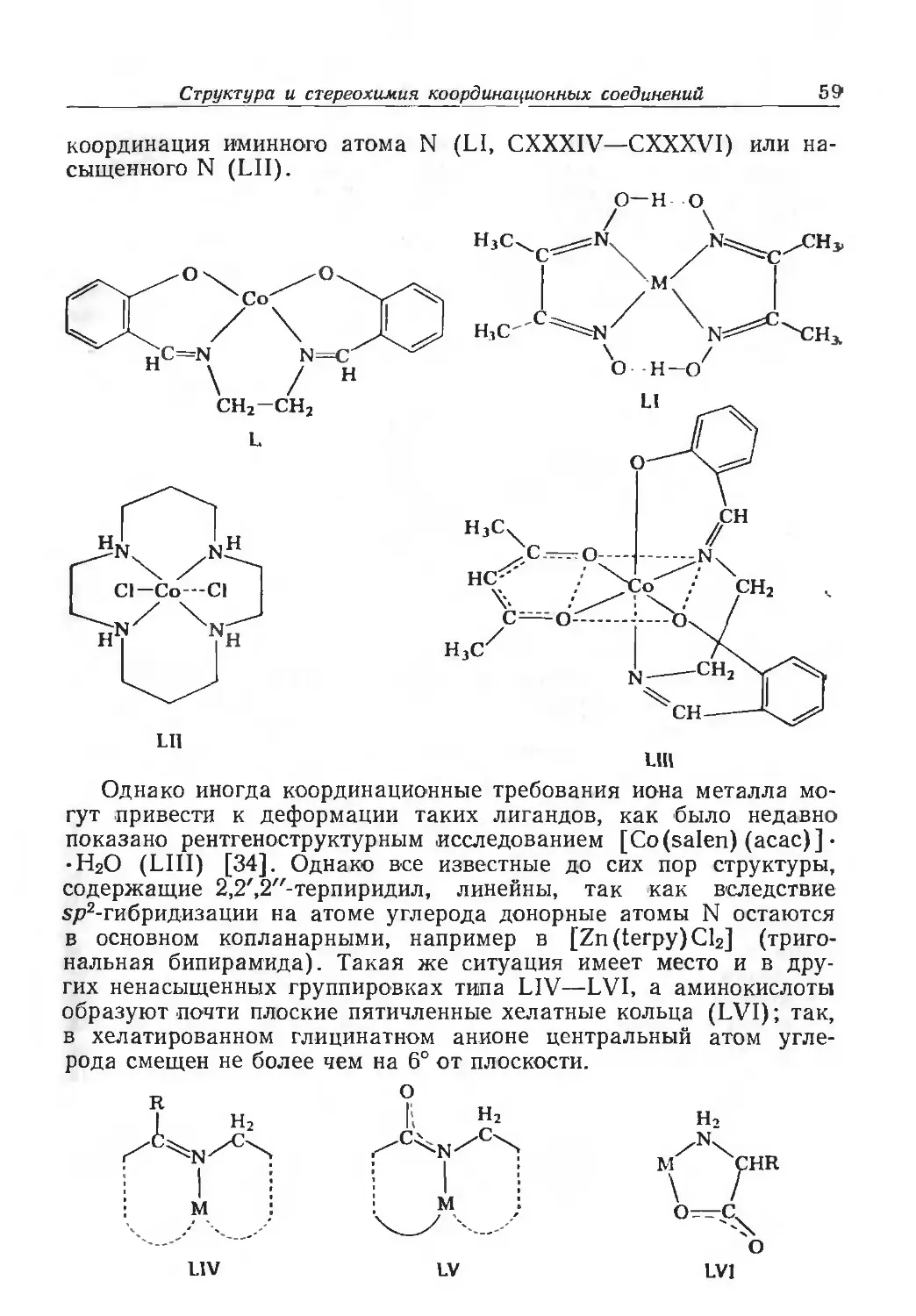
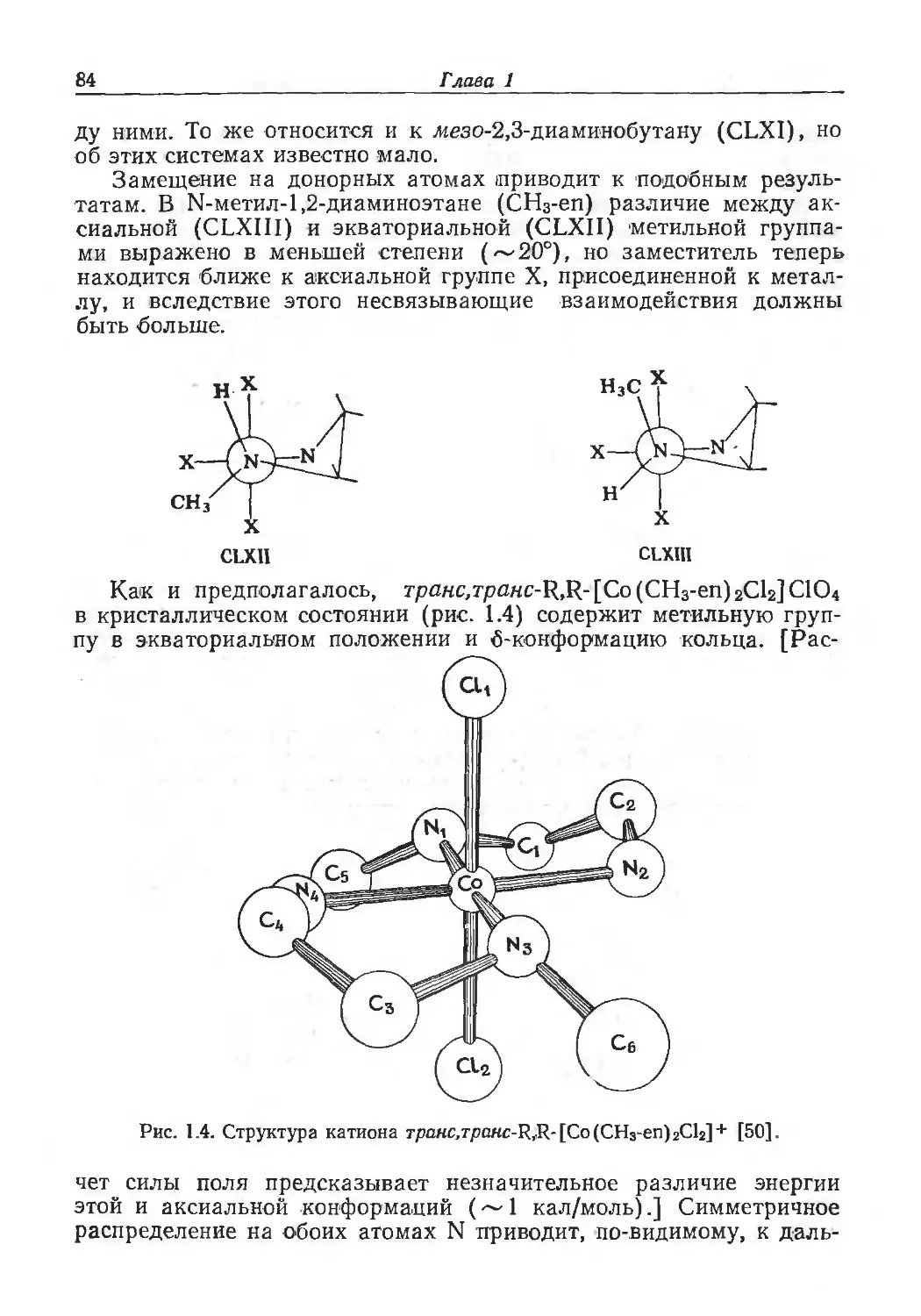
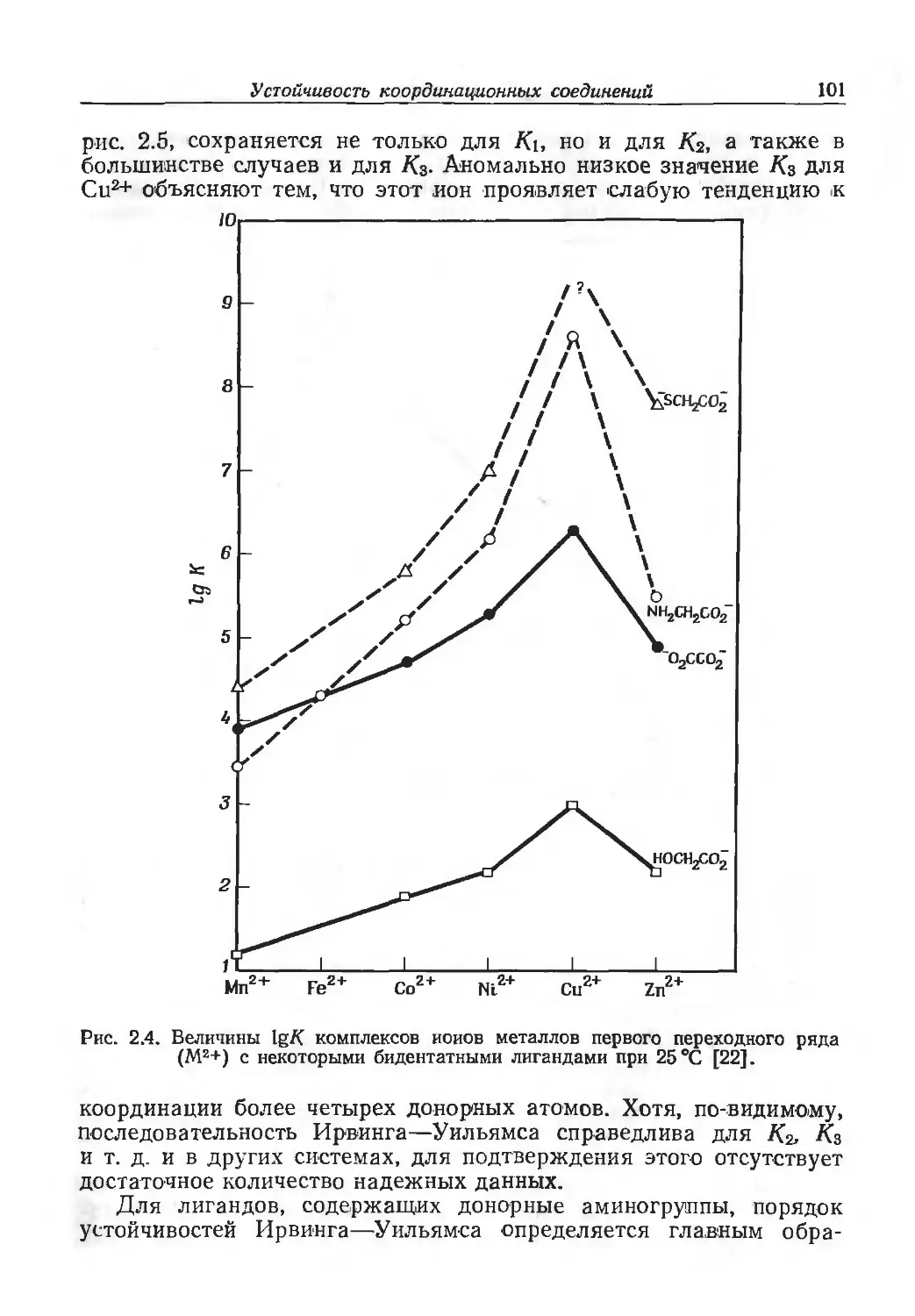
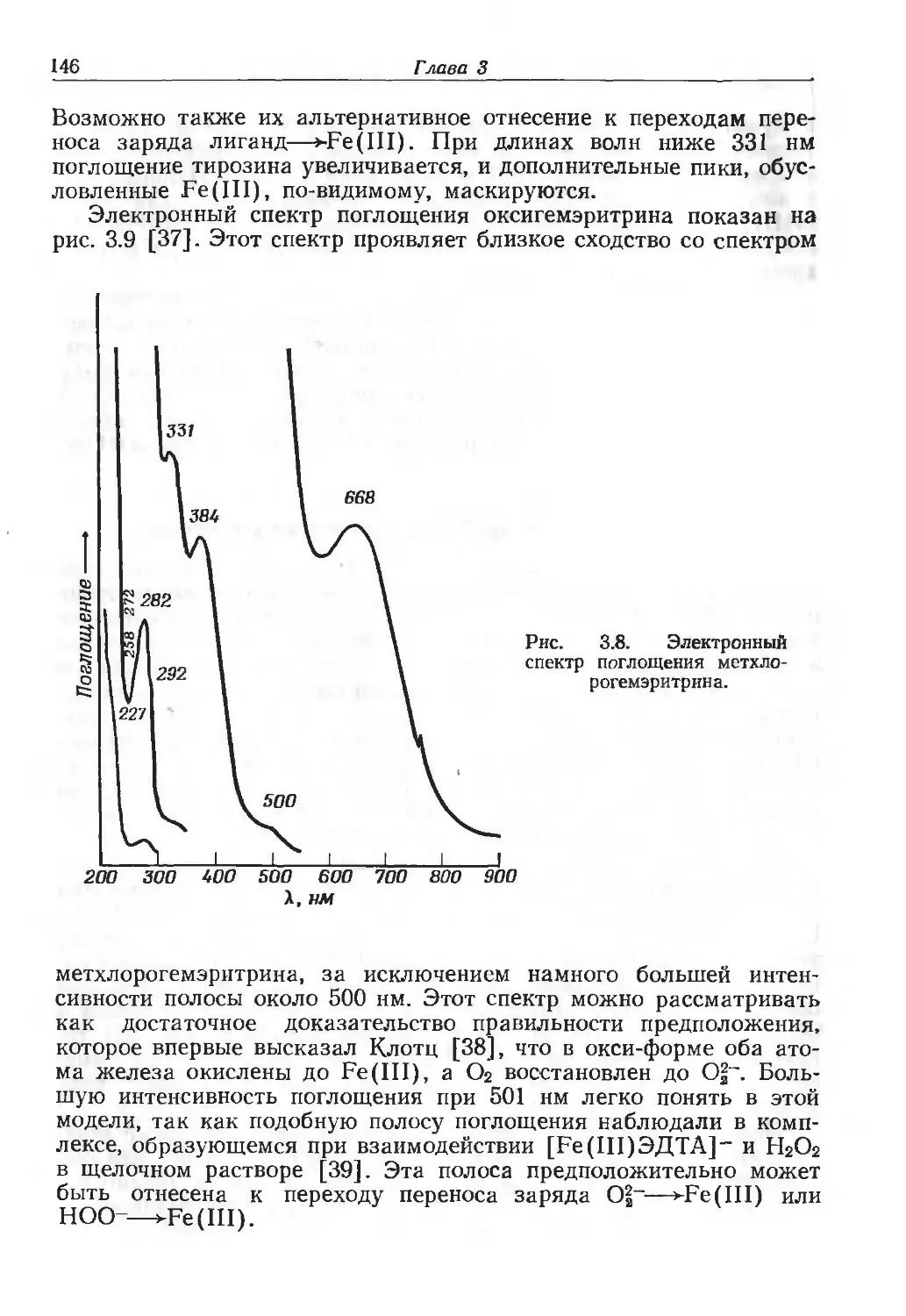
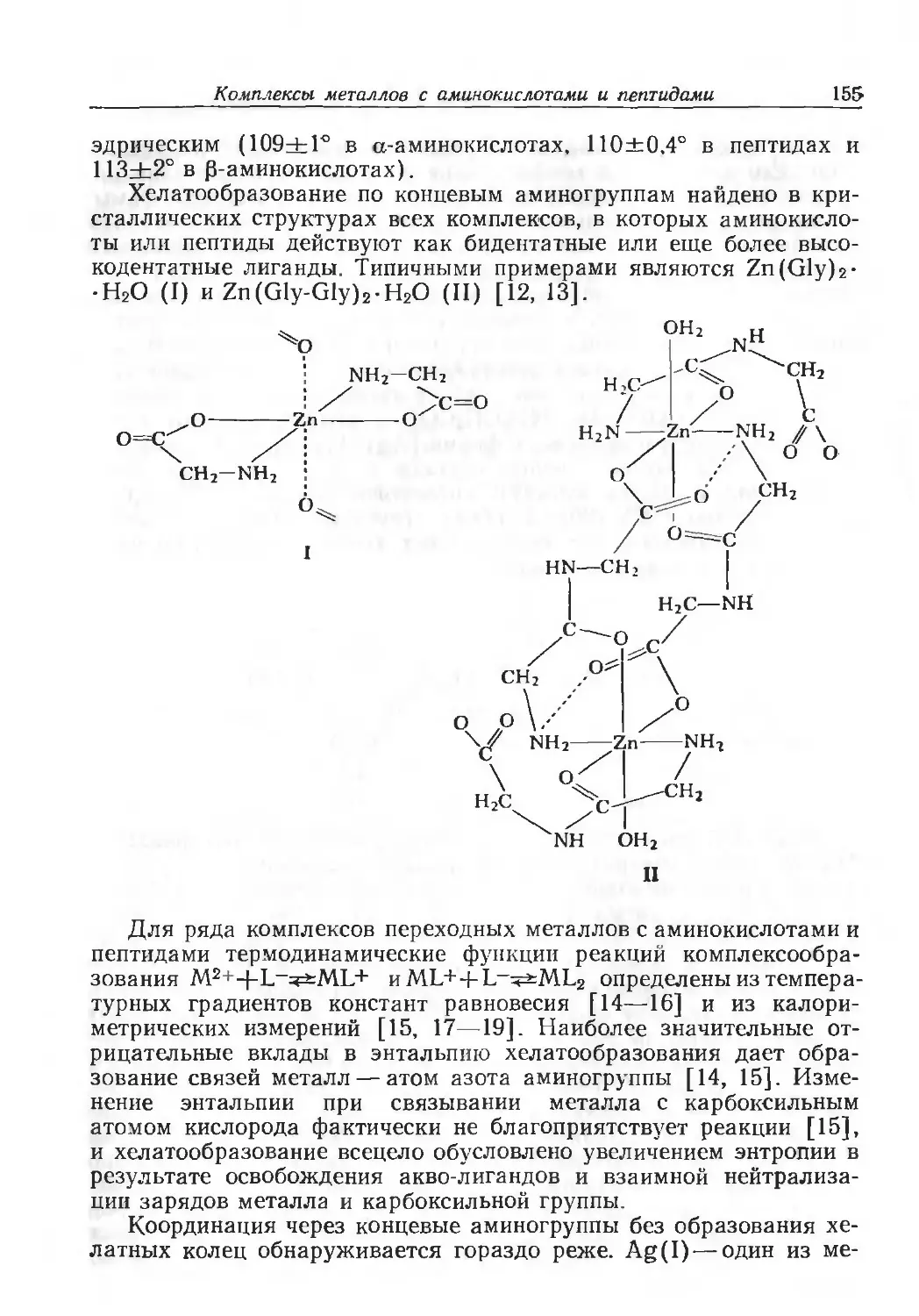
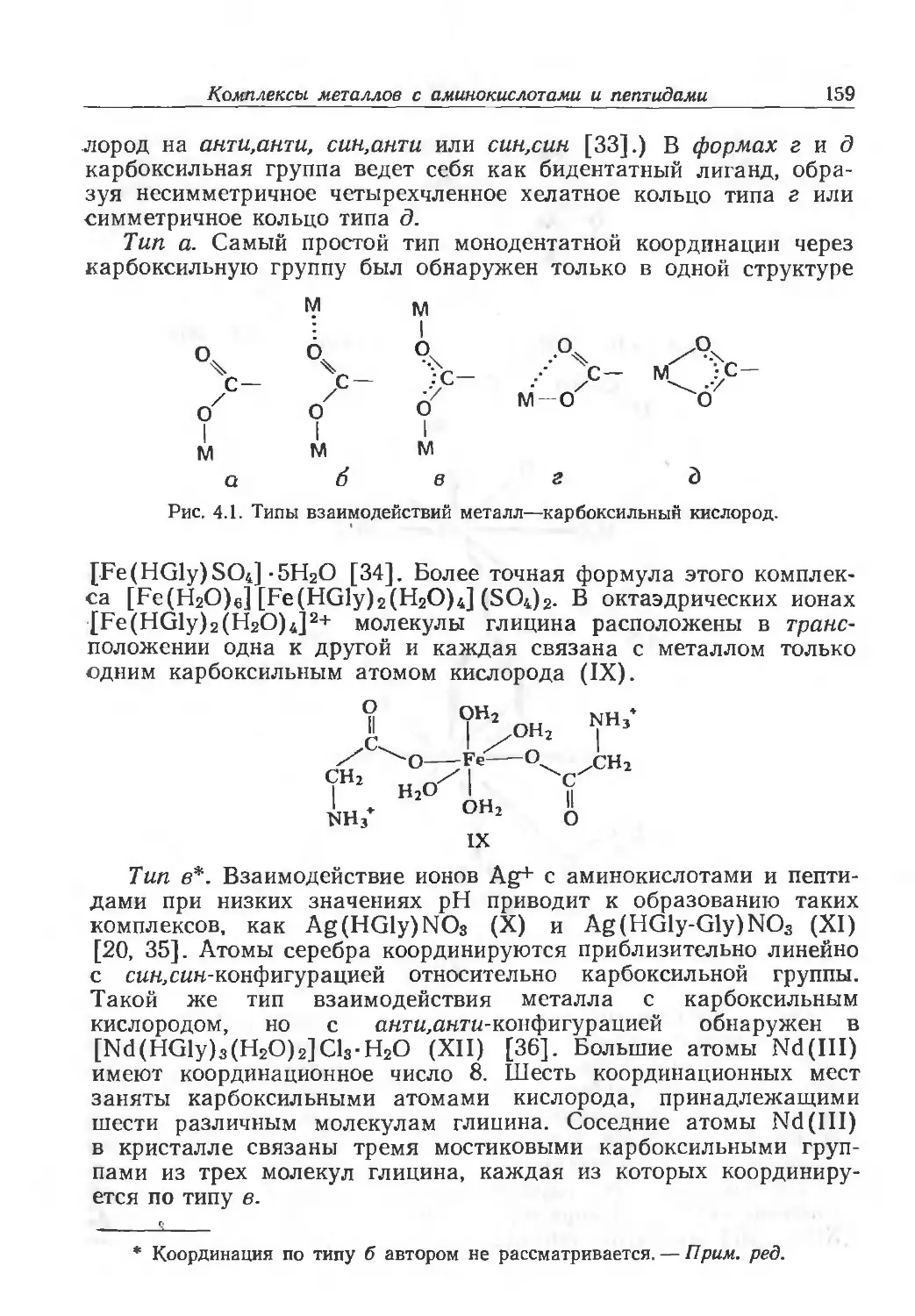
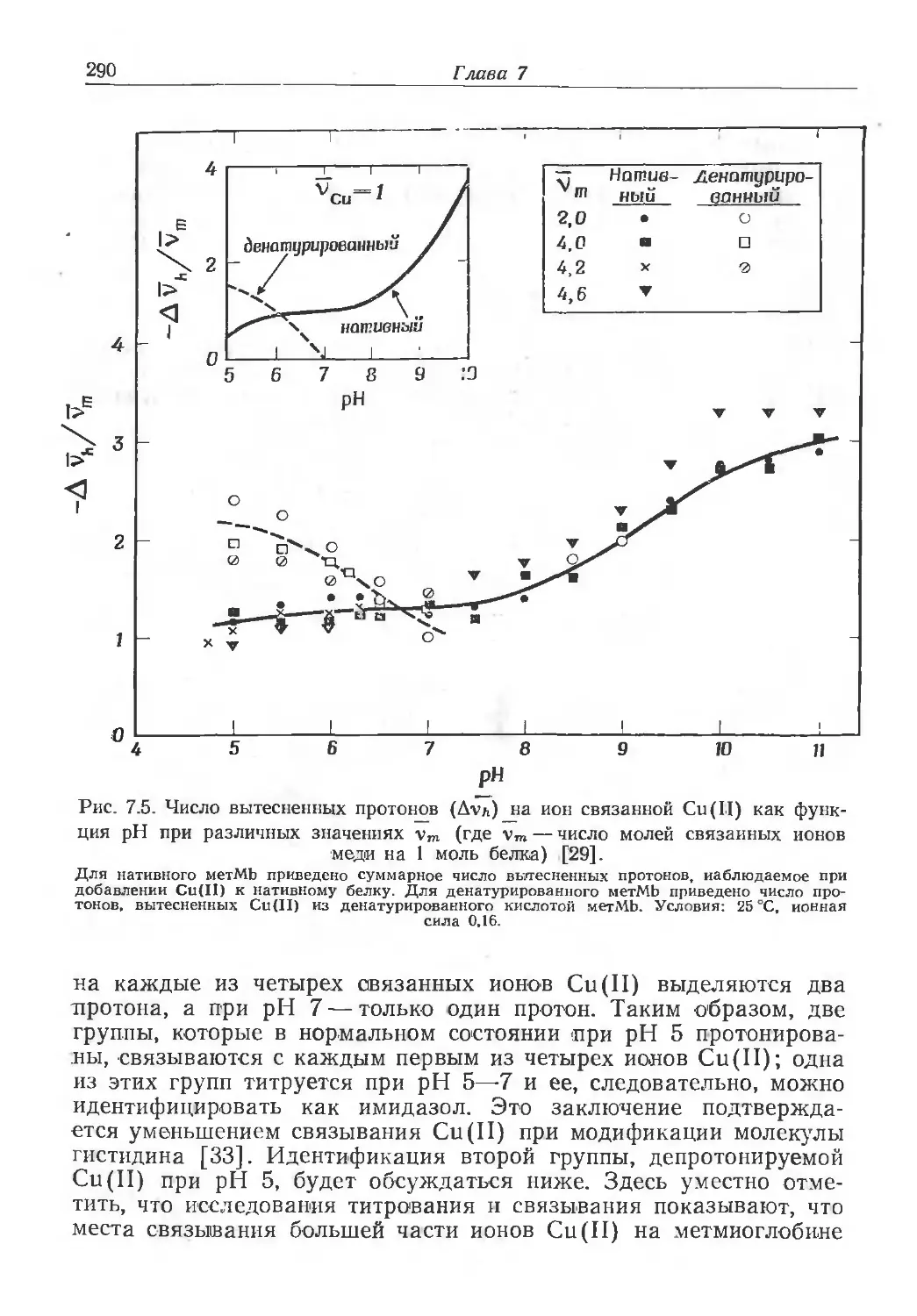
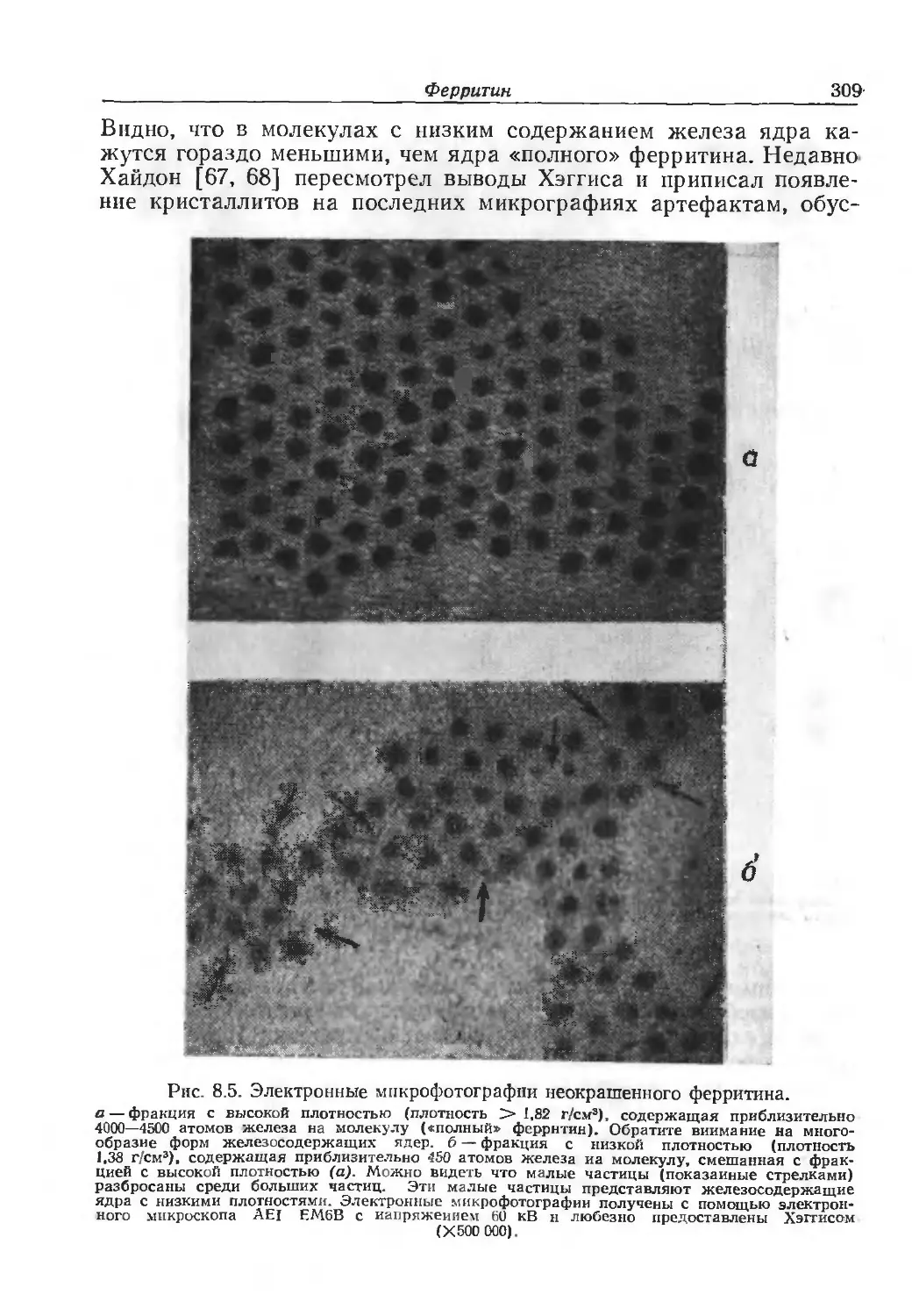
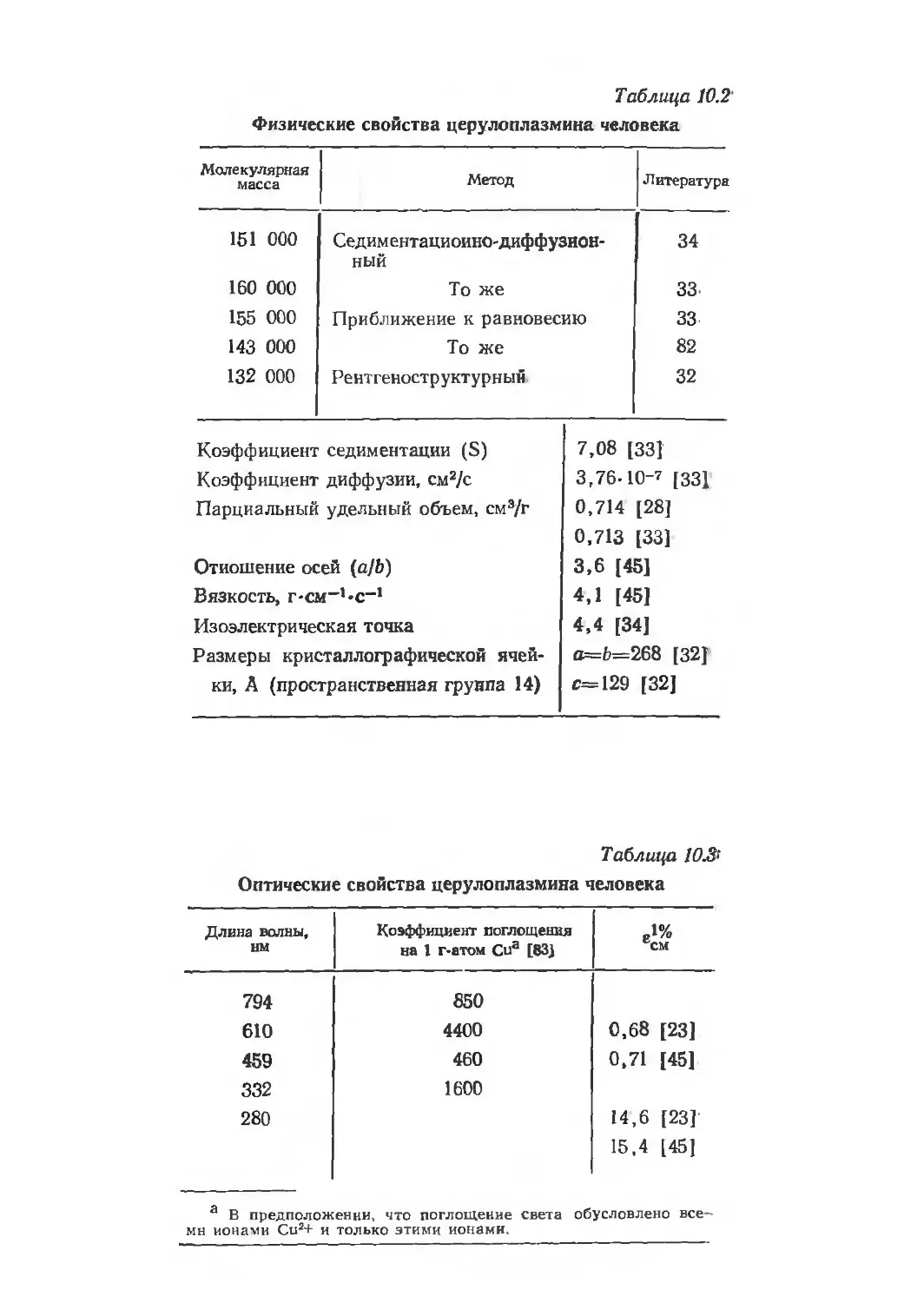
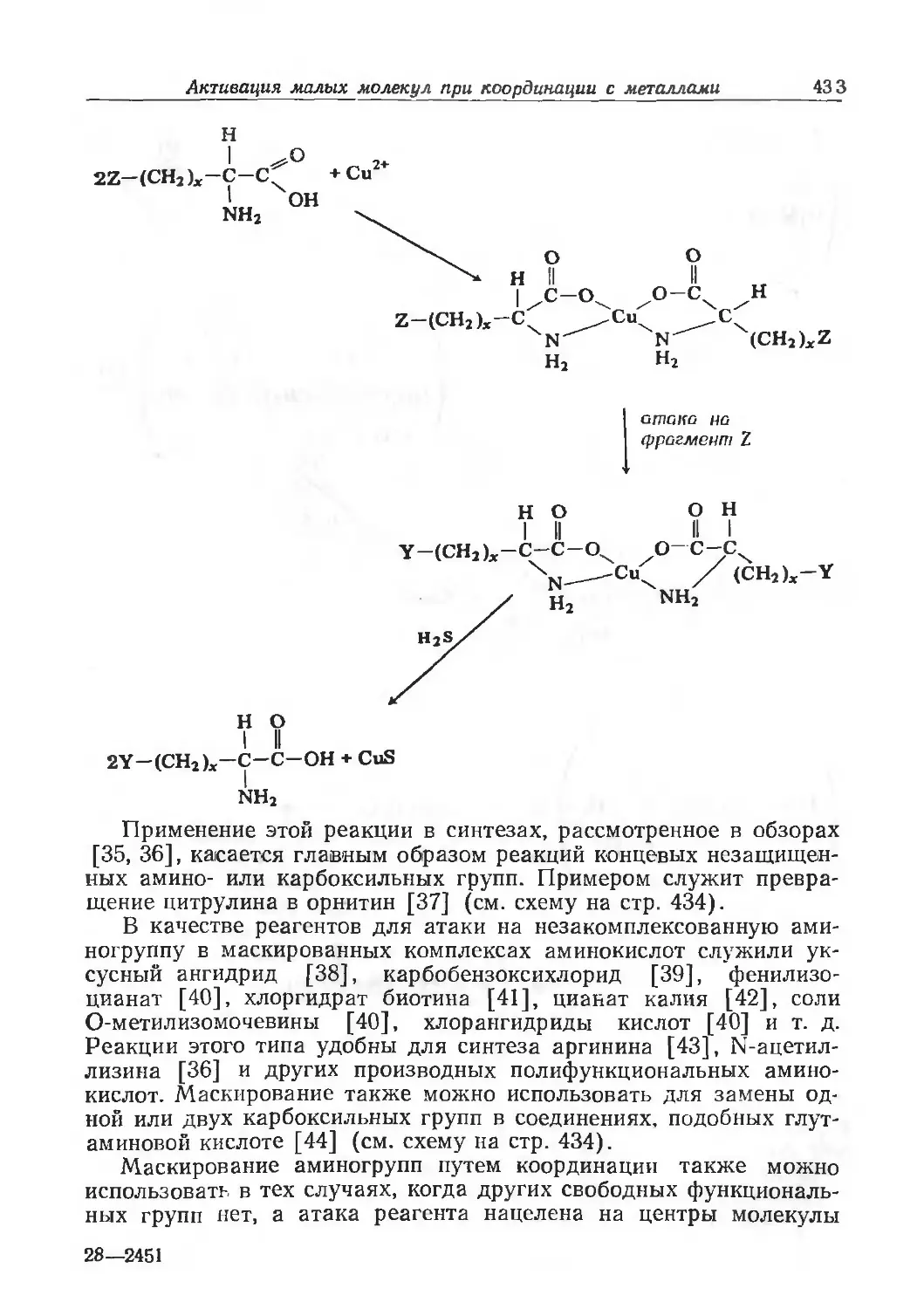
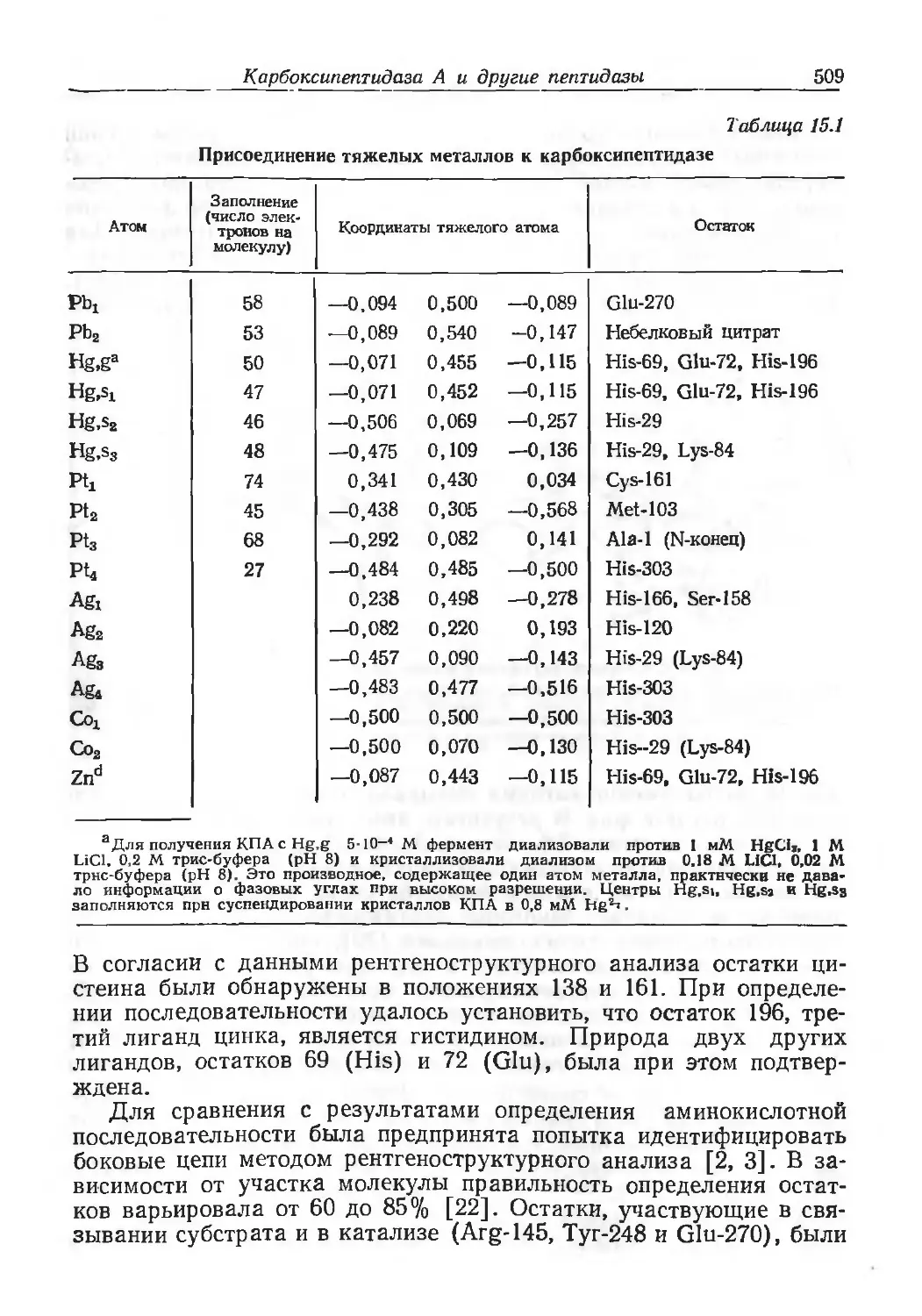
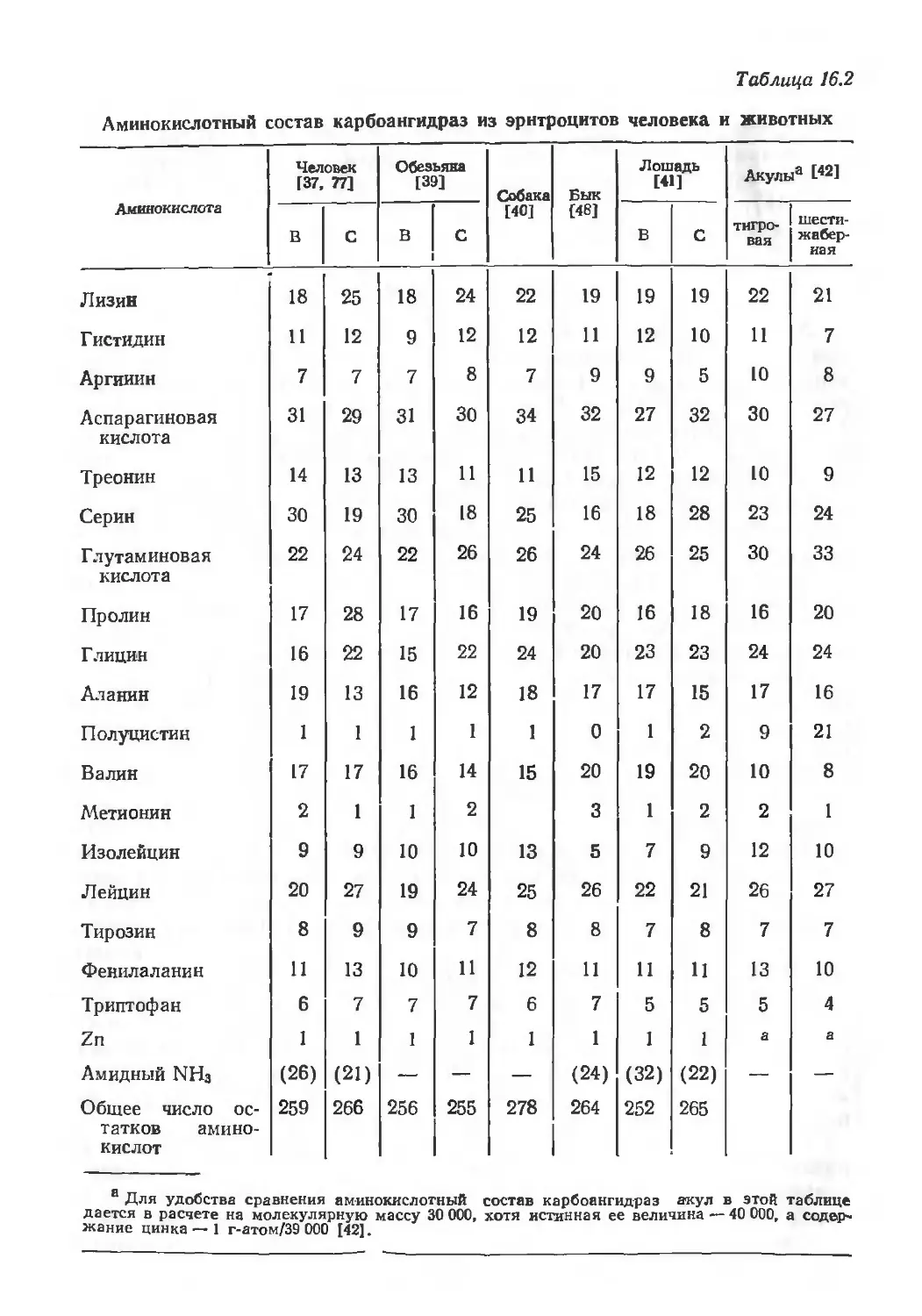
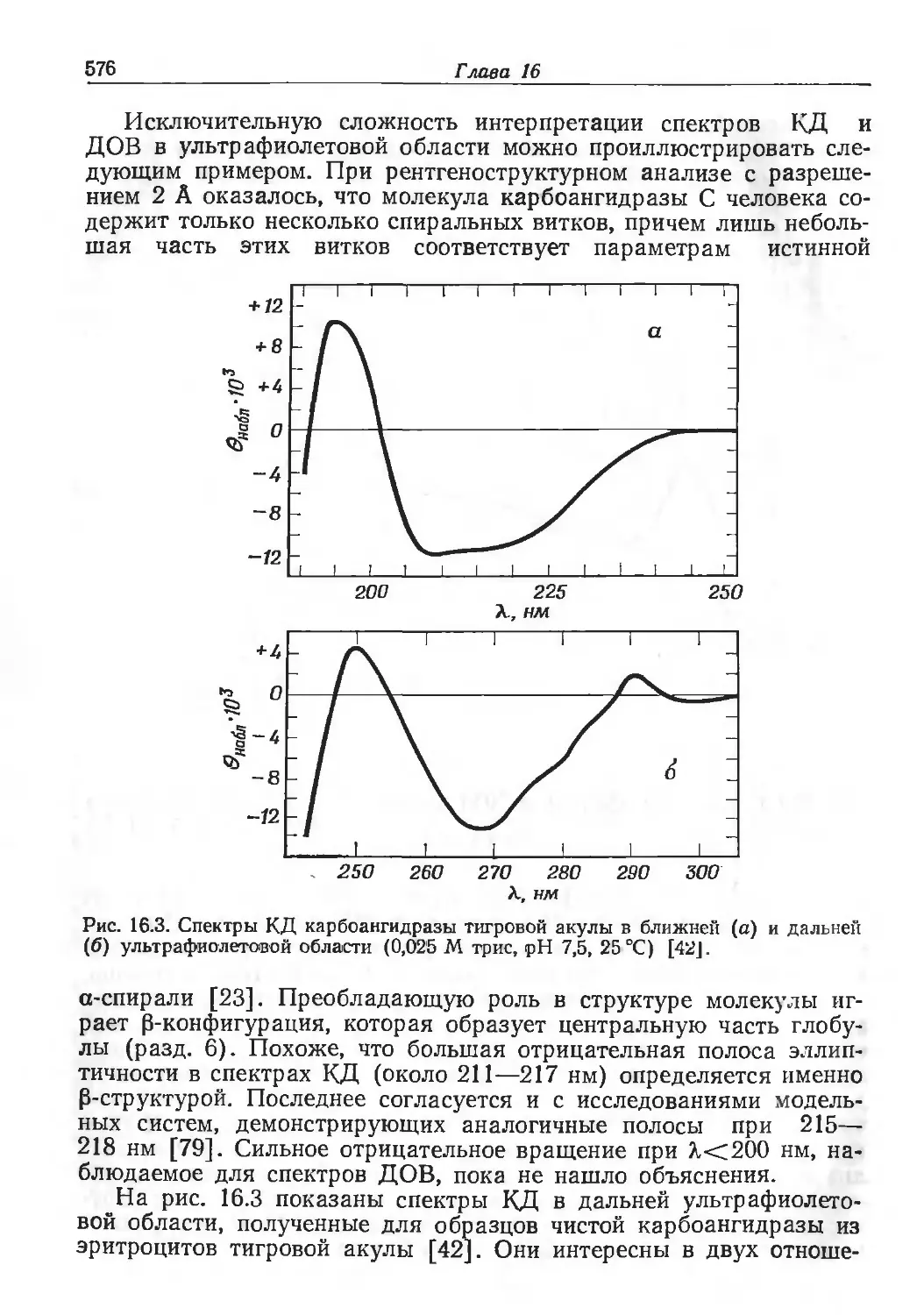
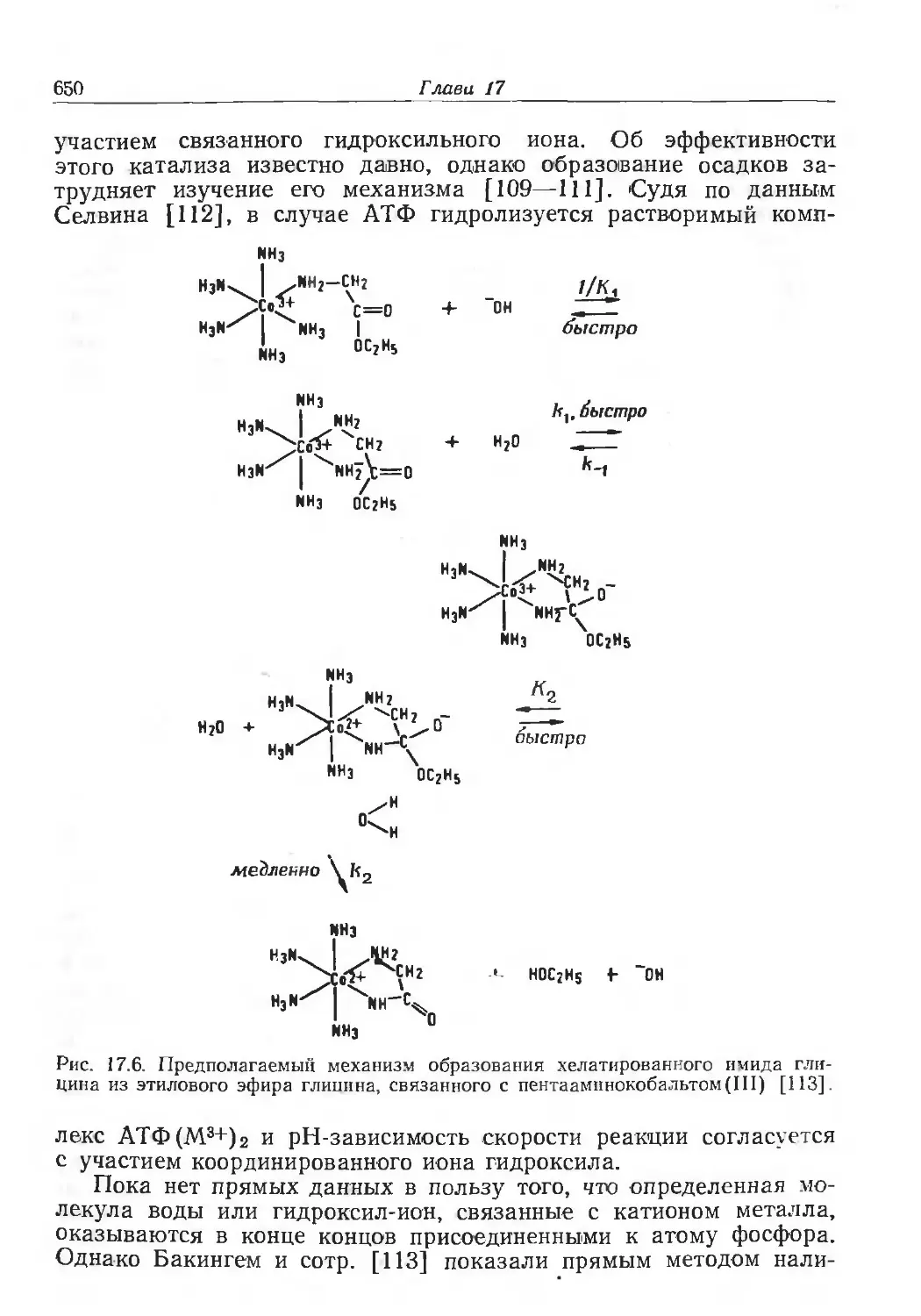
/

















































































































































































































































































































































































































































































































































































































































































































Author: Эйхгорн Г.
Tags: неорганическая химия материальные основы жизни биохимия молекулярная биология биофизика
Year: 1978




Text
НЕОРГАНИЧЕСКАЯ
БИОХИМИЯ
INORGANIC BIOCHEMISTRY
VOLUME 1
Edited by
GUNTHER L. EICHHORN
National Institutes of Health, Gerontology
Research Center, Baltimore City Hospitals,
Baltimore, USA
ELSEVIER SCIENTIFIC PUBLISHING COMPANY
AMSTERDAM — OXFORD — NEW YORK
НЕОРГАНИЧЕСКАЯ
БИОХИМИЯ
РЕДАКТОР
Г. ЭЙХГОРН
1
ПЕРЕВОД с английского
под редакцией
доктора хим. наук
М. Е. ВОЛЬПИНА
и акад. АН УССР
К. Б. ЯЦИМИРСКОГО
ИЗДАТЕЛЬСТВО «МИР» • МОСКВА 1978
УДК 546+577.1
Первый и пока единственный в мире фундаментальный труд в
области неорганической биохимии — одной из самых молодых отрас-
лей науки. Она охватывает такие вопросы, как исследование роли
металлов в биологических системах на молекулярном уровне, моде-
лирование биологических процессов, химическая бионика.
В т. 1 кратко обсуждены основные положения химии коорди-
национных соединений, устойчивость комплексов и методы опреде-
ления их структуры. Рассмотрены комплексы металлов с белками,
их роль в жизненно важных процессах и отдельные представители
класса металлопротеинов. Дан обзор металлофермеитов, их струк-
тур и механизмов, посредством которых ионы металлов участвуют
в ферментативной активности.
Книга предназначена как для исследователей, непосредственно
работающих в области бионеорганической химии, так и для пред-
ставителей смежных областей — химиков-органиков, физико-хими-
ков, биохимиков, медиков.
РедСГ^ия литературы по химии
© 1973, 1975 by Elsevier Scientific
Publishing Company, Amsterdam
20504-090
H 78—90-78 © Перевод на русский язык, «Мир», 1978
ПРЕДИСЛОВИЕ РЕДАКТОРОВ ПЕРЕВОДА
Предлагаемая вниманию читателей книга представляет собой
самую полную и исчерпывающую из всех существующих в настоя-
щее время монографий, посвященных проблемам неорганической
биохимии.
Вопросы, находящиеся на стыке двух областей науки — неор-
ганической химии и биологии, в последние годы привлекают все
больше внимания исследователей, работающих и в том, и в дру-
гом направлениях научных поисков. Не случайно в последнее вре-
мя появилось много книг, брошюр и статей, посвященных бионеор-
ганической химии, т. е. по сути тем же проблемам, но названным
несколько иначе. Повышенный интерес к этим вопросам вызван
не только развитием биологии, биохимии, неорганической химии,
но и необходимостью решения многих прикладных задач из об-
ласти медицины, сельского хозяйства, охраны окружающей среды
и т. д. Многие из поднятых в книге вопросов не могли быть реше-
ны раньше из-за отсутствия необходимых точных методов иссле-
дования и современной аппаратуры.
Отдельные главы книги, написанные разными авторами, неод-
нородны по содержанию. Первые главы в обоих томах посвяще-
ны по сути дела проблемам «чистой» координационной химии,
в других разделах изложены вопросы, относящиеся главным об-
разом к чисто биохимическим проблемам.
Для решения задач неорганической биохимии необходимо зна-
ние электронного строения природных комплексов, включающих
биометаллы (от одного до нескольких атомов) и соответствующее
окружение (ближайшие и более удаленные атомы). В книге рас-
смотрена электронная структура природных комплексов, содержа-
щих в своем составе железо (гл. 3). К сожалению, этого не сде-
лано по отношению к биокомплексам других жизненно важных
металлов.
Большое внимание в книге уделено вопросам окислительно-
восстановительных реакций, причем в двух главах (гл. 19 и 20)
рассматриваются общие вопросы протекания этих реакций в хи-
мии координационных соединений и при катализе ионами метал-
6 Предисловие редакторов перевода
лов. Специальная глава (гл. 23) посвящена сопоставлению хими-
ческой и биологической фиксации азота. Интересен материал
(гл. 14), касающийся функции металлов в металлоферментах.
Здесь дан обстоятельный критический анализ современных мето-
дов выяснения роли ионов металлов в ферментативных реакциях.
Отдельные очень важные классы бионеорганических соедине-
ний рассмотрены в книге достаточно подробно. К таким соедине-
ниям можно отнести сидерохромы, различные ионофоры, ферри-
тин, трансферрины, церулоплазмин, гемэритрин, гемоцианин, кар-
боксипептидазы и карбоангидразу, киназы, оксидазы, ферредокси-
ны, гемоглобин и миоглобин, цитохромы Ъ и с, цитохромоксидазы,
пероксидазы и каталазы, хлорофилл, корриноиды, комплексы ме-
таллов с витамином Вб, флавином, нуклеозидами, нуклеотидами,
полинуклеотидами и нуклеиновыми кислотами. Насколько нам
известно, такое детальное рассмотрение строения и функций пе-
речисленных соединений до сих пор нигде не проводилось.
Книга написана весьма квалифицированными авторами. Груп-
пировка материала производилась главным образом на основании
принципов, широко используемых собственно в биохимии.
Книга, которая представляется нам весьма полной и интерес-
ной, несомненно должна заинтересовать широкие круги специали-
стов в области молекулярной биологии, биохимиков и химиков-
неоргаников и особенно тех, кто занимается проблемами бионеор-
ганической химии и координационной химии применительно к
биологической химии.
М. Вольпин
К- Яцимирский
ПРЕДИСЛОВИЕ РЕДАКТОРА
АМЕРИКАНСКОГО ИЗДАНИЯ
Название «неорганическая биохимия» до недавнего времени
казалось большинству, да многим кажется и теперь, парадоксаль-
ным, так как биохимия звучит как «органическая химия». Синтез
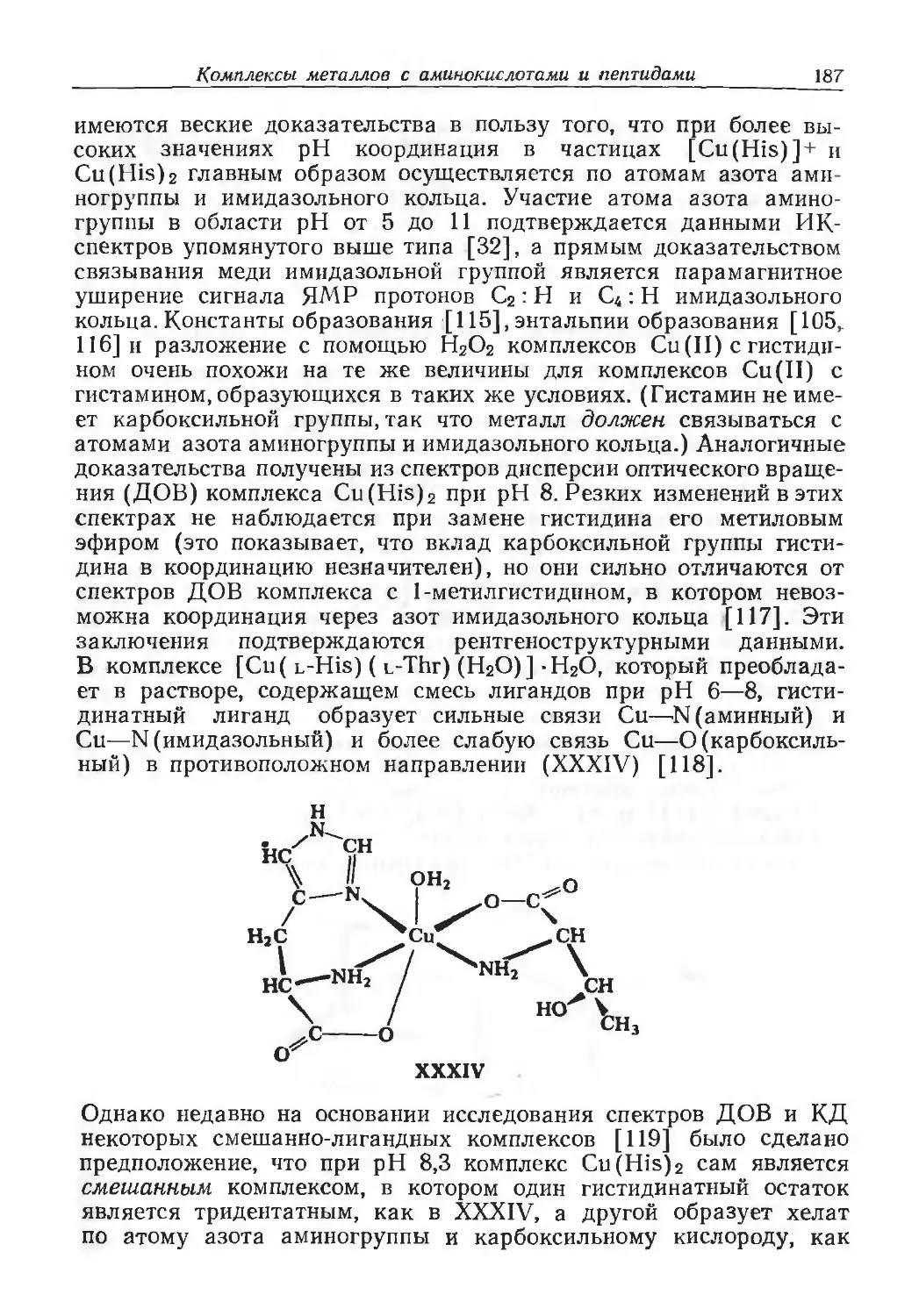
мочевины Вёлером в 1828 г. показал, что органическая химия не
обязательно должна быть биохимией, но еще целое столетие после
этого биохимические явления продолжали ассоциироваться пре-
имущественно с органической химией. В последние годы четкое
разграничение между классическими химическими дисциплинами,
так же как и между научными дисциплинами вообще, было нару-
шено. В ходе этого процесса стала очевидной полезность погра-
ничных областей между классическими разделами химии.
Общность интересов неорганической химии и биохимии была,
конечно, уже давно очевидна отдельным исследователям. Ученым,
занимающимся науками о жизни, приходилось сталкиваться в
своих исследованиях с проблемами неорганической химии и ре-
шать их. Химики-неорганики также иногда понимали, что их ра-
бота имеет отношение к биологическим процессам, и руководство-
вались этим в развитии своих исследований. Однако только
в 1950 г. были установлены контакты между химиками-неоргани-
ками и биохимиками благодаря созыву конференции по вопросам,
представляющим взаимный интерес, а в последние несколько лет
такие конференции стали обычными.
Широкое распространение недавно возникшего интереса к не-
органической биохимии делает эту монографию сейчас даже более
своевременной, чем это было тогда, когда издатели впервые пред-
ложили написать такую книгу. То, что предполагалось вначале
как монография одного или двух авторов, разрослось в двухтом-
ное издание, включающее 34 главы, написанные 45 авторами.
Большое число работ в этой области, опубликованных в печати,
позволяет дать исчерпывающее изложение вопроса.
Хотя материалы некоторых симпозиумов, посвященных этим
вопросам, были опубликованы, а также появилось несколько об-
зоров, необходимость исчерпывающей монографии по неорганиче-
ской биохимии стала очевидной; в связи с этим следовало опре-
8
Предисловие редактора американского издания
делить и ограничить круг вопросов, которые должна охватывать
такая монография. Очевидно, что наличие в биологических веще-
ствах таких элементов, как сера, азот или фосфор, не оправдыва-
ет их обсуждения здесь даже в том случае, когда химия этих
веществ, определяемая этими элементами, имеет «неорганиче-
скую» сущность, так как это привело бы к совпадению неорга-
нической биохимии с биохимией вообще. Термин «неорганическая
биохимия», по-ъидимому, должен ассоциироваться с идеей участия
ионов металлов в биологических процессах. Поэтому мы приняли
произвольное, но, надеюсь, оправданное и полезное определение
неорганической биохимии как приложения принципов координа-
ционной химии металлов к биологическим проблемам. Для того
чтобы еще больше ограничить круг рассматриваемых вопросов,
мы здесь не будем касаться применения координационных соеди-
нений в качестве лекарственных препаратов и пищевых продуктов,
за исключением краткого упоминания об этом во введении.
После определения круга рассматриваемых проблем 'встает
вопрос о том, как их скомпоновать. По-видимому, возможны раз-
личные схемы изложения. Одна из них — взять за основу атом
металла, имеющийся в соединении, т. е. все соединения меди
должны быть рассмотрены вместе, так же как соединения желе-
за, цинка и т. д. Такое рассмотрение представляло бы несомнен-
ную ценность. Однако мы предпочли в этой монографии взять за
основу структуру лигандов, полагая, что таким образом можно
добиться большей связности изложения. Материал по главам рас-
положен таким образом, чтобы обеспечить логическое развитие,
основанное на структурном сходстве.
Многие биологические координационные соединения пред-
ставляют собой макромолекулы. Мы предполагали уделить вни-
мание главным образом химии металла, но для ее понимания ча-
сто существенно рассмотреть, как влияет металлическая компо-’’
нента на конформацию макромолекулы, и поэтому значительное
внимание было уделено также «органической» части молекулы.
В то же время, поскольку основу этой книги составляет коорди-
национная химия, были включены некоторые существенно «неор-
ганические» главы. Таким путем могли быть установлены отноше-
ния между биологическими координационными соединениями и
«модельными» неорганическими комплексами. Во многих главах
биологические вещества и модельные соединения рассматривают-
ся вместе, но основное внимание уделяется биологическим веще-
ствам.
Часть I имеет своей целью дать биохимику достаточное пред-
ставление об основах неорганической химии и избавить его от
необходимости обращаться к учебникам или литературе по неор-
ганической химии для того, чтобы понять многие (но, конечно, не
все) неорганические явления, встречающиеся в последующих раз-
Предисловие редактора американского издания
9
делах этой монографии или в его собственных исследованиях.
Даны многие «ключи» для объяснения неорганических биохими-
ческих явлений. Гл. 1 представляет собой введение в химию коор-
динационных соединений и не ограничивается только ионами ме-
таллов, которые в настоящее время известны как «биологиче-
ские», так как структура и стереохимия могут быть лучше поняты
путем ссылок на наилучшие примеры независимо от того, какие
ионы металлов использованы для этой цели. Так, даже комплек-
сы платины могут иметь биологическое значение (см. «Введе-
ние»). В гл. 2 развиваются концепции, касающиеся структуры и
устойчивости, наиболее ярко представленные на примере сравне-
ния устойчивости различных биологических комплексов. Гл. 3
иллюстрирует полезность исследований, основанных на рассмот-
рении электронной конфигурации для выяснения строения неко-
торых веществ, описанных в последующих главах этой книги.
Многие биологические координационные соединения являют-
ся белками; они обсуждены в частях II—VI. Часть II начинается
с систематического рассмотрения (гл. 4) данных о комплексах
металлов с мономерами белков — аминокислотами и простыми
олигопептидами. Затем следует обзор некоторых встречающихся
в природе олигопептидов, которые в организмах обычно связыва-
ются с железом (гл. 5) и со щелочными металлами (гл. 6). Неко-
торые из этих веществ — олигопептиды, а другие — нет, но коор-
динационные свойства этих пептидных и непептидных соединений
во многом сходны. В гл. 7 рассматривается взаимодействие ионов
металлов с белками, для того чтобы продемонстрировать типы
координационных центров макромолекул, с которыми могут свя-
зываться ионы металлов.
В части III обсуждены некоторые биологические металлопро-
теины, принимающие участие в хранении и транспорте железа
(гл. 8 и 9) и меди (гл. 10), а также некоторые связывающие
кислород металлопротеины, обнаруженные в низших организмах
(гл. 11 и 12).
В части IV рассмотрены структуры металлоферментов и ме-
ханизмы, посредством которых ионы металлов принимают участие
в ферментативной активности, в частности, в разрыве связей.
В гл. 13 обсуждаются способы, которыми ионы металлов вызыва-
ют химические изменения в лигандах без помощи белков. В гл. 14
дан обзор металлоферментов, не рассматривающихся отдельно
в последующих главах. В гл. 15 и 16 некоторые металлоферменты
рассмотрены более подробно, частично из-за более обширной ин-
формации об этих металлоферментах. доступной в настоящее вре-
мя; другие ферменты рассмотрены отдельно в гл. 17 и 18 вследст-
вие относительной легкости классификации этих групп ферментов.
Те ферменты, которые принимают участие главным образом в ре-
акциях окисления — восстановления, а также ферменты, содержа-
10
П редисловие редактора американского издания
щие порфирины или другие простетические группы, рассмотрены
в т. 2 (гл. 19—34).
Ферментативные реакции окисления — восстановления специ-
ально рассматриваются в части V, которая начинается с обсужде-
ния (гл. 19) реакций окисления — восстановления координацион-
ных соединений, включающего изложение теории и ее применение
к биологическим системам. Гл. 20 содержит классификацию реак-
ций оксигенирования с помощью металлов в присутствии и в от-
сутствие ферментов. В гл. 21 обсуждены различные типы медьсо-
держащих оксидаз. Белки типа ферредоксина участвуют в пере-
носе электрона, они охарактеризованы в гл. 22, за которой следу-
ет гл. 23, посгященная фиксации азота и ферментам нитрогеназы.
В рассмотренных до сих пор белках ионы металлов были при-
соединены к боковым цепям аминокислот белков. В части VI мы
начинаем рассмотрение белков, в которых ионы металлов присо-
единены не непосредственно к белкам, а к «простетическим труп-
пам» или «коферментам». Наиболее распространенной простети-
ческой группой является порфирин; за обсуждением порфиринов
в гл. 24 следуют гл. 25—28, посвященные железопорфириновым
соединениям, гемопротеинам, и гл. 29, в которой обсуждается маг-
ниевое производное порфирина, хлорофилл. На основании струк-
турного сходства с порфиринами коррины, а также коферменты
и витамин В12 рассматриваются в гл. 30.
Часть VII посвящена комплексам металлов с другими просте-
тическими группами. Нельзя сказать с определенностью, что ком-
плексы металлов с витамином Вб (гл. 31) имеют биологическое
значение, но они представляют собой прекрасные модели фермен-
тативных процессов, катализируемых витамином Be- Комплексы
металлов с флавинами и флавопротеинами рассмотрены в гл. 32.
Часть VIII о взаимодействии ионов металлов с нуклеиновыми
кислотами начинается с гл. 33, в которой рассмотрены комплексы
металлов с нуклеозидами и нуклеотидами; они обладают некото-
рым сходством с металлофлавиновыми комплексами. Наконец,
в гл. 34 обсуждаются комплексы металлов с полинуклеотидами
и нуклеиновыми кислотами, а также их биологическое значение.
Хотя такой порядок изложения имеет ряд преимуществ, мож-
но было бы предложить и другую последовательность, также об-
ладающую достоинствами. Так, можно оспаривать целесообраз-
ность включения цитохромов при рассмотрении реакций окисле-
ния — восстановления до обсуждения порфиринов. Однако кажет-
ся предпочтительным обсудить порфирины перед цитохромами;
действительно, часть VI в основном касается реакций окисле-
ния — восстановления, и поэтому логически она следует за ча-
стью V. Могло бы быть оправданно помещение части VIII перед
частью IV, так как комплексы нуклеотидов, обсужденные лишь
в гл. 33, важны для изложения материала в гл. 18. Однако тогда
Предисловие редактора американского издания 11
гл. 34 была бы не на месте. Очевидно, не существует полностью
удовлетворительной последовательности, и поэтому некоторые
вопросы пришлось повторять в различных местах. Мы избегали
повторения во всех случаях, когда 'при этом не страдала ясность
изложения. Для того чтобы достигнуть этого, а также и для дру-
гих целей, поддерживались постоянные контакты между автора-
ми, а также между авторами и редактором.
Сотрудничество авторов (некоторые из них сделали ценные
указания и к тем частям, которые были написаны не ими) оказа-
лось очень полезным. Я получил ценные советы от очень многих
из них. Я обязан моим коллегам Натану А. Бергеру, Джеймсу
Дж. Бутсову, Патриции Кларк, Джейн Гейм, Джозефу Пита, Кар-
мен Ричардсон, Джозефу Рифкинду, Йонгу А. Шину и Эдварду
Тариену за их помощь во время подготовки и редактирования
этой книги.
Я благодарю Национальный институт здоровья, субсидировав-
ший написание этой книги.
Я очень обязан моему секретарю Жаклин Блейк за ее дели-
катную помощь на всех этапах издания этой книги. И наконец,
я должен поблагодарить мою жену и детей за их терпение во
время осуществления этого проекта, которое заняло значительно
больше времени, чем мы предполагали.
Г. Эйхгорн
ВВЕДЕНИЕ
Термин «бионеорганическая химия» впервые появился как на-
звание ряда недавно состоявшихся симпозиумов, а также как за-
главие нового научного журнала. Название настоящей моногра-
фии было задумано еще до появления «изомеров связи» (см.
гл. 1, разд. 5.1.1,б). Можно было бы последовать уже установив-
шейся тенденции, но вместо этого было решено сохранить пред-
полагавшееся ранее название этой книги. Одна из причин такого
решения заключалась в том, что существование двух названий
иллюстрирует различные ударения, которые биохимики и химики-
неорганики могут делать на различных составных частях этого
названия. Первые смотрят на неорганическую химию как на сред-
ство объяснения химического поведения комплексов металлов с
биологическими молекулами. Вторые рассматривают биохимию
как область, в которой могут найти подходящее применение их
открытия. Окончательный результат этих двух подходов, конечно,
один и тот же, аналогично тому как названия «изомеров связи»
имеют одно и то же значение. Однако исходная точка ученого
важна для определения того, что именно он должен узнать для
достижения своей цели, и поэтому для того, что он откроет на
этом пути.
Главное различие между химиками-неорганиками и биохими-
ками заключается в том, что первые обычно имеют дело с малы-
ми молекулами, которые иногда можно рассматривать как «моде-
ли» более сложных систем, которыми занимаются вторые. Фунда-
ментальный вопрос, с которым постоянно сталкиваются неоргани-
ческие биохимики, состоит в том, имеет ли смысл изучать модели,
если в наличии имеются «природные» вещества.
Для того чтобы ответить на этот вопрос, необходимо реалисти-
чески оценить методы, с помощью которых были выяснены био-
логические механизмы. Живая клетка так сложна, что ее функ-
ционирование можно понять только путем изолирования ее от-
дельных частей, причем всегда неизбежен риск, что изолированная
часть функционирует иначе, чем в клетке. Поэтому изолирован-
ные компоненты представляют «модели» этих компонентов в
14
Введение
клетке. Например, цитохром с in vitro в действительности пред-
ставляет модель цитохрома с в клетке. Но весьма вероятно, что
многие характеристики изолированного цитохрома с одинаковы с
характеристиками клеточного цитохрома с. Действительно, после
того как были выделены и изучены все другие молекулы, с кото-
рыми связан цитохром с в клетке, стало понятно поведение ци-
тохрома с в клетке.
Таким же образом неорганическое соединение, например про-
стой порфириновый комплекс, который может принимать участие
в переносе электрона и, возможно, с тем же окислительным по-
тенциалом, что и изолированный цитохром с, может служить мо-
делью цитохрома с, в то в.ремя как механизм переноса электрона
цитохромом с еще не понятен. Изучение простого порфиринового
комплекса может помочь пониманию изолированного цитохрома с,
а исследование изолированного цитохрома с в свою очередь мо-
жет помочь пониманию клеточного цитохрома с. Имея в виду та-
кую перспективу, можно полагать, что модели могут быть полез-
ны на всех уровнях. Конечно, при исследовании моделей необхо-
димо помнить об ограничении, которое представляет аксиому: мо-
дель системы не является самой системой. Химики-неорганики,
так же как и биохимики, могут внести положительный вклад,
если будут помнить об этом ограничении, и отрицательный вклад,
если они о нем забудут.
Хотя в данной работе основное внимание уделяется биологи-
ческим системам, в ней рассматриваются также многие модели,
иногда в отдельных главах, а иногда параллельно с природными
веществами, для конкуренции с которыми они предназначены.
Часто делаются корреляции, надеюсь — имея в виду приведенное
выше ограничение. Если существование неорганической биохимии
имеет какой-либо смысл, то такие корреляции должны приводить
к плодотворным результатам.
Тогда — в чем заключается важность этих результатов? В чем
состоит основная цель неорганической биохимии?
На эти вопросы можно ответить, рассмотрев пограничную об-
ласть этой части биологической науки. Существуют две цели
изучения биологической науки. Первая, как во всех открытиях, —
изучить явление, потому, что оно существует, понять, как устроены
вещи. Второе — добиться понимания нормальных и аномальных
процессов, происходящих в клетке, которое в конечном итоге
должно привести к победе над болезнью. Эти цели неорганиче-
ская биохимия разделяет с другими биологическими науками*.
Любознательность ученого и его желание быть причастным к дан-
* Неорганическая биохимия может иметь и другие прикладные аспекты,
в частности решение вопросов, связанных с сельскохозяйственным производством
и охраной от загрязнений окружающей среды. — Прим. ред.
Введение
15
ному вопросу в истории часто заставляют его руководствоваться
этими обеими целями.
В этой монографии не сделано никаких попыток дать перечень
применений неорганической биохимии в медицине, частично из-за
того, что мы считали такие попытки выходящими за рамки данной
книги. Ранее были опубликованы обзоры, касающиеся фармако-
логических и пищевых аспектов координационной химии [1, 2].
Тем не менее во введении мы хотим привести некоторые примеры,
иллюстрирующие практическое следствие изучения неорганиче-
ской биохимии.
Наиболее наглядное и широко распространенное использова-
ние комплексообразующих агентов в медицине — это выведение
нежелательных ионов металлов из организма. Такие лиганды, как
этилендиаминтетрауксусная кислота, пеницилламин и т. п., ис-
пользуются при лечении заболеваний, связанных с избыточным
содержанием железа или меди (гл. 10), а также при борьбе с ток-
сичным влиянием проникших в организм ионов металлов [3—5].
Недавно было обнаружено, что применение пеницилламина
может привести к резкому понижению остроты восприятия вкуса
[6—8]. Однако этого не наблюдается при лечении болезни Виль-
сона, при которой происходит накопление ионов Си(II) в организ-
ме (гл. 10). Была выдвинута гипотеза, что понижение остроты
восприятия вкуса обусловлено связыванием ионов меди в ком-
плекс и, следовательно, ионы меди каким-то образом принимают
участие в продуцировании вкусовых ощущений. Отсутствие пони-
жения остроты восприятия вкуса при болезни Вильсона обуслов-
лено столь высоким содержанием Си2+, что применение пени-
цилламина не может превысить влияние Си2+. Если эта гипотеза
правильна, то применение Си (II) должно восстановить остроту
восприятия вкуса, потерянную в результате действия комплексо-
образующих агентов. Эксперименты показали, что ионы меди дей-
ствительно восстанавливают восприятие вкуса; более того, ионы
Zn(II) и Ni(II) действуют аналогичным образом и восстанавли-
вают чувство вкуса, потерянное вследствие лечения пенициллами-
ном или из-за болезни. Таким образом, ионы металлов, по-види-
мому, имеют отношение к продуцированию чувства вкуса [4].
Недавно было обнаружено, что некоторые комплексы платины
обладают потенциальной антиканцерогенной активностью [9].
Активными являются комплексы 4«c-[Ptn(NH3)2C12], [PtnenC12],
4«c-[Ptn(NH3)2Cl4] и [PtIvenCl4]*. 'С другой стороны.
[Ptn(NH3)4]Cl2 и транс- [PtIV(NH3)2Cl4] неактивны. Активность
в ингибировании развития опухоли коррелировалась с ингибиро-
ванием репликации ДНК [Ю] (гл. 34). Нет необходимости гово-
еп — этилендиамин.
16
Введение
рить, что эти биологические явления представляют несомненный
интерес для химиков-неоргаников.
Следует ожидать, что такие открытия, как применение ионов
металлов для восстановления восприятия вкуса или применение
комплексов металлов для рассасывания опухоли, должны стиму-
лировать развитие неорганической 'биохимии.
СПИСОК ЛИТЕРАТУРЫ
1. Chaberek S., Martell А. Е., Organic Sequestering Agents, Wiley, New York, 1959,
p. 416.
2. Schulman A., Dwyer F. P., in F. P. Dwyer, D. P. Mellor (eds.), Chelating
Agents and Metal Chelates, Academic Press, New York, 1964, p. 383.
3. Seven M. J., Johnson L. A. (eds.), Metal Binding in Medicine, Lippincott, Phi-
ladelphia, 1960.
4. Johnson L. A., Seven M. J. (eds.). Federation Proc., № 3, 20 (1961).
5. Gross F. (ed.), Iron Metabolism, Springer Verlag, Berlin, 1964.
6. Henkin R. I., Keiser H. R., Jaffe I. A., Sternlieb I., Scheinberg J. A., Lancet,
1967, 1268.
7. Henkin R. J., Bradley D. F., Proc. Natn. Acad. Sci. U. S., 62, 30 (1969).
8. Henkin R. I., Graziadei P. P. G., Bradley D. F., Ann. Internal Med., 71, 791
(1969).
9. Rosenberg B., van Camp L., Trosko J. E., Mansour V. H., Nature, 222, 385
(1969).
10. Harder H. C., Rosenberg B., Int. J. Cancer, 6, 207 (1970). Earlier references
can be found in this paper.
ЧАСТЬ I
КООРДИНАЦИОННАЯ ХИМИЯ*
ГЛАВА 1
СТРУКТУРА И СТЕРЕОХИМИЯ
КООРДИНАЦИОННЫХ СОЕДИНЕНИЙ
Д. А. Букингем
Buckingham D. A., Research School of Chemistry, Australian
National University, Canberra, Australia
J, ВВЕДЕНИЕ
Знание структуры и стереохимии важно для всех отраслей хи-
мии, так как позволяет лучше понять свойства химических со-
единений в основном состоянии, а в некоторых случаях — ив воз-
бужденном состоянии. В неорганической координационной химии
такая информация имела огромное значение для развития теорий
координационной связи, т. е. связи между донорным атомом пли
донорными атомами лиганда и атомом металла. Однако знание
структуры само по себе недостаточно для определения химической
связи, и в настоящее время эта область координационной химии
находится на ранней стадии развития. Более результативным бы-
ло применение структурных исследований к реагентам, продуктам
и переходным состояниям химических реакций. В этой области
химической динамики структурные исследования сочетали с кв-
иетическими для того, чтобы понять механизмы химических ре-
акций, которых так много предложила неорганическая координа-
ционная химия. Такой подход особенно желателен по отношению
к предмету, рассматриваемому в этой книге, поскольку ионы ме-
таллов необходимы для многих биологических процессов, но ме-
ханизм их участия -в большинстве случаев неизвестен. В данной
* Сокращения, использованные в этой главе: еп — этилендиамин; рп — 1,2-
диамннопропан; dien — диэтилентрнамин; trien — триэтилентетрамнн; tetraen —
тетраэтиленпентамнн; асас — ацетилацетон; salen — салнцплальэтиленднамин;
dpm — дипнвалоилметан; Gly — глицин; Gly-Gly •— глицнлглицин; ру — пиридин;
dipy — дипнридил; terpy — терпириднл; diars — диарсин; ЭДТА — этнлендиамнн-
тетрауксусная кислота; СН3-еп — метилэтнлендиамин; tren — N(CH2CH2NH2)3;
urea — мочевина; ПДТА — пропилендиамннтетрауксусная кислота; ОЭДТА —
№-(2-оксиэтил)этилендиамин-Ь1,К,№-триуксусиая кислота; асат — ацетамид;
glyam — глицннамнд; sal — салицилальдегид; bzp — бензоилпируват: sarc — сар-
козннат; bdam — бутандиамин; tn — триметиленднамнн; ох — оксалат; sar — сар-
козин; ditn — днтриметилендиамин.
2—2451
18
Глава 1
главе будут кратко рассмотрены некоторые классические понятия,
развитые в неорганической 'координационной химии для описания
и обсуждения стереохимии неорганических комплексов, для того,
чтобы эти принципы были ясны при чтении последующих глав.
Во вступительной главе невозможно рассмотреть детально все
аспекты стереохимии неорганических комплексов, подробное их
изложение можно найти в учебниках [1—7, 53—55]. Поэтому мы
намерены дать не полный обзор, а лишь иллюстрации к тем
общим понятиям, которые, как мы полагаем, должны быть наи-
более полезны читателю этой книги: координационное число и
геометрия, тип лиганда, факторы, влияющие на стереохимию ком-
плексов, и различные типы изомерии координационных соедине-
ний. Стереохимические аспекты координационных соединений,
подобно другим областям неорганической химии, в настоящее вре-
мя интенсивно развиваются, в частности, в том, что касается оп-
ределения структуры, конформационного анализа, изучения тер-
модинамики и кинетики взаимопревращения структур. Здесь не
будет дано строгого обсуждения этих вопросов, за более подрой
ной информацией читатель отсылается к работам [2, 8—17].
Атомы или группы атомов, окружающие центральный атом
металла, называются лигандами, а атомы, яепосоедственно при-
соединенные к металлу, называются донорными стомами. Донор-
ные атомы обычно менее электроотрицательны, чем металл*, в ре-
зультате чего преобладающими чертами их взаимодействия
являются электростатическое притяжение и некоторое перераспре-
деление заряда к металлу (ковалентность). Лиганды — это обыч-
но нейтральные или отрицательно заряженные молекулы. Ком-
плексные соединения характеризуются сохранением их индиви-
дуальности в растворе, хотя возможна также заметная
диссоциация. Пространственное расположение лигандов вокруг
центрального иона называется конфигурацией комплекса, некото-
рые более детализированные аспекты стереохимии лиганда (без-
относительно к иону металла) обычно обсуждаются в терминах
конформаций лиганда. Общее число донорных атомов, присоеди-
ненных к центральному атому металла, называется координацион-
ным числом**. В это число включаются все донорные атомы, на-
ходящиеся от металла на расстоянии химической связи, хотя не-
которые из этих атомов могут располагаться дальше от металла,
чем другие, а некоторые из них .могут координироваться более чем
с одним атомом металла, как в кристаллах и полимерных коорди-
национных соединениях. Если известна стереохимия, то координа-
* Ошибка автора: донорные атомы (кислород, азот и др.) обычно более
электроотрицательны, чем атомы металлов. — Прим. ред.
** Это, конечно, слишком упрощенное определение, не затрагивающее, на-
пример, обширный класс л-комплексов. Следуя автору, пришлось бы приписать
атому хрома в Сг(СеН6)2 координационное число 12, — Прим.tped.
Структура и стереохимия координационных соединений
19
ционное число атома металла определяется однозначно, но стерео-
химия определяется не только координационным числом. Оба
свойства зависят от природы связи металл—лиганд, но обсужде-
ние этого более фундаментального свойства выходит за пределы
данной главы [18—22]. Структура комплексного соединения оп-
ределяется однозначно, если известны координационное число цен-
трального атома металла, стереохимия и конформации присоеди-
ненных к нему лигандов. В настоящее время такая детальная
информация ограничена твердым состоянием (методы дифрак-
ции), о структуре лигандов в растворе известно мало.
Возможно, наиболее важным понятием, связанным с коорди-
национными соединениями и контролирующим их, является льюи-
совская кислотность иона металла. Это понятие будет рассмотре-
но в гл. 2, а здесь достаточно сказать, что комплексы непереход-
ных металлов (Na+, К+, Са2+, Mg2+, Ва2+, А13+) удерживаются
вместе с электростатическими силами* и их стереохимия опреде-
ляется почти исключительно размером лиганда и зарядом на ионе
металла. Устойчивости комплексных ионов изменяются парал-
лельно с основностью протонов лигандов, и эффективная роль
иона металла подобна таковой протона. Стереохимия комплексов
переходных металлов 'более сложна, и в настоящее время не су-
ществует удовлетворительной эмпирической или теоретической
модели для детального описания всех аспектов их структуры или
даже стереохимии. Для многих из этих металлов ионная модель
усложняется тем, что их электронные облака не имеют сфериче-
ской формы (эффекты кристаллического поля), а также, что под-
разумевается в их названии, очень значительным отступлением от
ионного характера, связанным с переходом от ионной к ковалент-
ной связи. Для таких комплексов важна как нейтрализация за-
рядов, так и кислотность по Льюису, и для описания химической
связи в этих комплексах были развиты теория поля лигандов п
метод молекулярных орбиталей [2, 5].
2. КООРДИНАЦИОННОЕ ЧИСЛО И СТЕРЕОХИМИЯ
Начало развития структурной координационной химии относит-
ся к концу XVIII в. и связано с исследованиями двух замечатель-
ных химиков — датчанина С. М. Йоргенсена и швейцарца Альфре-
да Вернера. До этого времени было известно, что два или более
неорганических соединения могут соединяться в стехиометриче-
ских отношениях, образуя «комплексные соединения», но их обыч-
но писали как двойные соли, например ZnCl2-2CsCl, 2KCl-MgCl2,
A12(SO4)s K2SO4-24H2O, Fe(CN)2-4KCN, A1F3-3KF, CoC12-2KC1,
• В комплексах непереходных металлов (особенно А1’+, Ве*+ и др.) боль-
шую роль играет также ковалентная связь. — Прим. ред.
2*
20
Глава 1
хотя сегодня мы знаем, что формула некоторых из них должна
быть написана иначе, например КгСоС14, K?HgF4, K.2Fe(CN)fi*.
Вернер и Йоргенсен синтезировали сотни координационных
соединений, главным образом соединений Co(III), Pt(IV) и Pt(Il)
с аминами, а также изучили их превращения, степени ионизации
и наличие изомеров. В результате интенсивною со |?рничегтва
между этими двумя замечательными экспериментаторами были
быстро сформулированы структурные основы современной коор-
динационной химии. Вернер в 1893 г. правильно объяснил свои
результаты на основе понятий о первичной и вторичной валент-
ности. Первичная валентность рассматривалась, в сущности, как
нормальная электровалентность иона металла [например, 4 для
Pt(IV), 2 для Pt(II) и 3 для Со(Ш)] и определяла общее число
отрицательных зарядов, которые должны быть привнесены при-
сутствующими анионами, в то время как вторичная валентность
обусловливала пространственное расположение связей вокруг
иона металла с образованием октаэдрической ['Co(III), Pt(IV)]
или квадратно-плоскостной [Pt(II)] геометрии. Вторичная валент-
ность теперь называется координационным числом.
Вернером были развиты следующие два наиболее важных
принципа: во-первых, каждому иону металла может быть припи-
сано только одно координационное число, которое удовлетворяет-
ся присоединенными лигандами, и, во-вторых, эти координацион-
ные места имеют определенное стереохимическое расположение
в пространстве. Эти две идеи имели исключительное значение для
развития стереохимических аспектов координационной химии. Так,
praseo- и що/ео-формы CoCl3-4NH3 были правильно отнесены к
транс- и tyuc-изомерам [Co(NH3)4C12]C1 (группы внутри квадрат-
ных скобок образуют октаэдрическую координационную сферу,
а остающийся атом хлора удерживается электростатически-
ми силами и может быть легко оттитрован, например
[Co(NH3)4C12]+C1_). На основе координационной теории Вернера
удалось классифицировать все известные в то время данные о
комплексах металлов, а в последующие годы предположение Вер-
нера многократно подтверждалось рентгеноструктурными иссле-
дованиями. Однако в настоящее время известно, что их строгое
применение возможно не во всех случаях.
Вернером также были введены понятия хелатообразования
(или образование кольца) и оптической изомерии. Так, он обна-
ружил, что две функциональные аминные группы этилендиамина
* Часто невозможно отличить двойную соль и координационное соединение
по их поведению в водном растворе. Например, рентгеноструктурные данные
убедительно показывают, что в К2С0СЦ четыре иоиа С1_ расположены вокруг
иона Со(П) по вершинам тетраэдра, но в воде этот комплекс быстро диссо-
циирует с образованием розового раствора ноиа [Со(Н2О)6]2+, так что его пове-
дение в растворе сходно с поведением двойной соли.
Структура и стереохимия координационных соединений 21
могут замещать две молекулы NH3 в [Pt(NH3)4]C12 с образова-
нием [Pt(en)2]C12, при этом образуется пятичленное гетероцик-
лическое 'КОЛЬЦО.
H3N\ /NHj
H3N^ ^NH3
+ 2en
H2 H2
H2C"\ zN"CH2
Pt
H2C^N N CH2
H2 H2
4NH3
Подобным образом 'было принято, что соединение
[Со(еп)2(С2О4)]С1, которое образуется >при взаимодействии
[Со(еп)2С12]С1 с Na2C2O4, содержит хелатированный оксалатный
анион, замещающий два атома хлора, находившиеся в цис-поло-
жении.
С2О
Вернер также установил, что для комплексов с тетраэдриче-
ской и октаэдрической стереохимией возможна оптическая изо-
мерия, отличающаяся от геометрической. Так, цис-изомер
[Со(еп)2С12]С1 не имеет ни плоскости, ни центра симметрии и
должен существовать в виде пары соединений, имеющих строение
несовместимых друг с другом зеркальных изображений — право-
и левовращающего. Это положение Вернер доказал, разделив в.
I
(+)s89-A-[Co(en)2Cl2J*
(—)s89‘A"[Co(en)2Cl2]*
22
Глава 1
1911 г. с помощью d-бромкамфорсульфоната в качестве аниона
оптические изомеры иона [Co(en)2NH3Cl]2+, а годом позже — так-
же ионов [Со(еп)2С12]+, [Co(en)2(N02)2]+ и [Со(еп)3]3+.
Для того чтобы ответить на замечание Йоргенсена, что опти-
ческая активность этих комплексных ионов может быть обуслов-
лена асимметрией окружения углеродных атомов, Вернер синте-
зировал и разделил на оптические изомеры чисто неорганический
комплексный ион(III)—для того времени значительный подвиг.
1
Точка зрения Вернера на координационное число и стереохи-
мию составляет основу современной координационной химии [1],
и для того, чтобы обсудить даже наиболее тонкие детали струк-
туры комплексов металлов, необходимы лишь немногие дополни-
тельные понятия. Большое число известных в настоящее время
комплексов и их разнообразие потребовали некоторых незначи-
тельных изменений положений Вернера; наиболее существенное
из них то, что для некоторых ионов металлов существуют более
чем одно координационное число и стереохимия. Так, для Си(II)
возможны координационные числа 4, 5 и 6, а актиниды, такие,
как U, могут иметь координационные числа от 5 до 20. Кроме то-
го, стереохимия определяется не только лигандом и ионом метал-
ла. Так, существует несколько комплексов Ni(II) с координацион-
ным числом 4, которые могут иметь тетраэдрическую или квад-
ратно-плоскостную структуру в зависимости от температуры и
(или) природы растворителя. Известно, что некоторые комплексы
Ni(II), Zn(II) и Со(И) могут иметь, в зависимости от экспери-
ментальных условий, тетраэдрическую, квадратно-плоскостную
или октаэдрическую геометрию.
Со времени Вернера было накоплено большое количество
структурных данных с целью определения координационных чи-
сел и более тонких деталей стереохимии. Не вызывающие сомне-
ний прямые данные о структуре в твердом состоянии получают
главным образом методом дифракции рентгеновских лучей, и эта
область координационной химии за последние десять лет получи-
ла огромное развитие; в литературе по неорганической химии
публикуется от 20 до 30 рентгеноструктурных исследований каж-
Структура и стереохимия координационных ^единений
23
дый месяц, и это число быстро растет. Точность этих исследова-
ний также находится на высоком уровне. Для точного определе-
ния структуры координационного соединения в растворе в настоя-
щее время не существует какого-либо прямого метода, однако'
такие методы, как ЯМР (в частности, на ядрах *Н, 19F, 31Р, 13С)
и ЭПР (парамагнитные комплексы), часто дают надежные ре-
зультаты, а такие непрямые методы, как электронные спектры, ди-
польные моменты, магнитные моменты и измерение молекулярной
массы, интенсивно используются для предсказания структуры. Ни-
же мы приведем краткое общее обсуждение координационных
чисел и стереохимии координационных соединений. Возможно,
что одной из наиболее отличительных черт современной координа-
ционной химии является то, что численное превосходство структур
с координационными числами 4 и 6 серьезно поколеблено и что
«необычные» координационные числа (5, 7, 8) становятся все бо-
лее обычными.
2.1. Координационное число 2
Координационное число 2 не очень распространено, и оно
обычно характерно только для комплексов Cu(I), Ag(I), Au (I)
и Hg(II). Из двух возможных конфигураций — линейной (D^h)
и угловой (С2г1) — найдена только линейная форма, как полагают,
из-за того, что она обеспечивает минимум лиганд-лигандного от-
талкивания. В качестве типичных примеров можно привести
[CuCl2]_, [AuC12]_ и [Hg(CN)2]. Из примеров, представляющих
больший биохимический интерес, можно указать на [Ag(NHs)2]+,
Ag(Gly), Ag(Gly)-0,5Н2О и Ag(GIy-Gly) (гл. 4, формулы III, IV
и XI). В последних структурах Ag(I) прочно связывается с ами-
но- и карбоксильной группами глицина, но очень слабо с карбо-
нильным кислородом глицилглицина. По-видимому, и с другими
аминокислотами и пептидами Ag(I), Cu(I) и Au(I) будут свя-
зываться подобным образом.
Иногда необычные лиганды координируются с образованием
необычных конфигураций, например:
(CH3)3Sk zSi(CHs)s
/N—Со—ЬГ
(CHsJsSi/j »>Si(CH3)3
но это происходит довольно редко. Однако такие случаи показы-
вают, что ионы металлов даже в обычных состояниях окисления
могут иметь необычные координационные свойства.
2.2. Координационное число 3
Это координационное число встречается очень редко, за ис-
ключением молекул, содержащих относительно электроотрица-
тельные центральные атомы, например BF3, NO3 (плоский тре-
24
Глава 1
угольник); NH3, СЮз (тригональная пирамида). Так, KatCuCh]
и CssljHgCls] содержат бесконечные цепи тетраэдров МС14, в то
время как галогениды МС13 (М=Сг, Fe, Мп, V) кристаллизуются
в решетках, содержащих ионы металлов в октаэдрическом окру-
жении. Комплекс Cs[CuCl3] состоит из бесконечных цепей —С1—
—CuCh—Cl—CuCl2—Cl— с 4-кратной координацией иона метал-
ла, а АиС13 — димер, содержащий две плоские частицы АиСЦ со-
единенные общей гранью. Наиболее простые комплексы металлов
состава МХ2 или МХ3 в растворе либо легко диссоциируют, либо
образуют частицы МХ2(Н2О)4 или МХ3(Н2О)3, либо существуют
в виде полиэдров координационных чисел 4 или 6. Трис-(гексаме-
тилдисилиламинато) железо (III) (V) и анион Hgl3 в
(CH3)3S(HgI3) (VI) представляют примеры соединений, имеющих
•симметрию, близкую к плоскому равностороннему треуголь-
нику (D3h). Плоская 7-геометрия (C2v) обнаружена в катпоне
<CH3)3Se+ (VII).
[(CH3)3Si]2N\ zN[Si(CHs)sj2
Ре
N[Si(CHs)s]2
V
\ /Г
Hg
I
I
VI
CH3—Se—CH3
I
сн3
VII
2.3. Координационное число 4
2.3.1. Тетраэдр
Эту геометрическую конфигурацию имеют многие комплексы.
Юна обычна для координационных соединений непереходных эле-
ментов, для которых их устойчивость может быть частично объяс-
нена использованием ковалентных хр3-гибридных орбиталей
металла, а частично тем фактом, что при тетраэдрическом располо-
жении лигандов возникают наименьшие стерические и электро-
статические напряжения по сравнению со всеми другими способа-
ми расположения лигандов, возможными при координационном
числе 4. Так, Na(I) в жидком аммиаке существует в виде
[Na(NH3)4]+, а исследования методом рассеяния рентгеновских
лучей показывают, что первичное координационное число К(1)
в водном растворе равно четырем; комплексы В(Ш), Ве(П),
.Zn(II), Cd(II) и Hg(II) состава МХ4 (X = F, Cl, CN) имеют тет-
Структура и стереохимия коарс рационных соединений
25
раэдрическое строение, так же как многие комплексы Be(II) и
Hg(H) с бидентатными лигандами.
СН2ОН
Н2с- s S—СИ
I X I
нс—S S—сн2
О-С^СбН5
\ / \\
нс. Be СН
V 'У
СН2ОН
W
Тетраэдрическое строение бис-(бензоилпирувато)бериллия(П)
(IX) было установлено путем его разделения на оптические фор-
мы. Для тетраэдрических 'комплексов в принципе возможны толь-
ко оптические и невозможны геометрические изомеры. Однако
лабильность лигандных групп в противоположность инертности
связей углерода препятствует разделению на оптические изомеры
любого тетраэдрического комплекса, содержащего четыре различ-
ные монодентатные группы.
Тетраэдрические комплексы ионов переходных металлов обыч-
но устойчивы только при некоторых определенных условиях. Осо-
бое исключение представляют ионы [FeCl4]2~, [СоХ4]2~ (Х = С1, Вг,
I, NCS) и некоторые ионы [СоХ3(Н2О)]_; эти частицы сохраняют
в водном растворе тетраэдрическую геометрию, хотя можно было
бы ожидать их акватации с образованием октаэдрических ионов.
Причина их устойчивости неизвестна*. Тетраэдрические анионы,
такие, как [CuX4]2-, [NiX4]2-, [VX4]~ и [MnX4]2- (Х = С1, Br, 1),
стабилизируются в слабо координирующихся растворителях,
а также в твердом состоянии объемистыми катионами, например
[ (С6Н5)з(СНз)Р]+, [(СбНб^эр и [(C4H9)4N]+. Ни один из них
не сохраняет этой геометрии в координирующихся растворителях,
таких, как вода или спирты. Большинство тетраэдрических ком-
плексов— анионы или нейтральные молекулы, [МХ4]2-, [MLX3]~
или [ML2X2], где М — Со, Ni пли Fe, L — нейтральный лиганд
(Н2О, пиридин, POR3, AsOR3, PR3, AsR3, NR3), a X — анион, обыч-
но галогенид. Известны лишь очень немногие тетраэдрические
катионные комплексы, но Со(Н2О)4+ содержится в малых количе-
ствах в водном растворе в равновесии с Со(Н2О)б+, а тетраэдри-
ческий [Co(NH3)4]2+ найден в [Со(МН3)4] (ReO4)2. Иногда би-
дентатные лиганды могут образовывать с Со(11). а иногда и
с Ni(II) тетраэдрические комплексы под влиянием объемистых
заместителей, например бис- (N-изопропилсалицилальдимина-
* В случае комплексов типа [СоХ4]2- большую роль может играть стабили-
зация полем лигандов центрального иона с конфигурацией d7.— Прим. ред.
26
Глава 1
то) кобальт (I I) (X) и бис-(дипивалоилметанато)никель(П) (XI),
но обычно, если нет стерических препятствий, происходит полиме-
ризация с образованием октаэдрического окружения металла, как,
например, в [№(асас)2]з и [Со(М-СН3-5а1еп)2]2.
(СН3)3СЧ ZC(CH3)3
с—о о-с
/Г \ / 'х\
НС' Ni ;сн
\ч_ / V/
с—с о-с
(CH3)3CZ ЧС(СН3)3
XI
Кажется, что для тех комплексов, для которых существует
равновесие между квадратно-плоскостной и тетраэдрической гео-
метрией, как, например, для бис-(Ы-алкиламинотропониминато)-
никеля (II) (XII) и бис- (N-алкилсалицилальдиминато) никеля (II),
отталкивание между группами R при квадратно-плоскостном рас-
положении благоприятствует тетраэдрической структуре, но эта
причина не универсальна, и электронные эффекты также долж-
ны играть важную роль.
2.3.2. Плоский квадрат
Эта форма координации характерна для некоторых опреде-
ленных состояний окисления, но в других случаях она мало рас-
пространена. Обычно эту структуру имеют комплексы Pt (II),
Pd(II), Ag(II), Au(III), Rh(I), Ir(I), она встречается также в
комплексах Ni(II) и Си(II), в других случаях она практически
не наблюдается. Для квадратно-плоскостных структур возможна
геометрическая и невозможна оптическая изомерия. Замечатель-
ное исключение из этого правила представляет соединение XIII,
которое Миле и Квибелл в 1935 г. разделили на оптические изо-
Структура и стереохимия координационных соединений
27
меры и таким образом установили, что оно имеет квадратно-пло-
скостное, а не тетраэдрическое строение.
"С,НБХ
zC—h2n.
2+
н
С6н5.
-С—h2n
Pt.
nh2—сн2
сн3
XIII
nh2—с
СН3_
н
В этой молекуле асимметрия свойственна не атому металла
или лиганду самому по себе, а расположению хелатных колец.
Квадратно-плоскостная координация в комплексах Pt(II) и Pd(II)
была известна со времен Вернера и широко распространена, на-
пример бис-(глицинато)платина(II) (XIV) и (хлороглицин-Ё-ме-
тионинато)платина(П) (гл. 4, структура XXXIX). Известны также
XIV
многие комплексы Ni(II) и Си(II), имеющие почти плоское
строение. Обычно это нейтральные или анионные частицы, напри-
мер (тетраглицинато) металлаты натрия (XV) (M = Cu2+, Ni2+) и
(1-аминоциклопентанокарбоксилато)медь(П) (XV).
сг н2
hk.
С \ /
Си
Н2С—/
/ \ N
Н2С. .С Н2
схн2
Н2
н2
с—сн2
/ \
с ХСН2
О^С-^Н2
О
XV XVI
Примеры квадратно-плоскостных структур, представляющих
интерес для биохимии, ассоциируются, конечно, в первую очередь
28
Глава 1
с ядрами фталоцианина и порфирина [24]. Все металлофтало-
цианиновые красители (XVII) [M(II)=Be, Мп, Fe, Со, Ni, Си,
Pt] изоструктурны с исходным протонированным основанием
(пространственная группа P2j/a), а хлорофилл [Mg(II)], вита-
мин Bj2 [октаэдрический Сю(III)] и гемоглобин [Fe(II) 5-кратно
координированный] представляют важные примеры использова-
ния замещенных порфириновых ядер (XVIII).
XVII
Рентгеноструктурным методом было изучено большое число
разнообразных металлопорфиринов [Cu(II), Zn(II), Fe(III),
VO(IV), Ni(II), Rh(III), Co(III), Au(III), Sn(IV)]. Строение ком-
плексов металлов этого типа будет обсуждено в последующих
главах, а здесь достаточно лишь отметить, что плоское кольцо
тетрапиррольного ядра вынуждает металл координироваться в
квадратно-плоскостном или почти квадратно-плоскостном окруже-
нии.
2.4. Координационное число 5
Хотя исторически комплексы с координационным числом 5
имели относительно меньшее значение, в настоящее время они
известны намного лучше, главным образом в результате рентге-
ноструктурных исследований. Некоторые из них образуются без
явного сжатия лигандов, например Fe(CO)5, [MnCU]3-,
[Ni2Cls]4—, но образование большинства комплексов с координа-
ционным числом 5 вынуждено пространственным строением ли-
гандов, например {Со[ (CH3)6-tren] Вг}+ [ (СбН5)3Р]3КиС12,
TiBr3[N(CH3)3]2. Обычно такие частицы можно рассматривать
Структура и стереохимия координационных соединений
29
ОН2
n2|
н2с^\| он2
Си
ХО
о' \ /
с—с.
н2
XIX хх
как образованные из частиц с более высоким или более низким
координационным числом путем отщепления (XIX) или добавле-
ния (XX) лиганда. Для координационного числа 5 возможны два
довольно правильных геометрических расположения лигандов во
внутренней координационной сфере: тригональная бипирамида
(D3tl) (XXI) и квадратная пирамида (XXII). Обычно гео-
метрию тригональной бипирамиды имеют комплексы металлов в
низких валентных состояниях [Мп(—I), Fe(0), Со(1)] с лиган-
XXV, Fe(CO)s
ХХП, MnCls2“
дами, образующими л-связи (СО, CNR, CN), в то время как квад-
ратно-пирамидальное расположение лигандов характерно для
комплексов металлов правой стороны переходного ряда в состоя-
ниях окисления, имеющих более важное биологическое значение,
например Ni(II), Co(II), Zn(II), Cu(II). Комплексы меди обычно
содержат намного более длинную (более слабую) связь, направ-
ленную к вершине пирамиды.
Наконец, важно указать, что геометрии тригональной бипира-
миды и квадратной пирамиды действительно не сильно отличают-
ся одна от другой, и необходимы лишь незначительные измене-
ния углов для перехода одной в другую.
2.5. Координационное число 6
Это, несомненно, наиболее обычное координационное число,
встречающееся в комплексах металлов. Ионы всех металлов, за
исключением щелочных металлов Li+, К+ и, возможно, Na+ (коор-
30 Глг .а 1
динационное число 4), очень больших лантанидов (координацион-
ное число 9) л ионов группы урана (>10) в водных растворах
координируют шесть молекул воды. Как правило, при координа-
ционном числе 6 комплексы имеют октаэдрическую конфигурацию
(XXIII), но в ряде случаев с особыми лигандами обнаруживаются
и другие возможные правильные конфигурации, такие, как триго-
нальная призма (XXIV).
Октаэдр имеет высокую симметрию {Ок). Некоторые металлы
образуют комплексы с неискаженной или очень мало искаженной
октаэдрической симметрией. Так, комплексы Zn(II) и высокоспи-
новые комплексы Мп(П), а также комплексы Сг(Ш) и Со(Ш)
имеют симметрию, очень близкую к Ок. Комплексы ряда других
ионов металлов, например Си(II), Ni(II) и Со(II), также имеют
октаэдрическую геометрию со многими лигандами, включая во-
ду, но в некоторых случаях она сильно искажена. Обычно наблю-
даются два типа искажений — тригональное (XXV) и тетраго-
нальное (XXVI). В тригонально искаженной молекуле октаэдр
растянут или сжат вдоль одной из его осей 3-го порядка
XXV XXVI
(Ок—>-^зй) ; тетрагональное искажение происходит в результате
аналогичного растяжения или сжатия вдоль оси 4-го порядка
(Ок--->-^4л)-
Очевидно, что в пределе тетрагонально искаженная молеку-
ла полностью теряет два лиганда, находящихся в тракс-положе-
Структура и стереохимия координационных соединений
31
нии, и приобретает координационное число 4 и геометрию пло-
ского квадрата. Октаэдрические комплексы могут проявлять как
геометрическую, так и оптическую изомерию, этот вопрос обсуж-
дается в разд. 5.
2.6. Координационное число 7
Металлы первого переходного ряда редко имеют координаци-
онные числа больше шести, но некоторые примеры известны, на-
пример [(РеЭДТАЩаО)]- (XXVII) (пентагональная бипирами-
да) и [МпЭДТА(Н2О)]2_ (XXVIII) (гранецентрированная триго-
нальная призма). Более высокие координационные числа обычно
имеют металлы второго и третьего переходных рядов, а также
лантаниды и актиниды.
XXV11
Для координационного числа 7 известны три геометрические
расположения: пентагональная бипирамида (D5h) (XXVII), одно-
шапочная тригональная призма (C2v) (XXVIII) и тригональная
призма с тетрагональным основанием (Cs) (XXIX).
XXIX
32
Глава 1
2.7. Координационное число 8
Возможны разнообразные геометрии, но наиболее симметрич-
ное кубическое (О/,) расположение неизвестно ни для одной дис-
кретной молекулы МХ8 (такое расположение имеет место в крис-
таллической решетке CsCl). Это объясняют взаимным отталкива-
нием лигандов, в результате которого образуются более
благоприятные искаженные структуры. Из нескольких наблюдав-
шихся конфигураций наиболее обычны квадратная антипризма
(Du) (XXX) и додекаэдр (D2d) (XXXI). Они получаются в ре-
зультате соответствующих искажений куба.
XXX
Интересные случаи представляют [Со(NO3)4]2-и [Zr(C2O4)4]4-,
в которых наличие четырех- и пятичленных нитратного и оксалат-
ного хелатных циклов соответственно приводит к образованию
додекаэдрической конфигурации, в то время как [Th(acac)4] и
[Zr(acac)4], содержащие большие шестичленные хелатные коль-
ца, имеют структуру антипризмы.
2.8. Координационное число 9
При координационном числе 9 возможно несколько геометри-
ческих форм, и они наблюдались в некоторых случаях. Наиболее
распространена гранецентрированная тригональная призма (D3h)
(XXXII), которую можно получить из тригональной призмы путем
добавления трех атомов лигандов с наружной стороны над цент-
рами трех вертикальных граней.
XXXII
Структура и стереохимия координационных соединений
33
2.9. Координационные числа больше 9
Они встречаются только у ионов металлов большого размера
[например, Cs(I), La(III), U(III), Ce(III)], которые образуют
более слабые координационные связи, чем элементы первого пе-
реходного ряда (рис. 1.1). Во .многих случаях точное определение
координационного числа затруднительно и требует рентгенострук-
турных данных. Образующиеся координационные многогранники
часто неправильны.
Рис. 1.1. Структура аниона [La ЭДТА(Н2О)э]— (а) [49], содержащего 9-кратно
координированный La(III), и комплекса [LaОЭДТА(Н2О)4] (б), [48], содер-
жащего 10-кратио координированный La (III).
34
Глава 1
3. КООРДИНАЦИОННЫЕ ЧИСЛА И СТЕРЕОХИМИЯ
КОМПЛЕКСОВ ОБЫЧНЫХ ПЕРЕХОДНЫХ ЭЛЕМЕНТОВ
В табл. 1.1—1.10 приведены состояния окисления, координа-
ционные числа и геометрия комплексов наиболее распространен-
ных переходных металлов. Те, которые встречаются наиболее
часто, набраны курсивом. В примечаниях к таблицам обсуждает-
ся их распространенность и химическое поведение.
3.1. Титан (табл. 1.1)
~2В —0.1 В
Ti2+ ---► Ti3+ --> TiO2* (кислый раствор)
В водных растворах Ti2+ не существует; наиболее устойчивое
и обычное состояние окисления Ti(IV), а исключительная поля-
ризующая способность малого иона Ti(IV) приводит к значитель-
ной ковалентности образуемых им связей, вследствие чего Ti4+ не
Таблица 1.1
Состояние окисления Зб(-Электронная конфигурация Координацион- ное число Геометрия Примеры
Ti(-I) 6 Октаэдр [Tidipy3]-
Ti(0) 6 Tidipy3
Ti(II) 6 TiCl2
Ti(III) 6 [TiFe]3-[Ti(H2O)e]3+, [Ti(urea)e]3+
Ti(IV) 4 Тетраэдр TiCl4( л-С5Н5)2Т iCl 2
5 Искаженная K2T12O5
тригональная бипирамида
Квадратная TiO(acac)2
пирамида
6 Октаэдр TiO2, [TiFe]2-, Ti(acac)2Cl2, [Ti(OC2H5)4|4
8 Додекаэдр TiCl4(diars)2
существует. Соединения Ti(IV) легко гидролизуются в водном
растворе, образуя частицы со связями Ti—О; многие из них име-
ют октаэдрическую координацию. Титан предпочтительно коорди-
нируется через донорные атомы кислорода, образуя полимерные
октаэдрические структуры.
Структура и стереохимия координационных соединений 35
3.2. Ванадий (табл. 1.2)
0,255В —0,337В —1,00В
V2+ ----> V3+---------► VO2+ ----► [V(OH)4]+ (кислый раствор)
V(IV) и V(V) — наиболее обычные состояния окисления. Коор-
динируются предпочтительно с донорными атомами кислорода и
поляризующимися лигандами. Дискретные ионы V4+ и V5+ неиз-
вестны. Ион ванадила [VO(H2O)b]2+ и различные полимерные
частицы, содержащие пятивалентный ванадий — [УОз(ОН)]2-,
Таблица 1.2
Состояние окисления З^-Электронная конфигура- ция Координа- ционное число Геометрия Примеры
V(-I) 6 Октаэдр [V(CO)e]-, [V(CN)5NO]b-
V(0) d5(?) 6 » V(dipy)3, V(CO)e
V(I) d4 6 » [V(dipy)3]+, [V(CO)4(arene)]+
V(II) d3 6 [V(H2O)e]2+, [V(CN)e]4-
V(III) d2 4 Тетраэдр [VC14]-
5 Тригональная бипирамида mpaHc-{VCl3[S(CH3)2]i)
6 Октаэдр [V(NH3)e)3+, [V(C2O4)3]3-
V(/V) d* 4 Тетраэдр VC14
5 Т етрагональная пирамида VO(acac)2 [VO(SCN)4]2-
Тригональная бипирамида VOC12[N(CHs)3]2
6 Октаэдр УО2(рутил), VO(acac)2py
8 Додекаэдр [VCl4(diars)2]
V(V) d® 4 Тетраэдр VOC13, [VO4]3~ (ванадаты)]
5 Тригональная бипирамида VF6, полимерные ванадаты
6 Октаэдр VO6, октаэдрический в кислых ванадатах
[V2O6(OH)]3-, [VO2(OH)2]-—получаются из мономерного тет-
раэдрического иона ванадата VOi~, который существует только
в сильно щелочных растворах (рН>12), при pH ниже ~7 осаж-
дается V2Os. Все частицы V(V), по-видимому, представляют
5-кратно координированные производные иона перванадата
3
36
Глава 1
(VO*) и содержат гидроксильные мостики. Комплексы V(IV)
обычно содержат ванадил (VO2+) и имеют конфигурацию квад-
ратной или тетрагональной пирамиды, например [VO(acac)2]
(XXXIII).
гн о /СНз
СНз^с—о ° о—с
нс: /V\ >?сн
/С—О О-С^
СН1 снз
XXXIII
Это определяет основные черты стереохимии комплексов с хе-
латирующими лигандами, координирующимися через атомы кис-
лорода: [VO(C2O4)2]2-, [VO(C2O4) (Н2О)2]. Иногда более слабые
донорные группы могут координироваться в шестом положении с
образованием искаженных октаэдрических структур, например
[VO (асас)2ру], [VO (асас)2 (imid) ].
3.3. Хром (табл. 1.3)
0,41В
Сг2+ ---> Сг®+ (кислый раствор)
1,1В 0.I3B
Сг(ОН)2----► Сг(ОН)3----> СгО|- (щелочной раствор)
Полагают, что в водных растворах существуют только Сг(П)
и Сг(Ш); Cr(IV) и Cr(V) легко диспропорционируют на Сг(1П)
Таблица 1.3
Состояние окисления Sd-Электронная конфигурация Координа- ционное число Геометрия Примеры
Сг(0) d5s4(?) 6 Октаэдр Сг(СО)е, Cr(dipy)3
Сг(1) d5 6 » [Cr(CN)5NO]3-, [Cr(dipy)3]+
Сг(11) d4 6 Октаэдр (искажен- ный) CrCl2, CrS, [Cr(NCS)e]4-, [Cr(en)3]2+
Сг(ПГ) d3 6 Октаэдр [Cr(H2O)e]3+ [Cr(NH3)eJ3+ Cr(acac)3, [Cr(CN)e]3-
Cr(lV) d2 4 Тетраэдр Ba2CrO4
6 Октаэдр K2CrFe
Cr(V) d1 4 Тетраэдр ICrOJ3-
6 Октаэдр [CrOCl5l2-
Cr(VI) d° 4 Тетраэдр [CrO4]2-, CrO2Cl2. CrO3
Структура и стереохимия координационные соединений 37
и Cr(VI). В последнем состоянии окисления хром является силь-
ным окислителем и существует только в виде оксочастиц — СгО3,
CrOV, СгОгР2 и т. п.
Соединения Cr(II) —сильные и быстродействующие восстано-
вители, и в водных растворах они могут существовать только в
отсутствие кислорода. Для них характерно октаэдрическое строе-
ние*, например [CrfNCSJe]4-, [CrfCN)^2-. Однако можно легко
синтезировать очень устойчивый димерный ацетат хрома(II)
[Сг(ОСОСН3)2]2.
Комплексы Сг(Ш) представляют наиболее устойчивое и наи-
более важное состояние окисления, преимущественно они имеют
геометрию правильного октаэдра. Известны многие сотни ком-
плексов Сг(П1), главным образом с лигандами, координирующи-
мися через атомы О и N. Среди металлов первого переходного
ряда Сг(Ш) наряду с Со(III) образует кинетически наиболее
инертные комплексы.
3.4. Марганец (табл. 1.4)
—1,51В ЯЛ п2+ т. —0.95В ЯЛ «3+ —2,26В —0,564В
МПТ " * МП МПС/2 * AiiiC/д (кислый рдсгвор)
—0,1В 0.2В
Мп(ОН)2 >- Mn(OH)3 - >- МпО2 (щелочной раствор)
Таблица 1.4
Состояние З^-Электронная Координацией-
окисления конфигурация ное число Г еометрия
Мп(П) ? 4 (или 6) Квадрат [Mn(phtalocyanine)]2-
Мп(—I) ? 5 Тригональная [Мп(СО)Б]-
бипирамида
4 (или 6) Квадрат [Mn(phtalocyanine)]-
Мп(0) d’s1 6 Октаэдр Мп2(СО)10
Мп(1) d« 6 » [Mn(CN)e]5-, [Mn(CNR)„]+
Mn(CO)BCl
Мп(1Г) d6 4 Тетраэдр [MnClJ2-
4 Квадрат [Mn(H2O)4]SO4 • H2O
6 Октаэдр [MnfHaO).]2*
[Mn(SCN)e]«-
? {Mn[(CH3)Bdien]X2}
7 Структура [МпЭДТА(Н2О)]2-
[NbFy]2-
Мп(Ш) d4 6 Октаэдр Mn(acac)s, [Mn(C2O4)ds-
Mn(IV) d3 6 » MnO2, [MnClel3-
Mn(VII) d» 4 Тетраэдр [MnO4]-, MnO3F
Искаженное. — Прим. ред.
38
Глава 1
Химия водных растворов ограничивается исключительно хими-
ей Мп(II)*. Из-за большого размера иона Мп2+ по сравнению с
двухвалентными ионами последующих металлов (от Fe^ до Си2+)
и отсутствия стабилизации кристаллическим полем (высокоспино-
вая конфигурация d5) он образует очень слабые комплексы, кото-
рые легко диссоциируют в воде до [Мп(НгО)6]2+. Однако ком-
плексы с хелатирующими лигандами, такими, как этилендиамин,
оксалат и ЭДТА, могут быть выделены из водного раствора.
Соединения Мп(II) обычно имеют октаэдрическое строение,
например [Mn(NH3)6]2+, [Mn(en)3]2+, [Мп(СгО4)3]4-. Было пока-
зано, что некоторые соединения имеют более низкую симметрию
при координационном числеМп(II), равном 7 ([МпЭДТА(Н2О)]2-)
или 5 ([MnCls]3-), в то время как в Мп(асас)2 в результате об-
разования тримера достигается октаэдрическая координация. Из-
вестны некоторые тетраэдрические комплексы, например
[Мп(РН3РО)2Вг2] и (R4M)2[MnX4] (M = N, Р, As, а Х=галоген),
но они быстро диссоциируют в воде. Из комплексов Мп(III), по-
жалуй, наиболее известен октаэдрический ацетат Мп(С2,Н3О2) -
• 2Н2О, все они легко восстанавливаются в воде до Мп(II) и мож-
но ожидать, что при отсутствии влияния лигандов должны иметь
геометрию искаженного октаэдра.
3.5. Железо (табл. 1.5 )
—0,771В
Fe2+ -----► Fe3+ (кислый раствор)
0.56B
Fe(OH)2 --► Fe(OH)3 (щелочной раствор)
Комплексы Fe(II) и Fe(III) устойчивы в водном растворе и
при отсутствии влияния лигандов имеют октаэдрическое строение.
Fe(II) и Fe(III) связываются предпочтительно с лигандами, коор-
динирующимися через атомы кислорода и серы, по сравнению с
лигандами, координирующимися через атомы азота, особенно
Fe(III), для которого неизвестны простые аминаты (аммиакат из-
вестен). Однако координирующиеся через атомы азота хелатирую-
щие лиганды (en, phen и т. п.) образуют с Fe(II) очень устойчи-
вые комплексы, которые легко окисляются до Fe(III). Fe3+ в вод-
ных растворах при рН>3 полимеризуется, и [Fe(H2O)e]3+ суще-
ствует только в сильной кислоте, однако [Fe(H2O)6]2+ обладает
намного менее кислыми свойствами.
* В последнее время появилось значительное число работ, посвященных хи-
мии комплексов Мп (III) в водных растворах. — Прим. ред.
Структура и стереохимия координационных соединений
Таблица 1.5
Состояние окисления З^-Электронная конфигурация Координа- ционное число Геометрия Примеры
Fe(—II) ? 4 Тетраэдр [Fe(CO)4]2~, Fe(CO)2(NO)2
Fe(0) <Р(?) 5 Тригональная бипирамида Fe(CO)B, Fe(PF3)B
6 Октаэдр [Fe(CO)eH]+
Fe(/Z) de 4 Тетраэдр [FeFJ2-
5 Тригональная бипирамида Fe[(CH3)Bdien]X2
6 Октаэдр [Fe(H2O)e]2+, [Fe(CN)e]4~
& 4 Тетраэдр [FeClJ-, Fe3O4
6 Октаэдр Fe2O3, Fe(acac)3, [Fe(C2O4)3j3-
7 Пентагональ- ная бипира- мида [РеЭДТА(Н2О)1 -
Fe(IV) d* 6 Октаэдр [Fe(diars)2Cl2]2+
3.6. Кобальт (табл. 1.6)
—1‘,82В’
Со2+ ----*• Со3+ (кислый раствор)
—1.14В
Со(ОН)2 --->- Со(ОН)3 (щелочной раствор)
Наиболее обычные состояния окисления — Со(П) и Со(Ш),
хотя недавно было предположено, что синтетические «кобалок-
симные» комплексы Со(1) имеют некоторое биохимическое зна-
чение в связи с витамином Bj2 [25]. [Со(Н2О)б]3+ устойчив толь-
ко в сильных кислотах, и химия Со(III) ограничивается его ком-
плексными соединениями, которые все, за небольшим исключени-
ем, имеют октаэдрическое строение (d6, низкоспиновые).
Стереохимия комплексов Со(П) исключительно разнообразна;
наиболее распространены тетраэдрические, квадратно-плоскост-
ные и октаэдрические структуры, но встречаются также комплек-
сы, имеющие геометрию тригональной бипирамиды или квадрат-
ной пирамиды.
