/





















































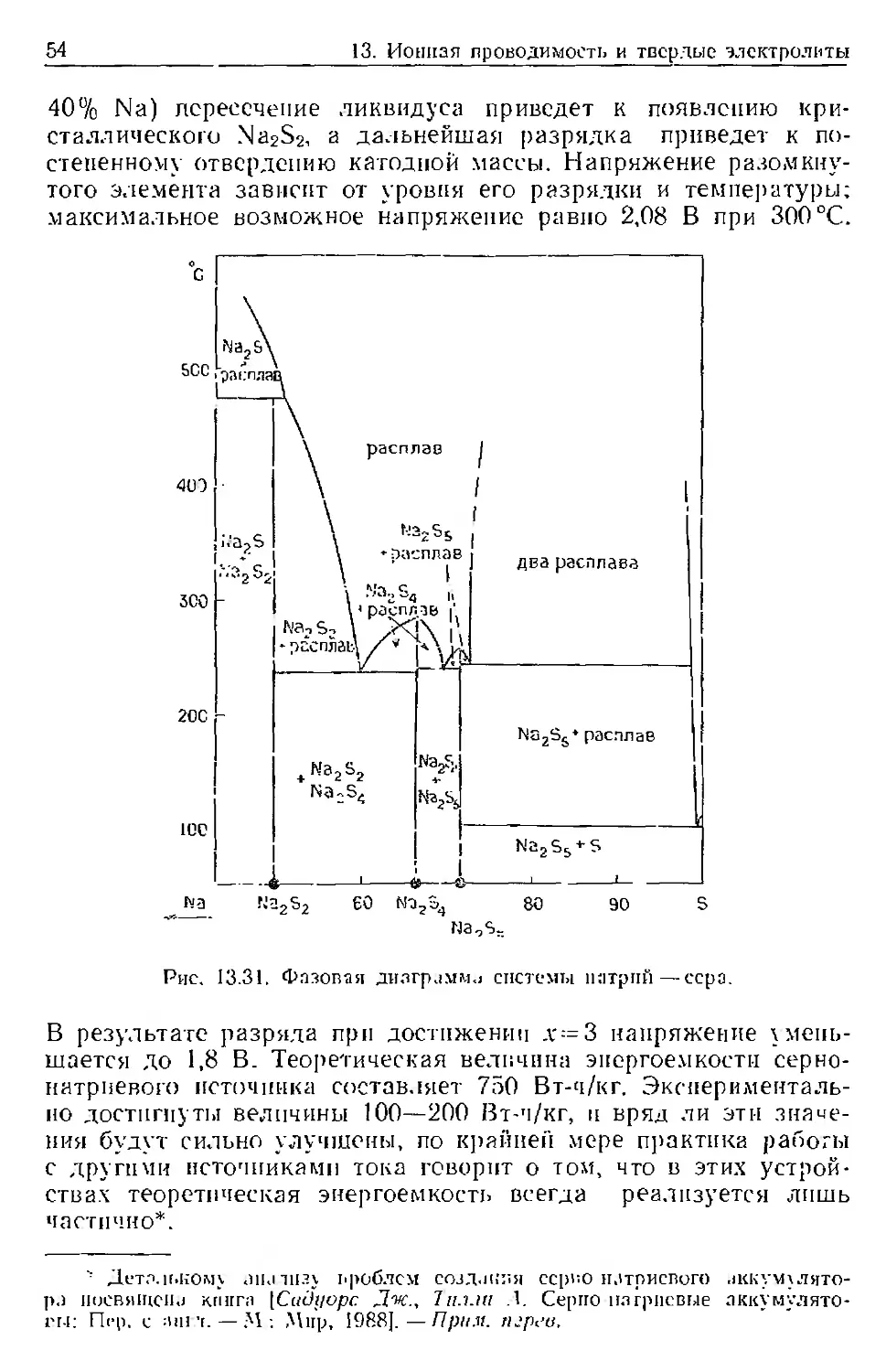























































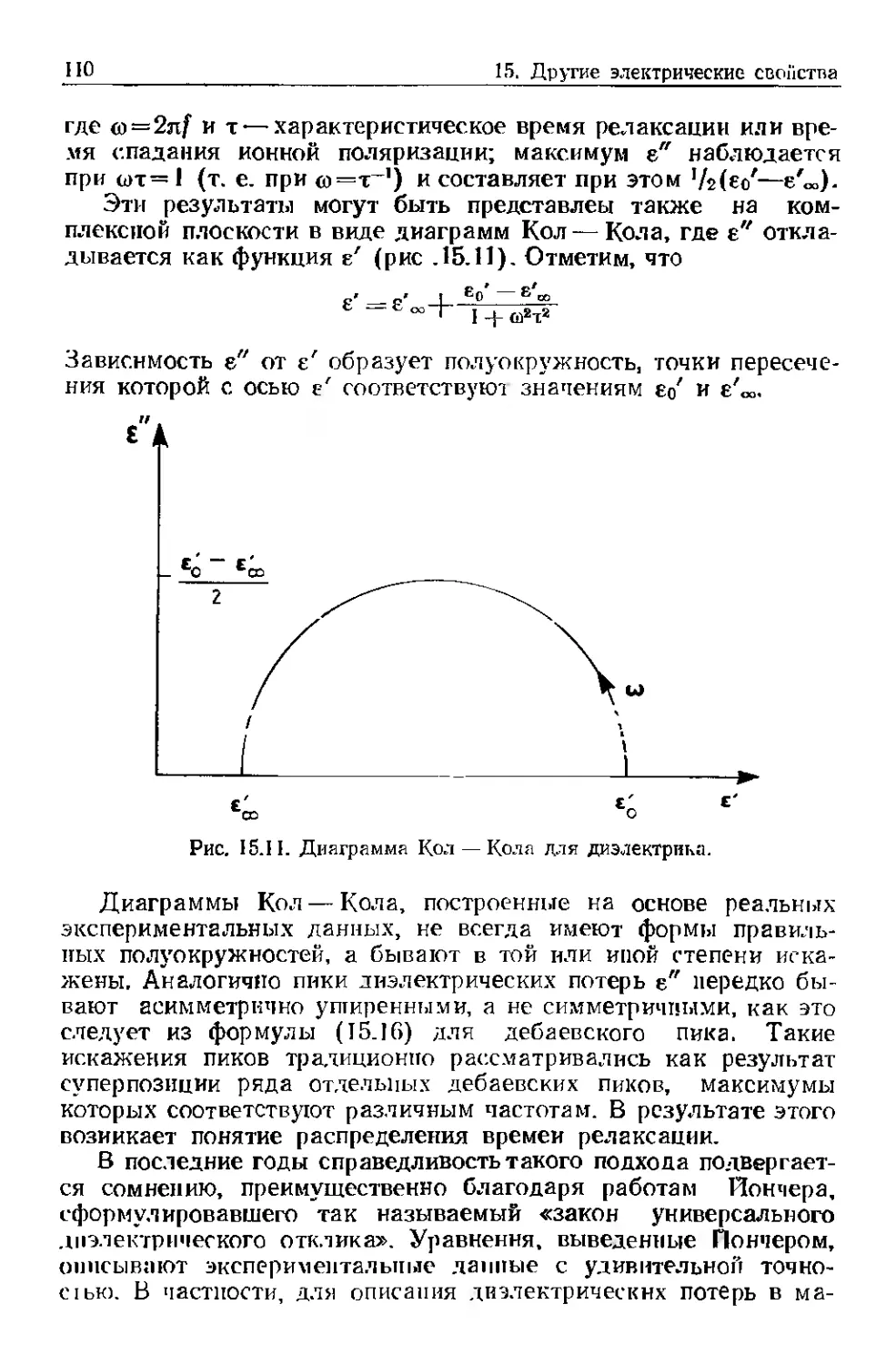









































































































































































































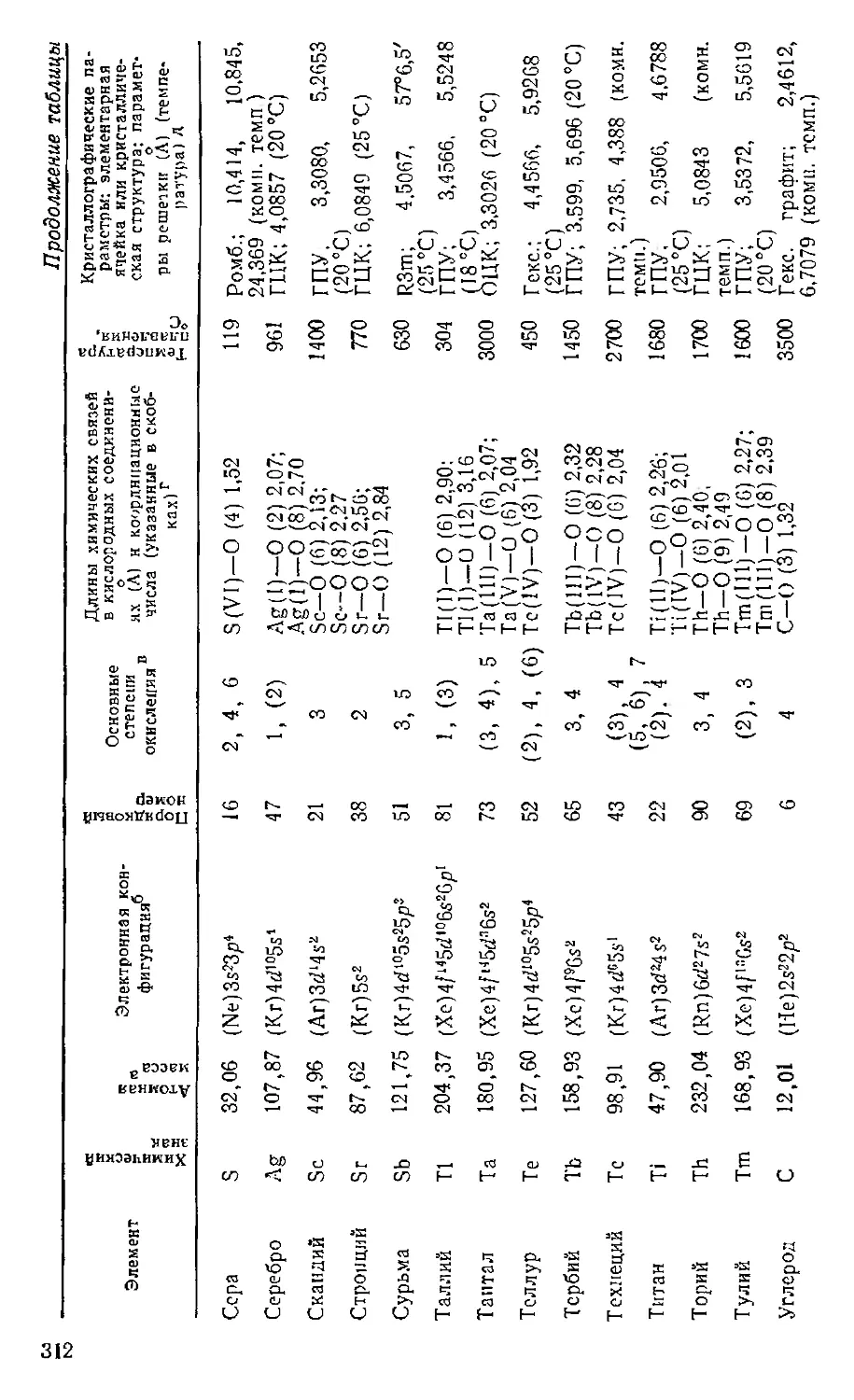



























Text
SOLID STATE
CHEMISTRy
AND
ITS APPLICATIONS
ANTHONY R. WEST
Department of Chemistry
University of Aberdeen
John Wiley & Sons
Chichester-New York
Brisbane • Toronto • Singapore
А. Вест
ХИМИЯ
ТВЕРДОГО
ТЕЛА
Теория
и приложения
В 2-х частях
Часть 2
Перевод с английского
канд. хим. наук Кауля А. Р.
и канд. хим. наук Куценка И. Б.
под редакцией
академика Ю. Д. Третьякова
Москва «Мир» 1988
ББК 24.5
В38
УДК 541.1
Вест А.
В38 Химия твердого тела. Теория и приложения: В 2-х ч.
Ч. 2: Пер. с англ. — М.: Мир, 1988. — 336 с, ил.
ISBN 5-03-000071-2
Лв ор книги — известный ученый из Великобритании А. Вест — оиределяе
химию твердого тела как науку о синтезе, структуре, свойствах и применениях.
твердых материалов. По содержанию и форме изложения книга может служить,
введением в предмет для всех, предполагающих специализироваться в данной
области или желающих получить о ней общее представление, что особенно вале-
валено в настоящее время, когда происходит оформление химии твердого тела в са-
самостоятельный раздел химической науки и возникает потребность подготовки кад
ров по этой специальности.
В часть 2 вошли гл. 13—21 и приложение английского издания, где излага-
излагаются практические приложения химии твердою тела.
Для студентов и преподавателей химических и химико-технологических ву
зов, а также для исследователей и инженеров различных отраслей, имеющих де
ло с химическими превращениями в твердом состоянии.
ББК
Редакция литературы по химии
ISBN 5-03-000071-2 (русск.) © 1984 by John Wiley & Sons Ltd
ISBN 5-03-000070-4 All right reserved
а Апл ПЛО77 c\ / \ Authorised translation from the
0 4/1 У1Ш/ У (англ.) English language edition published
by John Wiley & Sons Ltd.
%, перевод на русский язык, «Мир»
1988
Глава 13
ИОННАЯ
ПРОВОДИМОСТЬ
И ТВЕРДЫЕ
ЭЛЕКТРОЛИТЫ
Электрическая проводимость (электропроводность) твердых
тел осуществляется путем миграции на большие расстояния
электронов или ионов. Обычно доминирует электропроводность,
обеспечиваемая лишь одним из этих типов носителей заряда,
однако в некоторых неорганических материалах и электронная
и ионная проводимость проявляются одновременно.
Твердофазные материалы принято характеризовать удель-
удельной электропроводностью а, которая представляет собой элект-
электропроводность кристалла или таблетки с единичной площадью
поперечного сечения и единичной длиной. Удельную электро-
электропроводность обычно выражают в следующих единицах:
Ом^-см \ Ом ^м, См-м, 1 См {сименс) = 1 Ом.
Удельная электропроводность любого материала независимо
от природы носителя заряда определяется уравнением
A3.1)
где щ — число носителей заряда сорта i, ei и \xL — их заряд и
подвижность. Для электронов и однозарядных ионов в = 1,6•
•10~19 Кл (заряд электрона). В табл. 13.1 приведены типичные
величины удельной электропроводности проводников различных
классов. Обычно электрическая проводимость зависит от тем-
температуры и для всех материалов, за исключением металлов,
возрастает с повышением температуры. Проводимость метал-
металлов максимальна при низких температурах, а некоторые метал-
металлы при температурах, близких к абсолютному нулю, обладают
сверхпроводимостью.
В обычных условиях в большинстве твердых тел с ионной
и ковалеитной связью, например оксидах и галогенидах, мигра-
миграция ионов незначительна. Атомы обычно располагаются в опре-
определенных узлах решетки и передвижение их возможно только
13. Ионная проводимость и твердые электролиты
с участием дефектов кристалла. Лишь при высоких температу-
температурах, когда концентрация дефектов достаточно велика и атомы
обладают значительной тепловой энергией, проводимость ста-
становится заметной. Так, например, проводимость NaCI при
800 °С, т. е. чуть ниже точки плавления, составляет
10~3 Ом-см, тогда как при комнатной температуре хими-
химически чистый NaCI — изолятор.
Существует, однако, группа твердых тел, так называемые
«твердые электролиты», «ионные проводники» и «суперионные
Таблица 13.1. Типичные величины электропроводности
Тип проводимости Материалы о", Ом-1'см-1
Ионная Ионные кристаллы <Ч0~18—10~4
Твердые электролиты 10~3—101
Сильные (жидкие) электролиты 1СГ3—101
Электронная Металлы 101—105
Полупроводники Ю~5—102
Изоляторы <Ч0~12
проводники», в которых ионы одной из подрешеток могут дви-
двигаться достаточно быстро. Ионная проводимость таких мате-
материалов часто обусловлена особенностями их кристаллической
структуры, а именно наличием туннелей или слоев. Величины
проводимости этих материалов сравнимы с аналогичными ха-
характеристиками сильных жидких электролитов. Так, проводи-
проводимость р-глинозема при 25 °С, обеспечиваемая перемещением
ионов Na+, равна 10~3 Ом^-см. В настоящее время большое
внимание уделяется изучению свойств твердых электролитов,
разработке новых типов этих материалов и расширению обла-
областей их применения в твердофазных электрохимических устрой-
устройствах.
Эта глава посвящена только ионной проводимости; элект-
электронная проводимость рассмотрена в гл. 14. Обсудим вначале
более подробно поведение бинарных соединений, таких, как
NaCI и AgCl. Хотя обычно ионная проводимость этих соедине-
соединений рассматривается в работах, относящихся к физике твердо-
твердого тела, понимание связанных с ними явлений необходимо
также при изучении таких разделов химии твердого тела, как
твердые электролиты, дефекты в кристаллах и реакционная
способность твердых тел (см. также гл. 2 и 9). Последующие
13.1. Типичные ионные кристаллы
разделы главы посвящены твердым электролитам и их приме-
пению, а также экспериментальным методам изучения проводи-
проводимости.
13.1. Типичные ионные кристаллы
13.1.1. Галогениды щелочных металлов
В кристаллических галогенидах щелочных металлов, напри-
например NaCI, катионы более подвижны, чем анионы. На рис. 13.1, где
изображена одна из плоскостей структуры NaCI, показано, как
ион Na+ сдвигается на место примыкающей катионной вакан-
вакансии, оставляя таким образом свой собственный узел вакантным.
Далее этот ион Na+ не может мигрировать, поскольку вблизи
нет других вакантных мест, на которые он мог бы переместить-
переместиться, а межузельная миграция Na+ в NaCI чрезвычайно затрудне-
затруднена (см. разд. 13.1.2). В то же время катионная вакансия мо-
может продолжать свое движение, так как она всегда окружена
двенадцатью ионами-Na+, один из которых может скачкообразно
поменяться с вакансией местами. Таким образом, электропро-
Cl Na Cl Na Cl Na
Na Cl Na a Na Cl
Cl Na Cl v Cl Na
Na CL Na Cl Na Cl
Cl Na Cl Na Cl Na
Рис. 13.1. Миграция катионных вакансий (или ионов Na+) в NaCI.
водность NaCI обусловлена в основном наличием в нем катион-
катионных вакансий. Анионные вакансии также присутствуют в NaCI,
однако их подвижность много меньше.
Величина ионной проводимости NaCI зависит от числа име-
имеющихся катионных вакансий, которое в свою очередь чрезвычай-
чрезвычайно сильно зависит от химической чистоты и термической пре-
предыстории кристалла. Увеличения числа вакансий можно до-
добиться двумя способами. При нагревании кристалла экспонен-
экспоненциально возрастает число термодинамически равновесных ва-
вакансий [см. рис. 9.1 и уравнение (9.9)], присущих чистому бес-
беспримесному кристаллу. В то же время при введении гетерова-
8
13. Ионная проводимость и твердые электролиты
ю
сг>
миг
собственная
проводимость
Е.
лентных примесей могут возникать вакансии, компенсирующие
избыточный заряд примесных катионов. Так, например, добав-
добавление малых количеств МпСЬ приводит при достижении равно-
равновесия к образованию твердого раствора состава Nai_2*Mn*VNaCl,
где на каждый ион Мп2+ приходится одна связанная с ним ка-
тионная вакансия VNa. Такие вакансии называются примесны-
примесными, так как в чистом NaCl они не наблюдаются. При низких
температурах (~25°С) кон-
концентрация вакансий термиче-
термического происхождения настоль-
настолько мала, что, несмотря на вы-
высокую чистоту кристалла,
остается много меньше кон-
концентрации примесных вакан-
вакансий. При более высокой тем-
температуре, значение которой
определяется концентрацией
примеси, происходит переход
от примесной к собственной
проводимости.
Температурная зависи-
зависимость ионной проводимости
обычно выражается уравнени-
уравнением Аррениуса
примесная
проводимость
(О
Рис. 13.2. Зависимость ионной про-
проводимости NaCl от температуры
(схема). Параллельные линии в
примесной области соответствуют
различной концентрации легирую-
легирующих примесей.
= Аехр(—EIRT) A3.2)
где Е — энергия активации,
R — газовая постоянная, Т —
абсолютная температура. Пре-
дэкспоненциальный множи-
множитель А включает несколько констант, в том числе частоту ко-
колебаний потенциально подвижных ионов. Графическая зависи-
зависимость In о от Т должна выражаться прямой с углом наклона
—E/R. В некоторых случаях при обработке температурных за-
зависимостей электропроводности в предэкспоненциальный фак-
фактор вводят множитель 1/7\ При этом графическую зависимость
принято представлять в координатах In оГ—Т~1. Наклон по-
получающейся при этом прямой (—E/R) может несколько отли-
отличаться от наклона в аррепиусовских координатах. Аррениусов-
ская зависимость для NaCl схематически изображена на рис.
13.2. В низкотемпературной примесной области число вакан-
вакансий определяется концентрацией примеси и для каждого кон-
концентрационного уровня является величиной постоянной. На
рис. 13.2 этому соответствует ряд параллельных прямых, каж-
каждая из которых отвечает проводимости кристаллов с различ-
различным содержанием легирующей добавки.
13.1. Типичные ионные кристаллы
В примесной области зависимость о от температуры опреде-
определяется лишь температурной зависимостью подвижности катио-
катионов jx (см. уравнение 13.1), которая также подчиняется уравне-
уравнению Аррениуса:
\i = (i0 exp (—EMJRT) A3,3)
где /?миг — энергия активации миграции катионных вакансий.
Для того чтобы понять природу энергии активации миграции,
рассмотрим возможные пути скачкообразного движения иона
Na+ из узла решетки в близлежащую катионную вакансию.
-CI- --..--
Рис.
13.3. Путь миграции иона Na+
в NaCI.
Рис. 13.4. Треугольное междоузлие,
через которое должен проходить пе-
перемещающийся ион Na+ в NaCL
г' — радиус вписанной окружности;
окружности 1—3 изображают ионы
С1~ с радиусом х/2.
На рис. 13.3 изображен фрагмент структуры NaCI (Vs элемен-
элементарной ячейки) с одной незанятой вершиной. Один из трех
ионов Na+, расположенных в других вершинах куба, может за-
занять эту вакансию. Прямой перескок через грань куба («след»
его показан пунктирной прямой) оказывается невозможным
из-за ионов хлора 1 и 2, которые если и не касаются друг дру-
друга, то настолько близко расположены, что мешают иону Na"
протиснуться между ними. В итоге ион Na+ должен двигаться
не по прямой, а через межузельную позицию в центре куба,
равноудаленную от всех восьми вершин (эта траектория пока-
показана стрелкой). Четыре вершины заняты ионами С1~, образую-
образующими тетраэдр. Прежде чем передвигающийся ион Na+ до-
достигнет центрального междоузлия, он должен пройти через тре-
треугольное «окно», образованное ионами С1~ (/, 2 и 5). Оценим
размер этого «окна» для того, чтобы представить себе, насколь-
насколько трудно проходит сквозь него ион Na . Параметр а элемен-
10 13. Ионная проводимость и твердые электролиты
тарной ячейки NaCl равен 5,64 А. Длина связи Na—С1 состав-
составляет а/2 = 2,82 А. Табулированные ионные радиусы (величина
которых несколько меняется в зависимости от того, какая из
шкал используется) натрия и хлора равны —0,95 А и —1,85 А.
Длина связи Na—С1, рассчитанная как сумма этих ионных ра-
радиусов, оказывается —2,80 А, что близко к экспериментально
найденному значению.
В плотноупакованных структурах, таких, как NaCl, анионы
пли контактируют друг с другом, или находятся в непосредст-
непосредственной близости. Ионы хлора 1, 2 и 3 образуют фрагмент слоя
с плотнейшей упаковкой, и расстояние С1 (/)—С1C), равное
[(а/2J+(а/2J]1/2, составляет 3,99 А. Это на -0,3 А больше,
чем 2га-, и, следовательно, соседние ионы С1 в NaCl непо-
непосредственно не касаются друг друга.
«Радиус окна» f внутри треугольника, образованного ионами
хлора /, 2 и 3, может быть рассчитан следующим образом
(рис. 13.4):
cos 30° =
х/2 1 f995
У (ГСГ
Если rci- = 1,85 А то г'-0,45 А.
Подобным образом можно рассчитать радиус г" межузель-
ной позиции в центре куба. Длина объемной диагонали куба
(рис. 13.3) равна Bга- +2г"), т. е.
2 (гсг + О = [(^2J + (я/2J + (я/2J]1/2 = 4,88А
откуда г" = 0,59А.
Таким образом, очевидно, что миграция иона Na+ по решет-
решетке NaCl затруднена. Вначале ион Na должен протиснуться
сквозь узкое треугольное отверстие с радиусом вписанной ок-
окружности 0,45 А, после чего он попадает в маленькое тетраэд-
рическое междоузлие с радиусом вписанной сферы 0,59 А. Вре-
Время пребывания в этой позиции весьма невелико из-за находя-
находящихся на расстоянии 2,44 А двух ионов Na A и 2) и четырех
ионов С1~. Покидая межузельную позицию, ион Na+ вновь про-
протискивается через отверстие с г7 = 0,45 А, образованное С1~-
ионами 1У 2 и 4, и занимает вакантную октаэдрическую пози-
позицию. Вычисления, подобные сделанным выше, неизбежно стра-
страдают некоторой идеализацией, так как вблизи дефекта должны
происходить искажение и релаксация структуры, приводящие
к изменению значений расстояний по сравнению с расчетными.
Тем не менее расчет показывает, что миграция иона Na+ за-
затруднена и связана с преодолением значительного активацион-
13.1. Типичные ионные кристаллы и
ного барьера. В примесной области (рис. 13.2) проводимость
зависит, таким образом, и от концентрации вакансий, и от их
подвижности, что отражает уравнение A3.4), полученное комби-
комбинацией уравнений A3.1) и A3.3):
а = лв|л0 exp (—EMJRT) A3.4)
При более высоких температурах в области собственной
проводимости концентрация вакансий термического происхож-
происхождения превышает концентрацию вакансий, обусловленную леги-
легирующими добавками. Теперь уже число вакансий п зависит от
температуры и также подчиняется уравнению Аррениуса:
п = N const exp (—Eo6v/2RT) A3.5)
Это уравнение тождественно уравнению (9.9), в котором ЕОбР/2
есть энергия активации образования одного моля катионных
вакансий, т. е. половина энергии, требующейся для образования
одного моля дефектов Шоттки. Подвижность вакансий по преж-
прежнему описывается уравнением A3.3), и, таким образом, в целом
электропроводность в области собственной проводимости под-
подчиняется уравнению
а = N const ф0 ехр (—ЕШ1ШТ) exp (—Eo6v/2RT)
о = А ехр—E«w + *обр/2 A3.6)
На рис. 13.2 схематически показана аррениусовская зависи-
зависимость проводимости для кристаллов NaCl различной степени
чистоты. Серия параллельных прямых в примесной области со-
соответствует проводимости при различных концентрациях приме-
примеси, например Мп2+, в то же время единственная линия в соб-
собственной области показывает независимость проводимости от
содержания примесей. Последнее справедливо, если концен-
концентрация примеси очень мала (<1% Мн2 ). При таком уровне
примеси ионы Мп2 не влияют в заметной степени на энергию
активации миграции катионных вакансий. Наклон зависимости
в собственной области больше, чем наклон в примесной обла-
области, и если удается определить тот и другой, то это дает воз-
возможность рассчитать отдельно ?Миг и ЕовР.
Надежные экспериментальные данные, полученные на мо-
монокристаллах NaCl (рис. 13.5), показывают, что схематическая
зависимость проводимости от концентрации примеси, приведен-
приведенная на рис. 13.2, в значительной степени идеализирована; в
действительности картина несколько сложнее. Участки I и II
на рис. 13.5 соответствуют областям собственной и примесной
проводимости упрощенной схемы (рис. 13.2). Штриховые линии
па рис. 13.5 появляются при экстраполяции отрезков /и //, вы-
12
13. Ионная проводимость и твердые электролиты
полненной с целью обнаружения экспериментально наблюдаю-
наблюдающихся отклонений. Появление участка /' вблизи температуры
плавления (802 °С) приписывают двум различным причинам.
Во-первых, анионные вакансии становятся все более подвижны-
подвижными и дают значительный вклад в о. Во-вторых, с возрастанием
концентрации вакансий при высоких температурах становятся
заметными дальнодеиствующие дебай-хюккелевские взаимо-
взаимодействия между катионны-
ми и анионными вакансия-
вакансиями, подобные взаимодейст-
взаимодействию ионов в растворах.
При этом возникает при-
притяжение, которое частично
компенсирует энергию об-
образования вакансий. Таким
образом, образование ва-
вакансий облегчается, их кон-
концентрация увеличивается,
что приводит к увеличению
проводимости. Хотя неясно,
какое из этих объяснений
следует принять для NaCl,
вероятно, именно дебай-
хюккелевский эффект —
причина подобного высоко-
высокотемпературного отклонения
проводимости монокристал-
монокристаллов AgCl (см. обсуждение
рис. 9.4).
На участке /// (ниже 390 °С для данного образца NaCl)
электропроводность отклоняется вниз от идеальной зависимости
в примесной области. Это связывают с образованием комплек-
комплексов дефектов, таких, как пары «катиопная вакансия — анионная
вакансия» или «катионная вакансия — примесный (гетерова-
лентный) катион». Комплексы дефектов возникают при взаи-
взаимодействии простых дефектов, являющихся ближайшими сосе-
соседями или соседями второго порядка. Взаимодействие ближай-
ближайших соседей, имеющее место в данном случае, сильно
отличается от упоминавшегося выше дебай-хюккелевского вза-
взаимодействия, которое, будучи дальнодействующим, связано
с необходимостью сохранения электронейтральности. Для того
чтобы катионная вакансия, входящая в комплекс дефектов,
получила возможность двигаться, ей должна быть сообщена
дополнительная энергия, необходимая для диссоциации комп-
комплекса. В итоге энергия активации на участке /// оказывается
больше, чем ?миг на участке //.
1,0
1,2 1,4
ЮОО/ПК)
Рис. 13.5. Температурная зависимость
ионной проводимости «чистого» NaCl
[13].
13.1. Типичные ионные кристаллы 13
Несмотря на то что проводимость кристаллов NaCl множе-
множество раз исследовалась в различных лабораториях, в получен-
полученных величинах энергии активации миграции Na+ ?миг нет хоро-
хорошего согласия: эти величины лежат в интервале 0,65—0,85 эВ
F0—80 кДж/моль). В табл. 13.2 наряду с величинами ?миг
приведены значения энергий активации других процессов, свя-
связанных с ионной проводимостью в NaCl. Возможно, что наблю-
наблюдающееся непостоянство величин Емаг обусловлено неизбеж-
неизбежным присутствием в кристалле других дефектов, в особенности
дислокаций, и их влиянием на миграцию катионов. В области,
Таблица 13.2. Проводимость кристаллов NaCl
Процесс Энергия активации, эВ
Миграция ионов Na+ (?МИг) 0,65—0,85
Миграция ионов С1- 0,90—1,10
Образование пары Шоттки 2,18—2,38
Диссоциация вакансионной пары ^1,3
Диссоциация пары катиониая вакансия — ион Мп2+ 0,27—0,50
примыкающей к дислокации, кристаллическая решетка нахо-
находится в напряженном и искаженном состоянии, благодаря чему
миграция катионов вдоль «дислокационных трубок» может про-
проходить легче, чем в совершенных областях кристалла. Если это
так, то проводимость и энергия ?миг должны зависеть от числа
и распределения дислокации, а следовательно, и от термической
предыстории кристалла. Однако влияние дислокаций на элек-
электропроводность трудно подвергнуть экспериментальной провер-
проверке из-за невозможности точного определения плотности дисло-
дислокаций и их контролируемого введения.
Легче всего проследить зависимость о от концентрации при-
примесей, так как этим параметром можно управлять, и электро-
электропроводность в примесной области именно от пего зависит на-
наиболее существенным образом. Термодинамически равновесная
концентрация дефектов Шоттки в собственной области прово-
проводимости определяется законом действия масс [уравнение
(9.3)]
А -
где Vna иУс1 — вакансии в катионной и анионной подрешетках
соответственно. Принимается, что константа равновесия К не
14 13. Ионная проводимость и твердые электролиты
меняется от наличия малых количеств гетеровалентных приме-
примесей и что знаменатель — произведение концентраций занятых
узлов решетки — имеет примерно постоянное значение и равен
1. Тогда
-V
В собственной области хо= [V^a] = [Vci]. В соответствии с урав-
уравнением A3.8) увеличение числа катионных вакансий в примес-
примесной области за счет введения гетеровалентных катионных при-
примесей влечет за собой уменьшение концентрации анионных ва-
вакансий. Пусть ха и хс (причем хафхс) —концентрации анион-
анионных и катионных вакансий в примесной области, а с — концен-
концентрация двухзарядных примесных катионов. Тогда
х — х -4-с ИЗ Q)
что следует из общего баланса зарядов, а также из того, что
эффективный заряд катионов двухвалентной примеси и анион-
анионных вакансий равен +1, а катионной вакансии —1. Решение
квадратного уравнения, получаемого из системы уравнений
A3.8) и A3.9), дает следующие выражения для хс и ха (поло
жительные корпи):
1/2 1
A3.10)
A3.11)
Если А'о<Сс, то Хс-^с и Ха->0. Этот результат справедлив в
примесной области и тождествен выводу из уравнения A3.8).
Если хо^>с, то хс = Хо = Ха'—это область собственной проводи-
проводимости. Далеко не все примеси увеличивают электропровод-
электропроводность в примесной области. Примеси, имеющие тот же заряд,
например К или Вг в NaCl, обычно не влияют на а, по край-
крайней мере присутствуя в небольших концентрациях. Примеси,
вызывающие уменьшение концентрации подвижных частиц,
приводят к уменьшению а. Так, если двухзарядпые анионы мог
ли бы быть растворены в NaCl, то при этом возросла бы кон-
концентрация менее подвижных анионных вакансий, что вызвало
бы одновременное уменьшение концентрации более подвижных
катионных вакансий. Этот эффект не характерен для NaCl,
так как в этом веществе плохо растворяются двухзарядные
анионы, однако для AgCl это весьма существенно, что будет
показано ниже.
13.1. Типичные ионные кристаллы
15
13.1.2. Хлорид серебра
Доминирующим типом дефектов в AgCl являются дефекты
Френкеля, т. е. межузельные (внедренные) ионы Ag+, связан-
связанные с катионными вакансиями (гл. 9). Экспериментально по-
показано, что межузельные ионы Ag+ более подвижны, чем ва-
вакансии серебра. На рис. 13.6 схематически изображены два
принципиально возможных механизма миграции этих ионов.
Прямая «межузельная» миграция A) сводится к перескоку
внедренного иона Ag+ в соседнее пустое междоузлие. При кос-
Ад С1 Ад С1 Ад С]
С1
Cl Ag Cl Ag Cl Ag Cl Ag
-Д-Ад
Ag Cl Ag Cl Ag Cl Ag CJ
Cl Ag Cl Ag Cl *Ag Cl Ag
Ag Cl Ag Cl Ag Cl Ag Cl
Cl Ag Cl Ag Cl Ag Cl Ag
Ад
a
Рис. 13.6. Миграция межузелыюго иона Ag+: a — путем прямого перескока
из одного междоузлия в другое A) и с вытеснением регулярного иона в
междоузлие B); б— возможные направления межузельной миграции иона
Ag+ в AgCl.
венной миграции B) межузельный ион Ag+ вытесняет один из
четырех соседних ионов серебра в примыкающее междоузлие,
а сам занимает освободившийся при этом нормальный узел ре-
решетки.
Вопрос о том, какой из этих механизмов работает в дейст-
действительности, можно решить лишь на основе сопоставления на-
надежных количественных данных по диффузии и электропровод-
электропроводности. В диффузионных измерениях кристалл легируют ионами
радиоактивного серебра Ag+* и изучают миграцию этой метки.
В проводимость же вносят свой вклад не только радиоактив-
радиоактивные, но и все остальные ионы Ag . Взаимосвязь между коэф-
коэффициентом самодиффузии D и проводимостью а выражается
уравнением Нернста — Эйнштейна
fn {Zef
а
A3.12)
где Ze — заряд подвижных ионов, п — их концентрация. Вели-
Величина корреляционного множителя /, называемого фактором
Хейвеиа, зависит от механизма миграции и для двух механиз-
18 13. Ионная проводимость и твердые электролиты
мости. Ниже приведены значения энергии активации некоторых
процессов в кристалле AgCl с участием дефектов:
Е, эВ
Образование дефекта Френкеля 1,24
Миграция катионной вакансии 0,274=0,34
Миграция межузельных ионов Ag+ 0,05-^0,16
13.1.3. Фториды щелочноземельных металлов
В этой группе соединений доминирующими дефектами явля-
являются, очевидно, анионные дефекты Френкеля —¦ межузелытые
ионы F", занимающие центр куба, в вершинах которого нахо-
находятся восемь других ионов F~ (рис. 7.18). Измерения проводи-
проводимости свидетельствуют о том, что анионные вакансии более
подвижны, чем межузельные ионы фтора. Это противоположно
картине, наблюдающейся в AgCl, где межузельные ионы Ag
подвижнее катионных вакансий. В некоторых материалах со
структурой флюорита, например у PbF2, проводимость при вы-
высоких температурах становится весьма большой (разд. 13.2.3).
13.1.4. Простые стехиометрические оксиды
Изучение проводимости таких оксидов, как MgO, представ-
представляет собой трудную задачу, так как из-за высокой температу-
температуры их плавления (~2500°С) трудно вырастить чистые моно-
монокристаллы, которые не захватили бы примесей при этих темпе
ратурах. Другая причина, усложняющая исследования, состоит
в том, что в этих оксидах энергия образования и энергия
миграции дефектов в несколько раз больше, чем в NaCl. В ито-
итоге их проводимость чрезвычайно низка даже при высоких тем-
температурах и часто определяется в основном наличием приме-
примесей.
Между энергиями образования дефектов Шоттки (в галоге-
нйдах щелочных металлов), а также вакансий (в металлах) и
температурами плавления этих веществ существует линейная
корреляция, как это показано на рис. 13.8 для галогенидов ли-
лития. Такая корреляция вполне объяснима,поскольку температу-
температура плавления характеризует энергию, необходимую для полно-
полного разрушения кристаллической решетки, и, следовательно, зави-
зависит от энергии последней. В то же время энергию образования
дефекта Шоттки или вакансии можно приравнять энергии, не-
необходимой для разрушения чрезвычайно малой области крис-
кристалла и удаления атома пли пары противоположно заряженных
бонов из структуры. Таким образом, и в результате плавления,
и при образовании дефектов разрушаются связи. К сходству
между этими двумя процессами можно добавить, что некоторые
13.2. Твердые электролиты
19.
теории плавления основаны на образовании значительной кон-
концентрации точечных дефектов при приближении к температуре
плавления. Была выявлена столь же оправданная корреляция
между температурой плавления и энергией миграции дефектов,
например катионных вакансий. Справедливость такой корреля-
корреляции для оксидов позволяет оценить энергии образования и ми-
миграции дефектов. По этой оценке энергия образования дефектов
2,0
СП
с
.0
X
со
О
образование
дефектов
Шоттки
миграция
Li4-вакансий
Г Вг'СГ F"
¦ м—•—•-
500 1000
Температура плавления, К
Рис. 13.8. Корреляция энтальпий образования дефектов Шоттки и энтальпий
миграции катионных вакансий с температурами плавления галогеиидов ли-
лития [1].
Шоттки в MgO составляет 6,5 эВ, а энергия миграции катион-
катионных вакансий 2,5 эВ. Экспериментальное изучение самодиффу-
самодиффузии Mg2+ в MgO привело к значению энергии активации, рав-
равному 3,4 эВ. Сопоставление этого результата с оценочными ве-
величинами свидетельствует о том, что проводимость относится
скорее всего к примесной области. Доказано также, что на
проводимость оксидов сильно влияют дислокации и что кисло-
кислородные вакансии передвигаются предпочтительно вдоль дисло-
дислокационных трубок.
13.2. Твердые электролиты (ионные проводники,
суперионные проводники)
Большинство кристаллических материалов, подобно NaCI
или MgO, имеют низкую ионную проводимость, так как их ато-
атомы или ионы, несмотря на тепловые колебания, обычно не мо-
>*
13. Ионная проводимость и твердые электролиты
гут покинуть занимаемые узлы решетки. Исключение составля-
составляют немногие твердые электролиты, в которых один из струк-
структурных компонентов (катион или анион) не привязан к строго
¦определенным узлам решетки и может в значительной степени
свободно передвигаться по кристаллу. Твердые электролиты, та-
таким образом, представляют собой вещества, промежуточные по
структуре и свойствам между нормальными кристаллическими
твердыми телами с регулярной трехмерной структурой, пост-
построенной из неподвижных (в обычном смысле) атомов или
увеличение
¦концентрации
дефектов
вещество
в обычном
кристаллическом
состоянии
¦твердый
электролит"
¦жидкость
'фазовый
переход
Температура
Рис. 13.9. Положение твердых электролитов относительно обычных кристал-
кристаллических веществ и жидкостей.
ионов, с одной стороны, и жидкими электролитами, не имеющи-
имеющими регулярной структуры/ но обладающими подвижными иона-
ионами, с другой стороны. Нередко твердые электролиты устойчивы
только при повышенных температурах. При более низких тем-
температурах они могут претерпевать фазовый переход с образова-
образованием полиморфных модификаций, обладающих низкой ионной
проводимостью и более обычной кристаллической структурой
(рис. 13.9). Например, и Li2SO4 и Agl — плохие проводники при
25°С, но при 572°С (Li2SO4) и 146°С (Agl) они переходят в
другие полиморфные модификации — a-Li2SO4 и a-Agl, в кото-
которых ионы Li+ и Ag+ весьма подвижны (а~1 Ом^-см). Про-
Проводимость этих электролитов скачкообразно увеличивается при
достижении температуры фазового перехода.
Проводимость твердых электролитов может возрастать так-
также в результате постепенного увеличения концентрации дефек-
дефектов при повышении температуры. Например, в ZrO2 концентра-
концентрация анионных вакансий выше ~600°С настолько велика, что
диоксид циркония является хорошим высокотемпературным ки-
13,2. Твердые электролиты
слород-иоиным проводником*. Границу между обычными ион-
ионными кристаллами и твердыми электролитами зачастую трудно
провести, особенно для таких материалов, как ZrO2, свойства
и поведение которых меняются постепенно с ростом темпера-
температуры.
Как теоретические, так и экспериментальные исследования
очень многих веществ говорят о том, что максимальная ионная
проводимость, которая может быть получена в твердофазных
материалах, составляет 0,1—10 Ом~1-см~1; эти величины соот-
соответствуют такому состоянию, когда большая часть ионов одно-
одновременно находится в движении. По мнению ряда авторов,
именно к таким материалам следует относить термины «супер-
«суперионные проводники» и «быстрые ионные проводники». Эта
терминология получила широкое распространение, однако,
строго говоря, она не совсем правильна. Подвижные ионы не
обладают какими-либо «супер»-свойствами или суперподвиж-
суперподвижностью. Высокая проводимость этих веществ связана скорее
с большой концентрацией подвижных частиц.
Классификация твердых электролитов как веществ, проме-
промежуточных между обычными ионными твердыми телами и ион-
ионными жидкостями (рис. 13.9), подтверждается сопоставлением
энтропии полиморфных переходов с энтропиями плавления.
В обычных ионных кристаллах разупорядочение как катион-
ной, так и анионной подрешеток происходит при плавлении, и
для веществ, составленных из однозарядных ионов, таких, как
NaCl, типична энтропия плавления 24 Дж/(моль-К). Переход
Р~-^а в Agl при 146°С можно рассматривать как квазиплавле-
квазиплавление подрешетки ионов серебра; энтропия перехода составляет
14,5 Дж/(моль-К). При плавлении Agl разупорядочению под-
подвергаются лишь ионы I", вследствие этого энтропия плавления
оказывается существенно меньше, чем для обычных ионных
кристаллов; она составляет 11,3 Дж/(моль-К). Сумма энтропии
полиморфного перехода и плавления Agl близка к энтропии
плавления NaCl. Подобные соотношения наблюдаются и во
фторидах некоторых двухвалентных металлов, например в
PbF2. Энтропия плавления этого соединения составляет лишь
16,4 Дж/(моль-К), тогда как у MgF2 — типичного ионного соеди-
соединения с низкой проводимостью — она равна —35 Дж/(моль-К).
По-видимому, при нагревании PbF2 первоначально происходит
разупорядочение ионов F (при 7>500°С), а приведенное зпа-
* Строго говоря, в качестве кислород-ионного проводника выступает не
чистый, а легированный диоксид циркония, в котором концентрация анион-
анионных вакансий определяется уровнем легирования — содержанием гетерова-
лентной примеси: CaO, Y2O3, Sc2O3 и т. д. (см. книгу Чеботина В. Н. и
Перфильева М. В. в дополнительной литературе).—Прим. ред.
22
13. Ионная проводимость и твердые электролиты
чсние энтропии плавления соответствует разупорядочению толь-
только ионов свинца.
Удельная электропроводность различных твердых электро-
электролитов приведена на рис. 13.10 в форме аррениусовской зависи-
зависимости. Ниже обсуждаются некоторые наиболее важные твердые
электролиты; применению твердых электролитов посвящен
разд. 13.5.
13.2.1. ^-Глинозем
13.2.1J. Структура. Термин «C-глннозем» объединяет семейство
соединений с общей формулой МгО-яХгОз, где п может прини-
принимать переменные значения в интервале от 5 до 11, М — символ
однозарядных катионов, таких, как щелочные металлы, Си ,
Ag+, Ga , Tl+> In , NH4+, НзО+, а X — трехзарядные катионы
А13+, Ga3+ или Fe3'. Наиболее важным членом этого семейства
о
2
о
г" 2
о
\Q 3
СП
4
5
конц,Н2$04
Na-p-глинозем
Na3Zr2PSL2O12
12 3 4
tooo/rfK)
Рис. 13.10. Монная проводимость некоторых твердых электролитов. Для
сравнения показана также проводимость концентрированной серной кисло-
кислоты. Поиски новых твердых электролитов направлены на получение мате-
материалов с высокой проводимостью при низкой температуре (т. е. располага-
располагающихся в правом верхнем углу рисунка).
является натриевый р-глинозем (M — Na*, Х = А13+O который
был уже давно известен как побочный продукт при производст-
производстве стекла. Он образуется в футеровке стекловаренных печей
при взаимодействии оксида натрия, содержащегося в расплаве,
с оксидом алюминия, входящим в состав огнеупорных кирпи-
кирпичей. Название «ф-глинозем», укоренившееся за этими соедине-
соединениями, ошибочно, так как связано с тем, что вначале их прини-
принимали за одну из полиморфных форм оксида алюминия (глино
13.2. Твердые электролиты
23
зема). Теперь хорошо известно, что оксид натрия (или других
металлов) необходим для стабилизации структуры этих соеди-
соединений.
Интерес к р-глинозему как твердому электролиту возник
после пионерской работы сотрудников компании «Форд», кото-
которые в 1966 г. нашли, что ионы Na+ в нем чрезвычайно подвиж-
Рис. 13.11. Кислородные слои в р-глиноземе.
яы при комнатной и более высоких температурах. Этими же
исследователями было найдено, что ионы Na+ могут быть об-
обменены на другие катионы, которые также проявляют высокую
подвижность в структуре р глинозема. Возможность разработки
новых аккумулирующих систем с высокой плотностью энергии,
призванных сыграть важную роль в современной энергетике,
послужила главным стимулом дальнейшего развития исследо-
исследований этих твердых электролитов.
Высокая подвижность однозарядных ионов в р-глиноземе
является следствием его необычной кристаллической структуры
(рис. 13.11), которая построена из плотноупакованных кисло-
кислородных слоев, но в каждом пятом слое отсутствует 3/4 ионов
24
13. Ионная проводимость и твердые электролиты
кислорода, необходимых для плотной упаковки. Ионы Na+
располагаются в этих кислород-дефицитных слоях и имеют воз-
возможность легко перемещаться, так как, во-первых, мест, до-
доступных для ионов Na+, больше, чем самих ионов, и, во-вторых,
ионный радиус Na+ меньше радиуса кислорода. р-Глинозем су-
существует в двух структурных модификациях, известных как
[3- и ^"-глиноземы и отличающихся последовательностью чере-
чередования слоев (рис. 13.12). р"-Форма образуется при большем
А
В
А
В
А
С
А
В
С
А
В
А
С
В
А
С
А —
р
В —
А —
с —
в —
А —
с —
в —
А —
С —
в —
А —
С —
Q
А —
о О
ШБ
о О
ШБ
о О
ШБ
——
О-О-
ШБ
зеркального
отражения
- о О
ШБ
о О
ШБ
с
с —
о О
- о О
о Na4 О кислород ШБ шпинельный блок
Рис. 13.12. Последовательности упаковки кислородных слоев в структурах
р- и р"-глшюземов.
содержании щелочного оксида в кристаллах (/г^5-^7), тогда
как для р-формы п составляет 8-^11. Обе структуры тесно свя-
связаны со структурой шпинели MgA^O^ они построены из шпи-
нельных блоков, имеющих «в толщину» четыре кислородных
слоя с плотнейшей упаковкой, чередующиеся в последователь-
последовательности АВСА, характерной для кубических структур. Ионы
А13+ занимают некоторые из тетраэдрических и октаэдрических
позиций, образующихся между двумя примыкающими кисло-
кислородными слоями с плотнейшей упаковкой. Соседние шпинельные
блоки разделены слоями с дефицитом кислорода — плоскостя-
плоскостями проводимости, в которых локализованы ионы Na+. Гексаго-
Гексагональные элементарные ячейки характеризуются параметрами
а = 5,60 А и с = 22,5 А (р), 33,8 А (р"). В направлении оси с,
перпендикулярной кислородным слоям, элементарная ячейка
р-А12О3 содержит два шпинельных блока, а ячейка
[у'-А^Оз — три блока. Общая последовательность чередования
кислородных слоев, включающая и шпинельные блоки, и плос-
плоскости проводимости, в р"-фазе соответствует кубическому моти-
13.2. Твердые электролиты
25
ву ABC, тогда как в |3-А12О3 наблюдается более сложный мо-
мотив из десяти слоев (рис. 13.12).
Структуру шпинельных блоков надо рассматривать как де-
дефектную по сравнению с идеальной структурой шпинели. Так,
шпинель содержит ионы Mg2+ и А13+ в отношении 1 : 2, тогда
как шпипельные блоки ($- и ^"-глинозема содержат только
А13+, не считая малых количеств легирующих ионов, таких, как
обычно добавляемые Li+ и Mg2+. Вследствие этого и для под-
поддержания баланса зарядов в шпинельных блоках должны обра-
образовываться вакансии ионов А1ЗЧ\
т
Рис. 13.13. Плоскость проводимости в р-глиноземе.
Атомная структура плоскости проводимости р-глинозема
и примыкающих к ней областей была предметом многочислен-
многочисленных структурных работ, и тем не менее пока еще нет полного
понимания некоторых деталей этой структуры. На рис. 13.13
изображен слой плотпоупакованных ионов кислорода О2~ (А
и В), расположенный ниже плоскости проводимости. Заштрихо-
Заштрихованные ионы О2", обозначенные как С, принадлежат следую-
следующему слою (их называют мостиковыми или связующими иона-
ионами); они лежат в плоскости проводимости. Как видно, в этом
слое занята лишь 1/4 кислородных позиций, т. е. на каждый
С-ион кислорода приходятся три незанятых узла, обозначенных
символом т. В р-глиноземе плоскость проводимости является
плоскостью зеркального отражения (рис. 13.12), так что плот-
ноупаковаппые слои ионов А и В, расположенные снизу и свер-
сверху нее, неразличимы в проекции, показанной на рис. 13.13, так
как налагаются друг на друга. В ^''-модификации плоскость
проводимости не является плоскостью зеркального отражения,
а кислородные слои, лежащие выше и ниже нее, сдвинуты отно-
относительно друг друга*.
* Относительный сдвиг соседних кислородных слоев происходит путем
вращения вокруг оси с, являющейся осью 3-го порядка, и составляет 120°
(см. обзор по структуре р- и р^-А^Оз в кн. [Садуорса Дж. и Тилли А.]) —
Прим. перев.
26 13. Ионная проводимость и твердые электролиты
Ионы Na+ в р-глиноземе могут занимать позиции трех раз-
различных типов: 1) межкислородные позиции {т)\ 2) позиции
Биверса — Росса {br)\ 3) позиции анти-Биверса — Росса (abr).
Структурные исследования показывают, что ионы Na+ находят-
находятся преимущественно в позициях br и га, но любое их перемеще-
перемещение осуществляется через позиции abr. В позициях br, имею-
имеющих, как и га, большой размер, ион Na+ окружен тремя кис-
кислородными ионами А из нижнего слоя, тремя из верхнего слоя
и тремя (типа С) из плоскости проводимости. Длина связи
Na—О в этих позициях увеличена (~2,8 А) в сравнении
с нормальной величиной (~2,4 А). Позиции abr имеют
гораздо меньший размер, чем позиции br и га, из-за близ-
близкого расположения двух ионов кислорода, принадлежащих
верхней и нижней плоскостям. Расстояние Na—О в этом случае
составляет 2,3 А. Большинство других однозарядных катионов
также предпочитает занимать в C-глиноземе br- и т-позиции;
исключение составляют Ag+ и Tlh, которые проявляют заметное
предпочтение к позициям abr. Возможно, что это связано се
склонностью этих ионов к образованию ковалептпых связей
и к занятию узлов решетки, характеризующихся низкими коор-
координационными числами, таких, как позиции abr.
Оба соединения J}-A12O3 и р"-А12Оз являются нестехиоме-
трическими фазами, т. е. существуют в интервале составов.
Однако механизм возникновений пестехиометрии в этих фазах,
по-видимому, различен. Обычно считается, что идеальная струк-
структура [3-глпнозема отвечает формуле Na2O«ll AI2O3 или
NaAlnOi7, однако в действительности, р-глинозем содержит зна-
значительно больше оксида натрия, чем идеальный состав. В самой
первой работе по определению структуры [3-глинозема, выпол-
выполненной Биверсом и Россом, был сделан вывод о том, что каж-
каждая элементарная ячейка NaAlnOiz содержит в плоскости про-
проводимости один ион Na+, располагающийся в Ьг-позиции. Более
поздние исследования структуры, выполненные на образцах
состава, близкого к Na2O«8Al2O3, показали, что ионы Na+, со-
содержащиеся в количестве 1!/з на элементарную ячейку, распре-
распределены между позициями br и т. Электронейтральность дости-
достигается при этом за счет дополнительных ионов О2", занимаю-
занимающих некоторые из позиций типа га. Таким образом, по сравне-
сравнению со структурой NaAlnOi7 этот обогащенный щелочным окси-
оксидом C-глинозем «одержит внедренные ионы Na+.
В C"-глиноземе, для которого типична стехиометрия Na2O-
б,6А13О2, в каждой элементарной ячейке содержится ~12/з иона
Na+ на проводящую плоскость. Заряд дополнительных B/3)
ионов Na+ компенсируется не путем внедрения ионов в плос-
плоскости проводимости, а наличием вакансий АР в шпинельных
блоках. Описанные механизмы образования нестехиометриче-
13.2. Твердые электролиты
ских фаз р- и р -глиноземов в действительности могут сильно
усложняться при введении добавок, например VhO и MgO, ис-
использующихся для легирования чаще других оксидов. Оба этих
оксида внедряются в шпинельные блоки, при этом их катионы
(Li+ и Mg2+) замещают А13+ в его позициях.
Введение дополнительных количеств щелочного оксида в A- и
Р"-глиноземы по рассмотренному выше механизму приводит к
более полному соблюдению локальной электронейтральности.
В «идеальной» структуре NaAlndy в шпииельном блоке сосре-
сосредоточен слишком большой положительный заряд, тогда как
в проводящих плоскостях этот заряд слишком мал. В такой
структуре не выполняется правило Полинга, требующее соблю-
соблюдения локальной электронейтральности. В C-глииоземе, обога-
обогащенном оксидом натрия, введение каждого дополнительного
иона кислорода в плоскость проводимости приводит к образо-
образованию двух новых тетраэдрических узлов. Вследствие этого
ноны А13+ из шпинелыюго блока получают возможность пере-
перемещаться в эти новые позиции, благодаря чему уменьшается
отклонение от локальной электронейтральности. В р"-глииоземе
заряд избыточных ионов Na+ в проводящих плоскостях ком-
компенсируется либо возникновением в шпипельных блоках ва-
вакансий алюминия, либо частичным замещением последнего
ионами Li+ или Mg2 .
Как видно, любой из механизмов образования нестехиоме-
нестехиометрии уменьшает отклонение от локальной электронейтрально-
электронейтральности и тем самым повышает устойчивость структуры*.
13.2.1.2. Проводимость и механизм проводимости. Твердые
электролиты со структурой C-глпнозема относятся к числу дву-
двумерных проводников: щелочные ионы имеют возможность сво-
свободно двигаться вдоль плоскостей проводимости, но не могут
проникать сквозь плотноупакованные шпинельные блоки. Про-
Проводимость различных замещенных форм C-глинозема, изучен-
изученная на монокристаллических образцах в направлении, парал-
параллельном проводящим плоскостям, представлена на рис. 13.14.
До недавнего прошлого высококачественные монокристаллы jj"-
глинозема были недоступны, вследствие чего исследования про-
проводимости главным образом выполнены именно на C-фазс, не-
несмотря на то что по уровню проводимости она в 2—3 раза
уступает C"-глипозему. Максимальная величина проводимости
и одновременно минимальная энергия активации наблюдаются
у Na+ и А§+-проводящих C-глиноземов. Увеличение ионного
* Третьяков Ю. Д. Квазихимическая модель разупорядочения. Синтез и
свойства твердых электролитов со структурой глинозема. — Вестник МГУ,
сер. химия, 1974, № 6, с. 643—657. — Прим. ред.
28
13. Ионная проводимость и твердые электролиты
радиуса затрудняет проводимость, так как катионы большего
размера (К+, TI+) не могут двигаться столь свободно вдоль
проводящих плоскостей. Ионы Na4" n Ag ¦ имеют в этом отно-
отношении оптимальный размер. В то же время Li"-^-глинозем (со-
(соответствующие данные отсутствуют на рисунке) характеризует-
характеризуется и более высокой энергией активации, и меньшей проводимо-
проводимостью, чем для Na"- или
300 0 -100. -150 -180 °С Ag+-p-n'iHHO3eMOB. По-
видимому, маленькому
иону Li1', обладающему
высокой поляризующей
способностью, невыгодно
находиться в большом
междоузлии с высоким
координационным чис-
числом, что вынуждает его
смещаться к одному из
кислородных слоев, при-
примыкающих к плоскости
проводимости сверху и
снизу*.
Температурная зави-
зависимость проводимости ji-
глинозема в широком ин-
интервале температур
(вплоть до 1000°С) и
значений о (изменение
а составляет ~107 раз)
прекрасно описывается
Рис. 13.14. Проводимость монокристаллов уравнением Аррениуса.
некоторых замещенных форм S-глинозема. v rt j •
В скобках указаны соответствующие зна- 1акос поведение ОТЛИЧа-
чения энергий активации в элсм-ронво.п.- ется от обсуждавшейся
тач П2]. выше (рис. 13.5) зависи-
зависимости для NaCl, нес-
несколько раз меняющей наклон в интервале ~400°С. Монотон-
Монотонность изменения проводимости, наблюдавшаяся в случае 3-гли-
нозема, характерна для твердых электролитов вообще, а также
для множества сложных оксидов, силикатов и других соедине-
соединений, не отличающихся высоким уровнем ионной проводимости.
2 А б 8 10 12
10 00/Т" (К)
* Следует различать Li+'ji-глинозем и [3-глшюзсм, легированный оксидом
лития (о нем говорилось выше): первый получают методом ионного обмена
(гл. 2) при Г <--300 С, н ион лития в нем имеет высокую подвижность; вто-
второй получают, вводя литийсодержащис компоненты (ПгСОз или LiAl5O8)
па стадии высокотемпературного (~ 1200—1600 °С) синтеза; литии при этом
ъходит в состав ншннельных блоков и поэтому малоподвижен. — Прим.
черев.
13.2. Твердые электролиты
Обращает на себя внимание тот факт, что многие материалы с
простой кристаллической структурой, такие, как NaCl, прояв-
проявляют сложную зависимость проводимости, тогда как материалы
со сложной структурой и стехиометрией, в частности ^-глинозем,
напротив, демонстрируют простое поведение. Последнее наблю-
наблюдается вне зависимости от величины а. Так, для fj-глинозема
зависимость проводимости от температуры описывается одним
и тем же уравнением как при ~300°С (а~ 10 Ом^-см),
так и при —180°С (о~1()-« Ом-'-см).
Другой важной особепностью поведения твердых электроли-
электролитов является то, что в отличие от других ионных проводников,
подобных NaCl, их электропроводность, как правило, хорошо
воспроизводится от образца к образцу, даже если исследования
выполнены в различных лабораториях, что доказывает низкую
чувствительность проводимости этих материалов к присутствию
малых количеств примесей. Так, энергия активации проводи-
проводимости ^-глинозема по многочисленным данным различных ла-
лабораторий хорошо воспроизводится и составляет 0,16±0,01 эВ.
По ряду соображений при обсуждении суперионных провод-
проводников, в частности глинозема, нелегко применять концепцию
равновесия дефектов, в частости закон действия масс (см. вы-
выше примечания редактора). Во-первых, уравнение, устанавли-
устанавливающее концентрацию дефектов Френкеля и Шоттки, примени-
применимо лишь для очень малых концентраций (<0,1% узлов решет-
решетки могут быть дефектными). В то же время совершенно оче-
очевидно, что в {J-глинозсме значительная часть Na1', если не все
ионы, подвижна. Во-вторых, отсутствие влияния малых коли-
количеств примесей означает, что в jj-глиноземе энергию активации
проводимости нельзя разделить на энергию образования и энер-
энергию миграции дефектов и, более того, попытка осуществить та-
такое разделение ошибочна. Число подвижных ионов настолько
велико, что их следует рассматривать не как дефекты, а скорее
как нормальные узлы структуры. При таком рассмотрении ста-
становится понятно, что число подвижных ионов не зависит от
присутствия малых количеств примеси. Так, если добавка
МпС12 в NaCl приводит к возрастанию концентрации катион-
иых вакансий па несколько порядков и соответствующему «ка-
«катастрофическому» изменению проводимости кристалла, то вли-
влияние такой добавки па проводимость J5-глинозема будет прене-
пренебрежимо малым. Малые количества примесей могут влиять па
рабочие характеристики керамического 3-глино.чема, используе-
используемого в серпо-патриевых источниках тока, но это влияние осно-
основано на других явлениях.
Для понимания механизма проводимости в ^-глиноземе по-
полезно рассмотреть рис. 13.13 и 13.15. Мигрируя на большие рас-
расстояния, ион IVа' обязан проходить через позиции br, abr и т.
30 13. Ионная проводимость и твердые электролиты
образующие гексагональную решетку, в последовательности
—br—т—abr—m—br—т—. Энергия активации проводимости,
равная 0,16 эВ, представляет собой сумму энергетических барь-
барьеров, преодолеваемых ионом Na: при переходе из одной br-по-
br-позиции в соседнюю.
Результаты определения фактора Хейвена, рассмотрение пу-
путей проводимости, а также теоретические расчеты свидетельст-
свидетельствуют о том, что проводимость r ^-глиноземе осуществляется по
механизму выталкивания межузельиых ионов. Рассмотрим вна-
вначале проводимость идеализированного кристалла, характери-
О^_~ зующегося составом NaAlaOi;-.
т ( \ В нем ионы Na располага-
brV_y ются в позициях br; позиции
¦-'" '"abrm^~-^аЬг^— а^г " т остаются незапяты-
(~\ ( \ f^\ ми. Допустим теперь, что одни
\^)т К) 'п \^ ) из ионов Na1' покидает свою
lif DJjn t m br т т Г fcr-позицию. При этом он дол-
г/ ~\° / "\ жен проходить через примы-
lm I )т I ) каютую позицию abr и не-
^~"^ премепио натолкнуться иа
Рис. 13.15. Пути проводимости в следующий ион Na+, занима-
р глиноземе. ющий свою Ьг-познцпю, раз-
мер которой недостаточен для
того, чтобы два катиона могли в ней «разминуться». В пози-
позиции т двигающийся ион не может задержаться, так как при
этом ои оказался бы иа недопустимо близком расстоянии от
следующего нона Na!", занимающего позицию br. Таким обра-
образом, имеются лишь две возможности продолжения движения
рассматриваемого нами иона. Во-первых, он может вернуться
в собственную ftr-позицию, но в итоге такого «возвратно-посту-
«возвратно-поступательного» движения перенос электрического заряда не осу-
осуществляется. Во-вторых, движущийся ион может «выпихнуть»
поп Na+, препятствующий движению, из занятой последним
/^¦-позиции. Выбитый нон Na+ может сместиться в одну из
двух примыкающих m-позицпй, а «наступающий» нон Na+ при
этом займет третью из таких позиций, окружающих узел br.
Пели смещенный ион Na1 порывает со своей ?>г-позицией и ее
ближайшим m-окружепием, то это означает начало цепного
процесса выталкивания следующих катионов. При желании
уточнить и детализировать механику процесса выталкивания
эти рассуждения .можно продолжить, однако ясно, что ионы
Na"'" не могут двигаться в ^-глиноземе независимо друг от дру-
друга и перенос заряда — итог кооперативного процесса.
Интересное явление, так называемый полищелочной эффект,
удалось наблюдать при исследовании проводимости р-глпнозе-
мов, содержащих одновременно два различных катиона щелоч-
13.2 Тиерлые электролит))! 'il_
пого металла. Этот эффект состоит в том, что подвижность обо-
обоих щелочных катионов, например Na4" и КЛ оказывается в та-
таком смешанном J3-глиноземе (который можно рассматривать
как твердый раствор Na;--p-Al203 и К -li-AI2O3- — Прим. перев.)
меньше, чем подвижность тех же ионов в «чистых» компонен-
компонентах твердого раствора, т. е. Na:" в Na+-ji-глиноземе и К+ в Kh-p-
глипоземс. Для промежуточных составов твердых растворов
.этих компонентов проводимость проходит через минимум, а
энергия активации проводимости — через максимум. Для стек-
стеклообразного состояния поли щелочной эффект — хорошо извест-
известное, хотя еще недостаточно понятное явление (гл. 18). В кри-
кристаллических материалах этот эффект до исследовании ^-гли-
^-глинозема не встречался. Существование такого необычного явле-
явления и отсутствие его удовлетворительного объяснения показы-
показывают, насколько непросто достичь понимания природы сил, оп-
определяющих ионную подвижность, даже если речь идет о кри-
кристаллических телах, структура которых, казалось бы, хорошо
изучена.
Многие работы но (З-глппозему преследовали цель макси-
максимального увеличения проводимости керамических (полнкрп-
сталлических) материалов на его основе. Проводимость р"-гли-
нозема в несколько раз больше проводимости р фазы, поэтому
предпочтительны такие составы, которые сгабилизпруют ji''-фа-
зу (в системе Na2O — А12О3 без добавок эта фаза нестабильна)
и повышают се долю в смесях по отношению к ^-глинозему.
Ьмло найдено, что эти требования лучше всего удовлетворяют-
удовлетворяются путем легирования .малыми добавками U2O n MgO. Эти ок-
оксиды образуют твердые растворы с p''-глиноземом л стабили-
стабилизируют его кристаллическую структуру.
13.2.2. Иодид серебра и другие A g^-про водящие твердые
электролиты
13.2.2.1. Иодид серебра Agl. Давно известен фазовый переход
[5-Agl^a-Agl прн 146°С, приводящий к возникновению высо-
высокотемпературной модификации с исключительно высокой
(~1 O-vt'-cm) ионной проводимостью, которая примерно на
4 порядка превышает проводимость при комнатной температу-
температуре (рис. 13.10).
Структура a-Agl настолько благоприятна для свободного
движения ионов Ag+, что ионная проводимость при плавлении
E55 °С) не увеличивается, а даже несколько уменьшается.
Энергия активации проводимости в a-Agl составляет лишь
0,05 эВ.
?>-Agl, существующий ниже 146°С, кристаллизуется по
структурному типу вюртцита с гексагональной плотнейшей упа-
упаковкой ионов 1 " и ионами Ag+ B тстраэдрических пустотах меж-
.32
13. Ионная проводимость и твердые электролиты
ду ними. Другая низкотемпературная форма "(-Agl относится
к структурному типу сфалерита. Высокопроводящая полиморф-
полиморфная модификация a-Agl образует объсмпоцентрнрованный куб
из ионов 1~, а ионы AgJ при этом статистически распределяют-
распределяются между тетраэдричсскими позициями, которых насчитывается
в сумме 36 иа одну элементарную ячейку. Тетрагональные по-
позиции соединены друг с другом общими гранями, а тригональ-
ные позиции образуются
в центрах граней тетра-
тетраэдров Agl,}. Ионы иода
практически неподвижны,
тогда как ионы Ag+ мо-
могут передвигаться от по-
позиции к позиции насто-
настолько свободно, что это
напоминает перемещение
жидкости. Разупорядо-
ченное состояние ионов
Ag+ и их ускоренное дви-
движение от позиции к по-
позиции определяются при-
природой связи Ag—I. Ка-
Катион серебра обладает
высокой поляризующей
способностью, так как его
внешние 4с/-электроны
малоэффективно экрани-
экранируют заряд ядра. Боль-
Ag+ионная проводимость в шие иодид-ионы легко
Agl и RbAg4I5. поляризуются, и таким
образом между подом и
серебром образуется ковалентная связь, для которой характер-
характерно образование структур с низкими координационными числа-
числами. Участвуя в проводимости, серебро может свободно пере-
перемещаться из одной тетраэдрической позиции в соседнюю, про-
проходя через промежуточную позицию с треугольной координа-
координацией. Ковалентное связывание в промежуточной позиции ока-
оказывает стабилизирующее действие и уменьшает энергию акти-
активации проводимости. Интересно, что при высоких температурах
AgCl и AgBr имеют заметную проводимость, но ее величина
несравнимо меньше проводимости в a-Agl. Считается, что при-
причина такого поведения заключается в меньшей доле ковалент-
ностн связей Ag—С1 и Ag—Вг по сравнению с Ag—I. Впрочем,
другой безусловно важный фактор состоит в том. что кристал-
кристаллическая структура этих соединений (типа каменной соли)
сильно отличается от структуры a-Ag].
2,0
3.0 4.0 Ь.О
10О0/Г (К1)
6.0
Рис. 13.16.
13,2. Твердые электролиты
33
13.2.2.2. RbAgAlr>. При попытках стабилизировать высокопрово-
дяшуто фазу a-Agl использовались самые различные замеще-
замещения в катионной и анионной подрешетках. Наибольший успех
был достигнут путем частичного замещении серебра на руби-
рубидий при синтезе RbAg4l5. Этот материал превосходит все из-
известные кристаллические твердые тела по уровню ионной про-
проводимости при комнатной температуре — 0,25 Ом~1-см; энер-
энергия активапии проводимости — 0,07 эВ {рис. 13.16). Уровень
с
GOO
500
400
300
100
0
RL>I *- расплав
Rb J +
RbaAgI,
-
1 1 !_,
расплав
4
^ч расплав
\^ V /
Rb2Agl^AgI
i 1 1 1 : ,
/
/
/Agl*
/ расплав
Agl
, 1 J
Rbl 10 20 Rb2AgI3 50
Содержание Ag !,мол
70
Agl
Рис. 13.17. Фазовая диаграмма системы Agl—Rbl [11].
его электронной проводимости в этих условиях пренебрежимо
мал и составляет ~ 10~9 Ом"'-см").
Фазовая диаграмма системы Rbl—Agl приведена на
рнс\ 13.17. В системе образуются два соединения — Rb2Agl3 н
RbAg4I;; последнее распадается по перитектической реакции
при ~230°С, образуя Agl п жидкость. Эвтектоиднын распад
RbAg4I5 на Agl it Rb2AgI3 ниже 27°С кинетически заторможен,
однако влага и пары иода каталитически ускоряют этот про-
процесс. С соблюдением определенной осторожности RbAg^U можно
достаточно легко охладить без разложения ниже комнатной тем-
температуры и таким образом исследовать проводимость этого со-
соединения в широком иптепвп.к.' температур.
Для приготовления RL>.V_r.Is смесь Rbl n A^I в молыюм отношений
1 :4 плавят в п.1к>\ме при ~о1)!)"С и j;nc\i быстро о\ к)жд,иит до kdmh.it-
ион температуры. При охлаждении расплав кристаллизуется, давая мелко-
3—I42!i
34 13. Ионная проводимость и твердые электролиты
зернистое и микроскопически однородное тиердис тело. ГТи'.мсдлющнй деся-
десятичасовой отжиг при ~1С>5ОС приводит ь образованию RbAgJj. Можно oGoit-
тись и бе:< иг)еди;|ри'ге.1ы:о1'о п.i;m.к-ния, oi -пцет:'. 1Я.1 прямое тве;)Диф;|woo
взаимодействие Rbl и Agl в интервале 100 200 °С (реакция происходит
с дс1гт;1Т1>мисш см>ропью юл:.ко при исио.1.'.1ои.'1иии спрессованных тщатель-
тщательно гомогенизированных смесеп тошюдиснерснич рспент ов).
Кристаллическая структура RbAg.;Is значительно отличается
от структуры cc-Agl, no для нее также характерно статистическое
размещение ионов серебра по по.фгщетке [с-траэдричеекпх по-
позиций, касающихся своими гранями. В этой структуре, как и
в a-Agl, число позиций, доступных ионам серебра, намного
больше числа самих подвижных ионов. Ионы рубидия Rb+ за-
закреплены в искаженных октаэдрмческих позициях, образован-
образованных- окружением ионов иода.
Результаты измерения проводимости RbAgJs (рис. 13.16)
ложатся на пологую кривую, которая несколько отличается от
линейной арреннусовской зависимости.
13.2.2.3. Другие твердые электролиты — производные g
Разупорядочсппая структура, подобная cc-Ag], может быть ста-
стабилизирована при низких температурах путем введения многих
катионов п в первую очередь крупных катионов щелочных ме-
металлов, КН4±, замещенного аммония п некоторых органических
катионов. Примерами такого типа соединений, обладающих
проводимостью 0,02—0,2 Ом-'-см при 25°С, являются
[(СНзLМ]2Л?|з115, PyAgsIs [где Ру — поп пиридппия
(C5H5NH)"'"] и (NH4)Ag4l5. Иод в своей подрешетке может быть
также частично замещен, известны, в частности, такие фазы
с высокой ионной проводимостью, как AgaSI, Ag7I4PO4 и
AgeU^VO.j. Последнее соединение характеризуется неплохой
термической устойчивостью и не подвержено влиянию влаги или
паров пода. Среди этих соединений имеются твердые электро-
электролиты, пригодные для использования в различных электрохими-
электрохимических ячейках. Интересным новым направлением является
синтез стеклообразных твердых электролитов, которые получа-
получают быстрой зак-алкой до комнатной температуры распл-вов
смесей Agl, например, с Ag2Se04, Ag3As04 и Ag2Cr207. Ag1'-
иоппая проводимость этих стекол также очень велика (для
состава Ag7l4As04 она составляет 0,01 Ом~1-си). Структуру
иодпдноп и оксоапиоппой подрешеток в этих п подобных некри-
некристаллических материалах, естественно, установить не удается,
но есть основания считать, что и в них серебро проходит через
подрешетку, образованную тетраэдрами с общими гранями.
Одновременное частичное замещение в обеих подрешетка.х
Agl также даст ряд материалов, имеющих высокую проводи-
проводимость при комнатной температуре. К ним относятся, например.
13 2 Твердые электролиты 35
RbAgJ4CN и Agi RHgo45Seo.70I1.io, полученный замещением ионов
Ag+ па Hg1 и иоио"в I па Se~2 (o = 0,1 Ом-'-см-1 при 25 °С).
13.2.2.4. Фазы на основе халькогенидов серебра. Все описанные
выше соединения на основе Agl отличаются высокой проводи-
проводимостью ионов Ag"'¦ и низким уровнем электронной проводимо-
проводимости. Благодаря этому при их использовании в качестве элект-
электролитов практически молено не опасаться внутреннего шунтиро-
шунтирования ячеек электронным сопротивлением электролита. В то же
время для твердых электродных материалов чрезвычайно важ-
важным, если не главным качеством является одновременное нали-
наличие высокой ионной н высокой электронной проводимостей.
Халькогсниды серебра, такие, как устойчивый при высокой
температуре a-Ag2S, проявляют смешанную электронно-ионную
проводимость. Как и в случае Agl, путем разнообразных за-
замещений высокотемпературная разупорядочепная фаза халько-
геиида серебра может быть стабилизирована при более низких
температурах. Например, в системе Ag2Se — Ag^PO4 а-фа.ча
становится устойчивой вплоть до комнатной температуры при
растворении 5—10% Ag3PO4 в a-Ag2Se. Ионная проводимость
такого твердого раствора составляет 0,13 Ом-'-см-', а элект-
электронная— Ю4-н 105 Ом-'-см ' при комнатной температуре.
13.2.3. Г' а логенид-ионные проводники
К твердым электролитам можно отнести ряд галогеппдов,
имеющих структуру типа флюорита и проявляющих при высо-
высокой температуре галогенид-ионпую проводимость. Одним из
лучших- примеров таких соединений является PbF2, имеющий
при Г>500°С проводимость ~5 Ом~'-см~'. При комнатной тем-
температуре фторид свинца —типичный пониый кристалл с низкой
электропроводностью. С повышением температуры электро-
электропроводность монотонно и достаточно быстро увеличивается до
тех пор, пока при 500°С не достигнет предельной величины
~5 Ом-'-см ' (рис. 13.18). Выше этой температуры проводи-
проводимость возрастает очень слабо, а при плавлении (822 °С) практи-
практически вовсе не меняется. Интересно, что если материалы, род-
родственные Agl, переходят в высокопроводягцее состояние в ито-
итоге резкого изменения кристаллической структуры (рис. 13.16),
то материалы типа флюорита при нагревании постепенно при-
приобретают это качество. Подобно PbF2. ведут себя SrCl2. харак-
характеризующийся чрезвычайно высокой проводимостью в темпера-
температурном интервале or ~700°C до температуры плавления
(873°С), и CaF2, который переходит в высокопроводятсс состо-
состояние лишь в непосредственной близости к температуре плавле-
плавления (Ш8°С).
36
13. Иошия проводимость и твердые электролиты
Структуру флюорита можно представить построенной из
ионов F , образующих примитивную кубическую решетку, эле-
элементарные ячейки которой через одну центрируются ионами
Са2' (рис. 7.18). Позиции, доступные для внедрения ионов
F", совпадают с центрами незаполненных кубов. Эти позиции
окружены шестью нонами Са-' и ионами F~ в вершинах куба.
Мсжузельный ион F~ появляется при сдвиге регулярного нона
F" из занимаемой им вершины в объем куба. Возможно, что
при этом образуется
комплексный дефект, од-
однако детали этого меха-
механизма неизвестны, в осо-
особенности дли сильно ра-
зупорядочепного состоя-
состояния при высоких темпе-
температур ал.
13.2.4. Кислород-ионные
проводники
Высокотемпературная
кубическая фаза диокси-
диоксида циркония, имеющая
структуру типа флюори-
флюорита {рис. 7.18), становит-
становится стабильной гтрп ком-
комнатной температуре бла-
благодаря образованию тве-
твердых растворов с CaO, Y2O3 и др. Такой «стабилизированный
диоксид циркония» при высоких температурах имеет высокую
О2 -ионную проводимость, появляющуюся и результате на-
накопления кислородных вакансий, компенсирующих при образо-
образовании твердого раствора недостаток положительного заряда
катионов. Состав такого твердого раствора можно выразить
формулой (Ca.vZi-j л)СЬ-л-, где 0,1^x^0,2. Введение каждого
попа Са2г приводит, таким образом, к возникновению одной
кислородной вакансии (гл. 10).
Структура флюорита, очевидно, чрезвычайно «удобна» для
ночной проводимости, так как F--noHnue проводники, в част-
частности PbF2, также относятся к этому структурному типу. Про-
Проводимость диоксида циркония, стабилизированного, например,
15 мол.% СаО, составляет при 1000°С 5-Ю Ом-'-см и ха-
характеризуется энергией активации ~!,3 лВ. Естественно, что
при более низких температурах проводимость стабилизирован-
стабилизированного диоксида циркония па несколько порядков меньше прово-
проводимости Ма+- и Ag1-проводящих твердых электролитов. Прак-
1,0 2.D ЮОО/rfK
Рис. 13.18. Проводимость PbF2 [2].
13.2. Твердые электролиты 37
тическое значение стабилизированного ZrO2 определяется не
только его высокой 02~-иониой проводимостью, что само по се-
бе необычно, но еще и тем, что этот материал обладает огне-
огнеупорными свойствами и может быть использован при очень зы
еокой температуре (>1500°С) (в том числе как материал вы-
высокотемпературных нагревателей. — Прим, перев.). При высо-
высокой температуре, подобно ZrOj, кислород ионной проводимостью
обладают легированные диоксиды тория ТЪО2 и гафпия НЮ2.
13.2.5. Поиски новых твердых электролитов
Начиная с середины 60-х годов электролитические свойства
уже известных н вновь создаваемых материалов стали предме-
предметом интенсивных исследований. Многие из эти у работ были вы-
выполнены по принципу «попробуем, а там будет видно», т. е.
без предварительных структурных исследований.
В то же время для этих целей проводился также отбор на-
наиболее перспективных структур, имеющих открытые каналы,
слом и т. д.. которые могли бы быть путями облегченного ион
него транспорта. Несмотря па существование ряда теорий, объ-
объясняющих возникновение высокой ионной проводимости, в на-
настоящее время очень трудно сделать достоверные априорные
предсказания величины проводимости в конкретном материале,
даже с известной кристаллической структурой. Самое большее,
на что сейчас можно рассчитывать, зто сформулировать пред
посылки проявления веществом высокой ионной проводимости:
1) наличие большого числа подвижных ионов одного copra
(т. е. п в уравнении а = пси. должно быть большим);
2) наличие большого числа незанятых позиций, доступных
для подвижных ионов; это требование прямо вытекает нз пер-
первого, так как ионы могут быть подвижны, только если имеются
доступные незапятые позиции;
3) малое различие в энергиях незанятых и запятых позиций
и малая величина актпвацпопного барьера при перескоке иона
из одной позиции в соседнюю; наличие большого чиста свобод-
свободных мест окажется бесполезным, если движущиеся ионы не
смогут попасть в нп.\ из-за большой величины энергии актива-
активации;
4) наличие открытых каналов для миграции подвижных
ионов в структуре (предпочтительно построенной по тпп\ трех-
трехмерного каркаса);
5) анионная подрешетка (каркасного тина) должна быть
легкополярпз\ е.ма.
Б ^-глиноземе и стабилизированном диоксиде циркония вы-
выполняются первые четыре, а в Ag^-iioinibix проводниках — все
38 13. Ионная проводимость и твердые электролиты
пять условий. Материалы, ионная проводимость которых не
очень высока, могут удовлетворять лишь некоторым из перечис-
перечисленных требований. Например, многие из силикатов имеют кар-
каркасное строение, но подвижность катионов в iimy ограничена
из за достаточно глубоких потенциальных барьеров. Плохо
проводящие [3- п ¦y-Agl удовлетворяют пятому требованию, ноне
удовлетворяют третьему, и для ионной проводимости это оказы-
оказывается решающим фактором.
Поиск новых твердых электролитов привел к открытию не-
некоторых соединений с каркасной структурой, имеющих высокую
подвижность катионов. Одним из наиболее интересных и потен-
потенциально полезных соединений является Na3Zr2PSi2Oi2. назван-
названный его создателями (Хопг, Кафалас и Гуденаф) NAS1CON
(от англ. Na+-superionie conductor). Структурный каркас
NAS1CON образован сочленением вершин октаэдров Zi'Os и
тетраэдров (Р, Si)О4; при этом образуется трехмерная сеть ка-
каналов, в которых располагаются ионы Na1. Натриевая проводи-
проводимость NASICON сравнима по.уровню с проводимостью C-гли-
нозема.
Цеолиты, судя по большим пустотам в их каркасной струк-
структуре, могли бы быть твердыми электролитами, однако в дейст-
действительности их проводимость не достигает высокого уровня,
Катионы, находящиеся в структуре цеолитов, обычно гпдратп-
рованы, что повышает их способность к ионному обмену, но не
способствует высокой подвижности. Дегидратация цеолитов
приводит к тому, что нх каналы становятся слишком велики
и в итоге катионы смещаются в позиции, расположенные в
стенках канала. Подобный эффект проявляется и в Li'+-?> глино-
глиноземе: маленький ион Li+ занимает позицию вблизи стенки про-
проводящего слоя, благодаря чему проводимость Li+-3-AI2On на-
намного меньше, чем проводимость натриевого [З-глинозема. При
высоком давлении, однако, проводимость Li:-р-глинозема воз-
возрастает, так как размер каналов в проводящем слое уменьша-
уменьшается.
Потребность в новых материалах с высокой Li'-проводи-
Li'-проводимостью очень велика. Это связано с тем, что источники тока
с литиевыми анодами имеют более высокие э. д. с, чем анало-
аналогичные источники тока, скажем, с натриевыми анодами. Li2SO4
претерпевает фазовый переход при 572 °С. и проводимость вы-
высокотемпературной фазы составляет ~1 Ом^-см; ниже точки
фазового перехода проводимость намного меньше и Li2SO4 не
представляет интереса как низкотемпературный твердый элек-
электролит. Различного рода замещения в структуре Li2SO<, пред-
предпринимавшиеся для стабилизации высокотемпературной фазы,
так и не дали возможности снизить температуру фазового пере-
перехода ниже ~400°С.
13.2. Твердые электролиты
39
Li4Si04 и Li4GeO^ имеют умеренную Ы^-проводимость
(—10-" Om''-см в интервале ,300—400°С), а их структуры
представляют гобой удобную матрицу для разнообразных леги-
легирований. Эти соединения построены in изолированных тетраэд-
тетраэдров S1O4 и Gedi, образующих сетки полиэдрических пустот с
moo
500 500
100
1,0 2.0 3.0 4,0
1000/7 (К~!)
Рис. 13.19, Проводимост1> некоторых Li+-проводников.
общими гранями, внутри которых и размещаются
Путем ряда замещений, папркмер
ионы U-'.
Li-
Li*
Zn2+
Li1GeO4
удалось на несколько порядков увеличить ионную проводимость
матричных, фаз; эффект оказался особенно значительным при
невысоких температурах B5—300°С). Наилучшей проводи-
проводимостью в области умеренных температур среди материалов, по-
полученных таким образом, обладает LinZnGc^Oie (LISICON) —
а- 10"! Ом-'-ем при 300°С, а при комнатной — состав
Li3.fiVoRGeo.sO4 — а = 5- 10 Ом-'-см.
Весьма необычный эффект был обнаружен при изучении
смесей LiТ и А12О3. При том, что нет никаких свидетельств хи-
химического взаимодействия этих веществ, проводимость их экии-
40 13. Ионная проводимость и твердые электролиты
молярной смеси на несколько порядков выше проводимости
чистого LiI (чистый А1гО3 — практически изолятор) и при
25°С составляет ~10~~5 Ом-'-см >. Причина этого эффекта пе
вполне понятна, по возможно, что он связал с поверхностной
проводимостью вдоль границы раздела зерен Li 1 н АЬО3*.
В связи с тем что Lil обладает исключительной гигроскопич-
гигроскопичностью, нельзя забывать и о возможной роли воды в возникно-
возникновении высокой проводимости.
Множество разнообразных материалов было изучено с це-
целью обнаружения в них протонной проводимости. Наилучшие
результаты были получены на Н3О' -^-глиноземе и кислом ура-
нилфосфате НиС^РО^Н^О, имеющем при 25°С проводимость
-4-Ю-2 Ом-1-см-'.
13.3. Методы измерения проводимости
13.3.1. Измерения на постоянном токе
Точные измерения проводимости часто трудно выполнимы,
в особенности если речь идет о поликристалличеекпх материа-
материалах, В идеале следует измерять проводимость именно на по-
постоянном токе, чтобы иметь уверенность в том, что полученная
величина характеризует миграцию ионов на большие расстоя-
расстояния, а не диэлектрические потерн, возникающие при колебани-
колебаниях ионов внутри координационных полиэдров. Трудности осуще-
осуществления измерений на постоянном токе связаны с проблемой
нахождения электродного материала, совместимого с твердым
электролитом и не приводящего к поляризации на границе
электрод — электролит. Если, например, к кристаллу р-глипо-
зема прижать два золотых электрода и приложить к ним не-
небольшое (— 100 мВ) напряжение, то ионы Ма4" будут мигриро-
мигрировать преимущественно к катоду п, не разряжаясь, скапливаться
у границы электрод — электролит. При этом у анодной грани-
границы ^-глинозем/золото образуется слон, обедненный ионами Na .
Эта ячейка ведет себя как конденсатор: при коротком замыка-
замыкании в первое мгновение наблюдается импульс тока, величина
которого зависит от прилагавшегося напряжения и сопротивле-
сопротивления электролита, а затем экспоненциальное уменьшение тока во
времени (рис. 13.20,6).
Проблема поляризации границ может быть решена путем
использования обратимых электродов, т. е. электродов, которые
одновременно обладают как электронной проводимостью, так
* За материалами, в которых проводимость возникает в результате сме-
смешения ионных кристаллом с высокодисперснои фазой, закрепилось название
«дисперсоиды». — Прим. перев.
13.3. Методы измерения проводимости
41
и проводимостью по тем ионам, которые подвижны в твердом
*электролите. Жидкий натрий — обратимый электрод для р-гли-
р-глинозема; на его границе с электролитом устанавливается равно-
равновесие:
N;i+ -|- е ч > Nh
Поляризация не возникает, так как иои Na~ может легко пере-
пересекать границы электрод/электролит в обоих направлениях.
Весьма интересны электроды из материалов с широкой об-
областью гомогенности*, предложенные впервые Стилом. Эти об-
Аи
All
" глинозем
Время
Рис. 13 20. Поляризация блокирующих электродов при измерениях ня по-
постоянном токе, и—схема ячейки; б -¦ изменения тока по времени.
ратлмые электроды меняют свои состав при введении или уда-
удалении подвижных попов. Примером могут служить вольфрамо-
вольфрамовые бронзы NajsWO3) в которых ионы Na! весьма подвижны.
Материнская по отношению к этим гоедниеппям структура
\УОз образована октаэдрами WO6, которые сочленены друг
с другом вершинами (рис. 2.13). Такая каркасная структура
пронизана трехмерной сеткой взаимосвязанных каналов, вдоль
которых могут мигрировать катионы Na пли других щелочных
металлов. Химические реакции в электродал, происходящие при
введении или удалении катионов, связаны с изменением степени
окисления вольфрама:
,vNa+ Ь \\\\ 4- *е~ <—»- iVa^W VXWV\-*O3
Дпчалькогсниды переходных металлов со слоистыми струк-
структурами, в частности TiS2, благодаря своей способности образо-
образовывать соединения внедрения с широкими областями гомоген-
" Автор опюшт эти мнтсрил.ш к твердым р;1Створлм. Хотя в общем
c.-n'iai" пог.'и-дггпе дейсгвмтс-лмю мог\ т служить матери j,i<i ми для обрпти-
vi.i\ эль'Ктродои, приведенные ниже конкретные примеры и советской науч-
научной литературе принято относить не к твердым растворам, — Прим. перев.
42
13. Ионная проводимость и твердые электролиты
ности («интсркалаты» щелочных металлов) перспективны как
электродные материалы. Щелочные ноны входят в структуру,
раздвигая слои TiS2, и образуют при этом фа.чы переменного
состава типа LLTiS2 (рис. 2.9), где О^л'^!. Эта реакция обра-
обратима, и при удалении ионов I.iL структура TiS2 полностью вос-
восстанавливается.
