Chem., 19, 1706 (1954).
III. РАСЩЕПЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ СЕРЫ
617
При действий хлора на диацилднсульфпды расщепляется не S—S-,a С—S-связь
с образованием ацилдисульфенилхлорпдов [576]:
RC—SS—CR + Cla -* RC—SSC1-J-RC—Cl
Диацилмоносульфиды аналогичным путем расщепляются до ацилсульфенилхло-
ридов, дающих с меркаптанами алкилацилдисульфиды, из которых алкоголизом
получают алкилгидродисульфиды [577]:
RC-S-CR —* RC-SC1 RC-SS-R М- HSSR
Препаративно важным является хлорирование сероуглерода, приводящее
к перхлормотилмеркаптану [578], которому чаще всего приписывают формулу три-
хлорметпл сульфенил хлорида:
2CS2-L5C12 —> 2C13CSC1 + S<jC12
Однако по химическим свойствам перхлорметилмеркаптан не может быть одно-
значно охарактеризован ни как сульфенилхлорид, пи как тиокарбоннлтетрахлорид
(С12С<— SC12). С практической точки зрения важно его применение в качестве исход-
ного продукта для получения тнофосгена.
Перхлорметилмеркаптан [579]. В трехгорлую колбу емкостью 4 л,
снабженную термометром и трубками для ввода и отвода газа, помещает 1,8 кг
сероуглерода и добавляют 3 г иода. При охлаждении ледяной водой до 10—
20° С в смесь пропускают хлор до плотности смеси 1,61, что контролируется
отбором соответствующих проб. Продолжительность хлорирования 30—40 ч,
расход хлора около 3,5 кг. Полученную смесь перхлорметилмеркаптана и серо-
углерода разделяют перегонкой на колонне Видмера. Сначала при нормальном
давлении и температуре до 100° С отгоняют сероуглерод и далее При 12—13 мм
рт. ст. после небольшой первой фракции при 56—58° С получают чистый
перхлорметилмеркаптан. При перегонке необходимо заботиться о том, чтобы
трубки, и особенно сопло водоструйного насоса, ие засорялись выпадающей
серой. Поэтому целесообразно между насосом и аппаратурой включить напол-
ненную водой колбу, в которой будут разлагаться отсасываемые газы, и в на-
сосе не будет происходить выделение серы. Выход продукта 2,6 кг (60% от
’теоретичесиого). Перхлорметилмеркаптан — масло от желтого до красно-
коричневого цвета с очень неприятным запахом.
Так как сульфированпе ароматических соединений является обратимым про-
цессом, сульфокислоты гидролитическим расщеплением можно вновь превратить
в исходные продукты. Различную легкость отщепления сульфогрупп используют для
разделения веществ. Таким образом, например, можно отделить ле-кенлол от орто;
п пара-изомеров, так как л-ксилолсульфонислота гидролизуется наиболее легко [580],
пли выделить гомологи дифенила из тяжелого масла перегонки каменного угля.
В полисульфокислотах .частичное отщепление сульфогрупп часто происходит уже при
слабом нагревании в кислой среде [582]. Наконец, отщепление сульфогрупп
[576] Н. В б h m е, М. Clement, Ann., 576, 61 (1952).
[577] Н. В ohme, G. Zinner. Ann., 585, 142 (1954).
1578] Р. К 1 a s on, Вег., 20, 2376 (1887): G. Sosnovsky, Chem. Rev., 58, 509
(1958).
[579] G. M. Dyson, Org. Synth., Coll. v. 1, 1937, p. 507; P. КI a s о n,
Ber., 28, Ref. 942 (1896); герм. пат. 83124 (1894).
[580] N. Kishner, G. Wendelstein, C., 1926, I, 2681.-
[581] О. К г uber, Ber., 65, 1382 (1932).
[582] Англ. пат. 285488 (1928); С., 1929, II, 1470.
£18 ПОЛУЧЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ 8-ФУНКЦИИ
представляет препаративный интерес в том случае, если необходимо провести напра- ,
пленное замещение или активировать какие-либо заместители (атомы или группы j
атомов). Если, например, необходимо ввести нитрогруппу в иное положение, чем это «
возможно при прямом замещении, то замещают сначала нужное положение сульфо- ]
группой, затем вводят нитрогруппу, а сульфогруппу удаляют гидролизом. Так, из хлор- j
бензола через 4-сульфокислоту с последующими нитрованием, аминированием и отщеп-
леннем сульфогруппы можно получить 2,6-динитроанилнн [583] с выходом 30—36% от 1
теоретического. J
2. Расщепление S—Н-связи J
При хлорировании тиофенолов происходит расщепление S—Н-связи и обра- 1
зуются арплсульфенилхлориды [584—586], первый представитель которых был полу- |
чен Цинке [587] еще в 1911 г. J
RSH + CI2 -» RSCJ-l-HCl j
При хлорировании тиофенола вначале возникает арилсульфенилхлорид [588],
который далее реагирует со свободным тиолом и дает дисульфид. Третья стадия про-
цесса — хлорирование дисульфида с образованием 2 арилсульфенилхлорида.
Наличие электропоакцепторных заместителей повышает устойчивость арилсульфенил-
хлоридов, так как благодаря этому снижается электронная плотность на атоме серы
и тем самым затрудняется отщепление атома хлора. Во избежание гидролиза необхо-
димо полное исключение доступа влаги, а для уменьшения возможности С-хлориро-
вания процесс проводят в темноте при низких температурах.
Арилсульфенилхлориды. Общий способ получения [589]. В трехгорлую
колбу, снабженную мешалкой, капельной воронкой, хлоркальциевой трубкой •
и термометром помещают 0,1 молъ соответствующего тиофенола в 50 мл без- ;
водного ССЦ. При температуре не выше —1® С (в случае соединений с двумя
метильными группами в бензольном ядре реакцию проводят при —12° С) при
перемешивании добавляют по каплям раствор 0,1 лоль (7,09 г) хлора в 100 мл
безводного ССЦ и перемешивают еще 1 ч. Отгонку растворителя н перегонку -
сульфенилхлорида проводят в вакууме. Выход продукта 80—90% от теорети-
ческого.
Таким способом были получены фенил-, галогенф'енил-, толил-, ксилил- ,
и галогентолилсульфепилхлориды. :
Этот метод применим также для получения сульфенилхлоридов алифатического -
ряда. Известным ограничением метода является то, что алкилсульфеннлгалогениды 1
обычно неустойчивы и не могут быть выделены из раствора. Как и в случае аромати- ;
чсских представителей, алифатические сульфенилхлориды более доступны, чем суль- :
фенилбромиды. Сульфенилиодиды могут быть получены только в отдельных случаях,;
сульфенилфториды не известны вообще. Диметилдисульфид . при хлорировании
в сухом четыреххлористом углероде при —15° С превращается в дихлорид, мед-
ленное нагревание которого до комнатной температуры дает металсульфенилхло-
рид [590]: '
СНэ-S
Ц-С1г
СНз—S
С1‘ -> 2CH3SCI
[583] Ц. Р. Schultz, Org. Synth., 31, 45 (1951).
[584] N. Kharasch, S. J. Po tern pa, H. L. Wehrmeister, Chem. Rev.,./
39, 269 (1946).
[585] M. B. Spark e, J. L. Cameron, N. Kharasch, J. Am. Chem. Soc., ;
75, 4907 (1953). ;
[586] W. E. True e, M. F. Amos, J. Am. Chem. Soc., 73, 3013 (1951). ’
[587] T. Zincke, Bar., 44, 769 (1911).
[588] H. Ij с c h e 5, F. Holschneider, Ber., 57, 755 (1924). i
[589] L. A 1 m a s t, A. Han tz, Chem. Ber., 94, 728 (1961).
[590] E. Schneider, Chem. Ber., 84, 911 (1951). - •
Ш. РАСЩЕПЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ СЕРЫ
619
3. Расщепление S—S-связи
Для получения сульфенилхлоридов значительный препаративный интерес пред-
ставляет также расщепление S—8-связл [391] в дисульфидах. Об общих условиях
реакции было сказано в предыдущем разделе.
ч-Нитрофенилсульфенилхлорид [587]. о,о'-Динитродифенилдисульфид
заливают 10 ч. СС14 и пропускают до насыщения хлор. Через несколько часов
снова пропускают хлор и выдерживают реакционную массу в этой атмосфере.
Сульфенилхлорид выпадает в виде желтых игл; его отсасывают и перекристалли-
зовывают из бензина; т. пл. продукта 75° С.
Таким же образом получают п-нитрсфенилсульфенилхлорид [592] (т. пл.
52° С) и 2-нитро-4-метилфенилсульфенилхлорид [593] (т. пл. 90° С).
п-Хлор-о-нитрофенилсульфенилбромид [594]. Соответствующий дисульфид
в 10 ч. хлороформа нагревают с обратным холодильником с небольшим избыт-
ком брома до полного растворения, дают охладиться и выпавший бромид пере-
кристаллизовывают из бензина; т. пл. продукта 111° С.
При действии хлора на алифатические дисульфиды особеппо легко хлорируется
алкильный остаток. Так, например, при температуре от —15 до —20° С из диметил-
дисульфида образуется метилсульфенилхлорпд [595], а при 0° С — хлорметилсуль-
фенилхлорпд.
Хлорметилсульфенилхлорнд. В 23,6 г диметилдисульфпда прп температуре
от —15 до —20° С медленно пропускают сухой хлор до привеса примерно 17
Затем при постепенном нагревании до 0° С снова пропускают хлор до привеса
еще на 17 г. После непродолжительной выдержки при комнатной температуре
перегоняют продукт реакции в вакууме; т. кип. 50° С (40 .и.н рт. ст.).
Хлорметилсульфенилхлорнд может быть получен хлорированием бис-(хлорме-
тпл)-дисульфида хлором или хлорированием диметилдисульфпда сульфурилхлори-
дом. Су.тьфурилхлорид в качестве хлорирующего агента пригоден также для синтеза
х'лорэткисульфенплхлорида [596].
а-Хлорэтилсульфенилхлорид. К раствору 61,5 г диэтилдпеульфида в 150 мл
четыреххлористого углерода При —209 С и перемешивании медленно добавляют
по каплям 68 г сульфурилхлорида. Затем при —15° С постепенно добавляют
еще 123 г сульфурилхлорида, смесь медленно нагревают до комнатной темпе-
ратуры и оставляют на ночь. После отгонки четыреххлористого углерода оста-
ток перегоняют прн 38° С (27 мм рт. ст.). Многократной перегонкой первой
фракции может быть получено дополнительное количество основного продукта.
Хлорированием бис-(Р-хлорэтнл)-дисульфида хлором с выходом 79%
получают р-хлорэтилсульфенилхлорид [597].
На примере этилового эфира Х,№'-диацетнлцистина описай интересный спо-
соб расщепления дисульфидов при взаимодействии их с ароматическими соедине-
ниями в услопиях реакции Фриделя — Крафтса. Реакция ведет к образованию сме-
шанных алкцлароматпческих тпоафиров [598]:
ArH+BSSR 1 ArSH+BSH
[591] A. J. Parker, N. Kha газ ch, Chem. Rev., 59, 583 (1959).
[592] T. Zincke, Ann., 400, 9 (1913).
[593] T. Zincke, Ann., 406, 110 (1914).
[594] T. Zincke, Ann., 416, 95 (1918).
[595] II. В rint zi n ger, K. Pfannstiel, H. Roddebusch,
К. E. К ] i n g, Chem. Ber., 83, 89 (1950).
[596] H. В r i n t z i n g о r, H. E 11 wa n ger, Chem. Ber., 87, 307 (1954).
[•>97] R. C. Fuson et al., J. Org. Chem., 11. 471 (1946).
|598{ H. Behringer, K. Kachinka, Angew. Chem., 72J 348 (1960).
620
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ S-ФУНКЦИИ
Тиоэфиры получают также расщеплением дисульфидов литий- н натрийоргаяж-j
ческпми соединениями [599]. Из дифенилдисульфида н трифенилметилнатрия обра-;
зуется, например, трпфенилметилфенилсульфид. I
От сульфеяжлтиоцианатов под действием меркаптанов отщепляется остаток,
родана и возникают несимметричные дисульфиды [600]: ' ’j
BSH -<SCN),-> BSSCN -ASU BSSB' + HSCN
В некоторых случаях представляет интерес синтез сульфиновых кислот рас—’
щеплением S—S-связи в солях или эфирах тиосульфокислот. Этот метод используют^
когда наличие чувствительных заместителей исключает получение сульфиновых;
кислот другими способами. Арилтносульфонаты, образующиеся из арилсульфохлорн->
дов и сульфидов щелочных металлов, могут быть расщеплены действием кислот [601J"
и щелочей [602], восстановлением амальгамой натрия [603] или действием связыва-)
ющих серу агентов — сульфидов [602], цианидов [604] и арсенитов щелочных метал-»
лов [604]: ’
BSOsCl -f-Na3S BSOaSNa BSOjNa+S
n-Толуолсульфяжат калия [604] получают путем выпаривания смеси 4,5 г
n-толуолтиосульфояата калия, 2,5 г KCN, 2 г 20%-ного водного раствори!
КОН и 10 мл воды. j
Эфиры арилтиосульфокислот могут быть расщеплены до сульфинатов идпсуль*’
фидов -обработкой щелочами [605] или меркаптидами [606]. <
4, Расщепление связи S—С1 :
Расщепление сульфохлоридов
Омыление сульфохлоридов до сульфокислот часто используется для очистки-^
сульфокислот. J
Наиболее изящным способом превращения алкилсульфохлоридов в сульфо-'l
кислоты является алкоголиз; при этом в отсутствие веществ, связывающих кислотуЛ
этерификация не происходит [607]. 1
RSOaCl+R'OH -> RSO2OH + R’C1 -j
Арилсульфохлориды обычно можно прогидролизовать до сульфокислот KHHJ&1
ченпем с водой, водно-спиртовыми смесями, водной уксусной кислотой и концентри*-^
рованной муравьиной кислотой. При взаимодействии сульфохлоридов со спирташ-^
п фенолами в присутствии акцепторов кислоты образуются эфиры сульфокис-Ч
лот [608, 609]. 1 Л
Алкиловые эфиры бензолсульфокислоты [610]. При энергичном встряхи-ж
вании к смеси 1 ч. бензолсульфохлорида и I ч. спирта по каплям прибавляют^
20—30%-ный раствор NaOH до тех пор, пока даже после длительного встряхи*Я
599] A. Schonberg, A. Stephenson et al., Вег., 66, 237 (1933). J
600] R. G. Н i з к е у et al., J. Org. Chem. 26, 1152 (1961). -ja
601] J. Perl, Ber., 18, 67 (1885).
602] R. Otto, J. Triiger, Ber., 24, 494 (1891). |
603] H. Limprioht, Ann., 221, 347, 361 (1883). "я
604] A. Gutmann, Ber., 41, 3351 (1809). Я
605] E. Fromm, Ber., 41, 3409 (1908).
606] H. Gilman, L. E. Smith, H. H. Parker, J. Am. Chem. Soc., 47;iS
854 (1925); S. Smiles, D. T. Gibson, J. Chem. Soc., 125, 180 (1924).^
[607] Пат. США 2319121 (1940)- С. A., 37 , 5986 (1943); герм. пат. 742927 (19410
С., 1944, Т, 829. 1
[608] Герм. пат. 715846 (1938); С., 1942, Т, 2201. «
[609] S. S. R о s s а п Я е г, С. S. Marvel, J- Am. Chem. Soc., 50, 1493 (1928). $
[610] Z. Fol di, Ber., 53, 1836 (1920). 3
III. РАСЩЕПЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ СЕРЫ
621
вання реакция смеси остается щелочной, а отделившееся масло полностью
освобождается от хлора. К реакционной смеси добавляют бензол и воду и раз-
деляют слои; на бензольного слоя после промывки и высушивания отгоняют
в вакууме бенаол и непрореагировавший спирт при температуре водяной бани
не выше 30—40° С. Таким способом получают аллиловый эфнр (при пере-
гонке разлагается со взрывом), p-хлорэтиловый эфир (т. кип. 184° С
при 9лл рт. ст.), p-бромэтнловый эфир (т. кип. 185—1879 С при 16 хл рт.
ст.), О'-дихлоризопропиловый эфир (т. кип. 200—205° С при 20 мм рт. ст.;
т. пл. 50° С после перекристаллизации из бензола).
Описан общий способ синтеза простых алкиловых эфиров толуолсульфокис-
лоты [611]. Для получения эфиров н-алкансульфокислот в качестве акцептора нислоты
применяют пиридин [612], выходы эфиров в среднем 80% от теоретического.
Ариларилсульфонаты, как правило, хорошо кристаллизуются и пригодны для
идентификации фенолов и сульфокислот. Они могут быть легко получены по методу
Шоттена — Бауманна из сульфохлоридов и фенолов в присутствии щелочей [613}
пли третичных оснований J614].
Ариловые эфиры замещенных бензолсульфокислот [615]. При переме-
шивании 1 моль арилсульфохлорида нагревают с 1 моль фенола до 60—70° С
и в течение 30 мин при 60—80° С прибавляют 1 лоль 0,7 и. водного раствора
NaOH. Затем перемептивятот еще 1 ч при 60—80° С, охлаждают, отфильтровы-
вают эфир, промывают осадок водой и несколько раз перекристаллизовывают
из спирта с добавлением небольшого количества бензола. Выход эфира в среднем
90% От теоретического.
Ангидриды сульфокислот получают методами, аналогичными соответствующим
методам синтеза ангидридов карбоновых кислот. Так, ангидриды алифатических
сульфокислот могут быть получены действием сульфохлоридов иа алкилсульфонаты
серебра [616]. Ангидрид бензолсульфокислоты [617] получают медленным нагрева-
нием 2 моль бензолсульфохлорида с 1 моль безводной щавелевой кислоты до 200° С.
Амиды сульфокислот очень гладко образуются при взаимодействии сульфо-
хлоридов с аммиаком [618] или с сухим измельченным карбонатом аммония [619].
Реакцию с аммиаком проводят на холоду в водном растворе или, лучше, в индиффе-
рентном растворителе. Необходимо применение оптимального избытка аммиака для
предотвращения возможного омыления амидов до солей сульфокислот или образова-
ния дисульфоимидов RSOsNHSOaR.
Арилсульфамиды [619]. Общий способ получения. Нагревают в течение
10 мин 0,5 г арилсульфохлорида с 5 мл концентрированного раствора NHS
(d - 0,9), охлаждают до комиатИон температуры, смешивают с 10 мл воды
и отфильтровывают выпавший сульфамид. Осадок тщательно промывают,
перекристаллизовывают до постоянной температуры плавления из разбавлен-
ного этилового спирта и высушивают при 105’ С.
Этим методом получен ряд галоген- и алкилзамещениых бензол- н нафта-
линсульфамидов .
Чувствительные к влаге сульфохлориды превращают в сульфамиды пропуска-
нием сухого аммиака в охлаждаемый эфирный раствор сульфохлорида.
[bllJ А. Т. R о о si, Н. G 11 m а и, N. J. В е а b е г, Org. Synth., Coll. v. 1»
1937, p. 138.
[612] H. R. Williams, H. S. Mosher, J. Am. Chem. Soc., 76, 2985 (1954).
613] О. II ins berg, Ber., 23, 2962 (1890).
6t41 S. E. H a z 1 e t, J. Am. Chem. Soc., 59, 287 (1937).
61a] H. R. S 1 a g h, E. C. Britton, J. Am. Chem. Soc., 72, 2808 (1950).
6I6| О. С. В i 1 1 e t e r, Ber., 38, 2018 (1905).
617] R. G. Shepherd, J. Org. Chem., 12, 275 (1947).
618] F. H. Bergheim, W. В г a k e r, J. Аш. Chem. Soc., 66, 1459 (1944).
619] E. H. H u n t r e s s, H. Garten, J. Am. Chem. Soc., 62, .511 (1940);
E. H. H untress, J. S. Aut enr i eth, J. Am. Chem. Soc., 63, 3446
(1941).
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ S-ФУНКЩШ
Реакцию сульфохлоридов с аминами можно проводить в щелочной среде по
методу Шоттена — Бауманна 1620], либо, во избежание образования сульфимндов,
в слабощелочной, нейтральной или слабокислой среде [621], либо в присутствии
органических основании для связывания образующейся минеральной кислоты [622,
623]. N-Арилзамещениые 4-ацотиламинобензолсульфамиды [617] получают в присут-
ствии ацетата натрия и уксусной кислоты как растворителя.
Для синтеза большого числа N-гетероциклических замещенных сульфамидов
в качестве конденсирующего средства к одновременно растворителя был использован
пиридин [622].
N-Гетероциклические замещенные сульфамиды. К раствору 1 —2 моль
аминогетероциклического соединения в 500 мл пиридина порциями добавляют
1 моль замещенного бензолсульфохлорида и нагревают в течение 1—2 ч при
55—60Q С. Для выделения продуктов смесь выливают в холодную воду, в избы-
ток разбавленной холодной соляной кислоты или в разбавленный раствор хлори-
стого водорода в этиловом спирте.
Подробнее о методах получения сульфамидов см. [624—629].
Расщепление сульфинилхлоридов и сульфенилхлоридов
Сульфинилхлориды являются важными исходными продуктами для пол учения
различных производных сульфиновых кислот. Так, при нагревании сульфинилхлоридов
со спиртами образуются эфиры сульфиновых кислот [630, 631]. Этерификация может
проводиться также по Шоттену — Бауманну в присутствии пиридина или карбоната
калпя [632].
На примере эфира (—)-(р)-октплового спирта и толуолсульфпновои кислоты
показана возможность рацемизации оптически активных эфиров сульфиновых кис-
лот [633]. у
При взаимодействии сульфинилхлоридов с аммиаком п ачипами в эфирном
растворе образуются амиды сульфиновых кислот.
Бензолеульфинамид [634, 635]. Раствор бснзолсульфнихлорпда в пяти-
кратном количестве эфира обрабатывают 2 моль амина при 0° С. Полученный
амид отфильтровывают, промывают водой, растворяют в этиловом спирте и выса-
живают добавлением небольшого количества воды.
Этим методом получены бензолсульфинанилид (т. пл. 112—111° С),
бензолсульфинпиперидид (т. пл. 83° С), бензолсульфиндиметилаиилид (легко
растворим в эфире; т. кип. 90° С при 2—3 мм рт. ст.) и бензолеульфинамид
(т. пл. 121° С).
[620] L. Knorr, Р. Bossier, Вог., 36, 1279 (1903); О. Н i n s h е г g, J. Kes-
sler, Вег., 38, 906 (1905).
[621] Г. Rcverlin, Вег., 42, 1523 (1909).
[6221 М. Е. Hultquist et al., J. Am. Chem. Sue., 73, 2558 (1951)
[623] R. Adams, J. H. Looker, J. Am. Chem. Soc., 73, 1147 (1951).
[624] Ч. Сьютер, Химия органических соединений серы, пер. с англ., ч. 3,
Иадатинлит, 1951.
[625] Е. Н. N о г t h е у, The Sulfonamides and Alleid Compounds, Reinhold Publi-
shing Corp., New York, 1948.
[626] F. Hawking, J. S. Lawrence, The Sulfonamides, Loudon, I960.
[627] H. Herbst, Schwefelorganische Verbindungen und ihre Verwendung in der
Therapie, Akademiscbe Verlagsgesellschaft Geest a. Portig K. G., Leipzig,
1953.
[628] F. M i e t z s c h, R. В ehnisch, Therapciitisch verwendhare Sulfonaraid-
und Sulfonverbindungeo, 2 Aufl., Verlag Chemie, Weinhcim/BergstraBe, 1955.
[629] F. Mietzscli, Tberapentisch verwendhare Sulfonamid- und Sultonverbin-
dungen, Verlag Chemie, Berlin, 1945.
[630] J. v. Braun, K. We iBbach, Ber., 63, 2840 (1930).
631] R. О tto, A. Rossing. Ber., 18, 2506 (1885).
632] H. P h i I 1 i p s, J. Chem. Soc., 127, 2553 (1925).
633] K. Ziegler, A. W e n z, Ann., 511, 109 (1934).
634] T. I’. Hild’itch, S. Smiles, Ber., 41, 4115 (1908).
635] J. v. Brann, W. Kaiser, Ber., 56, 549 (1923).
ТП. РАСЩЕПЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ СЕРЫ
62
Таким же. образом могут быть синтезированы амиды п-толуолсульфиц
вий КИСЛОТЫ.
При взаимодействии чрезвычайно реакционноспособных: сульфецилхлорид<
с алкоголятамн или фенолятами натрия часто образуются эфиры сульфеновых кц
лот [552, 636, 637], хотя {реакция протекает не всегда гладко.
Метиловый эфир трифснилметилсульфеповой кислоты [638]. Раствор 3
сульфенплхлорпда в 50 мл метилового спирта кипятят непродолжительное врех
с раствором 0,3 г металлического натрия в 10 мл метилового спирта. При охла>
дении реакционной массы эфир выпадает в вице бесцветных игл. После разб
вления маточного раствора водой и экстракции эфиром получают вторую фра]
цию. Эфир перекристаллизовывают ив смеси хлороформ — метиловый спир
т. пл. эфира 124е С.
Значительно более гладко и однозначно происходит образование эфиров суд
феновых кислот из первичных, вторичных и третичных спиртов при взаимодействр
их г 2,1-динитробензолсульфенилхлорндом в присутствии пиридина. Эта реакция мож«
применяться для идентификации спиртовых гидроксильных групп [639].
Особенно легко и с хорошими выходами протекают реакции сульфеиилхлоридс
с аммиаком и аминами с образованием соответствующих сульфенамидов [640J. Реакцй
лучше всего идет в эфирных растворах [637],
Диметиламнд бензолсульфеновой кислоты. К раствору диметиламиц
(полученного из 9 г его гидрохлорида) в 100 мл сухого эфира при охлаждено
льдом с солью по каплям добавляют раствор 5 г бензолсульфенилхлорида в 30 л<
абсолютного эфира. Затем отфильтровывают выпавший гидрохлорид димети^
амина, полученный амид перегоняют в вакууме. Выход амида 4,9 г; т. кит
63,5—64° С (3 мм рт. ст.).
Этим методом синтезированы также 1-хлорнафталин-2-сульфенамид [636
га-нитробензолсульфепамид [592], 3-нитротолилсульфенамид [593] м п-хло|
о-нитробензолсульфенамид [594].
5. Расщепление S—О-связи
Расщепление S—О-связи в сульфокислотах и их солях под действием пеоргащ
чес.кцх галогеналгндридов является основным методом получения сульфохлоридо»
Судьфохлориды образуются при взаимодействии свободных кислот с тиоинлхлоре
дом [641, 642] или трихлоридом фосфора [523] или при взаимодействии солей сульфс
кислот с пятихлоридом фосфора [616 , 643].
Металсульфохлорид [616]. Хорошо высушенный метансульфонат натргг
постепенно смешивают с РС16 и отгоняют образующиеся летучие продукты
Дистиллят фракционируют, собирая чистый метансульфохлорид; т. кип. метац
сульфохлорида 161 —161,5° С (730 мм рт. ст.).
После добавления РС15 нужную температуру реакционной смеси обычж
необходимо поддерживать нагреванием. Устойчивые к гидролизу с^льфохлс
риды выделяют выливанием реакционной массы на лед и экстракцией сульфс
хлорида эфиром. Если реакционная масса имеет склонпость к спеканию, можн:
добавлять инертные растворители: бензол, толуол, ксилол, четыреххлористы.
углерод и др.
[636 [ Т. Zincke, К. Eismayer, Вег., 51, 751 (1918).
[6371 Н. Lecher, Вег., 58 , 409 (1925).
[638] D. Vorlander, Е. Mittag, Вег., 52, 413 (1919).
[639] N. К ha ra.?ch, I). Р. М с Q и а г г j е, С. М. В ues s, J. Am. Chen;
Soc., 75, 2658 (1953).
1640] J. H. Billman, E. O’ Mahony, J. Am. Chem. Soc,, 61, 2340 (1939}
J. H. Billman,J. Garrison ct al.,J. Am. Chem. Soc., 63, 1920 (1941}
|641| R. Noller, и. J. Hearst, J. Am. Chem. Soc., 70, 3955 (1948).
I(»42J P. J. Hearst, С. B. Noller, Org. Synth., 30, 58 (1950).
|643[ C. S. Marvel, M. D. Helfrich, J. P. В elsley, J. Am. Chem. Soc,
51, 1272 (1929).
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ S-ФУПКЦИИ
Этим же методом получены бензолсульфохлорид [644], нафтялин-1,5-
дпсульфохлорид [645], пиридин- и алкилпиридин-3-сульфохлориды [646].
Сульфохлориды с хорошими выходами можно получать также реакцией натри-
евых солей сульфокислот (высушенных в течение 3 ч при 130—200° С) с 93—96%-ной
хлорсульфоновой кислотой [647]. Полученную реакционную массу выливают на лед,
отделяют выделившийся сульфохлорид и сушат его в вакууме над NaOH. Этим путем,
в частности, получены бензол-, n-толуол-, 2-хлортолуол-, а-нафталпн- и 3-нитро-
бензолсульфо хлориды.
п-ХлорбензолсульфохлорВД [648]. При температуре не выше 60° С в тече-
ние примерно 15 мин в суспензию 335 г сухого n-хлорбензолсульфоната натрия
в 700 мл хлороформа при перемешивании вносят 370 г хлорсульфоновой кисло-
ты. Затем полученную плотную кашицу, перемешивая, нагревают еще 6 ч при
55—60° С, охлаждают и выливают в ледяную воду. Отделившуюся органиче-
скую фазу трижды промывают холодной водой, сушат над СаС12 и фракциони-
руют перегонкой в вакууме. Выход продукта 293 г (89% от теоретического);
т. кип. 140° С (12 мм рт. ст.); т. пл. 52—53° С.
Большое число примеров получения сульфохлоридов из сульфокислот
приведено в обзоре Сьютера [649].
Синтез ангидридов арилсульфокислот отщеплением воды от сульфокислот
может быть осуществлен при помощи тионилхлорида [650], а также пятиокиси фос-
фора [651] и ди-п-толилкарбодиимида [652].
Ангидриды ароматических сульфшювых кислот могут быть получены действием
уксусного ангидрида на суспензию безводных сульфиновых кислот в небольшом
количестве уксусной кислоты [653]. Катализаторами являются Н25О4илп 0,1%-ныв
раствор FeCl3 в ледяной уксусной кислоте или уксусном ангидриде. Цо этому спо-
собу были получены ангидриды бензол-, n-толуол-, n-ксилол-, псевдокумол- п мези-
тиленсульфиновых кислот, а также га-бром- и п-нодбеязолсульфиновых кислот.
6. Расщепление S—N-связи
Расщепление S—N-связи [654] в амидах сульфокислот с рбразованием сульфо
кислот приобретает препаративное значение в тех случаях, когда очистка сульфо
кислот через сульфохлориды затруднена, а полученный из сульфокислоты амид может
быть перекристаллизацией очищен лучше, чем сульфохлорид или сама кислота.
Гидролиз сульфамидов проводится соляной кислотой [655]: . 'J
RSO2NHR'-f- Н2О —-1 > RSO2OH + R'NH2 '
RSO3OH4-H2O -> RH+H2SO4
(побочная реакция)
[644] L. Gattermann, Н. W i е 1 a n d, см. [82], стр. 169.
[645] Р. D. Caesar, Org. Synth., 32, 88 (1952),
(6461 Герм. нат. заявка I 201/30 (1930); Frdl., If), 815 (1934).
[647] А. А. Спрысков, Н. В. А п а р ь ев а, ЖОХ, 20, 1043 (1950); С., 1952
2656.
[648] М. К ulka, J. Am. Chem. Soc., 72, 1216 (1950). ;
[649] Ч. Сыотер, см. [624]. '
[650] Н. Meyer, К. S с h 1 е g 1, Mh. Chem., 34, 561 (1913); Н. Meyer, Ann.;
433. 334 (1923); A. L. Bernoulli, H. Stauffer, Helv. chim. acta, 23
640 (1940).
{651] L. F i e 1 d, J. Am. Chem. Soc., 74, 394 (1952).
[652] H. G. Khorana, Canad. J. Chem., 31, 585 (1953).
{653] E. Knoe/enagel, L. P 0 1 a c k, Ber., 41, 3323 (1908).
[654] D. К I a m a n n, G. Hofbauer, Ann., 581, 182 (1952).
[655] R. S. Schreiber, R. L. Shriner, J. Am. Chem. Soc., 56. 1618 (1934)
IV, ПОЛУЧЕНИЕ ПРОИЗВОДНЫХ ТИОУГОЛЬНЫХ кислот
625
Общий способ гидролиза сульфамидов. Кипятят 25 г сульфамида со 125 г
25%-ной НС1 до полного гидролиза (в среднем 10—36 ч), что может быть в боль-
шинстве случаев установлено по растворению сульфамида. Затем для удаления
небольшого количества получившегося углеводорода смесь перегоняют с водя-
ным паром и от дистиллята после подщелачивания NaOH отгоняют с водя-
ным паром образовавшийся амин. Остаток от перегонки после фильтрования
упаривают почти досуха и многократной экстракцией горячим этиловым спир-
том отделяют сульфонат натрия от NaCl.
IV. Получение производных тиоугольных кислот
В этом разделе рассматриваются важнейшие методы синтеза производных
,тпоугольной кислоты. Подробные методикп приведены лишь для небольшого числа
соедипений, в основном даны ссылки на оригинальную литературу.
Для получения производных тпоугольной кислоты особенно часто используют
сероокись углерода и тпофосген, поэтому вначале приводится описание удобных
лабораторных способов их получения; о синтезе родана см. стр. 92-
Сероокись углерода [656—658]. В колбу емкостью 3 л вносят холодную
смесь 1000 г концентрированной IT2SO4, 500 г воды и 100 мл насыщенного
водного раствора KCNS (70 г KCNS). При нагревании до 20а С начинается
энергичное выделение газа; скорость выделения регулируют сначала охлажде-
нием колбы ледяпой водой, а затем нагреванием до 40° С. Влажный газ, загряз-
ненный СО2, H2S и CSg, пропускают для очистки через несколько пустых скля-
нок, охлаждаемых до 0° С, затем через концентрированную H2SO4, 30%-ный?
раствор NaOH (также охлажденный до 0° С), над СаС12 и Р2О5. О дальнейшей
очистке от следов CS3 и СО2 см. оригинальную литературу.
Сероокись углерода в чистом состоянии — бесцветный, без запаха,
легко воспламеняющийся газ, обладающий высокой токсичностью. Сероокись
углерода растворима в органических растворителях и в воде (приблизительно
1 : 1); ,в водных растворах постепенно разлагается па двуокись углерода и серо-
водород.
Тиофосген [659]. Для получения тиофосгена наиболее пригодным ока-
зался метод восстановления перхлорметилмеркаптапа тетралином. Колбу
емкостью 500 мл с насадкой Аншютца и шариковым холодильником, который
может обогреваться паром, соединяют через нисходящий холодильник с другой
колбой емкостью 500 мл, помещенной в смесь льда с солью. Ко второму шту-
церу реакционной колбы присоединяют капельную воронку емкостью 250 мл
с противодавлением. Отходящие газы поглощают в двух последовательно соеди-
ненных колбах Эрлснмеера емкостью по 750 мл. в каждой из которых нахо-
дится по 300 мл воды. Обе колбы охлаждают ледяной водой; отводная трубка
последней колбы направляется в отсос тяги. Реакционная колба равномерно
нагревается на воздушной бапе.
В реакционную колбу загружают 182 г сухого тетралина и нагревают
до 195—200^0. Затем из капельной воронки равномерно в течение 4—5 ч по
каплям прибавляют 465 г сухого перхлорметилмеркаптапа. Реакция гфотекает
спокойно и равномерно с выделением коричневых паров, содержащих примесь
хлористого водорода. Основное количество тиофосгепа конденсируется в ох-
лаждаемом приемнике, хлористый водород с остатком тиофосгена поглощается
водой в ловушках. Реакция считается закопченной, если после добавления
всего перхлорметилмеркаптапа при продолжающемся нагревании при 200° С
прекращается выделение коричневых паров. Из остающейся в колбе черной
массы после охлаждения кристаллизуется нафталин. Сконденсировавшийся
в обеих ловушках тиофосген отделяют в делительной воронке от водной НС1
и объединяют с основным количеством тиофосгепа в приемнике. Сырой продут
Р. К 1 a s о п, J. prakt. Cliem. 12]. 36, 64 (1887); R. J. Ferm, Chem. Rev.,
57, 621 (1957).
A. Stock, E. Knp, Ber., 50, 159 (1917).
Л. Stock, W. S i e c k e, E. P 0 h 1 a 11 d, Ber., 57, 719 (1924).
Пат. ФРГ 853162 (1950); С., 1953, 9314.
40 Заказ 1835.
£26 ПОЛУЧЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ S-ФУНКЦИИ
сушат небольшим количеством СаС13 и фракционируют перегонкой с дефлегш
тором. Выход тиофосгена 185 г (65% от теоретического); т. кип. 71—75° С
Тиофосген представляет собой красную с удушливым запахом ядовиту!
жидкость, довольно устойчивую к действию воды и разбавленных щелоче!
1. Тиоугольные кислоты
кислорода последовательно заме
Если в молекуле угольной кислоты атомы
щать серой, получится пять тиоугольных кислот:
zSII о=с< хон уон s=c< \он /SH s=c< \ОН /SH о=с< Чн ZSH S=C< XSII
тиолуголь- тионуголь- тиолтион- дитиолуголь- тритио-
ная кислота цая кислота угольная кислота (дитиоуголь- ная кислота) ная кислота угольная кислота
В свободном состоянии известна только тритиоугольная кислота (660], осталь
ные кислоты существуют в виде солей, хлорангидридов, эфиров и амидов.
2, Эфиры хлорангидридов тиоугольных кислот
Эфиры хлорангидридов тиоугольных кислот получают взаимодействием фос
гена пли тиофосгена со спиртами, фенолами или меркаптанами в присутствии водно!
щелочи в количестве, необходимом для связывания выделяющейся соляной кислоты
a-c-ci-^U BS-C-C1
Cl—С—Cl ВО-С-С1
S
S
S
О-эфир хлор-
ангидрида тиоугольиой
КИСЛОТЫ [661—663]
С1-С-С1 RS-C-C1
S
S-эфир хлор-
ангидрида дитиоуголь-
ной кислоты [665, 666]
О
О
S-эфир хлор-
ангидрида тиоугольиой
кислоты [664, 665]
[66$
О-4-Хлорфениловыи эфир хлорангидрида тиоугольиой кислоты „
К раствору 128,5 г n-хлорфенола и 115 г тиофосгена в 500 мл хлороформ!
в течение примерно 1 ч при 15—20<’С по каплям прибавляют раствор 4OJ
NaOH в 500 мл воды. После перемешивания 1 ч отделяют хлороформеннм
слой, промывают его водой, сушат над СаС12, затем отгоняют растворителя
и полученный продукт перегоняют в вакууме. Выход эфира 139,4 г (67% <д
теоретического); т. кип. 123—126° С (12 мм рт. ст.). Эфир представляв|
собой желтую жидкость с резким запахом.
О получении О-алкиловых эфиров хлорангидрида тиоугольиой кислотЯ
хлорированием бис-(алкилксантогенов) см. [667]. /1
660
661
662
663
664
665
666
667]
Н. М i 11 s, Р. L. R о b i п я о n, J. Chem. Soc., 1928, 2330.
М. Н. R i v i е г, Bull. Soc. chim. France [3], 35, 837 (1906).
H. R i v i e г, J. Schalch, Helv. chim. acta, 6, 612 (1923).
R. Philippson, Dissertation, Berlin, 1960.
R. Riemschneider, O. Lorenz, Mh. Chem., 64, 518 (1953)-
M. H. R ivier, Bull. Soc. chim. France [4], 1, 733, 737 (1907).
P. Klason, Ber., 6, 312 (1873).
Пат. ФРГ 1018054 (1957); С., 1958, 12244.
rv. ПОЛУЧЕНИЕ ПРОИЗВОДНЫХ ТИОУГОЛЬНЫХ кислот
62
3. Эфиры тиоугольных кислот
Полуэфиры тиоугольных кислот
Полуэфиры тиоугольных кислот получают взаимодействием алкоголятов ид
меркаптидов с сероуглеродом или сероокисью углерода:
COSH-ROMe —> RO—С—ОМе СЗг + ВОМе
RO-C—SMe
S
соль О-эфира тио-
угольной киолоты
(О-алкилтионкарбонат)
['668. 669]
CSaH-RSMe -
S
соль О-эфира дитио*
угольной кислоты (ксаитогенат,
О-алкилдитиокарбонат) [670]
RS —С —SMe
S
Соль эфира тритио-
Угольной кислоты (S-алкил-
тритиокарбонат) [671]
Образование соли О-эфира дитиоугольиой кислоты (ксантогеиата) из сер<
углерода и алкоголята калия происходит чрезвычайно легко, так что почти все спирт
могут быть подвергнуты такому взаимодействию.
Ксантогенаты. Общий способ получения [672]. Для получения ксантс
генатов калия раствор 1 .моль (56 г) тонко измельченного КОН в необходимо
для его растворения количестве спирта после охлаждения до комнатной темпе
ратуры смешивают (30 мин) с 1 моль (76 г) CS2. Затем в течение 1 ч перемет®
вают, добавляют эфир для полного отделения ксантогеиата, отфильтровываю
продукт и перекристаллизовывают его из этилового спирта. Выход ксантс
гената 80—95% от теоретического.
Полные эфиры тиоугольных кислот
Полные эфиры тиоугольных кислот образуются либо взаимодействием 2 мод
спирта с 1 моль тиофосгена или 2 моль меркаптида с 1 моль фосгена или тиофосгён®
либо взаимодействием алкоголятов и меркаптидов с эфирами хлорангидридов ти<
угольных кислот в молярном соотношении 1 : 1.
8|10Мц> но-С-OR <ROMc Cl-C-OR
Cl—С—Cl
s
S
О, О'-диэфир
тионугольной
кислоты [673, 674]
RO-C-C1 RS-C-OR
о
О
О, З-диэфир
тиолугольной
кислоты [675]
[668]
669]
670]
671]
'2]
С. Bender, Ann., 148, 137 (1868).
F. Salomon, J. prakt. Chem. (2], 5, 476 (1872).
M. Rar - oz oo
В. П ol____=
F. D r a w e г t,
(1960).
II. Eckenroth, K. Kock, Ber.. 27, 1368 (1894).
M. M. Delepine, Ann. Chimie [8], 25. 546 (1912).
F. Salomon, J. prakt. Chem. [2], 6, 433 (1873); E. M у 1 i u s, Ber., 6, 31.
(1873).
40*
eg, Chem. Ztg., 34, 83 (1910).
Im berg, J. prakt. Chem.
К. H. R e u t li о i
121, 73, 239
r, F. В о г n,
(1906).
Chem. Вег., 93, 306-
[673]
[674]
J675]
628 ПОЛУЧЕНИЕ СОЕДИНЕНИИ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ S-ФУНКЦИИ
Cl-C-Cl -2RSMC> RS—С—SR
I 'I
s s
диэфпр тритио-
угольной
кислоты [666, 676, 677]
Cl-С-Cl
2KSMC
II
RS—C—SR
RSMe
S, S'-диэфир
дитиолугольной
кислоты 1675, 678]
RO-C-SK -BC1; 'rN=JtCH> HO—C—SR
II II
s s
O, S-дияфир
дитиоугольной
кислоты [678,679,680]
Арилтнонкарбонаты [673, 676]. Раствор 2 моль одноатомного фенола
в небольшом избытке 10%-ного раствора NaOH разбавляют четырех-, пяти-
кратным количеством воды и к смеси при энергичном встряхивании постепенно
добавляют 1 моль тиофосгена. Если смесь сильно разогревается, необходимо
охлаждение. Образующийся фениловый эфир выделяется в виде белого осадка.
Осадок промывают холодной водой до исчезновения щелочи и перекристаллизо-
вывают (чаще всего из этилового спирта).
Этим способом, например, дифенилтионкарбонат получают с почти коли-
чественным выходом; т. пл. 106° С. ?
4. Производные тиокарбаминовой кислоты
Соли
N-Замещенные тиокарбаматы аммония и щелочных металлов в общем случае
получают взаимодействием первичных и вторичных аминов с сероокисью углерода
или с сероуглеродом в присутствии аммиака или гидроокисей щелочных металлов:
В отсутствие неорганических оснований при использовании спльпоосповдых ампноэ
образуются тиокарбаматы введенных в реакцию аминов [681, 682].
RNH2 + COS + MeOH —► RNII-C-SMe
соль монотиокарб-
амиковой
кислоты 1683, 684]
[676] W. Autenrieth, Н. Hefner, Вег., 58, 2151 (1925). 7
[677] J. A. Durden jr., Н. A. S t а п s b и г у jr., W. Н. С a t 1 е 11 о, J. Апц
Chem. Soc., 82 , 3082 (1960).
[678] R. L е и с k а г t, J. prakt. Chem. [2], 41, 179 (1890).
J679J W. Fomin, N. Sochanski, Ber., 46 , 244 (1913).
[680] L. Tschugaeff, Ber., 32, 3334 (1899); D. Martin, J. prakt. Chem. [4J*
9, 222 (1963).
[681] A. Rieche, G. H i 1 g c t a g et al., Arch. d. Pharm., 293 r
957 (1980).
[682] Герм. пат. 531007 (1929); Frdl., 18, 424 (1933).
[683] К. H. Slolta, H. Dressier, Ber., 63. 888 (1930).
[684] S. Kallcnberg, Ber., 56 , 320 (1923).
IV. ПОЛУЧЕНИЕ ПРОИЗВОДНЫХ ТИОУГОЛЬНЫХ кислот
62
RNII2+CS2+MCOH
RNH-C-SMe
S
соль дитиокарб-
аминовой
кислоты [683, 685 -687]
N-Фенилдитиокарбаминовокислый калии [688]. Получают смешение
эфирного раствора эквимолярных количеств сероуглерода и анилина со спирт*
вым раствором эквивалентного количества КОН. Продукт представляет собо
белые иглы, легко растворимые в воде и этиловом спирте и нерастворим^
в эфире.
Тиокарбаминоилхлориды
Для получения незамещенного тиокарбаминоилхлорида проводят присоедини
вне хлористого водорода к тиоциановой кислоте в среде безводного эфира [689
HN=C=S + HC1 —► H2N-C-CI
S
N-Замсщеппые соединения получают взаимодействием первичных или вторичны
аминов с тпофосгеном в абсолютном эфире [690—693]:
R-, 1\
>NH + Cl-С-Cl —>N- С- Cl
R'z II R'z ||
S S
Первичные амипы взаимодействуют с тпофосгеном только при полном отсутстви
влаги, тогда как гидрохлориды вторичных аминов можно ацилировать раствором тис
фосгена в»хлороформе [692] и в водной среде.
Эфиры тиокарбаминовой кислоты (тиоуретаны)
Эфиры тпокарбамиповой кислоты [694] получают различными способами
R'0-C-SR"
R-N--C=S
R.
/N—С— Cl R-ONa
R/ || -----
R'O— С—NHR
или R'O —С—NR2
1
8
R'o-c-ci
[685] С. Е. R edemann,R. N. I с k е, G. A. Alles, Org. Synth., 27, 73 (1947).
[686] F, В. D a ins, R. Q, Brews ter, С. P. О land er, Org. Synth., Coll,
v. 1, 1937, p. 446.
687] M. L. Moore, F. S. С г 0 s s 1 e y, Org. Synth., 21, 81 (1941).
688] g. M. Losanitsch, Ber., 24, 3021 (1891).
689] M. Ba tt egay, E. H ё g a z i, Helv. chim. acta, 16, 1003 (1933).
690] G. M. D у 3 о n, H. J. G e 0 r g e, J. Chem. Soc., 126, 1702 (1924).
691] О. С. В i 1 1 e t e r. Ber., 20, 1629 (1887).
692] О. С. В i 1 1 c t e r, H. R i v i e r, Ber., 37, 4317 (1904).
693| J. v. Braun, Ber., 36, 2274 (1903).
694] R. Zahradnik, Chem. Techn., 11, 546 (1959) (обзорная статья).
630
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ СЕРЫ ПУТЕМ ПРЕВРАЩЕНИЯ S-ФУНКЦИИ
Первый способ дает хорошие результаты, если R'-алкпл [695J, второй
способ дает хорошие выходы только в случае спиртов, но ие фенолов [696—698],
третий способ целесообразно использовать только с диэамещепными карбаминоилхло*
рядами [699], одпако четвертый способ универсален [697, 700].
О-Ариловые' эфиры N-замещенных тиокарбаминовых кислот ‘[700].
К 2 моль амина (растворенного в ацетоне или этиловом спирте) при 20—30° С
медленно прибавляют по каплям при перемешивании 1 моль О-арияового
эфира хлорангидрида монотиоугольной кислоты. Перемешивают еще 1 ч,
выливают массу в ледяную воду, отфильтровывают выпавший эфир и после
высушивания перекристаллизовывают из этилового спирта.
Эфиры тиокарбаминовых кислот (RaNCOSR) можно получать по описанному
выше методу из карбаминоилхлоридов и меркаптидов [701], из S-эфиров хлорангид-
рида монотиоугольной кислоты и аминов [702] и присоединением воды к эфирам тио-
{(Пановой кислоты [703].
Эфиры дитиокарбаминовой кислоты обычно могут быть легко синтезированы :
алкилированием или арилированием соответствующих дптиокарбаматов [704—708].
Другими методами являются присоединение меркаптанов к эфирам изотиоциановой
кислоты [704], взаимодействие эфиров хлорангидрида дитиоугольной кислоты с ами-
нами [704] и взаимодействие тиокарбаминонлхлоридов с меркаптидами [699J.
5. Бис-(алкилксантогены) и тнурамдисульфиды
При действии окислителей на ксантогенаты образуются бис-(алкилксантогены)
BO-C-SS-C-OR
II II •
S S
из солей дитиокарбаминовой кислоты получаются соответственно тпурамдисульфиды,
Бис-(адкнлксантогеяы) [709]. Через охлаждаемый льдом раствор алкил-
ксантогената калия в воде пропускают сильный ток воздуха, содержащего.’
примерно 5—10% хлора, до тех пор, пока из пробы реакционной смеси при
добавлении сульфата меди не прекратится выпадение ксантогената меди. Отде- <
ляют образовавшийся осадок или масло, растворяют его в эфире ипосяевысу- -
шивания отгоняют эфир.
Для окисления дитиокарбаматов наряду с другими окислителями предложены
хлорное железо [710], иод [711] и феррицианид калпя [712].
E. В i i 1 m a n n, Ann., 348, 140 (1906).
R. W. В о st, E R Andrews, J. Am. Chem. Soc., 65 , 900 (1943).
R. P. Mull, J. Am. Chem. Soc., 77, 581 (1955).
W. Schneider, F. Wrede, Ber., 47, 2038 (1914).
О. С. В ill e t er, A. S t rohl, Ber., 43, W (1910); 21, 102 (1888),
A, R ieche, G. H ilgetag et al., Arch. a. Pharm., 294 , 201 (1961).
Пат. США 2642428 (1953); С. A., 48, 5210 (1954).
R. Riemschncider, O. Lorenz, Mh. Chem., 84 , 520 (1953).
695]
696
697
698
699
700
701
702J ..._________ „ ....... ............. - . x----..
703] R. Riemschncider, F. Wojahn, G. О r 1 i c k, J. Am. Chem. Soc.
704]
705]
706]
707]
708]
[709]
[710]
[711]
[712]
73, 5905 (1951). 4
J. V. Braun, Вет., 35, 3369 (1902); 42, 4571 (1909).
Пат. США 1726648 (1928); С., 1929, II, 2938.
А. М. С 1 i f f о г d, J. G. Licht у, J. Am. Chem. Soc., 54, 1163 (1932). ;
J. C. Sauer, J. Org. Chem., 24, 1592 (1959).
G. R о s s i, V. Corradini, Chim. e Ind. (Milano), 42, 237 (1960); C., 1981
5043; A. R i ech e, G. H ilget a g, D. Mart i n, I. К г e у z i, Arch, • •)
d. Pharm., 296. 310 (1963). '
Герм. пат. 431752 (1924): С., 1926, II, 1460.
M. Freund, G. Bachrach, Ann., 285, 201 (1895). '
J. v. Braun, Ber., 35, 819 (1902).
R. Rothstein, К. В i n о vic, Rec. trav. chim., 73, 561 (1954).
IV. ПОЛУЧЕНИЕ ПРОИЗВОДНЫХ ТИОУГОЛЬНЫХ кислот
631
6. Изотиоцианаты (горчичные масла)
Исходными продуктами для получения изотиоцианатов являются соли дитио-
нарбаминовой кислоты, из которых действием солей тяжелых металлов [686], эфиров
хлормуравьиной кислоты [687], фосгена [683] или гипохлорита натрия [713] в боль-
шинстве случаев с хорошими выходами образуются изотиоцианаты. Широко распро-
страненным способом является также взаимодействие аминов [714, 715] или их гидро-
хлоридов с тиофосгеном [716].
р-Хлорэтилизотиоцианат [717]. К раствору 50 г хлорида р-хлорэтилам-
мония в 100 мл воды добавляют 200 мл хлороформа. К этой смеси при переме-
шивании и охлаждении льдом одновременно добавляют по каплям концентри-
рованный водный раствор 130 г Na2CO3 и раствор 50 г тиофосгена
в 50 мл СНС13. Реакционная смесь должна постоянно иметь щелочную реакцию.
После перемешивания в течение 30 мин отделяют хлороформенный слой, сушат
его над Na2SO4 и ректифицируют; т. кип. продукта 80° С (13 мм рт. ет.).
Изотиоцианаты легко могут быть также получены расщеплением диарнлтиомоче-
впн под действием кислот [718].
7. Солн изотномочевины
Тиомочевина и ее N-производпыо образуют таутомерные формы, в которых ти-
ольная группа почти всегда является активным центром взаимодействия. Поэтому
обычные алкилирующие агенты, например алкилгалогспиды [719—721], диметилсуль-
фат [722, 723] и эфиры сульфокислот [724], образуют с тиомочевиной соли S-алкилизо-
тиомочевины (изотиурониевые солп):
HjNk HaNy RX HaNx
>C=S ^C-SH-2--> ^C-SR-HX
ii2nz
тиомочевина
НГг
изотиомочевина
соли. Общий способ получения [551]. Молярные ноля-
смеси алкилгалогенида и спирта (объемное отношение
Изотиурониевые
чества тиомочевины и . _ .... _ ,
1 : 1) нагревают на водяной бане до тех пор, пока проба реакционной массы
не будет давать осадок сульфида серебра с аммиачным раствором нитрата
серебра.
При проведении реакции с этиленбромидом или этиленхлоридом необходимо
применять трек-, четырехкратное количество спирта; образующаяся соль 8,8'-этилен-
дппзотиомочевины нерастворима в спирте и может быть отфильтрована.
В некоторых случаях [725], особенно при получении солеи S-циклогексилтио-
мочевины [551], удается провести алкилирование соответствующим спиртом в присут-
ствии бромистоводородной кислоты.
Е. Schmidt, F. Z а 1 1 е г et aL, Ann., 585, 230 (1954).
G. M. Dyson, Chem. a. Ind., 1954, 934.
G. M. D у s о n, Org. Synth., Coll. v. 1, 1937, p. 158.
E. T i e t z e, S. Peterson, G. Domagk, Cnem. Ber., 86, 312 (1953).
H. Brintzinger, K. Pfannstiel, H. Koddebusch,
Chem. Bep., 82, 399 (1949).
L. Gattermann, H. Wieland, cm. [82], стр. 151.
E. Brand, F. C. Brand. Org. Synth., 22, 59 (1942).
A. Bertram, Ber., 25, 48 (1892).
J. D. Brooks, P. T. Charlton et al., J. Chem. Soc., 1950, 452.
F. Arndt, Ber., 54, 2237 (1921).
P. R. Shildneck, W. Wind us, Org. Synth., Coll. v. 2, 1943. p. 411.
D. Klamann, F. Drahowzal, Mh. Chem., 83, 463 (1952).
R. L. Frank, P. V. S in i t h, J. Am. Chem. Soc., 68, 2103 (1946).
632
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
(металлоорганические соединения) *
К металлоорганическим соединениям относятся вещества, которые содержат
органические радикалы, например алкильные или арильные группы, связанные
непосредственно с атомом металла, т. е. соединения с прямой металл-углеродной
связью. При этом понятие металла включает также некоторые неметаллы и элементы
со слабо выраженными металлическими свойствами **. Производные, в которых атом
металла связан с углеродом через другие атомы, например через азот пли кислород,
к металлоорганическим соединениям не относятся,
В этой исключительно широкой области органической химии имеется ряд обзор-
ных работ, например книга Краузе и фон Гроссе «Химия металлоорганических соеди-
нений» [1], и которой практически полностью охватывается литература по этому
классу соединений до 1936 г., а также книга Рунге «Металлоорганические соединения»
[2]. Основные начала химии металлоорганических соединений с многочисленными
ценными ссылками на оригинальную литературу можно почерпнуть из книг Коэйтса
[3] и Рочоу, Херда и Льюиса [4]. Небольшая обзорная статья Хейна [5] посвящена
развитию химии металлоорганических соединений, а в монографии Коэйтса [6] рассма-
триваются металлоорганические соединения первых трех групп периодической системы
элементов. В справочнике Кауфмана (6а] сопоставляются свойства многих металлоорга-
нических соединений и приводятся ссылки на оригинальную литературу
Алкил- или арилироизводные металлов по свойствам часто отличаются от обыч-
ных соединений металлов и во многих отношениях напоминают истинные органические
соединения. Так, например, многие из них летучи, нерастворимы в воде и растворимы
в органических растворителях, не проводят электрический ток. Напротив, органиче-
ские производные сильно электроположительных элементов, например щелочных
металлов, являются ярко выраженными лонными соединениями: они нелетучи, пред-
ставляют собой твердые вещества, практически нерастворимы в органических раство-'
рителях и, кроме того, являются хорошими электролитами.
Бее же классификация металлоорганических соединений по их свойствам на
строго различные типы, например па гомеополярные и гетерополярные, невозможна,
так как физические и химические свойства этих соединений внутри каждой группы
и каждого периода различны. Полярность металл-углеродной связи в каждой группе
элементов периодической системы возрастает сверху впиз, вместе с этим усиливается •
и солевой характер соединения, а в каждом периоде полярпость падает слева направо. * 1 2 3 4 5 6
Аналогичные закономерности наблюдаются в случае гидридов металлов. Гид-
риды щелочных металлов представляют собой соли, гидриды щелочноземельных метал-
лов солоподобны, а гидриды кремния, как и углеводороды, являются газами или
жидкостями.
Полярность металл-углеродной связи определяет не только физические, по 1
и в значительной мере химические свойствамоталлоорганическихспединений. Реакцпоп-
* Материал обработал Дж. Дапьманн. -
** Едва ли можно согласиться с таким широким определением металлоорганических :
соединений. Подобные вещества следует именовать «элемептоорганическими». —
Прим. ред. ‘
»** Исключительно-цепным изданием является серия монографий по металлоорганиче-i
ским соединениям, выпущенная издательством АП СССР под редакцией'
акад. А. Н. Несмеянова — Прим. ред. '.
[1] Е. Krause. A. v. G г о s s е, Die Chemie der metallorganischen Verbindungen,
Bomtraeger, Berlin, 1937. ?
[2] F. Runge, Organo-Metallvcrbindungen, Wissenschaftliche Verlagsgesellscliaft!
m. b. II., Stuttgart, 1944. J
(3] G. E. Coates, Organo-Metallic Compounds, Methuen Co. Ltd., London and5
John Wiley a. Sons. New York, 1956. !
[4] Ю. P ox о в, Д. X ер д, Р. Льюи с, Химия металлоорганических соедине-
ний, itep. с англ., Издатинлит, 1963.
[5] F. Hein, Chem. Techn., 9, 198 (1957).
[6] G. E. Coat e*s, Quart. Rev., 4, 217 (1950).
[6aJ H. C. Kaufman, Handbook of Organometallic Compounds, D. Van Norstrand
Company, JNC, Princeton, New Yersey, 1961.
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
633
пая способность этих соединений возрастает с увеличением полярности металл-угле-
родной связи, зависящей в первую очередь от различия в электроотрицательности
основного атома и связанного с ним атома углерода. Чем ниже электроотрицатель-
ность металла (по Полингу), том выше реакционная способность его органических
производных.
В соответствии с этим принципом при исследовании реакций присоединения
металлоорганических соединений к кратным С= О- и C^N-связям, а также к С=С-
снязям несимметричных олефинов установлено [ 7] > что реакционная способность органи-
ческих производных в каждом периоде для элементов главных подгрупп понижается
с увеличением порядкового номера элемента:
Li— R> Be—R > В—R
Na—R > Mg—R > Al—R > Si—R
И, наоборот, в главных подгруппах реакционная способность возрастает с уве-
личением атомного веса ключевого атома:
Li—R <С Na—R < К—R < Rb—R < С?—R
Be—R < Mg—R < Са — R и т. Д.
Для элементов побочных подгрупп наблюдается обратный порядок:
Си—R > Ag—R > Au-R
Кроме того, наименее реакционноспособный элемент данной или следующей главной
подгруппы более реакционноспособен, чем самый активный элемент соответствующей
побочной подгруппы:
Li—R > Си—R Be—R > Zn—R
Be—R > Си—R В—R > Zn—R
Для органических производных одного и того же металла реакционная способность
несимметричных соединений выше, чем реакционная способность его симметричных
производных.
Различия в реакционной способности металлоорганических соединений можно
проследить также на примере их гидролиза, приводящего к образованию углеводорода
и гидроокиси металла. При взаимодействии с водой алкильных производных щелочных
или щелочноземельных металлов протекает бурная реакция, а слабополярные
кремнийалкилы не вступают в эту реакцию. Аналогично драгируют с металлоорганиче-
скими соединениями другие полярные реактивы, например галогеноводороды. При
помощи этой реакции можно выявлять даже незначительные различия в полярности
четалл-углеродпых связей; так, например, водная соляная кислота отщепляет фениль-
ные группы, связанные с атомом кремния, не затрагивая метильных групп.
Полярность и реакционная способность металл-углеродной связи зависят,
таким образом, и от электроотрицательности атома углерода. Полярность такой связи
почти не зависит от строения углеводородного остатка, хотя различие между метиль-
ными и фенильными группами уже заметно. Значительно усиливается этот эффент
при введении в углеводородный остаток сильно электроотрицательного заместителя,
например хлора: при этом значительно повышается электроотрицательность атома
углерода и соответственно полярность металл-углеродпой связи. Противоположным
действием обладают электроположительные заместители у атома углерода или электро-
отрицательные у металла. Они понижают полярность металл-углеродпой связи и ее
реакционную способность.
Кроме того, при оценке реакционной способности металлоорганического соедине-
ния следует учитывать, что эти реакции обычно проводят в растворах. Реакционная
способность сольватированных и посольватированпых молекул различна. Так, напри-
мер, полярность металл-углеродной связи понижается в растворителе, содержащем
эфирный кислород, неподеленные электронные пары которого взаимодействуют с элек-
троположительным атомом металла.
Г] И. Gil man, Organic Chemistry, v. I, 2 ed., John Wiley a. Sons, New York,
1943, p. 520.
634
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
Непосредственной взаимосвязи между реакционной способностью и термической
устойчивостью металлоорганических соединений не существует. Никоим образом
нельзя делать общий вывод о том, что очень реакционноспособные металлоорганические
соединения особо чувствительны к нагреванию и что менее реакционноспособные г-оади-1
нения обладают повышенной термической устойчивостью. Правда, термическая ста-
бильность органических производных более тяжелых элементов главной подгрушод-
ниже, чем у производных более легких элементов. Так, тетразтилсилан (CtHs)4Si‘
устойчивее тетраэтилсвинца (CaHj)4Pb. Но, с другой стороны, органические проиэ-^
водные элементов побочной подгруппы Г группы гораздо менее устойчивы, чем'
более реакционноспособные соединения щелочных металлов. •
Аналогичная закономерность обнаружена в отношении чувствительности метал-*
лоорганических соединений к кислороду. Конечно, и в этом случае важную роль*
Играет полярность металл-углеродной связи; так, например, алкильные производный
щелочных металлов воспламеняются в присутствии кислорода воздуха, а алкильны^
производные ртути и кремния на воздухе не изменяются. Но кроме этого решающее;
значение могут иметь другие факторы, например наличие свободной пары электронов,’
или незаполненного октета у атома металла. Так, триалкильные производные метал*
лов третьей и пятой главных подгрупп, п которых металл-углеродная связь близка^
к ковалентной, исключительно бурно реагируют с кислородом. Низшие алкильные^
производные этих металлов на воздухе моментально воспламеняются. <5
Так же легко реагируют алкильные производные металлов третьей и пятой глав-?
ных подгрупп с другими донорами или акцепторами электронов. Алкильные производи}
пые металлов третьей группы (акцепторы электронов) дают с аналогичными производи
ными металлов пятой группы (доноры электронов) соответствующие комплексные соедн-i
пения, например (СН3),В • Аз(СН3)3 (см. обзор [8]).
Склонностью к образованию комплексов и продуктов присоединения с самыми!
различными органическими и неорганическими соединениями обладают и другие металл
лоорганические соедипепия. Известны также их аддукты друг с другом и олигомеры.?
Так, например, литийоргаяические соединения представляют собой ассоцниро-^
ванные продукты. Фениллитий в эфирном растворе в широких пределах концентраций^
находится в виде димера Li [Ы(СвН5)2]• ,£
При взаимодействии фениллитпя с металлоорганическими соединениями элй-
ментов, следующих за ним в периодической системе, т. е. с производными бериллия?
и бора, образуются хорошо кристаллизующиеся комплексы, устойчивость которых
возрастает в ряду:
Li[Li(С6Нб)2] < Li[Bc(CeH6)3] < Li[B(CeH6)4]
Свойства этих комплексов в некоторых случаях сильно отличаются от свойств;
составляющих их веществ. Если фениллитий и трифенилбор легко реагируют с кислой
родом, а фениллитий бурно разлагается в присутствии воды, то тетрафепилборат литий
устойчив к действию кислорода и растворяется в воде без разложения.
О комплексообразовании и реакционной способности металлоорганический
соединений см. [9J,
Способы получения металлоорганические соединений
В настоящее время известно большое число способов получения металлоорганя4
ческих соединений. Ниже приведены лишь важнейшие из них.
1) Прямое алкилирование или арилирование металлов алкил- или арилгал
гонидами:
2Me-f-RX RMe+MeX
Это одна из самых распространенных реакций в химии металлоорганичесш
соединений, поскольну таким способом прямо или косвенно можно получать почти в
металлоорганические соединения.
В тех случаях, когда сами металлы вступают в реакцию с большим труд
или вообще не реагируют с органическими галоидпропзводными, применяют их спла;
с щелочными или щелочноземельными металлами, в первую очередь с натрием:
, MeNa+RX RMe-f-NaX
[8] F. G. Stone, Chem. Rev., 58, 101 (1958).
[9] G. Wittig, Angew. Chem., 70, 65 (1958); 62, 231 (1950).
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
635
Аналогичная реакция расщепления простых эфиров металлами протекает лишь
с определенными эфирами и только под действием щелочных металлов:
ROR-]-2Me —> RMe + ROMe
2) Вытеснение одного металла другим в ранее полученном металлоорганическом
соединении:
Me + RMe' +2 RMe-j-Me'
Этот метод применяется для синтеза более реакционноспособных металлоорга-
нических соединений из менее реакционноспособных. Например, при взаимодействии
металлического натрия или лития с диалкил- или диарилпроизводными ртути обра-
зуются соответствующие литий- или натрийорганические соединения.
3. Взаимодействие металлов с углеводородами:
а) Замена водорода иа металл:
RH-f-Me —» RMe+H
Реакция может идти липп. с очень реакционноспособными металлами и с угле-
водородами, содержащими подвижный атом водорода.
б) Присоединение реакционноспособных металлов (щелочных металлов)
к С= С-связям.
в) Расщепление С—С-связей щелочпыми металлами.
4. Взаимодействие Солей металлов с другими металлоорганическими соеди-
нениями:
RMe + Me'X RMe' 4-МеХ
Реакция представляет собой важнейший и самый общий метод синтеза металло-
органических соединений. Таким методом можно получить практически все типы метал-
лоорганических соединений. При таком взаимодействии равновесие сдвинуто в сторону
образования менее реакционноспособного металлоорганического соединения. В каче-'
стве исходных веществ обычно применяют реактивы Гриньяра или литийоргапические
соединения.
5) Реакция галогенидов металлов и органических галогенидов с металлическим
натрием (по типу реакции Вюрца):
МеХ -f-RX 4- 2Na—>RMe4-2NaX
Основное применение этот метод находит для получения кремний-, олово-
и мышьякорганических соединений, а также органических производных сурьмы.
6) Взаимодействие солей металлов с углеводородами:
MeX + RH —» RMe + HX
, Так, например, самые разнообразные ароматические соединения подвергаются
мерйурированию под действием ацетата ртути, а соли меди и серебра реагируют с аце-
тиленами с образованием соответствующих ацетиленидов.
7) Реакция солей металлов с диазосоединениями. При разложении арилдиазо-
ниевых солей в присутствии солей металлов или при разложении комплексных солей
арилдиаэонля и металла образуются арилметаллические производные (реакция Несме-
янова), например:
RN2C1- HgCls + 2Cu—> RHgCl+Cu2a2 + N2
О механизме этой реакции см. статьи Реутова [10].
Аналогичная реакция протекает при разложении двойных -солей, полу-
ченных, из солей дифенилгалогония и галогенидов тяжелых металлов в присут-
ствии порошкообразных металлов (Реутов). Таким способом можно получать
арилпроизводные ртути, олова, сурьмы и висмута [10].
Важнейшим методом синтеза ариларсоповых кислот, представляющих опреде-
ленный химиотерапевтический интерес, является взаимодействие хлоридов арил-
диазояия с арсенитами натрия (реакция Варта), например:
CfjHgNaCl Na2HAsOa —* CeHgAsOaHNa -|- NaCl-|- N2
[10] О. A. R e и t о w, Angew. Chem., 72, 198 (I960); Tetrahedron, 1, 67 (1957).
636
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
Аналогичным методом можно получать арилпроизводные сурьмы.
8) Действие свободных радикалов на металлы. Взаимодействие триарилметиль-
ных радикалов с металлическим натрием [11] приводит к соединениям типа R3CNa.
При действии метильных и этильных радикалов на мышьяк, сурьму и висмут полу-
чаются соответствующие металлоорганические соединения [12].
Образование арильных производных металлов при разложении хлоридов арил-
диазония в присутствии таких металлов, как ртуть, сурьма, висмут, свинец или олово,
также протекает через свободные радикалы [13], в отличие от реакции Несмеянова.
9. Взаимодействие ранее полученных металлоорганических соединений с дру-
гими органическими или металлоорганическими соединениями.
а) Взаимодействие с металлоорганическими соединениями;
RMe-rH'Me' RMe'-|-R'Me
При этом в общем случае болео электроотрицательный органический остаток
переходит к более электроположительному металлу. Такой метод удобно применять
в тех случаях, когда один из продуктов реакции плохо растворяется в применяемом
растворителе и выпадает в осадок.
б) Взаимодействие с углеводородами. Исключительно реакционноспособные
металлоорганические соединения могут обмепивать атом металла на атом водорода:
BNa-| СбН6 —> CeII5Na-i-RH
Металлоорганические соединения щелочных металлов могут также присоеди-
няться к С—С-связям несимметричных олефинов.
в) Реакция с органическими галогенидами протекает с обменом галогена па
металл. Так, при взаимодействии я-бутиллития с некоторыми ароматическими броми-
дами или иодидами образуются ароматические производные лития:
«-C^HgLi |-RBr —* RLi-f-И-С4Н9ВГ
Кроме того, органические галогениды могут присоединяться к металлооргани-
ческим соединениям, содержащим металл в его низшей валентности, например:
RsAs |-R'I —> RgR'AsI
10) Присоединение неорганических или органических гидридов металлов
к С-С-связям:
>С-=С<+МеН —> >С-С<
Н Me
Примером может служить присоединение трихлорсилана к олефинам [14].
Особым случаем такого синтеза является разработанный Циглером и сотр. про-
мышленный метод получения алкильных производных алюминия из алюминия, водо-
рода (или из гидридов алюминия) и олефинов, особенно этилена.
Основные методы получения металлоорганических соединений и их обсуждение
приведены в обзоре [15].
Т. Органические производные элементов первой группы [16]
Щелочные металлы, являющиеся электроположительными элементами, обра-
зуют органические производные с сильно поляризованной металл-углеродной связью.
Вследствие этого органические производные щелочных металлов являются самыми
[11] W. Schlenk, Е. Marcus, Вег., 47, 1664 (1914).
[12] F. A. Paneth, Н. Loleit, J. Chem. Soc., 1935, 366.
[13] F. В. М a k i n, W. A. Waters,}. Chem. Soc., 1938, 843; W. A. W a t e r s,
J. Chem. Soc., 1937, 2007.
[14] E. W. P i e t r u s z a, L. H. Sommer, F. C. Whitmore, J. Аш. Chem.
Soc., 70, 484* (1948).
[15] R. G. Jo n es, H. Gilman, Chem. Rev., 54, 835 (1954).
[16] С. B. Wooster, Chem. Rev., 11, 1 (1932).
I. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ Г ГРУППЫ
637
реакционноспособными соединениями из всех известных в органической “химии. Ал-
кильные производные натрия и калия реагируют со всеми органическими соединениями,
за исключением парафиновых углеводородов. Широкое синтетическое применение
нашли менее реакционноспособные производные лития, что объясняется особым поло-
женном лития в периодической системе элементов, показывающей некоторые законо-
мерности в диагональных направлениях. Так, например, литийорганические соеди-
нения во многих отношениях похожи на магнийорганические соединения, но являются
более реакционноспособными, в то же время они не дают побочных реакций, часто
протекающих при использовании натрпй- или калпйоргапических соединений. Органи-
ческие соединения лития обычно хорошо растворяются в инертных органических рас-
творителях, тогда как аналогичные производные других щелочных металлов в них
не растворяются. Кроме алкилпропзводпых лития, алкил- и арилпроизводные щелоч-
ных металлов представляют собой солеподобные твердые соединения, которые не
удастся расплавить или перегнать без разложения. В специальных растворителях,
например в диметилццнке пли триметплалюминии, онп растворяются с образованием
комплексных анионов:
Me|[Zn(CHs)8R8]2'
Ути растворы обладают электропроводностью.
1. Литийорганические соединения
Литийорганические соединения чаще всего получают взаимодействием металли-
ческого лития с соответствующими органическими галогенидами:
2Li-(-RX — > LiR |-LiX
Циглер л Колоииус [17] впервые показали, что органические производные
щелочных металлов, образующиеся па первой стадпи реакции Вюрца, н определенных
условиях далее не реагируют и могут быть выделены в качестве основных продуктов
реакции. Этим методом поручено большое число литийорганических соединений [18].
Протекание реакции в значительной степени определяется применяемым галогенидом
и используемым растворителем. Легкость протекания реакции зависит также от содер-
жания патрия в исходном металлическом лптпи [19].
Для получения литийорганических соединений нельзя применять алкилиодиды,
за исключением метилиодпда, так как они слишком легко вступают в реакцию Вюрца
и поэтому дают плохие выходы литийорганических соединений. В алифатическом ряду
паилучпше результаты даст использование алкплхлоридов, а в ароматическом — арпл-
бромидов. Большое значение пмеет применение чистых галогенидов.
Хорошими растворителями для получения лпгийалкилов являются углеводо-
роды, например бензол, циклогексан, петролейный эфир; в тех случаях, когда образу-
ющиеся литийалкилы немедленно используют для дальнейших превращений, применяют
также эфир. Литийарилы заметно не взаимодействуют с эфиром, и для их получения
почти всегда используют эфир. Само собой разумеется, что растворители должны быть
абсолютно сухими и могут применяться только мосле продолжительного выдерживания
над металлическим натрием.
Реакцию следует проводить с измельченным литием для того, чтобы обеспечить
достаточно большую поверхность взаимодействия. Описаны различные методы измель-
чения лития [20].
Обычно получают литий в виде ленты шириной 1 см и толщиной 1 мм
с помощью пресса для натрия. Эту ленту выдавливают в абсолютный эфир
и затем разрезают ножницами на отдельные кусочки, сразу внося их в реакцион-
ную массу.
Для реакции не обязательно применять избыток металла, на 1 моль
галогенида берут 2—2,2 моль литпя.
[17] К. Ziegler, И. Colonius, Ann., 479, 135 (1930).
[18] Н. Gilman, Е. A. Z о е 11 n е г, W. И. Selby, J. Am. Chem. Soc., 54,
1957 (1932); 55, 1252 (1933).
[19] C. W. К a m i.e n s k i, D. L. Esmay, J. Org. Chem., 25, 1807 (1960).
[20] H. G i 1 m a n, W. L a n g h a m, F. W. M о о r e, J. Am. Chem. Soc., 62,
2327 (1940).
638
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
Получение и последующее взаимодействие литийорганических соединений нео^
ходимо проводить при полном отсутствии кислорода и двуокиси углерода. Обычно
синтез проводят в атмосфере азота. Азот очищают от кислорода пропусканием черв?
щелочной раствор пирогаллола, а затем сушат, пропуская через колонку с СаС1(
и трубку с РаО5. :
В качестве реакционных сосудов вначале использовали [17] трубки Шленк!
[21], а затем колбы Кьельдаля с газоотводной трубкой [22]. С успехом можно приме
нять также обычные приборы, состоящие из трехгорлой колбы, снабженной герметич1
ной мешалкой, обратным холодильником и капельной воронкой [20]. Воздух в прибо
рах вытесняют азотом и после загрузки растворителя и лития закрывают обратны!
холодильник ртутным клапаном. Можно проводить эту реакцию в пятигорлой колб
[23], снабдив ее дополнительно термометром для контроля темцературы и трубко!
для постоянного ввода азота.
Описана и другая аппаратура, применяемая для получения и дальнейшего
использования литийорганических соединений [24]. )
Литийалкилы. Общий метод получения в петролейном эфире [25]. В обыч^
ный прибор, состоящий из трехгорлой колбы с мешалкой, обратным холодил^--
ником и капельной воронкой, йомещают 50 мл сухого петролейного эфира
(т. кип. 28—38° С), не содержащего непредельных соединений, затем пропу-
скают азот и нарезают несколько более чем 0,7 г (0,1 г-атом) лития. PacTBOj
нагревают до кипекия и при интенсивном перемешивании прибавляют по каплям
в течение 1 ч раствор 0,05 моль алкилгалогенида в 50 мл петролейного эфира»
Перемешивание и нагревание продолжают до тех пор, пока заметно протеканий
реакции (0,5—1 ч). После охлаждения раствор в атмосфере азота переносят
в градуированный сосуд, из которого отбирают необходимое количество реак?
тива. Выход продукта от 50 до 85% от теоретического. -i
Фениллитий [34]. В трехгорлую колбу ёмкостью 2 л с герметично^
мешалкой, капельной воронкой и обратным холодильником помещают 500 жл
сухого эфира и пропускают азот. Затем добавляют 29,4 г нарезанного литий
и при перемешивании около 40 капель раствора 314 г бромбензола в 1 л сухого
эфира. Добавление остального раствора бромбензола продолжают после начал!
реакции, заметного по легкому помутнению реакционной массы. Как только
начинается интенсивное кипение, реакционную колбу охлаждают в бане с<5
льдом, следя за тем, чтобы кипение все же продолжалось. К концу реакция
баню со льдом удаляют и продолжают перемешивание смеси до прекращений
кипения. Продолжительность реакции составляет около 2 ч. Затем раствор
в атмосфере азота переливают через стеклянную вату в капельную воронку^
предварительно заполненную азотом. .
Для получения часто используемого в лабораторных целях н-бутпллития разра^
ботана методика, которая может использоваться также для синтеза других литий*
алкилов [26].
к-Бутнллнтий. В трехгорлую колбу емкостью 500 мл с мешалкой, низко-
температурным термометром и капельной йоронкой помещают 200 мл абсолют*
ного эфира. После заполнения прибора свободным от кислорода азотом в колбу
нарезают 8,6 г (1,25 г-атом) литиевой проволоки, продолжая пропускать
•азот. Затем при перемешивании из капельной воронки прибавляют около Зф
капель раствора 68,5 г (0,5 жолъ) н-бутилбромида в 100 мл эфира. Температуру:
реакционной массы понижают до —10° С, охлаждая ее смесью ацетона и сухого
льда. Начало реаиции определяется по легкому помутнению реакционной масс»’
21] W. Sc h 1 ей, A. Thai, Вег., 46, 2843 (1913).
22] G. Wittig, Angew. Chem., 53, 241 (1940).
23] Е. Т е г г е s, U. v. F е 1 d е et al., Erdol u. Kohle, 13, 84 (1960). : -i
24] R, Adams, H. Wolff et al., J. Am. Chem. Soc., 62. 1770 (1940). ' j
[25] H. Gilman, F. W. Moore, O. Baine, J. Am. Chem. Soc., 63^1
2479 (1941). ’
[26] H. G i 1 m a n, I. A. В e e 1 et al., J. Am. Chem. Soc., 71, 1499 (1949).
I. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ I ГРУППЫ QQQ
и ио появлению на поверхности лития светлых пятен. Остаток н-бутилбромида
равномерно прибавляют в течение 30 мин, поддерживая температуру внутри
колбы около —10° С. Затем реакционную массу перемешивают 1—2 ч, давая
ей нагреться до 0—10° С, после чего жидкость декантируют и фильтруют череа
узкую, набитую стеклянной ватой трубку в капельную .воронку, заполненную
предварительно азотом. Выход продукта 70—90% от теоретического.
Полученный таким образом эфирный раствор н-бутиллития не следует
длительно хранить и необходимо как можно скорее использовать для других
превращений.
Выделяющийся галогенид лития и непрореагировавший литий можно
отфильтровать в атмосфере азота. Описано большое число приспособлений,
применяемых для этой цели [20, 22, 23].
Содержание литийорганического соединения в реакционной массе определяют
разложением аликвотной части раствора водой и титрованием образующейся гидро-
окиси лития 0,1 п. НС1 по метиловому оранжевому. Затем можно рассчитать
количество раствора, необходимое для дальнейших превращений.
Для получения чистого литийорганического соединения, пе содержащего раство-
ритель, используют другой метод — взаимодействие металлического лития с ртуть-
органическими соединениями в инертных растворителях [27]:
R2Hg + 2Li —► 2RLi-]-Hg
Так как большая часть литийорганических соединений, особенно содержащих
высшие алкильные радикалы, достаточно хорошо растворима в углеводородах, их
растворы можно легко отделять от выделившейся ртути и после отгонки растворителя
получать чистые литийорганпческие соединения.
Этиллптпй [27]. К раствору 10 г дпэтилртути в 60 мл бензола нли
высококипящего лигроина прибавляют в атмосфере азота 5 г как можно
более мелко карезанного лития с блестящей поверхностью, после чего сосуд*
заплавляют и нагревают до 65° С. Через три дия реакция, поддерживавшаяся
частым встряхиванием, заканчивается. Реакционную смесь нагревают до 70° С
и фильтруют, не допуская контакта с воздухом. Из фильтрата при охлаждении
выделяется этиллитий в виде крупных кристаллов. Для более полного осажде-
ния раствор охлаждают льдом, продукт реакции отфильтровывают в атмосфере
азота, промывают холодным лигроином н сушат в токе азота.
Аналогичным методом в результате взаимодействия лития с бензилмагнийхло-
ридом удалось получить бензиллитий [28]:
CeH6CH2MgCl + 2Li —*- LiCI + Mg + CeH6CH2Li
Для синтеза литийорганических соединений используется также различная
растворимость некоторых металлоорганических соединений в инертных органических
растворителях. Например, при смешении растворов этиллития и диметилртути в бен-
золе или бензине образуется метиллитий, который очень плохо растворяется в этих
растворителях и выпадает в осадок [27]:
2C2H&Li + (CH3)2Hg —> 2CH3Li + (C2H5)2Hg
Металлирование некоторых реакционноспособных углеводородов литийоргани-
ческими соединениями представляет собой еще один метод получения отдельных труд-
нодоступных производных лития [29]. Так, при помощи литийорганичесиих соединений
можно замещать литием активный водород у атома углерода, активированного сосед-
ними арильными или винильными группами. {Кроме того, в некоторых ароматических
соединениях, например в эфирах фенолов или тиофенолов, атом водорода в орто-
положении может заменяться на литий под действием и-бутил- или фвииллития [30].
[27] W. Schlenk, J. Holtz, Вег., 50, 262 (1917)..
[28] К. Z i е g 1 е г, F. D ersch, Вег., 64, 448 (1931).
[29] Н. Gilman,.!, W. Morton, Org. Reactions, 8, 258 (1954).
[30] G. Wittig, U. Pock els, H. D roge, Ber., 71, 1903 (1938); H. G 11-
m a n, F. J. Webb, J. Am. Chem. Soc., 71, 4062 (1949).
640
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
2,6- Диметоксифениллитий [22]. Смесь 13,8 г (0,1 моль) диметилового
эфира резорцина и 0,1 моль фениллития в 400 мл абсолютного эфира оста-
вляют На 60 ч при комнатной температуре. Образующийся при этом 2,6-димет-
оксифениллитий выделяется в виде больших прозрачных кристаллов.
ОСНз ОСНз
1 1 Li
+c,H5Li —► /у +С,п„
Z'"'OCI|, ''^""'ОСИ,
Аналогичными свойствами обладают также гетероциклические соединения,
содержащие в цикле атомы кислорода, серы или азота [31]. Получаемые из них литий-
органические соединения могут, например, использоваться в качестве промежуточных
продуктов для введения других заместителей в гетероциклическую систему.
Литийорганические соединения реагируют с галогензамещенными органиче-
скими соединениями чаще всего по реакции, аналогичной реакции Вюрца — Фиттига,
с образованием соответствующих углеводородов. Но иногда, например в случае неко-
торых галогенированных гетероциклических соединений и ароматических простых
эфиров, атом галогена может замещаться на литий действием литийалкилов или литий-
арилов [32]. Так, при смешении эфирных растворов диметллового эфира 4-бромрезор-
цина и эквивалентного количества фениллития мгновенно с практически количествен-
ным выходом образуется 2,4-диметокспфениллитий [33]:
СН3О////ОСН3 СП3Очу, /ОСНз
|Х li -rCeH5Li —Y j| CeH5Br
'^Xet
Джонс л Джильмен [34] приводят обширный материал по этой теме.
2. Натрийорганические соединения *
Принципиально натрийорганические соединения можно получать теми же мето-
дами, что и литийорганические соединения.
В этом случае также нашло применение, особенно в последнее время, взаимо-
действие органических галогенидов (обычнохлоридов) с металлическим натрием в инерт-
ном растворителе:
RX+2Na —* RNa-|-NaX
Поскольку натрийалкплы и патрийарплы еще более реакционноспособны, чем
соответствующие органические производные лития (они значительно легче реагируют
с органическими галогенидами по реакции Вюрца — Фиттига или с некоторыми рас-
творителями), получение их должно проводиться при очень точном соблюдении опре-
деленных условий реакции. Кап л при получении литийорганических соединений,
реакцию осуществляют в атмосфере азота. В .качестве растворителей используют без-
водные и очищенные от олефинов углеводороды, иаггример пентан, петролейный эфир
л др. Образующиеся натрийалкплы и натрийарилы в этих растворителях нерастворимы.
* Классические исследования в области натрийорганических соединений проведены
П. П. Шорыгиным (Н.П. Ш о р ы г и н, Избранные труды, Изд. АН СССР,
1950 г., стр. 178—275). — Прим- ред.
[31] Н. Gilman, R. V. Young, J. Am. Chem. Soc., 56, 1415 (1934); 57, 1121
(1935); H. Gilman, R. L. В e b b, J. Am. Chem. Soc., 61, 109 (1939);
H, Gi Iman, A. L. Jacoby, J. Org. Chem., 3, i08 (1939); H. Gilman,
D. A. Shirley, J. Am. Chem. Soc., 71, 1870 (1949).
[32] W. Langham, R. Q. Brewster. H. Gilman, J. Am. Chem. Soc.,
63, 545 (1941); H. Gilman, H. B. Willis. J. Swis] owsky, J. Am.
Chem. Soc., 61, 1371 (1939); H. Gilman et al., J. Am. Chem. Soc., 62, 1843
(1940); 62, 2327 (1940).
[33] G. Wittig, U.fockels, Ber., 72, 89 (1939).
[34] R. G. Jones, II. Gilman, Org. Reactions, 6, 339 (1951).
I. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ I ГРУППЫ
641
поэтому взаимодействие должно проводиться с сильно измельченным натрием я при
интенсивном перемешивании. Обычно используют тонкую натриевую пыль или, еще
лучше, суспензию натрия (размер частиц мепсе 25 лк); применяемые для этого приборы
описаны различными авторами [35, 36].
Особенно подробно исследовано получение часто используемых для дальнейших
превращений и-амилнатрия [37] и фенилнатрия [38].
я-Амнлнатрий [39]. Натриевую пыль получают перемешиванием расплав-
ленного металла высокоскоростной мешалкой (10 000 об/мин) в течение 1 мин
в очищенном от олефинов декане при 110—130° С в атмосфере азота. Суспен-
зию охлаждают и передавливают жидкость через сифон азотом, остающуюся
натриевую пыль несколько раз промывают сухим пентаном.
В колбе емкостью 500 мл, снабженной скоростной мешалкой, к 23 г
(1 г-атом) натриевой пыли в 300 мл пентана в атмосфере азота при темпера-
туре от —10 до 0° С в течение 4 ч прибавляют по каплям 53 г (0,5 моль) чистого
амилхлорида. Выход н-амилнатрия около 80% от теоретического.
Во многих случаях реакцию можно проводить и с избытком натрия,
активируя его в начале реакции добавлением небольшого количества амило-
вого спирта (около 2 мл амилового спирта на 30 г патрия).
Фенилнатрнй [36]. В трехгорлой колбе в атмосфере азота к 54 г
(2,35 г-атом) натрия в мелкодисперсном состоянии (средний размер частиц
ниже 25 мк), суспендированного в 275 г толуола при 25—30° С, при легком
встряхивании прибавляют 10—15 мл раствора 112,6 г (1 моль) хлорбензола
в 100 г толуола. Реакция обычно начинается через 1—5 мин л сопровождается
быстрым повышением температуры. Остальной хлорбензол добавляют тогда,
когда начнется взаимодействие с первой порцией; скорость реакции контроли-
руют охлаждением реакционной смеси на бане с охлаждающей смесью (—20° С).
Температура реакционной массы не должна превышать 40° С. (Если взаимо-
действие через 20 мин не началось, то можно добавить 2—4 мл амилового
спирта. Сразу после этого реакционный сосуд надо помещать в охлаждающую
смесь, так как реакция начинается моментально.) Затем добавляют остальное
количество смеси хлорбензола и толуола. Скорость добавления и охлаждения
регулируют таким образом, чтобы образование фенилнатрия закончилось через
20—30 мин. Выход продукта 94—99% от теоретического.
Описана лабораторная аппаратура для непрерывного получения фенил-
натрия [40].
Ранее чаще всего применяли другой обшпй метод получения патрийорганиче-
ских соединений — взаимодействие металлического натрия с металлоорганическими
производными ртути, цинка, кадмия, свинца в инертных растворителях [27, 41}:
R2Hg + 2Na —> Hg + 2NaR
Этилнатрий [27]. В 40 мл сухого лигроина (т. кип. 100—150° С) вносят
такое количество нарезанного натрия, чтобы слой жидиости покрывал его.
Затем воздух полностью вытесняют сухим, тщательно очищенным от кислорода
[35] A. A. Morton, L. М. Redman, Ind. Eng. Chem., 40, 1190 (1948).
[36] I. F- Nobis,!.. F. Moormeie r, Ind. Eng. Cliem., 46, 539 (1954).
[37] A. A. Morton et al., J. Am. Chem. Soc., 58, 1697 (1936); 62, 1301 (1940);
64, 2242, 2247, 2250 (1942); 68, 93 (1946); 69, 172 (1947).
[38] M. В ockmiih 1, G. E h rh а г d, герм. пат. 62^875 (25/IV 1931); герм. пат.
633083 (30/IV 1931); С., 1937. II, 1082; герм. пат. 644486 (23/IX 1931): Н. G i 1-
m a n, H. A. Pacevitz, О. Baine, J. Am. Chem. Soc., 62, 1514 (1940);
A. A. M о r t о n, I. T. M a s s e n g a 1 c, J. Am. Chem. Soc., 62, 120 (1940).
[39] A. A. M о г t о n et al., J. Am. Chem. Soc., 62, 123 (1940); 72, 3785 (1950).
[40] H. R и s c h i g, R. Fugman n, W. Meixner, Angew. Chem., 70,
71 (1958).
[41] J. A. Wankly n, Ann., 107, 125 (1858); 108. 67 (1858); S. F. Acree, Am.
Chem. J., 29, 588 (1903); F. Hoin, H. Pctzchner et al., Z. anorg. allg.
Chem., 141, 161 (1924); F. H e i n, H. Schramm, Z. physik, Chem., A 151 r
234 (1930).
41 Заказ 1835,
642
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
азотом и добавляют 2 г диэтилртути. Сразу начинается реакция и через
1—2 ч на поверхности натрия осаждается прозрачный серо-зеленый осадок.
Реакционную массу охлаждают в течение 10 мин в эффективной охлаждающей
смеси и встряхивают, при этом продукт реакции отделяется от металла. Полу-
ченную суспензию в атмосфере азота сливают с избытка натрия, точнее с егс
амальгамы, этилнатрий отфильтровывают, несколько раз промывают сутям
лигроином и сушат в тоие азота.
Замещение водорода натрием, т. е. металлирование углеводородов натрийорга-
ническими соединениями, является еще одним из методов получения некоторых натрий-
органических соединений, синтез которых другими методами более сложен. Металли-
рование натрийорганическими соединениями протекает гораздо легче, чем с соответ-
ствующими литнйорганическими соединениями. Так, уже Шорыгин [42] наблюдал,
что при обработке бензола этнлнатрием образуется фенилнатрий:
CaHsNa + CeHe —► C2He-j-CeH5Na
О методах получения натрийорганических соединений, особенно о металлирова-
нии органических соединений натрийорганическимн соединениями, см. обзор [43].
Еще более реакционноспособными, чем натрийорганические соединения, яв-
ляются соответствующие производные калия. Если фенилнатрий можно получать
взаимодействием натрия с хлорбензолом в толуоле, то при реакции калия с хлорбензо-
лом в толуоле даже при низких температурах с хорошим выходом образуется бен-
зилкалпй [44]:
2К + С6Н5С1 —*- С6НбК + КС1
CeHgK-f-CeHsCHa —* СвШСНаК + СдНв
II. Органические производные элементов второй группы
Первый элемент второй группы — бериллий, как и литий в первой группе,
занимает особое положение. Диалкильные производные бериллия заметно отличаются
от аналогичных производных остальных щелочноземельных металлов, напоминая
во многом диалкильные производные элементов третьей группы. Пе содержащие
галоида диалкильные производные бериллия летучи и растворяются в неполярных
растворителях, например в бензоле, хотя они и представляют собой высокоассоцииро-:
ванные соединения; кроме того, все они, за исключением диметилбериллия, являются;
жидкостями. Соответствующие магнийорганпческио соединения имеют более ярко;
выраженный солевой характер — это неплавкие и практичесин нелетучие твердые,;
вещества.
О физических свойствах кальций-, стронций- и барийорганичесиих соединений^
в настоящее время известно мало.
В органических производных элементов главной’ подгруппы второй группы»’
в диалкилпроизводных цинка, кадмия и ртути металл-углеродная связь ковалентна;]
Эти соединения представляют собой обычные неассоцинроваппые жидкости с низкими^
температурами кипения, например, диметилцинк кипит при 46, диметилкадмий —.
при 105,5 и диметилртуть — при 92° С.
У органических производных элементов второй группы, как и у всех металле--
органических соединении, реакционная способность по отношению к кратным С—С-’
или С—N-связя’м уменьшается с увеличением электроотрицательности металла. Ана-w
логично ведут они себя по отношению к кислороду воздуха и воде; так, низшие алкиль-ч
ные производные, например метильные производные бериллия, магния и цинка, окис-';
ляются кислородом воздуха настолько бурно, что наступает самовоспламенение.']
Так же интенсивно протекает разложение их водой. Менее быстро реагирует диметнл-j
кадмий, а диметилртуть при комнатной температуре практически устойчива к дей-,’
ствию воды и кислорода воздуха.
[42] Р. Schorigin, Вег., 41, 2723 (1908).
[43] R. А. В е n k е s е г, D. J. F о s t е г et a]., Chem. Rev., 57, 867 (1957).
[44] Н. G i 1 m а и, Н. А. Расе vitz, О. Baine, J. Am. Chem. Soc., 62, 1514-
(1940].
II. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ II ГРУППЫ
64 3
1, Магнийорганические соединения
Магнийорганические соединения (реактивы Гриньяра) благодаря простоте
получения и легкости в обращении все еще наиболее часто используются в препаратив-
ной органической химии.
Практически единственным методом их получения является непосредственное
взаимодействие алкил- и арилгалогенидов с металлическим магнием *:
RX-j-Mg —► RMgX
Область применения этой реакции исключительно широка; практически все
алкил- и арилгалогениды, за исключением фторидов, могут реагировать с магнием,
образуя реактивы Гриньяра.
Магний обычно применяют в виде стружки («стружка Гриньяра»). Продажный
препарат, хранившийся в сухом месте, как правило, нс нуждается в дополнительной
очистке; при необходимости его можно промыть в реакционной колбе небольшим коли-
чеством сухого эфира. В отдельных случаях лучшие результаты получаются при
использовании порошкообразного магния с определенным размером частиц. Другие
металлы, содержащиеся в продажном препарате магния, реакции не мешают, они
остаются в виде черноватого порошка.
Количество магния, применяемого для реакции, обычно эквивалентно коли-
честву галогенида, хотя иногда следует применять избыток магния.
Из галогенидов чаще всего используют бромиды. Преимуществом их является
то, что они легче вступают и реакцию, чем аналогичные хлориды, хотя хлориды в неко-
торых случаях дают более высокие выходы магнийорганических соединений. Еще
более реакционноспособные подиды применяются редко, так как они более дороги
и чаще вступают в побочные реакции (например, в реакцию Вюрца). Применение йоди-
дов оправдано лишь в тех случаях, когда они более доступны, а также при получении
реактивов Гриньяра из высших алкил галогенидов, где стоимость иода уже не имеет
большого значения, или же при недостаточной реакционной способности соответству-
ющих бромидов.
Предварительная обработка различных галогенидов одинакова. Они должны
быть как можно более чистыми и не содержать кислот, спиртов и воды. Загрязнения
не только уменьшают выход магпийорганического соединения, по и могут привести
к тому, что взаимодействие не начнется или будет протекать с большим трудом. Гало-
гениды рекомендуется высушивать над безводным СаС12 и перегонять непосред-
ственно перед употреблением.
Как показали основополагающие работы Гриньяра [451 и последовавшие эа
этим, исключительно обширные исследования, важную роль при взаимодействии орга-
нических галогенидов с магнием играет растворитель. Обычно в качестве растворителя
применяется диэтиловый эфир. В отдельных случаях используют дипропиловый, дибу-
тиловый и диизоамиловый эфиры, а также диоксан и тетрагидрофуран. Эфир для
очистки от примесей спирта и воды длительное время выдерживают над натриевой про-
волокой. Иэоамиловый эфир рекомендуется несколько раз перегнать над металличе-
ским натрием**. Эфир обычно берется в таком количестве, чтобы к концу реакции
в 1 л раствора содержалось около 1—2 .моль магпийорганического соединения.
В этой реакции эфир играет, в первую очередь, роль растворителя магнийорга-
иических соединений, нерастворимых в большинстве растворителей; растворяя про-
дукты реакция, он освобождает^ поверхность магпия, что делает возможным дальней-
шее протекание реакции. Образующиеся магнийорганические соединения растворяются
в виде аддуктов с эфиром, образование которых сопровождается значительным выделе-
нием тепла, также способствующим протеканию реакции.
Эфираты магнийорганических соединений можно выделить в чистом виде. Они
представляют собой бесцветные кристаллические вещества состава: R2Mg-X2Mg’4RJO.
Кроме простых эфиров для ускорения реакции магния с алкилгалогенидом можно
применять другие соединения, способные к образованию продуктов присоединения,
* Иногда для этих целей применяют и реакцию металлирования, например в случае
ацетилена [Ж. И оц и ч, ЖРХО, 34, 242 (1902); 35,431 (1903); 38, 252 (1906)]. -
Прим. ред.
** Перед перегонкой простых эфиров во избежание взрывов следует убедиться
в отсутствии перекисей (проба с Nal и крахмалом). — Прим. ред.
[45] V. Grignard, С. г., 130, 1322 (1900).
41*
644
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
например дпалкильные производные других элементов шестой группы (серы, селена
Л теллура), а также третичные амины, например димстиланнлип, диэтцланилин, пири-
дин и хинолин.
Проводившиеся ранее попытки получения магнийоргапических соеди-
нений из магния и метил-, этил- и пропилиодпдов в отсутствие растворителя
дали неудовлетворительные результаты 1-46]. Позднее было показано [47], что
высшие иодиды, такие, как амилиодид, реагируют более гладко. Фенилмагний- -
хлорид образуется при нагревании хлорбензола с магнием в автоклаве при
150—170° С с выходом до 85% от теоретического [48].
Взаимодействие металлического магния с алкилгалогенидами можно
проводить также в среде углеводородов [49, 50], например в бензоле, толуоле •
и петролейном эфире, хотя образующиеся алкилмагнийгалогениды не дают
с этими растворителями аддуктов и не растворяются в них. Для ускорения
реакции можно добавлять небольшие количества эфира или третичного амина
(диметиланилин). "
Лучшие выходы [51] «индивидуальных реактивов Гриньяра» получают ‘
при добавлении к реакционной массе небольшого количества 10%-ного раствора ;
безводного Л1С19 в тетрагидрофуранс после проведения взаимодействия в бен-
золе. ',
Получение растворов магнийоргапических соединений проводят обычно в дву- '
гордой иолбе с обратным холодильником (широкий шариковый холодильник или У
холодильник Днмрота) п капельной воронкой. Некоторые преимущества дает исполь- <
зоваппр трехгорлой колбы — возможность перемешивания. Растворы магнийоргапиче- л
скпх соединений неустойчивы к действпю влаги, двуокиси углерода и кислорода,-
поэтому работы с ними следует проводить в соответственно защищенной аппаратуре. '
При простых непродолжительных операциях чаще всего можно обходиться только .<
защитой от влаги, закрывая обратный холодильник хлоркальциевой трубкой, лучшие
результаты дает комбинированная защита от влаги и С02 натронной известью.
Лишь в редких случаях принимаются особые меры по защите от кислорода *
воздуха. Находящийся над реакционной массой слой паров эфира служит обычно '
достаточной защитой, но все же иногда рекомендуется вытеснять воздух азотом, по t
крайней мере в начале реакции, п закрывать аппаратуру ртутным затвором. Описано ,
аппаратурное оформление получения магппйорганпческих соединений без доступа
кислорода воздуха [32].
• Реакция Гриньяра. Весь магний сразу загружают в реакционную колбу
и заливают абсолютным эфиром так, чтобы он был полностью покрыт слоем
жидкости *. Затем приливают небольшое количество галогенида; добавление
больших количеств галогенида нс рекомендуется, таи как поело начала взаимо-
действия скорость реакции быстро растет и наступает такое бурное кипение
яфира, что нормальный отвод тепла становится невозможным. Более тяжелый
органический галогенид необходимо осторожно приливать по стопке колбы
так, чтобы оп собирался под слоем эфира и па поверхности магния создалась
повышенная концентрация галогенида. После этого дожидаются начала реакции.
Ускорить начало реакции можно Добавлением кристаллика иода или нагрева-
нием на водяной бане до умеренного кипения раствора. При некотором навыке
[46]
[47]
[48]
[49]
[50]
[51]
[52]
Рекомендуется предварительно поместить под магний кристаллик иода,
колбу н магний нагреть голым пламенем до появления паров иода и охладить,
защищая от влаги. — Прим. ред.
Р. Lohr, Ann., 261, 48 (1891).
I. F. Spencer, Ber., 41, 2302 (1908).
" s m a n, R.E. Brown, J. Ara.
хх. х,х~,. ... .. _____ Chem. Soc., 52, 3330 (1930);
W. Issaguljanz et al., Her., 64, 2584 (1931).
z e f I, Ber., 37, 2081, 4534 (1904); 38, 3664 (1905); 40?
Н.
W. T s c h (
1487 (1907).
W. Schlenk jr., Ber., 64, 739 (1931).
F. I) r a h о w z a 1, II. К 6 n i g, H. Poll, Mh. Chem., 86, 419 (1955).
H. Gilman, P. Hewlett, Rec. trav. chim., 48, 1124 (1929).
И. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ II ГРУППЫ
645
начало реакции легко определяется по самопроизвольному кипепию эфира
и изменению внешнего вида металла, распадающегося на более мелкие кусочки.
Остальной галогенид добавляют затем из капельной воронки обычно в виде
эфирного раствора, регулируя скорость добавления таким образом, чтобы
эфир постоянно кипел, не перегружая обратного холодильника. Если насту-
пает слишком бурное кипение эфира, реакционную колбу охлаждают ледяной
или холодной водой. Проводя реакцию при охлаждении, можно также увели-
чить скорость добавления раствора галогенида и сократить тем самым время
реакции. Но слишком бурного протекания реакции следует избегать при всех
условиях. Для достижения возможно более полного взаимодействия оставшихся
небольших количеств металла и галогенида смесь после окончания реакции
еще некоторое время нагревают на водяной бане.
Если реакцию проводят с газообразными галогенидами, например с
GH3CI, СН3Вг или С2Н5С1, то капельную воронку в описанном приборе заменяют
трубкой для ввода газа. Галогениды отбирают из стального баллона или полу-
чают параллельно с основным опытом. Прежде чем вводить галогениды в реакцию,
их рекомендуется промыть в склянке Дрекселя водой или разбавленным раство-
ром щелочи и высушить, пропуская затем через две промывные склянки с кон-
центрированной H2SO4 и, если необходимо, через трубку с СаС12.
Получаемые таким образом растворы реактивов Гриньяра можно непо-
средственно применять для дальнейших превращений. Их можно хранить
в защищенной от света и воздуха посуде. Так, например, растворы этил- и фенил-
магнпйбромидов не изменяются даже через несколько месяцев [53].
Образование магнпйорганических соединений из арилиодидов и арилбромидов,
первичных алкилиодидов, алкилбромидов и алкплхлоридов протекает легко; хорошо
реагируют также галогениды бепзпльного и аллильного типа.
С большим трудом протекает взаимодействие с третичными алнплгалогени-
дами. Арилхлорццы реагируют с магнием настолько медленно, что применение их
для препаративных целей нецелесообразно. В этих случаях реакцию можно уско-
рить, либо проводя ее при более высокой температуре, т. е. используя более высоко-
кипящио растворители (дибутилопый эфир или диметиланилин), либо применяя
растворители с более ярко выраженным донорным эффектом, чем в случае диэтило-
вого эфира (тетрагидрофуран или диметиловый эфир этиленгликоля). Так, исполь-
зуя в качестве растворителя тетрагидрофуран, получают вицилмагнийгалоге-
1ш)(Ы [54].
Взаимодействие магнпя с мало реакционноспособными галогенидами, например
с хлорбензолом пли вигтилгалогенпдами, можно проводить, смешивая инертные гало-
гениды с более реакционноспособными галогенидами и проводя реакцию с таким коли-
чеством магния, чтобы его было достаточно для превращения обоих галогенидов;
прп этом получают смесь двух магпийорганическпх соединений. Таким ?ке образом
можно проводить реакцию со стерически затрудненными галогенидами типа пептамегил-
бромбсизола. Прибавляемый реакционноспособный галогенид подбирают так, чтобы
при дальнейших превращениях получались легко отделяемые побочные продукты;
чаще всего применяют этилбромид.
Реакция активируется также добавлением небольшого количества предвари-
тельно приготовленного раствора соединения Гриньяра к смеси эфира, магния и гало-
генида.
Еще одна возможность осуществления реакции магния с нереакционпоспособ-
ными галогенидами заключается в применении активированного магния, который полу-
чают нагреванием магниевых опилок с иодом [55]. Болес реакционноспособный продукт
образуется при активации иодом сплава магния с медью [56].
Синтез реактивов Гриньяра из соединений, содержащих более одного атома
галогена, во многих случаях ие удается проиестп и до настоящего времени. Если атомы
[53] Н. Gilman, С. Meyers, Ind. Eng. Chem., 15, 61 (1923).
[54] H. N о г ш а и t, С. г., 239, 1510 (1954); С., 1955, 2656.
[55] А. Ваеуог, Вег., 38, 2759 (1905).
[56] Н. Gilman, J. М. Peterson, F. Schulze, Rec. trav. chim., 47, 19
(1928).
646
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
галогена разделены двумя или тремя углеродными атомами, как, например, в эплен-
дибромиде пли триметилендибромице, единственной реакцией является л-ппеплапид.
галогена и образование галогенида магния. В то же время метилендибромнд или мети-
лендииодид дают соответствующие метилендимагнийгалогениды с очень хорошими
выходами [57]. Димагнийоргапические соединения можно получать также из дигало-
генидов, в которых атомы галогена разделены четырьмя и более углеродными атомами,
Так, при взаимодействии тетраметплепдибромида с магнием с хорошим выходом полу--
чается тетраметилендимагнийбромид [58]. Высшие гомологи показывают некоторую ,
склонность к образованию продуктов конденсации, например, пеитаметилендибромид.
частично превращается в декаметилендимагнийбромид.
Ароматические дигалогениды плохо реагируют с магнием, но реакцию можно/
инициировать, используя сплав магния и меди, активированной иодом (см. выше).^
Так, папример, из n-дибромбензола получают л-бромфенилмагнийбромид и п-фенилевк^
днмаглийбромид [59].
При низких температурах действием перфторалкилиодидов на магний можно?
получать фторсодержащпе магнийорганические соединения; так, при взаимодействие
гептафторпропилиодида с очень чистым магнием при —30° С образуется гептафторч.-'!
пропилмагнийиодид с выходом 55—60% от теоретического [60].
Побочцой реакцией, протекающей в большей или меньшей степени при синтез^:
магнийорганических соединений, является металлоорганический синтез:
RX-LRMgX —> R-R+MgX3
Эта нежелательная побочная реакция в гораздо большей степени протекает^
в случае иодидов; чем для соответствующих бромидов и еще менее реакциоццоспособ-
пых хлоридов. Кроме того, опыт показывает, что доля побочной реакции увеличивается•.
при форсированном проведении взаимодействия магния с галогенидом. Поэтому для?
получения возможно более высокого выхода магнийорганического соединения реакцию-,
необходимо проводить в самых мягких условиях. Органический галогенид следу»;
прибавлять с минимальной скоростью, достаточной для поддержания реакции. Реак-;.
циоиную массу интенсивно перемешивают, благодаря чему уменьшается опасностью
местного повышения концентрации органического галогенида и тем самым подавляется
побочная реакция, приводящая к образованию углеводорода. По этой же причин»
с лучшими результатами протекает взаимодействие в больших количествах рас-?/
творителя. I
Другая побочная реакция, чаще всего наблюдаемая при использовании третич-’ >
пых галогенидов, заключается в отщеплении галогеноводорода:
2НаС—CRg + Mg —> 2R2C=CR2 + H2-]-MgX2
Для качественного определения магнийорганических соединений предложена
общая цветная реакция (стр. 721).
Часто для дальнейшего препаративного использования необходимо определение,;
концентрации раствора реактива Гриньяра; с этой целью его титруют кислотой ПОч
методу Гильмена [61].
Определение концентрации раствора реактива Гриньяра. Аликвотную^
часть раствора реактива Гриньяра,"около 20 мл, отмеряют в мерном цилиндр»
с точностью до 0,1 мл и медленно выливают в коническую колбу емиостью.
400 мл, содержащую 50 мл дистиллированной воды. Мерный цилиндр промы-
вают сначала несколькими миллилитрами H2SO4 с точно известной концен*?
трацией (примерно 0,25 н.), затем несколько раз дистиллированной водой-
Все промывные воды помещают в коническую колбу с основным раствором.
После этого добавляют около 20 мл H2SO4 (до избытка) и нагревают снес»1
[57] G. Е m s с h w i 1 1 er, С. г., 183, 665 (1926).
[58] Т. v. Braun, W. S о b е с k 1, Вег., 44, 1918 (1911); Е. В п с h t а, Н. We К
dinger, Ann., 580, 109 (1953).
[59] Н. Gilman, J. М. Peterson, Proc. Iowa Acad. Sei., 33, 173 (1926);
C. A., 21, 3901 (1927).
[60] R. N HaszeldinXJ. Chem. Soc., 1952, 3423; 1953, 1748.
[61] H. Gilman et al., J. Am. Chem. Soc., 45, 150 (1923).
II. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ II ГРУППЫ
647
для завершения взаимодействия; затем раствор охлаждают и отгитровывают
избыток H2SO4 раствором NaOH по метиловому оранжевому. Расчет произво-
дится на основе следующего уравнения реакции;
2RMgX + H2SO4 —> 2RH + MgSO4-J-MgX2
В результате обычно получают несколько завышенные значения, ио
ошибки лежат в пределах, допустимых для препаративных целей.
Подробнее о синтезе реактивов Гриньяра см. в книге [62].
Самым простым методом получения «чистых» (свободных от галогенов) магний-
оргапических соединений является добавление диоксана к соответствующему раствору
реактива Гриньяра в эфире; при этом осаждается MgX3 и часть алкилмагнийгалоге-
япда. тогда как RgMg остается в растворе £63]:
R2Mg-MgXa —> RgMg + MgXa
При немедленном фильтровании выходы магнийоргапических соединений
составляют от 6% (при использовании этилмагнийиодида) и до 84% (в случае бутил-
магнийхлорида) [64]. Выходы повышаются при продолжительном встряхивании рас-
твора с осадком [65], а также при медленном прибавлении небольших порций диоксана
в течение трех дней [66].
Другим методом получения магнийдиалкилов и магпийдиарилов является взаи-
модействие магйия с соответствующими ртутьорганическими соединениями [67]:
RaHg + Mg —> R2Mg— Hg \
Диалкил- и диарилпроизводпые магния представляют собой бесцветные, кри-
сталлические, неплавкие, нерастворимые в углеводородах и практически нелетучие
вещества. В то же время все эти соединения растворимы в эфире или в других раство-
рителях, образующих комплексы за счет координационных связей, например в диок-
сане я пиридине. Эфират дифенилмагния растворяется также в бензоле, тогда как
несольватированное соединение в нем нерастворимо.
Алкильные производные магния, несмотря па их высокую реакционную способ-
ность, Цвляются сравнительно устойчивыми соединениями. Так, например, медленное
разложение диэтнлмагния начинается лишь при температурах выше 175° С; при этом
образуются гидрид магния, этилен и незначительные количества полимерного этилен-
магния (—CaH4Mg—)„ и этана [68]:
(CaHshMg 17—"° С:дта^ 2caH4+MgHa
2. Цинкорганические соединения
Цинкалкилы являются первыми из полученных металлоорганических соедине-
ний. Они были открыты в 1849 г. Франкландом [69] при попытках получить свобод-
ные этильные радикалы действием этилиодида на цинк. С этого открытия, по сути дела,
началась химия металлоорганических соединений.
Взаимодействие алкилиодидов с цинком [70] в несколько модифицированном
виде применяется и в настоящее время для синтеза алкилципкиодидов; вместо цинка
[62] М. S. Kharasch, О. R е in mu th, Grignard Reactions of Nonmetallic
Substances, Prentice-Hall, Inc., New York, 1954.
[63] W. Schlenk jr., Ber., 64, 734 (1931).
[64] G. 0. Johnson, H. Adkins, J. Аш. Chem. Soc,, 54, 1943 (1932).
[65] C. R. NoIler, W. R. White, J. Am. Chem. Soc., 59, 1354 (1937).
166] R. R ullman, C. r., 231, 866 (1950).
[67] H. Gilman.F. S c h u 1 z e, J. Am. Chem. Soc., 49, 2328 (1927); W. Schlenk
jr., Ber., 64 , 736 (1931).
[68] E. Wiberg, R. Bauer, Chem. Ber., 85, 593 (1952).
[69 E. Frank la nd, Ann., 71, 171, 213 (1849).
[70] E. Frankland, Ann., 95, 28 (1855); 111, 44 (1859).
648
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
используют цпнк-медную пару (нечто среднее между сплавом и тщательно гомогенизи-
рованной смесью), получаемую нагреванием смеси цинковой стружки с медным порош-
ком [71].
Еще более активный материал, чем цпнк-медпая пара, образуется при нагрева-
нии смеси цинковой пыли и цитрата меди до полного разложения последнего [73].
Описана [72] подробная методика получения диэтилципка взаимодействие»
цпп к-медном пары с этилиодидом:
RI + Zn(Cu) —► RZnI
. нагревание _ _ , „ т
2RZnI ----------* R2Zn-|-Znl2
Образующиеся по этой реакции алкилцинкгалогениды представляют собой
бесцветные кристаллические соединения, которые обычно пе выделяют, разлагая их
при нагревании до диалкильных производных. Хорошие результаты дает использо-
вание первичных или вторичных алкилиодидов (иногда в смеси с бромидами). Реакцию
проводят в атмосфере азота или углекислого газа.
Присутствие растворителя при получении цинкоргаппческих соединений не1
обязательно, но в некоторых случаях растворитель все же применяют для растворения
продукта реакции. Чаще всего в качестве растворителя используют этилацетат, обычно- ;
в смеси с толуолом, значительно ускоряющий взаимодействие. В качестве растворителя
можно применять диэтиловый эфир, хотя он и не оказывает ускоряющего действия;,
для более успешного протекапия реакции добавляют небольшое количество иода. Если :
необходимо проводить реакцию при более высоких температурах, используют диизо-
пропилопый пли дибутиловый эфиры |74] (см. реакцию Реформатского, стр. 751).
Растворы диарил- и диалкилпроизводных цинка, а также галогенсодержащих
цинкорганических соединений получают, кроме того, добавлением безводного
к растворам магштйорганических соединений в эфире:
ZnCh + RMgX —> RZnCl + MgXCl
ZnCl2-[-2RMgX —> R2Zn + 2MgXCl
Низшие, более легко летучие циякалкилы, например диметил- и диэтилцинкг
могут быть легко выделены из раствора перегонкой. Однако выделение этих соедине-
ний в чистом виде не всегда обязательно, обычно можпо применять их растворы.
Аналогично диалкилпроизводным магния цинкдиалкилы образуются при дей-
ствии металлического цинка на соответствующие органические производные ртутит
Zn |-R2Hg —>• R2Zn+IIg
Чистые цинкдиарплы лучше всего получаются взаимодействием цинка с ариль- ;
ними производными ртути в кппящем ксилоле [75] или взаимодействием ZnCl2 с фонил-
литие.м |76].
Цинкорганическпе соединения из-за более низкой реакционной способности и
сложности в обращении применяются гораздо реже, [чем соответствующие магнийорга-
нические соединения. Но они заменяют магнийорганические соединения в тех случаях,- 2
когда, например, появляется необходимость в применении менее реакционноспособ- ?
ного реактива. Так, взаимодействием хлорангидридов кислот с ципкорганическим® ?
соединениями получают соответствующие кетоны (стр. 761):
RCOCl + R'ZnI (пли R2Zn) —> RCOR' •
Прп использовании реактива Гриньяра образующийся па первой стадии кето» Vl
частично превращается в соответствующий третичный спирт. 3
[71] A. J о h, R. R е i с. h, Bull. Soc. chim. France [4], 33, 1414 (1923).
[72] C. R. Nollet, Org. Synth., Coll. v. 2, 1943, p. 184.
[73] R. C. Krug, P. 3. C. Tang, J. Am. Chem. Soc., 76, 2262 (1954).
[74] L. F. Hatch, G, Sutherland, W. J. Ross, J. Org. Chetn.. 14, 113(У
(1930),
[75] К. A. Kozeschkow, A. N. Nesmejanow, W. ]. Potrosow, Ber.r
67, 1138 (1934). ’
[76] W. S trohtneier, Chetn. Ber., 88, 1218 (1955),
II. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ II ГРУППЫ
G49
Цинкдиалкилы — бесцветные, устойчивые жидкости; без доступа кислорода
воздуха они могут храниться неограниченно долго и не разлагаться, в процессе пере-
гонки выдерживают температуры до 200’ С. Цинкдпарилы при комнатной темпера-
туре представляют собой твердые кристаллические вещества с характерной темпе-
ратурой плавления.
3. Кадмийорганические соединения
Лить после многих попыток был осуществлен синтез некоторых алкильных
производных кадмия с удовлетворительными выходами п в достаточно чистом виде.
Эти соединения обладают гораздо меньшей термической устойчивостью, чем все дру-
гие органические производные элементов второй группы. Из алкилпроизводных кадмия
только диметилкадмпй может храниться некоторое время. Высшие гомологи быстро
разлагаются уже при комнатной температуре, особенно под действием света. Более
устойчив твердый дифенилкадмий.
При действии алкилгалогенида на металлический кадмий [46, 77] на первой
стадии реакции, как и для всех других элементов второй группы, вероятно, образуется
соединение типа RCdX; но до настоящего времени такие соединения не получены в чис-
том виде и однозначно пе идентифицированы; изучены только соединения общего
состава R2Cd. Эти соединения рекомендуется получать взаимодействием соответству-
ющих реактивов Гриньяра или литийорганических соединений с безводным хлористым
кадмием [78J:
2RMgX-J-CdCl2 —> R2Cd-)-2MgXCl
Диметилкадмий. Обычным способом из метилбромида и магния получают
раствор 0,3 моль метилмагнийбромида. К охлаждаемому льдом реактиву Гринь-
яра медленно прибавляют 0.1G моль топко размолотого безводного CdCl2.
Баню со льдом удаляют и продолжают перемешивание реакционной массы
в течение 30 мин. Конец реакции определяют, убедившись при помощи цвет-
ной реакции, в отсутствии магнийорганического соединения (стр. 721).
Диалкильные производные кадмия можно выделять осторожной перегонкой
на водяной или масляной бане при пониженном давлении. Дифенилкадмий очищают
высоковакуумпой сублимацией [76]. Но в большинстве случаев избегают выделения
кадмийоргапических соединений и для дальнейших превращений непосредственно
попользуют реакционные массы, полученные по приведенному выше способу.
Кадмийорганпчебкие соединения обладают лишь незначительной реакционной
способностью по отношению к карбонильным группам и применяются поэтому, так же
как и цинкорганпческие соединения, для синтеза кетонов из хлорангидридов кислот
(стр. 760 сл., см. обзор [79]).
4. Ртутьоргаиические еоедииеиия
Ио ртутьорганическим соединениям проводилось большое количество исследова-
ний. в частности в поисках химиотерапевтических препаратов. Широкое применение
находят ртутьоргаиические соединения в качестве фунгицидов для защиты растений.
Число известных органических производных^ртути исключительно велико. Разрабо-
тано много методов получения самых разнообразных ртутьорганическпх соединений.
Исключительная токсичность органических производных ртути требует особой
осторожности прп синтезе и обращении с ними. Токсичность многих ртутьорганическпх
соединений превышает токсичность ртути. Особеппо опасны легколетучие алкильные
производные ртути.
Ртутьоргаиические соединения мало реакционноспособны. При нормальных
условиях они устойчивы к действию кислорода воздуха и воды, поэтому их получение
и использование могут проводиться в таких условиях, которые были бы недопустимы
для других, рассматривавшихся выше, металлоорганических соединений.
[77] L. A. W а и k 1 у и, J. Chem. Soc., 9, 193 (1857).
[78) И. Gilman, J. F. Nelson, Rec. trav. chim., 55, 518 (1936); E. Krause,
Ber., 50, 1813’ (1917).
[79] J. Cason, Chem. Rev., 40, 15 (1947).
650
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
Некоторые алкплподиды реагируют с металлической ртутью на солнечном свету
и в присутствии небольшого количества элементарного пода с образованием алкил-
ртутьиодидов [80]. Но эта реакция протекает слишком медленно.
Общеприменимым методом получения ртутьдиалкплов и ртутьдиарилов является
взаимодействие алкил- и арилгалогенпдов илн алкилсульфатов с амальгамой
натрия 181]:
2RX + 2Na + Hg —»- R2Hg ]-2NaX
Диметилртуть [82]. Смесь 10 вес. ч. метилиодида и 1 вес. ч. абсолютного
метилацетата обрабатывают 0,2%-ной амальгамой натрия (1,2 г-атом натрия
на 1 моль метилиодида). Реакцию проводят в круглодонной колбе с эффектив-
ным обратным холодильником; колбу время от времени встряхивают и охлаж-
дают водой, но не льдом, так как при охлаждении льдом реакция сильно замед-
ляется. Реакцию можно считать законченной, когда температура смеси пони-
жается и при кипячении нескольких капель реакционной смеси с азотной
кислотой выделяются лишь следы иода. Если реакционная масса слишком
густеет от выделяющегося KI, затрудняя доступ реагентов к амальгаме натрия,
рекомендуется непрореагировавший метилиодид и растворитель отогнать на
водяной бане и обработать в другой колбе свежей-, амальгамой натрия. После
окончания реакции к реакционной смеси добавляют воду и перегоняют на масля-
ной бане, температура которой не должна превышать 110° С. Похожий на эфир
дистиллят отделяют от воды и встряхивают со спиртовым раствором КОП для
удаления метилацетата, затем хорошо промывают водой и перегоняют; т. кип.
продукта 95° С.
Наиболее удобным методом получения простых диалкил- и диарилпроизводных
ртути служит взаимодействие соответствующих реактивов Гриньяра [83] или других
реакционноспособных металлоорганических соединений [84J с HgCl2:
RMgX + HgCl2 —> RHgCl + MgXCl
RMgX-f-RHgCl —> R2HgH-MgXCl ;
Те же самые продукты образуются, если реакцию проводят с солями одновалент-
ной ртути; при этом одновременно выделяется металлическая ртуть. '
Дифенилртуть [85]. Из 50 г бромбензола в 200 мл эфира получают^
реактив Гриньяра, Остатки магния отфильтровывают через стеклянную вату |
и соединяют реакционную колбу с небольшим экстракционным аппаратом.?
Сокслета. В экстракционную гильзу, предварительно высушенную в вакуум-$
эксикаторе, помещают 26 г тонко измельченного HgCl2; эфирный раствор кипя-^
тят на водяной бане до полного растворения сулемы. Спустя 1 ч смесь охлаж-^
дают и добавляют ледяную воду и разбавленную уксусную кислоту. Если про-1
дукт реакции выпадает в осадок, его переводят в раствор добавлением бензола;!
затем органический слой отделяют, сушат над Na2SO4 и отгоняют растворитель.1^
Остаток после отгонки растворителя состоит из дифснилртути и небольшого^
количества хлорида фенилртути. Эти соединения разделяют обработкой эфиром, d
в котором хлорид фенилртути растворяется очень плохо. Выход дифенилртутИ-ад
после перекристаллизации йз спирта 25 г (75% от теоретического, считая ид и
HgCI2); т. пл. 125° С; выход хлорида фенилртути 6 г. J
[80] J. L. М а у н а г d, J. Аш. Chem. Soc., 54, 2108 (1932). А
[81] К. Fuchs, J. prakt. Chem. [2] 119, 209 (1928). Ч
[82] W. Schlenk, в книге Ilouben-Woyl, Methoden dor organischen Chemie.,|
2 Aufl., 1924, S. 924. 1
[83] C. S. Marvel, H.O. Cal very, J. Am. Chem. Soc., 45, 820 (1923)f|
P. Pfeiffer, P. Truskier, Ber., 37, 1125 (1904); H. G i 1 in a n,-a
R. E. Brown, J. Am. Chem. Soc. 52 , 3314 (1930). ,!
[84] G. B. Buckton, Ann., 109, 218 (1859); R. C. Freidlina, A. N. Ne
mejanow, K. A. Kozeschkow, Ber., 68, 565 (1935). $
[85] W. S c h 1 e n 1», в книге Houhen-Wey], Methoden der organischea Chemie,J
2 Aufl., 1924, S. 927. 1
[Г, ОРГ4НИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ II ГРУППЫ
651
Хотя первая стадия этой реакции протекает намного быстрее, чем вторая, од.
нако получать таким образом моногалогеяиды органических производных ртути нецеле-
сообразно; известен более простой метод синтеза этих соединений:
R2Hg-| HgX2 —> 2RHgX
Несимметричные диарил-, диалкил- и жирно-ароматические производные ртути
образуются при взаимодействии галогенидов ртутьоргапическпх соединений с соответ-
ствующими магнийорганическими соединениями [86]:
RHgX+R'MgX —> RHgR'4-MgX2
Диарильные производные ртути можно синтезировать также восстановлением
арилгалогенидов ртути или других солей арилртути, которые легко получить описан-
ным ниже методом. В качестве восстановителей используют гидразингидрат [87],
станнит натрия [88], магний [89], медь в пиридине [90] или проводят электрохимиче-
ское восстановление; например, при использовании станнита натрия реакция протекает
по схеме:
2CeI-IsHgXH Na2SnO2 + 2NaOH —► (CeH6)2Hg-h Hg-f-2NaX+Na2SnO3+Н2О
Возможно также диспропорционирование галогенидов органических производ-
ных ртути на R2Hgn HgXs в присутствии соответствующих добавок, например, иодида.
калия, цианида или тиоцианата калия, связывающих выделяющийся галогенид ртутж
в виде комплекса K2HgX« [91 [:
2RHgI + 2KI —> R2Hg-f-K2HgI4
Для непосредственного получения солей арилртути в основном применяются
два метода: прямое меркурирование ароматических соединений и реакция Несмеянова.
, По реакции Несмеянова двойные соли хлорида ртути и солей диазония разла-
гают действием порошкообразной меди па холоду в ацетоне [92]:
: ArN2Cb HgCl2 + 2Cu —> ArHgCl + 2CuCl + N2
Если реакцию проводят с избытком медного порошка и после прекращения
выделения азота прибавляют концентрированный раствор аммиака, то образующийся
на первой стадии ArHgCl с хорошим выходом дает Ar2Hg.
Дифенилртуть. Сначала получают двойную соль хлорида диазония
и ртути, CflHjNjjCl • HgClz. Для этого обычным способом диазотируют анилин
в соляной кислоте при хорошем охлаждении. Если раствор диазосоединения
мутный, его быстро фильтруют через стеклянный пористый фильтр и при постоян-
ном перемешивании сразу приливают к приготовленному заранее эквимоляр-
ному раствору HgCl2 в равном весовом количестве концентрированной НС1
с тем же количеством льда, которое применялось для диазотирования. Осадок
отделяют на воронке Бюхнера, тщательно промывают водой, спиртом и эфиром
и сушат па воздухе. Смешивают 10 г полученной двойной соли с 8 г приготовлен-
ного по Гаттерману порошка меди и заливают 50 мл ацетона, охлажденного
смесью льда и соли. После окончания первой энергичной стадии реакция
к смеси при тщательном перемешивании добавляют равный объем 25%-ного
[86] S. Н i 1 р е г t, G. G г ii I t п е г, Вег., 48, 906 (1915).
[87] Н. Gilman, М. М. Barnett, Rec. trav. chim., 55, 563 (1936).
[88] 0. Dimroth, Ber.. 35, 2853 (1902); C., 1901, I, 449; W- Steinkopf,
W. Bielenberg, H. Augestad -J ensen, Ann., 430, 41 (1923);
J. L, Maynard, J. Am. Chem. Soc.. 46, 1510 (1924),
[89] F. R. J ensen, J. A. Landgrebe, J. Am. Chem. Soc., 82, 1004 (1960).
[90] F. Hein, K. W a g 1 e r, Ber., 58, 1499 (1925).
[91 ] F. C. W h i t ш о г e, R. J. Sobatzki, J. Am. Chem. Soc., 55, 1128 (1933);
R. W. В eattie, F. C. Whitmore, J. Am. Chem. Soc., 55, 1567 (1933).
192] A. N. Nesmejanow, Ber., 62, 1010, 1018 (1929); Org. Synth., Coll. v. 2,
432 (19431; A. N. Nesmejanow, N. Th. G1 uschnew et al., Ber., 67,
130 (1934k
652
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗКИ
водного аммиака и оставляют на 12—24 ч. Затем добавляют избыток воды для'
осаждения ртутьоргаппческого соединения. Осадок отфильтровывают и про-
мывают небольшим количеством эфира. Полученное симметричное ртутьорга-
иическое соединение извлекают бензолом и перекристаллизовывают. Выход
дифенилртути 65% от теоретического; т. пл. 125° С. Примесь галогена удаляют
кипячением с медью в пиридине.
'Одним из вариантов метода является разложение хлоридов арилдиазония непо—[
срсдствснпо мелкодисперсной металлической ртутью при 0—5° С [93]: *
ArN2C14-Hg —► ArHgCl + N2
К этому методу можно также отнести взаимодействие тстрафторборатов арил-
диазония с HgCl2 в присутствии SnCl2, при котором промежуточно образуется мелко-'
дисперсная ртуть [94].
Очень легко соли арилртути получаются при моркурпрованип ароматических *
соединений солями ртути, т. е. прямым замещением водорода на ртуть:
Arn + HgX2 —> ArHgX + HX
Ута реакция протекает не только с соединениями ряда бензола, по и с гетеро-'1
циклическими ароматическими соединениями, например с тиофеном, фураном и пири-
дином. Осуществлено прямое мернуриронание тиофена хлоридом ртути [95].
Таким образом, можно, например, отделять тиофен от бензола. При нагревании
технического бензола с иодным раствором ацетата ртути выделяется димеркурирован-
ный тиофен (а-ацетоксимеркурп-а'-оксимеркуритиофсн) [96]; примерно через 30 ми»
бензол, практически не реагирующий при этих условиях, освобождается от примеси
тиофена.
Образование ртутьорганичсских соединений может протекать также при дей-
ствии ацетата ртути на гомологи бензола [97]. О дальнейших исследованиях по мерку-
рированию ароматических соединений и теоретических аспектах реакции см. [98].
Скорости меркурцрования ароматических соединений различны; особенно легко-
реагируют амины, фенолы п ароматические простые эфиры, медленное — ароматические-'-
углеводороды, например бензол, и особеппо трудно меркурируются галоген- и нптро- :
замещенные производные бензола.
Взаимодействие ароматических углеводородов с ацетатом ртути может прово-
диться продолжительным кипячением их с обратным холодильником, причем ацетат
фенилртути образуется, например, с выходом 80% от теоретического. При нагревании;
бензола с ацетатом ртути в ледяной уксусной кислоте под давлением при Ц0° С про-
должительность реакции сокращается до 2 ч, а выход повышается до 92% от теорети-
ческого [99].
Ацетат фенилртути [100]. В колбе с обратным холодильником нагревают *
па водяной бане 15 г ацетата ртути, 80 мл бензола и 20 мл спирта. Через 5 ч -
добавляют еще 20 мл спирта. Образующийся желтый осадок растворяют
в нескольких миллилитрах ледяной уксусной кислоты. Через 55 ч раствор фильт- ’
руют, фильтрат упаривают и‘перекристаллизовывают ацетат фенилртути иа
спирта. Выход продукта 12,6 г (80% от теоретического); т. пл. 149D С. •
193] R. Е, М с С 1 и г е, Е. L о W у, J, Am. Chem. Soc., 53, 319 (1931).
[94] М. F. W. Dunker, E. B.’Starkey, G. L. Jenkin s, J. Am. Chem- «
Soc., 58, 2308 (1936),
[95] J. Volhard, Ann., 267, 172 (1892).
[96] O. Dimroth, Ber., 32, 759 (1899).
[97] O. Dimroth, Ber., 31, 2154 (1898); 35, 2853 (1902).
[98] M. S. Kharasch, L. C h a 1 k 1 e у jr., J. Am. Chem. Soc., 46, 1211 (1924)8
M. S. Kharasch, J. M. J a cofis oh n, J. Am. Chem. Soc., 43, 1894 [
(1921).
[99] K. A. Kobe, P.* F. Luclh jr., Ind. Eng. Chem., 34, 309 (1942).
[100] J. L. Maynard, J. Am. Chem. Soc., 46, 1510 (1924).
и. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ II ГРУППЫ
653
Дифенилртуть из этого соединения получают следующим образом. Сме-
шивают 30 г ацетата фенилртути в 300 мл воды, 125 мл 40%-ного раствора
NaOH и раствор 50 г SnCl2 • 2Н2О в 125 мл воды и перемешивают 1 ч. Отфильт-
ровывают серый осадок, состоящий из металлической ртути и дифенилртути,
добавляют для амальгамирования ртути цинковую пыль и дважды экстраги-
руют продукт 75 мл ацетона. Из полученного раствора дмфенилртуть осаждают
водой и перекристаллизовывают. Выход дифенилртути 13,2 г (95,6% от теоре-
тического); т. пл. 124,5° С.
Заместители, имеющиеся в ароматическом ядре, направляют ртутный остаток
прп реакции меркурирования так же, как и в случае других реакций электрофильного-
замещения: заместители первого рода — главным образом в орто- и пара-положение,
заместители второго рода — в мета-положение. Так, например, при меркурировании
толуола в первую очередь образуются орто- п пара-производные, а из нитробензола —
в основном мета-изомер. Все же замещение действием ацетата ртути, особенно при.
повышенных температурах, протекает не очень селективно, и образуется смесь трех
возможных изомеров. Гораздо более избирательным реактивом является перхлорат
ртути, меркурирование которым нитробензола в 60%-ной хлорной кислоте при 23 е* С
дает с 89%-ным выходом мета-пзомер |101[.
Перхлорат ртути является гораздо более эффективным меркурирующпм сред-
ством, чем являющийся ковалентным соединением ацетат ртути. Толуол мсркурируется
ацетатом ртути при комнатной температуре с незначительной скоростью, по н присут-
ствии небольшого количества хлорной кислоты взаимодействие значительно ускоряется
(в 2000 раз) [102J.
Как тиофен, так и фуран меркурируются исключительно легко. Ртуть при этом
вступает в положение 2, а если оно занято, то меркурирование протекает в положенно
3. При избытке ацетата ртути замещению на CH3COOHg-rpynny могут подвергнуться
несколько атомов водорода, например, в случае фурана — все четыре.
При проведении реакции с избытком ароматического соединения побочная реак-
ция полимеркурирования протекает в незначительной степени. Кроме того, в присут-
ствии избытка ароматического соединения, папримср бензола, по наблюдается также*
образования дифенилртути. Это можно объяснить тем, что выделяющаяся одновре-
менно кислота расщепляет возможный побочный продукт B2Hg на BHgX.
Другие органические производные ртути образуются в результате присоедине-
ния солей ртути к олефинам. Например, прп пропускании этилена и водный раствор-
ацетата ртути происходит присоединение основной соли ртути по двойной связи*
CH2=CH24-(CH3COO)2Hg4-H2O —> СПг—СН2 СНзСООН
НО HgOOCCHs
Еслп воду заменить метиловым спиртом, то протекает так называемое метокси-
меркурирование. Суспензия ацетата ртути в метаноле в течение 1 ч поглощает 1 моль
этилена с образованием ацетата метоксиэтилртути [103]:
СН2=СН2+(СП3СОода + СН3ОН —> СН2-СН2 4 СПзСООП
I I
СНзО HgOOCCHa
Для этой реакции можно применять ацетат, нитрат, сульфат и хлорид ртути
[104], При использовании всех этих реагентов, кроме ацетата ртути, реакцию необхо-
димо проводить в присутствии основания для связывания выделяющейся сильной
кислоты. В противном Случае образуется равновеспая система, ибо сильные кислоты
разлагают продукты присоединения на соответствующие олефины и соли ртути.
[Ю1]
[102]
[ЮЗ]
1Ю4]
W. J. К 1 а р р г о t h, F. Н. W е s t h о i hi е г, J. Аш. Chem. Soc., 72».
4461 (1950).
H. C. Brown, C. W. McGary jr., J.’ Am, Chem. Soc., 77, 2300, 2306,
2310 (1955).
W. Schoeller, W-Schrauth, W. Esser, Ber., 46, 2864 (1913).__
J. Chatt, Chem. Bev., 48, 7 (1951).
654
ОБРАЗОВАНИИ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
Реакция присоединения солей ртути к стиролу и этиловому эфиру коричной
кислоты [105], а также реакция ненасыщенных жирных кислот [106] с диацетатом
ртути изучались в поисках препаративного метода разделения насыщенных и не-
насыщенных жирных кислот.
Соли ртути могут присоединяться также к ацетиленовым углеводородам. В этом
случае присоединение идет даже в присутствии сильных кислот. Так, например, при
пропускании ацетилена в раствор HgCl2 в 15%-ной НС1 образуется хлорид транс-2-
хлорвинилртути [107].
Не следует путать эти продукты присоединения с так называемыми
ацетиленидами ртути. Соединения общего состава (RC = C— )2Hg получаются
из окиси ртути и соответствующих ацетиленов.
III. Органические производные элементов третьей группы
Для всех элементов третьей группы могут быть получены органические произ-
водные общей формулы R3Me. В этих соединениях металл-углеродная связь более
ковалентна, чем в соответствующих производных элементов второй группы, поэтому
они менее реакционноспособны. Например, при действии триалкильных производны^
алюминия на карбонильные соединения в реакцию вступает только одна алкильная
группа:
r3c«o+air; —► вас-oair;
R'
Очень большое влияние на химические свойства трехзамещенных органических-
производных элементов третьей группы оказывает наличие у всех этих соединений
незаполненной квантовой ячейки, вследствие чего такие триалкильные производные
исключительно легко реагируют с кислородом и другими донорами электронов.
1. Борорганические соединения
Бортриалкилы и бортриарилы получают обычно взаимодействием галогенида
бора или эфира борпой кислоты с реакционноспособным металлоорганическим соедине-
нием, 'чаще всего с раствором реактива Гриньяра; в качестве галогенида обычно
используют BF3, образующий устойчивый эфират [108J:
BF34-3RMgX —> R3B4-3MgFX
Соединения состава СН3ВС1г и (СНЭ)2ВС1 образуются при взаимодействии ВС1а
с соответственно рассчитанным количеством диметилцинка. Однако эти соединения
неустойчивы, легко диспропорционируютСя, давая (СНв)3В и ВС13 [109J, Аналогично,
с использованием диарильных производных ртути, синтезируют арилборгалогениды
[ПО], например:
ВС1з+ (CeHs)2Hg —► CflHsBCh-f- CeH6HgCl
CeH6BCl24-(CeHb)2Hg —> (CeH6).2BCl + CeH5HgCl
[105] G. Spengler, A. Weber, Brennstroff-Chem., 40, 22, 55 (1959).
[106] E, Jantzen, H. Andreas, Chem. Ber., 92, 1427 (1959).
]107] A. H. Несмеянов, P. X. Ф p e й д л и н а, Изв. АН СССР, ОХН. 1945, -
150.
[108] Е. Krause, R. N i t s с h е, Вег., 54, 2784 (1921); 55, 1261 (1922); J. R. J ohn- '
son, Н. R. Sn у d er, М. G, Van C a ni p e n jr., J. Am. Cnem. Soc., 60,
115 (1938); E. Krause, H. Polack, Ber., 61, 271 (1928); H. C. Brown, <
J. Am. Chem. Soc., 67, 374 (1945).
[109] E. Wiberg, W. R*uschmann, Ber., 70, 1583 (1937). ‘
[110] A. Michaelis, Ber., 27, 244 1894); Ann., 315, 19 (1901).
III. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ Ш ГРУППЫ
655
Сходным способом получают эфиры алкил- и арилзамещенных борных кислот —
взаимодействием триалкилборатов с соответствующими магнпйоргапическими соедине-
ниями (111):
B(OR)3 + R'MgX —> R'B(OR)2 + ROMgX
Известны еще два метода синтеза борорганических соединений: взаимодействие
углеводородов с галогенидами бора и присоединение гидридов бора к олефинам. Так,
например, разработан метод получения арилбордихлоридов действием ВС18 на арома-
тические углеводороды в автоклаве при 120—150° С в присутствии порошка алюминия
и хлористого алюминия [112]. Известен аналогичный метод синтеза фенилбордибро-
мида [113].
Триэтилбор образуется при нагревании диборана при 100° С с избытком эти-
лена [114]:
6С2Н4-ЬВ2Нв —► 2(С8Н5)зВ
Эта очень интересная реакция протекает и с другими олефинами [115], причем
боран может присоединяться при комнатной или несколько повышенной температуре
[116]. Соответствующие триалкильные производные бора образуются иногда уже при
комнатной температуре путем взаимодействия этилена, а также высших и циклических
олефинов со смесью боргидрида натрия и хлористого алюминия или с аналогичным
реактивом [117]. Таким Же образом протекает синтез триалкильных производных бора
[118] при нагревании N-трйалкилборазанов с олефинами в течение 4 ч при 200° С:
H3B-NR3A3R'CH=CH2 —> NR3 + (R'CH2CH2)3B
Бортриалкилы, за исключением газообразного при комнатной температуре
триметилоора, представляют собой бесцветные жидкости, тогда как триарнльные
производные бора являются кристаллическими веществами. В противоположность
алкильным производным других элементов третьей группы боралкилы мономерны.
Онй устойчивы к действию воды. С галогенами и галогеноводородпыми кислотами эти
соединения реагируют с образованием соответствующих органических галогени-
дов бора.
Исключительно чувствительны органические производные бора к действию
кислорода. Низшие бортрпалкилы воспламеняются на воздухе и горят зеленоватым
пламенем. При контролируемом аутоокислении получаются соответствующие эфиры
алкилборных кислот, R2BOR и RB(OR)2; в определенных условиях могут быть выде-
лены пероксипроизводные борорганических соединений [1191, R2BOOR или RB(OOR)4,
являющиеся первичными продуктами окисления.
Органические производные бора дают с донорами электронов самые разнообраз-
ные комплексные соединения; в качестве доноров могут служить нейтральные соеди-
нения, содержащие атомы во свободной парой электронов, например амины. При этом
возникают Продукты присоединения общей формулы RaB • NR3. При взаимодействии
борорганических соединений с эфирами при комнатной температуре в от.чичие от
алкильных производных алюмипия получаются неустойчивые эфираты.
Донорами электронов могут служить также отрицательные органические ионы:
при этом образуются комплексные попы. Так, например, триметилбор реагирует
с метиллитием в эфирном растворе с образованием тетраметилбората лития.
[Ill] Е. К h о t i n s к у, М. Melamed, Вег., 42, 3090 (1909); Н. R. Snyder,
J. А. К и с к, J. R. Johnson, J. Am. Chem, Soc., 60, 105 (1938).
[112] E. L. M u e tier tie s, J. Am. Chem. Soc., 81, 2597 (1959).
[113] Z. J. Bu jwid, W. Gerrard, M. F. Lappert, Chem. a. Ind., 1959,
1091.
[114] D. P. Hurd, J. Am. Chem. Soc., 70, 2053 (1948).
[115] R. S. В г о к a w, R. N. Pease, J. Am. Chem. Soc., 72, 3237 (1950)j
A. T. Whatley, R. N. Pease, J. Am. Chem. Soc., 76, 835 (1954).
[116] F. G. S t 0 n e, H. J. E m e 1 e u s, J. Chem. Soc., 1950. 2755.
[117] H. C. Brown et al., J. Am. Chem. Soc., 78, 5694 (1956); 82, 4233 (i960)',
J. Org. Chem., 22, 1136 1137 (1957).
[118] E. C. A s h b у,’ J. Am. Chem. Soc.', 81, 4791 (1959).
[119] M. H. Abraham, A. G. Davies, J. Chem. Soc., 1959, 429.
656
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
В ароматическом ряду практическое применение получил тетрафенплборат натрия,
Na(B(QH5)4J, используемый в качестве реактива для количественного определения
калия и ряда других ионов гравиметрическим методом. Тетрафенилбораты натрия '
п лития растворимы в воде, тогда как аналогичные соли калия, рубидия, цезия
п аммония практически нерастворимы.
Химия борорганических соединений получила в последнее время достаточно
широкое развитие. О методах получения этих соединений и их свойствах см. обзор
1120].
2. Алюминийоргаиические соединения
Удобным методом синтеза триалкил-, и особенно триарилпроизводных алюминия,
применявшимся для получения первых алюминийорганических соединений [121], ...
является взаимодействие металлического алюминия с соответствующими ртутьоргани- ’
ческими соединениями [122, 123]. Этот метод слишком дорог для синтеза больших коли-
чести алюминийорганических соединений. Однако его преимуществом является воз- *
мощность получения соединений, свободных от растворителей. ;
Действие алкилгалогенидов на металлический алюминий приводит к образова- *
ниЮ галогенидов органических производных алюминия: 7
2A1H-3RX — * R2A1X HRA1X2 <
Из этих галогенидов в некоторой степени устойчивы только хлориды [124], |
» то время как бромиды, и особенно иодиды, при нагревании легко диенропорциони-.д
руются до A1R3 и А1ХЯ. Так, например, взаимодействие метилиодида с алюминием сразу j
дает триметилалюминий [123]: 1
2А1 + ЗСН3! —*- А1(СНз)з-|-А11з
Синтезируемые упомянутым выше методом га.тогепиды органических производ- ;
пых алюминия при восстановлении их щелочными металлами также переходят в алю-.