Ион Со2+ — единственный имеющий значение ион с конфигу-
рацией d7 во всей серии переходных элементов. Для него наибо-
лее обычна геометрия тетраэдра, иногда — в равновесии с окта-
эдрической формой. Эта способность кобальта представляет инте-
рес и интенсивно изучалась в последние годы, благодаря чему
были исследованы комплексы Со(II) с большим числом разнооб-
40
Глава 1
Таблица 1.6
Состояние окисления М-Электронная конфигурация Координа- ционное число Геометрия Примеры
Со(—I) ? 4 Тетраэдр [Со(СО)4]~
Со(0) ? 4 » [Co(CN>4]4-
Со(1) da 4 > [Co(CN)3CO]2-
5 Тетрагональная пирамида (RsCS2)2CoNO
Тригональная бипирамида [Co(NCR)s]*
6 Октаэдр [Co(dipy)3]+
Со(П) <р 4 Тетраэдр [CoClJ2-
Квадрат {Co[(CH3)2-edt]}(C104>2
5 Тригональная бипирамида [Co(CHs-salen)2]2
6 Октаэдр [Co(H2O)e]s+, [Co(CN)e]‘-
Со(П1) d* » [Co(en)3F+ [Co(CN)ep-, [CoFe]3-
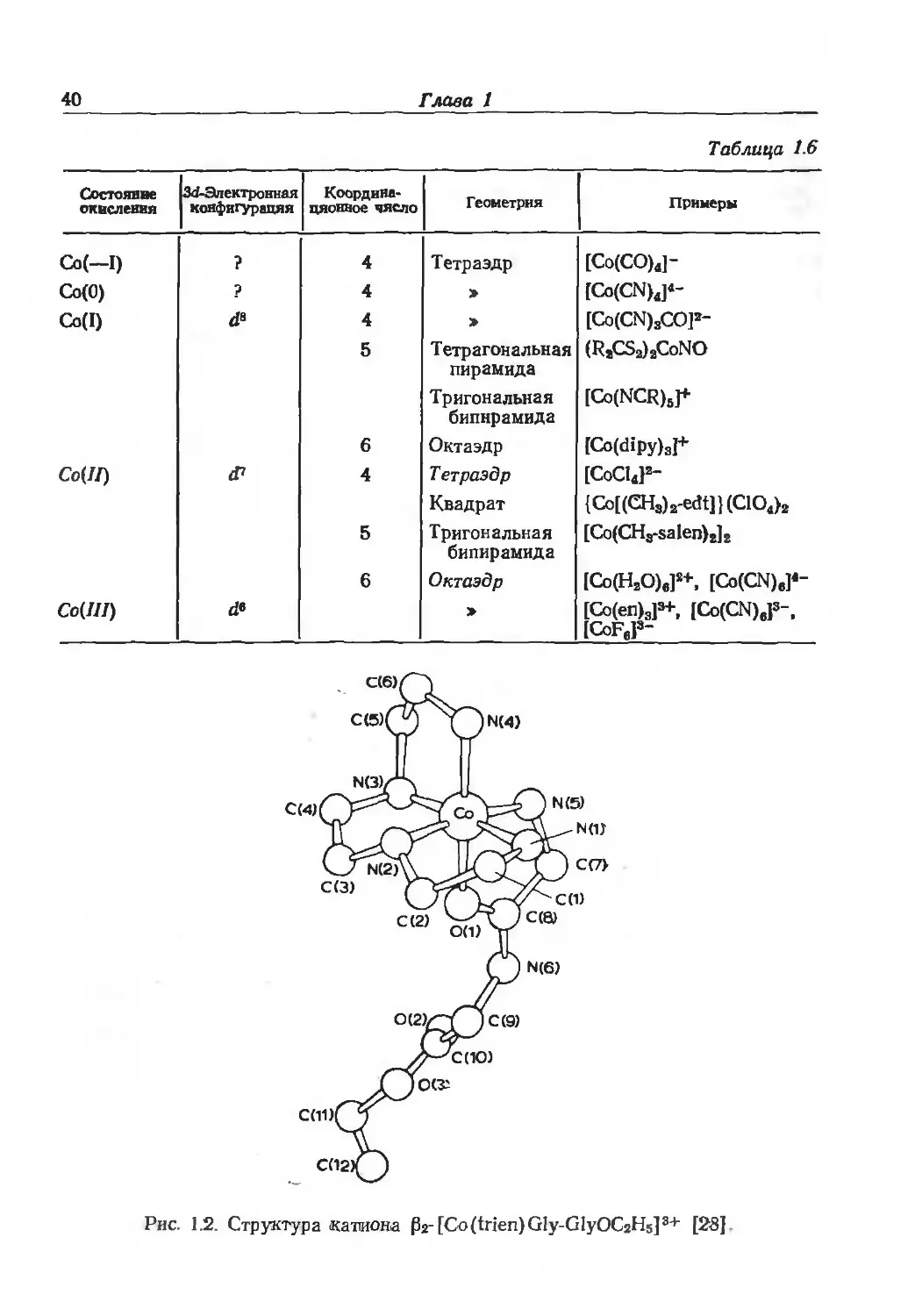
Рис. 1.2. Структура катиона P2-[Co(trien)Gly-GlyOC2H5]3+ [28]
Структура и стереохимия координационных соединений
41
разных лигандов в различных растворителях. Ясно, что геометрия
образующегося комплекса сильно зависит от стерических требо-
ваний, налагаемых на донорные атомы лигандными группами.
Тетраэдрическое строение имеют преимущественно комплексы с
монодентатными анионными лигандами (например, С1~, Br-, I-,
SCN-, N3, ОН-), как в [СоХ4]2~ или [CoL2X2] (L=PR3), а также
иногда с объемистыми бидентатными анионами (N-алкилсалицил-
аланинат, крупные р-дикетонат-анионы). Со(II) образует плоские
комплексы с диметилглиоксимом, аминооксалатом, о-аминофено-
лят-анионами и этилендитиолатом.
Инертность комплексов Со(1П) по отношению к реакциям за-
мещения лигандов использовалась во многих исследованиях,
в том числе и при исследовании механизма гидролиза эфиров ами-
нокислот и пептидов [27]. В последнем случае было показано, что
активная частица должна включать координацию N-концевого
остатка аминокислоты через атом азота аминогруппы и через
атом кислорода карбонильной группы (рис. 1.2).
3.7. Никель (табл. 1.7)
Из комплексов никеля наиболее важны, несомненно, комплек-
сы Ni(II), a Ni2+ — единственный простой ион, который находят в
водном растворе. Встречаются октаэдрическая, тетраэдрическая и
квадратно-плоскостная конфигурации, при этом комплексы с элек-
тронейтральными лигандами (Н2О, NH3, еп) имеют преимущест-
венно октаэдрическое строение, а комплексы с монодентатными
анионами и смешанно-лигандные комплексы с анионами и ней-
тральными монодентатными лигандами (галогениды и лиганды,
координирующиеся через атомы Р и As) образуют тетраэдриче-
ские структуры. Последние иногда существуют в равновесии
с частицами, имеющими квадратно-плоскостную структуру; в по-
следние годы эти равновесия интенсивно исследовались (L=ал-
кил, арилфосфины, алкилзамещенные салицилальдимины и ами-
нотроп олонимины) .
NiL2 < NiL2
(тетраэдр) (плоский
квадрат)
По-видимому, более открытую тетраэдрическую структуру пре-
имущественно имеют комплексы с более объемистыми лиганда-
ми — алкилзамещенным салицилальдимином и аминотропоними-
ном, в то время как менее объемистые заместители благоприятст-
вуют образованию комплексов с геометрией плоского квадрата.
Комплексы со смешанными лигандами этого типа могут иметь в
растворе как тетраэдрическую, так и квадратно-плоскостную кон-
фигурацию; иногда эти два типа геометрии встречаются вместе
в кристалле.
42
Глава 1
Таблица 1.7
Состояние окисления 3d-Электронная конфигурация Координа- ционное число Геометрия Примеры
Ni(0) 1ЧЙЗС dI0(?) 4 Тетраэдр Ni(CO)4, [Ni(CN)4]4-
Ni(II) d8 4 Квадрат {NiBr2[P(C2H5)3]2} t [Ni(CN)4]2-
4 Тетраэдр
5 Квадратная пирамида [Ni(CN)6]s-
Тригональная бипирамида Ni[5-Cl-salen ЬЦСгН*)^, {Ni[P(C3H7(CH3)2As)3]CN}+
То же Ni(CN)2[P(CeH6)(CH3)2]3
6 Октаэдр [Ni(H2O)e]2+, [Ni(SCN)e]4- [Ni(dipy)3]2+
Ni(III) d1 5 Тригональная бипирамида N iBr3[P(C6H5) (Ch3)2]2
6 Октаэдр [Ni(diars)2Cl2]+
Ni(IV) __ d* 6 [Ni(diars)iCl2]2+
Октаэдрическая форма комплекса также может находиться в
равновесии с квадратно-плоскостной, особенно при добавлении
лигандов, которые являются хорошими донорами электронов (А=
=Н2О, пиридины).
NiL2 + 2А < > NiL2A2
желтый голубой
(плоский (октаэдр)
квадрат)
где L=лиганды типа салицилальдимина, алкил- и арилзамещен-
ные этилендиамины, диалкилтиомочевины. Переход к октаэдриче-
ской симметрии может быть также осуществлен путем полимери-
зации, как в тримере [Ni(acac)2]3, или путем димеризации с об-
разованием частиц с 5-кратной координацией иона металла
(салицилальдиминатные комплексы при R=H, ОН). Таким об-
разом, стереохимия комплексов Ni(II) исключительно изменчива
и, возможно, это наиболее трудно предсказываемая область струк-
турных исследований химии переходных элементов.
3.8. Медь (табл. 1.8)
—0,153В
Си+-------> Си2+ (кислый^ раствор)
Наиболее важные геометрические формы — тетраэдр для Cu(I)
и сильно искаженный октаэдр для Си(II). Относительная устой-
Структура и стереохимия координационных соединений
43
Таблица 1.8
Состояние окисления 3d- Электронка я конфигурация Координа- ционное число Геометрия Примеры
Си(Г) dle 2 Линейная [Cu(NH3)2]+, Cu2O
4 Тетраэдр [Cu(CN)J3-, Cui
Си(1Г) 4 Тетраэдр (искажен- ный) Cs[CuCl4], Cu(N-C3H7salen)2
5 Тригональная бипирамида [Cu(dipy)2l]+
Квадратная пирамида [Cu(DMGH)2]2
4 Плоский квад- рат CuO, [Cu(py)4]2+
6 Октаэдр (иска- женный) [СиЭДТАр-, [Cu(H2O)„]2+
чивость комплексов Cu(I) и Cu(II) сильно зависит от природы
лигандов, и равновесие 2 Си(1)ч±Си(0)+Си(П) может быть сме-
щено в любом направлении в зависимости от условий. В водных
растворах Си+ неустойчива, но ее можно стабилизировать с по-
мощью мягких поляризующихся лигандов (CN_, I-, R2S) или пу-
тем образования очень малорастворимых комплексов — CuCN,
CuCl*.
В водных растворах изучена и описана главным образом хи-
мия соединений Си (II). Существует большое разнообразие кон-
фигураций — от плоского квадрата до октаэдра; их все можно
рассматривать как октаэдры с различной степенью удлинения
двух связей, находящихся в транс-положении (искажение вслед-
ствие эффекта Яна—Теллера); комплексы с четко выраженной
октаэдрической симметрией Oh, как и следовало ожидать, не об-
наружены.
Комплексы Си(II) в кристаллическом состоянии иногда, а пеп-
тидные комплексы обычно имеют геометрию квадратной пирами-
ды (координационное число 5) (гл. 4). По-видимому, в кристал-
лическом состоянии Си(II) может образовывать большое разно-
образие структур, соответствующих координационным числам 4,
5 и 6, точная геометрия которых определяется конформациями
лигандов и требованиями решетки; в растворе, по-видимому, пре-
обладает конфигурация искаженного октаэдра.
* CuCN и CuCl можно рассматривать и как простые соли меди(1).—
Прим. ред.
44
Глава 1
3.9. Цинк (табл. 1.9)
Известны только гидратированный ион Zn2+ и комплексы Zn (II).
Так как Zn2+ имеет полностью заполненную Зй-оболочку (d10),
то эффекты кристаллического поля отсутствуют. Таким образом,
стереохимия комплекса определяется только электростатическими
или ковалентными силами связывания и размерами лиганда. Бо-
лее всего распространена тетраэдрическая структура, за нею сле-
дует октаэдрическая и известно несколько примеров комплексов,
имеющих геометрию тригональной бипирамиды и квадратной пи-
рамиды.
Таблица 1.9
Координационное число Геометрия Примеры
4 Тетраэдр [Zn(CN)4p-, [Zn(NH3)4]2+
5 Искаженная тригональ- ная бипирамида или квадратная пирамида [Zn(acac)2H2O] [Zn(terpy)Cl2]
6 Октаэдр [Zn(H2O)ep+, [Zn(NH3)ep+
3.10. Молибден и вольфрам (табл. 1.10)
~о,ов
Мо3+ -----► МоО2,
0,15В
W3+ ------>- WO2 (кислый раствор)
Химические свойства Мо и W очень сходны, но значительно
отличаются от свойств Сг; для них важны состояние окисления
IV, V и VI, в то время как состояние окисления II практически
неизвестно. Для обоих элементов характерно большое разнообра-
зие геометрических конфигураций комплексов, причем имеется
много примеров комплексов с координационными числами больше
шести; например, [Мо(СЫ)8]4-, [Mo(CN)8]3- (искаженный тре-
угольный додекаэдр); полимерные окислы, сульфиды. Дискретный
[МоО4]2- существует в сильнощелочных растворах, а при подкис-
лении образуются разнообразные изополимолибдаты или изополи-
вольфраматы. Для этих соединений характерны октаэдрическая
координация металла с атомами кислорода и различные типы об-
щих углов или ребер. Относительно редки комплексы в состоянии
окисления III, а для Мо и W в состояниях окисления IV, V и
VI известны главным образом анионные комплексы.
Структура и стереохимия координационных соединений
45
Таблица 1.10
Состояние окисления 3 (/-Электронная конфигурация Координа- ционное число Геометрия Примеры
Мо(0) W(0) d3 6 Октаэдр Мо(СО)8, Mo(diphos)3
Мо(1) W(I) ds — «Сэндвичевая» связь [(СвНв)2Мо]+ [л-С5Н6Мо(СО)3]2
Мо(П) W(II) d4 4 Тетраэдр (?) Мо(ОСОСН3)2
Мо(Ш) d3 6 Октаэдр [Mo(SCN)6]3~, [WC1„]3-
W(III) 8 Додекаэдр [Mo(CN)7(H20)]‘-
Mo(IV) d3 6 Октаэдр [Mo(SCN)eJ3-
W(IV) 8 Додекаэдр [Mo(CN)8]4-
Mo(V) №(V) d1 5 Тригональная бипирамида MoClB
6 Октаэдр [WFe]-, [MoOFBp-
8 Додекаэдр [Mo(CN)8]3-, [W(CN)8J3-
Mo(Vl) d° 4 Тетраэдр [MoO4]2~, [WOJ2-
W(VI) 6 Октаэдр WC18, MoFe, MoO8, WOe (в поликислотах)
4. ФАКТОРЫ, ВЛИЯЮЩИЕ НА КООРДИНАЦИОННОЕ ЧИСЛО
И СТЕРЕОХИМИЮ
4.1. Эффекты ионов металлов
Ионы металлов на основании их способности к образованию
химических связей можно разделить на два довольно отчетливых
класса: 1) ионы щелочных (Li+, Na+, К+, Rb+, Cs+) и щелочно-
земельных* (Be24-, Mg2+, Са2+, Sr2+, Ва2+) металлов, 2) ионы пе-
реходных металлов, особенно металлов первого переходного ряда
(Ti2+, V2+, VO24", Сг2+, Cr3+, Mn2+, Fe24”, Fe3+, Co2+, Ni2+, Cu2+,
Zn24-).
4.1.1. Ионы щелочных и щелочноземельных металлов
Эти металлы характеризуются высокой электроположитель-
ностью и, за исключением Ве2+, который в виде дискретных ионов
неизвестен ни в кристаллическом состоянии, ни в растворе**, су-
* Бериллий вряд ли даже условно можно отнести к щелочноземельным ме-
таллам.— Прим. ред.
** Mg2+, а также в некоторой степени Li+ вследствие их малых размеров
проявляют сильные поляризующие свойства, и связи в их соединениях имеют
частично ковалентный характер.
46
Глава 1
ществуют в их кристаллических соединениях как отдельные ионы
(ионные решетки), а в растворе — как дискретные гидратирован-
ные ионы М+ или М2+. Составные части комплекса удерживаются
вместе почти исключительно электростатическим взаимодействием
между противоположно заряженными ионами или ион-диполями,
а так как ионы имеют электронную конфигурацию инертного га-
I
е
о
СР
Li Na+ К+ Rb+ Cs
5
Ве2+ Mgz+ Са2+ Sr2+ Baz+
Усиление ионного характера
за, то они сферически симметричны с точки зрения возможности
образования ими химических связей. Максимальные координаци-
онные числа (N) часто можно вычислить, предположив плотную
упаковку лигандов на поверхности иона металла и используя сле-
дующее выражение:
„____2л ( d\*( 1
(3)1/2 \ г ) —rz/8d?r
где d — расстояние металл—лиганд, г — вандерваальсов радиус
лиганда. Предполагается, что присоединенные лиганды имеют
правильную стереохимию, которая определяется только энергией
межлигандного отталкивания, т. е. что не существует стерических
требований самих ионов металлов, и в этом заключается их глав-
ное отличие от переходных металлов. Это означает, что меньшие
по размеру катионы (Li+, Na+, К+ и Ве2+), по-видимому, сущест-
вуют в водном растворе как тетраэдрические ионы М(Н2О)4,
в случае К+ это было подтверждено методом рассеяния рентгенов-
ских лучей. Однако, так как эти ионы очень малы, они имеют на
своей поверхности высокую плотность заряда, и приобретает зна-
чение вторичная сольватация. Так, щелочные металлы Li+, Na+,
К+ имеют большие эффективные радиусы гидратации (табл. 1.11).
При этом для Li aq он больше, чем для Csaq.
Найденное для Cstq число гидратации около 10, вероятно, це-
ликом относится к первой гидратной оболочке, вторичная гидра-
тация не имеет значения. Небольшие высокозаряженные ионы
щелочноземельных металлов приобретают в водном растворе вы-
Структура и стереохимия координационных соединений
47
Таблица 1.11
Ион КристэллохимнческиЙ Радиус гидратации Ион Кристаллохимический
радиус3, А (оцененный), А радиус, А
Li+ 0,60 3,40 Ве2+ 0,34
Na+ 0,95 2,76 Mg2+ 0,65
К+ 1,33 2,32 Са2+ 0,94
Rb+ 1,48 2,28 Sr2+ 1,1
Cs+ 1,69 2,28 Ва2+ 1,3
а3десь даны крнсталлохнмические радиусы по Л. Полингу; существует также часто при-
меняемая система кристаллохимнческих радиусов по Б. Голыпмидту. — Прим. ред.
сокое координационное число за счет прямого контакта катион —
анион. Так, Mg2+, хотя и имеет меньший ионный радиус, чем Na+,
существует в водном растворе в виде [Mg(H2O)e]2+. Однако сла-
боэлектроположительный ион Ве2+, существующий в виде тетра-
эдрического [Ве(Н2О)4]2+, проявляет большую кислотность по
сравнению с другими акватированными двухвалентными и однова-
лентными ионами, и молекулы воды в нем прочно связаны; так,
например, [Ве(Н2О)4]С12 не теряет воду над Р2О5.
Для кристаллического состояния имеют большое значение упа-
ковочные эффекты. Состояние с наиболее низкой энергией соот-
ветствует балансу между максимальным координационным чис-
лом, вычисленным по приведенному выше уравнению, и упаковоч-
ными силами, возникающими в результате упаковки в кристалл
повторяющихся структурных единиц. Для щелочных металлов в
ионных кристаллах это приводит к кубической 8-кратной коорди-
нации, если отношение радиусов аниона и катиона (г~/г+) мень-
ше 1,37 (структура CsCl), октаэдрической 6-кратной координации,
если т-]т+ меньше 2,44 (структура NaCl), и тетраэдрической
4-кратной координации, если г~/г+ меньше 4,4 (структура ZnS).
Твердые соединения ионного типа с иной стехиометрией, напри-
мер М2Х, имеют иные структуры решеток.
Химия водных растворов этих металлов почти исключительно
относится к ионам Мщ или М aq - Известно существование в вод-
ных растворах нескольких простых комплексов, содержащих дру-
гие лиганды (интересное исключение см. в гл. 6), а металлоор-
ганические соединения Na и К являются, по существу, ионными
и сильно гидролизуются водой. Только органические производные
Li+ и Mg2+ (чрезвычайно полезные как источник R+ в препаратив-
ной органической химии) имеют ковалентный характер и раство-
ряются в углеводородах и других неполярных растворителях. Бы-
ло показано, что реактив Гриньяра RMgX содержит тетраэдриче-
48
Глава 1
ски координированный Mg в кристаллах C6H5MgBr-2(C2H5)2O;
подобное строение имеет Na(acac)2 (XXXIV). Таким образом, 'во-
дородные связи с координированными молекулами воды и размер
СН3ч ZCH3
с=о р-с
/ \ / Л
Н2С Na 'СН
\ У X _,7
Jc=o О-Сх
СН3Х СНз
XXXIV
гидратированного ‘катиона — важные черты в ‘биологической функ-
ции Naaq И К ад.
Из щелочноземельных металлов только Ве2+ и в некоторой сте-
пени Mg2+ функционируют как достаточно сильные акцепторы
электронов, образуя довольно слабые ковалентные комплексы.
Благодаря малым размерам иона Ве2+ и отсутствию стереохими-
ческих требований, обусловленных электронным строением, пред-
почтительное координационное число равно 4 с тетраэдрическим
расположением лигандов, например BeFf- и Ве(асас)2 (XXXV).
СНз /СНз
с-о о-с
//' \ / “''X
НС Be ,'СН
X'-.- / \ У/
с-о о-с
СНз
СН3
XXXV
Однако если важна стереохимия лиганда, как, например, в пор-
фириновых ядрах, то образуются другие структуры, например
структура плоского квадрата в Mg(II)-хлорофилле (гл. 29,
рис. 29.1). Известен также октаэдрический комплекс
[Mg(NH3)6]Cl2, но он легко гидролизуется в воде. Из ионов дру-
гих щелочноземельных металлов только Са24- проявляет тенден-
цию к образованию комплексных соединений, координируя пред-
почтительно карбоксилатные лиганды. Так, октаэдрический ион
[СаЭДТА]2- имеет практическое значение (как «умягчитель» во-
ды), а связывание Са2+ и Mg2+ фосфатными группами АТФ и
АДФ играет важную роль в процессе переноса энергии в биоло-
гических системах*.
* В последнее время большое значение в химии, биологии и даже в промыш-
ленности приобретает распространение комплексов этих металлов с макроцикличе-
скими лигандами (см. также гл. 6). — Прим. ред.
Структура и стереохимия координационных соединений
49
4.1.2. Ионы переходных металлов
Координационные числа переходных металлов определяются
в основном их размерами и эффективными зарядами ядер, воз-
действующими на лиганды, а также свойствами самого лиганда.
Совокупность этих факторов обусловливает 'большие энергии
(50—500 ккал/моль) связей М—L и их постепенное повышение
Тетраэдр Свободный Октаэдр Тетрагональ- Плоский
ион ноя или квадрат
металла квадратная
пирамида
Рис. 1.3. Расщепление d-уровнен иона металла в кристаллических полях высокой
симметрии.
при переходе от более легких металлов (Ti2+) к более тяжелым
металлам (Zn2+). Неполное экранирование заряда ядра d-элек-
трона'ми приводит к более высокому эффективному заряду в слу-
чае нормальных расстояний связей и к меньшим ионным радиу-
сам; оба эти фактора приводят к понижению координационных
чисел в ряду Ti—Zn и к повышению ковалентного характера и
прочности координационной связи.
Однако стереохимия, а во многих случаях и координационные
числа часто отличаются от предсказанных только на основании
отношений размеров, зарядов и поляризационных свойств катиона
и аниона просто из-за того, что расположение электронов вокруг
иона металла больше не является сферически симметричным. Ион
металла не имеет конфигурации инертного газа, так как происхо-
дит заполнение d-оболочек (3d для Sc—Zn, 4d для Y—Cd, 5d для
Hf—Hg) и 4f- и 5^-оболочек для лантанидов и актинидов соответ-
ственно. Большое значение для стереохимии комплексов переход-
4—2451
50
Глава 1
Таблица 1.12
Энергии стабилизации кристаллическим полем для комплексов
d-переходных элементов (в единицах Dq*)
Октаэдр Тетраэдр Плоский квадрат
0) <u OJ
2 <и Примеры S 2 ? я Q) 3-g J я 3 = = « з Я Л _ x 2 CD X
X « A- = 8. OJ H 5 о ? & Oj О О СХ CL x о ? °-
о s* С X t x ax
Яз SB c 5 о л = S eg OJ E a rt el
d° Ca=+, Sc3* 0 0 0 0 0 0
d> Ti3+, LJ4+ 4 4 2,67 2,67 5,14 5,14
d2 •J'i2+j y3+ 8 8 5,34 5,34 10,28 10,28
d3 V2*, Cr3+ 12 12 3,56 8,01 14,56 14,56
d* Cr2+, Mn3+ 6 16 1,78 10,68 12,28 19,70
d5 Mn2+, Fe3+, Ru2+, Os2* 0 20 0 8,90 0 24,84
d« Fe2+, Co3*, Rh3+, It3* 4 24 2,67 7,12 5,14 29,12
d7 Co2*, Ni3*, Rh2+ 8 18 5,34 5,34 10,24 26,84
d8 Ni2+, Pd2+, Pt2+, Au3* 12 12 3,56 3,56 14,56 24,56
d9 Cu=+, Ag2+ 6 6 1,78 1,78 12,28 12,28
d10 Cu+, Zn2+, Hg2+, Ag* 0 0 0 0 0 0
а Dq (для тетраэдра) = "g- Dq (для октаэдра).
ных элементов имеет также тот факт, что d-электроны простира-
ются в область, находящуюся снаружи замкнутых оболочек бла-
городных газов, и поэтому от числа и расположения имеющихся
d-электронов очень сильно зависит их влияние на лиганды, а сле-
довательно, и на стереохимию основного состояния комплексов.
Мы не будем здесь обсуждать теоретические аспекты химиче-
ской связи в химии переходных элементов [18—22], но от нее
в значительной мере зависит стереохимия комплексов. Коротко
говоря, электростатическое поле лигандов расщепляет пять вы-
рожденных d-орбиталей газообразного иона металла на различ-
ные наборы уровней энергии (рис. 1.3). При заполнении этих ор-
биталей кристаллического поля вначале заполняются наиболее
низколежащие орбитали с соблюдением принципа Паули; при
этом в общем обычно получается выигрыш энергии. Он имеет
название энергии стабилизации кристаллическим полем (ЭСКП).
Некоторые характерные величины ЭСКП для различных конфи-
Структура и стереохимия координационных соединении
51
гураций приведены в табл. 1.12*. Учитывая энергии соответствую-
щих орбиталей лигандов и допуская, что орбитали металла и ли-
гандов могут взаимодействовать при условии близости их энергий
и соответствия по симметрии, можно сделать поправку на пере-
распределение электронов между металлом и лигандами, т. е. на
ковалентное связывание. Величина ЭСКП, имеющая порядок
10—50 ккал/моль, невелика по сравнению с общей энергией связи,
имеющей порядок 300—3000 ккал/моль, но стереохимия комплек-
сов во многих случаях определяется такими эффектами.
Наиболее важные стереохимические следствия теории крис-
таллического поля приведены в табл. 1.13.
Таблица 1.13
Стереохимические предсказания теории кристаллического поля [30]
Число иесвязыва- клцих d-электроиов Число неспаренных электронов Координационное число 4 Координационное число 6
Э; 0, 5, 10 4 или 9 3 или 8 2 или 7 1 или 6 кктроны не 0 или 5 1 2 3 4 '.парены (слабое поле — вь Тетраэдр Плоский квадрат Искаженный тетраэдр Тетраэдр Почти правильный тетра- эдр СОКИН спин) Октаэдр Тетрагонально искажен- ный октаэдр Октаэдр Почти правильный окта- эдр То же
Электроны спарены (сильное поле — низкий спин)
1 илн 2 3 1 Почти правильный тетра- —
4 0 эдр Тетраэдр Почти правильный окта-
5 1 Искаженный тетраэдр эдр То же
6 0 То же Октаэдр
7 1 Плоский квадрат Тетрагонально искажен-
8 0 То же ный октаэдр То же
Стереохимические следствия возмущений кристаллическим по-
лем полезно рассматривать совместно с другими свойствами
ионов переходных металлов.
* Случай слабого поля соответствует строгому соблюдению принципа
Паули**, случай сильного поля имеет место, когда разность энергий между на-
борами орбиталей (А) больше, чем энергия спаривания спинов. Вследствие этого
вначале полностью заполняется набор нижележащих орбиталей.
** Принцип Паули — один из основных законов природы, и поэтому, естествен-
но, он всегда соблюдается. — Прим. ред.
4*
52
Глава 1
а) Устойчивость состояний (степеней) окисления*. Для эле-
ментов первой половины первого переходного ряда, от Sc до Мп,
существуют все максимальные степени окисления: Sc(III),
Ti(IV)**, V(V), Cr(VI) и Мп(VII) (курсивом отмечены наиболее
устойчивые и распространенные). Однако из-за неполного экра-
нирования ядер d-электронами низшие состояния окисления по,-
степенно становятся более распространенными. Так, Cr(VI) и
Мп (VII) существуют только в виде окислов и оксо-анионов,
Сг(П1)—единственное состояние окисления, имеющее значение
в .водном растворе; Мп(II) и Мп(III) примерно одинаково важны,
Fe(II) несколько более распространен, чем Fe(III), а Со(II) —
более, чем Со(III) (который распространен только в комплексах,
где он находится в ниэкослиновом состоянии); единственное важ-
ное состояние окисления никеля—Ni(II), для меди распростра-
нены как Си(II), так и Cu(I), но двухвалентное состояние имеет
большее значение; цинк встречается только в виде Zn(II), имею-
щего конфигурацию d10.
б) Особенности комплекса, зависящие от природы иона метал-
ла. В общем комплексы металлов, находящиеся в начале ряда,
имеют ионный характер (Sc2+, Ti2+, V2+), в то время как комплек-
сы металлов, стоящих в конце ряда (Cu2+, Ni2+), более ковалент-
ны, особенно комплексы с поляризующимися лигандами (О, S) ***.
Такое изменение характера связи происходит вследствие повыше-
ния эффективного заряда ядра при переходе вдоль ряда. Однако
более ковалентная связь не обязательно означает повышенную ус-
тойчивость. Во втором и третьем переходных рядах элементов сте-
пень ковалентности также заметно повышается по аналогичной
причине, но здесь обычно она вызывает также повышение кине-
тической устойчивости.
Стереохимические выводы теории кристаллического поля мож-
но суммировать следующим образом.
1. Легкие переходные металлы предпочитают более высокие
координационные числа, а более тяжелые—'более низкие коор-
динационные числа. Так, комплексы ионов d°—d6 имеют преиму-
щественно октаэдрическую координацию, в то время как ионы
d7 [Co(II), Rh(II)], d8[Ni(II), Pd(II), Pt(II)] и d9[Cu(II),
Au (III)] предпочитают квадратно-плоскостные структуры. Это
особенно очевидно для второго и третьего рядов переходных эле-
ментов, где спаривание электронов редко имеет место (низкоспи-
новые комплексы).
* В русской терминологии термины «степень окисления» и «состояние
окисления» равнозначны. — Прим. ред.
** В оригинале Мп(IV), по-видимому, опечатка. — Прим. ред.
*** Большинство лигандов с донорными атомами кислорода поляризуется
слабо. — Прим. ред.
Структура и стереохимия координационных соединений
53
2. Высокие координационные числа обычно ассоциируются с
катионными комплексами ([Co(NH3)6]2+), а низкие — с анионны-
ми комплексами ([СоСЦ]2-). Так, комплексные фториды или
окислы в виде изолированных октаэдрических анионов в твердом
состоянии не образуются.
3. Более высокие координационные числа иногда ассоциируют-
ся с более высокими состояниями окисления, например [СиС12]“,
[CuCl4]2- [CuF6]3-, [FeCl4]2-, [FeCl6]3-
4. Координационное число иногда уменьшается с увеличением'
поляризующей силы атома металла, например [Ag(CN)2]~ (по-
тенциал ионизации 9,22 эВ) и [Cu(CN)4]3- (потенциал иониза-
ции 7,72 эВ).
5. На основе энергии стабилизации кристаллическим полем
(табл. 1.12) стереохимические требования иона металла при дан-
ном координационном числе могут быть предсказаны с хорошей
вероятностью. Так, действительно, квадратно-плоскостные ком-
плексы наиболее часто находят для ионов с конфигурациями
d& (Ni, Pd, Pt) и d9 (Cu2+) и в меньшей степени —с d7 [ионы
Au(III)*].
6. Энергия стабилизации кристаллическим полем для структу-
ры квадратной пирамиды (координационное число 5) имеет про-
межуточное значение между величинами ЭСКП для правильного
октаэдра и квадратно-плоскостной структуры. Так, для систем
d7[Co(II), Ni(III)] эта структура предпочтительна, например
[Ck^CNJs]3-, [Co(triars)I2], {Ni[ (C2H5)3P]2Br3}.
7. Системы, не имеющие ЭСКП (d°, d5, d10), легко образуют
тетраэдрические комплексы — Fe(III), Zn(II), Al (III), Cd (II),
Mn(VII).
8. Ион Co(II) (d7) в меньшей степени предпочитает октаэдри-
ческую координацию тетраэдрической, чем ионы с любой другой
^-конфигурацией.
9. Теория предсказывает, что искажения правильной октаэд-
рической геометрии можно ожидать в следующих случаях: высо-
коспиновые комплексы Сг(П) и Мп (III) (d4), низкоспиновые ком-
плексы Ni(III) и Со(II) (d7), комплексы Си (II) (d9), для кото-
рых искажение происходит за счет удлинения октаэдра вдоль од-
ной оси — тетрагональное искажение, например СгС12 (4С1~ на
2,39 А, 2С1- на 2,90 А).
10. Для хелатных структур с координационным числом 4 пред-
сказания теории кристаллического поля, кажется, сохраняют силу.
Так, для комплексов типа XXXVI имеет место следующая после-
довательность структур [31]: Cr(II) (d4)—плоскостная; Cu(II)
(d9) — плоскостная во всех комплексах за исключением систем со
стерическими затруднениями; Ni(II) (d3)—в большинстве случа-
Здесь ошибка: конфигурация Au(III) — d8. — Прим. ред.
54 Глава 1
•ев плоскостная, но для некоторых систем (О, NCH3; S, NCH3) име-
-ет место равновесие между плоскостной и тетраэдрической структу-
R'\
С-x Y-C
\ / 'Л
нс. м ;сн
\<_ / \ _-7
C-Y Х-С
r/ xr,
X, Y = O,S,NR
R = алкил, арил
XXXVI
,рами; Co(II)(d7)— в большинстве случаев тетраэдрическая, но
встречаются системы с равновесием между плоскостной и тетра-
эдрической структурами, а также чисто плоскостные (N, S);
F'e(II) (d6)—тетраэдрическая; Zn(II) (d10)—тетраэдрическая.
4.2. Свойства лиганда
Предполагается, что в определении стереохимии значительная
роль принадлежит лиганду, являющемуся партнером в образова-
нии координационной связи. Координационное число иона метал-
.ла .по отношению к монодентатным лигандам определяется глав-
ным образом размером лиганда и числом потенциальных донор-
ных атомов. Детальная стереохимия для данного координационно-
го числа в большинстве случаев зависит от требований иона
металла (эффекты кристаллического поля) и в изменяющейся сте-
пени от стереохимии лиганда, хелатообразующих свойств и при-
роды донорных атомов, принимающих участие в образовании
связи.
4.2.1. Донорные атомы
Для предсказания характеристик комплексов полезны понятия
о «жестких» и «мягких» кислотах и основаниях. Здесь термин
«кислота» относится к иону металла в его формальном состоянии
окисления (кислота Льюиса), а «основание» — к донорным ато-
мам лиганда. К «жестким» относятся трудно поляризующиеся
кислоты и основания, к «мягким» — более легко поддающиеся ис-
кажению. Следствие жесткости и мягкости ионов металлов и ли-
гандов рассматривается в гл. 2, табл. 2.2 и 2.3. Как там показано,
для жестких металлов, относящихся к классу (а), тенденция к
преимущественному взаимодействию с лигандами соответствует
ряду
F>Cl>Br
О )' S (> Se)
N > Р (> As)
Структура и стереохимия координационных соединений
55
в то время как для мягких металлов, принадлежащих
к классу б, порядок устойчивости образуемых ими комплексов
изменяется в следующей последовательности: S~C>I>Br>
>C1>N>O>F. Как правило, координационное число умень-
шается с увеличением поляризуемости лиганда, например
[FeF6]3-, [FeBr4]~; для зависимости координационного числа от
поляризуемости металла наблюдается такая же тенденция, на-
пример [МпС1б]4-, [ZnCl4]2-.
Было показано, что от изменения размера донорного атома
редко непосредственно зависит (если вообще зависит) изменение
координационного числа. Так, более вероятно, что высокие коор-
динационные числа, наблюдающиеся в комплексах с ионами F-
и Н- в IF5, [TaF8]3~, [ReH9]2- и т. п., в большей мере обусловле-
ны слабой поляризуемостью лиганда и .высоким состоянием окис-
ления акцептора [I(V), Ta(V), Re(VII)], чем малыми размерами
донорного атома. Однако ясно и то, что объемистые донорные ато-
мы или группы атомов будут препятствовать осуществлению не-
нормально высоких координационных чисел.
Имеют важное значение стереохимические требования донор-
ного атома. Это ясно 'видно на примере нескольких хелатов, обра-
зованных атомами N, S и О, в которых отчетливо проявляется
предпочтение атомов серы к координации по вершинам пирамиды,
в то время как требования N и О не столь отчетливы (угол связи
в H2S равен 92°, в Н2О — 105°, NH3 — 107°). Если У=Ы, то можно
наблюдать все три геометрические структуры (XXXVII—XXXIX),
но только две, если Y=S (XXXVII и XXXVIII).
XXXVII
Подобным образом с шестидентатным лигандом
возможна как изогнутая (XL), так и линейная (XLI) хелатная
связь, если Y='O, но только изогнутая (XL), если Y=S.
56
Глава /
4.2.2. Размер лиганда
Размер лиганда может быть очень важен для определения ко-
ординационного числа, а в некоторых случаях и стереохимии. При
прочих равных условиях объемистые лиганды предпочитают низ-
кие .координационные числа, например:
[Со(асас)2]4
(октаэдрическая
координация
ацетил ацетона)
{Co[N(CH2CH2NH2)3]Br2) и
(октаэдр)
и Co(dpm)2
(тетраэдрическая
координация
дипивалоилметана)
[Co(N[CH2CH2N(CH3)2]3)Br]Br
(тригональная бипирамида)
Стереохимия комплексов с шиффовыми основаниями изменяется
XLII
в зависимости от природы заместителя R: если R = H, то комплекс
имеет геометрию плоского квадрата, а при R = бутил — тетраэдра,
предположительно вследствие того, что стереохимия тетраэдра
более открытая. Кажется также, что увеличение размера кольца
в четырехдентатном лиганде МН2(СН2)ЖЫН(CH2)1/NH(CH2)a:NH2
от у (или х) =2 до 3 стабилизирует в его комплексах состава
[Со(амин)С1г]+ структуру XLIV по сравнению со структурой
XLIII, возможно, вследствие того, что при шестичленном кольце
возникают меньшие .напряжения валентных углов у пирамидаль-
ных центров атомов вторичного азота.
Структура и стереохимия координационных соединений
57
XL1I1
Необычная геометрия тригональной призмы (XLV), найденная
в трис-(цис-1,2-дифенилэтилен-1,2-дитиолате)Ке(У1), образуется,
по-видимому, вследствие стерического отталкивания между двумя
большими фенильными кольцами, которое дестабилизирует более
обычную октаэдрическую структуру. Известны также многие дру-
гие примеры влияния размера лиганда .на координационное число
и (или) стереохимию.
XLV
4.2.3. Стереохимия лиганда
Полидентатные лиганды иногда образуют геометрию, относи-
тельно не чувствительную к иону металла. В действительности
путем подбора подходящего лиганда можно получить почти лю-
бую геометрию [32, 33]. В качестве примера высокого координа-
ционного числа, вызванного требованиями лиганда, можно при-
вести 8-кратную координацию, найденную в [Со(Ь1Оз)4]2-, в кото-
ром угол между донорными атомами лиганда составляет всего
53° (XLVI). В качестве другого примера можно привести недавно
изученный лиганд 1,8-нафтиридин (XLVII), в комплексах с кото-
рым ионы двухвалентных металлов Мп, Fe, Ni, Си, Zn, Pd и Cd
имеют координационные числа 8, обусловленные, вероятно, огра-
ниченным углом связи лиганд—^металл—лиганд.
58
Глава 1
XLVI
XLVH
Комплексы лиганда N(CH2CH2NH2)3 (tren) с ионами метал-
лов, обычно предпочитающих геометрию плоского квадрата или
тетраэдра, имеют геометрию тригональной бипирамиды вследст-
вие тригональной природы третичного атома азота. Такую струк-
туру имеют, например, Cu(II) и Zn(II) в [M(tren)X]+ (Х=С1, Вг,
NCS) (XLVIII). Существует много примеров этого типа, в том
числе и четырехдентатных лигандов, содержащих кроме атома N
третичные атомы Р и As.
С подходящим образом сконструированным лигандом на-
блюдалась также необычная геометрия тригональной приз-
мы; было показано, например, что такую структуру имеет
[Zn(py3tach)](C104)2 (XLIX):
В результате ненасыщенности по лиганду может образоваться
комплекс с квадратно-плоскостной конфигурацией, как, например,
в [Co(salen)] (L), и многие подобные циклические соединения с
четырехдентатными лигандами этого типа, в которых имеет место
Структура и стереохимия координационных соединений
59
координация иминного атома N (LI, CXXXIV—CXXXVI) или на-
сыщенного N (LII).
LI1
LUI
Однако иногда координационные требования иона металла мо-
гут .привести к деформации таких лигандов, как было недавно
показано рентгеноструктурным исследованием [Co(salen) (асас)] •
•Н2О (LIII) [34]. Однако все известные до сих пор структуры,
содержащие 2,2',2"-терпиридил, линейны, так как вследствие
хр2-гибридизации на атоме углерода донорные атомы N остаются
в основном копланарными, например в [Zn(terpy)Cl2] (триго-
нальная бипирамида). Такая же ситуация имеет место и в дру-
гих ненасыщенных группировках типа LIV—LVI, а аминокислоты
образуют почти плоские пятичленные хелатные кольца (LVI); так,
в хелатированном глицинатном анионе центральный атом угле-
рода смещен не более чем на 6° от плоскости.
60
Глава 1
В качестве другого примера можно привести хелатное соединение
кобальта с тридентатным глицилглицинатным анионом [Co(Gly-
Glyh]- (гл. 4, структура XX). Из-за направленного характера
валентности карбонильного кислорода практически невозможно
более чем бидентатное хелатирование к одному и тому же атому
металла коротких пептидов, координированных амидной группой,
в том случае, когда в координации принимает участие атом кис-
лорода амидогруппы (LVII), так как амидная группа должна
LVII
оставаться в основном плоской; более чем одно хелатное кольцо
может образоваться только в том случае, когда в координации уча-
ствует депротонированный атом азота амидной группы. Подобная
ситуация должна наблюдаться и в более длинных пептидах, так
что, по-видимому, атом кислорода карбонильной группы не участ-
вует в координации, если образуется более чем одно хелатное
кольцо с одним и тем же центральным атомом металла.
Из этих примеров видно, что двухвалентные ионы переходных
металлов легко приобретают разнообразные координационные
числа и конфигурации с подходящими лигандами. Возможно, наи-
более поразительный результат новейших кристаллографических
исследований заключается в том, что при 4- и 6-кратной коорди-
нации редко достигается правильная геометрия, а искажение или
необычная стереохимия представляют в действительности обыч-
ное явление. Очевидно, это также в значительной степени имеет
место при связывании металлов с белками и в активных центрах
ферментных систем.
4.2.4. Хелатные кольца
Образование хелатных колец увеличивает стабильность комп-
лекса (гл. 2) и стабилизирует высокие координационные числа.
Это можно было бы предположить, исходя из внутримолекулярной
природы процесса замыкания кольца с /С1Ж2, а в некоторых слу-
чаях также из ограничения, связанного с малым расстоянием
Структура и стереохимия координационных соединений
61
между донорными атомами «пасти» хелатирующего лиганда на
металле.
Комплексы [Zn(en)3]2+ (октаэдр) и [Zn(terpy)Cl2] (тригональная
бипирамида) представляют примеры высоких координационных
чисел по сравнению с обычной тетраэдрической стереохимией при
4-кратной координации, например [ZnfNHs)^24-. Примерами вы-
соких «необычных» координационных чисел могут служить
[Co(NO3)4]2- (XLVI) и [Zr(С2О4)4]4~ (LVIII) (додекаэдр, коор-
динационное число 8) и комплексы Мп (II) и Fe(III) с шестиден-
татным лигандом этилендиаминтетраацетатом [МпЭДТА(Н2О)]2~
и [РеЭДТА(Н2О)]_ (координационное число 7) (XXVII и
XXVIII). Подобным образом в комплексе La (III) с ЭДТА, имею-
щем состав [ЕаОЭДТА(Н2О)4], осуществляется 10-кратная коор-
динация иона лантана (рис. 1.1,6).
их
Другое следствие хелатирования лигандов состоит в вынуж-
денной координации атомов, являющихся в других случаях сла-
быми донорами. Например, атом третичного азота в монодентат-
ных лигандах, как правило, не координируется к Со (III), но об-
наружена его координация в ионе [Co(tren) (Н2О)2]3+ (LIX).
Подобным образом атом эфирного кислорода в шестидентатном
лиганде
принуждается к координации расположением донорных атомов со-
седних хелатных колец (LX). Для простых координационных со-
62
Глава 1
единений известно много других подобных примеров, и можно
полагать, что та же тенденция будет иметь место при связывании
металлов координационными центрами белков, где фиксированная
геометрия потенциальных донорных атомов может быть достаточ-
ной для вынужденной координации.
5. СТЕРЕОИЗОМЕРИЯ
Стереоизомерия неорганических комплексов возникает в ре-
зультате различного пространственного расположения лигандов
или лигандных групп вокруг центрального иона металла. Все
комплексы, имеющие одинаковый химический состав и превра-
щающиеся друг в друга путем перестановки лигандов, называют-
ся конфигурационными изомерами. Было найдено полезным выде-
лить в пределах этого определения те изомеры, которые отличают-
ся только углами поворота вокруг связанных пар атомов в том
же лиганде, и назвать их конформационными изомерами.
цис траве
конфигурационные
изомеры
Н
конформационные
изомеры
Таким образом, конформационные изомеры, естественно, воз-
никают из конфигурационных эффектов, и иногда трудно прове-
сти четкую грань между этими двумя классами. Однако в данном
обзоре конформационные изомеры будут рассмотрены отдельно,
так как они не требуют перестановки донорных атомов, окружаю-
щих атом металла, а их взаимные превращения обычно происхо-
дят быстро и в основном, по-видимому, независимо от свойств ме-
талла.
Структура и стереохимия координационных соединений
63
5.1. Конфигурационные изомеры
Возможны два .класса конфигурационных изомеров — диасте-
реомеры и оптические изомеры. Диастереомеры имеют различные
физические и химические свойства и, по крайней мере теоретиче-
ски, могут быть разделены путем фракционированной кристалли-
зации или хроматографии или обнаружены в смеси физическими
методами (например, спектроскопия в ультрафиолетовой или ви-
димой области, ЯМР на ’Н, 19F, 13С, рентгеноэлектронная спектро-
скопия, электронно-эмиссионная спектроскопия). Они представля-
ют различные геометрические формы одной и той же молекулы
и могут существовать в виде нескольких изомеров. Оптические
изомеры отличаются только тем, что они являются несовмещае-
мыми зеркальными изображениями одной и той же молекулы; их
предельное число равно двум. Обычно их называют энантиомера-
ми или энантиомерными формами, а иногда катоптромерами.
5.1.1. Диастереомеры
а) Геометрические изомеры. Геометрическая изомерия являет-
ся следствием различного пространственного распределения донор-
ных атомов или хелатных колец вокруг центрального атома ме-
талла. Ее часто обнаруживают в квадратно-плоскостных или ок-
таэдрических комплексах, но она возможна и для других типов
комплексов. Классические примеры представляют квадратно-
плоскостные комплексы Pt (II), Pd(II) (LXI и LXII) и октаэдри-
LXI. транс(D2h)
LXII, цис (C2il)
LXffl, транс (D4h)
NH3
LX1V, цис (C2v)
ческие комплексы Сг(Ш) и Co(III) (LXIII и LXIV). Эти метал-
лы, так же как Ru(II), Rh(III), Cu(II), Ni(II), имеют много гео-
64
Глава 1
метрических изомеров различной степени сложности. Эти изомеры
обычно можно различить путем изучения их спектров (см. выше)
и разделить химическими методами. В качестве других примеров
можно привести цис- и транс-формы бис-(аланината) меди (II)
(LXV и LXVI) и дихлородитиоуреата никеля(II) (LXVII и
LXVIII), а также [Pt(NH3)pyClBr], который был разделен на три
возможные формы (LXIX—LXXI).
LXXI
Комплекс [PtClfSCzHs) (Р(СзН7)з]2 представляет пример ком-
плекса, содержащего соединенные мостиком атомы металла, для
него были охарактеризованы две симметричные формы (LXXII и
LXXIII). Примером октаэдрического комплекса, содержащего хе-
латные кольца, является граневое (LXXV) и реберное (LXXVI)
расположения донорных атомов в триглицинате кобальта(III).
СаН6 С2Н6
I I
Cl\ /С1 Cl\ /Р(С3Н-)3
zPtf JV ЛЧ zpt\
(CjHjJgP g/ Р(С3Н7)3 (CsHj)3P gZ Cl
I I
c2H& C2HB
LXXII LXXIII
Структура и стереохимия координационных соединений
65
Обе изомерные формы этого комплекса, так же как и соответст-
вующие изомеры аланината, были изолированы в твердом со-
стоянии.
l.XXV I.XXVL
Другой класс геометрических изомеров возникает в результа-
те различного расположения хелатных колец, образуемых поли-
дентатным лигандом вокруг иона металла. Известны многие при-
меры этого типа. Из последних примеров можно указать на ион
LXXX (аа-изомеру
66
Глава 1
[Co(trien) (Gly)]2+, для которого было получено три различных
расположения (LXXVII—LXXIX), и на ион [Co(tetraen)Cl]2+,
для которого возможны четыре геометрические формы (LXXX—
LXXXIII) в зависимости от стереохимии вокруг трех центральных
донорных атомов азота. 'Подобным образом «зеленой» и «корич-
невой» формам комплекса Со(III), содержащего шестидентатный
лиганд
были приписаны формулы LXXXIV и LXXXV соответственно; при
увеличении гибкости этого лиганда путем насыщения или увели-
чения хелатных колец становятся возможными еще две кон-
фигурации— LXXXVI и LXXXVII. Такие изомеры обычно легко
LXXXIV LXXXV LXXXVI LXXXVII
разделяются фракционированной кристаллизацией или ионооб-
менной хроматографией, но определение их строения часто оказы-
вается более трудной задачей; с помощью рентгеноструктурного
анализа не удается получить определенного отнесения.
Несколько иной тип изомерии получается в результате замеще-
ния в лиганде. Так, [Pt(L -рп)2]2+ существует как в цис-
(LXXXVIII), так и в транс-формах (LXXXIX), а граневая (ХС) и
реберная (XCI) формы Л-[Со( L-pnJg]^ были идентифицированы
после хроматографического разделения. Другие примеры этого
Структура и стереохимия координационных соединений
67
типа представляют различные хелаты несимметрично замещенно-
го ацетил ацетона, например [Co(tfa)3] (1Га = трифторацетилаце-
LXXX1X
XCI
тон). Такие комплексы, содержащие несимметричные хелаты, ока-
зались полезными при изучении механизма инверсии на асиммет-
ричных атомах металла.
Следующий класс изомеров, весьма сходных с описанными в
предыдущем параграфе, — это соединения, в которых ориентация
протона может привести к конфигурационным эффектам. Так, ион
[Co(dien) (en)Cl]2- может существовать в двух выделенных в ин-
дивидуальном состоянии геометрических формах (ХСП и ХСШ),
a p2-['Co(trien) (Gly)]2+ на катионообменной смоле дауэкс был
разделен на две формы — XCIV и XCV.
ХСП
хеш
Более сложный пример представляет изомерия [Co(tetraen)Cl]2+,
для которого возможно четыре расположения (XCVI—XCIX), за-
5
68
Глава 1
висящие от конфигурации вокруг двух атомов .вторичного азота,
соединяющих хелатные кольца, расположенные в той же плоскости.
В последних соединениях возникает также асимметрия на атоме
азота, но это явление будет обсуждено более детально в
разд. 5.1.2, б. В качестве других примеров изомерии этого типа
приведем комплексы с насыщенными соединениями, содержащи-
ми донорные атомы Р и As и образующими мостиковые хелатные
кольца в одной и той же координационной плоскости.
Для приведенных выше примеров при наличии атома N в ще-
лочных растворах, где происходит быстрый обмен протона NH,
легко достигается равновесие, но в кислых растворах изомеры
устойчивы, и во многих случаях их легко разделить и отличить
(с помощью спектроскопии в видимой области или ПМР-спектро-
скопии).
. ХСУИ
ХСУШ
XCIX
б) Структурные изомеры. Сюда относятся изомеры, которые
образуются в результате изомерии связей, 'возникающей при на-
личии альтернативных способов координации одного и того же
лиганда, и изомерии лигандов, которые сами отличаются по струк-
туре.
Существует много случаев изомерии связей для монодентат-
ных лигандов, содержащих два различных донорных атома (ам-
фифилы). Так, М—ONO (нитрито) и М—NOO (нитро), М—SCN
(тиоцианато) и М—NCS (изотиоцианате), М—OCN (цианато) и
М—NCO (изоцианато), М—CN (циано) иМ—NC (изоциано) пред-
ставляют классические примеры из химии Со (III). Подобным обра-
Структура и стереохимия координационных соединений
69
зом ион [Co(NH3)5(Gly)]2+ существует в виде двух форм: со
связью через атом азота (С) и через атом кислорода (CI). Ацет-
амид в [Со(МНз)5(асат)]3+ также может присоединяться через
атом О (СП) и через атом N (СШ); в этом случае различная
координация требует перестановки 'протона.
[(NH3)5Co—NH2CH2CO2]2+
G
СН3
2+
(NH3)5Co—ОС—nh2
СП
[(NH3)6Co—OCCH2NH2]2+
II
О
GI
Г сн3 у*
I
L (NH3)6Co—NH=C—ОН
CHI
В качестве примеров бидентатных лигандов можно указать на
изомеры хелатированного амида глицина, координированного по
атому азота или кислорода, который найден в [Co(en)2(glyam)]3+
(CIV в CV) (для последнего рКа енольного протона равен (при-
мерно 0,4), а также на своеобразный комплекс CVI, в котором
предполагают наличие двух хелатных колец различного размера
в одном и том же лиганде.
CIV
? /снз
Н3СС—C=N HN=C
О I zNi/ Jp-COCH3
_C=NH XO-N
H3C<
CVI
Лигандные изомеры также довольно многочисленны, так как
они просто образуются из совершенно различных лигандов, име-
ющих одинаковый химический состав. Классическими примерами
являются комплексы с 1,2-диаминопропаном (рп) и триэтиленди-
амином (tn), как в комплексах [Pt(tn)2]2+ (CVII) и [Pt(pn)2]2+
70
Глава 1
(CVIII) соответственно, и с
М—CNCH3 (метилизоцианид).
М—NCCH3 (метилцианид) и
CVIII
CVII
5.1.2. Оптические изомеры
Оптические изомеры возникают в том случае, когда молекула
и ее зеркальное изображение несовместимы друг с другом. Гово-
рят, что такая молекула диссимметрична, и диссимметрия возни-
кает во всех случаях, когда молекула не имеет ни центра симмет-
рии, ни плоскости симметрии, ни зеркально-поворотной оси сим-
метрии*. Таким образом, оптически активные формы всегда имеют
асимметричную структуру, но они могут иметь некоторые элемен-
ты симметрии, главным образом поворотные оси второго, третьего
или четвертого порядка.
Оптические эффекты в координационных соединениях по их
происхождению удобно классифицировать на три группы: а) воз-
никающие из геометрического расположения лигандных групп
вокруг металла (конфигурационные эффекты), б) обусловленные
асимметрией лиганда или его донорных атомов, существовавшей
или возникшей вследствие координации (вицинальные эффекты),
и в) происходящие из конформационных свойств координирован-
ного лиганда. Так как оптическая активность, возникающая из
конформационных эффектов, является следствием различных уг-
лов поворота вокруг связанных пар атомов, то она будет рассмот-
рена в разд. 5.2.
а) Конфигурационные энантиомеры. Впервые их существова-
ние наглядно показал Вернер с целью защиты своей теории коор-
динации. Так, он доказал октаэдрическую геометрию [Со(еп)3]3+
и цас-[Со(еп)2С12]+ путем их разделения на энантиомерные фор-
мы (CIX, СХ) и (CXI, СХП) соответственно, но ионы
[Co(en) (NH3)4]3+ (CXIII) и транс-[Со(еп)2С12]+ (CXIV), имеющие
плоскость симметрии, неспособны разделяться.
Со времен Вернера были разделены многие комплексы, в част-
ности комплексы Со(Ш) и Сг(Ш), а их абсолютная конфигура-
* Так как центр симметрии (i) эквивалентен зеркальио-поворотиой оси вто-
рого порядка а плоскость симметрии (о) можно рассматривать как элемент
симметрии Si, то можно дать следующее более краткое определение диссимметрии:
«если молекула не имеет зеркально-поворотных осей Sn с п^1».
Структура и стереохимия координационных соединений
71
CIX (Д)
СХ11 (Л)
ция была установлена рентгеноструктурным анализом [35, 51].
Изучение таких оптически активных комплексов существенно спо-
собствует лучшему пониманию двух главных областей координа-
ционной химии: а) стереохимия и механизм процессов замещения
и рацемизации и б) отнесение электронных полос поглощения пу-
тем исследования ДОВ (дисперсии оптического вращения) и КД
(кругового дихроизма) и усовершенствование нашего понимания
электронного строения комплексов. Обсуждение этих тем выхо-
дит за рамки данной главы, и читатель отсылается к другим
источникам [2, 36, 37].
Обычную процедуру разделения [10, 38] можно применить к
кинетически инертным комплексам, .стабильным или подвергаю-
щимся в водных растворах медленной рацемизации. Это относится
ко всем комплексам Со(Ш), Сг(Ш), Rh(III)) Ir(III), Os(II) и
Pt (IV) и в некоторых случаях к октаэдрическим комплексам
Fe(II), Ni(II) и Со(П). Для катионных и анионных комплексов
это обычно означает образование диастереомерных солей с оп-
тически активным органическим или неорганическим анионом или
катионом, но нейтральные комплексы разделить труднее; как пра-
вило, для этой цели используют методы хроматографии или зон-
ного плавления. Для кинетически лабильных октаэдрических или
тетраэдрических комплексов Fe(II), Fe(III), Ni(II), Co(II),
Mn(II), Cu(Il), Zn(II) и др. энантиомерные формы могут быть
выделены только в случае их исключительной стабильности.
Обычно их исследование проводят в асимметричном окружении,
таком, как активный спирт (бутанол-2), или в водном растворе,
72
Глава 1
содержащем добавленную асимметричную соль (аммоний-сТл-
бромкамфорсульфонат, натрийантимонил-4/-тартрат, цинхонин,
стрихнин и т. п.). Такие эффекты «конфигурационной активности»
возникают вследствие различий в активности энантиомерных
форм в асимметричном окружении.
Ни один из тетраэдрических комплексов типа M(ABCD), мо-
делирующих классическую химию углерода, не был разделен на
оптические изомеры, так как, во-первых, их трудно синтезировать,
а во-вторых, они, по-видимому, слишком лабильны для того, что-
бы их можно было изучать обычными методами. Были разделены
некоторые тетраэдрические комплексы типа М(А—гВ)2, например
[B(sal)2]+ (CXV) и [Be(bzp)2] (IX), но обычно эти комплексы
легко диссоциируют, и их также нельзя исследовать обычными
методами.
CXV
При квадратно-плоскостной координации оптические изомеры
обычно отсутствуют, но соединение XIII представляет необычный
пример, в котором имеет место скорее квадратно-плоскостная ко-
ординация, чем тетраэдрическая. При тетраэдрической координа-
ции комплекс имел бы плоскость симметрии.
Несомненно, наиболее интенсивно исследовались октаэдриче-
ские комплексы Со(III) или Сг(Ш) и Rh(III), содержащие хела-
тированные лиганды. Так, были разделены не только такие дис-
симметричные комплексы, как [Со(еп)з]3+, [Сг(еп)3]3+, [Со(ох)3]3-
и [Co(Gly)3], но и значительно менее симметричные молекулы.
CXVI
CXVII
CXV111
Структура и стереохимия координационных соединений
73
Например, из трех возможных геометрических форм [Co(dien)2]3+
(CXVI—CXVIII) форма CXVI, имеющая симметрию С2л, не дис-
симметрична, а формы CXVII и CXVIII ^симметрией С2 были раз-
делены на энантиомерные формы.
Интересный случай представляет изомер CXVIII, так как здесь
единственным элементом хиральности является отношение между
двумя находящимися в гране-положении связями N—Н вокруг
СХ1Х
ехх
CXXI
атома вторичного азота. Подобным образом были разделены две
диссимметричные формы [Co(trien))Cl2]+—CXIX и СХХ, но
транс-изомер неактивен относительно металлического центра.
Для [Co(tetraen)Cl]+ возможны четыре структурные формы
(LXXX—LXXXIII), но LXXXIII имеет ось второго порядка и по-
этому неактивна. Изомеры LXXX и LXXXI 'были разделены и их
конформации подтверждены рентгеноструктурным методом. Сим-
метричные плоские тридентатные лиганды, такие, как терпиридил,
образуют неактивные октаэдрические комплексы (СХХП), но
несимметричные тридентатные лиганды, такие, как глицилглицин,
образуют оптически активные комплексы типа аниона
[Co(Gly-Gly)2]- (СХХШ).
Были разделены некоторые .комплексы с шестидентатными ли-
гандами, из которых наиболее известны комплексы ЭДТА
(CXXIV), пропилендиаминтетрауксусной кислоты (ПДТА), а так-
74
Глава 1
же комплексы Со(III) с шиффовыми основаниями (LXXXIV и
LXXXV), синтезированные Двайером и Лайонсом [39].
CXX1V
б) Вицинальные эффекты. Этот тип диссимметрии обусловлен
лигандом, который либо сам по себе асимметричен, либо приобре-
тает при координации асимметричные донорные атомы. Существу-
ет большое число асимметричных лигандов; в качестве обычных
примеров можно указать на 1,2-диаминопропан (pn) (CXXV),
транс- 1,2-диаминоциклогексан (chan), аминокислоты (кроме гли-
цина), пропилендиаминтетрауксусная кислота (ПДТА) и асиммет-
ричные пептиды (CXXVI).
CXXV CXXVI
Очевидно, что если асимметричен лиганд, то асимметричен и
комплекс. Это приводит к асимметричным вкладам в d-электрон-
NH3 _ а*
H5N-—9==C-CH-NH2
/J^c° ; сн3
H3N--1: -~NH3
NH3
NH3 2*
1
H3N --- --•O^C
с^СНз
H3N——j NhT ’’H
NH3
CXXVII
CXXVIII
Структура и стереохимия координационных соединений
75
ные переходы металла. Ниже приведены два таких примера
(CXXVII и CXXVIII), содержащие монодентатный и бидентатный
аланин соответственно. Вклад асимметрического атома углерода
в круговой дихроизм, ассоциированный с ионом металла, сходен в
этих двух комплексах. Однако в некоторых случаях наличие двух
асимметричных лигандов может привести к внутренней компенса-
ции и поэтому к образованию неактивного комплекса. Так, ком-
плексы CXXIX и СХХХ неактивны, так как транс-изомер (CXXIX)
имеет центр симметрии, а СХХХ — плоскость симметрии. В таких
комплексах мутаротация асимметричных центров, соседних с ко-
сххх
ординированным карбонилом или карбонильными функциями,
сильно облегчается при координации, что может быть объяснено
повышенной кислотностью протона. Так, хелатированный аланин
в комплексе CXXXI подвергается быстрой мутаротации при pH 12,
в то время как ь-аланинглицин в СХХХП подвергается мутаро-
тации три pH 10.
CXXXI
схххп
схххш
76
Глава 1
Вицинальные эффекты, возникающие на донорных атомах при
координации, интенсивно исследовались лишь в последнее время,
хотя сам эффект был известен давно. Например, хелатированный
саркозин в ионе [Co(NHs)4(sar)]2+ (СХХХШ) асимметричен бла-
годаря четырехковалентному атому азота, и этот ион был разде-
лен на оптические энантиомеры.
Подобным образом на оптические изомеры была разделена
транс-форма комплекса (±.)-[14]-dieneNi(II) (CXXXIV), содер-
жащего четырехдентатное основание Шиффа, хотя он имеет в ос-
CXYXIV (rnponc-[Hl-diene) CXXXV (мезп-транс-[1^]- diene)
новном плоскую структуру. В .мезо-изомере (CXXXV), содержа-
щем вторичные протоны NH на противоположных сторонах от
плоскости координации, происходит внутренняя компенсация, и
поэтому он неактивен. Известна кристаллическая структура обоих
комплексов [40]. Подобным образом цис-[14]-diene (CXXXVI)
может быть и в активной и в .мезо-формах благодаря протонам
NH. Многие другие комплексы были разделены на энантиомерные
CXXXVI1
CXXXVIII
CXXXIX
CXL
Структура и стереохимия координационных соединений
77
формы, в том числе транс-[Со(trien)Cl2]+ (CXXXVIII), транс-
[Со(СН3-еп)2С12] + (CXXXIX), [Pt(CH3-en)2]2+ (CXXXVII),
[Ni(cyclam)]2+ (CXL).
Инертность N-центров по отношению к рацемизации резко от-
личается от быстрой инверсии в органических аминах или заме-
щенных ионах аммония, а изомеры комплексов оптически устой-
чивы в кислых растворах. Кажется, что рацемизация протекает
только в том случае, когда протоны N—Н удалены, и, более того;
что скорость рацемизации в 102—106 раз меньше, чем скорость об-
мена атомов водорода в комплексах Pt (II), Pt(IV) и Со (III) [26].
Это было объяснено большим фактором сохранения для основных
аминов с конъюгированными связями. В комплексах этого типа
как обмен водорода, так и инверсия азота подчиняются закону
СКОРОСТИ Анабл = А[ОН_].
.При координации посредством атомов Р, S и As также воз-
никает асимметрия. Как и можно было ожидать, здесь сохране-
ние конфигурации более выражено, чем в случае асим-
метричного атома азота. Так, соль Pt (IV) (CXLI) была
разделена еще в 1930 г. [41], а недавно мышьяковистое основание
As(CH3) (C2Hs) (СбН3) было разделено через комплекс Pt (II)
(CXLII) [42]. Подобный комплекс Pt(II) был использован для
разделения Р(СН3) (CeHs) (С4Н9) [43].
Cl
...--;NH2
:\s'—
Xch2ch2nh3+ci
CXLI
Cl...
/ ^Pt
CH3
3 As.....
/ \
C2HS CiHs
CXLII
Очевидно, что благодаря этим эффектам картина изомериза-
ции в инертных комплексах более сложна. Например, в то время
78
Глава 1
как ион a-[Co(trien)Gly]2+ имеет стереоспецифически направлен-
ный атом N вЛ- и Л-энантиомерах(СХЫП), ион Pi-[Co(trien)Gly]2+
cxliv (0,-д-шг)
существует в виде двух диастереомерных форм (имеющих одина-
ковую устойчивость) для каждой из Л- и Л-конфигураций вокруг
иона металла (CXLIV, CXLV). Подобное положение найдено и
для третьего геометрического изомера, p-[Co(trien) (Gly) ]а+ (с от-
ношением устойчивостей 9:1 в этом случае), так что для этого
комплекса реализуется десять возможных конфигурационных
изомеров. В щелочном растворе, где происходит быстрый обмен
протонов, внутренние диастереомерные пары легко подвергаются
взаимопревращению. Подобным образом ион CXLVI имеет
плоскость симметрии, если пренебречь асимметрическим атомом
азота и конформациями хелатного кольца, но этот ион также
Структура и стереохимия координационных соединений
79
можно разделить на зеркальные изомеры, что показывает важ-
ность изомерии этого типа.
Подобные рассмотрения часто указывают на существование
стереоспецифических эффектов. Это можно показать на примере
соединения CXLIII; конфигурация донорных атомов азота и фос-
фора, соединенных пятичленными хелатными кольцами в различ-
ных координационных плоскостях, зависит от ориентации колец.
Менее очевидный, но такой же специфический эффект проявляет-
ся для иона A-[Co(trien) (sarc)]2+ (CXLVII), в котором аминокис-
лотная часть имеет исключительно S-конфигурацию. Альтернатив-
ная R-конфигурация (CXLVIII) не найдена в этом ионе (она су-
ществует в Л-комплексе), так как несвязывающее взаимодействие
между метильной группой и направленным к вершине хелатным
кольцом дестабилизирует ее; вычисленная энергия равна
3,3 ккал/моль [12].
CXLVIII (A-02-RRR)
5.2. Конформационная изомерия
5.2.1. Общие положения
Конформационные изомеры возникают из несовместимости в
пространстве атомов или групп атомов в молекулах, имеющих
одинаковый химический состав и конфигурацию, но различные
углы поворота относительно связанных пар атомов. Уже давно
были известны примеры замещенных дифенилов с классичиским
затрудненным вращением в 2,2'-диамино-6,б'-диметилдифениле
(CXLIX). Оптически активные формы комплекса Си (II) с аддук-
том основания Шиффа с салицилальдегидом (CL) были синтези-
рованы путем использования оптических форм диамина. Вследст-
вие того что бензольные кольца размещены непараллельно, атом
Си (II) приобретает тетраэдрическую координацию. Однако обыч-
но энергия активации взаимного превращения конформационных
изомеров мала, так как она обусловлена только изгибающими
движениями, а скорость таких процессов поэтому велика. Так,
в кристаллическом состоянии существуют конформации как
80
Глава 1
6 (CLI), так и % (CLII) зеркальных антиподов хелатированного
этилендиамина, но в растворах в комплексах Со(Ш), Pt(II),
Ph (III) и Pt (IV) происходит быстрое взаимопревращение, и две
конформации до настоящего времени не были идентифицированы.
СП
5.2.2. Конформационная изомерия в хелатах
а) Пятичленные кольца. Несомненно, что большинство хелат-
ных колец являются пяти- или шестичленными. Многие из них,
образованные такими лигандами, как оксалат, аминокислоты, пеп-
тиды, четырехдентатные лиганды типа корринов, — плоские или
почти плоские, и для них отсутствуют многие конформационные
свойства. При квадратно-плоскостном или октаэдрическом типе
координации такие лиганды, как этилендиамин, содержащий че-
тыре тетраэдрических скелетных атома, приводят к образованию
хелатных колец с выраженными конформационными свойствами,
и это явление наблюдается для 'большинства систем с хелатными
кольцами, содержащими тетраэдрические атомы углерода. Такие
кольцевые системы отличаются от аналогичных карбоциклических
систем тем, что они имеют углы связей у металла, близкие к 90°.
Вследствие этого остающаяся молекула должна перекоситься, что-
бы воспрепятствовать нецелесообразному искажению тетраэдри-
ческих центров. Возникает несколько структур в зависимости от
Структура и стереохимия координационных соединений 81
точных углов металл—лиганды, длин связей, а также от размера
и типа участвующего донорного атома. Для этилендиамина обыч-
но находят структуры CLI и CLII, в которых два атома углерода
находятся на одинаковых расстояниях от плоскости М—N—N, но
на противоположных сторонах. В такой конформации полукресла
все атомы водорода более или менее смещены, и атомы водорода
при углероде являются почти полностью «аксиальными» или «эк-
ваториальными» в том смысле, как эти термины используются
для циклогексана; атомы .водорода, находящиеся при азоте, в кон-
формационном отношении отличаются в меньшей степени. Это
расположение найдено во всех структурах М—еп, исследованных
до сих пор кристаллографическим методом, а другие возможные
конформации, содержащие оба атома углерода на одной и той же
стороне плоскости М—N—N («заслоненная» форма) (СЫН), до
настоящего времени не идентифицированы [52].
Можно было бы ожидать стабилизации этой структуры по ме-
ре уменьшения длины связи М—N (~1,75 А) при условии, что
угол N—М—N будет оставаться неизменным. Для донорных ато-
мов большего размера, таких, как Р или As, можно ожидать
больших длин связей М—донор и преимущественного образования
конформации полукресла. Если в качестве донорного атома вы-
ступает атом S, то должна быть выраженная склонность к обра-
зованию меньших, чем тетраэдрические (~92°), углов М—S—С
и менее плоской конформации, что может быть измерено как уве-
личение диэдрального угла вокруг С—С связи. Подобного, но
меньшего эффекта следует ожидать, если донором является
атом О. Изгибание хелатных колец наблюдается также в структу-
рах, содержащих полидентатные пятичленные хелатные кольца,
таких, как диэтилентриамин (dien) (CXVI—CXVIII), триэтилен-
тетрамин (trien) и тетраэтиленпентамин (tetraen) (LXXX—
LXXXIII). В этих структурах конформации соседних хелатных
колец, находящихся в одной и той же координационной плоскости,
определяются конфигурацией вокруг общего асимметричного цент-
ра — атома азота, но направленные к вершине хелатные кольца
могут существовать в б- и ^-формах. Определенная недавно
структура кристаллического 02-[Со(trien) (Gly)] I2 подтвердила
6—2451
82
Глава 1
наличие в триэтилентетраминовом хелате конформаций ббЛ
(CLIV) и W, (CLV).
CLIV
CLV
Однако обычно устойчивость одной конформации превышает
устойчивость остальных в достаточной степени, чтобы исключи-
тельно (>99%) существовать в равновесных условиях. Подобным
образом очевидное отсутствие формы мезо- [Со (trien)X2]+ (CLVII)
может быть объяснено тем, что центральное кольцо имеет вынуж-
денную конформацию заслоненного «конверта»*. Подобные рас-
суждения применимы и к другим кольцевым системам, содержа-
щим донорные атомы N, О и S.
CLVI (предпочтительная
конформация)
Для тетраэдрических структур пятичленные хелатные кольца
этилендиаминного типа должны быть напряжены, так как угол
N—М—N увеличивается от 90 до 109°. Хотя хелатное кольцо бу-
дет предпочитать оставаться изогнутым, можно ожидать, что оно
будет расширяться и приобретать более плоское строение. При
структуре тригональной бипирамиды замыкание хелатных циклов
от экваториальной плоскости к вершине предпочтительно по срав-
* Дополнение, сделанное в корректуре. Недавно была получена мезо-форма
[Ni(trien)] (С1О4)2, в которой центральное хелатное кольцо имело заслоненную
конфигурацию [52].
Структура и стереохимия координационных соединений 83
нению с хелатированием в пределах экваториальной плоскости,
как в случае образования пяти-, так и шестичленных хелатных
колец.
Замещение атомов водорода у атомов углерода в этих пяти-
членных кольцах метильными группами может происходить либо
в «аксиальном», либо в «экваториальном» положении. Для R-1,2-
диаминопропана это дает б (CLIX) и X (CLVIII) конформации
соответственно.
6
С1ЛХ
В результате изучения кристаллических структур и теоретиче-
ского рассмотрения было установлено, что Х-конформация по
сравнению с б-конформацией стабилизирована на 2 ккал/моль.
Это, по-видимому, является следствием значительного 'взаимодей-
ствия между аксиальной метильной группой и аксиальной группой
X на металле. Для 5-1,2-диаминопропана подобным образом стаби-
лизируется б-конформация по сравнению с Х-конформацией. Как в
А-, так и в Д-формах [Co(R-(—)-рп3]3+ хелатные кольца приобре-
тают Х-конформацию. Более объемистые заместители будут усили-
вать этот эффект так же, как и симметричные дизаместители, такие,
как R,R-2,3-AnaMHHo6yTaH (CLX), и кажется, что здесь лиганд дол-
жен быть стереоспецифичен по отношению к иону Д-[Со(—)-2,3-
bdamjs]34' [45]. Однако если заместители находятся при одном и
CLX )-Ьл]
СНз
CLXI (RjS-мезо-Ьп)
том же атоме углерода, то всегда один из них будет аксиальным,
а один экваториальным или возникнет некоторый компромисс меж-
6*
84
Глава 1
ду ними. То же относится и к Д1езо-2,3-диаминобутану (CLXI), но
об этих системах известно мало.
Замещение на донорных атомах приводит к подобным резуль-
татам. В М-метил-1,2-диаминоэтане (СН3-еп) различие между ак-
сиальной (CLXIII) и экваториальной (CLXII) метильной группа-
ми выражено в меньшей степени (~20°), но заместитель теперь
находится ближе к аксиальной группе X, присоединенной к метал-
лу, и вследствие этого несвязывающие взаимодействия должны
быть больше.
CLXII
Как и предполагалось, трсяс,трсяс-Р,Р-[Со(СН3-еп)2С12]СЮ4
в кристаллическом состоянии (рис. 1.4) содержит метильную груп-
пу в экваториальном положении и б-конформацию кольца. [Рас-
Рис. 1.4. Структура катиона транс,транс-Я,В,- [Со (СНз-еп)2С12]+ [50].
чет силы поля предсказывает незначительное различие энергии
этой и аксиальной конформаций (— 1 кал/моль).] Симметричное
распределение на обоих атомах N приводит, по-видимому, к даль-
Структура и стереохимия координационных соединений
85
нейшей стабилизации 6-кольца, и в спектре ПМР иона транс-
[Со(СН3-еп)2С12]+ наблюдается хорошо разрешенный сигнал
АА'ВВ', которого следовало ожидать для замороженной конфор-
мации в растворе. Поэтому ясно, что замещение донорного атома
или атома лиганда в пятичленных хелатах приводит к преимуще-
ственной, а во многих случаях и к исключительной стереохимии
кольца.
б) Шестичленные кольца. Большинство шестичленных хелатов
проявляют выраженные конформационные свойства. Можно при-
вести такие примеры, как 1,3-диамино.пропан (триметилендиамин),
малонат-ион, р-аланин и 1,3-диарсенопропан или 1,3-дифосфино-
пропан с замещенными атомами As или Р. Хелатные системы
аналогичны циклогексану, и возможны конформации кресла
(CLXIV), ванны (CLXV, CLXVI) и скошенной ванны (CLXVII).
Структурный анализ A-[Co(tn)]Br-H2O [46] указывает, что
в противоположность Со—ей кольцам угол N—Со—N больше 90°
(92—96°) и это, по-видимому, оказывает влияние на угол
Со—N—С, который увеличивается до 117,4°. Угол С—С—С (112°)
ближе к тетраэдрическому. Все три кольца приобретают конфор-
CLX1V
CLXV1
CLXV1I
мации «искаженного кресла», и подобные конформации найдены
в [Co(tn)2CO3]ClO4. Тем не менее обнаружено, что малонат-ион
в [Со(еп) (mal)2]_ имеет конформацию уплощенной ванны. Ка-
жется, что предельная форма ванны для хелатов триметиленди-
аминового типа маловероятна, так как имеет место большое не-
связывающее взаимодействие между одним атомом Н централь-
ной метиленовой группы и аксиальным заместителем на металле
(CLXV), а также между атомами Н на N и С, а также на С и С
(CLXVI). Хотя для детального анализа этих систем еще мало
86
Глава 1
рентгеноструктурных данных, можно предположить, что вследст-
вие увеличенного размера кольца будут встречаться промежуточ-
ные конформации. Это предположение подтверждается структурой
одного из изомеров [Со(еп) (ditn)Cl]2+, в котором одно шести-
членное кольцо имеет конформацию кресла, а другое — искажен-
ной ванны (рис. 1.5) [47], По-видимому, важными факторами-
в таких кольцевых системах являются упаковочные эффекты ~
Рис. 1.5. Структура катиона [Co(en) (ditn)Cl]2+ [47].
катион-анионные взаимодействия, и во многих случаях конфор-
мации в кристаллическом состоянии отличаются от тех, которые
сохраняются в растворе.
СПИСОК ЛИТЕРАТУРЫ
1. Химия координационных соединений, под ред. Дж. Бейлара, ИЛ, М.,
1960.
2. Басоло Ф., Пирсон Р., Механизмы неорганических реакций, «Мир», М.,
1971.
3. Charberek S., Martell А. Е., Sequestering Agents, Wiley — Interscience, New
York, 1959.
4. Dwyer F. P., Mellor D. P. (eds.), Chelating Agents and Metal Chelates, Aca-
demic Press, New York, 1964.
5. Коттон Ф., Уилкинсон Дж., Современная неорганическая химия, «Мир», М.,
1969.
6. Современная химия координационных соединений, под ред. Дж. Льюиса и
Р. Уилкинса, ИЛ, М., 1963.
Структура и стереохимия координационных соединений 87
7. Martell А. Е., Calvin М., Chemistry of Metal Chelates, Prentice-Hall, New York,
1952.
8. Saito Y„ Pure Appl. Chem., 17, 21 (1968).
9. Freeman H. C., Adv. Protein Chem., 22, 337 (1967).
10. Sargeson A. M., Chapter 5 in ref. [4].
11. Sargeson A. M., in R. L. Carlin (ed.), Transition Metal Chemistry, Vol. 3,
Marcel Dekker, New York, 1966, p. 303.
12. Buckingham D. A., Sargeson A. M., in Eliel, Allinger (eds.), Topics in Ste-
reochemistry, Vol. 6, 1971, p. 219.
13. Edwards J. O., Inorganic Reaction Mechanisms, Benjamin, New York, 1964.
14. Basolo F„ Survey Progr. Chem., 2, 1 (1964).
15. Hunt J. P., Metal Ions in Aqueous Solution, Benjamin, New York, 1963.
16. Eigen M., Wilkins R. G., in «Mechanisms of Inorganic Reactions», Adv. in
Chem. Series, № 49, Am. Chem. Soc., Washington, D. C., p. 55.
17. Cartmell E., Fowles G. W. A., Valency and Molecular Structure, 2nd edn., But-
terworths, London, 1961.
18. Craig D. P., Nyholm R. S., Chapter 2 in ref. [4].
19. Gray H. B., J. Chem. Educ., 41, 2 (1964).
20. Dunn T. M., McClure D. S., Pearson R. G., Some Aspects of Crystal Field
Theory, Harper and Row, New York, 1965.
21. Грей Г., Электроны и химическая связь, «Мир», М., 1967.
22. Liehr A. D., J. Chem. Educ., 39, 135 (1962).
23. Bradley D. C., Hursthouse M. В., Rodesiler Р. F., Chem. Commun., 1969,
14.
24. Fleischer E. B., Accounts Chem. Res., 3, 105 (1970).
25. Schrauzer G. N., Accounts Chem. Res., 1, 97 (1968).
26. Buckingham D. A., Marzilli L. G„ Sargeson A. M., J. Am. Chem. Soc., 91,
5227 (1969).
27a . Buckingham D. A., Foster D. M., Sargeson A. M., J. Am. Chem. Soc., 90, 6032
(1968).
276. Buckingham D. A., Foster D. M., Sargeson A. M., J. Am. Chem. Soc., 92, 5571
(1970).
28. Buckingham D. A., Marzilli P. A., Maxwell I. E., Sargeson A. M., Fehlmann M.,
Freeman H. C„ Chem. Commun., 1968, 488.
29. Wells A. F., in «Structural Inorganic Chemistry», 3rd edn., Clarendon Press,
Oxford, 1962, p. 71.
30. Nyholm R. S., Proc. Chem. Soc., 1961, 273.
31. Gerlach D. H., Holm R. H. Inorg. Chem., 9, 588 (1970).
32. Lions F., Rec. Chem. Progr., 22, 69 (1961).
33. Lions F., Rev. Pure Appl. Chem. (Aust.), 19, 177 (1969).
34. Calliganis M., Nardin G., Randaccio L., Chem. Commun., 1969, 1248.
35. Saito Y., Pure Appl. Chem., 17, 21 (1968).
36. Gillard R. D., Progr. Inorg. Chem., 7, 215 (1966).
37. Gillard R. D., Mitchell P. R., Structure and Bonding, 7, 46 (1970).
38. Wilkins R. G., Williams M. J. G., Chapter 3 in ref. [6].
39. Dwyer F. P., Lions F., J. Am. Chem. Soc., 69, 2917 (1947); J. Am. Chem.
Soc., 72, 1546 (1950).
40. Bailey M. F„ Maxwell I. E., Chem. Commun., 1966, 908.
41. Mann F. G., J. Chem. Soc., 1930, 1745.
42. Bosnich B., Wild S. B., J. Am. Chem. Soc., 92, 459 (1970).
43. Chan J. H., Chem. Commun., 1968, 895.
44. Maxwell I. E., Thesis, Australian National University, 1969.
45. Woldbye F., Studier over Optisk Activet, Polyteknisk Forlog, Kobenhavn, 1969,
p. 176 et seq.
46. Nomura T., Marumo F., Saito Y., Bull. Chem. Soc. Japan, 42, 1016 (1969).
88 Глава 1
47. Ireland Р. R., House D. A., Robinson W. T., Inorg. Chim. Acta, 4, 137 (1970).
48. Lind M. D., Lee B., Hoard I. L., J. Am, Chem. Soc., 87, 1611 (1965).
49. Hoard I. L„ Lee B., Lind M. D., J. Am. Chem. Soc., 87, 1612 (1965).
50. Buckingham D. A., Chandler G., Marzilli L. G., Sargeson A. M, Chem. Com-
mon., 1969, 539.
51. Kennard O., Watson D. G. (eds.), Molecular Structures and Dimensions, In-
ternational Union of Crystallography, Vol. 2, 1970 and Vol. 3, 1971.
52. McPherson A., Rossmann M. G., Margerum D. W., James M. R., J. Coord.
Chem., 1, 39 (1971).
53. Гринберг А. А., Введение в химию комплексных соединений, «Химия»,
М. — Л., 1966.
54. Желиговская Н. Н., Черняев И. И., Химия комплексных соединений, «Выс-
шая школа», М., 1966.
55. Берсукер И. Б., Электронное строение и свойства координационных соедине-
ний, «Химия», Л., 1976.
ГЛАВА 2
УСТОЙЧИВОСТЬ
КООРДИНАЦИОННЫХ СОЕДИНЕНИЙ
Р. Дж. Ангеличи
Angelici R. J., Department of Chemistry, Iowa State
University, Ames, Iowa 50010, USA
1. ВВЕДЕНИЕ
Хотя слово устойчивость использовалось для описания различ-
ных свойств соединений, в данной главе оно применяется к кон-
станте равновесия реакции взаимодействия иона металла (Мто+)*
и лиганда (Ln~) с образованием комплекса
Мт+ + 1Д- —> ML(OT-n)+ (1)
Константа равновесия реакции (1) называется константой устой-
чивости (или иногда константой образования). Обычно для Мт+
zn= + l, +2 или +3. Так как гл. 6 специально посвящена комплек-
сам ионов щелочных металлов, здесь будут рассматриваться глав-
ным образом ионы переходных металлов (например, Fe2+, Fe34-,
Ni24', Cu2+, Zn2+ и т. д.) и в 'меньшей степени ионы лантанидов
(например, Се34-, Sm3+, Gd3+, Но3+, Tm3+, Lu3+ и т. д.). Как пра-
вило, лиганды имеют заряды 0, —1 или —2. Будет рассматри-
ваться только ограниченное число типов лигандов, в частности,
такие, как NiH3, Cl~, Br~, I-, РО4- , РгОГ, аминокислоты, малые
пептиды, нуклеотиды и порфирины. Комплексные соединения с
двумя ионами металла, которые соединены мостиком, состоящим
из одного или более лигандов, здесь также обсуждаться не будут,
хотя такие комплексы известны.
Цель данной главы — указать некоторые основные факторы,
влияющие на константы устойчивости, и дать читателю достаточ-
ное число ссылок на литературу, чтобы помочь ему ознакомиться
с обширной литературой в этой области. Мы не пытались дать
здесь исчерпывающее изложение предмета. Если не указано осо-
бо, использованные в данной главе константы устойчивости отно-
сятся к измерениям, проведенным при 25 °C.
Так как подавляющее большинство исследований реакции (1)
было проведено в водных растворах, то важно указать, что Мт+
* Автор использует обозначения М+т и L~". В отечественной и зарубежной
литературе значительно чаще заряженные ионы записывают как М™+ и L"~.—
Прим. ред.
90
Глава 2
существует в растворе как акво-комплекс. Ионы элементов пер-
вого переходного ряда существуют в растворе в виде [Мп (Н2О)б]2+>
[Fe(H2O)6]2+, [Со(Н2О)6]2+, [Ni(H2O)6]2+ и [Zn(H2O)6]2+. Ак-
тированные формы ионов лантанидов, ino-видимому, представляют
частицы с 9- или 10-кратной координацией иона металла, такие,
как [Ьа(Н2О)9]3+, [Рг(Н2О)9]3+, [Ег(Н2О)9]3+ или [Yb(Н2О)9]3+.'
Таким образом, реакции типа (1) более полно можно написать
как реакции замещения
М(Н2О)™+ + L«- М(НаО)ж_1(Ь)<«-«)+ + Н2О (2)
Если L — бидентатный или тридентатный лиганд, то из коорди-
национной сферы комплекса вытесняются две или три молекулы
Н2О.
Термодинамическую константу равновесия К° реакции (1)
{ML<m-n>+} [ML>-n>+] YML(m-n)+
= {Mm+} {Ln-} = -[Mm+] [Ln-] ’yLn-
можно выразить через активности частиц {ML<’n-’9+}, {М”1} и
{Ln-}. Заметим, что активность воды принимается постоянной и
включается в величину №, так как в разбавленных растворах ак-
тивность воды остается примерно постоянной независимо от поло-
жения равновесия (2). Так как активность частицы 'можно пред-
ставить как произведение ее концентрации на коэффициент актив-
ности, например {Mm+} = [Мт+] у мт+, то уравнение (3) можно
записать, используя концентрации и коэффициенты активностей,
так, как показано с правой стороны уравнения (3). Эксперимен-
тально обычно измеряют концентрации участвующих частиц. Тер-
модинамические константы устойчивости этих реакций можно
определить либо в том случае, когда известны коэффициенты ак-
тивностей, либо путем измерения констант равновесия при не-
скольких значениях ионной силы м последующей экстраполяции
к нулевой ионной силе, при которой коэффициенты активностей
принимаются равными единице. Эти методики либо невыполнимы,
либо очень трудоемки, либо и то и другое вместе, поэтому на
практике лишь относительно немногие исследователи пытались
оценить термодинамические константы устойчивости, и вместо это-
го в уравнениях равновесий просто используются концентрации.
Таким образом, подавляющее большинство опубликованных в
литературе констант устойчивости — это стехиометрические кон-
станты К (иногда их называют концентрационными константами
устойчивости)
[MLl"1-")-*-]
К = [Mm+] [L"-] (4)
в которых отношение коэффициентов активностей [в уравне-
нии (3)] включено в константу К- Единственное важное условие
Устойчивость координационных соединений
91
для определения К состоит в том, чтобы коэффициенты активно-
сти частиц 'были постоянными независимо от состояния равнове-
сия изучаемой реакции. Для поддержания постоянной ионной си-
лы добавляют нереатирующую ионную соль до высокой концент-
рации. Это позволяет поддерживать постоянство коэффициентов
активности частиц, реагирующих в соответствии с уравнением (1).
Желательно, конечно, чтобы эта соль сама не реагировала с час-
тицами, принимающими участие в реакции. Обычно для этой цели
используют LiC104, NaC104 или KNO3. Хлориды наименее удачны,
так как известна тенденция этого аниона координироваться с
ионами металлов в умеренно концентрированных водных раство-
рах. В связи с этим следует упомянуть, что основная компонента
.практически всех буферов также образует комплексы с ионами
металлов различной степени устойчивости.
Хотя постоянная ионная сила позволяет определить надежную
константу устойчивости, эта константа действительна только при
той ионной силе, при которой она была измерена.
Примером константы устойчивости, сильно зависящей от ион-
ной силы, может служить константа, полученная [7] для реакции
хелатообразования Си (II) с двухзарядным фталат-анионом
к
Cu2++ CeH4(CO2)!- [CeH4(CO2)2]Cu (5)
Как и следовало ожидать, стехиометрическая константа устойчи-
вости понижается с повышением концентрации фонового электро-
лита Na2SO4 (табл. 2.1). Зависимость IgK от (р)1/2 представляет
прямую линию в соответствии с предельным законом Дебая—
Хюккеля. Величины К, полученные с другими электролитами, та-
кими, как NaC104, NaNO3 или NaCl, почти идентичны тем, кото-
рые наблюдались при использовании Na2SO4. Так как лишь немно-
Таблица 2.1
Зависимость К для реакции (5)
от ионной силы (р)
Ц 10-з к
0,03 1,84
0,06 1,41
0,12 1,04
0,25 0,80
гие лиганды имеют отрицательный заряд более —2, то большинство
констант устойчивости [8] обладает менее выраженной зави-
симостью от ионной силы по сравнению с замеченной для реак-
92
Глава 2
ции (5). По сравнению с другими эффектами, рассматриваемыми
в настоящей главе, 'влияние ионной силы на константы устойчи-
вости в общем мало.
Так как ионы металлов обычно имеют четыре или более коор-
динационных мест, они часто координируют более чем один ли-
ганд. Последовательная координация лигандов ионом металла
происходит ступенчато, и ступенчатые константы устойчивости мо-
гут быть написаны и определены экспериментально для каждого
из равновесий. Для иона металла, имеющего четыре координаци-
онные места, такого, как Си2+, возможны следующие равновесия:
[ML*]
М2+ + L- ML*; К1 =
ML+ + L ч—ML2; К2 = [ML+] [L~]
МЦ -f- L МЦ"; К3 = [Ml2] [L“]
[ML2-!
МЦ" + L МЦ-; Kt = [ML-j fL-j
Для выражения этих последовательных равновесий использу-
ются также общие константы устойчивости р; они представляют
произведение ступенчатых констант устойчивости, т. е. Pi = Ki, р2=
= KiK2, Рз = Ki К2Кз и р4=К1К27<зК4.
Можно ожидать, что по статистическим [9], стерическим и
электростатическим причинам последовательное присоединение
отрицательно заряженных лигандов к М2+ должно становиться все
менее и менее благоприятным. Действительно, это в общем случае
верно, как, например, для равновесия с участием Си2+ и моноден-
татного лиганда ацетата: lg/Ci (1,67) >lgK2(0,98) >lg/C3(0,42) >
>lg/<4(—0,19). На рис. 2.1 показаны дополнительные примеры
этой тенденции: независимо от природы донорного атома Ki>K2
для реакции взаимодействия различных бидентатных лигандов
с Zn2+.
Различные аспекты устойчивости комплексов были рассмотре-
ны ранее. Обзоры общих экспериментальных методов даны не-
сколькими авторами [8-12]. Опубликованы также более специ-
альные обсуждения определения констант устойчивости методами
калориметрии [13], рН-потенциометрии [14, 15], полярографии
[16], ионного обмена [17—20], жидкостной экстракции [19] и рас-
творимости [21]. Очень полезную сводку констант устойчивости,
опубликованных в литературе до 1968 г., собрали Силлен и Мар-
телл [22], имеется также менее полная сводка Яцимирского и Ва-
сильева [23]. Большинство из опубликованных данных об устой-
чивостях относятся к ионам металлов, которые быстро (в преде-
Устойчивость координационных соединений
93
л ах секунд) реагируют с лигандами [24]. Значительно меньше
известно о равновесиях с участием инертных ионов металлов,
которые менее удобны для исследования. Анализ и обзор тенден-
ций в изменении констант устойчивости проводился многими ав-
торами [5, 8, 9, 23, 25—29]. Зигель и Мак-Кормик [29а] недавно
Рис. 2.1. Величины констант устойчивости
IgKi и IgKs комплексов Zn2+ с некоторыми
бидентатными лигандами при 25 °C [22, 23].
опубликовали краткое, но имеющее отношение к предмету данной
книги описание равновесий взаимодействия ионов металлов с ли-
гандами, имеющими биологическое значение. Энтальпии и энтро-
пии комплексообразования были недавно суммированы и обсуж-
дены в нескольких обзорах {8, 25, 30, 31].
2. ФАКТОРЫ, ВЛИЯЮЩИЕ НА УСТОЙЧИВОСТЬ
КОМПЛЕКСОВ ИОНОВ МЕТАЛЛОВ
2.1. Природа иона металла
2.1.1. Заряд и размер иона металла
Наиболее простое из возможных приближений для понимания
взаимодействия 'между ионом металла и лигандами заключается
в предположении, что связь металл—лиганд чисто ионная или
электростатическая. Так, например, координация иона фтора F-
94
Глава 2
ионом 'металла должна зависеть от зарядов на взаимодействую-
щих ионах и расстояния (г) между их ядрами. Предполагается,
что ионы представляют жесткие сферы
г
с зарядами, локализованными на их ядрах. Вычисленная электро-
статическая энергия такой связи равна £ = e2ZMm+ZF-/r, где е —
заряд электрона, a Z мт+ и Z F— — полные заряды на ионах.
Рис. 2.2. Увеличение величины IgK с увеличе-
нием заряда на ионе металла [22].
Из этого выражения следует, что высокий заряд на Мт+ и ма-
лое межъядерное расстояние (т. е. малый радиус Мт+) должны
благоприятствовать образованию устойчивого комплекса. Конеч-
но, эта простая модель игнорирует энтропию и эффекты сольва-
тации, а также какое-либо ковалентное взаимодействие*. Для
комплексов, образованных сильно электроположительными иона-
ми металлов, такими, как Li+, Mg2+, La3+ и т. д„ и сильно элек-
троотрицательными лигандами, например F" и О2~, ковалентное
взаимодействие мало и преобладающий вклад в энергию связи
дает электростатическое притяжение иона металла и лиганда.
* Эта модель игнорирует даже отталкивание электронных оболочек иоиов,
учитываемое в самых простых формулах. — Прим. ред.
Устойчивость координационных соединений
95
Ионы металлов и лиганды этого типа называются жесткими [или
принадлежащими к классу (а) ] кислотами и основаниями соот-
ветственно. Эта система классификации более детально обсужда-
ется в разд. 2.1.2.
Если рассматриваются только жесткие ионы металлов и ли-
ганды, то в общем верно, что наиболее устойчивые комплексы об-
разуют ионы металлов, имеющие наиболее высокие заряды, при
условии близости их ионных радиусов. Из рис. 2.2 видно, что IgA
для равновесия M’1,++Ln_^ML(m-n>+ увеличивается с увеличени-
ем заряда на ионе 'металла в следующем порядке: Li+(0,68 А) <
<Mg2+(0,65 A)<Fe3+(0,53 А), в скобках приведены ионные ра-
диусы. Следует также отметить, что соответствующее повышение
К с увеличением заряда на лиганде_отсутствует. Поведение ли-
ганда усложняется тем, что ион SO1- может быть как моноден-
татным, так и бидентатным, в то время как Р2О7 > по-видимому,
бидентатен.
Величины К повышаются также при уменьшении размера иона
металла. Так, например, при комплексообразовании ионов щелоч-
ноземельных металлов [22] с пирофосфатным ионом РгО*- вели-
чина К уменьшается при переходе в группе сверху вниз:
Радиус, А igK
Mg2+ 0,65 7,2
Са2+ 0,94 6,8
Sr2+ 1,10 5,4
Ва2+ 1,29 4.6
Эта тенденция иллюстрируется (рис. 2.3) также ионами лантани-
дов (Ln3+), ионные радиусы которых равномерно уменьшаются от
1,06 А для наиболее легкого элемента La3+ до 0,85 А для наиболее
тяжелого элемента Lu3+. На рис. 2.3 приведены кривые, относящие-
ся к константам устойчивости комплексов с анионом гликолевой
кислоты, который координируется бидентатно через атомы кисло-
рода карбоксильной и спиртовой групп, и комплексов с ацетат-
ным ионом. Простая электростатическая модель предсказывает
равномерное повышение IgA от La3+ до Lu3+. Точнее, зависимость
IgA от 1/г должна представлять прямую линию. Однако в дейст-
вительности такая зависимость не является прямой, и даже каче-
ственно отсутствует равномерное повышение IgA- Действительно,
лантаниды ведут себя так, как если бы они состояли из двух
отличающихся друг от друга групп: La3+—Eu3+ и Gd3+—Lu3+. Та-
кое изменение свойств при переходе к Gd3+ объясняли [5] различ-
ными причинами, такими, как энергия стабилизации кристалличе-
ским полем или стерические факторы, но полностью удовлетвори-
тельного объяснения не найдено. Такой же тип зависимости, как
для гликолята, обычно наблюдается для равновесий с участием
96
Глава 2
ионов лантанидов для большого числа других лигандов [5, 32].
Поведение ацетатпроизводных (рис. 2.3) во второй половине серии
лантанидов необычно.
Как наглядно показывает ряд лантанидов, относительно малые
изменения ионных радиусов не всегда приводят к повышению ве-
личин К даже в тех случаях, когда рассмотрение ограничивают
только жесткими ионами металлов и донорными атомами. Все же
Рис. 2.3. Величины lg/С гликолятных (□) и ацетатных (О) комплексов ионов
лантанидов (+3) (20°C, 0,1 М iNaClOj) [5].
значительное уменьшение радиусов, по-видимому, приводит обыч-
но к образованию более устойчивых комплексов. Влияние повы-
шения положительного заряда на ионе металла яснее: для жест-
ких ионов 'металлов и донорных атомов лигандов устойчивость
комплекса повышается при увеличении положительного заряда на
ионе металла.
2.1.2. Жесткая и мягкая природа ионов металлов
и донорных атомов лигандов
Были предприняты многочисленные попытки корреляции кон-
стант устойчивости со свойствами ионов металлов и лигандов. Как
было указано в разд. 2.1.1, ограниченная корреляция с зарядом и
размером иона металла наблюдается. Однако при рассмотрении
Устойчивость координационных соединений
97
широкого диапазона ионов металлов и лигандов становится ясно,
что существует очень мало корреляций. Более того, имеется мно-
го примеров обращения тенденции. Например, константы устойчи-
вости галогенидных комплексов Fe3+ понижаются в ряду F~>
>С1~>Вг_>1~. В противоположность этому Hg2+ образует более
устойчивые комплексы с 'более тяжелыми галогенидами, и К по-
нижаются следующим образом: I->Br->Cl~>F- Из этого
и многих других примеров Шварценбах [33], а затем Арланд,
Чатт и Дэвис [34] пришли к заключению, что по отношению к
ионам галогенидов некоторые 'металлы ведут себя подобно Fe3+,
а другие — подобно Hg2+. Первые были отнесены к классу (а),
а вторые — к классу (б) ионов металлов или кислот. Сравнивая
константы устойчивости комплексов других лигандов с различ-
ными ионами металлов, можно дать более полную характеристи-
ку двух классов ионов металлов. В табл. 2.2 приведены последо-
вательности констант устойчивости, которые обычно наблюдают-
ся для ионов металлов классов (а) и (б) с различными донорны-
ми атомами лигандов. Для классификации ионов металлов были
также важны термодинамические параметры [31] этих реакций.
В табл. 2.3 приведены ионы металлов, принадлежность кото-
рых к этим классам была установлена экспериментально [35].
Следует указать, что существуют металлы, подчиняющиеся неко-
торым тенденциям класса (а) и некоторым тенденциям класса (б)
ионов металлов; они располагаются в пограничной области этой
классификации. Можно заметить, что многие из ионов металлов,
находящихся в пограничной области, — это двухзарядные (+2)
ионы переходных металлов.
Таблица 2.2
Определение жестких [класс (а)] и мягких [класс (б)]
ионов металлов (в соответствии с порядком изменения
устойчивости комплексов с различными донорными
атомами лигандов)
Изменение устойчивости комплексов жестких, или класса (а), ионов металлов Изменение устойчивости комплексов мягких, или класса (б), ионов металлов
F>Cl>Br>I F<Cl<Br<I
О> S>Se>Te O<S~Se~Te
N»P>As>Sb N«CP>As>Sb
При рассмотрении ионов металлов класса (а) обнаруживает-
ся, что их свойства во многом сходны. Они имеют малые радиусы
и высокие положительные заряды, но обычно не имеют неподелен-
ных электронных пар в их валентной оболочке (а многие из них
7—2451
98
Глава 2
Таблица 2.3
Классификация ионов металлов
Жесткие, или класс (а)
Пограничная область
Мягкие, или класс (б)
Н+, Li+, Na+, к+
Be2*, Mg2*, Ca2+, Sr2*
Mn2+, Al3*, Sc3*, Ga3*
In3+, La3*, Gd3*, Lu3*
Cr3*, Co3*, Fe3+, Si4*
Ti4+, Sn4+, WO4*, VO2+
Fe2+, Co2*, Ni2*
Cu2+, Zn2+, Pb2+
Sn2+, Sb3+, Bi3*
Cu+, Ag+, Au+, T1+
Hg2+, Hg2+, Pd2+, Pt2*
Pt4*, Tl3*
обладают электронной конфигурацией благородного газа). Вслед-
ствие малых размеров и высоких положительных зарядов их
поляризуемость низка; по этой причине они были названы [35]
«жесткими». Наиболее прочные комплексы эти ионы металлов
образуют с очень электроотрицательными атомами, такими, как О,
N и F (табл. 2.2). Связь в этих комплексах преимущественно ион-
ная.
Ионы металлов класса (б) значительно больше; они имеют бо-
лее низкие положительные заряды и неспаренные валентные р-
или d-электроны. Благодаря этим свойствам ионы металлов клас-
са (б) в большей степени поляризуемы, вследствие этого они бы-
ли названы «мягкими». Наиболее устойчивые комплексы эти ионы
металлов образуют с донорными атомами лигандов (Р, S и I),
имеющими низкие электроотрицательности и высокую поляризуе-
мость. Связи в этих комплексах содержат значительный ковалент-
ный вклад.
Как было указано выше, жесткие ионы металлов образуют
наиболее прочные комплексы с донорными атомами (N, О и F),
которые обладают высокими электроотрицательностями, низкой
поляризуемостью, малыми радиусами и трудно окисляются. Бла-
годаря этим свойствам они были обозначены как жесткое основа-
ние Льюиса или жесткие донорные атомы. С другой стороны, те
атомы лигандов (Р, S и I), которые образуют наиболее устойчи-
вые комплексы с .мягкими ионами металлов, имеют большие раз-
меры, низкие электроотрицательности, высокие поляризуемости и
относительно легко окисляются; они были названы мягкими ли-
гандами. Наиболее типичные жесткие, мягкие и промежуточные
лиганды приведены [35] в табл. 2.4.
В терминах нашей новой номенклатуры тенденции табл. 2.2
можно суммировать следующим образом [40]: жесткие ионы ме-
таллов предпочитают координировать жесткие лиганды, а мягкие
Устойчивость координационных соединений
99
Таблица 2.4
Классификация лигандов
Жесткие, или класс (а) Промежуточные Мягкие, или класс (б)
Н2О, ОН-, F-, С1- СН3СО2“, РОГ. SO|- coj-, сюг, no; ROH, R2O, NH3 rnh2, n2h4 c6h5nh2, c6h5n n;, Br-, no; so2-, n2 R2S, RS, I- SCN-, S2O|- R3P, (RO)3P, R3As CN-, RNC, CO H-, R-, C2H4
ионы металлов предпочитают координировать мягкие лиганды.
Хотя были 'предприняты многочисленные попытки [35—-38] теоре-
тического обоснования этого правила, его следует рассматривать
как эмпирическое. Путем полуэмпирической обработки [39] с ис-
пользованием ионных радиусов, зарядов, потенциалов ионизации
и электроотрицательностей ионов металлов был получен количе-
ственный параметр мягкости иона металла. Наряду с другими
параметрами иона металла и лиганда он коррелируется с доволь-
но большим числом констант устойчивости комплексов металла.
В коррелировании свойств мягкости и жесткости было довольно
успешным и более теоретическое приближение [40] *.
Это правило полезно для качественного описания взаимодейст-
вий в биологических системах. Пирсон [41] указал, что биоло-
гические комплексы состоят преимущественно из жестких ионов
металлов и лигандов. Преобладающие донорные атомы лиган-
дов — кислород и азот, .а жесткие ионы щелочных и щелочнозе-
мельных металлов присутствуют в изобилии. Конечно, имеются
также относительно мягкие ионы металлов (например, Си2+)**
и лиганды (например, лиганды с донорными атомами серы), но
они относительно неподвижны и концентрация их низка. Пирсон
также заметил, что соединения, представляющие яды для живых
организмов, часто относятся к классу мягких, например такие ли-
ганды, как СО, CN- и H2S, и такие ионы металлов, как Hg2+. Ес-
ли эти мягкие ядовитые частицы присутствуют в относительно
высоких концентрациях, то они взаимодействуют с мягкими иона-
ми металлов и лигандами, препятствуя таким образом выполне-
нию их функций. Хотя несомненно, что эти обобщения не могут
быть применены ко всем аспектам рассмотрения живых организ-
* См. также Яцимирский К. Б., Ж- анал. хим., 6, 211 (1951); ЖФХ, 25, 221
(1951); Теор. эксп. хим., 6, 462 (1970). — Прим. ред.
** И в особенности Cu+ Cd2+, а также токсичные ионы Hg2+ и РЬ2+.—
Прим. ред.
7*
100
Глава 2
мов, они представляют еще одну исходную точку для исследова-
ния этих исключительно сложных систем.
Принцип жесткости и мягкости можно применить к координа-
ции ионов переходных металлов к нуклеотидди- и нуклеотидтри-
фосфатам [42]. Жесткие ионы металлов, такие, как щелочнозе-
мельные и Мп2+, координируются главным образом через атомы
кислорода фосфатных групп, в то время как более мягкий иои
Си2+ координируется как к мягкому атому азота ароматического
пуринового или пиримидинового основания, так и к_ фосфатной
группе. Подобным образом лиганд СНзСНгЭСНгСОг координи-
руется к относительно жестким ионам металлов Мп2+ и Zn2+ только
через атом кислорода, в то время как ион Си2+ образует хелат,
координируясь как через атом кислорода, так и через мягкий атом
серы [43].
Наше рассмотрение предполагает, что молекулы воды занима-
ют .координационные места, не занятые другими лигандами. Как
было указано [35], природа лигандов, присоединенных к иону
металла, влияет на его жесткость и мягкость. По-видимому, жест-
кие лиганды делают лиганд еще более жестким, а мягкие — более
мягким по отношению к дополнительным лигандам. Так, напри-
мер, ионы металлов, связанные с серусодержащими лигандами
или относительно мягкими порфириновыми кольцами, будут про-
являть большую тенденцию к образованию комплексов с допол-
нительными мягкими лигандами по сравнению с гидратированным
ионом металла ['М(Н2О)6]2+, связанным с молекулами воды, ко-
торые относятся к жестким лигандам. Именно влиянием лигандов
можно объяснить тот факт, что железо в геме столь прочно коор-
динирует такие мягкие лиганды, как СО и СЫ~, даже в присутст-
вии большого количества жесткого лиганда воды. Этим можно
объяснить такое высокое сродство Fe2+ (и, по-видимому, даже
Fe3+) к сере в негеминовых железо-серусодержащих белках
[44, 45].
2.1.3. Последовательность Ирвинга—Уильямса устойчивостей
комплексов ионов металлов первого переходного ряда
Как было отмечено в предыдущем разделе, ионы металлов пер-
вого переходного ряда по их жесткости и мягкости занимают про-
межуточное положение. По этой причине часто трудно предска-
зать устойчивости их комплексов. Однако в течение многих лет
было известно [46—48], что константы устойчивости двухзаряд-
ных ионов этих элементов с данным лигандом изменяются в соот-
ветствии с последовательностью Mn2+<cFe2+<Co2+<Ni2+<Cu2+>
>Zn2+. Эта последовательность часто называется порядком устой-
чивостей Ирвинга—Уильямса. Для нескольких лигандов она по-
казана на рис. 2.4. Эта последовательность, как можно видеть из
Устойчивость координационных соединений
101
рис. 2.5, сохраняется не только для Ki, но и для К2, а также в
большинстве случаев и для К3. Аномально низкое значение Кз для
Си2+ объясняют тем, что этот ион проявляет 'слабую тенденцию ,к
Рис. 2.4. Величины lg/С комплексов иоиов металлов первого переходного ряда
(М2+) с некоторыми бидентатными лигандами при 25 °C [22].
координации более четырех донорных атомов. Хотя, по-видимому,
последовательность Ирвинга—Уильямса справедлива для К2, Кз
и т. д. и в других системах, для подтверждения этого отсутствует
достаточное количество надежных данных.
Для лигандов, содержащих донорные аминогруппы, порядок
устойчивостей Ирвинга—Уильямса определяется главным обра-
102
Глава 2
зом энтальпиями (АД) реакций. Так, величины АН для этиленди-
амина и глицина отрицательны и ожидавшаяся последователь-
ность устойчивостей наблюдается (рис. 2.6). В противоположность
этому реакции лигандов с донорными карбоксильными труппами
Рис. 2.5. Величины IgXi, lgK2 и lg/C3 комплексов ионов металлов первого пере-
ходного ряда (М2+) с этилендиамином (NH2CH2CH2NH2) при 30 °C [22].
слегка эндотермичны, и энтропия (AS) вносит существенный вклад
в общий порядок Ирвинга—Уильямса для констант устойчивости.
Так как последовательность Ирвинга—-Уильямса определяет,
по крайней мере во многих случаях, АН взаимодействия между
ионом металла и лигандом, то попытки объяснения наблюдаемых
устойчивостей -были направлены на рассмотрение энергий связей
металл—лиганд. Хотя при использовании электростатической мо-
дели (разд. 2.1.1) для объяснения наблюдаемой шоследователь-
ности использовались изменения ионных радиусов ионов металлов,
эти изменения в действительности слишком малы для соответст-
вующего объяснения больших изменений величин К. Другое при-
ближение [51] включало корреляцию констант устойчивости с по-
тенциалами ионизации ионов -металлов.
Недавние успехи теорий кристаллического поля и поля лиган-
дов в объяснении термодинамики и кинетики реакций [24] комп-
лексов переходных металлов стимулировали использование этих
Устойчивость координационных соединений
103
теорий для объяснения порядка устойчивости комплексов Ирвин-
га—Уильямса [52]. Вычисленные энергии стабилизации кристал-
лическим полем вместе с энергиями электростатической связи ка-
чественно объясняют последовательность Ирвинга—Уильямса для
Рис. 2.6. Величины АД реакций где Ь=этилендиамин (•), глици-
нат (О) или малоиат (□), при 25°C [49, 50].
высокоспиновых комплексов ионов этих металлов. Хотя ковалент-
ное взаимодействие, энергии сольватации и энтропии в этих вы-
числениях не были учтены, возможность качественного согласия
не является неожиданной*.
Последовательность Ирвинга—'Уильямса справедлива для ог-
ромного большинства лигандов, однако существуют некоторые ис-
ключения. Во-первых, когда лиганд имеет донорные атомы, кото-
* Для оценки величины констант устойчивости комплексов, образованных
ионами близких размеров (г=0,8—1,0 А), можно использовать предложенную
нами линейную корреляцию логарифмов констант устойчивости (см. Яцимир-
ский К. Б., Введение в биоиеорганическую химию, «Наукова думка», Киев, 1976,
стр. 35). — Прим. ред.
104
Глава 2
рые могут все координироваться в октаэдрическом комплексе (на-
пример, Ni2-*-), ио не .все в квадратно-плоскостном ком-
плексе (Си2+). Так, например, К\ комплекса Си2+ с шести-
дентатным лигандом ЭДТА [этилендиаминтетраацетат
(^O2CCH2)2NCH2CH2N (СН2СОа )s] только немного больше, чем
для Ni2+. Медь неспособна прочно координировать все шесть до-
норных атомов. .Во-вторых, когда лиганды, характеризующиеся
высокой силой поля, изменяют состояние иона металла от высоко-
спинового к низкоспиновому. Это в основном имеет место для
комплексов Fe2+ с такими лигандами [53], как о-фенантролин,
2,2'-дипиридил и цианид. При этом образуются необычно устой-
чивые комплексы. Подобные изменения в порядке устойчиво-
стей наблюдаются для двух лигандов, представляющих интерес
для биохимии, — имидазола и гистамина. Тенденцию к образова-
нию исключительно устойчивых комплексов с Fe2+, кажется, наи-
более обычно проявляют лиганды, содержащие донорные атомы
ароматического гетероциклического азота.
2.2. Природа лиганда
2.2.1. Основность лиганда
Как было отмечено в разд. 2.1.1, электростатическая модель
химических связей в комплексах ионов металлов имеет ограничен-
ное применение для объяснения последовательности констант
устойчивости с различными ионами металлов и почти непримени-
ма для объяснения последовательностей изменения устойчивости
в зависимости от природы лиганда. Это не является неожиданным,
так как сольватация, энтропия и ковалентное взаимодействие
имеют большое значение. Поэтому были изучены корреляции дру-
гих свойств лиганда с константами устойчивости. Одной из наи-
более успешных была корреляция с основностью (рКа) лиганда
'[54]. При этом исходили из того, что и Н+, и ионы металла
Мт+ взаимодействуют как кислоты Льюиса с лигандами, пред-
ставляющими основания Льюиса. В качестве меры основности
лиганда (L~) по отношению к протону принят рКо (т. е. —1g Ка)
для реакции HL^H+-j-L" в водном растворе; чем выше рКа, тем
более сильным основанием является данный лиганд (L~) по от-
ношению к Н+ и, предположительно, также к М’п+. Действитель-
но, часто наблюдается линейная корреляция между рКа и лога-
рифмом константы К реакции комплексообразования.
На рис. 2.7 показано несколько таких корреляций ,[50], в
которых кислотами Льюиса являются [Cu(dipy)]2+, Cu2+,
[Zn(dipy)]2+ и Zn2+. Лиганды — монодентатные карбоксилатные
анионы RCO-. Как указано на рисунке, некоторые из этих ли-
Устойчивость координационных соединений
105
гандов — производные аниона ароматической бензойной кислоты,
в то время как другие (II, V и VII) к ним не относятся. Из ри-
сунка видно, что в случае Си2+ константы устойчивости хорошо
коррелируются с величинами рКа как ароматических, так и не-
ароматических карбоксилатов. С другой стороны, только арома-
тические бензоаты включены в корреляции с Zn2+ и [Zn(dipy)]2+
и только неароматические карбоксилаты — в корреляцию с
Рис. 2.7. Зависимость IgK для реакций M2++RCO7 =₽tRCO;iM+ от рКо RCOOH
[где Re=n-NO2C6H" (I); _ Н- (II); м-С1С6Н7 (III); C6H“(IV); СЩ (V);
п-СНзСеНГ (VI); СН3СНГ (VII)] в водно-диоксановом (50:50) растворителе
при 25 °C [55].
[Cu(dipy) ]2+. Это было сделано потому, что в случае ароматиче-
ских и неароматических карбоксилатов прямые имеют несколько
различные наклоны и обе группы карбоксилатов невозможно рас-
положить на одной и той же прямой. Для таких корреляций рК„
с 1g К в общем верно [56], что чем ближе структуры лигандов
в сериях, тем лучше корреляции. Для лигандов, имеющих одина-
ковые донорные атомы, но совершенно различное строение, как,
например, NH3, анилин, пиридин и имидазол, корреляции настоль-
ко неудовлетворительны, что не имеют никакой ценности для
предсказания неизвестных констант устойчивости из величин рКа.
Для лигандов, имеющих различные донорные атомы, таких,
как RNH2, RCO”, RS-, корреляция с рКа протонированного лиган-
да также отсутствует. С точки зрения приведенного в разд. 2.1.2
106
Плава 2
обсуждения, касавшегося сопоставления жесткости и мягкости
ионов металлов и донорных атомов лигандов, какой-либо корре-
ляции 1g К с рКа для различных донорных атомов и нельзя было
ожидать. Для того чтобы учегть колебания в жестком и мягком
характере ионов металлов и лигандов, в качестве эталона были
использованы и другие кислоты, кроме Н+. Так, в качестве меры
донорных свойств лигандов (L-) использовали константы устой-
чивости (1g К) их комплексов (ML) с ионом металла, выбранным
в качестве стандарта. Эти величины 1g К коррелировали [57, 58]
с константами устойчивости комплексов M'L, используя другой ион
металла (Мх) с сериями лигандов (L~) с различными донорными
свойствами. Такие зависимости, как показано на рис. 2.7, подчи-
няются уравнению
lgK = apKo + & (6)
Наклон а прямых для всех ионов металлов на рис. 2.7 примерно
равен 0,5. Хотя положительный наклон указывает, что более ос-
новные лиганды образуют более устойчивые комплексы, наличие
Н+ в водных растворах означает, что более основные лиганды
будут более сильно связываться с Н+.
Итак, в растворе существуют два равновесия:
Н+-J-L~ <=>: HL, 1/Ка (7)
Mm+ -J- L- ML/"1-1)-*-, К (8)
После вычитания уравнения (7) из уравнения (8) получаем урав-
нение (9), соответствующее равновесию, доминирующему в рас-
творе при низких pH, где концентрация L~ очень мала.
HL + Mm+ ML('"-1)+ + H+, КаК (9)
Таким образом, константа равновесия для реакции (9) равна
Ка/С При использовании величин_Ла и К из рис. 2.7 для реакции
Си2+ со слабым основанием НСО и сильным основанием СН3СО2
получаем следующие величины конста_нт равновесия для реакции
(9): (10-4-75) (102-8) = 10-1-95 для НСО2 и (IQ-601) (103-36) = IO-2-65
для СН3СО2. Таким образом, константы равновесия реакции
комплексообразования .[уравнение (9)] действительно меньше для
более основных лигандов, т. е. формиат будет связан в комплекс
с Си2+ в большей степени, чем ацетат. Необходимо помнить, что
количество комплекса, действительно присутствующего в водном
растворе, будет зависеть не только от константы устойчивости
комплекса, но также от рКа лиганда.
В случае сильноосновных лигандов иногда необходимо путем
добавления ОН- сделать раствор относительно щелочным для то-
го, чтобы обеспечить достаточную концентрацию непротонирован-
ного лиганда для комплексообразования с ионом металла. Иногда
необходимая концентрация ОН- превосходит произведение раство-
Устойчивость координационных соединений
107
римости гидроксида металла М(ОН)2 и он осаждается из раство-
ра, что исключает возможность образования комплекса. Это имеет
место в реакциях Ni2+ и Zn2+ с 1,1,7,7-тетраэтилдиэтилентриами-
ном; этот лиганд — очень сильное основание, но не очень сильный
лиганд. При тех значениях pH, при которых присутствует доста-
точное количество непротонированного триамина, пригодного для
координации с ионом металла, концентрация ОН- достаточно вы-
сока для осаждения гидроксида металла М(ОН)2. Осаждение
гидроксидов препятствует комплексообразованию этих ионов ме-
таллов с указанным лигандом в водном растворе. С другой сто-
роны, Си2+ образует с этим лигандом достаточно устойчивый
комплекс, чтобы его можно было исследовать [59]. Растворимо-
сти гидроксидов металлов приведены в работе {60].
Для серий лигандов, подчиняющихся уравнению (6), если
а<1, количество действительно присутствующего в растворе комп-
лекса иона металла с лигандом меньше для более основных ли-
гандов; если а=1, количество комплекса не изменяется с повыше-
нием рКа лиганда; если а>\, то при повышении рЛа лиганда
также увеличивается количество комплекса иона металла в рас-
творе.
Хотя представляло бы интерес узнать, какие свойства ионов
металлов и лигандов определяют величины а, в настоящее время
для этого недостаточно данных. Некоторые авторы {61, 62] пы-
тались объяснить те величины, которые известны, но, по-видимо-
му, не пришли к общим выводам. Величины а, кажется, лежат в
пределах [62] от 0,5 (как на рис. 2.7) до 1,5, многие близки
к 1,0. Как найдено для Си2+ и Zn2+, показанных на рис. 2.7, ве-
личины а для ионов металлов первого переходного ряда очень
близки, но больше, чем для Mg2+ или Са2+.
Такие графические зависимости 1g К от рКа были особенно
ценны для установления координации дополнительных групп ли-
ганда. Например, лиганды — производные ацетата (НОСН2СО2 и
CH3CH2SCH2CO2)— образуют с Си2+ более устойчивые комплек-
сы, чем можно ожидать, исходя из их величин рКа [43]. Это
указывает на то, что атом кислорода гидроксильной группы и
атом серы в этих двух лигандах координируются к иону металла,
образуя комплексы необычно высокой устойчивости. Подобным
образом было показано, что атом серы CH3CH2SCH2CO7 не ко-
ординируется к Мп2+ или Zn2+.
Получены корреляции констант устойчивости комплексов раз-
личных ионов металлов с производными иминодиуксусной кисло-
ты RN(CH2CO2)2 с рКа диссоциации Н+ от атома азота лиганда
[32, 63]. Для того чтобы установить зависимость между 1g К и
рЛа, использовали некоординирующиеся группы R, такие, как СН3,
(СН3)3С и С6Н5. Затем константы устойчивости комплексов с ли-
гандами, содержавшими потенциальные донорные атомы, были
108
Глава 2
сопоставлены с полученной зависимостью 1g Д’ от рДа. Были ис-
пользованы следующие группы R: НОСН2СН2—, СН3ОСН2СН2—,
HSCH2CH2—, -SCH2CH2— NH2CH2CH2—, -о2ссн2—
NH2(O)CCH2—, N=CCH2— и (CH3)3N+CH2CH2—. В зависимости
от природы иона металла и донорного атома в группе R некото-
рые константы устойчивости были намного больше, чем можно
было ожидать на основании значений рДо лиганда. Было предпо-
ложено, что это следствие наличия некоторого взаимодействия
между донорными атомами групп R и ионами металлов. Было
найдено, что группы R, содержащие мягкие донорные атомы (на-
пример, атом серы), координируются мягкими атомами металлов
(например, Си2+), а жесткие донорные атомы (например, О) —
жесткими ионами металлов (например, Mg2+) (разд. 2.1.2). В ре-
зультате подобного рассмотрения [64] констант устойчивости
комплексов C2HsO2CCH2N(CH2CO2)2 с различными ионами ме-
таллов был сделан вывод об отсутствии взаимодействия между
ионом металла и эфирной группой. Эти данные были использованы
для интерпретации механизма гидролиза эфиров, катализируемого
ионами металлов.
2.2.2. Хелатный эффект
Образование хелата. Экспериментально установлено, что комп-
лексы хелатообразующих лигандов более устойчивы, чем комплек-
сы аналогичных монодентатных лигандов. Например, константа
комплексообразования Ni2+ с этилендиамином (ей) больше, чем
константа комплексообразования с двумя молекулами NH3
Ni2+ + еп [Ni(en)]2+, 1gК = 7,5 (10)
NJ2+ + 2NH3 :<—> [Ni(NH3)2]2+, 1g К = 5,0 (11)
Комбинируя эти уравнения, получаем
[Ni(NH3)2]2++en [Ni(en)]8++2NH3, 1gЯ = 2,5 (12)
ДЯ°=—1,9 ккал/моль, Д5°=+6,2 кал-моль-1-град-1.* Этиленди-
амин и NH3 можно сравнивать, так как величины их рКо (9,6 для
H2NCH2CH2NH3 и 10,0 для NHJ) очень близки, и различие кон-
стант устойчивости нельзя объяснить их различной основностью
(разд. 2.2.1). Это повышение устойчивости хелатов по сравнению
с устойчивостью комплексов с аналогичными монодентатными ли-
гандами известно как хелатный эффект [65].
Ясно, что в случае реакции, включающей замещение двух ли-
гандов NH3 этилендиамином [уравнение (12)], как отрицательное
значение ЛН°, так и положительное значение Д5° дают вклад в
* Данные для ЛН° и Д5° автор приводит без ссылки на первоисточник. —
Прим. ред.