13.3.2. Измерения на переменном токе
Альтернативой метода измерения проводимости на посто-
постоянном токе являются измерения па переменном токе, выполнен-
выполненные в широком диапазоне частот. На основании этих измерений
в удачном случае получают, кроме проводимости на постоянном
токе, информацию об электродной емкости, емкостях н сопро-
сопротивлениях межкристаллитных границ, а также о вкладе элек-
электронной проводимости.
Рис. 13.21, Схема моста Уптстонч
для измерений R и С.
Рис. 13.22 Эквивалентная схема дли
по )шфист;п шчеекпго твердого
электролита. /?,-,, C-.s — сопротивле-
сопротивление и емкость грлнпп зерен; Rne,
Coi — объемные сопротивление и
емкость; R> — электронное сопротив-
сопротивление; С.-с — нриэлектршшаи емкость
двойного моя.
Измерения па переменном токе часто проводят, используя
электрическую схему типа моста Уитстона; при этом сопротив-
сопротивление R и емкость С изучаемого образца уравновешиваются
переменными резисторами и конденсаторами (рис. 13.21). Пе-
Переменные элементы R п С могут быть соединены параллельно
(так называемый мост адмнттанса, рис. 13.21) или последова-
последовательно {импедансный мост). Основная проблема измерений
проводимости на переменном токе заключается в правильной
13,3. Метилы измерения проводимости 43
интерпретации результатов, которая усложняется тем, что
эквивалентная схема ячейки (т. е. схематическое представление
последней в виде комбинации сопротивлений и емкостей), как
правило, неизвестна и, по сути, образец с примыкающими элек-
электродами представляет собой электрический «черный ящик». Это
означает, что величины R и С, найденные при уравновешивании
моста на какой-либо фиксированной частоте, совсем не обяза-
обязательно должны соответствовать реальным R и С образца или
ячейки. Поэтому необходимо проводить измерения в широком
интервале частот и выделять ту область, где измеряемые вели-
величины соответствуют истинному объемному сопротивлению об-
образца.
Многие измерения на переменном токе выполнены с приме-
применением блокирующих электродов из золота. В этом случае не
происходит разряда ионов или других реакций на границе элек-
электрод— электролит и, следовательно, эта граница может быть
эквивалентно представлена как емкость двойного слоя Сас, ти-
типичная величина которой составляет I • Ю~° Ф/см2. Емкость
двойного слоя последовательно соединена с сопротивлением
образца. В поликрнсталллческих материалах общее сопротив-
сопротивление образца представляет собой сумму объемного сопротив-
сопротивления зерен /?,,о и межзеренного сопротивления RTJ (иначе со-
сопротивления границ зерен) (рис. 13.22). Rri шунтировано ем-
емкостью границ зерен Сгз, величина которой обратно пропорци-
пропорциональна толщине межзеренного граничного слоя. Емкость плос-
плоского конденсатора определяется выражением
где А—площадь обкладок, d — расстояние между ними, е0 —
диэлектрическая проницаемость вакуума, равная 8,85- !0~и Ф/см,
е' диэлектрическая проницаемость среды, заполняющей прост-
пространство между обкладками. Обычно С„=10~9 Ф=1 нФ, тогда
как сопротивление границ зерен трудно характеризовать типич-
типичной величиной. Как правило, удельное сопротивление (т. е.
сопротивление, отнесенное к единичной длине) границ зерен
больше, чем объемное сопротивление кристалла, по так как
грашщы зерен могут быть на несколько порялкой тоньше самих
зерен, го в действительности А!,., может быть л меньше, чем /?,„-,.
Величины сопротивлений R,,,-. и Rr} почти всегда сильно зависят
от температуры, тогда как для емкостей эта зависимость не
характерна, Объемное сопротивление У?,,,- шунтировано объем-
объемной емкостью С,,,-, (рис. 13.22), связанной с геометрической ем-
емкостью* образца (или ячейки) Со и диэлектрической Пронина-
* Сл—геометрическая емкость ячейки, она тождестпенна емкости ячей-
е Ti'4 же расположением электродов, «заполненной» вакуумом.
44 ^ 13. Полная проводимость и твердые электролиты
емостью твердого электролита соотношением
е' = Соб/С0 A3.15)
Применяя представления и термины, принятые для диэлект-
диэлектриков (например, термин «диэлектрическая проницаемость»),
к ионным проводникам, следует делать это, отдавая себе отчет
в том, что эти группы веществ противоположны по электриче-
электрическим свойствам. Диэлектрическая проницаемость твердых элек-
электролитов характеризует их в отсутствии перемещения ионов на
большие расстояния. Экспериментально величина е' может
быть найдена из измерений па переменном токе, если частота
настолько велика, что направление приложенного электриче-
электрического поли меняется прежде, чем ионы смогут значительно
сдвинуться с места. Таким образом, как и в обычных диэлект-
диэлектриках, величины г' и С!Х- связаны с поляризацией атомов и
электронов. Типичные значения в' лежат в интервале от 5 до
20. Если принять геометрическую постоянную ячейки* равной
1, то е'=С„оА?О) откуда Со6~10-12 Ф (~! пФ).
Если твердый электролит характеризуется значительным
вкладом электронной проводимости, то в эквивалентной схеме
ячейки это отражают, включая параллельно контуру электрон
ное сопротивление Rj. Если электронное сопротивление доста-
достаточно мало, оно может закорачивать весь контур, включая ем-
емкость двойного слоя на границах электрод — электролит. Од-
Однако в большинстве твердых электролитов R^{R,Z + R,,o) и в
таких случаях закорачивание электронным сопротивлением
можно не принимать во внимание.
Моделирование процессов в ячейках с твердыми электро-
электролитами требует построения сложных эквивалентных схем типа
такой, как показана на рис. 13.22. Основная задача исследова-
исследования при это.м сводится к построению эквивалентной схемы, аде-
адекватно отражающей электрохимические процессы в ячейке, а
также к расчету различных омических и емкостных параметров
этой схемы. При проведении измерений по мостовой схеме на
постоянной частоте можно получить лишь результирующие зна-
значения R и С ячейки, отражающие в обобщенном виде всю со-
совокупность происходящих процессов. Гораздо большую инфор-
информацию можно получить при изучении частотных зависимостей
R и С. Не имея возможности полностью изложить теорию это-
этого метода, приведем ниже лишь краткое описание его возмож-
возможностей в применении к исследованию твердых электролитов**.
* То егть произведение A i!~' о формуле для емкости плоского конден-
конденсатор,'! (СМ. НЫ11И'). — Пр'А.Ц Hl'j):'ii
** Теирня методов исследования твердых электролитов с помощью пере-
переменного тока подробно изложим в кн. Графова и Укиш (доно.шитолмгая
литератора). — Прим. перса.
13.3, Меюды измерения проводимости
45
Напомним, что величина тока /, проходящего через сопро-
сопротивление R при приложении поля Е, определяется законом Ома
/ = ?/# A3.16)
и не зависит от частоты поля. Конденсатор блокирует прохож-
прохождение постоянного тока, а проходящий через него переменный
ток, описывается выражением
A3.17)
где со — угловая частота (т —2л/),
для R и С можно записать в виде
I=EIZ
/=У—1. Эти соотношения
A3.18)
где Z — полное сопротивление {импеданс) цепи. Емкостное со-
сопротивление является мнимой величиной, так как содержит мно-
множитель /. Это означает, что между синусоидальным напряжени-
напряжением и током имеется сдвиг по фазе на 90° (ток опережает напря-
напряжение на 90°). При последовательном соединении еопротнвле-
¦' Ех х Е 2—=
Рис. 13.23. Последон.пелыюо согли-
пенно сопротивления и емкости,
Е] и Е-> — cooTBcrcTKviormie падения
напряжений.
Рис. 13.24. Параллельное соединение
сопротивления п емкости.
ния и емкости (рис. 13.23) полное падение напряжения в цепи
Е складывается из падений на двух участках:
и, следовательно, полное сопротивление определяется выраже-
выражением
J_ „ L
A3.19)
Как видно, полное сопротивление включает в себя действи-
действительную п мнимую части (R и 1//оС) и потому называется
комплексным сопротивлением (пли импедансом), которое обо-
обозначается звездочкой Z*,
Z*^Z' — iZ", где Z'=R, Z" = l/coC A3.20)
46
13 Мойная проводимость и тиердие электролиты
При параллельном соединении R и С (рис. 13.24), склады-
складывая обратные величины омического и емкостного сопротивле-
сопротивлений, рассчитывают обратную величину импедлнса
A*=\/Z* = {\/R) + wC A3.21)
называемую адмиттансои цепи, который, так же как импеданс,
разделяется на действительную и мнимую части:
A*--=A'+jA",
где A3.22)
Л' = 1/Д и Л"=соС
Импеданс для схемы, изображенной нл рис. 13.24, можно пред-
представить как обратную величину адмпттанса:
Z* = (Л*) = |A IR) + /юС]-1 - /?/A + j<»RC)
RO— jvRC}R n
*Т7 A3.23)
Огк\'да
и Z*=
Формулы для вычисления импеданса и адмиттапса могут быть
выведены для любого сочетания омических сопротивлений и
t
ш
Я
Z'
Рис. 13.25. Эквивалентная схема
ячейки из твердого злекгрилита с
блокирующими электродамп без уче-
учета сопротивления границ череп.
Рис. 13.2G. Импеданс ячейки с по-
последовательным соединением R и С.
емкостей, однако сложность -лих соотношений резко возраста-
возрастает с увеличением числа составляющих /?(?-элемептов. Так, для
эквивалентной схемы, моделирующей электрическое поведение
монокристалла твердого электролита с двумя блокирующими
электродамп (рис. 13.25), импеданс Z* и аямпттанс А* могут
быть вычислены по формулам
A3.24)
j(oC,
А* =
A3.25)
13.3. .Методы измерения проводимости 47
Для эквивалентной схемы, изображенной па рис. 13.22:
(_1_ _ /@co6j ' +-
A3.26)
Представление этого уравнения в форме, позволяющей раз-
разделить действительную и мнимую части, существенно удлиняет
его запись; читатель может самостоятельно попытаться выпол-
выполнить это преобразование.
Обработка данных, полученных в экспериментах на пере-
переменном токе, выполняется путем их представления на комплекс-
комплексной плоскости (метод годографа) в координатах мнимая часть
(например, Z") — действительная часть (Z'). Будучи нанесены
на график в линейном масштабе, экспериментальные точки
обычно образуют полуокружности п/илп ..тучи. Например, им-
импеданс схемы последовательного соединения R и С (рис. 13.23)
дает в 7*-комплекспой плоскости вертикальный луч, так как
1' ¦ величина постоянная и равная У?, а Z" уменьшается с
ростом (I) (рис. 13.26). Параллельное соединение R п С
(рис. 13.24) дает в /*-плоекости полуокружность, которая на
рис. 13.27 построена для значений R = \0* Ом и С = 10~й Ф. По-
Полуокружность пересекает действительную ось Z' в ючках 0 и
/?, а ее максимум соответствует Z'^0,5 R и наблюдается при
частоте, удовлетворяющей условию otRC—l.
В более сложных эквивалентных схемах каждому парал-
параллельно подключенному /?С-элементу отвечает своя полуокруж-
полуокружность п комплексной 7*-плоскостп. Например, схеме, представ-
представленной па рис. 13.22, благодаря параллельному соединению
R,,t, С,,,-, п /?гз, С,-, должны соответствовать два полукруга.
В схеме, приведенной на рис. 13.25, элементам R и Ci отвечает
полуокружность, ?. последовательно включенной емкости С2 —
луч'(р.чс. 13.28).
Значения R определяют по пересечению луча или полуок-
полуокружности с осью 7/. Учитывая то, что каждая точка полуокруж-
полуокружности или луча соответствует определенной частоте, при изме-
измерениях чрезвычайно важно охватить достаточно широкий интер-
интервал частот. Напротив, результаты, полученные па единствен-
единственной частоте, трудно интерпретировать, так как неизвестно,
лежат ли они на луче (и тогда величина R найдена корректно)
пли па полуокружности (и тогда найденная величина Z' мень-
меньше действительного значения /?). Эквивалентная схема ячейки
с твердым электролитом может быть построена также только
но результатам экспериментов, выполненных в широком интер-
интервале частот. Например, наличие вертикального луча в комплек-
13. Ионная проводимость н твердые электролиты
сной плоскости Z* при низких частотах свидетельствует о нали-
наличии в эквивалентной схеме большой последовательно включен-
включенной емкости. Это в свою очередь может означать, что электроды
являются блокирующими, а электронная проводимость прене-
пренебрежимо мала по сравнению с ионной проводимостью. Если
экспериментальные данные целиком ложатся на единственную
полуокружность, как на рис. 13.27, то это может означать как
отсутствие последовательной емкости в эквивалентной с\еме,
так и то, что она проявляется только в области более низких
частот (рис. 13.28).
10"<Р
Z"
900
250
SO0
1000
г,'ом
Рис 13 27. Импеданс ячейки с па-
ря.-1ло.:П|11Ым соединением R п С.
Я/2
Рис. 13.28. Полуокружность и нертн-
кальпая прямая на графике импедан-
импеданса it комплексной плоскости Z*.
Напомним вкратце основную последовательность построений
в комплексной плоскости. Если для измерений использован пм-
педанспый мост, то полученные величины соответствуют после-
последовательному сопротивлению R< и последовательной емкости
С. Для построения зависимости Z" от Z' экспериментальные
результаты представляют в форме импеданса:
Z* = Rs-{- l//(oCs; Z! = Rs; Z" = l/o>Cs
и затем строят зависимость Z" от 7J. Для построения не требу-
требуется каких-либо априорных допущений или знания уравнений
импеданса ячейки. Этот метод «работает», потому что при
каждой частоте подбором переменных R и С производится ба-
балансировка моста и в нулевой точке 7/„щ.г,ка — Л\ a Z"h4Vt-lRllL-~
— \/шСа. Выполняя анализ в комплексной плоскости, строят за-
зависимость одной переменной от другой и подбирают уравне-
уравнение, выражающее объективную взаимосвязь этих переменных.
Все рассмотренные выше примеры анализировались путем
построения зависимостей в плоскости комплексного импеданса.
Б целом же существуют четыре основные способа предсчавле-
13.4. Другие экспериментальные методы
49
имя и анализа экспериментальных результатов, полученных на
переменном токе:
комплексный
импеданс
комплексный
адмиттанс
комплексная
проницае-
проницаемость
комплексный
электриче-
электрический модуль
1
(пли >¦*)--
,)->+jv>C1.
=A*/}<aC0--?'—je"
(индекс s означает из-
измерения при последова-
последовательном подключении)
(индекс р ошачаст n.v
ли'рпшя при параллель-
параллельном подключении)
A3.27)
A3.28)
A3.29)
A3.30)
Различные формы представления результатов соответствуют
различным способам записи уравнений для любой /?,С-е.\е.мм н
в принципе все содержат одну и ту же информацию. Однако
разные способы обработки результатов помогают выделить те
пли иные особенности исследуемой цепи. Для более сложных
цепей целесообразно обрабатывать результаты измерений, при-
применяя одновременно различные способы, чтобы извлечь из экс-
эксперимента максимум информации. Так, частотная зависимость
импеданса лучше выявляет участки цепи с наибольшими омиче-
омическими сопротивлениями. В поликристаддичеекпх материалах,
имеющих относительно большее сопротивление границ зерен и
малое объемное сопротивление кристаллитов, при исследовании
импеданса проявляются в первую очередь именно эти меж-
кристаллитные сопротивления, а объемные сопротивления мо-
могут быть на и\ фоне замаскированы. Напротпи частотная зп-
впенмость комплексного электрического модуля позволяет вы-
выявить элементы с минимальной емкостью. В этом случае на
результаты исследовании поликрисгаллических материалов в
основном илияют объемные свойства кристаллитов, а влияние
межкристаллтпых границ может быть малозаметным. Таким
образом, применяя оба способа обработки экспериментальных
данных можно раздельно анализировать явления, происходящие
па границе кристаллитов и в их объеме.
13.4. Другие экспериментальные методы
При исследовании твердых электролитов с различным успе-
успехом применяют широкую гамму экспериментальных методов.
В области высоких частот A09- 101J Гц) метод электропровод-
электропроводности дополняют спектроскопия дальней ИК-областн, спектро-
спектроскопия комбинационного рассеяния (КР) и микроволновая
-50 13. Ионная проводимость и твердые электролиты
спектроскопия. Широкие пики, наблюдающиеся в спектрах, по-
полученных этими метдами, связаны с колебаниями подвижных
ионов и дают некоторую информацию о механизме проводимо-
проводимости. Например, появление в КР-спектре пика при G9 см~' в
смешанном Na'¦/КЛглшюземе было объяснено возникновением
межузельпых пар ионов К/г, что, по-видимому, можно считать
еще одним свидетельством в пользу чежузельиого механизма
проводимости в ji-глпноземе (разд. 13.2.1). Часто, однако, ин-
интерпретация подобных результатов спектроскопических иссле-
исследований выбывает затруднения и оказывается неоднозначной.
Примеры использования метода ЯМР-епсктроскотш для изу-
изучения механизма миграции ионов рассмотрены в гл. 3.
Термодинамические исследования фазовых переходов в твер-
твердых электролитах (разд. 13.2.3) дают возможность оценить по
величине -штропип степень ионного разупорядоченпя. Измере-
Измерение коэффициентов диффузии весьма продуктивно дополняет
информацию, полученную при измерении электропроводности.
Мри радиометрическом изучении диффузии на поверхность ис-
исследуемого материала (например, кристалла \'а""-|3-глипозема)
наносится тонкая пленка вещества, содержащего радиоактивный
подвижный ион (например, Na'~*). При последующем отжиге
Na~* диффундирует в объем кристалла. После опреде-
определенного времени отжига кристалл извлекают из г.счп и разре-
разрезают па топкие пластинки параллельно той грани кристалла,
па которую вначале был нанесен радиоактивный изотоп. Изме-
Измерения активности каждой пластинки с последующим построени-
построением концентрационного профиля радиоактивной метки по глу-
глубине кристалла дают возможность определить коэффициент
диффузии Пхл+- В дальнейшем из этих результатов по уравне-
уравнению Периста — Эйнштейна можно рассчитать проводимость,
что является независимым способом ее оценки. В том случае,
когда есть возможность выполнить измерения электропровод-
электропроводности и диффузии с необходимой точностью, сопоставление
этих результатов дает величину фактора Хейвепа, несущего
информацию о механизме проводимости. Основные недостатки
этого метода связаны с необходимостью разрушения кристалла
при его исследовании и большой длительностью экспериментов.
Для исследования диффузии при каждой следующей темпера-
температуре необходим повий кристалл, что может- сказываться на
воспроизводимости результатов. Другой способ исследования
диффузии с помощью радиоизотопов, не требующий, однако,
разрушения образца, основан на ионном обмене изучаемого об-
образца с жидкой средой (раствор или расплав), в которую он
помещен. Радиоактивные гоны, введенные в эту жидкую среду,
диффундируют в изучаемый кристалл, а нерадиояктивпые ионы,
напротив, диффундируют из кристалла в жидкую среду. Через
13.5. Применение твердых электролитов
51
определенное время кристалл вынимают из жидкости, обмы-
обмывают с поверхности и измеряют радиоактивность всего
образца. Ионный обмен повторяют несколько раз и из ре-
результатов измерения радиоактивности рассчитывают, как
и в предыдущем способе, коэффициент диффузии и элект-
электропроводность.
13.5. Применение твердых электролитов
13.5.1. Принципы действия электрохимических ячеек
На основе твердых электролитов могут быть созданы элект-
электрохимические ячейки, предназначенные для решения разнооб-
разнообразных научных и технологических задач, что, безусловно, сти-
стимулирует интерес к исследованию материалов этого класса,
обладающих ионной проводимостью. Особую важность они
приобретают в тех многочисленных практических приложениях,
где жидкие электролиты оказываются неприменимыми.
Рассмотрим ячейку, схематически показанную на рис. 13.29,
которая состоит из твердого электролита, разделяющего два
электродных пространства, при-
причем последние могут содержать
твердные фазы, жидкости или
газообразные вещества одинако-
одинаковой или различной химической
природы. Например, по обе
стороны от твердого электроли-
электролита может находиться газообраз-
газообразный кислород при двух различ-
различных парциальных давлениях или
же с одной стороны — натрий, а
с другой — сера.
Э.д. с, возникающая в результате электрохимической реак-
реакции в такой ячейке, подчиняется уравнению Нерпста
эд.с.(?)
Рис. 13.29. Электрохимическая
ячейка с твердым электролитом.
RT |п 10,
пИ
Ked]
A3.31)
На каждой границе электрод — электролит устанавливается
окислительно-восстановительный потенциал; так, проходящему
па аноде процессу окисления
М > М+ -f- e
соответствует
A3.32)
52 13. Ионная проводимость и твердые электролиты
где ?°м/м+—-стандартный окислительно-восстановительный по-
потенциал этой реакции, [М~] н [М] — концентрации частиц,
/; — постоянная Фарадея, равная 96 500 Кл. Ионы М+, воз-
возникающие на аноде, двигаются через твердый электролит и
реагируют па катоде с анионами X", образующимися в резуль-
результате реакции восстановления
X -Ь е >- X"
которой соответствует окислительно восстановительный потен-
потенциал
^1п1ТТ A3.32а)
Алгебраическая сумма Е\ и ?2 дает э.д. с. цепи, связанную со
свободной энергией ДС суммарной (потенциалообразующей)
реакции ячейки М+Х->МХ соотношением
AG = —nEF A3.33)
Электрохимические ячейки с твердыми электролитами мож-
можно использовать для термодинамических исследований, в пер-
первую очередь для определения величин свободных энергий об-
образования различных соединений. Например, ячейка
Аятв | AgiTb | Ag2STb, S>k , Ств
была использована для измерения свободной энергии образо-
образования Ag2S. В этой ячейке проходит реакция
2А? + S >¦ Ag2S
Поскольку серебро и сера находятся в стандартном состоянии,
то
A3-34)
Получив из эксперимента температурную зависимость э. д. с.
ячейки, можно рассчитать энтропию и энтальпию потенциале-
образующей реакции.
13.5.2. Источники тока
Потенциальная возможность создания новых источников то-
тока на основе твердых электролитов — наиболее существенный
стимул для исследования этих материалов. Среди элементов
с твердым электролитом с технической точки зрения наиболее
важе!!, по-нпднмому, еерно-натрневый элемент, в котором ис-
используется N а '¦-? глинозем (рис. 13.30). Он представляет со-
собой вторичный источник тока (аккумулятор) с высокими удель-
удельными (т. е. рассчитанными на единицу массы) показателями
13.!i. Применение твердых электролитов
53
энергоемкости п мощности. Серно-натриевые аккумуляторы
разрабатываются в первую очередь для электромобилей и си-
систем выравнивания мощности электростанций, в настоящее вре-
время во многих странах эти аккумуляторы проходят стадию ис-
испытаний и усовершенствования. Очень упрощенно устройство
серио натриевого элемента можно описать следующим образом:
твердый электролит C-глипозсм разделяет расплавы металли-
металлического натрия (анод) и серы (катод). Обычно твердый элект-
электролит представляет собой полую трубку, закрытую с одного
Твердый ^-'
электролит
(р-глинозем)
-_^ —натрии
—-—^-изолятор (А1205)
сера (заряженное
сосюяние) или
"сульфид натрия
(разряженное
состояние)
Суммарная реакция ячейки- 2Na * bS^NSjS^ 2,08В
Рис. 13.30. Серно-н<1триевая ячейка с твердым электролитом.
конца, внутри которого содержится расплав натрия, а сама
трубка помещена в расплав серы (возможна и обратная схе-
схема). Поскольку расплавленная сера, будучи веществом с кова
лептиым типом связи, не проводит электрического тока, то ка-
катод изготавливают из графитового войлока, пропитанного се-
серой. Наружный корпус элемента, служащий одновременно
коллектором тока, изготавливается из нержавеющей стали.
При разрядке в элементе проходит реакция
В начальной стадии разрядки х~5. >по соответствует формуле
сульфида, наиболее богатого серой, Na2Sr,; no мере разряткп
х уменьшается. Диаграмма системы натрий — сера предстпп-
лена на рис. 13.31. Серпо-нятрпсвый элемент эксплуатируют
при 300—350°С; как видно из диаграммы (рис. 13.31), это ниж-
нижний порог температуры, обеспечивающей расплавленное состо-
состояние продуктов разряда (сульфидов натрия) в широком интер-
интервале составов. Диаграмма состояния показывает также, что
при достижении в холе разрядки значения лг<3 (т. е. 60% S,
54
13. Ионная проводимость и твердые электролиты
4О°/о Na) пересечение ликвидуса приведет к появлению кри-
кристаллического Nla2S2, а дальнейшая разрядка приведет к по-
постепенному отвердению катодной массы. Напряжение разомкну-
разомкнутого элемента зависит от уровня его разрядки и температуры;
максимальное возможное напряжение равно 2,08 В при 300°С.
расплав
I
* расплав, два расплава
< pacrwUe
20С
100
Na
Рие. 13.31. Фпзобяя дилгримм^ системы натрий—сера.
В результате разряда при достижении ,v~3 напряжение умень-
уменьшается до 1,8 В. Теоретическая величина энергоемкости серно-
натрпевого источника составляет 750 Вт-ч/кг. Экснерилченталь-
но достигнуть! величины 100—200 Вт-ч/кг, и вряд ли эти значе-
значения будут сильно улучшены, по крайней мере практика работы
с другими источниками тока говорит о том, что в этих устрой-
устройствах теоретическая энергоемкость всегда реализуется лишь
частично*.
': Детл.|г.иом\ .Нилину ъроблсм еозд.п;т;я еерпо натриевого ,1ккум\лято-
р.э ноевятсн.) ктггп [Cudt/орс Лж., Tn.i.itt А. Сергго-и.чгрпсвме пккумулято-
rr-i: Пер. с ;iiiri. — М: Д\пр, 1988]. — Прим. n.'psa.
13.5. Применение твердых электролитов 55
Другие типы ячеек с твердыми электролитами находят при-
применение в качестве миниатюрных первичных источников тока,
для которых характерен длительный рабочий ресурс при отно-
относительно малой мощности. Такие источники применяются для
питания электронных часов, кардиостимуляторов и некоторых
устройств военного назначения. Среди ряда успешно работаю-
работающих электрохимических систем для этих целей наиболее часто
используются два элемента:
Ag|RbA?4I0|I, @,65 В)
Li[LiI[I2 B,fiB)
В обоих элементах иод как таковой не может выполнять роль
катода, так как его электронная проводимость слишком низка,
чтобы обеспечить достаточный но величине разрядный ток.
13.место иода используются различные подпдпые комплексы, на-
например (СИ3)-i-NIi, содержащий полшюдмд-апиопы, п комплекс
с переносом заряда (поли-2-винплпирпдин)иодид. По рабочим
характеристикам элемент I.?JI^ пригоден для применения в кар-
кардиостимуляторах; при температуре 37°С п плотности тока 1-т-
-т- 10 мкА/см? достижима удельная энергия 0,8 Вт-ч/ом! и ра-
рабочий ресурс не менее десяти лет.
13.5.3. Кислородные концентрационные ячейки и датчики
Электрохимические ячейки с твердыми электролитами мо-
могут быть использованы для измерений парциальных давлений
газов или концентраций газов, растворенных в жидкостях.
На рис. 13.32 приведена схема устройств! концентрационной
ячейки такого назначения с твердым электролитом из стабили-
стабилизированного диоксида циркония, выполненная в форме трубки
с одним закрытым концом. Внутри трубки находится гач с из-
известным парцпгыьпь'м давлением кислорода, например воздух.
Порпеть'е металлические покрытия на трубке образуют элект-
электроды, проницаемые для кислорода в обоих направлениях. Па
рис. 13.,32. о схематически показано направление переноса
ионов кислорода через твердый электролит п электродные ре-
реакции, проходяшпе в том случае, если измеряемое парциальное
давление кислорода Л г,о меньше, чем давление над электродом
сравнения Р"п„ Э.д.с. ячейки связана с ра:шо'/ты<> давлений
кислорода
? = 1Г1пТ§- 03.35)
Поскольку стабилизированный ZrO2 обладает хорошей О5-
ионной проводимостью лишь при высоких температурах, то эта
56
13. Ионная проводимость и твердые электролиты
ячейка способна работать в интервале 500—1000°С и при этом
измерять парциальные давления кислорода вплоть до 106 атм.
При более низких /\-,2 вклад электронной составляющей в сум-
суммарную проводимость твердого электролита становится сущест-
существенным, что приводит к внутреннему закорачиванию ячейки.
Твердые электролиты на основе стабилизированного диоксида
тория ТЬОг сохраняют ионную проводимость в более широком
интервале давлений кислорода и могут быть использованы при
Ро2<Ю'F атм*. Кислородные концентрационные ячейки, по-
2е
Рис. 13.32. Кислородная концентрационная ячейкл с твердим
из стабилизированного диоксида циркония.
добные рассмотренному датчику ZrO^lCaO), находят широ-
широкое применение, в частности при анализе выхлопных га.чов, из-
измерениях поглощения кислорода при дыхании, изучении раз-
различных металлоксидных н газовых равновесий (СО/СО2,
Нг/Н;О), а также для измерения активности кислорода, раст-
растворенного в расплавах металлов. В последнем случае датчики
погружаются непосредственно в расплавленный металл, напри-
например в сталь. Приборы могут быть проградуироваиы сразу в ве-
величинах концентрации кислорода, что повышает удобство поль-
пользования датчиком. Важным качеством кислородных датчиков
является их способность, как правило, быстро откликаться па
изменение давления кислорода.
" Гранины так нлзывлемои электролитической области кпслиродирово-
ДЯЩИХ TliCpjUX ЭЛеКТрОЛНГОП (Т. С. O6.'I<ICTII, R КОТО'ЮН ЭЛекТрпп!!ЫП ПЛИ Д1.1-
рочыын вкллд м проводимость <1%) описывлюгея 4Kcnp]iu\ieni;i.r,iiu опре-
определяемыми зависимостями минимального (для улрктрщшоп проводимости)
и максимального (для .iLipo'inoi'i проводимости) д<;влемпй кислорода or тем-
температуры. Этот вопрос подробно рассмотрен п кпше Чоботипа В. Н. и Пер-
([;ильепа Л1. В. (см. дополнительную .штернгуру). — Прим. перев.
¦ 'прпжнгния
57
13.5.4. Топливные элементы
Стабилизированный диоксид циркония в форме трубок или
дисков находит применение в некоторых конструкциях высоко-
высокотемпературных топливных элементов. Одно из электродных
пространств содержит при этом воздух или кислород, а дру-
другое—горючий газ, например Н2 или СО. На циркониевый твер-
твердый электролит в этом случае также наносят пористые метал-
металлические электроды, на поверхности которых происходят ре-
реакции
н2 + VA »- Н-Р
или
СО 4- 1/2О2 >¦ СО,
Преимущества таких топливных элементов заключаются в
практическом отсутствии проблемы поляризации электродов, а
также в возможности получения высокой плотности тока
@,5 Л/см2) и высокой удельной энергии 0,5 Вт/см2. Однако
при практическом использовании таких элементов необходимо
соблюдать режим весьма постепенного разогрева их до рабо-
рабочих температур; конструирование таких элементов также связа-
связано с решением ряда проблем. Процесс, проходящий в топлив-
топливном элементе, обратим: изменив направление тока, можно раз-
разлагать водяной пар на водород и кислород. Такие высокотем-
высокотемпературные электролизеры могут быть использованы для
аккумулирования электроэнергии в химической форме. В рав-
равной степени их можно использовать для выделения кислорода
из СО?, например при регенерации атмосферы космических ко-
кораблей, и для электрохимического удаления кислорода из жид-
жидких металлов.
Упражнения
13.1. Окажет ли влияние (если д.|. то к.чкое) нл приводимость кристал-
кристаллов ХаС1 их легнров.пше небольшими количествами след\ ющих добавок:
,i) KC1: б) ХаПг; в) СлС12: г) Л«С1; д) N;i2O?
13.2. Каково влияние па проводимость кристаллов ЛрС1 следующих
примесей: я) Л^Вг; б) ZnCl2; в) Ag?O'
13.3. Используя данные по приводимости XnCl, прпиодеппiin.' ii.i рис. 135.
опепите: а) энт.имипо миграции клтпопных вакансий; б) читллмшю обра-
.чевгишя Дефектов Шотткп. Полученные дипчения должчи бып. ij.'iiijkh к при-
приведенным к т;|б.i. 13 2.
13.1. Некое твердое вешгетио имеет нроводи.мость ~1П 5 Ом^-см
при комн,п:к)й тгмпературо. Предложите способы определения носителя 3;i-
ряд/i, ответственного jn эту проводп.мость. К.и; можно рл/ппит;, провпди-
Moci'b. осуществляемую: а) электронами; б) ионами Na' ; в) попами О2"?
13.5. Используя длнные по проводимости для р.чз. шчпых л л мешенных
^-г.'щпочечпв (рис. 13.14), плесчитлйте для каждого из них прмэкспопешш-
,t.ii>ii'.)U"i м!'1)жнтс^1ь /1 и анергию лктив.пшп IS.
58 13. Ионная проводимость н твердые электролиты
13.6. Для эквивалентной схемы, представленной на рис. 13.25. выведите
уравнения а) импеданса, 6} ллмпттанса. Изобразите зависимость в комп-
комплексной плоскости при следующих параметрах: /?=10в Ом, С|--Н)-'2 Ф,
С2= 10" ь Ф. Какая область частот может быть наиболее полезной для оп-
определения R, С[ и С2У
13.7. С помошмо рис. 1331 объясните, почему режимы работы н рабо-
работоспособность серно-натрневого элемента при 200 ~С и 300 °С различны.
13.8. Одной из возможных сфер применений серно натриевого аккуму-
аккумулятора тока яиляется транспорт на электрической тяге. Какие преимуще-
преимущества и недостатки имеет этот элемент но сравнению с традиционным свин-
цово-кнелотным аккумулятором?
13.9. Предложите, конструкцию твердоэлектролнтного устройства —сен-
—сенсора газообразного фтора.
13.10. Сравните возможности методов измерений на постоянном и пере-
переменном токе при исследовании керамических материалов с ионной прово-
проводимостью.
Литература
1. Barr L. W., Lidiard А. В., In Physical Chemistry, An Advanced Treatise,
Vol. 10, Solid Stale, Academic Press, 1970.
2. Derrington C. ?., O'Kcefje M., Anion conductivity and disorder in lead
fluoride, Nature Phys. Sci., 24<>, 44—45, Nov. 19 A973).
3. Gcller S. (Ed.) Solid electrolytes, in: Topics in Applied Physics, Vol. 21,
Springer Verlag, 1977.
4. Hackwood S., Linfnrd R. G., Physical Techniques for ihe Study ol Solid
Electrolytes. Chem. Revs., 81, 327, 1981.
5. Hayes №'., Supcrionic conductors, Contemp. Phys., 19E). 469—480 A978).
6 Holzapi'el C, Rickcris П., Solid state electrochemistry— new possibilities for
research and industry. Die Nalunviss, 64B), 53—58 A977),
7. Hooper A., FaM ionic conductors. ConUrnp. Phys., 19, 147—168 A978).
8. O'Keeffe M., Hyde B. G., The solid electrolyte transition and melting in salts,
Phil. Mag., B3, 219—224 A976).
9. Rickert #., Solid ionic conductors—principles and applications, Angcw.
Chem. Int. Ed., 17, 37 -46 A978).
10. Steele В. С H., Electrical Conductivity in Ionic Solids, in: MTP International
Reviews of Science. Solid State Chemistry (Ed. L E. J. Roberts), p. 117,
Wiley, 1972.
11. Takahashi Т., Solid silver ion conductors, J. Appl. Electrochem., 3, 79—90
A973).
12. Whittingham M. S., Muggins R. A., Solid State Chemistry, NBS special
publication 3G4, 1972.
Дополнительная литература, ЧеОотии В. Н. Физическая химия твердого
тела. — М.: Химия, 1985; Чеботин В. Н., Перфильев М. В. Электрохимия твер-
твердых электролитов.—М.: Химия, 1978: Гуревич Ю, Я. Твердые электролиты.—
М: Наука. !986; Укше Е. А., Букцн И. Г. Твердые электролиты. — М.: Наука,
1977: Графов Б. М., Укше Г.. А. Электрохимические цепи переменного тока.—
М.: На\'ка, 1973; Сидуорс Дж„ Тилли А. Серио-натрпевые аккумуляторы Пер
с англ. — М: Мир, 1988.
Глава 14
ЭЛЕКТРОННЫЕ
СВОЙСТВА
И ЗОННАЯ ТЕОРИЯ:
МЕТАЛЛЫ,
ПОЛУПРОВОДНИКИ,
ТВЕРДЫЕ
НЕОРГАНИЧЕСКИЕ
СОЕДИНЕНИЯ,
ИХ ЦВЕТНОСТЬ
14.1. Введение — металлы, диэлектрики, полупроводники
Способность металлов проводить электрический ток извест-
известна уже многие годы. Открытие Бардиным, Шокли и Братейном
A948 г.) транзисторного эффекта и построение первого полу-
полупроводникового транзистора привели к бурному росту интереса
к электронным свойствам материалов, особенно в последние
годы в связи с созданием полупроводниковых интегральных
схем на основе кремния. В настоящей главе автор старался, не
используя строгого математического аппарата, дать представ-
представление об электронных свойствах твердых тел с позиции зонной
теории. Основное внимание уделено металлам и полупроводни-
полупроводникам, однако рассмотрены и другие твердые неорганические
фазы.
На первый взгляд кажется, что главное различие между
металлами, полупроводниками и диэлектриками состоит в ве-
величине проводимости. Металлы обладают высокой электропро-
электропроводностью (сяз Ю4-МО6 Ом-'-см-1), диэлектрики—низкой про-
проводимостью или практически полным ее отсутствием (с^
=STlO-1[3 Ом-1-см), полупроводники занимают промежуточное
положение (о» 10-5-f-l03 Ом-'-ем-1) (табл. 13.1). Приведенные
граничные значения проводимости весьма условны.
Однако существует и принципиальное различие механизмов
проводимости в металлах, с одной стороны, и в полупроводни-
полупроводниках и диэлектриках—с другой. Суть различия состоит в том,
что проводимость большинства полупроводников и диэлектриков
быстро возрастает с ростом температуры, в то время как
электропроводность металлов в этих условиях хотя и слабо, но
постоянно уменьшается.
Электропроводность а связана с концентрацией п, зарядом
е и подвижностью и. носителей заряда
а —пен A4.1)
60 14. Электронные свойства н зонная тоория
Поэтому температурная зависимость проводимости а различ-
различных материалов определяется температурными зависимостями
п, е и [I. Для всех электронных проводников заряд е постоянен
и не зависит от температуры. В большинстве материалов вели-
величина подвижности обычно слабо уменьшается с ростом темпе-
температуры из-за увеличения интенсивности столкновений между
движущимися электронами и фопонамн, т. е. из-за рассеяния
электронов на колебаниях кристаллической решетки. Поэтому
различное поведение металлов, полупроводников и диэлектриков
связано в основном с концентрацией носителей заряда и и с-е
температурной зависимостью:
1) в металлах концентрация носителей заряда п велика
и неизменна при изменении температуры. Ьдипствеиной пере-
переменной величиной, входящей в уравнение для о, является ц.
А поскольку \х слабо уменьшается с температурой, то также
уменьшается и о в этих условиях;
2) в полупроводниках и диэлектриках я обычно экспонен-
экспоненциально растет с температурой. Этот стремительный рост п
вносит более существенный вклад в изменение проводимости о,
чем уменьшение и, Следовательно, о быстро увеличивается с
повышением температуры. В этом смысле диэлектрики можно
рассматривать как некоторый предельный случай, так как при
обычных температурах величина п в этих веществах краппе
мала. При высоких температурах проводимость отдельных ди-
диэлектриков достигает полупроводникового уровня из-за роста
п. Наблюдается и обратное — при низких температурах некото-
некоторые полупроводники становятся диэлектриками.
Полупроводниковые материалы можно разделить па две
группы. Возможна следующая классификация полупроводни-
полупроводников.
1) Элементарные полупроводники. Это например такие полу-
полупроводники, как кремний и германии, которые можно назвать
классическими. Элементы Si и Ge находятся о IV группе перио-
периодической системы. С ростом атомного номера элементов IV
группы происходит переход от изоляторов (алмаз) к полупро-
полупроводникам (Si, Ge, серое олово) и далее к металлам (белое оло-
олово, РЬ). За исключением белого олова и свинца, все эти
вещества имеют структуру алмаза, в которой каждый атом
находится в тетрагональном окружении других атомов. Тетра-
Тетраэдры сочленяются друг с дрлгом общими вершинами и образуют
жесткий трехмерный каркас кубической симметрии (гл. 7).
По-видимому, структура алмаза особенно выгодна для прояв-
проявления полупроводниковых свойств.
2) Полупроводниковые соединения, например многие неор-
неорганические и некоторые органические соединения. Хороню из-
известны неорганические полупроводники типа A"'BV. В состав
14.2. 3,ivhT|>oii[[;in структура тиердыч тол. Зонппи теория 6!
утих Cf)eдnнeний входят элементы III и V групп периодической
системы и молярном отношении 1 : 1. Некоторые и:* этих веществ
изоэлектронны с находящимся между ними элементом IV груп-
группы. Например GaAs и InSb изозлектропны соответственно с гер-
германием и оловом. Другие соединения, имеющие электронную
концентрацию, отличную от концентрации элементов IV груп-
группы, например GaP, также являются полупроводниками. Боль-
Большинство соединений AmBv имеют структуру цинковой обманки
(разд. 7.1), которая родственна структуре алмаза.
Известны и другие классы соединении — оксиды, сульфиды и
т. л.--имеющие различные кристаллические структуры, кото-
которые также являются полупроводниками. Некоторые из них
будут рассмотрены позже.
14.2. Электронная структура твердых гел. Зонная теория
Электронную структуру металлов, полупроводников и мпо
гих других твердых тел можно описать в рамках зонной тео-
теории. В металлах, например в алюминии, внутренние электроны
(l.s, 2s и 2р) локализованы па дискретных атомных орбиталях
отдельных атомов алюминия. В то же время образующие ва-
валентную оболочку За'- и Зр-электроны занимают энергетические
уровни, которые делокатизовапы по всей кристаллической ре-
решетке металла. Эти уровни подобны гигантским молекулярным
орбиталям, па каждой из которых находятся по два электрона.
Практически в каждом твердом веществе должно существовать
огромное количество шкнх уровнен энергии, которые очень
близки друг к другу. Так, в кристалле алюминия, содержащем
.V атомов, каждый из которых имеет одну Зя-орбиталь, образу-
образуется зона, включающая Л' энергетически эквивалентных уров-
уровней. Такая зопл называется Зя-валептной зоной. Аналогично
3/7-орбитали атомов алюминия образуют при делокализации
3/?-зону энергетических уровней.
Аналогичным способом можно представить образование зон-
зонной структуры других материалов. Тогда металлы, полупровод-
полупроводники и диэлектрики различаются
1) по зонной структуре;
2) степенью заполнения валентных зон;
3) шириной энергетическом запрещенной зоны, расположен-
расположенной между полностью заполненной и пустой зонами.
Основные положения зонном теории построены на данных
репггепоспектральных исследовании и двух независимых теоре-
теоретических подходах. Согласно «химическому подходу», зонная
теория сводится к распространению теории молекулярных ор-
бнталей в том виде, в каком она обычно применяется для описа-
описания небольших молекул конечных размеров, на случай беско-
62 14. Электронные свойства и :юпчля теория
печных трехмерных структур. В рамках теории молекулярных
орбпталей двухатомных молекул рассматривается перекрыва-
перекрывание электронных орбиталей двух атомов, приводящее к обра-
образованию двух молекулярных орбиталей, которые делокализова-
ны в пространстве между обоими атомами. Одна из молекуляр-
молекулярных орбиталей — связывающая — характеризуется более низким
уровнем энергии, чем уровни атомных орбиталей. Энергия дру-
другой орбитали — антисвязыва-
Энергия ющей —¦ выше энергии атом-
} т антисвязывэгощая ПЫХ орбиталей (риС 14. i).
I орбитам Распространение такого
| атомная .А агомная подхода на молекулы, состоя-
i —1 — связывающая щие из большего чиста ато-
ор°бит1льрная ор6итзль мов, ведет к увеличению ко-
количества молекулярных орби-
Рис 14.1. Молекулярные орбит^ш талей. Из каждой атомной ор-
в двухатомной молекуле бптали, входящей в систему,
возникает одна молекулярная
орбиталь. Поскольку число молекулярных орбиталей возраста-
возрастает, энергетическое различие между соседними молекулярными
орбиталями должно уменьшаться (рис. 14.2). Энергетический
«зазор» между связывающей и антисвянывающей орбиталями
также уменьшается до тех пор, пока не достигается состояние,
когда образуется «континуум» энергетических уровнен.
Металлы можно рассматривать как бесконечно большие
«молекулы», в которых существует огромное число энергетиче-
7 2 3 4 N?.~cvcr-
Рис. 14.2. Расщепление энергетических уровней согласно теории молекуляр-
молекулярных орбитплс-й.
скнх уровней или «молекулярных» орбпталей. В одном моле
металла число таких уровнен —6-Ю23. В этих условиях пе име-
имеет смысла рассматривать каждый из энергетических уровней
как молекулярную орбнталь. так как каждый из них делокали-
зован в пространстве всех атомов кристаллической решетки
металла. Поэтому обычно говорят об энергетических уровнях
или энергетических состояниях.
14.2. Электронная структур;) твердых тел. Зонная теория
6.3
На рис. 14.3 приведена схема зонной структуры металли-
металлического натрия, рассчитанная согласно теории «плотной хими-
химической связи». Видно, что ширина каждой отдельной энерге-
энергетической зоны зависит от расстояния между атомами и, сле-
следовательно, от степени перекрывания орбиталей соседних ато-
атомов. Как показал расчет, при межатомном расстоянии г0 (эк-
(экспериментально найденное значение расстояния между атома-
атомами Na в кристаллической решетке металла) 3s- и Зр-орбигалн
о.
~ I
IS
Межатомное расстояние—э~
Pirc. 14.3. Влияние величины межатомного расстояния на положение энер-
энергетических \ровней н энергетических зон и натрии (рассчитано в рамк;)\'
теории «¦плотной химической связи»).
соседних атомов существенно перекрываются, образуя широкие
3s- и Зр-зопы (заштрихованные области па рис. 14.3). Верхние
уровни 35-зопы характеризуются той же энергией, что и нижние
уровни Зр-зопы. Следовательно, между 3s- и Зр-зонами не име-
имеется энергетической щели. Перекрывание энергетических полос
является важным моментом при объяснении металлических
свойств некоторых других элементов. например таких, как
щелочноземельные металлы (см. ниже).
При межатомном расстоянии r0 Is-, 2s- и 2р-орбитали сосед-
соседних атомов натрия не перекрываются друг с другом. Следова-
Следовательно, в таких условиях не происходит образования зон, а
сохраняются дискретные атомные орбытали, связанные с каж-
каждым отдельным атомом. Па рис. 14.3 эти атомные орбитали
изображены в виде тонких линий. Если бы было возможно
сблизить аго.мы до расстояния г'<г0, например путем воздейст-
воздействия высоких давлений, то 2s- и 2/з-орбитали перекрылись бы с
64 14. Электронные свойства и зонпля теория
образованием энергетических зон (заштрихованные области).
Однако ii'-уровпи и при г' остались бы дискретными. Предпо-
Предполагается, что аналогичные эффекты могут наблюдаться при
высоких давлениях и в других простых веществах. Например,
расчетным путем показано, что при давлениях 10е атм может
существовать металлический водород.
Запишем электронную конфигурацию атома натрия:
Is22s22pr'3sl; па каждый атом приходится один валентный
электрон. Поскольку 3s- и Зр-зоны перекрываются (рис. 14.3),
валентные электроны не принадлежат 3s-3one, а распределены
на более низких уровнях обоих Cs + 3p) зон. Это объясняем
воздух,вакуум ; — -металл ! — -- воздух,вакуум
работа
выхода ф
. энергия Ферми, Fr
Потенциальная ' =—'
энергия
Рис. 14.4. Теория свободного электронного гпза в метшляч; электроны на-
находятся в потенциальном яме.
наличие /(„-линий в спектре испускания металлического натрия;
/(„-линия отвечает переходу 3p->-\s.
Согласно «физическому подходу», зонная теория включает
анализ энергии и длины волны электронов в твердых телах.
В ранней теории свободного электронного газа Зоммерфельда
кристаллическая решетка металла рассматривается как некото-
некоторая потенциальная яма, в которой наименее прочно связанные
с остовом электроны могут двигаться свободно. Энергетические
уровни, которые могут занимать электроны, квантованы (как и
в квантовомехапическои задаче о движении частицы в потен-
потенциальном ящике). Энергетические уровни заполняются элект-
электронами попарно, начиная со дна потенциальной ямы. Наивыс-
Наивысший заполненный при 0 К уровень называется уровнем Ферми,
а отвечающая ему энергия — энергией Ферми EF (рис. 14.4).
Работа выхода ср— это энергия, необходимая для удаления ва-
валентных электронов с верхних уровней потенциальной ямы. Эта
величина соответствует потенциалу ионизации изолированного
атома.
Для наглядного представления плотности электронных сос-
состояний принято графически изображать число энергетических
уровней i\'(Iz) как функцию энергии Е (рис. 14.5). Согласно
гоорни Зоммерфельда, число доступных энергетических уровней
14.2. Электронная структура твердых тел. Зонная теория
G5
постоянно возрастает с ростом энергии. Хотя энергетические
уровни квантованы, их столь много, а разность энергии между
соседними уровнями так мала, что они расположены практиче-
практически непрерывно. При температурах выше О К некоторые элект-
электроны, находящиеся на уровнях пблизи Ег, получают достаточ-
достаточный запас тепловой энергии, чтобы занять уровни выше уровня
Ферми F.F. Следовательно, при некоторых конечных температу-
температурах отдельные состояния выше Еу заняты, а некоторые состоя-
состояния ниже Ер вакантны. Среднее заполнение энергетических
занятые
энергетические
уровни
Энергия-
Рис. И •>. Зависимость плотности электронных состояний о г анергии в тро-
рии свободного электронного газа.
уровнен при некоторой температуре 7">0 К показано па рис.
1-1.Г> в виде заштрихованной области.
Высокая электропроводпость металлов объясняется движе-
движением тех электронов, которые находятся на полузаполпенпых
уровнях вплнзи Et.: Электроны, попарно занимающие состояния
в нижней части валентной зоны, не могут передвигаться в ка-
каком-либо определенном направлении. Электроны же па одно-
однократно заполненных уровнях могут двигаться свободно. Таким
образом, переход электронов с заполненного электронного уров-
уровня ниже EF па пустой уровень выше Ev приводит к возникнове-
возникновению двух подвижных электронов.
Теория свободного электронного газа в общем весьма упро-
упрощенно отражает реальную электронную структуру металлов,
однако 'лга может служить полезной начальной моделью.
В более совершенных теориях потенциальная энергия внутри
кристалла или потенциальной ямы не постоянна, как в теории
Зоммерфельда, а меняется периодически (рис. 14.6). Положи-
Положительно заряженные ядра атомов расположены в кристалле
строго регулярно. Потенциальная энергия электронов прини-
принимает минимальные значения па узлах решетки (из-за кулонов-
ского притяжения) и максимальные значения в середине между
двумя соседними ядрами атомов. Используя фурье-преобразо-
.5-1426
66
[4. Электронные свойства и зоннля теория
ванне, Блох нашел решение уравнения Шрёдингера для случая
периодической функции потенциальной энергии (рис. 14.6).
Важным следствием подхода Блоха является вывод о юн, что
электронные уровни не могут непрерывно заполнять все энер-
энергетическое пространство, лишь некоторые энергетические зоны
(полосы) являются разрешенными для заселения электронами.
Запрещенная энергетическая зона отвечает длинам волн элект-
Потенциальная
энергия
Расстояние
Рис. 14.G. Потенциальная энергия электронов в твердом теле как функция
расстояния.
ронов. которые удовлетворяют условиям брэгговской дифрак-
дифракции вдоль некоторых особых направлений кристалла. Подроб-
Подробнее этот вопрос будет рассмотрен в разд. 14.3. Из рис. 14.7
видно, что, согласно теории Блоха, зависимость плотности
электронных состоянии от энергии должна быть дискретной.
Таким образом, как теория молекулярных орбиталей, так и
приближение периодического потенциального поля приводят к
выводу о существовании энергетических зон в твердых телах.
запреиеьнь с
энергетические
уровни
Эчгргия — -¦»
Рис. 14.7. Плотность состояний согласно зонной теории.
В некоторых материалах наблюдается перекрывание различ-
различных зон. В других материалах существует запрещенная зона
между разрешенными энергетическими зонами.
Экспериментальные подтверждения зонной структуры твер-
твердых тел получают из спектроскопических исследований. Элек-
Электронные переходы между различными энергетическими уровня-
уровнями можно наблюдать, используя спектральные методы исследо-
исследования твердых фаз (гл. 3). Наиболее эффективным методом
14.2. Электропил я структура твердых тел. Зонная теория
67
получения информации о строении внутренней и внешней
электронных оболочек атомов в твердых телах являются иссле-
исследования спектров испускания и поглощения рентгеновских
лучей. Определенную информацию о внешних валентных элект-
ptMiax атомов можно получить также из спектров в видимой и
УФ-области.
Рентгеновские эмиссионные спектры обычно содержат ли-
линии или полосы различной ширины. Переходы между внутрен-
внутренними электронными уровнями регистрируются в виде острых
72 74
Энергия, э.<
Рис. 14.8. Рентгеновский эмиссионный L-спсктр металлического алюминия.
пиков (например, переход 2p->ls в меди). Это означает, что
в металлической меди 2р- и ls-орбитали являются дискретными
атомными орбиталями. Переходы с участием внешних валент-
валентных электронов регистрируются в виде широких полос, особен-
особенно в спектрах металлов. Это означает, что валентные электро-
электроны характеризуются широким спектром энергий, и, следователь-
следовательно, электроны находятся па делокализованпых уровнях в энер-
энергетических зонах.
На рис. 14.8 приведен /,-эмиссионный спектр металлическою
алюминия. О'[ перекрывает энергетическую область в 13 чВ,
где происходят переходы с уровней с п=3 па уровни с я — 2,
т. е. УИ->-?-переходы. Край эмиссионной полосы в области
~73 эВ относится к переходу находящихся в энергетической
зоне Зр-электропов, энергия которых близка к EF. Форма L-
спектра (рис. 14.8) аналогична зависимости плотности состоя-
состояний, рассчитанной для алюминия. В области низких энергий
спектра видна широкая полоса, которая отвечает переходам из
3s-3OHbi. В области более высоких энергий она перекрывается
с другой полосой, которая отвечает переходам из Зр-зоны. Яс-
68 14. Электронные свойства и зонная теория
но, что лишь самые нижние уровни Зр-зоны заняты электро-
электронами.
Рентгеновские эмиссионные спектры содержат информацию
об энергетических уровнях ниже Еу. Для изучения структуры
энергетических уровней выше Ev проводят исследования рент-
рентгеновских спектров поглощения (разд. 3.2.3.6).
14.3. Усовершенствование простой зоннон теории.
fe-Пространство и зоны Бриллюэна
Дальнейшее развитие представлений о строении энергетиче-
энергетических зон в твердых телах основано на рассмотрении процессов
дифракции электронов. Свободно перемещающиеся валентные
электроны в металле могут в определенных условиях дифраги-
дифрагировать на периодической решетке атомов или ионов в кристалле.
Основной закон дифракции—это закон Брэгга (разд. 5.2.2.2).
Он связывает длины воли любой природы (для электронов см.
разд. 3.2.1.4, для рентгеновских лучей — 3.2.1 и 5.2, для нейтро-
нейтронов—3.2.1.5) с межплоскостиыми расстояниями d и углами
дифракции 8 [уравнение E.3)]:
пК = 2d sin 9
Проведение структурных исследований с помощью дифракцион-
дифракционных методов основано па воздействии излучением (конечно, от
внешнего источника) на вещество. Обычно этот источник'—
монохроматический (л = const), хотя иногда, как, например,
в некоторых нейтронографических методиках (разд. 3.2.1.5) или
при исследованиях методом ПТСРС, когда применяют сипхро-
тропное излучение (разд. 3.2.3.5) источник излучения имеет пере-
переменную длину волны.
При рассмотрении внутренней дифракции подвижных валент-
валентных электронов на кристаллической решетке твердого тела
было найдено, что условия возникновения дифракции накла-
накладывают ряд ограничений на длину волны, энергию и свободу
перемещения электронов. Более конкретно — запрещается,
чтобы на любой стадии движения свободных электронов вы-
выполнялся бы закон Брэгга. Свободные электроны в металлах
или полупроводниках имеют различную энергию и, следователь-
следовательно, различную длину волны. При оценке возможности дифрак-
дифракции электронов необходимо учитывать длину волны л, угол диф-
дифракции 6 и межплоскостное расстояние d.
Кинетическую энергию такой частицы, как свободный элек-
электрон, можно записать в виде
Е=^~ A4.1)
14.3. Усовершенствование простой зонной теории G9
Момент движения mv и длина волны X связаны друг с другом
уравнением Де Броиля:
mv
A4.2)
Для описания распространения волн вводят волновой вектор
к. Направление вектора к параллельно направлению распро-
распространения волн, а величина обратно пропорциональна длине
волны:
Л=-Х~ A4.3)
Комбинация уравнений A4.1) —{14.3) приводит к выражению
На рис. 14.9 изображена квадратичная зависимость энергии
электрона от величины волнового вектора.
Возвратимся к закону Брэгга ["уравнение E.3)] и заменим
К волновым вектором к [уравнение A4.3)]:
к— , . А' A4.о)
Таким образом, получены выражения для величин к и, следо-
следовательно, Е, для которых выполняется условие дифракции
Брэгга в любой кристаллической структуре.
Применим, например, уравнение A4.5) к примитивной ку-
кубической решетке с параметром элементарной ячейки а. Са-
Самым большим межплоскостпым расстоянием d в такой струк-
структуре является расстояние между плоскостями A00). Для него
с?юо = й- Рассчитаем значения к, при которых происходит диф-
дифракция электронов на плоскостях A00). Для отражений перво-
первого порядка (п=1) на плоскостях A00) к может принимать
любые значения, равные или большие, чем л/а. Для к — л/а
0 = 90°, a sin8=1. Это — критическое значение параметров
дифракции, при которых электронные волны направлены пер-
перпендикулярно плоскости отражения A00). Для к>п/а sin6<l
и 6<90°. Это — общее условие возникновения дифракции при
брэгговских углах 0<90° (рис. 14.10, а). Проекции плоскостей
A00) на рис. 14.10, а изображены вертикалями, поэтому ось х
элементарной ячейки горизонтальна.
Дифракционная картина, представленная на рис. 14.10, а,
является изображением в реальном пространстве. Ей
может быть поставлена в соответствие картина в обратном
пространстве или, как говорят, в ^-пространстве (рис. 14.10, о).
При таком изображении волновой вектор к величиной л/а sin 6,
70
14. Электронные сиойств;] и зонная теория
направленный под углом (90—0)° по отношению к оси х,
изображается отрезком такой же длины (в соответствующем
масштабе) и в том же направлении от некоторой произвольной
точки, т. е. от некоторого начала координат. Изображенные па
рис. 14.10, б волновые векторы направлены под углами 0 = 45.
60 и 90°, а также под некоторым произвольным углом 0. Мож-
Можно показать, что соблюдение соотношения A4.5) и геометриче-
геометрических :;акопов приводит к тому, что все векторы к закапчиваются
на вертикали XY. В трех-
трехмерном пространстве
линия XY преобразуется
в плоскость, которая об-
образует границу зоны
Бриллюэна A00). В к-
пространстве положение
этой границы представ-
представляет собой условие, при
котором соблюдается за-
закон Брэгга для отраже-
отражения первого порядка
(п=1) от плоскости
A00) при использовании
излучения с различной
длиной волны и разными
волновыми векторами к.
В трехмерном прост
ранстве для примитивной
кубической решетки сле-
следует рассматривать семейство пкоскостей {100}, включающее
плоскости A00), (ТОО), @10), (ОТО), @01) и @01). КаждоЛ
из плоскостей соответствует грань зоны Бриллюэна A00).
В трехмерном пространстве такая зона имеет форму куба
(рис. 14.11). Эта зона называется первой зоной Брпллюэна.
так как в примитивной кристаллической решетке она соот-
соответствует плоскостям с наибольшим межплоскосгным расстоя-
расстоянием и. Эта зона также соответствует наименьшему волновому
сектору к и наименьшим энергиям, так как k<xd~\ Необходи-
Необходимо также отметить, что грани зоны Бриллюэна параллельны
плоскостям дифракции {100}. Это также относится к другим
зонам Бриллюэна.
Как отмечалось выше, первая зона Бриллюэпа в примитив-
примитивной кубической решетке соответствует дифракции на плоскостях
{100}. Вторая зона Бриллюэна возникает при дифракции па
плоскостях {ПО}. В семейство {110} входят 12 плоскостей.
В этом случае зона Бриллюэна имеет форму правильного
о к
Рис. 14.9. Квадратичная зависимость
анергии свободных элоктронои Е от ве-
величины волнового векторл к.
14.3. Усоворшенствовлнпе простой зонной теории
71
додекаэдра (на рисунке не изображен), который окружает
первую зону Бриллюэна кубической формы. Плоские сечения
первой и второй зон Бриллюэна показаны на рис. 14.12.
Для веществ с объемпоцентрировапной кубической решеткой
(например, для натрия) первой зоной Бриллюэна является тот
самый правильный додекаэдр, о котором говорилось выше. Ол
соответствует дифракции на плоскостях {ПО}. Это объясня-
« PeajinHoe пространство 6 к -Просфансгоо
А-
/^
координа
1
//
оУ %
У в
- т /а, *
,bV
^°
ч.
= 90°
100
100
100 100
Л"
границл зоны
Бриллюэна(ЮО)
Рис. 14.10. Дифракция электронов на плоскостях A00) н реальном прост-
пространстве (а) и возникновение границы зоны Бриллюэна в /г-пространстве {б).
ется тем, что на рентгенограммах веществ с объемноцентриро-
ваннымп решетками рефлексы 100 погашаются.
Для веществ с грапецентрированной кубической решеткой
(например, для /y-Fe) первая зона Бриллюэиа имеет форму
усеченного октаэдра с 14 гранями. Причиной этого является
то обстоятельство, что дифракция идет как на семействе плос-
плоскостей {1П}, так и па семейстре плоскостей {200}. В набор
этих плоскостей входят восемь плоскостей {111} и шесть
{200}. Усеченный октаэдр можно рассматривать как октаэдр,
у которого каждая из шести вершин срезана так, что образо-
образовался квадрат.
Рассмотрим энергию валентных электронов и их положение
в ^-пространстве. Электроны, которые находятся вблизи дна
валентной зоны металлов нли полупроводников, имеют относи-
относительно низкую энергию и низкую величину вектора к. Поэтому
в ^-пространстве они находятся глубоко внутри первой зоны
Бриллюэна и их волновые векторы к закапчиваются, не дости-
достигая границы зоны. Поскольку энергия электронов возрастает по
72
14. Электронные свойства и зонная теория
мере заполнения более высоких энергетических уровней внутри
валентной зоны, то волновые векторы к постепенно удлиняют-
удлиняются, заполняя первую зону Бриллюэна. Однако условия, при
которых векторы к заканчиваются на границе зоны, являются
запрещенными. Когда энергия электронов становится еще боль-
больше, векторы к выходят за границу первой доны Бриллюэпа а
попадают во вторую.
Вопрос о запрещенных значениях волновых векторов к, ко-
которые закапчиваются на границе зоны, гораздо более сложен,
чем кажется на первый
взгляд. Дело в том,
что имеется мало запре-
запрещенных значений к, но
при этом может сущест-
существовать большое количе-
количество запрещенных энер-
энергетических уровней. Бо-
Более того, в области гра-
, ницы зоны энергия дви
* жущегося электрона не
связана с волновым век-
вектором уравнением A4.4)!
Вместо этого для значе-
значений к внутри границ зо-
зоны соответствующие эне-
Рис. 14.11. Первая зона Бриллюэна для
примитивной кубической решетки.
ргии меньше тех, кото-
которые можно рассчитать по
уравнению A4.4). В то
же время для значений
к, выходящих за границы зоны, величины энергий больше рас-
рассчитываемых по этому уравнению. Следовательно, хоти разры-
разрыва в значениях к на границе зоны не происходит, в значениях
энергии возникает разрыв. Поэтому квадратичная зависимость
Е от к на границах зон нарушается (рис. 14.13); кривые Е(к)
меняют свой ход и становятся горизонтальными, так как здесь
достигаются критические значения к. Следовательно, возникает
.юна запрещенных энергетических уровней.
Математическая интерпретация существования запрещенных
энергетических зон весьма сложна." Попытаемся, хотя бы ка-
качественно, попять, почему энергетические запрещенные зоны в
принципе существуют. В гипотетическом кристалле, внутри
которою потенциальная энергия постоянна (рис 14,-1). электро-
электроны двигались бы свободно, не подвергаясь дифракции на
кристаллической решетке. В реальном кристалле, однако, по-
потенциальная энергия внутри кристалла меняется периодически
(рис. 14.6). Форма кривой потенциальной энергии, глубина и
14.3. Уеокершепствсшание простой зонной теории
ширина минимумов зависят от величины заряда атомных ядер.
Речь идет о тех положительно заряженных ядрах, которые
обусловливают явление дифракции. Действительно, минимумы
потенциальной энергии действуют как вторичные источники,
которые повторно излучают сферические электронные волны.
Если ядра имеют большой положительный заряд, как, напри-
например, в случае многозарядных ионов, потенциальные минимумы
глубоки и электроны сильно дифрагируют на них.
ку
первая зона
Бриллгсэна
к . - -т/а
вторая зона
Бриллюэна
Рис. 14 12. Сечения первой и второй зон Брпллюэпа для примитивной к\бн-
ческой решетки.
Эти вторичные электроны интерферируют не только друг с
другом, но и с первичной волной движущихся свободных элект-
электронов. На краю границы зоны (где дифракционные явления
особенно существенны) вторичные электроны, генерируемые
соседними слоями положительно заряженных атомных ядер,
находятся в фазе и интерферируют друг с другом. Однако ре-
результат их интерференции с первичной волной валентных
электронов зависит от направления. Если вторичные электроны
распространяются в том же направлении, что и валентные
электроны, их интерференция приводит к увеличению амплиту-
амплитуды и энергии валентных электронов Если электроны распрост-
распространяются в направлении, противоположном движению валент-
валентных электронов, то происходит уменьшение амплитуды и
энергии последних. Поэтому влияние вторичной дифракции сво-
сводится к неизбежному изменению энергии валентных электронов.
Следовательно, возникает энергетическая запрещенная зона,
14 Электронные свойства п зонная теория
величина которой зависит от степени брэгговского рассеяния,
кристаллической структуры вещества и присутствующих в нем
элементов.
Теперь можно перейти к рассмотрению зошюй структуры
различных классов веществ и к анализу ее влияния на электри-
электрические свойства материалов. Не станем подробнее вникать в
суть зонной теории, поскольку и такого упрощенного и схема-
///у.
//запрещенная зона v
1 1 \
7 запрещенная лона У//С/
/ ' '
У 1 1
— 1 1
к,г К, о kt k2
Рис. 14.13. Влияние дифракции электронов на их энергию, приводящее к
возникновению запрещенных энергетических уровней. Границы первой и вти'
рой зоны обозначены соответственно kh /г_, и кг, к-г.
тичеокого описания строения разрешенных и запрещенных
энергетических зон, как здесь проведено, вполне достаточно для
объяснения многих явлении.
М.4. Зонная структура металлов
Для
металлов характерна такяч зонная структура, когда
высшая энергетическая зона, в которой находятся электро-
электроны,— валентная зона — заполнена лишь частично (рис. 14.14).
Занятые уровни схематически изображены заштрихованной
областью. Некоторые энергетические уровни, расположенные
несколько ниже уровня Ферми, не заселены, зато части уровней
выше уровня Ферми оказывается запятой. Электроны, находя-
находящиеся в однократно занятых состояниях вблизи ?V, способны
двигаться, и именно они ответственны за высокую электропро-
электропроводность металлов.
В некоторых металлах, таких, как натрий, энергетические
зоны перекрываются (рис. 14.3). Следовательно, как 35-, так и
14.5. Зонная структура диэлектриков
75
N(E)
(Ъленгная зона
свободные уровни
занятые уровни /'//,
/////-///У. /. -V
¦запоя- свободно
Рис. 14.14. Зонная структура металла.
Зр-зоно1 в натрии содержат электроны. Перекрывание зон объ-
объясняет металлические свойства, например, щелочноземельных
металлов. Зонная структура бериллия приведена на рис. 14.15.
Можно заметить, что зоны 2s и 2р, каждая из которых запол-
заполнена лишь частично, перекрываются. Если бы зоны не перекры-
Be
Is
2s
Pnc M.15. Перекрывание энергетических зон в металлическом бериллии.
вались, то 2s-3OHa была бы полностью заполненной, а 2р-зопа —
свободной. В этом случае бериллий не был бы металлом. Такая
ситуация наблюдается в изоляторах и полупроводниках.
14.5. Зонная структура диэлектриков
Валентная зона диэлектриков заполнена. Она отделена ши-
широкой запрещенной зоной от следующей энергетической зоны,
которая свободна (рис. 14.16). Алмаз — превосходный диэлект-
диэлектрик с шириной запрещенной зоны, равной ~6 эВ. Лишь неболь-
небольшое число электронов валентной зоны обладает достаточным
запасом тепловой энергии, чтобы переместиться в свободную
более высокую энергетическую зону. Следовательно, проводи-
76
14. Электронные свойства и зонная теория
N(E)
алмаз
запрошенная .
свободно
Рис. 14.16. Зонная структура диэлектрика, например углерода (алмаз).
мость пренебрежимо мала. Природа широкой запрещенной
'I'M 1-. алмазе обсуждается в разд. 14.6. Здесь наблюдается
ч'шая аналогия с кремнием.
14.6. Зонная структура полупроводников: кремний
Зонная структура полупроводников аналогична зопной струк-
структуре диэлектриков, и лишь ширина запрещенной зоны не так
велика. Обычно она составляет 0,5—3,0 эВ. По крайней мере
некоторая часть электронов обладает достаточным запасом
тепловой энергии, чтобы перейти в пустую зону.
Электрический ток в полупроводниках может переноситься
двумя разными типами носителей заряда (рис. 14.17). Любой
электрон, который может быть пе-
реброшен из валентной зоны в зо-
зону проводимости (верхняя пустая
зона), является носителем отрица-
отрицательного заряда. Под действием
внешнего электрического поля та-
такой носитель заряда переметает-
переметается к положительному электроду.
Освободившийся энергетический
уровень в валентной зоне называ-
называется дыркой. Дырка — носитель
положительного заряда Движение
дырок может происходить за счет того, что электрон с сосед-
соседнего энергетического уровня перескакивает в имеющуюся дыр-
дырку, оставляя незанятым свой собственный уровень, т. е. обра-
образуя новую дырку. Такое движение электронов можно рассмат-
рассматривать как движение дырок в направлении, противоположном
движению электронов.
зона проеоди^иыи
запрещенная зона
© © © © -
валентная зона
Рис. 14.17. Положительные и
отрицательные носители заря-
заряда.
6,0
1,1
0,7
0,1
0
0
Диэлектрик
Полупроводник
»
»
^Металл
14.6. Зоплпя структур;) полугфотюдпиков: кремний 77
Полупроводники можно разделить па два класса:
1) Собственные полупроводники — это чистые вещества, зонная
структура которых соответствует схеме, изображенной на
рис. 14.17. В таких веществах число электронов п в зоне про-
проводимости полностью определяется шириной запрещенной зоны
и температурой. Примером собственного полупроводника может
служить чисгый кремний. В табл. 14.1 приведены значения
ширины запрещенной зоны кремния и других элементов IV
группы.
Таблица 14.1. Ширина запрещенной зоны элемептии IV группы
~ Ширина запрещенной
Элемент ' -М11Ы эв Тпп материала
С (алмаз)
Si
Ge
Серое олово (>13°С)
Белое олово (<13°С)
РЬ
Зонная структура кремния и германия резко отличается от
той структуры, которую можно было бы ожидать, исходя из
зонной структуры натрия и магния. В последних элементах
энергетические уровни 3s и Зр перекрываются, что приводит
к возникновению двух широких зон, каждая из которых занята
лишь частично. Если бы эта тенденция распространялась на
другие элементы, то можно было бы ожидать, что и в кремнии
должны существовать две такие же зоны. Тогда эти зоны били
сы заполнены в среднем наполовину, и кремний был бы метал-
металлом. Известно, что это не так. На самом деле валентная зона и
зона проводимости в кремнии разделены запрещенной зоной.
В валентной зоне содержится по четыре электрона на один атом
кремния. Эта зона полностью заполнена. Пели бы запрещенная
зона просто разделяла s- и р-зоиы, то в s-зоие находилось бы
только два электрона па один атом кремния. Это не так. По-
Поэтому такая интерпретация зонной структуры неверна.
Зонная структура кремния может быть интерпретирована с
использованием представлений квантовой механики с учетом
того, что кристаллическая решетка кремния существенно отли-
отличается от кристаллической решетки натрия: натрий имеет
кубическую объемпоцептрированпую решетку (КЧ 8), и крем-
ний — кубическую гранецептрировапную решетку (КЧ 4).
Попытаемся все же упрощенно, без использования математи-
78 14. Электронные свойства и зонная теория
ческого аппарата, представить зонную структуру кремния.
Примем, во-первых, что каждый атом кремния образует четыре
одинаковые связи и находится в центре тетраэдра, образован-
образованного соседними атомами кремния. Каждую из орбиталей крем-
кремния можно рассматривать как гибридную ^р3-орбиталь. Каждая
из гибридных орбиталей перекрывается с аналогичной орби-
талыо соседнего атома кремния, так что возникает пара моле-
молекулярных орбиталей: одна связывающая'—о-орбиталь, другая
аптисвязывающая — о:<-орбнталь. На каждой из орбиталей
находятся два электрона — но одному от каждого атома крем-
кремния. И последнее приближение заключается в необходимости
предположить, что молекулярные о-орбитали перекрываются,
образуя о-зону. Эта зона станет валентной зоной. Аналогично
перекрываются и а?-орбигали, образуя зону проводимости.
о-Зона полностью заполнена, и, следовательно, в ней содержится
по четыре электрона на один атом кремния. о*-3опа свободна.
2) Примесные полупроводники — это материалы, уровень
проводимости которых определяется содержанием легирующих
компонентов. Кремний может стать примесным полупроводни-
полупроводником, если в него ввести элементы II1 или V групп периодиче-
периодической системы. Рассмотрим вначале влияние малых добавок
трехвалентных элементов (например, 10~2 ат. % галлия). Пусть
атомы галлия замещают атомы кремния в тетраэдрических
узлах решетки типа алмаза, образуя твердый раствор замеще-
замещения. Согласно теории валентных связей, в чистом кремнии все
связи Si —Si являются обычными ковалеитиыми связями, так
как у кремния четыре валентных электрона, н каждый атом
кремния окружен четырьмя другими атомами кремния (рис.
14.18). У галлия лишь три валентных электрона, и при введении
галлия в кремний должна образовываться одна электронодефн-
цитная связь Ga—Si. Согласно зонной теории, каждый энерге-
энергетический уровень, отвечающий одноэлектронной связи Ga—Si,
не будет попадать в валентную зону кремния. Вместо этого об-
образуется дискретный энергетический уровень или атомная
орбиталь чуть выше потолка валетной зоны. Такой уровень
называют акцепторным уровнем, поскольку он способен прини-
принимать электроны. Энергия этого акцепторного уровня отличается
от энергии потолка валентной зоны примерно на 0,1 эВ. Следо-
Следовательно, термически возбужденные электроны могут легко
покидать валентную зону и занимать акцепторные уровни.
Если концентрация атомов галлия мала, акцепторные уровни
дискретны. Поэтому электроны, находящиеся на акцепторных
уровнях, не вносят вклада в проводимость материала. Остав-
Оставшиеся в валентной зоне положительно заряженные дырки мо-
могут двигаться, и. следовательно, легированный галлием кремний
относится к полупроводникам р-типа.
14.6. Зонная структура полупроводников: кремний
При обычной (комнатной) температуре концентрация поло-
положительно заряженных дырок, возникших при введении атомов
галлия, намного выше концентрации дырок, возникших в ре-
результате термического возбуждения электронов и перехода их
в зону проводимости. Поэтому уровень примесной проводимости
существенно выше уровня собственной проводимости. Следова-
Следовательно, проводимость в этом случае определяется концентраци-
концентрацией атомов галлия. С ростом температуры концентрация собст-
зона проводимости
1,1 эВ
1 0,1 эВ у
e s e e
валентная зоне
~г~ акцепторные уровни
Рис. 14.18. р-Проводимость кремния, легированного галлием.
венных носителей заряда быстро растет. При достаточно
высоких температурах концентрация собственных носителей
заряда может превысить концентрацию примесных носителей.
При этом доминирующий вклад в проводимость будут вносить
именно собственные носители заряда (см. ниже).
Рассмотрим теперь, к чему приведет введение в кремний
атомов пятивалентного элемента, например мышьяка. Атомы
мышьяка также будут замещать атомы кремния в решетке типа
алмаза. Однако теперь каждый атом мышьяка дает один избы-
избыточный электрон по сравнению с теми электронами, которые
необходимы для образования четырех ковалентных связей
Si—As (рис. 14.19). В рамках зонной теории это означает, что
избыточный электрон занимает дискретный уровень, располо-
расположенный примерно на 0,1 эВ ниже дна зоны проводимости. Как
и в предыдущем случае, электроны на этих уровнях не могут
двигаться, поскольку их концентрация недостаточна, чтобы
образовать зону делокализованных состояний. Такие уровни
80 14, Электронные спойава и яолпи» теория
называются донорными уровнями, поскольку находящиеся на
них электроны легко переходят в зону проводимости за счет
термического возбуждения, где они могут свободно перемещать-
перемещаться. Такие материалы называются полупроводниками п-типа.
Сформулируем различия между собственными и примесными
полупроводниками.
I) Уровень проводимости в примесных полупроводниках при
комнатных температурах гораздо выше уровня проводимости
собственных полупроводников. Например, при 25 °С чистый
имеет собственную проводимость ~10~2 (Ом-см)".
0.1 эВ'
,- - один
/ избыточней
электрон
валенi
J0HJ
Рис. 14.19 /г-Проводимость кремния, легированного мышьяком.
При соответствующем легировании его проводимость возрастает
на несколько порядков.
2) Уровень проводимости примесных полупроводников мо-
может точно определяться концентрацией вводимой примеси. Это
используют при синтезе материалов с определенным уровнем
проводимости. Что касается собственных полупроводников, то
уровень их проводимости существенно зависит от температуры
и наличия случайных примесей.
Иа рис. 14.20 приведена температурная зависимость концен-
концентрации носителей и уровень проводимости полупроводниковых
материалов, которые при низких температурах являются при-
примесными полупроводниками, а при высокой температуре—соб-
температуре—собственными. При низких температурах (область /) концентрация
носителей зависит от температуры, так как относительно не-
небольшое различие в энергиях (~0,1 эВ) между акцепторными
(донорными) уровнями и краем валентной зоны (зоны прово-
проводимости) могут преодолеть лишь немногие электроны. При
возрастании температуры обе зависимости выходят на насыще-
насыщение (область 2), когда концентрация примесных носителей
принимает максимальное значение. На этом участке кривой кон-
концентрация носителей не зависит от температуры и проводимость
несколько уменьшается с ростом температуры за счет понижения
подвижности носителей из-за взаимодействия с кристаллической,
14.7. Полупроводники с контролируемой впле.нтностью
81
решеткой. В области высоких температур (область 3) концент-
концентрация собственных носителей превышает концентрацию примес-
примесных носителей. Здесь наблюдается резкий рост как концентра-
концентрации носителей, так и проводимости.
Рис. 14.20. Температурные зависимости электропроводности (б) и концент-
концентрации носителей заряда (а) в полупроводниках. 1—область примесной
проводимости; 2 — область насыщения; 3 — область собственной проводи-
проводимости.
Для надежного использования полупроводниковых приборов
было бы желательно, чтобы проводимость применяемых в них
полупроводниковых материалов была бы менее чувствительна
к изменению температуры (область 2). Чтобы область насыще-
насыщения 2 была как можно более широкой, на практике используют
два приема:
1) выбирают материалы с широкой запрещенной зоной;
2) вводят в полупроводники такие легирующие добавки,
чтобы возникшие акцепторные (или допорпые) уровни находи-
находились вблизи края валентной зоны (или зоны проводимости).
В этих случаях область / смещается в сторону более пгпких
температур, а область 3 — в сторону более высоких.
14.7. Полупроводники с контролируемой валентностью
Некоторые соединения переходных металлов, содержащие
элементы с несколькими степенями окисления, являются провод-
проводниками электрического тока. Хорошим примером таких веществ
может служить доокислепиый оксид никеля. Чистый оксид ни-
никеля NiO*— твердое вещество бледно-зеленого цвета, характе-
й Имеется в виду оксид никеля, полученный (или отожженный) :;рп
высоких температурах C>Ю00сС) или в среде с пониженным по сравпе-
парциальным давлением кислорода (Ро <SL0,2\ ,ггм.) - •
е воздухом
Прим ред.
426
14. Электронные свойства и зошия теория
рпзугощееся низкой электропроводностью. Это собственный
полупроводник. Его цвет определяется внутренними d—d-nepe-
ходами в ионах Ni2+, имеющих октаэлрическое окружение.
Доокислепие N40 на воздухе при температуре, например,
1000°С веде г к незначительному приросту массы благодаря
введению избыточного кислорода. Вещество приобретает чер-
черный цвет, а его электропроводность увеличивается. При такой
обработке ионы Ni2H" частично окисляются до Ni3' и состав
продукта окисления можно отразить формулой
Встраивающиеся из газовой фазы в кристаллическую решетку
ионы кислорода концентрируются вблизи поверхности кристал-
кристалла NiO, а затем часть никеля диффундирует к поверхности,
чтобы сбалансировать избыточный электрический заряд. При
этом в объеме кристалла возникают вакансии. Черный оксид
никеля проводит электрический ток*, так как электроны могут
легко переходить от Ni2~ к Ni3+:
Ni2+ *¦ Nis+ -V е
Ni3+ ^ Ni2+ + р
Благодаря этим электронным переходам происходит как бы
движение ионов Ni3+, и черный оксид никеля надо рассматри-
рассматривать как полупроводник р-типа. В отличие от механизма прово-
проводимости легированного галлием кремния, также полупроводни-
полупроводника р-типа, проводимость черного оксида никеля NiO имеет
прыжковый механизм. Причина этого заключается в том, что
обмен электронами в системе Ni?J/Ni3+ является термически
активируемым процессом и, следовательно, существенно зависит
от температуры. В рамках зонной модели это может быть объ-
объяснено тем, что d-орбнтали никеля перекрываются слабо, а
потому образуется узкая (i-зона (rf-орбитали локализованы на
отдельных ионах иикеля). Более подробное обсуждение этого
вопроса проведено в разд. 14.9.
Недостаточно широкое применение NiO в качестве полупро-
полупроводникового материала объясняется тем, что его проводимость
трудно задатт, контролируемым способом: она существенно за-
зависит как от температуры, так и от парциального давления
кислорода. Чтобы преодолеть эти трудности, Вервей A948 г.)
предложил полупроводники с контролируемой валентностью.
Тогда в оксиде никеля концентрация ионов Ni3+ будет в основ-
основном зависеть не от температуры, а от введения контролируемого
количества легирующих добавок.
* Правильнее назвлть его пол>лроводником с узкой полосой запрещен-
запрещенных энергий.—Прим. ред.
14.8- Применение полупроводников
Оксид лития может вступать в реакцию с оксидом никеля
и кислородом с образованием твердых растворов состава
В таких твердых растворах концентрация ионов Ni3;~, а следо-
следовательно, и проводимость зависят от концентрации ионов Li+.
При 25°С величина проводимоегп меняется очень резко с изме-
изменением состава: от —Ю-10 (Ом-см) (при * = 0), до
Ю-1 (Ом-см) "' (при л = 0,1).
14.8. Применение полупроводников
Главной областью применения полупроводников являются
такие устройства, пак транзисторы, интегральные схемы, фото-
фотоэлементы и т. п. В настоящем разделе основное внимание уде-
уделено такому явлению, как п—р-переход, использование которо-
которого лежит в основе работы многих полупроводниковых приборов.
е"
доиорные
уровни
р- тип
л- тип
акцепторные _е_
уровни
Рис. 14.21. я—р-Переход.
Так, полупроводниковый диод представляет собой полный ана-
аналог лампового выпрямителя. Предположим, что монокристалл
полупроводникового материала, например кремния, легирован
таким образом, что он является наполовину полупроводником
/i-типа, а наполовину полупроводником р-типа. Зонная структу-
структура такого полупроводникового материала изображена на" рис.
14.21. Положение уровня Ферми в различных половинах полу-
полупроводника различно. Следовательно, электроны имеют возмож-
возможность самостоятельно переходить из области с л-проводи.мостьга
34 14. Электронные свойства и зонная теория
в область с р-проводимостью через область п—р-перехода.
Энергия Ферми электронов в данном случае выполняет роль
электрохимического потенциала: до тех пор пока существует
разность уровней Ферми (электрохимических потенциалов
электронов), будет происходить самопроизвольный переход из
области с более высокой энергией Ферми (с более высоким
электрохимическим потенциалом) в область с более низкой
энергией Ферми.
В отсутствие внешнего поля в течение некоторого времени
будет наблюдаться движение части электронов слева направо
через область п—р-перехода, но вскоре зонная структура кри-
кристалла перестроится таким образом, что EF станет одинаковой
во всех частях кристалла. Другое объяснение этому явлению
можно дать с использованием представлений о слоях объемных
зарядов, которые быстро перемещаются в области п—р-перехо-
п—р-перехода благодаря тому, что электроны переходят из одной половины
кристалла в другую. После этого объемный заряд служит
барьером для дальнейшего движения электронов.
Совсем другие явления наблюдаются тогда, когда полупро-
полупроводниковый материал находится во внешнем электростатиче-
электростатическом поле, положительный полюс которого расположен со сто-
стороны р-области полупроводника, а отрицательный — со стороны
я-области. В этом случае в цепи течет постоянный электриче-
электрический ток. Электроны поступают в кристалл с правого электрода,
затем проходят через зону проводимости я-области, через об-
область п—р-перехода попадают в валентную зону р-области и
перемещаются здесь до границы кристалла за счет дырочной
проводимости, пока не попадут в левый электрод. В противопо-
противоположном направлении электрический ток течь не может, так как
при достаточно низком потенциале внешнего электрического
поля электроны не могут преодолеть барьер в области п—р-
перехода и, следовательно, не могут перемещаться слева напра-
направо. Такой кристалл работает как выпрямитель электрического
тока, пропускающий только ток определенного направления.
Его можно использовать для преобразования переменного тока
в постоянный. В настоящее время выпрямители на основе крем-
кремния все больше вытесняют ламповые диоды.
Более сложная картина наблюдается в случае р—п—р- или
п—р—/2-переходов. Полупроводниковые кристаллы с такими
переходами используются в качестве усилителей напряжения.
В современной технике полупроводниковые транзисторы все
больше вытесняют ламповые.
Полупроводники с контролируемой валентностью находят
применение в качестве термисторов, т. е. сопротивлений, чувст-
чувствительных к температуре окружающей среды. Их действие
основано на резком изменении проводимости с температурой,
14.9. Зонная структуре* неорганических твердых тел 85
поскольку в них механизм проводимости имеет прыжковый ха-
характер. Например, в полупроводниках Lio.osNio^O в широком
температурном интервале (до ~200°С) проводимость возра-
возрастает по закону Аррениуса с энергией активации ~0,15 эВ.
Если температурный ход проводимости хорошо воспроизводит-
воспроизводится, то такого типа полупроводники можно использовать для
контроля и измерения температуры. Для достижения хорошей
воспроизводимости необходимо использовать материалы, в ко-
которых уровень проводимости мало меняется при введении
легирующих добавок, например NiO, легированный Fe3O4,
Мп2О3 и Со2О3, а также некоторые шпинели.
Некоторые полупроводники обладают фотопроводимостью,
т. е. их проводимость сильно растет под действием света. Пре-
Прекрасным фотопр-оводником является аморфный селен. Его
используют как основной компонент в фотокопировальных уста-
установках (гл. 18). Классическая зонная теория не может быть
использована для объяснения свойств аморфных материалов,
таких, как аморфный селен, поскольку в них отсутствует даль-
дальний порядок.
14.9. Зонная структура неорганических твердых тел
В предыдущих разделах обсуждались материалы, которые
являются проводниками электрического тока. Однако зонная
теория с успехом может быть применена для объяснения пове-
поведения любых неорганических твердых тел независимо от того,
проводят они ток или нет. Зонная теория позволяет по-новому
взглянуть па структуру, природу химической связи и свойства
неорганических твердых тел. Она дополняет информацию, по-
получаемую при применении моделей ионной и ковалентиой свя-
связи. Большинство неорганических веществ имеют более слож-
сложную структуру, чем металлы и полупроводники. Им меньше
уделяют внимания и при проведении теоретических расчетов
зонной структуры. Следовательно, зонная структура многих
неорганических соединений известна весьма приблизительно.
Выше обсуждалась зонная структура элементов IV группы
периодической системы, в частности кремния, и ряда соединений
AniBv, таких, как GaP. Последняя группа веществ имеет то же
количество валентных электронов, что и элементы IV группы.
Сделаем шаг вперед и рассмотрим соединения типа AIBVI1
(например, NaCI) и AnBVI (например, MgO).
Характер связи в этих соединениях в основном ионный. Они
представляют собой вещества белого цвета с низким уровнем
электронной проводимости. Введение добавок приводит к повы-
повышению ионной, а не электронной проводимости (гл. 13). Будем
предполагать, что в NaCI реализуется чисто ионный характер
86 14. Электронные, свойства н эсштыя геория
связи и ионы имеют следующее строение электронных оболо-
оболочек:
Na\- Is2, 2s2, 2f
C1-: Is2, 2s2, 2p\ 3sa, 3p6
Следовательно, валентная оболочка иона С1" Cs. 3/э) заполне-
заполнена, а оболочка иона Na1" пустая. В \аС1 соседние ионы С1~
практически касаются друг друга и их Зр-орбитали могут пе-
перекрываться с образованием узкой валентной зоны (Зр-зона).
Эта зона состоит только из орбиталей анионов. 3s- и Зр-Орби-
тали Ха~ могут также перекрываться и образовывать зону,
которая является зоной проводимости. Эта /юна образована
только орбиталями катионов. В обычных условиях она полно-
полностью свободна, так как ширина запрещенной зоны велика
(~7 эВ). Зонная структура NaCI аналогична зонной структуре
изоляторов (рис. 14.16). Особенность зонной структуры NaCI
заключается в том, что валентная зона состоит из орбиталей
анионов, а зона проводимости — исключительно из орбиталей
катионов. Переход электрона из валентной зоны в зону прово-
проводимости можно рассматривать как обратную передачу заряда
от иона С1~ иону Na+.
Последний вывод дает основание ожидать, что существует
определенная корреляция между величиной ширины запрещен-
запрещенной зоны и разностью члектроотрицательностей аниона и кагио-
па. Большая разность электроотрицательностей способствует
образованию ионных связей. В таких случаях обратного пере-
переноса заряда от аниона к катиону ожидать трудно, и это хорошо
коррелирует с тем, что ионные твердые тела имеют широкую
запрещенную зону. Ширина запрещенной зоны ряда неоргани-
неорганических соединений приведена в табл. 14.2. Количественное соот-
соотношение между шириной запрещенной зоны и степенью ионности
связи предложено Филиппсом и Ван Фехтеном [уравнение
(8.31)]. Можно считать, что запрещенная зона состоит из двух
частей: «гомополярпая» часть запрещенной зоны — это такая
запрещенная зона, которая существовала бы в отсутствие раз-
разности электроотрицательностей составляющих данное вещество
компонентов, и часть, определяемая степенью ионности связи.
Подробнее такие корреляции обсуждаются в разд. 8.4.
В соединениях переходных металлов дополнительным факто-
фактором, имеюшим огромное значение, является наличие частично
заполненных rf-орбиталей ионов металла. В ряде случаев их
перекрывание приводит к образованию d-зоны или нескольких
rf-зон. Тогда материал имеет высокую электропроводность.
Возможно также, что rf-орбитали перекрываются весьма ограни-
ограниченно, т. е. они как бы локализованы на отдельных атомах.