минийтриалкилы. Эти зке соединения могут быть получены непосредственно при исполь-
зовании вместо чистого алюминия его сплава с более электроположильным металлом j’
(магний) [125]: t
Al2Mg3 + 6RX —► 2A!R3+3MgX2
Алюминийоргаиические соединения образуются также при действии реактива
Гриньяра на галогениды алюминия; однако последующее выделение этих соединений J
в чистом виде наталкивается па большое трудности, так как возникающие при этом' .'
эфираты очень стабильны [125].
Взаимодействие менее реакционноспособных органических производных крем- , i
ния пли свинца с А1С13 дает соответствующие хлориды органических производных °
алюминия [126].
Алюминийоргаиические соединения стали легко доступными только после соз- ®
даппя Циглером и сотр. методов их синтеза. Было установлено, что этилен и его выс- .
шие гомологи гладко присоединяются к гидриду алюминия с образованием триалкил-
производных, в которых алюминий связан только с первичными атомами углерода
[120] М. F, Lap pert, Chem. Rev., 56, 959 (1956).
[121 ] G. В. В uckt ом, W. О d 1 i n g, Ann., SuppL. 4, 109 (1965).
[122] E. Krause, P. D i 11 ш a r, Ber., 63, 2401 (1930); S. H i 1 p e r t,
G. Gruttner, Ber., 45, 2828 (1912); A. II. H e с м e я н о в, Н. Н. Н о в и-
к о в а, Изв. АН СССР, 1942, 372; С. Н. Bamf ord,D. L. L е v i, D. М. N е-
wett, J. Chem. Soc., 1946, 468. 'd
[123] K.S. Pit zer, H. S. Gutowski, I. Am. Chem. Soc., 68, 2204 (1946).'
[124] Л, v. Grosse, I, M. Ma vity, J. Org. Chem., 5. 106 (1940); V. Grig- y,
nard, R. J. Jenkins, C. r., 179, 89 (1924); V. F. Hnizda,
С. А. К г a u s, J. Am. Chem. Soc., 60, 2276 (19.38). J
[125| E. Krause, B. Wendt, Ber., 56, 466 (1923).
J126] W. F. E v i s о it, F. S. Kipping, J. Chem. Soc., 1931, 2774; H. Gil- (
man, L. D. Apperson, J. Org. Chem., 4, 162 (1939). i
in. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ Ш ГРУППЫ
657
[127]. На основании этого был разработан метод, позволяющий при определенных
легко контролируемых условиях проводить прямое взаимодействие олефпнов, водорода
и алюминия, приводящее непосредственно «к триалкильным производным алю-
миния [128]:
А1 + ЗС„Наи+1,5Н2 — - (С,Ниц)зА1
Однако в эту реакцию вступают только олефины, содержащие группировку
CHt=C<, т, е. этилен и его моно- и 1,1-дцзамещенные гомологи. 1.1-Дизамещениые
этилены, например изобутилен, непосредственно превращаются в триалкильные про-
изводные алюминия при взаимодействии их с активированным алюминием и водородом
в присутствии небольшого количества готового трпалкилалюмпния. Реакцию с другими
олефинами, в первую очередь с этиленом, нельзя проводить таким образом, так как,
при температуре реакции, составляющей около 12(гС, они могут реагировать далее,
давая смесь высших алюминпйалкплов. Это осложнение устраняется, если синтез
осуществляют в две стадии:
2(C,^+1)3A]4-A1-H,5H, — 3(C„HSw+1)3A]n
3(CBHa„+1)2Ani + 3CBHa„ - 3(СиН2»н)эА1
Первую стадию проводят прп 110—140° С и давлении водорода 50—200 атг
а вторую — присоединение диалкилалюмпнийгидрида к олефину — уже при 80—
100° С; еще более низкая температура применяется в случае этилена. При синтезе
триэтилалюминия максимальное давление этилена составляет 10 ат [129].
Таким образом, этим методом в качестве конечных продуктов можно получать
либо алюминийгриалкплы, либо гидриды дпалкилалюминия. Алюминнйдиалкилгид-
риды образуются также при нагревании алюмпнийтрпалкцлов с водородом при 140—
160° С под давлением [130]:
R3AI+II2 —* RH-' R2A1H
Еще один метод синтеза некоторых алюмиппйтрпалкилов и диалкилалюмпний-
гидридов основан на том, что триалкильные производные алюминия, например, легко
получаемый по приведенному выше методу триизобутллалюминий, при нагревании
с олефинами вступают в реакцию обмена [131}:
(иао-С4П9)зА1-|-ЗСяИ2я * (СяН;>я+1)зА1-[-Зизо^СцНв
Проведение реакции. Триизобутилалюминий смешивают с соответству-
ющим олефином и нагревают (иногда для достижения определенной темпера-
туры кипения к реакционной массе добавляют некоторое количество бензола,
толуола или гептана); образующийся изобутилен при этом улетучивается.
Для быстрого протекания реакции необходимы температуры выше 100° С.
Однако а-олефины общей формулы RCH=CH2 нельзя нагревать выше 110е С,
так как при этом частично происходит каталитическая димеризация.
В случае низкокипящих и веустопчцвых олефинов «реакцию вытеснения»
рекомендуется осуществлять в две стадии. Для этого сначала проводят первую
стадию реакции, протекающую при температуре выше 100° С, — расщепление
триизобутилалюминия на изобутилен и дипзобутилалюминийгидрид; во вто-
рой стадии в качестве исходного соединения используют диизобутилалюми-
пийгидрид, к которому при 70—80° С присоединяется 1 моль другого олефина.
Двукратным повторенном этих операции — отщеплением изобутилена при
[127] К. Ziegler, Angew. Chem., 64, 323 (1952); К. Ziegler, Н. G. Gel-
lert et al., Ann., 589, 91 (1954).
[128| K. Ziegler, H. G. Gellert et al., Angew. Chem., 67, 424 (1955).
[129] K. Z i eg 1 er. H. G. G e 1 1 e r t et al., Ann., 629, 1 (i960).
[130] R. Koster, Angew. Chem., 68, 383 (1956); R. К 6 s t e г, II. Lehmkuhl,
G. Bruno, пат. ГДР 16560; австр. пат. 202566.
[131] К. Z 1 е g I е г, Н. М а г t i n. F. Krupp, Ann., 629,14 (1960); K. Z i egl a rv
W. R. Kroll et al., Ann., 629, 53 (1960).
42 заказ 1835.
653
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
повышенной температуре (до 150° С) и присоединением нового олефина при более
низкой температуре можно, наконец, заменить все изобутильпые остатки на
другие алкильные группы.
В другом варианте метода олефин прибавляют с такой скоростью, чтобы
опа была меньше скорости выделения изобутилена, при этом в растворе по-
стоянно содержится свободный диалкилалюминийгидрид и доля вторичных
реакций понижается. К концу реакции температуру снижают до 70—80° С
и насыщают остаточные AI—Н-связи добавлением недостающего количества
олефина.
Если взаимодействие дпалкялалюмидийгпдридов с каким-либо олефином проте-
кает неколнчественно, достигая лишь равновесного состояния
R,A1H+C,H„ й Вг^Н^А!
то реакцию проводят с избытком олефина, отгоняя его затем в вакууме при возможно
более низкой температуре (ниже 50° С), чтобы избежать обратного расщепления про-
дукта реакции.
Как уже упоминалось, алюминнйтриалкилы могут реагировать с олефинами
общей формулы CHj^CHR, и особенно с этиленом, выше некоторой определенной
температуры таким образом, что они присоединяются к двойной связи олефина. В слу-
чае этилена образуются высшие алюминийалкнлы. Эта реакция, гладко протекающая
при температурах около 90—1202 С по уравнению [132]
R—а1-гпСгН« —•* R—(QH*)»—al
где al— Al/3
позволяет, исходя из этилена, алюминия и небольшого количества водорода, получать
практически все первичпые алифатические соединения с четным числом углеродных
атомов, например первичные спирты [133], нормальные парафины и а-олефины с чет-
ным числом углеродных атомов [134]. Прп аналогичном присоединении к этилену
трипропилалюминия можно синтезировать соответствующие соединения с нечетным
числом углеродных атомов.
Реакция наращивания углеродной цепи, т. е. гладкое последовательное присо-
единение этилена, всегда сопровождается «реакцией вытеснения», в результате
которой происходит отщепление олефина:
R—(СН2СН2)В-al+CjlU —> В-(СН2СП2)я.1-СН=СН2-(-С2Н5—al
Если давление этилена достаточно велико, эта реакция не имеет большого зна-
чения; рекомендуемое давление этилена около 100 am.
G высшими а-олофинами реакция присоединения приводит практически лишь
к каталитической димеризации, поскольку вслед за присоединением непосредственно
следует «вытеснение»:
RGH2GH2-al-i-RCH-=CH2 —* RCH2CH2CHCH2— al
R
RCH2CH2CHCH2—al |-RCH=CH2 —* RCH2CII2C=CH.2-j-RCH2CH2-al
A J
Новые алюмипийорганическпе соединения получаются также в результате
присоединения алюминийтриалкилов к производным ацетилена; например, 1 моль
триэтилалюминня уже при 40—60' С присоединяет 1 лолъ ацетилена с образованием»
диэтялбутснилалюмипия [135]:
(С2Н5)зА1 + СН=СН —* (С2Н5)гА1-СН=СН-С2Н5
[132] К. Z i е g 1 е т, Н. G. G с 1 1 в г t, et а)., Апл., 629, 121 (1960).
[133] К. Ziegler, F. Krupp, К. Zosel, Ann., 629, 241 (1960).
[134] К. Ziegle^r, Н. G. Gellert et al., Ann., 629, 172 (i960).
[135| G. Wilke. H. Muller, Ann., 629, 222 (i960).
IV. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ IV ГРУППЫ
65
С молекулами, обладающими электронодонорными свойствами, алюминийтриаз:
килы образуют комплексные соединения, которые гораздо более устойчивы, чем в cnj
чае бора. Исключительно устойчивые эфпраты в некоторых случаях могут перегонятьс
без разложения [136].
Донорами электронов могут быть и отрицательно заряженные органически
ионы; при взаимодействии соответствующих литий- и алюминийорганически
соединений образуются соли [137] типа Li [А1(СП3)41 и Li [А1(СвНв)«]. Комплексны
соединения, в которых координационное число алюминия равно четырем, могут дават
с алюминийтрпалкилами многие галогениды и цианиды щелочных металлов. Соста
таких комплексов |138] выражается формулами Me[AlRsX] и MeX-2AlRa, где Me -
щелочной металл, X — галоген пли циан. Точно так же из хлористого калия и диал
кплалюмиппйхлорида получается [139] К [А1ВгХа].
IV. Органические производные элементов четвертой группы
Органические производные кремния, германия, олова и свинца относятс
к самым устойчивым и наименее реакционноспособным металлоорганическим соедини
нпям. Они получили исключительно большое промышленное значение. Особенно шй
роко используются силиконы; некоторые оловоорганические соединения прнмеяяютс
в качестве стабилизаторов поливинилхлорида, тетраэтилсвинец — для повышена
октапового числа горючего для двигателей внутреннего сгорания и т. д.
О методах получения и химических свойствах кремний-, германий- и олово
органических соединений, особенно соединений с ненасыщенными углеводородным?
остатками, например с виппльпыми и этинильными радикалами, см. обзор [140]
1. Кремннйорганнческие соединения [141]
Область распространении кромцнйорганичоских соединений псключнтельн;1
широка. Наряду с тетраалкил- и тетраарилсилалами существуют самые разнообразны
органические производные кремния — галогениды, гидриды, алкоксиды, силанолы
силоксаны и другие соединения; особенно детально исследованы полисилоксаны
Сопоставление аналогичных органических производных кремния и углерод,
приводят Джильмен и Данн [142].
Кремнийорганическме соединения можно получать по реакции галогенидов крем
ния с другими металлоорганическими соединениями. Фридель и Крафтс [143] впервьк
синтезировали теграмртплсилан, действуя диметилцпнком на SiCl4 в запаянной трубк
при 200? С:
SiCl4 + 2(CH8)2Zn —(CH3)4Si + 2ZnCl2
Позже взаимодействием димстилшшка с хлоренлапом и дихлорсиланом npj
комнатной температуре были получены соответствующие метилсиланы [144], CHsSif£
и (CH3)2SiH2.
[136] Е. В. В a k е г, Н. Н. S i si er, J. Am. Chem. Soc., 75, 4828 (1953); G.Gei
s e 1 e r, W. Knothe, Chem. Ber., 91, 2446 (1958).
[137] D. T. Hurd, J. Org. Chem., 13 , 711 (1948); G. Wittig ct al., Ann., 56»
110 (1949); 573, 195 (1951).
[138] K. Ziegl er, R. Koster et al., Ann., 629, 33 (1960).
[139] R. Koster, W. R. Kroll, Ann., 629, 50 (1960).
[140] A. D. Petrow, V. F. Mironov. Angew. Chem., 73, 59 (1961).
[141] H. W. Post, Silicones and other Silicon Compounds, Reinhold Publishing
Corporation, New York, 1949; E. G. R о c h о w, An Introduction to the Chemi,
stry of the Silicones, 2 ed., John Wiley a. Sons, New York, 1951; Einfiihrung in di
Chemie der Silicone, Verlag Chemie, Weinheim/Bergstrape, 1952; A. Hunyae
Chemie der Silicone, Verlag Technik, Berlin, 1952; R. R. McGregor, Sili
cones and their Uses, McGraw-Hill Publishing Company, London, 1954; С. E %
born, Organosilicon Compounds, Butterworths Scientific Publications, London
1960.
Ц42] H. G i 1 m a n, G. E. Dunn, Chem. Rev., 52, 77 (1953).
]143 C. Friedel, I. M. Crafts, Ann., 136, 203 (1865).
[144] A. Stock, C. So mi eski, Ber., 52, 695 (1919).
42*
660
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
Реакцией диарилпроизводных ртути с SiCl4 можно получать арилтрихлор-
силаны [145]:
SiCl4+Ar2Hg —* ArSiCla+ArHgCl
Но самым общим методом синтеза кремнийорганических соединений является
взаимодействие реактивов Гриньяра с галогенидами кремния. В случае SiCl4 оно может
привести к образованию смеси продуктов, в которых на углеводородные остатки заме-
щены от одного до четырех атомов хлора:
Sid, KSida -2^ R^iCb RtSl
Разделение таких смесей затруднительно, но при соблюдении определенщ
условий и, главным образом, путем применения соответствующего количест
реаитива Гриньяра каждый из желаемых продуктов можно все же получать с хорош!
выходом. Например, таким методом впервые были синтезированы соединения [14
общего состава R4Si, R3SiCl, RaSiCla и RSiCl3. Взаимодействием этих хлорсодерж
щпх соединений с другими соединениями Гриньяра можпо получать смешанные кр'
нийалкилы и кремиийарилы.
Тетраметилсилан [147]. В колбу емкостью 1 л помацают 30 г маг:
в 500 мл абсолютного эфира и пропускают метилбромид. К полученному мет
магнийбромиду по каплям прибавляют 50 а четыреххлористого кремния в <а
с равным объемом абсолютного ефира и для завершения реакции реаициош
массу не менее 2 ч кипятят с обратным холодильником. Так как темперам
кипения тетраметилсилана ниже, чем температура кипения эфи^а, еле}
применять длинный шариковый холодильник или эффективный обрат
холодильник. Затем весь реакционный раствор перегоняют на водяной б
в приемник, охлаждаемый льдом. Полученная таким образом смесь тетрамет.
силана и эфира из-за близости температур кипения составляющих компов
(26 и 35° С) не разделяется перегонкой. Для разделения используют нераст
римость кремиийуглеводорода и концентрированной H2SO4 и приливают т
смесь при охлаждении льдом с солью к 500 мл HsSO4. При этом ефир раст
ряется в H2SO4, а в бесцветном верхнем слое остается тетраметилсилан. Э’
слой отделяют и перегоняют на слабо нагретой водяной бане, используя эффс
тивный холодильник, в приемник, охлаждаемый льдом. Дистиллят еще р
встряхивают с концентрированной H2S04 и очищают фракционироваг—
перегонкой; т. кип. тетраметилсилана 26—27° С (760 лл рт. ст.).
Дисилаиы, R3SiSiR8, н днсилоксаны, R3SiOSiR3, можно непосредственно ш
чать при помощи магний- или патрийорганических соединений из соответствую:
галогенидов [148], Cl3SiSiCl3 или Cl3SiOSiCl3.
Кремнийорганические соединения образуются также при взаимодейс
магнийорганических соединений с метиловыми или этиловыми эфирами ортокремь
кислоты [152] или же с фторсиликатом натрия [153].
Вместо реактива Гриньяра в этих реакциях можно применять литийорга!
ские соединения [149]. Кроме того, действием литий- и ватрийорганпческих сое/
[145] A. Ladenburg, Ann., 173, 143 (1874).
[146] F. S. Kipping, Proc. Chem. Soc., 20, 15 (1904); 21, 65 (1905); J. Ch
Soc.. 91, 209 (1907); 91, 717 (1907).
[147] A. Bygden, Ber., 44, 2640 (1911).
[148] W. C. Schumb, С. M. Saffer jr., J. Am. Chem. Soc., 61, 363 (193
63, 93 (1941).
[149] H. Rosenberg, J. D. Groves, C. Tamborski, J. Org. СЬвВ
25, 243 (1960).
[152] P.A.Di Giorgio, W. A. Strong, L. H. Sommer F. C. Whitmoi
J. Am. Chem. Soc., 68, 1380 (1946); E. Khotinsky, B. Seregenkol
Ber., 41, 2946 (1J08).
[153] E. M. Сошественская, ЖОХ, 10, 1689 (1940); С. A., 35, 3240 (194
IV. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ IV ГРУППЫ
661
ний можно заменять на органические остатки атомы водорода, связанные с нремнием
[150], например:
SiH4-HCeHsNa — * (CeH6)4Si4-4NaH
Таким ясе образом реагируют с магнийорганическими соединениями моно-,
ди- и трифенилсиланы [151].
Еще одна возможность получения кремнийорганичесних соединений заклю-
чается в аналогичной синтезу Вюрца конденсации тетрагалогенидов кремния с алкил-
□ а рил галогенидами в присутствии металлического натрия; в качестве растворителей
используют эфир или углеводорчды [154]:
SiX44-4RX4-8Na > R4Si-f-8NaX
Прямое взаимодействие элементарного кремния с газообразными алкил-
1155] и арилгалогенидами [156] протекает при высоких температурах в присутствии
подходящих твердых катализаторов, например меди или серебра (в виде сплавов ирем-
ния с медью или серебром). В результате реакции образуются в основном продукты
общей формулы R8SiXa, а также R3SiX, RSiXs, SiX4 и углеводороды. Этот метод
пмеет большое промышленное значение для получения диметилдихлор- и метил-
трихлорсиланов, применяемых в качестве промежуточных продуктов при синтезе
спликонов.
Кремнийорганические соединения образуются также при взаимодействии
углеводородов с некоторыми соединениями кремния. Например, фенплтрихлорсилан
[157] образуется при высоких температурах из бензола и четыреххлорнстого кремния.
Аналогичная реакция протекает и при взаимодействии ароматических углеводородов
с трихлорсиланом при повышенном давлении в температуре 400° С [158]:
ArH + SiHCls —> ArSiCls + Ha
Некоторые алкилхлорсиланы можно получать присоединением галогенсиланов
или их производных типа SiHX3, RSiHX» и RoSiHX к ненасыщенным углеводородам
[14, 159]:
СНа=СН,+SiHCla , o^sici,
GH=CH + SiHCla pt; Я1“м »““' > CHa=CHSiCls
О методах получения и свойствах кремнийорганичесних соединений, органиче-
ские остатки которых, связанные с атомом кремния, содержат функциональные группы,
например СЮН231(СН3)С1г, CH2=CHSiCl3, CH3SO2GHaSi(GH3)3 идр., см. обзор [160].
[150] J. S. Peake, W. Н. Nebergall, Yun Ti Chen, J. Am. Chem.
Soc., 74, 1526 (1952); R. N. Meals, J. Am. Chem. Soc., 68, 1880 (1946).
[151] H. Gilman, E. A. Zuech, J. Am. Ghem. Soc., 81, 5925 (1959).
[154] A. Polis, Ber., 18, 1540 (1885); F. S. К i p p i n g, L. L. L 1 о у d, J. Ghem.
Soc., 79, 449 (1901); F. T a u r k e, Ber., 38, 1661 (1905).
[155] E. G. R о ch о W, J. Аш. Ghem. Soc., 67, 963 (1945); D, T. Hur d, E.G.RO-
cbow, J. Am. Chem. Soc., 67, 1057 (1945).
[156] E. G. Rochow, W. F. Gilliam, J. Am. Chem. Soc., 67, 1772 (1945):
А. В. Топчиев, H. С. Наметкин, H. M. Шмухова, ДАН СССР,
78, 497 (1951); С. А., 46, 451 (1952).
[157] Н. С. Miller.R. S. S с h е i b е г, пат. США 2379821; С. А., 39, 4619(1945).
[158] A. J. Barry, J. W. Gilkey, D. Е. Hook, Metall-Organic Compounds,
Advances in Chemistry Series, № 23, American Chemical Society, Washington,
1959, p. 246.
[159] A. J. Barry, L. De P r e e, J. W. Gilkey, D. E. H о о k, J. Аш.
Chem. Soc., 69, 2916 (1947); J. L. Sp eier,R. Zimmermann,-1. A. Web-
ster J. Am. Chem. Soc., 78, 2278 (1956); J. L. S p e i e r, J. A. Webster,
G. H. Barnes, J. Am. Chem. Soc., 79, 974 (1957); J. L. S p e i e r,
J. A. Webster, J. Org. Chem., 21, 1044 (1956); P. X. Фрейдлнна,
E. 3. Чуковская, Цао И, ДАН СССР, 127, 352 (1959); В. A. Blue-
ston, J. Аш. Chem. Soc., 83, 1000 (1961).
[160] Р. D. G е or ge, М. Prob er, J. R. Elliot, Ghem. Rev., 56, 1065,(1956).
662
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
Органические производные германия [161] можно получать теми же мето-
дами, ято и кремнийорганическпе соединения.
2. Оловоорганические соединения [162]
Для олова известны практически тс же самые типы органических производных,
что п для кремния или германия. Однако олово-углеродная связь все же гораздо более
полярпа, чем соответствующие связи кремния и германия, поэтому органические
остатки в оловоорганических соединениях легче подвергаются реакциям отщепления
или обмена; более ярко выражена тенденция к комплексообразованию.
Самым общим методом получения оловоорганических соединений, применяемым
обычно в лабораторной практике, является взаимодействие тетрагалогенидов олова
с магнийорганическимп соединениями. Реакцию проводят так, чтобы она приводила
к тетраалкил- или тетраарилпроизводным олова [163]:
SnCU-HRMgX —ч. R4Sn + 4MgXCl
‘ Тетраэтилолово [164]. К эфирному раствору зтилмагнийбромида, полу-
ченному из 24,5 г магниевой стружки и 136 а этилбромида в 500 мл эфира,
при охлаждении медленно прибавляют по каплям 45 г SnC]4. Затем смесь кипя-
тят с обратным холодильником в течение 1 ч, после чего полностью отгоняют
эфир на водяной баие. Остаток нагревают иа кипящей водяной бане в течение
30 мин, после охлаждения к нему прибавляют отогнанный эфир и разлагают
реакционную массу водой и 5%-ной ДС1 до четкого разделения слоев. Эфирный
слой отделяют, сушат и отгоняют эфир. Остаток рекомендуется перегонять
в вакууме водоструйного насоса, так как температура кипения продукта реак-
ции довольно высока. Выход тетраэтилолова около 75% ог теоретического;
т. кип. 78° С (13 мм рт. ст.).
Из полученных таким образом тетраалкильпых производных олова можно
затем легко синтезировать соответствующие галогениды органических производных
олова, например, по реакции, известной под названием «диспропорционирование по
Кочешкову» [165]:
3R4Sn+SnC14 —> 4НзЗпС1
RiSn-j-SnCl* — + 2R2S11CI2
R4Sn + 3SnGI4 —* 4RSnCl3
Вместо магнийорганических соединений для синтеза некоторых оловооргани-
ческих соединений часто применяют органические производные лития [166].
Тетраалкил- и тетраарилстаннаны можно получать реакцией органических
шлогеппдов и SnCl4 с металлическим натрием [167, 168]:
SnCl4-[-4RX-f-8Na —* R4Sn + 4NaCl + 4NaX . -
[161] О. Н. Johnson, Ghem. Rev., 48, 259 (1951); Inorg. Synth., 5, 64 (1957);
E. Gastinge r, Fortschr. chem. Forsch., 3, 603 (M54—1958).
[162] G. J. M. van der Kerk, J. G. A, Luij t en, J. G. N ol t es, Angew.
Chem., 70 , 298 (1958); R. K. .Ingham, S. D. Rosenberg, H. Gil-
man, Chem. Rev., 60, 459 (1960).
[163] W. J. Popo, S.. J. Peachey, Proc. Chem. Soc., 19, 290 (1903); P. P f e i f-
fer, K. Schnurmann, Ber., 37, 319 (1904); G. J, M. van der К e r k,
J. G. A. Lui] ten, Org. Synth., 36, 86 (1956).
[164] Руководство по препаративной неорганической химии (ред. Г. Бра yep),
пер. с нем., Издатинлих, 1956.
[165] К. A. Kozeschkow, Вег., 62 , 996 (1929); 66, 1661 (1933); К.А.Ко-
zeschkow, М. М. Nad], Вег., 67 , 717 (1934); К. A. Kozeschkow,
М. М. N a d j, А. Р. Alexandrow, Вег., 67, 1348 (1934).
[166] Н. G i 1 m а и, С. Е. А г n t z е n, I. Org. Спет., 15, 994 (1950); Р. R; А и --
s t i n, J. Am. Ghem. Soc., 54, 3726 (1932),
[167] D. L. T a b e г n, W. R. О г n d о г f f, L. M. D ennis, J. Am. Chem.,
Soc., 47 , 2039 (1925).
[168] G. j, M, v a n d e г К erk, J, G, A. L u i j ten, J, Appl. Chem., 4, 301
(1954).
IV. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ IV ГРУППЫ
665
При прямом взаимодействии алкилгалогеыидов с металлическим оловом также
образуются оловоорганические соединения. Так были получены дниодид дпэтилодова
[169] я диодид диметилолова [170]. Описан синтез дихлоридов диметил- и диатилолова
по реакции расплавленного олова с метил- или этилхлоридом, а также галогенидов
трпметилолова при взаимодействии метилхлорида, метилбромида или метилиодида
со смесью SnO и Си при температуре выше 300° С [171]:
3SnO+2CH3Cl — (CH3)2Sn + SnOCl2-|-SnO2
(CH3)2Sn + CH3Cl — + (СНз)з5иС1
Галогениды трибензилолова образуются иногда с очень хорошими выходами
из беизилгалогенидов и суспендированного в воде порошкообразного олова при
100? С [172].
Для синтеза арильных и высших алкильных производных олова рекомендуется
вместо чистого олова использовать его сплавы с неблагородными металлами [173].
При взаимодействии с органическими галогенидами сплава олова и натрия образуются
соответствующие тетраалкил- или тетраарилстапнаны:
4SnNa-|-4RX----*• R*Sn4-4NaX -f-3Sn
В модифицированном методе вместо сплава олова с натрием используют гораздо
более удобный сплав магния и олова, Mg2Sn [174]:
Mg2Sn-[-4RX —* RaSn + 2MgX8
Олово-углеродные связи могут образовываться также в результате присоедине-
ния органических производных гидридов олова в реакционноспособным С=С-свя-
зям [175], например:
(CeH5)3SnH-|-CH2=CHCeH5 —> (CeHsJsSnCHsCHaCeHe
Органические гидриды олова способны присоединяться и к производным ацети-
лена. Реакция протекает при нагревании эквимолярной смеси реагирующих веществ
в течение короткого времени при 80—100”5 С. Выходы почти всегда близки к количе-
ственным.
Другой метод получения галогенидов арилпроизводных олова разработали
Несмеянов и сотр. Они обнаружили, что кристаллические двойные соли ароматических
диазониевых солей и SnCl4 разлагаются в кипящем этилацетате в присутствии порошко-
образных меди, цинка или олова [176]:
2ArN2Cl • SnCh 4- 2Ме —> Ar^nC^-b 2N84-2MeCl2
Этим методой ароматические группы можно вводить даже в тех случаях, когда
применение магнийорганических соединений дает отрицательные результаты.
[169] Е. Frankland, Ann., 85, 329 (1853).
[170] A. Gahours, Ann., Ц4, 354 (1860).
[171] А. С. Smith jr., Е. G. R ochow, J. Am. Chem. Soc., 75, 4103, 4015
(1953).
[172] K. Sisido, Y. Takeda, Z. Kinugawa, J. Am. Chem. Soc. 83 , 538
(1961).
[173] A. Polis, Ber., 22, 2915 (1889); T. Harada, Sci. Pap. Inst, physic, chem.
Res. (Tokyo), 35 , 290 (1939); C., 1939, II, 2912; J. Sci. Res. Inst. (Tokyo), 43,
31 (1948); C. A., 43, 4632 (1949).
[174] G. J. M. v a n der Kerk, J. G. A. L u i j t e n, J. Appl. Chem., 4, 307
(1954).
[175] G. J. M. van der К e г k, J. G. N о 11 e s, J. G. A. L u i i t e n, J. Appl.
Chem., 7 , 356 (1957); G. J. M. van der Kerk, J. G. Noltes, J, Appl.
Chem., 9, 106 (1959).
[176] A. N. Nesmejanow, K. A. Kozeschkow, W. А. К 1 i m о w a,
Ber., 68, 1877 (1935).
664
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
Дихлориды диари.тпроивводных олова можно получать также через окиси
дпйрялйроиэводных олова, образующиеся при разложении тетрафторборатов арилди-
азойля Действием SbCIa-2HaO И' цинковой пыля fl 77].
3. Органические производные синица {178]
Сравнение органических производных свинца с аналогичными производными
других элементов четвертой группы показывает, что они менее устойчивы и легче раз-
лагаются под действием света и нагревания; значительно легче расщепляются пип-
и под действием кислот и окислителей.
Органические производные свинца в лабораторной практике получают взаимо-
действием РЬС1а с магний- или литийорганическими соединениями. Реакция протекает
но следующему суммарному уравнению J179]; '
2PbCl2 + 4RMgX —> R4Pb + Pb + 2MgCl2 + 2MgX2
В результате такого взаимодействия наряду с тетраалкил- или тетрааридпроиэ-*
водными свинца получаются также соединения общей формулы RePb2.
В приведенной выше реакции половина вводимого в нее свинца восстанавли-
вается до металла и выводится из реакции. Во избежание этого вместо солей двухвалент*
иого свинца применяют соли четырехвалентного свинца, например тетраацетат свинца,
гексахлорплюмбат калия или аммония и др. [180].
Однако, даже используя соли двухвалентного свинца, можно перевести весь"
свинец в тетраалкил- или тетраарилпроизводные. Реакцию проводят в присутствии
алкилиоднда, содержащего тот же алкильный остаток, что и металлоорганическое'
соединение [181]. В результате первичной реакции выделяется мелкодисперсный-,
реакционноспособный металл, реагирующий с органическим иоДидом:
Ph + 2RI —» R2PbIa >
Образующееся соединение, RaPbla, реагирует далее с литий-или магнийорга-
ническими соединениями, давая К4РЬ. Таким образом получают, например, тетра-'
метилсвинец, добавляя метиллитий или метилмагнийиодид к кипящей суспензии'
РЬС1а в эфире в присутствии СН81:
РЬС124-ЗСНвЫ4-СНз1 —* (CH3)*Pb-b2LiCI4-LiI
f
Тетраалкил- или тетраарилпроизводные свинца образуются также при действии-
органических галогенидов на сплав натрия и свинца; катализаторами этой реакций.,
служат основания, например пиридин. Так, исходя из метилиодида, был получен тетра-
метилсвинец [182], а позднее из бромбензола — тетрафенилсвинец [183]. В промышлен,:
ном масштабе тетраэтилсвинец получают взаимодействием этилхлорпда со салаг***
натрия и свинца при низких или умеренных температурах [184].
Алкильные производные свинца образуются с высокими выходами при
ствии алкилгалогенидов па смеси свинца и магния [185].»
Синтезы тетраалкилпроизводных свинца из свинца или его сплавов подробил’
рассматриваются в обзоре 1186]. • &
А. Н. Несмеянов, Л. Г. Макарова, ДАН СССР, 87 , 421 (1952).
R. W. Leeper, L. Summers, Н. Gilman, Chem. Rev., 54, 101 (1954b
P. Pfeiffer, P. Huskier, Ber., 37, 1125 (1904); P. R. Austin
J. Am. Chem. Soc.. 54, 3726 (1932); G. Gruttner, E. Krause, Ber., 49;
1415 (1916).
F. W. Frey, S. E. Cook, J. Am. Chem. Soc., 82, 530 (1960). J
H. G i 1 m a n, R. G. J ones, J. Am. Chem. Soc., 72, 1760 (1950); H. G i I#
man, L. Summers, R. W. Leeper, J. Org. Chem., 17, 630 (1952).
A. Cab ours, Ann., 122, 67 (1862). ,
A. Polis, Ber., 20, 716 (1887). -
J. Edgar, Ind. Eng. Chem., 31, 1439 (1939).
G. Calingaert, H. Shapiro, пат.. США 2535190; С. A., 45, 3804 (1951)<
H. Shapiro, Metall-Organic Compounds, Advances in Chemistry S«rieft(
№ 23,4American Chemical Society, 1959, P- 290. J*
V ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ V ГРУППЫ
665
Циглер с сотр. [187] разработал специальный метод синтеза тетраэтилсвинца.
Исходным веществом служит триэтилалюминий; тетраэтилсвинец легко и с количе-
ственным выходом образуется при электролизе хорошо проводящих электрический ток
комплексных соединений NaF-2A](GaH5)s или foa[R3AlCaHb] с использованием свин-
цового анода.
Устойчивыми органическими производными элементов подгруппы титана яв-
ляются только циклопентадиенильные «сэндвичеобразные» соединения. Нормальные
алкильные производные почти неизвестны, они очень неустойчивы. Этилтитанхлориды,
например, содержатся в смесях триэтилалюминия и TiCl4, применяющихся в качестве
так называемых катализаторов Циглера, и играют, очевидно, важную роль при поли-
меризации олефинов [188].
V. Органические производные элементов пятой группы
Мышьяк, сурьма и висмут образуют два ряда органических производных с ва-
лентностями -]-3 и +5. Производные трехвалентных металлов цмёют недоделанную
пару электронов, поэтому многие из них исключительно бурно реагируют с акцепто-
рами электронов.
Низшие триалкильные производные этих элементов самовоспламеняются на
воздухе. Но в отличие от других металлалкилов в этом случае взаимодействие кислорода
с металл-угдеродной связью ве является первичным процессом; при осторожно прове-
денном аутоокислении чаще получают соединения общей формулы R3MeO. Триариль-
ные производные устойчивы к действию кислорода воздуха и дают соответствующие
оксиды только под действием более сильных окислителей.
Очень легко реагируют триалкил- и триарилпроизводные этих элементов
с хлором, бромом и иодом, превращаясь в галогендды R3MeXs. Аналогично реагируют
соединения общего состава R2MeX и ВМеХ2 с образованием соответственно производ-
ных RjMeX3 и RMeX4.
Триалкиларстты и триалкилстибияы, а также триариларсины могут присоеди-
нять алкилиодиды. При этом возникают соответствующие иодиды арсония и стибония,
R4MeI или R3R'MeI.
Из органических производных пятивалентных металлов известны в основном
следующие типы соединений: галогениды (RMeX4, RojfeX.,, R3MeXa в R*MeX), кисло-
родсодержащие соединения [RMeO(OH)s, RjMeO(OH), RjMeO, R4MeOH], а также
пентаалкил- и пентааридпроизводные (mMe).
Галогениды органических производных пятивалентных металлов отщецдяют
при нагревании алкил- или арилгалогенид и превращаются в соответствующие соеди-
нения трехвалентных элементов:
R3MeXa —► R,MeX + RX
Особенно легко протекает эта реакция с галогенидами общей формулы RMeX4
и RaMeX3, которые частично разлагаются уже при комнатной температуре.
1. Мышьякорганическне соединения
Органические производные мышьяка подвергались очень детальному исследо-
ванию, главным образом с целью изыскания химиотерапевтических препаратов.
Мышьякорганическне соединения можно получать взаимодействием трдгалоге-
нидов мышьяка с металлоорганическими соединениями. Триалкиларсины с хорошими"
выходами образуются из соответствующих цинкдиалкилов и треххлористого мышьяка
[189]. с галогенидами мышьяка реагируют в другие металлоорганические соединения;
этилдпхлорарсин получают, например, по следующей реакции [190]:
(CaH6)2Hg + AsCls —* С2Н6 AsGI2+C2HeHgCl
[187] К. Z i е g 1 е г et al., Angew. Chem., 67, 424 (1955); 71, 628 (1959).
[188] G. Beerman n, H. Beatian, Angew. Chem., 71, 618 (1959).
[189] A. W. Hofmann, Ann., 103, 357 (1857); R. R. R ensha w, G. E. H olm,
J. Am. Chem, Soc., 42, 1468 (1920).
[190] W- Steinkopf, W. M i e g, Ber., 53, 1013 (1920).
666
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
Этилдихлораргшт. К 150 в AsCl3 при охлаждении льдом добавляют
по каплям 150 е диэтилртути; реанция протекает с выделением значительного
количества тепла и с образованием кристаллического осадка. Затем смесь нагре-
вают в течение 30 мин на водяной бане, добавляют сухой эфир и отфильтровы-
вают выделившийся хлорид этилртути. После отгонки эфира полученный этил-
дихлорарсип отделяют от иабытка AsGl3 фракционной перегонной. Выход
этилдихлорарсина 90 г; т. кип. 145—150° С.
Самым удобным и чаще всего применяемым методом получения мышьякоргани-
ческих соедипений является взаимодействие галогенидов мышьяка с реактивами
Гриньяра (или же с литийорганическими соединениями) [191].
Триэтиларснн [192]. Раствор 50 г AsCl3 в 120 мл абсолютного эфира
прибавляют при охлаждении к раствору этилмагнийбромида, полученному
из 93 г этилбромида и 21 г магния. После окончания реакции к смеси добавляют
лед и разбавленную HG1 до четкого разделения слоев; эфирный раствор отде-
ляют и сушат над СаС1г. Затем осторожно отгоняют ефир в атмосфере углеки-
слого газа при температуре не выше 38° С, последние остатки эфира отгоняют
в вакууме. Полученный триэтиларснн перегоняют в вакууме, собирая дистиллят
в колбу для перегонки, из ноторой его затем перегоняют при атмосферном
давлении в тоне углекислого газа. Выход продукта 18 г (40% от теоретического);
т. нип. 140° С (736 мм рт. ст.).
Выход снижает высокая летучесть триэтиларсина с эфиром; отогнанный
эфир можно использовать для получения дигалогенида триэтиларсина.
При помощи ступенчатого синтеза Гриньяра или при действии магний- или
литийорганических соединений на полученные другим путем органические галогениды
мышьяка, RAsXa и R2AsX, можно синтезировать и смешанные арсины.
Вместо галогенидов в реакции с магнийорганическими соединениями можно
кспользовать As2O3; этим методом с хорошим выходом получают, например, триарил-
арсины [193].
Взаимодействие фениллития с дихлоридом трифениларсина или с бромидом
тетрафениларсония дает пентафенилмышьяк [194]. В аналогичной реакции иодида
тетраметиларсония или дибромида триметиларсина с метиллитием пентаметилмышьяк
выделить не удалось, но образование небольших иоличеств этого соединения наряду
с другими продуктами было все же доказано [195].
Конденсация трнгалогенидов мышьяка с алифатическими или ароматическими
галогенидами в присутствии металлического натрия приводит к триалкил- или соответ-
ственно триариларсинам. Так, например, трибензиларсин получают длительным кипя-
чением смеси бензилхлорида и AsGla в ефире с металлическим натрием в присутствии
небольшого количества этилацетата [196]. Аналогично из н-пропилхлорида, AsCls
и патрия образуется три-м-пропиларгин [197], а из арилбромидов, AsCl3 и натрия
в эфире, — соответствующие триариларсины [198].
Трифениларсин [199]. В колбе с шариковым обратным холодильником
к 146 а металлического натрия (нарезанного топкими пластинками; еще лучшие
результаты получаются при проведении реакции с проволокой диаметром 1 мм)
[191] Н. Н i b b е г t, Вег., 39, 160 (1906); Р. Pfeiffer, Вег., 37, 4620 (1904);
J. Seifter, J. Аш. Chem. Soc,, 61, 530 (1939).
[192] Ср. [1], стр. 456.
[193] F. F. Blicke.F. D. S m i t h, J. Am. Chem. Soc., 51,1558 (1929); F. S a c h s,
H. Kantorowicz, Ber., 41, 2767 (1908).
194] G. W i t ti g, К. C la u ₽, Ann., 577, 26 (1952).
195] G. Wittig, К. T о rs s ell, Acta Chem. Scand., 7, 1293 (1953).
196] A. Michaelis, U. Paetow, Ber., 18, 41 (1885).
197] W. M. Debn, Am. Chem. J.,'40, 115 (1908).
198] A. Michaelis, A. Reese, Ber., 15, 2876 (1882); Ann., 321, 160 (1902);
G. T. M organ, D. C. Vining, J. Chem. Soc., 117, 777 (1920).
[199] Ср. [1], стр. 497.
V. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ V ГРУППЫ
в 600 мл абсолютного эфира достаточно быстро прибавляют смесь 100 г As<
п 260 г бромбензола. Реакционную массу выдерживают не менее 12 ч, ча<
встряхивая, затем ее сливают с непрореагировавшего натрия и галогенид
натрия п отгоняют эфир. Оставшееся масло фраиционируют перегонкой в j
кууме; собирают фракцию с т. кип. 220—230® С (14 мм рт. ст.) и растра
со спиртом; т. пл. продукта 59° С.
Триалкпларсииы образуются также при действии алкилиодидов на сплавы и
соединения мышьяка с сильно электроположительными металлами, например натри
пли калием [2001.
Для синтеза важнейших арильных производных мышьяка, ариларсоновз
кислот, используют главным образом реакцию Барта [201, 202], т. е. взаимодейств
соли арилдиазония с арсенитом щелочного металла, например [203]:
CeH6N2C14-Na3AsO3 —> CeH5AsO(ONa)34-NaCl + Ns
Реакция Барта может применяться также для синтеза диариларсиновых кисл«
C6H5N2C14-CeH6As(ONa)8 —> (C6H5)2AsO(ONa) +NaCl + N2
Этим же методом получают диариларсиповые кислоты с двумя различны
ароматическими группами.
Арильные производные мышьяка можно синтезировать и по реакции Несм<
нова. Так, разложение двойных солей—галогенидов арилдиазопия и FeCl3 или Zn<
действием AsCls и металлического железа приводит к арилдихлор- и диарилхлс
арсинам [204]:
ArN2Cl • FeClg -f- AsCla -f- Fe —► ArAsCla-f-FeCla-f-FeCIaH-Na
2ArN2Cb FeCb+ AsCl3-h2Fe —> Ar2AsCl + 2FeC13-|-2FeCI2 + 2N2
Действием азотной кислоты полученные таким образом соединения легко nj
вращаются в соответствующие ариларсоновые или диариларсиновые кислоты.
С хорошими выходами триариларсины образуются при разложении хлррид;
арилдиазония или их двойных солей (ArN2)2ZnCl4 при действии AsCls и.
хлорарсицов и суспензии цинковой пыли в ацетоне [205]'.
3ArN2CI-|- AsClg-J-3Zn —► АгзАб-f-3N2-f-3ZnCl2
Удобным методом синтеза некоторых ариларсоновых кислот служит так назы«
емая реакция Бешана [202], хотя область ее применения сильно ограничена. Эта реа
ция заключается в непосредственном взаимодействии мышьяковой кислоты с аромат
ческимл соединениями, содержащими гидроксильные или аминогруппы.
например, пз анилина и мышьяковой кислоты при нагревании до температуры око
160° С получают п-аминофеннларсоиовую кислоту (арсаниловую кислоту):
СвНб~ NHa + HaAsO* —► H2N- С6Н4— AsO(OH)2-J-H2O
Аналогичная реакция с фенолом приводит к п-оксифениларсоновоЙ кислое
Общим методом получения арсоновых кислот, в первую очередь кислот алнфах
ческого ряда, является реакция Мейера [202, 206], которая заключается во взаимоде
ствии алкилгалогенидов с арсенитом натрия:
RX-f-NagAsOa —► RAsO(ONa)2-[-NaX
[200]
[201]
202
203
204
205
206
A. Cah ours, A. Riche, Ann., 92, 361 (1854); H. Landolf, Ann., 3
301 (1854).
II. Bar t, герм. пат. 250264; С., 1912, II, 882.
C. S. Hamilton, J. F. Morgan, Org. Reactions, 2, p. 415.
H. Schmidt, Ann., 421, 159 (1920).
О. А. Реутов, И. Г. Бундель, ЖОХ, 25, 2324 (1955).
W..E. Hanby, W. A. Waters, J. Chem. Soc., 1946, 1029.
G. Meyer, Ber., 16, 1439 (1883).
668
ОБРАЗОВАНИЕ МЕТАЛЛ-УГЛЕРОДНЫХ СВЯЗЕЙ
Метиларсоновая кислота. Сначала получают Na3AsO3 взаимодействием
99 г As2O8 в 250 мл воды с 3 моль NaOH. К полученному раствору добавляют
50 мл метилового спирта и 145 а йодистого метила и встряхивают реакционную
массу в течение 24 ч, периодически охлаждая ее в первые часы взаимодействия.
Иа образующейся с выходом 95% от теоретического натриевой соли метиларсо-
новой кислоты выделяют свободную кислоту, которая может быть очищена
через кальциевую соль.
Аналогично из натриевой соли алкиларсонистой кислоты и алкил галогенидов
получают натриевые соли диалкиларсиновых кислот:
RX + HAs(ONa)8 —> H2AsO(ONa) + NaX
2, Органические производные сурьмы '
Органические производные сурьмы можно получать, в общем, теми же методами,
, что и аналогичные производные мышьяка, например действием алкилиодидов на сплав
сурьмы и калия [207]:
ЗНХ + КзЗЬ —► RsSb-1-ЗКХ
Лучшие результаты, особенно при синтезе более высокомолекулярных триавил-
стибинов, дает метод, разработанный Михаэлисом и Реезе. Он заключается во взаимо-
действии арилбромидов, SbCla и избытка натрия в бензоле или исилоле [198, 208]
(реакция протекает при повышенной температуре, поэтому применение эфира неже-
лательно).
Три-м-ксилилстибин [208]. В 150 мл сухого бензола растворяют 56 s
бромксилола и 23 в SbCl8, добавляют 50 г нарезанного тонкими лиоточками
металлического натрия и смесь нагревают до начала реакции. После окончания
взаимодействия реакционную массу оставляют на ночь, фильтруют, продукт
трижды экстрагируют из остатка кипящим бензолом. Бензольные растворы
объединяют и отгоняют растворитель, маслянистый остаток выливают в абсо- ,
лютный спирт; при этом через неснолько минут начинается выделение стибина *
в виде длинных- игольчатых кристаллов. Выход ^продукта 25 г (57% от теоре-* 1-
тического).
Большое число органических производных сурьмы можно получить взаимодбй-
ствием галогенидов сурьмы с реакционноспособными металлоорганическими соедине-
ниями (главным образом с реактивом Гриньяра, а также с литий-и цинкорганическнми
соединениями). Так, из соответствующих магнийоргапических соединений и SbClj
можно синтезировать практически все триалкил- и триарилстибины [209].
В -то же время при помощи литийорганических соединений синтезируют
пентаарильныеи пентаалхильные производные сурьмы. Из дихлорида трифенилстибина
или бромида теТрафенилстибония и фениллития получается пентафенилсурьма [194]: '
[CeH6)aSbCls+2CeHeLi —> (CflHs)6Sb+2LiCl
(СеНб^ЗЬВг+СвНбЫ —* (CaH6)6Sb-[-LiBr
С избытком фениллития пентафенилсурьма легко реагирует с образованием
гексафенилантимоната лития:
(CeH^Sb-f-CeHeLi —> Li[(CeH5)eSb]
[207] Н. L a n d о 11, Ann., 78, 91 (1851): С. Lowig, Е. Schweizer Ann., i
75, 315 (1850). ’ ' i
[208] A. E. Goddard, J. Chem. Soc., 124, 2315 (1923).
[209] W. J. C. Dyke, W. C. Davies, W. I J ones, J. Chem. Soc., 1930, -
463; H. Hibbert, Ber., 39,160 (1906); P. Pfeiffer, Ber., 37, 4620 (1904);
L. H. Long, J. F. S a c k m a n, Trans. Farad. Soc., 51,1062 (1955); J. I. H a r- *
г i s, S. T. Bowden, W. J. Jones, J. Chem. Soc., 1947, 1568.
V. ОРГАНИЧЕСКИЕ ПРОИЗВОДНЫЕ ЭЛЕМЕНТОВ V ГРУППЫ
669
Аналогично приведенным выше превращениям получают пентаметилсурьму
действием метиллития на дибромид триметилстибина или тетраметалстибонийиодид
[195]. '
Различными методами можпо синтезировать арильные производные сурьмы
из солей арилдиазония. Так, арилстибоновые кислоты образуются при разложении
солей арилдиазония антимонитом щелочного металла (Sb2O3 + щелочь) [210, 211].
Другим вариантом этого метода является получение в кислой среде двойной соли
арилдиазония и SbCl3, например CgfigNjCl. SbCl3, которую затем разлагают рас-
твором NaOH.
Разложение солей арплдиазония раствором КОН в присутствии окиси арил-
стнбина дает диарилстибиилвьте кислоты [210]:
RNaX-[-RSbO + 2KOH —> RaSbOtOKJ + KX-l-HzO-l-Ns
Арильные производные сурьмы из солей арилдиааонин можно получать также
по реакции Несмеянова.
Известно большое число двойных солей различных галогенидов сурьмы с солями
арилдиазония, например ArNBCl«SbCl3, ArN2Cl-SbCU [212, 213], ArNaCLAr'SbCla
[214], ArN3Cl-Ar2SbCI3 [215] и др. Разложением этих соединений порошкообразными
металлами можно получать самые разнообразные арильные производные сурьмы.
При разложении двойных солей типа ArN2Cl-SbCl3 цинковой пылью в ацетоне
образуется смесь [211, 216], состоящая из ArSbC]., ArBSbCl, Ar8Sb и ArsSbCle.
А при разложении этих же солей в ацетоне порошкообразным железом при 0° С основ-
ным продуктом реакции становятся трихлориды диарильных производных сурьмы,
наряду с чаще всего незначительными количествами перечисленных выше продук-
тов [217]:
2ArN2Cl • SbCls + FeJ—»- Ат2ЗЬС1з+2N2+FeCl2+SbCb
Трихлорид бис-п-нитрофенилсурьмы [217]. К раствору 15 г (0,036 моль)
двойной соли хлорида n-иитрофенилдиазоиия и SbCl3 в 45 мл сухого ацетона
в течение 35—40 мин при 0° С и интенсивном перемешивании добавляют 2 г
(0,036 г-атом) порошкообразного желоза и перемешивают еще 40 мин. Неорга-
нический остатон отфильтровывают и упаривают фильтрат. К остатку добавляют
50 ли 5 н. НС1, а затем 25—30 мл 96%-него спирта. Полученный раствор
выливают в избыток 5%-ного аммиака со льдом, при этом выпадает бис-п-нитро-
фенилстибиновая кислота. Выход продукта 7 г (97% от теоретичесиого). Кис-
лоту переводят в соответствующий хлорид кристаллизацией из 5 и. НС1.
Соединения типа Ar2SbCl8 и Ar3SbCl2 образуются также прп разложении порош-
кообразным железом в ацетоне двойных солей состава ArNjCI-SbCl*. Эти двойные
соли получают из двойных солей арилдиаэонннхлорпда и FeCla действием SbCU в хло-
роформе или четыреххлористом углероде и осаждают пз раствора эфиром [212].
2ArN2Cl«SbCle+3Fe —> Ar2SbCl3-(-2Na+SbC13-|-3FeCl2
3ArN2Cl • SbCIe + SFe —> AraSbCl24-3N24-2SbC13 + 5FeCl2
[210]' H. Schmidt, Ann., 421, 174 (1920).
[211] А. Б. Брукер, ЖОХ, 18, 1297 (1948); С. A., 43, 4647 (1949).
[212] О. А. Реутов, В. В. Кондратьева, ЖОХ, 24, 1259 (1954);
О. А. Р ру то в, ДАН СССР 87, 73 (1952).
[213] О. А. Реутов, ДАН СССР, 87, 73 (1952).
[214] О. А. Реутов, А. Г. Марковская, ДАН СССР, 98, 979 (1954).
[215] О. А. Реутов, А. Г. Марковская, А. Н. Л о в ц о в а, ДАН СССР,
99, 269 (1954).
[216] А. Н. Несмеянов, К. А. Кочешков, Иав. АН СССР, ОХН, 1944,
416.
1217] А. Н. Н о с м е я н о в, О. А. Реутов, О. А. Птицына, ДАН СССР,
91, 1341 (1953).
670
ОБРАЗОВАНИЕ КРАТНЫХ С-С-СВЯЗЕЙ
Разложение двойных солей хлорида диазония и SbClg в ацетоне действием СнгС]г
протекает по следующему уравнению [218]:
ArN2Cl-SbCl5 + Cu2C12 —> ArSbCl4-f-N3-J-2CuCl2
По реакции Несмеянова можно синтезировать также смешанные арильные про-
изводные галогенидов сурьмы. Соединения общего состава ArAr'SbC]3 получают»,
например, разложением двойных содей общей формулы ArN2Cl-Ar'SbCl4 порошкооб-
разным железом в ацетоне [219]:
ArN2C] • Ar'ShCU-)- Ре ТрёсП"* ArNjCl. Ar'SbCI? “"* ArAr'SbCfo
Промежуточным продуктом в этой реакции является, вероятно, двойная соль
хлорида арилдиазония и арилстибица, образующаяся, по данным Реутова, также пз
арилдпхлорстибнна и двойной соли хлорида арилдиазония с FeC]3 [10]:
Ar'SbCl9 + ArNgCl - FeClg —> ArN2Cl-Ar'SbCbH-FcCh
Эта соль легко распадается с выделением азота, образуя ArAr'SbCl3.
При разложении солей арилдиазония порошком элементарной сурьмы в аце-
тоне или этилацетате в присутствии СаСО3 получают в зависимости от строения ариль-
ного остатка различные количества дихлорида триарилстибина (Ar3SbClg), а также
триарилстибип и диарилхлорстпбин [13].
Реутов с сотр. в последние годы исследовал еще одну очепь интересную реакцию,
при помощи которой можно получать арильвые производные сурьмы на основе солей
циарилгалогония. Обнаружено [220], что при разложении двойных солей диарил-
иодонийхлорида и SbCl3 общего состава (Ar2ICl)2- SbCJ3 и Ar2ICl-SbCl3 порошкообраз-
ной сурьмой в ацетоне или этилацетате при температурах до 120° С образуются соеди-
нения общего состава Ar3SbCl2, Ar3SbCl3 и Ar3SbCl.
3. Ввсмуторганические соединения [221]
Известны алкил- и арплпроизводпые трех- и пятивалентного висмута. Важней-
шие способы получения этих соединений, в общем, аналогичны методам, которые
используются для синтеза соответствующих производных мышьяка п сурьмы.
ОБРАЗОВАНИЕ КРАТНЫХ УГЛЕРОД-УГЛЕРОДНЫХ
СВЯЗЕЙ *
ЭТИЛЕНОВЫЕ СВЯЗИ
Этиленовые связи в углеродном скелете образуются путем отщепления замести-
телей у двух соседних атомов углерода. J [ри этом следует различать, является лл воз-
никшая двойная связь составной частью соиряжеппой, ароматической или гетероцикли-
ческой системы, или же она изолирована.
* Материал обработал И. Эйххори.
[218] А. Н. Несмеянов, О. А. Реутов, Р, Г. К н о л ь, Изв. АН СССР,
1954, 410.
[219] О. А. Реутов, А. Г. Марковская, ДАН СССР, 99, 543 (1954).
[220] О. A. R е u t о w, О. А. Р I i i. у и a, G. Е г t е 1, Chem. Techn., 10, 201
(1958k
[221] Н. Gilman, Н. L. Yale. Chem. Rev., 30, 281 (1942).
Г ИЗОЛИРОВАННЫЕ ЭТИЛЕНОВЫЕ СВЯЗИ
61
I. Изолированные этиленовые связи
1. Отщепление воды от спиртов
Для получения двойных связей наибольшее препаративное значение име
дегидратация спиртов. Дегидратация может осуществляться как действием водоотним
ющих средств, так и каталитически. Важнейшими дегидратирующими средствам
являются: концентрированная серная кислота, бисульфат калия, хлористый цин
фосфорный ангидрид, ангидрид борной кислоты, щавелевая и муравьиная кислота
ацетилхлорид, реактивы Гриньяра, иод и др.
Отщепление воды не всегда протекает однозначно. Часто в процессе дегидратацц
происходят изомеризация, перегруппировка или расщепление углеродной цепи. Таз
отщепление воды от н-бутанола [1] нагреванием'его с 85%-ной Н8РО4 или 60%-нс
H2SO4 приводит к образованию смеси бутена-1 и бутена-2. В некоторых случаях од
фпны из смеси изомеров, как показано на примере изомеров октена [2], можнр выдели^
фракционной перегонкой. При нагревании циклогексена и его гомологов до 470—480°
над окисью алюминия происходит изомеризация и образуются алкилциклопентаны [3
Спирты, содержащие разветвления у а-углероцного атома, например метил
щреш-бутилкарбинол, при дегидратации легко подвергаются перегруппировке [4, 5
2(СНз)3С-СНОН-СНз (СН3)2С=С(СН3).2^(СН3)2СН-С=СН2
СНз
Расщепление углеродной цепи происходит, например, ппи нагревании да-трет
бутилкарбинола с хлорнафталинсульфокислотой при 180° С [6]:
(СН3)зС-СНОН-С(СНз)з _н>о> (СНз)2С-СН-СНз + (СНз)2С=СН2
В общем случае направление отщепления воды приближенно определяется пра
видом Зайцева, согласно которому атом водорода отщепляется от углеродного атома
содержащего наименьшее число атомов водорода [7]. Так, например, при взанмодеа
ствии вторичного изоамцлового спирта с серной кислотой образуется триметилэтилен
а не жзопропилэтилен:
(СНз)2СН—СНОН-СПз “ГЩо-* (СНз)аС=СН-СНз
Как уже упоминалось (стр. 266), при обратной реакции водородных
атом воды присоединяется к углеродному атому, более богатому водородок
так что образуется ие вторичный изоамиловый спирт, а шретп-амиловый спир*
(СН8)аСОН—С2Н6.
Способность к дегидратации в значительной степени зависит от условий реак
ции и особенно от строения соответствующего спирта. Легкость отщепления воды ува
личивается в ряду: первичные спирты < вторичные спирты < третичные спирты
Для получения олефинов из третичных спиртов часто оказывается достаточным яагра
ванне в присутствии следов иода нли 15%-ной H2SO4 [8]. Например, 2-метилгексе*
[1] W. G. Young, Н. J. Lucas, J. Am. Chem. Soc., 52, 1964 (1930).
[2] F. C. Whitmore, H. E. Whitmore, N. C. Cook, J. Am. Chem. Soc.
72, 51 (1950).
[3] H. Adk ins, A. K. R о e buck, J, Am. Chem. Soc., 70 , 4041 (1948); Ho u-
ben-Weyl, Methoden der organischen Chemie, 4 Aufl., 4/2, Georg Thiem«
Verlag, Stuttgart, 1955, S. 14.
4] F. C. Whitmot e, P. L. Mcunic r, J. Am. Chem. Soc., 55, 3721 (1933i
5] P. L. Cramer, A. L. Glasebrook, J. Am. Chem. Soc., 61, 230 (1939}
6| F. C. Wh itmore, E. E. Stably, J. Am. Chem. Soc., 55, 4153 (1933).
7] A. Saytzeff, Ann., 179, 300 (1875).
8] J. M. Church, F. C. Whitmore, J. Аш. Chem. Soc., 56, 182 (1934).
672
ОБРАЗОВАНИЕ ЭТИЛЕНОВЫХ СВЯЗЕЙ
получается уже прп добавке к кипящему 2-метилгексанолу-2 следов иода [9]. При кипя-
чении тгеретге-амилового спирта [10] с 15%-ной H2SO4 образуется 2-метилбутен-2 и не-
большое количество 2-метидбутена-1:
ОН
СН8СН2-С—СНз —но-* СНзСН=С-СН8тСП3СП2-С^СН2
I ' I I
СНз СНз СНа
В тех случаях, когда третичное гидроксилсодержащее соединение синтезируют
по реакции Гриньяра, нагреванием продукта присоединения магнийорганического
соединения удается непосредственно получить соответствующий олефин, не выделяй
спирт. Для этого после взаимодействия алкнл- или арилмагпийгалогенпдов с карбо-
нильным соединением и отгонки растворителя остаток короткое время нагревают,
при этом отгоняется олефин [11]. Таким образом был синтезирован, например, дифе-
нилэтилен из этилацетата и фенилмагнийбромида [12]. 1-Метнл-1-фенилэтилен,;
С6Н5С(СН3)—СН2, был получен из ацетофенона и метилмагинйиодпда [13].
1-Метил-1-фенйлэтилен. Продукт, полученный обычным методом иа
2 моль метилмагнийиодида и 1 моль ацетофенона, после отгонки эфира*
нагревают при 100Q С в течение 6 ч. Затем разлагают образовавшийся продукт'
водой, экстрагируют алкен эфиром, сушат и перегоняют. Выход 1-метил-;
1-фенилзтилена 5—6 г (из 10 г ацетофенона); т. кип. 162° С.
Так как способность к дегидратации у первичных и вторичных спиртов довольно;
низка, для их дегидратации необходимо применять высококипящие минеральные кис-
лоты (Нг$О4 или Н3РО4) или такие сильные водоотнимающие средства, как KHSO^
или ZnCl2.
При взаимодействии кислот со спиртами, подвергающимися дегидратации, вна-
чале образуются соответствующие эфиры, от которых уже при-температуре около 2Q04 Cj
отщепляется олефин и регенерируется кислота:
RCH3CH2OH + H2SCU -н,о> HCH2CHaOSO3H -Htso/* RCH=CHa
Применение концентрированной серной кислоты препаративно просто, но, одй
нако, многие обстоятельства ограничивают возможность ее использования. При цовы^
шейной температуре серная кислота вызывает окисление, обугливание и изомеризацию^
кроме того, в этих условиях идет образование простых эфиров. Поэтому для дегидра-Д;
тации вместо H2SO4 часто предпочитают применять фосфорную кислоту. Таж^'*-
например, в лабораторных условиях синтез этилена из этилового спирта действием^
серной кислоты недостаточно удобен; его получают при помощи фоофорной кислого!^’
или лучше всего каталитически, пропуская пары спирта над нагретым гетерогенным^
катализатором [14]. Для синтеза циклогексена из циклогексанола вместо концентри*-
рованной серной кислоты, которая легко вызывает обугливание и загрязнение продукте
двуокисью серы, лучше применять бисульфат калия ,[15]. Быстро и с хорошим выходов
образуется циклогексен из циклогексанола, если в качестве дегидратирующего сред-1
ства используют фосфорную кислоту [16].
Циклогексен [16]. Помещают 1000 г циклогексанола и 375 мл 85%-ной
Н3РО4 в колбу емкостью 2 л, снабженную колонкой Вигре. соединенной,
с нисходящим холодильником. Смесь нагревают до кипения. При 95е С иачи»
[fl] G. Edgar, G. Calingaert, R. Е. Marker, J. Am. Chem. Soc., 51,
1483 (1929).
[10] F. C. Whitmore, C. S. Rowland et al., J. Am. Chem. Soc., 64, 297ft
(1942).
[11] J. M. C h r u c h, F. C. Whitmore, J. Am. Chem. Soc., 66, 183 (1934). a
[12] C. F. H. Allen, S. Converse, Org. Synth., Coll. v. 1, p. 219.
[13] A. Kl ages, Ber., 35, 2640 (1920).
[14] P. K. Saak min, Ber., 67, 392 (1934). -J
[15] Gait ormann - W i eland, Die Praxis des organischen Chemikers, 36 Aufl(j
Verlag Walter de Gruyter u. Co., Berlin, 1954, S. 100.
[16] В. B. Corson, V. N. Ipatieff, Org. Synth., Coll. v. 2, p. 152; E. L в
v a s, Ann. chim. [12], 3, 149 (1948). -<
1. ИЗОЛИРОВАННЫЕ ЭТИЛЕНОВЫЕ СВЯЗИ
673-
нается отгонка воды и циклогексена. В процессе отгонки температура пони-
жается до 72° С. Перегонку прекращают, когда в реакционной колбе образу-
ются два слоя жидкости, т. е. начнется полимеризация циклогексена.
Полученный дистиллят разделяется на два слоя. Нижний слой, состоящий
из воды и небольшого количества циклогексанола, отбрасывают, а верхний
после сушки над СаС12 перегоняют. Остаток после перегонки состоит на неболь-
шого количества непрореагировавшего циклогексанола. Выход циклогексена
690—700 г (до—91% от теоретического, считая по прореагировавшему цикло*
гексаподу); т. кип, 82—83° С.
Для дегидратации терпеновых спиртов н других соединений, которые могут-
легко подвергаться перегруппировкам, используют щавелевую кислоту [17]. При
кипячении терпинеола с водным раствором щавелевой кислоты получается в основном
терпинолен [18], в то время как при взаимодействии с серной кислотой происходит
изомеризация и образуется терпинеи.
Тетраметилэтилен [19]. Нагревают 408 з диметилизопропилкарбияола
с 1200 з сухой щавелевой ияслоты при 100° С в течение 8 ч; затем смесь пере-
гоняют. Дистиллят после многократной промывки ледяной водой сушат над
CaCIj и перегоняют над металлическим натрием. Полученный дистиллят раз-
деляют на две фракции фракционной перегонкой. Выход тетраметилэтгдека
80% от теоретического; т. кип. 72,9—73,2° С (760 -и* рт. ст.). •
В качестве побочного продукта образуется около 20% несимметричного^
метилизопропилэтилена: '
(СПз)2СОН-СН(СНз)2 —-> (СНз)гС=С(СНа)2+ (СНз)2СН-С=СН2
СНз
Для дегидратации терпеновых спиртов применяется также фталевый ангидрид*
[20]. Дегидратирующее действие фталевого ангидрида значительно усиливается при*
добавлении небольшого количества бензолсульфокислоты [21]. Например, при дей-
ствии фталевого ангидрида в присутствии бензолсульфокислоты пропанол-2, бутанол-2,
2-метилпропанол-1, циклогексанол и другие спирты легко дегидратируются до соответ-- _
ствующих алкенов.
С хорошими выходами и без примесей, обычно образующихся в результате пере-
группировок, получают ненасыщенные соединения пиролизом сложных эфиров соот-
ветствующих спиртов. Методика очень проста. Пиролиз проводят в жидкой или паро-
вой фазе при 300—500° С. Обработка продуктов реакции также упрощается, так как:
в них отсутствуют растворитель и другие реагенты. Пиролизом ацетата метилизопро—
пилкарбинола при 500° С получают чистый 2-метилбутен-З с 72%-ным выходом. Одно- •
значно протенает пиролиз ацетйта метилизобутнлкарбинола [22], в результате которого
получается 2-метилпентен-4. В этих случаях образование олефина происходит претив,
правила Зайцева:
(СНз)2СНСН2СНСНз (СНз)2СНСН2СН=СН2
ОАс
Пиролизом можно получить даже очень легко иаомеризуюпщйся 1,2-диметпл- -
иденциклогексен-4 из соответствующего ацетильного производного- [23]:
уСН2ОАс /ч zCH2
/у
_____________ zVh,ov
[17] N. Zelinsky, J. Zelikow, Ber., 34 , 3249 (1901).
[18] O. Wallach, Ann., 275, 106 (1893).
[19] 1. Schurman, С. E. В о о г d, J. Am. Chem. Soc., 55, 4932 (1933).
[20] J. Zelikow, Ber., 37, 1376 (1904); S. Nametkln, Ann., 440, 6& (1924);:
G. Komppa, G. A. Nyman, Ann., 535 252 (1938).
[21] H. Waldmann, F. Petru, Chem. Ber., 83, 287 (1950).
[22] W. J. В ailey, C. King, J. Am. Chem. Soc., 77, 75 (1955).
[23] W, J. В alley, J. Rosenberg, J. Am, Chem. Soc., 77/ 73 (1955).
43 Заказ 1835.
•Ч>74
ОБРАЗОВАНИЕ ЭТИЛЕНОВЫХ СВЯЗЕЙ
1,2-Диметилиденциклогексен-4. В вертикальную трубку для сжигания
(20 X 300 лл) с боросиликатной насадкой, нагретую до 500° С, в токе азота
по каплям вводят 300 г (1,33 моль) диацетата цис-1,2,3,6-тетрагидро-1,2-кси-
лиленгликоля со скоростью 1,5 г/мин,. Продукт реакции конденсируют, промы-
вают водой, сушат над К2СО3 и фракционируют. Выход 1,2-диметилпдеицикло-
гексена-4 составляет 68,1 г. В результате фракционирования получают также
исходное диацетильное производное и продукт пиролитического отщепления
одной ацетильной группы.
При обработке некоторых третичных спиртов ацилирующими средствами
в присутствии следов ZnCl, синтез олефинов происходит без промежуточного образова-
ния сложных эфиров [24]. Особенно легко подвергаются пиролизу эфиры высших
жирных кислот. Например, и-додедиловый эфир пальмитиновой кислоты при пере-
гонке при незначительном разрежении очень гладко образует додецилен-1 и пальмити-
новую кислоту [25].
Мягким дегидратирующим средством, которое в некоторых случаях весьма
пригодно для отщепления воды не только от первичных, но также от вторичных
п третичных спиртов, является борная кислота. Борная кислота вначале образует
со спиртом тримерный эфир метаборной кислоты, который разлагается до соответству-
ющего олефина с выделением борной кислоты; в этом обнаруживается некоторое сход-
ство с механизмом пиролиза сложных эфиров, хотя дегидратация борной кислотой
проводится при более низких температурах (300—350° С). Нагреванием эквимолеку-
лярных количеств н-гептанола, н-октанола, zt-гексанола, ментола, циклогексанола,
холестанола-ЗВ с борной кислотой можно легко получить соотвяте.твутопите оле-
фины [26, 27].
Так же, как и при пиролизе сложных эфиров, в результате термического разло-
жения эфиров ксантогеновой кислоты [28] (реакция Чугаева) образуются непасы
-щенные соединения, не содержащие примесей, возникающих в результате перегруп-
пировок *:
S
II
Н2СНС(Н2)ОСЗСНз —► RaC=CR2 + COS + CH3SH
Разложение происходит уже при 110—180° С и протекает, таким образом,
•в более мягких условиях, чем пиролиз сложных эфиров.
Ксантогенаты получают нагреванием соответствующих спиртов с серо-
углеродом и гидроокисями щелочных металлов [29], щелочными металлами
[30] или гидроокисью натрия [31] и взаимодействием образующегося ксанто-
гената щелочного металла с алкилиодидом. Большая часть применяемых ксан-
тогенатов является S-метилпроизводными.
О пиролитических реакциях, в результате которых образуются ненасы-
щенные соединения, см. обзор [32].
[24
[25
[26
[27
[28
[29]
[30]
[31]
.[32]
Другой важной особенностью реакции Чугаева является то, что она протекает
в большинстве случаев как реакция i/uc-элимннирования (Органические реак-
ции, сб. 12, Изд. «Мир», 19б5, стр. 71). — Прим. ред.
М. Н. Masson, С. г., 132, 484 (1901.
F. Krafft, Вег., 16, 3020 (1883).
G. L. O'Connor, Н. R. Nace, J. Am. Chem. Soc., 77, 1578 (1955).
W. Brandenberg, A. G a 1 a t, J. Am. Chem. Soc., 72, 3275 (19501.
L. Tschugaeff, Ber., 32, 3332 (1899).
F. C. Wh i t того, С. T. Simpson,]. Am. Chem. Soc., 55, 3809
(1933).
D. J. С га ш, J. Am. Chem. Soc., 71, 3887 (1949); E. R. Alexander,
A. Mudrak, J. Am. Chem. Soc. 72, 1810 (1950).
G. L. O’Conner, H. R. Nace, J. Am. Chem. Soc., 74, 5454 (1952).
С. H. DePuy, R. W. King, Chem. Rev., 60, 431 (1960).
I. ИЗОЛИРОВАННЫЕ ЭТИЛЕНОВЫЕ СВЯЗИ
67
Оиц&пление воды особенно легко происходит у спиртов, гидроксильная грущ
которых находится в ^-положении к кето-, карбалкоксп-, интро- или нитрильн»
группам. Так, при альдолизации производных ацетальдегида под действием имеющих»
в реакционной среде кислот наступает дегидратация с образованием а,р-ненасыщеннь
карбонильных соединений, т. е. соединений типа кротонового альдегида. Гептен-З-он
удается получить из гептанол-4-опа-2 уже при кипячении последнего в течение 1
с разбавленной серной кислотой L33J:
СНзСН2СН2СНСН2СОСНз -Zh^ СН3СП2СН2СН=СНСОСНз
он
Днаиетоновый спирт (СН3)2СОНСН»СОСН3 в присутствии следов иода дегидр»
тируется до окиси мезитила (СН3)гС=^СЙ'СОСН!|.
Окись мезитила [34]. К 700 г диацетонового спирта прибавляют 0,5
пода, смесь перегоняют с небольшой колонкой при атмосферном давлени]
После того, как около 2/з содержимого колбы перейдет в дистиллят, из остатг»
собирают фракцию, кипящую при 125 — 130° С и являющуюся чистой окись,
мезитила. Первые 2/з дистиллята образуют два слоя, из которых ннжний водны
слой отбрасывают, а верхний сушат над СаС12 и перегоняют. При этом получай;
ацетоновую фракцию (т. кип. 60—80е С), среднюю фракцию (т. кип. 80—125° <
и фракцию окиси мезитила (т. кип. 126—130° С). Из водного слоя, насыща
его К2СОа, можно выделить еще 11 г масла, состоящего из равных частей am
тона и окиси мезитила. Общий выход сырой окиси мезитила 578 г (98% от те<
ретического).
По другим данным [35], на 1100 г технического диацетонового спирх
достаточно 0,1 г иода.
р-Оксикислоты также легко отщепляют воду с образованием пепасыщеннщ
кислот. Однако этот способ не имеет большого значения, так как во многих случал
синтез ненасыщенных карбоновых кислот другими методами значительно легче, чв;
получение исходных оксмкислот.
У эфпров Р-оксикислот, образующихся по реакции Реформатского, очел
легко, часто уже при перегонке н омылении, происходит отщеплеине воды. Для полно
дегидратации необходимо нагревание с уксусным ангидридом, ацетилхлоридом
бисульфатом калия, муравьиной или серной кислотами. При этом в зависимости о
заместителей у 0-утлеродного атома образуются а.р- или р,у-ненасыщенные карбоне
выс кислоты, или их смесь [36]. В некоторых исключительных случаях из Р-оксикис
лот, выбрав соответствующее водоотнимающее средство, можно получить тольк
а,р- или Р ..у-ненасьпцеииые кислоты. Так, из цикпогсксанолуксусной [37] кислоту
(1) при действии KHSO4 или Р2О5 образуется циклогексенуксусная кислота (11)
а при действии уксусного ангидрида — изомерная а,р-ненасыщенная кислота (lllj
/у/ОН
I NcHsCOOH
* XHSCOOH
\/
II
/>Ч^СНСООН
III
При взаимодействии а-оксикислот с дегидратирующими средствами больше^
частью образуются не а,р-ненасыщенные кислоты, а в основном лактиды и ангидрида
пли в результате отщепления муравьиной кислоты — альдегиды. Однако а,р-иенасц
щенные кислоты легко можно получить омылением соответствующих а,р-ненасьпцен:
пых нитрилов, образующихся путем отщепления воды от а-оксинитрилов при помощи
[33] М. S m i t h, В. Chase, В. Rhodes, I. Am. Chem. Soc., 66/1548 (1944)
[34] H. Hibbert, J. Am. Chem. Soc., 37, 1755 (1915).
[35] B. Conant, N. Tuttle, Org. Synth., Coll. v. 1, 1941, p. 345.
[36] R. J,. Shriner, Org. Reactions, 1, 11 (19421.
[37] O. Wallach, Ann., 365, 257 (1909).
43*
*676
ОБРАЗОВАНИЕ ЭТИЛЕНОВЫХ СВЯЗЕЙ
тионилхлорида [38] или пятиокпси фосфора [39]. Примером может служить синтез
а-метплакрилонитрила.
а-Метил акрилонитрил [39]. В колбу емкостью 500 мл, снабженную
обратным холодильником с хлоркальциевой трубкой и капельной воронкой,
быстро отвешивают 75 г Р2О6; помещают колбу на водяную баню н при 10—15° С
по каплям прибавляют в течение 30 мин 42,5 г ацетонциангидрина при энер-
гичном перемешивании. Образующийся нитрил отделяют от вязкой массы
перегонкой. После повторной перегоняй выход а-метилакрилонитрила 11,5 г
(34,3% от теоретического); т. кип. 89—91” С.
Ненасыщенные кислоты типа малеиновой и фумаровой кислот можно легко
получить дегидратацией соответствующих оксикислот.
Фумаровая кислота [40]. Нагревают 500 а яблочной кислоты при 140—
150° С в течение 40 ч. Образовавшийся твердый плав растворяют в горячей
воде, отфильтровывают выпавшую носле охлаждения фумаровую кислоту,
фильтрат упаривают, а остаток, состоящий из непрореагировавшей яблочной
кислоты, вновь вводят в реакцию.
Малеиновую кислоту также можно получать дегидратацией яблочной кис-
лоты [41].
Каталитическая дегидратация спиртов имеет в основном большое значение ддя
промышленного синтеза олефинов, но применяется и в лабораторной практике, особенно
для получения низших олефинов: этилена, пропилена, бутилена. Подходящими ката-
лизаторами для этого процесса являются у-окись алюминия, окись тория и голубая
окись вольфрама. Первичные и вторичные спирты дегидратируются чаще всего в газо-
вой фазе при 250—450” С. Высококипящие спирты дегидратируются уже без катали-
затора при 150—200° С (стр. 672)-
2. Отщепление галогеноводородов
Отщепление галогеноводородов полностью аналогично отщеплению воды от
спиртов. Так как многие галогениды получают только из соответствующих гидроксиль-
ных соединений, то этот метод следует рассматривать в основном как обходный путь.
Однако известны многочисленные случав, когда ок обладает преимуществом. Дегидро-
галогенирование может проводиться двумя способами:
1. Термическое отщепление в присутствии катализаторов.
2. Отщепление действием органических или неорганических оснований.
Термическое отщепление галогеноводородов, которое протекает в большинстве
случаев по радикальному механизму, идет при довольно высокой температуре и боль-
шей частью в присутствие катализаторов. В качестве катализаторов применяют или
вещества с высокоразвитой поверхностью (каолин, кизельгур и др.), или металлы
[алюминий, никель, медь, цинк) и их безводные окиси. Так, из 1,2-дихлорэтана прн
гагревании до 600° С (20 ат) получают с хорошим выходом винилхлорид [42]. Анало-
ичная реакция происходит при 500° С в присутствии смеси SnO2 и FeCl2 на активиро-
занном угле [43].
Низкотемпературное дегидрогалогенирование под действием неорганических
тли органических оснований может протекать по двум механизмам [44] (в зависимости
[39
[40
[41
42
г43
44
[38] Л. Н. Cook, Н. Р. Linstead, J. Chem. Soc., 1934, 956; W. S. R a p s о n,
R. R obinson, J. Chem. Soc., 1935, 1538; L. E. King, R. Robinson,
J. Chem. Soc., 1941, 467.
D. Gotkis, J. B. Cloke, J. Am. Chem. Soc., 56, 2711 (1934).
A. Baeyer, Ber., 18, 676 (1885).
R. Anschutz, Ber., 14, 2791 (1881).
Пат. ФРГ 899191 (1951); Farbw. Hoechst, H. Krekeler; C., 1954, 7288.
J. C. G b о s h, S. R. p a s G u h a, Petroleum, 14, 261 (1951).
Houben-Weyl, Methoden der organischen Chemie, 4 Aufl., 5/4, Georg Thieme
Verlag, Stuttgart, 1960, S. 729.
I. ИЗОЛИРОВАННЫЕ ЭТИЛЕНОВЫЕ СВЯЗИ
677
от того, является ли первой стадией, определяющей скорость реакции, ионизация
соединения, содержащего галоген, или отщепление протона):
CHsCBjX -_к-' СНзСНг — > СН2=СН2 + Н2О
СН3СВ2Х -?!?•» СН2=СП»-г П,0 +Х-
В первом случае образование олефина зависит от полярности растворителя,
во-втором — от концентрации и силы основания.
Легкость отщепления, как и при дегидратации спиртов, тем больше, чем меньше
атомов водорода находится у галогенсодержащего углеродного атома, так что тенден-
ция к отщеплению увеличивается в ряду: первичные галогениды < вторичные гало-
гениды < третичные галогениды. Кроме того, она растет с увеличением атомного веса
галогена.
Отщепление галогеяоводорода, как и отщепление воды от спиртов, происходит
в соответствии с правилом Зайцева [45], согласно которому протон отщепляется пред-
почтительно от углеродного атома, содержащего меньшее число атомов водорода.
Благодаря этому обстоятельству возможно направленное перемещение двойной
связи посредством присоединения и обратного отщеплеиня галогеноводорода:
(СН3)2СН—СН=СН8+Ш —► (CHafcCH-CHI-CHa —> (СН3)2С=СН—СНз + Н!
Синт|р трнметилэтилена таким путем, конечно, не имеет практического значения,
так как его значительно удобнее получать из амиловых спиртов.
Шкала дегидрогалогенирующих средств достаточно обширна: от безводных
алкоголятов щелочных металлов, твердых щелочей или их растворов в воде или орга-
нических растворителях, солей щелочных металлов со слабыми кислотами до таких
органических оснований, как пиридин, аинлин, диметиланилин или хинолин.
Отщепление галогеноводорода часто сопровождается побочными реакциями:
омылением галогенсодержащего соединения неорганическими основаниями
RX-t-KOH —> ROH+KX
или обравованием простых эфиров при использовании алкоголятов:
НХ + КОСНз —► ROCHs+KX •
Отщепление галогеноводорода, легко протекающее в присутствии третичных
аминов — дцметилаинлина, пиридина или хинолина, сопровождается образованием
четвертичных аммониевых солей [46]. Хорошие результаты были получены [47] при
использовании третичных аминов, у которых образование четвертичных аммониевых
солей затруднено стерическими факторами, например в случае этилднизопропиламина,
этилдицшйгогексиламина. Так, при действии на 1,2-дибром-2-метилпропан КОН в эти-
ленгликоле [48] с очень хорошим выходом получается 1-бром-2-метилпропен-1:
СНзч СНзч
>С—СН2Вг —► Х=СНВг
СН3Х | ' CHZ
Вг
1-Бром-2-метилпропен-1. В трехгорлой колбе, снабженной мешалкой,
обратным холодильником и капельной воронкой, нагревают без доступа влаги
47 г (0,225 -ноль) этилдициклогексиламнна до 140° С н быстро прибавляют
по каплям 32,3 г (0,15 моль) 1,2-дпбром-2-метилпропана. Через 5,5 ч к колбе
присоединяют короткую колонку Вигре и при энергичном перемешикании
перегоняют реакционную массу, в результате чего получают 15,5 г дистиллята
(температура бани 140® С). При перегонке с водяным паром остатка, разбавлен-
ного 50 мл воды, выделяют еще около 1 г масла, которое объединяют с основ-
ным количеством и перегоняют еще раз. Выход продукта 5,5 е (77% от теорети-
ческого); т. кип. 91° С; ng 1,4623.
[45] A. Saytzeff, Ann., 179, 296 (1875).
[46] С. R. N о 1 1 е г, R. Dinsmore, J. Am. Cbem. Soc., 54, 1025 (1932).
{47J S. H ii n i-g, M. К i e s s 0 1, Chem. Ber., 91, 381 (1958). '
[48] E. A. Bra ude, E. A. E v a n s, J. Chem. Soc., 1955, 3328.
678
ОБРАЗОВАНИЕ ЭТИЛЕНОВЫХ СВЯЗЕЙ
Применяя этилднцпклогексиламин и этилдиизопропиламин, можно также
дегпдрогалотепировать а-галогенированные эфиры. Таким же методом получают с вы-
ходом 80—86% винилэтиловый и р-хлорвинилбутиловыи эфиры'[47].
Легко дегидрогалогенируются а,р-дигалогенкарбонильные соединения, давая
ненасыщенные а-галогенкарбонильные соединения. Например, а-бромкротоновая
кислота получается с выходом 90% при обработке а.Р-диброммаслянои кислоты вод-
ным раствором КОН [49].
При взаимодействии этилового эфира а.р-дибромиропионовой кислоты с этила- ' J
том натрия • в абсолютном этиловом спирте (охлаждение льдом) образуется <
с выходом 80% этиловый эфир а-бромакриловой кислоты [50]. Однако ненасыщенные ‘
карбоновые кислоты лучше получать отщеплением воды от оксикислот. Напротив,
ненасыщенные дикарбоновые кислоты типа малеиповой и фумаровой почти всегда '
синтезируют из галогензамещенных янтарных кислот. Фумаровая кислота легко обра-
зуется при кипячении мопобромянтарнои кислоты с водой [51] или нагревании ее '
выше точки плавления [52]; при этом отщепляется 1 моль НВг.
3. Отщепление галогена
Получение этиленовых сляттинений отщеплением галогена от 1,2-дигалогенидов
нецелесообразно, так как соответствующие дигалогениды могут быть синтезированы
почти исключительно путем присоединения галогена по двойной связи. Однако этот
метод важен тем, что продукты присоединения галогенов являются, большей частью,
хорошо кристаллизующимися, трудно растворимыми веществами, которые могут •
использоваться как для выделения веществ, так и для разделения смесей.
Дегалогенирующими средствами служат металлы: медь, магний, пипк, алюми-
ний, железо, щелочные металлы, а также гидроокиси или иодиды щелочных металлов, '
растворенные в этиловом спирте или ацетоне. Обычно отщепление галогена связано
со значительным тепловым эффектом, поэтому целесообразно применение растворителей '*
[53]. Общим методом дегалогенирования соединений, содержащих атомы галогена
у соседних углеродных атомов, является обработка цинковои пылью спиртового рас- >’
твора соединения.
4,4,4-Трифторбутен-1 [54]. Раствор 2,80 г 2-иод-1-хлор-4,4,4-трифтор- J
бутана в 3 мл этилового спирта медленно прибавляют к 10 г цинковой пыли
в 20 мл кипящего этилового спирта. Отгоняющийся продукт промывают водой
и перегоняют еще раз. Выход 4,4,4-трифторбутена-1 0,92 г (80% от теоретичен ?
ского); т. кип. 10,5° С.
F3C—СН2-СН1—СН2С1 Г:|С-СИ.- СН-СН,
Изолированные атомы галогена в этих условиях не отщепляются [55].
При дегалогопировапии 1,2-дигалогенопроизводных не происходят реакции С?
изомеризации и перегруппкровки. Одпако при дегалогенировании цинком или алюми- ’
нием 1,3-дигалогенопроизводных [56] и некоторых полигалогеносоединений в боль-
шинстве случаев нельзя избежать частичной циклизации. Для устранения циклизации
при дехлорировании полихлорсоединений применяют спиртовой раствор КОН в аце-
[49] N. Н. Cromwell, F. Pelletier, J. Org. Chem., 15, 881 (1950). -j
[50] H. Bretschneider, N. Karbitschka, G. Piekarski, Mh. Chem.r ’
84, 1084 (1952). r*
[51] R. F i 11 i g, L. D or n, Ann,, 188, 90 (1877); J- Volhard, Ann., 268, 256’
(1892); W. J. Muller, F. S uck er t, Ber., 37 , 2598 (1904).
[52] A. Kckulo, Ann., 180, 22 (1864); J. Volhard, Ann., 242, 158 (1887).
[53] Houben-Weyl, Mothoden der organischen Chemie, 4 Aufl., 5/4, Georg
Thieme Verlag, Stuttgart, 1960, S. 720. g
[54] R. N. H a s z e 1 d i n e, K. Leedham, B. R. Steele, J. Chenk-
Soc., 1954, 2040.
[55] C. Wilson, J. Chem. Soc., 1945, 50. U
[56] R. C, Fuson, H. Gilman, Organic Chemistry, v. I, 2 ed., Verlag J. Wi*1
ley a. Sons, New York, 1943, p. 74.
I. ИЗОЛИРОВАННЫЕ ЭТИЛЕНОВЫЕ СВЯЗИ
679
тоне [57]. Таким способом удалось получить с 84%-иым выходом а-перхлоргекса-
триен-1,3,5:
СС12=СС1—СС12—СС12—СС1=СС]2 К0П; ацртон> СС12=СС1—СС1=СС1—СС1=СС12
Дигалогепопроиаводпыс можно также дегалогенировать действием иодидов ще-
лочных металлон в ацетоне или других растворителях [58]. Образующиеся вначале
дииодиды неустойчивы и легко отщепляют иод, особенно в случае соединений, содержа-
щих карбонильную или карбоксильную группы.
Для дегалогенирования можно также применять смесь Mg и Mgl2 в абсолютном
эфире. Таким методом, например, получают диоксен из 2,3-дихлордиоксапа [59]:
О О
нас/ ' ( .нс, „, „ . н/'/ Хен
| j Mglg; Mg; эфир^ । ц
h2Cv zchci н2сх .CH
О О
Диоксен [59]. В трехгорлую колбу емкостью 5 л, снабженную капельной
воронкой, обратным холодильником с хлоркальциевой трубкой и мешалкой,
помещают 82,6 г (3,4 моль) магниевой стружки и 1200 мл абсолютного эфира.
Затем небольшими порциями добавляют 91,5 г (0,4 моль) иода (образование
Mglg происходит со значительным выделением тепла). К полученной бесцветной
смеси прибавляют по каплям при энергичном перемешивании 314 г (2 моль)
2,3-дихлордиоксана в 200 мл эфира с такой скоростью, чтобы окраска раствора
была светло-коричневой (продолжительность прибавления около 9 ч). После
перемешивания в течение 30 мин бесцветную смесь выливают в ледяную воду,
эфирный слой отделяют, сушат над Na2SO4 и перегоняют, собирая фракцию
, с т. ниц. 93—95’ С. Выход продукта 84,3 г (49% от теоретического).
Из возможных стереоизомеров этиленовых соединений при отщеплении галогена
пз дигалогенидов образуется в основном стабильный лгр««с-изомер, например из
дибром- и изодибромянтарной кислот — фумаровая кислота. Однако из эфира дибром-
коричной кислоты получается наряду с лгрляс-изомером около 10% эфира ifuc-коричной
(аллокоричной) кислоты [60].
Иногда дегалогенирование используют для получения производных неустойчи-
вых этиленовых соединений. Для этого сначала присоединяют к двойной связи галоген,
в результате чего образуется соответствующее производное, которое затем вновь
подвергают дегалогенированию. «
Метиловый или этиловый эфиры акриловой кислоты можно, например, получить
присоединением брома к акриловой кислоте с последующей этерификацией образовав-
шейся при этом а,р-дибромпропионовой кислоты й кипячением эфира в спиртовом
растворе с цинковой стружкой [61].
Интересный метод отщепления галогена основан на следующей реакции:
-СНХ-СН- —> —СН=СН—
I
ОСНз
Нагреванием p-галогенэфира с цинковой пылью [62] в 90—95%-ном этиловом,
н-пропиловом или изопропиловом спиртах можно отщепить галоген и алкоксигруппу
[57] A. Roedig, G. Voss, Е. Kucliinke, Ann., 580, 24 (1953).
[58] Н. Finkelstein, Вег., 43, 1528 (1910).
[59] R. К. Summerbell, R. R. Umnoef er, J. Am. Chem. Soc., 61, 3019
(1939).
[60] C. Liebermann, Ber., 24, 1108 (1891).
[61] Герм. пат.-555 933; C., 1932, 11, 1969.
[62] C. G. Schmi 11, С. E. В о о г d, J. Am. Chem. Soc., 54, 759 (1932).
680
ОБРАЗОВАНИЕ ЭТИЛЕНОВЫХ СВЯЗЕЙ
с образованием этиленовой связи. В основном применяют p-бромэфиры, которые полу-
чают по приведенной ниже схеме [63]:
CH3CI10 + CsH6OH-|-HCl —► СНз—СНС1 + Н2О
ОС2Н6
CH3-CHCI+ Вг2 —► СН2Вг-СНВг + НС1
I (
ОС2Н6 ОС2Н5
RMgBr+ Вт- СН-OCsH6 —► R—CH-OC2H5 + MgBr2
СН2Вг СН2Вг
Этим методом удалось синтезировать ненасыщенные жирные кислоты (олеиновую
и элаидиновую), идентичные природным соединениям [64].
Из хлорнонилового альдегида действием брома получают а-бром-со-хлорио
пиловый альдегид, который затем обрабатывают метанольным раствором .бромистого
водорода:
С1-(СН2)7-СНВг-СНО4 НВг+СНзОН —► С1-(СН2)7-СНВг—СНВт + Н2О
I
ОСНз
Полученный 8,9-дибром-9-метоксинонилхлорид дает с х-октилмагнийбройидом
8-бром-9-метоксигептадецилхлорид, при обработке которого цинковой пылью в кипя-
щем бутаноде образуется охлоргептадецен-8,9:
С1(СНг)7-СНВг-СН-(СН2)7-СНз —► С1(СНа)7-СН=СН-(СН2)7-СНз
ОСНз
Превращение его в нитрил и последующее омыление приводит к смеси олеиновой
и элаидиновой кислот.
4. Расщепление азотсодержащих соединений
В то время как элементы воды от спиртов можно отщеплять, прямо или косвенно,
при помощи водоотнимающих средств, аналогичное отщепление аммиака (или аминов)
от первичных, вторичных, а также третичных аминов протекает не всегда. Такую реак-
цию удается осуществить только в отдельных случаях: например, из галогеноводород-
ных солей аминов при отщеплении NH4C1 образуются олефины:
RCH2CH2NH2 • НС1 —> RCH=CHa + NH4Cl
Этот метод нашел препаративное применение прежде всего в гидроароматическом
ряду. Например, туйон, выделенный из масла туи, при нагревании с формиатом аммония
образует туйонамин[65], солянокислая соль которого при сухой перегонке даеттуйен.
Аналогично из 1,3-диамнио-1-метилцик*10гексана получили 2,3-дигидротолуол [66].
Основания Манниха [67, 68], которые образуются при конденсации соединений,-
содержащих активный водород, с формальдегидом и аммиаком или первичными или
вторичными аминами, являются во многих случаях ценными промежуточными продуй-,
тами при синтезе ненасыщенных соединений. При нагревании свободны* оснований иля
их солей происходит отщепление аминогруппы и образуются соответствующие неиасы-
[63]
[64
[65
[66
3397 (1930).
R. NoIIer, A. Bannerol, J. Am. Chem. Soc., 56,
0. Wallac h, Ann., 286, 99 (1895).
C. Harries, W. Antoni, Ann., 328, 88 (1903).
F. F. Blicke, Org. Reactions, 1, 303.
II о u b e n - W e у 1/ Methoden der organischen Chemie,
Thieme Verlag, Stuttgart, 1957, S. 731.
Am. Chem. Soc., 52,
1563 (1934).
4 Aufl., ц/1, Georgy
1. ИЗОЛИРОВАННЫЕ ЭТИЛЕНОВЫЕ СВЯЗИ
681
щенные соединения. Таким методом могут быть получены ненасыщенные кетоны [69],
альдегиды [70], кислоты [67] и ненасыщенные нитросоединения [71].
Метилвинилкетон [69]. Третичный амки, полученный кипячением аце-
тона, солянокислого диэтиламина и параформальдегида в изопропиловом спирте
в течение 6 ч, переводят в гидрохлорид и дезаминируют последний нагреванием
до 150° С; при этом образуется метилвинилкетон:
CHgCOCHa+CHsO+CCsIls^NH-HCl _Ht5> СНзСОСНаСН2^С2Н5)2 • НСЦ—>
—* CHaCOCH^CHa+^HsJaNH.HCI
Амины можно также превратить в ненасыщенные соединения через четвертичные
аммониевые основания. Из четвертичных аммониевых оснований, алкильные остатки
которых содержат два или более углеродных атома, при термической обработке
образуются наряду с я инном не спирТы, а олефниы и вода:
[(C3H7)(CsH5)(CH3)2N]+OH- —* CsH7N(CH3)2+CH2=CH2 + H2O
При этом из нескольких возможных олефинов всегда образуются олефины
с наименьшим числом атомов углерода [72]. Для получения олефинов таким методом
прежде всего синтезируют четвертичные аммониевые соли, в основном иодиды. Из
четвертичных аммониевых солей обработкой окисью серебра получают четвертичные
аммониевые основания, которые затем разлагают или термически или действием силь-
ных оснований. Таним образом, например, из гидроокиси пинаколилтриметиламмония
получают mpem-бутилэтилен [73].
mpem-Бутилэтилец, Метилируют пинаколиламин и из свободного амина
и нодйгстого Метила подучают четвертичную аммониевую соль. Обрабатывают
84 а пинаколилтриметиламмонийиодида избытком окиси серебра. Воду отго-
няют в вакууме (16—15 мм рт. ст.) до образования твердого белого вещества,
затем давление снижают до 0,01—0,005 жж рт. ст. и собирают продукт разло-
жения в ловушке, охлаждаемой жидким воздухом. Разложение длится около
трех недель. Конденсат промывают водой, отделяют углеводородный слой,
промывают его три раза 3%-ной НС1, два раза 2%-ным раствором К2СО8
и сушат над порошкообразным NaOH. Продукт очищают перегонкой. Выход
третп-бугилэтилена 13 г (50% от теоретического); т. кип. 41,1—41,6’С
(761 жж рт. ст.).
При исследовании механизма расщепления по Гофману было установлено
[74], что четвертичные аммониевые соединения могут*гакже образовывать соответству-
ющие олефины под действием металлоорганических соединений, например фениллития,
или при действии амида калия в жидком аммиаке в очень мягиих условиях.
Нитрилы с активированным водородом у p-углеродного атома могут переходить
в олефины под действием амида калия в жидком аммиаке с отщеплением синильной
кислоты [75]:
свн6-сн2-с(свн6)2 свнб-сн=с(свн6)а
CN
Трифенилэтилен, например, получают этим способом с выходом 94%'. Также
с хорошим выходом образуются 1,1,2-трифенилпропея-1 п тетрафснилэтплен.
[69] Н. J. Н a g е m е у е г jr., J. Am. Chem. Soc., 71, 1119 (1949).
[70] С. S. М а г v е 1, R. L. М у е г s, J. Н. S a u n d е г s, J. Am. Chem. Soc.,
70, 1694 (1948).
[71 ] А. Т. Blomquist, Т. Н. Shelley, J. Am. Chem. Soc., 70, 147 (1948).
{72j M. L. D h а г, E. D. Hughes et al., J. Chem. Soc., 1948, 2097.
[73] P. G. Stevens, J. H. R ich nt о n d, J, Am. Chem. Soc., 63, 3132 (1941).
[74] G. Wittig, R. Polster, Ann., 599, 13 (1956); 612, 104 (1958).
[75] C. R. Hauser, W. R. Brasen, J. Am. Chem. Soc., 78, 82 (1956).
ОБРАЗОВАНИЕ ЭТИЛЕНОВЫХ СВЯЗЕЙ
682
II. Сопряженные двойные связи
1. Диены
Методы, служащие для получения изолированных двойных связей, в принципе-
могут быть применены и для получения сопряженных двойных связей, причем образо-
вание сопряженных двойных связей происходит легче, так как оно часто энергетически
более выгодно. Важнейшими методами получения соединений с сопряженными связями
являются: отщепление воды от ненасыщенных спиртов; отщепление воды от гликолей;
отщепление галогеповодорода от ненасыщенных галогенидов; отщепление галогеноводо-
рода от дигалогенидов; дегалогенирование тетрагалогенидов; разложение четвертичных
аммониевых оснований (получаемых из ненасыщенных 1,4-диаминов исчерпывающим
метилированием).
Дегидратация ненасыщенных спиртов применяется очень редко, так как сами
олефиновые спирты являются трудно доступными соединениями. Отщеплением воды
от рицинолевой кислоты можно синтезировать октадекандиеповую кислоту [76],
которая благодаря наличию сопряженных двойных связей является цепным исходным
материалом для получения синтетических высыхающих масел типа древесного масла.
В случае дегидратации гликолей наиболее пригодны 1,2-, 1,3- и 1,4-диолы. Де-
гидратация пинакона 177] 48%-ной НВг дает с 60%-ним выходом 2,3-диметилбутадиеи,
более высокие выходы (86%) получают при пропускании паров пинакона над окисью
алюминия при 420—470° С.
В качестве примера дегидратации 1,3-диолов следует упомянуть синтез 1,3-
бутадиена. Такая дегидратация может быть осуществлена нагреванием с фосфорной
кислотой, фосфатами пли с хлорангидридом фосфорной кислоты [78].
Дпспы легко образуются также при пиролизе эфиров диолов, в основном ди-
ацетатов п диформиатов [79].
Отщепление галогеноводорода от ненасыщенных галогенидов находит приме-
нение при получении 2-фенилбутадиена-1,3 [80]:
СН2=С-СНС1-—СНз —> сн2=с—сн=сп8
Свн5 Свн5
2-Фенилбутадиен-1,3. Ненасыщенный хлорид растворяют в трехкратном
объеме пиридина и нагревают на кипящей водяной бане в течение 1 ч. После
охлаждения реакционной смеси прибавляют при перемешивании примерно
30%-ную охлажденную HoS04 и тотчас же небольшое количество гидрохинона.
Углеводород извлекают эфиром, промывают, сушат и перегоняют в вакууме.
Выход продукта около 50% от теоретического; т. кип. 66—67® С (13 мм рт. ст.).
Отщепление галогеноводорода легко достигается перегонкой ненасыщенного
галогенида с твердой гидроокисью щелочного металла. Таким образом синтезируют,,
например, 4-метилгексадиен-1,3 из 4-бром-4-метилгексена-1 [61].
В препаративных масштабах 1,4-дифенилбутадиен-1,3 легко получают отщепле-
нием галогеповодорода от дигалогенида. Для этого достаточно нагревать 2,3-дибром-
1,4-дифенилбутан С хинолином в течение 30 мин при 160? С [82].
Отщепление галогена от 1,2,3,4-тетрагалогепидов почти не имеет препаратив-
ного значения. Бутадиен-1,3~н лабораторной практике получают из 1,2,3,4-тетрабрОМ-
бутаца [83].
[76] J. Scheiber, Farbe u. Lack, 1929, 153; Angew. Chem., 46, 643 (1933).
[77] C. F. H. A I 1 e n, A. В e 1 1, Org. Synth., 22, 39, 40 (1942).
[78] J. Boeseken, W. J. v a n der Gracht, Rec. trav. chim., 56, 1206
(1937).
[79] L. D e n i v e 1 1 e, C. r., 208, 1024 (1939).
]80] K. A 1 (1 e r, J. II a у d n. Ann., 570, 208 (1950).
[81] N. A. Milas, A. McAlevy, J. Am. Chem. Soc., 57, 581 (1935).
[82] А. В. Домбровский, А. П. Терентьев, ЖОХ, 26, 2780 (1958).
[83] R. A. J а с о b s о n, J. Am. Chem. Soc., 54, 1545 (1932); E. H. Farmer,
C. D. Lawrence, j. F. Thorpe, J. Chem. Soc., 1928, 729.
и. СОПРЯЖЕННЫЕ ДВОЙНЫЕ СВЯЗИ
G83
2. Полиены
1 Гримером синтеза соединений с тремя сопряженными двойными связями является
синтез цик.тогсптатриена (тропилидена) по Вилынтеттеру [84].
Циклогептатриен. Исходным продуктом является циклогептанон IV.
Из циклогептанона сначала синтезируют цвклогептея V либо восстановлением
его до циклогептилового спирта с последующим превращением спирта в иодид
и отщеплением от иодида п! спиртовым раствором КОН, либо получением
циклогептаноноксима, восстановлением его до циклогептиламина, исчерпыва-
ющим метилированием последнего и отщеплением трпметиламана. Цикло-
гептен переводят потом в дибромид VI, при обработке которого диметиламином
в инертном растворителе одновременно отщепляется 1 моль НВг и образуется
диметиламиноциклогептен VII. Взаимодействие диметиламиноциклогептена
с иодистым метилом приводит к образованию четвертичной соли, которая дает
с окисью серебра основание, термически расщепляющееся на циклогептадиен
VIII, Триметиламин и воду. Присоединение брома к циклогептадиену происхо-
дит по «овцам сопряженной системы с образованием дибромциклогептена IX.
От дибромциклогептена при действии хинолина отщепляется 2 моль НВт,
в результате чего получается с теоретическим выходом циклогептатриен (тро-
иилиден) X:
Принципиально подобным же методом Вилыптеттеру удалось получить [85]
циклооктатстраен — соединение с четырьмя сопряженными связями *. Однако легко
доступным соединением циклооктатетраен стал только благодаря изящному синтезу
Реппе [86]: циклооктатетраен получают с 80%-ным вьцеодом полимеризацией ацети-
лена в автоклаве под давлением в тетрагидрофуранв в присутствии хлорида илн циа-
нида никеля и небольшого количества окиси этилена.
3. Ароматические и гетероциклические
непредельные соединения
Важнейшим методом получения ароматических и гетероциклических систем
двойных связей является дегидрирование. Реакции дегидрирования протекают в от-,
посительно мягких условиях, так как ароматические и гетероциклические соединения
являются энергетически наиболее выгодными системами. Легкость дегидрирования тем
больше, чем ближе соединение к ароматическому состоянию, т. е. чем больше оно уже
содержит двойных связей. Так, например, при нагревании эфира дигидролутидивдикар-
* Четыре двойные связи в циклооктатетраене, по существу, не являются сопряжен-
ными вследствие нецлоскостного строения его молекулы. — Прим. ред.
[84] R. W i 11 s t a t t е г, Ann., 317. 204 (1901).
[85] R. Willstatter, М. Heidelberg© г, Вег., 46, 517 (1913).
[86J W. Reppe, О. Schlichting, К. К I a g е г, Т. Т о е р е 1, Ann., 560,
1 (1948).
684
ОБРАЗОВАНИЕ ЭТИЛЕНОВЫХ СВЯЗЕЙ
боновой кислоты в присутствии палладиевой черни уже при 90“ С происходит отщепле-
ние водорода и образуется эфир лутидиндикарбоновой кислоты [87]:
СаНвООС^/ч /СОоСаНб СаНбООС^^^СООСаНв
-СНз
НзС/ NHXcH3 НзС'
Зелинский обнаружил [88], что уже при относительно низких температурах
(116—118° С) три изомера метилциклогексеиа при пропускании их над палладиевым ••
катализатором дегидрируются до толуола с одновременным образованкем метилцикло- -
гексана. Такое явление, при котором ненасыщенные соединения при относительно^
низких температурах образуют ароматические н насыщенные системы вследствие ;:
диспропорционирования, часто наблюдается при каталитическом дегидрировании,
в результате чего редко выделяется теоретически рассчитанное количество водорода.
Повышение температуры сдвигает эту реакцию в сторону полного дегидрирований.;1
Для дегидрирования алициклических соединений должны применяться гораздо^
более жесткие условия. В то время иак тетралин в присутствии платинового или пал-г
ладиевого катализатора дегидрируется в жидкой фазе при 185° С, для дегидрирования ,
декалина требуется температура не менее 300° С [89]. • ?
Процесс дегидрирования, так же, как и процесс гидрировании, зависит от тем<
пературы; между обоими процессами при дайной температуре существует равновесие^
которое при понижении температуры смещается в сторону гидрирования, а при новы--
шении температуры — в сторону дегидрирования. Катализаторы гидрирования часто.;
пригодны и для дегидрирования.
Наилучшими катализаторами дегидрирования являются платиновые и палла-
диевые катализаторы [90]. Никелевые катализаторы менее пригодны, так как из-за "
высокой температуры, необходимой в этом случае для дегидрирования, легко проис-*;
ходит обугливание. В качестве катализатора дегидрирования Иногда применяется-'
также осмий [91]. >
Высококипящие вещества могут дегидрироваться в жидкой фазе в результате
простого нагревания в присутствии катализатора. Более низкокипящие вещества де-
гидрируют в запаянной трубке или пропусканием их паров над катализатором [89]. л
Параллельно дегидрированию протекает ряд побочных реакций: отщепление,
заместителей, перемещение боковых цепей, сужение и расширение циклов, расщепление •
и замыкание колец. Насыщенные циклические соединения, которые не способны к об-'
разеванию ароматических систем (циклопентан, циклогептан), как правило, вообще
не дегидрируются. i
Реакцию дегидрирования с успехом используют для определения строения тер-<
ненов и камфор. Например, расщепление иарана или пинана до л-цимола становится-
возможным только потому, что при разрыве трех- или четырехчленных циклов в эти* •
случаях появляется возможность образования ароматической системы [92].
Окиси и сульфиды, например Сг2О3, MoS2, также катализируют реакцию дегял-
рпрования [93], однако при более высоких температурах. Кроме того, они катализи-,
[87] Е. Knoevenagel, j. Fuchs, Вег., 36, 2848 (1903).
[88] N. D. Zelinsky, Ber., 57, 2055 (1924).
[89] R. P. L i n s t e a d, К. О. A. M i c n a e 1 i s, J. Chem. Soc., 1940, 1127, 1134;
R. P. L ins tea d, A. F. M i 1 1 i d g e, S. L. S. Thomas, A. L. Wal-
pole, J. Chem. Soc., 1937, 1146. л
[90] N. D. Zelinsky, Ber., 44, 3121 (1911). *•’
[91] A. A. Balandin, Z. physik, Chem. Abt. B, 9, 49 (1930). n
[92] N. D. Zelinsky, R. J. Lewina, Ann., 476, 60 (1929). ,?
[93] S. G о 1 d w a ss er, il. S- Taylor, J. Am. Chem. Soc., 61, 1766 (1939);
Б. Молдавский; Г. Камушер, С. Лившиц, ЖОХ, 7 (69), 131 ;
(1937); С., 1938, 1, 4604. й
П. СОПРЯЖЕННЫЕ ДВОЙНЫЕ СВЯЗИ
685-
руют циклизацию парафиновых и олефиновых углеводородов в ароматические соеди-
нения [94].
Во многих случаях используют также химические методы дегидрирования. Хо-
рошие результаты получаются при дегидрировании частично ненасыщенных соедини
кий серой или селеном.
Дегидрирование серой [95]. Дегидрируемое вещество смешивают с рас-
считанным количеством серы и нагревают на металлической бане. При темпе-
ратуре около 180’ С начинается выделение сероводорода. До конца реанцию-
доводят нагреванием до 200—220° С и, если необходимо, повышают темпера-
туру до 260е С. Дегидрирование заканчивается через несколько часов; выделе-
ние вещества, если возможно, осуществляют вакуумной перегонкой.
1-Фенилнафталин [96]. В колбу Кланзеиа емкостью 200 мл вносят
смесь 6 г (0,18 леоль) порошкообразной серы и 35 а (0,17 молъ} 1-фенил-3,4-
дигидронафталииа и нагревают ее на металлической бане до 250—270’С. После-
нагревания в течение 30 мин выделение сероводорода прекращается. Образо-
вавшееся вязкое масло перегоняют в вакууме. Выход продукта 32—33 г (91—
94% от теоретического); т. кип. 134—135“ С (2 лл рт. ст.) или 189—190’С
(12 мм рт. ст.).
Селен в качестве дегидрирующего средства был впервые применен Дильсом [97].
Используя этот метод, удалось установить, что углеродный скелет никлопентанпфаиятт-
трена является основой стеринов, желчных кислот и стероидных гормонов. Пагипри-
ровапие при помощи селена требует применения высоких температур, что вызывает
иногда протекание побочных реакций.
Дегидрирование селеном. Соединение, подлежащее дегидрированию,
смешивают с рассчитанным количеством селена и нагревают при 250— 280’С
в колбе, снабженной воздушным холодильником. Температуру медленно повы-
шают, но обычно не выше 350° С. Продолжительность реакции 20—100 ч.
Продукты дегидрирования экстрагируют эфиром или бензолом и экстракт*
перегоняют.
Мягким дегидрирующим средством являемся хлоранил [98]. В качестве раствори-
теля в этом случае применяют ксилол, благодаря чем^ дегидрирование проходит при
относительно низких температурах (т. кип. ксилола*140° С). 2-Фенилнафталин, на-
пример, образуется с 90%-ным выходом кипячением 2-фенил-3,4-дигидронафталина1
и хлоранила в ксилоле в течение 15 ч [99].
Особенно пригоден хлоранил для дегидрирования тетрагидрокарбазолов.
Таким путем получили с 75—95%-ным выходом двадцать различных нарбазолов
[100]. При дегидрировании хлоранилом нитро- и карбокснгруппы не подвергаются
изменениям.
Кроме хлоранила для дегидрирования можно применять также другие хиноны,
например 2,4-дихлор-5,6-дициан-1,4-бензохинои или тетрахлор-1,2-бензохинон [103].
В некоторых случаях в качестве дегидрирующих средств могут служить также кислород
воздуха [101], двуокксь селена [102] или серная кислота. Быстро и полно протекающая
[94] Б. Молдавский, Г. Камушер, М. В. Кобыль’ская, ЖОХ,
7 (69), 169 (1937); С., 1937, II, 1546.
[95] L. Ruzicka, Fortschr. chem. Physik, physik. Chem., 19, 1 (1928);
L. Ru z icka, J. Meyer, Helv. chim. acta, 4, 505 (1921).
96] R. Weiss, Org. Synth., 24, 84 (1944).
97] O. D i e 1 s, Ber., 69, A 195 (1936).
98] R. T. А г n о 1 d, С. J. Collins. J. Am. Chem. Soc., 61, 1407 (1939).
99] N. Campbell, D. Kidd, J. Chem. Soc. 1954 2154
100] В. M. Barclay, N. Campbell, J. Chem. Soc., 1945, 530.
101 ] H. Lewis, G. Ramage. R. R obinson, I Chem. Soc., 1935, 1412.
102] E. Borgward t, E. Schwenk, J. Am. Chem. Soc., 56, 1185 (1934);
G. Stein, Angew. Chem., 54, 146 (1941).
[ЮЗ] E. А. В г a'n de, A. G. Brook, R, P. Linstead J. Chem. Sqc., 1954,
3569.
686
ОБРАЗОВАНИЕ ЭТИЛЕНОВЫХ СВЯЗЕЙ
реакция дегидрирования описана Бэке и Вюртеле [104]. Так, гексахлорциклогексан
и гексабромциклогексан могут дегидрироваться свободной трехокисью серы или ее
аддуктом, например хлорсульфоновой кислотой, до гсксагалогенбензола.
III. Кумулированные двойные связи
1. Аллены
Для синтеза соединений с алленовой структурой применяются те же методы,
что п для получения олефиновых структур. Важнейшими из них являются: отщеплений
1 алогена от ненасыщенных дигалогенидов RR'CX—СХ —CR’K"7; отщепление галогено-
водорода от ненасыщенных мопогалогенидов RR'CII — СХ — СВ "В"7; отщепление воды
от соответствующих а.Р-ненасыщсппых спиртов RR'COH —СН—СП2.
Метилаллен, например, легко получают кипячением 2,3-дибромбутеиа-1
<с цинком и спиртом [105]:
СН3-СНВг-СНВг-СН2Вг —> СН3-СНВт—СВт=СН2 —> СНз-СН=С=С112
2,3-Дибромбутен-1. В колбу емкостью 1 л, соединенную с нисходящим
• холодильником и приемником, помещают 419 г 1,2,3-трибромбутана, 20 мл
воды и 100 г NaOH. Содержимое колбы нагревают, время от времени встряхи-
вая. Вскоре начинается бурная реакция, сопровождающаяся отгонкой про- .
дукта реакции. Приемник охлаждают ледяной водой. После затихания перво-
начально бурной реакции реакционную массу нагревают до прекращения
отгонки дистиллята. Дистиллят после промывки водой перегоняют еще раз
я вакууме и таким образом отделяют от непрореагпровавшего трибромбу-
тана. Выход 2,3-дибромбутена-1 261 г (86% от теоретического); т. кип. 75° С
-(20 мм рт. ст.).
Метилаллен. Аппаратура: трехгорлая колба емкостью 1 л. снабженная
капельной воронкой, узкой, доходящей до дна стеклянной трубкой для про-
пускания СО2 и холодильником. Холодильник последовательно соединен с при-
емной колбой для спирта емкостью 100 мл, помещенной в нагретую до 30’ С .
водяную баню, с еще одним холодильником п конденсационным сосудом, охла-
ждаемым до —12° С смесыо льда и соли. В реакционную колбу помещают 300 мл
этилового спирта и 160 а цинковой пыли и медленно прибавляют по каплям
131 s 2,3-дибромбутена-1; при этом начинается бурное кипение спирта. Виде- .
ляющийся метилаллен улавливают в кондспсациониом сосуде. После оконча-
ния прибавления 2,3-дибромбутена-1 кипение поддерживают еще несколько
минут. Остаток метилаллена вытесняют пропусканием СО2. Сырой продукт
перегоняют. Выход метилаллена 33,6 г (72% от теоретического); т. кип. 10,3’ С.
Этот способ соответствует методике [106], предложенной для получения аллена —
простейшего представителя алленовых углеводородов.
Для синтеза тетрафенилаллена [107] исходят из бензальцетофенона XI, который.,
кипячением с фенилмаглийбромидом [108] превращают в тетрафенилпропиловый спирт
XII. Из последнего при кипячении его с кислотами образуется тетрафени.шропилен .
Х1П, обработка которого бромом приводит к мопобромтетрафенилпропплеиу XIV.
Отщепление бромистого водорода от монобромтетрафепплпропплена спиртовым раство- ,
ром КОН дает тстрафенплаллен XV: *
I
С6НбСН=СНСОС8Н5 (С6П6)2СН-СН2-С(СвНг,)2 —>
Xi хи
—> (С6Н6)2СН-СН=С(СвН5)г —► (СвН5)2СН-СВг -=С(Св1Т5)2 —>
XIII XIV
—> (СЬН5)2С=С=С(С0П5)2
XV
104] F. В е с k е, L. W и г t е 1 е, Вег., 91, 1011 (1958).
105] С. D. Hurd, R. N. М е i n в г t, J. Am. Chem., Soc., 53, 292 (1931).
106] N. Demjanoff„G. Gustavson, I. prakt. Chem., 38, 201 (1888).
107] D. VorlSnder, C. Siebert, Ber.. 39, 1025 (1906).
108] E. P. Kohler, Am. Chem. J., 31, 651 (1904).
Ш. КУМУЛИРОВАННЫЕ ДВОЙНЫЕ СВЯЗИ
687
Тетрафенилаллен получают также сухой перетопкой в вакууме бариевой соли-
дифенилуксусной кислоты [107]; при этом вначале, вероятно, образуется тетрафянил-
ацетон, который в результате отщепления воды превращается в тетрафенилаллен:
[(С6Н&)аСН—СОО]аВа .^67* (С8Н6)2СН~СО-СН(С6Н5)3
* (С0Нв)аС=С=С(СеНй)з
Однако в общем случае отщепление воды от ацетона или его производных осуще-
ствить нельзя.
В этом же разделе следует упомянуть о реакции, аналогичной реакции Виттита,
в результате которой из трифепилфосфиндифенилметилидена и дифенилкетена при
нагревании в бензольном растворе при 140Q С иод давлением в течение 2 ч с количе-
ственным выходом образуется тетрафенилаллен [109]:
(СвИ8)9Р=С(СвН6)а4-О=С=С(СвН»)8 (СвНй)аС=С==С(С6Н5)а4-(СеН5)3Р=О
Отщепление воды от некоторых а.р-ненасьпценных спиртов осуществляется-
нагреванием их с кислотами, например с щавелевой [НО] или уксусной ]111].
Аллены получают также из некоторых производных ацетилена, а именно из-
третичных хлоридов или спиртов, взаимодействием их со сплавами цинка и меди или-
с литийалюмиппнгидридом [112], например:
(СН3)2С—С=СН —► (СНз)2С=С=СНа
CI
2-Метил-бутадиен-2,3 [112]. К суспензии 79,7 г (2,10 молъ) литий-
алюминийтидрида в 1 л тетрагидрофурана прибавляют по каплям при переме-
шивании 108 г (1,05 моль) 2-хлор-2-метилбутина-3 с такой скоростью, чтобы
происходило слабое кипение растворителя. По окончании прибавления смесь
кипятят еще 1,5 ч, затем избыток гидрида разлагают водой и добавляют 10%-
ную НС1 до pH раствора около 3. Летучие углеводороды и тетрагидрофуран
отгоняют из смеси, подлежащей разгонке; смесь тщательно фракциони-
руют. Выход продукта 35 г (42% от теоретического); т. кип. 40,0—40,2° С.
(761 мм рт. ст.).
Алленовые соединения представляют интерес в стереохимическом отношении,
так как они могут образовывать оптические изомеры [113].
Кумулены [114], содержащие более чем две кумулированные двойные связи,
являются вполне стабильными соединениями. Для их синтеза конденсируют диацетилен
в виде дигрниьяровского соединения с кетонами (флуореноном, бензофеноном, ди-п-
толилкетоном и др.) до диацетилепгликолей, которые при восстановлении СгС1г,
и НС1 превращаются в кумулетты:
Ik ,П R, .R
BrMgCzsC—C=CMgBr —> >C—C=C-C=C-C< —> >C=C=C=C=C=C<
Rz I | \R Rz \R
OH HO
2. Кетены
Кетены являются очень реакционноспособным классом соединений. Старяйтпим*
методом синтеза кетенов является дегалогенирование галогенангидридов а-галогено-
кпслот цинком в эфире илн зтилацетате при температуре кипения растворителя [115].
[109] J. Meyer, Вег., 89, 842 (1956).
[110] Р. Mulliken, R. L. Wakeman, Н. Т. Gerry, J. Am. Chem. Soc.,
57, 1607 (1935).
[Ill] К. Ziegler, К. Richter, В. Schnell, Ann., 443, 178 (1925).
[112] W. J. В a 1 ey, Ch. R. P f e i f er, J. Org. Chem., 20, 95 (1955).
[113] W. II. M i 1 Is, P. M a i 11 a n d, Nature, 1935, 994; P. Kohler, T.’Wal-
ker, M. Tishler, J. Am. Chem. Soc., 57, 1743 (1935).
[114] R. Kuhn .et al., Ber., 71, 783, 1510 (1938); 73, 1410 (1940); 84, 566 (1951);.
86, 759 (1953).
[115] H. Staudinger, Ber., 38, 1735 (1905).
€88
ОБРАЗОВАНИЕ АЦЕТИЛЕНОВЫХ СВЯЗЕЙ
За исключением некоторых кетокетенов, которые можно получить с 90—95%-ямм
• выходом [116], и недокиси углерода, кетены по этому способу могут быть синтезиро-Ь
ваны лишь с незначительными выходами (не более 20%), у
Недокись углерода [117]. В колбе емкостью 1 л к 20 г цинковой стружки
прибавляют по каплям 30 в хлорангидрида диброммалоновой кислоты в 300 аЦу
абсолютного эфира с такой скоростью, чтобы жидкость энергично кипела. ..
Выделяющиеся вместе с эфиром пары недокиси конденсируют в холодильнике,!
и улавливают в приемнике, охлаждаемом- смесью льда и соли. Выход недокиси
4,8 8 (80% от теоретического). >
Недокись углерода является ядовитым, сильно раздражающим глаза''
и органы дыхания газом, который конденсируется при 7° С.
Другим способом получения кетенов является дегидрогалогенирование хлорзд
• ангидридов кислот при действии третичных аминов. Однако амины катализируют-
димеризацию кетенов, поэтому таким методом синтезируют только те кетены, которвим
не склонны к димеризации. Дифенилкетен, например, получают с почти количествен*^
ным выходом прн действии трипропиламнна на дифенилацетилхлорид [118]. <
Для синтеза алифатических кетокетепов наиболее пригодным основание»)
является триметиламин [119]. J
Дифенилкетен образуется также при термическом разложении диазодезоксибеи2^.
зоина [120].
Дифенилкетен. Моногидразон бензила действием желтой окиси ртути,
превращают в диазодезоксибензоин, из которого при нагревании до 100—110’Ес/
с выделением азота образуется дифенилкетен [121]:
CeH6-C-CO-CeH6-| HgO —C6HS-C-CO-CeH6 (CeH5)2C=C=O
n-nh2
+N=N-
Таким методом могут быть получены и другие кетены. Так как многие кетенъг*
легко димеризуются, расщепление их димеров является важным методом получении^
кетенов. При нагревании от 500 до 600° С происходит расщепление дикетенов до моно-
меров; при этом образуются кетеиы, свободные от побочных продуктов, вследствие чего,)
• этот метод может применяться для получения чистых кетенов [122].
АЦЕТИЛЕНОВЫЕ СВЯЗИ*
Образование тройных связей в углеродном скелете протекает почти исключи-* S
тельно из уже имеющихся или образующихся первоначально этиленовых связей,;^
Основным методом получения ацетиленовых соединений является дегидрогалогенирсн,,}'
ванне соответствующих галогенидов: хло'р-и бромолефипов (XVI, XVII) или дихлор*,-
Обзорные работы о получении ацетиленовых соединений см.: Органические;*
реакции, сб. 5, Издатинлит, 1951 г.; W. F г a n k е, W. L i е g е п Ь е i пЛ-
Н. Meister, Angew. Chem., 1960. — Ярил. ред. •.*
Н. Staudinger, Ann., 356, 51 (1908). А
Н. Staudinger, St. В е г е z а, Вег., 41, 4465 (1908). ’
Н. Staudinger, Вег., 44, 1619 (1911). *
Пат. США 2268169; С. А., 36, 2737 (1942). ?
G. Schroeter, Вег., 42, 2346 (1909)
L. I. Smith, Н. И. Hoehn, Org. Synth., Coll. v. 3, p. 356. '
A. B. Boes e, Ind. Eng. Chem., 32, 16 (1940). ‘
[116
117]
118
119
120
121
122]
ОБРАЗОВАНИЕ АЦЕТИЛЕНОВЫХ СВЯЗЕЙ
689
и дибромалканов (XVIII, XIX, XX), содержащих атомы галогена в 1,2-положениях
или у одного и того же углеродного атома.
RCX=CHR' RCH«CHX
XVI XVII
RCHX—СН2Х RCH2CHX2 RCX2CH3
XVIII XIX XX
Ацетиленовые соединения можно также получать дегалогенированием 1,2-
дигалогенолефинов, а в некоторых случаях разложением азотсодержащих соединений.
1. Дегидрогалогенирование дигалогенидов или галогенолефинов
Важнейшими соединениями, вызывающими отщепление галогеноводорода,
являются КОН и NaNH2, но применяются также и другие щелочные реагенты: алкого-
ляты, гидриды и карбонаты щелочных металлов, карбонаты и гидроокиси щелочно-
земельных металлов, а в некоторых случаях и металлоорганические соединения,
Если исходят из насыщенных галогеносоединений, то отщепление первой моле-
кулы галогеноводорода с образованием олефиновой двойной связи протекает значи-
тельно легче, чем дальнейшее отщепление галогеноводорода и образование ацетилене^
вого соединения. Так, например, при получении этоксиацетилена НС=С—ОСгН»
пз 1,2-дибромэтилэтилового эфира 1 моль НВг можно отщепить диэтиланилниом, в то
время как второй — только в вакууме при помощи КОН [123].
Для дегидрогалогенирования чаще всего применяют гидроокиси щелочных
металлов в растворителях или без них. В качестве растворителей используют главным
образом (расположенные по своему значению) этиловый, метиловый и бутиловый
спирты, гликоль и его простые эфиры и воду. В большинстве случаев употребляют на-
сыщенные при комнатной температуре растворы КОП в этиловом спирте (приблизи-
тельно 4 н. или 20%-ные, и всегда в избытке). Для дегидрогалогенирования арилгало-
геполефпнов часто бывает достаточно длительное кипячение в соответствующем рас-
творителе, в то время как для получения алифатических производных ацетилена иногда •
следует проводить реакцию при 170° С в автоклаве [124]. Продолжительность реакций
различна — от нескольких минут до нескольких часов. ,
и-Толилацетилен ]125]. Кипятят с обратным холодильником в течение
24 ч 85 г 1-п-толил-1-хлорзтилена с раствором 50 г КОН в 100 мл абсолютного
этилового спирта. Затем смесь выливают в 1 л ледяной воды, отделяют масло,
а продукт из водного слоя экстрагируют эфиром. Масло н экстракт объеди-
няют и сушат над КОН. После отгонки эфира остаток перегоняют в вакууме.
Выход п-толилацетилена 31 г (65% от теоретического); т. кип. 79—82е С (31—
33 мм рт. ст.).
ФеиилАпетилеи [126]. К смеси 200 мл хлороформа н 312 г стирола
прибавляют по каплям в течение 2 ч при охлаждении и перемешивании 410 г
брома. Затем реакционную массу перемешивают в течение 30 мин при 30е С
и отгоняют хлороформ. После сушки остатка на воздухе получают чистый
белый стиролдибромид. Выход продукта 653 г; т. пл. 73° С.
Нагревают с обратным холодильником 240 г КОН в 240 мл метилового
спирта и прибавляют при перемешивании небольшими порциями в течение
1,5 ч 264 а стиролдибромида. Смесь кипятят в течение 30 мин при перемеши-
вании, затем охлаждают и смешивают с 400 мл воды. Отделяют масло, сушат
его над КвСО3 и перегоняют в вакууме. Получают около 75 г масла с т. кип.
до 100° С (10 мм рт. ст.) и повторно перегоняют при нормальном давлении.
Выход фенилацетилепа 67 г (66% от теоретического); т. кип. 141—143е С.
[123] И. Н. Назаров, 3. А. К р а с н о в а, В. П. Виноградов, ЖОХ,
28, 460 (1958). • ,
[124] Т,. Pauling, Н. G. S pr i n ga 11, К. J. Palmer, J. Am. Chem.
Soc., 61, 928*(1939).
[125] L. J. Smith, П-H. Hoehn, J. Am. Chem. Soc., 63, 1175 (1941).
[126] W. Franke, W. Z i e g e n b e i n, H. Meister, Angew. Chem.,
72, 400 (1960).
44 Заказ I 835.
690
ОБРАЗОВАНИЕ АЦЕТИЛЕНОВЫХ СВЯЗЕЙ
Дегидрогалогенирование гидроокисями щелочных металлов можно осуществлять
и без растворителя [127], применяя твердую высушенную гидроокись щелочного ме-
талла. Твердую тонкоизмельчепиую гидроокись щелочного металла с успехом исполь-
зуют для получения ацетиленовых эфиров из алкокси- или арилоксибромэтиленов,
например, для синтеза феноксиацетилена [128] из бромфеноксиэтилена:
BrCH-CH-OCeHsI^£S* НС=С-ОС6Н5
При использовании расплавов гидроокисей щелочных металлов реакцию про-
видят при температуре около 200° С. Безводный КОН плавится урн 360° С, и поэтому
применяют или смесь из 2 ч. КОН и 1 ч. NaOH (т. пл. 200° С), или к КОН добавляют
небольшое количество воды. Но обычно КОН почти всегда содержит достаточное коли-
чество влаги и плавится при 200° С.
Фенилацетилен [129]. В колбу из йенского стекла илп стекла марка
«супремакс», снабженную капельной воронкой и дефлегматором, который со-
единен с нисходящим холодильником, помещают 80 г КОН. Колбу нагревают
на масляной бане до 200—215“ С и затем приливают из капельной воронки
25 з (о-бромстирола со скоростью примерно 1 капля/сек. Фенилацетилен кипит
при 142—143° С, а бромстирол — при 218—220° С, поэтому температура рас-
плавленного КОН вполне достаточна для отгонки образующегося фепилацети-
лена от непрореагировавшего бромстирола. В конце реакции температуру
бани медленно повышают до 2359 С. Если на поверхности расплава возникает
твердая корка КВг, ее разрушают стеклянной палочкой или встряхиванием
колбы. Слои конденсата разделяют, более легкий фенилацетилен отделяют»
сушат над КОН и фракционируют. Выход фенилацетилена 11 г (80% от теоре-
тического); т. кип. 142—144° С.
Дегидрогалогенирование действием NaNH2 проводят в инертных растворителях
при 110—160° С. Для реакции необходим избыток тонкоизмельченпого амида патрия.
Октилацетилен [130]. В фарфоровой ступке растирают 120 г NaNHj
с 200 мл чистого жидкого парафина, не содержащего фракций, кипящих ниже-
250° С (очень важно, чтобы NaNH2 был растерт как можно тоньше). Полученную ,
суспензию помещают в трехгорлую колбу, снабженную мешалкой с ртутным
затвором, обратным холодильником с хлоркальциевой трубкой и капельной *
воронкой. Ступку и пестик ополаскивают 250 мл парафина. Колбу нагревают >
иа масляной бане до 160—165° С и при перемешивании в течение 1—2 ч медленно ,3$
приливают 217 г 2-бромдецена-1. Выделяющийся аммиак поглощают водой-
Реакционную смесь нагревают еще 2 ч, затем охлаждают, растворяют в 500 мл ги*
эфира, выливают на 500 г измельченного льда и подкисляют 280 мл НС!. Эфир- ;
но-парафиновый слой сушат над СаС12, отгоняют эфир на водяной бане, а к-октил-
ацетилен перегоняют в вакууме. Выход w-октилацетилена около 68%; т. кип. а
80—82“ С (22 мм рт. cm.).
В особенно мягких условиях протекает отщепление галогеноводорода амидом ч
натрия в жидком аммиаке [131]. В этом случае реакцию проводят в открытом сосуде-
при температуре кипения аммиака или в автоклаве при комнатной температуре [132]. .И
[127] F. Krafft, L. Reuter, Ber., 25 , 2244 (1892).
[128] Т. L. Jacobs, R. С г а ш e г, F. F. Weiss, J. Am. Chem. Soc., 62, 1349- .4
(1940); T. L. Jacobs, R. Cramer, J. E. Hanson, J. Am. Chem. Soc., ;
64, 223 (1942). 'i
[129] J. Hessler, J. Am. Chem. Soc., 44, 425 (1922); Org. Synth., Coll. v. 1» 1
1941, p. 437. .!
[130] R. Lespieau, M. Bourguel, Org. Synth., Coll, v. 1, p. 185. ?
[131] T. H. Vaughn, R. R. Vogt, J. A. Nieuwland, J. Am. Chem. Soc.»
56, 2120- (1934). 1
[132] W. Franke, W, Zicgenhein, K. Meister, Angew. Chem., 72, 394 ’
(I960). Г’
ОБРАЗОВАНИЕ АЦЕТИЛЕНОВЫХ СВЯЗЕЙ
691
Амид натрия можно применять только при полном отсутствии влаги. В против-
ном случае дегидрогалогенирование отчасти протекает и под действием образующейся
гидроокиси натрия, что может вызвать миграцию ацетиленовой связи, поскольку
реакцию проводят при высоких температурах в инертных растворителях. Поэтому
NaNHj рекомендуется получать непосредственно перед применением из безводного
жидкого аммиака и натрия [133] в присутствии Fe(NO3)s или FeClg [131]. В качестве
примера приведено получение бутин-2-ола-4 из 2-хлорбутеп-2-ола-4 [134]:
СН3-С-СН-СН2ОН СНз-С=С—CHaONa NHtC1 > CH3-C=C-CH2OI1
I
Cl
Бутин-2-ол-4. В сосуд Дьюара (илп в обычную хорошо термоизолирован-
ную колбу) емкостью 4 л вводят через маленькое отверстие в пластмассовой
крышке 3 л сухого жидкого аммиака, 1,5 г гидрата Fe(NO3)3 и 65 г свеже-
нарезанного натрия. Смесь перемешивают до тех пор, пока весь натрий не пре-
вратится в NaNII2. Затем к NaNH2 прибавляют в течение 30 мин 134 г 2-хлор-
бутен-2-ола-4 и смесь перемешивают в течение ночи. После этого постепенно
добавляют 148 г твердого NH4C1 таким образом, чтобы реакция не была слишком
бурной. Смесь переносят в металлический сосуд емкостью 5 л и оставляют иа
ночь для испарения аммиака. Остаток пять раз экстрагируют 250 мл эфира,
затем отгоняют эфир и перегоняют полученный остаток в вакууме. Выход про-
дукта 66—75 г (75—85% от теоретического); т. кип. 55° С (8 л.ч рт. ст.).
Применение амида патрпя в жидком аммиаке особенно целесообразно для полу-
чения из диалкоксихлорацеталсй различных алкоксиацетиленов: метокси-, этокси-
и бутоксиацетилевов. При этом происходит отщепление галогеноводорода и спирта [135]:
С1СЩ-СН(0В)2 HGeC-OR
Относительно легко получаются ацетнленкарбоповые кислоты. Простейшую
ацетиленкарбоновую кислоту — пропиоловую — лучше всего синтезировать карбо-
ксилированием ацетиленида щелочного металла [138]; она легко образуется прн де-
карбоксилировании ацетилендикарбоповой кислоты [136, 137] уже прн кипячении
этой кислоты или ее калиевой соли с водой. Ацетилрндикарбоповую кислоту получают
из дибромянтарной кислоты действием раствора КОП в метиловом спирте [139].
Фенилпропиоловую кислоту можно получить действием спиртового КОН на этиловый
эфир а,0-дибромгидрокоричной кислоты и обработкой полученной калиевой соли
разбавленной серной кислотой [140]:
свп5снвгснвгсоос2н6-}-зкоп —► СвНбС^ссоок+СзИйОн-ьгквг-ьан.о
Описан несколько более простой метод получения небольших количеств
фенилпропиоловой кислоты из а,0-дибромгидрокоричнои кислоты [141].
сс,0-Дибромгидрокоричную кислоту получают с 95%-ным выходом действием
брома на коричную кислоту в кипящем четыреххлористом углероде [141].
1133] К. W. G г е е n 1 е е, A, L. Henne, Inorg. Synth., 2, 75, 79 (1946).
[134] Р. J. Ashworth, G. Н. Mansfield, М. С. Whiting, Org. Synth.,
35, 20 (1955J.
[135] G. E g 1 i n t о n, E. R. H. J on es, B. L. S h a w, M. C. Whiting,
J. Chem. Soc., 1954, I860.
(136] E. Baudrowski, Ber., 15, 2699 (1882).
[137] A. Bacycr, Ber., 18, 2269 (1885).
[138] F. St ra us, W. V о s s, Ber., 59, 1681 (1926); A. W. Johnson,. The che-
mistry of the acetylenic compounds, v. II, E. Arnold a. Co., London, 1950.
[139] T. W. Abbott, R. T. A г n о 1 d, R. В. T h о mp son, Org. Svnth., Coll,
v. 2. 1943, p. 10.
[140| T. W. Abbott, Org. Synth., Coll. v. 2, 1943, p. 515.
1141] M. Reims r, J. Аш. Chem. Soc., 64, 2510 (1942).
44*
692
ОБРАЗОВАНИЕ АЦЕТИЛЕНОВЫХ СВЯЗЕЙ
Фенилпропиоловая кислота. Помещают 25 г дибромида гидрокоричной
кислоты в чашку для выпаривания и к нему приливают 100 мл 25%-ного КОН
в метаноле. Смесь нагревают на энергично кипящей водяной бане до полного
испарения спирта*. К оставшейся густой массе прибавляют 75 мл метилового
спирта и снова упаривают для завершения реакции. После охлаждения бледно-
желтого зернистого продукта остатки метанола удаляют отсасыванием.
Сухое твердое вещество промывают несколькими миллилитрами охла-
жденного метанола и растворяют в 500 мл ледяной воды. Затем при перемешива-
нии медленно прибавляют до сильнокислой реакции охлажденную льдом HCI;
при этом отделяется масло, которое после стояния в течение ночи в бане со льдом
затвердевает. В результате получают с 80%-ным выходом бесцветную фенил-
пропиоловую кислоту; т. пл. 136—138° С.
Среди других дегидрогалогенирующих средств особого внимания заслуживает
гидрид натрия. Его применяют главным образом для синтеза эфиров замещенных арил-
пропиоловых кислот (<?-, х- пли п-хлорфенил-, метоксифенил- п ннтрофенилпропиоло-
еых кислот) из соответствующих производных а,₽-дибромпропионовой кислоты; реак-
цию проводят в бензоле в присутствии небольшого количества абсолютного спирта
[143]. Отщепление галогеноводорода вызывается при этом этилатом натрия, получа-
ющимся из гидрида натрия и этилового спирта. Этилат натрия при взаимодействии
с галогеноводородом вновь образует спирт, что дает возможность обходиться неболь-
шим количеством спирта:
C2H5OH-t-NaH —► C2H6ONa4-H2
CsiHgONa-f-HBr —>- C2H5OH-]-NaBr
В этом заключается преимущество такого метода дегидрогалогенирования, так
как при проведении синтеза с алкоголятами щелочных металлов или спиртовыми рас-
творами их гидроокисей легко протекает побочная реакция образования простых
эфиров.
И, наконец, для образования тройных связей с успехом применяют также
некоторые металлоорганические соединения, например бутиллитий и фениллитий.
Так, фенилацетилен получают с 70%-ным выходом при взаимодействии со-хлорстнрола
с фениллитием [144].
2. Дегалогенирование
Дегалогенирование не имеет большого значения для получения ацетиленовых
соединений. Только в некоторых случаях для этой цели применяют дегалогенирование,-
1,2-дигалогенолефипов металлами (большей частью тонко измельченными в эфирй
пли ацетоне). Так, например, дифенилацетилеп образуется с 85%-ным выходом при
дегалогенировании толандибромида цником в ацетоне [145]. Неоднократно делались
попытки синтезировать циклические ацетилены из 1,2-дигалогеициклоолефияов от-
щеплением галогенов натрием [146]. Таким способом был получен лишь циклооктив
пз 1-хлор-2-бромциклооктена [147].
Первые циклические ацетилены были синтезированы Ружичкой и сотр.
[148]. В качестве исходного материала они выбрали относительно легко доступ-
ные циклопентадецен и циклогептадецен и получили из них дибромиды. Из
дибромида отщеплением 1 моль НВг они выделили ненасыщенный циклический
бромид. Отщепление второй молекулы НВг удалось только лишь в результате
многочасового нагревания в запаянной ампуле при 150—180° С в присутствии
спиртового раствора КОН.
143
144
145
[148
Осторожно! Хорошая тяга! — Прим. ред.
М. S. Newman, S. П. Merrill, J. Аш. Chem. Soc., 77, 5549 (1955).
G. Wittig, H. Witt, Вег., 74, 1474 (1941).
F. Strauss, Ann., 342, 190 (1905).
A. E. Ф а в о p с к и й, ЖОХ, 6, 720 (1936).
Ц. Л. Домнин, ЖОХ, 8, 851 (1938).
L. R u z i с к а. *М. Н й г Ь i п, А. , В о е к е п о о g е n, Helv. chim.
acta, 16, 498 (1933).
ОБРАЗОВАНИЕ АЦЕТИЛЕНОВЫХ СВЯЗЕЙ
693
3. Расщепление азотсодержащих соединений
Ацетилены получают также разложением дичетвертичпых аммониевых со-
единений — реакцией, аналогичной расщеплению по Гофману, применяемому для
синтеза олефинов:
(CH3)BN—СНз-СНг—N(CH3)3 —> CH=CH4-2(CH3)3N + 2HaO
ОН' он-
Папример, ацетилен в лабораторном масштабе получают [149] с 80%-ным
выходом нагреванием с 40%-ным водным раствором КОН 1,2-дичетвертичной аммоние-
вой соли, образующейся из 1,2-дибромэтана и избытка триметиламида.
Таким способом можно также с хорошим выходом синтезировать метилацетилен
и гекссп-4-ин-2.
Диацетилен получают по этому методу из 1,4-дичетвертичных аммониевых солей,
полученных из 1,4-днбром-2-хлорбутена-2 или 1,4-дибромбутина-2 [150]:
R8N-CH3-CH=CC1-CHs-NR3-----------1
Вг Вг' NaOH
IIC=C-teCH
R3N-CHa-C=C-CH2-NR3----------1
Вг- Вг'
Здесь же следует упомянуть о синтезе тетроловой кислоты [151] действием
иодной гидроокиси щелочного металла на иодид триметил-(2,2,3,3-тетрахлорбутил)-
аммония, получаемый присоединением хлорак 1-диметиламинобутину-2 с последующим
метилированием образовавшегося 1-диметиламино-2,2,3,3-тетрахлорбутана:
!СП.;Л CH. -CJ: СНа — ► (СНз)2Н-СНг-СС1з-СС1!-СНз
—► (СНз)з^—СНа—СС1,—СС1,—СН, сНз-CsC-COOII
Г
1-Диметиламино-2,2,3,3-тетрахлорбутан. В раствор 48,5 г 1-диметил-
аминобутина-2 в 100 мл 34%-нои HCI пропускают 75 г хлора. Затем бмесь
нейтрализуют концентрированным раствором Na2CO8, Выпавший кристалли-
ческий амин отфильтровывают, несколько раз промывают водой и сушат. Выход
продукта 108 г (90% от теоретического); т. пл. 46—47° С.
Иодид триметил-(2,2,3,3-тетрахлорбутил)-аммония. Нагревают 23,9 а
1-диметилаиино-2,2,3,3-тетрахлорбутана и 21,3 г подпетого метила в 60 мд
ацетона на кипящей водяной бане в течение 3 ч. Кристаллический осадок от-
фильтровывают и промывают спиртом. Выход продукта почти количественный;
после перекристаллизации из метанола т. пл. 187° С.
Тетроловая кислота. К раствору 76,2 г иодида триметил-(2,2,3,3-тетра-
хлорбутил)-аммония прибавляют по каплям при —10’С в течение 20 мин
раствор 48 в NaOH в 40 мл воды. По истечении 1 ч реакция, идущая с выделе-
нием тепла, заканчивается. Выделяющийся триметиламин улавливают в про-
мывной склянке с НС1. Реакционную смесь промывают эфиром, водный слой
осторожно подкисляют НС! и несколько раз экстрагируют эфиром. Объединен-
ные эфирные вытяжки сушат над Na2SO4 и бтгоняют эфир. Остаток после отгонни
эфира полностью кристаллизуется. .Выход тетроловой кислоты 11,8 г (70,2%
от теоретического); после перекристаллизации из бензина т. пл. 78—78,5° С.
По Курциусу [152|, ацетиленовые соединения можно синтезировать также окис-
лением 1,2-дигидразонов желтой окисью ртути. Так, например, дифенплацетилен
[149]
[150]
[151]
[152]
И. М. Слоб один, II. А. С е л е з н е в а, ЖОХ, 26, 691 (1956); 27, 2478
(1957).
А. Т. Бабаян, Г. М. М р к я н, С. Л. Мвджоян, ЖОХ, 27, 604 (1957).
А. Т. Б а б а я н, А. А. Г р и г о р я н, ЖОХ, 26, 1945 (1956).
Т. Curtius, Вет., 22, 2161 (1889); Т. С и г t i и s, К. Т h и в, J. prakt.
Chem., 44, 168 (1891).
694
ОБРАЗОВАНИЕ АЦЕТИЛЕНОВЫХ СВЯЗЕЙ
получают с 75%-ным выходом из дигидразона бензила [1531. Диарилацетилены можно
получать с 80—85%-пым выходом по модифицированному методу Курциуса окислением
соответствующих дигндразонов трифторацетатом серебра в среде триэтиламина и аце-
тонитрила или триэтиламина и этилового спирта [154]:
СаН5-С—С-C6n5 + 4CF3COOAg + 4(C2Hd)3N —»•
II II
h2nn nnii2
—> CeH5-С=С-C6H5 + 4CF3COOH • (C2n5)3N^4Ag4-2N2
Иногда этим методом получают также циклические соединения. Циклодецин
[155], например, образуется с 36%-ным выходом при окислении дигидразона цикло-.
декандиона-1.2 желтой окисью ртути в бензоле в присутствии раствора КОН в этило- .
вом спирте и безводного Na2SO4:
(CH2)s C~NNHs нео; С.Н.: коН> (tn2)8
1------C=NNHi '----С
Таким же образом можно получить циклододецин [156].
[153] W. Schlenk, Е. Bergmann, Ann., 463, 76 (1928); • А. С. Соре, I.