Устойчивость координационных соединений
109
увеличение устойчивости хелатного комплекса. Образование 3 мо-
лей продукта по сравнению с 2 молями вступающих в реакцию
веществ должно давать положительную поступательную энтропию,
которая дает вклад в общую положительную величину Д5°. Та-
ким образом, хелатный эффект, по крайней мере частично, явля-
ется следствием благоприятного изменения энтропии реакции.
Следует, однако, заметить, что величина Д£° зависит от стандарт-
ного состояния, использованного для ее оценки. Если использу-
ются мольные доли вместо обычных одномоляльных стандартных
состояний, то вычисленные энтропии для такой реакции, как (12),
будут ниже, фактически — очень близки к нулю. Таким образом,
использование этого стандартного состояния приводит к умень-
шению эффекта поступательной энтропии, возникающего из обра-
зования 3 молей продукта из 2 молей реагирующих веществ. Бо-
лее того, зависимость Д5° от выбора стандартного состояния де-
лает трудной количественную интерпретацию величин Д5°.
Следует также отметить, что величина ДЯ, соответствующая
уравнению (12) (отрицательная), благоприятствует хелатообра-
зованию. Это обычно объясняют отталкиванием между двумя до-
норными атомами, когда они приближаются друг к другу при
образовании комплекса. Взаимное отталкивание двух групп NH2
этилендиамина имеет место уже в свободном лиганде до комплек-
сообразования, и при образовании комплекса возникает только
небольшое дополнительное отталкивание. С другой стороны, меж-
ду молекулами NH3 в растворе отталкивания нет, но они начина-
ют отталкивать друг друга при комплексообразовании. Это пре-
имущественно электростатическое отталкивание групп NH3 в
комплексе делает ДЯ° менее благоприятным. Независимо от того,
как объясняют изменение ДЯ° при комплексообразовании, оно
так же, как и Д5°, вносит существенный вклад в хелатный эф-
фект [66].
Размер хелатного кольца. В общем устойчивость хелатного
комплекса уменьшается [66] при увеличении размера хелатного
кольца от пятичленного к шести- и семичленному. Поэтому в ряду
комплексов дикарбоновых кислот с ионом металла наиболее
устойчивый комплекс образует оксалатный анион:
Данные, приведенные на рис. 2.8, иллюстрируют понижение
устойчивости пяти-, шести- и семичленных хелатных комплексов
ионов металлов первого переходного ряда с анионами щавелевой,
малоновой и янтарной кислот. Данные об устойчивости ряда
других пяти- и шестичленных хелатных комплексов приведены
в табл. 2.5.
110
Глава 2
В соответствии с данными этой таблицы можно отметить не-
сколько тенденций*. Во-первых, 1g Л для пятичленных колец на
1,1—1,5 единиц больше, чем для шестичленных. Во-вторых, раз-
личие величин 1g К. обусловлено как изменением ДЯ, так и Д5.
Рис. 2.8. Зависимость (при 25 °C) от размера хелатного кольца, образуемого
бидентатпым лигандом: оксалат (Ох2-), малоиат (Ма122-) и сукцинат (Sue2-) [8].
В-третьих, реакции становятся более экзотермичными в следую-
щем ряду лигандов: дикарбоксилат<аминоацидат<диамин. Ве-
личина АН, связанная с координацией Ni2+—карбоксилат, близка
к нулю или слегка положительна, в то время как координация
аминогруппы дает отрицательную величину АН. Таким образом,
ДЯ реакции более сильно стабилизирует комплексы с лигандами,
содержащими донорные аминогруппы, по сравнению с лигандами,
содержащими карбоксильные группы. В-четвертых, устойчивость
карбоксилатных комплексов определяется главным образом боль-
шими положительными энтропиями. Они заметно уменьшаются
при переходе от дикарбоксилатных лигандов к диаминам. Высо-
кие положительные значения Д5 для реакций карбоксилатов, по-
видимому, частично являются следствием отщепления сольвати-
рующих молекул воды от высокозарядных ионов Ni2+ и Ох2- (или
* Общая тенденция к увеличению прочности пяти- и шестичлениых циклов
была отмечена еще Л. А. Чугаевым и известна в отечественной литературе под
названием «правила циклов Чугаева». — Прим. ред.
Устойчивость координационных соединений
111
Таблица 2.5
Устойчивость пяти- и шестичленных хелатных комплексов*
при 25 °C [8, 50]
Ni2+ + Ln" NiL<2-n>+
Ni2+ + L«- IgK AAf, ккал/моль AS, кал моль—i • град—1
Ni2+ + Ox2- 5,2 0,15 24,2
Ni2+ + Mal2- 4,1 1,88 25,0
Ni2+-]-Gly- 6,2 —4,14 14,4
Ni2+ + p-Ala- 4,7 —3,81 10,2
Ni2+ + en 7,7 —9,05 4,0
Ni2+ -|- pn 6,3 — —
а Сокращения: оксалат (Ох*-); малонат (Mai2-); глицинат (Gly~); ₽-аланинат (Р-А1а—);
этилендиамин (еп); 1,3-пропилендиамии (рп).
Таблица 2.6
Устойчивость комплексов полидеитатных лигандов*
с Ni2+ при 25 °C
Ni2+ L«- <—> NiL<2-n>+
Ni2+ + l"“ IgK АН, ккал/моль AS, кал • моль—1 • град—1
Ni2+ + Gly- 6,2 —4,14 14,4
Ni2+ + IMDA2- 8,0 —5,05 20,0
Ni2+-]-NTA3- 11,3 —2,53 44,0
Ni2+ -|- en 7,7 —9,05 4,0
Ni2+ + dien 10,5 —11,85 8,5
Ni2+ + trien 13,7 —14,00 16,0
а Сокращенные обозначения лигандов см. в подписи к рис. 2.9.
Mai2-) при образовании нейтрального комплекса NiOx. В случае
моноаминоацидатов имеет место нейтрализация меньших зарядов.
Число хелатных колец. Если принять во внимание хорошо уста-
новленный хелатный эффект для бидентатных лигандов, то не
является неожиданным тот факт, что лиганд с большим числом
хелатных колец будет давать еще более устойчивые комплексы.
При этом предполагается, что геометрия лиганда и иона металла
разрешают координацию всех донорных атомов. Из рис. 2.9 вид-
112
Глава 2
но, что 1g Л для ряда аминокарбоксилатных лигандов повышается
по мере увеличения числа хелатных колец и донорных атомов:
GIy“<IMDA2-<NTA3-. Подобным образом для аминных лигандов
константы устойчивости повышаются в ряду en<dien< trien.
Рис. 2.9. Увеличение IgA (при 25 °C) с увеличением числа хелатных колец, образуе-
мых полидентатным лигандом: NH2CH2CO2 (Gly-), NH(CH2CO2) (1MDA2-),
N(GH2CO2")3 (NTA3-), NH2CH2CH2NHCH2CH2NHCH2CiH2NH2 (trien) [8].
В табл. 2.6 приведены значения АН и AS для комплексов этих
лигандов с Ni2+.
По мере увеличения числа донорных атомов в лиганде повыша-
ется AS комплексообразования. Этого следовало ожидать, так как
при этом увеличивается число молекул воды, которые лиганд вы-
Устойчивость координационных соединений
113
тесняет из координационной сферы Ni2+. В дополнение к стаби-
лизации комплекса энтропией с увеличением числа донорных
групп в амине величина АН становится более отрицательной.
В противоположность этому АН для аминокарбоксилатных комп-
лексов изменяется лишь слегка и немонотонно, что, возможно,
происходит вследствие отмеченной ранее тенденции карбоксилат-
ных лигандов давать близкие к нулю или положительные энталь-
пии координации. Эти два ряда лигандов также подтверждают
сделанное ранее наблюдение, что донорные атомы азота в аминах
дают намного большие вклады в АН комплексообразования, чем
карбоксильные донорные группы; с другой стороны, высокие по-
ложительные величины AS координации аминокарбоксилатных
лигандов обусловлены главным образом карбоксильными груп-
пами. Как будет отмечено ниже, эти тенденции особенно важны
для комплексообразования ионов металлов с аминокислотами.
2.2.3. Макроциклический эффект*
В то время как хелатный эффект известен уже в течение де-
сятилетий, устойчивость комплексов, содержащих макроцикличе-
ские лиганды типа порфиринов, была в сущности неизвестна.
Основная проблема при определении их констант устойчивости —
очень медленная скорость взаимодействия ионов металлов и мак-
роциклических лигандов.
Недавно Каббинесс и Марджерум [67] определили константу
образования комплекса Си2+ с нециклическим тетраминным ли-
гандом (2,3,2-tet, I) и аналогичным макроциклическим тетрамин-
ным лигандом (teta, II).
Н2
Н2С<
Н\ I
Н2С---N
н Н2 СН3
НЭС»>С<С^С—СН3
Н2С---N
Н2С---N
Н2
GH2
N---СН2
Н2С—-N
N---СН2
СН3—С.
.с.сн3
N^—СН2
СН3Н2 Н
I
2,3,2-tet
tet а
* В последнее время появилась обширная литература об устойчивости раз-
личных комплексов с макроциклическими лигандами. Gm., например, Lehn J. М.,
Structure and Bonding, 16, 3 (1973) и другие статьи в этом сборнике. — Прим,
ред.
8—2451
114
Глава 2
Из-за медленной скорости реакции с teta определение кон-
станты устойчивости было проведено при 50, 75, 100 и 130 °C.
Для сравнения с другими данными путем экстраполяции были
получены константы устойчивости при 25 °C. Эти константы для
2,3,2-tet и trien (триэтилентетрамин) даны в табл. 2.7.
Таблице 2.7
Величины 1g К комплексов Си2+
с тетраминиыми лигандами
при 25 °C [47]
Комплекс igK
Cu2+ -|- tet а 28
Cu2+ +2,3,2-tet 23,9
Cu2+ -|- trien 20,1
Константа устойчивости (1g К) [Cu(teta)]2+ на 4,1 логарифми-
ческой единицы больше, чем константа нециклического
[Си (2,3,2-tet) ]2+. Добавление мостика —СН2СН2СН2—, соединяю-
щего концевые группы —NH2 2,3,2-tet с образованием teta, повы-
шает устойчивость комплекса в большей степени, чем этого можно
было ожидать только из хелатного эффекта. Хотя о происхожде-
нии этой повышенной устойчивости известно мало, ее называют
макроциклическим эффектом. Почти с определенностью можно
сказать, что в высокую устойчивость [Cu(teta)]2+ по сравнению
с нециклическим лигандом, кроме благоприятного связывания в
комплекс с металлом, вносят вклад конфигурация и сольватация
свободного лиганда.
Из данных, приведенных в табл. 2.7, представляет также ин-
терес различие в константах устойчивости .[Си (2,3,2-tet) ]2+ и
[Си (trien) ]2+. Было указано [68], что геометрия лиганда пре-
пятствует образованию ненапряженной квадратно-плоскостной
координации trien к Cu(II). В отличие от trien, который имеет
группу —СН2СН2—, связывающую в лиганде два атома вторич-
ного азота, лиганд 2,3,2-tet имеет группу —СН2СН2СН2—, кото-
рая, по-видимому, уменьшает напряжение при координации с
Си2+. Это подтверждается существенным повышением 1g К для
2,3,2-tet по сравнению с trien.
Макроциклический эффект имеет особое значение для метал-
лопорфиринов. Хотя существует очень мало данных о константах
устойчивости этого типа комплексов, для взаимодействия Zn2+ с
дианионной формой диметилового эфира мезопорфирина была
оценена [69] величина lg/C=29. Такие высокие величины наблю-
Устойчивость координационных соединений
115
дались только для комплексов с макроциклическими лигандами.
Устойчивости металлопорфиринов более детально обсуждены в
разд. 3.4.
2.2.4. Смешанные (или тройные) комплексы
Как отмечено в разд. 1, при увеличении числа лигандов в
комплексе ступенчатые константы устойчивости, как правило, по-
нижаются (исключения, см. рис. 2.7). Таким образом, для реак-
ций комплексообразования
М + А^МА, К$А (13)
МА-J-A МАа, (14)
К ма больше, чем (верхний индекс у константы означает
реагент, нижний — продукт реакции). Именно такая тенденция
вытекает из рассмотрения статистических, стерических и электро-
статических факторов.
Если, кроме одного лиганда А, ввести также второй лиганд
В, то число возможных равновесий существенно увеличивается.
Кроме равновесий, включающих простое образование бинарных
комплексов, таких, как МА, МА2, МВ и МВ2, протекает также ре-
акция замещения
МА + В^=*МВ + А (15)
Этот тип реакции должен быть важен в тех случаях, когда один
или оба лиганда (А и В) занимают все или почти все координа-
ционные места иона металла, как, например, когда А=
= NH2CH2CO2, а В=ЭДТА4-. В данном случае шестидентатность
ЭДТА4- должна исключать одновременную координацию глици-
ната. Поэтому смешанный или тройной комплекс, содержащий
два или более различных лигандов, образовываться не будет. Кон-
станты равновесия реакций типа (15) просто равны отношениям
констант устойчивости МВ и МА. Факторы, от которых зависят
эти реакции, уже были обсуждены.
Теперь возвратимся к проблеме смешанных комплексов.
(В описании этих комплексов будут использоваться взаимозаме-
няемые термины: «смешанный» или «тройной».) Образование
смешанного комплекса возможно в том случае, когда А и В имеют
сравнительно мало донорных атомов, вследствие чего оба лиганда
могут координироваться к иону металла одновременно. Если
1 моль М реагирует с 1 молем А и 1 молем В, то возможное
равновесие имеет следующий вид:
MA + B^=tMAB, А$ав
(16)
116
Глава 2
Если известны А™дВ , и то можно рассчитать другую
смешанную константу, 1
f^MA izM
МВ+А^МЛВ, 4^в=~7м МЛ (17>
лмв
Константы равновесия таких реакций, как (16) и (17), изучены
сравнительно мало, но важность тройных комплексов для биохи-
мических систем представляет сильный стимул для интенсивных
исследований в этой области. В недавно опубликованном исчер-
пывающем обзоре, касающемся смешанных комплексов [70], сум-
мировано большинство опубликованных к настоящему времени
данных.
Особый интерес представляет решение вопроса о том, как влия-
ет координированный лиганд А [уравнение (16)] на тенденцию
к координации лиганда В; например, как изменяется в за-
висимости от природы А? В табл. 2.8 приведены некоторые полу-
ченные в последнее время данные о смешанных комплексах эти-
лендиамина, гистамина и аниона серина. Следует отметить не-
сколько тенденций.
Таблица 2.8
Константы устойчивости смешанных комплексов8 (при 37 °C
и ц=0,15 (KNO3) [71—73])
IgA
МА + В Co2+ Ni2+ Cu2* Zn2+
I М2+ + еп 5,30 6,98 10,18 5,53
[Меп]2+ -}• еп 4,28 5,81 8,77 4,75
[Mhm]2+ + еп 4,42 5,78 8,15 5,34
[MSer]+ + еп 4,84 6,57 9,31 5,39
II M2+4-hm 4,89 6,60 9,28 5,03
[Меп]2+ + hm 4,01 5,40 7,86 4,84
[Mhm]2+ + hm 3,54 4,84 6,30 4,78
[MSer]+ -|- hm 4,41 5,77 8,71 5,20
III M2+ + Ser~ 4,20 5,21 7,57 4,47
[Men]2+ -|- Ser- 3,74 4,80 6,70 4,33
[Mhm]2+ + Ser- 3,72 4,38 6,94 4,64
[MSer]+ -|- Ser- 3,36 4,38 6,45 3,84
еп=этилендиамин; Ьт=гистамин, Бег-=анион серила.
Устойчивость координационных соединений 117
Во-первых, подобно первым константам устойчивости (А^а)
все смешанные константы устойчивости изменяются в зависимости
от иона металла в соответствии с последовательностью Ирвинга—
Уильямса (разд. 2.1.3), т. е. Co2+<Ni2+<Cu2+>Zn2+. Этот поря-
док имеет место в любом ряду таблицы. Кажется, эта тенденция
выдерживается почти для всех констант устойчивости как двой-
ных, так и тройных (смешанных) комплексов.
Во-вторых, за исключением двух случаев—[Ni(en)]2+ и
[Cu(en)]2++en — константы устойчивости бинарных комплексов
(^мв2) меньше> чем константы устойчивости любого из смешан-
ных комплексов (К$дВ ) Например, в реакциях еп (в табл. 2.8
константа устойчивости [Со(еп)]2++еп (Лмв2) меньше, чем кон-
станта устойчивости для реакции [Co(hm)]2+ или [Co(Ser)]+ с
еп (К^щ). Поэтому кажется, что присоединение другого лиганда
В к комплексу МВ, который уже содержит лиганд В, происходит
труднее, чем присоединение В к комплексу МА, который содержит
другой лиганд А. Таким образом, эта группа данных (табл. 2.8)
наглядно показывает необычную устойчивость тройных комплек-
сов по сравнению с двойными. Хотя не совсем ясно, почему так
должно быть, это отношение наблюдается и для других систем,
и ниже оно будет обсуждено более детально.
В-третьих, константы устойчивости (ТСдщ) комплексов ионов
металлов (М2+) с различными лигандами (В) уменьшаются в
ряду лигандов en>hm>Ser~. Более того, тот же самый порядок
сохраняется в общем для присоединения В к комплексу МА. От-
сюда следует, что присутствие А не изменяет порядка комплексо-
образующей способности лигандов В. Эту тенденцию невозможно
объяснить многими факторами, определяющими комплексообра-
зующую способность лигандов. (В тех случаях, когда константы
образования бинарных комплексов Лмв2 необычайно низки, име-
ют место небольшие отклонения от этой тенденции.)
В-четвертых, для тех реакций, где В=еп, константы устойчи-
вости КмдВ в зависимости от МА понижаются в следующем по-
рядке: M2+>[MSer]+>[Men]2+>'[Mhm]2+. Исключения из этой
последовательности опять наблюдаются для низких констант
устойчивости бинарных комплексов (KJJf2). Почему выдержива-
ется указанная выше последовательность, также неясно. На осно-
вании простых электростатических соображений можно ожидать,
что [MSer]+ должен образовывать менее устойчивые смешанные
комплексы, чем любой из двухзарядных комплексов, однако даже
эта зависимость не наблюдается.
В другом исследовании [75] были определены константы устой-
чивости комплексообразования глицинатного аниона с рядом
комплексов Ni2+ (табл. 2.9). Здесь также, кажется, константы
118
Глава 2
не коррелируются с зарядом на комплексе. Джекобс и Мардже-
рум [75] использовали семь параметров для эмпирического описа-
ния изменения К в этих и подобных равновесиях и среди них чис-
ло связанных атомов азота, число связанных карбоксильных групп
в исходном комплексе Ni2+, заряды на комплексах и на Gly-. Все
эти три фактора дают важный вклад в величины констант.
Таблица 2.9
Координация глицинатного аниона
комплексами никеляа (при 25 °C, р.=0,5 (NaCl) [75])
Комплекс + Gly— *Cb
Ni2+ 4- Gly" 5,77
[Nidien]2+ + Gly" 5,13
[Ni(NTA)]- + Gly- 4,89
[NiGly]+ + Gly" 4,80
а Сокращения: GIy~=NH2CH2CO ; dien=NH(CH2CH2NH)2;
NTA=N(CH2CO2)3.
Распространение этого подхода могло бы быть полезным для вы-
яснения факторов, определяющих константы устойчивости смешан-
ных комплексов.
Хотя мы говорили об образовании смешанных комплексов как
о реакциях, не зависящих от других равновесий в растворе, сме-
шанные комплексы находятся, конечно, в равновесии с соответ-
ствующими бинарными комплексами. Это равновесие можно на-
писать следующим образом:
[МАВР
МАз+МВз^гМАВ, К = [МАзНМВзГ <18>
Для смешанных комплексов этилендиамина, гистамина и аниона
серина, приведенных в табл. 2.8, можно вычислить величины без-
размерных констант смешивания К. Они приведены в табл. 2.10.
Возможно, наибольший интерес представляет то, что все ве-
личины положительны, т. е. что все равновесия благоприятствуют
образованию смешанных комплексов. Необычная устойчивость
смешанных комплексов была уже замечена ранее и является, ка-
жется, общим правилом, хотя есть и исключения [70].
Несомненно, одна из причин смещения равновесия (18) впра-
во заключается в статистическом преимуществе образования МАВ
перед образованием МА2 или МВ2 (в отношении 2:1). Для урав-
нения (18) это приводит к выводу, что 1g К будет равен 0,60
Устойчивость координационных соединений
119
Таблица 2.10
Константы смешивания К для реакции (18)а
Комплекс IgK
Со2+ Ni2+ Си2+ Zn2+
(М(еп) (hm)]2+ 0,61 0,53 1,53 0,65
[M(en) (Ser)]+ 0,94 1,18 0,77 1,13
[M(hm) (Ser)]-»- 1,23 0,95 2,94 1,22
а Те же условия, сокращения и ссылки, как в табл. 2.8.
( = lg 4), если константы смешивания обусловлены только стати-
стикой. Таким образом, величины 1g К. в табл. 2.10, близкие к 0,6,
можно рассматривать как чисто статистические. Подобным обра-
зом были вычислены константы смешивания для более сложных
смешанных комплексов [76]. Однако, как правило, эти константы
отличаются от статистических величин.
Из-за малочисленности данных о константах устойчивости
смешанных комплексов и зависимости этих констант от ионной
силы раствора и других условий [70] эта область устойчивости
комплексов в настоящее время остается еще мало понятной.
2.2.5. Оптически активные лиганды
и стереоселективность
К обсуждению смешанных комплексов имеет близкое отноше-
ние рассмотрение смешанных комплексов с двумя лигандами, ко-
торые отличаются конфигурацией вокруг асимметрического атома
углерода. Например, можно сравнить константы равновесия сле-
дующих реакций, в которых участвуют оптически активные ами-
ноацидаты (т. е. анионы аминокислот), характеризующиеся раз-
личной хиральностью:
Cu(l-A)+ + l-A“ <—> Си(ь-А)а (19)
Cu(l-A)+-f-d-A“ < > Cu(l-A) (d-A) (20)
Экспериментально обнаружено [77], что константы устойчивости
реакций (19) и (20) одинаковы в случае аланина, фенилаланина,
валина и пролина. Для аспарагинатов меди(П) были опублико-
ваны [78] данные, в соответствии с которыми константа равнове-
сия реакции (19) больше (lg/C=6,45), чем константа равновесия
реакции (20) (1g К=5,85). Однако более позднее [78а] исследо-
вание аспарагинатов Си(II), Ni(II) и Со(II) показало, что кон-
станты равновесия реакций (19) и (20) одинаковы. Идентичные
120
Глава 2
константы устойчивости были также найдены для реакций опти-
ческих изомеров глутаминовой и аспарагиновой кислот и глутами-
на с Cu(II), Ni(II) и Со(П).
В противоположность этому определенная методом протонно-
го магнитного резонанса константа равновесия реакции [786]
взаимодействия Со(II) и о,ь-гистидина
Co(D-His)a + Co(L-His)a < -Ь 2Co(D-His) (L-His) (21)
равна lgK = l,l. Эта стереоселективность, благоприятствующая
образованию смешанного изомера, Со (o-His) (ь-His), позже была
подтверждена потенциометрическим методом [78в]. Наблюдалась
также подобная стереоселективность, благоприятствующая обра-
зованию Ni(ь-His) (D-His). Из раствора, содержащего Со(Н) и
d.l-гистидин, были выделены кристаллы Co(b-His)(D -His).
Рентгеноструктурным методом была установлена как структура
Со( D-His) (L-His) [79], так и структура Со( L-His)2 [80]. В обе-
их структурах гистидиновый лиганд тридентатен, но из-за разли-
чия в конфигурациях оптически активных лигандов возникают
существенные различия в относительных положениях молекул
гистидина в координационной сфере. Интересно, что из растворов,
содержащих Ni2+ и d,l-гистидин (гл. 4, разд. 4.6.2), могут быть
выделены только Ni(b-His)2 и Ni(o-His)2, но не Ni(o-His)( L-His)
[811-
Были также определены константы устойчивости комплексов
оптически активных аминоацидатов с п-валин-Н-моноацетатом
Cu(II), Cu(L-ValMA) [82]. Величины констант равновесия реак-
ции (22) даны в табл. 2.11.
О
с—О---------ОН2
(СНзИСН^ / : \
Н -----Q
Cu(L-ValMA) +l- или d-A" Cu(L-ValMA) (А)" (22)
Преимущественная координация L-аминоацидатов по сравне-
нию с D-энантиомерами должна быть, по-видимому, объяснена
геометрией комплексов Cu(b-ValMA) (А)-. Об их структуре из-
вестно мало, но величины констант устойчивостей указывают, что
аминоацидаты взаимодействуют как бидентатные лиганды. Хотя
можно предложить структуры, в которых координация l-A- по
стерическим соображениям должна быть предпочтительной по
сравнению с координацией d-А-, независимые данные для под-
тверждения таких структур отсутствуют. Было также показано
Устойчивость координационных соединений
121
Таблица 2.11
Константы устойчивости комплексов Cu(L-ValMA)
с оптически активными амииоацидатами в соответствии
с уравнением (22) при 25 °C [82]
А- IgK
L-A- D-A-
Leu- 5,74 4,93
PhAla- 5,52 4,92
Ala- 5,29 4,74
Ser- 5,55 5,03
[82], что Cu(L-ValMA) стереоселективно координирует эфиры
оптически активных кислот (по-видимому, только через атом N).
Вследствие этого Cu(b-ValMA) стереоселективно катализирует
гидролиз эфиров D- и ь-аминокислот.
Подавляющее большинство работ [83, 84] по стереоселектив-
ности комплексов ионов металлов относится к изучению комплек-
сов Со(Ш). Однако из-за инертности комплексов Со(Ш) лишь
немногие исследования были проведены в равновесных условиях,
и поэтому невозможно решить, являются многие примеры стерео-
селективности в химии Со(III) следствием кинетических или тер-
модинамических эффектов. Очевидно, предстоит еще много сде-
лать для изучения стереоселективности комплексов ионов метал-
лов, особенно лабильных ионов металлов.
3. НЕКОТОРЫЕ БИОХИМИЧЕСКИЕ ЛИГАНДЫ
3.1. Аминокислоты
Хотя константы устойчивости комплексов глицина с ионами
металлов были обсуждены выше в данной главе, представляет
интерес рассмотреть константы устойчивости других аминокислот.
В обзоре Гарди и Уилкокса [60] собраны данные ранних иссле-
дований в этой области.
Хотя нормальный способ координации аминокислоты состоит
в координации через амино- и карбоксильную группы, Чайлдс и
Перрин [85] сообщили константу устойчивости для координации
цвиттер-иона глицина к Си2+ только через карбоксильную группу.
Cu2+ + -O2CCH2NHs <—> Cu(O2CCH2NHj), 1g К = 1,53
Величина IgA 1,53 меньше, чем 1,84 для реакции Си2+ с СН3СО7;
можно полагать, что более низкая константа устойчивости объяс-
122
Глава 2
няется более низкой основностью (разд. 2.2.1) цвиттер-иона гли-
цина. Несомненно, это очень малые константы по сравнению с
теми, которые обсуждались выше. Действительно, другие иссле-
дователи [86, 87] не нашли никаких доказательств комплексооб-
разования ионов металлов с цвиттер-ионом аминокислоты.
Анионы аминокислот образуют устойчивые комплексы со мно-
гими ионами металлов, в частности с ионами переходных элемен-
тов. В большинстве случаев аминоацидаты взаимодействуют как
бидентатные лиганды, координируясь через амино- и карбоксиль-
ную группы. Такие аминокислоты, как глицин, аланин, валин, про-
лин, лейцин, саркозин, тирозин [88], серин [87], треонин [87],
лизин [89, 90], аргинин [22, 89, 91], триптофан, аспарагин и ме-
тионин, по отношению к Ni2+, Cu2+ и Zn2+ ведут себя как биден-
татные лиганды. Сходство констант устойчивости их комплексов
с Zn2+, Fe2+ и Fe3+ (табл. 2.12) указывает, что они все координи-
руются только через амино- и карбоксильную группы. Необычно
низкие константы устойчивости комплексов с метионином и арги-
Таблица 2.12
Константы устойчивости аминоацидатов Zn2+, Fe2+ и Fe3+
М^+Ч-А-ч^МА!”*-1)*
Аминокислота группы —NH2 IgK
Zn2+f22] Fe2+ [92] Fe3+ [93]
Аргинин 9,21 4,2 3,2 8,7
Метионин 9,13 4,4 3,2 9,1
Треонин 8,86 — 3,3 8,6
Аспарагин 8,79 — 3,4 8,6
Триптофан 9,43 — 3,4 9,0
Серин 9,12 — 3,4 9,2
Валин 9,59 5,0 3,4 9,6
Лейцин 9,62 4,9 3,4 9,9
Аланин 9,79 5,1 3,5 10,4
Саркозин 10,02 — 3,5 9,7
Глицин 9,76 5,0 3,8 10,0
Пролин 10,52 — 4,1 10,0
Глутаминовая кислота 9,54 5,4 3,5 12,1
Аспарагиновая кислота 9,56 5,8 4,3 11.4
Г истидин 9,20 6,6 5,9 4,7
Цистеин 10,78 9,9 6,2 —
Устойчивость координационных соединений
123
нином (содержащих протонированную гуанидиновую группу), по-
видимому, связаны с низкой основностью группы —NH2 в этих
аминокислотах.
Следует заметить, что метионин может координироваться к
мягким ионам металлов, например Pd(II) и Hg(II), через атом
серы. Перрин [92, 93] коррелировал константы устойчивости
комплексов Fe2+ и Fe3+ с аминокислотами (приведенные в
табл. 2.12) с величинами рКа их групп —NH2 (разд. 2.2.1). Эти
данные подчиняются уравнению (6) при а=0,4 для Fe2+ и а=1,8
для Fe3+. Это большое различие в наклоне а указывает, что ве-
личины 1g К комплексов Fe3+ являются значительно более чувст-
вительной функцией р/Са, чем величины 1g К комплексов Fe2+. На
основе отклонения зависимости 1g К — р/Са от линейной Перрин
предположил, что двухзарядный анион аспарагиновой кислоты в
его комплексах с Fe2+ и Fe3+ проявляет существенную тридентат-
ность. Из данных табл. 2.12, по-видимому, следует, что это отно-
сится также к комплексам с Zn2+. Другие данные [94, 95] ука-
зывают, что аспарагиновая кислота ведет себя как тридентатный
лиганд по отношению к Си2+ и Ni2+; рентгеноструктурные иссле-
дования [96] комплексов аспарагиновой кислоты с Со2+, Ni2+ и
Zn2+ ясно указывают, что в твердом состоянии она присоединена
как тридентатный лиганд. Рентгеноструктурные исследования
[96] показали также, что в противоположность этому глутамино-
вая кислота координируется через одну группу —NH2 и через
одну —СО2-группу к одному иону металла, в то время как дру-
гая группа —СО2 присоединяется к другому иону металла
(разд. 4.2, тип г). В растворе (на основании данных, полученных
в работах [94, 95]) вторая группа —СО остается несвязанной.
Как показывают константы устойчивости (табл. 2.12), глутами-
новая кислота ведет себя преимущественно как бидентатный ли-
ганд, за исключением комплексов с Fe3+.
Как следует из приведенных в табл. 2.12 констант устойчиво-
сти, две последние аминокислоты — гистидин и цистеин — коорди-
нируются иначе, чем другие аминокислоты. Сходство констант
устойчивости комплексов с цистеином и анионом меркаптоэтил-
амина NH2CH2CH2S- (например, lgK=9,9 для комплексов Zn2+)
позволяет утверждать, что ионы металлов первого переходного
ряда координируют цистеин через группы —NH2 и —S-, а карб-
оксильная группа —СО2 остается некоординированной.
На основании структурного исследования получены более опре-
деленные доказательства того, что такой способ координации
цистеина осуществляется в его комплексах с Со3+ [97]. Спектраль-
ные исследования [98] комплексов Fe3+ с цистеином показывают,
что при некоторых условиях карбоксильная группа, по-видимому,
также принимает участие в координации. Родственный лиганд,
пеницилламин _SC(CH3)2CH(NH2)CO2, подобно цистеину, коор-
124
Глава 2
динируется к Ni2+ и Zn2+ через группы —NH2 и —S~. Величины
1g К его комплексов очень близки к 1g К комплексов цистеина.
Пеницилламин и цистеин принадлежат к немногим лигандам
[99], для которых вторая ступенчатая константа устойчивости
(К2) больше, чем первая (Ki).
Таблица 2-13
Величины AG, А// и AS для реакции М2++А-я^МА+ при 25 °C
(А- — аминоацидат)
Амино- кислота Cu2+ Ni2+
AG, ккал/моль ан, ккал/моль AS, кал - моль—1 -град—1 AG, ккал/моль АН, ккал/моль AS, кал - моль—1 «град—1
Глицина —11,71 —6,76 16,6 —8,43 —4,14 14,4
Серин6 —10,71 —5,51 17,5 —7,43 —3,76 12,3
Треонин6 —10,83 —5,56 17,7 —7,45 —3,81 12,2
Тирозинв —10,80 —5,42 18.1
а Данные взяты из работы [50].
6 Данные взяты из работы [87].
в Данные взяты из работы [88].
Координация гистидина ионами переходных металлов пред-
ставляет несколько более сложную систему, которая рассматри-
вается в гл. 4. В этой главе обсуждаются комплексы Си2+ с ней-
тральным протонированным гистидином, а также другие, содер-
жащие в качестве лиганда анион гистидина.
Были определены энтальпии и энтропии реакций комплексо-
образования различных аминокислот. Некоторые из этих данных
приведены в табл. 2.13. Они указывают на сходство величин AG,
АЯ и АЗ для всех приведенных аминокислот. Как было отмечено
в разд. 2.2.2, энтальпии этих реакций отрицательны и определя-
ются главным образом координацией атома азота аминогруппы.
Приведенные в табл. 2.13 величины А// примерно равны
5,8 ккал/моль для комплексов Си2+ и 3,9 ккал/моль для комплек-
сов Ni2+. Если доминирующий вклад в эти величины дает коор-
динация группы —NH2, то они должны быть близки к Чъ&Н
для реакций образования комплексов с этилендиамином. Это
действительно имеет место, так как ЧъкН равна 6,5 ккал/моль
для [Cu(en)]2+ и 4,5 ккал/моль для [Ni(en)]2+ [8]. Подобным
образом было отмечено (разд. 2.2.2), что положительные значения
АЗ этих реакций определяются в основном координацией карбок-
сильных групп аминоацидатов. Действительно, энтропии образова-
ния СиА+ (17,5 э. е.) и NiA+ (12,9 э. е.) близки к ‘/sAS реакции
Устойчивость координационных соединений
125
образования оксалатных комплексов состава 1:1: Си (Ох) —
14,1 э. е. и Ni(Ox) —12,1 э. е.
Были проведены подобные корреляции термодинамических ха-
рактеристик образования бис-(аминоацидатных) комплексов
{100], М2+4-2А_^МА2. Кроме того, было найдено, что ЛЯ
(21,3 ккал/моль) образования комплекса Cu(His)2, в котором
предполагается координация как аминного, так и имидазольного
атомов азота, приближенно может быть представлена как сумма
Д.Н для Си(Gly)2, равной 12,8 ккал/моль, и ‘ЛАЯ для Cu(2,2'-Dip)2
(Dip=дипиридил), равной 8,3 ккал/моль. Это приближение оправ-
дывается также для аналогичных комплексов Ni2+ и Zn2+. И, на-
конец, было замечено, что энтальпия реакций взаимодействия
различных ионов металлов с аминоацидатами в общем следует
ряду Ирвинга—Уильямса (разд. 2.1.3).
3.2. Пептиды
Простые ди-, три- и тетрапептиды образуют комплексы с иона-
ми переходных металлов. Одна из наиболее изученных систем —
это Си2+ с глицилглицином. В результате интенсивных исследова-
ний было показано, что моноанион Gly-Gly- образует с Си2+ комп-
лекс состава 1 :1 при pH ниже примерно 4. Этот комплекс спосо-
бен диссоциировать с отщеплением протона (рАа=4,38). Эти два
равновесия приведены ниже:
Си2+ + Gly-Gly- <=* [Cu(Gly-Gly)]+, IgK = 5,42
[Cu(Gly-Gly)]+ Cu(Gly-Gly—H) -J-H+, 1gKa = — 4,38
Эти комплексы Си(II), их устойчивости и строение обсуждены
полностью в гл. 4 (см. также табл. 4.1—4.4). Изучены константы
устойчивости аналогичных глицинпептидных комплексов Со(II)
.[101], Ni(II) [102—104] и Pd(II) [90]. Легкость ионизации пеп-
тидного протона увеличивается в ряду Со(П) <Ni(II) <Cu(II) <
<Pd(II). Исследование [105] взаимодействия Zn2+ с синтетиче-
ским модельным пептидом, Ь1,Ь1'-диглицилэтилендиамином, пока-
зало, что Zn(II) не облегчает диссоциацию протона.
Были сделаны попытки определить места присоединения ионов
металлов к большим белкам путем сравнения констант устойчи-
вости комплексов модельных лигандов с теми же ионами метал-
лов. Пожалуй, наиболее характерный пример представляет ме-
таллофермент карбоксипептидаза А (КПА), который в естествен-
ном состоянии содержит ион Zn2+ и около 300 аминокислотных
остатков. Определены константы устойчивости комплексов Zn2+
и некоторых других ионов металлов с апоКПА (фермент, с кото-
рого удален ион металла) и сравнены с соответствующими кон-
стантами устойчивости комплексов модельных лигандов. На осно-
вании константы устойчивости, а также спектральных и других
126
Глава 2
химических данных [106] был сделан вывод, что Zn2+ координи-
руется к одной —NH2- и одной —S~-rpynne. Из-за большого числа
возможных мест координации и вероятности того, что различные
ионы металлов могут координироваться к различным местам
[107], было признано, что определенные таким путем константы
устойчивости могут быть ошибочными. Из полученных недавно
усовершенствованных рентгеноструктурных данных для КПА
[Ю8] стало ясно, что это опасение обоснованно. В действитель-
ности Zn2+ координируется к двум имидазольным группам гисти-
диновых остатков и карбоксильной группе остатка глутаминовой
кислоты. В то же время на основании исследования равновесий
в системах, содержащих ионы металлов и глицинпептиды, можно
было предположить, как указано выше, что ион металла присоеди-
няется к концевой —ЙН2-группе белка. Это также не наблю-
далось.
Трудности, связанные со сравнением констант устойчивости
комплексов с белками и модельными соединениями, были нагляд-
но показаны также при исследовании миоглобина кашалота [109,
ПО] и альбумина бычьей сыворотки [111, 112]. Отсутствие дан-
ных о константах устойчивости комплексов ионов металлов с ма-
лыми пептидами, содержащими координирующие боковые цепи
(т. е. гистидин, лизин или остатки глутаминовой кислоты), очень
задерживает понимание более сложных взаимодействий ионов ме-
таллов с белками.
3.3. Нуклеотиды и нуклеиновые кислоты
Нуклеотиды состоят из трех структурных единиц — пуринового
или пиримидинового основания, рибозы и моно-, ди- или трифос-
фатной группы. Структура нуклеотида аденозинтрифосфата (АТФ)
с обозначением каждой из этих единиц приведена ниже.
Хотя известно, что нуклеотиды образуют довольно устойчивые
комплексы с ионами металлов, места координации однозначно
Устойчивость координационных соединений
127
трудно установить, несмотря на большие успехи, которые недавно
были достигнуты в этом направлении. На основании слабых коор-
динационных свойств сахаров как лигандов по отношению к ионам
переходных, а также щелочных и щелочноземельных металлов
можно предположить, что рибозная часть нуклеотидов в общем
не будет сильным координационным центром. А так как известно,
что нуклеозиды (пуриновое или пиримидиновое основание, свя-
занное с остатком рибозы) по отношению к ионам металлов обла-
дают слабой координирующей способностью [42], то, следователь-
но, фосфатная группа должна быть наиболее сильным, местом
координации при связывании ионов металлов нуклеотидами. Де-
тально места координации обсуждены в гл. 33.
Наиболее широко был исследован аденозинтрифосфат (АТФ)
[ИЗ]; он содержит четыре способных к ионизации атома водоро-
да на фосфатной части молекулы.
Как четырехзарядный анион (АТФ4-), так и монопротониро-
ванная форма (АТФН3-) образуют в растворе комплексы с иона-
ми металлов. Константы устойчивости комплексов АТФ4- и
АТФН3- в зависимости от иона металла увеличиваются в следую-
щем ряду [114]: Ва(И) <Sr(II) <Mg(II) <Co(II) <Mn(II) <
<Zn(II) <Ni(II) <Cu(II). Эта зависимость представляет обычный
ряд Ирвинга—Уильямса, за исключением Со(Н) и Мп(П). Ре-
зультаты исследований с помощью многочисленных эксперимен-
тальных методов [42] показывают, что ионы металлов (за исклю-
чением Си2+) координируются с трифосфатной частью АТФ. Этот
вывод подтверждается константами устойчивости комплексов ана-
логичных нуклеотидов, содержащих различные пуриновые и пи-
римидиновые основания; эти константы мало зависят от природы
основания, содержащегося в молекуле. Более того, константы
устойчивости повышаются при увеличении числа фосфатных
групп. Так, величины IgA для комплексов Си(II) с аденозинмо-
нофосфатом (АМФ2-), аденозиндифосфатом (АДФ3-) и АТФ4-
увеличиваются в последовательности АМФ2- (3,2) <АДФ3- (5,9) <
<АТФ4- (6,1). Значения IgA для комплексов с АДФ3- и АТФ4-
аналогичны величинам IgA для комплексов с монопротонирован-
ными полифосфатами [115]: НР2О3- (5,4) <НР3О4^ (5,7).
Хотя тенденция в изменении констант устойчивости следует
известному ряду, различия в величинах IgA очень малы; напри-
мер. для комплексов с АТФ4- они равны: Mg(II) 4,2; Со(П) 4,7;
Мп(П) 4,8; Zn(II) 4,8; Ni(II) 5,0; Cu(II) 6,1. Сходство констант
устойчивости было объяснено [113, 114] тем, что фосфатные груп-
пы не координируются непосредственно к иону металла, а обра-
зуется гидратированная ионная пара, устойчивость которой за-
висит только от заряда на ионе металла. Несколько более высокая
устойчивость комплекса Си(II), а также спектральные данные
указывают, что Си(II) присоединяется и к адениновому кольцу
128
Глава 2
и к фосфатной цепи. Все другие ионы металлов связываются, ка-
жется, только с фосфатной цепью. Модельный лиганд, 2-пиридил-
метилфосфат, содержащий пиридиновую и фосфатную группы, по-
добным образом координируется ионами металлов только через
фосфатную группу, за исключением Си(II), к которой он присо-
единяется также через атом пиридинового азота [И6].
Взаимодействие ионов металлов с полинуклеотидами или нук-
леиновыми кислотами, такими, как РНК и ДНК, имеет в основном
тот же характер, что и с нуклеотидами [42, 117], как будет об-
суждено в гл. 34.
3.4. Порфирины
Порфирины [118] —это тетрапиррольные макроциклические
соединения, которые при координации с ионами металлов теряют
два протона. Тетрадентатный двухзарядный анионный лиганд Р2-
образует с ионами металлов комплексы [П9] , имеющие в основ-
ном плоское строение [120], но вследствие некоторой гибкости
они могут быть несколько искажены.
метоллопорфирин
В литературе опубликована только одна константа устойчиво-
сти металлопорфирина [69], а именно для реакции взаимодейст-
вия Zn(II) с диметиловым эфиром мезопорфирина (Р2~) с обра-
зованием ZnP. Величина 1g К приблизительно равна 29 — цифра,
которая достигается только макроциклическими лигандами
(разд. 2.2.3). Малочисленность данных для равновесий этих реак-
ций несомненно связана с исключительной медленностью их про-
текания, поскольку иногда необходимы месяцы для достижения
равновесия при комнатной температуре.
Из-за отсутствия данных о константах устойчивости металло-
порфиринов для установления качественного ряда устойчивостей
был использован [121] ряд других методов, таких, как реакции
замещения иона металла, реакции, включающие вытеснение иона
металла кислотой, и спектроскопические исследования. Этот ряд
имеет следующий вид: Ag(I)2<K2<Ba(II) <Na2<Li2<Sn(II) <
<Cd(II)<Mg(II)<Zn(II)<Cu(II)<Ag(II) < Co(II) < Ni(II)<
<Pd(II) <Pt(II). Предположили [121], что необычно низкая
Устойчивость координационных соединений 129
устойчивость комплекса Ва(П) обусловлена большим размером
этого иона, что препятствует его вхождению в плоское кольцо.
По какой причине устойчивости комплексов элементов первого
переходного ряда не следуют порядку Ирвинга—Уильямса
(разд. 2.1.3), неясно.
Квадратно-плоскостные металлопорфирины, так же как и род-
ственные им комплексы фталоцианинов [122], могут присоединять
один или два монодентатных лиганда, образуя комплексы с 5- и
6-кратной координацией иона металла. При исследовании равно-
весия а,₽,у,6-тетрафенилпорфириновых комплексов Zn(II), Cd (II)
и Hg(II) [123, 124] с замещенными пиридинами было установле-
но, что к иону металла присоединяется одна молекула пиридина.
Однако аналогичные комплексы Cu(II) и Ni(II) [125], кажется,
проявляют лишь незначительную склонность к присоединению пи-
ридина. Величины 1gК для Zn(II) и Cd(II) близки, но для Hg(II)
ниже; для реакции с пиридином они равны 3,78 (Zn), 3,51 (Cd)
и 1,21 (Hg). Для всех трех ионов металлов величины 1g К увели-
чиваются по мере повышения основности пиридина, и зависимость
1g К от рК линейна (разд. 2.2.1). К сожалению, подобная корре-
ляция не наблюдается [126] для бис-пиридинатных комплексов
нескольких различных железо (II) порфиринов.
Введение электронодонорных или электроноакцепторных групп
в порфириновое кольцо также изменяет тенденцию металлопор-
фиринов к присоединению лигандов. В общем [П8] электроно-
донорные группы уменьшают координацию аминов, в то время
как электроноакцепторные группы повышают координацию. Осо-
бенно наглядно это было показано [127] для ряда комплексов
Ni(II) е диметиловыми эфирами замещенных дейтеропорфири-
нов IX. Эти порфирины реагируют с 2 молями пиперидина в соот-
ветствии с уравнением NiP-p2L^NiPL2. Величины 1g Д' лежат в
пределах от —2,0 для 2,4-диэтилзамещенного металлопорфирина
до 0,15 для комплекса 2,4-диформилзамещенного порфирина. Ос-
новность (рЛа) диэтилпорфирина (5,8) намного больше, чем ос-
новность диформильного производного (2,8). Таким образом, чем
больше основность порфирина, тем меньше устойчивость комплек-
са NiPL2. Обратная линейная корреляция между 1g К и рКа пор-
фирина действительно имеет место.
СПИСОК ЛИТЕРАТУРЫ
1. Tapscott R. Е., Belford R. L., Paul I. С., Coord. Chem. Rev., 4, 323 (1969).
2. Fitzwater D. R., Rundle R. E„ Z. Kris , 112, 362 (1959).
3. Hunt E. B„ Rundle R. E, Stosick A. J., Acta Cryst., 7, 106 (1954).
4. Powell J. E., Rowlands D. L. G., Inorg. Chem., Б, 819 (1966).
5. Moeller T., Martin D. F., Thompson L. C., Ferrus R., Feistel G. R., Ran-
dall W. J., Chem. Rev., 65, 1 (1965).
6. Hinchey R. J., Cobble J. W., Inorg. Chem., 9, 917 (1970).
-2451
130
Глава 2
7. Graddon D. P., J. Inorg. Nucl. Chem., 5, 219 (1958).
8. Nancollas G. H., Interactions in Electrolyte Solutions, Elsevier Publishing Co.,
Amsterdam, 1966.
9. Jones M. M., Elementary Coordination Chemistry, Prentice-Hall, Englewood
Cliffs, N. J., 1964, p. 333.
10. Fronaeus S., in «Technique of Inorganic Chemistry», Vol. I, Interscience Pub-
lishers, New York, 1963, p. 1.
11. Poccwtu Ф., Россотти X., Определение констант устойчивости и других кон-
стант равновесия в растворах, «Мир», М., 1965.
12. Lingane Р. Л, Hugus Z. Z., Jr., Inorg. Chem, 9, 757 (1970).
12a. Бек M., Химия равновесий реакций комплексообразования, «Мир», М.,
1973.
13. Eatough D. J., Anal. Chem., 42, 635 (1970).
14. Goldberg D. E„ J. Chem. Educ., 39, 328 (1962).
15. Angelici R. J., Synthesis and Technique in Inorganic Chemistry, W. B. Saun-
ders Co., Philadelphia, 1969, p. 105.
16. Crow D. R., Westwood J. V„ Quart. Rev., 19, 57 (1965).
17. Schubert J., Methods of Biochemical Analysis, 3, 247 (1956).
18. Marcus Y., Ion Exchange, 1, 101 (1966).
19. Marcus Y., Kertes A. S., Ion Exchange and Solvent Extraction, John Wiley
and Sons, Inc., New York, 1969.
20. Helfferich F., Ion Exchange, McGraw-Hill, New York, 1962, p. 221.
21. Johansson L., Coord. Chem. Rev., 3, 293 (1968).
22. Sillen L. G., Martell A. E., Stability Constants of Metal-Ion Complexes, Spe-
cial Publication Nos. 17 and 25, The Chemical Society, London, 1964 and
1971.
23. Яцимирский К. Б., Васильев В. П., Константы нестойкости комплексных
соединений, Изд. АН СССР, М., 1959.
24. Басоло Ф., Пирсон Р., Механизмы неорганических реакций, «Мир», М.,
1971.
25. Россотти Ф., в «Современная химия координационных соединений» под ред.
Дж. Льюиса и Р. Уилкинсона, ИЛ, М., 1963.
26. Schwarzenbach G., Adv. Inorg. Chem. Radiochem., 3, 257 (1961).
27. Chaberek S., Martell A. E., Sequestering Agents, Wiley-Interscience, New
York, 1959.
28. Mellor D. P. in F. P. Dwyer, D. P. Mellor (eds.), Chelating Agents and Me-
tal Chelates, Academic Press, New York, 1965, p. 1.
29. Phillips C. S. G., Williams R. J. P., Inorganic Chemistry, Vol. II, Oxford Uni-
versity Press, New York, 1966, pp. 267—283, 517—549.
29a. Sigel H., McCormick D. B., Accounts Chem. Res., 3, 201 (1970).
30. Beech G„ Quart. Rev., 23, 410 (1969).
31. Ahrland S„ Structure and Bonding, 5, 118 (1968).
.32. Thompson L. G., Shafer B. L., Edgar J. A., Mannila K. D., Advances in Che-
mistry Series, No. 71, American Chemical Society, Washington, D. C., 1967,
169.
33. Schwarzenbach G„ Experentia Suppl., 5, 162 (1956).
34. Ahrland S., Chatt J., Davies N. R., Quart. Rev., 12, 265 (1958).
35. Pearson R. G., J. Chem. Educ., 45, 581, 643 (1968).
16. Williams R. J. P., Hale J. D., Structure and Bonding, 1, 249 (1966).
17. Ahrland S., Structure and Bonding, 1, 207 (1966).
38. Evans R. S., Huheey J. E., J. Inorg. Nucl. Chem., 32, 777 (1970).
39. Misono M., Ochiai E„ Saito Y„ Yoneda Y., J. Inorg. Nucl. Chem., 29, 2685
(1967).
40. Klopman G., J. Am. Chem. Soc., 90, 223 (1968).
41. Pearson R. G„ Science, 151, 172 (1966).
42. Weser U., Structure and. Bonding, 5, 41 (1968).
43. Sigel H., Griesser R., Prijs B., McCormick D. B., Joiner M. G„ Arch. Biochem.
Устойчивость координационных соединений
131
Biophys., 130, 514 (1969); Sigel Н., McCormick D. В., Griesser R., Prijs В.,
Wright L. D., Biochemistry, 8, 2687 (1969).
44. Kimura T„ Structure and Bonding, 5, 1 (1968).
45. Spiro T. G., Saltman P„ Structure and Bonding, 6, 116 (1969).
46. Mellor D. P„ Maley L., Nature, 159, 370 (1947); 161, 436 (1948)
47. Irving H„ Williams R. J. P„ Nature, 162, 746 (1948); J. Chem. Soc., 1953,
3192.
48. Calvin M., Melchior N. C., J. Am. Chem. Soc., 70, 3270 (1948).
49. Ref. [8], p. 183.
50. Boyd S., Brannan J. R., Dunsmore H. S„ Nancollas G. H., J. Chem. Eng.
Data, 12, 601 (1967).
51. Irving H., Williams R. I. P., J. Chem. Soc., 1953, 3192.
52. Ref. [24], p. 77.
53. Ref. [29], p. 269.
54. Martell A. E., Calvin M„ Chemistry of the Metal Chelate Compounds, Pren-
tice-Hall, Englewood Cliffs, N. J., 1952, p. 151.
55. Griesser R., Prijs B„ Sigel H., Inorg. Nucl. Chem. Lett., 4, 443 (1968); Gries-
ser R., Prijs B., Sigel H., Inorg. Nucl. Chem. Lett., 5, 951 (1969).
56. Ref. [9], p. 339.
57. Irving H., Rossotti H., Acta Chem. Scand., 10, 72 (1956).
58. Irving H„ da Silva I. I. R. F., J. Chem. Soc., 1963, 945.
59. Margerum D. W., Powell B. L., Luthy J. A., Inorg. Chem., 7, 800 (1968).
60. Gurd F. R. N., Wilsox P. E., Adv. Protein Chem., 11, 311 (1956).
61. Ref. [25], p. 56.
62. Jones I. G., Poole J. B„ Tomkinson J. C., Williams R. J. P., J. Chem. Soc.,
1958, 2001.
63. Schwarzenbach G., Anderegg G., Schneider W., Senn H., Helv. Chim. Acta,
38, 1147 (1955).
64. Angelici R. J., Leach В. E., J. Am. Chem. Soc., 90, 2499 (1968).
65. Martell A. E., in «Advances in Chemistry Series», No. 62, American Chemical
Society, Washington, D. C., 1967, p. 272.
66. Ref. [25], p. 57.
67. Cabblness D. K., Margerum D. W„ J. Am. Chem. Soc., 91, 6540 (1969).
68. Sacconi L., Paoletti P., Ciampolini M., J. Chem. Soc., 1961, 5115; Paoletti P.,
Ciampolini M„ Sacconi L., J. Chem. Soc., 1963, 3589.
69. Dempsey B., Lowe M. B., Phillips I. N., in J. E. Falk, R. Lemberg, R. K. Mor-
ton (ed.), Haematin Enzymes, Pergamon Press, London, 1961, p. 29.
70. Marcus Y., Eliezer I., Coord. Chem. Rev., 4, 273 (1969).
71. Perrin D. D., Sayce I. G., Sharma V. S., J. Chem. Soc. (A), 1967, 1755.
72. Perrin D. D., Sharma V. S„ J. Chem. Soc. (A), 1968, 446.
73. Perrin D. D., Sharma V. S„ J. Chem. Soc. (A), 1969, 2060.
74. Griesser R., Sigel H., Inorg. Chem., 9, 1238 (1970).
75. Jackobs N. E., Margerum D. W., Inorg. Chem., 6, 2038 (1967).
76. Sharma V. S., Schubert I., J. Chem. Educ., 46, 506 (1969).
77. Gillard R. D., Irving H. M., Parkins R. M., Payne N. C„ Pettit L. D., J. Chem.
Soc. (A), 1966, 1159; Gillard R. D„ Irving H. M„ Pettit L. D„ J. Chem. Soc.
(A), 1968. 673; Simeon V., Weber O. A., Croat. Chem. Acta, 38, 161 (1966).
78. Bennett W. E., J. Am. Chem. Soc., 81, 246 (1959).
78a. Ritsma J. IL, Wiegers G. A., Jellinek F„ Rec. Trav. Chim., 84, 1577 (1965).
786. MacDonald С. C., Phillips W. D„ J. Am. Chem. Soc., 85, 3736 (1963).
78b. Ritsma I. H„ Van de Grampel I. C., Jellinek F., Rec. Trav. Chim., 88, 411
(1969).
79. Candlin R.. Harding M. M„ J. Chem. Soc. (A), 1970, 384.
80. Harding Al. M., Long H. A., J. Chem. Soc. (A), 1968, 2554.
81. Fraser K. A., Harding M. M„ J. Chem. Soc. (A), 1967, 415.
82. Leach В. E„ Angelici R. I., J. Am. Chem. Soc., 91, 6296 (1969).
83. Dunlop J. H., Gillard R. D., Adv. Inorg. Chem. Radiochem., 9, 185 (1966).
132
Глава 2
84. Sargeson А. М, in F. Р. Dwyer, D. P. Mellor (eds.), Chelating Agents and
Metal Chelates, Academic Press, New York, 1964, p. 183.
85. Childs C. W„ Perrin D. D., J. Chem. Soc. (A), 1969, 1039.
86. Pearlmutter A. F„ Stuehr J., J. Am. Chem. Soc., 90, 858 (1968).
87. Letter J. E., Jr., Bauman J. E., Jr., J. Am. Chem. Soc., 92, 437 (1970).
88. Letter J. E., Jr., Bauman J. E., Jr., J. Am. Chem. Soc., 92, 443 (1970).
89. Greenstein J. P., Winitz M„ Chemistry of the Amino Acids, Vol. 1, John Wi-
ley and Sons, New York, 1961, p. 618.
90. Wilson E. W., Jr., Martin R. B., Inorg. Chem., 9, 528 (1970).
91. Clarke E. R., Martell A. E., J. Inorg. Nucl. Chem., 32, 911 (1970).
91a. Stephenson N. C„ McConnell J. F„ Warren R., Inorg. Nucl. Chem. Lett., 3,
553 (1967); Natusch D. F. S., Porter L. J., Chem. Comm., 1970, 596.
92. Perrin D. D„ J. Chem. Soc., 1959, 290.
93. Perrin D. D„ J. Chem. Soc., 1958, 3125.
94. Wellman К. M., Mecca T. G., Mungall W„ Hare C. R., J. Am. Chem. Soc., 90,
805 (1968).
95. Ho F. F. L., Erickson L. E., Watkins S. R., Reilley C. N„ Inorg. Chem., 9, 1139
(1970).
96. Freeman H. C., Adv. Protein Chem., 22, 258 (1967).
97. Kothari V. M., Busch D. H., Inorg. Chem., 8, 2276 (1969).
98. Tomita A., Hirai H., Makishima S., Inorg. Chem., 7, 760 (1968).
99. Perrin D. D., Sayce I. G., J. Chem. Soc. (A), 1968, 53.
100. Stack W. F., Skinner H. A., Trans. Faraday Soc., 63, 1136 (1967).
101. Michailidis M. S., Martin R. B., J. Am. Chem. Soc., 91, 4683 (1969).
102. Sarkar B., Wigfield Y„ J. Biol. Chem., 242, 5572 (1967).
103. Martin R. B., Chamberlin M„ Edsall J. T., J. Am. Chem. Soc., 82, 495
(1960).
104. Kim M. K., Martell A. E., J. Am. Chem. Soc., 89, 5138 (1967).
105. Bai K. S., Martell A. E„ J. Am. Chem. Soc., 91, 4412 (1969).
106. Vallee B. L., Williams R. J. P., Coleman J. E., Nature, 190, 633 (1961).
107. Dennard A. E., Williams R. J. P., Transition Metal Chem., 1966, 115.
108. Lipscomb W. N„ Accounts Chem. Res., 3, 81 (1970).
109. Gurd F. R. N„ Bryce G. F„ in Peisach J., P. Aisen, W. E. Blumberg (eds.).
The Biochemistry of Copper, Academic Press, New York, 1966, p. 115.
110. Gurd F. R. N., Falk K., Malmstrom B. G., Vanngard T., J. Biol. Chem., 242,
5724 (1967).
111. Bradsnaw R. A., Shearer W. T., Gurd F. R. N., J. Biol. Chem., 242, 5451
(1967); 243, 3817 (1968).
112. Sarkar B., Wigfield Y„ Can. J. Biochem., 46, 601 (1968).
113. Phillips R. S. J., Chem. Rev., 66, 501 (1966).
114. Khan M. M. T., Martell A. E., J. Am. Chem. Soc., 88, 668 (1966).
115. Watters J. I., Matsumoto S., Inorg. Chem., 5, 361 (1966).
116. Murakami Y„ Takagi M., J. Phys. Chem., 72, 116 (1968).
117. Eichhorn G. L., in «Advances in Chemistry Series», No. 62, American Chemi-
cal Society, Washington, D. C., 1967, p. 378.
118. Falk J. E., Porphyrins and Metalloporphyrins, Elsevier Publishing Co., Am-
sterdam, 1964.
119. Braterman P. S., Davies R. C., Williams R. J. P., Adv. Chem. Phys., 7, 359
(1964).
120. Fleischer E. B., Accounts Chem. Res., 3, 105 (1970).
121 Phillips J. N., Rev. Pure Appl. Chem.. 10, 35 (1960).
122 Lever А. В. P„ Adv Inorg. Chem. Radiochem., 7, 27 (1965).
123. Kirksey С. H., Hambright P., Storm С. B., Inorg. Chem., 8, 2141 (1969).
124. Kirksey С. H., Hambright P., Inorg. Chem., 9, 958 (1970).
125. Miller J. R., Dorough G. D„ J. Am. Chem. Soc., 74, 3977 (1952).
126. Falk J. E., Phillips J. N., Magnusson E. A., Nature, 212, 1531 (1966); Co-
le S. J., Curthoys G. C., Magnuson E. A., J. Am. Chem. Soc., 92, 2991 (1970).
127. McLees B. D., Caughey W. S., Biochemistry, 7, 642 (1968).
ГЛАВА 3
ЭЛЕКТРОННОЕ СТРОЕНИЕ
КОМПЛЕКСОВ ЖЕЛЕЗА
Г. Б. Грэй, Г. Дж. Шугар
Gray Harry В., Arthur Amos Noyes Laboratory of Chemical
Physics, California Institute of Technology, Pasadena,
Calif. 91109, USA
Schugar Harvey Department of Chemistry, Rutgers
University, New Brunswick, N. J. 08903, USA
1. ВВЕДЕНИЕ
Исследования электронного строения ионов переходных метал-
лов в молекулах, представляющих биологический интерес, в те-
чение последних нескольких лет проводились весьма интенсивно.
Хотя для исследования таких систем в принципе применимы
все спектроскопические и магнитные методы, известные химикам-
неорганикам [1—3], получение надежных экспериментальных
данных, касающихся ионов металлов, из-за сильного разбавления
часто представляет трудную задачу. Однако при благоприятных
условиях удается выяснить особенности геометрического и элек-
тронного строения, обусловленные координацией к металлу.
Для того чтобы сохранить разумные размеры этой главы, мы
решили обсудить здесь выбранную группу модельных соединений
и родственных им белков, содержащих в качестве центрального
металла железо (Fe2+ или Fe®+). Этот выбор обусловлен широким
распространением комплексов железа в биологии. Основное вни-
мание будет уделено моделям железосодержащих белков, исполь-
зующим в качестве лигандов соединения с донорными атомами
азота и кислорода.
2. ТЕОРИЯ ПОЛЯ ЛИГАНДОВ ДЛЯ ОКТАЭДРИЧЕСКИХ
КОМПЛЕКСОВ
Вначале мы рассмотрим электронное строение Fe2+ и Fe3+ в
октаэдрических комплексах. Для обсуждения электронных и маг-
нитных спектров этих и других ионов переходных металлов наи-
более полезной моделью несомненно является теория поля лиган-
дов [1]. Коротко говоря, валентные d-орбитали металла, находя-
щегося в молекуле в октаэдрическом окружении, расщепляются
на два подуровня. Подуровень, более высокий по энергии, назы-
вается eg и включает две d-орбитали (d^-^ и dz2), которые при-
нимают сильное участие во взаимодействии с лигандами по о-типу.
134
Глава 3
Три d-орбитали в подуровне t2g (dxy, dxz, d^) из-за их простран-
ственной ориентации могут принимать участие только в невзаимо-
действии с лигандами и вследствие этого имеют более низкую
энергию, чем е£-орбитали. Расщепление на уровни eg и t2g (рис. 3.1)
называется расщеплением в октаэдрическом поле лигандов и по
общему соглашению обозначается 10Лд или ДСкт-
Рис. 3.1. Расщепление d-орбиталей в октаэдрическом комплексе в соответствии
с теорией поля лигандов.
2.1. Комплексы Fe2+
Для построения конфигурации октаэдрических комплексов ме-
таллов в основном состоянии необходимо поместить соответству-
ющее число d-электронов на t2g- и ее-орбитали. Существуют два
различных случая, которые можно проиллюстрировать на комп-
лексах Fe2+: если 10Dq относительно мало, как в [Fe(H2O)6]2+,
то шесть валентных d-электронов распространяются на подуровни
t2g и еЁ с образованием максимальной спиновой мультиплетности
в соответствии с правилом Гунда. В том случае, когда КЮд боль-
ше, чем энергия спаривания электронов на ^-орбиталях, как в
[Fe(CN)6]4-, все шесть электронов будут оставаться на подуровне
t2g. Для описания этого различия в спиновой мультиплетности
основного состояния [Fe(H2O)6]2+ называют высокоспиновым
комплексом, a [Fe(CN)6]4-— низкоспиновым. На рис. 3.2 изобра-
жены конфигурации [Fe(H2O)6]2+ и [Fe(CN)6]4- в основном со-
стоянии в соответствии с теорией поля лигандов.
Полосы поглощения в близкой инфракрасной (ИК), видимой
и ультрафиолетовой (УФ) областях возникают в результате элек-
Электронное строение комплексов железа
135
тронных переходов между t2g- и eg-подуровнями октаэдрических
комплексов. Например, электрон может быть перенесен с t2g- на
«^-подуровень в [Fe(H2O)6]2+, при этом образуется конфигурация
возбужденного состояния (t2g)3(eg)3. В стандартном обозначении
электронного состояния* этот переход описывается следующим
образом:
B^2g (^zg)4(eg)2-► 6Eg (t2g)3 (eg)3
Спектр поглощения [Fe(H2O)6]2+ типичен для высокоспиновых
октаэдрических комплексов конфигурации d6-, широкая полоса по-
-ф®--°9
—@ФФ—Ч
—@@@—t2s
высокоспиновое основное низкоспиновое основное
состояние (tzgj^feg)2 состояние (tz^)e
5Ъ3 'А'д
Рис. 3.2. Электронное строение основного состояния [Fe(HjO)e]2+ и [Fe(CN)6]4—
в соответствии с теорией поля лигандов.
глощения, расположенная в близкой ИК-области примерно при
10 000 см-1, отнесена к переходу 5T2g—*-3Eg [1]. Эта полоса обыч-
но несколько расщеплена, вероятно, вследствие того, что возбуж-
денное 5£я-состояние имеет несколько искаженную октаэдрическую
* В обозначениях BT2g и 6Eg индекс с левой стороны вверху относится к
спиновой мультиплетности основного состояния, которая равна 2S+1. Для четы-
рех неспаренных электронов S=2 и 25+1=5. Т2в (или Eg) относится к симметрии
многоэлектронной пространственной волновой функции. Рассмотрение этих обо-
значений дано в нескольких книгах [1].
136
Глава 3
симметрию. Искажение основного состояния также может иметь
некоторое значение, особенно в случае неизбежно менее симмет-
ричных комплексов типа {Fe(II)L4X2].
Низкоспиновый комплекс [Ре(СН)6]^" имеет синглетное основ-
ное состояние (S=0; 2S-H = 1) Переход в поле лиган-
дов t2g—дает два возбужденных синглетных состояния, 'Лд
и которые вследствие различного межэлектронного отталки-
вания в этих состояниях имеют различную энергию. Энергию
межэлектронного отталкивания обычно выражают при помощи
параметров Рака —В и С [1]. Вычисленное расстояние между
состояниями 1Tie и 1T2g равно 16 В. Электронный спектр погло-
щения [Fe(CN)6]4- состоит из двух разрешенных по спину полос
поля лигандов при 31 000 и 37000 см-1, которые были отнесены к
переходам Mig—^Tlg и —*-lT2g соответственно [4].
2.2. Комплексы Ее3*
При этом же наборе лигандов расщепление в октаэдрическом
поле лигандов для Fe3+ больше, чем для Fe2+, поэтому также
больше и тенденция к образованию низкоспиновой структуры ос-
Рнс. 3.3. Уровни электронной энергии
для высокоспнновой конфигурации
d5 иона металла в поле октаэдриче-
ской симметрии [отношение парамет-
ров Рака (С/В) принято равным
3,00].
новного состояния. Тем не менее мономерные октаэдрические
комплексы типа [Fe(III)O6] всегда имеют высокоспиновые ос-
новные состояния.
Электронное строение комплексов железа
137
Высокоспиновое основное состояние конфигурации 3d5—
Mlg(f2g)3(eg)2. Наиболее низкие электронные возбужденные со-
стояния в порядке повышения энергии — 47'lg, 4?2g и вырожденная
пара (4£g, 4Alg). Схема этих и других важных уровней энергии
для октаэдрического комплекса ®Alg показана иа рис. 3.3.
Рис. 3.4. Электронные спектры поглощения.
а—[Ре(Н2О)б]3+в железоаммиачных квасцах Fe^SOJa-(NH4)2SO4-24H2O; б — тетраэдрический
[Fe(III)O4l в полевом шпате ортоклазе.
Как видно из рис. 3.3, все переходы поля лигандов в комплек-
се ®Alg запрещены по спину, и таким образом можно ожидать
появления только слабых полос. Спектр поля лигандов
[Fe(H2O)6]3+ изображен верхней кривой рис. 3.4 [5]. Три полосы
с наиболее низкими энергиями отнесены к переходам ®Atg—>4Tlg,
Mlg—>4T2g и ®Alg—4Alg) соответственно. Как и следует
ожидать для переходов, запрещенных по спину, молярные коэффи-
циенты поглощения этих полос очень низки. Результаты анализа
спектра обобщены в табл. 3.1.
В связи с этим важно указать на различие в спектре поля
лигандов октаэдрической [Fe(III)O6] и тетраэдрической
[Fe(III)O4] систем. Данные для последней системы были полу-
чены в результате исследования образца полевого шпата орто-
клаза (KAlSi3O8) с небольшим содержанием Fe3+, находящегося
в тетраэдрических ячейках А13+ [5]. Спектр поглощения поля ли-
гандов этого образца состоял из трех полос — при 444, 418 и
377 нм, как показано на кривой б на рис. 3.4. Теория поля ли-
138
Глава 3
гандов для тетраэдрического [Fe(III)O4| приводит к изображен-
ной на рис. 3.5 схеме уровней энергии. Теория предсказывает [6],
что в тетраэдрическом комплексе первые две полосы поля лиган-
дов должны иметь значительно более высокие энергии, чем в кота-
эдрическом.
Мы отнесли три полосы, проявляющиеся в спектре поглоще-
ния [Fe(III)O4], к переходам на возбужденные состояния 47’ь
Рис. 3.5. Уровни электронной энер-
гии для высокоспиновой конфигу-
рации d5 иона металла в поле те-
траэдрической симметрии (С/В
принято равным 7,83).
4Г2 и (4£, Mi) соответственно. Эти отнесения обобщены в
табл. 3.2. Анализ этого спектра дает значение 10£)9тетр=7350 см“>
и величину 540 см-1 для В. Как и предполагалось [1], найдено,
что величина lOZty меньше для [Fe(III)О4], чем для [Fe(III)O6].
Понятен также тот факт, что молярные коэффициенты поглоще-
ния полос поля лигандов для не имеющего центра симметрии
[Fe(III)O4] примерно в 10 раз больше, чем для соответствующих
полос, наблюдающихся в случае модели [Fe(III)O6],
Комплексы Fe(III) с лигандами, координированными через до-
норные атомы N, а также с CN- имеют низкоспиновое основное
состояние. Так, на основании исследования магнитных свойств
[Fe(cn)3]3+ (еп=этилендиамин) и [Fe(CN)e]3- было установлено,
что основное состояние обоих этих соединений 27’2g(f2g)5. Из наи-
более низкой возбужденной конфигурации поля лигандов
(4g)4(eg)’ возникает ряд дублетных (по спину) состояний, и
вследствие этого спектры [Fe(en)3]3+ и i[Fe(CN)e]3- усложнены.
Электронное строение комплексов железа
139
Таблица 3.1
Данные об электронных спектрах [Fe(H2O)6]s+
в Fe2(SO4)s-(NH4)2SO4-24 Н2О [б]
Ml-»- • V, СМ—1а е
«л 12 600 0,05
*ТХ 18200 0,01
24200
СА, <£) 24 600 1,3
25 400
‘Л 27 700 1
а Спектр удовлетворительно интерпретируется при использо-
вании следующих параметров: 10 ДАОКТ= 13 700 см-1; С/В=3,13;
В=945 см—1.
* Индексы в Т. , Т и т. д. автор, по-видимому, опу-
® ^Б 4 g
стил для простоты изображения термов. — Прим. ред.
Таблица 3.2
Данные об электронном спектре [Fe(III)O4]
в полевом шпате ортоклазе [5]
Mi —► V, см—1 е Вычислено* 3
«л 22500 0,73 22150
23 900 0,76 24550
(%. *Е) 26500 4,1 26550
*ТЛ 29200 0,1 28 940
а Для 10В<7тетр=7350, В=540, С=4230 см-1.
Последний комплекс поглощает свет также за счет переноса за-
ряда nCN—>Fe3+, характеризующегося низкой энергией. Тем не
менее были предложены отнесения ряда переходов поля лигандов
в [Fe(en)3]^ [7] и [Fe(CN)6]*- [8].
3. СПЕКТРЫ ПЕРЕНОСА ЗАРЯДА
Как было упомянуто выше, анализ спектров поля лигандов для
комплексов Fe3+ часто усложняется присутствием интенсивной
низкоэнергетической полосы переноса заряда лиганд—>Fe3+. По-
140
Глава 3
лагают, что происхождение этих полос связано с окислительными
свойствами Fe34-. Наоборот, для Fe2+ из-за его восстановительных
свойств можно ожидать низкоэнергетического перехода Fe2+—>
—>лиганд. Классический пример поглощения за счет переноса
заряда лиганд—►Fe3+ представляет кроваво-красный цвет высоко-
спинового комплекса [FeNCS]2+ (по-видимому, [Fe(H2O)5NCS]2+).
Полагают, что при этом происходит электронный переход с выс-
шего заполненного л-уровня NCS- на уровень /2g иона Fe34-
(nNCS—^2g) [9].
Систематическое исследование переноса заряда лиганд—>-Fe3+
было проведено в случае низкоспиновых систем [Fe(III) (CNJsX]3-
>[10]. Исходный комплекс [Fe(CN)e]3- имеет полосу около
24 000 см-1, которую можно отнести к переходу переноса заряда
oCN—>/2g [8]. При замещении CN~ на Из или NCSe- появляется
дополнительное поглощение за счет переноса заряда в области
16 000—19 000 см-1, которое было отнесено к переходу переноса
заряда лХ—>Z2g [10]. Найден следующий порядок энергии воз-
буждения переноса заряда X—>/2g: NCSe^<N3<:NCS_<CN_.
4. ДИМЕРНЫЕ КОМПЛЕКСЫ Fe3+
4.1. Оксомостиковые димеры
Для водных растворов комплексов Fe3+ характерна реакция
полимеризации путем образования димерных оксо- и дигидроксо-
мостиковых структур. Был выделен и охарактеризован ряд мо-
дельных димеров, а также выяснены их спектральные и магнит-
ные свойства [11].
Наиболее полно изученная оксомостиковая система — комп-
лекс [(РеОЭДТА)2О]2_ [ОЭДТА—№-(2-оксиэтил)этилендиамин-
Н.П.П'-триуксусная кислота] [11], содержащий примерно линей-
ный (~165°) мостик Fe(III)—О—Fe(III). На рис. 3.6 сопостав-
лены магнитные моменты этилендиаммониевой (епН|+) соли этого
димерного аниона и типичного высокоспинового мономерного
комплекса [Fe(pic)2(H2O)Cl] (pic=пиколинат) в интервале тем-
ператур 10—300 К- Уменьшение рэ$ф при понижении температуры
было истолковано как антиферромагнитное поведение, возникаю-
щее из спин-спинового взаимодействия двух ионов Fe3+ со спином
S = 5/2 [И]. Результаты могут быть интерпретированы на основе
обычной модели спин-спинового взаимодействия, которая описы-
вается гамильтонианом =—2AS]S2 (J— константа пропорцио-
нальности и при обработке экспериментальных данных рассмат-
ривается как параметр). При значении /=—86 см-1 получено
прекрасное согласование с экспериментальной кривой (рис. 3.6).
В результате нескольких подобных исследований [11] стало
ясно, что для примерно линейной частицы Fe(III)—О—Fe(III)z
Электронное строение комплексов железа
141
Рис. 3.6. Зависимость магнитных моментов от температуры для модельных моно-
мерного и оксомостикового димерного комплексов Fe3+.
Рис. 3.7. Электронный спектр поглощения [Fe ОЭДТА)2О]2—.
характерна величина J, находящаяся в пределах — 95±10 см-1.
Для оксомостиковых димеров иона Fe(III) (и других ионов пере-
ходных металлов) характерно сильное поглощение в ИК-области
спектра при ~850 см-1, обусловленное антисимметричными коле-
баниями Fe—О—Fe [И, 12].
142
Глава 3
Электронный спектр [ (РеОЭДТА)2О]2- был изучен детально
[11]. Он имеет вид, типичный для октаэдрического высокоспино-
вого комплекса Fe3+. Этот спектр показан на рис. 3.7.
Первый результат, который следует отметить,-—это тот факт,
что интенсивности полос поля лигандов намного больше (ХЮ2),
чем в случае аналогичного мономера. Это повышение интенсив-
ности не представляет исключительного явления для систем со
спаренным спином и наблюдалось для многих солей Mn2+ (MnF2
[13] и KMnF3 [14]). Возможно, более интересный факт представ-
ляет появление ряда интенсивных полос в УФ-области между
25000 и 42 000 см-1. Эти полосы трудно отнести к индивидуаль-
ным центрам Fe3+, и поэтому их появление было истолковано как
одновременное возбуждение пары электронов (ОПЭ) [11]. Два
антиферромагнитно спаренных иона Fe34 в поле лигандов одно-
временно возбуждаются одним фотоном, так что энергия перехода
с хорошим приближением представляет сумму двух одноцентро-
вых энергий. Детальное отнесение электронного спектра
[(РеОЭДТА)2О]2~ дано в табл. 3.3.
Таблица 3.3
Данные об электронном спектре
]епН2 [(РеОЭДТА)2О] [ -6Н2О [11]
Полоса '’макс- см^ Отнесение
а 11 200 «А —‘Л
ь 18 200 м,—‘Г2
с 21 000 «А —
d 24 400 6 А -— 1тй
е 29 200 (а 4-6) =29400
f 32 500 (а 4- с) = 32 200
g 36 800 (64-6) = зб 400
h 42 600 (6 4-d) = 42 600
4.2. Дигидроксомостиковые димеры
Дигидроксомостиковые димеры иона железа(III) также свя-
заны с химией этого иона в водных растворах. Они отражают
реакции гидролиза и полимеризации (или оляции), характерные
—н2о
[Fe(H20)5(OH)]2+
для «кислых» ионов металлов, например:
[Fe(HgO)ep+ Н+4-[Fe(H2O)6OH]2+
/0Н\
(HzO)4Fe Fe(H2O)4
\он/
акводимер
Электронное строение комплексов железа
143
Соли акводимера никогда не были выделены в кристалличе-
ском состоянии, но вывод о его существовании был сделан на
основе анализа данных титрования [15], появления полосы в
УФ-области при ~335 нм [16] и уменьшения парамагнетизма
[17]. Однако магнитные свойства акводимера не были объяснены
[18, 19].
Единственный хорошо обоснованный случай образования ди-
гидроксомостика Fe3+ — это желто-зеленое кристаллическое веще-
/0Н\
ство, по-видимому имеющее строение [(pic)2Fe<f yFe(pic)2]
\он/
[20] . Этот комплекс имеет пик УФ-поглощения при 342 нм и
проявляет слабые антиферромагнитные свойства (7=—8 см-1).
Был получен также ряд димерных комплексов Fe3+, содержащих
диалкоксимостики и проявляющих подобные свойства [21].
5. ЭЛЕКТРОННОЕ СТРОЕНИЕ МОДЕЛЬНЫХ
ХЕЛАТОВ ЖЕЛЕЗА И РОДСТВЕННЫХ СОЕДИНЕНИЙ,
ПРЕДСТАВЛЯЮЩИХ БИОЛОГИЧЕСКИЙ ИНТЕРЕС
5.1. Димеры железопорфирина
Известно [22], что парамагнетизм гемина в щелочном раство-
ре (т. е. гидроксида гемина или продуктов его полимеризации)
восстанавливается. Возможно, с этим наблюдением связаны дан-
ные полярографических исследований [23] и исследований окис-
лительно-восстановительных потенциалов [24], которые указыва-
ют на то, что в щелочных растворах существует димерный геми-
новый продукт неопределенного строения. После того как были
получены рентгеноструктурные данные о строении [(phtcMn)2O]
(ршс=фталоцианат) [25], было предположено [18], что указан-
ные выше результаты магнитных измерений могли быть обуслов-
лены оксомостиковыми димерами железопорфирина. В связи с
результатами электрохимических исследований представляет ин-
терес тот факт, что с помощью метода быстрого установления
равновесия наблюдалось двухэлектронное восстановление
.[(РеОЭДТА)2О]2- (или [ (РеЭДТА)гО]4-), находящегося в ла-
бильном равновесии с аналогичным высокоспиновым мономером
[РеОЭДТА(ОН)]- (или .[РеЭДТА(ОН)]2-) [26].
Оксомостиковый железопорфирин был впервые синтезирован и
охарактеризован Когей и сотр. [27], которые установили строение
соединения [(диметиловый эфир дейтеропорфирина)Ре(Ш)]2О
на основе поглощения в ИК-области при 840 см-1 и данных по
определению молекулярной массы. Позже были опубликованы
данные [28], согласующиеся с этой формулой. Синтезирован так-
144
Глава 3
же [(тетрафенилпорфин) Fe(III)]2O и его строение установлено
рентгеноструктурным методом [29]. В противоположность родст-
венному высокоспиновому мономерному комплексу этот димер
антиферромагнитен. Определенный для димера угол Fe—О—Fe
168° и расстояние Fe—О 1,76 А близки к соответствующим вели-
чинам 165° и 1,79 А, найденным [30] для [(РеОЭДТА)2О]2-.
В УФ- и видимой области спектра порфириновых димеров до-
минируют интенсивные полосы, которые обычно рассматривают
как возникающие исключительно за счет интралигандного воз-
буждения или возбуждения переноса заряда. Трудно было наде-
яться установить местонахождение нормально слабых переходов
поля лигандов, локализованных на ионе железа. Однако, как об-
суждалось ранее, в [(РеОЭДТА)2О]2- и подобных соединениях
наблюдалось усиление полос поля лигандов примерно в 100 раз.
Возможно, что такое же или даже большее усиление может иметь
место в оксомостиковых железопорфириновых димерах. Кроме то-
го, можно ожидать, что такие димеры могут иметь в УФ-области
довольно интенсивные полосы, возникающие из переходов, связан-
ных с ОПЭ. По этим причинам мы полагаем, что необходимо по-
вторное исследование электронных спектров димерных железо-
порфиринов, причем особое внимание должно быть обращено на
характерные спектральные особенности структурных единиц Fe2O.
5.2. Гидроксиды железо(1И)гемопротеинов
Магнитные свойства гидроксидов железо(III)гемопротеинов и
железо(III)порфиринов сходны в том отношении, что обе группы
соединений обладают пониженным парамагнетизмом. Магнитные
свойства гидроксидов железо(III)гемопротеинов были детально
рассмотрены в важной статье Георга, Битлестоуна и Гриффитса
[31]. Они объяснили наблюденное магнитное поведение как тер-
мическое равновесие между высоко- и низкоспиновыми состояния-
ми Fe3+. Сущность этого объяснения заключается в том, что ОН-
вызывает большее расщепление поля лигандов, чем Н2О, так как
известно, что аквомономеры имеют высокоспиновое состояние.
Это любопытно с точки зрения того факта, что в простых неор-
ганических гидроксо- и акво-комплексах обычно наблюдается
обратный порядок расщепления поля лигандов (Н2О>ОН-) [1].
По стерическим соображениям образование оксомостиковых
железо(III) гемопротеиновых димеров в щелочном растворе не ка-
жется возможным. Структурные исследования показывают, что в
кристаллических метгемоглобине и метмиоглобине [32] частицы
железо(III)порфирина окружены белком. Следует, однако, заме-
тить, что в железо (III) гемоглобине обмен гемовой группы в рас-
творе происходит довольно быстро. Скорость обмена, который мо-
жет обусловливать диссоциацию железо (III) порфиринового бло-
Электронное строение комплексов железа
145
ка, увеличивается на 52% при повышении pH от 6,4 до 7,7 [33].
Таким образом, не кажется невозможным то, что при pH 10 бе-
лок может диссоциировать с отщеплением железо(III)порфирина
и последний может димеризоваться (величина рК для желе-
зо (III) гемопротеина находится в пределах 8,2—11,3 [31]). Об-
суждалась также возможность оксомостиковых структур в цито-
хром-с-оксидазе [34].
Полученный в работе [28] спектр соединения [(диметиловый
эфир дейтеропорфирина)Ре(Ш)]2О в видимой области проявляет
сильное сходство со спектрами железо(III)гемоглобина, желе-
зо (III) миоглобина и гидроксида железо(III)пероксидазы [31].
Учитывая этот факт и приведенное выше обсуждение, мы прихо-
дим к выводу, что необходимо дальнейшее выяснение связи между
магнитными свойствами и строением гидроксидов железо(III) ге-
мопротеинов в растворах.
5.3. Гемэритрин и проблема связывания кислорода
Прекрасный пример определения структуры металлопротеина
в основном с помощью спектроскопических и магнитных исследо-
ваний представляет система кислород-транспортного гемэритрина.
Клотц и сотр. (гл. 11) [35] провели обширное исследование белка
из сипункулид Golfingia gouldii. Этот белок имеет .молекулярную
массу около 108000 и состоит из восьми соединенных субъединиц,
каждая из которых содержит два атома железа, способных обра-
тимо присоединять одну молекулу кислорода. Магнитные измере-
ния [35] показали, что деоксигенированная форма белка содер-
жит высокоспиновое Fe(II). Электронный спектр поглощения этой
формы относительно понятен до 300 нм; в УФ-области появляется
пик около 280 нм, характерный для тирозина.
При окислении до мет[Ре(Ш)]-протеина появляется пик по-
глощения в видимой области. Каждая субъединица метпротеина
связывает такие лиганды, как CI-, Вг~ и N з, в комплекс 1:2с
Fe(III); положение пика в видимой области спектров поглощения
этих производных слегка различно [36]. Электронный спектр по-
глощения метхлорогемэритрина показан на рис. 3.8. Полосы при
668 и 500 нм могут быть отнесены к полосам поля лигандов в
^-комплексе Fe(III). Их умеренные интенсивности [36, 37] со-
гласуются с моделью, в которой два иона Fe(III) име-
ют спаренные спины в оксомостиковом димере типа
[Cl—Fe(III)—О—Fe(III)].
Полосы при 384 и 331 нм в метхлоропроизводном также мож-
но отнести к электронным переходам, включающим один или оба
иона Fe(III). Положения и интенсивности этих полос можно по-
нять, если отнести их к переходам, соответствующим одновремен-
ному возбуждению пары электронов в оксомостиковом димере.
10—2451
146
Глава 3
Возможно также их альтернативное отнесение к переходам пере-
носа заряда лиганд—>Fe(III). При длинах волн ниже 331 нм
поглощение тирозина увеличивается, и дополнительные пики, обус-
ловленные Fe(III), по-видимому, маскируются.
Электронный спектр поглощения оксигемэритрина показан на
рис. 3.9 [37]. Этот спектр проявляет близкое сходство со спектром
А, нм
метхлорогемэритрина, за исключением намного большей интен-
сивности полосы около 500 нм. Этот спектр можно рассматривать
как достаточное доказательство правильности предположения,
которое впервые высказал Клотц [38], что в окси-форме оба ато-
ма железа окислены до Fe(III), а Ог восстановлен до Боль-
шую интенсивность поглощения при 501 нм легко понять в этой
модели, так как подобную полосу поглощения наблюдали в комп-
лексе, образующемся при взаимодействии [Ре(Ш)ЭДТА]_ и Н2О2
в щелочном растворе [39]. Эта полоса предположительно может
быть отнесена к переходу переноса заряда —>Fe(III) или
НОО~—>Fe(III).
Электронное строение комплексов железа
147
Полосы при 370 и 317 нм в оксигемэритрине сходны с поло-
сами при 384 и 331 нм в метхлоропроизводном, и, по-видимому,
возможно их аналогичное отнесение.
Спектральные данные для метхлоро- и оксипротеинов показы-
вают, что в образовании структуры координационного центра
принимают участие димерные блоки Fe(III). Проведенные недав-
но исследования метакво- и оксигемэритринов с использованием
А, нм
магнетометрической установки с ультрачувствительным датчиком
(сверхпроводящее квантовомеханическое устройство) в основном
установили природу этих димерных блоков [40]. Данные о маг-
нитных моментах в интервале 2—200 К для окси-формы (рис. 3.10)
убедительно устанавливают антиферромагнитное поведение. Более
того, эти данные интерпретируются при допущении спаривания
спинов высокоспиновой (S=5/2) пары Fe(III) с 1=—77 см-1. Ве-
личина настолько близка к соответствующей величине для димера
[(РеОЭДТА^О]2-, что наличие оксомостиковой структуры Fe(III)
в гемэритрине кажется весьма вероятным. Данные о магнитных
моментах, приведенные на рис. 3.11, показывают, что метакво-
форма также находится в спин-спаренном антиферромагнитном
10*
148
Глава 3
состоянии. Величина J=—135 см-1 может означать, что блоки
Fe(III)—О—Fe(III) в этом случае имеют структуру, более близ-
кую к линейной.
Спектроскопические и магнитные данные почти не оставляют
сомнения в том, что связывание Ог гемэритрином происходит по
Температура, К
Рис. 3.10. Магнитные моменты оксигемэритрииа в интервале температур 2—
200 К-
Рис. 3.11. Магнитные моменты метаквогемэритрина в интервале температур
2—200 К.
механизму окислительного присоединения. Два электрона, необ-
ходимые для восстановления О2, предоставляются двумя ионами
Fe(II), которые находятся в соответствии с магнитными и спект-
ральными данными в продукте окислительного присоединения
[О2-—Fe(III)—О—Fe(III)] в виде антиферромагнитно спаренных
ионов Fe(III). Двухэлектронное восстановительное отщепление
О2 от этого блока завершает обратимый процесс.
Электронное строение комплексов железа
149-
Модель окислительного присоединения для обратимого связы-
вания Ог металлопротеинами была предложена также для гемо-
цианина [6]. Гемоцианин — это медьсодержащий белок, который
присоединяет одну молекулу О2 на каждые два атома меди
(гл. 12). Деокси-Cu(I)-форма заметно не поглощает в видимой
области. При оксигенировании белок приобретает голубую окрас-
ку, а его спектр в видимой области имеет большое число полос,
расположенных при 700 (е75), 570 (е500), 440 (е 65) и 347 нм
(е 8900) [41]. Наличие полос около 570 нм почти не оставляет
сомнений в том, что оксигемоцианин содержит Си (II). Повышен-
ная интенсивность полос поля лигандов указывает на наличие в.
нем димерного комплекса Си(II) [6]. Таким образом, спектраль-
ные данные для оксигемоцианина полностью согласуются с мо-
делью связывания О2 по механизму окислительного присоедине-
ния типа гемэритрина. Для распространения этих представлений
на оксигемоглобин необходимо предположить структуру семикрат-
но координированного Fe(IV). Обсуждение этой структуры и дру-
гих моделей связывания молекулярного кислорода в гемоглобине
приведено в других работах [6].
СПИСОК ЛИТЕРАТУРЫ
1. Lever А. В. Р., Inorganic Electronic Spectroscopy, Elsevier, New York, 1968;
Sutton D., Electronic Spectra of Transition Metal Compounds, McGraw-Hill,.
New York, 1968; Schlafer H. L., Gliemann G., Basic Principles of Ligand Field
Theory, Wiley-Interscience, New York, 1969.
2. Earnshaw A., Introduction to Magnetochemistry, Academic Press, New York,.
1968; Figgis B. N., Lewis J., in H. B. Jonassen, A. Weissberger (eds.), Techni-
ques of Inorganic Chemistry, Vol. 4, Interscience, New York, 1965, pp. 137—
248.
3. Wertheim G. K„ Mossbauer Effect: Principles and Applications, Academic Press,.
New York, 1964; Herber R. H., in F. A. Cotton (ed.), Progress in Inorganic
Chemistry, Vol. 8, Interscience, New York, 1967, pp. 1—41 and references the-
rein.
4. Gray H. B„ Beach N. A., J. Am. Chem. Soc., 85, 2922 (1963).
5. Rossman G. R., Ph. D. Thesis, California Institute of Technology, 1971.
6. Gray H. B„ Adv. Chem. Ser., 100, 365 (1971).
7. Renovitch G. A., Baker W. A., Jr., J. Am. Chem. Soc., 90, 3585 (1968).
8. Alexander I. I., Gray H. B„ J. Am. Chem. Soc., 90, 4260 (1968).
9. Jorgensen С. K., Absorption Spectra and Chemical Bonding, Addison-Wesley,
Reading, Mass., 1962.
10. Gutterman D. F., Ph. D. Thesis, Columbia University, 1969.
11. Schugar H. I., Rossman G. R., Barraclough C. G., Gray H. B., J. Am. Chem.
Soc., 94, 2683 (1972) and references therein.
12. Wing R. M., Callahan К. P., Inorg. Chem., 8, 871 (1969).
13. Lohr L. L., McClure D. S., J. Chem. Phys., 49, 3516 (1968).
14. Ferguson I., Guggenheim H. I., Tanabe Y., J. Phys. Soc. Japan, 21, 692'
(1966).
15. Hedstrom В. O. A., Arkiv. Kemi, 6, 1 (1953).
16. Milburn R. M„ Vosburgh W. C., J. Am. Chem. Soc., 77, 1352 (1955).
17. Mulay L. N., Seiwood P. W., J. Am. Chem. Soc., 77, 2693 (1955).
150
Глава 3
18. Schugar Н. J., Walling C., Jones R. B„ Gray H. B., J. Am. Chem. Soc., 89, 3712
(1967).
19. Mathe J., Bakk-Mathe E., Revue Roumaine de Chemie, 11, 225 (1966).
20. Schugar H. J., Rossman G. R., Gray H. B., J. Am. Chem. Soc., 91, 4564 (1969).
21. Wu G.-H., Rossman G. R., Gray H. B., Hammond G. S., Schugar H. J., J. Am.
Chem. Soc., 11, 990 (1972).
22. Rawlinson W. A., Scutt P. B., Aust. J. Sci. Res., A5, 173 (1952).
23. Jordan J., Bednarski T. M„ J. Am. Chem. Soc., 86, 5690 (1964).
24. Shack J.. Clark W. №.. J. Biol. Chem., 171, 143 (1947).
25. Vogt L. H., Jr., Zalkin A., Templeton D. H., Inorg. Chem., 6, 1725 (1967).
26. Schugar H. J., Hubbard A. T., Anson F. C., Gray H. B., J. Am. Chem. Soc., 91,
71 (1969).
27. Sadasivan N., Eberspaecher H. 1., Fuchsman W. H., Caughey W. S„ Bioche-
mistry, 8, 534 (1969).
28. Cohen I. A., J. Am. Chem. Soc., 91, 1980 (1969).
29. Fleischer E. B., Srivastava T. S., J. Am. Chem. Soc., 91, 2403 (1969).
30. Lippard S. J., Schugar H. J., Walling C., Inorg. Chem., 6, 1825 (1967).
31. George P., Beetlestone J., Griffith J. S., Rev. Mod. Phys., 36, 441 (1964).
32. Dickerson R. E., Geis I., The Structure and Action of Proteins, Harper and Row,
New York, 1969.
33. Bunn H. F., Jandl J. H., Proc. Natn. Acad. Sci. U. S., 56, 974 (1966).
34. Caughey W. S„ Adv. Chem. Ser., 100, 248 (1971).
35. Okamura M. Y„ Klotz I. M., Johnson С. E., Winter M. R. C., Williams R. J. P.,
Biochemistry, 8, 1951 (1969); and references therein.
36. Garbett K, Darnall D. W., Klotz I. M., Williams R. J. P., Arch. Biochem. Bio-
phys., 103, 419 (1969).
37. Simon S., Grube B., Rossman G. R., Gray H. B., unpublished data.
38. Klotz I. M., Klotz T. A., Fiess H. A., Arch. Biochem. Biophys., 68, 284 (1957).
39. Walling C., Kurz M., Schugar H. J., Inorg. Chem., 9, 931 (1970).
40. Dawson J. W., Gray H. B., Hoenig H. E., Rossman G. R„ Schredder J. M.,
Wang R.-H., Biochemistry, 11, 461 (1972).
41. Van Holde К- E., Biochemistry, 6, 93 (1967).
ЧАСТЬ П
ВЗАИМОДЕЙСТВИЕ ИОНОВ МЕТАЛЛОВ
С АМИНОКИСЛОТАМИ, ПЕПТИДАМИ
И РОДСТВЕННЫМИ ИМ ПРИРОДНЫМИ
ХЕЛА ТИРУЮЩИМИ АГЕНТАМИ
И БЕЛКАМИ
ГЛАВА 4
КОМПЛЕКСЫ МЕТАЛЛОВ
С АМИНОКИСЛОТАМИ И ПЕПТИДАМИ *
Ганс К. Фриман
Freeman Hans С., School of Chemistry, University of Sydney,
Sydney 2006, Australia
Комплексы металлов с аминокислотами и пептидами в течение
многих лет служили химикам, занимающимся изучением комп-
лексных соединений, и биохимикам благодатным материалом в их
исследованиях. Твердые производные Cu(II) и Pt(II) с аланином
были выделены Стрекером в 1850 г. [1], а еще раньше темно-
красное окрашивание, возникающее в присутствии солей Си(II)
и щелочи, было использовано Видеманом (1847 г.) для того, что-
бы охарактеризовать только что открытое соединение биурет [2].
Упариванием раствора Видеман получил кристаллический, но не-
чистый продукт; при этом он отметил, что необходимо использо-
вать избыток биурета для возникновения цветной реакции, если
в качестве щелочи использовался гидроксид аммония. Прошло
более ста лет, прежде чем структура этих кристаллов была уста-
новлена методом дифракции рентгеновских лучей, и почти столько
* Сокращения, используемые в этой главе: HL* или HL — аминокислота или
пептид в форме цвиттер-иона, например: HGly — глицин +Ь1НзСН2СОО_; HSar —
N-метилглицин, саркозин; НА1а — аланин; HAsp — аспарагиновая кислота;
HCys — цистеин; HGlu — глутаминовая кислота; HHis — гистидин; HMet—ме-
тионин; HNle — норлейцин; HSer — серин; НТуг — тирозин; H(Gly-Gly)—глицил-
глицин +NH3CH2CONHCH2COO_. LH — нейтральная форма аминокислоты или
пептида, например: GlyH — глицин NH2CH2COOH; Gly-GlyH — глицилглицин
NH2CH2CONHCH2COOH; BiuH2 — биурет NH2CONHCONH2; ImH — имидазол
C3N2H4. Символы M(II) и M2+ используются, чтобы показать, что металл нахо-
дится в степени окисления II, и для ионных частиц М2+ соответственно. К\, Кг,
Кз — ступенчатые константы образования комплексных частиц ML<"-1>+, ML£n—2>+t
ML(«—3)+, где свободный ион металла=Мп+. PPgr= [M.pHgL^”₽+‘7—r,+ ]/[Мп+]р"
•[H+]«[L_]r — общая константа устойчивости частиц MpHgL{.n₽+9—
1152
Глава 4
же времени ушло на то, чтобы современная координационная хи-
мия объяснила равновесие между ионами [Cu(Biu)2]2~ и
[Cu(NH3)4]2+ и причины красной окраски первого и голубой
второго.
После этих первых открытий взаимодействие металлов с ами-
нокислотами и пептидами стало привлекать внимание как явле-
ние, связанное с координацией, как модель реакций металлов с
• белками и как модель биологических систем, в которых свойства
белка модифицированы присоединенными к нему атомами метал-
лов. Первый обзор литературы по этому вопросу был сделан Гур-
дом и Уилкоксом в 1956 г. [3], а исчерпывающее описание ос-
новных химических свойств вплоть до 1957 г. дано Гринштейном
и Виницем [4]. Интересные примеры комплексов металлов с ами-
нокислотами и пептидами как лигандами встречаются повсемест-
но в книгах Мартелла и Калвина [5] и Накамото и МакКарти
«Спектроскопия и структура хелатных соединений металлов» [6].
В последующих обзорах особое внимание уделено таким вопро-
сам, как стереоселективность и реакционная способность [7],
спектроскопическое поведение меди в комплексах с пептидами и
белками [8], анализ кристаллической структуры [9] и роль мо-
дельных соединений для понимания активности ферментов [10].
Исчерпывающий обзор новейшей литературы вплоть до конца
1968 г. дан Гиллардом и Лаури [11] в первой периодической се-
рии специальных сообщений.
В данной главе на примере взаимодействия с небольшим чис-
лом ионов металлов рассматриваются основные группы атомов
в аминокислотах и пептидах, потенциально способные к связыва-
нию металла. Прежде чем обсуждать эти взаимодействия, отме-
тим, что связывание металла функциональными группами белка
может отличаться от связывания металла такими же группами в
малых пептидах двумя важными особенностями.
Во-первых, функциональные группы, которые связывают атомы
металлов в белках, могут быть сближены в третичной структуре
белка, а в цепи белка разделены несколькими аминокислотными
остатками. Такие функциональные группы действуют сравнитель-
но независимо одна от другой. Каждая подчиняется геометриче-
ским требованиям, налагаемым цепью белка, находящейся в не-
посредственной близости к ней, и вследствие контакта с другими
функциональными группами координируется к тому же атому
металла. Эти ограничения входят в число свойств, которые мы
хотели бы изучить, но их трудно воспроизвести в малых модельных
соединениях именно потому, что лиганды являются малыми моле-
кулами. Даже в самой простой аминокислоте, глицине, расстояние
между концевыми NH2- и СОО~-группами составляет всего не-
сколько ангстрем, а в самом простом пептиде, глицилглицине, кро-
ме этих двух концевых, есть еще пептидная СО—NH-группа. Вся-
Комплексы металлов с аминокислотами и пептидами
153;
кий раз, когда полифункциональный лиганд действует полидентат-
но, одна молекула его замещает две или большее число молекул
воды или другого монодентатного лиганда из окружения иона
металла. Образующийся хелатный комплекс имеет дополнитель-
ную термодинамическую устойчивость вследствие того, что энтро-
пия (мера неупорядоченности) системы увеличилась*. «Хелатный
эффект» объясняет, почему существует лишь небольшое число
комплексов, в которых аминокислоты или пептиды действуют как
монодентатные или нехелатирующие лиганды. Следовательно,
рискованно переносить безоговорочно закономерности взаимодей-
ствия металлов с пептидами на взаимодействие металлов с бел-
ками, так как связь металла с донорным атомом, которая неустой-
чива, может стабилизироваться, если ее образование приводит к
замыканию пяти- или шестичленного кольца.
Во-вторых, все больше появляется доказательств того, что ак-
тивные центры ферментов, в том числе и металлоферментов, на-
ходятся в полостях или карманах белковой структуры, которые
выстланы главным образом неполярными боковыми цепями амино-
кислот и моделируют, таким образом, неводные растворы. Сле-
довательно, связывание металла таким активным центром или
вблизи него по сути осуществляется в неводных растворах, ди-
электрические проницаемости которых должны отличаться от ди-
электрических проницаемостей водных растворов электролитов, в
которых было исследовано большинство комплексов металлов с
пептидами. В то время как взаимодействие металлов с пептидами
в водных растворах может адекватно представлять условия на
поверхности раздела между белком и окружающей средой, оно1
не может быть хорошей моделью того, что происходит внутри бел-
ковой молекулы.
Значительная доля сведений о местах связывания аминокислот
получена анализом кристаллической структуры. Но это не значит,
что каждое взаимодействие, которое обнаружено в данной кон-
кретной структуре, сохраняется при растворении комплекса**.
По-видимому, некоторые межмолекулярные взаимодействия не со-
храняются, в противном случае не происходило бы растворения.
Нельзя также утверждать, что взаимодействие, которое не под-
тверждено анализом кристаллической структуры, не может иметь
место в растворе. Однако неточности, связанные с тем, что дан-
ным о кристаллической структуре придается слишком большое
значение, не больше, чем опасность, возникающая от попыток де-
лать выводы о структуре на основании данных, полученных с по-
* При образовании хелата происходит также дополнительное уменьшение
энтальпии (экзо-эффект), что существенно влияет на стабильность хелата (гл. 2,
разд. 2.2.2). — Прим. ред.
** Последние исследования американских ученых подтвердили это важное
предположение. — Прим. ред.
154
Глава 4
мощью приборов, которые не прямо измеряют структурные свой-
ства. По крайней мере можно быть уверенным, что любая комп-
лексная частица, существование которой подтверждено анализом
кристаллической структуры, была в растворе, из которого выпали
кристаллы. Эти частицы могли быть не единственными в растворе
и даже не преобладающими, но невероятно, чтобы новые комплек-
сы возникали как артефакт процесса кристаллизации. Наиболее
целесообразно использовать все увеличивающееся число структур-
ных данных для построения и проверки модели процессов или
свойств, обнаруженных другими методами.
1. ФУНКЦИОНАЛЬНЫЕ ГРУППЫ АМИНОКИСЛОТ
И ПЕПТИДОВ КАК МЕТАЛЛСВЯЗЫВАЮЩИЕ ЦЕНТРЫ
Участие данной функциональной группы в связывании металла
зависит от двух факторов, а именно: насколько успешно эта функ-
циональная группа конкурирует с другими соседними и насколько
успешно ионы металла конкурируют с протонами за потенциаль-
но донорные атомы. Часть ответа на первый вопрос можно полу-
чить из анализа констант диссоциации функциональных групп.
Чем ниже значение р/Са., тем больше способность донорного атома
к образованию связи металл—лиганд. В соответствии с этим
тенденция к связыванию металла будет изменяться в следую-
щем порядке: карбоксил > имидазол> аминогруппа (рКсоон —1.8.
pKimn2 — 6,5, pKNH3+ —9,0). Однако было бы рискованно исполь-
зовать один только этот критерий, так как порядок значений рК
может быть иным, чем порядок изменения энтальпии при комп-
лексообразовании, которая является мерой относительной термо-
динамической устойчивости связей металл—лиганд и протон—ли-
ганд. И наконец (как уже отмечалось), связи с низкой энталь-
пией образования могут тем не менее стабилизироваться благо-
приятными энтропийными факторами.
1.1. Концевые аминогруппы
Несмотря на высокие значения рКо, концевые аминогруппы —
наиболее обычные места связывания металла. Координации спо-
собствуют, во-первых, сильный электронодонорный (основный) ха-
рактер атомов азота аминогрупп, во-вторых, относительно силь-
ное влияние поля лигандов аминного атома азота в комплексах
переходных металлов и, в-третьих, тот факт, что атомы кислорода
карбоксильной или пептидной группы, способные образовывать
хелатное кольцо, никогда не бывают разделены более чем тремя
или четырьмя атомами. Единственное геометрическое требование
состоит в том, чтобы угол М—N(амино)—Са был почти тетра-
Комплексы металлов с аминокислотами и пептидами
155
эдрическим (109+1° в а-аминокислотах, 110+0.4° в пептидах и
113±2° в р-аминокислотах).
Хелатообразование по концевым аминогруппам найдено в кри-
сталлических структурах всех комплексов, в которых аминокисло-
ты или пептиды действуют как бидентатные или еще более высо-
кодентатные лиганды. Типичными примерами являются Zn(Gly)2-
•Н2О (I) и Zn(Gly-Gly)2-H2O (II) [12, 13].
П
Для ряда комплексов переходных металлов с аминокислотами и
пептидами термодинамические функции реакций комплексообра-
зования M2+-f-L-^ML+ и ML++L_4±ML2 определены из темпера-
турных градиентов констант равновесия [14—16] и из калори-
метрических измерений [15, 17—19]. Наиболее значительные от-
рицательные вклады в энтальпию хелатообразования дает обра-
зование связей металл — атом азота аминогруппы [14, 15]. Изме-
нение энтальпии при связывании металла с карбоксильным
атомом кислорода фактически не благоприятствует реакции [15],
и хелатообразование всецело обусловлено увеличением энтропии в
результате освобождения акво-лигандов и взаимной нейтрализа-
ции зарядов металла и карбоксильной группы.
Координация через концевые аминогруппы без образования хе-
латных колец обнаруживается гораздо реже. Ag(I) —один из ме-
156
Глава 4
таллов, с которым комплексообразование происходит по такому
типу. Его электронная конфигурация d10 и геометрия координаци-
онного полиэдра является линейной или тетраэдрической. Замы-
кание пяти- или шестичленных хелатных колец с образованием
линейного комплекса невозможно, а тетраэдрического комплекса
затруднено, поэтому координация Ag(I) с полидентатными ли-
гандами приводит к образованию либо полиядерных комплексов,
либо таких комплексов, у которых не все функциональные группы
участвуют в связывании металла. Образование комплексов пер-
вого типа подтверждается кристаллической структурой комплекса
Ag(Gly), который состоит из бесконечной цепи звеньев
—Ag—NH2CH2COO—Ag—NH2CH2COO— (III) [20]. Этот комп-
лекс кристаллизуется также в форме [Ag(Gly)] -‘/гНгО, в котором
чередующиеся атомы серебра связаны с двумя атомами азота
аминогрупп и двумя атомами кислорода карбоксильных групп
соответственно (IV) [20]. В обеих структурах лиганды должны
быть бидентатными, так как на один атом серебра приходится
только одна молекула лиганда.
о—
О—Ag—NH2
CH2-C.
/
CH2-C.
/ о
- Ag-NH2
—Ag—ЫН2
сн2-с
/ X
о
О—Ag—О
' \
C-CH2
//
о
NH2—Ag—NH2—
H
H
О
III
IV
Известно, что связи Ag—OH2 очень слабые, так что молекулы
НгО не могут вытеснить амино- или карбоксильные группы, чтобы
таким образом выполнялось требование связывания атома Ag, по
крайней мере с двумя донорными атомами. В водных растворах
и твердых комплексах с соотношением металл — лиганд 1 :2 име-
ется больше донорных атомов, чем необходимо, поэтому образую-
щиеся связи должны иметь наибольшую энтальпию образования.
Величины энтальпии образования неизвестны, но для аминокислот
с боковой цепью, не содержащей функциональных групп, констан-
ты устойчивости, соответствующие образованию комплексов AgL
и [AgL2]_ (lg₽ioi = 3,5—4,0; lgPio2 = 6,5—7,5 [21—23]), аналогич-
ны константам устойчивости комплексов Ag — аммин и увеличи-
ваются в таком же порядке, как константы кислотной диссоциации
ATnhs аминокислотных лигандов. Отсюда заключили, что серебро
связывается только атомом азота аминогрупп [23, 24]. К анало-
гичным выводам пришли в результате исследования ИК-спектров
твердых комплексов Li[Ag(Nle)2]. В комплексе асимметричная
полоса поглощения карбоксильной группы остается неизменной по
Комплексы металлов с аминокислотами и пептидами
157
сравнению с полосой поглощения свободного норлейцина (HNle),
что дает основание предположить, что карбоксильная группа сво-
бодна [25] (хотя аргументы такого типа можно подвергнуть кри-
тике, если отсутствуют наблюдения за частотами валентных коле-
баний связей металл — лиганд [26]).
Pt(II) — другой металл, к которому аминокислоты часто коор-
динируются только через атомы азота аминогруппы. Комплексы
[Pt(Gly)4]2- (V), [Pt(GIy)3]~ (VI), [Pt(GlyH)2Cl2] (VII) и
[Pt(GlyH)2(NH3)2]2+ (VIII) охарактеризованы либо только пре-
паративно [27—29], либо по величинам контактных сдвигов сиг-
налов протонов в спектрах ПМР [30]. Монодентатная координа-
ция в этих комплексах объясняется тем, что донорами являются
атомы азота, которые, находясь близко к концу спектрохимиче-
OOCCH2NH2 /Nh2ch2COO-
'OOCCH2NH2 xnh2ch2coo
V
[Pt(Gly)4p-
Ck >NH2CH2COOH
С1/ \nh2ch2cooh
VII
[Pt(GlyH)2Cl2]
H2C—NHa^ /NH2CH2COO~
O=C-----о/ \nh2ch2coo~
VI
[Pt(Gly)3]~
H3N. /NH2CH2COOH
\pt/
HsN-/ \nh2CH2COOH
VIII
[Pt(GlyH)2(NH3)2]2+
ского ряда (источники сильного поля), вызывают значительную
стабилизацию кристаллическим полем квадратно-плоскостных
комплексов Pt(II) [т. е. энтальпия связывания возрастает за счет
энергии стабилизации кристаллическим полем (ЭСКП) конфигу-
рации d8]. Этот эффект настолько выражен, что атомы азота ами-
ногрупп могут связывать Pt(II) даже тогда, когда карбоксильные
группы молекул лиганда протонированы, как в VII и VIII, в ре-
зультате чего порядок, выведенный на основании значений рКа,
становится обратным.
1.2. Концевые и находящиеся в боковых цепях
карбоксильные группы
Уже отмечена способность карбоксильных групп участвовать
в образовании хелатных колец. С металлами, имеющими подхо-
дящую «координационную геометрию», способность карбоксиль-
ных групп участвовать в хелатообразовании зависит от присут-
ствия второго донорного атома в подходящем месте для замыка-
ния пяти- или шестичленного кольца. Это условие выполняется в
а- и р-аминокислотах, так как они имеют концевую аминогруппу,
и этим объясняется хелатная структура большинства аминокис-
лотных комплексов. В пептидах, за исключением особых случаев
158
Глава 4
(разд. 4.7), концевая карбоксильная группа неблагоприятно рас-
положена по отношению ко второй координирующейся группе,
если только это не концевая СОО-группа гистидина. Следова-
тельно, взаимодействие металлов с атомами кислорода карбок-
сильных групп в пептидах должно протекать без хелатообразова-
ния или вовсе отсутствовать. Кроме того, при тех значениях pH,
при которых аминокислоты и пептиды находятся в форме цвиттер-
ионов +NH3CHR (CONHCHR)nCOO- (и при которых такая основ-
ная группа боковой цепи, как имидазол, протонирована), отри-
цательно заряженная карбоксильная группа является почти един-
ственной группой, которая может непосредственно связываться с
металлом. В этих условиях наиболее вероятно образование связи
металл — карбоксил для тех ионов металлов, на которые не влия-
ет ЭСКП. Ионы металлов, которые образуют прочные ковалент-
ные связи с сильными донорами электронов, а также те, которые
имеют большие значения ЭСКП в присутствии лигандов сильного
поля, могут успешно конкурировать с протонами за неподелен-
ную пару электронов атома азота концевой аминогруппы, как мы
видели это на примере комплексов Pt(II) (формулы VII и VIII).
Экспериментальным доказательством монодентатной координа-
ции через атомы кислорода карбоксильных групп служат кон-
тактные сдвиги в спектрах ПМР растворов гистидинового комп-
лекса Со(II) (растворы в D2O при низких значениях pD) [31],
а также частоты валентных колебаний карбоксильных групп в
ИК-спектрах комплексов 1 : 1 D.L-аланина и ь-гистидина с эле-
ментами первого переходного ряда [32]. Кроме того, взаимодейст-
вие металлов с атомами кислорода карбоксильных групп без хе-
латообразования было обнаружено для ряда комплексов, имеющих
кристаллическую структуру и выделенных при низких значениях
pH. Можно было бы подумать, что между положительно заряжен-
ными ионами металла и отрицательно заряженными карбоксиль-
ными атомами кислорода образуется простая связь, в которой
доли ковалентного и ионного вкладов зависят от электроотрица-
тельности металла. Однако такое предположение было бы слиш-
ком оптимистичным. На рис. 4.1 показаны пять типов взаимодей-
ствий металлов с атомами кислорода карбоксильной группы, ко-
торые можно обнаружить в кристаллической структуре комплек-
сов аминокислот и пептидов. При взаимодействии по типу а один
атом металла связывается с одним карбоксильным атомом кис-
лорода. При взаимодействии по типу б «свободный» карбоксиль-
ный атом кислорода связывается, но не так сильно со вторым ато-
мом металла. В форме в карбоксильная группа более или менее
одинаково связывает два атома металла, при этом она сама ста-
новится симметричным мостиком между ними. (Различия в ИК-
спектрах дают возможность дальше подразделить типы б и в в
соответствии с расположением относительно связей металл — кис-
Комплексы металлов с аминокислотами и пептидами
159
лород на анти,анти, син,анти или син,син [33].) В формах гид
карбоксильная группа ведет себя как бидентатный лиганд, обра-
зуя несимметричное четырехчленное хелатное кольцо типа г или
симметричное кольцо типа д.
Тип а. Самый простой тип монодентатной координации через
карбоксильную группу был обнаружен только в одной структуре
М м
О 6Ч о р
чс- tc- /С- <2 .jc-
/ / м-о о
I I I
м м м
а б в г д
Рис. 4.1. Типы взаимодействий металл—карбоксильный кислород.
[Fe(HGly)SO4]-5Н2О [34]. Более точная формула этого комплек-
са [Fe(H2O)6] [Fe(HGly)2(H2O)4] (SO4)2. В октаэдрических ионах
[Fe(HGly)2(H2O)4]2+ молекулы глицина расположены в транс-
положении одна к другой и каждая связана с металлом только
одним карбоксильным атомом кислорода (IX).
I Г>онг iH?
/ о Fe - С> сн
Тип в*. Взаимодействие ионов Ag+ с аминокислотами и пепти-
дами при низких значениях pH приводит к образованию таких
комплексов, как Ag(HGly)NO3 (X) и Ag(HGly-Gly)NO3 (XI)
[20, 35]. Атомы серебра координируются приблизительно линейно
с спщспн-конфигурацией относительно карбоксильной группы.
Такой же тип взаимодействия металла с карбоксильным
кислородом, но с анти, анга-конфигурацией обнаружен в
[Nd(HGly)3(H2O)2]Cl3-H2O (XII) [36]. Большие атомы Nd(III)
имеют координационное число 8. Шесть координационных мест
заняты карбоксильными атомами кислорода, принадлежащими
шести различным молекулам глицина. Соседние атомы Nd(III)
в кристалле связаны тремя мостиковыми карбоксильными груп-
пами из трех молекул глицина, каждая из которых координиру-
ется по типу в.
Координация по типу б автором не рассматривается. — Прим. ред.
160
Глава 4
О—Ай— Q КНэ
сн2-с^ ,7С-СНз
NH3 О——°
О— Ag—О /NH~C\
♦nh3-ch2 сн2-с^ |с-сн2 ch2-nh3+
C-NH О—Ag—О
XI
Тип г. Несимметричные четырехчленные хелатные кольца со
связями металл — карбоксил встречаются в комплексах, в которых
либо невозможно, либо энергетически невыгодно образование хе-
лата с участием карбоксила и второй функциональной группы.
Именно такое взаимодействие обнаружено в кристаллической
структуре комплексов, в которых атомы Си (II) и Zn(II) связаны
с концевыми карбоксильными группами пептидов или находящи-
мися в боковых цепях карбоксильными группами остатков глут-
аминовой кислоты. Например, в (Zn(Gly-Gly-Gly)] (SO4)i/2-4Н2О
(XIII) ,[37] молекула пептида связывает один атом Zn концом,
Комплексы металлов с аминокислотами и пептидами
161
содержащим аминогруппу, а другой — концом с карбоксильной
группой. Комплексы образуют бесконечные цепи. Каждый атом
Zn координирует одну молекулу пептида через концевую амино-
группу и первый пептидный атом кислорода, а другую — через
оба кислородных атома его карбоксильной группы. В комплексе
Zn(H-iGly)-2Н2О (XIV) [38] обнаружены три различных типа
взаимодействия Zn—О (карбоксильный): одна связь Zn—©(карб-
оксильный) находится в нормальном аминокислотном хелатном
кольце, вторая связь образуется со «свободным» кислородом карб-
хш
оксильной группы в соседнем хелатном кольце (тип в), а третья
и четвертая связи являются частью несимметричного хелатного
кольца с карбоксильной группой в боковой цепи третьего амино-
кислотного лиганда (тип г). В октаэдрических комплексах коор-
динация по такому типу приводит к искажению правильной гео-
метрии, так как менее прочно связанный карбоксильный кислород
неизбежно занимает «неправильное» координационное положение
по отношению к металлу. Замена двух монодентатных лигандов
одной карбоксильной группой, по-видимому, компенсирует пони-
женную энтальпию образования, обусловленную менее благопри-
ятной геометрией связывания.
Тип д. Четырехчленное хелатное кольцо с примерно равными
расстояниями металл — карбоксильный кислород встречается в
кристалле [Ca(HGly-Gly-Gly) (Н2О)2]С12-Н2О (XV) [39]. Один из
карбоксильных атомов кислорода находится в контакте со вторым
ионом кальция. Ион Са(П) имеет координационное число 7.
Кроме трех связей Са—О (карбоксильный), образуются две связи
11—2451
162
Глава 4
Са—О (пептидный) и две связи Са—ОН2. В этих взаимодействиях
участвуют четыре различные молекулы пептида и, наоборот, каж-
дая молекула пептида связывает четыре различных иона Са(П).
Хелатные кольца (типы а, б и в). Взаимодействия металл —
карбоксильный кислород, обнаруженные в хелатных кольцах,
представлены типами а, б и в на рис. 4.1. В хелатное кольцо
включен только один атом кислорода карбоксильной группы, вто-
рой карбоксильный кислород остается свободным и может обра-
зовывать водородные связи или связываться со вторым атомом
металла в соседнем комплексе (см., например, I). Взаимодействия
по типу бив часто встречаются в кристаллических структурах,
что дает основания предположить, что они также могут быть при-
чиной образования димерных и полимерных комплексов в рас-
творах.
Относительные порядки двух связей С—О в карбоксильной
группе можно получить из длин связей при условии, что они
определены с достаточной точностью. Значения длин связей обыч-
но лежат в интервале 1,25—1,30 и 1,22—1,27 А соответственно.
В пятичленном хелатном кольце угол М—О (карбоксильный)—С
всегда близок к 114°, а в шестичленном хелатном кольце обычно
составляет 123—126°. Нет никакого правила, в соответствии с ко-
торым атом металла должен находиться в плоскости карбоксиль-
ной группы; тем не менее отклонения редко бывают больше 0,5 А
в пятичленном кольце и 0,8 А в шестичленном кольце. В тех
случаях, когда есть вторая связь карбоксильный кислород — ме-
талл, ее направление относительно карбоксильной группы опреде-
ляется относительным положением соседнего комплекса. Для
угла М—О (карбоксильный)—С при втором атоме кислорода карб-
оксильной группы получены величины угла от 113 до 125° [9].
1.3. Пептидные группы
Хорошо известно, что пептидные группы в первом приближении
должны оставаться плоскими для того, чтобы сохранялся резонанс
между каноническими формами:
Пептидный кислород обладает слабыми основными свойствами.
Связывание металла с ним слабое, и если оно происходит, то
стабилизируется близостью хелатного кольца к соседней концевой
аминогруппе. Пептидный азот связывает металл только тогда,
Комплексы металлов с аминокислотами и пептидами
163
когда процесс сопровождается диссоциацией пептидного протона
[40]. В противном случае связывание металла с пептидным азо-
том подразумевает образование четвертой связи с изменением
тригональной гибридизации sp2 в тетраэдрическую sp3 и потерю
резонанса пептидной группы. Еще до того, как было установлено
это правило, были предложены структурные формулы, показы-
вающие связывание металла атомами азота протонированных
пептидных групп. Однако в свете изложенного все эти формулы
должны быть пересмотрены.
1.4. Атомы кислорода пептидных групп
Строго говоря, имеются только два комплекса, в которых пу-
тем анализа кристаллической структуры доказано отсутствие хе-
латообразования при взаимодействии ионов металлов с пептидны-
ми атомами кислорода. Первый —2 (HCys-Gly)-Nal {или
Na (HCys-Gly) 21], в котором ион Na+ удален на 2,23 А от каждого
XV
из двух карбоксильных атомов кислорода [41]. Второй—
{Ca(HGly-Gly-Gly) (Н2О)2]С12-Н2О (XV), который мы уже опи-
сали при объяснении связывания металлов с атомами кислорода
карбоксильных групп (разд. 4.2, тип д). В этом комплексе имеют-
ся две связи Са—О (пептидный) на молекулу пептида [39]. Пеп-
тидный лиганд имеет необычную конформацию, и сообщенное для
этого комплекса значение угла N—Са—С' в центральном остатке
глицина (120°) гораздо больше, чем среднее значение в других
пептидах (111°).
Отсутствие данных о структуре ощущается менее остро для
комплексов, в которых лигандами являются не пептиды, а «пепти-
доподобные» соединения. Биурет ведет себя как монодентатный
И*
164
Глава 4
лиганд в комплексах Co(BiuH2)2Cl2 и Hg(BiuH2)2Cl2 [42]. В обе-
их структурах молекулы биурета образуют по одной связи ме-
талл — амидный кислород. Атомы металла соединены в беско-
нечные цепи металл—С12—металл мостиками из двух атомов
хлора, которые дополняют октаэдрическую координацию. Форм-
амидный комплекс Cd(HCONH2)2Cl2 имеет аналогичную структу-
ру [43]. В N-метилацетамидном комплексе Li(CH3CONHCH3)Cl
все амидные связи С = О обращены к ионам Li+, а все
амидные связи N—Н направлены к ионам С1~ [44].
В Na(CH3CONHCH3)2Br амидные атомы кислорода молекул ди-
ацетамида отстоят на 2,3 А от ионов Na+ [45].
Взаимодействие пептидных групп с ионами щелочных и ще-
лочноземельных металлов, по-видимому, имеет в значительной
степени ионный характер, но получены доказательства того, что
это взаимодействие сохраняется и в растворе. Химические сдвиги
протонов в спектрах ядерного магнитного резонанса (ЯМР) ука-
зывают на то, что взаимодействие металл — амидный кислород
аналогично тому, которое описано для структур, существующих
в растворах N-метилацетамида и ионов Al3+, Th4,4-, Mg2+ и Li+;
в таком же порядке уменьшаются длины связей металл—-лиганд
[46, 47]. Не будучи специфическим свойством отдельных связей,
взаимодействия металл-—карбоксильный кислород и металл—
пептидный кислород доказываются также тем фактом, что раство-
римость аминокислот и пептидов в воде изменяется в присутствии
галогенидов щелочных и щелочноземельных металлов [48]. На-
пример, [Ca(HGly-GIy-Gly) (Н2О)2]С12-Н2О (XV)—это только
один из ряда стехиометрических комплексов, которые образуют с
аминокислотами и пептидами хлориды, бромиды и иодиды Са(П),
Sr(II) и Ва(П). Для всех выделенных комплексов найдено, что
растворимость пептида в растворе соли больше, чем в чистой воде
[48]. Дополнительным доказательством взаимодействия кальция
с пептидом в растворе служит наблюдение обратного факта —
растворимость йодата кальция в воде возрастает в присутствии
глицилглицина и некоторых других пептидов и аминокислот [49].
Увеличение растворимости йодатов щелочноземельных металлов
было использовано для определения констант устойчивости ком-
плексов металлов с пептидами в растворе [50]. И термодинамиче-
ская, и кинетическая устойчивость этих комплексов невелика.