Примером последнего является стехиометрический оксид нике-
14.9. Зошшя структурл неорганических твердых тел
87
ля:: MiO. Его слабо-зеленая окраска определяется электронны-
электронными переходами между отдельными ионами Ni2+. Он имеет край-
крайне низкую проводимость (~1(Н4 (Ом-см) при 25СС). Her
никаких доказательств того, что d-орбигали никеля заметно
перекрываются с образованием частично заполненной d-зоны.
Противоположным примером являются TiO и VO. Их кристал-
кристаллические решетки (как и N40) относятся к структурному типу
Таблща 14.2. Ширина запрещенной зоны (эВ) некоторых неорганических
твердых тел
Соединения
Li F
L-C1
NaF
NaCl
NaBr
KF
KC1
KBr
Kl
Л1 Bv"
11
9,5
11,5
8,5
7,o
11
8,5
7,5
5,8
Соединения
ZnO
ZllS
ZnSe
ZiiTc
CdO
CrlS
case
CdTe
PbS
PbSe
PbTe
A11 BVI
3,4
3,8
2,8
2,4
2,3
2,45
1,8
1,45
0,37
0,27
0,33
Соединения
Л1Р
AlAs
AlSb
GaP
GaAs
GaSb
lnP
hi As
InSb
?-SiC
a-SiC
Л111 bV
3,0
2,3
1,5
2,3
1,4
0,7
1,3
0,3
0,2
2,2
•V
Некоторые из приведенных величин (в частности, для галогенидов щелочных ме
тпллок) носят оценочный характер.
поваренной соли, однако в отличие от NiO <2-орбитали (dxy,
¦dxz, d,lz — см. разд. 8.6.1) ионов М2+ сильно перекрываются,
образуя широкую 4^-зону, Эта зона лишь частично заполнена
электронами. Следовательно, характер проводимости в TiO и
VO почти чисто металлический (при 25°С проводимость
~103 (Ом-см)-1).
Различие между TiO и NiO состоит еще и в том, что
/г^-зопа, в которой может содержаться до шести электронов
на один атом металла, в случае NiO должна быть полностью
* Строго говоря, монооксид никеля дэже в равновесии с металличе-
металлическим никелем кестехиометричен и имеет некоторый дефицит металла по от-
отношению к стехиометрическому составу NiO. См., например, Третьяков Ю. Д.
«Химии нестехиометрических окислов». — М.: Изд во МГУ, 1974. — Прим.
ред.
14. Электронные свойств;! и зончля теория
заполнена. Два других cf-электрона иона Ni2+ находятся на
^-уровнях {с1г2, dx2-yt). Эти е^-орбитали локализованы непос-
непосредственно над ионами кислорода (рис. 14.22, б). Поскольку
ионы кислорода находятся между ионами никеля, еуорбитали
соседних ионов Ni2+ не могут перекрываться и, следовательно,
не может образовываться соответствующая зона. Таким обра-
образом, <?5-орбитали остаются локализованными на отдельных
ионах Ni2
Рис. 14.22. а — сечение структуры TiO, параллельное грани элементарной
ячейки. Изображено только положение ионов Ti2+ (см. рис. 5.8). Перекры-
Перекрывание йда-орбнталей соседних ионов Ti2 + , а также соответственно dxz- и
с/4г-орбиталсй приводит к образованию ^?г-зоны. б—структура NiO. Изо-
Изображенные г/х2_г8-орбитали расположены над кислородными ионами, что>
препятствует их перекрыванию и образованию ея зоны.
Филиппе и Уильяме [5] сформулировали некоторые общие
правила возможности перекрывания fif-орбиталей. Согласно этим
правилам, образование rf-зоны весьма вероятно, если:
1) формальный заряд катиона невелик;
2) катион имеет небольшое число ^-электронов;
3) катион принадлежит группе переходных элементов 4- и
5-го периодов периодической системы;
4) электроотрицательность аниона не слишком велика.
Соображения, лежащие в основе этих правил, весьма про-
просты. Они абсолютно такие же, как рассуждения, используемые
в теории поля лигандов. Первое, второе и третье правила отра-
отражают необходимость того, чтобы cf-орбитали занимали как
можно большее пространство кристалла и «не ощущали» по-
положительного заряда ядер соседних переходных металлов.
Четвертое правило связано с необходимостью понижения степе-
14.9. Зонная структура неорганических твердых тел 89
ни ионностн химической связи и уменьшения ширины запрещен-
запрещенной зоны, о чем писалось выше в настоящем разделе.
Каждое правило можно ироиллюстрироиать ня ряде примеров:
Первое правило: TiO обладает металлической проводимостью, а
TiO2—диэлектрик, СигО и МоСЬ — полупроводники,
а СиО и .MoOi—диэлектрики.
Второе правило: 'ПО и VO обладают металлической проводимостью,
в то Бремя как уровень проводимости NiO и СиО
едва достигает полупроводникового уровня.
Третье правило: СггОл — плохой проводник, и то время как низшие
оксиды Мо и W — хорошие1, проводники.
Четвертое правило: NiO — плохой проводник, a NiS, NiSe, NiTe—хоро-
NiTe—хорошие.
Структура системы rf-орбиталей переходных металлов силь-
сильно зависит от кристаллической структуры твердых соединений
в состав которых входят данные металлы, а также от степени
окисления переходных металлов. Приведем некоторые интерес-
интересные примеры свойств сложных оксидов со структурой шпинели.
1) Хотя как FejO4, так и Mnjdi. имеют структуру шпинели, ЭД.П3О4 —
фактически диэлектрик, а РсьО4 обладает почти металлической проводи-
проводимостью. Структуру FejO4 можно изобразить в виде
lFes'-]reTp[Fe2-f-1 Fe3%KTO4 обращенная шпинель
я структуру MibOi можно представить
|Мп2+]ГС1р [Мп23+1окт04 нормальная шпинель
Поскольку Fe3Oj имеет структуру обращенной шпинели, ионы Гег+ п Fe3 +
распределены по октаэдрнческим позициям. Эти октаэдрпческис позиции
находятся близко Друг к другу, так как они принадлежат соседним сочле
ненпым ребрами октаэдрам. Следовательно, положнтельпо заряженные
дырки могут легко мигрировать от понок Fe2+ к ионам Fe3+ и FejO4 — хо-
pomnii проводник.
Мн^О) имеет структуру нормальной шпинели. Это означает, что рас-
расположенные по соседству друг с другом октаэдрические позиции занима-
занимают только ионы Мп3". Тетрачдрические позиции, занимаемые нонами Мп2+,
сочленяются лишь вершинами с октаэдрами. Расстояния между ионами
Мп2+ и Мп3 h больше, чем между Ге?+ и Г'е3;~ r мапютите, подточу про-
процесс обмена электронами затруднен.
2) Аналогичный пример—литиевые шпинели LiMnjO* и L1V2O4. Здесь
можно хороню проследить выполнение второго правила перекрывания с/ор-
битзлей. Структ\г])а этих нгпшюлей одинакова
[Ь!+1тегр [V3+ V*+UrO*
И в той и в другой шпинели в октаэдрически.ч позициях находятся как
ион1>1 со степенью окисления +3, так и поиы со стспепг.ю окпе.юпия +4.
Однако поскольку перекрывания rf-орбиталей в случае ванадия больше, чем
в случае марганца, это отражается пи электрических свойствах: LiMn?O4 —
полупроводник с прыжковым характером проводимости, a L1V2O4 характе-
характеризуется металлической проводимостью.
90 !4. Электронные свойства и зонная теория
14.10. Цветность неорганических твердых тел
Окрашивание твердых тел п тот или ипоп цвет связано с их взаимодей-
взаимодействием с видимой частью спектра электромагнитных волн. Чаще всего, ес-
если окрашенное твердое тело облучать белым светом, часть облучения из
видимой облети спектра поглощается этим веществом. Возникающая при
этом окраска вещества отвечает кеиоглощенной части облучения и соответ-
соответствующим ей длинам электромагнитных волн.
Часто, по далеко не всегда, окраска определяется присутствием в тиер-
дом тые ионон переходных металлов. В молекулярных соединениях с\ше-
с\шествуют две возможные причины появления окраски. Электронные переходы
между rf-орбиталямк попов переходных металлов (d—of-переходю) —основ-
—основная причина возникновения многих распространенных типов окраски неор-
неорганических соединений. С этими явлениями, например, связаны различные
оттенки голубого и зеленого цвета комплексных соединений меди(П). Эф-
Эффекты переноса заряда .между анионом и катионом также часто сказыва-
сказываются па интенсивности окраски соединений, например пермапганаты (фио-
(фиолетовый цвет) и хроматы (желтый цвет). В твердых телах существует сше
один источник возникновения окраски - - возможные электронные переходы
между энергетическими зонами.
Цветность соединении можно изучать количественно спектроскопичес-
спектроскопическими методами {гл 3). В ходе таких исследований регистрируются нснускл-
шк: или поглощение излччепня образцом в зависимости от частоты или дли-
длины полны. Этими методами можно также обнаруживать электронные пе-
переходы и другие эффекты, которые: проявляются за границами видимой
области спектра—-в инфракрасной и ультрафиолетовой частях спектра. Иа
рис. 14.23 схематически изображен спектр электромагнитных соли, который
охватывает ИК и УФ-облаетп. На нем видны пики поглощения в ИК-об-
ИК-области, которые отвечают колебаниям кристаллической решетки. В области
больших частот возможны электронные переходы, связанные с расщеплени-
расщеплением (/-уровня, присутствием примесных конов, дефектов кристаллической ре-
щетки и др. Многие такие спектральные эффекты можно обнаружить к ви-
видимой части спектра, и они определяют окраску соединений.
Важную роль играют электронные переходы через запрещенную зону.
Опи оказывают влияние не только па проводимость, но и на окраску твер-
твердых тол. Кроме того, отвечающие этим переходам частоты представляют
собой границу, за которой твердые, тела больше не прозрачны для облу-
облучения.
Приведенный на рис. 14.23 пример спектра поглощения относится к ве-
веществам, у которых' ширина запрещенной зоны велика (несколько элек! poir-
вольт) и переходи через нее регистрируются в УФ-областп спектря. Поэто-
Поэтому такие твердые тела — плохие электронные проводники. К тому же они
бесцветны, так как электронные переходы между дискретными энергетиче-
энергетическими уроинямп находятся пне видимой области спектра. В качестве t¦ ртi-
мера здесь можно привести TiCb, имеющий белую окраску (ширина запре-
запрещенной зоны 3.2 эВ); в то же время Сг-;О3 (ширина запрещенной зоны
3.4 эВ) зеленого цвета, так как для иопои Сг3+ характерны d—</ псре\од;.г.
Диалогично из-за d—d переходов зеленую окраску имеет и NiO (ширина
запрещенной зоны 3,7 эВ).
У некоторых твердил тел ширина запрещенной зоны составляет 1.7—
3.0 эВ, поэтому электронные переходы через нее отвечают линиям и види-
видимой части спектра, что находит свое отражение в окраске этих спеднко-
кгй. Примером таких веществ может служить CdS желтого цвета (ширина
запрещенной зоны 2,4.' эВ).
Если ширина запрещенной зоны меньше ~1,7 эВ, твердое тело имеет
темную окраску, так как оно поглощает белый свет, например PbS (шири-
(ширина запрещенной зоны 0,37 эВ) п СиО (ширина запрещенной зоны 0,G эВ)
14.11, Энергетические зоны или химический связь
91
чернот цвст.1. Такие вещества обладают заметной или даже очеш> нысокои
проводимостью.
Экспериментальное определение ширины запрещенной зоны проводят
путем изучения спектров соединений (рис. 14.23). Определенные трудности
здесь могут возникать (особенно при изучении твердых «ещечтв с широкой
запрещенной зоной), если экситоппые переходы лежат в области частот, не-
несколько меньших, чем электронные переходы через запрещенную зону. Это
относится к переходам на дискретные энергетические уровни, которые ле-
лежат чуть ниже дня зоны проводимости. В этом случае трудно точно от-
отличить экситонные переходы от переходов через запрещенную зону.
Поглощение
/
/ —светонепроницаемость
'запрещенную
зону
поглощение на
примесях.d-d -переходы и i n
ПК-область
видимоч
область
УФ-область
Частот
Рис. 14.23. Схематическое изображение спектра поглощения неметалличе-
неметаллических твердых веществ.
Некоторые нз упоминавшихся выше твердых веществ иепольз\гот и при-
приборах для обнаружения излучения (детекторы излучения). С помощью них
можно обнаружить излучение такой частоты, которая близка к положению
полос в спектре поглощения детектора, отвечающих переходам через запре-
запрещенную зону. Так, PhS, PbSe, PbTe, у которых ширина запрещенной зоны
~0,3 эВ (ИК-облпсть спектра), можно использовать в качестве детектора
И К излучения, а халькогениды кадмия, ширина запрещенной зоны которых
отвечает области видимого света. — для измерения интенсивности видимо-
видимого cbct;i.
14.11. Энергетические зоны или химическая связь.
Заключительные замечания
В неорганических твердых веществах существуют три типа
химических связей — ионная, ковалентпая и металлическая.
Можно привести множество примеров веществ, в которых пре-
преобладает тот или иной тип свя.чи. Однако часто химическая
связь в данном конкретном соединении не может быть с полной
92 14. Электронные свойства и зонная теория
определенностью отнесена к одному из этих типов. Не будем
здесь останавливаться на всех возможных вариантах химиче-
химической связи, имеющих место в твердых телах. Некоторые из них,
в частности образование смешанных ионно-ковалентных связей,
уже были обсуждены в гл. 8. Вместо этого сделаем некоторые
замечания относительно случаев, когда использование зонной
модели имеет определенные преимущества перед применением
модели химических связей при описании структуры твердых
тел.
Применение зонной модели более предпочтительно при опи-
описании тех систем, где существуют подвижные свободные элект-
электроны (например, металлы и некоторые полупроводники).
Экспериментальные методы измерения подвижности носителей
заряда в эгих веществах подтверждают их высокую подвиж-
подвижность и делокализованпость в кристаллической решетке. Для
других полупроводников, например, таких, как легированный
NiO, зонная модель, по-видимому, не слишком пригодна для
объяснения электрических свойств. Существуют доказательства
того, что механизм проводимости в таких веществах прыжково-
прыжкового типа и подвижность электронов здесь не очень высока. По-
Поэтому, видимо, более уместно в данном случае рассматривать
систему ^-электронов, занимающих дискретные орбитали ионов
никеля. Важно, однако, напомнить, что проводимость оксида
никеля не определяется положением лишь одного или двух
энергетических уровней. Оксид никеля, как и другие соединении,,
имеет несколько наборов энергетических уровней. Более низко-
лежащие уровни полностью заполнены электронами. Они дис-
дискретны и принадлежат отдельным анионам и ка шопам. Более
высокие энергетические уровни — это различные возбужденные
уровни, которые в обычном состоянии почти полностью не за
няты электронами. Однако эти уровни могут перекрываться с
образованием энергетических зон. Чтобы ответить на вопрос,
какая из теорий (зонная модель или модель химических связей)
лучше всего подходит для описания тех или иных твердых тел,
необходимо выяснить особые свойства отдельных энергетических
уровней или совокупности этих уровней. Так, многие твердые
вещества с ионным характером связи под действием УФ-пзлу-
чения проявляют электронную проводимость, которую л\ чше
всего описывать с использованием зонной теории.
Упражнения
14.1. Электронная конфигурация кальция Is22s22p63s23p4s2. Объясните,
почему кальцин обладает металлической проводимостью.
!4.2. Нарисуйте структуру энергетических зон кремния с шириной зап-
запрещенной зины 1,1 эВ. Какие элементы можно вводить в кремний, чтобы
он стал полупроводником р-типа? Нарисуйте получающуюся при этом
Литература 9?
структуру энергетических уровней с .-кцепторнымн уровнями, расположен-
расположенными на 0,01 эВ выше потолка валентной зоны. Какая доля акцепторных
уровней будет заполнена ври комнатной температуре? Предположим, что
вероятность возбуждения электронов пропорциональна ехр(—CikT), Какова
концентрация носителей заряд;!, образующихся в результате введения
1A пт. % примесей? Какова была бы концентрация собственных' носителей
заряда при комнатной температуре в отсутствие примесей? При какой тем
пературе концентрация собственных носителей заряда станет равной кон-
концентрации примесных носителей?
14.3. Ответьте нл предыдущие вопросы для германия, у которого шири-
ширина запрещенной зоны 0,7 эВ.
14 4. Почему в полупроводниковых приборах целесообразно использо-
использовать материалы с широкой запрещенной зоной межлу валентной зоной п зо-
зоной проводимости, но с близко расположенными у потолка валентной зоны
{или у дна зоны приводимости) примесными уринмями?
14.5. Все галогепиды ка.чия прозрачны для белого сиетп. Рассчитайте
длину полны, при которой они становятся прозрачными. Данные о ширине
запрещенной зоны соответствующих соединений приведены в табл. 14.2.
Литература
Существует множество мин. посвященных- вопросам зонной теории, физи-
физики н химии полупроводников. В приведенном списке литературы указаны лишь
те нз пп.х. в которых эти проблемы обс\ ждаются без привлечения серьезного
математического аппарата.
1. Barrett С, Massahki Т. В., Structure of Metals, Pcrgamou, 1982.
2 Gocdcnough I. В., Hong H. Y-P., Kafalas J. A., Fast Na+ ion Transport in
Skeleton Structures, Mat. Res. Bull., 11, 203 A97G).
3. Moore IV. /.. Seven Solid States, Benjamin W. A., 19C7.
4. Pascoe K. J., Properties of Materials for Electrical Engineers, Wiley, 1973.
5. Philips С S. G., Williams R. J. P., Inorganic Chemistry, Oxford,'1965.
15 Rose R. M, Shepard L. A., Wulff /., The Structure and Properties of Materials.
IV. Electronic Properties, Wiley, 1966.
7. Уэрт '/.. Тпнпсчн Р. Физика твердого тела. — M.: Мир, 1969.
8. Wright Г). A. Semiconductors, Alcilnicn, I966.
Дшю.'.нительная литература. Джпнг. Г. Теория зии Приллюэна и электрон-
электронные состояния в кристаллах. — Л1.: Мир, 1968: Иоффе А, Ф. Физика iwivnpo-
водпнкгж. — М — д.: Изд во АИ СССР, 1957: Сокг.лпвская Е. М., Л/лей 'Л. С.
Металлохимия.— ,VI.: Изд во .МГУ, 1986, гл. 1: Френкель Я. И. Введение в
теорию металлом. — Д.: Наука, 1972, ч. 2; Шцльцс Г. Металлофизика — ,М •
Мир, 1Я71, гл. 11.
Глава 15
ДРУГИЕ
ЭЛЕКТРИЧЕСКИЕ
СВОЙСТВА
Наряду с электропроводностью, которой были посвящены
гл. 13 и 14, твердые тела обладают рядом других электрических
свойств, наиболее важные из которых рассмотрены в настоящей
главе.
15.1. Термоэлектрические явления
В месте контакта двух разнородных металлов возникает
э. д. с, величина которой зависит от а) природы металлов и
6} температуры. Если металлический стержень находится в
зоне с градиентом температуры, то в нем возникает э. д. с,
величина которой также зависит от а) природы металла и
б) величины температурного градиента. Подобные явления
подразделяются на ряд разновидностей, составляющих в целом
группу термоэлектрических явлений, на которых основана рабо-
работа термопар, используемых для измерения температуры, термо-
термоэлектрических .холодильных установок и термоэлектрических
источников энергии.
15.1.1. Эффект Томсона
Наличие градиента температуры вдоль однородного провод-
проводника, например металлического стержня, приводит к возникно-
возникновению градиента потенциала W. Происхождение этого явления,
известного под названием эффекта Томсона, можно попять, рас-
рассматривая электроны проводимости в металле клк частицы, под-
подвижность и кинетическая энергия которых возрастает с темпе-
температурой. При установившемся температурном градиенте тепло-
тепловая энергия электронов на горячем конце стержня больше, чем
на холодном, и таким образом от горячего кочна к холодному
перечоднт большее количество носителей заряда, чем в обрат-
15.1. Термоэлектрические явления
95
ном направлении. В итоге на холодном конце возникает избыток
электронов и, следовательно, отрицательный полюс разности
потенциалов.
Эффект Тимсопа можно объяснить также на основе зонной
теории (рис. 15.1). На горячем конце электроны проводимости
распределены в некотором интервале энергетических уровней,
расположенных по обе стороны уровня EF (рис. 15.1, а); подоб-
подобное распределение устанавливается и на холодном конце, но
интервал энергетических уровней в этом случае уже (рис.
Рис. I5.I. Эффект Томсопя.
15.2, б). «Самые горячие» электроны (т. е. обладающие наи-
наибольшей энергией) занимают па горячем копне более высокие
уровни энергии, чем на холодном, поэтому от горячего к холод-
пому концу возникает дрейф электронов, величина которого
зависит от AT. Результирующая э. д. с. задается соотношением
? = (тДТ A5.1)
в котором коэффициент пропорциональности а носит название
коэффициента Томсона. Обычно Е по порядку величины состав-
составляет несколько милливольт.
Эффект Томсона наблюдается и в полупроводниках, По
знаку термо-э. д. с. можно различить полупроводники с /;- и
р-тином проводимости. В полупроводниках «-типа, как и в
металлах, «холодный» конец приобретает отрицательный знак.
Для полупроводников р-тииа в области собственной проводимо-
проводимости (рис. 14.20) носителями заряда являются положительные
дырки и холодный конец заряжен положительно. Происходит
это потому, что па горячем конце из валет ной зоны на акцеп-
акцепторные уровни переходит большее количество электронов, чем
па .холодном конце стержня. Избыток копнен грации положи-
положительных дырок, возникающий па горячем конце, приводит к
тому, что некоторые из них перемешаются к холодному концу
и сообщают ему положительный заряд.
/96 15. Другие электрические свойства
Как таковой эффект Томсона трудно использовать для полу-
получения энергии. В частности, с его помощью нельчя получить
электрический ток по замкнутому контуру, так как при созда-
создании разности температур в двух точках однородного металли-
металлического контура в двух его ветвях, соединяющих горячую и
холодную точки, создаются равные, по противоположно направ-
направленные э, д. с. (рис. 15.1, в). Если же ветви контура изготовлены
из различающихся металлов, то как показано ниже, в местах
их контактов возникают дополнительные термоэлектрические
эффекты.
15.1.2. Эффект Пельтьв
При пропускании электрического тока в определенном на-
направлении на границе двух разнородных проводников, например
железа и меди, выделяется теплота. При перемене направлении
тока происходит поглощение тепла. Когда два разнородных ме-
металла просто приведены в контакт друг с другом, то электроны
одного из этих металлов переходят r другой до тех пор, пока
не будет достигнута такая величина электрического поля (или
«пространственного заряда»), которая препятствует дальнейше-
дальнейшему переходу электронов. Процесс перехода электронов возни-
возникает вследствие разного положения уровней Ферми в различ-
различных металлах. Контакт разнородных металлов таким образом
становится источником э. д. с, называемой э. д. с. Пельтье п
обозначаемой буквой я; величина этой э. д. с. зависит от приро-
природы контактирующих металлов и температуры контакта. Э. д. с.
Пельтье, как правило, составляет несколько милливольт.
Наибольшие значения получены для пары олово —висмут и
некоторых полупроводниковых соединений.
Зонная структура контакта металл — полупроводник «-типа
схематически изображена па рис. 15.2. При установившемся
равновесии в каждом таком контакте уровни Фермн по обе сто-
стороны находятся па одинаковой высоте. Для достижения равно-
равновесия, однако, необходимо некоторое изменение зопион струк-
структуры па поверхности полупроводника (похожие явления
наблюдаются на поверхностях полупроводник — воздух: в при-
приповерхностном слое создается пространственный заряд, влияю-
влияющий на зонную структуру). В полупроводнике п-тппа уровень
Фе'.ши расположен па глубине U ниже дна зоны проводимости.
Для того чтобы электроны перемещались через контакт слева
направо, им необходима энергия, позволяющая перейти из ва-
валентной зоны металла в зону проводимости полупроводника, и
дополнительная энергия, равна zJ2kT, соответствующая кинети-
кинетической энергии свободных электронов. Таким образом, каждый
электрон, который осуществляет переход в указанном паправ-
15.1. Термоэлектрические явления
леции, «черпает» энергию из металла, что приводит в итоге
к охлаждению контакта. Наоборот, если электронный ток /
направлен справа палево, то на контакте выделяется теплота
+ ~-kT) A5.2)
15.1.3. Эффект Згебска
Рассмотрим замкнутый контур, образованный разнородными
проводниками А н В (рис. 15.3), контакты которых находятся
при температурах Т\ и 7';. Градиенты температуры устанавли-
устанавливаются в обои.х металлах, н соответственно в каждом из них
возникает э. д. с. Томсопа. В обоих контактах возникают э. д. с.
Пельтье, но они различны по величине вследствие различия
металл
|;С.'|упрОБОДШ1|<
зону ппо'.е
Рис. 15.2. Зонная структура па границе металл—полупроводник, приводя-
приводящая к возникновению э. Д. с. Псльтг.е.
температур контактов. Результирующая по всему контуру
э. д. с. равна алгебраической сумме двух э. д. с. Томсона
а двух э. д. с. Пельтье и, как правило, отлична от нуля
вГ2---ХавГг) A5.3)
Направления (знаки) отдельных составляющих э. д. с. указаны
на рис. 15.3. Ток будет идти по контуру все время, пока поддер-
поддерживается разность температур контактов двух проводников.
Это явление, называемое эффектом Зеебека, лежит в основе
7— 14:К
15. Другие электрические свойства
7Г
АВ,
Рис. 15.3. Эффект Пс,1ьтье и Томсона в термопаре.
действии термопар. Коэффициент термо-э. д. с. (или коэффици-
коэффициент Зеебека) а определяется соотношением
ее = л/Г A5.4)
Как правило, а~10 В/град, по для цепей с некоторыми полу-
полупроводниками значения а достигают ~ 1 мВ/град.
15.1.4. Термопары
Термопары являются незаменимыми устройствами при изме-
измерениях температуры. Они используются в чрезвычайно широком
температурном интервале, ограниченном сверху плавлением
используемых металлов (для платиновых сплавов, например,
предельные температуры достигают ~1700°С).
Термопара изготовляется из проволок двух разнородных
материалов, соединенных па концах и образующих замкнутый
контур, в который включают также прибор для измерения па-
пряжения. Если спаи (места контакта проволок) находятся при
разных температурах, то возникает э. д. с. (рис. 15.4). Во время
измерений температуру одного из спаев (спай сравнения) под-
поддерживают постоянной (обычно равной 0°С). Э. д. с. зависит,
15.1. Термоэлектрические явления
99
Pt/13%. Rh .
измерительный спаи
Рмс. 15.4. Схем;; цепи с термопарой.
таким образом, только от температуры другого (рабочего) спая
термопары. Для перевода термо-э. д. с. в значения температуры
пользуются справочными таблицами, а для измерения э. д. с,
составляющих, как правило, несколько милливольт, применяют
вольтметры с высоким входным сопротивлением или потенцио-
потенциометры. Чтобы напряжение на клеммах регистрирующего прибо-
прибора было равно э. д. с. термопары, необходимо проводить изме-
измерения в «бестоковом» режиме; это позволяет избежать возник-
возникновения в контуре потерь на нагревание, пропорциональных 12R.
7*
100 15. Другие электрические свойства
Схема цепи для измерения температуры изображена на
рис. 15.4. Металлами в данном случае являются платина и сплав
Pt/13% Rh. Для соединения в цепи использован еще и третий
металл, им, как правило, бывает медь; оба спая Pt—Си и
Pt/13% Ru—Си находятся при 0°С. Наличие медных проводни-
проводников не сказывается на э. д. с. термопары, поскольку их спаи
с металлами, образующими термопару, находятся при одной и
той же температуре (рис. 15.4, б). Электродвижущая сила та-
такой цепи, измеряемая на концах медных проводников, равна
э. д. с. пары Pt—Pt/13% Ph, спаи которой находятся при тем-
температурах 0 и Т°С.
Температурная зависимость э.д. с. достаточно хорошо описы-
описывается выражением
Е = аАВ(Т2-Т>) + ЧфАВ(Т2*-Т*) A5.5)
где а и Ь — термоэлектрические коэффициенты металлов А и В,
а Т\ и 7*2 — температуры спаев. Для сопоставления значений а
и b различных металлов, необходимо было выбрать некий ме-
Таблица 15 1. Термоэлектрические коэффициенты
Металлы или сплавы
Сурьма
Железо
Мель
Платина
Никель
Копстантан {60% Си, 40% Ni)
Висмут
а
+35,0
Н-16,7
+2,71
—3,03
—19,1
—38,1
—74,4
Ь
+0,145
—0,0297
+0,0079
—3,25
—3,02
—0,0888
+0,032
талл сравнения и принять его коэффициенты равными нулю;
таким металлом условились считать свинец. Подобным же
образом окислительно восстановительные потенциалы табулиро-
табулирован!.! по отношению к водородному электроду (V2H2/H1),
потенциал которого принят равным нулю. Термоэлектрические
коэффициенты некоторых металлов приведены в табл. 15.1.
Для любой пары АВ термоэлектрические коэффициенты равны
разностям соответствующих коэффициентов для индивидуаль-
индивидуальных металлов:
°дв = « а—ав и &лв = ЬА—Ьв
Тс термопары, которым соответствует большое значение коэф-
15.1. Термоэлектрические явления
101
фициепта aAn (например, для пары железо — копстантан а =
= 16,7—(—38,1) =54,8), характеризуются высокой чувствитель-
чувствительностью. Термопары па основе платины и ее сплавов характери-
характеризуются небольшими значениями а, однако они имеют то преиму-
преимущество, что их можно использовать при очень высоких темпе-
температурах.
Из уравнения A5.5) следует, что зависимость Е от Г2
является параболической (рис. 15.5). При Т2 = Т{ э. д. с. равна
Рис. 15.5. Зависимость э. д. с. термопары от температуры.
пулю, при Г2 = — а/b проходит через минимум, а при Т2~
= —2(а/Ь)—Т\ вновь становится равной нулю. Для большинст-
большинства практических применений термопар Т^Т^ так что зависи-
зависимость э. д. с. от температуры, как правило, соответствует высо-
высокотемпературной ветви параболы.
Применение термоэлектрических эффектов весьма разнооб-
разнообразно. Набор из п последовательно соединенных термопар об-
образует термобатарею, э. д. с. которой соответственно в п раз
больше, чем э. д. с. одной термопары. Такие термобатареи
используют для преобразования солнечной энергии, а также в
астрономии при исследованиях излучения звезд. Материалы,
имеющие большие коэффициенты Пельтье и Зеебека, могут
быть попользованы в термоэлектрических источниках энергии и
термоэлектрических системах охлаждения. Полупроводники
обычно характеризуются гораздо большими значениями коэф-
коэффициентов, чем металлы, однако материалы с большими а и л
102 15, Др;гис электрические свойства
имеют, как правило, низкую электропроводность а, что вызы-
вызывает затруднения при их использовании; при прохождении тока
через высокоомные полупроводники возникают большие потери
па выделение джоулева тепла, а это существенно понижает
к. п. д. полупроводниковых устройств. Оптимальные условия
для работы достигаются при максимальной величине 7.
где Л —теплопроводность материала. Найдено, что наибольшие
значения Z характерны для некоторых полупроводниковых сое-
соединений с большой молекулярной массой (например, Bi2Te3,
Ri2St'j и РЬТе). Одну ветвь термопары изготовляют из полу-
полупроводника л-типа, а другую — из полупроводника р-типа.
Таким образом, удается увеличить разность уровней Ферми
в .;иие контакта соединяемых материалов и достигнуть больших
значений л. Термоэлектрические охлаждающие устройства
с применением термопар из полупроводников системы Bi—Те—
—Se способны давать температуру примерно на 70°С ниже
комнатной, что используется в конструкциях небольших и бес-
бесшумных холодильников.
15.2. Эффект Холла
Важный источник информации о механизме проводимости в
твердых телах, и особенно полупроводниках, связан с изучением
эффекта Холла. Поскольку в уравнении A3.1) проводимость
определяется произведением числа носителей (электронов или
дырок) п на их подвижность \х, то из одних экспериментов по
электропроводности невозможно установить эти величины по
отдельности. Это удается сделать, комбинируя измерения элект-
электропроводности с измерением разности потенциалов, возникаю-
возникающей в эффекте Холла.
Обратимся к рис. 15.6. Если через образец в указанном
направлении проходит ток с силой /, а перпендикулярно ому
действует магнитное поле напряженностью И, то на противо-
противоположных сторонах образца возникает разность потенциалов,
направление которой перпендикулярно направлению тока и
.магнитного поля. Возникает это потому, что магнитное поле
вынуждает электроны отклоняться от прямолинейного пути и
к результате одна сторона проводника «обогащается» электро-
электронами по сравнению с противоположной стороной. Различная
концентрация электронов в поперечном сечении образца вызы-
вызывает возникновение электрического поля. Равновесие наступает
тогда, когда сила, отклоняющая электроны от прямолинейного
пути, уравновешивается холловской разностью потенциалов,
действующей в противоположном направлении. Коэффициент
15.2. Эффект Холла
103
Холла R, характеризующий данный материал, равегг градиенту
потенциала, возникающему при единичных значениях поля //
и тока /. Направление R определяется знаком носителей тока
и, следовательно, различно в случае электронной и дырочной
проводимости. На рис. 15,6 при потоке электронов, направлен-
направленном слева направо, полюс Л положителен. При действии маг-
Рис. 15.6. Схема, поясняющая возникновение эффекта Холла в проводнике.
нитного поля с напряженностью И на образец, через который
проходит ток с плотностью а, отклоняющая сила Fu действую-
действующая па подвижные заряды, определяется соотношением
Fl = Me\i A5.6)
где \х — подвижность (или средняя скорость дрейфа) электро-
электронов, а е— их заряд.
Холловская разность потенциалов Е действует на отклоняе-
отклоняемые электроны с равной и противоположно направленной силой
Fi, определяемой выражением
= Ee = HoRc = Hne\.iRe
Из равенства F\ и F2 следует
R=\lne
A5.7)
A5.8)
104 IS. Другие электрические свойстп,-.
Таким образом, измеряя э. д. с. Холла при известных / и Н,
можно рассчитать R и далее п. Если известна проводимость
о, то можно определить и подвижность ц.
Большинство металлов имеет отрицательные коэффициент
Холла R, а значит, носителями тока в них являются электроны;
однако некоторые металлы, r частности Zn и Cd, в этом отно-
отношении аномальны: для них коэффициенты Холла R— положи-
положительные ис.sичииы. Особое положение цинка и кадмия можно
качес"нешю объяснить па основе тонной теории; проводимость
в цинке и кадмии ос\¦щс-ci вляется главным образом положитель-
положительными дырками (электронными пакансиями), уровень которых
паходшея ниже уровня Ферми EF (подобно проводимости
в полупроводниках р-тииа). В собственных полупроводниках
число свободных электронов равно числу дырок и при равенстве
подвижностеи обоих типов носителей заряда результирующая
э. д. с. Холла должна быть равной пулю. В действительности
же электроны в большинстве случаев более подвижны, чем
положительные дырки, в результате чего в собственных полу-
полупроводниках возникает результирующая э. д. с. Холла, свиде-
свидетельствующая о проводимости «-типа. В этих случаях концент-
концентрация постелей п, получаемая из коэффициента R, ошибочна,
несмотря па то что при большом значении соотношения под-
вижностей электронов и дырок проводимость действительно
неликом определяется одним типом носителей. В области пере-
перехода к собственной проводимости температурные зависимости
R для полупроводников п- и р-типа различны. Эти факты гово-
говорят о необходимости осторожного отношения к интерпретации
величин R, наблюдаемых в полупроводниках.
Ионные проводники имеют обычно слишком низкие значения
п и it, что не дает возможности наблюдать в них эффект Холла.
Однако в некоторых электролитах, и частности в RbAg4I5,
проявляющем высокую Ag~ ионную проводимость (с«
za\ Om''-см при 25 DC), этот эффект был обнаружен.
15.3. Диэлектрики
Диэлектрики -- эго материалы, не проводящие электрическо-
электрического токп. Oirn находят применения при изготовлении конденсато-
конденсаторов н электрических и^ляторов. Основные гребоааппя, предъ-
предъявляемые практикой к диэлектрикам, сводятся к следующему:
1) высокая диэлектрическая прочность, т. е. способность
противостоять высоким напряжениям без деградации и перехо-
ча в проводящее состояние;
2) низкие диэлектрические потери, т. е. потери энергии
переменного электрического поля, выделяющейся в виде тепла.
Рассмотрим в первую очередь поведение диэлектриков в
15.3. Диэлектрики 10~>
переменном электрическом пиле. Несмотря па то что в диэлек-
диэлектриках смещение ионов или электронов на большие расстояния
невозможно, приложение разности потенциалов к противопо-
противоположным сторонам образца диэлектрика приводи г к поляриза-
поляризации зарядов внутри него. При выключении поля поляризация
исчезает. Сегьогоэлектрические материалы, которые наряду с
пиро- и ш,с.;очлектрнками более детально рассматриваются в
предам 'лвляюг eufiisf: осо-
особый iun [и--1 лскгрнклв г.
1 ом отношен:\\'. ч.о о;:-:
ео.храпу-hyv t'."! :,>чг. hi ov- ;.. ~~:." _~. _.\ .......
таточную пс....''р:;: ;';лпп ;>
рпчесК(Д\) :.ч.-':я ('i:':: ;.
15.4). ;
Диэлектрические свой- .-.-¦:.- ¦¦ .-._ - :.-
ства определяют ;>о по-
поведению материала r „'',.. .'' "' !.
плг;сК(;\1 \'.::;.\ен:': ¦¦¦гу^.
прс.Т'" n('.'!;ii0V;?.M cc,oi"/n Vi-.-;. 15.7. Кондспслтор с ппраллсльч,,п.'|;
дне параллель:::) раегто- ;.б:. ¦.¦¦ ал.и; ч .ни.-^.тмжом между iti:h:i
ложеш-гьгс ;?])():< ^.j,Hiune
пллстнп!,;, находящиеся друг от друга на расстоянии d, ком;.
рсо ::;:\;.ii-.':o мепыпе, !'ем 1/аз\:^р самих пластич (рис. 15.7j.
Дли K<.-M..;Ci:c;; :-о;>а. меж. у п.;;,''т1!нлми коюрого находится :;.п-
ку> \', ем|;-'>'ч:ть С^ рг;шк;
г.и- с",. ¦-.¦!.[[¦). 1?кг;-1.''-!е..-,'<ая проппцасмост:-. вакуума, равная
8.85 = ¦ !0-12 О/м, /1 ¦ - 1'.н;-.-:пд1. гнас~мп. Fimkoc;,, tjikov-'; ¦jcmci.1-
сатора ^а.чи-ч.'т то. ;ьг;о о,- его |"с;:ме"! рк^че^ких раз:'.:ероп. П*м
Ha.'icj/..'Cii/Пг па i-.'i'.icmhi;.-: ;)аз':;1>т:! п^лч^'чпд.югс !¦' х.слдег'.атог
¦¦.Янаjtior ".аьяд Q(,, равный
Если между пластинами пометен днэлстлрмк. vo npi; ла.чожениг
той же ;i<i.:!!осi и потенциалов :;аряд, ^акапли.^ае^гьп"! коиденса-
топом, ::;'^растает до Q.. ?. ег'-< емкость- -до С;. Ди^.-гектри'чсс-
кяя прг'мшлемосгь г', харакчс;".п ^'юситя .¦¦г;ломр1гк. связана i:
емкое; ;лг) соотпоыепием
i''---CJCa (I5.11)
f/ liUDHciir от сюпенн по.тя1)н?ацни или смещения :;нм:дов, про-
происходящего в материале. Для воздуха s-'•---/, для Солыпипстиа
ионных соединений е'~5~10, а для сегкетоэлекмркков, подоб-
подобных Ва'ПОа, ь =104-104.
НХ> 15. Другие электрические свойства
Поляризуемость диэлектрика а — коэффициент, связываю-
связывающий дипольпый момент р и локальное электрическое поле Е
р = аЕ A5.12)
Поляризуемость а можно записать как сумму
A5.13)
Электронная поляризуемость ае, возникающая в результате
смещения электронных орбиталей атома относительно положи-
положительного заряда ядра, присуща всем твердым телам; в тех ве-
веществах, в которых отсутствуют ионная ее,-, дипольная ad л
объемно-зарядная as поляризуемости (к таким веществам, в
частности, относится алмаз), ае — единственная составляющая
поляризуемости.
Ионная поляризуемость а.;, характеризующая возможность от-
относительного смещения или разделения катионов и анионов
в твердых телах, в основном определяет поляризацию в ионных
кристаллах.
Дипольная поляризуемость a,i возникает в таких веществах,
как НС1 или Н2О, имеющих постоянные электрические диполи,
которые могут удлиняться и менять свою ориентацию под дей-
действием внешнего электрического поля. Дипольная поляризуе-
поляризуемость, как правило, очень сильно зависит от температуры (при
пн:;ки\ температурах диполи могут быть «заморожены»).
Объемно-зарядная составляющая а, характерна для таких
материалов, которые не относятся к хорошим диэлектрикам
из-за возможности миграции носителей заряда на большие
расстояния. В NaCl, например, наблюдается преимущественная
миграция катионов по катионным вакансиям к отрицательному
электроду, в результате чего на границе электрод NaCl воз-
возникает двойной электрический слой. При таком поведении
материала его правильнее рассматривать как твердый электро-
электролит, чем как диэлектрик. При измерениях па таких веществах
можно полупить кажущиеся диэлектрические проницаемости,
равные ~ 106—107 (что соответствует емкости двойного электри-
электрического слоя •-—-I0 Ф), но эти величины не характеризуют
собственно диэлектрических свойав.
Величины составляющих поляризуемости обычно убывают в
ряду as>ccd>a;>«.>, хотя понятно, что в различных материалах
могут и не наблюдаться все виды поляризуемости. Перечислен-
Перечисленные составляющие поляризуемости а и диэлектрической прони-
проницаемости е' могут быть экспериментально найдены из емкост-
емкостных, микроволновых и оптических измерений, выполненных в
широком интервале частот переменного электрического поля
(рис. 15.8). При низких частотах (~103 Гц) все четыре состав-
составляющие (если они имеются) могут давать свой вклад в велипи-
15.3. Диэлектрики
ну а. В области радиочастот (~ 106 Гц) объемный заряд не
успевает образоваться у большинства материалов с ионной про-
проводимостью и потому не влияет иа релаксационные процессы.
В микроволновом диапазоне частот (~Ю9 Гц) перестает быть
существенной дипольпая переориентация, так как диполи не
успевают следовать за изменением внешнего поля. Время релак-
релаксации для ионной поляризации таково, что этот вид поляриза-
поляризации исчезает па частотах больших, чем соответствующие иифра-
ость t
тельн
проница
IX
га
se
ГГ
t;
,о6
ю5
ю3
ю2
1
— N
л-?а>
1
«, • «d •
4—
«s
1
«е. а,
. J
¦
1
\. 1
1 1
103 ЮЬ 109 1012
Часюта f
Рис. !5.8. Поляризационные явления в диэлектрике.
красной области спектра (~ 1012 Гц). При дальнейшем повы-
повышении частоты остается лишь электронная поляризация,
которая наблюдается еще в УФ-области, по пропадает при
частотах, соответствующих рентгеновскому диапазону. В хоро-
хороших диэлектриках, не имеющих а6 и сс0-, проницаемость па
низкой частоте ео' определяется в основном ионной и электрон-
электронной поляризацией. Величину ео' можно получить из изме-
измерений емкости с помощью моста переменного тока. Для
этого емкость измеряют дважды —без изучаемого вещества
между пластинами конденсатора и с веществом [уравнение
A5.11)] Величину е»', связанную только с электронной поля-
поляризуемостью, можно найти кл измерений показателя преломле-
преломления в видимой области спектра па основании простого соотно-
соотношения
108
J5. Другие электрические свойства
Для NaCl величины е0' и е'оо составляют 5,62 и 2,32. Эти значе-
значения достаточно типичны для ионных кристаллов в целом. Для
получения детальной информации о диэлектриках необходимы
измерения в широком интервале частот — от низких частот до
микроволнового диапазона. Результаты этих измерений пред-
представляют в ииде диаграмм комплексной диэлектрической
Рис. 15.9. Угол 90° между фазой тока и фазой напряжения в диэлектрике
(а и б), диэлектрические потери при 6VO (в) и tg6=e'7e' (г).
проницаемости, называемых диаграммами Кол — Кола, или в
виде тангенса диэлектрических потерь tg6.
Рассмотрим реакцию диэлектрика на переменное поле. При
наложении на диэлектрик синусоидальной э. д. с, меняющейся
с низкой частотой, в нем на половине периода колебания могут
возникать различные виды поляризационных процессов. Эти
процессы эквивалентны переменному току и приводят к пере-
переносу энергии через диэлектрик. При низких частотах перемен-
переменный ток опережает напряжение по фазе на 90° (рис. 15.9, а и б),
что для минимизации диэлектрических потерь идеально, так как
вектор произведения i-V при фазовом сдвиге в 903 между током
15.3. Диэлектрики
109
и напряжением равен нулю; энергия передается через образец
без диэлектрических потерь, соответственно пе происходит и
выделения джоулева тепла. При увеличении частоты наступает,
наконец, такое состояние, когда переменное поле вызывает
ионную поляризацию (полагаем, что <zs и ad равны пулю).
В результате ток будет опережать напряжение на фазовый
угол меньший, чем 90° (т. е. на угол 90 —б). Поэтому у тока
появляется компонента i sin 6, лежащая в одной фазе с напря-
напряжением (рис. 15.9, в), что приводит к рассеянию энергии в виде
тепла, т. е. к диэлектрическим потерям.
Рис. 15.10. Частотная зависимость е' и г".
Частоты, при которых фазовый угол б значителен, соответ-
соответствуют той области рис, 15.8, в которой е' не является посто-
постоянной величиной, а меняется от ео' до е'«. В этой области при-
принято представлять проницаемость в виде комплексной величины
8* = 8'
-/в'
A5.15)
где е' — вещественная часть г*, равная измеренной диэлектри-
диэлектрической постоянной, г" —фактор потерь, являющийся мерой
проводимости или диэлектрических потерь в материале; отноше-
отношение е"/в' равно tg6 (рис. 15.9, г). Частотные зависимости е' и
г" показаны на рис. 15.10. Для г" эта зависимость имеет харак-
характерный вид с острым максимумом, носящим название дебаевско-
го пика. Максимум г" наблюдается на частоте, при которой
происходит наиболее резкое падение величины е'. Профиль
дебаевского пика определяется выражением
8 —(8о —е'°
шт
A5.1G)
по
15. Другие электрические свойства
где o) = 2nf и т-—характеристическое время релаксации или вре-
время спадания ионной поляризации; максимум е" наблюдается
при ит= I (т. е. при оо=т~') и составляет при этом ЧгМ—е'„).
Эти результаты могут быть представлен также на ком-
комплексной плоскости в виде диаграмм Кол — Кола, где е" откла-
откладывается как функция г' (рис .15.11). Отметим, что
е' = е'оо-| ,°+ ш,тг
Зависимость е" от е' образует полуокружность, точки пересече-
пересечения которой с осью е' соответствуют значениям е</ и е'«>.
Рис. 15.11. Диаграмма Кол— Кола для диэлектрика.
Диаграммы Кол — Кола, построенные на основе реальных
экспериментальных данных, не всегда имеют формы правиль-
правильных полуокружностей, а бывают в той или иной степени иска-
искажены. Аналогично пики диэлектрических потерь в" нередко бы-
бывают асимметрично уширенными, а не симметричными, как это
следует из формулы A5.16) для дебаевского пика. Такие
искажения пиков традиционно рассматривались как результат
суперпозиции ряда отдельных дебаевских пиков, максимумы
которых соответствуют различным частотам. В результате этого
возникает понятие распределения времен релаксации.
В последние годы справедливость такого подхода подвергает-
подвергается сомнению, преимущественно благодаря работам Иончера,
сформулировавшего так называемый «закон универсального
диэлектрического отклика». Уравнения, выведенные Йончером,
онпсывшот экспериментальные данные с удивительной точно-
точностью. В частности, для описания диэлектрических потерь в ма-
15 4. Сегистоэлектрики
териалах, обладающих заметной электропроводностью, предло-
предложено уравнение
*"«^cc(V) +(^j A5.17)
где о) — угловая частота, равная 2л/, озр — частота перескока
носителей тока, a «i и пг-— константы. В основе этого уравнения
лежит представление о том, что отдельные поляризационные
явления, будь то перескоки ионов в проводниках или переориен-
переориентация диполей в диэлектриках, происходят не независимо друг
от друга, а в результате кооперативного взаимодействия. Это
означает, что если какой то отдельный диполь в кристалле
переориентируется, то он тем самым оказывает влияние па
окружающие его диполи. На теперешнем уровне понимания,
однако, неясно, как на основании закона Йончера прийти к
количественному описанию кооперативных явлений. Более
детально диаграммы в комплексной плоскости обсуждаются
в гл. 13 (но при этом сделан акцепт на описание проводимости,
а пе диэлектрических свойств),
15.4. Сегнетоэлектрики
Сегпетоэлектрические материалы o^inqaiOTCH от обычных ди-
диэлектриков двумя качествами: во-первых, они обладают чрез-
чрезвычайно большой диэлектрической проницаемостью и, во вто-
вторых, возможностью сохранения некоторой остаточной электри-
электрической поляризации после устранения внешнего электрического
поля. При повышении разности потенциалов, приложенной к
обычному диэлектрику, наблюдается пропорциональное увели-
увеличение наведенной поляризации Р или наведенного заряда Q
[упавмепие A5.10)]. В сегнетоэлектриках пет такой простой
линейной взаимосвязи между поляризацией Р и напряжением
V, а вместо нее наблюдается гистерезиспое поведение (рис.
15.12), т. е. поляризация, возникающая при повышении напря-
напряжения, пе воспроизводится при последующем его снижении.
Сегнетоэлектрики характеризуются наличием поляризации на-
насыщения Ps при высоких электрических напряжениях (для
BaTiCb Я3 = 0,26 К л/см2 при 23 °С) и остаточной поляризации
Рп, т. е. поляризацией, сохраняющейся после устранения внеш-
внешнего электрического поля. Для того чтобы уменьшить поляриза-
поляризацию до пуля необходимо приложить электрическое поле Ес
обратного знака, называемое коэрцитивным полем.
Некоторые из сегпетоэлектрических материалов приведены
п табл. 15.2. Все они обладают структурами, в которых один
гип катионов, например Ли в BaTiO.i может значительно
ц-j
Iо. Др\гис электрические
Р, Кл/М2
Рис. 15.12. Петля гистерезиса для типичного сегпстоэлееткша.
линия, проходящая через начало коордпгпт. пок.зыяает коь.'депп"
диэлектрика.
юы°:-'.о*, г.
смещаться (~0Д Л) относительно своего aiMionwiro ок!<\;ш.чы.
Это смещение .чар идо», приводи г к во:;;шкпоие1:':о :;nno. :e;i к
большому значению диэлектрической пролниас^ионг, что ',;!рлк-
терло для сеглетоэлектриков.
На рис. E.15 показана элементарная ячейка тптанатп строн-
стронция SrTiOa, имеющего, как и. BaTiO^, структуру типа перовскита.
Попы 'итана занимают порлтпы зroi'i кубической прммигииной
ячейки, ионы кислоро.ча расположоны п центрах ребе;>, а иоп
стронция — в центре куба. Элементарную ячейку можно выбрать
п иным образом, поместив се першш'у н позицию, занимаемую
сгронцием. В таком случае титан расположится в центре куба,
а ионы кислорода займут центры гранен. Однако пне .чагшен-
mocik от выбора элементарной ячейки структура nociроена из
октаэдров TiOe, образующих трехмерный каркас путем сочле-
сочленения общими вершинами; ионы стронция занимают и этой
каркасной структуре пустоты с К.Ч 12.
15.4. О-тпстозлсктрпки
Описанная идеальная кубическая структура перовскита, ста-
стабильная для ВаТЮз выше 120°С, не обладает собственным
дкпольпым моментом, 'iax как все заряды расположены сим-
симметрично. Следовательно, „чаюрна.ч с такой структурой будет
пссти себя клк" нормальный диэлектрик. _\отя г с очень высокой
диэлектрической проницаемостью. Ниже 120 "С и структуре
ВаТЮ.ч возникают искажения, выражающиеся в том, что окта-
октаэдры TiOs перестают бшь симметричными — ионы титана сме-
смещаются },.; цешральиых позиции но направлению к одной нз
1 аилли-' 1 о 2. Т;-ммс]>атур.>1 Кюри некоторых
;.[).-•; Тс, "С
Tim;ii!3T барля I3aTiO3 1*?0
C,i\,, юса t-n.ii. KN.-iGlbCViI+.O От — ['< до +24
Н;.обл . млил К\1Ч): 434
Дипглро||>ос(|,ит i-;;i.--:i/i KII2PO- —ПО
Тн--;-.-.1.-г г свг1оча РЬТЮ3 -100
I!i uu:i: лггии; LiNbO3 1JI0
'1 i:i;ii-.-:-r a:!Oi;i ra Lji.Ti.jO12 (i7"i
Л1ол[;блат галолнн'.1я Ос12(МоО4)з 15Ч)
Ui:pKoir;.r-;ла:..:г о;п:ц;;. (ЦТС)
!i'.:,)f!iin: окта-мрл. Г.слн тйк.чс Ол!е;ис:п-:я гтронехидят р
iio во всех окга.-лтраА ТЮс. то н .материале ио;-;л'.кает сиГи : в е * г -
1;ая с:11лг;агд:а;1 по.!я;-л.;ацпя. В се:г е10>..ект()Нке BaTi(.): каж-
дик пч октаэдров TiO.; пол'лр1.':юБ<1и; влияние здешнего j.icvipH-
ческого лсли сводная к <-пр.ч:'\дщслык^Ь opireniaiihii uv..::;mi-
.;у<!льн!>:х диполе-''!. После ныеграяьа;:!-!;! и:ч;\ дн|;олеЛ рлоль
нигп;авлгп'.:;1 ~г:;у достигаете-' (О'.'тоя.чне по [ярн^апии H3''!t;'.p-
::ti-,t. !';-сетоя1-;1л\ иа лочорое смсньнотся помы ттана п.ч центров
;жч:1-:)дроп к о .чьим у и.; нлелоро ^ов, по оценкам, сделанным па
ocii^rio 'jKci;e:Hi\iCi'TaLibiio наблюдаемой величины Рх, госгаилясг
--П,i Л, '-то подтв;'рих'даеюя также данными penireiiocr:)yKiy;)-
пего аналнча. Как видно, лп [)асстояннс оказывается достаточ-
достаточно малым в сравнении со средней длиной связи Ti—О и окта-
-ui'iax TiOr., равной 1,95 А. Упорядоченная ориентация диполей
схематически изображена на рис. 15.13, а, где каждая стрелка
соответствует одному ik кажепиому октаэдру TiO0-.
В сегнею^локгрнках, подобных' BaTiO3, образуются домен-
доменные структуры благодаря тому, что соседствующие диполи TiOc
114
15. Другие электрические свойства
самопроизвольно выстраиваются параллельно друг другу
(рис 15.14). Размер образующихся доменов различен, но, как
правило, может достигать в поперечном сечении десятков —
сотен ангстрем. В пределах одного домена диполи поляризова-
поляризованы в одном кристаллографическом направлении. Собственная
по"дризация какого-нибудь образца сегнетоэлектрика равна
век.орной сумме поляризаций отдельных доменов.
а
i
j
А
J
t
\
/
\
/
\
/
\
Pik\ 15.13. Схематическое изображение ориентации диполей в сегнсто-
электрикс (а), аитисегнетоэлектрике (б) и сегнетиэлектрике (в).
рассматриваемого
все диполи в доме-
домена параллельную
Наложение внешнего электрического поля приводит к изме-
изменению собственной поляризации образца сегнетоэлектрика; при-
!шюй таких изменений могут быть следующие процессы:
1) изменение направления поляризации доменов. Это про-
изойдеч, если все диполи TiOB в пределах
домела сменят свою ориентацию; например,
не B) (рис. 15.14) изменяют ориентацию
диполям домена (Г);
2) возрастание поляризации в пределах каждого домека, что
особенно вероятно, если до наложения поля имелась некоторая
неупорядоченность в ориентации диполей;
3) движение ,'юменных стенок, в результате которого разме-
размеры доменов, ориентированных вдоль поля, увеличиваются за
счет уменьшения юменов с неблагоприятной ориентацией. На-
Например, домен / (рис. 15.14) может вырасти при сдвиге юмеп-
ной стенки на один шаг вправо. Для осуществления такого
сдвига диполи на границе домена 2 должны принять ориента-
ориентацию, показанную штриховыми стрелками.
Сегнетоэлектрическое состояние обычно наблюдается при
ни .ких температурах, поскольку тепловое движение, усиливаю-
усиливающееся с ростом температуры, нарушает согласованный харак-
характер смещения в соседних октаэд.рал, а следовательно, нарушает
15,4. Сегнетоэлектрики l_l?
и доменную структуру. Температура, при которой происходит
это разрушение, называется сегнетоэлектрической точкой Кюри
Тс (табл. 15.2). Выше Тс материалы становятся параэлектрика-
ми (т. е. «несегиетоэлектриками»), их диэлектрические прони-
проницаемости имеют по-прежнему высокие значения (рис. 15.15),
но остаточной поляризации в отсутствие внешнего поля уже не
1 t f t f t
i t f t
<D
\ 1 i t ! X / /
! t
Рис. 15.14. Сегнетоэлектрические домены, разделенные доменной стенкой
(границей).
наблюдается. Выше Тс величина е' обычно описывается законом
Кюри — Вейсса
е'-С/(Г—6)
где С — постоянная Кюри и 0—-температура Кюри—Вейсса.
Как правило, Тс и 0 совпадают или же различаются всего лишь
на несколько градусов. Переход из сегнетоэлектрического в
параэлектрическое состояние при Тс — пример фазовог.. пере-
перехода порядок - беспорядок. Однако в отличие от переходов
порядок — беспорядок, наблюдающихся, скажем, в брон::а>:. при
этом не происходит диффузионного смещения ионов на большие
расстояния. Ниже Тс упорядочение осуществля гея путем пре-
преимущественного искажения или согласованного наклона
потиэдров и относится, таким образом, к фазовым пере\одам
со смещением (гл, 12). В высокотемпературной параэлектриче-
.116
15. Другие электрические свойства
ской фазе искажения и наклон полиэдров, если и присутствуют,
то во всяком случае носят случайный характер.
Необходимое условие спонтанной поляризации и сегнето-
электрических свойств в кристалле состоит в том, что послед-
последний должен относиться к пространственной группе, не обладаю-
ег
шооо Ь
8000
€000
4000
2000
40
80
120
160
Рис. 15.15. Температурная зависимость диэлектрической проницаемости кера-
керамического ВаТЮз.
щей центром симметрии (гл. 6). Параэлектрические фазы,
устойчивые выше Тс, часто бывают центросимметричными, а
упорядочение, происходящее при охлаждении, сводится к пони-
понижению симметрии до нецентросимметричиой пространственной
группы.
В настоящее время известно несколько сотен сегнетоэлектри-
ческих материалов, среди которых выделяется большая группа
оксидных соединений со структурой искаженного (некубическо-
(некубического) перовскита. Эти соединения содержат такие катионы, кото-
которые «чувствуют себя» удобно в искаженном октаэдрическом
окружении —Ti, Ni, Та; неравноценность связей внутри таких
15,4. Сегнетоэлектрики
искаженных октаэдров МО6 является причиной возникновения
поляризации и дипольного момента. Далеко не все перовскиты—
сегнетоэлектрики, так, например, в противоположность ВаТЮз
и РЬТЮз СаТЮз не проявляет сегпетоэлектрических свойств,
что, по-видимому, связано с различием в размерах двухзаряд-
ных катионов. Большой радиус иона Ва2+ вызывает расширение
элементарной ячейки по сравнению с СаТЮ3, что в свою оче-
очередь приводит к большим длинам связей Ti—О в ВаТЮ3 и
большему смещению ионов Ti4+ внутри октаэдров ТЮб. В состав
других оксидов с сегнетоэлектрическими свойствами входят
катионы, связи которых с ионами кислорода неравноценны
из-за наличия свободной электронной пары на внешней обо-
оболочке; это могут быть катионы тяжелых р-элемеытов, отвечаю-
отвечающие степеням окисления, на две единицы меньшим, чем предель-
предельная для данной группы, такие, как Sn2i', Pb2+, Bi3+ и т. д.
Сегнетозлектрические оксиды используются для изготовле-
изготовления конденсаторов благодаря высокой диэлектрической прони-
проницаемости, которая особенно велика вблизи Тс (рис. 15.15).
Поэтому, преследуя практическую цель увеличить г', следует
создавать материалы с точками Кюри, близкими к комнатной
температуре. В частности, температура Кюри, составляющая
для ВаТЮз 120 °С (рис. 15.15), может быть значительно сни-
снижена, а температурный интервал перехода расширен путем
•частичного замещения Ва2+ или Ti4+ другими катионами: заме-
замещение Ва2+ на Sr2+ вызывает сжатие элементарной ячейки
структуры и уменьшение 7с; замещение «активных» ТИ^-ионов
другими «неактивными» четырехзарядными ионами, в частности
Zr4+ и Sn4+, приводит к резкому падению Тс.
В антисегнетоэлектриках также наблюдается спонтанная по-
поляризация, сходная по природе с поляризацией сегиетоэлектри-
ков. Индивидуальные диполи антисегнетоэлектриков упорядочи-
упорядочиваются относительно друг друга таким образом, что каждый
диполь оказывается антипараллельным соседним диполям
{рис. 15.13, б). В результате собственная спонтанная поляри-
поляризация материала оказывается равной нулю. Выше антисегне-
тозлектрической точки Кюри материал становится нормальным
параэлектриком. Цирконат свинца PbZrO3 B33°С), ниобат нат-
натрия NaNbO3 F38°С) и дигидрофосфат аммония NH4H2PO4
(—125°С) представляют собой примеры веществ с антисегпето-
электрическими свойствами (цифры в скобках означают соот-
соответствующие точки Кюри).
Электрические свойства антисегнетоэлектриков и сегнето-
электриков значительно различаются. Так как в антисегнето-
электрическом состоянии спонтанная поляризация отсутствует,
то не наблюдается и гистерезисных явлений (петли гистерезиса)
в переменном электрическом поле, хотя для этих материалов
118
15. Другие электрические свойства
также характерно сильное повышение диэлектрической прони-
проницаемости вблизи Тс (для PbZrO3 е'^100 при 200°С и
л;3000 при 230°С). Иногда антипараллельное упорядоченное
расположение диполей в антисегнетоэлектриках и параллель-
параллельное в сегнетоэлектриках оказываются весьма близкими по
устойчивости. В таких случаях небольшие изменения внешних
условий могут приводить к фазовому переходу, например под
30
20 -
10 -
сегнетодлсктрик
ачтисегнегоэлектрик \ параэгектрик
220
225
230
235
I С
Рис. 15.16. Зависимость температуры перехода антисегнетоэлектрик — сегне-
тоэлектрик в PbZrCb от приложенного напряжения {а) и поведение поля-
поляризации при этом переходе (б).
действием внешнего электрического поля PbZrO3 переходит из
гштисегнетоэлектрического в сегнетоэлектрическое состояние
(рис. 15.16, а); электрическое напряжение, вызывающее пере-
переход, зависит от температуры. Соответствующее этому случаю
поведение поляризации во внешнем поле показано па рис.
15.16, б: при низких напряжениях гистерезис отсутствует, так
как PbZrCb в этих условиях — антисегнетоэлектрик, а при более
сильных полях (обоих направлений) PbZrCb переходит в сегне-
сегнетоэлектрическое состояние и возникает петля гистерезиса.
Известен также такой вид поляризационных явлений, когда
ангисегиетоэлектрическая структура возникает лишь в каком-то
одном направлении (или направлениях). Доменная структура
такого типа показана па рис. 15.13, в: собственная поляризация
равна нулю в направлении л', как у аптнсегиетоэлектрика, но не
равна нулю в направлении г. Материалы, обладающие такой
структурой, называются сегнетиэлектриками; к ним относятся,
например, Bi4Ti3Oi2 и моногидрат двойного тартрата лития и
аммония.
Важным фактором, определяющим сегнето- и антисегнето-
электрические свойства некоторых веществ, является наличие
1Г).4. Ссгпетоэлектрики 119
в них водородной связи. Рассмотрим это Eia примерах сегнето-
элекгрнка КН2РО4 (рис. 15.17, «) и антисегпетоэлектрика
NH4H2PO4 (рис 15.17, б), основное структурное различие кото-
которых заключается в положении атомов водорода, образующих
водородные связи. Структура и того и другого соединения по-
построена из изолированных тетраэдров РО4, соединенных ионами
К1 или NH<r (эти ионы на рисунках не показаны) и водород-
водородными связями между атомами кислорода, принадлежащими
соседним тетраэдрам. Оба соединения имеют большие элемен-
элементарные ячейки ромбического типа, фрагменты которых показа-
показаны на рис. 15.17, а и б, в проекции па плоскость, перпендику-
перпендикулярную оси с. В таком ракурсе тетраэдры РО4 видны со стороны
ребер и выглядят как квадраты. Атомы кислорода расположены
в углах этих квадратов, а диагонали соответствуют ребру тетра-
тетраэдра, соединяющему два атома кислорода и обращенному вверх.
Расположение тетраэдров относительно плоскости рисунка по-
казапо на рис. 15.17, а с помощью соответствующих цифр при
атомах фосфора; для структуры NH4H2PO4 (рис. 15.17, б) все
изображенные атомы находятся в плоскости рисунка. Вдоль
направления с упаковка тетраэдров такова, что верхнее ребро
(например, XX') одного тетраэдра находится примерно на оди-
одинаковой высоте с нижними ребрами {YY') двух соседних тетра-
тетраэдров. Каждый тетраэдр РО4 образует с соседними тетраэдрами
четыре водородные связи, отличающиеся несимметричным поло-
положением водорода в них: атомы водорода находятся ближе к од-
одному из связываемых тетраэдров, чем к другому. На каждый
тетраэдр приходится по два близко расположенных и по два
ба-'ее удаленных атома водорода. В высокотемпературных нара-
мск'трических формах КН2РО4 и NH4H2PO4 взаимное располо-
расположение «дальних» и «ближних» атомов водорода в каждом
тетраэдре хаотично и структура разупорядочена. В низкотем-
низкотемпературной форме КН^РС^, обладающей сегпетоэлектрическнмн
свойствами, атомы водорода самопроизвольно упорядочиваются
тамгм образом, что оба «ближних» атома становятся связанны-
связанными с верхним ребром каждого тетраэдра. Атомы фосфора внут-
внутри тетраэдров смещаются при этом вниз подальше от атомов
водорода, что приводит к образованию диполей,ориентированных
вдоль оси с. Таким образом атомы водорода косвенно ответст-
ответственны за возникновение спонтанной поляризации. Для того
чтобь- перевернуть направление диполя, совсем необязательно
поворачивать весь тефаэдр; для достижения такого же эффек-
эффекта достаточно простого сдвига атомов Н вдоль водородных
связей. Два водорода, расположенные близко к верхним кис-
лородам тетраэдра (рис. 15.17, а), сметаются от них по гори-
горизонтали и примыкают к нижним атомам кислорода соседних
тетраэдров. Одновременно к нижним атомам кислорода дан-
120
15. Другие электрические свойств.;
ного тетраэдра приближаются два других атома водо;;ода.
В итоге такого перемещения атомов водорода в плоскости, пер-
перпендикулярной оси с, происходит поворот диполей, параллель-
параллельных с, на 1803.
Расположение атомов водорода при каждом тетраэдре анти-
сегнетоэлектрической фазы NH4H2PO4 иное (рис. 15.17, б):
15.5. Пироэлектрики
121
группы Р ОН
Рис. 15-17. Структуры сегнетоэлектрика КН2РО4 (о) и антисегнетоэлектрика
NH4II2PO4 (б) [проекция на плоскость @01)].
один из двух «ближних» атомов водорода присоединяется к
верхнему атому кислорода, а второй — к нижнему, ".то приводит
к возникновению диполей, направленных перпендикулярно оси
с [т. е. лежащих в плоскости @01)]. Эти направления па
рис. 15.17, б обозначены стрелками; видно, что суммарная по-
поляризация всего кристалла равна нулю.
35,5. Пироэлектрики
Пироэлектрические кристаллы родственны сегнетоэлектри-
кам в том отношении, что у них отсутствует центр симметрии и
возникает спонтанная поляризация Р«. Однако от сегпстоэдект-
риков их отличает то, что направление Р, не может бить изме-
122 15. Другие электрические свойства
нено внешним электрическим полем. Поляризация пироэлектри-
ков зависит от температуры
APs = nAT A5.18)
где л.— пироэлектрический коэффициент. Возникновение пиро-
пироэлектрической поляризации объясняется тем, что при нагрева-
нагревании происходит расширение кристаллической решетки и при
этом меняются длины диполей. Типичный пример пироэлектри-
пироэлектрических кристаллов — оксид цинка ZnO со структурой вюртцита.
Это соединение состоит из слоев
ионов О2~ с гексагональной плот-
пейшей упаковкой и ионов Zn2+, за-
занимающих один тип тстраэдричег-
ких позиций, например Т+-тип (гл.
7). Все тетраэдры ZnO4 ориентиро-
ориентированы в одном направлении, а так
как каждый из них обладает ди-
польным моментом, то кристалл в
целом оказывается поляризован-
„ vp. ,„ _ н:>гм. На противоположных гранях
И' %о8сфор^МТНтеСтраэдР°ё "P"™"a ZnO С индексом @01)
РО2(ОНJ, приводящее к воз- должны находиться только ионы
пикиовению спонтанной поля- О2" или Zn2+. Однако обычно па
ризации. поверхностях кристалла адсорби-
адсорбируются полярные молекулы при-
примесей, маскирующие поверхностные заряды. В результате пи-
пироэлектрический эффект в кристаллах часто не обнаружива-
обнаруживается в исследованиях при постоянной температуре, а прояв-
проявляется только при нагревании, приводящем к изменению Р,..
15.6. Пьезоэлекгрики
В пьезоэлектрических кристаллах поляризация и электричес-
электрический заряд противоположных граней возникают под действием
приложенного механического напряжения. Как и в случае сег-
пето-и пироэлекгриков, для возникновения пьезоэлектрических
свойств, необходимо, чтобы кристалл относился к одной из
ноцентросимметричных точечных групп. Появление пьезоэлект-
пьезоэлектрического эффекта определяется кроме кристаллической струк-
структуры материала еще и направлением приложенного напряже-
напряжения. Так, например, в кварце поляризация возникает при
сжатии вдоль направления [100] и отсутствует, если механиче-
механическое напряжение направлено вдоль [00!]. Возникающая поля-
поляризация Р и напряжение а связаны соотношением
P^do A5.19)
где d—пьезоэлектрический коэффициент.
15.7. Сегнето-, гтьезо- и пироэлектрические свойства
123
Пьезоэлектрическими свойствами обладают многие кристал-
кристаллы, структура которых содержит тетраэдрические фрагменты
(например, ZnO, ZnS), так как приложение сдвиговых напря-
напряжений искажает тетраэдры. Одну из наиболее важных групп
пьезоэлектриков образуют материалы типа ЦТС, представляю-
представляющие собой твердые растворы циркоиата и титаната свинца
кубический перовсквш (параэлемрик:
100 I
— 00 I
ромбоэдрическая
!0О j_ —i— зшисегнетсэлемрическоя
\
тетрагональная
qjjs3 ; ее г hi', гоэлекгрик),
PbZrO,
0,2
0,4 U -JT) 0,6
0,8
PbTiOj
Рис. 15.19. Фазовая диаграмма системы ЦТС [4].
и РЬТЮз). Как видно из фазовой диаграммы этой
системы (рис. 15.19), при некоторых составах твердые растворы
ЦТС обладают также сегнето- и антисегнетоэлектрическими
свойствами. Наилучшие пьезоэлектрические свойства достига-
достигаются при соотношении компонентов 1 : 1.
15.7. Взаимосвязь между сегнето-, пьезо-
и пироэлектрическими свойствами
Сегнето-, пьезо- и пироэлектрические свойства обусловлены
возникновением в некоторых кристаллах поляризованных обла-
областей, и совершенно очевидно, что между материалами, прояв-
проявляющими эти свойства, должно быть много общего. Все эти
материалы относятся к классу диэлектриков, на их свойства,
124 15. Другие электрические свойств»
в первую очередь электрические, влияет электрическое поле.
Пьезоэлектрики составляют подкласс диэлектриков; при
механических напряжениях в них возникает электрический за-
заряд, а в электрическом ноле в них возникают механические
напряжения. Пироэлектрики образуют подкласс пьезоэлектри-
ков; в этих материалах имеется спонтанная поляризация к
суммарный дипольный момент. Некоторые пироэлектрики обла-
обладают и сегнетоэлектрическими свойствами; это проявляется, и
частности, в том, что направление спонтанной поляризации в
них можно развернуть в противоположную сторону, воздейст-
воздействуя внешним электрическим полем. Следовательно, по опреде-
определению сегнетоэлектричеекие материалы являются также и
пиро- и пьезоэлектрнками. Обратное утверждение однако невер-
неверно, т. е. далеко не все пьезоэлектрики могут проявлять пиро-
пироэлектрические свойства и т. д.
15.8. Применение сегнето-, пьезо- и пироэлектриков
Естественно обсуждать применение этих материалов в одном
разделе, поскольку, во-первых, часто возможна взаимная заме-
замена этих материалов и, во-вторых, в некоторых случаях необхо-
необходимо, чтобы материал обладал не одним, а одновременно
двумя или даже всеми тремя перечисленными свойствами. Воз-
Возможности применения этих материалов чрезвычайно разнооб-
разнообразны, в особенности для сегнетоэлектриков, и потому здесь
ограничимся лишь кратким перечнем наименее экзотических
приложений, порекомендовав читателю, проявившему интерес
к этому вопросу, познакомиться с прекрасной и исчерпывающе
полной на сегодняшний день книгой Барфута и Тейлора [1].
Основная сфера промышленного применения сегнетоэлектри-
сегнетоэлектриков— это конденсаторы. Поскольку у этих материалов диэлект-
диэлектрическая проницаемость г' высока (как правило, в интервале
102—104), то их можно использовать для создания конденсато-
конденсаторов с большой емкостью [см. уравнения A5.9) и A5.Л)]. Для
этих целен используются в основном BaTiOrj и ЦТС (твердый
раствор цнрконата и титаната свинца) в виде плотной поликри-
поликристаллической керамики. Для сравнения заметим, что е' таких
общепринятых диэлектриков, как ТЮг или MgTiO3, находится
в пределах от 10 до 100. Следовательно, при одинаковом объеме
конденсатор из ВаТЮз обладает емкостью, в 10—1000 раз пре-
превышающей емкость диэлектрического конденсатора.
Важную область применения некоторых сегнетоэлектриков,
в частности ВаТЮз и РЬТЮ^, вообще говоря, непосредственно
не связанную с сегнетоэлектрическими свойствами, составляет
15.8. Применение сегнето-, пьезо- и пироэлектриков
125
изготовление так называемых позисторов (термочувствительных
резисторов с положительным температурным коэффициентом).
Известно, что удельное электросопротивление р большинства
неметаллических материалов уменьшается с ростом темпера-
температуры, т. е. сопротивление имеет отрицательный температурный
BaTiO3,легированный Mnz
10'
о Ю5
10*
10'
10
BaTlOj,легированный Fe2
BaTiO3
100
200
500 "С
Рис. 15.20. Тсмггературнля зависимость удельного сопротивления керамиче-
керамического полупроводник.! ВаТЮз, легированного различными доблвкамп [5].
коэффициент. В то же время в некоторых сегнетоэлектриках,
в том числе ВаТЮз, происходит аномальное увеличение сопро-
сопротивления, когда температура приближается к точке перехода
сегнетоэлектрпк — параэлектрик Тс (рис. 15.20). Рост сопро-
сопротивления происходи! одновременно с ростом диэлектрической
проницаемости р/; однако причина возрастания р вблизи Тс>
по-видимому, пока еще по-настоящему не раскрыта. Позисто-
ры используются в качестве выключателей; при прохождении
тока по сопротивлению последнее разогревается за счет выде-
выделения джоулева тепла, которое пропорционально PR. Сопро-
Сопротивление термнггора на основе ВаТЮ,< при его нагревании
возрастает чрезвычайно сильно, в результате ток резко падает.
Это находит применение в ограничителях температур и устрой-
126 ^ 15. Другие электрические свойства
ствах для предупреждения токовых перегрузок (позистор играет
здесь роль плавкого предохранителя многократного пользова-
пользования), а также в выключателях временной задержки. Описание
термисторов с отрицательным коэффициентом сопротивления,
например на базе полупроводников МпО и NiO, см. в гл. 14.
Пироэлектрические кристаллы используются преимуществен-
преимущественно в инфракрасных детекторах излучения. При необходимости
им можно придать спектральную чувствительность, покрывая
рабочую поверхность кристалла материалом, поглощающим из-
излучение в нужной спектральной области. Для детекторов жела-
желательно иметь как можно большее отношение зх/е', а это означа-
означает, что сегнетоэлектрнки, обладающие большой диэлектрической
проницаемостью, для этой цели не подходят. Наилучшим мате-
материалом для детекторов, найденным к настоящему времени,
является триглиципсульфат.
Пьезоэлектрические кристаллы используются уже много лет
как преобразователи механической энергии в электрическую и
наоборот. Технические приложения ихт чрезвычайно разнообраз-
разнообразны- микрофоны и наушники, динамики и стереопроигрыватели,
предохранители, системы зажигания в двигателях и бытовые
зажигалки, звуковые генераторы, приборы ультразвуковой очи-
очистки поверхностей, а также более сложные устройства-—преоб-
устройства-—преобразователи, фильтры и осцилляторы. В большинстве перечислен-
перечисленных случаев используют ЦТС-керамику, кварц, сегнетову соль
К\гаС4Нг,О6-4Н2О или Li2SO4-H2O.
Упражнения
15.1. Назовите два экспериментальных метода, с помощью которых мож-
можно различить дырочную и электронную проводимость в полупроводниках.
15.2. Постройте температурную зависимость э. д. с. термопары железо —
константан в интервале ЮО-г-500 °С.
15.3. В чем состоит различие между параэлектриками (т. е. диэлектри-
диэлектриками). 1:егпетоэлсктриками, сегнетпэлектриками и антисегнетоэлектриками?
15.4. Каким должен быть порядок величины кажущейся диэлектрической
проницаемости: а) газообразного ар гоня: б) поды; в) льда; г) монокрис-
монокристаллов чистого кремния: д) монокристаллов чистого КВг. е) монокрнстпл-
лов КВг. легированного СаВг2; ж) Na ^-глинозема; з) BaTiO-j?
1Г>.Г>. Какие различия должны наблюдаться ii температурных зависимо-
зависимостях' проводимости СаТЮ3 и PbTiOn?
15.G. Кристаллы каких из перечисленных веществ могут обладать пьезо-
пьезоэлектрическими свойствами: а) NaCI; б) C;iF?; и) CsCI; г) ZnS (вюртцкт);
д) NiAs: e) TiO2 (рутил)?
15.7. Справедливо ли утверждение, что ппроэлектрпкп—это вешестна, в
которых при п.иренанин возникает спонтанная поляризация?
1">8. Что тако.^ диэлектрические потери, чем они вызваны и как можно
-14 ; ¦•.eir-.LPUTb 1! материалу, которой предполагается использовать в к<гге-
v I ik' ¦•),!.¦;;п.юизоля1! ора?
Литература 127
Литература
1. Burjoot I С. Taylor G. \V., Polar Dielectrics and Their Applications, Macmil-
lan, 1979.
2. ionschar A. K-, Dielectric Relaxation in Solid, Chelsea Dielectrics Press, Lon-
London 1983.
3. Newnkam R. ?., Structure — Property Relations. Springer-Vctiag, 1975.
4. Sawaguchi ?., J. Phys. Japan, 8, G15 A953),
5. Uaoka H., Ferroclectrics, 7C), 351 A974).
Дополнительная литература. Лайнс М,, Гласе Л. Сегнетоэлектрики и род-
родственные им материалы, — М.: Мир, 1981; Кенциг В. Сегнетоэлектрики и аити-
сегнетоэлектрики.—М.: ИЛ, 19G0: Окадзаки К. Пособие по электротехниче-
электротехническим материалам: Пер. с япон. — М.: Энергия, 1979; Богородицкий Н П., Па-
Пасынков В. В., Тареев Б. М. Электротехнические материалы: Для студентов ву-
вузов.— Л.: Энергоатомиздат, 1985; Майофис И М. Химия диэлектриков. — М,:
Высшая школа, 1970; Фесенко Е, Г. Семейство перовскита и сегнетоэлектриче-
ство. — М: Атомиздат, 1972; Оксидные материалы в электронной технике-
(Сборник статей), серия «Химия», № G, ;Ч.: Знание, 1983.
Глава 16
МАГНИТНЫЕ
СВОЙСТВА
16.1. Введение
Помимо диамагнетизма, который присущ всем веществам,
некоторые неорганические твердые тела проявляют особые маг-
магнитные свойства, связанные с наличием в них псспаренных элек-
электронов. Эти неспаренпые электроны обычно локализованы на
катионах металлов. Поэтому те магнитные свойства, о которых
пойдет речь в настоящей главе, характерны главным образом
для соединений переходных металлов лантаноидов, так как
многие из них имеют иеспаренные d- и /-электроны. Магнитные
материалы можно классифицировать следующим образом.
Спины неспаренпых электронов различных атомов мо]ут
быть ориентированы случайным образом. Такие материалы на-
называются парамагнетиками. Если почти все спины иеспаренных
электронов выстраиваются параллельно, то материал характе-
характеризуется общим магнитным моментом. Такой материал называ-
называют ферромагнетиком. Возможен и противоположный случай- ¦
антипараллельное упорядочение спинов неспаренных электро-
электронов. Общий магнитный момент при этом равен нулю. Такие ве-
вещества называются антиферромагнетиками, Если спины элек-
электронов антипараллельны, по количество спинов, ориентирован-
ориентированных в противоположных направлениях, разнос, то общий маг-
магнитный момент отличен от нуля. Эти вещества называют ферри-
магнетиками.
Ил сказанного ясно, что существует близкая аналогия между
описанными магнитными свойствами и соответствующими элек-
электрическими свойствами, в частности сстиетоэлектрическими
свойствами (гл. 15). Единственное различие состоит в отсутст-
отсутствии у магнитных материалов единичного магнитного заряда (мо-
нополи), который можно было бы поставить в соответствие еди-
единичному электрическому заряду, т. е. заряду иона или элек-
электрона.
16.2. Теория магнетизма 129
Магнитные материалы на основе оксидов, например ферриты
(MgFe^Oii и др.). находят в настоящее время широкое примене-
применение при изютовлепии сердечников трансформаторов, устройств
для магнитной записи и хранения информации и т. п. Теория
магнетизма, к сожалению, довольно сложна. В ней используется
большое число терминов, символов, единиц измерений, в кото-
которых легко запутаться. Ситуация усугубляется тем, что на прак-
практике используют два разных (и к тому же противоречащих друг
другу) способа оценки магнитных моментов ионов, имеющих не-
спаренпые электроны. В настоящей главе изложен самый необ-
необходимый минимум теоретических положений, чтобы имелась
возможность представить себе различные типы магнитных мате-
материалов и выявить связи с кристаллической структурой веществ.
16.2. Теория магнетизма
16.2.1, Поведение веществ в магнитном поле
Прежде всего рассмотрим, как ведут себя различные материа-
материалы в магнитном ноле. Если вещество помещается в магнитное
ноле напряженностью Н, то плотность силовых линий в образце,
называемая магнитной индукцией, равна сумме напряженности
поля Н и некоторого вклада самого образца
В=*Н+4я1 A6.1)
где / — магнитный момент образца единичного объема. Маг-
Магнитная проницаемость Р и магнитная восприимчивость -л опре-
определяются следующими соотношениями:
с A6.2)
A6.3)
Мольную магнитную восприимчивость % можно представить
в виде
A6.4)
где Г — молярная масса образца, а А — ого плотность.
Различные типы магнитных материалов отличаются друч от
друга значениями Р, у., % и их зависимостью от температуры и
напряженности поля (табл. 16.1).
Вещества, характеризующиеся малыми отрицательными зна-
значениями у, и у. и Р<1, называются диамагнитными. Парамаг-
Парамагнитные вещества имеют противоположные значения этих пара-
параметров (Р>-1, у., х положительные). Если вещество поместить
в магнитное поле, то число силовых линий, проходящих через
вещество, будет больше (в случае парамагнетика) или несколь-
несколько меньше (в случае диамагнетика) числа силовых линий, кого-
9—H2G
130
16. Магнитные свойства
Таблица 16.1. Магнитная восприимчивость различных веществ
Тип вещества
Типичное Изменение X с ро Зависимость or
значение % сю» температуры напряженности поля
Диа магнетики
Парамагнетики
Ферромагнетики
Антиферромагнетики
— МО"» Нет Нет
0—10~2 Уменьшение »
10—10й » Зависит
0—10~2 Увеличение (Зависит)
рые проходили бы в вакууме (рис. 16.1). Следовательно, пара-
парамагнитные вещества притягиваются магнитным полем, а диамаг-
диамагнитные испытывают слабое отталкивание.
Для ферромагнитных веществ характерны большие значе-
значения х н %, а Р^§>1. Такие материалы сильно притягиваются маг-
магнитным полем. У анти-
антиферромагнетиков Р>1.
к и 7 также положитель-
положительные величины, причем х
и у сравнимы по величи-
величине со значениями для па-
парамагнитных веществ.
Рис. 16.1. Поведение диашгнитргых (а) и
парамагнитных (б) веществ в магнитном
поле.
16.2.2. Влияние
температуры.
Законы Кюри
и Кюри — Вейсса
Магнитная восприим-
восприимчивость различных типов
магнитных материалов
различается температу-
температурной зависимостью, а
также своими абсолют-
абсолютными значениями. Для
многих парамагнитных веществ выполняется простой закон
Кюри (особенно в области высоких температур), согласно ко-
которому магнитная восприимчивость обратно пропорциональна
температуре
У, = С/Т A6.5)
где С — постоянная Кюри. Однако часто лучшее согласие с экс-
экспериментальными данными даст закон Кюри-Вейсса
A6.6)
16.2. Теория магнетизма
131
где 0—константа Вейсса. На рис. 16.2 приведены два типа за-
зависимостей магнитной восприимчивости в координатах y~'=
=f(T).
Для ферро- и антиферромагнитных веществ температурная
зависимость % не удовлетворяет законам Кюри и Кюри — Вейс-
Вейсса (рис. 16.3). Ферромагнитные материалы обладают очень
большой магнитной восприимчивостью при низких температу-
температурах, которая уменьшается все более резко по мере роста тем-
лон - С
в Т.К.
Рис. 16,2. Зависимость -^~l=f(T), отпечающпе законам Кюри и Кюри —
Всйссл.
пературы (рис. 16.3,6). Выше некоторой температуры (называе-
(называемой температурой Кюри Тс) материал теряет ферромагнитные
свойства и превращается в парамагнетик, для которого справед-
справедлив закон Кюри—Вейсса. Магнитная восприимчивость анти-
антиферромагнетиков (рис. 16.3, в) возрастает с ростом температу-
температуры. Температура, при которой магнитная восприимчивость при-
принимает максимальное значение, называется температурой
Нселя 7\. Выше 7"v материал превращается в парамагнетик.
Величины х различных материалов и их зависимость от тем-
температуры можно объяснить следующим образом.
Положительные значения магнитной восприимчивости пара-
парамагнитных материалов х объясняются тем, что в веществе име-
имеются нсспаренные электроны, причем существует тенденция
к выстраиванию их спинов в направлении магнитного поля.
В ферромагнитных материалах спины электронов выстроены
параллельно из-за кооперативного взаимодействия спинов со-
соседних попов в кристаллической решетке. Большие величины %
132
16. Магнитные свойства
парамагнетики
парамагнетики
I
парамагнетики
антиферромагнегики
Рис. 16.3. Температурная зч-
висимость jM.TrHHTHoii воспри-
восприимчивости парамагнитных (а),
ферромагнитных {б) и анти-
ферромагннтны.х (в) псщсств.
объясняются именно этим парал-
параллельным расположением многих
спинов. Вообще говоря, не все спи-
спины в данном веществе параллель-
параллельны, даже если вещество находится
в сильном магнитном поле при низ-
низкой температуре. В антиферромаг-
антиферромагнитных материалах электронные
спины выстраиваются антипарал-
лельно и взаимно компенсируются,
Поэтому для этих материалов мож-
можно ожидать небольших величин %.
Остаточное значение %, видимо,
связано с разупорядоченпем в рас-
расположении спинов.
В любых материалах рост тем-
температуры приводит к увеличению
тепловой энергии ионов и электро-
электронов. Поэтому, естественно, сущест-
существует тенденция к увеличению
структурного разупорядочения с
повышением температуры. В пара-
парамагнитных материалах тепловая
энергия электронов и ионов спо-
способствует частичной компенсации
упорядочения, возникающего под
действием внешнего магнитного
поля. Действительно, как только
внешнее магнитное поле исчезает,
ориентация электронных спинов-
становится беспорядочной. Следо-
Следовательно, в парамагнитных матери-
материалах магнитная восприимчивое! >>
X уменьшается с ростом температу-
температуры по закону Кюри или по закону
Кюри-— Вейсса.
аптиферромагнитных материалах влияние тем-
к увеличению разупорядоченности в идеаль-
или антипараллельном распо-
В ферро- и
пературы сводится
но упорядоченном параллельном
ложении спинов. В случае ферромагнитных материалов это ве-
ведет к быстрому понижению % с ростом температуры, В антифер-
антиферромагнетиках это приводит к уменьшению степени антипарал-
лелыюго упорядочения, т. е. к увеличению числа «разупорядо-
ченных» электронных спинов и, следовательно, к росту -/•
Часто магнитные свойства материалов удобно характеризо-
характеризовать с помощью магнитного момента ц, так как этот параметр
16.2. Теория магнетизма 133
непосредственно связан с числом неспаренных электронов. За-
Зависимость между /_ИA имеет следующий вид:
Х-Лфу/3/еТ A6.7)
где Л'—число Авогадро, р — магнето и Бора, к — постоянная
Больцмана. Подставляя вместо Л', ) и k их численные значения,
получим
jjt = 2,83Vx7i A6.8)
Магнитную восприимчивость и магнитный момент веществ
часто определяют экспериментально с помощью магнитных ве-
весов Гуи. Образец помещают между полюсами электромагнита
и измеряют массу образца в зависимости от напряженности при-
приложенного поля. В парамагнитных веществах неспарспные элек-
электроны притягиваются магнитным полем, что регистрируется по
возрастанию массы образца при отключении электромагнита.
Коррекцию полученного значения магнитной восприимчивости
ведут с учетом различных факторов, в частности диамагнетизма
образца и держателя образца.
16.2.3. Расчет величины магнитного момент и
Опишем подход, используемый обычно спектроскопистами и
химиками при расчете величины магнитного момента ионов, ко-
которые имеют песпаренные электроны. Считается, что имеются
две основные причины возникновения магнитного момента: спнн
электрона и орбиталыше движение электронов. Наибольший
вклад в суммарный магнитный момент вносит спин электрона.
Грубо можно представить электрон как точечный отрицатель-
отрицательный заряд, вращающийся вокруг своей оси, Величина спинового
момента ц,- в такой модели составляет 1,73 магнетона Бора
(|хв), где
liiB = eft/4 лше A6.9)
где е — заряд электрона, h — постоянная Планка, пг — масса
электрона, с — скорость света. Для расчета спинового момента
одного электрона используют формулу
где s=lJ2 — спиновое квантовое число, g^2,00 — гиромагнитное
отношение. Подставляя численные значения s и g, получаем для
спинового магнитного момента электрона |л<г=1,73 р-в-
Для атомов или ионов, у которых число неспаренных элек-
электронов больше единицы, общий спиноьой момент равен
) A6.11)
где 5 — сумма спиновых квантовых чисел отдельных неспарен-
134 16. Магнитные свойства
ных электронов. Так, для высокоспинового иона Fc3+, содержа-
содержащего пять неспаренных электронов, 5 = 5/2, a jis —5,92ц.В-
В табл. 16.2 приведены значения jis для ионов с разным числом
неспаренных электронов.