D. S. Smith, R. J. Cotter, Org. Synth., 34, 42 (1954).
[154] M. S. Newman, D. E. Reid, J. Org. Chem., 23 , 665 (1958).
[155] A. T. Blomquist, R. E. Burge, A. C. S u c s y, J. Am. Chem. Soc.,
74, 3636 (1952).
[156] V. Prelog, M. Speck, Helv. chim. acta, 38, 1790 (1955).
ОБРАЗОВАНИЕ
НОВЫХ
УГЛЕРОД-
УГЛЕРОДНЫХ
СВЯЗЕЙ*
Углеродные связи могут образовываться
в результате реакций присоединения или обмена.
Предпосылкой к этому является в общем случае
соответствующая поляризация связывающихся
углеродных атомов, т. е. наличие карбкатиона
и карбаниона. Благодаря присоединению сво-
бодной электронной пары карбаниона к карбка-
тиону возникает новая связь. Это взаимодей-
ствие в какой-то мере аналогично реакции ней-
трализации.
нн НН
II II
R-C’-b+C-R —> R-C-C-R
II II
НН н п
ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ
В РЕЗУЛЬТАТЕ РЕАКЦИЙ
ПРИСОЕДИНЕНИЯ
В следующем разделе рассматриваются реак-
ции присоединения, причем речь идет о реакциях
присоединения к кратным связям (например,
к С=С, С=О, C=N). Очень часто при этом
речь идет об анионоидных реакциях присоеди-
нения, прежде всего о присоединении к карбо-
нильной или антрильной труппе. Присоединя-
емая группа является при этом карбанионом,
а акцептор, содержащий кратную связь, играет
роль катиона, так как л-электропная пара его
двойной связи сдвигается в направлении элек-
троотрицательного*ключевого атома.
Н
I
R-C <— *
II
О
II
I
R-C+
I
о-
Аниолоидное присоединение происходит так-
же к С=С-связям, если они сопряжены с кар-
бонильной или антрильной группами.
Образование карбаниона достигается
большей частью отдачей протона. Одиано
во многих случаях карбанион возникает
лишь под действием оснований (NaOH,
RONa):
H-C-COOB-5§g>
н
~н
I
:С—COOR
I
_н
Na+
Соседний электроотрицательный
гетероатом, например карбоксильный
кислород в приведенном выше уравнении,
облегчает отщепление протона. Аромати-
ческие или гетероциклические углеводо-
роды не вступают в эту реакцию, так как
подвижность водорода С—H-связи у них
очень мала. Образование карбаниона в этих
* Материал обработали X. Гросс, X. Бишофф,
Е. Гефт, Е. Грюндемав.
596 ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
случаях возможно лишь при переводе таких углеводородов в металлооргани-';
ческие соединения, как, например, при реакции Гриньяра.
Если С=С-связь не активировала соседними группами, то обычно возможной
только катионопдное присоединение. При этом карбанион акцептора взаимодействует?
с карбкатионом адденда с образованием новой связи. Все реакции присоединения’^
алкилгалогенидов, ацилгалогенидов и насыщенных углеводородов к олефинам^
в присутствии сильнокислых катализаторов происходят по приведенному ниже!
катионоидному механизму присоединения: *
НН ПН |
II II -I
(СНз)зС* Х- + С--С* —> (СН3)3С-С-С-Х ;
II II i
НН пн
Наряду с такими анпоноидными и катпоноиднымп реакциями присоединения-
возможны реакции присоединения к С=С-связям, протекающие по свободпоради-*
кальному механизму. Катализаторами таких реакций служат перекиси или азосоедич
нения. Образование новой С—С-связи происходит при этом благодаря вэаимодей-;
ствию адденда с двумя «вспаренными электронами гомолитически расщепленной
л-электронной пары двойной связи акцептора:
В—СП- СИз + ССЦ —> П-СН-СН3-СС1з ’ |
01 ' Я
7. Присоединение по С-С-связям ||
1. Присоединение углеводородов по этиленовым связям я
Присоединение алифатических углеводородов к этиленовым связям происходили
в присутствии катализаторов [1] (серной и фосфорной кислот, треххлористого алкнЦ
миния). Из изобутана и пропилена получают, например, три изомерных гептаналя
В лабораторией практике этот метод особого значения не имеет. Напротив, присоедини
неиие олефинов к ароматическим соединениям является часто применяемым методом^
алкилировакия последних [2]. Так, из этилена и бензола, под действием А1С13 йляя|
катализатора на основе двуокиси кремния и'окиси алюминия получают этилбензодЛя
из бензола и гексена-1 — 2-фенил-«-гексан [3]. Образование С—С-связи происходит®
всегда у углеродного атома двойной связи, содержащего наименьшее количеством
атомов водорода. Из бензола и пропилена образуется кумол, используемый дляй'Я
синтеза фенола. Особенно легко бензол реагирует с циклогексеном, давая циклоге- ’м
ксилбензол [4J. .
Циклогекеилбензол. К охлажденной льдом смеси 400 г бензола и 92
концентрированной H2SO4 добавляют в течение 2 ч при перемешивании 164
циклогексена. Получается 200 г циклогексилбензола (т. кил. 239—245° С).<
При обработке реакционной массы очень важно промыть маслянистый слой^
перед перегонкой сначала дважды 50 мл холодной концентрированной H2SOt,^
потом водой, разбавленной щелочью и опять водой. '
В тех же условиях взаимодействием аллилхлорида с бензолом можно получить^
ш-хлоркумол.
zCHs
СвНб + СН2=СНСН2С1 —► СвП5СН< ,
_______________ хСНаС1
[1] N. 1 р a t i в f f, A. G г о ss е, Н. Р i n е s, V. Komarewskf J Am. Chem.
Soc., 58, 913 (1936). ;
[2] cp. A. A. D’K oily, J. К e 1 1 e t, J. Plucker, Ind. Eng. Chem., 39, 154J
(1947). .
[3) A. Brochot.’C. r., 117, 115 (1893). i
[4] B. Corson, N. Ipatieff,!. Am. Chem. Soc., 59, 645 (1937). '
I. ПРИСОЕДИНЕНИЕ ПО С=С-СПЯЗЯМ
Полученное соединение содержит реакционноспособный атом хлора, который
может участвовать в дальнейших превращениях.
Применение в качестве катализатора металлического алюминия прн повышенной
температуре дает возможность осуществить алкилирование олефинами фенолов и аро-
матических аминов [5]. Алкилирование происходит предпочтительно в орто-поло-
жение,
2. Полимеризация производных этилена
Полимеризация производных этилена в настоящее время очень широко исполь-
зуется для получения пластических масс. Определенные заместители в этилене, которые
вызывают повышенную поляризацию молекулы, увеличивают как степень, так и ско-
рость полимеризации. Такими заместителями являются ароматические радикалы
(стирол), кислородсодержащие группы (акролеин, эфир акриловой кислоты, простые-
и сложные виниловые эфиры) и галогены (винилхлорид). По прн накоплении этих
заместителей в молекуле способность производных этилена к полимеризации умень-
шается или исчезает совсем. Стильбен, например, дает при освещении в бензоле только-
димер [6].
Полимеризацию можно ускорить с помощью УФ-облучения, действием перекисей,
а также кислотных или основных катализаторов. ,
В присутствии перекисей или при УФ-облучении полимеризация протекает по
радикально-цепиому механизму:
В--.- СЩ-СП —► R-СПа-СН. CH.-CHCl^ ц_(;н2—СН—СЩ-СТГ. и т. д.
II II
С| С1 С1 С1
Образовавшиеся в начале реакции активированные мономеры начинают рост-
цепи. В присутствии большого количества активных центров получаются короткие-
цепи. Если же их образуется немного, то возникают бойее длинные цепи. Антиокисли-
тели, такие, как гидрохинон или тиофенол, препятствуют радикально-цепной полиме-
ризации. Обрыв цепи вызывают также многие неорганические восстановители (гидро-
сульфит натрия, сульфит натрия и т. д.). Используя подходящее сочетание окислителей
(перекисей) и восстановителей — так называемую окислительно-восстановительную
систему, можно оказывать требуемое влияние на процесс полимеризации.
Рост цепи может прекратиться, когда свободнорадикальный центр молекулы
полимера переходит на другую молекулу, например в результате отщепления атома
водорода. Присоединение атомарного водорода к молекуле мономера приводит к воз-
никновению нового активного центра:
R—/СН2-СН\—СП2-СН- +СН2=СН —»В-/СНг— СН\— СН=СН+СНэ-СН.
\ ci /„ ci ci \ ci /„ ci а
Одним из видов радикальной полимеризации является так называемая теломери-
зацил [7], В результате этой реакции при участии, например, галогенметанов получа-
ются полимеры с малой длипой цепи и заданными начальной и концевой группами.
В приведенном ниже суммарном уравнении показана реакция таксогена (например, эти-
лена) с телогеном (например, четыреххлористым углеродом) с образованием теломера:
sCH2=CH54-CCh —> С1-(СН2~СН2)Л-СС13
Теломеризация [8]. В автоклав из нержавеющей стали емкостью около
350 мл, снабженный штуцером для ввода газа и термопарой для измерения
[5J R. Stroh,' J. Ebersberger, Н. Н a b erland, W. Hahn, Angew-
Chem., 69, 124 (1957); R. Strob,R. Seydel, W. Hahn., Angew. Chem,»
69, 699 (1957).
[6] G. Cimician, P. Silber, Ber., 35, 4129 (1902).
[7] Ф. А з и и г с p, Химия и технология моноолефинов, пер. с нем., Гостоптехиздат,
1960; см. также Е. Muller, Angew. Chem., 64, 246 (1952).
[8] R. М. Joyce, W. E. Hanford, J. Harmon, J. Am. Cbemi Soc.*
70, 2529 (1948).
L.
f)98 ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
температуры, загружают 210 г (1,36 моль) свежеперегнанного четыреххлори-
стого углерода, 35 г воды и 0,47 в (0,00194 моль) перекиси бензоила. После
удаления воздуха из автоклава в него подают этилен до давления 35 ат. Авто-
клав покачивают в горизонтальном положении и нагревают. Когда температура
реакционной смеси достигнет 70° С, давление нагнетанием этилена повышают
до 100 ат. Реакционную смесь выдерживают 5 ч при 95° С. Подачей этилена
давление поддерживают в пределах 85—100 ат. После охлаждения реакцион-
ную смесь выгружают из автоклава, отделяют воду, органический слой сушат
над безводным MgSO4. Непрореагировавший четыреххлористый углерод отго-
няют. Выходы и температуры кипения теломеров (после перегонки) приведены
ниже:
X Пределы выкипания фракций, °C Выход, г Т. кип. чистых веществ, °C
1 90 (при 12 мм рт. ст.) 11.7 159 (при 760 мм рт. ст.)
2 90—115 (при 11 лл рт. ст.) 97.0 112 (при 24 рт. ст.)
3 115—145 (при 11 мм рт. ст.) 36.0 143 (при 24 мм рт. ст.)
4 145—175 (при 11 мм рт. ст.) Остаток 12.5 5.0 168 (при 20 мм рт. ст.)
Чем выше давление таксогена (в приведенном примере — этилена), тем больше-
молекулярный вес образующихся теломеров. При низком давлении х может быть
равным 1.
При большом избытке телогена (100 : 1) происходит не теломеризация, а при-
соединение (х = 1) (см. также стр. 708). ___
При использовании четырехбромистого углерода уже при мольном соотношении
от 4 : 1 до 2 : 1 полимеризация практически не идет; происходит лишь присоединение
СВг4 к олефину.
Кроме упомянутых выше галогенметанов [7] функции телогенов могут выполнять
спирты [9], альдегиды и кетоны [10], эфиры [11] и амины [12]:
яСН2=СН2 + СНаОН —> Н-(СН2—СНаЬ-СНОН
В случае альдегидов происходит присоединение водорода альдегидной группы^
в результате чего образуются кетоны.
При использовании кислотных катализаторов (А1С18, BF3, кислот) полимери-
зация олефинов протекает по катионному ионно-цепному механизму. В технике поли-
меризацию изобутилена проводят при температурах от —70 до —100° С в присутствии
0,05% В F3 в течение нескольких минут. При этом с количественным выходом получйот
каучукоподобный полимер [13]. Температура реакции сипьно влияет на величину
молекулярного веса получающегося полимера. Чем ниже температура реакции, тем
выше молекулярный вес. Например, При полимеризации а-метилстирола в этилхло-
[9] Esso Research Engeneering, цат. ФРГ 1058981, К1. 120; С., 1960, 297; J. D. La-
ze г t е, R. J. К о a h а г, J. Am. Chem. Soc., 77, 910 (1955); W. H. U г г у,
F. W. Stacey, Е. S. Н и у s е г, О. О. J uveland, J. Am. Chem. Soc.,
76, 450 (1954).
[10] M. S. Kharasch, W. H. U г г у, В. M. Kuderna, J. Org. Chem., 14,
248 (1949); M.S. К h а г a s c h, J. Kud erna, W. N ud enb erg, J. Org.
Chem., 18, 1225 (1953).
[11[ W. H. Urry, E. S. Huyser, J. Am. Chem. Soc., 75, 4876 (1953).
[12] W. H. Urry, O. O. J и v eland, F. W. Stacey, J. Am. Chem. Soc.,
74, 6155 (1952); W. H. Urry, О. O. Ju vel-an d, J. Am. Chem. Soc., 80,
3322 (1958).
[13] K. Hamann, Aftgew. Chem., 63, 234 (1951).
[14] D. Pepper, Nature, 158, 789 (1946); Trans. Farad. Soc., 45 ,397 (1949).
Г. ПРИСОЕДИНЕНИЕ ПО С-С-СЛЯЗЯМ
699
ряде в присутствии А1С13 прп 23° С молекулярный вес полимера составляет около
1000, а при —130° С — 84 000. Растворитель также влияет на величину молекулярного
веса. При исследовании полимеризации а-метилстирола в присутствии SnCl4 в различ-
ных растворителях найдено, что молекулярный вес тем выше, чем больше диэлектри-
ческая проницаемость применяемого растворителя.
При использовании менее активных катализаторов [15] (SnCl4 и РЬС14) для
начала реакции необходимо присутствие сокатализаторов, например воды, трет-
бутилового спирта, уксусной, трихлоруксусной или серной кислот. Вероятно, катали-
затором полимеризации в этом случае является кислота Льюиса. Изобутилен в гек-
сане при —80° С в присутствии TiCl4 реагирует очень медленно, но при пропускании
слабого тока влажного воздуха начинается бурная реакция.
Сокатализаторы необходимы и при использовании активных катализаторов.
Таи, реакция безводного изобутилена с безводным BF3 в колбе, высушенной в глубо-
ком вакууме при 400° С, не начинается при —80° С в течение 2 ч [16]. Если же колбу
сушить в глубоком вакууме без нагревания, то реакция начинается сразу. При этом
достаточны уже очень небольшие количества влаги, чтобы вызвать полимеризацию.
Реакция протекает по следующей схеме:
BFa+HX —> H+[BF3X]-
CHg СНз СНз . СНз
H+[BF3X]~+CH0=C —>СН3-С+ [BF3X]- Gn,~c(CI*,),-> СН3-^-СН2-С* [BF3X]*
II II
СНз СНз СНз СНз
Обрыв цепи происходит в результате присоединения аниона или отдачи протона:
СНз / СНЭ\ СН8
I / I \ I
СНз—С— СН2-С- -СН2-С-Х
1 \ I / I
СНз > СНз/я СНз
СНз / СПз\ СНз
СН3—С— [ СН2—С— |—СН2—С* [BF8X]-
I \ I / I
СНз СНз'в СНз
СНз/ СНз\ СНз
I / I \ I
—СНз—С—| СН2—С — -СН=С
-H[bf4xi । I j I 1
CH3\ CH3/„ СНз
Полимеризацию этилена раньше удавалось осуществить только при высоком
давлении и высокой температуре. Но благодаря исследованиям Циглера н сотр. [17]
стало возможным проводить полимеризацию этилена при нормальном давлении под
действием катализатора на основе триэтилалюмииия и TiCl4 в подходящем раствори-
теле. Методику лабораторного получения полиэтилена низкого давления приводят
Циглер и Мартин [18].
Анионная ионно-цепная полимеризация протекает под действием щелочных
катализаторов. При полимеризации стирола в присутствии амида натрия в жидком
[15] Р. Н. Pies ch, Nature, 160, 868 (1947).
[16] A. Evans, G. Maedow, Trans. Farad. Soc., 46, 327 (1950). »
[17] K. Ziegler, E. H о 1 z k а ш p, H. Broil, H. Martin, Angew. Chem.*
67, 541 (1955).
[18] K. Ziegler, H. M а г t i n, Makromol. Chem., 18/19, 186 (1956).
. 700
ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
аммиаке начальная реакция состоит в присоединении аниона амида к двойной связи
с образованием карбаниона:
NHa+CH=CH2 —> NH2-C.H-CH, с-н»~9н-сн^
С«Н6 с6нБ
NH2-CH-CH2—СН-СН2 И т. д.
I I
свн6 Свн8
Этот механизм подтверждается тем, что молекула полистирола содержит амино-
группу. В эфире стирол в присутствии амида натрия не полимеризуется.
По катионному типу реагируют преимущественно такие соединения, замести-
тели у двойной связи которых вызывают смещение л-электропов двойной связи в на- ,
правлении к p-углеродному атому, например, виниловый эфир:
СН,=СН-OR <--♦ СЩ-СН—о—R > СНг—СН—OR
Соединения, полимеризующиеся по анионному тику, имеют у двойной связи.'
сильно электроноакцепторные группы, которые притягивают л-электроны двойной":
связи, например акрилонитрил:
СНа=СН-CN СНз-СН-CN
3. Диеновый синтез *
Диеновым синтезом, по Дильсу [19] и Альдеру [20], называют реакции присоеди-
нения этиленовых соединений (диенофилов) к сопряженным системам (диенам):
R R
[ CH CH
/ \
HC CH—R HC CH-R
i + II II 1
HC CH-R HC CH-R
диенофил \ /
CH СП
ll 1 R
диен
Присоединение диенофила к диену происходит так, что образуется шестичлениов
кольцо (1,4-присоединение). В реакцию вступают три двойные связи, однако продукт
реакции содержит только одну связь, да и то в измененном положении. Перегруппиро-
вок атомов или атомных групп, в общем, не происходит. Диеновый синтез протекает
стереоспецифично. Например, при присоединении малеиновой кислоты к диену про-
дуктом реакции является tyuc-дикарбоновая кислота, а при присоединении фумаровой
кислоты — транс-дикарбоновая кислота. Не последнюю роль при этом процессе при-
соединения играют заместители в диенах и дйенофилах. Так, например, 1,2,3,4-
тетрафенилбутадиец не склонен к присоединению диенофила, в то время как 1,2,3,4-
тетраметилбутадиен реагирует гладко [21]. Объемистые заместители в положениях
2,3 диена могут полностью исключить возможность присоединения, поскольку они
* См. монографию А. С. Онищенко, Диеновый синтез, Изд. АН СССР,
1963 г. — Прим. ред.
[19] О. Diels, Angew. Chem., 42, 911 (1929).
[20J К. А 1 d о г, в книге W. Foerst, Neuere Methoden der organischen Chemie,
Verlag Chemie, Weinheim.
[21] H. Backer, J. £ t r a t i n g, L. Huisman, Rec, trav. chim., 53, 525
(1934); 58, 761 (1939); 60, 557 (1941).
[. ПРИСОЕДИНЕНИЕ ПО С=С-СВЯЗЯМ
701
ограничивают свободное вращение и тем самым препятствуют установлению необходи-
мой для присоединения цисоидной (б) конфигурации [22].
R СН2
С
I
С
Н2С^''R
(а)
R СН2
С
I
С
(б)
Так, 2,3-ди-трет-бутилбутадиен [23] не реагирует с диенофилом, тогда как
в случае 1,3-ди-трет-бутилбутадиена [24] присоединение происходит нормально.
Особенно реакционноспособным является циклопентадиен, у которого благодаря
устранению возможности свободного вращения фиксируется особенно благоприятная
конформация.
Ацетиленовые соединения типа енинов (С=С—С=С) и диенииов
(С=С—С=С—С —С) также обладают способностью к присоединению. Как и при реак-
ции Дильса— Альдера, происходит 1,4-присоединенке, однако с одновременным
перемещением атома водорода:
R R
I I
R СН С
zO \ ,о
С СН-С<^ R-С СН-С^
\ 1 I >
С СН-С< НС СН-С<
. X) \ / ^0
СН сн
Кроме ациклических 1,3-диеиов в реакцию Дильса — Альдера вступают али-
циклические и некоторые ароматические соединения. Простейшие ароматические си-
стемы, такие, как бензол, дифенил и фенантрен, не реагируют по Дильсу — Альдеру,
в то время как более сложные конденсированные системы, например, перилек и бенз-
антрен, могут вступать в эту реакцию. Сопряженные двойные связи принадлежат при
этом различным ядрам (стрелками показаны места присоединения диенофила).
В случае антрацена присоединение диенофила протекает по сопряженным двой-
ным связям среднего ядра с образованием углеродного мостика.
СН-R
II
CH-R
При реакции циклических диенов, таких, как циклопептадиен или цикло-
гептадиен, например, с ангидридом малеиновой кислоты, также возникают цикличе-
ские соединения с углеродным мостиком. Из двух возможных стереоизомерных форм
[22] К. Alder, М. Schumacher, Ann., 571, 87 (1951).
[23] Н. Backer, Rec. trav. chim., 58, 643 (1939).
124] H. Backer, J. S t r a t i n g, Rec. trav. chim., 56, 1069 (1937).
702 ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
всегда образуются соединения с эндо-конфигурацией. Одиако реакция удается только
в том случае, когда образование соединения не противоречит правилу Бредта.
эндо -цис
При замене циклопентадиена фураном получают соответствующее эндоокса-
соедипение.
НС- R
II
НС—R
Однако при взаимодействии фурана с менее активными диенофилами происходит
заместительное присоединение. Для пиррола оно является основным направлением
реакции [25]:
II й+сна=снн ГП
NH^cHjCHiR
Для тиофена не характерны ни 1,4-присоединение, ни заместительное присоеди-
нение; он вообще не вступает в реакцию Дильса — Альдера. В качестве дненовой
компоненты тиофен, подобно бензолу, выступает только в более высококопдепсиро-
вапных системах.
Способностью к реакции с диенами обладают не только этиленовые двойные
связи. В качестве диенофильной компоненты наряду с нитрил-, азо-, нитрозогруппами
может реагировать даже молекулярный кислород, и особенно пригодны ацетиленовые
тройные связи. Реакционная способность как диенов, так и диенофилов зависят от
их строения. Особенно благоприятно влияют на процесс присоединения ак пивные
полярные группы. Наиболее реакционноспособными соединениями являются а,р-
ненасыщенные карбонильные соединения, например акролеин, акриловая кислота,
малеиковая кислота и ее ангидрид, ацетилендикарбоновая кислота, n-хнион, коричный
альдегид, а также а,р-ненасыщенные нитрилы и а,₽-некасыщенные нитросоединения.
У диенофилов, содержащих такие активные группы, сопряжение двойных связей не
является обязательным; при благоприятных условиях происходит присоединение дие-
нов к диенофилам с изолированными двойными связями. Диенофилами этого типа могут
служить виниловый эфир и винилуксуспая кислота. Реакция кетенов с диенами про-
текает пс по Дильсу —-Альдеру, а с образованием производных циклобутанона [26]:
В случае бензохинона протекает двойной диеновый синтез. Например, при реак-
ции бензохинона с двумя молекулами циклогексадпена образуется диэндозтиленовая
[25] I. D. Webb, G. Т. В о г с h е г d t, J. Am. Chem. Soc., 73, 752 (1951); К. A 1-
der, С. H. Schmidt, Ber., 76, 183 (1943).
[26] H. Staudinger, E. S u ter, Ber., 53, 1092 (1920); В. T. Brooks,
G. Wilbert, i. Am. Chem. Soc., 63, 870 (1941); K. Aider, H. F. R i c-
kort, Ann., 543, 15 (1940).
I. ПРИСОЕДИНЕНИЕ ПО С=С-СВЯЗЯМ
70;
циклическая система, которая легко теряет четыре атома водорода и при нагревант
переходит в результате отщепления этилена в антрахинон:
Используя производные циклогексадиена, можно получить гомологи антрахв
нона. В качестве исходных соединений пригодны также 1,4-нафтохиноны; этиленовы
мостик образующегося при этом соединения может содержать заместители, причё]
его способность к элиминированию не теряется. Так, взаимодействием 1,4-нафтс
хинона с а-федлаидреном получают р-метнлантрахинон:
сн2=сн
сн
Н3С сн
Некоторые соединения, например циклопентадиен, часто реагируют и как диень
и как диенофилы. Примером служит реакция их димеризации *:
Практическое осуществление такого рода диеновых синтезов исключительв
просто. Реакция протекает уже при смешении рассчитанных количеств диеиа и диен«
фила в органических растворителях с выделением тепла (17—19 ккал/моль). С бол»
медленно реагирующими веществами ее можно вести при нагревании (например, не
давлением или в расплаве). Однако экзотермическая реакция Дильса — Альдер
является в то же время равновесной, поэтому с повышением температуры легко пр
исходит обратное расщепление на компоненты. Большой избыток одного из двух комп
нентов сдвигает равновесие реакции в сторону образования продукта црисоединени.
Ниже приведены методики проведения диеновых синтезов.
Ангидрид ^ис-эндометилеитетрагидрофталевой кислоты [27]. К суспе:
зии 1 моль ангидрида малеиновой кислоты в пятикратном количестве бензол
при охлаждении постепенно прибавляют 1 моль цпклопентадиена, при этс
* Димеризация диенов изучена впервые С. В. Лебедевым, задолго до раб
Дильса и Альдера. — Прим. ред.
[27] О. D i е 1 з, К. А 1 d е г, Ann., 460, 111 (1928).
ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
малеиновый ангидрид переходит в раствор с большим выделением тепла. В ре-
зультате с почти теоретическим выходом образуется ангидрид цис-эндометилентет-
рагидрофталевой кислоты; после перекристаллизации из лигроина т. пл. 164—
165° С.
Несколько трудное с ангидридом малеиновой кислоты реагирует бутадиен [28].
Ангидрид ifuc-тетрагидрофталевой кислоты. В 10 мл бензола растворяют
2—2,5 г, бутадиена, раствор нагревают с 4 г ангидрида малеиновой кислоты
в запаянной трубке, оставляют на 12 ч и затем нагревают 5 ч при 100° С.
В результате с количественным выходом получается ангидрид t^uc-тетрагидро-
фталевой кислоты; после перекристаллизации из лигроина т. пл. ЮЗ—104° С.
Норкантаридпн [29]. Б эфире суспендируют 2 г ангидрида малеиновой
кислоты и прибавляют рассчитанное количество фурана. При слабом нагрева-
нии постепенно начинается реакция. Через несколько часов большая часть
продукта присоединения — ангидрида эндооксатстрагидрофталевой кислоты
выделяется при охлаждении в кристаллическом виде. После отгонки эфира
продукт получают с теоретическим выходом. Это вещество плавится при 125° С,
вспениваясь и разлагаясь на исходные компоненты. При гидрировании полу-
ченного соединения коллоидным палладием в содовом растворе образуется
норкантаридин.
Эндоэтилентетрагидробензальдегид [30]. В течение 3,5 ч нагревают
в запаянной трубке при 100° С 7 г циклогексадиена и 10 8 акролеина. После
фракционирования получают 3 г альдегида; т. кип. 84—85° С (12 мм рт. ст.).
Его семикарбазон имеет т. пл. 176—177° С.
Если в качестве диеновой компоненты применяют антрацен, то присоединение
происходит у 9-го и 10-го углеродных атомов с образованием 9,10-дигидроантрацен-
9,10-эндоэтилендикарбоповой-Г,2' кислоты.
9,10-Дигидроантрацен-9,10-эндоэтилендикарбоновая-1',2' кислота [31].
На парафиновой бане нагревают 12 в антрацена и 8 г ангидрида малеиновой
кислоты до начала реакции, которая сопровождается кристаллизацией реак-
ционной массы, начинающейся у стенок колбы. Нагревают еще 15________20 мин,
Следя за тем, чтобы температура бани не превышала 260° С, охлаждают и непро-
реагировавший малеиновый ангидрид экстрагируют эфиром из тонкоизмель-
ченной реакционной массы. После перекристаллизации из малонового эфира
получают бесцветные кристаллы, которые при быстром нагревании плавятся
при 262—263° С.
Дегидробелзол также может применяться как диенофил. Примером может слу-
жить синтез триитицена по Виттигу и Людвигу [32].
Триптицен. При взаимодействии 42 ммоль антрацена с 30 ммоль о-фтор-
бромбензола и магнием в тетрагидрофуране получают смесь углеводородов,
из которой непревращенный антрацен извлекают малеиновым ангидридом.
Хроматографированием получено 11% трифенилена (т. пл. 194—195° С>
и 28% триптицена. Образование триптицена объясняется следующей реакцей;
[28] О. D i е I в, К. Alder, Ann., 460. 113 (1928).
[29] О. Diels, К. Alder, Вег., 62, 557 (1929).
[30] О. Diols, К. Alder, Ann., 478, 144 (1930).
[31] О. Diels, К. der, Ann., 486, 196 (1931).
[32] G. Wittig, R. Ludwig, Angew. Chem., 68, 40 (1956).
I. ПРИСОЕДИНЕНИЕ ПО С=С-СВЯЗЯМ
705
4. Присоединение соединений с активированной С Н-связью
(реакция Михаэля)
Но Михаэлю [331 и Клапзену [34], можно осуществить присоединение соедине-
нии с активным атомом водорода к а,р-ненасыщенным альдегидам, кетонам, эфирам,
нитрилам, сульфонам и нптросоединелиям, причем присоединение происходит па
углеродному атому внннльной группы, находящемуся в ^-положении к карбониль-
ной группе:
СН3СО—СН2 +СН2=СН—СОСНз СНзСО—СН—СН2—СН2—СОСНз
I I
COOR COOR
Из ацетоуксусного эфира п метилвивилкетопа получают этиловый эфир гептан-
дион-2,6-карбоновоя-З кислоты [35].
Этиловый эфир гептандион-2,6-карбоновой-3 кислоты. К 780 г ацето
уксусного эфира добавляют 10 мл раствора метилата натрия (23 г натрия
в 400 мл метилового спирта), затем при перемешивании и периодическом охлаж-
дении, чтобы поддерживать температуру 30—35° С, прибавляют по каплям
210 г свожеперегнанного метилвинилкетона. Смесь оставляют на ночь, реакцион-
ную массу разбавляют метиленхлоридом, нейтрализуют разбавленной уксусной
кислотой и после отгонки метиленхлорида перегоняют в вакууме. Получают
бледно-желтое масло; т. кип. 127—129° С (4,5 мм рт. ст.). Выход продукта
85—88% От теоретического.
Так как полученный этиловый эфир гептандион-2,6-карбоновой-3 кислоты со-
держит еще один подвижный атом водорода, то это соединение можно взаимодействием
со вторым молем метилвинилкетона превратить в эфир 5-ацетнлнопандион-2,8-карбоно-
вои-5 кислоты:
СН3СО-СП-СН2—СНг-СОСНз Н- СП2=СН-СОСНз •—>
COOR
.СНа—СН2-СОСН3
—> СНзСО—С—CHg-CHa—СОСНз
\ COOR
Это соединение можно получить в одну стадию по указанной выше мето-
дике, если подействовать на ацетоуксусный эфир двойным количеством метил-
винилкетона. Выход продукта 92% от теоретического; т. кип. 160е С (1,3 мл*
рт. ст.).
По Форлендеру и Кнетчу [36], можно получить эфир а-ацетилглутаровои
кислоты из ацетоуксусного эфира и этилового эфира акриловой кислоты.
Скорость реакции зависит как. от подвижности С—Н-связи аддепда, так и от
поляризуемости двойной эталеповой связи акцептора. Поэтому в роли адденда высту-
пают чаще всего только метиленовые или метиновые соединения, активированные
двумя соседними карбонильными или иитряяьными группами, например малоновый
эфир, динитрил малоновой кислоты, циануксусный эфир, 1,3-дикетосоединення, эфиры
1,3-дикетокарбоновых кислот, а также их моноалкилцроизводные. Поляризуемость
двойной этиленовой связи акцептора активируется сопряжением этой связи с одной иа
полярных кратных связей. Поэтому в качестве этиленовой компоненты используются,
в основном, а,p-ненасыщенные кетоны, а также а,р-ненасыщенные эфиры и нитрилы.
Присоединение катализируется основаниями, например едким кали, этилатом на-
трия, аминами и др.
[33] A. Michael, J. prakt.' Chem. |2], 35, 349 (1837).
[34] Т. Komnenos, Ann., 218, 161 (1883).
[35] Н. JI е n е с k a, Chem. Вег., 81, 189 (1948).
[36] D. Vorlander, A. Knotzsch, Ann., 294, 317 (1897).
45 3ai<a.s 1885.
706
ОБРАЗОВАНИЕ НОВЫХ G—С-СВЯЗЕИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
При использовании молярных количеств катализатора полученные'
продукты присоединения могут подвергнуться вторичной циклизации (реакция
Михаэля, сопровождаемая конденсацией). Если продуктами присоединения
являются 1,5-дикетоны, то вследствие кротоновой или альдольной конденсации
образуются производные циклогексенона или циклогексанолона:
воос
R- R'
J I
сн Rooc сп
\ \/ \
сн нс сн2
I —► 11
с=о с с=о
/ /\ /
СНз Н3С О СНз
Rz
I
ROOC СН
\ / \
НС сн2
I I
с с=о
НзС СН
Rz
I
ROOC СН
\/\
ПС СН2
или НО. | |
>С С=О
Н3(У \ /
СН2
Если аддуктом являются не дикетоны, а эфиры 6-кетокапроиовой кислоты,
то в результате сложноэфирной конденсации образуются производные дигидро-
резорцина:
СНз ' СНз СНз
1'1 I
С2Н6ООС сн С2Н5ООС СН С2Н5ООС СН
НС сн2 НС СНа
=С С=О -GaHb0H О=С С-0
\ / \ /
НзС OC2Hs СН2
СНа ^Ьн
I 4- I
О=С С=О
\ /
НзС 0С0Н5
Этиловый эфир метилдигидрорезорпинкарбоновой нислоты [37]. К рас-
твору 6,9 г натрия в 100 мл абсолютного спирта прибавляют 42 а ацетоуксус-
ного ефира, затем 34 е этилового эфира кротоновой кислоты. После нагревания
в течение 2 ч на водиной бане выделяется натриевая соль эфира метилдигидро-
резорцинкарбоновой кислоты. Эфир в виде масла осаждают разбавленной
H2SO4 из водного раствора его соли. Масло быстро затвердевает. После пере-
кристаллизации из горячей воды т. пл. продукта 89—90е С.
Четвертичные основания Макниха в условиях реакции Михаэля реагируют
так же, как а,р-ненасыщенные кетопы. Очень реакционноспособный вниилкетон, обра-
зующийся из основания Манниха в момент выделения, может под влиянием амида
натрия присоединять вместо обычных 1,3-дикарбоиильных соединений даже простые
кетоны с одновременным замыканием цикла:
N(CH3)3
I
/ NaNH8
I -N(CHj)», H+’
(371 R. v. Schilling, D. Vorland er, Ann., 308, 195 (1899).
Т. ПРИСОЕДИНЕНИЕ НО С==С-СВЯЗЯМ
Этот метод, метод Робинзона — Манниха, особенно часто применяют для синтляя
стероидов [38].
Здесь следует также упомянуть о цианэтилировании ненасыщенными нитри-
лами [39]. Например, по данным Байера и сотр. [40], можно присоединить пиколил-
2-кетопы к акрилонитрилу:
— A ™2CHsCN
N CH2COCH3 + 2CH2=CHCN N С-СОСНз
CH2CH2CN
Описано присоединение метилового эфира флуорен-9-карбоновой кислоты,
к аллилцианиду [41]:
I II II I +CH2 = CHCH?CN — I [I || I
Н СООСНз СН3СН СООСНз
I
ch2cn
Хотя аллилцианид является р,у-ненасыщенным соединением, С—С-связь
возникает и в этом случае у Р-углеродного атома аллилцианида. Присоединение этило-
вого эфира вышеупомянутой кислоты к акрилонитрилу также происходит в р-положе—
пне [42].
2-Питропропан присоединяется к бенаальацетофенону в метиловом спирте в при-
сутствии гидрида кальция [43]:
(CH8)2CNO2
I
С6Н5СОСП=СНС6Н5 + (СНз)2СНХ02 —> СбНБСОСН2СНСвН5
При взаимодействии нитрометана с этиловым эфиром коричной кислоты в при-
сутствии гидроокиси бензилтриметнламмония (трилон Б) образуется этиловый эфир
p-фепил-у-нитромасляпой кислоты с выходом 76% от теоретического [44] [см. также-
Org. Syntheses, 32, 86 (1952)].
5. Присоединение формальдегида (реакция Прайса)
Под действием кислых агентов формальдегид может присоединяться к этилено--
ним соединениям [45]. Так, при взаимодействии стирола с формальдегидом в присут-
ствии серной кислоты образуется 4-фенил-1,3-диоксан:
СНа
С„Н6-СН=СНа + 2СП2О С„Н5-СН СН2
О О
CH2
Ber., 90, 592 (1957).
[38] V. A. Mondon, Angew. Chem., 64, 123 (1952).
[39] Org. Reactions, v. 5, p. 79.
[40] H. Beyer, W. L as s i g, G. S c hud y, Chem.
[41] S. H. Tucker, J. Chem. Soc., 1949, 2182.
[42] A. Campbell, 8. П. Tucker, I. Chem. Soc., 1949, 2623; см. также
T. II о I b о, E. T a g m a n, Helv. chim. acta, 33, 2187 (1950); M. C. Kloe t-
7. el, F. L. Chubb, J. Am. Chem. Soc., 72, 150 (1950).
[43] N. Fi shman, S. Z u f f a n t i, J. Am. Chem. Soc., 73, 4466 (1951); см. также
Th. Rogers, J. Chem. Soc., 1943, 590; W. С. Kloetzel, J. Am. Chem.
Soc., 72, 4786 (1950).
[44] N. j. Leonhard, A. B. S i m о n, D. L. F e 11 e y, J. Am. Chem. Soc., 73,
857 (1951).
[45] II. J. P r i n s, Chem. Weekblad, 16, 1072, 1510 (1919).
45*
708
ОБРАЗОВАНИЕ НОВЫХ С—G-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
4-Фенвл-1,3-диоксан *. Смесь 675 г 37%-ного формалина, 48 г H2SO4
(d = 1,84) и 312 г стирола осторожно нагревают с обратным холодильником
при перемешивании в течение 7 ч. После охлаждения к смеси добавляют при
перемешивании 500 мл бензола. Затем бензольный слой отделяют и дважды
промывают по 750 мл воды. Бевзол удаляют перегонкой, а остаток фракциони-
руют. Выход 4-фенил-1,3-диоксана 353—463 а (70—88% от теоретического):
т. кип. 96—103Q С (2 мм рт. ст.).
В присутствии уксусной кислоты образуется в основном дпацетильное производ-
ное 1-фенилпропандиола-1,3 [46]:
С3Н5СН=СП2+СН2О + Н2О -3^21^ СвН6СНСН2СНг СН|СООН> СвН5СНСН8СН2
НО ОН СНзСОО ОСОСНз
В эту реакцию вступают и другие олефины, однако арилолефины дают лучшие ;
выходы [47].
6. Присоединение органических гапогенпроизводиых
В противоположность насыщенным галогенидам галогснпроиэводные ненасыщен-
ных углеводородов при обработке хлористым алюминием ведут себя подобно аромати-
ческим углеводородам [48]. В качестве присоединяющего компонента можно исполь- .
аовать галогенпроизводные этилена. Присоединяются преимущественно хлорметаны, *
особенно легко хлороформ, а также метиленхлорид; способны присоединяться также ;
хлорэтаны, однако реакция протекает с меньшей скоростью. В качестве примера
приведен синтез!,1,2,3,3-пентахлорпропапа [49] из хлороформа и си.ил1-дихлорэтилена:
СНС1з+С1СП=СНС1 —> СНС12-СНС1-СНС12 >
1,1,2,3,3-Пентахлорпропан [49]. Кипятят в течение нескольких минут '
120 г чистого СНС13 с А1С13 (1%). Такой же обработке подвергают 97 г дихлор- •
этилена, но нагревают до температуры не выше 40° С. Оба реагента, насыщенные
А1С13, после охлаждения смешивают и нагревают при 50° С в течение 5—10 мин >
до тех пор, пока температура не начнет самопроизвольно подниматься. Охлаж- _!
дением проточной водой поддерживают температуру около 50—55° С. Если она
поднимется выше, то реакция выйдет из-под контроля. По истечении 30 мин •
температура начинает падать, после чего смесь еще около 10 мин нагревают ;
при 55—60° С. Затем отгоняют с водяным паром непрореагировавшие исходные
продукты, следя за объемным соотношением отходящих водяных паров и масла.
Когда содержание масла на 10 мл общего объема дистиллята падает до 3 мл, t
приемник следует заменить. Теперь отгоняется пентахлорпропан в практически
постоянном количестве (2,5 .мл на 10 мл дистиллята). Отгонку заканчивают, ,
когда в 10 мл дистиллята будет содержаться только 0,5 мл масла. Выход v
продукта составляет 70—75% от теоретического.
В оригинальной статье указаны условия, тормозящие реакцию, причем
причина этого явления до настоящего времени не выяснена.
Описана реакция изопропилхлорида с трихлорэтиленом и аналогичные реакции >
[50]. Атом галогена при этом присоединяется к углеродному атому с меньшим числом
атомов водорода. В качестве катализатора кроме А1С13 можно применять FeCl3 [51]. >
* См. Org. Syntheses, 33, 72 (1953).
[46] R. W. Short ridge, J. Am. Chem. Soc., 70, 873 (1948).
[47] E. Drukker, M. В e e t s, Roc. trav. chim., 70, 29 (1951); Org. Synth., 33, »
72 (1953).
[48] H. J. Prins, F. E ngelhard, Rec. trav. chim., 54, 307 (1935); 57, 659
(1938).
[49] H. J. Prins, F. Engelhard, Rec. trav. chim., 54, 309 (1935).
[50] L. S c h га e r 1 i n £, J. Ara. Chem. Soc., 68, 1655 (1946).
J51] L. Schmerling, J. Am. Chem. Soc., 71, 701 (1949).
I. ПРИСОЕДИНЕНИЕ ПО С^С-СВиЗЯМ
709
Осуществлена также реакция этилена с различными разветвленными алкилгалогени-
дами (52]:
(СНз)3ССЦ-СН2^СН2 А1СЬ^ (СНз)зССН2СН2С1
При взаимодействии mpem-бутилхлорида с этиленом получается 4-хлор-2,2-
диметилбутап с выходом 75% от теоретического.
Свободнорадикальное присоединение галогенированных метанов к олефинам
можно осуществить (531, нагревая при 70—90° С смесь олефина, например, с четырех-
бромистым углеродом или с избытком четыреххлористого углерода в присутствии
1 — 5 мол. % диацилперекиси:
СН3(СН2)5СН=СН8 + СВГ4 —> CH3(CH2)8CHBrCH2CBr3
Без перекисного катализатора тетрабромметан может присоединяться под дей-
ствием света [54].
Удалось также осуществить [55] катализируемое перекисями присоединение
к олефинам эфиров а-бромзамещенпых жирных кислот:
С8Н1зСН=СНа + ВгСНСООСаН5 —> СвН13СНСНаСНСООС2Н5
f I I
СНз Вг СНз
Таким способом получают эфиры у-бромзамещенных жирных кислот.
Исключительно легко протекает присоединение а-галогенированных простых
эфиров в присутствии хлористого цинка [56]:
CH3OCH2C1 + RCH=CHR' —> RCHCHC1R'
СП2ОСН3
Образующиеся при этом у-галогенвамещенные простые эфиры далее уже ие мо-
гут реагировать, так как подвижность галогена уменьшается с увеличением расстоя-
ния от эфирного кислорода.
Под действием хлористого алюминия могут присоединяться по двойной связи
также хлорангидриды кислот * с образованием ^-галогенкетонов [57]:
/\/СОСНз
( 1+сп3сос1 \ Г
* Реакция И. Л. Кондакова; см. Bull. Soc. Chim. France, [3], 7, 576 (1892). —
Прим. ред.
[52] L. Schmferling, J. Am. Chem. Soc., 67, 1152 (1945).
[53] M. S. Kharasch, 0. R e i n m u t h, W. H. U г г у, J. Am. Chem. Soc.,
69, 1105 (1947); обзор в книге Г. Ч. Уоллинг, Свободные радикалы
в растворах, пер. с англ., Издатиилит, i960.
[54] М. S. К h а г а в с h, Е. V. J е п в е n, W. Н. U г г у, J. Am. Chem. Soc.,
68, 154 (1946).
[55] М. S. К. h £ г а в с h, Р. S. Skell, Р. F i s h е г, J. Am. Chem. Soc., 70,
1055 (1948); см. также обзор в иниге Г. Ч. У о л л и н г, Свободные радикалы
в растворах, пер. с акгл., Издатиилит, 1960.
[56] F. S t г a u s, W. Т h i е 1, Ann., 525, 151 (1936).
[57] Н. Wieland, L. Bettag, Вег., 55, 2246 (1922); A. Bremer,
H. Schinz, Helv. chim. acta, 35, 1621 (1952); W. S c h о e 11 er, C. Z 5 11-
n e г, пат. США 1737203; С. A., 24, 626 (1930).
710 ОБРАЗОВАНИЕ НОВЫХ С—С-СИЯЗЕИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
7. Присоединение диазометана и синильной кислоты [58]
Реакцией диазометана с ацетиленом можно получить пиразол:
нс=сн
HCZ2CH + CHSN2 —> I I
z ПС NH
N
Аналогично из этилена получают пиразолин. С диазометаном могут реагировать
также замещенные этилены. Так, из бензальацетофенона и диазометана образуется
4-фепил-5-бензоилпиразолин [ 59 ]:
СвН5СН=СНСОСвПб -j- CH2N2
СбН5СП-СНСОСбН5
I 1
НС NH
Так же протекает взаимодействие с диазометаном эфира акриловой кислоты.--'
В подобных реакциях СН2-группа диазометана присоединяется всегда к 0-углеродному
атому «.(^-ненасыщенных кетонов или эфиров с одновременным перемещением одного •
атома водорода.
Пиразолины при нагревании обычно разлагаются с отщеплением азота,
иногда с образованием производных циклопропана*, например [60]:
Так ясе, как диазометан, могут присоединяться и другие диазосоединения:
с-соосн,
< III
С-СООСНз
N С-СОоСН,
II II 3
N — С-СООСНз
Соединение I было получено с количественным выходом [61] при прове-
дении реакции в абсолютном эфире в течение одного дня. Следует упомянуть
также о получении эфира индолил-3-уксусной кислоты из нидЬла й диазо-
уксусного эфира, протекающее с орцеплением азота [62].
Взаимодействие синильной кислоты с ацетиленом протекает с образованием акри-
лонитрила. Олефины же, как правило, с синильной кислотой не реагируют. Но если
двойная связь олефина активирована соседними электропоакцепторными группиров-
* Превращение пиразолинов в производные циклопропана открыто II. М. Киж-
нером [ЖРХО, 44, 165 (1912); 45, 1770 (1913)]. — Прим. ред.
[58] Обзорная статья В. Е i s t е г t, в книге W. Foerst, Methoden der prapara-
tiven organischen Chemie; L. Summers, Chem. Rev., 55, 301 ‘(1955); обзор-
ная статья Houben-Weyl, Bd. 5, S. 265.
[59] L. I. Smit b, W. B.’P i n g s, J. Org. Chem., 2, 23 (1937).
[60] H. Wieland, О. P/obst, Ann., 530, 277 (1937).
(61 ] J. v. Alphen, Rec. trav. chim., 62, 491 (1943). 4
[62] R. W. Jackson, R. H. M a n s k e, C., 1936, I, 4005.
I. ПРИСОЕДИНЕНИЕ ПО С=С-СВЯЗЯМ
711
ками, то присоединение синильной кислоты к двойным связям протекает с хорошим
выходом. При взаимодействии виниловых эфиров с синильной кислотой образуются
а-алкоксинитрилы [63]:
СН2=СНОС2Н5 СН3СНОС2Н5
Присоединение происходит под действием щелочных катализаторов (цианидов
щелочных металлов, пиридина). Такие реакции были проведены с метил-, этил-, бутил-,
циклогексил- и декагидронафтилвиниловыми эфирами.
Нитрнн а-бутоксипропиоиовой кислоты. Во вращающемся автоклаве
нагревают в течение 6 ч при 250° С 200 а к-бутнлвннилового эфира, 200 g тет-
рагидрофурана и 54 г синильной кислоты. После перегонки реакционной смеси
получают 145 а нитрила а-бутоксипропионовой кислоты.
Еще легче происходит присоединение сложных виниловых эфиров. С выходом
выше 80% образуются питрилы сложных эфиров а-оксипропионовой кислоты. Удается
также осуществить присоединение синильной кислоты к эфирам а,Р-пенасыщенных
кислот [64] (например, к этиловому эфиру кротонской кислоты, днэтиловому эфиру
алкилидепмалоновой кислоты):
/COOCsHfi Kcw+faci^
Ч2ООС2Н5
yCOOCaHs
RCHCH
\cOOCjHs
Наличие в винильной группировке алкильных или арильпых заместителей
снижает способность к присоединению.
Диэтиловый эфир (j-цианбензплмалоповой кислоты [65]. В 250 мл спирта
растворяют 24,5 г дивтилового эфира бензальмалоновой кислоты и к нему
прибавляют раствор 6,5 г K.CN в 100 мл воды. К прозрачной жидкости при
охлаждении очень медленно добавляют по каплям 11 г 30%-ной НС1, разба-
вленной, 50 мл воды. Реакционную смесь оставляют на несколько дией. После
этого к слабокислой, почти не пахнущей синильной кислотой реакционной
массе добавляют воду до появления мути. Через 24 ч выпадает достаточно
большое количество игольчатых кристаллов, которые отфильтровывают и про-
мывают разбавленным спиртом; т. пл. 48,5° С.
В то время как присоединение синильной кислоты к двойной связи а,|}-ненасы- •
щенных кислот и эфиров происходит не всегда, присоединение ее к двойной связи а, р-
ненасыщеняых кетонов удается в большинстве случаев. При Этом образуются нитрилы
у-кетокарбоиовых кислот, карбонильная группа которых может присоединять вторую
молекулу синильной кислоты. Ненасыщенные альдегиды присоединяют синильную
кислоту только по карбонильной группе. Из бепзальацетофенона и синильной кислоты
получается нитрил а-фенил-р-бепзоилпропионовой кислоты [66] с выходом более 90%:
CeH6GH=CHCOCeH5 + HCN —> СеН5СНСНгСОСвНв
I
CN
Возможно такое присоединение синильной кислоты к а,р-ненасыщенпым нитри-
лам, сульфонам и нитросоединениям [67].
[63] 1. G. Farbenindustrie A. G., фрапц. пат. 892870, С., 1948, I, 170; ср. также
Houben-Weyl, Bd. 8, S. 267.
[64j L. Higginbotham, A. Lapworth, J. Chem. Soc., 121, 51 (1922);
см. также Org. Synth., 26, 54 (1947).
[65] J. Bredt, J. Kallen, Ann., 293, 343 (1896).
[66] A. Hann, A. Lapworth, E. Wechsler, J. Chem. Soc., 85, 1358 (1904);
97, 41 (1910); см. также Org. Synth., Coll. v. 2, 1943, p. 498.
[67] P. Kurtz, Ann. 572, 35 (1951).
712 ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
II. Присоединение по С=^0-связи !
1. Альдольная конденсация
Образование альдолей (альдегидоспиртов) или кетоспиртов происходит под дей- J
ствием щелочных агентов. Так, при димеризации двух молекул ацетальдегида полу- ;;
чается ацетальдоль: .. i
СНэСНО -ЬСНзСНО —> СН3СНСН2СНО \
•I "
ОН J
1
Ацетальдоль [68] (по Клайзену). В 200 мл охлажденной льдом воды J
медленно прибавляют 100 г свежеприготовленного ацетальдегида (из параль-
дегида). Смесь охлаждают до —12° С и осторожно приливают ледяной раствор •
2,5 г KCN в 100 мл воды с такой скоростью, чтобы температура ке поднималась
выше —8° С. Реакционную массу охлаждают смесью льда и соли в точбйие 4
еще 2 ч и оставляют на холоду на 30 ч. Затем реакционную массу насыщают Д
NaCl, продукт несколько раз экстрагируют эфиром, сушат и фракциони- •
руют. Выход альдоля 40% от теоретического; т. кип. 80—90° С '
(20 мм рт. ст.); т. кин. чистого альдоля 83° С.
Взаимодействие остальных альдегидов приводит к получению соответствующих .
продуктов присоединения, причем присоединение происходит всегда к СНа-группе,
находящейся в а-положепии к карбонильной группе:
ВСН2СНО+СН2СПО —> RCH2CHCHCHO
I II
R НО R •
Примером реакции альдегида с кетоном может служить получение пентапол- '
2-она-4 из ацетальдегида и ацетона [69].
Пентанол-2-он-4. К 105 г ацетона, охлажденного до —12° С, приливают •#
раствор 5 г KCN в 100 мл воды и при дальнейшем охлаждении прибавляют 40 s
свежеприготовленного ацетальдегида. Затем смесь оставляют стоять на холоду
8 ч, после чего продукт экстрагируют эфиром; вытяжки встряхивают с насыщен-
ным раствором NaCl, сушат над Na2SO4 и после отгонки эфира перегоняют у
в вакууме. Т. кип. продукта 77—78° С (19 мм рт. ст.).
Из 2 моль ацетона в присутствии гидроокиси бария [70] или ионообменных
смол [71] получают днацетоновый спирт:
ОН
2СНзСОСН3—>СНз-С1-СН2СОСН3
I
СНз
При такой альдольной конденсации происходит присоединение свободной пары
электронов «метиленовой компоненты» к атому углерода поляризованной карбониль-
ной группы, в электронной оболочке которого находится только шесть электронов:
Na+ -Уон- сн8снсн2сно
он
CH3—C+ + [‘CH2CH=O]Na+ —> CH3CCH2CH^O
• Более правильный термин — альдольное уплотнение. — Прим. ред.
[68] L. С 1 a i s е n, Ann., 306, 324 (1899); см. также L.-P. Kyriakides,
J. Am. Chem. Soc., 36, 532 (1914).
[69] L. C 1 a i s e n, Ann., 306, 324 (1899).
[70] B. Conant, W. Puttie, Org. Synth., 1, 45 (1921).
[71] С. J. Schmi die, R. C. Mansfield, Ind. Eng. Chem., 44, 1389 (1952).
П. ПРИСОЕДИНЕНИЕ ПО С=О-СВЯЗЯМ
713
Предпосылкой для этой реакции, таким образом, является наличие активиро-
ванной С—Н-связи. В указанных выше реакциях роль'метиленовых компонент кроме
альдегидов и кетонов могут играть также другие соединения.
Пропионовый альдегид может реагировать с нитрометаном с образованием 1-
питробутанола-2 [72]:
CH3CH2CHO + CH3NO2 —* CH3CH2CHCH2NO2
I
он
1-Нитробутанол-2. Через колонку диаметром 18 мм', заполненную
на 40 см амберлитом марки IRA-400 (ОН"-форма) и снабженную рубашкой,
при 27—30° С в течение 3 ч медленно пропускают смесь 58 s пропвояовогс
альдегида, 91 г нитрометана и 50 г этилового спирта. Выход продукта 70%
от теоретического; т. кип. 107—115° С (25 мм рт. cm.); ng 1,4432.
Реакцию нитропарафинов с альдегидами можно осуществить тремя спо-
собами [73]:
1. В присутствии такого количества щелочи, чтобы взаимодействие протекалс
с достаточной скоростью и в то же время без побочных процессов дегидратации или по.
димеризации. Реакция протекает медленно, и в случае более сложных исходных со-
единений выходы продуктов сильно уменьшаются.
2. В присутствии эквимолярных количеств 10 н. NaOH. Хорошие выходы про-
дуктов получаются только при взаимодействии нитрометана с ыеразветвленнымк
альдегидами при температуре ниже 10° С. Для вторичных нитроиарафинов этот метод
непригоден.
3. Прп нагревании раствора бисульфитного соединенна альдегида и растворе
натриевой соли нитронарафина. Первичные нитросоединения реагируют с выходам?
70—80% от теоретического.
2-Нитро-1'фенилпропанол-1 [74] получают постепенным прибавлением
раствора нитроэтана в водной гидрорклси натрия к продукту присоединение
бисульфита к бензальдегиду, перемешивая реакционную массу при иомнатноё
температуре.
При взаимодействии этилового эфира глицина с п-нитробензальдегидом обра.
зуется этиловый эфир 0-(п-нитрофенил)-серина [75J:
CH2C00CjHs
I II +1
NH,
o2n
^ ^H-CHCOOCjHs
J! он nh2
Аналогично реагируют глицин и тиофен-2-альдегид с образованием Р-тпофен
серика [76].
При реакции бензилцианида с фенилглиоксалевой кислотой в пиперидине полу-
чается с выходом 40% от теоретического мононитрил дифенилоксиянтарно4
кислоты [77]:
ОН
(
СвН5СН2 + СвНб-СО-СООН —* СвН6СН-ССвН6
I 'II
CN CN СООН
[72] С. J. S с h m 1 d 1 о, R. С. Mansfield, Ind. Eng. Chem., 44, 1388 (1942>
[73] C. A. Sprane, E. F. D eger ing, J. Am. Chem. Soc., 64» 1063 (1942>
[74] Пат. США 2151517.
[75] G. С а г г а г a, G. Cristian i et al., Gazz, chim. ital., 82, 325 (1952).
[76] G. Weitnauer, Gazz. chim. ital., 81, 162 (1951).
[77] P. С a r d i e r, J. Moreau, C. r„ 214, 621 (1942); C., 1943, Ii, 898.
714
ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Реакция присоединения может происходить также п между ацетиленом и кар-
бонильной группой*:
СНзч СПз-,
>С=О+НС=СН —> >с-с=сн
СНз7 CH3Z I
on
Диметилэтинилкарбинол [78]. К 300 мл жидкого аммиака при —50° С
прибавляют маленькими кусочками 4,6 г натрия. В полученный раствор про-
пускают сухой ацетилен со скоростью 15—20 л/ч до исчезновения голубой
окрасив. Затем к суспензии ацетиленида натрия прибавляют по каплям при
перемешивании в течение 1 ч 14,6 мл (11,6 з) сухого ацетона (содержание
воды не более 0,042%). Реакционную массу перемешивают еще 4 ч при —50° С
и дают испариться аммиаку.' Бесцветный остаток растворяют в 120 мл воды
и подкисляют 90 мл 50%-ной уксусной кислоты. Карбинол из раствора шесть
раз экстрагируют эфиром. Объединенные эфирные вытяжки сушат над Na2SOv
После фракционирования на колонке Вигре получают продукт с т. кип. 100—
102° С, ng 1,4202; выход 86% от теоретического.
Хороших выходов при этой реакции можно добиться только в том случае,
если удается полностью избежать попадания влаги в реакционную массу. При
проведении реакции с амидом патрия в этиловом спирте выход продукта соста-
вляет только 40—60% от теоретического [79].
В присутствии КОН к карбонильной группе может присоединяться хлороформ
с образованием производного трихлорметилкарбипола:
СН3ч
>С=О + СНС1з
СНз/
СНз^ /ОН
СНа/ \СС13
Трихлорметилдйметилкарбннол [80]. К 500 мл сухого диметилацеталя
формальдегида (метилаль) прибавляют прп энергичном перемешивании 102 г
свежеплавленного, растертого в порошок КОН, поддерживая при этом темпе-
ратуру —4° С. Затем при той же температуре прибавляют в течение 2 ч смесь
215 г хлороформа и 116 з ацетона и перемешивают еще 2 ч, после чего реакцион-
ную массу выливают в смесь разбавленной серной кислоты и льда. Верхний
слой отделяют, а продукт экстрагируют из водного слоя метилалем. Раствори-
тель отгоняют, а остаток перегоняют с водяным паром. Продукт реакции полу-
чают в виде игольчатых кристаллов; т. пл. Ul° С.
Альдегиды в присутствии едкого кали реагируют по приведенной выше схеме
лишь в том случае, если карбонильная группа связана с третичным атомом углерода.
Можно осуществить взаимодействие бензальдегида с хлороформом в присутствии водно-
спиртового раствора едкого натра [81]. Взаимодействие хлороформа с нитропроизвод-
ными ароматических альдегидов и алифатическими альдегидами с неразветвлвнной
цепью протекает с хорошим' выходом в безводной смеси тетрагидрофурана и трет-бути-
лового спирта в присутствии mpem-бутилата калия при температуре от 0 до —20° С [82].
* Реакция А. Е. Фаворского [ЖРХО, 37, 643 (1905)]. — Прим. ред.
[78] С. D. Hurd, W. D. McPhee, J. Аш. Chem. Soc., 69, 239 (1947).
[79] D. D. Coffman, Org. Synth,, 20, 40 (1940).
[80] C. W e i z m a n n, E. В e г g m a n и, M. Sulibacher, J. Am. Chem.
Soc., 70, 1189 (1948); E. Bergmann, D. G i n s b a r g, D. Lavie,
J. Am. Chem. Soc., 72. 5012 (1950).
[81] W. Rapson, D. launder, E. Stewart, J. Chem. Soc., 1944, 74.
[82] E. Kaspar, R. Wiechert, Chem. Ber., 91, 2664 (1958).
]J. ПРИСОЕДИНЕНИЕ ПО С---О-СВЯЗЯМ
715
2 s Бензоиновая конденсация *
Так называемая бензоиновая конденсация протекает по схеме:
2ВСНО —>- RCHOHCOR
Наибольший интерес в препаративном отношении эта конденсация имеет в аро-
матическом ряду; впервые опа была проведена с бензальдегидом. Алифатические аль-
дегиды обычно не вступают в эту реакцию. Одним из немногих исключений, когда
бензоиновая конденсация гладко протекает не с чисто ароматическими альдегидами,
является конденсация ароматических глиоксалей типа фенилглиоксаля [83].
Бензоин. Кипятят с обратным холодильником, обычно с добавкой воды,
раствор альдегида с цианидом калия (0,1 от массы альдегида). После охлаждения
реакционной массы выпадает кристаллический бензоин. Образующийся бензонн,
как правило, окрашен в слабо-желтый цвет и очень плохо очищается от при-
месей. Бесцветный бензоин [84] с т. пл. 137® С получается при перекристалли-
зации из диизоамилового эфира. Вместо цианида калия можно использовать
цианиды натрия и бария, но чистую синильную кислоту и комплексные цианиды
применять нельзя. Поэтому считают, что катализатором бензоиновой конден-
сации служит цианид-ион, который в случае синильной кислоты и комплексного
цианида не образуется в достаточном количестве из-за их незначительной дис-
социации.
Подобно бензальдегиду, с образованием соответствующих ацилоинов реаги-
руют также его гомологи и эфиры оксиальдегидов, например анисовый альдегид и фур-
фурол. Но при конденсации свободных оксиальдегидов, а также галогенированных
нитро- и аминобензальдегидов бензоины не образуются. Конденсация коричного
альдегида проходит с ничтожно малым выходом.
3. Пинаноны
Получение пинаконов из кетопов происходит под действием самых разнообраз-
ных восстановителей:
Н1 R\ /R
2R-C-H' —4 /С-С<
II R'Z I I XR'
НО он
О
Бензпинакон [85]. Медленно кипятят в течение 15 мин при энергичном
встряхивании 1 ч. бензофенона, 2 ч. цинка и 10 ч. 80%-ной уксусной кислоты.
Выпавшие после охлаждения кристаллы отфильтровывают, фильтрат еще раз
кипятят с цинком и обрабатывают, как указано выше. Выделенный бензпинакон
промывают разбавленной уксусной кислотой и перекристаллизовывают из кипя-
щей ледяной уксусной кислоты. Продукт плавится при 185—186° С, разлагаясь
на бепзофенон и бензгидрол.
С 95%-ным выходом бепзпинакоп был синтезирован фотохимическим восстано-
влением бензофенона в изопропиловом спирте на солнечном свету [86].
* У Вейганда (Органические реакции, сб. 4, Издатпнлит, 1951 г., стр. 229) бензоино-
вая конденсация названа ацилоиновой (о последней см. там же, стр. 214). —
Прим. ред.
]83J G. So derbaum, Р. W. Abenius, Ber., 25, 3468 (1892).
[84] A. WeiBberger, Н. Mainz, Е. Strasser, Вег., 62, 1952
(1929).
[85] A. Zagumenny, Вег., 14, 1402 (1881); ЖРФХО, 12, 429 (1880);
Vanino, Handbuch der . praparaiiven organischen Chemie, Bd. II,
Stuttgart, 1937, S. 560.
[86] Org. Synth., Coll. v. 2, 1943, p. 71; см. также J. W. Lynn, J. English,
J. Org. Chem., 16, 1547 (1951); M. В. К e g с 1 ш a n n, E. V. Brown,
J. Am. Chem. Soc., 75, 5961, 4649 (1953).
716
ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Большое число ароматических пинаконов [87, 88] было получено с хорошими
выходами при использовании в качестве восстановителя бинарной смеси магния с его
бромидом или иодидом.
Реакция протекает по схеме:
MgMgb RaCOMgl R2COH
2R2C=O w’ g ‘ > ( — > I
R2COMgI H2COH
Вначале возникает соответствующий металлкетил, который находится в равно-
весии с йпнаконатами;
R2C-OMgI
| 2R2C— OMgl
R2C—OMgl
Дифенилди-0-нафтилнинакон. Полностью исключив доступ воздуха
и влаги, встряхивают 2,32 г фенил-0-иафтилкетояа с 1 г магния и 3 г Mgla
л 30 хл смеси эфир — бензол (1 : 1). Раствор окрашивается в красно-коричне-
вый цвет. В присутствии кислорода воздуха раствор тотчас же обесцвечивается
Окраска раствора восстанавливается вновь, если доступ воздуха прекращается-
Реакционную смесь отфильтровывают от избытка магния и разлагают смесью
льда и NH4C1. Органический слой промывают водой, сушат над Na2SO4 и раство-
ритель отгоняют в вакууме. После перекристаллизации продукта из смеси
хлороформа и спирта образуются игольчатые кристаллы. Выход дифеиилди-0-
иафтилпинакона 91% от теоретического; т. пл. 175° С. Безводную бинарную
смесь восстановителя (Mg — Mgl2) получают из 4 г порошка магния и 12 е
иода в 75 мл абсолютного эфира и 75 мл сухого бензола [87].
Описано восстановление ненасыщенных альдегидов до пинаконов действием
инк-медной пары [89]. Реакция протекает уже при 0° С в разбавленном растворе
ксусной кислоты. Дипропонилгликоль [90] с помощью ципК-медной пары [91] полу-
ен из кротонового альдегида с 67%-ным выходом.
Восстановлением смесей кетонов можно синтезировать несимметричный
инаконы [92].
При восстановлении метидэтнлкетона амальгамой магния (из магния и хлорида
гути) получили 3,4-диметилгександиол-3,4 [93]. При восстановлении натрием или
•о амальгамой [94] щелочь, выделяющаяся в результате гидролиза пинаконата щелоч-
тго металла, расщепляет пинакон с образованием кетона и соответствующего спирта:
. R2C—ONa „ n К2С-ОН
2R.C-ONa —> | |
R2C-ONa R2C—OH
2NaOH
R2C=O + R2CHOII
Поэтому гидролиз следует вести в уксусной кислоте.
Об электролитическом восстановлении см. [95. 96], о восстановлении амальга-
iii алюминия см. [97]. z
7J М. Go mberg, W. F.. Bachmann, J. Am. Chem. Soc., 49, 236
(1927).
8] W. E. Bachmann, R. V. S ha nk 1 a n d, J. Am. Chem. Soc., 51. 306
(1929).
9] R. Kuhn, O. Rebel, Бег., 60, 1565 (1927).
0] W. G. Young, L. Levanas, Z. J esait is, J. Am. Chem. Soc., 58,
2274 (1936).
1] J. Gladstone, A. Tribe, J. Chem. Soc., 31, 561 (1877).
2] G. Laud e, J. Wiemann, Bull. Soc. chim. France [5], 13, 256 (1946).
3] D. W. D a v i s, C. S. Marvel. J. Am. -Chem. Soc., 53, 3843 (1931).
4] W. E. Bachmfittn, J. Am. Chem. Soc., 55, 1183, 2829 (1933).
5] M. 1. A 11 e п, I. Am. Chem. Sac., 72, 3797 (1950); 73, 3503 (1951).
6] I. A. Pearl, J. Am. Chem. Soc., 74. 4260 (1952).
7] K. S i s i d о, H. Nozaki, J. Am. Chem. Soc., 70, 776 (1948).
II. ПРИСОЕДИНЕНИЕ ПО С=О-СВЯЗЯМ
717
4. Реакция Кольбе — Шмитта
По Кольбе [98], салициловую кислоту получают пропусканием сухого COt
над сухим фенолятом натрия, постепенно повышая температуру до 110—200° С:
^\/0Na ^\/0Na
21 II +СО4 | || +CeH5OH
'^''"•COONa
•При реакции образуется фенол, который не реагирует с двуокисью углерода.
По Шмитту, для полного превращения фенолята (промышленный метод) взаимодей-
ствие двуокиси углерода и фенолята ведут под давлением и затем нагревают смесь до
120—150° С.
Если реакцию проводят с фенолятом калия, то получают n-оксибензойпую кис-
лоту. В интервале температур 100—150° С образуется также дикалийсалицилат, ко-
торый, однако, изомеризуется (при 200° С полностью) в калиевую соль п-оксибензой-
ной кислоты. В случае многоатомных фенолов п аминофенолов взаимодействие с дву-
окисью углерода протекает уже в водном растворе. Эту реакцию можно провести также
с а-нафтолом.
5. Циангидриновые синтезы
Цпангидриновые синтезы состоят в присоединении синильной кислоты к кар-
бонильным соединениям по схеме:
/ОН
R2CO4-HCN в2с<
XCN
При этом реакция синильной кислоты с альдегидами проходит, в общем случае,
быстрее и полнее, чем с кетонами. Смешанные жирно-ароматические кетоны дают лишь
незначительные выходы циангидринов. Невозможно получение циангидринов из арома-
тических, а также из алициклических кетонов, содержащих и кольце более десяти
атомов углерода.
Реакция образования циангидринов является равновесной, причем равновесие
при отсутствии катализатора устанавливается очень медленно. Каталитическим дей-
ствием обладают незначительные количества щелочных реагентов, например, цианид
калия [100], карбонат калия [101], аммиак [102] и др. Циангидрины являются тер-
мически нестойкими веществами и при повышенной температуре разлагаются на исход-
ные компоненты. При циангидриновых синтезах проводят взаимодействие карбониль-
ного соединения с концентрированным'раствором сппильной кислоты, а в некоторых
случаях к с безводной синильной кислотой или же смесью цианидов щелочных металлов
и кислот.
Реакцию с синильной кислотой рекомендуется проводить следующим образом*.
К эквимолекулярной смеси сниильной кислоты и ацетальдегида (в случаи
кетона необходим 10%-иый избыток синильной кислоты) осторожно прибавляют
несколько капель концентрированного раствора KCN. Смесь вскоре закипает,
При этом следует применять эффективный обратный холодильник, а в некоторых
случаях реакционную массу необходимо охлаждать. По окончании реакции
к реакционной массе при хорошем охлаждении прибавляют H2SO4 до ярко
выраженной кислой реакции, отгоняют непрореагировавшие синильную кислоту
и альдегид (или кетон) и остаток фракционируют в вакууме.
* Осторожно! Синильная кислота и многие циангидрины сильно ядовиты! —
Прим. ред.
[98] Н. К о 1 b е, R. S с h m i t t, J. prakt. Chem. [2], 10, 89 (1874); 31, 397 (1885);
cmi также A. S. Lindsey, H. Jeskey, Chem. Rev., 57, 583 (1957).
[99] cp. Houben-W-eyl, Bd. 8, S. 274.
[100] A. Lapworth, J. Chem. Soc., 85, 1206 (1904).
[101 ] M. v. Romburgh, M, U I t e e, Rec. trav. cnim., 28, 2 (1909). 1
[102] IT. Kiliani, Ber., 21. 916 (1888).
718
ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕИ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
При получении больших количеств циангидрина целесообразно при-
бавлять по каплям синильную кислоту к смеси карбонильного соединения
и катализатора. Если реакцию проводят с низкокипящими альдегидами, то
смесь альдегида и синильной кислоты прибавляют по каплям к смеси предвари-
тельно полученного циангидрина и катализатора.
Нитрил миндальной кислоты [103]. Немного более 1 моль KCN увлаж-
няют небольшим количеством воды, прибавляют 1 моль бензальдегида и при-
бавляют по каплям при частом встряхивании и охлаждении реакционного
сосуда 1 моль НС1 (дымящая соляная кислота). Полученное коричневое масло
декантируют от солей.
Иногда для этой реакции [104] используют бисульфитные соединения кетонов
или альдегидов и водный раствор цианида калия, причем обычно выделение бисуль-
фитного соединения не обязательно.
Ацетонциангидрин. Встряхивают 1 моль ацетона с концентрированным
водным раствором 1 моль бисульфита натрия. Смесь охлаждают и прибавляют
по каплям при энергичном встряхивании концентрированный водный раствор
1 лоль цианида калия, при этом раствор снова разогревается. Его охлаждают,
отделяют маслообразный ацетонциангидрин от водного слоя; дополнительное
количество ацетонциангидрина несколько раз экстрагируют из водного слоя
эфиром. Эфирные вытяжки объединяют с основным количеством продукта,
встряхивают для удаления ацетона с раствором бисульфита, потом с насыщен-
ным раствором NaCl и отгоняют эфир. Совершенно не содержащий щелочи
ацетопциангидрин имеет т. кип. 82° С (23 мм рт. ст.) и т. пл. — 19° С.
При перегонке циангидринов для устранения щелочи их стабилизируют
прибавлением следов кислот [105]. Можно применять добавки 1—2% органических
кислот (тио- или хлоруксусной кислот), которые перегоняются вместе с циангидри-
нами; используются также серная и фосфорная кислоты и иод.
Следующим методом получения циангидринов является взаимодействие циан-
гидринов (в основном кетонциангидринов) с карбонильными соединениями (в основ-
ном альдегидами) в присутствии незначительных количеств щелочных катализаторов:
R х /CN
>С< +R"-СНО Н"—СИ-CN + RCOR'
Н'х ^ОН I
ОН
Равновесие устанавливается сразу, причем преобладают более устойчивые
циангидрины.
Особое значение циангидриновый сиптев имеет в химии углеводов. Этим .
способом по методу Фишера через лактоны альдоновых кислот при их дальнейшем
восстановлении можно переходить от пентоз к гексозам и далее к.высшим сахарам.
Правда, возникающие в качестве промежуточных продуктов нитрилы, которые обычно
лучше всего получать отщеплением воды от соответствующих оксимов, большей
частью при этом выделить нельзя. Напротив, соответствующие амиды кислот выде-
ляются, как правило, в чистом виде.
Амид гептоновой кислоты [102] (по Килиани). Смешивают 30 г тонко
размельченной галактозы с 5 мл воды и рассчитанным количеством примерно
50%-ного раствора HCN, прибавляют каплю аммиака, плотно закрывают реак-
ционный сосуд и хорошо встряхивают. При стоянии углевод окрашивается
в желтый цвет и постепенно переходит в раствор. Через 6—8 ч начинается
выпадение бесцветных игольчатых кристалликов, при этом смесь иногда довольно
сильно разогревается. Еще через 12 ч добавляют равный объем воды, встряхи-
вают, отфильтровывают или отжимают массу на пористой пластинке. Выход
амида гептоновой кислоты 40—50% от теоретического.
103] A. Spiegel, Вег., 14, 235 (1881).
104] Н. Bncherer, A. G г о 1 е е, Вег., 39, 1225 (1906).
105] М. U 1 t е е, Rec. trav. chim., 28, 5 (1909).
И. ПРИСОЕДИНЕНИЕ ПО С-О-СВЯЗНМ
719
Модификацией циангидринового синтеза является синтез аминокислот по
Штреккеру, который основан на реакции синильной кислоты и аммиака с альдегидами
или кетонами с образованием соответствующих а-аминонитрилов *:
R—СНО —NH3 + HCN —► R-CH-CN-[-H2O
I
nh2
При омылении нитрильной группы минеральными кислотами получают «-амино-
кислоты.
Другой вариант этого метода предложен Бухерером [106], который в качестве
исходных соединений использовал бисульфитные производные альдегидов; реакцию
этих производных с синильной кислотой и карбонатом аммония проводили в горячем
спиртовом растворе:
R-CH-CN + (NH4)2CO3 —> R-CH-CO
i I I
NH2 HN NH
\ /
CO
Возникающие в результате этой реакции хорошо кристаллизующиеся гидан-
тоины горячими минеральными кислотами можно также гидролизовать до аминокислот.
6. Присоединение диазометана к карбонильной группе
Первой ступенью реакции диазометана с альдегидом или кетоном является,
по данным Арндта и Айстерта, присоединение диазометана к поляризованной карбо-
нильной группе:
К '>C+ + CHJ-N=N —К ^С-СНа—N=N
В'/ | В'/ |
о- о-
где R = H, алкил, арил.
Возникающий при этом диазонийбетаин в некоторых случаях довольно устой-
чив. О происходящей с выделением азота перегруппировке, приводящей к гомологич-
ным кетопам или альдегидам, будет подробно сообщено па стр. 884. Ниже будет рас-
смотрено только получение а-окисей, протекающее также с выделением азота:
R ч + R ч + R х
>С - CH8-N=N — *• >С-СНз —> >с—сн2
R'/ | Д'/ | Д'/ \ /
О- О- о
Образование эпоксисоединений происходит почти исключительно в тех случаях,
когда R' является электроноакцепторной группой. Так, например, при реакции хло-
раля с диазометаном получают 1,1,1-трихлорпропиленоксид-2,3 [108], а из о-нитро-
бензальдегида — о-нитрофенилэтиленоксид [109].
* Штренкер обрабатывал циангидрины аммиаком; указанный же Вейгандом метод
разработан Н. Д. Зелинским и Г. Л. Стадниковым [ЖРХО, 38, 722 (1906)]. —
Прим. ред.
[106] Н. Bucherer, J. prakt. Chem., 140, 291 (1934); 141, 5 (1934).
[107] Обзорная статья В. Е i s t е г t, в книге W. Foerst, Neuere Methoden der
praparativen organischen Chemie, Bd. I, 3 Auff., Ver]ag Chemie, Weinheim,
1949.
[108] F. Arndt, В. E i s t о г t, Ber., 61, 1118 (1928).
1109] F. Arndt, В. E i s t e r t, W. P a r t a 1 e, Ber., 61, 1107 (1928); 60, 446
(1927).
720
ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
о-Нитрофенилэтиленоксид. При —10° С Добавляют 10 j о-питробензаль-
дегида к эфирному раствору диазометана, содержание которого в 1,5 раза
превышает эквимолекулярное. После того как альдегид растворится и прекра-
тится интенсивное выделение азота, смеси дают нагреться до комнатной тем-
пературы и оставляют на 2 ч. После отгонки эфира (под конец в вакууме) остает-
ся масло, которое затвердевает на холоду почти полностью. Расплавленный;
продукт смешивают с двойным объемом теплого метилового спирта и постепенно,
охлаждают до —10° С. При этом большая часть соединения выкристаллиаовы-,
вается в довольно чистом виде. Маточный раствор упаривают и остаток пере-
гоняют с водяным паром. Перегонка идет медленно и продукт чаще всего кристал-
лизуется уже в холодильнике. Общий выход о-нитрофенилэтиленоксида 6—8 г..
Для полной очистки его несколько раз перекристаллизовывают из метанола^
до тех пор, пока раствор ег» в спирте не будет больше давать окраски со ще-.;
лочыо. Получают почти бесцветный и не обладающий запахом кристаллический,
продукт с т. пл. 65° Сит. кип. 144° С (10 мм рт, ст.).
Если R’ является электронодонорной группой, то образования а-окиси не про-й
•исходит, а идет перегруппировка, рассматриваемая на стр. 884, 885. i
7. Присоединение металлоорганических соединений
к карбонильной группе '
Реакция карбонильной группы с соединением- Грипьяра представляет собой'-
«восстановление с увеличением углеродного скелета», причем углеводород RH, лежа-^
щий в основе гриньяровского соединения RMgX, присоединяется к карбонильной^
группе:
Rs + _ 7-2+ R\ н8о ';|
>С—О-l-R~MgBr—> >0— OMgBr —*—> >С—OH + MgOHBr S
Rz Rz I Rz I
R R *
В качестве растворителя чаще всего используют диэтиловый эфир и в некоторых^
•особых случаях — дибутиловый или диизоамиловый эфир. >
Работа с небольшими количествами магнийорганических соединений,',
в эфире не представляет никаких трудностей, одиако необходимо отметить,-.’
что при проведении реакции с большими загрузками могут возникнуть значи- '
тельные осложнения: добавлять воду к реактиву Гриньяра, не вступившему.'
полностью в реакцию, рискованно не в меньшей мере, чем лить воду в концевт-"
рированную серную кислоту.
Эфирные растворы магнийорганических соединений чувствительны к влаге^
двуокиси углерода и кислороду. Для проведения реакции при полном отсутствии кис- Л
лорода воздуха разработана специальная аппаратура [110]. Концентрацию растворов."
Веактивов Гриньяра целесообразно определять титрованием кислотами [111], стр. 646.„а
случают растворы магнийорганических соединений непосредственно перед-
употреблением.
В результате реакции не всегда сразу образуется то конечное соединение, •
которое постулируется в суммарном уравнении реакции. Так, например, прн
взаимодействии описи этилена с алифатичесними магнийорганическими соеди-
нениями сначала, очевидно, образуется оксониевое соединение [112].
В каждом конкретном случае реакция с реактивом Гриньяра проводится нрИ"
определенной оптимальной температуре, чаще всего при охлаждении, поэтому целесо-
образно в каждом случае предусмотреть возможность измерения температуры.
[110] Н. Gilman, Р. Hewlett, Rec. trav. chim., 48, 1124 (1929). •
[111] H. G i I m a n, P. D. W i Iki n so n, W. P. F i s h e ), С. H. M e у ers,
J. Am. Chem. Sdc., 45, 150 (1923).
J112] E. E. D г e g e r, Org. Synth., 6, 56 (1926), Note 5.
If. ПРИСОЕДИНЕНИЕ ПО С«О-СВЯЗЯМ
721
Соединения Гриньяра обычно полностью растворяются в эфире, однако при
последующих реакциях легко образуются твердые осадки. Это следует учитывать в тех
случаях, когда второй реагент является газообразным веществом (например, двуокись
углерода, окись этилена, формальдегид и др.). Реакцию с такими соединениями про-
водят, пропуская их непосредственно над поверхностью раствора магиийорганнческого
соединения, так как иначе возможна закупорка барботера.
Во многих случаях реакцию необходимо закончить нагреванием. Конец реакции
определяют по отсутствию гриньяровского соединения в растворе.
Цветная реакция на соединения Гриньяра [ИЗ]. К 0,5 мл 1%-ного рас-
твора кетона Михлера в сухом бензоле прибавляют равное количество анализи-
руемого раствора. Потом смесь осторожно гидролизуют 1 мл воды и, наконец,
прибавляют несколько капель 0,2%-ного раствора иода в ледяной уксусной
кислоте. В присутствии соединения Гриньяра возникает голубовато-зеленое
окрашивание. Если окрашивание раствора не наблюдается, то это говорит
об отсутствии металлоорганического соединения. t
После получения соединения Гриньяра и его присоединения к реакционно-
способному веществу проводят последнюю операцию — разложение первичного про-
дукта реакции. Так как этот процесс также всегда эк.зотермичен, то неосторожное
его проведение может не только поставить под вопрос успех операции, но и сделать ее -
опасной. В качестве гидролизирующих средств применяют в первую очередь соляную
и серную кислоты, раствор хлористого аммония. При разложении только водой обра-
зуется гидроокись магния, которая усложняет дальнейшую обработку.
Если реакционная масса легко удаляется из колбы, то ее рекомендуется выли-
вать на лед, добавляя затем соответствующую кислоту подходящей концентрации
и, при необходимости, лед. В том случае, иогда под влиянием кислот возможна поли-
меризация или другие побочные реакции, используют раствор хлористого аммония.
Но если реакционную смесь из-за ее консистенции нельзя удалить из реакцион-
ной колбы, то гидролизующие добавки вносят непосредственно в колбу. При этом
необходимо постоянно заботиться о быстром разбавлении и охлаждении, добавляя до-
статочное количество льда и воды, чтобы вследствие местных перегревов не испарялся
эфир, не было выброса реакционной массы и связанных с этим пожаров. Дальнейшие
операции по окончательному выделению конечных продуктов реакции слишком разно-
образны и общие рекомендации здесь излишни.
Взаимодействие реактива Гриньяра с кетонами
При взаимодействии кетонов с соединениями Гриньяра обычно образуются
третичные спирты:
СбНб\ СвНзч
С6Нб-С-CH2F-CeH5MgBr —> >C-CH2F —> >C-CH2F
II CeHZ I CeH5z I
О OMgBr OH
Таким образом из фенилмагнийбромида и ш-фторацетофенона был получен ди-
фенилфторметилкарбинол [114].
Дифенилфторметилкарбинол. К раствору фенилмагнийбромида, получен-
ному из 20 г бромбензола и 2,5 г Магния в 50 мл эфира, прибавляют по каплям
при о° С раствор 14 г фторацетофенона в 25 мл эфира в течение 15 мин. Затем
медленно прибавляют охлажденный раствор NH4C1 в воде. Эфирный слой отде-
ляют и продукт трижды экстрагируют из водного слоя эфиррм. Объединенные
эфирные растворы сушат над Na2SO4 и после отгонки эфира фракционируют
в вакууме; т. кип. продукта 70—75° С (0,2 мм рт. ст.). Дистиллят быстро
кристаллизуется. Из лигроина выпадают бесцветные призмы с т. пл. 71—72° С.
Выход дифенилфторметилкарбинола 65% от теоретического.
[ИЗ] Н. Gilman, F. S с h u 1 z е, L. Heck, J. Am. Chem. Soc., 47, 2002
(1925); 52, 4949 (1930).
[114] F. Bergmann, A. Kalmus, J. Am. Chem. Soc., 76, 41'37 (1954).
46 Заказ 1835.
ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
При взаимодействии стерически затрудненных кетонов с реактивом Гриньяра,
имеющим атом водорода в ^-положении к связи углерод — металл, происходит вос-
становление карбонильной группы до вторичного спирта [114]. Из реактива Гриньяра
при этом образуется олефин:
(CH3)2CHv (СНз)2СНх ,Н
>C-O4-(CH3).2CH-MgBr —> >С< Ч-СН3СН=С1Г
(СНз^СН/ (СН3)2СН/ xOMgBr
Если с n-пропилмагнийбромидом реагирует диизонропилкетон, то наряду
с его восстановлением до вторичного спирта (63%) происходит также нормальное при-
соединение реактива Гриньяра к карбонильной группе с образованием (30%) третичного
спирта [115]. Выход третичного спирта можно существенно повысить (до 65%) добав-
кой бромида магния к исходной реакционной смеси [115}.
Такое повышение выхода объясняют тем, что с участием растворителя образуется
циклический комплекс, состоящий из одной молекулы кетона и двух молекул реактива
Гриньяра [116]:
СНз
I
CH2 Br CH2CH3
4.--' \ / I сольв.
R2C Mg<—сольв. B2c 1
I -I - Mg—Br
"О СНг-СНз о I
/ I CH2CHS
MgBr
T T
сольв. сольв.
Благодаря образованию комплекса гетерополярпость реактива Гриньяра и поля-
ризация карбонильной группы усиливаются в такой мере, что становится возможным
нормальное протекание реакции. У стерически затрудненных кетонов образование
комплекса, необходимого для протекания реакции, невозможно из пространственных
соображений и происходит, как описано выше, восстановление карбонильной группы.
Однако, в присутствии бромида магния соответствующий циклический комплекс обра-
зуется из одной молекулы кетона, бромида магния и реактива Гриньяра, так что реак-
ция Гриньяра, по крайней мере отчасти, может идти по нормальному пути.
Присоединение реактива Гриньяра к кетенам протекает по карбонильной
группе. При реакции дцмезитилкетена и мезитилмагнийбромида получается три-
мезитилвиниловый спирт [117]:
(СНз)зСвН? (СНз)зСйН2х /СвНг(СПз)з
>С=С-О-]-(СПз)зС6На-MgBr —>С=С<
(СНз)зСвН2/ (СНэ^С^На^ ^ОН
Двойная углеродная связь в этой реакции не участвует.
По данным Колера [118], при реакции <х,|3-непасБпцеппых кетонов с соедине-
ниями Гриньяра Происходит как 1.2-присоединенне к карбонильной группе, так
и 1,4-присоединепие к сопряженной системе двойных связей. Так, из бензальацетофе-
нона и фенилмагнийбромпда получают ш;ш-дифепи.тпропиофенон:
CeH5CH-CH-C-CeH5 c,TUMgB.^. (С6Н5)2СН-СП=С.-СвНо —
II I
О OMgBr
—> (СвП5)2СП~СН=С-СвН5 (С6Нб)2СН-СН2-С-С6Н3
I II
он о
[115] А. Н. В 1 a t t, J. F. Stone, I. Am. Chem. Soc., 54, 1495 (1932);
C. G. Swaiu, H. В. В о у 1 es, J. Am. Chem. Soc., 73, 870 (1951).
[116] V. Franzen, H. Krauch, Chem. Zig., 79, 137 (1955).
[117] R. C. Fusion, R. H. Chadwick, M. E. War d, J. Am. Chem. Soc.,
68, 389 (1946).
[118] E. P. Kohler, Am. Chem. J., 31, 642 (1904); 29, 352 (1903).
TJ. ПРИСОЕДИНЕНИЕ ПО С=О-СВЯЗЯМ
723
о),со-Дифенилпропиофенон. К реактиву Гриньяра, приготовленному
обычным образом из 12 г магния в 750 мл эфира и 80 г бромбензола, постепенно
прибавляют 54 г бензальацетофенона. Полученную смесь вносят в разбавлен-
ную охлажденную серную кислоту. При этом выделяется дифенплпропиофенон|
который кристаллизуется пз спирта в виде бесцветных игольчатых кристаллов.
Выход продукта 90% от теоретического; т. пл. 96° С.
Дпфенилпропиофенон реагирует с фенилмагнийбромпдом по нормальной схеме
с образованием а,а.у,у-тетрафенилпропилового спирта [119| (т. пл. 95—96° С):
(CeH6)3CH-CH2-C—CeH5+CeH5MgBr —* (СвН3)2СН—СНз—С(СвН,-,)2
Некоторые алифатические кетоны не присоединяют мстилмагнийгалогенпдов,
а реагируют как соединения с активным водородом [120]:
(c2Hr,);lc-c-CT-T3+cn3Mgi —> (C2H5)3C-c-cii2-;-ch4
I I
О OMgl
Взаимодействие реактива Гриньяра со сложными эфирами
При взаимодействии магнийорганических соединений с эфирами карбоновых
кислот также образуются третичные спирты:
О
R'Mgx R \ /OMgX
R'/ '"'OR
R'MgX R \ /OMgX к x zon
-ROMEx-> — B-/C\n.
Возможности применения этой реакции нс столь широки, как в случае взаимо-
действия с кетонами, поскольку образующиеся соединения должны содержать по край-
ней мере два одинаковых радикала, вводимых с реактивом Гриньяра. Одпако в ряде
случаев предпочитают этот синтез, так как сложные эфиры более доступны и дешевы.
Побочные реакции здесь почти не протекают, а выходы, как правило, удовле-
творительные.
Трифенилкарбипол [121]. К фенилмагнийбромпду в сухом эфире приба-
вляют в течение 1 ч при охлаждении водой этиловый эфир бензойной кислоты
в сухом бензоле с такой скоростью, чтобы смесь умеренно кипела. Затем смесь
нагревают еще 1 ч с обратным холодильником на паровой бане. После охла-
ждения раствора продукт гидролизуют смесью льда и серной кислоты. Выход
трифенилкарбшюла 90% от теоретического; т. пл. 161 — 162' С.
Задержать реакцию на первой стадии удается только у эфиров муравьиной
кислоты; гидролизом соответствующих продуктов присоединения можно получить
альдегиды:
xOMgX н чо ,он
HCOOC^Hs-l-RMgX —> ВСН< ...........с--> всн< н nH> RCHO
Х0С2Н3 \ОС2Н5 СгИ‘ои
Ортоэфиры очень гладко реагируют с реактивом Гриньяра с образова-
нием кетонов:
/ОСгНз .ОСгПэ Y
R-C^OC^R'MgX _c,H,OMgX -> R-C(-R' н-C-R'
чосгн5 хос2н.-,
[119] К. Р. Kohler, Am. Chem. J., 31, 651 (1904).
[120] F. C. Whit m о re, С. E. Lewi s, J. Am. Chem. Soc., 64, 2964 (1942).
[121 ] W. E. В a c h in a n и, II. P. Hetzne r, Org. Synth., 23, 98 (1943).
46*
724 -
ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
iij этилового эфира ортомуравьиной кислоты получают соответствующий аль-
дегид •, панример тиофенальдегид:
СН(ОС2И6)з-Н1 II
В Mgl
Тиофенальдегид
S СН(ОСаН6)3 s Ью
[122]. К реактиву Гриньяра, полученному из 6,6 г
магния в 65 мл абсолютного эфира и 53 г 2-иодтиофена, прибавляют при охла-
ждении 38 г этилового эфира ортомуравьиной кислоты и смесь нагревают в тече-
ние 6 ч с обратным холодильником. После охлаждении смесь переносят в дели-
тельную воронку, содержащую раствор хлористого аммония, хорошо встряхи-
вают; эфирный слой еще раз промывают раствором хлористого аммония, затем
водой и сушат над СаС1г. При перегонке в вакууме вначале отгоняется непро-
реагировавший ортоэфир, затем при остаточном давлении 15 мм рт. ст,
и 97—102° С отгоняется диацеталь тиофенальдсгида в виде бесцветного масла.
Выход продукта 24 г (51% от теоретического).
Из ацеталя кипячением его с разбавленной НС1 получают тпофеналь-
дегид. Его отгоняют с водяным паром, экстрагируют эфиром, промывают водой,
сушат и перегоняют. Тиофенальдегид кипит при 192“ С.
Особым случаем являются эфиры угольной кислоты, взаимодействием которых
с реактивом Гриньяра можно получить третичные спирты с тремя идентичными
остатками:
О=с/ Ч-ЗСгНбМвВг ^^с/щомвВг, (СзНб)аС—ОН
\ОС2П6 -MgOHJBr
Триэтилкарбинол удобнее получать из эфира пропионовой кислоты и этилмагний-
бромпда, однако чистый, свободный от прпм'есей гомологов эфир пропионовой кислоты
отнюдь не дешев. Поэтому триэтилкарбинол обычно получают из диэтилового эфира
угольной кислоты и этилмагнийбромида [123].
Триэтилкарбинол. В трехгорлую колбу емкостью 3 л, снабженную
капельной воронкой, мешалкой и обратным холодильником с хлоркальциевой
трубкой, вносят 107 г магниевой стружки и 800 мл сухого эфира, затем, ие
перемешивая, прибавляют 5 мл этилбромида и после начала реакции прилИ-.
вают смесь 480 г этилбромида и 1 л эфира с такой скоростью, чтобы смесь уме-’
ренно кипела. Для этого необходимо около 2 ч. После окончания прибавления'
этилбромида реакционную смесь перемешивают еще 15 мин и затем при энер<-.
гичном перемешивании в течение 3 ч приливают смесь 156 г диэтилового эфира
угольной кислоты и 200 мл эфира, по-прежнему поддерживая реакционную
массу в состоянии кипения. Для завершения реакции реакционную массу
нагревают при перемешивании на водяной бане еще 1 ч и выливают, непрерывно
встряхивая, в колбу емкостью 5 л,-содержащую смесь раствора 300 з NH(CI.
в 600 мл воды и 1500 з.льда. После отделения эфирного слоя продукт из водного.
слоя экстрагируют двумя порциями эфира, по 500 мл каждая (можно при-
менять эфир, отогнанный от основного иоличества). Затем отгоняют раство-
ритель, сырой продукт сушат с 10 г безводного К2СО3 и перегоняют, собирая
отдельно фракцию, кипящую при 139—142° С. Из первой фракции после обра-
ботки безводным КаСО3 и повторной перегонки получают сначала 25 г, а при
повторной обработке еще 20 г фракции, кипящей при 139—142° С. Общий
выход триэтилкарбинола составляет 125—135 г (80—82% от теоретического).
* Этот метод предложен А. Е. Чпчибабпным [Вег., 37, 480 (1904)]. —Прим. ред.
[122] Е. Grischkewit sch-Trochimowski, I. Mazurewitsch,
С., 1911, I, 1852; 1913; II, 1561.
[123] W. W. Moye r, C. S. Marvel, Org. Synth., Coll. v. 2, p. 602.
II. ПРИСОЕДИНЕНИЕ ПО С=О-СВЯЗЯМ
725
Таким же образом с хорошим выходом получают высшие гомологи —
три-н-гептилкарбинолов.
Реакция лактонов с реактивом Гриньяра протекает следующим образом:
Подробнее об этой реакции см. обзор [124].
Взаимодействие реактива Гриньяра с альдегидами
При реакции альдегидов с магнийорганическими соединениями образуются,
согласно общей схеме присоединения, соответствующие вторичные спирты:
RCHO +R'MgX
Rx /Н
>С<
R'/ XJMgX
R\ ,Н
R'/^OH
Так, например, по Коберну [125], пентен-2-ол-4 [126] синтезируют, добавляя
при хорошем перемешивании и охлаждении эфирный раствор Кротонового альде-
гида к раствору метилмагнийхлорида, полученного в сухом эфире из магния
и метилхлорида:
CH3MgCl 4-СНз—СН-=СН—СНО —* СНз—СН=СН—СП—СНэ
ОН
При реакции формальдегида с реактивом Гриньяра образуются первичные
спирты:
CHaO-]-RMgX —> RClhOH
Таким способом был получен гептин-2-ол-1 [126] после предварительного взаимо-
действия гекснна-1 с этилмагнийбромидом:
CH3(CH2)3C^CH-J-C3H5MgBr —► СН3(СН2)зС=СМ?Вг-|-С2Нв
Из получающегося при этом гексинилмагнийбромида при реакции с формальде-
гидом образуется гептин-2-ол-1:
CH3(CH.2)3C^CMgBr-CH2O —>- СНз(СН2)зС^ССН2ОН
[124] II. К a g 1, К. Miescher, Helv. chim. acta, 32, 2489 (1949).
|125] Е. R. Coburn, Org. Synth., 27, 65 (1947).
1126] M. 8. Newman, J. H. W о t i z, }. Am. Chem. Soc., 71, 1294 (1949),
образование новых с—с-связей в результате присоединения
Гептин-2-ол-1. К 1,1 модъ этилмаггтпйбром(гда в 300 мл эфира медленно
прибавляют 82 г гиксипа-1 (1 моль) в 300 мл эфира. Смесь нагревают 6 ч с обрат-
ным холодильником и после этого охлаждают в атмосфере азота. Затем про-
пускают газообразный формальдегид в токе азота с такой скоростью, чтобы
реакционная смесь умеренно кипела. После пропускания эквивалентного коли-
чества формальдегида экзотермическая реакция заканчивается. Реакционную
массу обрабатывают обычным образом. Выход гептин-2-ола-1 92 г (82% от тео-
ретического), т. кип. 93,Gf С (22 мм рт. ст.).
Формальдегид для этой реакции получают из параформа. Пропускают
сухой азот через колбу, нагретую на бане до 180° С и содержащую параформ
(выдержанный не менее двух недель над пятиокисью фосфора). Этот метод
более удобен, чем старый способ [127[, основанный на взаимодействии три-
оксиметилипа с реактивом Гриньяра: реакция продолжается очень долго, чаще
всего при нагревании в течение нескольких дней, и выход неудовлетворитель-
ный. В отличие от триоксиметилена паральдегид для реакции Гриньяра при-
менять нельзя. Его предварительно, незадолго до употребления, необходимо
деполимеризовать, перегоняя с 5% серпой кислоты.
Магнийорганические соединения, как правило, разлагаются соедине-
ниями, содержащими активный водород, например гексином-1. Так как при
этом каждый реакционноспособный атом водорода дает 1 моль углеводорода,
можно, используя метилмагнийподид и измеряя количество выделивше-
гося метана, количественно определять число активных атомов водорода
(Церевитипов):
ROH-|-CH3MgI —► ROMgl + CIh
Взаимодействие магнийорганических соединений
с окисью этилена и СО
Реакции магнийорганических соединений с окисью этилена протекают по схеме:
Н2С--CHs + RMgX —>- RCH2CH2OMgX —* RC.H2C.H2OH
\ /
О
Так получают первичные спирты с удлинением цепи на два атома углерода.
Таким образом можно, на тример, получить с выходом 60% от теоретического «-гекси-
ловый спирт из а-бутилмагнийбромида п окиси этилена [128].
При взаимодействии окиси изоамилена с этилмагнийбромидом образуется
пропилизопропилкарбинол [129]:
СЛ3х СНз\
УНСН— Clh + CsHsMgBr —> >СНСНСН2(’2Н5
СНз7 \ / CIV I
о он
Для синтеза первичных спиртов можно применять также окись триметилена,
причем происходит удлинение цепи на гри углеродных атома:
СН2
/ \
Н2с C.Tb + RMgX —*- rch8cii2ch2oh
\ /
о
L. Tissier, С. г , 134, 107 (1902).
@rg. Synth., 6. Г>4 (1926).
в с к п и. В. Н. К о и с в и ч е в, ЖОХ, 18. 1833 (1948);
1127] V. Grignard,
[128] Е. Е. Dreger,
[129] М. С. М а л и и о
С. Л., 43. 3776d.
1Г ПРИСОЕДИНЕНИЕ по (д<н;ннзлм
727
Общая методика работы с окисью триметплена [130]. К холодному эфир-
ному раствору реактива Гриньяра прибавляют прп перемешивании раствор
окиси триметилепа в трехкратном количестве сухого эфира. После окончании
реакции, протекающей вначале с небольшим выделением тепла и сопровожда-
ющейся выделением белого осадка, реакционную смесь нагревают еще 1 ч
с обратным холодильником, затем добавляют бензол, эфир отгоняют с колонкой
Впгре и смесь нагревают еще 4 ч. После охлаждения реакционную смесь гидро-
лизуют насыщенным раствором ГЧП4С1.
При взаимодействии магнииорсанических соединений с двуокисью углерода об-
разуются соответствующие карбоновые кислоты с выходом 50—85% от теоретического:
BMgX + COa —► RCOOH
Важными условиями при проведении этих реакций являются хорошее перемеши-
вание и низкая температура, При этом следует обрабатывать эфирный раствор магний-
органичсского соединения 1азообразной двуокисью углерода [1311 при температуре
от 10 до —10° С или приливать реактив Гриньяра к избытку безводного размельченного
сухого льда [132]. Реакцию третичных гряньяровскпх реактивов с двуокисью углерода
проводят иногда под давлением [133].
8. Синтезы с литинорганическими соединениями [134]
Литийоргапические соединения реагируют с карбонильными соединениями
таким же образом, как и реактив Гриньяра, но они значительно более реакционно-
способны и поэтому применяются в тех случаях, когда магнийорганические соедине-
ния не реагируют нужным образом. Например, кетоны, не взаимодействующие
с реактивом Гриньяра из-за стеричсских затруднений, могут реагировать с литййорга-
ническпшг соединениями, как, например, диизопропилкетон с изопропиллитием [136]:
(СНз)2СНССН(СНэ)2-(СНз)2СНи —> [(СНз)2СН]зС-ОЫ
Недостатком литийорганических соединений является их большая чувстви-
тельнощь к кислороду, что вынуждает проводить процесс в атмосфере азота. Литий-
алкилы или лмтийарилы обычно получают взаимодействием органических галогенидов
с щелочным металлом в различных растворителях (стр. 637). Эфирные растворы литий-
оргапических соединений, как правило, неустойчивы и должны применяться тотчас
же после получения. При длительном стоянии они разлагаются с образованием алко-
голятов лития. Одиако растворы литийорганических соединений в бензоле или цикло-
гексане можно использовать даже после хранения в течение нескольких месяцев.
При хранении их следует исключить доступ кислорода, влаги и углекислого газа. Титр
раствора определяют, приливая аликвотную часть в воду и титруя выделившуюся
гидроокись лития 0,1 и. НС1 по метиловому оранжевому. Следующая пропись служит
примером практического проведения присоединения литийорганических соединений
к С=0-связям.
Циклогексенилдифенилкарбинол [135]. К суспензии тонкоизмсльченного
лития в сухом эфире медленно прибавляют в атмосфере азота при перемешива-
нии свежеперегнаняый чистый 1-хлорциклогексен и перемешивают в течение
ночи.
[130]
1131]
[1.32
[133
[134
И 35,
[136;
8. S е а г 1 е s, J. Am. Chem. Soc., 73, 124 (1951); L. Bemejo,
V. G. Aranda. C., 1930, I, 2382.
H. Gilman, N. J ohn, F. Schulze, Org. Synth., Coll. v. 2, 1943,
p. 425.
T. L. J aco bs, S. W insteia et al., J. Org. Chem., 11, 233 (1946).
C. Lester, J. Proffitt, J. Am. Chem. Soc., 71, 1878 (1949).-
Обзорная статья G. Wittig, Angew. Chem., 53, 241 (1940).
E. A. Braude, J. A. Coles, J. Chem. 8oc,, 1950, 2014. ,
G. V a v a n, П. Colin, С. r.. 222, 801 (1946); E. Muller, E. Hertel,
Ann., 555, 157 (1944).
728
ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Затем прибавляют при 0° С эфирный раствор бензофенона, перемешивают
18 ч и гидролизуют насыщенным раствором NH4C1.
2,6-Диметоксибензальдегид [137]. Смесь 13,8 е диметилового эфира
резорцина (0,1 жоль) с 0,1 моль фениллития в 100 мл абсолютного эфира выдер-
живают при комнатной температуре в течение 60 ч; диметиловый эфир литий-
резорцина выпадает в виде бесцветных кристаллов. После этого добавляют
раствор 13,5 г N-метилформанилида в 100 мл абсолютного эфира. Реакция
происходит с выделением тепла и эфир вскипает. После окончания реакции
смесь оставляют еще на 0,5 ч при комнатной температуре, затем приливают
к избытку разбавленной серной кислоты. Отделяют эфирный слой, сушат его,
отгоняют растворитель; остаток перегоняют в вакууме при 13 мм рт. ст..
отгоняя фракцию, кипящую до 130° С. Остаток кристаллизуют пз циклогек-
сана или из большого количества воды и получают бесцветные иглы. Выход
продукта 55% от теоретического; т. пл. 98—99° С.
При взаимодействии окиси стирола с фениллитием был получен фенилбензил-
карбивол [138]:
СвН6СН—CHa4-C6H5Li —> СбН9СНСН2СвН5
он
Как и в случае реакции Гриньяра, при реакциях с литийорганическими соеди-
нениями конец реакции определяют по пробе Джильмена (стр. 721).
III. Присоединение по кратным С—N-сеязям [140]
1. Присоединение магнийорганичееких соединений
Присоединение реактива Гриньяра к нитрилам протекает по схеме:
R\
R-C=N4-R'—MgX —> >G=N-MgX
R'z
При кислотном гидролизе приведенных выше продуктов присоединения обычно
образуется соответствующие кетоны:
Вч R.
>С=N-MgX4-2H2O —> >C=O4-NH3-[- MgOHX
R'Z R'Z
В некоторых случаях, если гидролиз проводят в достаточно мягких условиях,
например водным раствором хлористого аммония при низкой температуре, в качестве
промежуточных продуктов образуются кетимины [141]. Кетимины выделяют в форме
гидрохлорида, обрабатывая сухой эфирньпграствор высушенным хлористым водородом.
Если кетимины обладают повышенной чувствительностью к кислотам, разложение
проводят жидким аммиаком [142].
[137] G. Wittig, Angew. Chem., 53, 243 (1940).
[138] S. Crist ol, J. Douglass, J. Meek, J. Am. Chem. Soc., 73, 816 (1951).
[139] K. Ziegler, H. Colon iu s, H. Zeiser, Ann., 479, 147 (1930); 485.
185 (1931); Ga Hermann, Die Praxis des organischen Chemikers, 35 Aufl..
Veriag de Gruyter u. Go., Berlin, 1953, S. 320.
[140] M. К h a r a s c h, O. R ei nmu t h, Grignard Reactions of nonmetallic Sub-
stances, Prentice Hall, Inc., New York, 1954.
[141] G. Moureu, G. Mignonac, C. r., 156, 1801 (1913).
/142] E. F. Cornell, J.*'Am. Chem. Soc., 50. 3311 (1928); J. С I о k e, L. Baer,
J. Robbins,G. Smith, J.Am. Chem. Soc., 67, 2155 (1945); 62, 117 (1940).
II). ПРИСОЕДИНЕНИЕ ПО КРАТНЫМ C-N-СВЯЗЯМ
729
Этилциклопронилкетимин. К эфирному раствору этилмагнийбромида,
полученному из 42,3 ч этилбромида и 9 а магния в 150 мл безводного эфира,
медленно прибавляют по каплям раствор 10,35 г циклопропилциаиида в трех-
кратном объеме эфира. Затем реакционную смесь кипятят с обратным холодиль-
ником в течение 12 ч, переливают в делительную воронку, из которой медленно
прибавляют к 300 мл жидкого аммиака, защищая от проникновения влаги
осушительной колонкой, заполненной эффективным поглотителем основного
характера. В течение последующих 20 ч реакционную массу периодически
встряхивают, затем прибавляют 200 мл сухого эфира и фильтруют на воронке
Бюхнера, защитив всю аппаратуру от проникновения влаги. Аммиак удаляют
током сухого воздуха. Остаток немного разбавляют эфиром и обрабатывают
сухим хлористым водородом. Гидрохлорид кетимина очищают растворением
в смеси 50 мл ледяной уксусной кислоты и 5 мл ангидрида уксусной кислоты
с последующим осаждением сухим эфиром. Выход гидрохлорида этилцинло-
пропилкетимина 10 г (48% от теоретического).
Описан также синтез довольно устойчивого кетимина [143].
Реактив Гриньяра, полученный из 66 а 1-бром-8-метилнафталина и соот-
ветствующего количества магния, смешивают с 26 г о-хлорбензонитрила в бен-
золе и нагревают в течение 16 ч с обратным холодильником. После добавления
разбавленной соляной кислоты кристаллический гидрохлорид кртимпття отфиль-
тровывают, промывают холодной водой, спиртом и эфиром. Выход продукта
60 г. Этот кетимин устойчив к действию горячей соляной кислоты различных
концентраций.
Реакция магнийорганических соединений с нитрилами протекает в общем труд-
нее, чем реакции с Кислородсодержащими компонентами, так что часто необходимо
длительное нагревание или применение высококипящих растворителей. Было описано
получение алкилфенилкетонов из алифатических нитрилов и фенилмагнийбромида
[144], причем наилучшие результаты дает использование четырехкратного избытка
реактива Гриньяра.
Алкилфенилкетоны. К фенилмагнийбромиду, полученному из 25 г магния
и 160 г бромбензола в 300 мл сухого эфира, в течение 15жпн прибавляют при
перемешивании раствор 0,25 моль нитрила в 100 мл сухого эфира. Реакцион-
ную массу перемешивают еще 1 ч и оставляют на ночь, а затем выливают на
смесь 500 г льда и 300 мл концентрированной НС1. Водный слой отделяют
и нагревают при интенсивном кипении с обратным холодильником. После охла-
ждения встряхивают четыре раза с 200 мл эфира. Эфирные вытяжки сушат над
безводным СаС12 и после отгонки эфира перегоняют в вакууме.
Присоединение реактивов Грвньяра к альдаминам (основаниям Шиффа) приво-
дит к образованию вторичных аминов:
Нч /В' Бч
RCH = NR'-]-R’MgX —> >СН—N< —> >СН—NIIR’
R-Z \MgX В"/
Этил-1-фенилбутиламин [145]. К 1 моль м-пропнлмагннйбромида в 250 мл
сухого эфира прибавляют в течение 1,5—2 ч 66,5 г (0,5 моль) бензилидеп-
этиламина в 50 мл сухого эфира. Реакционную смесь несколько часов нагре-
вают с обратным холодильником и оставляют на ночь, затем гидролизуют,
выливая на смесь льда и соляной кислоты.
[143] L. F i е s е г, A. Seligma n, J. Am. Chem. Soc., 61, 141 (1939). ’
J144] R. S h г i и е г, T. Turner, J. Аш. Chem. Soc., 52. 1267 (1930).
[145] К. Campbell, C. Helbing, M. Florkowski, B. Campbell,
J. Am. Chem. Soc., 70, 3868 (1948); H. Gilman, J. Mort in, там же, 70,
2514 (1948); R. Moffett, W. Hoehn, там же, 69, 1792 (1947).
730
ОБРАЛОИАШ.Ч: НОВЫХ С —С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Взаимодействие молярных количеств альдимипа и реактива Гриньяра приводит
к образованию целевого продукта с выходом 60—90% oi теоретического только в слу-
чае простейшего азомстина (бензилиденметиламип) и очень реакционноспособных
соединений Гриньяра (метилмагнийбромпд). Во всех других случаях рекомендуется
применять двукратный избыток реактива Гриньяра (см. приведенную выше методику).
К кетиминам реактив Гриньяра присоединяется не так, как это описывалось
выше для альдпминов [146}. Алифатические или жирно-ароматпческие кетоанилы
реагируют с магнийорганичосклмп соединениями таким образом, что можно предполо-
жить их существование в форме таутомерного енамина:
/СвН5
CH<,-C=N-CeH5 + CoHB—MgBr —> СНг=С-М< 4-С2Нв
| I 'MgBr
СНз СНз
При реакции анпла бензофенона с фенилмагнийбромпдом происходит 1,4-
присоединение (выход продукта 42% от теоретического) [147]:
СвНб-C=N-СвН5 _£»н‘м^
MgBr
I
С9н5-С-N—С9Н&
Л/’1
uXCSHi
CeH5-CII-NH-CeH5
Присоединение реактива Гриньяра к изоцианатам [148] и горчичным маслам
[149] гладко протекает с выходом 80—90% от теоретического. При этом образуются
соответствующие амиды кислот или их тиоаналоги:
R—NCO + R'MgX —> R— N=C—R' —> R-NH-C-R'
I II
OMgX о
К C—N-саязи могут присоединяться и литийорганические соединения. По Циг-
леру [1391, присоединение фениллитин к хинолину протекает по схеме:
I ll l + CfiH5bi
N
Дегидрирование на последней стадии осуществляется нитробензолом.
[146] W. F. Short, J. S. Watt, J. Chem. Soc., 1930, 2293.
[147] H. Gilman, J. Kirley, C. Kinney, J. Am. Chem. Soc., 51, 2252
(1929).
[148] R. С а г I i n, L. Smith, J. Am. Chom. Soc., 69, 2007 (1947).
[149] G. Alliger, G. Smith, E. Carr, H. Stevens, J. Org. Chem., 14,
962 (1949).
Ш. ПРИСОЕДИНЕНИЕ ПО КРАТНЫМ С—N-СВЯЗЯМ
731
2. Присоединение нптросоедииеиий
Присоединение нитрометана к бензилиделапилппу протекает по схеме [150]:
CeIIr-CH=N— СвН5-ЬСНзЬ'О2 — > CeH5-CH-NH-CeH5
CH2NO2
1-Фениламино-2-нитро-1-фенилэтан. В течелйе 6 ч нагревают с обратным
холодильником 100 г (0,55 моль) бензальавилипа 100 г (1,64 моль) нитро-
метана и 50 мл абсолютного этилового спирта. Растворитель и избыток нитро-
метана удаляют при комнатной юмперачуре сухим воздухом в вакууме, осадок
нагревают с 95%-ным этиловым спиртом, отсасывают, несколько раз промывают
95%-ным этиловым спиртом и высушивают на воздухе. Выход продукта 106 г
(79% от теоретического); т. пл. 85—87° С. После перекристаллизации
из 95%-ного этилового спирта получают желтые призмы; г. пл. 86—86,5° С.
1-Фениламино-2-нитро~1-фенилпропан. Если эту реакцию проводят
с нитроэтаном, то нагревают в течение 15 мин с обратным холодильником 5 а
бензальанилииа (27,6 ммоль), 6,2 г нитроэтана (82,6 ммоль), 5 мл 95%-ного
этилового спирта и 0.6 г диэтиламина (8.2 ммоль). После обработки, как в при-
веденном выше примере, продукт реакции получают в виде желтых призм.
Выход продукта 2,1 г (30% от теоретического); т. пл. 100—102е С.
1-Фениламино-2-нитро-1-фенилбутан. Аналогично протекает реакция
с нитропропаном, нагревание проводят в течение 2 ч. 1-Фениламино-2-витро-
1-фенилбх’тан получают в виде желтых прпзм с выходом 55% от теоретического:
т. пл. 128 — 130° С.
1-Нитро-2-фениламино-1,2-дифеннлэтан [151]. Смесь 10.3 г фенилнитро-
мстана, 9,1 г бензальанилииа, 0,5 мл диэти.тамина выдерживают в течение
10 дней в холодильнике, в результате чего выкристаллизовывается 1-нитро-
2-фениламино-1,2-дифенилзтан. Выход продукта 91% от теоретического; после
двукратной осторожной перекристаллизации из лигроина т. лл. 124° С (разд.).
Фснилнитрометан может реагировать с бензнлиден-п-толуидином, лг-нит-
робензилидсп-п-толуидином, а также с ж- и п-нитробензнлиденанилином.
Была Исследована реакция присоединения этилового эфира нитроуксусной
кислот/J к основаниям Шиффа [152]; в присутствии эквимолярных количеств диэтил-
амина получены аддукты типа:
C6HsCH = NC6H5 + CIl2-NO2+(C2TI5)2NH - *
СООС2Н5
CgHgCH—С—NO2 (CsHshNHa
—* I I
CellsNH COOC2II5
Одпако 3i и аддукты неустойчивы и могут существовать некоторое время только
в сухом состоянии. Из двух молекул такого первичного аддукта очень легко образуется
этиловый эфир 1,3-динитро-2-арилглутаровой кислоты.
При взаимодействии ряда ароматических изоцианатов с нитрометаном или этило-
вым эфиром нитроуксусной кислоты получаются соответствующие амиды кислот [153J:
Аг—Х=С^О + СН2—СООС2Н5 —> Ar—NH—С—СН—СООС2Н6
I II I
no2 о no2
[150] К Leonhard, G. Lenbner, В. Burk, J. Org. Chem., 15, 979 (1950);
G. Hurd, J. Strong, J. Am. Gliein. Soc., 72, 4813 (1950).
[151] A. Doinow, F. Boberg, Ann., 578, 94 (1952).
[152] A. D о г n о w, A. F r e c e. Ann., 578, 112 (1952).
[153] R. Boyd, R. Leshin, J. Am. Chein. Soc., 75, 2762 (1953).
32
ОБРАЗОВАНИЕ НОВЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
Реакция с этиловым эфиром нитроуксусной кислоты протекает в присутствии
юлярного количества карбоната калия в сухом бензоле. В случае взаимодействия
итрометана с о-хлорфенил- и о-толилизоцианатами реакция протекает только
натриевой солью нитрометана, которую получают из нитрометана и метал-
:ического натрия.
3. Циклизация нитрилов
Метод циклизации нитрилов по Циглеру, Эберли и Олингеру [154] особенно
ригоден для получения макроциклических систем. Циклизацию динитрилов проводят
атмосфере азота в присутствии натрийметиланилина:
,C=NNa
C.H.NNaCH, . -
-------------* (СПг),
HtO
-NH» *
^СН—ON
^CH-CN
Реакцию проводят в эфирном растворе (диэтил-, диизопропил-, ди-н-пропиловый
^»пр) при температуре не выше 100’ С. Для предотвращения полимеризации внутри-
олекулярную циклизацию ведут, медленно и непрерывно добавляя динитрил в реак-
ионную среду при перемешивании (принцип разбавления Ругли — Циглера). Реша-
щим фактором при этом является поддержание постоянной концентрации, обеспечи-
зющее постоянство скорости взаимодействия реагирующих веществ. Большое раз-
авление благоприятствует внутримолекулярной циклизации и препятствует межмоле-
улярной конденсации; оно достигается постепенным введением в реакционную среду
гносительно небольших количеств циклизующегося материала.
Катализаторы этой реакции лучше всего получать по способу, описанному
.иглером и сотр. [155]:
2СвНбКнСНз-)-СвН6СН=СНг4-2Ыа —► 2С$НвММаСНз + СвН6СН2СНз
Образующийся этилбензол не мешает дальнейшей реакции. Вследствие чувстви-
шьности катализаторов к воздуху реакцию проводят в атмосфере чистого азота.
Циклизация нитрилов [156]. Смесь 0,1 моль дииитрила и 1,5 л 0,6— .
0,67 М раствора натрийметиланилниа, содержащего избыток (25%) метнл-
анилииа, кипятят с обратным холодильником в аФмосфере азота, перемешивая
и непрерывно в течение 72 ч вводя динитрил в стекающий из обратного холо-
дильника растворитель. После гидролиза получают соответствующие цикли-
ческие кетоны, согласно приведенному выше уравнению.
54] Houben-Weyl, Methoden der organischen Chemie, Bd. 4/2, Georg Thieme
Verlag, Stuttgart, 1955, S. 758.
.55] K. Z i e g 1 e r, L. J a k о b, H. W о 1 1 t h a n, A. W e n z, Ann., 511, 69
(1934). *
.56] k. Ziegler, R, A urn ha m mor, Ann., 513, 55 (1934).
П1. ПРИСОЕДИНЕНИЕ ПО КРАТНЫМ С—N-СВНЗЯМ
733
Более простым способом {157] можно осуществить циклизацию ди-(0-циан-
этил)-амина в З-циан-4-иминопиперидин с 70%-ным выходом:
N III NH
/
H2C CH2—C=N - -> II2C CII-C==N
H2C CH2 \ / NH । H2C CH2 \/ NH
З-Циан-4-иминопиперидин. В трехгорлой колбе, снабженной барботером,
обратным холодильником с хлоркальциевой трубкой и хорошо действующей
мешалкой, смешивают 400 мл перегнанного над натрием диоксана с 25 а нафта-
лина, 2 г натрия и , 50 г дн-(р-цианэтил)-амина. Воздух в колбе вытесняют
азотом. Реакционную смесь нагревают на паровой бане при перемешивании
в течение нескольких часов. Горячую реакционную массу выливают в 1 л* бен-
зола; циклическое соединение в бензоле нерастворимо. После перекристалли-
зации из этилового спирта т. пл. З-циан-4-иминопнпоридина 187—188g С (разл.).
4. Присоединение сниильиой кислоты
Миллер и Плёхль [158] осуществили присоединение синильной кислоты к анилу:
HCN4-CeHgN=CHR —> CeH6NHCHR
I
CN
Анилин растворяют в избытке эфира и при охлаждении прибавляют
альдегид, например пропионовый. Реакция протекает очень энергично с выде-
лением воды, которую затем отделяют. При прибавлении к раствору чистой
синильной кислоты начинается бурная реакция. Через некоторое время из
реакционной массы испаряют эфир и избыток синильной кислоты. Остается
густое масло, которое кристаллизуется через несколько дней. Кристаллизация
ускоряется при охлаждении. Синильную кислоту можно также прибавить
к раствору анилина в эфире, а затем по каплям добавлять альдегид (при этом
происходит бурная реакция с выделением воды) или же проводить взаимодей-
ствие анилина с синильной кислотой в эфире. При омылении образующихся
нитрилов получают соответствующие а-феннламиноалкановые кислоты.
Таким же образом можно осуществить присоединение синильной кислоты
к гидразонам и оксимам [158]. Установлено [159], что при взаимодействии пропионо-
вого и масляного альдегидов в водных растворах с гидрохлоридом гидроксиламина,
цианидом натрия и NaH2PO4 с хорошим выходом получаются а-цнаипропилгидрокснл-
амин Пли соответственно а-цианбутилгидроксиламин:
CH3CH2CH«NOH + HCN —► CH3CH2CHNIIOH
CN
Однако эта реакция применима не во всех случаях.
Взаимодействие ацетальдоксима или гептанальоксима с синильной кислотой
лучше всего осуществляется с избытком синильной кислоты и эквимолярным количе-
ством пиридина.
[157] G. В ас h man, R. Barker,]. Am. Chem. Soc., 69, 1535 (1947).
[158] W. v. M i I I e r, J. P I б c h I, Ber., 25, 2020 (1892); 26, 154o (1893); 27, 1281
(1894); H. Y a 1 e, K. L о s e e et al., J. Org. Chem., 16, 761 (1951).
[159] C. Hurd, J. Longfellow, J. Org. Chem., 16, 761 (1951).
734
ОБРАЗОВАНИЕ НОВЫХ С— С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ПРИСОЕДИНЕНИЯ
В присутствии следов основных катализаторов, из которых наиболее пригоден
пиридин, можпо также присоединить синильную кислоту к изоцианатам:
CeII5N-C=O + HCN —> CeHbNII—C-CN
А
Образующийся при этом циапформанилид со второй молекулой фснплизо-
цианата легко дает 1,3-дифенил-4-иминопарабановую кислоту [160]:
QH5
I
HN----С=О СвНб-N-0 = 0
-г I —* I !
0-С C=N 0—0. C=NH
\
I I
С6Нй С6Н5
Петерсен [1611 осуществил присоединение синильной кислоты к диизоцианатам
и обнаружил, что цианформамиды легко разлагаются на исходные компоненты ужо
при 120—130° С. Реакция гексаметилендиизоцианата с сннильпой кислотой протекает
по уравнению:
OCN(CH2)eNCO + 2HCN NC-C—Nll(CH2)6NU—C— CN
О О
В 660 мл толуола растворяют 252 г гексаметилендиизоциапата п сме-
шивают с 120 мл безводной синильной кислоты. Смесь хорошо охлаждают
и добавляют 10 мл пиридина. Сначала температура смеси повышается медленно,
а когда начинается выделение продукта, происходит ее быстрый подъем до
45° С. На следующий день полученную плотную кашицеобразную массу вымы-
вают из колбы толуолом, отсасывают и промывают толуолом от синильной
кислоты. Для очистки продукт растворяют в 750 мл горячего этилацетата
и добавляют примерно двойное количество толуола. Образуются белые иголь-
чатые кристаллы; т. пл. 102° С. Выход продукта 290 г.
5. Присоединение нетоиов к бензилиденанилину
Присоединение кетонов к бсизплидепанилину протекает под действием трех-
фтористого бора по схеме [162]:
СеН5-СН—N-СвН6 —CeH5-CH=N-СвПБ СН»СОС1Ь.^
BF3
СН2СОСНз • СН2СОСН3
[ I
—> CeHs-CH-NH-С6Нг, Cells—CII-NII—Cells
ir,
1-Фспиламино-1-фенилбутанон-3. К 2 86 г бензплпденанплина в 155 мл
сухого эфира прибавляют 2 мл эфирата трифторида бора. Образующийся
осадок отделяют. После многочасового стояния в закрытой колбе выпадают
[160] W. Dieckmann, Н. К а ш ш о г е г, Вег., 40, 3737 (1907).
1161] S. Р о I е г s р n, Aniv, 562, 211, 220 (1919).
[162] Н. Snyder, Н. Kornberg, J. Romig, J. Am. Chem. Soc.. 61/3556
(1939).
1. ОПРАЗОНАН11Е С—С-СВЯЗЕЙ ПРП ОТЩЕПЛЕНИИ ВОДОРОДА
735
желтые игольчатые кристаллы; т. пл. 135 — 145° С (разл.). Затем 1,5 г этого
продукта растворяют в 25 мл ацетона, причем раствор слабо разогревается.
Через несколько минут добавляют иоду и охлаждают- Кристаллизующийся
п виде бесцветных игл 1-фениламино-1-фенилбутанон-3 после трехкратной
нерекристаллизадпи из разбавленного спирта имеет т. пл. 87—88ь С. Выход
продукта 0,5 г.
Выделение борфторидного комплекса не является обязательным. Реакцию
можно проводить также следующим образом: 3,6 г бензиляденанилина раство-
ряют в четырехкратном избытке кетона и при охлаждении льдом перемешивают.
К раствору добавляют 2,8 г эфирата трифтОрида бора, выдерживают еще 5 лип
ири охлаждении льдом и затем выливают на смесь 100 мл льда и воды; выде-
лившееся масло быстро кристаллизуется.
Кроме ацетона в этой реакции могут участвовать этилметилкетон, иаобутилме-
тилксгон, к-амилметилкегон, 2-метллгексапон-4, ацетофепон и циклопентапон.
ОБРАЗОВАНИЕ ПРОСТЫХ С-С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ
РЕАКЦИЙ ОБМЕНА
/. Образование С—С-связей в результате отщепления водорода
1. Пирохимические реакции
В промышленности пирохимические реакции (коксование, крекинг и т. д.)
имеют большое значение, однако в препаративной органической химии такие реакпип
в настоящее время применяются лишь в редких случаях [163].
Одной из самых старых пирохимических реакций является образование дифенила
из бензола. По старым прописям пары бензола пропускают через раскаленную стллъную
трубку; при этом поряду с дифенилом в качестве побочных продуктов образуются
водород, углерод и дифопилбензолы. Оптимальная температура [164] реакции 750° С,
при ее повышении образуется много побочных продуктов. Наряду с температурой варь-
ировали скорость подачи бензола, длину и диаметр стальной трубки, а также исследо-
вали влияние добавок двуокиси бария, окиси алюминия, окиси цинка и окиси свинца
1165]; применяли раскаленную металлическую проволоку [166]. На том же принципе
основана лабораторная аппаратура для получения дифенила [1б7|. Описана аппара-
тура для проведения реакции непосредственно в нагретой свинцовой бане [168, 163/.
Проведение цирохимического синтеза дифенила под давлением в присутствии
катализаторов (Ni, I) позволяет снизить температуру реакции [ 169]. Правда, при этом
на передний план выступают побочные реакции, например образование метана при
использовании в качестве катализатора никеля. Сообщалось о получении дифенила,
антрацена и нафталина из бензола, толуола и н-бутилбензола в присутствии хром-
алюминиевых дегидрирующих катализаторов [170].
Полностью аналогично образованию дифенила протекают пирохимические пре-
вращения дифенмлметана в флуорен и дибензила в фенантрен, которые впервые были
[163] Н. Koch. Allgemeine Methoden zur Ausfiihrung pyrocheinischer Reaktionen,
в книге H о и h e ц - W e у I, Methoden der organischen Chemie, Bd- 4/2,
4 Aufl., Georg Thieme Verlag, Stuttgart, 1955, S. 435.
[164] J. E. Zane,tti, G. E g I о f f, Ind. Eng. Chem., 9, 350 (1917); C., 1918,
II, 1026.
[165] C. Smith, W. Lcwcock, J, Chem. Soc., 101, 1453 (1912).
[166] A. J. G г a n t, C. J a m e s, J. Am. Chem. Soc., 39, 934 (1917).
[167] С. H. Lowe, C. James, J. Am. Chem. Soc., 45, 2666 (1923).
[168] A. W. Hixson, L. T. Work et al., Ind. Eng. Chem., Analyt. Edit., 3,
289 (1931); см. также Honben-'Weyl, Methoden dor organischen Chemie,
Bd. 4/2, 4 Aufl., Georg Thieme Verlag, Stuttgart, 1955, S. 453.
[169] J. P. Wibaat, H. M. Rom ijn, H. D. Tjeenk W i 11 i n k, Rec.
trav. chim., 53-, 584 (1934).
1170J W. ]. Mattox, A. V. Grosse, J. Am. Chem. Soc., 67, 84 (1945).
736
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
доведены Гребе [171]. Зелинский и сотр. [172] получили карбазол из дифениламина
при 300° С в присутствии платинированного угля. Сообщалось [173] также о получении
флуорена и 4-метилфлуорена при многократном пропускании 2-метилдифенила
а 2,2'-диметилдифенила над палладированвым углем при 450° С.
2. Каталитическое отщепление водорода
Каталитическое отщепление водорода с образованием новых углеродных свя-
1ей протекает прежде всего под действием А1С13. Основное значение эта реакция имеет
утя синтеза конденсированных циклических систем ароматического ряда. При этом
•ледует различать межмолекулярную реакцию, как, например, получение перилена из
гафталина, и внутримолекулярную — образование перилена из 1,1'-динафтила [174]:
Эту реакцию исследовал Шолль, и поэтому она называется также реакцией
Долля [175].
Если исходное соединение является углеводородом, то прп внутримолекулярные
реакциях, например при уже упоминавшемся выше образовании перилена из 1,Г-
(инафтила, выходы продуктов незначительны. Лучшие результаты получаются в тех
щучаях, когда отщепляющийся водород, по крайней мере временно, имеет возможность
присоединиться к какому-либо другому месту молекулы, например к карбонильной
'руппе кетона или хинона. Водород в этих случаях удаляется окислением кисло-
)одом воздуха при выделении продукта. Так, из а-бензоилнафталниа получают
«ензантроп [176]:
Реакция носит общий характер. Она может проводиться с ароматическими
«оно- и поликетонами, имеющими по нрайней мере одно свободное пери-положение
ю отношению к карбопильноп группе кетона.
Бензантрон [176]. Тщательно измельченную смесь 1 ч. а-бензоилнаф^а-
лина и 5 ч. безводного А1С13 защищают от влаги хлоркальциевой трубкой
и нагревают в течение 0,5 ч от 60 до 115° С, 1 ч — от 115 до 130° Сив течение
еще 1ч — от 130 до 150° С. Затем образовавшийся плав разлагают разбавлен-
ной HCI, фильтруют, промывают спиртом н эфиром. Остаток состоит на
относительно чистого бензантрона. Выход бензантрона 76% от теоретического.
Перекристаллизацией из ледяной уксусной кислоты с добавкой активиро-
ванного угля получают чистый бензантрон в виде светло-коричневых игл;
т. пл. 167—168° С (неисправл.).
171] С. G гае be, Ann., 174, 194, 177 (1874); Ber., 7, 48 (1874).
172] N. D. Zelinsky ITitz, M. Gaverdowskaja, Ber., 59, 2591
(1926).
173] M. Orchis, J. Am. Chem. Soc., 67, 499 (1945); M. Orchin, E. O. W о о 1- -
folk, J. Am. Chem. Soc.. 67, 122 (1945).
174] R. Scholl, Ch. Seer, R. W e i t z e n b б c k, Ber., 43, 2202 (1910).
175] Ч. Томас, Безводный хлористый алюминий в органической химии, пер.
с англ., Издатинлит, 4949.
176] R. Scholl, Ch. Seer, Ann., 394, 111 (1912).
I. ОБРАЗОВАНИЕ С—С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ ВОДОРОДА
737
Более высокие выходы получают, если через реакционную смесь пропускают воз-
дух или кислород. Дальнейшее улучшение этого процесса состоит в том, что в рас-
плав AlCIy и хлорида щелочного металла пропускают ток сухого воздуха или кислоррда.
Примером может служить получение пирантрона из 3,8-дпбензоилпирена [177]:
О О
Пирантрон [177]. В расплав 500 г смеси А1С!з и NaCl (266 ч. AIGI3 -|-
4- 56 ч. NaCl) добавляют при перемешивании и температуре около 120° С 50 ?
3,8-днбензоилпирена. Постепенно повышая температуру до 140—160° С и хо-
рошо перемешивая, в смесь пропускают ток сухого кислорода со скоростью 60 л/ч,
Расплав, имеющий вначале фуксниово-красную окраску, постепенно (в течение
30—45 мин) становится голубым, а затем фиолетовым (длительность переход»
зависит от эффективности перемешивания). При выливании в воду получают
светлые коричневато-оранжевые хлопья почти чистого пирантрона, который
перекристаллизовывают из а-хлорнафталина. Выход продукта 40 г (80% от
теоретического).
Так как полициклические кетоны, применяемые для реакции Шолля, синтезируют
большей частью по реакции Фриделя — Крафтса, оба процесса можно проводить
в одну операцию. Так, 3-метилпирантрон [178] получают сплавлением 3-бензоилпи-
рена и n-толуилхлорида со смесью А1С13 и Nad при одновременном пропускании кис-
лорода:
О
О
[177 ] Н. Vollmann, Н. Becker, М. Corel I, Н. Streeck, Ann.,
118 (1937).
[178 ] R. Scholl, К. Meyer, J. Donat, Ber., 70, 2180 (1937).
47 Заказ 1835.
38
ОБРАЗОВАНИЕ ПРОСТЫХ С — С-СВЯЗЕП В РЕЗУЛЬТАТЕ ОБМЕНА
Обычно при реакции Шолля образуется шестичлсппое кольцо. Но в отдельных
лучаях, особенно у ароматических кетонов, карбонильная группа которых находится
пара-иоло;кенпи к гидроксильной или алкоксильной группам, образуется пягичлеп-
оо кольцо. Так, из 4-окси- или 4-тет&кс1!-1-бензоилнаф7ИЛИиа получается 6-окси-
,8-бензфлуореяон, а не 3-оксибензантрон [179, 180]:
Еслп пери-положение замещено, то также возникает пятичленное кольцо,
апример, из 1-бензоил-8-бензилнафталина образуется бепзилфлуоренон |181]:
Межмолекулярная конденсация двух ароматических ядер при помощи А1С13
©ответствует рассмотренным выше пирохимическим реакциям с той лишь разницей,
то в этом случае реакция протекает при более низкой температуре.
В то время как для ароматических углеводородов под действием А1С]3 наряду
аутокопдеясациой протекают побочные реакции (сужение кольца, частичное гидриро-
апие и др.), эфиры фенолов и нафтолов реагируют однозначно. Так, при обработке
-зт оксинафталина AIC13 в нитробензоле с хорошим выходом был получен4.4'-диэтокси*
,1'-дипафтил [182].
До сих пор речь посюянно шла о примерах конденсации молекул одного вида
ли внутримолекулярных реакциях. Удалось провести конденсацию и двух различных
оединений 1183]. Наиримор, из хинопа и бензола был получен ге-дифенилхинон;
иободный водород при этом нс выделялся, так как хпион частично гидрировался,
бедующей реакцией такого вида является образование a-амнподифеяила из азобензола
бензола [184] (выход 70—80% от теоретического):
S-N --N—/~S + 2Ci|H, >2^ NH3
3. Окислительное отщепление водорода
Под действием окислителей может проходить связывание ароматических flflep,v
также алифатических метил-, метилен- или метиновых групп с отщеплением водорода-
'акую реакцию можно рассматривать как окислительную димеризацию.
179] Н. Е. F i е г z - D a v i d, G. Jaccard, Helv. chim. acta, 11, 1046 (1928).
180J J. W. Cook, R. W. G. Preston, J. Chem. Hoc., 1944, 553.
181] K. D 7, i c w о ti s k i, J. M о s z e w, Rocznifei Chem., 11, 169 (1931); C., 1931,
I, 2875.
182] R. S c h о I I. Ch. Seer, Ber., 55, 330 (1922).
183] R, P и m in e г e r, Bef, 56, 3105 (1922); 60, 1439, 1442 (1927).
184J R. Pnmmorer, J. Bi napfl, Ber., 54, 2768 (1921).
1. ОБРАЗОВАНИЕ С-С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ ВОДОРОДА
739-’
Уже давно известно, что при окислении нафталина двуокисью марганца и сер-
ной кислого» образуется 1,Г-дицафтил [185, 186].
1,1'-Динафтил. Кипятят нафталин с MnOs и 50%-ноп IlaSOi, смесь раз-
бавляют кипящей водой, фильтруют, исчерпывающе экстрагируют остаток на.
фильтре спиртом и продукт перегоняют. Собирают отдельно фракцию с т. кип.
выше 360° С; продукт несколько раз перекристаллизовывают из спирта и лигро-
ина; т. пл. 1,Г-динафтила 160,5° С.
Эта методика была успешно применена для синтеза полиарилкетона [187]г.
2-метилбензантрон превращается под действием двуокиси марганца в 80%-ноД сер-
ной кислоте при 0—5° С с 78%-ным выходом в 2,2'-диметил-3,3'-дибензантронил,.
который под действием КОП и небольшого количества спирта при 120—130° С дает
16,17-диметилдибензантрон:
Н3С СНз
Окислительная димеризация 1-амино-2-метилнафталина в А ,4'-диамино-3,3'—
диметилдинафтил-1,1' может протекать [188] под действием Fe2O3 или HgSO4.
При использовании в качестве окислителя перманганата калия реакции препят-
ствует щелочная среда, образующаяся при раскислении окислителя. Этот эффект
устраняется при проведении реакции в присутствии буферных солей или в среде уксус-
ного ангидрида.
Чаще всего окислительное олцепление водорода с успехом применяют к феполам
и их эфирам. Например, 6-метил-2-нафтол |189| в присутствии солей трехвалентного
железа, а метплопый эфир а-нафтола [190] под действием надмуравъиной кислоты дают
соответст пующие а,а'-динафтилпроизводные.
4,4'-Днметоксвдинафтил-1,1'. К раствору 2,37 г (0,015 моль) метилового-
эфира а-нафтола в 75 мл 98- 100%-ной муравьиной кислоты прибавляют
по каплям в течение 90 мии при 40° С и перемешивании смесь 1,65 -w.i пергид-
роля (0,0165 моль Н2О2) и 25 мл муравьиной кислоты. Реакционная масса
через несколько минут окрашивается в красный цвет и в течение 30 мин начи-
нает обраэоиыисиься кристаллический осадок. После окончания прибавления
смеси пергидроля п муравьиной кислоты реакционную массу перемешивают
еще 15 мин. Надмуравьиная кислота после этого л реакционной массе не обна-
руживается. Смесь выдерживают 12 ч при комнатной температуре, затем про-
дукт отсасывают и промывают небольшим количеством спирта. Выход 4.4'-
днметоксидинафтила-1,1' 1,35 г; т. пл. 256—257° С. Упариванием маточных
растворов выделяют еще 0,3 г того же продукта. Общий выход 70% от теорети-
ческого.
[185] F. Lessen, Ann., 144, 78 (1867).
[186] W. Smith, J. Chem. Soc. 35, 225 (1879).
[187] D. H. Hey R. J. Nicholls, C. W. Pritchett, J. Chem. JJoc.,.
1944. 97.
[188] H. E. Fi er z -D a v i d, L. В la ri gey, II. Dubendorfe r, Helv. chim..
acta. 29, 1661 (1946).
[189] R. Royer, Ann. chim. [12], 1, 429 (1916).
[190] II. Fernholz, G-’Piazolo, Chem. Ber., 87, 578 (1954).
47*
740
ОБРАЗОВАНИЕ ПРОСТЫХ С—G-СВЯЗКП В РЕЗУЛЬТАТЕ СЕМЕНА
Окисление метилового эфира р-нафтола, напротив, дает хинон — 4-(2'-меток-
синафтил-1')-нафтокицон-1,2.
Окислительную димеризацию аптрона и его производных можно проводить
под действием кислорода [191], ароматических иитросоединений [192], диазометана
[193} или кислородом в присутствии палладия [194]. В качестве примера приведено
окисление антрона нитробензолом [192].
Диантрон. Раствор 5 г антрона и 3 мл нитробензола в 30 мл сухого
ксилола, содержащий небольшое количество хлористого водорода нагревают
в течение 5 ч с обратным холодильником. В холодильнике образуются кри-
сталлы гидрохлорида анилина и капельки воды. Из раствора, который окраши-
вается в оранжевый цвет, получают при охлаждении 4,8 г (86% от теорети-
ческого) почти чистого диантрона*.
Окислительная димеризация имеет значение не только для получении диарил-
производных. но и для образования новых С—С-связей в алифатическом ряду. При по-
мощи перекисей, особенно перекиси ацетила, можно провести такие реакции димери-
зации. как, паиример, синтез дихлорянтарной кислоты из хлоруксусной и 2,3-диметил-
2,3-дифедилбутана из изопропилбензола|195]. Для подобных реакций рекомендуется
также применение перекиси водорода [196] и смеси персульфата аммония и ацетата
натрия [197].
В некоторых случаях окислительное образование С— С-связн может применяться
также для получения производных дифенилэтапа, например для синтеза п,п'-динитро-
дибепзила из п-нитротолуола [198].
п,п'-Динитродибензил [ 198]. В трехгорлую колбу емкостью Зле мешал-
кой и трубкой для ввода газа, доходящей до дна колбы, помещают 2 л 30%-ного
раствора КОН в метиловом спирте. Колбу ставят в баню со льдом и после
охлаждения раствора до 10° С прибавляют 100 е (0,729 моль) п-нитротолуола.
Содержимое колбы энергично перемешивают и пропускают сильный ток воздуха.
Через 3 ч ледяную баню удаляют и еще в течение 5 ч пропускают сильный
ток воздуха при энергичном перемешивании. Затем реакционную смесь быстро
фильтруют и остаток на фильтре промывают 2 л кипящей воды и потом 300 мл
95%-ного этилового спирта. Продукт сушат на воздухе и перекристаллизовы-
вают из бензола. Выход продукта 73—75 г (74—76% ог теоретического); т. пл.
179—180° С.
Окисление кислородом воздухом в присутствии солей меди часто применяется
для синтеза диацетиленовых соединений из производных ацетилена. Так, например,
из метилэтинилкарбинола в присутствии CuCl, NH4C1 и водно-спиртового раствора
HCI с очень хорошим выходом получают смесь стереоизомеров октадиин-3,5-
диолов-2,7 [199]:
2СИз—СН—С=СН 1. > сНз-СН-СяС—CsC-CH-СНз
I I I
он он он
♦ О номенклатуре таких соединений см. Chem. Вег., 86, 294 (1953).
[191] В. В. Козлов, К. И. Куделина, ЖОХ, 17, 302 (1947).
[192] V. М. Ingram, J. Chem. Soc., 1950, 2246.
[193] F. Arndt, J. M. Schlatter, Chem. Ber., 87, 1336 (1954).
[194] A. S t о ] ], B. Becker, А. Ц e 1 f e n s t e i n, Helv. chim. acta, 33, 313
(1950).
[195] M. S. К h а г a s c h, E. V. J ensen, W. H. U г г у, J. Org. Chem., 10,
386 (1945); M. S. Kha rase h, H. С. M с В a y, W. H. Urry, там же, 10,
. 394 (1945).
[196] H. J. Barber, R. S 1 a c k, J. Chem. Soc., 1944, 612.
[197J H. Shechter, R. B. Kaplan, J. Am. Chem. Soc., 75, 3980 (1953).
[198] H. O. House, Оrg^Synth., 34, 35 (1954).
[199] K. Bowden, I. H e i 1 b г о n, E. R. H. Jones, К. H. Sargent,
J. Chem. Soc., 1947, 1579.
I. ОБРАЗОВАНИЕ С—С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ ВОДОРОДА
741
Аналогичный метод использован в случае различных производных ацетилена,
в том числе и высших [200|.
Окислительной димеризации могут подвергаться также гетероциклические со-
единения, Например, из пиридина можно получить а,а-дипиридил [201].
а,а-Диниридил. В автоклаве со стеклянным вкладышем емкостью 750 .ил
нагревают 165 г пиридина и 135 г сублимированного FeCI3 в течение 4 ч при
320— 340° С. Темно-красный реакционный продукт, который не должен выгля-
деть как обугленная масса, грубо размельчают, смешивают с водой, доводят
концентрированным раствором щелочи до сильпогцелочной реакции и пере-
гоняют с перегретым водяным паром. Перегонку продолжают до тех пор,
пока дистиллят не будет давать с FeCls только очень слабое красное окрашивание,
что длится довольно долго. Дистиллят подкисляют концентрированной H2SOt
и концентрируют до объема 500 мл. После охлаждения и подщелачивания
выделяется в виде масла основное количество дипиридила вместе с непроре-
агировавпшм пиридином. Это масло сушат над КОН» а продук! из водного
слоя экстрагируют эфиром. Экстракт также сушат над КОН, объединяют с основ-
ным количеством и фракционируют. Фракция, перегоняющаяся при 265—280° С,
быстро затвердевает. Ее отжимают на пористой пластинке. Для очистки про-
дукт перекристаллизовывают из низкокипящего петролейного эфира с живот-
ным углем. При сильном охлаждении осаждается дипиридил в виде бесцветных
кристаллов; т. пл. 71—72° С. Из маточного раствора выделяют дополнитель-
ное количество практически чистого продукта.
Описана также димеризация пиридина, хинолина и их производных при помощи
особо приготовленного контактного катализатора на основе никеля Ренея [202].
4. Фотохимическая дегидродимеризация [203]
Образование С—С-связи с отщеплением водорода может также осуществляться
фотохимическим методом. Такие реакции проводят большей частью в присутствии
акцепторов водорода, например, кислорода, карбонильных соединений (кетоны или
хиноны) или в присутствии красителей, которые превращаются при этом в лейко-
соединенпя.