Еще недостаточно данных, которые подтверждали бы взаимо-
действие металлов с кислородными атомами пептидов с образо-
ванием хелатов. Из комплексов, структура которых уже обсужда-
лась, Zn(Gly-Gly)2-2H2O (II) [19] и Zn(Gly-Giy-Gly) (Sb4)i/2-
-4Н2О (XIII) [37] служат примерами комплексов с хелатными
кольцами, в которых донором помимо пептидного кислорода яв-
ляется аминный азот. Координация Cu(II) в Cu(Gly-Gly-Gly)Cl-
-11/2Н2О похожа на координацию Zn в XIII [51], и нет сомнений
Комплексы металлов с аминокислотами и пептидами
165
в том, что такой способ хелатообразования -общий в тех слу-
чаях, когда концевая аминогруппа участвует в связывании с ме-
таллом; исключение составляют комплексы, в которых пептидные
атомы азота депротоннрованы (см. ниже). Аналогично этому пеп-
тидоподобный лиганд биурет ведет себя как бидентатный хела-
тирующий агент за счет обоих своих амидных атомов кислорода
в Zn(BiuH2)2Cl2 (XVI) [52], а также в аналогичных комплексах
с Ni(II), Мп(П) (данные ИК-спектров [53, 54]) и Cu(II) (дока-
зано путем структурного анализа [55]).
Ряд методов подтверждает существование в растворах метал-
лов с пептидами пятичленных хелатных колец, в которых донора-
ми являются атомы азота аминогрупп и пептидные атомы кисло-
рода. Например, в ПМР-спектре глицилглицина в D2O имеются
два сигнала протонов, обусловленных двумя неэквивалентными
группами —СН2—. При добавлении ионов Cd2+ к раствору один
сигнал сдвигается сильнее, чем другой. Более чувствительный
сигнал должен принадлежать СН2-группам, которые расположены
ближе к донорным атомам, т. е. СН2-группам, находящимся меж-
ду NH2- и пептидной группами. Оказалось также, что при добав-
лении к раствору малых концентраций ионов Си2+ этот сигнал
исчезает первым (вследствие селективного парамагнитного уши-
рения линии). Это доказывает, что первоначальные места хелато-
образования для Cd2+ и Си2+ одни и те же. До сих пор экспери-
мент лишь идентифицировал протоны, которым соответствуют
определенные частоты в спектрах ЯМР, при этом предполагалось,
что донорные группы известны. Распространяя эти подходы на
комплексы Cd(II) с аминокислотами и пептидами с боковыми
цепями, можно дать расшифровку, которая не зависит от этого
предположения. Таким способом были подтверждены места коор-
динации в глицилглицине [56]. В спектрах три- и тетрапептидов
при низких значениях pD сигналы, которые исчезают в присутст-
вии ионов Си2+, всегда принадлежат метиленовым протонам остат-
ка аминокислоты с концевой NH2-rpynnofi; это вновь приводит к
заключению, что хелатообразование осуществляется по атому азо-
та аминогруппы и первому пептидному кислородному атому [57].
Такое же заключение получено из исследования ИК-спектров
комплексов металлов с пептидами в D2O. Когда НЬ=глицилглицин,
частота пептидной группы С = О смещается от 1645 см-1 в свобод-
ном лиганде до 1625 см-1 в [CuL]+. Когда НЬ=ди- или тригли-
цилглипин, [CuL]+ содержит и свободные и координированные
пептидные атомы кислорода и в спектре наблюдаются обе ожи-
даемые частоты С = О [58—60].
И наконец, тщательное калориметрическое изучение системы
Си(II) — пептид по-новому осветило реакцию образования этих
хелатов [18]. Некоторые результаты этого исследования приведе-
ны в табл. 4.1. Константы равновесия являются функциями как
166
Глава 4
Таблица 4.t
Термодинамические функции3 некоторых реакций Си(П) с глицином
и пептидами глицина (25 °C, /=0,1 М) [18]
Реакция AG, ккал/моль AH, ккал/моль AS, к ал моль-1 - град—1
HGG* GG- + Н+ —8,09(1) + 11,03(1) +10,6(1) +1,5(4)
HGGG GGG" 4- Н+ —7,87(1) + 10,73(1) + 10,1(1) -2,1(4)
Cu2+ + G- [CuG]+ +8,62 —11,71(4) —6,76(4) + 16,6(3)
Cu2+ + GG" [CuGG]+ +5,56(1) —7,58(1) -6,1(2) +5,0(7)
Cu2+ + GGG- <—» [CuGGG]+ —5,04(1) —6,68(1) —6,3(2) +2(1)
[CuGG]+Ci+LjGG + H+ —4,06(1) +5,54(1) +6,9(2) +4,5(7)
[CuGGG]+;«=tCiiH_1GGG + H+ —5,06(1) +6,90(1) +7,5(2) +2,0(7)
CuH.jGGG [CuH_2GGG]--|-H+ —6,78(2) +9,25(2) +7,4(2) -6,2(8)
а HG = HGly—, HGG = HGJy-GJy“, HGGG = HGIy-Gly-GIy“-
Число в скобках — стандартное отклонение, вносящее поправку в последнюю значащую
цифру.
энтальпии, так и энтропии реакции (—RTInK=AG=AH—TAS).
Величина АН определяется главным образом образованием и раз-
рывом связей металл — лиганд* и в меньшей степени изменением
стерических факторов, таких, как напряжение в хелатном кольце.
Три комплекса [CuL]+ (L-=Gly-, Gly-Gly- и Gly-Gly-Gly-) име-
ют фактически одинаковые энтальпии образования, что указывает
на то, что со всеми тремя лигандами образуются однотипные
комплексы. Это согласуется со структурными данными, свиде-
тельствующими о том, что с аминокислотами и пептидами обра-
зуются пятичленные хелатные кольца. Из данных табл. 4.1 видно,
что понижение ступенчатых констант устойчивости от [Cu(GIy)]+
к [Cu(Gly-Gly-Gly)]+ целиком обусловлено энтропийным эф-
фектом.
1.5. Пептидные атомы азота
Процессы лабилизации пептидных протонов и образования
связей металл — пептидный азот вместо связей металл — пептид-
ный кислород характерны только для металлов, имеющих такие
ЭСКП, которые могут значительно увеличиваться при замещении
лиганда слабого поля •— кислорода лигандом сильного поля —
* Величина АН зависит также от изменения степени связывания молекул
воды исходными комплексами и продуктами их диссоциации. — Прим. ред.
Комплексы металлов с аминокислотами и пептидами
167
азотом. В настоящее время известно, что это имеет место для ме-
таллов Co(III) (d6), Co(II) (d7), Ni(II), Pd(II), Pt(II) (все d8)
и Си(II) (d9). Влияние поля лигандов пептидного атома азота
само по себе проявляется неодинаково для разных ионов метал-
лов: путем стабилизации Со(1П) по сравнению с Со(П) в случае
кобальта, переходом от голубых парамагнитных октаэдрических
комплексов в желтые диамагнитные квадратно-плоскостные комп-
лексы в случае Ni(II), очень низкими значениями pH, при которых
Рис. 4.2. Титрование гидроксидом натрия тетрапептида аланилглнцилглицилгли-
цнна и его комплекса с Си (II) (-----------) (ионная сила=0,16, температура
25 °C) [61].
протоны пептида замещаются Pd(II) или Pt(II) (свидетельствуя
о жадности, с которой оба металла связывают азотсодержащие
доноры), и характерным изменением окраски от голубой через
фиолетовую к розовой («биуретовая реакция»), когда пептиды
титруют щелочами в присутствии Си(II).
Пептидные комплексы Си(II) и Ni(II) исследованы особенно
большим числом экспериментальных методов, так как они в одно
и то же время термодинамически относительно устойчивы и кине-
тически лабильны. В системах Cu(II)—пептид и Ni(II)—пептид
равновесия устанавливаются достаточно быстро [особенно в слу-
чае Си(II)], что позволяет для определения их термодинамиче-
ских параметров успешно применять метод потенциометрического
титрования и калориметрический метод. Кинетика реакции была
исследована методом остановленной струи и релаксационным ме-
тодом.
168
Глава 4
Результаты типичного потенциометрического изучения приве-
дены на рис. 4.2 для титрования тетрапептида без иона Си2+ и
в присутствии эквимолярной концентрации иона Си2+ [61]. При
pH 6 пептид находится в форме цвиттер-иона, и требуется только
1 экв ОН~, чтобы оттитровать протон из концевой \Н3-группы
в интервале pH 7—9. В присутствии Си2+ протон группы NH3
титруется при значительно более низких pH, так как ион Си2+
конкурирует с ионами Н+ за NH2-rpynny. Затем в интервале pH
от 5 до 10 титруются еще более трех протонов. Если частицы,
образуемые по реакции Cu2+ с L-, обозначить [CuL]+, последова-
тельная диссоциация трех протонов приведет к CuH_iL,
[CuH_2L]_, [CuH_3L]2-. Диссоциация с образованием CuH_iL не
имеет места, если остатком второй (считая от концевой NH2-rpyn-
пы пептида) аминокислоты является саркозил или пролил, т. е.
если первая пептидная группа не имеет диссоциирующих протонов.
Аналогично этому [CuH_2L]~ не образуется, если один такой
остаток занимает третье место в пептидной цепи. Это показывает,
что пептидные группы являются местами дополнительной диссо-
циации протонов. Депротонирование приводит к тому, что пептид-
ные атомы приобретают способность связывать металл без потери
пептидом резонансной энергии. (После того как депротонируются
все пептидные группы, способные связывать металл, дополнитель-
но еще могут лабилизоваться протоны координированных моле-
кул воды.)
Среди систем металл — пептид наиболее подробно методом по-
тенциометрического титрования была исследована система
Cu(II) — диглицилглицин [62]. Титрование проводили со стеклян-
ным электродом в 3 М растворе NaClO4 для пяти отношений
металл—лиганд при каждой из пяти общих концентраций металла
(включая нулевую). Полученные данные охватывают следующие
интервалы: 1,5<рН<11,0, общая концентрация металла от 1 до
100 мМ и общая концентрация лиганда от 2 до 250 мМ. Второй
набор данных был получен для серий растворов, в которых кон-
центрация лиганда варьировалась, а концентрация свободных
ионов водорода и общая концентрация металла сохранялись по-
стоянными. Концентрация свободных ионов металла измерялась
с помощью медь-амальгамного электрода. Полученные данные бы-
ли обработаны двумя методами — графическим и численным [62].
В результате этого исследования было обнаружено не менее
13 комплексных частиц, входящих в состав системы. Состав этих
комплексов и их константы образования приведены в табл. 4.2.
Константы образования могут быть использованы для расчета со-
става равновесной смеси, присутствующей в растворе при любых
заданных условиях. Такой расчет показал, что при низких кон-
центрациях (~10“3 М) существенно важны только моноядерные
частицы, причем главными являются три частицы: [CuL]+,
Комплексы металлов с аминокислотами и пептидами
169
Таблица 4.2
Комплексы Cu(II) с диглицилглицином [CupH5Lr]<гр+«-г)+ и ло-
гарифмы их констант устойчивости (L_ = Gly-Gly-Gly-, 25 °C, 3 М NaC10<) [62]
Состав комплекса le$Pgr Состав комплекса Состав комплекса
[CuHL]2+ [CuL]+ CuH^L [CuH_2L]_ 10,13 5,66 —0,13 —6,86 [CuH2L2]2+ CuL2 [СиН_Л2]- [СиН_2Ц]2- 19,0 10,17 3,91 —4,81 [Cu2H2L2]*+ [Cu^UP* [Cu2L2]2+ CllgH—21-<2 [Cu2H_4L2[-~ 21,0 17,3 13,12 1,44 —13,4
CuH-^L и [CuH_2L]- (табл. 4.3). При более высоких концентра-
циях (2>1О-2 М) возрастает роль полиядерных частиц с отноше-
нием металл : лиганд 1:2. При титровании раствора 10-1 М при
любых значениях pH концентрация комплекса состава 1 : 1 никог-
да не превышает половину от общей концентрации Си(II)
(табл. 4.4).
Химические выводы ясны. При низких концентрациях ряд
[CuHL]2+ ч> [CuL]+ ч—>- CuH_xL ч > [CuH_2L]-
представляет большинство присутствующих частиц; при этом под-
разумевается, что по мере повышения pH комплексы последова-
тельно депротонируются. Имеются данные в пользу образования
при более высоких концентрациях металла и лиганда следующих
серий комплексов:
[CuH2L2]2+ <-=> CuL2 <—> [CuH-iU]- ч—> [CuH_2L2]2-
и
[Cu2HL2]3+ [Cu2L2]2+ «==> Cu2H_2L2 [Cu2H_4L2]2~
Для удобства комплексы расположены в такой же последова-
тельности, в какой обсуждались их структурные соотношения, но
при этом следует помнить, что все комплексы состава 1:1, 1:2
и 2 :2 находятся в равновесии друг с другом, а также со свобод-
ным лигандом и ионами металла и водорода.
К сожалению, такое детальное описание требует накопления
большого числа данных о равновесии, а наши сведения о других
системах металл — пептид часто основаны на гораздо более огра-
ниченном числе данных. Стало обычной практикой подгонять
константы устойчивости для постулированного ряда комплексных
частиц к небольшому числу кривых титрования или даже к одной
кривой титрования. Это допустимо только с оговоркой, что при
170
Глава 4
Таблица 4.3
Состав раствора Си(П)-дкглицилглицин (концентрация ~ 10~3 М) [62]
pH Приблизительное содержание Си(П)а (%), присутствующей в виде
свободный Си2+ LCuL]+ CuH-tL [СиН-2Ц-
5 72 20 3
6 16 24 47 9
7 — 2 28 60
8 — — 4 85
9 •— — — 90
а Второстепенные частицы: [CuHL]2+ при рН<6, CuL2 при pH 6—1, [СиН—iL2]— при
pH 7—9 н [СиН—sL2]2— при pH > 9.
Таблица 4.4
Основные частицы, присутствующие в растворе Си(11)-глицилглниина
(концентрация ~10-1 М) [62]
pH Приблизительное содержание Си (II) (%), присутствующей в виде
свободный Си2+ комплексы 1 : 1 комплексы 1 : 2я комплексы 2: 2
3 60 [CuHL]2+ (35) — —
4 15 [CuHL]2+ (30) — [Cu2HL2]3+ (20)
5 [CuL]+ (20) — [Cu2L2]2+ (50)
6 CuH_1L (20) CuL2 (20) Cu2H_2L2 (30)
7 [CuH_2L]- (30) СиН-тЦ (35) Cu2H_2L2 (15)
8 [CuH_2L]“ (45) CuH_iL2 (35) [Cu2H_4LJ2- (10)
9 [CuH_2L]- (50) [CuH_2L2]- (30) [Cu2H_4L2]2- (10)
’Второстепенные частицы: [CuHjLrJH- прн pH 4. Бблыная часть приведенных частиц
дает значительный вклад в равновесие также при значениях pH на 1 выше или ниже, чем
те, при которых онн являются доминирующими.
Комплексы металлов с аминокислотами и пептидами
171
-обработке результатов не могли быть учтены все присутствующие
в данной системе частицы.
В табл. 4.1 включены некоторые термодинамические данные
для реакций депротонирования в системах Cu(II)—дипептиды и
Си(II) — трипептиды. Лабилизация пептидных протонов, по-види-
мому, в основном обусловлена энтальпийным эффектом. Депро-
тонирование ([CuL]+—>CuH-iL—>[CuH_2L]_)— процесс эндотер-
мический (Д77 положительна), но в значительно меньшей степени,
чем диссоциация протонов в незакомплексованном лиганде
(HL—*-L~). Энтальпии реакций диссоциации протонов из [CuL]+
и CuH-i'L примерно постоянны, и это приводит к поразительному
результату: более положительный энтропийный вклад сам по себе
приводит к тому, что легче удалить протон из [CuL]+, чем из
CuH_iL. Частичным объяснением этого различия является то, что
депротонирование [CuL]+ дает нейтральный комплекс, причем по-
ложительное изменение энтропии главным образом обусловлено
«хелатным эффектом»; потеря следующего протона приводит к
разделению заряженных ионов (Н+ и [CuH_2L]^), при этом мо-
лекулы воды входят в гидратную оболочку этих ионов и неупо-
рядоченность молекул воды в них понижается [18].
Большинство прямых доказательств того, что диссоциация пеп-
тидных протонов сопровождается образованием связей металл—
пептидный азот, получено из данных рентгеноструктурного анали-
за. Структура частиц [CuL]+, в которых металл хелатируется
только атомом азота аминогруппы и пептидным кислородом, уже
обсуждалась на примере [Cu(Gly-Gly-Gly)]Cl- 1!/2H2O (разд.4.4).
Аналогично этому анализом кристаллической структуры получе-
ны данные о строении комплексных частиц, в которых Си(II)
связана с депротонированным пептидным лигандом. Примерами
служат CuH-iL (XXI, XXII) [63, 64], [CuH_2L]- (в [Cu2H_4L2]2-,
XVII [65]), [CuH_3L]2- (XVIII) [66] и ,[CuH2L2]2- (XIX) [67].
Существует корреляция между структурой и окраской этих
частиц.
По мере того как число донорных пептидных атомов азота,
связанных с ионами Си(II), увеличивается, главный d—d-переход
Си(II) сдвигается в более коротковолновую область. Одновре-
менно постепенно понижаются суммы порядков связей для двух
аксиальных связей металл—лиганд до тех пор, пока в [CuH_^L]2~
аксиальные лиганды не отойдут настолько, что координационное
число меди станет 4 [9].
В то время как координированные пептидные атомы кислорода
геометрически не могут образовать более чем одно пяти- или шес-
тичленное хелатное кольцо, координация по депротонированному
пептидному атому азота создает идеальную конформацию лиган-
дов для сближения двух смежных хелатных колец, имеющих об-
щую связь металл — пептидный азот. Таким образом, в этом слу-
172
Глава 4
nh2
C^NH
n___7t,_n NH2
H2N~_c<*° °
CI
HN-C
Cl
h2n
XVI
xvn
[Cu( H _ jGly-Gly-Gly )] ?-
6 Na[Cu(H_2Gly-Gly-Gly)]- H2O
XVIH
[MlH-jGly Gly-Gly-Gly)]2-, M - Cu(II), Ni(II)
чае возможен термодинамический выигрыш в форме повышенного
хелатного эффекта, так как дополнительно еще одна монодентат-
ная молекула воды замещается пептидным лигандом. Донорными
атомами, которые в хелате чаще всего соседствуют с пептидным
азотом, являются атомы азота аминогруппы или пептидные атомы
азота на конце с NHa-группой, а также пептидный азот, пептидный
кислород, карбоксильный кислород, атом азота гистидина (имид-
азольный) или атом серы метионина (тиоэфирный) на конце с
COO-группой. В качестве примеров в дополнение к уже перечис-
ленным приведем комплексы XX, XXXVI и XXXIX.
H2C
«=? сн, с '°
I
---NH2--Си—nh2-
Н2С
CH
*1 9H*
с •
//\;
М = Ni(II), Со(Ш)
XIX
Ъимерный комплекс в CuQJ-ALa-L-H-jHlsJ^HaO
XXII
174
Глава 4
Участие пептидного атома азота в полидентатном хелатообра-
зовании определяется двумя геометрическими условиями. 1. До-
пускаются только небольшие искажения нормальной тригональной
конфигурации связей у атома азота. Этим условием объясняется
тот факт, что пептидный атом азота может быть окружен двумя
пятичленными хелатными кольцами или пяти- и шестичленным
кольцами, но не двумя шестичленными кольцами [63]. 2. Металл
и атомы пептидной группы должны быть почти копланарны. Вслед-
ствие этого, если пептидный атом азота и два соседних с ним до-
норных атома тридентатного или тетрадентатного пептида связа-
ны с одним и тем же атомом металла, они должны занимать ко-
планарное координационное положение (как в XVIII, XX и XXII).
Размеры пептидной группы слегка (но это существенно) изме-
няются при координации по атому азота пептидной группы [9].
При координации Cu(II) длина связи С = О изменяется от 1,24
до 1,26 А, а длина связи С—N от 1,325 до 1,30 А. Это свидетель-
ствует о том, что образование связи металл — пептидный азот
I I
уменьшает вклад резонансной структуры —N—С=О и увеличи-
I !
вает вклад структуры —(N+ —С—О-, т. е. сдвиг электронов к
металлу меньше, чем к протону, который в комплексе замещен
медью. Такое же заключение получено на основании понижения
частоты валентных колебаний группы С = О в ИК-спектре системы
Си(II)—пептид в D2O. По мере того как p.D повышается, частоты
свободной и координированной группы С = О (1645, 1625 см-1)
заменяются полосой 1600—1610 см-1 [58—60].
Изучение взаимодействия между пептидами и Ni(II) помогает
получить представление о связывании металла пептидными ато-
мами азота в комплексах Си(II)—пептид. Существование комп-
лекса Ni (II), в котором пептидные группы депротонированы, под-
тверждено препаративным [68], потенциометрическим [60, 69—
71], ИК-спектроскопическим [60] и кристаллографическим [72]
исследованиями; все они дают результаты, очень похожие на те,
что получены для системы Си(II)—пептиды. Анализ кристалли-
ческой структуры был выполнен для комплексных частиц
[NiH_3L]2- (XVIII) и [NiH_2L2]2- (XX) ,[72]. Термодинамическая
устойчивость комплексов Ni(II)—пептиды несколько ниже, чем
устойчивость соответствующих Си (II)-пептидных комплексов. На-
пример, константы образования некоторых комплексов Ni(II) с
HGly-Gly-Gly [в скобках даны значения констант устойчивости
комплексов Си (II), взятые из табл. 4.2] равны lg [hoi = 3,7 (5,7),
lg₽i-n = —5,1 (—0,1) и lg₽i-2i=—12,8 (—6,9). Так как различия
между ступенчатыми константами устойчивости отражают значе-
ния р/Со пептидных групп, из приведенных данных можно сделать
Комплексы металлов с аминокислотами и пептидами
175
вывод, что пептидные протоны лабилизируются Ni(II) (рКа~8)
менее эффективно, чем Си(II) (рКо~6).
Однако комплексы пептидов с Ni (II) кинетически более устой-
чивы, чем аналогичные комплексы с Си(II). В ходе потенциомет-
рического титрования необходимо выдерживать раствор по мень-
шей мере 5 мин для того, чтобы в нем после добавления каждой
порции основания наступало равновесие [69]. Методом останов-
ленной струи была исследована кинетика обратной реакции, а
именно протонирование [МН_2Ь]~. Скорость этой реакции в 25—
50 раз меньше для [Ni(H_2Gly-Gly-Gly)]_, чем для
[Cu(H_2Bly-Gly-Gly)]~ [71, 73]. С обоими комплексами реак-
ция протекает гораздо медленнее, чем процесс, контролируемый
диффузией, и стадией, определяющей скорость, является пере-
группировка из состояния с координацией М-—N (пептидный) в
состояние с координацией М—О (пептидный).
При низких значениях pH растворы комплексов Ni(II) с пеп-
тидами имеют голубовато-зеленую окраску, что свидетельствует
о том, что комплексные частицы октаэдрические и парамагнитные.
Правильная октаэдрическая геометрия позволяет Ni(II) образо-
вывать с бидентатными пептидами комплексы с отношениями ме-
талл— пептид 1:1, 1 :2 и 1:3. С тридентатными или с еще более
длинными лигандами диссоциация двух или трех протонов по ме-
ре повышения pH приводит к образованию желтых квадратно-
плоскостных диамагнитных комплексов, в которых отношение
металл — лиганд 1 : 1. Неожиданным следствием приведенных кон-
стант устойчивости комплексов Ni(II) с глицилглицином является
тот факт, что второй пептидный протон диссоциирует легче, чем
первый протон (p/<a!! = ₽i-ii—Pi-2i = 7,7; рКО1 ='₽ioi—₽i-n=8,8).
Другими словами, частицы NiH_i>L (октаэдрические) термодина-
мически неустойчивы по сравнению как с [NiL]+ (октаэдрически-
ми), так и [NiH_2L]_ (квадратно-плоскостными). Депротонирова-
ние является так называемой кооперативной реакцией, и хотя
комплекс NiH-iL должен быть интермедиатом, его содержание
никогда не превышает 5% содержания Ni(II) .в растворе [71].
Кооперативная природа перехода от [NiL]+ к i[NiH_2L]~ обус-
ловлена изменением координации от октаэдрической к квадратно-
плоскостной, при этом изменение координационной геометрии
обусловлено усилением поля лигандов по мере того, как пептид-
ные атомы азота связываются с металлом. Комплексы Ni(II)
либо октаэдрические и парамагнитные, либо квадратно-плоскост-
ные и диамагнитные. Для них в отличие от Си(II) не наблюда-
ется постепенного изменения геометрии. ЭСКП квадратно-плос-
костного комплекса Ni(II), выраженная в единицах А(10Д7),
больше, чем эта величина для октаэдрического комплекса Ni(II).
Более сильному полю лигандов в квадратно-плоскостной конфи-
гурации способствует повышение ЭСКП. Координации с одним
176
Глава 4
пептидным донором недостаточно, чтобы вызвать изменение гео-
метрии, однако возможность образования квадратно-плоскостной
конфигурации с ее более высокой ЭСКП способствует диссоциа-
ции еще одного пептидного протона, с тем чтобы получились до-
норы с более сильным полем. Кроме того, в кв а др атно-плоскост-
ных комплексах Ni (II) с пептидами донорные атомы находятся на
0,2 А ближе к иону металла, чем в октаэдрических ,[72], так что
влияние поля лигандов еще больше усиливается. Энтропийный
эффект (освобождение аксиальных лигандов) действует в этом
же направлении.
Реакция между Ni(II) и глицилглицином, по-видимому, ано-
мальна. При pH —10 Ni(Gly-Gly)2( = NiL2) теряет два протона,
образуя голубовато-зеленый ион i[Ni(H_iGly-Gly)2]2~
( = [NiH_2L2]2-) [69]. Натриевая соль этого иона кристаллизуется
в двух гидратированных модификациях, кристаллическая структу-
ра которых показывает, что комплекс является бис-тридентатным
и октаэдрическим (XX). Но, когда HL — глицинамид, частицы
[NiH_2L2]2~ желтые и квадратно-плоскостные.
Pt и Pd в периодической системе расположены под Ni и также
имеют электронную конфигурацию d8. Подобно Ni(II), двухва-
лентные ионы этих металлов вызывают ионизацию пептидных ато-
мов водорода. Они образуют квадратно-плоскостные комплексы,
в которых местами связывания металла являются депротонирован-
ные пептидные атомы азота. По мере продвижения сверху вниз
в группе периодической системы стабилизация кристаллического
поля донорами более сильного поля увеличивается и, следова-
тельно, повышается эффективность ионов металлов в лабилизации
пептидных протонов. В присутствии Pd(II) пептидные протоны
титруются при pH 3,5 [74] по сравнению с pH 8—9 для Ni(II).
Когда в растворе происходит смешивание [PtCl4]2~ с пептидами,
депротонирование пептидных групп осуществляется даже при еще
более низких значениях pH. Это демонстрируется структурой
комплекса Pt (Gly-ь-Met) С1-Н2О, который кристаллизуется при
pH 2,5 (см. формулу XXXIX [75]). Положение атомов водорода
в этом комплексе установлено методом дифракции нейтронов, так
что нет сомнения в том, что пептидные группы депротонированы,
в то время как карбоксильная группа еще нейтральна. (Отсюда
не следует, что в связывании [PtCl4]2~ с белками всегда участ-
вуют депротонированные пептидные группы, так как пептидные
атомы азота в белках обычно менее доступны, чем в растворенных
молекулах пептидов.)
Недавно было опубликовано сообщение [76], что Со(П) так-
же промотирует диссоциацию пептидных протонов. Для реакции
требуется pH — 10 и отношение металл — лиганд 1 :2. Образуют-
ся октаэдрические комплексные частицы, в которых связующие
атомы включают два аминных и два пептидных атома азота —
Комплексы металлов с аминокислотами и пептидами
177
четыре донора сильного поля. В поле сильного донора становится
предпочтительной более высокая степень окисления кобальта.
В соответствии с этим растворы, содержащие частицы
'[Con(H_iGly-Gly)2]2-, легко окисляются (через оксигенированное
промежуточное состояние, которое является обратимым носителем
молекулярного кислорода) в [Coni(H_iGly-Gly)2]~. Комплексные
частицы Сога нельзя прямо приготовить по реакции
[Coin(Gly-Gly)2] + с основанием, так как для комплексов Со(Ш)
особенно характерно затруднение разрыва связей металл —
лиганд.
Кинетическую устойчивость связей Со(Ш)—лиганд можно ис-
пользовать в экспериментах, которые невозможны с более ла-
бильными комплексами .Ni(II) и Си(II) с пептидами. Структура
иона i[Coni(H_iGly-Gly)2]~ в четырех различных солях такая же,
как структура [Ni (H_iGly-Gly)2]2- (XX) [77, 78]. Единственным
элементом симметрии для обоих комплексных ионов является ось
симметрии второго порядка, и они должны существовать в виде
энантиомеров. Это можно доказать для комплексов Со(Ш) [но
не для комплексов Ni(II)] разделением энантиомеров хромато-
графически на колонке с крахмалом [79]. При добавлении силь-
ной кислоты к раствору [Co(H_iGly-GIy)2]_ анион быстро и
обратимо протонируется, образуя [Co(GIy-Gly)2]+ [76, 77, 79].
Структурный анализ Co(Gly-Gly)2(ClO4) был использован для
доказательства того, что протонирование не затрагивает связи
металл — лиганд, а добавленные протоны присоединяются к пеп-
тидным атомам кислорода [78]. Протонированные катионы, в ко-
торых Со(III) еще связан с пептидными атомами азота, термо-
динамически не устойчивы и, по-видимому, медленно восстанав-
ливаются до Со(П) [77].
1.6. Имидазольные атомы азота
Боковые цепи, содержащие остатки гистидина, являются важ-
ными группами, связывающими металл как в природных металло-
протеинах, например карбоксипептидазах (гл. 15), миоглобине и
гемоглобине (гл. 25), так и в комплексах металла с белком, по-
лученных в лаборатории (гл. 7). При таких взаимодействиях ме-
таллов с белками функциональные группы, присоединенные к ме-
таллу, в большинстве случаев принадлежат аминокислотным
остаткам, которые не находятся по соседству друг с другом в бел-
ковой цепи. Следовательно, остатки гистидина являются типич-
ными группами, не образующими хелатов.
1.6.1. Взаимодействия металл—имидазол
без образования хелатов
Моделирование такого взаимодействия металлов с белками
более простыми системами затруднено из-за того, что гистидин и
гистидилпептиды очень склонны действовать как хелатирующие
12—2451
178
Глава 4
агенты. Единственным комплексом, в котором, как было показано,
имидазольная боковая цепь ведет себя не как хелат, является
Cu(p-Ala-L-H-iHis)-2Н2О («медный карнозин», XXI) [63]. Осо-
бенно интересно в этом комплексе то, что Cu(II) связывается
с атомом азота имидазольного кольца в положении 3 (как в ме-
тилмиоглобине кашалота [80]), в то время как в хелатах, образуе-
мых гистидином и гистидилпептидами, всегда участвует атом азо-
та в положении 1 имидазола. Еще не доказано, что структура,
обнаруженная в кристаллическом состоянии, сохраняется в раство-
ре, но такое предположение удовлетворительно объясняет данные
ранее выполненного потенциометрического титрования [63]. Су-
ществование димерных частиц было доказано по уширению линий
в спектрах ЭПР замороженных растворов комплекса независимо
от какой-либо структурной модели [81].
Одним из средств, которое было использовано для того, чтобы
нарушить склонность гистидина к хелатообразованию, было при-
готовление смешанно-лигандных комплексов, в которых содержа-
щие гистидин боковые цепи смоделированы молекулами имидазо-
ла. Существование смешанного комплекса такого типа в растворе
при pH от 5,8 до 7 впервые было доказано тем, что скорость
катализируемого имидазолом гидролиза n-нитрофенилацетата по-
нижается в присутствии Си(II) и глицилглицина [82]. Смешанные
комплексы имидазола и глицина, глицилглицина или диглицил-
глицина с Си(II), Ni(II) и Cd(II) были последовательно выде-
лены и охарактеризованы [64, 83, 84] и кристаллическая струк-
тура некоторых из них была определена [64]. Примером может
служить [Cu(H-iGly-Gly-Gly) (ImH) (Н2О)] -Н2О (XXII). Наконец,
имеется много спектроскопических и магнитных данных [85—87],
относящихся к комплексам, которые содержат только имидазол
и не содержат аминокислотных или пептидных лигандов. Четыре
комплекса такого типа, структура которых была определена, при-
ведены ниже: это [Cu(ImH)4]I2 (катионный) [88], [Zn(ImH)6]Cl2-
•4Н2О (катионный) [89], [Zn(ImH)2]Cl2 (молекулярный комп-
лекс, образованный водородными связями) [90] и [Zn(Im)2]oo
(полимерный) [91] (XXIII—XXVI).
Наиболее полезной информацией, полученной из структурного
исследования модельных соединений — смешанно-лигандных и
чисто имидазольных комплексов, а также хелатов с гистидином,—
было обнаружение гибкости имидазольной группы как лиганда.
Связь металл — имидазольный азот отклонена на 30° от плоско-
сти имидазола, как и в [Cd (ь-His) 2]-5Н2О [92], и угол
М—N (имидазольный)—С при донорном атоме лежит в интервале
121—131°. Анализом структуры [Cu(H-iGly-Gly) (ImH) (ОН2)] -
•1V2H2O и [Cu(H-iGly-Gly-Gly) (ImH) (ОН2)]-Н2О (XXII) [64]
получены доказательства, что стерически незатрудненные имид-
азольные кольца стремятся быть копланарными со связью
Комплексы металлов с аминокислотами и пептидами
179
/ HC—NH
II CH
HC-NZ/
HN—CH
I w
HC^ /N
CH
// N
HCZ II
\ _^CH
HN
Hfi >Н
/N—CIV
HC' II
hVch
,CH
'N Лзн
\\ /
HC—NH
XXV
Zn(ImH)iC12
XXIV
[Zn(ImH)6p*
XXVI
[Zn(Im)2]
N
M—N (имидазольный) и тремя другими связями металл—лиганд
(и, следовательно, в случае Си перпендикулярными к единствен-
ной занятой dzz -орбитали). Два имидазольных кольца, занимаю-
щих смежные координационные положения в октаэдрическом или
квадратно-плоскостном комплексе, соприкасались бы одно с дру-
гим, если бы они были копланарны, поэтому кольца так повернуты
относительно связи М—N (имидазольный), чтобы между ними не
было контакта [64]. В целом вращение вокруг связи М—N (имид-
азольный), по-видимому, определяется не электронными фактора-
ми, а стерическими требованиями остатка гистидина, находяще-
гося в состоянии покоя, которые возникают за счет контактов
между имидазольными группами и соседними лигандами, а также
за счет связывания водородными связями свободного (пиррольно-
го) атома азота имидазола.
12’
180
Глава 4
Гибкость связей имидазол—металл может быть одной из при-
чин эффективности гистидиновых боковых цепей как центров свя-
зывания металла в белках. Другой причиной может быть тот
простой факт, что имидазольные группы являются достаточно хо-
рошими донорами электронов для того, чтобы образовать сильные
ковалентные связи. Калориметрические измерения показали, что
связи металл—N( имидазольный) дают только немного меньшие
вклады в изменение энтальпии в реакциях комплексообразования
[116, 157], чем связи металл—^(аминный). Изменения энтальпии,
связанные с комплексообразованием ионов М2+ в растворе с
образованием [M(ImH)4]2+, составляют —23,0, —18,4 и
—16,2 ккал/моль, когда M=Cu, Ni и Zn соответственно. Измене-
ния энтальпии на первой стадии комплексообразования тех же
ионов металлов с одной молекулой имидазола равны —7,6, —5,8
и —3,8 ккал/моль [158]. Наиболее важные вклады в энергию
связей металл — имидазольный азот дает a-связывание. Возможно
также л-связывание и влияние ЭСКП. Данные о d —р -связыва-
нии изменяются от комплекса к комплексу (см. ниже), а когда
ЭСКП вообще есть, она играет меньшую роль в стабилизации
связей с имидазольным азотом, чем с атомами азота амино- и
пептидных групп. Положение имидазольного азота в спектрохи-
мической серии, по-видимому, ближе к воде (т. е. ниже аминного
и пептидного атомов азота [9, 93]).
Удивительно, что не найдено никакой корреляции между дли-
нами связей металл — имидазол и вращением имидазольных колец
относительно связей металл — азот [64], но химические, магнит-
ные и спектроскопические свойства свидетельствуют о делокали-
зации л-электронов. Во-первых, в [Cu(IrnH)4]I2 (XXIII) металл
стабилизирован в степени окисления II, так что он не претерпе-
вает обычного восстановления в присутствии ионов I- (Си2++
+I-—►CU++V2I2). Несомненно, что исключительная стабильность
Си(II) в этом комплексе обусловлена взаимодействием с четырь-
мя имидазольными лигандами. Они лежат в плоскости, которая
перпендикулярна плоскости четырех связей Си—N, и эта ориен-
тация способствует перекрыванию dxy—рж-орбиталей. Заметная
делокализация неспаренного электрона от атома Си на имидазоль-
ные кольца проявляется как сверхтонкое расщепление сигнала
меди, а также как сверхтонкое расщепление, обусловленное ато-
мами азота, в спектре ЭПР комплекса при 80 К [88]. Во-вторых, ши-
рокая полоса в ультрафиолетовом спектре Со(СО3) (ImH)2(OH2)2
подобным же образом была приписана делокализации л-электро-
нов. Структура комплекса — искаженный октаэдр, и каждый
имидазольный лиганд расположен примерно в перпендикулярной
плоскости, образуемой им самим и тремя другими связями ме-
талл—донор [94]. В-третьих, в полимерном комплексе i[Co(Im)2]00
атомы Со(II) имеют тетраэдрическую координацию. Магнитный
Комплексы металлов с аминокислотами и пептидами
181
момент аномально низкий, и это объясняется сверхобменным взаи-
модействием атомов Со(II), т. е. взаимодействием через подвиж-
ные л-электронные системы имидазольных лигандов, которые соеди-
няют атомы металла [86]. В-четвертых, комплекс [Cu(Im)2]oo
кристаллизуется в голубой, зеленой и коричневой полимерных мо-
дификациях [95]. (Известно, что голубая форма содержит четы-
рехкоординированные атомы Си (II), связанные в трехмерный по-
лимер бидентатными имидазольными лигандами [96].) В голубой
и зеленой модификациях имеются сильные взаимодействия между
атомами Си через имидазольные кольца, обнаруживаемые в каж-
дом случае по большой отрицательной константе Вейса (абсолют-
ная температура, при которой обратная магнитная восприимчи-
вость равна нулю) [95].
Третьим свойством имидазольных групп, объясняющим их по-
пулярность как центров связывания металла, является их способ-
ность к координации при физиологическом значении pH. В ряду
гистидилпептидов pKimHJ лежит между 5,5 и 7,2, т. е. на три еди-
ницы рК ниже, чем pKnhJ [97]. ЭСКП для иона переходного ме-
талла, связанного с имидазольным атомом азота, меньше, чем
ЭСКП для иона, связанного с атомом азота аминогруппы, но при
этом в случае имидазола ион металла не так жестко конкурирует
с протоном за электронную пару азота. В случае самого гистидина
равновесие между имидазолом и аминогруппами как донорами
создает интересную проблему в координационной химии.
1.6.2. Хелатообразование с гистидином
Частично протонированная форма лиганда НгН1з2+ представле-
на формулой XXVII. Константы кислотной диссоциации для ряда
H3His2+ ^=> H2His+ «=> HHis* «=> His~ <=> H^His2-
рЭВНЫ рКсоОН=1,8, рАимидазолий = 6,0, рКмнз =9,1 И рАимидазол =
= 14. Очень соблазнительно допустить, что последовательность, в
которой удаляются протоны при титровании, соответствует поряд-
ку, в котором потенциально донорные атомы используются для
связывания металла по мере повышения pH. Согласно этой после-
довательности, при комплексообразовании металлов с гистидином
должны быть такие стадии: (1) монодентатная координация по
карбоксильному кислороду при низких значениях pH; (2) связы-
вание металла имидазольным атомом азота, приводящее к обра-
зованию семичленного хелатного кольца; (3) связывание по атому
азота аминогруппы, делающее гистидин трехдентатным лигандом,
и, возможно, (4) диссоциация второго протона из имидазольной
группы.
182
Глава 4
XXVII
H3His2*
Опубликованные данные о кристаллической структуре комп-
лексов металлов с гистидином не проливают свет на порядок дис-
социации протонов и порядок реакций связывания металлов. За
одним исключением (см. ниже), все они относятся к области pH,
в которой гистидин ведет себя как трехдентатный хелатирующий
агент [стадия (3)]. Комплексы Со(П) [98, 99] и Ni(II) i[100] с
гистидином, как и ожидали, являются октаэдрическими (XXVIII).
Комплекс Zn(II) имеет тетраэдрическую структуру с сильными
связями Zn—N (аминный) и Zn—N (имидазольный); имеются еще
более слабые связи Zn—О (карбоксильный) в направлении от двух
граней тетраэдра (XXIX).
Кристаллические производные Zn(II) с d.l-гистидином имеют
структуру, состоящую из равного числа комплексов Zn(L-His)2 и
Zn( D-His)г- Однако частицы Zn( ь-His)2 в комплексах Zn(t-His)2-
-2Н2О и Zn (D-His) ( ь-His) -5Н2О имеют различные конфигурации
[101а]. Это явление уже обсуждалось ранее и было названо «сте-
реоселективность» [9]. Комплекс Cd(II) с гистидином ведет себя
подобно комплексу Zn(II), но имеет более правильную геометрию
[92].
Доказательства того, что последовательность реакций согласу-
ется с порядком значений рКо, во многих случаях были выведены
из ИК-спектров (хотя, к сожалению, наблюдения не были рас-
пространены на область, в которой можно измерять частоты свя-
зей металл — лиганд) [32]. При очень низких значениях pH
(pD=2,2—3 в растворе D2O) комплексообразование с Мп(П),
Fe(II), Со(П), Ni(II), Zn(II) и Cd(II) при отношениях 1 : 1 вызы-
вает в ИК-спектре одинаковые сдвиги полосы антисимметричного
колебания карбоксильной группы как а-аланина (1615 см-*’), так
и гистидина (1625 см-1). Это доказывает монодентатную коорди-
нацию по СОО--группе. При значениях pD выше 3 в спектрах
комплексов с гистидином появляется полоса, приписываемая ва-
Комплексы металлов с аминокислотами и пептидами
183
ZnfL-HisJg в (a) Zn(L-His)z-2Hz0 и (d) Zn(D-His)(L"His)-5H20
лентным колебаниям имидазольного кольца (1490—1501 см-1).
Другая полоса, отнесенная к антисимметричным валентным коле-
баниям связи NCa—С (карбоксильный) (1110 см-1), при значениях
pD от 4 до 7 замещается новой полосой (1100 см-1), соответствую-
щей координации по азоту аминогруппы. [Для Си(II), Zn(II) и
Cd(II) выпадение осадка мешает наблюдению при значениях pD,
достаточно высоких для связывания металла по азоту амино-
группы.]
Литература изобилует константами образования [101—104]
и величинами энтальпии и энтропии [17, 105, 106] для комплексов
металлов с гистидином, но большинство этих данных и в этом
случае относятся только к образованию частиц [M(His)]+ и
M(His)2 [стадия (3)]. Система Со(П) — гистидин является одной
из немногих, которые были изучены в широкой области pH и при
помощи большого числа экспериментальных методов.
384 Глава 4
Интерес к этой системе отчасти возникает из-за способности
Con(His)2 образовывать оксигенированные производные, из кото-
рых можно снова получить молекулярный кислород [76, 107—
109], но эта проблема здесь обсуждаться не будет. Измерения
контактных сдвигов в ПМР-спектрах растворов Со(II) — гистидин
[31] показали, что монодентатная координация по карбоксильно-
му кислороду имеет место при рН<4, а октаэдрические комплек-
сы состава 1:1 и 1:2, в которых гистидин тридентатен, сущест-
вуют при 4<рН<11 и что тетраэдрический комплекс образуется
даже при более высоких значениях pH. Существование октаэдри-
ческого комплекса подтверждается анализом кристаллической
структуры обоих комплексов: Со( L-His)2-H2O (XXVIII) [98] и
Co(b-His) (D-His)-2Н2О [99], что находится в согласии с выво-
дами, сделанными на основании сдвигов в спектрах ПМР [31]
и данных потенциометрического титрования [НО] о том, что в
растворе эти частицы более устойчивы, чем оптически чистые.
Предложенная тетраэдрическая структура голубого комплекса,
существующего при высоких значениях pH, [Со( i.-H_1His)2]2“,
подтверждается как потенциометрическим титрованием, так и от-
несением полос поглощения в УФ- и видимой частях спектра
[111]. Эти частицы образуются после диссоциации второго («пир-
рольного») протона имидазольной группы. Гистидиновые лиганды
бидентатны; донорами являются азот аминогруппы и имидазоль-
ный азот, а тетраэдрическая конфигурация объясняется отталки-
ванием зарядов.
Методом ПМР-спектроскопии было исследовано образование
комплекса 1:2 Pt(II) с ь-гистидином. Химические сдвиги пока-
зали, что, как и ожидали для Pt(II), координация квадратно-
плоскостная и донорными атомами являются азот аминогруппы
и имидазольный азот во всей области pH: 1^рН^12. Большие
XXX
координация в [Pt(HHis)2]z*
XXXI
координация в [Pt(H_, Н18)2)‘
сдвиги в сильное поле для протонов Са : Н и С2 : Н при повыше-
нии pH отчетливо указывают на диссоциацию протонов как из
Комплексы металлов с аминокислотами и пептидами
185
карбоксильной, так и из имидазольной групп. Таким образом,
гистидин образует хелаты с Pt(II), которым соответствуют фор-
мулы XXX и XXXI при низких и высоких значениях pH соответ-
ственно. Комплексы существуют в виде двух групп изомеров, и
для обоих групп был сделан полный анализ спектров. Химические
сдвиги протонов С2: Н дают возможность идентифицировать один
ряд изомеров как цис, а другой как транс. И, наконец, на осно-
вании спин-спиновых взаимодействий *Н—'Н и 195Pt—*Н можно,
дать отнесение конформаций хелатного кольца во всех частицах.
Для аналогичного комплекса Pd(II), Pd(L-His)2, длина волны
максимума УФ-поглощения и характер спектра кругового дихро-
изма (КД) (и то и другое по сравнению с аналогичными данными
для комплексов с другими аминокислотами) показывают, что
структура комплекса квадратно-плоскостная со связями
Pd—N(аминный) и Pd—N(имидазольный) [74].
Таким образом, Pd(II) и Pt(II) служат примерами, когда по-
рядок связывания металла не всегда такой, как порядок, в кото-
ром депротонируются потенциальные донорные атомы в отсутствие
ионов металла. Нет сомнения, что причину этого следует искать
в значительном увеличении ЭСКП при связывании Pd(II) и Pt(II)
с аминным и имидазольным атомами азота вместо карбоксильного
кислорода и имидазольного азота. Этот эффект может оказывать
влияние только на те ионы металлов, которые «чувствуют» стаби-
лизацию кристаллического поля, но есть и вторая причина того,
почему порядок значений рКа не всегда соблюдается при связы-
вании металлов. Стадия (2) в предложенной выше последова-
тельности приводит к обпазованию семичленного хелатного коль-
ца (XXXII), хотя известно, что энтропийный фактор не благопри-
семичленное хелатное кольцо 2+
в предложенной структуре [M(HHis)]
ятствует его образованию. Семичленные хелатные кольца редко
находят в других координационных соединениях, и (несмотря на
данные ИК-спектров [32]) еще предстоит объяснить, почему они
186
Глава 4
стабилизируются, когда гистидин и гистидилпептиды образуют
хелаты с металлами по атомам карбоксильного кислорода и имид-
азольного азота.
В случае взаимодействия Си(II) с гистидином существует
прямое и неразрешенное противоречие между данными ИК-спект-
ров для комплекса состава 1 : 1 в D2O [32] и анализом кристалли-
ческой структуры комплекса 1 :2. ИК-спектры показывают, что
за связыванием Си(II) с карбоксильным кислородом при очень
низких значениях pH следует при немного более высоких pH об-
разование семичленного хелатного кольца, в котором вторым до-
норным атомом является имидазольный азот. Исследования мето-
дами температурного скачка и остановленной струи реакции меж-
ду Си2+ и HHis* в интервале pH от 2,5 до 4,0 согласуются с
образованием комплексов [Cu(HHis)]+ и [Cu(HHis)2]2+; в этих
реакциях стадией, определяющей скорость, является замыкание
семичленных хелатных колец [О(карбоксильный), N(имидазоль-
ный)]. Авторы этих работ не могут воспроизвести наблюдаемую
кинетику, если их расчеты основываются на образовании пяти-
членных хелатных колец (по карбоксильному кислороду и амин-
ному азоту) [112]. По-видимому, кинетические расчеты не учи-
тывают существования других частиц, кроме [Cu(HHis)]2+ и
[Cu(HHis)2]2+. Новейшее и наиболее обширное исследование си-
стемы Си(II)—гистидин методом потенциометрического титрова-
ния показало, что в условиях, при которых измерялась скорость
некоторых реакций, частицы [Cu(HHis)]+ и [Cu(HHis) (His)] +
должны составлять до 7 и 24% соответственно от общего количе-
ства Си. В противоположность данным ИК-спектроскоиии и кине-
тическим данным анализ кристаллической структуры комплекса,
кристаллизующегося при pH 3,7, [Cu(b-HHis)2(H2O)2] (NO3)2,
показал, что молекулы гистидина координируются через атомы
азота аминогруппы и карбоксильного кислорода и что оба атома
в каждой имидазольной группе протонированы и не участвуют в
хелатообразовании (XXXIII) [113].
ск
.СН2 I
''С-
н
HC-NH
он2 сн2
°\J -'NH2"CH
NHj | --
он2
нсАсн
HN
Какой бы ни была
при низких значениях
хххш
структура комплексов Си(II) с гистидином
pH—[Cu(HHis)]2+ или [Cu(HHis)2]2+,—
Комплексы металлов с аминокислотами и пептидами
187
имеются веские доказательства в пользу того, что при более вы-
соких значениях pH координация в частицах [Cu(His)]+ и
Си (His) 2 главным образом осуществляется по атомам азота ами-
ногруппы и имидазольного кольца. Участие атома азота амино-
группы в области pH от 5 до 11 подтверждается данными ИК-
спектров упомянутого выше типа [32], а прямым доказательством
связывания меди имидазольной группой является парамагнитное
уширение сигнала ЯМР протонов С2 : Н и С4: Н имидазольного
кольца. Константы образования [115], энтальпии образования [105,
116] и разложение с помощью Н2О2 комплексов Си (II) с гистиди-
ном очень похожи на те же величины для комплексов Cu(II) с
гистамином, образующихся в таких же условиях. (Гистамин не име-
ет карбоксильной группы, так что металл должен связываться с
атомами азота аминогруппы и имидазольного кольца.) Аналогичные
доказательства получены из спектров дисперсии оптического враще-
ния (ДОВ) комплекса Cu(His)2 при pH 8. Резких изменений в этих
спектрах не наблюдается при замене гистидина его метиловым
эфиром (это показывает, что вклад карбоксильной группы гисти-
дина в координацию незначителен), но они сильно отличаются от
спектров ДОВ комплекса с 1-метилгистидином, в котором невоз-
можна координация через азот имидазольного кольца [Н7]. Эти
заключения подтверждаются рентгеноструктурными данными.
В комплексе [Си ( ь-His) ( ь-Thr) (Н2О)] -Н2О, который преоблада-
ет в растворе, содержащем смесь лигандов при pH 6—8, гисти-
динатный лиганд образует сильные связи Си—N (аминный) и
Си—N (имидазольный) и более слабую связь Си—О (карбоксиль-
ный) в противоположном направлении (XXXIV) [118].
Однако недавно на основании исследования спектров ДОВ и КД
некоторых смешанно-лигандных комплексов [119] было сделано
предположение, что при pH 8,3 комплекс Cu(His)2 сам является
смешанным комплексом, в котором один гистидинатный остаток
является тридентатным, как в XXXIV, а другой образует хелат
по атому азота аминогруппы и карбоксильному кислороду, как
188
Глава 4
в ХХХШ. Ко времени написания этого обзора эти результаты
были подвергнуты сомнению [114, 117]. Ясно, что последнее слово
по поводу этой интересной проблемы еще не сказано.
За исключением комплекса Си (И) с карнозином [63, 120—
122], по поводу хелатов металлов с гистидилпептидами возникает
гораздо меньше противоречий и страстей по сравнению с гистиди-
ном. Мы отсылаем читателя к оригинальным работам [93, 97].
1.7. Гидроксильные атомы кислорода боковых цепей
Гидроксильные группы боковых цепей серина, треонина и ти-
розина, по-видимому, не являются местами связывания металла.
ПМР-исследование серина в присутствии ионов Cd2+ не показало
изменения химического сдвига сигнала протонов, обусловленного
связыванием Cd—ОН [56]. Энтальпия образования Cu(Ser)2 при
pH от 9,1 до 10,1 (от —12,6 до —13,4 ккал/моль) хорошо согласу-
ется со значениями А// для Cu(Gly)2 и Си(А1а)2 (от —12 до —13
и от —11 до —12 ккал/моль соответственно), в случае которых
можно не сомневаться в том, что нет взаимодействия с функцио-
нальными группами боковых цепей [106]. (При pH 12 ДЯ для
Си (Ser) 2 резко возрастает до —23 ккал/моль, возможно, вслед-
ствие образования полиядерных частиц с мостиковыми гидроксо-
группами [106].) Для других металлов первого переходного ряда
константы устойчивости комплексов с серином и треонином, с од-
ной стороны, и комплексов с а-аланином и ц-аминомасляной
кислотой—с другой, настолько похожи, что предполагается, что
им всем свойствен один и тот же тип хелатообразования [16].
Анализ кристаллической структуры Ni(b-Ser)2(OH2)2 [123],
Cu(b-Ser)2 [124] и Zn( L-Ser)2 [125] подтверждает, что коорди-
нация осуществляется только через атом азота аминогруппы и
карбоксильный кислород. В Zn( L-Ser)2 имеет место взаимодействие
XXXV
координация в Zn(L-Ser)2 ;
без образования хелата между боковой цепью серина в одном
комплексе и атомом Zn соседнего комплекса (XXXV), но приве-
Комплексы металлов с аминокислотами и пептидами
189
денные термодинамические данные заставляют предположить, что
это взаимодействие не играет важной роли в растворе.
Уместно упомянуть здесь о неожиданном типе контакта между
атомами Си(П) и боковой цепью тирозина, обнаруженного струк-
турным анализом Cu(Gly-L-Leu- ь-Н_]Туг) -4Н2О- 1/2(С2Н5)2О
[126]. Координация Си пятью атомами лиганда в этом комплексе
XXXVI
Ъшиер Си (Gly-lrLeu-L-H-i Туг) '4 Н20 • 2(C2Hs)20
квардатно-пирамидальная (XXXVI). Фенольное кольцо располо-
жено примерно параллельно основанию пирамиды, а несколько
ароматических атомов углерода осуществляют связи с атомом Си,
которые значительно короче (3,2—3,3 А), чем рассчитанные по
сумме вандерваальсовых радиусов. Вторая структурная особен-
ность этого комплекса представляет особый интерес в связи с ре-
акцией депротонирования пептидных групп (разд. 4.5). Это комп-
лекс такого же типа, как (CuH_]L)2, но депротонированная пеп-
тидная группа лиганда находится по соседству с концом, содер-
жащим карбоксильную группу, а не аминогруппу. Трипептиды
образуют пятичленные хелатные кольца с двумя атомами Си, в
которых каждый атом Си связан с двумя молекулами лиганда;
этим объясняется их димерный состав.
1.8. Сульфгидрильные атомы серы*
Металлы, которые явно предпочитают серусодержащие доноры
(по сравнению с кислородсодержащими), занимают треугольник
* Обсуждение серусодержащих лигандов содержит полезные замечания
профессора Ливингстона.
90
Глава 4
в центре периодической таблицы и относятся к «классу (б)»
[127]. Они характеризуются способностью образовывать не только-
сильные о-связи с легко поляризуемыми лигандами, но также и
л-связи посредством обратного переноса электронов с Дя-орбита-
лей металла на dn- или рл-орбитали лиганда. Даны три объясне-
ния этой способности серусодержащих лигандов [128].
1. Электроотрицательность серы низкая (гораздо ниже, чем
брома, и примерно равна электроотрицательности углерода), а ее
поляризуемость высокая. Атом серы сильно поляризуется в поле
маленького иона металла с высокой плотностью заряда, даже
если ион металла имеет конфигурацию d10 i[Cu(I), Ag(I), Hg(II)].
2. Расчеты показали, что для ионов с конфигурацией d10 ни
поляризуемость, ни теплота образования простой ковалентной
связи не могут объяснить даже порядков устойчивости сравни-
ваемых связей металл—О и металл—S [128]. Поэтому пришли
к заключению о существовании dn—^п-связи металл—>-лиганд,
хотя следует признать, что в настоящее время еще нет прямых
доказательств этого.
3. Возможно, в случае связей с низкоспиновыми ионами с кон-
фигурацией d8 [Pd(II), Pt(II), Au(III)] такая дополнительная
стабилизация вызвана увеличением ЭСКП. Этот вопрос оконча-
тельно еще не решен, так как серусодержащие лиганды, по-види-
мому, должны находиться в различных местах спектрохимической
серии. Некоторые занимают место ближе к более низким значе-
ниям спектрохимического параметра вблизи иона Вг, другие, на-
пример тиоэфиры, занимают положения с промежуточными и вы-
сокими значениями спектрохимического параметра [129].
Хорошо известно особое сродство цистеинильных боковых це-
пей к Ag(I) и Hg(II); это свойство широко использовалось для
приготовления производных белков с тяжелыми атомами. Устой-
чивость [Ag(H_iCys)2]3- примерно такая же, как устойчивость
[Ag(CN)2]~. В растворах с низкими значениями pH доминируют
частицы [Ag(HCys)2]+ и комплексообразование в этой системе
заметно даже при pH 1,3 [130]. Однако связывание металлов
ионизированными тиоловыми группами боковых цепей цистеина
не ограничивается ионами металлов «класса (б)». Константы об-
разования цистеинатных комплексов Мп(П), Fe(II), Со(П),
Ni(II), Zn(II) и Pb(II) [131] значительно выше, чем константы
образования глицинатных [131—134] и гистидинатных [102, 104]
комплексов тех же металлов (табл. 4.5). Многие другие свойства
подтверждают, что повышенные устойчивости во всех случаях
сопровождаются связыванием металла с серой.
Например, в ИК-спектрах твердых образцов ряда цистеиновых
комплексов Zn(II), Cd(II), Hg(II) и Pb(II) отсутствует погло-
щение, обусловленное группой S—Н [108]. Два из этих комплек-
сов — Na2[Zn(H-iCys)2] -4Н2О и [Zn3(Cys)4] [ZnCl4],— очевидно,
Комплексы металлов с аминокислотами и пептидами
191
Таблица 4.5
Логарифмы констант устойчивости некоторых комплексов ML+ и ML2
с глицином [132—134], гистидином [102, 104] и цистеином [131]
L tePioi lg₽102
Gly- His- Cys— Gly— His- Cys-
Мп(П) 3,2 3,2 5,8 5,5 6,1 10,3
Fe(H) 4,1 — 7,7 7,6 — 14,2
Со(П) 4,8 6,7 9,4 8,6 12,1 17,4
Ni(II) 5,7 8,4 10,8 10,6 15,1 23,5
Cu(II) 8,2 10,1 — 15,2 18,1 —
Zn(II) 5,0 6,3 10,3 9,2 11,7 20,0
Pb(II) 5,3 6,0 13,8 8,6 9,0 18,1
получены при высоких и низких значениях pH соответственно.
В спектре первого появление полосы НН2-группы указывает на
связывание металла с азотом аминогруппы, в то время как час-
тоты карбоксилатных групп (1588 и 1404 см-1) такие же, как для
свободной СОО~-группы. Анализ кристаллической структуры под-
тверждает, что геометрия координации является тетраэдрической
и что донорными атомами действительно являются азот амино-
группы и сера сульфгидрильной группы [136]. Существование и
структура полиядерного иона [Zn{Zn(Cys)2}2]2+ во втором комп-
лексе еще находятся под сомнением, но дублетная полоса (1510,
1600 см-1) и сильно асимметричная полоса валентных колебаний
карбоксилатной группы (1642 см-1) интерпретируются как доказа-
тельство того, что NHs-группы цистеина свободны, а СОО_-груп-
пы координированы. В соответствии с этим цистеин координирует-
ся к Zn(II) атомами азота и серы при высоких значениях pH и
атомами кислорода и серы при низких значениях pH [135]. При
отсутствии эффектов кристаллического поля такая координация
соответствует ожидаемой.
Качественное доказательство связывания металла с серой полу-
чают также из сравнения спектров в ультрафиолетовой и видимой
областях цистеинатных и других аминоацидатных комплексов.
Молярный коэффициент поглощения Pd(Cys)2 (в 1300 М^'-см-1
при ХМакс 337 нм) в 3 раза больше, чем молярные коэффициенты
поглощения соответствующих полос в УФ-спектрах комплексов
Pd(II), для которых известно, что аминокислоты координируются
через атомы азота аминогруппы и атомы кислорода карбоксиль-
ной группы [74]. Сравнение спектров комплексов Со (III) с цистеи-
192
Глава 4
ном NH2—СН2—СН2—SH и НООС—СН2—СН2—SH показало, что
в K3[Coin(H_iCys)3]-ЗН2О (зеленый, высокие pH) координация
осуществляется через атом азота аминогруппы и атом серы сульф-
гидрильной группы, а в Co(III) (Cys)3-3H2O (красный, низкие
pH) — через карбоксильный кислород и сульфгидрильную серу.
ИК-спектр зеленого комплекса подтверждает, что карбоксильные
группы в нем не координированы [138].
Комплексы Fe(III) с цистеином особенно важны, так как они
имеют отношение к изучению ферредоксина (гл. 22). Fe3+ (подоб-
но Си2+) катализирует окисление цистеина. Чрезвычайно лабиль-
ные комплексы с соотношениями металл—лиганд 1 : 1 (голубой),
1:2 (красный) и 1:3 (фиолетовая и зеленая формы, термически
равноценные) были выделены при —78 °C. Координация осущест-
вляется через кислород и серу в голубой, красной и фиолетовой
формах и через азот и серу в зеленой форме. Это доказывается
спектрами поглощения, ДОВ и КД, а также сравнением этих
спектров со спектрами аналогичных тиогликолятных комплексов
[139]. В родственном комплексе [Fen( L-Cys)2(CO)2] координация
осуществляется через атомы азота и серы. Тиоловая группа обла-
дает большим сродством к иону Ag+, чем к иону Fe2+, и ионы
Ag+ превращают комплекс в [Fe( L-CysAg)2(CO)2], в котором
Fe(II) связывается с цистеинатным лигандом теперь уже посред-
ством атомов азота аминогруппы и карбоксильного кислорода
[140].
Взаимодействие Си(II) с цистеином также осложняется вос-
становлением иона металла. Аминокислота окисляется до цисти-
на. Взаимодействие Cu(I) с меркаптидами широко исследовалось
титрованием в водно-ацетонитрильных растворах [141].
Упоминавшиеся до сих пор комплексы служили примерами
монодентатной координации только по атому серы и бидентатно-
го хелатообразования по N и S, N и О, О и S. Выводы о триден-
татном хелатообразовании со всеми функциональными группами
цистеина основаны на детальной интерпретации спектров ДОВ
растворов, содержащих ионы Со(II) и цистеинатные ионы [142].
Такая координация обнаружена также в кристаллической струк-
туре Na2[Moy (L-H_1Cys)2O4]-5H2O (XXXVII) [143]. Другими
XXX VII
Комплексы металлов с аминокислотами и пептидами
193
интересными особенностями этой структуры являются мостики
Mov—О—Mov, двойные связи Моу=Оисвязи металл—металл
Mov—Mov (2,57 А). Атомы Mo(V) семикоординированы. .Перво-
начально этому комплексу была приписана формула Na2[Mo2O4( -l
H-i (Cys)2(H2O)2]-ЗН2О, а полоса 1590 см-1 в ИК-спектре была
отнесена к несимметричному колебанию некоординированной
СОО~-группы [144].
1.9. Тиоэфирные атомы серы
Предыдущее обсуждение поведения тиоловых (—S—) групп
также в значительной степени применимо к тиоэфирным группам
(—SR). Тиоэфирные атомы серы обладают меньшей поляризуе-
мостью и являются более слабыми донорами, чем сульфгидриль-
ные атомы серы, но они имеют меньше неспаренных электронов и,
следовательно, должны быть лучшими акцепторами dn-электронов
{128]. С боковой цепью метионина —CH2CH2SCH3 связывается
только небольшая группа ионов [«класса (б)»] с конфигурациями
d8 и d10: Pd(II), Pt(II), Ag(I), Cu(I) и Hg(II).
Связывание Pt(II) боковой цепью метионина было доказано
анализом кристаллической структуры нескольких производных
белков, a [PtnCl4]2- была предложена как специфическая метка
на наружные остатки метионина [145]. Кроме того, известны че-
тыре комплекса с метионином, анализ кристаллической структуры
которых подтверждает, что и Pd(II) и Pt(II) координированы
группами —SCH3 (как и было предсказано на основании их хи-
мических свойств [146] и ИК-спектров) [147]. Шестичленные хе-
латные кольца между атомами азота аминогрупп и серы тиоэфир-
ных групп встречаются в Pd(o,L-MetH)Cl2 [148], Pt(D,L-MetH)Cl2
и Pt( L-MetH)Cl2 [75] и между атомами пептидного азота и серы
тиоэфирных групп в [Pt(Gly-L-Met)Cl]-Н2О (XXXVIII и XXXIX)
XXXVIH
XXXIX
M(L-MetH)Cl2 в Pt(L-MetH)CT2,
Pt(4L-MetH)Cla и Pd(D,L-MetH)Cl2
13—2451
194
Глава 4
i[75]. Все четыре комплекса получены при низких значениях pH.
Эти комплексы являются примерами координации через амино-
группы (а в XXXIX—через пептидные), в то время когда карб-
оксильные группы лигандов еще протонированы (разд. 1.2). О ко-
ординации Pd (II) метионином через донорные атомы азота и серы
при более высоких значениях pH заключили на основании высо-
кого молярного коэффициента поглощения Pd(Met)2 в ультрафио-
летовой области (е 2220 при Хмакс 315 нм) [74].
В каждой из этих метионильных боковых цепей связывающие
металл донорные атомы серы занимают вершину тригональной
пирамиды, в углах основания которой находятся атомы углерода
метиленовой и метильной групп и атом Pt. Координация, таким
образом, создает новый хиральный (асимметрический) центр при
тиоэфирном атоме серы. Оба хиральных центра обнаружены в
кристаллической структуре хелатов Pt (II)—метионин: например,
в кристаллах Pt(b-MetH)Cl2 имеются два кристаллографически
независимых комплекса, в которых донорные атомы серы проти-
воположны в оптическом отношении <[75]. Это согласуется с на-
личием двух альтернативных мест связывания [PtCl4]2- остатка-
ми метионина в цитохроме с по одному с каждой стороны от ато-
ма серы [145].
Доказательством связывания Ag(I) через атомы серы являют-
ся высокие константы устойчивости иона [Ag(Met)2]- [149], а так-
же то, что положение полос валентных колебаний NH2-rpy.nn
Li[Ag(Met)2] и Li[Ag(Nle)2] отличается на 100 см-1 [25].
В [Ag(Met)2]_ в координации Ag(I) участвуют только атомы серы
метионина, что позволяет свободным амино- и карбоксильным
труппам образовывать хелат с атомами металлов «класса (а)».
Таким путем можно получить смешанно-металлические комплексы
с Сг(Ш), Fe(III), Co(II), Ni(II) и Cu(II) [25].
Образование комплекса Cu(I) с метионином можно продемон-
стрировать потенциометрическим титрованием смеси метионина и
Си1 (CH3CN) 4(0104) в водно-ацетонитрильном растворе. Известно,
что Cu(I) не координируется через .кислород, поэтому металл дол-
жен быть связан с метионином через атомы серы тиоэфирных
групп и, возможно, азота аминогрупп [141]. Важно также то, что
Си(II) не относится к металлам «класса (б)» и не взаимодейст-
вует с группами —SCH3. Анализ кристаллической структуры ком-
плекса Cu( D,L-Met)2 показал, что Cu(II) образует хелат с ме-
тионином обычным способом, т. е. посредством амино- и кар-
боксильной групп [150].
Еще недавно полагали, что Hg(II) в реакции с метионином
ведет себя только как металл «класса (а)» (см. ниже). Однако
при очень низких значениях pH добавление ионов Hg2+ к раство-
ру метионина вызывает в спектре ПМР большие сдвиги сигналов
наиболее близко расположенных к сере протонов в сторону ела-
Комплексы металлов с аминокислотами и пептидами 195
бого поля. Эти сдвиги максимальны, когда отношение металл —
лиганд равно 1 :2, что свидетельствует о том, что главные обра-
зующиеся частицы имеют состав [Hg(H2Met)2]4+. Эти частицы
термодинамически устойчивы и кинетически лабильны [151].
Однако группы —SCH3 метионина в Hg(Met)2, а также в лю-
бом из комплексов M(Met)2 [М=Мп(П), Co(II), Ni(II), Cu(II),
Zn(II), Cd(II), Pb(II)] и M'(Met)3 [M'=A1(III), Cr(III), Bi(III),
Fe(III), Rh(III)] не связаны с атомом металла [25]. Данные
ИК-спектроскопии являются непрямыми (т. е. они доказывают
образование связей через атомы азота и кислорода, а не отсутст-
вие связи через атом серы), потому что полоса валентных коле-
баний связи С—S не наблюдалась в условиях описанного экспе-
римента. Доказательством того, что труппы S—СН3 в таких ком-
плексах, как Cr(Met)3, не участвуют в хелатообразовании,
является их 1способность связывать Ag(I), что приводит к обра-
зованию такого же типа смешанно-металлических комплексов,
как упомянутые ранее [25]. В случае Hg(Met)2 измерения сдви-
гов в спектрах ПМР растворов, содержащих Hg(II) и метионин,
при pH 9 подтверждают выводы, полученные из ИК-спектров
[151].
1.10. Мостиковые (дисульфидные) атомы серы
Мостики —S—S— цистина в одно и то же время являются
важным структурным звеном белков и очень реакционноспособ-
ным центром для связывания некоторых металлов. Ag(I) в вод-
ном растворе вызывает диспропорционирование, в результате ко-
торого образуются цистеин [стабилизированный в виде комплекса
с Ag(I)] и сульфиновая кислота [152]:
2RSSR + 2Н2О --> 3RS“ + RSO2H + ЗН+
Cu(I) связывается цистином, а Си(II) —нет. Комплекс может
содержать Си в неопределенной степени окисления. К равнове-
сию можно приблизиться с обеих сторон и в присутствии специ-
фических для Си(II) лигандов его можно сдвинуть дальше к ци-
стеину [141].
2Cu+ + RSSR
R
I
S
<- > Cu+|Cu+
S
I
R
R
-<—>- Cu2+///Cu2+ «=► 2Cu2++2RS“
Sz
I
R
Это объясняет, почему восстановление цистеином Си(II) в Си(1)
ингибируется в присутствии гистидина [специфического лиганда
13*
196
Глава 4
для Си(II)]. Фиолетово-черный комплекс стабилизируется в рас-
творе [153]. Его окраска, несомненно, обусловлена переносом за-
ряда, как показано центральными формулами в приведенном вы-
ше уравнении.
2. ЦЕННОСТЬ ИНФОРМАЦИИ, ПОЛУЧАЕМОЙ
ПРИ ИССЛЕДОВАНИИ «МОДЕЛЬНЫХ СОЕДИНЕНИЙ»
Часто утверждается, что комплексы такого типа, как обсуж-
даемые в этой главе, представляют «биологический интерес».
Иногда это верно, так как некоторые комплексы встречаются в
биологических системах. Однако гораздо чаще фраза «биологиче-
ский интерес» выражает надежду, что систематическое изучение
многих комплексов поможет понять закономерности, в соответст-
вии с которыми осуществляется взаимодействие между ионами
металлов и 'встречающимися в природе лигандами. Многие встре-
чающиеся в природе лиганды — чрезвычайно сложные молекулы.
Исследовать систематически можно только комплексы с относи-
тельно простыми лигандами. Комплекс металла с пептидом может
иметь некоторые общие свойства с комплексом металла с белком.
Когда речь идет только об этих общих свойствах, простой ком-
плекс является «модельным соединением» для биологического
взаимодействия. Но мы должны помнить, что это модель, а не ко-
пия [154].
В качестве примера полезности и ограниченности модельных
соединений рассмотрим, в какой степени данные о кристалличе-
ской структуре простых комплексов Zn(II) помогли нам понять
один вопрос — геометрию центра, связывающего Zn(II) в кар-
боксипептидазе А. Этот пример выбран потому, что мы можем ис-
пользовать известные данные о кристаллической структуре белка
для контроля за правильностью наших предсказаний. Кроме того,
модельные структуры дают некоторые, но не все числовые данные,
которые одним только анализом структуры белка пока еще точно
получить нельзя.
Активный центр карбоксипептидазы (гл. 15) содержит aTOMZn.
Лигандами являются остатки His-69, His-196 и Glu-72, так что
известными донорными атомами являются два имидазольных ато-
ма азота и один или оба (карбоксильных атома кислорода из кар-
боксильных групп глутаминатной боковой цепи. В комплексах
Zn(II) с 'аминокислотами и пептидами координационное число
атома Zn почти всегда равно четырем (тетраэдрическая конфигу-
рация) , иногда — шести (октаэдрическая конфигурация) или име-
ет промежуточное значение (неправильная координация). Если
анализом структуры сложного белка [155, 156] показано, что ко-
ординация тетраэдрическая, молекула Н2О (или, возможно, га-
Таблица 4.6
Некоторые структурные параметры комплексов Zn(II)
А. Комплексы, описанные в данной главе
Комплекс Номер формулы Координа- ционное число Геометрия координации Донорные атомы
Z п(1т)2 Zn(ImH)2Cl2 Zn(L-His)a-2HaO Zn(D,L-His)a -5Н2О Zn(L-Ser)a Zn(Gly-Gly-Gly)(SO4)i/ • •4HaO Zn(L-H_1Glu)2HaO Zn(ImH), Cla-4H2O Zn(Gly-Gly)a-2HaO ZnfL-H-pAsp) -ЗН2О Zn(BiuH2) aCla XXVI XXV XXIX XXIX XXXV XIII XIV XXIV II XVI 4 4 4+2 | 4+2 J 5 6 6 6 6 6 6 Тетраэдр Тетраэдр плюс две сла- бые связи Искаженная квадратная пирамида Искаженный октаэдр Октаэдр 4 N (имидазоль- ный) 2 М(имидазоль- . ный), 2С1 2 N (аминогруп- пы) 2 N (имидазоль- ный) 2 ©(карбок- сильный) 2 N (аминогруп- пы) 3 □(карбок- сильный) 1 N (аминогруп- пы) 2 ©(карбоксиль- ный) 1 ©(пептид- ный), 2НгО 1 N (аминогруп- пы) 4 ©(карбок- сильный), 1Н2О (гб N (имидазоль- ный) 2 N (аминогруп- пы) 1 ©(карбоксиль- ный) 2 ©(пептид- ный), 1Н2О 1 N (аминогруп- пы) 3 ©(карбо- ксильный), 2Н2О 4 О (амидный), 2С1
Продолжение табл. 4-6
Б. Длины связей Zn(Il) —лиганд6
Связь Геометрия координации О1 Длина, А Число образцов
среднее значение интервал
Zn—N (имидазоль- ный) Тетраэдр 2,00 1,96—2,04 12
Октаэдр 2,20 2,15—2,26 6
Z—О (карбоксиль- ный) Искаженная 2,8 2,66—2,9 4
Квадратная пирамида 2,08 1,96—2,16 3
Октаэдр 2,09 2,03—2,20 9
Zn—О (пептидный) » 2,18 2,18—2,19 2
Zn—О (амидный) » 2,04 2,03—2,05 2
Zn—ОН2 2,14 2,07—2,19 6
В. Углы между связями лиганд — Zn — лиганд*
Угол Геометрия координации Угол, град Число образ- цов
среднее значе- ние интервал
N (имидазольный)—Zn—N (имида- зольный) Тетраэдр 109 105—117 13
Искаженная 115 112—117 2
Октаэдр 90 87—93 12
N (имидазольный)—Zn—О (карбо- ксильный) Искаженная — 77—164 2
О (карбоксильный) —Zn—О (карбо- ксильный) » — 81—139 2
Октаэдр 94 89—103 6
» — 876—149б 2
О (карбоксильный) —Zn—ОНг » 93 88—100 6
» — 776 1
аЦанные взяты из работы [9], за исключением значений для Zn(L-Ser)i [1251 я
Zn (Gly-Gly-Gly) (SOJ i/a-4HsO [37].
^Параметр относится к необычной связи, когда оба атома кислорода карбоксильной
группы взаимодействуют с одним и тем же атомом Zn.
вНе приводятся величины углов для случая, когда обе связи находятся в одном и том
же хелатном кольце.
Комплексы металлов с аминокислотами а пептидами
199
логенид-ион или другой ион) должна занимать четвертое коорди-
национное место. В процессе гидролиза молекула пептидного суб-
страта прикрепляется к атому Zn через атом кислорода той
пептидной группы, которая гидролизуется. Структуры модельных
соединений не позволяют предсказать, будет ли субстрат образо-
вывать дополнительную связь с .атомом Zn или он будет замещать
Рис. 4.3. Предсказанное окружение атома Zn (II), лигандами которого являются
две гистидильные и одна глутамильная боковые цепи и молекула Н2О.
один из имеющихся лигандов (например, молекулу воды). До-
вольно определенно только то, что, если связь Zn—О (пептидный)
не стабилизирована хелатообразованием, она должна стабилизи-
роваться с помощью других контактов между ферментом и моле-
кулами субстрата.
Для описания окружения атома Zn нам необходимы два типа
данных о структуре: длины связей Zn — лиганд и наиболее веро-
ятные значения углов между связями. Данные, которые можно
было отобрать из всех опубликованных данных структурных ана-
лизов соответствующих «модельных соединений», представлены в
табл. 4.6.
Данные табл. 4.6 приводят к следующим выводам: (а) связи
Zn—N (имидазольный) имеют длину 2,0 А, если предположить, что
координационная геометрия ближе к тетраэдрической, чем к окта-
200
Глава 4
эдрической; (б) длины связей Zn—О (карбоксильный), Zn—ОН2 и
(или) Zn—О (пептидный) равны 2,1—2,2 А. Мы должны учиты-
вать возможность того, что карбоксильная группа может коорди-
нировать Zn посредством обоих своих атомов кислорода. Если это
действительно имеет место, длина второй связи равна примерно
2,6 А, а угол между двумя связями Zn—О (карбоксильный) равен
примерно 60°; (в) единственные известные данные об углах
N (имидазольный)—Zn—О (карбоксильный) относятся к двум ком-
плексам Zn(II) с гистидином, в (которых связи Zn—О (карбоксиль-
ный) слабые и карбоксильные атомы кислорода занимают особые
координационные места (координационный полиэдр искажен).
В лучшем случае эти углы можно использовать как пределы (77
и 164°), (между которыми лежат углы в других комплексах;
(г) еще меньше данных имеется об углах между связями Zn—ОН2
и Zn—О (пептидный); (д) геометрия связывающих металлфунк-
циональных групп легче предсказуема, так как на примерах боль-
шого числа комплексов с разными металлами показано, что она
остается довольно постоянной. Таким 'образом, мы можем с уве-
ренностью сказать, что угол С—О—Zn при донорном атоме кар-
боксильного кислорода должен быть близок к 104° и что углы
С—N—Zn при донорных атомах имидазольного азота должны ле-
жать в интервале 121—131° и т. д.
Картина активного центра, которая, наконец, вырисовывается,
показана на рис. 4.3. Она такая, какую мы предсказали бы, если
бы наша информация об активном центре, как это часто случает-
ся, ограничивалась знанием только связывающих групп. Предска-
занную геометрию следует сопоставлять с детальной интерпрета-
цией структуры самого протеина (гл. 15).
СПИСОК ЛИТЕРАТУРЫ
1. Strecker А., Ann., 75, 27 (1850).
2. Wiedemann G„ J. Prakt. Chem., 42, 255 (1847); 43, 271 (1848).
3. Gurd F. R. N., Wilcox P. E., Adv. Protein Chem., 11, 311 (1956).
4. Гринипейн Дж., Виниц M„ Химия аминокислот и пептидов, «Мир», М., 1965,
гл. 6.
5. Martell А. Е„ Calvin М„ Chemistry of Metal Chelate Compounds, Prentice-
Hall, New York, 1962.
6. Nakamoto K., McCarthy P. J., Spectroscopy and Structure of Metal Chelate
Compounds, Wiley, New York, 1968.
7. Gillard R. D., Inorg. Chem. Acta. Rev., 1, 69 (1967).
8. Brill A. S., Martin R. B., Williams R. I. P„ in B. Pullman (ed.), Electronic
Aspects of Biochemistry, Academic Press, New York, 1964, p. 519.
9. Freeman H. C., Adv. Protein Chem., 22, 257 (1967).
10. Vallee B. L„ Williams R. J. P., Proc. Natn. Acad. Sci. U. S., 59, 498
(1968).
11. Gillard R. D., Laurie S. FL, in «Amino Acids, Peptides and Proteins», Vol. 1,
The Chemical Society, London, 1969, p. 262.
Комплексы металлов с аминокислотами и пептидами
201
12. Low В. W„ Hirshfeld F. L„ Richards F. М., J. Am. Chem. Soc., 81, 4412
(1959).
13. Sandmark C., Lindqvist I., personal communication, 1965, quoted in ref. [9].
14. Sharma V. S.. Mathur H. B., Indian J. Chem., 3, 475 (1965).
15. Boyd S., Brannan J. R„ Dunsmore H. S., Nancollas G. H., J. Chem. Eng. Da-
ta, 12, 12 (1967).
16. Raju E. V., Mathur H. B., J. Inorg. Nucl. Chem., 30, 2181 (1968).
17. Stack W. F., Skinner H. A., Trans. Faraday Soc., 63, 1136 (1967).
18. Brunetti A. P., Lim M. C., Nancollas G. H., J. Am. Chem. Soc., 90, 5120
(1968).
19. Letter J. E., Bauman J. E., J. Am. Chem. Soc., 92, 437 (1969); 92, 443
(1969).
20. Acland С. B., Freeman H. C., Chem. Commun., 1971, 1016.
21. Datta S. P., Grzybowski A. K., J. Chem. Soc., 1959, 1091.
22. Alner D. J., J. Chem. Soc., 1962, 3283.
23. Азизов Ю. M„ Мифтахова A. X., Торопова В. Ф., ЖНХ, 12, 661 (1967).
24. Rossotti F. J. C., in J. Lewis, R. G. Wilkins (eds.), Modern Coordination Che-
mistry, Interscience, New York, 1960, p. 57.
25. McAuliffe C. A., Quagliano J. V., Vallarino L. M., Inorg. Chem., 5, 1996
(1966).
26. Cotton F. A., in J. Lewis, R. G. Wilkins (eds.), Modern Coordination Che-
mistry, Interscience, New York, 1960, p. 387.
27. Гринберг А. А., Стеценко A. FL, Инкова E. FL, ДАН СССР, 136, 821
(1961).
28. Вольштейн Л. М., Володина И. О., ЖНХ, 5, 35 (1960).
29. Вольштейн Л. М„ Мотягина Г. Г., ЖНХ, 5, 1730 (1960.
30. Erickson L. Е., McDonald J. W., Howie J. К., Clow R. P., J. Am. Chem. Soc.,
90, 6371 (1968).
31. McDonald С. C., Phillips W. D„ J. Am. Chem. Soc., 85, 3736 (1963).
32. Carlson R. H., Brown T. L., Inorg. Chem., 5, 268 (1966).
33. Nakamoto K, Infra-Red Spectra of Inorganic and Coordination Compounds,
2nd edn., Wiley-Interscience, New York, 1970, p. 222.
34. Lindqvist L, Rosenstein R„ Acta Chem. Scand., 14, 1228 (1960).
35. Rao J. К- M„ Viswamitra M. A., Acta Cryst., B28, 1484 (1972).
36. Беляева К. Ф., Порай-Кошиц М. А., Малиновский Т. И., Асланов Л. А., Су-
ханова Л. С., Мартыненко Л. И., Ж. структ. хим., 10, 557 (1969).
37. Van der Helm D., Nicholas H. B., Abstracts, Am. Cryst. Assoc. Meeting,
Tucson, Paper F5, 1968; Acta Cryst, B26, 1858 (1970).
38. Gramaccioli C., Acta Cryst., 21, 600 (1966).
39. Van der Helm D., Willoughby T. V., Acta Cryst, B25, 2317 (1969).
40. Rabin B. R., Biochem. Soc. Symp., 15, 21 (1958).
41. Dyer H. B„ Acta Cryst., 4, 42 (1951).
42. Cavalca L., Nardelli M., Fava G„ Acta Cryst, 13, 594 (1960).
43. Mitschler A., Fischer J., Weiss R., Acta Cryst., 22, 236 (1967).
44. Haas D. L, Nature, 201, 64 (1964).
45. Roux J. F., Boeyens J. C. A., Acta Cryst., B25, 1700 (1969).
46. Hinton J. F., Amis E. S., Chem. Commun., 1967, 100.
47. Hinton I. F., Amis E. S„ Mettetal W., Spectrochim. Acta, A25, 119 (1969).
48. Pfeiffer P„ Organische Molekillverbindungen, Enke, Stuttgart, 1927, quoted
in ref. [4].
49. Davies C. W., Waind G. M., J. Chem. Soc., 1950, 301.
50. Monk С. B., Trans. Faraday Soc., 47, 285, 292, 297, 1233 (1951).
51. Freeman H. C., Robinson G., Schoone J. C., Acta Cryst., 17, 719 (1964).
52. Nardelli M., Fava G., Giraldi G., Acta Cryst., 16, 343 (1963).
53. Kedzia В. B., Armendarez P. X., Nakamoto K., J. Inorg. Nucl. Chem., 30, 849
(1968).
54. Nuttall R. H„ Melson G. A., J. Inorg. Nucl. Chem., 31, 2979 (1969).
202
Глава 4
55. Freeman Н. С., Smith I. Е. W. L„ Acta Cryst, 20, 153 (1966).
56. Li N. C„ Scruggs R. L., Becker E. D., J. Am. Chem. Soc., 84, 4650 (1962).
57. Kim M. K., Martell A. E„ J. Am. Chem. Soc., 91, 872 (1969).
58. Kim M. K., Martell A. E., Biochemistry, 3, 1169 (1964).
59. Kim M. K, Martell A. E„ J. Am. Chem. Soc., 88, 914 (1966).
60. Martell A. E., Kim M. K-, personal communication.
61. Hartzell C. R„ Gurd F. R. N., J. Biol. Chem., 244, 147 (1969).
62. Osterberg R., S joberg B., J. Biol. Chem., 243, 3038 (1968).
63. Freeman H. C„ Szymanski J. T., Acta Cryst., 22, 406 (1967).
64. Bell J. D., Freeman H. C., Wood A. M., Driver R„ Walker W. R., Chem. Com-
mun, 1969, 1441.
65. Freeman H. C., Schoone J. C., Sime J. G., Acta Cryst., 18, 381 (1965).
66. Freeman H. C., Taylor M. R„ Acta Cryst., 18, 939 (1965).
67. Sugihara A., Ashida T„ Sasada У., Kakudo M„ Acta Cryst., B24, 203
(1968).
68. Manyak A. R., Murphy С. B., Martell A. E„ Arch. Biochem. Biophys., 59, 373
(1955).
69. Martin R. B., Chamberlin M., Edsall J. T., J. Am. Chem. Soc., 82, 495
(1960).
70. Kim M. K, Martell A. E„ J. Am. Chem. Soc., 89, 5138 (1967).
71. Billo E. I., Margerum D. W., J. Am. Chem. Soc., 92, 6811 (1970).
72. Freeman H. C., Guss J. M., Sinclair R. L., Chem. Common., 1968, 485.
73. Pagenkopf G. K., Margerum D. W„ J. Am. Chem. Soc., 90, 501 (1968).
74. Wilson E. W„ Martin R. B., Inorg. Chem., 9, 528 (1970).
75. Freeman H. C„ Golomb M. L., Chem. Common., 1970, 1523.
76. Michailidis M. S., Martin R. B„ J. Am. Chem. Soc., 91, 4683 (1969).
77. Gillard R. D., McKenzie E. D., Mason R., Robertson G. B„ Nature, 209, 1347
(1966).
78. Barnet M. T., Freeman H. C., Buckingham D. A., Hsu I. N„ Van der Helm D.,
Chem. Common., 1970, 367.
79. Gillard R. D., Harrison P. M., McKenzie E. D„ J. Chem. Soc. (A), 1967,
618.
80. Banaszak L. J., Watson H. C., Kendrew J. C„ J. Mol. Biol., 12, 130 (1965).
81. Boas J. F., Pilbrow J. R„ Hartzell C. R., Smith T. D., J. Chem. Soc. (A), 1969,
572.
82. Koltun W. L., Fried M., Gurd F. R. N., J. Am. Chem. Soc., 82, 233 (1960).
83. Rao G. N.. Li N. C„ Can. J. Chem., 44, 1637 (1966).
84. Driver R.. Walker W. R„ Aust. J. Chem., 21, 671 (1968).
85. Edsall ]. T., Felsenjeld G., Goodman D. S., Gurd F. R. H., J. Am. Chem. Soc.,
76, 3054 (1954).
86. Eilbeck W. J., Holmes F„ Underhill A. E., J. Chem. Soc. (A), 1967, 757.
87. Goodgame D. L., Goodgame M., Hayward P. I., Rayner-Canham G. W„ Inorg.
Chem., 7, 2447 (1968).
88. Akhtar F., Goodgame D. M. L., Goodgame M., Rayner-Canham G. W.,
Skapski A. C., Chem. Common., 1968, 1389.
89. Sandmark C„ Acta Chem. Scand., 21, 993 (1967).
90. Lundberg В. K. S„ Acta Cryst, 21, 901 (1967).
91. Strandberg B„ Svensson B., Branden C. L, personal communication in ref.
[9].
92. Candlin R., Harding M. M., J. Chem. Soc. (A), 1967, 421.
93. Bryce G. F., Gurd F. R. N., J. Biol. Chem., 241, 122 (1966).
94. Baraniak E., Freeman H. C„ James J. M., J. Chem. Soc. (A), 1970, 2558.
95. Inoue M., Kishita M., Kubo M„ Bull. Chem. Soc. Japan, 39, 1352 (1966).
96. Jarvis J. A. J., Wells A. F„ Acta Cryst., 13, 1028 (I960); see also ref. [9],
p. 413.
97. Bryce G. F., Roeske R. W., Gurd F. R. N„ J. Biol. Chem., 240, 3837 (1965).
98. Harding M. M., Long H. A., J. Chem. Soc. (A), 1968, 2554.
Комплексы металлов с аминокислотами и пептидами
203
99. Candlin R„ Harding М. М., J. Chem. Soc. (А), 1970, 384.
100. Fraser К. A., Harding М. М., J. Chem. Soc. (А), 1967, 415.
101. Valladas-Dubois S., Compt. rend., 237, 1408 (1953); Bull. Soc. Chim. France,
1954, 831; Compt. rend., 246, 2619 (1958).
101a. Harding M. M., Cole S. J., Acta Cryst., 16, 643 (1963); Kretsinger R. H.,
Bryan R. F., Cotton F. A., ibid., 651.
102. Perrin D. D., Sharma V. S., J. Chem. Soc. (A), 1967, 724.
103. Perkins D. J., Biochem. J., 55, 649 (1953).
104. Freeman H. C., Martin R. P., J. Biol. Chem., 244, 4823 (1969).
105. Raju E. V., Mathur H. B., J. Inorg. Nucl. Chem., 31, 425 (1969).
106. Thornton A. C. R„ Skinner H. A., Trans. Faraday Soc., 65, 2044 (1969).
107. Burk D„ Hearon J. Z., Caroline L., Schade A. L., J. Biol. Chem., 165, 723
(1946).
108. Earnshaw A., Larkworthy L. F., Nature, 192, 1068 (1961).
109. Sitnplicia Wilkins R. G., J. Am. Chem. Soc., 89, 6092 (1967).
110. Morris P. J., Martin B. R., J. Inorg. Nucl. Chem., 32, 2891 (1970).
111. Morris P. J., Martin R. B„ J. Am. Chem. Soc., 92, 1543 (1970).
112. Pearlmutter A. F., Stuehr J. E., J. Am. Chem. Soc., 90, 858 (1968).
113. Evertsson B„ Acta Cryst., B25, 30 (1969).
114. Sigel H., Griesser R., McCormick D. B., Arch. Biochem. Biophys., 134, 217
(1969).
115. Doran M. A., Chaberek S„ Martell A. E„ J. Am. Chem. Soc., 86, 2129 (1964).
116. Meyer I. L., Bauman J. E., J. Am Chem. Soc., 92, 4210 (1970).
117. Sigel H., McKenzie R. E., McCormick D. B„ Biochim. Biophys. Acta, 200, 411
(1970).
118. Freeman H. C., Guss J. M., Healy M. J., Martin R. P„ Nockolds С. E., Sar-
kar B., Chem. Commun., 1969, 225.
119. Wellman К. M., Wong В. K., Proc. Natn. Acad. Sci. U. S., 64, 824 (1969).
120. Martin R. B., Edsall J. T„ J. Am. Chem. Soc., 82, 1107 (1960).
121. Lenz G. R., Martell A. E„ Biochemistry, 3, 745 (1964).
122. Kustin K-, Pasternack R. F., J. Am. Chem. Soc., 90, 2295 (1968).
123. Van der Helm D., Hossain M. B., Acta Cryst., B25, 457 (1969).
124. Van der Helm D., Franks W. A., Acta Cryst., B25, 451 (1969).
125. Van der Helm D., Nicholas A. F„ Fisher C. G., Acta Cryst., B26, 1172
(1970).
126. Van der Helm D., Franks W. A., J. Am. Chem. Soc., 90, 5627 (1968);
Franks W. A., Van der Helm D., Acta Cryst., B27, 1299 (1970).
127. Ahrland S., Chatt J., Davies N. R.. Quart. Rev., 12, 265 (1958).
128. Livingstone S. E., Quart. Rev., 19, 386 (1965).
129. Jorgensen С. K, J. Inorg. Nucl. Chem., 24, 1571 (1962); Inorg. Chim. Acta
Rev., 2, 65 (1968).
130. Valladas-Dubois S., Compt. rend., 231, 53 (1950).
131. Doornbos D. A., Pharm. Weekblad, 103, 1213 (1968).
132. Gergely A., Acta Chim. Acad. Sci. Hung., 59, 309 (1969).
133. Evans W. P., Monk С. B., Trans. Faraday Soc., 51, 1244 (1955).
134. Albert A., Biochem. J., 47, 531 (1950).
135. Shindo H„ Brown T. L., J. Am. Chem. Soc., 87, 1904 (1965).
136. Lum R. P. K, Eriks K-, Abstracts, Am. Cryst. Assoc . Ottawa, Paper D4,
1970.
137. Gorin G., Spessard J. E., Wessler G. A., Oliver I. P„ J. Am. Chem. Soc., 81,
3193 (1959).
138. McCormick B. J., Gorin G., Inorg. Chem., 2, 928 (1963).
139. Tomita A., Hirai H„ Makishima S., Inorg. Chem., 7, 760 (1968).
140. Tomita A., Hirai H., Makishima S., Inorg. Nucl. Chem. Lett., 4, 715 (1968).
141. Hemmerich P. in J. Peisach, P. Aisen, W. E. Blumberg (eds.), The Bioche-
mistry of Copper, Academic Press, New York, 1966, p. 15.
204
Глава 4
142. Urry D. IF., Miles D., Caldwell D. J., Eyeing H., J. Phys. Chem., 69, 1603
(1965).
143. Knox J. R., Prout С. K.. Acta Cryst., B25, 1857 (1969).
144. Kay A., Mitchell P. С. H„ Nature, 219, 267 (1968).
145. Dickerson R. E„ Eisenberg D., Varnurn J., Kopka M. L„ J. Mol. Biol., 45, 77
(1969).
146. Вольштейн Л. M., Могилевкина M. Ф., ЖНХ, 8, 597 (1963).
147. McAuliffe C. A., J. Chem. Soc. (A), 1967, 641.
148. Stephenson N. C„ McConnell J. F., Warren R., Inorg. Nucl. Chem. Lett., 3,
553 (1967); Warren R. C„ McConnell J. F., Stephenson N. C., Acta Cryst.,
B26, 1402 (1970).
149. Lenz G. R„ Martell A. E„ Biochemistry, 3, 750 (1964).
150. Veidis M. V., Palenik G. J., Chem. Commun., 1969, 1277.
151. Naitusch D. F. S., Porter L. Chem. Commun., 1970, 596.
152. Ref. [4], p. 645.
153. Hallman P. S.. Proc. XII. I.C.C.C., 1970, 127.
154. Malmstrom B. G., Plenary Lecture, XII Int. Conf. Cordination Chem., Sydney,
1969, Butterworth, London, 1970.
155. Lipscomb W. N., Accounts Chem. Res., 3, 81 (1970).
156. Lipscomb W. N., Hartsuck I. A., Quiocho F. A., Reeke G. H., Proc. Natn.
Acad. Sci., 64, 28 (1969).
157. Holmes F., Williams D. R., J. Chem. Soc. (A), 1967, 1702.
158. Bauman J. E., Wang I. C„ Inorg. Chem., 3, 370 (1964); see also Williams,
J. Chem. Soc. (A), 1968, 2965.
ГЛАВА 5
ТРАНСПОРТ СОЕДИНЕНИЙ ЖЕЛЕЗА
МИКРОБАМИ (СИДЕРОХРОМЫ)
Дж. Б. Нейлендс
Neilands I. В., Biochemistry Department, University of
California, Berkeley, California, USA
1. ОБЩИЙ ОБЗОР
1.1. Введение
Из гл. 4 мы знаем, что 'функциональные группы пептидов могут
связывать ионы металлов. В данной главе мы обсудим структуры
некоторых специфических пептидов и других комплексообразую-
щих агентов, которые продуцируются микроорганизмами и, по-
видимому, специально предназначены для связывания железа.
В ходе эволюции переход от анаэробной к аэробной атмосфере*
потребовал мощных специальных агентов для очень малораство-
римого иона Fe(III) [Kso гидроксида железа(Ш) ПР^Ю-36].
Вследствие этого большинство аэробных и «случайно» аэроб-
ных микроорганизмов, по-видимому, способны накапливать и
транспортировать железо и механизмы этих процессов примерно
эквивалентны тем, которые показаны схематически на рис. 5.1.
В этой схеме можно выделить следующие основные черты: а) био-
синтез специфического лиганда для иона Fe(III) в ходе фермен-
тативных реакций; б) регуляция при помощи железа генетическо-
го аппарата, производящего описываемые здесь ферменты; в) диф-
фузия лиганда к мембране, поверхности клетки пли в среду, свя-
зывание иона Fe(III) и активный транспорт иона металла в виде
его комплексов в клетку; г) накопление комплекса Fe(III) в клет-
ке, последующее восстановление и доставка атома железа в тре-
буемое место биосинтетическим аппаратом клетки. В условиях
низких концентраций железа перепроизводство лиганда 'может
привести к образованию значительного количества не связанных
с металлом молекул лиганда, которые должны быть выведены из
клетки в среду.
* «Аэробная» и «анаэробная» атмосфера — термины неудачные: лучше было
бы «содержащая» и «не содержащая» кислорода атмосфера. — Прим. ред.
206
Глава 5
Внешняя
Внутренняя
железосодержащий фермент и йелок
ген-репрессор оператор- гена eeng еенв и т.й.
ныа ген
Рис. 5.1. Схема микробной железотраиспортирующей системы и ее оперой.
Информация, содержащаяся в генах, транскрипируется в молекулы матричной (информа-
ционной) РНК и передается от них для синтеза ферментных белков. Серии генов, таких, как
обозначенные буквами а, б и в, могут находиться под контролем операторного гена. При
ингибировании этого операторного гена геном-репрессором прекращается синтез всех белков,
которые соответствуют всем генам, контролируемым этим операторным геном.
1.2. Терминология
Содержащим железо метаболитам красно-коричневого цвета,
характеризующимся полосой поглощения при 420—440 нм, было
дано общее название «сидерохромы» [1]. Метаболиты, обладаю-
щие способностью промотировать рост, были названы «сидерами-
нами», а метаболиты с антибиотическими свойствами —«сидеро-
мицинами». Такая терминология не вполне удовлетворительна, так
как ряд соединений нельзя отнести ни к факторам роста, ни к ан-
тибиотикам. Более того, некоторые антибиотики этого класса в
зависимости от используемого организма могут служить и факто-
рами роста (табл. 5.2). К тому же феноляты железа(III) (класс
соединений с такой же биофункцией, как сидерохромы) также
имеют максимум поглощения вблизи 500 нм. >По этой причине, по-
видимому, желательно отказаться от названий «сидерамины» и
«сидеромицины» и оставить термин «оидерохромы» или «железо-
форы» для всех микробных железотранспортирующих факторов.
Транспорт соединений железа микробами (сидерохромы)
207
В настоящем обзоре будет .использоваться название «сидерохром»
{sidero — железо, chrome — цвет); работающие в этой области
должны будут разработать удовлетворительную номенклатуру.
1.3. Классификация
В данном обзоре основное внимание будет уделено химии мик--
робных железотранспортирующих соединений. Но, кроме тогог
будет приведено достаточно данных, полученных в результате
биологических исследований, чтобы показать, что в действительно-
сти главной ролью этих веществ является роль «капсул» или «обо-
лочек». Таким образом, область на рис. 5.1, обозначенная как «ак-
с =0
I
N-O-
✓
R1
Рис. 5.2. Фенолятные (внизу) и гидроксаматные (вверху) комплексы железа (III),
найденные в микробных железотранспортирующих соединениях.
тивная транспортная зона», слишком нехарактерна для настояще-
го обзора. Понятно, что особое внимание должно быть уделено
структуре соединений, так как какой бы ни была биологическая
функция, она всегда будет зависеть и определяться химическим
строением молекул.
В настоящее время мы различаем два главных типа сидеро-
хромов—феноляты и гидроксаматы. Оба лиганда представляют
собой слабые кислоты (р/Са — 9), и координационная сфера свя-
занного иона металла состоит только из атомов кислорода
(рис. 5.2). На этом рисунке показаны структуры комплексов со-
става 3:1, в которых заняты все шесть октаэдрически располо-
женных координационных мест иона Fe(III). Вещества, ближе
других связанные с метаболизмом железа, представлены энтеро-
бактином (I) [2] и феррихромом (Va) [3], относящимися соответ-
ственно к фенолятной и тидроксаматной сериям; их молекулы со-
держат по 6 атомов кислорода каждая. Кроме того, здесь будет
описан ряд соединений, которые, как полагают, также участвуют
в транспорте железа, но содержат меньше шести связующих ато-
мов в молекуле; но главное внимание будет уделено системам ти-
па энтеробактина и феррихрома.
208
Глава 5
Классификация на феноляты и тидроксаматы является одной
из наиболее четких, но, к сожалению, она 'несовместима с разно-
образием структур, представленных факторами микробного транс-
порта железа. Так, микобактерия синтезирует множество соедине-
ний, называемых 'микобактинами (XI) [4], которые содержат две
гидроксаматные связи (т. е. четыре атома кислорода), одну фено-
лятную группу и остаток замещенного оксазолина, причем шестым
металлсвязывающим атомом является атом азота оксазолина.
Следовательно, микобактины можно рассматривать как гибрид
чистых тригидроксаматной и трифенолятной систем. Кроме того,
и аэробактин (IX) [5] из Aerobacter aerogenes и деферришизоки-
нины (Ха) [5а] из Bacillus megaterium включают две гидроксамат-
ные группы и внутренний остаток лимонной кислоты, имеющий
свободную карбоксильную и гидроксильную группы, каждая из
которых может координироваться к связанному с гидроксаматом
иону Fe(III) (X). В общем можно сказать, что тидроксаматы
встречаются в грибах, дрожжах и бактериях. Трифенолят до сих
пор обнаруживали только в истинных бактериях.
1.4. Биосинтез
Прежде чем начать обсуждение структур соединений, транс-
портирующих железо, полезно рассмотреть некоторые факторы,
регулирующие их биосинтез, главный из которых — железо. Роль
железа в этом процессе впервые была описана Гарибальди и Ней-
лендсом около 15 лет назад. Они продемонстрировали накопление
большого количества свободного от металла лиганда при культи-
вировании головневых грибов Ustilago sphaerogena и других мик-
роорганизмов при низких концентрациях железа [6]. Полное ис-
ключение железа из среды, естественно, будет препятствовать
росту всего организма, и, очевидно, данные, приведенные на
рис. 5.3, следует соотносить с выходами клеток. Поэтому незна-
чительное стимулирование железом образования деферрисидеро-
хрома может объяснить высокие выходы клеток, или же можно
предположить участие железа в ферментативных биосинтетиче-
ских процессах. При концентрации соли железа примерно 10-5 М
обычно происходит усиленный рост клеток и не происходит выде-
ление транспортных лигандов. Тот факт, что регулирующее влия-
ние иона металла можно наблюдать уже при концентрации при-
мерно 10-7 М, заставляет предположить репрессивный механизм,
но более убедительное доказательство этому получают в резуль-
тате эксперимента, в котором железо добавляют к культуре, ак-
тивно синтезирующей гидроксамат, — синтез продолжается не-
сколько часов, на основании чего исключается предположение
о ферментативном ингибировании добавленным железом. Изменяя
содержание железа в питательной среде некоторых дрожжей и
грибов, создают возможность для образования гидроксамата в
Транспорт соединений железа микробами (сидерохромы)209
количестве, составляющем несколько граммов на литр среды [7].
Столь высокие выходы никогда не наблюдают для фенолятов из
бактерий, даже когда уровень железа «оптимальный», хотя обра-
зование фенолята аналогичным образом подавляется железом. Это
отражает различия в оперонах и ферментах, которые управляют
синтезом гидрокса матов и фенолятов. .По аналогии с биосинтезом
малых пептидов, подобных грамицидину, мы допускаем, что фраг-
менты деферрисидерохрома собираются посредством некодиро-
ванного процесса.
Рис. 5.3. Общая зависимость, от-
ражающая подавление железом
производства деферрисидерохро-
мов, наблюдаемое в различных ви-
дах микробов.
1g (количество добавленного
железа, г-атом/л)
Гарибальди [7а] сделал интересное открытие, что флюорес-
цирующая псевдомонада, которая была выделена из испорченного
куриного мяса при 2 °C, при 28 °C разрушается с образованием
сидерохрома гидроксаматного типа. Для роста при более высоких
температурах требуется или больше железа, или следовые коли-
чества железотранспортирующих соединений.
Природа связывающих центров деферрисидерохрома показана
на рис. 5.4. Эта схема иллюстрирует тот факт, что биосинтезы фе-
нолята (энтеробактина) относительно хорошо прослеживаются
вплоть до ферментативного уровня [8]; железо не участвует в
этом биосинтезе. Таким образом, путь а на рис. 5.4 представляет
переход хоризмата в обычные ароматические соединения и за-
канчивается энтеробактином; единственной целью этого стимули-
рования, по-нидимому, является синтез сидерохрома (энтеробак-
тин, I). В случае гидроксаматного сидерохрома гидроксилирова-
ние атома азота может осуществляться либо до (путь б, рис. 5.4),
либо после (путь в, рис. 5.4) ацилирования. Первая реакция, бо-
лее выгодная термодинамически, осуществляется в случае ферри-
14—2451
210
Глава 5
хрома [9] и хадацидина [10], в то время как с аспергилловой
кислотой [11] (и, возможно, с .микобактином [4]) свободное гид-
роксиламинопроизводное может и не образовываться в качестве
промежуточного соединения.
С практической точки зрения сведения о способности железа
подавлять некоторые биосинтетические процессы оказались чрез-
вычайно важными для получения достаточных количеств железо-
транспортирующих соединений для биологических исследований.
Если такие соединения не удается обнаружить, то это может
означать, что: а) организм имеет альтернативный механизм для
< ароматический путь
—^2,3-Ъиоксибензоат -* -* энтеробактин
он о
। 11 ,
R—N-C—R
Рис. 5.4. Пути, посредством которых биосинтезируются атомы, связывающие
металл, в фенолятных (вверху) и гидроксаматных (внизу) деферрисидерохромах.
усвоения железа; б) соединения не выделяются; в) они произво-
дятся в количествах, меньших того предела, когда их можно обна-
ружить; г) организму требуются для синтеза особые концентра-
ции железа или д) природные или создаваемые искусственно ве-
щества в среде 'могут выполнять не только транспортную 'функ-
цию. Углеродные субстраты, которые могут окисляться только
через цикл трикарбоновых кислот (ТК.К), должны иметь макси-
мальную потребность в железе [12].
К сожалению, очень мало известно о метаболизме и распаде
сидерохромов. Почвенные микроорганизмы Pseudomonas Fc-1 спо-
собны расти на феррихромах как на единственном источнике уг-
лерода и азота. Первоначальная атака осуществляется пептидаза-
ми, которые расщепляют циклический пептид при карбоксильном
атоме углерода 6-М-ацил-6-Ь1-оксиорнитинтрипептида [13]. Желе-
зо можно заменить алюминием, но деметаллированные соединения
не расщепляются.
1.5. Биологическое действие
С точки зрения структуры феррихромные соединения (V) от-
носятся к наиболее интенсивно исследованным в биохимии. Пол-
ное кристаллографическое представление тщательно разработано
Транспорт соединений железа микробами (сидерохромы)211
для феррихрома A (V6) [14]. Феррихром (Va) [15] был синте-
зирован и исследован методами протонного магнитного резонанса
[16], электронного парамагнитного резонанса и спектроскопии
Мессбауэра [17]. Из фенолятпроизводных только энтеробактин
(I) не был синтезирован химически. Теперь полезно рассмотреть,
насколько структура этих довольно полно изученных соединений
соответствует их биологической функции, иллюстрируемой рис. 5.1.
Первоначальное предположение о том, что феррихром (Va)
[18] и итоевая кислота (IV) [19] действуют как агенты, перено-
сящие железо, в значительной мере основывалось на том факте,
что комплексы железа(III) чрезвычайно стабильны (константа
образования ~ 1030), в то время как аналогичные комплексы же-
леза (II) сравнительно неустойчивы. Эти данные подтверждают
механизм, предполагающий освобождение железа в ходе реакции,
и в то же время исключают возможность переноса электрона в
процессе окисления—восстановления центрального иона металла.
Было установлено, что продукты, образовавшиеся при недостатке
железа, должны иметь в клетке определенные эволюционные пре-
имущества, и это также согласуется с тем, что оба соединения —
и феррихром (Va) и итоевая кислота (IV) —действуют как аген-
ты, переносящие железо. Доказательство другого типа основано
на том, что некоторые грибы и бактерии, живущие главным обра-
зом в почве и навозе, требуют для роста сидерохромы (табл. 5.2).
Впоследствии обнаружили, что синтетические хелатирующие аген-
ты в надлежащих концентрациях могут промотировать рост при-
родных сидерохромных ауксотрофов [20]. Как показал Эмери
[21], феррихром играет роль непосредственного переносчика же-
леза в U. sphaerogena— головневых грибах, которые, как извест-
но, славятся своей необычайно высокой способностью синтезиро-
вать сидерохромы. Для Bacillus megaterium, дикий тип которой
продуцирует шизокинины [22], можно получить мутанты, нужда-
ющиеся в сидерохромах [23]. Стабильный комплекс микобактина
с хромом(III) конкурнрующе подавляет активность фактора роста
микобактина в некоторых микобактериях [4]. Это означает, что
физиологически активной формой метаболита является комплекс
микобактина с металлом, по-видимому, комплекс железо(Ш)ми-
кобактин (XII). Наконец, роль природного гидроксамата желе-
за(Ш) как переносчика железа может быть сопоставлена с экспе-
риментами по выявлению его антагонизма токсичности родствен-
ных ему антибиотиков класса сидерохромов. Так, сидерохромы,
активные как факторы роста, 'блокируют проникновение таких ан-
тибиотиков в клетки Staphylococcus aureus [24]; по-видимому, ме-
ханизм этого процесса — конкуренция с транспортной системой
такого же типа. Иногда внутри клетки сидерохромный антибиотик
может оказаться токсичным для различных процессов [25] (в за-
висимости от его конкретной структуры), которые могут не иметь
14*
212
Глава 5
никакого отношения к метаболизму железа и которые соответст-
венно не обращаются аналогами сидерохромов.
Так как биологическая активность гидроксаматов и фенолятов
железа(III) может 'быть одинаковой — первые характерны для
грибов и дрожжевых частиц, а вторые функционируют (в соответ-
ствии с последними данными) только в истинных бактериях, —
по-видимому, вполне приемлемо для выяснения механизма их био-
логического действия использовать такие микроорганизмы как Ва-
рне. 5.5. Поглощение железа штам-
мом Bacillus subtilis dhb-4 при до-
бавлении 50 мкг/мл итоевой кислоты
(----------) и без нее (------------
[Pe/ers, Warren, Biochim. Biophys.
Acta, 165, 255 (1968)].
cillus sp. и особенно группу энтеробактерий типа Escherichia colt и
Salmonella typhimurium. Например, из Ustilago sp. невозможно
получить феррихромный ауксотроф; такие мутанты были бы же-
лательны, поскольку у дикого типа высока концентрация сидеро-
хрома. Таким образом, весомые доказательства активного транс-
порта сидерохромов, впервые полученные Петерсом и Уорреном
[26] в опытах с Bacillus subtilis, были вскоре подтверждены дру-
гими исследователями, использовавшими Е. coli [27] и S. typhimu-
rium [28].
Выделение из культуры В. subtilis первого фенолятного дефер-
рисидерохрома, итоевой кислоты [19] (название которой проис-
ходит от /гоп Transferring Ortho-phenol), охарактеризованной как
2,3-диоксибензоил глицин (IV), последовало после того, как из
других видов бактерий был выделен его сериновый аналог (III)
[29], бис-лизиновое производное (II) [30] и энтеробактин (I) [2].
Было показано, что, за исключением бис-лизинового соединения,
все другие фенолятные деферрисидерохромы транспортируют же-
лезо в организмах В. subtilis, Е. coli и S. typhimurium соответст-
венно.
Транспорт соединений железа микробами (сидерохромы)
213
Как показано на рис. 5.5, Петерс и Уоррен [26] установили,
что итоевая кислота стимулирует поглощение железа мутантны-
ми штаммами В. subtilis, частично блокированными в биосинтезе
этого деферрисидерохрома. Поглощение железа зависит как от тем-
пературы, так и от энергии и, по-видимому, повышено в клетках,
накапливающих железо. Аналогичные данные были получены Ван-
гом и Ньютоном [27], которые, работая с Е. colt, показали, что
для вхождения железа требуется активная транспортная система,
а также 2,3-диоксибензойная кислота или ее эквивалент; неизвест-
но, проводились ли такие исследования с энтеробактином.
8 час
Рис. 5.6. Иллюстрация феррихромного антагонизма альбомицина и метод отбора
мутантов Salmonella typhimurium, не активных в феррихромном транспорте.
Исследование биохимической генетики S. typhimurium убеди-
тельно продемонстрировало участие сидерохромов в переносе же-
леза. Этот организм образует серию мутантов, которые способны
расти в среде с минимальным содержанием цитрат-иона только
при добавлении солей железа, энтеробактина или других синтети-
ческих или природных связующих агентов [28]. По-видимому,
в среде с минимально низкой концентрацией этих веществ цитрат
связывает железо и делает его бесполезным для клетки. Энтеро-
бактин (I) при низкой концентрации облегчает усвоение S5Fe сус-
пензией клеток ауксотрофов S. typhimurium; при этом в .молярном
отношении он в 100 раз активнее, чем III. Мутанты делятся на
два класса: те, которые в процессе биосинтеза блокируют 2,3-ди-
оксибензойную кислоту, и те, которые не способны соединять эту
кислоту с серином. Эксперименты с трансдукцией при участии Фа-
га Р-22 дают основание предположить, что гены для биосинтеза
энтеробактина собраны в «железном опероне». S. typhimurium LT2
хорошо растет на сложной естественной среде и вообще чувстви-
телен к альбомицину (рис. 5.6). Высказывалось мнение, что штам-
мы, устойчивые к антибиотикам, не имеют сидерохромной транс-
214
Глава 5
портной системы; эта гипотеза подтверждена с помощью ферри-
хрома, меченного тритием (рис. 5.7) [31]. Таким образом, даже
если S. typhimurium не синтезирует тидроксаматы, он содержит
транспортную систему для группы широко распространенных в
природе сидерохромов. Поскольку некоторые энтеробактерии мо-
Рис. 5.7. Поглощение меченного три-
тием феррихрома клетками Salmo-
nella typhimurium (Pollack I. R„ Do-
ctoral Dissertation, University of Ca-
lifornia, Berkley, 1970).
гут создавать как фенолятные, так и гидроксаматные оидерохро-
мы [5], способность S. typhimurium продуцировать последние мог-
ла исчезнуть ,в ходе эволюции.
1.6. Назначение структуры сидерохромов
Определив функцию сидерохромов как переносчиков железа,
целесообразно рассмотреть в свете их признанной биологической
роли химическую структуру и свойства наиболее типичных соеди-
нений— фенолята железа(Ш) [железо(III)энтеробактин (1а)] и
гидроксамата железа(III) [феррихром (Va)].
Энтеробактин растворим в эфире, но его комплекс с желе-
зом (III) имеет отрицательный заряд, равный —3, и растворим
только в полярных растворителях. Феррихром умеренно раство-
рим в метаноле и нерастворим почти во всех неполярных средах.
Таким образом, растворимость в воде комплекса железа, по-ви-
димому, присуща всем этим системам. С другой стороны, мико-
бактины с ионом металла или без него растворимы только в ли-
пофильных средах, и именно этим можно объяснить тот факт, что
отличные от микобактина сидерохромы неактивны в ауксотрофных
микобактериях.
В феррихроме три гидроксаматных аниона соединяются с
ионом железа(III), образуя нейтральный комплекс; в энтеробак-
тине комплекс железа имеет суммарный заряд —3 (так же как в
Транспорт соединений железа микробами (сидерохромы)
215
феррихроме А, заряд обусловлен остатком дикарбоновой кислоты
в ацильном заместителе гидроксамовой кислоты). Все низшие фе-
ноляты, такие, как Реш(|итоевая кислота) з, также имеют большой
отрицательный заряд. Однако родоторулиевая кислота (VI)
[32] —деферрисидерохром, обычно обнаруживаемый в некоторых
дрожжах, в комплексе с железом состава 1 : 1 имеет общий поло-
жительный заряд, равный единице. Таким образом, функции си-
дерохромов, по-видимому, в значительной степени определяются
общим зарядом.
Как уже было отмечено, присутствие исключительно атомов
кислорода в качестве координирующих центров во всех деферри-
сидерохромах будет гарантировать высокое отношение устойчи-
востей Fe(III)/Fe(II). В земной коре железо, окисляясь кислоро-
дом, выделяемым в процессе фотосинтеза, находится главным об-
разом в состоянии окисления +3. И поэтому для эффективного
комплексообразования требуются лиганды, обладающие высоким
сродством именно к состоянию окисления +3. Исследование ком-
плексообразования некоторых ионов металлов с синтетическими
и природными гидроксамовыми кислотами показало, что наибо-
лее прочно с ними связывается ион Fe(III) [lg/G деферриоксами-
на В с Fe(III) равен 30,6] (табл. 5.1) [33]. Такие высокие кон-
станты связывания сопоставимы с константами лучших синтети-
ческих связывающих реагентов для железа(III), используемых в
неорганической аналитической химии. Железо в феррихроме на-
ходится в «ионном», высокоспиновом [34] состоянии и довольно
Таблица 5.1
Константы устойчивости железо(Ш)тригидроксамат-
ных сидерохромов®
Лиганд IgK Увеличение по сравнению с триацетилгид- роксаматом
Деферриферрихром 29,1 0,8
Деферриферрихризин 30,0 1,7
Деферриферриоксамин В 30,5 2,2
Деферриферриоксамин Di 30,8 2,5
Деферриферрихром А ~32,0 3,7
Деферриферриоксамин Е 32,4 4,1
aAnderegg G. et al., Helv. Chim. Acta, 46, 1409 (1963); Wetlands J. B.. Experentla Suppl.,
IX, 22 (1964).
216
Глава 5
быстро обменивается [35]. Комплекс с железом(III) образуется
быстро и устойчив при высоких значениях pH; его можно переве-
сти в гидроксид только действием разбавленной щелочи. Ферри-
хром А, в котором сконцентрированы три отрицательных заряда,
удерживает железо еще прочнее, чем феррихром, и гораздо более
устойчив к действию щелочи [36]. Меньше известно о свойствах
фенолятных сидерохромов железа; известно лишь, что в условиях,
при которых железо (III) гидроксаматы образуются быстро [37],
фенолятный комплекс образуется медленнее. Известно также, что
железо(III) феноляты, производные пирокатехинов, устойчивы и
используются в промышленном производстве чернил.
Как показано на рис. 5.1, предполагается, что в условиях нор-
мального питания железом ([Fe]^10~7 М) транспортные лиган-
ды пассивно направляются к поверхности клетки, где они могут
захватывать железо из среды. Стенка клетки должна действовать
как матрица, ограничивающая диффузию таких молекул; некон-
тролируемое выделение в среду было бы расточительным для ор-
ганизмов, живущих в воде, но может быть полезно для питания
организмов, обитающих в почве. Однако при низкой концентра-
ции железа нарушение регуляторного контроля над биосинтезом
деферрнсидерохрома привело бы к его перепроизводству и, сле-
довательно, накоплению в среде. Это могло бы обусловить образо-
вание очень небольшого количества железа в окружении, доступ-
ном для клетки; возможно, это очень важно экологически для
роста смешанных популяций в почве.
Глубокие конформационные изменения лиганда, сопровождаю-
щие комплексообразование в водной среде, были исследованы в
ряду феррихромов с помощью метода ПМР [16]. Можно ожидать,
что аналогичное конформационное изменение будет наблюдаться
и в случае энтеробактина. Возможно, благодаря этому конформа-
ционному изменению поверхность клетки приобретает способность
селективно удалять деферрисидерохромы. Интересно отметить, что
для ионофоров одновалентных катионов щелочных металлов так-
же характерны конформационные различия между свободным и
комплексным состояниями ионофора [38].
К сожалению, мы еще слишком мало знаем о химии микроб-
ных мембран для предсказания детального механизма включения
сидерохромов в клетку. Однако основным процессом, посредством
которого сидерохромы попадают в клетку, является активный
транспорт [26, 27]. Устойчивость к антибиотическому сидерохрому
альбомицину была использована для отбора мутантов, неактив-
ных в транспорте сидерохромов, но это нельзя установить, если
мутантам недостает специфических связывающих белков [31].
Транспорт железа, подобно транспорту других важных питатель-
ных веществ, может быть очень сложным и многостадийным про-
цессом.
Транспорт соединений железа микробами (сидерохромы)
217
В случае гидроксаматных сидерохромов кажется очевидным,
что комплекс не может диффундировать назад через мембрану.
Так, феррихром .первоначально был выделен из клеток U. sphaero-
gena, и опыты с применением меченного радиоактивным изотопом
лиганда показали, что деферрисоединение не выходит из клетки
до тех пор, пока все железо не будет израсходовано за счет свя-
зывания в железосодержащие ферменты и белки [21]. И опять
следует учитывать конформационные различия между свободными
и комплексными частицами и (или) привлечь интактную энергию
метаболизма системы для объяснения причины сохранения хелат-
ного соединения. Высокий уровень содержания кобальта в среде
для роста организмов вызывает накопление сидерохромов в клет-
ках или мицелии грибов [40]. Механизм такого влияния кобальта
не ясен, но можно представить конкуренцию с Fe(II) за репрес-
сорные белки, предполагая при этом, что комплекс кобальт—белок
не способен присоединяться к операторному гену.
Флавопротеины из U. sphaerogena катализируют восстановле-
ние феррихромного железа под действием TPNH или DPNH*, при-
чем более предпочтителен первый [41]. Специфичность этой реак-
ции вызывает сомнения; в любом случае вряд ли клетке выгодно
освободить железо из сидерохрома раньше, чем потребуется ме-
талл. Такая реакция может и иметь место, если железо необходи-
мо сделать восприимчивым к гидроксилированию и осаждению.
Таким образом, гораздо убедительнее выглядит гипотеза, которая
позволяет мысленно представить прямую передачу железа акцеп-
тору, например порфириногену или порфириноген-апоферментному
комплексу. В культурах с низким содержанием железа при ста-
рении некоторые полифункциональные сидерохромы как фенолят-
ного, так и гидроксаматного типов подвергаются распаду, но еще
не установлено, действительно ли для освобождения железа тре-
буется такой «деструктивный» механизм. Высокая активность
сидерохромов должна означать, что молекулы-носители сохраня-
ются и посредством каталитического цикла вводят железо в
клетку.
Известно, что феррохелатаза связана с липидом и что ферри-
хром— эффективный донор железа для этого фермента [42]. Фер-
рихром проявляет универсальную активность по отношению ко
всем сидерохромным ауксотрофам (за исключением микобакте-
рий) , которую можно объяснить способностью этого особого
транспортного агента растворяться ib неводных растворителях ти-
па спиртов. >В случае Arthrobacter JC-9 сидерохромы могут быть
полностью заменены синтетическими липидорастворимыми связы-
* TPNH — трифосфопириднннуклеотид, восстановленная форма, DPNH —ди-
фосфопиридиннуклеотид, восстановленная форма — устаревшие названия. Теперь
первое соединение называют никотинамидадениндинуклеотид (НАДН), второе —
никотинамиддинуклеотидфосфат (НАДФН). — Прим, перев.
218
Глава 5
вающими агентами, которые нужно использовать в подходящих
концентрациях [20]. Все эти данные заставляют предположить,
что по крайней мере у Arthrobacter сидерохромы не являются спе-
цифическими «коферментами», участвующими во внутреннем ме-
таболизме железа в клетке, а просто служат для передачи иона
металла в то место, где он может быть превращен в физиологиче-
ски функциональное производное.
Следует упомянуть еще об одной проблеме, связанной с транс-
портом железа. Деферриферрихромы обычно готовят посредством
ферментации в условиях низкой концентрации железа и последую-
щей кристаллизацией с добавлением солей Fe(III) и Fe(II) (при
насыщении воздухом). Вследствие особого уникального располо-
жения связей от трех молекул гидроксамовых кислот шесть ато-
мов кислорода лиганда приобретают особенное специфическое
расположение вокруг иона металла [43]. В области длин волн,
соответствующих максимуму поглощения тригидроксамата желе-
за (III) (около 420—440 нм) [3], проявляется эффект Коттона,
а кристаллографический анализ феррихрома А показывает, что
абсолютная конфигурация окружения иона железа является кон-
фигурацией левостороннего пропеллера [14]. Интересно отметить,
что построение молекулы железо (III)энтеробактина с помощью
пространственных моделей показало, что для этого сидерохрома
возможна такая же абсолютная конфигурация [2].
1.7. Распространение
Возможно, что все аэробные и «случайно» аэробные мик-
робные клетки продуцируют сидерохромы или нуждаются ib них.
Рассмотрение очень большого числа дрожжей и дрожжеподобных
организмов показало, что родоторулиевая кислота (VI) является
общим деферрисидерохромом для всех этих частиц; кроме того,
обнаружено образование культурой Crytococcus melibiosus ново-
го аланинсодержащего феррихрома [компонент 'С (Уж)] [7].
В общем случае сидерохромы, являющиеся производными
б-М-ацил-Ь-б-Ы-оксиорнитина, обычно встречаются в грибковых
культурах ‘актиномицетов, базидиомицетов и несовершенных гри-
бов [13]. Один из актиномицетов, Streptomyces griseus, образует
антибиотики феррихромного типа [альбомицин, гризеин (Уи—Ул),
a Pseudomonas fluorescens выделяет циклический декапептид, фер-
рибактин [44], содержащий и о- и ь-формы б-М-оксиорнитина.