В некоторых веществах движение электронов вокруг ядра
приводит к возникновению орбитального момента, который вно-
Таблица 16.2. Экспериментальные и теоретические значения
магнитных моментов попов некоторых переходных металлов [1]
Ион электронов "s (patci.) M-5_, (pai-сч.) ц (эксп )
V'1+ 1 1,73 3.00 ~l,8
V!+ 2 2,83 4,47 -2,8
Cr3+ 3 3,87 5,20 ~3,8
,Mn2+ 5 (высокосшшовое состояние) 5,92 5,92 ~5,9
Fc'+ 4 (высокоспиновое состояние) 5,92 5,92 —/5,9
Fe2+ 4 (высокоспиновое состояние) 4,90 5,48 5,1-—5,5
Co3+ 3 (иысокоспиновое состояние) 4,90 5,48 ~5,4
Со2+ 3 (аысокоспиновое состояние) 3,87 5,20 4,1-—5,2
Ni2+ 2 2,83 4,47 2,8—4,0
Cu2+ I 1 73 3,00 1,7—2,2
сит вклад в полный магнитный момент. Для таких веществ пол-
полный магнитный момент равен
(J6.12)
где /, — орбитальное квантовое число иона. Уравнения A6.10) —
A6.12) применимы для описания магнитного момента изолиро-
изолированных атомов или ионов. К твердым веществам уравнение
A6.12) неприменимо, так как орбитальный момент либо частич-
частично, либо полиостью подавляется. Это происходит из-за того, что
электрическое поле, создаваемое окружающими атомами или
ионами, ограничивает орбитальное движение электронов.
Экспериментально определяемые значения магнитных момен-
моментов многих иолов приведены в табл. 16.2 наряду со значениями,
рассчитанными по уравнениям A6.11) и A6.12). В большинстве
случаев экспериментально найденные значения магнитных мо-
моментов примерно равны или несколько больше величин, рассчи-
рассчитанных исходя из существования лишь спинового момента.
Описанные в общих чертах методы расчета магнитных мо-
менюь основываются па кваптовомехапическом подходе. Де-
та.чыюе рассмотрение этих методов весьма затруднительно из-за
16.2. Теория магнетизма 135
сложности математического аппарата. Согласование теории и
эксперимента в общем не слитком хорошее (табл. 16.2). Часто
специалисты, занимающиеся изучением свойств и применением
ферро- и антиферромагнитных материалов, используют другой,
гораздо более простой метод расчета величины магнитных мо-
моментов. В рамках этого метода принимается, что магнитный мо-
момент одного неспаренного электрона составляет 1 f.iB- В таком
случае магнитный момент иона, имеющего п неспареппых элек-
электронов, равен пив. Например оба высокоспиновых иона Мп2~ и
Fe'!+ имеют магнитный момент 5цв. Более точное соотЕЮшение
для расчета магнитных моментов имеет вид
ii = gS A6.13)
где я^2,00, a S=n/2 — спиновое состояние иона. Значения маг-
магнитных моментов \1и рассчитанные этим методом, несколько
ниже истинных и.ЭКсп (табл. 16.2); тем не менее метод полезен
для расчета ц. Чтобы уравнение A6.13) хороню описывало экс-
экспериментальные данные, варьируют g, рассматривая его в каче-
качестве «подгоночного» параметра. Уравнение A6.13) учитывает
лишь спиновый магнитный момент. Если принять, что g может
быть больше двух, то это отвечает введению поправки на нали-
наличие орбитального момента. Например, для Ni2+ принимают, что
$"=2,2-^2,3. При обсуждении магнитных свойств ферритов
(разд. 16.3) для оценки величины ц используется уравне-
уравнение A6.13) в связи с его простотой.
16.2.4. Механизм ферро- и антиферромагнитного
упорядочения, обменное взаимодействие
В парамагнитных веществах магнитные моменты отдельных
ионов, содержащих неспаренные электроны, ориентированы
случайным образом. Их упорядочение происходит лишь под дей-
действием внешнего магнитного ноля. Энергия взаимодействия
между диполем и магнитным полем может быть легко рассчита-
рассчитана. Детали этого расчета в настоящем разделе не приводятся,
но в целом можно сказать, что величина этой энергии больше
тепловой энергии ионов или диполей, равной kT.
В ферро- и антиферромагнитных веществах упорядочение
ориентации магнитных диполей происходит самопроизвольно.
Поэтому такое упорядочение должно характеризоваться некото-
некоторой положительной энергией взаимодействия соседних спинов,
которые упорядочиваются либо параллельно, либо антипарал-
лелыю. Спаривание спинов илн их кооперативное взаимодейст-
взаимодействие имеет кваптовомсхапичсскук) природу. Качественно легко
представить себе происходящее явление, хотя окончательной и
136 16. Мапштные свойства
ясной картины ферромагнетизма, например для железа или ко-
кобальта, до сих пор нет.
Взаимодействие спинов, приводящее к антиферромагнетиз-
антиферромагнетизму, называется обменным взаимодействием. Этот процесс для
оксида никеля схематически представлен па рис. 16.4. Ион Ni2"
имеет восемь с?-электронов. Если ион Ni2+ находится в октаэдри-
ческом окружении иопов кислорода, то два из этих электронов
распределяются по одному на <?в-орбиталях (dz-z- и dX2-y2-op6n-
гали). Такие орбитали ориентированы параллельно осям эле-
элементарной ячейки, и поэтому они ориентированы в направлении
соседних ионов кислорода. Неспареиные электроны на ее-ор6в-
талях ионов Ni24" могут взаимодействовать с р-орбиталями
Рис. 16.4, Вяанмолейстпио спинов «/-электронов ионов Ni2' через р-орби'ш-
ли ионов кислорода, и[>иводящее к возникновению антиферромагнетизма.
иопов О-~. В результате такого взаимодействия возникает неко-
некоторое возбужденное состояние, в котором осуществляется пере-
перенос электрона с <?г-орбитали иона КЧ2т на р-орбиталь кислород-
кислородного иона. На каждой р-орбптали иона О2'" имеются два элек-
электрона, которые спарены аптпиараллелыю. Следовательно, в слу-
случае, если ионы Ni2~ и О2" находятся настолько близко друг
к другу, что возможно взаимодействие их электронов, то во всем
кристалле образуется цепь взаимодействующих электронов
(рис. 16.4). В результате спины всех соседних иопов Ni2"*", раз-
разделенных ионами О2~, ориентируются антииараллельпо.
16.2.5. Некоторые другие понятия
Ферромагнитные материалы имеют доменную структуру, ана-
аналогичную доменной структуре сегнетоэлектриков (гл. 15). Внут-
Внутри каждого домена все спины ориентированы параллельно.
Однако, несмотря на то что в материале в целом имеется пол-
полное магнитное насыщение, различные домены имеют разную
ориентацию спинов.
Под действием внешнего магнитного поля в ферромагнитных
веществах возникают явления, аналогичные тем, которые воз-
возникают в сегнетоэлсктрнках при действии внешнего электриче-
электрического поля (разд. 15.4). На кривой зависимости намагниченно-
намагниченности М или магнитной индукции В от напряженности магнитного
поля Н наблюдается гистерезис. Аналогичный гистерезис
15.2. Теория магнетизма 137
имеется и на зависимостях поляризации от напряженности поля
в ссгнетоэлектрических материалах (рис. 15.12). При достаточ-
достаточно больших значениях напряженности магнитного поля достига-
достигается Ha.viai пичснность насыщения, когда спины всех доменов
ориентированы параллельно друг к другу. При намагничивании
и размагничивании веществ в магнитных полях противополож-
противоположного направления происходит диссипация энергии, обычно в ви-
виде рассеянного тепла. Величина магнитной энергии, потерянной
в течение полного цикла (намагничивание — размагничивание),
пропорциональна площади петли гистерезиса. Эта энергия носит
название магнитные потери на гистерезис. Для некоторых обла-
областей применения ферромагнитных материалов необходимо, что-
чтобы магнитные потери на гистерезис были минимальными. Это
значит, что площадь, охватываемая петлей гистерезиса, должна
быть как можно меньше.
Материалы с низкой коэрцитивной силой Пс называют маг-
магнитно-мягкими материалами. Коэрцитивная сила — это напря-
напряженность обратного магнитного поля, которое необходимо при-
приложить, чтобы достичь полной размагниченности материала.
Магнитно-мягкие материалы характеризуются низкой магнит-
магнитной проницаемостью, и, следовательно, их петля гистерезиса
должна быть «узкой в талии» и небольшой площади. Магнитно-
жесткие материалы характеризуются высокой коэрцитивной си-
силой и большой остаточной намагниченностью М,. Последняя ве-
величина отвечает намагниченности, которая остается после того,
как напряженность внешнего магнитного поли становится рав-
иой нулю (рис. 15.12). Магнитко-жесткис материалы трудно
полностью размагнитить, поэтому их часто используют в каче-
качестве постоянных магнитов.
В кристаллах ферромагнитных материалов результирующий
магнитный момент располагается вдоль некоторых преимущест-
преимущественных направлений. У железа, например, такие направления
параллельны осям элементарной ячейки кубической решетки.
Их называют осями легкого намагничивания (рис. 16.5, о).
Энергией магнитной кристаллографической анизотропии назы-
называют эперппо, необходимую для поворота направления резуль-
результирующего магнитного момента в сторону от оси легкого намаг-
намагничивания.
Дополнительным источником потерь энергии при изменении
напряженности внешнего магнитного поля являются электриче-
электрические токи, так называемые вихревые токи, которые индуцируют-
индуцируются в материале. Переменное* магнитное поле индуцирует пере-
переменное электрическое поле, в котором потери, связанные с вих-
вихревыми токами, равны PR или V2/R. Поэтому в материалах
с высоким сопротивлением вихревые токи малы, Одно из пре-
преимуществ большинства магнитных оксидов по сравнению с ме-
138
16. Магнитимр свойству
таллами заключается в их заметно большем электрическом со-
сопротивлении.
Многие магнитные материалы обладают магнитострищией,
т. с. способностью к изменению формы при намагничивании, На-
Например, никель и кобальт сжимаются в направлении намагни-
намагничивания и растягиваются в перпендикулярном направлении.
В слабых магнитных полях железо ведет себя противоположным
образом. В сильных магнитных полях форма железа меняется,
так же как у никеля и кобальта. Происходящие изменения раз-
размеров невелики. Коэффициент магнитострикции Xs, определяе-
определяемый как Я.,1 = Д///0, возрастает с ростом напряженности поля от
1 до 60-10~5 при намагниченности насыщения. Поэтому весь эф-
эффект изменения размеров образца под действием магнитного
поля сравним по величине с эффектом изменения формы при
нагревании на несколько градусов.
16.3. Примеры магнитных материалов, их структуры и свойства
16.3.1. Металлы и сплавы
Пять переходных металлов Cr, Mn, Fe, Co, Ni и большинство
лантаноидов проявляют ферро- и антиферромагнитные свойства.
Для очень многих сплавов и интерметаллических соединений
также характерен тот или иной тип магнитного упорядочения.
a-Fe, 7c = 1043 К
Рис. 16.5. Ферромагнитное упорядочение в объемноцентрированной струк-
структуре a-Fc, гранецентрироваиной структуре № и гексагональной плотноупако-
вашгой структуре Со.
Как видно из рис. 16.5, железо, кобальт и никель ферромаг-
нитны. В объемноцептрироваппой кубической структуре a Fe
спины ориентированы в направлении [100], параллельно ребру
элементарной ячейки. В гранецентрнроваппой структуре никеля
ориентация спинов соответствует направлению [111], т.е. парил
лельпо объемной диагонали к\'ба. Кобальт имеет гсксагопаль-
16.3. Примеры магнитных материалов 139
иую плотпейшую упаковку, и спины электронов ориентированы
параллельно оси с элементарной ячейки. Эти примеры наглядно
демонстрируют, что существование ферромагнетизма не связано
с каким-либо особым типом кристаллической структуры!
Хром и марганец при низких температурах—аптиферромаг-
нетики. Температуры Нееля 7V для Мп и Сг равны 95 и 313 К.
Кристаллическая решетка марганца весьма сложна, хром же
имеет объемноцентрпрованную кубическую решетку, аналогич-
аналогичную сс-г'с. В решетке хрома спипы ориентированы антипарал-
лелыю одной из осей кубической элементарной ячейки.
Некоторые характеристики ферромагнитных материалов при-
приведены на рис. 16.6. На рис. 16.6, а приведена температурная за-
зависимость величины магнитной восприимчивости или магнитно-
магнитного момента. Обозначение осей, правда, по соответствует этим
физическим величинам. По вертикальной оси отложена намагни-
намагниченность насыщения железа, отнесенная к ее максимально воз-
возможной величине (при О К). По горизонтальной оси отложена
так называемая «приведенная температура», т. е. отношение
реальной температуры к температуре Кюри. Поэтому в точке
Кюри приведенная температура равна единице G/Гс=1).
Использование приведенных координат позволяет сравнивать
материалы с различными температурами Кюри и различными
магнитными моментами. Из рис. 16.6, а видно, что по магнитным
свойствам железо и никель очень сходны между собой: с повы-
повышением температуры от О К. при малых Т/Тс намагниченность
насыщения металлов остается постоянной и начинает быстро
падать при приближении к температуре Тс.
Выше температурь: Кюри железо, кобальт и никель стано-
становятся парамагнетиками. При температурах существенно выше
7'с выполняется закон Кюри—Вейсса, вблизи точки Кюри наблю-
наблюдается отклонение от прямолинейности (рис. 16.5,6). Это откло-
отклонение связано с существованием ближнего упорядочения спинов.
Дальний порядок, существовавший в ферромагнитном состоянии
(ниже Тс), исчезает, а ближний порядок сохраняется и несколь-
несколько выше 7с. Поэтому температура Э (константа Вейсса) не-
несколько отличается от Тс. Приведенные па рис. 16.6, С данные
относятся к никелю, однако аналогичные результаты были полу-
получены также для железа и кобальта.
Переход из ферромагнитного в парамагнитное состояние при
температуре Тс описывается такими же параметрами, как и лю-
любой фазовый переход второго рода или А-образпып фазовый пе-
переход (гл. 12). Это классический пример перехода типа поря-
порядок — беспорядок. Абсолютно упорядоченное состояние может
существовать лишь при абсолютном нуле. При любой реальной
температуре идет процесс разугюрядочения, причем с ростом
температуры разупорядочение быстро усиливается. Зависимость
140
16. Магнитные свойства
теплоемкости от температуры проходит через максимум при
температуре 7с (рис. 16.6, в).
Здесь следует обратить внимание на до сих пор нерешенную
проблему теории ферромагнетизма. Почему ферромагнитные
вещества сгруппированы так плотно в периодической системе
элементов, а главное, сколько неспарснных электронов атома
могут вносить вклад в магнитные характеристики ферромагнети-
10-КГ -
8-Ю4
6-Ю4
4'104 -
-
\. /
"Н/
. >| 1 1 1
б
/
/
1
400
С00
800 I С
J6.3. Примеры мигпитпых материалов
141
200 400 600 800 1000
г, к
Рис. 16.6. Некоторые свойства ферромагнитных материалов, а — зависимость
намагниченности насыщения, отнесенной к намагниченности насыщения при
абсолютном нуле, от приведенной температуры; б—зависимость обратной
величины магнитной восприимчивости от температуры (закон Кюри— Вейс-
са). Показано отклонение от прямолинейной зависимости вблизи Тс', в —теп-
—теплоемкость железа как функция температуры [J4].
ков? Фактический материал приведен ниже. А какова ваша точ-
точка зрения на эту проблему?
Для трех ферромагнитных элементов первого ряда переход-
переходных металлов электронная конфигурация приведена в табл. 16.3.
Из таблицы видно, что у изолированных атомов всех этих эле-
элементов в основном состоянии 4s-ypoBHH полностью укомплекто-
Таблица J6.3. Электронное строение железа, кобальта, никеля
Ферромагнитное состояние
'Металл
Электроники конфигура-
конфигурация изолированного иона
число неепаренныч
конфигурация
Fc
Со
2,2
1,7
0,6
ваны. В то же время в ферромагнитном состоянии 45-зопа не за-
заполнена, так как некоторые из s-электронов переходят в З^-зо-
ну. Доказательства такого распределения электронов основыва-
основываются на расчетах зонной структуры и величинах намагничен-
намагниченности насыщения, которые пропорциональны числу неспаренных
электронов. Так, суммарный магнитный момент атома железа
142 16. Магнитные свойства
составляет 2,2,ив, следовательно, на 1 атом железа в среднем
приходится 2,2 неспаренного d-э.чектрона. Это значит, что из 7,4
d-электрона 4,8 имеют один спин, а 2,6—другой. В таком под-
подходе пет ничего необычного; химики привыкли к тому, что элек-
электроны могут переходить с 4s уровня на 3^-уровень и обратно
в зависимости от обстоятельств. В данном случае намного боль-
больше интересен вопрос, какое максимальное число неспаренпых
электронов может вносить свой вклад в ферромагнитные свой-
свойства З^-элсментов или их сплавов. Как показали расчеты, по-ви-
по-видимому, максимально па один атом может приходиться в сред-
среднем 2,4 электрона. Однако в настоящее время не существует
сколько-нибудь удовлетворительного объяснения именно этой
величины. У З^-элементов аффективное число неспаренпых элек-
электронов меняется с изменением общего числа электронов. Мак-
Максимальное значение 2,4 электрона на i атом было эксперимен-
экспериментально найдено у сплава состава Feo,»Coo.a- С увеличением об-
общего количества электронов число неспаренных спинов посте-
постепенно уменьшается. У кобальта и никеля они меньше, чем 2,4,
а в сплаве Nio.Xuu.c равно нулю. Чистая медь парамагнитна.
В другую сторону от сплава Ftn.sCon 2 (в ряду железо —»- марга-
марганец —*¦ хром) число неспаренных электронов также систематиче-
систематически понижается. Марганец и хром становятся аптиферромагпе-
тиками при низких температурах.
По всей видимости, необходимо учитывать такие факторы,
как степень перекрывания rf-орбиталеи и ширина af-зоны, кото-
которые непосредственно связаны с межатомными расстояниями
в металлах, возрастающих с увеличением атомного номера
З^-элемсита. При небольших межатомных расстояниях перекры-
перекрывание iZ-орбиталей очень велико и силы кваитовомехапического
взаимодействия приводят к возникновению аптииараллельно
ориентированных спинов. Именно это имеет место у хрома и
марганца. При увеличении межатомных расстоянии перекрыва-
перекрывание по-прежнему достаточно велико, однако магнитное взаимо-
взаимодействие спинов приводит к их параллельной ориентации. Это
происходит в ферромагнетиках — железе, кобальте и никеле.
При еще больших межатомных расстояниях магнитное взаимо-
взаимодействие ослабевает, что приводит к появлению парамагнетизма
у меди.
Лантаноиды также имеют упорядоченную магнитную струк-
структуру, которая связана с наличием неспаренных 4/-э.г1ектронов.
Исключение составляют те элементы, у которых 4[-оболочка
пуста (La 4f°) или заполнена (Yb 4/14, Lu 4fM). При температу-
температурах ниже комнатной большинство лантаноидов антиферромаг-
нитны. Некоторые лантаноиды, в основном в конце этого семей-
семейства, в зависимости от температуры могут быть как ферро-, так
и антиферромагнетиками. Для них характерна следующая по-
16.3. Примеры магнитных материалов ^__ ИЗ
следов а тел ьпость превращений при понижении температуры:
парамагнетик —*- антиферромагнетик *¦ ферромагнетик
Соответствующие температуры Нселя и Кюри приведены
в табл. 16.4, только гадолиний при любых условиях не может
быть аытиферромагнетиком.
Некоторые аптиферромагнитные лантаноиды проявляют так
называемый яи'тамагнетизм. Это явление заключается в том, что
Таблица 1C.4. Температуры II ем я (шгтпферромагптнос превращение)
и Кюри (ферромагнитное превращение) лантаноидов [11]
Элемент Темперлу|>а Поели /'v, К Темнературп Кюри Тс, К
Се 12,5
Pi 25
Nd 19
Sin 14,8
F.u 90
Gd — 293
Tb 229 222
Dy 179 85
Ho 131 20
Er 84 20
Tm 56 25
под действием магнитного поля достаточно высокой напряжен-
напряженности такие вещества переходят в ферромагнитное состояние.
Например, ниже 85 К диспрозии ферромагиитен, а при более
высоких температурах он превращается в антиферромагиитпую
фазу. Под действием внешнего магнитного поля ферромагнитное
состояние может быть сохранено при температурах выше 85 К
вплоть до точки Нееля A79 К).
16.3.2. Оксиды переходных металлов
Для оксидов переходных металлов четвертого периода перио-
периодической системы характерны весьма заметные систематические
изменения свойств с изменением порядкового номера и числа
of-электронов. Об изменении электропроводности оксидов двух-
двухвалентных металлов МО уже упоминалось в гл. 14. Там отмеча-
отмечалось, что оксид TiO обладает металлической проводимостью,
а оксид NiO является диэлектриком. В ряду этих оксидов на-
144 16. Магнитные cuoficiu.]
блюдается систематическое изменение и магнитных свойств
симбатпо с изменением электрических свойств. Оксиды первых
трех элементов этой группы (TiO, VO и СгО) диамагнитны.
В них J-электроны не локализованы на отдельных ионах М2~,
а делокализовапы по всей решетке. Они находятся в частично
заполненной /2й-зоие. По-видимому, магнитные моменты делока-
лизованных электронов не взаимодействуют друг с другом. Сле-
Следовательно, эти вещества диамагнитны и обладают высокой
электропроводностью. Оксиды следующих элементов (МпО,
FeO, СоО и NiO) при высоких температурах парама! питны,
а при низких характеризуются упорядоченной магнитной струк-
структурой, в которой J-элсктроны локализованы на отдельных ионах
М-~. Именно локализация неспаренпых электронов является
причиной как упомянутых магнитных свойств, так и отсутствия,
электрической проводимости.
Оксиды МпО, FeO, СоО, .NiO при низкой температуре анти-
ферромагнитпы, а с ростом температуры переходят в парамаг-
парамагнитное состояние. Температуры Нееля 7\ этих оксидов таковы:
МпО —153 СС, FeO — 75 °С, СоО --2СС, N40 +250°С. Кристал-
Кристаллические структуры всех оксидов как в антиферромагпитпом,
так и в парамагнитном состоянии похожи друг на друга. При
высоких температурах они имеют структуру тина KaCI
(рис. 5.9). Эту структуру можно представить и описать различ-
различными способами. Здесь удобнее рассмотреть кристаллическую
структуру вдоль любого из четырех направлений [111]. парал-
параллельных объемным диагоналям грапенентрированной кубической
элементарной ячейки. Например, кристаллическая структура
NiO состоит из последовательных слоев ионов N'i21 и О2'.
Ниже 250 °С кристаллическая структура NiO претерпевает
ромбоэдрическое искажение: вдоль оси третьего порядка, парал-
параллельной направлению [III], происходит слабое сжатие решетки.
Аналогичное сжатие имеет место и в структуре МпО. В структу-
структуре FeO, наоборот, вдоль этой оси происходит растяжение. Сим-
Симметрия структуры понижается, так как все оси четвертого по-
порядка и три из числа осей третьего порядка перестают быть эле-
элементами симметрии. Остается лишь одни элемент симметрии —
ось третьего порядка. Степень искажения кубической решетки
невелика и па аорошкограммах (например, монооксида никеля)
едва заметна.
Причина ромбоэдрического искажения кристаллической ре-
решетки NiO связана с антиферромагнитпым упорядочением ионов
NF\ В пределах одного катиошюго слоя спины всех ноиов Ni2+
ориентированы параллельно друг другу, но антипараллельно
спинам ионов Ni^~ соседних слоев (рис. 16.7). Магнитные сверх-
сгруктуры, подобные описанной, лучше всего исследовать мето-
методом нейтронографии (разд. 3.2.1.5). При этом нейтронное рас-
16.3- Примеры магнитных материалов
145
сеяние будет происходить как на ядрах атомов, так и на неспа-
ренных электронах. Первый тип рассеяния даст дифракционную
картину, подобную картине рассеяния рентгеновских лучей. Ин-
Интенсивности рефлексов на пейтроиограммах и рентгенограммах
могут несколько отличаться. Антиферромагнитиая структура
(второй тип рассеяния) проявляется r виде дополнительных ли-
линий на нейтронограммах порошкообразных веществ. Существу-
Существуют по меньшей мере две причины появления этих линий: I) ко-
порядка
1
i
-i
м '*-,
Рис. 16.7, Лптиферромагтгшля структура МпО, FeO, NiO Изображена
псевдокубическая элементарная ячейка. Параметр а (суперъячейки) •= 2а (субъ-
(субъячейки). Позипип ноноз кислорода не обозначены.
оперативное взаимодействие неспарениых электронов может
привести к появлению сверхструктуры; 2) нейтроны сильно рас-
рассеиваются иа неспаренных электронах, в то время как рентге-
рентгеновские лупи пс рассеиваются.
На рис. 16.8 приведены нейтронограммы МпО, снятые выше
и ниже температуры Нееля, а также рентгенограмма порошко-
порошкообразного образца МпО при комнатной температуре. Сопостав-
Сопоставление двух дифракционных картин, полученных выше Г.у
(рис. 16.8, бив) показывает, что положение линий аналогично,
по они резко отличаются по интенсивности. Структура типа
NaCl такова, что индексы рефлексов /г, k, l должны быть либо
только четными, либо только нечетными. Следовательно, первые
четыре линии па обеих дифрактограммах имеют индексы 111,
200, 220, 311, по на нейтронограмме линии 220 и 200 отличаются
малой интенсивностью (рис. 16.8,6). Последнее объясняется
тем, что рассеяние нейтронов на нонах Мпг+ и О2' противопо-
10—1426
16. Магнитпью свойства
ложно по знаку и слабо отличается по величине. Совершенно
другая картина наблюдается при дифракции рентгеновских лу-
лучей. Фактор рассеяния всех элементов имеет один и тот же знак,
и рефлексы 200 и 220 имеют высокую интенсивность, так как
рентгеновские лучи, рассеянные ионами Мп2+ и О2", распростра-
распространяются синфазно.
дифракция
неитрсмов
8ОК,
а--8,85А
222
111
622
351
511 440
дифракция
нейтронов
293 к, ^
Э-4.43А
Ш
200
К
220
рентгенография
295 к
111
200
220
311
20
40
60
80 26
Рис. 16.8. Нсйтронограммы и рентгенограмма порошкообразного МпО
(Л= 1,542 А). Для каждой линии указан шаекс Миллера для кубической
элементарной ячейки. Данные нейтроиографического исследования взяты из
работы [9]; рентгенограммы—из картотеки порошкограмм (карточка №7—
230),
Сравнение рис. 16.8, а и б показывает, что ниже 7\v на ди-
фрактограммах появляются дополнительные линии (отмечены
звездочками). Эти дополнительные линии относятся к антифер-
антиферромагнитной сверхструктуре. Хотя, как отмечалось выше, истин-
истинная симметрия антиферромагнитной фазы ромбоэдрическая,
в первом приближении ее можно рассматривать как кубическую
с удвоенными параметрами элементарной ячейки высокотемпе-
высокотемпературной парамагнитной фазы. При 80 К (<7\-) а = 8,85 А, а
при 293 К (>Tv) a-=4,43 А (рис. 16.7). Объемы элементарных
ячеек относятся, следовательно, как 8: !. Дополнительным ли-
16.3 Примеры мапштпых материалов 147
ниям па нейтропограмме аптиферромагиитпой фазы можно при-
приписать определенные индексы (рис. 16.8,а). Все значения h, k,
I в этих индексах нечетные.
16.3.3. Шпинели
Некоторые важные в практическом отношении оксиды имеют
структуру шпинели. Рассмотрим ее особенности на примере
MgAl20.4. Структура типа MgA!2O,i представляет собой кубиче-
кубическую плотпоупакованную решетку ионов кислорода, в тетра-
эдрических и октаэдрических пустотах которой находятся соот-
соответственно ионы Mg2¦'¦ и А13+. В настоящее время известно бо-
более era соединений со структурой шпинели*. Большинство из
них — оксиды. Известны также сульфиды, селеннды и теллури-
ды со структурой шпинели. Такую же структуру имеют некото-
некоторые галогениды. В образовании структуры типа шпинели могут
участвовать многие разнозарядные катионы. Ниже приведены
примеры различных шпинелей и заряды образующих их катио-
нов-;
Li.MTiO,
1, 3, 4
Li
1.
o.:-AI25(
3
D4 LiNiV
1,2.,
o.
5
Na2WO,
1, (i
Заряд 2. 3 2. A
катиона
Подобные комбинации катионов разного заряда характерны и
для сульфидов: ZnAl2S.j B, 3) и Cu2SnS4 B, 4). В галогенидных
шпинелях заряд катионов может быть равен только 1 и 2, чтобы
отношение числа катионов к числу анионов было равно 3 : 4,
например как у l^NiF,).
В табл. 16.5 приведены кристаллографические параметры
структуры шпинели. На рис. 16.9 изображена проекция элемен-
элементарной ячейки MgAl2O4. Последняя содержит восемь формуль-
формульных единиц (Z—8) и отвечает формуле MgeAlieC^. Ионы рас-
располагаются в позициях трех разных типов (табл. 16.5). Напри-
Например, машин находится в позициях, кратность которых равна
восьми (обозначены 8а). Для двух ионов магния в таблице при-
приведены координаты: 0, 0. 0, и 'Д, '/ч, 'А- Позиции остальных шести
ионов магния можно получить, имея в виду, что шпинель имеет
кубическую грапецентрироваппую решетку. Таким образом, если
имеется позиция с координатами х, у, z, то существуют три дру-
другие эквивалентные позиции с координатами: я+'/г. y+!/2i z\ x-\~
+ 7г, у, г+'/г; х, у+'/г, -с+'/г- Таким образом, координаты ос-
остальных шести ионов Mg: '/г, 7г,0; '/г, 0, '/г: 0, 7г, У2; 3/4, 3/4, lU;
* Число их гораздо больше, если принять в расчет халькошпинели и
.¦loi'iiiHiitvni. — Прим, ред.
гал-
10*
148 16. Магнитные свойства
Таблица 16.5. Кристаллографические параметры структуры шпинели
Пространственная группа Fd3m, N? 227, грапецентрироваппая
кубическая решетка
Кои|>дипаты аточчи в MrAI/Oj (в долях
Атом Тин позиции периода решетки)
_ 1 I
О 32е иии; и и и; и и и ; и и и; — — и, — — и,
4 4
1 11 11
—- — и; —- + ", — — и, — + и; — — и,
4 4 4 4 4
1 1 1,1,1
4 4 4 4 4
+гранецентричсское преобразование
_5__5__5_ _5__7_^_ 7 5 7 7 7 5
AI 888'888'888'888
-j- гранецеитрич-сское преобразование
Mg 8а 0 0 0; +грапецентрическое преобра-
4 4 4
зование
Координационные числа
Mg 8a тетраэдр MgO4
AI 16d октаэдр АЮе
О 32е тетраэдр OMgAl3
3Д, 'А, 3А; 'А, 3А, 3А- Аналогично можно определить координаты
ионов А13'- и О2- (табл. 16.5).
На рис. 16.9 показано координационное окружение ионов
Mg2+ и А13+. Ион Mg2+ находится внутри тетраэдра, а ион
А13+ — внутри октаэдра. Ионы кислорода образуют идеальную
кубическую плотную упаковку, если параметр и (табл. 16.5)
принимает значение %. Чтобы увидеть это, элементарную ячей-
ячейку, изображенную на рис. 16.9, следует разделить на октанты
(на проекции видны лишь квадранты), а начало координат
сдвинуть вдоль объемной диагонали куба так, чтобы оно совпа-
совпадало с положением кислородного иона, а не с положением иона
AF+ (рис. 16.10).
Осложняющим фактором является то, что распределение ка-
катионов по позициям 8а и 16d может меняться. Необходимо раз-
различать два крайних случая такого распределения. В нормальной
16.3. Примеры магнитных материалов
149
1/6.5/8
1/8.5/8
©
Рис. 16.9. Проекция структуры шпинели.
'/8 элементарной
ячейки шпинели
грань гранецентриро-
ванной кубической
элементарной ячейки
1/8@) 3/8A/2) 1/8@)
Рис. 16.10. Фрагмент структуры шпинели, демонстрирующий плотнейшую
упаковку ионов кислорода.
шпинели катионы занимают позиции, которые определяются
формулой АВгО4, т. е. ионы А находятся в тетраэдрических по-
позициях 8а, а ионы В — в октаэдрических позициях 16d.
М^А1гО4 и MgTi2O4 •—¦ примеры нормальной шпинели.
В обращенной шпинели половина ионов В находится в тетра-
эдрических позициях 8а, а ионы А и оставшиеся ионы В зани-
150
16. Магнитные CRoftcrna
Таблица 16.6.
Соединение
MgM2O4
СоА12О4
CuCr2S4
CuCrjSi'4
CuCr2Tc4
MgTi2O4
C,o2GcO,
Fe2Gc04
MrFc2O»
NiFc-O,
¦MRlnzO,
MgIn2S4
Mg2TiO4
Zii2SnO4
Zn2TiO4
LiAlTiO4
J.iMi:TiO4
LiZnSbO4
LiCoSbO4
Кристаллографические параметры некоторых шпинелей
Тип шпинели
2,3
2,3
2,3
2,3
2,3
2,3
2,4
2,4
2.3
2,3
2,3
2.3
2,4
2,4
2,4
1,3, 1
1,3,4
1,2,5
1,2,5
О
а. А
8,0800
8,1068
9,629
10,357
11,051
8,474
8,318
8,411
8,389
Н,3532
8,81
10,708
8,44
«,70
8,467
8,34
8,30
8,55
8,56
и
0,387
0,39
0,38!
0,380
0,379
—
—
—
0,382
0,381
0,372
0,384
О.ЗУ
0,390
0,380
—
—.
—
—
Структура
Нормальная
»
»
»
»
Обращенная
»
»
»
»
»
Li is 8a
Li п 8а
Li J! 8a
I.i ii 8a
мают позиции 16d. Обычно эти ионы распределены по позициям
lGd статистически. Примеры обращенных шпинелей — MgFe^O-i
и Mg^TiO^
В табл. ]6.6 приведено довольно много веществ, имеющих
структуру нормальной и обращенной шпинели, а также пара-
параметры их элементарных ячеек и параметр и (если последний из-
известен).
Чтобы различить нормальную и обращенную шпинель, фор-
формулы этих веществ записывают следующим образом:
нормальная шпинель [A]ti-i рШаЬктО*
обращенная шпинель [В]гстр[А, В]0щ04
Кроме крайних случаев (нормальная и обращенная шпине-
шпинели) возможно промежуточное распределение катионов по пози-
позициям. Иногда распределение катионов меняется с температурой.
Катиопыое распределение можно легко рассчитать с помощью
16.3. Примеры магнитных матсриялов UH
параметра v (степень обращенности), который соотиетствует
доли катионов А, находящихся в октаэдрических позициях:
нормальная шпинель [ЛЬетр^г^и.О* t=0
обращенная шпинель [В]ТетР[А, В]ш;,О4 '{-1
смешанная шпинель [Во.втАо.ззЬспЛЛо^В^ззЬитО^ y -0,67
В ряде работ детально изучены распределение катионов но
различным узлам шпинели и степень обращенности у. На вели-
величину v влияет ряд факторов. Так, степень предпочтения ионов
к тому или иному типу позиций определяется размером ионов,
степенью ковалентности связи, энергией стабилизации кристал-
кристаллического поля. Более подробно, на конкретных примерах это
рассмотрено в разд. 8.6.1. В каждой конкретной структуре ве-
величина у определяется совместным действием всех перечислен-
перечисленных параметров.
Наиболее важные в практическом отношении соединения со
структурой шпинели —это ферриты MFe2C>4, где М —двухза-
рядный ион {Fe2+, Ni2+, Cu2^, Mg2-1")*. Ферриты, как правило,
являются частично или полностью обращенными шпинелями.
Причина этого состоит, видимо, в том, что ион Fc3'1' с пятью
^-электронами, согласно теории кристаллического поля, не име-
имеет предпочтения к октаэдрическим позициям. Следовательно,
двухзарядные ионы большого размера занимают в основном
октаэдрические позиции, а ионы Fe3+ распределяются по тетра-
эдрическим и октаэдрическим узлам.
Весьма интересна магнитная структура ферритов, которые
могут быть либо ферримагнетиками, либо антиферромагнети-
антиферромагнетиками. Это объясняется тем, что ионы, находящиеся в тетраэдри-
ческих узлах 8а, имеют магнитные спины, направленные антипа-
раллельно спинам ионов, занимающих октаэдрические 16с1-узлы.
Магнитная структура шпинели представлена на рис. 16.11. Здесь
изображены четыре октанта элементарной ячейки; начало коор-
координат, как и на рис. 16.9, совпадает с положением иона, нахо-
находящегося в узле 8а. Кислородные ионы, находящиеся в позициях
1ъ, Ч&, 3/в, не изображены па рисунке. Ориентация элемен-
элементарной ячейки такая же, как на рис. 16.9. Изображенную в та-
таком виде элементарную ячейку можно представить как кубиче-
кубическую упаковку ионов, находящихся в узлах 8а. Эти ионы зани-
занимают вершины п центры граней кубической решетки. Кроме
того, ионы этого сорта располагаются в центрах половины ок-
октантов ячейки. В целом в одной элементарной ячейке содер-
содержится 8 ионов, занимающих позиции 8а. Ионы, находящиеся
в позициях типа 16d, располагаются в вершинах октаэдров
¦ На практике ершшиголыю pt-дко используют индивидуальные соеди-
соединения. Техническое значение имеют твердые раствори па основе этих лмш-
пелей. — Прим. ред.
152
16. Магнитные свойств;:
"!лло координат
0 позиции Й а С/) nojvuHkf
Рис. 16.П. Магнитная структура антиферромлгнитной и ферримагнитноГг
шпинелей.
в смежных октантах элементарной ячейки; на одну элементар-
элементарную ячейку приходится 16 попов такого типа. Как показано на
рис. 16.11, магнитные спины ионов, находящихся в позициях 8а
и 16C, антипараллельпы. Рассчитаем магнитные моменты раз-
различных шпинелей по уравнению A6.13).
Для начала хорошим примером будет феррит цинка ZnPe2O4. При очон.
низких температурах он имеет структуру обращенной шпинели
[l--e3+jTeip[Zn2+',Fe3+j0KT04
Поскольку полозинл ионов Fe3 + находится в тетраэдрических позициях 8а,
а другая половина — п октаэдричеекпх позициях 16d и спины их Лмтинл-
раллельны, то суммарный магнитный момент ионов Fe3 ¦ равен нулю. Ион
7л\- '•¦ имеет нулевой магнитный момент, поэтому и общий магнитный момонч
равен нулю. Это пещество - аптнферромагнетик. Экспериментальные .'шпик-
подтверждают эти выводы (Гм=9.5 К)*.
К. аналогичному результату можно было бы прийти, видимо,
и в случае феррита магния MgFe2CXi. Однако было найдено, что
* Более общепринятая точка зрения о причине нулевого магнитного мо-
момента феррита нипка изложена, например, в книгах [Смит Я-, Вейн X. Фер-
Ферриты. Физические свойства и практические применения." М.: ИЛ, 1962;
Крупинка С. Физика ферритов и родственных им магнитных окислов'
В 2-х т. — М.: Мир, 19761. — Прим. ред.
16,3. Примеры магнитных материалов 153
это вещество ферримапштпо, т. е. обладает некоторым магнит-
магнитным моментом. Возможны два объяснения такого явления.
Во-первых, возможно, что феррит имеет частично смешанную
структуру, в которой большая часть ионов Fe::~ находится
в октаэдрпческих узлах, а меньшая — в тетраэдрпческих. Поэто-
Поэтому аптипараллельные спины этих ионов лишь частично компен-
компенсируют друг друга. Во-вторых, эффективные магнитные момен-
моменты ионов Fe3" в разных позициях могут быть неодинаковы.
Экспериментальные исследовании подтверждают первое
предположение. При высоких температурах Mg):e204 none пенно
приобретает структуру нормальной шпинели. Степень обращен-
обращенности шпинели при комнатной температуре сильно зависит от
термической предыстории, в первую очередь от скорости, с ко-
которой образец охлаждался от высоких температур. Так, в быст-
быстро закаленных образцах степень обращенности шпинели невели-
невелика и магнитный момент таких веществ выше, чем образнов, мед-
медленно охлажденных до комнатной температуры.
Феррит марганца MnFe2Oi представляет собой смешанную
шпинель, степень ее обращенности составляет 20%. Поскольку
оба катиона Мп'2 ~ и Fc3+ имеют одинаковую электронную кон-
конфигурацию (dc), общий магнитный момент не зависит от степе-
степени обращенности и термической предыстории образца. Можно
ожидать, что МпРегО4 будет ферримапштным веществом, а его
общий магнитный момент ~5ив- Эксперимент это подтвердил.
Исключительно большое значение распределения катионов по
узлам различного типа хорошо иллюстрируется на примере фер-
ферритов состава Mi.-*ZrbFe2O4, где M = Mg, Ni, Co, Fe, Мп. При
х = 0 такие ферриты в основном имеют структуру об-
обращенной шпинели, т. е. [Fe3H'].iCTp[Fe3+, M2+]OKTO4. Для пол-
полностью обращенной шпинели можно ожидать, что магнит-
магнитные моменты и. различных ферритов имеют следующие зна-
значения: M = Mg u = 0; M=Ni u. = 2; M=Co u = 3; M = Fe jx = 4;
M=Mn ц = 5. Экспериментально найденные значения магнитных
моментов несколько выше (см. величины (л, отложенные на ле-
левой вертикальной оси рис. 16.12). Чистый феррит цинка Z11FC2O4
(х—1) при комнатной температуре имеет структуру нормальной
шпинели. Однако спины ионов Fe3'1', находящихся в октаэдпиче
ских позициях решетки ZnFe2O4, ориентированы случайным об-
образом, а вовсе не упорядочены вдоль какого-либо направления.
Поэтому ZnFe.^C1! — парамагнетик. Его намагниченность насы-
насыщения равна нулю. При образовании смешанных ферритов пу-
путем частичного замещения ионов М2+ па попы Zn2+ происходит
постепенный переход от структуры обращенной шпинели к струк-
структуре нормальной шпинели. Введение попов Zn2 в тстраэдрпче-
ские позиции приводит к вытеснению ионов Fe:i+ из них в окта-
эдрические узлы, т. е. [Fe3-!-i-AZnc2][]
154
16. Магнитные свойства
Если бы твердый раст-
раствор оставался антифер-
ромагнитпым, как и
MFe2O4 (л- = 0), то маг-
магнитный момент ]х должен
был линейно расти с по-
повышением х, и при ,v--=l
(ZnFe^C^) должен был
стать равным — 10ац.
Однако задолго до сос-
состава х — 1 антиферромаг-
ннтное взаимодействие
сшшоб 1-iohor, находя-
находящихся в позициях 16tl и
8а, нарушается и вели-
величина намагниченности
насыщения начинает па-
падать (рис. 16.12). При
малых значениях х экс-
экспериментально найден-
найденная величина намагни-
намагниченности насыщения воз
растает, что находится в
согласии с предположе-
предположением об антиферромаг-
пптном (феррнмагниг
пом) упорядочении. При
.V-0,4-^0,5 зависимость
намагниченности касы-
шеппя от состава твер-
твердого раствора проходит
через максимум.
Магнитные свойства
феррпюв и их изменения
определяются не только величиной их моментов, по и другими
параметрами. К ним относится намагниченность насыщения
М„м, постоянная магпитострикции Я*, магнитная проницаемость
Р и постоянная магнитной кристаллографической анизотропии
К]. Достаточно сказать, что значении этих параметров у раз-
различных ферритов существенно отличаются. Имеется возмож-
возможность для практических целей выбрать феррит с желаемым
значением того или иного свойства. Дальнейшее изменение маг-
магнитных параметров может быть достигнуто путем создания
смешанных ферритом — твердых растворов двух или более чис-
чистых ферритов. Например, при замещении ионов Мп21" на ноны
Fe2+ в MnFe2O4 образуется твердый раствор
о
0,2
Рис. 16.12. Зависимость изменения намаг-
намагниченности насыщения от состава твердых
растворов
о..
16.3. Примеры магнитных материалов 155
с параметром магнитной анизотропии, равным пулю. Этот па-
параметр характеризует легкость переориентации магнитного мо-
момента вещества во внешнем магнитном поле. Уменьшение маг-
магнитной анизотропии приводит к возрастанию магнитной про-
проницаемости, что очень важно для практических применений.
Нежелательный побочны!! эффект, однако, связан с. увеличе-
увеличением электропроводности материала с ростом содержания Fe2+
в нем.
16.3.4. Гранаты
Гранаты — название обширного семейства сложных оксидов.
Некоторые из гранатов являются важными ферримагпитными
материалами. Их общая формула A3B2X3Oi2. A — ион с боль-
большим ионным радиусом (~1 А) и КЧ 8. В структуре типа грана-
граната ион Л находится в центре искаженного куба. В и X — ионы
небольших размеров, занимающие соответственно октаэдриче-
ские и тетраэдрические позиции. К веществам с интересными
магнитными свойствами относятся гранаты, у которых A=Y или
РЗЭ (Sm, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu), В и X=Fe3+. Одним
из наиболее важных представителей гранатов является железо-
иттриевый гранат (ИЖГ) Y3Fe5Oi2. Возможны и другие сочета-
сочетания ионов А, В и X, например
Гроссуляр
Уваровит
Пироп
Лнлрядит
А
Са3
Са3
Mg,
Са,
Саз
Са3
NajCa
NaCa2
в
А12
Сг,
ли
iv2
Cs7r
le2
Ti2
Zn2
X
Si3
Si3
Si3
Si»
Gc3
Znj
Gc3
V,
о
0,2
O,2
0,2
0,2
o,2
0,2
0,2
0,2
В табл. 1G.7 приведены кристаллографические параметры
железо-иттриевого граната (ИЖГ). В объемноцентрированнон
кубической элементарной ячейке (а— 12,376 А) содержится во-
восемь формульных единиц. Мы не решились изобразить здесь
кристаллическую решетку граната. Ее можно рассматривать
как каркас, построенный из сочлененных вершинами тетраэдров
Х04 и октаэдров В06. Крупные ионы А занимают пустоты, ок-
окруженные восемью соседними ионами. В железо-иттрисвом гра-
гранате и гранатах редкоземельных элементов позиции ионов В и
X занимают ионы Fe3+.
ИЖГ и все гранаты редкоземельных элементов — ферримаг-
нетики с температурой Кюри от 548 до 578 К. Чтобы оценить
общий магнитный момент этих гранатов, необходимо учитывать
156 16. Магнитные свойстпа
магнитные моменты трех типов ионов, находящихся в позициях
24с, 24d и 16а. По-видимому, спины ионов, находящихся в узлах
типа 24d, ориентированы антипараллелыю спинам ионов, кото-
которые занимают позиции 24с и 16а.
Магнитные момемты ионов Fc:i , занимающих два разных ти-
типа позиций, частично компенсируют друг друга, так что на одну
формульную единицу M3Fcii0i2 нсскомпенсированным остается
Таблица 16.7. Кристаллографические параметры структуры граната
Пространственная группа: Ia3d, „Yv 230, объемноцентриронанпая
кубическая решетка
Координаты атчтои u Y-.Ke-,Ou (ИЖП
Атом Iitn поящий (и долях период:, решетки)
Y 24с Т ° Т
3 I
Fed) 21d — 0 —
О Ч.
FcB) 16а 0 0 0
О 9fih и, v, и1: и--—0,0275, у =0,0572, а; =0,1495
Координационные числа
Y 24с. КЧ 8 искаженный куб
Fe(I) 24d КЧ 4 тетраэдр
Ft:B) Ilia КЧ G октаэдр
О yfib КЧ 4 искаженны:! ткгразчр: 2\i+, ]Fei+(l),
1Гс--B)
магнитный момент одного иона Fe3"*". Таким образом, суммар-
суммарный магнитный момент ионов Fe3' в ИЖГ должен быть равен
5Aв. Ионы Y3;" характеризуются нулевым магнитным моментом,
так как их электронная конфигурация d°. Ожидаемый магнит-
магнитный момент ИЖГ, равный 5цв, находится в отличном согласии
с экспериментальными данными. Магнитные моменты гранатов
редкоземельных элементов можно рассчитать по формуле
(Зим —5)цв
где им — магнитный момент иона, находящегося в позиции 24с.
Ион Gd3+ имеет семь /-электронов и, следовательно, jug<i = 7{.ib.
Поэтому суммарный магнитный момент железо-гадолиниевого
граната должен быть равен 16цв> что также хорошо совпадает
с экспериментально определенным значением. У иона Lu3+
(электронная конфигурация /и) ulu = 0 и, следовательно, сум-
16.3. Примеры магнитных материалов
157
10
-5
Cd
марный момент равен 5|лв. Такая же величина была получена и
экспериментально. Орбитальный момент ионов от ТЬ до Yb, ви-
видимо, не полностью подавляется, поэтому значения цм несколь-
несколько выше, чем рассчитанные по формуле A6.11) (g = 2,00) с уче-
учетом лишь спинового магнитного момента. На рис. 16.13 для
сравнения нанесены эк-
экспериментальные и рас-
рассчитанные значения ц.
Для последних приведе-
приведены две кривые, соответ- 2Ь
ствующие уравнениям
A6.11) и A6.12). В ос- 20
новном эксперименталь-
экспериментальные значения находятся is
между двумя теоретиче- ^
скими кривыми, из чего
ясно, что орбитальный
момент подавляется
лишь частично.
Температурная зави-
зависимость магнитных мо
ментов гранатов редко-
редкоземельных элементов
имеет интересный и не-
необычный ход. Магнитные
моменты гранатов при
0 К, приведенные на
рис. 16.13, уменьшаются
с ростом температуры и
при температуре компенсации становятся равными нулю. Зач-
Зачтем они вновь возрастают, но приобретают противоположный
знак. После этого снова понижаются до нуля при температуре
Кюри. На рис. 16.14 приведена температурная зависимость
спонтанной намагниченности железо-дпепрозпевого граната.
Причина такой зависимости состоит в том, что спины ионов
в подрешетке редкоземельных металлов разупорядочиваются
быстрее, чем спины ионов в подрешетке железа.
Катионы, входящие в состав гранатов, можно легко заме-
заместить другими катионами и тем самым систематически изменять
магнитные свойства этих материалов. Например, ионы трехва-
трехвалентных элементов с большим ионным радиусом, занимающие
позиции 24с, можно частично замещать па ионы Са2", а чтобы
сохранялась электронейтралыюсть, некоторую часть ионов Fe3+,
занимающих тетраэдрические узлы, заменяют ионами V3". Фор-
Формулу такого граната можно записать в виде
[ Y3+s-2* <^2+2х1 Pe.,s+ [Fe34-.,-* VA6+1012
Pirc. 16.13. Магнитные моменты гранатов
редкоземельных элементов нрп 0 К [10].
/ —расчет ло уравнению AС.12); 2—
расчет по уравнению A6.11); 3— экспери-
экспериментальные данные.
358
!6. Магнитные свойства
16.3.5. Ильмениты и перовскиты
Ильмениты — название группы фаз с общей формулой АВО3,
где A = Fe, Co, Ni, Cd, Mg; В = Ti, Rh, Mn. Их структура близка
структуре полуторного оксида хрома Сг2О3, гематита a-Fe2O3
или корунда а-А12О3. Ильмениты имеют ромбоэдрическую ре-
решетку, но более удобно рассматривать при обсуждении их
|
12
10
8
6
4
2
0
-2
-4
температура
компенсации
температура
Кюри
300
400
500
100
200
600 Г. К
Рис 16.14. Спонтанное намагничивание железо днепрозисного
функция температуры.
граната как
структуры гексагональную элементарную ячейку больших раз-
размеров (табл. 16.8). Кристаллическая структура в упрощенном
виде может быть представлена в виде плотнейшей гексагональ-
гексагональной упаковки ионов кислорода, две трети октаэдрических узлов
которой заняты катионами. Катионпая подрешетка представ-
представляет собой чередующуюся последовательность слоев ионов А и
В, расположенных вдоль оси с. Структура ильменита может
быть представлена и в виде производной от структуры типа
NiAs, в которой одна треть октаэдрических позиций вакантна.
Структура псровскита SrTiO3 уже была описана ранее в гл. 6.
Некоторые оксиды, содержащие ионы Fe3'1", Mn3K 4+, имеют
структуру перовскита и являются ферромагнетиками. В качест-
качестве примера можно привести смеси ЬазхМп3+0з и А2+Мп3 '¦ 4+Оз,
которые образуют двойные твердые растворы замещения;
Fe
Ti
0
6c
6c
18f
16.3. Примеры магнитных материалов 159
Таблица 16 8. Кристаллографические параметры ильменита
Пространственная группа FcTiO3: R3 {Ст-2Оу. КЗс).
Параметры ромбоэдрической ячейки: а = Г>,538 А, а-54,4Г.
Параметры гексагональной ячейки: а = 5,048 А, с= 14,026 А.
Атом Tiut позиции Координаты агочои
О, 0 и: « = 0,358
0, 0, и: м-0,142
и, V, со; « = 0,305, у= 0,015, ге = 0,25
В ннх ионы больших ])азмеров La3' и А2 занимают позиции
с К.Ч 12, а ноны Мп3"-4" расположены в октаэдрических узлах.
В качестве ионов А могут выступать Са21, Sr2+, Ba2f, Cd2~,
Pb2 . В результате более подробных исследований таких систем
было обнаружено, что их фазовые диаграммы, магнитные свой-
свойства и кристаллохимическис характеристики весьма сложны.
16.3.6. Магнетоплюмбиты
Магнетоплюмбит — это минерал PbFe12Oi9; соединение с той
же структурой, в которой свинец заменен барием, BaFe^Om (гек-
саферрит бария), является важным компонентом постоянных
магнитов. Структура машетоплюмбита родственна структуре
C-глинозема «NaAlnOi7» (гл. 13). Последняя представляет со-
собой плотноупаковапную кристаллическую структуру. В одной
субънчепке помещается пять кислородных слоев. Каждым слой
содержит по четыре иона кислорода в одной элементарной ячей-
ячейке. Основная особенность структуры состоит в том, что в каж-
каждом пятом слое три четверти ионов кислорода отсутствует. Сле-
Следовательно, в каждой пятислойной субъячейке имеется D-4) +
+ 1 = 17 ионов кислорода. В ,3-глиноземе пустые позиции в пя-
пятом слое заняты ионами Na\ Магпстоплюмбит имеет такую же
пятисложную ячейку, как и ji-глипозем; четыре слоя пред-
представляют собой плотпоупаковапные с.чом ионов кислорода. В пя-
пятом слое ионы кислорода занимают лишь три четверти общего
количества кислородных узлов. В остальных позициях кислород-
пых ионов находятся большие двухзарядные катионы РЬ2' или
Ва-л Следовательно, в одной субъяченке имеется D-4) +
+ A-3) —19 кислородных иолов и один ион Ва2: или Pb2f, до-
дополняющие пятый кислородный слой.
Гсксаферрит бария имеет весьма сложную магнитную струк-
структуру, так как ионы Fe!f занимают пять различных кристалле»-
16. Магнитные свойства
графических позиций. Однако суммарный магнитный момент
BaFe^Oio складывается из магнитных моментов восьми ионов
Fe' -, ориентированных в одном направлении, и магнитных мо-
моментов четырех ионов Fe-3~, ориентированных антипараллельно.
В результате суммарный магнитный момент равен ~20гив.
16.4. Применение. Взаимосвязь структуры и свойств
Магнитные свойства материала зависят от многих парамет-
параметров. В настоящее время возможно «конструировать» материалы
с необходимым набором свойств путем тщательного контроля
состава и выбора методов получения. В данном разделе кратко
обсудим некоторые области применения магнитных материалов
и факторы, влияющие на подбор материалов для особых целей.
16.4.1. Сердечники трансформаторов
В основном ферро- и ферримагнитные материалы исполь-
используются в качестве сердечников трансформаторов и электромото-
электромоторов. Для этих целей необходимо иметь магнитно-мягкие мате-
материалы с высоким коэффициентом преобразования энергии и
низкими магнитными потерями. Магнитно-мягкие материалы об-
обладают высокой магнитной проницаемостью (они легко намаг-
намагничиваются в слабых магнитных полях) и низкой коэрцитивной
силой. Это обеспечивает низкие потери па гистерезис. Все эти
свойства характерны для материалов с низким коэффи-
коэффициентом магнитострикцни ks и низким коэффициентом магнит-
магнитной кристаллографической анизотропии Ki. Магнитно-мягкие
материалы — это такие материалы, в которых доменные стенки
могут легко перемещаться. Все вышеперечисленные параметры
легко варьируются путем подбора состава материала и методов
его получения.
Помимо уменьшения потерь на гистерезис серьезную пробле-
проблему представляет собой борьба с вихревыми токами в материа-
материалах с низким электрическим сопротивлением. Особое значение
это имеет при использовании магнитных полек высокой частоты,
так как сила вихревых токов пропорциональна квадрату часто-
частоты. Вихревые токи в таких металлах, как железо, можно пони-
понизить при сплавлении железа, например, с никелем или крем-
кремнием. Понижение силы вихревых токов происходит из-за того,
что сплавы, как правило, имеют сопротивление гораздо выше,
чем сопротивление чистых металлов. Одно из основных преиму-
преимуществ ферримагннтных оксидов, таких, как маргансц-цнпковые
В
16.4. Применение. Взаимосвязь структуры и свойств \Ы
ферриты, никель-цинковые ферриты и железо-иттриевый гра-
гранат, состоит в том, что, будучи правильно синтезированными,
они обладают высоким электрическим сопротивлением и вихре-
вихревые токи в них пренебрежимо малы. Особенно высокое электри-
электрическое сопротивление имеет железо-иттриевый гранат, поскольку
в его состав входят катионы
только трехвалентных метал-
металлов, что затрудняет электрон-
электронную проводи мость. Проводи-
Проводимость этого материала при
комнатной температуре со-
составляет \0~'-2 (Ом-см).
При использовании ферритов
необходимо специально .чаио-
титься о том, чтобы все ионы
железа имели заряд +3. Ина-
Иначе возможен перенос электро-
электронов в ходе окпслительно-ROc-
стзновительной реакции:
Fe2~^Fe3 + 4-e ¦, что приводит
к высокой электронной прово-
проводимости. Например, проводп-
Рис. 16.15. Прямоугольная петля ги- мость магнетита Fe:;04 --=
стерезиса. необходимая для работы — Fe2+Fe23+O4 составляет
устройств храпения информации.
Нп
И
10г—Ю3 (Ом-см)
при ком-
комнатной температуре, что па
— 15 порядков выше проводимости железо-иттриевого граната.
Высокая проводимость Fe3O.j объясняется присутствием в нем
железа в разных степенях окисления.
16.4.2. Запоминающие устройства
Магнитные материалы, используемые в устройствах для хра-
хранения информации, должны обладать определенным набором
магнитных свойств. Это должны быть магнитно-мягкие материа-
материалы, вихревые токи в которых малы, и главное они должны ха-
характеризоваться петлей гистерезиса определенной формы—
прямоугольной или квадратной (рис. 16.15). Материалы с пря-
прямоугольной или квадратной петлей гистерезиса замечательны
тем, что, если намагниченный образец поместить в магнитное
поле обратной полярности, в нем не будет происходить никаких
изменений до тех пор, пока напряженность магнитного поля не
превысит величины коэрцитивной силы Нс. В этот момент на-
намагниченность резко изменит свой знак. Два направления ориен-
Н—1426
162 16. Магнитные свойства
тации намагниченности (+ и —) используют для обозначе-
обозначения 0 и 1 в двоичной системе исчисления. Некоторые магнитные
ферриты обладают этими необходимыми характеристиками, при-
причем время переключения составляет ^10~s с. Такие материалы
находят применение в современной вычислительной технике.
16.4.3. Элементы памяти на цилиндрических магнитных
доменах
Не так давно были разработаны новые типы элементов памяти па ос-
основе тонких слоев гранатов (толщиной в несколько микрометров)i энитак-
еиально выращиваемых на немагнитных подложках (гл. 2). Пленки осажда-
осаждают при высоких температура. Путем тщательного подбора состава гранлта,
и в особенности получая гранаты с определенным параметром решетки, до-
добиваются небольшого различия коэффициентов термического расширения
пленки и подложки. При охлаждении до комнатной температуры сжатие
п.чслки и подложки идет в разной степени. Возникающие в результате это-
этого напряжения достаточны для появления в пленке граната направлений
наиболее предпочтительной намагниченности, которые перпендикулярны пло-
плоскости пленки. Образующаяся при этом доменная структура пленки имеет
результирующую намагниченность, направленною строго ьверх или линз. Ес-
Если эту пленку рассматривать я поляризационный микроскоп, можно уси-
усидеть, что домены имеют форму цилиндров (или пузырьков). Как отмечл-
.¦;ос"> в рлзд. 16.4.2, такие магнитные материалы можно использовать как
элементы памяти в ЭВМ, использующих двоичною систему исчисления.
16.4.4. Постоянные магниты
Чтобы материалы можно было использовать в качестве по-
постоянных магнитов, они должны обладать высокими значениями
намагниченности насыщения, магнитной энергии (т. е. В-И),
коэрцитивной силы, остаточной намагниченности, температуры
Кюри и магнитной кристаллографической анизотропии. Имеются
две группы материалов, используемых в качестве постоянных
магнитов: материалы па основе таких металлов, как Fe, Co, Ki,
п магпитио-жесткие оксиды типа гексаферрита бария или магне-
томлюмбита бария. Рассмотрим пути оптимизации магнитных
характеристик этих материалов.
Жесткость магнитов может быть повышена, если будут най-
найдены способы закреплении доменных стенок или уменьшения их.
подвижности. Этого можно достичь, например, путем введения
в сталь специальных добавок, таких, как хром или вольфрам,
которые вызывают при охлаждении выделение карбидной фазы
или мартенситное превращение. Одно из последних достиже-
достижений— создание сплава алнико—ферромагнитного материала,
представляющего собой матрицу па основе алюминия, в кото-
которую внедрено большое количество мелкокристаллических обла-
областей В состав этих областей входят кобальт и никель. Намагни-
Намагниченность исех областей имеет одно и то же направление, причем.
Упражнения 163
оказывается очень трудно размагнитить такой магнит или изме-
изменить гго магнитную ориентацию.
Оксидные магниты, такие, как гексаферрит бария, относи-
относительно легки и дешевы. Хотя их собственные магнитные свойст-
свойства уступают магнитным характеристикам сплава алнико, их
можно улучшить путем создания на стадии синтеза магнитпо-
улорядочешюй текстуры. Это достигается следующим образом:
исходный материал помещают во внешнее магнитное поле и
хфидают ему соответствующую форму, а затем спекают при вы-
высокой температуре. Влияние внешнего магнитного поля состоит
в магнитном упорядочении зерен, и что повышает остаточную
намагниченность материала.
Упражнения
!6.1. Как, используя магнитные весы Гун, можно определить, является
ли данное вещество парамагнетиком, ферромагнетиком или пнтиферромлгке-
тпком?
16.2.Мопоксид ванадия диамагнитен, он — хороший проводник электри-
честпа. Оксид никеля — парамагнетик при высокой температуре и питифср-
рсмэгнетик при низкой температуре. Он плохо проводит электрический ток.
Объясните эти факты.
16..3. Объясните следующие магнитные свойства шпинелей' a) Z;iFe:.Oi —
амтнферромлгпстик; б) MgFe2O4 — ферримагнетик, причем его магнитный мо-
момент возрастает с температурой; в) МпРегО.! — феррнмагиетпк, его магнит-
магнитный момент не зависит от температуры.
16.4. Почему магнитные материалы, используемые в качестве элемен-
элементов памяти, должны k\icti> квадратную пли прямоугольную петлго шпере-
^ИС,1.
16.5. Почему чистое железо нельзя использовать в качестве материала
для млпштлих сердечников?
16.6. Покажите, что приведенные ниже величины магнитной восприим-
восприимчивости никеля удовлетворяют закону Кюри — Всйсса:
т, к
800
3,3
900
2,1
1000
1,55
1100
1,2
1200
1,0
Оцените значения Тс или 0 и С.
Литература
). Коттон Ф., Уилкинсон Док., Современная неорганическая химия.—М.:
Мир, 1970.
2. Craik D. I. (ed.). Magnetic Oxides, Wiley, 1975, part 1 and 2.
3. Earnshaw A., Introduction (o Magnt-tochemistry, Academic Press, 1968.
4. Gnodennugh J. В., Magnetism and the Chemical Bonds, Wiley, 1963.
5. Gorier L. IF., Nature (London), 165, 798 A450).
G. Hibst /¦/., Hexagonal ferrites from melts and aqueous solutions, magnetic;
recording materials, Angew. Chcmie, hit Ed. Engl., 21, 270 A982).
11*
164 16. Магнитные свойства
7. Кингери У. Д. Введение в керамику. — М.: Стройиздат, 1967.
8. Schieber M. M., Experimental Magnetochemistry, North Holland, 1967.
9. Skull С. G., Strauser W. A., Wollan E. 0., Neutron diffraction by paramag-
paramagnetic and aiitiferromagnetic substances, Phys. Rev., 83, 333 A951).
10. Standley K. J., Oxide Magnetic Materials, Clarendon Press, 1972.
11. Taylor K. hi. R., Contemp. Phys., 10, 423 A970).
12. Tebble R. S., Graik D. J., Magnetic Materials, Wiley, 1979.
13. Wernick J. H., Structure and composition in relation to properties, in Treatise
on Solid State Chemistry, N. B. Hannay (ed.), Plenum Press, vol. 1 A973).
14. Уэрт Ч., Томсон Р. Физика твердого тела.'—M.: Мир, 1969,
Дополнительная литература. Бобак Э., Делла-Торр Э. Цилиндрические
магнитные домены. — М.: Энергия, 1977; Вонсовский С. В. Магнетизм. — М.:
Наука, 1971; Кринчш: Г. С. Физика магнитных явлений. — М.: Изд-во МГУ,
1976; Мишин Д. Д. Магнитные материалы. — М.: Высшая школа, 1981; Преоб-
Преображенский А. А., Бишард Е. Г. Магнитные материалы и элементы. — М.: Выс-
Высшая шкода, 1986; Смоленский Г. А., Леманов В. В. Ферриты и их техническос-
применсние. — Л.: Наука, 1975; Третьяков Ю. Д. Термодинамика ферритов.—
Л.: Химия, 1967.
Глава 17
ЛЮМИНЕСЦЕНЦИЯ
И ЛАЗЕРЫ
Явление поглощения света и природа окраски твердых тел
уже обсуждались в гл. 14. В настоящей главе внимание концент-
концентрируется в основном на свойствах и применении материалов, ос-
основанных на испускании света.
17.1. Люминесценция и люминофоры
17.1.1. Определения и вводные замечания
Люминесценция — общее название явлений, выражающихся
в испускании света веществом после поглощения им энергии.
Существуют различные типы источников возбуждения излуче-
излучения. Тип источника возбуждения включается в термин «люми-
«люминесценция» (часто в виде соответствующей приставки). Так, фо-
фотолюминесценция— это люминесценция, возникающая под дей-
действием фотонов света (УФ-облучсние), При электролюминес-
электролюминесценции для возбуждения излучения используют электрическую
энергию, а при катодолюминесценции— катодные лучи или
электроны достаточной энергии. Различают два типа фотолюми-
фотолюминесценции. Если длительность свечения после прекращения воз-
возбуждения составляет <10~8 с, то процесс называется флуорес-
флуоресценцией; таким образом, флуоресценция прекращается практи-
практически сразу после того, как удаляется источник возбуждения.
Если свечение после удаления источника возбуждения длится
долго (секунды, минуты и даже часы), то такой процесс назы-
называется фосфоресценцией.
Фотолюмннесцентные материалы содержат два компонен-
компонента — кристаллическое вещество основу (например, ZnS, CaW04,
Zn2Si04 и др.) и активатор, добавляемый в небольших количе-
количествах к основе (например, катионы Mn2+, Sn2+, Pb2+, Eu2H);
иногда вводят вторую добавку — сенсибилизатор. Механизм ге-
:G6
17. Люминесценция
возбуждение
К Н ; Н Н Н Н
испускание
Н Н \Н Н К_-/ Н
Н Н
Н Н
Н
возбуждение
Н\ И Н Н
Н \ Н Н Н
/ перенос
н s'"~V н
Рис. 17.1. Механизм возникновения люминесценции [2]. а — в кристалличе-
кристаллическую решетку основы Н введен активатор А; б —а кристаллическую ре-
решетку основы Н введены активатор А и сенсибилизатор S.
перации светового излучения неорганическими люминофорами
(кристаллофосфорами) схематически изображен на рис. 17.1.
Заметим, что в общем случае энергия испускаемого света мень-
меньше энергии возбуждающего облучения, т. е. свечение твердого
тела обычно смещено в длинноволновую область по сравнению
с возбуждающим лучом. Это возрастание длины волны назы-
называется стоксовским сдвигом. В люминесцентных лампах —наи-
слекляннзя трубка
покрытия- из люминофора
,'//7T7~/r77r777r7r7,' ¦
\\ / \
Ослый свет
Рис. 17.2. Схема устройства люмаиесиентиоП /шмиы.
более важной области применения неорганических люминофо-
люминофоров— возбуждающим изучением является УФ-свст (ртутные ис-
источники излучения). На рис. 17.2 схематически изображено
устройство люминесцентной лампы, которая представляет со-
собой стеклянную трубку, внутренняя поверхность которой покры-
17.1. Люминесценция и люминофоры
167
та люминофором. Трубка заполняется смесью паров ртути и ар-
аргона. При пропускании электрического тока через лампу элек-
электроны сталкиваются с атомами ртути, последние возбуждаются,
и их электроны переходят на более высокие энергетические
уровни. Обратный переход в основное состояние сопровождается
испусканием УФ-лучей двух характеристических длин воли 2540
и 1850 А. Эти лучи попадают па фосфоресцирующее покрытие
А
/ N
lr
/лз
гогу*
l
Со
/
/
\ /
\ ,
зелеь'ый
\/
V '
А
\
-—.
йглт
Ми
\
\
красный
4000 ЬООО .6000
Длинэ волны. А
Рис. 17.3. Ci/ектры люминесценции люминофоров на основе ZnS, содержа-
содержащих различные активаторы, после их облучения УФч-ветом.
внутренней поверхности стеклянной трубки, которое испускает
белый свет.
На рис. 17.3 приведены примеры спектров испускания люми-
люминофоров на основе ZnS, активированных различными катионами.
Каждому активатору соответствует характеристический спектр,
что придает кристаллу ZnS определенный цвет. В ходе фотолю-
фотолюминесценции ноны активаторов переходят в возбужденные со-
состояния:
Ион
Основное
состояние
Возбужденное
состояние
Sb-
Eu2+
4/7
4/<*5d>
Материалы основы, используемые для получения кристал-
лофосфоров, можно разделить на две группы:
168
17. Люминесценция
1) диэлектрики, характеризующиеся ионным типом связи,
например Cd2B2O5, Zn2SiO4, апатит 3Ca3(PO4J-Ca(Cl,FJ. При
введении активаторов в такие вещества возникает серия дис-
дискретных энергетических уровней, положение которых модифици-
модифицируется локальным окружением кристаллической решетки ос-
основы. Для качественного описании люминесценции в этих ион-
ионных соединениях служит модель конфигурационных координат;
2) полупроводниковые сульфиды с ковалеитным характером
связи, например ZnS. В них зонная структура основы модифи-
модифицируется путем добавления локализованных энергетических
уровней, возникающих при введении ионов активаторов.
17.1.2. Модель конфигурационных координат
Изобразим график зависимости потенциальной энергии ос-
основного и возбужденного электронных состояний центров люми-
несценшш от некоторой обобщенной координаты (чаще всего
межатомного расстояния); на рис. 17.4 такая зависимость при-
приведена для основного сос-
состояния. Кривая потенциаль-
потенциальной энергии позволяет ка-
качественно оценпчь измене-
изменение потенциальной энергии
в зависимости от межатом-
межатомного расстояния. При неко-
некоторой равновесной величине
длины химической связи
гс. кривая потенциальной
энергии проходит через ми-
минимум. В данном основном
электронном состоянии воз-
возможны различные дискрет
ные колебательные состоя-
состояния иона, которым отвеча-
Межатомное расстояние
Рис. 17.4. Изменение тлотенцил.шноп
энергии основного состояния центра
люминесценции п зависимости от меж
атомного расстояния в с.пч.и1 полного
кристалла.
ют уровни энергии Vo, У\
н т. д. (горизонтали на рис.
17.4).
Каждому электронному
состоянию центра люминес-
люминесценции соответствует кривая потенциальной энергии, аналогич-
аналогичная том, которая приведена па рис. 17.4. Типичное положение
кривых потенциальной энергии основного н возбужденного со-
состояний показано на рис. 17.5. Воспользуемся рис 17.5 для
объяснения некоторых особенностей люминесценции.
Во-первых, пропесс возбуждения представляет собой пере-
переход активного центра с энергетического уровня А основного со-
17.1. Люминесценция и люминофоры
169
стояния на одни из более высоких колебательных уровней, на-
например уровень В, возбужденного состояния. Во-вторых, в ходе
быстрой релаксации в возбужденном состоянии (переход на
более низкий колебательный уровень С) происходит некоторая
диссипация энергии. Энергия теряется в виде тепла, рассеивае-
рассеиваемого кристаллической решеткой основы. В-третьих, активный
центр возвращается в свое основное состояние (на уровни А
ге Межатомное расстояние
Рис. 17.5. Изменение потенциальной энергии основного п возбужденного со-
состояний центра люминесценции в зависимости от межатомного расстояния.
или D), испуская при этом свет. Поскольку энергия возбужде-
возбуждения перехода А-+В выше, чем энергия испускания при переходе
C-+D, то испускаемое излучение характеризуется большей дли-
длиной волны по сравнению с возбуждающим облучением. Это объ-
объясняет стоксовский сдвиг.
С помощью рис. 17.5 можно также объяснить явление термиче-
термического тушения, проявляющееся в том, что выше определенной
температуры свечение люминофора заметно уменьшается.
Из рис. 17.5 видно, что в точке Е кривые потенциальной энер-
энергии основного и возбужденного состояний пересекаются. В этой
точке ион, находящийся в возбужденном состоянии, может пере-
переходить обратно в свое основное состояние с той же энергией.
Затем он может перемещаться на более низкие колебательные
уровни основного состояния путем ряда последовательных пере-
переходов. Таким образом, точка Е является своеобразной перевалъ-
170
17. Люминесценция
нон точкой. Если ион при переходе в возбужденное состояние
приобретает достаточно энергии и достигает точки Е, он может
попасть на один из колебательлых уровней основного состояния.
Если это произойдет, то вся высвобождающаяся энергия выде-
выделится в виде колебательной энергии, и люминесценции ие на-
наблюдается. Поэтому энергия, отвечающая точке Е, называет-
называется критической энергией. Такой критический уровень энергии
может быть достигнут путем нагревания, так как при этом
увеличивается тепловая энергии ионов и становится возможным
их переход па более высокие колебательные уровни.
основное
состояние
сенсибилизак;.
перенос
энергии
ч
испускание
основное
состояние
активатора
Рис. 17.6. Безызлучательный перенос энергии в сл\час люминофоров, содер-
содержащих сенсибилизатор.
Описанный выше для объяснения явления термического ту-
тушения тип перехода — один из примеров так называемых белыз-
лучательных переходов. При таких переходах возбужденный
ион теряет часть своеГт избыточной энергии путем передачи ко-
колебательной энергии своему окружению в кристаллической ре-
решетке основы. В этом случае возбужденный v.uw может веркуть-
си па более низкий энергетический уровень, не испуская элек-
электромагнитного излучения, т. е. света.
Другой тип безызлучательпых переходов имеет место в слу-
случае сенсибилизированных люминофоров. Этот переход называют
безыз.гучателмшм переносом энергии (рис. 17.6). Бсзызлуча-
тельпый перепое энергии осуществляется, если: 1) энергетиче-
энергетические уровни ионов активатора и сенсибилизатора в возбужден-
возбужденном состоянии близки между собой; 2) ионы активатора и сен-
сенсибилизатора занимают относительно близкие позиции в кристал-
кристаллической решетке основы. В этом случае под действием возбуж-
возбуждающего облупсипя в возбужденное состояние переходят прежде
всего ноны сенсибилизатора. Затем происходит перенос энергии
на соседние ионы активатора. При этом в ходе переноса не про-
17.1. Люминесисипмя и люминофоры 175
исходит потери энергии (или теряется лишь очень незначитель-
незначительная доля энергии), ионы же сенсибилизатора переходят обрат-
обратно в основное состояние. И наконец, ионы активатора переходят
в свое основное состояние, излучая свет (люминесцируя).
Безызлучатсльный перенос энергии лежит в основе отрав-
отравляющего действия некоторых примесей. Механизм отравляюще-
отравляющего действия заключается в переносе энергии от сенсибилизатора
или активатора на примесный атом. При этом энергия рассеи-
рассеиваете;-! в кристаллической решетке основы, переходя в колеба-
колебательную энергию. При получении кристаллофосфоров необходи-
необходимо удалять ионы, склонные к бе:шзлучатслышм переходам п ос-
основное состояние (например. Со'-'', Fe2'', Ni2+).
17.1.0. Некоторые фосфоресцирующие митериалы
К настоящему времени экспериментально изучено множества
систем тина «основа — активатор» е целью определения их ос-
основных характеристик как люминофоров. Многие из этих систем
оказались весьма перспективными для практического использо-
использования. Однако дальнейший прогресс в создании новых люминес-
люминесцентных материалов связан с теоретическим выяснением соот-
соотношения между кристаллической структурой и положенном
эперге!ических уровнен легирующих ионов.
Приведем некоторые примеры применения люминофоров.
В люминесцентных лампах широко используются люминофоры
па основе апатита с добавками ионов Мп-' и Sb11'. Сам фторо-
анапгг имеет формулу Са5(РО4)зР. При введении ионов Si<r" on
светится синим цветом, а добавки Мп2'" дают оранжево-желтое
свечение. Совместное же присутствие ионов Мп2+ л Str+
приводит к широкому спектру испускания фосфоресцеитиогс
материала, который охватывает почти всю область белого света,
Путем частичного замещения ионов F~ во фтороапатите нп
ионы С)" можно достигнуть некоторого изменения распределе-
распределения длин волн в спектре испускания. Такой эффект объясняется
тем, что подобное замещение ведет к изменению положения
энергетических уровней ионов активатора и, следовательно,
к изменению длин волн испускаемого излучения. Такп.м обра-
образом, парьировапие химического состава люминофора -- вполне
эффективное средство достижения желаемой окраски при све-
свечении. В табл. 17.1 указаны люминофоры, используемые в лю-
люминесцентных лампах.
Важное значение как ион-активатор имеет ион Ьиз:'. Его вве-
введение в люминофор даст красное свечение, что используют, на-
например, при изготовлении экранов цветных телевизоров. В лю-
люминофоре YVO< '. Ни3'' группа [VO4]3~ поглощает энергию катод-
катодных лучей в электронно-лучевых трубках, эмптером (иснускатс-
172 ^ ^^ 17. Люминесценция
л ем) же энергии является ион Ей3. Перенос заряда от группы
[VO4]3~ па ион Ей3""", по-видимому, безызлучательный процесс,
протекающий по обменному механизму через кислородные ионы.
Таким же .механизмом обменного взаимодействия объясняется
возникновение аптиферромагннтпоги упорядочения ионов Ki-X
в NiO (гл. 16). Перенос энергии в люминофорах по обменному
механизму будет особенно эффективным, если химические свя-
связи металл — кислород—металл будут направлены вдоль одной
линии. Именно в этом случае достигается максимальное пере-
перекрытие атомных орбиталей. В люминофоре YVO4 : Eu3+ угол
Таблица 17.1. Некоторые люминофоры, используемые п люминесцентных
лампах [1]
Люминофор AK-iHHtH'Jii Циог
Виллемит
Y2O3
Диопсид
Волластоииг CaSiOj
(Sr, ZnK(PO4J
Фтороапатит Са5(РО4)з(Р, С1)
Mil2*
Eu
1:
Pb
Sn
Sb
, Mn
. Mn
Зеленый
Красный
Синий
Оричжево-
желтый
Оранжевый
«Белый?
между связями ванадий — кислород-—европий составляет 170°,
поэтому перенос энергии осуществляется весьма быстро.
При добавлении в качестве активатора ионов Ей31" в принци-
принципе возможны энергетические переходы па различные /-уровни и
с различных /-уровней. Эти разные переходы действительно на-
наблюдаются, и поэтому окраска таких люминофоров зависит от
кристаллической структуры основы, и в особенности ст симмет-
симметричности позиции, которую занимает пои EuJl. Если ион Еи-3^
попадает в центр симметрии (как в случае легированных евро-
европием кристаллов NaLuO2 n Ba^GdNbOe), то наиболее вероятен
переход типа 5D0^-7Fl. В результате свечение этих люминофоров
имеет оранжевый цвет. Если ион Eu:i4- находится в пецептросим-
мстричной позиции (как в случае NaGdO2 : Eu31"), то наиболее
вероятен переход типа &Dt)^7F2 и цвет свечения красный. Таким
образом, кристаллическая структура основы имеет первостепен-
первостепенное значение для окраски люминофора.
При изготовлении экранов цветных телевизоров используют
катодолюминофоры трех цветов:
1) красный (обычно используют упоминавшийся ранее
YVO4: Eu3+);
17.1. Люминесценция и люминофоры.
173
2) синий (ZnS : Ag ');
3) зеленый (ZnS:Cu:).
При изготовлении экранов
смеси «синего» (ZnS : Ag1)
долюмипофоров.
черно-белых телевизоров используют
и «желтого» ((Zn, Cd)S : Ag") като-
17.1.4. Антистиксовские люминофоры
Антнстоксовские люминофоры—-относительно новый класс
светящихся материалов, к которым в последние годы проявля-
проявляется повышенный интерес. Отличительная особенность этих ве-
веществ состоит в том, что они испускают поток фотонов боль-
большей анергии (меньшей длины волны), чем первичное возбуж-
возбуждающее облучение. Применение антистоксовских люминофоров
позволяет преобразовывать инфракрасное излучение в электро-
электромагнитное излучение большей энергии, в видимый свет. Закон
сохранения энергии, конечно, не нарушается. Дополнительная
энергия, естественно, поглощается люминофором, но процесс
возбуждения протекает в две или более стадии (рис. 17.7).
йторая
стадия
nepn.iH
¦ладил
возбуждения
испускание. возбуждение
i испускание
Рис 17.7. Схематическое изображение процессов, протекпющих в антисток
совских (а) и обычных F} люминофорах.
В настоящее время наиболее подробно исследованы анти-
антистоксовские люминофоры па основе YF3, NaLa(WO4b Ha-NaYF4,
которые легированы ионами Yb3+ (сенсибилизатор) и Ег3+ (ак-
(активатор). При облучении таких люминофоров инфракрасным
светом возникает свечение зеленого цвета. При этом осущест-
осуществляется перенос двух фотонов, поглощенных нонами Yb3+ в хо-
ходе ИК-облучения материала, на соседние ионы Ег3+. После
двухстадпшюго процесса возбуждения активатора (рис. 17.7)
начинается испускание электромагнитных волн видимой части
спектра.
174 17. Люминесценция
17.2. Лазеры
Работа твердотельных лазеров основана на использовании
люминофоров, удовлетворяющих некоторым особым требова-
требованиям. Термин лазер составлен из первых букв английских слов
Light Amplification by Stimulated Emission of Radiation (уси-
(усиление света стимулированным испусканием излучения). Процесс
возбуждения заключается в переводе активных центров в воз-
возбужденное состояние (так называемая накачка), в котором они
могут пребывать достаточно длительное время. Состояние, ха-
характеризующееся тем, что большая часть активных центров на-
находится в возбужденном состоянии, называется «инверсной за-
заселенностью». В лазерах испускание фотона света одним актив-
активным цстром стимулирует испускание свеча другими центрами,
причем все пзл\ чаемые электромагнитные волны распростра-
распространяются в одной фазе. Таким обрлзом, возникает интенсивны/)
пучок или импульс когерентного излучения.
В 1960 г. Мспман сообщил об изобретении первого рубино-
рубинового лазера. В настоящее время можно говорить о существова-
существовании самостоятельного направления пауки и техники, связанного
с исследованием и применением лазеров в различных облапнх
человеческой деятельности: фотографии, хирургии, средствах
связи, для точных измерений и многих других. Открыты многие
типы лазерных систем. Все используемые лазеры можно разде-
разделить на три типа: газовые лазеры, лазеры на красителях с пол
страиваемой длиной волны и твердотельные лазеры. В цели ав-
автора не входило дать общий обзор всех лазерных систем; кос
немея лишь некоторых вопросов, связанных с работой твердо
тельных лазеров.
17.2.1. Рубиновый лазер
Рубиновый лазер был первым оптическим квантовым генера-
генератором. И сегодня, более двадцати лет спустя, он находит широ-
широкое применение. Наиболее важная часть рубинового лазера ¦-
монокристалл А12О.-„ легированный небольшим количеством
( — 0,05 масс. %) Cr3J\ Ионы Сг?+ замещают ионы АГ"<" в иска
жепных октаэдрах кристаллической решетки корунда (структу-
(структура корунда близка структуре ильменита FeTiOs; см. разд. 16.3.5).
При введении Сг2О3 в монокристаллы АЬОз белого цвета по-
последние окрашиваются в красный (при малой концентрации
ионов Сг31) или зеленый (при больших концентрациях ионов
Cr:t+) цвет. Твердые растворы системы А12Оз—Cr2O:t обсужда-
обсуждались ранее в гл. 10.
На рис. 17.8 схематически показано положение энергетиче-
энергетических уровнен попа Сг:~ в рубине. Накачку лазера производят
17.2. Лазеры
175
с помощью мощной вспышки света, например ксеноновой лам-
лампы. При этом d-электроиы ионов Сг3+ могут переходить из ос-
основного состояния Мг в более высокие состояния 4/72 и '^ь
Затем быстро протекает безызлучательный переход на уро-
уровень 2Е. Время пребывания электронов в возбужденном состоя-
состоянии 2Е весьма велико и составляет ~5-10~3 с. Это значит, что
успевает образоваться состояние инверсной заселенности. Ла-
Лазо
20
10
поглощение
\ перехолм
лазерное
излуче^т:
G934A
* основное состояние
Рис. 17.8. Энергетические уровни попа Сг3+ и лазерное излучение в кристал-
кристалле рубина.
зерпос излучение возникает при переходе электронов из возбуж-
возбужденного состояния 2Е в основное состояние. Этот переход стиму-
стимулируется одновременно для многих ионов, причем излучаемые
электромагнитные волны распространяются в одной фазе. Дли-
Длина волны мощного когерентного импульса красного спета состав-
составляет 6934 А.
На рис. 17.9 изображена схема устройства рубинового лазе-
лазера. Основная часть лазера — рубиновый стержень длиной не-
несколько сантиметров и диаметром 1—2 см. Как показано на
рис. 17.9, рубиновый стержень окружает в виде спирали газо-
газоразрядная импульсная лампа. Возможно и другое, расположенно
лампы рядом со стержнем. В этом случае лампа и рубиновый
стержень помещаются внутри отражающего кожуха так, чтобы
стержень интенсивно облучался со всех сторон. С одного торца
176
17. Люмиксптешеп!
ИМПуЛиС-lii'H ,¦ i-/.:id
О О О О
зеркало
пере-
ИЛЮЧ.Т"
лазер;-
О О О
Рис. 17.9. Схема устройства рубинового лазера.
стержня устанавливается зеркало для отражения импульса ис-
испускаемого света обратно в сторону рубинового стержня. С дру-
другого конца стержня ставят специальное устройство — так на-
называемый Q-переключатель, который может либо выпускать луч
лазера из системы, либо отражать его обратно для нового цик-
цикла. Q-Псреключатель обычно представляет собой вращающееся
зеркало, которое в строго определенное время выпускает лазер-
лазерный луч из системы. Такая конструкция позволяет добиться из-
излучения оптимальной интенсивности, так как импульс испус-
испускаемого света, прошедший после отражения в зеркале вперед
и назад, вдоль стержня, увеличивает интенсивность излучения.
z
"о
10
У?"
поглощение
8800 А
лазерное
излучение
10 600 А
'Л,,.
П/2
Рис. 17.10. Энергетические уровни иона Nd3+ в неодимовыч лазерах
Упражнения 177
Это связано с тем, что лазерный луч стимулирует испускание
новыми активными центрами, причем стимулированное излуче-
излучение когерентно начальному импульсу света.
17.2.2. Нсодимовые лазеры
В качестве основы люминофоров, используемых в неодимо-
вых лазерах, служит либо стекло, либо алюмо-иттриевый гра-
гранат Y3A!r,Oi2 (свойства алюмо-иттриевого и других гранатов об-
обсуждались в разд. 16.3.4). Активными центрами в этих материа-
материалах являются ионы Nd3+. На рис. 17.10 показано положение
энергетических уровней и схема энергетических переходов, реа-
реализующихся при работе неодимовых лазеров. Возбуждающее
облучение люминофора проводят с помощью мощной лампы.
При поглощении энергии возможны переходы на несколько
энергетических уровней (на рисунке изображен переход лишь
в одно возбужденное состояние). Далее следует безызлучатель-
ный переход на возбужденный уровень 4F3/2, с которого осу-
осуществляется переход в состояние 4/ц/2- Последний перолч^д со-
сопровождается лазерным излучением с длиной волны 10 600 А
(для пеодимового стекла) или 10 640 А (для УзА15О12: Nd3+).
Время пребывания в состоянии 4F3/2 достаточно велико
(~Ю~'' с) и зависит от концентрации ионов Nd34'. Это позволя-
позволяет возникать состоянию инверсной заселенности. На основе ма-
материалов, содержащих ионы Nd:': в качестве активаторов, соз-
созданы лазеры высокой мощности.
Упражнения
17.1. Какие общие требования предъявляются к твердым материалам,
используемым в лпзерах?
17.2. Лптнстоксовские люминофоры испускают свет меньшей длины вол-
волны, чем длима волны нозб уж дающего нз.т, чеиия. Объясните, почему не на-
нарушается при агом алкоп сохранения энергии.
Литература
1. Burr us H. L., Lamp Phosphors, .Mills am) Boon, 1972.
2. Deluca J. А., Ли introduction to luminescence in inorganic solid?, J. Chcm
Ы., 57, 5-41 A980).
3. Evans T. /?., Applicaiions of Lasers lo Chemical Problems. Wiley, 1982.
4. Maiman Т. Н., Stimulated oplica! radiation in ruby. Nature, 187, 493 (l%0).
5. Мпоге С. В., Chemical and Biochcmicnl Applications of Lasers, Academic
Press, 1974.
6. Newnham R. П., Structure — Property Relations Springer-Verlag, 1975.
Дополнительная литература. Башкип А. С. Игошин В. И., Орасвский А. П.,
Щеглов В. А Химические лазеры./Под ред. Басова II. Г. — М/ Наука, 19Н2;
Казанкин О. Н., Марковский Л. Я-, Мирпнов И. А. и др. Неорганические лю-
люминофоры.— Л.; Химия, 1975; Левшин В. Л., Левшин Л. В. Люминесценция
\\ се применение. — М.: Паука, !972.
12—1426
Глава 18
СТЕКЛО
Стекло — один из самых древних синтетических материа-
материалов, используемых человечеством. Сведения о стекле накапли-
накапливались в течение многих веков, но научный подход к исследо-
исследованию стекла зародился лишь в начале XIX в. благодаря рабо-
работам Фарадея и его современников. К настоящему времени
исследованием стекол занимается самостоятельное научное
на правление, которое быстро развивается благодаря привлече-
привлечению самых современных методов изучения структуры и свойств
стекол. Исследование стекол имеет и отчетливые практические
приложения, поскольку постоянно создаются новые стеклообраз-
стеклообразные материалы со специальными характеристиками. Известно
два основных определения стекла. В одном определении акцен-
акцентируется внимание на наиболее распространенном способе полу
чения стекла. Этот способ заключается в охлаждении расплава
без его закристаллнзовывания до того момента, пока за счет
резкого повышения вязкости не возникает упругое твердое тело.
Coi.'iacHO второму определению, первостепенное значение имеет
структура стекла: стекло — это аморфное твердое тело, в кото
ром отсутствует дальний порядок и периодичность в располо-
расположении атомов. По-видимому, ни то ни дру! ос определение нель-
нельзя считать идеальным. Ведь в настоящее время помимо ох
лаждения расплавов известны и другие методы получения сте-
стекол, например высушивание гелей или осаждение из irapoBoii
фазы. Известно, что аморфный твердый материал, аналогичный
но свойствам кварцевому стеклу, можно приготовить путем гид-
гидролиза тетрамстилортосиликата Si(OCH:iL и последующего на-
назревания желеобразного продукта до ~о50°С. В свяли с этим
вполне правомочны и такие вопросы: любое ли аморфное твер-
твердое тело можно считать стеклом или относится ли стекло про-
просто к одному из типов некристаллических твердых тел.
Существует широкий круг материалов, который образуют
стекла. Наибольшее значение для практики имеет ряд неоргапи-
18. t. Факторы, влияющие на стеклообразование _ Г79
чсских силикатов и боратов. Однако стекла также можно полу-
получат)) из некоторых водных растворов, смесей расплавленных со
.'•ей, металлических и полупроводниковых расплавов, жидких
органических соединений. В данной главе речь пойдет в основ-
основном о структуре, свойствах и применении оксидных (главным
образом силикатных) стекол, а также о некоторых новых стек-
стеклах, имеющих интересные свойства и области применения.