Из дигидроантрацена этим методом получают диантрацен [204]; тиоксантен
и антрон превращаются на воздухе под действием солнечного света в соответственно
дитиодиксантил и диантрон [205]. Сообщалось [206] о фотохимической дегидродимери-
аации 2-окси-1,4-нафгохинона в ди-0-оксиЕафтохпнон. Ряд даарилметанов без доступа
воздуха в присутствии хинона или кетона удалось перевести в соответствующие тетра-
арилэтаны [207].
Дитиодиксантил [207]. Раствор 1 г тиоксантена с эквивалентным коли-
чеством ксантона в 15 мл бензола выдерживали в течение 6 ч на свету (в июле,
в Каире). Бензол нс содержал тиофена и был высушен над натрием. Реакцию
проводили в запаянной трубке (стекло Монаке), которую перед запаиванием
[200] Ю. С. Залкинд, Н. Иремадзе, ЖОХ, 18, 1554 (1948); Ju. S. S а ]-
k i n d, Fr. В. Fundyler, Ber., 69, 128 (1936); J. D. Rose, В. C. L. W e-
e d о n, J. Chem. Soc., 1949, 782; H. К. В ] а с к, В. C. L. Weedon, там же,
1953, 1785; R. A h m a d, В. C. L. Weedon, там же, 1953, 3286; A. W. N i-
neham, там же, 1953, 2601; В. L. S h a w, M. C. Whiting, там же, 1954,
3217; А. И. II о г а й д e л и, Р. Ш в а н г и р а д з е, ЖОХ, 24, 1025 (1954).
[201] F. Hein, Н, Schwedler, Вег., 68, 682 (1935); F. Н е i n, W. R е 11 е г,
там же, 61, 1790 (1928).
[202] G. М. В a d g е г, W. Н. F. S a s s е, J. Chem. Soc,, 1956, 616.
[203] А. Шенберг, Препаративная органическая фотохимия, пер. с нем., Издат-
инлит, 1963.
[2041 Н. Meyer, A. Eckert, Mh. Chem., 39, 243 (1916).
[205] A. Schonberg, A. Mustafa, J. Chem. Soc., 1945, 657.
[206] S. C. 11 о о k e r, J. Am. Chem. Soc., 58, 1212 (1936).-
[207] A. Scb on berg, A. Mustafa, J. Chem. 8oc., 1044, 67; 1945, 657.
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
заполняли двуокисью углерода. Образовавшийся дитподиксантил отфильтро-
вывали. промывали бензолом и перекристаллизовывали из ксилола, Выход
продукта 80% от теоретического; т. пл. 325n С.
Была исследована возможность дегидродимеризации алифатических спиртов,
юнокарбоповых кислот, альдегидов и простых эфиров при ультрафиолетовом облуче-
[ии [208], и и большинстве случаев удалось, хотя и с незначительным выходом, выдв-
инь соответствующие продукты димеризации.
II. Образование С—С-связей в результате отщепления Л
галогена в виде галогенида
1. Отщепление галогена щелочными металлами
Реакция Вюрца и Фшптига 2g
Синтез по Вгорцу основан на отщеплении галогена от алкилгалогенида действием ..
{елочного металла, причем два алкильных остатка соединяются с образованием угле- й
одорода: -
RX-?2Na—> RNa-j-NaX
RNa-FR'X —> R—R' + NaX :
Фпттиг перенес эту реакцию на ароматические соединения и проводил конден-
ацпю алкилгалогенидов с арилгалогенидами.
В алифатическом рядуудовлетиорительные результаты получают большей частью - <
элько при связывании одинаковых радикалов. Гладкое же протекание реакции Фит- •
ага основано на том. что ароматический галогенид значительно быстрее реагирует ;
металлом, чем алифатический, а арилнатрий, в свою очередь, быстрее реагирует
алки.тгалогенидом, чем с арилгалогенпдом (подробнее этот вопрос рассматривается Лчг
элементарных курсах органической химии).
Синтезы по Вюрцу практически применяются только в очень редких случаях.
днако, они все ясе служат одной из немногих возможностей для получения очень длин-
ых углеродных цепей определенного молекулярного веса. 'гы.
Гексаконтан [209] пз мирицилиодида *. Мирицплиодид не реагирует ..
с металлическим натрием более или менее энергично ни в петролейном эфире, '
ни в бензоле. Быстрее протекает реакция в расплаве, но при такой высокой
температуре, что при этом легко происходит выделение йодистого водорода . ;
и иода. Однако наилучшие результаты получаются при проведении этой реакции
с тонко нарезанным калием. При нагревании 1 ч. калия и 10 ч. мирицилиодида
при 139—140° С реакция заканчивается за 2 ч. Выделение продукта реакции
представляет значительные трудности. Реакционную массу последовательно 'л'Яяр
кипятят с водой, спиртом, петролейный эфиром и ледяной уксусной кислотой,
потом перекристаллизовывают пз бензола и снова кипятят с петролейный эфн- жй®
ром. Гексаконтан (см. прим, ред.) остается в вице неотчетливо кристалличе-
ского, бесцветного порошка; т. цл. • 101 —102° С.
Мортон и сотр. [210], а также Циглер и сотр. [211] подробно исследовали реак-
но Вюрца и дали важные общие рекомендации д.тя ее проведения.
* Мприцилиодид имеет состав С.^Н^! и по может, естественно, дать по реакции
Вюрца углеводород состава С6рН122> а только состава СвгНгав-
Прим. ред.
!08] G. Leuschner, К. Р f о г d t е, Ann., 619, 1 (1958); К. Р f о г d t е,
G. Leuschner, там же, 622, 1 (1959); G- Leuschner, К. Pfordte,
там же, 622, 6 (1959); К. Р I о г d I е, там же, 625, 20 (1959).
109] С. Н е 1 I, С. Il a ge ] е, Вег., 22, 502 (1889).
,10] A. A. Morton, J, В. Davidson, II. A. No w е у, J. Am. Chem. Soc.,
64, 2240 (1942).
11] К. Z i eg] е г, Н. Weber.H. G. Gellert, Ber., 75, 1715 (1942).
II. ОБРАЗОВАНИЕ С—С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ Hal В ВИДЕ ГАЛОГЕНИДА 743
По Мортону, выход продукта реакции повышается при использовании избытка
топкопзмельченного металла и дсрсмсшивапии мешалкой с возможно большим числом
оборотов. Протеканию реакции мешает образование твердой и непроницаемой пленки
на поверхности металла, которая возникает в том случае, когда реакция со слишком
реакционноспособным галогенидом протекает очень быстро, особенно в случае вторич-
ных и третичных алкилхлоридов. Третичные алкильные остатки вообще можно вводить
по Вюрцу только с трудом, но для их введения можно с успехом использовать метод
Фриделя — Крафтса.
Циглер показал, что для синтеза Вюрца иодиды более пригодны, чем бромиды,
а натрий лучше калия. В качестве растворителя следует применять абсолютный диэти-
ловый эфир или, если соединение трудно растворимо, дибутиловый эфир. Температура
реакции должна быть как можно ниже, а концентрация галогенида — как можно
выше, хотя даже и в таких случаях, когда растворимость в кипящем эфире составляет
около 0,1 л»оль/л, можно получить выход продукта до 80%.
1,32-Бис-(«-метоксифенокси)-дотриаконтян. В течение Ю ч кипятят
11,9 е метилового эфира иодгексадецилгидрохинона с 1,8 г натрия в смеси
40 лл диэтилового и 20 мл дибутилового эфира. После окончания реакции
эфир испаряют, к остатку добавляют метиловый спирт для разложения непро-
реагировавшего натрия и фильтруют. Остаток на фильтре затем растирают
с метиловым спиртом, водой, опять с метиловым спиртом и, наконец, с эфиром
и отсасывают. Промывкой эфиром удаляют легко растворимые побочные
продукты. Выход практически чистого 1,32-бис-(я-мстоксифснокси)-дотриакон-
тана 6.3 я; т. пл. 124° С.
О более часто применяемом синтезе Фиттига даются некоторые общие сведения.
Галогениды, так же как и разбавитель, должны быть тщательно высушены. Натрий
следует применять в виде проволоки или нарезать тонкими листочками. Если реакция
протекает слишком медленно, то лучше всего использовать порошкообразный натрий
или калий.
Порошкообразный натрий. Лабораторный метод получения [212J. В колбу
для сульфирования емкостью 1,5 л помещают 100 г натрия и заливают 400 мл
абсолютного ксилола. Воздух из колбы вытесняют азотом. Содержимое колбы
нагревают до 120° С и расплавленный натрий возможно быстрее перемешивают
при помощи быстроходной мешалки К11Г. В отверстие на нижнем конце мешалки
протянута тонкая проволока так, что образуются петли диаметром 3—5 см.
Образовавшийся пучок проволоки закрепляют п равномерно распределяют
вокруг оси вращения. Перемешивать нужно непродолжительное время. Если
перемешивать до тех пор, пока температура содержимого колбы не понизится
до температуры плавления натрия, то натрий снова собирается в большой сли-
ток. Операцию в этом случае следует повторить. В конце операции декайтируют
ксилол и заливают натрий эфиром или другим растворителем.
О получении тонко распыленного натрия см. [210].
В качестве разбавителя наряду с бензолом или петролейным эфиром пригоден
сухой эфир. Реакционная способность галогенидов увеличивается от хлоридов к бро-
мидам н Йодидам. Для проведения реакции натрий и разбавитель помещают в кругло-
донную колбу, снабженную капельной воронкой и обратным холодильником с хлоркаль-
циевой трубкой, и медленно прибавляют по каплям смесь галогенидов (если необходимо,
в растворе). В некоторых случаях начало реакции сильно задерживается, даже на
несколько часов, но лотом она начинается очень интенсивно. Реакцию можно иницииро-
вать добавлением в реакционную смесь небольшого количества этилацетата, который
активирует поверхность натрия. Образующийся при этом натрийацетоуксусный эфир
легко удаляется. Часто в качество катализатора применяют также ацетонитрил.
Обычно уже через некоторое время начинается кипение разбавителя. Смесь
галогенидов прибавляют таким образом, чтобы поддерживать гладкое течение реакции,
а после прекращения самопроизвольного кипения нагревают еще некоторое время на
масляной бане (но нс ла водяной) или на плитке с закрытой спиралью. По внешнему
[212] Н. G г о р, Dissertation, S. 62, Jena 1957.
744
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
виду натрия можно определить степень протекания реакции: в начале реакции металл
покрывается серо-голубыми пятнами, позже куски вспучиваются, образуется галоге-
нид натрия, который еще содержит в кристаллической решетке металлический натрий
и поэтому окрашен. Можно также исследовать отдельный кусочек на содержание в нем
металла. Иногда синтез длится в течение 2—3 дней.
О соотношении количеств реагирующих веществ нельзя дать общих указаний.
В большинстве случаев можно применять незначительный избыток металла (1/20 моль).
Если начало реакции между ариляатрием и алкилгалогенидом задерживается, как,
например, с вторичными бромидами, то в качестве побочного продукта образуется
диарил. Превращение иногда протекает более быстро, если арилгалогенид и соответ-
ственно натрий применяют в избытке, С другой стороны применение избытка алкилгало-
генида имеет то преимущество, что при последующей обработке он легко удаляется.
Продукт выделяют отгонкой непосредственно из реакционной колбы, прп необ-
ходимости в вакууме, продолжая перегонку до тех пор, пока она идет без разложения;
остаток в колбе экстрагируют. Осадок NaCl на дне колбы может значительно затруд-
нить перегонку, поэтому рекомендуется, особенно при применении высококипящих
углеводородов, после окончания реакции осторожно прибавлять спирт и потом воду
до тех пор, пока весь непрореагировавший натрий не растворится. Органический слой
промывают водой, высушивают н перегоняют.
Для удаления последних остатков галогенида перед последующим более тщатель-
ным фракционированием всегда прибавляют небольшое количество натрия.
м-Процилбензол [213] (по Фиттигу, модификация Вейганда). В кругло-
донную колбу с обратным холодильником и капельной воронкой помещают
44 г натрия, нарезанного тонкими листочками, и 300 мл сухого эфира, затем
, смесь охлаждают до 0° С и приливают к ней в течение 5 -ими охлажденную до
0° С смесь 100 г свежеперегнаиного бромбензола (т. кип. 156—157° С) и 98 г
н-пропилбромида (промытого раствором карбоната натрия, высушенного и све-
жеперегнанного; т. кип. 70—71° С) и после этого охлаждение прекращают.
Реакция начинается через 45 мин и заканчивается через 3 ч, причем в случае
необходимости колбу периодически охлаждают. Через сутки на водяной бане
отгоняют эфир и затем продолжают перегонку на масляной бане. Сухой остаток
несколько раз промывают декантацией отогнанным эфиром. Основное. коли-
чество продукта и эфирный экстракт перегоняют с колонкой Видмера. Выход
к-пропилбензола 60 г (79% от теоретического, считая на н-пропилбромид);
т. кип. 157—158° С. Дистиллят совершенно не содержит брома и поэтому пере-
гонка его над натрием не нужна. Выход продукта не меняется, если после
окончания реакции добавлять спирт и обрабатывать, как указано выше.
Таким же образом можно получить к-бутилбензол с выходом 65—70%
изн-бутилбромида и бромбензола [214]-Цетилбензол [215] был получен кипячением
бромбензола и цетилбромида с натрием в течение 6 ч в отсутствие растворителя.
Выход продукта 40% от теоретического.
Алкилгалогениды можно также заменять алкилсульфатами. Так, например, при
взаимодействии 2-метнл-1-бромнафталина с литием в эфире с последующей обработкой
полученного продукта диметилсульфатом получают 1,2-диметилнафталин с выходом
76% от теоретического [216J.
В некоторых случаях при реакции Вюрца — Фиттига вместо образования С— С-
связи происходит обмен галогена на металл [217, 218J. Например, Виттиг получил
из фениллития и бензилбромида вместо днфеннлметана бромбенэол и дибеняил [219],
[213] R. Fittig, Ann., 149, 324 (1869).
[214] R. R. Read et al., Org. Synth., Coll. v. 3, 1955, p. 157.
[215] J. P. W i b a u t, J. О verhof f, E. W. J о n к e r, Rec. trav. chim.,
62, 31 (1943).
[216] P. A. P ] a 11 n e r, A. Ronco, Helv. chim. acta, 27 , 400 (1944).
[217] G. Wittig, U. Pockels, Ber., 72, 89 (1939).
[218] R. G. J on es, Й. Gilman, Org. Reactions, 6, 339 (1951).
[219] G. Wittig, H. Witt, Ber., 74, 1474 (1941).
11. ОБРАЗОВАНИЕ С —С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ Hal В ВИДЕ ГАЛОГЕНИДА 745
а из фениллития и диметилового эфира 4-бромрезорцина — диметиловый эфир 4-литий-
розорцина:
СНзО^^/ОСНз СНзО^^ОСНз
I || -|-LiCeHg —|Z || +СвН5Вг
'V'Xbf
Это обстоятельство, с другой стороны, может быть использовано для некото-
рых синтезов. Например, из о,о'-бис-бромметилдифенила и фениллития был получен
9,10-дигидрофенантрен с выходом 86% от теоретического [220].
Натрийорганические соединения, полученные из конденсированных аромати-
ческих соединений, таких, как нафталин или антрацен, взаимодействием с натрием
в жидком аммиаке иди других средах, также реагируют с алифатическими галоге-
нидами [221J, особенно с изоамил- или изобутилхлоридом. При этом образуются дву-
замещенные углеводороды.
1,4- Диизобутилтцтралин. В 1 л жидкого аммиака растворяют 45 s
натрия, 30 г нафталина и 160 г изобутилхлорида. После испарения аммиака
остаток исчерпывающе экстрагируют бензолом и при его фракционировании
получают некоторое количество тетралниа, 10 г непрореагировавптего нафта-
лина и 1,4-диизобутилтетралин (т. кип.. 170—175° С при 12 мм рт. ст.).
9,10-Диизоамил-9,10-дигидроантрацен. Встряхивают 60 г натрия с 200 в
антрацена в 2 л эфира в атмосфере азота в течение 48 ч. К полученному фиоле-
тово-красному раствору осторожно прибавляют при охлаждении льдом и про-
пускании азота 239 г пзоамилхлорида и после обесцвечивания раствора прили-
вают по каплям воду для удаления избытка натрия. Затем отгоняют эфир,
. а маслянистый остаток растворяют в небольшом количестве петролейного
эфира. Через несколько дней отфильтровывают непрореагировавший антрацен,
отгоняют петролейный эфир и остаток фракционируют в высоком вакууме.
Вначале отгоняется антрацен, затем при 134—138° С — диизоамилдигидро-
антрацен в виде вязкой жидкости, из последних порций которой он частично
выкристаллизовывается. Выход продукта 75% от теоретического.
Реакции ацетиленидов щелочных металлов
с галогенидами
При реакции ацетилена или алкилацетилена с щелочным металлом или его
амидом в жидком аммиаке легко происходит непосредственное металлирование
[222—225]. Взаимодействие полученных ацетиленидов с алкилгалогенидами или алкнл-
сульфатами приводит к соответствующим алкил- или диалкилацетиленам;
HC=CNa + R'X —> HteC-R'
R-feCNa + R'X —> R-C=C-R'
При использовании алкилбромидов моноалкилацетилены [226, 230] получают
с 70—90%-ным выходом, а диалкилацетнлены [225] — с выходом 30—70% .прячем выход
снижается с ростом цепи алкилбромида. Алкилсульфаты [227] и алкилсульфонаты [228]
дают моно- или диалкилацетилепы с 60—85%-ным выходом.
220) D. М. На]], М. S. Lesslie, Е. Е. Turner, J. Chem. Soc., 1950, 711.
221] G. Huge], M. Lerer, C. r., 195, 249 (1932).
222] O. Neunhoeffer, V. Georg i, Ben, 92, 649 (1957).
223] O. Neunhoeffer, V. Georg i, Ben, 92, 791 (1959).
224] T. L. Jacobs, Synthese von Acetylenen, Org. Reactions, 5, 1, 25 (1949).
224] K. N. Campbell, M. J. O’C оплот, J. Am. Chem. Soc., 61, 2897 (1939).
226] K. N. Campbell, В. K. Campbell, Org. Synth., 30, 15 (1950).
227J T. F, R u t ledge, J. Org. Chem., 24, 840 (1959).
228] A. L. К r a n z f e I d e r, F. J, Sowa, J. Am. Chem. Soc., 59. 1490 (1937).
229] M. S. Newman, J. H. W о t i z, J. Am. Chem. Soc., 71, 1292 (1949).
230] A. L. Hcnne, K. W. Greenlee, J. Am. Chem. Soc., 67, 484 (1945).
ОВРАЗОНАНИГ ПРОСТЫХ С С-СВЯЗЕЙ В РЕЗУЛЬТАТ!: ОБМЕНА
Ацетиленид натрия 1223] (см. также [222]). В трсхгорлый сосуд с мешал-
кой КПГ и барботером, помещенный в сосуд Дьюара с охлаждающей смесью
из метилового спирта и сухого льда, конденсируют 200 лл аммиака и насыщают
его при —70° С чистым ацетиленом. Затем при дальнейшем пропускании ацети-
лена прибавляют большими порциями 6 г натрия. Получают суспензию ацети-
ленида натрия, которую, как правило, можно примени ,ь для дальнейших пре-
вращений без выделения ацетиленида.
6-Хлоргексин-1 [229]. К смесп 5 моль ацетиленида натрия и 3 л жидкого
аммиака прибавляют в течение 4 ч 858 г (5 моль) тетраметиленхлорбромида
и прп перемешивании «кипятят» в течение 7 ч с обратным холодильником.
После испарения основного количества аммиака (8 ч) медленно прибавляют
2 л коды. Органический слой разбавляют эфиром, промывают разбавленной
кислотой и водой, сушат и перегоняют. Выход продукта 430 г (74% от теорети-
ческого); т. кип. 143—144° С.
Прн реакции ацетиленидов с эфиром хлормуравьиной кислоты образуются эфиры
'етиленкарбоновых кислот [231]:
R—С=CNa—C1COOR' —> R—CsC—COOR'
2. Отщепление галогена другими металлами
Отщепление галогена магнием
Получение углеводородов при помощи синтеза Гриньяра (стр. 643)
RMgX + R'X —► R-R'-] MgX2
лесообразно проводить только в некоторых случаях, прежде всего для введения не-
сыщенных боковых цепей в ароматические углеводороды, например при реакции
илмагнийгялогеиида с аллилгалогенидом. Эта реакция экзотермична и протекает
tCTpo уже при комнатной температуре. Однако аллилхлорид рекомендуется медленно,
каплям, прибавлять к раствору реактива Гриньяра при температуре около 0° С
(ЛЯ доведения реакции до конца смесь нагревать непродолжительное время. Следует
еть в виду, что аллилбензол под действием щелочей переходиi в проценилбенэол.
Аллилбензол получают по прописи Хершберга [232], переработанной
едерском.
Аллилбензол. Пз 50,3 г магния, 324 г бромбензола и 800 мл эфира полу-
чают раствор фенилмагнийбромида. К этому раствору при охлаждении ледяной
водой прибавляют ио каплям в течение 1,5 ч смесь 250 г аллилбромида с 300 мл
эфира. Затем смесь кипятят 2 ч на водяпой бане, охлаждают колбу, содержа-
щую два слоя, ледяной водой и прибавляют по каплям 500 мл воды. Фазы
разделяют, водный слой два раза экстрагируют эфиром, объединенные эфирные
вытяжки сушат пад Ха,8О4 и после отгонки эфира перегоняют над натрием.
Выход углеводорода 213 г (87% от теоретического); т. кип. 150—170° С. Выход
чистого аллилбензола после фракционирования с колонкой Видмера над нат-
рием 153 г (62.7% от теоретического); т. кип. 158—160° С.
Аллилбензол можно получать также с выходом 85% от теоретического,
используя вместо аллилбромида в реакции с фенилмагнийбромидом диаллиловый
эфир |233].
Этот метод пригоден не только для введения олефиновых боковых цепей в аро-
гпческпе соединения. Алифатические соединения Гриньяра реагируют с р,у-ненасы-
нными алкилгалогенидами с образованием олефинов. В качество растворителя для
гктива Гриньяра в этом случае рекомендуется дибутиловый эфир [234]. Так, напри-
11] Ch. Moureu, R. D е I a n g е, С. г.. 136, 552 (1963).
12] Е. В. Hersh berg, Helv. diim. acta, 17. 3o2 (1934).
13] A. Liittringhans *ot al., Ann., 557, 46 (1947).
14] Cb. D. Hur d, G. II. Good ye a r, A. R. G olds b y, .1.
Soc., 58, 235 (1936).
Am. Chem.
II. ОБРАЗОВАНИЕ С. С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ Hal В ВИДЕ ГАЛОГЕНИДА 747
мер, получают 1-пентен из этилмагнийбромида н аллилбромида (выход 54% от теоре-
тического). Из аллилбромида и магния можпо получить с 55—65%-ным выходом диал-
лил (гексадиен-! ,5) [235],
В эту реакцию вступают также а,|3-ненасыщенные алкилгалогсниды. Например,
фенилмш иийбромид с 1-бромпропеном-1 в присутствии хлорида кобальта дает пропе-
нилбенлол с 51%-ным выходом [236], а с дихлорацетиленом — 1-фенил-2-хлорацети-
лен с 70%-ным выходом [237].
Ацетиленовые углеводороды образуются также с хорошими выходами из реак-
тива Гриньяра п замещенного пропаргилгалогенида R —С=С—СН2Х [238).
Возможность получения насыщенных углеводородов реакцией магнийоргаипче-
ских соединений с алкилгалогенидами используют лишь d отдельных случаях, так как
реакция редко протекает с хорошим выходом.
н-Амилбензол [239]. Раствор бепзилмагпийхлорида, приготовленный
из 24,3 г магния, 126,5 г бепзилхлорида и 500 мл эфира, нагревают в течение
15 мин до кипения и потом охлаждают. К нему в течение 2 ч приливают раствор
456 г бутилового эфира n-толуолсульфокислоты в двойном объеме эфира с такой
скоростью, чтобы эфир все время умеренно кипел. Нс охлаждая, смесь пере-
мешивают еще 2 ч, выливают получспную густую массу на лсд, прибавляют
около 125 мл концентрированной НС1 и растворяют n-толуолсульфонат магния
в 2 л воды. Затем эфирный слой отделяют, из водного слоя углеводород экстра-
гируют 200 мл эфира. Объединенные эфирные растворы промывают 100 мл
воды и сушат, встряхивая с 10 г К2СО3. После фильтрования отгоняют эфир
на водяной бане и остаток кипятят около 2 ч с 5 г свежспарезанного натрия
(при этом связывается бензиловый спирт, образовавшийся в результате частич-
ного окисления кислородом воздуха бензилмагнийхлорида). Жидкость, декан-
. тированную с натрия, перегоняют на эффективной колонке. Выход «-амилбен-
зола 74—88 г (50—60% от теоретического); т. кип. 198—202° С (основное
количество перегоняется при 199—201° С). z
Удлинение углеродной цепи галогенида (RX) па три СН2-группы рекомен-
дуется осуществлять взаимодействием полученного из него магнийоргапическЗго
соединения (RMgX) с З-хлорпропил-л-толуолсульфонатом (выход 50—60% от теорети-
ческого) [240]:
RMgX4-TsO(CII2)sCl —> В(СН2)зС] + XMgOTs
Аналогичная реакция протекает, например, с |3-хлорэтил-п-толуолсульфонатом
(выход 71% от теоретического) [241].
Подобно эфирам сульфоновых кислот реагируют с арил-или бензилмагнийгало-
гспидами диалкилсульфаты. При этом получаются соответствующие алкилароматиче-
ские соединения [242, 243].
Точно так же как синтез Вюрца дает обычно отрицательные результаты с тре-
тичными алкилгалогенидами, так и метод Гриньяра при получении высокоразвствлен-
ных парафиновых углеводородов не дает удовлетворительных выходов. Описан синтез
1ексамстплэтана из mpem-бутилхлорида и магния [244, 245]. Однако конденсация
[235] A. Turk, Т]. Chanan, Org. Synth., 27, 7 (1947).
[236] М. S, Kharasch, C. F. Fuchs, J. Am. Chem. Soc., 65, 504 (1943).
[237] E. О t t, \V. Bossallcr, Ber., 76, 88 (1943).
[238] K, N. Campbell, L. T. E b y, J. Am. Chem. Soc.. 62, 1798 (1940).
[239] TI. Gilman, L. L. H e c k, J. Am. Chem. Soc., 50, 2223 (19^8); Org.
Synth.. Coll. v. 1, p. 47.
[240] S. S. Rossander, C. S. Marvel. J. Am. Chem. Soc., 50, 1491 (1928);
J. ГГ a r m о n, C. S. Marvel, J. Am. Chem. Soc., 54, 2515 (1932).
[241] F. F. BJicke, F. Leonard, J. Am. Chem. Soc.. 68, 1934 (1946). '
[242] H. Gilman, R. E. Hoyle, J. Am. Chem. Soc., 44, 2621 (1922).
[243] С. E. Boord et al.. Ind. Eng. Chem., 41, 609, 613 (1949).
[244] F. C. Whitmore et al., J. Am. Chem. Soc., 55, 3807, 1561 (1933).
[245[ G. С a I i и g a e r t et al., J. Am. Chem. Soc., 66, 1389 (1944).
7-48
ОБРАЗОВАНИЕ ПРОСТЫХ С —С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
соединений Гриньяра, получсппых на основе третичных алкилгалогенидов или гало-
генароматических соединений, может осуществляться [246] под действием CuBr2
или AgBr:
2RMgBr-f-2AgBr —► R—R+2MgBr2 + 2Ag
1,1 '-Динафтил [247]. Смесь 2,4 г (0,1 моль) магниевой стружки,
18 мл сухого эфира, 20,7 е (0,1 моль) а-бромнафталина и один кристалл иода
умеренно нагревают на водяной бане до начала реакции (около 30 мин) и затем
баню отставляют. Выпадающий вскоре осадок растворяют прибавлением 25 мл
сухого бензола, высушенного над натрием. Нагревание продолжают до тех
пор, пока не израсходуется основное количество магния. Раствор а-нафтил-
магнийбромида при охлаждении и отсутствии влаги приливают в суспензию
13,5 г (0,1 моль) безводного СиС12 в 50 мл эфира. Реакционную смесь нагревают
1 ч с обратным холодильником и потом встряхивают с водой и разбавленной
НС1 до растворения хлорида меди. Органический слой отделяют и промывают
разбавленной НС1, разбавленным NH3 и водой. После сушки, над СаС12 эфир
отгоняют. Выход 1,Г-динафтила 10 а (80% от теоретического); т. пл. 154° С.
Соединения Гриньяра гладко взаимодействуют с а-алкоксиалкилгалогенидами
(а-галогенэфирами). При этом в большинстве случаев с хорошими выходами получаются
разветвленные эфиры [248, 249]:
/R'
RO-CHCl-R'f R'MgX —*- RO-CH<
XR"
В случае а,Р-дигалогенэфиров в реакцию вступает только а-атом галогена.
Образующиеся 0-галогеизамещенные эфиры служат промежуточными продуктами
для протекающего с хорошими выходами синтеза а-олефинов [249]:
RMgX-i Вг-СН-СН2Вг —> R—СН—CH2Br -Zn: Ж-> R-CH=CH2
OR' OR'
Циклические галогенэфиры также реагируют с магнийорганическими соедине-
ниями. Из смеси 2,3-дихлортетрагидропирана и пропилмагнилбромида получают
З-хлор-2-пропилтетрагидропиран с выходом 73% от теоретического [250]. Соединения
такой структуры при взаимодействии с натрием расщепляются с образованием соответ-
ствующих ненасыщенных спиртов [250, 251]:
/\/С1 /\
! J I I иос112с11.,спасн сш1
О Nab
В качестве примера ниже приведена реакция дихлортетрагидропирана с пропил-
магннйбромидом [250].
[246] J. В. Conant, А. Н. Blatt, J. Am. Chem. Soc., 50, 551 (1928);
J. H. Gardner, P. Bergstrom, J. Am. Chem. Soc., 51, 3377 (1929).
[247] E. Sakellerios, Th. Kyrimis, Ber., 57, 322 (1924); J. L. H a-
m о n e t, Bull. Soc. chim. France [4], 3, 254 (1908); C. r., 138, 813 (1904).
[248] H. 1. Waterman et al., Rec. trav. chim., 58, 437 (1937); C. D. Hurd,
M. A. Pollack, J. Am. Chem. Soc., 60, 1905 (1938); L. Malm, L. Sum-
mers, J. Am. Chem. Soc., 73, 362 (1951).
[249] L, C. S wallen, С. E. В о о г d, J. Аш. Chem. Soc., 52, 651 (1930);
H. B. Dykstra, J. F. Lewis, С. E. В о о r d, J. Am. Chem. Soc., 52,
3396 (1930); В. H. Shoemaker, С. E. Boord, J. Am. Chem. Soc., 53,
1505 (1931).
[250] M. Jacobson, J. Am. Chem. Soc., 72, 1489 (1950).
[251] L. Crombit.S. H. П ar per, J. Chem. Soc., 1950, 1707. 1714; R.C.Bran-
don, J. M. D e r f e г, С. E. Boord, J. Am. Chem. Soc., 72, '2120 (1950).
Л. ОБРАЗОВАНИЕ С—G-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ На! В ВИДЕ ГАЛОГЕНИДА
З-Хлор-2-н-пропилтетрагидропиран. К эфирному раствору и-проц
мшнийбромида, полученного из 600 г (4,96 моль) сухого свежеперегнанн
н-цропилбромида и 127 г (5,22 моль) магниевой стружки, в трехгорлой ко
емкостью 5 л, снабженной обратным холодильником с хлоркальциевок тр
кой, мешалкой и капельной воронкой, прибавляют по каплям при перемета
нип 482 г (3,11 моль) 2,3-дпхлортетрагидроиирана в 1 л безводного эфт
с такой скоростью, дтобы раствор умеренно кипел (при необходимости реакци
ную массу охлаждают в бане со льдом). Смесь постепенно густеет и приобрет
серую окраску, так что для облегчения перемешивания периодически необ
димо добавлять эфир. После окончания прибавления 2.3-дихлортетрагид
пирана (4,5 ч) смесь охлаждают льдом и осторожно гидролизуют смесью Л]
и соляной кислоты (1 : I). Эфирный слой отделяют, промывают водой и суп
иад Na2SOj. После отгонки эфира и фракционирования остатка в вакуз
получают 367,6 г (73% от теоретического) бесцветной Жидкости с мятным за.
хом; т. Кип. 72—80° С (13 ммрт. ст.), пг^ 1,4550.
Гриньяровские соединения ацетиленового ряда можно получить взаиыодейства
этилмагнийбромида с ацетиленом, алкил- или арилацетиленами. Эти соединения мож
гладко алкилировать алкилгалогенидами, алкилсульфатами или алкилсульфоната
[224, 252; 253, 254]:
И- С_СП K-CsC-MgX В1~01-> R-С=С-Н'
1-Фенил-2-бензилацетилен [252, 253]. Эфирный раствор фенилэтинв
магнийбромида получают кипячением с обратным холодильником 63 г (0,6 мо*
фенилацетилена с незначительным избытком этилмагнийбромида (в приблиа
тельно молярном растворе). Этот раствор в течение 2,5 ч прибавляют по капл^
при перемешивании к умеренно кипящему раствору 330 г (1,25 моль) бензи
n-толуолсульфоната в 900 мл абсолютного эфира. Затем смесь кипятят в
с обратным холодильником и гидролизуют разбавленной охлажденной НС
Эфирный слой промывают водой, карбонатом натрия и опять водой. Пос.
удаления эфира маслянистый остаток обрабатывают 15%-ным раствором КО
и потом перегоняют с водяным паром. Дистиллят, содержащий бензилброил
и бензиловый спирт, отбрасывают. Маслянистый остаток извлекают эфире»
тщательно промывают водой, сушат над безводным карбонатом калия и пер
гоняют в токе азота. Выход продукта 84 г (72% от теоретического); т. ка:
150—160° С (4 мм рт. ст,). После второй перегонки получают светло-желту!
сильно преломляющую свет жидкость с т. кип. 128—129° С (I — 2 мм рт. ст,
= 1,0273, п2о 1,5946.
Отщепление галогена серебром и медью
Синтезы Вюрца и Вюрца — Фиттига основаны, как известно, па конденсацЦ
алкил- или арилгалогепидов в углеводороды под действием металлического натри
(стр. 742). Однако применение натрпя невозможно, если галогенид содержит еще ода
функциональную группу, способную реагировать с натрием. В этом случае вместо Из
трия используют другие металлы, которые индифферентны по отношению к соответствч
ющим функциональным группам.
В алифатическом ряду для отщепления галогенов пригодно так называемо:
молекулярное серебро [258]. Таким образом получены, например, адипиновая кйслох
[252] J. В. J ohnson, А. М. Schwartz, Т. L. J а с о b s, J. Am. Chen:
Soc., 60, 1882 (1938).
[253] J. R. J ohnson, T. L. Jacobs, A. M. Schwartz, J." Аш. Chen:
Soc.. 60, 1885 (1938).
[254] Ю. Нью ленд, P. Фогт, Химия ацетилена, пер. с англ., Издатинли%
1947; Т. L. J а с о Ь я, S. S i n g е г, J. Org. Chem., 17, 475 (1952).
1258] М. Gomberg, L. II. Cone, Вег., 39, 3274 (1906).
750
О БРА 30 Р.ШПЕ ПРОСТЫХ С — С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
и.» р-иодпропиоповой кислоты [255], эфир 1.2-димет11ля11тарпой кислоты из эфира
7.-бро.мпрошюновой кислоты [256], производные янтарной кислоты, содержащие вне-
шне алкилы [257]. Цо синтезы с помощью молекулярного серебра я настоящее время
представляют лишь исторический интерес, так как эти вещества лучше получать при
помощи малонового эфира или сложпоэфирной конденсацией.
В ароматическом ряду в качестве отщепляющего галоген сродства применяется
медь. Таким способом получили фенидуксусный эфир взаимодействием бромбензола
и эфира хлируксусной кислоты [259].
Позднее [260] разработан метод синтеза диарилов из 2 моль арплгалогенида я
меди в качестве отщепляющего галоген средства [281]. Лучше всего с медью реагируют
иодиды. Хлориды вступают в реакцию только тогда, когда заместители, такие, как
бензоил- [262] или пптрогруппа. ослабляют связь галогена с ядром. Например,
•о,о--дииитродифенил легко получается из о-.\лорнигробензола {263J. С другой стороны,
реакция соединений, содержащих особо активные атомы галогена, может протекать
со взрывом; в этих случаях взаимодействие необходимо проводить в инертной среде
(разбавитель) [263]. В качестве примера приведено получение о,о'-динитродифенила
[263, 264].
о,е'-Динитродифенил. Смесь 30 г о-хлорнитробензола с 50 г сухого
песка нагревают до 200—210'’ С и медленно прибавляют при перемешивании
термометром 30 г порошка меди. Температура при этом не должна превышать
250s С. После прибавления каждой порпии меди необходимо выждать, пока
температура не снизится до 220° С. Когда вся медь прибавлена, смесь нагре-
вают еще 30 мин при 240—245° С. охлаждают, исчерпывающе экстрагируют
бензолом п упаривают экстракт. Выход 2,2'-динитродифенила 60% от теорети-
ческого: т- пл. 124° С.
Отщепление галогена цинком
В некоторых особых случаях в качестве га.чкгенотщепляющего средства при
•реакции Вюрца может применяться цинк [265]. Он особенно пригоден для евптеза али-
циклических соединений с малыми циклами, например циклопропанов из 1,3-дигало-
геппроизводных [266].
1,1-Диметнлцнклопронан [2116}. В трехгорлую колбу емкостью 2 л,
снабженную капельной воронкой, мешалкой с обратным холодильником (со-
единенным с приемником, охлаждаемым смесью ацетона и сухого льда), поме-
щают 900 мл 95%-ного спирта, 90 мл дистиллированной воды и 628 г (9.6 моль)
цинковой пыли (смесь сильно перемешивают, чтобы препятствовать спеканию
цинка). Затем смесь нагревают до умеренного кипения и прибавляют ио каплям
562 г (2,4 моль) 1,3-дибром-2,2-димотилпропана, после чего продолжают нагре-
вание и перемешивание в течение 24 ч. Основное количество углеводорода
собирается в приемнике. Остатки 1,1-диметилциклопропана отгоняют (с неболь-
шим количеством спирта) в приемник. Технический продукт (162 г) промывают
ледяной водой и сушат. Для дальнейшей очистки его перегоняют с охлаждаемой
колонкой; т. кип. продукта 19,9—20,6° С (подробнее см. [266]).
[255J W. Wislicenus, Лип.. 149, 220 (18в!)).
[256] С. Heil, М. Rothberg, Вег., 22, 60 (1889).
[257] D. В. Jones, J. Am. Chem. Soc., 37, 586 (1915).
[259 Th. Zinke, Ber., 2. 738 (1869).
[ 260J F. Ullmann, G. M. M eyer, O. Loewenthal, E. G i 11 i, Ann.,
.332, 38 (1904).
[2(J [ Литературный обзор по реакции Ульмана, Р. Е. Fanta. Chem. Rev., 38,
139 (1946).
[262] Е. М <i 11 о г. Е. Hertel. Aon., 555, 157 (1944).
[263] F. Ullmann, J. Bi eleek i, Ber., 34. 2174 (1901).
[264] R. C. Fuson, E. A. Cleveland, Org. Synth., Coll. v. 3, 1955. p. 339.
[265] M. Gomberg, Ber., .33. .3150 (1900).
[266] F. C. Whit in о rt> et al., J. Am. Chem. Soc., 63, 124 (1941); R. W. S h о r-
t г i cl g a et al., J. Am. Chem. Soc., 70, 946 (1948).
1Г. ОБРАЗОВ\HI1E Г- С-СВЯЗЕН ПРИ ОТЩЕПЛЕНИИ Hal В ВИДР ГАЛОГЕНИД \ 754
ТТррпаратИпный интерес представляет применение цинка в реакции Реформат-
ского [267] — взаимодействие а-галогенэфпров с карбонильными соединениями [268].
Эта реакция родственна синтезам Гриньяра.
QZnBr ОН
I I
Н—СО—СПз l-Zn+BrCH2C’OOCsH5 —* R — С—СН2СООС2ТТ5 —> R—С—СН2СООС2Н6
СНз СНз
Образующиеся вначале из цинка и а-бромэфпра металлоорганические соедине-
ния нестабильны, поэтому реакцию проводят ера sy в присутствии карбонильного-
соединения. Па первой стадии образуется эфир р-оксикислоты, который часто уже-
самопроизволыто переходит с отщеплением водорода в а,Р-ненасьпценный эфир.
При взаимодействии а-бромэфира с кетоном получаются соединения типа I,
с альдегидами — типа П, а эфиры замещенных а-бромкнелот с кетонами дают соеди-
нения типа III {269]:
R' R' R"
I I I
R—С^СН—СООС2НЬ R—СН=СН—СООС21Тб R—С —C-COOC2Hft
[ П ill
Реакцию Реформатского лучше всего проводить при 90—105° Цинк можпо-
применять и виде фольги, поверхность которой обработана наждачной бумагой. Неко-
торые авторы предла!ают использовать гранулированный цинк, активи руя его соляной-
кислотой [270] или нагреванием в круглодонной колбе с 0,1% иода до тех пор. пока
поверхность цинка не потеряет металлический блеск [271]: рекомендуют также цинк
амальгамировать * [272] или погружать в раствор сульфата меди для омеднения
поверхности. Если вместо эфира бромуксуспой кислоты применяют более дешевый эфир
хлоруксусной кислоты, то рекомендуется промотировать реакцию добавлением неболь-
шого количества порошкообразной меди [273].
В качестве примера приведены синтезы этиловою эфира р-окси-0-фенил-
пропионовой кислоты [274] и этилового эфира 1-оксицпклогсг>-сплуксусной кислоты
Г268, 275].
Этиловый эфир р-окси-Р-фенилпропионовой кислоты. В трехгорлую-
колбу емкостью 500 лл, снабженную мстпалкоп, капельной воронкой и обрат-
ным холодильником с хлоркальцневой трубкой, помещают 40 г (0.82 моль)
очищенной цинковой пыли или гранулированного цинка. К цинку прибавляют
15 мл раствора, приготовленного пз 83,5 г (0.5 моль) этилбромацетата, 85 г
(0,61 холь) беплалг.дегида в 80 мл сухого бензола ц 20 мл абсолютного эфира,
и нагревают колбу до начала реакции. Смесь начинают перемешивать и посте-
пенно прибавляют оставшийся раствор так. чтобы смесь умеренно кипела (около-
1 ч). затем кипятят еще 0,5 ч. охлаждают колбу в ледяной бане п ее содержимое
выливают при энергичном перемешивании в 300 мл охлажденной льдом 10%-ной
* Для этой реакции можно использовать также амальгамированный магний. Тан,
например, из гептепоца-4 и эфпра а-бромпротюповой кислоты получили-
эфир 1-метпл-2,2-длпроплл-2-оксипроттоновой кислоты с 70%-пым выхо-
дом [273].
]267[ S. Reformatsky, Вег., 20. 1210 (1887).
|268] R. L. Shrinpr, lire Reformatsky-Reaktion. Org. Reactions. 1. 1 (1942).
[269] TT. Rupe, et al., Ber.. 47, 68 (1914); J. v. H r a u n, Ann.. 451, 1, 47 (1927).
[270] E. Schwenk, D. Pap a. J. Am. Chem. Soc., 67, 1432 (1945).
1271] A. Lcwinsolin, Perfum. a. assent. Oil Rec., 15, 45 (1924).
[272] J. A. Nieuwland. S. F. Daly. J. Am. Chem. Soc., 53. 1842 (1931):
G. A. R. К on, K. S. Nargnnd. J. Chem. Soc., 1932. 2461.
[273] J. Colonge, D. Joly, Ann. Chimie fll], 18, 306 (1943).
[274] Ch. R. Hauser, D. S. Breslow, Org. Synth., 21, 51 (1941);
см. также [268].
]27.’>] S. N a f el son, S. P. G о t t f r i e d, J. Am. Chem. Soc., 61, 970 (1939);-
см. также [268].
752
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
H2SO4. Бензольный раствор промывают дважды 50 мл охлажденной 5%-ной
H,SO4, один раз 25 мл охлажденного 10%-ного раствора NaaC05, потом 25 мл
охлажденной 5%-ной H2SO4 и, наконец, дважды промывают водой (по 25 мл).
Объединенные кислотные экстракты встряхивают два раза с 50 лл эфира,
объединяют эфирные вытяжки с бензольным раствором и сушат смесь над 5 г
безводного сульфата магния или над перхлоратом магния. После отгонки раство-
рителя остаток фракционируют в вакууме. Выход этилового эфира р-окси-
В-фенилпропионовой кислоты 59—62 г (61—64% от теоретического); т. кип.
151 — 154° С (11 —12 мм рт. ст.) или 128—132° С (5—7 мм рт. ст.).
Этиловый эфир 1-оксициклогексилуксусной кислоты. Смешивают 800 мл
бензола и 700 мл толуола с 334 а (2 моль) этилбромацетата и 196 е (2 моль)
циклогексанона. В трехгорлую колбу емкостью 5 л, снабженную мешалкой,
обратным холодильником с хлоркальциевой трубкой и капельной воронкой,
помещают 300 мл полученного раствора и прибавляют 130 г (2 моль) цинковой
tольги, зачищенной наждачной бумагой и нарезанной полосками. Затем при-
авляют несколько кристалликов иода и нагревают при перемешивании на кипя-
щей водяной бане. При этом начинается бурная реакция. Оставшуюся часть
раствора прибавляют по каплям с такой скоростью, чтобы кипение продолжа-
лось, после чего перемешивают d течение 2 ч. Цинк растворяется почти полно-
стью. Смесь охлаждают и обрабатывают разбавленной H2SO4. Бензол-толуоль-
ный слой сушат над Na3SO4 и перегоняют в вакууме. Выход этилового эфира
1-оксициклогекснлуксусной кислоты 219—278 г (56—71% от теоретического);
т. кип. 86—89° С (2 мм рт. ст.).
Таким же образом можно получать этиловый эфир р-окси-р-февил-р-ме-
тилпропионовой кислоты из ацетофенона, бромуксусиого эфира и цинка [276].
В большинстве случаев при помощи реакции Реформатского получают эфиры
акриловой, а не р-оксипропионовой кислоты. Если ненасыщенные соединения в про-
цессе синтеза сами не образуются, проводят дегидратацию эфиров оксикпслот методом,
описанным на стр. 675. Для этой цели часто бывает не обязательно выделять эфиры
оксикислот в чистом виде. Продукт реакции непосредственно нагревают с хлор-
•окисью фосфора или перегоняют над пятиокисыо фосфора.
Эфир р-метнлкоричной кислоты [277]. К продукту реакции, полученному
из 72 г ацетофенона, 100 г эфира бромуксусной кислоты, 250 г бензола и 41 г
цинковой стружки, после отгонки бензола прибавляют 20 капель хлор-
окиси фосфора. Эфир p-метилкоричной кислоты отгоняют в интервале 155—165°С
(28 мм рт. ст.)', выход 80 а.
Низкие выходы продуктов, часто наблюдаемые- при реакции Реформатского,
-объясняются протеканием побочных реакций. Например, основная соль цинка может
вызвать альдольную конденсацию альдегидов. Возможно также взаимодействие 2 моль
-эфира а-бромкислоты под действием цинка с образованием эфиров янтарной кислоты.
Рекомендации для уменьшения доли побочных реакций см. [278].
Вместо сс-бромэфйров можно применять также другие галогениды. Например,
при взаимодействии пропаргилгалогенида с различными карбонильными соединениями
с хорошими выходами образуются Р,у-ацетиленкарбинолы [279]:
ОН
R—С—СН2—С=СН
I
R'
[276] 8. Lind enbaum, Ber., 50. 1270 (1917).
*1277] R. Stoermer, F. Grimm, E. La age, Ber., 50, 959 (1917).
[278] D. L i p k i n, T. D. Stewart, J. Am. Chem. Soc., 61, 3295 (1939).
[279] H. B. Hen be s t, E. R. H. Jones, I. M. S. W a 11 s, J. Chem. Soc.,
1949, 2696.
II. ОБРАЗОВАНИЕ С—С-СВЯЗЕЙ ПРП ОТЩЕПЛЕНИИ Hal В ВИДЕ ГАЛОГЕНИДА 75
Прп использовании в качестве карбонильной компоненты эфиров ортомуравьд
ной кислоты можно получать ацетали альдегидных производных малонового эфир]
с выходом 44—58% 1280]:
СООС2Н4
I
R—С—СП(ОС2Н5)2
<]оОС,Н5
Реакцию Реформатского применяют при синтезах различных важных нрярод
ных соединений, например фитола (281], полиенов [282], витамина А (из р-ионнлидев
ацетальдегида) [283].
3. Отщепление галогена алкоголятами, амидами
и гидридами щелочных металлов и другими основаниями
Эфиры rx-глициднои кислоты
Получение эфиров глицидной кислоты * по Дарзану—Клайзену [284, 285] про-
текает по схеме {286]:
ArCllO + CICHaCOOCaHs + CaHsONa’—> ArCH-CHCOOC2H5 + NaCl-C2H6OH
\/
Реакцию можно проводить с сухим этилатом без растворителя, а также с амидом
натрия в эфире или других растворителях.
Реакцию следует проводить при полном отсутствии воды и в определенных
случаях в атмосфере инертного газа. Целесообразно применять на 1 моль кар-
бонильного соединения 1,6 моль эфира хлоркислоты и 1,6 моль алкоголята
[286]. Начальная температура реакции должна быть низкой (до —80° С) [287],
а для завершения процесса реакционную массу короткое время нагревают
па водяной бане. Реакционную смесь обрабатывают разбавленной соляной
кислотой ифракционируют в вакууме.
Вместо ароматических альдегидов можно с успехом применять алифатические
альдегиды или ароматические ксюны, однако алифатические альдегиды дают очень
низкие выходы продуктов реакции. Вместо эфиров а-хлоруксусной кислоты могут
найти применение эфиры других а-хлоркислот.
Изучена также реакция ароматических галогепкетонов с ароматическими аль-
дегидами. Например, при взаимодействии со-бромацстофеыопа с п-нитробензальдегидои
с 88%-ным выходом получается а-(п-нитрофенил)-р-бензоилэтиленоксид [288]:
o3n -сн-сн-со—у
О
В качестве примера следует привести две прописи [289, 290].
* Обзор, см. «Органические реакции», сб. 5, Издатинлит, 1951, стр. 319.—1
Прим. ред.
[280] N. С. D сп о, J. Am. Chem. Soc., 69, 2233 (1947).
[281] F. G. Fischer, Ann., 475, 183 (1929).
[282] R. Kuhn, M. Hoffer, Ber., 65, 651 (1932).
[283] P. Karr er et al., Helv. chim. acta, 15, 883 (1932).
|284] G. Darzens, C. r., 139, 1214 (1904).
/285] L. Ciaisen, Ber., 38, 693, 699 (1905).
[286] Обзорная статья M.S. Newman, B. J. Magerlein, Org. Reactions,
5, 413 (1949).
[287] W. A. Yarnall, E. S. W a 1 1 i s, J. Org. Chem., 4, 270 (1939).
]288] H. H. Wasserman N. E. A u brey, J. Am. Chem. Soc., 77, 590 (1955).
[289] C, F. H. A 1 1 e n, J. van Allan, Org. Synth., Coll. v. 3, .1955, p. 727.
[290j R. H. Hun t, L. J. Chinn, W. S. Johnson, Org. Synth., 34, 54 (1954).
48 Заказ 1835.
7Ы
ОБРАЗОВАНИЕ ПРОСТЫХ С — С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
Эфир феннлметилглициднон кислоты, К смеси 120 г (118,5 лм, 1 моль)
ацетофенона, 123 г (109 мл, 1 моль) этилхлорацетата и 200 мл сухого бензола,
находящейся в трехгорлой колбе емкостью 1 л с мешалкой и термометром для
низких температур, прибавляют в течение 2 ч 47,2 г (1,2 моль) тонко растертого
амида натрия. Температуру посредством внешнего охлаждения поддерживают
в интервале 15—20° С. После добавления амида патрия смесь перемешивают
в течение 2 ч при комнатной температуре п затем, встряхивая, выливают на 700 а
раздробленного льда. (Органический слой отделяют, а водный встряхивают один
раз с 200 мл бензола. Объединенные бензольные растворы сушат над 25 г NanSO4,
после отгонки растворителя фракционируют остаток в вакууме. Продукт при
первой перегонке имеет т. кип. 107 —113° С (3 мм рт. ст.), а после второй
перегонки 111 — 114° С (3 мм рт. ст.). Выход продукта 128—132 г (62—64%
от теоретического).
Этиловый эфир р,р-пентаметиленглицидной кислоты*. Реакцию проводят
в трехгорлой колбе емкостью 500 мл, снабженной мешалкой, термометром
и капельной воронкой с противодавлением. Верхняя часть капельной воронки
соединена с системой для создания вакуума ** и введения азота. Аппаратуру-
сугпат в вакууме, обогревая пламенем. В колбу помещают 14,5 е (0,148 моль)
свсжепсрегпанпого циклогексанона и 18,15 г (0,148 моль) свсжеперегнанного-
этилхлорацстата. В капельную воронку помещают раствор трет-бутилата
калия, полученный из 6 г (0,153 .моль) калия и 125 мл сухого mpem-бутилового
спирта, систему вакуумируют и заполняют азотом. Колбу охлаждают льдом
и при перемешивании прибавляют по каплям в течение 1,5 ч раствор трет-
бутилата калия, поддерживая температуру смеси 10—15° С. Затем смесь пере-
мешивают еще 1 —1,5 ч при 10° С. Основное количество mpem-бутилового спирта
удаляют в вакууме водоструйного пасоса при температуре бани около 100® С.
Маслянистый остаток переносят в эфир, промывают водой и насыщенным раство-
ром NaCl и сушат над Na2SO4. Остаток, образующийся после отгонки эфира,,
перегоняют с колонкой Вигре. Выход бесцветного продукта 22,5—26 г (83—
95% от теоретического); т. кип. 134—137° С (21 мм рт. ст.), п-$ 1,4568—1,4577.
C-Алкилирование и С-ацилирование соединений
с подвижным атомом водорода
Алкилирование или ацилирование малонового эфира, ацетоуксусного эфира
или других соединений, метиленовые группы которых активированы сильными электро-
аоакцепторными группами, часто используется в препаративных целях для синтеза •
Золыпого числа промежуточных соединений, применяемых для различных дальней-
них превращений:
RHal+CH2X2 НСНХ2
RHaI + RCHX2 -^‘наТ* Н2СХ2
je X~.CN, COR'. COOR'. NOS и т. д.
Алкилирование обычно проводят следующим образом. К раствору или суспензии
1атрисвого производного алкилируемого соединения при соответствующей температура
(сбавляют алкилпрующее средство, обычно алкилгалогедид, причем выделение|на-
риевого производного в чистом виде по обязательно. Натриевое производное получают
]заимодействием активных метиленовых соединений с натрием, гидридом или амидом
[атрия в инертных растворителях, например в эфире; спиртовые растворы натриевого
гроизводного получают взаимодействием метиленовых компонент с рассчитанным коли-
есгвом раствора алкоголята натрия.
Моноалкилировапие натриймалоповых эфиров первичными или некоторыми вто-
шчными алкилгалогенидами обычно протекает с выходом от 75 до 90% . Как правило,.
* Более простой и удачный метод разработан В.М. Родионовым и Т. С. Кисе-
левой (Синтезы органических препаратов, сб. 2, Изд. АН СССР, 1952,.
стр. 58). — Прим, ред*
* См. Org. Synth., 30, 18 (1951).
ГГ. ОБРАЗОВАНИЕ С —С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ На! В ВИДЕ ГАЛОГЕНИДА 755
на выход очень благоприятно влияет применение избытка малонового эфира [291].
При замещении второго водородного атома выходы в большинстве случаев снижаются.
При моноалкилировании низшими алкилгалогенидами следует учитывать, что реакция
обычно протекает с образованием диалкилированных продуктов 1292], отделение котц-
рых представляет значительные трудности. Точно так же при одностадийном синтезе
диалкилированных продуктов получаемые соединения всегда содержат в виде примесей
моноалкильные производные [293]. Моноалкилированные продукты можно отделить
кипячением в течение 2ч с 50%-ным раствором гидроокиси калия [2941, при этом омы-
ляются только моноалкилпроизводные. Для алкилирования малонового эфира
применяют также диэтилсульфат [295], этиловый эфир [296] и другие эфиры п-толуол-
сульфокислоты [297].
В качестве примера алкилирования алкилгалогенидами приведено получение
эфира н-бутилмалоновой кислоты [601].
н-Бутялмалоновый эфир. Охлажденный раствор алкоголята натрия, полу-
ченный из 2,5 л абсолютного спирта и 115 г натрия, постепенно смешивают с 825 г
малонового эфира (фракция, кипящая в интервале 5J С). Затем прибавляют
по каплям в течение 1 ч 685 з к-бутилбромида, охлаждая реакционную смесь,
если необходимо. После этого смесь нагревают на водяной бане до нейтральной
реакции на лакмус, обычно в течение 2—3 ч. Наконец, отгоняют на водяной
бане 2 л спирта, остаток встрихивают с равным объемом воды, органический
слой отделяют и перегоняют в вакууме, выделяя головную фракцию, которая
содержит помимо других примссой пспророагировавший малоновый эфир.
Выход н-бутилмалопового эфира 960—990 г (89—92% от теоретического);
т. кип. 144—145° С (40 мм рт. ст.).
Для алкилирования эфиров р-кетокислот, а-цианокпслот и малоновой кислоты
рекомендуется добавлять алкилкарбонаты [298]. При таком методе работы раствор
метиленового соединения в алкилкарбопате смешивают с соответствующим количеством
•алкоголята натрия и отгоняют освобождающийся спирт. Вследствие этого равновесие
сдвигается в сторону образования натриевого производного соединения е активной мети-
леновой группой. При последующем алкилировании этого производного получают
с хорошими выходами продукты алкилирования даже в тех случаях, когда другие ме-
тоды дают неудовлетворительные результаты например при алкилировании моно-
тор-алкилзамещенных малоновых эфиров. Дополнительным преимуществом этого
способа является уменьшение возможности алкоголиза или отщепления карбоксильной
группы, что наблюдается иногда при проведении реакции в среде чистого спирта.
В качестве примера приводится алкилирование етор-бутилмалонового эфира [298].
Этил-етор-бутилмалоновый эфир. Получают алкоголят натрия растворе-
нием натрия в избытке спирта и упариванием в вакууме досуха. К образовав-
шейся твердой-массе прибавляют при охлаждении эквимолекулярное количество
етпор-бутилмаионового эфира и избыток диатилкарбопата (4—6 моль) и пере-
мешивают при комнатной температуре до растворения алкоголята. Затем пол-
ностью отгоняют спирт, используя колонку с электрическим обогревом (с голов-
кой полной конденсации и контролируемым отбором дистиллята: внутренний
диаметр колонки 13 мм, длина 44 см\ колонка заполнена насадкой из стеклян-
ных спиралей): отгонку заканчивают в вакууме. Затем колбу снабжают обратным
холодильником, мешалкой и капельной воронкой и в течение 45—20 мин доба-
вляют этилбромпд (избыток 10—15%). Потом температуру хорошо перемеши-
ваемой смеси поднимают до 95—105° С и поддерживают на этом уровне до
J. В. Cohen et'al., J. Chem. Soc., 1915, 887.
L. T. C. S c b о y. Rec. trav. chim., 16, 332 (1897).
E. Fischer, A. D i 1 t li e y, Ber., 35, 844 (1902).
J. F. Norris, H. F. Tucker, J. Am. Chem., Soc., 55, 4697 (1933).
C. D. H u г d, R. N. Jones, E. H. В lunch J. Am. Chem. Soc., 57,
2033 (1935).
D. H. Peacock, P. T h a, J. Chem. Soc., 1928, 2303.
W. В г a k e r, E. J. P г i b у 1. W. A. Lott, J. Am. Chem. Soc., 69, 866
(1947). ,
V. H. W a 1 1 i n g f о r d, M. A. Thorpe, A. H. Homeyer, J. Am.
Chem. Soc., 64, 580 (1942).
48*
756
ОШ’АЗОГИНПЕ ПРОСТЫХ С—ОСВЯЗЕИ В РЕЗУЛЬТАТЕ ОБМЕНА
полного превращения патриевого производного (около 54 ч). т. е. до отсутствия
щелочной реакции среды по фенолфталеину. После охлаждения реакционную
массу выливают в равный объем холодной воды, нейтрализуют уксусной кисло-
той и продукт экстрагируют изопропиловым эфиром. Эфирный раствор cvihat
и перегоняют. Выход продукта 95% от теоретического; п. кип. 69—72° С (1 .w.w
рт. ст.), nty 1.4331.
Синтез а,ш-дикарбоновых кислот осуществляют взаимодействием а,со-поли-
метилендибромида с избытком малопового эфира:
Вг(СНг)„Вг —дНг(С00^.-Ль > (C2H5OOC)2CH(CIl2)„CH(COOC2HB)2 — >
—> НООС(СН2)И+2СООН
Так, например, 1,12-додСкандикарбоновая кислота получена пз 1,10-дибромде-
кана с 64%-пым выходом [299].
При реакции этилендибромида пли триметилендибромида с натриймалоновым
эфиром прп избытке дибромида может происходить замыкание кольца с образованном
производных циклопропана [300] или циклобутана [301]:
Br(CH2),Br -CH-tcooc.H,),^ (сн2)„ С(СООСгЩ)2
При помощи ацетиламиномалонового эфира можно синтезировать аминокислоты
[302]. Так, из хлорметнлтиофена и ацетиламиномалонового эфира в присутствии алко-
голята натрия был получен тиенилметилацетиламипомалоновый эфир (выход 87%),
который при кипячении с бромистоводородпой кислотой переходит в р-тиенилаланив
(выход 78%):
Г“1 +ПС^Ш1СОСН3 Г%1 /соос2щ
Х^СЩС! ХСООС2Н5 X-^CHs-CcNnCOCHs —►
° Ь \COOC2HS
, ii il
\у/\сп3-сн-соон
nh2
В аналогичных условиях протекает алкилирование алифатических [303] или
жирно-ароматических [304] кетонов. При определенных обстоятельствах в этом случае
могут замещаться все а-водородные атомы. Например, прп взаимодействии ацетофе-
нона с 3 моль аллилбромида и дгретп-амилатом нагрия образуется [305] триаллилацето-
фепоп с выходом 70% . Метилирование ацетофенона в присутствии гидроокиси калия
проводилось Нефом [306]. Состав продуктов реакции при алкилировании кетонов
иногда сильно зависит от температуры реакции [307]. В качестве конденсирующего
средства особенно рекомендуется амид натрия [308]. Бензольный раствор кетона ки-
пятят с обратным холодильником с рассчитанным количеством тонко измельченного
[299] R. G. Jones, J. Ага. Chem. Soc., 69, 2350 (1947).
[300] A. W. Dox, L. Yoder, J. Am. Chem Soc., 43, 2097 (1921).
|301] G. B. Heisig, F. H. Stodola, Org. Synth.; 23, 16 (1943); A. W. D о x,
L. Yoder, J. Am. Chem. Soc., 43, 680 (1921).
[302] K. D i 11 m e r, W. H e r t, J. S. C h a m b e r s, J. Biol. Chem., 166, 541
(1946).
[303] I. N. Nasarow, Ber., 70, 594 (1937).
[304] A. Haller, Bull. Soc. chim. France |4), 31, 1073 (1922).
[305] G. Vavon, J. Conia, C. r., 223, 157, 245 (1946).
[306] J. U. N ef, Ann., 310, 316 (1900).
[307] G. Wash, B. Shi ve, H. L. Loch te, J. Am. Chem. Soc., 63, 2975 (1941).
[308] A. H a 1 1 e г, E. В a u e r, C. r., 148, 70 (1909).
л. (ii;i'.\3OB4HJii; <: с-свил.П npir о'пцгплгнш! Hal J! вид); г ч.’ютшд\ 75
амида натрия до прекращения выделения аммиака. Для моноалкплпровапия испод
uyioi laK/KC изопропплат натрия. Применение треш-ампла1 а натрия рекомендуете
в тех случаях, когда с амидом натрия получаются плохие выходы [309J; иногда пр]
меняют также гидрид натрия.
Дикетоны в эфирном растворе могут непосредственно реагировать с натрие
[310, 311]; последующее алкилирование проводят в ацетоне пли дпоксапе. В некоторь
случаях, когда применение натрия дает отрицательные результаты, с успехом испод
зуют калий [312].
Метилбен8оила{(егон |310] (см. также [311]). Растворяют беиаоплацетс
в шестикратном количестве сухого эфира и прибавляют рассчитанное кодич
ство натриевой проволоки. Таким образом получают рыхлый желтоваты
порошок натриевого производного бензешлацетона. Для дальнейшего превр,
щения [313] растворяют при нагревании 3 а высушенного над Р2Ог, натри]
бензонлацетона в 50 мл сухого ацетона, прибавляют 1,9 а метилиодида и зате
нагревают смесь с обратным холодильником в течении 4 ч. После отгони
ацетона остаток растворяют в эфире, встряхивают с водой, сушат и фракц]
оиируют. Выход метилбепзоплацетона 88% от теоретического; т. кип. 130'
134е С (11 3f.u pm. cm.).
Циклогексаидпон-1,3 можно легко проалкилнровать в положение 2. Глдролх
получаемых алкцлпроизводных щелочью дает б-кетокарбоповые кислоты (см. обзе
[314]).
О О
J' %/н
\ I —У —Н-> rCH!CO(CH,)sCOOH
\/\0 \/^0
Алкилирование эфиров р-кетокислот проводят аналогично алкилированию кет<
лее. Конденсирующие средства, олпсаппыо для алкилирования кетонов, лригодн
и в этом случае. Была исследована [315] возможность применения различных коцдег
спрующих средств; для введения вторичных алкильных групп в а-замещенный ацетх
уксусный эфир особенно рекомендуется трет-бутилат калия, причем выходы достиге
ют 80%.
О внутримолекулярном алкилировании 0-кето-и-галогенэфиров до циклически
Р-кетоэфиров см. [316].
В случае эфпров а.сс-диалкилзамещоппых уксусных кислот третий алкильиы
остаток можно ввести при помощи трпфенилмотилпатрия или трифенилметилкали
с последующим взаимодействием полученного металлированного производного с алки?
галогенидом [317]:
В. Rx
(C6n6)3CNaH >СН-СООСаН5 >С-С00С2НБ
д/ -(CeHjJjGH J|Z |
Na
R
>С-СООС. Н
R/ [
R'
С выходами свыше 90% протекает алкилирование производных барбитурово
кислоты [318], а эфиры циапуксусной кислоты алкилируются с выходами 60—78% Q
теоретического [298].
[309] М. J. С о n i a, Bull. Soc. chim. France [5], 17, 537 (1950).
[310] E. Fischer, C. Bulow, Ber., 18, 2|31 (1885).
|3U] C. Weygand, J. prakt. Client. 12], 116, 293, 297, 306 (1927).
[312] C. Weygand, C. Bischoff, Ber., 61, 688 (1928).
[313] C. Weygand, H. Forkel, Ber., 61, 687 (1928).
[314] H. S t e t t e r, Angew. Chem. 67, 769 (1955).
[315] W. B. Renfrow, A. Renirow, J. Am. Chem. Soc., 68, 1801 (1946).
[316] H. Hu usd iecker, Ber., 76, 142 (1943).
[317] R. Levine, E. Baumgarten, C. R. Hauser, J. Am. Chem. Soc.
66, 1230 (1941) и более ранние работы.
[318] О. Rosen, Е. Sandberg, Acta Chem. Scand., 4, 675 (1950).
758
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕП Б РЕЗУЛЬТАТЕ ОБМЕН \
По описанному способу можно также алкилировать нитрилы [319, 320], содер-
жащие в а-положенпп атом водорода. Конденсирующим средством прп атом обычно
является амид натрия. Реакцию проводят в инертных растворителях (эфир, бензол)
или я жидком аммиаке.
Циклогексилфснилацстопптрил [321J. Трехгорлую колбу емкостью 1 л
с мешалкой, барботером и обратным холодильником, который соединен через
колонку, заполненную патронной известью, с сосудом для абсорбции газа, тща-
тельно высушивают и защищают от влаги воздуха. Колбу охлаждают смесью
сухого льда и трихлорэтилена и конденсируют в ней 200 мл безводного аммиака.
Охлаждающую баню удаляют, газоподводящую трубку заменяют пробкой
и добавляют около 0,2 г кристаллического гидрата нитрата железа. Затем
быстро вносят маленький кусочек (около'5 мм) из предварительно взвешенной
порции натрия (8,1 г, 0,35 моль). Раствор перемешивают до исчезновения
голубого окрашивания, затем небольшими порциями прибавляют остаток
натрия, энергично перемешивая раствор. После того как голубая окраска
раствора изменится па серую, реакционную массу взбалтывают, смывая частич-
ки натрия, приставшие к стенкам колбы. Колбу вновь охлаждают в бане с сухим
льдом и в течение 10 мин прибавляют из капельной воронки 41 ? (0,35 моль)
бензмлциапида. Затем охлаждающую байю удаляют, раствор перемешивают
в течение 15 мин, после чего прибавляют по каплям 200 мл высушенного над
натрием и не содержащего серы толуола и 25 мл сухого эфира; аммиак при
этом испаряется. Остаток аммиака удаляют нагреванием на водяной бане,
причем большая часть эфира также выделяется через псохлаждаемый обратный
холодильник. Па холодильник надевают новую осушительную трубку, опя,ть
подставляют охлаждающую баню и к теплому раствору прибавляют 65,2 г
(0,4 доль) сухого бромциклогексана в течение 20 мин. Так как реакция про-
ходит с выделением тепла, необходимо охлаждение. Затем реакционную смесь
в течение 2 ч кипятят с обратным холодильником па масляной бане, охлаж-
дают и промывают 300 мл воды. Продукт из водпого слоя дважды экстрагируют
50 мл бензола. Объединенный бензол-толуольный раствор промывают дважды
50 мл воды и перегоняют в вакууме. Выход а-циклогексилфонилацетопитрила
45—53 г (65—77% or теоретического), т. кип. 174—176° С (13 мм рт. ст.),
т. пл. 50—53.5° С. Нитрил можно перекристаллизовывать пз пентана (потери
15%). После перекристаллизации т. пл. продукта 56—58° С.
Алициклические нитрилы можно получать внутримолекулярным алкилирова-
нием галогеннитрилов. Например, циклопропилцианид образуется из у-х.юрбутиропи
трила с выходом 74—79% [322].
Алкилирование циклонентадиена протекает аналогично алкилированию ацето-
уксусного эфира, а эфир глутаконовой кислоты (С,Н5ООССН — СНСН»СООС2П8)
который можно считать винилогом малонового эфира, реагирует подобно малоновому
эфиру [323[.
Извесшо также алкилирование пиколинов в боковую цепь [324]. Для
С-алкилиропанпя в качестве конденсирующих средств подобно трифснилметилнатршо
(см. стр. 757) могут найти иримснспио и литийорганические соединения (чаще фенил-
или бутмллитий), так как опи в определенных случаях реагируют с активированной
С—Н-связыо по типу металлирования. Например, при реакции диметилового эфира
резорцина с фепиллитием легко образуется с 70%-ным выходом диметиловый эфир
2-литийре.зорципа. Такие литицоргапичсские соединения легко реагируют с алкил-
[319] Обзорная статья F. W. В е г g s t г о m, W. С. Fernelius, Chem. Rev., 12,
135 (1933); 20, 451 (1937).
[320] Обзорная статья А. С. Cope, П. L. Holmes, H. O. Hous e, Org. Rea-
ctions, 9, 107 (1957).
[321] E. M. Hancock, А. С. С о p c, Org. Synth., Coll. v. 3, 1955,
p. 219.
[322] С. M. McCloskey, G. H. Coleman, Org. Synth., 24, 36 (1944).
[323] F. Henrich, Ber., 31, 2103 (1898).
[324] G. A. Knight, B. D. Shaw, I. Chem. Soc., 1938, 682; F. Brody,
M. T. В о g e r t, J. Am. Chem. Soc,, 65, 1075 (1943).
II, ОБРАЗОВАНИЕ С G-СВЯЗИЙ ПРИ ОТЩЕПЛЕНИИ H.1I В ВИДЕ ГАЛОГЕНИДА 75
1 алогснидамц пли алкилсульфатами с образованием С—С-связп 1325]. Напримех
при действии диметилсульфата названное выше литийрезорциннроизводвое npeept
ищется в 2,6-диметокситолуол [326]:
ОСНз ОСНз ОСНз
О получении фениллития и металлировании диметилового эфира резорцина ц
Виттиту [327] см. стр. 638, 640.
Через стадию непосредственного металлирования углеводорода протекает, в<
роятно, также сиптез w-гексилбензола из толуола и я-амилхлорида под действие,
металлического натрия в присутствии изопропилата калия (выход 76%) [329]:
CeHsCIIs-?Cl(CH2)4CHs Сг|.н,(Снг)5С.Нз
Кроме простых алкилгалогенидов с реакционноспособными метиленовыми соедв
нениями может реагировать ряд других галогенсодержащих соединении. Обычно гладк
реагируют галогенкегопы, эфиры галогенкарбоновых кислот и другие га.югенсодержа
щие соединения с функциональными группами, например (5-дпметиламиноэтилхлс
рид [330].
Введение ацильной группы в активные метиленовые соединения затрудняе
операцию, так как ацилированные продукты являются более Кислыми, чем исходны
материал, и поэтому дальше реагируют легче, чем непрореагировавшие исходные пре
дукты (см. [331 [).
В качестве примера приведено получение бензоилацетоуксусного эфира.
Бснзоилацетоуксуспый эфир [331], Растворяют 35,4 г натрия в спирт
(общий объем 600 мл), смешивают'300 мл полученного раствора этилата натри,
на холоду со 100 г ацетоуксусного эфира. Смесь охлаждают до 5° С и при постояв
ном перемешивании прибавляют по каплям в течение 15 мин 45 мл бенуоилхлори
да, причем температура не должна подниматься выше 10° С. Смесь оставляют ж
30 мин и затем добавляют постепенно, но достаточно быстро половину остав
шегося количества раствора этилата (150 мл) и 22,5 мл бензоилхлорлда. Чере
некоторое время эти операции повторяют, прибавляя каждый раз половин)
от использованного ранее количества этилата и бенаоилхлорида. Между теа
из спиртового раствора выделяется смесь NaCl и натрицбевзоилацетоуксусноГ'
эфира. Смесь оставляют стоять еще около 12 ч па холоду, отсасывают и пре
мывают эфпром. Из фильтрата промывным эфиром высаживают дополнительно
количество енолята натрия и объединяют с основным количеством. После непра
должительной сушки этот продукт растворяют в тройном количестве воды и прт
охлаждении льдом добавляют уксусную кислоту до тех пор, пока не закончите^
выделение масла. Масло растворяют в эфире, сушат над СаС12 и перегоняют
Продукт кипит при 175—176° С (12 мм рт. ст.).
Таким же образом можно провести бензоилирование бензоилацетона:
С8Н5СО С6Н5СО
\СП2 _ с*”^ос]^ >сн-сосвп3
СНзСО ~ СНзСО
[325] G. Wittig, Synthesen mit Li-organischen Verbindungen, в книге W. F о e г s t
Neuere .Methoden der praparativen organischen Chemie, Bd. I, 3 Aufl., Verlaj
Chemie, 1949, S. 469.
[326] I. P. Lamboo у, I. Am. Chem. Soc., 78, 771 (1956).
[327] Cm. [325], стр. 473, 476.
[328] II. Gilman, J. W. Morton jr., Org. Reactiofls, 8, 258 (1934).
[329] A. A. Morton, A. E. Brachman, J. Am. Chem. Soc., 73, 4363 (1951)
[330] J. A. Barltrob, J. Chem. Soc., 1946, 958; О. E i s 1 e b, Ber., 74, 1435
(1941).
[331] L. Claisen, Ann., 277, 196 (1893); 291, 53, 67 (1896).
GO
ОБРАЗОВАНИЕ ПРОСТЫХ U--C-C И 1'КЗУ. НУГА'ПГ ОБМЕН \
Однако образующийся при этом ацетплдпбеи.зоилметан значительно удобнее,
равда, с менее хорошими выходами можно получать но способу Клайзева [3311,
редложенному им ранее для бензоилирования ацетоуксусного и малонового эфиров.
1етод заключается и обработке смеси бензоплхлорида и бензоилацетона в кипящем
фирс сухим Na2CO3.
Ацетилдибснзоилметан. В 100 .ил сухого эфира растворяют 16,2 г бензоил-
яцетопа и 14 г бензоилхлорида; к раствору прибавляют 32 г топко измельчен-
ного сухого Na.,CO3 и оставляют смесь в колбе, снабженной обратным холодиль-
ником и изолированной от доступа влаги. Через некоторое время начинается
кипение эфира. Реакционную массу выдерживают 16 ч прп комнатной темпера-
туре, затем отсасывают смесь NaCl, Na2CO3 и енолята натрия п прозшвают
ее небольшим количеством эфира. Содержащий эфир остаток растворяют н поде,
удаляют эфир током воздуха и осаждают продукт уксусной кислотой. При этом
получают 10 г чистого ацетилбензоплметапа. Из эфирного маточного раствора
и Промывной жидкости можно 10%-ным раствором Na2CO3 извлечь еще неко-
торое количество енолята, так что выход продукта увеличивается до 14 г (87%
от’теоретического). Чистый продукт имеет т. пл. 140° С [332].
При взаимодействии натриевой соли дибензоилметана в ацетоне с бепзоилхлорп-
эм был получен трнбензоилмеган (выход 45% от теоретического) [313].
Для введения различных ацильных остатков в малоновый эфир пригодны это-
;имагнийпроизводные[333], в то время как кетоэфиры могут быть ацилированы кипя-
нием их с ацилхлоридом и магнием в бензоле (ныход 46—52%) |334].
а,а-Диалкллзамещенныо 0-кетоэфнры без примесей моноалкилпрованных
родуктов получают ацилированием натриевых производных эфиров а.а-диалкил-
ксусной кислоты, которые образуются (стр. 757) при взаимодействии эфиров диалки.т-
чсусной кислоты с трифенплметилнатрием |335].
Описан метод удлинения углеродной цепи па пять атомов углерода [336].
Реакция протекает с высокими выходами по схеме:
i [ + C2H3MgBr —I I _rcoci_^ i j
OMgDr /\^°
COOC2H5 COOC2H5 ROC COOC2II5
RCO(CH2)1COOH
4. Синтезы кетопов
при помощи металлоорганических соединений
Реакция галогенидов карбоновых кислот с соединениями Гриньяра обычно дает
езичные спирты. Однако она останавливается на стадии кстопа. если образование
етшшого спирта затруднено стерическими факторами:
RCOCl-1-R'MgCl —* RCOR'-ф VgCb
32] W. D 1 е с k щ а п л. Вег., 49. 2203 (1916); С. Weygan d, Z. anorg. allg.
Chem., 205, 411 (1932).
33] G. W. Andorso n et al., ,T. Am. Cheffl- Soc.. 67, 2197 (1945); B. Riegel,
W. M. L i 1 i e H f e 1 cl, J. Am. Chem. Soc., 67. 1273 (1945); ТГ. Lund,
A. Voigt, Org. Synth., Coll. v. 2, p. 594-
341 A. Spassow, Org. Synth., 21, 46 (1941); J. П. Hunte r, J. A. Hog g,
J. Am. Chem. Soc., 71, 1922 (1949).
35] C. R. Hauser, W. B. Renfrew jr., Org. Synth., Coll. v. 2, 1943,
p. 268.
36] J. Plesok, Coll, czoah. chem. Comm., 21, 1312 (1956); 22, 49, 1661 (1957).
. ОБРАЗОВАНИЕ С С-СВЯЗЕП ПРИ ОТЩЕПЛЕНИИ Hal Б ВИДЕ ГАЛОГЕНИДА 76[
Например, ди-7??рг/н-бутплкотоп получен [337] с 71.6%-ны.м выходом из трет-
бутплмагпийхлорида и хлорангидрида триметплуксусной кислоты и присутствии СиС12.
Взаимодействие хлорангидрида кислоты с- реактивом Гриньяра может быть
остановлено на стадии кетона при соблюдении особых условий реакции. Для этого
изменяют порядок смешения реагентов: реактив Гриньяра добавляют к х.юрангидриду,
тогда как обычно применяют обрагнын порядок [338]. В атом случае отсутствует
избыток реактива Гриньяра, который, очевидно, легче реагирует с хлорангидридом,
чем с уже образовавшимся кетоном,поэтому реакция останавливается на стадии кетона.
Скорость прибавления магнилорганического соединения илн температура реакции
не оказывают влияния на выходы продуктов, которые, как правило, невысоки. Этот
метод обычно использую! для синтеза смешанных кетонов.
Выходы продуктов зпачптельио увеличиваются, если соединение Гриньяра
перевести в цинкир1аническое сордппспир. для чего к нему прибавляют по каплям
ZnClj в эфире с такой скоростью, чтобы раствор постоянно кипел.
RMgCl -|- ZnCls —► BZnCJ-r-MgC]2
Полученный хлорид цппкорганическото соединения может ротировать с хлор-
ангидридами кисло г, причем выходы кетонов достигают 93% [339]. Известен, например,
синтез кетонов при помощи этилципкиодпда [340}.
Прп реакции хлорангидридов кислот с кадмпйалкнламн, получаемыми из соеди-
нений Гриньяра и CdCL [3411, образуются кетоны с выходом около 50—84% . однако
использование дп-лтор-длкилкадмпевых соединений дает плохие результаты [342].
2BMgX-HCdCl2 — > CdR3 -2MgXCl
Лри.ч- или алкплбромиды для этой реакции более пригодны, чем йодиды [343].
Вообще, кадмийалкилы следует предпочесть ципкалкилам. так как они обладают
незначительной реакционной способнос!ыо по отношению к карбонильной группе.
В качестве примера описано получение 1-фепилэйкозанопа-З из хлорангидрида
стеариновой кислоты и бис-р-фенилэтилкадмия [344]:
2СН8(СН4)]вСОС1 + Сб(СП;СП2СбП5)2 —> 2СНз(СП2)1еСОС1Г2С[Т2СвНп - Cd С12
1-Фенплэнкозанон-З. Раствор р-фонилэтплмагнпйбромида получают из
1,78 .ноль (42,7 г) магния и 1,755 моль (325 г) Р-фенилэтилбромида в безводном
эфире. К хорошо перемешиваемому и охлаждаемому льдом раствору Гриньяра
прибавляют в течение 10 .чип 0,936 моль (171,5 г) С<1С12. Зачем смесь нагревают
до комнатной температуры. Растворитель отгоняют при перемешивании па подя-
пой бане до затвердевания остатка, после чего к нему прибавляют 1,5 л сухого
бензола п повторяют процесс упаривания. Затем снова прибавляют сухой бен-
зол, смесь быстро перемешивают и кипятят с обратным холодильником до пол-
ного размельчения твердого остатка. К хорошо перемешиваемой суспензии
добавляют в течение 2 ч раствор 1,405 моль (400 г) хлорангидрида стеариновой
кислоты в 1 л сухого бензола и кипятят в течение 5 ч с обратным холодильником.
Затем смесь обрабатывают льдом и соляной кислоюй. Органический слой отде-
ляют, встряхивают с раствором NajCOj и отюнягот бензол. Остаток омыляют
избытком спиртовой щелочи обычным образом. Продукт выделяют в виде
пеомыляемой фракции экстракцией бензолом, тщательно промывают водой
[337] N. С. С о о k, W. С. Percival,}. Am. Chem. Soc., 21, 1315 (1956); 22, 49,
1661 (1937).
[338] H. Gilman, M. L. Mayhu e, Rec. trav. chim., 51, 47 (1932).
1339] G. A. Sell m id t, D. A. S h i г I e y, J. Am. Chem. Soc., 71, 3804 (1949).
[340] J. Colnnge, D. Joly, Ann. Chimie [11], 18, 306 (1943).
[341] Г. L. de BenneviHe, J. Org. Chem., 6, 462 (1941); G. W. Stacy,
R. M. M с C u r d y, J. Am. Chem. Soc., 76, 1914 (1954).
[342] J. Cason, J. Am. Chem. Soc., 68, 2078 (1946); W. G. Dauben, J. Org.
Chem., 13, 313 (1948).
[343] W. G. Daub e n, T i И о s, I. Org. Chem., 15, 785 (1950); R. P. J acob-
sen. J. Am. Chem. Soc., 73, 3463 (1951).
[344] K. W. Sherk, M. V. A u g u r. M. D. S о f f e r, J. Am. Chem.- Soc., 67,
2239 (1945).
762
ОБРАЗОВ UHIH ПРОСТЫХ (Г-С-СБЯЗЕЙ Ji РЕЗУЛЬТАТЕ ОБМЕЛА
и насыщенным раствором NaCl и сушат над MgSO4. Затем отгоняют раствори-
тель, а остаток перегоняют в вакууме. Выход продукта 65,5% от теоретического:
т. кип. 219е* С (1 л,н put. ст.); т. пл. 56° С.
О получении кетонов этим методом см. обзоры [345, 346].
При взаимодействии ангидридов карбоновых кислот с кадмийалкилами образу-
ются кетоны с выходом 50—70% [341]. Так же гладко ангидриды карбоновых кислот
реагируют с реактивом Гриньяра, особенно при более низких (—70° С) температурах
1347]. Реакция кадмий- или цинкалкилов с циклическими ангидридами приводит
к кетокарбоповым кислотам [341, 348, 349]. Кетокарбоновые кислоты образуются
также при взаимодействии хлорангидридов моноэфиров дикарбоповых кислот с изо-
алкнлмагнийбромидом [350]. Эфир о-толилглиоксалевой кислоты можно получить
с 50%-ным выходом по реакции хлорангидрида моноэфира щавелевой кислоты с ди-
о-толилкадмисм [341].
Эфир о-толилглиоксалевой кислоты. Раствор Гриньяра получают обычным
образом из 45 г (0,26 моль) о-бромтолуола, 5,75 г (0.24 моль) порошка магния
и около 150 мл эфира, декантируют от магния и прибавляют по каплям к пере-
мешиваемой суспензии 22 г (0,12 моль) безводного CdClg в 150 мл эфира с такой
скоростью, чтобы эфир умеренно кипел. Затем перемешивапио продолжают
до отрицательной пробы Джильмена. Для удаления солей магния реакционную
смесь, исключив доступ влаги, фильтруют через стеклянный пористый фильтр.
Фильтрат прп сильном перемешивании и охлаждении смесью сухого льда и аце-
тона прибавляют к раствору 54,4 г (0,40 моль) этоксалилхлорида в 70 мл
эфира, поддерживая температуру от —30 до —403 С. Затем температуру реакци-
онной массы поднимают до комнатной и выливают реакционную массу на смесь
70 г льда и 70 мл воды. Смесь энергично встряхивают до исчезновения запаха
этоксалилхлорида. Эфирный слой отделяют и промывают два раза 70 мл 10%-
ного раствора Na2CO3 и один раз 35 мл воды. Эфирный слой сушат пад безвод-
ным NaoSOa и после отгонки растворителя фракционируют на колонке длиной
15 см. Выход эфира о-толилглиоксалевой кислоты 23,4 г (50% от теоретиче-
ского); т. кип. 136—137° С (8 мм рт. ст.); 1,5178; = 1,1016.
Метилкетопы образуются с превосходным выходом при реакции хлорангидридов
карбоновых кислот с дихлоридом метилалюминия [351]. Лцетиленкетоны типа
ПС=ССОСН3 легко возникают при взаимодействии уксусною ангидрида с ацетилен-
магнийгалогенидом.
Октин-З-он-2 [352]. Получают этилмагнийхлорид взаимодействием этил-
хлорида в двойном количестве эфира с магнием. К нему прибавляют при пере
мешипании в атмосфере азота рассчитанное количество алкилацетилепа в эфире
и кипятят с обратным холодильником до прекращения выделения этана. Про-
должительность реакции прп загрузке 0,25 моль около 2 ч. Потом аппаратуру
вновь заполняют азотом.
Полученный таким образом гексинилмагнийхлорид (0.25 м-оль) охлаж-
дают до —30° С. К нему при энергичном перемешивании в течение 2,5 ч при-
бавляют 0,5 моль уксусного ангидрида в эфире с такой скоростью, чтобы темпера-
тура не поднималась выше —25° С. Затем реакционную массу перемешивают 2 ч
ври —30° С и затем 2 ч при —53 С в бапе со смесью льда и соли, разлагают
[345] D. A. Shirley, Org. Reactions, 8, 28 (1954).
[346] J. Cason, Chem. Rev., 40, 15 (1947); J. Cason, F. S. P г о u t, Org.
Synth., 28, 75 (1948).
347] M. 8. h ewman, A. S. Smit h, J. Org. Chem., 13, 592 (1948).
348] L. F. Fieser, F. C. No vello, J. Am. Chem. Soc., 64, 802 (1942).
349] G- Komppa, W- R ohrmann. Ann., 509, 259 (1934).
3501 C. R. Fordyce, J. R. I olin son, J. Am. Chem. Soc., 55, 3368 (1933).
351] II. Adkins, C. S с a n 1 e y, J. Am. Chem. Soc., 73, 2854 (1951).
[352] J. W. Kroeger, J. A. N i e u w I a n d, J. Am. Chem. Soc., 58, 1861
(1936).
11. ОБРАЗОВАНИЕ С—С-СВНЗЕИ ПРП ОТЩЕПЛЕНИИ Hal В ВИДЕ ГАЛОГЕНИДА
ледяной водой и фракционируют. Получают 8 г гек<инг-1 и 18 г октмн-3-она
с г. кип. 76—77° С (15 лл рт. ст.). Выход, считая на гексин-1, составил
98% ОТ теоретического.
Для синтезов ацетпленкетонов ароматического ряда применяют обычно ацетил
ниды натрия, которые могут реагировал. как. с хлорапглдридами кислот [353], те
и с их ангидридами [354|.
Бензоилфенилацетилен [355J. Фенилацетилен растворяют в пяти-или десj
тикратном количестве сухого эфира и прибавляют к рассчитанному количеств
натриевой проволоки, находящейся под слоем эфира. При этом тотчас выдел:
ется водород, а на поверхности натрия образуется зернистая пленка натри!
фенилацетилен^. Реакционную смесь время от времени встряхивают и осторожв
отгоняют эфир без доступа влаги, однако в большинстве случаев это ‘не o6j
аательно.
К эфирной суспензии патрийфснилацетилена добавляют ряссчитлинг
количество бензоилхлорида и смесь оставляют на несколько часов. Потом пр»
бавляют ледяную воду, отделяют эфирный слой, сушат его над СаС12 и фракци»
нируют в вакууме прп возможно низком давлении, так как бензоилфеннз
ацетилен довольно неустойчив. Выход продукта 74% от теоретического; т. кит
200—202“ С (15 мм рт. ст.).
Ацетилфепилацетилен [354]. Из 76 г (0,75 моль) фенплацетплеиа в 50 л*
эфира и 16,8 г натрия в 100мл эфира получают натри1(фйнилацетилеп[з54а]. Поел
добавки 100 мл эфира натрипфенилацетнлен медленно прибавляют при пере
мепнтании и охлаждении льдом к 76 с (0,75 молъ) ацетапгидрпда в 200 л
эфира. Смесь перемешивают несколько часов, потом добавляют холодную раг
баллонную НС1 и оставляют стоять до полного растворения ангидрида. Эфирнц
слой тщательно промывают раствором Na«CO3, сушат над MgSO4 и перегоняю
в вакууме. Выход продукта 58 г (55% от теоретического); т. кип. 120—125° i
(14 мм рт. ст.).
5. Синтезы нитрилов [356]
При реакции алкилгалогенидов с цианидами щелочных металлов легко обра.
зуются нитрилы:
RX + NaCN —> RCN--NaX
Высокие выходы нитрилов получают в случае первичных алкилгалогенидон
'Реакция с вторичными алкилгалогенидами протекает хорошо лишь в тех случаях, когде
н качестве растворителя применяют диметилсульфокспд |357]. Третичные алкилгало
гениды в эту реакцию не вступают.
Нитрил ундекановой кислоты [358J. В колбе с обратным холодильнике»,
нагревают на водяной бане в течение 10 ч смесь 475 г децилбромида, 150 г КСУ
1500 мл 06%-ного спирта и 250 мл воды. После охлаждения отсасывают КВг
спирт отгоняют на водяпой бане и оставшееся масло перегоняют при понижен
ном давлении. Для очистки нитрила его омыляют концентрированной НС(
[353] С. D. llnrd F. L. С о h е n, J. Am. Chem. Soc., 53, 1068 (1931).
[354] D. Nightingale, F. Wadsworth, J. Am. Chem. Soc., 67, 416 (1945).
[354a] H. Gilman. R.V. Young, J. Org. Chem., 1, 315 (1936).
[355] J. U. Nef. Ann., 308, 275 (1899).
[356] II. Brintzinger, A. Scholz, Ber., 83, 141 (1950); см. также D. T. M о w-
г у, Chem. Rev., 42, 189 (1948); V. M i grd ichian, The Chemistry of organic
Cyanogen Compounds, Reinhold Publishing Corp., New York, 1947.
[357] R. A. S m i I e у, C. Arnold, J. Org. Chem., 25, 257 (i960).
[358] R. Merckx, I. V e r h u 1 s t, P. В г u у I a n t s, Bull. Soc. chim. Belgi-
que, 42, 180 (1943); P. К u rtz, в книге Houben-Weyl, Methoden de»
organischen Cnemio, Bd. 8, 1932, S. 293.
0ВРЛ.ЮЛЛ1ШЕ ПРОСТЫХ С - Ь-СПЯ.'!I!fl II РЕЗУЛЬТАТ !•'. ОБМЕНА
ка холоду до амида, перекристаллизовывают амид из спирта п затем отщепляют
от амида иоду действием РгО.ч с образованием нитрила (II о и b <• n-W е у 1,
Mi'tliodoii tier organisclien Chemie, Bd. 8. 1952. S. 330),
Цяаппд натрия менее реакционноспособен, чем цианид калия, ио может с успе-
хом применяться во многих случаях. Например, дипитрил глутаровой кислоты
NC(Cir2)3CN получают из 1,3-дибром11ронаиа и NaCN с 80%-пым выходом [359].
Реакционная способность повышается в ряду хлорид <j бромид <j иодид.
Поэтому реакцию можно проводить селективно. Так, пз тримстилснхлорбромпда и
KCN в количестве, немного меньшем рассчитанного, получают исключительно нитрид
у-х'.тормасляпои кислсныс ггы\щом 70% от теоретического [360].
Бензмлгалогенлды очень ыадко превращаются в нитрилы. Например, из хлор-
метплмезитлена образуется мезитплацетонитрил с 89—93%-ным выходом [361].
Применение и качестве растворителя днметилсульфоксида [357] дает исключи-
тельно хорошие результаты; даже и случае вторичных галогенидов выходы достигали
70%.
2-Цианбутан. Смесь 30 г NaCN и 150 мл днметилсульфоксида нагревают
до 90° Сив течение 30 мин добавляют 0,5 моль 2-хлорбутана. При этом темпе-
ратура смеси повышается. З.ыем смесь кипятят в течение 3 ч с обратным холо-
дильником (температура поднимается до 150" С). Затем колбу охлаждают,
реакционную смесь выливают в вору и экстрагируют нитрил хлороформом
пли эфиром. Экстракт несколько раз промывают раствором NaCl, сушат над
СаСЬ п перегоняют. Выход продукта 79% от теоретического; г. кип. 125—
126° С.
Вместо алкиигалогепидов можно использовал, кислые и средние эфиры серной
кислоты [358, 364].
Для получения ароматических нитрилов из арилгалогенидов применяют, по
Роземупду — Брауну, вместо цианида щелочного металла цианид меди. Для этого
нагревают галогенид с цианидом меди, если необходимо — и присутствии небольшого
количества пиридина, при 150—250° С. Реакцию можно ускорить добавкой небольшого
количества нитрила или сульфата меди |362].
а-Нафтонитрил [363] получают из о-бромпафталппа и CuCN. В сухую
колбу емкостью 200 мл, снабженную обратным холодильником с хлоркалтщиевой
трубкой, помещают 66 г (0,32 моль) а-бромиафталпна. 35 г (0,39 моль) сухого
порошкообразного CuCN и 30 мл пиридина (высушенного над ВаО), причем
температура смеси повышается. Затем смесь нагревают в течение 15 ч прп 215—
2202 С. Темный коричневый раствор еще горячим (около 100е С.) выливают
в колбу, содержащую 150 мл водного аммиак^ = 0,90) и 1500 мл воды. Затем
прибавляют 140 мл бензола, колбу закрывают и встряхивают до полного
растворения смеси, после чего смесь охлаждают до комнатной температуры,
прибавляют 100 мл эфира и фильтруют через стеклянный мористый фильтр
(смесь разъедает бумагу). Эфирно-бензольпый слой четыре раза промывают
разбавленным водным NHS, порциями по 100 мл. Если при последней промывке
промывная жидкость окрашена, то промывают еще 100 мл NII4OH. Затем дважды
промывают 6 н. IIC1. Если образуется осадок, то его можно удалить фильтро-
ванием. Иод конец экстракт дважды промывают водой (по 100 мл) и два раза
насыщенным раствором НС]. Эфир и бензол отгоняют, а остаток перегоняют
в вакууме. Выход продукта 40—44 г (82—90% от теоретического); т. кип.
173—174е С (27 мм pm., cm.), 166—169° С (18 мм рт. ст.).
[359] С. В. Marvel, Е.-М. McColm, Org. Synth.. 5, ЮЗ (1925).
[360] С. F. П. Allen, Org. Synth., 8, 52 (1928).
[361] R. C. Fuson, N. R a bjohn, Org. Synth., Coll. v. 3, 1955, p. 557.
[362] C. F. К о e 1 s c li, A. G. Wb ilne y, J. Org. Chora., 6, 795 (1941); C. F. К o-
elsch, I. Am. Chem. Soc., 58, 1328 (1936).
[363] M. S. Newman, Org. Synth., 21, 89 (1941).
|364] M. Z i о I, IT. G. F-1 о r c h e г jr.. H. К. К i rshen, J. Am. Chem.
Soc., 68, 2743 (1946)f V. C. Spkera, C. S. Marvel, J. Am. Chem.
Soc., 55, 345 (19.33),
Ш. ОБРАЗОВАНИЕ С—С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ Hal В ВИДЕ HHal
765
Такой жо выход получают при использовании а-хлорнафталпна (нагре-
вание в течение 24 ч прп 245—250° С).
При сплавлении цианидов щелочных металлов с арилсульфонатами щелочных
металлов в результате обмена сульфогруппы образуются ароматические нитрилы. Эта
реакция применяется в основном в ряду нафталина [3(35].
Для получения ненасыщенных нитрилов, например аллилцианида из аллил-
хлорида, вместо цианида щелочного металла также лучше применять CuCN. Например,
прп кипячении указанных веществ с обратным холодильником в течение 7 ч в присут-
ствии KI (защита от попадания влаги) образуется аллилцианпд (выход 79—84%)
1366]. Для реакций винплгалогенидов можно рекомендовать такие же условия, как
и для реакций ароматических галогенидов. Например, фумаронитрил образуется
с выходом 74% из дииодэтилена [367]. Пропиолонитрил получают из ацетилениодида
действием цианида щелочного металла в ацетоне или цианида меди в ксилоле [368].
Реакции такого типа применимы также для синтеза оксииитрилов из хлор-
гидринов (выход около 80% от теоретического) [369] или а-алкоксииитрилов иа
•а-галогенэфиров и CuCN (выход 55—80% от теоретического) [370].
Ацилцианмды обычно получают нагреванием ацилгалогенидов с CtiCN без
растворителя (выход 60—87%) [371, 372], причем бромиды реагируют лучше, чем
хлериды.
Ацетилцианид [371]. Цианид меди получают из CuSO^ добавлением к нему
порциями KCN при охлаждении и энергичном перемешивании. Образовавшийся
CuCN отфильтровывают, тщательно промывают п сушат при 60—70° С. В кругло-
донной колбе с обратным холодильником на водяной бане нагревают без раство-
рителя в течение 1,5 — 2 ч 1 моль ацетилбромида и 1 моль CuCN. При добавке
ацетилбромида к CuCN реакционная масса вскипает. Ацетилцианид отделяют
от образовавшегося прп реакции CuBr перегонкой на масляной бане. Выход
продукта 85—87% от теоретического; т. кип. 93° С.
III. Образование С—С-связей в результате отщепления галогена
в виде галогеноводорода
1. Синтез Фриделя — Крафтса [373]
Реакция Фриделя — Крафтса [373] протекает по следующей схеме:
Аг-Н + ПХ Аг-Н + НХ
где R — алкил или ацил.
Этот метод имеет большое препаративное значение для введения различных
углеродсодержащих остатков в ароматические соединения.
Растворители. При проведении синтезов Фриделя—Крафтса значительную
роль играет среда. Наиболее подходящим растворителем является сероуглерод, иногда
применяют петролейный эфир. Используя особые растворители, например нитробензол,
[365] Е. С. Whitmore, A. L. Fo х, I. Am. Chem. Soc., 51, 3363 (1929).
[366] Е. R i е I г, Org. Synth., 24, 96 (1944).
[367] С. A. Ilochwalt, пат. США 2399349 (1946); С. А., 40, 4744 (1946).
|368] М. S. Newman, J. Н. W о t i z, J. Am. Chem. Soc., 71, 1292 (1949).
1-369] E. C. Kendall, B. McKenzie, Org. Synth., 3, 57 (1923);
С. H. G. Hands, B. Y. Walker. J. Soc. Chem. Ind., 67, 458 (1948).
[370] H. R. Henze, G. W. Benz, G. L. Sutherland, J. Am. Chem. Soc.,
71, 2122 (1949).
[371] W. Tschelinzef f, W. Schmidt, Ber., 62, 2210 (1929).
[372] T. S. Oakwood, C. A. Weisgerber, Org. Synth., 24, 14 (1944).
[373] Обзор: Groggins, Unit Processes in Organic Synthesis, McGraw-ШИ Book Co.,
New York, 1947, p. 759; Ч. T омас, Безводный хлористый алюминий в орга-
нической химии пер. с апгл., Издатиплит, 1949. G. Kranzle i.n, Alumi-
niumchlorid in der organischen Chemie, 3 Aufl., Verlag Chemie, Berlin, 1939;
С. C. Price, Org. Reactions, 3, 1 (1946); E. Berliner, Org. Reactions, 5,
229 (1949); A. Schaarschmidt, Angew. Chem., 37, 286 (1924);
W. В о г s c h e el al., Arm., 553, 260 (1942); 549, 238 (1941).
766
-< ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
можно изменять соотношение изомеров, если их образование возможно при конденса
ции. Выбор растворителей не очень велик, так как многие из них неустойчивы в усло-
виях данной реакции. Поэтому при проведении реакции с дешевыми ароматическими
углеводородами, нанример с бензолом или толуолом, в качестве разбавителя приме-
няют избыток углеводорода. ,
Катализаторы конденсации. А1С1з- Вопросам наиболее целесообразного со-
става А1С13 для реакции Фриделя — Крафтса посвящено большое число исследований
1374—378]. Химически чистый А1С13 часто вызывает совсем другие реакции, чем ката-
лизатор, содержащий некоторое количество основного хлорида, в то время как при-
менение значительно гидратированного А1С13 не дает удовлетворительных результатов.
Так, например, по обычно применяемым для синтеза Фриделя — Крафтса прописям
1379] из стильбендибромида и бензола под действием А1С13, который находился 1 —
2 ч на воздухе, образуется тетрафенилзтан с лучшим выходом [377], чем в случае чи-
стого А1С13. В то же время из трнхлорацетилхлорида и бензола в присутствии чистого-
AlClg с хорошим выходом образуется трифепилвиниловый спирт или (о,о)-дифенилаце-
тофенон, тогда как с влажным, уже почти расплывшимся А1С13 возникает только
ш-трихлорацетофснон [380].
Раньше большое внимание уделялось описанию удобной аппаратуры
для лабораторного получения А1С13 [381]. В настоящее время применяют исклю-
чительно продажные препараты. При открывании ампул с А1С13 следует соблю-
дать осторожность, так как опи большей частью находятся под давлением,
создаваемым хлористым водородом. Ампулу следует обернуть полотенцем,
поставить в стеклянный стакан, сделать надрез напильпиком и вскрыть при
помощи нагретой стеклянной палочки. Взяв необходимое количестбо А1С13,
ампулу нужно вновь запаять. Крупные куски продажного препарата исполь-
зуют обычно без размельчепия, так как реакция протекает настолько бурно,
что ее необходимо сдерживать, а крупные куски реагируют более' спокойно.
Топкоизмельченный А1С13 рекомендуется в тех случаях, если проводится раз-
дельное получение комплексных соединений, применяемых в реакции, или
если реакция проводится при перемешивании из-за пассивности некоторых
взаимодействующих соединений. Качество препарата проверяют по его способ-
ностп^сублнмироваться без заметного остатка.
TiCI4, SnCI4. Важным преимуществом этих более мягко действующих катализа-
торов является их растворимость в органических растворителях, например в сероугле-
роде, что позволяет проводить реакцию в гомогенной среде. Для алнилирования лучше
всего использовать TiCl4 [382], так как при этом не наблюдается полиалкилирования.
В случае ароматических углеводородов с заместителями первого рода алкильный ради-
кал под влиянием TiCL вступает главным образом в пара-положение. Для получения
циклических кетонов [383] часто применяют SnCl4. Реакцию с производными фурана-
также проводят в присутствии SnCI4.
Вопросам применимости других катализаторов для реакции Фриделя — Крафтса
посвящены многие исследования. Однако до сих пор пе найден катализатор, который
мог бы серьезно конкурировать с А1С13. Хотя реакция с хлоридом бериллия, протека-
ющая при более высоких температурах, дает хорошие выходы чистых продуктов,
этот катализатор слишком дорог [384].
[374
[375
376
377
378
379
F. Stockhausen, L. Gattermann, Вег., 25, 3521 (1892).
М. Gomberg, Вег., 33, 3144 (1900).
V. Меуег, Вег., 29, 847 (1896).
Н. В i i t z, Вег., 26. 1960 (1893).
R. Scholl, Ber., 32, 3492 (1899).
R. A n s C'hii t z, Ann., 235, 207 (1886).
___ II. Blitz, J. prakt. Chem., 142, 196 (1935).
[381] G. A, Dawson, J. Am. Chem. Soc., 50, 133 (1928)-
[382] N. M. CuiJinane, D, M. Leyshon, J. Chem. Soc., 1954, 2942.
»[383] W. E. Bachmann et al., J. Am. Chem. Soc., 64, 94 И942У 62 824 "(1940V
J. Org. Chem., 6, 36 (1941). k '
[384] H. Bredere6k et ah, Angew. Chem., 52, 445 (1939); Ber., 72, 1414 (1939)». -
in. ОБРАЗОВАНИЕ С—G-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ Hal В ВИДЕ HHal
7
По Меервейну [385, 386], можно применять BF3, который образует с простым
•алифатическими карбоновыми кислотами молекулярные соединения. Напрнмв]
уксусным ангидридом и BFS можно гладко и с хорошими выходами ацетилирова-]
ароматические и гетероциклические углеводороды и простые эфиры фенолов. О п
лучении BFg см. [386а].
Сублимированный FeCl3 вызывает различные побочные реакции, наприме
окисление, хлорирование или отщепление НС1. Его с успехом используют для дв^
кратного введения ацильных остатков в многоатомные фенолы [387], а также дл
введения mpem-бутильного остатка в о-ксилол (выход 73%) [388,. Хлористый циц
пригоден в качестве катализатора для введения ацетильной групсы в Многоатомны
фенолы [389]. Проведено сравнительное исследование эффективности различны
металлгалогенидов в качестве катализаторов [390].
Вместо хлоридов металлов в определенных случаях можно применять так»
другие катализаторы. Например, с 90%-ным выходом протекает реакция нафталив
•с бензоилхлоридом при 180—200° С в присутствии каталитических количеств пята
окиси фосфора [391]. В ряду тиофена хорошие результаты дает этот метод [392
а также применение фосфорной кислоты (выходы свыше 90%) [393, 394]. Хлорокис
фосфора катализирует реакцию' многоатомных фенолов с ацетилхлоридом [395
В некоторых случаях для алкилирования используют фтористый водород [396
применяется также амальгамированный алюминий [397].
Ароматические илн гетероциклические компоненты. Реакция Фриделя -
Крафтса легко протекает при взаимодействии хлорангидридов и ангидридов карбоновы
кислот со всеми ароматическими и многими гетероциклическими соединениями, особенн
•с производными тиофена, фурана и пиррола. Пиридин в эту реакцию по вступав
[398, 399]. Углеводороды, содержащие заместители второго рода, реагируют с тр\
дом, а некоторые из них, например иитропроизводные углеводородов, вообще а
вступают в эту реакцию. При попытках осуществить реакцию в более жестких услс
виях происходит восстановление нитрогруппы или хлорирование ядра [400]. Не
оборот, ароматические углеводороды с заместителями первого рода (алкилпроизвод
ные 1алогениды, эфиры фенолов, замещенные амины и фенолы) реагируют легкс
Эфиры фенолов и карбоновых кислот, получаемые из фенолов без применения ко®
денспрующего средства (стр. 349, 351), могут перегруппировываться по Фрису (стр. 865
в ацилфенолы. Пассивирующее действие одного заместителя второго рода снимаете
другими заместителями; так, 4-нитроанизол [373] или эфир анисовой кислот]
[38 .5] Н. Meer wei п, Вег., 66, 411 (1933); Н. MeeJwein, D. Vossen
I. prakt. Chem. [2], 141, 149, 156 (1934); H. D. Hartough, A. I. Kosak
J. Am. Chem. Soc., 70, 867 (1948); H. G. Walker jr., I. J. Sanderson
C. R. Hauser, I. Am. Chem. Soc., 75, 4109 (1953).
[386] Обзор: D. Kastner, Neuere Methoden der prSparativen organischen Chemia
Veriag Chemie. 1949, S. 413.
[386a] E. Krause, R. Nits ch e, Ber., 54, 2784 (1921); H. S. Booth
K. S. Willson, J. Am. Chem. Soc., 57, 227,3 (1935).
[387] M. Nencki, Ber., 30, 1766 (1897); 32, 2418 (1899).
[388] B. W. Larner, A. T. P et ers, J. Chem. Soc., 1952, 680; E. I i i i n £
worth, A. T. Peters, J. Chem. Soc., 1952, 2730-
S. R. Cooper, Org. Synth., 21, 103 (1941).
О. C. Dermer, J. Am. Chem. Soc., 63, 2881 (1941).
S. Gru carevic, V. Merz, Ber., 6, 1240 (1873).
W. Steinko pf, Ann., 413, 343 (1917); 424, 1 (1921).
II. D. Hart ou gh, A. I. Kosak.'L. G. Conley, J. Am. Chem. Soc., 60
3093, 3096, 3098 (1947).
A. I. Kosak, H. D. Hartough, Org. Synth., 28, 1 (1948).
H. v. Euler, H. Hasselqnist, В. H 6 g b e r g, Ark. Kerni, 20 A.
№ 20 (1945).
J. H. Simons, S. Archer, J. Am. Chem. Soc., 60, 2952, 2953 (1938).
L. I. D i u g u i d, J. Anf. Chem., Soc., 63, 3527 (1941); W. W. Hartman.
R. Phillips, Org. Synth., Coll. v. 2, 1955, p. 232.
С. M. I e p h с 0 11, J. Am. Chem. Soc., 50, 1189 (1928).
K. Thomas, D. Jerchel, Angew. Chem., 1958, 719.
П. Gilman et al., J. Am. Chem. Soc., 57, 907 (1935).
7(58
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
в отличие от их аналогов, не содержащих алкоксшрупп, способны к ацилированию
пли алкилированию.
Редким исключением из этого правила являются некоторые очень реакционно-
способные ароматические соединения, которые несмотря на наличие в их молекулах
заместителей второго рода легко вступают в реакцию Фриделя — Крафтса, напри-
мер, эфиры пироелпзевой [401] или а-нафтойной кислот.
Образование изомеров. При ацетилировании ароматических соединений с за-
местителями первого рода получаются почти исключительно о- и п-пропзводные,
а при алкилировании — также ж-производные. Уже рассматривавшееся выше влия-
ние растворителя на образование изомеров отчетливо проявляется при ацетилиро-
вании нафталина: используя в качестве растворителя нитробензол, получают 0-про-
изводные, в сероуглероде, особенно при низких температурах,—а-производные,
в лигроине — смесь а- и 0-производпых [402].
Естественно, что у замещенных нафталинов и более высококонденсировапных
соединений число изомеров возрастает. Более подробные сведения об ориентации
заместителей при реакции Фриделя — Крафтса приведены в обзоре [373].
При введении достаточно длинных боковых цепей получают изоалкилпроиз-
водные даже тогда, когда в качестве исходного соединения используют нормальные
галогениды. Например, из zt-пропилхлорида и бензола образуется изопропилбензол.
Заведомо чистые соединения получают из максимально разветвленных галогенидов,
например трепг-амилбепзол из бензола и трет-амялхлорида.
Проведение реакции. Порядок добавления реагентов. Три
основных реагента — катализатор, галогенид (или ангидрид кислоты) и ароматиче-
ское соединение могут вводиться в реакцию различным образом. Например, раство-
ряют или смешивают оба органических соединения и прибавляют порциями А1С13.
Этот старый способ, неудобный из-за чувствительности к влаге А1С1Э, почти не имеет
преимуществ перед другими. Лучше смешивать А1С1Я с одним из органических
реагентов и растворителем и приливать в эту смесь второй органический реагент
или его раствор. При этом следует установить, что целесообразнее смешивать с ката-
лизатором: углеводород или галогенид.
Если катализатор смешивают с галогенсодержащим соединением, то следует
учитывать возможность образования комплексов, главным образом с хлоранги-
дридами кислот. Иногда такой способ проведения реакции даст преимущества, таю
как образующиеся комплексные соединения менее энергично реагируют с угле-
водородом (модификация Перрье [403]). И наоборот, лучше смешивать катализатор
с углеводородом, если углеводород одновременно является растворителем, как это
часто бывает в случае бензола.
Обычно, если необходимо использовать инертный растворитель, рекомендуется
следующий способ проведения реакции: суспендируют или растворяют конденси-
рующее средство в разбавителе и прибавляют постепенно смесь или раствор обоих
реагентов. Этот способ целесообразно применять главным образом тогда, когда одно
из реагирующих соединений неустойчиво. Так, по Вейганду апетилфуран получают
из фурана, ацетилхлорида и SnCl4 в потролейном эфире со значительно лучшим
выходом, чем по методу Рейхштейна [404], т. е. приливанием ацетилхлорида к смеси
фурана, петролейного эфира и А1С13. Это объясняется тем, что в последнем случая
в присутствии А1С1Я фуран, находящийся вначале в избытке, частично осмоляется.
Если же эквимолекулярные количества фурана и ацетилхлорида прибавлять к ка-
тализатору, осмоления но происходит. Образовавшийся ацетилфуран или его мо-
лекулярное соединение с А1С1Я устойчивы.
Оптимальное количество катализатора указано пе всегда. Для синтеза кетонов,
например, применяют не менее эквимолекулярного количества А1С13, чаще 30%-ный
избыток.
Протекание реакции. Хорошие результаты получаются, если
скорости реакции и прибавления реагентов согласованы так, что в реакционной среде
[401] И. G i 1 m а п, N. О. С а 11 о w а у, J. Am. Chem. Soc., 55, 4197 (1933).
[402] G. Baddeley, J. Chem. Soc., 1949 Suppl., 99; N. P. Buu-Hoi,
P. Cagnia n t, Bull. Soc. chim. France [5], 12, 307 (1945); M. Chopin,
Bull. Soc. chim. Franco [4], 35, G13 (1924).
[403] D. T. Mo wry, M. Benoll, W. E. H u b e r, J. Am. Chew. Soc., 68,-
1105 (1946). ,
[404] T. R oichst ein, Helv. chim. acta, 13, 357 (1930).
Ш. ОБРАЗОВАНИЕ С С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ Hal В ВИДЕ HHal
нет большого избытка исходных соединений. Протекание реакции контроля
по выделению хлористого водорода. Если проводят неизвестную реакцию, то о,
чинают при охлаждении льдом или даже охлаждающей смесью. (Но при этом сл<
убедиться в том, что реакция идет и при низких температурах, для чего на i
торос время прекращают охлаждение и следят за выделением газа или повыше
температуры,) В противном случае реакция может начаться внезапно и возм<
выброс реакционной массы. Иногда реакцию приходится начинать при повыше
температуре и проводить затем при охлаждении. Хорошие результаты могут
получены также, если реакционную массу к концу реакции нагревать. Это возм<
при использовании в качестве растворителя низкокипящего сероуглерода. Во
других случаях нагревания следует избегать.
Часто бывает достаточно периодически встряхивать содержимое колбы, од
механическое перемешивание практически более удобно. Если в конце реакции е
по нужно нагревать, то встает вопрос, как долго после окончания прибавления
агентов следует выдерживать или перемешивать смесь. Этот период не следует слиц
растягивать, во всяком случае не оставлять па ночь. Часто рекомендуют выждад
прекращения выделения хлористого водорода. Однако полное прекращение выдел,
хлористого водорода, особенно прп повышенных температурах, почти никогда н<
стигается. При исследовании условий синтеза кегопов [405] установлена возможн
протекания нежелательных побочных реакций. Так, для ацетофенона длительное
гревание реакционной смеси с обратным холодильником приводит к образова
высококипящих продуктов, в основном к дипнону, [СвН5—С(СНЭ)=СН—СО—Сч
Образование дипнона из двух молекул ацетофенона, вероятно, аналогично сиа
бензальацетофенона из смеси ацетофенона, бензальдегида и А[С13 в сероугле
(выход 90% от теоретического). Имеются данные о том, что при пспользов?
недостаточных количеств А)€13 доля побочных реакций повышается, напрх
упомянутое выгпе образование дипнона.
Разложение реакционной массы. После окончания реаь
Фриделя — Крафтса продукты реакции находятся большей частью в форме ад,
тов с Л1С13. При встряхивании аддуктов с водой происходит разложение их на j
понепты. Смесь сильно разогревается, поэтому вместо воды обычно применяют
Если продукты реакции выделяют перегонкой с водяным паром, то в перегон:
колбу вносят мелкораздробленныи лед, прибавляют еще неразложенную реакц]
ную массу и подкисляют НС) до полного растворения гидроокиси алюминия. Мо
также сразу внести в колбу смесь льда и дымящей НС), причем возникает сила
охлаждение. Если после разложения смеси продукт экстрагируют эфиром или j
гими растворителями, то лучше всего добавлять лед в широкогорлую делитель:
воронку. Во всяком случае, перед добавлением льда и ПС1 к реакционной св»
следует убедиться в том, что этот процесс, связанпый со значительными места:
перегревами, не вызывает разложения продукта реакции. Если же высокая j
кость реакционной массы исключает возможность выделения продукта реакции х
гпм способом, то, чтобы избежать бурного разложения, лсд прибавляют в один пр
к заранее охлажденной смеси.
Ацилирование
Ацилирование ароматических соединений. Ацплнровацпс по Фриделю—Kpaq^
подробно описано в элементарных руководствах, здесь приведено только
примера.
н-Амилацетофенон (по Вейганду). В колбу, снабженную мешалкой и
пельиой воропкой, помещают 20 г А1С13, 100 мл C.S2 и прибавляют по кап^
при охлаждении льдом и перемешивании смесь 15 г н-амилбензола и 10.
ацетилхлорида. Образовавшийся светло-желтый раствор Тотчас, несма’
на возможно продолжающееся выделение хлористого водорода, вылив«
нэ смесь льда и концентрированной НС1 п добавляют эфир до отделения мае?
нистото слоя; маслянистый слой промывают водой до нейтральной реакэ
и сушат над СаСЯг- Затем отгоняют смесь растворителей и фракциояиру-
Выход и-амилацето^енона 16 г (83?4 от теоретического); т. кип. 132—133
[405] N. О. С а 11 о w а у, L. D. G г е е n, J. Am. Chem. Soc., 59, 809 (1937).
49 Заказ 1S35
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕИ В РЕЗУЛЬТАТЕ ОБМЕНА
(4 мм рт. ст.). Если реакционную массу после добавки смеси реагентов оста-
вить на длительное время, то выходы сильно уменьшаются.
трачс-Дибензоилэтилен (из хлорангидрида фумаровой кислоты и бен-
зола) [406]. К смеси 31 г тонко размолотого A1C1S и 250 мл бензола медленно
при перемешивании (с обратным холодильником) Прибавляют 18 г хлорангид-
рида фумаровой кислоты. После перемешивания в течение 1 ч при комнатной
температуре смесь выливают на лед, бензольный слой промывают раствором
NagCOj, сушат и концентрируют. Дибензоилэтилен выделяется из концентри-
рованного бензольного раствора в виде светло-желтых игл. При дальнейшем
испарении бензола получается продукт, окрашенный имеющимися примесями
в красноватый цвет. Кристаллы перекристаллизовывают из спирта (кипячение
с животным углем). Выход продукта 20,5 г (74% от теоретического); т. пл.
111° С.
Из пирокатехина и хлорацетилхлорвда кипячением в течение 6 ч с хлорокисью
фора в бензоле можно получить с выходом 85% 3,4-диокси-ю-хлорацетофенон
5]. Описано также ацилированне ацетанилида в пара-положение (выход 79—83%)
рацетилхлоридом и А1С13 в сероуглероде [407].
Дикетоны легко образуются нз дихлорангидридов двухосновных карбоно-
: кислот. Так, например, из дихлорангидрида адипиновой кислоты получают
ензоилбутан (выход 31% от теоретического) [408]. Образование дикстонов из
еров кетенов протекает по следующей схеме [409]:
сн2=с—о о о
1 1 A1G], A fl
СН2-С=О + СвНб ---’* С8Н6-С-СН2-С-СНэ
73%
Для введения ацильных радикалов в фенолы и иу эфиры используют
гез Губена — Геша [410]. В присутствии НСГ нитрилы реагируют с фенолами по
ге:
RCN4- СвН4(ОН)2 —К-С-СвН3(ОН)2 ' R-CO-CSH3(OH)2
NH • НС1
В качестве промежуточных продуктов образуются гидрохлориды кетиминов,
>олиз которых дает соответствующие кетоны. Растворителем' обычно служит
). В некоторых случаях рекомендуется применять в качестве катализатора ZnCl2.
Синтез циклических кетонов [4111, Замыкание кольца в орто-положение про-
>дит легко и часто с очень хорошими выходами, если может образовываться
|- или шестичленный цикл. Получающийся при этом цикл может содержать также
] J. В. С о и a n t, R. Е. L u t z, J. Am. Chem. Soc., 45, 1303 (1923); N. Camp-
bell, N. M. Khanna, J. Chem. Soc., 1949, Suppl. 33.
]J. L. Leiserson, A. W e i ss b erger, Org. Synth., 28, 26 (1948);
N. J. Leonard, A. M. Hyson, J. Am. Chem. Soc., 71, 1961 (1949).
] R. C. Fuson. J. T. Walker, Org. Synth., Coll. v. 2, 1955, p- 169.
] А. В. В oese jr., Ind. Eng. Chem., 32, 16 (1940).
] К. II о e s c h, Ber., 48, 1122 (1915); К. H о e s c h, Th. v. Zarzecki,
Ber., 50, 462 (1917); J. Houben, Ber., 59, 2878 (1926); обзорная статья
P. E. S p о e г r i, Л. S. Du В о i s, Org. Reactions, 5, 387 (1949).
J Обзорная статья: W. S. Johnson, Org. Reactions, 2, 114 (1944).
III. ОБРАЗОВАНИЕ С—С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ Hal В ВИДЕ HHal 77J
гетероатомы. Из имеющихся в литературе многочисленных примеров здесь приво-
дятся только некоторые.
Исходное вещество
а-Нафтилацетил хлорид
Хлорангидрид фенилтиогликолевой кислоты
Хлорангидрид 3-фенилпимелиновой кис-
лоты
Анилид 0-хлорпропионовой кислоты
2-(4'-Метилбенэил)-бензойная кислота
2-Б ром-5-метоксигидрокОричная кислота
Бромангидрид о-бромметилбензойной кис-
лоты + бензол
Продукт реакции
Ацетонафтепоп (412]
Тиоиндоксил [412]
р-(4-Тетралон-1)-пропионовая
кислота [413]
Тетрагидрохинолон-2 [414]
2-Метплантрон-9 [415]
4-Бром-7-метоксииндапон-1 [416
Антрон [417]
В иачестве примера приведен синтез бензоциклооктапона [418], что одновре-
менно является примером синтеза по методу разбавления.
Бензоциклооктанон. В колбу емкостью 4 л, снабженную мешалкой и обрат-
ным холодильником, помещают 100 г (0,75 моль) тонко растертого А1С13 и 1750 мл
CS2, перегнанного над А1С13. Затем при перемешивании и энергичном кипячении
с обратным холодильником прибавляют ио каплям в течение 40 ч 45,6 г (0.216 моль)
хлорангидрида е-фенилкапроновой кислоты в 1250 мл CS2 через обратный
холодильник таким образом, чтобы стекающий растворитель разбавлял рас-"
твор хлорангидрида. Через некоторое время реакционная смесь окрашивается,
а на стенках колбы образуется темио-коричневая корка. После окончания
прибавления хлорангидрида смесь кипятят еще 2 ч, затем отгоняют раствори-
тель и гидролизуют остаток в колбе, добавляя 1 кг ледяной кашицы. Продукт
исчерпывающе отгоняют с водяпым паром, дистиллят (около 5 л) насыщают
NaCl и экстрагируют продукт эфиром. После отгонки эфира кетоп перегоняют
в вакууме при 146—148® С. Выход продукта 25,9 г (68% от теоретического).
При более тонкой разгонке получают бензоциклооктанон в виде бесцветной
жидкости с т. кип. 148—148,5° С (12 мм рт. ст.).
В качестве катализатора при синтезе подобных циклических соединений при-
меняют также фтористый водород [415], полифосфорную кислоту [416], хлоронись.
фосфора [419] или хлорид цинка [420].
Ацилирование алициклических соединений или олефинов. Подробно исследо-
вана реакция Фриделя — Крафтса циклопарафинов с ацетилхлоридом [421]. Уста-
новлено, что в зависимости от условий реакции образуются насыщенные или не-
насыщенные кетоны, а При использовании в качестве исходного соединения цикло-
гексана протекают перегруппировки с образованием производных метилциклопен-
тапа (см. также [422—424]).
[412]
[413]
414
415
416
417
418
419
420
421
[422
[423
[424
49*
Friedlaender, Fortschritte der Teerfarbenfabrikation, 10, 199
(1910—1912); 9, 562 (1908-1910).
R. H. Manske, J. Am. Chem. Soc., 53, 1104 (1931).
F. Mayer et al, Ber., 60, 858 (1927).
L. F. F i e 3 e г, H. Heymann, J. Am. Chem. Soc., 64, 376 (1942).
H. R. Snyder, F. X. Werber, J. Am. Chem. Soc., 72, 2965 (1950).
F. Mayer, W. Fischbach, Ber., 58, 1251 (1925).
R. Huisgen, W. Rapp, Ber., 85, 826 (1952).
L. Goldberg, R. Robinson, J. Chem. Soc., 1941, 575.
A. T. M a p ч ев с кий, M. И. У ш а к о в, ЖОХ, 10 (72), 1369 (1940).
N. D, Z е 1 i n s к у, E. M. T a r a s s о w a, Ann., 508, 115 (1934).
C. D. N e n i t z e 3 c u et a]., Ann., 491, 189 (1931); Ber., 66, 1097 (1933).-
C. D. Nenitzescu, I. P. Can t un iari, Ber., 65, 1449 (1932).
H. Hopff, Ber., 65, 482 (1932).
772 ОБРАЗОВАНИЕ ПРОСТЫХ С-С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
реакция Фриделя — Крафтса возможна также для циклических олефинов,
при этом в качестве катализатора применяют SnCI4 [425, 426]:
СвН5СН.2СО
С1Н„СПгСОС1 + f'''i---> Y)
Реакция хлорангидрида фенплуксусной кислоты с циклогексеном проходит
с выходом ниже 30%. Более высокие выходы достигаются при использовании ацетил-
хлорида и хлористого цинка в качестве катализатора (например, 1-ацетилцикло-
гексен-1 образуется с выходом 60% от теоретического [427, 428]) пли уксусного ан-
гидрида и хлористого цинка [429].
1-Ацетилциклогексен-1. В колбу с мешалкой Хершберга, обратным холо-
дильником с хлоркальциевой трубкой, капельной воронкой и термометром, по-
груженным в реакционную смесь, вносят 123 г (1,5 моль) циклогексена и 260,5 г
(1 моль) SnCl4. Колбу охлаждают льдом и прибавляют по каплям в течение
30 мин 102 г (1 моль) 99—100%-ного уксусного ангидрида таким образом, чтобы
температура реакционной смеси колебалась в интервале 25—35° С. Затем реак-
ционную смесь в течение 15 мин перемешивают на ледяной бане, выливают
на 300—400 г раздробленного льда и экстрагируют продукт эфиром. Эфирный
раствор промывают раствором NaHCO3 и водой и сушат над CaClo. Эфир отго-
няют, а остаток фракционируют (с колонкой) в вакууме. Выход продукта 6.7 г
(54% от теоретического, считая на ацетангидрид); т. кип. 65—69° С при 5 мм
рт. ст. (202,5° С при атмосферном давлении); т. пл. семикарбазона 220° С;
т. пл. оксима (из водного спирта) 58—59° С. Побочными продуктами являются
хлорциклогексан (т. кип. 142° С) и циклогексилацетат (т. кип. 174—175° С).
С хлорангидридами кислот реагирует также ацетилен [430], давая р-хлор-
винилкетоны с выходом 62—80% :
RCOCl-f-CHsCH —> НСОСН=СНС1
Алкилирование [431, 373]
Алкилирование по Фриделю — Крафтсу не имеет такого препара гнилого зна-
чения, как ацилирование. В качестве катализатора вместо более ангинного А1С13 *
используют TiCl4 [382], который хорошо растворяется в органических средах, не
вызывает полиалкилирования и способствует введению алкильного остатка только
в пара-ноложопие к уже имеющемуся заместителю. Алкилирующими средствами
могут служить аллилхлориды или олефины, а также спирты. При использовании
спирта необходимо эквимолекулярное количество TiCJ4, а и случае алкилхлоридов
тли олефинов достаточно его следов. Можно использовать также FeCl3, например,
грп введении пгретв-бутильиою остатка в о-ксилол [388] выход продукта достигает
73%. В качестве алкилирующих средств наряду с галогенидами применяют также
’ См. также Ч. Томас, Безводный хлористый алюминиь в органической
химии, пер. с англ., Издатиилит, 1949.
425] И. Borgs, Вег., 67, 238 (1934).
'426] W. С о о k, L. Ilowott, J. Chem. Soc., 1933, 1098.
427] R. E. Christ, R. C. Fuson, J. Am. Chem. Soc., 59, 893 (1937).
428] L. Ruzicka, D. R. К о о 1 1> a a s, Л. II. W i n d, Helv. chim. acta,
14, 1157 (1931).
429] E. E а г 1 Royals, С, M. Hoiidr y, J. Org. Chem., 15, 1147 (1950).
430] С. С. P г i c c, J. A. 1’appalardo, J. Am. Chem. Soc., 72, 2613 (1950).
431] Обзорная статья: С. C. Price, Org. Reactions, 3, 1 (1946); A. W. Fra uc is,
Chom. Rev,, 43, 257 0948); O. Calloway, Chem. Rev., 17, 327 (1935);
С. C. Price, Chem. Rev., 29, 44 (1941).
1П. ОБРАЗОВАНИЕ С— С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ Hal В ВИДЕ НПа! 773
>фиры n-Tiwijолсулт.фокмслоты [432 — 434[. Реакции олефинов, также применяемых
вместо галогенидов, протекают таким образом, как будто в процессе реакции про-
исходит присоединение хлористого водорода. Так, пропилеи реагирует как изопро-
пплхлорид, а изобутилен — как трет-бутилхлорид.
При алкплировапии соединениями, содержащими более двух атомов углерода,
всегда образуются мропзподныс с разветвленной боковой цепью. Например, при ре-
акции н-пропилхлорида с бензолом получают изопропилбензол, а не н-пропил-
бензол. Однако в некоторых случаях алкилирование протекает без изомеризации:
если реакцию проводят при низких температурах или с соединениями, содержащими
длинные углеродные цепи |432. 433].
Обычно для синтеза алкплароматических соединений с неразветвленной боко-
в ой цепью применяют реакцию Вюрца — фиттига. Оба метода дополняют друг друга.
Октадецилбепзол [432]. От 156 г бензола отгоняют для его осушки 39 г; оста-
ток смешивают с 42,5 г (0,1 .чоль) н-октадецил-л-толуолсульфопата и 13,3 з
(0,1 лола) А1С13 и перемешивают 15 ч при комнатной температуре, 10 ч при 50—
55° С и 14 ч при комнатной температуре. Темно-оранжевую смесь переносят в смесь
льда (избыток) л НС1. Органический слон отделяют, промывают теплой разбав-
ленной НС1, сушат над СаС12 п отгоняют бензол. Остаток перегоняют в вакууме
водоструйного насоса и затем при остаточном давлении 0,25 м.ч рт. ст. Выход
продукта 24.2 г (73% от теоретического); т. кин. 155—160^ С (0,25 лич рт. ст.).
Для синтезов алкплбензолов и алкилнафталивов по Фриделю — Крафтсу
в качестве катализатора можно применять также амальгамированный алюминии
[397], который непосредственно грред реакцией активируют алкилхлоридом. В не-
которых случаях эта реакция протекает с хорошим выходом, причем образования
смол и побочных продуктов по наблюдается.
Внутримолекулярным алкилированием можно получить циклические соеди-
нения. Прп циклизации хлорацетилдифениламина нагрсвапием с Л1С13 получается
N-фенилоксиндол с выходом 90% от теоретического [435]:
С1-СН2
I
с6н5
Св^б
Прп действии формальдегида и хлористого водорода на фепилэтиловый спирт
образуется изохроман с выходом более 90% от теоретического [436]. Возникающий
в качестве промежуточного продукта хлорметилфеппловый эфир циклизуется с от-
щеплспием хлористого водорода даже без добавки специальных катализаторов:
СН2
Il I Z
\Z он сн2
При реакции ароматических слсцппонпй с циклическими С1ерически напря-
женными эфирами образуются соответствующие оксиялкилпроизнодные. Из окиси
1432] D. Л. S h i г 1 о v, J. R. Z i е I z jr., J. Am. Chem. Soc., 75, 6333 (1953).
[433] ]j. Gil m an, J. Л. V. T u г c k jr., J. Am. Chem. Soc., 61, 47S (1939).
[434] W. J. 11 ic]i 1 в bo t t о га, N. W. Roger s, J. Chem. Soc.t 1957, 4124.
[435] P. Chovin, J. Ciintha r 1, Bull. Soc. chim. France [5], 12, 100 (1945).
[436] Л. llf'echo, E. Schmit z, Rer., 89, 1254 (1956).
774
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
атилена и бензола с выходом 70% от теоретического получается фенилэтиловый спирт
[437]
/СНгСНзОН
| 11+п3с-сн3 z
ох
в то время как из триметиленокслда и бензола образуется гидрокоричный спирт [438]-
Лактоны также вступают в аналогичную реакцию. Например, из бутиролактона-
и бензола в присутствии 0,3 моль А1С13 получают с выходом 73% от теоретического
у-фонилмасляную кислоту, а в присутствии 0,6 моль А1С13 — тетралон с выходом
66% от теоретического [439], Описан также синтез замещенных пропионовых кислот
при помощи р-лактонов [440].
Особенности реакции Фриделя — Крафтса
Замена ацилгалогепидов другими соединениями. Вместо хлорангидридов кар-
боновых кислот иногда применяют также соответствующие ангидриды карбоновых
кислот или даже сами карбоновые кислоты. Ракция с ангидридами протекает по-
схеме:
СвНв+(СН8СО)2О —> СвН5СОСПз+СНзСООН
Очень часто, например в случае ацетофенона, выходы продуктов при этом зна-
чительно выше, чем при реакции с соответствующими хлорангидридами кислот. В ка-
чество мягкого конденсирующего средства здесь особенно пригоден трифторид бора
[385, 386].
2-Ацетилтиофен, К охлажденной льдом до 10° С смеси 252 г (3 .ноль) тио-
фена и 107 е (1 моль) 95%-ного уксусного ангидрида прибавляют 4 г комплекса-
BF3 с метиловым спиртом (60% BFg). При этом температура сразу по-
вышается до 13° С, а через 5 мин понижается до 5’ С, поело чего ледяную баню
удаляют и колбу нагревают в течение 2 ч при 50* 5 С. Затем к реакционной смеси
прибавляют 200 мл воды и перемешивают 15 мин. Нижний органический слой
отделяют, промывают 10%-ным раствором Na2CO3 до нейтральной реакции
и перегоняют. Получают 180 г непрореагировавшего тиофена и 84 г (70%
от теоретического) 2-ацетилтиофспа; т. кип. 87 —88° С (8 .«.к рт. ст.)\ п£
1,5666.
2-Ацетилтиофен можпо получать в присутствии 85%-ной фосфорной
кислоты [441]. В качестве катализатора применяют также чистую фосфорную-
[394] или серную [442] кислоты.
При реакции циклических ангидридов дикарбоновых кислот (см. обзор [373])
получаются соответствующие котокислоты. Например, из ангидрида фталевой кис-
лоты и бензола образуется о-бензоилбензойная кислота с выходом более 90% от
теоретического [443]. Взаимодействие янтарного ангидрида с бепзолом дает 0-бен-
зоилпропионовую кислоту [444], а с анизолом в присутствии А1С13 — р-(п-анизоил)-
[437] Н. Hopff, К. Koulen, Вег., 85, 897 (1952).
[438] S. Searles, J. Am. Chem. Soc., 76, 2313 (1954).
[439] W. E. T г u с e, С. E. О 1 s о n, J] Аш. Chem. Soc., 74, 4721 (1952); С. E. 0 1-
s о n, A. R. В a d e r, Org. Synth., 35, 95 (1955); W. L. M о s b y, J. Org. Chem.-.
18, 964 (1953).
[440] J. Ilarley-Mason, Chem. a. Ind., 1951, 886; J. Chem. Soc., 1952, 2433;
T. L. Gresham et al., J. Am. Chem. Soc., 73, 2345 (1951).
[441] J. Kellett, H. E. Rasmussen, Ind. Eng. Chem., 40, 384 (1948).
[442] S. S. I s г a e 1 s t a in, H. Stephen, J. South African Chemical insti-
tute, 26, 41, 49 (1943); 27, 15 (1944).
[443] C. Friedel, J. M. Crafts, Ann. Chinlie [6], 14, 446; Frdl., 16, 376 (1931);
G. Heller, Angew.» Chem., 19, 669 (1906); Ber., 41, 3631 (1908).
[444] S. Gabriel, J. Colman, Ber., 32 , 395 (1899).
Ш. ОБРАЗОВАНИЕ С—С-СВЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ Нл1 В ВИДЕ ННа!
775
пропионовую кислоту с выходом 93% от теоретического [445]. Реакция малеинового
ангидрида с .и-ксилолом легко проходит в тетрахлорэтапе в присутствии А1С18 при
О3 С с образованием р-(2,4-диметилбензоил)-акриловой кислоты (выход 91% от тео-
ретического) [446].
Реакция с двуокисью углерода протекает не однозначпо; при взаимодействии
с бензолом в присутствии А1С13 наряду с бензойной кислотой образуется бензофенон.
Ацилирование можно осуществлять также непосредственно карбоновыми
кислотами. Многоатомные фенолы реагируют уже в присутствии ZnCl2 [389]. Прн
взаимодействии избытка толуола и уксусного ангидрида в присутствии избытка А1С1а
выход кетона выше, чем следовало бы ожидать по уравнению:
С6Н5СН3+(СНЯСО)2О —> СН3СвН4СОСНз+СНзСООН
11а основании этого был сделан вывод, что образовавшаяся уксусная кислота
с избытком А1С1а дает ацилирующий агент. Такое предположение было подкреплено
наблюдением, что из смеси уксусной кислоты и А1С13 отгоняется с 20%-ным выходом
ацетилхлорид [447]. Вследствие этого оказалось возможным осуществлять ацети-
лирование уксусной кислотой в присутствии А1С13.
Ацетофенон [447]. Нагревают при 86° С (т. кип. бензола) в течение 5,5 ч 390 г
(5 моль) бензола, 60 г (1 моль) ледяной уксусной кислоты и 400 г А]С1Э. Выход
ацетофенона 77 г (64% от теоретического).
Этот метод можно распространить на ароматические карбоновые кислоты [447].
Так, из мезптилена п уксусной кислоты в присутствии смеси фосфорного ангидрида
и кизельгура [394, 448] получают ацетомезитилен с выходом до 82,5%.
Следствием относительно высокой температуры реакции при использовании
этеге метода может быть образование побочных продуктов. Например, при ацетили-
ровании нафталина образуется дипафтил.
При выборе метода следует принимать во внимание его экономичность.
При дешевых исходных веществах можно применять уксусную кислоту, при-
миряясь с незначительным выходом продуктов, чего не следует делать при
ацилировании трудно доступных веществ, например тиофена. В тех случаях,
когда ацилируемое соединение или ацилирующий агент является цепным соеди-
нением, рекомендуется метод, дающий высокие выходы.
Реакция с галогенидами более сложного строения. Фосген может реагировать
с отщеплением одного или двух атомов хлора. При этом образуются хлорангидриды
карбоновых кислот или карбоновые кислоты, или же кетоны [449]. Например, нз
диметнланилина и фосгена без катализатора получают п-диметилампнобензойн$то
кислоту с выходом 50% от теоретического [450].
При использовании оксалплхлорида образуются дикетоны (бензол —► бен-
зил). Однако в условиях реакции оксалилхлорид частично разлагается с выделением
фосгена, что может привести к хлорангидриду или монокетону [451]. С несимметрич-
ным дитолилэтиленом, например, оксалилхлорид реагирует с образованием 0,р-ди-
толилакрплог.ой кислоты (выход 80% от теоретического) [4521.
[44.?1 D. G. Thomas. А. Н. Nathan, ,Т. Am. Chem. Soc., 70. 331 (1948).
[446] D. Papa et al., J. Am. Chem. Soc., 70, 3356 (1948); О. Grummitt,
К. 1. В с с к е г, С. М i е s s е, Or?. Synth., 29, 11 (1949).
j 447] Р. Н. Groggi п s, R. IT. N я g е 1. A. J. S t i г t о n, Ind. Eng. Chem.,
26, 1313, 1317 (1934).
[448] G. М. К о s о 1 а р о f f, J. Am. Chem. Soc., 69, 1651 (1947).
[449] C. Liebermann. M. Z sulfa. Ber., 44, 202 (1911); 45, 1186 (1912);
TI. Staudinger. Ber., 41, 3558 (£908).
[450] D. ч. в rosl о w, J. Am. Chem. Soc., 72, 4244 (1950).
1451] II. G. Latham jr., E. L. Mav, E. Mosettig, J. Am. Chem. Soc., 70,
1079 (1948).
]452] F Bergmann et al., J. Аш. Chem. Soc., 70, 1612 (1948).
776
ОВРЛЗОЛАШП: ПРОСТЫХ 0—С-СВИЗЕЙ в РЕЗУЛЬТАТЕ or.MElU
Интересно применение оксалпдхлорида для синтеза мстнлтлопзатина с хорошим
ВЫХОДОМ
п3сх
С0С1
НзС.
СО cl
ИзСх
С0С1
о
Хлорангидриды эфиров дикарбоновых кислот также могут реагировать по Фри- ,
делю Крафтсу. Так, из эфира монохлорапгидрида щавелевой кислоты образуется
эфир арплглиоксалевой кислоты [454, 455]
АгН + С1СОСООС2Н5 —* ЛгСОСООС2Н5
который можно использовать, например, для получения арплуксуспых кислот [455}
или альдегидов (стр. 816).
При синтезе ароилбензойных кислот или их эфиров метиловый эфир хлоран-
гидрида фталевой кислоты обладает рядом преимуществ по сравнению с фталевым
ангидридом (более короткое время реакции, более низкая температура, хорошие
выходы [456] и более удобное выделение продуктов реакции, гак как они являются
нейтральными соединениями).
В случае галогенциаиов с хорошими выходами образуются нитрилы [457,
458}. Описано также цианирование дицианом [459]. Хлорашидрид циануровой
кислоты реагирует с заменой всех трех атомов хлора.
Карбамоплхлорид в форме его комплекса с А1С13 может найти применение для
введения карбамидной группы [460]:
ArH+ClCONH2 ArCONHa
Из дифенила и карбамоилхлорида получают с выходом 83% 4-фенилбепламид
[461]. Взаимодействие имидхлоридов с фенолами приводит к фсколкетопам [462]:
дг—С-С14 НАг'ОН 41СЬ- Аг— С—Аг'ОН —> Аг—СО—Аг'ОН.
N-CsH5 N-CeH6
2. Синтезы альдегидов и карбоновых кислот [463, 4641
При взаимодействии ароматических соединений со смесью окиси углерода
и хлористого водорода в присутствии смеси Л1С13 и CuCl получают соответствующие
ароматические альдегиды. Из окиси углерода и хлористого водорода, вероятно, об-
разуется формилхлорид, который, однако, стабилен только в форме комплекса
с АК33. Этот комплекс реагирует как хлорангидрид:
АтИ-р[нс/° 1 А1СЧ АгСНО | НС1
L C1J
[453] D. Papa, Е. Schwenk, П. F. G i n s Ь е г g, J. Org. Chem., 14, 723
'(1949).
1454] L. Roser, Ber., 14, 940, 1750 (1881): L. В о u v e a u 1 t, C- r., 122, 1062,
1207 (1896); L. Bouveanlt, Bull. Soc. chim. France |.3], 15, 1014 (1896).
[455] K. Kindler, W. M e I i e n d о r f, D s c h i - у i u - К w о k, Ber., 76,
308 (1943).
[456] C. Dufraisse, A. A 1 1 a i s, Bull. Soc. chim. France [5], Ц, 531 (1944).
[457] P. К а г г e г, А. КеЬшапо, E. Zeller, Helv. chim. acta, 3, 267 (1920);
2, 482 (1919).
[458] G. W. Gray, B. Jones, J. Chem. Soc., 1954, 678.
[459] G. .1. Jan z„ J. Am. Chem. Soc., 74, 4529 (1952).
[460] L. Gattermann, Ann., 244. 50 (1888). .
[461] H. H о p f f, II- Ohlinger. Angew. Chem., 61, 183 (1949).
[462] H. Pbadke, R. C. Sha h, J. Indian Chem. Soc., 27, 349 (1950).
[463] Обзорная статья: N.* N. Cronnse, Org. Reactions, 5, 290 (19521.
[464] Honben-Weyl, blethoden der organischen Chemie, Bd. 7/1, 1Й52, S. 16;
Bd. 8, 1952, S. 377.
III. ОБРАЗОВАНИЕ С, -С-С11ЯЗЕЙ ПРИ ОТЩЕПЛЕНИИ Hal В ВИДЕ НИл! 777
Этот синтез, называемый синтезом Гаттермана — Коха, в некоторых случаях
применяете» в промышленности. Для лабораторных целей он менее пригоден, так
как неудобен в препаративном отношении, длителен и в некоторых случаях должен
проводиться под давленном. Его применении ограничивается, в основном, реакцией
с алкплбензоламп. Реакцию осуществляют пропусканием сухой смеси СО и НС],
выделяемой из хлорсульфоновой п муравьиной кислот |465]. в раствор углеводо-
рода в эфире или нитробензоле в присутствии А1С13 и CuCl. Выход составляет около
30- 50%, а при проведении реакции под давлением в отсутствие CuCl — 80 — 90%
[446]. О получении п-толилальдегида ем. [467].