Следующий, более высокий гомолог этой аминокислоты, ь-е-Ы-ок-
силизин, найден в семействе деферрисидерохромов из микобакте-
рий [микобактины (XI)] [4] и в аэробактине (IX) [5] из Aerobac-
ter aerogenes. Сидерохромы ‘актиномицетов отличаются тем, что
содержат основания, которые можно рассматривать как продукты
декарбоксилирования а-амино-со-оксиаминокислот, а именно а-ами-
Транспорт соединений железа микробами (сидерохромы)
219
Таблица 5.2
Сидерохромо-зависимые ауксотрофные микроорганизмы
Источник Активные факторы3 Литера- тура
А. Природный 82
Pilobolus kleinii Экстракт навоза, копроген, феррихром, ге- мин
Arthrobacter terregens Террегенс-фактор, гемин, копрогеи, ферри- хром, аспергилловая кислота, экстракты почвы и печени, синтетические хелатирую- щие агенты, микобактин 83
Arthrobacter JG-9 (A. fla- vescens ATCC 25091) Террегенс-фактор, ферриоксамин В, ферри- хром, аспергилловая кислота, копрогеи, гризеин, микобактин, гемин, высшие фуза- ринииы, родоторулиевая кислота, а цетил- гидроксамовая кислота и синтетические хелатирующие агенты 20, 84
Microbacterium lacticum ATCC 8181 Феррихром, копроген, террегенс-фактор, ге- мовые соединения, гликозил глицин 85
Mycobacterium johney Микобактины 4
Б. Индуцированный
Bacillus megaterium, ATCC 19213 (Sk-му- танты) Шизокиннны, ферриоксамин В, ферримицин, А, феррихром, микобактин, 2,3-диоксибен- зойная кислота, ацетилгидроксамовая кислота, природные и синтетические хела- тирующие агенты 23
Escherichia coli (Chr 2-му- 2,3-Диоксибензоилсерин, цитрат 27
тант)
Salmonella typhimurium LT2 (ЕпЬ-мутанты) Диоксибеизоилсерин, эптеробактин, ферри- хром, шизокинин, деферриферриоксамин Е, родоторулиевая кислота, аскорбат- ион и ЭДТА 28
® Деферрисидерохромы в большинстве случаев были бы так же активны, как комплексы с
железом, если бы не предпринимались меры для удаления загрязняющих количеств железа
нз среды. Большинство сидерохромных ауксотрофов обладают способностью расти в присут-
ствии высоких концентраций неорганического железа.
ночо-оксиамино'пента'н или а-амино-щ-оксиаминогексан; такие про-
дукты, называемые ферриоксаминами или ферримицинами (XV,
XVI), найдены во всех детально исследованных актиномицетах
[45].
Сидерохромы фенолятного типа также широко распространены
в микробах, особенно в истинных бактериях. Выделены и охарак-
теризованы следующие деферрисидерохромы: 2,3-диоксибензоил-
глицин [итоевая кислота (IV)] [19] из В. subtilis, 2,3-диоксибен-
зоилсерин (III) юз Е. colt [29] и A. aerogenes [46], бис-2,3-диокси-
бензоиллизин (II) [30] из Azotobacter vinelandii и энтеробактин
220
Глава 5
(I) [2] из 8. typhimurium. Специфический пирокатехин этих де-
феррисидерохромов, 2,3-диоксибензойную кислоту, можно обнару-
жить в актиномицетах, грибах и высших растениях [8].
Сидерохромиую активность можно обнаружить ib почве и на-
возе, откуда можно заключить, что такие соединения имеют общее
значение в метаболизме железа микробами. Степень их важности
определяется существованием обоих как .природных, так и инду-
цированных ауксотрофов для таких метаболитов (табл. 5.2). Ге-
мин активен для нативных иуксотрофов; нет сомнения, что такие
штаммы адаптировались к использованию нескольких природных
железосодержащих соединений. Тот факт, что сидерохромные ан-
тибиотики, такие, как альбомицин (Уз) и ферримицин (ХУд), обла-
дают «широким спектром» действия, свидетельствует о повсемест-
ном распространении транспортных механизмов для этих веществ
[47]. Возможно, Fe(II) достаточно растворимо в культурной сре-
де анаэробных частиц для того, чтобы отпала необходимость в си-
дерохромных носителях, но эта точка зрения нуждается в экспе-
риментальной проверке. Специфические транспортные системы
для ионов металлов, отличных от железа, могут и не требоваться,
поскольку такие ионы обычно гораздо более растворимы, чем же-
лезо, и, кроме того, они требуются в меньших количествах.
1.8. Заключение
В схеме, показанной на рис. 5.1, имеются некоторые детали,
которые напоминают 'метаболизм железа у млекопитающих, не-
смотря на то, что высшие организмы, ,по-видимому, не содержат
сидерохромов [48]. Так, у животных не существует никакого ме-
ханизма для выведения железа. Этот элемент всасывается через
стенки кишечника по мере необходимости. В этом много общего
у микробов и животных; однако, возможно, только животные име-
ют сложные устройства для запасания железа и накопления его
в ферритине, хотя этот белок, по-видимому, встречается и у неко-
торых Phycomyces sp. [49]. Сидерохромы должны присутствовать
в кишечнике высших животных, но значение этих веществ для ме-
таболизма железа в организме-хозяине пока неясно. Способность
получать железо, по-видимому, является ключом к пониманию
изменчивости и патогенности при микробном заражении [50].
С медицинской точки зрения представляет значительный инте-
рес роль пацифарина [51], сидерохрома фенолятной группы [52].
Не считая нескольких интересных работ, а именно по гемовым
требованиям некоторых нематод [53], пока нет ни одного система-
тического исследования сидерохромов всех животных. Ткани жи-
вотных обладают очень ограниченной способностью преобразовы-
вать ароматические соединения; следовательно, можно ожидать,
что их сидерохромы, если они существуют, будут относиться к гид-
Транспорт соединений железа микробами (сидерохромы)221
роксаматной группе. Предполагают, что некоторые животные .мем-
браны .содержат 'низкомолекулярные вещества, образующие хела-
ты с железом [54].
Мезилат деферриферриоксамина В выпускается компанией Ci-
ba Pharmaceutical под торговым названием «десферал» и находит
важное применение для удаления патологических отложений же-
леза, а также для нейтрализации случайно проглоченного железа
[55]. Оксимочевина, синтетическая гидроксамовая кислота, ис-
пользуется 1в химиотерапии рака; она контролирует синтез ДНК,
очевидно, за счет связывания железа в белок Вг риботидоредук-
тазного комплекса [56].
Гидроксамовые кислоты и (или) сидерохромы были найдены
в морских водорослях [57] и некоторых высших растениях [58],
но их роль в этих жизненных формах не установлена. Высшие
растения, произрастающие при низких концентрациях железа, вы-
деляют вещества, напоминающие деферрисидерохромы [59]. Син-
тетические хелатирующие агенты делают железо доступным для
растений без присоединения другого лиганда [60]; было бы инте-
ресно систематически исследовать растительный метаболизм из-
вестных микробных сидерохромов, обычно встречающихся в поч-
ве. Синтетические гидроксамовые кислоты применяются ib агро-
технике для того, чтобы сделать железо более доступным. Такие
вещества .могут, ингибируя уреазу, препятствовать .потере аммиа-
ка из почвы [61].
В заключение можно сказать, что сидерохромы играют актив-
ную роль в транспорте железа в микробах, которая в некоторых
отношениях параллельна роли мембраноактивных «ионофоров»
для катионов щелочных металлов [38]. Очевидно, небольшое
уменьшение диффузной подвижности, возникающее из-за увели-
чения молекулярной массы комплексного иона, более чем компен-
сируется преимуществом органической оболочки. Синтез последней
можно точно регулировать и, кроме того, специфические транс-
портные системы для комплекса могут быть локализованы в кле-
точной мембране. При низких .концентрациях железа происходит
увеличение внутренней плотности деферрисидерохромных молекул,
благодаря чему организм может выделить больше лиганда и за-
хватить больше ионов металла из среды. .При высоком дефиците
железа лиганд выделяется в среду в значительном количестве и
служит для растворения нерастворенных остатков железа. Этим
транспортная система защищает клетку от вторжения токсических
количеств неорганического железа и выбрасывает лиганд, не на-
грузив его специфическим ионом металла.
В последние годы появился ряд обзоров, касающихся различ-
ных аспектов химии сидерохромов. Читатель отсылается к стать-
ям, в которых рассматриваются в основном феррихромы [3, 13],
ферриоксамины [1], сидерохромные антибиотики [47] и микобак-
222
Глава 5
тины [4]. В данной главе особое внимание будет уделено фенолят-
ным сидерохромам, так как .сведения о них до сих пор не собраны
в литературе.
2,. СТРУКТУРА И СВОЙСТВА
2.1. Методы исследования
Методы идентификации сидерохромов довольно хорошо .разра-
ботаны в химии природных продуктов. Однако первоочередная за-
дача состоит в том, чтобы увеличить их выход; этого можно до-
биться, регулируя соответствующим образом концентрацию железа
в среде. Возможно, иногда необходимо, как в случае Rhodotorula
pilimanae, для (получения максимального выхода добавлять в сре-
ду больше углерода и азота [32]. Лучше всего выделить вещество
без железа, но если это не удастся, железо можно удалить из
комплекса с помощью различных методов [3].
Все известные фенолятные деферрисидерохромы растворимы в
органических растворителях при значении pH, которое примерно
на 2 единицы ниже значения рКо карбоксильной группы. Хотя эн-
теробактин (I) не имеет свободной кислотной группы, он может
присутствовать в среде в виде комплексов с ионами и перед экс-
тракцией должен быть отделен от них путем подкисления. Гидро-
литические фрагменты, полученные с 6 н. НС1, .а также 2,3-диокси-
бензойную кислоту и а-аминокислоту можно обнаружить по мето-
ду Арноу [62] и по реакции с нингидрином соответственно. Кроме
того, многоосновные фенолокислоты при ультрафиолетовом облу-
чении обнаруживают интенсивную и характеристичную флуорес-
ценцию [19].
Гидроксамовые сидерохромы более разнообразны по структуре,
но и у них прослеживаются некоторые общие свойства. Эти соеди-
нения обычно можно экстрагировать либо смесью хлороформ—фе-
нол (1:1 по весу), либо бензиловым спиртом [3]. Из этих орга-
нических растворителей активное начало можно вернуть в воду
добавлением больших количеств эфира. При гидролизе не содер-
жащих железа молекул под действием 6 н. НС1 из гидроксамат-
ной части образуются положительно заряженные частицы тетра-
золийгидроксиламина. Гидролиз с 6 н. HI превращает гидроксил-
аминогруппу в первичную аминогруппу [4]. Гидрлиз под действи-
ем НС1 в присутствии железа приводит к образованию артефактов
с отличными выходами. Перйодат является реагентом для изби-
рательного окисления связей в гидроксамовой кислоте, поскольку
при такой обработке и сложноэфирные, и амидные связи в моле-
куле остаются интактными [3].
Гидроксаматные сидерохромы являются всегда вторичными,
так как в них к атому азота (гидроксиламина присоединена ал-
Транспорт соединений железа микробами (сидерохромы)223
кильная группа [—CON (ОН) R]. Все гидроксаматы такого типа
гладко превращаются при обработке перйодатом в легко обна-
руживаемый димер quc-нитрозоалкана (амм при 267 нм ~ 10)
[3]. Можно предполагать, что карбоксилатные группы, образую-
щиеся после окисления перйодатом, присоединены к гидроксил-
аминогруппам по связям гидроксамовой кислоты в первоначаль-
ной молекуле. Изменения в спектре гидроксамата железа (III) при
изменении pH могут помочь детально установить структуру же-
лезосодержащего центра.
И диоксибензоильные кольца, и свободные гидроксиламино-
группы неустойчивы в щелочной области pH до соответствующих
им значений рКа-
2.2. Феноляты
2.2.1. Энтеробактин
энтеробактин (I)
железо (III) энтеробактин (1а)
Энтеробактин (I) был получен из культуры Salmonella typhi-
murium LT2 с низким содержанием железа, к которой были до-
бавлены ионы марганца [2]* в относительно высокой концентра-
ции. Соединение кристаллизуется из смеси этанол—вода, т. пл.
202—203 °C.
* Идентичное соединение, названное энтерохелином, было выделено из
бульона Escherichia coli и других кишечных бактерий Гибсоном с сотр. [62а].
S. typhimurium образует приблизительно только 6 мг на 10 л среды. Для получе-
ния большего количества этого соедииення можно использовать штамм 62-1
Гибсона Aerobacter aerogenes.
224
Глава 5
Энтеробактин практически нерастворим .в воде, но легко рас-
творяется в ацетоне, диоксане, диметилсульфоксиде или метаноле.
Его растворимость в воде увеличивается при значениях pH 7—8,
при которых гидроксильная группа в .положении 2 начинает иони-
зоваться. Эта группа нейтральна при электрофорезе на бумаге
при pH 5 и обнаруживает яркую голубую флуоресценцию в
ультрафиолетовом свете, характерную для 2,3-диоксибензоильных
колец. Гидролиз под действием 6 н. НС1 в качестве единственных
идентифицируемых фрагментов дает ь-серин и 2,3-диоксибензой-
ную кислоту; элементный анализ на С, Н и N позволяет предпо-
ложить, что структура энтеробактина представляет собой ангид-
рид, состоящий из этих двух фрагментов.
ИК- и ПМР-спектры энтеробактина напоминают спектры
2,3-диоксибензоилсерина. Сильное поглощение при 1760 см-1 мож-
но отнести к деформационным колебаниям сложноэфирной карбо-
нильной группы. ПМР-спектры при 220 МГц в дейтеродиметил-
сульфоксиде с тетраметилсиланом в качестве внутреннего стан-
дарта имеют следующие характеристики: 6 = 8,90, дублет (амид),
6 = 7,30, дублет (ароматические протоны), 6=6,96, дублет (арома-
тические протоны), 6=6,71, триплет (ароматические протоны),
6 = 4,91, сложная полоса (а-протоны), 6=4,64, сложная полоса
(метиленовые протоны) и 6=4,41, сложная полоса (метиленовые
протоны). В масс-спектре главный ион имеет т/е 223, что соот-
ветствует мономеру; метилирование энтеробактина диазометаном
дает ионы, Miacca которых соответствует димеру или более высоко-
молекулярному соединению. Седиментационное равновесие в воде
и D2O показало, что энтеробактин является тримером с .молеку-
лярной массой 669 и структурой, изображенной формулой I. Та-
ким образом, энтеробактин является циклическим триэфиром
u-серина, в котором все три аминогруппы ацилированы остатками
2,3-диоксибензойной кислоты.
Железо(Ш)энтеробактин (1а) проявляется при электрофорезе
на бумаге при pH 6,5 как трехвалентный анион; предполагается,
что его структура такая, как показано формулой Га. Три пирока-
техиновые группы расположены по одну сторону от плоскости
12-членного циклического полиэфира и образуют левосторонний
пропеллер вокруг иона Fe(III). Это абсолютно такое же распо-
ложение, как d-конфигурация, обнаруженная в феррихроме А
кристаллографически [14].
Мутанты S. typhimurium, не способные синтезировать ком-
плекс I, реагируют с ним при молярных концентрациях на два
порядка меньших, чем те, которые требуются, когда сидерохро-
мом является 2,3-диоксибензоилсерин. Последнее соединение, по-
видимому, может использоваться для транспорта железа, хотя ме-
нее эффективно у Salmonella и, возможно, в других видах. Одна-
Транспорт соединений железа микробами (сидерохромы)225
ко вполне вероятно, что энтеробактин является главным
активным сидерохромом в этих микроорганизмах*.
Энтеробактин проявляет пацифариновую активность в мыш-
цах [626]; очевидно, она идентична ингибированию колипина В,
которое обнаружено в кишечных бактериях [62в].
2.2.2. 2-№£-№-Ди(2,3-диоксибензоил)-1.-лизин (II)
Это соединение было получено в виде белого аморфного по-
рошка с выходом приблизительно 90 мг/л при росте Azotobacter
vinelandii О в среде с низким содержанием нитрата железа [30].
Оно отличается от соединения, полученного химически путем аци-
лирования обоих атомов азота ь-лизина 2,3-диоксибензойной кис-
лотой. Хотя комплекс с железом не был исследован, интересно
отметить, что точно такое же число атомов, а именно 5, разделяет
две ацилированные аминогруппы в энтеробактине. Это позволяет
предположить, что соединение II участвует в транспорте железа
у A. vinelandii.
2.2.3. 2,3-Диокси-^-бензоил-ь-серин (III)
III
* Мутации, влияющие на транспорт железа у Escherichia coli, сравнимы
с теми, которые имели место у Salmonella typhimurium с предполагаемым заме-
щением в хромосоме. Однако в отличие от Salmonella. Е. coli, по-видимому,
имеет индуцируемую систему для усвоения цитрата железа (III) [Сох G. В.,
Gibson F., Luke R. К., Newton N. A., O’Brien J. G., Rosenberg H., J. В act., 104,
219 (1970)].
15—2451
226
Глава 5
Брот с сотр. [29] установили, что при росте ауксотрофа .метио-
нин— витамин Bi2 Е. coli К12 на среде из солей глюкозы без до-
бавления железа с выходом 75 мг/л образуется соединение, выде-
ляемое в виде комплекса с Fe(III). Раствор этого соединения
обесцвечивается при pH 2 или в присутствии 0,1 М ЭДТА. Лиганд
был идентифицирован как 2,3-диокси-Ы-бензоилсерин (III); его
структура подтверждена О’Брайном с сотр. [46] посредством син-
теза d- и d,l-конфигураций. Полагают, что конфигурация при-
родного соединения должна быть ь, хотя синтетический продукт,
содержащий d-серин, по активности сопоставим с мутантным
штаммом Escherichia coli. Железо действует как мощный угнета-
ющий агент как для фермента, соединяющего 2,3-диоксибензойную
кислоту с ъ-серином в Е. coli [63], так и для всей ферментативной
системы, превращающей хоризмат в 2,3-диоксибензоат у A. aero-
genes [8]. Соединение III было получено из A. aerogenes [46] и
S. typhimurium [31, 50], так что можно было провести корреляцию
вирулентности S. typhimurium с его способностью извлекать же-
лезо из сыворотки человека [50].
2.2.4. 2,3-Диоксибензоилглицин (итоевая кислота) (IV)
с=о
I
H-N
I
н-с-н
I
о=с-он
итоевая кислота (IV)
Итоевая кислота (IV) —глицин, конъюгированный с 2,3-диок-
сибензойной кислотой, был первым описанным фенолятным сиде-
рохромом [19]. Он был выделен в количестве около 50 мг/л из
культуры Bacillus subtilis с низким содержанием железа. Химиче-
ски это соединение было синтезировано взаимодействием 2,3-диок-
сибензойной кислоты с эфиром глицина с последующим омылени-
ем. Было показано, что образование этого соединения полностью
подавлено у В. subtilis при концентрациях железа 3-10_6 г-атом/л
[64]; при этом железо(Ш)шизокинин более активен, чем неор-
ганическое железо [65]. Было обнаружено, что фенолокислоты
способствуют транспорту железа в В. subtilis [26]; это подтвер-
ждает предположение, согласно которому такие «мономерные»
сидерохромы, как IV, могут играть роль, эквивалентную роли эн-
теробактина.
Транспорт соединений железа микробами (сидерохромы)
227
2.3. Тидроксаматы
2.3.1. Семейство феррихрома
Феррихром (Va) R = R' = R" = Н; R'" = СН3—
Феррихром A (V6): R=R'=HOCH2—; R"=H;
у /СН3
R"' = '=/ (транс)
Vh2co2h
Феррихризин (Vb) : R = R' = НОСН2—; R" = Н; R” = СН3—
Феррикрокин (Vr): R=R”=H; R'=НОСН2—; R"'=CH3-a
Феррирубин (Уд): R=R'=HOCH2—;
у /СН3
R" = Н; R" = (транс)
'СН2СН2ОН
Ферриродин (Ve): R = R'=HOCH2—;
ЧСН2СН2ОН
(цис)
сн3
* Кристаллическая структура, установленная Залкинымссотр. [14] для фер-
рихрома А. Нумерация а-углеродных атомов дана Залкиным с сотр. [14]. Об-
суждение вопроса о конформации феррихромных соединений см.: Emery, Bio-
chemistry, 6, 3858 (1967); Llinas М., Klein М. Р., Neilands J. В., J. Mol. Biol., 52,.
399 (1970); Llinas M., Doctoral Dissertation, University of California. Berkeley,
1971.
228
Глава 5
Феррихром С (Уж): R = R’ = Н; R' = R"' = СН3—а
Краситель Сейка А (Уз): R = НОСН2—; R' = R'" = СН3—; R" = На
СН,
N-Z
Альбомнцин 62 (Уи): R = Ацил—N=/ 'N—SO2—О—СН2—;
R' = R" = НОСН2—; R'" = СН3—
СН,
1 //°
N—
Альбомицин е (Vk): R=H—N=^ —SO2—О—CH2—;
R'= R" =НОСН2—; R'"=CH3—
сн3
Альбомицин 6i (Ул): R = О=/ 'N—SO2—О—CH2—;
R' = R" = HOCH2—; R"' = CH3—
Гризеин: этот антибиотик может быть идентичен одному из компонентов альбо-
мицина.
Феррибактин: циклический декапептид, содержащий и в- и L-формы S-N-оксиор-
нитина.
Характерной структурной особенностью семейства феррихро-
ма является то, что все его члены — циклические гексапептиды,
содержащие трипептид из б-П-ацил-ь-б-И-оксиорнитина и трипеп-
тид, состоящий из маленьких нейтральных аминокислот. Послед-
ний может быть построен либо из одного глицина, серина или
аланина, либо из некоторой определенной комбинации этих ами-
нокислот. Так, феррихром (Va) содержит триглицин, в то время
как альбомицин (Уи—Ул) состоит из трисерина. Последователь-
ность серилсерилглицин является общей для феррихрома А (V6),
феррихризина (Ув), феррирубина (Уд) и ферриродина (Ve). Кра-
ситель Сейка А [86] и феррихром С [7] содержат аланин. Таким
образом, конформации альбомицинов могут быть отличными от
конформаций других феррихромов, так как только они из всего
ряда не содержат остатков глицина в положении R" в формуле V.
Соединения ряда феррихрома образуют устойчивые нейтраль-
ные комплексы с ионами металлов с координационным числом 6,
преимущественно с октаэдрической конфигурацией. Из всех био-
элементов ион Fe(III) образует наиболее прочные комплексы;
логарифмы констант устойчивости триацетилгидроксамата желе-
за, феррихрома и феррихрома А равны соответственно 28,3, 29,1 и
32 (табл. 5.1). Несмотря на прочное связывание, неорганическое
а Последовательность аминокислот определена методом ПМР (Llinas М.,
частное сообщение)
Транспорт соединений железа микробами (сидерохромы)229
железо наполовину обменивается в феррихроме при нейтральных
значениях pH всего за несколько минут [35]. В ряду феррихрома
положение максимумов поглощения и миллимолярные коэффици-
енты поглощения зависят от таких факторов, как число связанных
ионов железа, pH и природа 'ацильного заместителя. Как прави-
ло, при нейтральных значениях pH амМ равно 3—4 в области
420—440 нм [3]. Гидроксаматные комплексы Fe(II) бесцветны и
относительно неустойчивы. Свойства железо(П1)гидроксаматов
несовместимы с окислительно-восстановительным механизмом для
железа, но, с другой стороны, идеально подходят для выполнения
биологической роли — транспорта металла.
Полная кристаллографическая структура феррихрома A (V6),
установленная Залкиным с сотр. [14], подтвердила формулу, ос-
нованную на разложении и синтезе феррихромов, и, кроме того,
позволила определить конформацию молекулы. Пептидные части-
цы примерно планарны, антипараллельны («кросс-0») углам с
двумя гидроксаматными остатками (и, следовательно, иону ме-
талла), расположенными на другом конце молекулы. В кристалле
определено положение только одной трансаннулярной водородной
связи, образуемой остатком орнитина [3]; вторая водородная
связь образуется между орнитином [2] и гидроксаматным кисло-
родом того же остатка. В растворе комплексы имеют четыре мед-
ленно обменивающихся атома водорода, которые с помощью
ПМР-исследования отнесены следующим образом: алюмихром—-
орнитин [1] («внутренний»), орнитин [2] (участвующий в обра-
зовании водородной связи), орнитин [3] (участвующий в образо-
вании водородной связи) и глицин [3] (связанный водородной
связью или «внутренний»). Пептиды, не содержащие железа, ни-
когда не образуют кристаллов и не имеют медленно обмениваю-
щихся протонов в водной среде; в диметилсульфоксиде для двух
протонов в деметаллированной молекуле обнаружена пониженная
температурная зависимость химического сдвига. Методом ПМР
было установлено, что эти протоны принадлежат глицину [3] и
орнитину [3]. Таким образом, в неводной среде не содержащая
металла пептидная частица, по-видимому, имеет конформацию,
соответствующую конформации циклогексапептида типа Швайце-
ра с двумя трансаннулярными водородными мостиками.
Было найдено, что поликристаллический феррихром А пара-
магнитен до по крайней мере 10 К, и для него были рассчитаны
значения g-фактора, собственные значения энергии и собственные
значения функции. Были измерены также параметры Мессбауэра
для феррихрома А [17] и синтетических железо (III) гидроксама-
тов [67]. В трех железо(III) гидроксаматных кольцах длины свя-
зей гидроксиламин—кислород составляют 1,97, 1,96 и 2,00 А;
в этих же трех кольцах длины связей с карбонильным кислородом
равны 2,02, 2,03 и 2,06 А соответственно [14]. Эти измерения по-
230
Глава 5
называют, что все три гидроксаматные группы могут подходить к
центральному иону металла и, следовательно, все они увеличива-
ют устойчивость комплекса (табл. 5.1).
Предполагают, что феррихромы образуются при окислении ор-
нитина по 6-атому азота с последующим ацилированием углерод-
ного фрагмента, связанного с 3,5-диокси-З-метилвалериановой кис-
лотой, что приводит к производному кофермента А. Феррихромы
обычно продуцируются аскомицетами, базидиомицетами и несо-
вершенными грибами; они представлены формулами V и были
выделены из культур Aspergillius, Penicillium, Neurospora, Usti-
lago и других головневых грибов [3]. Кроме того, одна дрожже-
вая Crypotococcus melibiosus 'форма феррихрома С (Уж) [7] и
альбомииины (Уи—Ул) продуцируются Actinomyces griseus.
Независимо от вида организма феррихром эффективно обра-
щает токсичность альбомицина и обычно активен как фактор
роста при понижении концентрации до нанограммов на милли-
литр. С другой стороны, феррихром А обычно лишен биологиче-
ской активности и поэтому может рассматриваться как специаль-
ный сидерохром, специфичный для первичных организмов.
2.3.2. Семейство родоторулиевой кислоты
м 1 О ОН Г Гн *-с-сн> И 1 к! 1 п 1 •* 1 н роЪопюрулиевая кислота (VI) %F«m + н о о VXT^-CCH, н железо (ЦТ) родоторулат (УШ) н НО о 0YN'si<?c-n-r । ц н J Гн » । «-н-ОЧЛо 0 °и н решрорддоторулиевые кислоты (VII) VII<4L, R - Н) f VII^(L, R - CH,) VIIB (D, R - CH3) VII e (L, R - CHjCHjCHj)
Циклический дипептид или дикетопиперазин б-М-ацетил-ь-(5)-
6-Ц-оксиорнитина, химически идентифицированный как N,N',3,6-
диоксопиперазин-2 (S) ,5(S) -ди (триметилен) - бис-ацетогидроксамо-
Транспорт соединений железа микробами (сидерохромы)
231
вая кислота или родоторулиевая кислота (VI), был выделен из
лишенной железа культуры Rhodotorula pilimanae и родственных
ей дрожжей [32]. Это соединение было охарактеризовано 'восста-
новлением известного дикетопиперазина 6-М-ацетил-ь-орнитина,
а также с помощью ряда физических ‘методов, таких, как ИК-,
ЯМР- и масс-спектрометрия. Как и ожидали, в ИК-спектре соеди-
нения VI цпс-пептидные связи кольца проявляются по отсутствию
либо полос «амид II», либо полос валентных колебаний связей
NH в области выше 3250 см-1 и по отчетливым плоским деформа-
ционным колебаниям вблизи 1450 см-1. Предполагаемая структу-
ра соединения VI подтверждена сравнением характеристичных
сигналов кольцевых протонов в ПМР-спектре с аналогичными сиг-
налами модельного дикетопиперазина. Исследования дисперсии
оптического вращения в дальней УФ-области показали, что это
соединение должно иметь конфигурацию ь,ь.
Несмотря на то что нет никаких сомнений относительно струк-
туры родоторулиевой кислоты, химически она не была синтези-
рована. Однако был получен ряд ее ь- и о-аналогов, представлен-
ных общей формулой VII; все они имеют более короткие боковые
цепи, чем соединение VI [68]. Эти соединения называются «ретро-
родоторулиевыми кислотами», поскольку в них и относительное
положение дикетопиперазиновых колец, и ацильных заместителей
относительно звена гидроксамовой кислоты обратно их располо-
жению в VI.
Из спектра поглощения родоторулата железа (III) при низких
значениях pH можно заключить, что обе гидроксаматные группы
могут быть связаны с расположенным в центре ионом металла
(VIII). Растворы, содержащие % моля соли Fe(III) на 1 моль
соединения VI, ведут себя как полиядерные комплексы, молеку-
лярная масса которых составляет несколько тысяч. По-видимому,
родоторулиевая кислота претерпевает такой же путь биосинтети-
ческих превращений, как и феррихром.
Обзор 142 различных культур, представляющих 19 классов
дрожжей, показал, что гидроксамат образуют два неклассифици-
рованных штамма и 52 штамма из Aessosporon (3 из 3), Crypto-
coccus (1 из 43), Leucosporidium (3 из 11), Rhodosporidium (4 из
14), Rhodotorula (27 из 39), Sporidiobolus (2 из 2) и Sporobolomy-
ces (12 из 13). За исключением одного штамма Cryptococcus, ко-
торый образует феррихром С, большинство других дрожжей обра-
зует родоторулиевую кислоту в количествах, позволяющих выде-
лить ее в виде кристаллов [7].
В то время как родоторулиевая кислота является потенциаль-
ным фактором роста для Arthrobacter JG-9 (половинный рост при
3-10~9 М), она не может обращать токсичность альбомицина для
Е. coli или В. subtilis [32]. Дезоксиродоторулиевая кислота, ди-
кетопиперазин fi-N-ацетил- ь-орнитина, неактивна. Интересно, что
232
Глава 5
ее аналоги VII6 и VIIb (проявляют одинаковую активность с
Arthrobacter JG-9 и обеспечивают (половинный рост при 10-5 М;
ретрородоторулиевая кислота Vllr .всего на порядок менее актив-
на [68]. Эти эксперименты ясно показывают, что родоторулиевая
кислота не может служить в Arthrobacter sp. агентом для перено-
са ацильной группы и что ее биологическая активность должна
быть связана со способностью связывать железо.
Димерумовая кислота (Villa), дикетопиперазин транс-изомера
фузаринина, была получена в конце ферментативного периода из
культур Fusarium dimerum [68а]. В этом соединении уксусная
кислота в ацильной части феррирубина (Уж) замещена родото-
рулиевой кислотой. Однако, по-видимому, не это соединение яв-
ляется предшественником родоторулиевой кислоты [686].
Ьимерумовая кислота (Villa)
Копроген (VIII6) —один из первых выделенных агентов, пере-
носящих железо, продуцируемый Neurospora и другими грибковы-
ми частицами, недавно был охарактеризован как комплекс желе-
Транспорт соединений железа микробами (сидерохромы)
233
за (III) с молекулой димерумовой 1кислоты, этерифицированной по
одной первичной гидроксильной группе остатком N-ацетилфузари-
нина [68в]; копроген В является его диацетильным аналогом
[68а].
2.3.3. Аэробактин
Aerobacter aerogenes 62-1, росший 20 ч на среде из минераль-
ных солей глюкозы без добавления железа, образует сырой аэро-
бактин (IX) с выходом 1 г/л [5]. Восстановительный гидролиз с
HI дает лизин, в то время как разложение с НС1 приводит к об-
разованию лимонной кислоты и двух остатков аминокислот, найден-
ных в микобактине, L-e-iN-оисилизине. Анализ спектров ПМР, элек-
трометрическое титрование, перйодатное окисление, ИК-спектр и
спектр комплекса Fe(III) при разных значениях pH подтвердили
структуру, представленную формулой IX. Это соединение, возмож-
но, играет роль в транспорте железа в A. aerogenes 62-1 —штам-
ме, который также образует 2,3-диоксибензоильное соединение.
СН3 СНз
1 ।
с=о с=о
। ।
NOH NOH
4 I
<СН2)« СООН (СН2)<
I I I
CH-NH-C0-CH2-C-CH2-C0-NH-CH
I I I
СООН ОН СООН
CHS СНз
I Ш '
С=О----------Fe*“---------О = С
I I
NO" О О '-ON
1 \ 1 I
(СН2)4 \ С = О (СН2)«
СН- NH- СО - СН2- С - СН2- СО - NH- СН
I I
СООН СООН
аэробактин (IX)
жепеза(Н!)йзроб^1:тин (X)
Другие штаммы Aerobacter sp. не продуцируют ни гидроксаматов,
ни фенолятов; этот факт подтверждает концепцию о многообразии
путей микробного транспорта железа. В любом случае рассмот-
рение молекулярной модели комплекса железа(III) с аэробакти-
ном показывает, что свободные карбоксильные и гидроксильные
группы остатка лимонной кислоты, а также две гидроксаматные
группы могут связываться с ионом Fe(III). Однако структура,
изображенная формулой X, нуждается в экспериментальном под-
тверждении.
Предполагают, что лиганд шизокинин (Ха) близок к аэробак-
тину, в котором центральный остаток лимонной кислоты симмет-
рично связан замещенными амидными связями с 2 молями 1-ами-
но-3-ацетилгидроксамидопропана [5а].
234
Глава 5
/с 'HN''CHz'CH^'CH2~' N—I
сн/ in.
НО-Ь-С02Н он
C^NH
II
О
Ъеферри шизокинин (Ха)
С-СНЭ
II
О
о
. II
^,N—с—СН3
сн2 2^сн2
2.3.4. Семейство микобактина
микобактин (XI)
железо (III) микобактин (XII}
Микобактин Ri Ra R3 r4 R= а ь с d е Г
А6 (ХНа) Fa (ФПб) 13Д 17,15,13,11,9Д СН3 Н Н СН3 сн3 сн3 Н Н трео L S () L
Н (ХПв) : 19.17Д СН3 СН3 сн3 Н R L S () L
М (ХПг) N6 (ХПд) Р (ХПе) 1 2 19,17,15 цисД’п Н Н СН3 СН3 сн3 н 18,17,16,15® 18,17, 16,15в с2н6 сн3 сн3 СН3 ( ) L L L R2 S S2 R L
R (ХПж) 19Д Н н с2н5 сн3 ( ) L L R S L
S (ХПз) 19,17,15,13 чысД Н н СН3 Н ( ) L S ( ) L
Т (ХПи) 20,19,18,17 | 20,19,18,17Д J Н н сн3 н ( ) R ( ) L
Общая структура комплексов железя(Ш) с микобактинами. Боковые цепи Ri
являются алкильными группами и имеют указанное число атомов углерода;
там, где известно, показаны двойные связи. Цифры указывают основные типы
Транспорт соединений железа микробами (сидерохромы) 235
боковых цепей; некоторые из них встречаются много раз в каждом микобактине.
Асимметрические центры обозначены буквами а — f. Пробелы показывают, что
конфигурация не определена. Обозначения: ( ) — отсутствие асимметрического
центра; а структура выведена для смеси с ХХв; ® предположительная структура;
в насыщенные алкильные группы, имеющие указанное число атомов углерода;
г относительная конфигурация при d и е — эритро, абсолютная конфигурация не
определена [Snow G. A., Bact. Rev., 34, 100 (1970)].
Микобактинам посвящен недавно появившийся обзор Сноу [4].
В 1911 г. Творт и Инграм показали, что Mycobacterium johnei, тре-
буемый как фактор роста, находится в экстрактах органическими
растворителями из других микобактерий. Тридцатью годами позд-
нее в связи с попытками создать специфический антиметаболит
для Mycobacterium tuberculosis вновь возродился интерес к этому
соединению, впоследствии названному микобактином. Было най-
дено, что, комбинируя надлежащим образом питание железом и
время роста, можно получить максимальные выходы микобакти-
нов, из которых наиболее полно изучен компонент Р (ХПе) из
Mycobacterium phlei. В отличие от других гидроксаматов микобак-
тины и их комплексы с железом растворимы в липидах и прони-
кают в клетки; лучший метод экстракции — вымачивание в эта-
ноле. Есть сообщения о .выходах, составляющих до 2% сухого ве-
са клеток в зависимости от природы организма.
Второй необычной особенностью микобактинов является их
микрогетерогенность, обусловленная наличием гомологов жирных
кислот в боковых цепях. Каждый индивидуальный микобактин
обозначается буквой, за которой следует число эквивалентных
атомов углерода в боковой цепи. Для нескольких микобактинов
были установлены длины боковых цепей, их относительное содер-
жание, присутствие или отсутствие ненасыщенных связей и т. д.,
но до сих пор полностью охарактеризован только компонент Р,
главная частица имеет здесь н-ч«с-октадецен-2-оильную боковую
цепь и обозначается микобактин Р 18-tfwc-A2 (ср. ХПе).
Выделены два основных класса микобактинов, примерами ко-
торых могут служить соединения типа М (ХПг) и Р (ХПе). Ми-
кобактины типа Р содержат в микобактиновой кислотной части
салициловую или 6-метилсалициловую кислоту, серин или треонин,
смесь жирных кислот с длинными цепями (обычно с двойной
связью рядом с карбоксильной группой, как в феррихроме А) и
L-e-N-оксилизин. Кобактиновая часть содержит 0-оксикислоту
(3-оксимасляную или З-окси-2-метилвалериановую) и вторую мо-
лекулу L-e-N-оксилизина. Вариации в ряду типа Р возникают в
результате замещения в бензольном или оксазолиновом кольце
или во фрагменте 0-оксикислоты, а также от присутствия различ-
ных оптических изомеров в последней частице [ср. микобактины S
(ХПз) и Т (ХПи)]. Микобактины типа М имеют только одну ко-
роткую боковую цепь К в XI, т. е. ацетил и пропионил в микобак-
тинах М (ХПг) и N (ХПд) соответственно. Конфигурация окси-
236
Глава 5
кислоты по-прежнему эритро:, однако ,в микобактинах М эта части-
ца имеет длинную алкильную боковую цепь.
Микобактины образуют нейтральные лилидорастворимые ком-
плексы с железом(III) такого типа, как показано для соединения
XII, которые превосходят по стабильности ферриоксамии В. Ос-
новное различие между тригидроксаматами железа(III) и ком-
плексами железа (III) с микобактинами состоит в том, что в по-
следних одна гидроксаматная группа замещается бидентатным
лигандом, содержащим фенольную гидроксильную группу и атом
азота в оксазолиновом кольце. Исследование молекулярных мо-
делей показывает, что гексадентатная структура, приведенная для
соединения XII, возможна, но точное расположение атомов вокруг
железа кристаллографически не установлено. Вероятно, осуще-
ствимы только некоторые из 16 изомерных расположений. Ком-
плексы микобактина Р с алюминием, галлием и хромом были вы-
делены в виде кристаллов; комплекс с алюминием достаточно
летуч для масс-спектрометрического изучения, с помощью которо-
го обнаруживают присутствие гомологов. Было найдено, что ком-
плекс с хромом, который диссоциирует очень медленно, противо-
действует эффекту микобактина, промотирующему рост М. johnei;
следовательно, физиологически важной формой микобактина яв-
ляется его комплекс с железом(III). Несмотря на очевидную
структурную аналогию между микобактином и тригидрокоамат-
ными сидерохромами, ни один из последних не обладает никакой,
даже самой незначительной активностью по отношению к зависи-
мым от микобактина штаммам. Эту специфичность можно объяс-
нить особыми структурными требованиями М. johnei и (или) уни-
кальной растворимостью микобактина и его комплекса с Fe(III).
Еще очень мало известно о биосинтезе микобактинов. Было ус-
тановлено, что все штаммы микобактерий, которые могут размно-
жаться в искусственной среде, продуцируют хелатирующие желе-
зо соединения, родственные лиганду XI. Доказано, что распреде-
ление микобактинов по различным штаммам полезно в таксономи-
ческом отношении.
2.3.5. Семейство фузаринина
Fusarium roseum АТСС 12822 и другие виды Fusarium, культи-
вируемые при низких концентрациях железа (<~10-7 М), про-
дуцируют ряд основных гидроксаматов, дающих .положительную
реакцию с нингидрином и названных фузарининами (XIII) [69J.
НО—
" О NH21 НО О
II I I II
—С—С-(СН2)3—N-C.
фуэарннии (XIII)
/СН2=СН2—О—
^СН3
Транспорт соединений железа микробами (сидерохромы)
237
Фузаринин (ХШа): п = 1
Фузаринин А (ХШб): п = 2
Фузаринин В (ХШв): п = 3
Фузаринин С (деферрифузиген) (ХШг):
п = 3, цикло
'/з Реш
о о
। и
О NH,
г II I . ..
Цикло 4-с - C-(CHz)3-N — С
н
^CHj-CHg-O^-
н
Н сн3
фузиген (XIV)
Накопление фузаринина, мономера (ХШа), достигает максималь-
ной концентрации ~ 1 мМ после 5 суток роста, и с этого момента
он 'быстро исчезает из среды. Всего 25—30 мг компонента А
(ХШб) и еще меньше компонента В (ХШв) можно выделить из
1 л среды. Было показано, что фузаринин С — циклическое соеди-
нение, идентичное деферрифузигену [70]. Все эти соединения со-
держат частицы ь-б-ЬЬоксиорнитина и цыс-5-окси-3-метилпентен-2-
овую кислоту, этерифицированную по типу «голова к хвосту». Та-
ким образом, его ацильная часть идентична ацильной части
ферриродина.
Фузаринины железа при подкислении ведут себя так, как это
и ожидают, т. е. спектр комплекса Fe(III) с ХШв довольно нечув-
ствителен к такой обработке. Фузиген, выделенный из Fusarium
cubense, проявляет примерно такую же активность по антагони-
стическому критерию по отношению к Staphylococcus aurens, как
копроген, т. е. его активность составляет примерно 1 % от актив-
ности ферриоксамина В [70]. Хотя фузаринин не может поддер-
живать рост Arthrobacter JG-9, фузаринины А, В и С проявляют
небольшую активность по отношению к этому организму.
2.3.6. Семейство ферриоксамина
Сидерохромы группы ферриоксамина содержат ацетатную,
сукцинатную и а-амино-о-оксиаминоалкильную части и разделя-
ются на два типа — линейные (XV) и циклические (XVI) [45].Не-
которые производные ферриоксаминов проявляют антибиотиче-
скую активность и обозначаются как ферримицины. Нет никакой
структурной связи между этими антибиотиками и альбомицином,
за исключением того, что в обоих случаях токсическим началом
является выделяющийся позднее сложный агент, действующий как
переносчик железа. Таким образом, если замещенный урацил от-
щепить от альбомицина, это соединение будет действовать как
фактор роста для сидерохромных ауксотрофов. Антагонистический
238
Глава 5
тест (рис. 5.6) был использован сотрудниками Свисса для того,
чтобы выделить длинный ряд ферриоксаминов, которые являются
одновременно и факторами роста и антибиотиками [45].
R
।
H-N CONH CONH
\ / \ / \
<СН2)В (СНг)2 (СНг)в (СНг)г (СНг)п .
\ / \ / \ I
N-C N-C N-C
линейные ферриоксамины (XV)
R п R'
Ферриоксамин В (XVa) Н 5 CHS—
Ферриоксамин Dr (XV6) СН3СО— 5 CHS—
Ферриоксамин G (XVb) н 5 HOjQCHs),—
Ферриоксамин Ar (XVr) н 4 НО2С(СН,)а—
Ферримицин At (ХУд) 5 CHS-
но
(СНг)5 (СНг)г (снг)в
\ / \
N-C N-C
/ \ I
(СН2)г (СНг)г, (СНг)2
циклические ферриоксамины
(XVI)
Ферриоксамин(XVIa): п = 5
Ферриоксамин D2 (XVI6): п — 4
Метаболит «С» — А-производное ферриоксамнна В, в котором концевая
аминогруппа окислена до СООН. Возможно, ферриоксамин В идентичен терре-
Транспорт соединений железа микробами (сидерохромы)
239
генс-фактору. Ферриоксамии А2 содержит один остаток уксусной кислоты, два
остатка малоновой кислоты, один остаток 1-амино-5-оксиа'минопентана и два ос-
татка 1-амино-4-оксиамннобутана. Компоненты С и F—основные феррнокса-
мины.
Ферриоксамины — относительно простые .молекулы, лиганды
которых лишены оптической активности. Ряд их был синтезирован
химическим путем. Константы комплексообразования для них ле-
жат в той же области, что и для феррихрома (табл. 5.1). Фер-
риоксамины были найдены во всех актиномицетах, которые были
исследованы на их присутствие.
2.4. Другие соединения
Некоторые из сидерохромов еще не изучены; наиболее извест-
ный из них, по-видимому, феррибактин [44] из Р. ftuorescens и
некоторые железосодержащие антибиотики. Несколько других
микробных продуктов, возможно, содержат одно или большее чис-
ло гидроксаматных звеньев, но неизвестно, имеют ли они отно-
шение к транспорту и метаболизму железа. Некоторые из них,
такие, как сукциноминин [71], даноминин [72] и ASK [73], яв-
ляются антибиотиками.
Антибиотики семейства аспергилловой кислоты исторически ин-
тересны как первые микробные метаболиты, содержащие связан-
ную гидроксамовую кислоту. Сама по себе аспергилловая кислота
(XVIIa) действует как фактор роста для Arthrobacter JG-9, но об-
разование некоторых членов ряда (XVII6, ХУПд) культурой As-
pergillus sclerotiorum не регулируется железом [68]. Одна молеку-
ла связанной гидроксамовой кислоты в этих соединениях образу-
ется в результате окисления предшествующего циклического
амида, флавакола, т. е. обратным путем, по сравнению с ферри-
хромом.
аспергилловые кислоты (XVII)
Аспергилловая кислота (XVIIa): R = Н, R' = R’ = СН3
Неоаспергилловая кислота (XVII6): R = R" = Н, R' = СН(СН3)2
Мутааспергилловая кислота (XVIIb) : R = ОН, R' = R" = СН3
Оксиаспергилловая кислота (XVIIr): R = ОН, R' = QHj, R* = СН3
Неоокснаспергилловая кислота (XVIIa) : R = OH, R" = СН(СН3)2, R"=H
Мицелианамид (XVIII) [74], дикетопиперазин с двумя гидро-
ксилированными атомами азота в кольце, образуется в немного
240
Глава 5
большем количестве при низких концентрациях железа; другими
словами, ничто не говорит о его связи с метаболизмом железа.
Третий тип соединения, подобного дикетопиперазину, пулчер-
риминовая кислота (XIX) [75], образуется внутри- и внеклеточно
в среде с высоким содержанием железа, в которой она присутст-
вует в виде очень мало растворимого железного комплекса пул-
черримина. Это соединение синтезируется Candida pulcherrlma и
родственными ему дрожжами. Оно не активно по отношению к си-
дерохромозависимым ®идам Arthrobacter и, следовательно, по-ви-
димому, не связано с метаболизмом железа [76]. 1В соединении
XIX кольцо окисляется дальше, чем в мицелианамиде, и хотя ему
можно приписать структуру с одной молекулой связанной гидро-
ксамовой кислоты, сопряженная форма XIX, возможно, является
доминирующей формой частиц. Оно получается биосинтетически
из ь-лейциндикетопиперазина.
пулчерриминовая кислота (XIX)
Противоопухолевый агент П-формил-П-оксимицин, или хадаци-
дин (XX), идентичен асимметрину, ингибитору роста растений,
имеющему грибковое происхождение. В Penicillium aurantio-viola-
сеит F-4070b хадацидин [10] возникает в результате окисления
глицина с последующим формилированием и опять, по-видимому,
железо не подавляет этот синтез [68].
О ОН
II I
н—С—N—СН2—СО2Н
хадацидин (XX)
Транспорт соединений железа микробами (сидерохромы)241
Актинонин (XXI), первичная гидроксамовая кислота (Н вме-
сто R при атоме азота), завершает 'Перечень микробных продук-
тов, содержащих ацилированную гидроксиламиногруппу. Было
найдено, что О-бензилактинонин токсичен, так что значение гид-
роксамового звена для проявления антибиотической активности
находится под сомнением [77].
актинонин (XXI) '
Циклическая гидроксамовая кислота ХХП была выделена из
высших растений [58], но ее биологическая роль неизвестна. Од-
нако по биопробе с Arthrobacter JG-9 была обнаружена сидеро-
хромная активность в водных экстрактах из листьев томата, ла-
тука, цветной капусты, лука-порея и капусты и в экстрактах кор-
ней моркови [59]. Ферриоксамин В переходит в листья томата
при выращивании в питательном растворе [78].
I
ОН
2,4-д«окс«-7-метокси-1.4-бензоксазинон-3 (ХХП) (DIMBOA)
И наконец, два реагента, связывающих ионы Fe(II) и проду-
цируемые микроорганизмами, заслуживают внимания, хотя сейчас
эти соединения и нельзя связать с метаболизмом железа. Ферро-
вердин (XXIII), зеленый пигмент из мицелия Streptomyces sp.,
охарактеризован кристаллографически (ХШа) [79]. Каждый
атом железа связан с фенолятным кислородом и азотом нитрозо-
группы в питрозофенольном лиганде. Pseudomonas GH и Р. roseus
fluorescens образуют пиримин (XXIV [80, 81], представляющий
собой пиридиновое кольцо, замещенное в положении 2 у-кар-
боксильной ь-глутаминовой кислотой. При всех разумных значе-
ниях pH пиримин существует в иминной форме и в такой форме
16—2451
242
Глава 5
образует стабильный, дающий окраску с фуксином комплекс с
Fe(II) (XXV).
O-N
Ъеферроферровердин (XXIII)
ферровердип, кристаллическая
структура (XXUla) [79J
пироман (XXIV)
ферропиримин (XXV)
3. ЗАКЛЮЧЕНИЕ
Практически все аэробные и «случайно» аэробные микро-
организмы. которые были исследованы, будь-то бактерии, дрожжи
или грибы, имеют транспортную систему для железа или нужда-
ются в ней. Путь железа в микробном метаболизме в общих чер-
тах был описан на примере исследования мутантов Salmonella
typhimurium и других кишечных бактерий. Эта система содержит,
во-первых, специфический низкомолекулярный носитель для иона
Fe(III) (сидерохром) фенолятного типа (энтеробактин) или гид-
роксаматного типа (феррихром) и, во-вторых, связанный с клет-
кой активный транспортный механизм для введения специфиче-
ского сидерохрома.
Несмотря на то что механизм введения сидерохромов в клетку
детально не изучен, составлен каталог транспортных соединений,
который тотчас же обнаружил некоторые определенные общие
принципы. Биосинтез транспортных соединений сильно подавляет-
ся железом. Эти транспортные соединения обладают высокой спе-
цифичностью связывания Fe (111) /Fe (11) и встречаются и действу-
ют либо как одиночные частицы, либо .как димеры или тримеры,
связанные амидными или сложноэфирными связями, при этом
Транспорт соединений железа микробами (сидерохромы)243
они претерпевают выраженные конформационные изменения при
комплексообразовании с Fe(III).
Таким образом, мы приходим к выводу о том, что сидерохро-
мы выполняют уникальную и необходимую функцию в регуляции
питания железом микробных клеток.
СПИСОК ЛИТЕРАТУРЫ
1. Keller-Schierlein W., Prelog V., Zahner И., Fortsch. Chem. Org. Natur., 22, 279
(1964).
2. Pollack J. R., Neilands J. C., Biochem. Biophys. Res. Commun., 38, 989
(1970).
3. Neilands J. B., Structure and Bonding, 1, 59 (1966).
4. Snow G. A., Bact. Rev., 34, 100 (1970).
5. Gibson F., Magrath D. I., Biochim. Biophys. Acta, 192, 175 (1969).
5a. Mullis К. B., Pollack J. R., Neilands I. B., Biochemistry, 10, 4894 (1971).
6. Garibaldi J. A., Neilands J. B., Nature, 177, 526 (1956).
7. Atkin C. L., Neilands J. B., Phaff H„ J. Bact., 103, 722 (1970).
7a. Garibaldi J. A., J. Bact., 105, 1036 (1971).
8. Gibson F., Pittard J., Bact. Rev., 32, 465 (1968).
9. Entergy T., Biochemistry, 5, 3694 (1966).
10. Emery T., in D. Gottlieb and P. D. Shaw, Antibiotics, Vol. 2, Springer, Berlin,
1967, p. 439.
11. MacDonald I. C., ibid., pp. 43—51.
12. Garibaldi J. A., personal communication.
13. Neilands J. B., Science, 156, 1443 (1967).
14. Zalkin A., Forrester J. D„ Templeton D. H., J. Am. Chem. Soc., 88, 1810
(1966).
15. Keller-Schierlein W., Maurer B., Helv. Chim. Acta, 52, 603 (1969).
16. Llinas Al., Klein M. P., Neilands J. B., J. Mol. Biol., 52, 399 (1970).
17. Wickman H. H., Klein M. P., Shirley D. A., J. Chem. Phys., 42, 2113 (1965).
18. Neilands J. B„ Bact. Rev., 21, 101 (1957).
19. Ito T., Neilands J. B., J. Am. Chem. Soc., 80, 4645 (1958).
20. Morrison N. E., Antoine A. D., Dewbrey E. E„ J. Bact., 89, 1630 (1965).
21. Emery T., Abstr. 158th Am. Chem. Soc. Meeting, New York, 1969, Sept. 8—12.
22. Byers B. R., Powell M. V., Lankford С. E., J. Bact., 93, 286 (1967).
23. Arceneaux I. L., Lankford С. E., Biochem. Biophys. Res. Commun., 24, 370
(1966).
24. Kniisel F., Schiess B., Zimmerman W., Arch. Mikrobiol., 68, 99 (1969).
25. Zimmerman W„ Knusel F., Arch. Mikrobiol., 68, 107 (1969).
26. Peters W. J., Warren R. A. J., Biochim. Biophys. Acta, 165, 225 (1968).
27. Wang С. C„ Newton A., J. Bact., 98, 1135, 1142 (1969).
28. Pollack I. R.. Ames B. N„ Neilands J. B„ J. Bact., 104, 635 (1970).
29. Brot N., Goodwin J., Fates H., Biochem. Biophys. Res. Commun., 25, 454
(1966).
30. Corbin I. L., Bulen W. A., Biochemistry, 8, 757 (1969).
31. Pollack J. R„ Ames B. N„ Neilands J. B., Abstr. Fed. Proc., 29, 3132 (1970).
32. Atkin C. L., Neilands I. B., Biochemistry, 7, 3734 (1968).
33. Anderegg G., L’Eplattenier F., Schwarzenbach G., Helv. Chim. Acta, 46, 1409
(1963).
34. Ehrenberg A.. Nature, 178, 379 (1956).
35. Lovenberg W., Buchanan В. B., Rabionwitz I. C., J. Biol. Chem., 238, 3899
(1963).
36. Emery T., Neilands J. B„ J. Am. Chem. Soc., 82, 3658 (1960).
16*
244
Глава 5
37. Neilands J. B„ in J. E. Falk, R. Lemberg, R. K. Morton (eds.), Haematin En-
zymes, Vol. 1, Pergamon Press, Oxford and New York, 1961, p. 194.
38. Shemyakin M. M„ Ovchinnikov Y. A., Ivanov V. T., Antonov V. K- et al.,
J. Membrane Biol., 1, 402 (1969).
39. Neilands J. B„ J. Am. Chem. Soc., 74, 4846 (1952).
40. Komai H., Neilands I. B„ Science, 153, 751 (1966).
41. Komai H., Doctoral Dissertation, University of California, Berkeley, 1968.
42. Jones О. T. G., personal communication.
43. Gillard R. D„ Mitchell P. R., Structure and Bonding, 7, 46 (1970).
44. Maurer B., Muller A., Keller-Schierlein IF., Zahner H., Arch. Mikrobiol., 60, 326
(1968).
45. Prelog V., Pure Appl. Chem., 6, 327 (1963).
46. O’Brien L. G., Cox G. B., Gibson F., Biochim. Biophys. Acta, 177, 321 (1969).
47. Niiesch J., Knilsel F., in D. Gottlieb, P. D. Shaw (eds.), Antibiotics, Vol. 1,
Springer, Berlin, 1967, p. 499.
48. Spiro T. G., Saltman P., Structure and Bonding, 6, 116 (1969).
49. Bergman K-, Burke P. V., Cerda-Olmedo E„ David C. N. et al., Bact. Rev., 33,
99 (1969).
50. Wilkins T. D., Lankford С. E., J. Infect. Dis., 121, 129 (1970).
51. Schneider H. A., Science, 158, 597 (1967).
52. Wawszkiewicz E. J., Schneider H. A., Abstr. Fed. Proc., 29, 3256 (1970).
53. Hieb E. F., Stokstad E. L. R., Rothstein M., Science, 168, 143 (1970).
54. Larkin E. C., Weintraub L. R., Crosby W. H., Am. J. Physiol., 218, 7 (1970).
55. Hedenberg L., Scand. J. Haemat. Suppl, 6, 86 pp. (1969); see also Clin. Phar-
macol. Then, 10 (4), 595 (1969).
56. Krakoff I. H., Brown N. C., Reichard P., Cancer Res., 28, 1559 (1968).
57. Zahner H„ Bachmann E., Hutter R„ Niiesch I., Path. Mikrobiol., 25, 708
(1962).
58. Tipton C. L., Klun J. A., Husted R. R„ Pierson M. D., Biochemistry, 6, 2866
(1967) (see also ref. [3]).
59. Page E. R„ Biochem. J., 100, 34P (1966).
60. Price C. A., A. Rev. Plant Physiol., 19, 239 (1968).
61. Pugh К. B., Waid J. S., Soil Biol. Chem. Biochem., 1, 195, 207 (1969).
62. Arnow L. E., J. Biol. Chem., 118, 531 (1937).
62a. O’Brien I. G„ Gibson F., Biochim. Biophys. Acta, 215, 393 (1970).
626. Wawszkiewicz E. J., Schneider H. A., Starcher B., Pollack I. R„ Neilands J. B.,
Proc. Natn. Acad. Sci. U. S„ 68, 2870 (1971).
62b. Guterman Sonia K-, Biophys. Res. Commun., 44, 1149 (1971).
63. Brot N„ Goodwin L, J. Biol. Chem., 243, 510 (1968).
64. Peters W. J., Warren R. A. J., J. Bact., 95, 360 (1968).
65. Downer D. N., Davis W. B„ Byers B. R., J. Bact., 101, 181 (1970).
66. Tadenuma N., Sato S., Agric. Biol. Chem., 31, 1482 (1967).
67. Epstein L. M., Straub D. R., Inorg. Chem., 8, 453 (1969).
68. Atkin C. L., Doctoral Dissertation, University of California, Berkeley,
1970.
68a. Diekmann H., Arch. Mikrobiol., 73, 65 (1970).
686. Akers H., Llinas M., Neilands J. B., Abstr., Am. Soc. Biol. Chem. Meeting, San
Francisco, 1971.
68b. Keller-Schierlein W., Diekmann H., Helv. Chim. Acta, 53, 2035 (1970).
69. Sayer J. M., Emery T. F., Biochemistry, 7, 184 (1968).
70. Diekmann H„ Zahner H., Eur. J. Biochem., 3, 213 (1967).
71. Haskell T. H., Bunge R. H., French J. C., Bartz Q. R„ J. Antibiot., 16, 67
(1963).
72. Tsukiura H., Okanishi M., Ohmori T., Koshiyama H., Miyaki T., Kitazima H..
Kawaguchi H., J. Antibiot., 17, 39 (1964).
73. Shimi L R., Imam G. M„ Haroun B. M„ J. Antibiot., 22, 106 (1969).
74. Bates R. B., Schauble J. H., Soucek M., Tetrahedron Lett., 1963, 1683.
Транспорт соединений железа микробами (сидерохромы)
245
75. MacDonald J. С., Biochem. J., 96, 533 (1965).
76. Burton М. О., Sowden F. I., Lochhead A. G., Can. J. Biochem. Physiol., 32, 400
(1954).
77. Attwood М. М., J. Gen. Microbiol., 55, 209 (1969).
78. Stutz E„ Experientia, 20, 430 (1964).
79. Candeloro S., Grdenic D., Taylor N„ Thompson B., Viswamitra M., Crowfoot-
Hodgkin D., Nature, 224, 589 (1969).
80. Shiman R., Neilands J. B., Biochemistry, 4, 2233 (1965).
81. Pouteau-Thouvenot M„ Choussy M., Gaudemer A., Barbier M„ Bull. Soc. Chim.
Biol., 52, 51 (1970).
82. Hesseltine C. W. et al., Mycologia, 45, 7 (1953).
83. Burton M. O., Lochhead A. G„ Can. J. Bot., 31, 145 (1953); Morrison N. E.„
Dewbrey E. E., J. Bact., 92, 1848 (1966).
84. Burnham B. F., Neilands I. B., J. Biol. Chem., 236, 554 (1961).
85. Hendlin D., Demain A. L., Nature, 184, 1894 (1959).
ГЛАВА 6
ИОНОФОРЫ— ХЕЛАТИРУЮЩИЕ АГЕНТЫ
ДЛЯ ЩЕЛОЧНЫХ МЕТАЛЛОВ
В. К. Прессман
Pressman В. С., Department of Pharmacology, University
of Miami School of Medicine, Miami, Florida, USA
Ионофоры (т. e. переносчики ионов) — это соединения с такой
молекулярной структурой, которая обусловливает их способность
переносить небольшие ионы через липидные барьеры. Они вызы-
вают 'большой интерес из-за своей способности изменять прони-
цаемость для катионов как природных, так и искусственных мем-
бран. С одной стороны, они предоставляют возможность изменять
проницаемость и градиенты биологических мембран, нарушая тем
самым связанные с ними метаболические процессы. С другой сто-
роны, ионофоры — это аналоги компонентов природных мембран,
и с их помощью можно придавать искусственной мембране селек-
тивную проницаемость и электрические свойства типичных био-
логических мембран.
Известные ионофоры устойчивы как химически, так и метабо-
лически. Они либо имеют готовую макроциклическую структуру,
либо способны образовывать макроциклические кольца при помо-
щи водородных связей. Их молекулярные массы лежат в .преде-
лах от 200 до 2000. Они содержат большое число атомов кислоро-
да, регулярно распределенных в молекулярном остове. Замеча-
тельным свойством ионофоров является способность образовывать
липидорастворимые катионные комплексы, играющие важную
роль в транспорте ионов.
Ионофоры 'можно разделить на две группы. К первой относят-
ся нейтральные ионофоры, не содержащие способных к диссоциа-
ции протонов; образуемые ими катионные комплексы имеют об-
щий положительный заряд. Остов ионофоров второй группы по-
строен из цепи атомов углерода, соединенных ковалентными свя-
зями. Они содержат одну карбоксильную группу, которая посред-
ством водородной связи по типу «голова к хвосту» включена в
кольцевую структуру. Как будет показано ниже, наличие или от-
сутствие этой единственной карбоксильной группы оказывает
большое влияние на реакции, катализируемые ионофорами*.
* По-видимому, термин «катализ» здесь употреблен автором недостаточно
строго. — Прим. ред.
Ионофоры — хелатирующие агенты для щелочных металлов
247
1. ИСТОРИЧЕСКАЯ СПРАВКА
Валиномицин — первое соединение, признанное ионофором; он
был выделен [2] и охарактеризован [3] Брокманном. Впоследст-
вии его синтезировал и идентифицировал как циклический доде-
кадепсипептид Шемякин с сотр. [4], который изучил анти-
биотические характеристики валиномицина и нескольких его
синтетических аналогов [5, 6]. МакМюррей и Бегг [7] обнаружи-
ли, что валиномицин является мощным агентом, нарушающим
окислительное фосфорилирование в митохондриях; как установи-
ли Мор и Прессман [8], это свойство сильно зависит от присутст-
вия ионов К+ но не Na+. С .помощью ион-селективных электродов
было показано, что гвалиномицин в первую очередь влияет на
транспорт ионов К+ через митохондрии и его воздействие на про-
цесс фосфорилирования обусловлено тем, что он вызывает рас-
сеивание энергии за счет циклического перемещения ионов К+
[8, 9]. Детально описана серия наблюдений, с помощью которых
было установлено, что валиномицин обладает свойствами ионофо-
ра [10]; кроме того, опубликованы более общие обзоры по транс-
портным свойствам других ионофоров [11—13].
Первоначально на основании зависимости между структурой
и активностью неправильно полагали, что валиномицин взаимо-
действует с уже существующими высокоселективными транспорт-
ными рецепторами в митохондриях [8]. Однако опытным путем
было обнаружено, что валиномициновая группа ионофоров уве-
личивает катионную проницаемость искусственных липидных би-
молекулярных слоев; на основании этого пришли к заключению,
что транспортная активность и ион-селективность внутренне при-
сущи самим молекулам валиномицина [14, 15]. В результате де-
тального исследования установили, что валиномицин обладает
комплексообразующими свойствами по отношению к ионам щелоч-
ных металлов и транспортной активностью в обычном состоянии,
из чего в конечном счете заключили, что способность ионофоров
служить мобильными носителями ионов обусловлена их собствен-
ными свойствами [1, 16—19].
Способность нигерицина придавать растворимость в липидах
комплексам с солями щелочных металлов впервые была обнару-
жена Харнедом с сотр. [20]. Было найдено, что это соединение
вместе с родственным ему дианмицином ингибирует дыхание и
фосфорилирование в интактных, но не в поврежденных митохонд-
риях [21]. На основании способности нигерицина обращать инду-
цируемое валиномицином поглощение К+ митохондриями [22, 23]
Ларди с сотр. [16] заключили, что ионофоры нигерициновой груп-
пы блокируют механизм ионного транспорта в митохондриях. Од-
нако, исходя из его транспортных свойств в обычных системах
вода—масло и эритроцитах и его способности образовывать ком-
248
Глава 6
плексы с ионами щелочных металлов, Прессман и сотрудники при-
шли к выводу, что нигерицин и другие родственные ему карбок-
сильные ионофоры, так же как нейтральные ионофоры, действу-
ют как мобильные носители ионов. Различие в поведении
ионофоров разных групп объясняется наличием или отсутствием
остаточного заряда на катионном комплексе [1, 18].
Были сделаны попытки объяснить селективность комплексооб-
разования ионофоров типа валиномииина соответствием между
формой молекулы — изображением их известной формальной
структуры в виде плоского кольца — и радиусом входящего ка-
тиона [14, 24]. Для того чтобы объяснить одинаковые комплексо-
образующие свойства 'макроциклической молекулы валиномици-
на (36 атомов) и меньшей по размеру циклической молекулы эн-
ниатина (18 атомов), предположили, что связываемый ион может
приспосабливаться к кольцу ионофора, десольватируясь в различ-
ной степени [14]. Методом рентгеноструктурного анализа крис-
таллов [25, 26] была установлена структура К+-комплекса нонак-
тина (32 атома в кольце) и показано, что его кольцо неплоское,
а образует заметно изогнутую макроциклическую полость вокруг
несольватиров,а1нного катиона. Главная особенность образующих
комплексы молекул ионофоров — обволакивание ими несольвати-
рованного катиона >в комплексе — распространяется на все изве-
стные ионофоры.
2. ОСНОВНЫЕ СТРУКТУРНЫЕ ОСОБЕННОСТИ
ИОНОФОРОВ
Ионофоры — это лиганды, способные образовывать ион-диполь-
ные комплексы с катионами посредством соответствующим обра-
зом повернутых атомов кислорода, равномерно встроенных в их
циклический молекулярный остов. Нейтральные ионофоры — ти-
пичные циклические ковалентные соединения, кольца которых
включают от 18 до 40 атомов. Кольца обычно содержат повторяю-
щиеся субъединицы с регулярным чередованием центров оптиче-
ской асимметрии. Валиномицин и энниатин — депсипептиды, коль-
ца которых .содержат чередующиеся амидные и сложноэфирные
группы. Повторяющиеся единицы ‘макротетралидных актинов [28]
соединены .между .собой исключительно сложноэфирными связя-
ми. Несколько соединений, .подобных по активности ионофорам,
содержат только пептидные связи, хотя, например, грамицидин не
является ковалентным циклическим соединением [29] и не обра-
зует высоколипофильных катионных комплексов. Синтетические
полиэфиры, «короны»* [30], еще более инертны и содержат в мо-
лекулярном остове только эфирные атомы кислорода.
* Некоторые авторы не переводят английское слово «crown» на русский язык
и называют соответствующие соединения «краун-эфирами». — Прим, ред.
Ионофоры — хелатирующие агенты для щелочных металлов 24&
Известные карбоксилсодержащие ионофоры представляют со-
бой формально линейные цепи, содержащие гетероциклические
кольца с карбоксильными группами с одного конца молекулы и
одной или двумя гидроксильными группами с другого конца.
В комплексах карбоксил депротонируется и образует кольцо по
типу «голова к хвосту» при помощи водородных связей с проти-
воположной гидроксильной группой. Из-за необходимости депро-
тонпрования комплексообразование с карбоксильными ионофора-
ми зависит от pH, причем более высокие значения pH благопри-
ятствуют процессу [1]. Такие комплексы являются электроней-
тральными цвиттер-ионами в противоположность «нейтральным»
ионофорным комплексам, которые в действительности образуют
не чувствительные к pH заряженные комплексы.
3. СПЕЦИФИЧЕСКИЕ ГРУППЫ ИОНОФОРОВ
3.1. Макротетралидные актины
Четыре гомологических макротетралидных актина, содержа-
щих 18-членные кольца (рис. 6.1,Л), продуцируются соответству-
ющими штаммами актиномицетов. Самый низший гомолог — но-
нактин— образован симметричным чередованием четырех d- и
l -энантиомеров нонактиновой кислоты (других диастереомеров не
было обнаружено), представляющей собой оксикарбоновую кисло-
ту, содержащую эфирный кислород в тетрагидрофурановом коль-
це примерно в середине ее цепи. В более высоких гомологах — мо-
нактине, динактине и тринактине — остатки нонактиновой кис-
лоты соответственно замещаются одним, двумя или тремя
остатками монометилированной нонактиновой кислоты — гомоно-
нактиновой кислотой [28]. Хотя при беспорядочной последова-
тельности нонактиновых и гомононактиновых остатков возможны
различные рацемические смеси, природные более высокие гомо-
логи нонактивна все оптически активны. Известны еще более вы-
сокие гомологи актинового ряда, содержащие диметилированную
нонактиновую кислоту (бпс-гомононактиновую кислоту); однако
имеется пока мало данных о конформациях и ионной селективно-
сти этих соединений [31].
Представление о сложной трехмерной структуре нонактина
было получено рентгеноструктурным анализом тиоционатной соли
К+-комплекса. В комплексе существуют ион-дипольные взаимо-
действия между катионом и четырьмя эфирными и четырьмя кар-
боксильными атомами кислорода, расположенными по вершинам
куба: расстояние от негидратированного иона К+ до кислорода
(от центра до центра) составляет 2,7—2,9 А. Положение тиоцио-
натных групп рентгенографически определяется не четко. Пола-
250
Г лава 6
Рис. 6.1. Структуры ионофоров и атомы в лигаидах, участвующие в образовании
связи с катионом.
~ макролидные актины; Б — энниатин В; В — валиномицин; Г — циклогексиловый эфир;
Д — моненснн; Е — нигернцин; ЛС —Х-537А (без штриха); 3 —Х-537А (со штрихом).
Ионофоры — хелатирующие агенты для щелочных металлов
251
гают, что они «ногда довольно свободно ориентированы внутри
кристаллической решетки .вокруг .сферического катионного ком-
плекса [26].
Строгое определение конформации этого комплекса, впервые
полученное для ионофора, позволило в значительной степени вы-
яснить молекулярное поведение этих комплексов. Для поддержа-
ния молекулярного остова в конформации, требуемой для ком-
плексообразования, необходимо точное пространственное распо-
ложение компонентов остатков. Эта конформация была образно
описана «...как напоминающая бороздку на теннисном мяче»
[25]. Тот факт, что катион в комплексе не сольватирован, свиде-
тельствует, что при образовании комплекса в полярной среде зна-
чительная доля свободной энергии затрачивается на десольвата-
цию катиона. Для того чтобы такой комплекс образовывался в
присутствии воды, т. е. в условиях, которые имеют место на по-
верхности биологической мембраны, эта свободная энергия долж-
на быть компенсирована ион-дипольным взаимодействием между
катионом в комплексе и атомами кислорода лиганда. Высокая сте-
пень ион-селективности объясняется различием свободных энергий
дегидратации и комплексообразования, которое в свою очередь
зависит от гибкости остова ионофора. Полное экранирование ка-
тиона (катион в «клетке») показывает, что для того, чтобы катион
мог войти внутрь полости, ионофор до комплексообразования дол-
жен чаще бывать в несколько иной, «открытой» конформации.
Кристаллографическое исследование показывает, что свободный
нонактин образует элементарные ячейки различного типа и его
конформация отличается от конформации в К+-комплексе [26];
однако детальные данные о строении свободного ионофора отсут-
ствуют.
Приведенные выше заключения основываются на допущении,
что в растворе ионофор сохраняет конформацию, подоб-
ную наблюдаемой в кристаллическом состоянии. Это допу-
щение можно проверить ЯМР-исследованием, с помощью ко-
торого обнаружены различные конформации комплексов К+,
Na+ и Cs+ и свободного ионофора [32, 33]. При описании валино-
мицина будет дан другой пример применения метода ЯМР для
изучения ионофоров. Ионная селективность макролидных актинов
(смешанные образцы из нонактина и .монактина), впервые обна-
руженная по зависимости стимуляции дыхания в митохондриях
от природы иона, изменяется в ряду K+>Rb+>Cs+>Na+>Li+
[9]. Такой же ряд селективности был получен на основании
АТФ-азной индукции, которая также зависит от связанного с
транспортом рассеяния энергии митохондриями [34, 35]. У более
высоких членов гомологического ряда ионофоров способность
индуцировать К+-зависимую АТФ-азу увеличивается, но отноше-
ние селективностей К+ : Na+ уменьшается. У более низких гомо-
252
Г лава 6
логов по электрометрическим изменениям .в объемной фазе полу-
чена такая же последовательность [36]; однако сообщалось [14],
что в липидных двуслойных мембранах наблюдается обратная
селективность по отношению к iK+ и Rb+ (т. е. Rb+>-iK+).
Из констант устойчивости в метаноле, полученных осмометри-
чески [12, 36], и в водном ацетоне, полученных методом ЯМР
[33], можно заключить, что К+ заметно предпочтительнее Na+;
однако в менее полярном сухом ацетоне константы устойчивости
комплексов с К+. Na+ и Cs+ все одного порядка (табл. 6.1). Та-
ким образом, в сухом ацетоне, в котором различие в свободных
энергиях десольватации ионов заметно не сказывается на равно-
весии комплексообразования, внутреннее сродство нонактина к
Na+, К+ и Cs+ примерно одинаково, что свидетельствует о зна-
чительной гибкости скелета ионофора, 'позволяющей ему приспо-
сабливаться к ионам с различными радиусами. В более полярных
растворителях получают такие же ряды селективности, как и в
многофазных системах, что подтверждает влияние сольватации
на ионную селективность. Для десольватации меньшего по разме-
ру иона Na+ требуется больше энергии, чем для десольватации
иона К+, в то же время еще больший по размеру Cs+, хотя и
требует для десольватации меньше энергии, чем К+, но при этом
заставляет полость ионофора сильнее открыться, делая тем самым
и катион и ионофор более доступными для сольватации. Система-
Таблица 6.1
Влияние растворителя на селективность макролидных актинов
Ионофор Растворитель Анион Относительная селективность Лите- ратура®
Na+ к+ Cs+
Нонактин Ацетон сю? 1 1 0,20 33
Ацетон — Н2О сю? 0,012 1 0,024 33
СН.ОН SCN- 0,025 1 — 44
С4Н9ОН — толуол SCN- 0,004 1 0,77 51
СН2С12 Пикрат 0,017 1 0,061 76
Монактин CHSOH SCN- 0,004 1 — 44
С4Н,ОН —толуол SCN- 0,010 1 0,42 51
СН2С12 Прикрат 0,009 1 0,029 76
Динактин C4HjOH — толуол SCN- 0,022 1 0,21 51
СН2С12 Прикрат 0,013 1 0,023 76
Тринактин С4Н2ОН — толуол SCN- 0,042 1 0,087 51
СН2С12 Прикрат 0,011 1 0,019 76
Методы, используемые при определении констант сродства:
метрия; 151] и [761 —- исследование распределения между двумя
133] — ЯМР; [44] — осмо-
фазами.
Ионофоры — хелатирующие агенты для щелочных металлов 253
тическое определение способности к комплексообразованию в рас-
творителях с умеренной полярностью показало уменьшение селек-
тивности К+ : Na+ у более .высоких гомологов макротетралидных
актинов (табл. 6.1) (ор. митохондриальный критерий в предыду-
щем разделе).
3.2. Энниатин
Энниатины относятся к группе циклических гексадепсипепти-
дов, содержащих кольца из 18 атомов; их получают из некоторых
штаммов Fusarium [37, 38]. Входящая в их состав D-оксиизовале-
риановая кислота чередуется с N-метил-ь-аминокислотой. В со-
став энниатинов А, .В и С входят аминокислоты изолейцин, .валин
и лейцин соответственно. Структура энниатина В показана на
рис. 6.1, Б. Еще одна разновидность энниатина — боварицин, в со-
став которого .входит аминокислота N-метилфенилаланин [39].
Так как энниатин содержит кольца небольшого размера, то
для его комплексов возможно лишь ограниченное число конфор-
маций. ’Пространственные модели показывают, что, когда карбо-
нильные группы направлены внутрь, наблюдается хорошее соот-
ветствие между ионом К+ и полостью, и такая структура для
комплекса с KI подтверждена рентгеноструктурным анализом
[40]. Шесть образующих связи атомов кислорода находятся на
расстоянии 2,6—2,8 А от К+ и подразделяются на две триады, од-
на из которых на 1,5 А выше, а вторая соответственно ниже сред-
ней плоскости кольца. «Верх и низ» этого дискообразного ком-
плекса более четко выражены, чем в комплексах макротетралид-
ных актинов или валиномицина, но не настолько, чтобы был
возможен тесный контакт между катионом и его противоионом.
Можно полагать, что анионы не дискретно расположены <в крис-
таллической решетке.
Конформация энниатинов в растворе изучена более детально
методами дисперсии оптического вращения, кругового дихроизма
и ядерного магнитного резонанса [41, 42]. Энниатин В постепенно
раскрывается, и его карбонильные группы выворачиваются нару-
жу по мере увеличения размеров катионов, входящих в ком-
плекс. В этом отношении он обладает меньшей селективностью,
чем более жесткая молекула валиномицина, хотя порядок селек-
тивности K+>Rb+>Cs+>Na+>Li+, определенный разными ме-
тодами, остается таким же, как и для других типичных нейтраль-
ных ионофоров [43, 44]*.
Одним из интересных представителей энниатинового ряда яв-
ляется синтетический энантиоморф энниатина В. Это соединение
* Здесь речь идет, по-видимому, только о природных ионофорах. — Прим,
ред.
254
Глава 6
обладает такой же активностью по отношению к бактериям и ми-
тохондриям, как природное соединение [43, 45]. Это свидетель-
ствует о том, что при взаимодействии этого ионофора с мембран-
ной системой селективный рецептор может не включаться. При-
родные рецепторы главным образом должны содержать оптически
активные белковые компоненты, которые, как можно ожидать, от-
личают компетентный триггер от его энантиоморфа.
3.3. Валиномицин
Брокманн [3] правильно определил, что валиномицин постро-
ен из остатков d- и ь-валина, ь-лактата и D-изовалерата; однако
он считал, что эта последовательность повторяется только дваж-
ды. Впоследствии Шемякин и сотрудники показали, что эта после-
довательность повторяется три раза с образованием додекадепси-
пептида [4] (рис. 6.1, В) и подтвердили это синтезом [5]. Они
синтезировали обширный ряд аналогов валиномицина и показали,
что даже небольшие изменения молекулы приводят к потере био-
логической активности, измеренной по интенсивности процессов,
происходящих в митохондриях, либо по их антибиотической ак-
тивности [5, 6, 9]. И увеличение, и уменьшение повторяющейся
последовательности из четырех остатков приводит к потере ак-
тивности, так же как и инверсия любого оптически активного
центра. Для того чтобы активность сохранилась, можно либо
только один оптически неактивный глицин заменить на валин, ли-
бо произвести инверсию всех оптически 'активных центров. Рас-
крытие цикла приводит к потере активности, если даже концевые
группы защищены формильной (свободный аминный конец) или
этаноламидной (свободный карбоксильный конец) группами.
Исследование химических сдвигов в спектре ЯМР показало,
что каждой катионной комплексной частице присуща своя уни-
кальная конформация валиномицина, причем наибольшие изме-
нения в спектре ЯМР наблюдаются в области поглощения коль-
цевого остова [42, 46—50]. На основании соотношения между
константами спин-спинового взаимодействия протонов амидных
групп с а-протонами валина и двугранным углом при амидной
связи получено представление о трехмерной конформации связан-
ного в комплекс валиномицина. Методом ИК-спектроскопии по-
казано, что карбонильные атомы кислорода сложноэфирных групп
связываются водородными связями с амидными протонами, пред-
ставляя свободным карбонильным кислородам амидных групп
возможность образовывать координационные связи с катионом.
Водородные связи создают упорядоченную вторичную структуру,
в которой валиномициновый остов делает петлю и образует «брас-
лет» высотой 4 А и диаметром 8 А [48]. Алкильные боковые цепи
направлены от граней «браслета», которые становятся асиммет-
Ионофоры — хелатирующие агенты для щелочных металлов 255
ричными из-за того, что все остатки .молочной кислоты группиру-
ются с одного конца «браслета». В комплексной структуре амид-
ные карбонилы обращены внутрь «браслета», по три на каждой
грани, дополняя ячейку вокруг находящегося в ней катиона. В не-
закомплексованной форме карбонилы обращены наружу от грани
противоположно остаткам молочной кислоты, открывая таким об-
разом катиону путь внутрь ячейки. Часть водородных связей сво-
бодного валиномицина может разрываться, что приводит к по-
явлению некоторого количества молекул с еще более открытой
структурой [42, 45]. Это заключение было подтверждено после-
дующим ЯМ.Р-исследованием на спектрометре с еще более высо-
ким разрешением (220 МГц); зависящие от температуры химиче-
ские сдвиги амидных протонов в слабое поле и их медленный
обмен с D2O подтверждают образование водородных связей [49,
50]. Результаты дальнейшего исследования методом ЯМР валино-
мицина, а также макролидных актинов энниатина В и дицикло-
гексил-18-короны-6 приведены в диссертации Ханнеса [51].
По существу, такую же структуру ячейки получили независимо
рентгеноструктурнЫ|М анализом комплекса валиномицина с хло-
рауратом калия (KAuCh) [52]. Это снова подтвердило справедли-
вость положения о сохранении в растворе конформации, обнару-
женной в твердом состоянии. Положение аниона внутри решетки
четко установлено; следовательно, анион строго ориентирован от-
носительно комплекса ионофора (ср. нонактин). Основные
участвующие в связывании карбонильные атомы кислорода нахо-
дятся на расстоянии 2,7—2,8 А от катиона (от центра до центра),
в то время как карбонильные атомы кислорода, связанные водо-
родными связями, находятся иногда значительно дальше от ка-
тиона.
Метод ядерного магнитного резонанса дает также информацию
о динамике комплексообразования. Дублетный сигнал метильных
групп молочной кислоты валиномицина особенно отчетлив и иде-
ально подходит для измерения химических сдвигов и анализа
форм линий в процессе титрования катиона, на основании чего
можно получить данные о скорости обмена катион — ионофор.
В растворителе с низкой диэлектрической проницаемостью, CDC13,
скорость обмена лишь едва измерима; это указывает, по-видимо-
му, на отсутствие диссоциации комплекса внутри липидной мем-
браны, которая характеризуется низкой диэлектрической прони-
цаемостью. Повышение диэлектрической проницаемости посредст-
вом постепенного добавления CD3OD приводит к измеримому уве-
личению скорости обмена, что согласуется с легкой диссоциацией
комплекса на внешней стороне мембраны в областях с высокой
диэлектрической проницаемостью [46]. Так как внутримолеку-
лярные водородные связи придают жесткость ячейке валиноми-
цина, специально приспособленной для К+, ее трудно сжать до
256 Глава 6
размера Na+. Поэтому К+: ^^-избирательность валиномицина по
всем критериям является самой .большой среди всех известных
ионофоров [8— 15, 41 —44].
3.4. Грамицидин
Грамицидины — это линейные пентадекапептиды, продуцируе-
мые штаммом В. brevis [53]. Возможны структурные варианты:
в начале цепи — валин или изолейцин, а фенилаланин, тирозин
или триптофан на двенадцатом месте в цепи, состоящей из 15 ос-
татков [29]. Наблюдается также типичное для многих ионофоров
чередование d- и ь-конфигураций, концевые группы блокируются
Н О
I II
формильной (R—N—СН2О.Н) или этаноламидной (R—С—NH—
—СН2—СН2ОН) группами. Точная конформация молекулы не ус-
тановлена, хотя известна ее склонность к димеризации [29]. Хотя
грамицидин похож на описанные выше ионофоры по структуре и
влиянию на мембраны, он не образует легко обнаруживаемых
липидорастворимых комплексов и может и не выполнять функции
истинного подвижного «ионофороподобного» носителя. Осущест-
влен синтез грамицидинов [29].
Иногда возникает путаница между выше описанными грами-
цидинами «Дюбо» [53] и грамицидином S [54], который является
циклическим основным декапептидом, продуцируемым другим
штаммом В. brevis. Грамицидин S, который по структуре также
родствен тироцидину [55], нарушает окислительное фосфорилиро-
вание в митохондриях, так же как и другие ионофоры, хотя этот
эффект не зависит от ионов. В грамицидине S внутри молекулы
образуется большое число водородных связей, так что не происхо-
дит раскрытие цикла, необходимое для полилигандного комплек-
сообразования [50]. Грамицидин содержит также основные орни-
тиновые остатки, которые придают молекуле положительный за-
ряд, тем самым препятствуя комплексообразованию с катионом
вследствие электростатического отталкивания. Таким образом,
нарушающие обмен в митохондриях свойства этой молекулы, по-
видимому, обусловлены ее поверхностной активностью. Тироциди-
ны А, В и С, очищенные методом противоточного диализа, не толь-
ко не вызывают вхождения иона в митохондрии, а даже наруша-
ют этот процесс. Однако промышленный тироцидин не обладает
ионофорной активностью, главным образом из-за загрязнения гра-
мицидином, который одновременно продуцируется теми же самы-
ми организмами. Возможно, этим объясняются сообщения об ин-
дуцируемом тироцидином транспорте'через мембраны [56].
Ионофоры — хелатирующие агенты для щелочных металлов
257
3.5. Полициклические эфиры
Ионофоры этого ряда получены Педерсеном конденсацией
эпоксиэтилена с резорцином и его производными с последующим
кипячением со щелочью [30, 57, 58]. Наиболее доступный предста-
витель этого ряда, дибензо-18-корона-6, получается с высоким вы-
ходом при проведении конденсации в присутствии КОН; если вме-
сто едкого кали использовать NaOH, получают много побочных про-
дуктов. Ароматические короны (краун-эфиры) умеренно раствори-
мы как в водных, таки в органических растворителях и их эффектив-
ность как ионофоров увеличивается при гидрировании ненасыщен-
ных колец. При этом образуется ряд геометрических изомеров,
два из которых составляют основную часть неочищенной дипик-
логексил-18-короны-6 (рис. 6.1,Г). Полиэфиры типа корон наибо-
лее интересны тем, что у химика появляется возможность синте-
зировать различные полиэфиры, размер кольца у которых систе-
матически изменяется. Таким образом, ионную селективность
можно коррелировать с размером кольцевого отверстия (табл. 6.2).
Как и следовало ожидать, соединения с меньшими размерами
кольца (14- и 15-членные) предпочитают Na+, для К+ наиболее
подходящими являются 19-членные кольца, в то время как для
Cs+ — кольца с 21—24 атомами. Увеличение констант ассоциа-
ции с К+ (Кл) при переходе от 24- к 30-членным кольцам обус-
Таблица 6.2
Влияние размера кольцевого отверстия на ион-селективность полиэфира®
Полиэфир Размер кольцевого отверстия, о А для К* Относительное сродство
Na (D = 1,9A) К (D=2,66A) Cs (D=2,34A)
Дициклогексил-14-корона-4 1,5 1,30 7,6 1 0,16
Циклогексил-15-корона-5 2,2 2,18 1,3 1 0,16
Дициклогексил- 18-корона-6 3,2 6,01 0,016 1 0,040
(изомер А)
Дибензо- 18-корона-6 3,2 5,00 0,23 1 0,036
Д ибензо-21 -корона-7 4,3 4,30 0,012 1 0,80
Дибензо-24-корона-8 3,49 1 0,19
Дибензо-30-корона-10 4,60 0,002 1
18-Корона-6 3,2 6,10 0,021 1 0,033
21-Корона-7 4,3 4,41 1 4,0
24-Короиа-8 3,48 1 4,7
аКонстанты селективности определены в метаноле с помощью ион-селективных электро-
дов [59]. кольцевые отверстия измерены иа моделях Фишера—Хиршфельдера—Тейлора.
17—2451
258
Глава 6
ловлено главным образом тем, что последние скручиваются и им
легче принять более сферическую конформацию [59].
Дициклогексил-18-корона-6 достаточно растворима в воде для
того, чтобы комплексообразование можно было исследовать либо
с помощью ион-селективных электродов [59], либо калориметри-
чески [60]. При прочих равных условиях комплексообразование в
10 000 раз эффективнее в метаноле, чем в воде, по-видимому,
вследствие того, что ионофору трудно вытеснить воду из гидрат-
ной оболочки катиона [60]. Дициклогексил-18-корона-6 образует
комплексы состава 2:1 и 3 : 2 с Cs+ и Rb+ [58].
Проведен рентгеноструктурный анализ кристаллических ком-
плексов дибензо-18-1короны-6 с Rb+ и Na+, а также свободного
ионофора. Катионы находятся в одной плоскости с атомами кис-
лорода* (ср. с энниатином). Расстояния Rb—О и Na—О состав-
ляют 2,86—2,93 и 2,74—2,89 А соответственно. Открытая структура
этих ионофорных комплексов становится очевидной на основании
того, что анион SCN- находится на расстоянии вандерваальсова
радиуса от Rb (расстояние Rb—N 2,94 А), т. е. образует ионную
пару, в то время как расстояние Na—N заметно больше 3,32 А.
Конформация свободного ионофора в решетке менее симметрична,
чем конформация связанного в комплекс ионофора [61, 62].
Спектр протонного магнитного резонанса раствора, по крайней
мере при 60 МГц, недостаточно разрешен для того, чтобы на его
основании можно было провести простой конформационный ана-
лиз [51].
Криптанды** относятся к синтетическим полиэфирам, содержа-
щим азот, например N(—С—С—О—С—С—О—С—C)3N [63]. Это
довольно широкий набор комплексующих агентов, однако, по-
скольку их полярность слишком велика, чтобы они могли входить
в липиды, их нельзя полностью отождествлять с мембранными
ионофорами. Рентгеноструктурный анализ комплекса RbSCN с
упомянутым выше соединением показывает, что расстояние Rb—О
составляет 2,9 А и что атомы азота также находятся в контакте
со связанным в комплекс катионом (Rb—N 3,00 А) [64].
3.6. Другие соединения
Циклический декапептид антаманид, выделенный из гриба
Amanita phalloid.es [65], действует как антидот против сопутству-
ющего ему токсического олигопептида фаллоидина. Антаманид
образует липидорастворимые комплексы с катионами щелочных
металлов (причем предпочитает натрий калию) и промотирует
активный перенос К+ в митохондриях [11, 66]. Он весь состоит
* Это не совсем точно — катионы несколько отклонены от плоскости, в кото-
рой лежат атомы кислорода короны. — Прим. ред.
** Автор называет их «dentates». — Прим. ред.
Ионофоры — хелатирующие агенты для щелочных металлов 259
из остатков с ь-конфигурацпями; однако четыре пролиновых
остатка могут значительно искривить кольцо, оптимально фоку-
сируя карбонильные группы для участия в комплексообразовании,
и из-за этого отпадает потребность в d-конфигурации.
Аламетицин— другой полностью состоящий из ь-конфигура-
ций циклический полипептид, продуцируемый Trichoderma [67].
Семь .из 17 его остатков состоят из необычной аминокислоты —
ь -аминоизомасляной кислоты. Семнадцать остатков образуют
кольцо, а один остаток глутаминовой кислоты со свободной кар-
боксильной группой подвешен к кольцу [68]. Хотя по способности
стимулировать вхождение ионов в митохондрии аламетицин ведет
себя подобно типичным ионофорам и образует липидораствори-
мые катионные комплексы [18], он имеет отличительные электро-
метрические свойства. Аламетицин, как и другие антибиотики ти-
па валиномипина, индуцирует катионный ток в двухслойных ли-
пидных мембранах. Однако в том случае, когда потенциал превы-
шает критический пороговый уровень, начальное сопротивление
мембраны падает до более низкого значения; это явление анало-
гично поведению возбудимых мембран нервов и мускулов [69,
70]. Аналогичным образом ведет себя моназомипин, но в этом
случае сопротивление мембраны возрастает с повышением транс-
мембранного потенциала [69, 70]. Моназомицин также стимулиру-
ет транспорт ионов в митохондриях [16], однако он не солюби-
лизирует ионы щелочных металлов в толуоле [71] и поэтому его
нельзя классифицировать как истинный ионофор. Молекулярная
масса моназомицина 1050—1200, он содержит 20 атомов кислоро-.
да и один основной атом азота; его точная структура в настоящее
время не известна. Но, возможно, он относится к антибиотикам
полиенового ряда [72]. Антибиотик LL-491 (мол. масса 1500)
[72а] по активности похож на моназомицин, но по отношению к
митохондриям его активность составляет всего около 20% от ак-
тивности моназомицина [71].
Монамицины [73] — группа циклических октадекапептидов с
чередующимися d- и ь-конфигурациями в кольце. Несколько ами-
нокислот являются производными пиперидазина или хлорпипе-
ридазина [74]. Они образуют комплексы с К+ [75] и переносят
ионы через липидные двойные слои, следовательно, являются на-
стоящими ионофорами. Как и в случае аламетицина и моназоми-
цина, вольт-амперные характеристики проводимости липидных
двойных слоев не линейны и резко изменяются с изменением по-
тенциала [75].
3.7. Катионная селективность
Для оценки катионной селективности ионофоров типа валино-
мицина было использовано несколько критериев, например:
1) зависимость нарушения окислительного фосфорилирования в
17*
260
Глава 6_______________________________
митохондриях от катиона; 2) проводимость двойных липидных
слоев; 3) биионные потенциалы, возникающие в двойном липид-
ном слое при разделении различных катионных частиц; 4) спо-
собность образовывать липидорастворимые комплексы в двухфаз-
ных системах органический растворитель — вода; 5) прямое тит-
рование ионофора в одном растворителе, используя некоторые
Cs Rb К Na Li
Рис. 6.2. Селективность валиномицина, измерен-
ная различными методами [46].
Шкалы рис. 6.2 и 6.3 логарифмические, значение се-
лективности для наиболее предпочитаемого иона обозна-
чено значением 2, так что различие между двумя катио-
нами на 1 означает, что их селективности отличаются
в 10 раз. а — анализ дыхания; б — бииониый потенциал;
в — электропроводность; г — комплексообразование.
физические измерения в качестве .индикатора. Как ранее указыва-
лось, комплексообразование с грамицидином и моназомицино,м
трудно обнаружить, так как, например, критерий (4) для этих со-
единений применить нельзя. Критерий (4) можно оценить двумя
способами: прямым измерением миграции комплексообразующего
катиона в органическую фазу [1, 10, 18, 46] и косвенно — измере-
нием концентрации анионов, сопутствующих .катиону, например
SCN- [10] или пикрат-аниона [76]. Некоторые методы использо-
вались для титрования ионофоров с 'катионами .в однофазной си-
стеме с применением ион-селективных электродов [59], осмомет-
рии [44], электропроводности [41, 42, 51] и ЯМР [33, 42, 51].
Ионная селективность валиномицина, оцененная по нескольким
критериям, показана на рис. 6.2. Можно видеть, что все методы
обнаруживают очень сходную картину разделения ионов.
Нейтральные ионофоры, согласно всем критериям, обнаружи-
вают обычный ряд селективности: К+—Rb+>Cs+>Na+>Li+.
Степень избирательности весьма различна; биологическая оценка
по соответствующему отношению К+: Na+ изменяется от 10 000: 1
для валиномицина, 25 : 1 для актинов и энниатинов и близка к 1
для грамицидина [критерий (1)] [43]. Внутри каждого ряда ионо-
форов-аналогов наблюдаются заметные отклонения, которые за-
висят от природы растворителя.
Так как карбоксилсодержащие ионофоры обычно электрически
никак себя не проявляют, критерии (2) и (3) нельзя использовать
Ионофоры — хелатирующие агенты для щелочных металлов 261
для их оценки. Они вызывают эндогенное выделение К+ в обмен
на Н+ в митохондриях, что можно использовать для измерения
их динамической способности переносить К+ через мембраны; од-
нако такого рода измерения нельзя просто .распространить на дру-
гие ионы. Вместо нарушения процессов в митохондриях, как это
делают ионофоры типа валиномицина, карбоксилсодержащие
Kjj для
комплексообразо-
Cs Rb К Na Li вопия с К+
Ионный мМ
Х-537А
(589)
Валиномицин
(1110)
Х-206
(841)
Миверицин
(736)
Моненсин
(670)
Диаммиццн
(956)
Рис. 6.3. Селективность различных ионофоров, изученная методом двухфазной
экстракции [18].
ионофоры ингибируют дыхание в митохондриях, очевидно, косвен-
ным образом препятствуя вхождению субстрата. Ингибирование
можно предотвратить, обеспечив высокий экстрамитохондриаль-
ный уровень К+ [23]. Равновесие комплексообразования с этими
ионофорами можно изучать методом двухфазной экстракции. Ре-
зультаты такого исследования (рис. 6.3) показывают, что, несмот-
ря на структурное сходство, карбоксилсодержищие ионофоры про-
являют гораздо более широкий диапазон селективностей по срав-
нению с родственными им ионофорами типа .валиномицина [10,
18, 46].
Ионофоры могут образовывать комплексы и с другими ионами
•подходящего диаметра и заряда, однако имеющиеся о них дан-
ные отрывочны. Согласно критерию митохондриального взаимо-
действия, NH4 связывается валиномицином и энниатином иесколь-
262
Глава 6
ко менее эффективно, чем Cs+, а актинами иногда более эффек-
тивно, чем К+ [76]. Возможно, это обусловлено наличием других
мостиковых атомов кислорода, образующих водородные связи с
NH4; такого взаимодействия с ионами металлов нет. О комплек-
сообразовании Т1+ [76] и Ag+ [59] с актинами и полиэфирами
также сообщалось, и это не удивительно с точки зрения близости
их атомных* радиусов и зарядов к зарядам и радиусам ионов ще-
лочных металлов.
4, КАРБОКСИЛАТНЫЕ ИОНОФОРЫ
Хотя нейтральные ионофоры подразделяются на несколько
различных групп, известные структуры карбоксильных ионофоров
близки по характеру. Ковалентные структуры моненсина, нигери-
цина, Х-537А и гризориксина характеризуются наличием цепей,
содержащих эфирные атомы кислорода в пяти- или шестичлен-
ных гетероциклических кольцах; в остове бывают и дополнитель-
ные атомы кислорода. Единственная карбоксильная группа с од-
ного конца цепи посредством водородной связи образует кольцо
с одной или двумя гидроксильными группами на противополож-
ном конце молекулы.
Первым карбоксильным ионофором, структура которого была
установлена, был моненсин (рис. 6.1,Д), выделенный из штамма
Streptomyces [77]. Рентгеноструктурный анализ его серебряной
соли показал, что шесть атомов кислорода связаны ион-диполь-
ным взаимодействием с катионом и находятся от него на расстоя-
ниях 2,40—2,68 А. Однако карбоксилатные атомы кислорода не
являются сильными лигандными группами, так как они находятся
от катиона на расстоянии 3,8 А. Кольцо, состоящее из 22 атомов,
вокруг катиона уложено нерегулярно, но с приблизительным со-
блюдением плоского расположения [78, 79]. Моненсин обладает
умеренной селективностью, предпочитая ионы Na+ ионам К+ в от-
ношении 10 : 1 [10, 18].
Серебряная соль нигерицина (рис. 6.1, Е) [20] содержит
24-членное кольцо. Два атома кислорода, соответствующие ион-
дипольным лигандным группам моненсина, заменены карбокси-
латным атомом кислорода, который теперь входит в координа-
ционную сферу катиона (Ag—О 2,25 А) и образует сильную ион-
ную связь. Четыре лиганда, образующих ион-дипольные связи
(Ag—О 2,45—2,69 А), и карбоксилатный кислород образуют пяти-
членную координационную ячейку иона серебра [80, 81]. Сродст-
во нигерицина к К+ в 400 раз сильнее, чем моненсина, хотя изве-
стно, что их структуры почти идентичны. Структура гризориксина
Точнее ионных радиусов. — Прим. ред.
Ионофоры — хелатирующие агенты для щелочных металлов 263
[82] идентична структуре нигерицина, за тем исключением, что он
не содержит гидроксильной группы в концевом кольце напротив
кольца, содержащего карбоксил. Атомы кислорода, образующие
связь с солью серебра, такие же, .как у нигерицина; однако из-за
отсутствия гидроксильной группы образуются не две, а одна во-
дородная связь по типу «голова к хвосту» [83]. О катионной се-
лективности этого ионофора еще не сообщалось. Расстояния
Ag—О составляют 2,4—2,7 (ион-дипольные) и 2,2 А (с карбокси-
латным кислородом).
Большее число атомов водорода, химически .похожих, но не
идентичных, делает спектры TIM.P карбоксилсодержащих ионофо-
ров более сложными для интерпретации, чем спектры нейтраль-
ных .ионофоров. Однако поле лигандов, образуемое цвиттер-ион-
ной структурой карбоксильных .ионофоров, можно исследовать
другим вариантом метода ЯМР. Вследствие того что ядро 23Na
обладает ядерным спином 3/2, оно «чувствует» асимметрию окру-
жающего его поля, реагируя на нее уширением ЯМР-линий. Та-
ким образом, большая асимметрия, наблюдаемая для комплексов
Na+ с нигерицином по сравнению с моненсином, согласуется с 'бо-
лее близким подходом отрицательно заряженного карбоксильного
атома кислорода к катиону [47], .как это следовало из кристалли-
ческой структуры его серебряной соли [79, 80].
Наиболее существенное отличие ионофора Х-537А (обозначе-
ния по Гоффману—Ла Рошу) [84] от других карбоксилатных
ионофоров состоит в том, что в нем имеется ароматическое коль-
цо, к которому присоединена концевая карбоксильная группа.
Кроме того, кольцо содержит фенольный гидроксил, благодаря
чему оно легко поддается химическим превращениям. Второй его
уникальной особенностью является наличие кетонной карбониль-
ной группы в области прямолинейного участка цепи антибиотика.
Так как, по-видимому, эта группа участвует в образовании ион-
дипольных связей, комплексообразующие свойства молекулы та-
ковы, что она очень чувствительна к реагентам, образующим ад-
дукты с кетонами.
Х-537А — до сих пор единственный карбоксильный ионофор,
избирательный по отношению к Cs+ [10, 18]. Его относительно
низкая степень селективности, примерно следующая липотропно-
му ряду, по-видимому, свидетельствует о ‘большей гибкости его
образующего комплекс кольца по сравнению с антибиотиками с
более выраженной селективностью.
Структура Х-537А 'была установлена рентгеноструктурным
анализом его бариевой соли [85]. Комплекс с двухвалентным ка-
тионом состоит из электронейтрального цвиттер-иона, содержаще-
го два аниона ионофора. Один «без штриха» (unprimed) анион
координируется к катиону атомами кислорода с образованием пя-
ти ион-дипольных связей (Ва—О 2,7—3,1 А) и шестой связи с за-
264
Глава 6
ряженной карбоксильной группой (Ва—О 2,8 А) (рис. 6.1, Ж).
Другой, «со штрихом» (primed) анион хотя и имеет почти такую
же конформацию, как молекула ионофора «без штриха», коорди-
нируется только через карбоксилатный анион (Ва—О 2,6 А) и
концевой гидроксил (Ва—О 2,8 А) (рис. 6.1, 3). Молекула ионо-
фора «со штрихом», кроме того, связана .водородной связью с од-
ной молекулой воды, (которая в свою очередь связана с барием
(Ва—О 2,7 А), так что координационное число катиона равно 9.
Мы можем также показать, что Х-537А в растворе образует
комплексы с Sr2+, Са2+, Mg2+, а также Ва2+ и ионами щелочных
металлов. Изотермы связывания с Ва2+ согласуются с таким со-
ставом комплекса: (ионофор)2Ва2+ [71]. Х-537А действует как
ионофор для двухвалентных ионов; мы подтвердили его способ-
ность переносить ,33Ва2+ и 90Sr2+ в объемную фазу, состоящую из
толуола и бутанола [71].
Имеются сведения и о структуре других моненсинов (моненси-
нов В и С), но и этих данных недостаточно, чтобы охарактеризо-
вать их селективность при комплексообразовании. Есть предва-
рительные данные, что дианмицин (селективный к Na+) также
имеет сходство с этим рядом [80], как, по-видимому, и Х-206 [84]
Молекулярные свойства ионофоров
Таблица 6.3
Ионофор Соль О Расстояния кислород—катион, А Селективность комплексообразования
ион-дипольные с карбоксилом
Ноиактин KSCN 2,7—2,9 [26] NH4>K>Rb>Cs>
Энниатин В KI 2,6—2,8 [40] >Na>Li [44] K>Rb>Cs>
Валиномицин КАиС13 2,7—2,8 [52] > Na> Li[51] K>Rb>Cs>
Дибензо- 18-ко- RbSCN 2,7—2,8 [62] >Na>Li [18] Ag>K>Na>NH4>
рона-6 Криптанд Моненсин RbSCN Ag* 2,9 [64] 2,4—2,7 [79] 3,8 > Cs > Li [59] Na>K>Rb>
Нигерицин Ag* 2,4—2,7 [80] 2,2 >Li>Cs[18] K>Rb>Na>
Х-537А Ba2* 2,7 [85] 2,8 >Cs>Li [18] Cs>Rb =
Гризориксин Ag* 2,4—2,7 [83] 2,2 = K>Na>Li [18]
Ионофоры — хелатирующие агенты для щелочных металлов
265
(селективный к К+). Дианмицин напоминает Х-537А тем, что его
спектр разделения щелочных ионов относительно широкий [10,
18]. Поэтому, может быть, важно то, что дианмицин также скло-
нен образовывать комплексы с двухвалентными катионами [71].
Расстояния кислород—катион, определяющие наличие связи .в
комплексах с этим ионофором, найденные рентгеноструктурным
методом, приведены в табл. 6.3.
5. ДИНАМИКА КОМПЛЕКСООБРАЗОВАНИЯ
С ИОНОФОРАМИ
Хотя на основании простого подбора соответствующих молеку-
лярных моделей и можно сделать некоторые предположения о
причинах ионной селективности данного ионофора, равновесие
комплексообразования зависит также и от других факторов. Энер-
гии сольватации свободных ионов щелочных металлов сильно от-
личаются между собой. Когда комплексообразование происходит
в присутствии полярного растворителя, процесс десольватации
соответственно сам по себе будет сильно зависеть от природы ка-
тиона. Свободная энергия комплексообразования, следовательно,
определяется различием между процессами десольватации иона и
ассоциации иона с ионофором. Таким образом, наблюдаемая се-
лективность данного ионофора может меняться с изменением рас-
творителя. Ион-дипольное взаимодействие между ионофором и
ионом имеет много общего с сольватацией иона; в процессе ком-
плексообразования ионофор действительно сольватирует ион. Из-
менение энергии в процессе комплексообразования в свою очередь
можно разделить на два слагаемых: энтальпия, возникающая при
взаимодействии между ионом и ионофором, и энтропия, опреде-
ляемая изменением конформации ионофора от той, которая наи-
более предпочтительна в свободном состоянии, до той, которая
требуется в комплексе для наиболее выгодного расположения
соответствующих атомов кислорода возле катиона*.
Следующая последовательность стадий выведена для динамиче-
ского, катализируемого ионофором транспорта ионов через био-
логические мембраны и другие липидные барьеры. Мембрану
можно рассматривать как тонкий липидный слой, помещенный в
водную среду, причем несвязанный катион первоначально концен-
трируется в водной фазе, а гидрофобный ионофор — в липидной
фазе. Силы взаимного отталкивания внутри электроотрицательной
кислородной системы должны вынуждать несвязанный в комплекс
ионофор принять конформацию, отличную от его конформации в
* Общее изменение энтропии при образовании комплекса включает также
изменение энтропии при десольватации исходного сольватированного иона —
Прим. ред.
266
Глава 6
комплексе. Таким образом, свободный ионофор, по-видимому,
«чувствует» присутствие катиона еще до начала перегруппировки
в конформацию комплекса. Это означает, что в «интерфазе» об-
разуется слабый промежуточный комплекс ионофор—катион. За-
тем ионофор может изменять свою конформацию так, чтобы его
кислородная система вытеснила сольватную воду катиона. Про-
цесс заканчивается образованием стабильного липофильного ионо-
форного комплекса, который диффундирует через мембрану, после
Рис. 6.4. Последовательность стадий, происходящих в процессе транспорта, ин-
дуцируемого нейтральным ионофором.
Начиная сверху, ионофор I диффундирует через липидный барьер внутренней поверхности,
где он встречается с гидратирован ныйи катионом, М+-Н2О. В интерфазе образуется слабый
комплекс М+-Н2О-1. Затем I претерпевает конформационное превращение в I*, после чего он
вытесняет сольватную оболочку М.+, образуя лнпидорастворимый комплекс M+-I*. который
легко диффундирует через мембрану. В другой интерфазе комплекс атакуется растворителем,
при этом катион вновь гидратируется, а освободившийся I диффундирует назад в противо-
положную интерфазу и начинает новый транспортный каталитический цикл.
чего комплекс атакуется водой и происходит его диссоциация. Те-
перь ионофор снова свободен и может диффундировать в обрат-
ном направлении к другой стороне мембраны, чтобы участвовать
в новом каталитическом цикле транспорта катиона. Эта последо-
вательность реакций иллюстрируется рис. 6.4.
6. ТРАНСПОРТНЫЕ РАВНОВЕСИЯ,
катализируемые ионофорами*
Нейтральные ионофоры переносят ионы через липидные барье-
ры в форме заряженных частиц, затем незаряженный свободный
ионофор возвращается, чтобы закончить каталитический цикл. Та-
ким образом, такие ионофоры способны к общей транслокации
зарядов и выражение для равновесия, катализируемого этими
* Неточное выражение, так как равновесия не могут быть каталитическими,
катализируются отдельные этапы равновесия. — Прим. ред.
Ионофоры — хелатирующие агенты для щелочных, металлов 267
ионофорами, содержит электрическую составляющую; равновесие
достигается, когда электрохимическая активность транспортируе-
мого иона по обе стороны 'мембраны становится одинаковой. Рав-
новесие описывается уравнением Нернста для диффузионного по-
тенциала:
[М+]1 го п.- tM+Ji
А£_ nF ln [М+]2 ~59 MBlg [М+]2
(1)
где [M+]i и [М+]г — концентрации катионных частиц М+ на сто-
роне 1 и 2 соответственно.
Когда больше чем одна из присутствующих катионных частиц
может образовать комплекс с ионофором, применимо уравнение
Гольдмана
ДЕ = 59 мВ 1g
[M+h + Р mn
[М+]2 +Pmn [N+]2 + Pmo [O+]2.
(2)
Множители Pmn и Pmo представляют относительные проникающие
способности ионных частиц М+ и N+ и М+ и 0+ соответственно.
Эти факторы являются функцией концентрации ионофорного ком-
плекса [IM+] в интерфазе, которая при отсутствии большой насы-
щающей концентрации катиона связана с равновесием выраже-
нием
[I] [М+]
- [IM+]
(3)
Следовательно, биионный потенциал, возникающий вдоль мем-
браны или липидного барьера между растворами двух связываю-
щих ионы частиц, выражает относительное сродство этих проти-
вопоставляемых ионных частиц к ионофору и подвижности ком-
плексов при прохождении через мембрану. Второе назначение тех
же коэффициентов проницаемости — выражать относительные про-
водимости мембран для одинаковых .концентраций различных
ионов. Корреляции мембранных проводимостей и равновесий ком-
плексообразования для ряда щелочных ионов даны па рис. 6.2;
там же приведены соответствующие значения селективности, по-
лученные по митохондриальному критерию.
Для того чтобы облегчить миграцию катиона из водной среды
в липофильную фазу, содержащую ионофор, нужно дать возмож-
ность липидосовместимому аниону мигрировать вместе с катио-
ном. Это мешает созданию достаточного межфазового потенциала
[ср. уравнение (1)] для уравновешивания электрохимического по-
тенциала катиона в обеих фазах до того, как сможет осуществить-
ся измеримая миграция катиона. При .фиксированной концентра-
ции таких анионов, как SCN~, и при более высоких концентраци-
ях катиона миграция катиона в идеальном случае достигает
изотермы насыщения Ленгмюра, начиная с которой можно опре-
268
Глава 6
делять сродство ионофора к определенному катиону:
I_________!__ , _ 1 ,,,
[М1+]оРг - [М+]н2О + П1орг + [1]орг W
где [М1+]орг—концентрация катиона в органической фазе (при
отсутствии контроля за концентрацией ионофора), [М+]
Н2О
концентрация М+, добавленного к -воде, и [I] орг— концентрация
ионофора, добавленного в органическую фазу. Кажущаяся кон-
станта диссоциации Kd является функцией концентрации липидо-
совместимого аниона, при этом она заметно не изменяется при из-
менении pH (ср. ниже карбоксильные ионофоры).
Карбоксильные ионофоры могут входить -в липидные фазы ли-
бо как электронейтральные цвиттер-ионные комплексы, либо как
незаряженная недиссоциированная карбоновая кислота. При нейт-
ральных значениях pH только небольшое количество ионофора
проходит липидный барьер в виде заряженных частиц; следова-
тельно, этот класс ионофоров фактически не проявляет себя элек-
трически по отношению к мембранам. При более высоких значе-
ниях pH несвязанная в комплекс карбоксилатная форма этих
ионофоров, по-видимому, становится достаточно преобладающей,
чтобы проводить ток и вызывать электрометрически детектируе-
мые эффекты на мембранах. Хотя в выражении для трансмем-
бранного равновесия нет никакой электрической составляющей,
появляется преобладающий протонный градиент:
[H+h [М+]2 [N+h [О+]2
[Н+]2 - [М+]х ~ [N+]x - [O+]j
Идентичные градиенты в конечном счете достигаются для каж-
дого иона, способного к прохождению сквозь мембраны с ионо-
фором, независимо от его сродства к ионофору.
Однако в экспериментах по экстракции катионные частицы
конкурируют с протонами за ионофор:
I.H +М+ Г.М+ +Н+ (6)
Таким образом, при фиксированном значении pH можно полу-
чить идеальную изотерму насыщения Ленгмюра как функцию кон-
центрации катиона и его сродства. Так как общего переноса за-
ряда не происходит, не возникает никакого межфазного потен-
циала и на это равновесие не влияют присутствующие анионные
частицы. По мере того как pH влияет на кажущееся сродство
к катиону карбоксильного ионофора, на кажущуюся константу
диссоциации карбоксильной группы (рКа) влияют катионные час-
тицы и концентрации. Равновесия, описывающие взаимодействие
между карбоксильными ионофорами и двухвалентными катиона-
ми, иногда более сложные.
Ионофоры — хелатирующие агенты для щелочных металлов
269
7. ПРИМЕНЕНИЕ ИОНОФОРОВ j
Несмотря на то что большинство ионофоров при выделении'
рассматривали как антибиотики, их терапевтическое применение
сводится лишь к случаям, когда необходимо нарушить процессы'
накопления энергии митохондриями высших организмов [8, 9]..-
Антибиотическая активность ионофоров, по-видимому, также обус-
ловлена тем, что они нарушают метаболизм энергии и ионный
метаболизм в микроорганизмах [86—88]. Хотя грамицидин систе-
матически токсичен, он находит ограниченное медицинское при-
менение как антибиотик местного назначения. Токсичность ионо-
форов по отношению к морской креветке Artemia salina использо-
валась как процедура отбора («скрининг») для их обнаружения
[89].
Некоторые карбоксильные антибиотики имеют ограниченную
способность 'проникать в кишечник птиц, а моненсин, как сообща-
лось, эффективен против кокков и кишечных паразитов птиц [90].
Валиномицин и актины были применены для получения ион-селек-
тивных электродов [см. уравнения (1) и (2)] [91—93]. Срок экс-
плуатации таких электродов ограничен, но благодаря экстремаль-
но высокой К+ Ыа+-селективности валиномицинового электрода
он может найти широкое применение для контроля за концентра-
цией К+ в присутствии высоких концентраций Na+, например в
крови «ли моче. Возможно с его помощью можно осуществлять
контроль за содержанием К+ в сыворотке во время лечения сер-
дечных кризов или диабетической комы.
Ионофоры широко применялись как средство, позволяющее
экспериментально нарушать биоэнергетику и ионный метаболизм
субклеточных органелл, например митохондрий, как изолирован-
ных [1, 7—11, 13, 16, 18, 21, 24, 34, 35, 46], так и in situ (асцитная
опухоль) [94—96], а также хлоропластов [97—100] и хромато-
форов [100—105]. Их также использовали для селективного уве-
личения проницаемости других биологических мембранных систем,
например эритроцитов [106—108] и цист [109].
Одним из наиболее поразительных свойств ионофоров являет-
ся их способность индуцировать постоянную селективную проводи-
мость в искусственных двойных липидных слоях, которая копиру-
ет проводимость природных мембран. Электрометрически эта по-
стоянная селективность параллельна селективности биологических
мембран. Липидные мембраны, обработанные некоторыми ионо-
форами, например аламетицином, используются для изучения мо-
лекулярной природы электровозбуждения, которое характерно
для мембран нервов и мышц [69, 70].
Так как все описанные свойства ионофоров обусловлены их
способностью действовать в качестве мобильных переносчиков
ионов, основное внимание было сосредоточено на изучении их как
270
Глава 6
модели системы связанных с мембраной носителей. В действитель-
ности, их специфичность к субстрату и числа оборотов (например,
валиномицин — 200 с-1, нигерицин — 500 с~* в мембране мито-
хондрий) достаточны для истинных ферментов, хотя способность
валиномицина сублимироваться без разложения необычна для
биологических катализаторов. Маленький размер ионофоров об-
легчает изучение молекулярной структуры и конформационных
превращений, ответственных за их свойства, разнообразными
сложными физическими методами. Такие же структурные особен-
ности (а именно катион, находящийся в полярной внутренней
части циклической молекулы с липофильной наружной оболочкой)
имеют мембранопроницаемые железосвязывающие агенты биоло-
гического происхождения (гл. 5). Поэтому проблема мембранного
транспорта в природе имеет к ним прямое отношение.
Следует ожидать, что природные связанные с мембранами но-
сители должны обладать дополнительными свойствами, помимо
тех, которыми обладают ионофоры. Они должны быть прикрепле-
ны внутри данной мембраны так, чтобы они находились в равно-
весии со всеми целлюлярными мембранными системами. Это мо-
жет сопровождаться тем, что ионофороподобная структура при-
крепляется к большой объемистой молекуле белка. Для
построения активной транспортной системы необходимо участие
природного носителя, способного реагировать с источником энер-
гии, таким, как АТФ. Можно думать, что для того, чтобы оказать
аллостерический эффект на конформацию активной ионофорной
части носителя, который в свою очередь обратимо изменяет срод-
ство транспортируемого субстрата, необходимы связывание и по-
следующий гидролиз АТФ. Таким образом, нужно перевести носи-
тель в конформацию с высоким сродством для того, чтобы связать
субстрат в области низкой электрохимической активности. По ме-
ре того как носитель переносит субстрат в область высокой хими-
ческой активности субстрата, сродство должно быть понижено
аллостерически, посредством связывания или гидролиза АТФ, что
приводит к освобождению субстрата. Такая система циклического
изменения сродства части субстрата за счет внутренней энергии
системы напоминает схему транспорта, предложенную Вилбранд-
том и Розенбергом [110].
Ионофоры стали применять и как средство для изменения
функции мембран, и как модель для понимания поведения мем-
бран. Мы можем надеяться, что в будущем из мембран будут вы-
делены природные эквиваленты ионофоров, что значительно по-
могло бы нашему проникновению в глубины их более простых
прототипов.
Ионофоры — хелатирующие агенты для щелочных металлов 271
ДОПОЛНЕНИЯ, ВНЕСЕННЫЕ В КОРРЕКТУРУ
Недавно методом рентгеноструктурного анализа были установ-
лены структура и конформация еще двух карбоксильных ионо-
форов— Х-206 [111] и дианмицина [112].
СПИСОК ЛИТЕРАТУРЫ
1. Pressman В. С., Harris Е. J., Jagger W. S., Hohnson J. Н., Proc. Natn. Acad.
Sci., U. S„ 58, 1949 (1967).
2. Brockmann H., Schmidt-Kastner G., Chem. Ber., 88, 57 (1955).
3. Brockmann H., Springorum M., Traxler G., Hofer I., Naturwiss., 50, 689
(1963).
4. Shemyakin M. M., Aldanova N. A., Vinogradova E. I., Feigina M. Yu., Tetra-
hedron Lett., 1963, 1921.
5. Shemyakin M. M., Vinogradova E. I., Feigina M. Yu., Aldanova N. A., Logi-
nova N. F., Ryabova N. F., Pavelenko I. D., Experientia, 21, 548 (1965).
6. Шемякин M. M., Виноградова E. И., Фейгина M. JO., Алданова H. А., Шве-
цов Л. Б., Фонина Л. А., Ж0Х, 36, 1391 (1966).
7. McMurray W., Begg R. W„ Arch. Biochem. Biophys., 84, 546 (1959).
8. Moore C., Pressman В. C., Biochem. Biophys. Res. Commun., 15, 562 (1964).
9. Pressman В. C., Proc. Natn. Acad. Sci. U. S., 53, 1076 (1965).
10. Pressman В. C., in «Mitochondria, Structure and Function», 5th Meeting, Fed.
Europ. Biochem. Soc., Academic Press, New York, 1969, p. 315.
11. Pressman В. C., in E. Racker (ed.), Membranes of Mitochondria and Cho-
lorplasts, Reinhold Van Nostrand, New York, 1970, p. 213.
12. Simon W., Pioda L. A. R., Wipf H. K-, in T. Biicher, H. Sies (eds.), Inhibitors,
Tools in Cell Research, Springer-Verlag, New York, 1969, p. 356.
13. Pressman В. C., Antimicrobial Agents and Chemotherapy-1969, 1970, 28.
14. Mueller P., Rudin D. O., Biochem. Biophys. Res. Commun., 26, 398 (1967).
15. Лев А. А., Бучинский E. П., Цитология, 9, 102 (1967).
16. Lardy H. A., Graven S. N., Estrada-O. S., Federation Proc., 26, 1355 (1967).
17. Eisenman G., Federation Proc., 27, 1249 (1968).
18. Pressman В. C., Federation Proc., 27, 1283 (1968).
19. Wipf H. K., Pache W., Iordan P., Zahner H., Keller W„ Schierlein, Simon W.,
Biochem. Biophys. Res. Commun., 36, 387 (1969).
20. Harned R. L., Hidy P. H., Corum C. J., Jones K. L„ Antibiot. Chemother., 1,
594 (1951).
21. Lardy H. A., Johnson D., McMurray W. C., Arch. Biochem. Biochys., 18, 587
(1958).
22. Graven S. N., Estrada-0 S., Lardy H. A., Proc. Natn. Acad. Sci. U. S., 53,
1076 (1965).
23. Graven S. N., Estrada-0 S., Lardy H. A., Proc. Natn. Acad. Sci. U. S., 56,
654 (1965).
24. Chappell J. B„ Crofts A. R., Biochem. J., 95, 393 (1965).
25. Kilbourn В. T„ Dunitz J. D„ Pioda L. A. R., Simon W., J. Mol. Biol., 30, 559
(1967).
26. Dobler M., Dunitz J. D., Kilbourn В. T., Helv. Chim. Acta, 52, 2573 (1969).
27. Shemyakin M. M., Angew. Chem., 72, 342 (1960).
28. Beck J., Gerlach H„ Prelog V., Voser W., Helv. Chim. Acta, 45, 621 (1962).
29. Serger R., Witkop B., J. Am. Chem. Soc., 87, 2011 (1965).
30. Pederson C. J., J. Am. Chem. Soc., 89, 7017 (1967).
31. Keller-Schierlein W„ in T. Biicher, H. Sies (eds.), Inhibitors, Tools in Cell
Research, Springer-Verlag, New York, 1969, p. 365.
32. Prestegard J. H., Chan S. I., Biochemistry, 8, 3921 (1969).
33. Prestegard J. H„ Chan S. I., J. Am. Chem. Soc., 92, 4440 (1970).
'272
Глава 6
34. Graven S. N., Lardy H. A., Johnson D.. Rutter A., Biochemistry, 5 (1966).
35. Graven S. N., Lardy H. A., Rutter A., Biochemistry, 5, 1735 (1966).
36. Pioda L. A. R., Wachter H. A., Dohner R. E., Simon W., Helv. chim. Acta, 50,
1373 (1967).
37. Plattner P. A., Vogler K., Studer R. O., Quitt P., Reller-Schierlein W„ Helv.
Chim. Acta, 46, 927 (1963).
38. Quitt P„ Studer R. O., Vogler K., Helv. Chim. Acta, 46, 1715 (1963).
39. Hamill R. L„ Higgens С. E., Boaz H. E., Gorman M., Tetrahedron Lett., 1969,
4255.
40. Dobler M., Dunitz J. D., Krajewski J., J. Biol. Biol., 42, 603 (1969).
41. Shemyakin M. M., Ovchinnikov Yu. A., Ivanov V. T., Antonov V. K. et al.,
Biochem. Biophys. Res. Commim., 29, 834 (1967).
42. Shemyakin M. M., Ovchinnikov Yu. A., Ivanov V. T., Antonov V. K. et al.,
J. Memb. Biol., 1, 402 (1969).
43. Pressman В. C., Harris E. I., Abstracts of Seventh International Congress of
Biochemistry, Tokyo, August 1967, Vol. V, p. 900.
44. Pioda L. A. R., Dissertation, Eidenossischen Technische Hochschule, Zurich,
1969.
45. Shemyakin M. M., Ovchinnikov Yu. A., Ivanov V. T., Evstratov A. V., Nature,
213, 412 (1967).
46. Pressman В. C., Haynes D. H„ in D. C. Tosteson (ed.), The Molecular Basis
of Membrane Function, Prentice-Hall Inc., Englewood Cliffs, N. J., 1969,
p. 221.
47. Haynes D. H., Kowalsky A., Pressman В. C., J. Biol. Chem., 244, 502 (1969).
48. Ivanov V. T„ Laine I. A., Abdullaev N. D., Senyavina L. B. et al., Biochem.
Biophys. Res. Commun., 1969, 34.
49. Ohnishi M., Urry D. W„ Biochem. Biophys. Res. Commun., 36, 164 (1969).
50. Urry D. W., Ohnishi M., in D. W. Urry (ed.), Spectroscopic Approaches to
Biomolecular Conformation, American Medical Association, Chicago, III, 1970,
p. 263.
51. Haynes D. H., Dissertation Thesis, University of Pennsylvania, 1970.
52. Pinkerton M„ Steinrauf L. K-, Dawkins P., Biochem. Biophys. Res. Commun.,
35, 512 (1969).
53. Dubos R., Hotchkiss R., J. Exp. Med., 73, 629 (1941).
54. Stern A., Gibbons W., Craig L. C., Proc. Natn. Acad. Sci. U. S., 61, 734
(1968).