18.1. Факторы, влияющие на стеклообразование
18.1.1. Оксидные стекла. Электроотрицательность
и run химической связи
Перечень стеклообразующих оксидов включает в основном
следующие: SiO2, B2O;i, GeO2, P2Oj. Элементы, образующие эти
оксиды, находятся в довольно узкой области периодической си-
С1смы (рис. 18.1) и характеризуются средними значениями элек-
троотрицатслыюсп:, т. е. элсктроотрицателыюсть этих элемен-
элементов не настолько мала, чтобы они образовывали ионные соеди-
соединения (такие, как MgO или NaCl), по и не настолько всличл.
чтобы на их основе возникали соединения с молекулярное
структурой н ковалентнымн связя-
связями (типа СО2). Характер связей в
соединениях таких элементов— I^J^
смешанный (ионный и ковалент- <- |Al\Si P) S
пып). а их структуры представля- ^ Zn GcAOa'As Se
k;v собой обычно трехмерный по- r q ci -
димерный каркас. Оксиды других л л"
элеуентив, находящихся по сосед- Ti Pb Bi
ству в периодической системе, так- РйС ,8л элементы, оши«
же проявляют склонность к стек- которых склонны к стеклооб-
ло!)бразоваиию. Некоторые из них, рпзовапию.
такие, как AS2O3 и Sb2O3, перехо-
переходят в стеклообразное состояние лишь при очень большой ско-
скорости охлаждения расплава. Другие, например А1:>О^
Ga?O3, Bi?O3, SeO2, TeO2, называют условными сгек-
лообразователями, так как сами по себе они стекол не образу-
образуют, но в присутствии некоторых других псстеклообразующих ок-
оксидов все-таки возникают стеклообразные фазы. Например,
в системе СаО—A!2O-,j существует область составов, в которой
стекла образуются при охлаждении расплавов, хотя ни д-1Я СаО.
пи для А12О3 не известны стеклообразные формы. Стеклообра-
зующис оксиды (SiO2, В2О3 и др.) могут образовывать стекла
как в чистом виде, так и при добавлении к ним значительных
количеств нестеклообразующих оксидов. Например, стекла лег-
легко образуются при добавлении к SiO2 или В2О^ до 20—40%
оксидов щелочных металлов.
12*
J80 18 Стекло
18.1.2. Вязкость
Вязкость расплава несколько выше температуры плавления
несомненно является важным фактором в процессе стеклования.
Все епклообразующне оксиды (рис. 18.1) в расплавленном со-
состоянии характеризуются очень высокой вн.чкосчыо. Так, вяз-
вязкость оксида кремния выше температуры плавления (при
171,з~С) составляет ~ 10; П (разд. 18.6). В противоположноеil
этому многие неорганические соединения, не склонные к стекло-
образованию, в жидком состоянии характеризуются низкой вяз-
вязкостью. Так. вязкость воды при О"С или расплавленного хлори-
хлорида лития при (з13°С составляет -—-2-10 2 П. Вязкость расплава
определяется структурой и типом химических связей. Расплав-
Расплавленный иксид кремния имеет аморфную полимерную структуру
с прочными связями Si—О, благодаря этому и высокую вяз-
вязкость. Для того чтобы такая жидкость закристаллизовалась, не-
необходимо разрушить и переориентировать прочные химические
.связи и провести перегруппировку атомов. Ясно, что подобные
процессы в вязком полимерном расплаве протекают с большим
¦трудом, чем в ионных или молекулярных жидкостях. Однако
существуют и некоторые исключения в таком общем подходе
к вязкости как к критерию склонности расплава к стсклообразо-
ванию. Вот два примера жидкостей, имеющих низкую вязкость
в жидком состоянии и тем не менее образующих при охлаж-
охлаждении стекло: а) водные растворы ZnCl2; б) расплавленная
смесь LiXO3 и Са(МО3J.
18.1.3. Влияние структуры. Правила Захариассна
Захарпасен A932 г.) изучил относительную склонность про-
простых оксидов к стеклообразованию и сделал вывод о том, что
важнейшим условием стек.тообразьвания является способность
вещества образовывать непрерывную трехмерную сетчатую
структуру, в которой отсутствует дальний порядок. На основа-
основании такого подхода Захариассп сформулировал ряд правил, ко-
которые играют большую роль при изучении стеклообразного со-
состояния твердых тел. Поскольку строение стекол обычно неиз-
неизвестно, эти правила оперируют с кристаллическими структура-
структурами соответствующих оксидов, из которых образуются или не об-
образуются стекла. Такое допущение вполне разумно, так как
кристаллические и стеклообразные модификации оксидов ха-
характеризуются, видимо, одинаковым типом химической связи,
одними п теми же координационными полиэдрами и т. п. При
обсуждении этих правил необходимо различать кислородные
атомы и «другие» атомы в бинарных оксидах. Правила Заха-
риасеиа формулируются следующим образом:
18.1. Факторы, влияющие на стсклообразоцант? _____ 181
Ij атом кислорода может быть связан максимально с дву-
двумя «другими» атомами;
2) координационные числа «других» атомов невелики;
3) образуемые кислородными атомами координационные по-
полиэдры, в которых находятся «другие» атомы, сочленяются друг
с другим вершинами, а не гранями или ребрами;
4)полиэдры объединены друг с другом в трехмерную сетку.
Четвертое правило уже обсуждалось выше. Наличие сетча-
сетчатой структуры, особенно в совокупности с высокой вязкостью
расплава, существенно способствует стеклообразованию.
Выполнение первого и третьего правил открывает возмож-
возможность существования в стекле пли в расплаве трехмерной сетча-
сетчатой структуры, в которой отсутствует дальний порядок. Так,
кварцевое стекло построено из сочлененных вершинами тетра-
тетраэдров SiO4, в которых каждый кислородный атом связан только
с двумя атомами кремния. Такая нежесткая структура способ-
способствует тому, что валентные углы между связями кислородного
атома Si—О—Si могут изменяться в широких пределах, не при-
приводя к разрушению самих тетраэдров. Таким образом, возмож-
возможна реализация трехмерной сетчатой структуры, в которой отсут-
отсутствуют дальний порядок и периодичность расположения атомов.
В каждой полиморфной модификации кристаллического оксида
кремния (кварц, крнсюбалит и др.) величина валентных углов
Si—О—Si строго постоянна, что приводит к регулярному распо-
расположении) тетраэдров SiO4. И кристаллический оксид кремния, и
кварцевое стекло имеют сетчатые структуры, по сама структура
сетки у них различная: одна сетка — регулярная, упорядочен-
упорядоченная, другая — нерегулярная, разупорядочетшая. Если структура
такова, что кислород имеет высокое координационное число (см.
первое правило), то в особенности при сочленении полиэдров
ребрами или гранями (см. третье правило) возникают затруд-
затруднения с образованием неупорядоченной сетки (естественно при
условии, что внутри полиэдров существует полный порядок).
Второе правило тесно связано с первым. Для соединении оп-
определенного состава координационные числа различных атомов
взаимно обусловливают друг друга. Так, в SiO2 КЧ атома крем-
кремния равно 4 и, следовательно, для кислорода КЧ должно состав-
составлять 2 (гл. 8).
Посмотрим, насколько приведенные выше правила примени-
применимы к оксидам элементов других групп периодической системы.
В оксидах щелочных н щелочноземельных металлов, не обра-
образующих стекол, очевидно, эти правила не выполняются. Напри-
Например, в Na2O КЧ кислорода равно 8, а в MgO — 6 (см. первое
правило), и полиэдры, составляющие структурную основу их
кристаллических решеток (NaO4 и MgO6), сочленены ребрами,
а не вершинами.
182 ___ 18. Стекло
К оксидам третьей группы правила Захариасена применимы
только в том случае, если «другие» элементы имеют КЧ 3. Так,
если исходить из формулы М2О3, «другие» элементы должны ко-
координировать вокруг себя три атома кислорода, чтобы коорди-
координационное число кислородного атома было равно двум (первое
правило). В В2О3 это условие выполняется (атом бора находит-
находится в центре трсуюльииков из кислородных атомов), а в А12О3
не выполняется (алюминий занимает положение в центре окта-
октаэдра нз кислородных атомов). Такая структура оксидов бора п
алюминия хорошо коррелирует с их способностью к стеклооб-
разованию.
Для оксидов элементов IV и V групп правила выполняются
в том случае, если «другие» элементы находятся в тетраэдрите-
ском окружении атомов кислорода. Лишь такой вариант струк-
структуры позволяет кислороду иметь КЧ 2 (в случае элементов
IV группы) и в среднем несколько меньше двух (элементы
V группы). Дробные координационные числа, естественно, не-
невозможны. Например, в Р^О.-, часть кислородных атомов имеет
КЧ 2, а остальные атомы кислорода — КЧ I.
Используя сформулированные правила, Захариасеп смог про-
прогнозировать склонное1ь некоторых оксидов к стеклообразова-
нию,* иод1вердив чатом свои ожидания .экспериментально (для
Y2O5, Nb2O5. Ta2Os, Sb2O5, P2O3, Sb2O3).
В последние годы установлено, что правила Захариасепа
все же имеют некоторые ограничения. Например, были получе-
получены оксидные стекла, не удовлетворяющие этим правилам.
В частпосш, существуют стекла, не имеющие трехмерную сетча-
сетчатую структуру. 3ih примеры вовсе не свидетельствуют о беспо-
бесполезности правил Захариассна, а лишь еще раз напоминают
о затруднениях, которые ожидают теоретиков и экспериментато-
экспериментаторов при создании общей структурной теории стеклообразоваппм.
18.1.4. Критерии Сана и Роусона
Известны и другие корреляции между особенностью кристал-
кристаллической структуры и склонностью простых оксидон к стеклооб-
разовапию. Сап предположил, что весьма важной характеристи-
характеристикой является прочность связи кислорода с «другими» элемента-
элементами. Он обратил внимание на то, что энергия связи в стеклооб-
разующих оксидах составляет >330 кДж/моль, в то время как
энергия связи кислорода с ионами модификаторов, которые по-
поучаствуют в образовании сетчатой структуры, существенно
меньше.
Роусон модифицировал критерий Сапа и связал склонность
к стеклообразовакию с отношением энергии связи к температу-
температуре плавления. Это отношение должно характеризовать как проч-
18.2. Термодинамика процесса стеклования 183
ность связи, так и тепловую энергию, необходимую для разрыва
связей, которая зависит от температуры. Практически невоз-
невозможно закристаллизовать стеклообразный В2Оз, и это полно-
полностью соответствует критерию Роухона, так как температура
плавления В2О3 невелика и составляет —400 °С. С помощью
этого критерия можно также объяснить, почему в бинарных си-
системах область стеклообразования часто находится вблизи низ-
низкоплавкой эвтектики. В этом отношении показательна система
СаО—АЬОц: пн СаО, пи ЛЬО^ не способны образовывать стек
ла (если не учитывать получение тонких пленок методом
•сверхбыстрой закалки), по в области составов между CaA!2O,j
(СА) н Са3А12О{, (С3А) (рис. 19.8) стекло легко образуется. Эти
составы лежат вблизи ннзконлавкой эвтектики (температуры
ликвидуса ~ 1400—1600°С).
Дополнительно к вышесказанному необходимо учитывать,
что в бинарных и многокомпонентных стеклообразующих систе-
системах состав стекла может существенно отличаться от состава со-
соответствующей кристаллической фазы. Поэтому для кристалли-
кристаллизации охлаждаемого расплава необходимо, чтобы диффузия
атомов и ионов осуществлялась в достаточно больших областях
расплава. Это ведет к уменьшению скорости кристаллизации и
к возрастанию вероятности стеклообразования.
18.2. Термодинамика процесса стеклования.
Поведение жидкостей при охлаждении
При охлаждении в расплавах могут протекать два различ-
различных процесса. Либо расплав кристаллизуется при температуре
плавления или несколько ниже этой температуры, либо он зна-
значительно переохлаждается и без кристаллизации переходит
в стеклообразное состояние. На рис. ]8.2 приведены температур-
температурные зависимости объема в системах, где идет кристаллизация
или стеклообразованнс. Изменение объема при охлаждении
большинства нестсклообразующих расплавов может быть описа-
описано кривой abed. При температуре У'Пл начинается кристаллиза-
кристаллизация (отрезок be), хотя иногда перед началом кристаллизации
из-за кинетических затруднений может происходить некоторое
переохлаждение расплава. Больший наклон отрезка ab по срав-
сравнению е наклоном cd указывает на то, что коэффициент терми-
термического расширения жидкости обычно больше, чем твердой
фазы.
Изменение объема при охлаждении стеклообразующей систе-
системы показано кривыми abef и abgh. В области be существует пе-
переохлажденная незамороженная жидкость. В этой области при
любой температуре жидкость быстро достигает состояния внут-
внутреннего равновесия, и оно успевает устанавливаться вслед за
184
18. Стекло
изменением температуры окружающей среды. Тем не менее со-
состояние переохлажденной жидкости является метастабильным по
отношению к кристаллическому состоянию. При понижении тем-
температуры вязкость жидкости постепенно возрастает до тех пор,
пока не достигнет значения, при котором внутреннее равновесие
жидкости уже не устанавливается. Расположение атомов в та-
'У Ып
Темпер?iура
Рис. 18.2. Зависимость объема от температуры для кристаллических, жид-
жидких и стеклообразных веществ.
кой переохлажденной жидкости как бы замораживается, и при
последующем охлаждении вещество становится таким же упру-
упругим и жестким, как и кристаллическое твердое тело. Образо-
Образовавшееся твердое тело, однако, характеризуется отсутствием
трехмерной периодичности в расположении атомов, присущей
кристаллическим веществам. Такое изменение свойств при пе-
переходе из расплава в стеклообразное состояние происходит при
некоторой температуре (или в некоторой области температур),
называемой температурой стеклования Tg.
Для любого данного состава в принципе возможно синтези-
синтезировать стекла с разной степенью стабильности, т. е. характери-
характеризующиеся различными значениями Ts. Если расплав не закри-
сталлизовывается, то при медленном охлаждении жидкость мо-
18,2. Термодинамика процесса стеклования
185
жет пребывать в состоянии внутреннего равновесия до более
низких температур, чем в случае быстрого охлаждения распла-
расплава. Следовательно, температура Т-, может быть несколько пони-
понижена (ср. точки е и g на рис. 18.2). Переход в стеклообразное
состояние может осуществляться тогда, когда время измерения
какого-либо свойства переохлажденной жидкости, например ее
вязкости (при температуре s^Tg —- 30'3 П), становится соизме-
соизмеримым со временем внутренней перестройки структуры жидко-
жидкости, происходящей при измене-
изменении температуры. Время струк-
структурной перестройки жидкости
составляет при Т5 примерно не-
несколько минут. Отрезок eg (рис.
18.2), вообще говоря, слишком
удлинять невозможно, так как
при этом существенно увеличи-
увеличивается время, необходимое для
достижения внутреннего равно-
равновесия в жидкости.
В 1970 г. Эпжелл показал,
что с позиций теоретической
термодинамики должна сущест-
существовать нижняя температурная
граница, где с неизбежностью
из переохлажденного расплава
образуется стекло. Эта темпе-
температура называется температу-
температурой стеклования идеального стекла То. Существование такой
температуры становится попятным при сравнении теплоемко-
сгей и энтропии жидкой и кристаллической фаз одинаково-
одинакового состава. Энтропия вещества S связана с теплоемкостью С:,
соотношением
ср
При
-//.S
\ // /
Ш1
^ч~^кр' ^>лл
0
ж
/ //*
^^
кристаллы
У 1
ЖИДКОСId f
Ln Г
Рис. 18.3. Зависимость теплоемко-
теплоемкости и энтропии от температуры.
ы Л
A8.1)
Из температурной зависимости теплоемкости (рис. 18.3) видно,
что при заданной температуре Т\ энтропия определяется пло-
площадью под кривой Cp = f(\n T) в интервале изменения аргумента
от 7 = 0 до T = Ti. В случае если при некоторой температуре
(ниже температуры, до которой ведется интегрирование) про-
происходит фазовый переход, в уравнении A8.1) появится дополни
телыюс слагаемое, учитывающее энтропию этого фазового пре-
превращения. Например, при температуре Tn.i (рис. 18.3) энтропии
18f>
18. Стек.чо
жидкости 5Л4 и кристаллов SKV описываются разными уравне-
уравнениями
A8.2)
A8.3)
В интервале температур между 7"й и Г„л (рис. 18.4) переохлаж-
переохлажденная жидкость (кривая be) характеризуется более высоким
значением теплоемкости, чем соответстьующая кристаллическая
фаза (кривая eg). Поэтому при охлаждении жидкости ниже
Тпл энтропия жидкости уменьшается быстрее, чем энтропия кри-
кристаллической фазы, охлаждаемой до той же температуры. При
температуре стеклования Tg заштрихованная область на
рис. 18.4 характеризует суммарную избыточную энтропию пере-
переохлажденной жидкости
по отношению к энтро-
энтропии кристалла. Тем не
менее эта избыточная
энтропия меньше энтро-
энтропии плавления, и поэто-
поэтому энтропия переохлаж-
переохлажденной жидкости или
стекла выше энтропии
кристалла. Предполо-
Предположим, что Та смещается
в область более низких
температур из-за умень-
уменьшения скорости охлаж-
охлаждения жидкости. Это
приведет к тому, что из-
Рис. 18.-1 Оценка температуры стеклоиа- быточпая энтропия жид-
пня идеального стекла Гя. кости или стекла увели-
увеличится. В предельном слу-
случае при некоторой температуре То площадь, ограниченная кри-
кривой bdfe, станет равной энтропии плавления кристалла. При
этой температуре энтропия переохлажденной жидкости или
стекла должна понизиться на величину избыточной энтропии
и стать равной энтропии кристаллической фазы. Поэтому тем-
температура То является нижней границей перехода в стеклооб-
стеклообразное состояние, так как, если бы можно было бы переохла-
переохладить жидкость ниже То, возникла бы физически абсурдная си-
1мГ
18.3. Кинетика кристаллизации и стекловании 187
туацня, когда энтропия аморфной фазы была бы ниже энтро-
энтропии термодинамически стабильной упорядоченной кристалличе-
кристаллической фазы.
Действительно, было установлено, что всегда Га>70, т. е.
при охлаждении стеклование начинается раньше достижения
продельной температуры То. Чтобы стекло образовалось при
Таблица 18.1. Экспериментальная температура стеклования Те
и температура стеклования идеального стекла Го
Си-к.чо /'„. "С. То. "С
Пи реке
Оконное стекло (NajO, СлО, SiO?)
Хрусталь (РЬО, SiO?)
250
550
550
440
—50
60
350
270
150
—70
температуре 7"о. необходимо было бы охлаждать расплав с бес-
бесконечно малой скоростью. В табл. 18.1 приведены значения То и
Ts для некоторых стекол.
18.3. Кинетика кристаллизации и стеклования
Для перехода расплава при охлаждении в стеклообразное
состояние необходимо, чтобы скорость кристаллизации переох-
переохлажденной жидкости была крайне низкой. Поэтому для опи-
описания процесса стсклообразования полезно использовать кине-
кинетические соображения, а также структурные и термодинамиче-
термодинамические критерии, о которых речь шла выше.
Кристаллизация переохлажденной жидкости включает две
стадии: 1) образование кристаллических зародышей; 2) после-
последующий рост кристаллов, Благоприятными кинетическими усло-
условиями стеклообразования являются малые скорости зародыше-
образовапия и (или) роста кристаллов. В некоторых переохлаж-
переохлажденных жидкостях процесс зарождения кристаллов облегчен
из-за наличия в них большого количества центров кристаллиза-
кристаллизации: инородные частицы, поверхности контейнера и т. п. Поэто-
Поэтому л имитирующей стадией кристаллизации является стадия рос-
роста кристаллов. На рис. 18.5 приведена типичная температурная
зависимость скорости кристаллизации. В точке плавления ско-
скорость кристаллизации равна нулю, затем она возрастает до мак-
максимального значения при некоторой промежуточной степени пе-
переохлаждения и вновь понижается до нуля при низких темпера-
температурах.
188
18. Стекло
Кривую, аналогичную изображенной на рис. 18.5, можно
получить и экспериментально, измеряя скорость кристаллизации
в системах, где этот процесс идет достаточно медленно. Боль-
Большинство же жидкостей замерзает при температуре ^Тпл, и ско-
скорость их кристаллизации настолько велика, что она не может
Скорость кристаллизации »-
Рис. 18.5. Зависимость скорости крнсталлизаиии переохлажденной жидкости
от температуры (но Тамчану).
быть измерена. Форму кривой, представленной на рис. 18.5,
можно объяснить следующим образом.
При температурах, близких к точке плавления Тпл, свобод-
свободные энергии кристаллов и жидкости примерно одинаковы. По-
Поэтому движущая сила процесса кристаллизации очень мала,
и скорость кристаллизации почти равна нулю.
При температурах <ТПЛ свободная энергия кристаллов
меньше свободной энергии жидкости. Принимая, что энтропия
плавления ASn.q не зависит от температуры, можно записать сле-
следующее выражение для разности свободных энергий жидкой
и кристаллической фаз:
AG = Atf — TASn,,
18.3. Кинетика кристаллизации и стеклования 189
Поскольку же при температуре Г„л
то A8.4)
AG-AS^T^-T) для Т<Тпл
Возрастание скорости кристаллизации ниже температуры плав-
плавления Тип (рис. 18.5) отвечает увеличению разности свободных
энергий кристаллов и жидкости и, следовательно, объясняется
ростом движущей силы процесса кристаллизации. В области
еще более низких температур важную роль, особенно для стек-
лообразующих веществ, приобретает вязкость переохлажденных
расплавов. С ростом вязкости диффузия ионов и атомов в объ-
объеме жидкости к поверхности растущего кристалла становится
все более затрудненной и скорость кристаллизации начинает
уменьшаться.
Таким образом, при понижении температуры начинают про-
проявляться два конкурирующих процесса. Если увеличение разно-
разности свободных энергий кристалла п жидкости благоприятствует
процессу кристаллизации, то повышение вязкости переохлаж-
переохлажденной жидкости уменьшает склонность к кристаллизации. Мак-
Максимум па кривой скорости кристаллизации (рис. 18.5) соответ-
соответствует ситуации, когда оба конкурирующих процесса уравнове-
уравновешивают друг друга. Ниже температуры максимума доминирую-
доминирующим фактором является вязкость, а выше максимума кинетика
процесса кристаллизации определяется разностью свободных
энергий кристаллов и жидкой фазы.
На графике зависимости скорости кристаллизации от темпе-
температуры (рис. 18.5) существует область температур, так называе-
называемая «опасная зона стеклования», где скорость кристаллизации
максимальна. Если удается быстро переохладить жидкость до
температур ниже этой зоны, то можно не опасаться последую-
последующей кристаллизации (расстекловываиии), поскольку образуется
кинетически стабильное стекло.
Все вышеприведенные рассуждения относятся к кинетике
роста кристаллов Предполагалось, что зародышеобразоваиие
либо уже произошло, либо происходит сравнительно легко. Что
же касается самого зародышеобразования, то необходимо раз-
различать два механизма этого процесса: гомогенное и гетероген-
гетерогенное заролышеобразовапие. Если гетерогенные зародыши (на-
(например, инородные частицы) отсутствуют в переохлажденной
жидкости, то может происходить гомогенное зародышеобразо-
вапие. Этот процесс протекает в объеме жидкости, когда пег ис-
искусственно введенных центров кристаллизации. Например, вода
18., Стекло
обычно замерзает при температуре 0°С или чуть ниже, по-
поскольку в ней существуют центры кристаллизации. Если же
вода очень чистая, то ее можно переохладить до — 20 СС, и кри-
кристаллизация может быть задержана кинетически на значитель-
значительное время. Однако при охлаждении до —40 СС, несмотря на все
меры предосторожности по поводу чистоты воды, она всегда
очень быстро кристаллизуется. Дело в том, что при такой стс
пени переохлаждения быстро идут процессы гомогенного заро-
зародышеобразования.
Зависимость скорости гомогенного зародышеобразования от
температуры имеет тот же вид, что и зависимость скорости рос-
роста кристаллов (рис. 18.5), но вся кривая несколько смещена
тз сторону более низких температур. Существуют две причины
такого смещения. Во-первых, зародышеобразование идет в объ-
объеме жидкости (трехмерный процесс), в то время как рост кри-
кристаллов—на поверхности растущего зародыша (т. е. не более
чем двумерный процесс). Следовательно, энергия активации
зародышеобразоваппя заметно выше, чем в случае роста кри-
кристаллов. Во-вторых, поверхностная энергия небольших зароды
uicii, а значит и повышенная свободная энергия процесса заро-
зародышеобразования, связанная с созданием новых поверхностей,
должны быть скомпенсированы благодаря уменьшению свобод-
свободной энергии в процессе кристаллизации, т. е. благодаря пониже-
понижению объемной свободной энергии зародышей (гл. 12). При ма-
малом переохлаждении, когда суммарное понижение свободной
энергии при кристаллизации невелико, зародыши нестабильны
и повторно растворяются в жидкости из-за относительно высо-
высоко:! поверхностной свободной энергии. Поэтому при температу-
температурах чуть ниже точки плавления стабильными будут лишь очень
большие гомогенные зародыши, у которых поверхностная сво-
свободная энергия почти равна пулю. Альтернативная, хотя и близ-
близкая по смыслу точка зрения о причинах смещения максимума
скорости зародышсобразовапия в область более низких темпера-
температур сводится к тому, что небольшие зародыши имеют более низ-
низкую температуру плавления, чем большие зародыши, и, следо-
следовательно, лишь последние могут устойчиво существовать вбли-
вблизи Гпл-
Критический размер зародыша определяется соотношением
двух упоминавшихся выше величин — поверхностной и объем-
объемной свободной энергии. Зародыши устойчивы, если понижение
объемной свободной энергии превосходит рост поверхностной
свободной энергии. Критический размер зародыша отвечает си-
ситуации, когда эти величины сравнимы между собой. Следова-
Следовательно, если размеры зародышей меньше этого критического
значения, то они нестабильны п могут повторно растворяться,
в то время как зародыши г размером больше критического ста-
18,4. Структура стекол 191
билыш и продолжают расти. Значения критических размеров
зародышей сильно зависят от температуры. При температуре
плавлении Гпл {и несколько выше) критический размер зароды-
зародышей велик, однако с ростом переохлаждения (<СТПЛ) он быстро
уменьшается. При достаточно большом переохлаждении крити-
критический размер становится весьма малым и зародыши легко об-
образуются при случайных термических флуктуациях или флук-
туациях состава жидкости. На этой стадии идет спонтанное го-
гомогенное зародышсобразованис.
Таблица 18.2. Классификация стсклообразующпх материалов
по типу химической связи [б]
Тип сияни Примеры
Ковалентная связь Оксиды (силикаты и др.), халькон1-
пидьг, органические полимеры
Ионная связь Галогеппди, ни граты и др.
Ппратнрованпая ионная связь Водные растворы солей
Молекулярная связь Органические жидкости
Металлическая связь Быстро закаленные сплавы
Таким образом, для некоторых жидкостей имеется область
температур ниже 7*пл, в которой {в отсутствие гетерогенных за-
зародышей) они кинетически стабильны (не кристаллизуются); их
можно рассматривать как гомогенные переохлажденные жидко-
жидкости. При дальнейшем понижении температуры может спонтанно
идти зародышеобразование и кристаллизация (вспомним приме])
с водой).
Математический аппарат теории кристаллизации, развитый
в основном Турнбуллом и сотр., приводит в принципе к таким
же зависимостям скоростей зародышеобразования и роста кри-
кристаллов от температуры, как и наблюдаемые экспериментально.
Некоторые переменные, входящие в теоретические уравнения,
весьма сложно оценить количественно (например, поверхност-
поверхностную энергию зародышей), поэтому пока можно говорить лишь
о качественном согласии теории и эксперимента.
18.4. Структура стекол
Стекла образуются не только в неорганических силикатных
системах, они характерны и для других классов веществ. Как
видно из табл. 18.2, вещества с любым типом химической связи
способны переходить в стеклообразное состояние. Каждая груп-
группа материалов обычно имеет различный тип структуры, и, еле-
*92 18. Стекло
довательно, факторы, влияющие на стеклообразовапис, несколь-
несколько изменяются от группы к группе. В настоящем разделе опи-
описаны структуры некоторых наиболее важных оксидных стекол.
J8.4.1. Кварцевое стекли
Кварцевое стекло — простейший пример стекол на основе
SiO2. Изучение его структуры и свойств очень важно для пони-
понимания строения и характеристик химически более сложных си-
силикатных стекол. Общепринятая точка зрения на структуру
кварцевого стекла во многом совпадает со взглядами Захариа-
сепа A932 г.), которые были подтверждены рентгеновскими ис-
исследованиями Уоррена A936 г.). Основу структуры кварцевого
стекла составляют сочлененные вершинами тетраэдры SiO.j,
которые образуют непрерывную сетку. Эта сетка не симметрич-
симметрична, в пей отсутствует дальний порядок. Поскольку соблюдается
условие электроиейтралыюсти, каждый атом кислорода, нахо-
находящийся в вершине, принадлежит лишь двум тетраэдрам, поэто-
поэтому структура носит довольно открытый характер.
Рентгенограммы порошков стеклообразных веществ пред-
представляют собой весьма размытую картину, состоящую из широ-
широких диффузных рефлексов, а не из острых пиков. На рис. 18.6
для сравнения представлены порошкограммы кварцевого стекла
и кристобалита. Поскольку для стеклообразного вещества не-
невозможно выделить элементарную ячейку структуры, единствен-
единственной информацией, которую можно получить рентгеновским ме-
методом, является функция радиального распределения (ФРР).
ФРР отражает вероятность обнаружения второго атома в зави-
зависимости от расстояния до данного атома. На рис. 18.7 приведена
ФРР кварцевого стекла. Вероятность обнаружения второго ато-
атома выражается ординатой функции парного распределения при
данном значении межатомного расстояния. Прямая на рнс. 18.7
соответствует ФРР гипотетического вещества, состоящего из бес-
беспорядочной совокупности невзаимодействующих друг с другом
атомов. Первый и самый высокий максимум па кривой для БЮг
находится при межатомном расстоянии, равном 1,62 А, второй
(меньший по высоте) максимум — при 2,65 А. Эти межатомные
расстояния отвечают соответственно длинам связей кремний —
кислород и кислород— кислород в тетраэдрах SiO4. Такие зна-
значения межатомных расстояний близки к соответствующим вели-
величинам в кристаллическом SiO2 и в силикатах. Два первых мак-
максимума па рис. 18.7 весьма узки. Это вполне попятно, поскольку
соответствующие им межатомные расстояния примерно постоян-
постоянны как в стекле, так и в кристаллах, т. с. тетраэдры SiO: не раз-
упорядочепы. Однако следующие пики все больше расширяются,
так как в стекле существует разброс значений межатомных рас-
18.4. Структура стекол
193
стеклообразный оксид кремния
dO 2B
Рис. 18.6. Порошкограммы кристобалита (а) и стеклообразного SiO2 (б)
(СиКа-излучение).
4000
S 3000-
2000 -
1000 -
Межаюмное расстояние, А
Рис. 18.7. Результаты рентгеновского исследования кварцевого стекла [11].
стояний. Третий пик при ~3,12 А отвечает минимальному рас-
расстоянию между атомами кремния, т. с. расстоянию между цент-
центрами двух тетраэдров SiO4. Угол между валентными связями
кислорода (угол Si—О—Si) несколько меняется, поскольку тет-
тетраэдры поворачиваются и вращаются в разные стороны друг
относительно друга. Поэтому существует и разброс значений
межатомных расстояний Si—Si. Другие пики отвечают расстоя-
расстояниям между следующими атомами: —4,15 А — расстояние меж-
13—1426
194 18. Стекло
ду кремнием п кислородом во второй координационной сфере;
~3,1 А—расстояние между атомом кислорода и другим ато-
атомом кислорода во второй координационной сфере или между
атомом кремния и другим атомом кремния во второй коорди-
координационной сфере. При межатомных расстояниях, больших, чем
Ь—7 А, па ФРР отсутствуют явно выраженные пики. Этот факт
вполне соответствует модели неупорядоченной сетки для струк-
структуры кварцевого стекла.
Несмотря па то что резулыаш исследований Уоррена мож-
можно ле1 ко описать в рамках простой физической модели неупоря-
неупорядоченной сетки, в течение многих лег выдвигаются и обосно-
обосновываются аргументы и пользу другой модели, описывающей
строение стекол — так называемой кристаллнтиой модели
структуры кварцевого стекла. В рамках этой модели
предполагается, что в стекле существуют очень маленькие упо-
упорядоченные области (или мнкрокрисгаллиты), которые связаны
друг с другом неупорядоченными участками. Для объяснения
ширины линий на порошкограммах (рис. I8.G) необходимо при-
принять, что размер кристаллитов очень мал (не больше 8—10 А).
Поэтому всегда возникает вопрос, насколько можно применять
термин «кристаллит» к таким небольшим образованиям. Совер-
Совершенно определенно, что один отдельно взятый кристаллит тако-
такого размера был бы неустойчив, так как он обладает очень высо-
высокой поверхностной энергией. Однако в стеклообразном SiO;,
который характеризуется определенной сетчатой структурой, на
границе раздела между упорядоченной и неупорядоченной об-
областями отсутствуют атомы кислорода или кремния, потенциаль-
потенциально способные образовывать новые химические связи, поэтому
и границы кристаллитов не характеризуются высокой поверх-
поверхностной энергией.
Доказательства применимости кристаллитной модели для
описании строения стекол основываются на наблюдениях за их
кристаллизацией и расстекловыванием. Согласно правилу Ост-
Оствальда, первый продукт кристаллизации стекла может соот-
соответствовать тем структурным образованиям, которые присутст-
присутствуют в самом стекле. Кварцевое стекло получают плавлением
либо кристаллического кварца, либо кристобалита; при кристал-
кристаллизации кварцевою стекла в зависимости от условий проведе-
проведения этого процесса образуются либо кварц, либо кристобалит.
Это является доказательством того, что в стекле присутствуют
кварцеподобные или кристобалитоподобпые микрокристалли-
микрокристаллиты. Расплавленный SiO2 является очень вязкой жидкостью, и
даже при 1800 °С нельзя быть вполне уверенным, что процесс
плавления и гомогенизации полностью завершен. Это утвержде-
утверждение справедливо, несмотря на то что образующееся стекло рент-
гепоаморфпо и оптически однородно. Таково в настоящее время
18,4- Структура стекол
состояние кристаллитной модели стекла. Несмотря па то что эта
модель, возможно, и не вполне корректна, с се выводами нельзя
не считаться.
18.4.2. Силикатные стекла
Структура и свойства двойных силикатных стекол в сильной
степени зависят от природы второю оксида. При введении моди-
модификатора {оксида, который модифицирует исходную сетчатую
структуру), например оксидов щелочных или щелочноземельных
металлов, силикатная сетка все больше разрушается по мере
добавления этих компонентов. Это проявляется, например,
в том, что вязкость расплавов двойных силикатов существенно
меньше вязкости расплавленного SiCb. При введении в структу-
структуру SiO2 посторонних ионов мостиковые связи Si—О—Si разру-
разрушаются и возникают иемостиковые кислородные атомы
{рис. 18.8, а).
Если соотношение количеств модифицирующего оксида и ок-
оксида кремния увеличивается до 1 : 2 (например. Na2O-2SiO2 =
•=Na2Si2O5), то отношение числа атомов кремния к числу ато-
атомов кислорода уменьшается до 1 : 2,5. Это означает, что в вер-
вершинах каждого тетраэдра SiO4 в среднем один атом кислорода
немостиковый. В кристаллических силикатах того же состава,
т. е. NagS'^Oj, силикатные анионы обычно имеют структуру бес-
бесконечных двумерных слоев; в стеклах могут также присутство-
присутствовать небольшие участки этих слоев. Однако более вероятно, что
существует открытая трехмерная каркасная структура, а катио-
катионы Na~' и им подобные размещаются в относительно больших
пустотах этого каркаса.
Распределение таких катионов, как Na", по указанным пусто-
пустотам не вполне беспорядочно, так как из рентгеновских данных
известно, что катионы могут объединяться в кластеры. Причина
этого не до конца ясна. Может быть это обусловлено небеспо-
небеспорядочным расположением доступных для ионов Na ; пустот в си-
силикатной сетке. Другое возможное объяснение заключается
в том, что существует специфическое катион-катионное взаимо-
взаимодействие, возможно, с участием кислородных ионов, которое
приводит к кластерообразованию.
Если вводить оксид второго компонента в структуру стекла
в еще больших количествах, то силикатная сегка разрывается
еще больше, расплав становится все более текучим и при ох-
охлаждении возрастает тенденция к расстекловываппю. Обычно
при отношении оксид второго компонента: оксид кремния =1:1
трудно, да и едва ли возможно сильно переохладить расплав и
получить стекло.
Эффект от добавления к кремнезему других сгеклообразую-
щпх оксидов или условных стеклообразующих оксидов заметно
13*
196
18. Стекло
о/Ч
\
\
о
Рис. 18.8. Структура силикатных стекол (а) и две бороксоловые группы,
связанные мостиковым кислородным атомом (б). В структуре силикатов
изображены только по три кислородных атома вокруг каждого атома крем-
кремния.
отличается от эффекта введения модификаторов. Стеклообра-
зующие оксиды обычно замещают оксид кремния, так что бес-
беспорядочная трехмерная сетчатая структура сохраняется. Сле-
Следовательно, при охлаждении расстекловывание практически не
идет, а область стеклообразования существенно расширяется.
Так, стекла на основе силиката свинца могут содержать до
80 мол.% РЬО (РЬО —условный стеклообр азующий оксид),
а в системе В2Оз—SiO2 стекла существуют во всем интервале
составов от чистого SiCb до чистого В2Оз-
18 4 Структура стекол 197
18.4.3. Стеклообразный В2ОЛ и йоритные стекла
Хотя боратные стекла имеют ограниченное практическое зна-
значение из-за их растворимости в воде, В2О3 —важный компонент
боросиликатных стекол, таких, как пирекс. В отличие от квар-
кварцевого и силикатных стекол, в которых атомы кремния находят-
находятся внутри тетраэдров SiO^, структура стеклообразного В2О3 по-
построена из треугольных структурных элементов ВО.ч, а в борат-
пых стеклах присутствуют треугольники ВО3 и тетраэдры ВО4,
соотношение которых зависит от состава стекла. Важную струк-
структурную роль в стеклообразном В2О3 играют бороксоловые груп-
группы (рис. 18.8, б) -— это плоские шестичленпые кольца, образо-
образованные атомами бора и кислорода. Такие группы сочленяются
в трехмерную сетку посредством мостиковых кислородных ато-
атомов. Однако, поскольку в стеклообразном В2О3 основные струк-
структурные элементы имеют плоскую форму, его структура более
открытая по сравнению со структурой кварцевого стекла, по-
построенной из трехмерных тетраэдров SiO,(. Поэтому расплавлен-
расплавленный В2Оз менее вязкий, чем расплавленный SiO2.
Тригоаальная координация бора в структуре стеклообразного
В2О3 была установлена рентгенографическими и спектроскопи-
спектроскопическими методами. Особенно убедительные данные получены
методом ЯМР на ядрах В11. При рентгенографических исследо-
исследованиях обнаружено, что кривая радиального распределения
стеклообразного В2Оз имеет максимумы при 1,37 и 2,40 А; эти
значения соответствуют длинам связей бор — кислород и кисло-
кислород— кислород в треугольниках ВО3. Полученные значения
межатомных расстояний отличаются от межатомных расстояний
в кристаллических боратах, содержащих тетраэдры ВО^, в кото-
которых расстояния между бором н кислородом больше A,48 А).
Введение оксидов щелочных металлов в стеклообразный
В2О3 приводит к иным последствиям, чем в силикатных стеклах.
В боратных стеклах наблюдается так называемая борная ано-
аномалия. Например, в системе Na2O— B2O3 вязкость расплава воз-
возрастает с ростом содержания Na2O. Она максимальна при
~16 мол. % Na2O. Коэффициент термического расширения
стекла понижается с ростом содержания Na2O. Он принимает
минимальное значение при 16 мол. % Na2O. На зависимостях
других свойств стекол от состава также наблюдается экстрему-
экстремумы в этой области составов. В отличие от этого силикаты ще-
щелочных металлов с увеличением содержания оксида щелочного
металла становятся менее вязкими, а коэффициент их термиче-
термического расширения возрастает. Никаких минимумов и максиму-
максимумов на зависимостях свойств от состава обнаружено не было.
По-видимому, существуют несколько факторов, приводящих
к возникновению борной аномалии, т. е. к наличию максимумов
198 18, Стекло
и минимумов на зависимостях свойств от состава при
~16% №гО, Но и после многолетних дискуссий и экспери-
экспериментальных исследований природа этого явления так и осталась
не до конца выясненной.
Используя данные исследовапнй методом ЯМР на ядрах В11,
Брей показал, что добавление оксидов щелочных металлов
к В2Ол ведет к постепенному изменению координационного чис-
числа бора от трех до четырех. Когда добавка оксида щелочного
металла достигает ~30%, то приблизительно 40% атомов бора
имеют четверную (тетраэдрическую) координацию, причем это
не зависит от того, оксид какого щелочного металла использу-
используют. Если атом бора находится в центре треугольника из атомов
кислорода, то в спектре ЯМР присутствуют широкие резонанс-
резонансные линии из-за квадрупольного расщепления. При тетраэдриче-
ской координации бора квадрупольное расщепление является
слабым, а резонансные линии — узкими. Данные ЯМР свиде-
свидетельствуют о том, что на зависимостях координационного чис-
числа от состава боратных стекол особые эффекты при 16% Na2O
отсутствуют; доля тетраэдрически координированных атомов
бора линейно растет в области составов от 0 до 30% Na2O.
Борная аномалия в какой-то степени может быть объяснена.
При малых добавках оксидов щелочных металлов некоторые
атомы бора оказываются в тетраэдрическом окружении атомов
кислорода, и это приводит к их «связыванию» в сетку и. следо-
следовательно, к возрастанию вязкости. Таким образом, при введении
оксидов щелочных металлов соотношение между атомами бора
и кислорода меняется от 1 : 1,5 (в В2О3) до 1 : 2 (как в квар-
кварцевом стекле). Пространственная сетка, целиком состоящая из
тетраэдров, теоретически может возникнуть при содержании
оксида щелочного металла 50% (т. с. при составе Na2B2CM.
Однако известно, что задолго до этого вязкость боратпого рас-
расплава начинает вновь понижаться. Вероятно, это связано с тем,
что сетка, содержащая большое количество тетраэдрически ко-
координированных атомов бора, не является такой прочной и жест-
кон, как соответствующая силикатная сетка. Поскольку фор-
формальный заряд бора (+3) ниже, чем кремния (+4), химические
связи В—О в тетраэдрах существенно слабее связей Si—О
в тетраэдрах SiO4.
18.5. Несмешиваемость жидкостей и ликвация в стеклах
Для многих практических нужд необходимо, чтобы стекла
представляли собой однофазные гомогенные материалы, т. с.
при сплавлении компонентов стекла должен образовываться го-
гомогенный расплав, который сохраняет свою однофазпость при
переохлаждении. Однако иногда, например при получении стек-
18.5. Несмешиваемость жидкостей н ликпация в стеклах 199
ла викор, краппе необходимо, чтобы в стекле присутствовали
области микрорассланвания. Чтобы понять природу несмешивае-
несмешиваемости жидкостей, необходимо рассмотреть соответствующую
диаграмму состояний (см. рис. 11.9 и 11.10). В области соста-
составов, богатых оксидом кремния, например в системе СаО—SiO;,
на диаграмме состояния (рис. 11.8) при температуре ~1700°С
видна область расслаивания в жидком состоянии. Поэтому за-
заведомо можно утверждать о невозможности получения одно-
однофазной жидкости, например состава 20% СаО+80% SiO2. Хотя
теоретически однофазная жидкость такого состава существует
при ~2000°С, при охлаждении этот расплав неизбежно прой-
пройдет через область расслаивания и очень быстро распадется на
две фазы. Относительно небольшой купол расслаивания, сущест-
существующий при высоких температурах в системе СаО—БЮг, фак-
фактически представляет собой лишь верхнюю часть более широкой
метастабилыюй области расслаивания при более низких темпе-
температурах (рис. 11.10). Для точного определения положения гра-
границы купола расслаивания при более низких температурах не-
необходимо, чтобы переохлаждение жидкости происходило без
кристаллизации, т. е. чтобы жидкость при охлаждении вела себя
так, как будто на фазовой диаграмме отсутствуют линии ликви-
ликвидуса и солидуса.
В системе СаО—SiO2 широкая метастабнльная область не-
несмешиваемости, которая появляется при переохлаждении жид-
жидкости, как бы накрыта сверху куполом расслаивания (он изо-
изображен па диаграмме состояния). В таких системах, как
Li2O—SiO2 или Na2O—SiO2, метастабильная область несмеши-
несмешиваемости целиком находится ниже линии ликвидуса, и купол
расслаивании отсутствует на равновесной диаграмме состояний
(рис. 11.10,6).
Появление или отсутствие в стеклах ликьационных областей
зависит от состава и температуры, при которой охлаждаемая
жидкость попадает в область купола расслаивании. Если это
происходит при очень высоких температурах (например,
1500 °С), то скорость диффузии велика, и даже при очень
быстром охлаждении в жидкой фазе идет процесс расслаива-
расслаивания. Структура ликвационных областей в стекле зависит от
объемной доли каждой из двух жидких фаз; области ликвации
могут иметь форму капель (когда одна жидкая фаза вовлека-
вовлекается в матрицу другой жидкой фазы) или состоять из взаимно
проникающих жидких сеток (если объемные доли обеих фаз
примерно одинаковы). Если процессы расслаивания начинаются
при гораздо более низких температурах (например, 800°С), то
скорость диффузии существенно меньше, так как, во-первых,
запас тепловой энергии атомов и ионов недостаточен для интен-
интенсивного перемещения и, во-вторых, жидкость более вязкая. Это
200 18. Стекло
приводит к образованию гораздо более мелких ликвационных
областей.
Возможность визуального наблюдения за образованием не-
неоднородных областей зависит от соотношения размеров этих
областей и длины волны белого света. Если эти области явля-
являются более крупными, чем длина волны света, или, особенно,
если показатели преломления двух стеклообразных фаз силь-
сильно различаются между собой, то получаемое стекло непрозрач-
непрозрачно и кажется, что оно покрыто эмалью. Это объясняется тем,
что свет не проходит сквозь стекло, а отражается на границах
раздела двух фаз. Можно ожидать, например, что из расплава
состава 30% СаО, 70% SiO2 будет образовываться при охлаж-
охлаждении матовое стекло.
Если размеры областей неоднородности примерно равны дли-
длине волны белого света и разность показателей преломления двух
жидких фаз мала, то в результате может получиться опалесци-
рующее стекло. Так, можно приготовить опалесцирующее стек-
стекло на основе силиката лития, содержащее ~25% Li2O. Такое
стекло в отраженном свете голубое, а в проходящем оранжевое.
В этом случае свет частично рассеивается на внутренних грани-
границах раздела фаз.
Возможен и другой крайний случай: размеры областей неод-
неоднородности гораздо меньше длины волны белого света, а показа-
показатели преломления обеих жидких фаз практически неразличимы.
Тогда свет не рассеивается на границах фаз, и стекло кажется
оптически прозрачным и гомогенным. Чтобы исследовать неод-
неоднородность стекла, в этом случае необходимо использовать элек-
электронный микроскоп с высоким разрешением. Примером стекла,
имеющего такую структуру, является стекло состава
20% Na2O+80% SiO2.
18,5.1. Структурные теории расслаивания жидкостей
Ликвация— характерная особенность силикатных стекол, содержащих
большое количество оксида кремния. Это явление, например, наблюдается в
стеклах системы оксид щелочного металла — SiO;, пли оксид щелочнозе-
щелочноземельного металла—SiO2. В некоторых системах купол расслаивания появ-
появляется на диаграммах состояния (например, для систем SiO2 с MgO, СаО,
SrO; рис. 11.10, о), с других — область расслаивания полностью метаста-
бильна {системы SiO2 с ВаО, оксидами щелочных металлов; рис. 11 10, б).
В этих двух группах систем склонность к расслаиванию максимальна в си-
системе MgO—SiO2 (купол расслаивания наибольший) и минимальна п си-
системе Cs2O—SiO2. Уоррен и Пипкус A940 г.) изучали тенденцию изменения
склонности к расслаиванию стекол в упомянутых системах и обнаружили
корреляцию этой характеристики с увеличением прочности химической связи
между кислородом и катионом модификатора. Величина, характеризующая
прочность химической связи, определяется отношением г/г (где г — заряд
катиона, г — его ионный радиус). Очевидно, что отношение z/r максимально
у Mg2+ и минимально у Cs+. Качественно несмешиваемость жидкостей
можно рассматривать как результат «конкурирующего» стремления катионов
18-5. Несмешиваемость жидкостей и ликвация в стеклах 2СИ
модификатора и кремния образовывать химические связи с атомами кисло-
кислорода. Катионы модификатора склонны к координации немостиковых кисю-
родных атомов, и это ведет к возникновению в расплаве двух областей: обла-
области, богатой оксидом кремния, и области с недостатком оксида кремния.
Если отношение zlr для катионов модификатора велико, то вероятности
расслаивания жидкостей увеличивается.
Описанная выше качественная корреляция между прочностью химиче-
химической связи и склонностью к разделению фаз в некоторых системах вполне
очевидна; однако имеются системы, в которых этой закономерности не об-
обнаруживается. В последнем случае более существенную роль играют дру-
другие факторы, которые определяют несмешиваемость жидкостей или, точ-
точнее, отсутствие несмешиваемости. Одним из таких факторов является коор-
координационное число второго компонента. Если он склонен к образованию
тстраэдрического окружения, то может замещать атомы кремния в струч-
турной сетке стекла, что существенно понижает склонность к расслаиванию.
Поэтому на диаграммах состояния систем, образованных SiCb и такими ок-
оксидами, как БеО, АЬОз, GeC>2 и P^Oj, отсутствуют области расслаивания, не-
несмотря на то что отношение zlr у jtiix катионов велико. Этот факт обьяс-
пяегся тем, что перечисленные оксилы встраиваются в существующие тет-
тетраэдры и образуют часть сетчатой структуры. В отличие от атого ионы,
имеющие, высокие координационные числа (-~6), такие, как Mg2+, разру-
разрушают сетку оксида кремния.
Левин и Блок A957 г.) изучали координационные числа атомов и при-
применили правило Полипга, предложенное для ионных кристаллов (гл. 8). к
стеклам. Прочность электростатических связен (ПЭС) определяется соот-
соотношением:
заряд катиона г
координационное число катионов
Ленин и Блок обратили внимание на то, что склочность к расслаиванию в
бинарных силиюпны.х и боратных системах зависит от значений ПЭС. вто-
второго катиона. При малых значениях ПЭС, например V8 для К+ (КЧ 8),
Че для Na'1, Li+ (КЧ 6), имеет место лишь расслаивание мечастабилыгаго
переохлажденного расплава ниже линии ликвидуса. Для более высоких
значений ПЭС С/4 Для SH+, Ва ; КЧ 8) h.i диаграммах состояния возни-
возникает купол расслаивания в жидкой флзо, в случае же еще больших зпачр-
iinii ПЭС (>'/з как для Fe2~, Zr.2'; КЧ 6) области несмешиваемости су-
существуют как в силикатных, так и в боратиых расплавах. Исключение со-
составляют ионы BeJ + и А13+, которые замещают атомы кремния в сетчатом
каркасе стекла, и поэтому расслаивания в таких системах не происходит.
18.5.2. Термодинамика расслаивания в жидкой фазе
Рассмотрим термодинамические факторы, которые влияют
(или не влияют) на расслаивание в системе из двух жидкостей
А и В. Важным параметром, безусловно, является свободная
энергия. Если при данном составе смесь двух жидкостей имеет
свободную энергию более низкую, чем однофазная жидкость, то
в такой системе идет расслаивание. Выделим энтропийный (AS)
и энтйльпийпын (Д#) вклады в свободную энергию:
AG = АН — TAS
При смешивании двух жидкостей А и В и образовании гомоген-
гомогенной жидкой смеси происходит увеличение энтропии, т. е. AS>0.
202 18. Стекло
Смешиваются или не смешиваются две жидкости между со-
собой— это зависит от энтальпии смешения, которая в свою оче-
очередь зависит от соотношения энергий взаимодействия между
молекулами А—А, А—В и В—В. При этом возможны два слу-
случая:
1) суммарное изменение энтальпии смешения компонентов
А и В либо отрицательно, либо равно нулю, т. е. энергия гетеро-
атомного взаимодействия А—В больше или равна энергии взаи-
взаимодействия одинаковых атомов А—А и В—В. Тогда Д//^:0.
Поскольку Д5>-0, то ДС<0 и жидкости смешиваются между
собой при любых температурах;
2) суммарная энтальпия смешения положительна, т. е. ато-
атомы А и В отталкиваются друг от друга. В этом случае наблю-
наблюдается тенденция к сегрегации атомов одного сорта. Положи-
Положительное значение энтальпии смешения АН уравновешивается
отрицательным вкладом в ДС члена TAS, который, что совер-
совершенно очевидно, зависит от температуры. Поэтому если АИ^>
>ГД5, то происходит расслаивание, так как AG>-(). Расслаи-
Расслаивание наблюдается при низких температурах (когда Д//>ГД5),
однако с ростом температуры область расслаивания уменьшает-
уменьшается и выше некоторой критической температуры Тс происходит
полное смешивание жидкостей (AH<iTAS).
18.5.3. Механизм расслаивания
Расслаивание — это процесс разделения гомогенной жидко-
жидкости на две жидкие фазы, когда однофазная жидкость при ох-
охлаждении попадает в область несмешиваемости диаграммы со-
состояния. Процесс расслаивания представляет несомненный инте-
интерес, и в настоящее время известны два механизма его протека-
протекания:
1) образование и рост зародышей и 2) сиииодальный распад
жидкости. Кан и Хиллерт показали, что эти два механизма от-
отличаются друг от друга тем, как изменяется свободная энергия
первоначально гомогенной однофазной жидкости при флуктуа-
флуктуации ее состава.
На рис. 18.9, а показана зависимость свободной энергии от
состава при двух температурах 7\ и Т2, причем эти температуры
выбраны таким образом, что при Г2 в системе происходит рас-
расслаивание, а при Ti расслаивания лет (рис. 18.9,6). При темпе-
температуре Т\ жидкость однофазна при всех составах к зависимость
свободной энергии от состава описывается кривой, вогнутой
вниз. Таким образом, при смешении компонентов А и В в любых
соотношениях при температуре 7\ происходит уменьшение сво-
свободной энергии. Если в жидкой смеси происходит флуктуация
состава, это приводит к общему повышению свободной энергии.
18.5. Несмешиваемость жидкостей и ликвация в стеклах
203
и:
3.
сх
о;
X
г>
одная
о
б
сх
~>^
сх
и;
t_
—
h
i
1
у
л . .
/
/
1 /
/
fa с
1 ;
i спи1|С
/
X
\
\
г* * ¦
d\b
\ хупол
у расслои
. \83НИЯ
•""¦ \
Состав, х
ft
Рпс. 18.9. Зависимости свободной энергии от состава в системе с расслаи-
расслаиванием.
18. Стекло
Поэтому такие флуктуации не сопровождаются устойчивым из-
изменением состава и со временем жидкость вновь станет гомоген-
гомогенной (однородной). Например, если жидкость состава х в резуль-
результате флуктуации состава переходит в смесь двух жидкостей со-
составов у и г, то это выбывает увеличение свободной энергии (по-
(показано стрелками на рис. 18,9, а).
При температуре 7'2 происходит расслаивание жидкости па
две фазы составов а и Ь. Следовательно, при любом составе
между а и b устойчива только гетерогенная смесь двух жидко-
жидкостей этих, составов. На кривой свободной энергии в точках а и
b имеются минимумы. Между точками а и b свободная энергия,
относящаяся к мстастабилыюн и неустойчивой жидким фазам,
повышается и проходит через максимум. В этой области проис-
происходит разделение гомогенной жидкости на две фазы, которое со-
сопровождается понижением свободной энергии (показано стрел-
стрелками на рис. 18.9, а). Возникающая двухфазная смесь характе-
характеризуется свободной энергией, величина которой отвечает пря-
прямой, соединяющей точки а и Ь.
Рассмотрим теперь, к чему приводит флуктуация состава
однофазной гомогенной жидкости при температуре Т2. Жидко-
Жидкости, составы которых находятся между точками с и d, ведут
себя иначе, чем жидкости в интервале составов а—с и d—Ь, Не-
Небольшие флуктуации в жидкости состава е. находящегося меж-
между точками а к с (рис. 18.9, в), приводят к ее распаду на две
жидкости составов / и g. Этот распад сопровождается ростом
суммарной свободной энергии, так как точка с лежит ниже ли-
линии, соединяющей точки / и g. Поэтому такое небольшое изме-
изменение состава неустойчиво во времени и возникшие фазы со-
составов / и g вновь образуют однофазную жидкость. Если, одна-
однако, в результате флуктуации состав жидкости изменится сильно,
то это может привести к понижению свободной энергии и обра-
образованию смеси фая составов а и Ь. Таким образом, жидкость
состава е устойчива при небольших флуктуация* состава и не-
неустойчива при сильных флуктуациях. Расслаивание жидкости
состава с па две фазы идет по механизму образования и роста
зародышей. Разделение фаз в этом случае не происходит спон-
спонтанно, и жидкость состава е является метастабильной фазой.
Для распада такой жидкости на две фазы нужно преодолеть
определенный потенциальный барьер, связанный с необходимо-
необходимостью возникновения сильной флуктуации и, следовательно,
с диффузией ионов на большие расстояния. Часто процессы об-
образования и роста зародышей кинетически идут весьма медлен-
медленно, и необходима значительная энергия активации для образова-
образования зародышей.
Жидкости, составы которых принадлежат области с—d, ве-
ведут себя совершенно иначе. Например, жидкость состава h при
18.5. Несмешиваемость жидкостей и ликвация в стеклах 205
небольшой флуктуации состава распадается па две жидкости
составов / и k. Этот процесс сопровождается понижением сво-
свободной энергии. Поскольку флуктуации состава возникают при
обычном термическом движении атомов или ионов в жидкости,
фаза состава h неустойчива и спонтанно распадается на две
жидкости. В отличие от жидкости состава е в данном случае
отсутствует какой-либо потенциальный барьер для такого рас-
распада. Процесс спонтанного разделения фазы состава h пазы-
Таблпца 18.3. Некоторые различия в микроструктуре стекол,
образующихся в результате енпнодалыюго распада
и возникновения и роста зародышей [3]
Образование и рост зародыше" Спшюдлльпый распад
Состаи второй срази лри лосюяппои Состаи обеих фаз меняется со вре-
Ti-\ntcpaT\pc не меняется со времс мспем, пока не будет достигнуто
нем равновесие
Между двумя фазами всегда сущ ест- Границы между двумя фазами сна-
вуют редкие граплцы чала размыты, но со временем ста-
становятся достаточно резкими
Часищи второй фазы, как правило, Вторая ф.иа характеризуется рпу-
разпых размерен ляриыы распределением частиц по
размеру, которые определенным спо-
способом pac"o.no/i;ein>i в образце
Частицы второй фазы, как правило, Вторая фраза, как правило, пред-
лредстаоляюг собой сферические кап- аавляет coooii соединенные ,ipyv с
ли, не связанные друг с другом другом лесферн'тскис области
вают спинпдальныи распадом. Он обычно протекает чрезвычай-
чрезвычайно быстро; поэтому гомогенную жидкость или стекло состава h
весьма трудно охладить до комнатной температуры без их рас-
расслаивания.
Спинодальнып распад представляет собой обратный процесс
по отношению к процессу смешения жидкостей, сопровождаю-
сопровождающемуся ростом разупорядоченности и возрастанием энтропии.
Если обычно процессы диффузии ведут к выравниванию соста-
составов в различных частях системы, то при спинодалыюм распаде
происходит обратное — диффузия увеличивает градиент состава
в системе. Движущей силон такого понижения энтропии являет-
является значительное уменьшение энтальпии, происходящее при раз-
разделении фаз, которое приводит к общему понижению свободной
энергии.
Сиииодальнып распад характерен для жидкостей, составы
которых находятся между точками с и d. Геометрическое место
точек end при разных температурах (штриховая кривая на
рис. 18.9,6) называется спинодалыо. Точки end являются точ-
206 18. Стекло
ками перегиба на кривой зависимости свободной энергии от со-
состава, т. с. здесь d2(jjdx2 = 0.
Вследствие различных механизмов расслаивания (спинодалй-
пый распад или образование и рост зародышей), протекающего
в системе, образующиеся продукты распада имеют различную
структуру (табл. 18.3).
18.6. Вязкость
Вязкость является важным параметром, который необходи-
необходимо учитывать при получении и эксплуатации стекол.
Во-первых, от этого параметра зависит выбор условий плав-
плавления стеклообразующих компонентов, т. е. температуры
расплава и времени, в течение которого сырье нагревается
для получения однородного расплава. Ксли при температуре
синтеза расплав очень вязок, необходимо длительное время, что-
чтобы растворить или расплавить кристаллы исходной смеси. В та-
таких случаях более предпочтительно проводить плавление при
более высоких температурах, где жидкость менее вязкая и гомо-
гомогенность расплава достигается быстрее.
Во-вторых, чтобы качество продукции было высоким, необ-
необходимо «рафинировать» расплав, г. е. удалить из него пузырь-
пузырьки газа. Чем ниже вязкость, тем легче идет «рафинирование».
В-третьих, заметим, что после стадий плавления и рафиниро-
рафинирования для получения стекла расплав охлаждают, а в области
температур стеклования обычно проводят дополнительную тер-
термическую обработку стекла. Это делают прежде всего для того,
чтобы придать стеклу желаемую форму. Кроме того, отжиг стек-
стекла необходим для устранения механических напряжений, кото-
которые могут возникнуть в ходе охлаждения. Если стеклянное из-
изделие недостаточно отожжено, оно может неожиданно раско-
расколоться в процессе эксплуатации. При выборе температуры обра-
обработки и отжига стекла один из главных факторов, на который
следует обращать внимание,—-это вязкость переохлажденного
расплава.
В-четвертых, температура размягчения стекла и (или) тем-
температура расстекловывапия (кристаллизации) обычно представ-
представляют собой максимальные температуры его эксплуатации, при
которых стекло сохраняет свои основные свойства. Вязкое!ь
является важнейшим параметром, определяющим температуру,
при которой происходят эти процессы.
Вязкость можно определить следующим образом. Пели на
жидкость действует внешняя сила /\ то жидкость начинает течь.
Вязкость т| — есть отношение величины приложенной силы
к скорости течения. Если жидкость находится между двумя па-
параллельными пластинами, площадь поверхности которых' А и
18.6. Вязкость 207
расстояние между которыми d, а внешняя сила F приложена
к этим пластинам, то
y\ = FdlAv A8.5)
где v — скорость движения жидкости относительно пластин.
Единицей измерения вязкости является пуаз (П).
Вязкости обычных жидкостей в нормальных условиях состав-
составляют -~10~2 П. Различают несколько характеристических уров-
уровней вязкости, отвечающих различным стадиям получения
стекла;
Л. П
Температура обработки 101
Температура размягчения 107-6
Температура отжига 101'1-4
Температура снятия напряжений 10HiC
Температур;) стеклования 10';—1015
Теория вязкого течения стекла и переохлажденной жидкости
пока еще, видимо, далека от совершенства. Для простых жид-
жидкостей при описании температурной зависимости вязкости в ши-
широком интервале температур используют уравнение Аррениуса
где Q-—энергия активации вязкою течения. Однако часто зави-
зависимость Ig ц—К 1/7") отклоняется от прямолинейной. Экспери-
Экспериментальные данные лучше описывает уравнение Фулчера (осо-
(особенно в случае многокомпонентных сисгем)
Л =->;осхр f' T __ Т( ) A8.7)
где В—константа, То—некоторая идеальная предельно низкая
температура, при которой вязкость становится бесконечно боль-
большой величиной. На практике уравнение Фулчера хороню опи-
сынаст экспериментальные данные при температург'х Т^>Т0.
При Т—гТо наблюдаются значительные отклонения до данной
зависимости.
На рис. 18.10 в координатах lgri = f(I/7") приведена зависи-
зависимость вязкости переохлажденного расплава В2Оз в области тем-
температур выше Тя. Видно, что температурная зависимость вязко-
вязкости представляет собой кривую, которую нельзя описать урав-
уравнением Аррениуса.
208
18. Стекло
18.7. Электрическая (ионная) проводимость стекол
и полищелочной эффект
Большинство боратпых и силикатных стекол при комнатной
температуре являются изоляторами с уровнем проводимости
~ 10~10—10~20 (Ом-см) '. С ростом температуры, однако, под-
подвижность ионов щелочных металлов возрастает, и проводимость
стекол па основе силикатов щелочных металлов при температуре
стеклования Ts (~ 500 °С) становится равной .~ 10~5 (Ом-см) ].
Интересный и убедительный опыт для демонстрации под-
подвижности ионов Na+ при высоких температурах в обыч-
обычном стекле типа Na2O—CaO--SiO2 описан Бертом
в 1925 г. Электрическая лампа (негазопаполненпая, ва-
вакуумного типа) была ча-
частично погружена в рас-
расплавленную солевую ван-
ванну, содержащую ионы
Na7". Когда был включен
электрический ток, элект-
электроны начали покидать
вольфрамовую нить на-
накала п нейтрализовать
ноны Na+ на внутренней
поверхности стекла. Под
действием внешнего по-
постоянного электрического
поля, приложенного ме-
между расплавом к питью
накаливания, ионы нат-
натрия из расплава прохо-
проходили через стеклянную
оболочку лампы. В ре-
результате образовывалась
пленка металлического
натрия на внутренней поверхности стекла лампы. Этот экспе-
эксперимент свидетельствует о том, что ионы Na'" являются носите-
носителями электрического заряда в стекле.
Путь, по которому движутся носители заряда, например
иопы Na+, не известен, так как обычно имеются сведения лишь
о строении областей ближнего порядка в стекле. В кристалличе-
кристаллических твердых телах ионная проводимость, видимо, характери-
характеризуется прыжковым механизмом, при котором ионы преодоле-
преодолевают «энергетические барьеры» одной и тон же высоты (см. при-
приложение 8). В стеклах, однако, по-видимому, имеются потен-
потенциальные барьеры разной высоты вследствие разунорядочеипой
структуры материала и из за того, что не все позиции в стекле
Рис. 18.10. Вязкость ВгОз выше темиер.м-
туры стеклования.
18.7. Электрическая проводимость и по.шщелочной эффект 209
имеют одинаковые размеры и форму. Таким образом, при дви-
движении ионов Ка+ им приходится преодолевать то высокие, то
низкие потенциальные барьеры (рис. 18.11,а).
Проводимость стекла сильно зависит от его состава.
На рис. 18.11,E представлена зависимость проводимости натрий-
силикатного стекла от состава при 400 °С. Проводимость квар-
кварцевого стекла очень низка, и ее уровень существенно зависит
от присутствия примесей. При введении карбоната натрии
в кремнезем проводимость возрастает на несколько порядков.
Однако рост проводимости не прямо пропорционален концентра-
концентрации ионов натрия, присутствующих в единице объема. При уве-
увеличении в два раза содержания Na2O (от 10 до 20%,
рис. 18.11,6) проводимость растет почти в 10 раз. Причины
такого роста не до конца ясны. Частично это может быть связа-
связано с изменением энергии активации проводимости при измене-
изменении состава: когда содержание Na2O в стекле возрастает, энер-
энергия активации уменьшается н ионы Na~ легче передвигаются
в объеме образца. Согласно более современным взглядам, стек-
стекло относится к слабым электролитам. В натрийсиликатных
стеклах, как и в других слабых электролитах, подвижна лишь
небольшая доля ионов NaT, причем эта доля растет с ростом
температуры. Такой подход объясняет нелинейность зависимо-
зависимости Igo7" = / A/7") (т. е. закон Аррениуса не выполняется) тем,
что в предэкспоненциальпый множитель входит величина кон-
концентрации подвижных ионов, которая зависит от температуры
(см. приложение А.8). Таким способом можно объяснить слож-
сложную зависимость проводимости от состава.
Рассмотрим вкратце такое необычное явление, как полище-
полищелочной эффект. Несмотря па многочисленные исследовании, это
явление не получило еще полного и достаточно удовлетворитель-
удовлетворительного объяснения. Полищелочной эффект состоит в том, что си-
силикатные стекла, содержащие оксиды щелочных металлов, на-
например стекло состава 33,3% Li2O, 66,7% SiO2, резко уменьша-
уменьшает свою проводимость при последовательном замещении I.i>0
на оксид другого щелочного металла, например Ка2О, Зависи-
Зависимость проводимости стекол от состава проходит через минимум
при некоторых средних содержаниях одного из оксидов щелоч-
щелочного металла. Как показано на рис. 18.11, я, в трех системах
силикатных стекол общего состава 33,3% А2О, 66,7% SiO, (где
А2О — оксид щелочного металла) такие явления наблюдаются
при 150°С. Чистое литийсиликатпое стекло характеризуется
уровнем проводимости — 6-10 ?0 (Ом-см) (чисто литиевая
проводимость), а проводимость натрийсиликатпого стекла при-
примерно на два порядка выше (~10 9 (Ом-см). Однако а сме-
сметанных стеклах происходит уменьшение проводимости почти
па три порядка. Аналогичный, но еще более ярко выраженный
14-Н2Г.
О Ю 20 30 40 50
Ю
Na
И
К
Ц
Риг. 18.11. я — схематическое представление "птенци.ин.пьг; ;гм и б.нтыпои
и стекле; б — изменение проводимости натрпйсиликатного стекла с изме-
изменением состава при 400 "С; в—полигцслочной эффект в стеклах на основе
двойных силикатов, содержащих иол Li+ п ион другого щелочного метплла.
18.tf. Силикатные и боратпые стекла 211
эффект наблюдается в двойных силикатных стеклах Li2O —¦
К2О—SiO2, Li2O— Rb2O— SiO2, Li2O—Cs2O—SiO2. При
более низких температурах полищелочной эффект проявляется
еще сильнее, так как энергия активации проводимости в лоли-
щелочиых силикатных стеклах выше, чем в силикатных стеклах,,
содержащих оксид одного щелочного металла. Измерения ко-
коэффициента диффузии показали, что в нолищелочных силикат-
силикатных стеклах подвижность щелочных катионов любого сорта
уменьшается. Кроме того, обнаружено, что уменьшение прово-
проводимости, которое наблюдается в полищелочных стеклах, тем
сильнее, чем больше различие в размерах катионов щелочных
металлов. Несмотря на многолетние исследования, полищелоч-
полищелочной эффект так и не нашел удовлетворительного объяснения.
Он проявляется не только на зависимостях проводимости от со-
состава. Аномалии наблюдаются и на таких свойствах, как вяз-
вязкость и объем, однако не такие сильные, как в случае проводи-
проводимости.
В i972 г. Хсндриксон и Брей создали новую теорию полище-
полищелочного эффекта. В основе ее лежит учет энергии взаимодей-
взаимодействия разнородных катионов, расположенных в соседних пози-
позициях. Такое взаимодействие объясняется тем, что различные
ионы характеризуются различными частотами колебании у.
С помощью развитых теоретических представлений было пред-
предсказано, что в стеклах, содержащих различные изотопы одного
и того же щелочного металла, также должен иметь место по-
полищелочной эффект. Позже такой эффект был действительно
обнаружен.
Проводимость большинства стекол при низких (комнатной)
температурах мала; однако в последние годы резко увеличилась
потребность в стеклообразных материалах с высокой как кон-
конной, так и электронной проводимостью. Свойства таких материа-
материалов обсуждаются в разд. 18.9. Стекла с высокой ионной проводи-
проводимостью, например Ю (Ом-см)~' при 25 °С, как правило, со-
содержат соединения серебра; для получение этих стекол готовит
смеси Agl с кислородсодержащими солеобразпыми веществами,
такими, как Ag2MoO4 или Ag3As04. Например, стекло
состава 3AgI-Ag2MoO4 имеет при комнатной температуре прово-
проводимость ~10~2 (Ом-см)~'. Такие материалы относятся к но-
новому классу веществ — твердым электролитам; принципиально
они могут использоваться в твердотельных источниках тока.
Этот вопрос обсуждается в гл. 13.
18.8. Промышленные силикатные н боратные стекла
Чистое кварцевое стекло характеризуется целым набором
важных- эксплуатационных свойств — высокой температурой
стеклования (~1200°С). высокой температурой размягчения,
14*
212 18. Стекло
устойчивостью к расстскловыванию и действию агрессивных
сред, высокой прозрачностью для видимого и УФ-свста; однако
производство такого стекла весьма дорого. Причиной этого яв-
является высокая температура плавления A713СС) и высокая вяз-
вязкость образующегося расплава. Так, для получения гомогенно-
гомогенного расплава необходимо нагревать материал до температур су-
существенно более высоких, чем 170U°C, и выдерживать расплав
при этих температурах длительное время. Поэтому кварцевое
стекло не применяют для изготовления оконного стекла, буты-
бутылок, ламп и др., т. е. для изделий массового употребления. Для
понижения температуры плавления при сохранении высокой хи-
химической стойкости и устойчивости к расстекловыванию к окси-
оксиду кремния добавляют различные оксиды.
Добавки Na^O ( — 25 мол. %) к SiO2 понижают температуру
ликвидуса (плавления) от 1700 до 800 °С (рис. 20.3). Однако
получаемое стекло растворимо е воде и склонно к расстекловы-
ваиию. Поэтому к силикатному сгсклу (Na2O—SiO2) добавля-
добавляют другие оксиды (CaO, MgO, Л12О3), которые при сохранении
низкой температуры плавления стекла повышают его химиче-
химическую стойкость и устойчивость к расстекловыванию. Таким об-
образом, оконное стекло — многокомпонентный материал. Чтобы
стекло было прозрачным, из него необходимо удалять примеси
оксидов переходных металлов. Для бутылочного стекла корич-
коричневого цвета такая предосторожность не обязательна, и в каче-
качестве добавки к кремнезему можно использовать песок, содержа-
содержащий оксид железа.
Важной операцией при производстве стекла является его
правильный отжиг. Объем и показатель преломления стекла су-
существенно зависят от режима охлаждения, используемого на
практике. Отжиг проводят путем изотермической выдержки
стекла в течение определенного времени при температурах не-
несколько ниже температуры стеклования с целью его стабилиза-
стабилизации, гомогенизации и доведения температуры стеклования до
примерно постоянной величины. Отжиг особенно важен при
производстве высококачественных оптических стекол, для кото-
которых весьма существенны однородность состава и постоянство
показателя преломления. Отжиг также служит для снятия ме-
механических напряжений, поскольку иногда иеотожжепные стекла
раскалываются из-за внутренних напряжений. Для большинства
стекол время отжига составляет несколько часов и лишь для
некоторых специальных марок стекла или для стекол, исполь-
используемых в особых условиях, необходим более длительный отжиг.
Например, огромное зеркало телескопа, установленного на горе
Паломар, отжигалось при охлаждении от 500 до 300°С в тече-
течение десяти месяцев!
Поскольку изготовление стекла с высоким содержанием ок-
18.8. Силикатные и боратиые втекла
213
сида кремния обычными методами весьма дорого, для получения
стекла марки викор (до ~96% SiC^) используют специальный
методический прием, который помогает обойтись без нагрева-
нагревания до очень высоких температур, что кажется на первый взгляд
совершенно необходимым для производства стекол такого соста-
1600
140С
1200
юоо
800
600
ЛП Л
-
_
жидкость
-
— у
i
/
/
/
/
/
\
\
\
1 '
кугоп рас-
расслаивания
^етастабильной
жидкости
20 40 60
Na2B8O,3
t t
викор пиреке
масс,/о
Si О,
Рис. 18 12 Область расслаивания метастабплыюй переохлажденной жидко-
жидкости в системе Na2BaOi3—SiO2 [16].
ва. Этот прием заключается в получении первоначально натрий-
боросиликатного стекла примерного состава 10% Na2O+
+ЗОп/о В2О3+60% SiO2. В процессе его получения переохлаж-
переохлажденная жидкость попадает в область метастабильного расслаи-
расслаивания и претерпевает, вероятно, спиподальный распад на две
жидкие фазы. Одна из образовавшихся жидкостей имеет состав,
близкий к чистому оксиду кремния, другая — обогащена Na2O
и особенно ВгО3. На рис. 18.12 представлена часть диаграммы
состояния системы борат натрия Na2B8O[3 — оксид крем-
214 18. Стекло
ния, на которую нанесена область расслаивания переохлажден-
переохлажденной жидкости. Состав стекла викор лежит на этом разрезе (илы
вблизи него) тройной системы Na2O—В2О3— SiO2 в середине об-
области расслаивания. Обе жидкости образуют взаимно прони-
проникающую структуру. На следующей стадии получения стекла ви-
викор оксиды натрия и бора выщелачиваются путем обработки ма-
материала кислотой. Остаток представляет собой хрупкий ячеис-
ячеистый стеклообразный почти чистый оксид кремния. В заключение
технологического цикла проводится термическая обработка при
-~- 1000 °С, благодаря чему материал переходит в текучее состоя-
состояние и образуется беспористое прозрачное стекло, похожее на
плавленый SiO2.
Стекло иирекс— другой пример стеклообразного материала,
свойства которого улучшаются в результате фазового распада.
Это стекло также готовят из смеси Na2O, В2О3 и SiO2, но
с иным начальным составом — 4% Na2O, 16% В2О3, 80% SiO2
(этот состав показан стрелкой на рис. 18.12). Содержание SiO2
здесь даже выше, чем в стекле марки викор. Согласно диаграм-
диаграмме состояния системы Na2BsOi3-—SiO2, состав стекла пирекс так-
также попадает в область расслаивания переохлажденной жидко-
жидкости. Однако объемная доля тстрабората натрия мала ( — 10—f-
4-20%), и поэтому в матрице на основе оксида кремния обра-
образуются изолированные капли жидкости, богатой оксидами нат-
натрия и бора. В отличие от стекла викор такие капли невозможно
выщелачивать, так как они как бы обволакиваются матричной
фазой. Таким образом, химическая устойчивость такого стекла
определяется свойствами богатой оксидом кремния жидкой фа-
фазы. Следовательно, химическая стойкость стекла иирекс замет-
заметно повышается в результате протекающего в системе процесса
разделения фаз.
18.9. Халькогенидные и другие полупроводниковые стекла
В настоящее время заметно возрос интерес к стеклообразным
или аморфным полупроводникам, изготовляемым из халькоге-
нов — серы, селена теллура — или из их соединении с другими
элементами. Аморфный селен, например, используют в фотоко-
фотокопировальных устройствах. Другие традиционные полупроводни-
полупроводники, такие, как германий и кремний, могут быть также получены
в виде топких аморфных пленок. В настоящее время существует
возможность легировать их таким же образом, как и кристалли-
кристаллические германий и кремнии. Аморфные германии и кремний
смогут найти свое применение в устройствах, где необходимо
иметь большую поверхность полупроводникового материала, на-
например в преобразователях солнечной энергии.
18.9. Ха.чькогенлдиые и другие полупроводниковые стекла 215
18.9.1. Халькогенидные стекла
Сера и селен при плавлении образуют вязкий расплав, ко-
который при охлаждении проявляет склонность к стеклообразова-
иию. Химическая связь в этих веществах —ковалснтная, и
¦структура жидкости характеризуется наличием колец и цепей
атомов серы и селена. Этим жидкие сера и селен отличаются от
жидкого кислорода, температура плавления которого очень низ-
низкая. Жидкий кислород — молекулярная жидкость, несклонная
к образованию стекол.
18.9.1.1. Сера. Сера имеет сложную структуру. Элементарная
сера существует в виде нескольких полиморфных модификаций
(ромбическая, моноклинная и др.), однако наиболее не-
необычным является то, что структура жидкой серы сильно зави-
зависит от температуры. При температуре плавления A14 °С) се-
сера—весьма текучая жидкость (вязкость ~10"~2 П), состоящая
главным образом из колец Sg. С ростом температуры до 160 °С
текучесть и характер структуры не меняются. В интервале от
160 до ~180°С вязкость резко возрастает (примерно на пять
порядков), а затем (>180°С) снова начинает понижаться
(рис. 18.13). Причиной повышения вязкости, которое происходит
выше 160 °С. является полимеризация серы. Многие кольца Sg
разрываются и соединяются друг с другом, образуя полимерные
цепи, состоящие в среднем из 105 — 10б атомов. С ростом темпе-
температуры идут два конкурирующих процесса: с одной стороны, все
больше колец S« раскрывается и иолимеризуется, с другой—¦
средняя длина образующихся полимерных цепей быстро умень-
уменьшается. Выше 180 °С доминирующим становится второй процесс
и, следовательно, вязкость расплава понижается.
Изменения в структуре жидкой серы, происходящие с изме-
изменением температуры, носят обратимый характер. Весь процесс
можно выразить следующим равновесием: jcSs^S^. При охлаж-
охлаждении системы равновесие смещается влево. Однако быстрой
закалкой от температур >16()°С можно получить продукт, со-
содержащий при комнатной температуре очень большую долю
полимерных молекул. Такой материал — пластическая сера —
на самом деле представляет собой метастабпльиую переохлаж-
переохлажденную вязкую жидкость, которая после длительно» выдержки
при комнатной температуре переходит в кристаллическое со-
состояние. Пластическая сера при охлаждении до температур бо-
более низких, чем комнатная, превращается в стекло с температу-
температурой стеклования — 27 °С.
Присутствие примесей оказывает заметное влияние на струк-
структуру жидкой серы. Присутствие небольших количеств иода ве-
ведет к существенному понижению вязкости, например, при 180 °С.
216
18. Стекло
960 -
150 200 Z50 300 °С
Рис, 18.13. Зависимость вязкости расплавленной серы от температуры [2J.
Видимо, иод разрушает цепи атомов серы:
{—S—S—S—}+ 12 * {—S—S—I + F—S—S—}
В то же время фосфор оказывает противоположное действие,
сшивая полимерные цепи. Это ведет к увеличению вязкости.
18.9.1.2. Селен. Кристаллический селен, структура которого со-
состоит из длинных спиралей атомов селена, плавится при 217 DC.
Расплав имеет весьма высокую вязкость C0 П), которая с рос-
ростом температуры постепенно уменьшается. Никаких аномалий
на температурной зависимости вязкости не наблюдается. В рас-
расплавленном селене также имеются длинные полимерные цепи,
по их средняя длина меньше, чем в расплавленной сере при той
же температуре. Следовательно, вязкость жидкого селена при
тех же условиях меньше. При охлаждении жидкий селен легко
переохлаждается и при комнатной температуре образует стекло
(ТВ-3!°С).
18.9. Хплькогенидные и другие полупроводниковые стекла 217
18.9.1.3. Теллур. Кристаллический теллур плавится при 453 °С.
Вязкость расплава быстро понижается до значений, типичных
для расплавленных металлов. Теллур пе образует стекол, за ис-
исключением тех случаев, когда проводят его быструю закалку
или когда осаждают топкие слои теллура из газовой фазы.
18.9.1.4. Стекла более сложного состава. Термическая стабиль-
стабильность стеклообразных серы и селена может быть повышена пу-
путем добавления к ним некоторых элементов IV и V групп пе-
периодической системы, в частности мышьяка н германия. Напри-
Например, атомы селена и серы можно заместить на 60% атомами
мышьяка. Таким образом, в системах As—S и As—Se существу-
существуют широкие области стеклообразоваиия. При введении мышья-
мышьяка в серу температура стеклования материала повышается от
—27 °С (чистая сера) до ~160°С (стеклообразный As2Ss). По-
Повышение Ts связано с увеличением вязкости расплава из-за
того, что атомы мышьяка как бы сшивают отдельные цепочки
серы. Структурными элементами, присутствующими в стеклооб-
стеклообразном As2S3, являются складчатые слои ковалеитио связанных
атомов. Поскольку Т5 стеклообразного As2S3 лежит достаточно
высоко, это стекло весьма устойчиво к расстекловыванию, и оно
находит применение в качестве прозрачного для ИК-излучения
материала.
При одновременном введении двух других элементов в халь-
когены область стеклообразующих составов может быть сущест-
существенно расширена. Например, при добавлении к сере фосфора
или германия в системах Р—S или Ge—S возникает ограничен-
ограниченная область стеклообразования. В то же время в тронной систе-
системе Р—Ge—S область существования стекла весьма обширна.
Еще более интересна тройная система Si—As—Тс: пи один из трех
элементов сам по себе, пи одна из трех двойных систем Si—As,
Si—Те, As—Те не обнаруживают склонности к стеклообразова-
пию. Тем пе менее в области, богатой теллуром, тройной систе-
системы Si—As—Те устойчиво существует стеклообразная фаза. Эти
стекла, а также стекла в системе Ge—Р—S обладают важными
эксплуатационными свойствами: они устойчивы на воздухе, мо-
могут быть использованы до 500 °С и обладают хорошими оптиче-
оптическими характеристиками. Такие стекла находят применение
в навигационных приборах самолетов.
18.9.2. Электрические свойства
Стекла на основе халькогенидов обладают полупроводнико-
полупроводниковыми свойствами и, как правило, являются электронными полу-
полупроводниками с уровнем проводимости от ]0~3 до 10~13
(Ом-см). Проводимость чистых халькогенов растет с ростом
218 18. Стекло
атомной массы халькогена. Например, расплавленный теллур
по своим электрическим свойствам полностью соответствует
жидким металлам. Кроме различия структуры существуют дру-
другие принципиальные отличия аморфных полупроводников, таких,
как селен, от кристаллических полупроводников, таких, как
кремний и германии.
Во-первых, аморфные полупроводники существуют в широ-
широкой области составов, и они не соответствуют в точности соста-
составу какпго-лпбо стехиомстрического соединения. Состав кристал-
кристаллических полупроводников, как правило, точно отвечает составу
стехиометрического соединения или близок к нем}1. Возможность
систематического варьирования состава аморфных материалов
позволяет получать материалы с различными свойствами.
Во-вторых, аморфные полупроводники могут легко изменять
свою форму, например в аппаратах для ксерокопирования ис-
используют тонкие аморфные пленки селена. Монокристаллам го-
гораздо сложнее придавать нужную форму, поэтому непрактично
(и дорого!) изготавливать монокристаллы в форме тонких пле-
пленок.
В-третьих, аморфные полупроводники, как правило, не чув-
чувствительны к наличию примесей, что принципиально отличает
их от кристаллических полупроводников. В последние годы,
однако, были найдены методы легирования аморфных германия
и кремния.
Чтобы понять влияние примесей па свойства аморфных полу-
полупроводников, рассмотрим структурные отличия между кристал-
кристаллическим и аморфным германием. Кристаллический германий
имеет структуру алмаза. В его решетке каждый атом распола-
располагается в тетраэдрическом окружении четырех других атомов
германия, так что возникает трехмерная каркасная структура.
При легировании германия, например, мышьяком, пятивалент-
пятивалентный мышьяк вынужден занимать тетраэдрическую позицию вме-
вместо атома германия. Поэтому атом мышьяка образует четыре
связи с соседними атомами германия. Поскольку у атома
мышьяка имеется пять валентных электронов и лишь четыре из
них участвуют в химической связи, пятый электрон может сво-
свободно двигаться в кристалле и участвовать в переносе электри-
электрического заряда. (Согласно зонной теории этот электрон нахо-
находится в зоне проводимости; см. гл. 14.) На рис, 18.14, а схема-
схематически изображена (в плоскости) кристаллическая решетка
германия, легированного мышьяком.
Структура аморфного германия представляет собой прост-
пространственную сетку, в которой большинство атомов германия так-
также находится в тетраэдрическом окружении других атомов гер-
германия. Однако некоторые атомы имеют лишь три ближайших
соседа и один валентный электрон остается у атома германия
18.9. Хальк'огемидные и другие полупроводниковые стекла
219
неспарснным. Образуется ненасыщенная связь. Два таких слу-
случая схематически представлены на рис. 18.14,6. В аморфной сет-
сетчатой структуре легированного мышьяком германия атомы
мышьяка предпочитают занимать такие позиции, которые коор-
координируют вокруг себя лишь три атома германия. Три электрона
каждого атома мышьяка участвуют в образовании ковалентных
связей, а два оставшихся электрона образуют несвязывакнцую
(неподслепную) пару. Поэтому атом мышьяка электронейтрален
ненасыщенные связи
О
As
Рис. 18.14. Изображение на плоскости структуры кристаллического герма-
пин (а) п аморфного германия (б), легированных мышьяком [4]. В кристал-
кристаллическом германии каждый ;пом, и том числе н .ттч мышьяка, занимает
регулярный узе.i решетки и окружен четырьмя соседними .itom.'imh.
и не имеет свободных неспаренных электронов, участвующих
в проводимости. Итак, эффективность легирования аморфного
германия мышьяком с целью создания проводимости я-типа
гораздо меньше, чем в случае кристаллического германия.
Существуют две причины, препятствующие проявлению эф-
эффекта легирования в аморфных германии и кремнии, если леги-
легирующие примеси вводить обычными методами. Псриая состоит
в том, что добавление мышьяка обычно не приводит к появле-
появлению свободных электронов, так как атомы мышьяка часто за-
занимают позиции, окруженные лишь тремя ближайшими соседя-
соседями. Вторая причина заключается в том, что если бы свободные
электроны удалось ввести в структуру стекла, то они вскоре
оказались бы «пойманными в ловушки» ненасыщенных связей
атомов германия, координирующих вокруг себя лишь три со-
соседних атома. Поэтому эти свободные электроны скоро переста-
перестали бы участвовать в переносе электрического заряда. Для полу-
220 18. Стекло
чения тонких пленок используется наряду с другими методами
метод катодного распыления в атмосфере аргона (гл. 2). Ионы
аргона выбивают из материала мишени (кристаллического гер-
германия) атомы германия, которые переносятся Fia подложку, где
и осаждаются в виде тонких аморфных пленок.
Недавно был развит новый метод получения аморфных гер-
германия и кремния. Этот метод, видимо, исключает образование
ненасыщенных связей и, следовательно, позволяет эффективно
легировать аморфные полупроводники. Газообразный моногер-
ман GeHU, содержащий небольшие количества фосфина РН3 и
диборана В2Н5, подвергается плазменному разложению в усло-
условиях тлеющего разряда. Атомы Ge, Si, P и В, получаемые при
разложении, осаждаются па подложке в виде тонкой пленки,
в которой, по-видимому, отсутствуют дефекты типа ненасыщен-
ненасыщенных связей, поскольку, вероятно, любая ненасыщенная связь,
которая могла бы существовать, способна образовывать кова-
лептную связь с атомарным водородом и, следовательно, она
перестает действовать как ловушка свободных электронов. Хотя
некоторые атомы фосфора и бора, введенные в аморфный крем-
кремний, и занимают позиции с координационным числом, равным
трем, другие атомы этих элементов должны попадать в тетра-
эдрическне позиции, что приводит к росту проводимости по та-
такому же механизму, что и в кристаллических полупроводниках
(я- и р-тииа).
18.9.3. Фотокопировальный процесс
В фотокопировальном процессе используются специфиче-
специфические свойства аморфного селена. Во-первых, селен может быть
приготовлен в виде тонкой полупроводниковой пленки. Во-вто-
Во-вторых, селен является фотопроводником, т. е. его электронная про-
проводимость резко возрастает под действием света. В состав фото-
фотокопировального аппарата входят два цилиндрических металли-
металлических барабана, на которые методом вакуумного напыления
наносится тонкая пленка аморфного селена. Фотокопировальный
процесс включает в себя несколько этапов (рис. 18.15).
Поверхность селена заряжается положительно под действием
коронарного разряда, индуцируемого проволокой, к которой
приложено высокое напряжение. Эта проволока движется па-
параллельно поверхности селена (рис. 18.15,а). Затем копируемый
лист документа оставляет свое «изображение» па селеновом
экране под действием видимого света: светлая поверхность стра-
страницы отражает фотоны света на селеновый экран, в результате
чего на поверхности пленки аморфного селена образуются па-
пары электронов и дырок (рис. 18.15, б). Под действием электри-
электрического поля эти пары диссоциируют, и дырки движутся внутрь
когпшдлыс л другие полупроводниковые стек.'];]
а
Заряд
проволока, около
которой возникает
[Г\ коронарный
разряд
пленкэ
¦металлическим оарзоан
б
Съемка
Разряд
Проявление
краска
д
Перевод
отполированного
рисунка на бумагу
Рис. 18.15. Стадии фотокопировальчого процесса [4J.
222 __ 18. Стекло
слоя селена к металлическому барабану, а электроны—к по-
поверхности селена, где они частично нейтрализуют индуцирован-
индуцированный положительный заряд (рис. 18.15, в). Таким образом, па се-
селеновой пленке остается изображение в виде положительных
зарядов, расположение которых отвечает темным участкам по-
поверхности документа, который копируется. После этого па селе-
селеновый экран наносится отрицательно заряженная угольная пыль
в виде черной типографской краски. Краска прилипает к тем
положительно заряженным участкам поверхности пленки, кото-
которые не разрядились (рис. 18.15, г). Эта краска переводится на
чистый лист бумаги под действием второго коронарного разряда
(рис. 18.15, д). Затем бумага убирается и нагревается, чтобы
изображение сохранилось надолго.