Для формилпрования фенолов и эфиров фенолов реакция Гаттермана — Коха
не пригодна. Однако, но Гаттерману (см. [468]), хорошие результаты получают,
если формулируемое вещество обрабатывают смесью хлористого водорода и синильной
кислоты в присутствии ZdCl2. Вначале образуется гидрохлорид альдимина, омыле-
ние которого дает альдегид:
АгН — ИСК 4 НС1 ArCH — NH-HC1 —> АгСНО
Удобным лабораторным методом является модификация этого метода, разра-
ботанная Адамсом и сотр. [469], которые вместо смеси синильной кислоты и хлори-
' стого водорода применили смесь цианида цинка и хлористого водорода: Выходы иногда
превышали 90% •
Резорциловый альдегид [469]. Колбу емкостью 500 мл снабжают хорошей
мешалкой, широкой трубкой для подвода газа и обратным холодильником,
конец которого соединен с промывной склянкой с H2SO4. К пей присоединя-
ется предохранительная склянка, от которой отходит трубка, опущенная над
раствором NaOH. К 20 г резорцина в 150—200 ш сухого эфира прибавляют
1.5 моль сухого цианида цинка па 1 люль фенола. Затем при перемешивании
быстро пропускают сухой IIC1. Продукт реакции выделяется в виде густого
масла, которое затвердевает через 10—30 мин. После 1,5 ч эфир обычно насы-
щается. Медленно пропускают ток НС] н течение еще 0,5 ч. Затем эфир сливают,
к гидрохлориду имида прибавляют 100 мл воды, нагревают раствор до кипения,
фильтруют и охлаждают. Выделяется около половины всего количества альде-
гида. после отделения которого остаток альдегида выкристаллизовывается
через 10—15 ч. Выход продукта около 95% от теоретического; т. пл. 135—
136° С (перекристаллизация из воды с углом).
О полученан безводного цианида цинка см. в оригинальной работе,
В эту реакцию вступают также ароматические уысводороды, особенно по мо-
дифицированному способу Хпнкеля [470]. который использовал аддукты А1С13 и HCN.
Дальнейшее улучшение модификации Адамса — повышение температуры ре-
акции до 70° С, применение в качестве растворителя тетрахлорртапа дает очень хо-
рошие результаты при реакции с ароматическими соединениями [508].
Имеются сообщения о формилпрования с очень хорошим выходом производных
пиррола смесью синильной кислоты и хлористого водорода при использовании в ка-
честве растворителя сноси эфира и хлороформа пр методу Фишера и Цервека [472].
Вариант реакции Гаттермана — Коха, применяемый для формилироваиия
ароматических углеводородов, фенолов, эфиров фенолов и гетероциклов состоит
[465] L. Bert, С. г., 221, 77 (1945).
1466] J. Л. Holloway, N. W. Erase, Ind. Eng. Chem., 25, 497 (1933).
[467] G. II. Coleman, D. С г a i g, Org. Synth., Coll. v. 2\ 1943, p. 583.
[468] W. E. Truce, Org. Reactions, 9, 37 (1957); Houben-Weyl, Methodon
der organischen Chemie, Bd. 7/1. S. 20.
[469] R..A d a ms, I. Levine, J. Am. Chem. Soc.. 45, 2373 (1923); R. Adams,
E. Montgomer y, J. Afli. Chem. Soc., 46, 1518 (1924).
1470] L. E. Il i n k e I, E. E. А у 1 i n g, J. И. В e у n 0 n. J. Chem. Soc., 1936, 339.
[471] H. Groft, J. Ru%che, M. Mirsch. Chem. Ber., 96, 1382 (1963).
1472] S. F. McDonald, J. Chem. Soc., 1952, 4184.
778
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СБЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
во взаимодействии алкилдихлорметиловых эфиров с ароматическими соединениями
в присутствии SnClj или А1С13 (выход 70—90%) [473]:
ArH-. BOCHCI, А-'С'*^ [Аг— СНС1—О1<| -И- АгСНО
О формулировании соедипений с активированной метиленовой группой см.
[474]; ацетилацетон, например, реагирует следующим образом:
(CHsOO^CHz + HCN + HCl —»- (CH3CO)2CH-CH=NH-HC1
Ароматические карбоновые кислоты в некоторых случаях были получены
но реакции типа Фриделя — Крафтса (стр. 774) [464]. Удобный лабораторный
способ непосредственного карбоксилирования ароматических соединений состоит
во взаимодействии ароматических соедипений с пирокатехиндихлорметилеповым
эфиром в присутствии А1С13 или SnCl4 £471]. Этот способ применим для карбоксили-
рования ароматических углеводородов,, конденсированных ароматических соедине-
ний, фенолов, эфиров фенолов и ароматических гетероциклов. Выход обычно 80—
96% от теоретического.
О
О II
CL / \/\ ’• AJC1, Аг—С— Оч .
А,н [7,Л J J Агсоон
по/\^
Реакция Реймера — Тимана [475] основана на взаимодействии фенолов с хло-
роформом в щелочной среде с образованием альдегидов:
Этот синтез пригоден для формилирования многих соединений, однако дае1
невысокие выходы, и поэтому обычно применяют другие методы, например метод
Гаттермана.
Получение 2-оксипафтальдегида-1 протекает с 38—48%-ным выходом [476];
пиррол по Реймеру — Тиману также можно формулировать в а-положение [477].
Ппрролальдсгид. К смеси 210 г хлороформа, 600 .ил спирта, 60 а свежопсро-
гнанного пиррола (т. кип. 124° С) и 200 мл воды прибавляют л течение 45 мин
при 40° С и энергичном перемешивании раствор 240 г КОП в 480 мл поды.
Температура смеси повышается до 45—60° С. Перемешивают еще 1 ч п выдер-
живают смесь 15 ч при 55—60° С. Затем как можно быстрее отгоняют ciuipi
и продукт перегоняют с перегретым до 200" С водяным паром до тех пор, пока
в колбе не останется лишь небольшое количество вязкого остатка (5—8 л ди-
стиллята). Дистиллят насыщают NaCl, продукт экстрагируют 2—3 раза эфиром
(порциями по 1 л), экстракт сушат над К2СО3 и упаривают до объема 100 мл.
Остаток перегоняют в вакууме. Пирролальдегид (т. кип. 108—109J С при
14 мм рт. ст.) отгоняется в виде бесцветного масла, которое быстро затверде-
вает. При дальнейшей очистке быстрой перекристаллизацией из петролейного
эфира получают 16—18 г продукта; т. ил. 50° С.
[473] A. R 1 е с 11 е, Н. G г о 0, Е. II б f t, Chem. Вег., 92, 88 (1960); Н. G г о 0,
A. R I с с h е, G. Matthey, Chetn. Вес., 96, 308 (1963).
[474] И. W i е 1 a n d, Е. Dorrer, Вег., 58, 818 (1925); 63, 404 (1930).
[475] Н. Wy n berg, Chern. Rev., 60, 169 (1960); Houben-Weyl, Bd. 7/1,
S. 36.
[476] A. Russell, L. B. Lockhart, Org. Synth., Coll- v. 3, 1955, p. 463.
[477] Г. Фншер, Г. О p т, Химия пиррола, пор. с нем., ОНТИ, Хнмтеорет, 1937.
V. ОТЩЕПЛЕНИЕ ПОДЫ С ОБРАЗОВАНИЕМ НОВОЙ С—С-СВЯЗИ
779
JV. Образование С—С-связей в результате отщепления металла
* в виде его галогенида
Отщепление металла галогеном имеет практическое значение только в особых
«случаях. Например, 1,4-днкетоны типа ацетонилацетона получают следующим об-
разом [478]:
CH3COCII-COOR
2CII3COCH-COOR + I2 | —> (CH3COCH2)a
| CH3COCH-COOR
Na
Эфир диацетилянтарной кислоты. К 125 г ацетоуксусного эфира в сухом
эфире прибавляют 5 г натриевой проволоки; при этом водород вначале выделяется
очень бурно, но вскоре натрий обволакивается натрийацотоуксусяым эфиром,
f Проводить операцию удобнее всего в толстостенном сосуде с пробкой, снабжен-
, пом холодильником с хлоркальциевой трубкой. Через 1—2 ч, когда основная
' реакция закончится, холодильник удаляют, а скляпку закрывают и хорошо
- встряхивают. После многократного встряхивания натрий растворяется; затем
прибавляют порциями при непрерывном встряхивании раствор 20 г иода в эфире,
причем нод мгновенно расходуется и выделяется Nal. После появления устой-
Г чивой окраски нода раствор фильтруют и испаряют эфир. Оставшееся масло
Г. скоро затвердевает. Его отжимают на пористой пластинке и перекристаллизо-
•' вывают из 50%-ной уксусной кислоты. Выход, по данным Кнорра, переменный:
при обработке маточпых растворов он составляет 40% от теоретического. При
больших загрузках маслянистую часть можно также встряхивать с раствором
соды и при этом получать дополнительные количества эфира диацетилянтарной
кислоты. О температуре плавления (85—90° С) см. оригинальную литературу.
Из этого соединения может быть получен ацетопилацСтон [479].
Описано получение эфира этантетракарбоновой кислоты из натриймало-
нового эфира и пода, которое протекает с почти количественным выходом [480].
V. Отщепление воды с образованием новой С—С-связи
1. Алкилирование
Углеродную цепь высших алифатических спиртов можно удлинить ио реак-
ции Гербе:
RONa-1-H—R'OH —► R—R'OTT-HNaOH
, ' Механизм этой реакции не выяснен. Она приводит к образованию самых разно-
образных продуктов; побочной реакцией прежде всего является получение различ-
•вых жирных кислот. Поэтому реакция не пашла практического применения, хотя
щоздпее была подробно исследована [481].
Спиртами можно алкилировать соединения, содержащие активированные ме-
тиленовые группы; прп этом также отщепляется вода. Из ацетоуксусного эфира и
«зопропилового спирта в присутствии BF3 с 60—70%-ным выходом образуется изо-
щропилапетоуксусный эфир ]482]. Описана реакция малонового эфира с ксаптгидро-
•лом [483].
Реакция с отщеплением воды гладко протекает между спиртами и ароматиче-
скими углеводородами. Для конденсации особенно пригодны спирты типа бензило-
вого и высшие алифатические спирты с длиной цепи пе менее трех атомов углерода.
[478] L. Knorr, F. Haber, Пег., 27, 1155 (1894); Е. Fischer, Ann., 306,
356 и примечание (1899); Anleitnng znr Darst. org. Praparate, Verlag Vieweg,
Braunschweig, 1930, S. 53.
(479] L. Knorr, Ber., 22, 2100 (1889).
4480] С. A. В i s c h о f f, C. Rach, Ber., 17, 2781 (1884).
[481] Ch. Weizmann, E. Borgmann, M. Sulzbacher, J. Org. Chem.,
15, 54 (1950).
(482] J. T. Adams, R. L e v i n e, C. R. Hanse r, Org. Synth., 27, 35 (1947).
J483J R. G. J 0 n e s. et al. J. Am. Chem. Soc., 70, 2843 (1948); K. Ziegler,
Ann., 434, 60 (1923).
780
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТ!-! ОБМЕНА
При взаиа «действии бензилового спирта с бензолом и смесыо уксусной и сер-
ной кислот получается уже при обычной температуре дифенилметэн 1484), а из бенз-
гидрола прп кипячении с толуолом и пятпокисью фосфора с почти теоретическим
выходом образуется дифенилтолилмстан [485].
При реакции с алифатическими спиртами ле удастся ввести нормальные цепи,
а всегда получаются, как и при синтезе Фриделя — Крафтса с алкилхлоридами,
разветвленные соединения. Алкилирование ароматического ядра действием спирта
в некоторых случаях используется для промышленного получения алкпларомати-
ческих соединений, папримор, кумола из бензола и изопропилового спирта или
тимола из .к-крезола и изопропилового спирта [486]. Обычно в качестве конден-
сирующего средства применяют сорпую кислоту [487], по пригодны и другие ката-
лизаторы,- например SnCl4, ZnCl3 [488], n-толуолсульфокислота [489] и Д1С13 [490,
491]. При алкилировании первичными спиртами с длиной цепи от 4 до 12 углеродпых
атомов в присутствии BF3 и фосфорного ангидрида [492, 493] выходы достигают
80%. Особенно хорошим катализатором для алкилирования фенолов и эфиров фе-
нолов является фосфорная кислота [494].
Кумол. В течение 3—4 ч при 65° С нагревают 50 г бензола с 19 г изопро-
пилового спирта и 400 мл 80%-ной H2SOd. После обычной обработки получают
кумол с выходом 65% от теоретического.
Таким же образом реагируют толуол, ксилолы, крезолы, фенолы, резор-
цин, о-иитротолуол, хлорбензол и нафталин с изопропиловым спиртом, втор-
бутиловым и изобугиловым спиртами и циклогексанолом.
л-Цимол можно получить из толуола и изопропилового спирта в присутствии
НоР04 с выходом 80% от теоретического [495]. дтор-Бутилбензол в присутствии
BF3 и Р2О5 синтезируют следующим образом [492].
$тор-Бутилбензол. Раствор 456 г (2 моль) бензола в 37 г (0,5 моль) и-бу-
тплового спирта перемешивают и охлаждают водой, пропуская в это время RF3,
пока не поглотится 34 г (0,5 .моль) (тяга!). Затем быстро прибавляют 17,7 г
(0,125 моль) Р2О5 и медленно при перемешивании доводят до кипения. Раствор
становится мутным и разделяется па два слоя, после чего перемешивание
прекращают. Нагревание продолжают еще 3 ч. Затем верхний слой отделяют,
промывают разбавленной водной щелочью и фракционируют, собирая продукт
с т. кип. 170—172° С (744 мм рт. ст.). Выход продукта 50 г (75% от теоре-
тического).
В качестве примера синтеза при помощи А1С13 приведено получение трет-
бутплбензола [491].
тердаг-Бутилбензол. Суспензию 66,5 з (0,5 моль) AICI3 в 390 г (5 моль) бен-
зола быстро перемешивают и при 20—30е С прибавляют по каплям 74 з (1 моль)
трет--бутилового спирта- После выдерживания в течение ночи раствор выли-
вают в смесь мелко раздробленного льда if IIC1. Бензольный слой отделяют,
а ил водного слоя продукт экстрагируют эфиром. Объединенный эфнрпо-бен-
эольпый раствор упаривают и остаток фракционируют, применяя колонку
длиной 30 см. Выход mpe/rt-бутилбепзола 65—70% от теоретического; т. кии.
168—170° С (740 мм рт. ст.).
V. Meyer, С. Wurst е г. Вег., 6, 964 (1873).
W. Н е m i 1 i а п, Вег., 7, 1203 (1874).
А. V в г 1 е у, С., 1929, I, 697; англ. пат. 288122, I6/VI 1927.
Н. Меуег, К. Beruliauer, Mh. Chem., 53/54, 721 (1929).
И. И. Ц у к е р в а ни к, В. Сергеева. ЖОХ, 17, 1005. 1009 (1947).
Е. F. Pratt, R. К. Presto и, J. 1). Drape г. J. Am. Chem. Soc., 72,
1367 (1950).
R. С. II ns to и, I. А. К a у e, J. Am. Chem. Soc., 64, 1576 {1942).
R. С. II 11 s t о n, W. B. F о x, M. N. Binder, J. Org. Chem., 3, 251 (1939).
N. F- T ou 55a in t, G. F. H en nio n, J. Am. Ghem. Soc., 62, 1145 (1940).
Ch. E. Weis h, G. F. Henni 0 11. J. Ain. Chem. Soc., 63, 2603 (1941).
A. E. T s c h i t s c li i b a b i 11, Bull. Soc. chim. France [5], 2, 497 (1935).
И. П. Ц у к о p в а н и к, Т о м б о и ц о в а, ЖОХ, 45, 699, 820 (1945).
V. ОТЩЕПЛЕНИЕ ВОДЫ С ОБРАЗОВАНИЕМ ПОБОИ С—С-СВЯЗИ
781
Фенолы алкилируются в основном в пара-положение [4961.
Толуидин можно метилировать в ядро действием метанола и хлористого водо-
рода под давлением [497]. Описано также алкилирование анилипа спиртами в ядро-
[498] и алкилирование индола спиртами при 220° С л присутствии алкоголятов
под давлением [4991.
Циклодегидратацией [500] 1-арила.тканолов-З или 1-арилалкаполои-4 легко-
получаются производные индана или тетралина. Изомеризации при этом не происхо-
дит. Например, из 4-фенилбутилового спирта образуется тетралин, а не метилгид-
риндан. В гидрпндановом ряду из соответствующих третичных спиртов получают
только 1,1;диалкилпроизводные, например 1,1-диалкил-З-фенилпропанол
/R
.CHa-CHs-a
/V IXn
V он
в то время как первичные и вторичные гидрокоричные спирты не циклизуются, веро-
ятно, вследствие их полимеризации.
Описано также получение 1,1,6-триметилтетралииа (ионена) [501].
1.1-Диметилгидриндан [500]. К 1,2 объемн. ч. 85%-ной H2SO4 медленно
при энергичном перемешивании прибавляют по каплям 1 объемн. ч. 1,1-диметил-
3-фенилпропанола, поддерживая температуру реакционной массы 10° С. Реак-
ция, очевидно, протекает мгновенно, однако на всякий случай смесь следует
перемешивать еще 1 ч. при комнатной температуре. Затем ее разбавляют 10—
15 объемами воды и перегоняют. От дистиллята отделяют маслянистый слой,
а водный слой переносят обратно в перегонную колбу н снова перегоняют.
Маслянистый продукт реакции отгоняют еще раз с паром из смеси, в которую
добавлена гидроокись щелочного металла до щелочной реакции. Масло
сушат и перегоняют. Выход 1,1-диметилгидрипдала 65% от теоретического;
т. кип. 191° С.
По такому же способу были получены 1,1,2-триметилгидрипдан (т. кип.
208° С) с выходом 90%, тетралин с выходом 55% и 1,1,4-триметилтегралин
[502] с выходом 86%.
При перегопке реакционной смеси с водяным каром в колбе остаются
продукты полимеризации в виде вязкого масла или смолы.
Циангидрины могут реагировать с углеводородами; при этом происходит отщеп-
ление воды и образование нитрилов. Например, при взаимодействии диметилового
эфира пирокатехина' с 3,4-диметокснбензальдегидциангидрияом с хорошим выхо-
дом образуется бис-(3,4-диметоксифенил)-ацетонитрил [503].
ОН
Cli'aO-Z Ч-| СН-<^\-ОСН3 —► CIISO-Z Ч-СП-/ S-ОСНз
\=/ I \=/ /=='г' I
СН О7 CN ХОСНз СНзО/ CN ХОСН3
[496] R. С. Huston et al., J. Am. Chem. Soc., 58, 439 (1936); 59, 2001 (1937).
[497 R. W. Cripps, D. H. Hoy, J. Chem. Soc., 1943, 14.
[498] I. J. R i и k e s, Rec. trav. chim., 62. 557 (1943).
[499] R. H. С о r ii f о г t h, R. Robinson, J. Chem. Soc., 1942, 680.
[500] M. T. Bogert, D. Davidso n, J. Am. Chem. Soc., 56, 185 (1934).
[501] M. T. Bogert, D. Da vidson, P; M. A p f e I b a u m, J. Am. Chem.
Soc., 56, 959 (1934).
[502] M. C. Kloetzel, J, Am. Chem. Soc., 62. 3105 (1940).
[503] A. M u 1 1 с г, M. V a j (1 a, J. Org. Chem., 17, 800 (1952).
782
1 ОБРАЗОВАНИИ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
Взаимодействие циануксуспого эфира с циангидрином ацетальдегида в присут-
,-ствии алкоголята натрия дает эфир 2,3-дицианмасляной кислоты (выход 80% от
•теоретического) [504).
Все описанные выше методы приводят к образованию только одной новой
С—С-связи. Если же вместо спиртов применять карбонильные соединения, то одновре-
менно могут образоваться две новые С—С-связи:
R\
2RH4-R-CHO —> >СН-R |-НйО
R'z
Примерами таких реакций могут служить получение диарилмстанов из аль-
.дегидов и 2 моль ароматического углеводорода
2С1—С13ССПО —> Cl—^>-СН-^\-С1
СС1з
или получение продукта конденсации типа бакелита из фенола и формальдегида.
С образованием двух С—С-связей протекают также реакция ароматических
-альдегидов с 2 моль третичного амина, давая производные трифенилметана
/Ar'NR2
ArCHO + 2HAr'XR2 —* ArCH< +Н2О
xAr'NR2
•и взаимодействие 1 лсо/ь фталевого ангидрида и 2 моль фонола или резорцина с об-
разованием фталеинов:
СО СО
^O-ISHArOH —> Ъ ' +H2O
V\ / ^/\ /
co c
HOA^^ArOH
Из 1 лмь фталевого ангидрида и 1 -нефснольных ароматических соединений, молъ гидрохинона или пирокатехина, или
напротив, образуются антрахиноны:
СО он 1 о он II 1
^\/\ Z\ —^W\d-Hao
! IJ О
\/\/ СО м 1 он \Z\Z\Z 11 1 О он
2. Хлорметилированне [5051
Хлорметильные группы можно непосредственно ввести в ароматическую сп-
•стему по схеме:
АгН-|-НСЦ-СН2О —► RCHaCl-1-НгО
В промышленном масштабе этот синтез проводят без растворителя [506], в то
время как для препаративных целей в лаборатории применяют наряду с хлорофор-
мом и четыреххлористым углеродом, петролейный эфир или уксусную кислоту,
насыщенную НС]. В_качестве конденсирующего средства обычно используют ZnCl2.
(504] А. II i gs о о, J. F. Т h о г р е, J. Chem. Soc., 89, 1455 (1906); R. Р. L i n-
stead, M. Whalley, J, Chem. Soc., 1954, 3722.
[505] Обзорная статья: R. C. Fuson, С. H. McKeever, Org. Reactions, 1,63
(1942).
[506] G. В ed d e] i n, II. Lange, герм. пат. 508890; С., 1931, I. 1830.
V. ОТЩЕПЛЕНИЕ ВОДЫ С ОБРАЗОВАНИЕМ НОВОЙ С—С-СВЯЗИ 783
В некоторых случаях выходы продуктов увеличивают добавкой фосфорной
кислоты. По Вейганду смесь бензилхлорида и кснлилендихлорида получают следу-
ющим образом.
Ксилилендихлорид. В колбу для сульфирования емкостью 1 л, снабженную
газоподводящей трубкой, обратным холодильником н мешалкой, помещают
84 г ZnCl2, 90 г параформальдегида, 200 мл хлороформа (или четыреххлористого
углерода) и 78 г бензола. При перемешивании пропускают сильный ток НС],
причем реакционная смесь разогревается. Путем охлаждения в начале реак-
ции или умеренного нагревания в конце поддерживают температуру 50—60’С.
Через 2 ч смесь промывают несколько раз водой, разбавленным раствором
соды и опять водой, сушат над СаС12 и отгоняют' основное количество хлоро-
форма. Остаток перегоняют в вакууме и получают 84 г (66% от теоретического)
бензилхлорида и 55 г (30% от теоретического) ксплплендихлорида (в основном
пара-изомер).
Ксилилендихлорид аналогичным образом можно получить также из бен-
зилхлорида (42 г ZnCl2, 45 г триоксиметилена, 127 г бензилхлорида в 100 мл
чотыреххлористого углерода). После окончания реакции продукт экстрагируют
эфиром, промывают водой и разбавленным раствором соды, далее обрабаты-
вают, как указано выше. Выход ксилилендихлорида 145 г (83% от теорети-
ческого). Из полученной смеси изомеров кристаллизуется 100 г пара-соединения.
Нафталин можно хлорметилировать в а-положенве [507] с выходом
Вместо смеси формальдегида и хлористого водорода можно также применять
монохлорднметиловый эфир. Например, 2,4,6-триизопропилбензол переводят в 2,4,6-
триизопропилбензилхлорид [508], используя в качестве растворителя сероуглерод,
,а в качестве катализатора — SnCl-4:
СНаС1
(СЦ3)3 НС^/^СН (СНз)г (СН3)2 нС\/\/Сн(СНз)2
( I СНаОСНгС! || |
-GH.OH; ~НгО Ху/
С'Н(СНа)з CH(CHs)2
Описано со-хлорметилированис стирола [509].
Введение а-хлорэтильной группы в ароматические соединения происходит
по схеме [510]:
АгН + СН3СНО 4- НС1 —> АгСНС1СНз + Н2О
Описана также реакция бромметилирования [511, 512], например получение
бензилбромида с выходом 86,5% из бензола, формальдегида и бромистою водорода
в присутствии смеси уксусной и серной кислот.
3, Оксиметилпроваиис
При реакции фора альдегида с соединениями, содержащими реакционноспо-
собные атомы водорода, в щелочной среде происходит окенметплирование:
ВН + СН2О —► ВСНаОН
[507] О. Gru mini 11, A. Buck, Org. Synth., 24, 30 (1944).
[508] R. C. F u s о n ct al., J. Аш. Chem. Soc., 64, 30 (1942); Org. Synth., 23, 5/
[509] Wichterle, J. Cerny, Coll. Czech. Chem. Comm., 20, 1288 (1955).
[510] R.L. Frank et al., J. Am. Chem. Soc., 68, 1365 (1946); R. Q u e 1 о t,
J. Ducasse.C. r., 208, 1317 (1939); Bull. Soc. chitQ. France [5], 7, 196, 205
[511] G. Kubiczek. L. Neugebauer, Mh. Chem., 81, 917^(1950).
(512] G. Darzens, C. r., 208, 818 (1939).
784
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕИ В РЕЗУЛЬТАТЕ ОБМЕНА
Так, при взаимодействии этилового эфира циклогексанон-1-карбоновой-2
кислоты с водным раствором формальдегида в присутствии СаО получают практи-
чески с количественным выходом этиловый эфир 2-оксиметилциклогексапои-1-кар-
боновой-2 кислоты [513]. При реакции метилэтилкетона с формальдегидом в при-
сутствии NaOH образуется с выходом 70% 2-метилбутаиол-£-оп-3, который при от-
щеплении воды превращается с выходом 74% в 2-метилбутен-1-он-3 [514].
Описано также оксиметилирование ацетона [515] и фенюшитрометана [516].
Легко протекает в слабощелочных средах введение оксиметильных групп в фенолы.
Например, можно получить замещенные 2,6-диоксиметилфеяолы с выходами 50—
96% от теоретического [517]. Эта реакция имеет большое значение для синтеза фс-
ноло-формальдегидных смол.
Из ненасыщенных соединений взаимодействием их с формальдегидом в кислой
среде получают, по Прилсу [518], в зависимости от исходных продуктов п условий
.реакции 1,3-гликоли [519], или лг-диоксаньт, пли же ненасыщенные спирты [519]:
RCH2CH=CH2 + CH2O —> НСН2СН(ОН)СН2СН2ОН
_______________________________I_______
~Н!° I |снго
ВСИ=СНСН2СН2ОН ЛНг-СНа
RCHsCH^ N)
%—сн2
4-фснил-.и-диоксан [520]. К смеси 1350 г 37%-пого раствора- формалина
(16,6 .ноль) и 96 г H2SO4 прибавляют 624 г (6 моль) стирола. Смесь перемеши-
вают при температуре около 93° С (чуть ниже т. кип.) в течение 7 ч. После
охлаждения до комнатной температуры прибавляют бензол и верхний слой
промывают до нейтральной реакции. Растворитель отгопяют, а продукт фракцио-
нируют в вакууме. Получают 32 г стирола. Выход 4-феннл-лг-диоксана 805 г
(86% от теоретического); т. кип. 99° С (2 мм рт. ст.).
Имеются и другие примеры этой "реакции [520, 521, 522]. При взаимодействии
углеводородов с метоксиметнлацетатом происходит ацетоксиметилирование. Напри-
мер, пл мезигилена образуется с выходом 53% ацетоксимезитилен [523].
1зС\/\/ СНз 3 \/ч/ 3
||. I l-CHsCOOCH.OCHs—gjj-gg» II I
у ' ’ уЧщососнз
СПз СНз
[513] Kl-Wei Hioug, Ann. Cluimo (И], 17, 26!) (1M2).
[514] J. Colonge, L. Cumet, Bull. Soc. chim. France [5], 14, 838 (1947).
[515] S. Olsen. Chem. Ber., 88, 205 (1955).
[516] L. F. Fiesor et al., J. Am. Chem. Soc., 68, 2248 (1946).
[517] J. S tra t ing, II. J. Backor, Rcc. trav. chim., 62, 57 (1943).
[518J H. J. Prins Proc. Acad. Sci. Amsterdam, 22, 51 (1919); J. Chem. Soc., 118,
I, 42 (1920).
[519J Обзорная статья: E. A r u n d a I e, L. A. Mikeska, Chem. Rev., 51, 505
(1952)- N. C. Yang, D. D. H. Yang, С. B. Ross. J. Am. Chem. Soc.,
81, 133 (1959).
[520] M. G. J. Beets et al., Rec. trav. chim., 70, 29, 20, 25 (1951).
[521} M. G. J. Beets et al., Rec. trav. chim., 71. 343 (1952).
[522] R. W. Shortridge, J. Am. Chem. Soc.. 70, 873 (1948).
1523] L. Summers, J- Am. Chem. Soc., 76, 3481 (1954).
V. ОТЩЕПЛЕНИЕ ВОДЫ С ОБРАЗОВАНИЕМ НОВОЙ С—С-СВЯЗИ
785
1 4. Аминометилирование (реакция Манниха) [5241
С помощью формальдегида н аммиака или первичных или вторичных аминов
? подвижный водород может замещаться на аминометильную или замещенную амино-
г метильную группы. Эта реакция, систематически разработанная Манпихом [525]
> я протекающая по приведенной ниже схеме, нашла в последние годы широкое пре-
> саративное применение:
RCOCHg + CHaO + HNRa . HCI - > RCOCH3CHaNRs • НСЦ-НаО
?..? Кроме кетонов [526] аналогично могут реагировать также другие соединения
! с подвижным атомом водорода, например альдегиды [527], ацетилены [528], карбо-
новые кислоты [529], сложные эфиры или нитропарафипы [530]. В эту же реакцию
вступают фенолы [531], а в некоторых случаях и гетероциклические соединения [532].
Вместо формальдегида, который применяется в виде водного раствора или
в форме параформальдегида, в отдельных случаях можно использовать также дру-
гие альдегиды, например ацетальдегид [533].
В качестве аминной компоненты можно применять аммиак и различные пер-
I. [винные н вторичные амины, большей частью в виде солей.
1р Ниже приведено получение гидрохлорида Р-диметиламинопропиофенона [534]
кв 2.4.6-три-(диметиламинометил)-фенола [531].
$ ' Гидрохлорид p-диметиламинопропиофенона. В круглодонную колбу ем-
?! костью 500 мл с обратным холодильником помещают 60 г (58,5 мл, 0,5 моль)
г - ацетофенона, 52,7 г (0,65 моль) гидрохлорида днметиламина и 19, 8 а (0,22 моль)
к параформальдегида. После прибавления раствора 1 мл концентрированной
к? HCI (d — 1,19) в 80 мл 95%-ного спирта смесь кипятят в течение 2 ч с обратным
к: холодильником. Желтоватый раствор, если он мутный, фильтруют через воронку
|f' для горячего фильтрования и еще горячим разбавляют 400 лл ацетона. Охла-
!ждают медленно до комнатной температуры я оставляют на ночь в холодильнике.
Кристаллы отфильтровывают, промывают 25 мл ацетона и сушат в течение
2,5 ч при 40—50° С. Выход сырого продукта составляет 72—77 г (68—72% от
теоретического); т. пл. 138—141° С. Этот продукт гигроскопичен. После сушки
в течение еще 4 ч т. пл. повышается до 152—153° С. Продукт можно перекри-
сталлизовать растворением в 85—90 мл горячего 95%-ного спирта и медленным
прибавлением 450 мл ацетона. Выход при перекристаллизации 90%; т. пл.
дат". 155—156 С.
№f524] Обзорная статья: F. F. ВИске, Org. Reactions. 1, 303 (1942); Н. II е I 1-
fcr mann, G. О р i t z, Angew. Chem., 68, 265 (1956); B. Reichert, Die
By.. Mannich-Reaktion, Springer-Verlag, . 1959.
№(525] G Mannich et al., Atch. d. Pharm., 250, 647 (1912); 264, 65, 164 (1926);
265, 684 (1927): Ber., 55, 3510 (1922); 53, 1874 (1920).
Щ526] E. C. Spaeth, T. A. Geissman, T. L. J а с о b s, J. Org.* 1 Chem.,
& 11, 399 (1946); D. R. H о w t о n, J. Org. Chem., 12, 379 (1947); C. Mannicb,
fe O. Hieronimus, Ber., 75, 49 (1942); A. W. Ruddy,!. S. Buckley,
f:- jr., J. Am. Chem. Soc., 72, 718 (1950).
| (527] C. Mannich, B. Lesser F. Silten, Ber., 65, 378 (1932).
1528] C Mannich, F. T. Chang, Ber., 66, 418 (1933).
1 1529] C. Mannich, E. Ganz, Ber., 55, 3486 (1922).
(530] G. B. Butler, F. N. McMil [an, J. Am. Chem. Soc., 72, 2978 (1950);
Г. A. T. Blomquist, T. H. S h e 1 1 e у jr., J. Am. Chem. Soc., 70, 147
(1948).
f. (531] G. F. G r i 11 о t, W. T. Gormley jr.. J. Am. Chem. S'oc., 67, 1968 (1945);
: . H. A. Brnson, C. W. McMullen, J. Am. Chem. Soc., 63, 270 (1941).
4532] H. Kuhn. O. Stein, Ber., 70, 567 (1937); W. Herz, K. Dittmer,
St. J. C r i s t о 1, J. Am. Chem. Soc., 69, 1698 (1947); H- D. Hartougb
" et al., J. Am. Chem Soc., 70, 4013, 4018 (1948).
4533] C. Mann ich, P. M о h s, Ber., 63, 608 (1930); P.Petrenko-Krit-
schenko, Ber., 42, 3683 (1909).
. <534] С. E. M a x we 1 I, Org. Synth., 23, 30 (1943).
50 Заказ 1835.
786
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
2,4,6-Три-(диметиламинометил)-фенол. Смесь 94 г (1 моль) фенола а 720 г-
(4 моль) 25%-ного водного раствора диметиламина охлаждают до 20° С в трех-
горлой колбе емкостью 2 л, снабженной мешалкой, термометром и капельной
воронкой. При перемешивании прибавляют по каплям 350 а 30%-ного водного
раствора формальдегида в течение 0,5 ч, поддерживая температуру 25—30° С,
и перемешивают еще 1 ч при этой температуре. Затем капельную воронку за-
меняют на обратный холодильник и смесь при перемешивании нагревают 2 ч
на водяной бане. К горячему раствору прибавляют 200 е NaCl и еще около
20 мин нагревают и перемешивают. Органический слой отделяют от горячего
раствора, переносят в колбу Клаизена емкостью 500 мл и перегоняют в вакууме.
Выход продукта 228 г (86% от теоретического); т. кип. 130—450° С (1 — 2 мм
рт> ст.). Слабая красная окраска устраняется при повторной перегонке (г. кип.
130—135° С при 1 мм рт. ст.).
Побочной реакцией является замещение второго подвижного атома водорода
на аминометильную группу [524]:
C6H5COCH2CH2NR2 • HCI + CH2O + IINR2 —► CeH5COCH(CHaNR2)2-HCH-H2O
При взаимодействии с аммиаком или первичными аминами образующиеся
па первой стадии основания Манниха могут снова реагировать с формальдегидом
и исходным соединением, давая третичное основание-[535]г
СООН СООН / COOII \
I I / । ]
СНз—C-CH2-NHR.HCI + CH2O+CH3-CH —> CH3-C-CH2 NR-HC1+H2O
I I \ । /
СООН СООН X СООН /2
Об алкилировании основаниями Манниха см. стр. 796.
5. Сульфометилирование [536]
При взаимодействии р-нафтола, формальдегида и сульфита натрия с выходом
95% был получен 2-оксинафтил-1-метнлсульфонат натрия
СН2—SOgNa
^\/ч/он ^\А/0Н
1 I +HCHO + Na2SO3 —>1 II |
VI. Образование С—С-связи в результате отщепления спирта,
карбоновой кислоты и др.
1. Сложиоэфирные конденсации [537]
Под сложноэфирными конденсациями в общем случае понимают взаимодей-
ствие эфиров карбоновых кислот или эфиров угольной кислоты с соединениями, со-
держащими активированные метиленовые или метиновые группы. Реакция протекает
в щелочной среде с отщеплением спирта и образованием новой С—С-связи по общей
схеме:
.В' zR'
ПСООС.Н5 I CHS Cc7h?<S’ HCOOi
\Н" \R„
[535] С. Mannicli, В. Kather, Вог., 5з, 1368 (1920).
[536] С. М. S u.t е г, R. К. В a i г, F. G. Bordwel 1, J. Org. Chem., 10, 47<:
[537] Обзорная статья: Н. Н enecka, в книге Н о u b е n - W е у 1, Methodeii
der organischen Chemie, Bd. 8, 1952. S. 564; Ch. R. Hauser, В. E. И и d-
s 0 n jr., Org. Reactions, 1, 266 (1942); Ch. R. Hauser, F. W. S w a m e r,
J, T. Adam s, Org. Reactions, 8, 59 (1954).
VI. ОБРАЗОВАНИЕ С—С-СИЯЗИ С ОТЩЕПЛЕНИЕМ СПИРТА И др.
787
Ниже приведена лишь часть возможных реакций (некоторые из этих реакций
не имеют большого препаративного значения, так как образующиеся вещества с луч-
шими результатами могут быть получены другими методами).
1) Конденсация двух молекул эфира с образованием (3-кетоэфиров:
КСООС2Н5 + И'СНгСООСзНб —> RCOCHGOOC2H54-C2H5OH
I
R
2) Конденсация эфира с алифатическим, ароматическим или гетероцикличе-
ским кетоном с образованием 1,3-дикетона:
CH3COOC2H5 + RCH2COCH8 —► СН8СОСНСОСНз + С3Н5ОН
I
R
3) Конденсация эфира муравьиной кислоты с кетоном до 1,3-кетоальдегида
НСООС2Н5 + СвН6СОСНз —► С6Н5СОСН2СНО + С3НбОН
4) Конденсация эфира щавелевой кисноты с кетоном с образованием дикето-
эфиров;
ВСОСПз + (СООСзЩ)з —> RCOCH2COCOOC2H5 + C2H5OH ’
5) Конденсация алкилкарбоната с нитрилом до эфира сс-цианкарбоновой кис-
лоты:
RCH2CN + O=C(OC2H6)2 —> RGHGNCOOC2H5 + CsH5OH
В качестве катализаторов конденсации применяют щелочные металлы и их
производные, чаще всего натрий, ямид натрия, алкоголят натрия, а также гидрид
патрия [538]. Описано использование диэтилмагнийбромида в качестве конденси-
рующего средства для сложноэфирной конденсации [539]. Натрий применяют в виде
Проволоки или листочков. При работе с большими количествами натрия не рекомен-
дуется проводить реакцию на водяной бане, следует применять индифферентный по
отношению к натрию теплоноситель.
При сложноэфирной конденсации большие загрузки часто дают лучшие вы-
ходы, чем небольшие, что пока осталось без объяснения. Применение металлического
натрия исключается в том случае, когда реагирующая молекула содержит группы,
-которые могут реагировать с водородом в момент выделения. В таких случаях реко-
мендуется применять алкоголят натрия.
, При обращении с амидом натрия следует соблюдать осторожность, так как
он часто взрывается. Поэтому препараты, хранившиеся долгое время, нельзя при-
менять. Ниже приведен удобный и безопасный метод получения амида натрия [540].
Амид натрия. К 500 мл жидкого аммиака в колбе емкостью 2 л прибавляют
0,3 г порошкообразного Fe(NO3)s - 6Н2О. К смеси добавляют при перемешивании
около 1 г натрия. Перемешивание продолжают до тех пор, пока голубая окраска
не перейдет в серую. Затем но мере исчезновения голубой окраски небольшими
порциями прибавляют патрий. Всего прибавляют 26 г (1,13 моль) натрия.
После того как голубое окрашивание исчезает, смесь перемешивают еще 15 мин.
Полученный таким образом амид натрия нельзя храпнть в сухом виде
(сухой NaNH2 получается при испарении аммиака), так как при этом образу-
ются взрывоопасные соединения.
[538] F. В. La F о г g е, S. В. S о 1 о w а у, J. Аш. Chem. Soc., 69, 2932 (1947);
69, 2677 (1947); N. G-reen, F. В. La Forge, J. Am. Chem. Soc., 70, 2287
(1948).
1539] G. R. Hauser, H. G. Walker jr., J. Am. Chem.. Soc. 69, 295 (1947).
1540] T. H. Vaughn, R. R. Vogt, J. A. Nieuwland, J. Am. Chem. Soc..
56, 2120 (1934); A. С. Cope, E. M. H a n с о c k, J. Am. Chem. Soc.. 60, 2644
(1938); cxr. также «Preparation of org. Intermediates», John Wiley a. Sons, Inc.,
New York, 1951, p. 267; Org. Synth., 25. 25 (1945); 30, 72 (1950).
50*
788
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕИ В РЕЗУЛЬТАТЕ СЕМЕНА
Для нормального протекания сложноэфирной конденсации необходимо при-
сутствие спирта, причем достаточно даже его следов. Поэтому часто для начала ре-
акции необходимо добавлять соответствующее количество абсолютного спирта; даль-
нейшее ее протекание обеспечивается спиртом, выделяющимся в результате реакции.
Причиной отрицательных результатов, получаемых с плохо очищенными эфирами,
является, очевидно, не спирт, а содержащаяся в них вода.
Для проведения сложноэфирной конденсации нельзя дать общих рекоменда--
ций. В начальном периоде в некоторых случаях реакционную массу следует умеренно
нагревать, в процессе реакции часто рекомендуется охлаждение, чтобы избежать
побочных реакций, например восстановления по Вуво — Блану (стр. 83). Конец
реакции не всегда легко обнаружить, если образуются твердые осадки, покрыва-
ющие натрий и препятствующие, таким образом, дальнейшему протеканию реакции.
Поэтому при последующей обработке, которую большей частью начинают с под-
кисления реакционной массы, следует соблюдать осторожность. Если возможно,
уксусную кислоту, применяемую для подкисления, не прибавляют в реакционную
колбу, а переносят сначала реакционную массу в открытую фарфоровую чашку
и механически удаляют большие кусочки натрия. Для конденсации Часто исполь-
зуется этилацетат.
Очистка этилацетата. Этилацетат промывают два раза водой или раствором
соды, если он обладает кислой реакцией, и снова водой до исчезновения щелоч-
ной реакции, и сушат небольшим количеством СаС12. Потом оставляют стоять
24 ч' над большим количеством'СаС12, причем потери из-за образования моле-
кулярных соединении неизбежны. Инглис и Робертс [541] промывали этил-
ацетат только водой и затем сушили над поташем. Обработанный таким образом
эфир содержал спирт и поэтому перегонялся в интервале 2—3° С. Высушенный
этилацетат можно не перегонять. Он очень гигроскопичен и его не следует
фильтровать, а нужно слить с осушителя.
Для лабораторного получения ацетоуксусного эфира разработан удобный
и дающий очень хорошие выходы метод [542]. Промышленный синтез ацетоуксус-
ного эфира проводится в автоклаве с натриевой пылью, причем имеются данные
о количественном выходе ацетоуксусного эфира в этой реакции [543].
Реакция этилового эфира бензойной кислоты с ацетофеноном приводит с вы-
ходом 70% к дибензоилметану [544]. При конденсации о-оксиацетофонопа с эти-
ловым эфиром пропионовой кислоты и натрием в эфире получается «в-пропионил-о-
окскацетофенон, который циклизуется под действием смеси соляной и уксусной кис-
лот в 2-этилхромон (выход 70—75%) [545]:
А/ОН +<А-СЛ _ /уОН Нв _ Агоч/сн6
\^\сосн3 0СаНб ^^СО-СНй
о
При помощи эфиров можно, как уже упоминалось, ацилировать и другие
соединения с подвижным атомом водорода. Например, при обработке нитрилов эфи-
рами в присутствии этилата натрия образуются соответствующие кетонитрилы [54б|:
ВСН2 + О=С-R' —-> RCHCOR' + CsHsOH
I I I
CN ОС2Н6 CN
Из фенилацетонитрила, сложных эфиров и амида натрия в эфире получают
с 60— 80%-ным выходом кетонитрилы [547].
541) J. К. Н. I n g 1 i s, К. С. R о b е г t s, Org. Synth., 6, 36 (1926).
542) H. Scheibler, Ann., 565, 176 (1949).
543) 0. D. Frampton, J. F. Nobis, Ind. Eng. Chem., 45, 404 (1953).
5441 A. Magnani, S. M. M с E 1 vain, Org. Synth., 20, 32 (1940).
545) R. Mozingo, Org. Synth., 21, 42 (1941).
546) J. B. D о r sc h, S. M. M с E 1 va In, J. Am. Chem. Soc., 54, 2969 (1932);
R. 8. Long, J. Am. Chem. Soc., 69, 990 (1947).
[547) R. Lev in e, C. R. H a u s e r, J. Am. Cbem. Soc., 68, 760 (1946).
VI. ОБРАЗОВАНИЕ С—С-СВЯЗИ С ОТЩЕПЛЕНИЕМ СПИРТА и др.
789
Конденсация с эфирами муравьиной кислоты приводит к соответствующим
формилпроизводным. В метиленовую группу диэтилового эфира 4-метоксифенилянтар-
ной кислоты при помощи эфира муравьиной кислоты можно ввести одну альдегидную
группу .(выход около 50%) [548]. Образующиеся из кетоиов и эфиров муравьиной
кислоты 0-кетоальдегиды находятся обычно в таутомерной оксиметиленовой форме.
Для получения а-оксиметиленциклопентадеканона из цннлопентадеканона в ка-
честве катализатора применяют этилат натрия (выход до 66%) [549]. Описано формили-
рование ацетофенона [550].
Общий метод синтеза хромонов основан на конденсации с эфиром муравьиной
кислоты (выход около 80%) [551].
/0П -/\ /0Н
iTV +НСООС2Н6 9Н0
^^СОСНа '^"Ч'СО-СН2
З-Метилхромон [551]. Смесь 20 г е-оксипропиофенона и 300 г этилформиата
охлаждают до 0’С к при встряхивании медленно прибавляют маленькими
кусочками 16 г натрия. После замедления реакции массу оставляют на три
дня при номиатной температуре. Колбу от проникновения влаги воздуха защи-
щают ртутным затвором. Реакционную массу часто и подолгу встряхивают.
Через три дня ее выливают на лед, смешанный с эфиром. Водный слой отделяют
и несколько раз экстрагируют эфиром до полного удаления окрашенных про-
дуктов. Объединенные эфирные растворы промывают несколько раз насыщен-
ным раствором (NH4)2SO4, сушат над NaaSO4 и испаряют эфир. Остаток, содер-
жащий значительное количество воды, перегоняют в вакууме. Выход продукта
18 г; т. кип. 156’С (15—16 мм рт. ст)-, т. пл. 68° С (после перекристаллиза-
ции из бензола и петролейного эфира).
Альдегидную группу удается также ввести при помощи эфиров ортомуравьи-
ной кислоты [552]:
сн8соснвсоос2нБ-|- нс(осан5)3 _2САащОН-
СНзСОССООСзНй СИ3СОСНСООС2Н5
I! I
СН-ОС2Нб СПО
Эта, открытая еще Клайзеном, реакция протекает с очень хорошим выходом.
Например, этоксиметилбновые соединения получаются с выходом до 98% [553[.
При помощи эфира щавелевой кислоты можно заменить реакционноспособный
атом водорода на кетоэфирную группу [554]. Например, из эфира пропионовой кис-
лоты и эфира щавелевой кислоты получают с выходом 70% эфир а-оксалилпропио-
новой кислоты [555]. При взаимодействии эфира циклопентанон-а-карбоновой
[548] G. Swain, A. R. Т о d d, W. S. W a r i n g, J. Chero. Soc., 1944, 548.
[549] V. P r e 1 0 g, U. Geyer, Helv. chim. acta, 28, 1677 (1945).
[550] L. Panizzi, Gazz. chim. ital., 77, 549 (1947).
[551] C. Mentzer, P. Meunier, Bull. Soc. chim. France [5], 11, 302 (1944).
[552] L. Claisen, Ber., 26, 2729 (1893); Ann., 297, 1 (1897).
[553] R. G. Jones, J. Am. Chem. Soc., 74, 4889 (1952).
[554] L. Claisen, N. Stylos, Ber., 20, 2188 (1887); C. S. Marvel,
E. E. D г e g e r, Org. Synth., Coll. v. 1, p. 232.
[555] R. F. J. Cox, S. M. M с E 1 v a i n, Org. Synth., Coll., v. 2, 1943, p. 272;
E. A. S t e c k, L. L. Ha Hock, A. J. II 0 11 a n d, j. Am. Chem. Soc.,
68, 131 (1946).
790
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
кислоты с эфиром щавелевой кислоты легко образуется метиловый эфир 3-метокса-
дяЛт2гкетоциклопентан-1-карбоновой кислоты (выход 90%) [556]:
___ СООСНз
II +1
\/\0 СООСНз
СООСНз
•СОСООСНз
СООСНз
Из 2 моль ацетофенона и эфира щавелевой кислоты получают 1,6-дифенилгек-
<антетраон-1,3,4,6 (выход 62—74%) [557]. Эфир кротоновой кислоты в качестве вини-
лога этилацетата также вступает в сложноэфирную конденсацию. Т^ак, из эфира кро-
соновой кислоты и смеси эфира щавелевой кислоты и этилата натрия получают эфир
вдавелевокротоновой кислоты [556]:
(СООС2Н5)2 + СНзСН=СНСООС2Н5 —> С2Н6ООС-СО-С112СН=СНСООС2Н5
В качестве примера приведено получение эфира щавелевосорбиновой нислоты
х<559J С2Н6ООС — СО — СН2СН = СНСН = СНСООС2НВ.
Эфир щавелевосорбиновой кислоты. К алноголяту, полученному из 13 г
калия в 85 мл эфира к 58 мл спирта, прибавляют при охлаждении и встряхивании
вначале смесь 24,3 г диэтилоксалата и 10 мл эфира и через 15 мин смесь 23,3 г
этилового эфира сорбиновой кислоты и 10 мл эфира; при этом реакционная
смесь тотчас окрашивается в темно-красный цвет. Колбу хорошо закрывают
и оставляют на 24 ч, затем как можно быстрее отсасывают образовавшуюся
желтую калиевую соль енольной формы эфира щавелевосорбиновой кислоты,
промывают ее холодным эфиром и сушат в эксикаторе на пористой пластинке.
Получают 27—28 г енолята, растворяют его в воде п подкисляют 2 н. уксусной
кислотой, в результате чего высаживается быстро застыв&ющяй свободный
эфир, который после перекристаллизации из этилацетата или лигрониа имеет
т. пл. 100ф С.
Превращение сложных эфиров карбоновых кислот в эфиры а-кетокарбоновых
цислот нагреванием с диэтилоксалатом и этилатом калия в пиридине при 709 С про-
текает следующим образом [560]:
' ВСНаСООСзНа-ИСООСаНбЬ —> RCH2COCOOH + CO2 + 3C2H5OH
Эфиры угольной кислоты реагируют с образованием нарбалкокспльной группы
1561]. При реакции октанона-2 с диэтилкарбонатом и амидом натрия получают с 52% -
щым выходом этиловый эфир гептаноилуксусной кислоты [562]. Из нитрилов, соответ-
ственно, получают эфиры а-цианкарбоновой кислоты с выходом 87% от теоретиче-
ского [563]:
ВСН2СХ-|-О=С(ОСгН5)2 —> RCII(CN)COOC2H8
3556]
557
558
559
560
561
V562]
•1563]
Р. S. Pinkney. Org. Synth., Coll. v. 2, 1955, p. 116; G. К 0 m p p a,
A. T a 1 v i t i e, Ann. Acad. Sci. fenn., Ser. A., 57, № 15, 3 (1941); C., 1943,
If, 1279.
I. L. Finar, J. Chem. Soc., 1955, 1205.
A. Lapworth, Proc. Chem. Soc.. 16. 132 (1900).
W. Borsche, R. Manteuffel, Ber., 65, 868 (1932).
F. Adickes, G. Andresen, Ann., 555, 41 (1944).
J. C. Hessler, Am. Chem. J., 32, 120(1904); W.L.Nclson, L. H. ,C ret-
cher, J. Am. Chem. Soc., 50, 2758 (1928).
B. Levine, C. R. Hanse r, J. Am. Chem. Soc., 66, 1758 (1944); G. W. A n-
derson et al., J. Am. C.hpm. Soc., 67, 2197 (1915).
V. H. Wa 11 ing f 0 rd, D, M. J ones, A. H. 11 oineyr, J. Am. Chem.
Soc., 64, 576 (1942).
VI. ОБРАЗОВАНИЕ С—С-СВЯЗИ С ОТЩЕПЛЕНИЕМ СПИРТА И Др.
793
С хорошим выходом протекает синтез 4-оксикумаринов из производных о-окс®-
ацетофенона и диэтилового эфира угольной кислоты [564]:
СНдО. л /ОН СНзО\ а ,он
YY +0=6(0^-^ YY “оад
учосп, у\с04н,
СНдО СН3О
CHS°\/\/0^°'
CH3O
ОН
С отщеплением спирта протекает реакция между ортоэфирами и синильной,
кислотой в присутствии ZnCl2; при этом с очень хорошим выходом образуются а,а-
диалкоксинитрилы [565]:
RC(OR')3-i- HCN
RC(OR')2-I-R'OH
1
CN
При использовании в качестве эфирной компоненты этоксиизоформанилидг.?
можно с хорошим выходом (80%) получить азометины [566]:
CH=NC6H5
-ОН
он
+ CoH50-CH=NCeHs
Важный вариант сложноэфирной конденсации разработал Дикман [567/..
.Он состоит во внутримоленулярной конденсации эфира двухосновной кислоты. Так,,
например, из диэтилового эфира адипиновой кислоты получается эфир циклопентанов-
карбоновой кислоты [556].
]/'Ч-СООС2Н5
-СООС2Н5
-----р-СООСзНб
Эфир циклопентанонкарбоновой кислоты. В трехгорлую колбу емкостью/
3 л, снабженную мешалкой, капельной воронкой на 250 мл и обратным холо-
дильником с хлоркальциевой трубкой, помещают 23 г (1 моль) натрия и 250 мл
сухого толуола и при перемешивании прибавляют по каплям 202 г (1 моль)
диэтилового эфира адипиновой кислоты в течение 2 ч. Во время прибавления
и ещо в течение 5 ч температуру масляной бани, в которую помещена колба»
поддерживают около 100—115® С. Периодически прн перемешивании через
обратный холодильник добавляют сухой толуол, для того, чтобы смесь была
достаточно жидкой; необходимо от 750 мл до 1 л толуола. После охлаждения
смеси льдом ее медленно выливают в 1 л охлажденной до 0® С (смесью льда»
и соли) 10%-ной уксусной кислоты. Толуольный слой отделяют, промывают
один раз водой, два раза холодным 7%-ным раствором Na2CO3 и опять водой-
Толуол отгоняют при нормальном давлении, а остаток перегоняют в вакууме-.
Выход продукта 115—127 г (74—81% от теоретического); т. кип. 83—88’ СТ
(5 мм рт. ст.) или 79—84° С (3 мм рт. ст.). х
[564] J. Boyd, A. Robertson, J. Chem. Soc., 1948, 174.
[565] J. G. Erickson, J. Am. Chem. Soc., 73, 1338 (1951).
[566] E. В. К n 0 t t, J. Chem. Soc., 1947, 976.
[567] W. Diockmann, Ann., 317, 27 (1901).
792
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
Конденсацию Дикмана с успехом применяют при полном синтезе алкалоидов,
например тропина (см. [568]). С помощью конденсации Дикмана получают также
циклические кетоны с большим числом членов [569].
2. Отщепление карбоновых кислот
0-Дцкетоны, которые, как уже упоминалось, легко образуются при конден-
сации сложных эфиров с кетонами, можно получить еще и другим путем, а именно
конденсацией метилкетонов с ангидридами кислот в присутствии BFS:
П-СО-СП3 + (СН3СО)аО Н-СО-СН2-СО-СН3 + СНзСООП
Бензоилацетон [570]. Смешивают 20 г ацетофенона с 34 г (2 моль) уксусного
ангидрида и при охлаждении льдом пропускают до насыщения BFS. К концу
реакции реакционная масса почти полностью затвердевает с образованием жел-
товатых кристаллов. Их переносят в раствор 45 г кристаллического ацетата
натрия в 100 мл воды и перегоняют с водяным паром. Вначале выделяется
незначительное количество масла, затем в холодильнике начинает отделяться
кристаллический бензоилацетон. Его экстрагируют эфиром, промывают бикар-
бонатом, сушат над Na2SO4 и перегоняют в вакууме. Выход бензоилацетона
22,5 8 (83,3% от теоретического).
Имеются и другие примеры этой реакции [571].
VII, Образование С—С-связи в результате отщепления азота,
аминов и аналогичных соединений
1. Отщепление элементарного азота
Образование новой С—С-связи возможно при действии диазосоединений на
альдегиды и другие классы соединений, а также через диазониевые соединения.
Как алифатические, так н жирно-ароматические азосоедииения отщепляют
при нагревании азот 'по общей схеме:
R—N=N-R' —> R—R' + N2
Эти реакции представляют только теоретический интерес. Таким методом Гом-
бергу [572] удалось получить из трифевилметаназобензола тетрафЬнилметан, иоторый
однако легче может быть синтезирован другими методами [573].
Альдегиды или кетоны могут-реагировать с диазометаном с образованием повой
С—С-связи различным образом в зависимости от их строения. Подробнее об этой
реакции см. стр. 719.
Диазоуксусный эфир реагирует с бензолом с отщеплением азота и образова-
нием ф-фенилуксусного эфира (эфира норкарадиенкарбоновой кислоты) [574]:
j j + NjCHCOOtiHs | 1/CIICOOCaHs
[568] R. W 111 s t a 1t e r, M. Bommer, Add., 422, 15 (1921).
[569] M. Stoll, Chimia (Zurich), 2, 223 п примечание 31 (1948); E. A. Prill,
S. M- McElvaia, J. Am. Chem. Soc.. 55, 1233 (1933); N. J. Leonard
et al., J. Am. Chem. Soc., 71, 3098 (1949); 74, 1704, 4620, 6251 (1952).
[570] H. Meerwein, D. V о s s e n, J. prakt. Chem., 141, 157 (1934).
[571] C. R.,Hauser, J. T. Adams, J. Am. Chem. Soc., 66, 345 (1944);- 67,
284 (1945).
[572] M. Gomberg, II. W. Berger, Ber., 36, 1090 (1903); 30, 2043 (1897).
[573] M. Gomberg, О. К a m m, J. Am. Chem. Soc., 39, 2009 (1917).
[574] N. Braren, E. Buchner, Bor., 34, 989 (1901).
VII. ОБРАЗОВАНИЕ С—С-СВЯЗИ С ОТЩЕПЛЕНИЕМ АЗОТА, АМИНОВ
793
Замещенные эфиры уксусной кислоты получают освещением смеси диазо
уксусного эфира и алициклических соединений [575]:
' Г-i+N2CHCOOC2H6 Г“|
>/ ' '/'СНгСООСШ.,
Эфиры алкилзамещениых а-галогенуксусных кислот легко образуются из алкнл-
галогенидов и диаэоуксусного эфира, например, эфир а-аллил-а-бромуксусной нис-
лоты—из аллилбромида (выход'68—74%) [576].
Диазосоединения могут различным образом реагировать с образованием С—С-
связи. Так, из диазотированного п-нитроанилина и бутадиена получают с 88%-ным
выходом 4-хлор-1-(п-нитрофенил)-буген-2 [577] в присутствии хлорида меди и ацетата
натрия в водном ацетоне
O!N-<^y^>-N.Cl + CH2=CH-CH=CH2 —>
—► O2N-<^^>-CH,-CH=CH-CH2Cl
в то время как при тех же самых условиях в случае циннамилиденакриловой кис-
лоты получается 1-фенил-4-(п-нитрофенил)-бутадиен-1,3 [578]
O.N—+ НООС-СН=СН-СН=СН-^2/ —-
—> O2N-^ СН=СН-СН=СН-<^2^>
Последняя реакция является особым случаем: арилирования по Меервейну,
j., при помощи которого можно получить с хорошим выходом стильбены нз солей диазо-
f ния и коричной . кнслоты [579]:
' ArN2Cl + HOOC-СН=СН-Аг' —>• Ar-CH=CH—Ar'-pHCl + CO2-f-N2
п-Хлорстильбен [580]. При 0°С диазотируют раствор 26 г га-хлоранилина
в 100 мл воды н 80 мл НС1 раствором 15 г NaNOa в 30 мл воды. Прозрачный
•; (обязательное условие!) раствор диазосоединения прибавляют к охлажденному
у раствору 30 г коричной кислоты в 250 мл ацетона. После добавления 44 г
ацетата натрия прибавляют раствор 8,5 г СиС12 в 20 мл воды. Когда начнется
выделение газа, температура может повыситься до 16° С. Смесь перемеши-
вают 3 ч при 14—16е С. К концу реакции образуются 2 слоя. Верхний слой —
темио-золеное масло, нижний — смесь воды и ацетона (окрашен в светло-зеле-
1 ный цвет). Реакционную массу перегоняют с водвиым паром. Остаток раство-
• ряют в бензоле и промывают несколько раз 3 н. раствором NHS и водой. После
отгонки бензола остается кристаллическое вещество, которое кристаллизуется
из изопропилового спирта в виде блестящих листочков. Выход продукта 40%
от теоретического; т. пл. 129’ С.
[575] W. ч. £. D о ет i n g, L. Н. К п о х, J. Am. Chem. Soc., 78, 4947 (1956).
[576J И. А. Д ь я н о в а, Н. Б. Виноградова, ЖОХ, 21, 851 (1951): С. А.,
46, 440d (1952). •
[577] Е. С. С о у п е г, G. A. R о р р, J. Am. Chem. Soc., 70, 2283 (1946).
< [578] F. В er g т a n n, Z. Weinberg, J. Org. Chem., 6, 134 (1941).
! [579] H. M eerwein, E. Buchner, K. van В mst er, C., 1939, II, 1665;
J. prakt. Chem. [N. F. ], 152, 237 (1939).
[580] F. Bergmann, J. Welzman, D. Schapiro, J. Org. Chem., 9,
408 (1944). B
794
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕИ В РЕЗУЛЬТАТЕ ОБМЕНА
При взаимодействии солей диазония с акрилонитрилом с отщеплением азота
образуется а-фенил-а-хлорпролионитрил (выход 80% от теоретического) [581], а при
.реакции с оксимом формальдегида — альдегид [582]:
С1-О“ NaCI + H'ic=N0H —* [ci— S—сн=кон] —>
—*“ С1—\_/~СН0
По способу Зандмейера антрилы легко получаются из диазониевых соединений
яри нагревании их с цианидом меди [583]. В качестве примера приведена пропись
получения о-нитробензонитрила из о-нитроанилина [584].
o-Нитробензонитрил. Смесь 13,8 г (0,1 моль) порошкообразного о-питроани-
липа перемешивают с 17,5 мл концентрированной НС1 до превращения о-нитро-
анилина в бесцветный гидрохлорид. Затем быстро прибавляют 500 мл воды
и к полученной суспензии при перемешивании прибавляют по каплям в течение
1 ч раствор 7 г (0,1 моль) NaNOz в 40 мл воды. Охлаждать прн этом не обяза-
тельно. Олесь перемешивают еще 1 ч, после чего отстаивают и декантируют
от непрореагировавшего о-нитроанилина. К нагретому до 90° С раствору
K.CN и CuCN (из 25 в гидрата CuSO4, 150 мл воды и 28 г 96%-ного KCN) очень
медленно прн перемеютванин приливают полученный, как указано выше,
раствор соли диазония. Смесь выдерживают при 90—100° С, затем нагревают
несколько минут до кипения и фильтруют в горячем состоянии. (Синтез проводят
в вытяжном шкафу, так как может образоваться некоторое количество ди-
циана.) При охлаждении фильтрата выделяются кристаллы. Остаток на фильтре
экстрагируют кипящей водой до тех пор, пока из экстракта при охлаждении не
перестанет выделяться осадок. Выделившиеся из экстракта кристаллы отфиль-
тровывают, промывают водой, сушат и экстрагируют кипящим четыреххлористым
углеродом, причем неорганические соли остаются в остатке. Раствор охлаждают
и отфильтрованный осадок дважды перекристаллизовывают из разбавленной
уксусной кислоты. Выход продукта 9,7 г (65% от теоретического); т. пл. 109,5° С.
Известны и другие примеры использования реакции Зандмейера в препара-
•фявных целях [585].
Диазоеоединения разлагаются уже в водных растворах в присутствии порошка
меди и спирта, причем образуются производные дифенила [586]. Реакция исполь-
зуется только в отдельных случаях. Вариантом этого способа является разложение
солей диазония раствором тетрамминсульфата меди и гидроксиламина, в результате
чего • получают с 84%-ным выходом диарилы [587].
С почти количественным выходом протекает реакция диазотированного га-ами-
воацетофенона с хиноном с образованием 2-(л-ацетилфенил)-хинона [588].
Очень ценным синтезом является внутримолекулярная конденсация ю-фенили-
фованных о-аминокоричных кислот с образованием производных фенантрена [589,
[581] W II. В г u п н е г et al., Mh. Chem., 79, 187 (1948); 82, 100 (1951)/
[582] W. F. Beech, J. Chem. Soc.. 1954, 1297.
<583] T. S an d mey er, Ber., 17, 2650 (1884).
{3841 M. T. В о g e г t W. Fl H а н d, i. Am. Chem. Soc., 24, 1035 (1902).
(585] H. T. C i a r k e, R. R. Re'ad, J. Am. Chem. Soc., 46, 1001 (1924);
H. T. Clarke, R. R. Read, Org. Synth., 4, 69 (1925).
1586] L. Gattermann, R. Ehrhardt, Ber., 23, 1226 (1890).
(5871 L. Chardonnens, A. Wiirmli, Helv. chim. acta, 29, 922 (1946);
E. R. Atkinson et al., J. Am. Chem. Soc., 63, 730 (1941); 67, 1513 (1945),
(588] G. A. Heynoldg, J. A. Van Allan, Org. Synth., 34, 1 (1954).
M589] Обзорная статья: De Les E. de Tar, Org. Reactions, 9, 409 (1957).
VII. ОБРАЗОВАНИЕ С—С-СВЯЗИ С ОТЩЕПЛЕНИЕМ АЗОТА, АМИНОВ
795
590]. Эта реакция, известная под названием фенантренового синтеза Пшорра [591],
протекает по схеме:
Таким способом можно получать замещенные фенантренкарбоновые кислоты,
декарбоксилирование которых приводит к производным фенантрена.
Аналогичным способом возможно замыкание флуоренонового кольца. Например,
при нагревании диазотированного 4-метил-2'-амино-5'-нитробензофенона при 50е С.
в течение 3 ч получают с выходом 75% З-метил-7-нитрофлуоренон 1587, 592):
Диазокетоны могут циклизоваться с отщеплением азота. Например, при обр&-
£ , ботке 2-(2'-<в-диазоацетилфенил)-нафталина смесью уксусной и оерной кислоты обра-
ауется хризен од [593]:
ОН
2. Отщепленне аминов [600]
Наряду с описанными здесь методами образования С—С-связи с отщеплениявв
элементарного азота известны также способы, при которых отщепляются производвые-
аммиака. Так, из этилового эфира пиррол-ГЧ-карбоновой кислоты и нитрозоацет-
анилида был получен этиловый эфир 2-фенилпиррол-М-карбоновой кислоты [594]г
____ NO ____
; ! JI+CsHsN<r00c н ~\ Л
\ Z соисана \ /\г и
ь N N beHs
СООС^Нб СООС2Н5
F, С отщеплением амина протекает ряд синтезов, имеющих препаративное значение..
, Так, при кипячении гидрохлорида Р-диметиламинопрониофенона в течение-
30 мин с водным раствором K.CN отщепляется диметнламин и образуется с 67%-ньид
•• выходом ₽-бен8оилпропионитрил [595]:
£ СвН6СОСНаСНаМ(СНз)3 - HC1 + KCN —> CeHfiCOCH2CH2CN + HN(CH3)2
V. [590] Р. Н. Leake, Chem. Rev., 1956, 27.
[591] R. P schorr, Ber., 29, 496 (1896); 39, 3106 (1906).
[592] L. Chardonnens, C. Perriard, Helv. chim. acta, 28, 593 (1945/
[593] J. W. Cook, R. Schoental, J. Chem. Soc., 1945 288.
. [594] I. J. Rinkes, Rec. trav chim., 62, 116 (1943); C., 1943; П, 1363- E. Bam-
berger, Ber., 30, 366 (1897).
. [595] E. B. Knott, J. Chem. Soc., 1947, 1190; E. Haggett, S. Archer,
J. Am. Chem. Soc., 71, 2255 (1949).
796
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕ ИА
Аналогично с отщеплением триметиламина протекает реакция иодметилата
грамина и KCN с образованием индолил-3-уксусной кислоты (выход 76% от теорети-
ческого) через соответствующий нитрил (596, 597]. Этот нитрил можно получать также
нз индола в диэтиламиноацетоиптрила (выход 33—44%) [598]:
|| 4- (C2H6)2NCHaCN
NH
NH
CH,CN + HN(C2HS)2
Алкилирование соединений с подвижным атомом водорода с отщеплением
амина наблюдается и в других реакциях. Например, при нагревании кетонов с 0-ди-
метнламинопропиофеноном в течение 20 мин при 160° С легко получаются 1,5-дике-
тоны (выход 95%) [599]
^^.СНаСНгСОСвНз
| I -|-(СНз)2МСН2СН2СОСвП6 —>1 [ +(CH3)2NH
\Z\q
в то время нак при кипячении этого же аминокетопа с 2-ннтропропаном и NaOH
в течение 6,5 ч образуется с выходом 82% 2-нитро-4-бензоил-2-метилбутан [599J.
При нагревании грамина до 115° С с эфиром метантрикарбоновой кислоты получается
продукт конденсации, который без выделения в чистом виде превращают далее щелоч-
ным омылением и частичным декарбоксилированием в скатилмалоновую кислоту
(выход 67%) £[602]:
А-----S-CH!N(CHJ), СНаС(СООС2Н„)3 —
I! I II -|-НС(СООСзг15)з —► ||
—СН3СП(СОЭН)2
При взаимодействии грамина с эфиром нитромалоновой кислоты вначале полу-
чают этиловый эфир а-нитро-а-карбэтоксн-0-(индолил-3) -пропионовой кислоты (выход
96,5%), который частичным омылением и декарбоксилированием и последующим
восстановлением и омылением может быть превращен в триптофан [603]:
/Ч------ CH,N(CH,),
М\ Z
NH
+HC(COO<iH,), — TCH4(COOC^
U 7 N0’
-Г—CIbCHCOOCiHs „ —CHjCHCOOH
« । Ji
Z NOa / NHa
[596] T. A, Geissman, A. Armen, J. Am. Chem. Soc., 74, 3916 (1952).
[597] H. R, Snyder, F. J. Pilgrim, J. Am. Chem. Soc., 70. 3770 (1948).
[598] E. L, E 1 i e I, N. J. Murphy, J. Am. Chem. Soc., 75, 3589 (1953).
[5991 N, S. G i 11 et al., J. Am. Chem. Soc., 74, 4923 (1952).
[600] Обзорные статьи: H. Hellmann, Angew. Chem., 65, 473 (1953);
J. H. Brewster, E. L. Eli •!,’Org. Reactions, 7, 100 (1953).
[601] R. Adame, C. S. Marvel, J. Am. Chem. Soc., 42, 310, 316 (1920).
[602] H. R. Snyder, E. L E liel, J. Am. Chem. Soc., 71, 663 (1949).
VII. ОБРАЗОВАНИЕ С—С-СВЯЗИ С ОТЩЕПЛЕНИЕМ АЗОТА, АМИНОВ
797
> Для синтеза различных аминокислот с большим успехом применяют ацилиро-
ванные аминомалоновые эфиры 1604]. Например, реакцией грамина с ацетиламино-
• малоновым эфиром [605] получают триптофан с выходом 73%, считая на грамин.
I, Й,Л-Трнитофан. К кипящей смеси 1200 мл толуола (или ксилола) и 17 г
порошкообразного NaOH в трехгорлой колбе емкостью 5 л, снабженной мешал-
кой, обратным холодильником и барботером, прибавляют 250 а (1,43 моль)
грамина и 311 г (1,43 моль) этилового эфира ацетиламиномалоновой кислоты.
Смесь нагревают 5 ч при энергичном перемешивании и пропускании азота. Бурное
। вначале выделение диметилаланина и концу реакции ослабевает. Реакционную
смесь фильтруют на воронке для горячего фильтрования и фильтрат охлаждают
до иомнатнон температуры. После выдерживания в течекие нескольких часов
» при 5° С продукт отфильтровывают и промывают сначала холодным толуолом,
затем петролейным эфиром. Выход продукта 446 г (90% от теоретического);
т. пл. 158—159® С.
В течение 4 ч кипятят с обратным холодильником 33,62 г (0,097 моль)
полученного эфира с раствором 19,2 г (0,48 моль) NaOH в 192 мл воды. После
' обработки активированным углем раствор охлаждают льдом и прибавляют
50 мл холодной концентрированной НС1 (0,6 моль) так, чтобы температура
не превышала 25° С. Раствор охлаждают в течение 4 ч, отфильтровывают
светло-розовый осадок, и сушат его в вакуум-эксикаторе над СаС1» в течение
15 ч. Сырой продукт, который содержит небольшое количество NaCl, плавится
при 136—139° С. Выход продукта 32 г.
j. Смесь 28 г полученной кислоты и 120 мл воды в течепие 2,5 ч кипятят
£ с обратным холодильником. Образующийся N-ацетилтриптофан менее растворим,
чем кислота, и выкристаллизовывается в процесс^ реакции, Затем прибавляют
16 г (0,4 моль) NaOH в 40 мл воды п кипятят раствор с обратным холодильии-
ь ком в течение 20 ч, после чего обрабатывают углем и подкисляют 24 г (0,4 моль)
, ледяной уксусной кислоты. При этом тотчас же образуется белый осадок, кото-
рый после выдерживания в течение 12 ч в холодильнике отфильтровывают
и растворяют в 200 мл воды, содержащей 5 г NaOH. Раствор обрабатывают
u активированным углем, прибавляют 100 мл 95%-ного этилового спирта, нагре-
вают до 70° С, подкисляют 7,5 мл ледяной уксусной кислоты и медленно охла-
j ждают. Выпавшие кристаллы отфильтровывают, промывают двумя порциями
1 (по 40 мл) воды, 40 мл этилового спирта и 30 мл эфира. Выход триптофана
J 14 г (81% , считая на эфир); т. пл. 273—280° С (разл.); перекристаллизация
из 33%-ного спирта.
С отщеплением амина происходит также ациламипометилирование ацетиламино-
ь малонового эфира;
г NHCOCH3
« ^H6CONHCH2N(CH3)2 + CH(COOC2H5)2 —>
* NHCQCHg NH2 NH2
1 I I I
) —> CeH5CONHCH2C(COOC2H&)2 —*• CH2— CH-COOH
Гидролизом продукта конденсации, образующегося из диметиламинометил-
k ^бензамида и ацетиламиномалонового эфира с выходом 87% , была получена а,0-диами-
вопропионовая кислота (выход 94%) (606}.
1603} D. I. We isblat, D. A. L у t 11 е, J. Am. Chem. Soc., 71, 3079 (1949).
1604] A. Oalat, J. Am. Cliem. Soc., 69, 965 (1947); H. R. Snyder et al., J. Am.
Chem.'Soc., 66, 200 (1944); C. Schopf, J. Thesing, Angew. Chem., 63,
377 (1951); A. Butenandt, W. W ei d el, W. v. D e г j u g i n, Natur-
wiss., 30, 51 (1942); N. F. Albertson, B. F. Tnllar, J. Am. Chem.
Soc., 67, 502 (1945); H. Hellm ann, E. Brendlo, Hoppe-Seyler's Z. phy-
siol. Chem., 287, 235 (1951).
1605] E. E. Howe, A. J. Zambito, H. R. Snyder, M. Tisher, J. Am.
Chdm. Soc., 67, 38 (1945); H. R. Snyder, C. W. Smith, J. Am. Chem.
вое., 66, 350 (1944); H. Hellmann, Hoppe-Seyler’s Z. physiol. Chem., 884,
163 (1949).
(6Й61 Hellmann, G. Haas, Ber., 90, 1357 (1957).
798
ОБРАЗОВАНИЕ ПРОСТЫХ С—С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
Исходным соединением для синтеза азулена с хорошим выходом является
производное фульвена [607], ноторое получают с количественным выходом ив цикло-
пентадиена и монометиланилида глутакоиового альдегида;
'^/^-СНСН-СНСН=СНЛ/СНЗ —► Г~/ \
Если вместо аминов применять диалкиламиды ароматических карбоновых
кислот, то можно ввести ацильные группы. Например, при реакции пиридина с диме-
тилбензамйдом образуется смесь, содержащая в основном 2-оензоилпиридин и неболь-
шое количество 4-бенаоилпирвднна [608]:
Л+СвН6С(Ж(СНз)з
V' VZoc,hs
По такой же схеме протекает синтез альдегидов по Вильсмейеру [610, 611],
который заключается во введении формильной группы при помощи алкилированных
производных формамида:
0 R
ArH+HC<R -^-Агсно + hn/
\R, к
Эта реакция имеет широкую область применения. При взаимодействии аромати-
ческих углеводородов, ариловых зфиров [611], конденсированных ароматических
соединений [612а] и гетероциклов (например, производные тиофена [613] или пиррола
[614]) с N-метилформанилидом и хлорокисью фосфора с превосходными выходами
образуются соответствующие альдегиды. При проведении реакции с высокоплавящи-
мися конденсированными ароматическими соединениями рекомендуется использовать
растворители, например днхлорбензол.
В качестве примера синтеза Вяльсмейера описано получение 2-этокси-1-нафт-
альдегида [612а].
2-Этокси-1-нафтальдегид. В круглодоииой колбе емкостью 500 мл,
снабженной воздушным холодильником, нагревают па водяной бане в течение
6 ч смесь 45 з (0,33 моль) N-метилформанилида' [6126],- 51 г (0,33 моль) РОС1з
и 43 з (0.25 моль) р-иафтплэтилового эфира. Горячий раствор • выливают
при энергичном перемешивании тонкой струйкой в 700 мл холодной воды,
чтобы избежать образования сгустков. Альдегид выделяется в виде крупинок.
Его отсасывают и тщательно промывают 1 л воды. Влажный альдегид раство-
ряют в 450 мл этилового спирта, прибавляют 4 е активированного угля, кипятят
15 мин н горячий раствор фильтруют па воропке для горячего фильтрования
с двойным фильтром. Фильтрат охлаждают, выпавший продукт отсасывают'
[607] К, Ziegler, К. Hainer, Angew. Chem., 67, 301 (1953).
[608] G. В. В а с h m a n, R. М. S с h i s 1 a, J. Org. Chem., 22, 858, 1302 (1957).
[609] R. H. Glauert, F. G. Mann, J. Chem. Soc., 1952, 2127.
[610] O. Bayer, в книге Houben-Weyl, Methoden der organischen Chemie,
Bd. 7/1, S. 29; A. v a n Dormael, Ind. chim. beige, .16, 433 (1951);
A. Vilsmeier, Chem. Ztg., 75, 133 (1951); H. II. Bosshard, Hch.
Zollinger, Helv. chim. acta, 42, 1659 (1959).
[611] A. Vilsmeier, A. Haack, Ben, 60, 119 (1927).
[612] L. F. Fieser, J. L. Hartwell, J. E. J 0 n e s, Org. Synth., Coil,
v. 3. 1955, а) стр. 98; б) стр. 590.
[613] A. W. Westo n, B. J. Michaels jr., J. Am. Chem. Soc., 72, 1422 (1950);
Org. Synth., 31, 108 (1951).
[614] A. C. Shabica et al., J. Am. Chem. Soc., 68, 1156 (1946).
I. ОТЩЕПЛЕНИЕ ВОДОРОДА С ОБРАЗОВАНИЕМ НОВЫХ С=С-СВЯЗЕЙ
799
и промывают 40 мл холодного этилового спирта. Альдегид кристаллизуется
в виде светло-желтых игл. Выход продукта 37—42 г (74—84% от теоретиче-
ского); т. пл. 111—112’С.
Вместо N-метилформапнлида можно применять также другие формамиды* [615].
Хлорокись фосфора можно заменить фосгеном или тионилхлоридом (616]. Введение
«ето- или альде'гидной группы возможно также с помощью амидинов. Например,
«из тиооксиидола и дифенилацетамидина был получен анил 3-ацетилтиооксиядола
/выход 64%) (609], который с 90%-ным выходом можно перевести в 3-ацетилтио-
•оксиндол:
СНз
Таким же образом действием днфенилформамидина получают апил тиооксин-
дол-3-альдегида (выход 70—78%), омыление которого дает альдегид с выходом 72%.
k По реакции Даффа получают феполальдегиды (617] нагреванием соответствующего
•фенола с уротропином, борной кислотой и глицерином в точение 30 мин при 150—
160’0. И хотя выход здесь пе превышает 15—20% от теоретического, однако этот
Лспособ проще и требует меньше времени, чем, например, синтез Реймера — Тимана.
£ Таким же образом можно формилировать диалкиланилин [618]. Вдетом случае обра-
зуются «-альдегиды, в то время как фенолы дают о-соединепия.
; ОБРАЗОВАНИЕ ДВОЙНЫХ С=С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ
> РЕАКЦИЙ ОБМЕНА
,7. Отщепление водорода с образованием новых этиленовых связей
< . Классическим примером образования этиленовой связи с отщеплением водорода
является синтез нндиго. Хотя в качестве дегидрирующего средства здесь достаточно
кислорода воздуха, реакция протекает лучше в присутствии окислителей. Например,
производные стильбена могут быть получены окислением иодом замещенных толуолов.
п,п'-Дибром-а,а'-дицианстильбен [619]. Смешивают эфирный раствор иода
к с раствором п-бромбензилцианида в спирте и к смеси при охлаждении приба-
вляют по каплям этилат натрия в этиловом спирте. Выход продукта 78—82%
£ от теоретического. Эта пропись основана на ранней работе Чаланан и Кневс-
£•_ нагеля (620].
• В другом методе в качестве дегидрирующего средства применяют серу. Еще
«.^Циглер [621] получал тетрафенилэтилен нагреванием дифенилметапа с серой при
!|240—250° С. Сообщалось также о синтезе с 57%-ным выходом п,п'-стильбендикарбо-
-'-новой кислоты из л-толуиловой кислоты и серы [622].
'• Особенно рекомендуется более доступный диметилформамид. — Прим. ред.
[615] Е. Campaign е, W.L. Archer, J. Am. Chem. Soc., 75, 989 (1053);
S. Akabori, Y. Senoh, Bull. chem. Soc. Japan, 14, 166 (1939).
i’-[616J N. Roh, G. Kochendorfer, герм. лат. 677207 (1937); Frdl., 25, 171;
К. Hafner, C. Bernhard, Angew. Chem., 69, 533 (1957).
t. (61*1] J. C. D u f f, J. Chem. Soc., 1941, 547; L. M. L i g g e t, H. Diehl, Proc.
Iowa Acad. Sci., 52, 191 (1945).
; 618J J. C. Duff, J. Chem. Soc., 1945, 276.
i- 619] M. Weizmann, S, Patai, J. Am. Chem. Soc., 71, 2587 (1949).
620] L. Chalanay. E. Knoe venagel Ber., 25, 285 (1892).
( 621] H. Ziegler, Ber., 21, 779 (1888).
[62SJ W. G. Toland jr., J. B. Wilkes F.-J. Brutschy, J. Am. Chem.
Soc., 75, 2263 (1953); 76, 307 (1954).
I
800
ОБРАЗОВАНИЕ ДВОЙНЫХ С=С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
В подобных синтезах с успехом использовали также монохлористую серу [623].
Так, с выходом 78% получали М,М-дифеннлизоиндпго из N-фенилоксиндола, а пз
оксиндола — пзонндиго с выходом 90%.
II. Отщепление галогена с образованием новых
эти леновых связей
1, Отщепление галогена действием металлов
Эта реакции, расширяющая синтезы Вюрца — Фиттига, имеет некоторое зна-
чение только для синтеза углеводородов, да н то лишь в ряду производных стильбена.
Так, из дифени.чдихлорметана и порошка меди с выходом 70% был получен тетра-
фенилэтилен [624].
Достойным упоминанием является синтез бис-1,3-дикетоияданилиденов, явля-
ющихся углеродными аналогами индиго:
О 0 0
Бис-1,3-дикетоинданилиден [625]. Кипятят с обратным холодильником 2 г
2,2-дихлориидандиона-1,3 в присутствии медной бронзы в сухом бензоле в те-
чение 6 ч. Выход продукта 1,2 з.
2. Отщепление галогена действием щелочей
Удаление ^галогеноводорода щелочами для С =С-связывания фрагментов удается
только в том случае, если связь углерод — галоген ослаблена каким-либо заместите-
лем, например нитрогруппой, в ряду алифатических соединений. Так, можно получить
3,4-динитрогексен-3 из 1-хлор-1-нитропропана [626] обработкой его водным КОН.
Эта реакция значительно чаще применяется в случае жирно-ароматических соедине-
ний. Ослабление связи углерод — галоген вызвано здесь одной или несколькими
фенильными группами. Правда, оказалось, что применение гидроокисей щелочных
металлов часто дает неудовлетворительные результаты и что лучших выходов можно
достичь, используя органические основания. Кроме того, имеет значение выбор раство-
рителя. Например, бис-(2,7-дибромфлуореиилиден) был получен с 67%-пым выходом
из 2,7-дибром-9-хлорфлуорена обработкой его триэтвлампяом в нитрометане [627];
применение спиртового раствора NaOH или триэтиламина в изоамиловом эфире дает
плохие результаты.
В начестве отщепляющих средств с успехом используют также холин (628]
и амид калия [629]. При обработке спиртовой гидроокисью щелочного металла нитро-
или циэнбеязилхлориды очень гладко переходят в соответствующие стильбены.
4,4'-Дннитростнльбен [630]. Растворяют 50 г n-иитробензилхлорида в 150 г
теплого этилового спирта и медленно смешивают после охлаждения со спирто-
вым раствором КОН, полученным из 17,5 г КОН, 15 мл воды и 60 г этилового
спирта. При прибавлении первых капель раствор окрашивается в зеленый
[623J Р. Chovin, Bull Soc. chim. France, 12, 100, (1945); 11, 82 (1944).
[624] R. E. Buckles, G. M. Ma t lack, Org. Synth., 31, 104.
[625] A. Schonberg, R. Moubasher, J. Chem. Soc., 1949, 212.
[626] D. E. В i s g г о v e, J. F. Brown jr., L. B. Clapp, Org. Synth., 37, 23.
[627] L. A. Pine k, G. E. Hilber t, J. Am. Chem. Soc., 68, 2014 (1946).
[628] F. Bergmann, Bull. Soc. chim. France, 1952. 78.
[629] C, R. Hauser, J. Am. Chem. Soc., 78, 1653 (1956).
JII. ОБРАЗОВАНИЕ НОВЫХ С = С-СВЯЗЕЙ ПУТЕМ ДЕГИДРАТАЦИИ
801
цвет, который по мере протекания реакции переходит через синевато-красный
в желтый. Одновременно выделяются желтые кристаллы. Кристаллы после
полного охлаждения раствора отсасывают, промывают горячей водой и теплым
неразбавленным этиловым спиртом до отсутствия ионов хлора в промывной
жидкости. Выход продукта 36 г (90% от теоретического). Сырой продукт явля-
ется смесью стереоизомеров 4,4'-динитростильбена, и поэтому ие имеет отчетли-
вой температуры плавления. Экстракцией ацетоном удаляют более пизко-
плавкий изомер и перекристаллизацией из ледяной уксусной кислоты, аце-
тона и нитробензола получают чистый высокоплавкий изомер с т. пл. 280—
III. Образование новых С~С-связей путем дегидратации
Образование двойной связи путем отщепления воды может проходить главным
образом по реакциям двух типов:
2RaCHOH —> R2C^CR24-2H2O (1)
НгСО4-СЩН5 —> В2С=СЩ-Н2О (2)
Реакция, выраженная первой схемой, имеет небольшое препаративное значение.
Например, ангидрид дифенилмалениовой кислоты [631] получают нагреванием мин-
дальной кислоты при остаточном давлении 500 мм рт. ст.
Важнейшие методы конденсации основаны на второй схеме. Однано метильная
или метиленовая группы способны участвовать в конденсации только в том случае,
если они активированы соседними карбонильными, карбоксильными, нитрильными
или нитрогруппами или некоторыми, замещенными ароматическими остатками. Эти
реакции известны под различными названиями, в зависимости от вида функциональных
групп и применяемых конденсирующих средств.
1. Синтез «,Р- и Р,у-ненасыщенных карбоновых кислот
Синтез Перкина
Перкии в 1868 г. синтезировал кумарин нагреванием ацетата натрня, салицило-
вого альдегида и уксусного ангидрида [632]. Эта реакция получила особое значение
для синтеза «.^-ненасыщенных кислот. Ее осуществляют нагреванием ароматиче-
ского альдегида с ангидридом алифатической кислоты и ее натриевой или калиевой
солью. Реакцию Леркина можно проводить также с коричным альдегидом, но чисто
алифатические альдегиды для этой реакции не применяются.
Коричная кислота [633]. В течение 16 ч без доступа влаги кипятят с обрат-
ным холодильником 20 г свежеперегнанного бензальдегида, 30 г уксусного ангид-
рида и 10 г свежерасплавленного ацетата натрня. Затем после охлаждения
прибавляют воду, отгоияют с водяным паром непрореагировавший бензальдегид
и часть уксусной кислоты, остаток нейтрализуют разбавленной щелочью, филь-
труют в горячем виде и осаждают коричную кислоту соляной кислотой. Выход
продукта 15—17 г (57% от теоретического). При более длительном нагревании
(24 ч) выход повышается до 70—75% от теоретического [634].
Большое влияние оказывает на выход наличие заместителя в молекуле бензаль-
дегида. Например, при реакции нитро- или галогензамещеиных бензальдегидов [634,
635] с очень хорошим выходом образуются замещенные коричные кислоты. Выход
630] Р. Walden, A. Kernbaum, Вег., 23, 1959 (1890).
631] С. Bischoff, Р. Walden, Ann., 279, 120 (1894).
632] W. Н. Р er ki n, J. Chem. Soc., 21, 53, 181 (186&).
633] W. H. Perkin, J. Chem. Soc., 31, 389 (1877); Ber., 10, 299 (1877).
634] F. В б c k, G. L о c k, K. Schmid t, Mh. Chem., 64, 401 (1934).
[635] Org. Synth., 5, 83 (1925).
51 Заказ 1835.
«02
ОБРАЗОВАНИЕ ДВОЙНЫХ С = С-СБЯЗЕЙ Б РЕЗУЛЬТАТЕ ОБМЕНА
4-онсикоричной кислоты из 4-оксибепзальдегида выше (62%), чем выход коричной
кислоты из незамещенного бензальдегида. Алкил- или оксиалкилбензальдегиды
реагируют с незначительными выходами, а диметиламииобензальдегид не реагирует
вообще [636]. Для реакции Перкииа с успехом применяют также гетероциклические
.альдегиды. Так, из тиофен-2-альдегида была получена тиенилакриловая кислота
[637], описан синтез фуран-2-акриловой кислоты [638].
Конденсацией ангидридов других жирных кислот с ароматическими альдеги-
дами в присутствии соответствующих солей щелочных металлов получают а-замещен-
,ные ‘коричные кислоты.
а-Метплкоричнаянислота[639] образуется с выходом 82% при нагревании
бензадьдегида, ангидрида пропионовой кислоты и пропионата натрия в запаян-
ной трубке при 150® С в течение 24 ч.
В обычных условиях реакции Перкина коричный альдегид с очень хорошим
выходом превращается в 0-стирилакриловую кислоту [640]. При нагревании корич-
ного альдегида с фекилуксусной кислотой в присутствии уксусного ангидрида про-
исходит декарбоксилирование и образуется с 30%-ным выходом 1,4-днфенилбута-
диен [641]. В таких же условиях образуется 1,8-дифенилоктатетраен [642] из 2 моль
коричного альдегида и 1 моль янтарной кислоты.
Особое место занимают синтезы Перкина, при которых используется эквимоле-
кулярное соотношение ароматических или алифатических альдегидов и янтарной
кислоты. Прп этом получаются с хорошим_[выходом у-фенил- и у-алкилпараконовые
кислоты:
СН2-СООН СвНб-СН-СН-СООН 1ппЪг
свн5-сно +1 —* I i Чзпг
СЙ2-СООН НО СНг-СООН
Св1-1б-СН-СЕ£-С00Н
—> | I 150 Свн5—СН=СН-СП2-СООН4-СО2
,0 6н2
Этн кислоты при нагревании отщепляют двуокись углерода с образованием
-Р.у-ненасыщенных^кислот и незначительного количества соответствующего у-бутиро-
лакюна.
Фенилпараконоваякислота[643]. Нагревают при 100° С в течепие 15—20 ч
смесь бензальдегида, сукцината натрия и ацетангидрида. Затем реакционную
массу растворяют в кипящей воде, охлажденный раствор встряхивают с эфиром
и экстрагируют фенилпараконовую кислоту из эфнрпого раствора раствором
соды. Из щелочного экстракта свободную кислоту осаждают соляной кислотой.
Экстрагированием сероуглеродом можно выделить еще небольшое количество
фенилизокротоновой кислоты. По Фиттигу [644], фенилизокротоновая кислота
получается с лучшим выходом при 125—130° С или при нагревании фенил-
параконовой кислоты до 300° С. Синтез параконовых кислот по Фиттигу удается
также и с моноэамещенными янтарными кислотами. Взаимодействие фенил-
янтарной кислоты и бензальдегида при 125° С приводит к дифенилизокротоно-
[636]
[637]
[638]
[639]
640
641
642
643
644
Н. Meyer, R. Beer, Mh. Chem., 34, 649 (1913).
A. В iedermann, Ber., 19, 1855 (1886).
Org. Synth., Coll. v. 3, p. 426.
M. Conrad, C. A. Bischoff, Ann., 204, 187 (1880); R. F i t t i g,
Ber., 16, 1436 (1883).
W. H. Perkin, J. Chem. Soc., 31, 388 (1877).
В. B. Corson, Org. Synth., Coll. v. 2, p. 229.
R. Kuhn, A. Winters tei n, Helv. chim. acta, 11, 103 (1928).
R. Fit tig, H.W. Jayne, Ann., 216, 99 (1883).
R. Fittig, Ann., 216, 113 (1883).
III. ОБРАЗОВАНИЕ НОВЫХ С=С-СВЯЗЕИ ПУТЕМ ДЕГИДРАТАЦИИ
803
вой кислоте [645] СвН6—CH = C(CeHs)—СН2—СООН. При термическом-
декарбоксилировании параконовых кислот наряду с изокротоновыми иислотами
образуются также ангидриды итаконовых кислот (алкилиден- или арилиденян-
тарные кислоты) и цитраконовые кислоты (замещенные малеиновые кислоты).
Реакция Кневенагеля
Активированная метиленовая группа малоновой кислоты или циануксусиой
кислоты в мягких условиях конденсируется с алифатическими и ароматическими
альдегидами, а также с кетонами. Вначале эти реакции проводили, как и синтез Пер-
кина, в растворе уксусной кислоты с добавкой уксусного ангидрида, присутствие
которого в этой реакции пе обязательно.
Бензилцдеималоновая кислота [646].Смесь равных количеств малоновой кис-
лоты н свежеперегнанного бензальдегида нагревают 8—10 ч на кипящей водя-
ной бане с ледяной уксусной кислотой (50% от количества малоновой кислоты).
При охлаждении выкристаллизовывается с выходом 55% от теоретического
бензилиденмалоновая кислота, которую отфильтровывают и промывают хлоро-
формом. Если эту реакцию проводят в кипящей ледяной уксусной кислоте,
то одновременно происходит декарбоксилирование.
Кротоновая кислота[647].Кипятят три дня с эффективным обратным холо-
дильником смесь эквимолекулярных количеств малонового эфира и ледяной
уксусной кислоты с избытком паральдегида и затем фракционируют.]
Кневенагель [648] применил для реакций такого типа в качестве катализатора
слабое основание (аммиаг? или амины). В этом случае конденсация и декарбоксилиро-
вание образующихся на первой стадии арилиден- или ялкилидепмялиповых кислот
протекали при более низкой температуре, благодаря чему повышался выход целевых
продуктов *.
п-Метокснкоричная кислота [649]. Раствор 13,6г анисового альдегида и 12,6 г
малоновой кислоты в небольшом количестве 95%-ного спирта приливают к
раствору 21 г 8% -ного аммиака в 95% -ном спирте и смесь нагревают на паровой
бане. После того как отгонится спирт, маслянистый остаток нагревают на кипя-
щей водяной бане до прекращения выделения СО2 и затвердевания остатка.
Продукт обрабатывают теплой водой и растворяют при добавлении небольшого
количества воды. Раствор несколько минут кипятят с 1—2 г активированного
угля н затем фильтруют. Теплый фильтрат приливают к избытку охлажденной
20%-ной H2SO4; при этом выкристаллизовывается свободная кислота.
Большое значение имеет также использование в качестве растворителя пири-
дина, в результате чего становится возможным синтез кислот с несколькими двой-
ными связями. Дебнер [650] смог получить таким способом сорбиновую кислоту
(гексадиен-2,4-овую кислоту) из кротонового альдегида и малоновой кислоты. С хоро-
шим выходом проходит реакция фурфурола и малоновой кислоты с образованием
* Изучая в 1925 г. реакцию Кневонагеля с применением в качестве катализатора
спиртового аммиака, В. М. Родионов нашел, что прп определенных условиях
главным продуктом становятся p-аминокислоты. Это позволило ему разработать
общий метод получения последних (реакция Родионова; см. В. М. Р о д и о-
нов, Избр. труды АН СССР, 1958 г., стр. 327). — Прим. ред.
[645] Fr. Fichte г, W. L a t г к о, J. prakt. Chem. [2], 74, 330 (1906).
[646] L. Claisen, L. Chrismer, Ann., 218,135 (1883).
[647] T. Komnenos, Ann.,218, 149(1883); R. Fittig, Ber., 26, 2088 (1887);
Ann., 283, 82(1894); J. v. Braun, Mh. Chem., 17, 213 (1896).
[648] E. Knoevenagel, Ber., 31, 2604 (1898).
[649] J. R. Johnson, Org. Reactions, v. 1, p. 249, John Wiley a. Sons,
Inc., New York, 1942.
[650] O. D oebner, Ber., 33, 2140 (1900); 35, 1137 (1902); см. также Org. Synth.,
24, 92 (1944). ' ’ ’
51*
ОБРАЗОВАНИЕ ДВОЙНЫХ С = С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
-2-фурилдкриловой кислоты [654]. По методу Дебнера часто применяют добавку пипе-
ридина. Так, например, были получены изопренкарбоновая кислота [651], 2,3-димет-
онсикоричная кислота [652] и 2-нитро-4,5-метилендиоксикоричная иислота [653].
/1-Диметиламииобензальдегид, который не реагирует в условиях реакции Перкииа,
чпо методу Дебнера превращается в соответствующую коричную кислоту с выходом
.80% от теоретического [655]. По такому же способу была получена додекапентаекил-
иденмалоновая кислота [656], СН3—(СН=СН)5—СН=С(СООН)2, декарбокси-
лирование которой кипячением со смесью уксусной кислоты н уксусного ангидрида
Д1:1) дает с хорошим выходом тетрадекагексаен-2,4,6,8,10,12-овую кислоту.
С помощью синтеза Кневенагеля удалось также осуществить взаимодействие
нетоиов с малоновой кислотой. Например, нагревание малоновой кислоты с ацетоном
в пиридине в присутствии небольшого количества пиперидина приводит с выходом
-60% к р,0-диметилакриловой кислоте [657]. При такой же реакции с диэтилкетоном
выход снижается до 35%, а для циклогексанона он составляет всего 5% от теоретиче-
ского. В этих случаях, однако, можно осуществить конденсацию нетона с малоповой
кислотой, используя в качестве катализатора ацетат пиперидина.
Кун [658], исследуя конденсацию кротонового альдегида (2 моль) до окта-
триеналя, показал, что катализаторами этой реакции могут быть только соли вторич-
ных аминов:
2СНз—СН=СН-СНО:—»-[СНз—(СН=СН)з-СНО
Исходя из своих наблюдений, а также данных других авторов, он сделал вывод, что
•эффект рёакции?Кневенагеля основан не столько на каталитическом действии амикои,
сколько на каталитическом действии их солей.
Коуп [659] для конденсации альдегидов с циануксусным эфиром применял
ацетат пиперипипя и осуществил взаимодействие кетонов с циануксускым эфиром,
используя в качество катализатора ацетат аммония. Растворителем служил бензол,
а реакционную воду удаляли при помощи ловушки Днна — Старка. Ацетон по этому
.способу реагирует только в том случае, если вместо бензола в качестве азеотропной
.компоненты используется хлороформ [660].
Алкилидёнциануксусный эфир. Смесь 0,5 моль циануксусного эфира, 0,6 моль
кетона, 0,05 моль (3,85 г) ацетата аммония, 0,1 моль (6 з) ледяной уксусной
кислоты и 50 мл бензола нагревают до кипения с ловушкой Дина — Старка
на масляной бане при 130—160’ С. Постепенно отделяется 10—12 мл воды,
которая содержит незначительное количество ацетата аммония и ацетамида.
После отделения воды смесь кипятят еще 1 ч.
В иачестве катализаторов реакции Кневенагеля с успехом использовались
Л аминокислоты [661]. Из метилэтилкетона и этилового эфира циануксусной кислоты
в присутствии р-аланнна с выходом 87% получают этиловый эфир еигор-бутилиден-
-циануксусной кислоты [662]. Катализаторами могут служить также бензиламин
[663], добавленный к соли вторичного амина, и ионообменные смолы [664].
В качестве карбонильных компонент могут применяться эфиры 0-кетокарбоно-
вых кислот [665]. Напротив, если хотят осуществить конденсацию альдегидов с эфи-
651]
652
653
654
655
657
658
659
660
661
662
663
664
665]
Т. L е n п artz, Вег., 76, 1009 (1943).
Org. Synth., 31, 35 (1951).
К. Schofield, J. С. Simpson, J. Chem. Soc., 1945, 512.
Org. Synth., Coll. v. 3, p- 425.
Ch. W. Shop pee, J. Chem. Soc., 1930, 968.
R. Kuhn, Ch. GruBdmann, Ber., 70, 1326 (1937).
S. Dutt, Quart. J. Indian Chem. Soc., 1, 297; C., 1925, II, 1853.
R. Kuhn, W. В a dstubner, Ch. Grundmann, Ber., 69, 98 (1936).
A. C. Cope, J. Am. Chem. Soc., 59, 2327 (1937); 63, 733, 3452 (1941).
S. Wiedequist, Acta Chem. Scand., 3, 304 (1949).
F. 3. Prout, J. Org. Chem., 18, 928 (1953).
Org. Synth., 35, 7 (1955).
8. Deo, J. Indian Chem. Soc., 30, 665 (1953).
M. J. As tie, W. C. G erg el, J. Org. Chem., 21, 493 (1956).
R. Grewe, A. Mondon, Chem. Ber., 81, 283 (1948).
III. ОБРАЗОВАНИЕ НОВЫХ С=С-СВЯЗЕЙ ПУТЕМ ДЕГИДРАТАЦИИ
805
рами 0-кетоиарбоновых кислот, то используют метод Кневенагеля; хорошие резуль-
• таты получались также по методу Коупа [666].
Алкилиденацетоуксусный эфир. Смесь 0,5 лоль ацетоуксусного эфира
. и 0,55 моль альдегида охлаждают до 5° С н затем прибавляют в течение 5—10 мин
0,5 г пиперидина в 1 г спирта таким образом, чтобы температура не поднималась
?, выше -}-5—|-10Q С. Реакционную смесь затем охлаждают до 0Q С, выдержи-
вают в холодильнике 12—20 ч и промывают водой, к которой добавлено несколь-
К. ко капель ледяной уксусной кислоты (3 раза по 100.ил), Промывные воды экстра-
гируют эфиром, эфнрный слой объединяют с продуктом н фракционируют
а в вакууме, используя колонку Видмера. Выход 80% от теоретического.
Описана аналогичная реакция, которая в одну стадию приводит к эфирам
<х,р-ненасыщеииых карбоновых ннслот [667]; в качестве метиленовой компоненты
использовали моноэфир малоновой ннслоты.
Метиловый эфир п-диметиламинокоричной кислоты [667]. Нагревают с обрат-
ным холодильником на паровой бане в течение 4 ч 3 г (0,02 моль) п-диметилами-
побепзальдегида, 4,8 мл (около 0,04 моль) монометилового эфира малоновой
кислоты, 0,25 мп пиперидила н 10 мл сухого пиридина. Пиридин затем отго-
няют в вакууме, а кристаллический остаток растворяют в 10—15 мл горячего
ацетона. В этот раствор при перемешивании медленно прибавляют по каплям
воду до полного выпадения осадка. Кристаллы отсасывают, промывают ацето-
ном н сушат при 90—100° С. Выход продукта 3,9 г (95% от теоретического).
Синтезы эфиров коричной кислоты по К лай гену
Конденсация Клайзена имеет небольшое значение для синтеза «^-ненасыщен-
ных карбоновых кислот, так как в большинстве случаев предпочитают применять
указанные выше способы. По Клайзену эфиры коричной кислоты получают из арома-
тических альдегидов и эфиров жирных кислот под действием металлического натрия.
Таким способом с выходом от 70 до 80% были синтезированы метиловый эфир п-мет-
оксикоричной кислоты [668] и этиловый эфир у-фурфурилиденкротоновой кислоты
[669]. Из гомологов эфиров жирных кислот этим методом получают эфиры «-замещен-
ных коричных кислот [670].
Конденсация Штоббе
В то время кан в описанном выше синтезе параконовой кислоты исходят из ян-
, тарной кислоты, при конденсации Штоббе реакцию проводят с эфирами янтарной
кислоты или продуктами их замещения:
COOR COOR
I I
R2C=O4-H2C—CHg—COOR4-NaOR —> R2C=C-CII2-COONa + 2ROH
К карбонильным соединениям, которые могут вступать в эту реакцию, отно-
сятся алифатические, ароматические и «.^-ненасыщенные альдегиды, алифатические,
.алициклические и ароматические кетоны, дикетоны, кетоэфиры и цианкетоны, В ка-
честве конденсирующего средства применяют большей частью этилатднатрия или
.pipem-бутилат калия.
р-Карбэтокси-у,у-дифенилвинилуксусная иислота [671]. К охлажденному
раствору алкоголята, полученному из 2,15 г (0,055 лоль) калия в 45 мл сухого
[666
[667
[669
[670
4671]
А. С. С о р е, С. М. Hofmann, J. Am. Chem. Soc., 63, 3457 (1941).
A. G a 1 a t, J. Am. Chem. Soc., 68, 376 (1946).
A. H oreau, Bull. Soc. chim. France, 1948, 414.
A. H i n z, G. Meyer, G. Schiicking, Ber., 76, 683 (1943). .
L. С 1 a i s e n, Ber., 23, 978 (1890); T. Posener, J. prakt. Chem. [2], 82,
435 (1910).
W. S. Johnson, J. Petersen, W. P. Schneider, J. Am.
Chem. Soc., 69, 76 (1947).
I
806
ОБРАЗОВАНИЕ ДВОЙНЫХ С=С-СБЯЗЕЙ В РЕЗУЛЬТАТЕ СЕМЕНА
mpem-бутилового спирта, прибавляют 9,11 г (0,05 моль) бензофенона и 13,05 а
(0,075 .моль) диэтилового эфира янтарной кислоты. Воздух вытесняют сухим
азотом, смесь умеренно кипятят в течение 30 мин и после охлаждения подкисля-
ют разбавленной НС1. Сначала отгоняют в вакууме rnpem-бутиловый и этиловый
спирты, а остаток смешивают с водой и встряхивают с эфиром. Из эфирного
раствора моноэфир извлекают 20%-ным раствором NaOH и после подкисления
щелочного раствора получают 13,94 г (90% от теоретического) указанного
выше соединения с т. пл. 119—123° С. После перекристаллизации из смеси
бензола и петролейного эфира т. пл. 125—126° С.
Другие примеры см. [672].
2. Синтез а,р-иенасыщеиных альдегидов и кетонов
При реакции двух карбонильных соединений (альдегида или кетона), по мень-
шей мере одно из ноторых содержит в a-положении метильную или метиленовую
группы, образуются, обычно в очень мягких условиях, альдоли (0-оксикарбоншгь-
ные соединения). Альдоли самопроизвольно или при нагревании до 60—100° С отщеп-
ляют воду и переходят в а,[J-нснасыщенные альдегиды пли кетоны.
Катализаторами этой реакции служат спиртовые и водные растворы щелочей,
алкоголяты алюминия, ацетат пиперидина, минеральные кислоты и ионообменные
смолы.
Смешанная альдольная конденсация между альдегидом и кетопом теоретичес-
ки может привести к четырем продуктам реакции. Практически она протекает только
в одном направлении, так как из-за большей реакционной способности альдегида
кетон всегда выступает в роли метиленовой компоненты. При реакции метилкетонов
типа мегилэгилкетона в зависимости от условий опыта реагируют либо метильные
группы, находящиеся в a-положении, либо метиленовые группы, стоящие в а'-поло-
жении. Так, из метилэтилкетона и бензальдегида в присутствии 10%-ного раствора
NaOH был получен 1-бензилиденбутанон-2, а с газообразным IIC1 — 3-бензилиден-
бутанон-2[673]:
СвН5СН-СНСОСН2СНз СНзСО—С-СНз
II
снс6н5
. Если в качестве метиленовой компоненты применяют ацетон, необходим его-
избыток, так как обычно с 1 моль ацетока реагируют 2 моль альдегида [674].
Бензилпденацетон 1675]. В круглодонной колбе емкостью 2 л с мешалкой
смешивают 635 г (около 11 моль) ацетона с 420 г (4 моль) свежеперегнанного, не
содержащего кислоты бензальдегида л 400 мл воды. К этой смеси при пере-
мешивании и охлаждении водой медленно (0,5 ч—1 ч) приливают 100 мл 10%-
ного раствора NaOH, так, чтобы температура в колбе поддерживалась в интер-
вале 25—31° С. Затем перемешивают при комнатной температуре еще 2,25 ч
и, наконец, подкисляют (по лакмусу) разбавленной НС1. Отделяют более тяже-
лый водный слой, встряхивают его со 100 мл бензола; бензольные вытяжки
объединяют с бензилиденацетоновым слоем, промывают 100 мл воды, слои раз-
деляют и отгоняют бензол на водяной бане, а остаток перегоняют в вакууме
(вода почти полностью отгоняется с первой фракцией). Собирают фракцию
с т. кип. 148—160° С (25 мм рт. ст.). После повторной перегонки получают
от 375 до 459 г продукта с т. кип. 137—142° С (16 мм рт. ст.). Выход про-
дукта 65—78% от теоретического.
По аналогичным методикам с хорошим выходом получают фурфурил-
иденацетон [676] и бензилйденацетофеноп [677].
[672]
[673
{674
[675
[676
J677
Org. Reactions, 6, 1—73 (1951).
С. Harries, G. Н. М ii 11 е г, Вег., 35, 968, 970 (1902).
С. R. С о п а г d, М. Л. Dolliver, Org. Synth., Colt. v. 2, 1943, p. 167.
N.L. Drake, P. A 11 e n jr., Org. Synth., 3, 17 (1923).
G. J. Leuck, L. C e j k a, Org. Synth., 7, 42 (1927).
E. P. Cohler, R.C. Leavitt, Org. Synth., 2, 1 (1922).
Ш. ОБРАЗОВАНИЕ НОВЫХ С = С-СБЯЗЕЙ ПУТЕМ ДЕГИДРАТАЦИИ
807
Конденсация оксиацетофенонов с оксибопзальдегидами приводит к замещенным
халконам (678], из?которых обработкой минеральной кислотой могут быть легко
получены соответствующие флаваноны. Кроме того, альдольная конденсация с успе-
хом применяется для введения боковой цепи в циклопентаноновое кольцо стеро-
идов [679].
Псевдоионон (680] — важнейший продукт для синтеза витамина А, легко
получается с выходом 59% из цитраля и ацетона в присутствии этилата натрия в эти-
ловом спирте, «я
г Альдольная конденсация чисто алифатических альдегидов протекает одно-
значно только в тех случаях, когда из двух молекул одного альдегида одна фигурирует
в качестве активной С—Н-компонентЫ, а другая — в роли акцептора протонов:
С2Н5-С1Ь—СнО-СЩ-СНО N-P^ с2н5-сн2-сн=с-сно
I I
С2Н5 с2н6
2-Этилгексен-2-аль-1 [681]. К 750 мл 1 н. раствора NaOH, нагретого до
80°С, при энергичном перемешивании в течение 1,5 ч прибавляют 2520 г свеже-
перегнанного н-масляного альдегида (скорость прибавления определяется
эффективностью обратного холодильника и должна быть как можно выше).
Температура при этом повышается до 93° С. Затем смесь кипятят еще 1 ч. После
охлаждения верхний слой отделяют и без дальнейшей обработки перегоняют
с колонкой Вигре высотой 150 см. Выход чистого 2-этилгокссналя 1880 г (86%
от теоретического); т. кип. 59,5—60° С (10 мм рт. ст.); пг£ 1,4556.
Для альдольной конденсации с успехом применяют также дикетоны. Напри-
мер, тетрафенилциклопептадиенон (682J получают с выходом 95% из бензила и дп-
бензилкетона в присутствии КОП. Обработка соответствующих 1,4-дикетонов водным
NaOH приводит к производным циклопептепопа [683]. Если по соседству с одной
из двух карбонильных групп находится метильная группа (CH3COCHSCH2COCH2R),
то она не принимает участия в конденсации. Сам ацотоиилацетон в такую реакцию
не вступает. Из 1,5-дикетопов образуются соответствующие циклогексеноны [684].
Их можно получить также из альдегидов и ацетоуксусного эфира (685]. Для синтеза
макроциклических кетонов можно исходить из дикетонов. Например, конденсацией
гексадекапдиопа-2,15 в присутствии магиийоргакического соединения N-метилани-
лина с последующим гидрированием был получен мускон [686].
Важный синтез полиеновых альдегидов разработал Кун (687]. Он синтезировал
с выходом 50% 7-феиилгептатриеналь конденсацией коричного и кротонового альде-
гидов в 70%-ном спирте в присутствии ацетата пиперидина. Таким же способом полу-
чают 11-фенилундекапентаеналь и фенилпентадекагептаеналь. При помощи ацетата
пиперидина удалось осуществить конденсацию 4-метоксидезоксибензоииа с формаль-
дегидом до метилеи-4-мстоксидезоксибеизоина [688]:
С11зО_\2/-СО~СПз“'О+СШ° CH3O-^J>-CO-C-^_^)
сн2
[678]
[679
(680
[681
[682
[683
[684
[685]
[686]
[687]
[688]
L. R е i с h е 1, W. Burkart, К. Muller, Ann., 550, 146 (1942).
W. С. J. Ross, J. Chem. Soc., 1945, 25.
A. R n s s e 1, R. L. Kenyon, Org. Synth., Coll. v. 3, 1955, p. 747.
M. Hausermann, Helv. chim. acta., 34, 1487 (1951).
J. R. Johnson, O. Grummitt, Org. Synth., 23, 92 (1943).
H. Hunsdiocker, Ber., 75, 455 (1942).
V. Prelog, L. Ruzicka, P. Barman, L. Frenkiel, Helv. chim.
acta, 31, 92 (1948).
E. C. Horn ing, M. O. Denekas R. E. F i e 1 d, J. Org. Chem., 9
547 (1944); Org. Synth., 27, 24 (1947).
M. S t 0 1 1, A. R ou ve, Helv. chim. acta, 20, 525 (1937); 30, 2019 (1947).
R. Kuhn, A. Wint erst ein, Helv. chim. acta, 12, 493 (1929).
H. Fisselmann, J. R ibka, Chem. Ber., 89, 27 (1956).
808
ОБРАЗОВАНИЕ ДВОЙНЫХ С==С-СВЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
а-Метиленальдегиды легко получаются также через основания Манниха.
Например, а-этилакролеин [689] образуется из w-масляного альдегида, гидро-
хлорида диметиламина и водного формальдегида.
Очень* изящно проходит конденсация кетонов при использовании в качестве
катализатора сульфированных ионообменных смол. Так, окись мезитила [690] полу-
чена с почти 80% -пым выходом пропусканием даров ацетона над ионообменной смолой
дауэкс 50. Из метилэтилкетона получается смесь обоих изомеров (З-метилгептен-З-
он-5 и 3,4-диметилгексен-3-он-2). Аналогично реагирует диэтилкетон с образованием
5-этил-4-метилгептен-4-она-3. Из циклогексанона образуется. 2-( Г-цнклогексенил) -
циклогексанон, из ацетофенона наряду с основным количеством дипнона — 1,3,5-три-
фенилбенздл.
3. Прочие конденсации
Конденсация метильных групп, связанных с ядром. Метильные группы, свя-
занные с ароматическим ядром, реакционноспособны лишь в том случае, когда аро-
матическое ядро содержит другие заместители в определенных положениях или же
если метильная группа связана с гетероциклическим кольцом.
Наличия одного заместителя обычно недостаточно для протекания конденсации.
Реакции мононнтротолуола с бензальдегидом проходит очень плохо, в то время как
конденсация дннитротолуола с бензальдегидом приводит к соответствующим произ-
водным стильбека [691]. Гораздо чаще для конденсации используют реакционно-
способные метильные группы гетероциклических систем. Так, из а-пиколнна и ацет-
альдегида при 250Q С образуется а-пропеннлпиридин [692]. у-Пиколин также всту-
пает в реакцию конденсации [693].
Способность метилпиррола к конденсации с пнрролальдегидом многократно-
использовали Фишер и сотр. [694] при синтезе порфиринов. В конденсацию с альде-
гидами могут вступать метильные группы более высококонденснрованных гетеро-
циклических систем. При взаимодействии гидрохлорида хинальдина с 2-этилгексан-
алем-1 в присутствии ZnCl2 образуется 2'-(3-этилгептен-1-ил)-хинолин с выходом
87% от теоретического [695]. С хорошими выходами конденсируется с м- или п-нитро-
бензальдегидом 9-метилакрндин, превращаясь в соответствующие стирильные произ-
водные акридина [696].
Метиковые конденсации. Разработано много методов получения метиновых
красителей, имеющих исключительное значение в качестве сенсибилизаторов фото-
материалов. Для синтеза триметиновых красителей служит введенный Кенигом [697]
«ортоэфирный метод». Этим методом, например, конденсацией двух молекул иодэти-
лата хинальдина с одной молекулой этилового ортоэфира муравьиной кислоты полу-
чен иодид пинацианола:
rm ОА1
VV^CH-ch=ch/V^/ I-
I I
C2H5 C2Hs
[689]
690
691
692
693
694
695
[696]
[697]
C. S. Marvel, R. L. Myers, J. H. Saunders, J. Am. Chem.
Soc., 70, 1694 (1948).
N. B. Lorette, J. Org. Chem., 22, 347 (1957).
J. Thiele, R. E scales, Ber., 34, 2843 (1901).
A. Ladenburg, Ann., 247, 26 (1888).
K. Friedlander, Ber., 38, 2838 (1905).
Г. Ф и ш e p, Г. Орт, Химия пиррола, пер. с нем., ОНТИ, Хпмтеорет,' 1937.
Е. Graef, J. М. Frederickson, A. Burger, J. Org. Chem., 11,
261 (1946).
W. Sharp, M. M. J. Sutherland, F. J. Wilson, J. Chem. Soc.,
1943, 5, 344.
W. Koning, Ber., 55, 3293 (1922).
III. ОБРАЗОВАНИЕ НОВЫХ С=С-СБЯЗЕЙ ПУТЕМ ДЕГИДРАТАЦИИ
809
Иодид пинацианола. В колбе Клайзена с капельной воронкой и термо-
метром нагревают 6 а иодэтилата (хинальдина и 50-ил уксусного ангидрида.
К слабо кипящему раствору в течение 5жин прибавляют 2 г этилового эфира
ортомуравьиной кислоты. Содержимое колбы, имеющее вначале желтый цвет,
скоро приобретает зеленую окраску, переходящую к концу реакции в темно-
синюю: при этом выделяется кристаллическая масса и отгоняется смесь этил-
ацетата. йодистого этила и небольшое количество уксусного ангидрида. Затем тем-
пературу повышают к отгоняют примерно половину загруженного уксусного ан-
гидрида. Из остатка при охлаждении выделяется кристаллическая масса уже
достаточно чистого иодида пииацианола. Прн перекристаллизации можно полу-
чить краситель в виде маленьких призм с красивым -зеленым блеском, которые
после высушивания при 140° С имеют, т. разл. 278° С. Выход продукта 2,4 г
(50% от теоретического).
При использовании ортоэфиров других карбоновых кислот получают тримети-
п новую цепь с ответвлением в виде алкильного остатка. Этот метод подвергался много-
шсленным модификациям, ио пригоден только Хля получения симметричных краси-
телей. Практическое значение имеет замена ортоэфиров на формамид, алкилизоформ-
аннлид (698] или N.N'-дифенилформамиднн [699], с помощью которых реакцию
можно проводить в две стадии. Таким образом становится возможным синтез несим-
метричных красителей.
4 Синтезы аминонислот по Эрледмейеру. Этотскптез [700] основан на конденса-
? цпн альдегида с гиппуровой кислотой. В качестве промежуточного продукта образу-
ется алкилиденазлактон, который восстановлением н гидролизом может быть превра-
^щен в а-аминоиислоту.
H2C-COOH 1 - H2C-CO RCH-C—CO -> 1 1 , !
HN 0 \ c NO NO \ / "W C C
CeH6 СбНб СаНб
Алкилиденазлактоны образуются при кипячении с обратным холодильником
• смеси гиппуровой кислоты, альдегида, ледяной уксусной кислоты к ацетата патрия.
£:Эти соединения могут подвергаться многочисленным превращениям. При омылении
?. их щелочами образуются а-кетокислоты, а при окислении перекисью водорода в щелоч-
ной среде — карбоновые кислоты, гомологичные исходному альдегиду. Таким обра-
?зом была получена 4-метокси-2-метилфенилуксусная кислота [701]. '
j Часто вместо гиппуровой кислоты применяют N-ацетиламиноуксусную кислоту.
!'-’Из бензальдегида и ацетилглицппа синтезирован З-бенэилпден-5-метилоксазолоп
' с выходом 75% от теоретического [702].
В качестве метиленовой компоненты использовали также креатинин. Таким
^способом получали Р-замещенпые N-метилаланины [703]. Описана конденсация
’ ванилина с креатинипом, протекающая с 95%-ным выходом [704].
< Кроме того, для синтеза соответствующих «.^-ненасыщенных соединений
;.примепяли также гидантоин, тиогидантоин, дикетопиперазнн и роданин.
4. Получение ненасыщенных нитросоединений
Первичные алифатические нитросоединенпя гладко конденсируются с альде-
гидами и кетонами. Начальный продукт реакции обычно представляет собой соот-
вётствующий ннтроспирт, из которого чаще всего последующей этерификацией
698
699
700
702
703
704
Е. В. Knott, J. Chem. Soc., 1946, 120.
S. Hu nig, Ann., 574, 99, 106, 112 (1951).
E. Erlenmeyer jr., Ann., 271, 164 (1894); 307, 138 (1899); 337, 265 (1904).
J. F. Hudson, E. Walton, J. Chem. Soc., 1946, 86.
R. N. Herbst, D. Sheniin, Org. Synth., Coll. v. 2, p. 1.
B. N. N i с о 1 e t, E. D. Campbell, J. Am. Chem. Soc., 50, 1158 (1928).
V. D e u 1 о f e u, T. J. Guerro, Org. Synth., 22, 89 (1942).
701;
810
ОБРАЗОВАНИЕ ДВОЙНЫХ С = С-СВЯЭЕЙ IJ РЕЗУЛЬТАТЕ ОБМЕНА
и отщеплением кислоты можпо получить питроолефин. При взаимодействии с арома-
тическими альдегидами образование двойной связи может протекать в процессе
конденсации;
АгСНОН—CHNO2 —> ArCH=CNO2
В качестве побочных продуктов иногда образуются динитросоединения, в ре-
зультате присоединения ннтропарафина к нитроолефину.
Катализаторами могут служить соединения основного характера, например
карбонаты щелочных металлов, их гидроокиси, бикарбонаты и алкоголяты, первич-
ные алифатические амины, а также Са(ОН)2. В случае ароматических альдегидов
применяют и ZnCl»,
Лернер предложил использовать для синтеза ненасыщенных нитросоедипений
ароматического ряда этилендиамин [709]. В его работе приводится большое число
примеров, когда выходы продуктов реакции составляли около 95%.
Взаимодействие ароматических альдегидов с первичными нитросоединениями
в Присутствии первичных аминов сразу приводит к нитроолефинам, но при использо-
вании других катализаторов реакция может протекать иначе.
2-Нитро-1-фенилпропен-1 [706]. В колбе емкостью 1л в течение 8 ч нагре-
вают с обратным холодильником 1 моль бензальдегида, 1моль нптроэтана, 5 мл
н-бутиламина и 100 мл абсолютного спирта. При охлаждении образуется жел-
тая кристаллическая масса, которую перекристаллизовывают из абсолютного
спирта. Выход продукта 105 г (60% от теоретического); т. пл. 65® С'.
Так же из соответственно замещенного бензальдегида и нитрометана в при-
сутствии метиламина получают 4-окси-З-метокси-ш-нитростирол [707].
2,3-Диметокси-о)-нитростирол[705]. Из 2,3-диметоксибензальдегида, нитро-
метана и спиртового раствора КОН после подкисления реакционной массы
50%-ной уксусной кислотой получают соответствующий а-нитроспирт; прп
обработке реакционной массы 1 н. H2SO4 с выходом 93% от теоретического
образуется 2,3-диметокси-со-питростирол [705].
Чисто алифатические нитроолефины обычно по могут быть получены в одну
стадию, так как при конденсации образуется нитроспирг. Ею ацетилируют, а из
полученного эфира действием КПСОд снова отщепляют уксусную кислоту, в резуль-
тате чего получается питроолефин [708].
5. Получение ароматических систем
Взаимодействие соответствующих соединений с отщеплением воды часто исполь-
зуется для синтеза ароматических ядер. Так, при помощи серной кислоты из ацетона
получен мезитилов [710]. Описай общий метод синтеза замещеппых 1,3,5-трпарид-
бепзолов [711].
1,3,5-Трнарилбензолы. К 50 мл насыщенного газообразным IIC1 абсолют-
ного спирта прибавляют Юг замещенного ацетофенона. Через 30 дней твердый
трпарилбензол отфильтровывают и промывают холодным спиртом. Повышение
[705]
706]
707
708
709
710
711
Е. Spath, К. Riedl, G. Kubiczek, Mh. Chem., 79, 72’(1918);
L. Canonic a, Gazz. chim. itaL, 79, 192 (1949).
H. B. Hass, A. G. Susie, R. L. H e i d e r, J. Org. Chem., 15, 8 (1950)
F. A. R a m i г e z, A. Burger, J. Am. Chem. Soc., 72, 2781 (1950).
E. Schmidt, G. Rutz, Ber., 61, 2142 (1928).
О. M. Лернер, ЖПХ, 31, 663 (1958).
R. Adams, R. W. Hufferd, Org. Synth., 2, 41 (1922).
R. E. Lyle, E. J. De Wi t t, N. .M. Nichols, IV. Cleland, J- Am.
Chem. Sue., 75, 5959 (1943).
IV. ОТЩЕПЛЕНИЕ АЗОТА С ОБРАЗОВАНИЕМ НОВОЙ С-тС-СВЯЗИ
811
выхода достигается осаждением твердого продукта из маточных растворов
водой. Получаемая при этом вторая фракция несколько загрязнена и ее много
раз промывают на фильтре этиловым спиртом. Выход зависит от исходного
производного ацетофенона и лежит в пределах от 20 до 85% от теоретического.
Для получения ароматических систем использовали также синтез Кневенагеля.
Так, из диацетила и о-фекилендиацетонитрила в безводном пиперидине с выходом
>65% от теоретического получают [712] 1,4-дициан-2,3-диметилиафталин (реакцион-
ную массу сначала охлаждают, затем оставляют на 3 ч).
Диарилкетоны, содержащие в орто-положении к карбонильной группе метиль-
ную пли метиленовую группу, при пиролизе часто отщепляют воду с замыканием
кольца (реакция Эльбса): • ч
СО
//'\/
I 11 II -Н.0 III
I Эта реакция имеет некоторое значение для получения производных антрацена
С[713], 1,2,5,6-дибензантрацена [714], 1,2-бензантрацена [715] и холантрена [716].
? Обычно реакцию проводят, нагревая кетон без катализатора или раство-
' рителя при температуре кипения или при 400—450° С до прекращения выделе-
ния воды. Лишь в редких случаях выходы превышают 50% от теоретического.
[. Подробнее о реакции Элбсэ см. обзор [717[.
С IV. Открепление азота с одновременным образованием
новой С=С-связи '
F
Разложение диазосоединений. При каталитическом разложении диазоуксус-
й ного эфира в присутствии порошкообразной меди образуется, как известно, эфир
К' фумаровой кислоты [718]. Грундман распространил эту реакцию на диазокетоны
? [719]. Из диазоацетона он получил с умеренным выходом гексен-З-дион-2,5. Ана-
\ логичные соединения были синтезированы из о-диазоацетофенона и 1-диазонона-
деканона-2.
Расщепление оснований Шиффа. В описанных ранее (стр. 801) реакциях
; альдегидов с соединениями, содержащими активные метиленовые группы, часто можно
i заменять альдегиды полученными из пих основаниями Шиффа. В некоторых случаях
*,- втот метод дает определенные преимущества, так как, например, соединения типа
i о-аминобепзальдегида труднодоступны. Так, Борще и сотр. [720] в хинолиновом
К синтезе Фридлендера заменили о-аминобензальдегид 2-аМипобензаль-п-толуидином
и с хорошими выходами получили этиловый эфир хинальдипкарбоновой-3 кислоты.
[712] Н. Moureu, Р. С h о v i п, G. R i v о а 1, Bull. Soc., chim. France, 1946,
Г 106.
E- [713] G. T. Morgan, E. A. Coulson, I. Chem. Soc., t929, 2203.
Г [714] J. W. Cook. J. Chem. Soc., 1931, 489; E. Clar, Ber., 62, 1574 (1929);
Kj W. E. Bachin a n tt, L. П. Репс e, J. Auk Chem. Soc., 59, 2339 (1937).
Г [715] J. W. Cook, J. Chem. Soc., 1932, 432; L.P, Fieser, E. B. Hersh-
g berg, J. Am. Chem. Soc., 59, 2502 (1937).
К [716] L. F. Fieser, A. M. Scligma n, J. Am. Chem. Soc., 57, 942, 2174 (1935);
I, W. E. Bachmann, J. Org. Chem., 3, 431 (1939).
F, [717] Org. Reactions, v. 1, John Wiley a. Sons, Inc., New York, 1942, p. 129.
К [718] A. Loose, J. prakt. Chem. [2], 79, 507 (1909).
E' [719] Ch. Grundmann, Ami., 536, 30 (1938).
SC (720j W. Borsclio, W. Doellc r. M. Wagner-Roemmick, Ber., 76,
f 1099 (1943); W. Boreche. J. В a r t he nhcicr, Anu., 548, 50 (1941).
812
ОБРАЗОВАНИЕ ДВОЙНЫХ С = С-СБЯЗЕЙ В РЕЗУЛЬТАТЕ ОБМЕНА
Аналогичным образом реагирует с малоновым эфиром группировка —CH = N—
в бнс-(-и-хлорфенил)-формамидине (721J:
/-\ //-X /г-\ /СООС2Н5
/ Ч-NH-CH-==N—)>—NH—CH = C<f
х=< у=/ хсоос2нв
С1 С1 С1
V. Отщепление серы с образованием новой С—С-связи
Взаимодействие между этиловым эфиром тритиоортомурайьиной кислоты
и малоковым эфиром полностью аналогично реакции, протекающей в тех же условиях
между этиловым эфиром ортомуравьиной кислоты и малоновым эфиром, и приводит
к образованию серного аналога дпэтилового эфира этоксиметилепмалоновой кислоты:
НС(ЗС2Н8)з+ НаС(СООСаН5)2 —> С2НвЗ-СН=С(СООСаНб)2
Диятиловый эфир этилмеркаптометиленмалоновой кислоты [722]. На мас-
ляной бане при 130—140° С в течение 10 ч нагревают с обратным холодильником
30 г (0,15’лоль) этилового эфира тритиоортомуравьиной кислоты, 24,8 г малоно-
вого эфира, 31,5 г уксусного ангидрида и 2,5 с безводного ZnCI2. После охлажде-
ния отделяют смесь от небольшого количества смолы и перегоняют в вакууме; при
этом в конце перегонки наблюдается незначительное разложение. Диэтиловый
эфир этилмеркаптометиленмалоповой кислоты перегоняется в виде светло-
желтого масла с т. кип. 170—172° С (19 мм рт. ст.); выход 17,67 г (49%
от теоретического).
Эфир тритиоортомуравьиной кислоты применялся в аналогичных конденсациях
при получении цианиновых красителей [723].
VI. Отщепление фосфора с образованием новой С=С-связи
(реакция Виттига)
Первое применение этой реакции описано Виттигом и Гейслером [724]. полу-
чившими при взаимодействии трифенилфосфинметилидена с бензофеноном 1,1-дифспил-
этилен с выходом 84% от теоретическою:
(СвНв)зР=СН2-]-О=С(СьН5)2 —*• (СеН5)зР = О4-СН2=С(СбНб)2
Известны взаимодействия фосфориленов с ароматическими альдегидами [725],
С альдегидами этиленового ряда [726] и с полиеновыми альдегидами, а также
с ароматическими и алифатическими кетонами.
о-Дивинилбензол[727]К бОллоль NaNHeB 300 мл жидкого NH3 прибавляют
55 ммоль бромида трифеннлметилфосфония и дают испариться аммиаку. Остаток
кипятят с 200 мл эфира в колбе с обратным холодильником. К оранжево-
желтому раствору трифенилфосфинметилидена добавляют при перемешивании
в течение 15 мин 28 ммоль диальдегида фталевой кислоты в 100 лм эфира п
в течение 2 ч нагревают смесь с обратным холодильником. Реакционную массу
[721
[722
[723
[724
[725
[726]
[727J
С. С. Price, R. М. Roberts, J. Am. Chem. Soc., 68, 1255 (1946).
К. D. Gun d ermann, Ann, 578, 48 (1952).
I. D. Cendall, I. R. M a i e r, J. Chem. 80c., 1948, 687.
G. W i t t i g, G. G e i s s 1 0 r, Ann., 580, 44 (1953).
U. Schollkopf, Dissertation, Tubingen, 1955; G. Wittig, U. Scholl-
kopf, Chem. Ber., 87, 1318 (1954); F. Bohlmann, Chem. Ber., 90, 1519
(1957).
G. Wittig, U. Schollkopf, Chem. Ber., 87, 1318 (1954).
G. W i t t i g, H. E g g e r s, P. Du ff n er, Ann., 619, 22 (1958).
VI. ОТЩЕПЛЕНИЕ ФОСФОРА С ОБРАЗОВАНИЕМ НОВОЙ С=С-СВЯЗИ gjj
фильтруют, фильтрат упаривают до объема 50 мл и снова отфильтровывают
трифенилфосфиноксид; из фильтрата получают 2,7 г о-дивннилбензола с т. кип.
75—78° С (14 мм рт. ст.); выход 75% от теоретического.
Описан удобный метод получения ликопина и Р-каротина [728]; о новом методе
синтеза витамина А см. [729]-
Исключительно важное значение имеет взаимодействие алициклических кетонов
с нленами, так как эта реакция представляет собой единственный метод превращения
циклического кетоиа' в соответствующий циклоолефин с экзоциклической двойной
связью. При взаимодействии циклогексанона с трифенилфосфинметилиденом обра-
зуется свободный от изомеров метиленциклогексап [726]’ а с трифеннлфосфинбензил-
иденом аналогичным образом можно подучить бензилидеициклогексан.
Метиленциклогексан. К раствору 100 ммоль трнфенилфосфинметилидена
в 200 мл абсолютного эфира прибавляют 9,8 г (100 ммоль) циклогексанона,
при этом сразу образуется белый осадок. Смесь нагревают в течение 3 ч прн
65° С, затем осадок отделяют центрифугированием и промывают его несколько
раз эфиром. Эфирные растворы промывают водой, отгоняют растворитель
и перегоняют маслянистый остаток с колонкой Вигре высотой 30 см. Выход
метиленциклогексана 5,4 г (52% от теоретического); т. кип. 100—103° С (744 мм
рт. ст.).
Трифеннлфосфинметилиден. В утку для гидрирования помещают 150 мл
эфира и 40 мл 1 н. раствора бутиллития в эфире и при интенсивном встряхивании
медленно прибавляют 14,3 г тщательно измельченного бромида трифенил-
метилфосфония. Реакционный сосуд встряхивают в течение 3 ч. На дне сосуда
остается лишь незначительное количество нерастворсннои соли, а трифенил-
фоофинметилиден переходит в раствор, окрашивая его в желто-оранжевый
цвет. В некоторых опытах продукт реакции выделялся в виде бледио-желтых
кристаллов.
Реакция Виттига с успехом применялась и в Химин витамина D- [730], так как
с ее помощью можно вводить метиленовую группу по углеродному атому Сщ- Описано
олефинирование тринадцати стероидных кетонов [731].
Подробный обзор по реакции Виттига см. [732].
[728
[729
[730
[731
[732
О. I з 1 е г et al., Helv. chim. acta, 39, 463 (19o6).
Пат. ФРГ 950552; С., 1957, 5362.
H. H. Inhoffen et al., Chem. Ber., 9f, 2309 (1958).
F. Sondheimer, R. Mechoulani, J. Am-, Chem. Soc., 79, 5029 (1957)-
U. Schollkopf, Angew. Chem., 71, 260 (1959).
Реакции, ведущие к разрыву углерод-
углеродной связи, имеют несколько меньшее
препаративное значение, чем реакции, прнводя-
щие к образованию углерод-углеродных связей.
Однако онк широко применяются для устано-
вления строения органических соединений.
В этом разделе описывается возможность
разрыва углерод-углеродных связей в результате
реакций термического распада, деструктивного
окисления и гидролиза.
ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ
РАЗРЫВ
УГЛЕРОД-
УГЛЕРОДНЫХ
СВЯЗЕЙ*
I. Отщепление
двуокиси углерода
В большом числе случаев термическое рас-
щепление углерод-углеродной связи заключается
в отщеплении двуокиси _ углерода от соответ-
ствующих соединений. Способность карбоновых
кислот к термическому отщеплению двуокиси
углерода всегда предопределяется электронным
состоянием углеродных атомов, смежных с кар-
боксильной группой. Отщепление двуокиси yj -
лерода протекает относительно легко и гладко,
если электронная плотность на углеродном атоме,
находящемся в a-положении к карбоксильной
группе, мала и тем самым высоко его сродство
к электрону (тип. I), или если на атоме углерода
в р-положенки к карбоксильной группе может
возникнуть катиоиоидиый центр, который при-
нимает связанную карбокснгруппой электронную
пару с образованием а,Д-двойной связи и таким
образом облегчает отщепление двуокиси угле-
рода (тип II).
Эти крайние случаи можно наглядно
пояснить следующими примерами:
—* С1,СН +сог
С1ЯС-СООП -1-*- С13С’ + СОг*Н
Тип '
1. Декарбоксилирование по типу I
Реакции декарбоксилирования, протека-
ющие по типу I, иллюстрируются следующими
примерами.
Для а-галогсиированных жирных кислот
и их солей существует правило, что «разрых-
ляющее действие» атома галогена на карбоксиль-
ную группу увеличивается с увеличением атом-
ного веса галогена. Например, трихлоруксусная
кислота может перегоняться без разложения
и медленно разлагается только при температурах
свыше 200 °C. Трииодуксуспая кислота, напротив.
Материал обработал Г. Хильгетаг.
I. ОТЩЕПЛЕНИЕ ДВУОКИСИ УГЛЕРОДА
815
разлагается уже при умеренном нагревании. Еще легче разлагаются соли кислот.
Трихлоруксусиая кислота при нагревании с анилином в бензольном растворе пре-
вращается f в хлороформ и двуокись углерода; калиевая соль трибромуксусной
кислоты распадается уже ори растворении в ацетоне на бромоформ и бикарбонат;
соли трииодуксусной кислоты, очевидно, вообще неустойчивы. Такие реакции
получили в последнее время препаративное значение для получения дигалоген-
карбенов.
Аналогично ведут себя а-нитро- и а-аминокарбоновые кислоты. Особенно
легко декарбоксилируются а-ннтрокарбоновые кислоты. Нитроуксусная кислота,
например, очень неустойчива и уже при кипячении с водой или растворении натриевой
или калиевой соли в разбавленной H2SO4 превращается в -нитрометан. Значение таких
реакций декарбоксилирования для получения нитроуглеводородов алифатического
ряда было уже рассмотрено (стр. 442). Общий метод получения первичных нитро-
соедииений из нитрилов кислот [1, 2] выражается схемой:
R-CHa-CN-l-CaHfiONOs -^-H°0Na^ R-C-CN >
NO2Na
—> R-C—COO Na R -CH2
NO2Na NO2
Реакция протекает через натриевое соединение ai/u-нитронитрила с образова-
нием a-нитрокарбоновой кислоты, пз которой путем отщепления двуокиси углерода
получается китрососдинение.
Гораздо слабое склонность к декарбоксилированию выражена у а-аминокарбо-
новых кислот. Нагреванием a-амннокислот при 200—250° С в таких индифферентных
растворителях, как глицерин, дифепилмстан или дифениламин, могут быть получены
низшие первичные амины. Таким путем, например,из тирозина был получен тирании [3].
Модификация этого метода по Вада [4] заключается в предварительном пре-
вращении «.-аминокислоты реакцией с мочевиной в гидантоин и кипячением послед-
него с кислотами или щелочами с одновременным гидролизоми Отщеплением двуокиси
углерода. Однако данные Вада не подтвердились.
Относительно легко протекает декарбоксилирование N- или О-гетероцикличс-
скпх «.-карбоновых кислот, например a-диколиновой. хинальдиновой или хелидоно-
вой; причем образуются пиридин, хинолин п у-пирон. Лучшим методом получения
фурана является также термическое отщепление двуокиси углерода от пирослизевой
кислоты:
НС—СН
нс-сн
о
Эту реакцию раньше проводили, нагревая ппрослизевую кислоту в запаянной
трубке или с натроииой известью; аппаратурно просто и удобно и с очень хорошими
выходами (72—78% от теоретического) реакцию удается провести по способу Виль-
сона 15].
Еще лучшие выходы (85—91% от теоретического) получают, если разложение
пирослизевой кислоты проводят в примерно 3,5-кратном количестве высококипящсго
сырого пиридиниевого основания в присутствии окиси меди при 170° С. При этом
используется тот факт, что декарбоксилирование такого рода особенно гладко
[1] W. Wislicenus, A. Endres, Вег., 35, 1757 (1902).
[2] Gattermann- Wieland, Die Praxis des organischen Chemikers, 33 Aufl.,
Walter de Gruyter a. Co., Berlin, 8. 231.
[3] E. Abderhalden, E. Gebelem, Hoppe-Seyler’s Z. physiol. Chem., 152,
126 (1926).
[4] M. Wada, Biochem. Z., 260, 47 (1933); Houben-Weyl, Methoden der
organischen Chemie, Bd. 11/1, S. 992.
[o] W. C. Wilson, Org. Synth., 7, 40.
«16
ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ
происходит в присутствии высококипящих третичных аминов и может быть каталити-
чески ускорено добавлением порошка меди или окиси меди.
Легко проходит также декарбоксилирование «.^-ненасыщенных карбоновых
кислот. Так, например, из коричной кислоты получают стирол [6]:
СН=СН-СООН СН=СН2
’ Стирол. В аппаратуре, состоящей из круглодонпом колбы, колонки и холо-
дильника точно указанных размеров, нагревают до энергичного кипения коричную
кислоту в присутствии небольшого количества гидрохинона в качестве анти-
оксиданта. Температура верха колонны должна повыситься до 120° С. Собрав-
шийся в приемнике стирол, стабилизованный небольшим количеством гидро-
хинона, перегоняют с водяным паром и полученное бесцветное масло после
высушивания над СаС12 перегоняют в вакууме; т. кип, 44—46° С (40 л.чрт. ст.).
Выход стирола 38—41% от теоретического.
Механизм реакции декарбоксилирования а,^-ненасыщенных кислот ке может
быть однозначно отнесен к типу I. Имеются данные [7], что «^-ненасыщенные кислоты
«лутем миграции двойной связи могут превращаться в ^.^-ненасыщенные кислоты,
декарбоксилирование которых проходит по типу II (стр, 822).
Большое препаративное значение имеет также декарбоксилирование а-ксто-
кислот И ГЛИЦИДНЫХ КИСЛОТ.
а-Кетокислоты распадаются при высоких температурах, правда неоднозначно,
на альдегид и двуокись углерода; с этой реакцией конкурирует отщепление окиси
углерода с образованием карбоновых кислот, имеющих на один атом углерода меньше,
-чем исходная кислота:
CO + RCOOH ч— R-СО—СООН —> RCHO + CO2
В алифатическом ряду до сих пор не найдено препаративного метода проведения
этой реакции для получения альдегида, хотя такая реакция представляет наиболь-
ший интерес. Более благоприятно отношение к декарбоксилированию ароматических
нетокислот. Их термическое расщепление протекает также неоднозначно (отщепление
СО и С02). Однако его можно преимущественно направить в сторону образования
.альдегида, если декарбоксилировать шиффовы основания кетокарбоновых кислот
[8], причем промежуточно образуются шиффовы основания конечных альдегидов.
ZN-R'
R—-СО—СООН —> R—С< —> R—CH«NR' —> RCHO
ХСООН
На практике а-кетокислоты обычно нагревают с обратным холодильником
-с избытком анилина до окончания выделения воды. Затем отгоняют воду и анилин,
остаток растворяют в минеральных кислотах, причем шиффово основание гидроли-
зуется, и выделяют образовавшийся альдегид. В качестве примера приводится пропись
.получения 4-н-Пропил-бензальдегида [9].
4-и-Пропил-бензальдегид. Неочищенную кислоту, полученную омылением
водным едким натром 44 г этилового эфира 4-н-пропил-фенилглиоксалевоИ
кислоты (с хорошим выходом образуется из м-пропилбензола и хлорангидрида
моно&фира щавелевой кислоты по Фридолю — Крафтсу), заливают в колбе
Клайзена небольшим избытком свежеггерегнанного анилина (около 20 г), ири-
[6] Т. W. Abbot, I. R. Johnson, Org. Synth., 8, 84.
L7J R. Т. А г п о 1 d, О. С. Elmer, R. М. Dodson, J. Аш. Chem. Soc., 72,
4359 (1950).
[8] L. Bouvault, Bull. Soc. chim. France [3], 15, 1020 (1896); 17, 363 (1897).
j9] Th. Siebcnmark, Dissertation, Leipzig, 1940.
I. ОТЩЕПЛЕНИЕ ДВУОКИСИ УГЛЕРОДА
817
тем при подогревании образуется твердая анилиновая соль. При дальнейшем
нагревании происходит слабое вспенивание с отщеплением воды и двуокиси
углерода. Остающуюся в колбе желтоватую жидкость тотчас же по окончании
реакции перегоняют в вакууме; при 205° С (12 мм рт. ст.) перегоняется -
32 е шиффова основания (72% от теоретического). К 32 г этого основания до-
бавляют 150 мл 25%-ной HgSOj и нагревают 15 мин на водяпой бане; после ох-
лаждения продукт трижды обрабатывают эфиром, экстракт промывают раствором
Na2CO3, водой, высушивают над Na2COs и перегоняют. Выход 4-н-проппл-бенз-
альдегида 14 г (65% от теоретического); т. кип. 112—114° С (13 мм рт. ст.).
В более высококипящей конечной фракции содержится еще значительное коли-
чество альдегида.
По другому способу декарбоксилирование а-кетокнслот проводят через бисуль-
фитные соединения. Так, например, фталоновую кислоту превращают в формилбеп-
зойную кислоту (стр. 842).
Изомерные а-кетокислотам глицидные кислоты термически или под действием
кислот также расщепляются до альдегидов и двуокиси углерода:
R—СН—СН—СООН _GOi-> R—СН=СНОН —> R—СН2—СНО
хо
Глицидные эфиры обычно получают из карбонильных соединений и эфиров
хлоруксусной кислоты (стр. 753) по методу Дарзана, поэтому после омыления эфира
и декарбоксилирования глицидной кислоты в качестве конечного продукта из перво-
начального альдегида или кетона можно получить ближайший высший Альдегид:
R4 TRX R4 1 R4
>С-0 —> >С СП—COOR —> >С СП—СООН —> >СН—СНО
Я/ R/ \ / R/ \ / Rz
L О О J
Превращение глицидных эфиров в альдмиды проводят, как правило, в две
Стадии. Сначала омыляют эфир водным или спиртовым раствором щелочи и затем
расщепляют образовавшуюся глицидную кислоту нагреванием с разбавленной НС1
или нагреванием в вакууме в присутствии меди или хромита меди.