55. Hotchkiss R„ Adv. Enzymol., 4, 153 (1944).
56. Goodall M. C., Biochim. Biophys. Acta, 203, 28 (1970).
57. Pederson С. J., Federation Proc., 27, 1305 (1968).
58. Pederson C. J., J. Am. Chem. Soc., 92, 389, 391 (1970).
59. Frensdorff H. K., J. Am. Chem. Soc., 93, 600 (1971).
60. Izaat R. M., Rytting J. H., Nelson D. P., Haymore B. L., Christensen J. J.,
Science, 164, 443 (1969).
61. Bright D„ Truter M. R., Nature, 225, 176 (1970).
62. Bright D., Truter M. R., J. Chem. Soc. (B), 1970, 1544.
63. Dietrich B., Lehn I. M., Sauvage J. P., Tetrahedron Lett., 34, 2889 (1969).
64. Metz B., Moras D., Weiss R., Chem. Commun., 1970, 217.
65. Wieland T., Luben G-, Ottenheyn H., Faesel J. et al., Angew. Chem., 80, 209
(1968).
66. Wieland T., Faulstich H„ Burgermeister W„ Otting W. et al., FEBS Letters,
9, 89 (1970).
67. Meyer С. E.. Reusser F., Experientia, 23, 85 (1967).
68. Payne J. W., Jakes R., Hartley B. S., Biochem. J., 117, 757 (1970).
69. Mueller P„ Rudin D. 0., Nature, 217, 713 (1968).
70. Mueller P., Rudin D. O., J. Theor. Biol., 18, 222 (1968).
71. Pressman В. C., unpublished observations.
72. Akasakim K, Karasawa K„ Watanabe M., Yonehara H., Umezawa H„ J. An-
tibiot. Ser. A, 16, 127 (1963).
Ионофоры — хелатирующие агенты для щелочных металлов 273
72а. Mitcher L. A., Shayand A. J., Bohonos N., Appl. Microbiol., 15, 1002 (1967).
73. Hassall C. H„ Magnus К. Е., Nature, 184, 1223 (1959).
74. Bevan К-, Davies J. S., et al., Experientia, 26, 122 (1970).
75. Goodall M. C., Nature, 225, 1258 (1970).
76. Eisenman G., Ciani S., Szabo G., J. Memb. Biol., 1, 294 (1969).
77. Haney M. E., Hoehn M. M„ Antimicrobial Agents and Chemotherapy-1967,
1968, 349.
78. Agtarap A., Chamberlin J. W., Pinkerton M., Steinrauf L. K-, J. Am. Chem.
Soc., 89, 5737 (1967).
79. Pinkerton M., Steinrauf L. K-, J. Mol. Biol., 1970, 49.
80. Steinrauf L. K, Pinkerton M„ Biochem. Biophys. Res. Commun., 33, 29
(1968).
81. Kubota T., Matsutani S. et al., Chem. Commun., 1968, 1541.
82. Gachon P., Kergomard .4.. Veschambre H„ Esteve C„ Staron T., J. Chem. Soc.
(D), 1970, 1421.
83. Alleaume A., Hickel D., J. Chem. Soc. (D), 1970, 1422.
84. Berger J., Rachlin A. I., Scott IE. E., Sternbach L. H., Goldberg M. W., J. Am.
Chem. Soc., 73, 5295 (1951).
85. Johnson S. M., Herrin J., Liu S. J., Paul I. C., J. Am. Chem. Soc., 92, 428
(1970).
86. Pressman В. C., in «Wirkungsmechanismen von Fungiziden und Antibiotika»
(Biologische Gesellschaft der D. D. R., Sektion Microbiologie), Academie Ver-
lag, Berlin, 1967, p. 3.
87. Harold F. M., Baarda J., Bacteriology, 94, 53 (1967).
88. Harold F. M„ Bact. Rev., 1969, 45.
89. Higgens С. E., Westhead J. E., Gorman M., Hamill R. L., Antimicrobial
Agents and Ghemostherapy-1968 (1969).
90. Shumard R. F., Callender M. E., Antimicrobial Agents and Chemotherapy-1967,
1968, 369.
91. Stafanac Z., Simon W., Microchem. J., 12, 125 (1967).
92. Pioda L. A. R., Simon W., Chimia (Switzerland), 23, 72 (1969).
93. Frant M. S., Ross J. W., Science, 167, 987 (1970).
94. Wenner С. E., Harris E. J., Pressman В. C., J. Biol. Chem., 242, 3454 (1967).
95. Gordon E. E.. Nordenbrand K., Ernster L„ Nature, 213, 82 (1967).
96. Gordon E. E., Bernstein J., Biochim. Biophys. Acta, 205, 464 (1970).
97. Packer L., Allen J. M., Starks M., Arch. Biochem. Biophys., 128, 142 (1968).
98. Shavit N., San Pietro A., Progress in Photosynthesis Research, 3, 1392 (1969).
99. Karlish S. J. D„ Avron M., FEBS Letters, 1, 21 (1968).
100. Karlisch S. J. D., Avron M., Eur. J. Biochem., 9, 291 (1969).
101. Shavit N., San Pietro A., Biochem. Biophys. Res. Commun., 28, 277 (1967).
102. Shavit N., Thore A., Keister D. L., San Pietro A., Proc. Natn. Acad. Sci. U. S.,
59, 917 (1968).
103. Jackson J. B., Crofts A. R., Von Stediggk L. V., Eur. J. Biochem., 6, 3499
(1968).
104. Junge W., Witt H. T. Z. Naturforsch., 23b, 244 (1968).
105. Nishimura M., Pressman В. C., Biochemistry, 8, 1360 (1969).
106. Chappell J. B., C'ofls .4. R., in J. M. Tager et aL, (eds.), Regulation of Meta-
bolic Processes in /Aotochondria, Biochim. Biophys. Acta Library, Amsterdam.
Vol. 7, 1966, p. 293
107. Harris E. J.. Pressman В. C., Nature, 216, 918 (1967).
108. Tosteson D. C., Andreoli T. E., Tieffenberg E., Cook P„ J. Gen. Physiol., 51,
373S (1968).
109. Wyssbrod H. R. Biochim. Biophys. Acta, 193, 361 (1969).
110. Willbrandt W., Rosenberg T„ Pharmacol. Rev., 13, 109 (1961).
111. Blount J. F„ Westley J. W„ Chem. Commun., 1971, 927.
112. Czerwinsky E. W„ Steinrauf L. K., Biochem. Biophys. Res. Commun., 45, 1284
(1971).
18—2451
ГЛАВА 7
КОМПЛЕКСЫ МЕТАЛЛОВ
С БЕЛКАМИ
Е. Бреслоу
Breslow Esther, Department of Biochemistry, Cornell
University Medical College, New York, USA
1. ВВЕДЕНИЕ
Взаимодействия ионов металлов с белками, естественно, от-
личаются от взаимодействий ионов металлов с аминокислотами
и пептидами, поскольку в белках группы a-NH2 и a-СООН длин-
ных полипептидных цепей разделены ковалентными связями ряда
расположенных между ними остатков. Эти взаимодействия отли-
чаются также из-за влияния конформационного состояния пептид-
ной цепи, в результате которого потенциальное место присоеди-
нения может блокироваться, а удаленная боковая цепь может
оказаться в подходящем месте для образования хелатного кольца.
Примерами подходящего расположения боковой цепи лиганда,
делающего возможным образование прочного хелата со специфи-
ческим ионом металла, могут служить металлопротеины и метал-
лоферменты, в которых сильное взаимодействие между металлом
и белком играет решающую и специфическую биологическую
роль. Металлопротеины и металлоферменты будут рассмотрены
в последующих главах. В этой главе в основном будет обсуждено
поведение белков in vitro в присутствии ионов металлов, с кото-
рыми они не обязательно реагируют в природе. Биологическая
функция двойных и других описанных здесь комплексов металлов
с белками не известна, за исключением комплексов иона меди(II)
с альбумином и ионов цинка с инсулином, для которых было по-
стулировано участие в транспорте и хранении соответственно.
Интерес к изучению неспецифических взаимодействий метал-
лов с белками возник исторически в связи с использованием ионов
металлов для очистки белков на основании известных эффектов
ингибирования ионами металлов некоторых ферментов, а также
возможности того, что неспецифические комплексы ионов металлов
с белками послужат моделями металлопротеинов. Учитывая спе-
цифичность окружения иона металла в металлопротеинах [1],
в данной главе, посвященной исследованию неспецифических
взаимодействий ионов металлов с 'белками, будут главным обра-
Комплексы металлов с белками
275
зом обсуждены механизмы денатурации белков ионами металлов-
посредников и ингибирования ферментов, а также 'проблема ис-
пользования ионов металлов в качестве структурных зондов, с
помощью которых может быть определена доступность и реакцион-
ная способность потенциальных лигандных групп белка. Повы-
шенная реакционная способность потенциально способного к коор-
динации атома в белке по сравнению с его реакционной способ-
ностью в модельных системах заставляет предположить наличие
рядом с ним в молекуле белка второго донора электронов, кото-
рого нет в используемых моделях. И наоборот, относительно по-
ниженная реакционная способность потенциального места связы-
вания свидетельствует о стерических затруднениях и (или) кон-
формационных изменениях, сопутствующих связыванию. Для тех
белков, которые, будучи изолированными, находятся в состоянии
с самой низкой свободной энергией, все такие конформационные
изменения будут связаны с положительными вкладами в свобод-
ную энергию связывания, и поэтому будет наблюдаться более низ-
кая кажущаяся константа связывания, чем та, которая была бы
найдена в отсутствие конформационного изменения.
2. РАССМОТРЕНИЕ КОМПЛЕКСОВ МЕТАЛЛОВ С БЕЛКАМИ
И МЕТОДЫ ИХ ИССЛЕДОВАНИЯ
Методы, используемые для изучения взаимодействия металлов
с белками, зависят от специфики системы и информации, которую
хотят получить. Однако главным образом задаются следующими
вопросами: сколько связывающих центров имеется для данного
иона металла, какова их сила и через какие атомы — остова или
боковых .цепей —происходит координация?
Число связывающих центров и их сродство можно определить
прямыми методами, такими, как равновесный диализ [2] или хро-
матографическая гель-фильтрация [3]. Во всех исследованиях
такого типа число связанных ионов металла определяется непо-
средственно анализом концентрации комплекса металла с белком
как функции равновесной концентрации свободного иона металла.
При изучении связывания с металлом особенно важны значения
pH как для получения достоверных данных, так и для их интер-
претации (разд. 3), и в зависимости от цели исследования значе-
ния pH могут поддерживаться постоянными или изменяться. По
возможности следует избегать большинства буферов из-за конку-
ренции между белком и буфером за металл, а также из-за воз-
можности образования тройного комплекса, включающего металл,
белок и буфер [4]. Со способами обработки первичных данных
с целью определения числа различных центров связывания и оцен-
ки их сродства можно ознакомиться в специальных руководствах
[5].
18’
276
Глава 7
Определить число имеющихся связывающих центров и оценить
их относительное сродство, кроме того, можно непрямыми мето-
дами исследования, в которых за связыванием наблюдают по из-
менению некоторого свойства системы при координации металла
с белком. Такие методы, из которых наиболее типичными являют-
ся потенциометрическое титрование (разд. 3) и различные спек-
троскопические методы, требуют знания некоторых количествен-
ных соотношений между (Величиной эффекта и степенью связыва-
ния. И преимущества этих методов и их ограничения зависят от
степени различия в свойствах комплексов, образующихся при ко-
ординации металла к различным центрам. Например, таким пу-
тем можно четко идентифицировать слабый связывающий центр,
который можно и не обнаружить с помощью прямого метода ис-
следования; для этого 1необходимО' лишь, чтобы свойства этого
центра были достаточно характерными.
К числу прямых методов, используемых для идентификации
атомов или боковых цепей, к которым присоединяются ионы ме-
таллов, относятся рентгеноструктурный анализ (см. исследование
миоглобина) и ядерный магнитный резонанс (см. изучение ион
металла — РНаза, разд. 4.6). Кроме того, для этой цели использу-
ются различные непрямые методы. К числу последних относятся:
исследование влияния модификации боковой цепи специфического
белка на связывание с ‘металлом, титриметрические определения
значений рКа. участвующих в координации лигандных групп
(разд. 3), и, по мере 1возможности, корреляция спектров комплек-
са иона металла с белками со спектрами подходящих модельных
систем. Хотя в случае металлопротеинов это последнее и риско-
ванно, оно оказалось особенно плодотворным для изучения неопе-
цифических комплексов ионов металлов с белками. Исследование
влияния модификации белка на связывание с металлом несомнен-
но играет полезную роль, однако при этом необходимо учитывать
возможность того, что модификация боковой цепи, которая ранее
не участвовала в связывании, может повлиять на него ib результа-
те изменения либо общего заряда на белке, либо его конфор-
мации.
3. pH И ДРУГИЕ ФАКТОРЫ, ВЛИЯЮЩИЕ
НА СРОДСТВО ИОНА МЕТАЛЛА К ЛИГАНДУ
Потенциальными местами присоединения ионов металлов к
белку являются главным образом способные к ионизации боковые
цепи и группы —NH и —С=О остова пептидной цепи. В неко-
торых особых случаях нельзя не учитывать вклада в связывание
за счет координации металла с гетероатомами боковых цепей се-
рина, треонина, цистина и метионина (в цитохроме с одним из ато-
Комплексы металлов с белками
277
мов, связывающих железо, ио-видимому, является атом серы
метионина), хотя участие этих групп в образовании неспецифи-
ческих комплексов экспериментально еще не было продемонстри-
ровано. В рамках этой широкой проблемы вероятность того, что
данный ион металла 'будет -взаимодействовать с данным центром,
определяется, помимо конформации белка, еще рядом других
факторов. Главные из них—относительное внутреннее сродство
иона металла к различным способным к координации атомам и
pH. Роль первого фактора подробно обсуждена в работе [6]. Ис-
следование взаимодействия между ионом Н+ и ионами металлов
при связывании с белками имеет непосредственное отношение как
к -проблеме доступности для ионов металлов различных мест свя-
зывания при различных значениях pH, так и к интерпретации кри-
вых титрования комплексов металлов с белками. Поэтому имеет
смысл суммировать главные аспекты этих взаимодействий.
Предположим, что L~ — связывающий центр, который может
реагировать или с одним ионом Н+, или с ионом металла М2+.
Собственные константы ассоциации этого центра с ионом Н+ и
с ионом металла соответственно определяются следующими вы-
ражениями:
L- + H+ = LH Kh = (L-^h+) (О
(LM+)
L" + М2+ = LM+ Km = (11)(МЦ' (2)
Для макромолекулы, содержащей п таких независимых цент-
ров, упомянутые выше равновесия с Н+ и М2+ в наиболее общем
виде можно переписать так:
vh e+2“z
(3>
- +*»z (Zm)
где Vh и vm — число центров, занятых ионами Н+ и М2+ соответ-
ственно, а Кь и Кт — внутреннее сродство каждого из этих цент-
ров к ионам Н+ и М2+ соответственно. Члены e+2coZ и e+2coZ(Zm)—
факторы электростатического взаимодействия, представляющие
взаимодействие зарядов на макромолекуле [7], где Z — остаточ-
ный заряд белка, отнесенный к иону металла, и <в — электроста-
тический фактор, который обратно пропорционален ионной силе
и размеру молекулы. Комбинируя соответствующим образом урав-
нения (1) и (2) или уравнения (3) и (4), можно определить ка-
жущуюся зависимую от pH константу Кт, которая выражает от-
278
Глава 7
ношение числа связывающих центров, присоединивших металл,
к числу центров, не присоединивших металла:
—2<oZ (ZA
(LM+)_________Кт_________Кте 1 т’
т - (L- + LH)J(M2+) - 1 + Kh (Н+) - ! + Kh (Н+) е~2coZ
Из уравнения (5) следует несколько важных заключений. Во-
первых, кажущееся сродство к иону металла будет меньше, чем
внутреннее сродство, до тех пор, пока pH приблизительно на
2 единицы или больше выше значения рКа атома, координирующе-
го металл. Это приводит к тому, что в случае связывающих
центров с очень высокими значениями рКа, таких, как боковые
цепи аргинина, вклады от их участия в связывании при нейтраль-
ных значениях pH будут очень незначительными. Во-вторых, для
связывающего центра, содержащего единственную ионизируемую
труппу, изменение кажущегося сродства иона металла при изме-
нении pH можно использовать для определения значения рКа
этой группы. И, наконец, следует ожидать, что для белков, кото-
рые содержат несколько потенциальных связывающих центров,
обладающих различным сродством к иону Н+ и иону металла, от-
носительное распределение ионов металлов по этим центрам будет
зависеть от pH. Например, внутреннее сродство карбоксильных
групп к иону Н+ и иону Си(II) соответственно составляет 104-5 и
IO1-8; внутреннее сродство имидазольных боковых цепей к иону
Н+ и иону Си(II) составляет 107 и 104. Рассчитанное по уравне-
нию (5) при pH 4 кажущееся сродство карбоксильных групп к
Си(II) немного больше, чем сродство имидазола к Си(II), в то
время как при pH 7 наблюдается обратное. Таким образом, мож-
но ожидать, что при низких значениях pH ионы Си(II) будут
главным образом связаны с карбоксильными группами, а по мере
повышения pH до нейтрального значения ионы Си(II) будут пе-
ремещаться к имидазольным группам, как это и наблюдали в дей-
ствительности [4].
Второе следствие, вытекающее из конкуренции между ионами
Н+ и ионами металла, состоит в том, что связывание металла
центром, который в отсутствие металла в нормальном состоянии
протонирован при тех значениях pH, при которых происходит свя-
зывание, сопровождается замещением протона; при значениях pH,
гораздо более высоких, чем рКа, протоны не замещаются (за ис-
ключением некоторых примеров незначительного замещения, обус-
ловленного изменением общего заряда белка, сопровождающимся
электростатическим влиянием на кислотность). Легко показать,
что для любой одиночной координирующей группы отличие в свя-
зывании иона Н+ незакомплексованной группой по сравнению с
полностью закомплексованной определяется ее кривой титрова-
ния. Этот факт имеет практическое следствие, заключающееся в
Комплексы металлов с белками
279
том, что в связанном состоянии, в котором связывающие центры
не изменяются с изменением pH, можно определить рКа коорди-
нирующей группы (и, следовательно, ее идентифицировать), изме-
ряя уменьшение в замещении протонов как функцию pH. Однако
в тех случаях, когда место связывания изменяется при изменении
pH, титриметрические соотношения гораздо более сложные. На-
пример, изменения при повышении pH в сторону более высоких
значений рКа (относительно pH) будут приводить к увеличению
в замещении протонов как функции pH. Такие случаи будут ил-
люстрироваться специальными примерами. Характерные примеры
констант ассоциации ионов Н+, Cu(II) и Zn(II) с типичными
координирующими группами белков приведены в табл. 7.1.
Таблица 7.t
Ионизирующиеся группы белков и их «нормальные» константы ассоциации
с Н+, Cu(II) и Zn(II) при 25°О
Группа IgKft igKm
Cu(II) Zn(Il)
а-СООН 3,1—3,5 — —
Р, у-СООН (Asp, Glu) 4,4—4,7 1,8 (ацетат) [55J 1,0 (ацетат) [55]
Имидазол (His) 6,4—7,2 3,1—4,4 [10] 2,5 (имидазол) [55]
a-NH2 7,8—8 4,9—5,5 [9]
e-NH2 (Lys) 9,6—10,5 4,3 (NH3) [55] 2,6 (NHS) [55]
Фенольная ОН (Туг) 9,6—9,8 — —
Сульфгидрильная (Cys) 9 — 7 [6]
Гуанидиновая (Arg) >12 — —
а3начения 1g представляют нормальные значения, найденные для белков. Значения
lg Km определены для модельного комплекса состава 1:1, указанного в скобках и (или) в-
работе, на которую дана ссылка.
4. ВЫБРАННЫЕ СИСТЕМЫ
Неспецифические взаимодействия ионов металлов с белками
изучены главным образом на примере Cu(II) и Zn(II), которым
и будет уделено здесь основное внимание. Свойства Си(II) и
Zn(II), которые представляют особый интерес при рассмотрении
их взаимодействий с белками, можно суммировать следующим;
280
Глава 7
образом: 1) комплексы Zn(II) в основном тетраэдрические или
октаэдрические, в то время как комплексы Си(II) квадратно-пло-
скостные или искаженные октаэдрические; 2) координирующиеся
атомы по способности связываться и с Cu(II) и с Zn(II) распо-
лагаются 'В ряд S>N>0, но при этом каждый из этих атомов
взаимодействует с Cu(II) примерно в 10'раз сильнее, 4eMcZn(II);
3) ассоциация Cu(II) [но это не характерно для Zn(II)] с не-
большими пептидами характеризуется тенденцией к связыванию
с атомами азота пептидной связи с одновременным замещением
пептидного протона [8]. При высоких значениях pH эта тенден-
ция проявляется в образовании комплекса биуретового типа,
в котором Си(II) координирована только с атомами азота пеп-
тидной связи. При значениях pH от 5 до 9 она проявляется -в ко-
ординации Cu(II) с а-МН2-группами и (или) с внутренней цепью
имидазола и следующим соседним атомам азота пептидной свя-
зи, как в комплексах состава 1 : 1 Си(II) с Gly-GIy-Gly-GIy-Gly и
ацетил-GIy-Gly-GIy-His соответственно [9, 10]. Таким образом,
Cu(II) координируется с а-Ь1Н2-группами Gly-GIy-GIy-Gly-GIy и
замещает протоны трех соседних пептидных связей; соответствую-
щие кажущиеся значения р/С равны 5,85, 6,95 и 8,1. С ацетил-GIy-
Gly-GIy-His Си(II) координируется по атому Ni имидазола и за-
мещает протоны пептидных связей; кажущиеся значения р/Са рав-
ны 6,5, 7,35 и 8,8.
Следует также отметить, что имеется зависимость между силой
поля атомов*, связанных с Си(II), и положением главной полосы
поглощения в видимой области для комплексов Си(II) [8]. Эти
полосы сдвигаются в более коротковолновую область по мере то-
го, как увеличивается число доноров электронов, создающих
«сильное поле», сдвигаясь от выше чем 700 нм для комплексов,
подобных комплексу состава 1:1с имидазолом, до 515 нм для
комплексов, в которых донорами электронов являются a-NH2-
группа и три атома азота пептидных связей. В общем координи-
рующиеся группы в соответствии с силой поля лигандов можно
классифицировать следующим образом: NH2>N(пептидной свя-
зи) >имидазол> кислород.
4.1. Ионы цинка и сывороточный альбумин
Сывороточный альбумин — белок с молекулярной массой при-
близительно 69 000, который содержит (наряду с полным набором
других аминокислот) 16 остатков гистидина. Проведенное ранее
методом равновесного диализа исследование связывания с Zn(II)
и влияние этого связывания на равновесие сывороточного альбу-
* Речь идет, по-видимому, о «силе поля лигандов», создаваемой данными
атомами. — Прим. ред.
Комплексы металлов с белками
281
мина с ионом Н+ было вызвано использованием Zn(II) для разде-
ления белков крови и привело к заключению, что основной способ,
связывания Zn(II) при pH от 5,5 до 7,5—взаимодействие по ти-
пу 1 : 1 между ионом Zn(II) и каждой из 16-ти имидазольных бо-
ковых цепей [2]. Были получены следующие доводы в подтверж-
дение такого типа взаимодействия: 1) значительное понижение
pH, которым сопровождается присоединение Zn(Il), показывает,
что Zn(II) связывается с центрами, которые при pH 5,5 обычно,
протонированы. Так как карбоксильные группы и остатки лизина
можно исключить из числа наиболее вероятных мест связывания
(карбоксильные группы имеют слишком низкие значения р/Са>.
а гуанидирование лизина не влияет на связывание), наиболее ве-
роятными местами связывания остаются имидазольные группы;
2) равновесные значения vm и (Н+) показывают, что число моле-
кул имидазола (п—vm), участвующих в равновесии с ионами Н+
по уравнению (3), уменьшается на единицу на каждый моль свя-
занного Zn(II). И наконец, предположение о взаимодействии
Zn(II) с имидазолом по типу 1 : 1 приводит к хорошему согласию
между значениями Кт, рассчитанными по уравнениям (3) и (4),
и известной первой константой ассоциации Zn(II) с имидазолом
при тех же условиях.
Рао и Лал [11, 12] усомнились в справедливости этих выводов,
отметив, что первоначальное значение Кт получено исходя из
ошибочного значения Kh и что исправленное среднее значение Кт
гораздо выше для взаимодействия 1 : 1 Zn(II) с имидазолом. Бо-
лее того, их собственные исследования с применением электрофо-
реза и полярографии [11, 12] показали, что приблизительно в
двух местах Zn(II) связывается сильнее, чем в остальных, и что
величины сродства только более слабых связывающих центров
совместимы с образованием комплекса Zn(II)—имидазол соста-
ва 1:1. На основе наблюдаемого выделения одного протона при
связывании каждого из двух первых ионов Zn(II) при pH 6,6 эти
авторы предположили, что каждый из двух наиболее сильных
центров связывания Zn(II) состоит из имидазольной группы и
карбоксильной группы боковой цепи или из имидазольной группы
и карбонильного кислорода пептидной связи. Здесь уместно за-
метить, что эти два наиболее сильных центра связывания Zn(II),
по-видимому, не идентичны двум наиболее сильным центрам свя-
зывания Си(II) (разд. 4.5), так как вытеснение протона Си(II)
и Zn(II) из этих наиболее сильных центров связывания протекает
по-разному [12] и высокая концентрация Zn(II) не блокирует
взаимодействие Си(II) с местом, где она прочнее всего связыва-
ется.
Другие аспекты взаимодействия Zn(II) с сывороточным аль-
бумином также не совсем понятны. Главный из них — спорное вли-
282
Глава 7
яние этерификации карбоксильной группы на связывание с Zn(II)
[12, 13] и влияние температуры. Инкубация сывороточного мер-
каптальбумина при 37 °C приводит к увеличению числа связываю-
щих центров, при этом их число превышает число имидазольных
групп [14]. Новые центры не были идентифицированы.
4.2. Взаимодействие цинка с инсулином
Гормон инсулин выделяют из веществ биологического проис-
хождения в виде комплекса с Zn(II). Этот комплекс можно пере-
кристаллизовать, в нем содержится как минимум два атома Zn(II)
на элементарную ячейку в 36000 дальтон [15]. Инсулин, не со-
держащий цинка, биологически активен, и, по-видимому, повтор-
ная ассоциация с Zn(II) in vivo не обязательна для его биологи-
ческой активности.
Мономер инсулина (молекулярная масса 6000) содержит две
неодинаковые пептидные цепи А и В, связанные дисульфидными
мостиками; каждая молекула мономера содержит два остатка
гистидина в положениях Bg и Вю соответственно. В растворе не
содержащий Zn(II) инсулин существует в виде смеси частиц с
разными молекулярными массами, из которых главной, по-види-
мому, является димер [16]. Добавление 0,7 атома Zn(II) на моле-
кулу димера приводит к монодисперсному гексамеру [16], что
находится в согласии с кристаллической структурой. Хотя с мо-
лекулой гексамера может быть связано больше двух атомов
Zn(II), первые два атома Zn(II) являются особыми в том смыс-
ле, что они представляют тот минимум, который необходим для
кристаллизации, и их нельзя удалить диализом [17].
Проведенное ранее Тэнфордом и Эпштейном [18] титрование
инсулина и комплекса Zn(Il) с инсулином показало, что ком-
плекс Zn(II) — инсулин при титровании ведет себя двояко — кри-
вая «прямого» титрования будет обратимой при значениях pH от
3,5 до 8, а метастабильная кривая «обратного» титрования полу-
чается после выдерживания при значениях pH ниже 2 или вы-
ше 11, как это показано на рис. 7.1. Кривая «прямого» титрования
представляет титрование кристаллического вещества и имеет не-
посредственное отношение к настоящей дискуссии. Было показа-
но, что из ее рассмотрения следуют два очевидных вывода [18].
Во-первых, в комплексе Zn(II) — инсулин по сравнению с инсу-
лином между pH 8 и 12 титруются дополнительно два протона на
молекулу димера; это было приписано диссоциации воды, нахо-
дящейся в координационной сфере связанного Zn(II). Во-вторых,
две боковые цепи в молекуле димера, которые в инсулине титру-
ются около pH 6,5 (идентифицированные как имидазольные),
в присутствии Zn(II) депротонируются при pH 4. Наиболее прямо
это можно наблюдать, исследуя различие в связывании Н+ инсу-
Комплексы металлов с белками
283-
лином и кристаллическим комплексом Zn(II) —инсулин в интер-
вале pH от 4,5 до 7,5. При pH 4,5 связывание Zn(II) приводит
к освобождению двух протонов из инсулина; число освобождае-
Рис. 7.1. Кривая титрования ионами водорода комплекса цинка с инсулином при
25 °C и ионной силе 0,075 [18].
О «прямое» титрование; J) «обратное» титрование, кислотное; ; J «обратное» титрование,
основное; — ——кривая титрования инсулина в отсутствие цинка. Области частичной не-
растворимости на кривых «прямого» и «обратного» титрований показаны стрелками ниже
и выше соответственно.
мых протонов уменьшается до нуля в интервале pH 5,5—7,5, это
соответствует области титрования имидазола в отсутствие цинка.
Содержание Zn(II) составляет 0,7 атома на молекулу димера; на
284
Глава 7
этом основании можно заключить, что каждый атом Zn(II) свя-
зан с тремя остатками имидазола и тремя молекулами воды [19].
(Уместно отметить, что первоначальное заключение — два имид-
азола на один атом цинка — основывалось на ошибочном опре-
делении содержания цинка, которое предполагалось равным одно-
му атому Zn на молекулу димера.)
Марккер с сотр. [20, 21] выразили сомнение в том, что цинк
координируется с имидазольными группами, и предложили вме-
сто этого гексамерную модель, в которой каждые два атома Zn(II)
связаны с а-МН2-группами из В-цепи каждого из трех димеров.
Их модель основывалась на имеющихся рентгеноструктурных
данных, согласно которым тредполагали, что каждая элементар-
ная ячейка содержит три молекулы димера, имеющие ось враще-
ния третьего порядка [21], и на химических данных, которые по-
казали, что модификация a-NH2-rpynn В-цепей сохраняет 'молеку-
лярную массу постоянной при связывании Zn(II) [20]. Однако
Брилл и Венейбл [19], впоследствии повторно анализируя дан-
ные прямого титрования Тэнфорда, указали, что а-МН2-группы не
могут быть центрами связывания Zn(II), так как при pH 7,3 (при
этом значении pH а-МН2-группы в отсутствие цинка ib значитель-
ной степени протонированы) не меньше протонов связывается
кристаллическим комплексом Zn(II)—инсулин, чем самим инсу-
лином (рис. 7.1). Кроме того, они показали, что кривая прямого
титрования комплекса Zn(II)—инсулин удовлетворительно соот-
ветствовала модели, в которой каждый из двух атомов цинка в
гексамере связан с тремя имидазольными группами, причем кон-
станта связывания второго атома цинка (1013) была больше, чем
константа связывания первого (109).
Последние рентгеноструктурные данные, полученные при раз-
решении 2,8 А [22], хорошо согласуются с химическими данными.
Эти данные показали, что каждая элементарная ячейка комплек-
са Zn(II) —инсулин содержит три димера инсулина, которые рас-
положены в виде шестимерной структуры вокруг оси симметрии
третьего порядка и связаны двумя ионами цинка. Каждый ион
Zn(II) связан с тремя Вю-имидазольными группами (по одной от
каждого димера) и еще с тремя дополнительными группами, пред-
варительно идентифицированными как молекулы воды; располо-
жение координирующих атомов не создает правильного октаэдра.
Теперь стала также понятна причина, по которой а-МН2-группы
В-цепи 'были ошибочно определены как лиганды цинка [20]. По-
видимому, она состоит в том, что в положении 1 В-цепи димеры
гексамера очень тесно контактируют между собой, почти сплетены
вместе; такой контакт был бы затруднен при модификации a-NH2-
групп.
И, наконец, в овете имеющихся рентгеноструктурных данных
полезно вновь рассмотреть полученные константы связывания
Комплексы металлов с белками
285
Zn(II) с инсулином. Кооперативность связывания легко объясня-
ется тем, что уменьшение энтропии, связанное с переводом беспо-
рядочно агрегированных димеров не содержащего Zn(II) инсули-
на в гексамерную конфигурацию, обусловлено связыванием пер-
вого атома Zn(II) [19], второй же атом Zn(II) практически
присоединяется к только что возникшему т/шс-имидазольному
центру. Хотя еще не имеется удовлетворительной модели второго
центра связывания Zn(II), наблюдаемую константу связывания
1013 разумно сопоставить со значением 1010, найденным для свя-
зывания Zn(II) с бис-имидазольным лигандом [23].
4.3. Комплексы иона меди с окситоцином и вазопрессином
Окситоцин и вазопрессин близки по структуре к полипептид-
ным гормонам (рис. 7.2), которые обнаружены в гипоталамусе и
задней доле гипофиза млекопитающих и некоторых низших орга-
12 3
S—Cys—Туг—I leu
I I
S—Cys—Asn—Gin
| 6 5 4
Pro—Leu—Gly-амид
7 e
ОКСИТОЦИН
12 3
S—Cvs—T yr—Phe
I ' I
S—Cys—Asn—G1 n
| 6 6 4
Pro—L ys—G1 у-амад
7 8
лизинпроизводное вазопрессина
Рис. 7.2. Первичные структуры окситоцина и лизинпроизводного вазопрессина.
низмов. В то время как можно спорить по поводу того, относить
ли гормоны к большим пептидам или малым белкам, в их реак-
ционной ‘способности по отношению к Си(II) проявляются особен-
ности, свойственные белкам.
Оба гормона обладают высоким сродством к Си(II) [24, 25],
образуя при pH 8 комплекс розового цвета с единственной поло-
сой поглощения в видимой области при 525 нм. Титрование
Си(П)-комплексов окситоцина (рис. 7.3) и вазопрессина [25] по-
казывает, что связывание иона меди сопровождается выделением
четырех протонов — одного из а-МН2-группы (положение 1) и
трех протонов из других групп, которые при pH 8 обычно прото-
нированы. Участие а-Г4Н2-грулпы в связывании подтверждается
тем, что Си(II) не образует никакого комплекса с дезаминиро-
ванным окситоцином, в котором ia-NH2-rpynna замещена водоро-
дом, а также с N-ацетиллизинвазопрессином, в котором a-NH2-
группа ацетилирована [25]. То, что фенольные гидроксилы не
участвуют в связывании, было доказано тем, что замена тирозина
на фенилаланин никак не влияла на комплексообразование
(рис. 7.3). Три дополнительные группы, депротонируемые Си(II),
были идентифицированы как атомы азота из пептидных связей
286
Глава 7
(или атомы азота амидных групп). Основанием для такой иден-
тификации послужило сходство спектров комплексов гормонов
с Си(II) и комплекса, образуемого Cu(II) с Gly-Gly-Gly-Gly, в ко-
тором Си(II) координируется к a-NHs-rpynne и трем соседним
Рис. 7.3. Непрерывные кривые кислотного титрования окситоцина и 2-феиилала-
нинпроизводного окситоцина (в котором тирозин в положении 2 замещен фенил-
аланином) и их комплексов с Си (И) [24].
А окситоцин; ▲ окситоцин4-СиС1я; О 2-фенилаланинокситоцин: ф 2’фенилаланииокситоцИн+
CuCI2; 25 °C, ионная сила 0.16.
атомам азота пептидных связей [24]. Для комплекса Си (II) с ва-
зопрессином была предложена специфическая структура, вклю-
чающая координацию Cu(II) с a-NH2-rpynnoft и тремя следующи-
ми пептидными связями, точно так же, как в комплексе Си(II)
с Gly-Gly-Gly-Gly [25].
Однако маловероятно, что комплексы Си(П) с гормонами бы-
ли в точности аналогичны комплексам простых линейных пепти-
дов. Кажущиеся значения рК для диссоциации протона из третьей
пептидной связи в комплексах Cu(II)—вазопрессин и Cu(II)—•
окситоцин равны 6,8 и 7,4 соответственно [24, 25] (рис. 7.3). Эти
величины значительно ниже значения 8,1, найденного для ком-
плекса с пентапептидом, таким, как тетратлпцилглипин [9]. Кро-
Комплексы металлов с белками
287
ме того, наклон кривых титрования как комплекса Cu(II) с окси-
тоцином (рис. 7.3), так и комплекса Cu(II) с вазопрессином [25]
при pH примерно 6,5 указывал на кооперативное участие трех
пептидных атомов азота противоположно их ступенчатому вовле-
чению в комплексообразование, наблюдаемому для линейных пеп-
тидов [9]. Принцип «все или ничего» в комплексах с гормонами —
это факт, установленный на основании спектральных данных,
.которые показывают, что частицы, образующиеся при всех значе-
ниях pH выше 6, спектрально почти идентичны конечному ком-
плексу [24, 25]. Возможное объяснение этих данных состоит в
том, что в кольцевую структуру этих гормонов входит один и
более пептидный (или амидный) атом азота, который по отноше-
нию к a-NH2-rpynne расположен благоприятнее, чем в линейных
пептидах. Такое влияние конформации означает, что атомы азота
пептидных связей, участвующие в координации вместе с a-NH2-
грулпой, не обязательно должны быть смежными с ней. Так, не-
давно выполненные ЯМР-исследования показали, что комплекс
•Си(П) с окситоцином — трансаннулярный, в котором в координа-
ции с Cu(II) участвуют a-NH2-rpynna и атомы азота пептидных
связей из положения 6 цистеина, из положения 2 тирозина и из
положения 5 аспарагина [26].
4.4. Взаимодействие метмиоглобина кашалота
с ионами меди и цинка
Метмиоглобин (метМЬ) кашалота был первым белком, трех-
мерная структура которого была установлена методом рентгено-
структурного анализа [27]. Вследствие этого он вскоре стал мо-
делью для интерпретации взаимодействий металлов с белками.
Несколько аспектов химии миоглобина непосредственно касаются
его взаимодействия с ионами металлов.
Единственная полипептидная цепь метМЬ (рис. 7.4) представ-
ляет высокоупорядоченную структуру с молекулярной массой
17 000. содержащую 75% a-спирали, которая легко и обратимо
раскручивается под действием ионов Н+ при pH около 4,5. Связь
с гемом осуществляется посредством координации железа к гисти-
дину (Fg), ионного взаимодействия между карбоксильными груп-
пами боковых цепей гема и положительно заряженными группа-
ми белка, а также посредством неполярного взаимодействия меж-
ду п'ротопорфириновым кольцом гема и неполярными боковыми
цепями аминокислот. Денатурация сопровождается изменением
спектра гема и ориентацией в сторону растворителя 4—6 гистиди-
новых боковых цепей, которые в нативном состоянии нереакцион-
носпособны ни по отношению к ионам Н+, ни к карбоксиметили-
рованию [28].
Совершенно очевидно, что метмиоглобин претерпевает обра-
тимую денатурацию в присутствии ионов Cu(II) и Zn(II); это
288
Глава 7
6
Комплексы металлов с белками
289
явствует из изменения в спектре гема [29], из выраженной нерас-
творимости комплекса иона металла с белком [29], из размаски-
ровки имидазольных остатков [29] и, наконец, из уменьшения со-
держания а-спирали ИЗО]. Движущей силой денатурации, по-ви-
димому, является то, что денатурированная форма имеет более
высокое ородство к металлу, чем нативная 'Структура [29]. Одна-
ко денатурация проявляется только при значениях vm выше 1
[29] и первый ион металла, по-видимому, связывается с центром
нативной структуры [29, 31].
Места присоединения первых ионов Си(II) и Zn(II) к натив-
ной структуре при pH 6 'были определены 'кристаллографически
[31]. Интересно, что хотя связывание Си (II) и Zn(II) конкуриру-
ющее [21], первые ионы Cu(II) и Zn(II) присоединяются к час-
тично совпадающим, но не идентичным центрам (рис. 7.4). Так,
очевидно, что когда Zn(II) присоединяется к имидазольному ос-
татку His-GHi, он находится очень близко (но не прямо связы-
вается) с Lys-Аи и с Asn-GH4. Си(II) же связывается с His-Аю и
при этом соседствует с Lys-Ац и Asn-GH4, причем различия меж-
ду этими двумя металлами главным образом обусловлены их раз-
личными стереохимическими требованиями. Несмотря на то что
в отношении координации с иными группами, чем имидазольные,
результаты рентгеноструктурных исследований не однозначны,
данные о связывании [29] заставляют предположить участие в
связывании [по крайней мере с Си (II)] других доноров электро-
нов. Данные о связывании при pH 6,8 можно интерпретировать
следующим образом: значение Кт для первого иона Си(II) равно
2,5-105, в то время как внутреннее сродство Си(II) к имидазолу
равно только 104. Маловероятно, чтобы вся дополнительная стаби-
лизация возникала только за счет карбонильных атомов кислоро-
да аспарагина, к тому же результаты титрования (рис. 7.5) неод-
нозначны в отношении участия в связывании лизина. Эта неод-
нозначность еще больше усиливается, если учесть, что центры
связывания первых ионов Си(II) при pH выше 7 изменяются, что
следует из увеличения вытеснения протонов первыми ионами
Си(II) и также из данных ЭПР [32].
Изучение титрования особенно полезно для выяснения природы
мест связывания Си(II) в денатурированном состоянии [29]. Вы-
теснение протонов Си(II) из денатурированного кислотой метмио-
глобина показано на рис. 7.5. Согласно этим данным, при pH 5
Рис. 7.4. (а) Изображение молекулы миоглобина н виде диаграммы по Дикер-
сону [56] и (б) увеличенное изображение части молекулы миоглобина, ограни-
ченной прямоугольником иа рис. а, показывающее отношения между ионами
Cu(II) и Zn(I'I) и потенциальными связывающими группами [31].
19—2451
290
Глава 7
РН
Рис. 7.5. Число вытесненных протонов (Ava) на ион связанной Си(II) как функ-
ция pH при различных значениях vm (где — число молей связанных ионов
меди на 1 моль белка) [29].
Для нативного метМЬ приведено суммарное число вытесненных протонов, наблюдаемое при
добавлении Cu(II) к нативному белку. Для денатурированного метМЬ приведено число про-
тонов, вытесненных Cu(II) из денатурированного кислотой метМЬ. Условия: 25 °C, ионная
сила 0,16.
на каждые из четырех связанных ионов Си(II) выделяются два
протона, а при pH 7 — только один протон. Таким образом, две
группы, которые в нормальном состоянии при pH 5 протонирова-
ны, связываются с каждым первым из четырех ионов Си(П); одна
из этих групп титруется при pH 5—-7 и ее, следовательно, можно
идентифицировать как имидазол. Это заключение подтвержда-
ется уменьшением связывания Си(II) при модификации молекулы
гистидина [33]. Идентификация второй группы, депротонируемой
Си(II) при pH 5, будет обсуждаться ниже. Здесь уместно отме-
тить, что исследования титрования и связывания показывают, что
места связывания большей части ионов Cu(II) на метмиоглобине
Комплексы металлов с белками
291
и до и после кислотной денатурации остаются одними и теми же.
Таким образом, вытеснение меньшего числа протонов Си(II) из
нативного метМЬ при pH 5 (рис. 7.5) можно объяснить присоеди-
нением иона Си(II) к двум группам, которые в нормальном со-
стоянии при pH 5 протонированы, а также обычным поглощением
протона, сопровождающим размаскировку при денатурации ос-
новного имидазола [29].
Вторая группа, из которой Cu(II) вытесянет протон при pH 5—
6, по-видимому, представляет собой атом азота пептидной связи,
хотя кажущееся значение рК диссоциации этой группы в комплек-
сах метМЬ ниже, чем в модельных комплексах [34]. Лизины мож-
но не считать источником протонов, так как их гуанидирование
не влияет на связывание с Си(II) [29]. Участие в 'связывании
атомов азота пептидных связей особенно подтверждается спектра-
ми поглощения в видимой области комплексов Си(II) с апомио-
глобином, которые показывают, что при pH 5,5 вместе с каждым
имидазольным остатком в связывании должен участвовать по
крайней мере еще один лиганд сильного поля [35]. Кроме того,
и данные титрования (рис. 7.5), и спектральные данные [35] по-
казывают, что число пептидных связей, участвующих в связыва-
нии с Си(II), увеличивается по мере того, как pH повышается
выше 8. Однако следует отметить, что по крайней мере при pH 11
одно из первых четырех мест связывания Си(II), по-видимому,
должно содержать концевую a-NH2-rpynny [34].
Возможное связывание Си(II) с имидазолом и атомом азота
пептидной связи при нейтральных значениях pH предполагает еди-
ный механизм для индуцируемой Си(II) денатурации метМЬ, так
как было показано, что структурные требования a-спирали несов-
местимы с таковыми в аналогичных комплексах Си (II) [36]. Од-
нако Zn(II), который, как известно, мало склонен к координации
с атомами азота пептидных связей, также денатурирует метМЬ,
вызывая изменения и в спектрах гема [37], и в содержании а-спи-
рали [30]. Кроме того, исследование модифицированного миогло-
бина предполагает, что изменения в спектрах гема и уменьшение
содержания a-спирали не обязательно должны быть связаны [33].
В этом отношении уместно отметить, что связывание ионов Си(II)
и Zn(II) с имидазолом гемового звена Fs считается критической
стадией ib денатурации миоглобина [33, 37].
Не решены также другие аспекты взаимодействия металлов:
с миоглобином. Среди них следует назвать взаимозависимость
между очевидным максимальным числом центров связывания при
нейтральных значениях pH (приблизительно 7 как для меди, так
и для цинка) и присутствием 12 гистидиновых остатков в белке.
Таким образом, некоторые имидазольные группы, по-видимому,
недоступны для реакции с ионом металла, несмотря на то, что
белок денатурируется, или по крайней мере они очень мало реак-
292
Глава 7
ционноспособны по сравнению с другими. В этом примере такое
подразделение по реакционной способности на два класса, воз-
можно, отражает локальную последовательность аминокислот
[38].
4.5. Комплексы ионов меди с сывороточным альбумином
Оптические свойства Си (II) позволяют получить более точную
картину взаимодействия сывороточного альбумина с Си(II), чем
та, которая получена для его взаимодействия с Zn(II), включая
и явно сложную картину основного центра связывания Си(II).
'Вскоре после начала исследований взаимодействия Си(II) с
сывороточным альбумином [4, 39] стало ясно, что первые ионы
Си(II) связываются более прочно, чем последующие [40]. Пред-
положение о том, что в состав сильного центра, связывающего
Си(II), входит а-МН2-группа, было подтверждено работой, пока-
завшей, что один эквивалент Си(II) блокирует реакцию a-NH2-
групп с 2,4-динитрофторбензолом [41]. Выполненные позднее тит-
рометрические исследования показали, что одна координирующая
группа наиболее сильного связывающего центра имеет рКа при-
близительно 8 [35]. Кроме того, тот факт, что комплекс Си(П)
с сывороточным альбумином состава 1 : 1 имеет полосу поглоще-
ния при 525 нм, а также то, что первый ион Си(II) при pH 9 вы-
тесняет два протона, показывают, что наряду с а-МН2-группой
в состав сильного центра связывания входят еще два атома азота
пептидных связей и, возможно, еще какой-то четвертый координи-
рующий атом. Расположение сильного центра связывания Си (II)
среди первых 24 аминокислотных остатков и расшифровка их по-
следовательности на N-конце как Asp-Thr-His-Lys позволила иден-
тифицировать четвертую лигандную группу как имидазол [42].
На этом основании предположили, что, так же как в спектрально
идентичном .комплексе Си(II) с Gly-Gly-His, в сильном связыва-
нии Си(II) с сывороточным альбумином участвуют также имид-
азольная группа His-3, a-NH2-rpynna и два расположенных между
ними атома пептидных связей. Предложенная модель сильного
центра связывания Си(II) в сывороточном альбумине показана на
рис. 7.6. Корректность отнесения координирующих групп, по-ви-
димому, подтверждается тем, что первый связанный нон Си(II)
защищает при алкилировании и a-NH2-rpynny, и His-3 [43], а так-
же тем, что комплекс Си(II) с сывороточным альбумином состава
1 : 1 имеет такие же оптические и вращательные свойства, как
комплекс с пептидом Asp-Thr-His-Lys [43]. Нет никаких данных,
подтверждающих участие в комплексообразовании е-МН2-группы
Lys-4.
Меньше известно о связывании последующих ионов Си (II)
с сывороточным альбумином при нейтральных значениях pH. При
Комплексы металлов с белками
293
pH выше 10 все связанные ионы Си(II) проявляют свойства, со-
гласующиеся с участием атомов азота нескольких пептидных свя-
зей [35]. Однако титриметрические и спектральные изменения,
ассоциируемые с координацией второго, третьего и четвертого
ионов Си (II), подтверждая участие в координации имидазольных
Рис. 7.6. Модель связывающего центра для первого иона Cu(II) в бычьем сыво-
роточном альбумине: концевая последовательность Asp-Thr-His показана в кон-
формации, предложенной для комплекса с Си(II), но ион металла не пока-
зан [42].
групп, в то же время предполагают, что при pH 8—9 в координа-
ции принимают участие меньше атомов азота из пептидных свя-
зей, чем это найдено в сравнимых условиях для модельного гисти-
динсодержащего пептида [35]. Эти данные заставляют предполо-
жить, что в сывороточном альбумине либо затруднен доступ к
центру, состоящему из атомов азота пептидной связи, либо имеют-
ся вторичные хелатные центры, имеющие промежуточную устой-
чивость между устойчивостью самого сильного центра и центра
в состав которого входят гистидин и атом азота пептидной связи.
Данные, подтверждающие состав второго связывающего центра,
аюлучены исследованием методом электрофореза [12].
Особенно интересные данные относительно одного из более
слабых центров связывания Си(II) получили Клотц и сотр. [44,
45] в присутствии четырех или более эквивалентов Си(II). Этот
центр характеризуется сильным максимумом поглощения в обла-
294
Глава 7
сти 375 нм, его .интенсивность со временем уменьшается. Образо-
вание этого центра 'блокируют реагенты, взаимодействующие с
сульфгидрильными группами, а реагенты, взаимодействующие с
дисульфидными связями, способствуют его исчезновению. Предло-
жена структура этого центра, в котором Си(II) связана с сульф-
гидрильной группой и находится очень близко к дисульфидной
связи.
4.6. Взаимодействие ионов металлов с рибонуклеазой
Панкреатическая рибонуклеаза (РНаза) катализирует гидро-
лиз рибонуклеиновой кислоты, расщепляя 3',5'-фосфодиэфирные
связи, соединяющие соседние нуклеотиды. Предполагают, что
реакция протекает через образование промежуточного 2',3'-цикли-
ческого нуклеозидфосфата, который затем гидролизуется до
З'-фосфата. И 2'-, и З'-пиримидиннуклеотиды являются потенци-
альными ингибиторами фермента.
Структура РНазы образована одной полипептидной цепью
с молекулярной массой 13 683, в состав которой входят четыре
дисульфидные связи, способствующие стабилизации нативной
конформации. На основании химических рентгеноструктурных
данных было показано, что из четырех остатков гистидина два —
His-12 и His-119 — особенно пространственно сближены и необхо-
димы для проявления ферментативной активности. Каталитическую
роль выполняет, по-видимому, также остаток Lys-41.
Активность РНазы ингибируется ионами и Си(П) и Zn(II)
[46]. Росс, Матиас и Рабин [47] показали, что связывание одного
только Zn(II) оказывает ингибирующее, но все же относительно
слабое влияние. Однако в присутствии цитидин-З'-фосфата
(З'-ЦМФ) ингибирование наблюдалось при очень низкой концент-
рации Zn(II) и было приписано образованию сильного «тройного»
комплекса между Zn(II), РНазой и З'-ЦМФ. Для иона Cu(II)
вначале предположили, что ингибирование обусловлено хелатиро-
ванием одного иона Си(П) между His-12 и His-119 [48]. Однако
исследования последних лет показали, что Си (II) также образует
«тройной» комплекс с РНазой и З'-ЦМФ [49] (см. ниже) и что
связывание Си(II) в отсутствие нуклеотидов более сложное, чем
ранее предполагали. На основании результатов нескольких иссле-
дований можно сделать следующие заключения о взаимодействии
Си(II) с РНазой в отсутствие нуклеотидов.
1. В интервале pH 5—7 РНаза имеет пять центров с близким
сродством к Си(П).
Наличие большого числа центров с близким сродством впервые
было предположено на основании спектрофотометрического и по-
тенциометрического исследований [49], которые показали, что
при условии практически полного связывания добавление каждых
Комплексы металлов с белками
295
четырех молярных эквивалентов Си(II) к РНазе вызывает практи-
чески одинаковые изменения в спектре поглощения в видимой об-
ласти (рис. 7.7) и протонном равновесии. Методом гель-фильтра-
ции было обнаружено пять наиболее важных центров, сродство
которых к Си (II) резко возрастает между pH 5,5 и 7. И при
pH 5,5, и при pH 7 сродство одного центра к свободному иону меди
w
Л, нм
о
СО
С
Рис. 7.7. Спектры РНазы в присутствии СиС12 [49].
Концентрация белка 3—4%; ионная сила 0,16. А—молярное отношение СиСЬ к РНазе рав-
но 1; Б — молярное отношение CuCI2 к РНазе равно 3. Приведены молярные коэффициенты
поглощения на 1 моль РНазы.
приблизительно в 10 раз больше, чем каждого другого из четырех
[3, 52]. Исследуя скорость релаксации протонов при pH 5,1, уста-
новили, что из трех центров, связывающих Си(II), один центр
в 11 раз сильнее, чем два оставшихся [50].
2. Три центра содержат имидазольные боковые цепи, а один из
них содержит a-NН^-группу; атомы азота пептидных связей, входят
в состав всех четырех центров, причем степень их участия в связы-
вании увеличивается при повышении pH.
При pH 7 наиболее сильным центром является а-МНг-группа,
но при pH 5,5 самым активным центром становится имидазол. Нет
296
Глава 7
никаких данных, подтверждающих хелатирование одного иона
Си(II) между His-12 и His-119.
Участие одной имидазольной группы вместе с соседними к ней
пептидными атомами азота в каждом из четырех центров впервые
Рис. 7.8. Вытеснение протонов Си (II) из РНазы в присутствии и в отсутствие
ЦМФ [49].
Условия: 25 СС, ионная сила 0,16. Концентрация белка 1%. Экспериментальные точки: ф про-
тоны, вытесненные (—A/i) из РНазы 1 экв Си(II); О протоны, вытесненные нз комплекса
З'-ЦМФ—РНаза 1 экв Си(II); □ протоны, вытесненные из комплекса 2'-ЦМФ—РНаза 1 экв
Си(П).------теоретическая кривая, рассчитанная для распределения Cu(II) между че-
тырьмя независимыми имидазольными центрами в предположении, что каждый содержит
по два атома азота из пептидных связей при pH ниже 8.
было предположено на основании того, что и изменения в спект-
ре поглощения в видимой области комплекса Си (II) с РНазой
(рис. 7.8) при изменении pH, и титриметрические свойства этого1
комплекса аналогичны свойствам комплексов Си(II) с ацетил-Gly-
Gly-Gly-His [49]. Ниже приведены факты, свидетельствующие об
участии в связывании также а-МНг-групп, на основании которых
пришли к выводу, что данная выше интерпретация результатов
титрования нуждается в доработке. Однако методом ЯМР были
получены дополнительные доказательства взаимодействия Си(II)
Комплексы металлов с белками
297
с имидазолом [51], согласно которым при pH 5,5 Си (II) уширяет
сигналы протонов С-2 His-12, His-105 и His-119. Еще нет никаких
доказательств участия His-48 в связывании с Cti(II). Невозмож-
ность одновременной координации одного иона Си(П) между
His-12 и His-119 следует из данных титрования [49] и ЯМР-иссле-
дований, которые показали, что низкие концентрации Си (II) силь-
нее влияют на His-12, чем на His-119 [51].
Участие а-МНг-групп в связывании Си(II) было продемонстри-
ровано следующими фактами: дезаминирование а-МНг-групп при-
водит к исчезновению самого сильного центра связывания Си(II)
при pH 7 и к спектральным изменениям к комплексе Си(II) —бе-
лок, которые свидетельствуют об исчезновении центра, содержа-
щего а-МНг-группу и атомы азота пептидных связей [52]. Однако
при pH 5,5 дезаминирование приводит только к незначительным
изменениям в связывании Си (II) [52], в то время как взаимодей-
ствие РНазы с 2'-ЦМФ (который присоединяется к активному
центру), по-видимому, блокирует связывание с наиболее сильным
центром [53]. На основании того, что карбоксиметилирование
гистидина также влияет на сильный центр связывания Cu(II) при
pH 5,1 [50], можно предположить, что при pH 5,5 наиболее силь-
ным центром связывания Си(II) может быть His-12.
К другим интересным аспектам связывания Си(II) с РНазой
в отсутствие нуклеотидов относятся быстрое образование комплек-
са биуретового типа при pH выше 8 [отчасти это явствует из вы-
теснения Си (II)большого количества протонов при высоких зна-
чениях pH, рис. 7.8], взаимодействия между связывающими цент-
рами [49, 50, 52] и связывания РНазой как свободного иона меди,
так и моноацетатного комплекса меди [3]. Кроме того, было пока-
зано, что РНаза претерпевает значительное агрегирование в при-
сутствии Си(II) при pH около 7 [54]. Условия, при которых про-
исходит агрегация, позволяют предположить, что причина ее —
образование межмолекулярных мостиков между группами связан-
ный белок — СиОН [54].
В присутствии З'-ЦМФ связывание Си(II) с РНазой заметно
изменяется. Впервые это обнаружили по большим сдвигам в длин-
новолновую область полосы поглощения комплекса Си(II) —
РНаза в присутствии З'-ЦМФ [49], а также по изменениям при ти-
тровании (рис. 7.8), которые показали, что З'-ЦМФ увеличивает
силу взаимодействия Си(II) с РНазой [49]. Интересно, что влия-
ние З'-ЦМФ заключается в облегчении взаимодействия Си (II) с дву-
мя действующими кооперативно центрами связывания, как это осо-
бенно отчетливо было показано методом гель-фильтрации [53].
Природа связывающих центров в комплексе З'-ЦМФ—РНаза не
установлена. Однако примечательно то, что тройной комплекс
Zn(II) с РНазой и З'-ЦМФ, стереохимия которого должна быть
отлична от стереохимии комплекса с Си (II), также облегчает свя-
298
Глава 7
зывание двух ионов металла [49, 54]. Уместно также отметить,
что 2'-ЦМФ не образует аналогичного тройного комплекса
с Си(П) и РНазой (рис. 7.8), а вместо этого конкурирует с Си (II)
[49, 53]. Различия между 2'-ЦМФ и З'-ЦМФ в этом отношении
позволяют предположить, что нуклеотидфосфат может координи-
роваться со связанным ионом металла.
5. ЗАКЛЮЧЕНИЕ
Из вышеизложенного ясно, что идентификация центров связы-
вания металла осуществляется наиболее просто в том случае,
когда все они относятся к одному классу и (или) когда они в точ-
ности совпадают со связывающим центром в известных модельных
системах. Для идентификации уникальных центров связывания
в данном конкретном белке, по-видимому, необходимы новые ме-
тоды. Ясно, что даже полное представление о трехмерной структу-
ре белка (см. миоглобин) в настоящее время не дает возможности
предсказать детально ни природу связывающих центров, ни влия-
ние связывания на свойства белка. Для того чтобы делать какие-
либо предсказания в этой области, необходимо получить более
количественные представления о влиянии потенциальных лиганд-
ных атомов на конформацию белка.
СПИСОК ЛИТЕРАТУРЫ
1. Vallee В. L., Williams R J. Р., Proc. Natn. Acad. Sci. U. S., 59, 498 (1968).
2. Gurd F. R. N., Goodman D. S., J. Am. Chem. Soc., 74, 670 (1952).
3. Girott A. W„ Breslow E., J. Biol. Chem., 243, 216 (1968).
4. Klotz I. M„ Fiess H. A., J. Phys. Coll. Chem., 55, 101 (1951).
5. Edsall J. T., Wyman J., Biophysical Chemistry, Vol. 1, Academic Press, New
York, 1958.
6. Gurd F. R. N., Wilcox P. E., Adv. Protein Chem., 11, 311 (1956).
7. Linderstrom-Lang K-, Compt. rend. trav. lab Carlsberg, 15, № 7 (1924)
8. Freeman H. C., Adv. Protein Chem., 22, 257 (1967).
9. Hartzell C R„ Gurd F. R N J. Biol. Chem., 244, 147 (1969).
10. Bryce G F., Roeske R. W., Gurd F. R. N., J. Biol. Chem., 240, 3837 (1965).
11. Rao M. S. N., Lal H„ J. Am. Chem. Soc., 80, 3222 (1958).
12. Rao M. S. N., Lal H., J. Am. Chem. Soc., 80, 3226 (1958).
13. Perkins D. J., Biochem. J., 80, 668 (1961).
14. Gurd F. R. N., J. Phys. Chem., 54, 788 (1954).
15. Schlichtkrull J., Acta Chem. Scand., 10, 1455 (1956).
16. Marcker К, Acta Chem. Scand , 14, 194 (I960)
17. Cunningham L. W., Fischer R. L., Vestling C. S., J. Am. Chem. Soc., 77, 5703
(1955)
18. Tanford C., Epstein J., J Am. Chem. Soc., 76, 2170 (1954).
19. Brill A. S.. Venable J. H„ Jr., J. Am. Chem. Soc., 89, 3622 (1967).
20. Marcker K-, Acta Chem. Scand., 14, 2071 (I960).
21. Marcker K., Grae J., Acta Chem. Scand., 16, 41 (1962).
22. Adams M. J., Blundell T. L., Dodson E. J., Dodson G. G. et al.. Nature, 224, 491
(1969).
Комплексы металлов с белками
299
23. Drey С. С., Fruton J. S., Biochemistry, 4, 1258 (1965).
24. Breslow Е., Biochim. Biophys. Acta, 53, 606 (1961).
25. Campbell В. J., Chu F. S., Hubbard S., Biochemistry, 2, 764 (1963).
26. Walter R., Havran R. T., Schwartz I. L„ Johnson L. F„ in E. Scoffane (ed.),
Proc. 10th European Peptide Symposium, North Holland Publishing Co., Amster-
dam, in press.
27. Kendrew J. C., Watson H. C., Strandberg В. E„ Dickerson R. E., Phillips D. C.,
Shore V. C., Nature, 190, 666 (1961).
28. Banaszak L. J., Andrews P. A., Brugner J. W., Eylar E. H., Gurd F. R. N.,
J. Biol. Chem., 238, 3307 (1963).
29. Breslow E., Gurd F. R. N., J. Biol. Chem., 238, 1332 (1963).
30. Hardman K. D., Ph. D. Thesis, Indiana Univ., 1965.
31. Banaszak L., Watson H. C., Kendrew J. G., J. Mol. Biol., 12, 130 (1965).
32. Gurd F. R. N., Falk К. E., Malmstrom B. G., Vanngard T., J. Biol. Chem., 242,
5724 (1967).
33. Hartzell C. R., Hardman K. D., Gillespie J. M., Gurd F. R. N., J. Biol. Chem.,
242, 47 (1967).
34. Bryce G. F„ Roeske R. W., Gurd F. R. N„ J. Biol. Chem., 241, 1072 (1966).
35. Breslow E., J. Biol. Chem., 239, 3252 (1964).
36. Shearer W. T„ Brown R. K-, Bryce G. F., Gurd F. R. N., J. Biol. Chem., 241,
2665 (1966).
37. Cann J. R., Biochemistry, 3, 714 (1964).
38. Gurd F. R. N., Bryce G. F„ in J. Peisach, P. Aisen, W. E. Blumberg (eds.), The
Biochemistry of Copper, Academic Press, New York, 1966.
39. Klotz I. M., Curme H. G., J. Am. Chem. Soc., 70, 939 (1948).
40. Kolthoff I. M„ Willeford B. R., Jr., J. Am. Chem. Soc., 79, 2656 (1957).
41. Peters T., Jr., Biochim. Biophys. Acta, 39, 546 (1960).
42. Peters T., Blumenstock F. A., J. Biol. Chem., 242, 1574 (1967).
43. Bradshaw R. A., Shearer W. T., Gurd F. R. N., J. Biol. Chem., 243, 3817
(1968).
44. Klotz I. M., Urquhart J. M., Fiess H. A., J. Am. Chem. Soc., 74, 5537 (1952).
45. Klotz I. M., Urquhart J. JVL, Klotz T. A., Ayers J., J. Am. Chem. Soc., 77, 1919
(1955).
46. Davis F. F., Allen F. W., J. Biol. Chem., 217, 13 (1955).
47. Ross C. A., Mathias A. P., Rabin B. R., Biochem. J., 85, 145 (1962).
48. Crest field A. M., Stein W. H., Moore S., J. Biol. Chem., 238, 2421 (1963).
49. Breslow E., Girotti A. W., J. Biol. Chem., 241, 5651 (1966).
50. Joyce B. K„ Cohn M., J. Biol. Chem., 244, 811 (1969).
51. Ihnat M., Bersohn R., personal communication. Similar data were cited in ref.
50 as a communication from G. Roberts and O. Jardetzky.
52. Girotti A. W., Breslow E., J. Biol. Chem., 245, 3066 (1970).
53. Breslow E„ Girotti A. W., J. Biol. Chem., 245, 1527 (1970).
54. Breslow E., Girotti A. W., unpublished observations.
55. Martell A. E., Sillen L. G., Stability Constants, Special Publication Ns 17, The
Chemical Society, London, 1964.
56. Dickerson R. E., in H. Neurath (ed.), The Proteins, Vol. 2, Academic Press, New
York, 1964.
ЧАСТЬ JU
РОЛЬ МЕТАЛЛОПРОТЕИДОВ
В НАКОПЛЕНИИ И ТРАНСПОРТЕ
ГЛАВА 8
ФЕРРИТИН
77. 7И. Харрисон, Т. Г. Хой
Harrison Р. М., Ноу Т. G., Department of Biochemistry,
University of Sheffield, Sheffield, Gt. Britain
1. ФЕРРИТИН КАК СОЕДИНЕНИЕ,
накапливающее ЖЕЛЕЗО
Ферритин — красно-коричневый водорастворимый содержащий
железо белок, который очень широко распространен в раститель-
ном н животном мире. Он был найден в организмах позвоночных
[1—3] и беспозвоночных [4—8], в цветковых растениях [9—11]
и даже в грибах [12, 13]. Его повсеместное распространение сви-
детельствует о древнем эволюционном происхождении и почти
универсальной функциональной роли, которую он играет в мно-
гоклеточных организмах. Впервые ферритин был выделен Лауф-
бергером [14] в 1937 г. из селезенки лошади. Его роль в организме
животных как соединения, накапливающего железо, была уста-
новлена Граником [15, 16], и, по-видимому, такую же роль он
играет и в растениях.
Установлено, что в организме человека большая часть находя-
щегося в крови железа непрерывно циркулирует — очень мало
железа поступает из пищи и очень мало выводится [17]. Около
70% содержащегося в крови железа находится в гемоглобине,
20—25%—-в запасном депо. В нормальных условиях большая
часть запасенного железа входит в состав ферритина, а остаток
в виде богатых железом нерастворимых гранул известен как ге-
мосидерин [16, 18]. При разрушении гемоглобина его железо вре-
менно может храниться в виде ферритина и в дальнейшем исполь-
зоваться для получения гемоглобина [17]. Некоторая часть содер-
жащегося в организме железа может сохраняться в виде ферри-
тина или гемосидерина в течение длительного времени. Эти запасы
могут быть использованы после потери крови или если в пище
содержится мало железа, а также для роста плода при беремен-
ности.
Ферритин
301
Роль ферритина [16] состоит в том, чтобы сохранять Fe(III)
в нетоксичной растворимой и легко доступной форме. Fe (III) очень
мало растворимо при нейтральных значениях pH и токсично даже
в очень низких концентрациях. Ферритин накапливается главным
образом в печени, селезенке и костном мозге. При кормлении жи-
вотных пищей, богатой железом, или при введении больших доз
железа ферритин находят в слизистых оболочках желудочно-ки-
шечного тракта [19]. Первоначально полагали, что присутствие
ферритина в слизистых оболочках блокирует поглощение железа
из кишечника [16, 19]. Хотя и не очень долго существовало мне-
ние, что в слизистой оболочке есть специальный блок для погло-
щения железа, образование ферритина в слизистых оболочках
может прямо или косвенно играть роль регулятора поглощения
железа [20].
Какого типа должна быть молекула ферритина, если этим ис-
черпываются его функции? Данная глава будет посвящена в ос-
новном описанию структуры ферритина в связи с этой его функ-
цией.
Для того чтобы ферритин как соединение, накапливающеее
железо, был эффективен, отношение в нем железа к белку долж-
но быть высоким; при этом белок, окружающий железо, должен
обеспечивать его растворимость. Если это необходимо для того,
чтобы предотвратить аккумулирование свободного железа, белок,
который, как можно ожидать, играет активную роль в поглощении
железа, нормально должен присутствовать в клетке в состоянии,
которое ненасыщенно по отношению к железу, и его синтез дол-
жен зависеть от железа. Поглощение железа должно быть обра-
тимым, если оно тратится только на кругооборот в организме.
2. ПОЛУЧЕНИЕ И ХАРАКТЕРИСТИКА ФЕРРИТИНА
Для получения ферритина до сих пор широко используется ме-
тод Граника [21], основанный на относительной устойчивости
ферритина к нагреванию. Водный экстракт измельченной ткани
нагревают до 80 °C на водяной бане. После охлаждения и удале-
ния денатурированных белков ферритин осаждают сульфатом ам-
мония и выделяют центрифугированием. Его можно очистить
перекристаллизацией из 5%-ного раствора CdSO4. Добавление
CdSO4 и последующий диализ при pH 4,6 иногда приводят к осаж-
дению нерастворимого продукта. Это вещество и коричневый
маточный раствор, окружающий кристаллы ферритина, иногда назы-
вают «некристаллизующимся» ферритином. Ферритин можно так-
же получить, минуя стадию нагревания [22, 23], и перекристалли-
зовать в присутствии различных ионов двухвалентных металлов
[И].
302
Глава 8
Ферритин из селезенки лошади может содержать различные
количества железа, обычно 17—23% от его сухой массы [1], при
отношении Fe/N приблизительно 2,0 [24]. Ферритин, выделенный
из других органов и видов организмов, может содержать меньше
железа, обычно 12—20%. Такие отличия заставляют предположить
гетерогенность этого соединения. Показанная на рис. 8.1 шлирен-
диаграмма ферритина [25] подтвердила это предположение. На
рис. 8.1 можно видеть медленный пик частиц, не содержащих
Рис. 8.1. Шлирен-диаграмма ферритина из селезенки лошади в 0,1 и. ацетатном
буфере прн pH 4,4.
Картина снята через 20 мин после достижения скорости 33 450 об/мин. Седиментация справа
налево. Обратите внимание на неокрашенный пик от частиц, не содержащих железа (около
18S), и широкий пик, относящийся к молекулам ферритина с различным содержанием же-
леза (примерно 62S).
железа (~ 18S), представляющий примерно 20—25% общего ко-
личества препарата, и широкий (примерно '60S) пик, относящийся
к частицам с различным содержанием железа. Главная компонен-
та не содержащего железа белка, полученная после удаления
железа (см. ниже), имеет молекулярную массу около 450 000 [26].
Изопикническое центрифугирование позволяет разделить ферритин
на частицы, плотности которых, измеренные поплавковым методом,
изменяются непрерывно от 1,25 до 1,81 [26]. Максимально ферри-
тин содержит 4500 атомов железа на молекулу с молекулярной
массой около 900 000 [26]. Спектры поглощения ферритина
в ультрафиолетовой и видимой областях главным образом обус-
ловлены присутствием железа [16]. Так, поглощение при 280 нм,
обусловленное белком, составляет только около 7% от общего по-
глощения, а при 310 нм — меньше 1%.
Граник [27] показал, что ферритиновое железо находится
в «мицеллах», образуемых комплексом гидратированного оксида
железа с фосфат-ионом, приблизительный состав которого выра-
жается формулой (FeOOH)fi(FeO : ОРО3Н2). Мицеллы, содержа-
Ферритин
303
щие железо, могут быть почти полностью отделены от белка обра-
боткой растворами сильных щелочей [28], 5%-ным гипохлоритом
натрия [29], перекисью водорода [30], додецилсульфатом натрия
(SDS) [31] или уксусной кислотой [32]. При такой обработке
мицеллы теряют большую часть содержащегося в них фосфата
[28] и агрегируют, образуя темный красно-коричневый осадок. Под
Рис. 8.2 Электронная микрофотография ферритина, полученная методом нега-
тивного контрастирования с помощью натриевой соли фосфорномолибдеповой
кислоты, любезно предоставленная Д. X. Хаггисом. Железосодержащие ядра
ферритина окружены белковыми оболочками, которые кажутся светлыми на фоне
негативной окраски (X 500 000).
электронным микроскопом они выглядят как непроницаемые для
электронов частицы диаметром 55—60 А [33]. Каждая такая «ми-
целла» образует центральное ядро внутри белковой оболочки. Это
было показано методом электронной микроскопии с негативным
контрастированием препаратов [34] (рис. 8.2), посредством рас-
сеяния электронов под малыми углами на молекулах ферритина
в растворе [26] и методом дифракции рентгеновских лучей на
кристаллах ферритина и апоферритина [35, 36] и денатурирован-
ного ферритина [37]. Диаметр ядра, определенный рентгенострук-
турным методом, составляет 73 А [26, 36, 37].
3. УДАЛЕНИЕ ЖЕЛЕЗА ИЗ ФЕРРИТИНА; ПОЛУЧЕНИЕ
И СВОЙСТВА НЕ СОДЕРЖАЩЕГО ЖЕЛЕЗА БЕЛКА — АПОФЕРРИТИНА
Методы, используемые для выделения железосодержащих ядер,
разрушают белок. Для получения интактных частиц белка железо
нужно удалять действием восстанавливающих агентов.
304
Глава 8
Апоферритин удобно получать, используя дитионит натрия
в ацетатном буфере при pH 4,6—5,0 [38]. Белок можно отделить
от Fe(II), либо превращая последнее в хелат с а,а'-дипиридилом
[38], либо осаждением белка сульфатом аммония [39], либо гель-
фильтрацией [22]. Восстановление дитионитом проходит медленно
в течение нескольких часов и особенно медленно при pH 7,0.
Подобно ферритину, апоферритин может быть очищен пере-
кристаллизацией из 5%-ного раствора CdSO4. Белок с очень
низким содержанием железа можно отделить от препаратов фер-
ритина центрифугированием с высокой скоростью.
Установлено, что молекулярная масса апоферритина лошади
изменяется от 430 000 до 480 000 [25, 36, 40—42]. Рентгенострук-
турные исследования кристаллов и растворов показали, что моле-
кула апоферритина имеет вид ровной сферической оболочки, сред-
ний внутренний диаметр которой составляет 73 А, а внешний —
122 А [26, 36, 40]. Внутренний диаметр точно равен диаметру
ядра.
Из симметрии дифрактограммы рентгеновских лучей [35, 36]
получены доказательства существования субъединиц в апоферри-
тине. Эти данные были подтверждены разрушением агрегатов до
приблизительно 2S субъединиц [32, 43]. Молекулярная масса
субъединиц, определенная рядом методов, равна приблизительно
23 000, что соответствует 20 субъединицам на молекулу [42, 44,
45]. Самая последняя оценка молекулярной массы методом миг-
рации в полиакриламидном геле, содержащем додецилсульфат
натрия, дала значение только 18 000+1100, что соответствует
24 субъединицам при молекулярной массе 450 000 [47]. Имеющие-
ся химические данные заставляют предположить, что субъединицы
идентичны [2, 48]. Аминокислотный состав апоферритина из се-
лезенки лошади был определен в нескольких работах [22, 44,
48—50] и приведен в табл. 8.1.
Эта довольно простая картина осложняется неоднородностью
в содержании как белка, так и железа. Содержащиеся в неболь-
ших количествах компоненты, обнаруживаемые при ультрацент-
рифугировании [22, 23, 25] и при гель-электрофорезе [51—53],
были идентифицированы как мономер, димер, тример и т. д., при-
чем содержание мономера составляло 80—90% от массы всего
препарата [22, 23, 41, 49]. Биологическое значение этого полимера
не выяснено. Неоднородность ферритина и апоферритина наблю-
дали также посредством изоэлектрического фокусирования [54,
55]. По-видимому, она не связана с содержанием железа или по-
лимера, а, возможно, обусловлена изменениями в ацетильной,
амидной, ионизированной карбоксильной или других присоединен-
ных группах или ионах. Средняя изоэлектрическая точка феррити-
на лошади 4,4. Кроме того, недавно было показано, что ферри-
тины из различных органов одного и того же животного имеют
Ферритин
305
Таблица 8.1
Аминокислотный состав апоферритина из селезенки лошади
Аминокислота Число остатков в расчете иа молекулярную массу 22 500
по данным работы [48] по данным работы [49]
Цистеин а 2,39
Аспарагиновая кислота 21,05 21,03
Треоиин 6,67 6,70
Серин 10,91 11,55
Глутаминовая кислота 29,01 29,19
Пролин 3,43 2,51
Глицин 12,01 11,92
Аланин 16,99 17,26
Валин 8,43 8,59
Метионин 3,36 3,59
Изолейцин 4,25 4,56
Лейцин 30,35 30,52
Тирозин 6,07 6,33
Фенилаланин 8,92 8,87
Гистидин 7,03 7,14
Лизин 10,62 10,90
Аргинин 11,49 11,98
Триптофан 1,066 0,98в
а т т
Не определено.
® Данные работы [45].
в Данные работы [44].
различную электрофоретическую подвижность [56—58] и амино-
кислотный состав [59]. Ферритины из канцерогенных и неканцеро-
генных тканей также имеют различные электрофоретические под-
вижности [52, 54].
4. ЧЕТВЕРТИЧНАЯ СТРУКТУРА АПОФЕРРИТИНА
Структура ферритина формально аналогична структуре неболь-
ших сферических вирусов; молекулы обоих состоят из большого
числа белковых субъединиц, внутри которых содержится непротеи-
20—2451
306
Глава 8
новое «ядро», хотя ферритин гораздо меньше по размеру, чем лю-
бой вирус, и имеет в своем составе меньше белковых субъединиц.
Тем не менее можно ожидать, что его основные структурные свой-
ства будут напоминать свойства икосаэдральных вирусов.
Рис. 8.3. Возможные варианты моделей четвертичной структуры апоферритина.
а — 20 субъединиц расположены по вершинам пеитагонального додекаэдра; б — 24 субъеди-
ницы расположены по вершинам искаженного куба. Субъединицы изображены в виде ма-
леньких соприкасающихся сфер. Отметим, что в обеих моделях имеется пространство между
субъединицами, благодаря которому центральное пространство молекулы доступно для ато-
мов железа. Размерь^ этих пространств в центре молекулы будут зависеть от истинной фор-
мы субъединиц, которая вияд ли является точно сферической.
Ферритин лошади был получен в двух основных кристалличе-
ских формах — кубической и орторомбической, а апоферритин —
в кубической и тетрагональной формах [35, 36]. Кубические фор-
мы, имеющие форму октаэдра, изоморфны. Это дает возможность
прямо наблюдать влияние железа на дифрактограмму. Молекулы
плотно упакованы, межмолекулярные расстояния составляют 130 А.
Пространственная группа F432 формально указывает на то, что
каждая молекула содержит 24п идентичных субъединиц, но их
расположение в кристаллах орторомбической структуры свидетель-
ствует о более низкой молекулярной симметрии. Дифрактограммы
как кубических, так и орторомбических кристаллов показывают,
что молекулы могут иметь псевдоикосаэдральную симметрию. Чет-
вертичная структура, предложенная для объяснения этой симмет-
рии и большинства химических данных, состоит из 20 белковых
субъединиц, занимающих 20 граней икосаэдра '[36, 60]. Эта струк-
тура изображена на рис. 8,3, а. Если такое расположение коррект-
но, симметрия кубических кристаллов должна статистически воз-
никать из нескольких молекулярных ориентаций. Предложенная
ранее модель [35] с октаэдрической, а не с икосаэдральной сим-
метрией показана на рис. 8.3, б. В этой модели 24 субъединицы
Ферритин
307
расположены по вершинам деформированного куба. Эта модель
объясняет многие особенности дифракционной картины как на
кубических, так и на тетрагональных кристаллах и их векторные
карты Паттерсона. Для точного определения четвертичной структу-
ры белка необходим более детальный анализ кристаллической
структуры, который в настоящее время пытаются осуществить.
Полное понимание функций белка всецело зависит от знания тон-
ких особенностей его структуры, но даже не зная их, очень многое
можно понять о природе железного ядра, его связи с белком и
биосинтезе молекулы ферритина. Для последующего обсуждения
необходимо лишь знать, что белковая оболочка представляет собой
правильный ансамбль из субъединиц, охватывающий железосо-
держащее ядро и позволяющий железу входить и выходить из
молекулы по каналу между субъединицами.
5. МОРФОЛОГИЯ И АТОМНАЯ СТРУКТУРА
ФЕРРИТИНОВЫХ ЯДЕР
Приведенные выше данные показывают, что в молекуле апо-
ферритина имеется центральная полость диаметром приблизитель-
но 73 А, в то время как ферритин содержит непостоянное число
атомов железа вплоть до 4500. В молекулах с максимальным со-
держанием железа пространство внутри белковой оболочки долж-
но быть довольно плотно заполнено. В молекулах с низким содер-
жанием железа частицы железа должны быть либо беспорядочно
распределены по всей полости, либо находиться в изолированных
областях, как это показано на рис. -8.4. Можно думать, что веще-
ства, изображенные на рис. 8.4, а и г, аморфны, а на рис. 8.4, в, д
и е — кристаллические. Если частицы железа распределены бес-
порядочно, ядра могут отличаться по плотности, но при этом они
все равно должны иметь одинаковый внешний вид и одинаковую
форму, независимо от содержания железа. Если же железосодер-
жащая компонента кристаллическая, ядро может дополнительно
делиться, как это изображено на рис. 8.4, е, и иметь множество
форм, как на рис. 8.4, б, в, д и е. Природа вещества, из которого
состоит ядро, была исследована несколькими методами.
5.1. Электронная микроскопия
Первые электронные микрофотографии показали, что железосо-
держащие ядра ферритина разделены на ряд дискретных частей,
«тетрад», которые иногда видны [33]. Было предложено несколько
структур с альтернативным симметричным расположением четы-
рех или шести частиц [61—64]. Хаксли (на которого ссылаются
в работе [2]) и Хэггис [65] нашли, что ядро при рассмотрении
его на очень тонкой поддерживающей пленке имеет внешний вид
20*
308
Глава 8
Рис. 8.4. Схематическое изображение способов, которыми железосодержащая
компонента может распределяться в молекулах с различным содержанием же-
леза.
а — белок изображен как внешняя почти сферическая оболочка; а, б ив— молекулы с низ-
ким содержанием железа: г, д и е — молекулы с довольно высоким содержанием железа.
Если железосодержащая компонента аморфна и беспорядочно распределен по всему про-
странству внутри белковой оболочки, ядра с различным содержанием железа должны
иметь одинаковую форму, но различную плотность, как в а и г. Если железо находится
в локализованных участках, как в б, в, д или е, возможно многообразие форм в зависи-
мости от числа, расположения и формы различных частиц внутри белка (бив содержат
по одной частице, д — одну частицу, но она разделена расщепляющей линией; в случае
е — трн отдельные частицы). Электронная спектроскопия и данные рентген ©структурного
анализа подтверждают такие структуры-, как б, в, д и в, и показывают, что заштрихован-
ные области имеют почти кристаллическую структуру.
«классической» субъединицы только при грубой фокусировке.
Хэггис [65] наблюдал ядра различной формы в различных моле-
кулах, иногда они кажутся одинаковыми по плотности, но имею-
щими различную форму многогранниками (треугольники, ромбы,
квадраты, пятиугольники и шестиугольники), иногда они разделе-
ны на две или более плотные области. Случается, что расщепляю-
щая линия проходит через середину, тогда она имеет U-образную
форму. Хэггис пришел к заключению, что «железо находится
в форме небольших кристаллитов», возможно, в составе несколь-
ких молекул одного небольшого кристалла, находящегося внутри
почти сферической оболочки». Это заключение позднее было под-
тверждено изучением фракций ферритина с высоким и низким
содержанием железа [66], как это показано на рис. 8.5, а и б.
Ферритин
309'
Видно, что в молекулах с низким содержанием железа ядра ка-
жутся гораздо меньшими, чем ядра «полного» ферритина. Недавно
Хайдон [67, 68] пересмотрел выводы Хэггиса и приписал появле-
ние кристаллитов на последних микрографиях артефактам, обус-
Рис. 8.5. Электронные микрофотографии неокрашенного ферритина.
а — фракция с высокой плотностью (плотность >1,82 г/см3), содержащая приблизительно
4000—4500 атомов железа на молекулу («полный» ферритин). Обратите внимание на много-
образие форм железосодержащих ядер, б — фракция с низкой плотностью (плотность
1,38 г/см3), содержащая приблизительно 450 атомов железа иа молекулу, смешанная с фрак-
цией с высокой плотностью (а). Можно видеть что малые частицы (показанные стрелками)
разбросаны среди больших частиц. Эти малые частицы представляют железосодержащие
ядра с низкими плотностями. Электронные микрофотографии получены с помощью электрон-
ного микроскопа AEI ЕМ6В с напряжен нем 60 кВ н любезно предоставлены Хэггисом
(Х500 000)
310
Глава И
в
Рис. 8.5. в — образец неразделенного на фракции ферритина, снятый на сканирующем
электронном микроскопе высокого разрешения. Видны различия в форме, структуре и плот-
ности железосодержащих ядер. Фотографии опубликованы с разрешения 4. В. Греве и
Д. Уолла (Х480 000).
ловленным зернистостью фона. Тот факт, что Хайдон не видел
таких изображений, как Хэггис, возможно, объясняется использо-
ванием слишком толстой поддерживающей пленки [69]. Изобра-
жения, полученные одним из самых новых методов с помощью
сканирующего трансмиссионного электронного микроскопа высо-
кого разрешения [70], так же как показано на рис. 8.5, в, по-ви-
димому, подтверждают мнение Хэггиса. В некоторых случаях на-
блюдается субструктура ядер, особенно для образцов с довольно
низким содержанием железа; в других случаях ядра имеют вид
полиэдров.
Ферритин
31*
5.2. Рассеяние рентгеновских лучей
под малыми углами
Дифрактограммы рентгеновских лучей промытого монокристал-
ла неразделенного на фракции ферритина (в котором преоблада-
ли молекулы с высоким содержанием железа) показали, что же-
лезосодержащие ядра ферритина не состоят из правильно располо-
женных четырех, шести или восьми дискретных субъединиц [36].
В среднем ядро, по-видимому, должно быть полиэдром одного типа.
Более детально ферритиновые железосодержащие ядра были
исследованы Фишбахом и Андереггом [26] с использованием рент-
геновских лучей с малыми углами рассеяния. Они исследовали
серии ферритиновых фракций, в каждой из которых содержание
железа изменялось в узком интервале, в 53%-ном растворе саха-
розы. В этих растворах рассеяние на белковой части ферритина
незначительно, так как она имеет такую же электронную плот-
ность, как и среда, в то время как рассеяние на ядрах, обладаю-
щих высокой электронной плотностью, относительно велико.
Наблюдаемое рассеяние сравнивали с кривыми рассеяния, рассчи-
танными для нескольких моделей. Никакое сочетание маленьких,
идентичных, примерно сферических частиц не дало так хорошо
совпадающего с экспериментальными результатами распределения
рассеяния, как рассеяние на единственной частице. «Полный» фер-
ритин (плотность больше 1,80) был представлен в виде сферы
диаметром 73 А с молекулярной массой 418 000. Средние размеры
рассеивающего объекта становятся меньше, по мере того как со-
держание железа уменьшается. Модели, которые обладают наи-
лучшим соответствием, — это одиночные параллелепипеды или
цилиндры, размер которых такой, что они всегда закрывают дырку
в апоферритине. Фракция с наиболее низкой молекулярной массой
из всех изученных, которая содержала приблизительно 800 атомов
железа, была аппроксимирована параллелепипедом с размерами
17X34X68 А. Теоретическая кривая была получена для единст-
венного объекта, и она не представляла возможности варьировать
форму и размер частиц, из которых состояло ядро, в молекулах
с одинаковой плотностью. Тем не менее эти модели, возможно,
представляют какую-то усредненную частицу ядра. Во фракции,
примерно содержащей 450 атомов железа на молекулу, электрон-
ная микрофотография которой показана на рис. 8.5, б, многие
ядра кажутся примерно одинаковыми по размеру, хотя некоторые
из них вытянуты. Это может быть отражением действительных
отличий в форме или результатом рассмотрения одной и той же
формы с разных точек зрения. Большая асимметрия частиц, о ко-
торой заключили на основании результатов рассеяния рентгенов-
ских лучей под малыми углами, возможно, обусловлена большим
числом атомов железа, имеющихся в данной фракции.
312
Глава 8
5.3. Исследование дифракции рентгеновских лучей
с большими углами рассеяния
и дифракции электронов на железосодержащих ядрах
ферритина и их аналогах
Диаграммы Дебая — Шеррера, полученные с помощью элек-
тронов или рентгеновских лучей, ясно показали, что ферритиновые
железосодержащие ядра состоят из кристаллического материала,
содержащего кристаллиты небольшого размера [29, 65, 71]. Ана-
логичные результаты дает рентгеноструктурный анализ сырого и
высушенного на воздухе вещества [71] как для изолированных
ядер, так и для интактных молекул [71, 72] (хотя для непромытых
ядер, полученных в результате обработки щелочью, были сообще-
ны дополнительные линии [72]). Атомная структура ядер, по-види-
мому, не зависит от присутствия фосфата [71]. Фосфат отделяется
при обработке щелочью [28]. Он может быть связан поверхностью,
возможно также его участие в связывании ядра с белком.
Дифрактограммы железосодержащих ядер отличны от таковых
для хорошо известных оксидов или оксигидроксидов железа (III)
(а- или у-Ре2О3, а-, {J-, у- или 6-FeOOH) [29, 65, 71]. Однако не-
давно выполненные исследования «гидрата оксида железа(Ш)»,
полученного гидролизом соли Fe(III) и ранее считавшегося аморф-
ным, показали, что этот продукт дает дифрактограмму Дебая —
Шеррера, которая во многом похожа на таковую для ферритина
[29, 73, 74]. Характеристики интенсивных линий от продуктов,
один из которых получен гидролизом или добавлением аммиака
к нитрату железа(III) [73], а другой нагреванием нитрата желе-
за (III) до 85 °C [29], показаны в табл. 8.2 вместе с характеристи-
ками линий, полученных от ферритина [71]. На основании этих
дифрактограмм можно заключить, что по своей атомной структуре
все три вещества во многом сходны.
Формула продукта гидролиза — FeOOH-nH2O, где п равно
приблизительно 0,3 или 0,4 [29, 74]. Диаметр частиц, измеренный
с помощью электронного микроскопа, для продукта, полученного
добавлением аммиака, составляет 20—30 А; было рассчитано, что
эти частицы содержат приблизительно 1000 атомов железа ( в рас-
чете на молекулярную массу 100 000) [74], в то время как про-
дукт, полученный нагреванием, имеет молекулярную массу
200 000—250 000 [29]. Полимерное соединение железа, образую-
щееся при добавлении К'НСО3 к водному раствору Fe(NO3)3. было
изучено также Спиро, Залтманом и сотр. [75—78]. Было установ-
лено, что этот полимер имеет состав FeOojsOH (N03)o,5(H20)0,36
и содержит около 1200 атомов железа в молекуле. Его молекуляр-
ная масса после ультрацентрифугирования была 150 000, а диа-
метр, измеренный по электронной микрофотографии, 70 А. Эти
авторы отметили сходство в диаметрах между этим соединением
Ферритин
313
Таблица 8.2
Дифракционные данные для ферритиновых железосодержащих ядер
и их аналогов
Параметры наблюдаемых линий3, А Ферритин (дифракция электронов) [65] феррмтми (дифракция рентгеновских лучей) [71] Гидрат оксида железа(Ш) (рентгенострук - турные данные) [73J Гидролизат нитрата железа(Ш) (рентгеноструктурные данные) [29]
4,70 Неопределенная (U)
4,20 U (широкая)
3,30 То же
2,55 | s 41 1 100 8 1 неразрешен-
2,47 J 73 J М J ная
2,35 и
2,24 S 33 20 М—S
1,98 м 18 10 М (широкая)
1,73 W 27 15 W
1,50 VW 40 М
1,47 S 100 60 S
1,40 и
1,34 VW VW
1,25 . 7 VW
1,23 I широкая
1,18 W средняя и
1,12 VW
1,06 м широкая 10 и
' слабая
0,96 2
0,92 W
0,88 VW 1
0,85 м 3
а Средние значения.
и ферритиновым железосодержащим ядром. Однако число атомов
железа, содержащихся в этом полимере, гораздо меньше, чем
в плотно заполненных молекулах ферритина.
5.4. Магнитная восприимчивость и спектры Мессбауэра
ферритина и гидролизатов Fe (III)
Высокоспиновый ион Fe(III) с пятью неспаренными электрона-
ми характеризуется магнитным моментом 5,9 магнетона Бора.
Свободный ион Fe3+ в сильнокислом растворе имеет момент
134
Глава 8
5,8 магнетона Бора [79]. Измерения магнитной восприимчивости
ферритина дали значение 3,8 магнетона Бора для магнитного мо-
мента [27, 28, 80]. Подобные значения были получены также для
растворов полимерного гидролизата железа(III) [77] и мостико-
вого гидрокси-димера [81] [Ре2(ОН)г]4+ (который может быть и
полимером) [78]. Для объяснения низкого магнитного момента
ферритина Михаэлис, Кориэлл и Граник [27] предположили, что
атомы железа могут находиться в довольно необычном состоянии,
имея только три неспаренных электрона и кислород в качестве
лиганда в квадратно-плоскостной конфигурации. Альтернативно
ферритин может содержать смесь ионов Fe34* с пятью и одним не-
спаренным электроном. Недавно Блайз и сотр. [82, 83] обнаружи-
ли заметную зависимость магнитной восприимчивости ферритина
от силы поля и получили магнитный момент 5,08 магнетона Бора
при высокой силе поля. Они также показали, что тонкие зернистые
частицы ядер ферритина проявляют суперпарамагнетизм. Получе-
ны также доказательства суперпарамагнетизма гидроксида желе-
за (III) [74, 77].
Исследование спектров Мессбауэра ферритина было выполнено
при нескольких температурах [77, 82, 84, 85]. При 77 К или выше
в спектре наблюдаются две линии, а при 4 К — шесть линий.
Между 20 и 45 К по мере повышения температуры наблюдался
постепенный переход от спектра из шести линий к спектру из двух
линий. Такое поведение свидетельствует о суперпарамагнетизме
частиц, размеры которых отличаются не больше чем на 100 А.
Шесть линий в спектре обусловлены расщеплением энергетических
уровней под действием магнитного поля на ядрах железа. Сверх-
тонкая структура спектра и магнитные измерения показали, что
железосодержащие ядра ферритина антиферромагнитны [82, 84].
Наблюдаемый магнитный момент, возможно, обусловлен неком-
пенсированным спином, возникающим из-за статистического раз-
броса в числе занятых мест за счет двух магнитных подрешеток
в очень маленьких частицах [86]. Квадрупольное расщепление,
приводящее к спектру из двух линий, свидетельствует о градиенте
электрического поля, вызванном несимметричным распределением
атомов кислорода вокруг ядра железа.
Если ядра железа находятся в двух значительно отличающихся
состояниях, их спектры Мессбауэра могут быть уширены, несим-
метричны, а иногда разрешены в виде двух групп линий, как это
видно в спектрах y-Fe2O3 [87] и 6-FeOOH [88], которые содержат
как октаэдрически, так и тетраэдрически координированные ато-
мы железа. Спектры железосодержащих ядер ферритина довольно
симметричны, поэтому нет оснований предполагать присутствие
более чем одного центра, разве только что их поля и изомерные
сдвиги очень похожи. Величины [82, 84] квадрупольного расщеп-
ления изменяются от 0,60±0,10 до 0,74±0,04 мм/с, изомерные
-------1--------------1.---------1____I____
-3-2-1 О 1 2 3 Цлми/с
Рис. 8.6. Спектры поглощения Мессбауэра железосодержащих ядер ферритина
при различных температурах. Показано соответствие со спектрами, рассчитан-
ными на ЭВМ (Д. М. Вильямс и Т. Н. Харрисон, в печати).
Железосодержащие ядра выделены посредством дезагрегации в 67%-ной уксусной кислоте
с последующей обработкой белковых субъединиц пепсином в 1 М. уксусной кислоте при
pH 2,4. а — температура 10 К; изомерный сдвиг (относительно Fe в Pd) равен 0,30±0,04 мм/с;
напряженность внутреннего магнитного поля составляет 473±10 кЭ. б — температура 77 К, изо-
мерный сдвиг равен 0,29±0,04 мм/с; квадрупольное расщепление (28) 0,79±0,04 мм/с. в—темпе-
ратура 295 К; изомерный сдвиг равен 0,18±0.03 мм/с; квадрупольное расщепление 0»7±0,04 мм/с.
316 Глава 8
сдвиги относительно нержавеющей стали от 0,47+0,05 до 0,50±
±0,05 мм/с, а внутреннее магнитное поле 493±10 кЭ аналогично
полю, наблюдаемому для продукта гидролиза железа(III) [74, 77]
и для оксида и гидроксида железа(III) [89, 90]. Значения изомер-
ных сдвигов довольно близки к таковым для железа в октаэдриче-
ском окружении, в то время как внутреннее магнитное поле до-
вольно близко к значению, найденному для железа в тетраэдри-
ческом окружении.
Спектр Мессбауэра железосодержащего ядра ферритина, кото-
рый почти не содержал белка, показан на рис. 8.6.
6. АТОМНАЯ СТРУКТУРА ЯДЕР ФЕРРИТИНА
Атомные структуры минеральной части ферритина и продуктов
гидролиза железа(III), по-видимому, одинаковы или очень сходны.
Гидратированный оксид железа(III) имеет в ИК-спектре полосу
поглощения при 1620—1630 см~*, приписываемую воде, которая
почти полностью исчезает при высушивании [74]. Не исчезает
сильная полоса при 3400 см-1, возможно, она обусловлена
ОН-группами, находящимися внутри решетки. Такое отнесение
подтверждается также спектрами ядерного магнитного резонанса
[74].
Для объяснения наблюдаемой дифрактограммы был предложен
ряд различных элементарных ячеек и атомных расположений
[29, 71—73]. Наличие сильных полос в области 2,54 и 1,47 А, таких
же, как от образцов 6-FeOOH, позволяет предположить структуру,
образованную системой близко расположенных атомов кислорода,
отстоящих друг от друга на 2,94 А. Действительная форма элемен-
тарной ячейки зависит как от способа укладки последующих слоев
из атомов кислорода, так и от положения иона металла в проме-
жутках между ними. Как показано на рис. 8.7, атомы кислорода
могут занимать положения А, В или С. При расположении типа
АВАВ... образуется гексагональная плотная упаковка, а
АВСАВС... — кубическая решетка. Возможно и более сложное
расположение, например АВАСАВАС..., как в ТЮ2 [91]. Для фер-
ритина Харрисон и др. [71] предложили простую гексагональную
элементарную ячейку с а=2,94 А и с = 9,40 А, в то время как Жи-
рарде и Лоренс [72] считали, что это ячейка большего размера
с а=11,79 А и с=9,90 А. Похожую ячейку Toy и Брэдли [29]
предложили для продукта гидролиза нитрата железа(III) с а=
= 5,08 А и с = 9,40 А, в то же время Гиссен [73] считает, что гид-
рат оксида железа имеет кубическую решетку с а=8,37 А. В рас-
положении атомов, предложенном Харрисом с сотр. [71], атомы
кислорода в слоях имеют последовательность АВАСАВАС..., а ато-
мы железа беспорядочно распределены по всем имеющимся окта-
Ферритин
317
эдрическим и тетраэдрическим положениям. В структуре Toy и
Брэдли [29] последовательность кислородных атомов АВАВ.., при
этом все атомы железа находятся в октаэдрических положениях,
как в гематите, но с более низкосимметричным расположением,
в
Рис. 8.7. Изображение одного слоя тесно упакованных атомов кислорода и очер-
тания двух возможных элементарных ячеек (а), предложенных для железосодер-
жащих ядер ферритина.
В маленькой элементарной ячейке 171] длина стороны (расстояние между атомами кисло-
° ° Vai-
рода) равна а=2,94 А, в то время как в ячейке большего размера 129] а=5,08 А [2,94-(3)
о
В третьей предложенной элементарной ячейке [72] 11,79 А (2,94-4), Для того, чтобы
плотная упаковка сохранялась во всех трех направлениях, последующие слои атомов кис-
лорода должны располагаться выше или ниже пространства между кислородными атомами
слоя. Имеются три возможных положения кислородных слоев — А, В и С, но при этом
последовательности слоев могут несколько различаться. Две нз таких последовательностей,
предложенные как альтернативные расположения ядер ферритина, показаны на схемах б
и в. На схеме в показана элементарная ячейка, предложенная Toy н Брэдли [29]; в этой
о
ячейке упаковка — гексагональная типа АВАВ... с с=9,40 А, что соответствует четырем
кислородным слоям. Большая элементарная ячейка возникает при вхождении атомов железа.
На схеме б показано расположение, предложенное Харрисоном с сотр. [71]; кислородные
слои располагаются в последовательности ABAC. . образуя четырехслойную последова
о
тельность с с=9,40 А. Атомы железа могут размещаться в пространстве между кислород-
ными слоями, при этом они либо октаэдрически, либо тетраэдрически координированы ато-
мами кислорода Два таких положения в кристаллической решетке показаны справа на
схеме е, атомы железа находятся в центрах изображенных октаэдров. В расположениях,
предложениьих Toy и Брэдли, все атомы железа находятся в октаэдрических положениях,
в то время как в структуре, предложенной Харрисоном и сотрудниками, могут быть беспо-
рядочно заняты и октаэдрические и тетраэдрические положения.
приводящим к образованию четырехслойной ячейки. В случае ку-
бической решетки никаким расположением атомов железа нельзя
объяснить наблюдаемые интенсивности [74]. Не была предложена
также структура для очень большой гексагональной ячейки, хотя
и было сделано заключение, что атомы железа должны находиться
и в октаэдрических, и в тетраэдрических положениях [85].
Брэди с сотр. [77], исходя из функции радиального распреде-
ления в полимерном соединении железа, полученном добавлением
318
Глава 8
КНСОз к водному раствору Fe(NO3)3, предложили совсем другой
тип атомной структуры. Атомы железа тетраэдрически координи-
рованы к ионам О2- или ОН- и связаны .ими в плоскую ленту. Эта
лента может быть свернута в сферическую частицу.
Исследование различных расположений железа и кислорода
было выполнено Хойем с помощью ЭВМ.
7. СВЯЗЬ БЕЛКОВОЙ ОБОЛОЧКИ С ЯДРОМ,
СОСТОЯЩИМ ИЗ ГИДРАТА ОКСИДА ЖЕЛЕЗА
Внешняя геометрическая форма макроскопического кристалла
зависит от его атомной структуры. Хорошо развитые грани обычно
характеризуются высокой атомной плотностью заселения, хотя
кристаллы, росшие в условиях каких-либо ограничений, по разви-
тию граней могут отличаться от тех, которые росли свободно.
Обычно макроскопические кристаллы представляют собой мозаику
из кристаллитов гораздо меньшего размера, расположенных почти
параллельно. Условия кристаллизации могут влиять на размер
и форму как макроскопических кристаллов, так и микроскопиче-
ских кристаллитов и на порядок их расположения в мозаике.
Если ферритиновые ядра кристаллические, их внешний вид
должен быть близок геометрически их атомной структуре. Если
кристаллиты ядра растут в виде правильного полиэдра, точно со-
ответствующего пространству внутри белка, атомная структура
ядра будет всегда специфически ориентирована относительно бел-
ка. Такое положение можно было бы ожидать, если бы ядра об-
разовывались независимо от белка и действовали как матрицы для
связывания субъединиц. Если же гидрат оксида железа растет
в специфическом месте внутри возникшей ранее белковой оболоч-
ки, направление роста кристалла может определяться белком.
Альтернативный путь — кристаллизация внутри белка — осущест-
вляется беспорядочно в разных направлениях.
Электронно-микроскопические исследования привели к заклю-
чению, что морфология ядра неодинакова, различные молекулы
имеют разное число и расположение кристаллитов. В частично
заполненных молекулах иногда обнаруживают пустоту в центре,
при этом предполагается, что кристаллиты прикреплены к белку;
плотно заполненные молекулы чаще имеют более единообразный
внешний вид [65, 66]. Методом дифракции рентгеновских лучей
с малыми углами рассеяния на кристаллах ферритина также было
показано, что ядра не одинаковы [92]. Дифрактограммы фракций
с низкой плотностью подтверждают различие в форме, положении
и ориентации кристаллитов, образующих ядра. Фракции с высокой
плотностью имеют более правильную форму. Возможно, в этих
фракциях полость внутри белка в значительной мере или полно-
Ферритин
319
стью заполнена гидратом оксида железа. Дифракция рентгенов-
ских лучей с большими углами рассеяния дает как отпечатки
одиночных кристаллов, благодаря наличию правильной решетки
у молекул белка, так и дифрактограммы Дебая—Шеррера от ядер.
Это означает, что кристаллическая структура вещества, из кото-
рого построены ядра, не имеет правильного расположения отно-
сительно кристаллической структуры белка [92].
Размер кристаллитов ферритиновых ядер, оцененный по шири-
нам линий Дебая — Шеррера, равен 75+30 А [71] или 68 А [85].
Рис. 8.8. Схематическое изображение молекул ферритина, содержащего почти
полный комплект железа, на основании результатов электронной микроскопии и
дифракции рентгеновских лучей.
Ядра построены из кристаллического материала. Некоторые молекулы могут содержать
один кристаллит, как в а и б, а другие — несколько кристаллитов, как в в. Ядерные кри-
сталлиты растут в различных направлениях по отношению к белку в разных молекулах
(а и б) или внутри одной и той же молекулы (в). В ферритине с высоким содержанием же-
леза имеются все типы частиц (а, б и в), но преобладающими могут быть а и б.
Так как размер отверстия в белковой оболочке составляет 73 А,
вещество ядра должно представлять собой либо один кристаллит,
либо небольшое число кристаллитов в нескольких молекулах [93].
Таким образом, полный ферритин, по-видимому, должен быть
смесью молекул, ядра которых для заполнения полости начинают
расти из различных центров и поэтому в различных молекулах
получаются неодинаково ориентированными относительно белка,
как это показано на рис. 8.8.
8. ВЛИЯНИЕ ЖЕЛЕЗОСОДЕРЖАЩЕЙ КОМПОНЕНТЫ
НА СВОЙСТВА И СТРУКТУРУ БЕЛКА
Ферритин более устойчив к атаке некоторыми протеолитически-
ми ферментами, чем апоферритин [24, 48]. При pH 8,5 апоферри-
тин скорее и в большей степени расщепляется трипсином, химо-
трипсином и субтилизином, а при pH 7,0 и 5,0 — папаином [94].
Пепсин при pH 3,0 гидролизует апоферритин и ферритин с одина-
ковой скоростью, но при pH 2,5 он расщепляет апоферритин в 2—
3 раза быстрее, чем ферритин [94]. Влияние катепсина D на фер-
320
Глава 8
ритин одинаково при этих двух значениях pH [94]. Крихтон [48]
объяснил различия в расщеплении трипсином ферритина и апофер-
ритина конформационными различиями в белке. Возможно также,
что железо ингибирует протеолиз, препятствуя развертыванию
белка.
Исследование дисперсии оптического вращения ферритина и
апоферритина в водных растворах показало сравнительно высокое
содержание спиралей: 40—50% в ферритине и 50—60% в апофер-
ритине [95]. Среднее остаточное вращение при 233 нм для ферри-
тинов, содержащих от 100 до 4000 атомов железа и отделенных от
нативного ферритина центрифугированием в градиенте плотности,
было для всех одинаковым, но при этом более низким, чем для
апоферритина, полученного из ферритина восстановлением дитио-
нитом [95]. По-видимому, удаление атомов железа вызывает кон-
фигурационные изменения в молекулах, если только дитионит не
оказывает какого-либо другого влияния. Возможно, покажется
довольно странным, что повышенное содержание спиралей нужно
связывать с большей легкостью атаки протеолитическими фермен-
тами. Доказательства того, что различия между ферритином и
апоферритином обусловлены либо изменением конформации по-
верхности, либо связыванием иона, были получены при помощи
хроматографии на DEAE-целлюлозе (диэтиламиноэтилцеллюлозе).
С помощью этого метода было обнаружено, что ферритин гетеро-
генен, в то время как апоферритин дает одиночный пик [22]. Об-
работка дитионитом также может вызвать полимеризацию моно-
мерных молекул ферритина [22, 49]. С другой стороны, ферритин
и апоферритин обладают одинаковой электрофоретической по-
движностью и одинаковыми серологическими реакциями [96].
Данные рентгеноструктурного анализа показывают, что кон-
формация белка в ферритинах с различным содержанием железа
и в апоферритине, полученном восстановлением с дитионитом,
в кристаллическом состоянии по существу одна и та же [92]. Ди-
фрактограммы, снятые с разрешением от 2,5 до 10 А, очень мало
зависят от присутствия железа. По-видимому, очень маловероятно,
по крайней мере для кристаллических белков, чтобы содержание
спиралей в ферритине и апоферритине отличалось более чем на
10%- Относительно небольшие отличия в структуре поверхностей,
такие, как расположение или химическая модификация некоторых
боковых цепей, могут остаться незамеченными.
9. БИОСИНТЕЗ ФЕРРИТИНА
И ЕГО СИНТЕЗ IN VITRO
Уже давно было показано, что ферритин появляется в клетках
слизистых оболочек при наличии железа в пище и что этот процесс
сопровождается de novo синтезом ферритинового белка [19]. Такие
Ферритин
321
же данные получены и для других тканей; при этом с помощью
меченых атомов было показано, что первые образующиеся моле-
кулы либо вовсе не содержат железа, либо содержат его очень
мало [31, 97—106]. Они постепенно за период свыше 72 ч накап-
ливают железо, как это показано на рис. 8.9.
Рис. 8.9. Распределение радиоактивного ферритина, выделенного из печени крыс,
по градиенту плотности сахарозы, через различные промежутки времени после
инъекции 5 мкК лейцина-**С на 100 г веса тела.
В опытах на схеме А каждой крысе вводили 400 мкг железа на 100 г веса тела за 2 ч до
введения меченой аминокислоты. В контрольном опыте (Б) каждая крыса получала инъек-
цию 0,9% NaCl вместо железа. Заметьте, что вначале в обеих группах максимальное коли-
чество метки перешло во фракции с наименьшим содержанием железа (наименьшей плот-
ностью), а в последующие промежутки времени наибольшее количество метки было найдено
в ферритинах с повышенным содержанием железа. Сравнение этих двух групп ясно пока-
зывает индуктивное влияние железа на синтез ферритового белка [воспроизводится с раз-
решения Дж. Дрисдейл и X. Н. Мунро [103] из J. Biol. Chem., 241, 3630 (1976)].
Механизм стимуляции железом синтеза ферритинового белка
не был понятен. Обнаруженная в большинстве экспериментов не-
способность актиномицина D ингибировать синтезы послужила
указанием на пост-транскрипционный эффект [103, 105, 107]. Же-
лезо может подавлять трансляционный ингибитор [107], может
промотировать образование белковых субъединиц .[103] или дей-
ствовать каким-либо другим путем. Было выдвинуто предположе-
ние, что общее повышение выходов при биосинтезе происходит от
стабилизации обогащенных железом молекул до их разрушения
внутриклеточными ферментами [103], но этот оборот совершается
слишком медленно, чтобы его можно было считать единственной
причиной, в то время как in vitro катепсин D атакует ферритин и
апоферритин с одинаковой скоростью '[94].
21—2451
322
Глава 8
Предположение о том, что железо в виде внутриклеточного гид-
ратированного полимера оксида железа (III) диаметром 70 А про-
мотирует образование свободных субъединиц было выдвинуто
Залтманом с сотр. [108]. Такой механизм только в том случае
согласуется с образованием in vivo апоферритина, если последний
распадается, а затем вновь образуется вокруг ядра [109]. Измен-
чивость размера и формы железосодержащих ядер ферритина за-
ставляет усомниться в том, чтобы такие неправильные объекты
контролировали сборку белковой оболочки '[109]. Кроме того, если
это единственный механизм, которым стимулируются биосинтезы,
он должен требовать значительного предварительного накопления
железа в клетке.
Эксперименты по синтезу in vitro апоферритиновых субъединиц
показали, что присутствие железа (полиядерного или моноядерно-
го) не обязательно для образования полных оболочек [32]. Моле-
кулы распадаются в 67%-ной уксусной кислоте и вновь образуют-
ся при постепенном повышении pH в присутствии тиолов, образуя
частицы, напоминающие нативный апоферритин [32] по виду под
электронным микроскопом, по коэффициенту седиментации, элек-
трофоретической подвижности и иммунологическим реакциям; они
дают такие же, как апоферритин, кристаллы с растворами CdSO4.
Молекулы ферритина распадаются под действием уксусной кисло-
ты или додецилсульфата натрия и вновь образуют агрегаты, давая
апоферритин, но не ферритин, тогда как ядра образуют агрегаты
и с уксусной кислотой, и с додецилсульфатом натрия [32, ПО].
Полные оболочки апоферритина могут накапливать железо
in vitro, образуя продукт, напоминающий ферритин. Воссоздание
ферритина из железоаммиачных квасцов и апоферритина в бикар-
бонатном буфере при pH 7,6 в присутствии атмосферного кислоро-
да впервые осуществили Билих и Байер [111]. Лоэвус и Файнберг
'[И2] подтвердили этот результат и показали, что захват соли
железа(Ш) апоферритином может регулироваться кипящим экст-
рактом печени.
«Ферритин», собранный по методу Билиха и Байера, по-види-
мому, не может накапливать свой полный набор железа [71, 108,
111]. Коэффициент седиментации этого продукта в среднем со-
ставляет примерно 45S (рис. 8.10, с), а его дифрактограмма и раз-
мер кристаллитов почти такие же, как у ферритина с низким со-
держанием железа. С имидазолом или трис-буфером с pH 6,7—
7,3 вместо бикарбонатного буфера получается широкий интервал
содержания железа и некоторое количество почти полных молекул
(рис. 8.10, б). Третий способ воссоздания ферритина из апоферри-
тина и железоаммиачных квасцов в незабуференном растворе при
pH 5,8—7,4 с использованием в качестве окислителя КЮ3 в при-
сутствии Na2S4O6 впервые применили Орме-Джонсон, Джилхрист
и Коллинс [71]. В этом случае процесс воссоздания, по-видимому,
Ферритин
323
должен быть процессом по принципу «все или ничего». Прочный
железосодержащий пик имеет S приблизительно 60. Его дифракто-
грамма похожа на дифрактограмму нативного ферритина, что сви-
детельствует о сходстве их структуры и размеров кристаллитов,
несмотря на отсутствие фосфата [71]. В отсутствие апоферритина
получаются различные соединения железа [71]. Окисление Fe2+
кислородом в бикарбонатном буфере приводит к образованию
Рис 8.10 Шлирен-диаграммы «ферритина», воссозданного из Fe2+ и апоферри-
тина в условиях окисления.
Картина получена через 20 мни после достижения скорости 33 450 об/мин. Седиментация
справа налево. Растворы в 0,1 н. NaCl. а — воссоздание в бикарбонатном буфере при pH
6,9—7.9 с О2 в качестве окислителя. Главная железосодержащая компонента имеет S при-
близительно 45. хМаленькое медленное плечо — апоферритин, а маленькая быстрая компо-
нента представляет собой воссозданные димерные молекулы, б — реконструкция в трис-бу-
фере при pH 6.7—7,3 с О2 в качестве окислителя. Содержание железа в продукте колеблется
в широком интервале с наиболее быстрым плечом 65 S. в — воссоздание в незабуференном
растворе при pH 5,8—7,4 с К1О3 в качестве окислителя. Бесцветная не содержащая железа
компонента с S= 18 — апоферритин. Железосодержащая компонента имеет S около 59. Апо-
ферритин. использованный в в, был выделен из ферритина седиментацией с высокой ско-
ростью. Аналогичный результат был получен прн восстановлении ферритина днтноннтом.
Апоферритин, использованный в а и б, был получен восстановлением днтноннтом.
ct-FeOOH, тогда как при окислении КЮ3 в растворе Na2S4O6 об-
разуется y-FeOOH. Совершенно очевидно, что белок оказывает
влияние на продукт. В то время как в процессе воссоздания фер-
ритина первым способом некоторое количество железа может вы-
пасть в осадок вне белка, в третьем способе фактически все железо
попадает в белковую оболочку. В третьем способе исходят из Fe2+
и получают продукт, напоминающий гидрат оксида железа (III)
внутри белка. Попытки воссоздать ферритин гидролизом Fe3+
в присутствии апоферритина пока были безуспешными.
Известно, что сывороточное железо, связанное с трансферри-
ном, in vivo переносится в ферритин. Миллер и Перкинс [ИЗ] ис-
следовали этот перенос in vitro. Ферритин, находящийся в ячейке
для диализа, был погружен в раствор трансферрина, меченного 59Fe,
21*
324
Глава 8
в трис-буфере при pH 7,4. Испытывалось влияние различных по-
средников на перенос железа в отделении, в котором находится
ферритин. Было обнаружено, что АТФ и аскорбиновая кислота
регулируют перенос, что подтверждает результаты, полученные
Рис. 8.11. (а) Общее поглощение железа из 59Ре2-трансферрина ферритинами с
различным содержанием железа (полученными из ферритина восстановлением с
Na2S2O4 в течение различных промежутков времени), (б) Зависимость поверх-
ность/объем для ферритинов с различным содержанием железа, рассчитанная в
предположении, что во всех молекулах ферритина все железо входит в состав
одного кубического кристаллита. Сравните со схемой а. Заметьте, однако, что
ие вся площадь поверхности каждого кристаллита может быть использована для
присоедииеиия атомов железа, так как кристаллиты, вероятно, прикреплены к
белку. Площадь поверхности может уменьшиться, когда полость внутри белка
более чем наполовину заполнена железосодержащей компонентой.
В опытах 1' мл 0,2%-ного по железу насыщенного ферритина был подвергнут дналнзу про-
тив 10 мл 0,2%-иого “Ез-трансферрина, 1,0 мМ аскорбиновой кислоты, 1,0 мМ АИФ в 0,1 М
трис-буфере, pH 7,4 [воспроизводится с разрешении Дж. П. Г. Миллера н Д. Дж. Перкинса
[113] из Eur. J. Biochem., 10, 149 (1969)].
ранее [114, 115]. При этом АТФ действует не как источник метабо-
лической энергии, а как хелатирующее соединение [113, 114]. Ее
можно заменить АМФ, АДФ, лактатом, глюкозой и другими хела-
тирующими агентами, но более сильные хелатирующие агенты
типа ЭДТА или нитрилтриуксусной кислоты удаляют железо из
трансферрина, но не переносят его в ферритин. В присутствии
АТФ повышение концентрации аскорбиновой кислоты увеличивает
перенос. Очень мало железа переносится в отсутствие восстанавли-
вающего агента. В процессе переноса участвует атмосферный
кислород, и этот процесс прекращается, когда весь восстанавли-
вающий агент израсходован. По-видимому, роль хелатирующего
Ферритин
325
агента состоит в удалении железа из трансферрина, так как экспе-
рименты по воссозданию показали, что хелатирующее соединение
не требуется для вхождения железа в ферритин.
Миллер и Перкинс [113] показали, что ферритин поглощает
железо против его градиента концентрации. Используя ферритины,
из которых восстановлением были удалены разные количества
железа, они обнаружили, что вхождение железа зависит от его
содержания, как это показано на рис. 8.11, а. Из этого рисунка
видно, что очень мало железа поглощается ферритинами с низким
содержанием железа по сравнению с ферритинами, содержащими
более 3% железа.
10. МЕХАНИЗМ ОБРАЗОВАНИЯ ФЕРРИТИНА
ИЗ АПОФЕРРИТИНА
Структурные данные и эксперименты по воссозданию дают ос-
нования полагать, что ферритин образуется из апоферритина и
Fe2+ посредством процесса, включающего окисление железа, по-
следующий гидролиз и кристаллизацию. Железо попадает в белок
через пространство между его субъединицами. В какой момент
происходит его окисление — неизвестно. Возможно, белок участву-
ет в удалении электронов от Fe2+ или хелатирует атомы железа.
Мазур, Баез и Шорр [116, 117] предположили, что небольшое ко-
личество Fe2+ связывается в хелат тиоловыми группами, находя-
щимися на поверхности белка. Однако в экспериментах in vitro
предварительная обработка сульфгидрильными реагентами не
влияла ни на вхождение, ни на удаление железа. При нейтраль-
ных значениях pH гидроксильные группы обладают большим срод-
ством к Fe3+, чем к Fe2+. Если железо вошло в молекулу как
хелатное соединение Fe(II), хелатирующий агент может быть заме-
щен в процессе гидролиза и окисления до Fe3+. Протоны образу-
ются при гидролизе ионов Fe(III). Белок может промотировать
гидролиз, присоединяя эти протоны. Возможно, это имеет значе-
ние на начальных стадиях образования ядра.
В растворах концентрация ионов Fe(III) меньше той, при кото-
рой полиядерные комплексы неустойчивы, и железо находится
только в моноядерной форме. Для иона Fe(III) при pH 7 этот
«моноядерный барьер» соответствует концентрациям меньше
IQ-3-7 М [118]. В молекуле апоферритина эта концентрация пре-
вышена уже при наличии одного атома железа внутри централь-
ной полости. Следовательно, можно думать, что при дальнейшем
добавлении ионов Fe(III) будут образовываться полиядерные
комплексы, которые вскоре должны становиться слишком боль-
шими, чтобы пройти через пространство между субъединицами. Так
как полиядерные комплексы кристаллические, на их образование
могут влиять факторы, обычно влияющие на образование ядер и
326
Глава 8
рост кристаллов. Число ядер, скорость роста и число и размер
кристаллов, которые в конечном счете образуются, зависят от сте-
пени перенасыщения, температуры и наличия центров зарождения
ядер. В случае ферритина повышение pH и концентрации железа
может вызвать увеличение числа ядер и быстрое осаждение гидра-
та оксида, что, по-видимому, приводит к образованию менее плот-
но заполненных ядер. Возможно, различия в значениях pH — это
один из факторов, объясняющих различия в процессе воссоздания,
иллюстрируемые рис. 8.10,а-—в. Понижение температуры от 25
до 0 °C ухудшает процесс воссоздания по методу Билиха и Байера.
Кристаллическое ядро характеризуется высоким отношением по-
верхности к объему и большой поверхностной энергией. Если бы
оно не могло расти дальше критического размера, оно бы раство-
рялось. Только при размерах больше критического энергетически
выгоднее добавлять атомы к этому ядру, чем начинать образова-
ние других ядер, чем можно объяснить результаты, показанные на
рис. 8.10, в и 8.11, а. В условиях эксперимента, показанного на
рис. 8.11, а, критическая стадия достигается при концентрации при-
мерно 2—3% железа (200—300 атомов Fe на молекулу). Сравнение
рис. 8.11, а с графиком зависимости отношения поверхность/объем
от содержания железа, рассчитанным в предположении, что все
железо находится только в кубических кристаллитах (рис. 8.11,6),
показывает, что поверхностная энергия кристаллитов, возможно,
является важным фактором в связывании железа. Согласно
рис. 8.11, а, молекулы, содержащие свыше 10% железа, захваты-
вают меньше железа. Возможно, это объясняется тем, что ком-
понента, содержащая железо, включена в белок и теперь белок
уже наполовину заполнен, а поверхность, к которой могут добав-
ляться новые ионы, уменьшилась. Из рис. 8.10, в также следует,
что в условиях такого воссоздания добавление атомов железа
к частично заполненным молекулам является более выгодным, чем
образование новых ядер внутри новых молекул апоферритина.
Теория роста кристаллов показывает, что атом, добавленный к су-
ществующему слою, в основном будет удерживаться сильнее, чем
атом, образующий новый слой. Результаты Фишбаха и Андерегга
[26] по рассеянию под малыми углами на ферритине с низким
содержанием железа свидетельствуют о поперечном росте кристал-
лов. Когда железо медленно осаждается в ферритине, может полу-
читься небольшое число кристаллитов, возможно только по одному
в большом числе молекул заполненного ферритина [93]. И наобо-
рот, быстрое накапливание может привести к образованию боль-
шого числа ядер и возникновению менее заполненных молекул. Из
таких молекул железо легче удалить. Возможно, биологически вы-
годно, чтобы ферритин содержал смесь молекул так, чтобы из не-
которых молекул железо можно было бы взять быстрее, чем из
других.
Ферритин
327
11. МОБИЛИЗАЦИЯ ЖЕЛЕЗА ИЗ ФЕРРИТИНА
Механизм мобилизации железа из ферритина in vivo все еще
остается спорным. Железо может освобождаться из интактных
молекул, из ядер ферритина после разрушения белка или из тех
и из других. Выделение железа может осуществляться либо путем
восстановления до более растворимых ионов Fe2+, либо посредст-
вом образования хелатных соединений Fe3+, либо и тем, и другим
путем.
Предположили, что гемосидерин состоит главным образом из
ядер ферритина, белковая оболочка которого «снята» в результа-
те протеолиза [30]. Гемосидерин дает такие же дифрактограммы,
как ферритиновые ядра, хотя размер его кристаллитов в среднем
меньше [42]. Однако исследование методом меченых атомов пока-
зало, что хотя некоторая часть входящего в гемосидерин железа
получается из ферритина, при очень высоких концентрациях желе-
за он образуется независимо [119].
Способность восстанавливающих агентов, цистеина, аскорбино-
вой кислоты и глутатиона выделять железо из ферритина при pH
7,4 свидетельствует о вероятности восстановительного механизма
in vivo [116, 120]. Опыты Грина и Мазура [121] показали, что
ферритиновое железо может действовать как акцептор электронов
по отношению к восстановительной ксантиноксидазе и что этот
процесс может приводить к выделению железа из ферритина
в плазму в условиях низкого напряжения кислорода.
Предложен также механизм с участием хелаторов, однако био-
логический хелатирующий агент не был идентифицирован [78,
122]. Хелатирующие агенты, обладающие высоким сродством
к железу, медленно выделяют его из ферритина. В качестве хела-
тирующего агента был успешно применен дезферриоксамин [123],
1,10-фенантролин [124] и нитрилуксусная кислота [125]. В срав-
нимых условиях ЭДТА освобождает мало железа '[125]. Выделе-
ние железа с помощью хелатирующего агента — медленный про-
цесс, требующий нескольких недель. Для того чтобы хелатирую-
щий агент мог конкурировать с гидроксилом, он должен иметь
высокую константу связывания. Остается неясным, является ли
этот механизм единственным механизмом освобождения железа
in vivo или, возможно, играют роль и восстановление, и хелатиро-
рование. Для решения этой проблемы требуются дальнейшие
исследования.
ДОПОЛНЕНИЯ, ВНЕСЕННЫЕ В КОРРЕКТУРУ
Подтверждение того, что ферритиновый белок построен из
24 субъединиц, было получено в результате определения молеку-
лярной массы методом седиментационного равновесия и гель-
фильтрации в 6 М гидрохлориде гуанидина [126, 127].
328
Глава 8
Недавно было сообщено, что частично очищенный фермент,
ферриредуктаза из печени млекопитающих, способен катализиро-
вать восстановление связанного ферритином Fe(III) в свободное
Fe(II) [128]. Свойства этого фермента указывают, что он не яв-
ляется ксантиноксидазой.
СПИСОК ЛИТЕРАТУРЫ
1. Michaelis L., Adv. Protein Chem., 3, 53 (1947).
2. Harrison P. M., Iron Metabolism: an International Symposium, Springer-Ver-
lag, Berlin, 1964, p. 40.
3. Kato T„ Seikagaku, 41, 61 (1969).
4. Roche J., Bessis M., Breton-Gorius J., Compt. rend. Acad. Sci., 252, 3886
(1961).
5. Towe К. M., Lowenstam H. A., J. Ultrastruct. Res., 17, 1 (1967).
6. Towe К. M., Lowenstam H. A., Nesson M. H„ Science, 142, 63 (1963).
7. Heneine I. F„ Gazzinelli G„ Tafuri B7. L., Comp. Biochem. Physiol., 28, 391
(1969).