18.10. Металлические стекла
Жидкие металлы, как правило, не склонны к переохлажде-
переохлаждению с образованием стекла. Однако недавно были открыты не-
некоторые металлические составы, которые удалось перевести
в стеклообразное состояние. Структура жидких и кристалличе-
кристаллических металлов характеризуется отсутствием направленных свя-
связен между атомными ядрами и электронами, участвующими
в переносе электрического заряда. Таким образом, чтобы жид-
жидкий металл закристаллизовался, нет никакой необходимости
разрывать прочные ковалентные связи. Сказанное можно по-
пояснить па примере халькогепов: в жидком селене существуют
ковалентные связи —Se—Se—, и селен легок образует стекло,
а в жидком теллуре характер связи более металлический и тел-
теллур при обычных условиях не переходит в стеклообразное со-
состояние. Аналогичная ситуация наблюдается и в ионных мате-
материалах: в расплавленном NaCl ионные связи между Na1" и
С1~ ненаправленные. Это существенно понижает высоту потен-
потенциальных барьеров процессов кристаллизации и перемещения
атомов. В результате NaCI, как и многие другие простые соли,
не образуют стекол.
Для получения металлических стекол необходимо использо-
использовать специальные методы сверхбыстрой закалки. В методе рас-
распылительной закалки капли расплава под действием высокого
давления газа выстреливаются с большой скоростью на охлаж-
охлажденную поверхность, и жидкость эффективно охлаждается за
доли секунды. В методе закалки на валки расплав направляет-
направляется па охлаждаемый вращающийся барабан, как показано на
рис. 18.16. В результате получается тонкая лента металлическо-
металлического стекла. Возможен также вариант, когда расплав впрыски-
впрыскивается в узкий зазор между двумя быстро вращающимися ох-
18.10. Металлические стекла
223
лаждаемьпш валками. Таким методом можно достичь скоростей
охлаждения — 10ь — 10s К/с.
Металлические стекла образуются при закалке расплавов
лишь в некоторых системах, причем обычно в состав расплава
входят не менее двух различных элементов. Одним из компонен-
компонентов расплава является типичный металл, например переходный
металл, такой, как железо или палладий. Другой компонент —
это обычно элемент, занимающий промежуточное положение
между металлами и диэлектриками, например полупроводник,
нагреватель
охлаждаемый
вращающийся
медный ¦—
бараосэч
лемл аморфного
Рис. 18.1 G. С\сма осуществления мстила злк.ъткм им валки.
такой, как кремний или фосфор. В каждой системе, как прави-
правило, возможно получить стекла в целой области составов, напри-
например в системе Pd—Si область стсклообразовання находится
в интервале составов от 15 до 25% Si. Способность к стеклооб-
разовапию обычно связывают с наличием на равновесной диа-
диаграмме состояния системы пизкоплавкоп эвтектики (разд. 18.1.4).
Принимается, что структура металлических стекол представля-
представляет собой беспорядочную плотную упаковку шаров двух различ-
различных размеров. Такое расположение шаров можно получить, на-
например, если взболтать внутри какой-нибудь емкости два сорта
шаров.
Металлические стекла обладают некоторыми необычными и
ценными свойствами по сравнению с кристаллическими метал-
металлами.
Во-первых, металлические стекла обычно имеют гораздо бо-
более высокую прочность, чем кристаллические металлы. В неко-
некоторых аморфных сплавах практически был достигнут теоретиче-
теоретический предел прочности. Многие чистые металлы являются
относительно мягкими из-за наличия в них дислокации (гл. 9),
которые могут легко перемещаться под действием внешнего па-
224
18. Стекло
пряжения. Дислокации легко перемещаются в кристалле частич-
частично из-за того, что структура кристалла строго периодична: дис-
дислокации перемещаются из одной области решетки в идентичную
соседнюю область. Не существует однозначного ответа на во-
вопрос, могут ли существовать дислокации в непериодических
структурах, например в стекле. (Имеется, правда, и другая
точка зрения, согласно которой структуру стекла можно рас-
рассматривать как целиком состоящую из дислокаций.) Если ди-
дислокации в металлическом стекле все же существуют, то вряд
ли они могут так легко
перемещаться, как в кри-
кристаллическом материале.
Поэтому металлические
стекла характеризуются
высокой прочностью. В
то же время это очень
пластичные материалы, в
чем они совершенно не
похожи на такие твер-
твердые и прочные материа-
материалы, как литая сталь, ко-
которая обычно весьма
хрупкая. Металлические
стекла могут выдержи-
выдерживать 50%-ную (и более)
деформацию сдвига,
стекло
кристалл
в
Рис. 18.17. Кривые намагничивания
FersPisCio (а и б) и Со78р22 (в, г). В слу-
случае аморфных материалов (а и в) гисте-
гистерезис меньше, петля гистерезиса имеет бо-
.¦ice прямоугольную форму, коэрцитивная
сила меньше, а магнитная проницаемость
выше.
прежде чем они оконча-
окончательно разрушатся по механизму пластичного излома. Весьма
вероятно, что металлические стекла в будущем могут исполь-
использоваться в качестве армирующих волокон.
Во-вторых, металлические стекла более устойчивы в химиче-
химически агрессивных средах. Они более коррозионно стойки, чем по-
поликристаллические металлы. Химические процессы особенно ак-
активно протекают на границах зерен м на поверхностях с повы-
повышенной энергией, например в местах выхода дислокаций или
других дефектов. Поскольку в стеклообразных образцах отсут-
отсутствуют границы зерен и дислокации в обычном смысле этого
слова, они химически более инертны.
В-третьих, некоторые металлические стекла характеризуются
интересными магнитными свойствами. Кобальт- и железосодер-
железосодержащие стекла характеризуются низкой коэрцитивной силой, они
легко намагничиваются и размагничиваются (рис. 18.17), Эти
свойства также связаны с отсутствием границ зерен в стеклах.
Металлические стекла могут найти применение в элементах па-
памяти в тех случаях, когда необходима быстрая регистрация и
перезапись информации.
18 11. Стеклокерамика 225
18.11. Стеклокерамика
Стеклокерамика — это кристаллические вещества, получае-
получаемые из стеклообразных исходных материалов. Они обладают
комбинацией наиболее важных свойств как стекла, так и кера-
керамики. Стеклокерамика сохраняет механическую прочность до
гораздо более высоких температур, чем стекло, которое размяг-
размягчается при 500 °С. Регулируя состав стеклокерамических мате-
материалов, можно варьировать их свойства, например коэффициент
термического расширения. Стеклокерамические материалы полу-
получают путем термической обработки некоторых стекол в специаль-
специально подобранном и строго контролируемом режиме. Такая обработ-
обработка приводит к появлению кристаллических зародышей и их рос-
росту в матрице стекла. Часто в результате этого происходит пол-
полная кристаллизация образца, иногда в образце присутствует ос-
остаточное количество стеклообразной фазы.
Расстекловывание или кристаллизация стекла обычно озна-
означает окончание его использования, так как стекло при этом те-
теряет свою механическую прочность, прозрачность и другие ра-
рабочие характеристики. Суть развитого в 1957 г. фирмой Stookey
of Corning Glass метода получения стеклокерамики сводится
к преднамеренному проведению расстекловываиия в контроли-
контролируемых условиях. Возникающий при этом материал представ-
представляет собой мелкодисперсное керамическое вещество с набором
важных параметров, в частности с высокой механической проч-
прочностью. Дли проведения расстекловывания в контролируемых
условиях необходимо создать большую концентрацию кристал-
кристаллических зародышей (обычно 1012—101Г> зародышей в 1 смэ),
равномерно распределенных в объеме образца. Кроме тою,
важно исключить возможность начала кристаллизации на не-
нескольких зародышах, находящихся на поверхности. Для полу-
получения зародышей используют различные методы:
1) готовят коллоидный раствор таких металлов, как Си, Ag,
Аи и Pt в стеклообразующем расплаве. Коллоидные частицы
этих металлов не растворяются полностью и, следовательно,
могут служить центрами кристаллизации при отжиге стекла при
более низких температурах (например, 500°С). Поскольку стек-
стекла фоточувствительны, осаждению металлических зародышей
может способствовать облучение их УФ-светом;
2) в исходную смесь компонентов при приготовлении стекла
добавляют такие оксиды, как TiO2, P2O5 и ZrOj, которые рас-
растворимы в расплаве при высоких температурах, но выпадают
в осадок в результате отжига при более низких температурах.
Их осаждение, видимо, является следствием расслаивания
в жидкой фазе. Осаждающиеся частицы этих оксидов и образу-
образуют центры кристаллизации стекла;
15-1426
226
18. Стекло
3) с целью проведения гомогенного зародышеобразования
(разд. 18.3) стекло отжигают при температурах вблизи темпера-
температуры стеклования. При этом в объеме стекла возникают заро-
зародыши кристаллов.
Успешное осуществление гетерогенного зародышеобразова-
зародышеобразования (первый и второй методы) зависит от двух факторов.
мегаста0иЛ1>»дя область
переохлаждения
скоробь роста кристаллов
скорость гомогенного
засодъш:гобразованк я
Скорость зородь1Ш?о5рпзовуния л роста кристаллов
Рис. 18.18. Скорости гомогешюго зпродышеобразовашш и роста кристаллов
is вязкой жидкости (по Таммаму).
Во-первых, межфазное натяжение между зародышами и кри-
кристаллизующейся фазой должно быть мало, т. е. должен быть
мал краевой угол а (рис. 20.2) между подложкой, на которой
образуется зародыш, и зарождающимся кристаллом. Малый
угол а во многом определяет низкое значение свободной энер-
энергии зародышеобразовапия.
Во-вторых, необходимо, чтобы кристаллическая структура
зародышей была родственна структуре зарождагощейся фазы,
причем в первую очередь важно, чтобы величины расстояний d
между плоскостями с малыми индексами hkl обеих струк-
структур были близки. В случае выполнения этого условия возможен
направленный или эпитаксиальный рост, если размеры элемен-
элементарных ячеек зарождающейся фазы и зародыша не отличаются
больше, чем на 10—15%.
18.11. Стеклокерамика
227
После этапа зародышеобразования, который обычно прово-
проводят при температурах, близких к температуре стеклования, когда
вязкость расплава высока (Ю11—10'2 П), а скорость роста кри-
кристаллов мала, стекло нагревают до более высоких температур.
Здесь начинается рост кристаллов иа поверхности образовав-
образовавшихся зародышей. Вследствие того что концентрация зароды-
зародышей велика и они равномерно распределены в объеме стекла,
каждый из них вырастает очень незначительно, после чего он
сталкивается с соседним растущим зародышем. Следовательно,
СО
г>
>ч
ь-
О.
с
Плавление
и формо
вакие
\
\
кристаллизация
зародышеобразо-/ \
блние / ¦ 1
Время
Рис. 18.19. Двухстэднйпый процесс нагревания образца при производстве
стеклокерлыических материл.чои.
размер кристаллов в стеклокерамических материалах невелик.
Обычно он составляет 0,1—I мкм A0~7—10" G м) в диаметре.
Температура, при которой проводится рост кристаллов, выше
температуры, при которой ведут зародышеобразовапие, так как
максимальные скорости того и другого процессов отличаются
друг от друга (рис. 18.18). Поскольку зародышеобразованис—•
более сложным процесс, чем рост кристаллов, необходимо су-
существенное переохлаждение расплава, чтобы возникла доста-
достаточная разность свободных энергий зародышей и расплава и
началось образование зародышей.
Таким образом, технологический процесс получения стекло-
керамических материалов должен включать несколько стадий.
Первоначально необходимо приготовить стекло путем сплавле-
сплавления оксидных компонентов совместно с веществами, которые
впоследствии станут центрами гетерогенного зародышеобразо-
вация. Расплавы заливают в емкости желаемой формы. Затем
15*
228 18. Стекло
расплав охлаждают и подвергают двухстадийной термической
обработке, проводя стадию образования зародышей и стадию
роста кристаллов (рис. 18.19).
18.11.1. Наиболее распространенные сгек.юкеримичсскис
системы
Стеклокерамические композиции имеют сложный химический
состав и содержат несколько компонентой. Однако обычно их
свойства можно свести к свойствам более простых систем. Наи-
Наиболее важным прототипом счеклокерамнческнх систем является
система 1л2О—SiO2, диаграмма состояния которой представле-
представлена па рис. 18.20. Добавление ~30 мол. % Li2O к S1O2 приводит
к резкому понижению температуры ликвидуса от 1713 до
1П30°С. При охлаждении жидкости образуется прозрачное стек-
стекло. Из расплавов, содержащих менее ~25% Li2O, при охлаж-
охлаждении образуются мутные стекла с оиалесценциеи из-за рас-
расслаивания переохлажденной мстастабильной жидкости. При
кристаллизации в интервале температур между температурой
стеклования (-~500°С) и температурой солидуса (~1030°С)
основным продуктом является Li2Si20s с небольшими добавками
либо SiO2, либо Li2Si03, либо и той и другой фазы. Для полу-
получения топкодиснерсиой стеклокерамики на основе силиката ли-
лития обычно проводят двухстадийпую кристаллизацию: первая
стадия—• зародышеобразовамие при 450—500 °С, следующая
стадия — рост кристаллов f-i2Sia05 при 650—700 °С.
Одной из важнейших для промышленности стеклокерамиче-
ских систем является система Li^O—А12Оз—SiO2. Для проведе-
проведения процесса гетерогенного зародышеобразования в нее вводят
металлы (например, Аи) или оксиды (например, ТЮ2 или
РгОг,). Для проведения процесса зародышеобразования под дей-
действием света используют составы с низким содержанием AI2O3,
в которых основными кристаллическими фазами являются
Li2Si20.-, и (или) Li2SiOa. В более богатых оксидом алюминия
материалах основными продуктами кристаллизации будут |}-сно-
думсн Li2AlSi206 или твердые растворы на основе |}-кварца. По-
Последние характеризуются необычными свойствами, в том числе
очень низким отрицательным коэффициентом термического рас-
расширения (<10"ь).
Стеклокерамика на основе силиката лития с добавками MgO
находит применение в связи с высоким коэффициентом терми-
термического расширения (~ 1,4 ¦ 10~5). В ее состав входят различные
кристаллические фазы, в том числе Li2MgSi04. Аналогичные
материалы, но с добавкой ZnO, имеют не только высокий коэф-
коэффициент термического расширения, но и высокую механическую
18.П. Стеклокерамика
229
прочность. В состав этого материала входят такие кристалли-
кристаллические фазы, как Li2ZnSi04 и виллемнт Zn^SiO/i.
Стеклоксрамическис материалы, получаемые в системе
MgO—AI2O3—SiO2 с добавками TiO2 и РгО^ в качестве затрав-
затравки для гетерогенного зародышеобразования, являются хороши-
хорошими электрическими изоляторами, поскольку в их состав не вхо-
!700
1600
1500
1400
1300
1200
1100
1000
900
-
-
1Л2О + жидк \
1255 \ / '
Li2O + U4Si04
-
i i
VI
\/Ю24
Li4SiO,
Li 2 Ы О5
1
+ /
ЖИДК -V /
/ 1470
Г
?5i0j +жидк. /
">ч/ /5Ю2(тридимит)
N. / ЖИДК.
1033 \ / ,028
Ц2Ы03
Li25t205
Li2Si20b
SiO2
I i i
О 1° 20 30 40 50 60 70 80 90 SiO2
Рис. 18.20- Диаграмма состояния системы Li2O— SiO2.
дят оксиды щелочных металлов. Они же отличаются высокой
механической прочностью при высоких температурах. Основная
кристаллическая фаза в иих — сх-кордеирит 2MgO-2AI2O3-5SiO2
(Mg^AUSisOig) с небольшими добавками (в зависимости от со-
состава стеклокерамики) кристобалита SiO2, клиноэнстатита
MgSiO3 и форстерита Mg2Si04.
18.11.2. Свойства стеклокерамичсских материалов
Перечислим наиболее важные свойства стеклокерамических
материалов.
1) Стеклокерамика обладает, как правило, более высокой
прочностью к внешним деформирующим и ударным нагрузкам,
230 18. Стекло
чем обычное стекло. Если прочность на разрыв стеклянных тру-
трубок обычно составляет 210—700 кг/сма, то соответствующая ве-
величина для стеклокерамических изделий равна 2800—
4200 кг/см5. Это, вероятно, связано с тем, что имеющиеся в стек-
стеклокерамических материалах кристаллы ограничивают размер
возможных дефектов, и это понижает скорость распространения
трещин. Кроме того, стеклокерамические вещества обычно отли-
отличаются повышенной по сравнению с обычными стеклами устой-
устойчивостью к истиранию.
2) Коэффициент термического расширения стеклокерамики
(как и стекла) можно легко регулировать ее химическим со-
составом. При этом могут быть достигнуты как крайне низкие
значения коэффициента термического расширения (около нуля),
так и весьма высокие (до 2-10~5). Поэтому возникает возмож-
возможность подбирать коэффициент термического расширения стекло-
керамического материала таким же, как, например, у металлов.
Это обстоятельство оказывается очень важным при создании
герметичных сочленений металла с изделием из стеклокерамики.
Стеклокерамические образцы с низким или даже нулевым ко-
коэффициентом термического расширения устойчивы к тепловым
ударам. Это означает, что такие материалы не разрушаются
под действием больших и резких колебаний температуры.
3) Стеклокерамика характеризуется существенно более вы-
высокими температурами деформации, чем стекла того же хими-
химического состава. Например, многие оксидные стекла имеют тем-
температуру стеклования Тйл?450сС, а размягчаются при 600—•
700°С. Стеклокерамические материалы того же состава сохра-
сохраняю г свою механическую прочность и жесткость до более высо-
высоких температур (~ 1000—1200 °С).
4) Стеклокерамические материалы, как правило, — хорошие
изоляторы, особенно, если они не содержат оксидов щелочных
металлов.
5) Внешний вид стеклокерамики зависит от присутствующих
в них кристаллических фаз. Она может быть прозрачной на
просвет или непрозрачной в зависимости от размеров кристал-
кристаллов, возможного двулучепреломления в кристаллах и разности
показателей преломления у различных кристаллов или у кри-
кристаллов и оставшейся стеклообразной матрицы.
6) Стеклокерамические материалы имеют пулевую порис-
пористость в отличие от большинства керамических изделий, спрессо-
спрессованных обычными методами. Пористость последних составляет
в среднем 10%. Высокая плотность стеклокерамики объясняется
тем, что в процессе кристаллизации стекло может течь, «зале-
«залечивая» тем самым поры, возникающие при изменении объема.
18.11. Стеклокерамика 231
18.11.3. Применение стеклокерамики
Устойчивые к резким перепадам температуры марки стекло-
стеклокерамики используют при производстве изделий для высокотем-
высокотемпературной обработки материалов, например контейнеров или
оболочек печей с встроенными внутренними электронагревате-
электронагревателями. Их используют также в качестве несущих конструкций,
так как их износостойкость в несколько раз выше, чем износо-
износостойкость стали, а также в качестве покрытий па металлах (ти-
(типа эмалей) и для герметичного сочленения металла и керамики.
В связи с высокой термостабильностью и устойчивостью к теп-
тепловым ударам стеклокерамика применяется в качестве теп-
теплозащитной оболочки носовой части ракет.
Шлакокерамика — это стеклокерамика, получаемая из шла-
шлаковых отходов доменных печей. Она может найти применение
в качестве строительного материала. Шлаки — это сложные бо-
богатые оксидом кальция вещества, которые можно перевес™
в стеклообразное состояние путем сплавления с SiO2. Получен-
Полученное стекло затем кристаллизуется, переходя в стеклокерамику.
Основными кристаллическими компонентами такой стеклокера-
стеклокерамики являются волластонит CaSiO3, диопсид CaMgSi2O6 и анор-
анортит CaAl2Si2O8.
«Химически обработанная» стеклокерамика используется как
основа при изготовлении печатных монтажных плат в электрон-
электронной технике. В процессе химической обработки фоточувствитель-
фоточувствительные стекла, такие, как стекла системы Li2O—AI2O3—SiO2,
содержащие небольшие количества меди, серебра или золота, под-
подвергаются воздействию УФ-излучспия. В результате в них обра-
образуются кристаллические зародыши. При соответствующей тем-
температуре кристаллы Li2Si03 растут. Затем их удаляют путем
травления плавиковой кислотой. Важным обстоятельством при
этом является то, что кристаллический силикат лития гораздо
лучше растворяется в плавиковой кислоте, чем окружающая его
стеклообразная матрица. Если до облучения на стекле находил-
находился какой-либо шаблон, препятствующий попаданию света, то
после такой процедуры можно получить фотоизображение шаб-
шаблона внутри стекла. Таким образом, соответствующий темпера-
температурный отжиг и травление позволяют получить стекло, которое
содержит сложную систему отверстий для элементов электри-
электрических цепей. Если впоследствии необходимо из оставшейся
стеклянной матрицы получить стеклокерамическое изделие, про-
проводят обычный двухстадийный процесс зародышеобразования и
роста кристаллов.
При химической обработке стекол и стеклокерамических ма-
материалов для инициирования кристаллизации широко применя-
применяют облучение. Процессы, аналогичные процессам образования
232 18. Стекло
зародышей под действием света, происходят в фотохромных
стеклах. В объеме таких стекол равномерно распределены кри-
кристаллы AgCl, имеющие размеры долей микрона. Под действием
облучения видимым светом некоторые ионы Agf превращаются
в атомы, которые коагулируют, образуя небольшие кластеры ме-
металлического серебра. Наличие таких кластеров вызывает по-
потемнение стекла. Механизм этого процесса, видимо, аналогичен
механизму процесса потемнения фотопленки под действием све-
света. Однако в фотохромных стеклах процесс потемнения обра-
обратим, т. с. при удалении источника света кластеры серебра рас-
растворяются и стекло вновь становится прозрачным. Фотохромное
стекло используют в качестве материала для линз в солнечных
очках. Возможны и другие области его применения.
Упражнения
18.1. Основываясь на правилах Захариасеца, выскажите свои предполо-
предположения относительно склонности к стеклообразованию следующих веществ:
a) ZrO2; б) BeF2; в) MgP2.
18.2. Объясните, почему экспериментально определяемая температура
стеклования Ts всегда выше температуры стеклования идеального стек
л а 7о-
18.3. Образование стекла иногда считают фазовым переходом второго
рода. Справедлив ли такой подход?
18.4. Почему трудно получить информацию о структуре стекла? Какую
информацию можно получить, используя дифракционные, спектральные и
микроскопические методы исследования?
18.5. Хотя полищелочной эффект в стеклах широко изучен эксперимен-
экспериментально, до сих пор он не нашел удовлетворительного объяснения. Предло-
Предложите эксперименты, которые могли бы способствовать лучшему пониманию
природы полищелочного эффекта.
18.6. Сделайте то же самое применительно не к полищелочному эффек-
эффекту, а к борной аномалии.
18.7. Почему сера и сечен легко образуют стекло, а другие халькогены
(кислород, теллур) нет?
18.8. Стекла находят разнообразные сферы применения. Какие специ-
специальные свойства стекол необходимы и какой химический состав стекол опти
мален для их использования: а) в качестве электролита в электрохимиче-
электрохимическом элементе Ag/U; б) в качестве материала для окон; в) в качестве ма
териала контейнера, устойчивого до высоких температур (900 °С); г) в ка-
качестве изолирующего стсклокерамического материала; д) в качестве воз-
возможного заместителя стали в упрочняющих волокнах; е) в качестве полу-
полупроводниковой пленки, проводимость которой чувствительна к облучению
видимым светом; ж) в качестве материала для магнитных элементов па-
памяти?
18.9. Гипотетическим примером неустойчивого равновесия (гл. II) яв-
является гомогенное стекло или жидкость, состав которой при данной темпе-
температуре попадает в область спиподалыюго распада. Объясните эту ситуа-
ситуацию.
Литература
1. Angell С. A, The data gap in solution chcmislry. The ideal glass transition
puzzle, J. Chem. Ediic, 47. 583 A970)
2. Bacon R. F., Fanelli R., The Viscosity uf Sulphur. J. Ашсг. Chcm Soc. 65,
639 A943).
Литература ____ ?^
3. Cahn R. Г., Metallic glasses, Contemp. Physics, 21, 43 A980).
4. Dauis E. A., Non crystalline materials, Endeavour, 1, 103 A977).
5. Doremus R. #., Glass Science, Wiley, 1973.
6. Gilman J. J., Metallic glasses, Physics Today, May 1975, 46.
7. Jones G. 0., Glass, 2nd cd., Chapman and Mall, 1971.
8. Mackenzie J. D., Modern Aspects of ihc Vitreous Stale. Buttenvorths, 1964.
9. McMillan P. ft1'., Glass ceramics, 2nd «d., Academic Press, 1979.
JO. Murcy G. IF., The Properties of Glass. Reiiihold, 1954.
11. Mozzi R. L., Warren B. ?., The Structure of Vilreuus Silica, J. Appl. Cryst.,
2, 164 A969).
12. Paul A., Chemistry of glasses. Chapman and Hall, 1982.
13. Purlhasarathi/ R,, Rao K- J.. Rao C. .V. R., The Gla?s Transilion: Salient
Facts and Models, Chnn. Soc. Revs, 12, 361 A984).
14. Роусон Г. Неорганические стеклообразные системы —M: Мир, 1970.
15. Ransson H, Properties and Applications of Glass, in: Glass Science and
Technology, Elsevier, 1980. vol. 3.
16. Rockvtt J. J., Foster U". R., J Amer. Ccrain. Soc, 49, 31 A966).
17. Uhlmaim D. R , Inorganic amorphoii.s solids and glass-ceraraic materials in
Treatise on Solid State Chemistry, Hannay N. B. (cd.). Plenum Press, 1976,
vol. 3, p. 293.
18. Weyl W. A., Marboc E. С The Constilulion of Glasses, WileyTntcrscience,
1902.
Дополнительная литература. Аппен А. Л. Химия стекла.— Л.: Химия,
1974; Борисова 3. У. Химия полупроводниковых стекол. — Изд-во ЛГУ, 19й4;
Мазурин О. В. Стеклование и стабилизация неорганических стекол. — Л.: Нау-
Наука, 1978; Тагаринива Я. И. Структура твердых аморфных и жидких ве-
веществ.— М.- Наука, 1983; Шульц М. М. О химическом строении стеклообра-
зующнх расплавов и стекол. — В сб. Стеклообразное состояние. Л.: Наука,
1982; Фельц Л. Аморфные и стеклообразные неорганические твердые тела. —
М.: Мир, 1986.
Глава 19
ЦЕМЕНТ И БЕТОН
Цемент-—это собирательное название большой группы вя-
вяжущих материалов, которые обладают свойством схватываться
и твердеть в результате химической реакции, идущей при сме-
смешивании этих веществ с водой. С древних времен людям были
известны различные типы вяжущих веществ. Древние египтяне
использовали жженный гипс CaSCv У2Н2О с различными до-
добавками; древние греки и римляне применяли прокаленный из-
1лсг!!як CaCOj. Позже для получения иеюна они вводили в него
такие заполнители, как песок и щебень. Однако известковые
строительные растворы неравномерно взаимодействовали с во-
водой и получался некачественный продукт. Позже римляне улуч-
улучшили качество этих вяжущих веществ, введя в них вулканиче-
вулканический пепел—источник активных кремнезема и глинозема. По-
Получаемый таким образом материал в настоящее время известен
как пуццолановый цемент. Современный портландцемент был
изобретен в Великобритании в XIX в., когда впервые стали при-
применяться высокотемпературные методы получения цементов. Си-
Силикаты кальция Ca2SiC>4 и Ca3Si05, отличающиеся высокими вя-
вяжущими свойствами, получаются в результате высокотемпера-
высокотемпературных химических реакций. Первоначально портландцемент
получали путем прокаливания смеси глины и мела. В настоящее
время используют различные исходные вещества: источниками
оксида кальция могут служить мел, известняк или гипс, кроме
того, н шихту добавляют песок, глину, оксиды железа.
Смесь портландцемента с заполнителями и водой схватыва-
схватывается с образованием бетона. Роль заполнителей двояка. С од-
одной стороны, они уменьшают стоимость бетона, с другой — уве-
увеличивают прочность на излом бетонных конструкций. В отсут-
отсутствие заполнителей затвердевший цемент легко растрескивает-
растрескивается. Для уменьшения стоимости портландцемента в него вводят
и другие добавки, такие, как пуццолан, топливные и доменные
19.1. Портландцемент 235
шлаки и др. Некоторые из этих добавок необходимы для связы-
связывания Са(ОНJ, выделяющегося при гидратации цемента. По-
Поэтому введение таких компонентов повышает химическую стой-
стойкость схватившегося цемента.
19.1. Портландцемент
19.1.1. Производство, Портландцемент—наиболее важный и
распространенный вид цементов. На рис. 19.1 в самом общем
виде приведены отдельные стадии его производства. Свое на-
название портландцемент получил из-за схожести его внешнего
вида и цвета с камнем портланд, найденным в Дорсете (Анг-
(Англия).
Первая стадия промышленного производства портландцемен-
портландцемента заключается в приготовлении сырьевой смеси путем дробле-
н2о
сырьевая смесь —-—;->¦ клинкер __, >¦ портландцемент
(CaO, A12O3, (Ta-jSiOg, (пираты сили-
SiQ, Fe2O3 и т. д.) Са3А12О6, катов кальция)
Р-Са2Ы04ит.д.)
Рис. 19.1. Стадии производства и применения портландцемента.
ния исходных материалов, их помола и тщательной гомогениза-
гомогенизации либо в сухом виде, либо в виде цементного теста. Этот ма-
материал вводят в верхнюю холодную часть (загрузочная часть)
длинной вращающейся цементной печи, представляющей собой
слегка наклонный (под углом несколько градусов к горизонту)
барабан. Противоположный конец печи (выгрузочная часть) на-
нагревается до температуры 1300—1500 °С путем сжигания угля,
нефти или природного газа. По мерс прохождения сырьевой
смеси по печи она постепенно разогревается. При этом вначале
происходит потеря И2О и СО2, а затем начинается твердофазная
реакция, которая полностью закапчивается в самой горячей зо-
зоне печи. Здесь материал частично плавится. Присутствие жид-
жидкой фазы существенно увеличивает скорость протекающих реак-
реакций, поскольку в жидкой среде облегчается перепое реагирую-
реагирующих веществ. Как отмечалось в гл. 2, взаимодействие твердых
веществ в отсутствие жидкой фазы протекает, как правило,
весьма медленно. Окислительная атмосфера в цементной печи
гарантирует существование лишь одного оксида железа —
Fe2O3. Частично подплавленный и спеченный материал чергюго
цвета — клинкер-—выгружается из нижней части печи, быстро
охлаждается струей воздуха и дробится. Обычно на этой стадии
к клинкеру добавляют гипс, чтобы воспрепятствовать мгновенно-
мгновенному схватыванию немента при взаимодействии с водой. Получив-
236 19. Цемент и бетон
шийся порошок представляет собой широко известный портланд-
портландцемент, который при смешивании с водой и соответствующими
заполнителями гидратируется и превращается в бетон.
В табл. 19.1 приведено содержание различных оксидов в наи-
наиболее распространенных марках портландцемента. Здесь же
представлен примерный фазовый состав этого материала. Глав-
Главные компоненты портландцементов-—высокоосновные силикаты
кальция p-Ca2Si04 и Ca3Si05. Кроме того, в портландцементе
Таблица 19.1. Химический ir фазоиый состав портландцементов
наиболее распространенных марок
. Содержание. Содержание.
Оксид ма(.с. % Ф.-u-.i масс_ %
5—12
50—70
20—30
,) '•—12
СаО
SiO2
Л12Оз
Fc;03
SOj
MgO
К2О
Ки2О
Прочие
)
1
63
20
6
3
2
2
1
3
QA (С;,3А12О6)
C3S (Ca3Si(M)
j}-C3S (Cn3Si04
C4AF (Ca4Al2F
присутствуют алюминат кальция Са3А12Об и алюмоферрит каль-
кальция примерного состава Ca4Al2Fe2Oio. В технологии вяжущих
веществ принята несколько иная система обозначения компонен-
компонентов, входящих в состав цемента. Каждый оксид обозначается
одной буквой: CaO^^C, SiO2=S, А12О3=А, Ре2Оз=Р, Н2О^^Н.
Формулы различных фаз в этих обозначениях выглядят следую-
следующим образом:
Ca2Si04 ~2CaO.SiO2 = C2S
Ca3Si06 =3CaO-SiO2 = C^S
Ca,Al2Og = ЗСаО-А1гО3 = С, А
19.1.2. Особенности диаграммы состояния. Фазовый состав це-
цементного клинкера и химические реакции, протекающие в це-
цементной печи, могут быть поняты при рассмотрении соответст-
соответствующей диаграммы состояния (гл. 11). Полная фазовая диаг-
диаграмма системы, включающей оксид железа и другие микроком-
понепты (такие, как оксиды щелочных металлов, MgO и др.),
19.1. Портландцемент
237
весьма сложна. Поэтому обсуждение шести- иди семикомпопент-
ной системы вряд ли целесообразно. Для практических оценок
часто ограничиваются рассмотрением диаграммы состояния
тройной системы CaO—SiO2—Al2O:i (рис. 19.2 и 19.3). Точка Р
на этих рисунках отвечает составу портлаидцемептного клинке-
cs
siO,
МОЛ.%
c,s.
CaO
Рис. 19.2. Субго.чпдхсная честь рцштвчсиой диаграммы состояния системы
С;'О— А12О..—SiO;. Р — примерный состав портллняпсмеитоп; Q—состав
глшклемноих цементов. С = СаО, Л =ЛЬОз, S = SiO?. Например, СзЛ =
3COMO CAlO
pa. Из приведенной диаграммы состояния можно получить сле-
следующую информацию.
Точка Р находится внутри треугольника с вершинами Ca^SiC^
(C2S), CaaSiOi (C3S) и Са3Л12О6 (С3А) (рис. 19.2). Поэтому
в равновесных условиях эти три фазы являются составными
частями цементного клинкера состава Р. На практике как в пе-
печи, гак и при охлаждении клинкера в системе успевает уста-
устанавливаться равновесие. Относительные количества каждой из
фаз можно определить по правилу рычага (гл. 11).
Как видно из рис. 19.3, трехфазная смесь CyS+CjS+СзЛ
устойчива лишь при температурах ниже 1455 3С. Выше этой
температуры в системе появляется жидкая фаза и, следователь-
следовательно, согласно правилу фаз, должна исчезнуть одна из кристалли-
кристаллических фаз. Такие изменения в системе следуют из формы по-
238
19. Цемент и бетон
верхности ликвидуса и распределения полей первичной кристал-
кристаллизации на диаграмме состояния (рис. 19.3). Поля первичной
кристаллизации C2S, C3S и СзА (т. е. области температур и со-
составов, для которых в равновесии с жидкостью существует от-
отдельно каждая из этих фаз) пересекаются в перитектической
точке Y. Твердые фазы C2S, C3S и СзА могут находиться в рав-
МОЛ /С
Сао
Рис. 19.3. Участок диаграммы плавкости системы ОаО—Al.,0;- SiOv и обла
сти, богатой оксидом калышя, с обозначением полей первичной кристаллиза-
кристаллизации (например, СаО). Поля первичной кристаллпз.шии разграничены моио
варисштиымк кривыми и инвариантными точками. X — иеритектпческая вч-
влрилнгпая точка, принадлежащая треугольнику С i-C2S+C-,A: У ¦ перитек-
тическая точка, относящаяся к тро\голышьу CSjCSdA
новесии друг с другом лишь при температурах ниже перитекти-
перитектической температуры (<1455°С). В самой горячей части цемент-
цементной печи температура достигает ~1500°С, поэтому материал
состава Р частично расплавлен. При этом возникает новое трех-
трехфазное равновесие: С25+С35+жидкость состава В. Точка В
расположена на пересечении моновариантпой кривой, отвечаю-
отвечающей равновесию фаз C2S и СзЭ, и изотермы 1500 °С. При 1500 °С
состав Р лежит внутри фазового треугольника, ограниченного
фазами: C2S, C3S и жидкость состава В. Следовательно, при
этой температуре в материале состава Р в равновесии находятся
три фазы: Сг^ + СзЭ+жид кость состава В, Соотношение между
количествами отдельных фаз, а значит, и количество жидкой
фазы можно рассчитать по правилу рычага.
19.1. Портландцемент 239
Как уже отмечалось, одним из следствий присутствия жид-
жидкой фазы в обжигаемой смеси является заметное ускорение хи-
химического взаимодействия исходных веществ и образования C2S
и OS. Таким образом, время пребывания материалов в горячей
зоне печи составляет не более нескольких часов. Если бы жид-
жидкая фаза отсутствовала (т. е. при чуть более низкой темпера-
температуре), то та же степень превращения достигалась бы за не-
несколько суток.
Хотя присутствие фазы С3А, по-видимому, слабо влияет на
конечную прочность схватившегося цемента или бетона, роль
этой фазы существенна для повышения экономичности процес-
процесса производства цемента. Дело в том, что глинозем играет роль
флюсующего материала. Из рис. 19.3 и 11.8 видно, что темпе-
температура плавления фаз в системе оксид кальция — оксид крем-
кремния в области, где в равновесии существуют C2S и C3S, лежит
гораздо выше 2000°С. Введение же А12О:) понижает температу-
температуру солидуса примерно па 600 °С. Это позволяет весьма эффек-
эффективно вести обжиг сырьевой смеси в цементных печах при гораз-
гораздо более низких температурах. В отсутствие глиноземного флю-
флюса и, следовательно, и жидкой фазы при температурах 1400—
1500 °С образование C2S и C3S из исходных веществ шло бы не-
несколько суток.
С помощью рис. 19.3 можно также объяснить текстуру це-
цементного клинкера, т. е. размеры и распределение частиц раз-
различных фаз. При 1500 °С в равновесии находятся три фазы:
C2S, CsS и жидкость. Размеры кристаллов C2S и C3S относи-
относительно велики (обычно 10—50 мкм в диаметре), так как в при-
присутствии жидкой фазы лх рост длится достаточно долго. Когда
клинкер покидает горячую зону, еще до выгрузки, он несколько
охлаждается. Кристаллизующиеся при этом фазы C-S и C-S,
вероятно, осаждаются па поверхности уже существующих кри-
кристаллов. Эта вторичная кристаллизация отвечает движению фи-
фигуративной точки из В в Y. При температуре <С1455°С остав-
оставшаяся часть жидкости обычно полностью закристаллизовывает-
ся, образуя мелкодисперсную матрицу, внутри которой присут-
присутствуют зерна C2S и C3S больших размеров. Текстура и фазоиын
состав матрицы изменяются в зависимости от скорости охлаж-
охлаждения. Обычно матрица состоит по меньшей мере нз двух крис-
кристаллических фаз, одна из которых С3А. Предполагается, что при
охлаждении имеют место и другие процессы, например если не-
небольшая часть расплава не успевает закристаллнзовываться, то
происходит образование сгекла.
Следующим по значимости после СаО, А12О3 и SiO2 компо-
компонентом портландцемеитпого клинкера является Fe2O3, Из соот-
соответствующей диаграммы состояния можно сделан, вывод, что
Fe2Cb играет ту же роль, что и А12О3. При 1500 °С оксид железа
240
19. Цемент и бетон
J
X CC
О а;
-Л. 670
55
а4
SiO4
5Ь
\
Fe2O3 находится в жидкой фазе. В процессе выгрузки из цемент-
цементной печи ои кристаллизуется, в результате чего образуются
ферриты и алюмоферриты состава, близкого к CiAF. Ферриты
и алюмоферриты можно рассматривать как твердые растворы
переменного состава, образуемые Ca2Fe206 (C2F) и гипотетиче-
гипотетической фазой Ca^AisOs (С2А) (см. рис. 19.7).
19.1.3. Полиморфизм силикатов кальция. Прежде чем перейти
к рассмотрению процессов гидратации цемента, обсудим некото-
некоторые особенности кристаллической структуры и полиморфных
превращений безводных силикатов
кальция.
Силикат кальция состава C^S
устойчив от температуры его кон-
конгруэнтного плавления B070°С) до
~'1250°С (рис. 11.8). Ниже 1250°С
C3S должен распадаться на C2S и
окенд кальция. На практике, одна-
однако, скорость разложения этого ве-
вещества ниже 1250 °С мала, поэто-
поэтому C3S легко сохраняется в це-
цементной клинкере до комнатной
температуры, C3S кристаллизуется
в структуре ортосиликатов, кото-
которая содержит изолированные тет-
тетраэдры SiO41~. Однако в структуре трикальцийсиликата име-
имеются свободные ноны кислорода. Поэтому более правильно
формулу этого соединения записать в виде Ca:i(SiO4)O.
В структуру C3S можно внедрить небольшие количества таких
конов, как Ali+ или Mg2~:. Возникающая при этом фаза, не-
несколько отличного от C3S состава, называется алитом. Именно
оиа входит в состав клинкера.
Обсудим полиморфные превращения силиката кальция со-
состава C2S, обратившись к температурной зависимости свободной
энергии (рис. 19.4). C2S имеет три стабильные полиморфные мо-
модификации, называемые а-, а- и "^-фазами (а—наиболее высо-
высокотемпературная фаза, ^—низкотемпературная фаза). Прн
охлаждении переход a'-*-j (при 735 °С) идо г весьма медленно,
а при быстром охлаждении a'-фаза склонна к переохлаждению.
При 670 "С из a'-фазы образуется мстастабильпая $-фаза.
Именно эта модификация C2S, обычно присутствующая в це-
цементном клинкере, обладает наиболее цепными вяжущими свой-
свойствами. Хотя чистый f5-C2S метастабилен прн комнатной тем-
температуре но отношению к f-C2S, присутствие различных посто-
посторонних ионов, способных внедряться в структуру i5-C_»S, способ-
способствует стабилизации этой фазы.
Температура —*¦
Рис. 19.4. Зависимость сио
бодной энергии различных по-
полиморфных модификаций C2S
от температуры.
19.1. Портландцемент
24 i
10000
1
6000
2000
При производстве цемента из сырьевой смеси необходимо
удалять все добавки, ускоряющие фазовые переходы типа а'-+~\
и JJ-+-T, так как t-CaS не обладает вяжущим действием. Необхо
димо также предотвращать превращения нсгидратированно1'>
^-CmS в f-QS » затвердевшем цементе или бетоне, так как свя-
связанное с Р->Ч-превращсиием заметное изменение объема может
привести к разрушению цемента.
19.1.4. Гидратация портландцемента. Присутствующие в цементе
силикаты и алюминаты взаимодействуют с водой, образуя про-
продукты гидратации. В то же время именно а тот процесс веде г
к затвердеванию цементной мас-
массы. Как видно из рис. 19.5, раз-
различные безводные фазы харак-
характеризуются неодинаковыми вя-
вяжущими свойствами. CaS быстро
взаимодействует с водой и дос-
достигает максимальной прочности
уже иа раиией стадии гидрата-
гидратации. j3-C2S твердеет гораздо мед-
медленнее. Продукты гидратации
С3А и C4AF имеют низкую проч-
прочность. Процесс гидратации раз-
различных промышленных марок
цемента обычно рассматривают
как совокупность процессов гид-
гидратации отдельных составных
частей цемента. Фаза C3S обеспечивает начальное твердение це-
цемента; а фазы C3S и f5-C2S придают схватившемуся цементу и
бетону прочность в течение длительного времени.
Гидратацию цемента следует рассматривать как фнзпко-хи
мтеекпй процесс, причем определенные затруднения, возникаю
тис при его изучении, связаны с тем, что основные продукты
гидратации либо плохо закристаллизованы, либо находятся в ге-
леобразном состоянии Это крайне усложняет проведение рент-
рентгеновских исследований. Главным продуктом гидратации, обес-
обеспечивающим высокую прочность затвердевшего цемента, являет-
является плохо кристаллизующийся гидрат силиката кальинч
(С—S — Н). Ею иногда называют С—S—Н-геле\1 или, чго но
совсем верно, тоберморит гелем. Химический состав этого веще
ства точно не установлен. Вероятно, содержание как оке им
кальция, так и оксида кремния в нем может быть весьма раз-
различно. От образца к образцу меняется также п соотношение
между оксидом кремния и водой В состав С—S--H-rcvui могут
входить также попы А1';+, Fe3! и SO;2". Затвердевший цемент
кроме С— S-—И геля содержит нспрорсатиринавшую с во до О
180 .560
время, сут
Рис. 19.5. Увеличение прочнею и
па сжатие различных компонен-
компонентов цемента при гидратации [-'1
16—H2G
242 19. Цемент и бетон
часть клинкера, Са(ОИJ, гидраты алюминатов и алюмосульфа-
тов, а также воду.
Многие физические и механические свойства затвердевших
цемента и бетона, по-видимому, в большей степени зависят от
физической структуры продуктов гидратации и образующихся
коллоидных растворов, чем от их химического состава. В па-
стоящее время благодаря применению современных методов ис-
исследования, в том числе электронной микроскопии в сочетании
с микроанализом существующих фаз, достигнут определенный
успех в объяснении механизма процесса гидратации. Процесс
гидратации, видимо, протекает в две стадии. Первая — образо-
образование С—S—Н-геля на поверхности частиц безводного цемента;
вторая — утолщение образовавшегося покрытия как путем рос-
роста наружу, так и путем проникновения геля внутрь частиц без-
безводного цемента. Таким образом, в течение нескольких часов
тонкие слои, покрывающие отдельные частицы, соединяются
между собой и цемент затвердевает или схватывается.
На свойства цемента заметное влияние оказывает соотноше-
соотношение между массой воды и массой цемента (вододементнос отно-
отношение). Когда цементная паста схватывается, ее объем остает-
остается примерно постоянным, причем этот окончательный объем тем
больше, чем выше водоцементное отношение исходной смеси.
Затвердевший цемент представляет собой пористый материал,
содержащий как очень маленькие заполненные водой поры
(~10—20 А в поперечнике; гелевыс поры), так и большие кана-
каналы (~ 1 мкм в поперечнике; капиллярные поры). Именно соеди-
соединенные между собой капиллярные поры в основном ответствен-
ответственны за водопроницаемость затвердевшего цемента и разрушение
бетонных конструкций па морозе. Поэтому очень желательно,
чтобы соединения капиллярных пор не происходило. Этого мож-
можно достичь путем длительной выдержки бетона в период схваты-
схватывания во влажной атмосфере и путем уменьшения водоцемспт-
ного отношения. Так, при водоцементном отношении —0,4 необ-
необходимо в течение грех суток поддерживать цемент влажным,
чтобы разрушить соединения между капиллярными порами
в цементе, при водоцементном отношении ~0,7 для этого тре-
требуется не менее одного года.
В связи с быстрым протеканием реакции между С3А и водой
возникает проблема так называемого мгновенного схватывания.
По-видимому, образованию гидратов алюмината кальция пред-
предшествует стадия быстрого растворения С3А, которая сопровож-
сопровождается значительным выделением тепла. Хотя реакция и проте-
протекает весьма быстро, механические свойства цемента подвергше-
подвергшегося мгновенному схватыванию, заметно ухудшаются. На прак-
практике мгновенного схватывания можно избежать путем введения
и цементный клинкер !—2% гипса. В результате сложных xhvh-
19.1. Портландцемент
243
нсских реакций гипс в присутствии Са(ОНJ тормозит гидрата-
гидратацию С3А, На поверхности кристаллов С3А, видимо, образуется
защитное покрытие па основе алюмосульфатных фаз состава
Ca6Al2(OH)[2(SO4h-26H2O или Ca4Al2(OHI2SO4.6H2O.
Существуют несколько безводных силикатов кальция, но
лишь два из них — C3S и fi-C2S — обладают вяжущими свой-
свойствами, необходимыми для их применения в составе гидравли-
гидравлических вяжущих материалов (т. е. таких веществ, которые взаи-
взаимодействуют с водой с образованием нерастворимого продукта,
схватывающегося с образованием твердой массы). Причины та-
такого распределения свойств силикатов кальция не до конца
S
с;
пз
x
cf
о
1С
О
О
c3s
"^" -- ^
С С+ Г
¦—
J7CT >¦
a
so" v
C2S^<i.l2b0
4 ^\
дс I
C-S-H+ca(OHJ
Температура
Рис. 19.6. Зависимость свободной энергии образцов состава CrS от темче-
pfiTyphi (но.шморфшле модификации Спз5Юз не \клзаны) (а) и ехгма из-
изменения свободной энергии гидратации CiS (б).
понятны. Видимо, на вяжущие свойства материалов оказывает
влияние целый ряд факторов.
Одной из особенностей C3S и |3-C2S, отличающей эти фазы от
других силикатов кальция, является их метастабильность. Ниже
670 "С |b-C?S мета стабилен по отношению к ^f-C2S. При темпера-
температурах ниже 1250 °С C3S метастабилеп по отношению к смеси
CaO-f-C2S. Последнее видно из зависимости свободной энер-
энергии образцов состава C3S от температуры (рис. 19.6, а). Сплош-
Сплошная линия на рис. 19.6, а отвечает изменению равновесной ве-
величины свободной энергии с температурой. Как видно, при ком-
комнатной температуре фаза C:(S метастабильиа по отношению
к смеси двух фаз ib-C^S+CaO, которая в свою очередь также
метастабильна (по отношению к смеси *f-C2S+Ca0). Поскольку
и CiS, и '^-C5S метаетабильны, изменение свободной энергии
реакции их гидратации должно быть больше, чем при гидрата-
гидратации соответствующих равновесных фаз, т. с. -f-C^S+CaO н
"f-C^S. При этом, естественно, предполагается, что продукты
гидратации 3-C2S п ^-C^S характеризуются одной п топ же ве-
величиной свободной энергии. Однако, хотя C3S и ,3 C2S являются
метастсюильпыми фазами, из этого факта вовсе и пс следует, что
16*
244 19. Цемент и бетон
их гидратация протекает быстрее, чем гидратации соответствую-
соответствующих равновесных фаз. На рис. 19.6,6 приведена схема измене-
изменения свободной энергии реакции гидратации. Символом АЕ обо-
обозначена энергия активации этого процесса, величина которой
определяется легкостью взаимодействия молекул воды и безвод-
безводной фазы. Следовательно, величина АН зависит от кристалли-
кристаллической структуры безводной фазы. Предполагается, что C3S
и в некоторой степени 1$-C2S имеют достаточно открытую кри-
кристаллическую структуру, что облегчает проникновение молекул
«оды в объем вещества. Это облегчает взаимодействие молекул
воды с кристаллами упомянутых фаз и, следовательно, пони-
понижает величину АЕ. Другим фактором, который может понижать
энергию активации процесса гидратации C3S, является присутст-
присутствие в кристаллической решетке областей, подобных СаО. Такие
области в первую очередь вступают во взаимодействие с во-
водой и инициируют последующий процесс гидратации. Трудности
при сравнении вяжущих свойств двух фаз состоят в том, что
оценить различные факторы, влияющие па величину энергии
активации процесса гидратации, не так уже просто.
Существенное влияние на кинетику гидратации оказывает
размер частиц цемента. Чем он меньше, тем больше площадь
поверхности кристаллов и тем выше скорость реакции гидрата-
гидратации, которая протекает главным образом на поверхности кри-
кристаллов. Этот фактор необходимо учитывать па практике при
дроблении цементного клинкера. Очень мелкие частицы способ-
способствуют более быстрому нарастанию прочности при схватывании
материала. Для обычных марок портландцемента, производи-
производимого в Великобритании, нормативно установлена минимальная
площадь поверхности в 225 м2/кг, хотя на практике эта величи-
величина, как правило, составляет —300 м2/кг.
19.1.5. Разновидности портландцемента. Варьированием состава
портландцементпого клинкера или количеств разных добавок
достигается изменение свойств материала. Наиболее распрост-
распространен обыкновенный портландцемент (портландцемент общего
назначения). Однако эта разновидность портландцемента не
устойчива к действию сульфатов, поэтому его нельзя использо-
использовать, например, если изделие или конструкция будут иметь кон-
контакт с морской водой. Сульфаты могут взаимодействовать с дву-
двумя компонентами цемента: во-первых, с гидратироватшыми алю-
алюминатами, образуя сульфоалюминаты кальция низкой плотности
(этому процессу сопутствует расширение, что ведет к разруше-
разрушению конструкции) и, во-вторых, с Са(ОНJ с образованием гип-
гипса (что ведет к растрескиванию затвердевшего цемента).
В сульфатостоиких цементах содержание СзА заметно ниже,
чем в обыкновенном портландцементе. На практике этого доби-
19.2, Глиноземистые и оысокоглиноземистыс цементы 245
ваются путем увеличения соотношения Fe2O3 : А12О3 в исходном
сырье. В результате в клинкере возрастает содержание алю-
моферритной фазы C4AF. По не вполне попятным причинам при-
присутствие CjAF повышает устойчивость гидратированного цемен-
цемента к действию сульфатов.
Быстротверденщий портландцемент можно получить двумя
способами: путем увеличения содержания C3S по сравнению
с содержанием C2S или путем более тщательного измельче-
измельчении клинкера. Поскольку быстрое твердение связано с высокой
скоростью тепловыделения, такой цемент нельзя использовать
при изготовлении монолитных конструкций из-за возможности
растрескивания. В то же время эту разновидность портландце-
портландцемента можно применять в условиях низкой температуры окру-
окружающей среды, так как выделяемое при гидратации тепло мо-
может предохранять цемент от разрушающего воздействия низких
температур на ранней стадии гидратации. Еще больше повысить
скорость твердения цемента можно путем совместного измельче-
измельчения быстротвердеющсго клинкера с 1—2% СаСЬ.
Для изготовления монолитных бетонных конструкций обыч-
обычно используют портландцемент с умеренной жзотермией, в кото-
котором понижено содержание С3А и QS. Нарастание прочности
в нем замедленно, хотя предельная величина прочности остается
неизменной.
По своим свойствам шлакопортландцемент очень похож на
обыкновенный портландцемент. Его получают путем смешения
нортлапдцементпого клинкера с доменным шлаком (побочным
продуктом процесса восстановления железной руды; он обра-
образуется при взаимодействии известняка с кремнеземом, глинозе-
глиноземом и другими компонентами руды), Возможен и иной способ
получения шлакопортлаидцемента. Шлаки с модифицированной
структурой и составом можно использовать совместно с извест-
известняком в качестве сырья при производстве портландцемента.
Модифицирование шлака заключается в зака„1ке расплавленно-
расплавленного шлака. Образующийся при этом продукт содержит значи-
значительное количество стеклообразной фазы, которая быстро взаи-
взаимодействует с водой.
19.2. Глиноземистые и высокоглиноземистые цементы
Глиноземистые цементы были впервые получены во Фран-
Франции в начале нашего столетия при изучении свойств сульфато-
стонкнх цементов. Основные компоненты этих цементов —- оксид
кальция и оксид алюминия, присутствующие примерно в одина-
одинаковых количествах. Кроме того, в состав глиноземистых цемен-
цементов входят небольшие количества оксидов железа, кремния, маг-
магния, шелочных металлов и титана. Сырьем для их производства
246 19. Цемент и бетон
служит известняк (или мел) и боксит. Исходные вещества пла-
плавят в обжиговой печи при температурах 1500—1600 °С, Стои-
Стоимость получаемого при охлаждении клинкера выше стоимости
портландцемента. Это объясняется высокой стоимостью бокси-
бокситов, высокой температурой плавления сырьевой смеси и высо-
высокой твердостью клинкера, дробление которого представляет
весьма трудную задачу. Важнейшие достоинства глиноземистых
цементов связаны с их быстрым твердением (высокая прочность
достигается уже через 24 ч), устойчивостью к действию сульфа-
сульфатов и жаростойкостью.
Высокоглиноземистые цементы содержат гораздо больше ок-
оксида алюминия, чем обычные глиноземистые цементы
(~80 масс. % А12Оз), а оксидов железа, кремния и других при-
примесей в них очень мало. Эти материалы можно использовать
в качестве жаростойких цементов до температур ~1800°С.
Главный комионепт обычных глиноземистых цементов — алю-
алюминат кальция СаА12О4 (СА). Это вещество обладает ценными
вяжущими свойствами. Присутствует и другой алюминат каль-
кальция— С12А7. Однако на практике состав цемента стараются по-
подобрать так, чтобы ограничить количество этого алюмината.
Объясняется это тем, что алюминат С12А7 быстро гидратируется,
что ведет к мгновенному схватыванию цемента. Уменьшение со-
содержания Ci2A7 достигают путем добавления в сырье Fe2O3-
Роль Fe2O3 можно понять, если обратиться к соответствующей
области диаграммы состояния системы СаО—Al2O.i—Fe2O:i
(рис. 19.7). Большинство фаз в данной системе, в том числе СА,
С12А7 и феррит СгР, образуют твердые растворы путем взаимно-
взаимного замещения ионов АР4 и Fe!+. Заштрихованная область па
диаграмме отвечает составам типичных глиноземистых цемен-
цементов. Эта область принадлежит различным треугольникам, па ко-
которые распадается диаграмма состояния после ее триангуляции.
В одной из вершин каждого из этих треугольников находится
твердый раствор на основе фазы СЛ; в других вершинах —фа-
—фазы, содержащие различные количества оксидов кальция и же-
железа. Алюминат С12А7 не попадает ни в один из этих треуголь-
треугольников. Таким образом, в равновесных условиях, которые практи-
практически достигаются при охлаждении клинкера, в глиноземистых
цементах алюминат С12А7 должен отсутствовать.
Содержание кремнезема в г.чшоземиетьк цементах невелико, оно nf
превышает нескольких процситон. Эго синмко г тем. что при Сол миом см-
держании SiO.. образуется члюмоенлнкатнля ф:;з;< состав;) СагЛЬБЮ/
(C^AS)—геленпт. Относительно небольшого количеств.! кремнезема доста-
достаточно для образования значительного количества геленнта. К такому вынп-
ду можно прийти, анализируя диаграмму состояния системы СлО SiCV—•
.\ljO~, (рис. 19.2). Прогссдем прямую нч точки Q, отвечающей" составу глино-
глиноземистого цемента, к иершине SiOV Видно, что присутствие уже ~I0% S1O2
(точка R) приводит к исчелниченшо бо.плпеГ! ч.-юти cpa:ti.i СЛ и возпшшоие-
19.2. Глиноземистые и высоког.шночемнстые
247
пию гелрнито. Поскольку геленит плохо затвердевает при гидратации и
разуется за счет ценного вяжущего материала — алюмината кальция,
держание оксида кремния в глиноземистых цементах ограничивают до
масс. (''и. Небольшое количество кремнезема в глиноземистых цементах
Же весьма желательно, так как при этом вместо геленнта образуется р1
(точка S на рис. 19.2). Кроме упомянутых выше фаз в глиноземистых
б РО)
А1-0,
GA
масс.%
Ci2A
z:\o
Г 0,0.
Риг 10 7. Обсолплуспля часть диаграммы состояния системы СаО—А!2Оз—
Fe2O-.. Зшнтричовпиная облпеп. отвечает сост;там глиноземистых цементов.
Пгрочеркпутне линии соответствуют областям твердых растворов. Цифра
ми 2 и 3 обозначены количества фаз. находящихся и данной обллгтн диа
грлччы состояния.
Взапмо.чгпствие глнноземистмч цементов с водой при темчерлтурах
'525 СС перчог'ячяльно приводнт к образованию гидрата алюмината кальция
состава СГ A111..., а также небольших количеств С? АН в и геля гидроксидя
алюминия. CAHio—метастабилькая фаза, отвечающая за прочность отпер-
дспгр.его цемента. При более высоких температурах C0—40 °С), особенно во
влажном атмосфере, С'АНщ Си СгЛН«) превращается в С..ЛНи. гель гндрок-
сила :>люм:и:!1я н мес^яини1 ю :;оду. (Ге.т, гид^'/ьтнда алюминия постепен-
постепенно кристаллизуется с образованием гиббента Л1(ОПK.) Поскольку С3АП0
характеризуется большей плотностью, чем СЛЧ]0 и С2АН8, такое «превра-
«превращение» ведет к увеличекню пористости, проницаемости и некоторой потере
прочности при сжатии. В 1973 г, в Великобритании было зарегистрировано
повреждение нескольких зданий из-за разрушения бетонных строительных
конструкций, наполненных из глиноземистого цемента. Наиболее дпам.чтнч-
ные последствия имел обкил крынш плавательного бассейна в одной из лон-
лондонских школ. К счастью, а в я пня произошла через несколько мнн\т после
того, как бассейн нокипчлн люди. Хотя упомянутое выше превращение, по-
248 19. Цемент и бетон
видимому, является наиболее распространенной реакцией, протекающей в
глиноземистых цементах, оио приводит к уменьшению прочности отвердеи-
шего цемента лишь при очень высоком води цемент нон отношении. Для пол
пои гидратации СА с образованием САНщ расчетное значение водоцемешно
ю отношения состапляет 0,5. Если это отношение меньше 0,5, то в отвер-
отвердевшем цементе присутствует некоторое количество негидратированного СА.
(Заметим кстати, что при водоцемептном отношении 0.35 не возникает пи
каких проблем с разрушением бетона.) Видимо, вида, выделяющаяся при
превращении САПц, в СзА116 и А1(ОНK, вступает во взапмодепетние с ие-
гидрлтировапным СА. При атом Заполняются поры, возникшие в ходе пред
шествующего превращения. Поэтому глиноземистые цементы можно считать
вполне безопасными материалами при условии, что выдержано нужное .«ia-
чецис водоцемептиого отношения. Обычно всегда существует соблазн доба
иить больше воды к цементу, гак как цементное тесто при водешемеитном
отношении 0,35 имеет слишком сухой вид, особенно по сравнению с порт -
лмицеусптным тестом. Именно Ошибки строителен, видимо, и яии.'шсь
причинами повреждений зданий в Великобритании Во мношх странах мн-
рл существует запрет на использование глиноземистых цементов при изго-
изготовлении некоторых типов конструкций.
Стойкость отвердевшего глиноземистого цемента к действию
сульфатов объясняется отсутствием в нем в отличие от порт-
портландцемента Са(ОНJ (.образующийся в процессе гидратации
Са(ОНJ тут же вступает ъ реакцию с гелем гидроксида алю-
алюминия). Кроме того, гель гидроксида алюминия создает защит-
защитное покрытие па кристаллах алюминатов и их гидратов. При
гидратации цемента вначале схватывается С^А;, а затем СА.
Поэтому для более быстрого схватывания цемента необходимо
несколько увеличить в нем соотношение между оксидом каль-
кальция и оксидом алюминия и тем самым увеличить содержание
С|2А7. В то же время присутствие в клинкере стеклообразной
фазы, по-видимому, замедляет протекание реакции гидратации.
Вследствие того что высокая прочность цемента достигается от-
относительно быстро (80% конечной величины прочности за 24 ч),
быстро происходит и тепловыделение. Поэтому глиноземистый
цемент не следует использовать при сооружении массивных
конструкции. Его почти всегда используют для изготовления
топких профилей.
Бетон, получаемый из глиноземистого цемента (особенно из
высокоглиноземистого цемента), является важным огнеупорным
материалом с высокой механической прочностью и термостойко-
термостойкостью в сухой атмосфере. Бетонную смесь готовят и укладывают
обычным способом, а через 24 ч ее нагревают. Продукт гидрата-
гидратации теряет воду, и при 900—11003С его прочность достигает
минимума. При более высоких температурах продукты дегидра-
дегидратации начинают реагировать с заполнителями — раздробленным
шамотовым кирпичом, А12Оа или SiC. При этом связи, возник-
возникшие при затвердении бетона, разрушаются, а вместо них обра-
образуются прочные связи керамического типа. Таким образом,
прочность материала вновь повышается. Большинство жаростоп-
19.3. Пуццоланы и пуццолаповые цементы
249
ких бетонов, пригодных для использования до температуры
1800°С, готовят из белого глиноземистого цемента и корунда
(AI2O3) в качестве заполнителя. Бетон почти целиком состоит
из алюмината кальция и А12О3 и не содержит примесей, пони-
понижающих температуру плавления, таких, как Ре2Оз. На рис. 19.8
приведена диаграмма состояния системы СаО—А12О3. Взаимо-
Взаимодействие СА из дегидратированного цемента (температура
2000 -
isoo -
1600
1400 -
1200
СаО 3:1 12:7 1:1 Г. 2 116 А1203,мол^
Рис. 19.8. Диаграмма состояния системы СгЮ—¦Л1.Hз.
плавления 1608°С) с заполнителем А12О3 приводит к образова-
образованию СА2 и, что более важно, САе (температура плавления
1860°). Таким образом, жаростойкость материала заметно по-
повышается. Для сравнения отмстим, что портландцемент редко
используют при температурах выше 500 °С.
19.3. Пуццоланы и пуццолановые цементы
Пуццолан представляет собой природную (вулканический
пепел) или синтетическую (размельченные топливные шлаки и
золы) форму высокорсакционноспособного оксида кремния. При-
Причина высокой реакционной способности такого кремнезема со-
состоит в метастабильности и (или) высокой дисперсности. Кри-
Кристаллический кварц в обычных условиях химически не активен.
В то же время плохо закристаллизованный оксид кремния, со-
состоящий из частиц малых размеров, матовое (т. е. ликвирован-
пос) кварцевое стекло, а также силикагель взаимодействуют
250 19. Цемент и бетон
с НгО и Са(ОНJ при комнатной температуре с образованием
продуктов с вяжущими свойствами. Уже древние римляне ис-
использовали известково-пуццолановыс строительные растворы,
и некоторые из этих составов находят применение до сих пор.
Продуктом взаимодействия извести с пуццоланом является гель
гидрата силиката кальция, аналогичный гелю С—S—Ы, который
образуется при гидратации портландцемента. Особенность такого
взаимодействия заключается в том, что весь процесс протекает
при комнатной температуре. Это отличается от условий приме-
применения портландцемента, так как для использования последнего
прежде всего необходимо получить при высоких температурах
силикаты кальция C3S и j}-C2S.
Пуццоланы можно также смешивать с портландцементом.
При этом получается пуццолановый цемент. Портлапдцемептная
составляющая пуццолапового цемента взаимодействует с водой
по обычному механизму. Возникающий при этом Са(ОНJ реа-
реагирует с пуццоланом с образованием С—S—Н-геля. Удаление
Са(ОНJ в результате реакции с пуццоланом имеет два преиму-
преимущества: во-первых, пуццолапоиып цемент обладает высокой хи-
химической стойкостью к сульфатам, во-вторых, прочность конеч-
конечного отвердевшего продукта повышается по сравнению с порт-
портландцементом, так как Са(ОНJ, обладающий плохими механи-
механическими характеристиками, заменяется на С—S—Н-гель. Пуц-
Пуццолановый цемент твердеет медленнее, чем портландцемент, и,
следовательно, скорость тепловыделения невелика. Поэтому
пуццолановый цемент можно использовать для изготовления
массивных конструкций.
19.4. Продукты автоклавного синтеза
Большинство форм кремнезема, за исключением реакционно-
способного пуццолана, практически не взаимодействует при ком-
комнатной температуре с известью п водой. Однако при 175—200°С
взаимодействие с водяным паром и известью протекает весьма
интенсивно. Б результате такой реакции получают автоклавный
силикат кальция. Обычно в состав исходной смеси для изготов-
изготовления силикатных кирпичей входят негашепная известь D —
12%), кварцевый песок и вода. Смесь формуется под давлением
35 МПа и обрабатывается водяным паром под давлением 8.5—
14 МПа при 175—200 °С в течение 12—15 ч. Взаимодействие
песка, извести и воды ведет к образованию С—S—Н-геля. Пер-
Первоначально соотношение СаО; SiO2 в геле достаточно велико
(--— 1,75). Когда вся известь прореагирует, это соотношение по-
понижается до величины ~ 0,8 за счет взаимодействия геля с пе-
вступившим в реакцию SiO2. Если увеличить время пребывания
сырьевой смеси в автоклаве, С—S—Н-гель частично кристалли-
кристаллизуется с образованием тоберморита С55бН5. В настоящее время
19.6. Последние достижения — беспорисшй бетон
остается неясным вопрос о том, что является главным источни-
источником прочности цемента: С—S—Н-гель или тоберморит.
Водяной пар под высоким давлением используют также для
ускорения твердения бетона, изготовленного из портландцемен-
портландцемента. Для производства готовых бетонных блоков портландцемент
смешивают с мелкораздробленным песком и заполнителем; ко-
количество добавляемого песка весьма существенно для достиже-
достижения высокой прочности бетона. В процессе выдержки в автокла-
автоклаве (8—15 ч при 180 "С и давлении 1 МПа) цемент взаимодейст-
взаимодействует с песком. Отношение СаО : SiO2 в образующемся С—S—Hi-
геле невелико (~0,8). При кристаллизации геля возникает то-
тоберморит. Получившиеся после обработки в автоклаве бетонные
блоки характеризуются высокой прочностью при сжатии и
устойчивостью к действию сульфатов. Последнее свойство связа-
связано с тем, что в бетоне отсутствует известь, так как присутствую-
присутствующий в цементе Са(ОНJ вступает в реакцию с песком.
19.5. Магнезиальный цемент
Магнезиальный цемент (по химическому составу представляет собой
основной хлорид магния) достаточно широко используется (особенно в
США) для залипки полов, декоративной внутренней отделки помещении, на-
наружной штукатурки здппий. Конструкции из такого цемента характеризу-
характеризуются хорошими акустическими и упругими свойствами, они не наклп.шва-
ют статического электричества, а по своему внешнему виду напоминают
мрамор. Все эти свойства весьма важны для заливки полов.
Цемент готовят путем смешивания мелко раздробленного активного по-
порошка MgO с водным раствором MgCl2. В результате смешения возникает
гомогенный тиксотроппый гель, который кристаллизуется с образованием
плотного твердого алгомерата оксохдоридных фаз 5:1:8 EМц(ОНK-
•MgC12-8H2O) и 3:1:8 CMg(OHJ-MprCJ2-8H2O). Главное требование к ис-
исходным веществам касается мелкодисперсное™ MgO: в противном случае
растпоргние MgO идет медленно, вод? испаряется, возникает градиент со-
состава, нспрореагировавший MgO выпадает в осадок. Образовавшийся про-
продукт характеризуется разными свойствами в разных участках материал;).
Изделия из пего подвержены атмосферному и коррозионному воздействию
Как и в случае портландцемента, в магнезиальном цементе процесс
схватывания длится весьма долго. Устойчивость к атмосферному воздейст-
воздействию, вилимо, связана с поглощением атмосферного СО2> реакция с котоои.м
приводит к образованию нерастворимой фазы состава Mg(OHJ-2MgCOn'
-AlgCb'(iIl2O. Это соединение присутствует по крайней мере в виде поверх-
поверхностной фазы. После длительного промежутка времени .хлорид иыщелачп-
вастся и в поверхностном слое остается гпдромдглезит БМО^СОЯНО
19.6. Последние достижения — беспористый бетон (MDF)
Недавно в Великобритании разработан новин вид цемента,
механические свойства которого намного превосходят свойства
обычного портландцемента и бетона. Этот новый вид цемента,
получивший название «беспористый цемент», характеризуется
важными эксплуатационными свойствами: средней прочностью
па изгиб ( — 70 МПа по сравнению с 10 МПа в портлаидцемеп-
252
19. Цемент и бетсш
Рис 19 9 Растяжение пружины, изготовленной из нового беепорнстого це-
цемента A]. (Воспроизведено с разрешения Royal Society oi Chemistry-)
тс), умеренной энергией растрескивания A кДж/м2 но сравне-
сравнению с 0,02 кДж/V в обычных марках цемента). Эти величины
более чем на порядок меньше соответствующих величин в высо-
высококачественных сталях. Тем не менее в перспективе эти харак-
характеристики материала могут быть улучшены.
В настоящее время сведения о химическом составе и струк-
структуре беспористого цемента все еще весьма ограниченны. Важной
характеристической особенностью нового вида цемента является
отсутствие крупных пор или пустот в микроструктуре твердого
материала. В обычных цементах имеются большие «дыры» (до
1 мм в диаметре), которые, по крайней мере частично, отвечают
за плохие механические свойства цемента. При производстве
беспористого цемента смешение компонентой и формование из-
Упражнения 253
делий осуществляются в присутешии поверхностно-активных ор-
органических веществ, реологические свойства которых способ-
способствуют улучшению упаковки частиц цемента.
На рис. 19.9 приведена убедительная иллюстрация интерес-
интересных механических свойств беспорпстога цемента. Обратите вни-
внимание па возможность растяжения пружины из цемента.
Упражнения
19.1. 11спол>зуя рис. 19 2, укажите, к;жие фазы находятся в равновесии
в цементах состава P. Q, R и S при темт-ратурлч ниже температуры ео-
-илуса. По правилу рычаги (гл. ! I) оцените содержание р шновеспмх фа.}
для каждого из составов.
19.2. Используя диаграмму состояния (рис. 19.3), определите, какие фа-
фалы существуют при l.iflO "С ч цементе следующего состава: а) 80% СаО. 10%
AI,Ol 10% SiO,; б) 60% СаО, 20% Л\2О?„ 20% SiO2; в) 70% СаО, 25%
А1;О3, 5% SiO2.
19.3. Какие виды цемента (бетон;-)] можно использован, для а) строи-
строительства длчбы и водоеме с пресной водой; б) строительства свай мост.к
в) строительных работ и условиях Арктики; г) изготовления конструкции,
которые должны быть устойчивы до шлсокнх температур (~ЮО0°С)?
19.4. Механизмы процессов, лежащих в основе твердения цемента м
бетона, изучены пока еще плохо. Как иы думай с, почему сложилась та-
такая ситуация? Предложите эксперимент, который пролил бы свет на приро-
природу протекающих процессом.
19.5. Используя рис. 19 8, определите, какие фазы возникают при натре-
в.пши образцов следующих составон: л) 50% СаО. 50% АЬОз; б) 70% СаО.
3U% АЬОз; к) 25% СаО, 75% ЛШ;; г) 10% СаО, 90% AI2Oj. Н.повнте.
температуры, при которых начинается n.iaiuenne- каждого из перечисленных
образцов.
Литература
1. Birchall D., Howard А. /., Kendall К., New cements — Inorganic plastics nf
the future, Clieni. Brit., 18. 860 A982).
2. Rogue R. //., Lonh W., hid. Eng. Clieni., 26, 837 A934)
3. Lea F. M., Desch C. #., The Chenibtry of CenienL and Concrete, 2nd ed ,
Arnold, London, 1956.
4. Mindess S., Young I. Г., Concrete, Pienlice Hall, 1981.
5 Taylor H. F. IV-'. (ed.), 'Hie Cliemisiry ol" Cements, Academic Press, Luiulon
and New York, 1964, 2 vol.
6. Taylor H. /-. li".. The Chemistry of Cements, RfC Monograph, No 2, Ro\a!
Institute of Chemistry, London, 1966.
7. Taylor H. Г, IV'., Modern сЬешЫту of ccmcuis, Cliem. and !nd., 1981, 620
A981).
Дополнительная литература. Будников П. П. Химия и технология строи-
строительных материалов и керамики. — М.: Стройиздат, 1965; Бутт Ю. М., Рши
коиич Л. Н. Твердение вяжущих веществ прл повышенных температурах.—
М.: Стропиздат, 1965; Бутт Ю. М., Тимишев В. В. Портландцеыентнын клин
кср. — М.: Стройиздат, 1967; Куприянов В. П. Технология производства сили-
силикатных изделии.—М.: Высшая школа, 1975; Мчсдлов Петросян О. П. Химия
неорганических строительных материалов. — М.: Стройиздат, 1971: Пащен-
Пащенко А. Л., Сербии В, П , Стричеиская Е. Л. Вяллщие .материалы./Под ред. Па-
Пащенко А. А. — Киев: Вита школа, 1985.
Глава 20
ОГНЕУПОРНЫЕ
МАТЕРИАЛЫ
Огнеупоры — это материалы, характеризующиеся высокой
прочностью, механической стабильностью и химической инерт-
инертностью при температурах ^1400°С. Их применение имеет пер-
первостепенное значение для многих технологий, например в про-
производстве чугуна и стали, стекла, цемента и т. п., где из огне-
огнеупорных материалов выполнена футеровка печей. Многие из со-
современных производств были бы невозможны по крайней мере
в том виде, в котором они существуют сегодня, если нельзя
было обеспечить эту инертную футеровку.
Огнеупоры обычно мало интересуют химиков, поскольку они
заметно отличаются от типичных химикатов. Из-за их особых
свойств — химической инертности и высоких температур плав-
плавления— считается, что исследование этих материалов не входит
в задачи химической науки. На самом деле, конечно, такое мне-
мнение ошибочно. Для описания свойств огнеупоров необходимо
располагать сведениями о природе химической связи, фазовых
равновесиях, кинетике протекающих процессов и поверхностном
натяжении в этих веществах.
В настоящей главе рассмотрены некоторые общие вопросы
получения и свойств огнеупоров, а также примеры конкретных
огнеупорных материалов.
20.1. Микроструктура или текстура
Огнеупорные материалы представляют собой поликристалли-
поликристаллические твердые вещества, которые содержат одну или несколько
кристаллических фаз, а также весьма часто жидкую или стек-
стеклообразную фазу. Физические свойства этих материалов, напри-
например прочность, зависят от размеров и формы отдельных кри-
кристаллов, природы связи между ними, способа распределения
присутствующей жидкой фазы. Совокупность этих характери-
20.2. Размеры зерен и их рост 255
стик называют текстурой или микроструктурой огнеупорного ма-
материала.
Основными методами исследования текстуры являются опти-
оптическая микроскопия н сканирующая электронная микроскопия
(разд. 3.2.2). Микроскопические исследования, как правило,
проводят па шлифах. Для приготовления шлифа делают срез
или скол огнеупорного материала, поверхность которого поли-
полируют до тех пор, пока она не становится гладкой. Затем произ-
производят травление поверхности соответствующим реактивом, кото-
который либо преимущественно взаимодействует с некоторыми из
фаз, входящими в состав огнеупора, либо разрушает границы
между фазами. В результате текстура материала становится как
бы «рельефной» и все ее особенности размером более ~1 мкм
A04 А) можно легко различить под оптическим микроскопом.
Имея определенный опыт и навыки, можно путем беглого ви-
визуального наблюдения провести определение присутствующих
фаз, а также оценить размеры частиц, объемное содержание
каждой из фаз, степень пористости структуры, влияние имею-
имеющейся жидкой фазы на текстуру.
Аналогичную информацию можно получить методом скани-
сканирующей электронной микроскопии (СЭМ). Благодаря гораздо
большей разрешающей способности методом СЭМ удается уста-
установить также ряд дополнительных характеристик материала.
Например, этим методом можно исследовать грубую поверх-
поверхность скола, не заботясь о возможности разрушения образца
или некоторых «искусственных эффектах», которые могут воз-
возникнуть при его полировании. Если электронный микроскоп
снабжен приставкой для проведения микроанализа, с его по-
помощью можно изучать и химический состав отдельных кристал-
кристаллов или областей образца. Если такая приставка отсутствует,
шлиф материала можно дополнительно исследовать е помощью
электронного микроапализатора.
20.2. Размеры зерен и их рост
При нагревании иоликристаллического материала до высоких
температур средний размер кристаллитов постепенно увеличи-
увеличивается. Небольшие кристаллиты и зерна исчезают, а большие
растут. Движущая сила процесса роста зерен возникает вслед-
вследствие того, что поверхность твердого тела, состоящего из не-
нескольких больших зерен, меньше поверхности тела топ же мас-
массы, в состав которого входит большое число мелких зерен.
Рост зерен — важный фактор уменьшения пористости огнеупор-
огнеупорных материалов или полного удаления пор. Размеры зерен (гра-
(гранулометрический состав образца) влияют на такие свойства
огнеупоров, как прочность.
256
20. Огнеупорные материалы
20.3. Спекание
Спекание — общее название процесса уплотнения поликри-
поликристаллических веществ. Иногда спекание ведут в присутствии
жидкой фазы, которая облегчает перенос вещества в образце.
Для облегчения спекания исходная смесь должна представлять
собой мелкодисперсный порошок желательно в спрессованном
виде. Само спекание проводят при нагревании вблизи или чуть
ниже температуры солидуса. Иногда температура обработки не-
несколько превышает температуру солидуса, в этом случае про-
происходит частичное плавление образца. Наличие жидкой фазы
способствует переносу вещества из одного зерна в другое.
На первой стадии спекания при температурах ниже температу-
температуры солидуса происходит увеличение поверхности контакта меж-
между отдельными частицами. Образуются «перешейки» между зер-
зернами, которые становятся вес шире. Это приводит как бы
к стягиванию кристаллитов и увеличению плотности. По мерс
спекания с ростом температуры сжатие образца продолжается,
промежутки между частицами становятся все меньше, связи
между порами нарушаются. Если при росте зерен поры сужают-
Рис. 20.f. Последовательные стадии пронесся спекания, а — исходный мате-
материал— спрессованный порошок; б — возникновение контакта между зернами;
в — образование пористой трехмерной структуры при соединении частиц;
г — образооание компактного материала с изолированными порами. H;i
последней стадии спекания (па рисунке не изображена) я ходе роста зе-
зерен изолированные поры могут «выталкиваться» па поверхность.
ся до нулевых размеров или «выталкиваются» на поверхность
образца, то плотность материала приближается к теоретической
(кристаллографической) плотности. На рис. 20.1 схематически
изображены различные стадии процесса спекания.
Наличие небольшого количества жидкой фазы обычно силь-
сильно ускоряет процесс спекания, что даст возможность проводить
спекание при гораздо более низких температурах, чем в отсут-
отсутствие жидкости. Однако слишком большое количество жидкой
фазы может привести к изменению формы керамических изде-
изделии и потере прочности. В процессе производства и эксплуата-
эксплуатации огнеупорных материалов весьма важно также, чтобы коли-
20.4. Поверхностные явления
257
чество присутствующей жидкой фазы tic увеличивалось слиш-
слишком быстро, когда температура превысит температуру солидуса.
Таким образом, область стеклования, т. е. интервал температур
между температурой начала уплотнении в результате появления
жидкой фазы и температурой начала проседания образца в при-
присутствии слишком большого количества жидкой фазы, должна
быть как можно шире. Ширина области стеклования су-
существенно зависит от состава материала и вида диаграммы
состояния. Хуже всего в этом отношении случай, когда состав
материала отвечает эвтектическому составу, а полное плавле-
плавление материала происходит в интервале нескольких градусов,
И наоборот, для огнеупорных материалов наиболее подходят
такие составы, которые находится далеко от эвтектики.
20.4. Поверхностные явления
Поверхностное натяжение жидкостей и кристаллических тел
имеет важное значение для кинетики спекании и для процессов
взаимодействия шлаков и огнеупорных материалов. Кинетика
роста зерен и текстура твердого тела зависят от диэдрического
угла, под которым понимают угол между двумя кристаллически-
кристаллическими зернами, находящимися в жидкой фазе (рис. 20.2). При не-
небольших диэдрических углах контакт между зернами недостато-
недостаточен и большое количество жидкой фазы попадает в пустоты
Рис. 20.2 Диэдрнчсскйй
угол и влияние его величины
между зернами.
пл степень контакта
между зернами. Было установлено, что при малых диэдрических
углах скорость роста зерен увеличивается, и в результате обра-
образуются зерна больших размеров. Кинетика роста зерен зависит
также от их начальных размеров. Спекание образцов, состоя-
состоящих из мелких зерен, идет более быстро, чем в случае образцов
с крупными зернами. На практике при необходимости получе-
получения плотного материала требуется прежде всего тшателыю раз-
размельчить исходную шихту, увеличив тем самым поверхность кон-
контакта между частицами. Зависимость скорости роста зерен от
температуры выражается следующим уравнением:
— = — B0 1)
17—1426
258 20. Огнеупорные материалы
где D — диаметр зерни, к- -константа скорости роста. Величина
диэдрического угла влияет не только на скорость роста зерен,
по и на жаропрочность огнеупоров и степень воздействия на них
шлаков. Для обеспечения хорошего контакта между твердыми
частицами в керамике необходимо, чтобы диэдрическне углы
были большими (рис. 20.2,6). Если диэдрическип угол мал или
равен нулю, жидкий шлак легко проникает внутрь огнеупорного
материала, затекая в пустоты между зернами. Это приводит
к вымыванию кристаллов и разрушению твердого тела.
20.5. Воздействие шлаков на огнеупоры
Прежде чем жидкий шлак начнет взаимодействовать с огнеупорно]!
футеровкой, необходимо, чтобы он смочил керамику. В противном случае на
поверхности огнеупорною материала возникают изолированные кайли шла-
шлака. В данном разделе пе будем подробно рассматривать теорию поиермюст-
ного натяжения, а ограничимся лишь самими общими замечаниями но дон-
донном} вопросу. Смачивание происходит тона, когда поверхностное матяжг-
пне жидкоои (т. е. границы раздела жидкость-•-воздух) больше, чем по-
поверхностное натяжение границы раздела между огнеупорным материалом
и жидким шлаком. Это условие, как правило, выполняется для жидки ч
шлаков и lie выполняется для жидких металлов. Последние редко смачи-
смачивают огнеупорную футеровку.
Интенсивность озалмодепстпня между расплавленным шлаком и огне-
огнеупорным материалом зависит от флюсующей иктивности шлака, т. е. от
способности шлака понижать температуру плавления огнеупорного мате-
материала. Оксиды кальция и алюминия имеют различную фшосуюшую актив-
активность по отношению к дппасовоыу кирпичу. Как тшдпо из соответствующих
участков диаграмм состояния систем СаО—SiO2 (рис. 11.8) и AI-.Oj—SiO>
1ркс. 20.3, а), добавление ~4 мол. % ЛЬОз достаточно для понижения тем-
температуры плавления SiO? (температуры ликвидуса) от 1720 до 1.~>93СС.
К такому же понижению температуры плавления SiO2 приводит добавле-
добавление 5=30% СаО. Еще более резко изменяется температура плавления SiO2
при виеденни добавок Na2O или КзО (рис. 20.3,6). Кривая ликимдуса в
этих системах круто понижается до ~800°С. Приведенные па рис 20.3. С>
данные относятся к системе Na2O—SiO2. В области, богатой оксидом крем-
кремния, система КзО—SiO2 полностью аналогична приведенпоп.
Используемый для футеровки доменных; ночей дппасоньш кирпич до икса
быть устойчив к действию больших количеств раатлапленных оксидов же-
железа. В условиях окислительной газовой с]>еды устойчивыми при высоки*
температурах оксидами железа являются фазы FeoO4 (магнетит) н (ч^СН
(гематит). Рассмотрим, например диаграмму состояния chctcvtы Fe.Oj—SiO;
(рис 20.3, <•>). Видно, что она .характеризуется высокими темнопатчрач,!
ликвидуса л наличием широкой области расслаивания в жидкой фазе
В восстановительной же атмосфере устойчивым оксидом железа являет о;
1'еО (вюстнт), который в.чпимолспствует с SiOo с образопяипем нпзкпл.'.ав-
кг.го силиката leiSiOi. (,'ог.;;;сио диаграмме ci4-ifi:mr,:i C'ictcmm FcO—SiO-
(рис. 20 3, ы), температур;] плавления чтот соо.пнкнпя составляет ~]200°С
20.6. Прочность
При комнатной температуре огнеупоры, как правило, пред-
представляют собой хрупкие материалы с невысокой прочностью па
разрыв. При высоких температурах они склонны к пластической
20.7. Виды огнеупорных материалов 259
деформации. Несмотря на то что огнеупоры обладают высокой
пористостью, па холоду они характеризуются высоким коэффи-
коэффициентом прочности на раздавливание (до нескольких сотен ки-
килограммов на квадратный сантиметр, т. е. ~ 10 МПа). Важное
значение для прочности материала имеет его микроструктура.
Хотя в общем случае связь между микроструктурой и прочно-
прочностью довольно сложная, с определенностью можно сказать, что
прочнойь материала возрастает с уменьшением размеров зерен
и уменьшением его пористости. На прочность огнеупоров оказы-
оказывают также влияние те изменения формы и объема отдельных
зерен, которые происходят в ходе термических циклов нагрева-
нагревания и охлаждения материалов. Эти изменения объема связаны
либо с термическим расширением и сжатием (которые могут
быть как изотропными, так и анизотропными), либо с поли-
полиморфными превращениями. Ярким примером вещества, в кото-
котором проявляется последний эффект, является чистый ZrO2. Это
вещество не используют в качестве огнеупорного материала
из-за того, что при ~1000°С оно подвергается полиморфному
превращению (моноклинная фаза ^^ тетрагональная фаза), что
приводит к разрушению материала.
20.7. Виды огнеупорных материалов
Поскольку огнеупорные материалы имеют высокие темпера-
температуры плавления, они, видимо, характеризуются прочными хими-
химическими связями. Причем химические связи могут быть как
ионного, так и ков а ленч по го типа. Огнеупорные вещества с ион-
ионным характером связи должны иметь высокую энергию кристал-
кристаллической решетки. Из уравнения (8.19) следует, что энергия
кристаллической решетки пропорциональна следующему отно-
отношению
U.X——=-
где Z \. и Z_ — заряды ионов, а ге — расстояние между анионом
и катионом. Величина энергии U в основном определяется про-
произведением зарядов ионов Z+Z-. Например, энергия кристалли-
кристаллической решетки оксида щелочноземельного металла примерно
в четыре раза выше энергии решетки галогенида щелочного ме-
металла, имеющего ту же кристаллическую структуру (например,
типа поваренной соли) п близкие значения rL. Качественно эта
же ч-.1 коном ер пост ь проявляется и в величинах температур плав-
плавления, например NaCl плавится при 800°С, a MgO — прн
2S0U С. Таким образом, общим условием существования ионных
17*
260
20. Огнеупорные материалы
материалов с высокими температурами плавления является при-
присутствие в них одного, а еще лучше двух мпогозаридных
ионов. Примерами таких материалов являются А12О3, Сг2О3 и
ZrO2.
Влияние ге на жаростойкость может быть прослежено при
сравнении температур плавления оксидов щелочноземельных
металлов. Все они (за исключением ВеО) имеют кристалличе-
2ЙЮ -
1800
1600
1400
жидкость
2060.
1850
SiO2> муллит
К .
муллит
-муг.лит
SiO2
20 40 60
мол "/„
1700
1S00
1500
1100 -
Si02
Рис. 20.3. Диаграммы состояния систем А12Оз—SiO2 (a), Nn2O—SiO2 (б),
FeO—SiO2 (e), Fe-O4—SiO2 (г) и MgO— SiO2 (д). [Phase Diagrams for
Ceramists, American Cer.imic Society, 1964, 1969, 1975 гг.].
[еупорных материалов
1800
<еоо
1400
120П
8
/две жидкости ¦
\ жидкость
- 5'°2*жидк \
1180 1. L.
StO - SiO
i i i
' ^/FeO
Fe2Si04^
FeO
Si О,
20
40 60
мол.%
80
FeO
1800 -
1600 -
1400 .
1200 -
i две
-
жидкости
2 "Жчдк
м
1
\
гне
1
тит + ol07
m'-SiOj
г
1455
1590
20 40 60 80
мол °/„
1800
1GO0 -
1400 -
1200
510.
262 20, Огнеупорные материалы
скую решетку типа NaCh
Оксид MgO Ca() SrO ВаО
?„.-,, °С 2800 2580 2430 1923
Наибольшее значение гс н, следовательно, наименьшее значе-
значение U в этом ряду имеет ВаО. Это коррелирует с тем, что тем-
температура плавления ВаО самая низкая.
Аналогичные рассуждения применимы к огнеупорным веще-
веществам с ковалентным характером связи. Прочные химические
связи в этих веществах — причина образования ими каркасной
структуры. Соединения одновалентных элементов, например га-
галогенов, по-видимому, не могут быть использованы в качестве
огнеупорных материалов либо из-за их летучести, либо из-за
низких температур плавления. Например, в А!С13 {^пл 190°С)
каждый атом хлора связан с шестью атомами алюминия и каж-
каждая из связей А1—С! слабая. Поэтому А!С13 и подобные ему
вещества имеют низкие температуры плавления. Ковалеитные
соединения, пригодные для использования в качестве огнеупо-
огнеупоров, должны отличаться прочными химическими связями. Они,
как правило, содержат многовалентные элементы с примерно
одинаковой электроотрицательностью и небольшими координа-
координационными числами {обычно КЯ 4). Эти соединения имеют кар-
каркасную структуру. Ниже приведены примеры возможных огне-
огнеупорных материалов и их температуры плавления:
SiC
2700
HfN
3305
Si2N4
-1900
HfC
3890
BN
— 3000
TaN
33G0
B4C
-2350
TaC
3880
NhB2
-2900
ZrC
3540
(В перечень включен и HfC — одно из наболее тугоплавких ве-
веществ, известных в настоящее время.)
Выше шла речь в основном о материалах, которые в прин-
принципе можно использовать в качестве огнеупоров. Широкое про-
промышленное применение в настоящее время имеют главным об-
образом кремнеземистые, хромомагнезитовые, шамотовые и высо-
высокоглиноземистые огнеупорные материалы. Другие тугоплавкие
вещества, такие, как SiC и Si3N4, имеют более специфические
области применения.