Например, ct-фенилпропионовый альдегид [10] получают из легко доступного
(из ацетофенона и хлоруксусного эфира) фенилметилглицидного эфира [11] омылением
его алкоголятом и разложением полученной натриевой соли фенилметилглицидной
кислоты осторожным нагреванием в разбавленной НС1; при этом выделяется двуокись
углерода и выпадает продукт декарбоксилирования;
СН3
<^_^>-СО-С11з+С1-СП2-СООИ ---СП—COOR
О
СН3 СН3
—> ^“>-С--CH-COONa —^у-СН-СНО-СО,
О
Высококонденснрованные ароматические карбоновые кислоты часто очень
легко декарбоксилируются до углеводородов прн нагревании их в хинолине
[10] С. А 1 1 a n, J. v a n Allan, Org. Synth., 24, 87.
(11 ] С. A 1 1 a n, J. v a n Allan, Org. Synth., 24, 82.
52 Заказ 1835.
в присутствии порошка меди. Таким образом, например, из 4-метоксихризен-1-кар-
боновой кислоты при 230—250е С получают 4-метоксихризен [12]:
m. па
гуу^
.' СН3о/'^/Ч'!/ СООН СНаО^ ''/’'-.^
Трудно декарбоксилируемые карбоновые кислоты, радикал которых не содер-
жит функциональных групп, способствующих расщеплению, могут быть декарбо-
ксилированы лишь в форме сойей в присутствии избытка гидроокиси щелочного ме-
талла или СаО. В этом случае только добавочный отрицательный заряд карбоксиль-
ной группы посредством индукции создает возможность отщеплеиня остатка R в виде
аинона:
ОН ОН
R-c/° R-C-O- —> R- + C-O- —> RH + COg"
АО' | II
О- О
Для разложения используют солк карбоновых кислот с щелочными или ще-
лочноземельными металлами, ио более удобно применение серебряных, ртутных или
медных солей. Иногда такое декарбоксилирование удается провести в высококипящем
растворителе, найример этиленгликоле. Главной областью применения этого метода
является расщепление таких ароматических и гетероциклических карбоновых кислот,
подвижность карбоксильной группы которых не облегчена наличием соответствующих
гместителей в орто- или пара-положении или соседством с гетероатомом в кольце.
Коллидин. Сухую калиевую соль коллидин-3,5-дикарбоновой кислоты сме-
шивают с двойным весовым количеством Са(ОН)а и нагревают в трубке для про-
каливания до светло-красного каления, улавливая отгоняющийся при этом
коллидин [13].
При декарбоксилировании других карбоновых кислот, особенно алицикличе-
ских, часто протекают побочные реакции. Так, из бариевой соли циклогексанкарбо-
иовой кислоты образуется не циклогексан, а смесь дигидро- и тетрагидробензолов [14].
Из солей жирных кислот таким способом углеводороды получают только с не-
высоким выходом; побочной реакцией в этом случае является частичное декарбокси-
лирование, приводящее к кетонам. Если кальциевые соли карбоновых кислот подверг-
нуть разложению не избытком- СаО, а только сухой перегонкой, то образование кето-
нов становится основной реакцией:
(ВСОО)2Са —*- RCOR + СаСОз
При сухой перегонке кальциевых солей]дикарбововых кислот гладко получаются
циклические кетоны; из солей щелочноземельных металлов адипиновой кислоты
[15] образуется циклопектанон, из соответствующих солей пимелиновой кислоты
[16] — циклогексанон. По данным Блана [17], такие же кетоны образуются из сво-
бодных 1,6- и 1,7-дикарбоиовых кислот нагреванием их при температуре около
240’ С с уксусным ангидридом, в отличие от 1,4- и 1,5-дикарбоновых кислот, которые
в этих условиях дают только циклические ангидриды. «Правило Блана» особенно
часто использовали в химии стероидов, как один из способов установления
12] J. W. Cook, R. Schonta], J. Chem. Soc., 1945, 288.
13] G a 11 e г ш a n n-Wi el a n d, см.2, стр. 329.
14] N. Zelinsky, J. G u 11, Ber., 41, 2074 (1908).
15] J. F. Thorp e, G. A. R. К о n, Org. Synth., 5, 37.
16] L. Ruzicka et al., Helv. chim. acta, 9, 515 (1926).
17] H. G. В 1 a n с, C. r.. 144, 1356 (1907); A. Windaus, Hoppe-Seyler’s.
Z. physiol. Chem., 130, 116 (1923).
величины кольца; иногда в указанных условиях и из 1,6-дикарбоновых кислот
вместо циклических кетонов образовывались ангидриды [18]. z
Применяемый в технике способ котонизации жирных кислот, по которому
пары карбоновой кислоты при температуре около 300° С пропускают над кальциевыми,
бариевыми или ториевыми окисными катализаторами, является в принципе тем же
«самым методом сухой перегонки кальциевых солей.
’ Предполагают, что эта реакция в большинстве случаев протекает через про-
межуточно образующиеся (5-кетокарбоновые кислоты [19]:
(СН3СОО)2Са —► СНз—СО—СНг—СООСаОН —> СНз—СО—СН3+СаСО3
Для декарбоксилирования жирных кислот в соответствующие углеводороды
с наибольшим успехом используют так называемое расщепление солей. Серебряные
или ртутные соли преимущественно алифатических и алициклических карбоновых
.ннслот подвергают взаимодействию с бромом в индифферентных растворителях., В ре-
зультате этой, в большинстве случаев экзотермической, реакции наряду с двуокисью
углерода и бромидом серебра образуются галогенированные углеводороды (реакция
Бородина — Хунсдикера):
RCOOAgH-Br2 —> RBr + CO2 + AgBr
которые далее легко дегалогенируются каталитически или через гриньяровское
^соединение. В качестве побочного продукта вследствие алкилирования серебряной
холи карбоновой кислоты галогенидом в незначительных количествах получается
^соответствующий сложный эфир:
RCOOAg + RBr —> RCOOR + AgBr
><•: При использовании в этой реакции иода в зависимости От молярного соотно-
шения серебряной соли карбоновой кислоты и иода основной реакцией может стать
„образование эфира [19а].
й. Таким способом из лауриновой кислоты был получен ундецилбромид:
С СН3(СН2)1оСООН —> СНз(СН2)9СН2Вг
* ' Ундецилбромид [20]. Горячие растворы 50 г AgNO3 в 100 мл воды и 59 г
: лауриновой кислоты в 200 мл 1,45 и. раствора NaOH одновременно при переме-
шивании вливают в 100 мл горячей воды. После охлаждения в темноте отсасывают
образовавшуюся серебряную соль лауриновой кислоты, промывают ее водой,
ацетоном и высушивают в темноте сначала на воздухе, а затем в высоком вакууме
над Р2О6 при 60° С. Выход соли 85 г. В трехгорлой колбе, снабженной мешал-
кой, обратным холодильником и капельной воронкой, к 46 г полученной сереб-
ряной соли и 200 мл СС14 медленно при охлаждении и сильном перемешивании
добавляют раствор 7,5 мл брома в 20 мл СС]4; затем реакционную смесь осто-
. . рожцо нагревают и для полного выделения двуокиси углерода непродолжитель-
ное время кипятят. Отфильтровывают выпавший AgBr через экстракционную
i гильзу и экстрагируют продукт фильтратом в течение 1—2 ч. Промывая
экстракт разбавленным раствором NaOH и водой, удаляют непрореагировавшую
' лауриновую кислоту (5,5 г, 18% от теоретического). Поело отгонки СС14 унде-
". цилбромид перегоняют в вакууме. Выход продукта 24 а (67% от теоретического);
т. кип. 131—134® С (15 мм рт. ст.).
118] Е. Н. Farmer, J. Kracovsky, J. Chem. Soc., 1927, 680; F. Vock e,
‘ . Ann., 508, 1 (1934).
[19] O. Neunhoeller, Ber., 72, 919 (1939).
[19a] J. W. H. О Idbam, J. Chem. Soc., 1950, 100.
[20] A. L ii 11 r i n g h a u s, Ber., 74, 1565 (1941); J- Kleinherg, Chem.
Rev., 40, 381 (1947); R. G. Johnson, R, К. I n g h a m, Chem. Rev.,
56, 219 (1956); С. V. Wilson, Org. Reactions, 9, 332 (1957).
52*
820
ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ
2. Декарбоксилирование по типу II
Кетонное расщепление $-кетокислот
Декарбоксилирование 0-кетокислот не зависит от способности кетогруппы
к енолизации, вследствие чего возможно декарбоксилирование и <х,а-дизамещенных
Р-кетокислот. Термостабильны только такие Р-кетокислоты, декарбоксилирование
которых приводило бы к кетонам, неспособным к образованию енольной формы- Так,
например, не может быть декарбоксилирована кетопиновая кислота, поскольку в этом
случае, согласно правилу Бредта, из-за стерических затруднений невозможно образо-
вание еиольной формы продукта декарбоксилирования.
Фо
соон
Декарбоксилирование р-кетокпелот катализируется как кислотами, так и,
прежде всего, малыми количествами слабых оснований. Ацетондикарбоновая кислота
бурно разлагается при добавлении небольшого количества анилина с образованием
ацетона и двуокиси углерода. Щавелсвоуксусная кислота, декарбоксилирование
которой до пировиноградной кислоты кипячением в спиртовом растворе происходит
очень медленно, при добавлении небольших количеств пиридина уже при комнатной
температуре отщепляет двуокись углерода. Кетонное расщепление Р-кетокис.ют
имеет большое практическое значение для синтеза кетонов общей формулы
/В*
В—СО-СН<
\r"
В качестве исходных продуктов применяются соответствующим образом заме-
щенные эфиры p-кетокарбоновых кислот, которые вначале омыляют и затем декарбо-
ксилируют. Ацетоннлацетон, например, но Кпорру [21] получают из эфирадиацетил-
янтарной кислоты:
СЩСОСП-СНСОСПз CHsCOCH,-СН3СОСН3
[ 1 —2KU11:
ROOC COOR -2С0'
Ацетоннлацетон. К 10 г эфира диацетилянтарпой кислоты добавляют 100 мл
оттитрованного точно 3%-ного раствора NaOH, закрывают колбу трубкой
с натронной известью и 2—3 ч нагревают на водяной бане, до тех дор, пока
при подкислении пробы реакционной массы ке прекратится выделение эфира
диацетилянтарной кислоты. Затем массу охлаждают, насыщают К2СО3, отделяют
выделившееся масло, остаток продукта из щелочного слоя дважды экстраги-
руют эфиром, сушат и перегоняют. С парами эфира переюняется значительное
количество ацвтонилацетона. Общий выход продукта 4 г (90% от теоретиче-
ского); т. кии. 194° С.
Декарбоксилирование малоновых кислот
При декарбоксилировании алкилированных малоновых кислот, также катали-
зируемом кислотами и слабыми основаниями, образуются замещенные уксусные
кислоты:
R СООН Ik /Н
R'/Cx-COOH Н'/^СООН
Отдельные примеры этой реакции были приведены ранее (стр. 803). Ниже
приводится пример получения «-капроновой кислоты из бутилмалоновой кислоты
[21] L. Кпогг, Вег., 22, 2100 (1889).
I. ОТЩЕПЛЕНИЕ ДВУОКИСИ УГЛЕРОДА
821
по Адамсу и Марвелу [22]. Авторы исследовали реакцию декарбоксилирования при
различных условиях. По их данным лучшие результаты получаются прн проведении
этой реакции одновременно с омылением бутилмалонового эфира.
^-Капроновая кислота. В круглодонную колбу емкостью 5 л, снабженную
холодильником и капельной воронкой, помещают раствор 500 г КОН в равном
количестве воды. Затем при нагревании и частом встряхивании постепенно
добавляют 500 г н-бутилмалового эфира (получение см. стр. 755), который
быстро омыляется.. Основная реакция полностью проходит после добавления
всего эфира; раствор становится прозрачным. Для полного гидролиза и отгонки
образующегося спирта открытую колбу нагревают в'течение 1 — 2 ч на водяной
бане, затем вновь устанавливают обратный холодильник и капельную воронку
и подкисляют массу 1500 мл HCI (d =- 1,19); необходимо Избегать слишком
бурного иснениваиия. После подкисления смесь кипятят в течение 4—5 ч и затем
перегоняют с нисходящим холодильником до почти полной отгопки «-капроно-
вой кислоты, выделяющейся в дистилляте в виде масла. Кислоту в дистилляте
отделяют от водного слоя. Около 400 мл последнего переносят опять в колбу
и перегоняют вновь. Отделяют вновь н-капроновую кислоту, объединяют водные
слои и высаливают кислоту раствором СаС12. капроновую кислоту сушат над
СаС1„ и перегоняют; выход продукта 200 г (74% от теоретического); т. кил,
200—205" С.
, Омылением й декарбоксилированием ацилированпых налоговых эфиров, реат-
рующих одновременно как р-кетокислоты, получают метилкетокы:
rcoch(coor'), нсосн3 |' гсо2н 2r'oh
Из ангидридов диалкилмалоиовых кислот нагреванием при 100—200° С прп
пониженном давлении можно получать также диалкллкетены [23[:
СО
(СН3)2С// X) — > (СНз)2С=СО4-СО2
СО
Диметилкотен. В сухую колбу Клайзена вносят 25 г уксусного ангидрида,
одну каплю концентрированной H2SO4 и 6,5 г диметилмалоновой кислоты Смесь
встряхивают до полного растворения и оставляют раствор на двое суток при
комнатной температуре. Затем добавляют немного порошкообразного ВаСО3
и при слабом нагревании на масляной бане отгоняют в вакууме уксусную ки-
слоту и уксусный ангидрид. Устанавливают новый приемник, охлаждаемый льдом.
При постепенном повышении температуры реакционной массы до 100° С проис-
ходит разложение ангидрида диметилмалоновой кислоты с образованием диме-
тилкотена п двуокиси Углерода. Выход диметилкетена 2,3 г (65% от теоретиче-
ского); т. кип. 34° С.
Этим способом могут быть также получены диэтил-, дипроппл- п диизо-
пропидкетены.
Так как ашидрцды малоповых кислот часто трудно доступны, Штаудипгер
рекомендует проводить разложение смешанных ангидридов, которые, например,
могут быть получены взаимодействием днзамещенпых малоновых кислот с дифенил-
кетеном [24].
[22] R. Adams, С. S. Marvel,']. Am. Chem. Soc.. 42. 317 (1920).
[23] H. Staudinger, Helv. chim. acta, 8, 306 (192j).
[24 ] H. Staudinger. H. S c he i der, P. Schot г, P. M. Strong, Helv.
chim. acta, 6, 291 (1923).
822
ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ
Декарбоксилирование $ ^ненасыщенных кислот
< Примером этой реакции служит декарбоксилирование циклогексенилуксуспой
кислоты при ее перегонке с образованием метиленциклогексана [25]:
Декарбоксилирование р,у-непасыщенных кислот, следовательно, связано с ми-
грацией двойной связи.
Декарбоксилирование ароматических окси-
и аминокарбоновых кислот
Ароматические о-оксикарбоиовые кислоты декарбоксилируются при нагрева-
нии в пиридине или диметиланилиие, а в некоторых случаях даже при кипячении
с водой. На основании этого можно сделать вывод, что ароматические о-оксикарбоно-
вые кислоты реагируют в таутомерной кето-форме:
ОН
'''/'cooe
Так, например, 0- и у-резорциловые кислоты кипячением с водой можно легко
декарбоксилировать до резорцина [26].
Аналогичным образом декарбоксилируются пиридон- и о-оксихинолинкарбопо-
вые кислоты. 4-Оксихинолин-З-карбоновую кислоту можно превратить в 4-окси-
хинолин двумя методами [27] (см. пропись):
ОН
ОН
Z\/^ZC00H
I ft I
4-Окснхинолин. 1. Сухую измельченную 4-оксихинолик-З-карбоновую ки-
слоту нагревают прп температуре плавления на металлической бане до прекра-
щения выделения двуокиси углерода. Сырой 4-оксихинолин растворяют в этило-
вом спирте и обесцвечивают раствор углем. После отгонки спирта полученный
продукт очищают перекристаллизацией.
' 2. 4-Оксихинолин-З-карбоиовую кислоту добавляют к пятикратному
количеству кипящего даутерма и нагревают около 30 мин до полного растворе-
ния кислоты. После охлаждения раствора п разбавления его двойным объемом
гексана выделяется 4-окскхинолин, который очищают, как описано выше.
о-Аминокарбоновые кислоты также могут декарбоксилироваться. Гак, например,
антраниловая кислота мрй быстром нагревании приблизительно до 210° С полностью
разлагается ка двуокись углерода п аннляи:
NHa .
J^yCOOlI
I II
nh2
[251 F. Sorm, J. Beranek, Chem. Listy, 47, 708 (1953).
[26] F. v. Hemmelmayer, Mh. Chem.. 46, 144 (1925).
[27] B. Riegel, J. Am. Chem. Soc., 68, 1264 (1946).
If. ОТЩЕПЛЕНИЕ ОКИСИ УГЛЕРОДА
823
Аналогичное разложение происходит и при кипячении с водой [28].
л-Окси и л-аминокарбоновые кислоты декарбоксилируются в сходных условиях,
то время как соответствующие мета-производные значительно более устойчивы.
II. Отщепление окиси углерода
Декарбонилирование ряда карбоновых кислот и их производных, например
лорангидридов или эфиров, возможно в том случае, если под действием очень сильных
ислот или других реагентов происходит промежуточное образование ацильного
атиона, легко отщепляющего затем окись углерода:
zO w+ zO ______________ -+ + - +
. R-C< R-C<f[+ R—С=О * R—С=О —> R+-|-C&0
ХОН \о-н
: ’ I
н
Остаток R в ацильном катионе должен быть построен так, что возникающая
юле отщепления окиси углерода недостача электронов каким-либо образом может
иь восполнена. Катион может стабилизоваться также путем отщеплеиня протона.
По этому механизму декарбокилирования протекают следующие реакции:
щавелевая кислота расщепляется с образованием окиси углерода, двуокиси
щерода и воды при нагревании в сухом виде при температуре свыше 100° С, особенно
присутствии концентрированной H3SO4.
Хлорангидрид щавелевой кислоты под действием А1С13 при слабом нагре-
яик разлагается с образованием фосгена и окиси углерода [29]:
>/О А1Г1. Г Ох +1
С-С<с1 -------- [ci>C-C^oj[AlCld-
—> [О=С—C1J[A1C1J-—>А1С1з + СОС12
а-Оксикислоты при нагревании или в присутствии концентрированной H2SO4
^сщепляются с выделением окиси углерода н воды; при этом образуются альдегиды
ди кетоны. При декарбонилировании жирных кислот (Се и выше) выходы продуктов
Ьстигают 90% от теоретического. Из лимонной кислоты может быть получена ацетон-
якарбоновая кислота [30]:
ОН О
НООССНа—С—СНзСООН —> НООССНа—С—CHsCOOH
I
СООН
Ацетондикарбоновая кислота. Тонко измельченную лимонную кислоту при
0—10° С порциями вносят в дымящую H2SO4, содержащую 20% SO3; лимонная
кислота полностью растворяется. При медленном нагревании'реакционного
раствора до 30е С выделяется [окись углерода. Когда выделение газа прекра-
щается, массу охлаждают до 0° С и добавляют раздробленный лед, причем темпе-
ратура медленно возрастает до 25—Зб° С. Затем охлаждеттный снова до 0° С
раствор отсасывают через пористый стеклянный фильтр; полученный продукт
размешивают с небольшим количеством этилацетата и вновь отсасывают, повто-
ряя эту операцию несколько раз. Выход ацетондикарбоновой кислоты 92—97%
от теоретического. Кислота устойчива только в течение нескольких часов,
и поэтому сразу должна перерабатываться далее.
Если расщепление сс-оксикарбоновых’кислот до альдегидов, содержащих на
’[Один атом углерода меньше, чем исходная кислота, происходит неудовлетворительно,
?(28] L. McMaster, R. L. S h г i п е г, J. Am. Chem. Soc., 45, 751 (192.3).
129] ,H. Staudinger, Ber., 41, 3566 (1908).
130] R. Adams, H. M. Chiles. C. F. Rassweller, Org. Synth., 5, 5.
824
ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ
применяют ацетоксикарбоновые кислоты или, по Дарзапу [31], а-алкоксикарбоновые
кислоты:
RCHCOOH —> RCHO + CO + CH3OH
I
ОСНз
Этим способом, например, а-бромстеариповую кислоту, получаемую бромиро-
ванием стеариновой кислоты, обработкой метилатом натрия превращают в а-метокси-
стеариновую кислоту, при нагревании которой до 300° С с почти теоретическим выхо-
дом образуется маргариновый альдегид.
а-Бромкарбоновые кислоты, необходимые для синтеза а-алкоксикарбоповых
кислот, часто получают также с помощью малонового эфира. Алкилгалогепид под-
вергают вначале взаимодействию с малоновым эфиром. Образовавшийся алкилмалоно-
вый эфир омыляют в свободную алкилмалоновую кислоту, которую бронируют при
низкой температуре и затем декарбоксилируют полученную а-бромдикарбоновую
кислоту [32]. Этим путем, имеющим препаративное значение для синтеза альдегидов,
алкилгалогенид RX можно превратить в альдегид RCHO, содержащий на один атом
углерода больше, чем исходный алкилгалогенид:
RX-|-CH2(COOR')2 —> R—CH(GOOR')3 —► R—CII(COOII)2 —>
/СООН
—> R—C< “сб”*’ R—CH—COOK —> R—СП—СООЦ —►
I XCOOII ’ 1 I
Br Br OR"
—> RCHO + CO-LR'OH
Этот же альдегид можно получить более коротким путем, если алкилгалогенид
подвергнуть взаимодействию с алкокспмалоновым эфиром [33[:
/COOR' /СООН
RX4 HC(COOR')., —> R—С< —> R—С/ —>
| [ XCOOR' | ХСООН
OC2II5 ОС2Нб ОС2Н5
—> R—СН-СООН —> RCHO + CO + C2H5OII
ос2н5
К реакции докарбонплирования а-оксикислот очень близко примыкает рас-
щепление по Волю, когда из а-оксиальдегидов образуются альдегиды, имеющие па
один атом углерода меньше, чем исходный оксиальдегид. Исходят при этом из оксима,
который с отщеплением воды превращается в оксииитрил. Отщеплением HCN, анало-
гичным отщеплению окиси углерода и воды ота-оксикислот, из оксинитрилов получают
соответствующие альдегиды:
/9 nh.oh х^ОП
R-CH-C^--------R— СН-C<f
| | ^Н
ОН он
/О
—> R—СН—CN .—► R—С<^ +HCN
011
Эта реакция с особенным успехом применяется в ряду углеводов. Так, например,
при нагревании оксима глюкозы с уксусным ангидридом и ацетатом натрия образуется
нитрил пентаацстилглюконовой кислоты, из которого, по старой прописи Воля [34],
либо действием окиси серебра и аммиака, либо обработкой метилатом натрия в хлоро-
[31] G. Darz ens, С. г.. 196, 348 (1933).
[32] Е. Fischer, W. Schmitz, Вес., 39, 2208 (1906).
[33] G. Darzens, \I. М е v е г. С. г., 196, 489 (1933).
134] A. W о 11 1, Вег., 26, 734 (1893).
II. ОТЩЕПЛЕНИЕ ОКИСИ УГЛЕРОДА
825
форме при —12° С отщепляют цианистый водород. После омыления разбавленной
; H2SO4 получают ^-арабинозу с выходом примерно 65% от теоретического [35].
Равным образом склонны к декарбопилироваиию эфиры щавелевожирных
у кислит и кетоны, легко получаемые сложноэфирной конденсацией диалкилоксалатов
с соответствующими реагентами (стр. 789). Такие эфиры щавелевожирных кислот
термолабильпы и нагревание их прп температурах выше 120° С приводит с отщопле-
I. нием окиси углерода к монозамещенным малоновым эфирам:
R—СН—COOR’ R4 .COOR"
I —> CO- >c<
f CO—COOR' Hz 4COOR'
Монометилмалоновый эфир получают с 97%-ным выходом нагреванием эфира
щавелевопропионовой кислоты при 130“ С до' прекращения выделения окиси углерода
. [36].
” От енол-лактона левулиновой кислоты прп нагревании его с кусочками кремие-
_ вема до 570° С отщепляется окись углерода и образуется ненасыщенный кетон с выхо-
дом 80% от теоретического [37|.
i нс-сн2
/ | /СО со^сн3-со-сн=сн2
СНз—С—О
При титровании бромом о- и n-окси- или аминобензальдегидов в водном растворе
. при 40—45° С получают 2,4.6-трибромфенол или 2,4,6-трибромапилин; альдегидная
группа при этом замещается бромом с отщеплением окиси углерода [38]:
ВГ
I
ЗВГ,. 40-45° С;
"У^СНО Вг-/у/Ч-Вг
OH(NH2) 0H(NH2)
При температурах ниже 10—15J С о- и тг-окся(амино)-бензальдегиды могуг
быть превращены в 4,6-дибромпроизводные с сохранением альдегидной группы.
’ В диеновом синтезе при применении в качестве диеновой компоненты цикло-
пентадиенона получают первичные продукты, эндокарбонпльный мостик которых
5 отщепляется при нагревании в виде окиси углерода. В зависимости от того, прово-
' дится ли диеновый синтез с олефиновыми или ацетиленовыми компонентами, при
.этом образуются производные дигидробепзола или бензола [39|:
36
37
38
39
G. Braun, Org. Synth.. 20. 4.
R. Сох, S. М. McElvaiii. Oig. Svnth.. 17, 56.
Англ. пат. 601922; С. А., 42, 7319 (1948).
A. W. F г a n с j з, A. J. Н i 1 L, J. Am. Chem. Soc., 46, 2498 (1924).
W. Foerst, Neuere Methoden der praparativen organischen Chemie, Bd. 1,
Verlag Chemie, Weinheim, 1949, S. 311.
826
ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ
Если олефиновая компонента содержит на обоих углеродных атомах двойной
связи атомы водорода, то нередко наряду с декарбонилированнем происходит дегидри-
рование с образованием ароматических систем,
•Тетрафенилциклопентадиенои (тетрациклон) с малеиновым ангидридом в кипя-
щем бензоле почти количественно образует нормальный аддукт, ангидрид 3,6-эндо-
карбонил-3,4,5,6-тетрафенил-1,2-дигидрофталевой кислоты. При 'нагревании в хлор-
бензоле этот аддукт с отщеплением окиси углерода превращается в ангидрид тетра-
фенилдигидрофталевой кислоты. Если далее реакцию ведут в кипящем нитробензоле,
происходит дегидрирование и сразу же образуется ангидрид тетрафенилфталевой
кислоты. Таким образом, в зависимости от рода растворителя при диеновом синтезе
тетрафенилциклодентадиенояа с малеиновым ангидридом могут быть получены три
соединения:
СйНк СяНк
С6На
СвН5
гы I СО
СвН5\//\/ \
свн5
III. Отщепление других составных частей молекулы
Термический разрыв углерод-углеродиой связи без выделения окиси или дву-
окиси углерода и, следовательно, с отщеплением других частей молекулы в лаборатор-
ной практике используется относительно редко.
1. Расщепление рицинолевой кислоты
Пиролитическим расщеплением эфиров рицинолевой кислоты с хорошими
выходами получают ундециленовую * иислоту и энанталь:
CH3(CH2)SCHCH2CH=CH(CH2)7COOR —> CH3(CH2)6CHO + CH2=CH(CH2)8COOR
ОН
Эта реакция напомидает декарбоксилирование Д,у-ненасыщеины кислот.
Процесс практически проводят следующим образом: неочищенный метиловый эфир
рицинолевой кислоты, полученный переэтерификацией касторового масла смесью
метилового спирта и серной кислоты, прибавляют по каплям в соответствующий тепло-
носитель, нагретый до 500—600° С; собранный дистиллят перегоняют в вакууме [40].
. 2. Расщепление алкоголятов
Свойства, в принципе аналогичные эфирам рпцинолевой кислоты, могут про
являться также и у других спиртов, если их в виде алкоголятов в присутствии щело-
чей подвергают действию высоких температур. Однако для успешного протекания
этой реакции молекула спирта должна соответствовать опредёленным требованиям,
чтобы поляризация, индуцированная отрицательным зарядом аниона алкоголята,
[40] Франц, паг. 952985; С., 1959, II, 464.
1П. ОТЩЕПЛЕНИЕ ДРУГИХ ЧАСТЕЙ МОЛЕКУЛЫ
827
i действительно привела к отщеплению радикала, связанного со спиртовым углеродом,
в виде карбаниона.
Такие предпосылки, обусловленные строением, имеются в p-кетоспиртах с тре-
тичнымц спиртовыми группами. .Так, йапример, диацетоновый'спирт в жестких усло-
; виях в присутствии щелочей может расщепляться на две молекулы ацетона [41]:
сн3ч
>С-СН2СОСНз —* 2СН3СОСН3
СН37 I
ОН
< В этом случае речь идет о процессе, обратном альдольной или кетольной
5 денсацин. Определенное препаративное значение такое расщепление имеет для
..раля, который как а.Р-ненасыщекный альдегид при кипячении с раствором карбоната
у, калия превращается в метилгептенон н ацетальдегид [42], предположительно с про-
V-межуточным присоединением воды и образованием третичного спирта:
кон-
цит-
СНах | О СНзх I zO
•г >с=снсн2сн2-с=сн-се —> >С=СНСН2СНа-С-СН2-С<7 —>
Сн/ \н СНз7 I Хн
, ОН
J ' СНз о
—> СН9ч>С=СНСНоСНй-С=О^СН3-с/
СНз7 ХН
Как правило, неоднозначно протекает в тех же условиях расщепление вторич-
' пых спиртов, так как первично образующиеся альдегиды легко подвергаются дальней-
„ шим превращениям. Удобный препаративный способ расщепления углеводов заклю-
чается в обработке гидразином продуктов окисления тиоацеталей углеводов —сульфо-
нов; при этом расщепление вторичных спиртов под действием гидразина протекает
v с хорошими выходами [43]:
8 ,CH(SC2Hs)2 C(SO2C2H5)2 CH(SO2C2H2)2
" 1 ' О !1 |
I СНОАс —► СП спои —► CH2(SO2C2Hs)2 + RCHO
t Я R в
t . а,р-Ацетилсновыо спирты расщепляются при нагревании с концентрированными
‘щелочами по реакции, обратной синтезу таких соединений по Реппе, с образованием
'? ацетилена и карбонильного соединения. Пропаргиловый спирт, получаемый присое-
§;динеиием формальдегида к ацетилену, расщепляется при нагревании с концентриро-
>' ванными щелочами на исходные вещества; правда, формальдегид тотчас же превра-
, щается далее в формиат или карбонат щелочного металла:
носн2-с==сп СП2О-СП~СН
Аналогичному расщеплению подвергаются также 0-оксикислоты и а,0-пенасы-
t щекные кислоты, причем последние, предположительно, вначале превращаются
•_ в Р-оксикпслоты. При расщеплении наряду с уксусной кислотой образуется кар-
бонильное соединение, содержащее на два углеродных атома меньше, чем исходная
К; кислота. Если образуется альдегид, то он в щелочной среде окисляется до
КК" [41] L. В о u v е а п 1 t. R, Locquin. Ann. Chimie [8], 21, 415 (1910);
E‘ ,.л c- c- French, i. Am. Chem. doc., 51, 3215 (1929).
E? [42] M. A. Verley, Bull. Soc. chim. France ]3], 17, 176 (1897); F. T iemann,
F Ber., 32, 107 (1899).
E [43J D. L. M c D on a ld.II. 0. L- F i s c h e Г, J. Am. Cliein. Soc.. 74, 2087 (1952).
828
ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ
соответствующей кислоты. Такое расщепление олефиновых двойных связей концентри-
рованными щелочами до ненасыщенных карбонильных соединений, являющееся также
окислительным, обсуждается поэтому при рассмотрении возможностей окислительною
расщепления углерод-углеродных связей (стр. 832).
3. Расщепление ацетона
Кетен наряду с метаном получают пирогенетическим расщеплением ацетона:
СН3СОСН3 —> СНа=С=О + СН4
Аппаратура [44], пригодная для лабораторного получения кетена, показана
на рис. 5.
В колбе 1 емкостью 1 л ацетон нагревают до кипения. Пары проходят
в реакционную эону 2, в которой помещена нагревательная проволока, и затем
поступают в холодильник 4, где конденсируется непрореагировавший ацетон,
который возвращается в реакционную колбу. Кетен проходит через второй
холодильник и отделитель в приемник, где его либо конденсируют (т. кит?,
“41° С), либо непосредственно подвергают дальнейшему взаимодействию.
Рис. 5. Прибор для лабора-
торного получения кетена:
1 — колба; 2 — кетенный генератор;
3 — делительная воронка; 4 — холо-
дильник .
Рис. 6. Кетенный генератор:
1 — медные проводчики (один из них
до места оплавления в стекло изолиро-
ван стеклянной трубкой), 2 — отрезки
проволоки из сплава «ковар» (для
подавления в стекло), прочно припаян-
ные к медным проводникам, 3 — клеммы
из стали V»A; 4 и 6—стеклянные
крючки для укрепления нагревательной
пяти; в — пружины ив стали V«A для
натяжения нагревательной проволоки,
7 — крючки из стали V,A; в — нагре-
вательная проволока.
Улучшение этой аппаратуры по сравнению с более ранними прописями
в основном заключается в усовершенствовании (производительность и стойкость)
греющего агрегата (рис. 6). Используют проволоку диаметром 0,4 .чл из
[44] . G. Quad beck, Chem. (ng. Tcchn., 24, 210 (1952); см. также Angew. Chem.,
68, 361 (1956).
Ш. ОТЩЕПЛЕНИЕ ДРУГИХ ЧАСТЕЙ МОЛЕКУЛЫ
высокопрочной хромоникелевой стали F extra фирмы Heraeus, натянутую
на стеклянные крючки, термическое расширение которой 1{омпенсируется пру-
жинами из стали V2A. Подвод тока осуществляется с нижнего конца греющего
агрегата, что значительно снижает нежелательное осаждение угля. Сила тока
регулируется реостатом сопротивлением около 30 ом или автотрансформатором.
Производительность кетенного генератора сильно зависит от напряжения.
Для того чтобы избежать осаждения угля, необходимо поддерживать оптималь-
ную силу тока.
Осторожно при обращении с кетеном! Это чрезвычайно ядовитый газ
по токсичности соответствующий фосгену; вызывает примерно аналогичные
фосгену явления (отек легких).
4. Расщепление производных циклогексена
г Ряд производных циклогексена может термически, особенно при пропускании
’ их в виде паров над раскаленной металлической проволокой, расщепляться па олефин
? и молекулу диена. В этом случае речь идет о реакции, обратной диеновому синтезу
. Дильса — Альдера. Наиболее ранним примером является получение изопрена из
дипептена [45]:
зоо ®С
дипентен изопрен
В качестве нагревателя использовали так называемую изопреновую лампу,
которая в дальнейшем применялась для проведения целого ряда других процессов.
В этой лампе пары дипентепа омывали нагреваемую электрическим током платиновую
проволоку, натянутую на стеклянный стержень.
Аналогичным способом [46J можно получать бутадиен из циклогексена [47]:
/СН2
/X нс сн3
I II —* ’I -I I!
\/ НС сн2
Хп,
Используя подобную аппаратуру, термическим разложением ангидрида дйаце-
тилвинной кислоты [48] на слабо раскаленной платановой проволоке можно получать
недокись углерода, С3О2 (механизм реакции не установлен).
5. Превращение лумистерина в тахиетернп
1 В некоторых случаях лабильные углерод-углеродные связи расщепляются
; не только под действием высоких температур, но и при облучении ультрафиолетовым
Р светом. Тйк протекает фотохимическое превращение, например, эргостерина в вита-
мин D,. При этом пз системы лумистерина, содержащей четыре кольца, образуется
I [45] С. D. Harries, К. G о И 1 о Ъ, Ann,, 383, 228 (10Ц).
[46] С. D. Harries, Unlersuchungen uber die natiirlichen und kiinstlichen Kauts-
chukarten, Springer-Verlag, Benin., 1919, S. 142.
[47] F. B. Herschberg, J. R. R uhoff, Org. Synth., 17, 25.
f [48] E. О t t, K. Sclj tn i d I, Ber., 55, 2126 (1922).
830
.ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ С-С-СВЯЗЕЙ
с разрывом связи между углеродными атомами Сд и Сю тахистерин, который в отличие
от лумистерина имеет три цикла и дополнительную двойную связь *:
Различные другие стерины, имеющие аналогичное расположение двойных
связей в кольце В стероидного скелета могут подвергаться такому же фотохимиче-
скому расщеплению {49].
ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ С-С-СВЯЗЕЙ
I. Окислительное расщепление углерод-углеродных связей
в насыщенных алифатических или алициклических
углеводородах
Окислительное расщепление углерод-углеродных связей в алифатических
углеводородах^ как правило, не имеет препаративного значения, так как при этом
может происходить разрыв связи в любом месте углеродной цепи с образованием
смеси карбоновых, дикарбоновых, оксикарбоновых кислот и других продуктов рас-
щепления. Только при точно установленных условиях проведения реакции высоко-
молекулярные парафины можно, например по Фишеру — Трошпу — Гачу, катали-
тическим окислением кислородом воздуха относительно однозначно расщеплять
до жирных кислот со средней Длиной цепи Сю — С т8. Такой способ приобрел большое
техническое значение в мыловаренной промышленности.
Эту реакцию прн использовании в качестве катализаторов солей Mapiaa-
ца проводят следующим образом: через нагретый примерно до 100° С парафин
пропускают тонко распределенный ток воздуха. Реакция заканчивается после
окисления около К'з исходного парафина.
Циклопарафины, как правило, е большим трудом и с плохими выходами оки-
сляются до неразветвлениых дикарбоновых кислот. Из циклогексана, например,
окислением азотной кислотой получают смесь 29% адипиновой кислоты и 36% нитро-
циклогексана {50].
II. Окислительное отщепление боковых цепей,
связанных с ароматическим или гетероциклическим ядром
1. Окислительное отщепление длинных боковых ценен
ароматических и гетероциклических соединении
РЯ ‘ W Ароматическое ядро в обычных условиях устойчиво'к действию окислителей,
однако алифатические боковые цепи ароматических углеводородов относительно легко
онисляются до карбоксильных групп. Подобное окисление метильной группы было
* Формулы лумистерина и тахнстерина уточнены (см. Л. Физ ер, М. Физ ср.
Стероиды, Изд. Мир. 1964, стр. 144 л 149). — Прим. ред.
|49j. Lettre-Inhoffen-Tscliesche, Uber Storine, Gallcnsauron und verwandte Naturstolle,
Bd. 1, 2 Aufl., Ferdinand Knke Verlag, Sturrgart. 1954, S. 184 If.
{50J. Пат. США 2343594; С. Л.. 38, (1, 2971 (1944).
II. ОКИСЛИТЕЛЬНОЕ ОТЩЕПЛЕНИЕ БОКОВЫХ ЦЕПЕЙ
83
уже подробно рассмотрено (стр. 299). Более длинные разветвленные и неразретвленны
алкильные боковые цепи, как правило, также окисляются до карбоксильной группь
Если в ароматическом углеводороде имеется несколько боковых цепей, то в больший
стве случаев вначале расщепляются более длинные цепи, а разветвленные цепи с тре
тичным углеродным атомом окисляются легче, чем нормальные. В противополож
ность этому, например, торет-бутильная группа, содержащая четвертичный углерод
ный атом, чрезвычайно устойчива к окислению.
Наиболее употребительными окислителями для окисления боковых цепе!
замещенных ароматических соединений являются разбавленная азотная кислота
хромовая кислота или перманганат калия. Направление окисления ароматически:
соединений, содержащих несколько заместителей, зависит от применяемого окисли
теля. Так, например, из n-цимола действием разбавленной азотной кислоты получаю-
n-толуиловую кислоту, а действием перманганата калия — оксиизопропплбензойнуи
кислоту [51]: <
СПз СНэ СООН
I I I
А А А
Y । ।
СООН (СНз)гСН (СНз)гС—ОН
Азотная кислота, применяемая для окисления боковых цепей, для подавления
нитрующего действия должна быть разбавлена водой в соотношении 1 : 3. Тольк<
в случае нитроуглеводородов, например 2-хлор-6-нитротолуола, можно использоватг
концентрированную азотную кислоту [52]. Способность к окислению боковых алкиль-
ных цепей, как правило, сильно снижается при наличии в орто-положенин атома
галогена. В отличие от описанного вигне превращения n-цимола в n-толуиловую ки-
слоту хлорцимол азотной кислотой окисляется до 3-хлоркуминовой кислоты [53]
СНз СООН
I I . '
Al ^2^ А
у\а у\С1 ’
(СНз)зСН (СН3)2СН ।
При окислительном отщеплении галогенсодержащих боковых цепей азотной
кислотой необходимо добавление нитрата серебра для связывания затрудняющего
окисление галоюпа.
Более энергично, чем азотная кислота, действуют хромовая кислота и перман-
ганат калия. Посредством этих окислителен, как правило, все алкильные группы
окисляются до карбоксильных. Алкилпиридины, аналогично гомологам бензола,
также окисляются с образованием пиридинкарбоновых кислот, напркмер, 4-этил-
пиридин окисляется до ияопик оптовой кислоты:
с3н5 соон
I I
А ™^А
Изоинкотииовая кислота. Смесь 160 г 4-этилпиридина и 300 лм воды на-
гревают до 80° С и прибавляют к ней также нагретый до 80J С раствор 960 г КМпО4
в 4 л воды. Прибавление ведут с такой скоростью, чтобы обеспечить постоянное
кипение реакционной массы. Затем кипятят еще 15 мин и в горячем виде отса-
сывают от образовавшейся двуокиси марганца. Осадок хорошо промывают
[51] A. R. Leeds, Вег., 14, 484 (1881).
[52] A. W. 3 1 a g е г, S. М. Me Е 1 vain, Org. Synth., 20, 79.
[53] Е. v. Gerichteii, Ber., 11, 364 (1878).
832
ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ С—С-СВЯЗЕЙ
горячей водой и объединенные фильтраты упаривают в вакууме до объема 1 л.
Затем подкисляют при 60° С до pH 3—4 концентрированной НС1, охлаждают до
20° С и отсасывают выделившуюся изоникотиновую кислоту. Выход продукта
165 г (90% от теоретического).
2. Расщепление гомологов фенола до фенолкарбоновых кислот
путем щелочного плавления
Гомологи фенола с помощью названных выше окислителей могут быть расщеп-
лены до фенолкарбоновых кислот только в том случае, если гидроксильная группа
защищена, например, алкилированием. Прямое получение свободных фенолкарбоно-
вых кислот из гомологов фенола удастся окислением боковой цепи щелочным плавле-
нием в присутствии кислорода воздуха. Так, например, щелочным плавлением из ти-
мола наряду с другими продуктами получают о-окси-п-толуиловую кислоту [54].
III. Окислительное расщепление олефинов
Соединения с одной олсфиповой двойной связью при действии окислителей,
как правило, легко расщепляются на два фрагмента. Однако очень редко можно на-
блюдать гладко протекающее расщепление под действием кислорода воздуха;
к таким иенлючениям относятся окисление углеводорода Гребе, бис-(флуоренилидспа),
который в спиртовом или эфирном растворе на воздухе расщепляется на две молекулы
флуоренона [55, 56]:
О о о
IXJ -* 21_>°
. о о о
В большинстве же случаев необходимо применение более сильных окислителей.
Для расщепления олефинов наряду с обычными окислителями, такими, как азотпая
кислота, перекись водорода, перманганат калия, хромовая кислота и высококонцентри-
рованные щелочи, используют также озон. Расщепление углерод-углеродной связи
олефинов под действием названных окислителей приводит к карбонильным соедине-
ниям. При этом часто в качество промежуточных продуктов образуются 1,2-гликоли.
Конечными продуктами такого окислительного расщепления являются в основном
альдегиды или кетоны, в зависимости от того, имеется ли у углеродного атома двойной
связи водород или нет.
Rx zH Rx
>C=C<f --> >C=O-LO=C<
R'Z \r” R'Z \R"
Если у углеродного атома двойной связи отсутствует атом водорода, то в любом
случае в качестве продукта реакции образуется только кетон. Но если у такого угле-
родного атома находится по крайней мере один атом водорода, то очень часто первично
образующиеся альдегиды под действием окислителя превращаются далее в карбоно-
вые кислоты. Альдегиды этим путем можно получить только через озониды с использо-
ванием специальных способов проведения расщепления. Превращение олефинов
в альдегиды под действием других окислителей удается только при наличии опреде-
ленных предпосылок в строении олефина.
В исключительных случаях даже в жестких условиях окисление олефинов
не приводит к разрыву углерод-углеродной связи. Сообщалось |57|, например, о полу-
чении с хорошим выходом бензои.тмуравьиной кислоты окислением стирола перман-
ганатом калия.
[54] L. Barth, Вег., 11, 1571 (1878).
[55] Л. Hantzsch, VV. И. Glover. Вег.. 39, 4i56 (1906).
[56] С. Gia be, Ber.. 25, 3146 (1892).
[57] С. D. II u r c[, R. \\ . McNamee, F. О. (; r e e n, J. Ain. Chem. Soc., 61,
2979 (1939).
III. ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ ОЛЕФИНОВ
83
1. Окислительное расщепление ненасыщенных карбонильных
соединений посредством щелочного плавления
Окислительное расщепление олефиновых двойных связей помимо его ирепаратив
ной ценности имеет еще большое значение как метод определения положения двои
ной связи. Однако, следует заметить, что прп щелочном плавлении пенасыщенны?
карбонильных соединений расщеплению молекулы предшествует перемещение двои
ной связи непосредственно к кярботтльной группе (стр. 847)- Так, при сплавлсню
олеиновой кислоты со щелочью с хорошим выходом образуется пальмитиновая кислота
CH3(CH2)7CH=CH(CH2)7COOH —* С15Н31СООН + СН3СООН
Поэтому для установления строения этот метод окислительного расщепления
олефинов часто неприменим. Однако с препаративной точки зрения он иногда очень
удобен, например для получения пеларгоновой кислоты из легко доступной ундеци
леновой кислоты [58]:
СН2=СН(СН2)8СООН [СНз(СН2}7СН-СНСООП] —>
—► СН3(СН2)7СООН^СНзСООН
Пеларгоновая кислота. В стальном котле смешивают 1 ч. ундецилеповой
кислоты с 3—4 ч. КОН и небольшим количеством воды и нагревают при пере-
мешивании. После прекращения выделения водяных паров при более высокой
температуре образуется однородный расплав. Нагревание продолжают еще
2—3 ч, причем выделяется значительное количество водорода. После охлажде-
ния плав растворяют в воде, фильтруют, фильтрат нагревают с HCI В промы-
вают выделившееся масло водой. Ректификацией сырой кислоты в вакууме
наряду с содержащей влагу первой фракцией получают почти совсем чистую
пеларгоновую кислоту; т. кип. 186° С (100 мм рт. ст.); т. пл. 12,5° С.
2. Окислительное расщепление олефинов до альдегидов
обычными окислителями
Обычно при расщеплепии олефинов с помощью перманганата калия в качестве
промежуточных продуктов образуются 1,2-гликоли, которые при соответствующих
экспериментальных условиях могут быть выделены из реакционной массы. Для полу-
чения альдегидов рекомендуется проводить окисление олефинов перманганатом только
до стадии гликолей, которые затем подвергаются действию специфических-окислите-
лей (стр. 835)- Метод получения альдегидов из олефинов действием чистого перман-
ганата через стадию образования гликолей представляет препаративный интерес
лишь при наличии определенных структурных предпосылок. Например, расщепление
арилэтиленовых производных останавливается на стадии альдегидов только в том
случае, если образующиеся альдегидные группы как бы «защищены» наличием элек-
троноакцспюрного заместителя в орто-положении. Из 4,4г-Динитростильбеп-2,2'-
дисульфокислоты можно действием перманганата калия с очень хорошим выходом
получить 4-нитробенэальдегид-2-сульфокислоту [59[:
SO3n SO3H ЗОзН
_Z \ Z
°!NZZ—сн=сн—ZZn°! —” 2°2n—\ Zcho
Иногда альдегиды могут быть получены окислением олефинов хромовой или
азотной кислотой, например, ацетилванилин пз ацетилизоэвгенола [60].
о- и n-Оксибензальдегиды оказалось возможным синтезирова! ь расщеплением
соответствующих арилэтиленов с помощью нитробензола и щелочи. Ванилин, например,
158] F. Krafft, Вег., 15, 1691 (1882).
(59J Герм. пат. 115410; Frdl., 6, 128.
[60] J. Schwyzer, Chem. Ztg., 1930, 840-
53 заказ 1835
834
ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ С—С-СВЯЗЕЙ
получают нагреванием изоэвгенола в течение' 2 ч при 180° С с нитробензолом
и 2 в. раствором NaOH под давлением [61J:
СНзО
СНзО
ВО— < >-СН=СН-СНз —>- СПО
3. Окислительное расщепление олефннон до кетонов
и карбоновых кислот обычными окислителями
Окислительное расщепление олефинов обычными окислителями пспользуетСя
для препаративного получения кетонов и, главным образом, карбоновых кислот.
Из рицинолевой кислоты окислением перманганатом калия получают азелаиновую
кислоту [62].
Азелаиноваи кислота. Раствор 240 г рицинолевой кислоты и 64 г КОН
в 1600 мл воды в один прием вносят в нагретый до 35° С раствор 625 г КМпО4
в 7,5’ л воды. Температура при этом повышается до 75° С. После перемешивания
в течение 30 мин исчезает окраска перманганата, затем массу подкисляют HsSO4,
в горячем состоянии отсасывают от двуокиси марганца, еще раз кипятят осадок
с водой, фильтруют и упаривают объединенный фильтрат до объема 4 л. При
охлаждении раствора выкристаллизовывается азелаиновая кислота, которую
можно очистить перекристаллизацией из воды. Выход продукта 48—56 г (32 —
36% от теоретического); т. пл. 104—106° С.
Большое синтетическое значение имеет также окислительное расщепление
циклоолефинов,- при этом с расщеплением кольца обычно образуются (в,(в'-дцкарбо~
новые кислоты. Например, нз циклогексена окислением КМпО4 получают адипиновую
кислоту (63]. Если у одного нз атомов углерода двойной связи не имеется атомов водо-
рода, то окисление таких циклоолефинов приводит к кетокарбоновым кислотам, на-
пример, при окислении сс-пинена образуется а-пиноновая кислота [64]:
а-Питтоппнэп кислота. Энергичным перемешиванием эмульгируют в 600 лм
воды 100 г а-пинена (т. кип. 155—160® С), полученного из французского ски-
пидара. К этой эмульсии при охлаждении льдом добавляют но каплям теплый
раствор 233 з КМпО4 в 2 л воды до тех пор, пока перманганат не перестанет обес-
цвечиваться. Смесь оставляют на 12 ч, затем обесцвечивают небольшим коли-
чеством метилового спирта и отфильтрованный от двуокиси марганца раствор
упаривают до объема 2 л; примеси исчерпывающе экстрагируют эфиром. Вод-
ный раствор подкисляют H2SO4, насыщают (NH4)3SO4 и снова тщательно экстра-
гируют эфиром. Полученный из кислого раствора эфирный экстракт нейтрали-
зуют NaHCO3, растворенным в небольшом количестве воды, причем выпадает
труднорастворимая натриевая соль. Осадок отфильтровывают и фильтрат исчер-
пывающе экстрагируют эфиром. Затем водную часть вновь подкисляют, насы-
щают (NH4)2SO4 и вновь экстрагируют эфиром. При отгонке эфира из объеди-
ненного экстракта получают' 150 з сырого продукта, из которого наследующий
день выкристаллизовывается 10 з вещества. Незакристаллизовавшуюся часть
[61 ] В. Leopold, Acta Chim. Scand., 4, 1523 (1950).
[62 ] J. W. Hill, W. L. McEwen, Org. Synth., 13, 4.
[63 ] K. Vogt, Mitteilungen aus dem schlesischen Kohlenforschungsinstitut; C., 1926,
I, 2341.
[64 ] А. В a e у e r, Ber., 29, 22 (1896).
III. ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ ОЛЕФИНОВ
835
фракционируют. Выделяют три фракции с т. кип. (14 мм рт. ст.) 180—187,
187—200, 200—230° С, Из этих фракций кристаллизуется 30—40 г пиноновой
кислоты (из 300 в пинена). После перекристаллизации из воды т. пл. ЮЗ—
' 4. Окислительное расщепление олефинов озоном
Лучшие выходы карбоновых или дикарбоновых кислот получают, если окисли-
/, тельное расщепление соответствующих олефинов или циклоолефинов проводят
у с помощью озона. Вследствие того, что к олефиновой двойной связи озон присоеди-
' няется гораздо легче, чем к ароматической системе, арилолефины можно без особых
£ затруднений подвергать озопированню, заметно не затрагивая ароматическое ядро.
Ч Разложение образующихся на первой стадий озонидов водой протекает не однозначно,
д; При этом образуется перекись водорода, альдегиды и продукты их окисления.
' пен сии' ВСИ О СИП' RCHO + R'CIIO + П2О2
Для получения карбоновых кислот из олефинов озонированием рекомендуется
проводить дополнительное окисление продуктов расщепления озонидов горячей
J суспензией окиси серебра [65J или надуксусной кислотой {66]. Этим путем удалось
из циклогексена получить адипиновую кислоту с примерно 90% -ным выходом от
• теоретического [66].
t /'ll НООС(СН2)4СООН
Адипиновая кислота. Циклогексен озонируют в метил- или этилацетате, хло-
роформе или метилендихлориде при температуре от —50 до —20° С или в несколько
разбавленной уксусной кислоте при 0° С. По окончании озонирования реакцион-
ную массу смешивают с равным объемом ледяной уксусной кислоты и над-
уксусной кислотой в количестве, соответствующем одному атому активного
кислорода на каждую двойную связь. Если при этом в смеси еще остается зна-
чительное количество нерастворенного озонида, смесь осторожно нагревают
при 50° С до полного растворения и в вакууме при 50° С отгоняют низкокипящнй
растворитель. К оставшемуся уксуснокислому раствору добавляют надуксусную
кислоту, количество которой соответствует двум атомам активного кислорода
на двойную связь, и оставляют на 12 ч при 50—60° С. Затем кипятят еще 1 ч
с обратным холодильником для полного разложения непрореагировавших
озонидов и надуксусной кислоты и после отгонки ледяной уксусной кислоты
в вакууме получают продукты расщепления, обычно уже в виде твердого кри-
сталлического соединения.
Для получения альдегидов из озонидов последние подвергают восстановитель-
ному расщеплению либо действием цинка в ледяной уксусной кислоте, либо каталити-
ческим гидрированием [67, 68J над катализатором палладий — карбонат кальция
(5% Pd, 95% СаСО3) [69]. Этим путем можно из циклогексена получить адипиновый
альдегид с выходом 60 — 70% от теоретического [70J:
СПО
сно
65] F. Asinger, Вог., 75, 656 (1942).
66] Н. Wilms, Ann., 567, 96 (1950).
67] F. G. F i s с h е г, H. Dull, L. E г t e I, Ber., 65, 1467 (1932).
68 J A. L. Henne, W. L. P erilst ein, J. Am. Chem. Soc.. 65, 2183 (1943).
69] M. В u s c h, H. Sto w e, Ber., 49, 1064 (1916).
70] Gattermann - Wieland, Die Praxis des organischen Clienukcrs, 32 Aufl.
Walter de Gruyter u. Co., Berlin, S. 352.
53*
836
ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ С—С-СВЯЗЕЙ
5. Укорочение боковой цепи по Виланду *
Особый случай окислительного расщепления олефинов представляет собой
укорочение боковой цепи по Виланду. Эта реакция позволяет ступенчато проводить
укорочение алифатической цепи с концевой карбоксильной группой на один атом
углерода. Виланд, Якоби и Шлихтинг [71], применив метод Барбье [72], подвергли
такому ступенчатому расщеплению боковую цепь холановой кислоты. Вначале эфир
желчной кислоты взаимодействием с гриньяровским соединением** превращали в тре-
тичный спирт, из которого отщеплением воды получали олефин. Окисление олефина,
например, хромовой кислотой приводило к карбоновой кислоте, содержащей на один
углеродный атом меньше, чем исходный продукт:
СНз СНз
I 2CHsMffX । /СНз
C19II3iCIICH2CII2COOR --?-=-* CigHgiCHCHaCHaC^
I ^СНз
ОН
СНз СНз
I /СНз „ „ I /СНз
—> С19Н31СНСНоСН=С< СгО’^ С19Нз1СНСН2СООН^О=С<
ХСН3 хсн3
Этим способом из холановой кислоты получили порхолановую. Операцию
можно повторить еще дважды и тем самым сократить боковую цепь до стоящей у ядра
карбоксильной группы. Метод постепенного упрощения молекулы был использован
для точного выяснения строения боковой цепи желчных кислот и места присоединения
боковой цепи к кольцевой системе. В определенной модификации (применение бром-
сукцннимида) метод и в настоящее время имеет препаративное значение. По этому
методу эфир желчной кислоты вначале также действием гриньяровского соединения
с последующим отщеплением воды превращают в олефин, который подвергают взаимо-
действию с N-бромсукцинимидом. Бром вступает в аллильное положение, но сразу
же с отщеплением бромистого водорода образуется диен. Одностадийным окислением
диен превращается в кетосоедивсние, часто являющееся исходным продуктом для
частичных синтезов стероидных гормонов [73]:
СНз СНз
J /СНз I /СНз
Ci,HslCHCn!Ctt=C< —> с1,нясиснсн=с( —
СП3 I XCHs ~HBr
Br
СНз СНз
I /СНз I
— -> С19Нз1С=СНСН=С< его». CJ9n31C=O
^СНз
IV. Окислительное расщепление С—С-связей
в соединениях с гетерофуикцией в месте расщепления
Из большого числа реакций окислительного расщепления углерод-углеродных
связей, рассматриваемых в этом разделе, препаративное значение имеют главным обра-
зом реакции расщепления вторичных спиртов и кетонов, в частности метнлкетопов
и гликолей, и аналогичных соединений, протекающие под действием как обычных, так
и специальных окислителей.
* Этот метод обычно называют методом Барбье — Виланда. — Прим. ред.
* * Прп расщеплении боковой цепи по Барбье — Виланду применяют обычно [фе-
нилмагнийгалогепиды. — Прим. ред.
[71] Н. Wieland, О. S с h 1 i с h t i n g, R. J akobi, Hoppe-Scyler's Z. phy-
siol. Chem., 161, 80 (1926).
[72] Ph. Barbier, R. Locquin, C. r.. 156, 1443 (1013).
[73] F. Bohlmann, Arznciroiltel-Forsrlj., 4, 28 (1934).
IV. ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ С-С-СВЯЗЕЙ В МЕСТЕ ГЕТЕРОФУНКЦШ1 8,-
1. Окислительное расщепление вторичных спиртов н кетонов
Алифатические вторичные спирты и кетовы с метиленовыми или метинпыь
группами в «-положении к карбонильной группе расщепляются, как правило, не одн
значно/а с образованием смеси карбоновых кислот или карбоновых кислот и кетоно
так как окислению подвергаются оба углеродных атома, находящиеся в а-положент
к карбонильной группе. Поэтому, например, из симметричных кетонов с дву^
«-метиленовыми группами при окислительном расщеплении получаются две карбон
вне кислоты, различающиеся по составу на один углеродный атом. Из несимметричны
кетоиов образуется смесь до четырех карбоновых кислот*, разделение которой част
представляет большие трудности.
> RCHaCOOII-f-HOOCR'
RCH2COCH2R'
-* RCOOH-HIOOCCII2R'
Окислительное расщепление протекает более однозначно, если в а-положепи
к карбонильной группе находится метильная группа (см. ниже) или если у а-углеро;
ного атома не имеется атомов водорода. В этом случае окисление происходит тольк
со стороны метиленовой или метинной группы. Ди-трет-кетопы, например бенз(
фенон, вследствие этого почти совершенно устойчивы к окислению.
Большое синтетическое значение имеет окислительное расщепление кольц
алициклических спиртов и кетонов, которые превращаются в ю,й>'-дикарбоновь]
кислоты с тем же числом атомов углерода. Так, например, из циклогексанола ил
циклогексанона в присутствии солей ванадия и меди действием 60% -пой HNO3 полу
чают адипиновую кислоту с выходом до 90% от теоретического [74].
Адипиновая кислота. Нагревают до 55° С 0,32 ч. ванадата аммония и 0,4 ч
медных стружек с 301 ч. 60% -ной IINO3 и при 55—60° С в течение 5 ч посгепенн
добавляют 100 ч. циклогексанола. Затем массу в течение часа выдерживаю
при этой температуре, после чего охлаждают до 17° С, фильтруют, осадок н,
фильтре дважды промывают 100 ч. охлажденного до 17° С насыщенного вод
ного раствора адипиновой кислоты и сушат при 90—95° С. Выход 129,9 г (89?!
от теоретического).
Действием перкислот, например кислоты Каро, на циклические кетоны полу
чают со-оксикарбоновые кислоты или пх циклические лактоны. Последние при взаимо
действии со спиртами в присутствии кислот образуют эфиры о-оксикарбоновы;
кислот. Этим путем, например, из циклогексанона получают эфир <и-оксикапроново1
кислоты [75, 76];
2. Окислительное расщепление метилкетоиов
В лабораторной практике окислительное расщепление метилкетонов исполь-
зуется довольно часто. В случае алифатических метилкетонов вследствие большой
устойчивости метильной группы к окислению обычными окислителями реакция про-
текает достаточно однозначно. Например, окислением неразветвлепных алифатиче-
ских метилкетонов хромовой смесью наряду с уксусной кислотой получают жирную
* Правило А. Н. П о п о в а. — Прим. ред.
[74J Франц, пат. 919755; С., 1947, II, 745.
[75] А. В а е у е г, V. Villager, Вег., 32, 3625 (1899).
[76J S. L. F г i е з з, J. Am. Chem. Soc., 71, 2571 (1949).
838
ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ С—С-СВЯЗЕЙ
кислоту, имеющую на два атома углерода меньше, чем исходный кетон; из октадека-
вона-2 образуется пальмитиновая кислота J77J:
С1вНззСОСНз —> С15Н31СООН + СН3СООН
Окислительное расщепление метилкетонов было использовано Краффтом
для выяснения строения стеариновой кислоты [78]. При сухой перегонке
(стр. 818) смеси бардовой соли стеариновой кислоты и ацетата бария был получен
соответствующий метилкетон, окислительное расщепление которого дало на-
ряду с уксусной карбоновую кислоту, имеющую на опил атом углерода меныпс.
чем стеариновая кислота. Эти операции повторялись до тех пор, пока стеарино-
вая кислота не была расщеплена до уже тогда синтетически доступной «-каприно-
вой 1 кислоты.
Если в таких кетонах у соседнего с ацетильной группой углеродного атома
отсутствуют атомы водорода, то при окислительном расщеплении они дают карбоновую
' кислоту, имеющую на один углеродный атом меньше, чем исходный кетон [79]. Пина-
колин хромовой кислотой в серной кислоте окисляется до тркметилуксусной кислоты:
(СНз)зССОСН3 —> (СНа)зССООН + СО2
При окислении в других условиях, например щелочным раствором перман-
ганата, пинаколин может бытЬ превращен в триметилпировиноградную кислоту.
Эту e-кетокислоту действием щелочного раствора перекиси водорода затем можно
окислительно декарбоксилировать до триметилуксусной кислоты [80]:
КМпО«; HtO,:
(СНз)зССОСНз - К"°” - (СНз)зССОСООН -Na—(СНз)зССООН
Такое двухегадийное расщепление метилкетонов до карбоновых кислот, име-
ющих на один углеродный атом меньше, чем исходный кетон, возможно только в том
случае, если карбонильная труппа метилкетока связана с четвертичным углеродным
атомом, как, капример, у пинаколина и ряда ароматических метилкетонов типа аце-
тофенона.
Легче и часто тагеже в более мягких условиях протекает расщепление алкил-
и арилметилкетонов до карбоновых кислот, имеющих на один атом углерода меньше,
чем исходный кетон, по так называемой галоформной реакции, причем в качестве
окислителей применяют гипогалогениты. Этим способом из пинаколина получают
триметилуксуспую кислоту с 70%-ным выходом от теоретического [81]. При действии
гипогалогенитов концевая метильная группа промежуточно превращается в тригало-
генметильную группу; полученный продукт легко гидролизуется щелочью до три-
галотенметана и соответствующей карбоновой кислоты (стр. 842):
HC0CH3+3NaC10 —> [RCOCCls] —> RCOOH + CHCI3
При проведении галоформной реакции с 2,4,6-трихлорацетофенояом удалось
выделить промежуточно образующийся а,а,а-трибром-2,4,6-трихлорацетофенон [82].
а,а,а-Трябром-2,4.6-трихлорацетофенон. Перемешивают 5 г 2,4,6-трихлор-
ацетофенона (полученного с 50%-ным выходом из 2,4,6-трихлорбензоилхлорида
и метилмагпийбромпда) в течение 30 ч при 50° С и затем еще 50 ч при 70° С
с 500 .мл раствора гипобромита натрия, приготовленного растворением прп
[77
[78
[79
[80
[81]
[82[
F. Krafft, Бег., 15, 1707 (1882).
F. Krafft, Вег., 12, 1672 (1879).
S. Reformatzky, Вег., 23, 1596 (1890).
М. J. В б s е к с п, Rec. trav. chim., ЗО. 142 (1911).
L. Т. Sandborn, Е. W. Bousquet, Org. Synth., 8, 108.
R. C. Fuson, J. W. B'ertelti, W. E. R 0 s s, J. Am. Chem.
4380 (1932).
Soc.,
54,
IV. ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ С—С-СВЯЗЕЙ В ‘МЕСТЕ ГЕТЕРОФУНКЦИИ j
0° С 50 г брома в 500 мл 10%-ного раствора NaOH. При этом выделяется -
трибромкетока (80% от теоретического; т. пл. 126—127° С), длительное ки<
чение которого с 50%-ным раствором NaOH с почти количественным выход
даст трнхлорбенаойную кислоту.
Для гладкого протекания галоформной реакции желательно, чтобы в оста!
R мстилкетонов общей формулы RCOCHa не имелось водородных атомов, легко за!
щаемых на галоген. Однако при проведении реакции в соответствующих услови
замещение таких водородных атомов затрудняется; например, из 2-метил-2-фен<
пентаиона-4 с выходом 84% от теоретического гладко образуется (3-фенилизовале(
ановая кислота [83]:
СН8 х ' СНзх
>С-СН2СОСН3 —► >С-СН2СООН
СНз 7 I CH3Z I
С6Н6 свн5
К аналогичным превращениям склонны также этил- и пропилкетоны общ
формулы RCOC2H5 или RCOCsH7, еслк R является ароматическим или гетероцикл
ческим остатком [84]. Например, из пропиофенона получают бензойную кисло
с выходом 64% от теоретического:
СвН6СОС2Нб —> CeHsCOOH
Этим путем могут быть синтезированы также ненасыщенные кислоты, ecj
окислению гипогалогенитами подвергают а,0-нонасыщенные метилкетоны. Та
иэ окиси мезитила можно получить р,р-диметилакриловую кислоту [85], а из цин
.амилиденацетона — циннамилиденуксусную кислоту с выходом 70% от теорет
ческого [88].
СеНаСН=СНСН=СНСОСН3 —> СвНйСН=СНСН=СНСООН
Недостатком этого метода является необходимость проведения реакции в гет
рогенной среде. Весьма целесообразно добавление устойчивых к окислению эмул
-^гаторов, а также соответствующих растворителей, например диоксана, к водны
растворам гипохлоритов или гипобромитов.
Описан довольно гладкий способ расщепления метилкетонов до карбоновы
кислот, содержащих на oJjhh атом углерода меньше, чем исходное вещество [87
Принцип метода заключаете^ в том, что монобромпроизводные, полученные действие
•брома на метилкетоны, обрабатывают пиридином, причем в большинстве случае
образуются хорошо кристаллизующиеся пиридинийбромиды. Последние в водп<
спиртовом растворе гидролизуются NaOH до карбоновых кислот и метилпнридини!
•бромида:
RCOCH, — >• НСОСН2Вг —► [всосна-й^-^]вг- —►
—> RCOOH + [СНз-^>]вг
Весь цикл превращений проводят без специальной очистки промежуточны
продуктов,[при этом желаемые карбоновые кислоты образуются с хорошими выходами
В качестве’ примера можно привести получение а-нафтойной кислоты.
а-Нафтойная кислота. а-Ацетилнафталин растворяют в 5—10 ч. ледяно)
уксусной кислоты, раствор смешивают на холоду с рассчитанным количество!
брома и затем умеренно нагревают на водяной бане. Реакционную смесь выливаю
в воду, содержащую небольшое количество двуокиси серы; выпавшие кристалл!
[83] J. Colong е, L. Pi chat, Bull. Soc. chim. France [5], 16, 180 (1949).
[84] R. Levine, J. R. Stephens, J. Am. Chem. Soc., 72, 1642 (1950).
[85] L. J. Smith, W. Richard, L. J. Spillane, Org. Synth., 23, 27.
[86] J- T. P 1 a t i, W. II. S t г a i n. S. L. W arren, J. Am. Chem. Soc., 65
1273 (1943).
[87[ F. Krohnke, Ber.. 66, 604 (1933).
840
ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ С—С-СВЯЗЕЙ
отфильтровывают и отжимают на пористой пластинке, Полученный а-нафтацил-
бромйд смешивают с избытком пиридина; мосле затихания энергичной реакции
смесь короткое время нагревают ка водяной бане, извлекают продукт этиловым
спиртом и осаждают из раствора эфиром. а-Нафтацилпиридинийбромид можно
перекристаллизовать из 10 ч. воды (т‘. пл. 170° С). Затем 0,5 е бромида раство-
ряют в смеси 40 мл воды и 12 мл этилового спирта и в течение 12 мин нагре-
вают на водяной бане с 2 мл 10%-ного раствора NaOH. При подкислении выпа-
дает а-нафтойная кислота.
Соответствующим образом синтезируют и ^-нафтойную кислоту; при
этом из 0,5 з пиридинийбромида получают 0,25 г ^-нафтойной кислоты.
Благодаря предложенному Кингом 188} упрощению этот метод получил еще
более широкое распространение. Метил- или метигхенкеток примерно в течение часа
нагревают на водяной бане с кодом в сухом пиридине, при этом в очень мягких усло-
виях непосредственно образуется пиридинийиодид, который легко расщепляется
при нагревапии с водно-спиртовым раствором NaOH. Суммарный выход кислот соста-
вляет 60—83% от теоретического.
3. Окисление тетраацетатом свинца
Соединения, имеющие у соседних углеродных атомов функциональные группы,
особенно легко расщепляются по связи, соединяющей эти атомы. К ним относятся
1,2-диолы, кетолы, а-оксикислоты, 1,2-оксиамины, 1,2-диамины, а-аминокислоты
и щавелевая кислота. Расщепление протекает однозначно под действием тетраадетата
свинца [89] или иодной кислоты [90]. Продуктами расщепления являются карбониль-
ные соединения или соответственно имины, которые, однако, обычно дегидрируются
до нитрилов:
\с-он \с—6
о — С +
">с-он ^с-^о
ПСНОН-СООН —> RCIIO -со2
R\ R\
/СОН— СООН —> Ъс=О —СОч
R'/ R’z
RCH— СООН -Тсб;? RCH—NH —» RC^N
NH3
Тетраацетат свинца ввиду его растворимости в оргатгческнк растворителях
используется наиболее часто. Тетраацетат свинца с успехом может применяться также
в водных эмульсиях или в смесях растворителей с водой [91]. Окисление иодной
кислотой ведут в водных средах, часто с применением веществ, повышающих раство-
римость, — эмульгаторов, диоксана или ледяной уксусной кислоты, В некоторых
случаях используют также тетраокись осмия [92].
Тетраацетат свинца и иодная кислота действуют в общем примерно одинаково
[93], и хотя тетраацетат свинца является более дешевым реагентом, необходимо отме-
тить, что иодная кислота отличается большей селективностью. Так, третичные гликоли,
а также а-оксикислоты не окисляются подпой кислотой. Поэтому, например, р-фенил-
[88] L. С. К, i и g, J. Am, Chem. Soc., 66, 894, 1612 (1944)
[89] R. Criegee, Z. angew. chem., 53 , 324 (1940).
[90] L. Malaprade. C. r., 186, 382 (1928).
[91 ] E. Baer, J. M. Grossheint z, H. O. Fischer, J. Am. Chem. Soc.,
61, 2607. 3379 (1939).
[92] R. Criegee, Ann., 522, 75 (1936).
[93/ P. Caro, R. Hiroluta Helv, chim. acta, 16, 959 11933).
V. ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ КОЛЬЦЕВЫХ СИСТЕМ
&
молочную кислоту можно превратить в фенилацетальдегид только действием тстраац
тэта свинца [94]:
С0Н6СН2СНСООН —> СвН6СН2СНО
I
он
Фенилапятальдягид, Кипятят 10'г 8-фенилмолочной кислоты (получение
из фенилаланина) со 150 мл бензола до растворения большей части кислот,
и затем постепенно при перемешивании добавляют 26 г тетраацетата свинца
Выделившийся ацетат свинца отфильтровывают и промывают бензолом. Обт
единенный бензольный раствор перегоняют в вакууме. Выход фенилацетал!
дегида 4,2 г (58% от теоретического); т. кип. 84—86° С (17 мм рт. ст.)
Наоборот, превращение винной кислоты в глиоксалевую возможно тояьк
по реакции с иодной кислотой, так как при действии тетраацетата свинца винна;
кислота расщепляется по всем трем углерод-углеродным связям [95]. Напротив, и
эфиров винной кислоты по разработанному Вейгандом способу действием тетраацетат,
свинца можно получить эфиры глиоксалевой кислоты, так как этсрифицированные п<
карбоксильной группе а-оксикислоты, аналогично сложным или простым эфира»
гликолей, таким способом полностью не расщепляются:
' ROOCCH—CHCOOR —> 2ROOC—СНО
I I
НО он
Эфир глиоксалевой кислоты. Растворяют 232 г диэтилового эфира винцо»
кислоты в 1200 мл высушенного над натрием бензола, охлаждают льдом и в те-
чение часа при перемешивании вносят в раствор 500 г тетраацетата свинца,
причем температура пе должна превышать 10° С. Затем перемешивают еще
12 ч, отсасывают осадок, промывают его бензолом. От бензольного раствора
отгоняют бензол при уменьшенном давлении. После отгонки первых 500 мл
меняют приемник и испытывают дистиллят на наличие эфира глиоксалевой
кислоты нагреванием пробы с равным объемом 2 н. раствора NH3. При наличии
эфира образуется усиливающееся от желтого до иссиня-красногб окрашивание.
В этом случае ведут отгонку до остаточного объема 300 мл; остаток раство-
ряют в 900 г абсолютного этилового спирта и раствор фракционируют на
колонке в вакууме. После отгонки спирта при 40—48° С (17 .мл рт. ст.) пере-
гоняется в основном бензол, при}54—55° С (16 мм рт. ст.) — эфир глиоксалевой
кислоты в виде алкоголята. Выход алкоголята эфира 146 г (43% от теоре-
тического).
V. Окислительное расщепление кольцевых систем
ароматических и гетероциклических соединений
1. В конденсированных ароматических системах с помощью окислителей воз-
можно расщепление отдельных колец. Наиболее известным примером этого является
синтез фталевой кислоты из нафталина, причем хорошие выходы получают при окисле-
нии олеумом в присутствии HgSO4 при 250—300° С [96]. В настоящее время нафталип
окисляют кислородом воздуха при 400—500° С в присутствии ванадиевых ипи молиб-
деновых окисных катализаторов [97]. Наличие определенных заместителей, например
окси- или аминогрупп, в ароматическом ядре конденсированной системы, как правило,
облегчает-окисление. Поэтому из нафтолов и нафтиламинов относительно легко полу-
чают фталевую кислоту:
NH2
^/соон
II! I —> I II
А/\Соон
[94] Н. О е d a, Bull, chem. Soc. Japan, 9. 13 (1934).
f95] F r e u г у, В on - В e r n a t e t s, J. Pharm. Chim. [8]. 23, 85 (1936).
[96] Герм. пат. 91202; Frdl., 4 164.
[97] Герм. пат. 347610, .379822; Frdl., 14. 450.
842
ОКИСЛИТЕЛЬНО® РАСЩЕПЛЕНИЕ С-С-СВЯЗЕЙ
Так называемые «отрицательные заместители», как нитрогруппа и галогены,
повышают устойчивость к окислению замещенных колец в конденсированных системах.
Из а-нитронафгалина при окислеини хромовой кислотой получают поэтому только
нитрофталевую кислоту 198]:
NOa
Хл°он
^z\cOOH
Если , расщепление нафталина проводят действием перманганата калия, то
в тщательно контролируемых условиях реакции в качестве промежуточного продукта
можно Получить также фталоиовую кислоту, которая далее может быть декарбокси-
лирована в формилбензойную кислоту [99] (ср. стр. 817).
zx ух zx ,СОСООН /СНО
zZ х / Xj. KMnO« ; zZ у NaHSO»; HCI । zZ 'у'
^/\соон XZ\COOH
2. Часто препаративное значение имеет окислительное расщепление конденси-
рованных гетероциклических систем. Хинолин, например, окислительным расщепле-
нием бензольного ядра под действием щелочного перманганата можно превратить
в пйридин-2,3-дикарбоновую кислоту [100]. Из изохиколина в таких же условиях
иолучают пириднн-3,4-дикарбоновую кислоту. Но в других условиях может окисляться
также и гетероциклическая часть молекулы. При окислепии перманганатом в кислой
среде из изохинолина, например, образуется фталимид [101]:
НООС\У\
ч I N ОН-; КМаОд
I II *
uooc/^Z
Н+;- КМпО<
3. В качестве примера окислительного расщепления одноядерных гетероцик-
лов следует упомянуть превращение фурфурола в фумаровую кислоту [102]:
[I Д
осно
УаСЮ»
НООС— СН
II
НС—соон
+ COs
Возможность окислительного расщепления фуранового кольца была исполь-
зована для синтеза триалкилуксуспых кислот. Эфир фуран-2-карбоновой кислоты
вначале алкилировали триалкилгалогенидами по Фриделю — Крафтсу и полученные
далее омылением 15-алкилфуран-2-карбоновые кислоты окислительно расщепляли
до триалкилуксуспых кислот действием перманганата калия в щелочной среде [103]:
НС-СН НС—СП
НС J-COOCaHs В,СХГ А1с1^ R3c-С С-СООСаНй -а0Н; КМп0< - RSCCOOH
О о
98] F. Beilstein, A. Kurbatow, Апо., 202, 217 (1880).
99] J. Н. Gardner, С. A. Naylor, Org. Synth., 16, 68.
100] W. v. Muller, Ber., 24, 1900 (1891).
101] G. Goldschmied, Mh. Chem., 9, 676 (1888).
102] N. A. Milas, 3. Am.-Chem. Soc.. 49, 2007 (1927); Org. Synth., 11, 46.
103] T. Reichstcin et al., Helv. chim. acta, 18, 721 (1935).
ГИДРОЛИТИЧЕСКОЕ РАСЩЕПЛЕНИЕ
84:
ГИДРОЛИТИЧЕСКОЕ РАСЩЕПЛЕНИЕ
Гидролитическое расщепление углерод-углеродных связей ограничено относи-
тельно немногими, ио важными реакциями. Происходящее, как правило, под дей-
ствием концентрированных щелочен гидролитическое расщепление предполагав!
наличие в исходном соединении карбонильной группы*. К карбонильной группе
впачале присоединяется акион гидроксила и образующийся промежуточный продук!
расщепляется затем на карбанион и карбоновую кислоту:
-С-+С-:
он
В случае простийтпих альдегидов и кетонов такого гидролитического расщепле-
ния ожидать нельзя, так кан реакция протекает в относительно жестких условиях
и прежде, чем наступит расщепление, чаще всего происходят другие реакции.
Гидролитически расщепляться под действием щелочей при высоких темпе-
ратурах могут только те кетоны, для которых исключены побочные реакции.
Фенхон, например, в молекуле которого оба углеродных атома, находящиеся в a-поло-
жении к карбонильной группе, не имеют атомов водорода, под действием раствора
КОН при нагревании до 220° С гладко превращается в фенхоловую кислоту [104]:
,СН-СН-СН8
н2с I i
• | СН2 СНз
Н2с |
\с-соок
СНз
В более мягких условиях происходит щелочной гидролиз С—С-связей Р-дикето-
нов и эфиров Р-кетокислот. В этом случае находящийся в ^-положении к карбониль-
ной группе катионный центр облегчает растепление углерод-углеродной связи,
исходящей от карбонильной группы:
°' сн3 о- СНа
R—С—СН2—С—R П1Г_ Л—с/ ^С-R R—с/ ^С+—К
11 II I II —> I I
0 0 ОН О О О'
W
он
I
—> R-COO- + CH2=C-R
It
о
СНз-С-R
• Это утверждение Вейганда пе совсем точно. Работами М. М- Шемякина и сотр.
показано, что при кипячении в щелочном растворе а, ^-непредельных карбо-
нильных соединений, кислот, ннтросоедпнеинй и т. д. С=С-связь претерпевает
гидролитическое расщепление [М. М. Ш е м я к и н, И. А. Р е д ь к и я, ЖОХ,
, И, 1142 (1941) и его, последующие статьи в ЖОХ]. — Прим. ред.
[104] О. Wallach, Wienhaus, Ann., 369, 72 (1909).
844
ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ С—С-СВЯЗЕЙ
При гидролизе Р-дикетонов происходит, следовательно, расщепление дикетонз
на молекулу кислоты к молекулу кетона. При гидролизе несимметричных Р-дикетонов
в большинстве случаев возможно образование смеси двух кислот и двух кетонов,
так как расщепление, согласно следующему общему уравнению, может идти по двум
направлениям:
RCOCHs ! R'COOH RCOCHjCOK' —- RCOOH+ СН3СОН'
Однако если R — ароматический, a R' — алифатический остатки, то гидролиз
идет по реакции (я) и получается почти исключительно жирно-ароматический кетон,
например ацетофенон из бензоилацетона:
С6Н&СОСН2СОСН3 —> СвН6СОСН3+СН3СООН
Из эфиров р-кетокислот в результате гидролиза образуются две молекулы
карбоновых кислот:
R' R’
I I
RCO-C-COOR"’ —> RCOOH+HC-COOH^ R"'OH
I I
R" R"
Такое расщепление, происходящее, как правило, под действием концентриро-
ванных щелочей, называют «кислотным расщеплением» эфиров р-кетокислот в отличие
от протекающего под действием кислот или разбавленных щелочей «кетонного расщеп-
леиня» с одновременным декарбоксилированием (стр. 820). Кетонное расщепление
часто происходит более гладко, чем кислотное, успех которого в сильной степени
зависит от выбора оптимальной концентрации щелочи. Кислотное расщепление не
нашло очень большого препаративного применения уже потому, что образующиеся
при этом карбоновые кислоты обычно более изящно и удобно могут быть получены
путем соответствующих синтезов с малоновым эфиром. Однако этот метод оказался
пригодным для синтеза а-замещенных адипиновых кислот и пимелиновой кислоты.
В первом случае эфир циклопентаион-2-карбоновой кислоты, полученный конден-
сацией Дикмана (стр. 791) из диэфира адипиновой кислоты [105], вначале путем
взаимодействия его енольной соли с алкилгалогенидами алкилируют по а-углеродному
атому и затем подвергают «кислотному расщеплению», причем обычно с почти количе-
ственными выходами образуются а-замещенные адипиновые кислоты:
COOR'
I
COOR COOR ,СН— R’
j 1 R'X; NaOH , p—J—R' > П2С
\/^>O \/^O H2C соод
CH2
Пимелиновую кислоту также очень гладко получают кипячением эфира цикло-
гексанон-1-карбоновой-2 кислоты с избытком КОП в метиловом спирте [106]. Пиме-
линовая кислота может быть также легко синтезирована восстановлением сали-
циловой кислоты водородом в момент выделения. При этом несомненно, что реакция
протекает через промежуточное образование циклогексанонкарбоновой кислоты [107]:
’ /\/0Н /\/0Н /\
f t -УЬ» I II | |'Z Уаон- ПООС(СНа)5СООП
^/^соон х^'\соон \/ХСООН
В аналогичных условиях n-аминосалициловая кислота пе превращается в р-амипо-
пимелиновую кислоту [108].
[105] Р. S- Pinkney, Org. Synth., 17, 30.
[106] W. Dieckmann, Ann., 317, 100 (1901); Org. Synth., 11, 44.
[107] А. МйИег, E. R о 1 t z, Mh. Chem., 48, 734 (1927).
[108] L. В i г k о f e г, I- Storch, Ber., 86 749 (1953).
ГИДРОЛИТИЧЕСКОЕ РАСЩЕПЛЕНИЕ
845
Кислотное расщепление особенно легко протекает в случае ацилированных
эфиров р-кетокислот. Исходя из ацетоуксусных эфиров, относительно легко можно
синтезировать любые эфиры р-кетокислот. Продукт ацилирования ацетоуксусного
эфира подвергают гидролизу или алкоголизу. При этом, как Правило, происходит
отщепление ацеткльнои группировки, а не введенного в ацетоуксусный эфир ациль-
ного остатка. Так, например, из а-бензоил-а-метилацетоуксусного эфира слабым
нагреванием в сильно разбавленном метанольном растворе метилата натрия получают
эфир а-метилбензоилуксусной кислоты [109]:
СНз СНз
CH3CO-C-COOR CH3COOR4-CeH5CO-CH-COOR
I
сосвн5
Точно так же из бензоилацетоуксусного эфира слабым нагреванием его в водном
растворе NII3 и NH4C1 получают бенз онлуксу оный эфир [110].
Бензоилацетоуксусный эфир. В колбе емкостью 5 л с мешалкой и обратным
холодильником в течение 24 ч кипятят 4,5 л сухого бензола с 250 а ацетоуксус-
ного эфира и 46 г натрия. Затем в течение 3 ч прибавляют по каплям 350 г
бепзоилхлорида и вновь кипятят 8 ч. Содержимое колбы выливают на 500 мл
измельченного льда, отделяют бензольный раствор, отгоняют бензол и остаток
перегоняют в вакууме. Выход продукта 350 г (74,8% от теоретического); т. кип.
177 —181° С (20 мм рт. ст.).
Бензоилуксусный эфир. К нагретой до 40° С смеси 32 г NH4CI, 10 мл кон-
центрированного NH3 и 150 мл воды добавляют 58,5 г бензоилацетоуксус-
ного эфира и нагревают в течение 10 мин цри 40° С. Затем массу быстро охла-
ждают и экстрагируют продукт 100 мл эфира. Эфирный раствор сушат над
безводным MgSO4, отгоняют эфир и остаток перегоняют в вакууме. Выход
бензоилуксусного эфира 37,5 г (78,1% от теоретического); т. кип. 165—169° С
(20 мм рт. ст.).
а,а-Дизамещенные эфиры р-кстокислот иногда могут быть расщеплены также
путем катализируемого щелочами алкоголиза. Из соответствующего эфира ₽-кето-
кислоты при этом образуются два эфира жирных кислот, например этилацетат и этил-
изобутират из а,а-диметилацетоуксусного эфира [111]:
СН3
еН3СО-С-СООС2Н5 СгНб0Н-^ СН3СООС.-Н5^-СН3СНСООС2Н6
I I
СНз СНз
Аналогично кислотному расщеплению p-кетоэфиров под действием щелочей
протекает гидролиз хлораля с образованием хлороформа и муравьниок кислоты:
/Н он- /О
С13С-С< ——* CI3C-CH-0- —► снс1з+нс<г
X) | \О-
он
Трихлорметилкетоны также претерпевают аналогичное разложение до хлоро-
форма и карбоновой кислоты (Стр. 838):
Л COCCI, Ха0П- RCOONa + CHCl,
Если на хлораль действуют первичными или вторичными аминами, то с хоро-
шими выходами получаются формильные производные аминов (стр. 511):
R2N-CH-CCI3 —> r2n—спо-; снс1з
[109] W. Dieckmann. A. Wittmann. Ber., 55, 3344 (1922).
[110] R. L. S h г 1 n е г A. G. Schmidt. J. Ain. Chem. Soc., 51, 3636 (1929).
[HI] W. Dieckmann, Ber.. 33, 2672 (1900).
ПЕРЕГРУППИ-
РОВКИ
УГЛЕРОД-
СОДЕРЖАЩИХ
СОЕДИНЕНИИ
(ЗА ИСКЛЮЧЕНИЕМ
ПРОСТРАНСТВЕН-
НЫХ ПЕРЕГРУППИ-
РОВОК) *
Под перегруппировками понимают не-
которые «ненормально» протекающие реакции.
Однозначного во всех отношениях определе-
ния понятия перегруппировки ке 'имеется.
Предпосылкой для него явилось бы такое
же однозначкое определение понятия нор-
мально протекающих реакций.
Общей характеристикой большинства
перегруппировок является следующее.
Любая реакция начинается с разрыва
или со значительного’ ослабления связи между
атомами. При нормально протекающих реак-
циях новая связь с находящимся вне молекулы
реагентом образуется в том же месте, где*
произошел разрыв старой связи. В реакциях
перегруппировок, напротив, вначале в месте
разрыва старой связи образуется новая
.связь с атомом той же молекулы. Это приводит
к миграции заместителей, миграции или об-
разованию двойных связей или к замыканию
кольца. Только после этого все еще ненасы-
щенный скелет молекулы стабилизуется по-
средством отщепления или присоединения
атома или группы атомов.
Если же двойная связь образуется
с соседним атомом и стабилизация происхо-
дит в результате отщепления атома или группы
атомов от того же соседнего атома, то проте-
кает нормальное 1,2-отщепление.
Например, неопентиловый спирт при
замещении гидроксильной группы кодом может
реагировать либо нормально, когда новый
заместитель встает на место старого, либо
после отрыва гидроксила метильная группа
мигрирует к соседнему углеродному атому
и новый заместитель вступает на ее первона-
чальное место. Обе реакции происходят парал-
лельно [1]:
(СН8)3ССНаОН—>(СН3)зССНа —^(СН3)3ССНаТ
(СН3)2ССН2 —> (СНз)2С1СН2
I I
СН3 С1Т3
Приведенное выше определение пере-
группировок справедливо почти для всех
рассматриваемых в этом разделе реакций.
Кроме того, оно охватывает также все реакции
замыкания кольца. Однако нецелесообразно и не
принято все случаи замыкания кольца рассмат-
ривать как перегруппировки, поскольку многие
реакции замыкания кольца в принципе не отлп-
чаютей от реакций, происходящих без замыкания
кольца. Например, внутримолекулярная цикли-
зация фенилэтилциклогексена с образованием
* Материал обработал Е. Шмптц.
[1] F. С. W h i t m о г е, Е. L. Witt! о,
В. R. Harrimann, J. Am. Chem.
Soc., 61, 1585 (1939).
I. ПЕРЕМЕЩЕНИЕ КРАТНЫХ СВЯЗЕЙ
847
октагидрофенайтрена [2] соответствует образованию связи между бензольным
кольцом к циклогексеновым:
Реакции такого рода уже рассматривались в предыдущих разделах.
Напротив, циклизация дихлорангидрида фталевой кислоты в дяхлорфталид
(стр. 859) не имеет аналогии в ациклическом ряду. Эта реакция является настоящей
Ик?.' перегруппировкой и будет рассмотрена нйЖе.
И&' Рассматриваемые здесь перегруппировки разделены пами на четыре типа по
Ир- следующим признакам:
НКг перегруппировки с сохранением углеродного скелета;
перегруппировки с построением углеродного скелета;
Изд перегруппировки с расщеплением углеродного скелета;
перегруппировки с перестройкой углеродного скелета.
В каждом подразделе вначале рассматриваются перегруппировки углеводоро-
’ дов. а затем перегруппировки’соединений, содержащих кислород, азот, галоген и серу.
* Соответственно основной концепции этой книги при изложении материала
Жданного раздела авторы исходили только из того, какие связи разрываются и обра-
- зуются при образовании конечных продуктов из исходных веществ, и не учитывали,
’ протекают ли реакции фактически внутри- или межмолокулярно и происходит ли
^ разрыв связей по ионному или радикальному механизмам. Механизм перегруппировок
г'подробно рассмотрен в руководствах по теоретической органической химии.
* ПЕРЕГРУППИРОВКИ С СОХРАНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
* 1 I. Перемещение кратных связей
1. Перемещение двойной связи
i В ненасыщенных соединениях более или менее легко происходит перемещение
л двойных связей, особенно легко в присутствии кислотных или основных веществ.
. Это наблюдается утке при введении двойных связен в молекулу. При достаточно продол-
гЖятельном действии условий, вызывающих изомеризацию, часто устанавливается
J-.равиовесное состояние, при котором образуется смесь всех возможных по положению
•^щойной связи изомеров [3]. Так, из н-додецена изомеризацией под действием карбо-
,;нила кобальта при 150° С получили смесь всех возможных изомеров с разным положе-
книем двойной связи [4]. Соответствующие смеси олефинов образуются при отщопло-
НИИ хлористого водорода от н-додецилхлорида и при отщепленни воды от первичных
^высших нормальных спиртов.
При взаимодействии с соляной кислотой бензоата днгидрозимостерика были
.•получены бензоаты а- и р-холестенола [5].
[2] D. Perlman, D. Davidson, M. T. Bogert, J. Org. Chem., 1, 288
(1936).
I1! [3] F. Asinger, Ber., 75, 1247 (1942).
gj [4] F. Asinger, Chem. Ber., 88, 445 (1955).
К [5] II. Wieland, L. Gomhardt, Ann., 557, 248 (1947).
ПЕРЕГРУППИРОВКИ G СОХРАНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
До некоторой степени однозначное и полное перемещение двойной связи и нуж-
ное положение может произойти, как правило, только в том случае, если новая двой-
ная связь будет находиться в сопряжении с С—С-, С—О- или С—N-кратнымн связями.
Известно большое число примеров таких направленных перегруппировок, вызываемых
действием щелочей, кислот или простым нагреванием.
Так, при получении пеларгоновой кислоты из ундециленовой посредством
щелочного плавления происходит перемещение двойной связи в а-положенио к карб-
оксильной группе и затем отщепление двух атомов углерода (стр. 833).
СН2^СН(СН2)8СООН —> СН3(СН2)7СН=СНСООН —► СН3(СНг)7СООН
Изоэнгенол (по Тиману [6]). Для превращения эвгенола в изоэвгенол 12,5 ч.
КОН растворяют в 18 ч. кипящего амилового спирта, декантируют раствор
от нерастворимого карбоната и нагревают его с 5 ч. эвгенола при 140° С в тече-
ние 16—20 ч. После отгонки амилового спирта с водяным паром реакционную
массу подкисляют H2SO4, промывают выделившееся масло раствором соды
й перегоняют с водяным паром. После соответствующей обработки получаю г
пзоэвгенол в виде бесцветного вязкого масла; г. кип. 261° С.
СН3Ох ,СП2— СН=СН2 СН3О. /,хуСН=СН—СН3
A Y —* А "
р.р-Диметнлакрнловэя кислота [7]. Смесь 10 г jJ-метнлвинплуксусной
кислоты в 20 мл сухого эфира |И 10 г 90%-ной H2S04 оставляют на 24 ч при
комнатной температуре. Затем разбавляют водой и выделяют продукт. Получают
9,7 г (97% от теоретического) 0,р-диметилакриловой кислоты; т. пл. 68—70е С.
СН3\ снзх
/с—сн2—соон —> >с=сн- соон
сн/ сн/
Термическое перемещение двойной связи происходит прн перегруппировке
ангидрида итаконовой кислоты в ангидрид цитраконовой кислоты [81:
сн2=с—со СНз—с—со
I I >
СИ2-СО СИ-СО
Ангидрид цитраконовой кислоты. При атмосферном давлении 250 г ангид-
рида итаконовой кислоты возможно быстрее перегоняют с колонкой Вигре длиной
15 ем. До 200° С перегоняются продукты разложения и небольшое количество
воды. При 200—215° С перегоняется 170—180 г ангидрида цитраконовой ки-
слоты. Выход продукта 68—72% от теоретического.
Сопряженные двойные связи только в том случае могут перемещаться в изоли-
рованное положение, если образующееся в незначительном количестве песопряжепное
соединение непрерывно удаляется пз равновесной смеси. Благодаря этому, например,
возможно превращение окиси мезитила в изомерный 2-метиллевтел-1-он-4 [9):
СН3к СН-к
)С-СНСОС.Н3 —* z/С-СН2СОСН3
CH3Z СНг^
[6] F. Tipmann, Вег., 24, 2870 (1891).
[7] R. В. Wagner, J. Am. Chem. Soc., 71. 3214 (1949).
]8] R. L. Sil riner, S. G. Ford. L. J. Roll, Org. Synth., 11, 28 (1931).
[9] F. H. Stress, I. M. Monger, H. De V, Fine k, J. Am. Chem. Soc.,
69 1627 (19'17).
I. ПЕРЕМЕЩЕНИЕ КРАТНЫХ СВЯЗЕЙ
849
2-Метилпентен-1-он-4. Продажную окись мезитила, содержащую около 9%
несопряженного изомера, предварительно очищают (см. [9]) и при пормальпом
давлении в присутствии 0,2% п-толуолсульфокислоты перегоняют с колонкой
в. 85 теоретических тарелок. Кислота способствует установлению равновесия,
а низкокипящий изомер отгоняется в более высокой концентрации. Фрак-
цию с коэффициентом преломления 1,4210—1,4230 перегоняют на такой же
колонке с флегмовым соотношением 1 : 40. Окись мезитила имеет т. кип. 129,8° С,
изомерный несопряженный кетон — т. кип. 121,5° С.
2. Аллильная перегрупппробна
К аллильным перегруппировкам [10] относятся реакции, протекающие по схеме:
R—СН=СП—СНХ—R' —> K-CHX-CII=CH-R'
i Заместитель X мигрирует из положения 1 в положение 3 с одновременным
г перемещением двойной связи. Эта миграция в случае аллилгалогенидов происходит
так легко, что можно говорить о подлинной таутомерии. Например, равновесие между
: обоими изомерными метилаллилбромидами
СН3СН=СН-СН2Вг СНз-СНВг-СИ-С1Ь
>s- устанавливается при 75° С за несколько часов [11]. Поэтому недопустимо определе-
2:ние структуры аллилгалогепида на основании способа его получения. ,
< Точно так же аллильные перегруппировки происходят при замене галогена
^другими остатками. Исходя из аллилгалогенидов, получают не только изомерные
j.: спирты, но и изомерные сложные или простые эфиры или смеси изомерных продуктов
* ’ с продуктами, образующимися в результате нормального взаимодействия [12].
Аллильная перегруппировка чрезвычайно затрудняет выяснение строспия
Э' многих соединений аллильного типа. Однако известен ряд случаев препаративного
./использования этой перегруппировки.
v Например, путем двойной аллильной перегруппировки удалось превратить
| декадиен-2,8-ИН-5-ДИОЛ-4,7, полученный из комплекса Иоцича (гриньяровское соеди-
х Пение ацетилена) и кретонового альдегида, в изомерный декадиен-3,7-ин-5-диол-2,9
Г[13]:
: СНз-СН=СН-СНОН-С==С-СНОН-СН=СН-СН3 —
—> СНВ~СНОН-СН=СН-С==С-СП-СН-СНОН-СН»
Декаднеп-3,7-ин-5-диол-2,9. Раствор 5 г декадиен-2,8-ин-5-диола-4,7 в
20 мл эфира при 20° С в течение 24 ч встряхивают в токе азота с 30 мл 10%-ной
H2S0.. Из промытого и высушенного эфирного раствора получают 4,3 г дека-
диен-о,7-ин-5-диола-2,9 в виде вязкой жидкости, перегоняющейся при 10-4 мм
рт. ст. и температуре водяной бани 65—70° С.
Перегруппировка третичных спиртов в первичные через их ацетаты исполь-
зуется в синтезе гераниола, фарнезола (R = CI0Hi7) и фитола:
ОН
R-CH2-C-CH-CTT2 —> R—СН2—С=СН—СНгОН
СНз СНз
[10] R. II. D е Wolf е, W. G. Y о п и g, Chem. Rev.. 56, 753 (1956).
[lip W. G. Young, S. Winsl ein, J. Am. Chem. Soc., 57, 2013 (1935).
[12] Более подробные теоретические п практические данные см. В. X ю к к е л ъ,
Теоретические основы органической химии, т. I, пер. с нем., Издатпнлит, 1958.
[13] J. М. II е i 1 b г о п. Е. В. JI. J о n е s, В. A. R а р h а ₽ I, J. Chem. Soc.,
1943. 268.
51 Заказ 1835.
850
ПЕРЕГРУППИРОВКИ С СОХРАНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
Фарнезол [14]. Нагревают 1ч. неролидола с 1,5 ч. уксусного ангидрида
в течение 20 ч в атмосфере двуокиси углерода при 120—140° С. После удаления
в вакууме при 100° С образовавшейся уксусной кислоты и избытка уксусного
ангидрида остаток омыляют в токе водорода спиртовым раствором КОН. При
вакуумпой перегонке после первой фракции, состоящей из образовавшегося
при отщеплении воды фарпезена и пецрореагировавшего неролидола, прп
115—130° С (0,5 мм рт. ст.) перегоняется сырой фарнезол; его очищают через
кислый фталевый эфир. Фарнезол имеет т. кип. 120—121° С(0,3 мм рт. ст.).
Понятие аллильных перегруппировок может быть значительно расширено,
благодаря чему появляется возможность для формального рассмотрения целого ряда
изомеризаций с одной общей точки зрения. Эти изомеризации протекают по общей
схеме:
где А, В, С — атомы углерода, азота или кислорода, X — мигрирующий при перегруп-
пировке остаток. Например, если А, В и С — углерод, а X — Н, то миграция двойной
связи является наиболее простым случаем аллильной перегруппировки (трехуглерод-
ная таутомерия):
-СН=СН-СН2- -СН2-СН=СН-
Далее необходимо назвать следующие перегруппировки:
Х':С—С—О—Н 5—
>СН—с=о
кето-еиольная таутомерия
-NH—1
превращение ааоеоедипений в гидразоны
^СП—N=O
>C=N-O-H
превращение питрозосоединеннй в оксимы
\?=N-OH \сН—N=O
таутомерии питро- и изонитроеоединении
3. Перемещение тройной связи
Факт перемещения тройных связей был открыт Фаворским [15]. Он установил,
что н-пропилацетилен (пентин-1) под действием раствора КОН в зтиловом спирте
при 170° С превращается и метилэтилацетилен (пеитин-2)
НС=С—СП2—СН2—СНз •—> СН3—С==С—СэЩ
в то время как в тех же условиях из изопропилацетилена образует ся диметилаллен:
НС-С-СП(СН3)2 •—> СН2=С=С(СНз)2
По Джекобсу, Акавп и Кунеру [16], из пентипа-1 и ил пгптипа-2 в спиртовом
растворе КОН при 175° С через 3 ч получают идентичные равновесные смеси, содер-
[14] L. Ruzicka, Helv. chim. acta, 6, 489 (1923).
|15J Al. Faworski, J. prakt. Chem., 44, 208 (1891).
j 16] T. L. Jacobs, R. A k a w i e, R, G. Cooper, J. Am. Chem. Soc., 73,
1273 (1951).
)i. НЕРЕГРУШГИРОВКИ С ИЗМЕНЕНИЕМ КИСЛОРОДНОЙ ФУНКЦИИ
•жащпе 95,4% пентина-2, 1,3% пентина-1 и 3,3% нентадиена-1,2. Равновесие, следов
тсльно, практически полностью сдвинуто в сторону дизамещенного -ацетилена.
Однако можно осуществить и обратную перегруппировку, превращение д
замещенного ацетилена в монозамещенпый, так как монозамещенные я дети лег
способны к солеобразованию и тем самым могут непрерывно выводиться из равн
весной смеси. Днзамещенные ацетилены при нагревании до 100° С с металлическг
натрием превращаются в монозамещенные. При этом лучше использовать амид натри
так как под действием натрия происходит частичное восстановление тройной связ
Последовательное проведение метилирования ацетиленида натрия и перетру
шгровки с амидом натрия является изящным способом удлинения цепи;
NaC=-C-H —> СН3-С=:С-П - > NaC=C-CH2-R
Этот цикл можно многократно повторить. Таким способом Бургслю удалог
из З-циклогексилпропина-1 получить 6-циклогексилгексин-1.
Мы ограничимся здесь описанием собственно стадии перегруппировки по Бу|
гелю [ 171:
С6НиС1ЬСН2С=ССН3 —* СвПцСН2СП3СН2С=-СХа
5-Циклогексилпентип-1. Смесь 107 г 5-циклогексилпентипа-2, 39 а амид
натрия и 300 мл парафиновой фракции (т. кип. 125—140° С при 15 мм рт. ст
нагревают до 160е С. Через 2,5 ч оканчивается выделение аммиака. Зато
в вакууме отгопяю! часть растворителя, причем вместе с ним удаляется ненре
реагировавший исходный продукт, и остаток разлагают льдом. После выдедени
получают 81 г (89% or теоретического) 5-циклогсксилпентинач-1; т. кип. 84-
85,5е' С (1Г> мм рт. ст.}.
Наблюдается и препаративно используется также миграция тройных связег
через несколько метиленовых групп [18J.
It. Перегруппировки с изменением кислородной функции
1. Превращение сс-окисей в карбонильные соедггнения
При изомеризации а-окисей в ^карбонильные соединения происходит превраще-
ние эфирной функции в карбонильную, причем обратного превращения не наблюдя
ется. Окись этилена при 400° С превращается в ацетальдегид, окись пропилена прп
500° С — в смесь пропионового альдегида (2/8) и ацетона (1/'з) [19]. При перегруппи-
ровке эфиров фенилглицидола, происходящей при надевании с небольшим коли-
чеством хлорида цинка 120], образуются бензилкетоны; раскрытие эпоксидного кольца
происходит, следовательно, но соседству с фенильной группой:
СеН5СН—СНСН2ОК —> СвНбСПгСОСНзОН
\ /
О
При перегруппировке алифатических эпоксисосдпнений раскрытие кольца
ведет к кетонам с карбонильной группой, расположенной ближе к началу цеди [21],
например:
CHSCH-CIIC2H5 —> СН3СОСН2С2Н5
О
[17J М. Bonrguel. Ann. Cliirn [10], 3, 363 (1925).
[18| ’I’li. II. Vaughn, j. Ara. Ciram. Soc., 55. 3153 (1933).
[19] J. Г. NP(, Ann., 335, 2Щ (1904).
[20] M. D.irmon, C. r., 197, 1649 (1933).
[21) M. F a w о r s k i. M. T s ch itsc h onki n, ). I w a n о w, C. r., 199,
1229 (1934).
54*
852
ПЕРЕГРУППИРОВКИ С СОХРАНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
Перегруппировка осуществляется пропусканием а-окиси над нагретым до
320° С ZnCJ2.
2-0-Феннлэтнлциклогексанон [22]. Нагревают с обратным холодильником
в течение нескольких часов 10 г р-фенилэтилциклогексепоксида с 10 г концен-
трированной НС1 и пятикратным объемом метилового спирта. Образовавшийся
кетон осаждают водой, извлекают эфиром и очищают перегонкой в вакууме.
Выход продукта 60% от теоретического: т. кип. 174" С (12 мм рт. ст.).
Для перегруппировки эпоксисоедннений можно применять щелочные ката-
лизаторы [23,24].
З-Ицтромезнтоилбензилкетон [23]. Спиртовой раствор 1-(3'-нитромезитоил)-
2-фенилэтилеяоксида нагревают в течение 1,5 ч с раствором КОН (1.5 моль).
Выход продукта 75% от теоретического.
2. Реакция Вильгеродта [25]
Неразветвленные арилалкилкетоны при нагревании до 200° С с полисульфндом
’аммония превращаются в амиды кислот [26]:
СаНзСОСНз ——C6H5CH2CONH.2
При этом перемещается не арильный остаток, как при перегруппировке Вольфа
(стр, 882), а кислород мигрирует па конец цепи при сохранении углеродного скелета.
В реакцию вступают не только арилметилкетоны. Например, из дрониофепона обра-
зуется амид гидрокоричнои кислоты [27]:
СбН5СОС2Н5 —>.CeH6CH2CII2CONH2
Эпоксисоединения, ацетиленовые соединения, олефины и карбинолы жирно-
ароматического ряда, если они содержат неразветвленную цепь, также всту-
пают в реакцию Вильгеродта. Особенно хорошие результаты получаются в случае метил-
кетонов высших конденсированных ароматических систем, таких, как фенантрен или
пирен.
Физер и Килмер [28] дают следующую пропись для превращения 1-ацетил-
аценафтена в 1-аценафтилуксусную кислоту.
[22] R. Grewe, Вег., 72, 1314 (1939).
[23] R. Р. Barnes, L. A. Gist jr., J. Am. Chem. Soc . 72, 2509 (1950).
[24] А. С. С о p e, B. D. Tiffany, J. Am. Chem. Soc., 73, 4158 (1951).
[25] Обзорная статья: M. Carmack, M. A. Spielman, Org. Reactions, 3, 83
(1949).
[26] C. Willgerodt. Ber., 21, 534 (1888).
[27] C. W i 1 1 g e г о cl I, F. H. Mor k, J. prakt. Chem. |2J. 80. 192 (1909).
[28] L. F. Fieser, G. W. К i I m e r, J. Am. Chem. Soc., 62. 1351 (1940).
II ПЕРЕГРУППИРОВКИ С ИЗМЕНЕНИЕМ КИСЛОРОДНОЙ ФУНКЦИИ
85;
1-Аценафтилуксусная кислота. Раствор 1 г серы в 10 г раствора (NH4)2S (по-
лученного насыщением концентрированного раствора аммиака сероводородом;
нагревают при 160° С с 2 г кетона и 8 г очищенного диоксана в течение 12 ч
в .запаянной трубке. Полученный 1-аценафтилацетамид омыляют кипячением
в течение 4 ч с 50 мл 15% -лого раствора NaOH. Выход кислоты 57% от теорети-
ческого, считая на введенный в реакцию кетон.
СОСНз
/АА/СНгС00Н
Прп таком способе проведения реакции температуру 160—170° С можно считать
стандартной температурой. Однако, используя предложенную Киндлерои (29] моди-
^фнкацию, можно применять и более низкие температуры. По Киндлеру, метнлкетон
&; взаимодействием с примерно эквивалентными количествами серы и амина (как правило,
К, морфолин) превращают в тиоамид, который затем омыляют кислотой. Реакция про-
|/ текаст при температуре кипения морфолина (128° С) с достаточной скоростью {30].
2-Нафтилтиоацетоморфолид, Нагревают смесь 373 г (2,2 моль) 2-ацетил-
нафталииа, Ю5 г (3,3 моль) серы и 290 г (3,3 моль) морфолина. Во избежание
сильного вспенивания и бурного выделения сероводорода нагревание сначала
ведут медленно. Через час нагревание усиливают. В общем кипячении с обрат-
ным холодильником проводят в течение 10—15 ч. Затем разделившуюся па два
слоя реакционную смесь вливают в 1200 мл горячего этилового спирта и оста-
вляют для кристаллизации. Получают 534 г (90% от теоретического) достаточно
чистого для дальнейшей переработки 2-нафтйлтноацстоморфолида: т. пл. 102 —
108° С. Кипячением со смесью ледяной уксусной и разбавленной серной кислот
в течение 5'ч тиоамид омыляют до свободной кислоты.
3. Кето-енольная таутомерия
Карбонильные соединения находятся в равновесии с производными винило-
вого спирта, что связано с миграцией протона и перемещением двойной связи. У аль-
|детидов и кетонов доля епольной формы неизмеримо мала. Однако кето-и енольпая
формы существуют в сравнимых количествах, если образующаяся при енолизацни
двойная С—С-связь сопряжена со второй карбонильной группой, как, например,
в случае 13-дикетонов и эфиров р-кетокислот:
RCOCTl2COR' RC=CHCOR'
I
. ОН
Д'шалвил-юш алкоксил
О таутомерии вообще говорят в тех случаях, когда при быстро протекающей
и обратимом реакции перегруппировки количества обеих форм в равновесном состоя-
, нии соизмеримы.
) Это равновесие может быть сдвинуто только в сторону енола и нет никакого
химического средства для с-мещения равновесия в обратном направлении. Прсвращр-
ние енольном формы в кетонпую может быть лишь ускорено действием катализатора.
[29] К. Kindler, Ann.. 431, 193, 222 (1923).
130] М. С а г m а с k, М. A. Spielman, Org. Reactions, 3, 97 (1949).
854
ПЕРЕГРУППИРОВКИ С СОХРАНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
Вейганду и Коху [31] удалось получить в чистом виде енольную форму метил-
бензоилацетова следующим путем:
СеЩСОСНСОСНз •—* Енолят —> Енол
I
СН3
Енольная форма метилбензоилацетона. К раствору 0,15 г натрия в 3 дм
метилового спирта прибавляют 1 г маслянистого метилбензоилацетояа, тотчас же
фильтруют темно-желтый раствор и по каплям прибавляют его к 30 мл 5 н.
H2SO4, охлажденной смесью льда с солью. Образующуюся вначале эмульсию
тщательно разбивают интенсивным перемешиванием стеклянной палочкой,
что способствует кристаллизации основного количества продукта в виде тонкого,
хорошо отфильтровывающегося порошка. Если масло собирается в большие
капли, то продукт кристаллизуется с трудом и не выдерживает длительного
хранения- Осадок сразу же отфильтровывают, промывают водой подкисленной, со-
ляной кислотой, небольшим количеством охлажденного льдом этилового спирта,
петролейным эфиром и тотчас же перекристаллизовывают из низкокипящего
петролейного эфира в кварцевой посуде. Выход продукта почтя количественный;
т. пл. 51° С.
4. Изомеризация кетоспиртов
Описана [32] перегруппировка а-кетоспиртов *, -происходящая но схеме:
RCOCHB' BCIICOR'
I ।
ОН он
Но в то время как таутомерные кето- и енольные формы могут быть обнаружены
только п кристаллическом состоянии, а в растворе или расплаве они быстро переходят
друг в друга, метилбензоилкарбинол С.НЧСНОНСОС6Н5 и фенил ацетил карбинол
СН3СОСНОПСвН5 не только получены в чистом виде, по и охарактеризованы хими-
чески как различные вещества. Взаимная перегруппировка этих соединений катали-
зируется щелочами.
Особым случаем является равновесие меящу кегоспиртами с конечными функ-
циональными группами и соответствующими а-оксмальдегпдами. Оно полностью сдви-
нуто в сторону кетола:
RCHOHCHO —> всосщон
Например, миндальный альдегид (Н =- CfiH5) тотчас же после выделения его
из ацеталя превращается в бепзоилкарбинол [33].
Частичный синтез кортикостерона 134] основан па легко происходящем превра-
щении а-оксиальдегида в соответствующий кстол:
* Эта перегруппировка открыта впервые А. Е. Фаворским и Т. И. Темниковой
|А. Е. Ф а п о р с к я й, Т. 1). Тем я и к о в а, ЖОХ, 4, 745 (1934)]. —
Прим- ред-
[31] С. Weygand, Р. Koch, Вег., 68, 237 (1935).
[32J К. V. Anwars, И. Ludewig, A. Muller, Aon.. 526, 143 (1936).
[33] W. G. Danbe n. W. L. E v a n s, R. I. M e I t z о г. J. Am. Chem. Soc.,
63, 1883 (1941).
[34] D. T aub et al., J. Am. Chem. Soc., 76, 4094 (1951).
П. ПЕРЕГРУППИРОВКИ С ИЗМЕНЕНИЕМ КИСЛОРОДНОЙ ФУНКЦИИ §51:
Кортикостерон. Из Д4-прегпеидио.т-11р,20р-он-3-аля-21 с бисульфитом нат-
рия в водно-метанольпом растворе получают бисульфитное соединение. Оно явля-
ется производным альдегидной формы, так как при разложении кислотой снова
превращается в альдегид. Разложение бисульфитного соединения раствором
метилата натрия приводит с изомеризацией к образованию кетола — кортико-
стерона. Выход 60% от теоретического.
5. Перегруппировка Канниццаро
а-Кетоальдсгиды. в общем случае,'под влиянием щелочей перегруппировы-
ваются в а-оксикислоты;
НСОСНО нснопсоо-
Например, из фенил глиоксаля количественно образуется миндальная кис-
лота |35].
Эта реакция имеет препаративное значение только в том случае, когда фенил-
глиоксаль, образующийся из соответствующих исходных соединений в результате ще-
лочного гидролиза, подвергается дальнейшему превращению без выделения его в чис-
том виде.
Например, таким способом можно получить 4-бромминдальнуЮ кислоту из 4,
со, со-трибромацетофенопа [36J:
.СОСНВг2 /х /СПОНСОО-
A -он~- fY
Вг/^/ Вг-7^/
4-Еромминдальная кислота. Оксикислота образуется с выходом 69—85% от
теоретического при многосуточиом стоянии трибромсоединения в водном рас-
творе NaOH при 5° С.
Оппсан [37] изящный способ получения а-оксикислот из карбоновых кислот
с меньшим на один числом атомов углерода. По этому способу на диазокетоны, легко
доступные реакцией хлорангидридов и дпазометана, действовали этилсульфенилхлори-
дом и обрабатывали полученный продукт водной щелочью:
ZCI
• CeH3COCHN8 СгНбЯС1+ СеН5СО-СН СЙН5СНОНСОО-
^SCsIIs
Миндальная кислота. К раствору 12 г диазоацстбфенона в 75 мл абсолют-
ного эфира при 0—5° С. медленно добавляют 7,9 г этилсульфенхлорида, причем
происходит бурное выделение азота. После двухчасовой выдержки при комнат-
ной температуре отгоняют в вакууме эфир. Полученный с почти количествен-
ным выходом со-хлор-со-этилмсркаптоацетофенон необходимо тотчас же пере-
работать далее. Для этого 3 г сырого продукта в течение 2 ч встряхивают с 1 н.
раствором КОН и затем в течение 1 ч нагревают на водяной бане. После охла-
ждения из реакционной массы эфиром экстрагируют побочные продукты. Обра-
зующуюся с 65%-ным выходом миндальную кислоту выделяют подкислением
и извлекают эфиром.
|3э] II. V. Pechmann, Вег., 20, 2904 (1887); 22. 2558 (1889).
[36] J. J. К 11 n g е n Ь е г g. Org. Synth., 35, 11 (1955).
[37 j F. W e у g а и d, II. J. Bestman n, Chem. Ber., 88, 1991 (1955).
856
ПЕРЕГРУППИРОВКИ С СОХРАНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
6. Прочие перегруппировки
Перегруппировка а-оксиацетиленов в внниянетоны,* происходящая при нагр
вании сконцентрированными муравьиной или щавелевой кислотами, или с пятиокись
фосфора, названа по имени открывателя перегруппировкой Рупе [38].
4-Окси-1-ацетилциклогексен [39]. 1-Этинилхинит кипятят в течение 2,5
с 85%*нон муравьиной кислотой, затем отгоняют в вакууме кислоту и остатс
для омыления муравьиного эфира в течение 30 мин нагревают с водой. Выл
4-окси-1-ацетилциклогексона 62% от теоретического.
НО\ НО. ..
f |/он I II
\/\С0СН8
Тетрагидрофуриловый спирт [40] при пропускании над окисью алюминия пр
370—380° С с дегидрированием и одновременным расширением кольца превращаете
в дигидропиран. Экспериментальные подробности см. [41].
। —► А
'OZ' CHgOH XQ/
III. Перегруппировки азотсодержащих соединений
1. Перегруппировка Амадори
Под перегруппировкой Амадори ** подразумевают, ио Куну и Вейгапду [43]
изомеризацию N-гликозидов в соответствующие N-замещенные дезоксиаминокетоаы
например, превращение п-толуидин-7)-глюкозида в 1-п-толиламино-1-дезокси-£>-фрув
тозу:
NH- СвП4СП3 NH-СвН4СН3
I I
Н-С------j сн2
I _ I '
н-с-он- с=о
1 А ।
НО—С—Н О —> но—с—н
н-с-он н-с-он
Н-С----1 н-с-он
I I
сн2он сн2он
Производное ZJ-глюкозы превращается, следовательно, в производное 7)-фрук
тозы. Реакция аналогична рассмотренной на стр. 854 изомеризации кетОлов.
* Фундаментальные, исследования в близкой области выполнены И. И. Назаровы»
и его школой. — Прим. ред.
** М. Амадори^посредстпом конденсации Р-глюкозы с ариламинами наряду с N-глю
коэидами получил продукты их перегруппировки, но описал их как осповапи»
Шиффа [42].
[38] H. Rupe, Helv. chim. acta, 11, 454 (1928).
[39] E. R. H. J ones, F. S on d h о i m er, J. Chem. Soc., 1949, 615.
40] R. Paul, С. r., 196, 1409 (1933).
[41] R. L. Sawyer, D. W. Andrus, Org. Synth., 23, 25 (1943).
[42] M. Amadori, Atti Reale Accad. Lincei Roma [6], 2, 337 (1925).
[43] R. К и h ii. F. W e у g a n d. Ber., 70, 769 (1937).
HI. ПЕРЕГРУППИРОВКИ АЗОТСОДЕРЖАЩИХ ^СОЕДИНЕНИЙ
857
Недавно было показано, что перегруппировка Амадори имеет биологическое
^'значение [44J.
По Вейганду, перегруппировка Амадори катализируется ионами водорода [45|.
ЕКятализаторами перегруппировки могут служить также натриевые соли соединений
Кё подвижным (кислым) водородом у углерода [4G].
ЬДнбензиламино-Ьдезокси-О-фруктоза- Смешивают 5 г а-Л-глюкозы с 8,1 г
дибензиламина и добавляют 150 10%-ного раствора натрийэтнлмалоната
в абсолютном отиловом спирте. Смесь 2 ч нагревают на водяной бане. Через
j:.- полчаса сахар растворяется. Золотисто-желтый раствор охлаждают, фильтруют
и в течение 20 ч выдерживают при 0° С- Получают 8,9 г (89% от теоретического)
йг 1-дибонзяламино-1-дезокси-0-фруктозы; т. пл. 161—162*0.
2. Перегруппировка Чепмена
.* Изящным методом получения трудно доступных несимметричных диарилами-
является перегруппировка Чепмена. При нагревании O.N-диарилпрованяых бенз-
(Ыдадов «арильный остаток мигрирует с одновременным перемещением двойной связи
кислорода к азоту. Например, замещенный бензимид количественно персгруппи-
^фовывается при натреванин в течение 2 ч при 250—270° С [47):
3. Перегруппировка окисей аминов
Было найдено, что при нагревания окисей аминов в водном растворе NaOH
Й)к 100° С аллильные и бензильные группы могут перемещаться от азота к кислороду
^образованием О-эфиров замещенных гидроксиламинов [48]:
СВН6\
CH3->N->O
сн2=снсн</
с6н6,
>noch2ch=ch2
сн/
Реакция позднее была подробно изучена (49) и распространена на окиси чисто
(тических третичных аминов.
[49
R. Kuh n. Angpw. Chem.. 69. 27 (1957).
F. Weygnnd, Пег.. 73, 1259 (1940).
J. E. Hodge, С. E. Rist, 5. Am. Chem. Soc.. 75, 316 (1953).
A. W. Chapman, J. Chem. Soc., 1929, 569.
J. Meisenheimer, Ber., 52, 1667 (1919); 55, 513 (1922).
А. С. С о p e et al., J. Am. Chem. Soc., 71, 3423 (1949); 72, 4896 (1950).
§58
ПЕРЕГРУППИРОВКИ С СОХРАНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
4. Псевдоосноваиня
При обработке щелочами четвертичных аммониевых солей гетероциклических
основании наряду с аммониевыми основаниями образуются так называемые псевдо-
основания 150]:
.0 NR I I NR
\/\^+ \/\/
I
ОН- он
Пссндооснования пиридинового ряда неустойчивы. В ряду хинолина и изохи-
нолина они могут быть получены при определенных условиях. В ряду акридина и не-
ароматических гетероциклических соединений с большей вероятностью образуются
псевдооснования, причем в некоторых случаях они являются единственно устойчивой
формой [51].
Большее препаративное значение имеет открытая Фройндом [52] способность
солей четвертичных ароматических оснований реагировать с гриньяроискими соеди-
нениями таким способом, что алкильный остаток гриньяровского соединения вступает
к соседнему с азотом углеродному атому. Б этой реакции, следовательно, четвертич-
ная соль также реагирует в лсевдоформс. Например, из иодметилата изохинолина
и метилмагпийиодида образуется 1,2'ДиметПл-1,2-днгидроизохинопин:
АА АА
I I CHsMgl I
I II NCH3 -------| NCH3
i- сн8
1,2- Диметил--1,2-дигидроизохинолпы. В раствор соединения Гриньяра, при-
готовленный из 28.4 г метилиодида, 5 а магния и 100 мл абсолютного эфира,
порциями уносят 27 г иодметилата изохинолина. Соль растворяется в резуль-
тате бурной реакции. Реакционную массу разлагают льдом с добавкой соляной
кислоты, доводят до щелочной реакции добавлением NH40H и извлекают про-
дукт эфиром. Полученный 1,2-диметил-1,2-дигидроизохинолнн перегоняют
при 150° С (20 мм рт. ст.).
Эта реакция, например, нашла применение прп синтезе N-метилморфинала [53].
IV. Перегруппировки галогенсодержащих соединений
1. Перемещение галогена
Установлено, что 1-бром~и-бутаи при 248° G постепенно, но полностью, пре-
вращается в 2-бром-н-бутан [54]. Препаративного значения реакция не имеет.
В значительно более мягких условиях происходит перегруппировка а-бром-р-ке-
тоэфиров в у-бром-р-кетоэфиры [55J:
СН3СОСНВгСООС2Н6 —► СН2ВгСОС11гСООС2Нй
Реакция катализируется бромистым водородом.
[50] Обзор предыдущих работ: Н. Decker, Л. Kaufmann, J. prakt. Chom.,
84, 220 (1911).
(51] Литературные данные гм. F. Krohnke, К. Е I I a g a s t, Arm., 600, 17G
(1936).
[52] М, Freund. G. Bode, Ber., 42, 1758 (1909).
[53] R. Grewe, A. Mondon, Chem. Ber., 81, 279 (1948).
|54] H. Lucas, A. Jameson, J. Am. Chem. Soc., 46, 2476 (1924).
[55] Л, Hantzscb, Ber., 27, 3168 (1894).
TV. ПР ГЕГРУППДРОВКИ ГАЛОГЕНЦОЛР’.РЖЛЩИХ СОЕДИНЕНИЙ 8,59
Но Риду и Фортенбауху [56], бронируют а-метил-у-иаопропилацето-
уксусный эфир в хлороформе и оставляют па ночь. При атом из получающегося
вначале а-бромэфира образуется у-бромэфчр.
Особенно хорошо изучена взаимная перегруппировка обеих изомерных форм
дихлоранглдрида фталевой кислоты [57]:
^\/С0С1
I И
^^-СОС!
CI С1
При температуре кипения равновесие полностью сдвинуто в сторону симметрич-
ного фталоилхлорида; в присутствии А1С13 симметричный хлорангидрид перегруппи-
ровывается в несимметричный дихлорфталид, так как последний образует с AICI3
стабильное комплексное соединение. О проведении синтеза см. пропись [58].
Аналогично происходит перегруппировка а-галогепбен.зиловых эфиров [59J.
1-Хлоризохромап при нагревании превращается в 2-({}-хлорэтил)-бензальдегид:
XH2CH2CI
^/х-сно
2-(|3-Хлорэтил)-бензальдогид. В течение 4 ч при охлаждении охлаждающей
смесью и без доступа влаги в 134 г (1 моль} иаохромана пропускают 71 г хлора.
Дозировку хлора проводят путем его конденсации в ловушке, охлаждаемой
смесью метилового спирта с сухим льдом, взвешивания и испарения током азота.
В начале реакции необходимо следить за тем, чтобы изохромаи (т. пл. 3° С)
не закристаллизовался. После указанного времени смесь в течение 1 ч выдер-
живают в вакууме, затем нагревают па масляной бане до 180° С и 10 м-ии выдер-
живают при этой температуре.
При фракционировании в вакууме с колонкой Впгре длиной 50 см вна-
чале при 49—51° С (0,5 ,ил« рт. ст.} перегоняется 28 г непревращонного
иаохромана и затем при 80—85° С (0,8 мм рт. ст.) 118,4 г 2-(р-хлорэтил)-
бензальдегпда. Выход продукта 89% от теоретического, считая на вступивший
в реакцию изохроман [60].
Обе описанные выше перегруппировки протекают по схеме:
I
—с—О—С---> С=О | —С--С1
С1
56
57
58
59
60
Е. В. Reid, В. Fo t i с и 1) а и с li. J. Org. Chvrn., 16. 33 (1951),
E. О t t, Ann., 392, 2i'> (1912).
E. О t t, Org. Synth., 11, 88 (1931).
A. Rieche, E. Scumitz, Chem. Ber.. 89. 12,54 (195(1).
E. Sc limit z, Chem. Ber., 91, 1133 (1%S).
860
ПЕРЕГРУППИРОВКИ С СОХРАНЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
протекает реакция, при которой
С—N-связи;
В известной степени аналогично этому
с 1,3-сдвигом галогенид-иона происходит разрыв
-t—iLc---► -C=N4—C-Cl
Например, в случае циклических аминов расщепление по Брауну бензоилирован-
ных амидов кислот пентахлоридом фосфора происходит как перегруппировка:
CeH6CO-N(' У CfiH5C->
cfci
CeH5C=N(CH2)5Cl CeH&CONH(CH3)6Cl
I
С1
1-Бензоиламино-5-хлорпентан [61]. Бензоилпиперидин без доступа влаги
нагревают с обратным холодильником с 1 -ноль РС1а. После того, как закончится
бурная вначале реакция, массу еще 15 мин- нагревают при слабом кипении.
После охлаждения образовавшийся в результате перегруппировки имидхлорид
разлагают ледяной водой, нейтрализуют основную часть кислоты и с водяным
паром отгоняют побочные продукты (бензонитрил и 1,5-дихлорпентан). Оста-
ющийся в виде масла 1-бонзоиламино-5-хлорпентан при охлаждении затверде-
вает. Его очищают перегонкой в вакууме. Выход продукта 50% от теоретиче-
ского; т. кип. 210—220° С (12 мм рт. ст.); т. пл. 66° С.
В общем случае бензоильные производные вторичных аминов при нагревании
с РС15 до 130° С с отщеплением 1 моль алкилхлорида превращаются в бензимидхло-
риды, из которых при более сильном нагревании (175° С) отщепляются в виде алкил-
хлорида также и второй алкильный остаток и образуется бензонитрил [62]:
C8H5CONR8 рС1>: 130° с-* CeH5C=NR CeHsCN-j-RCI
I
Cl
2. Перегруппировки при реакциях
галогенсодержащих соединений
Замощение галогена другими группами атомов часто ведет к перегруппировкам.
Приведено несколько примеров этих перегруппировок, особенно при реакциях гало-
генкарбонильных соединений, протекающих без изменения углеродного скелета.
При взаимодействии З-хлор-пентандиояа-2,4 с формиатом щелочного металла
формильный остаток не вступает на место галогена, а в результате перегруппировки
образуется 2-формилокси-пентандион-3,4 [63]:
СНзСОСНСОСНз —ГС?—- СНзСНСОСОСНз
I
G1
I
о-сно
[61] J. V. Braun, Вег.,"37, 2915 (1904).
[62] J. v. Braun, Вет., 37, 2812 (1904).
[63J G. Hesse, II- Stahl, Chem. Ber., 89, 2414 (1956).
IV. ПЕРЕГРУППИРОВКИ ГАЛОГЕНСОДЕРЖАЩИХ СОЕДИНЕНИИ
861
Гидролиз 2,6-дибромциклогексанона дает циклогександион-1,2 [64]:
Вг
''-/'''Вг //^О
а,а'-Дибромкетоны при взаимодействии с алкоголятом натрия или щелочами
превращаются в замещенные акриловые кислоты [65]. Эта реакция (перегруппировка
Фаворского) часто протекает с перестройкой углеродного скелета. С нажущимся
[65а] сохранением углеродного скелета происходит реакция с щелочными агентами
а,а'-дибромметилизопропилкетока *:
СН3ч СНзч
>СВг-СО-СН2Вг —> >с=сн-соон
СНз7 CH3Z
р.р-Диметилакриловая кислота [65]. Фаворский к 35 г днбромкетона прп
охлаждении и перемешивании добавлял раствор 26 г КОН в 60 мл безводного
этилового спирта (3 моль КОН на 1 моль кетона). Тотчас же исчезал запах
кетопа и жидкость окрашивалась в оранжевый цвет. Перогонка реакционной
массы давала две фракции (всего около 10 г) с т. кип. 103—105 (170 мм рт. ст.)
п 147—150° С (170 мм рт.. ст-)- Первая фракция являлась метиловым эфиром
p.p-диметилакриловой кислоты, более высококипящая фракция затвердевала
в кристаллы и была идентифицирована как ^ф-диметилакриловая кислота
Позднее реакция была далее исследована Вагнером и Муром [66]. Вагнер [67]
описал также перегруппировку а,0-дибромкетонов, превращающихся под действием
метилата натрия в метиловые эфиры ^.^-ненасыщенных кислот:
СИзСНВгСВгСОСНз —► CHSCH=CCH2COOCHS
I I
СНз СНз
Превращения галогенкарбонильных соединений следует применять для дока-
зательства строения веществ лишь с-крайней осторожностью.
Перегруппировка такого рода происходит также и при омылении дибромалка-
нов с третичным атомом углерода. В некоторых случаях эта реакция имеет препара-
тивное значение, Из дибромизобутана таким способом получают изобутнральдегид
[b8. 69]:
сн8 - Вг сн8ч сн3ч
\СОН-СНз >СВг— СН2Вг —>- >СН-СНО
СП3/ ChZ СН3/
Способные к перегруппировке дибромиды можно получать бромированием тре-
тичных спиртов. Особенно гладко происходит образование н.юбутиральдегнда £08].
Изобутиральдегид. В колбе, соединенной с ректифпкациопиой колонкой дли-
ной 90 см, нагревают 2 моль дибромизобутана с 2 л воды. Вначале, нагревание
* К перегруппировке Фаворского способны не только дибромкетоны, во и другие
а-галогепопроизводные кетонов, в том числе и монозамещеиные. Подробнее
см. Органические реакции, сб. 11, Изд. Мир, 1965, стр. 267. —Прим. ред.
[64] О. Wallach, Ann., 437, 173 (1924).
[65] Al. Faworski, J. prakt. Chem., 88. 665 (1913) и более поздние работы.
[65а] Механизм реакции см. R. В. L о f t f i e 1 d, J. Am. Chem. Soc., 73, 4707
(1951).
166 J R. B. W a g a e г, I. A. Moore, J. Ara. Chem. Soc., 72, 974 (1950).
[67] R. B. Wagner, J. Am. Chera. Soc., 71, 3214 (1949).
[68] F. C. Whitmore et al., J. Am. Chem. Soc., 55, 1136 (1933).
[69] A. H. Blatt, Org. Reactions, 1, 342 (1947).
862
ПЕРЕГРУППИРОВКИ С ПОСТРОЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
ведут так, чтобы колонка работала как обратный холодильник; спустя некото-
рое время температуру повышают. При повышении температуры верха колонки
до 59—64е С начинается отгонка изобутиральцегида. Черед 50 ч слой дибро-
мида в колбе исчезает и в приемнике собирается 185 е дистиллята. Повторной
перегонкой получают 108 г (75% от теоретического) изобутиральдегида: т. кии.
58—65° С. (Перемешивание значительно ускоряет гидролиз.)
ПЕРЕГРУППИРОВКИ С ПОСТРОЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
I. Перемещение от кислорода к углероду
1. Перегруппировка Фрнса
Перегруппировкой Фриса [70] называют довольно универсальную перегруппи-
ровку феноловых эфиров в фенолкетоны по схеме:
ОН ' OCOR
ОН
I
COR
Перегруппировка Фриса имеет большое экспериментальное значение; на этой
реакции основано много синтезов практически важных фенолкетонов. Реакция важна
еще и потому, что, как заметил уже Фрис, прямое владение ацильной группы в фе-
нолы, особенно в замещенные, проходит далеко не так гладко, как через сложные
эфиры фенолов, поскольку при прямом С-ацилировашти действие А1С13 па фенолы
иногда приводит к получению ряда нежелательных продуктов. Кроме того, проведение
реакции Фрисй примерно однотипно для большого числа эфиров.
Реакция Фриса проводится в условиях, аналогичных реакции ацилирования
по Фриделю — Крафтсу, однако, как правило, требует более высоких температур.
В качестве растворителя обычно применяют нитробензол, реже сероуглерод, петро-
лейный эфир пли хлорбензол. В качестве катализатора чаще всего используют А1С13
в стехиометрическом соотношении. При этом следует отметить, что в определенных
условиях под действием A1CL, может происходить перемещение или отщепление свя-
занных с ядром алкильных групп. Меервейн п Ксстпер изучили каталитическое дей-
ствие трифторида бора [71J; проведение перегруппировки Фриса в безводной плавико-
вой кислоте разработало Данном и Милиусом [72].
Перемещение ацильного ос>атка может происходить D орто- или пара-ноложепия.
Соотношение орто- и пара-изомеров зависит от структуры применяемого эфира фе-
нола, однако па него в известных пределах можно влиять посредством изменения тем-
пературы, растворителя, вида и количества катализатора. Низкие температуры (ниже
60° С) способствуют перегруппировке в пара-положение, более высокие — в орто-но-
ложение. Особенно ярко выражен этот эффект в случае л-крезплацетата, из которого
при 25° С образуется 80% пара-соединения, а при 165° С, напротив. — 95% орто-со-
единения [73]. Однако получение желаемого изомера возможно нс do всех случаях.
Разделение обоих изомерных фенолкетонов часто можно провести перегонкой с водя-
ным паром. Как правило, с водяным даром перегоняются только орто-изомеры, пара-
изомеры не летучи.
Описана перегруппировка фепилацетата в о-оксиацещфенон [74].
о-Оксиацетофенон. Фепилацстат (100 г) порциями смешивают с 200 г А1С13,
причем смесь сначала охлаждают, а затем после окончания довольно бурной
[70] К. Fries, G. Fink, Вег.. 41. 4271 (1908).
[71 ] D. Kastner, в книге W. Foerst, «Хспсге Melhoden der organischen Che-
mie», Verlag Chemie, Weinhcin, 1949, S. 444—448.
[72] Q. Dan n, G. M у 1 i ii s. Ann., 587, 1 (1954).
173] K. R о s e ri m u n d, W. Sclinur r, Ann . 460. 56 (1928).
[74] K. Fre n d ₽ n b er g, L. Orthne r, Ber.. 55. 17 48 (1922).
I. ПЕРЕМЕЩЕНИЕ ОТ КИСЛОРОДА Н УГЛЕРОДУ
86;
реакции нагревают еще 5 ч при 120° С на масляной бане. Продукт реакция
разлагают льдом и затем перегоняют с перегретым до 150° С водяным паром.
Обычпой переработкой дистиллята получают 37 г о-оксиацстофенона; т. кип.
91 — 92е С (13 л.и рт. ст.).
Данн и Милиус получили хороший выход n-оксиацетофедона при использовании
безводного фтористого водорода [75].
п-Оксиацетофеяон. При 0° С в литую стальную бомбу загружают 10 г фенил»
ацетата и 20 мл технического безводного HF и, периодически встряхивая,
в течение 3 ч нагревают смесь при 100° С. После 'охлаждения основную часть
HF упаривают на водяной бане, остаток нейтрализуют концентрированным
раствором NaOH и экстрагируют продукт эфиром. Поело отгонки эфира реак-
ционную смесь подвергают перегонке с водяным паром. Из остатка от перегонки
при охлаждении кристаллизуется n-оксиацетофенон; т. пл. 105—107е С; выход
80% от теоретического.
Реакция Фриса может быть применена также к ацильным соединениям гетеро-
циклов, содержащих кислые имнногруппы. Например, из N-ацетилкарбазола получают
З-ацетилкарбазол наряду с небольшим количеством 1-ацетилкарбазола [76].
2. Перегруппировка Клайзеиа [77]
Клайзен в 1912 т. открыл гладко протекающую перегруппировку снол-аллило-
вых’эфиров в С-аили л соединения, папример, в случае ацетоуксусных эфиров или аце-
тилацетона ]78]:
СН3СОСН=ССН3 —* СИзСОСНСОСНз
I I
ПСП = СПСН2О RCH—СНСПз
Еще более важной является также открытая Клайзеном перегруппировка фе-
пилаллиловых эфиров в а.глилфенолы:
осн2сн=снв
он
снсн=сн
I
R
2
Если в молекуле имеется хотя бы одно незамещенное орто-пол ожепие, то про-
исходит орто-сдвиг, причем аллильный остаток претерпевает аллильную перегруппи-
ровку. Если замещепы оба орто-положения, то аллильный остаток сдвигается в пара-
положепие, причем, как правило, аллильной перегруппировки не происходит.
och2ch=chr он
I
ch2cii=chr
Реакция идет без катализатора. В большинстве случаев процесс проводят при
температуре около 200‘' С, целесообразнее в отсутствие кислорода. Эфир фонола нагре-
вают без растворителя илп применяют кипящие диметплапилин (т. кип. 193° С) пли
диэтиланмлин (т. кип. 215° С). Заместителя в ядре почти по оказывают влияния на
реакцию. Перегруппировка Клайзена может быть проведена в присутствии как нитро-,
так и ацетиламиногруппы.
[75] О. Dann, G. М у 1 i u s, Ann., 587, 1 (1954).
[76] Е. Meitzner, J. Am. Cbem. Soc., 57. 2327 (1935).
|7j D.S. Tarbell, Org. Reactions, 2, 1 (1948).
78] L. Claisen, Ber., 45, 3157 (1912).
864
ПЕРЕГРУППИРОВКИ С ПОСТРОЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
о-Аллилфенол. Фенилаллпловый эфир, по Кламзену [79], кипятят в атмосфе-
ре двуокиси углерода до тех пор. пока температура массы пе повысится от 190° С
до примерно 220° С; для этого необходимо около 6 ч. Если за это время темпе-
ратура не поднимется до 220° С, то дальнейшее нагревание не имеет смысла,
так как в этом случае образуется несколько большее, чем обычно, количество
побочного продукта — 2-метилкумарана, присутствие которого снижает темпе-
ратуру кипения о-аллилфенола. Для удаления постоянной примеси 4—6%
2-метилкумарана продукт растворяют в двойном объемном количестве 20% -ною
раствора NaOH и дважды извлекают примесь петролейным эфиром. Затем
подкисляют, экстрагируют продукт эфиром, сушат экстракт пад СаС12 и пере-
гоняют. Выход о-аллилфенола 'выше 80% от теоретического; т. кип. 99е С
(12 мм рт. ст.}. Добавление гидрохлорида пиридина способствует повышению
выхода 2-метилкумарана до 60%.
11 11 LJ\ Jcn-cii,
^^ОСНгСН-СН! ^/ХОН С>
Тенденция к перемещению аллильной группы в ядро столь сильна, что при повто-
рении вышеописанных операций (этерификация о-аллилфенола и перегруппировка)
получают 2,6-диаллилфенол и далее 2,4,6-триаллилфенол, При перегруппировке
аллилового эфира 3,5-диаллилсалициловой кислоты аллильная группа даже вытесняет
карбоксильную группу с образованием 2,4,6-триаллилфепола и двуокиси углерода:
/х/СООН
II k II
Y^OCsHs уЧн
C3H5 C3H5
3. Перегруппировка Бекера — Венкатарамана
Перегруппировка ароилированных 2-оксиарилметилкетонов в <о-(2-окснбензоил)-
ацетофеионы была одновременно описана Бекером [80] и Венкатараманом [81]:
у. /СОСНз хчуСОСНаСОСиЩ
। Y —
^\)СОСвН6
Перегруппировка происходит под действием основных катализаторов (карбонат
калия, амид натрия, этилат натрия и др.). Ее можно рассматривать как внутримоле-
кулярную конденсацию Клайзена. Реакция имеет большое препаративное значение,
так как продукты перегруппировки легко циклизуются, например, под действием
бромистого водорода в ледяной уксусной кислоте с образованием флавонов:
О
/х/сосигсос«н5
ГI ч i "п
|| Ml С—Cells
f79J L. С la iso n. Ann.. 418, 79 (1919).
[80] W. Baker, J. Chem. Soc., 1933. 13S1.
[81] K. Venkataram an, Current Scj. 2, 214 (193S).
II. ПЕРЕМЕЩЕНИЕ ОТ АЗОТА К УГЛЕРОДУ
865
2-Окси-2'-метилдибеизоилмеган [82]. К раствору 10 г эфира 2-о-толуил-
оксиацетофеяона в 200 -мл сухого бензола добавляют 10 г этилата натрия и
20 мин кипятят на водяной бане. После охлаждения массу встряхивают с раз-
бавленной НС1, бензольный слой сушат и отгоняют бензол. Остаток перокристал-
лизормвают из метилового спирта о добавлением активированного угля. Полу-
чают 4,2 г (42% от теоретического) 2-окси-2'-метилдибепзоилметана в виде
желтоватых кристаллов; т. пл. 96—97° С.
4. Перегруппировка бензиловых эфиров
Шорыгин в 1925 г. длительным нагреванием при 100° С дибензилового эфира
с металлическим натрием получил фенилбензилкарбпнол [86|:
СоН5СН3ОСП2СвНб —> CeH5CHOHCH2C6Il5
Вначале эта реакция не нашла препаративного применения, так как жесткие
условия нс позволяли проводить перегруппировки сложных соединений, а простые
фенилалкилкарбиполы были более доступны другими путями.
Благодаря применению в качестве металлирующего средства фениллития Вит-
тнгу удалось позднее изомеризовать бензиловые эфиры при комнатной температуре.
В качестве примера препаративного применения этой перегруппировки следует при-
вести синтез фенантрена [84]:
Фенантрен. К раствору 0,4 г (2 ммоль) циклической окиси в 15 мл абсолют-
ною эфира добавляют 2 мл 1.3 н. раствора фениллития и оставляют смесь
на педелю. Постепенно наступающее красное окрашивание затем бледнеет
и выпадают бесцветные кристаллы. После добавления воды п оггонки эфира
остается 0,3 г (77% от теоретического) дигидрофенантрола. Отщеплением воды
посредством обработки продукта смесью ледяной уксусной п серной кислот
гладко получается фепантрен.
ZZ. Перемещение от азота к углероду
1. Бензидиновая перегруппировка
Перегруппировка гидразобензола в бензидин под действием сильных кислот
является прототипом многочисленных аналогичных реакций, которые поэтому в целом
рассматриваются как бензидиновые перегруппировки:
<^_^>-ИН-МП-ф~ф —> H2N—
Бензидины являются промежуточными продуктами для получения субстантив-
ных красителей для хлопка.
[82] F, С г a m е г, G. Н. Е 1 s с h n i g, Chem. Ber., 89, 8 (1956).
[83] Р. Schorl gin. Ber., 58, 2031 (1925).
[84] G. Wittig, P. Davis, G. К 6 n i g, Chem. Ber., 84, 630 (1951).
55 Заказ 1835.
866
ПЕРЕГРУППИРОВКИ С ПОСТРОЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
Гидрохлорид бензидина [85]. При прибавлении по каплям раствора
гидразобензола в возможно малом количество эфира в охлажденную льдом раз-
бавленную HC1 (1 : 1) выпадают кристаллы гидрохлорида бензидина.
Бензидиновые перегруппировки в ряду тиазола были описаны Байером и Кройтц-
бергером 186]. Перегруппировка осуществляется только в том случае, если образую-
щийся диамин действием фталевого ангидрида связывается в виде фталимида:
2,2'-Дифталимидодитиазолил-5,5'. Тщательно растирают 10,9 г дигидрохло-
рида 2,2'-дигидразодитназола с 11,9 г фталевого ангидрида и смесь нагревают
на масляной бане. При темпе,ратуре выше 100° G начинается спекание массы.
Часть сублимированного фталевого ангидрида периодически возвращают
в колбу. Смесь еще 1 ч периодически перемешивают при 180—200° С. Темные
продукты разложения удаляют кипячением с пиридином п раствором карбоната
натрия. В осадке в виде желтовато-серого нерастворимого во всех раствори-
телях порошка остается 2,2'-дифталимцдодитиазолил-5,5'. Фталоильные остатки
отщепляются при нагревании с дымящей НС1.
2. Индольный синтез по Э. Фишеру *
Индольный синтез по Э. Фишеру по своему механизму похож на бензидиновую
перегруппировку [87]. Фенилгпдразопы* или замещенные фенилгидразоны альдеги-
дов, кетопов и кетокислот при нагревании с отщепляющими аммиак средствами —
ZnCl2, SnCl2 или минеральными кислотами, превращаются в замещенные индолы. Ре-
акция протекает по схеме [88]:
A A-R'_A—
^\h-A-R
Реакция приобрела большое значение в синтезе природных веществ; например,
полный синтез стрихнина [89J начинается с получения 2-веритрилиндола из ацето-
вератрона и фенилгидразппа.