8. Baba A., J. Biochem., 65, 915 (1969).
9. Hyde В. B„ Hodge A. J., Kahn A., Birnstiel M. L., J. Ultrastruct. Res., 9, 248
(1963).
10. Robards A. W., Humpherson P. G„ Planta, 76, 169 (1967).
11. Seckbach J., J. Ultrastruct. Res., 22, 413 (1968).
12. Peat A„ Banbury G. H.. Planta, 79, 268 (1968).
13. David C. N„ Dissert Abstr., 29, 2733 (1969).
14. Laufberger V., Bull. Soc. Chim. Biol., 19, 1575 (1937).
15. Hahn P., Granick S., Bede W., Michaelis L., J. Biol. Chem., 150, 407 (1943).
16. Granick S., Chem. Rev., 38, 379 (1946).
17. Bothwell T. H., Finch C. A., Iron Metabolism, Little, Brown & Company,
Boston, 1962.
18. Cook S. F.. J. Biol. Chem., 82, 595 (1929).
19. Granick S., J. Biol. Chem., 164, 737 (1946).
20. Conrad M. E., Crosby W. H„ Blood, 22, 406 (1963).
21. Granick S., J. Biol. Chem., 146, 451 (1942).
22. Suran A. A., Tarver H., Arch. Biochem. Biophys., Ill, 399 (1965).
23. Harrison P. M., Gregory D. W., J. Mol. Biol., 14, 626 (1965).
24. Mazur A., Litt L, Shorr E., J. Biol. Chem., 187, 473 (1950).
25. Rothen A., J. Biol. Chem., 152, 679 (1944).
26. Fischbach F. A., Anderegg J. W„ J. Mol. Biol., 14, 458 (1965).
27. Michaelis L., Coryell C. D., Granick S., J. Biol. Chem., 148, 463 (1943).
28. Granick S., Hahn P. F., J. Biol. Chem., 155, 661 (1944).
29. Towe К- M., Bradley W. F„ J. Colloid Interface Sci., 24, 384 (1967).
30. Matioli G. T., Baker R. F„ J. Ultrastruct. Res., 8, 477 (1963).
31. Drysdale J. W., in «Regulatory Mechanisms for Protein Synthesis in Mamma-
lian Cells», Academic Press, New York, 1968, p. 431.
32. Harrison P. M., Gregory D. W„ Nature, 220, 578 (1968).
33. Farrant J. L„ Biochim. Biophys. Acta, 13, 569 (1954).
34. Richter G. W., J. Biophys. Biochem. Cytol., 6, 531 (1959).
35. Harrison P. M., J. Mol. Biol., 1, 69 (1959).
36. Harrison P. M., J. Mol. Biol., 6, 404 (1963).
37. Kleinwachter V., Arch. Biochem., 105, 352 (1964).
38. Granick S., Michaelis L., J. Biol. Chem., 147, 91 (1943).
39. Behrens M., Taubert M., Hoppe-Seyler's Z. Physiol. Chem., 290, 156 (1952).
40. Bielig H.-L, Kratky 0., Rohns G., Wawra H., Biochim. Biophys. Acta, 112,
110 (1966).
Ферритин
329
41. Richter G. W., Walker G. F., Biochemistry, 6, 2871 (1967).
42. Harrison P. M., in «Iron Metabolism and Anemia», Pan American Health Orga-
nization, Scientific Publications № 184, 1969, p. 2.
43. Hofmann T., Harrison P. M., J. Mol. Biol., 6, 256 (1963).
44. Harrison P. M., Hofmann T., Mainwaring W. I. P., J. Mol. Biol., 4, 251
(1962).
45. Spies J. R„ Anal. Chem., 37, 1412 (1967).
46. Harrison P. M„ Hofmann T., J. Mol. BioL, 4, 239 (1962).
47. Crichton R. R„ Bryce C. F. A., Febs. Letters, 6, 121 (1970).
48. Crichton R. R., Biochim. Biophys. Acta, 194, 34 (1969).
49. Williams M. A., Harrison P. M., Biochem. J., 110, 265 (1968).
50. Shiniyo S., Seikagaku, 39, 23 (1967).
51. Kopp R., Vogt A., Maass G., Nature, 198, 892 (1963).
52. Richter G. W„ Lab. Invest., 12, 1026 (1963).
53. Theron J. J., Hawtrey A. O., Schirren V., Clin. Chim. Acta, 8, 165 (1963).
54. Makino У., Konno K-, J. Biochem., 65, 471 (1969).
55. Righetti P. G., Drysdale J. W., Malt R. A., Federation Proc., 29, 813 (1970)
Abs.
56. Alfrey С. P., Lynch E. C„ Whitley C. C., J. Lab. Clin. Med., 70, 419 (1967).
57. Gabuzda T. G.. Gardner F. H., Blood, 29, 770 (1967).
58. Gabuzda T. G., Pearson J., Biochim. Biophys. Acta, 194, 50 (1969).
59. Crichton R. R., Millar J. A., Cumming R. L. C„ Biochem. J., 117, 35P (1970).
60. Hoy T. G., Harrison P. M., Acta Cryst. Congress. Suppl., S261 Abstr. (1969).
61. Richter G. W., J. Exp. Med., 109, 197 (1959).
62. Bessis M., Breton-Gorius Compt. Rend. Acad. Sci., 250, 1360 (1960).
63. Muir A. R., Quart. J. Exp. Physiol., 45, 192 (1960).
64. van Bruggen E. F. J., Wiebenga E. H., Gruber M., J. Mol. Biol., 2, 81 (I960).
65. Haggis G. H., J. Mol. Biol., 14, 598 (1965).
66. Haggis G. H., Sixth International Congress for Electron Microscopy, Maru-
zen Co., Ltd., Kyoto, 1966, p. 127.
67. Haydon G. B., J. Microsc., 89, 251 (1969).
68. Haydon G. B., J. Microsc., 91, 65 (1970).
69. Haggis G. H„ J. Microsc., (1970) in press.
70. Crewe A. V., Wall J., J. Mol. Biol., 48, 375 (1970).
71. Harrison P. M., Fischbach F. A., Hoy T. G., Haggis G. H„ Nature, 216, 1188
(1967).
72. Girardet J.-L., Lawrence J. J., Bull. Soc. Fr. Mineral Cristallog., 91, 440
(1968).
73. van der Giessen A. A., J. Inorg. Nucl. Chem., 28, 2155 (1966).
74. van der Giessen A. A., Thesis, Technical University, Eindhoven, 1968.
75. Spiro T. G„ Allerton S. E., Renner J., Terzis A., Bills R., Saltman P., J. Am.
Chem. Soc., 88, 2721 (1966).
76. Spiro T. G„ Pape L., Saltman P., J. Am. Chem. Soc., 89, 5555 (1967).
77. Brady G. W„ Kurkjian C. R., Lyden E. F. X., Robin M. B., Saltman P„ Spi-
ro T„ Terzis A., Biochemistry 7, 2185 (1968).
78. Spiro T. G., Saltman P„ Structure and Bonding, 6, 116 (1969).
79. Mulay L. N., Seiwood P. W., J. Am. Chem. Soc., 77, 2693 (1955).
80. Schoffa G., Z. Naturforsch., 20b, 167 (1965).
81. Schugar H., Walling C., tones R. B., Gray H. B., J. Am. Chem. Soc., 89, 3712
(1967).
82. Blaise A., Chappert J., Girardet J. L., Compt. rend. Acad. Sci., 261, 2310
(1965).
83. Blaise A., Feron J., Girardet J. L., Lawrence J. J., Compt. rend. Acad. Sci.,
265B, 1077 (1967).
84. Boas J. F„ Window B., Aust. J. Phys., 19, 573 (1966).
85. Girardet J.-L., Thesis, University of Grenoble (1969).
86. Neel L., J. Phys. Soc. Japan, 17, 676 (1962).
330
Глава 8
87. Armstrong R. J., Merrish A. H., Sawatzky G. A., Phys. Lett., 23, 414 (1966).
88. Dezsi I., Keszthelyi L., Kulgawizuk D., Molnar B., Eissa N. A., Phys. Stat.
Sol., 22, 617 (1967).
89. Kundig W., Bommel H., Constrabaris G., Lindquist R. H., Phys. Rev., 142, 327
(1965).
90. Rossiter M. J., Hodgson A. E. M., J. Inorg. Nucl. Chem., 27, 63 (1965).
91. Pauling L„ Sturdivant I. H., Z. Krist., 68, 239 (1928).
92. Fischbach F. A., Harrison P. M., Hoy T. G., J. Mol. Biol., 39, 235 (1969).
93. Harrison P. M., Hoy T. G., J. Microsc., 91, 61 (1970).
94. Crichton R. R., Biochim. Biophys. Acta, 229, 75 (1971).
95. Listowsky I., Betheil J. J., Englard S., Biochemistry, 6, 1341 (1967).
96. Mazur A., Shorr E., J. Biol. Chem., 182, 607 (1950).
97. Fineberg R. A., Greenberg D. M., J. Biol. Chem., 214, 97 (1955).
98. Fineberg R. A., Greenberg D. M., J. Biol. Chem., 214, 107 (1955).
99. Loftfield R. B., Eigner E. A., J. Biol. Chem., 231, 925 (1958).
100. Richter G. W., Nature, 190, 413 (1961).
101. Matioli G. T., Eylar G. H., Proc. Natn. Acad. Sci. U. S., 52, 508 (1964).
102. Saddi R., von der Decken A., Biochim. Biophys. Acta, 90, 196 (1964).
103. Drysdale J. W., Munro H. N., J. Biol. Chem., 241, 3630 (1966).
104. M'dler A. O. A., Biochim. Biophys. Acta, 155, 262 (1968).
105. Smith J. A., Drysdale J. W., Goldberg A., Munro H. N„ Br. J. Haemat., 14, 79
(1968).
106. Yoshino У., Manis J., Schachter D., J. Biol. Chem., 243, 2911 (1968).
107. Chu L. L. H., Fineberg R. A., J. Biol. Chem., 244, 3847 (1969).
108. Pape L., Multani J. S., Stitt C„ Saltman P., Biochemistry, 7, 606 (1968).
109. Drysdale J. W., Haggis G. H., Harrison P. M„ Nature, 219, 1045 (1968).
110. Smith-Johannsen H., Drysdale J. W„ Biochim. Biophys. Acta, 194, 43 (1969).
111. Bielig H.-J., Bayer E., Naturwiss., 42, 125 (1955).
112. Loewus M. W., Fineberg R. A., Biochim. Biophys. Acta, 26. 441 (1957).
113. Miller I. P. G., Perkins D. J., Eur. J. Biochem., 10, 146 (1969).
114. Mazur A., Green S„ Carleton A., J. Biol. Chem., 235, 595 (1960).
115. Mazur A., Carleton A., Carlsen A., J. Biol. Chem., 236, 1109 (1961).
116. Mazur A., Baez S., Shorr E., J. Biol. Chem., 213, 147 (1955).
117. Mazur A., in «Essays in Biochemistry», John Wiley & Sons, New York, 1956,
p. 198.
118. Schubert J., in «Iron Metabolism: An International Symposium», Springer-
Verlag, Berlin, 1964, p. 466.
119. Sturgeon P., Shoden A., in «Iron Metabolism: An International Symposium»,
Springer-Verlag, Berlin, 1964, p. 121.
120. Bielig H.-J., Bayer E., Naturwiss., 42, 466 (1955).
121. Green S., Mazur A., J. Biol. Chem., 227, 653 (1957).
122. Saltman P., J. Chem. Educ., 42, 682 (1965).
123. Wohler F., Acta Haemat., 30, 65 (1963).
124. Jones M. M., Johnson D. O., Nature, 216, 509 (1967).
125. Pape L., Multani J. S., Stitt C., Saltman P., Biochemistry, 7, 613 (1968).
126. Bryce C. F. A., Crichton R. R., J. Biol. Chem., 246, 4198 (1971).
127. Bjork I., Fish W. W., Biochemistry, 10, 2844 (1971).
128. Osaki S., Sirivech S., Federation Proc., 30, 1292 Abstr. (1971).
ГЛАВА 9
ТРАНСФЕРРИНЫ (СИДЕРОФИЛИНЫ)
П. Айсен
Alsen Р., Department of Biophysics, Albert Einstein
College of Medicine, New York, USA
1, ИСТОРИЯ и НОМЕНКЛАТУРА
Еще в 1920 г. [1] в сыворотке крови была обнаружена негемо-
вая фракция, которую нельзя было выделить ни ультрафильтра-
цией, ни диализом. Вторая мировая война послужила толчком
к исследованию белковой фракции крови, однако в то время еще
нельзя было установить точную физико-химическую природу этой
железосодержащей компоненты крови. Холмберг и Лаурелл в Шве-
ции доказали наличие в крови i[2] железосодержащего белка,
который они назвали трансферрином [3]. В это же время Шэйд и
Каролин в США показали, что бактериостатическая активность
сыворотки крови частично обусловлена белком, проявляющим
чрезвычайную жадность к железу, который поэтому был назван
ими сидерофилином {4, 5]. Позднее Шэйд и Каролин [6] устано-
вили, что этот белок по способности связывать железо похож на
кональбумин — белок, присутствующий в яичном белке. Ранее
в 1939 г. железосодержащий белок был обнаружен в коровьем мо-
локе. Ему дали точное, но прозаическое название «красный белок
молока» [7]. Впоследствии было замечено сходство этого белка
с трансферрином и кональбумином. С тех пор возникали разногла-
сия и споры относительно выбора подходящего названия для этого
связывающего железо белка, который, как было показано, присут-
ствует во многих жидкостях тела и тканях.
Название «трансферрин» общепринято в медицинской литера-
туре. Поскольку это название точно отражает, по-видимому, наи-
более важную биологическую функцию сывороточного белка, его
сохранение представляется и резонным, и полезным. Белок, выде-
ленный из яичного белка, по своей первичной полипептидной
структуре, по-видимому, идентичен белку, выделенному из сыво-
ротки крови тех же видов ,[8], так что, вероятно, оправданно и на-
звание «овотрансферрин», используемое Фини и его сотрудниками;
тем не менее были и возражения против этого названия на том
332
Глава 9
основании что еще не установлена функция этого белка как пере-
носчика железа [9]. Поэтому термин «кональбумин» будет со-
хранен в этом обзоре как признание его близкого сходства с сыво-
роточным белком. С другой стороны, за исключением своих цент-
ров связывания, железосвязывающие белки молока и других сек-
реторных жидкостей структурно, по-видимому, совершенно отличны
от белков сыворотки крови [10, 11], так что желательно иметь
свое название для этого класса белков, связывающих железо. Так
как первым и наиболее тщательно изученным источником этого
белка было молоко, для него было предложено и получило всеоб-
щее признание название «лактоферрин»; используется также тер-
мин «красный белок молока». Чтобы подчеркнуть общие свойства
этого железосвязывающего белка, Фини и сотр. [9] предложили
для молочного белка название «лактотрансферрин». Это вносит
некоторую путаницу, так как, кроме белков, характерных для
внешней секреции, молоко некоторых видов содержит еще железо-
связывающие белки, почти не отличимые и, возможно, производ-
ные от белков крови [12]. Кроме того, в молоке кролика преобла-
дающий и, возможно, единственный железосвязывающий белок
идентичен (за исключением того, что он еще содержит сиаловую
кислоту) сывороточному белку, хотя, вероятно, его выделяют мо-
лочные железы [13]. Поэтому разумно, по-видимому, сохранить
новый термин «лактоферрин» для белков, характерных для молока
и внешних секреций, а найденный в молоке белок, который по
своей структуре напоминает сывороточный трансферрин, называть
«молочным трансферрином».
Недавно Шэйд и сотр. [14] предложили термин «эккриносиде-
рофилин» для белка лактоферринового типа, выделенного из жид-
кости внутренней секреции. Хотя сообщения о том, что идентичный
белок найден во многих тканях [15], делают это предложение
иногда неуместным, в этом обзоре будет использоваться название
«лактоферрин».
Таким образом, в соответствии с первичной структурой белка
можно выделить два обширных класса железосодержащих белков:
сывороточный трансферрин и кональбумин из яичного белка,
с одной стороны, и лактоферрины молока, слез, желудочного сока,
бронхиальной и тканевой секреции, — с другой. Общее отличитель-
ное свойство этих белков — их уникальная способность связывать
металл, поэтому общее название «сидерофилины» для всех белков,
обладающих такой способностью, наиболее точно их описывает.
Однако термин «трансферрин» получил широкое распространение
и служит для того, чтобы объединить все члены этого класса со-
единений с наиболее тщательно изученным прототипом этих бел-
ков — сывороточным трансферрином. Поэтому пока не достигнуто
согласие между всеми работающими в этой области, для облегче-
ния общения следует сохранить оба термина.
Трансферрины (сидерофилины)
333
2. ВЫДЕЛЕНИЕ И ОЧИСТКА
2.1. Сывороточный трансферрин
Первые препараты сывороточного трансферрина были получе-
ны по методу Кон [16], а также фракционированием с помощью1
сульфата аммония в сочетании с осаждением этанолом [I]. Впо-
следствии стали использовать ионообменную хроматографию,
которая проще и, вероятно, мягче, чем методы, в которых исполь-
зуется спирт [17]. Последнее замечание, по-видимому, имеет неко-
торое значение, так как Шэйд [18] указывал, что некоторые пре-
параты трансферрина могут быть физиологически неактивны, хотя
их способность связывать железо остается неизменной. Самой
главной проблемой при очистке трансферрина было отделение его
от связанного с гемом сывороточного белка, гемопексина. Кристал-
лизацию сывороточного трансферрина проводили следующим об-
разом: постепенно добавляли спирт к концентрированному раство-
ру белка [19] с одновременной диффузией спирта в раствор белка
во время его концентрирования в аппарате для ультрафильтрации
[20]; кристаллы были также выделены из концентрированных
растворов при низкой ионной силе и низких значениях pH [13].
2.2. Кональбумин
Кональбумин был получен из белка куриных яиц фракциониро-
ванием с сульфатом аммония [21] или этанолом [22], а также
с помощью ионообменной хроматографии с использованием суль-
фометилцеллюлозы (СМ-целлюлоза) или сульфометилсефадекса
(СМ-сефадекс) [23]. Упрощенный метод для получения относи-
тельно больших количеств кональбумина, в котором используется
осаждение сульфатом аммония и ионообменная хроматография,
был предложен Фини и Коматчу [9]. Оба соединения — и апоко-
нальбумин и железосодержащий кональбумин — были выделены
в виде кристаллов из водно-этанольных растворов [21].
Следует отметить, что поступающие в продажу препараты ко-
нальбумина и трансферрина обычно гетерогенны и могут содер-
жать переменные количества низкомолекулярных веществ, в част-
ности хелатирующих агентов, используемых для их получения
[24].
2.3. Лактоферрин
В 1959 г. Гровс '[25] детально описал процесс выделения лакто-
феррина из осажденной кислотой казеиновой фракции коровьего
молока. Этот метод состоит в использовании фракционирования
334
Глава 9
с сульфатом аммония и хроматографии на диэтиламиноэтилцел-
люлозе и позволяет выделить приблизительно 1 г белка из 60 л
молока. Гораздо более простой метод выделения лактоферрина из
женского молока был предложен Юханссоном [26]. Этот метод
состоит в прямой адсорбции протеина на СМ-сефадексе, который
добавляют к молоку. Выходы составляют от 0,5 до I г белка
на 1 л молока; такое повышение выхода по сравнению с тем, ко-
торый был достигнут при использовании коровьего молока, воз-
можно, связано и с различием в методике, и с большей концентра-
цией лактоферрина в женском молоке. Кристаллический лакто-
феррин можно выделить из разбавленного фосфатного буфера без
использования спирта [26].
3. ХИМИЧЕСКИЙ СОСТАВ
3.1. Аминокислотный состав
В основном данные об аминокислотном составе и определении
N-концевой аминокислоты в трансферрине и кональбумине, по
сведениям из ряда лабораторий, находятся между собой в хоро-
шем согласии (табл. 9.1) и не содержат ничего необычного. Так
как было сообщено, что кональбумин и птичий сывороточный
трансферрин имеют одинаковую белковую часть [8], можно пола-
гать, что различия в аминокислотном составе между сывороточным
трансферрином человека и кональбумином отражают видовое раз-
нообразие. Оба — и трансферрин и кональбумин, — по-видимому,
содержат только по одному концевому N-аминокислотному остат-
ку в молекуле. Иногда сообщают о присутствии небольшого коли-
чества остатков второй аминокислоты; возможно, это обусловлено
недостаточной чистотой анализируемого препарата белка или ча-
стичным гидролизом чувствительных к нему пептидных связей
[30].
По аминокислотному составу лактоферрин человека и коровий
лактоферрин заметно отличаются от соответствующих сывороточ-
ных белков. По-видимому, лактоферрин человека должен содер-
жать мало или совсем не содержать триптофана, который благо-
даря его свойствам можно обнаружить спектрофотометрическим
методом (см. ниже). Один N-концевой остаток аланина был обна-
ружен в коровьем лактоферрине [29]. Еще никак не объяснено от-
сутствие аминокислотного остатка со свободной аминогруппой
в лактоферрине человека [31]*.
* Массон (частное сообщение) недавно показал, что лактоферрин человека
содержит 10 остатков триптофана на молекулу.
Трансферрины (сидерофилины)
335
Аминокислотный состав сидерофилинов
Таблица 9.1
го 600 г [8])
Аминокислота Сывороточный трансферрин человека/76 700 [27] Лактоферрин человека/77 ООО 1 [28] Коровий лакто- феррнн/77100 г [29] Коиальбумин/76i (данные работы |
Лизин 52 45 42 62
Гистндии 19 10 10 13
Аргинин 24 43 32 33
Аспарагиновая кислота 83 66 71 74
Треонин 31 28 39 35
Серин 41 42 45 42
Глутаминовая кислота 59 71 73 70
Пролин 31 34 31 31
Глнцнн 55 53 43 58
Аланин 60 58 59 52
Цистеин (‘/г цистина) 22 13 28 22
Валин 44 40 43 46
Метионин 4 5 4 11
Изолейцин 14 16 17 24
Лейцин 58 53 61 48
Тирозин 24 23 19 20
Фенилаланин 29 30 25 25
Триптофан 9 1 или 0 9 18
Для сравнения взята молекулярная масса 77 000. Единственное сообщенное значение
молекулярной массы лактоферрина человека равно 95 000 [9].
3.2. Пептидные карты
Дальнейшие доказательства структурных различий между
трансферрином и лактоферрином были получены в результате ис-
следования пептидных карт, полученных путем протеолитического
расщепления этих белков. Интересно отметить, что когда металл
присоединен к белку, наблюдается гораздо большая устойчивость
к протеолитическому расщеплению, чем в отсутствие металла [32].
Исследование белка человека показало, что в двумерных картах
336
Глава 9
триптического расщепления обнаруживается гораздо меньше пеп-
тидов, чем можно было бы ожидать на основании известного
числа имеющихся остатков лизина и аргинина [27]. Значение это-
го наблюдения будет обсуждаться в разделе, посвященном субъ-
единичной структуре трансферрина.
3.3. Углеводный состав и структура
Все известные сидерофилины правильно классифицировать как
гликопротеины. Трансферрин содержит около 5% углевода, кото-
рый расположен в двух разветвленных цепях, идентичных по со-
ставу и оканчивающихся остатком сиаловой (N-ацетилнейрамино-
вой) кислоты и, возможно, присоединенных к полипептидной цепи
аспарагинильными связями [33]. Концевые остатки сиаловой кис-
лоты могут быть полностью удалены обработкой нейраминидазой
[34]. Сообщалось, что трансферрин, дефицитный по нейраминовой
кислоте и, возможно, другим углеводам, находится в крови пупо-
вины и цереброспинальной жидкости [35]. После удаления сиало-
вой кислоты из трансферрина с помощью нейраминидазы белок
может служить субстратом для галактозоксидазы, что указывает
на присоединение нейраминовой кислоты концом к галактозе с об-
разованием углеводных целей [36].
Хотя, по-видимому, кональбумин и сывороточный трансферрин
домашних птиц идентичны по своему аминокислотному составу,
иммунохимической реакционной способности и структуре пептидов,
в их углеводных компонентах замечены отчетливые различия.
В гликопептиде яичного белка не обнаружено ни сиаловой кисло-
ты, ни галактозы. В отличие от сывороточного белка человека
в белках птиц углевод, по-видимому, должен находиться в одной
цепи [37].
Было показано, что лактоферрин человека содержит около
6,6% углевода, расположенного в трех цепях, каждая из которых
оканчивается остатком сиаловой кислоты и содержит один или два
остатка фукозы, шесть остатков гексозы и четыре остатка N-ацет-
глюкозамина [38]. Представлены также доказательства того, что
углеводные цепи связываются с белковой частью при помощи гли-
козидной связи к гидроксильной группе треонина. Углеводный со-
став коровьего лактоферрина, по-видимому, аналогичен со-
ставу белка человека, за исключением того, что в нем обнаружен
только один остаток сиаловой кислоты на молекулу [25].
Функциональная и структурная роль углеводных частей сидеро-
филинов еще не выяснена. По-видимому, удаление сиаловой кисло-
ты из трансферрина не влияет на ее металлсвязывающие свойства
и на ее способность служить донором железа для ретикулоцитов
Трансферрины, (сидерофилины)337
3.4. Иммунохимия
Все сидерофилины — антигены. Кональбумин и трансферрин,
полученные из яичного белка и сыворотки домашних птиц, имму-
нохимически идентичны, хотя они синтезируются в разных местах
{8]. В отличие от этого нет перекрестной иммунореактивности
между трансферрином из сыворотки человека и лактоферрином
человека [40]. Это почти определенно указывает на то, что белки
структурно не родственны один другому, за исключением их спо-
собности связывать металл, и что белки молока не являются про-
изводными белков крови.
4. ОСНОВНЫЕ ФИЗИЧЕСКИЕ СВОЙСТВА
4.1. Молекулярная масса
Более ранние определения молекулярной массы сывороточного
трансферрина с помощью ультрацентрифугирования дали значе-
ния, изменяющиеся от 68 000 до 03 000. Недавно проведенные ис-
следования показали, что молекулярная масса изменяется от 75 000
до 80 000 дальтон [27]. В растворах нативного трансферрина не
наблюдается тенденции к образованию агрегатов или диссоциации
[27]. Молекулярная масса не зависит от того, насыщен ли транс-
феррин железом, наполовину насыщен или вовсе не содержит же-
леза [41].
Кональбумин и лактоферрин изучены в меньшей степени, чем
трансферрин. Наиболее точная оценка молекулярной массы ко-
нальбумина, определенная по его способности связывать железо,
дала значение около 76 000 дальтон [42]. Методом седиментацион-
ного равновесия для коровьего лактоферрина получена молекуляр-
ная масса 77 100 [29], для лактоферрина человека исследованием
связывания железа получено значение 80 000 [43], а методом оп-
ределения скорости седиментации при ультрацентрифугировании
[11] —значения 95 000 [11] и 82 000 [14].
4.2. Оптические свойства
В табл. 9.2 приведены молярные коэффициенты поглощения
комплексов трансферрина в ультрафиолетовой и видимой обла-
стях. Особенно интересным является увеличение поглощения в уль-
трафиолетовой области при связывании ионов металла ,[44];
причина этого увеличения будет рассмотрена ниже.
При связывании с ионами металла наблюдается гашение флуо-
ресценции триптофана в трансферрине [46].
22—2451
338
Глава 9
Таблица 9.2
Молярные коэффициенты поглощения сывороточного трансферрина человека
Белок X., нм .1% см Литература
Сывороточный апотраисферрин человека 280 11,4 9
Комплекс сывороточного трансферрина
человека
с Fe3+ 280 14,3 44
470 0,57 9
с Сг3+ 280 12,75 44
440 0,22 45
615 0,18 45
с Со3+ 280 14,3(45)
405 1,2 9, 44
с Мп3+ 280 14,3 44
330 1,3 45
430 1,2 45
4.3. Гидродинамические свойства
Основываясь на измерении диэлектрической дисперсии и внут-
ренней вязкости, молекулу трансферрина в водном растворе ап-
проксимируют вытянутым эллипсоидом вращения с отношением
главных осей 3,0 :4,5 ;[27, 47]. Так как связывание с ионами ме-
таллов либо вообще не вызывает, либо вызывает небольшое из-
менение внутренней вязкости и совсем не изменяет диэлектриче-
ские свойства [28, 48], можно предположить, что геометрия и
гидратация молекулы при связывании с ионом металла существен-
но не изменяются.
Измерения зависимости скорости протонной релаксации от
частоты в растворах трансферрина позволили заключить, что при-
близительно только 13 (или 2%) молекул воды в первой гидрат-
ной оболочке белка связаны относительно прочно (невращатель-
но); это число увеличивается на два, когда белок насыщен метал-
лом [49, 50].
На том основании, что скорость обмена водорода в апокональ-
бумине больше, чем в комплексе белка с металлом, Улмер [51]
предположил, что белок, насыщенный металлом, имеет более ком-
пактную конформацию.
Трансферрины (сидерофилины) 339
4.4. Субъединичная структура
Можно ожидать, что белок с двумя специфическими центрами
и двумя идентичными углеводными цепями построен из двух субъ-
единиц. Для выяснения этого было поставлено множество экспе-
риментов в ряде лабораторий. Джеппсон [52] сообщил, что вос-
становленный алкилированный трансферрин после высушивания
и повторного растворения диссоциирует на субъединицы с молеку-
лярной массой 39 000—42 000, как было определено методом рав-
новесного центрифугирования. Исследование восстановленного
алкилированного трансферрина независимо в лабораториях
Безкоровайного [53] и Фини [54] не подтвердили диссоциацию на
субъединицы. Впоследствии Безкоровайный [55] при изучении
восстановленного алкилированного и сульфитолизированного
трансферрина и кональбумина вновь не получил данных, свиде-
тельствующих о субъединичной структуре этих соединений. Дейст-
вительно, измерения вязкости обработанного трансферрина
в присутствии 8 М раствора мочевины показали только тенденцию
к образованию агрегатов [55]. Недавно проведенное тщательное
исследование вопроса о субъединичной структуре трансферрина
с использованием методов седиментационного равновесного цент-
рифугирования, измерения внутренней вязкости и гель-фильтрации
в 6 М гидрохлориде гуанидина подтвердило одноцепочечную при-
роду сывороточного трансферрина человека [27]. Если бы субъ-
единицы действительно существовали, они должны были бы соеди-
няться необычными и еще неизвестными связями. В свете имею-
щихся данных заманчивым выглядит предположение о том, что
структурный ген для трансферрина в ходе эволюции подвергся
дубликации и последующему делению [27, 54].
5, МЕТАЛЛСВЯЗЫВАЮЩИЕ ЦЕНТРЫ
5.1. Существует ли взаимодействие
между связывающими центрами?
После выделения относительно чистого трансферрина и кональ-
бумина удалось установить, что каждый белок способен специфи-
чески и прочно связывать два атома железа [1]. Было также по-
казано, что для развития характерной красной окраски требуется
двуокись углерода или одна из ее ионизированных форм [5]. Шэйд
и сотр. [5] смогли показать, что для полного развития окраски на
каждый связанный атом железа требуется один бикарбонатный
ион. При этом два или три протона выделяются в раствор.
Количественное изучение механизма комплексообразования
железа с кональбумином было предпринято Вернером и Вебером
22*
340
Глава 9
[42], которые установили, что связывание каждого атома железа
при pH от 7,5 до 9,5 сопровождается освобождением в раствор
трех протонов, а связывание с медью вызывает выделение двух
протонов. Хотя трудно определить, не освобождаются ли эти про-
тоны в результате гидролиза иона металла, претерпевающего свя-
зывание, представляется довольно вероятным, что они действитель-
но вытесняются из белка в процессе связывания, в частности
изучение равновесия связывания при несколько различающихся
значениях pH согласуется с этой точкой зрения [56]. Так как при
связывании двухвалентного катиона освобождаются два протона,
а при связывании трехвалентного иона — три, общий заряд белка
не изменялся бы, если бы никакие другие заряженные частицы не
участвовали в связывании. Однако измерение свободной электро-
форетической подвижности обнаружило повышение анионной
подвижности, соответствующее увеличению общего отрицательного
заряда белка на единицу на каждый связанный атом железа [42].
На основании этого факта, а также опытов Шэйда с сотрудника-
ми, пришли к заключению, что бикарбонатный ион является той
формой, в которой двуокись углерода принимает участие в металл-
связывающей функции трансферрина и кональбумина. Впослед-
ствии Массон и Хереманс [43] показали, что аналогичные явления
происходят при связывании железа и меди лактоферрином.
Таким образом, связывание первого и второго атомов железа
сидерофилинами можно представить следующими уравнениями:
Fe3+ + HeTfn + НСОГ ч—> [Fe^HgTfn -НСО3]“ + ЗН+ (1)
Fe3+ + [Fe3+H3Tfn-HCO3]_ + HCOr [(Fe3+)2Tfh-(HCO3)2]2-+ ЗН+ (2)
Такая формулировка послужила поводом к одному из наиболее
интересных и горячих споров в химии и физиологии сидерофили-
нов. Используя цитрат в качестве агента, конкурирующего за же-
лезо с сидерофилинами, и измеряя окраску растворов кональбуми-
на, в которых термодинамическая активность железа изменялась
при изменении концентрации цитрата, Вернер и Вебер [42] при-
шли к заключению, что связывание железа кональбумином осу-
ществляется в две стадии, причем вторая константа устойчивости
гораздо больше, чем первая. Шэйд [57], исходя из нетермодина-
мических наблюдений за скоростью поглощения железа из белка
клетками печени, предложил такой же способ освобождения желе-
за из трансферрина. И, наконец, Вудворт [58] на основе измере-
ния кинетики связывания железа получил новые аргументы в поль-
зу двухступенчатого механизма связывания железа кональбуми-
ном. Следовательно, до последнего времени преобладало мнение,
что все время два атома железа входят и выходят из молекулы
сидерофилина, так что стабильные частицы в растворе должны
Трансферрины (сидерофилины)341
либо вовсе не содержать, либо содержать 2 атома железа на мо-
лекулу сидерофилина.
В 1963 г. Мальмстрём с сотр. [56] опубликовал серию работ,
в которых изложил результаты тщательно выполненных экспери-
ментов с использовании классического термодинамического метода
равновесного диализа, дополненного методом электронного пара-
магнитного резонанса. Уделяя особое внимание тому, чтобы пока-
зать, что равновесие действительно достигается, и обрабатывая
свои данные новым методом графического анализа, эти исследова-
тели заключили, что связывание железа трансферрином можно
описать, предположив участие в связывании двух эквивалентных
невзаимодействующих центров. Если это так, то напрашивается
вывод, что трансферрин в растворах не полностью насыщен желе-
зом и должны сосуществовать три типа белковых частиц: частицы
с двумя связанными атомами железа, частицы только с одним свя-
занным атомом и частицы, не содержащие металла [59]. Вернер
и Вебер [21] на основании электрофоретических диаграмм, скорее
интуитивно, различили три типа молекул в неполностью насыщен-
ных железом растворах кональбумина. Однако они не сопоставили
это наблюдение со своими заключениями, сделанными на основа-
нии исследования равновесия. Позднее это предположение было
детально подтверждено для трансферрина с помощью фронтально-
го электрофореза методом движущейся границы [59], а оконча-
тельно для кональбумина методом изоэлектрического фокусирова-
ния [60]. Было высказано предположение, что частицы, обладаю-
щие промежуточной подвижностью при электрофорезе и в опытах
по изоэлектрическому фокусированию, могут представлять собой
димер апопротеина и белка, насыщенного железом [61]. Однако
позднее было доказано, что это предположение несостоятельно
[41].
Вся сумма имеющихся в настоящее время данных подтвержда-
ет мнение о том, что термодинамические константы, описывающие
связывание железа с трансферрином, внутренне идентичны. По-
вторное изучение методом равновесного диализа связывания желе-
за кональбумином также показало большое сходство в связыва-
нии металла этим белком и трансферрином [62]. По-видимому,
более ранние данные Вернера и Вебера были получены до дости-
жения равновесия, так как в этих опытах проходило только 18 ч
между добавлением железа к раствору апокональбумина и време-
нем наблюдения; проведенное позднее исследование зависимости
от времени показало, что для достижения истинного равновесия
требуется по крайней мере несколько дней [56]. Физиологическое
исследование Шэйда не содержало термодинамических измерений
и поэтому не противоречит предшествующим результатам. Коли-
чественных данных о связывании железа с лактоферрином в на-
стоящее время не имеется.
342
Глава 9
5.2. Значение констант связывания
Существуют некоторые разногласия по поводу значения чисел,
полученных при измерении связывания металла с сидерофилинами
методом равновесного диализа. Константы связывания Ki и Кг
характеризуют химические реакции, представленные выше уравне-
ниями (1) и (2), и записываются в следующем виде {55]:
[Fe^HgTfn-НСОД [Н+]3
«1 - [Fe3+] [HeTfn] [НСО3]
„ l(Fs3+)2Tfn(HCO3)2~] [H+J^
л2 - [Fe3+] [Fe3+H3Tfn-HCO3] [HCO3]
Так как на каждый атом связанного железа освобождаются три
протона, а связывается один бикарбонатный ион, каждая из истин-
ных термодинамических констант прямо пропорциональна третьей
степени концентрации ионов водорода и обратно пропорциональна
концентрации бикарбонат-иона в растворах, в которых выполняют-
ся измерения. Статистическое рассмотрение показывает, что, если
два специфических связывающих центра эквивалентны и независи-
мы, отношение Ki к Кг должно быть равно четырем.
При фиксированных значениях pH и активности бикарбонат-
иона вклады этих составляющих в уравнения связывания можно
учесть в выражении для кажущихся констант равновесия следую-
щим образом [56]:
К, [НСО3] К1РСО2КСо2
(const. pH, рСО2) — [Н+]3 = [Н+]4 (5)
где Ксо2 — соответствующая константа гидратации СО2 или иони-
зации карбоновой кислоты. Так как активность бикарбонат-иона
находится в обратной зависимости от pH, в этом выражении
должна быть отражена зависимость кажущейся константы от чет-
вертой степени концентрации ионов водорода, при которой иссле-
довалось равновесие, и прямая зависимость от парциального
давления двуокиси углерода в атмосфере, в равновесии с которой
они находятся. При относительно фиксированных значениях pH и
рСО2, которые преобладают в кровеносной системе человека, ка-
жущееся значение Ki составляет примерно 1024 М-1. Учитывая все
изложенное, можно показать, что атому железа потребуется при-
близительно 10 000 лет, чтобы самопроизвольно отделиться от мо-
лекулы трансферрина в крови [63]. Из величин, которые исполь-
зуются для количественного описания связывания металлов
с трансферрином, по-видимому, наиболее важны с точки зрения
физиологии кажущиеся константы устойчивости.
Трансферрины (сидерофилины)
343
Если принять во внимание значения р/С протонодонорных групп
в белке, внутреннюю константу связывания металла с образова-
нием хелата можно записать в следующем виде:
К1 (хелат) = Кг Ю * (6)
При значениях pH, равных или больших 9,5, выделяются три
протона, так что значения р/С протонодонорных групп должны
превышать эту величину. В соответствии с этим внутренняя кон-
станта связывания железа трансферрином в хелат должна превы-
шать 1032; возможно, она составляет 1036 [56].
После того как была обнаружена идентичность констант свя-
зывания металла с двумя центрами белка, тотчас же возник во-
прос, идентичны ли сами связывающие центры. Эту проблему осо-
бенно трудно решить для белков, построенных из одной полипеп-
тидной цепи. Й физиологические, и физические исследования про-
лили свет на эту еще не до конца решенную проблему, к которой
мы ниже вернемся.
5.3. Типы и состояния ионов металлов,
специфически связанных с трансферрином
Кроме железа и меди, комплексы с которыми хорошо изучены,
белок способен связывать и многие другие многовалентные катио-
ны. Цинк, хром, кобальт, марганец, кадмий, никель и галлий об-
разуют комплексы такого же стехиометрического состава, как
железо [9, 44, 64, 65]. Связывание хрома, марганца, кобальта и
меди также сопровождается связыванием бикарбонат-иона [45].
Еще не исследовалось, участвует ли бикарбонат-ион в связывании
других ионов металлов. Индий также способен связываться
с трансферрином, однако еще не было показано, те ли же самые
специфические центры принимают участие в связывании металла
[66].
По-видимому, из-за гидролиза первоначальные исследования
связывания железа сидерофилинами были выполнены с использо-
ванием ионов Fe(II). На вопрос о валентном состоянии связанно-
го железа нельзя было ответить до тех пор, пока Эренберг и Лау-
релл [67] не измерили при комнатной температуре статические
магнитные восприимчивости и не показали, что специфически свя-
занный металл находится в высокоспиновом состоянии (da)
Fe(III). Парамагнитная восприимчивость иногда даже больше, чем
было предсказано для невзаимодействующих ионов Fe(III), так
что было выдвинуто предположение, что имеет место слабое взаи-
модействие между ионами металлов, связанными с одной и той же
молекулой. Однако последующий анализ спектров электронного
парамагнитного резонанса [57] и исследование статической вос-
приимчивости при низкой температуре [45] не подтвердили нали-
344
Глава 9
чие такого взаимодействия. Эти данные находятся в хорошем со-
гласии с законом Кюри, наблюдаемом при температурах между
1.5 и 77 К.
Все комплексы металлов с трансферрином имеют отчетливую
окраску и полосы поглощения в видимой области. Инман [9] на-
блюдал усиление характеристической окраски при образовании
комплексов с марганцем и кобальтом при добавлении перекиси во-
дорода и предположил, что, по-видимому, эти ионы металлов окис-
ляются при связывании до трехвалентного состояния. Такое пред-
положение подтверждается измерениями статической восприимчи-
вости при низких температурах, которые также показали, что
кобальт связан в нпзкоспиновой диамагнитной форме [45].
С точки зрения физиологии интересно знать, могут ли связы-
вающие центры трансферрина присоединять и ионы Fe(II) и ионы
Fe(III) или ионы Fe(II) должны быть окислены в Fe(III) до того,
как произойдет связывание. Неопубликованное исследование Габе-
ра, проведенное с применением дифференциальной спектрофото-
метрии и изотопного обмена, показало, что Fe2+ если и связывает-
ся, то очень слабо [68].
5.4. Роль бикарбоната
Характеристическая красная и желтая окраски комплексов же-
леза и меди с сидерофилинами не развиваются в отсутствие бикар-
боната. Отсюда следует, что этот ион играет главную роль
в комплексообразовании металлов с белками [5]. Прямое измере-
ние количества двуокиси углерода, выделяющейся при кислотной
денатурации комплексов с железом [42], медью [69], хромом,
марганцем и кобальтом [45], подтвердило сделанное ранее пред-
положение Шэйда [5] о том, что на каждый связанный ион метал-
ла связывается один бикарбонатный ион. Связывание бикарбоната
не является обязательным, и это было продемонстрировано серией
исследований связывания металла с трансферрином методом спек-
троскопии электронного парамагнитного резонанса, которые пока-
зали, что специфическое связывание, по крайней мере железа и
меди, может происходить и в отсутствие бикарбоната [70]. Обра-
зующиеся при этом комплексы были бесцветны и поэтому недетек-
тируемы до появления метода ЭПР. Очевидно, в отсутствие бикар-
боната связь железо — белок гораздо слабее, чем в его
присутствии, так как при стоянии не содержащего бикарбоната
комплекса железа с трансферрином при нейтральных или более
высоких значениях pH наблюдается гидролиз железа с образова-
нием нерастворимого гидроксида железа (III). Возможная физио-
логическая роль этого эффекта будет обсуждена в разделе, посвя-
щенном биологическим функциям сидерофилинов.
Трансферрины (сидерофилины)345
Центр, связывающий бикарбонат-ион, может также связывать
и многие другие анионы, в том числе этилендиаминтетраацетат,
нитрилтриацетат и оксалат [70, 71]. Общим свойством этих анио-
нов является наличие двух или более карбоксильных групп; еще
не известно, существенно ли это для того, чтобы анионный центр
связывания могли занять ионы, отличные от бикарбоната. О соот-
ношении и взаимном расположении связывающих центров бикар-
боната и металла также ничего не известно, хотя они должны
быть очень близки, так как место присоединения аниона может
быть занято только тогда, когда есть центр, связывающий металл.
Кроме того, было показано, что связыванию бикарбонат-иона соот-
ветствующей группой препятствует доступ воды как растворителя
к специфически координированному иону металла [72]. Когда свя-
зывание произошло, бикарбонат нельзя ни удалить длительным
откачиванием раствора белка, ни заменить на другой анион,
способный занять его место. Из изложенного следует, что, по-ви-
димому, бикарбонат связывается не только с ионом металла, но
еще каким-то образом присоединяется к одной или большему числу
групп в белке.
5.5. Кинетика связывания железа
Изучение кинетики комплексообразования металла с трансфер-
рином осложняется двумя факторами: скорость связывания может
быть такой большой, что превышает разрешающие способности
цбычных приборов; необходимо принять во внимание наличие по-
бочных реакций, включая реакции гидролиза и реакции замещения
лигандов в тех случаях, когда металл взаимодействует с белком
в виде хелатного соединения. Для исследования этой проблемы
Залтман и сотр. [24] для случая взаимодействия ионов Fe(III)
с белком в виде нитрилтриацетатного комплекса разделили про-
цесс связывания на две фазы: на более раннюю первую фазу,
которую они не смогли измерить, и вторую, более медленную фазу,
которая имеет кинетический первый порядок по концентрации
комплекса железа. Последнее наблюдение согласуется с термоди-
намическим изучением, показавшим независимость связывания
ионов Fe(III) [55]. Эти исследования расходятся с результатами,
сообщенными Вудвортом [58], который обнаружил простую зави-
симость второго порядка для скорости комплексообразования от
концентрации железа, которое и в этом случае присутствовало
в виде нитрилтриацетатного комплекса железа (III). Временные
пределы используемых физических методов и временная шкала
спектрометра таковы, что можно надеяться разрешить это противо-
речие, только используя специальные методы для изучения быст-
рых реакций, в частности спектрофотометрию с остановкой струи.
346
Глава 9
5.6. Спектроскопические исследования
5.6.1. Оптические исследования
Большинство комплексов металлов с сидерофилинами имеют
интенсивные полосы поглощения в видимой области, которые обус-
ловлены их характерной окраской (табл. 9.2). Детальный кванто-
вомеханический анализ хромофорных центров не проводился, так
что еще нет объяснения природы полос поглощения в видимой
области. Простой перенос заряда, вероятно, исключается [73].
По-видимому, разумно предположить, что эти полосы обусловлены
переходами между главным образом Зй-орбиталями центрального
иона металла, разрешенными вследствие гибридизации с 4р- и
4д-орбиталями, возникающей из-за напряженной геометрии коор-
динирующихся лигандных групп белка. Было показано, что это
действительно имеет место в случае координации меди с церуло-
плазмином [74]. Имеет смысл упомянуть здесь и о других заслу-
живающих внимания особенностях полос поглощения в видимой
области комплексов металлов с сидерофилином: в присутствии
бикарбоната комплекс меди имеет необычную глубокую желтую
окраску, обусловленную поглощением с максимумом при 430 нм,
в то время как в спектре комплекса хром—трансферрин—бикар-
бонат в видимой области обнаруживаются две отдельные полосы.
Методом дифференциальной спектрофотометрии в ультрафио-
летовой области были обнаружены изменения в спектрах при свя-
зывании ионов металлов с сидерофилинами [45, 64, 70]. В спектрах
растворов комплексов металлов по сравнению со спектрами рас-
творов апофермента максимумы поглощения наблюдаются вблизи
245 и 295 нм [64]. В литературе имеются некоторые разногласия
относительно формы и интенсивности дифференциальной полосы
при 295 нм [46]; возможно, они связаны с различиями в концент-
рации исследуемых растворов, которые приводят к разным вкла-
дам в приборную ошибку, связанную со светорассеянием или
с общими трудностями в измерении небольших различий между
большими величинами поглощения. По-видимому, дифференциаль-
ная полоса поглощения при 295 нм обусловлена воздействием свя-
занного иона металла на тирозильные остатки [76]. Остатки трип-
тофана также могут давать вклады в дифференциальные спектры
[63]. Если это так, то спектры лактоферина человека должны от-
личаться от спектров сывороточного трансферрина, так как послед-
ний либо вовсе не содержит, либо содержит мало триптофана.
Спектрофотометрическое титрование комплексов железа и меди
с сидерофилином также показало значение тирозильных остатков
в металлсвязывающей функции белка [75, 76]. В дифференциаль-
ных спектрах поглощения белка в ультрафиолетовой области
в присутствии щелочи по сравнению со спектрами при нейтраль-
Трансферрины (сидерофилины)
347
ных значениях pH обнаружены полосы поглощения при 295 нм,
которые очень похожи на полосы в спектре п-ацетил-ь-тирозина
при pH 10,8 по сравнению с его спектром при pH 7, т. е. на полосы
ионизированной фенольной ОН-группы в сравнении с неионизиро-
ванной [64]. Таким образом, различия в спектрах белков объяс-
няются ионизацией тирозина. Молярный коэффициент поглощения
дифференциальной полосы можно получить из спектра поглощения
полностью денатурированного щелочью белка по сравнению со
спектром нативного белка, приняв во внимание число тирозильных
остатков в молекуле или из измерений, выполненных на модель-
ных соединениях. Существуют некоторые расхождения между зна-
чениями, полученными дифференциальным методом, и значениями,
сообщенными для различных белков, и эти расхождения трудно
объяснить [64, 76]. Кроме того, значение молярного коэффициента
поглощения, сообщенное для случая щелочной денатурации, воз-
можно, нельзя прямо применять к измерениям на нативном белке
[76]. По-видимому, изменения, наблюдаемые при связывании ме-
талла с сидерофилинами, действительно, связаны с изменениями
в тирозильных остатках, несмотря на то, что не существует зави-
симости от числа этих остатков в молекуле белка. Тан и Вудворт
[64] объяснили свои данные, предположив, что в связывании
с железом участвуют два остатка тирозина, а в связывании
с медью — один; Ааса и Айзен [76] сообщили, что при связывании
с медью оказываются затронутыми 1,3 остатка тирозина, а Виш-
ниа, Вебер и Вернер [75] установили, что в связывании с железом
участвуют три таких остатка. Следует указать, что различие
в числе спектрофотометрически титруемых остатков тирозина
в присутствии металла и без него не обязательно должно рассмат-
риваться как свидетельство их прямой координации к металлу.
Возможно также, что наблюдаемые различия вытекают из раз-
личий в значениях рК, изменяющихся в результате конформацион-
ных изменений или электростатических эффектов, вызываемых свя-
зыванием металла.
5.6.2. Исследование дисперсии оптического вращения
и кругового дихроизма
Как для трансферрина, так и для кональбумина в видимой об-
ласти наблюдается эффект Коттона, который зависит от присутст-
вия специфически связанного иона металла [77]. Полоса поглоще-
ния комплекса железа при 470 нм характеризуется отрицательным
эффектом Коттона, в то время как полоса комплекса марганца —
положительным эффектом Коттона. В отличие от этого комплексы
кональбумина н трансферрина с медью, имеющие максимальное
поглощение при 430 нм, не проявляют аномальной дисперсии
оптического вращения в этой области. Тем не менее природа опти-
348
Глава 9
ческой активности, ассоциируемой с этой полосой, была установле-
на исследованием кругового дихроизма [78].
Необычная способность металлсвязывающего центра трансфер-
рина связывать и ионы меди, и ионы железа заслуживает обсуж-
дения. При связывании меди окружение иона металла приобретает
симметрию, очень близкую к аксиальной [76]. Небольшое откло-
нение от аксиальности явственно вытекающее из спектров ЭПР,
снятых при 35 °C, согласуется с оптической активностью, наблю-
даемой Наги и Лерером [78]. В отличие от этого спектры ЭПР
[55] и оптическая активность комплекса железа свидетельствуют
о гораздо более низкой симметрии металлсвязывающего центра.
По-видимому, лиганды, предоставляемые белком, обладают очень
большой гибкостью, и этим объясняется легкая приспособляемость
этого центра* к иону металла.
6. СПЕКТРОСКОПИЯ МАГНИТНОГО РЕЗОНАНСА
По-видимому, наиболее ценные сведения о природе металлсвя-
зывающих центров сидерофилинов были получены методами маг-
нитного резонанса, особенно методом электронного парамагнитного
резонанса (ЭПР). Спектр ЭПР нативного комплекса Fe-трансфер-
рин — НС'О3 является характеристичным для иона Fe^ в высоко-
спиновом состоянии в ромбически искаженном октаэдрическом
окружении [56, 79]. Отчетливый сигнал с g=4,14 почти несомнен-
но возникает от ионов Fe3+ одного и того же типа, при этом не на-
блюдается никаких различий в форме линии как функции степени
насыщения белка железом [56]. Ааса [80] недавно произвел де-
тальную теоретическую обработку этого спектра, проанализировав
результаты в виде спин-гамильтониана следующего типа:
Н = Ц- D [3S| - S (S + 1)] + Е (S® - SJ) -f- g₽B-S.
Главные линии с g, приблизительно равным 4, возникают от
среднего из трех крамеровских дублетов со спином 5/2. Экспери-
ментально полученные спектры замороженных растворов интерпре-
тируются как спектры порошка и представляют собой суперпози-
цию спектров беспорядочно ориентированных кристаллитов. Ранее
в несколько различной форме Блюмберг [81] и Доусинг и Гибсон
[82] показали, что экспериментально наблюдаемые линии возни-
кают из переходов в главных плоскостях, а также вдоль главных
осей тензора кристаллического поля. Значения параметров спин-
гамильтониана, выведенных Ааса, приведены в табл. 9.3; в ней
также даны параметры комплексов меди и хрома.
* В данном случае автор, по-видимому, подразумевает под binding site «свя-
зывающие полости» в белке. — Прим. ред.
Трансферрины (сидерофилины)
349
Таблица 9.3
Параметры спин-гамильтониана для комплексов трансферрина
Комплекс
Щ|,
см—1
|E/D|
UN,
сверх-
тонкое
расщеп-
ление
Число
N-ли-
гандов
Fe3+ — бикарбонат
Сг3+ — бикарбонат
тип I
тип 2
Си2+ — бикарбонат
Си2+ (без бикар-
боната)
0,27
0,55
0,55
0,31—0,32
0,32—0,33
0,27
2,312
2,205
gx=2,042
gy=2,059
2,05
156
210
1
4
При отсутствии связанного бикарбоната спектр ЭПР комплекса
Fe—трансферрин значительно изменяется, но все еще характери-
зует специфически связанный металл [70]. При переходе от ней-
тральных к кислым значениям pH в спектре наблюдаются изме-
нения, сопровождающиеся появлением новой компоненты, свиде-
тельствующей о различии между связывающими центрами в таких
условиях [76]. При отсутствии бикарбоната в спектрах обнаружи-
ваются различные сигналы от тройного комплекса железа, транс-
феррина и многих других хелатных соединений металлов [70].
Спектр ЭПР комплекса железо—кональбумин—бикарбонат по
своим главным особенностям идентичен спектру комплекса с транс-
феррином; однако он содержит еще дополнительную компоненту
в области низкого поля с g'=4,3 {56]. Такую же компоненту на-
блюдали и другие авторы в спектрах комплекса с лактоферрином
(рис. 9.1). Детально эта дополнительная полоса еще не исследова-
на, но, по-видимому, она может быть обусловлена ионом Fe3+,
связанным иначе, чем в комплексе с трансферрином. Таким обра-
зом, вполне вероятно, что связывающие центры этих белков могут
несколько отличаться.
Спектр ЭПР комплекса трансферрина с медью также зависит
от связывания бикарбоната [75]. В обоих случаях спектры явля-
ются характеристичными для атома меди, находящегося в аксиаль-
ном окружении. Наблюдаемая сверхтонкая структура находящейся
в слабом поле линии желтого комплекса медь—трансферрин—би-
карбонат обусловлена взаимодействием с одним ядром атома азо-
та. В отсутствие бикарбоната сверхтонкая структура в спектре
бесцветного бинарного комплекса меди с трансферрином удовлет-
350
Глава 9
ворительно интерпретируется на основе взаимодействия неспарен-
ного спина меди с четырьмя ядрами азота (рис. 9.2). Хотя и не
совсем понятно, почему наблюдаются изменения при связывании
Рис. 9.1. Спектр ЭПР трансферрина (а) н коровьего лактоферрина (б).
а — концентрация по Fe3+ 1гЗ-10—3 М; температура 77 К; микроволновая частота 9090 МГц;
усиление 1,5-104, б — концентрация по Fe3^ 6,9-10-4 М; температура 77 К; микроволно°яя
частота 9090 МГц; усиление 10-10*.
бикарбоната, ясно, что места связывания металла трансферрином
должны иметь четыре азотных лиганда. Это прямое подтверждение
участия азотных лигандов в координации иона металла трансфер-
рином подкрепляет и дополняет выводы, сделанные ранее на осно-
ве изучения титрирования ионами водорода, которое показало уча-
стие в связывании каждого иона металла двух имидазольных
групп гистидина [83].
Трансферрины (сидеррофилины)
3S1
Изучение спектров ЭПР специфических комплексов хрома
с трансферрином также оказалось очень информативным. При фи-
зиологических значениях pH в спектрах обнаруживают два типа
похожих, но не одинаковых ионов хрома, которые с различными
скоростями замещаются ионами Fe3+ {48]. Возможно, это и есть
Рис. 9.2а. Спектр ЭПР комплекса 6!Си—трансферрин, не содержащего бикарбо-
нат-иона, при 77 К.
Микроволновая частота 9149-МГц. Пунктирная кривая рассчитана теоретически для слабо-
польной линии, имеющей сверхтонкую структуру, в предположении, что эта линия имеет
гауссову форму (ширина 10 Гс) и что сверхтонкое расщепление на ядрах четырех эквива-
лентных атомов азота равно 12 Гс; параметры меди g =2,205, g =2,05, А =210 Гс
и А , =21 Гс.
спектроскопическое проявление различия между поставляющими
железо полостями связывающих центров трансферрина при физио-
логических исследованиях, которое обнаруживается, несмотря на
беспорядочную природу связывания железа [84, 85].
Дальнейшие доказательства того, что в связывании ионов
металлов сидерофилинами принимают участие тирозильные остат-
ки, были получены при изучении спектров ядерного магнитного ре-
зонанса (ЯМР) высокого разрешения {65]. Специфическое связы-
вание галлия (Ga3+) приводит к сдвигу части сигнала ароматиче-
ских протонов в сильное поле, в это же время связывание Fe3+
уменьшает интенсивность полосы в этой области спектра вследст-
352
Глава 9
вие либо парамагнитного уширения, либо сдвига. Применение
этого метода, по-видимому, открывает большие возможности в изу-
чении связывания металла сидерофилинами.
Рис. 9.26. Спектр ЭПР комплекса 6!Си—трансферрин—бикарбонат.
Микроволновая частота 9151 МГц. Пунктирная кривая рассчитана теоретически в предполо-
жении, что линия имеет гауссову форму (ширина 9,5 Гс); расщепление на одном ядре
атома азота составляет 9,5 Гс; параметры: g
II
=2,312, g L =2,05, А
= 156 Гс и A t =16 Гс.
7. ИЗУЧЕНИЕ ХИМИЧЕСКОЙ МОДИФИКАЦИИ
Дополнительные сведения о металлсвязывающих центрах си-
дерофилинов были получены путем изучения влияния химической
модификации белка на его металлсвязывающую функцию. Моди-
фикация менее 50% свободных аминогрупп не содержащего ме-
талла сидерофилина существенно не сказывается на его способно-
сти связывать железо, в то время как более обширная модифика-
ция приводит к постепенной потере способности связывать железо
[86]. В отличие от этого алкилирование остатков гистидина со-
провождается потерей железосвязывающей активности, если 14 из
17 остатков гистидина в молекуле модифицированы [87]. Когда
изменены только 10 остатков гистидина, способность связывать
железо практически не теряется. Эти данные объяснили тем, что
Трансферрины (сидерофилины)
353
в связывании каждого атома железа участвуют два остатка гисти-
дина, но этот вопрос остается открытым, так как с карбоксимети-
лированными гистидинами не получено доказательств кооператив-
ного эффекта; связывание, скорее всего, осуществляется беспоря-
дочным образом, хотя способность связывать металл резко теряется
при изменении более чем 10 остатков. По-видимому, отсутствие
эффекта кооперативности при модификации гистидина должно
отражать постепенную, но при этом ускоряющуюся потерю железо-
связывающей активности в зависимости от числа измененных ги-
стидинов. Возможно, также, что потеря способности связывать же-
лезо отражает изменения в третичной структуре белка при увели-
чении числа модифицированных гистидиновых остатков.
Опубликовано всего несколько работ о влиянии модификации
тирозина на связывающие центры сидерофилинов. При сравнении
чувствительности к химической модификации тирозильных остатков
белка, не содержащего металла, и насыщенного железом белка
установили, что число тирозильных остатков, претерпевших моди-
фикацию, уменьшается примерно на 6 в случае, когда белок свя-
зан с железом, по сравнению с белком, не содержащим железа.
Это было верно и для случая, когда процесс модификации вклю-
чал ацетилирование гидроксильных групп N-ацетилимидазолом
[88] с последующим нитрованием тетранитрометаном [87]. Фе-
нольные остатки содержащего металл белка оказались также
более устойчивыми к иодированию [88]. Согласие между этими
результатами и результатами спектрофотометрического титрова-
ния, проведенного в лаборатории Вернера, служит веским доказа-
тельством участия тирозильных остатков в комплексообразовании
с металлом. Но, как указывали Баткус, Кларк и Фини [86], необ-
ходимым условием связывания является наличие полости: «Так
как соответствующая ориентация металлсвязывающего центра,
по-видимому, стабилизируется водородными связями и взаимодей-
ствием зарядов, внутренняя конформация молекулы играет очень
важную роль. При химических модификациях эти межмолекуляр-
ные взаимодействия изменяются, происшедшие изменения могут
привести к изменениям в устойчивости хромогенного комплекса,
спектральным сдвигам и в конечном счете к потере белком поло-
сти, связывающей металл».
8. ОБЗОР СОВРЕМЕННОГО СОСТОЯНИЯ ВОПРОСА
О МЕТАЛ Л СВЯЗЫВАЮЩИХ ЦЕНТРАХ СИДЕРОФИЛИНОВ
При физиологическом значении pH и в присутствии бикарбо-
нат-иона два связывающих железо центра трансферрина идентич-
ны по параметрам спектров ЭПР. Однако при низких значениях
pH и в отсутствие бикарбоната становятся очевидными отличия
23—2451
354
Глава 9
между этими двумя центрами. Каждый центр содержит четыре
азотных лиганда, но не все из них могут действительно связывать-
ся с металлом при всех обстоятельствах. Два из этих атомов
азота могут находиться в имидазольных кольцах гистидина, источ-
ник же двух других —неизвестен. Два или три тирозильных остат-
ка, по-видимому, непосредственно участвуют в связывании железа,
и один или два остатка — в связывании меди, хотя, по-видимому,
прямая координация фенольных атомов кислорода к иону металла
недоказуема. Молекула воды прямо координируется со связанной
медью и, по-видимому, также легко связывается и железом.
Константы устойчивости для этих двух центров трансферрина
и кональбумина одинаковы или почти одинаковы. Тем не менее
в комплексе Fe3+ с трансферрином в отсутствие бикарбоната при
низких значениях pH или когда металл связан с Сг3+ обнаружи-
ваются различия между этими центрами. Наблюдались также раз-
личия между двумя центрами трансферрина по физиологической
способности служить донорами железа.
Несмотря на то что Fe3+ и Си2+ обычно имеют различные сте-
реохимические требования, оба этих иона захватываются (прини-
маются) металлсвязывающими центрами сидерофилинов. Возмож-
но, это объясняется значительной гибкостью полипептидных боко-
вых цепей в области связывающих центров, так что каждый ион
может выбрать такое лигандное окружение и симметрию, которые
ему наиболее подходят.
9. БИОЛОГИЧЕСКИЕ ФУНКЦИИ СИДЕРОФИЛИНОВ
9.1. Потребность в физиологическом
переносчике железа
Элементарные термодинамические расчеты показали, что при
физиологических значениях pH и парциальном давлении газооб-
разного кислорода наиболее предпочтительным для железа в рас-
творе в условиях равновесия является степень окисления +3
(Fe3+). Приняв растворимость гидроксида железа (III) равной
Ю-36 М* [4], получим, что равновесная концентрация свободных
ионов железа(III) в крови не может превышать 10-14 М. Отсюда
вытекает необходимость в переносчике для ионов железа.
Однако, возможно, что трансферрин выполняет еще и более
важную, чем транспорт, функцию. Этот белок способен специфиче-
ски узнавать синтезирующие гемоглобин ретикулоциты, и эта его
способность к узнаванию обеспечивает доставку содержащегося
* Здесь, по-видимому, идет речь о произведении растворимости Fe(OH)3.—
Прим. ред.
Трансферрины (сидерофилины)355
в нем железа только к таким клеткам, которые специфически нуж-
даются в железе [89]. Кроме того, сами ретикулоциты могут эф-
фективно акцептировать железо только из специфических носите-
лей [90], наилучший из которых, по-видимому, трансферрин.
Таким образом, способность трансферрина к узнаванию имеет
двойственную роль, включающую и это специфическое взаимодей-
ствие белка с клеткой.
9.2. Взаимодействие трансферрин — ретикулоцит
Для того чтобы ретикулоцит мог взаимодействовать с трансфер-
рином и акцептировать из него железо, на его поверхности должны
быть специальные рецепторы. Обработка поверхности клетки тиро-
зином, который, как известно, гидролизует сиалогликопептиды из
клеточных мембран [91], в значительной степени лишает поверх-
ность способности акцептировать железо из белка [89].
Морган [92] предположил, что взаимодействие между транс-
феррином и ретикулоцитами осуществляется в следующие четыре
стадии:
1. Адсорбция трансферрина рецепторами на поверхности рети-
кулоцита.
2. Образование прочной связи между трансферрином и ретику-
лоцитом. На этой стадии белок может действительно проникать
внутрь клетки [93].
3. Перенос железа из белка в клетку.
4. Удаление белка.
Трансферрин, связанный с Fe3+ или Сг3+, обладает большим
сродством к рецепторам ретикулоцитов, чем апотрансферрин [94,
95]. Этот эффект частично зависит от природы закомплексованно-
го иона металла и, по-видимому, обусловлен более высокой кон-
стантой скорости реакции диссоциации комплекса ретикулоцит —
трансферрин в том случае, когда белок не содержит металла.
Таким образом, молекулы трансферрина, содержащие Сг3+ или
Fe3+, имеют более продолжительное время жизни на поверхности
ретикулоцита, при этом среднее время жизни молекулы белка на
поверхности клетки, вероятно, составляет 5—10 мин [4]. Трансфер-
рины, содержащие марганец, медь или цинк, ведут себя подобно
апотрансферрину [93]. Яндл и Катц [96] и Корнфельд [94] рас-
считали, что на поверхности ретикулоцита имеется около 50 000 ре-
цепторных центров, так что в условиях насыщения около 2% пло-
щади поверхности клетки занято трансферрином. Бейкер и Морган
[97] подсчитали, что с ретикулоцитом может быть связано
500 000 молекул белка.
Скорость поглощения железа ретикулоцитами не зависит от
степени насыщения железом трансферрина, с которым они инкуби-
руются при условии, что общая концентрация комплекса железо —
23*
356
Глава 9
трансферрин постоянна. Это указывает, что апотрансферрин не-
значительно препятствует процессу переноса железа [57, 98].
В литературе имеются противоречивые сообщения о влиянии кон-
центрации трансферрина на поглощение железа ретикулоцитами
[57, 96, 98, 99]. Было сделано предположение, что некоторые рас-
хождения в результатах можно объяснить различиями в физиоло-
гической однородности используемых препаратов трансферрина
[18, 99]. Этот автор также подчеркнул, что при изучении эффек-
тов концентрации трансферрина и его насыщения нужно принимать
во внимание тот факт, что, хотя обмен- между молекулами транс-
феррина самопроизвольно не протекает вследствие огромной устой-
чивости комплекса металла с белком, он может быть промотирован
присутствием таких связывающих железо агентов, как цитрат [62].
Влияние структурных модификаций на биологическую актив-
ность трансферрина было изучено Корнфельдом [100]. По-видимо-
му, эти исследования можно разделить на исследование влияния
модификации на связывание белка с поверхностью клетки рети-
кулоцита и на исследование последующего специфического погло-
щения железа клеткой. Ферментативное удаление большинства
углеводных связей в трансферрине не вызывает никаких значи-
тельных изменений ни в связывании белка, ни в переносе железа.
Механизм переноса железа из молекул трансферрина в синте-
зирующий гемоглобин аппарат ретикулоцита не известен. Этот
механизм не может быть простой диссоциацией из-за очень боль-
шой устойчивости комплекса железа с белком. Присутствие хела-
тирующих агентов в инкубационной среде не влияет на поглоще-
ние железа in vitro. Так как устойчивость комплекса железо —
трансферрин значительно понижается в отсутствие бикарбоната,
предположили, что в этом взаимодействии может участвовать
центр белка, связывающий бикарбонат [101]. Молекула трансфер-
рина, как было показано, способна проникать внутрь ретикулоцита
[93]; возможно, это сопровождается понижением локального зна-
чения pH внутри клетки, достаточным для того, чтобы железо
могло отделиться от белка.
На концентрацию трансферрина в крови не влияет поглощение
железа из кишечника, хотя последующее распределение поглощен-
ного железа зависит от того, насколько трансферрин как носитель
насыщен им [102].
9.3. Трансферрин как буфер для ионов железа
Так как циркулирующий трансферрин только на ’/з насыщен
железом, он обладает способностью приспосабливаться к изме-
няющимся концентрациям железа, так что он является эффектив-
ным буфером для железа, а также его поставщиком. Насколько
важна эта функция, хорошо видно тогда, когда она не может про-
Трансферрины (сидерофилины.)
357
явиться. В условиях хронической перегрузки железом, когда транс-
феррин насыщен железом или когда имеется врожденный дефицит
белка [ЮЗ], железо откладывается в тканях в такой степени, что
возможно отравление им.
9.4. Биологическая функция кональбумина
и лактоферрина
Специфическая роль кональбумина и лактоферрина в транс-
порте железа не доказана. Кональбумин защищает яйцо от мик-
робной инфекции [6], соединяясь с железом и препятствуя росту
микроорганизмов. В этом может заключаться одна из его биоло-
гических функций. Повсеместное распространение лактоферрина
в выделениях дыхательных, пищеварительных, мочевых и слезных
трактов, возможно, связано с бактериостатической активностью
этого белка [15]. При насыщении железом антимикробные свой-
ства кональбумина и лактоферрина исчезают [6].
Лактоферрин может участвовать в транспорте железа из крови
в молоко кормящей матери, но это еще не точно установлено.
Имеется также несколько предположений о роли этого белка в ре-
гуляции поглощения железа из желудочно-кишечного тракта [104].
9.5. Генетическая разнородность трансферринов
Обнаружено не меньше 18 генетических разновидностей транс-
феррина человека [105]. Биологическое значение этой изменчиво-
сти не известно (если оно вообще имеется).
Ю. ЗАКЛЮЧЕНИЕ
Трансферрины (сидерофилины) — группа белков из разнообраз-
ных источников, характеризующихся способностью специфично,
прочно и обратимо связывать ионы Fe(III) и ионы других пере-
ходных металлов. Функция сывороточного трансферрина, который
наиболее подробно изучен, состоит в том, чтобы транспортировать
ион железа в кровь и направлять его к ретикулоцитам, которым
он нужен для биосинтеза гемоглобина. Система трансферрин —
ретикулоцит до сих пор является уникальной для изучения взаимо-
действия металла с белком и белка с клеткой.
ДОПОЛНЕНИЯ, ВНЕСЕННЫЕ В КОРРЕКТУРУ
Гибсон и Прайс [Biochem. Biophys. Res. Commun., 46, 646
(1972)] недавно сообщили, что специфическое связывание Fe3+
с трансферрином не осуществляется в отсутствие подходящего
358
Глава 9
аниона. Тем самым было высказано сомнение в существовании
бинарного комплекса Fe — трансферрин. Результаты, полученные
ранее Ааса и Айзеном [76] и Айзеном с сотр. [70], были объясне-
ны присутствием цитрата, используемого для получения апотранс-
феррина в образцах, изученных этими исследователями.
СПИСОК ЛИТЕРАТУРЫ
1. Laurell C.-В., Ingelman В., Acta Chem. Scand., 1, 770 (1947).
2. Holmberg C. G., Laurell C.-В., Acta Physiol. Scand., 10, 307 (1945).
3. Holmberg C. G., Laurell C.-В., Acta Chem. Scand., 1, 944 (1947).
4. Schade A. L„ Caroline L., Science, 104, 3420 (1946).
5. Schade A. L., Reinhart R. W., Levy H., Arch. Biochem., 20, 170 (1949).
6. Schade A. L., Caroline L., Science, 100, 14 (1944).
7. Sorenson M„ Sorenson S. P. L., Compt. Rend. Trav. Lab. Carlsberg, 23, 55
(1939).
8. Williams J., Biochem. J., 83, 355 (1962).
9. Feeney R. E., Komatsu St. K., Structure and Bonding, 1, 149 (1966).
10. Bron C., Blanc B., Isliker H., Helv. Physiol. Acta, 25, 337 (1967).
11. Montreuil L, Tonnelat J., Mullet S„ Biochim. Biophys. Acta, 45, 413 (1960).
12. Groves M. L., Biochim. Biophys. Acta, 100, 154 (1965).
13. Baker E., Shaw D. C., Morgan E. H., Biochemistry, 7, 1371 (1968).
14. Schade A. L., Pallavicini C„ Wiesmann U., Protides of the Biological Fluids,
Proc. Colloq. Bruges, 16, 619 (1968).
15. Masson P. L., Heremans J. F., Schonne E., Crabbe P. A., Protides of the Bio-
logical Fluids, Proc. Colloq. Bruges, 16, 633 (1968).
16. Koechlin B. A., J. Am. Chem. Soc., 74, 2649 (1952).
17. Gelotte B., Flodin P., Killander J., Arch. Biochem. Biophys. Supp., 1, 319
(1962).
18. Schade A. L., Sonderdruck Behringwerk-Mitteilung., 1961, 39.
19. Laurell C.-В., Acta Chem. Scand., 7, 1407 (1953).
20. Leibman A., Aisen P., Arch. Biochem. Biophys., 121, 717 (1967).
21. Warner R. C., Weber L, J. Biol. Chem., 191, 173 (1951).
22. Bain J. A., Deutsch H. F., J. Biol. Chem., 172, 547 (1948).
23. Woodworth R. C., Schade A. L., Arch. Biochem. Biophys., 82, 78 (1959).
24. Bates G. W., Billups C., Saltman P., J. Biol. Chem., 242, 2810 (1967).
25. Groves M. L., J. Am. Chem. Soc., 82, 3345 (1960).
26. Johansson B. G., Acta Chem. Scand., 23, 683 (1969).
27. Mann K- G„ Fish W. W., Cox A. C., Tanford C., Biochemistry, 9, 1348 (1970).
28. Blanc B., Bujard E., Mauron J., Experientia, 19, 299 (1963).
29. Castellino F. J., Mann K. G., submitted to Biochemistry.
30. Jeppsson J.-O., Ph. D. Dissertation, Department of Microbiology, University of
Umea, Umea, Sweden, 1967, p. 18.
31. Montreuil J., Biserte G„ Mullet S., Spik M., Leroy N., Compt. Rend. Acad.
Sci., Paris, 252, 4065 (1961).
32. Azari P. R., Feeney R. E., J. Biol. Chem., 232, 293 (1958).
33. Jamieson G. A., Biol. Chem., 240, 2914 (1965).
34. Jamieson G. A., Biochim. Biophys. Acta, 121, 326 (1966).
35. Bearn A. G., Parker W. C., in F. Gross (ed.), Iron Metabolism: An International
Symposium, Springer-Verlag, Berlin, 1964, p. 67.
36. Robinson J. C., Pierce J. E., Arch. Biochem. Biophys., 106, 348 (1964).
37. Williams J., Biochem. J., 108, 57 (1968).
38. Got R„ Goussault Y., Font J., Carbohydr. Res., 3, 157 (1966).
39. Morgan E. H., Marsaglia G., Giblett E. R., Finch C. A., J. Lab. Clin. Med., 69,
370 (1967).
Трансферрины (сидерофилины)
359
40. Montreuil J., Tonnelat J., Mullet S., Biochim. Biophys. Acta, 45, 413 (1960).
41. Aisen P., Koenig S. H., Schillinger W„ Scheinberg I. H., Mann K. G., Fish W.,
Nature, in press.
42. Warner R„ Weber L, J. Am. Chem. Soc., 75, 5094 (1953).
43. Masson P. L., H eremans J. F„ Eur. J. Biochem., 6, 579 (1968).
44. Perkins D. J., Protides of the Biological Fluids, Proc. Colloq. Bruges, 14, 85
45. Aisen P„ Aasa R., Redfield A. G., J. Biol. Chem., 244, 4628 (1969).
46. Lehrer S. S„ J. Biol. Chem., 244, 3613 (1969).
47. Bezkorovainy A., Rafelson M. E., Jr., Arch. Biochem. Biophys., 107, 302
(1964).
48. Verbruggen R., Blaton V., Rosenau-Motreff M. Y„ Peelers FL, Protides of the
Biological Fluids, Proc. Colloq. Bruges, 16, 101 (1968).
49. Koenig S. H., Schillinger W., J. Biol. Chem., 244, 3283 (1969).
50. Koenig S. H., Schillinger W., J. Biol. Chem., 244, 6520 (1969).
51. Ulmer D. D., Biochim. Biophys. Acta, 181, 305 (1969).
52. Jeppsson J.-O., Acta Chem. Scand., 21, 1686 (1967).
53. Bezkorovainy A., Grohlich D., Biochim. Biophys. Acta, 147, 497 (1967).
54. Greene F. C., Feeney R. E., Biochemistry, 7, 1366 (1968).
55. Bezkorovainy A., Grohlich D., Gerbeck С. M., Biochem. J., 110, 765 (1968).
56. Aasa R„ Malmstrom B. G., Saltman P., Vanngard T., Biochim. Biophys. Acta,
75, 203 (1963).
57. Schade A. L„ Farmaco, 19, 185 (1964).
58. Woodworth R., Protides of the Biological Fluids, Proc. Colloq. Bruges, 14, 37
(1966).
59. Aisen P., Leibman A., Reich H. A., J. Biol. Chem., 241, 1666 (1966).
60. Wenn R. V., Williams J., Biochem. J., 108, 69 (1968).
61. Woodworth R. C., Tan A. T., Virkaitis L. R., Nature, 223, 833 (1969).
62. Aisen P., Leibtnan A., Biochem. Biophys. Res. Commun., 30, 407 (1968).
63. Aisen P„ Leibtnan A., Biochem. Biophys. Res. Commun., 32, 220 (1968).
64. Tan A. T., Woodworth R., Biochemistry, 8, 3711 (1969).
65. Woodworth R. C., Morallee K- G., William R. J. P„ Biochemictry, 9, 839
(1970).
66. Hosain F., McIntyre P. A., Poulose K-, Stern H. S., Wagner H. N., Jr., Clin.
Chim. Acta, 24, 69 (1969).
67. Ehrenberg A., Laurell C.-В., Acta Chem. Scand., 9, 68 (1955).
68. Gaber B„ Aisen P., unpublished observation.
69. Schade A. L., Reinhart R. W., Protides of the Biological Fluids, Proc. Colloq.
Bruges, 14, 75 (1966).
70. Aisen P„ Aasa R., Malmstrom G., Vanngard, J. Biol. Chem., 242, 2484 (1967).
71. Young J. W„ Perkins D. J., Eur. J. Biochem., 4, 385 (1968).
72. Gaber B., Koenig S. H., Schillinger W., Aisen P., J. Biol. Chem., in press.
73. Blumber W. G., Eisinger J., Aisen P., Morell A. G., Scheinberg J. FI., J. Biol.
Chem., 238, 1675 (1963).
74. Brill A. S.. Bryce G. F„ J. Chem. Phys., 48, 4398 (1968).
75. Wishnia A., Weber L, Warner R. C., J. Am. Chem. Soc., 83, 2071 (1961).
76. Aasa R., Aisen P., J. Biol. Chem., 243, 2399 (1968).
77. Ulmer D„ Vallee B. L., Biochemistry, 2, 1335 (1963).
78. Nagy B„ Lehrer S. S., Abstracts, Third International Congress of Biophysics,
Cambridge, Mass., 1969, p. 11.
79. Windle J. J., Wiersema A. K., Clark J. R., Feeney R. E., Biochemistry, 2, 1341
(1963).
80. Aasa R., J. Chem. Phys., in press.
81. Blumberg W. E., in A. Ehrenberg, B. G. Malmstrom, T. Vanngard (eds.),
Magnetic Resonance in Biological Systems, Pergamon Press, Oxford, 1967
p. 119.
82. Dowsing R. D„ Gibson J. F., J. Chem. Phys., 50, 294 (1969).
360
Глава 9
83. Hazen Е. Е., Jr., Ph. D. Thesis, Harvard University, 1962.
84. Dern R. J., Monti A., Glynn M. F., J. Lab. Clin. Med., 61, 280 (1963).
85. Fletcher J., Huehns E. R., Nature, 215, 584 (1967).
86. Buttkus H., Clark J. R., Feeney R. E., Biochemistry, 4, 998 (1965).
87. Line W. F., Grohlich D., Bezkorovainy A., Biochemistry, 6, 3393 (1967).
88. Komatsu S. K., Feeney R. E., Biochemistry, 6, 1136 (1967).
89. Jandl J. H„ Inman J. K-, Simmons R. L., Allen D. W., J. Clin. Invest., 38, 161
(1959).
90. Princiotto J. V., Rubin M., Shashaty G. C., Zapolski E. J., J. Clin. Invest., 43,
825 (1964).
91. Cook G. M. W., Heard D. H„ Seaman G. V. F., Nature, 188, 1011 (1960).
92. Morgan E. H., Br. J. Haematol., 10, 442 (1964).
93. Morgan E. H., Appleton T. C., Nature, 223, 1371 (1969).
94. Kornfeld S., Biochim. Biophys. Acta, 194, 25 (1969).
95. Baker E., Morgan E. H., Biochemistry, 8, 2954 (1969).
96. Jandl J. H., Katz J. H., J. Clin. Invest., 42, 314 (1963).
97. Baker E., Morgan E. H., Biochemistry, 8, 1133 (1969).
98. Katz J. H„ Jandl J. H„ in F. Gross (ed.), Iron Metabolism, Springer-Verlag,
Berlin, 1964, p. 112.
99. Morgan E. H., Laurell C.-В., Br. J. Haematol., 9, 471 (1963).
100. Kornfeld S., Biochemistry, 7, 945 (1968).
101. Aisen P., Mount Sinai J. Med., in press.
102. Wheby M. S., Umpierre G., New Engl. J. Med., 271, 1391 (1964).
103. Heilmeyer L., in F. Gross (ed.), Iron Metabolism An International Symposium,
Springer-Verlag, Berline, 1964, p. 201.
104. DeLacy P., Masson P. L., Heretnans J. F., Protides of the Biological Fluids,
Proc. Colloq. Bruges, 16, 633 (1968).
105. Giblett E. R„ Genetic Markers in Human Blood, Blackwell Scientific Publi-
cations, Oxford, 1969, p. 138.
ГЛАВА 10
ЦЕРУЛОПЛАЗМИН*
И. Г. Шейнберг, А. Дж. Морелл
Scheinberg I. Н., Morell A. G., Department of Medicine,
Albert Einstein College of Medicine, New York, USA
1. ВВЕДЕНИЕ
Медь, как и железо, оказывает двоякое действие на большин-
ство живых организмов, в том числе и на человека: с одной сторо-
ны, она необходима для жизнедеятельности биологических систем,
с другой стороны, может быть для них токсична [1—4]. О значе-
нии меди для нормальной жизнедеятельности различных биосистем
говорят следующие примеры. Овцы, выпасаемые на почвах, бедных
медью, дают потомство, у которого развиваются нейроны, не по-
крытые миелиновой оболочкой. Это явление устраняется при вне-
сении солей меди в почву пастбищ. В условиях дефицита меди
заметно ухудшается захват железа трансферрином из депо организ-
ма, что является необходимой стадией включения железа в гемо-
глобин [5, 6]. Медь входит в качестве необходимого элемента
в состав цитохромоксидазы, тирозиназы и других белков [7, 8].
Недавно получены данные, свидетельствующие о влиянии содер-
жания меди в организме на развитие нормальных вкусовых ощу-
щений [9], на рост костных [10] и соединительных тканей [11,
12]. Физиологические последствия дефицита меди проявляются
исключительно у животных; у человека случаи дефицита меди
вообще неизвестны, так как в любой пище человека медь содер-
жится в большом избытке.
Несмотря на поступление большого количества меди в организм
человека с пищей, концентрация этого металла в тканях здорового
человека поддерживается строго постоянной в течение всей его
жизни: общий долговременный баланс по меди равен нулю. По-
скольку ионы меди обладают слабой способностью к диффузии и
поэтому могут легко усваиваться в желудочно-кишечном тракте,
в организме должны существовать какие-то системы, препятствую-
щие непрерывному накоплению меди в тканях путем ограничения
ее адсорбции или стимуляции ее выведения при физиологических
•условиях. Примерно четыре человека из миллиона страдают из-за
нарушения или полного отсутствия системы удаления меди, и
♦ © by I. Herbert Scheinberg.
362
Глава 10
в этих случаях твердо установлено, что избыточная медь, накапли-
вающаяся в живых тканях, вызывает токсикоз [13]. Ранним приз-
наком этого заболевания, известного под названием болезни Виль-
сона, является уменьшение содержания в плазме церулоплазмина
медьсодержащего гликопротеина, который в настоящее время изу-
чен лучше всех других биологических соединений меди.
2. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
ЦЕРУЛОПЛАЗМИНА
Церулоплазмин обнаружен в плазме человека, обезьяны,
свиньи, лошади, осла, коровы, оленя, козы, собаки, кошки, мор-
ской свинки, кролика, мыши, крысы и курицы [14—18]. В лягуш-
ках он обнаруживается только после метаморфозы [19]. Синяя
окраска и каталитическая активность церулоплазмина обусловле-
ны содержащейся в ней медью и характерны (но не идентичны)
Рис. 10.1. Кристаллы церулоплазмина человека (Х75). Микрофотография Миль-
тона Куртца и Ирмин Штернлиб.
для всех его разновидностей, выделенных из разных объектов.
Церулоплазмин человека, а2-глобулин, содержит 0,3% меди и
около 8% углеводов. Он проявляет оксидазное действие по отно-
Таблица 10.1
Аминокислотный состав церулоплазмина человека
Аминокислота Количество остатков, отнесенных к молекулярной массе 151 000а [79]
Аспарагиновая кислота 143
Треонин 89
Серин 73
Глутаминовая кислота 132
Пролин 60
Глицин 89
Алании 58
’/г цистина 10
Цистеин —
Валин 73
Метионин 19
Изолейцин 61
Лейцин 80
Тирозин 57
Фенилаланин 66
Лизии 77
Гистидин 50
Аргинин 44
Триптофан 23
N-Концевые аминокислоты на молекулу белка [57]
Валин 0,9
Лизин 0,25
Количество SH-групп, обнаруживаемых в молекуле
церулоплазмин 1 [45, 57, 80]
.апоцерулоплазмин 5 [45]
4 [81]
а В предположения, что молекулярная масса белка равна
151 000 и что приблизительно 9% от нее составляет масса угле-
лзодов и меди, на долю аминокислотных остатков приходится
масса от 137 000 до 138 000.
Таблица 10.2'
Физические свойства церулоплазмина человека
Молекулярная масса Метод Литература
151 000 Седиментациоино-диффузион- 34
ный
160 000 То же 33.
155 000 Приближение к равновесию 33
143 000 То же 82
132 000 Рентгеноструктурный 32
Коэффициент седиментации (S) 7,08 [33]
Коэффициент диффузии, см2/с 3,76-10-7 [зз1
Парциальный удельный объем, см3/г 0,714 [28] 0,713 [33]
Отношение осей (а/Ь) 3,6 [45]
Вязкость, г-см_,'С-1 4,1 [45]
Изоэлектрическая точка 4,4 [34]
Размеры кристаллографической ячей- 0=6=268 [32]
ки, А (пространственная группа 14) с=129 [32]
Таблица 10.3:
Оптические свойства церулоплазмина человека
Длина волны, нм Коэффициент поглощения на 1 г-атом Cua [83J е1% "см
794 850
610 4400 0,68 [23]
459 460 0,71 [45]
332 1600
280 14,6 [23]
15,4 [45]
а В предположении, что поглощение света обусловлено все-
мн ионами Си2+ и только этими ионами.
Церулоплазмин
365
Таблица 10.4
Электрофоретические свойства церулоплазмина человека3
Буфер Т/2 pH Подвижность, 105 см2*В—1-с—1
Барбитуратиый 0,1 8,76 —5,20(33]
Трис-буфер 0,1 8,41 —5,26(33]
Фосфатный 0,1 7,01 —4,72(33]
Ацетатный 0,1 5,48 —2,74(33]
0,2 5,40 —3,50(44]
а Электрофорез с движущейся границей. С помощью электрофореза на крахмальном ге-
ле [84], на бумаге [85] или микроиммуноэлектрофореза [861 удается выделить две и более
компонент, обладающих оксидазной активностью.
шению к ряду полиаминов, полифенолов и иону железа(II). Его
легко можно получать в количестве граммов из объединенных
фракций плазмы человека, в 100 мл которой обычно содержится
20—40 мг церулоплазмина, либо из обогащенной им фракции, на-
пример фракции IV Кон. Разработано несколько препаративных
методов его выделения с помощью хроматографии на диэтилами-
ноэтилцеллюлозе или гидроксиапатите либо путем осаждения суль-
фатом аммония, этанолом или смесью этанола с хлороформом
[20—27]. Степень очистки обычно оценивают по отношению свето-
поглощения растворов образца при 610 и 280 нм, которое для наи-
более чистых образцов достигает значения 0,046. Из таких раство-
ров церулоплазмин человека выкристаллизовывается в виде тетра-
гональных призм [28] (рис. 10.1).
Аминокислотный состав церулоплазмина человека приведен
в табл. 10.1. О последовательности аминокислот в его белковой
цепи данных пока не имеется. Вторичная структура имеет р-кон-
фигурацию и беспорядочную укладку с небольшой примесью а-спи-
ралей или при их отсутствии. Судя по гидродинамическим свой-
ствам церулоплазмина (табл. 10.2), третичная структура его
молекулы представляет собой очень плотную упаковку. Недавно
Саймонс и Берн [29] на основании своих исследований предложи-
ли тетрамерную модель четвертичной структуры белка, которая
включает две полипептидные цепи с молекулярной массой 15 900
и N-концевыми аминогруппами валина и две полипептидные цепи
с молекулярной массой 59 000, в которых в качестве N-концевых
выступают остатки лизина. В табл. 10.3 и 10.4 представлены неко-
торые оптические, электрофоретические и кристаллографические
свойства церулоплазмина человека.
366
Глава 10
3. СОСТОЯНИЕ МЕДИ В ЦЕРУЛОПЛАЗМИНЕ
Атомы меди являются неотъемлемой частью молекулы церуло-
плазмина, и в ходе всех процессов с его участием они постоянно
соединены с белком [30]. При введении неорганической соли
радиоактивной меди она не обменивается с медью, содержащейся
в церулоплазмине in vivo. Следовательно, сразу отпадает гипоте-
за о том, что церулоплазмин способен обратимо диссоциировать,
выделяя ионы меди, и что физиологическая функция его в связи
с этим может якобы состоять в поддержании определенного гра-
диента концентрации ионов меди и регулировки их усвоения из
пищи [31].
Большая прочность связывания меди белком в церулоплазмине
обусловлена строением всей молекулы белка. Диссоциация моле-
кулы на субъединицы или ее протеолитическое расщепление приво-
дит к разрыву или значительному ослаблению связи меди с остав-
шимися компонентами. Особый характер этих связей можно понять
только при изучении исходного белка в целом, либо путем приго-
товления апопротеина с последующим переведением его в холопро-
теин при добавлении солей меди.
3.1. Число атомов меди в молекуле церулоплазмина
Эта величина остается до сих пор неопределенной из-за отсут-
ствия точных данных о молекулярной массе церулоплазмина и со-
держании меди, которое стало известным в самое последнее время.
Величина молекулярной массы церулоплазмина, по-видимому, на-
ходится между 150 000 и 160 000, хотя по рентгеноструктурным
данным она составляет всего 132 000 [32]. Содержание меди по
данным ранних исследований было 0,32—0,34%, однако позже
анализ кристаллическил образцов показал, что содержание меди
в них близко к 0,28% [35]. Исходя из последних данных, можно
считать, что в молекуле церулоплазмина содержится шесть или
семь атомов меди.
3.2. Свойства церулоплазмина,
обусловленные присутствием в нем меди
Способность ионов меди (II) обратимо восстанавливаться суб-
стратом и окисляться кислородом обусловливает оксидазную
активность церулоплазмина [36]. Все ли атомы меди, входящие
в состав белка, ответственны за его ферментативную активность,
не известно. Интенсивная синяя окраска церулоплазмина обуслов-
лена ионами меди (II), и белок может полностью (и обратимо)
обесцветиться под действием восстановителей. В этом отношении
Церулоплазмин
367
также неизвестно, все ли атомы меди, входящие в состав церуло-
плазмина, являются хромофорами, обусловливающими синюю
окраску.
3.3. Степень окисления меди в церулоплазмине
Степень окисления меди в церулоплазмине первоначально была
определена химическими методами с помощью избирательных реа-
гентов на ионы меди (II) [38], а позже — методами полярографии
и ЭПР. Различные методы дали хорошо согласующиеся результа-
ты, согласно которым примерно половина атомов меди находится
в церулоплазмине в степени окисления II (табл. 10.5). Эти ионы
меди(II) неэквивалентны между собой, поскольку при последова-
тельном их восстановлении аскорбиновой кислотой интенсивность
синей окраски уменьшается не линейно с изменением эффективно-
сти ионов меди в магнитной релаксации протонов [39]. Ионы
меди(1) церулоплазмина окисляются только при разрушении бел-
ка, хотя ионы Си(II) могут восстанавливаться обратимо. Послед-
ние могут реокисляться только кислородом, и не могут реокислять-
ся такими окислителями, как Cu2+, Fe3+ или феррицианид [34].
Так же как и ионы Си (II), ионы Cu(I) церулоплазмина неравно-
ценны между собой: из трех или четырех ионов Cu(I), входящих
Таблица 10.5
Характеристика меди, содержащейся в церулоплазмине
Количество атомов Си в молекуле церулоплазмина 8 [33, 34]
7(35 , 87 , 88] 6 [32]
Доли ионов Си2+ по отношению к общему количеству атомов Си, определенная методом:
ЭПР 0,52 [89] 0,29 [83] 0,43; 0,48 [90] 0.40 [91]
магнитной восприимчивости 0,40 [92] 0,44 [87]
полярографическим 0,54 [93]
Параметры спектра ЭПР
2,048 [94]
£ц 2,214 [94]
А а 0,008 см-1 [94]
368
Глава 10
в молекулу белка, только два связаны с атомами серы. В исходном
синем церулоплазмине ни Си (II), ни Cu(I) не взаимодействуют
сколь-нибудь заметно с молекулярным кислородом [40].
3.4. Обмен меди церулоплазмина на медь
из растворов ее солей
Ионы радиоактивной меди(1) из раствора могут замещать
атомы меди церулоплазмина только in vitro при условии полного
восстановления меди(П) до меди(1)* [31, 41]. Различные атомы
меди в белке обмениваются с неодинаковыми скоростями. В при-
сутствии цистеина медь церулоплазмина не обменивается на рас-
творенную. Вероятно, разбросанные по всей молекуле церулоплаз-
мина остатки цистеина могут служить по крайней мере одной из
причин того, что in vivo меченые атомы б4Си или 67Си, введенные
в церулоплазмин, не обмениваются с ионами меди из раствора
[30].
4. АПОЦЕРУЛОПЛАЗМИН
Если церулоплазмин подкислить до pH ниже, чем его изоэлек-
трическая точка, или обработать цианидом [42], из него можно
удалить медь и получить бесцветный белок. Однако при добавле-
нии к такому белку ионов меди реставрировать исходный фермент
не удается. Обратимое удаление меди из церулоплазмина дости-
гается при обработке его диэтилдитиокарбаматом (ДТК) [43].
Предварительно медь(П), содержащаяся в белке, восстанавли-
вается цистеином при pH 5,0 и высокой ионной силе. Образующий-
ся затем коллоидальный комплекс ДТК с вышедшей из белка
медью с помощью ультрацентрифуги отделяют от раствора апофер-
мента. Последний может быть очищен от всплывающих примесей
путем осаждения сульфатом аммония, повторного растворения и
диализа. В замороженных растворах апофермент устойчив в тече-
ние нескольких месяцев. При добавлении к такому апоферменту
ионов меди(1) и реокисления на воздухе из него можно вновь
получить синий церулоплазмин, который по всем своим свойствам
неотличим от исходного. В процессе перевода церулоплазмина
в его апофермент и обратно не было обнаружено никаких форм,
в которых содержание меди было бы промежуточным, т. е. отлич-
ным от содержания ее в апоферменте и холоферменте [44].
Согласно последним данным, полученным в лаборатории, где
работают авторы, обратимое удаление меди из церулоплазмина
* Первоначально сообщалось об обмене только половины атомов меди бел-
ка [31]. Однако при увеличении периода обмена и концентрации соли меди(1)
можно достигнуть обмена всех атомов меди.
Церулоплазмин
369
сопровождается заметными изменениями конформации белка,
а в нативной форме его два иона меди(1) связаны с атомами серы
[45].
При инъекции апоцерулоплазмина экспериментальным живот-
ным он быстро выводится из физиологического кругооборота.
5. ОКСИДАЗНЫЕ СВОЙСТВА ЦЕРУЛОПЛАЗМИНА
Ферментативная активность церулоплазмина была впервые
описана Холмбергом и Лоуреллом [46], которые показали, что это
единственный фермент, катализирующий окисление полиаминов и
полифенолов в плазме. Катализируемая церулоплазмином реак-
ция окисления п-фенилендиамина и других аналогичных соедине-
ний используется обычно для количественного определения белка
в растворе [31, 47, 48]. Однако не было замечено, чтобы церуло-
плазмин играл сколь-нибудь существенную физиологическую роль
при окислении таких важных биологических субстратов, как адре-
налин, норадреналин, серотонин, катехоламин или допамин [49].
Относительно ферментативных свойств церулоплазмина имеют-
ся противоречивые данные, в особенности это касается реакции
окисления аскорбиновой кислоты [25, 50]. В настоящее время счи-
тают, что ионы меди (II) церулоплазмина попеременно восстанав-
ливаются и окисляются, взаимодействуя то с ионами железа(II),
то с молекулярным кислородом [51]. В процессе этих превращений
могут окисляться некоторые соединения, в том числе ДОПА и
аскорбиновая кислота. Осаки и сотр. [52] для обозначения этого
свойства ввели термин «ферроксидазная активность». По их пред-
положению церулоплазмин способствует окислению железа(II),
хранящегося в печени, после чего железо (III) может быть связа-
но в трансферрин. Действительно, с его помощью можно вызвать
мобилизацию железа из депо печени, что было проделано in vivo
на опытах со свиньями, в организме которых был дефицит меди,
хотя пока не доказано, связано ли это явление с какими-либо
физиологическими или патологическими последствиями. Так, на-
пример, у больных, страдающих болезнью Вильсона, годами может
сохраняться дефицит церулоплазмина или даже наблюдается пол-
ное его отсутствие, что особенно часто бывает во время лечения.
Тем не менее, хотя к моменту начала лечения анемия может до-
стигать средней стадии, почти у всех таких пациентов содержание
гемоглобина и гематокрита вскоре достигает нормального уровня,
а в метаболизме железа не наблюдается каких-либо видимых от-
клонений от нормы. Эти парадоксальные явления нашли объясне-
ние после того, как Тофам и Фриден [54] сообщили об идентифи-
кации и выделении в чистом виде из сыворотки крови человека
нового медьсодержащего белка, отличающегося от церулоплазми-
24—2451
370
Глава 10
на и обладающего также ферроксидазной активностью. Кроме
того, предполагается, что третьим компонентом, обладающим фер-
роксидазной активностью и присутствующим в сыворотке крови
человека, может быть цитрат-ион [55].
6. НЕОДНОРОДНОСТЬ И ГЕНЕТИЧЕСКИЙ ПОЛИМОРФИЗМ
ЦЕРУЛОПЛАЗМИНА
Бромэн [56] первым показал, что церулоплазмин человека
представляет собой смесь двух форм, отличающихся между собой
хроматографическими свойствами [57, 58] и подвижностью при
электрофорезе [59]. Причины различий не были ясны, за исключе-
нием догадок о том, что это явление связано с механизмом на-
следственности. Указанные две хроматографические фракции (изо-
ферменты) были идентичны по содержанию меди, аминокислотно-
му и углеводному составу, обладали одинаковыми спектральными
характеристиками и проявляли одинаковую ферментативную
активность. Рихтерич [60] обнаружил различия в относительном
содержании двух изоферментов у новорожденных, взрослых людей
и людей, страдающих болезнью Вильсона, однако Хиршман [61]
и Бромэн [58] не смогли воспроизвести эти данные. Хольцман, со-
ставив пептидные карты кристаллических образцов церулоплазми-
на от здоровых людей и больных болезнью Вильсона, пришел
к выводу, что образцы между собой не отличаются [62]. По не-
опубликованным данным, полученным в лаборатории авторов
данной главы, оба изофермента синтезируются с одинаковой ско-
ростью, а медь для их синтеза используется из одного и того же
источника.
Шреффлеру с сотр. [63] удалось зафиксировать истинный гене-
тический полиморфизм церулоплазмина, который проявляется
в пяти фенотипах: СрА, СрВ (обычная форма), СрАВ, СрАС и
СрВС. Рассчитанная ими частота появления фенотипа оказалась
для американских кавказцев следующей: СрА = 0,006, СрВ = 0,994
и СрС = 0, а для американских негров — СРА=0,053, СрВ = 0,944
и СрС = 0,003. По мнению авторов, полученные данные свидетель-
ствуют о том, что в аутосоматическом локусе сосуществуют три
гена, каждый из которых управляет синтезом одного изофермента,
отличающегося своими электрофоретическими характеристиками.
7. ХАРАКТЕРИСТИКА ЦЕРУЛОПЛАЗМИНА
КАК ГЛИКОПРОТЕИДА
В табл. 10.6 приведены данные Джеймайсона об углеводном
составе церулоплазмина. В его молекуле имеется девять или де-
сять гетеросахаридных цепей, каждая из которых состоит из
Ц ерулоплазмин
371
Таблица 10.6
Углеводный состав церулоплазмина человека
Содержание углеводов, % 7—8
Число гетеросахаридных цепей на молекулу 9 нли 10
Число остатков сиаловой кислоты на молекулу 9
Число остатков N-ацет илглюкозамина на молекулу 18
Число остатков фукозы на молекулу 2
Суммарное число остатков маннозы и галактозы на молекулу 36
Отношение числа остатков маннозы к числу остатков галак- [3:2
тозы I 1:1(62]
шести-семи углеводных остатков. Эти цепи очень схожи между
собой, но по своему составу все же, по-видимому, не идентичны
[64]. Установлено, что на свободном конце каждой цепи находит-
ся остаток N-ацетилнейраминовой кислоты (сокращенное обозна-
чение NANA или реже фукоза), а на другом конце находится оста-
ток N-ацетил-о-глюкозамина, который, вероятно, связан с амид-
ным атомом азота аспарагинового остатка белка.
Сиаловая кислота может быть выделена из церулоплазмина
с помощью гидролиза при умеренном подкислении или путем
ферментативного расщепления. Полученный по второму способу
церулоплазмин, лишенный остатков сиаловой кислоты, при дейст-
вии р-галактозидазы теряето-галактозу, а при последующей обра-
ботке гексозаминидазой отщепляет N-ацетил-о-глюкозамин. Ли-
шенный остатков сиаловой кислоты асиалоцерулоплазмин
содержит то же количество меди и обладает теми же иммунохими-
ческими, спектральными и ферментативными свойствами, что и на-
тивный белок [35], хотя, разумеется, потеря остатков сиаловой
кислоты, несущих отрицательный заряд, сказывается на подвижно-
сти белка при электрофорезе. Из раствора асиалоцерулоплазмина,
содержащего 0,10 моль/л ацетата натрия с примесью 1% NaCl при
pH 5,45, можно выделить синие кристаллы, имеющие форму гекса-
гональных призм, на основании которых находятся невысокие
пирамидки (рис. 10.2). Эти кристаллы отличаются по форме от
кристаллов исходного церулоплазмина [65].
Хикман и сотр. [66], исходя из полностью десиалилированного
церулоплазмина, сумели при помощи специфической сиалилтранс-
феразы, добытой из печени крыс, и CMP-NANA-14C ввести сиало-
вую кислоту в белок и реставрировать исходный церулоплазмин
на 75%. Авторы установили интересный факт: десиалилированный
апоцерулоплазмин оказался лучшим акцептором NANA, чем де-
сиалилпрованный церулоплазмин. Отсюда возникает предположе-
24*
372
Глава 10
ние, что в процессе биосинтеза церулоплазмина присоединение
углеводов к белку предшествует вхождению в него ионов меди.
Протоны концевых спиртовых групп асиалоцерулоплазмина мо-
гут быть замещены на тритий, что достигается последовательной
обработкой белка галактозоксидазой и боргидридом, замещенным
Рис 10.2. Кристаллы десиалированного церулоплазмина четовека [651 (Х400).
Микрофотография Ирмин Штернлиб
на тритий. Так же как и церулоплазмин, асиалоцерулоплазмин, ме-
ченный тритием, может быть также помечен радиоактивной медью,
для чего применяется реакция обмена или реставрация из апофер-
мента [67]. Меченые белки были использованы при выполнении
интересных исследований, описанных ниже.
При инъекции кроликам гомогенного радиоактивного аспалоце-
рулоплазмина он удаляется из биологического кругооборота в те-
чение нескольких минут. В то же время известно, что полупериод
физиологической деятельности нативного церулоплазмина кролика
составляет 55 ч. Оказывается, асиалоцерулоплазмин захватывается
паренхимальными клетками печени и не поглощается ретикуло- эндо-
телиальной системой [67]. Такой быстрый перенос асиалоцеруло-
плазмина из плазмы в печень возможен только в том случае, если
на концах двух или большего числа полисахаридных цепей сохра-
нились остатки галактозы [68]. Если же эти остатки или окислить
Церулоплазмин 373
до производных альдегидов (в присутствии галактозоксидазы),
или удалить (при помощи 0-галактозидазы), или вновь к ним
присоединить остатки сиаловой кислоты, то период циркулирова-
ния белка в организме становится примерно таким же, как и в слу-
чае нативного белка [67].
Асиалоцерулоплазмин, захваченный печенью, быстро подвер-
гается разложению, которое происходит главным образом в лизо-
сомах [69]. В течение 20 мин белок теряет почти половину своей
меди, распад галактозы происходит еще быстрее. Быстрый захват
печенью гликопротеидов, не покрытых остатками сиаловой кислоты,
был продемонстрирован еще на семи примерах, когда в каче-
стве белков использовались оросомукоид, тироглобулин, «2-макро-
глобулин, гаптоглобин, лактоферрин, фетуин, а также гонадотроп-
ные гормоны человека [70]. Единственное исключение в этом от-
ношении составляет асиалотрансферрин, который циркулирует
в организме столько же, сколько исходный трансферрин. Указыва-
ют ли эти эксперименты на действительный путь катаболизма
церулоплазмина и других гликопротеидов в обычных условиях,
этот вопрос остается выяснить.
8. БИОСИНТЕЗ ЦЕРУЛОПЛАЗМИНА
Как и многие белки плазмы, церулоплазмин синтезируется
в печени [71], и скорость этого процесса контролируется гормона-
ми [72]. Концентрация церулоплазмина в плазме довольно ста-
бильна на протяжении всего периода жизни человека, за исклю-
чением того, что у новорожденных эта концентрация меньше. При-
сутствие церулоплазмина обнаружено не только в плазме, но также-
и в спинно-мозговой жидкости [73], лпмфе, межсуставной жид-
кости, слезах, в выделениях из носа и пищеварительного тракта1
[74]. Снижение скорости его синтеза у человека наблюдается толь-
ко в случае болезни Вильсона или дефицита меди [3].
Медь включается в молекулу церулоплазмина только в про-
цессе его биосинтеза. Внутриклеточным (хотя и очень бедным)
источником меди для биосинтеза служат, по-видимому, микросо-
мы [75]. Причиной очень малой скорости включения радиоактив-
ной меди в церулоплазмин у больных болезнью Вильсона могут
быть увеличенные запасы обычной меди в печени и малая скорость
собственно синтеза церулоплазмина. Гетерозиготные носители
одного из генов, ответственных за проявление болезни Вильсона,
обычно характеризуются нормальной скоростью синтеза церуло-
плазмина, однако скорость включения радиоактивной меди у них
понижена, что может быть вызвано небольшим увеличением содер-
жания меди в печени.
374
Глава 10
Таким образом, биосинтез церулоплазмина, вероятно, происхо-
дит в такой последовательности: синтез нескольких полипептидных
цепей, их сборка, присоединение полисахаридов и, наконец вклю-
чение меди. Недавно было установлено, что при дефиците меди
в плазме крыс присутствует апоцерулоплазмин, который обнару-
живается иммунохимическими методами [76]. Он был также об-
наружен в сыворотке крови здоровых людей и людей, страдающих
болезнью Вильсона, у которых было отмечено пониженное содер-
жание церулоплазмина [77, 78]. Химическая природа апоцеруло-
плазмина и его физиологическая роль пока изучены слабо.
СПИСОК ЛИТЕРАТУРЫ Т
1. Elvehjem С. A., Physiol., Rev., 15, 471 (1935).
2. Marston Н. R., Physiol. Rev., 32, 66 (1952).
3. Schneinberg I. H„ Sternlieb L, Pharmacol. Rev., 12, 355 (1960).
4. Underwood E. J., Trace Elements in Human and Animal Nutrition, 2nd edn.,
Academic Press, New York, 1962.
5. Lee G. R., Cartwright G. E„ Wintrobe M. M., Proc. Soc. Exp. Biol. Med., 127,
977 (1968).
6. Lee G. R., Nacht S., Lukens J. N., Cartwright G. E., J. Clin. Invest., 47, 2058
(1968).
7. Peisach J., Aisen P., Blumberg W. E. (eds.). The Biochemistry of Copper, Aca-
demic Press, New York, 1966.
8. Malmstrom B. G., Neilands J. B., A. Rev. Biochem., 33, 331 (1964).
9. Henkin R. L, Keiser H. R., laffe I. A., Sternlieb L, Scheinberg 1. H., Lancet,
ii. 1268 (1967).
10. Henkin R. I.. Keiser H. R., Kare M., Proc. Soc. Exp. Biol. Med., 129, 516
(1968).
11. Nimni M. E., Biochim. Biophys. Acta, 111, 576 (1965).
12. laffe I. A., Merriman P., Jacobus D., Science, 161, 1016 (1968).
13. Scheinberg L H., Sternlieb L, A. Rev. Med., 16, 119 (1965).
14. Garattini S., Giachetti A., Pieri L„ Arch. Biochem. Biophys., 91, 83 (1960).
15. Seal U. S., Comp. Biochem. Physiol., 13, 143 (1964).
16. Van Den Hamer C. J. A., Buyze G., van der Heyden M. С. M., in H. Peeters
(ed.). Protides of the Biological Fluids, Vol. 11, Elsevier, Amsterdam, 1964,
p. 382.
17. Martin G. M., Derr M. A., Benditt E. P„ Lab. Invest., 13, 282 (1964).
18. Bingley J. B., Dick A. T„ Clin. Chem. Acta, 25, 480 (1969).
19. Inaba T., Frieden E J. Biol. Chem., 242, 4789 (1967).
20. Sanders В. E., Miller О. P„ Richard M. N., Arch. Biochem. Biophys., 84, 60
(1959).
21. Curzon G., Vallet L., Nature, 183, 751 (1959).
22. Curzon G., Vallet L„ Biochem. J., 74, 279 (1960).
23. Deutsch H. F.. Arch. Biochem. Biophys., 89, 225 (1960).
24. Deutsch H. F., Kasper С. B„ Welsch D., Arch. Biochem. Biophys., 99. 132
(1962).
25. Morell A. G., Aisen P„ Scheinberg I. H„ J. Biol. Chem., 237, 3455 (1962).
26. Sgouris J. T., Coryell F. C., Gallick H., Storey R. W„ McCall К B„ Ander-
son H. D„ Vox Sang., 7, 394 (1962).
27. Broman L„ Kjellin K., Biochim. Biophys. Acta, 82, 101 (1964).
28. Morell A. G., Van Der Hamer C. J. A., Scheinberg I. H., J. Biol. Chem., 244, 3494
(1969).
Церулоплазмин
375
29. Simons К., Bearn A. G., Biochim. Biophys. Acta, 175, 260 (1969).
30. Sternlieb I., Morell A. G., Tucker W. D., Greene M. W., Scheinberg I. H., J. Clin.
Invest., 40, 1834 (1961).
31. Scheinberg I. H., Morell A. G„ J. Clin. Invest., 36, 1193 (1957).
32. Magdoff-Fairchild B., Lovell F. M., Low B. J. Biol. Chem., 244, 3497
(1969).
33. Kasper С. B., Deutsch FL F., J. Biol. Chem., 238, 2325 (1963).
34. Holmberg C. G., Laurell C.-В., Acta Chem. Scand., 2, 550 (1948).
35. Morell A. G., Van Den Hamer С. Л A., Scheinberg I. H., Ashwell G., J. Biol.
Chem., 241, 3745 (1966).
36. Broman L., Malmstrom B. G., Aasa R., Vanngard T., Biochim. Biophys. Acta,
75, 365 (1963).
37. Andreasson L.-E., Vanngard T., Biochim. Biophys. Acta, 200, 247 (1970).
38. Felsenfeld G., Arch. Biochem. Biophys., 87, 247 (I960).
39. Morell A. G., Aisen P„ Scheinberg I. H., Blumberg W. E„ Eisinger J., Federa-
tion Proc., 22, 595 (1963).
40. Morell A. G., Aisen P., Blumberg W. E., Scheinberg L H., J. Biol. Chem., 239,
1042 (1964).
41. Marriott Perkins D. J., Biochim. Biophys. Acta, 117, 387 (1966).
42. Kasper С. B., Deutsch H. F., J. Biol. Chem., 238, 2343 (1963).
43. Morell A. G., Scheinberg I. H., Science, 127, 588 (1958).
44. Aisen P., Morell A. G., J. Biol. Chem., 240, 2974 (1965).
45. Van Den Hamer C. J. A., Morell A. G., Scheinberg I. H., unpublished results.
46. Holmberg C. G., Laurell C.-В., Acta Chem. Scand., 5, 476 (1951).
47. Rice E. №.. J. Lab. Clin. Med., 55, 325 (1960).
48. Ravin H. A., Lancet, i, 726 (1956).
49. Laurell C.-В., in F. W. Putnam (ed.), The Plasma Proteins, Vol. 1, Academic
Press, New York, 1960, p. 349.
50. Osaki S., McDermott J. A., Frieden E„ J. Biol. Chem., 239, 3570 (1960).
51. Gurzon G., O’Reilly S., Biochem. Biophys. Res. Commun., 2, 284 (1960),
52. Osaki S., Johnson D. A., Frieden E„ J. Biol. Chem., 241, 2746 (1966).
53. Ragan H. A., Nacht S., Lee G. R., Bishop C. R., Cartwright G. E., Am. J. Phy-
siol., 217, 1320 (1969).
54. Topham R. W.. Frieden E„ J. Biol. Chem., 245, 6698 (1970).
55. Lee G. R., Nacht S„ Christensen D., Hansen S., Cartwright G. E., Proc. Soc,
Exp. Biol., Med., 131, 918 (1969).
56. Broman L., Nature, 182, 1655 (1958).
57. Deutsch H. F„ Fisher G. B., J. Biol. Chem., 239, 3325 (1964).
58. Broman L., Acta Soc. Med. Upsal. SuppL, 69, 7 (1964).
59. Morell A. G., Scheinberg I. H„ Science, 131, 930 (1960).
60. Richterich R., Gautier E., Stillhart H., Rossi E„ Helv. Paediat. Acta, 15, 424
(1960).
61. Hirschman S. Z., Morell A. G., Scheinberg L H., Ann. N. Y. Acad. Sci., 94, 960'
(1961).
62 Holtzman N. A., Naughton M. N., Iber F. L., Gaumnitz В. M., J. Clin. Invest.,
46, 993 (1967).
63 Shreffler D. C., Brewer G. J., Gall J. C., Honeyman M. S., Biochem. Genet.,
1, 101 (1967).
64. Jamieson G. A., J. Biol. Chem., 240, 2019 (1965).
65. Morell A. G., Sternlieb I., Scheinberg I. H., Science, 166, 1293 (1969).
66 Hickman J., Ashwell G., Morell A. G., Van Den Hamer C. J. A., Scheinberg I. H„
J. Biol. Chem., 245, 759 (1970).
67 Morell A. G., Jrvine R. A., Sternlieb J., Scheinberg I. H., Ashwell G., J. Biol.
Chem., 243, 155 (1968).
68. Van den Hamer C. J. A., Morell A. G., Scheinberg J. H., Hickman J., Ashwell G„
J. Biol. Chem., 245, 4397 (1970).
376
Глава 10
69. Gregoriadis G., Morell A. G., Sternlieb l„ Scheinberg I. H., J. Biol. Chem., 245,
5833 (1970).
70. Morell A. G., Gregoriadis G., Scheinberg I. H., Hickman J., Ashwell G., J. Biol.
Chem., 246, 1461 (1971).
71. Owen C. A., Jr., Hazelrig I. B„ Am. J. Physiol., 210, 1059 (1966).
72. Evans G. Ж, Cornatzer N. F., Cornatzer W. E., Am. J. Physiol., 218, 613
(1970).
73. Jensen K„ Acta Neurol. Scand., 39, 237 (1963).
74. Wilson J. F., Heiner D. 'C., Lahey M. E., J. Pediatrics, 60, 787 (1962).
75. Sternlieb I., Scheinberg I. H., Trans. Ass. Am. Phys., 75, 228 (1962).
76. Holtzman N. A., Gaumnitz В. M., J. Biol. Chem., 245, 2350 (1970).
77. Carrico R. J., Deutsch H. F., Beinert H., Orme-Johnson W. H., J. Biol. Chem.,
244,4141 (1969).
78. Carrico R. J., Deutsch H. F., Biochem. Med., 3, 117 (1969).
79. Edsall J. T., Spahr P.-F., personal communication (1960).
80. Witwicki J., Zakrzewski K„ Eur. J. Biochem., 10, 284 (1969).
81. Erickson J. O., Gray R. D., Frieden E., Proc. Soc. Exp. Biol. Med., 134, 117
(1970).
82. Poillon W. N., Bearn A. G., Biochim. Biophys. Acta, 127, 407 (1966).
•83. Blumberg W. E„ Eisinger J., Aisen P., Morell A. G., Scheinberg I. H., J. Biol.
Chem., 238, 1675 (1963).
84. Morell A. G., Scheinberg I. H., Science, 131, 930 (1960).
85. Siva Sankar D. V., Federation Proc. 18, 441 (1959).
86. Uriel J., Gotz H„ Grabar P., J. Suisse Med., 87, 431 (1957).
87. Aisen P., Koenig S. H., Lilienthal H. R., J. Mol. Biol., 28, 225 (1967).
88. Kasper C. B„ Biochemistry, 6, 3185 (1967).
89. Vanngard T„ in A. Ehrenberg, B. G. Malmstrom, T. Vanngard (eds.), Magne-
tic Resonance in Biological Systems, Pergamon, Oxford, 1966, p. 213.
90. Broman L„ Malmstrom B. G., Aasa R„ Vanngard T., J. Mol. BioL, 5, 301
(1962).
91. Kasper С. B., Deutsch H. F„ Beinert H., J. Biol. Chem., 238, 2338 (1963).
92. Ehrenberg A., Malmstrom B. G., Broman L., Mosbach R., J. Mol. Biol., 5, 450
(1962).
93. Kalons V., Hobzova H., in H. Peeters (ed.), Protides of the Biological Fluids,
Vol. 14, Elsevier, Amsterdam, 1966, p. 173.
94. Vanngard T„ Aasa R„ in W. Low (ed.), Paramagnetic Resonance, Academic
Press, New York, 1963, p. 509.
ГЛАВА 11
ГЕМЭРИТРИН *
М. И. Окамура, И. М. Клотц
Okamura М. Y., Klotz 7. М., Biochemistry Division,
Department of Chemistry, Northwestern University,
Evanston, Illinois 60201, USA
1. ВВЕДЕНИЕ
У беспозвоночных вместо хорошо известного гемоглобина роль-
переносчиков кислорода часто выполняют пигменты негемовой
природы — гемэритрин и гемоцианин. Несмотря на сходство функ-
ций всех трех названных белков, они сильно отличаются между
собой по молекулярной структуре и химической природе активного,
центра, ответственного за перенос кислорода. Некоторые характер-
ные свойства этих белков даны в табл. 11.1. Как в гемоглобине,
так и в гемэритрине кислород связывается атомом железа, однако-
в последнем (а также и в гемоцианине) отсутствует гемовая груп-
пировка, несмотря на то, что в самих терминах содержится корень-
«гем». Кроме того, гемэритрин соединяется с кислородом в соот-
ношении 2Fe : О2, тогда как гемоглобин характеризуется соотноше-
нием Fe : О2. Молекулярная масса и число субъединиц, входящих,
в макромолекулу нативного белка, тоже существенно разнятся для
двух указанных белков. И все же, несмотря на эти различия, и ге-
моглобин, и гемэритрин с одинаковым успехом выполняют роль-
переносчиков кислорода. На этом примере было бы интересно про-
следить, до какой же степени можно варьировать строение моле-
кулы без нарушения ее функций как переносчика кислорода.
* Большая часть нз рассматриваемых здесь работ выполняется при поддерж-
ке (№ НЕ-08299) со стороны Национального института исследования сердца
при Службе здоровья США.
В главе употребляются следующие сокращения: salen — М.Ы'-бис-салицили-
денэтилендиамин; phen — 1,10-фенантролин; bipy — 2,2'-дипнридин; terpy — 2,2',2"-
трнпириднн; Ас — ацетат; ЭДТА — этилендиамннтетрауксусная кислота; (—)-
ПДТА — левовращающая пропнлендиамннтетрауксусная кислота; ОЭДТА —
N-окснэтилэтиленднамннтриуксусная кислота; В — 2,13-диметнл-3,6,9,12,18-пеита-
азабнцнкло-[12,3,1]-октадекапентаен-123, 2, 12, 14, 16; еп — этнлендиамин.
.378
Глава И
Таблица 11.1
Сопоставление некоторых свойств белков — переносчиков кислорода
Свойства Гемоглобин Гемэритрин Гемоцнаннн
Металл Fe Fe Си
Степень окисления металла II П I
в деоксигенированном белке
Стехиометрия взаимодейст- Fe: О2 2Fe : О2 2Си : О2
вия с кислородом М : О2
.Цвет оксигенированного белка Красный Розово-фноле- товый Синий
Двет дезоксигенированного белка Пурпурно-крас- ный Бесцветный Бесцветный
Координация железа нлн Порфириновое Боковые цепи Боковые цепи
меди кольцо белка белка
Молекулярная масса 65 000 108 000 400 000— 20 000 000
Число субъединиц 4 8 Много
2. РАСПРОСТРАНЕНИЕ БЕЛКА СРЕДИ ЖИВЫХ
ОРГАНИЗМОВ И ЕГО ВЫДЕЛЕНИЕ
Гемэритрин был обнаружен у представителей четырех типов
•беспозвоночных: сипункулидов, полихетов (многощетинковых),
приапулидов и брахиаподов (плеченогих) [1]. Большая часть
работ, рассматриваемых здесь, выполнена на препаратах гемэрит-
рина, выделенных из сипункулида Golfingia gouldii.
Белок Golfingia gouldii может быть выделен из внутриполост-
ной жидкости этих морских червей. Для этого вскрывают полости
100—200 червей, дают стечь жидкости и после окончания коагуля-
ции фильтруют ее через стеклянную вату. Затем эритроциты мно-
гократно промывают морской водой (или 2,5%-ным раствором
NaCl), каждый раз отделяя их центрифугированием. Небольшое
количество бесцветных клеток, образующих слой над красными
кровяными клетками, удаляют отсасыванием. После этого для
разрушения красных кровяных клеток к ним добавляется дистил-
лированная вода в объеме, равном примерно половине объема ис-
ходной внутриполостной жидкости. Через 12 ч с помощью высо-
коскоростного центрифугирования отделяют осколки лизирован-
ных клеток, а полученный темно-красный раствор подвергают
>быстрому диализу по отношению к 0,4%-ному раствору NaCl.
Гемэритрин
379
Полученный раствор можно еще раз подвергнуть диализу по отно-
шению к раствору, содержащему 20% этанола и 0,4% NaCl, что
позволяет выделить вещество в кристаллическом виде. Все эти
процедуры проводятся на холоду (4 °C) и занимают 3—4 дня.
Получаемые таким способом кристаллы оксигемэритрина довольно
устойчивы при хранении при 4 °C, хотя белок все же медленно-
превращается в метгемэритрин или форму, содержащую желе-
30(111) [2].
3. ХИМИЧЕСКИЕ ФОРМУЛЫ ГЕМЭРИТРИНА
На рис. 11.1 схематически показана взаимосвязь различных
форм гемэритрина. Физиологическое значение имеет реакция об-
ратимого связывания гемэритрина с молекулярным кислородом,
[HrFe“ L
K3Fe(CN)e
реагент,
блокирующий [HrR
SH-группы -.Х£
▼
8 [HrFe“ 0SR 5
Н+
_Х*^ дипиюниш или
. формалтидинсульфиноеая
8 кислота
[НгГе"'он]6
Рис. 11.1. Взаимные превращения химических форм гемэритрина.
однако в присутствие определенных реагентов возможны и другие-
его превращения. При добавлении к оксигенированному гемэрит-
рину таких окислителей, как КзРе(СМ)б, образуется метаквогем-
эритрин, в котором железо находится в степени окисления +3.
Метгемэритрин в свою очередь может взаимодействовать с разно-
образными лигандами малых размеров, образуя производные
с характерными спектрами поглощения [3]. Молекула нативного-
гемэритрина состоит из восьми субъединиц, на которые она может
диссоциировать при добавлении реагентов, связывающих SH-груп-
пы [4].
4, РАВНОВЕСИЕ ВЗАИМОДЕЙСТВИЯ С КИСЛОРОДОМ
Обратимое связывание молекулярного кислорода гемэритрином
является его физиологической функцией. Кривая связывания кис-
лорода гемэритрином из Siputiculus nudus в отличие от аналогии-
380
Глава 11
ной характеристики гемоглобина имеет слабо выраженный сигмои-
дальный характер без эффекта Бора (т. е. равновесие связывания
кислорода не зависит от pH*) [5]. Сигмоидальный характер кри-
вой и эффект Бора в случае гемоглобина особенно ярко выражены
при низких значениях pH среды Различие в характере указанных
зависимостей двух переносчиков кислорода можно объяснить раз-
личиями в их физиологических функциях [6]. Гемэритрин обычно
находится в главной полости организма и служит в основном депо
кислорода, тогда как гемоглобин находится в постоянно циркули-
рующей системе и выполняет главным образом функцию перено-
счика кислорода. Именно при транспорте кислорода становятся
особенно ценными такие качества переносчика, как сигмоидальная
характеристическая кривая равновесия связывания кислорода и
смещение этого равновесия при изменении pH [7].
Другой важной особенностью гемэритрина является его высокое
сродство к кислороду, которое характерно для связывающих кис-
лород пигментов, содержащихся в главной полости организмов.
Такое сродство к кислороду обусловлено большим выигрышем
в энтальпии, сопровождающим процесс связывания кислорода: от
—12 до —20 ккал/моль [6].
Но, пожалуй, самое существенное отличие между гемэритри-
ном и гемоглобином заключено в стехиометрии реакции этих бел-
ков с кислородом. Для того чтобы связать одну молекулу кислоро-
да в гемэритрине, требуются два атома железа [2], тогда как ге-
моглобину достаточно одного.