Основным сырьем для производства динасового кирпича является квар-
кварцит, в который для облегчения спекания добавляют 2—3% СаО. При этом
из сырья необходимо удалить такие примеси, как оксиды щелочных мотал
лов, так как оии обладают высокой флюсующей активностью (рис. 20.3,6).
Одна из проблем, возникающих при эксплуатации динасовых кирпичей, —
их частичное разрушение из-за изменения объема, которым сопровождается
полиморфное превращение кварца (а*±$) при 573 "С. Это разрушение про-
20.7. Виды огнеупорных материалов 263
является п виде растрескивания. Выше 573 СС кирпичи вполне устойчивы и
механически прочны вплоть до температур плавления (~1700°С). Из-за
высокой вязкости расплавленного оксида кремния за время эксплуатации а
кирпичах может накапливаться значительное количество жидкой фазы.
После обжига многие виды глин становятся жаростойкими. Чистый као-
каолин (известный тзкже как фарфоровая глина) имеет примерный состав
Al2O3-2SiO2-2H2O. Его свойства при высоких температурах можно объяс-
объяснить с помощью диаграммы состояния системы Л12О3— SiO2 (рис. 20.3. а).
После потери воды и последующей рекристаллизации материала к-мппп
превращается в смесь муллит;! и кремнезема состаиа, примерно отвечающе-
отвечающего точке К. Плавление этой смеси начинается при температуре ~1595 °С,
закапчивается при ~1800°С. В монтмориллоннтовых же глинах, таких, как
бентонит, где отношение оксида кремния к оксиду алюминия гораздо выше
(~4:1), содержится значительное количество других катионов, в том чис-
числе и катионы щелочных металлов. Комбинация этих двух факторов приво
дит к понижению температуру,] плавления м.тгериа.чп до <—1300°С и, следо-
следовательно, к понижению жаростойкости бентонита.
Высокоглпноземистые огнеупорные материалы обычно содержат
>Я5 масс. % А12Оз; SiO2 присчтетвует в них и виде главной примеси Эти
материалы изготавливают из диаспора или боксита. Согласно диаграмме
состояния системы ЛЬСЬ—SiO2 (рис. 20.3, а), в равновесных условиях ниже
температуры солидуса (<1840°С) в них сосуществуют две фазы- корунд
АЬО3 и муллит 3AI2O3-2S1O2. Выше 1840 °С происходит частичное плавле-
плавление смеси фаз с образованием АЬСЪ и жидкости. Известны две различные
технологии изготовления высокоглиноземистых огнеупорных кирпичей. Пере-
Переплавленный оксид алюминия получают путем плавления в электрических
печах при ~2000°С. Футеровкой печей служит сама загружаемая шихта.
Спеченные глиноземистые кирпичи изготавливают путем обжига при 1700—
1800°С. При таких пониженных температурах проводят спекание оксида
алюминия, содержащего значительное количество прлмесей, причем спекание
происходит с участием жилкой фазы Получающийся продукт отличается
низкой пористостью и устойчивостью к истиранию. Спекание высокочистого
оксида алюминия, содержащего >99,8% AkOj. протекает по твердофазно-
твердофазному механизму. При этом в А12Оз обычно вводит ~0,2% /М<*0. Хотя дейст-
действие этой добавки не вполне понятно, получающийся в результате продукт
характеризуется нулевой пористостью и оптической прозрачностью. Способы
получения жаростойких нысокоглииоземпстнх бетонов описаны в гл. 19.
Магнезиальные и хромомлгнезнтопые огнеупорные материалы широко
используются в качестве футеровки металлургических печей. Это объясня-
объясняется их устойчивостью к действию расплавленных ш.члков. MgO плавится
при 2800 °С. его можно получить из брусптп М(т(ОП)г. магнезита .MgCCb
или доломита СаСОз-М^СОз. Сырье обжигают (т. е. нагревают) при 500—-
1000°С. В ходе термического разложения выделяются СО2 и Н2О
и образуется высокодисперсный порошок «активного» MgO. «Активный»
MgO характеризуется большой удельной поверхностью н гигроскопично-
гигроскопичностью. Образующийся при взаимодействии М«О с водой Мр(ОН)-. имеет бо-
более низкую плотность, чем оксид магния. Если гидратация MgO идет в
процессе его эксплуатации, то увеличение объема пядешя ведет к разру-
разрушению последнего. Такое разрушение можно предотвратить путем предва-
предварительного прокаливания MgO, т. е. путем пагпеиания MgO до достаточно
высоких температур с целью увеличения средних размеров зерен и получе-
получения хорошо спеченного материала с низкой пористостью. Температуру пред-
предварительной прокалки варьируют от 1400 до 1700 "С. в зависимости от со-
содержания гтгшмесей в МсО.
Для получения плотной прозрачной магнезиальной керамики и для
улучшения спекания к ней добавляют MF. В процессе спекания LiF улету-
улетучивается, а оставшийся чистый оксид магния вполне пригоден для исполь-
использования при высоких температурах. Промышленные магнезиальные огне-
264 ^ 20. Огнеупорные материалы
унорные материалы содержат в виде примесей CaO, AhOj, Ре»О3 и SiO2.
поэтому в ходе прокалки происходит частичное плавление огнеупоров. За
метмос облегчение спекания MgO (которое проводят чрп умеренных темпе-
температурах), видимо, связано с появлением жидко") фазы, а т;мже с образо
ваипем твердых растворов па основе иерпклаза (MgO), и которых часть
атомов магния замещается атомами алюминия и железа.
Использование в качестве исходного сырья доломита приводит к неко-
некоторым неприятным последствиям. Дс.ю в том, что после обжига доломита
образуемся весьма гигроскопичная смесь оксидов с высоким содержанием
оксида кальция. Прокаливание образовавшегося материала лишь незначи-
незначительно снижает его склонность к гидратации. Для борьбы с этим явлением
пелольз} ют два метода: во-первых, обожженный продукт можно покрывать
специальной смолой, надежно изолирующей поверхность материала, во-вто-
во-вторых, и керамику вводят добавки, которые при нагревании связыиаю-i сво-
свободный оксид кальция. В качестве такой добавки можно исмользпиаги
серпентин JMgO -2SiO2'2H2O, который взаимодействует с СаО с образова
пнем СазБЮб (и форстерита i\lg2SiO4).
Хромомагнезптовые огнеупорные материалы применяют для футеровки
доменных печей. Оксид хрома Сг2О3 является жаростойким соединением с
температурой плавления 2275 °С. В природе в состав хромовых руд обычно
"ходит ряд других оксидом. Как правило, хроч присутствует в виде шпине
лей АВ^О4, где Л — магний и железо (II), а В — алюминий, железо (III), a
также хром. Второй сопутствующей фазой в хромовых рудах является фа-
фаза, близкая по составу серпентину. При нагревании хромовых руд до
~ 1400 'С серпентин теряет воду, а все железо (II) окисляется до Fe2O:;.
Последний может взаимодействовать с MgO пз серпентинм с образованием
дополнительного количества шпинелъной фазы. При этом серпентин обед-
обедняется оксидом магния и возникает менее жаропрочная богатая SiO2 жид-
жидкая фаза. Поэтому добавление MgO к хромо.чагпезиговьш огнеупорам
прежде всего необходимо для удаления атой богатой S1O2 жидкой фачы
путем се взаимодействия с MgO, в результате образуется более жаропроч-
жаропрочная фаза — форстерит M^SiOj (рис, 20.3,C).
Кроме перечисленных выше областей применения огнеупорных материа-
материалов, некоторые из них, имеющие счрого контролируемое содержание приме-
примесей, находят применение в особых отраслях техники. Так, упоминавшийся
ранее снеченшлй оксид алюминия ислолизуют. например, при изготовлении
свечей задет ания, гнезд клапанов, обжимных устройств для волочения про-
проволоки п т. п. .Муллнт, получаемый но реакции ЛЬОз (боксит или байерит)
с S1O2 (кварц или фарфоровая глина), используют для ироизнодства тиг-
тиглей, трубок пирометров и т. п. Форстерит, плавящийся при 1890 °С. приме-
применяют в качестве формовочного песка при разливке стали. Форстерит встре-
встречается в виде минерала, в котором магний частично заметен на желе-
зо(П). Он может быть получен путем прокаливания MgO с тальком 3MgO-
4SiO2-H2O или более часто с серпентином 3MgO-2SiO2-2H2O. Изделия in
стеатита получают из смеси талька и глины. Их используют в качестве
электроизоляционных материалов. Количество глины, необходимое для полу-
получения стеатита, невелико, и основным продуктом прокалки является клино
енстатит — одна из полиморфных модификаций MgSiO3. Другой ценный
электрпкерамический материал—кордиерит 2A\gO-2АЬОз-ЗБЮг, имеющий
очень низкий коэффициент термического расширения и, следовательно, про-
проявляющий устойчивость к резким изменениям температуры. Его можно так-
также получить из смеси талька с глиной путем введения псболынил добавок
А12Оз или силлиманита Al>SiOr,.
Диоксид циркония ZrO2 — весьма перспективный огнеупорный материал,
температура плавления которого составляет ~2700°С. Однако при нагрева-
нагревании он часто растрескивается из-за изменения объема, которое сопровожда-
сопровождает переход моноклинной модификации ZrO2 в тетрагональную. Моноклинный
7лО-2 имеет плотность 5,56 г/см3, а более высокотемпературная тетрагональ-
20.8. Сналомы — питридные огнеупорные материалы 265
ная фаза — 6.10 г/см3. Поэтом)" при нагревании происходит сжатие- мате-
материала на 9% его объема. Фазовый переход и, следовательно, растрескива-
растрескивание можно предотвратить н\тем добавления 10—20% CtiO, MgO пли \?Оз.
Эти добавки стабилизируют кубическую высокотемпературную модификацию
ZrOj (>стойчниую в отсутствие добавок лини, при температурах выше
2400°С). Образующиеся твердые раствори стабильны при гораздо более
низких температурах, чем чистый ZrO2. Формулу твердого ряствпра на ос-
основе оксплон циркония и кальипя можно зг.писать в виде Ca.Zr._,()>_,.
Такая запись предполагает, что замещение Zr'1- па Са2+ идет с образова-
образованием кислородных вакансий (гл. 10). При циклическом нагревании- ох-
охлаждении стабилизированный .¦шокенд циркония обратимо расширяется п
сжимается. Фазовых переходов, разрешающих керамику, при этом не про-
происходит. Такой диоксид циркония широко применяется в качестве твердого
электролита в высокотемпературных электрохимических источниках тока и в
кислородеслектпвпых электродах: применение стабилизированного диоксида
цирконии в качестве твердого электролита связано с высоком подвижностью
в нем кислородных вакансий (ы. 13).
Циркон ZrSiO4 имеет низкий коэффициент термического расширения,
поэюму он выдерживает резкие колебания температуры. Этот материал пс-
нользиот л-'П1 ф\теровки сгсклонаренныл печей и и качестве формовочно-
формовочного песка. ,5-1 лнпозем— полиалюминат натрия состава NaoO- G-=-9) А^Оз —
образуется при взаимодействии Al^Oj. входящего в состав футеровки сто.с-
доваренных печен, с NajO, содержащимся в расплавленном стекле. В на-
настоящее время Р-глипозем вмзывает повышенный интерес в связи с высокой
подвижностью в нем ионов N;ik. ^-Глинозем используется в качестве твер-
твердого электролита в твердофазных электрохимических источниках тока но-
нового типа (г:). 13).
Многие неоксидиые материалы имеют весьма высокие температуры
плавления. Тем не менее их использование в качестве огнеупоров ограни-
ограничено возможным окислением и пысокимп финансовыми затратами на про-
производство. О,-1ип из наиболее перспективных и этом отношении материал--
нитрид кремния S10N4, который можно использовать до температур
— 1400'С. При высоких температурах он сохраняет высокую прочность, что
позволяет изготавливать из пего лопасти турбин и подшипники, работаю-
работающие при высоких температурах. Карбид кремния SiC—широко распрост-
распространенный материал, устойчивый па воздуле до ~ 1700'С (благодаря обра-
образованию защитной поверхностной нлепкп SiOJ. Его используют ъ качест-
качестве абразивных материалов (карборунд), нагревательных элементов электрн-
ческих иечои, при изготовлении массинных огнеупорных конструкции Кар-
Карборундовые огнеупорные материалы отличаются низкой пористостью, высо-
высокой теплопроводностью, они прекрасно выдерживают резкие колебания
температуры. Отличный огнеупорный материал —синтетический графит, ко-
который весьма устой чип к действию шлаков и расплавленных ме7Л«1лои, зн
исключением металлов группы железа Он обладает высокой прочностью,
хорошей тепло- и электропроводностью. Эти свойства позволяют пепо.-пво-
вать синтетический графит в качестве электродов промышленных печей.
Дисилнцнд молибден.'! MoSIj используют для изготовления нагревательных
элементов электрических печей, в которых создаются температуры до
1600 "С. Устойчивость этого материала к окислению связана с образовани-
образованием на воздухе защитной поверхностной пленки SiOj.
20.8. Последние достижения. Сиалоны — нитридные огнеупорные
материалы
Нитрид кремния Si3N4 — инертный материал, который устой-
устойчив при более высоких температурах, чем многие металлические
сплавы. Поэтому в последние годы он находит широкое приме-
266 20. Огнеупорные материалы
пение как весьма эффективный высокотемпературный конструк-
конструкционный материал для изготовления керамических лопаток га-
газовых турбин. Одной из трудностей производства изделий из
нитрида кремния как соединения с ковалентным характером хи-
химической связи является получение плотноспечеппой керамики.
В начале 70-х годов в США при изучении процессов спекания
Si3N4 открыт совершенно новый класс химических веществ, ко-
который назван сиалонами. В состав этих веществ входят четыре
элемента Si, A1, О, N (отсюда и название — сиалоп), а также ряд
других элементов, внедряющихся в кристаллическую решетку
сиалопов. Сиалоны—оксонитридпые фазы, структура которых
построена из тетраэдров (Si, Л1) (О, NL, сочлененных и трех-
трехмерный каркас. Некоторые сиалоновые фазы характеризуются
прекрасными механическими свойствами и устойчивостью в ус-
условиях Действия химических агрессивных сред при высоких тем-
температурах. Спекание сиалонов осуществляется более просто, чем
спекание нитрида кремния. Несомненно, в ближайшее время эти
материалы могут найти широкое применение.
Сиалопы представляют интерес и для кристаллохимиков.
Многие из этих фаз изоструктурны известным силикатам, напри-
например:
1) YSiO2N изоструктуреп волластониту CaSiO3. Его кристал-
кристаллическая решетка состоит из бесконечных цепей (SiO2NK-, ана-
аналогичных метасиликатным цепям (SiO3J~;
2) Y2O3-Si3N4 или Y2Si[Si203N4] изоструктурен акерманиту
Ca2MgSi207;
3) Yo(Si04KN изоструктурен апатиту Са5(РО4)зОН;
4) Li2SiA103N— кристобалитоподобная фаза, в которой
ионы Li+ занимают междоузлия в каркасе кристобалитоподоб-
пой структуры.
Совсем недавно было найдено, что некоторые композиции
в системе MgO—Si3N4 могут быть получены в стеклообразном
состоянии. Поскольку добавление небольших количеств нитрида
в оксидные стекла ведет к повышению температуры стеклова-
стеклования Tg, новые стеклообразные материалы, содержащие большое
количество нитрида кремния, могут также иметь весьма инте-
интересные свойства, например могут быть устойчивыми при высо-
высоких температурах.
Упражнения
20.1. Какими свойствами должны обладать огнеупорные материалы, ис-
используемые в качестве футеровки доменных и мартеновских печей? Объяс
•нитс, почему динасовый кирпич пригоден в этих целях, несмотря n.i сильно
восстановительную атмосферу в печах.
20.2. В чем заключается значение диаграмм состояния для практиче-
практического использования огнеупорных материалов^1
Литература 267
20.3. Объясните, почему динасовый кирпич не используют для футеровки
печей, в которых варят оконное стекло.
20.4. Объясните, почему ценные огнеупорные материалы можно полу-
получить при обжиге фарфоровой глины (каолина) и нельзя — при обжиге
монтмориллонитовой глины.
Литература
1. Alpcr A. AT. (ed.). High Temperature Oxides, vol. 1—4, Academic Press,
New York. 1971.
2. Bell /., The ceramic age dawns. New Scientist, 1983, Jan. 26, 10 A984).
3. Chandler ,\[., Ceramics in the Modern World, Aldus, 1967.
4. ford IV'. ?.. The Effect of Hcaf on Ceramics, MacLaren and Sons. 1967.
5. Jack K. II., Nitrogen ceramics, Trans. J. Brit. Сргаш. Soc, 19, 376 A973)
6. Jack A'. N.. Sialnns and related nitrogen ceramics, J. Materials Science, 11,
1135 A976).
7. Кингери У. Д. Введение в керамику.— .4.: Стройнздат, 1967.
8. McColm I. J., Ceramic Science lor Materials Technologists, Chapman and
Hall, 1983.
9. Pwnpuch R., Ceramic Material*, F.lscvier, 1976.
10. Ryshkewiich E., Oxide Ceramics, Academic Press, I960.
11. Searlr Л. В., Refractory Materials, Griffin and Co. 1940.
12. Sosman R. В., The Phases oi Silica, Rutgers University Press, 1965.
13. While .'., New perspective and aspirations of ceramic science and technology
Trans. J. Brit. Ceram. Soc. 1983, 108 A983).
Дополнительная литература. Бакунов В. С, Балкепич В. Л., Взаюи А. С.
и др. Керамика и.^ высокоогиеупорпых окис.юв./Под ред. Д. Н. Полубояри-
нова и Р. Я. Попильского—М.; .Металлургия, 1977; Бибкопа Н. М. Физиче-
Физическая химия силикатов и тугоплавких соединении. — Минск: Высшая школа,
1984; Будншов II. П. Химия и технология оксидных и силикатных мате-
материалов. — Киев': Наукова думка, 1970; Гсгузин Я. Е. Физика спекания. — М.:
Наука, 1967; Гордеев С. Я. Физико химические основы керамической техно
логии. — Иваново: Изд-во ИХТИ, 1979; Рутман Д. С, Торопов Ю. С, Пли-
нер С. Ю. и др. Высокоогнеупорные материалы из диоксида циркония. — М..
Металлургия. 1985; Стрелков К. К. Теоретические основы технологии огне-
огнеупорных материалов —М.: Металлургия, 1985.
Глава 21
ОРГАНИЧЕСКАЯ
ХИМИЯ
ТВЕРДОГО ТЕЛА
Эта книга была бы незавершенной без рассмотрения, хотя бы
вкратце, органических материалов. Обычно тот факт, что мно-
многие органические вещества при комнатной температуре находят-
находятся в твердом состоянии не рассматривается как существенный
для их химии. Однако из этого правила есть и исключения, и
ниже рассмотрены два специфических проявления именно твер-
твердого состояния органических соединений. Во-первых, некоторые
органические реакции в твердом состоянии приводят к образо-
образованию совсем иных продуктов, чем реакции тех же веществ, осу-
осуществляемые по традиции в растворах. Это обстоятельство
открывает новые направления в синтетической химии и производ-
производстве новых материалов. Во-вторых, специфика твердофазных ор-
органических соединений состоит в том, что среди них обнаруже-
обнаружены вещества с интересными физнческими свойствами, в частно-
частности «органические металлы», такие, как пилиацетилеи.
21.1. Топохимический контроль органических реакций
в твердом состоянии
В традиционной органической химии реакционная способ-
способность веществ и состав продуктов пх взаимодействия опреде-
определяются структурой молекул реагентов. Однако для некоторых
реакций в твердом состоянии важен также топохимический
контроль, т. е. влияние кристаллической структуры исход-
исходных веществ и в особенности способа упаковки отдельных мо-
молекул в кристаллическую решетку. В большинстве кристаллов
упаковка молекул характеризуется регулярностью и высокой
степенью порядка, допускающего лишь строго ограниченный на-
набор ориентации молекул. Подвижность последних также сильно
ограничена. В итоге соседние молекулы могут взаимодейство-
взаимодействовать друг с другом лишь при выполнении некоторых вполне
21.1. Топохимичсский контроль органических реакции 269
определенных требований. В первую очередь соседние молекулы
должны обладать соответствующими реакционными центрами,
благоприятно ориентированными и достаточно близко располо-
расположенными друг к другу. Например, для того чтобы была воз-
возможной реакция полимеризации, необходимо, чтобы пространст-
пространственно сблизились олефиновыс группы >С = С< соседних моле-
молекул. Установлено, что для прохождения полимеризации в твердом
состоянии двойные связи должны быть примерно парал-
параллельны друг другу и расстояние между ними не должно превы-
превышать 4 Л. Взаимодействие двух олефиновых групп приводит
к образованию цнклобутанового фрагмента молекулярной струк-
структуры (рис. 21.1, я).
^ "- 7 i> ? ! l
а б^ е
Рис. 21.1. Димеризация с образопанием циклобутанового кольца (а) и мо-
молекула цпклогексанл r конфирмации кресла (б) и ванны (в).
Органические реакции в твердом состоянии, как правило, на-
начинаются не спонтанно, а лишь при инициирующем воздействии
УФ-излучения. Продукты топохимически контролируемых реак-
дип в твердом состоянии часто значительно отличаются от про-
продуктов реакций в растворах, что открывает возможности синте-
синтеза новых полимеров, оптически чистых соединений и т. д. Ряд
пионерских работ в этой области был выполнен в 60-х годах
Шмидтом с сотр. [6].
Роль стереохимического фактора в органических реакциях
чрезвычайно важна и хорошо знакома химикам-органикам. Рас-
Рассмотрим здесь влияние только тех дополнительных стерических
факторов, которые возникают вследствие различных способов
расположения молекул в кристаллических структурах. При этом
молено выделить I) внутримолекулярные эффекты, которые
включают перегруппировки, образование циклических молекул,
реакции отщепления и другие процессы, проходящие в пределах
одной молекулы, и 2) меж молекулярные эффекты, проявляю-
проявляющиеся при взаимодействии молекул, занимающих соседние по-
положения в молекулярном кристалле. Среди рассмотренных ниже
примеров преобладают межмолекулярные эффекты, отличаю-
отличающиеся особенным многообразием.
270
21. Органическая химия твердого тола
21.1.1. Внутримолекулярные реакции. Конформационные
эффекты
Молекулы большинства органических соединений, находя-
находящихся в жидком и газообразном состояниях, обладают гибко-
гибкостью и могут менять свою форму. Пример такого поведения де-
демонстрирует циклогексан, который может существовать в двух
формах—кресла и ванны (рис. 21.1,6 и в). Молекулы перехо-
переходят из одной формы в другую достаточно легко, так как при
этом не происходит разрыва связей; такие эффекты носят наз-
название копформационных. В отличие от жидкостей и газов в твер-
твердом состоянии подобная гибкость и перестройка молекул прак-
практически исключаются. В результате какая-то из конформацип
молекулы оказывается «замороженной» и все последующие
реакции ограничиваются возможностями именно этой конфор-
B)
R=CH3COO
C)
C)
_j- другие
1 продукты
Рис. 21.2. Реакции элиминирования диметилового эфира -
адипинооой кислоты.
мации. Пример, поясняющий различие реакций в твердом и
жидком состояниях, приведен на рис. 21.2. Молекулы днмети-
лового эфира игезо-^'-дибромоадипиновой кислоты в кристал-
кристаллическом состоянии принимают копформацию A). При обработ-
обработке кристаллов этого соединения газообразным аммиаком каж-
каждая такая молекула отщепляет две молекулы НВг и превраща-
превращается в молекулу диметилового эфира транс, транс-муконовои
кислоты B), также находящегося в твердом состоянии. Важно
подчеркнуть, что молекулы и исходного вещества, и продукта
21.1. Топохнмичсский контроль органических реакции 271
реакции обладают центром симметрии. В растворах молекулы 1
образуют набор разнообразных конформаций, дающих при от-
отщеплении НВг различные стереоизомеры C, 3', 3" и др.).
21.1.2. Межмолекулярные реакции. Эффекты молекулярной
упаковки
В жидкостях молекулы непрерывно совершают вращатель-
вращательные и поступательные движения, в результате чего взаимодейст-
взаимодействующие молекулы (одинаковые или разные) сталкиваются друг
с другом, находясь в самых различных ориентациях. В твердых
телах молекулы, напротив, занимают фиксированные друг отно-
относительно друга положения при очень небольшом наборе возмож-
возможных ориентации, что накладывает жесткие ограничения на
характер и возможности их взаимодействий. Используя это об-
обстоятельство, можно получить стереохимически чистые продук-
продукты, а в некоторых случаях благодаря более близкому располо-
расположению молекул в твердом теле осуществить такие реакции, ко-
которые в жидком и газообразном состояниях вообще не проходят.
21.1.3. Фотодимеризация о-этокси-транс-коричной кислоты
Структура молекулы этого вещества изображена па рис. 21.3
D). В кристаллическом состоянии могут существовать три по-
полиморфные формы а, {1 и y> которые отличаются взаимной
ориентацией молекул в решетке. Под действием ультрафиолето-
ультрафиолетового облучения эти три формы ведут себя по-разпому.
21.1.3.1. а-Форма. Кристаллы а-формы образованы молекулами,
расположенными по принципу «голова к хвосту» и образующи-
образующими пары с центром симметрии D'). При УФ-облучении происхо-
происходит димеризация, приводящая к образованию молекул траксил-
линовой кислоты E), также обладающих центром симметрии.
В этом превращении происходит сшивание соседних молекул по
двойным связям и образование в молекуле 5 циклобутанового
кольца. Очевидно, что эта реакция может происходить только
при таком благоприятном взаимном расположении молекул (и
двойных связей), какое имеется в а-форме. Относительное сме-
смещение реагирующих молекул, необходимое для их слияния в мо-
молекулу продукта, чрезвычайно мало.
21.1.3.2. §-Форма. В кристаллах р-формы, молекулы, уложенные
«голова к голове» F) и связанные зеркальной плоскостью, так-
также способны димеризоваться путем присоединения по двум
двойным связям. Продуктом димеризашш являются молекулы
траксиповоп кислоты, сохраняющие зеркальную симметрию рас-
расположения исходных молекул.
272
2). Органическая химия твердого тела
D)
D)
а-форма,
кристалгизирушаяся
из эфира
E)
x=chsch2c-
со2н
Рис. 21.3. Фотодимсризация а- и J5 форм о-этоисп-тринс-коричиой кислоты
13].
21.1.3.3. ч-Форма. В кристаллах -/-формы, кристаллизующейся
из этанола, соседние молекулы расположены так, что их двой-
двойные связи оказываются удаленными друг от друга. В результате
реакция фотодимс-ризацни в ¦у-форме не происходит. В раство-
растворах этот процесс также не идет, по наблюдается цис-транс-мзо-
меризация.
В результате изучения большого числа реакций димеризаиии
и полимеризации, подобных описанным выше, установлено, что
определяющую роль в жтих процессах играет относительное рас-
расположение двойных связен в соседних молекулах. Расстояние
<4 Л благоприятствует протеканию реакции, а при больших
21.1. Топохимичеекий контроль органических реакций
273
г.—л
(8)
(9)
A0)
Рис. 2!.4. Фотополимеризация 2,5-днстирилпиразина [8].
расстояниях (~5 А) молекулы оказываются слишком далеко
друг от друга, чтобы могло происходить диклоприсоединение.
Требование параллельного расположения двойных связей и рас-
расстояния между ними, не превышающего 4 А, известно как кри-
критерий Шмидта, определяющий возможность циклизации по
двойным связям.
21.1.4. Фотополимеризация 2,5-дистирилпиразина
В кристаллах 2,5-дистирилпиразииа (8), выращенных из рас-
раствора, под действием УФ-облучепия при —ВО °С происходит об-
образование высокомолекулярного соединения 9 (рис. 21.4). Этот
процесс также относится к числу топохимически контролируе-
контролируемых, так как его протекание в сильнейшей степени зависит от
взаимного расположения молекул в образованном ими кристал-
кристалле. На рис. 21.4 показано, как отдельные молекулы (8) уклады-
укладываются параллельно в горизонтальный ряд A0); каждая моле-
18-1426
274 21. Органическая .химия твердого толп
кула при этом оказывается несколько смещенной относительно
соседней. Расстояние между двойными связями соседних моле-
молекул составляет всего лишь 3,94 А, что способствует легкому про
теканию фотоциклоприсоединения. Поскольку каждая молекула
содержит но две двойные связи, образующийся продукт (9)
характеризуется большой длиной цепи и стереорегулярпон
структурой. Полимеризация молекул (8) происходит также и
в растворах, однако, получающиеся при этом полимеры отли-
отличаются меньшей длиной цепей и аморфной структурой.
21.1.5. Фотополимеризация диацетиленов
Молекулы углеводородов диацетилеповогоряда 11 (рис. 21.5)
содержат по две реакционноспособные тройные связи. При об-
образовании кристаллической решетки эти молекулы закономер-
закономерным образом смещаются относительно друг друга (как это
показано на том же рисунке). При выборе подходящего заме-
r—= — ^ — r
A2)
Рис. 21.5. Фотополимеризация замещенных дпацстиленов.
стителя R можно добиться настолько близкого расположения
тройных связей, что становится возможной реакция полимериза-
полимеризации. В результате такого процесса, идущего под действием
УФ-облученин, получаются полимеры со стерсорегулярноп
структурой.
21.1.6. Асимметрический синтез
В описанных выше примерах молекулы продуктов реакции
сохраняли симметрию молекул исходных веществ. Однако при
взаимодействии молекул двух разных веществ возможно обра-
образование асимметрических соединений, некоторые из которых мо-
могут обладать оптической активностью. Получение таких соеди-
21.1. Топохимический контроль органических реакций 275
нений в твердом состоянии основано на использовании твердых
растворов исходных реагентов.
Ранее (гл. 10) были рассмотрены твердые растворы метал-
металлов и других неорганических веществ. Органические твердые
растворы образованы молекулами двух (или большего количе-
количества) соединении, неупорядоченно расположенными в узлах кри-
кристаллической решетки. На рис. 21.6 приведены примеры соеди-
соединений A3, 14), которые могут образовывать бинарный твердый
раствор — замещенные бутадиепы, различающиеся одним из за-
заместителей; эти соединения могут образовывать кристалличе-
A5)
А г = 2,6-дихлорофенил C5H3CI2
Ph = фенил С6И5; Th-тиенил C4H3S
Рпс. 21.6. Образование молекул с оптически активными центрами по реак-
реакции фотоирисоедннения в твердых растворах.
скис твердые растворы с упорядоченной (а не хаотической) упа-
упаковкой молекул. В таких твердых растворах под действием об-
облучения и при тщательном контроле других условий между со-
соседними разнородными молекулами A3, 14) проходит реакция
присоединения по двойной связи с образованием продукта A5),
содержащего два асимметрических атома углерода А и В. Воз-
Возможности осуществления подобных реакций синтеза асимметри-
асимметрических молекул весьма широки. Такой подход позволяет также
получить важную информацию о механизмах образования опти-
оптически активных молекул, особенно в тех случаях, когда энан-
тиомерные продукты образуются в неравных количествах. Эта
информация становится доступной благодаря тому, что для изу-
изучения геометрии молекул реагентов и продукта можно приме-
применить рентгеноструктурпый анализ.
21.1,7. Димеризация антрацена. Роль дефектов
кристаллической решетки
Топохнмичсски контролируемые реакции фотодимеризации
проходят в замещенных антраценах. Часто, однако, исследова-
исследование этих реакций приносит неожиданные результаты. Так, в не-
некоторых соединениях наблюдается фотодимеризация, которую
нельзя было ожидать исходя из геометрии упаковки молекул;
18*
276
21. Органическая химия твердого тела
в других случаях димеризация дает продукты с симметрией, от-
отличающейся от ожидаемой. Томас, Мореси и Девернь [9], вы-
выполнив тонкие и остроумные микроскопические исследования,
показали, что эти непредсказуемые эффекты возникают благо-
благодаря наличию линейных и пленарных дефектов органических
кристаллов (дислокаций, дефектов упаковки, аптифазных гра-
границ).
Пример соединения, димеризующегося неожиданным обра-
образом, приведен на рис. 21.7. 9-Цианоантрацеп A6) образует кри-
CN
A6) A8) A7)
Рис. 21.7. Фотодимеризация антрацена.
сталлическую структуру, в которой соседние молекулы обраще-
обращены друг к другу «головами». В соответствии с этим следовало
бы ожидать, что будет образовываться (^нс-димер A7); в дейст-
действительности же образуется транс-изомер A8). Происходит это
потому, что в кристаллах 9-цианоантрацена имеются дефекты
упаковки, вдоль каждого из которых одна половина кристалла
фактически смещена относительно другой. Такое боковое сме-
смещение приводит к тому, что молекулы, находящиеся по разные
стороны плоскости дефекта, расположены теперь по принципу
«голова к хвосту». При УФ-облучении молекулы, лежащие в об-
области дефекта упаковки, проявляют большую реакционную спо-
способность, чем молекулы в объеме кристалла, и димеризуются,
образуя транс-кзомер A8). По не вполне понятному механизму
(возможно, что он связан с порождением новых дефектов упа-
упаковки) следом за этим и весь кристалл димеризуется в транс-
изомер. При микроскопическом исследовании похожей реакции
фотодимеризации ацетонафтилена Томасу [8] удалось непосред-
непосредственно наблюдать образование димеров на дислокациях.
21.1.8. Регулирование способа упаковки молекул
в кристаллах
Открытие первых топохимически контролируемых органи-
органических твердофазных реакций всецело зависело от случайных
благоприятных совпадений во взаимном расположении молекул
21.1. Топохимическни контроль органических реакций 277
в кристаллах исходных соединений. Однако чаще всего в орга-
органических кристаллах взаимная ориентация молекул, их реакци-
реакционных центров, а также расстояния между последними не спо-
способствуют успешному протеканию реакций. Следовательно, же-
желая расширить возможности метода топохимического контроля,
следует научиться в первую очередь модифицировать структуру
кристаллов-реагентов и управлять ею. На возможность такого
регулирования структуры впервые обратил внимание Шмидт
[6].
о
A9) B0)
R = 3,4-C6H3CL2
Рис. 21.8. Образование трициклнпеской структуры при фотодимеризации дие-
нона.
К настоящему времени в этом синтетическом направлении
достигнуты определенные успехи. В частности, было показано,
что в результате галогенирования (в особенности хлорирования)
ароматических колец молекулы ароматических соединений вы-
выстраиваются параллельно друг другу в слои, в которых плоско-
плоскости соседних молекул перекрываются н располагаются под
углом к плоскости слоя; благодаря такому расположению
в кристаллах образуется плотная упаковка. Межслоевые рас-
расстояния составляют ~4 А. Чем обусловлено такое строение ве-
вещества еще не ясно, однако, не исключено, что но крайней мерс
частично это вызвано притяжением между атомами хлора, вхо-
входящими в молекулы соседних слоев.
На основе этого открытия был осуществлен синтез широкого
круга хлорзамещенных соединений, кристаллизующихся подоб-
подобным образом и, следовательно, позволяющих провести реакции
фотоприсоединения. Интересна в этом отношении восьмицент-
ровая димеризация (рис. 21.8); циклоприсоединение по двум
парам двойных связей в кросс-сопряженных диенонах A9) при-
приводит к образованию необычной трициклической структуры ди-
кетона B0). Сейчас есть все основания полагать, что роль этого
направления синтеза, получившего название «инженерия кри-
кристаллов», в будущем должна неуклонно возрастать.
278 21. Органическая химия твердого тела
21.1.9. Органические реакции в неорганических матрицах
Как показано выше, сущность топохимического контроля
твердофазных органических реакций сводится к обеспечению
правильного взаимного расположения молекул, обеспечивающего
возможность прохождения данной реакции между ними. Один
нз вариантов топохимического контроля реакций осуществляет-
осуществляется с использованием неорганических слоистых соединений (та-
(таких, как силикаты или халькогениды переходных металлов).
Органические молекулы, внедряясь между неорганическими
слоями, принимают там определенную ориентацию, благодаря
чему реакции с их участием становятся высокоселективными.
Используя в качестве матрицы глинистый минерал моптморило-
нит (слоистый силикат), удалось синтезировать пептиды из ами-
аминокислот и осуществить перегруппировку бензидипа. Этот прием
регулирования реакций, пока еще не получивший широкого рас-
распространения, имеет большие потенциальные возможности в ор-
органическом синтезе и катализе.
21.2. Электропроводящие органические твердые тела.
Органические металлы
Получить электропроводящие органические полимеры (или
органические металлы) весьма заманчиво, но до сих пор это
было труднодостижимой целью. Такие материалы должны были
бы сочетать в себе механические свойства полимеров (гибкость
и легкость получения тонких пленок) с высокой электропровод-
электропроводностью, которая обычно считается прерогативой металлов.
На создание подобных материалов в последние годы были на-
направлены большие усилия исследователей; однако до настояще-
настоящего времени A983 г.) ни одно из этих веществ не удалось полу-
получить в промышленных условиях, так как еще не решены про-
проблемы сохранения свойств продукта при перенесении экспери-
эксперимента из лаборатории в промышленность, в частности проблема
нестабильности продукта в атмосферных условиях. Рассмотрим
основные виды исследованных материалов.
21.2.1. Сопряженные системы
21.2.1.1. Легированный полиацетилен. Органические твердые те-
тела обычно не проводят электрического тока, поскольку элек-
электроны не могут свободно перетекать пи вдоль ковалептпых свя-
связей в пределах молекулы, ни от одной молекулы к другой в пре-
пределах кристалла. Исключение составляют сопряженные систе-
системы, в которых атомы углеродного скелета связаны чередующи-
чередующимися двойными и одинарными связями подобно тому, как это
21.2. Электропроводящие органические твердые то.1
u a
279
происходит в графите. В противоположность полиэтилену, ко-
который является насыщенным соединением, содержащим только
одинарные связи, полиацетилен — полимер с длинной цепью
сопряженных связей — представляет собой потенциальный про-
проводник электричества. (Образование этих полимеров из соответ-
соответствующих мономеров — этилена, содержащего двойные связи, и
а
этилен
п (н-С=С-н
ацетилен
н
1
н
(
\
У
V
1
н
н
/
н
н
1
с
н н
\
ь
\г
с
/
1
1
н
н
\
н
н
J
с
/
/
к
полиэтилен
ч
1
г
\
>с
1
и
н
[
с
\
н
"с
1
н
транс
\
цис
н н
полиацетилен
Рис. 21.9. Образование полиэтилена (а) и полиацетилепа (б).
ацетилена с тройными связями — схематически показано на
рис. 21.9.) Однако в действительности полиацетилен обнаружи-
обнаруживает весьма «скромную» электропроводность, лежащую в ин-
интервале от Ш"э (Ом-см) (цис-форма) до 10"Е (Ом-см)
(транс-форма), что сравнимо с электропроводностью такого по-
полупроводника, как кремний.
Уровень проводимости полиацетилепа оказывается низким
потому, что в отличие от графита в его сопряженной системе
электроны ие полностью делокализованы. (Ширина запрещен-
запрещенной зоны составляет 1,9 эВ.) Однако введение электроактив-
электроактивных неорганических добавок в полиацетилен вызывает резкое
увеличение его проводимости. Вещества — акцепторы электро-
электронов (Вг2, SbFs, WF6, H2SO4) или типичные доноры (щелочные
металлы) —• повышают электропроводность гранс-полиацетиле-
280 21. Органическая химия твердого тела
на до 103 (Ом-см)-1. Эта величина проводимости сравнима
с проводимостью многих металлов, что дало основание назвать
эти материлы «синтетическими металлами». На начальном
участке зависимость проводимости от концентрации добавки
имеет чрезвычайно резкий характер: переход от полупроводни-
полупроводникового уровня к металлическому происходит при содержании
добавки ~1-г5 мол. %. Дальнейшее увеличение содержания
добавки (до ~ 10 мол. %) вызывает более плавное увеличение
проводимости.
Синтез. Полиацетилеи получают путем каталитической поли-
полимеризации ацетилена в отсутствие кислорода, используя для
этого катализатор Циглера — Натта, представляющий собой
смесь А1(СН2СН3)з и Ti(OC4HaL. Для этого газообразный аце-
ацетилен барботируют через раствор катализатора; полиацетилеи
выделяется в виде осадка. В некоторых случаях полимеризацию
проводят, пропуская ацетилен по стеклянной трубке, внутрен-
внутренняя поверхность которой покрыта тонкой пленкой катализатора;
при таком способе полиацетилен образуется в виде слоя на ка-
каталитической поверхности.
Соотношение цис- и гранс-форм в полимере зависит от тем-
температуры полимеризации. Транс-форма, будучи более устойчивой,
преобладает при более высоких температурах (~100°С), цис-
форма — при более низких (около — 80 °С); при комнатной тем-
температуре продукт представляет собой смесь этих изомеров.
Обычно стремятся получить траяс-форму, поскольку она имеет
более высокую проводимость, и достигают этого либо путем
проведения полимеризации ацетилена при 100 °С, либо путем
нагревания цис-транс-смесн до <~-150°С (в этих условиях
ц«с-форма легко переходит в гракс-изомер).
Введение добавки можно осуществить просто, выдерживая
полиацетилен в среде газообразного или жидкого легирующего
реагента. Происходящие при этом реакции подобны хорошо из-
известным реакциям образования соединений включения графита
(гл. 2), при которых молекулы или ионы внедряются между
слоями атомов углерода; электропроводность графита меняется
в зависимости от того, увеличивают или понижают внедренные
частицы концентрацию электронов в зоне проводимости графи-
графита. По отношению к полиацетилену такая добавка, как бром,
играет роль акцептора электронов, что можно отразить с по-
помощью формулы (СН)л6+Вг°~. В этом соединении, по-видимому,
происходит частичный или полный перенос заряда от двойных
связей полиацетилена к атомам брома, но следует заметить, что
до конца электронное строение полиацетилеповых пленок еще
не выяснено. В первую очередь не понятен механизм переноса
электронов от одной полиацетиленовой молекулы к другой. Этот
вопрос необходимо рассматривать, учитывая морфологию пле-
21,2. Электропроводящие органические твердые тела 281
нок полимера, которая весьма сложна: полиацетиленовые цени
сворачиваются в плоские диски, наслоения которых формируют
волокна. Потребуется еще очень большая работа по изучению
текстуры полиацетиленовых пленок, чтобы в дальнейшем, конт-
контролируя ее, можно было бы сказать, какая морфологическая
организация пленки необходима для достижения высокой элек-
электронной проводимости.
Применение. Потенциальное применение проводящего поли-
ацетилена весьма разнообразно, хотя пока еще (до 1983 г.) он
не используется в каких-либо производимых промышленностью
устройствах. По аналогии с традиционными полупроводниками
его можно использовать для изготовления диодных выпрямите-
выпрямителей, работающих на р — я-переходе. В этих устройствах исполь-
используют контакт двух пленок, одна из которых легирована акцеп-
акцептором электронов и, следовательно, имеет проводимость р-типа,
а другая — донором (л-проводник). Технология изготовления та-
таких элементов должна быть простой, а возможность получения
пленок с большой площадью поверхности позволяет планиро-
планировать их применение для преобразования солнечной энергии
в электрическую. Однако, прежде чем это произойдет, предстоит
решить проблему чувствительности нолиацетилена к кислороду.
Возможно, что в будущем появятся замещенные или модифи-
модифицированные аналоги полиацетилена, которые, сохранив высокую
проводимость, не будут подвержены агрессивному воздействию
атмосферы.
До сих пор обсуждалась только электронная проводимость
полиацетилена. Однако недавно было показано, что в таком
полимере возможна также и ионная проводимость, благодаря
чему некоторые легированные полиацетилены можно использо-
использовать в качестве материала обратимых электродов в химических
источниках тока нового поколения. Легирование нолиацетилена
в таком случае осуществляют электрохимически. В одном из ва-
вариантов с этой целью полиацетиленовую пленку погружают
в жидкий электролит, например в раствор LiC104 в пропилен-
карбонате. Другим электродом, который также погружен в элек-
электролит, является металлический литий. При зарядке этой ячей-
ячейки от внешнего источника тока напряжением 1,0 В при комнат-
комнатной температуре перхлорат-ионы «входят» в полиацетиленовын
электрод и реагируют с ним, образуя продукт состава
(С\ 1)у+(СЮi),, , где О^г/^0,06. Ионы Li: в это время восста-
восстанавливаются на литиевом электроде, сохраняя тем самым элек-
электронейтральность жидкого электролита. Введение перхлорат-
ионов в структуру полиацетилена обратимо; это означает, что
прн последующем разряде ячейки они могут диффундировать
из полимера в электролит. Полиацетилеп проявляет в этих про-
процессах свойства электрон-ионного полупроводника.
282
21, Органическая химия твердого тела
//Л //Л //\ //
Возможность использования полимерных электродов в хими-
химических источниках тока, в особенности полностью твердотель-
твердотельных, является чрезвычайно привлекательной по двум причинам:
1) полимеры имеют малую плотность (по сравнению со свиниом
в свинцово-кислотных аккумуляторах) и 2) полимеры обладают
большой пластичностью, которая создаст предпосылки уменьше-
уменьшении контактного сопротивления па границе электрод—твер-
электрод—твердый электролит.
21.2.1.2. Полипарафенилен. Другим перспективным длинноцеп-
ным полимером с полупроводниковыми свойствами является по-
а лнпарафепилен (рис.
21.10, а), длинные це-
цепи молекул которого
содержат бензольные
кольца. Хотя это ве-
вещество и не изучалось
столь широко, как по-
полиацетилен, тем не ме-
менее его проводимость
удалось сильно улуч-
улучшить путем легирова-
легирования. Например, полу-
чаемый при добавле-
добавлении FeC!3 продукт с
ориентировочным составом (С6Н4(РеС!з)о.1б)х обладает при
комнатной температуре проводимостью 0,3 (Ом-см)-1.
21.2.1.3. Полипиррол. Пиррол представляет собой гетероцикли-
гетероциклическое соединение с пятичленными кольцевыми молекулами
C4H5N. При его полимеризации образуется длинноцепочечный
полимер, в котором чередование двойных и одинарных связей
обеспечивает делокализацию л-электронов (рис. 21.10,6). Про-
Проводимость полипиррола как такового невелика, но при окисле-
окислении перхлоратом она возрастает до 102 (Ом-см)-' и имеет ды-
дырочный характер. Полипиррол имеет по сравнению с другими
проводящими полимерами преимущества, так как он устойчив
на воздухе и выдерживает нагревание до 250 °С.
21.2.2. Органические комплексы с переносом заряда.
Новые сверхпроводники
В течение целого ряда лет химики проявляли большой инте-
интерес к двухкомпонентиым органическим системам, содержащим
донор я-электронов и акцептор. Некоторые из таких систем об-
обладают высокой проводимостью, а в некоторых других (впро-
Рис. 21.10. Полипярафсиилои
пиррол (б).
(а) и поли-
21.2. Электропроводящие органические твердые тела 283
чем, немногочисленных) обнаружена сверхпроводимость при
очень низких температурах. Стремление получить материалы,
обладающие сверхпроводимостью при 50—100 К, служило
одним из главных стимулов развития исследований комплексов
с переносом заряда.
Примерами сильных акцепторов л-электроиов являются тет-
рацианохинодиметан (ТЦХ) и хлоранил, а примерами сильных
доноров я-электронов— парафенилендиамин (ПД), тетраметил-
парафенилендиамип (ТМФД) и тетратиофулвалеи (ТТФ)
а б
a а
R = Н.СНз
Рис. 21.11, Тетрацианохииодиметан (ТЦХ) (а), х.юранш (б) и /г-фениленди-
амины (е) и тетратиофулвалеи (г),
(рис. 21.11). В кристаллических я-комплексах чередующиеся
(как правило) молекулы донора и акцептора образуют подобие
штабеля. При этом электронные я-облака соседних разнородных
молекул перекрываются, что позволяет осуществить перенос за-
заряда вдоль направления укладки штабеля. Например, ТМФД н
хлоранил, чередуясь образуют молекулы вида: (ТМФД)+ (хло-
(хлоранил)- (ТМФД)^ (хлоранил)- и т. д. Существуют, кроме того,
комплексы, в которых только один из компонентов содержит
ароматическую я-электронпую систему. Так, ТЦХ образует
комплексы с переносом заряда со щелочными металлами, на-
например (TUXKCs2, а ТТФ —с галогенами: (ТТФJВг.
Большинство органических комплексов с переносом заряда
обладает полупроводниковыми свойствами, однако встречаются
н такие, проводимость которых очень велика, в частности в си-
системах ТТФ —ТЦХ и NJV№ — ТЦХ (КТМФ = N-метилфеназин)
в~\03 (О.М'См), а комплекс (ТТФJВг при низкой температу-
температуре (<4,2 К) и высоком давлении B5 кбар) проявляет сверх-
сверхпроводящие свойства.
284 21. Органическая химия твердого тела
Литература
1. Bloor ?>., Plastics (hat conduct cleciricity, New Scientist, 1982, 577—580.
2. Cohen M. D., The photochemistry of organic solids, Angcw. Cheniie Int. Ed.,
14, 386—393 A975).
3. Cohen M. D., Green B. S., Organic chemistry in the solid state, Chem. Brit.,
9, 490—497 A973).
4. Jones W., Thomas I. AI., Applications of electron microscopy to organic solid
state chemistry, Progr. Solid St. Chem., 12, 101—124 A979).
5. Nigrey P. J., Maclnnes D.. Kairns D. P., MacDiarmid A. G., Lightweigt rechar-
gable storage batteries using polyacetylcne (CH2), as the cathode active
material, J. Electrochera. Soc., 128, 1651—1654 A981).
6. Schmidt G. M. J., Photodimcrisafion in the solid state, Pure and Applied
Chemistry, 27, 647—678 A971).
7. Seymour R. В., Conductive Polymers, Plenum Press, 1981.
8. Thomas J. M., Topography and topology in solid state chemistry, Phil. Trans.
Roy Soc, 277, 251—286 A974).
9. Thomas J. M., Moresi S. E., Desvcrgne J. P., Topochemical phenomena in
organic solid slate chemistry, Adv. Phys. Org. Chem., 15, 64—151 A977).
Приложение
А1. Геометрические подходы в кристаллохимии
А 1.1. Замечания по геометрии тетраэдров и октаэдров
А.1.1.1. Тетраэдр и куб. На рис. А1.1,я изображен куб с разметенным
внутри тефаэдром; псптри тетраэдра и куба совпадают, i вершины тетра-
тетраэдра являются одновременно четырьмя противоположными вершинами ку-
куба А'ь Xi, А'а и Xi. Используя эти сведения, сравнительно легко проводит:)
расчеты, связанные с геометрией тетраэдров.
Л1.1.2. Расстояния М—X а X—X в тетраэдре. Обозначим ребро куба /. По
теореме Пифагора отрезок X—X — диогональ грани куба—имеет Длину
1}!2. Отрезок М—X равен половине объемной диагонали куба, т. е. М—X —
/УЗ/2. Следовательно, отношение ХХ/МХ=^2~-Л/2уЗ~=У8/3=1,633. В структу-
структуре оксида крсмчия основным структурообразующим фрагментом является
тетраэдр SiO4. При этом расстояние Si—О обычно составляет -—-1,62 Д.
Тогда расстояние О—О, совпадающее с ребром тетраэдра, равно
~l,633(Si—О) «2,65 А.
А1.1.3. Угол ХМХ тетраэдра. Согласно теореме косинусов (рис. Al.l.c и б),
(Xj — Хпу = (М — Хху + (/И - X,J — 2 (М — А-,) (М — Х2) cos г. Хг
Учитывая предыдущий резулглат, запишем
[1,633 (М—XJ]2 =2 (.И — ХХУ~ — 2 [М — Х^чаа Z
откуда Z А' МХ= 109,48°.
ALIA. Симметрия тетраэдра. Тетраэдр имеет оси вращения третьего поряд-
порядка, проходящие через отрезки М—А", т. е. вдоль каждой объемной диаго
пали куба. Таким образом, и куб, и тетраэдр обладают четырьмя осями
третьего порядка. У куба также есть оси четвертого порядка, проходящие
через каждую пару противоположных граней. Для тетраэдра эти оси 4-го
порядка становятся инверсионными осями 4 (т. е. иращение на 90° сопро-
сопровождается инверсией относительно центра тетраэдра).
Л1.1.5. Центр масс тетраэдра. Во-первых, необходимо знать высоту центра
масс (точки М) над треугольным основанием тетраэдра. Рассмотрим се-
сечение Х\Х%М (рис. Al.1,6). Точка У, продолжение линии Xi—AJ, лежит в
центре основания X}XiXt (на рис. А 1.1, а не изображено). Следовательно,
286
Приложение
Рис. А 1.1. Взаимосвязь между тетраэдром и кубом.
расстояние М—Y дает иысоту центра масс над основанием. Поскольку
L.XxbAX2-= 109,48°, то
', =-35,26°
ZMX,X2 =•
и соь
Значит,
2, т. е.
Х1У= 1,155'
УМ = А',У - Л,,М =0,259/
YM/yX: = 0,2891/1,155! = 0,25
т. е. центр масс делит высоту тетраэдра в отношении 1 : 3.
At.1.6. Октаэдр и куб. На рнс. Л1.2 изображен октаэдр, вписанный в куб,
при этом их центры совпадают, а вершины октаэдра находятся в центрах
Рис. Л 1.2. Взаимосвязь между кубом и октаэдром.
А2. Построение моделей
287
граней куба. Если длину ребрл кубп обозначить /, то расстояние М—.Y=
= 1/2. По теореме Пифагора А'—A' = /j.
Октпэдр имеет три оси вращения 4то порядки, параллельные каждому
из ХМХ-иаирав.чеина. Эти оси совпадают с осями 4то порядкп куба, про-
лодящими через его противоположные грани. Оси третьего порядка, про-
лодящие ъ кубе через противоположные вершины вдоль объемных диаго-
диагоналей, в октаэдре пронизывают попарно противолежащие грани.
А 1.2. Гексагональная элементарная ячейка.
Доказательство того, что с/а — 1,633
При ге;;с.1го"олышй плотной упаковке атомы рпеполагяютгя ь верши-
/ 1 2 1\
нах элементарной ячейки и пиутрп пес в точке с координатами ['%> ~"> ")
,Рнс. 7!ij. Вдоль репер а и Ь атомы касаются, так как величина а ранна
Рис. AL3. Гскспгопальнпя v
ячейка; с/а~ 1,633.
диаметру атома. Как показано па рис. AL3, два тетраэдра имеют общую
/ 1 2 1\
вершину-—атом, расположенный б точке =", -тт, тг . Следовательно, вели-
\ 6 л г ]
чипа с в точности равна удвоенной высоте такого тетраэлря, т, с.
_? 9.ХjY 2-1,155/ _
а ~ Х,Х> ~
Л2. Построение моделей
Рассмотрим изготовление дву.ч типов моделей, основанных на шарах
и полиэдрах.
Необходимые принадлежности.
Шары ivi полистирола A00—200 шт\к) любого диаметра, по диаметр 30 мм
наиболее \добсп.
Растворитель E — 10 мл) в бутылочке с капельницей или кисточкой (мож-
(можно испемьзоиать хлороформ).
Плотная бумага B—3 м2), желательно рязнотгнотная; клей.
288
Приложение
А2.1. Характер упаковки шаров
Для соединения полистиролышх шариков можно использовать клей,
однако работать с ним труднее, чем с растворителем, в котором растворя-
растворяется полистирол. Небольшое количество растворителя наносят на поверх-
поверхность шарика, после чего к этому месту прижимают второй шарик. Если
нанесенная капля растворителя слишком велика, она может растекаться
ло поверхности и испортить ее внешний вид. Обычно шарики слипаются
сразу же при соприкосновении, однако полностью контакт затвердевает
Рис. А2.1. КПУ и элементарная ГЦК-ячейка.
лишь через несколько часов. Поэтому большие трехмерные модели лучше
собирать в несколько стадий.
Сравнительно просто первоначально склеить из шариков плотноупако-
ванные слои (рис. 7,1) и затем наложить их друг на друга в гексагональ-
гексагональную (рис. 7.2) и кубическую (рис. 7.3) последовательности. Более деталь-
детальные указания, по-видимому, излишни; лучше опишем два полезных упраж-
упражнения.
А2.1.1. Демонстрация связи между КПУ-структурой и элементарной ГЦК-
ячейкой. Склейте вместе 6 шариков, чтобы получить изображенный на рис.
А2.1,и фрагмент плотпоуиаконанного слоя. Поместите один шарик вп вто-
второй слой, как показано па рис. А2.1,б.
Повторите операции а л б так, чтобы полу-
получить две идентичные группировки атомов.
После высыхания клея сориентируйте одну
модель, как показано m e, и расположите
лад ней вторую, как показано на г, В рс-
зулыгие получится кубическая гранецент-
рированная элементарная ячейка с шарами
в углах и центрах граней (рис, 7.5, а). Сле-
Следовательно, ГЦК ячейка содержит плотно-
упакованные слои, параллельные плоско-
плоскостям A11) элементарной ячейки. При ус-
установке модели па одну из вершин так, что
плотноупакованные слои располагаются го-
горизонтально, становится ясно, что порядок
укладки слоев соответствует последователь-
последовательности ЛВС.
Рис. А2.2. Плотноупоноваши
слои п КПУ структуре.
А2.1.2. Демонстрация четырех ориентации плотноупакованных слоев в эле-
элементарной ГиК-ячейке. Основное различие между КПУ и ГПУ заключа-
заключается в том, что в последней только один из плотноулаковамных слоев p?w-
положен параллельно плоскости основания гексагональной элементарной
ячейки. В КПУ плотиоупаковаиные слои встречаются в четырех ориент;:-
1шях. Это можно показать путем сооружения пирамиды (рис. А2.2). Сна-
A3. Как узнать плотноупакованные структуры? 289
чала сложите из 6 шаров треугольное основание, как на рис. А2.1, затем
добавьте три тара в слой, расположенный н;|Д ним, и один шар в верши-
вершину. Проверьте, что последовательность слоев ABC, т. е. соответствует КПУ.
Пирамиду можно теперь повернуть так, чтобы любая из трех ее других
граней легла в основание, в результате чего возникает идентичная струк-
структура. Четыре эквивалентные ориентации пирамиды соответствуют, таким
образом, четырем ориентациям нлотноупакованпых слоев в КПУ-структурс.
А2.2. Полиэдрические структуры-
Тетраэдры, октаэдры и любые другие полиэдры можно изготовить из
плотной бумаги. Пользуясь выкройками (рис. А2.3), скопируйте их, вы-
вырежьте, согните и склейте полиэдры. Для изготовления достаточно проч-
Рис. А2.3. «Выкройки» для изготовления тетраэдра и октаэдра,
ны\ полиэдров длину ребра следует сделать равной -~5 см. Полиэдры
можно соединять друг с другом, склеивая их вершинами, ребрами или
гранями. Склейка полиэдров вершинами не обеспечивает достаточной проч-
прочности, однако ее все же можно сделать при наличии времени н терпения.
Различные варианты компоновки полиэдров изображены на рис. А2.4.
A3. Как узнать плотноупакованные (эвтактические)
структуры?
Когда встречается новая или незнакомая структура, обычно возникает
вопрос: является ли она плотиоупакованной? Определенного ответа часто
нельзя дать, пока не построена трехмерная модель. Ниже даются некоторые
рекомендации для определения того, является ли структура плотпоунако-
ваммой; предложить более четкие и строгие правила затруднительно.
1) Для неметаллических плотноупакованных (или эвтактических) струк-
структур с общей формулой A.vB,y атомы одного из элементов (А или В) должны
образовывать плотную упаковку.
2) Координационные числа, определяемые числом атомов В, окружаю-
окружающих атом А, и наоборот, часто равны 4 » 6, однако возможны величины
от 2 до 8.
Первое правило должно иметь силу п эвтакгичсской плотноупаконин-
поii структуре; чтобы понять расположение, одинаковых атомов часто на-
надо представить себе более одной элементарной ячейки, что бывает од-
однако трудно. Наилучший, но мнению автора, тест заключается в рассмот-
рассмотрении одного плотноупаковлшюго слоя, т. с. следует выбрать центральный
атом и определить шесть равноудаленных от пего атомов этого же э-ie-
19—1428
290
Приложение
Рис. А2.4. Некоторые способы соединения
полиэдров.
мента, образующих гексаго-
гексагональное окружение в рассмат-
рассматриваемом слое (рис. 7.1). Пос-
После того как такой слой иденти-
идентифицирован, обычно довольно
легко найти шесть других ато-
атомов к слоях с каждой сторо-
стороны. Таким образом, будут рас-
рассмотрены 12 соседей, что и
требуется в плотпоупнкиваиной
структуре. Часю в структуре
можно видеть разные слои, из
которых часть является плот-
поупаковаппыми, а часть —
пет. После удовлетворения вы-
вышеуказанного критерия о числе
идентичных соседей в гглотно-
упакованном слое, часто мож-
можно выделить два типа слоев,
в которых ненлотноупакован-
пые слои могут содержа п. как
атомы А, так и В, тогда как
плочноупаковапные слои содер-
содержат атомы только одного сор-
сорта (если не обра эуются сме-
смешанные плотноупакованные
слон, как в перовектгте). Так,
грани элементарной ячейки в
структуре каменной соли могут
рассматриваться как слои ато-
атомов, по эти с.™и не. характери-
характеризуются плотной упаковкой, так
как. во-первых, каждый ион
Na + (и.чи С1~) соседствует
лишь с четырьмя ионами Na'
(или Ct~) в слое и, во-вторых,
слои содержат как ноны NaH,
так и (",] -. Напротив, слои, па-
рал.1елын>:е плоскостям {111},
в структуре каменной соли ян
ляются плотноупагловапныыи,
так как каждый ион С1~ (или
Na + ) имеет шесть таких же
соседних ионов, эти слои со-
содержат ионы только одного
пида и чередуются со слоями
nvnoii другого вида,
чш при плотной упаковке лто-
п октаэдрпчеекпе межузелыше
А или 6.
Вторие правило следует из того фактп,
мов А атомы В занимают тстраэдрптеские
позиции. Следовательно, К11 ато.чои В ранно
Возможность др\гих координационных чисел возникает, когда рассматри-
рассматривается координационное число ;пимов Л, k;iic это п. гцоетрируют несколько
следующих примеров.
1) Na2O (антифлюорпт) имеет КПУ кислородных ионов с тетраэдричс-
ской коорднпанпен для Na'1'. Кислородный поп находится н
восьми ионов \'п + .
Л4. Положительные и отрицательные координаты атомов
291
2) Ni,\s имеет ГПУ ионов As2- с октаэдрически координированными
ионами Ni2+. Мышьяк координирует вокруг себя б ионов \Ч2+, образую-
образующих тригоняльную призму.
3) Си2О (куприт) содержит КПУ ионов Си+ и тетраэдрические О2~-
ионы. КЧ Си+ (окружение из ионов О2-) равно 2.
Итак, для того чтобы болсц полно понять и шлссифициронить. струк-
структуру /УхЪу, необходимо проверить:
1) характер упаковки атомов А;
2) характер упаконки атомов В;
3) координационное число атомов А и
•1) координацию атомов В.
А4. Положительные и отрицательные координаты атомов
Координаты атомов в кристалле могут быть выражены через соответ-
соответствующие параметры кристаллической решетки. Обычно считают, что ато-
атомы, находящиеся внутри некоторой данной элементарной ячейки, имеют
атомные координаты от 0 до 0,999. Если координаты какою-либо атома
отрицательные или >1, то такой атом принадлежит соседней элементарной
ячейке. Рассмотрим рис. А4.1, где изображены четыре элементарные ячей-
ячейки. Например, выделим правую верхнюю элементарную ячейку; начало ко-
i -
¦ 2 2 1
о
о
т
I
о
1 I
2 2
О
г-t-
I
о
1 1 .
2 2 "'
О
Рис. А4.1. Положительные и отрицательные координаты атомов.
ординат в ной обозначено с помощью черного кружка, а стрелки указы-
указывают положительные направления соответствующих' осей. Указаны коорди-
координаты о.-шоп пары узлов, расположенных в центрах противоположных грл-
19*
292
Приложение
ней каждой из четырех ячеек. Видно, что все узлы, принадлежащие двум
правым элементарным ячейкам, имеют координату х— -п~ относительно выб-
выбранного начала координат. В левых элементарных ячейках аналогичные
узлы имеют координату х——-к-. Координата у всех изображенных на
рмс. А4.1 ячеек одна и та же (+1/2). Зато значения координаты z в зави-
зависимости от принадлежности узла к топ пли иной ячейке могут быгь I, О
или —1. Исходя из вышесказанного, можно сделать вывод о том, что к
выбранной элементарной ячейке относится лишь один узел с координата-
координатами 2~^' ^се осталыше изображенные позиции принадлежат соседним
элементарным ячейкам.
А5. Кристаллографические точечные группы
Как указывалось в гл. 6, всего имеется 32 точечные кристаллографиче-
кристаллографические группы. На рис. А5.1 изобрлжены стереографические проекции всех
этих точечных групп. Использованные обозначения отвечают тем, которые
приняты в «Интернациональных таблицах» (International Tables lor Crys-
Crystallography). Для точечных групп нскубнческих кристаллографических си-
систем на каждую из проекций нанесены как элементы симметрии, так п
система эквивалентных позиций. Что касается точечных групп кубической
системы, то для их изображения используют по два разных рисунка — один
с элементами симметрии, второй с системой эквивалентных позиций.
триклииные
моноклинные
ромбические
А5. Кристаллографические точечные группы
293
теграгональные
4 mm
42 т k/mmm
фигональные
кубические
Рис. А5.1. Кристаллографические точечные группы.
Аб. Межплоскостные расстояния и объемы элементарных ячеек
Значения межплоскостных расстояний d — кратчайших расстояний между
соседними плоскостями данного семейства плоскостей (hkl), а также объем
294 Приложение
элементарной ячейки V можно рассчитать по следующим формулам:
1 /г2 + к1 4- P
Кубическая ячейка ~м = ~г ; V = а3
1 h2 -Ь к2 I2
Тетрагональная ячейка -jj~ = -j -j- -jjf V=a:c
1 /i2 ?2 ^
Ромбическая ячейка -рГ = ~г~ + ТГ" + Г" V-=abc
1 4 //i2 -f /г/г -;-?2\ Р
Гексагональная ячейка *~рт =¦• —^- ; + "Тг~
A2 A-2 sin2 p /2 2WcosE
у ^ + р + ~Г ~ ^
V = aic sin E
Триклинния ячейка -^~ = -pj- [/гг62с2 sin2 а + Pate2 sin2 (i -f /2а2й2 sin2 у
+ Ihka'-c1 (cos a cos 8 — cos 7) -|- 2k!a2bc (cos 8 cos 7 — cos a)
-f- 2hla'j2c (cos a cos у — cos P)]
V = doc A — cos2 a — cos2 6 — cos2 7 -J- 2 cos ct cos 6 cos
A7. Обратная решетка
В гл. 5 обсуждалось рассеяние рентгеновских лучей кристаллическими
веществами; при этом привлекались такие понятия, как элементарная ячей-
ячейка, тип кристаллической решетки, симметрия кристалла, закон Брэггп и др.
Концепция плоскостей кристаллической решетки предусматривает сущест-
существование нескольких семейств плоскостей, различным образом ориентирован-
ориентированных в пространстве и характеризуемых разными значениями межплоскост-
межплоскостных расстояний. При выводе закона Брэгга принималось, что рентгеновские
лучи дифрагируют или отражаются от этих плоскостей решетки. Таком
подход обеспечивает описание дифракции рентгеновских лучей в реальном
пространстве. Согласно закону Брэгга, направления распространения пер-
первичного и отраженного лучей связаны друг с другом величиной межтос
костного расстояния и длиной волны рентгеновского излучения.
Во многих случаях, однако, удобно и полезно описывать явление ди-
дифракции па языке представлений обратной решетки. Обратное пространстве;
и обратные решетки не являются некоторыми физическими рсалпями. Это
скорее система представлений, позволяющая успешно описывать явления
дифракции в кристаллах. Цель настоящего приложения состоит в следую-
следующем:
во-первых, необходимо показать, как можно получить обратную ре-
решетку из соответствующей кристаллической решетки, существующей в ре-
реальном пространство'.
во-вторых, следует шляеппть. как выглядят в обратном пространства
разные типы кристаллических решеток, открытые элементы симметрии, раз-
различные типы кристаллографических систем н др.;
в-трегьих, нужно составить представление о той, как можно описы-
описывать явленно дифракции в терминах обратной решетки.
А7. Обратная решетка 295
A7.I. Изображение кристаллической решетки в реальном
и обратном пространствах
Связь между изображением кристаллической решетин в реальном и об-
обратном пространстве удобно рассмотреть на примере моноклинной элемен-
элементарной ячейки (рис. A7.i). 11а рис. А7.1,к изображена проекция элемен-
элементарном ячейки на плоскость ас. Поскольку ось b япчястся особой осью, то
по определению угол 13^90°. Ось Ъ перпендикулярна плоскости рисунка,
так как а=^ = 90*°. На рис. А7.1.а изображены плоскости решетки A00)
и @01) и соответствующие ыежплоскостиые расстояния ^юэ и cf0oi. Отме-
Отметим, что поскольку р^90°, то йюоч'-а и doo\??c. Начало координат выбран-
выбранной элементарной ячейки находится в точке А'.
Как показано на рис. А7.1.6 каждое из семейств плоскостей (ЮП),
(ОП!) и т. д. изображается в виде одной-едпнетсешюй точки. Эги точки
па.\одя1ся ни расстоянии l/d (для каждой из рассматриваемых плоскостей)
от начала координат в обратном пространстве. По отношению к началу ко-
ординг.т они расположены в направлении, перпендикулярном направлению
расположения семейства кристаллографических плоскостей в реальном про-
пространстве. Направления, перпендикулярные плоскостям A00) и @01),
обозначены нп рисунке а' и с" п называются осями обратной решетки
Чтобы отличать оси и углы в реальном и обратном пространстве, для обо-
обозначения этих элементов в последнем случае используют звездочки. Заме-
Заметим поп\тко, что в данном конкретном примере оси обратной решетки а"
и :' непараллельны осям а а с кристаллической решетки в реальном про-
пространстве. Причиной этого является то, что C=^90°. В то же время оси b
и b параллельны друг Другу (н перпендикулярны плоскости рисунка), так
как а=ч =90°.
Кажлос семейство плоскостей кристаллической решетки имеет спонм
отображением точку в обратной ренк-ткс. Можно показать, что r случае
большого числа семейств плоскостей в обратном пространстве возникает
совокупность точек, образующих трехмерную решетку (обратную решетку).
На рис. А7.1,« изображено сечение обратной решетки, состоящее из боль-
большего числа отдельных точек. Элементарная ячейка в обратной решетке ны-
делена сплошными п штриховыми линиями. Вершинами выбранной ячейки
га проекции А7.1 служат точка начала коордикит м узлы обратной решег-
ки с координатами 100, 001 и 101. Вдоль каждого in направлений обратной
решетки па равном расстоянии друг от дрчга расположены \уы атой ре-
шеткп. Поскольку duc2 = "rfy.ii. то узел 0П2 \дилен от пятила координат па
расстояние, в два раза превышающее расстояние от начала координат до
узла 001. Координаты узлов обратной решетки могут бьпч. как положитель-
положительными, т л "С п отрицательными, и, следовательно, точка начала координат рас-
расположена и центре решетки.
До с,ix пор рассматривались лить такие семейства плоскостей кристал-
кристаллической решетки, которые параллельны Ь, т. с. с индексами типа {liQl).
Узлы обратной решетки, отвечающие этим плоскостям кристаллической ре-
решетки, лежат в одном и том же сечении обратной репктки (рис. А7.1,о).
Чтобы представить себе полную картину обратной решетки, необходимо
дополнить ее другими сечениями, индексы (hid) которых будут иметь нену-
ненулевые значения k. Б этом случае возникает трехмерная решетка в обрат-
обратном пространстве, состоящая из \злов, отвечающих плоскостям кристалли-
кристаллической решетки. Таким образом, сечение обратной решетки, изображенное
на рис. А7.1.е, представляет собой нулевой слой (/с=0). Следующий слой,
расположенный над данным сечением и имеющий в примитивной решетке
точно такое же изображение, будет иметь индексы lill; по аналогии со
сказанным слой, находящийся под данным сечением, будет иметь индексч
lill. Эти слои называются первым слоем л т. д. Таким образом, обратную
296 Приложение
, A00)
1100)
а*= /rf
loo
(?* = (8О"-C
• 201
• 201
201
201
зоо
20?
102
102
Рис. Л7.1. а —проекция моноклинной элементарной ячейки на плоскость ас;
С — точки обратной решоткн, отиечаюше'л i.ie\ioiiT-'j|>iion ячоичс. пзопрлжсп-
ной на рпс. а; о — большее число узлов той же др\ мерной обратной ро
П1СТКЛ.
А7. Обратная решетка
297
решетку можно рассматривать как бесконечную во всех направлениях. На
практике размеры обратной решетки ограничивают некоторым минималь-
минимальным значениям межплоскостного расстояния d (максимальной величиной
\jd).
А7.1.1. Доказательство того, что обратная решетка является трехмерной
совокупностью узлов. Чтобы доказать сформулированное положение, доста-
достаточно показать, что всем семействам плоскостей hkl с одним и тем же зна-
значением h отвечают такие точки обратного пространства, которые образуют
один слой, перпендикулярный а. Каждому иному значению h соответствует
другой (причем каждому свой) слой узлов обратной решетки. Поскольку
Рис. А7.2. Построение обратной решетки.
такое же доказательство может быть применено к другим семействам пло
гкостпй с постоянными значениями k и I, то узлы обратной решетки долж-
должны образовывать периодическую трехмерную структуру.
Рассмотрим рис. Л7.2. Пусть одна и та же точка X будет центром ре-
решетки как в реальном, так и обратном пространстве. Пусть также ось х
кристаллической решетки вертикальна. Никаких предположений относитель-
относительно величины параметра а элементарной ячейки делать нет необходимости.
Показаны также две плоскости (/ и 2) одного семейства плоскостей hkl.
Плоскость / проходит через начало координат. Плоскость 2 пересекает ось
х в точке S. По определению индексов Миллера имеем
XS = а/А
Пусть точка 7.—узел обратной решетки, отвечающий семейству плоскостей
hkl. Тогда отрезок прямой XZ перпендикулярен плоскостям hkl, а его дли-
длина равна \}d:
XZ = \jd
В прямоугольном треугольнике XYZ (/_XYZ=§0°) катет XY равен
XY — XZ cos ф = -j- cos ф
298 Приложение
Величину cos <i> можно определить из треугольника XSF:
с us ф - А Т 'А Л' .-- d (a. h)
Поэтому
А'У ,_ 1.d\.i-(a h}\ =-. U а
Поскольку дли семейства плоскостей hid. при данном значении // обе вели-
величины, стоящие в правой тети данного уравнения (а и Л), постоянны, то
р.-.гстоянне XY также постоянная величина. Отрезок YZ перпендикулярен
отрезку XY, поэтому псем плоскостям hid с одним и тем же значением h
отвечают узлы Z обратной решетки, которые обр^.пют слой, перпендику-
перпендикулярный .г. Разным значениям h соотвелствмот разные слон узлов обратной
решетин. Так, например, дли к — О точка .S должна совпала! >> с точкой X. и,
следовательно, пулевой слон обратной решетин ОМ проходит через начало
координат А". Первый слой (Ш) пересекает ось х на расстоянии 1/п от па-
чала координат. Второй слой BН) пересекает ось х па расстоянии 21а от
точки и т. д.
До енх нор обсуждение не затрагивало расположения, отдельны\ илов
внутри одного и того же слоя обратной решетки. Однако эта задач., легко
решаема при рассмотрении семейства плоскостей с постоянными значения-
значениями к или /. Так, плоскостям Ш1, h\i, h2i и т. д. отвечают узлы обратной
решетки, находящиеся в слоях, перпендикулярных оси у. Эти \члы распо-
расположены друг от друга на расстоянии \jb. Аналогично узлы обратной ре-
решетки, отвечающие плоскостям МО, ЛИ и др., образуют слои, перпенди-
перпендикулярные оси z. Расстояния между узлами в этих слоях равны ]/с. Каждо-
Каждому из семейств плоскостей отвечает одна точка, которая как бы принад-
принадлежит сразу трем слоям обратной решетки. Например, точка 20( находится
во втором слое для плоскостей, перпендикулярных оси л\ в нулевом слое
для плоскостей, перпендикулярных оси //, и в перпом слое для плоскостей,
перпендикулярных осп г. "Поскольку узлы обратной решетки находится па
пересечении различных слоев и поскольку эти слои расположены регуляр-
регулярно в обратном пространстве, то обратная решетка представляет еооон ре-
регулярную трехмерную конструкцию.
Л7.1.2. Соотношение между параметрами кристаллической решетки и пара-
метрами обратной решетки. В предыдущем разделе было показано, чго уз-
узлы обратной решетки, отвечающие семействам плоскостей ОН, Ш, 2kl и
т. д., расположены в слоях, перпендикулярных осп Л" в реальном простран-
пространстве или оси а элементарной ячейки кристалла. Н зчих же слоя.ч
(Old. \kl и т. Д.) находятся осп Ь* и с* обратной решетки К такому выводу
можно прийти, рассмотрев, например, рис. А7.), if, па котором клобрпже
по гечеппе /id/ обратной решетки. Впчпо, что н этом сечешш раеппло/.и-пы
оси tr и с Поэтому
ось а перпендикулярна плоскости Ь'с*
аналогично ось Ъ перпендикулярна плоскости а*с*
а ось с перпендикулярна плоскости п^Ь*
и наоборот ось а"" перпендикулярна плоскости he
ось Ь* перпендикулярна плоскости не
ось с* перпендикулярна плоскости ah
Если все \глы кристаллической pcineiKit и обратной решетки прямые, т е.
а=Р"ру = а*--р ^"(*=90°, то соответствующие пари осей к реальном и об-
обратном пространстве параллельны, т. е. n'}a*, b']b\ с']с. В общем пиле со-
соотношения между имами и кристаллической решетке и в обратном прост-
Л7, Обратная решетка 299
рнпстве можно записать следующим образом: а+а*^180°, р+р* = 180°,
А7.2. Систематическое погасание рефлексов и обратная
решетка
В гл. 5 показано, что существование центрированных (непримптивных)
кристаллических решеток — причина систематического погасания определен-
определенных рефлексов на рентгенограммах кристаллических веществ. Каждый тип
решетки характеризуется собственной совокупностью отсутствующих реф-
рефлексов. Кроме того, наличие в кристалле открытые элементов симметрии - ¦
плоскостей скользящего отражения или нинтовых осей — тлкже ведет к ха-
характерному систематическому погасянию рефлексов. Рассмотрим, как это
систематическое погасание проявляется в обратной решетке. Прежде псеш
сведем одно допущение.
Как было показано ранее, рентгенограммы, получаемые при съемке мо-
монокристаллов, представляют собой непосредственно изображение обратной
решетки. Однако интенсивности каждой точки обратной решетки пропор-
пропорциональны интенсивности рентгеновских лучей, рассеянных на разных семей-
семействах плоскостей кристаллической решетки. Поскольку интенсивности рас
сеянии\ рентгеновских ;(\пен существенно меняются при отражении от раз-
различных плоскостей, то и интенсивности точек обратной решетки заметно
отличаются друг от друга. Чтобы ч обратной решетке выделить точки раз-
разной интенсивности, последние изображают в виде пятен различного размера
(см., например, рис. 3.2). Такое изображение называют взвешенной обратной
решеткой. Для настоящего изложения представляет интерес лишь сам факг
существования или отсутствия рефлексов на рентгенограммах вне зависимо-
зависимости от их интенсивности. Поэтому вес разрешенные рефлексы будут изобра-
изображаться в виде точек одинакового размера. Для того чтобы продемонстриро-
продемонстрировать трехмерность обратной решетки, на рис. Л7.3 предпринята попытка изоб-
изобразить па одной плоскости различные сечения обратной решетки. С этой
целью используются разные обозначения: маленькие темные кружки, большие
светлые окружности и крестики, которые относятся к различным сечениям
обратной решетки. Большинство обсуждаемых" вопросов рассматривается на
примере ромбических кристаллов, поскольку именно они характеризуются
наибольшим разнообразием типов кристаллических решеток (табл. 5.2).
H;i рис. А7'Л.ч изображена обратная решетка. <>гвсч;пош;'И примитив
поп npnei :).1лическоп решетке Р. В эгом c-i>час систематическое погаелнш'
рефлексов отсутствует. Следовательно, пулевой слом обратной решетки,
видимо, ничем не должен отличаться от первого слоя: более высокие сс-
чепия обратной решетки также имеют тот же вид, что и нулевой слон.
Начало координат обратной решетки имеет координаты 000.
H,i рис. А7.3,0 приведена обратная решетка, отвечающая объемпоцеш ¦
рпроп'п:нон решетке I. Условием появления рефлексов дли такой решетки
является: h-\-k-]-l=2n. Следовательно, в нулевом слое (темные кружки)
присутствуют такие рефлексы, как 200, 020, ПО и 220. В то же время та-
такие рефлексы, как ТОО, 010, 120 п 2!0, отсутствуют. В первом слое (свет-
лыс окружности) условия систематического погасания иные: в ->то.м сеч.'
mm обратной решетки такие рефлексы, как 01!. 10!. 121. 21!, присутству-
присутствуют, а рефлексы 20!, 021, 001, 111, 221 отсутствуют. Второй слой пмеег
тг.коп ;ке внешний вид, кяк и нулевой слон; третий слой аналогичен пер-
первому И Т. Д.
После внимательного изучения рис. A7 3.fi становится очевидным, что
возникающие па дифракционной картине рефлексы образуют граненептрп
ронянную решетку. До[ктнптелыю, легко мысленно представить себе, что
рефлексы 000, 200, 020, 220. 002, 202, 022 п 222 относятся к вершинам
• по *юо «по *Тго а) р
X 6*
• 010 *ооо »ою *ого -» • ^-а 1 г
• 1 Го *юо *по • I г о
• 2 Го *гоо • •
!• *
• т То
ООП
• ПО
О2Т1
СЮ
X
• 000
О501
• 2 00
• 110
Col 1
• 110
0211
0
• 020
О1 г 1
.•220
Ь'
6I
tiki h^k'l- гп
• 1= 0. 2 ¦¦¦
О1- \з
с
oTTi
ОМ!
О
X
• ооо
• 200
О
оТп
О'"
о
•
6"
• 020 ->
•
Q
либо
0 1--
'!
все четной.
0,2 ••
', 3 ¦
0111 «ТОО 0 111 »121
г) А
* Ь
ООП *000 ООП »020 -»¦
hkl кн- гп
ciTi вюо еш
О «200 0211 «220 • I- 0.2
О ( = 1,5
О • О •
Рис. А7.3 (см. продолжение).
В)Р
• •I0O «2 2 Э
• oic »Тк
• flOOO #T2O
• ПО I 'eOiO
• *ioo ' «ого
•210 «ПО
• 200 •
• = '.ВО - X ¦ 6 О"
» I ¦ 0.1.2 ¦
e)R
О О
XXX
•По »гю •
оог| оГо! огг)
хоТг XI iг х
• #ооо «2То
ОМ5 ' "ФОП С
X ХЮ2 ; X
#гТо 'впо «озо
О5?1 0201 C121
X X X
• «300 •
с с о
hkl -h.k-l^n
I - г
I • I
I» о
Рис. А7.3 (см. продолжение).
приложение
b Hi) с-плоскость скользящего
•00° • •- отражения 1 Ь
/• о.г
h 0.1.5
о
-i* 3} л-плоскость скользящего
~* отражения 1Ь
• i> о,г
Obi, 3 ¦
Рис. А7.3, Изображение различных типов систематического погасания в об-
обратных решетках.
грансцснтрироилиной ячейки, рефлексы 110 и 112 — к центрам верхней и
нижней граней, а рефлексы 011, 101, 121 и 211—к центрам боковых граней
этой ячейки. Существование такой гранецептрированноп ячейки в обратном
пространстве не имеет важного самостоятельного значения. Тем не менее
обнаруженный факт ее появления лает основание сформулировать не ппол-
не очевидное, но часто повторяемое утверждение: объемноцентрированной
решетке в реальном пространстве соответствует гранецентрированная ре-
решетка в обратном пространстве.
На рис. А7.3, о изображена обратная решетка, отвечающая грапецепт-
рировянной кристаллической решетке F. Условием существования рефлексов
на рентгенограммах веществ, имеющих такую решетку, является либо чет-
четность, либо нечетность всех индексов hkl для каждого из рефлексов. В ну-
нулевом слое присутствуют рефлексы 020, 200 и 220 и отсутствуют рефлек-
рефлексы 100, 010, 110, 120,210 и др. В первом слое присутствуют рефлекаы 111,
311, 131 и др., а рефлексы 101, 011, 121, 211 и др. отсутствуют. Имеющие
ся рефлексы образуют объемноцснтрированную решетку в обратном про-
пространстве. Следовательно, можно утверждать, что гранецентрированной ре-
решетке в реальном пространстве отвечает объемноцентрированная решетка
в обратном пространстве.
На рис. А7.3, г показана обратная решетка, отвечающая кристалличе-
кристаллической решетке, в которой центрирована только одна пара граней. Условием
Л7. Обратная решетка 303
с\ществовипия рефлексов на рентгенограммах веществ, имеющих кристал-
кристаллическую решетку А-типа, является четность суммы индексов k-\-l каждо-
каждого отдельного рефлекса. Выполнение этого условия приводит к тому, что
D каждом слое обратной решетки (рис. А7.3, г) отсутствует половин;) рядов
рефлексов, параллельных оси а*.
Единственным типом кристаллической решетки, который осталось обсу-
обсушить, является ромбоэдрическая решетка. Ее можно рассматривать как од-
одну из разновидностей гексагональной решетки. На рис. А7.3, д изображена
примитивная гексагональная обратная решетка. Она отличается от решет-
решетки, изображенной на рис. А7.3, я, только тем, что угол f* в ней равен 60°,
в то время как на рис. А7.3, a f =90°. Ось с", как и во всех предыдущих
рассмотренных здесь примерах, расположена перпендикулярно плоскости
рисунка.
Нл рис. Л7.3, е изображена ромбоэдрическая решетка, построенная па
тех же осях гексагональной ячейки а' и 6", что и в случае, приведенном
пл рис. А7.3, д. Однако две трети всех рефлексов рентгенограммы отсутст-
виот, так как условие существования рефлексов hid в данном случае за-
запишется так: —А + Л + /=Зя. Чтобы представить себе нею ромбоэдрическую
решетку и целом, необходимо рассмотреть три сечения обратной решетки
(при /-0, I, 2).
Ромбоэдрическая ячейка и обратном пространстве может быть построе
i:a на осях не гексагональной, а ромбоэдрической решетки. Вершины такой
ячейки образуют след\'ющие узлы обритой решетки:
нулевой слой: 000 _
первый слой: 101, 011,_П1
UTOport слой: 012, И2, Т02
третий слон: 003
На рис. 5.35,G приведен пример влияния наличия винтовых осей на ус-
условия систематического погасания. Если винтовая ось 2, параллельна
оси с, то единстненпымн рефлексами, на которых проявляется систематиче-
систематическое погасание, будут рефлексы типа 00/. Те из них, у которых I четное,
существуют в обратной решетке.
На рис. А7.3, ж и з приведены обратные решетки, отвечающие решет
им в реальном пространстве, которые содержат два типа плоскостей сколь-
скользящего отражения. Так, в случае, изображенном па рис. А7.3, ж плоскость
скользящего отражения с расположена перпендикулярно оси Ь. При отра-
отражении в плоскости с происходит смешение на с/2. Условие систематическо-
систематического погасания в этом случае проявляется в том, что в обратной решетке
(•рпсутстиуют те рефлексы с индексами /тО?, у которых L четное. Поэтому
в плоскости hOl отсутствует половина рядов рефлексов, параллельных оси
а'-. Если плоскость скользящего отражения с перпендикулярна оси а крис-
кристаллической решетки, то па рентгенограмме проявляются лишь те из реф-
рефлексов 0W, у которых 1-=2п.
На рис. А7.3, з показано влияние наличия диагональной плоскости
скользящего отражения п на системашческое погасание рефлексов. Пло-
Плоскость отражения перпендикулярна оси 6, период трансляции равен (а/24-
-fc/2). Условие существования рефлексов htil в этом случае запишется
так: h-\-l—1n, т. е. в плоскости ММ половина рядов рефлексов отсутствует.
Третьим типом плоскостей скользящего отпажения является плос-
плоскость d. Такие плоскости встречаются нечасто и только в системах с ром-
ромбической, тетрагональной и кубической симметрией. В ромбических кристал-
кристаллографических системах систематическое погасание проявляется лишь на
рефлексах типа hOl (и/или W0, 0к1). Условием существования рефлексов
bQt является: h-\-1=4п. В тетрагональных и кубических системах наличие
плоскостей скользящего отражения d типа сказывается на рефлексах hhl.
Для них условие существования рефлексов: 2ft+/ = 4«. Схемы обратных ре-
решеток, отвечающих решеткам в реальном пространстве, которые содержат
304 Приложение
плоскости скользящего отражения третьего типа, здесь не приведены. Они
могут быть построены тем же методом, что и другие обсужденные выше
обратные решетки (рис. Л7.3, а—з).
А7.3. Дифракция и обратная решетка.
Сфера отражения Эвальда
Результаты дифракции можно обсуждать, рассматривая отражение
рентгеновских лучей от плоскостей кристаллической решетки в реальном
пространстве или дифракционную картину, возникающую в обратном про-
пространстве. Первый из этих подходов был обсужден в гл. 5. Ниже развит
подход, основанный на применении представлений об обратной решетке.
На рис. А7.4, а показано семейство плоскостей hkl, характеризующееся
межплоскостным расстоянием d. Пусть точка X — начало координат обрат-
обратной решетки, а точка 7.—точка обратного пространств;), отвечающая пло-
плоскостям hkl. Отрезок YZ перпендикулярен XZ, угол ATZ=6:
sin G = XZ/XY = —
XY '
— dsinO
Согласно закону Брэгга, 7rfsin6 — 2Д, следовательно,
XY = 2Д
Таким образом, треугольник XYZ можно исиользовлть для иллюстрации
выполнения условий дифракции и закона Брэгга. Поскольку этот треуголь-
треугольник прямоугольный, то точки X, У и Z расположены па окружности с диа-
диаметром XY (рис. А7.4, б). Пусть исследуемый образец помещен в центр
окружности (точка 0). Тогда ОУ и 0Z соответствуют направлениям распро-
распространения первичного и отраженного рентгеновских лучей, н 0Z паралле-
параллелен XY'. Начало координат обратной решетки не совпадает с положением
самого образца. Однако совершенно непоггятпо, почему они должны совпа-
совпадать.
Дифракционные эксперименты организуются таким образом, что иссле
дуемый образец либо вращается, либо в нем присутствуют кристаллы, раз-
различным образом ориентированные в пространстве. Следовательно, обрат
ная решетка также вращается или принимает различные ориентации. За-
Закон Брэгга выполняется (т. е. дифракция наблюдается) только в случае
реализации тех условий, которые вытекают из рис. А7.4, б, т. е. когдя точ-
точки обратной решетки попадают на изображенную на рис. А7.4, б окруж-
окружность. В трехмерном пространстве при анализе распространения отражен-
отраженных лучей во всех направлениях такая окружность превращается в сферу.
Эту сферу называют сферой отражения Эвальда. Цель методических уси-
усилий экспериментаторов, занимающихся рентгенографическим исследованием
монокристаллов, состоит в ориентации или вращении кристалла таким об-
образом, чтобы различные точки обратной решетки в определенные моменты
съемки попадали бы на сферу отражения. Каждый раз, когда точки обрат-
нон решетки оказываются па сфере отражения, пучок отраженных лучен
распространяется в выбранном направлении 0Z. Фиксируя этл различные
отраженные лучн, можно получить картину обратной решетки. В рамках
некоторых методов, в частности в рамках прецессионного метода, непосред-
непосредственно получают неискаженную картину обратной решетки. В рамках дру-
других методов исследования, например при съемке вайссепбергограмм, полу-
получают искаженную картину обратной решетки.
.7, Обратная связь
Рис. А7.4. Возникновение сферы отражения Эвальда.
-1426
306
Приложите
А8. Уравнение Аррениуса для ионной проводимости
Перенос ионов « кристаллических твердых веществах, стеклах или рас-
расплавах осуществляется путем активируемых перескоков иопоп [гз одного
положения в другое. На рис. А8.1.Я схематически показала зависимость
энергии от пространственной координаты при движении ионов \л + в кри-
кристаллическом К'а+-проводящем материале (показано движение вдоль одно-
одного из направлении). Пополнения А а В в кристалле отвечают позициям, ко-
которые ионы натрия занимают большую наси> времени. Они .чарлкте.рип юг-
ся наименьшими значениями потенциальной энергии. .Между позициями ,4
к В питенцпальпая энергия принимает максимальные значения. Через ъуч
седлоиидные точки новы Ха + перескакивают и процессе своего движения
по кристаллической решетке. Потенциальная энергия катионов в позициях
Лий одинакова.