2-Фенилиндол [90]. Смесь 53 г свежеприготовленного фелилгидразона аце-
тофенона и 250 г порошкообразного безводного ZnCl2 при перемешивании поме-
щают в нагретую до 170° С масляную баню. Через 3—4 мин. масса разжижается
* Обзоры см. П. Н. Суворов, В. П. Мамаев, В. М. Родионов, Реакции
и методы исследования органических соедипений (РиМИОС), Госхпмиздат,
9, 7 (1959); Ю. П. Китае в, Усп. хим., 28, 336 (1959); J. Robinson, Chem.
Rev., 63, 373 (1963). — Прим. ped.
[85] Gatterman n-W i elan il, Die Praxis des organischen Chemikers, 33
Aufl., S. 172.
[86] H. Beyer, A. Kreatzherger, Chem. Ber., 84, 518 (1951).
[87] G. M. R о h i n s о n, R. Robinson, J. Chem. Soc.. 1918, 639.
[88] Герм. лат. 38784, Farbwerke Hoechst, Erfinder Emil Fischer; Friedl., 1, 151 (1886);
E. Fischer, О. Il e ₽, Ber., 17, 559 (1884).
[89] R. B. Woodward et al., J. Am. Chem. Soc., 76, 4749 (1954).
[90] R. L. S h r i и e r, W. C. Ashley, H. Coonrad t, Org. Synth.,
22, 98 (1942).
II. 11ЕР1:М1с!ЦЕНИЕ ОТ АЗОТА К УГЛЕРОДУ 8(17
и начинается выделение белых паров. Масляную бапю удаляют и перемешивают
массу еще 5 мин. После соответствующей обработки получают с выходом 72—
80% ог теоретического 2-фенилиндол; т. пл. 188—189° С.
Особенно приюдсн этот синтез для получении тетра1идрокарбазолов из цикло-
гексанона и фенплгидразина и продуктов их замещения; в качестве конденсирующего
средства в этом случае используют ледяную уксусную кислоту:
C'i К4) — f ( ]
Тетрагидрокарбазол [91]. К кипящей смеси 98 г циклогексанона и 360 г ледя-
ной уксусной кислоты в течение 1 ч при перемешивании добавляют 108 г фепил-
ыэдразина, кипятят еще 1 ч. при перемешивании охлаждают до 5J С и отсасы-
вают выкристаллизовавшийся тетрагидрокарбазол. Продукт промывают водой,
75%-ным этиловым спиртом и перекристаллизовываю г из метилового спирта.
Выход продукта 75 — 85% от теоретического; т. пл. 115—116° С.
3. Перегруппировка Стивенса
Соответствующим образом замещенные четвертичные аммониевые соли иод дей-
ствием сильных оснований подвергаются перегруппировке с перемещением замести-
теля от азота к а-углеродному атому по схеме (например, при R = Ph, см. [92]):
R—СН2—N(CIIg)a 2!^ К— СН-N(CH3)2 — > R—СН—N(CII3)2
I I I
CH2C6H6 CH2CeH.-t CH2C6H5
По Виттшу [93], под действием основания (алкоголят натрия, фениллитий
или амид натрия) от а-атома уъгерода отрывается протон с образованием неустойчи-
вою «илида», в котором затем бензильный остаток перемещается от азота к углероду.
В качество препаративного примера приводится пропись [94] для превращения
дифенациламмопнйбромндов в дибензонлэтилены. Реакция происходит уже с относи-
тельно слабыми основаниями, водными растворами диметидамина или едкого натра
по следующему уравнению:
ПзС СНз N(CH3)2
С6ИйСО-С1Т2-1Х-СПг-СОС6Н5 —> С6Н5СО-СП-СП3-СОС6Н.г,
—> C6H6CO-CH-CH-COC6HS
Дибензоилэтилси. Дифенацилдиметиламмонийбромид прп перемешивании
добавляют к 10 ч. 33% -иого водного раствора дпметиламнна. Через непродолжи-
тельное время желтая окраска смеси бледнеет, происходит сильное помутнение
раствора. Массу охлаждают до 0” С и затравкой вызывают кристаллизацию.
После 8 ч стояния при 0е С осадок отфильтровывают и промывают небольшим
количеством воды. Выход продукта 78% от теоретического; после перекристал-
лизации пз метилового спирта т. ил. 65° С.
Нагреванием полученного продукта,, в нейтральном или слабокислом
растворе с отщеплением диметиламина получают тра«с-дпароилэтплси.
[91] С. U. Rogers, В. В. Colson, Org. Synth., 30, 90 (1930).
[92] Th. Thomson, Th. St. 8 t e v e n s, J. Chem. Soc., 1932, 1932.
[93] G. Wittig, Angew. Chem., 66, 10 (1954).
[94] W. II e f f e, F. Krohnke, Chem. Der., 89, 835 (1956).
55*
868
ПЕРЕГРУППИРОВКИ С ПОСТРОЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
4. Перегруппировка Соммле
В качестве побочнрй реакции при перегруппировке Стивенса часто происходит
алкилирование бензольного ядра в орто-положение [95]. Эта реакция становится ос-
новной при низких температурах. Она получила свое название по имени Соммле,
впервые наблюдавшего перегруппировку такого типа [96]. Благодаря работам Кан-
тора и Хаузера [97] эта реакция приобрела препаративное значение. Например, при
применении в качестве металлирующего средства амида натрия перегруппировка
происходит почти мгновенно при температуре кипения аммиака (—33° С).
Пропись для получения 2-метилбевзилдиметиламина иэ бензилтриметиламмоний-
подида см. [98]; выходы составляют 90—95% от теоретического.
CHg ртт
/j3 '/ с11.А!С11я)а
Путем ряда последовательных превращений o-замещенпого бензилдиметиламииа
в четвертичное основание и перегруппировои Кантор и Хаузер [97] получили пол-
ностью метилированный бензиламин
CH2N(CH3)2
СНз
III. Перемещение от серы к углероду
Аналогично двум последним рассмотренным выше перегруппировкам под вли-
янием сильных оснований происходит превращение сульфониевых солей.
Как и при перегруппировке Стивенса, фенацилбензилметилсульфонийбромид
изомеризуется в и-бензил-о-метилмеркаптоацетофенон:
СвНбСОСН2-8—СНз СвНбСО—СН—SCHa
СН2С0НВ СНаСвПб
Эта реакция также описана Стивенсом [99].
<в-Бензил-®-метилмеркаптоацетофенон[100[- Раствор метилата натрия, при-
готовленный из 8 г патрия и 580 мл метилового спирта, смешивают с 40 г суль-
фонийбромида и в течение 1 ч нагревают на водяной бане. После отгонки с водя-
ным паром метилового спирта оставшееся светло-желтое масло растворяют
в эфире, сушат над СаС12 и отгоняют эфир. со-Бензил-а-метилмеркаптоацето-
феион выкристаллизовывается при потирании стеклянной палочкой; после
перекристаллизации из этилового спирта т. пл. 55—56Q С. Выход продукта
81% от теоретического.
95] G. Wittig, Ann., 560, 116 (1948).
96] М. Sommelet, С. г., 205, 56 (1937).
97] S. W. С an t о г, С. R. Н а и з е г, J. Am. Chem. Soc., 73, 4122 (1951).
98] W. R. В г a s e n, C. R. Hauser, Org. Synth., 34, 61 (1954).
99] Th. Thomsen, Th. St. Stevens, J. Chem. Soc., 1932, 69.
100] F. Krollpfeifer, H. Hartmann, Chem. Ber., 83, 97 (1950).
IV. ПЕРЕГРУППИРОВКА ТИФФЕНО
869
Описаны перегруппировки сульфопиовых солей по типу реакции Соммле [101].
Бензилдиметилсульфонийбромид, например, под действием амида натрия в кипящем
эфире превращается в 2-метилбензилметилсульфид:
СН8
^\/ \+
1 || S—СН»
СНз
СНз
CH2SCH3
IV. Перегруппировка Тиффено
Реакция бснзилмагнийхлорида с формальдегидом приводит к получению не
р-фенилэтилового спирта, а 2-метилбензилового спирта [102]:
zCH2MgCl zCHa
'^/'XCH2OH
2-Метилбензиловый спирт [103]. К профильтрованному эфирному раствору
бензилмагнийхлорида (из 3,4 моль бензилхлорида) добавляют 165 г пара-
формальдегида, в течение нескольких суток высушенного в вакууме над пяти-
окисью фосфора. Смесь при перемешивании нагревают в течение 18 ч, выливают
иа лед и подкисляют разбавленной H2SO4. Затем органический слой промывают
водой, 5%-ным раствором соды и вновь водой. Из водных вытяжек извлекают
продукт эфиром и промывают экстракт таким же образом. Перегонкой с наса-
дочной колонкой получают 248—263 г 2-метилбензнлового спирта; т. кип.
106—109° С (12 мм рт. ст.). Выход 59—63% от теоретического.
Полученный спирт можно снова превратить в хлорид и подвергнуть перегруп-
пировке. Трехкратной перегруппировкой Тиффено был получен 2,3,4-триметилбен-
зиловый спирт "[104]. На последних стадиях, однако, были неудовлетворительные
рыходы.
Высшие альдегиды иногда реагируют с бензилмагнийхлоридом с образованием
гликолей [105]:’
CH2MgCl
2RCH0
'^/CHaCHOHR
I II
'''СПОНЕ
[101] C. R. Н a u s е г, S. W. Cantor, W. R. Brasen,!. Am. Chem. Soc., 75,
2660 (1953).
[102] Tif f eneau, Delange, C. r., 137, 573 (1903).
[103] L. I. S mi th, L. J. Spillane, J. Am. Chem. Soc., 62, 2639 (1940).
[104] T. Reichstein et al., Helv. chim. acta, 19, 412 (1936).
[105] S. Siegel, S. K. Coburn, D. R. Levering, J. Am. Chem. Soc., 73,
3163 (1951).
870
ПЕРЕГРУППИРОВКИ С РАСЩЕПЛЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
ПЕРЕГРУППИРОВКИ С РАСЩЕПЛЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
I. Перемещение от углерода к кислороду
1. Перегруппировка Криге
Криге установил [106], что продукты бензоилированияр идроперекиси декалина
при нагревании перегруппировываются в изомеры, для которых доказано строение бен-
зоилированных полукеталей:
Кипятят в течение 1 мин 0,28 г бензоата гидроперекиси декалина (т. пл.
67—68° С) в 2 .й.г пиридина с 0,2 г гидрохлорида пиридина. После охлаждения
выливают массу в разбавленную H2SO4 и для кристаллизации выделившегося
масла ставят смесь в холодильник. Выход изомерного бензоата 0,15 г; после
перекристаллизации из разбавленною метилового спирта т. пл. 96—97° С.
Эта реакция представляет собой пример уже давно известного и распространен-
ного для органических перекисей расщепления С—С-связи. Она протекает, например,
при широко применяемом в промышленности расщеплении кислотами гидроперекиси
кумола [107] на фенол и ацетон. Реакция идет по следующему уравнению:
С6Н5С(СН3)2 —> СбН5С(СН3)2 —> С(С11з)з
I I ‘I
ООН о* с8н5о
он
1
СвН5ОС(СН3)2
Посредством ионогенного расщепления перекисной группы образуется катион
кислорода, который перетягивает к себе фенильную группу с электронной парой,
трежде связывавшей ее с атомом углерода. К образующемуся таким образом карбка-
гиону присоединяется вода с образованием неустойчивого полукеталя, тотчас же раз-
дающегося на фенол и ацетон.
Еще одним примером такой перегруппировки является превращение гидропере-
киси метилтетралина при нагревании с серной кислотой в 1-(2'-оксифепнл)-пента-
1он-4[108|:
Н3С ООП
4 (СТЫзСО си.
Расщепление проводят преимущественно ледяной уксусной кислотой в присут-
твии каталитических количеств хлорной кислоты [109].
Промежуточный сдвиг перекпеного кислорода между а- и p-углероднымп ато-
!ами объясняет также часто наблюдаемое аномальное протекание озонирования оле-
шков [110]. Известны многочисленные примеры тою, что ненасыщенные соединения,
106] R. Criegee, Вег., 77, 724 (1944).
107] Н. II о с k, S. L а о g, Вег., 77. 257 (1944).
108] Н. II о с к, F. I) е р к о, G. К n a u е 1, Chem. Вег., 83, 238 (1950).
109] М. S. К h а г a s с Ii, A, F о п о, W. Nudenber g, J. Org. Chem., 15,
748 (1950).
110] J. E. Lef lie r. Chnm. Rev., 45, 385 (1919).
II. ПЕРЕМЕЩЕНИЕ ОТ УГЛЕРОДЛ К АЗОТУ
871
имеющие функциональные группы по соседству с двойной связью, при озонировании
наряду с расщеплением двойной связи претерпевают расщепление простых связей,
которые соединяют углерод двойной связи с этими группами. Например, при озони-
ровании кротонового альдегида образуется муравьиная кислота 1111 ];
СПз— СН=СПуСНО
2. Перегруппировка гипохлоритов
При попытке перегонки в вакууме 1-метилциклопентилгипохлорита, получен-
ного хлорированием 1-метилциклопентанола в щелочном растворе, с количественным
выходом был выделен изомерный 6-хлоргексанон-2 {112J:
сн2—СП2 ОС1
СН2-СН2 СНз
СН2СН2С1
СН2СН2СОСНз
Реакция открыла возможность синтеза со-галогенк₽тонов, трудно доступных иными
способами.
II. Перемещение от углерода к азоту
1. Гофмановское расщепление амидов кислот [113]
При гофмановском расщеплении происходит дегидрирование амидов кислот
по азоту. Промежуточный предполагаемый продукт дегидрирования с одновалентным
атомом азота стабилизуется, превращаясь в изоцианат, путем перемещения радикала,
связанного с соседним атомом углерода, в виде аниона к азоту. Изоцианат в водных
растворах гидролитически распадается на двуокись углерода и амин, содержащий
на один атом углерода меньше, чем исходный амид:
HCONHs —> [BCO-fNJ —► BNCO RNH,
Реакция в общем случае проводится путем быстрого нагревания амида кислоты
в водном растворе гипогалогенита примерно до 70° С. Приводится следующая пропись
для превращения амида никотиновой кислоты в 3-ямпттопиридии [114].
З-Амннопиридин. К раствору гипобромпта натрия, приготовлеппому при
охлаждении смесью льда и соли из раствора 75 г NaOH в 800 мл воды и 95,8 г бро-
ма, прибавляют лри0° С в один прием 60 г тонкоизмельченного амида никотино-
вой кислоты. При эпергичпом перемешиванип растворение происходит в течение
15 мин. Реакционный сосуд помещают затем в нагретую до 75° С водяную
баню и выдерживают 45 мин при 7U— 75° С. Соответствующей переработкой
(подробности в оригинале) получают З-амипопиридин с выходом 60—65% от тео-
ретического.
Прп проведении реакции Гофмапа н водной среде для алифатических амидов
с шестью и более атомами углерода возможно получение алкилацилмочевин, образо-
вание которых объясняют взаимодействием щелочных солей N-галогенированных
амидов с промежуточно образующимися изоцианатами. В этом случае предпочти-
тельнее вести реакцию в спиртовом растворе [115] и образующиеся при этом уретаны
омылять далее до аминов.
Ill] W. G. Young et a]., J. Am. Chem. Soc., 68, 293 (1946).
112] T. L. Cairns, В. E. Englund, J. Org. Chem., 21, 140 (1956).
113] E. S. Wallis, S. F. Lane, Org. Reactions, 3, 267 (1949).
114] C. F. H. A 1 I e n, C. N. W о 1 f, Org. Synth., 30, 3 (1950).
115] E. Jeffreys, Ber., 30, 898 (1897).
872
ПЕРЕГРУППИРОВКИ С РАСЩЕПЛЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
Метиловый эфир пентадецилкарбаминовой кислоты. При слабом нагрева-
нии 25,5 г амида пальмитиновой кислоты растворяют в 65 г метилового сиирта,
раствор смешивают с метилатом натрия (из 4,6 г натрия в 115 г метилового
спирта) й тотчас же по каплям прибавляют 16 г брома. Порядок прибавления
реагентов ие имеет значения; сначала можно прибавить бром и затем раствор
метилата. После смешения компонентов массу в течение 10 мин нагревают
на водяной бане, нейтрализуют уксусной кислотой, отгоняют спирт, промы-
вают остаток водой. Отфильтровывают уретан (метиловый эфир пиптадецил-
карбамииовой кислоты), промывают его, высушивают и при нагревании раство-
ряют в лигроине (т. кип. 70—\80э С), причем непревращенпый амид нс раство-
ряется, Из лигроина получают чистый уретан с выходом 84—93% от теорети-
ческого; т. пл. 60—62° С.
• Пентадециламин. Уретан тщательно смешивают с четырехкратным количе-
ством Са(ОН)3 и перегоняют смесь. Прп атом почти количественно отгоняется
образующийся пентадециламин. Его растворяют в лигроине, сушат над КОН
и перегоняют над натрием; т. кип. 298—301° С; т. пл. 36,5° С.
При больших загрузках дорогой гипобромит натрия может быть заменен гипо-
хлоритом.
Из а-окси- и а.р-шнасыщенных кислот при гофмаповском расщеплении обра-
зуются альдегиды, имеющие на один атом углерода меньше, чем исходная кислота.
Эта реакции может служить, например, для получения фепилацетальдегида:
С0П5СН=СНСООН —> [СвН6СН=СНКН2] —> С6Н5СП2СНО
В случае диамидов дикарбоновых кислот с соответствующей длиной цели после
превращения одной амидной группы происходит замыкания кольца вследствие обра-
зования ацилмочевинной группировки. Например, диамнд имидазол-4,5-дикарбоно-
вой кислоты превращается в ксаптин [116],
h2nc-c-nh
II I
H2NC—с сн
II \/z
О N
H2NC-C-NH
II I
OCN — с СН
J
о
II
с
— > ПгГ \1-NH
I II I
с с сн
^\/'W
О NH N
О
о
2. Расщепление по Курциусу [117]
Такое же препаративное значение, как и гофмановское расщепление амидов,
имеет термическое расщепление азидов кислот, называемое расщепленпем по Кур-
циусу* :
RCON3 —>RNCO-]-N2
В отличпе от реакции Гофмана при проведении процесса в инертном раствори-
теле, например бензоле, может быть выделен изоцианат.
Феиилизоцианат [118]. Осторожно и медленно нагревают до 60—70° С 12 а
абсолютно сухого бензазида в 40 мл высушенного над натрием бензола, причем
происходит бурное выделение азота. После окончания основной реакции повы-
шают температуру до 80е С, Отгоняют цри нормальном давлении бензол и затем
при 60° С (20 мм рт. ст.) перегоняют изоциапат. Выход продукта 7—8 ?.
* Осторожно! При проведении реакции Курциуса с большими загрузками может
произойти взрыв. — Прим. ред.
[116] R, A. Baxter, Г. S. 8 р г i n g, J. Chem. Soc,, 1945, 2.32.
[117] Р. A. S. S m i t h, Org. Reactions. 3, 337 (1949).
[118] Ga tt er ma n n-VV island, Die Praxis ties organtsclien Chemikera,
35 Aufl., S. 138.
Ц. ПЕРЕМЕЩЕНИЕ ОТ УГЛЕРОДА К АЗОТУ
873
Если разложение азидов проводят в спиртовом растворе, тополучают уретаны,
которые действием концентрированной НС1 могут быть гидролиаонаны до аминов,
например:
CON3 ( NHCOOR NH2
I 3 _ Л A >11 — > t ।
CON3 NHCOOR Nib
1,4-Диамипоциклогексаи, дигидрохлорид [119]. Раствор 5 г диазида транс-
гексагидротерефталевой кислоты в 200 мл абсолютного этилового спирта нагре-
вают в течение 2 ч с обратным холодильником, фильтруют и упаривают в ваку-
уме, Прп охлаждении кристаллизуется 5,2—5,5 г (90—95% от теоретического)
уретана; т. пл. 236° С.
Смесь 4,8 г уретана и 50 мл концентрированной НС1 нагревают в запаян-
ной трубке 7 ч. при 120° С (или 24 ч с обратным холодильником) я затем выпари-
вают досуха. Выход дигидрохлорида 1,4-диамин Оциклогек сана 95—98% от
теоретического.
Для проведения реакции Курциуса не всегда является необходимым выделение
азидов в виде индивидуальных веществ. Часто можно галогеиангидрид кислоты
подвергнуть взаимодействию с азидом натрия и в одну стадию получить изоцианат.
Этот вариант способа использован, например, для синтеза метилизоцианата из аце-
тилхлорида 1120].
Метилизоцианат. При охлаждении смешивают 14,5 г азида натрия в 100 мл
амилового эфира с 15,5 г ацетилхлорида. Реакция начинается при нагревании
смеси до 60—70° С и вскоре становится очень бурной. Основная часть метилизо-
цианата отгоняется через обратный холодильник и конденсируется л Приемнике,
охлаждаемом охлаждающей смесью. В конце процесса отключают охлаждаю-
щую воду и сильнее нагревают реакционную массу. Из 10 г дистиллята фракцио-
нированием получают 8 г чистого метилизоцианата; т. кип. 42—43’С.
' Согласно данным Брауна [121], расщепление по Курциусу а-бромкарбоновых
кислот является хорошим методом получения альдегидов или кетонов, имеющих на
один ато.м углерода меньше, чем исходная кислота. В случае хлорангидрида а-бром-
кцелоты реакцию проводят по следующей схеме:
ВСНВгс.оа — BCHBrCONs BCHBrNCO BCHBrNHs —> BCHO
Промежуточные продукты не выделяют. Выходы карбонильных соединений
'Составляют около 60% от теоретического.
у-Фенил-н-бутиралъдегид. Раствор хлорангидрида а-бром-б-фенилвале-
риановой кислоты в бензоле смешивают с 1,5 моль азида натрия. Азид натрия
рекомендуется активировать гидразингидратпм (122). Реакцию проводят путем
кратковременного нагревания на водяной бане. Для омыления образующегося
изоцианата после охлаждения смесь обрабатывают холодным раствором 2,5 моль
КОН в этиловом спирте и оставляют иа некоторое время. Затем подкисляют
соляной кислотой и отгоняют у-фепил-м-бутпральдегид с водяным паром. Выход
альдегида 58% от теоретического; т. кип. 120—122° С (16 мм рт. ст.).
СвПаССНгЬСНВгСОС! —> С6Н5(СН2)3СНО
[119] Th. Curtius. Е. Darmstiidter,
Chem.. 91, 1 (1915).
[120] G. Sc,hroter, Ber.. 42, 3356 (1909).
[121] J. v. Braun, Ber., 67, 218 (1934).
[122] J. N ell es, Ber., 66, 1315 (1933).
H. P г i и g s li e i m, J. prakt.
874
ПЕРЕГРУППИРОВКИ С РАСЩЕПЛЕНИЕМ УГЛЕРОДНОГО СКЕЛЕТА
3. Перегруппировка Бекмана
Кетоксимы при обработке их в пиридине пентахлоридом фосфора, серной кисло-
той, поллфосфорной кислотой или сульфохлоридами превращаются в амиды кислот;
при этом оксигруппа обменивается местами с-mpawc-расположспным остатком [123J:
R—С—R' —> HO-C-R' —> R'CONHR
I' II
NOH NR
Бекман f 124] при обработке бснзофопон оксима н хлорокиси фосфора пятихло-
ридом фосфора выделил имидхлорид, гидролизом которою водпым этиловым спиртом
был получен бензанилид:
СвН5-С-С6Н6 — > С1-С-С6Н6 —> C6H5NIICOQH6
Г I’
NOH N-C6H5
Описано проведение реакции с РС15 в эфире или бензоле.
2-Пиридилацетанилид [125]. К раствору Зг оксима 2-фенацилпиридина
в абсолютном эфире при «стряхивании постепенно прибавляют 3 г PC1S и встря-
хивают массу еще 3 ч. Затем слегка подщелачивают раствором К2СОЭ и выде-
ляют продукт. Выход 2-пиридилацетанилида 90% от теоретического.
'а -а
N СН2-С-СбП5 СН-2СОКПСеНз
II
HON
Технически лажной является реакция расширения кольца циклогексанон-
оксима с образованием капролактама [126]:
СН2
/\
vnw Н2С NH
/wN0H | I
fl —CO
H2C CH2
\ У
СПз
Перегруппировку оксима 4-этилциклогсксанона в у-этилкапролактам проводят
по следующей прописи [127].
у-Этилкапролактам. К 30 г концентрированной H3SO4 при перемешивании
прибавляют по каплям 19 г оксима 4-этилциклогоксанона. Температура смеси
не должна подниматься выше 125° С. Затем массу охлаждают до 90° С и одно-
временно медленно добавляют еще 150 г оксима и 150 г 25%-ного олеума.
Перегруппировка оканчивается через 45—60 мин. В заключение массу еще
некоторое время нагревают при 130° С. Затем реакционную массу нейтрали-
зуют, прибавляя ко каплям в водный раствор аммиака, продукт извлекают
1123J J. Meisenheimer, Ann., 468, 202 (1929); 495, 249 (1932).
[124] Е. Beckmann, Вег.. 19. 988 (1886).
[125] F. Galin о wski, G. К a i в z, Mh. Chem.. 77. 137 (1947).
1126] Лабораторные данные: J. J. D о n I е a v у, М. А. К i s е, Org. Synth., f7,
63 (1937).
[127] W. Z i e g e n b e i n, A. S c Ii a f f I e r, R, К a 11 f h о I d, Chem. Bor., 88,
1906 (1955).
II, ПЕРЕМКГЦЕНПЕ ОТ УГЛЕРОДА К АЗОТУ
875
большим количеством мстиленхлорида и пере.гоняют в вакууме. Получают
с выходом 90% от теоретического сырой у-этилканролактам; i. кип. 1(50 —165° С
(15 мм рт. ст.).
Еще один пример расширения кольца описан Кауфманом [128]:
NO11
Ацетат лактама дегидроэпиандростерона. К раствору 10 г окспма ацетата
дегидроэпиандростерона в 100. мл пиридина прибавляют раствор 10 г 2-ацстил-
амикобензолсульфохлорида н 50 мл пиридина. Реакционную массу выдержи-
вают 3 ч при комнатной температуре, затем выливают на лсд и нейтрализуют
разбавленной IICI, Из осадка продукт экстрагируют хлороформом, промывают
экстракт водой и сушат пад NajSO^. Затем отгоняют растворитель, остаток
перекристаллизовывают из метилового спирта. Выход ацетата лактама дегидро-
эпиандростерона 50% От теоретического; т. пл. 295 — 298° С.
Особенно хорошие результаты получают при проведении бекмановской пере-
группировки с полпфосфорной кислотой [129].
Анизонланизидин. Смесь 2 г оксима п.я'-диметоксибензофенона и 60 г поли-
фосфорпой кислоты нагревают при перемешивании до 130° С и 10 мин выдержи-
вают при этой температуре. Затем массу выливают в воду и продукт экстраги-
руют смесью эфира с этилацетатом (1 : 1). Экстракт промывают водой, насы-
щенным раствором NaCl и сушат над Na..SO4. Выход аинзоиланизидина 91%
от теоретического; т. пл. 199 —203е С.
4. Реакция Шмидта [130, 131]
Реакция Шмидта включает все реакции карбонильных соединений с азотисто-
водородпой кислотой в присутствии сильных минеральных кислот.
Реакция карбоновых кислот с азотистоводородной кислотой, приводящая к по-
лучению обедненных на один атом углерода аминов, аналогична расщеплениям по
Гофману и по Курциусу:
RCOOIT + HN3 —* RNH2-CO2 ' ^2
Преимуществом этой реакции является возможность синтеза аминов в одну
стадию, в то время как для расщепления по Гофману и по Курциусу необходимо пред-
варительное получение производных кислоты. Недостатком реакции Шмидта является
большая ядовитость азотнетоводородной кислоты * и. кроме того, необходимость
использования в качестве катализатора концентрированной H«SO4, что ограничилае.т
применение реакции в случае веществ, неустойчивых к H2SO4. Хорошие выходы по-
лучают при использовании для реакции высших алифатических и гидроароматических
карбоновых кислот.
а-Бензилэтиламин [132]. Растворяют 5.5 г бензилметилуксусной кислоты
в 8 мл концентрированной H3SO4 и при 40° С по каплям прибавляют 10%-ный
раствор азотпетолодородной кислоты (1,3 моль) в хлороформе. По оконча-
нии выделении азота массу выливают на лед. Затем отгоняют хлороформ.
Осторожно! Азотпстоводородиая кислота шльпо взрывчата! — Прим. ред.
128] S. Kaufman, J. Am. Chem. Soc., 73, 1779 (1951).
129] Е. С. II о г и i n g. V. I/. S t г о in b е г g, J. Am. Ghem. Soc., 74, 2681 (1952).
130] H. Wolff, Org. Reactions. 3, 307 (194'9).
131] K. F. Schmidt, Ber. 57, 704 (1924).
132] J. v. Brann, Ber., 66, 684 (1933).
876
ПЕРЕГРУППИРОВКИ С ПЕРЕСТРОЙКОЙ УГЛЕРОДНОГО СКЕЛЕТА
а а-бензилэтпламин отгоняют с водяным паром. После нейтрализации дистиллята
НС1 и выпаривания получают 4 г (73% от теоретического) чистого гидрохлорида;
т. пл. 146° С.
Прн реакции котонов с азотистоводородной кислотой формально NH-rpyuna
встает менаду карбонильным и смежным с ним атомами углерода:
R-CO-R > B-C0-NH-R + N3
В этом случае получают такой же амид кислоты, что и из кетона (через оксим)
посредством перегруппировки Бекмана. Б случае несимметричных кетонов теорети-
чески могут образоваться два изомера, в зависимости от того, какой из двух остатков
присоединяется я азоту. О попытках проведения реакции в нужном направлении
см. [133].
Из левулиновой кислоты, раствора азотистоводородяой кислоты в хлороформе
и концентрированной H2SO4 в результате почти взрывоподобно протекающей реакции
в качестве одного из основных продуктов получили метиламин с выходом 73% от теоре-
тического [134]:
CHsNHCOCH2CH2COOK CH3COCH2CH2COOR
; J.HN.
CH3NH2 CH3CONHCH2CU2C00H
X
nh2ch2ch2cooh
Биркоферу и Шторху при более низкой температуре и с применением вместо
серной кислоты соляной кислоты удалось получить ^-аланин [133].
З-Алаянн. Через раствор 3,4 г этилового эфира левулиновой кислоты в 6 мл
хлороформа, содержащего 8,1% азотисговодородной кислоты при перемешива-
нии и 3—5° С пропускают слабый ток хлористого водорода, причем тотчас же
начинается бурпое выделение азота. Через 3 или 6 ч добавляют в реакционную
массу 5,5 мл 8,1%-ного раствора HN3 в хлороформе; общая продолжительность
пропускания НС[ 9 ч. Затем массу в два приема экстрагируют 200 мл концен-
трированной НС) и после добавления 30 мл воды в течение 6 ч нагревают
с обратным холодильником. Соответствующей обработкой получают [1-аланин
с выходом 48,5% от теоретического.
ГГЕРЕГРУ1ШИРОВКИ С ПЕРЕСТРОЙКОЙ УГЛЕРОДНОГО СКЕЛЕТА
I, Перегруппировка Вагнера—Меервейна
и близкие к ней реакции
1. Перегруппировка Вагнера—Меервейна
Перегруппировка Вагнера — Меервейна охватывает реакции, протекающие
с перестройкой углеродного скелета при получении олефинов отщеплением аиноиа,
при присоединении по кратным связям и прп нуклеофильном замощении. Общим при-
знаком этих реакций является перемещение алкильного пли арильного остатка к об-
разовавшемуся у соседнего углеродного атома катионному центру молекулы.
Образование этого центра может происходить:
а) посредством отщепления гидроксил-иона или аниона сильной кислоты:
[133] L. Birfeofer. I. S t о г с 11, Сбега. Вег.. 86, 719 (1953).
[134] М. О е s t е г I i n, Angew. Chora., 45, 536 (1932).
I. ПЕРЕГРУППИРОВКА ВАГНЕРА — МЕЕРВЕЙНА И БЛИЗКИЕ К НЕЙ
877
б) посредством присоединения протона по С = С-связи:
)СН-С-С-
и) посредством раскрытии
эпоксидного кольца:
К катиопному центру смещается заместитель от соседнего атома углерода. Вновь
образовавшийся катион' стабилизуется в результате присоединения ёпиопа или отще-
пления протопа, например:
ОН
СНЭ\ I СНзх +
)С-С1Г-СНз —> >С- СН-СПз —*
СНзХ I СН3/ I
Сйз СПз
СНвк +
>С-СН-СНз
CH3Z ' I
СПз
СНз-.
>С-СН-СЩ
chZ । 1
CI СНз
Формально осуществляется обмен между алкилом и галогеном или гидроксилом.
Перегруппировка Вагнера — Меервойпа очень часто происходит у развет-
вленных алифатических или жирно-ароматических соединений. Особенное значение
она имеет в ряду терпенов и камфоры [135].
! Примером (случай а) является перегруппировка камфенгидрохлорида в изо-
'борнилхлорид:
С)
Изоборнилхлорид [136]. Раствор 300г камфена в 150 г этилбромида насы-
щают при 10—20° С хлористым водородом и в течение шести суток непрерывно
нагревают с обратным холодильником при 55° С. Затем основную часть этил-
бромида удаляют током сухого воздуха, отсасывают изоборинлхлорид и сушат
в вакууме над КОП. Выход продукта 271 г, т. пл. 145° С. После перекри-
сталлизации из амилового спирта т. пл. 161,5° С.
Пример перегруппировки олефина (случай б) описали Криге и Рибелт, [137].
Сообразно с этим бис-циклоионтилиден путем двойной перегруппировки Вагнера —
Меервейпа черев катион превращается в Д*>М-окталин;
ехз - оа - 00
[135] Литературный обзор в книге В. X ю к к е л ь, Теоретические основы органи-
ческой химии, пер. с нем., т. I, Издатиилит, 1955.
[136] II. М е е г w е 1 и. К. van Е m s t е г, Вег.. 55, 2526 (1922).
[137] В. С г i е g е с, A. R i е b е 1, Angew. Chera., 65, 136 (19эЗ).
ПЕРЕГРУППИРОВКИ С ПЕРЕСТРОЙКОЙ УГЛЕРОДНОГО СКЕЛЕТА
д0’1О-октадин. В течение .3 ч нагревают при перемешивания при Н0п С 2,5 г
бис-циклопептилидена с 1 г Р2О5. Затем смесь охлаждают и продукт акстраги-
руют эфиром. Экстракт промывают сначала разбавленным раствором соды,
затем водой, сушат и перегоняют- Выход продукта 1,2 г (50% от теоретиче-
ского); т. кип. 190° С.
Описана также перегруппировка а-окисей [138], вызываемая действием BF3:
CeHsCOCH-CIIC6H6
\ /
о
C6HftCOCIICHO
СеН5
Формилдезоксибензоин. Раствор 0,5 г трлне-халконоксида в 25 мл абсо-
лютного эфира 30 мин кипятят с 5 мл эфирата BF-,. Реакционную массу затем
встряхивают с водой it количественно образующийся формилдезоксибензоин
осаждают в виде медной соли.
2. Пииаколнновая и ретропинаколиновая перегруппировки *
Наиболее давно известным случаем перемещения алкильной группы в насы-
щенных системах является открытая фиттцгом перегруппировка двутретичного гли-
коля — нинакопл — в пинаколгш {139];
СНзх /СПз
сн/f fX'Jh
ОН он
сн3х
>С-СОСП3
СПя in
СНз
Перегруппировка имеет препаративное значение, так как с ее помощью можно
получить нс только трудно доступные иными путями кетоны, но и исходя из них,
трехзамещенпые уксусные кислоты. Катализаторами перегруппировки могут служить
как органические кислоты средней силы, так и минеральные кислоты.
Перегруппировка пинакона в пинаколин осуществляется нагреванием пина-
кона с б я. H2SO4. Выход пинаколина 65—72% от теоретического |140].
Принципиально таким же способом из ароматических гликолей типа бепзппна-
кона получают ароматические пннаколины:
СбПй\ /С6н0
>С—С< — > (СвНб)3ССОС6Но
Сйн./1 |\свП5
он он
Описан способ превращения беияиинакопа в бензпинаколин 1142] кипячением
с пятикратным количеством ледяной уксусной кислоты и 1% иода; выход 95—96% 1,1
теоретического.
Приведенное здесь подразделение на перегруппировки Вагнера — Меервейна н
пинаколиновые или соответственно ретропинаколиновые перегруппировки
основывается на классификации насыщенных перегруппировок по Ипго.1Ду{[141 I-
Часто многие из приведенных в подразделах 1 и 2 перегруппировок носят общее
название пинаколиновых плп ретропинаколпновых, а к перегруппировка')
Вагнера — Меервейна относят лишь некоторые особые случаи, например пере-
группировку гидрохлорида камфена в изоборнилхлорид (стр. 877).
ГГ ’п LI Т А™ Г1,лт С,.'. -1С1 /IO'Z>
[138] Н. О. House, J. Am. Chem. Soc., 76, 1235 (19,54).
[139] R. F i t t i g. Ann., 114, 56 (1860).
1140] G. A. IJ i 1 1, E. W. Flosdorf. Org. Synth., 5. 91 (192,5).
1141] С. K. Ingold, J. Chem. Soc., 1953, 2845.
[142] M. Gomberg, W. E. Г1 acli man n, J. Am. Chem. sor.. 49. 246 (1927);
cp. Org. Synth., 14, 2 (1934).
J, ПЕРЕГРУППИРОВКА ВАГНЕРА — МЕЕРВЕЙНА И БЛИЗКИЕ К НЕЙ
87!)
Осуществлена перегруппировка. смешанных жирно-ароматических пинако-
нов £143]:
СНз\ /С6Н6 /СвНэ
>С—С/ —> СНзСО—с<
сн/ | I хСвНь I ХСвВ5
ОН ОН СНя
2,2-Дифенилбутанон-З. Перемешивая, при 5° С к 300 мл концентрированной
Н2УО4 прибавляют 50 г 1,1-димстил-2,2-дифснилэтиленгликоля. После двух-
часового выдерживания при комнатной температуре смесь выливают на лед,
разбавляют водой до объема 3 л и очищают выпавший продукт перегонкой
в вакууме. Получают 38 г 2,2-дифепилбутанона-З; т. кип. 101 —103° С (0,3 мм
рт. ст.)\ т. пл. 40 — 41° С.
Образование из двух соседних третичных атомов углерода одного четвертичного
: и одного вторично, о называют ципаколиновой перегруппировкой, обратное превра-
f щение — ретропипаколиновой перегруппировкой:
Сч X С\
>С-С< C/1-C-C
/ с/
Классическим примером этой реакции является отщепление воды от пипаколи-
нового спирта, ведущее к образованию тетраметилэтилена:
СН3ч СН3ч /СНз
>С— СЛОН-СПз —> /С-С<
СН3Х ! сн/ \СНз
СНз
Теграметилэтилен, однако, никогда пс получается в чистом виде; в нем всегда
имеется примесь изомеров [144[.
Бепзиипаколиновый спирт (1,1.1.2-тетрафенилэтанол-2) также легко перегруп-
пировывается в тетрафснилэтилен. По Орехову [145], 2 г спирта на1ревают с 25 г
ацетилхлорида в течение 5 ч на водяной бане и выделяют углеводород выливанием
реакционной массы в воду.
3. Диеион-феиолытая перегруппировка
4,4-Дизамещенные циклогексадиепоны при обработке кислотами превращаются
Со смещением алкильной группы в замещенные фенолы:
Эга перегруппировка имеет много общего с перегруппировкой семпхннонов,
описанной Ауверсом и Циглером [146|. Ес можно рассматривать как аналог ретропи-
наколиновой перегруппировки, протекающей через мезомерный аллильный катион.
[143] IT. Е. Z a u g g, М. F г е i f е I d е г, В. W. И о г г о и, J. Org. Chem., 15,
1191 (1950).
[144] С. W li i t m о г е. L. Meunie г, J. Am. Chem. Soc., 55, 3721 (1933).
[145] A. 0 rpkhof f. Bull. Soc. chim. France [41, 25, 189 (1919).
[146] K. v. A ii w e r s. K. Ziegler, Ann., 425, 217 11921).
880
ПЕРЕГРУППИРОВКИ С ПЕРЕСТРОЙКОЙ УГЛЕРОДНОГО СКЕЛЕТА
Описано превращение 4,4-Диметил-1-кето-1,4-дягидронафгэлина в 3.4-диметил-
яафтолацетат [147J:
О ОСОСНз
3,4-Диметил-1-нафтолацетат. Смешивают 1,72 г диенона с раствором 0,5 г
H2SO4 в 35 мл ацетангидрида и оставляют на 5 ч при комнатной температуре;
за это время происходит перегруппировка. Реакционную массу затем осторожно
вливают в 250 мл холодной воды, выпадает 2,01 г бесцветных кристаллов',
т. пл. 88—90° С.
В аналогичных условиях Ингоффену удалось осуществить ароматизацию
кольца А в Д1’4 -холестадиеноце-З [148]:
4. Изомеризация углеводородов [149]
Нсницеску и Драгапом [150] изучено влияние Alds на алифатические угле-
водороды. н-Гексан уже прп кипячении в основном превращается в смесь изомерных
метилпентанов, незначительная часть дегидрируется до циклогексана.
Определенным завершенней многочисленных работ о влиянии катализатора
аа перегруппировку циклических углеводородов являются исследования Неницеску
и Кантуннари [151]. Ими показано, что чистый безводный Л1С1Я практически не дец-
ствует на циклогексан; только в присутствии воды может быть достигнута изоме-
ризация.
Метилцяклопентан. В течение 3 ч 1500 мл циклогексана кипятят с 500 г све-
жесублпмировавного А1С13 и 13,6 мл воды. Жидкую часть реакционной массы
промывают и сушат. При фракционировании получают 330 мл фракции с т. кип,
71,3—72,3° С, представляющей практически чистый мотилциклопентан.
В таких же условиях метилциклопентан, наоборот, даст смесь циклогексана
и метилциклопентана. В этом случае, следовательно, речь идет о равновесии между
обеими циклическими системами; на основании преломляющей способности равновес-
ной смеси установлено, что она содержит 23% метилциклопентана.
Превращение метилциклопентана в циклогексан использовали цля получения
14С-бенэола [152):
[147] R. Т. Arnold, J. S. Buckley, J. Richter, J. Am. Chem. Soc., 69,
2322 (1947).
[148] H. И. Inhoflen, Angew. Chem., 53, 171 (1940).
[149] Обзорная статья: L. Schmer] ing, Ind. Eng. Chem., 45, 1147 (1953).
[150] C. D, N e n i t z esc n, A. Dragan, Ber., 66. 1892 (1933).
[151] C. D. N e n i t 2 e s c u, P. Cantuniari, Bor., 66, 1097 (1933).
[152] H. 8. Turner, R. J. W e г n e, J. Chem. Soc., 1953, 789.
IU ПРОЧИЕ ПЕРЕГРУППИРОВКИ
881
14С-Бензол. Отщеплением воды от циклопептплметплоцого спирта при 320° С
пад окисью алюминия получают смесь олефинов, из которой гидрированием
получают 14С-метилциклопентан, содержащий 34% 14С-циклогексана (выход
94% от теоретического). Смесь цнклопарафинОв изомеризуют по Неницеску
и Кантуниари [1511 до 14С-циклогексана. Дегидрирование до 14С-бензола про-
водят на платиново-угольном катализаторе при 360’ С.
При ароматизации циклоолефинов может происходить миграция заместителей.
Например, дегидрирование 1-фонил-3,4-дигидроыафталина на силикагеле при 350е С
дает 2-фенилнафталин f 153]:
С9П5
х\А
и । - ►
V\/
z/X/X/^
Обнаружена также перегруппировка стероидного скелета в антраценовую
систему [154].
II. Прочие перегруппировки
1. Реакция Якобсена
В производных бензола, содержащих не менее четырех алкильных групп,
при сульфировании концентрированной серной кислотой или ^олеумом .происходит
перемещение алкильных групп [155]. Реакция не имеет существенного препаративвого
значения. Она применяется, например, для получения из смеси дурола и изодурола
(образующейся алкилированием по Фриделю — Крафтсу смеси изомерных ксилолов)
чистого пренитола [156]:
СНз СНз
HsC\zX\/CHs HsC\^\/CHs Н3СЧ А/СНз н,сД
II! + I 1| —> ( J —> 1| I
ПзС/^/^СНз НзС/у уХСП3 ^^СНз
СНз SO3H
2. Перегруппировка типа бензиловой кислоты
<. Одпой из наиболее давно известных перегруппировок является превращение
бензила в бензиловую кислоту под действием сильных щелочей [157]:
* , Cgllsx
) СдЩсососвЩ —> ;с-соон
Г CeH5Z I
г ОН
Бензиловая кислота (по Гаттерману — Виланду [158]). Кппятят на водяной
бане 5 г бензила в 15 мл этилового спирта с раствором 5 s КОН в 10 мл воды
• в течение 10 жаи. После охлаждения быстро отсасывают кристаллическую
[153] F. М а у е г, R. Schiffner, Бег., 67, 67 (1934).
[154] W. R. N е s, Е. М о s е t t i g, J. Am. Chem. Soc., 75, 2787 (1953); K. Alder,
B. Kruger, Chem. Ber., 86, 985 (1953).
[155] L. J. Smith, Org. Reactions, f, 370 (1947).
[156] См.»*, стр. 381.
[157] О. K. N e v i 11 e et al., J. Am. Chem. Soc., 77, 3280 (1955).
[158] Gatermann-Wieland, Die Praxis des organischen Chemikers, 33 Aufl.,
S. 204.
56 Заказ 18.45.
882
ПЕРЕГРУППИРОВКИ С ПЕРЕСТРОЙКОЙ УГЛЕРОДНОГО СКЕЛЕТА
кашицу бензиловокислого калия, промывают этиловым спиртом и растворяют
в 20—30 мл воды. После фильтрования бензиловую кислоту осаждают кипяче-
нием разбавленной H3S04. Выход продукта 4 а.
Эта реакция является общей для всех ароматических кетонов. В алифатическом
ряду она применяется лишь в отдельных случаях [159]. При проведении бензиловой
перегруппировки необходимо обращать внимание на то, чтобы бензил был свободен
от цианида. Установлено, что бензил в присутствии циан-ионов гидролитически рас-
щепляется иа^бензальдегид и бензойную кислоту [160].
3. Перегруппировка при окислении диалкнлацетиленов
При окислении дпалкилацетпленов иадуисусной кислотой наряду с расщепле-
нием тройной связи происходит образование диалкилуксусных кислот [161]:
С4Н9х
с4нвсоон <— с4н9-с=с-с4ив —> уснсоон
CiH9y
Эта перегруппировка является известной аналогией перегруппировки Вольфа-
Шрётера.
Ди-«-бутилуксусная кислота. Смешивают 20 г ди-н-бутилацетилена с 12%-ным
раствором надуксусной кислоты (3 моль) в ледяной уксусной кислоте. Смесь
оставляют на 10 сутои, охлаждая ее сначала. После удаления в вакууме уксус-
ной кислоты остаток подвергают переэтерификации смесью метилового спирта
и ЫС1 и перегоняют. Фракцию с т. ЦИП. 90” С (9 мм рт. ст.) омыляют в щелоч-
ной среде. Выход ди-и-бутилуксусной кислоты 12 г (54% от теоретического);
т. кип. 137е С (12 лм рт. ст.).
111. Перегруппировки при разложении диазосоединении
1. Перегруппировка Вольфа—Шрёгера
Шрётер установил, что при нагрсв'акии диазодезоксибензоина отщепляется азот
н образуется дифенилкетен [1624:
С0Н5-CNa-СО—С6Н5 (СвН5)йС=С«О
В диазокетонах после отщепления азота остаток R', находящийся у карбониль-
ного углерода, перемещается к соседнему атому углерода:
R-CNa—С—R' —> R—С=С=О
1) I
О R'
Дифенилкетен [163]. Раствор диазодезоксибензоина в бензоле медленно при-
бавляют по каплям в колбу КлаЙзена, помещенную в нагретую до НО3 С. баню.
При этой температуре отгоняется бензол, а дяазодезоксибензопв претерпевает
перегруппировку с выделением азота. Выход днфеяилкетена 64% от теоре-
тического.
[159] В. Хюккель, Теоретические основы органической химии, пер. сном
г. I, Издатиплит, 1955.
[160] W. D i i t h е у, Р. Scheldt, J. prakt. Chem., 142, 125 (1953).
1161} V. Franzen. Chem. Ber., 87, 1223 (1954).
J162] G. Schroter, Ber., 42, 2346 (1909).
16 3] L. I. Smith, H. H. Hoehn, Org. Synth.. 20, 47 (1940)
III. ПЕРЕГРУППИРОВКИ ПРИ РАЗЛОЖЕНИИ ДИАЗОСОЕДИПЕНИЙ
883
Описано |164] разложение диазокетонов путем УФ-облучения в очень мягких
условиях при 0° С.$Выходы около^90% от теоретического.
В качестве источника'' света используется лабораторная погружная
лампа (S 81), которая вмонтирована в охлаждаемый ледяной водой кварцевый
штуцер. Сосуд для облучения (емкостью 170 мл) помещают в сосуд Дьюара
со льдом или охлаждающей смесью.
Дифенилкетен. Раствор 4,4 г диазодезоксибензоина в 170 мл абсолютного
эфира облучают в течение 3 ч, одновременно пропуская через раствор слабый
ток азота. По окончании реакции удаляют эфир в токе азота и перегоняют
в вакууме дифенилкетен. Выход^продукта 3,5 г.
Мономерные кетены в большинство .случаев неустойчивы. Однако значенне
перегруппировки Вольфа — Шрётора заключается в том, что разложение диазокето-
нои можно проводить в присутствии воды, спирта или аммиака и связывать образу-
ющийся кетой в виде кислоты, эфира или амида.
Как было показано еще Вольфом [165], со-диазоацетофепон при нагревании
в присутствии соответствующих катализаторов и аммиака превращается, в фенил-
ацетамид.
После того как и-диазокетонЫ стали легко доступным классом соединений [166,
167), эта перегруппировка с образованием производных кислот приобрела большое
препаративное значение. По разработанному Арндтом и Эйстертом [168] способу
почти все карбоновые кислоты через их хлорангидриды могут быть превращены в со-ди-
азокетопы, легко перегруппировывающиеся в производные кислот, гомологичных
исходной кислоте:
> RCOCl -CH,N*-> RCOCHN, —► RCH,COOH
По Арндту иуЭйстерту [168], дИазокетоп в присутствии тонкораспыленного
серебра, меди или платины нагревают с водой, спиртом, аммиаком или амином до
окончания выделения азота, в результате чего образуется’Производное гомологичной
.кислоты. Как правило, предпочитают вначале получить соответствующий эфир или
амид и омылить его до кислоты.
Способ является общим, если в молекуле не имеется функциональных групп,
реагирующих^ диазометаном, например фенольных гидроксилов или карбонильных
групп.
2-Питрофеннлацетамид[168]. Прп50~г60сС 16г 2-нитробензоилдназометана
растворяют в 150 мл этилового спирта, смешивают е равным объемом 2 п.
раствора NH3, прибавляют 10 мл 10%-ного водного раствора AgNO3 п переме-
шивают смесь при 70° С до окончания выделения азота (2—3 ч). Затем ее раз-
бавляют водой, охлаждают и отсасывают .образовавшийся 2-нитрофенилацет-
амид. После перекристаллизации из этилового спирта т. пл. продукта 160—
161° С. Выход 10 з (66% от теоретического).
В случае а-нафтоилдиазометана [169] осуществлена с приемлемым выходом
прямая перегруппировка в кислоту.
а-Нафтилуксусная кислота. Раствор Зе а-нафтоилдиазометапа в 25лсд диок-
сана при 50—60° С добавляют к раствору 2 г окиси серебра и 3 г тиосульфата
натрия в 100 мл воды. Массу перемешивают до окончания выделения азота
(1 ч), охлаждают и подкисляют разбавленной HNO3. После перекристаллиза-
ции из воды выход а-нафтилуксусиой кислоты 1,3 г (49% от теоретического);
т. пл. 130° С.
[164] L. Horne г, Е. Spietschka, A. Gross, Ann., 573, 17(1951).
[165] L. Wolff, Ann., 394, 43 (1912).
[166] В. Eister t, в книге W. Foerst, Neuere Methoden der nraparativen orga-
nischen Chemie, Verlag Chemie, Weinheim, 1949, S. 378—388.
[167] W. E. В a c h m a n n, W. S. S tru we, Org. Reactions, 1, 38—63 (1947).
[168] F. Arndt, В. E i s t e r t, Ber., 68 , 204 (1935); 69, 1805 (1936).
[169] F. Arndt, B. Eister t, Ber., 68, 200 (1935).
56*
884
ПЕРЕГРУППИРОВКИ С ПЕРЕСТРОЙКОЙ УГЛЕРОДНОГО СКЕЛЕТА
Перегруппировка Вольфа — Шрётера была распространена [170] иа о-хинонди-
азиды, легко получаемые диазотированием о-аминофенолов с последующей обработкой
щелочью. Под действием света хинондиаэиды с отщеплением азота и сужением кольца
образуют кетены, которые сразу превращаются в кислоты, так как реакцию всегда
проводят в присутствии воды:
О
Реакция имеет большую область применения. Как хиноидное, так и аромати-
ческое ядро могут нести самые различные заместители, любое из колец быть гетеро-
циклическим. Например, из хиколинхиноидиазида получают индол-3-карбоиовую
кислоту:
О
Недостаток способа заключается в том, что реакцию кеобходимо проводить
в очень разбавленных растворах, иначе хинондиазид вступает в реакцию сочетания
с продуктом перегруппировки н образуется краситель. Хниондиазиды растворяют
в 100—1000 ч. смеси растворителей, состоящей из ледяной уксусной кислоты, воды
и дноксана в перемекком соотношении. Раствор затем освещают при охлаждении
льдом до отрицательной реакции на диазосоединение (реакция с флороглюцином).
6-Нитроннден-З-карбоновая кислота [171]. Раствор 15 г 6-нитронафтохинон-
1,2-диазида-2 в 4,5 л ледяной уксусной кислоты и 240 мл воды фильтруют
с Углем и в закрытых чашках Петри освещают при охлаждении льдом солнечным
светом или дуговой лампой. Если в растворе больше не обнаруживается диазо-
соединекие (примерно через 4 ч), его вновь фильтруют с углем и тотчас же при
' температуре бани 35—40° С упаривают до объема 500 мл. Выпадающую при
этом в виде кристаллов 6-нитроииден-З-карбоновую кислоту отсасывают и про-
мывают петролейным эфиром. Выход продукта 6,5 а, еще 2 г можно получить
из фильтрата. После перекристаллизации из ледяной уксусной кислоты т. пл.
продукта 188—189° С (раэл.).
, 2. Перегруппировки прп взаимодействии
карбонильных соединений с диазометаном [172]
Альдегиды и неспособные к онолизациИ кетоны реагируют с диазомётаном по
карбонильной группе. Из образующихся при этом «диазонийбетаинов» отщепляется
азот, и молекула может стабилизоваться двумя способами: а) «нормально» с образова-
нием эпоксида; б) с миграцией остатка от соседнего атома углерода; при этом в резуль-
тате перегруппировки образуется карбонильное соединение:
R\ CH.N. + (а}
>C-CH3-№N _Д >с—сн2
R'/ II В'-7 I -N, R'/ \ /
ОО- о
-N, j (б)
R'COCH2R
[170] О. Sils et al., Ana., 556, 65, 85 (1944); 579, 133 (1953); 583, 150 (1953); 593.
91 (1955).
[171] О. Sus, Ann., 579, 133 (1953).
[172] В. Ei start, cm. [166], стрАЗбб.
HI. ПЕРЕГРУППИРОВКИ ПРИ РАЗЛОЖЕНИИ ДИАЗОСОЕДИНЕНИЙ
885
В данном разделе рассмотрены только реакцни, протекающие по пути б. Эта
реакции могут идти при взаимодействии альдегидов с дказометаном либо путем пере-
мещения гидрид-иона с образованием метилкетонов, либо путем перемещения карба-
нионного остатка с образованием гомологов альдегида или продуктов его дальнейших
' взаимодействий:
R-CH2-CHO *— R—СН—СН2 —> R—СО—СНд
I
0"
1Какой из возможных продуктов реакции преобладает в реакционной массе,
зависят от природы остатка R и условий реакции. Присутствие метилового спирта,
например, способствует образованию метилкетонов [173].
Взаимодействие циклогексанона с диазометаном ведет к расширению кольца
до циклогептанона [174]:
Диазометаи получают непосредственно в реакционной массе из нитрозометил-л-
' толуолсульфамида.
Циклогептанон[174]. Смесь 49 г циклогексанона, 125 г нитрозометил-л-то-
луолсульфамида, 150 мл 95%-ного этилового спирта и 10 мл воды охлаждают при
перемешивании до О’С и к ней медленно прибавляют по каплям раствор 15 г
КОН в 50 мл 50%-ного этилового спирта. Скорость прибавления регулируют
так, чтобы температура не повышалась выше 20° С. Через 2 ч реакция окан-
чивается. Массу нейтрализуют 2 н. НСЭ по лакмусу и образовавшийся цикло-
гептанон выделяют через бисульфитнос соединение. Выход 33—36% от теоре-
тического.
3. Перегруппировки при взаимодействии аминов с HNOq
Реакция первичных аминов (как правило, с концевыми аминогруппами) с азо-
тистой кислотой часто приводит к перегруппировкам. Например, при взаимодействии
с азотистой кислотой н-дропиламина образуется смесь нормального и изопропило-
вого спиртов.
Эти реакции часто протекают с перестройкой углеродного скелета. Циклобутил-
метиламин при взаимодействии с азотистой кислотой превращается в циклопентанол
(расширение кольца по Демьянову [175]):
СН2—CH-CH2NH"
I I
СН2-СН2
I X CHs-CII-CJfeOH
хспон+ I I
I / сп,-сн2
СНз—Gtis
При
С—С-связь
Кроме того
реакции
этом к углеродному атому, несущему аминогруппу, перемещается
находившаяся в исходном соединении при соседнем атоме углерода,
в переменном количестве получают образующийся в результате нормальной
первичный спирт,
а также олефины.
Препаративное использование реакции
[173]
[174]
[175]
Е. Mosettig, Вег., 61, 1391 (1928); 62, 1271 (1929).
Th. J. de В oer, Н. J. В acker, Org. Synth., 34, 24 (1954).
H. Демьянов. М. Л у ч н к к о в, ЖРФХО, 35, 26 (1903).
88Q
ПЕРЕГРУППИРОВКИ С ПЕРЕСТРОЙКОЙ УГЛЕРОДНОГО СКЕЛЕТА
ограничено еще и тем, что при наличии в гидроароматическом ядре заместителя
образуются два изомера, например [176]:
Согласно давним Альдера с сотр. эта реакция расширения кольца с успехом
использована в ряду бициклических аминов. Альдер, Хартунг и Хаусман [177] при-
водят для получения бицикло [2,3,3]деканола следующую пропись.
Бицикло[2,3,3]деканол. К раствору 13 г гидрохлорида амина в 50 мл воды
при пропускании сильного тока водяного пара последовательно прибавляют
по каплям раствор 4,5 г NaNO3 и 4 г ледяной уксусной,кислоты. По окончании
реакции операцию повторяют с 2 г нитрита и 1,8 г ледяной уксусной кислоты.
Продукт реакции из дистиллята извлекают эфиром, промывают экстракт до
отсутствия амина разбавленной НС1, нейтрализуют NaHCOa и после отгонки
эфира вновь перегоняют с водяным паром в присутствии 10 мл 10%-ного рас-
твора NaOH. Отсасывают выпадающий из дистиллкта в виде хлопьев цикличе-
ский спирт. Выход продукта 7,5 г (71% от теоретического).
[176] Н. Arnold, Вег., 76, 777 (1943).
- [177] К. Alder, S. Hartung, G. Hausman, Chem. Ber., 89, 1977 (1956).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Ширным шрифтом выделены страницы, на которых приведены методики синтеза
^Адамса катализаторы 34, 54
? — синтез 777
Идажса и Мареела синтез 821
адипиновая кислота 749, 830, 834, 835,
F 837
| дегидратация 358, 359
Е декарбоксилирование 818
I дигидразпд 459
г- диимидазолид 506
Г дихлорангидрид 166, 236
Ь — ацилирование 770
¥ диэфир, реакции 85, 791, 844
ь '«-замощенная 844
F моноэфир хлорангидрида 236
Ьщпкновьт альдегид 835
Бипиновый ангидрид 359
Ё0кинеа катализаторы 39, 309
Евелаиновая кислота •834
иидоспир'ты 501
виды 443, 510, 541
Е восстановление 530
| реакции 373, 376, 507
Квины 407, 474
Г восстановление 519, 530
Ьлактоны, реакция с меркаптанами 552
Ьоаминобензол, аэосочетанне 406
шобевзол 545
Е восстановление 514, 529
реакции 738
la'-Азоизомасляная кислота 138
Г диметиловый эфир 136, 146
5 динптрил 136, 138, 146
^оксибепзол, восстановление 532, 545
Еаоксясоединения 412, 514, 534
• восстановление 532
Дометан, восстановление 515
йометины 412, 470, 791
йОсОедкнспия 497, 495
восстановление 514, 529
изомеризация 850
окисление 534
отщепление азота 792
аосочетание 400 сл.
окислительное 407
. с разрывом С —С-связи 510
Азотсодержащие соединения 372 сл.
алкилирование 463, 465
расщепление 680 сл., 693 сл.
Азоттрикарбоновая кислота 438
Азулен 798
Акридин
восстановление 23
псевдооснования 858
Акриловая кислота 368
бромирование 679
производные 861
реакции ИЗ, 552
хлорангидрид 237
эфиры 679, 705
— реакции 130, 280, 710
Акрилонитрил 710
реакции 103, 120, 124, 281, “07, 794
Акролеин
диэтилацеталь 355
реакции 118, 124, 280, 281, 704
Аланилглицип 425
Аланины 327 , 367 , 418, 485, 876
реакции 245
Ализарин 287, 328
N-Алкиланилипы 462
Алкилариламины 519
Алкиларялкетоны 307
изомеризация 852
Алкилариловые эфиры, расщепление 361
Алкилацилдисульфиды 617
Алкилацилмочевины 871
2-Алкплбензпмидазолы 449
Алкилбромнды 203, 207, 208, 209, 210
очистка 209
оптически активные 212, 213
Алкилгалогенвды 225, 242, 245
Алкилгвдразины см. Гидразины
Алкилгидродисульфиды 617
Алкилгипоиодит 131
Алкилгипохлориты 127
О-Алкилдитиокарбонат 627
Алкиленоксиды 262
Алкиленсульфиды 593
Алкилеятиомочевины 379
Алкилиденазлактоны 809
888
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Алкилиденацетоуксусный эфир 805
Алкилиденйалоновый эфир, реакции 711
Алкидиденциануксусный эфир 804
О-Алкилизомочевины 463
Алкилизотиоцианаты см. Изотиоцианаты
Алкилизоформанилид, реакции 809
Алкилизоцианаты см. Изоцианаты
Алкялиодиды 210, 213, 217, 220
Алкилирование металлов 634
i С-Алкилирование
алкилгалогенидами 754
— по Фриделю — Крафтсу 765, 772 сл.
олефинами 696
пиколинов 758
снйртамп 779 сл.
N-Алкилироваиие
алкилгалогенидами 413
алкилсульфатамп 463
арилсульфокатами 466
восстановительное 485
диаэометаном 510
кетоиами 490
иптрамидов 537
спиртами 492
уретанов 537
фосфорнокислыми эфирами 465
.О-Алкилирование
алкилгалогенидами 333, 348
диазоиетаном 342, 349
диметилсульфатом 340, 348
карбоновых кислот (солей) 348
толуолсульфонатами 341
фенолов 331, 335, 340, 697
S-Алкялирование 598, 630
Алкилмеркаптохиноны 552
2-(Алкилмеркапто)-циклогексилсульфиды
615
К-Алкил-К-нитрозо-ЬГ-гуанидипы, реак-
. ции 547 ‘
Алкилпараконовые кислоты 802
Алкилпиридилы, окисление 831
Алкилроданиды 201
Алкилстаннаны 662, 663
О-Алиилтионкарбопат 627
б-’-Алкилтиопурин 585
Алкилтиосульфаты 582
реакции 614
S-Алкилтритиокарбонат 627
Алкилфенилкетоны 729
Р-Алкил-8-фенилмеркаптопропяоннтрнды
552
Алкилфториды 196, 202, 218
5-Алкилфуран-2-карбоновые кислоты
842
Алкнл~р-хлорэтилкетоны 124
Алкилциклогексиламины, дезаминирова-
ние 319
Алкилциклопентаны 671
Алкииолы, галогенирование 148
Алкины (см. Ацетиленовые углеводороды)
Алкоголка сложных эфиров 352
Алкоголяты 333, 334
расщепление 826
Алкоксиальдегиды 280
галогенирование 178
Алкоксиацетали 280
Алкоксиацетилены 691
гидратация 271
Алкоксибромиды 130
Алкоксибромэтилены, дегидробромирова-
пке 690
Алкоксигалогенирование 130
а-Алкоксиизомасляиая кислота 318
а-Алкоксикарбоновые кислоты 824
8-Адкоксикетоны 281
р-Алкоксимеркаптаны 615
а-Алкоксинитрилы 711, 765
0-Алкоксипропионитрилы 281
р-Алкоксипропионовая кислота, эфирь
280
0-Алкоксистирол 284
2-Алкоксиэтиловые спирты 334
Аллены 686 сл.
Аллигера метод 549
Аллкламип 468
алкилирование 422
реакции 553
Аллилариловые эфиры 336
Аллилбензол 746
присоединение НВг 115
Аллилбромид 219
бромирование 96
гвдрооромирование 111
окисление 293
Аллилбромирование 130, 137, 138
а-Аллил-а-бромуксусная кислота, эфн
793
Аллилиодцд 205, 210
Аллилиатрий 365
Аллиловый спирт 52
бромирование 99, 100
восстановление 29, 31
обмен ОН на галоген 205
реакции присоединения 101, 553, 5.
Аллиловый эфир, дихлоргидрин 126
Аллилфеяиловыи эфир 336
Аллилфенолы 863, 864
N-Аллилфталимид 130
Аллилхлорид, реакции 96, 333, 696
Аллилхлорирование 136
Аллилцианид 765
реакции 707
Аллильная перегруппировка 137, 204, 41
849
Аллокоричная кислота, эфир 679
Аллопрегнандион-3,20, восстановлен
79
Аллопрегнанон-20 79
Альдазлны 475
2-Альдегидобензимидазол 317
Альдегидоспирты 712
гидрирование 69
Альдегиды 74, 88, 294 сл., 303 сл., 316 с
322 сл., 798, 824, 832, 833, 835, !
ароматические 75, 318, 776 сл., 81
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
889
льдегиды
восстановление 55, 56, 62, 63, 66, 68 сл.,
78 сл., 716
галогензамещенные 176, 297
дегидродимернзация 742
димеризация 313
диэтилацетали 99, 118, 176,177, 228, 423
ненасыщенные 318, 369, 681, 806 сл.
окисление 288, 292, 310
терпенового ряда 304
льдимины 323, 470, 777
реакции 556, 729, 730
льдозы, окисление 311
льдопьная конденсация 314, 706, 712 сл.
льдоновые кислоты 311
лактоны 718
дюминийоргаяические соединения 656
сл.
люмогидрид лития 28 сл.
приготовление раствора 61
мадори перегруппировка 856
МВД натрия 787
мпднны 375, 438, 460, 462, 468
гидрохлориды 462
реакции 799 ,
циклические 463
идокспмы 375
идразоны 408, 463
иды
галогензамещснных кислот 169
карбоновых кислот 327, 730, 731
— галогенирование 101, 163
— гидрирование 86, 88, 89
— омыление 321
— реакции 464, 468, 537
сульфокислот см. Сульфамиды
иЛазид 444
миламин 526
реакции 493, 543
ьмилацетофепон 769
1МИлбензол 747
реаиции 769
ет-Амилбромпл 2U3
чМИ.тгидразин 543
кМИлен 671
реакции 267, 553
йлмеркиптаи 608
1МИл-2~метплшп1орпдцн 493
щилнатрий 641
em-Амиловый спирт 267, 671
дегидратация 672
обмен ОН на галоген 203, 214
згилхлорид 203
инали 481, 482, 483
Жнирование 408 сл.
восстановительное 487
карбонильных соединений 480, 485
лин ©азобензол
восстановление 530
диазотирование 539
1*иноаэосоедпненпя 544
Амниоакриловая кислота, эфир 473
п-(Аминоалкил)-бензойные кислоты 418
Аминоа льдегиды
восстановление 62
галогенирование 178
1-Аминоантрахинон 511
2-сульфокислота 569
Аминоарилсульфохлориды 576
Аминоацетальдегид, диэтилацеталь 423
Аминоацетон 527
м-Аминоацетофенон 418
Аминобенза льдегиды
галогенирование 151, 248, 715
реакции 321, 825
2-Аминобензаль-п-толуидин, разложение
811
Аминобензойные кислоты
галогенирование 151, 171, 172
.гидрирование 48
реакции 255, 539
1-Аминобензол-4-сульфо-2-карбоновая кис-
лота 566
.и- Аминобензотрифторид 193
Аминобензофеноны, восстановление 79,
81
2-Аминобутап 486
Амниогептапы 486, 487
Аминогуанидин 376
2-Амино-3,5-Дниодбензойная кислота 172
п-Аминодиметпланилин 528
3-Амино-2,4-диметилпентан 487
Аминодифенилметаны 79, 81
Аминодифенилы 738
замена NH2 на галоген 253
2-АминО-4,6-диэтоксипиримидин 335
1 а-Аминододецилбензол 491
3-Амино-1~еиамяны 482
Аминоизомасляная кислота, нитрил 484
2-Амиио-5-иодбензойная Кислота 172 z
2-Амино-5-иодпиридин 189 /
2-Амино-5-иодпиримидин 189
е-Аминокапроновая кислота 76
бензоилирование 433
4'Амино-4-карбметоксистильбен 327
2-Амино-б-карбэтоксибензтиазол 191
Аминокетоны 415, 418, 498
реаицни 453, 492
Аминокислоты 327, 418, 425, 484, 527, 719,.
756, 797, 809
ацилирование 433, 452
галогенирование 169
защитные группы 369
ненасыщенные 473
реакции 244, 251, 457, 822
тиоэфиры 552
хлорангидриды 233
эфиры 85
— гидролиз 366
а-Аминокислоты 395, 402, 416, 485
декарбоксилирование 815
окисление 840
р-Аминокпслоты, производные 457
ю-Аминокислоты 417
о-Аминокоричные кислоты, реакции 794
890
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Р»Аминокротоновая кислота
реакции 474
эфир 473
Аминомагннйгалогениды 457
Аминомалоновый эфир
ацилирование 797
бензоилирование 434
а-Аминомасляная кислота 425
Аминомеркаптаны 415
2-Амино-1-метилбензол-5-сульфокислота
566
2-Амиио-6-метилбензтназол 191
Аминометилеищавелевоуксусный эфир
498 '
Аминометилировавие 785 сл.
1-Амино-2-метилнафталин, димеризация
739
4-Амияо-2-метилпентан 487
2-Аминометилфлуорен, реакции 323
2-Амино-4-мвтоксифенол 529
Аминонафтални, замена NH, на галоген
253
1-Аминокафталиндисульфокислоты, дезами-
нирование 320
2-Амиио-З-пафтойная кислота 497
1,4-Аминонафтол-2-сульфокислота 562
Аминопитрилы 483, 484, 719
•омыление 327
а-Амиво-со-нитроалканы 442
2-Амино-5-нитроанизол, дезаминирование
320
1-Амино-5-нитронентан 442
Аминопитропроизводные ароматические
389
2-Амино-4-нитрофенол, восстановление
524
5-Амиао-6-нитрохияолин 409
Аминооксибензолсульфокислоты 567
1-Амино-2-оксинафталиисульфокислота-4
573
З-Амино-4-окситолуод 523
2-Аминооктан 526
2-Аминопентан 526
2-Аиииопентанон-2 527
2-Аминопиридин 408
алкилирование 494
З-Аминопиридин 871 '
4-Аминопиридии 505
Аминопиридины, окисление 392, 535
Р-Аминопропионитрил, омыление 327
а-Аминопропяоновый альдегид 423
5-Амниорезорцин 495
Аминосалццпловая кислота
восстановление 844
роданировапие 257
Аминосахара ацилированные, реакции
224
Аминосппрты 69, 85, 415, 500
обмен ОН па галоген 215
реакция с HNO* 319
Аминосульфокислоты 483, 523 ।
галогенирование 171
5-Амииотетразол 376
п-Аминотиофенол 580
3-Амино-2,4,6-трибромбепзойная кислота
140
3-Амипо-2,4,6-трииодбенэойная кислота
171
1-Амино-3-трифтопметилбензол-4-сульфо-
кислота 572
Аминоуксусная кислота, хлорангидрид
233
Аминофенантрен, замена NH2 па галоген
. 253
п-АМинофепиларсоновая кислота 667
2-А1мино-1-фенилпропапы 414
а-,Амнно-а-фенилпропиойовая кислота 327
/ превращение в а-аминонитрил 484
It- Аминофенилуксусная кислота, эфирь
346
12-Аминофеаол-4~сульфокислота 567
1 Аминофенолы 527
N-алкилирование 464
диазотирование 884
реакции 309, 567, 600, 717
2-Аминофлуорен, N-алкилированис 465
5-Аминохинолин 521
реакцик 321
8-Аминохинолин 521
Амиио-о-хиноны 309
N-Амниохлорирование 550
Аминоэтиловый спирт, обмен ОН па гало
ген 207
Р-Аминоэтилсульфиды 602
2-Амино-4-этокси-6-метоксипиримпцин 33!
Аминоэфиры 335
Амины 680 сл., 513, 516, 520, 525, 53'
ароматические 427 сл.
— азосочетание 405
— алкилирование 465, 697
— галогенирование 156, 157, 158, 171
247, 249, 253
— гидрирование 49 сл.
— диазотирование 248 сл., 539
— реакции 190, 297/ 320, 545
окисление 533, 535
отщепление аммиака 680 сл.
первичные 417, 815
реакции 242 сл., 319, 320, 375, 492
508, 538, 630, 782, 785, 845, 885
третичные 87, 677, 688
— окисление 533
• формильные производные 845
Аммониевые четвертичные основания 411
423, 693, 706, 858
восстановительное расщепление 87
разложение 681, 682
реакции 603
соли 243, 858, 867
Ангидриды карбоновых кислот 282, 358 сл
как ацилирующие агенты 351
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
891
£ Анетол
? нитрование 382
присоединение родана 101
' ж-Анизидин 429
i нитрозирование 400
& п-Анизидин
N-алкилирование 464
; ацетилирование 451
- Анизидины, реакции переаминирования
Г 503
’ Анизоиланизидин 875
, n-Анизоилбензоилметан, нитрозопроиз-
• водное димерное 396
0-(п.-Анизоил)-пропионовая кислота 774
Анизол 340
галогенирование 154, 156
расщепление 363, 365
: реакции 774
Анилиды 446, 457
£ арилсульфокислот, алкилирование 465
бензоилуксусной кислоты 456
диэтилмалоновой кислоты 457
капроновой кислоты 457
фталевой кислоты 451
Анилин 623, 530, $22
алйилирование 463, 465, 486, 493, 494,
781
галогенирование 156, 157, 158
гидрирование 50
реакции 191, 472, 473, .503, 733
' сульфирование 565, 572
фосгенирование 439
Анилиновый черный 546
4-Анилинопиридин 500
Анилинсульфокислота 565
диазотирование 539
-Анилы 323, 422, 470, 472, 603, 730
реакции 733
Анисовая кислота, дезалкилирование 363
Анисовый альдегид, реакции 389, 715, 803
'Антраниловая кислота 302, 566
галогенирование 142, 169, 172
декарбоксилирование 822
эфир гераниола 353
Антрахинон 298, 703, 782
сульфирование 562, 569
сульфохлориды, восстановление 607
Антрахинонсульфокислоты 562, 570
реакции 328
Антрацен 735
восстановление 22, 23
галогенирование 106, 107, 142, 143
нитрование 386
окисление 298
• производные 811
реакции 191, 701, 704
сульфирование 569
Антрацен-9,10-дихлорид 106
Антролы, простые эфиры 331
Антрон 771
дегидродпмерпзация 741
Арабиноза 82.5
восстановление 66
Аралкилгидразины 426
Арахидоновая кислота, присоединение бро-
ма 102
Аргинин 463
Арилазиды 542
реакции 544
Арилалкилкетоны 307
изомеризация 852
0-Арилакриловые кислоты, восстановле-
ние 28
N-Ариламниометилфталимиды 483
N-Ариламиноииридины 502
Ариларсоновые кислоты 667
Арилбензойные кислоты 776
Арилборгалогениды 654
Арилгидразины см. Гидразины
Арилгидразоны см. Гидразоны
Арилгядроксиламины см. Гидроксиламиньь
замещенные
Арилглиоисалевая иислота, эфир 776
Арилиэотиоцианаты см. Изотиоцианаты
Арилимидоэфиры см. Имидоэфиры
Арилирование
аминов 427 сл.
дитиокарбамат^в 630
металлов 634
Арилмасляные кислоты 80
N-Арил-а-нафтиламины 503
Арилоксибромзтилены, дегидробромиро-
вание 690
Арилоксиметансульфокислоты, галогени-
рование 258
Арилоксиметансульфохлориды, отщепле-
ние SO2 250
Арилолефины
озонирование 835
присоединение формальдегида 707, 708
N-Арилпиперидины 512
Арилпропиоловые кислоты, эфиры 692
Арилроданиды см. Роданиды
Арилстанианы см. Станнаны
Арилсульфонилгидрохиноны 556
Арилсульфохлориды, отщепление SO8 258
Арилтиосульфонаты 556
расщепление 620
Арилтиоцианаты см. Роданиды
Арилтрихлорсиланы 660
Арилуксусвые кислоты 776
N-Арилформимидоэфиры, перегруппиров-
ка в форманилиды 462
Арилфториды 253
Арилэтилены, окислительное расщепление
833
Арины 429
Аристол 152
Арндта — Эйстерта синтез 883
Ароматические углеводороды 810, 881
алкилирование 696, 779, 780
ацилирование 768 сл.
бромметплирование 783
галогенирование в ядро 139, 140, 142,
154, 155, 156
— в боковую цепь 134,141, 142, 145 сл.
892
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Ароматические углеводороды
— присоединением 104 сл.
> гидрирование 47 сл.
нитрование 385
оиислеиие 830 сл.
окислительное расщепление 841
разделение смесей через сульфокисло-
ты 89
сульфирование 89
фторирование 253
хлорметилирование 782
хлорэтилирование 783
Арсаинловая кислота 667
Арсиновые кислоты 667
Арсины 665 сл.
Арсониевые соединения 665
Арсонистые кислоты 668
Арсоцовые кислоты 635, 667
Аскаридол 261
Z-Аспарагин, дезаминирование 319
Аспарагиновая кислота 417
Аутроиисление 260, 261, 289, 290
борорганических соединений 655
Аценафтен 29
окисление 285, 297
Аценафтеиол 285
Аценафтенхинон 257
1-Аценафтилуксусная кислота 853
Ацетали 99, 118, 160, 228, 275, 280, 283,
353 сл., 423
галогенирование 176, 177
гидроксилирование 275
гидролиз 369
окисление 292
реакции с хлорангидридами 228
N,N-Ацетали 481, 482, 483
Ацеталирование 353 сл.
Ацетальдегид 269, 827, 851
ацетали 283, 354
галогенирование 174, 175
дяизоамилацеталь 283
димеризация 314, 712
реакции 241, 313, 404, 785, 808
Ацетальдегидсульфокислоты 555
Анетальдегпдцияпгидрин, реакции 782
Ацетальдоксим, реакции 733
Ацетальдоль 712
Ацетамид 446, 455
Ацетамидин 461, 462
2-Ацетамидо-1-фенилбуганон-3 454
Ацетанилид
алкилирование 420
ацилирование 770
нитрование 387
Ацетилампноацетоуксусный эфир 526
Ацетиламкнобензойная кислота 302
4-Ацетиламинобензолсульфокислоты,
N-арилзамещенные 622
Ацетиламацомалоновый эфир 526
ациламипометилирование 797
п-Ацетиламино-п'-питродифенилсульфон
691
п-Ацетиламинотиофенол 607
N-Ацетиламиноуксусная кислота, реакции
809
4-Ацетидамино-4-цианстильбен, омыление
327
1-Ацетилаценафтен, изомеризация 852
Ацетилацетон
изомеризация 863
реакции 606, 778
Ацетилбензоила перепись 293
Ацетилбромид 236
реакции 765
. Ацетилванилин 833
8-Ацетилгексанон 324
р-Ацетилгликозиды 337
Ацетилгликозилгалогениды 224
N-Ацетилглицин 451
реакции 809
а-Ацетилглутаровая кислота, эфир 705
N-Ацетилглюкозамин, реакции 224
Ацетилдибенэоилметан 760
Ацетилен 693
галогенирование 103, 148 сл.
гидрогалогепироваиие 120 сл.
полимеризация 683
производные 272, 745, 785 *
реакций 101, 283, 710, 714, 745, 772,
827
а,Р-Ацетиленамиды 271
Ацетилеидикарбоновые кислоты 691
гидрирование 52, 55
галогенирование 104
Ацетилениды металлов 654, 745, 746
разложение 53
Ацетилениарбоиовые кислоты 691
галогенирование 148
гидратация 270
эфиры 746
Ацетиленкетоны '762, 763
гидратация 271
а,0-Ацетиленнитрилы, гидратация 271
Ацетиленовые спирты 752, 827
галогенирование 162, 219
гидратация 271
расщепление 827
этерификация 347
Ацетиленовые углеводороды 688 сл.
галогенировапие 148
гидратация 269
гидрирование 52 сл.
гидрогалогенирование 120 сл.
реакции присоединения 123, 124, 131,
654, 658, 663
циклические 692
Ацетилизоцианат 445
Ацетплиэоэвгенол, окисление 833
N-Ацетилимидаэол 458
Ацетилкарбазолы 863
'Ацетилмалоновый эфир в синтезах амино-
кислот 756
а-Ацетилмеркапто-а-фепилацетои 581
2-Ацетилмеркацто-З-хлорбутан 554
N-Ацетилморфолпн 458
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
893
’ 2-Ацетилнафталпн, реакции 839, 853
L М-Ацетил-а-пафтвдамин, азосочетание 406
, 5-Ацетилноцапдион-2,8-карбоновая-5 кис-
лота, эфир 705
i 2-Ацетилпиррод
восстановление 64
? сульфирование 571
' Ацетилтиазол, восстановление 60
г З-Ацетилтиооксиндол 799
2-Ацетилтиофен 774
восстановление 64
[. N-Ацетил-3,4,6-три-О-ацет гл-a-D-r лк>-
коааминилгалогенвд 224
Адетилфенилацетилен 7бЗ
. 2-(п-Ацетилфенил)-хинои 794
Адетцлфуран 768.
2-Ацетил-5-фурансульфоиат бария 571
. Дцетилхлорид, реакции 237, 768, 769,
г 771 сл., 873
* Ацетилцианид 765
i 1-Ацетидциклогексанол 272
Д-Ацетилциклогексеи-1 772
N-Ацетилцистеамин 602
Адетилэтилеидиамин 421
Ацетимидобенэиловый эфир, реакции 463
. р-Ацетнафталид, металлирование 420
Адетобромглюкоза 223, 259
гидролиз 315
, _ превращение в глюкозид 337 '
?: S-глюкозид 585
"Адетовератрон, реакции 866
а-Ацетогалогенозы 222
синтез гликозидов 337, 338
^Ацетоины, восстановление 78
Адетоиодсахара 224
о-Адетоксиацетоуксусныи эфир 285
нН-Ацетоксибеязальбромид 147
!Адетоксикетоны, гидролиз 315
[Ацетоксималоновый эфир 285
:Ацетоксимезитилен 784
^Ацетоксиметилирование 784
(tt-Ацетоксимеркури-а'-оксимеркуритиофен
652
Ацетоксициклодеятены 286
Ацетомезитилен 775
, Ацетон
анил 472
ацетали 346, 354
/ гидразон 474
2,4-динитрофенилгидрааоя 431
оксим 536
расщепление 828
К реакции 68, 178 сл., 285, 404, 486, 681,
I/ 712, 714, 718, 784 , 804 , 806 807,
F 810
К тиосемикарбазон 477
кАцетонафтеноп 771
f Ацетондикарбоноаая кислота 823
реакции 394, 404, 820
( эфиры 346, 511
[ Ацетониды см. Изопропилидоновые произ-
водные
Ацетоннлацетон 779, 820
Ацетонитрил, трнмеризация 376
Ацетоноксим 480, 536
Ацетонциангидрин 718
реакции 383, 385, 484, 676
Ацетотолуидниы, окисление 302
Ацетофенон 774, 775
разложение 768
реакции 285, 295, 302, 390, 479, 490,
560, 595, 672, 754, 756, 785, 788,
790, 792; 810, 817
со-сульфокислота 560
фенилгидрааон 475
— реакции 866
Ацетофгорсахара 22
Ацетохлоргалактоза 223
Ацетохлорглюкоза 222, 223
Ацилазиды 542
Ацилгалогениды 176, 231 сл., 687
Ацилгидразины, алкилирование 426, 427
Ацилгипогалогениты 140, 149
N-Ацилимидазол, переамидирование 506
Ацилирование
аминов 431, 432, 445, 450, 510
аминокислот 433, 452
ангидридами кислот 450, 774
ароматических соединений 768 сл.
гидродсиламина 437
имидоэфирами 462
карбоновыми кислотами 445, 775
меркаптанов 589
ортоэфираыи 460
по Фриделю — Крафтсу 769 сл.
спиртов 349
фенолов 349, 767, 770, 775
хлорангидридами 349, 359, 770 сл.
циклоолефинов 772
циклопарафинов 771
эфирами 454, 463
Ацилоиновая конденсация 715
Ацилоины 274, 715
реакции 492
Ацилоксибромиды 130
N-Ацилпиридинин, соли 437
N-Ацилпиразолы, восстановление 89
Ацилсульфенилхлориды 617
N-Ацилфтацимйды 438
Ацилцианиды 765
ацилирование аминов 510
омыление 327
Барбитуровая кислота 459
эфиры, алкилирование 757
Барбье — Виланда метод окислительного
расщепления олефинов 836
Барта реакция 635, 667
Баудиша реакции 398
Бекера — Венкатарамана перегруппи-
ровка 864
Бекмана
окислительная смесь 303
перегруппировка 526, 874
Бензазид, расщепление 872
Бенаальазин 475
894
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Бенаальанилин (Бензилиденанилин) 472
восстановление 519
нитрование 388
реакции 556, 731, 734
Бензальаитраниловая кислота, реакции
556
Бензальацетофенон (Бензилиденацетофе-
нон) 769. 806
реакции 373, 686, 707, 710, 711, 722, 723
эпоксидирование 265
Бензальбромид 147, 247
Бензальгалогениды, гидролиз 316
1,3-Бензальглицерин 358
Бензальдегид 75, 89, 323, 882
ацетали, гидролиз 371
«-бензолазофеяилгидразоп 404
восстановительное аминирование 486,
487
восстановление 80
гликольацетали 292
ди-га-бромфеиилмеркапталь 596
ди-«-бутилацеталь 356
Диэтилацеталь 355
замена кислорода на га-логен 240 сл.
нитрование 389
окисление 264, 293
оксим, восстановление 526
O.N-полуацетали 378
реакции 357, 471, 472, 481, 483, 490,
713, 714, 715, 718, 801—803, 806,
808—810
сульфирование 567
фенилгидразон, окисление 534
Бензальдегид-3,5-дисульфокислота 567
Бенвальдегид-о-сульфокислота 590
р-Бензальдоксим 396 .
Бензальмалоновая кислота 803
эфиры 280, 711
Бензальметиламин 471, 518
Бензаль-а-метилглюкозид 371
Бенаальфлуорен, восстановление 24
БензальхлОрид 145, 146, 147, 240
Бензамид, реакция 507
Бензамидоксим 375
Бензанилид 433, 446, 874
1,2- Бензантрацен, производные 811
Бенааитрен, реакции 701
Бензантрон 736
Бензгидразид 507
Бевзгидрил-Р-хлорэтиловый; эфир^ЗЗО
Бензгидроксамовая кислота 460
Бензгидрол 59, 715
реакции 329, 780
Бенздиоксап, галогенирование 186
Бензедрин 414
Бензидин 865
гидрохлорид 866
Бензидиновая перегруппировка 865
Бензил 307. 324
бромистый 142, 147, 258, 783
гидразон 474, 686
изомеризация 881
хлористый 145, 14G, 147
Бензилазоксибензол 534
Бензиламины 416, 486, 490, 526
ацилирование 450
метилированный 868
реакции 323, 493
Бензиламин-4-карбоповая кислота 517
2-Бензиламинопкридин 494
N-Бензиланилии 418, 519
N-Бензилацетамид 450
Бензилацетат 285
Бензилбензоат 314
Бензилгалогениды
гидролиз 315
реакции 318, 764
О-Бейзилгидроксиламин, ацилирование 449
Бензилгликольальдегвд 315
Бензилдйметилсульфонийбромид, изо-
меризация 868
З-О-Беизил-1,2,5,6-ди~О-изопрО1гилиден-
а-Д-глюкоза 339
Бензилиден хлористый 145, 146, 147, 240
Бензилиденанилин см. Бснзальанилин
Бензилиденацетон 806
восстановление 60
реакция с тиофенолом 551
Беняилидеыацетофенон см. Бензальацето-
феноп
Бензилиденбутанопы 806
4,6-Бензилиденглюкоза 357
4,6-Бензилиден'2-дезокси-1-галактонитрил,
гидролиз 370
Бензилиденмалоновая кислота 803
З-Бензилиден-5-метилоксазолон 809
Бензилиденовые производные углеводов
356
Бензилидеициклогексан 813
Бензилинден 24
Бензилкалий 642
Бензилкарбаминовая кислота 369
Бензилкетоны 851
Бензиллитий 639
Бензилмагнийбромид, реакции 869
Бензилмагнийхлорид, реакции 639, 869
Бензилмеркаптан 608
реакции 588, 596
Бензилмеркаптолы ароматических кетонов,
расщепление 616
S-Бензнлметилизотиуронийхлорид 563
Бензилметиловый эфир, расщепление 362
бензнлметилуксусная кислота, реакции 875
N-Бензилморфолин 467
N-Бенаил-а-нафтпламин 494
Бензиловая кислота 881
восстановление 76
Бензиловые снирты 285
окисление 303, 304
эфиры 331
Бензиловые эфиры, изомеризация 865
Бензиловый спирт 315
восстановитель 31
восстановление 77
реакции 205, 780
ИР ЕД М СТ 11ЫЙ У К Л 3АТЕ ДЬ
85
1-Бейзилпиридин 493
Бензилсульфинат. реакции 591
Бензилсульфоцнслота 590
Беязилсульфохлорид, отщепление SO2 258
Л-Бепзил-л-толуидин 487
Бецзилтрпметиламмопий, гидроокись 707
Бензилтриметиламмонийиодид, изомери-
зация 868
Бензилфениловые эфиры, расщепление 365
Бензилфенилсуяьфид, окисление 612
Бспзилфенилсульфоксид 612
' Бензилфлуоренон 738
Бензилфталпмид 416
Бензилхлорид 258, 783
замена хлора алкоксилом 333
Бензилцианампд 436
Беизилцианид
гидрирование 323
реакции 713
а-Бензилэтиламия 875
Бензимидотиофениловый эфир 557
Бепэимидхлориды 860
’ Бензкарбазолы 497
Бензоила перекись 288
перекристаллизация 263
6-Бензоилакриловая кислота, реакции 119
. Бепзоиламин 242
2-Бензоиламино-«-бутанол 432
' е-Бспзоиламинокапроновая кислота, га-
логенирование 162
Г- Бензоиламипомалоновая кислота, эфир 434
е-Бензоиламино хлорка ироновая кислота
162
1-Бенэоиламино-5-хлорпептан 860
Бензоилацетилен, гидратация 271
Беизоилацетон 792
алкилирование 757
• ; бензоилирование 759
• ' нитрозопроизводное димерное 396
реакции 760
. Бензоилацетоуксусный эфир 759, 845
!,1-Бензоил-8-бензилпафталин, дегидриро-
вапие 738
о-Бензоилбензонпая кислота 774
‘ тргт-Бутиловый эфир 279
!‘ВенэоилглИцил-Р, Z-лейцинамвд, омыле-
ние 321
Бензоилизоцианат 445
Бензоилирование 434
аминокислот 433
оксисоединений 350
Бензоилкарбияол 854
ацетат 285
Бензоил-2),Z-лейцин 321
а-Бензоил-а-метилацетоуксусный ЭФИР,
расщеплопие 845
<В-Бензоил-(о-мстилмеркаптоацетофснон
868
1-Бспзоил-4-ме гилппперпдин 243
Вензоплмуравышая кислота 832
Бензоилнафталин
восстановление 80
дегидрирование 736
N-Бензолл-а-нафти ламин, азосочетани
406
Бензоилнитрат 382
Бензоплииперидины 798 *
изомеризация 860
реакции 415
Р-Бензоилпролиопнтрпл 795
гидрирование 488
р-Бензоилпропионовая кислота 774
восстановление 80
Бензоплуксусная кислота, анилид 456
Бепзоилуксусш.ги эфир 845
нптрозирование 394
Бензоилфенилацетиден 763
гидратация 271
И-Бензоил-К'-фенилмочевина 378
Бензоилфторяд 231
Бензоилхлориц
реакции 363, 760
сульфирование 577
1'Беизоил-2-циан-1,2-Дигидрохинолин 71
75
Бензоилциапид, реакции 511
S-Бензомлцистеамни 589
К-Бензоил-Л-эфедрин 511
Бензойная кислота 299, 302, 319, 775, 839,
882
ангидрид 359, 360
галогенирование 173
«оксипитрование» 380
сульфирование 567
эфиры 78, 84
— фениловый 347
— этиловый 345, 363, 723, 788
Бензоин 715
оксим, восстановление 526
реакции 592
фенилгидразон, расщепление 324
Бензоиновая (ацилоиновая) конденсация
715
Бензоины, окисление 307
Бензол
алкилирование олефинами 696
галогенирование замещением 142, 143,
144, 145
— присоединением 105, 106
гидрирование 25, 45, 47
меркурирование 653
меченный 14С 880, 881
нитрование 382, 383
окисление 298
3-Бензолазо-1,5-дифенилформазап 404
2-Беизолазо-4-метоксифенол, восстановле-
ние 529
Бснзолазоформазаны 404
Бензол-1,3-дисульфокислота 564
реакции 328
Бензол-1,4-дпсульфокислота 602
Бонзолсульфеновая кислота, диметила мп л
623
Бензолеульфинамид 622
Бепзолсульфиноная кислота 578, 601, (ИО
ангидрид 624
896
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
n-Бензолсульфоарсоновая кислота 602
Й-Бензолсульфо-РГ-ацетилатилендиамин
421
Брнзолсульфодибромамид 130
Бензолсульфокислота 563, 673
ангидрид 621
алкиловые эфиры 620
ариловые эфиры замещенных 621
галогенирование 151
Бензолсульфонат, реакции 328
Бенэолсульфохлориды 576, 601, 624
Бензолтрцсульфокислота, реакции 328
Бензонитрил 860
нитрование 381, 390
реакции 557
тримеризация 377
Бензонитрилы о-дизамещенные, гидролиз
326
Бензотрифторид, гидролиз 319
Бензотрихлорид .302
Бензофенон 715, 775
азин 474
восстановление 57, 59
' гидразон 474, 548
дииетклкеталь 355
оксим, восстановление 526
оксимирование 479
реакции 470, 472, 687, 806, 812
Бензофенонанил 472, 603, 730
Бензофеноноксим, перегруппировка Бек-
мана 874
' Бензофенонхлорид,' обмек хлора на серу
586
о-Бензохинон
восстановление 65
реакции 702
Бензохиноны 298, 309
Бензоциклооктанон 771
Бензпинаколип 878
Бензпинакон 715
изомеризация 878
Бензпинацоиовый спирт, дегидратация
679
Бензпирен, роданнрование 191
Бензтиазол-2-сульфокпслота 611
Бензтриазолы 541
галогенирование 143
Бензтрифторид 192
Бензтрихлорид 192
Бензфуран, галогенирование 186
Бергманна карбобензоксиметод 369
Бериллнйорганпческие соединения 642
Бетаин 719
Бетаиихлорид 232
Бешана реакция 667
Бис-(алкилксантогепы) 630
хлорирование 626
1,5-Бис-(бензоилиеркапто)-пентан 589
о,о'-Бис-бромметилдифепил, дебромпрова-
ние 745
Бис-диазоаминосоединения 544
2,3-Бис-диб1)Ом.мст11лнафталин, гидролиз
317
Бие-(2,7-днбромфлуоренияидед) 800
Бнс-(а,0-дибромзтиловый) эфир 222
Бис-1,3-дикетоиндаинлиден 800
4,4'-Бис-(диметиламино)-ааобензол 528
Бис-(3,4-диметонснфенил)-ацетонитрил
781
Бис-изонятрозоацетон 394
2,2'-Бис-(Р’Карбоксиаинилфенил)-дисуль-
фид 600
1,2,4,5-Бис-(о-ксилилен)-гексагидро-си.м.м-
тетрааин 483
1,32-Бис-(п-метоксифецокси)-дотриакоп-
тап 748
Бис-(р-нафтилметил)-сульфид 583
Бис-п-ннтрофенилсурьма, трихлорид 669
К,Ь7-Бис-П1-оксиэтил)-гидроксиламин 501
Бисульфитное сульфирование 592
Бисульфитные соединения 481, 555, 558
реакции 483
Бис-сульфоновые эфиры гликолей, алкили-
рующие агенты 467
Бис-(тиобензил)-метан 588
Бис-(флуоренилиден), окисление 832
Бис-(п-хлорбензил)-сульфид 584
Бис-(хлорметил)-дисульфид, хлорирова-
ние 619
Бис-(а-хлорметиловын) эфир 221, 222
Бис-(о-хлорфенил)-формамидин 461
Бис-(л-хлорфенил)-формамидин, реакции
812
Бис-(Р-хлорэтил)-ами1г, гидрохлорид 215
Бис-(р-хлоратил)-дясульфид, хлорирова-
ние 619
Бпс-(а-хлорэтиловый) эфир 221
Бпс-циклопентилиден, изомеризация 877
878
Бирненбаха — Губо — Уотерса способ
галогенирования 140, 141, 145
Бицикло (2,3,3/деканол 886
Блана правило 358, 818
Бланксма синтез дисульфидов 582
Бобру реакция 457
Бородина — Хунсдикера реакция 140, 819
Боргидриды
комплексные 65 сл.
щелочных металлов 86
Борнеол 65, 279
Борорганические соединения 654 сл. ‘
Брауна реакция 242
перегруппировка 860
Бринтцингера метод 516
а-Бромадипиновая кислота, диэфиры 168
а-Бромакриловая Кислота, эпгловып эфир
678
а,со-Бромалканы 208
а-Бромалкиловые эфиры 228
1-Бромалкины 149
Бромаль, окисление 166
Бромальдегиды 160, 176 сл.
М-((|)-Бром-в-амил)-анилпн 244
З-Бром-4-аминотолуол, реакции 321
N-Бромамииы 550
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
897
г Броманглдрпды карбоновых кислот 236
К Бромапизолы 156, 249
№ о-Броманилин, N-алкплированпе 464
Е п-Броманилин 156, 157, 158
к Бромантрахиноны 257
к- 9-Бромантрацен 143
В Бромацотальдегид 176
к диэтилацеталь 99, 176
к. сульфокислота 555
К N-Бромацетамад 128, 136
Е реакции 308
Г а-Бро.чацетилбромнд, реакции 435
В 1-я-Б])Ом-№-ацетил-3,4.6-три-О-ацетил-
> 1>-глюкозамип 224
К Бромацетон 179, 181, 182
К Бромацетонитрил 244
Г Бромацетоуксусные эфиры 179
В w-Бромацетофемон 179, 183
I’ . реакции 586, 753
Еха-Бромбензальацетофенон, реакции 281
п-Бромбемзальбромид, гидролиз 316
В м-Бромбензальдегид 248
к- и-Бромбензальдегид 147, 316
Р дегидрирование 799
к- а-Бром-р-бензоилпропионовая кислота 119
। я-Бромбензоилуксуспая кислота 269
К Бромбензойные кислоты 173, 319
R- Бромбензол 143, 744
к реакции 721
Г' хлорирование 199
К- п-Бромбензолсульфиновая кислота 578
|; ангидрид 624
Е л-Бромбензолсульфокислота 564
к; п-Бромбензолсульфохлорид 576, 601
в п-Бромбензотрлхлорид, гидролиз 319
К- o-Бромбензофеноц, восстановление 78
t’ Бромбутаны 114
L изомеризация 858
В 1-Бромбутанол-2 125
Е. 2-Бромбутаноп-З 181
В'БрОмбутплсны 116, 205
| а-Бромбутдролактон' 166
К- у-Бромбутпронитрил, алкилирующий агент
Г 424
Г а-Бромвалеральдсгид 176
F 6-Бромгексанол-1 206
В Бромгоксены 122
Г 1-Бромгексин 1-149
f а-Бромгептаналь 176
Г, Бромгептапы 112, 199
[ а-Бромглутаровая кислота, диэфир 168
® 2-Бромдсцен-1, реакции 690
Р-Броадигидроэвгенол 118
; Бромдикарбоновые кислоты, диэфиры 165,
168
о-Бромднметпл'анилин 464
n-Бромдиметпланилин, реакции 406
а-Бром ипн'тплацеталь 177
о-Бромдпфеннлмвтан 78
З-Бром-М-дифепилпропаион 246
а-Бромдодекановая кислота 164
а-Бромизовэлеральдегид 176
а-Брэмпзовалерпаповая кислота 164
57 Заказ 1835.
р-Бромизомасляная кислота, метиловый
эфир 119
а-Бромизомасляный альдегид, алкилиру-
ющий агент 424
а-БромизокапронОвая кислота 164
Бромирование
аминов 249, 252
гетероциклических соединений 186 сл.
кетонов 178 сл.
КИСЛОТ 161 сл,
лактонов 166
нитрилов 169
• нитросоединений ароматических 173
оксикислот 169, 186 сл.
полуацеталей 221
простых эфиров 156, 164, 165
спиртов 203, 204, 205, 207, 212. 213,
216, 218 сл.
сульфокислот 257 сл.
углеводов 223, 224
фенолов 150, 151
циклепов 683, 692
е-Бромкапронптрпл, алкилирующий агент
424
2-Бром-З-карбметоксипиразин ,252
n-Бромкарбобензокснаминокислоты 434
Бромкарбоновые кислоты 119, 824
расщепление 873
эфиры 709
Бромкетоны 245, 246
Бромкетоэфиры 858
х Бромкротоновые кислоты 678
эфиры 166
Бромксилолы 143, 321
реакции 317, 668
Броммалоновая кислота 167
Броммасляные кислоты 119, 165
гидролиз 315
эфиры 165, 224
Броммезнтилен 143
о-Бромметилбензойная кислота, реакции
771
N-Бромметилбутиламип, циклизация 410
З-Бром-З-метилгексеп-5, дегидрогалогени-
рование 682
Бромметилироваиие 147, 783
2-Бром-2-метилпептанон-4 118
Бромметилпиридины 230, 252
1-Бром-2-метилпропен-1 677
З-Бромметилтиофен 187
4-Бромметилхииолин 190
З-Бром-4-метоксиацетофенон 183
а-Бром-р-метоксивалерпановая кислота
130
8-Бром-9-метоксигспталецилхлорид 680
2-Бром-5-метоксигидрокоричиая кислота,
реакции 771
4-Бром-7-метоксииядапон-1 771
а-Бром-р-метоксимасляпая кислота, эфир
130
4-Бром-1-метоксипаф1алин 144
а-Бром-6-метокснпропионовая кислота,
эфир, алкоксибромироваппе 130
898
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
4-Бромминдальная кислота 855
Броммуконовая кислота, окисление 104
а-Бромнафталиц 144
реакции 764
Вромнафгалипсульфохлориды 576
4-Бромнафтол 144
а-Бромнитрилы 169
О'Бромнитробензол 249
«-Бромнитробензол 173
2-Бром-4-нитротолуол 174
2-Бром-6-нитрофенол-4-сул1.фокпслота 257
а-Бром-0-оксимасляная кислота 128
Бромоктаны 199, 216
Бромоформ 200
15-Бромиектадеканол-1 117
Бромпентаны 112, 114, 203, 219
4-Бромпентен-2~овая кислота, эфиры 166
2-Бромпиридин 188, 252
•4-Бромлиридин 252, 230
•а-Бромпробковая кислота, диэфиры 168
1-Бромпропапол-2 125
Бромпропаны 112
(О'Бромпропилбензол 115
•а-Бромпропионилбромид, ацилирование
аминов 435
1У-(а-Б£омпрО1И10пил)-г лицин, аммонолиз
Бромпропионовые кислоты 207,"245
эфиры 119, 164
З-Бромпропионовый альдегид, диметил-
ацеталь 118
4-Бромрезорцип 151
эфир 640
Бромртутные соединения 130
ct-Бромсебациновая кислота, диэфиры 168
и-Бромсорбиновая кислота 166
ы-БрОмспирты 206
а-Бромстеариновая кислота 824
<о-Бромстпрол, реакции 690
N-Бромсукцинимвд 122, 136, 137
реакции 303, 309, 836
1-Бром-5,6,7,8'Тетрагидроизохиполин 252
2-БромтиоФен 186
п-Бромтиофенол 608
'<х~Бромтиоэфиры 259
Бромтолуолы 144, 249
галогенирование 146, 199
Р*Бром-у,у,у-трифтормасляная кислота
113
Бромуглеводы 224, 259
Бромуксуспая кислота 165
li-Бромундекаповая кислота 119
Бромуидеканолы 117
Бромфенантрены 143, 253
п-Бромфенацилбромид 183
а-Бромфенилацетальдегид 176
а-Вром-о-фенилвалериановая кислота,
хлорангидрид 873
п-Бромфенилиро1тиоловая кислота, гид-
ратация 269
а-Врои-В-феиилироппоиовая кислота 167
а-Бромфенилуксусная кислота 164
эфиры 168
2-Бром-1-фештлэтиловый спирт 127
Бромфеноксиэтилеп 690
Бромфенолы 150, 151
а-Бромхлорангидриды 165
3-Вром(хлор)бензальдегид 178
2-Бром-4-хлорбензтиазол 253
а-Бром-со-хлорпониловый альдегид 680
Бромциан 243
реакции 436
Бромцианамиды N-замещенные 243
2-Бромциклогексапоп 182
1-Бромцикдогексец, реакции 115
а-Бромциклогексилуксусная кислота, эфи-
ры 168
6-Бромциклогептадиен-1,3 ИТ
2-Бромциклогептанон 182
Бромэтансульфокислота 589
р-Бромэтиламин 415
гпдробромид 207
2-Бромэтиловый спирт 228
р-Бромэтилфталимид 415
а-Бром-р-этоксипропиоповый альдегид,
диэтилацеталь 177
' Бромэфиры 164, 165
•дигалогенирование 680
реакции 751
Буво — Блана реакции 84
Бунте соли 582
Бутадиен-1,3 55, 682, 829
галогенирование 98, 122, 125, 126
гидрирование 45
гидрогалогенирование 116
реакции 704, 793
Бутандиол-1,4, реакции 209, 331, 493
Бутандисульфокислота 590
м-Бутанол, дегидратация 671
н-Бутансульфохлорид 614
Бутены 671, 676
гидратация 266
трг’го-Бутила
гидроперекись 138, 266, 276
перекись 289
Бутиламины 413, 514, 536, 810
N-Бутиланилин 519
Бутилат алюминия 58
трегп-Бутилацетат 283
w-Бутилацетоуясусный эфир 935
Бутилбснзолы 744, 780
галогенирование 146
реакции 735
н-Бутил-б -бромбутплцианамид 244
н-Бутилбромвды 204, 209, 210
Бутилвинпловый эфир, реакции Ц8, 711
Бутплдиметилсульфонкйподцд 586
mpem-Бутилгипохлорит 127
Бутилен, галогенирование и гидрогало-
гепирование 114, 125
н-Бутилидеп-к-бутилампп, конденсация
471
emop-Бутплиденцпануксусная кислота,
этиловый эфир 801
трепг-Бутилизопропиловый эфир 329
н-Бутилиодид 227
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
89»
Бутнлкарбитол 585
к-Бутиллитий 638
реакции 692
Бут илмагнийхлорид 647
«-Бутилмалоновая кислота
декарбоксилирование 820
эфир 820
Бутилмалоновые эфиры 755
н-Бутилметилкетон, диметилацеталь 283
н-Бутилмеркаптан 607, 608, 609
трет-Бутилнитрит 396
Бутиловые спирты
ацетилирование 283
галогенирование 207, 209
эфиры 329
1-н-Бутилпирролидин 244
1-Бутилпирролидон-З, восстановление 87
N-Бутил-п-толуидин 519
N-н-Бутил-га-толуолсульфамид 468
Бутилфеноксиацетилен, гидратация 271.
N-mpem-Бутилфталимид 452
Бутилхлорвды 114, 206, 207, 709
реакции 709
mpem-БутилхрОмат, окислитель 303
mpem-Бутилэтилен 681
третп-Бутилэтиловый эфир 329
Бутины, реакции 120, 693
Бутинолы 691
восстановление 53
Бутин-1-он-З, реакции 474
Бутирамид 431
н-Бутирилмочевина 438
н-Бутирилхлорид 235
«-Бутироин, реакции 492
Бутиролактон
галогенирование 166, 224
реакции 458, 774
Бутлерова — Элътекова реакция 124
2-н-Бутокси-4-аминохинолии 335
Бутоксиацетилен 691
Р-м-Бутоксипропионитрил 281
а-Бутоксипропионовая кислота, нитрил
Бухерера
синтез аминокислот 719
реакция 321, 495, 497, 503
Бухерера — Хенце синтез 485
Вагнера — Меервейна перегпуппиповкп
116, 876 сл. »
Валерианован иислота 77
Балеролактон, восстановление 77
Валин, реакции 449
Ванилин 833
восстановление 63
реакции 809
Вейганда синтез 590
2-Бератрилицдол, реакции 866
Вератровып альдегид, восстановление' 63
Вератрол 364
галогенирование 142, 156
Виланда метод 449
— реакции 836
57*
Вильгеродта реакция 852
Вильсмейера синтез 798
Вильсона метод 815
Вилыитеттера синтез 683
Вильямсона метод 334
Винилакриловая кислота, гидрирование 45
Винилацетат
переэтерификация 353
реакции 260, 552
Винилацетилен
гидрирование 55
• реакции 121, 122, 284
Винилбромвд 121
реакции 112
Винилбутиловый эфир, реакции 118, 711
Винилгалогениды, реакции 765
Винилирование 283, 372
Винилкарбшголы, восстановление 77
Вииилкетоны 856
реакции 281, 706
Винилмагнийгалогениды 645
Виниловые эфиры
простые, расщепление 362
сложные, присоединение HCN 711
Винилфторид 120
Винилхлорид 121, 676
присоединение галогеноводородов 11
реакции 552, 553
Бинилэтиловый эфир 678
бромирование 99
реакции 711
эпоксидирование 262
Винная кислота
ацетилирование 352
окисление 841
эфиры 344, 851
Бисмуторганические соединения 670
Витамин А 63, 753, 813
Витамин D2 829
Витта метод 539
— реакция 812
Виттига —- Людвига синтез 704
Вольфа — Шретера перегруппировка,
882 сл.
Воля метод 479, 824
Воля — Циглера реакция 136
Восстановительное
алкилирование аммиака и аминов 485v
аминирование 486
расщепление 87, 89, 368
Восстановление
азидов 530
азинов 530
азоксисоединений 532
азосоединепин 529
альдегидов 716
алюмогидридами 61, 517, 527
амальгамами 716
боргидридамп комплексными 65 сл._
галогензамещеипых 70 сл.
гидразонов ц гидразинов 530
кетонов 715 сл.
металлами 22 сл., 55 сл., 518
wo
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Галогеппропионовые кислоты 245
0-Галогенстпролы 135 1
Галогенсульфинаты 211
а-Галогентиоэфиры 558
а-Галогенфенилацетальдегиды 176
а-Галогенэфиры 159 сл., 334
реакции 748 , 751, 765
Р-Галогенэфиры, дегалогенирование 679
Галоформная реакция 838 сл.
Гаттерлана — Виланда синтез 881
Гаттермана — Коха синтез 777
Гекса-О-ацетилмапнит, девацилированпе
368
Гексабромциклогексан, дегидрирование 686
Гексагалогенбензолы 686
Гексагидро-п-аминобензойная кислота 48
Гексагидробензойная кислота 49
Гексагидроиарбазол 30
Гексагидропиримидин 482
Гексагидротерефталевая кислота, диазид,
расщепление 873
Гексагидротриазины 471
___н-Гексадекаи 71
Гексадекаидиол-1,2 276
Гексадекандион-2,15, конденсация 807
Гексадеканол, окисление 311
Гексадецен-1, гидроксилирование 276
Гексадиен-1,5 747
Гексаднен-2,4, бромирование 100
Гексадиен-2,4-овая кислота 803
Гексаконтан 742
Гексаметилацетои 307
Гексаметилбензол, окисление 299
Гексаметиленгликоль 85
Гексаметилендиамин, фосгенирование 439
Гексаметилеидиизоцианат 439
реакции 734
Гексаметилентетрамин см. Уротропин
w-Гексан, изомеризация 880
Геисандиолы, реакции 206, 493
Гексанитритокобальтиаты диазония 508
Гексанон~2 270
Гексатриен, галогенирование 98
Гексафенилантимонат лития 668
Гексахлорантимонат карбоксония 437
Гексахлорацетон 178
Гексахлорбензол 106
Гексахлорциклогексан 105
дегидрирование 686
Гексен-З-циоя-2,5 811
Гексея-4-йн-2 693
а-Гексеновая кислота 46
Гексены
гидрохлорирование 114
реакции 696
w-Гексилбенэол 759
ч-Гексилизоциапат 444
N-н-Гексиллактаыид 456
н-Гексилоксиметилсульфон 559
н-Гексилфторид 196
Гексин-1
галогенирование 149
гидратация 270
Восстановление
нитрилов 517
нитрозосоединепий 527
яитросоединений 520
а-окисей 531
оксимов 527
по Буво — Блану 83
— Вольфу 82
— Кижнеру 80, 81
— Клемменсену 78 сл.
— Меервейну — Понндорфу — Верлею
57 сл.
фотохимическое 715
S-фуякций 606
химическое 21 сл., 52, 55
электрохимическое 28, 87, 716
Вюрца реакция 635, 637
Вюрца — Фитпгига реакция 640, 742 сл.
Габриэля метод 414 сл.
Галагептоза 325
Галаитоза
восстановление 66
простые эфиры 332 1 ~ ~
З-Галлоилглюкоза 365
Галогеналкиловые эфиры 118, 221
присоединение к олефинам 122
а-Галогенальдегиды 99
N-Гадогенамины 549
Галогеиаягидриды карбоновых кислот 176,
' 231 -сл., 687
М-Галогенариламины 550
Галогеиацетоны 178
З-Галогенацетофенон 178
<х-Галогенбензиловые эфиры, изомериза-
ция 859
Галогенбензойные кислоты 312
Галогенгидрины (см. также Хлоргидрины)
63, 125, 228
Галогенирование 90 сл.
«аномальное» 111
замещением 92, 132 сл.
«нормальное» 111
окислительное 613
присоединением 92, 94 сл.
а-Галогенкарбоняльные соединения, гид-
ролиз 315
Галогенкарбоновые кислоты 161 сл.
галогенангидриды 162 сл., 687
Галогенкетоны 119, 245, 709, 871
реакции 492, 753
В-Галогенкетоэфиры 553
р-Галогенмаслянан кислота, эфиры 11'3
Галогенметаны при теломеризации 697
а-Галогенозы 222
Галогеннроизводные до сл.
восстановление 70 сл.
дегалогенирование 135
дегидрогалогенирование 682
изомеризация 858 сл.
ненасыщенные 708 сл.
— присоединение галогена 96
реакции с аммиаком и аминами 413
предметный указатель
90
Гексин-1
гидрогалогенирование 120
Гексоген 539 .
Гелиантин 406
Гемеллитол 78
Генри синтез 221
Гептадиеи-1,6 53
Гептанальоксим, реакции 733
^ис-Гептандиол-1,2 276
Гептандион-2,6-карбоновая-3 кислота, эфир
705
Геитаноилуксусная кислота, эфир 790
ГептаноЛ'4-он-З, дегидратация 675
Гептанон-2 270
. восстановительное аминирование 486
Гептафторнитрозопропан, окисление 535
Гептафторпропилмагннйиодид 646
Гептен-1, присоединение НБг 112
Гептен-2-ин-6-ол-1 52
' Гептен-З-он-2 675
. Гептиловый спирт 56, 62
н-Гептиламин 486, 526
4-Гептилнитрит 443
• Гептин-2, гидратация 270
Гептин-2-ол-1 725, 726
Гептеновая кислота, амид 718
Гераниол 46, 849
антраниловый эфир 353
дегидрирование 306
Герца реакция 580
Гваякол 364
расщепление 227, 361
сульфирование 565
Германийорганические соединения 662
Гетероциклические соединения
галогенирование 139, 154, 155, 173
186 сл., 229, 252, 258, 259
гидрирование 50
непредельные 683 сл.
окисление 300, 831
Гидантоины 483, 485, 815
реакции 809
фторирование 256
Гидразиды 230, 437, 452, 459, 477, 507
восстановление 89
реакции 83, 542
2-Гидразии-7-оксинафталин 497
Гидразииоспирты 426, 501
а-Гидрааинпипидины 409
Гидразинсульфокислоты 515
а-Гидразинуксусная кислота, эфир 427
Гидразины 519, 528, 543
алкилирование 426, 427
окисление 546
реакция 378, 380, 541
Гидразобензол 514
диспропорционирование 546
изомеризация 865, 866
Гидразодикарбоновые кислоты, эфиры, де-
гидрирование '546
Гидразониевые соли 543
Гидразоны 82, 371, 401, 474, 504, 508, 850
арилированио 430
Гидразоны
восстановление 402, 519, 530
окисление 547, 693, 694
расщепление 324
реакции с HCN 733
Гидракриловая кислота 267
диметиламид 458
Гидратроповый спирт 268
Гидрпнден, окисление 289
Гидрирование 40 сл. (см. также Восстано-
вление)'
аппаратура 40
каталитическое 32, 44, 51, 54, 68,
323, 516
нитрилов 323, 516
сложных эфиров с расщеплением 368
химические методы 52
шиффовых оснований 518 сл.
Гидробензамид 470
нитрование 389
Гидрогалогенирование 111 сл.
алкинов 120 сл.
олефинов 111
Гвдрокоричная кислота 23
амид 852
дибромид 692
нитрил 51
тиоамид 598
ГидрокоричнЫй спирт 63, 75, 774
Гидроксамовые кислоты 437, 449, 460
хлорангидриды 393
Гидроксиламин-М,М-дисульфонат натрия
480
Гидроксиламии — пиридин, оксимиру-
ющий агент 479
Гидроксиламин-О-сульфокислота 543, 544
Гидроксиламины замещенные 427, 525
восстановление 525
окисление 535 ,
реакции 375, 546
О-эфиры 857
Гидроксилирование 272 сл.
Гидролиз
ацеталей 369
восстановительный 368
галогенидов 314, 316, 318
карбонильных соединений 843, 844
кеталей 369
меркапталей 371
нитрилов 326, 675
ортоэфиров 372
сложных эфиров 365 сл.
Гидроперекиси 288 сл.
удаление из растворителей 291
Гидрохинон 300
ацетилирование 282
окисление 309
реакции 782
эфиры, расщепление 364
Гидрохлорированпе 114, 117 сл.
Гипогалогепиты 140, 871
как окислители 838 сл.
реакции 125, 148, 149
902
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Гиппуровая кислота
реакции 809
фениловый эфир 350
Гликозиды 222, 337, 338, 350
N-Гликозиды 222
изомеризация 856
S-Гликозиды 585
Гликозиламипилгалогениды 224
Гликолевая кислота 315
диацетат ангидрида 285
Гликоли 272 сл., 314, 784, 832, 833. 869
дегидратация 682
окисление 309, 840
реакции 203, 205, 208, 224
Гликоциамин 513
Глиоксалевая кислота 841
эфиры 841
Глиоксали 295, 318
ароматические 715
Глицерин
галогенирование 205, 207
0-мопоэфиры 371
сложные эфиры 345
тритиловые эфиры 340
хлоргидрины 207, 334
Глицериновый альдегид 369
ацеталь 275
Глицвдлловый спирт, реакции 593
Глицидные кислоты
декарбоксилирование 817
эфиры 753 сл., 817
Глицилхлорид 233
Глицин 713
ацетилирование 451
ацилирование 452
бензоилирование 433
галогенирование 233
карбобензоксипроизводное 434
эфир 346, 713
Глутаровая кислота
галогенирование 167
дегидратация 358, 359
динитрил 167, 764
— омыление 326
моноэфир хлорангидрида 236
Глутаровый ангидрид 358, 359
Глутаровый диальдегвд 324
оксим 505
тетраэтилацеталь, галогенирование
161
Глутаконовая кислота, эфир 758
Глутаконовый альдегид, производные, ре-
акции 504, 798
Глюкоза
ацеталироваппе 357
ацетилирование 351
восстановление 66
диэтплмеркапталь 597
изопропплпденовос производное 357
метилирование 341
оксим, реакции 824
простые эфиры 332
реакции 857
Глюкоза
тетраацетат 315
тритильное производное 339
Голбергд реакция 125
Горчичные масла см. Изотиоцианаты
Готье метод 445
Гофмана расщепление 681, 871
Гофмана -- Леффлера реакция 410
Грамин, реакции 796, 797
Графа — Риттера реакции 374
Гребе углеводород 832
Грипьяровские реактивы 644
окисление 289
определение концентрации 646
Грунди метод 307
Грундмана реакция 811
Гуанидины 375, 512
Гуанидоуксуснал кислота 513
Гуанин, превращение в ксантин 321
Губена — Геша метод 770
Дакина — Вести реакция 453
Дарзана метод 214
Даффа синтез 799
ДДТ 175
гидролиз 319
Дебнера метод 804
Дегалогенирование 70 сл., 678 сл., 742 сл.,
765 сл., 800 сл.
Дегидратация 801 сл.
гликолей 682
карбоновых кислот 358
оясикислот 675
спиртов 671 сл., 682, 686, 687
Дегидрирование 683 сл., 735 сл., 799 сл.
каталитическое 305, 684, 736 сл.
окислительное 738 сл.
пирохимическое 735 сл.
спиртов 303 сл.
химическое 685 сл., 799
Дегидробенаол 704
Дегидрогалогенирование 135, 676 сл.,
682, 689 сл., 763 сл.
Дегидродимсризация фотохимическая
741 сл.
Дегидрокортикостерон 185
Дегидрорезорцин, моноэтиловый эфир 330
Дегидроэпиандростерон, ацетат лактама
875
Дезаминирование 319
восстановительное 87 сц.
Дезилтиогликодеиая кислота 592
Дезокспамтюкетозы N-замещенпые 85С>
5-Деэокси-Л-арабл)Юза 371
Дезоксикортпкостерон 185
1-Дс'Зоксп'В-ксилит 83
Дезоксисахара 83, 371, 856
Дскади₽н-3,7-пп-5-диол~2,9 849
Декалин 48
гидроперекись 289, 870
дегидрирование 684
сс-Декалоп, восстакоплеппе 60
ПРЕДМЕТНЫЙ указать ttl
90
Диалкилсульфаты
алкилирующие агенты 463, 598
пергидролиз 289
Диалкилсульфиды см. Тиоэфиры
Диаллил 747
Диаллиламин 423
Диаллиловый эфир, расщепление 365
3,5-Диаллилсалициловая кислота, эфир 86‘
2,6-Диаллилфенол 864
Диаллилцианамид 423
Диальдегиды 317
.сцб-Диаминоадипиновая кислота 417
’Диамицоалтрлхипоны 511
Диаминоацетон 527
3,5-Дпаминобензопиая кислота 172
4,4'-Диамино-3,3'-диметилдинафтил-1,1'73(
2,2-Диаминодифенил, реакции 503
4,4-Диаминодифенилсульфид 579
1,3- Диамино-1-метилциклогексан, расще-
пление 680
1,3-Диамипопафталины 512
1,5-Диаминоиеитан 415
2,6-Диаминопиридин 408
а,Р-Диаминопропиоповая кислота 797
3,5-Диамино-2,4,6-трииодбензойная кисло-
та 172
1,4- Диаминоциклогексан, дихлорид 873
1,2- Диамины, окисление 840
1,2'-Диаитрэхппоилсульфид-2-карбоновая
кислота 600
Диантрацен 741
Диантрон 740, 741
Диариларсиновые кислоты 667
Диарилацстилопы 694
1,3- Диарилпмидазолидпны 482
Диарилкетокспмы 479
Диарилкетоны, реакции 811
Диарилмотапы 782
дегидродимеризация 741
№,№-Диарилэтиленднамин, реакции 482
Диарилы 794
Диароилэтилен 867
Диацетил 294
монооксим 393
п,га'-Ди-(ацетпламинофенил)'Сульфоксдд
611
w-Диацотилбензол 296
Длацетнлвилпая кислота, ангидрид 352
расщепление 829
о,о'-Диацетилдифенилдисульфид 609
Диацетилен 693
реакции 687
Диацетилспглпколи, восстановление 687
М,К'-Диацетилцпстеин, эфир 619
Диацетплянтарпая кислота, эфир 779
реакции 820
2,4-Диацстокспмеркурифенол 155
Диацетонимил 373
Диацетонгидроксиламин 373
Диацетоновый спирт 712
дегидратация 675
разложение 827
Диацплсульфпды, хлорирование 617
Декаме гилендимагпийбромпд 646
Декарбоксилирование 814 сл.
Декарбопилировапис 823 сл.
Дексаметазон 129
Делепини метод 417
Демьянова реакция 885
и-Децилбромид 209
реакции 763
N-w-Децилникотинамид 456
Джепа — Клингемана реакция 402, 510
Диазоалканы 539
реакции 373
Диазоаминосоединения 544
2-[(2'-а>-Диазоацетил)-фенил]-няфтялин,
циклизация 795
Диазоацстоп, разложение 811
Диазоацетофепон
разложение 811, 883
реакции 855
Диа зодезоксибензош!
отщепление N2 882, 883
реакции 688
Диаяокарбононые кислоты, эфиры 245
• Дпазокетоны 245, 855, 883
отщепление азота 89, 811, 882, 883
циклизация 795
o-Диазокоричная кислота, реакции 600
Диазомстап 536, 548, 885
разложение 548
реакции 342, 349, 510, 710, 719, 792
884
Диазониевые соли
восстановление 515
гсксаиитрокобальтиаты 508
ксантогенаты 599
пербромиды, реакции 541, 542
стабилизация 540
трифторацетаты 321
фторбораты 321, 541
«Диазониибйтаияы» 884
1-Диазононадеканон-2, разложение 811
1-Диазо-2-оксинафталинсульфокпслота,
реакции 602
Диазосоединения 411, 512, 546, 722
разложение 88, 670, 794, 811, 882 сл.
реакции 12.3, 320, 508, 541, 542, 544,
600—602, 635, 651, 652, 663, 664,
667, 669, 710, 792-795
Диазосульфаниловая кислота, реакции 602
Йиазосульфонаты 515
иазотаты, окисление 538
Диазотирование 320, 508
ароматических аминов 539
протеинов 541
прямое 411
Диязоуксуспый эфир 549
разложение 548, 811
реакции 710, 792, 793
Диазоцпаппды 400
Р-Дпалкнламиноэтаполы 501
Диалкплборан 52
Дналкплгалогенамины 549
Диалкилкетены 821
9(14
ПРЕДМЕТНЫЙ указатель
Дибензамид 437
1,2,5,6-Дибензантрацен, производные 811
Дибензил 744
реакции 735
Дибензиламин 423, 487
реакции 857
1-ДибензиламинО-1-дезокси-£)-фруктоза 857
'Дибензилдисульфид 582
Йибензилкетон, реакции 807
ибензиловый эфир, изомеризация 865
Дибензилсульфид, окислительное хлори-
рование 614
Дибензилсульфоксид 611
1,4-Дибензоплбутан 236, 770
К,М'-Дибензоилгидразин 437
метилирование 465
Дибензоилметан 271, 788
натриевая соль 760
нитрозопроизводное димерное 396
3,8-Дибензоилпирен, дегидрированйе 737
Дибензоилэтилен 770, 867
Ди-(бензтиазолпл)-дисульфид 610
Дибензфуран, галогенирование 186
с^а'-Дибромадининовая кислота 167
диальдеглд 176
эфиры 168
М,М-Дибром-4-амипогеитан 410
2,6-Дибромаиилин 157 -
9,10-Дибромантрацен 143
Дибромацетилен 148
1,3-Дибромацетон 179, 180, 182
2,5-Дибромбензолсульфохлорид 576
Дибромбензолы 137, 249
1,4-Дибромбутап 209, 228
реакции 590
Дибромбутены 98, 686
1,6-Дибромгексан, реакции 334
2,5-Дибромгексен-З 100
<х,Р-гДибромгидрокоричная кислота 102, 691
этиловый эфир 69
а,а'-Дибромглутаровая кислота 167
1,10-Дибромдекан, реакции 756
9,10-Дибром-9,10-дигидроантрацен 107
9,1О-Дибром-9,Ю-дигидрофенантрен 107
2,5-Дибромдигидрофуран 160
2,3-Дибром-2,3-диметилбутаи 134
1,3-Дибром-2,2-диметилпропан, дебромп-
рованпе 750
2,3- Дибром-1,4-дифенилбутап, дегидро-
галогенирование 682
Дибромдифспилы 143, 253
1,2-Дибром-1,2-дифенилэтан 97
п,п'-Дибром-а,а'-дицианстильбен 799
1,12-Дибромдодекаидион-2,Н 246
Дибромизобутан, гидролиз 861
а,а'-Дибромизоцроиила перекись 293
Дпбромнетоны 246
реакции 861
Дпбромкорпчный эфир, дегалогенирование
^679
л-Дибром-л-ксилол, окисление 301
а,а'-Дибром-о-ксилол 147
3,5-Дибром левулиновая кислота 163
Диброммалеиновая кислота 104
Диброммалоновая кислота 167
хлорангидрид 688
а,р-Диброммасляная кислота 101
дегидрогалогенирование 678
сс,у-Диброммасляная кислота, бромангид-
рид 166
Дибромметан 72
а,а'-Дибромметилизопропилкетон, реак-
ции 861
1,5-Дибром-З-метилпентан 243
1,2-Дибром-2-метилпропап, дегидрогало-
генирование 677
8,9-Дибром-9-метоксинонилхлорид 680
1,3-Дибром-1-метоксипропан 118
Дибромнафталины 253
3,5-Дибром-4-оксиацетофенон 183
3,5-Дибром-4-оксибензойная кислота, га-
логенирование 172
3,5-Дибром-4-оксипирвдин 189
1,5-Дибромпентан 226, 243
реакции 419
а,а -Дибромпимслиновая кислота 167
3,6-Дибромпиридазин 230
3,5-Дибром-4-пиридон, галогенирование
173
4,5-Дибромнирокатехин 142, 1,51
1,3-Дибромпроцап 210
реакции 764
1,3-Дибромпропанол-2 213
2,3-Дибромпропанол 99^100
2,3-Дибромлропилен-1 135
а,в-Дибромпропионитрил 102, 103
а,р-Дибромпропионовая кислота
дегидробромирование 692
эфиры 101, 678, 679
2,4-Дибромрезорцин 151
а,а'-Дибромсебациновая кислота 168
а,р-Дибромстирол 104
а.а'-Дибромсукциндиальдегид 176
Дибромтерефталевая кислота 301
2,5-Дибромтиофен 186
2,6-ДибрОм-л-толуидия, диацетилирова-
ние 451
Дибром-п-толуиловая кислота 301
Дибромуксусная кислота 166
2,3-ДибрОм-1-фенилпропиловый спирт 100
1,2-Дибром-1-фенилэтан 97
2,6-Дибромфенол 150
9,9-Дибромфлуорен, гидролиз 316
Дибромфума роняя кислота 104
2,7-Дибром-9-хлорфлуоре„оп, реакции 800
цис-1,2-Дпбромцнклогексап 115
2,6-Дибромцпклогексанон, гидролиз 861
Дибромциклогептен, реакции 683
7,8-Дибромцикло [0,2,4]октадиен-2,4 95
Дпбромцнклопентемы 97
Дибромэтапы 112, 121
реакции 693
1,2-Дибромэтплеп 103
а, Р-Дибромэтилмстиловый эфир 99
а,р-Дибромэтилэтиловый эфир 161
дегидрооромпровапис 689
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
•1,2^ибромэтокспэтан 99
Диоромянтарная кислота 167
дебромирование 679
дегидробромирование 691
Дибутиламин 87
М,М-Дибутиланилин 420, 465
Ди-н-бутилацетилен, окисление 882
Ди-тр<?пг-бутилбутадиепы, реакции 701
Ди-/Н5утиддисульфид 582
восстановление 609
Ди- трет-бутилкарбинол
дегидратация 671
окисление 307
ДИ'пгрет-бутилкетон 761
Дибутилмеркаптали 596
Дибутилмеркаптолы 596
Ди-ч-бутиловый эфир 227
Ди-н-бутилсульфид, окислительное хлори-
рование 614
Ди-н-бутилуксусная кислота 882
N^N-Дибутилформамид 441
Ди-горего-бутилэтилен, озонирование 262
Дивинилацетилены, гидратация 272
о-Дивинилбенаол 812
Дигалогеналканы 225, 227, 362
Дигалогеналкиламины 549
3,5-Дигадогенаминобензойная кислота 169
а,₽-Дигалогенгидрокоричные кислоты, де-
карбоксилирование 135
Дигалогенднкарбоновые кислоты, диэфи-
ры 167
ге.н-Дигалогениды, гидролиз 316
3,5-Дигалоген-4-оксибензойная кислота 169
1,2-Дигалогенолефины, дегалогенирование
3,5-Дигалогенсалициловые кислоты 169
галогенирование 172
1,2-Дигалогенциклоолефины, дегалогени-
рование 692
2,2-Дигидраэодитиазол, реакции 866
Дигидроантрацеп, дегидродимерязация 741
9,10-Дигидроа нтра цен-9,10-эндоэтиленди-
кариоиовая-1,2-кислота 704
Дигидрозимостерин, бензоат, реакции 847
Дигидродутидиндикарбоновая кислота,
эфир 683
5,8-Дигидроиафтол-1 25
Дигидроциран 856
3,8-Дигидропирен 25
Дигидрорезорцин 49
производные 706
1,3-Двгидротолуол 680
9,10-Дигидрофенантрен 48,. 745
Дигидрофанантрол 865
Диглим 65
1,8-ДидеэоксИ'1,6-дииод-2,4;3,5-димети-
лен-2?-сорбит 220
Диеновый синтез 700 сл.
Дпенон-фанольная перегруппировка 879
Диены 682 сл.
аутоокисление 260
реакции 700 сл.
9,10-Диизоампл-9,10-днгидроантрацен 745
Диизобутилкетоп, восстановительное at
нирование 486
1,4-Диизобутилтетралпн 745
Г^,М-Диизопропилгидразин 488
1,2;5,6-Диизопропилидепглюкоза 357
бензилирование 339
Диизопропилкетен 821
реакции 722, 727
Динзопропиловый эфир
правила.обращения 291
расщепление 227
• Диизоциаиаты, реакции 734
а,а-Дииодалканы 210
2,6-Дииод-4-аминоанизол, реакции 4и2
2,4-Динодаиизол 156, 157
2,4-Дииоданилин 154
Дииодацетилеи 149, 200
п-Дииодбензол 145, 250
1,4-Дииодбутан 227
1,6-Дииодгексаи 211
3,4-ДинОд-2,5-диметилфуран 189
Дииодкетоны 246
Дииодметап 72
2,5-Дииод-З-метилтиофси 189
2,6-Дииод-4-нитроаиилии 158
3,5-Дипод-4-оксипиридин 189
^-(3,5-Дииод-4-оксифенил)-пропионовая кт
слота 153
3.5-Дииод-4-окснфенилуксусная кислота
152
5,7-Дииод-8-оксихинОлпн 154
3,5-Динодсалициловая кислота 154
3,5-Дииодтирозин 152
3.5-Дииодтиронин 152
2,4-Дииодфеяол 155
1,2-Дииодэтилен 104
реакции 765
2 5-Ди-(калии-ацг/-питро)-циклопС[{тано11
’ 385
о,о'-Дикарбоксидифенялдисульфид 60(
Дикарбоновые кислоты 756, 834, 835
галогенирование 166 сл.
дегидратация 358
дигалогенангидриды 167, 168, 259
динитрилы 167, 199
Дикарбэтоисикарбамяновая кислота,
эфир 438
1,1'-Ди-(карбэтоксинафтол)-4,4'-дисуль-
фид 609
3,5-Дикарбэтоксп-4-оксиизоксазол 394
. Дикетены
расщепление 688
реакции 770
Дпкетоны 194, 271, 295, 779, 787, 792, 796
алкилирование 757
восстановление 27, 59, 79
гидролиз 843, 844
реакции 706, 802
Дикетоциперазин, реакции 809
Дпкетоэфиры 787
Дпкетоянтарная кислота, эфир 296
Дикмана метод 791
Ди-.к-ксилилсульфоц 577
906
ПРЕДМЕТНЫЙ указатель
Дильса— Альдера диеновый синтез 700 сл.
Димезилкстен, реакции 722
Димсзитилкарбпнол 63
Димезитилкетон, восстановление 63
Димеризация 293
альдегидов 313, 712
диенов 703
кетонов 688
окислительная 738 сл.
1,2-Дп-(меркаптометил)'ЦНКЛО1 ексаны,
окисление 610
1,5-Димеркаптопептан, З-бензоилирова-
пие 589
Димеркурптиофсй 652
р,Р-Диметилакриловая кислота 804, 839,
848, 861
реакции 128, 130
Ь^М-Диметил-р-аланип 459
Диметилаллен 850
Диметллампн 464
нитрование 538
реакции 494, 785, 786, 867
п-Диметиламипоазобснзол 406
сульфокислота 406
п-Диметиламиноацстанплид, нитрование
388
Дпметиламиноацстон 424
н-Диметнламинобепзальдегпд, реакции
804, 805
п-Диметиламинобензойная кислота, азосо-
четание 406
2,4-Диметил-1-аминобепзол-6-сульфокис-
лота 566
Диметиламиноборан, восстановитель 519
1-Димстилаьпгаобутаноп-З 494
1-Диметиламинобутин-2, хлорирование 693
п-Диметиламинокоричная кислота, эфир 805
0-Диметиламипокротоновая кислота, эфир
473
Диметиламинометплбепзамид, реакции 787
4-Диметиламиноциридип 505
2,4-Диметил-б-аминопиримндин 376
0-Диметиламппопропиофепон 785
реакции 795, 796
1-Диметпламино-2,2,3,3-тетрахлорбутан 693
Диметиламнпоциклогептен, реакции 683
р-Диметиламииоэтилхлорид, алкилиру-
ющий агент 759
М,М-Днметил-п-анизидин, нитрование 388
Димстилавплин 465, 493
нитрование 388, 536
нитрозпронание 399
реакции 503, 775
сульфокислоты 5(56
а.а-Дгшетплацетоуксусный эфир, алкого-
лиз 845
Димстилбепзампд, реакции "98
Димотилбеизиламин 489
2,3-Диметилбензиловый спирт, гидрирова-
ние 78
п.п'-Димети.чбензоила перекись 105
р-(2,4-Димет11лбепзоил)-акриловая кислота
775
п-Димстилбснзойвая кислота 775
4,5-Диметилбепзохинон-1,2 299
2,3-Димегилбугадисн 682
2,3-Диметилбутан, бромирование 134
Диметил-н-бутиламип 489
3,4-Диметилгександиол-3,4 716
Диметилгептиламип 490
5,5-Диметилгидантоип 485
М,М'-Диметилгидразип 465, 476, 515, 528
дегидрирование. 546
1,1- Диметилгидриндан 781
16,17-Диметилдибснзантрон 739
1,2- Диметил-1,2~дигидроизохинолин 858
1,3- Диметил-4,6-динптробензо.т, реакции
295
Диметилдисульфид, хлорирование 619
1,1-Диметил-2,2-дцфснил этиленгликоль,
изомеризация 879
2,2'-Диметилдифенил, реакции 736
ДиметилДихлорсилан 661
1,2-Диметилвделциклогексеп-4 673
а-Диметилизомасляный альдегид 424
Диметилизопропиламин 490
Диметилизопроцилкарбинол, дегидратация
673
Диметилкадмпй 649
Диметплкетазин 474
Диметилкетсп 821
сополимеризация с 03 260
4,4-Диметил-1-кето-1,4-дпгидронафталин,
изомеризация 880
Днметплмалоновая кислота 302
декарбоксилирование 821
2,2-Диметил-З-метиламинОпропиоцовый
альдегид, восстановление 63
Диметплнафталины 744
окисление 298, 300
Диметил-а-нафтиламип 244
реакции 436
Диметил-р-нафтиламин 463
3,4-Диметил-1-нафтилацетат 880
2,3-Диметил-1,4-нафтохпноп 298
2,6-Диметплпикотиновая кислота, этило-
вый эфир 474
Диметилнитрометан, реакции 796
Диметилолово, дигалогениды 663
2,6-Диметвлпирпдип, нитрование 392
2,6-ДиметилпирОн-4, расщепление амином
Димстилпропиламин 490
Диметилртуть 650
Р,р-Димстилсерин 130
Диметилсульфат, реакции 225, 340, 348,
463, 599
Диметилсульфлд, хлорирование 618
Дпметплсульфоксид, реакции 442, 763,
764
2,5-Димстплтетраглдрофуран, расщепление
225
2,6-Диметплтиоурацпл, аллилхлорпрованпе
136
Диметилтиохромоп 596
п.п'-Дпметилтолан, гидратация 269
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
о-Динитробензол 509
реакции 603, 604
п-Динитробензол 509
Динитробензолдиазонийхлорид 601
2,4-Динитробензолсульфиновая кислота (
1,4-Динитробутап 385
3,4-Дииитрогексен-3 800
п,п'-Динитродибензил 740
2,4-Дипитродиметиланилин 388
4,5-Динитро-1,2-диметилбензол 535
о,о'-Динитродифенил 750
о,о'-ДпнитродифенилДисульфид 583
окислительное хлорирование 619
2,4-Дипитродпфениловый эфир, расщепл
ние амином 499
о,о'-Динитродифенилсульфид 603
2,4-Динитродифенилсульфон, щелочное
расщепление 616
2,4-Динитрозорозорцип 398
4,6-Дпнитроизофталевый альдегид 295
2,4-Динитроиодбензол 387
3,5-Динитро-1-метилпиридон-2 510
Динитропафталины 509
2.4-Динитро-1-нафтиламия 468
2,4-Динитро-1-нафтол-7-сульфокислота 31
2,4-Динитро-М-(р-оксяэтил)-анилин 429
2,5-Дииитропиррол 391
4,4'-Динитростильбен 800
дисульфокислота, окислительное pat
щеплеиие 833
2,4-Динитротиофепол, реакции 584
3,3'-Дп-(иитротолпл)-4,4'-дисульфид, окт
лительнос хлорирование 614
Динитротолуолы 386, 510
реакции 808
2,4-Динитрофенилалкилсульфоны 613
2,4-Динитрофенплампп 499, 505
2,4-Динитрофенилгидразин 430, 500
реакции 475, 497
2,4-Динитрофенилгидразоны 475
ацетона 431
расщрпление 324
Д|<-(о-нитрофспил)-дисульфид, восстано-
вление 609
2.4-Динитрофенил-8-нафтиловый эфир, рас-
щепленние 499
2,4-Динитрофенилйиридиниевые соли, ре-
акции 504, 505
2,4-Динитрофепил-н-пропиловый эфир 336
2,4-Динитрофепол 380
восстановление 524
2,4-Динитрофторбензол 197 , 386
в анилизе пептидов 429
реакции 336, 429
Дппитрохиполин 382
Диннтрохлорбензолы 386
реакции 197, 198, 430
Диоксаны 784
аддукт с SO, 560
дибромид 100, 158
правила работы 291
реакции 853
хлорирование 160
Диметил-п-толилсульфониймстиадульфат
599
Диметил-п-то^уидип
ааосочетание 406
нитрование 382, 388
Димстилфенацилсульфонийбромид 586
^ДТС-Диметил-М'-фенилацетамидип 469
1,1-Диметил-З-фенилпропалол, дегидрата-
ция 781
Дпметилфенилсульфопийпсрхлорат 592
№,1\-Димстил-М'-фенилформамидин 469
3,4-Димстилфенол, окисление 299
Диметилформамид 65
реакции 442, 469, 799
1,1-Диметилфталан, гидроперекись и пере-
кись 292, 293
1,2-Диметилциклогексадиеп-1,4 25
цис-Диметилциклогексен, гидроксилирова-
ние 274 .
1,2-ДиметиЛЦИКЛОгекссп-4 674
Х,1Ч-Диметилциклогексиламин 491
1,1 -Диметилциклопропан 750
’ Диметилэтипилкарбинол 714
Диметоксибензальдегид 728
реакции 810
.3,4-Диметоксибензальдегидциангпдр1ш,
реакции 781
п,п'-Диметоксибензофеноноксим, изоме-
ризация 875
З.З'-Диметоксидибензил, галогепированпе
157
1,4-Диметокси-2,6-дибензоксибеизол, гид-
ролиз 362
4,4'-Диметоксидинафтил-1 ,Г 739
п,п'-Диметоксидифениловьги эфир, дезал-
килирование 361
2,3-Диметоксикоричная кислота 804
2,3-Диметокси-(о-нитростирол 810
Диметокситетранитродифенилсульфид 603
2,6-Диметокситолуол 759
Ди-(метоксифенил)-дисульфиды 610
2,6-Диметоксифениллитии 640
4.5-Диметоксифталальдегид 303
1,3-Диметоксифталап 354
4,5-Диметоксифталиловый спирт, окисле-
ние 303
Диморфолинофенилметан 481
1,Г-Динафтнл 739, 748, 775
дегидрирование 736
Ди-(Р-нафтил)-амин 497, 503
а.Р-Динафтилсульфид 585
Ди-а-нафтилтиомочевнна 440
2,4-Динитроанизол, реакции 500
Динитроанилины 89, 430, 499, 618
диазотирование 539
9,10-Динитроантрацеп 386
2,4-Динитробензальдегид, п-дпэтиламтшо-
анил 412
2,6-Динитробензальдегид, реакции 313
3,5-Дииитробензойная кислота 390
л-Динитробензол, восстановление 523
908
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Диоксен 679
1,4-Диоксиантрахинон-2-сульфокислота
573
1,5-Диоксиантрацен, простые эфиры 331
Диокскацетон, окисление 311
Диоксибензойные кислоты
галогенирование 151
гидрирование 49
3,6-Диоксигексагидропиридазин 230
1,2-Диокси-1,2-дигидроиафталин 65
4,4'-Дпоксидифениламин 503
4,4'-Диоксидифеняловый эфир 361
2,6-Диоксиметилфенолы 784
2,5-Диокси-6-метоксиацетофенон 287
<1,8-Диоксииафталип 328
Диоксинафталины 65
окисление 299
Ди-р-оксинафтохинон 741
3,6-Диоксипиридазин 229, 230
2,5-Диоисипиридии 287
9,10-Диоксистеарииовая кислота 274
3,4-Диокси-(1>-хлорацетофенон 770
1,3-Диоксициклогексантрисульфокис-
лота-1,3,5 573
Диолы 84, 86 (см. также Гликоли)
Дипентен, расщепление 829
а,а-Дипиридпл 741
Дипнон 769
Дипропенилгликоль 716
Ди-м-пропиланилии 465, 466
нитрозирование 399
4,5-Ди-н-пропилимидазол 492
Дипронилкетен 821
Дипропионамид 451
Диродан см. Родан
Дисилапы 660
Дисилоксаны 660
Диспропорционировапио
по Кочешкову 662
прй гидрировании 684
Дистирилдисульфиддикарбоновая кислота
609
Дисульфиддпмалоновая кислота 583
Дисульфиддифенилдигликолевая кислота
Дисульфиды 136, 579, 580, 582, 606, 609,
620
восстановление 608
окисление 611
окислительное хлорирование 614
реакции 614, 619 сл.
1,5- Дисульфофталевый ангидрид 570
1,итетралилсульфон 577
,2-Дитнагидриндан 610
!,3-Дитиадекалины 610
.,3-Дитиатетралин 610
Дитизон 380
{птиогликоли, моноэфиры- 615
{итиодигликолевая кислота, эфир 582
1итиодиксантил 741
Дитиокарбаминовая кислота
соли 379, 629
— окисление 630
эфиры 629 сл.
Дитиокарбоиовые кислоты 605
гем.-Дитиолы 594
Дитиосалициловая кислота 600
Дитиоугольиая кислота 626
эфир хлорангидрида, реакции 630
Дитиоуретановый метод 581
0,р-Дитолилакриловая кислота 775
Ди-п-толилкарбоднимид, дегидратирующий
агент 624
Ди-п-толилкетон, реакции 687
Ди-п-толилсульфоксид 577
Ди-п-толилсульфон 577
Дитолилэтилен, реакции 775
Дифенил 735
галогенирование 143, 145
реакции 776
сульфирование 570
Дифениламин 409, 502
арилирование 428
ацилирование 451
окисление> 544
реакции 440, 736
N.N-Дифеиилайетамид 457
ДкфенилацетамиДин, реакции 799
Дифенилацетил, перекись .105
Дифенилацетилен 692, 693
• Дифенилацетилхлорид, реакции 688
Дпфепнлацетон, восстановление 57, 63
ю,о>-Дифенилацетофенон 766
ГчЛ'-Дифенилбензамид 457
1,4-Дифенилбутадиен 682, 802
2,2-Длфенилбутанон-З 879
Дифенилбутатриен 54
Дифенилгалогенметан, реакции 333
Дифенилгалогоиий 635
1,4-Дифенилгексагидротетразин 475
1.6-Дифенилгексантетраон-1,3,4,6 790
Дифенилгидразин несимметричный 528
а,р-Дифенилглицидная кислота, амид 266
Дифенилдиааометан 548
реакции 601
Дифенилдианизилэтиленсульфид 616
Дифенилдиацетилен, гидрирование 54
Дифенилди-6-нафтилпинакон 716
Дифенил-4,4'-дитиол 607
Дифенилдихлорметан, дехлорирование 800
N.N-Дифенилизокпдиго 800
Дифенилизокротоновая кислота 802
1.3- Дифенилимидазолидины 482
1,3-Дифенил-4-иминопарабановая кислота
734
Дифенилкадмий 649
Дифенилкарбаминовая кислота, х.чоран-
гидрид 440
Дифенилкарбинол, ацетат 285
Дифенилкетен 437, 688, 882, 883
восстановление 29
реакции 687, 821
Дпфенцлкетимин 470
ПРЕДМЕТНЫЙ указатель
909
Дифенилмалеиновая кислота, ангидрид 801
Дифенклметан 780
* дегидрирование 799
окисление 285
i реакции 579, 735
Дифеиилметан-4,4'-диизоцианат 439
Дифенилметандиметилдигидразин 476
< К-Дифенил-И'-метилмочевииа 440
Дифенилмочевина 506
реакции 439, 446
Дифениловый эфир 336
расщепление 364, 365
1 Дифеннлоксияитарная кислота, моно-
нитрил 713
1,8-Дифеяилоктатетраен 802
со.со-Дифенилпропиофенон 722, 723
1 Дифенилртуть 650, 651, 653
Дифенилсульфид 579, 585, 600, 604
окисление 612
Дифенилсульфоксид 577, 611
; Дифенилсульфон 577, 612
- Днфеяилтионкарбонат 628
; Дифенилтолилметан 660 • ’
L Дифелилуксусная кислота 76
бариевая соль 687
Дпфенилуксусный альдегид 29
; N.N'-Дифенилформамидин 469
реакции .799, 809
- Дифенилфторметилкарбинол 721
п-Дифенилхинон 738
Дпфепилетилен 672, 812
. бромирование 97
гидратация 268, 269
перекись 260
сополимеризация 1 с О2 260
1\.\'-Дифенилэтилендиа_>.шн, реакции 482
2,2-Дифенилэтиловый спирт 268
а, <о-Дифеноксиалканы 227
2.2- Днфталимидодвтиаэолил-5,5 866
1,5-ДифталпмндОпентан 416
, К,ГГ-Диформилгидразин 507
дигидразон 508
(й.со-Дифторалканы 196
Дифтордихлорметан 192
Дифтор-1,2-дихлорэтилен 135
2.2-Дифтороктан 120
1,2- Дифтортетрахлорэтан, дегалогенирова-
ние 135
а,а'-Дихлорадипиновые кислоты 166, 167
I а,|3-Дихлоральдегиды, реакции 334
: N.N-Дкхлорамины, нитрование 537
; 2,5-Дихлораннлины 157, 521
Дпхлорантрахиионы 257
। 9,10-Дихлораятрацен 1Q6
г тетрахлорнд 106
I Дихлорацетальдегид 175
I Дихлорацетнлек 148
? Дихлорацетилхлорнд, ацилирование ами-
нов 435
1,3- Дихлорацетон, восстановление 63
Дихлорацетонптрил, трпмеризация 376
3,4- Дихлорацетофенон, гидрирование 69
п-Дихлорбеизо.г 250
3,4-Дихлорбензолсульфохлорид 576
7,8-Дихлорбицикло [0,2,4]октадиен-2,4 95
Дихлорбуте'ны 98
гидролиз 314
4,4-Дихлорбутиловый эфир 226
3,3-Дихлоргексафторбутилен-2 194
Дихлордиметкловый эфир 160, 212
2,3-Дихлордиоксан, дегалогенирование 679
п,тг-Дихлордифевилсульфон 576
Дихлордкфенилтрихлорэтан см. ДДТ
Дихлордифеиилы 253
1,2-Дихлор-1,2-дифенилэтан 97
Дихлордиэтиловые эфиры 99, 159, 160
2,2-Дихлориндандион-1тЗт дехлорирование
800
2,4-Дихлоркарбэтоксипиримидин 258
Дихлоркетоны 246
Дихлормалеииовая кислота 104
2,5-Ди-(хлормеркури)-3-метилтиофен 169
а,а-Дихлорметилалкиловые эфиры 241
2-(Дихлорметил)-бензимидазол, гидролиз
316
Днхлорметидгексахлорциклогексан 106
Дихлорметплметилсульфон 612
а,сс-Дихлорметилметиловый эфир, реакции-
239
Дихлорметиловые эфкры 16Q
4,6-Дихлор-2-метилниримидин 229
Дихлормуконовая кислота, окисление 104
а.а-Днхлорнитрилы 167
3,5-Дяхлор-4-оксибензойная кислота 151
2,6-Дихлор-8-оксипурии 229
3,6-Дихлорпирндаэии 229
Дихлорпиридин 188
1,3-Дихлорпропанол 207
1,2-Дихлорпропеи-2, гндрогалогенирова-
ние ИЗ
а,6-Дихлорпропилметнловый эфир 221
а.р-Дихлорпропионитрил 103
2,о-Дихлортетрагидропиран, реакции 748,
749
2,5-Дихлортиофенол 607
Дихлоруксусная кислота 175
Дихлорфенантрентетрахлорнд 106
1,1-Ди-(п-хлорфенил)-2,2-дихлорэтилец 319
Ди-п-хлррфенилсульфон 577
1,1-Ди-(в-хлорфенил)-2,2,2-трихлорэтан,
гидролиз 319
Ди-к-хлорфенклуксусная кислота 319
1,2- Дихлор-1-фенилэтилен, дегидрохлори-
рование 135
Дихлорфеноксиуксусная кислота 155
2,6-Дихлорфенол 150
1,4-Дихлорфталазин 229
Дихлорфталид 847
1,2-Дихлор-2-фторпропан 113
2,4-Дихлорхинолин, гидролиз 315
1,2-Дихлорциклопентан 129
1,1-Дихлорэтаи 121
дегидрогалогенирование 676
1,2-Дихлорэтилен 96, 103, 135
реакции 708
1,2-Дихлорэтиленкарбонат 174
‘910
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Дихлорэтоксиуксусный эфир 241
Дихлорянтарная кислота 740
Ди-(о-цианбензил)-дисульфид 609
Ди-(п-цианбеизил)-дисульфед 609
Дициандиамид, реакции 376
1,4-Дициан-2,4-димстилпафталин 811
2,3-Дицианмасляная кислота, эфир 781
2,6-Дицианпиридин, омыление 328
Ди-(р-цианэтил)-амин, циклизация 733
Н,Н'-Дициклогексилкарбодиимид 448
К^'-Дициклогексилмочевина 449
, Дициклопептендиолы 278
Дициклопентепиловый эфир 330
Дициклопентилсульфид, окисление 612
Дициилопеытилсульфоксид 612
-о,о'-Дициннамиламидофенилдпсульфид 609
Диэндоэтиленовые соединения 702
Диэтаноламин, обмен ОН иа галоген 215
Диэтилалюминийгидрид 29
Диэтилампды 432
Диэтиламин солянокислый 681
Диэтиламиноацетонитрил 484
реакции 796
у-Диэтиламинобутиронитрил 424
^Диэтиламиногептаяоп^, восстановитель-
ное аминирование 487
Р,Р'-Диэтилампнодиэтиловый эфир 467
е-Диэтиламинокапропитрил 424
"у-Диотиламипомасляная кислота, эфир 426
2-Диэтиламннофлуорен 465
'р-Диэтилампноэтанол 418, 500
N-6-Диэтиламиноэтиламидины 463
ЬТ,К-Диэтил-п-анизидин 464
Диэтиланилип 465, 494
нитрозироваиие 399
прямое диазотирование 411
Диэтилацетали альдегидов 99, 118, 176,
177, 228, 423
Н,М-Диэтилглицин, анилиды этилового
эфира 457
Диэтилдйсульфид, хлорирование 619
Диэтилизопропила мин 418
Дйэтилкарбонат, реакции 459, 790, 791
Диэтилкетен, сополимеризация с О2 2(50
Диэтгыгкетон
окисление 286
реакции 804
Диэтилмагний 647
Диэтилмалоновая кислота, дианилид 457
Диэтиловый эфир 329
гидроперекись 291
хлорирование 159
Диэтилоксалат, реакции 790
Диэтилолово, дигалогениды 662
Диэтилпропилкетен 821
Диэтилртуть, реакции 639, 666
Диэтилсульфид 598
Диэтилсульфоксид 611
а,а-Ди-(этилсульфонил)-пропионовая кис-
лота, эфир 613
Диэтил-п-толуидин 464
нитрование 382
М’^-Диэтил-.и-фенетидшг 464
Диэтилцинк 648
2,4-Диэтокси-6-бромппримидпн 335
4,4-Диэтокси-1,1-дииафтил 738
Диэтоксидисульфид, восстановление 609
1,5-Диэтокси-1,2,4,5-тетрабромпентан 161
Ди-(этоксифснил)-дисульфиды 610
к-Додекан, хлорирование 133
1.12-Додеканкарбоновая кислота 756
Додекансульфохлорид, отщепление О2 258
Додекапептаенилиденмалоновая кисло га
804
Додекахлорциклогексан 106
Додецен-1 674
изомеризация 847
окисление 265
Додецилбромид 208
н-Додецилмеркацтан 582, 608
Додецнлхлорид 258
дегидрохлорирование 847
Дурол 881
нитрование 385
Енамины 456, 481, 482, 730
ацетилированные 453
Енол-аллиловые эфиры, изомеризация 863
Енолацетаты 184
эпоксидирование 264
Еполы,'восстановление 77
ЕнолэфПры, расщепление аминами 498
499
Жирара реактивы 324, 426, 478
Жиры
омыление 368
определенке С = С-связей 132
Зайцева правило 671, 677
«Запекание» 565, 569
Зандмейера реакции 247, 257, 794
Зелинского и Стадникова метод 719
Изатин, полумеркапталь 558
Изоамилен, присоединение НС] 114
Изоамиловый спирт 283
дегидратация 671
Изоамилоксиметилсульфон 559
Изоборнеол 65, 279
Изоборнилацетат 279
Изоборнилбромид 116
Изоборнилхлорид 116, 877
Изобутан, хлорирование 133
Изобутилбромнд 204, 212
Изобутилен 368
гидрогалогенирование 111, 114
полимеризация 698 сл.
присоединение галогонэфиров 122
Изобутиленгликоль 268
Изобутиловый спирт
дегидрирование 303
обмен ОН на галоген 204, 212
окисление 303
Изобутилэтиловый эфир 334
Изобутирамид 431
а,а'-Изобутиронитрил 136, 138, 146
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
911
Изоцианаты 438, 444, 445, 466, 510, 871
ди- и тримеризация 378
реакции 377, 730, 731, 734
Изоциануровая кислота, эфиры 378
Изоэвгенол 848
нитрование эфиров 382
окислительное расщепление 834
Имидаз.олидины 482
Имндазолиды 448, 506, 507
Имидазолины 449, 463
Имидазол-4,5-карбоновая кислота, диамид,
расщепление 872
Имидазолы 440, 492
N-ацетилирование 458
галогенирование 187
фосгенирование 435
Импдоэфиры 327, 461, 465
ацилирование 462
реакции 603
циклические 463
Имидхлорнды 75, 322
реакции 240, 776
Имиды циклические 452
Ингерсолла реактив 490
Инголда классификация перегруппировок.
878
Индан, проивводные 781
Иидиго, дианил 440
Индол
алкилирование 781
производные 866
реакции 710, 796
сульфирование 571
Илдолиды р-аминокислот 457
Ипдолил-З-уксусная кислота
нитрил 796
эфир 710
Иидол-З-карбоновая кислота 884
Иодалканы 150, 362
а-Йодалкиловые эфиры 228
Иодалкины 150
а-Иод-р-алкоксикарбоновые кислоты 131
Иодангпдриды карбоновых кислот 237
4-Иоданизол 154, 156
4-Иоданилин 156, 159
Иодацетальдегнд, диэтилацеталь 177
Иодацетилены 149
4-Иодбензойная кислота 155, 173
Иодбензол 144, 145, 201
п-Иодбенэолсульфиповая кислота,
ангидрид 624
Иодбензолсульфокислота 564
1-Иод-4-бромбеязол 145
1-Иодгексадекан 214
Иодгексадецилгидрохинон, деиодирование
743
Иодгептапы 199
Изовалериановая кислота, галогенирова-
ние 163, 164
Изодибромяптарная кислота, дегалогени-
рование 679
' Изодурол 881
Изокапролактон 286
Изокапроновая кислота
галогенирование 163, 164
окисление 286
Изокро'гоновые кислоты 803
Изомасляная кислота
галогенирование 163
окисление 286
хлорангидрид 232
Изомасляный альдегид 303, 861
оксим, восстановление 526
Изоникотиновая кислота 301, 831
N-окись 534
Ийонитрилы 440, 445
присоединение аминов 375
Изонитроэоацетоуксусный эфир
восстановление 526
• окисление 443, 535
Изонитрозобензоилуксусный эфир 394
Изонитрозокетоны 294
Изонитрозомалоновый эфир
восстановление 526
окисление 535
Изонитрозосоединения 527
Изопрен 829
Изопренкарбоновая кислота 804
Изопропеннлацетат 184, 458
Изопропиламин 486, 526
окисление 536
Изопропилат алюминия 58
Изоиропилацетилен, изомеризация 850
Изопропилацетоуксусный эфир 779
Изопропилбензол см. Кумол
Изопропилиденовые производные углево-
дов 356, 357
гидролиз 370
Изопропилиодид 205, 227
Изопропилцнклопентадиен 29
Изосафрол, нитрование 362
Изотетралин 25
Изотиомочевипы 614, 631
Изотиоцианаты 201, 257, 440, 591, 631
восстановление 90
реакдни 377, 605, 606, 630
Изотиурониевые соли 512, 581, 631
превращение в сульфогалогениды 614
Изофоронокснд 265
Изофталевая кислота, дииитрил, омыление
328
Изофталевый альдегид, ацеталй*354
Изохинолиния соли, восстановление 27
Изохинолины 374, 858
реакции 443
Иодгидрины 127, 228
3-Иод-2,5-диметилфуран 189
4-Иод-1,2-диметоксибензол 156
1-Иод-2,4-динитробензол 198
4-Иоддпфегпгл 145
аминированке 409
гидрирование 51
окисление 842
реакции 496, 858
Иаохроман 773
аутоокисление 292
293
912
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
З-Иодиндол 201
Иодирование
। замещением 140, 141, 144, 145, 148, 149,
150, 152, 157. 197
присоединением 93, 95, 97 }
хлоридом иода 170
углеводов 224
Иодкетоны 185
2-(3-)-Иодметилбензойные кислоты 251
-2-Иод-Ы-метилпиридинийиодид, гидролиз
316
.2-Иод-З-метилтиофен 189
1-Иод-2-метокси(этокси)яафталнп 154
1-Иоднафтални 145
о-Иоднитробензол 252
Иодное число 102
'2-Иодоктан 199
Иодониевые •соли 670
Иодофоры 200
Йодоформная проба 186
21-Иод~Д-прегненол-36-он-20 184, 185
р-Иодиропионовая кислота ИЗ
дегидрирование 750
Иодродан 101
N-Иодсукцинимид 184, 185
Иодстеронды 184, 185, 194
Иодтиофен 144, 167
реакции 724
Иодуксусная кислота 198
ИодфенЪлы 155, 252
1-Иод-4-хлорбензол 145
а-Иод-В-хлоркарбоновые кислоты 131
2-Иод-1-хлор-4,4,4-трифторбутан, реакции
678
Иодциклогексап 117
Нонен 781
р-Ионилидеиацетальдегид 323
реакции 753
,р-Ионилвденкарбоновая кислота, о-толу-
идид 457
р-Ионилидепуксусная кислота, о-толуидид
457
Иоцича Комплекс 849
Исчерпывающее метилирование углеводов
338, 341
Итаконовые кислоты
ангидрид 803
— изомеризация 846
хлорангидрид 232
Кадаверин 415
Кадмийорганические соединения 642, 649
Камфан 82
Камфен, реакции 279
Камфенгидробромпд, изомеризация 877
Камфенгидрохлорид 116
Камфора
восстановление 65, 82
оксимы 479
— восстановление 526
разделение антиподов 478
реакции 490
сульфокислота 560
Канниццаро реакция 312, 313
перегруппировка 855
КалрилОнитриЛ, гидрирование 517
w-Каприновая кислота 838
Капролактам 874
алкилирование 465
нитрозирование 536
Капролактон, восстановление 77
Капроновая кислота 76, 77, 820, 821
ангидрид 282
анилид 457
фениловый эфир 271
Каран, расщепление 684
Карбазиновая кислота, Z-мептиловый
эфир 478
Карбазол 409, 503, 736
арилирование 428
ацилирование 451
гидрирование 30
сульфирование 570
Карбазолиды ^-аминокислот 437
Карбаминовая кислота
хлорангидриды 435
— реяйции 630, 776
Карбидсульфаты 554
Карбитолы 444, 585
Карбббепзокси-Л-алапилглицин, этиловый
эфир 447
Карбобензоксиаминокислоты 369, 433, 434
Карбобензоксигидразии 437
N-Карбобенэоисиглицкн 434
Карбодиимиды 448
реакции 344, 376
Карбоксибеизолсульфиновая кислота 601
Карбоксибенэодсульфохлорид 577, 601
Карбоксилирование ароматических соеди-
нений 778
Карбокснметилбутилсульфид 584
/г-Карбоксифенилгвдразин 476
о-Карбоксифенилтиогликолевая кислота
584
Карбоксоиия гексахлорантимонат 437
Карбонилбромфторид 202
ЬГ^'-Карбонилдиимидазол 435, 440, 448,
506
реакции 347
Карбоновые кислоты 185, 310, 326, 834 835
амипометплироваипе 785
ангидриды 282, 358 сл.
ароматические 778
восстановление 25
галогенирование 141, 161 сл., 173
гидрирование 85
дегидратация 358
дегидродимерпзация 742
а,р-дихлорзамещенные 166
ненасыщенные см. Ненасыщенные кар
боновые кислоты
полиеновые, галогенирование 98
этерификация 343
Карбостирил 287
Р-К.арбэтокси-у.'у-дифенилвпнилуксусная
кислота 805
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
913
3-Карбэтокси-1,5-дифенилформазан 404
1-(Г<)-Карбэтоксипиррол. гидрирование 51
р-Каротин 813
окисление 278
Каротиноиды 63
Катехин 1Ц
Квазцфосфониевые соединения 216, 217
Кватерннзация 411 сл.
третичных аминов 423
шиффовых оснований 422
Кенига синтез 808
Кстаинвы 474
Кетали 278, 283, 355
гидролиз 369
реакции 473
циклические 354
Кетены 437, 687 сл., 828. 882 сл.
димеризация 688
реакции 29, 132, 260, 282, 702, 722,
770, 821
Кетимины 470, 728, 729, 770
реакции 730
а-Кетоадиппновая кислота, фенплгндразон
402
Кетоальдегиды 787, 789
изомеризация 855
Кетоамиды 271
Кетоанплы, реакции 730
З-Кето-12-а-ацетокспхолановая кислота,
эфир 179
З-Нетобутанол-1. реакции 494
1-(у-Кстс>бутил)-ш1Перидин 494
у-Кстовалериановая кислота см. Левули-
новая кислота
Р-Кето-ш-га.тогенэфиры, алкилирование
(57
а,а-Кетокарбинолы, окисление 311
Кетокарбоновые кислоты 270, 308 327,
395, 757, 774. 809, 834
восстановление 79
. декарбоксилирование 816, 820 сл.
нитрилы 711
эфиры 308, 352, 755, 757, 790 820
843. 845
•- гидролиз 367
б-Кетокапродовая кислота, эфиры 706
Потоке гены 688
Кстоксимы, изомеризация 874
Китолы 246
окисление 840
6-Кего-1-метнл-1 .б-дишдронико типовая
кислота, нитрование 510
Кетонитрилы 271. 788
Кетонцпангндрииы. реакции 718
Кетоны 269, 294 сл.. 306 сл.. 323. 369,
648, 728, 760 сл., 769 сл., 820 сл.
832. 834 сл.. 851 сл.
восстановление 56, 57 сл.. 63 61 67.
68 сл.. 78 сл., 715
галогенирование 163, 178
гидролш ячеекое ра< гиеплеиие 819
а.р-пенасыщенные см. а.Р-Ненасыщен-
ные кетоны
58 Заказ 1835.
Кетоны
окисление 285 сл., 288, 292, 294
окислительное расщепление 837 сл.
реакции 687, 717, 719, 721, 727, 734,
736, 751, 756, 785, 787, 789. 792,
796, 804, 812, 813, 884
циклические 22, 25, 732, 766, 770,
792, 813, 818
— восстановление 60, 81
— галогенирование 182
— расщепление 837
2.-Кетопиверидпн-3-карбоновая кислота,
азосочетание 402
Кетоспирты 712
гидрирование 69
а-Кетоспирты
изомеризация 854
окисление 840
p-Котоспирты, разложение 827
Кетостероиды, реакции 179, 180, 181,
182, 184, 195, 813
а-Кетосульфиды 553
у-Кетосульфоны 556
восстановление 60
Кетоэфиры 298, 789, 757, 760, 787
Кианметин 376
Киафенин 377
Кижнера восстановление 80, 81
реакции 474
Килиани синтез 718
Кинуренин 417
Клайзена конденсация 789
перегруппировка 863
синтезы 336, 712, 760, 805
Клауса метод 539
Клемменсен а метод восстан овлеипя
78 сл.
Кневенагеля реакция 803 сл.
синтез 811
Кнорра син геи 820
Коллидин 817
Коллидпн-3,5-дикарбоновая кислота, де-
карбоксилирование 817
Кольбе метод 442
Кольбе — Шмидта реакция 717
Кондакова реакция 709
Коричная кислота 801
галогенирование 94. 102, 691
гидрирование 23, 11
декарбоксилирование 816
нитрил 51
реакции 132, 262. 552, 793
хлорангидрид 235
— восстановление 75
эфиры 805
— реакции 3/3, 654
Коричный алъдетд 306
бисульфитное соединение 559
восстановление 29. 46. 56, 63, 67
дифенил меркапталь 59(‘>
реакции 118. 802. 807
Коричный спирт 29. 46, 6,3, 67. 73
иосстамов.чгмпе 76
914
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Коричный спирт
дегидрирование 306
реакции 205, 374, 553
Корн-блюма реакциц 442, 443
Кортизон 185
Кортикостерон 854, 855
Кочешкова реакция диспропорционирова-
ния 662
«Красный Лаутемана» 152
Креатинин, реакции 809
КрезолсульФокислош 561, 572
Крезолы • 328
азосочетапие 405
гидрирование 49
нитрование 382
окисление 299
сульфирование 565
Кремнпиорганические соединения 659
Кронке метод, 304
Криге перегруппировка 870
Кротпламин 55
Кротилиодид 116
Кротиловый спирт 53, 58
реакции 205 ,
Кротилфталимид 55
Кротоновая кислота 803
галогенирование 101, 119, 128
гидрирование 46
реакции 262, 267, 551, 555
хлорангидрид 236
эфмр^г 711
— реакции 113, 130. 265. 790
Кротоновая конденсация 706
Кротоновый альдегид
восстановление 716
озонирование 871
реакции 260, 281, 725, 803, 804,
807
Ксантгпдрол, алкилирование эфиров 779
Ксантилиевые соли 502
Ксантин 321, 872
Ксаптион 616
Ксантогенаты 599, 627
. окисление 630
пиролиз 674
Ксантодибензмлмернаптол. пиролиз 616
Ксантон, восстановление 57
л-Ксиленол, окисление 302
Ксплилеидихлорпд 783
Ксилилсульфенилхлорид 618
Ксилоза, восстановление 66
Ксилолсульфиновая кислота 578, 601
ангидрид 624
Ксилолы 84
алкилирование 881
бромирование 143
нитрование 385
окисление 289. 299. 30О
разделение изомеров 89
Кумариловая кислота 102
Кумарин 801
дибромид 102
Куминовыи спирт, восстановление 76
Кумол 696, 780
галогенирование 146
гидроперекись 294, 870
нитрование 385
окисление 289
Кутиулены 687
гидрирование 45
Куна синтез 607, '
Курциуеа метод 693 сл.
расщепление 872
Лактамы, реакции 86, 458, 464, 510
Лактоны
альдоновых кислот 718
восстановление 77, 86
галогенирование 166
окисление 308
полиоксикарбоновых кислот, восстано-
вление 76, 77
реакции 137, 458, 725, 774
Лантпонин 417
Лаурпда пёрекйс’ь- 112
Лаурилдимстилсульфонийиодид 586
Лауриловый спирт 85
Лауриновая кислота, реакции 85, 819
Левомицетин 511
Левулиновая кислота
галогенирование 163
снол-лактон 825
реакции 876
Лейкарта — Валлаха реакция 485, 489
Лсйкооснования 286
Леперка реакция 442
Леппдплбромид 190
Лепидин 73
галогенирование 190
Либена йодоформная проба 186
Лизин 485
Ликопин 813
Лимонная кислота
декарбонилировапие 823
этерификация 346
Линалоол, восстановление 77
Линалоолен 77
Линдлара катализатор 33, 45, 55
Линолевая кислота, присоединение брома
102
Линоленовая кислота, присоединение бро-
ма 102
Лптийалкилы 638
Литийорганичесяие соединения 637 сл.,
727 сл.
реакции 606, 660, 662, 664. 666, 668,
730, 758, 759
Литийрезорцин, диметиловый эфир 728,
745, 758
Лумистсрнн, реакции 829
Лугидипдикарбонивая кислота, эфир 684
Магпиядиалкилы 647
Магппйдиарплы 647
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
911
Магнийорганические соединения 643 сл.,
720 сл.
реакции 72, 513, 604, 650, 651 660,
662, 664, 666, 668, 720, 727
Майласа реагент 276
Малахитовый зеленый 287
Малеиновая кислота 676, 678
бромирование 94
гидравиды 230 , 453
гидратация 267
игдрдрованпе 44, 47
имид 452
реакции с диенами 700
— с надкислотами 262
эфир 296
Малеиновый ангидрид 358, 701
реакции 703 сл., 775, 826
Малонгидроксамовая кислота 4б0
Малоновая кислота 327
галогенирование 167
декарбоксилирование 820
диамиды 436
реакции 404, 803, 804
- эфиры см. Малоновые эфиры
Малоновые эфиры 84, 167, 754 сл., 825
алкилирование 755, 779
альдегидацетали 753
ацилирование аминов 454
гидрирование расщепптельное 368
гидролиз 367
нитрование 381, 383, 385
пптрозирование 395
реакции 402, 459, 460, 756, 803, 812,
824
Маннит 368
пзопропилидецовое производное 357
— — гидролиз 370
окисление 310
Манииха основания 69, 87, 680 706
786, 808
расщепление 680
реакции 503, 504, 785 сл.
Манноза 310
восстановление 66
Маргариновая кислота 327 '
Маргариновый альдегид 82!
Марковникова правило 550, 551
u-Маслийая кислота 46
ангидрид 359
бромангпдрид 165
i алогенирование 162, 163. 165
«-Масляный альдегид, реакции 471. 807.
808
Меервейна реакция 123. 793
Меервейна — Понндорфа — Нерлея —
Оппеиауэра реакция 304
Мейера реакции 515, 667
Мезитила окись 472
реакции 118, 551 1
япоксид 265
Меаитилацетонитрмл 764
Мезитплеп 810
ацилирование 775
58*
Мезитилен
галогенирование 143
реакции 400, 781
Мезитиденсульфиновая кислота 578
ангидрид 624
Мезитилмагнлйбромпд, реакции 722
Мезоксалевая кислота
эфир 294, 295
— фенилгйдразои 402
Меламин 376
Меллитовая кислота 299
Ментилампн, N-формальное производное
456
£-д-Ментилсемикарбазпд 478
Ментол 64
дегидрирование 307
Ментон 307
восстановление 64
Меркаптали 83. 588, 596
восстановительное расщепление 89
гидролиз 371
Меркаптаны 580, 592, 606
ацилирование 589
окисление 609
пиролиз 615
реакции 328, 550 сл., 602,‘ 614
Меркаптиды
окисление 611
реакции 584, 600, 630 .
Р-Меркалтоалкнламины 602
2-Меркапто-4-аминониримидип, обмен SH
на ОН 328
р-Меркаптоантрахипон, расщепление С— S-
связп 615
2-Меркаптобензтиазол 74
окисление 610, 611
реакции 259
p-Меркаптовалериановая кислота 554
МеркаптОгпдрохинон 556
Меркаптокарбоновые кислоты см. Тиокар-
боповые кислоты
а-Меркаптокетоны 553
присоединение альдиминов 556
Меркаптолы 558, 588, 596
расщепление 371, 616
Р-Меркаптомаслянаи кислота 551
2-Меркапгонентанон-З. реакции 553
6-Меркаптопурин, S-алкплнрование 585
Меркаптоуксусная кислота см. Тпокксус-
ная кислота
а-Меркапто-а-фенплацетон 581
2-Мсркапто-4-хлорапн.'тпны 580
Мсрнурировяние ароматических соедине-
ний 651 сл.
Метакриламид 455
Метакриловая кислота, эфир
аминолнз 455
эпоксидирование 265
Металлилбромид, изомеризация 849
Металлиловый спирт, гидратация 268
Металлирование
ацетилена маптем 643
углеводородов 636, 639, 642
916
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Моталлилхлорид, хлорирование 96
Металлкетнлы 57, 716
Металлоорганические соединения 632, 634
ацетиленового ряда 148, 149
комплексы 634, 637
реакции 364, 513. 720 сл., 754 сл.,
760 сл.
элементов I группы 636
- IJ группы 642
— Ill группы 654
— IV 1 руппы 659
— V. г руины 665
Метан-иульфонат бария 599
Метансульфоновая кислота, эфиры.
першдролиз 289
расшеплепие 78
Метансульфохлорид 623
Метантрикарбоноеая кислота, эфир, реак-
ции 796
9-Метилакридин, реакции 808
Метилакрилат, реакции 119, 368
а-Метилакрилонптрпл 676
Метилаланнны 425. 809
Метилалкнлкетоны. восстановительное ами-
нирование 486
Метилаллен 686
Метилаллпламин 422
К-Метил-ТЧ-аляил-п-толуолс) льфамид 422
Метилаль 354
реакции 714
N-Метиламиды 432, 458
Мети ламп лкотон, восстановительное ами-
нпрованпе 486
Метиламин 485. 876
реакция с бензальдегидом 471
1-Метиламиноаптрахинон 512
В-Метиламинокротоповая кислота, эфир
473
а-Метиламиномасляная кислота, этиловый
эфир 425
1-Мегил-2' -амнно-б '-ни гробенэофонон,
реакции 795
х-Метиламлноиропяоновын альдегид 423
N-Метил-п-анизидип 465
N-Метмлапилнды
^-аминокислот 457
карбоновых кислот, восстановление 68,
323
\-Мстиланплин 519
сульфокислота 567
форматирование 446
р-Метилантрахинон 703
Метилантрон 771
окисление 286
Метпларсоновая кислота 668
N-Метилацетанилид 420
сульфирование 567
Метилацетилон 693
гидробромировалие 122
о-Метллбепзальдегид, восс1ановител|.ное
аминирование 487
2-Метилбепзантрон. реакции 739
N-Метилбснзиламнн 488. 518
2-(4'-Метилбонэил)-бснзойная кислота, ре-
акции 771
2-Метилбензнлдиметиламин 868
2-МетнлбРпзидметилсульфид 869
2-Метилбепли.товьш спирт 869
Мотплбензплсульфон 591
Метилбензоилацетоп 754
енольная форма 854
Метилбензоилкарбннол, изомеризации 854
а-Метилбензоилуксуспый эфир 845
нптрозированпе 397
о-Мот илбепзолкарбоновая кислота, ани-
лид 323
4-Мегилбензолпнон-1,2 299
(З-Метплбензтиазолил)-2-азино-1 -(2-кет о-
1,2-дигпдрояафталин) 408
М-Матилбеизтпазолон-2, азосочетание 408
2-Метил-1-бромнафталин, дебромиронанне
744
2-Метн.'>бутадиен-2.3 687
2-Метилбугандпол-1,3 86
2-Метилбутанол-1-он-3 784
2-Метнлбуганол-2-он-3 271
Метплбутапон, галогенирование 180
2-Метнлбутен-1 672
2-Метилбутеп-2 672
нитрозохлорид 132
2-Метилбутен-З 673
2-Метнлбутсн-ЗгОл-2 55
2-Метилбутон-1-он-3 784
Мстил-глретп-бутилкарбинол, дегидратация
671
Метил-н-бутиловий эфир 343
2-Метилбутиц-.3--ол-2, гидратация 271
Метплвимийкарбинол, обмен ОН па га.ло-
. ген 205
Me гйлвииилкетон 681
реакции 266, 281, 551, 705
Мсттгявиниловый эфир, реакция с IICN 711
Р-Метилвиинлуксуспзя кпслота, изомери-
зация 848
3-Метнлгоксадиен-3,5 682
2-Метнлгоксанол-2, дегидратация 672
2-Мети л гексен 671
З-Метилгептанол-2 56
2 Метилгептанол-2-он-6 267
Метилгептепон 827
2-Метнлгептен-2-он-6, гидратация 267
Метилгндразин 476
О -Мстилгидроксиламин 514
Метилглноксаль. 1-феип.чигдразон 402
а-Метил! лнцидпая кислота, мет иловый
эфир 265
се-Метил-Л-глюкоэид 332
2-Метилдекалпн 72
3-Метил~2,4-днбромгексан 213
2-Мет»л-2.4-Дйбродшептан 213
Метилдпгидрорезорципкарбоповая кислота-
этп.гоный эфир 706
1-Мет11Л-2.2-Дппропи.1-2-оксипропиоиовая
кислота, эфир 751
2-Метплд1н|)еш1.1, реакции 736
М етплднфенилс>льфпд 585
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
91'
3,5-Метилен-Л-ксилоза 325
Мстилендима mini галогениды 646
Мотилсн-4-метокси дезокси бензоин 807
Метилепцпк.югексан 813. 820
N-Метилизобутпрамид 433
О-Метилизомочевина. реакции 463 1
а-Метил-у-нзопропилацето уксусный эфур,
бромирование 859
Метилпзопропенллкетон, эпоксидирование
266
Метилизопропилэти.1еп 673
Метичизоциаиат 440, 444. 873
З-Метилиидо.ч, сульфирование 571
Метилиодид 225
М-Метмл-2-подпирпдпнийподид, гидролиз
316
Метилирование
емипов 489
углеводородов 337, 338, 341
фенолов 851
Метилкетоны 270. 762, 812
галогенирование 179
окпелриие 295, 302
окпелителыюс расщепление 837 сл.
реакции 792
а-Метилкоричная кислота 802
В-Метилкоричная кислота, эфир 752
р-Метплкротоновый альдегид 370, 371
2-Метилкумаран 864
N-Метиллейцин 425
Метиллитпй 639
2-Метилмезобсизаптрон, галогенирование
142
Метилмеркаптан, реакции 551
4-Метилмеркаптобутанон-2 551
Метил-0-меркаптоизобутилкотон 551
2-М етилмеркапто-1,4-нафт охпнон 553
р-Метилмеркаптопропиоповый альдегид 552
1-(п-Метилмсркаптофепил)-этанол 60
N-Метилметакрпламид 433
Метилметакрилат, гидробромиронание 119
N-Метил-п-метокспацетанилид 420
4-Метил-6-метоксихинолин 300
N-Метилморфинан 858
Метилмочрвина 506
2-Метплнафталпн. окисление 295
N-Мотил-р-нафтиламин 420
М-Мстил-ТЧ-а-ыафтилцпапамид 244, 436
6-Мотил-2-пафтол. окислительное дегидри-
рование 739
Ь-Метилпикотиновая кислота, гидразид
478
Метп.тпнтрамвп 537
N-Метил-п-питреацетанплид 421
N-Мети л-Ы-иитрозо-л-толуолсульфамид,
реакции 547
З-Мегил-7-питрофлуореноп 795
2-Метпл-5-нитрофуран 391
Метплнониламил 422
Мстнл-н-нонилкетоп, восстановление 80
1()-МетиЛ-10-окспаптрон 286
N-Мотплоксиндолы. восстановление 28
4-Метп.г-5-(а-<>ксиэг11Л)-тиа.|Ол 60
N-Метилол-М'-фенилмочевина, «-бутило-
вый эфир 330
2-Метилпентен-4 673
З-Метилпентен-2, окисление 266
3-Метилпентен-2-ол-4 286
2-Метилнентен-1-он-4 848. 840
а-Мстилштмелмповая кислота, эфии. гид
релиз 367
N-Метилпиперидпн 51, 489
З-Метилппрантрон 737
З-Мстилпирен. 82, 83
Метилииридинийбромид 839
ТИетилпирндины 532
галогенирование 190
гидрирование 51
N-окиси, восстановление 532
сульфирование 570
5\-Метилпирндоц-2 316
Метилппррол 83
реакции 808
2-Метилпирролпдпи 410
N-Метилиирролидин 410
Метилпропилкетон, окисление 285
М-Метил-2-про1шлпирролидин 410
Метилсиланы 659
4-Метилстильбен, реакции 317
сс-Метилстирол
гидратация 268
полимеризация 698 сл.
Метплсульфенилхлорид 618, 619
М-Меигл-со-сульфокпслоты ароматические,
азосочетание 406
1-(«-МетилсульфопилфенИл)-атанол 60
Метилтетралииа гидроперекись, изомери-
зация 870
4-Метилтиазол-5-альдр1 ид 322
Метнлтиолзатин 776
Метплтиофены, галогенирование 187
N-Метил-и-толуолсульфамид 468
нитрование 537
N-Мети л-2,4.6-тринитрофснилнитрамин
388, 538
N-Мотилтриптофап, реакции 453
Метилтрпфеноксифосфонпйиодпд 217
Метилтрпхлорсилан 661
5-Метилурацил, аллплхлорировлние 136
Метилфенилгидразин 528
4-Ме гил-4-фенилпентано)1-2, галоформная
реакция 839
МетШ|фепплсул1.фпд, алкилирование 592
1-Метил-1-фепилэтилен 672
2-Метнлфе1гол-4-сульфокислета 565
4-Метилфлуорен 736
N-Метцлформаиилпд, реакции 798
Метилформиат 469
З-Метпл-4-фторбензо.тсульфохлорид 576
2-Метилфуран 81
нитрование 391
1 -Метпл-4-хлор-н-ами лацетат 225
2-Метпл-4-хлорб('Изолсульфо\лорид 576
З-Метил-З-хлорбутн ион-2 180
З-Метилхромои 789
Мсти тщпклогекс m 684
918
предметный указатель
Метилциклогексаноны
восстановление 68
галогенирование 178^180
реакции 472
1-Метилциклогексен-1 114
дегидрирование 684
присоединение меркаптокетона 553
N-Метвлциклогексиламип 491
Метилциклопентан 880
производные 771
1-Метплциклопентаиол, хлорирование 871
2-Метилциклопентанол 269
1-Метилциклоиен'1ен, гидратация 269
1-Метилциклонентплгипохлорит 871
Метилцпклопропилкарбинол 69
Метилциклонропилкстон, восстановление
69
S-Метплцпстеамин 602
Метилэтиланилин, N-окись 533
Метплэтилацвтилен, изомеризация 850
Метилэтилендиамин 421
Метилэтидкетон
восстанопление 716
галогенирование 181
реакции 486, 784, 804, 806
Метилэтинилкарбипол, окисление 740
Метлноиые конденсации 808
Метионин 166, 485
Метионинсульфонийподид 586
Метоксиацетилен 691
4-Метоксиацегофенон, галогенирование 183
л-Метокспбепзиловъш спирт 68
4-Метоксл-1-бензоилнафталин, реакции 738
Метоксибензолсульфиновая кислота 601
Метоксибензолсульфохлорид 601
2-Метокспбензофенон, реакции 363
1-Метокси-6-бромгексан 334
Метоксибромпрованпе 130
i-Метоксибутанон-З 281
В-Метоксибутпральдегид 280
2-Метоксидезоксибензоин, реакции 363
Метокеидпфенплсульфпд 585
0-Метоксикетоны, превращение в 0-амино-
кетоны 498
п-Метоксикоричная кислота 803
метиловый эфир 805
Метоксимеркурирование олефинов 653
Метоксиметилацетат, реакции 784
Метоксиметнлизоциаиат 202
4-Метокси-2-метилфенНлуксусная кислота,
реакции 809
Метоксинафталины, галогенирование 142
4’(2-Метоксинафтил)-нафтоАПион-1,2 740
Р-Метокснпропионовый альдо ид 280
а-Метоксистеариновая кислота 824
1-Метоксиуксуспая кислота 334
2-Метокспфенпловый эфир. омыление 364
Метокснфенилпропиоловая кислота, афирьг
692
4-(п-Метокснфеш1Л)-тетразол 376
•и-Метоксофепилуксусная кислота, дезал-
килирование 361
4-Метоксифенилянтарная кислота, эфиры,
реакции 789
2-Метоксифенол-4-сульфокислота 565
2-Метокси-З-фталимидпропилгалогснид 130
2-Метокси-З-фталимидпропилрт утьиодид
130
4-Метокснхриасп 818
Метоксиэтилртуть, ацетат 653
Метоксиянтарная кислота, диметиловый
эфир 280
Миндальная кислота 855
дегидратация 801
нитрил 718
этиловый эфир 215
Миндальный альдегид, изомеризация 854
Мирпцилиодлд, деиодпрование 742
Михаэлиса метод 546
Михаэля реакция 705, 706
Михлера кетон 721
Молочная кислота
аминодиз эфира 455
этерификация 346
Моноацетилэтилендиамин 421
Морфолин
N-алкилирование 466, 490
N-ацетилирование 458
реакции 372, 481, 853
1-Морфолиноциклопентен-1 481
Мочевина /
ацилирование '438
производные 377, 435, 506, 511
реакции/446, 459, 495, 499, 815
хлорирование 129
Муравьиная кислота 871
ортоэфир, реакции 355
реакции 30, 368, 489
хлорангидрид диметиламида 240
эфиры 344, 460, 723
— гидролиз 366
— реакции 353, 455, 787, 789, 808, 809
Муравьиноуксусныи ангидрид 360
Мускон 807
Мьдпьякорганические соединения 665 сл.
Надкислоты 288, 292, 837
мононадфталевая, реакции 262, 264
надбензойная 262, 263, 264, 293
надуксусная 264
правила работы 263
реакции 262, 272, 273, 533, 534, 835,
882
Патрийацетоуксусный эфир, реакцнп 606
Натриймалоновый эфир, реакции 606, 756,
779, 857
Натрииорганические соединения 640, 745
реакции 364, 606, 754 сл.
Нафталин 735
ацетилирование 768, 775
восстановление 22, 25, 28
галогенирование 105, 107, 112 сл.
гидрировайие 48
конденсация в перилси 736
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
91
Нафталин
нитрование 386
окисление 298, 739, 841, 842
сульфирование 567, 572
хлорметилирование 783
На<Ьталин-2,3-диальдегид 317
Нафталин-! ,6-дикарбоновая кислота 300
Нафталиндисульфокислоты 568, 602
Нафталин-1,5-дисульфохлорид 624
Нафталинди^лорид 106
Пафталинсульфиновые кислоты 578( 601
Нафталипсульфокислоты (а и 0) 568
Нафталинсульфохлорид 576, 601, 624
1,2,3,4-Нафгалинтетрахлорид 106
Нафталинтрисульфокислоты 568
Нафтальдегид 295, 317, 322
^-Нафтиламин 496
а-Нафтиламинсульфокислоты 496, 569
диазотирование 539
нитрозирование 400
0-Нафтиламип-1Ч-формаль 456
Нафтиламины
азосочотание 406
N-алкилирование 463, 465, 493, 494
аминирование 408 1
N-арилирование 496, 497
врсстановление 22
дезаминирование 321
диазотирование 540
нитрозирование 400
окисление 841
переаминировапие 503
превращение в а-нафтол 495, 496
сульфирование 569
0-Нафтилацетампд 431
а-Нафтилацетилхлорид, реакции 771
б-Нафтилдиазоппйхлорид 540
р-Нафтилдиметилкетон 80
2,3-Нафтиленгодразин 497
а-Нафтилизотиоцианат 440
а-Нафтилизоцианат, реакции 377
S-a-Нафтилмети.тизотиуронийхлорид 563
а-Нафтплметиловый эфир 331
2-(а-Нафтпл)-проиаиол-1 56
2-Нафтилтиоацетоморфолид 853
Нафтилтиогликолевые кислоты 600
а-Нафтилтиомочевипа. реакции 440
0-Нафтилтолилсулъфон 577
а-Нафтилуксусная кислота 883
0-Нафтилэтиловый эфир 342
реакции 798
Нафтионовая кислота, нитрозирование 400
а-Нафтоилдиазометан, реакции 883
р-Пафтоилпроппоновые кислоты, восста-
новление 80
а-Иафтойная кислота 839
эфиры, реакции 708
?-Нафтойная кислота 840
Гафтоловыи желтый S 512
1,8- Нафтолсульфокпслота, ангидрид 328
Нафтолсульфокислоты 561, 568, 572
реакции 496
Нафтолы 328
азосочетание 405
алкилирование 342
аминирование 408
восстановление 25, 49
нитрозирование 398
окисление 298, 841
реакции 495, 496, 717, 786
сульфирование 561, 568
эфиры простые 331
— сложные 351
«-Нафтонитрил 764
Нафтосультон, щелочное плавление 328
Нафтохиноны 298
восстановление 65
реакции 553, 703
Недокись углерода 688, 829
Нейрин 372
а,0-Непасьпценные кетоны 194, 272 681,
806 сл.
восстановление 79
гидрогалогенирование ИЗ, 118, 119
реакции 137, 262, 265, 266, 281
Ненасыщенные карбоновые кислоты
801 сл.
галогенирование 101 1
гидратация 267
гидрирование 46, 47, 84
декарбоксилирование 816, 822
разложение 827
реакции 872
родановое число 101
эфиры 861
Неоментол 64
Неопентилбромид 220
Неопентилиодид 217, 220
Неопентиловый спирт, реакции 203, 217,
Неопептилхлорид 220
Неролидол, реакции 850
Неролин 342
Несмеянова реакции 636, 651, 667, 669, 670
— синтез 663
Нефа реакция 323
Никотиновая кислота
амид 327
- — дегидрирование 871
N-окись 534
хлорангидрид 238
эфир, аминолиз 456
Никотиновый альдегид 89
Нитрампды 537
Нитрамины 537
Нитрилы 479, 733, 763, 776, 781, 840
алициклические 758
алкилирование 445, 758
ароматические 764, 765
N-ацилирование 445
восстановление 516
дикарбоновых кислот, галогенирование
167, 169
ненасыщенные 707, 765,
— аллилбромпрованпе 137
920
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Нитрилы
ненасыщенные
— галогенирование 123, 167, 169
— реакции 112, 118, 120, 266, 552, 707,
711. 795
омыление 326, 675, 733
разложение 681
реакции 322, 323. 327, 373, 375, 376,
557, 728, 729, 770, 788, 790, 795, 815
циклизация 732
Питроазосоединения, реакции 515
З-Нптроакриловая кислота, гидратация 267
Нитроаминогуапидин 477
5-Нитрп-2-аминотолуол, окисление 536
6-Питро-4-аминохинолин 500
и-Ннтроапнзол 509
Нптроанилипы 387, 388, 523
ацетилирование 450 '
бензоилирование 451
га лорнирование 158
диазотирование 539
реакции 320. 402, 483, 516, 793, 794
Нитроантрахпнои, реакции 604
9-Нитроантрацен 386
Нитроацетанплпд 387, 450
алкилирование 421
арилирование 428
Нитроацстплхлорид 132
Нитроацетофононы 390
оксимы 395
Нитробензальдегиды 389
аниды 390, 412
ацетали 354
восстановление 522
галогенирование 715
диацетат 297
реакции 480, 713, 719, 720, 753, 808
сульфокислоты 590, 833
тиосемикарбазон 477
4-Нитробепзилбромид 147
п-Нитробенэилмеркаптан 597
Нитробензилхлорид, реакции 333, 413, 800
ле-Нитпобензоилазид 444
п-(л'-Нитробензоиламипо)-бензойная кис-
лота, метиловый эфир 434
2-Нитробензонлдиазометан. реакции 883
2-Нптро-4-бензоил-2-метилбутан 796
п-Нитробензоилхлорид 232. 240
Нлтробензойные кислоты 301, 302, 312,
390
хлорангидриды 240
Нитробензол 382
аминирование 409
восстановительное алкилирование 488
восстановление 523, 525, 545
галогенирование 173, 174
мсркурировапле 653
реакции с аридогилелямл 833
л-Ндтробензолсульфенпламид 623
Иптробензолсульфокислоты 567
Ийтробензолсульфохлорпд 601. 614, 624
Нитробензоиитрилы 390, 480. 794
1-1 [птробутапол-2 713
Нитрование 380 сл.
алифатических соединений 383
альдегидов ароматических 389
амидов 537
аминов 387
ароматических соединений 385
гетероциклических соединений 391
с разрывом С —С-связи 510
сульфокислт ароматических 567
фенолов 387
4-Нитрогваякол 320
4-Нитрогеп1ан 443
2-Ннтрогндрохпнон 287
Питрогуанидпн 513, 539
переамидирование 507
2-Нитро-4.6-дибромфенол 257
4-Нитро-2.6-дпиодапииин, диазотирование
539
Нитродпметиланилины 388, 430
3-Нитро-2.6-димотилпиридип 392
л-Нитродифсниламип 428
Нитродифсиилсульфид 585
Питродифенилсульфоны 591
4-Нптродиэтиланилип 430
N-Нитродпэтилмочевина, восстановление
528
Питро-к-додекан 384
Нмтрозамнны 536
восстановление 528
окисление 537, 538
расщепление 531
реакциц 544
Нитрозилмртлл-и-толуолсульфамнд, реак-
ции 885
Нитрозирование 393. 396, 397 с,п,
с разрывом С—С-связи 510
фотохимическое 396
Х-Нптроэо-К’-алкнлапилпны, перегруппи-
ровка ;399
Ншрозоалкйлмочевины» реакций 528, 547
Нитрозоалкйлурстаны, реакции 546
п-Нитрозоанилпн 494
Нш ровоацстанилпд, реакции 795
^-Пптрозо-Я-ацетилглициноный эфир,- ре-
акции 549
Лптрозоацйлалкиламины, реакции 516
Питрозобепзол 536
реакции 413
Нитрозобутап 536
N-Нитрозоглпцип 536
п-Нитрозодиалкнланплины, дезаминирова-
ние 398
4-Нит розо-2,6-дибромфснол 510
Ннтрозодпметиламид 536
п-Ннтрозодпметиланилин
восстановление 528
реакции 294, 295, 318. 390
4-11итрозо-1,2-дпметилб1-Ч1.1о.1, окисление
535
Пнтрозодисульфоцат калия, реакции 298,
2t>9
ГГитрозодиэтпланплин, реакции 112
г
предметный указатель
Нитрозокаиролактам 536
а-Питрозокапроновая кислота 397
2-Нитро.зо-2-карбэтоксицпклопентанон ди-
мерный 396
а-Нитрочомасляная кислота 397
Н-Нлт|)озо-Н-мет1глампноизобутилметил-
кетон. реакции 547
а-Ннгрозо-р-метпламинонафтол 494
_\-Пгтр<.13ом('тнланилин 536
Х-Нптрозо-Х-метилантраииловая кислота,
перегруппировка 399
а-Нитрозо-а-метплбензоплуксусный эфир
397
51-Питрозо-М-метил-л-бром(хлор)аиилин,
перегруппировка 399
Нитрозометилмочевина 536
Нитрозометилуретан 536
Нвтрозинафтолы 397
восстановление 528
реакции 494
о-Г1птрозонитросо₽дин₽йия, окисление 535
2-Нитрозо-5-яитротолуол 536
окисление 535
' Нитрозопиридины. окисление 535
а-Нитрозопропионовая кислота 397
4-Нитрозорезорцип 398
Цитрозосалициловая кислота 399
Пптрозосоедпнения 396, 520, 535
восстановление 527
изомеризация 850
окисление 534
реакции 545
сульфирование 573
Нитрозотимол 398
Нитрозофенол 397
реакции 494
4-Ннтрозо-1\-этил-сб-нафтиламин 399
Н-Нптрозо-Н-этмл-о-толуидин, перегруп-
пировка 399
а-Пптрозоэфиры 502
6-Питроипден-З-карбоновая кислота 884
л-Н|>трокарбобензоксиаминокислоты 434
а-Нмтрокарбоновые кислоты 385
пнтрозированис 397
а-Ннтро-а-карбэтокси-Р-(пндолил-З) -про-
пионовая кислота, эгпловый эфир
796
Питрокорнчиые кислоты 389
о-Нитрокорпчныи альдегид 389
Питрокрезолы 512
Питромалоновый эфир 385, 535
реакции 796
а-Пйтромасляная кислота, эфир 443
3-Пптроме:и1Т(>]1лбен;ш.1кетон 852
1-(3'-Н|1троме-ппоил)-2-феннл»тиленокспд,
изомеризация 852
Нитрометан 442, 815
реакции 707. 713, 730. 810
З-Питро-Х-метилапилин 531
5-Нптро-2-метилбен юлсульфокис.чота 5(57
2-Н|1тр<»-’/1,о-метилендпокспкор11чиая кис-
лота 804
N-II п । ро-]\-метил »-толуолс> льфамид 537
2-Нптро-4-метилфснилсул1.фенплхлорид
619
Нитромочевйиа
амид 506
восстановление 524
Нитронафталинсульфокислоты
восстановление 569
галогенирование 257
Нитронафталины 257, 386, 509
аминирование 409
окисление 842
Нптронафтиламин 409
' диазотирование 539
реакции 509
6-Нитронафтохиион-1,2-д1таз11д-2, отщепл!
ние азота 884
Нитронитриты и нитронитраты 374
Нитроны 318, 376, 412
З-Нитро-4-окситолуол, восстановление 52
1-Нитрооктан 442
Нитроолефины 809,810
восстановление 132
реакции 552, 555
Нитропарафины, реакции 318, 713, 78
5-Нптропентанол-2 67
N-Нптропиразолы 538
Нптроппрпдины 383, 392, 532, 535
N-окись, восстановление 532
Нитропирролы 391
4-Нитропиррол-2-карбоновая кислота, д(
карбоксилирование 391
Нитропропан, реакции 318, 707, 731
Нитросалпцйловая кислота 391
Нитросоедппения 88. 89, 441, 508, 512, 53
восстановление 520, 545
галогенирование 173 '
ненасыщенные см. Нитроолефины
реакции 322, 323. 711, 731
Нитроспирты 809, 810
са-Нитростиролы, восстановление 28
Питротетралины 386
7-Нитро-а-тетралон. оксимпрование 479
о-Нитротиофснол 609
Пнтрошофены 392
З-Нптротолилсульфеыампд 623
З-Нитротолуол-4-сульфохлорид 614
Нитротолуолы 509
галогенирование 146, 147
окисление 297. 301, 389, 390
реакции 390, 740
n-Нитротрифенилам ип 409
п-Нитротрпфонилкарбииол 315
п-НктротриЛепилметпл.хлорнд. гидролиз
315
Нитроуисусиля кислота
деI;а рбоьспли реванш* 815
эфир ИЗ. 535
— реакции 731
ПлipoypcTaiiH 537
о-Пптро-л-фенетидин. ди аэоч провалпе ”3!
и-Пи 1 рофепнл ал к n.i(apiuO сульфоны 613
1-Нит ро-2-фспп.1<1 •1'1 и<>-( ,2-днфепйлшап 731
, 922
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
'и-Нитрофенилацегазид 542
2-Нигрофенилацетамид 883
а-(п-Нитрофенил)-В-бензоилэтилеиокспд
753
га-Ннтрофенил-н-гептиловый эфир 335
п-Нитрофеиилгпдразин 516
п-Нитрофенилдиазопиевые соли, реакции
669
М-(п-Ни-грофенил)-карбазол 409
п-Нитрофснилметплсульфон 612
1-(4'-Нитррфенил)-пиррол-2-альдегпд 499
Я-Нигро-г-фепцлгропанол-! 713
2-Нитро-1 -фенилпроиен-1 810
Нптрофеннлпропполовая кислота, эфиры
692
Р’(л-Нитрофенлл)-сер1ш, этмлодый эфир
713
Нитрофенилсульфенилхлорид 619
о-Нцтрофенилтиогликолевая кислота 604
п-Нитрофенилуксусная кислота, гиДразид
459
о-Нитрофенилэтиленоксид 719, 720
2-Нитрофенол-4,6-дисульфокислота, гало-
генирование 257
Нитрофенолы 320, 387, 512
окисление 287
реакции 499, 500
эфиры простые 335
9-Нитрофлуорен, калиевая соль дг/у-формы
386
Нитрофталевые кислоты 391, 842
Нитрофураны 391
5-Нитрофурфурол 391
ацетат 392 \
5-Нитро-2-хлоррензальдегид, реакции 590
Нитрохлорбензрлы 386
аминирование 430
восстановление 521
реакции 584'
Нитрохинолин ьй
аминирование 409
восстановление 521
нитрование 382
Нитроцпклогексан 830
реакции 318
2-дуи-Нптроциклогексанон 385
Нитроэтан 442
реакции 713, 731
2-Нитроэтапсульфокислота 555
Нитроэтилбензол, нитрозирование 395
2-Ннтроэтилнитрат 501
2-Нитроэтпловыи спирт 501
3-(1-Нитроэтил)-цпклогексанон, реакции
324
Нитрующие агенты 380
Нонантиол 580
Нонилампн, алкилирование 422
н-Нонпловый альдегид 306
н-Нониловый спирт, дегидрирование 306
Норнантарпдпн 704
Норкарадпенкарбоновая кислота, эфир
792
Норхолановая кислота 836
Озазоны 475
сахаров, расщепление 325
Озониды 261, 262, 835
Озонирование 835
Окиси
алкенов, реакции 593, 594
бензалг,ацетофенона 265
изоамилена, реакции 726
мезитила 472, 675
— реакции 266, 839. 848, 849
пропилена, реакции 726. 727, 851
стирола 263
— реакции 728
циклогексена 273
— реакции 593
этилена, реакции 500, 501, 593, 720,
726, 774, 851
а-Окисн 719
изомеризация 851, 878
N-Окиси 427, 502, 533
восстановление 531
изомеризация 857
Окисление
ацилирующее 286
гиногалогенпгами 838 сл.
по Элбсу 287
тетраацетатом свинца 308, 840
«фотосепсибилизированное» 291
S-функций 609
электрохимическое 298
Оксазцраны. расщепление 397
Оксазоллдины 379
Оксазолпдон-2 449
Оксазолины 374
Оксалаты 344
Оксалплбромид 239
3-Оксалил-1,5-дифенилформазап 404
а-Оксалплпропионовая кислота, эфир 789
Оксалилхлорид 232
реакции 775, 776
Оксанилид, реакции 388
Оксетаны 331
Оксиальдегпды 309
галогенирование 178
гидрирование 81
изомеризация 854
расщепление 824
эфиры 715
Р-Окси-а-аминокпслоты 102
2-Окси-4-аминопиримпдпв 328
6-Оксиаптрахинон окисление 287
Оксиантрацены, эфиры 331
2-Оксиарилметилкетоньг, изомеризация
864
«-Окспацетилены, изомеризация 856
4-Окси-1-ацетилциклогексен 856
Оксиацетон 272, 285
Оксиацетофеноны 182, 862, 863
галогенирование 183
реакции 788, 791, 807
Окспбензальдегпды 147, 321, 833
галогенирование 151
реакции 802, 807, 825
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Оксибензальдегиды
эфиры 335
о-Оксибепзилмстиловый эфир 331
Оксибензиловые спирты 63
дегидрирование 304
й-(2-Оксибензопл)-ацетофеноны 864
4-Оксн-1-бензоилкафталин, реакции 738
Оксибензойные кислоты 312, 363, 713, 717
азосочетание 405
галогенирование 142, 151, 153, 169
N-метпламид 458
окисление 287
л-Оксибснзолсульфокислота 328
Оксибензолсульфохлориды 576
о-Оксибензофенон 363
6-Окси-7,8-бензфлуоренон 738
Оксивалериановая кислота, этерификация
346
Р-Оксиглутаровая кислота, дпэтиловый
эфир 69
4-Окси-3,5-дибромбензойная кислота, ни-
трование 510
pt-Оксипзомасляная кислота 286
Оксиизопропплбензойная кислота 831
Оксикапроновая кислота
лактон 76
эфир 837
Оксикетоны 271
восстановление 76, 79, 81
21-Окси-20-кетостероиды 185
4-Окси-1 -кето-1,2,3,4-терагидронафталин
65
Оксикислоты 286
гидрирование 49, 76
декарбоксилирование ароматических
822
производные 837
реакции 214
этерификация 346
а-Ок сикпслоты
окисление 308, 840
реакции 283, 823, 872
р-Оксикислоты
дегидратация 675
расщепление 827
эфиры, дегидратация 675
4-Оксикоричная кислота 802
4-Оксикумарины 791
<х-Оксимасляная кислота 315
этерификация 346
Р-Оксимасляная кислота 267
р-Оксимеркаитаны 593
о-Оксиметплбензойная кислота, N-метил-
амид 458
2-Оксн-2' -метилдибензоилметан 865
а-Оксиметнленциклопентадеканон 789
Оксиметилирование 783 сл.
N-Окснметплкарбонамнды 379
N-Оксиметплпицеридин, реакции 592
а-Оксиметпдсульфоны 559
N-Оксиметилфталнмид 379
2-Оксимеги лцпклогексанон-1-карбоновая-2
кислота, эфир 784
2-Окси-6-метоксиацетофенон, окисление 25
4-Окси-3-метокси-(1)-нитростпрол 810
а-Оксимипоадипиновая кислота, эфир 3£
сс-Оксиминоалкилуксусные эфиры 395
у-Окспминоацетоуксусная кислота 394
а-Оксиминоацетоуксусный эфир 394
а-Оксиминокалроновая кислота 395
а-Оксиминокетояы 393, 394
а-Оксиминонптрилы 395
З-Окспминонентанон-2 394
а-Оксиминоэфиры 395, 442, 443, 502
Оксимирование 479, 510
• Оксимы 132, 371, 393 сл., 524, 536
восстановление 525, 527
окисление 534
расцепление 324
реакции 411, 733
Оксивафталпндисульфокислоты 320, 60
Окспнафталинеульфохлориды 576
2-Оксинафтальдегид-1 778
2-Оксинафтил-1-метилсульфонат натрия
786
Оксинафтойные кислоты, реакции 391, 49
Оксинафтохиноны 298
дегидродимеризация 741
Оксинитрплы 765
дегидратация 675
отщепление HCN 824
2-Окси-З-нитропропионовая кислота 26
Оксипиридины 328
галогенирование 189, 230
окисление 287
2-Оксипиримидин 229
Оксипировияоградная кислота 311
3-Оксипропансульфокислота 555
2-Оксипропйнсульфокислота 594
а-Оксипропилбензойная кислота 299
Р-Оксицропиопитрил 207
а-Оксяпропмоновая кислота, эфиры 71
В-Оксипропионовая кислота 267
Оксипроппофенопы, реакции 183, 789
Оксисульфокислоты 554, 558, 594
а-Оксисульфоны 559
Окситолуиловые кислоты 302, 832
Оксифенантрены, простые эфиры 331
8-Окси-2-фениламинонафта.|Ин-6-сульфо-
кислота 503
п-Оксифениларсоновая кислота 667
2-Оксифениловый эфир 364
1-(2'-Оксифенил)-пентанон-4 870
В-(4-Оксифепил)-проиионовая кислота 15;
р-Окси-Р-фенилпронпоновая кислота, эфи>
751
Оксифенилуксусная кислота 361
галогенирование 152
4-Окснхинолпн-З-карбоновяя кислота, де
карбоксилирование 822
Оксимшолины 287, 321, 498. 822
восстановление 28
галогенирование 154
1-ОксициклогексилуксУсная кислота, эфир
752
924
предметный указатель
2-Окспциклодеканон. дегидрирование 307
0-Окспэтилциклогексен-1 26
З-Оксопентил-2-цпклогексилсульфид 553
1,2,3,4,5,6,7,8-Октагидроалграцея-9-суль-
фокислота, галогенирование 258
Октагмд рофе!i ai(трен 847
Октадекандиеловая кислота 682
Октадеканон-2, окислительное расщепле-
ние 838
Октадецллбензол 773
и-Октадецилподид 211
«-Октаденил-л-толуолсульфонат, реакции
773
Октадиин-3,5-диолы-2,7 740
ДвДО-Окталин 877, 878
Октамешлслхароза 339
Октаиоп-2, реакции 790
Октатриеналь 804
Октахлорантрацен 106
Октахлорфенантреи 106
Октены, изомеры 671
Октиламнн 517
н-Октилацетплен 690
Октилд и моти. । сульфонийи одпд 586
н-Октилмагпийбромид 680
Октллнитриг 442
н-Октилхлорид 466
Октпн-1, реакции 120
Октип-З-он-2 762 х
Олеиловый спирт 84
Олеиновая кислота 680
реакции 101, 274, 446\
щелочное плавление 833
эфир, восстановление 84
Олефины 52 сл. 198, 333, 670 сл., 674,
680, 081, 722, 746, 748
алкилирование 696 сл.
ацилирование 772
галогенирование 92 сл.. 122 сл., 131 сл.,
135, 136
гидратация 266, 268, 269
гидрирование 21 cjc. , 44
гидрогалогенироваыие 111 сл.
гидроксилирование 272 сл.
действие надкислот 262
изомеризация 847
образование алюмпнийорганическнх со-
единений 657
озонирование 261 сл., 835
окисление 272 сл., 286, 288, 290, 832 сл.
окислительное расщепление 261, 832 сл.
окенметилирование 784
полимеризация 260. 665, 697 сл.
присоединение бисульфита 555
— гйлокщов 92. 97, ill сл., 122 сл.,
131 сл.
— гидридов бора 655
— кислот и спиртов 278 сл.
— мрркаптосоедпнеиий 551, 553
— солей ргутп 653
— сульфиновых кислот 555
— SO2 и SO3 554
— тиокарбоновых кислот 553
Олефины
— N-триалкилборазаиов 655
циклизация 684, 685
Оловоорганические соединения 662 сл.
Омыление см. Гидролиз
Олдтема - Резерфорда правило 219
Оппенауэра реакция 308
Орнитин 417
реакции 463
Ортокремневые эфиры.ацеталпронаипе кар-
бопни.ных' соединений 355
Ортомуравьиные эфиры, реакции 460, 498,
724, 753. 789
Ортотиомуравьиный эфир 597
Ортотиоэфиры 597
Ортоуксусный эфир, ацилирующий агент
460
Ортоэфпры, реакции 355, 372, 460, 723,
791, 808
Пааля — Кнорра реакцця 473
Пальмитиновая кислота 311, 674, 833, 838
амид, расщепление 872
эфир, гидролиз 674
Папаверин, дезалкилирование 363
Паральдегид
галогенирование 161, 176
реакции 712, 803
Пеларгоновая кислота 833, 848
Пента-О-ацетилглюкоза 222 сл., 351
Пентаацетилглюкоповая кислота, ширил
824
Пентабензоил-|3-4)-глюкоза 223
Пентабромпиридон 173
ТТентадецилампп 872
Пентадеднлкарбаминовая кислота, метило-
вый эфир 872
Пентадиен, гидрирование 45
Нс'ита-О-мрп ил-р-Д-глюкоэа 341
0,[3-11ентаметиленгл11цидная кислота, эфир
754
Пептамотилендпбромид 646
Пентаметилмышьяк 666
Пентаметилсурьма 669
Пентандиол 274
бис-бензолсульфопат 467
Псптанол-2-оя-4 712
Пентанолы, реакции 203
Пентапопоксим, гидрирование 526
н-Пеитапсульфохлорид 614
Пентафепилмышьяк 666
Пенгафенилсурьма 668
1,1,2,3.3-Пептахлорпропан 708
Пентаэритрит 313
Пентаэритритбромпд 213. 219
Пенген-1 747
I пдробромирование 112, 114
Пегпеновая кислота 45
Т|ентец-2-ол-4 725
Прпгии-З-ол-2, окисление 307
Пентин-3-д>н-2 307
Пепсины 850
Iпдрофторирование 120
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
92
Пептиды 369, 447, 448, 506
реакции 5U7
Переамидировапие 505
Пореамлнирование 502
Перегруппировки 846 сл.
.1 мадори 856
<| |Кплроданпдов 201
аллильная 137. 204, 4(3, 849
Бекера — Веньагпарамаиа 864
Бекмана 526, 874
Брауна 860
бензидиновая 865
Вагпера — Меервейна 876 сл.
Вольфа — Шретера 882 сл.
ишохлоритов 871
дибромциклопептапов 97
диенон-фенольная 879
Канниццаро 855
Клайзена 863
Криге 870
нитрози.рслоридов в хлорокснмы 131
плнаколиновая 878
ретропинаколиновая 879
Рупе 856
Соммле 868
Стивенса 867
Тиффено 869
Фаворского 861
Фаворского и Темниковой 854
Фриса 862
Чепмена 857
Перекиси 260 сл., 288 сл.
борорганические 655
Переэтерификация 352 сл., 368
Перилен 736
и диеновом синтезе 701
Перкина синтез 801 сл.
Перфторсоедпнешгя 132, 646
а-Перхлоргексагриел-1,3,5 679
Перхлорметплмеркаптап 617
восстановление 625
реакции 591
Перхлорнафталин 106
Пиколилкетоны, реакции 707
Пиколиновая кислота 301
декарбоксилирование 815
Пиколины
аминирование 408
окисление 300. 301
реакции 808
Пикрамид 495
диазотирование 539
Пикриловые эфиры 337
•Никрплхлорпд 230
реакции 430
Пикриновая кислою 380, 512
ред.цпи 230. 493
11 и мели новая кислота
декарбоксилирование 818
а-замгчценная 844
Ппнаколитшая перегруппировка 878
Пннаколиновый спирт. до|идрагацпн
СТО
Пияаколины 878
алкилирование 758, 681
окисление 838
Пинаконы 56, 715 сл.
дегидратация 682
изомеризация 878
Пинан, расщепление 684
Липацианол, -иодид 808, 809
Пинен
окисление 834
реакции 2.79
а-Пиноновая кислота 834
'Пиперазин, реакции 490
Пиперидиды лития и натрия 409
4-Пинеридил-1-Н11тронафталпн 409
Пиперидпнацетонитрил 244
у-Пиперпдип-N-бутиронитрил 424
2-(Пппериди(|метил)-т11офсп 187
Пиперидины 23, 28, 29, 503
Малкилировамис 424
арилирование 428
реакция 490, 494, 804
Пиперонал
дезалкилирование 363
дифенилмеркапталь 596
Пиперониловый спирт 59
Пиразолальдегнд-3 373
Пиразолал1.дегид-3-карбоновая-4 кислот?
373
Пиразолпны 504, 710
Пиразолы 373, 475, 710
Пиран 331
Ппрантрон 737
Пирен
восстановление 25
изомеризация 852
сульфирование 569
Пнрен-З-альдегид, восстановление 83
Пирна реакция 573
2-Пиридилацетанилид 874
ГТиридилпиридиниевые солп, реакции 504
4-Ппридплфепиловые эфиры, расщепление
500
М-Ппрпцпл-4-пиридинпйхлорпд 188, 4(1
З-Пирпдплпропчоповая кислота 30
Пиридин 501, 815
алкилирование 423
ацилирование 437
восстановление 23. 28, 29
галогенирование 187, 188
гомологи, окисление 300
комплекс с СгОп 309
— с SO3 560, 571
меркурировяпие 652
нитрование 382. 383. 392
N-пкпсь 502, 533, 534
реакции 741. 798. 859, 810
сульфирование 570
3-Пиридинальдо| пд 323
ПириднндикарбоНовые кислоты 842
Ииридцнипбромплы 8,39
пербромид 99, 100
Пирпдипийподид S1O
926
предметный указатель
Пирпдинкарбоновые кислоты 831
декарбоксилирование 822
окисление 534
этерификация 345
Пиридинпикрат 230
Пиридинсульфокислоты 610
реакции 328
Пиридинсульфотриоксид 560, 571
Пиридивсульфохлориды 624
Пирилиевые соли, реакции 501
Пировиноградная кислота 820
азосочетание 404
диэтилмеркаптол, окисление 613
полумеркапталь 558
Пирогаллол
галогенирование 151
триметиловый эфир, галогенирование
142
Порфирины 808
Пирокатехин 65, 361
галогенирование 142, 151
дегидрирование 309
реакции 770, 782
эфир, реакции 781
у-Пироны 815
реакции 499, 5Й6
Пирослизевая кислота 310, 312
декарбоксилирование 815
эфиры, реакции 768
Пирохимпческпе реакции 735
Пиррол
восстановление 27, 50
галогенирование-Т87
нитрование 391
производные, реакции 798
расщепление 505
реакции 702
сульфирование 571
формилпрованпе 777, 778
Пирролальдегпд 778
реакции 808
Ппрролизпдпн 410
Пирролидины‘50 , 410 , 500 , 503
Пирролии 27
Пиррол-М-карбоновая кислота, эфир 795
Пирролы 473
Полиазоксисоедпненпя 534
Полиазосоединения 545
Полиалкилбензолы. сульфирование 89
Полиарилкетоны 739
Полиены 683, 753
галогенирование 97
Полисульфоны 554
Полиэтилен 699
хлорирование 133
О,N-Полуацетали 470. 480
Полукеталя бензоилированные 870
Полумеркаптали 558
бромпроизводные 259
Прево реактив 276
Д4-Прегпендпол-110,20(3-он-3-а ль-21, реак-
ции 855
Проиитол 881
Принса метод 784
реакция 707
Пробковая кислота, динитрпл
галогенирование 167
омыление 328
Пропан, сульфохлорирование 575
Пропандпон-1.3, галогенирование 206
Пропанол-1 29, 31
окисление 310
Пропаргилбромпд 212
Пропаргилгалогениды 752
Пропаргиловая кислота, галогенирование
- 104
Пропаргиловый альдегид 303
реакции 373
Пропаргиловый спирт 827
гидратация 272
окисление 303
реакции 212
Пропенилбензол 76
а-Пропенилппрпдин 808
н-Проппламин, реакции 471
я-Пропплаиплин 466, 493
N-Пропплацетальдимин 453
N-н-Пропилацетамид 447
я-Пропплацстплен, изомеризация 850
я-Проппл-я-бензальдегид 816
я-Пропилбензол 76, 80, 744
реакции 816
N-Пропилбутиральдпмин 471
М-Проппл-М-винилацетамид 453
Пропилен 676
алкилирование бензола 696
гидрогалогенпрование 111, 112, 114
Пропиленхлоргидрпн 206
Пропилиденуксусная кислота, реакции
554
Пропплизопропилкарбпнол 726
Пропилкетоны, галоформная реакция 839
н-Пропилмагиийбромид, реакции 722
2-н-Пропплпнрролидин 410
я-Цропил-я-фенилглиоксалевая кислота,
эфир 816
н-Пропилхлорнд 206
я-Пропилциклобутаи 82
Пропин, гидрофторпрованпе 120
Пропиоловая кислота 691
восстановление 52
Пропиолонптрпл 765
(о-Пропионил-о-оксиацетофенои 788
М-Прошюнилфталимид 451
Пропионилхлорид, хлорирование 162
Пропионовая кислота 310
ангидрид, реакции 802
эфир, реакции 788
Пропионовый альдегид 303, 85 Г
реакции 713
феиилгидразон, реакции 403
Пропиофенон
восстановление 80
галоформная реакция 839
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Пропиофенон
изомеризация 852
Простые эфиры 278, 279, 329
галогенирование 117 сл., 159 сл.
расщепление 225 сл., 36-1 сл., 635
реакции 288, 291. 742
Протеины, диазотирование 541
Протокатеховая кислота 287
Псевдоионон 807
Псевдокумолсульфнновая кислота 578
ангидрид 624 ,
Пурпурин, сульфирование 573
Пшорра синтез 795
Рашига метод 543
Резорциновый альдегид 777
Рморцпловые кислоты, декарбоксилирова-
ние 822
Резорцин 328, 822
галогенирование 142
гидрирование 49
нитрозирование 398
реакции 282, 496, 640, 777, 782
сульфирование 573
эфир 758
Реймера — Тимана реакция 778
Рейссерта соединение 74, 75
Реппе синтез 683
Ретропинаколиновая перегруппировка 879
Реформатского реакция 751
Рида метод 573
Рицидолевая кислота
дегидратация 682
окисление 834
Робинзона — Манниха метод 707
Родан 92, 101, 191, 625
реакции 591 у
4-Родапанилпн 191
Родангвдрины, 201
Роданиды 201, 377, 591
ароматические 257
расщепление 616
реакции 614
Роданин, реакции 809
Роданирование 92, 101, 201, 190, 257
Родановое число 101
5-Родансалициловая кислота 257
Роземунда — Брауна метод 764
Ртутьацетамид 270
Ртутьорганические соединения 649 сл.
реакции 639, 647
Ртуть-п-толуолсульфамид 270
Рубрена перекись 261
Рупе перегруппировка 856
.©-Сабинол, восстановление 77
Салигенин, простой эфир 331
Салициловая кислота
восстановление 844
галогенирование 154, 169, 171
дикалиевая соль 717
нитрование 391
Салициловая кислота
реакции 801
эфиры, реакции 49, 570
Сахарин 302
Сахароза, исчерпывающее метилировавг
338, 341
Свартса реакция 193
Свиисцорганическпе соединения 664 сл.
Себацнновая кислота
динитрил, галогенирование 167
дихлорангидрид 246
моноэфир'хлорангидрида 236
• Семикарбазид 507, 524
реакции 497
Семикарбазоны 323, 474, 477
расщепление 83. 324, 325
Семихиноны, перегруппировка 879
Серенсена метод 416
.О,£-Серин 130
Сероокись углерода 625
реакции 380, 605, 627, 628
Сероуглерод, реакции 379, 617, 627
Сероорганичсскпе соединения 550 сл.
Силаны 659
Симонипи комплекс 149
Спналар 129
Синильная кислота, реакции 710 сл.,
717 сл,, 733, 791
Скатилмалоновая кислота 796
Сложноафпрная конденсация 706
Сложные эфиры 83, 271, 278, 313, 342
347, 348,- 786 сл.
восстановление 83 сл.
галогенирование 101, 163
омыление 365 сл.
пиролиз 673
реакции 137, 723 сл., 785
третичных спиртов 347
Соммле реакция 317, 823, 869
Сорбиновая кислота 803
гидрирование 46, 47
9,9-Спиробифлуорен-2,2-дпсульфокислота
572
Спиро{3,4]октанол-5 57
Спирты 55 сл., 83 сл., 285
алкилирование 329 сл.
ацетиленовые см. Ацетиленовые спир
ты
ацилирование кетенами 282
восстановление 75 сл. \
галогензамещенные 225
галогенирование 99, 202 сл., 217 сл.
дегидратация 671 сл., 682, 686, 68'
дегидрирование 303 сл.
дегвдродпмерпзация 742
ненасыщенные 286, 784
окисление 136, 288, 306, 309, 31р
окислительное расщепление 836 сл.
яергидролиэ 289
этерификация 329, 343 сл.
Станнаны 662, 663
Стеариновая кислота 838
хлорангидрид, реакции 761
928
предметный указатель
Стероидные '
гормоны ЯЗВ
спирты 268
— реакции 260
Стероиды 129
введение С—С-связей 198
восстановление 27
галогенирование 98, 179, 203
гидроксилирование 274
N-хлорирование 550
эпоксидирование 266
Стибиновые кислоты 669
Стибипы 668, 669
Стибониевые соединения 665, 669
Стибоновые кислоты 669
Стивенса перегруппировка 867
Стпльбен
галогенирование 94, 96, 97, 132
димеризаиия 697
производные 793, 800, 808
роданнрование 101
Стильбен-4-альдегид 317
Стильбендибромид, реакции 766
га,п'-Стпльбендпкарбоновая кислота 799
Р-Стиракриловая кислота 802
Стирол 816
бромирование 97. 100
гидробромцрование 111, 112
окисление 832
оксиметплирование 784
полимеризация 699
реакции 101, 260, 263. 553, 654, 689.
707, 708 - - - - . ...
хлорметилирование 783
Стлролбромлд 100
Стиролдибромид 97
реакции 689
Стрихнин 866
Сукцинимид, переамидпроваппе 506-
Сукцинимидоуксусная кислота 452
Сультоны 575
Сульфамиды 421, 621
алкилирование 465
N-гетероциклические 622
гидролиз 624. 625
Сульфаниловые кислоты 523
Сульфаниламиды, галогенирование 157
Сульфанилдиазонипсульфат 546
Сульфаниловая кислота 565
реакции 257, 387. 408
Сульфонамиды 623
Сульфепплбромиды 259
Сульфенилтиоцианаты. расщепление 620
Сульфенилхлориды 591, 617, 618, 619
реакции 553. 583. 604, 623
Сульфоновые кислоты, эфиры 623
Сульфонамиды 622
Суиьфинопыс кислоты 578, 606. 604, 606,
607, 616. 620
амиды 622
восстановление 608
реакции 555, 559
эфиры 622
Сульфинилхлориды, реакции 622
Сульфирование 559 сл.
бисульфитом 592
Сульфобензойная кислота 567
ангидрид, галогенирование 173
Сульфокислоты 592, 610, 611, 620
алифатические 589
ангидриды 621
ароматические 561
— производные 341. 563
— реакции 302, 511
выделение 562
галогенирование 257 сл.
Iидролиз 89
фторангидриды 196
(Сульфоксиды 611
ароматические 577
окисление 612
Сульфометилирование 786
Сульфониевые соедпнения 585. 592, 598, 605
Сульфоиилмочевины 378
Сульфоны 258, 577, 590, 601, 611, 612 613
расщепление 616
реакции 711
циклические 554
Сульфоокисленне 560
Сульфосалициловая кислота, метиловый
эфир 570
4-Сульфофталевый ангидрид 570
Сульфохлориды 613, 623
ароматические 601
восстановление 606
реакции 258, 620
Сульфохлорирование 573 сл.
Сурьмы органические производные 668
Сэндвичсобразные соединения 665
Таксогены 697
Тапсневая кислота, тетрабромдинптрил 169
D-Тартрамидгидразид 477
Тахистерии 830
Твитчелла реактив 368
Телогены 697
Тсломерпзация 697
Терефталевая кислот а
восстановление 23
диимидазолид 506
динитрпл. омыление 328
Терефталевый альдегид 147
ацетали 351
Терпеновые спирты, дегидратация 673
Терпены, реакции 116. 131, 279. 304,
396. 573
Терпивен 673
мутоокислсчию 261
Терпинеол. реакции 673
Терпинолен 673
Тестостеронацетат 182
Тетраазосоединенпя, сочетание 407
2.2,3,3-Тетраалкилэ1пленимипьг 375
Тотра-я-аппзплэтиленсульфцд 616
Тетраарплэтаны 741
Тетраарнлэтнленсульфпды б 16
предметный указатель
Т?|раарилэтилоны, эпоксидирование 266
Тетраацетат свинца 285
как окислитель 308, 840
Тетра-О-ацетмл-Р-беняилглюкоэид 338
Тотраацетилглюкозиламин- 224, 530
Тетраацетилглюкозплазид, восстановление
530
2,3,4,6-Тетраацетил-а-0-глюкоэо-1-изотио-
цианат 202
Tcipa-O-ацетил-р-метилглюкозид 337
2,3,4,6-Тетраацетил-а,[}-этилтио-£)-гл10КО-
зид, реакции 259
i,1.1,3-Тетрабромацетон 182
1,2,3,4-Тетрабромбутан, дегалогенирование
682
Тстрабромдикарбоновые кислоты 168
Тетрабромдинитрилы 169
Тетрабромксилолы 147
гидролиз 317
Тетрабромметан 199
реакции с олефинами 709
2,3,5,6-Тетрабром-4-оксибензойиая кислота
172
хлорангидрид 235
2,3,5,6-Тетрабромпиридон 173
1,2,3,4-Тетрабром-1,2,3.4-тетрагид ропафта-
лин 107
Тетрабромтиофеп 186
1,1,2,2-Тетрабромфенплэтап 104
Тетрабромфталевая кислота, ангидрид 173
1,1.2,2-Тетрабромэтан 104
Тетрагидрокарбазол 867
дегидрирование 685
Тетрагидроксилиленгликоль, реакции 674
Тетрагидронафтолы-2 49
1,2,3,4-Тстрагидро-8-оксг1хинолпн 28
To'ipai идроииран
расщепление 225
реакции «500
Тетрагидрофталазпн. конденсация с форм-
альдр| и,том 483
Тетрапц^офталевая кислота, ангидрид
Теграгпдрофуран 50, 208, 331
।идронерекись 242
правила обращения 291, 292
расщепление 225, 226, 362
Тетрагидрофурилаллиловый спирт 46. 50
изомеризация 856
Гетрагидрофурфурплбромпд 212
Тетрагпдрофурфурилевый спирт
окисление 306
реакции 212, 213
Тетрагпдрохиполин-2 771
Тетрадекатексаен-2,4,6,8,10,12-овая кис-
.лота 804
Тетразен 404
Тетралины 483
Тетразолы 376
Тетраподметап 200
2.3.4.5-Tei раиодпиррол 187
Тетраиодфта.1гвая кислота, ангидрид 173
Тетралллто.’ныо льфон 577
59 Заказ 1835,
Тетралин 48. 781
гидроперекись 290
дегидрирование 684
нитрование 386
окисление 290. 296
производные 781
реакции 51. 625
Р-(4-Тетралон-1)-пронионовая кислота 7'
а-Тетралон-З-сульфокислота 497
Тетралоны 296. 774
восстановление 60
Тетрамоти ламмошшбромид, бромаддукты
100
Тетраметилборат лития 655
1,2,3,4-Тетраметилбутадпен, реакции 7(
п.п'-Тетраметплдиаминодифенилэтилен-
сульфид 616
Тетраметплондпмагнлйбромид 646
Те । ра-О-метил-а-метилглюкозид 338
Тетраметилсвинец 664
Тетраметплсилан 659, 660
Тетраметнлстибонийиодид, реакции 669
Тетраметилэтилсн 673
эпоксидирование 262 1
Тетраметилянтарная кислота, деметилирс
ванне 368
Тетранитробечзолы 386
2,4,2',4'-Тетранптродпфепилсулг.фпд 584
Тетранитрометап 382
Тетрафенилаллен 686, 687
Тетрафениларсонпй 666
Тетрафенилацетон. реакции 687
Тетрафенилборат натрия 656
Тетрафрнилбутадпены 45. 700
Тетрафенилгексанентаеп 45
Тетрафепилгидразип 544
2,2,4.6-Тетрафепил-2.3-дигпдро-силл-трп-
азин 377
Тетрафенилметан 792
Тетрафенилпропплен, реакции 686
а.а.у.у-Тотрафенилпропнловый спирт 72.
реакции 664
Тетрафенилспипец 66i
Тетрафенилфталевая кислота, ангидрид 82i
Тетрафенилциклоиентадиеноп 294, 295, 80
реакции 826
Tei рафенилатан 766
Тетрафенплэтилен 681, 799. 800, 879
гидрирование 44
Тотрафенилэтиленсульфид 616
Тетрафон л лэтилрнсульфон 601
Тетраформаль-трпс-азпн 475
Тетрафторборат арилдиазония, реакции 65.’
Тетрафторгексахлорциклогрксан 105
Тетрафторэтплен, иодирование 97
Тетрахлоралюминийсульфинат 578
Тетрахлорацетальдегнд см. Хлораль
4.5,6,7-Тетрахлорбопзтиазол 143
Тетрахлордикарбоновые кислоты 167
динитрилы 167
Тетрахлордииито илы 169
Тетрахлор [0,2.4|о ктен 95
Тотрахлортолуол ы 106
930
предметный указатель
1,1,2, 2-Тетрахлорэтан, дегалогеп ирон апис
135
Тетрахлорэтилен, бромирование 94
Тетраэгилолово 662
Те»раэтилпирофосфит, реакции 449
Тетраэтилсвинец 664, 665
Тетрил 388, 538
Тетроловая кислота 693
Тиазолидоны-4 556
да-Тиааолины 556
Тиазолы, изомеризация 866
Тиазтиовийхлориды 580
1,3,5- Тпациклогексян 595
Тненилакриловаи кислота 802
^-Тиенил-2-аланин 756
Тиенилацетилмалоновыи эфир 756
1-(2-Тиенил)-этиловый спирт 64
Тимана синтез 848 ‘
Тимол 780
галогенирование 152
гидрирование 49
нитрозировапие 398
роданид 191
щелочное плавление 832
Тиоальдегиды 594
'Тиоамиды 598
Тиоангидриды карбоновых кислот, цикли-
ческие 259
6-Тиоантрахпноил-1-антрахинон-2-карбо-
новая кислота 600
Тиоацетали 827
реакции 259
Тиоацетамид 598
Тиоацетон 595
Тпобензамцд 598
Тиобензанилид 598
'гиобензойяая кислота 588, 605
реакции 580
Тиобензофенон 587, 596, 603
Тиогидаптоин, реакции 809
Тяогидрохпнон 600
Тиогликолевые кислоты 328, 580, 582
реакции 592
эфиры, реакции 556
Тиоглицерин 593
Тиодигликоль, простые эфиры 330
8,Р'-Тиодидроплоновая кислота 551
Тиопмины 557
Тиоиндокспл 771
Тиокамфора 596
Тнокарбаминовая иислота
N-производные О-ариловые эфиры
630
соли 628
эфиры 629
Тиокарбаминоилхлорпды 629
реакции 630
ЭД,№-Тиокарбоннлдипмадазол 436, 448
реакции 440
Тиокарбоновые кислоты 588, 605
амиды 557
реакции 553, 580
Тиокарбоновые кислоты
эфиры 597
— реакции 89
Тиокетоны 579, 586, 594, 603, 615
ароматические, реакции 616
восстановительное расщепление 89
Тиокрезолы 30, 115, 607
реакции 592, 614, 615
эфир 598
Тиоксантен, дегидродимеризацпя 741
Тиоксантион 587
Тиоксантов, восстановление 57
Тиолкарбоновые кислоты 589
эфиры 553
Тиолтионугольная кислота 626
Тиолугольная кислота 626
Тиолы см. Меркаптаны
Тиомочевины 377, 379
арилиронанные, расщепление 631
реакции 446, 513, 581
токсичность 74
Тионамиды 557, 605, 606
Тионафтендпальдегид-2,3 318
8-Тионафтол 607
Тионилариламины, реаиции 546
Тионилбромпд 216
М'.Х'-Тионилдппмидазол 448
Тионилхлорид
очистка 215
реакции 799
Тионкарбонаты 628
Тпонкарбоповые кислоты 603, 605
Тпононановая кислота 588
Тионугольная кислота 626
эфир, реакции 605
4-Тиолфлавон 603
Тионы см. Тиокетоны
Тпооксиндол-З-альдегид, анил 799
Тиопиридоны 587
4-Тио-у-пирон 596
Тпопропионовая кислота 605
Тш'салпциловая кислота 580,. 584,
608
Тиосемикарбазид 377
Тиосемикарбазоны 477
Тиосульфиновые ипслоты, эфиры 611
Тиосульфокислоты
реакции 620
эфиры 611
Тяотолуйловые кислоты 605
Тиоугольная кислота, хлорангидрид
б-4-хлорфениловый эфир 626
S-эфир, реакция 630
Тпоугольные кислоты 625 , 626
эфиры 627
Тиоуксусная кислота 588
реакции 554, 557, 580, 581, 602
эфиры 597
Тиоуретаны 629
Тиофен
галогенирование 186, 187
меркур'ированпе 652, 653
нитрование 392
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
93
Тиофен
реакции 774, 798
элиминирование серы 90
Тиофенальдегиды 187, 317% 724
дпацеталь 724
реакции 713, 802
Тпофенолы 111, 115, 599, 606
реакции 551 сл., 557, 583
хлорирование 614, 618
эфиры, реакции 639
р-Тиофеисерин 713
Тпофен-2-сульфиновая кислота 607
Тиофенсульфонилхлорид 392
восстановление 607
4-Тпофлавон 596
Тиофлуоренон 596
Тиоформальдегид 595
Тиофосген 617, 625
реакции 436, 587, 627, 629, 631
Тиохпнальдонр 587
Тиоцианаты ей. Роданиды
Тиоциановая кислота, реакции 629, 630
Тиоэтиленгликоль 593, 609
Тиоэтилтпобензилметан 588
Тиоэфиры 328. 551 ел., 579, 583 сл., 585,
592, 598, 603, 604, 615 сл., 619
восстановительное расщепление 89, 90,
608
галогенирование 259
окисление 611
хлорзамещеиные 558
Тирамин 815
Тирозин, реакции 815
Тироксин 152
Тиурамдисульфиды 630
Тиурониевые соли 513
Тиффено перегруппировка 869
Тищенко реакция 312
Тобиаса кислота 569
Тозиламинокарбоновые кислоты, реакции
239
Тозилаты см. Толуолсульфокислота, эфиры
N-Тозилнафтиламины, азосочетание 406
Толандибромид, дебромпрование 692
Толилальдегпды 322, 777
реакции 313
1-ге-Толиламино-1-дезокси-£>-фруктбза 856
о-Толиларилкетопы, оксимирование 479
Толилацетилеп 689
о-Толплбензойвая кислота 326
о-Тилилглиоксалевая кислота, эфир 762
n-Толилглиоксалевая кислота 302
п-Толи.ткарбинол 313
»-Толпл-л-ксилилсульфон 577
Толплметилкетон 299
окисление 302
Толилнафтилсульфиды 585
Х-(о-Толил)-ц|1перпдин 419
Толилсульфенилхлорид 618
реакции 604
п-Толплтрифенилметил сульфид 592
1-л-Толпл-1-хлорэтплен, реакции 689
п-Толуидпн-£>-глюкозид, изомеризация 856
59*
Толуидины 410
реакции 191, 400, 472. 503, 781
Толуиловые альдегиды 227, 317, 318
Толуиловые кислоты 23, 299, 300, 831
дегидрирование 799
2-(о-Толуилокси)-ацетофенон, эфир, изом<
ризация 865
Р-Толуплпроиионовые кислоты, воссташ
вление 80
о-Толунитрпл, омыление 326
Толуол 84, 684
аминирование 410
галогенирование 106, 141, 142, 145, 30
меркурирование 653
нитрозирование 396
окисление 285
реакции 735
сульфирование 564
Толуолсульфамиды
окисление 302
расщепление 422
п-ТолуолсульФинамид 623 .
n-Толуолсульфинат калия 620
n-Толуолсульфиновая кислота 578, 60
ангидрид 624
октиловый эфир 622
п-Толуолсульфобромид 615
п-Толуолсульфокислота 561
выделение 562
реакции 330, 414
эфиры 218
— реакции 220, 341, 466, 755, 773
Г<-(п-Толуолсульфонил)-фталпмид 438
п-Толуолсульфохлорид 601, 614, 624
алкилирующее средство 342
Траубе реакция 459
£>,-£-Треонии 130
Трпазены 406
Триазины 376. 377, 463. 508
Триазолиды 506
Трпалкиларсины 665
Триалкилациламмониевые соли 437
N-Трпалкилборазаны, .реакции 655
’"риалкилстибины 668
Триалкилуксусные кислоты 842
Триаллилацетофснон 756
2,4,6-Триаллилфенол 864
2,4’б-Триаминопиридин 408
Трпамсинолон 129
Триариларспны 666
1,3,5-Триарилбензолы 810
Триарилкарбинолы, восстановление 77
Триарилмстильные радикалы, реакцш
636
Триарилстибпны 668
Три-О-ацетил-Л-глюкаль 223
Трибензиларсин 666
3,5,6-Трибепзил-а-глюкоза, ацетониды,
гидролиз 370
Трибепзилолово, галогениды 663
Трибензоилметан 760
2,4,6-Трлброманилия 140, 151, 257, 82.
1 Д 3-Трибромацетон 182
932
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Триб ромацетофепон ьг 183
изомеризация 855
2,4.6-Трибромбепзо1П1ая кислота. 140
Трлбромбензол 88, 139
1,2,3-Трибромбутан, дегпдробромироваиие
888
3,4,5-Трибромпирид1ш 230
2,4,6-Трпбр<;>мпирпмидшг, реакции 335
3,4,5-Триб рочпирокатехин 151
1.2.3-Трмбромпропан. дегидробромирова-
ни? 135
2,4.6-Трпбромреаорцпн 142, 151
. Трибромтлофеп 186
а,<Х,а-Трмбром-2,4,6-трихлорацетифснон
838
Трибромуксусная кислота 166
декарбоксилирование 815
1,2,3-Трибром-1-фспилпропан 100
2,4,6-Трибромфепол 151, 825
Трпбромфеполбромлд 151
Трибутилацетамид, омыление 322
Трибутилуксусная кислота 322
Три-трет-бутоксиалюмогпдрпд лития 75
1,1,1-Тр1палогенацетон, гидролиз 318
ТригалогснМетилкетоньг, расщепление 185
w-Тридекановая кислота, глицерид 345
2,4,6-Три-(дпбромметпл)-1,3,5-триазип 169
2,4,6-Три-(диметилампнометил)-фенол 786
Тринлзобутилалюминий 657
Триизопронаполамин, реакции 419, 420
2,4,6-Триизопроиплбепзилхлорид 783
2,4,6-Триизопропнлбепзол, хлормотилиро-
вание 783
Трииоданилин, реакции 402
2.3,5-Трииодбензойная кислота 251
Трииоднптробензолы 251
Трииодсткрол 104
Трпиодуксуспая кислота, декарбоксили-
рование 814
2,4,6-Трииодфенол 152, 154
Т'ри-.н-ксплплсгибпн 668
Трилоп Б 707
Тримезитилппнилопый спирт 722
Триметилалюмппий 656
Триметилаыип 485
гидрат окиси 533
окись 533
реакции 688
Триметилбензиламмоннй 266, 281
Триметилбензиловый спирт 869
Триметилбетаиншдразнд, хлорид 426
Триметилбор 655
Триметилбутан 77
2,2,3-Триметплбутанол-1, восстановление
77
2,4.6-Трич(>тн..1П'ксагидро-1,3 5-грпатн
470
1,1,2-Тримотп пиди ;цдап 781
2,2,4 -Три метилди гид ро\ оно. i ин 172
Триме । и. юиокепд
производные 33!
реакции 500. 774
'Гриме-i ил-малкаиы 313
Триметилолово, галогениды 663
2,4.6-Триметплпиридин, ши рование 302
1.2.6-Триметилпирпдон-4 499
Триметилпировнноградная кислота 838
Триметилстибипдибромид, реакции 669
Трпметилтетралип 781
Тримстил-(2,2,3,3-тетрахлорбутил)-аммо-
нийиодид 693
2,4,6-Три.четилтриазин 463
Триметилуксусная кислота 838
окисление 302
хлорангидрид, реакции 761
2,4,6-Трпметилфенол, азосочетание 105
Триметилэтилен 671
реакции 267, 553
Триметоксиборгадрид натрия 30
2.2,4-Триметоксибутан 284
Тринптроапи.юл, реакция 603
Тринитробеизойная кислота 302
Тринитробензол 385
2,4,6-Тринитробонзолазомсзйтилен 400
Трияптрозофлороглюцип 398
2 4 6-Тринптро-З-оксибензойпая кислота
380
Тринитротолуол 385
окислении 302
реакции 390
2,4,6-Трин11трофсиилметнлнитрам11я 538
1,2,4-Триокспантрахицоп-З-сульфок ио-
лота 573
Трппропиламин, реакции 688
Трм-и-цроппларсин 666
Триптамин 30, 402
Триптан 77
Трпптицен 764
Триптофаны 796, 797
2.4,6-Трис-(бензолазо)-резорцин 405
2,4.6-Трис-(бензолязо)-фенол 405
2,4,6-Трис-(ди.хлорметил)-1,3,5-триазин
377
Трпс-(0-оксиэт11л)-амлноксид 501
1,3,5-Трис-(трифторметил)-силл-триазип
193
я-Тритианы 595
Тритил 339
б-О-Трптил-а-ТЭ-глюкоза 339
Тритиловые эфиры глицерина 340
Трптиоацеталвдогид 595
Тритиобензальдегпд 595
Тритиомуравьиный эфир, реакции 812
Тритиоугольная кислота 626
Тритиоформальдегид 595
Трптпоциклогексавон 595
Тритиоциклопентанон 595
Три-п-толплкарбинол. сульфирование 593
Трп-п-толплмртапсульфок и слота 593
Т ри-и-тридеци. и л и церин 343
Гритон Г> 266, 28 Г
Грифк ни.типип 427
Грифеии [ампп-о-карбнновая кислота 128
Трифен илами ногуакпдпн 376
Трмф('НП.|Арс11н 666
Трифопилацетампд. омыление 322
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
9
Трифенил виниловый спирт 766
Трифенилгалогенметан, реакции 333
Трнфенплгуаиидин 376
Трпфепилен 704
Трифепилизоамиловый эфир 330
Трифенилкарбинолы 286, 723
реакции 329. 592
сульфирование 593
эфиры, расщепление 362
Трифепллметан 77
галогенирование 146
окисление 286
производные 782
Трифеиилметаи-о-карбоновая кислота 77
Трифон пл метановые красители 286
Трифенплметансульфокпслота 593
Трмфенилметпла перекись 293
Трифенилметплсульфоновая кислота, эфир
623
Три^юнилметилфепилсульфид 620
Тркфенклмотилфосфонийбромит;, реакции
812, 813
2.4,6-Трифецилпирпдин 502
2,4,6-Трифенплпирилий, реакция 502
1,1,2-Трифенилпропен-1 681
2.4,6-Трифенилтриазил 377
Трифенилуксусиая кислота 322
Трифенилфосфии
как восстановитель 516
окись 530
'Грифснплфосфинбензилиден, реакции 813
Трпфен»глфосф|’ндифенилметилиден, реак-
ции 687
Трифенилфосфипимин, реакции 470
Т ряфенилфосфинметплнден 813
реакции 812
Трифенилфосхрит 216, 217
Трифенплхлормстап 146
алкилирующий агент 339, 340
Трифенилэтилеп 681
бромирование 97
Трифторацетаты
диазония 321
серебра 140
трпэтиламмонпя 273
Трифторацогилирование аминокислот 457
4.4,4-Трпфторбутен-1 678
Трифториодметан 140
Трифтрркроюновий эфир, реакции 113
Трлфторладуксусняя кислота, реакции 262,
273. 538
Трифторуксусная кислота 193, 194
аш идрид 194
фениловый эфир, реакции 457
1,1,2-'Е[»ифт(>р-2-.\лор-3'фепилциклобут11-
-теп. гидролиз 316
Трихлорацотнлхлорид. реакции 766
Т рпхлорацетонитрил. фторп рование 193
2,4,6-Трихлорацетофенон, галоформная ре-
акция 838
со-ТрН.хлорэцетоф печ 766
Трпхлорбромметан 199
Трихлордимегилопыи эфир 210
Трихлорметилдиметилкарбипол 714
4-Трихлорметилппридин 190
1,2,3-Трихлор-2-иетилпропан 96
Три хлорметилсульфенилхлорнд 617
1,1,1-Трихлорпропилснокспд-2,3 719
2,6,8-Трихлорпурип 229
Трихлоруксусная кислота 166
декарбоксилирование 814
хлоран|идркд 237
Трпхлорфе1гоксиуксуеиая кислота 155
Трпхлорэтилен, реакции 71 >8
Трнхлорэтиловый спирт 59, 175
Трнэтиламин, реакции 800
Триэтил аммоний, трифгорацегат 273
Триэтиларснн 666
Триэтилбор 655
Триэтйлбутепилалюмппий 658
Триэтилкарбинол 724
Трпэтилнеопентоксисилан 220
Трпэтокси । риз гиламип 335
Тромановыс алкалоиды, реакции 366
Тродплиден 117, 683
Тропин 792
Тропинов, нитрозирование 393
Тростниковый сахар, исчерпывающее М(
тидпрование 338
Туйоп 77, 680
Туйон 680
Туйонампд 680
Углеводороды 70, 75 сл., 78 сл.
ароматические см. Ароматические угле
водороды
металлирование 636, 639, 612
окисление 285 сл., 288, 289. 297
Углеводы
ацетаты, дезацилированпе 367
ацилирование 357
бензилиденовые производные .356
восстановление 66, 69
галогензамещенные 222 сл.
изопропилиденовые производные 356
357
— гидролиз 370
меркаптали 596
- расщепление 371
метилирование 337, 338
— исчерпывающее 338, 341
окисление альдегидных групп 311
оксимирование 479
трпфепилметплоные эфиры 339
трифторуксусиыо эфиры, расщеплонИ!
368
установление структуры 2'9
фенрлгидраяопы 478
— расщепление 325
Угольная кислота, эфиры 724
реакции 790
Уксусная кислота, хлорирование 162, Ilk'
Уксусный ашпдрид 282. 359_
реакции 360. 762. 772. 77л. 792. 801
809, 821. 821. 850
Ул,.у/1'1<7 реакция 310, 427
934
предметный указатель
Ундекадиин-1,7, восстановление 53
в-Ундекан 80
Ундекановая кислота 46
нитрил 763
Ундеценнлацетат, присоединение НВг 117
Ундецен-7-ин-1 53
Ундеценовая кислота, гидрирование 46
Уидеценол, присоединение НВг 117;
Ундецилбромид 819
Ундецилеповая кислота 826
присоединение НВг 119
щелочное плавление 833, 848
Ундецилпзоцианат 444
Уреидокислоты 377
N-Урепдопиридпны 502
Уреидостпльбенальдегид 371
Уретаны 435, 506, 873
алкилирование 537
ацилирование 438
расщепление фталевым ангидридом 452
Уретдионы 378
Уроновые кислоты 311
Уротропин 470, 486
алкилирование 417, 464, 468
реакции 317, 323, 799
Фаворского
перегруппировка 861
реакция 283, 284, 714
Фаворского и Темниковой перегруппировка
854
Фарнезол 849, 850
а-Фелландреп, реакции 703
Фенамин 414
Фенантрен 78, 735, 865
галогенирование 105 сл., 142, 143
гидрирование 48
изомеризация 852
окисление 298
производные 794, 795
сульфирование 569
Фенантренкарбоновые кислоты замещен-
ные 795
9,10-Фенантренхинон 298
полумеркапталъ 558
Феиантрол, восстановление 78
Фенацилбензилметилсулъфонийбромид,
изомеризация 868
2-Фенацилпирпдпнокслм, изомеризация
874
р-Фенетиламины 418
Фенетол 342
> расщепление 365
Фенилазид 542
восстановление 530
Фенйлаланин 167, 485, 526 •
реакции 454
Фенплалкплкетоны. 729
Феиилаллпловые эфиры, изомеризация
863, 864
1-Фенилаллиловый спирт бромирование
100
а-Фснилампноалкановые кислоты 733
1-Фениламино-2,4-динитронафталпн 468
1-Фениламино-2-нитро-1-фенилбутан 731
1-Фениламнно-2-нитро-1-фенилпроиан 731
4-Фениламинопиридин 505
1-Фениламино-1-фенилбутанон-3 734
2-Феннл-7-аминохинолин 497
N-Фенилантраниловая кислота, арплиро-
вание 428
фенилацетальдегпд 306, 323, 841, 872
окисление 295
Фенилацетамид 883
Фенилацетат, изомеризация 862, 863
Фенплацетилен 97, 689, 690, 692
восстановление 53
галогенирование 104, 123, 132
гидратация 269
реакции 284, 763
Фенилацетнлкарбинол, изомеризация 854
Фенилацетон, восстановительное аминиро-
вание 487
Фенилацетонитрил
омыление 326
реакции 788
4-Фенилбензампд 776
1 -Фенил-2-бепзилацетилен 7 49
Фенплбенэялкарбинол 728, 865
Фенилбензиловый эфир, расщепление
365
Фенилбенаилсульфид 604
Фенплбензплсульфон 591
Фенилбензоат 347
4-Фенилбензоилппразолины 373, 710
а-Фенил-Р-бензо1глиропионовая кислота,
нитрил 711
Фенилбордибромид 655
Фенилбутадиены 682
галогенирование 98
гидрирование 46
Фенилбутилены 46
4-Фенилбутиловый спирт, дегидратация.
781
у-Фенил-н-бутиральдегид 873
а-Фенилбутирамид 431
2-Фенпл-н-гексан 696
7-Фенплгеитатрненалъ 807
Фенплгпдразпн 515
алкилирование 427
гидрохлорид 516
реакции 483, 542. 866, 867
Фепилгидразопы 322, 497
ацетофенона 475
бензальдегида, окисление 534
а-кетоадипиновой кислоты 402
метилглпоксаля 402
отщепление аммиака 866
формальдегида 475
циклопенз андиона-1,2 402
эфира мезоксалепой кислоты 402
Фенилгидроксиламин 525
окисление 536
Фенилгликозпды 338
Фенилглиоксалевая кислота, реакции
713
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
9с
Фенилглиоксаль 295
изомеризация 855
конденсация 715
Фенилглиоксиловая кислота 302, 367
Феийлглицидол, эфиры, изомеризация 851
Фенилгорчичное масло 74
Фенилдиазопийхлорид 540
Фенил-3,4-дигидронафталины, дегидриро-
вание 881
4-Феппл-1,3-диоксан 707, 708. 784
Р-Фепплдитиокарбазицовая кислота 380
N-Фенилдитиокарбамат калия 629
Фепилендиамипы 530
ацилирование 449
диазотирование 541
сульфирование 573
о-Фенплендиацетонитрил, реакции 811
р-Фенплизовалериановая кислота 839
Фенилпзокротоновая кислота 802
Фенилизонптрил 440
Фенилизоцианат 439, 444, 872
реакции с HCN 734 -
N-Фенплимидомуравьиный эфир 461
2-Фенилиндол 866
Фенилиодидхлорид 99
N-Фенилкарбазол 428
с-фенилкапроновая кислота, хлорангид-'
рид, реакции 771
Фенил-у-кислота 503
а-Фепилкоричная кислота, нитрил, эпо-
ксидирование 266
Фенпллптий 638, 730
димер 634
реакции 364, 606, 692, 728, 744, 745,
758, 759, 865
Фепплмагнийбромид 721 сл.
реакции 686, 672, 721
фенилмагнийляорид 644
У’фенилмасляная кислота 774
хлорангидрид 236
Фенилмеркаптан 608
реакции 596
Р-Фенилмеркаптобензплацетон 551
Р-Фенилмеркаитогидрокорпчная кислота
552
Фенилметансульфокислота, отщепление
SO2 258
Фенилметилгпдразин 476
Фенилметилглицидная кислота, эфиры
754, 817
Фенплметилдикетон 324
р-фенплмолочная кислота, окисление 841 ’
фенмлмочевина 506
фенилнатрпй 641, 642
реакции 606
Фенилнафталнн 685, 881
N-Фенил-а-нафтплампи 503
Фенпл-р-нафтилкетон, реакции 716
Фенилнафтилсульфиды 585
Феннлнитрампн 538
Фенил-ацы-нитроацетонитрпл 384
Фенллпитрометан 384
оксиметилирование 784
реакции с бензальдегидом 731
1-Фенпл-2-иитроиропая 29
1-Фенил-4-(л-питрофенил)-бутадиеп-1,3 73
N-ФенплоксинДол 773
дегидрирование 800
2-Фенил-7-оксихино.чпи, реакции 497
Фенил-(Р-оксиэтил)-сульфид 593
фенилпараконовая кислота 802
Фени.цпентадекагептаеналь 807
З-Фенилпимелпноная кислота, хлоран-
гидрид, реакции 771
М-Фенплпшюридин 428, 500
4-Фенилппразолип-3-карбоновая кислоп
эфир 373
Феяилпнровиноградная кислота, оксш
восстановление 526
М-Фениллиррол, гидрирование 51
2-Фенилппрролидпл 488
2-Фенилш1ррол-1\'-карбоновая кислота,
эфир 795
1-Фенилпропандиол-1,8, ацетильное пре
нзводное 707
1-Фенилпролаядион-1,2 271
Фенилпропаргиловый альдегид 869
восстановление 52
р-Фенилпропилалкиламяны 422
1-фенилпропин-2-ол-1, окисление 308
1-Фенилпцопин-2-он-1 308
Фенилиропиоловая кислота 691, 692
Фенилпропионовая кислота, хлорангидри,
235
а-Фенплиропионоцый альдегид 817
Фенилртуть, ацетат 652
N-Фенилсемикарбазид 507
окисление 541
Фенилсмланы 661
N-Фенилсукцинимнд 446, 452
Фенилсульфенплхлориды 618
реакции 553, 604
Фенилсульфиновая кислота, реакции 55!
р-Фспилсульфомасляная кислота 555
Фенилтиоацетамид 598
Фенилтиогликолевая кислота, хлорангид
рид, реакции 771
Фенил-ге-толплсульфид 604
Фенилтолилсульфон 577
Фенплтрпхлорсилан 661
Фенилуксусная кислота 326
бензиловый эфир 344
галогенирование 164, 234
реакции 802
хлорангидрид 234
— реакции 772
ф-Фенплуксусный эфир 792
Н-Фенплундекапентаеналь 807
N-Фенил-р-фенилэтпламин 372
Фенил-р-фенилэтилсульфон, щелочное рас
щеиленпе 616
N-Фенплфталимпд, нитрование 388
2-Фенилхпнолин-4-карбоновая кислота,
эфир 347
936
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
1-Фенил-1-хлор-2-окспмино-3-ацстоксп-
проиап 374
- а-Фенил-а-хлорпроп»юпитрил 794
Фенил-п-ллорфенилсульфон 577
2-Фенил-1-хлорэтиловый эфир 221
Фенил цикл обутендиол 316 _
1-Фенилэйкозанон 761
р-Фонплэтилазпд, восстановление 530
Феиплэтнламин 487, 517, 530
реакции 511
N-формильное производное 456
N-a-Феяплэтилацртамид 374
Фенплэтилбромиды 112, 208
Фснидятпловый спирт 774
дегидрирование 306
реакции 773
Фенилэтиловый эфир 342
5-(а-Фснилэтил)-семиоксамгидразид 477
2-р-Фенилэтилциклогексанон 852
Фенилэтилциклогексеи, циклизация 846
р-Фепилэтплциклогексеноксид, изомериза-
ция 852
Феноксиацетилен 690
р-(п-Феноксибепэоил)-проиионовая кис-
лота, восстановление 83
4-Феноксипиридин, реакции 500
Фенокспуксусная кислота, галогенирова-
ние 155
4>-(п-феноксифенил)-масляная кислота 83
Фенол 328. 365. 717, 870
гидрирование 49
гомолонг, щелочное плавление 832
сульфирование 565, 572
Фенолальдегиды 799
восстановление 63
Феполднсульфокислоты, галогенирование
150
Фенолкарбоновые кислоты 832
Фенолкетоиы 776, 862
Феноловые эфиры
галогенирование 154, 155
изомеризации 862
реакции 639
Фенолсульфокислоты 565
Фенол-л-сульфокислый натрии 573
Фенолы 314, 316. 320. 361, 879
алкилирование 331, 335, 310 сл.,
696. 781
ацилирование 282. 349 сл., 770
iTi.noi сяпровааие 150 сл..
диизо тронание 412
Miioi отимиыс 151. 287. 717
Окисление 287. 298. 309
реакции 78, 190. 229, 405. 495. 770,
776, 778. 784. 785.
т рил одза мещеппые 153
эфиры, расщнн.чеппе 364
Фенотиазин 579
Фенхоловая кислота 813
Фенхон, гидролиз 843
Феофилактова метод 530
Фиттига синтез параконовых кислот 802
Фитол 753, 849
Фишера синтезы 515, 528, 718, 866
Фишера — Хеппа перегруппировка 399
Флаваноны 807
Флавонбензнлимин, реакции 603
Флавоны 864
Флороглюцин 328
реакции 282 , 337 , 442 , 495
триоксим 495
эфиры 331
Флуорен 735, 736
нитрование 386
нитрозирование 396
окисление 289, 295
Флуорен-2-альдегид 323
Флуорен-9-карбоновая кислота, эфиры
707
Флуореконпмин 470
Флуоренонкарбоновыс кислоты, восста-
новление 57
Флуореноны 295, 316, 795, 832
восстановление 57
реакции 470, 472, 687
Формазаны 401 сл., 508
Формазилглиоксалонам кислота 404
N-Формальанилин 456
Формальдазпн 475
Формальдегид 726
ацетали 354, 714
оксим 794
О, N-полуацетали 378
реакции 221, 222, 312. 313. 482. 483,
681, 707 сл.. 807, 808, 827, 869
фенплгпдразон 475
N-Формальментиламип 456
N-Формаль-р-нафтиламип 456
N-формаль-!-фенплэтпламин 456
формамидин 450. 461, 469, 508
метилсульфат 469
N.bi’-производпые 375
Формамиды 440. 505
реакции 446, 409, 491. 492. 798, 809
Форманилпды 462
алкилирование 421
реакции 469
Формиаты 344
Формилбензойная кпелоза 8|7, 812
Формилде.юкуибснзопи 878
Формпли|1(пи1пие 446. 789. 798. 799
N-Формплпрование 511
2-Формилокси-3.4-днкетопентап 860
2-Фо рмплпнр рол. восстановление 83
Формилфторид 196, 231
Формилхлорид 776
Формпмид<|беи.Н1лоц1>гй эфир, тримери.кшля
463
Формимидоэфпрьт 469
Фосгсп 823
амидирование 435
реакции 439. 627, 631, 775, 790
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Фосфннимины 530
реакции 470
Фосфоразопроизводные 447
Фосфорилены, реакции 812
•Фосфорилирование 195
Фотонптрозироваиие .396
Фотохлорирование 133
Фриделя — Крафтса реакция, 587, 765,
774 сл.
Фридлендера синтез 811
Фриса перегруппировка 350, 862
/J-Фруктоза, восстановление 66
Фгалазон 414
о-Фталальдегнд 317
Фталан 331, 332. 354
перекиси 292, 293
Фталаипл, нитрование 388
Фталевая кислота 300, 841
анилид 446, 451
бнс-амидин 452
гидразиды 453
диальдегнд, реакции 354, 812
дифениловый эфир 350
дихлорангидрид, реакции 847, 859
нитрование 391
окисление 264
тиоапгндрид 259
эфир, восстановление 84
Фталевый ангидрид 147
галогенирование 173
реакции 673, 774, 782, 866
сульфирование 570
Фталеины 783
восстановление 77
Фталилгидразпд 229
Фталиллейцилвалин, этиловый эфир 449
Фтал1гл-£-.цейцин, реакции 449
Фталпловый спирт, эфиры 331, 332
Фталплхлорид, реакции 438
Фталимид 452, 842, 866
алкилирование 414
калия 415
нитрование 391
переамидированпе 506
Фталимидмалоновый эфир 416
натриевое производное 417
«-фталимидо-ы-галогеналканы 442
Фталпмидокарбоновые кислоты 415
4-Фталимидомасляная кислота 458
о-(фталимпдомгтил)-бензойная кислота
• 458
Фталоилампиокпслоты 452
реакции 239
л-фталоплхлорид 233. 234, 238, 259
Фгалоновая кислота 842
декарбоксилирование 817
Фторапглдрпды ИИ, 196. 23!
Фторацетамид 455
ш-фгорацетофепон 246
реакции 721
о-Фторбепзальбромид. гидролиз 316
с-Фторбензальдегид 316
п-Фторбензойиая кислота 255
Фторбензол 255
нитрование 197, 386
4-Фторбензолсульфохлорид 576
Фторбораты диазония 254, 321, 5’<
о-фторбромбецзол, реакции 704
10-Фтордеканол, окисление 310
Фторирование 132
ароматических аминов 253
— углеводородов 253
гетероциклов 256
замещением 132, 139. 191
присоединением 93, 105
спиртов 202
углеводов 224
электрохимическое 132
Фторкетоны 246
Фтормуравьиная кислота, эфиры 202
фторпиридины 256
Фторспирты 228
21-Фторстероиды 194
Фторуксусный эфир 196
амлнолиз 455
л-Фторфснилуксусная кислота 256
Фторфенол 256
реакции 329
Фторэтилены, галогендровапис 97
2-Фторэтиловый спирт 197. 228
Фульвены 798
восстановление 24, 28
Фумаровая кислота 676, 678, 67'
842
дихлорангидрид 238
нитрил 765
реакции 94, 262, 267, 358, 700
эфпр 811
Фуран 331, 815
ацетилирование 768
галогенирование 160, 186, 187
гидрирование 50
меркурмрование 652
реакции 702, 704
Фураи-2-акрнловая кислота 802, 804
фуран-2,5-дикарбоновая кислота, нитрс
ванне 391
Фуран-2-карбоновая кислота, эфир, алки
лированпе 842
ф\ рап-2-сумьфокислота 571
а-Фурйлакролеин, гидрирование 46
фурплаллилопый спирт 46
Фурфуральфлуорен, восстановление 24
Фурфурилидспацетои 806
ч-Фурфурплпденкротоновая кислота, этп
лоный эфпр 805
фурфуриловый спирт 77. 312
фурфурплхлорпд, реакции 333
ФурФЯЮЛ
гидрирование 50. «1
дегидрирование окислительное 812
нитрование 391
938
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
ФУРФУРОЛ
окисление 310, 312
реакции 499, 715, 803
Халкон 28
оксид, изомеризация 878
эпоксидирование 266
Хасса, правило 575
Хекера метод 497
Хелидоновая кислота, декарбоксилирова-
ние 815
Хинальдин
гидрохлорид, реакции 808
иодэтилат. реакции 808, 809
Хинальдинкарбоновая-3 кислота, этило-
вый эфир 811
Хинальдиновая кислота, декарбоксили-
рование 815
Хинизарин, сульфирование 573
Хннизарин-6-сульфокпслота 570
Хинкеля синтез ароматических альдегидов
777
п-Хпнолацетат 497
Хинолин 815
аминирование 408, 409
гидрирование 28, 29. 51
псевдооснования 858
окислительная димеризация 741
окислительное растпеилспие 842
реакции 287. 496, 730
сульфирование 570
Хпнолин-S 74
Хинолинкарбоновые кислоты, этерифика-
ция 345
Хинолинхпнондиазид, реакции 884
о-Хинондиазиды 884
п-Хиноноксимы, гидролиз 324
Хиноны 298, 299, 309
реакции 552, 556, 685, 794
а-Хлоракрплонитрил 103
1-Хлоралкины 149
Хлораль 175
восстановление 59
гидролиз 318, 845
окисление 166
полумеркапталь 558
реакции 378. 719
5-Хлор-н-амилацетат 225
Хлорамин Т 142
4-Хлор-2-ам11но-6-метоксиплримпдпн, ре-
акции 335
2-Хлор-4-аминохинолин, реакции 335
Хлорамфеникол 511
Хлорангидриды карбоновых кислот 162,
’ 232,235. 236, 239
Хлоранизолы 156
аминирование 429
Хлоранил, реакции 685
Хлоранилины 521 ’
реакции 461. 503. 793
а-Хлор-р-арилпроппонолая кислота, эфиры
а-Хлор-р-арилпропионовые альдегиды 123
Хлорацетальдегяд 174
сульфокислота 555
Хлорацетилдпфенпламин, циклизация 773
Хлорацетилхлорид, реакции 770
Хлорацетон 180
алкилирующий агент 424
Хлорбспзальдегиды 147, 248, 306
аминирование восстановительное 487
галогенирование 176
реакции 590
n-Хлорбензальхлорпд, гидролиз 316
Хлорбензиловые спирты 86
дегидрирование 306
а-Хлорбензиловый эфир 228
а-Хлор-Р-бензоплпропионовая кислота Д19
2-Хлорбензоилхлорид 176
Хлорбензойиыс кислоты 301
эфиры, восстановление 86
Хлорбензол 142, 199
нитрование 386 '
омыление 316
Хлорбензолдисульфокислота 564
n-Хлорбензолсульфиновая кислота 578
п-Хлорбензолсульфохлорид 576, 601, 624
2-Хлорбензтиазол 259
реакции 335
Хлорбромбензолы 199, 249
1-Хлор-4-бромбутап 226
2-Хлорбутан, реакции 764
4-Хлорбутапол-1 225. 362
Хлорбутапсульфокнслоты 575
Хлорбутансульфохлорпды 575
2-Хлорбутен-2, реакции 554, 693
Хлорбутенолы 126, 314
дегидрохлорирование 691
4-Хлор-н-бутилацетат 225
4-Хлорбутиловые эфиры 363
а-Хлор-я-бутиловый спирт 226
а-Хлорбутирилхлорид 162
у-Хлорбутиронптрил, алкилирующий
агент 424, 758
В-Хлорвпнплбутиловый эфир 678
р-Хлорвпнилкетоны 772
2-Хлорвинилртуть, хлорид 654
6-Хлоргексанон-2 871
1-Хлоргексен, реакции 727
6-Хлоргексин-1 746
и-Хлоргентадецен-8.9 680
Хлоргептины 214, 215
Х.’1оргидрины_125, 128, 206 сл.
аллилового эфира 126
ацетона 83
глицерина 207, 334
реакции - 765
«-Хлоргидрокоричная кислота 51
сс-Хлордикарбоповые кпелоты. дпэфиры 168
3-Хлор-2,4-дицетопентан, реакций 860
2-Хлор-4,6-д>1метилпприм11Д1)ны 229
Р-Хлорпаобутирплхлорид 163
а-Хлорпзовалорлановая кислота, аылд-
рпд- 162
а-Хлорнзсмасляпая кислота 163
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
9
а-(р)-Хлоризопропилбензолы 146
1-Хлоризохромап, изомеризация 859
З-Хлорлпдазол 229
а-Хлор-р-иодэтилацетат 177
Хлорирование
аминов 157, 247, 253
алканов 133 сл.
альдегидов 118, 174 сл.
ароматических углеводородов 105, 106,
139, 145
ацетиленов 148
гетероциклических соединений 187, 188,
258
замещением 133 сл., 139, 145, 148 сл.,
155, 157, 159
карбоновых кислот 161
кетонов 178, 180
нитрилов 167, 169
окислительное 614, 619
олефинов 94, 96, 135
присоединением 94 сл., 99, 105, 106,
114, 116 сл., 121 сл.
простых эфиров 159
спиртов 99, 119, 203, 205, 206, 211
сульфидов 614
сульфокислот 257 сл.
углеводов 222
фенолов 150
эфиров 99. 199
N-Хлорирование 550
сс-Хлоркарбоновая кислота, хлорангидрид
162
4-Хлоркарбостирил 315
2-Хлор-5-карбэтоксипиримлдин 258
Хлоркетоны 245, 246
n-Хлоркоричная кирлота, гидрирование 51
а-Хлоркротоновый альдегид, реакции 553
л-Хлор-л-ксилол, окисление 301
З-Хлоркумиловая кислота 831
(о-ХлоркумВл 696
Хлормалоновая кислота 831
Хлормасляная кислота 161, 162, 167
нитрил 764
Хлормезитилсн ИЗ
л-Хлормеркурибспзойная кислота 155
2-Хлормеркури-З-метплтиофен 189
о-Хлормеркурифенол 155
Хлорметаны, реакции 708
Хлорметплбензилсульфпд, реакции 588
2-Хлор-3-метплбутадисн-1,3 122
2-Хлор-2-метилбутанои-3, оксим 132
2-Хлор-2-метилбутш1-3. реакции 687
Хлорметилгексахлорциклогексан 106
Хлорметплиропание 147, 782 сл.
Хлорметнлмезитилен, реакции 764
З-Хлор-2-метилмезобензант рок 112
6-Хлорметил-2-метилураци.'1 136
1-Хлоряртилнафталин, реакции 317
2-Хлор-4-метилпиримидин 229
Хлорметплсулъфенплхлорнд 619
реакции 583
2-Хлорметилтилфен 187
реакции 756
5-Хлорметилурацил 136
2-Хлор-4-метилхинолин 229
1-Хлор-1-метилциклогексан 114
2-Хлор-2-метилциклогексанон 180
Хлорметилэтилсульфид 558
Хлормочсвина 129
, Хлормуравьиныс эфиры, реакции 435, 6
1-Хлорпафталип 106, 142
1-Хлорнафталин-2-сульфенамид 623
Хлорнафталинсульфохлориды 576
N-Хлор-Х-нитрамины 537
а-Хлорнитрплы 169
' о-Хлорнитробензол, дехлорирование 7.
л-Хлор-о-нитробензолсульфенампд 623
1-Хлор-1 -нитропропан, дегидрохлориро-
вание 800
а-Хлоршгтросоедпнения 132
2-Хлор-6-нитротолуол, окисление 831
4-Хлор-1-(л-нитрофенил)-бутен-2 793
а-Хлор-В-(п-нптрофенил)-цропионитрпл
124
л-Хлор-о-питрофенилсульфенилброьшд б!
1-Х лор-1-нитроциклогексан 396
Хлорнониловый альдегид, бронирован!
680
ct-Хлор-В-оксиизовалерианоиая кислота
128
а-Хлорокспмы 131, 132
2-Хлороктан 199
Хлоропрен 121
Хлороформ 845
галогенирование 199
реакции 689, 708, 714, 778
1-Хлорпентен 214
4-Хлорииридин 188, 230
2-Хлорпиримидин 229
2-Хлорироиан 114
З-Хлорпропандпол-1,2 207
1-Хлорпропан-2-сульфохлорид 613
2-Хлорпроппловый спирт 125, 128
2-Хлор-2-пропплтетраг1!дропиран 748, 74
0-Хлорпропиональ, окисление 311
а-Хлорпропионилхлорид 162, 163
З-Хлорпропионитрил 120
Хлорпропионовая кислота 245, 311
амид 771
6-Хлорпурип, реакции 585
Хлорсиланы 660, 661
ш-Хлорсппрты 206
Хлорстильбены 123, 793
Хлорстиролы 136
реакции 692
Х-Хлорсукцинимид 136
реакции 550
л-Хлортпофенол 607
Хлор-.м-толуилопая кислота 301
2-Хлортолуолсульфохлорид 624
Хлортолуолы 106, 248
окисление 301
Хлоруксуспая кислота
гидролиз 315
реакции .328
эфир 817
940
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Хлорфеваш роны 253
2-Хлор-1-фенилпропаи, аминирование 414
Хлорфенилиропполовые кислоты, эфиры
692
Хлорфенилпронионовыи альдегид 124
диэтилацеталъ 118
а-Хлорфепилуксусная кислота 215
метиловый эфир 215
Хлорфонолы 150. 250. 321
азосочетание 405
5-Хлорф> рфгральдоксим, восстаиоцлепш
526
5-Хлорфурфурпламин 526
Хлорциан, трлмеризяция 376
1-Хлорциклогексеп-1, реакцн и 553
Хлорциклонен, анолы 129
2-Хлорциклонентанон, реакции 317
З-Хлорцшслопентен-1 116
гидролиз 314
реакции 317
2-Хлорлтпюксалин-З-карбоиовая кислота,
эфир 335
Хлорэтан 363
2-(6-Хлорэтил)-беизалг.дегид 859
а-л ли рэтилбекзол 146
а-Хлорэтилбутиловый эфир 118
Хлорэтилспкарбонат 174
Р-Хлорэтилизотиоцианат 631
(й-Хлор-со-этилмеркаптоацетофеноп 855
В-Хлоротплпропилкетон 124
Хлорэтплсульфенилхлориды 6(9
6-Х.ТорЭТИЛ10ЛИЛсуЛЬфон 612
р-Хлорэтилфспилсульфид 553
а-Хлорэтиловый эфир 221, 228
Хлорэфиры 159
Холановая кислота, расщепление 836
Холантреи, производные 811
Холестаднен 364
ЛМ-Холсстадненон-З, изомеризация 880
Холестаноп 179
Л4-Холестендиоп-3,6 297
Холестенолбензоаты 847
Холестенон 307, 308
Холестерин
дегидрирование 307, 308
окисление 297
хлорирование 99
эфиры, разложение 364
Холин, реакции 800
Хризенол 795
Хризенсульфокнслота 572
Хромай, галогенирование 186
Хромиллевые соли 502
Хромилхлорид 128
Хррмоны 789
Хюннига метод 481
Целлюлоза, ацетат, дезацилировапнр 368
Церевитинова метод 726
ЦетилдиметилсульфонииНодид 586
Цетиловый спирт
ацеталь 356
реакции 214
Цетнлциатшд, омыление 327
Цианалкнлирование 552
1-Циан-2-а.1|Иилмеркаптоииклогексаны 552
Цианамид
алкилирование 422, 464
ацилпропппие 438
димеризация и тримеризация 376
производные 436
реакции 375, 376
Цианаты 377
алкилирование 445
Цпанацетилмонсвина 448
Р-Цпапбензплмалоповый эфир 7(1
и-Циапбспзплмеркаптаны, де: идрпрование
609
4-Цианбензойная кислота, гидрирование
517
2-Цпанбутан 764
а-Циапбутилгидроксиламин 733
Циангидрины 481
реакции 484, 485, 717 сл., 781
З-Циап-4-иминопиперидин 733
Цианпповые красители 812
Цианкарбоповые кислоты 328
эфиры 787, 790
а-Цпанпропилгидроксилампн 733
Цианпнридины, реакции 323. 327
Циапстпльбен, омыление 327
Цпануксусная кислота, омыление 327
Циануксусный эфир
алкилирование 757
иминолиз 455
гидрирование 367
гидролиз 367
нитрозирование 395
реакции 459, 782. 804. 805
Цианурхлорид 376
Цианформамиды 734
1 -Цианциклогексен-1, реакции 552
Цианэтилирование 707
Циглера синтезы 268. 656
Циклиммопиевые соли, гидрирование 30.
31
Циклобутан 72
производные 756
Циклобутанон, производные 702
Циклобутилампн, реакции 885
Цпклогексадисн. реакции 260. 702. 704
Циклогексан 47 , 880
аллилбромпрование 137
нитрозирование 396
окисление 830
реакции 132, 829
Циклогександнол-1.2 273, 277
Циклогександион-1,2 861
моноокспм 395
Цпклогександиоп-1,3. алкилирование 757
Цпклогександион-3,5-карб<>но1>ая кисло га
49
Циклогексанов ‘49
дегидратация 672. 675
др| ил рирование 3(19
окислительное риг’цеп.'нчтИе 837
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Циклогсксанол
реакции 206. 492. 804
Циклогексаиодовы 706
простые эфиры, расщепление 362
Циклогексанол-2-сульфбкпслота 554
Циклогексанон 309, 807, 818
цнмстилкеталь 356
пнтрозировапие 393
окислительпое расщепление 837
реакции 472, 595, 813, 867, 885
Цпклоггксапонкарбоновыс кислоты 844
эфиры 395. 784
Цпклогексанопоксим 396, 480
изомеризация 874
Циклогексен 672
ацилирование 772
галогенирование 96
гидрогалогеиирование 114, 117
дке-гпдроксилированпе 277
изомеризация 671
озонирование 835
окись 273
окисление 834
расщепление 829
реакции 553, 696
Циклогексепплдифенилкарбинол 727
Цпклогексенплуксуспая кислота, декар-
боксилирование 822
Циклогексенов
производные 706
эпоксидирование 266
Циклогексенсульфпд, реакции 615
Ццклогоксен-1-сульфокислота 554
Цпклогексепуксусная кислота 675
Цпклогсксиламин 50, 491, 492
арилировапие 429
N-Циклогексиланилип 429
Цпклогексилбензол 606
ЦнклогексилбрсЯипд 208
6-1 |иклогексплгексип-1 851
Цпклогексил-1,4-диизоцианат 439
Циклогексилдифгниламип 429
Цмклогексиленсульфид 593
Циклогексилизотноцпанат, реакции 605
Циклогексилизоцианат 440
5-Циклогексилпентин-1 851
N-Циклогексилшшерпдин 467, 491
N-Циклогрксплпирролндин 51
З-Цпклогекснлиропин-1,удлинение цепи 851
N-Цпклогексилтиобензампд 605
Б-Цпклогексилтломочс'вина 631
Циклогексилфенилацетонитрил 758
Циклтекспл.хлорпд 114, 206
Цик 'нмептадецен, бромирование 692
Цпк.югептадиен. бромирование 683
Цнклоюиганон 885
восстановление 683
оксим, реакции 683
Цпклогептатрпен 683
гидрогалогенировапис 117
Цик.тогевтеи
гидрокси, птронание 276
peaKniiii 1>8.'1
Циклогеитиламин, реакции 683
Циклогептиловый спирт, реакции 683
а-Циклогерапиол, реакции 304
Циклодекапдпоп-1,2 307
Циклодецсн 55
Циклодеипн 694
Циклооктатетраеп 28. 683
галогенирование 95
Ццклооьтак 692
Циклоолефины 319. 813
ароматизация 881
озонирование 835
окисление 834, 835
окислительное расщепление 834
I (иклопарафипы
ацилирование 771
окисление 830
Ццклопевтадопапоп, реакции 789
Циклопентадецеи, бромирование 692
Циклопентаднен
алкилирование 758
галогенирование 97
гидрогалогенирование 116
гидроксилирование 278
нптрозированме 396
реакции 260. 701-, 703, 798
Цпклопсптэдиенильные производные
таллов 665
Циклопентадиопоп, реакции 825
Цпклоиентандион-1,2 317
монофенилгидразон 402
Циклолентанол 885
Циклоиентанон 358, 817
производные 807
реакции 481, 595
Циклопентанонкарбоновая кислота
азосочетапие 402
питрозирование 396
эфир 791
— реакции 395. 789, 790
Циклопептен
окисление 286
присоединение HCIO 129
Циклоиентепилкумила перекись 294
Циклоиентеи-1-ол-З 314
эфир 330
ЦНклопентен-1-он-З 317
Циклопентилам пн 526
Цпклопентплбромнд 213
Циклоиентилметиловый спирт, дегидрат
ция 881
Цикловен гилхлорид 205
Циклопропаны 710, 750, 756
Цикломропплцианпд 758
а-Цпклоцптралк 304
п-Цимол 76, 684, 786
нитрование 510
окисление 299. 831
Цпнкдпалкплы 618
реакции 659
Цинкдпарилы 648
Цинке расщепление 501. 505
Ципкорганическио соединения 647 с.
«42
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
циннамплиденацетон, галоформная реак-
ция 839
Циннамилиденуксусная кислота 839
Цистеамин 602
S-бензоилирование 589
Цистеин 608, 609
Цистин, восстановление 609
Цитозин 328
Цитраконовая кислота 803
ангидрид 848
Цитраль 306
бисульфптное соединение 559
гидрирование 46
конденсация с ацетоном 807
расщепление 827
Цитронеллаль, бИсульфитное соединение
559
Цитронеллол 46
Чепмена перегруппировка 857
Четыреххлористый углерод
при теломеризации 697 сл.
реакции с олефинами 709
Четырехбромистыи углерод, реакции
с олефинами 1709
'"Четвертичные аммониевые основании см.
Аммониевые четвертичные основа-
ния
Четвертичные пиридиниевые соли, релк-
цгпг с аминами 504
Чичибабина метод 724
Чугаева реакция 674
Шеффера кислота 568
Шимана реакция 253
Шиффовы основания 75, 294, 295, 371,
422, 470, 483, 816
апилированпе 453
гидрирование 518 сл.
замена азота серой 603
расщепление 325, 811
реакции 518, 529. 730, 731
Шмидта реакция 875
Шолля реакция 736, 738
Шоттена — Баумана
‘ реакция 350, 433, 437, 589, 597, 621,
622
Штааба синтез 507
Штоббе конденсация 805
Штреккера синтезы 484, 590. 719
Штреккера — Тимана синтез 484
Щавелевая кислота
декарбонилироваяпр 823
окисление 840
Хлорангидрид, мояозфлр 236, 241
— реакции 816, 823
эфиры 344
— реакции 787, 789. 790
Щавелевокротоновая кислота, эфир 790
Щавелевопропионовая кислота, эфир, ре-
акции 825
Щавелевосорбиновая кислота, эфир 790
Щавелевоуксусная кислота, декарб-
оксилирование 820
Щелочное плавление
гомологов фенола 832
ненасыщенных карбонильных соедине-
ний 833, 848
сульфокислот 328
Эвгенол
изомеризация 848
присоединение НВг 118
Эйнгорна метод 350, 434
Элаидиновая кислота 680
Элбса метод 287
— реакция 811
Электрофильное присоединение галогоно-
водородов 118
Электрохимическое '
восстановление 29, 516
галогенирование 132
Элсментоорганическис соединения 633
Элиминирование серы из сульфосоедпне-
пий 89
Энантальдоксим 479 ,
Энантамид 446
Энантовая кислота 311
Энантол 826
окисление 311
реакции 479, 486
3,6-Эпдокарбонил-3,4,5,6’-тетрафепил-1,2-
догидрофталевая кислота, ангидрид
826
Эндооксасосдннения 702
Эидооксатетрагидрофталевая кислота, ан-
гидрид 704
tyuc-Эпдометилентетрагиднофталевая кис-
лога, ангидрид 703
Эндоэтилснтетрагидробепзальдегид 704
Эпихлоргидрин, реакции 333, 593
Эпоксиамиды 266
2,3-Эпоксибутан 228
1.2-Эпоксндодекап 265
Эпоксикетоны 265
2.3-Э|гоксч1пентан 228
Эпоксисоединсния 719
гидролиз 272, 273
расщепление 228
реакции 318
2,3-Эиоксп 3,5.5-гриметнлциклогекеанон
265
Эргостерин
перекись 261
реакции 829
Эрленмейера синтез 809
Эрлиха — Закса реакция 412
Эстрогены амипозамещенные 497
Этапдисульфокислота 589
Этаноламин, восстановительное алкилиро-
вание 488
Этаноламины 500
циклизация сернокислых эфиров 16!
Этапсульфокислоты 589
Этапсульфохлорид 614
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
9
Этаптетракарбоповая кислота, эфир 779
Этерификация 224, 329, 343 сл.
Этионин 417
Этилакрилат, бромирование 101
а-Этилакролеин 808
Этпланилин 372, 419, 519
М-Эталанилин-4-сульфокпслота 566
Этилат алюминия 58
Этилацетат 314. 845
очистка 788
реакции 672
а-Этилацетоуксусхгая кислота, нитрозиро-
лание 394
-и-Этилацетофенон 298
n-Этплацетофенон. окисление 296
Этилбензиланплин, сульфирование 567
Этплбензилсульфон 591
Этплбензоат 345
2-Этплбонзойная кислота 76
Этилбензол 696
галогенирование 146
нитрование 385
окисление 289
Этилбромид 209, 210
Этил-атор-бутялмалоповый эфпр 755
Этил-»-бутиловый эфир 342
Этилбутират 845
2-Этилтсксаноль-1, реакции 808
2-Этилгексем-2-аль-1 807
2'-(3-Этилгептен-1-пл)-хинолиц 808
Этплгидразин 528
3-Этил-1,5-дифенилформаэап 403, 404
Этилдихлорарсин 665, 666
Этилом 672, 676
алкилирование бензола 696
окись см. ОтТись этилена
производные, полимеризация 697 сл.
реакции 101, 114, 124. 128, 653, 657,
697 сл.. 709, 710
Этиленбромшдрии 128
Этиленгликоль, диметиловый эфир 65,
334
Этилендламин, реакции 810
8,8'-Этиленди11зотномочев1тна 631
Этиленимины 464
расщепление 505
реакции с меркаптанами 602
производные 411
Этиленкарбонат 174
Этиленмагний полимерный 647
Этнленсульфенилхлорид, реакции 855
Этилрпсульфпды. расщепление 615
Этилен.хлорптдрин 125. 126
Этилидена перекись 291
N-Этплигобутирамид 433
Этнлпзонитрил 445
у-.Этилкапролэктам 874
I' нтлкетоны. галоформная реакция 839
Этилкротплбарбптуровая кислота 116
Этиллитий 639
Этплмагнийбромид в синтезе тетраэп
олова 662
Этилмалоновая кислота
азосочетапие 404
диамид 445
эфир 167 ’
Этилмеркаптометилепмалонопая кислота
диэтиловый эфир 812
Этилмеркаптоииримлдим, реакции 258
N-Этилморфолин 466
Этпдмочевина 506
Этплнатрий 641
N-Этпл-Р-нафтиламин 493
Этилнитрат 382
Этиловый спирт, дегидратация 672
Этпл-(Р-окси-у-хлорлроиил)-сульфмд 59,
1-Этилпиперидин 244
4-Этилпиридпн, окисление 831
1-Этплпирролидпн 244
Этилроданид, реакции 614
Этилсульфецилхлорпд, реакции 583
Этил-(л-сульфобензил)-анили1Г 567
Этилтиогликозиды, реакции 259
Этилтитанхлориды 665
N-Этил-п-толуолсульфамид 468
N-Этил-и-толуолсульфанилид 468
Этил-1 -фепилбутиламип 729
Этилфенмлдисульфид 583
N-Этилформавилид 462
Этилформпат 344
реакции 789
1-Этилхинит, изомеризация 85G
Этилхлорацетат, реакции 754
2-Этилхромои 788
З-Этилциклогексанол 49
2-Этилциклогексанон, оксим. Птдриров
ние 526
4-Этилциклогексанон, оксим, изомсриз
ция 874
Этилциклоироиилкетимин 729
S-Этилцистеамин 602
Д6-17-Этинпландростспдиол-3,17 272
Этпнилкарбмнолы
галогенирование 149
стероидного ряда, дегидратация 2'
1-Этннилциклогексанол, гидратация 2'
Этоксиацетальдегид, ацеталь 335
Этоксиацстилен 691
Этоксиацстон 689
п-Этокспбснлгидразид 4Q0
Этокс’ибепзолсульфиновая кислота 601
2-Этокспбензтиа.зол 3,35
Этоксиизоформанилпд. реакции 791
Этоксиметилснацетплацетон, реакции 4f
Этокспметилснацетоуксуспый эфир, роат
ции 498
Этокспметиленмалоповая кислота
эфиры 812
- реакции 498
Этого нметнлепы 789
Этонслметилпзогпоцпанат 202
2-Эт оксп-1 -нафта тьдст и я 798
‘944
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
fl-Этоксипропионовая кислота, этиловый
эфир 334
га-Этоксифенилбутилсульфид 600
2-Этоксихиноксалин-3-карбоновая кис-
лота, эфир 335
(З-Этоксиэтилбромид 212
Р-Этоксиэтиловый спирт, реакции 212, 215
2-Этоксиэтилхлорид 215
L-Эфедрпн, реакции 511
-Эфираты магнийорганических соединений
643
Юрьева реакция 500
Якобсена реакция 881
Янтарная кислота
дегидратация 359
динитрил, омыление 327
дихлорангидрид 234
реакции 802, 805
Янтарный ангидрид 359
реакции 774
Янтарный диальдегид 324
диоксим 505
Вейганд-Хильгетаг
Методы эксперимента в органической химии
Издательство «Химия», М., 1968 г.
944 с.
УДК 542.9 : 547
Редакторы Ф. В. Рабинович. М. И. Пастушенно
Технический редактор Л. А. Пантелеева
Художник Е. В. Венетов
Корректор В. А. Асикаева
Подписано к печати 8/Х 1968 г. Формат бумаги 70 X 1001 /1в.
Усл нет. л. 77,7. Уч.-изд л. 96,75. Тип>гр. бум. Л» 2.
Тираж 1-го завод., 12000 эка БЗ Л5 20—1968 г.—4. Цена 6 р. 96 к.
Зак. 1835.
Типография Л'а 14 Главполиграфпрома Комитета по печати
прп Совете Министров СССР. Ленинград, Московский проспект, 9 I.