Все ионы в твердом веществе облпдлот определенным запасом тепло-
тепловой энергии. Вследствие этого они колеблются около своего равновесииго
положения с частотой v. рапной ~1012—1013 Гц. В отсутствии впешяпо
электрического ноля энергия системы колеблющихся ионов лароктеризупся
определенным распределением по различным энергетическим \ровням. При
температуре Т (в кельяннах) вероятность того, что нон NaH- обладает ко-
колебательной энергией, большей, чем Е, и можег перепрыгнуть in позиции А
а
Потенциальная
энергия
Расстояние
h d Ч •
Рис. А8.1. Схема движения иона в твердом теле.
в позицию В или обратно, пропорционально величине ехр (—FJkT) (где к —
постоянная Больцман;|). Число прыжков (в секунду) каждого иона в сред-
среднем пропорционально v е.\р (—?/кТ). Таким образом, и и отсутствие внеш-
внешнего электрического ноля некоторые катионы способны случайно перепры-
перепрыгивать из одной позиции и другую (естественно, если соседняя позиция не
зйпятл). Поскольку вероятиисть прыжков в рпзных направлениях одина-
одинакова, в целом в кристалле отсутствует электрический ток.
Если к кристаллу приложено внешнее электрическое ноле, то схема за-
зависимости потенциальной энергии от пространственной коорияп.пы не-
несколько меняется (рис. A8.I.6). Потении ильные барьеры, находящиеся пч
пути движения ионов Nap'r к отрицательному электроду, слегкт понижаются,
а барьеры, находящиеся па пути движения к положительному электроду,
слегка повышаются. Величину возникающее! разности высоты потенциаль-
потенциальных блрьероо можно рассчитать следующим образом.
Пусть напряжение внешнего поля равно V, тогда для перемещешг.;
иона с зарядом е на расстояние d и направлении действия ноля необходимо
Л8. Уравнение Арренпуеа для ионной проводимости 307
яафатпть работу, равную Ved. Величина произведенной работы соответст-
соответствует разности потенциальной энергии между позициями А и В. Энерютн-
ческий барьер па пути движения ионов в двух- направлениях А -В и В 'А
становится равным (Е—MiVed) и (E-\-\j2Ved). Следовательно, более ве-
вероятен перескок иола из положения А в положение В, я не наоборот.
Оценим общую вероятность перескока Р иона из позиции А и пози-
позицию /?¦
—(?4- 1 liVed)'
P'-Xv exp
kT
ccvexp) —-jpj.
e.xp
ve.xP
eVd \ I eVd
2kT
Выражение в квадратных скобках можно разложить в степенной ряд; мож-
можно покачать, что при <?Vrf<<W в разложении останется лишь первый член
ряда. Тогда
eVdv I E
Средняя скорость движения иона и — функция Р и </:
u = Pd
а подвижность иона равна
р, = u/V
Величина электропроводности о связана с подвижностью иона уравнением
а = A'ej-i
где N-—число движущихся ионов. Откуда имеем
Ne*d*v ! AS
а= gkT w[-F
где exp(AS//c) —коэффициент пропорциональности, AS — энтропия актива-
активации, a g — геометрический фактор, позволяющий учитывать вероятность
того, что данный ион может перескакивать в нескольких направлениях Вы-
Выведенное уравнение часто записывают в ином виде
AS \ I E
ГЬсле логарифниров.-шнч это ураииепие удобно использовать для построе-
построения лависииости lg а'/' от Т~'; график должен изображаться прямой, нл-
kjoh которой дает величину —Elk, а координата точки пересечения прямой
AS
'\
ных ионов N меняется с температчрой по закону
j AS \
с oci.io (/ — величину (.\'e2d2x/gk)cxpl—т—j. Иногда концентрация нодг.нж-
JV = Л'„ e.xp \—-j— j exp ^ -
где Л'о ^общее число потенциально возможных подвижных ионов в струк-
структуре, forvj и ASoup — энергия активации ц энтропия образования подниж-
ны.х попои.
Объединяя дна последних уравнения, получаем
AS -b ASo6p
1—
20*
A9. Некоторые свойства элементов
Элемент
Электронная кон-
конфигурация6
Основные
степени
II
Длины химических связей
а кислородных соединени-
соединениях (Л) и координационные
числа (указанные в скоб
ка\)'"
Азот N 14,01 (HeJs22p3
Актиний Ac
Алюминий Al
B27) . .
26,98 (NeKs23p1
Америций Am B43) (RnM/77s2
39 95 (NeKs23p6
B10) (XeLfu5du
137 34 (XeNs2
Apron
Астат
Барий
Бериллий
Берклий
Бор
Бром
Ванадий
Висмут
Водород
Вольфрам
Гадолиний
Аг
At
Ва
Be
Bk
В
Вг
V
Bi
Н
W
Gd
9,01 (HeJs2
B47) (RnM/86rf'7
10,81 (HeJs22p'
79,91
50,94
208,98 (XeL/l45rfl06s26p3
1,01 Ь1
183,85 (XcLf45J<Gs2
157,25
7 B,3,4), 5 N(V)-0 C) 1,28
89
13
95
18
85
56
4
97
5
35
23
83
1
74
64
3
3
3,4
Д1-О D) 1,79;
A1--O F) 1,93
Am(lll)-0 FJ,40;
Am (IV)—О (8J,35
2 Ва—О F) 2,76;
Ва—О A2) 3,00
2 Вс-0 C) 1,57;
Be—О D) 1,67
3, D) Bk (III)—О F) 2,36;
Bk(lV)--O-(8) 2,33
3 В—О C) 1,42;
В-0 D) 1.52
1, C,5,7) Br(VH)-0 D) 1,66
2—5 V(ll)-0 F) 2,19;
V(V)—О D) 1,76
1,3, E) Ri(lll) -О F) 2,42;
Bi(Ill)—О (8) 2,51
1 Н—О 1,02 до 1,22
D), 6 W(VI)-0 D) 1,81;
W(V1)—О F) 1,98
3 Gil-0 G) 2.44
Кристаллографические па-
параметры: элементарная
ячейка или кристалличе-
кристаллическая структура- иарамег-
о
ьы решетки (А) (темпе-
(темпера гурн)л
—210 Геке; 4,03, 6,59
(—234 °С)
1050 ГЦК; 5,311 (комн. темп.)
660 ГЦК; 4,0495 B5°С)
850 Гскс. ABAC; 3,642, 11,76
(коми, темп.)
—189 ГЦК; 5,42 (—233 °С)
710 ОЦК; 5,019 (коми.
темп.)
1280 ГПУ; 2,2856, 3,5832
B5 °С)
2300 Тетр; 8,73, 5,03 (комн.
гемп.)
—7 Ромб; 4,48, 6,67, 8,72
(-150 °С)
1920 ОЦК; 3,028 C0 °С)
271 R3m; 4,7457, 57°14,2'
C1 °С)
—259 Геке; 3,75, 6,12
(—271 °С)
3380 ОЦК, 3,1650 B5 °С)
1320 ГПУ; 3.6315, 5,777
B0 °С)
Галлий
Гафний
Гелий
Германий
Гольмий
Диспрозий
Европий
Железо
Золото
Индий
Иридий
Иттербий
Иттрий
Иод
Кадмий
Калий
Калифорний
Кальций
Кислород
Кобальт
Кремнии
Ga
Hf
Не
Ge
Но
Dy
Eu
Fc
Аи
In
Ir
Yb
Y
I
Cd
К
CI
Ca
О
Co
Si
69,72
178,49
4,00
72,59
164,93
162,50
151,90
55,85
196 97
114,82
192,22
173,04
88,91
126,90
112,40
39,10
B49)
40,08
16,00
53,93
28,09
(ArKrf'°4s24p*
(XeL/l45d26s2
Is2
(ArKrf'04s24p2
(XeLfs2
(XeL/l0Gs2
(XcLf~Gs2
{ArKd4s2
(XeL/'J5J'°6s1
(KrLrf'05s25pI
(XeL/K5rf76s2
(XeLfl46s2
(KrL<f'5s2
(KrLrf'°5s25p3
(KrL<f'°5s2
(ArLs'
(RnM/107s2
(ArLs2
(HeJs!2p*
(Ar) 3d4s2
(NeKs23p2
31
72
2
32
67
66
63
26
79
49
77
70
39
53
48
19
98
20
8
27
14
A). 3
4
B), 4
3
3, D)
2,3
2,3, D,6)
A), C)
(I), 3
ЗЛ F)
2,3
3
1, C,5,7)
2
1
3
2
<'.). 2
2,3
4
Ga—О D) 1,87;
Ga—О F) 2,00
Hf-0 (G) 2,11;
Hf—О (8) 2,23
Gc—О D) 1,79;
Gc—О F) 1,94
По—О (G) 2,30;
Ho-0 (8) 2.42
Dy(IIl)-O FJ,31
Eu A11)—О F) 2,35;
P 11 / 111 \ (~\ i1\ О Л1
¦uU(lllJ—U (/) <i,4o
Fe(II)-0 FJ,18;
Fo(lll)—О D) 1,86
Аи(Ш)—О D) 2,10
In—О (G) 2,18;
1П-—D [b) Z,oZ
lr(lll)—О F) 2,13;
Ir(IV)—О F) 2,03
Yb(IIl)-0 F) 2,26;
Yb(lII)— О (9) 2,38
Y—С) @) 2.29;
Y--0 (9) 2,50
I(V)—О C) 1,83
Cd—О D) 2 19-
Cd—О (G) 2.35
K- -O F) 2,78
К—О A2K,00
CF—О F) 2,35
Са—О F) 2,40;
Са—О (8) 2,47
Со(П)—О F) 2.054-2,14
Co(lll)-O F) 1,93-=-
-4-2,01
Si—О D) 1,66;
Si—О F) 1,80
30 Ромб; 4,520, 7,661, 4,526
B0 °C)
2000 ГПУ; 3,1946, 5,0511
B4 °C)
—270
958 Алмаз; 5,6575 B0 °C)
1500 ГПУ; 3,5761, 5,6174
B0 "С)
1500 ГПУ; 3,5923, 5,6545
B0 °C)
900 С)ПК; 4,578 B0°C)
1539 ОЦК; 2,8664 B0 °C)
1063 ГЦК; 4,0783 B5 °C)
156 Гетр; 3,2512, 4.9467
B0°C)
2443 ЩК; 3,8389 (комн.
темп.)
824 ГЦК; 5,481 B0 °С)
1500 ГПУ; 3,6451 5,7305
B0 °С)
114 Ромб; 4,792, 7,271, 9,773
(комн. темп.)
321 ГПУ; 2,9788, 5,6167
B1 °С)
63 ОЦК; 5,32 B0 °С)
850 ГЦК; 5,582 A8°С)
—219 Куб; 6,83 (—225 °С)
1492 ГПУ; 2,507, 4,069 (комн.
тема.)
1410 Алмаз; 5,4305 (комн.
темп.)
Продолжение таблицы
Элемент
Криптон
Ксенон
Кюрий
Л питан
Литий
Лоурснсий
Л [отгний
Магний
Марганец
Менделеешш
Медь
Молибден
Мыи{ьяк
Натрий
Неодим
Неон
Нептуний
и
V
h
Кг
Хс
Cm
La
Li
Lr
Lii
Mg
Mil
Md
Си
Mo
As
Na
Nd
Ne
Np
«
С 'w>
83 80
131,30
B47)
138,91
(j.94
B57)
174,97
24,31
54,94
B56)
63,54
95,94
74,92
22,99
144,24
20 18
B37)
Электронная кон-
конфигурация
<ArKdl04s24p6
(KrLrfI05s25p6
(Rn)of~Gd[7s*
(XeMrf!6s2
(HqJs1
(RhM/l4Gd'7s2
(XeL/ilf5d'Gs2
(NcKs=
(ArK<Nsa
(RnM/'-7s2
(\vKdl0-lsl
(KrLt/:o5'
(ArKd<4s4p-
(NeKs'
(XcLf>6.s--
(HcJs22/)°
(RnM/°7s2
Порядко
нимер
36
54
96
57
3
103
71
12
25
101
2<)
42
33
11
60
10
93
Основные
степени
окисления
2, 4, 6
3, D)
3
1
3
3
2
2, 3, 4, :
3
1, 2
C, 4, 5),
3, 5
1
3, D)
B,3), 4,
F,7)
Д.шны .химических снязен
и кислородных соедингин-
о
ях iA) и ьоирдшищнопные
числа (указанные в скоб-
скобках) |%
Кристаллографические па-
параметры: элементарная
seiciWa или кристалличе-
кристаллическим структура: парачет-
о
ры решетки (Л) (гечпе-
parjpa)д
Cm(Hl)-0 (G) 2,38;
Cm(IV)-0 (8) 2,35
Ln--0 (G) 2,46;
La—O A0) 2,68
I.i—O D) 1,99;
Li—О ((i) 2,1 1
I.li—О ((i) 2,25;
Lu -0 (8) 2,37
Мк—О D) 1,89;
\\g- О ((i | 2,12
Mn(II)—О (fi) —2,10;
Мп(\'П)-О D) 1,GG
Cn(l)--0 B) 1,80;
Cn(Il) О (()) 1,97-7-2,60
Mo(Vl)-O A) 1,82;
A\o(Vl)-O F) 2,00
As(V) -O D) 1.74;
Щ\)—О ((j) 1.90
N'a ¦<) D) 2,39;
N;i—О (У) 2,72
\d —О (В) 2,40;
Nd -О (8) 2,52
;\])(ID—О (С) 2,50;
ХрA\) -О (8) 2,38
— 1Г>7 ГЦК; 5,С8 (-191 °С)
— 112 ГЦК; 6,24 ( -18.Г>°С)
920 1'скс; ABAC; 3,770,
12,131 B0сС)
180 ОЦК; 3,5092 B0 °С)
1700 ГПУ- 3,5050, r),548G
B0 °С)
650 ГПУ; 3,2094, 5,2105
B5СС)
1250 Куб; 8,914 B5 °С)
1083 ГЦК; 3,0147 B0°С)
2620 ОИК: 3,1109 B0 °С)
814 R3m; 4,131, 5-1° 10'
BоТ.)
98 ОЦК; -1.290E B0 СС)
1024 Гскс: ЛВЛС: 3,6582,
11,802 B0 СС)
—249 ГЦК: 1,52 (-208°С)
640 ромб. 4,723, 4,887, 6, 663
B0 СС)
Никель
Ниобий
Нобелий
Олово
Осмий
Палладий
Платина
Плутонии
Полоний
Празеодим
Прометий
Протактиний
Радий
Радой
Рений
Родий
Рп-гь
Рубидий
Рутений
Самарий
Снши-ц
Селе)!
Ni
\'b
No
Sii
Os
Pd
PI
Pu
Po
Pi
Pm
Pa
Ra
Rn
Re
Rh
Hg
Rb
Ru
Sra
Pb
Sc
58,71
92,91
B53)
118,69
190,20
106,40
195,09
B42)
B10)
140,91
A47)
B31)
B2G)
B22)
186,23
102,91
200,59
85,47
101,07
150,35
207,19
78,96
(ArKrf84s2
(KrL^5s'
(RnM/ss
(KrLrfi;i5i-25p2
(XcL/I4o<№s2
(KiLrfiy
(XcL/I45(/ERi-(
(Rn) 5f e7&2
(XeL/145Jl06.v26p4
(XcL/36s2
(XeL/5Gs2
(RnM/26(p7s2
(RuOs2
(Xo) 4/I'15t/inGs26n6
(Xc'L/'H5?/56s2
(Ki"J 4t/®5s1
(XeL/|;5c/'n6s2
(Kr)Ss1
(KrLars1
(XcL/eGs2
(XcL/'45dI(l6s26p2
(ArKrfIO4s-4p4
28
41
102
50
76
46
78
94
84
59
61
91
88
86
75
45
80
37
44
62
82
34
2, C)
D), 5
3
2,4
4, F), 8
2, D)
4, (G)
3 4,6
2, 4
3 D)
3
4, 5
2
3, 4, 5, 7
3, D, 6)
1, 2
1
4, F), 8
B). 3
2, 4
B). 4, 6
Ni(II)-0 (G) 2,10;
Mi(lll) - О F) 1,98
Nh(V)-0 (G) 2,04;
Nb(V)—0 G) 2,06
Sn(ll)--() (8) 2,62;
Sn(IV)-O F) 2.00
Os(IV)—О F) 2,03
Pd(ll) —0 D) 2,04;
Pd(IV) -() F) 2,02
Pt (IV)— О F) 2,03
Ptif III)—О (G) 2 40;
Pu(IV)--0 (8) 2,36
Po(lV) -O (8) 2,j0
PiAII)-0 F) 2,41;
Pr(lV)-0 (8) 2,39
Ptn(III)—О (G) 2,38
Pa (IV) - -() (8) 2,41;
Pa(V)—О (9) 2,3:3
Re(IV)-O (G) 2,03;
Re(VII)- О (G) 1.97
Rh(III)-0 FJ,07;
Rh(lV)-O F) 2,02
llg(l) 0CJ,37:
11^A1) -0 B) 2,09
Rb -O (IS) 2,89;
Rb 0 A2) 3,13
Ru(lll)-O F) 2,08;
Ru(IV)--0 (G) 2,02
SmA11) — О (G) 2,36,
Sin(Ill)— О (8) 2,49
Pb(II)-0 D) 2,34;
Pb(IV)-0 ((i) 2,18
Se(Vl)-0 (¦!) 1.G9
1453
2420
232
2700
1552
17G9
254
935
3000
700
¦—71
3170
1960
—39
39
2400
1052
327
217
ГЦК; 3,524 A8 °C)
OUK; 3,000 B0 °C)
1Ч-тр; 5,831Г), 3,1814
Bo°C)
ГПУ; 2,7353, 4,3191
/On i-' Г* \
ГЦК; 3,8907 B2 °Q
ГЦК; 3,9239 B0 CC)
Мшюкл.; 6,18,4,82, 10,97,
101,81° B1 °C)
Куб., 3,345 A0qC)
IVkc, ABAC, 3,6702,
11,828, B0 °C)
To-p.; 3,935, 3,238 (комн.
Tl'xHI.j
ГПУ; 2,760, 4,458 (комн.
ieMn.)
ГЦК; 3,8044 B0 °C)
R3in; 3,005, 70°32'
(-46 °C)
OUK; 5,70 B0CC)
ГПУ; 2,7058, 4,28F
B5 °C)
R; 8,996, 23°13' B0 °C)
ГЦК; 4,9.502 B5 °C)
Гекс.;4,3656, 4,9590B5°(;)
SIC
•о
О
it
s
Н
S
н
*^
X
н
о
X
^
J3
К
о
о>
н
о
1—
п о п о
la tr О 3
S S = 1а
» 3 я к
¦о
о
о
н
— о-
СЛ
о
I
¦о
I
ев
ся со
СЛ^ООСЛСОГО-^'—
ЮСО^- — 00^-—JCT>
;--оо5г <<5 <5Ет
I 1\ со
^1 I
- -^ °
о ел--
f
О
слк>*-.°2
<^. -J СО
2°> ~<=>*ю*со
„ ^- ~ N3 —
S S S
54
О
3
->
о
S
р
тз
Сэ
•в-
н
¦•!°
со
СЛ
СО
^-J
ю
5,5
СЛ
,08
со
п
со
00
00
о
S
-• п-
СО—'
СЛ
3^
СЛ
ъ
Химический
знак
Атоикая
Порядковый
номер
¦ой m J
Ч
Температура
плавления,
-В 3
^>».°;;
я «о*
? н ? a g
"а
о
§
3
г
Уран
Фермий
Фосфор
Франций
Фтор
Хлор
Хром
Цезий
Церий
Цинк
Цирконий
Эйнштейний
Эрбий
и
Fm
Р
Кг
F
С1
Сг
Cs
Се
Zn
Zr
Es
Er
238,03
B53)
30,97
223
19,00
35,46
52,00
132,91
140,12
65,37
91,22
B54)
167,26
(RnMf!27s2
(NeKs23p3
(RnOs'
(HeJs22p5
(ArKd4s>
(XeNsl
(XeLpGs2
(ArKdl04s2
(KrLd25s2
(RnMp7s2
92
100
15
87
9
17
24
55
58
30
40
99
68
Щ, 4-
E), 6
3
3, 5
1
1
!, C, 5,
3, 6
1
3, 4
2
4
3
3
U (IV)—О (9) 2,45;
U(V1)-O D) 1,88
P(V)~O D) 1,57
7) Cl(V)-0 C) 1,Г>2,
Cl(VlI) -O t.4) 1,60
Cr(lll)—О F) 2,02;
Cr(Vl)—О D) 1,70
Cs-0 (8) 3,22;
Cs—О A2) 3,30
Се(Ш) -О (9) 2,55;
Ce(lV)—О (8) 2,33
Zn—О D) 2,00;
Zn—О F) 2,15
Zr—О F) 2,12:
Zi- -О (8) 2,24
Er A11)-О FJ,29
1133 Ромб.; 2,854, 5,869, 4.955
B7 °C)
44 Ромб.; 3,32, 10,52, 4,39
(черный) (комн. теми.)
220
-101 Тетр.; 8,56, 6,12
1900 OUK, 2,8846 B0 °С)
29 О ЦК; 6,14 (—10 °С)
804 ГЦК; 5,1604 B0 °С)
419 ГПУ; 2,0649, 4,9468
B5 °С)
1850 ГПУ; 3,2312, 5,1477
B5 "С)
1525 ГПУ; 3,559, 5,592 B0 °С)
со
со
Атомная масса выражена о углеродных единицах. Они птвечиот естественному распространению изотопов данного элемента и приро-
природе. В скобках даны значения атомной массы наиболее долго живущего изотопа без учета еп> распространении « природе.
Приведены электронные конфигурации элементов в основном состоянии У некоторых переходных элементов, лантаноидов п акти
ноидов разность энергий основного н первого возбужденного состояний очень мала.
Многие элементы наряду с устойчивыми степенями окисления характеризуются и большим числом неустойчивых степеней окисления.
При составлении данной таблицы основное внимание обращалось на степени окисления элементов в кристаллических оксидах и галогепидах.
г Приводимые значения взяты из [Shannon. PrewiH, Acta, Cryst., B2o, 925 A969); B26, 1046 A970I. Заметим, что длины химических
связей, как правило, увеличиваются с ростом координационного числа и уменьшаются с возрастанием степени окисления. Длины химических
связей во фторидах обычно на 0.05—0,10 Л короче длин связей в оксидах; в хлоридах длины химических связей обычно на 0,20—0,40 А боль-
больше, чем в оксидах.
л Данные позаимствованы в основном из «Интернациональные таблиц» (i. Ill, с. 278). Приняты следующие сокращения. ГЦК~1'ра
иеиентрированная кубическая решетка (плотнейшая кубическая упаковка); Геке.--гексагональная решетка; ГПУ — гексагональная плот-
нейтая упаковка; R3m, R3m — ромбоэдрическая решетка; ОЦК — объемноцентриропанная кубическая решетка, Тетр. — тетрагональная ретет
ка-, Ромб. — ромбическая решетка; Куб. — кубическая решетка.; Монокл — моноклинная решетка; Алмаз — структура алмаза.
ФОРМУЛЬНЫЙ УКАЗАТЕЛЬ
Ag 1. 150, 275. 357, 425: 2, 55. 225 AmO2 1, 297
AgjAsCU 2. 34. 211 ArCl 1, 35!
AgBr 1. 294, 350, 390: 2, 32 As 1, 357
AgC) I, 294, 350, 360, 390, 392, 395, As2O, 2, 179
396- 2. (i, 12. 14—18, 21, 22, 32, 232 Au 1. 275. 425; 2, 225, 228
AgF 1, 294, 350, 353 AuCu 1, 275
AgF2 1. 353 AuMg 1, 31 1
Agl 1, 296, 306, 350, 512, 520, 521, AuZn 1, 311
545 546' 2, 20—22, 31—35, 38, 211
AgrI4As04 2. 34
Agrl4PO4 2, 34 В 1, 357
AgsI4WO4 2, 34 BAsl. 296
Ag2MoOt 2, 211 B4C 2, 2C2
AgNO.i 1, 44 BN 1, 296; 2, 262
Ag2O 1, 361 В2Оя 1, 68. 361; 2, 179, 182, 183, 187,
Ag2S 2. 35, 52 196—198, 207, 213
AgjSi2O3 1, 44 BP 1. 296
AgZu 1. 311 Ba 1 275. 375
Al 1, 48, 275, 321, 357, 425 BaCl2 1, 2U7, 360
AlAs 1. 296; 2. 87 BaF2 1, 297. 365
Al.Br6 1, 282, 286. 328 BaCrO, 1, 60
AIC13 1. 328, 351; 2. 262 BaFc.2OI9 2, 159, 160
A1F3 t, 328 Ba2GdNd06 2, 172
A1I3 1, 36, 328 Bab 1. 360
A1N 1. 306 BaO 1, 20. 294, 327, 347, 361, 365,
/MNi 1. 311 473; 2. 200, 262
Л12О3 1. 14 — 16. 19. 23—26, 36, 47, BaS 1, 291
50, 53. 59, GO, 66, 303, 115, 276, BaSc I, 294
279, 329. 361 436, 440, 441, 444— BaTe 1. 294
446. 457, 458. 480. 481; 2. 24—26. BaTiO3 1, 142. 232. 233; 2. 111 — 113.
31. 38—40, 174, 179, 182, 183, 201, 117. 124. 125
212. 228, 236, 239, 246. 248. 249, BaTiSi3O3 1. 320. 322
258. 260 263—265 Be 1, 275. 357, 363, 425, 429
Л1(ОНK 1. 24; 2, 248 Be,A!2SiG0,8 1. 320
AI2(OHLS2O5 1. 322 BeCl> 1. 3G0
A!P 1. 29G- 2 87 RcF, 1. 328, 365, 386
A!2S, 1 36 37 BeO 1, 30C, 328. 361. 365. 390; 2. 260
AI,(Si<O,n(OH)8 1. 138 BeS 1. 296
AlSb 1. 296; 2, 87 BcSc 1. 296
Формульный указатель
315
ВеТс 1. 296
Ш 1. 357. 385; 2, 102
ВьО, 2, 179
Bi,Scj 2. 102
Bi Tc^ 2, 102
Bi,Ti3Oi2 2, 113. 118
С 1, 59. 357. '185
QBr 1. 38, 40
CF 1. 38, 39
OF Т. 39
С1ЬС12 1. 244. 245
СчК 1. 38—40
C24K 1. 38
СИК 1. 38
С а К 1, 38
С60К 1, 38
СОг 1. 134. 328, 34], 361, 365, 366
Са I, 357
Ca.MjOi 2. 183. 246
Ca2,\l2Oi 2, 240
Са3Л1-.О8 2. 183. 235. 236, 237
СаА12ЭиО, 1. 321, 443. 477; 2. 231
Ca2Al3Si0; 2. 246
Ca^AljFejO,;, 2. 236
Ca4Al4(OH).2SOrGH2O 2. 243
CasAlj(OH)i2(SaK-26H2O 2. 233
Ca,AbSi-,Oi2 1. 386
G;]Al2@11b(Si2Ali)Oio 1. 322
OiC2 I. Hia
CaCb 1. 360, 399: 2, 245
CaCO3 1, 96, 134; 2. 234
СяСгОз 1. 60
CaF2 1, 200, 206, 279, 297, 299, 300.
325. 328, 365. 38G, 390. 393. 441,
451; 2, 22, 35
Ca.Fc2O5 2, 240
Cal2 1, 316
CaMg(SiO3)s 1, 322; 2. 172, 231
Ca2MgSt,OT 1. 322: 2. 266
Ca,Mg-,SisO22(OH, FJ 1, 322
CaNb-A 1, 52
CaO i, 36. 55. 58, 294. 328. 347, 362,
365, 377. 437. 441. 450. 456, 458.
459. 471- -473. 182: 2, 21, 36, 179,
183. 200. 212, 236, 258. 262, 264
Ca(OH), 2. 235, 242—244. 248. 250,
251
Ca,@HJfSi60,7) I. 105
3Ca,(P(),)?-Ca(Cl. F), 2. 168
o,,po,ci i. no, in
Са,(РСЦK(Г. С1) 2, 171, 172
Ca5(PO4KOH 2. 266
CaS 1, 204
CaS0,'2H2O 1. 96
CaSe 1. 294
CaSiO3 1, 320, 457, 471, 482, 498;
2. 172, 231, 266
Ca2Si04 1, 142. 322, 455, 457, 459,
471, 498, 521. 522: 2, 234—237
Ca3SiOo 1, 60, 457, 459, 471, 498;
2. 234—237, 243
CaiSi,Or 1, 320, 457, 471, 482, 498
CacSieO,7(OHJ 1, 58
Ca,SnO4 1, 36
Cafe 1, 294
<".аТЮ3 I, 47. 279, 281, 325; 2, 117
CaTi-A, 1, 47
CiAVOi 2, 1G5
Cd 1. 275. 357. 428, 429
Cd2R2O5 2, 168
CdBr2 1, 318
CdCU 1, 279, 316—319, 326, 328, 329,
339, 341. 3E0; 2, 17
Ceil., 1, 278, 279, 314—318, 328
(VI6 1, 294. 361; 2, 87
ОМОН), 1, 316
OIS 1. 59. 291, 30C. 541; 2, 87
OlSe 1, 296. 306; 2, 87
CdTc 1. 296; 2, 87
CiITiCb 1, 519
002 1. 297. 311. 390
CmO, 1. 297
CoAI2O4 2. 150
CoBr2 1, 316
CoCU 1. 318
CuCr2O4 I, 28
СоГ2 1, 312. 350
CoiGeOj 2, 150
Col. 1, 316
CoO 1, 294, 361, 386; 2, 144
СсьОз 2, 85
Cu3O4 1. 384
Cn(OHJ 1, 3Hi
CoS I. 309
CoSb 1. 309
CuSe 1. 309
Co Те 1. 309
Cr 1. 150, 27o. 425
ОСЬ I. 279. 282. 285, 328
OF, 1, 381
CrO I. 381; 2. 144
CrOs 1. 312
CrO; 1. 36. 3R1
Cr.,O3 1. 36. 101. 436, 447, 455, 457:
458, 481: 2, 89. 90. 174, 265
Cr,S., 1, 519
CrSb I, 309
CrSc 1, 309
Ci\Si 1, 47
CrTc 1. 309
Cs 1. 357
CsBr 1, 311
316
Формульный указатель
CsCl I, 59, 96, 109, 200, 298, 308, 310,
311, 322, 346, 347, 360, 390, 507,
508, 512, 519, 541, 544, 546
CsCN 1, 311
Cs2CuCl4 1, 383
CsF 1, 96, 341, 347, 365
Csl 1, 311
Cs2O 1, 318, 319, 328, 361, 365
Cu 1, 150, 275, 425; 2, 99, 10O, 225
CuAu 1, 412
CuBr 1, 296
CuBr2 1, 351
CuCl 1, 35, 117, 118, 296, 360
CuCl2 1, 351, 380
CuClr2H2O 1, 117, 118
CuCr2O4 1. 384
CuCr2S4 2, 150
CuCr2Se4 2, 150
CuCr2Te4 2, 150
CuF 1, 96, 353
CuF2 1, 351, 353, 380, 383
CuFe2O4 3, 384
CuFeS2 1, 279
Cul 1, 296
Cul2 1, 351, 353
CuO 1, 312, 361, 380, 381; 2, 89, 90
Cu2O 1, 35, 361; 2, 89, 291
CuPd 1, ЗП
Cu.,TaSe, 1, 37
CuZn I, 313, 406, 449
Cu«;/rSi 1, 119, 120
Eu 2, 143
HuF2 1, 297
Fe 1, 150, 176, 181, 200, 275, 402, 425,
485, 486, 542; 2, 141
FeBr, 1, 316
Fe3C 1, 91, 485, 542
Fe(C2H.,K 1, 27
FeCI2 1, 318
1;l-C13 I, 38: 2, 282
FeCr2O., 1, 28, 519
PeF2 1, 312
Fe5Gc04 2, 150
Fcb 1. 310
Fe2O3 1, 16, 47, 363; 2, 235, 239, 246,
249. 258, 264
FcO 1, 294, 311, 361, 386, 402, 456;
2, 141, 145, 247, 258
Fe,O4 1, 384; 2, 85, 89, 258
FcOCl 1, 43
Fe(OHb 1, 316
FeP 1, 47
lreS 1, 309
FeSb 1, 309
FeSc 1, 309
Fe2SiO, 2, 258
FeTc 1, 309
FeTi 1, 310
FeTiO3 2, 159, 174
FeV2O4 1, 47
Gn 1, 357
GaAs 1, 52, 53, 296; 2, 61, 87
GaNT 1, 306
G;bO, 1, 361; 2, 179
GaP 1, 29G; 2, 01, 85, 87
GuSh 1, 296; 2, 87
Gd 2. 143
(id2(MoO,,K 2, 113
Tic 1, 49, 52, 53, 101, 357
Ge(J 1, 120, 312, 341, 342, 361, 541;
2. 179, 202
GeH 1. 49
GeH,, 2, 220
1ЬО2 1, 366
Н2П4О0-П,О 1, 46
HiC 2, 262
HfN 2, 262
НЮ2 1, 98, 99, 341, 361; 2, 37
Hg 1, 357
HgF2 1, 297
Hfi-0 1, 32 7, 3G3
HgS 1, 296
3IqSc 1, 296
HgTc 1, 296
Hn 2, 14.3
HUO2PO4-4H2O 2, 40
InAs 1, 296; 2, 87
InBi 1. 30G
inN 1, 306
In-O.i 1. 363
InP 1, 296; 2, 87
inSb 1, 296; 2, 61, 87
IrO2 1, 312
KA1F4 1, 285
iai2(OHJ(Si3AlH,o 1, 321
KAlSiO4 1, 321
K.AlSi3Os 1, 106, 107, 115, 321
KBr 1. 200, 294, 519; 2, 87
KC.M 1, 546
KCI 1, 59, 96, 200, 210, 227, 294, 360,
398. 519, 341; 2, 87
s 1, 123, 124
Формульный указатель
317
KF 1, 96, 200, 227, 294, 347, 365;
2, 87
W;oS, I, 129
КН2РО, 2, 113, 119
К.1 I, 200, 227, 294; 2, 87
KMg3(UHJ(Si3.\l)O,0 I, 321
KNUj 1, 46
KNa3(AlSi04L 1. 106, 107
KNbO3 2. 113
K2O 1, 46, 279, 280. 297, 318, 361,
3E5, 473; 2, 236, 258
K2S 1, 297
K?Se 1. 297
K2Tc 1, 297
K2Ti409 1, 45, 46
.LaX 1, 294
La2O3 1, 361
Li 1, 41, 42, 357
Li.Ag 1, 311
LL\IO2 1, 283, 284, 438, 442
LiA]5OR 1, 26; 2, 28
LiAlSiO4 1, 107, 108, 321, 442, 483
LiAlSijOe 1, 321, 442; 2, 228
LiAlTiO* 2, 147, 150
Li2BeSi04 1, 280
LiBr 1, 294, 347
Li2CO3 1, 26; 2, 28
LiCI 1, 29-1, 347, 360; 2, 87
LiCoSbO4 2, 150
LiCrO2 1, 438
LiF 1, 47, 73, 96, 294, 326, 330, 331,
335, 347, 3(i5, 422, 437; 2, 87
LiFeO2 i, 507, 509, 510, 545
.LiGaO2 1, 279, 280
Li4Ge04 2. 39
liH 1, 204
LiHg 1, 311
l.il 1, 294, 343, 347; 2, 39, 40
Li2Mf>Si04 2, 228
LiiMn-O, 2, 89
LLMnTiOi 2, 150
Li:Mo04 1, 59
l.i3N S, 108. 109
LiNbO3 1, 97—99, 445; 2, 113
LiNiF, 2, 147
LiXiVO* 2, 147
Li,0 I. 26. 98, 99. 120, 297, 361, 365;
2. 27. 33. 200. 209, 228
L13PO, 1. 279, 280, 283 284
\.\2S 1, 297
l.i-Se 1, 297
Li;SiAIO3K 2, 266
Li2Si0j 1. 233, 439; 2. 228, 231
ki,Si2O5 1, 521, 522, 524; 2, 228
Li4Si04 1, 233, 320, 449; 2, 39
LiTaO3 1, 96
I.ijlc 1, 297
Li/1'Юз 1, 444, 510
Li,TiS2 1, 41, 42; 2, 42
Li^SUj 1, 512; 2, 20, 38
LiV2O4 2, 89
Li14ZiKie,Oi6 2, 39
LiZnSbO, 2, 150
LbZnSiO, 1, 218, 279, 280, 449, 450,
533, 534, 538, 539; 2, 229
Mg 1, 275, 375, 426, 429
MgALO, 1, 14—16, 19, 23, 53, 60,
2Г9, 285, 325, 337. 384, 406, 440,
441, 445, 480; 2, 24, 147—150
Mg2.'\l4Si-,O,8 2, 229
Mg8AI,6O32 2, 147
MgCO3 1, 23, 25; 2, 263
MgCI2 1, 338, 351, 352, 360; 2, 251
MgCr2O4 1, 28
MgF2 1, 312, 328, 365, 386; 2, 21
MgFe2O4 1, 17, 384; 2, 129, 150, 152,
153
Mgl2 1, 316
.Mghio(L 2, 150
MgIii2S.i 2. 350
MgO I, 14—21, 23—26, 60, 294, 326,
328, 346, 347, 361, 365, 441, 446,
450; 2, 18, 19, 27, 31, 126, 179, 183,
200. 212, 228, 236, 251, 259,
2G2 -2(,4
Mg(OHh 1, 316; 2, 263
ASg.i(OHJSi40iu 1, 321
MgS 1, 294
MgSe 1, 294
AigSiOj 1. 320, 455; 2, 229, 264
MgaSiO* 1, 105, 279, 339, 320, 438,
455, 480; 2, 229
MgSr 1, 311
MgTiO3 2, 124
•MgTt2O4 2, 147, 149, 150
Mg2'n04 1, 406; 2, 150
AinAs 1, 309
MnBi 1, 309
Мя.Вг2 1, 316
MnCIj 1, 318; 2, 29
MnP, 1, 312
MnFc-,0, 2, 153, 154
A5nl2 1, 316
MnO 1, 78. 294. 361, 386; 2, 144—146
MnO2 1. 43. 283, 284, 312, 361
Mn2O3 2, 85
Л\пА 1. 384; 2, 89
AlnS 1, 294, 296, 306, 541
318
ФормульнмЧ \к;..;и!сль
MnSb I, 309
MnSc 1, 296, 306
МпТс 1. 309
Д\о 1, 50, 150, 275, 425
МоО2 1, 47, 312, 2, 89
МоОз 1, 43, 47, 408; 2, 89
MoSi? 2, 265
X 1, 357
ХИ4Вг 1, 311, 512, 541
NH4C1 1, 311. 512, 54С
NH.CX 1, 311
\H4F 1, 306
NH4HjPO4 I, 507, 510
NaAlO2 t, 59. GO
NaAl.,0,7 1, 281; 2, 159
XaAISi3O8 1, 321, 443, 477
Na2Al2Si3O,p-2H2O I, 106, 107
Na2BbO[3 2, 213
XaBi 2, 87
NaCI 1 11, 21. 59, 78, 158, 165, 166,
169 181, 199, 208. 209, 227, 261,
262 276, 278, 279, 284, 285, 289—
294, 296. 298, 301,308.310,311,316,
322, 327—330, 338-341, 343—351,
364, 381, 390, 391, 394. 397—399,
4 И 440, 468, 507, 508, 510, 512,
519, 541; 2. 6—14, 16, 19, 28, 29,
85, 86, 87, 106, 108, 144, 145, 179,
222, 259. 262
NaF 1, 47, 96, 294, 347, 358, 359, 365;
2, 87
KaGriO, : Eu3^ 2, 172
Nail 1, 294
Nal 1, 294
NaLa(\V04b 2, 173
XaLu(J 2, 172
NaKOr, 1. 9G, 166
NaNbO., 2. 1 17
NaNiO* 1, M
Xn2O 1. 276, 290, 292, 299, 322, 361,
365, 393. 444. 45E; 2, 179, 181, 200,
209, 212, 213, 258, 265, 290
Nn2S 1, 297
NasSO4 1, 47
Na?S?O3 1. 123
Na2Sc 1. 297
КагЯГО3 1. 30, 320, 438
Na2Si204 1, 44, 320, 459; 2, 195
\"a3Si-,O7 1. 321
No-.Te 1, 297
ХаДУО, 1, 43, 47; 2. 41
\'пЛУО< 1, 46, 47; 2, 147
W-iYF, 2, 173
Na-)Zi-2PSi?0,2 2, 22, 38
Mb 1, 48, 150, 425
Bo 2, 262
Nbl4 1, 36
Nb\" 1, 309
NbO 1, 36
NbO2 1, 312, 541; 2, 182
NIj2O3 1, 98, У9, 409; 2, 182
Nb25Oc2 1, 409
1, 409
j.eij 1, 409
Mb,Si3 1, 36
NcCl I, 351
Ni 1, 150, 275, 425; 2, 141
NiAI2O4 1, 15, 16
XiAs 1, 279 300, 306—309, 311, 312,
315, 316, 322; 2, 290
NiBr2 1, 318
NiCI2 1, 318
NiCr2O; 1. 28, 36, 384
.\IF2 1, 312
NiFc2O. 1, 27, 28; 2, 150
XijGc 1, 47
Nil2 1- 318
XiO 1, 16, 36, 47, 60, 78. 294, 361.
381, 386; 2 82, 85, 87—89, 92, 126,
143—145, 172
\UOHJ 1. 316
XiPS, 1, 43
X'iS 1. 309; 2, 89
NiSb 1, 309
NiSe 1, 309: 2, 89
XiSn 1. 30!)
XiTc 1, 309; 2, 89
NpO2 1. 297
()s I, 275
GiO, 1, 312
P20j 2. 182
P5O5 2, 179, 182, 201. 225, 228, 229
P-dO2 1, 297
PI) 1, 150, 275, 357, 385
PbCiOn 1, 60
PbFj 1, 108, 297; 2, 18, 21. 35, 36
РЫ;е,2Ог9 2, 159
Pbl2 1, 31 f>
PbO 1. 312, 381; 2, 196
PbO2 1, 297, 312, 328, 341, 361, 385
PbS 2. 87, 90. 91
PbSe 2, 87. 91
PbTc 2. 87, 91, 102
PbTiO.i 1, 343; 2, 113, 117, 123, 124
PbZrO.i 2, 117, 118, 123
Pb(Zr.,Ti, .,)O3 1, 60; 2, 113
Pd 1, 275
PdF, 1, 312
Форм) льни ft. указатель
319
PdO I, 311. 361, 382
Pr 2, 143
PrO2 1. 297, 415
Pr3O, 1, 115
Pt I. 275, 425; 2, 99, 100, 225
PiB 1, 309
PlBi 1, 309
РЮ 1, 382
PlS 1, 311
PiSb 1, 309
PtSii 1. 309
PuO2 1, 297
"Rb 1, 357
RbAg4I6 1, 60, 521; 2, 22, 32—34, 55,
101
RbBr 1, 294
RbCl 1, 9G, 294, 360, 519, 541
RI>F 1, 9G, 294, 347, 3G5
Rb] 1, 294, 341; 2, 33, 34
HbN'O3 1, 512
Rb3O 1, 297, 318, 319, 3C1, 365
Rb2S 1, 297
Re 1, 275
ReO3 1, 45, 46, 124, 280, 285, 328, 408
Rli 2, 99
Ru 1, 275
b 1, 312
S 1, 35Г; 2, 215
SI) 1. 357. 385
SbF5 2, 279
Sb2O3 2, 179, 182
Sb2Oi 2. 182
ScN 1, 294, 326
Sf,Os 1, 361; 2, 21
Sc,Si3O- 1, 320
50 1. :;57; 2, 222
SeC 2. 172
51 1, 50. 52. 59, 101, 357; 2. 223
Sit: 1. 281, 286, 296, 30C; 2, 87, 262,
265
Sb\'i 2. 262
Si,\4 1. 443; 2, 265, 2GC
SiO 1. 36, 457
S;O2 1. 25, 36, 49. 53. 58, 59, 68, 70,
79, 84, 106, 140, 230, 285, 320, 325,
328, 341, 342, 361, 365 367 442,
446, 457, 458, 463, 471—-173, 475,
481, 482, 505, 512, 519, 521—523,
52R. 5-11- 2, 179, 193—196. 200, 201,
209, 212. 228, 229, 231 23G 246,
250. 258, 262, 2G4, 265
•SiP.,O- 1. 319
Sn 1, 357, 385
SnAs 1, 294
SnBr, 1. 283
SjiO 1, 36, 312, 361
SnOj 1, 312, 341, 361
S»S 1, 386
Sr 1, 357
Si-Cl? 1, 297, 360; 2, 35
SrC\'O3 1, 60
SrF, l, 297, 365
SrO I, 262. 294, 327, 347, 361, 3G5;
2. 200, 2G2
SrS 1, 294
SrSc 1, 294
SrTe 1, 294
SrTiO3 1, 259—261, 322, 2, 112, 158
Та 1, 48, 275
T;iB2 1, 47
TaC 2, 2G2
TaN 1, 306: 2, 262
TnO2 1, 312
Ta2O-; 1, 47; 2, 182
Ta2S2C 1, 43
Tc 1, 357, 385; 2, 102
TcO2 2, 179
Thl, 1, 316
Tli62 1, 53, 278, 297, 341, 390; 2, 37,
124
Ti 1, 48, 275, 425, 429
TiBr2 1, 31G
TiC 1, 294, 326, 328
TiCl2 1, 316
Tib 1, 31fi
TiMgO4 1, 384
TLMgA 1, 384
TiN 1, 294
TiO l, 294, 377; 2, 87—89, 143, 144
TiO,, 1, 43, 45, 46, 262, 265, 266, 279,
283—285, 312. 325, 328, 341. 361,
413, 444; 2, 89, 90. 225, 228, 229
TiS2 1, 37, 38, 40—42; 2, 41, 42
TI 1, 357
TIBr 1, 311
T1C1 1, 311, 390
T1F 1, 311
Til 1, 311
T1,O3 1, 361
Tm 2, 143
Tml, 1. 316
UC 1, 294
I'N 1, 294
l.'O2 !, 297, 404
320
Формульный указатель
V 1, 425
V8r2 I, 316
VC12 1, 316
VI2 1, 316
VO 1, 541; 2, 87, 89, 144
V2Ob 1, 43, 47, 341; 2, 182
W 1, 275, 425
U^NbjeCb 1, 409
WO2 1, 35, 312
WO3 1, 37, 43, 45—47, 124, 408
3WO3-H2O 1, 46
W02l2 1, 35
VVS2 1, 47
XeF 1, 351
XePtFe 1, 351
Y,Ai5Ol2 2, 177
YF3 1, 441, 451; 2, 173
Y3Fe5012 1, 60; 2, 155
Y2Si303N4 1, 60
Y2O3 1, 361; 2, 21, 36, 172
YSi<Si2O3N4) 2, 266
YSiO2N 2, 266
Y5<SiO4KN 2, 266
YVO4 : Eu3+ 2, 171, 172
Ybl2 1, 316
Zn 1, 275, 357, 428, 429
ZnAl2S4 2, 147
ZnBr2 1, 318
Zn{COOJ 1, 27
ZnCr2O4 1, 28
ZnF2 1, 312
ZnFe2O4 1, 27; 2, 152, 153
Znl2 1, 316, 318
ZnO 1, 37, 276, 279, 280, 296, 306,
361, 519, 541; 2, 87, 123, 228
ZnS 1, 279, 282, 285, 289, 290, 292,
296, 300, 306, 328, 347, 390, 541;
2, 87, 123, 165, 167, 168, 173
ZnSe 1, 296, 306; 2, 87
Zn2Si04 1, 438, 449, 480; 2, 165, 168V
172, 229
Zn2Sn04 2, 150
ZnTe 1, 296, 306; 2, 87
Zn2Ti04 2, 150
ZnWO., 1, 37
Zr 1, 48, 275, 425, 429
ZrC 2, 262
ZrO2 1, 25, 98, 99, 342, 361, 393, 441,
450, 511, 541, 544; 2, 20, 22, 37, 55,
225, 259, 260, 264, 265
ZrSiO4 2, 265
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адмиттапс 2, 46, 49
Акерманит 2, 266
Аккумуляторные батареи 1, 42
Алмаз 1, 59, 60, 504; 2, 60
Алннко 2, 162
Альбит 1, 321, 443, 447, 478
Алюмо-иттриевый гранат 2, 177
Алюмосиликаты 1, 198, 373, 442
Алюмофсрриты 2, 240
Аморфные вещества 1, 46, 83
Анализ качественный 1, 65
— количественный 1, 66
— нейтронографический 1, 77
— рентгенофазовый 1, 64, 65, 66, 112
— термический 1, 63
Анатаз 1, 46
Андрадит 2, 155
Аномальная дисперсия 1, 180
Анортит 1, 321, 443, 477, 478
Антисегнетоэлектрики 2, 117, 118
Антистоксовские люминофоры 2, 173
Антиферромагнетики 1, 78; 2, 128—
154
Антифлюорит 1, 288, 291, 297, 318,
393; 2, 290
Антрацен 2, 276
Апатит 1, 107; 2, 266
Асимметрический синтез 2, 275
Атомная функция рассеяния (форм-
фактор) 1, 196, 198, 203
Аустенит 1, 486, 542
Бетон 1, 58; 2, 234, 236, 241, 248
— беспористьш 2, 251
Боксит 2, 263, 264
Борная аномалия 2, 197, 198
Ьороксоловые группы 2, 197
Бронзы 1, 47, 124, 439, 449
Брукит 1, 46
Брусит 1, 263
Брэгговская дифракция 2, 66, 74
Брэгговский угол 1, 89, 156, 182—184,,
201, 210, 213, 220
Вакансии 1, 22, 69; 2, И, 14
— анионные 2, 12, 13, 18, 20, 33
— катионные 1, 388, 402; 2, 7, 9,
И—13, 17, 33, 106
Валентная зона 1, 101; 2, 61, 65, 71,
75—77, 82—86, 98, 97
Валентность 1, 326, 329, 385
Вектор Бюргерса 1, 418, 424, 430
Виллемит 1, 438; 2, 172
Винтовая ось 1, 72, 168, 177, 178,.
192, 193, 247, 252, 255, 257—259
Вихревые токи 2, 137
Вода 1, 57, 58
Волластонит 1, 70; 2, 172, 266
Вольфрам 1, 35, 50, 52
Вюртпит 1, 59, 296 и ел., 307, 311,.
368, 450, 519; 2, 31, 122
Вюстит 1, 402, 404, 440, 456
Бадделеит 1, 342
Безызлучательные переходы 2, 170,
171
Бензодин 2, 278
Бенитоит 1, 320
Бентонит 2, 263
21—1426
Гексаферрит бария 2, 159, 162, 163
Геленит 2, 246
Гели 1, 30, 31, 46, 68
Гематит 2', 158
Гиббсит 2, 247
sp-Гибридизация Hg и РЬ 1, 327, 385-
322
Предметный указатель
Гипс 2, 235
Гистерезис 1, 139; 2, 136
Г.-шнозсм 1, 113; 2, 158
^-Глинозем 1, 37, 43, 44, 281; 2, 2, 6,
22 и ел., 37, 38, 40, 41, 50, 52, 159,
265
Гониометрия 1, 82
Гранат 2, 155, 157, 102
Границы зерен 1, 85, 411
Графиг 1. 37—40, 47, 59, 60; 2, 53,
265, 279, 280
Гроссуляр 1, 386; 2, 158
Дебай-хюккелсвскос притяжение 2,
12, 17
Дсфектообразоваиие 1, 22, 396, 400,
406
Дефекты кристаллической решетки 1,
74, 90, 100, 144
— линейные 1, 390
— пеетехиометрические 1, 390
— органических кристаллов 2, 276
— плапарпые 1, 391
— примесные 1, 400
— протяженные 1, 391
— стехиометрические I, 390
— точечные 1, 181, 390, 401, 405, 413,
451; 2. 6. 19
— У одели I, 410, 413
— упаковки 1, 74. 275, 399. 411
— Френкеля 1, 390, 392—397, 412;
2. 15, 18. 29
— Шоттки 1. 390—400, 402, 412;
2, 11, 13, 18, 29
Диаграммы Кол—Кола 2, 108, 110
— Мумра — Пирсона 1, 354, 368
— - состояния 1, 53
— ТТТ 1, 535
•— фазовые см. Фазовые диаграммы
Диамагнетизм 1, ПО, 382; 2, 128—
130, 133
Диаспор 2, 263
Диацетилены 2, 274
Динасовый кирпич 2. 262
Диопенд 2, 172
Дислокации 1, 85, 80, 91, 415. 416,
424, 430 и ел.
— винтовые 1, 22
Дислокационные трубки 1, 22; 2, 13
Дисперсоиды 2. 40
Дихалькогсниды переходных метал-
лип 1. 10. 43
Дифрпктометрпя 1. 66. 70, 73, 185.
209. 212, 216. 217
Дифракционная решетка I, 152—155
Дифракция 1, 26, 68, 74-76, 90, 148,
154, 155, 177, 182, 184, 187, 201,
220, 449; 2, 68. 71—73, 304
Дифференциальная термопара I, 136
Диффузионный барьер I, 36
Диффузия 1, 15 — 17, 23; 2, 15, 50
— скорость 1. 19, 22
Диэдрическцй угол 2, 257, 258
Диэлектрики 2, 59, 60, 104—108, 113,
123, 124, 168
Доломит 2. 263, 264
Доменная структура сегпетоэлектри-
ков 1, 91
— -¦ ферромагнетиков 1, 91; 2, 136
Доменные стенки 2, 160, 162
Доменные шлаки 2, 234
Дополнение Ван-дср-Ваальса 1, 479
Железо-иттрневый гранат (ИЖГ) 2,
155, 156, 161
Жидкие электролиты 2, 6, 21. 51
Жидкость переохлажденная 1, 140,
141
Закон Брэгга 1, 77, 155, 1Г>6, 171,
174, 175, 201; 2, 68—70, 304
— Вульфа — Брэгга 1, 446
— Иончсра (универсального диэлек-
диэлектрического отклика) 2, ПО, 111
— Кулона 1, 335
— Кюри 2. 130. 131
— Кюри — Всйг.саЧ, 115, 130, 131,
139. 141
— Моз.ги I, 114, 116, 150
— Розсбома 1, 479
Запрещенная зона 1, 101; 2, 66. 72—
77. 81. 86—90, 279
ЗародышепОразонннке 1, 14. 15, 17,
19, 20, 529 и ел.; 2, 187, 190, 202,
206, 226
Зеркальная плоскость 2, 270
Зона проводимости 1, 39, 100; 2, 76,
77. 8(>
Зонная структура 2, 168
— - — а.шази 2. 75, 78
Зонная теория 1. П. 39; 2, 59. 74, 78,
85. 95. 104
Зоны Бриллюяна 2, 70—73
—• Гинье — Престона 1, 120
Июли гор I, 50; 2, 208
И.-ллк-imr 1. 519: 2. 158, 159
Импеданс 2. 42. 45. 46, 48. 49
Инверсионная ось 1. 163, 164., 238
Предметный указатель
323.
Инверсия I, 163, 249
Индексы Миллера 1, 20, 66, 171—173,
175, 179, 188, 191, 199, 202, 223
Инженерия кристаллов 2, 277
Пнконгруэптиие плавление 1, 485, 494
Иптеркалаты 1, 37; 2. 42
Ннтсрметалличесшю соединения 2,
138
Иттрий-жслезнып грешат (ИЖГ) 2,
155, 156, 161
Кильсн.шт 1, 321
Камера Гандольфи 1, 230
— Гиньв 1, 184—186, 212, 217, 225,
233
— Девая — Шерера У, 230
— дифракционная 1, 71
— Краттки 1, 69
— прецессионная 1, 71
¦ ¦ Хегга 1, 216
Каолин I, 138, ИЗ; 2, 263, 264
Картотека порошкограмм 1, 186, 197
Катализатор Цигяера — Натта 2, 280
Катодолюминесценция 2, 165, 172,
173
Квадраты повторяемости 1, 158, 159
Кварц 1. 58. 59, 70, 442, 443, 452,
463, 464, 472. 519; 2, 122, 126, 181
Кварцит 2, 202
Керамика 1, 50. 265; 2, 258, 263, 266
Классификация Бюргера У, 505, 5№,
511
— Уббр.юОв 1, 517. 524,
Кластер Коха I. 403
Кластерообразоиание 1, 120
Кластеры дефектов 1, 440
Клинкер I. 142; 2. 235 и ел.
Клинпэпстатит 2, 229, 264
Колебательные моды 1. 95
Кочптогювское рассеяние I, 196, 226
Коннода 1. 476
Коноскопическпе наблюдения 1. 82
Константа Вейсса 2, 131
-- Мадслунга У, 345. 347
Коцформапионпые эффекты 2, 270
Координаты Аррениуса 1. 16
Координационное число 1, 39. 59, 73,
102. 114, 120, 126. 270, 283, 290
п ел.. 296. 318, 325—329, 332, 333,
335. 336. 3-11. 342, 350. 373 508;
2. 32. 156. 181, 182. 198, 262, 289,
290
Корднсрпг 2. 229. 264
Корунд 1, 59, 455; 2. 263
Коэрцитивная сила 2. 137, 160, 161
Коэрцитивное поле 2, 111
Коэзит I, 403, 519
Коэффициент термического расшире-
расширения I. 132
Кристалл анизотропный 1, 81, 83
— затравочный 1, 51—53, 55, 58
— двуоснып 1, 81, 84
— изотропный 1, 81, 83
— идноосный I, 81, 83
Кристаллиты 1, 211; 2, 255
Кристаллографическая система 1,
159. 108, 187, 194, 237. 241, 243,
244. 240, 247, 254
-- -- гексагональная I, 106, 239
кубическая 1, 239
— — моноклинная I. 84, 239
— — ромбоэдрическая 1, 166, 179,
239. 242
— — тетрагональная 1, 239
— — тригональпая 1. 165. 166, 239
— триклинная 1, 84, 239
Кристаллографические точечные груп-
группы 1, 164. 237, 238. 241, 2411 246
Крнстобалю I. 142, 230, 442, 443,
463, 4G-1. 472. 505; 2, 181. 192. 194,
22!)
Критерий Сана 2. 182
— Pot/сона 2, 182. 183
— Шмидта 2, 273, 277
Ксонитлит 1. 105
Купол кристаллизации 1. 489
— рясоланнаипя 1. 472; 2. 199
Куприт 2. 291
Лазеры 1. 52, 103; 2, 174
Латуни 1. 406. 434
Легиропанис 2. 162, 171. 174. 219,
279. 282
Легирующие добавки 2, 8. 21. 25, 27.
28. 31 36. 39. 78. 81, 82. 85, 125
Лпкнапия I, 477; 2. 199. 200
Ликвидус 1, 405, 475. 489, 490; 2. 199,
238
Люминесценция 2. 165, 168
Люминофоры 2. 166, 167, 169—174,
177
Мягнезит 2. 263
.Магнетит 1. 100; 2, 161
Магнетон Бора У 109; 2, 133
Мяпчч'онлюмбт бария 2. 159. 162
Магнитная кристалл oipa фи ческая
анизотропия 2. 137, 160
Магнитно-жесткие материалы 2, 137,
160. J 62
21'
324
Предметный указатель
Магнитно-мягкие материалы 2, 137,
160
Магнитные весы Гуи 2, 133
Магнитные материалы 2, 128, 129,
НЮ—162
Магнитный момент 2, 128, 132, 137,
139, 111, 144, 156, 160
.Магнитные потери на гистерезис 2,
137, 160, 161
Мапштостриышя 2, 138, 160
Мартенсит 1, 486, 487, 542
Мартснсптные превращения 1, 542,
513, 544; 2, 162
Массонсренос 1, 23, 31, 32, 34, 37
Мгновенное схватывание 2, 242
Межатимпыс расстояния 1, 269
Межплоскостные расстояния 1, 64,
65, 156, 171, 174, 175, 178, 179, 185,
189, 192, 202, 205, 210, 215, 218,
222, 223, 227, 231
Металлообработка 1, 48
Метастабилыюе состояние 1, 455,
460, 474, 520, 522, 523; 2, 184, 199,
200
Метастабильные фазы 1, 138
Метод атомыо-адсорбционный 1, 27
— Вайссинберга 1, 187, 189
— вращения или качения 1, 187
— Гинье 1, 182
— годографа 2, 47
— Дсоая — Шерсра 1, 182, 183, 184,
211
— дилатометрии 1, 69
- дифракции 1, 10, 63, 89, 94
— ДТА 1. 523, 544
— Лауз 1, 182
— малоуглового рассеяния 1, 69
— наименьших квадратов 1, 66
- ггрецисснонный 1, 187, 189, 191,
192
— рептгено.пюминесцентный 1, 27
— эмиссионный 1, 112
Механизм Вагнера 1, 16
Микроликвация 1, 91
Микроскопы 1, 79, 80, 82, 84, 86, 87,
90, 91
Минерализатор 1, 58
Мишень 1, 49
Модель конфигурационных коорди-
координат 2, 168
Модели Сандсрсона 1, 354—356,
362—367, 386
Модификатор 2, 195, 200
Монипил 2, 128
Монокристалл 1, 17, 18, 26, 31, 34,
51, 58, 70
Мопотектическос равновесие 1, 473
Монтмориллонит 2, 263, 278
Морфология кристаллов 1, 79, 82, 84
Мост Уитсона 2, 42
Муллит 2, 263
Мусковит 1, 321
«Мягкая» химия 1, 45, 444
Наклеп 1, 68, -116
NAS1CON 2, 38
Пейтронограммы 2, 145
Пеуиругое рассеяние 1, 78
Область стеиования 2, 257
Область расслаивания 2, 213
Обменное взаимодействие 2, 136
Опеидиан 1, 526
Огнеупоры 1, 142; 2, 37, 254 и ел.
Оже спектроскопия 1, 88
Окраска неорганических твердых тел
2, 90
— хроматов 1, 100
Оливпн 1, 278, 443
Оптическая пндпкатоиса 1, 81; 2,
274
Оптическая микроскопия 1, 84—86
Оптические оси 1, 81, 83, 84
Орбитальный момент 2, 134, 157
Органические металлы 2, 268, 278
Органические твердые растворы 2,
275
Орт.жлла 1. 321
Остаточная намагниченность 2, 137
Ось симметрии 1, 72, 83, 153, 165—
167, 188
Отравляющее действие примесей 2,
171
Очистка зонная 1, 52
— но способу Ван-Арксля 1, 35
—• улыразпуковая 1, 51
Парамагнетики 1, ПО, 331; 2, 128—
131, 133, 135, 139, 142-144
Парафенилеидиамии 2, 283
Парачлектрик 1, 507, 510
Переохлажденная жидкость 2, 184,
1R6, 187, 191, 213
Переохлажденный расплав 2, 207
Переход парамагнетик — ферромаг-
петш; 1, 85
— париэлектрнк — сегнетоэлектрик 1,
83: 2. 125
— порядок — беспорядок 1, 276, 510,
545; 2, 139
Периклаз 2, 264
Предметный указатель
325
Пери тактическая температура 1, 485,
493
Перитектоидная реакция 1, 470, 485;
2, 33
Исрнтсктоидная точка 1, 4G8, 485
Перлит 1, 48G
Перовскит 1, 259, 325, 519; 2, 112,
113, 116, .117, 158, 290
Петля гистсрезпел 2, 117, 137
Пикномстрия 1, 452
Пирскс 2, 187, 197, 214
Пироп 2, 155
Пнроэлектрики 1, 24G; 2, 105, 121,
124
Плап.-icniie 1, 144
— инкошруэнтное 1, 32, 4G9. 481
— коигру.-жтное 1, 31, 4G9, 4У2, 493,
.y?9
Плагиоклазы 1. 412, 443
Носики 2, 162
— нитридов 1, 48
— оксидные 1, 48
— тонкие кристаллические 1, 47, 49,
50, 7G
— эппгаксиальиые 1, 49
Плоскости глбптусл 1, 542
Плоскость зеркальною отражения 1,
162 238, 241, 242, 244, 247, 253,
255
— решетки 1, 170, 171
— сдвига 2, 85
— симметрии 1, 163, 164, 1G6, 167
Поворотная ось 1, 161, 164 —1G6, 238,
•J47. 251, 252, 25G
Пчд.ю,кка 1, 19, 48, 49, 53; 2, 162
J ifbHcrop 2, 126
Позиции тпи-Биверса — Росса 2, 2G
— Бит'рса — Росса 2, 26
— oiu лэдричеекгге I, 14
¦ тстраэдрнчегкпе 1, 14
регулярные 1, 60
Показатели преломления 1, 84, 85
Покрытие никелевое 1, 48
— оксидное 1, 48
Полппцетилсп 2, 2G8, 278—280
Полимеры аморфные 1, 142; 2, 180,
274
— электропроводящие 2, 278—280
Полиморфизм 2, 14, 31, 44, 53, 105,
218, 3/5, 382
Полиморфные превращения 1, 218,
505, 526, 529, 534, 537
ПолAпарафенн.1си 2, 282
По.иширрил 2, 282
Полтиппя 1. 271, 275, 411
Пилшцелочной эффект 2, 30, 31, 211
Пплнщс.ючиые отекла 2, 211
Полоски Бекке I, 81
Полупроводники 1, 68, 112; 2, 59—61,
95, 96, 101, 104, 125, 126, 168, 281,
282
Поляризация 2, 106, 109, 113—118,
121, 122, 137
Поправочный температурный множи-
множитель 1, G8
Пористость I, 181
Порошкограммы 1. 62, G5—G7, 98,
148, 171, 179, 180, 185, 18G, 194,
198, 200, 210, 214, 217—219, 222,
225 и ел., 231—233, 445; 2, 144—
1-16, 192
Портландцемент 2, 234 и ел., 241, 248
Портланд цементный клинкер 2, 498
Порядок реакции 1, 29
Постоянная Болъцшна 1, 545; 2, 133,
306
•• • Планка 1, 92
— Фирпдся 2, 52
Правила Зихариасена 2, 180—182
— Полита 1, 28G, 330, 362, 304; 2,
27
Привило Всгарда 1, 67, 446, 447
— моментов 1, 491
— рычага 1, 446, 447, 448, 449, 4GG,
47G, 491
— ХцнОа 1, 374
— фаз I. AM, 455, 484, 487, 490, 497,
524. 525, 540
Принцип Ли Шите.лье 1, 519, 520, 540
— неопределенности Гсйзенберга 1,
219
-- орпешацпонпого соответствия 1,
53
Проводимость диэлектриков 2, 143
-- дырочная 2, 5G, 84, 103, 282
— ионная 1, 108: 2, 5, 6, 7, 19, 21,
31, 34—39, 48. 104, 281, 300
— примесная 2, 11, 16, 78—80
— собственная 2, 11, 16, 79, 82
— электронная 2, 5, 6, 35, 42, 48, 5G,
103
Пространственные группы 1, 72, 75,
168, 187, 194, 237, 246—253, 256,
257
Пссвдоволластопят 1, 70
Пьезо-мектрнки 1, 24G; 2, 105, 122,
124
Равновесие 1, 49
Радиусы атомные 1, 350
— ионные 1, 74, 330, 332, 333, 437
— по Гольдш.мндту 1, 330, 332
Пплши-.у I, 330, 332, 437
— термохимические 1, 348
Разуморядоченне 1, 74, 144
Распил пермектоидиып 1, 471
326
Предметный указатель
— -шгектсидныи 1, -4/1
Рассеянно электронов 2, 60
Расстеклсвнванпе I, 141; 2, 194, 195,
212
Растепление Зеемана ), 128
Неакции внедрения 1, 48
- гетерогенные I, 48
-¦ гидре [ермальиые h 31 _
— димеризации 2, 2/1, 2,5, 277
— ионного обмена 1, 38, 43, 44
— полимеризации 2, 273, 274. 280
— разложения 1, 145
-¦ сополимернзации 1. 3D
— твердофазные 1, 23, 27, 29
— топитакгическне 1, 19, 20
— транспортные 1, 33. 3G, 49
¦- фотоприсоедипепня 2, 277
— эвтектические 1, 467, 484, 485
-¦- „штектиидпые 1, 484, 485
эшгт аксиальные 1, И), 20
Рентгенограмма 1, 62, 64, 447; 2, 70,
145
Решетка базоцептрнроканпан 1, 169,
170, 176
— гексагональная 1. 1/3, 228
— грапецентриронанпая кубическая
(ГЦК) 1, 78 169, 176, 199,2U2.204,
257, 328, 411, 425, 486; 2, 77
пепрнммтииная 1, 176
— обратная 1, 71
— объемпоцентрирсванпая кубиче-
кубическая (ОЦК.) 1, 169, 170, 176, 194,
259. 328. .}()!, 425, 439, 496; 2, 32,
77, 138
¦¦ - примишнпая кубическая 1, 69, 83;
2, 36, 147
— ромбоэдрическая 1, 176
Решетки Право 1, 168, 170, 237, 246
Puc-i- .icptn 2, 255, 257
Рост кристаллов .метод Бриджмена
1, 52
— ¦ Всрнсйлл 1, 55
— — — гидротермальный 1, 55, 56,
57, 58
— ¦¦ — — Стпкбаргери 1, 52
— -- — «холодного затвора» 1, 52
— Чохральского 1, 51
— — механизм I, 85
— топотнксичеекип 1. 53
— •лштакспальнмй 1, 53
Рубин 1. 55, 59
Рутил 1, 46. 59, 262. 278, 283, 312,
336. 380. 381, 386. 407, 409, 536
Санидин 1. 107
Сапфир 1, 55
Снерхпроводпмсеть 2, 283
Свер\с1-руктура 1. 78; 2. 145
Связь н керамике 2, 266
— — огнеупорах 2, 262
— водородная 1, 96
— длина 1, 73, 114
— ионная 1, II, 74. 326—329, 3G8;
2, 85, 86, 89, 91. 168
— ковалентная 1, 74, 96, 326—328,
351; 2, 1Ь8
— прочность 1, 10; 2. 20Г)
— тип 1, 73, 269; 2, 179. 130, 191,
262
— энергия 1, 10, И, 362, 365, 366;
2, 182
Сегнетсэлектрпки 1, 91, 97, 142, 343,
504. 507, 510. 2, 105, 111 — 113. 115,
117, 118, 124. 126, 136, 137
Сенсибилизаторы 2, 165, 170, 171, 173
Серпентин 2, 261
Сиалон 1, 443; 2, 2(>(i
Силлиманит 2, 264
Символика Гер.чина—Могена 1, 161
— Uh-нфлиса 1. 161, 162
«Синтетические металлы» 2, 28IJ
Синхротрон 1,117
Система As—Те 2. 217
. в,О—SiOo 2. 196
— СаО -А!2Ь3 2. 183
_ Ge—P—S 2, 217
— Li2O-Al?O3- SiO2 2, 231
— MgO—A12O3 SiO2 2, 229
— MgO- S3M, 2. 258
¦ \a2O—)^O, 2, 197
— Na2O—B2O3—SiO2 2, 214
_ P-.Se— S 2. 217
-¦¦¦ -Si—Al 2, 217
— Si Ла—Те 2, 217
¦ Si—Те 2. 217
Система '^мпшаленткых позиций 1,
239. 240
(лпаллы 1, 33
Скорость тоердефазпей реакции 1,
16. 17, 22
Смачивание керамики 2, 258
Соединения внедрения 1, 38, 40
Солпдус 1, 466, 475, 490
Спекание 2, 256, 262, 261, ?66
Спектр с переносом заряда 1, 100
Спинодаль 2. 205. 213, 460
Сплавы 1, 69, 78; 2, 265
Сподумен I, 321, 442; 2, 228
Способ счистки Каы-Лрксля 1, 35
Стали 1, 485, 487, 504, 542; 2, 254
Стандартные таблицы оптических
данных 1, 84
Стекло 1, 33. 48, 50, 152, 171, 522;
2. 177, 178. 254
— Пиратпое 2, 179, 197, 208
Предметный указатель
327
— боросплнкатпое 2, 179
— кизкость 2. 180, 189, 195, 197, 198,
206 207, 212. 235—217
— впкор 2, 1У9, 21-4
-- ионная прокодпмостъ 1, 68; 2, 208,
21 1 и ел.
— кварцевое t. 1-41, 523; 2, 178, 181,
192. 104. 197. 211. 212
— керамика J. 522; 2, 34, 225
— кристаллизация 2, 183, 187 и ел.
— металлическое 1, 68, 142
— пеодимовое 1, 68
— оконное 2, 187, 212
— окраска 1, 102; 2. 200. 212
— оксидное 1, 120; 2. 266
— ипалесцепцпя 2, 200, 228
— основность 1, 102
— - получение 1, 140
— расслаивание в жидкости 2, 200,
2П2
— силикатное 1, 84, 85, 120; 2, 179,
191. 1ЯГ). 147, 200, 208
— структура 1, 102
— фотохромнпе 2, 231
— хальксгенидкое 1, 101, 214
Стоклообралсвание 1, 110, 2, 179—
183 187, 189, 192. 223
Стеклооиразователп условные 2, 179
Сгеклсобразующие оксиды 2, 179,
1Я0. 182, 196
Степень иенности 1, 327
Сюрсографические позиции 1, 239
Ciepeope. улярные структуры 2, 274
GiHumispif 1, 59, 463, 519
Г.тиксиискип сдвиг 2, 166
Структура кристаллическая, опреде-
определение 1, 73. 76
— магнитная 1, 78
— rc-i'iaiasi 2, 181, 194
— скшоая 1, 108, 328. 411
Структурная амплитуда I, 20-1, 207,
231
Структурный фактор 1. 204
Суперпсппые проводники 2, 6
Сфалерит 2, 32
Счетчик Гейгера 1. 185, 210
Тиердыс электролиты 1, 43, 2, 6, 19,
21 и гл.
Твердый раствор 1, 27, 67, 14-4, 400,
1116. 43о п ел. 486, 489, 500, 501,
542; 2. 2, 8, 36, 41, 83. 123, 154,
158. 17:). 275
Текстура 2. 210. 231. 239. 255. 257
— м.чпштпп-уиорядоченнаи 2, 1G2
Темпераiура компенсации магнитных
моментов 2. 157
— критическая води 1, 57
-¦¦ Кюри 1, 98, 2, 115, 139, 143
— Кюри — Вейсса 2, 215
— II ее ля 1, 139, 143, 145
— стеклования 1, 141, 523; 2, 184,
185, 207, 227, 266
Тенортит I, 380
Теория Блоха 2, 66
— ^ом.черфельда 2, 64
— кристаллического поля 1, 374, 378
Теплопроводность 1, 39; 2, 265
Термисгоры 2, 84, 125
Термический анализ (ДТА) 1, 134
Термическое тушение люминесценции
2, 169
Тетраметидор-юсиликат 2. 178
Тетраметилпарафеиплепдиамин 2, 283
Тетратиофулвален 2, 283
Тетрацнапохинсдиметап 2, 283
Тлеющий разряд 1, 49
Тсберморит 2, 251
Тонкие пленки, анализ 1, 80
— — получение 1, 47
Топотакгические превращения 1, 538
Точечная симметрия 1, 164
Точечные группы 1, 241—247
Точка Ван Рейна I, 492
Травление 1, 51, 85, 86; 2, 255
Тридимпт 1, 442, 443, 452, 463, 464,
472
Трубки газоразрядные 1, 48
Увировпт 2, IM
Угловой множитель 1, 206, 207
Упаковка гексагональная плотней-
шая (ГПУ) 1, 269 и ел, 279, 282,
288, 300, 315, 336, 425; 2, 31, 138
— кубическая плотпейшая (КПУ) 1,
14. 269. 271 и ел, 396, 403 406,
111
— объемноцентрированпая (ОЦУ) 1,
примитивная чеграгональная I,
266. 283, 336
Уравнение А в рами 1, 533
— Арречиуси 1. 527; 2, 8, У, II, 85,
207. 306
— Борна — Майера 1, 346, 363, 378
— Дс Бройля 2, 69
— Капустинского 1, 347, 348. 386
-- Клаузиуса — Клапейрона 1, 520
— JIat/э 1, 155
— Нррнгта—Эйнштейна 2, 15, 50
— скорости реакции 1, 29
— Томсона 1, 195
— Фи 14,'jm 2, -207
— Шрсдиигера 2, 6С
328
Предметный указатель
Уровень Ферми 2, 65, 74, 83, 84, 96,
102, 104
Фаза, определение 1, 455
Фазовая диаграмма 1, 32
анортит — альбит (плагиокла-
(плагиоклаза) 1, 477
воды 1, 462
однокомпонентной системы 1,
461
углерода 1, 60
Agl—Rbl 2, 33
А12О3—CaO 2, 249
А12О3—Cr2O3 1, 438, 447, 448,
457, 458, 481; 2, 174
А12О3—SiO2 2, 258, 260
CaO—А12О3—Fe2O3 2, 247
CaO—А12О3—SiO2 2, 237, 238
CaO—SiO2 1, 472, 473, 482,
199; 2, 258
Fe—С 1, 485
Fe—SiO2 2, 260
Fe3O4— SiO2 2, 258, 260
K2O—SiO2 2, 258, 260
Li2O-Nb2O5 1, 99
Li2O—SiO2 2, 199, 229
MgO—A12O3 1, 441, 445, 446
MgO—SiO2 2, 258, 260
MgAl2O4—A12O3 1, 438, 480
Mg2Si04—Zn2Si04 1, 438, 480
Na—S 2, 54
Na2B8O13—SiO2 2, 213
Na2O—SiO2 2, 199, 258, 260
PbCrO3— РЬТЮз (ЦТС) 2, 123
SiO2 1, 463
SiO2—LiAlO2 1, 442
SiO2—UAIS1O4 1, 481
Фазовые переходы порядок — беспо-
беспорядок 1, 218, 230, 405, 413
Фазовый переход I рода 1, 149, 219
II рода 1, 149
классификация Эренфеста 1,
511
Фазовый сдвиг 1, 199, 200, 201, 202
Фазы Магнели 1, 407
— Судзуки 1, 413
Фактор повторяемости 1, 178, 179
— расходимости 1, 207
— спектроскопического расщепления
1, 109
— Хейвена 2, 15, 16, 30, 50
Фенацит 1, 59
Ферримагнетики 1, 78, 91; 2, 128, 151,
160
Ферриты 1, 27; 2, 129, 135, 151, 152,
162, 240
Ферромагнетики 1, 504; 2, 128, 130,
131, 135—144, 158, 160, 162
Флогопит 1, 321
Флуоресценция 2, 165
Флюорит 1, 393, 404, 441; 2, 35, 36
Флюс 1, 53
Флюсующая активность шлака 2, 258
Фононы 1, 78
Формула Больцмана 1, 389
— Уоррена 1, 221
— Шерера 1, 221
Формульные единицы 1, 180, 181
Форстерит 1, 438, 455; 2, 228, 264
Фосфоресценция 2, 165
Фотолюминесценция 2, 165
Фотопроводимость 1, 101; 2, 85
Фракционная кристаллизация 1, 476
Функция радиального распределения
(ФРР) 1, 68; 2, 192
Футеровка доменных печей 2, 263,
264
Халькогениды 1, 142, 294, 296, 332;
2, 35, 91, 278
Химические сдвиги, эффект Мёссбауэ-
ра 1, 126, 127
ЯМР 1, 105, 107, 109, 123
Хлоранил 2, 283
Хрусталь 2, 187
Шкала Полинга 1, 437
Шлаки 2, 258, 265
Шлакокерамика 2, 231
Шлакопортландцемент 2, 245
Шпинель 1, 16, 19, 23, 27, 43,47, 59, 229,
278, 285, 325, 384, 385, 406, 440,
445, 446, 519; 2, 24, 25, 85, 89,
147—149, 153
Цемент 1, 58, 522; 2, 234, 254
— высокоглиноземистый 2, 446
— глиноземистый 2, 245
— пуццолановый 2, 234, 249, 250
Цементит 1, 91, 485
Цементный клинкер 1, 142; 2, 235
и ел.
Центр симметрии 1, 180, 206, 243, 246
и ел.; 2, 116, 121, 271
Центры окраски 1, 100, 112, 397, 398,
399
Цеолиты 1, 30, 37; 38, 2, 38
Цикл Борна — Габера 1, 348—351,
378
Цинковая обманка 2, 61
Предметный указатель
329
Цирконат-титанат свинца (ЦТС) 2,
123, 126
Циркония диоксид 2, 36, 37, 57
Эвкриптит 1, 321, 442, 481
Эвтектика 1, 143, 466, 484, 490, 491,
493, 542; 2, 183, 257
Эвтектоидный распад 1, 484; 2, 33
Экситонный переход 2, 91
Экситонная полоса 1, 100
Электродвижущая сила ячейки 1, 42
Электролюминесценция 2, 165
Электронные переходы I типа 1, 102
Электропроводность 1, 39; 2, 5 и ел.,
50, 59, 65, 82, 102, 265
Элементарная ячейка 1, 164 и ел.
Элементы симметрии 1, 159, 161, 163,
164, 166—168, 177, 253, 257, 259
Энантиомеры 2, 275
Энергия активации 1, 16
— Гиббса 1, 511, 520
— Ферми 2, 64
Энстатит 1, 443
Эпитаксиальный рост 1, 49; 2, 162,
226
Эффект Допплера 1, 126
— Зеебека 2, 97, 101
— Киркендаля 1, 17
— Пельтье 2, 96, 101
— Томсона 2, 94—96
— Холла 2, 102
Ячейки электрохимические 1, 41
ОГЛАВЛЕНИЕ
Глава 13. Ионная проводимость и твердые электролиты .... 5
13.1 Типичные ионные кристаллы 7
13.1.1. Галогениды щелочных металлов 7
13.1.2. Хлорид серебра 15
13.1.3 Фториды щелочноземельных металлов 18
13.1.-4. Проем ые С1схномстр|[']еские оксиды 18
13.2. Твердые электролиты (ионные проводники, супериоппые провод-
проводники) 19
13.2.1. р-Глинозсм 22
13.2.1.1. Структура 22
13.2.1.2. Проводимость и механизм проводимости .... 27
13.2.2. Иодид серебра и др>~1 не Ag+-проводящие твердые ллектро-
литы 3J
13.2.2.1. Иодид серебра Agl J1
13.2.2 2. Rb.Ag4b, 33
13.2.2.3. Другие твердые электролиты — производные Agl . 34
13.2.2.4 Фазы на основа халькоюнидов серебра .... 35
13.2.3. Галогемид-иопиые проводники 35
13.24 Кислород-иоипьге проводники . 36
13 2.5. Попеки новых твердых электролитов 37
13.3. Методы измерения проводимости 40
13.3.1. Измерения на постоянном токе 40
13.3 2. Измерения на переменном токе 42
13.4. Другие экспериментальные методы 49
13.5. Применение твердых электролитов 51
13.5.1. Принципы действия электрохимических ячеек ... 51
13.5.2. Источники тока 52
13.5.3. Кислородные концентрационные ячейки н датчики ... 55
13.5.4. Топливные элементы 57
Упражнения 57
Литература 58
Глава 14. Электронные свойства и зонная теория: металлы, полупровод-
полупроводники, твердые неорганические соединения, их цветность. . 59
14 1 Введение —металлы, диэлектрики, полупроводники ... 59
14.2. Электронная структура твердых тел. Зоилая теория . . 61
14.3. Усовершенствование простои лонцоп теории. /г-Простракство и
зоны Бриллюэпа 68
14 4 Зонная струмлря металлов . 74
Оглавление 331
1-1.5. Зонная структура диэлектриков 75
14.6, Зонная структура полупроводников: кремнии 76
U.7. Полупроводники с контролируемо)"! валентностью .... 81
14.8. Применение полупроводников 83
14.9. Зонная структура неорганических тиердьгх тел . 85
1-1.10 Цветность неорганических твердых ту л . 90
1-111. Энср!етпчеекп,.* зоны и.1ч химическая связь. Заключительные
замечания 91
Упражнения . . 92
Литература ... 93
Глава 15. Другие электрические соойства 94
15 1. Термоэлектрические явления 9-1
l.i. 1.1. Эффект Томсона 94
15 1.2. Эффект Псльтье 96
!.~! I 3. Эффект Зесбека 97
15.1.4. Термопары 98
15.2. Эффект Холла 102
15.3. Диэлектрики 104
15.4. Сстнетоэлсктрикп Ill
15.5 Пироэлектрикп 121
15.6. Пьезоэлектрикн 122
15.7. Взаимосвязь между сегнето-, пьезо- н пироэлектрическими свон-
стнпми 123
15.8. Применение сегнето-, пьезо- и пироздектриков 124
Упражнения 126
Литература 127
Глава 16. Магнитные свойства 128
16.1. Введение 128
16.2. Теория магнетизма 129
16.2.1. Поведение вешестп в магнитном поле J29
16.2.2. Влияние температуры. Законы Кюрин Кюри — Beiioca . . 130
16 2.3. Расчет величины магнитного момента 133
16.24 Механизм ферро- и антиферромапштного упорядочения, об-
обменное взаимодействие '35
16.2.5. Некоторые другие понятия 136
16.3. Примеры магнитных материалов, их структуры и свойства . . '38
16.3.1. Металлы и сплавы '38
16.3.2 Оксиды переходных меааллов '43
16.3.3. Шпинели . . . . . , 47
16.3.-1. Гранаты . . . . • '55
16.3.5. Ильменитьт и перовскиты 158
16.3 6. МагпетоплюмГжты '59
16.4. Применение. Взаимосвязь структуры и свойств '60
16.4.1. Сердечники трансформаторов '60
16.4.2. Запоминающие устройства 161
16.-1.3. Элементы памяти на цилиндрических магнитных доменах 162
16.4.4 Постоянные магниты '62
Упражнения 163
Литература 163
Глаиа 17. Оптические свойства. Люминесценция и лазеры .... 165
17.1. Люминесценция и люминофоры 165
332 Оглавление
17.1.1. Определения и вводные замечания 165
17.1.2. Модель конфигурационных координат 168
17.1.3. Некоторые фосфоресцирующие материалы 171
17.1.4. Антистоксовгкие люминофоры 173
17.2. Лазеры 174
17.2.1. Рубиновый лазер 174
172.2. Неодимопые лазеры 177
Упражнения 177
Литература 177
Глава 18. Стекло 178
18.1. Факторы, влияющие на стеклообразоваиие 179
18.1.1. Оксидные стекла. Электроотрицательность и тип химической
связи 179
18.1.2. Вязкость 180
18.1.3. Влияние структуры. Правила Захариасена 180
18.1.4. Критерии Сана и Роусона 182
18.2. Термодинамика процесса стеклования. Поведение жидкостей прл
охлаждении 183
18.3. Кинетика кристаллизации и стеклования 187
18.4 Структура стекол 191
18.1.1. Кварцевое стекло !92
18.4.2. Силикатные стекла '95
18.4.3. Стеклообразный В2О3 и боратные стекла 197
18.5. Несмешиваемость жидкостей и ликвидация в стеклах . . . 198
18.5.1. Структурные теории расслаивания жидкостей .... 200
18.5.2. Термодинамика расслаивания в жидком фазе .... 20!
18.5.3. Механизм расслаивания 202
18.6. Вязкость 206
18.7. Электрическая (ионная) проводимость стекол и полищелочной
эффект 208
18.8. Промышленные силикатные и боратные стекла ;г:'
18.9. Халькогепидпыс и другие полупроводниковые стекла . . . ^|?
18.УЛ. Халькогенидпые стекла ~!^
18.9.1.1. Сера ^15
18.9.1.2. Селен 216
18.9.1.3. Теллур 21?
!89 1.4. Стекла более сложного состаиа 217
18.9.2. Электрические свойства 217
18.9.3. Фотокопировальный процесс 220
18.10. Металлические стекла . 222
18.11. Стеклокерамика 225
18.11.1. Наиболее распространенные стеклоксрамичсскне системы 228
18.11.2. Свойства стеклокерамических материалов 229
18.11.3. Применение стеклокерамики 23!
Упражнения 232
Литература 232
Глава 19. Цемент и бетон 234
19.1. Портландцемент 235
19.1.1. Производство 235
19.1.2. Особенности диаграммы состояния 236
19.1.3. Полиморфизм силикатов кальция 240
19 1.4. Гидратация портландцемента 241
19.1.5. Разновидности портландцемента 244
Оглавление 333'
19.2. Глиноземистые и высокоглнноземистые цементы .... 245-
19.3. Пуццоланы п пуццолановне цементы 249
19.4. Продукты автоклавного синтеза 250
19.5. Магнезиальный цемент , 251
19.6. Последние достижения — беспористый бетон (MDF) . . . 251
Упражнения 253
Литература 253
Глава 20. Огнеупорные материалы 254
20.1. Микроструктура или текстура 254
20.2. Размеры зерен и их рост 255
20.3. Спекание 256
20.4. Поверхностные явления 257
20.5. Воздействие шлаков на огнеупоры 258
20.6. Прочность 258
20.7. Виды огнеупорных материалов 259'
20.8. Последние достижения Сиалоны — питридпые огнеупорные ма-
материалы 265
Упражнения 266
Литература 267
Глава 21. Органическая химия твердого тела 268
21.1. Топохимичсскин контроль органических реакций в твердом со-
состоянии 268
21.1.1. Внутримолекулярные реакции. Конформациоппые эффекты 270'
21.1.2. Межыолекулярные реакции. Эффекты молекулярной упаковки 271
21.1.3. Фогодимсризация о-этокси-транс-коричной кислоты . . 27!
21.1.3.1. а Форма 271
21.1.3.2. Р-Форыа 271
21.1.3.3. у-Форма 272
21 1.4. Фотонолнмеризация 2.5 дистирилпиразипа 273
21.1.5. Фотоиолпмеризация диацетилепов 274
21.1.6. Асимметрический синтез 274
21.1.7. Димеризация антрацена. Роль дефектов кристаллической ре-
решетки 275
21.1.8. Регулирование способа упаковки молекул в кристаллах 6
21.1.9. Органические реакции в неорганических матрицах . . . 278
21.2. Электропроводящие органические твердые тела. Органические
металлы 278
21.2.1. Сопряженные системы 27»
21.2.1.1. Легированный полиацетилен 27S
21.2.1.2. Полипарафениле.н 282
21 2.1.3. Полипиррол 282
2J.2.2. Органические комплексы с переносом заряда. Новые сверх-
сверхпроводники 282
Литература 284
Приложение 2^5
А1. Геометрические подходы в кристаллохимии 285
А1.1. Замечании по геометрии тетраэдров и октаэдров .... 285
А1.1.1. Тетраэдр и куб . 285
А1.1.2. Расстояния М~~ X и Х~ X в тетраэдре 285
А1.1.3. Угол ХМХ тетраэдра 285
А!. 1.4. Симметрия тетраэдра 285
334 Оглавление
Л 1.1.5. Центр масс тетраэдра ... 285
AI.16. Октаэдр и куб 286
AI.2. Гексагонялышя элементарная ячейка. Дока^псльетви тогм.
что с'щ= 1,633 287
А2. Построение моделей 287
A2.I. Характер упаковки шаров 288
A2.1.I. Демонстрация связи между КИУ-струк ryjioii и элементар-
элементарной ГПК ячейкой 288
.•\2.!.2. Демонстрации четырех ориентации л.'ттпо;, пакин.'шш )\
сто с и и a-EvMfiiтripf;oii ГЦКячсйке 288
А2.2. Полиэдрические структуры 289
A3. Как узнать ллотноупакованние (эвтакткчесьпе) структуры? . . 289
А4. Положительные н отрнцтельпые координаты атомов . . . 291
А5. Кристаллографические точечные группы 292
А6. Л\ежплоског1пые расстояния и объемы осмешарных ячеек . . 293
А7. Обратная решета .... ....... 294
А7.1. Изображение кристаллической решежн в реальном и обратном
пространствах "°
A7.I.1. Доказате.м,стпо того, что обратная решетка является трех-
трехмерной совокупностью узлов 297
A7.I.2. Соотношение между параметрами кристаллической решет-
решетки I! параметрами обратной решетки 298
А7 2. Систематическое ноккапие рефлексов п обратная решетка 299
Л7.3. Дифракция и обратная решетка. Сфера отражения ,'-)вальда 30-4
А8. Уравнение Арренпуса для ионной проводимости ... - 306
А9. Некоторые свойства элементов 308
Формульный указатель 314
Предметный указатель 321
СОДЕРЖАНИЕ 1-й ЧАСТИ
Предисловие редактора перевода
Предисловие.
Глава 1. Что такое химия твердого тела?
Глава 2. Препаративные методы
Глава 3. Применение физических методов для ис-
исследования твердых неорганических ве-
веществ
Глава 4. Термический анализ
Глава 5. Дифракция рентгеновских л\ чей
Глава 6. Точечные группы, пространственные груп-
группы, кристаллическая структура
Глава 7. Описательная кристаллохимия
Глава 8. Некоторые факторы, влияющие на струк-
структуру кристаллов
I лава 9. Дефекты и кристаллах и нсетехиометрия
Глава 10. Твердые растворы
Глава 1 1. Интерпретация фазовых диаграмм
Глава 12. Фазовые переходы