Изучена также кинетика оксигенации и деоксигенации гемэрит-
рина из Sipunculus nudus [5]. Оба процесса оказались быстрыми,
но они не могут быть описаны с помощью только одной констан-
ты скорости. Более того, константы скорости деоксигенации окси-
гемэритрина дитионитом, полученные методом температурного
скачка и методом резкой остановки струи, отличаются между со-
бой. Эти результаты указывают на то, что процесс присоединения
кислорода к белку происходит не по простому механизму для би-
молекулярных реакций, а сопровождается, по-видимому, еше и
конформационными изменениями.
5. ПРОЦЕССЫ ОКИСЛЕНИЯ И РЕАКЦИИ
С МАЛЫМИ МОЛЕКУЛАМИ
При длительном хранении даже при 4 °C оксигемэритрин мед-
ленно переходит в метгемэритрин и теряет свою способность свя-
зывать кислород. Добавление же окислителей, например
Тем не менее у гемэритрина из Lingula эффект Бора обнаруживается [6].
Гемэритрин 381
K3Fe(CN)6, вызывает быстрый переход оксигемэритрина в метгем-
эритрин. Превращению оксигемэритрина в метгемэритрин способ-
ствует также относительно высокая концентрация (~0,2 моль/л)
какого-либо лиганда в растворе [3]. При этом в качестве одного
из продуктов реакции обнаруживается перекись водорода* [8].
Эта реакция имеет первый порядок по концентрации лиганда, ка-
тализируется кислотой в области pH от 6 до 9, и, вероятно, проте-
кает путем вытеснения перекисного (или гидроперекисного) иона:
HL+HrFe2O2 ---> HrFel+L+НОГ
Самыми эффективными катализаторами в этой реакции являются
ионы F-, NO2, N3 и_ НСЮ2 L менее эффективным оказывается
ион SCN-, а ионы SC? , С2Н3О и СН не проявляют никакой актив-
ности. Как видно, эффективность понижается с увеличением кис-
лотных свойств аниона, так что, возможно, здесь мы имеем дело
с общим кислотным катализом.
В нейтральной среде в отсутствие лигандов окисление гем-
эритрина приводит к образованию метаквогемэритрина. При дости-
жении рН>9 метаквогемэритрин переходит в метгидроксигемэрит-
рин, что сопровождается характерными изменениями в спектре
поглощения белка**. Кажущееся значение рКа для этого перехода
значительно возрастает в присутствии специфически действующих
анионов СЮ? и NO3, которые связываются с белком, но железом
непосредственно не координируются [9]. Указанные два иона
влияют не только на равновесие перехода метаквогемэритрина
в метгидроксигемэритрин, но также и на другие свойства белка,
в частности на реакционную способность его сульфгидрильных
групп [9].
Атом железа метгемэритрина также может координировать са-
мые разнообразные лиганды (например, N3, SCN-, СН, F-). В тех
случаях, когда удалось определить стехиометрию (например, при
комплексообразовании с N и SCN-), оказалось, что одна молеку-
ла лиганда связывается с двумя атомами железа [3, 4]. Однако
данные, полученные из спектров поглощения и спектров кругового
дихроизма, указывают на то, что с такими лигандами, как F",
НгО и ОН", метгемэритрин может взаимодействовать в отношении
один лиганд на один атом железа [10].
* В присутствии ионов F~ и NO 7 кинетика протекания реакции соответству-
ет псевдопервому порядку. В присутствии же N3 вначале протекает быстрая
реакция, за которой следует более медленная двухстадийная реакция.
** Обозначения «метакво» и «метгидрокси» относятся к окисленным формам
гемэритрина, существующим в растворе при pH около 7 и около 9 соответствен-
но. Возможно, что в метаквогемэритрине атом железа связан не с молекулой
воды, а с гидроксил-ионом, как это предполагается в случае гемоглобина.
382
Глава 11
6. ОБРАЗОВАНИЕ СУБЪЕДИНИЦ
Молекула гемэритрина состоит из восьми субъединиц, на кото-
рые она распадается при действии разнообразных средств. Наи-
более мягким из них является действие реагентов, блокирующих
меркаптогруппы, например, таких, как ртутьсодержащие соедине-
ния или цистин [4, И]. Различные признаки указывают на то, что
образующиеся при этом субъединицы отличаются своей конформа-
цией от субъединиц нативного белка. Спектр поглощения в уль-
трафиолетовой области и спектр кругового дихроизма цистеинат-
ного производного субъединиц отличаются от соответствующих
спектров октамера или субъединиц, полученных простым разбав-
лением раствора белка [12].
Кроме того, субъединицы, полученные путем блокирования
SH-групп, отличаются от нативных субъединиц или октамера своей
способностью к связыванию с различными бидентатными хелато-
образующими лигандами типа о-фенантролина или тайрона и об-
разованию комплексов, не поддающихся диализу [13]. Так, о-фе-
нантролин взаимодействует с гемэритрином в форме, содержащей
Fe(II), тогда как тайрон образует комплекс с той формой гемэрит-
рина, в которой содержится Fe(III). Последний комплекс гемэрит-
рина с блокированными SH-группами проявляется в спектре ЭПР,
давая сигнал с g-фактором, равным 4,2, характерным для желе-
за(Ш).
7. КООРДИНАЦИЯ ЖЕЛЕЗА В БЕЛКЕ
Поскольку в гемэритрине отсутствует гемовая группировка,
часть координационных мест у атома железа должны занимать
боковые группы полипептидной матрицы белка. Первичная струк-
тура гемэритрина известна [14]. Она показана на рис. 11.2. Только
небольшая часть из указанных на рисунке 113 остатков аминокис-
лот может рассматриваться в качестве потенциальных лигандов,
способных связывать железо. Из рассмотрения можно сразу ис-
ключить все неполярные боковые цепи (такие, как Phe, lie, Vai
и т. п.), а также остатки треонина, серина, аспарагина, глутамина
и, по-видимому, аргинина. Напротив, остатки цистеина, лизина,
гистидина, тирозина, метионина, аспарагиновой и глутаминовой
кислот могут координироваться железом. Всего таких остатков 43
(табл. 11.2). Этого более чем достаточно, если учесть, что субъ-
единица содержит два атома железа, которые могут связывать 8—
10 лигандов*. За исключением, пожалуй, атомов серы цистеина и
* Одно из препятствий на пути определения мест связывания железа с по-
липептндиой цепью — это невозможность обратимого удаления железа из гем-
эритрина
Гемэритрин
383
метионина, все потенциальные лиганды, входящие в состав белка,
относятся к числу неполяризующихся «жестких» лигандов [15].
Долгое время наиболее вероятными лигандами, связывающими
железо в белке, считались сульфгидрильные группы остатков ци-
Рис. 11.2. Первичная структура гемэритрина.
стеина. Однако блокировка последних при обработке ртутьсодер-
жащими соединениями не приводит к каким-либо изменениям
в спектре поглощения железа [4], что дает основание отбросить
такое предположение. Аналогичным образом можно исключить из
рассмотрения е-аминогруппы лизина, а также концевую а-амино-
группу, которые можно блокировать, например, тринитросульфо-
бензолом, что также не приводит к изменениям в спектре погло-
щения [16].
С другой стороны, четыре остатка гистидина в белке прояв-
ляют пониженную реакционную способность по отношению к ди-
384
Глава 11
Таблица 11.2
Возможные места связывания железа с белком
Группа
Остаток
аминокислоты
Порядковый
номер в белке
Вывод о связи с Fe
на основании опытов
по химической
модификации
SH
Cys
Lys
NH2 (концевая)
Туг
His
Glu
Asp
Met
6
11
1
He участвует
To же
Возможно, участ-
вуют 2 группы
Возможно, участ-
вуют 4 группы
азоний-1Н-тетр азолу, что может быть обусловлено их связыванием
с атомами железа [16]. Кроме того, обнаружено, что взаимодейст-
вие двух остатков тирозина с тетранитрометаном сопровождается
высвобождением одного атома железа из субъединицы, что указы-
вает на возможность связывания атомов железа и с остатками ти-
розина [17]. Местами связывания железа могут быть также кар-
боксильные группы остатков глутаминовой или аспарагиновой кис-
лот и тиоэфирная группа остатка метионина (табл. 11.2). Однако
в отношении этих потенциальных лигандов нет никаких экспери-
ментальных данных, подтверждающих или опровергающих такое
предположение.
8. МАГНИТНЫЕ И СПЕКТРАЛЬНЫЕ СВОЙСТВА
АКТИВНОГО ЦЕНТРА
Поскольку исследование электромагнитных свойств железа,
входящего в состав гемэритрина, направлено на выяснение строе-
ния активного центра, полученные данные необходимо сопостав-
Гемэритрин
385
лять с результатами исследований модельных соединений, строение
которых известно. Учитывая еще и тот факт, что в субъединице
гемэритрина содержатся два атома железа, в рассмотрение полез-
но включить как биядерные, так и моноядерные его комплексы.
8.1. Модельные соединения
Железо(III) в нейтральных или щелочных растворах проявля-
ет сильную склонность к полимеризации. Число охарактеризован-
ных биядерных и полимерных комплексов железа (III) с неоргани-
ческими и биологическими лигандами непрерывно растет [8, 19].
Железо(III) в биядерных комплексах обладает аномальными маг-
нитными свойствами, которые обусловлены антиферромагнитным
обменным взаимодействием (—2/515г) между двумя атомами же-
леза (/ — отрицательная величина). При таком взаимодействии
двух высокоспиновых ионов железа(III) основное состояние систе-
мы— диамагнитное синглетное (5=0), а возбужденные состояния,
которых имеется целый набор (5=1, 2, 3, 4, 5), расположены не
слишком высоко и могут частично заселяться при повышении тем-
пературы. При понижении температуры заселенность их умень-
шается, и магнитные свойства образца приближаются к свойствам
диамагнитных комплексов. Это проявляется в уменьшении экспе-
риментально измеряемого магнитного момента [18, 20] и интенсив-
ности сигнала ЭПР [21, 22]. При низких температурах спектр
Мессбауэра такого комплекса становится похожим на спектры
диамагнитных комплексов и в нем отсутствует уширение, обуслов-
ленное сверхтонким взаимодействием, которое не проявляется
даже при наложении магнитных полей высокой напряженности
[22—25].
В спектрах поглощения биядерных комплексов железа (III)
с антиферромагнитным взаимодействием центральных атомов на-
блюдается увеличение интенсивности d—d-полос, расположенных
в длинноволновой области (800—900 нм). Это явление, по крайней
мере отчасти, объясняется тем, что для указанных переходов сни-
мается «запрет по спину» [26].
В биядерных мостиковых комплексах железа(III) антиферро-
магнитное взаимодействие происходит по механизму сверхобмена
и зависит от характера перекрывания орбиталей атомов железа
с орбиталями мостиковых атомов. В табл. 11.3 приведены магнит-
ные характеристики некоторых мостиковых биядерных группиро-
вок.
Биядерные комплексы железа (III) с линейным оксомостиком
(тип I в табл. 11.3) встречаются очень часто и характеризуются
сильным обменным взаимодействием, которое обусловлено коротким
25—2451
386
Глава 11
расстоянием Fe—О*, свидетельствующим о существенном вкладе
л-связывающих орбиталей в образование связей в этой мостико-
вой группе. Второй тип биядерных комплексов (тип II в табл. 11.3)
характеризуется наличием двух гидрокси- или алкокси-мостиков.
В комплексах этого типа обменное взаимодействие намного сла-
Таблица 11.3
Строение и магнитные свойства некоторых биядериых
комплексов железа(П1)а
Строение Тип —J, см—1 Р<эфф при комнатной температуре, отнесенный к одному атому Fe, , магнетоны Бора
Fe —О —Fe I 80—100 1,28—2,0
ГР Цб 8 5,3
О Fe/ \ре 120° III 30 3,6s
Низкоспиновое желе- зо (III) 0 2,0—2,5
Высокоспиновое желе- зо (III) 0 5,7—6,0
а
б
в
См. работу [10] и литературу к ней.
R=H, salen.
В расчете на g—2\ не зависящий от температуры парамагнетизм отсутствует.
бее, что частично обусловлено меньшей прочностью одинарных
связей Fe—О, а частично тем, что обменное взаимодействие под
углом 90° менее эффективно**. Третий тип мостиковых комплек-
сов, представителем которых является тримерный ацетат желе-
за (III), по эффективности обменного взаимодействия занимает
промежуточное положение между комплексами типа I и II.
* В оксомостиковой группе расстояние Fe — О равно 1,8 А, тогда как обыч-
ная связь F — О имеет длину 2 А [26а].
** Меньшая эффективность обмена при угле мостика Fe—О—Fe, равном 90°,
объясняется тем, что спиновый момент может передаваться только через одинар-
ную о-связь Fe — О, тогда как передача спинового момента через л-перекрываю-
щиеся орбитали в данном случае малоэффективна.
Гемэритрин
387
8.2. Гемэритрин
8.2.1. Спектры поглощения и кругового дихроизма (КД)
Спектральные характеристики гемэритрина были недавно рас-
смотрены в обзоре [10]. Они сопоставимы с аналогичными харак-
теристиками модельных биядерных оксомостиковых комплексов,
что видно из данных, приведенных в табл. 11.4.
Спектры поглощения и КД всех форм гемэритрина, за исклю-
чением его акво-, гидрокси- и фторидного комплексов, не чувст-
вительны по отношению к изменению pH среды. Особый интерес
представляют полосы в спектре поглощения гемэритрина, располо-
женные в области 600—700 нм, которые были отнесены к d—d-пе-
реходам. Молярные коэффициенты поглощения этих полос в ра-
счете на атом железа (е«20—50) намного больше тех значений,
которые следовало бы ожидать в случае «спин-запрещенных»
d—d-переходов мономерных высокоспиновых комплексов желе-
за (III). Увеличение интенсивности этих полос объясняется обмен-
ным взаимодействием между двумя атомами железа.
Полосы переноса заряда с лиганда на атом железа, а также
другие полосы переноса заряда примерно при 480, 380 и 330 нм,
наблюдаемые в спектре поглощения гемэритрина, похожи на ана-
логичные полосы простых комплексов [Fe(H2O)sL]3+.
Спектры КД комплексов метгемэритрина с анионами С1~, Вг_,
NCO-, CN~, SCN- и N3, за исключением полосы переноса заряда
с лиганда на металл, весьма сходны между собой. Два комплекса
метгемэритрина — с НгО и с ОН- — резко отличаются своими
спектрами КД от группы перечисленных выше комплексов. Спек-
тры КД фторидных комплексов в зависимости от условий экспери-
мента могут быть похожи как на спектры первой группы комплексов,
так и на спектры акво- и гидрокси-комплекса. Такое явление мож-
но объяснить различным составом образующихся фторидных
комплексов: в первом случае спектр КД соответствует, по-види-
мому, комплексу с соотношением лиганд: металл, равным 1 : 2,
а во втором — комплексу с соотношением, равным 1:1 [10].
Примечательно, что как спектры поглощения, так и спектры КД
оксигемэритрина очень близки с соответствующими спектрами
метгемэритрина, на что впервые обратили внимание Надь и Клотц
[3]. На основании этого сходства спектров можно предполагать,
что железо в гемэритрине находится в степени окисления +3.
8.2.2. Магнитная восприимчивость
Магнитная восприимчивость различных форм гемэритрина при
комнатной температуре измерялась во многих лабораториях и
с использованием различных методов [25, 28, 29]. Результаты всех
25*
Спектры поглощения комплексов гемэритрина и модельных оксо-мостиковых
Комплекс Лиганд® Стехио- метрияв Поло
X, нм Ем 1, нм ем 1, нм
Оксигем— Oa 1 750 пл. 200 — — 500
эритрин sh- 1(?) (не набл.) — — 510
Na’ 1 680 190 — — 446
NCS- 1 674 200 — — 452
NCO- 1 650 166 — — 480 пл.
Br- 1 677 165 — — 505 пл.
ci- 1 656 180 — — 490 пл.
CN- 1 695 140 — — 493
F~ 1 или 2 595 пл. 200 — — 480 пл.
OH- 2 597 160 — —• 480 пл.
HaO 2 580 пл. 200 — —“ 480 пл.
HaO/I- 550 пл. 800 — — 480 пл.
[Fe3O(Ac)e(HaO)sl2- — — 973 15,7 515 пл. 124 464
[(Fe(HaO)B)aOl«- — — 876 7,2 495 160 465
[(РеЭДТА)аО]«- — — 878 6,6 540 пл. 113 475
[(Fe(—)-ПДТА)аО]4- — — 880 8,4 540 пл. 115 475
[(РеОЭДТА)аО]2- — — 875 6,5 540 пл. 104 474
I(Fe(phen)a)aO]4+ — — -970 — 580 пл. — —
[(Fe(bipy)a)aO]«- — — 960 — 575 пл. — 525 пл.
[(Feterpy)aO]*+ — — —950 — 590 пл. — 525
t(Fesalen)aO] — — —1200 — — — 500 пл.
I(Fc(salen)Cl)a] — — —1250 — — — 525
Данные взяты из работы [10]. См. также библиографию.
б Лиганды, образующие комплексы с гемэритрином. Иодид ие координируется непосред-
в Указанная стехиометрия точно установлена только в случае кислорода, амида и
константы образования.
г Молярные коэффициенты поглощения даны в расчете на одну субъединицу, содержа -
мостиковым лигандом, пл. означает «плечо».
Таблица 11.4
комплексов железа(111)а
сыг
ем X, нм Ем им Ем X, им Ем X. нм Ем X, нм ЕМ
2200 360 пл. 5450 330 6800 — — — — — —
—4000 ~370 пл. —5000 —330 ~7000 — — — — — —
3700 380 пл. 4300 326 6750 — — — — — —
5100 370 пл. 4900 327 7200 — — — — — —
700 377 6500 334 6550 — — — — — —
950 387 5400 331 6500 — — — — — —
750 380 6000 329 6600 — — — — — —
770 374 5300 330 6400 — — — — — —
400 362 5000 317 5600 — — — — — —
550 362 5900 320 6800 — — — — — —
€00 — — 355 6400 — — — — — —
1200 — — 340 6300 — — — — — —
199 406 пл. 630 335 5010 — — — — — —
172 — — 364 9360 — — 274 15756 — —
199 405 пл. 810 335 10700 303 14100 267 14100 240 17200
197 400 пл. 920 335 10600 305 13200 267 14700 240 16500
196 405 пл. 830 335 пл. 11400 305 15400 270 16400 237 17600
— — — 352 пл. — 310 пл. •1 f. 273 — 225 —
— — — 345 пл. — 310 пл. — 281 — 234 —
— 410 — — — — — — — —
— — — 377 — 290 пл. — 256 пл. — 226 —
- 435 — — — 322 — 265 — 225 —
ственно атомом железа, но находится поблизости к нему.
тиоцииата. В случае цианида, хлорида и гидроксида стехиометрию выводили исходя из
щую два атома железа. Курсивом обозначены полосы переноса заряда, ие связанные с оксо-
390
Глава 11
этих измерений качественно согласуются между собой. Деокси-
генированный гемэритрин содержит железо (II) в высокоспиновом
состоянии. Оксигенированный гемэритрин, а также все изученные
комплексы гемэритрина (с N3, SCNH, F-, CN~, Н2О и CNO-) обла-
дают более низкой магнитной восприимчивостью. Разность квад-
ратов эффективных магнитных моментов деоксигемэритрина и
окси- или метгемэритрина равна примерно Д|4фф=18±2 (магнетон
Бора)2 на атом Fe, хотя точной поправки на диамагнитную со-
ставляющую при этих расчетах не вводилось.
Низкую магнитную восприимчивость оксигемэритрина и метгем-
эритрина при комнатной температуре можно в общем объяснить
спариванием спина, обусловленным сильным расщеплением
d-уровней иона железа (III) в кристаллическом поле или сильным
антиферромагнитным взаимодействием соседних атомов железа
(см. комплексы типа I в табл. 11.3).
Измерение магнитной восприимчивости при низких темпера-
турах и данные гамма-резонансной спектроскопии подтверждают
последнее предположение. При низких температурах магнитная
восприимчивость оксигемэритрина и метгемэритрина уменьшается
[30, 31]. В области температур 1,4—4,2 К железо в этих формах
белка диамагнитно [31]. Если бы ионы железа(III) были изолиро-
ваны друг от друга, то диамагнитное состояние никоим образом не
могло быть достигнуто ввиду отсутствия дополнительных неспарен-
ных электронов. Таким образом, по крайней мере в случае мет-
гемэритрина, антиферромагнитное взаимодействие двух атомов
железа можно считать твердо установленным фактом* [7].
8.2.3. Спектры Мессбауэра
Спектры Мессбауэра производных гемэритрина [25, 32] дают
информацию об электронной плотности на ядре железа и о гра-
диенте электрического поля, что отражает электронное состояние
атома железа. Полученные параметры спектров Мессбауэра гем-
эритрина сопоставлены в табл. 11.5 с соответствующими данными
о некоторых модельных биядерных мостиковых комплексах желе-
за(Ш).
а) Деоксигемэритрин. Спектр Мессбауэра деоксигемэритрина
состоит из одного дублета, обусловленного квадрупольным рас-
щеплением (рис. 11.3). Сильное квадрупольное расщепление
и большой изомерный сдвиг (табл. 11.5), наблюдаемые в этом
спектре, характерны для высокоспинового железа(II), что согла-
суется с данными по измерению магнитной восприимчивости деок-
* Можно допустить, что в случае оксигемэритрина диамагнетизм основного
состояния обусловлен присутствием двух атомов Fe(II) в низкоспиновом состоя-
нии. Однако кажется более правдоподобным, что диамагнетизм здесь обусловлен
той же причиной, что и в случае метгемэритрина.
Гемэритрин
391
Таблица П.5
Параметры спектров Мессбауэра комплексов гемэритрина
и модельных систем
Комплекс Лиганд * Параметры спектра Мессбауэра2
изомерный сдвиг О, ММ/С квадрупольное расщепление AFKB, мм/с
Оксигемэритрин O2 0,46 1,87
0,47 0,94
NCS- 0,55 —1,92
ci- 0,50 2,04
Метгемэритрин F- 0,55 1,93
w 1 0,50 1,91
H2O 0,46 1,57
(FeaO(Ac)«(H2O)3l2- — 0,43 0,43
f(Fe(H2O)B)2O]«- — 0,54 0,85
((РеЭДТА)2О]«- — 0,46 —1,44
((Fe(—)-ЦЦТА)2О]4- — 0,44 1,74
((РеОЭДТА)2О]2~ — 0,46 —1,68
((Fe(phen)2)2O]«- — 0,49 1,68
[(Felbipy^Ol" — 0,48 1,51
[(Feterpy)2or — 0,59 2,35
f(Fesalen)2O] — 0,46 4-0,78
[(FesalenCl)2J — 0,51 —1,40
Деоксигемэритрин H2O 1,15 2,86
а Спектры снимались при 77 К по отношению к железной фольге. Знак при Д£кв приве-
ден для случаев, где он определялся. Данные взяты из работы 1131.
сигемэритрина. Одинаковые параметры спектра Мессбауэра от
двух ядер железа, входящих в субъединицу, свидетельствуют об
одинаковой координации обоих атомов.
б) Метгемэритрин. В спектрах акво-, тиоцианатного, фторидно-
го, хлоридного и азидного комплексов метгемэритрина при сниже-
нии температуры до 4,2 К (рис. 11.4) наблюдается один дублет.
Изомерные сдвиги для всех указанных комплексов (табл. 11.5)
находятся в пределах от 0,45 до 0,55 мм/с (по отношению к метал-
лическому железу), что характерно для высокоспинового состоя-
ния железа (III). Величины квадрупольного расщепления для этих
392
Глава 11
комплексов очень близки между собой; они больше, чем обычная
величина квадрупольного расщепления для простых солей желе-
за (III) в высокоспиновом состоянии. Однако такие большие ве-
личины квадрупольного расщепления наблюдаются в случае бия-
дерных мостиковых высокоспиновых комплексов железа(III) или
Х<19зк
*• " * • * *+
' «Л* 77 к
-А0
2Р ОР 2,0
Скорость, мм/с
1----1____I____Г----1___L____1----1___I
-Ар -гр qo гр 4,0'
Скорость, jkm/c
Рис. 11.3. Спектры Мессбауэра де-
оксигемэритрина при разных темпе-
ратурах.
Рис. 11.4. Спектры Мессбауэра тио-
цианатогемэритрина (/) и аквогемэ-
ритрина (2) при 77 К.
мономерного высокоспинового комплекса железа (III) с
салицилиденэтилендиамином (salen) (табл. 11.5). Эти параметры
спектров Мессбауэра соответствуют высокоспиновым атомам же-
леза (Ш), связанным между собой антиферромагнитным взаимо-
действием.
в) Оксигемэритрин. Спектр Мессбауэра оксигемэритрина со-
стоит из двух полос, квадрупольно расщепленных на дублеты
(рис. 11.5). Оба дублета определенно отличаются от дублета
в спектре железа(II) деоксигемэритрина. Полученный спектр
свидетельствует о том, что в оксигемэритрине оба атома железа
принимают участие в связывании кислорода и что электронное
окружение у этих двух ядер железа различное. Наблюдение двух
дублетов можно было бы объяснить существованием равновесия
между двумя электронными состояниями одного и того же атома
железа или присутствием двух геометрических изомеров. Однако
эти предположения отпадают ввиду того, что относительная ин-
тенсивность указанных двух дублетов остается неизменной и при
4,2 К- Изомерный сдвиг обоих дублетов очень близок к сдвигу
дублетной полосы у производных метгемэритрина. В спектре окси-
Гемэритрин
393
гемэритрина пара крайних линий расщеплена так же сильно, как
и в случае комплексов метгемэритрина, а расщепление между
двумя внутренними линиями меньше. Изомерный сдвиг обоих
дублетов свидетельствует о том, что степень окисления обоих ато-
мов железа оксигемэритрина равна +3.
г) Влияние магнитных полей. Парамагнитные состояния атомов
железа могут вызвать магнитное сверхтонкое расщепление их
Рис. 1.1 Л Спектры Мессбауэра ок-
сигемэритрина при разных темпера-
турах.
Jo,5’/.
<---1___।---1---1---1__।___1---1___।__।
-Ар -гр ср гр ар
Скорость, лии/с
спектров Мессбауэра [23, 24]. Величина такого расщепления про-
порциональна усредненному магнитному моменту <ц>, создавае-
мому электроном на ядре за период, соизмеримый с временем су-
ществования возбужденного состояния 57Fe. Это время имеет
порядок 10~7 с, и если время релаксации магнитного момента элек-
трона намного короче этого периода (за счет спин-решеточного или
спин-спинового взаимодействия), то усредненный магнитный мо-
мент электрона в масштабе времени гамма-резонансного перехода
будет равен нулю и сверхтонкого расщепления не будет наблю-
даться. Напряженность поля сверхтонкого взаимодействия имеет
порядок нескольких сотен килоэрстед. Это поле можно индуциро-
вать при понижении температуры и наложении внешнего магнит-
ного поля высокой индукции.
На рис. 11.6 показаны спектры Мессбауэра парамагнитного
моноядерного (а) и биядерного (в) комплексов для сравнения их
поведения в случае наложения внешнего магнитного поля. Ведут
они себя по-разному, и расщепление линии спектра во втором
случае можно объяснить воздействием приложенного внешнего
магнитного поля. В спектре Мессбауэра оксигемэритрина, а также
акво- и тиоцианатного комплексов метгемэритрина, снятых при
394
Глава 11
Скорость, Л1Л1/С Скорость, мм/с
Рис. 11.6. Спектры Мессбауэра модельных комплексов железа (III) при темпе-
ратуре 4,2 К и различной напряженности магнитного поля.
а — мономерный комплекс Fe® (salen)CI; б— димерный комплекс [Fe ®(sa!en)hO (мо-
номер а, по-видимому, содержит примесь димера).
Скорость, лыи/с
Рис. 11.7. Спектр Мессбауэра тио-
цианатогемэритрина при 4,2 К в маг-
нитном поле напряженностью 30 кЭ.
4,2 К, внешнее магнитное поле с индукцией 5 кЭ не вызывает ника-
ких изменений. Такое поведение спектра показательно для диамаг-
нитного состояния железа. В поле 30 кГс линии в спектре тиоциа-
натного комплекса метгемэритрина расщепляются [32], что можно
полностью отнести за счет внешнего магнитного поля (рис. 11.7).
Отсутствие индуцированного поля сверхтонкого взаимодействия
в этих условиях определенно указывает на то, что основное состоя-
ние комплекса — диамагнитное, которое может быть следствием
антиферромагнитного взаимодействия между атомами железа(III).
Гемэритрин
395
8.2.4. Электронный парамагнитный резонанс
Спектры ЭПР гемэритрина изучались при разных температу-
рах [25, 31]. В этих спектрах ни разу не было обнаружено сигна-
ла, который однозначно можно было бы отнести к сигналу желе-
за. В некоторых образцах окси- и метгемэритринов был зафикси-
рован сигнал при g=2, который, по-видимому, обусловлен какой-то
примесью. Кроме того, при восстановлении оксигемэритрина дитио-
нитом натрия наблюдался сигнал при g=l,94. Однако этот сигнал
не появляется, если деоксигемэритрин получать из оксигемэритри-
на путем деоксигенации [29].
Некоторые модельные биядерные антиферромагнитные комп-
лексы железа (III) дают сигналы ЭПР [21, 22]. Интенсивность их
уменьшается при понижении температуры, как и следовало ожи-
дать при наличии парамагнитных возбужденных состояний и диа-
магнитного основного состояния. Однако у других аналогичных
комплексов сигналов ЭПР не было обнаружено. Возможно, в этих
случаях спектр ЭПР не наблюдается из-за большого расщепления
в нулевом поле или из-за быстрой релаксации [33]. Спектр ЭПР
антиферромагнитно спаренных ионов железа(III) в биологических
образцах легко можно было бы охарактеризовать по температур-
ной зависимости его интенсивности. Однако, как мы убедились,
этот спектр не всегда может наблюдаться.
9. СТРОЕНИЕ АКТИВНОГО ЦЕНТРА
До тех пор пока структура гемэритрина не будет однозначно
установлена с помощью кристаллографических методов, о природе
связывания железа в его активном центре можно судить по таким
косвенным показателям, как параметры спектров поглощения,
спектров Мессбауэра и магнитная восприимчивость. Интерпрета-
ция этих характеристик по отношению к структурным характери-
стикам не всегда однозначна, и хотя с ними может быть связан
критерий той или иной структуры, они обычно не дают возмож-
ности полностью расшифровать структуру вещества, как это
удается сделать с помощью дифракции рентгеновских лучей.
Итак, строение гемэритрина, которое мы хотим себе предста-
вить, должно объяснять следующие его свойства:
1) малая величина магнитной восприимчивости окси- и метгем-
эритринов и диамагнетизм их основного состояния при низких
температурах;
2) неэквивалентность окружения атомов железа в оксигемэрит-
рине и эквивалентность их окружения в метгемэритрине и диокси-
гемэритрине, что следует из соответствующих спектров Мессбау-
эра;
396
Глава tl
3) отсутствие эффекта Бора, т. е. выделения протона при окси-
генации;
4) отсутствие сигнала ЭПР от ионов железа в окси- и метгем-
эритринах.
5) спектры поглощения и КД.
9.1. Метгемэритрин
Спектры Мессбауэра метгемэритрина в условиях низкой темпе-
ратуры и сильного магнитного поля, а также величина его магнит-
ной восприимчивости при низкой температуре указывают на то,
что железо в этой форме гемэритрина находится в состоянии анти-
ферромагнитного обменного взаимодействия. Малая магнитная вос-
приимчивость при комнатной температуре и величина изомерного
сдвига в спектре Мессбауэра, характерная для высокоспинового
состояния железа(III), ближе всего подходят к картине сильного
(|/|^30 см-1) антиферромагнитного взаимодействия между дву-
мя атомами железа(III), находящимися в слабом поле лигандов.
Это антиферромагнитное обменное взаимодействие, по-видимому,
обусловлено сверхобменным взаимодействием через мостиковый
лиганд.
Поскольку остатки цистеина, как было показано, не координи-
рованы атомом железа, это обменное взаимодействие происходит,
по-видимому, без участия серусодержащих лигандов (хотя пока
не исключена возможность участия в мостиковой связи атомов серы
остатка метионина). Среди биядерных неорганических комплексов
железа(III) встречаются примеры мостиковых связей через кис-
лород гидроксил-иона или алкоксигруппы, который подобен фе-
нольному кислороду в остатке тирозина. Однако обменное взаи-
модействие в такого типа мостиковых неорганических комплексах
слишком мало и оно не могло бы настолько сильно снизить магнит-
ную восприимчивость метгемэритрина при комнатной температуре.
Оксомостиковая структура Fe—О—Fe наилучшим образом объяс-
няет как данные, полученные из спектров Мессбауэра, так и вели-
чину магнитной воприимчивости метгемэритрина.
По мнению Гарбетта и сотр. [10], стехиометрия взаимодействия
гемэритрина с азидом и тиоцианатом (лиганд : металл = 1 : 2) и
эквивалентность окружения атомов железа (о чем свидетельствует
вид спектров Мессбауэра) могут быть объяснены образованием
симметричной двухмостиковой структуры I:
О
^е\ /Fe
xz
I
X = СГ, Вг“ F^,
:n=c=s,
=N=N,
;c=n,
•N=C=O
Гемэритрин
397
Указанные выше комплексы метгемэритрина обладают близкими
спектрами КД.
Комплексы метгемэритрина с водой, гидроксил-ионом и фторид-
ионом при большой концентрации последнего отличаются от пре-
дыдущего ряда комплексов своими спектрами КД. Как предпола-
гают Гарбетт и сотрудники, эти комплексы соответствуют другому
соотношению лиганд: металл, а именно 2:2. Это отражено в
структуре II:
О
Fe^ 'ре
X X
И, Х=Н2О, НО-, F"
Оксомостиковые структуры, подобные структуре I, в которой
из-за отталкивания между атомом кислорода и вторым лигандом
угол Fe—О—Fe должен приближаться к 90°, в неорганических
комплексах железа (III) до сих пор не встречались. Так что в дан-
ном случае трудно оценить ожидаемую степень обменного взаимо-
действия. Мостиковая группировка Fe—О—Fe образует угол, близ-
кий к 90°, только в том случае, если кислород связан с третьим
партнером, например протоном. Это сопровождается ослаблением
связи Fe—О и значительным уменьшением эффективности обмен-
ного взаимодействия. Возможно, что протонированию мостикового
этого высказывались предположения о том, что обменное взаимо-
ковой группы в белке либо оксо-мостик связан с белком слабой
водородной связью. В таком случае в оксомостиковой структуре I
могло бы сохраниться короткое расстояние Fe—О и сильный анти-
ферромагнитный обмен, присущие оксомостиковой приблизительно
линейной структуре Fe—О—Fe.
Однако, как было замечено Дунитцем и Оргелем [27], связь
Fe—О, прочная в линейных мостиковых структурах, при больших
отклонениях от линейности может значительно ослабиться. Помимо
этого высказывались предположения о том, что обменное взаимо-
действие через мостик Fe—О—Fe при угле 90° должно быть намно-
го ослаблено по сравнению с обменом при угле 180° [18, 34]. Если
высказанные предположения верны, то для объяснения экспери-
ментально полученных значений магнитной восприимчивости при-
ходится предположить такое строение комплекса, в котором имеет-
ся один приблизительно линейный оксо-мостик (структура III):
Fe—О—Fe
I
X
in
Хотя по своему виду структура III противоречит сделанному
ранее выводу об эквивалентности окружения атомов железа, мож-
398
Глава 11
но допустить, что присутствие лиганда X вблизи одного атома
железа в точности компенсируется на другом атоме железа за счет
координации какого-то остатка белка. Учитывая, что изомерный
сдвиг в спектре Мессбауэра высокоспиновых комплексов желе-
за (III) сравнительно мало изменяется в зависимости от лигандного
окружения, приведенное выше объяснение вполне допустимо. Ве-
личина квадрупольного расщепления линий в спектре Мессбауэра
может определяться главным образом кислородным мостиком и
не быть чувствительной к остальным лигандам, если они не отно-
сятся к сильно поляризуемым лигандам и не искажают геометрию
комплекса.
9.2. Оксигемэритрин
Спектр Мессбауэра оксигемэритрина совершенно отличен от
спектра деоксигемэритрина и состоит из двух дублетов. Вид спек-
тра свидетельствует об участии обоих атомов железа в связывании
кислорода, хотя по характеру связи с кислородом атомы железа
между собой различаются. Появление двух дублетов в спектре
Мессбауэра нельзя объяснить наличием равновесия между двумя
электронными состояниями или геометрическими изомерами, так
как в широком интервале температур соотношение интенсивностей
линий в спектре не изменяется, а совпадение энергий этих предпо-
лагаемых состояний маловероятно.
Магнитные свойства оксигемэритрина можно было бы объяс-
нить присутствием в нем изолированных низкоспиновых атомов
железа(II), однако близость изомерных сдвигов железа в окси-
гемэритрине и метгемэритрине свидетельствует о том, что в обеих
этих формах гемэритрина присутствуют атомы железа(Ш), свя-
занные сильным антиферромагнитным обменом. Таким образом,
связанная гемэритрином молекула кислорода по своему состоянию
близка к пероксо-иону.
Исходя из предполагаемой структуры I для комплексов мет-
гемэритрина и по аналогии с модельными оксигенированными
комплексами кобальта, Гарбетт и сотр. [10] предположили, что
активный центр оксигемэритрина имеет следующее строение:
IV
Асимметричная координация мостиковой пероксо-группы в этом
комплексе, которая может быть связана водородной связью с ка-
кой-то группой белка, объясняет различие между двумя атомами
Гемэритрин
399
железа в величине квадрупольного расщепления линий спектра
Мессбауэра.
Если исходить из предполагаемой структуры метгемэритрина II,
то молекулу кислорода в оксигемэритрине можно представить
в виде пероксо-мостикового лиганда:
V
Различие в величине квадрупольного расщепления у разных
атомов железа может быть обусловлено различием в прочности их
связи с пероксо-ионом*, которое в свою очередь может возникнуть
из-за образования водородной связи между пероксо-ионом и бел-
ком.
Наконец, активный центр оксигемэритрина может иметь строе-
ние, соответствующее предполагаемой структуре III метгемэритри-
на, причем пероксо-ион может координироваться только одним
атомом железа по типу координации VI, обнаруженному Гриффи-
сом, или по типу VII, обнаруженному Полингом:
Fe—О—Fe >. Fe—О—Fe
/ \ Л I
°---° °\0
VI VII
В структуре VI молекула кислорода связана за счет л-взаимодей-
ствия, что существенно отличает ее от других лигандов, например
хлорида или азида, связанных с метгемэритрином. Кроме того,
в этом случае координация кислорода должна сопровождаться вы-
теснением лигандов, входящих в состав белка. Оба эти эффекта
должны сказываться на величине квадрупольного расщепления на
атоме железа, связанного с кислородом. В случае структуры
VII различие в величине квадрупольного расщепления между
двумя атомами железа можно объяснить большей поляризуе-
мостью пероксо-иона. В обоих случаях (VI и VII) предполагается,
что окружение атома железа, не связанного с кислородом, остается
точно таким, каким было в метгемэритрине. Это как раз и следует
из экспериментальных данных: один из дублетов в спектре Месс-
* Такая неравноценность связей с кислородом была недавно обнаружена
при определении строения оксигенированного комплекса кобальта [34а].
400
Глава 11
бауэра оксигемэритрина по величине квадрупольного расщепления
и положению почти совпадает с дублетом в спектре метгемэрит-
рина.
9.3. Деоксигемэритрин
Данных о строении активного центра в деоксигемэритрине еще
меньше, чем в отношении других форм гемэритрина. Атомы желе-
за в деоксигемэритрине, очевидно, находятся в одинаковом окру-
жении. Он обладает магнитной восприимчивостью, которая типич-
на для высокоспиновых соединений железа (II), что, однако, не
исключает возможности существования мостиковых групп, посколь-
ку обменное взаимодействие между атомами железа (II) должно
быть слабым.
Во всяком случае, предполагаемое строение деоксигемэритрина
должно соответствовать тому свойству гемэритрина, что равнове-
сие связывания его с кислородом не зависит от pH среды, т. е. от-
сутствует эффект Бора [5, 27]. Более того, измерения pH в про-
цессе оксигенации и деоксигенации гемэритрина показали, что этот
процесс не связан с выделением или поглощением протонов [8].
Так что если в оксигемэритрине присутствует оксо-мостик, то и
деоксигемэритрин должен содержать оксо- или гидроксомостико-
вый лиганд, в противном случае образование оксо-мостика при
оксигенации должно сопровождаться образованием протонов, кото-
рые могут, не выделяясь в раствор, передаваться либо белку, либо
присоединенному молекулярному кислороду.
Из сказанного выше видно, что точное расположение атомов
в активном центре гемэритрина к настоящему моменту нельзя
считать твердо установленным.
9.4. Строение и функции гемэритрина
Одна из главных задач биохимии вообще и неорганической био-
химии в частности — раскрытие механизма действия биологиче-
ских молекул в связи с их строением и электронными свойствами.
При рассмотрении механизма действия гемэритрина как перенос-
чика кислорода мы должны выяснить причины устойчивости (и об-
ратимого образования) оксигемэритрина по отношению к диссоциа-
ции на деоксигемэритрин и молекулярный кислород, а также по
отношению к распаду оксигемэритрина на метгемэритрин и пере-
кись водорода [35]. Устойчивость в первом отношении обусловле-
на термодинамикой процесса и должна быть объяснена исходя из
прочности связей в оксигенированном комплексе. Устойчивость во
втором отношении, по-видимому, обусловлена кинетическими осо-
бенностями системы, поскольку необратимый процесс образования
метгемэритрина все же происходит, хотя и очень медленно. Этот
Гемэритрин
401
процесс, вероятно, связан с большой энергией активации
(рис. 11.8).
Для всех известных комплексов — переносчиков кислорода —
справедлива следующая закономерность: прочность связывания
кислорода увеличивается при увеличении донорных свойств лиган-
дов, координированных центральным ионом. Это наблюдается, на-
пример, в оксигенированном комплексе Васки (гл. 20, разд. 4.1),
Рис. 11.8. Изменение потенци-
альной энергии в процессе ок-
сигенации и окисления гемэри-
трина.
когда иодид замещает хлорид [36], или в аддуктах состава 1 : 1
молекулярного кислорода и комплексов Со (II) с основаниями
Шиффа, когда в качестве оснований используются производные
пиридина [37]. Аналогичные эффекты наблюдаются и при варьи-
ровании заместителей в порфириновом кольце производных гемо-
глобина [38, 39]. В молекуле же гемэритрина не содержится таких
хороших (и мягких) лигандов, как, например, порфириновая груп-
па, поэтому его способность связывать кислород остается несколь-
ко загадочной. Отсутствие таких лигандов в молекуле гемэритри-
на, возможно, и обусловливает необходимость присутствия в ней
двух атомов железа.
Прочность связи кислорода с гемэритрином, как было предло-
жено ранее [40], можно объяснить тем, что два атома железа пе-
редают два электрона на антисвязываюшие орбитали молекулы
кислорода. Очевидно, прочность связи возникающего таким обра-
зом пероксо-иона с атомом железа больше, чем у супероксо-иона,
который образуется при передаче на О2 только одного электрона.
Прочность связывания О2 может быть увеличена также за счет
двойной координации молекулы кислорода двумя атомами железа.
С другой стороны, гемэритрин устойчив по отношению к окисле-
нию, что, по-видимому, обеспечивается главным образом природой
белковой цепи, окружающей активный центр. Судя по тому, что
26—2451
402
Глава 11
в гемэритрине с трудом удается произвести обратимое удаление
железа и что оно не может быть извлечено из белка большими
хелатирующими лигандами [12], атомы железа находятся в глу-
бине белковой глобулы. Факторами, замедляющими разложение
оксигемэритрина, могут быть малая диэлектрическая проницае-
мость и отсутствие кислотных групп вблизи железо-кислородного
комплекса, что препятствует разделению зарядов или кислотному
катализу окисления гемэритрина [2, 41]. Другой причиной устой-
чивости оксигемэритрина может быть специфическое строение са-
мого комплекса железа с кислородом, которое диктуется белковым
окружением и при котором затруднена перегруппировка с выделе-
нием продуктов разложения. Это можно рассматривать как про-
явление эффекта «ловушки» [42].
ДОПОЛНЕНИЯ, ВНЕСЕННЫЕ В КОРРЕКТУРУ
Во время подготовки этого издания к публикации вышло из пе-
чати несколько работ, продолжающих главным образом исследо-
вание спектров Мессбауэра гемэритрина [43, 44] и его магнитной
восприимчивости при низких температурах [45, 46]. Общее мнение
сходится на том, что по крайней мере в окси- и метгемэритринах
атомы железа располагаются в непосредственной близости друг
к другу. Однако детали строения активного центра остаются неяс-
ными. Безусловно, еще необходимо приложить немало усилий,
прежде чем мы сможем окончательно выяснить функции гемэрит-
рина на уровне его молекулярной структуры.
СПИСОК ЛИТЕРАТУРЫ
1. Ghirett F., in О. Hayaishi (ed.), Oxygenases, Academic Press, New York, 1962,
pp. 517—553.
2. Klotz I. M., Klotz T. A., Fiess H. A., Arch. Biochem. Biophys., 68, 284 (1957).
3. Keresztes-Nagy S., Klotz I. M„ Biochemistry, 4, 919 (1965).
4. Keresztes-Nagy S., Klotz I. M., Biochemistry, 2, 923 (1963).
5. Bates G., Brunori M., Amiconi G., Antonini E., Wyman J., Biochemistry, 7,
3016 (1968).
6. Manwell С., N. Rev. Physiol., 22, 191—244 (1960).
7. Wald G„ Allen D. W„ J. Gen. Physiol., 40, 593—608 (1957).
8. Okamura M. Y., Klotz I. M., unpublished observations.
8a. Caughey W., in B. Chance, R. Estabrook, T. Yonetani (eds.), Hemes and He-
moproteins, Academic Press, New York, 1966, p. 276.
9. Darnall D. W„ Garbett K., Klotz I. M., Biochem. Biophys. Res. Commun., 32,
264 (1968).
10. Garbett K., Darnall D. W., Klotz I. M., Williams R. I. P„ Arch. Biochem. Bio-
phys., 103, 419 (1969).
11. Klotz I. M., Keresztes-Nagy S., Biochemistry, 2, 455 (1963).
12. Darnall D., Klotz I. M., unpublished observations.
13. Garbett K, Darnall D. W„ Klotz I. M., in preparation.
Гемэритрин 403
14. Klippenstein G. L„ Holleman J. W., Klotz I. M., Biochemistry, 7, 3868 (1968).
15. Pearson R. G., Survey of Progress in Chemistry, Vol. 5, Academic Press, New
York, 1969.
16. Fan С. C., York J. L., Biochem. Biophys. Res. Commun., 36, 365 (1969).
17. Rill R- L., Klotz I. M., Arch. Biochem. Biophys., 133, 103 (1969).
18. Ball P. W., Coord. Chem. Rev., 4, 361 (1969).
19. Spiro T. G., Saltman P., in P. Hemmerich et al. (eds.), Structure and Bonding,
Vol. 6, Springer-Verlag, New York, 1969, 116—156.
20. Kam.be K., J. Phys. Soc. (Japan), 5, 48 (1950).
21. Mulay L. N., Hofmann N. L., Inorg. Nucl. Chem. Lett., 2, 189 (1966).
22. Okamura M. Y.. Hoffman В. M., J. Chem. Phys., 51, 3128 (1969).
23. Johnson С. E., in A. Ehrenberg (ed.), Magnetic Resonance in Biological
Systems, Pergamon, New York, 1966, p. 405.
24. Johnson С. E„ Hall D. O., Nature, 217, 217 (1968).
25. Okamura M. Y„ Klotz I. M„ Johnson С. E., Winter M. R. C., Williams R. J. P„
Biochemistry, 8, 1951 (1969).
26. Reiff W. M„ Long G. J., Baker W. A., J. Am. Chem. Soc., 90, 6347 (1968).
26a. Lippard S. J., Schugar H„ Walling C., Inorg. Chem., 6, 1825 (1967).
27. Dunitz J. D., Orgel L. E„ J. Chem. Soc., 1953, 2593.
27a. Owen J., Thoruley J. H. M., Rep. Progr. Phys., 29 (2), 675 (1966).
28. Kubo M„ Bull. Chem. Soc. Japan, 26, 246 (1953).
29. York L„ personal communication.
30. Gray H. B„ personal communication.
31. Moss T., personal communication.
32. Garbett K-, Johnson С. E., Klotz J. M., Okamura M. Y., Williams R. J. P. (1969),
in preparation.
33. Marchant L„ Hoffman В. M., personal communication.
34. Gilleo M. A., Phys. Rev., 109, 777 (1958).
34a. Wang B., Schaefer W. P., Science, 166, 1404 (1969).
35. Basolo F., Pearson R. G., Mechanism of Inorganic Reactions, 2nd edn., John
Wiley and Sons, Inc., New York, 1968, pp. 641—-648.
36. McGinnety J. A., Doedens R. J., Jbers J. A., Science, 155, 709 (1967).
37. Grumbliss A. L., Basolo F., J. Am. Chem. Soc., 92, 55 (1970).
38. Rossi-Fanelli A., Antonini E., Arch. Biochem. Biophys., 80, 299, 308 (1959).
39. Rossi-Fanelli A., Antonini E., Cupato A., Arch. Biochem. Biophys., 85, 37
(1959).
40. Klotz J. M., Klotz T. A., Science, 121, 477 (1955).
41. Wang J. H., Accounts Chem. Res., 3, 90 (1970).
42. Vallee B. L., Williams R. J. P., Proc. Natn. Acad. Sci. U. S., 59, 498—505
(1968).
43. York J. L., Bearden A. J., Biochemistry, 9, 4549 (1970).
44. Garbett K-, Johnson С. E., Klotz I. M., Okamura M. Y., Williams R. J. P., Arch.
Biochem. Biophys., 142, 574 (1971).
45. Moss T. H., Moleski C., York J. L., Biochemistry, 10, 840 (1971).
46. Dawson J. W., Gray H. B„ Hoenig H. E., Rossman G. R., Schredder J. M.,
Wang R., Biochemistry, 11, 461 (1972).
ГЛАВА 12
ГЕМОЦИАНИН
Р. Лонтье, Р. Виттерс
Lontie R., Witters R., Laboratorium voor Biochemie,
Katholieke Universiteit te Leuven, В 3000, Louvain, Belgium
1, ВВЕДЕНИЕ
В 1847 г. Харлесс [1] сообщил о том, что в голубой крови
улитки Helix pomatia присутствует медь. В 1878 г. Фредерик [2]
описал бесцветный внеклеточный белок, выделенный им из крови
Octopus vulgaris, который при взаимодействии с кислородом да-
вал неустойчивое соединение, окрашенное в темно-синий цвет. Им
и был впервые введен термин «гемоцианин», который составлен
из корней греческих слов aip,a (кровь) и zoavog (синий). Это,
конечно, не означает, что в белке содержится гемовая группа.
К настоящему моменту опубликовано несколько обзоров, по-
священных гемоцианину, в которых рассматриваются его распро-
страненность в живых организмах, состав, молекулярная масса,
его диссоциация, связывание с кислородом и имунные свойства
[3-14].
2. РАСПРОСТРАНЕНИЕ В ЖИВОТНОМ МИРЕ
Гемоцианины обнаружены в артроподах (членистоногих) и
моллюсках и характеризуются различным содержанием меди в за-
висимости от биологического типа (табл. 12.1).
Типичными представителями членистоногих, в крови которых
содержатся гемоцианины, являются краб-наездник из класса ме-
ростомовых, лангуста, омар и некоторые крабы из класса рако-
образных (отряд декаподов). Гемоцианины найдены в таких мол-
люсках, как блюдечко, волокнистый рожок, пульмонатная улитка
из класса гастроподов, из класса головоногих — кальмар, карака-
тица (отряд декаподов) и осьминог (отрядоктоподов) (табл. 12.1).
Гемоцианины обнаружены также в моллюсках хитонах из класса
амфинейров [33—35]. Кроме того, они найдены в скорпионах [33,
36] (класс паукообразных, см. в табл. 12.1 Androctonus australis),
пауках [17, 37, 38], в некоторых ракообразных из отрядов изопо-
дов [39—41] и амфиподов [39, 41] и, наконец, в сороконожках
(класс хилоподов) [42].
Г емоцианин
405'
Таблица 12.1
Коэффициенты седиментации (S20) основного компонента, содержание меди
и минимальная молекулярная масса в расчете на один атом меди типичных
гемоцианинов
Организм S20 Литература Cu, % Литература М/Си
Членистоногие Мер остом аты
Limulus polyphemus Паукообразные 56,6 21 0,170 16 37 400
Androctonus australis Ракообразные 33,0 17 0,180 18 35 300
Jasus lalandii 17,1 19 0,176 20 36 100
Palinurus vulgaris 16,4 21 0,170 16 37400
Homarus americanus 24,5 22 6,175 23 36 300
H. vulgaris 22,6 21 0,169 16 37 600
Eriphia spinifrons 24,0 24 0,167 16 38000
Cancer magister 25,0 25 0,166 25 38300
Carcinus maenas 23,3 21 0,173 7 36 700
Maja squinado Моллюски Г астроподы 23,4 26 0,182 7 Среднее: 34 900 36 700
Busycon canaliculatum 102,0 15 0,245 27 25 900
Helix pomatia 104,3 28 0,250 7 25 400
Murex truculus Г оловоногие 102,7 29 0,257 16 24 700
Sepia officinalis 55,9 21 0,256 7 24 800
Loligo pealei 59,0 30 0,260 27,31 24 400
Octopus vulgaris 49,3 21 0,250 16,32 25400
Eladone moschata 49,1 21 0,252 16 Среднее: 25 200 25 ЮО
3. МОЛЕКУЛЯРНАЯ МАССА И ЧЕТВЕРТИЧНАЯ
СТРУКТУРА
Содержание меди в гемоцианине антроподов колеблется от
0,166 до 0,180%, в гемоцианине моллюсков — от 0,245 до 0,260%.
Минимальная молекулярная масса соответственно равна 36 700 и
и 25 100 (см. табл. 12.1, а также [16]).
Поведение гемоцианинов артроподов при ультрацентрифугиро-
вании соответствует нескольким коэффициентам седиментации:.
165, 245, 335 и 575 (см. табл. 12.1 и работы [21, 43]). В гемо-
406
Глава 12
цианине из Limulus polyphemus, например, в области устойчивости
обнаруживаются все четыре указанных компонента [21]. Эти ком-
поненты после негативного окрашивания можно изучать с по-
мощью электронного микроскопа [44—46]. Их рассматривают как
соответственно мономеры, димеры, тетрамеры и октамеры [17,47].
Например, мономеры (коэффициент седиментации 16S, разме-
ры 10X10 нм) наблюдались у Palinurus vulgaris [45] и изопода
Рис. I2.I. Предполагаемая модель мономеров с коэффициентом седиментации
16S гемоцианинов членистоногих.
а — искаженное октаэдрическое расположение шести функциональных субъединиц; б —12
субъединиц, расположенные попарно (вид сверху): в —то же. вид сбоку.
Oniscus asellus [46], димеры (24S, 22X9 нм) —у Cancer pagurus
[45] и Homarus vulgaris [44, 46, 48], тетрамеры (333, 22Х
Х20 нм) —у паукообразных Nemesia cementaria [17, 46, 47] и
Avicularia metallica [45], октамеры (57S, 21X24 нм) —у Limulus
polyphemus [45, 47].
Гемоцианин из Homarus vulgaris, рассматриваемый как димер,
имеет молекулярную массу 825 000 [22]. Каждый мономер состо-
ит, .вероятно, из 12 субъединиц с молекулярной массой 34 400,
которая соответствует нефункционирующим субъединицам, вы-
деленным после сукцинилирования [23].
Предполагаемая модель мономера показана на рис. 12.1. С по-
мощью октаэдрического расположения шести сфер можно объяс-
нить все типы контуров мономеров, наблюдаемые через электрон-
ный микроскоп: правильный шестиугольник, квадрат, прямоуголь-
ник и ромб. Если молекулярная масса действующих субъединиц
достигает 68000—70 000 [23], то, согласно октаэдрической модели
Левина [44], для объяснения наблюдаемых контуров мономера
необходимо расположить 12 более мелких субъединиц попарно
в вершинах октаэдра. Здесь возникает вопрос, обладает ли пеп-
тид одного типа, образующий одну субъединицу, стерическими
возможностями для создания предполагаемой четвертичной струк-
туры.
Гемоцианин
407
Гемоцианины моллюсков также изучались методами ультра-
центрифугирования [21, 33, 43, 49] и электронной микроскопии
[44, 45, 50]. Формы частиц этих белков, наблюдаемые с помощыо
электронного микроскопа после негативного окрашивания, пока-
заны .на рис. 12.2. Видимые в микроскоп прямоугольники и кру-
жочки можно интерпретировать как различные ракурсы частиц
Рис. 12.2. Форма и размеры частиц гемоцианинов моллюсков, наблюдаемые через
электронный микроскоп после негативного окрашивания, и соответствующие им
коэффициенты седиментации.
цилиндрической формы, обладающих осью симметрии пятого по-
рядка.
В гемоцианинах моллюсков ультрацентрифугирование выявля-
ет два типа частиц: с коэффициентом седиментации 59S (напри-
мер, у организмов Loligo pealei) и с коэффициентом седиментации
105S (например, Helix pomatia) (рис. 12.2, а и б и табл. 12.1).
Частицы гемоцианина из Cymbium neptuni [46] и Buccinum unda-
tum [51], по данным электронной микроскопии, также имеют ци-
линдрическую форму. Частицы гемоцианина из Busycon canalicu-
latum проявляют, кроме того, тенденцию к линейной полимериза-
ции [45] (рис. 12.2,в). Модели этих частиц, предложенные на
основании данных гидродинамических измерений [52], подтверди-
лись данными, полученными при их ультрацентрифугировании:
коэффициенты седиментации частиц варьируются от 59S до 178S.
Гемоцианин из Kelletia kelletia в условиях изоэлектрической точ-
ки существует в виде длинных линейных полимеров [53]
(рис. 12.2, г).
Некоторые гемоцианины с коэффициентом седиментации 105S,
как, например, a-компонента гемоцианина из Helix pomatia, в пре-
делах области стабильности в присутствии 1 моль/л хлоридов ще-
лочных металлов делятся надвое [28, 54, 55] (p-компонента в этих
408
Глава 12
же условиях не диссоциирует). При более высоких значениях pH
в области стабильности эти половины могут распадаться еще на
более мелкие, равные 1/10 и 1/20 долям исходной субъединицы
[28, 56]. При помощи измерения светорассеяния [57] было пока-
зано, что при добавлении небольших количеств иона Са2+, сопо-
ставимых с содержанием этого иона в крови, область стабильно-
сти очищенного гемоцианина из Helix pomatia может быть рас-
ширена за счет смещения ее (верхней границы pH от 8 до 10 [58].
Одна двадцатая доля субъединицы гемоцианина из Helix po-
matia, по-видимому, является его наименьшей функциональной
субъединицей, которую можно получить при подщелачивании [56]
или сукцинилировании [59]. Наименьшие субъединицы, наблюдав-
шиеся при pH 12,5 после сукцинилирования, восстановления и ал-
килирования имеют молекулярную массу 22 300 [60].
4. СПЕКТРЫ ПОГЛОЩЕНИЯ
Голубой цвет крови некоторых беспозвоночных, .который вызы-
вал удивление еще у первых исследователей [1, 2], нельзя путать
с цветом действительно синих медьсодержащих белков, к каким
относится, например, церулоплазмин (ХмаКс 610 нм). Например,
синяя окраска гемоцианина из Helix pomatia в области стабиль-
ности, где молекулярная масса его частиц равна 9-106, представ-
ляет собой суммарный эффект поглощения и интенсивного рассея-
ния света. В проходящем же свете концентрированный раствор ге-
моцианина выглядит пурпурным.
В спектре поглощения оксигемоцианинов, кроме обычной для
белков полосы при 280 нм, наблюдаются полосы при 346 и 580 нм
[4], обусловленные присутствием меди (табл. 12.2). Моляр-
Таблица 12.2
Молярные коэффициенты поглощения (л-моль-1-см-1) полос, обусловленных
присутствием меди гемоцианинов моллюсков, в расчете на 1 атом меди
Организм SnaKc Емакс Литература
Helix pomatia 346 8800 28
580 540
.Loligo pealei 345 8900 30
580 370
"Omma(to)sprephes sloani pacificus 347 6100 61
580 390
Octopus vulgaris 347 8900 62
580 500
Гемоцианин
409-
ный коэффициент поглощения деоксигенированного гемоцианина
находится в обратной зависимости от четвертой степени длины
волны (в видимой области [5], а также в видимой и ближней
ультрафиолетовой областях спектра [63]). Вклад рассеяния в
кажущийся коэффициент поглощения особенно велик для гемо-
цианинов с наибольшей молекулярной массой из гастроподов [55].
Значения молярных коэффициентов поглощения, приведенные в
табл. 12.2 для гемоцианинов из Helix pomatia и Octopus vulgaris,
получены с учетом поправки на рассеяние, а для гемоцианинов из
Loligo pealei и Omma(to)strephes sloani pacificus — с учетом по-
правки на рассеяние и на остаточное поглощение путем вычита-
ния спектров апогемоцианина и деоксигенированного гемоциани-
на соответственно.
При понижении температуры до —195° С в спектрах поглоще-
ния гемоцианинов происходят обратимые изменения. В спектре
гемоцианина из Cancer magister появляются две новые полосы с
максимумами поглощения при 420 и 652 нм, а полоса, находив-
шаяся в обычных условиях при 585 нм, смещается в область-
566 нм [64]. В спектре гемоцианина из Octopus vulgaris при та-
кой же пониженной температуре появляется плечо при 440 нм и
полоса при 700 нм, а бывшая при 580 нм полоса смещается до-
570 нм [62]. В спектре гемоцианина из Omma(to)strephes sloani
pacificus при комнатной температуре, кроме указанных в
табл. 12.2 полос, имеется также плечо при 800 нм [61].
Две основные полосы меди, наблюдаемые при комнатной тем-
пературе, проявляют оптическую активность. При измерении дис-
персии оптического вращения (ДОВ) ib области этих полос был
обнаружен общий отрицательный эффект Коттона [30, 65]. Из-
мерения кругового дихроизма (КД) выявили две полосы с отри-
цательным эффектом Коттона, а две — с положительным
(табл. 12.3). Ван Хоулд [62] обратил внимание на удивительное
сходство спектров КД в области 490 и 590 нм гемоцианинов мол-
люсков и комплекса меди(II) с пептидом ацетил-Gly-Gly-His
[67].
В отличие от синих медьсодержащих белков в спектрах погло-
щения гемоцианинов полосы меди проявляются только в присут-
ствии кислорода. Предполагается, что активный центр оксигени-
рованного гемоцианина можно представить в виде следующих ре-
зонансных форм [12,68]:
Cu+O2Cu+ ч—► Си2+ОГСи+ ч—> Си+ОГСи2+ ч—> Cu2+O^-Cu2+
из которых последняя наименее вероятна.
Основные полосы меди в спектрах поглощения гемоцианинов,,
находящиеся при 346 и 580 нм, были отнесены к переходам с пе-
реносом заряда [69], причем подчеркивалось [71] сходство поло-
сы при 346 нм (lge=3,9) с так называемой «пероксо-специфиче-
410
Глава 12
Таблица 12.3
Полосы в спектре КД гемоцианинов моллюсков
Организм Хмакс- “• Литература
— + — +
Loligo pealei 347 440 570 700 62
Octopus vulgaris 347 440 570 Heonp. 62
350 450 570 >700 66
Helix pomatia 346 485 578 710 a
8 Не публиковавшиеся ранее данные.
ской» полосой комплекса декаммино-р,-пероксокобальт(Ш)—ко-
бальт(1У) (Амане=341 нм, lge=3,5) [70]. В этом комплексе,
а также в оксигенированном комплексе кобальта (II) с бис-(3-
фторсалицилальдегид) этилендиимином расстояние О—О равно
1,31 А, что близко к расстоянию О—О в супероксо-ионе Ог
(1,28 А) [72].
Появление двух дополнительных полос в спектрах поглощения
гемоцианинов при —195 °C или в их спектрах КД свидетельствует
о сходстве спектральных свойств меди в этих белках со свойства-
ми меди(II) в синих белках типа церулоплазмина или лакказы
[62, 73]. Указанные полосы в спектрах гемоцианинов мало отли-
чаются по положению от соответствующих полос в спектрах си-
них белков, но значительно отличаются по своей интенсивности.
По-видимому, их можно было бы отнести к d—d-переходам ме-
ди (II) [62]. Однако свежеприготовленные образцы оксогемоци-
анина при комнатной температуре не дают никаких сигналов
ЭПР в области g-фактора, близкого к 2,0. Это было проверено
на образцах из Cancer magister [74], Limulus polyphemus [75],
Jasus lalandii [76], Octopus vulgaris [77], Busycon canaliculatum
[75] и Helix pomatia [28]. Гемоцианин из Jasus lalandii при 77 К
дает сигнал ЭПР, интенсивность которого соответствует присутст-
вию 10% парамагнитных частиц от общего содержания меди [76].
Сигнал ЭПР от Си (И), наблюдаемый у свежеприготовленного
гемоцианина из Homarus americanus, соответствует ’Д общего ко-
личества меди [78].
5. СВЯЗЫВАНИЕ КИСЛОРОДА
Еще первые исследователи гемоцианина отметили связь меж-
ду его синей окраской и поглощением кислорода [1, 2]. Его зна-
чение в процессе дыхания было показано на примере Sepia offi-
Гемоцианин
411
cinalis [79] и Octopus vulgaris [80]. Хотя по поводу этой функции
гемоцианина иногда высказывались сомнения, она 'была оконча-
тельно установлена в работе Редмонда [81].
В условиях, которые существуют в крови, для большинства
гемоцианинов характерен кооперативный (положительный) эф-
фект связывания кислорода [5, 75, 82], причем величина п в урав-
нении Хилла достигает значений 3,5—4,0.
Если считать, что одна молекула кислорода связывается в ге-
моцианине двумя атомами .меди [83—85], то в мономере гемо-
цианина из артроподов содержится 6 связывающих кислород
центров (рис. 12.1), а в гемоцианинах моллюсков — 90—100
(рис. 12.2, а) или 180—200 (рис. 12.2,6) таких центров. Таким об-
разом, гемоцианины должны описываться общей теорией алло-
стерических переходов помимо представлений о так называемом
гем-гемовом взаимодействии [86].
Кооперативный .механизм поглощения кислорода активными
центрами гемоцианина из Helix pomatia теряется при диализе
[87]. При этом кривая оксигенации имеет вид гиперболы, что ука-
зывает на изолированность активных центров. Такой же характер
имеет кривая оксигенации гемоцианина в области pH от 4,0 до 9,0.
Если же в растворе свежеприготовленного гемоцианина создать
концентрацию ионов Са2+ порядка 6-10-3 г-ион/л (какая бывает
в крови), то при pH 7,8—8,2 можно вновь получить S-образную
кривую оксигенации, характерную для кооперативного механизма
поглощения кислорода [88]. Гемоцианин из Homarus americanus
теряет свойство кооперативного связывания кислорода при удале-
нии ионов Са2+ в щелочной среде при низкой ионной силе раство-
ра, в результате чего происходит диссоциация белка на субъеди-
ницы, содержащие по одному активному центру [23].
S-образный характер кривых равновесия юксигенирования ге-
моцианинов связан, очевидно, с конформационными изменениями
функциональных субъединиц. Известен интересный пример влия-
ния оксигенации на четвертичную структуру гемоцианина из Loli-
go pealei [89]. Этот деоксигенированный гемоцианин в среде, 'со-
держащий Mg2+ в количестве 1-10-3 г-ион/л, при pH от 6 до 10
обладает коэффициентом седиментации 59S, но при частичной ок-
сигенации при pH 7 он диссоциирует на частицы размером в Vs
или Vio от размеров исходной субъединицы. Наблюдались также
небольшие отличия в спектрах КД (в области 250—280 нм) окси-
и деоксигемоцианина.
В опытах с гемоцианином из Levantina hierosolima было заме-
чено, что при деоксигенации сильно увеличивается флуоресцен-
ция гемоцианина [90]. Это явление может послужить новым сред-
ством изучения процесса связывания кислорода гемоцианином.
412
Глава 12
6. СВЯЗЫВАНИЕ ДРУГИХ ЛИГАНДОВ
Гемоцианины обратимо взаимодействуют с окисью углерода,
образуя бесцветные соединения (не поглощающие цвета ни в ви-
димой, ни в ближней ультрафиолетовой областях). Гемоцианины,
по крайней мере моллюсков, образуют такие же соединения с CN-,
этилизоцианатом SCN- и тиомочевиной (при кратковременной об-
работке) .
Гемоцианин из Limulus взаимодействует с СО в том же соот-
ношении, что и с Ог, однако относительно высокое сродство к оки-
си углерода проявляют только частицы размером в V20 исходной
субъединицы [91]. Гемоцианин из Octopus связывает СО в соот-
ношении 1 СО : 2Си, причем это соединение разрушается CN-
[32]. В случае Palinurus vulgaris никакого видимого взаимодейст-
вия гемоцианина с СО не было обнаружено [85]. Связывание
гемоцианина из Octopus vulgaris в комплекс с окисью углерода
было зафиксировано по ингибированию его псевдокаталазной и
псевдопероксидазной активности, а также непосредственным опре-
делением СО, выделяющейся при обработке этого комплекса ци-
анид-ионом [92]. Соединение СО с гемоцианином из Cancer ma-
gister было изучено изотопным методом с помощью 14СО [93].
Давление полунасыщения гемоцианина меченой окисью углерода,
измеренное в этой работе, оказалось равным 134 мм рт. ст. Спек-
трофотометрическое изучение конкурентного взаимодействия ге-
моцианина из Helix pomatia с окисью углерода и кислородом при
pH 9,2 дало величину константы равновесия, равную 1,00 [94]. Все
исследователи указывают на то, что гемоцианин связывает окись
углерода <в стехиометрическом отношении 1СО:2Си, однако нет
прямых доказательств того, что она образует связь с обоими ато-
мами меди.
Исследование реакции гемоцианина из Limulus с цианид-ионом
при малых концентрациях последнего показало, что эта реакция,
по-видимому, частично обратима [95]. В работе [96] наблюда-
лось восстановление синей окраски при действии кислорода на
гемоцианин из Octopus vulgaris, обработанный цианидом. Эти дан-
ные трудно интерпретировать. Эксперименты, проводимые в лабо-
ратории авторов, показали, что гемоцианины из Helix pomatia об-
ратимо взаимодействуют с CN- при изученных концентрациях по-
следнего вплоть до 0,2 ммоль/л.
Гемоцианин из волнистого рожка Murex trunculus также обра-
тимо взаимодействует с этилизоцианатом [97].
При изучении реакции SCN- с оксигенированным гемоциани-
ном из Helix pomatia [98] было показано, что при постоянном pH
равновесие замещения кислорода на SCN- описывается следую-
щим уравнением:
k'alk°-x =X(lSCN-]/Po2)Va,
Гемоцианин
413
где ka — оптическая плотность три 346 нм (где поглощает оксиге-
нированный медьсодержащий активный центр), концентрации бел-
ка 1 г/л и толщине поглощающего слоя 1 см, к — максимальное
значение ka при полной оксигенации всех активных центров. По-
лученные данные указывают на то, что один ион SCN- вытесняет
одну молекулу О2, но при этом происходит обесцвечивание двух
активных центров, связывающих кислород [99].
Быстрая реакция вытеснения тиомочевиной кислорода из ге-
моцианина протекает обратимо. Однако ей сопутствует медленная
вторичная реакция, которая сопровождается поглощением кисло-
рода и исчезновением оставшихся в спектре полос меди. Эта реак-
ция напоминает процесс старения белка [100].
Окись азота с гемоцианинам из Helix pomatia образует зеленое
соединение (с гемоцианином из Homarus никакого видимого
взаимодействия не наблюдалось) [101]. Аналогичное соединение
упоминалось Мэзоном (см. [93], обсуждение в гл. 15, разд. 3.4).
Поскольку это соединение дает отчетливый спектр ЭПР, оно за-
служивает дальнейшего изучения.
7. СТАРЕНИЕ И РЕГЕНЕРАЦИЯ
Исследование спектров поглощения гемоцианинов из Helix po-
matia [55, 63], Eriphia spinifrons и Octopus vulgaris [102] пока-
зало, что в области полос, обусловленных присутствием меди, они
меняются от образца к образцу. При хранении твердого гемо-
цианина, полученного путем осаждения сульфатом аммония, ин-
тенсивность этих полос монотонно убывает со временем. Особен-
но это характерно для гемоцианина из Helix pomatia [103].
В противоположность свежим растворам постаревшие препа-
раты дают сигнал ЭПР, характерный для Си(II) (Jasus lalandii
[76], Octopus vulgaris [77], Helix pomatia [28]). Правда, старые
препараты гемоцианина из Limulus и Busycon не дают таких сиг-
налов [78].
Интенсивность полос поглощения меди старого препарата ге-
моцианина из Helix pomatia может быть восстановлена при до-
бавлении цистеина [104], перекиси водорода и гидроксиламин?
[104, 105].
В случае гемоцианина из другого моллюска Busycon canalicu-
latum обработка деоксигенированного образца небольшим количе-
ством Н2О2 приводит к частичному ослаблению полос меди и со-
ответствующему переходу Сп(1) в Си (II). При добавлении
большего количества Н2О2 интенсивность указанных полос увели-
чивается до прежнего уровня. С другой стороны, деоксигениро-
ванный гемоцианин из артропода Limulus polyphemus необратимо
обесцвечивается при добавлении стехиометрического количества
414
Глава 12
Н2О2, и никакими восстановителями регенерировать его окраску
не удается [106]. Молекулярный кислород предохраняет оба на-
званных гемоцианина от воздействия Н2О2.
Метгемоцианин, полученный действием перекиси водорода на
гемоцианин из Cancer magister, проявляется в спектре ЭПР [74].
При облучении аналогичную инактивацию претерпевает гемо-
цианин из Limulus, а под действием перекисных соединений на-
блюдается инактивация и реактивация гемоцианина из Busycoty-
pus [107]. Помимо указанных свойств гемоцианины проявляют
каталазную активность, которая подавляется молекулярным кис-
лородом. Эта активность у гемоцианинов из моллюсков проявля-
ется намного сильнее, чем у гемоцианинов из ракообразных. На-
пример, если принять каталазную активность гемоцианина из
Octopus vulgaris за 100, то относительная активность гемоциани-
нов из других организмов будет меняться в следующем порядке:
Helix legata — 41, Maja squinado — 3, Palinurus vulgaris — 3
[108].
Самым простым объяснением процесса старения гемоцианинов
моллюсков является тепловое равновесие [109]:
Cu+O2Cu+ + 2H+ 4—>• Cu2+Cu2+ + Н2О2
Однако поскольку метгемоцианин дает сигнал ЭПР, ионы ме-
ди (II) в нем, очевидно, не вступают в обменное взаимодействие и,
следовательно, находятся на значительном расстоянии друг от
друга. В аналогичном процессе образования метмиоглобина из
оксигенированного миоглобина также предполагается образование
супероксо-иона 07 [НО].
Каталазную активность гемоцианинов моллюсков также мож-
но объяснить способностью двух атомов меди одновременно изме-
нять свою степень окисления:
Cu+O|-Cu+ 4- 4Н+-> Cu2+Cu2+ + 2НаО
Эта реакция ингибируется молекулярным кислородом
Cu2+Oi~Cu2+ ---------------------> Cu+O2Cu+
Второе уравнение отражает также способность перекиси водорода
регенерировать старый гемоцианин, которая, по-видимому, и в
этом случае взаимодействует одновременно с двумя соседними
атомами меди (образование промежуточного состояния Cu+Cu2+>
вероятно, затруднено). Мы также предполагаем, что и регенера-
ция цистеином протекает на самом деле за счет перекиси водоро-
да, которая образуется в небольших количествах при медленном
окислении цистеина.
Предложенный механизм объясняет также и то явление, что
процесс старения не идет бесконечно, а останавливается на ка-
ком-то определенном уровне: образующаяся в этом процессе пе-
Гемоцианин
415
рекись водорода может захватываться «состарившимися» актив-
ными центрами и регенерировать их.
Гемоцианины не сохраняются при лиофилизации, однако име-
ется довольно эффективное средство, предохраняющее их от раз-
рушения, а именно добавка невосстанавливающих сахаров, на-
пример сахарозы [105, 112, 113]. Хранение под атмосферой СО
также предохраняет гемоцианины от старения: кривая оксигена-
ции остается S-образной [94]. При старении именно частичное
окисление меди в активных центрах до степени окисления +2 пре-
пятствует такому изменению конформации, которое обеспечивает
кооперативное действие активных центров.
В качестве критерия для оценки качества препаратов гемо-
цианина следует рекомендовать величину е при 346 нм либо по
крайней мере отношение оптических плотностей О346 нм/О280 вм-
8,АПОГЕМОЦИАНИН И РЕСТАВРАЦИЯ ГЕМОЦИАНИНА
Медь можно удалить из гемоцианина (Octopus vulgaris) при
подкислении соляной кислотой [114]. Однако при этих условиях
нарушается структура самого белка. В тех же условиях, когда не
происходит разрушения белка, медь может быть извлечена из ге-
моцианина только с помощью цианида, что было показано Кубо-
вичем [32]. С помощью CN~ количественное извлечение меди, по-
видимому, легче осуществляется в случае гемоцианинов из антро-
подов, чем из моллюсков (например, в одних и тех же условиях
извлекаются соответственно 100 и 85%; см. также [32, 105]).
Частичная реставрация гемоцианина из Octopus vulgaris была
достигнута путем обработки апофермента в анаэробных условиях
медью (I) в виде хлоридного комплекса (реставрация на 50%
[32] и на 60% [113]), в виде аммиака (65% [115]) и Си2О (80%
[115]). Обработка ацетонитрильным комплексом меди(1) [116]
приводит к количественной реставрации гемоцианина [105, 117],
на что указывает восстановление его полос поглощения, обуслов-
ленных медью, и его способности обратимо связывать кислород.
Интенсивность полос меди при последовательном увеличении ее
содержания в частично реставрированном холоферменте возрас-
тает в квадратичной зависимости от количества связанной меди.
Этот факт указывает на то, что активные центры гемоцианина,
связывающие кислород, содержат по два эквивалентных и неза-
висимых атома меди [105].
Фиксация меди представляет собой также одну из интересных
проблем биосинтеза гемоцианина. Известно, что гепатопанкреас,
особенно у головоногих, богат медью [118]. Кроме того, функцио-
нирующий гемоцианин удалось получить из апогемоцианина и
плавающего на поверхности раствора гомогената гепатопанкреаса
омара шипастого Panulirus interruptus [119].
416
Глава 12
9. ЦЕНТРЫ СВЯЗЫВАНИЯ МЕДИ
Одинаковы ли все медьсодержащие группы в гемоцианине, оди-
наковы ли центры связывания меди, каково число и природа
окружающих медь лигандов и какова геометрия этого окружения?
Эти вопросы остаются пока нерешенными (обсуждение их см.
в работах [109, 120]). Различия в аминокислотной последователь-
ности различных полипептидных цепей определяют неоднород-
ность гемоцианина, которая проявляется при ультрацентрифуги-
ровании [43, 54, 121] и при зонном электрофорезе [122—124]. Мо-
гут ли эти различия быть настолько велики, что благодаря им
меняется даже место связывания меди? Для объяснения четвер-
тичной структуры гемоцианина более или менее часто приходится
предполагать существование в одном гемоцианине различных
субъединиц. Количество пептидов, обнаруженных в гемоцианине
из Helix pomatia, например, указывает на то, что в нем присутст-
вуют по крайней мере два типа наименьших субъединиц с моле-
кулярной массой 22 300 [125]. Константы устойчивости при свя-
зывании меди в гемоцианине из Limulus polyphemus, которые име-
ют порядок 101Э, соответствуют устойчивости комплекса меди(1)
с цистеином.
Поскольку медь не может быть удалена из гемоцианина ника-
кими классическими реагентами, кроме CN~, можно предполагать,
что прочность связывания меди в нативном белке возрастает еще
благодаря соответствующей конформации последнего [127]. Медь,
входящая в гемоцианин, может вступать в комплексообразование
с дитиокарбаматом только после денатурации гемоцианина моче-
виной [128], она может высвобождаться также и при частичном
гидролизе белка [129, 130].
В качестве возможных лигандов, координируемых атомом ме-
ди в гемоцианине, рассматривались различные группы, имеющие-
ся в белке: тиоловые группы [131], имидазольные остатки гисти-
дина [132], аминогруппы [133] и дисульфидные мостики [134].
Между прочим, известно, что комплекс меди с аргенином прояв-
ляет отчетливую каталазную активность [135].
При удалении меди из гемоцианинов, выделенных из Cancer
magister [25] и Octopus vulgaris [16, 136], с помощью CN- уста-
новлено, что диссоциация четырех атомов меди сопровождается
высвобождением только одной группы — SH. Таким образом, уча-
стие этих групп в связывании меди маловероятно. Показано так-
же, что блокировка тиоловых групп в апогемоцианине из Мигех
trunculus не препятствует реставрации активного гемоцианина
[137]. Однако в случае Jasus lalandii после обработки апогемо-
цианина ионами серебра(I) реставрация частично ингибируется
Гемоцианин
417
Из потенциальных лигандов, связывающих медь в гемоциани-
не, наиболее вероятным остается остаток имидазола. Здесь можно
снова отметить уже упоминавшееся сходство спектров КД оксиге-
моцианина и комплекса меди(II) с Ac-Gly-Gly-His [62, 67]. Этому
предположению не противоречат данные ЭПР, а также результа-
ты изучения процесса фотоокисления, поскольку исчезновение по-
лос, обусловленных атомом меди, происходит с такой же скоро-
стью, с какой исчезают полосы гистидина [138, 139].
10. ВЫВОДЫ
Гемоцианины делятся на два типа в соответствии с двумя ти-
пами живых организмов, в которых они обнаружены: членистоно-
гих и моллюсков. В гемоцианинах первого типа содержание меди
равно 0,173%, в гемоцианине второго типа — 0,253%. Молекуляр-
ная масса наименьших субъединиц этих белков равна соответст-
венно 36 700 и 25 100.
В состав деоксигенированного гемоцианина входит медь(1), на
что указывает легкость реставрации холофермента при помощи
ацетонитрильного комплекса Cu(I), а в случае гемоцианинов мол-
люсков — регенерацией старых препаратов при помощи Н2О2.
Вместо известной ранее полосы поглощения при 580 нм в ви-
димой и ближней ИК-области в спектре КД гемопротеида при
—195 °C проявляются три полосы.
Гемоцианины обратимо взаимодействуют с окисью углерода,
образуя бесцветное соединение, которое имеет большое значение
для предотвращения разрушения препаратов. Окись азота образу-
ет с гемоцианином зеленое соединение. Гемоцианины моллюсков
обратимо взаимодействуют с цианидом, этилизоцианатом, SCN-
и тиомочевиной, образуя бесцветные соединения.
Физиологические функции гемоцианина связаны с транспор-
том кислорода. В условиях, которые создаются в крови соответст-
вующих организмов, гемоцианины при связывании кислорода про-
являют кооперативный эффект.
Конформация белка стабилизирует центры связывания меди.
Тиоловые группы вряд ли могут быть единственными лигандами,
связывающими медь. Наиболее вероятным лигандом в этом отно-
шении является имидазольный остаток гистидина.
ДОПОЛНЕНИЯ, ВНЕСЕННЫЕ В КОРРЕКТУРУ
Сигнал ЭПР, наблюдаемый в старых препаратах гемоцианина,
соответствует такому количеству парамагнитных ионов меди(П),
которое составляет всего несколько процентов от общего количе-
27—2451
418
Глава 12
ства «постаревших» медьсодержащих центров, что свидетельству-
ет о спаривании спинов в большинстве пар ионов Си2+. Появление
сигнала ЭПР у гемоцианина, обработанного окисью азота, обус-
ловлено окислением меди(1) до меди (II); соединение NO с гемо-
цианином, содержащим Си(II), не дает заметного сигнала ЭПР.
СПИСОК ЛИТЕРАТУРЫ
1. Harless Е., Arch. Anat. Physiol., 1847, 148.
2. Fredericq L., Compt. Rend. Acad. Sci. Paris, 87, 996 (1878).
3. Quagliariello G., in H. Winterstein (ed.), Handbuch der vergleichenden Phy-
siologie, Vol. 1/1, G. Fischer, Jena, 1925, p. 597.
4. Dhere C., Rev. Suisse Zool., 35, 277 (1928).
5. Redfield A. C.. Biol. Rev., 9, 175 (1934).
6. Haurowitz F., in C. Oppenheimer, L. Pincussen (eds.), Tabulae Biologicae Pe-
riodicae, Vol. IV, W. Junk, Berlin—Den Haag, 1935, p. 18.
7. Roche J., Essai sur la biochimie generale et comparee des pigments respira-
toires, Masson, Paris, 1936.
8. Dawson C. R., Mallette M. F„ Adv. Protein Chem., 2, 179 (1945).
9. Redfield A. C., in W. D. McElroy, B. Glass (eds.), Symposium on Copper Me-
tabolism, Johns Hopkins Press, Baltimore, Maryland, 1950, p. 174.
10. Haurowitz F„ Hardin R. L., in H. Neurath, K. Bailey (eds.), The Proteins,
Vol. IIA, Academic Press, New York, 1954, p. 336.
11. Biellg H. J., Bayer E., in K. Lang, E. Lehnartz (eds.), Hoppe-Seyler/Thierfel-
der, Handbuch der physiologisch- und pathologisch chemischen Analyse,
Vol. IV/1, Springer, Berlin — Gottingen — Heidelberg, 10, 1960, p. 748.
12. Manwell C., A. Rev. Physiol. 22, 191 (1960).
13. Ghiretti F., in O. Hayaishi (ed.), Oxygenases, Academic Press, New York and
London, 1962, p. 517.
14. Ghiretti F., in К. M. Wilbur, С. M. Yonge (eds.), Physiology of Mollusca,
Vol. II, Academic Press, New York and London, 1966, p. 233.
15. Stanley W. M„ Anderson T. F., J. Biol. Chem., 146, 25 (1942).
16. Ghiretti-Magaldi A., Nuzzolo C., Ghiretti F., Biochemistry, 5, 1943 (1966).
17. Feytmans E., Wibo M., Berthet J., Arch. Int. Physiol. Biochim., 74, 917 (1966).
18. Goyffon M„ Compt. Rend. Soc. Biol., 162, 1123 (1968).
19. Joubert F. J., Biochim. Biophys. Acta, 14, 127 (1954).
20. Moore С. H., Henderson R. W., Nichol L. W., Biochemistry, 7, 4075 (1968).
21. Eriksson-Quensel I. B., Svedberg T„ Biol. Bull., 71, 498 (1936).
22. Lauffer M. A., Swaby L. G„ Biol. Bull., 108, 290 (1955).
23. Pickett S. M., Riggs A. F., Larimer J. L„ Science, 151, 1005 (1966).
24. Di Giamberardino L„ Arch. Biochem. Biophys., 118, 273 (1967).
25. Thomson L. C. G., Hines M„ Mason H. S., Arch. Biochem. Biophys., 83, 88
(1959).
26. Svedberg T„ J. Biol. Chem., 103, 311 (1933).
27. Hernler E, Philippi E., Z. Physiol. Chem., 216, 110 (1933).
28. Lontie R., Witters R., in J. Peisach, P. Aisen, W. E. Blumberg (eds.), The Bio-
chemistry of Copper, Academic Press, New York and London, 1966, p. 455.
29. Wood E. J., Bannister W. H., Oliver C. J., Lontie R., Witters R., Comp. Biochem.
Physiol., 40B, 19 (1971).
30. Cohen L. B., Van Holde К. E., Biochemistry, 3, 1809 (1964).
31. Van Holde К. E., Cohen L. B., Biochemistry, 3, 1803 (1964).
32. Rubowltz F„ Biochem. Z., 299, 32 (1938).
33. Svedberg T„ Hedenius A., Biol. Bull., 66, 191 (1934).
34. Manwell C., J. Cell. Comp. Physiol., 52, 341 (1958).
Гемоцианин
419
35. Redmond J. R., Physiol. Zool., 35, 304 (1962).
36. Padmanabhanaidu B., Comp. Biochem. Physiol., 17, 167 (1966).
37. Boyd W. C., Biol. Bull., 73, 181 (1937).
38. Cloudsley-Thompson J. L„ J. Linn. Soc., 43, 134 (1957).
39. Berthet J., Berthet P., Arch. Int. Physiol. Biochim., 71, 124 (1963).
40. Berthet J., Baudhuin P., Wibo M„ Arch. Int. Physiol. Biochim., 72, 676
(1964).
41. Wieser W., Naturwiss., 52, 133 (1965).
42. Sundara R. G„ Current Sci. (India), 38, 168 (1969).
43. Svedberg T„ Pedersen К. O., The Ultracentrifuge, Clarendon Press, Oxford,
1940, p. 363.
44. Levin 0., Acta Univ. Upsal., 1963, 24.
45. van Bruggen E. F. J., in F. Ghiretti (ed.), Physiology and Biochemistry of
Haemocyanins, Academic Press, London, 1968, p. 37.
46. Baudhuin P„ Rev. Questions Sci., 1964, 77.
47. Wibo M„ Baudhuin P., Berthet J., Arch. Int. Physiol. Biochim., 74, 945
(1966).
48. Van Bruggen E. F. J., Schuiten V., Wiebenga E. H„ Gruber M., J. Mol. Biol.,
7, 249 (1963).
49. Brohult S., J. Phys. Colloid Chem., 51, 206 (1947).
50. Ferndndez-Mordn H., van Bruggen E. F. J., Ohtsuki M., J. Mol. Biol., 16, 191
(1966).
51. Eskeland T., J. Ultrastruct. Res., 17, 544 (1967).
52. Bloomfield V. A., Science, 161, 1212 (1968).
53. Condie R. M., Langer R. B„ Science, 144, 1138 (1964).
54. Brohult S., Borgman K., in «The Svedberg», Almqvist and Wiksell, Uppsala —
Stockholm, 1944, p. 429.
55. Heirwegh K., Borginon H., Lontie R., Biochim. Biophys. Acta, 48, 517 (1961).
56. Konings W. M., Siezen R. J., Gruber M., Biochim. Biophys. Acta, 194, 376
(1969).
57. Putzeys P., Brosteaux J., Trans. Faraday Soc., 31, 1314 (1935).
58. Brosteaux J., Naturwiss., 16, 249 (1937).
59. Konings W. N., Dijk J., Wichertjes T., Beuvery E. C., Gruber M., Biochim.
Biophys. Acta, 188, 43 (1969).
60. Cox J., Witters R., Lontie R., Int. J. Biochem., 3, (1972) in press.
61. Omura T., Fujita T., Yamada F., Yamamoto S., J. Biochem. (Tokyo), 50, 400
(1961).
62. Van Holde К- E., Biochemistry, 6, 93 (1967).
63. Moring Claesson L, Arkiv. Kemi, 10, 1 (1956).
64. Mason H. S„ in F. Dickens, E. Neil (eds.), Oxygen in the Animal Organism,
Macmillan, New York, 1964, p. 117 (discussion).
65. Foss J. G., Biochim. Biophys. Acta, 79, 41 (1964).
66. Takesada H.r Hamaguchi K., J. Biochem. (Tokyo), 63, 725 (1968).
67. Bryce G. F., Gurd F. R. N„ J. Biol. Chem., 241, 1439 (1966).
68. Orgel L. E„ in E. M. Crook (ed.), Metals and Enzyme Activity, Cambridge
University Press, London, 1958, p. 19.
69. Frieden E., Osaki S„ Kobayashi H., J. Gen. Physiol., 49, 213 (1965).
70. Yamada S„ Shimura Y„ Tsuchida R., Bull. Chem. Soc. Japan, 26, 72 (1953).
71. Bannister W. H., Wood E. J., Nature, 223, 53 (1969).
72. Wang В.-C., Schaefer W. P„ 166; 1404 (1969).
73. Tsuchida T„ Biophysics, 1, 42 (1961).
74. Nakamura T., Mason H. S., Biochem. Biophys. Res. Commun., 3, 297 (1960).
75. Manwell C„ in F. Dickens, E. Neil (eds.), Oxygen in the Animal Organism,
Macmillan, New York, 1964, p. 49.
76. Boas J. F., Pilbrow J. R„ Troup G. J., Moore C., Smith T. D., J. Chem. Soc.,
1969, 965.
77. Bayer E„ Fiedler H„ Ann., 653, 149 (1962).
27*
420
Глава 12
78. Blumberg W. E., in J. Peisach, P. Aisen, W. E. Blumberg (eds.), The Bioche-
mistry of Copper, Academic Press, New York and London, 1966, p. 472.
79. Bert P., Compt. Rend. Acad. Sci., Paris, 65, 300 (1867).
80. Winterstein H., Biochem. Z., 19, 384 (1909).
81. Redmond J. R., in F. Ghiretti (ed.), Physiology and Biochemistry of Haemo-
cyanins, Academic Press, London and New York, 1968, p. 5.
82. Wolvekamp H. P., Waterman T. H., in T. H. Waterman (ed.), The Physiology of
Crustacea, Vol. I, Academic Press, New York and London, 1960, p. 35.
83. Redfield A. C., Coolidge T., Montgomery H., J. Biol. Chem., 76, 197 (1928).
84. Guillemet R„ Gosselin G., Compt. Rend. Soc. Biol., Ill, 733 (1932).
85. Rawlinson W. A., Aust. J. Exp. Biol. Med. Sci., 18, 131 (1940).
86. Wyman J., J. Am. Chem. Soc., 89, 2202 (1967).
87. Stedman E., Stedman E., Biochem. J., 22, 889 (1928).
88. Lontie R., Meded. Vlaam. Chem. Ver., 16, 110 (1954).
89. DePhillips H. A., Nickerson K. W., Johnson M., Van Holde К. E., Biochemistry,
8, 3665 (1969).
90. Shaklai N., Daniel E., Biochemistry, 9, 564 (1970).
91. Root R. W„ J. Biol. Chem., 104, 239 (1934).
92. Rocca E„ Ghiretti F., Boll. Soc. Ital. Biol. Sper., 39, 2075 (1963).
93. Vanneste W., Mason H. S., in J. Peisach, P. Aisen, W. E. Blumberg (eds.),
The Biochemistry of Copper, Academic Press, New York and London, 1966,
p. 465.
94. De Ley M„ Lontie R„ FEBS Lett., 6, 125 (1970).
95. Pearson О. H. J. Biol. Chem., 115. 171 (1936).
96. Craifaleanu A., Boll. Soc. Nat. Napoli, 32, 141 (1919).
97. Wood E. J., Bannister W. H., Nature, 215, 1091 (1967).
98. Rombauts W., Lontie R., Arch. Int. Physiol. Biochim., 68, 695 (1960).
99. De Ley M„ Lontie R„ Arch. Int. Physiol. Biochim., 76, 175 (1968).
100. Rombauts W., Lontie R., Arch. Int. Physiol. Biochim., 68, 230 (1960).
101. Dhere C„ Schneider A., Compt. Rend. Soc. Biol., 82, 1041 (1919).
102. Ghiretti-Magaldi A., Ghiretti F., Nardi G., Parisi V., Zito R„ Boll. Soc. Ital.
Biol. Sper., 38, 1851 (1962).
103. Heirwegh K., Lontie R., Nature, 185, 854 (1960).
104. Heirwegh R, Blaton V., Lontie R., Arch. Int. Physiol. Biochim., 73, 149
(1965).
105. Ronings W. N., van Driel R., van Bruggen E. F. J., Gruber M., Biochim. Bio-
phys. Acta, 194, 55 (1969).
106. Felsenfeld G., Printz M. P., J. Am. Chem. Soc., 81, 6259 (1959).
107. Schubert J., White E. R., Science, 155, 1000 (1967).
108. Ghiretti F., Arch. Biochem. Biophys., 63, 165 (1956).
109. Morpurgo G., Williams R. J. P„ in F. Ghiretti (ed.), Physiology and Bio-
chemistry of Haemocyanins, Academic Press, London, 1968, p. 113.
110. George P., Siratmann C. J., Biochem. J., 57, 568 (1954).
111. Slater E. C.. Nature, 170, 970 (1952).
112. Heirwegh R„ Borginon H„ Lontie R„ Arch. Int. Physiol. Biochim., 67, 514
(1959).
113. Ghiretti-Magaldi A., Nardi G„ Ghiretti F., Zito R., Boll. Soc. Ital. Biol. Sper.,
38, 1839 (1962).
114. Henze M„ Z. Physiol. Chem., 33, 370 (1901).
115. Gl.irctti-Magaldi A., Nardi G., in H. Peeters (ed.), Protides Biol. Fluids, Proc.
11th Colloquium, Bruges, 1963, Elsevier, Amsterdam, 1964, p. 507.
116. Hemmerich P., Sigwart C., Experientia, 19, 488 (1963).
117. Lontie R., Blaton V., Albert M., Peeters B., Arch. Int. Physiol. Biochim., 73,
150 (1965).
118. Rocca R., Comp. Biochem. Physiol., 28, 67 (1969).
119. Johnston W., Barber A. A., Comp. Biochem. Physiol., 28, 1259 (1969).
Г емоцианин
421
120. Bayer Е.. Schretzmann Р., in С. К. Jorgensen et al. (eds.). Structure and
Bonding, Vol. II, Springer, Berlin, 1967, p. 236.
121. Witters R., Lontie R., in F. Ghiretti (ed.), Physiology and Biochemistry of
Haemocyanins, Academic Press, London, 1968, p. 61.
122. Woods K. R., Paulsen E. C., Engle R. L., Jr., Pert J. H., Science, 127, 519
(1958).
123. Elliott F. G., Hoebeke J., in F. Ghiretti (ed.), Physiology and Biochemistry of
Haemocyanins, Academic Press, London, 1968, p. 81.
124. Wood E. J., Salisbury С. M„ Formosa N., Bannister W. H„ Comp. Biochem.
Physiol., 26, 345 (1968).
125. Gruber M., in F. Ghiretti (ed.), Physiology and Biochemistry of Haemocyanins,
Academic Press, London, 1968, p. 49.
126. Felsenfeld G„ J. Cell. Comp. Physiol., 43, 23 (1954).
127. Zuckerkandl E„ Bull. Soc. Chim. Biol., 41, 1629 (1959).
128. Griffe M., Lontie R„ Arch. Int. Physiol. Biochim., 69, 594 (1961).
129. Brauns G., Lontie R., Arch. Int. Physiol. Biochim., 68, 211 (I960).
130. Nardi G., Ghiretti-Magaldi A., Caserta G., Zito R., Ghiretti F., Boll. Soc. Ital.
Biol. Sper., 38, 1845 (1962).
131. Klotz 1. M„ Klotz T. A., Science, 121, 477 (1955).
132. Lontie R., Clin. Chim. Acta, 3, 68 (1958).
133. Zito R„ Nardi G., Caserta G., Ghiretti-Magaldi A., Boll. Soc. Ital. Biol. Sper.,
38, 1855 (1962).
134. Hemmerich P., in J. Peisach, P. Aisen, W. E. Blumberg (eds.), The Bioche-
mistry of Copper, Academic Press, New York and London, 1966, p. 15.
135. Rocca E„ Boll. Soc. Ital. Biol. Sper., 39, 2077 (1963).
136. Ghiretti-Magaldi A., Nuzzolo C., Comp. Biochem. Physiol., 16, 249 (1965).
137. Wood E. J., Salisbury С. M., Bannister W. H., Biochem. J., 108, 26P (1968).
138. Wood E. J., Bannister W. H„ Biochim. Biophys. Acta, 154, 10 (1968).
139. Engelborghs Y., Witters R„ Lontie R., Arch. Int. Physiol. Biochim., 76, 372
(1968).
ЧАСТЬ IV
РЕАКЦИИ ЛИГАНДОВ,
ОБУСЛОВЛЕННЫЕ КООРДИНАЦИЕЙ
С МЕТАЛЛОМ, И МЕТАЛЛОФЕРМЕНТЫ
ГЛАВА 13
АКТИВАЦИЯ МАЛЫХ МОЛЕКУЛ
ПРИ КООРДИНАЦИИ С МЕТАЛЛАМИ
М. М. Джонс, Дж. Э. Хикс мл.
Jones М. М., Department of Chemistry, Vanderbilt University,
Nashville, Tennesse 37203, USA
Hix J. E., Jr., Department of Chemistry, East Carolina
University, Greenville, North Carolina 37834, USA
1. ВВЕДЕНИЕ
Координация малой молекулы с ионом металла или другой
льюисовской кислотой существенно изменяет ее реакционную спо-
собность. Во-первых, малая молекула, которая представляет со-
бой независимую частицу, имеющую по крайней мере одну сво-
бодную пару валентных электронов, становится частью более
крупной молекулы, связь с которой осуществляется по крайней
мере за счет одной из этих электронных пар. Во-вторых, она пре-
вращается из нейтральной или отрицательно заряженной частицы
в группу, несущую положительный заряд. В результате этого ак-
тивируются обычно нереакционноспособные группировки атомов,
находящиеся вблизи положительно заряженного центра. Таким
образом, реакционная способность лигандов должна зависеть от
координации. Более детальное рассмотрение зависимости кон-
кретных реакций лигандов от координации и составляет предмет
настоящей главы. Как мы увидим, существует большое число
экспериментальных данных, которые можно использовать для
установления взаимосвязи между такими процессами и для про-
гнозирования. Целью нашего исследования является как более
глубокое понимание природных процессов, в которых ионы метал-
лов действуют как катализаторы, так и поиск более эффективных
катализаторов реакций различных лигандов. В связи с этим сле-
дует заметить, что ионы металлов могут активировать некоторые
ферменты не только в результате возникновения координационных
мест для ферментативного процесса, но и вследствие создания
определенной конформации белковой цепи. Так, ион металла мо-
жет находиться довольно далеко от активного центра фермента,
Активация малых молекул при координации с металлами 423
но тем не менее быть необходимым для проявления его активно-
сти. Таким образом, наряду с ферментами, у которых ион метал-
ла является составной частью активного центра, может наблю-
даться и рассмотренный выше тип активации, при котором ион
металла удален от активного центра.
2. ОСНОВНЫЕ ЭФФЕКТЫ координации
В результате присоединения лиганда к положительно заряжен-
ному иону металла электронная плотность смещается в направ-
лении этого иона. Теория молекулярных орбиталей для комплек-
сов, в которых л-взаимодействие не играет существенной роли,
приводит, по существу, к такому же результату. С наибольшей
уверенностью этот вывод применим к комплексам жестких осно-
ваний с жесткими кислотами [1]. Если л-связывание становится
существенным, следует учесть, что эффект будет уменьшаться за
счет «обратного допирования» от металла к лиганду или увели-
чиваться при прямом л-донировании.
При взаимодействиях типа жесткое основание — жесткая кис-
лота (гл. 2) поляризация лиганда может вызвать следующие ха-
рактерные последствия:
1) увеличение кислотности атомов водорода при донорном
атоме;
2) облегчение атаки лиганда нуклеофилами и затруднение ата-
ки электрофилами;
3) «маскирование» некоторых реакций свободного лиганда,
зависящих от стерических или электронных взаимодействий, кото-
рые затрудняются или становятся невозможными при координа-
ции.
Помимо перечисленных выше эффектов при координации воз-
можно изменение окислительно-восстановительных свойств лиган-
да или в случае лигандов, способных к таутомерии, смещение
таутомерного равновесия в сторону таутомера, образующего бо-
лее устойчивые комплексы. Большинство этих общих следствий
было впервые четко сформулировано Меервейном в конце 20-х го-
дов [2].
Увеличение кислотности атомов водорода при донорном атоме
впервые наблюдал Вернер [3], который обнаружил, что кислотные
свойства координированных молекул воды или аммиака увеличи-
ваются в зависимости от природы центральных атомов в ряду
Co3+<Cr3+<Ru4+<Pt4+. Универсальность этого явления была
продемонстрирована на большом числе комплексов. Сравнение
свойств свободных и координированных молекул воды можно про-
вести на примере [Си(Н2О)4]2+. Константа равновесия, равная
1,07-10-3 для реакции
[Cu(H2O)4]2+ [Си(Н2О)3(ОН)]+ + Н+
424
Глава 13
примерно в 107 раз превышает аналогичное значение для чистой
воды [4]. Подобное повышение кислотности обнаружено также
для органических лигандов, у которых атомы водорода соединены
с донорными атомами [5, 6].
Тот факт, что нуклеофильная атака координированной частицы
происходит легче, был использован в конце 40-х — начале 50-х
годов [7] для объяснения роли ионов металлов в некоторых фер-
ментативных реакциях и еще раньше был замечен Геллерманом
[8]. Реакции этого типа с участием воды наиболее многочислен-
ны. Сюда относятся реакции гидролиза простых частиц, таких,
как,. ЗгО?' [9], и конденсированных фосфатов, а также более
сложных соединений [10]—пептидов, производных аминокислот
и фосфорных эфиров. Ниже эти реакции будут рассмотрены бо-
лее подробно.
Маскирование реакций лигандов — одно из первых свойств
комплексов, которое привлекло внимание исследователей. Тот
факт, что координированный органический лиганд менее чувстви-
телен к действию типичных окислителей, был замечен еще в 1857 г.
[11]. Имеется много примеров подобного маскирования, причем
в большинстве из них донорные атомы участвуют в окислительно-
восстановительных процессах. Есть данные по маскированию ко-
ординированного оксалата при действии на него брома [12], по
защите ЭДТА от действия перманганата [13] и ОЭДТА в реакции
с ванадатом [14]. Также давно замечено, что координированные
анионы не дают характерных для них реакций осаждения или
теряют присущие им основные свойства (например, NH3 в
[Co(NH3)6]3+). Наконец, маскирование определенных реакций при
координации может играть существенную роль в биологии.
Для больших полидентатных доноров особенно важен еще один
эффект координации — фиксация донорных атомов в определен-
ных положениях друг относительно друга. Создание определенной
конформации лиганда при координации стимулирует реакции, тре-
бующие именно этой конформации, и затрудняет или делает невоз-
можным протекание других реакций.
Последний эффект координации, на котором следует остано-
виться, сводится к активации малых молекул в реакциях с соот-
ветствующими реагентами. Этот эффект часто связан с нуклео-
фильной атакой и поэтому может стимулировать некоторые окис-
лительно-восстановительные реакции.
3. КАТАЛИЗ НУКЛЕОФИЛЬНОЙ АТАКИ
Основным следствием координации субстрата с положительно
заряженным ионом металла является катализ нуклеофильной ата-
ки. Причина его кроется в более благоприятных электростатиче-
Активация малых молекул при координации с металлами 42
ских взаимодействиях в комплексе. Согласно Лейдлеру [15], для
моделей, активированный комплекс которых состоит из двух сфе-
рических реагентов, можно оценить электростатический вклад
(в энтропийных единицах) в энергию активации следующим об-
разом:
AS^ =—10ZaZb (для воды^как растворителя)
Если ZA— заряд атакующего реагента, ZB1— заряд лиганда, а
Zb2 — заряд иона металла до координации, то
^лиганда ^А^
^комплекса = Ю^А (^В1 +-^В2)
Тогда изменение энтропии в процессе комплексообразования
A AS* = -10ZaZb2
Можно записать альтернативное выражение для активированного
комплекса, состоящего из одной сферы, однако его использование
потребует знания радиусов активированного комплекса и реаген-
тов. Рассчитанное значение изменения энтропии активации зави-
сит от природы растворителя и заряда частиц, атакующих суб-
страт. Если растворителем является вода и атакующие частицы
несут положительный заряд, то при координации энтропия примет
более отрицательное значение и достижение переходного состоя-
ния будет затруднено. При атаке отрицательно заряженными час-
тицами эффект будет обратным.
Используя аналогичные модели, можно рассчитать отношение
констант скоростей реакций лиганда и комплекса. Для простой
двухсферной модели результат можно записать так:
kc Z^Z^p1
ln kf
где kc — скорость реакции комплекса, kf — скорость реакции сво-
бодного лиганда, Zm — заряд комплекса металла без учета ли-
ганда, Zb — заряд атакующей частицы, е — диэлектрическая про-
ницаемость реакционной среды, rfAB — расстояние между субст-
ратом и атакующей частицей в активированном комплексе (одина-
ковое для реакций комплекса и свободного лиганда), k — посто-
янная Больцмана и Т—абсолютная температура. Аналогичное
уравнение можно вывести для ион-дипольных реакций.
Рассматривая такую модель, мы всегда ожидаем, что нуклео-
фильная атака ускоряется в результате координации с катионом,
и полагаем, что это ускорение тем сильнее, чем больше заряд ка-
тиона и чем ниже диэлектрическая проницаемость растворителя.
Большой недостаток этой модели состоит в том, что она не учиты-
вает жесткой или мягкой природы взаимодействующих частиц и
426
Глава 13
поэтому неадекватно отражает специфику каталитических процес-
сов этого типа. Можно вывести более совершенное уравнение, од-
нако для этого нужно более детально описать переходное состоя-
ние.
Перечисляя типы таких реакций, следует назвать все примеры
катализа нуклеофильной атаки ионом металла, приведенные в
обзоре Бендера [10] и иллюстрирующие их широту и общность.
Тут и реакции гидролиза ангидридов кислот, эфиров и амидов
карбоновых кислот, фосфорных эфиров и оснований Шиффа, реак-
ции карбоксилирования и декарбоксилирования, переаминирова-
ния, гидратации, гидрирования, расщепления простых эфиров и
различные процессы, сходные с более тщательно исследованной
группой реакций, в которых протон катализирует атаку ряда дру-
гих нуклеофилов (кроме ОН-). Большое преимущество ионов ме-
талла перед протоном во многих реакциях этого типа заключает-
ся в том, что они могут эффективно катализировать процесс в рас-
творах со значительно большим pH. По этой причине такие ионы
металлов были названы «суперкислотами» [16]. Это название
справедливо особенно в тех случаях, когда ион металла образует
хелат, значительно более эффективный в катализе, нежели протон
[17]. Если же ион металла координирует лиганд только по одному
месту, то в этом случае его поляризующее действие и каталитиче-
ская активность обычно не выше, чем у протона; однако ион ме-
талла может проявить свойства «суперкислоты», действуя в обла-
сти pH, не доступной для протона.
4. НЕКОТОРЫЕ ВАЖНЫЕ ГИДРОЛИТИЧЕСКИЕ ПРОЦЕССЫ
В качестве примера того, как координация способствует нук-
леофильной атаке лиганда, рассмотрим некоторые гидролитиче-
ские процессы, служащие «модельными» системами для ряда фер-
ментативных реакций. Эти хорошо изученные системы включают
каталитический гидролиз эфиров аминокислот, пептидных и фос-
фатных связей.
Во всех реакциях термодинамическая устойчивость образую-
щихся комплексов играет главную роль в проявлении катализа.
Если ион металла и молекула лиганда не образуют комплекса, то
экспериментально наблюдается только реакционная способность
свободного лиганда. В водном растворе ион металла конкурирует
с ионом водорода за координацию с лигандом. При увеличении
кислотности реакционной среды уменьшается концентрация не
только нуклеофильного агента — ионов гидроксила (молекулы
воды, хотя и в меньшей степени, также могут выступать в роли
нуклеофила), но и образующегося комплекса. С другой стороны,
Активация малых молекул при координации с металлами 427
при увеличении основности среды происходит гидролиз ионов
металла, приводящий к образованию нерастворимых, а следова-
тельно, и неактивных гидроокисей. Поэтому в большинстве слу-
чаев для проведения каталитического процесса используют огра-
ниченную область pH.
Сравнивая наблюдаемые скорости гидролиза и истинные ско-
рости гидролиза комплексов, содержащих различные лигандные
частицы, нужно учитывать состав раствора. Лиганд в каждом
комплексе подвергается щелочному гидролизу с характерной для
него скоростью, а наблюдаемая общая скорость складывается из
частных скоростей реакций всех присутствующих в реакционной
системе частиц:
^набл. — ^своб. лиганда + ^комплекса^ 4~ 1'комплекса2
Теперь рассмотрим каталитическую активность комплексных
соединений в реакциях гидролиза лигандов различных классов.
4.1. Эфиры аминокислот
Впервые о катализе щелочного гидролиза эфиров аминокислот
ионами металлов сообщил Кролл в 1952 г. [18]. Впоследствии
многие исследователи изучали эти каталитические реакции и
полностью выяснили роль ионов металла в ускорении гидролиза
[19].
С точки зрения электростатических эффектов следует ожидать,
что координация эфира аминокислоты с положительно заряжен-
ным ионом металла или комплексом будет увеличивать скорость
щелочного гидролиза, так как эта реакция включает нуклеофиль-
ную атаку лиганда. В первом приближении скорость каталитиче-
ского процесса будет определяться главным образом электроста-
тическим зарядом реагирующего комплекса. Обычно это действи-
тельно так: большинство трехзарядных комплексов ускоряют
гидролиз в растворах с pH 3, двухзарядные комплексы — при
pH 5—7, а однозарядовые — в более щелочных растворах, при
рН>7 [17, 20, 21].
Для более точной оценки влияния ионов металла на гидролиз
эфира следует более детально разобраться в процессе взаимодей-
ствия лиганда и металла. В большинстве случаев эфир координи-
руется с металлом, используя в качестве донорного атома только
аминный азот. Карбонильная группа эфира, как и карбоксильная
группа соответствующей аминокислоты, обладает слабыми нук-
леофильными свойствами и не склонна к хелатообразованию. Од-
нако, если в качестве катализатора использовать более электро-
фильный ион металла или комплекс, между металлом и карбони-
лом наблюдается слабое взаимодействие, сопровождающееся
428
Глава 13
поляризацией карбонильной связи. Эффект хелатообразования
изображен на приведенной ниже схеме:
6* O-R
R-CH-C
।
HzN\ У°&~
Заметим, что при образовании хелата карбонильная группа поля-
ризуется, причем ее углеродный атом становится более электро-
фильным. Теперь уже не только облегчается нуклеофильная ата-
ка комплекса, но и активированные центры лиганда становятся
местом атаки. В случае Си (II), катализирующей гидролиз этил-
противоположная конфигурация
Активация малых молекул при координации с металлами 42&
глицината, увеличение скорости реакции почти в 200 раз больше,
чем допускают электростатические соображения [22]. Буш [20]
продемонстрировал аналогичный эффект для Со(III), катализи-
рующего гидролиз координированных эфиров аминокислот.
Из-за асимметрии а-углеродного атома всех аминокислот и их
производных (за исключением глицина) в структуре и реакциях
комплексов, образованных этими лигандами, может проявляться
стереоселективность. Стереоселективность этого типа была обна-
ружена только в тех случаях, когда лиганд первоначально имел,
кроме потенциально координируемой эфирной группы, по крайней
мере два донорных атома, как, например, у гистидина [23]. При
гидролизе эфиров гистидина в присутствии d- и ь-изомеров гис-
тидинатоникеля(П) для непосредственного взаимодействия кар-
бонильных групп с ионом металла эфир гистидина и гистидинат
в никелевом комплексе должны иметь противоположные конфи-
гурации. Если они имеют одинаковую конфигурацию, то в этом
случае эфирная группа удалена от иона металла и их непосредст-
венного взаимодействия не происходит.
4.2. Пептидные связи
Полипептиды и амиды аминокислот также подвергаются ка-
талитическому гидролизу. Однако каталитические эффекты часто
выражены не так ярко, как в случае гидролиза эфиров. Широко
используются четырехвалентные металлы типа тория (IV) и це-
рия (III), а также ряд более тяжелых трехвалентных металлов
[24].
Было показано, что при гидролизе пептидных связей цис-гид-
роксиаквотриэтилентетраминкобальтом(Ш) избирательно отщеп-
ляются N-концевые аминокислоты [25]. Комплексы этого типа
координируют концевую аминогруппу и за счет индуктивного эф-
фекта помогают разорвать связь с остатком следующей аминокис-
лоты. Они широко используются при определении аминокислотной
последовательности пептидных фрагментов.
4.3. Фосфатные связи
Было также обнаружено каталитическое расщепление фосфор-
ных эфиров [26], ацилфосфатов [27] и полифосфатов [28]. Связи
в этих соединениях гидролизуются тоже не так легко, как в эфи-
рах аминокислот, но при использовании высокозаряженных
комплексов и (или) при повышенной температуре эти системы
гидролизуются довольно быстро. В гл. 17 такого рода реакции
будут рассмотрены детально.
430
Глава 13
5. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ
СВОЙСТВА ЛИГАНДОВ
При координации окислительно-восстановительные свойства
лигандов меняются незначительно, однако имеются данные о том,
что ион металла, обладающий окислительными или восстанови-
тельными свойствами, может изменить это общее положение. Изу-
чение поведения таких частиц, как галогенид-ионы, показало, что
координация делает их более устойчивыми по отношению к внеш-
ним окислителям; примером могут служить рекации окисления 1~
В [Hgl4]*~ или В г- в [HgBr4]2-. В значительной степени это яв-
ление можно отнести за счет уменьшения доступности электрон-
ных пар лиганда внешним реагентам. Если ион металла сам яв-
ляется окислителем, то координация будет способствовать окисле-
нию лиганда, что подтверждается на примере многоступенчатых
окислителей.
Так, при окислении ионами МпО4 или СгО^- скорость первой
стадии реакции часто меньше, чем последующих стадий, в кото-
рых участвуют ионы марганца и хрома в низших степенях окис-
ления, способные координировать окисляемый лиганд. Сам факт
координации, при котором происходит смещение электронной
плотности лиганда в сторону льюисовской кислоты должен уве-
личивать окислительную способность комплексных частиц. Хотя
эту гипотезу трудно проверить, но для Cu(II), Tl(III), Fe(III) и
им подобных окислителей общепринят внутрисферный механизм
окисления и есть данные, подтверждающие это.
Особый интерес представляют системы, в которых лиганд свя-
зан с легко восстанавливающимся ионом металла. В большинстве
случаев восстановление металла в таких системах сопровождает-
ся превращением лиганда в реакционноспособный свободный ра-
дикал, который участвует в последующих реакциях и в образова-
нии конечных продуктов. Иногда при этом образуются важные
продукты, как, например, при окислении Си (II) а-оксикетонов в
а-дикетоны:
Н
R—i—С—R + 2Си«------->- R—С—С—R -f- 2Cu+ -f- 2Н+
I || пиридин || ||
он о о о
Эта реакция может быть использована для превращения бензоина
в бензил [29] и фуроина в фурил [30]. Как было показано Брок-
маном [31], при синтезе нитрилов в присутствии кислорода про-
цессы окисления протекают глубже и за счет повторного окисле-
ния ионов Сп(1) становятся каталитическими. В общем виде реак-
ция выглядит так:
RCHO + NH3 -f- Оа -> RCN + НаО + НаОа
Активация малых молекул при координации с металлами
431
Си (И)
НаО2 + субстраты ------->- Н2О -f- продукты
и включает следующие стадии:
комплекс СиИ
RCHaOH ----------> RCHO-f-HaO
RCHO+NHS-------> RCH=NH-f-H2O
медленно
RCH=NH -|- комплекс Cull ---->- иминный радикал -f- H+ + комплекс Cui
иминный радикал-f-O2 -» RCsN4-HO2-
НО2- +Н+-----> Н2Оа
Активной формой в этой реакции считают комплекс меди
[Cu(NH3)4(OCH2R)]+. В отсутствие кислорода этот комплекс
реагирует в стехиометрических количествах, как и следует ожи-
дать, если допустить промежуточное образование комплекса с ос-
нованием Шиффа.
Существует множество подобных автокаталитических процес-
сов, протекание которых зависит от легкости окисления субстра-
та комплексным ионом металла и возможности регенерации окис-
ленной формы последнего кислородом воздуха. Еще одним приме-
ром реакций этого типа является катализ ионами Си (II) авто-
окисления аскорбиновой кислоты [32] (гл. 20).
Замечательным примером использования координации для
ускорения процессов окисления служит реакция окисления этиле-
на в ацетальдегид тетрахлорпалладатом [33]. Этот процесс ста-
новится каталитическим, если в реакционной среде имеются соли
Си (II), соляная кислота и кислород. Суммарное уравнение реак-
ции следующее:
CjH, + PdClJ- + ЗН2О--> CHSCHO -f- Pd -f- ЗН3О+ + 4СГ
Вначале происходит координация и окисление олефина:
PdCl»- -f- ед =г=^ [PdCl3(CaH4)J- + СГ __
[PdCls(C1H<)r -f-HaO PdCla(HaO) (QHJ +СГ
PdCla(HaO) (C2H4) +H2O [PdCla(OH) (C2H4)]- + H3O+
[PdCla(OH) (CaHjr ------> CHSCHO-f-Pd°-f-2СГ
затем
Pd° -f- 2CuCla -f- 2СГ -> PdClj- -f- 2CuCl
И
4CuCl + 4HC1 -f- O2 ----> 4CuCIa -f- 2HaO
Реакции восстановления лигандов лучше всего изучены на при-
мере комплексов Pt(II) с ненасыщенными молекулами. В таких
комплексах олефиновые лиганды восстанавливаются значительно
432
Глава 13
легче и быстрее, чем свободные лиганды [34]. Примеров восста-
новления других типов лигандов известно довольно мало.
Таким образом, окислительно-восстановительные свойства ли-
ганда изменяются при координации. Если ион металла сам не под-
вергается окислительно-восстановительным превращениям, то он
обычно препятствует окислению лиганда внешними реагентами.
6. МАСКИРОВАНИЕ РЕАКЦИОННОЙ СПОСОБНОСТИ
ЛИГАНДОВ В КОМПЛЕКСАХ
Под маскированием реакционной способности лиганда понима-
ют затруднение или полную невозможность протекания характер-
ных для лиганда реакций в результате координации. Мы уже от-
мечали, что координация препятствует реакциям осаждения га-
логенид-ионов. Последствия координации в таких простых случа-
ях можно предсказать заранее, зная ее влияние на растворимость
продуктов. Для более сложных реакций предугадать эффект мас-
кирования значительно труднее.
Ниже приведены возможные причины появления эффекта мас-
кирования.
1. Образование координационной связи с использованием элек-
тронной пары, необходимой для реакции.
2. Приобретение лигандом в результате координации конфор-
мации, неблагоприятной для реакции.
3. Резкое изменение окислительно-восстановительного потен-
циала лиганда в результате координации, препятствующее само-
произвольному протеканию реакции.
4. Изменение заряда активного центра, затрудняющее ход ре-
акции.
5. Переход лиганда в результате координации в другую форму
(таутомерную или ионную), которая неспособна к реакции или
реагирует значительно труднее.
Маскирование может явиться следствием комбинации двух или
более таких факторов, однако для объяснения этого эффекта в
каждом конкретном случае обычно ищут частные причины, кото-
рыми легче оперировать.
Одним из наиболее ценных приложений маскирования являет-
ся защита карбоксильных и а-аминогрупп в реакциях аминокис-
лот. Образование хелата с Си(II) позволяет весьма надежно за-
щитить эти группы от большинства характерных для них реакций.
При этом другие, незамаскированные функциональные группы
становятся активными центрами реакции. Затем медь можно от-
делить от аминокислоты в виде нерастворимого сульфида, дейст-
вуя сероводородом или сульфидом натрия. Общая схема реакций
этого типа такова:
Активация малых молекул при координации с металлами
43 3
атака на
фрагмент Z
2Y—(СН2)Х—С—С—ОН + CuS
nh2
Применение этой реакции в синтезах, рассмотренное в обзорах
[35, 36], касается главным образом реакций концевых незащищен-
ных амино- или карбоксильных групп. Примером служит превра-
щение цитрулина в орнитин [37] (см. схему на стр. 434).
В качестве реагентов для атаки на незакомплексованную ами-
ногруппу в маскированных комплексах аминокислот служили ук-
сусный ангидрид [38], карбобензоксихлорид [39], фенилизо-
цианат [40], хлоргидрат биотина [41], цианат калия [42], соли
О-метилизомочевины [40], хлорангидриды кислот [40] и т. д.
Реакции этого типа удобны для синтеза аргинина [43], N-ацетил-
лизина [36] и других производных полифункциональных амино-
кислот. Маскирование также можно использовать для замены од-
ной или двух карбоксильных групп в соединениях, подобных глут-
аминовой кислоте [44] (см. схему на стр. 434).
Маскирование аминогрупп путем координации также можно
использовать в тех случаях, когда других свободных функциональ-
ных групп нет, а атака реагента нацелена на центры молекулы
28—2451
434
Глава 13
I
2H2N-C-N-(CH2)3-CH-C<
+ CuS
+ C6HSCH2C1
с низкой реакционной способностью. Хорошим примером этого мо-
жет служить конденсация Си (II)-комплексов аминокислот по
Кнёвенагелю [45]. В одной из таких реакций бис- (глицина-
те) медь(II) конденсируется в щелочном растворе ацетальдегида
с образованием б«с-(треонинато)меди(П):
Активация малых молекул при координации с металлами
435
СН3СНО
+ но’
I \ X
СН3-С-С-Н Си
он Хс-(У
II
нс-ссн
°-сХ он
3
Другие реакции позволяют получить р-оксилейцин [46], серин и
фенилсерин [47].
В дипептидных комплексах Со(III) активируются реакции об-
мена протонов в концевых метиленовых группах, в то время как
протоны аминогруппы не обмениваются [48]. Этот эффект дока-
зан для комплексов с глицилглицином, аланил глицином и глицил-
аланином и согласуется с типом реакционной способности, уста-
новленным для таких молекул в комплексах. Время обмена про-
тона в подобных метиленовых группах составляет менее полупе-
риода конформационного сдвига.
В случае комплексов, образованных лигандами, несущими не-
координированную гидроксильную группу, проявляется другой тип
маскирования. Гидроксильные группы таких лигандов иногда ока-
зываются устойчивыми к этерификации, хотя и неясно почему.
Так, бцс-(2-оксиэтилиминодиацетато)хром(1П)
очень устойчив к действию типичных ацетилирующих агентов, хо-
тя в кипящей уксусной кислоте ацетилируется с небольшим вы-
ходом [49]. Причина этой инертности неизвестна, хотя ее можно
связать с действием факторов, которые затрудняют этерификацию
некоторых спиртов.
Наконец, еще один аспект маскирования, который следует от-
метить, состоит в использовании координации в целях уменьшения
силы некоторых очень сильных кислот Льюиса (для удобства их
использования). Например, аддукт SO3— пиридин при диссоциа-
ции будет выделять лишь небольшие количества SO3.
QK:s0-’^O;:+S01
Равновесия этого типа можно использовать для снижения актив-
ности каталитической реакции с участием кислоты Льюиса (вклю-
чая ионы металлов) в тех случаях, когда введение чистой кислоты
приводит к неконтролируемому течению реакции или к образова-
28*
436
Глава 13
нию нежелательных побочных продуктов. При этом исчезают
проблемы, связанные с высокой локальной концентрацией кисло-
ты Льюиса.
Маскирование таит в себе огромные возможности в синтезах
с участием молекул, имеющих много реакционных центров, часть
из которых связана в хелатном кольце, а часть — свободна.
7. АКТИВАЦИЯ МАЛЫХ МОЛЕКУЛ ПОСРЕДСТВОМ
КООРДИНАЦИИ [50—55]
Значительное число двухатомных и некоторых более крупных
молекул можно активировать с помощью координации так, чтобы
они реагировали вполне определенным образом. Другими спосо-
бами достичь этого очень трудно. В качестве примера приведем
недавно обнаруженные кислородные комплексы на основе изонит-
рильных комплексов Ni(II) и Pd(II), катализирующие следующий
процесс [56, 57]:
Ni(CNR)4
RNC + О2-------»- RCNO
Координацию можно использовать для активации N2, Н2, О2,
С12, Вг2, C1CN, Н2О, СН2=СН2, СО, NOCI, NO2C1, Н2О2, а также
более крупных молекул. Многие из этих молекул могут принимать
участие в реакциях внедрения с комплексами низковалентных пе-
реходных металлов. Имеется также большое число других про-
цессов, в которых происходит специфическая активация таких мо-
лекул. В данном разделе мы коснемся главным образом актива-
ции Н2 и СО.
Использование координации для активации малых молекул
наиболее ярко прослеживается на примере оксо-реакций и сход-
ных процессов введем . о 'иси углерода и водорода в подходящие
органические соедине, .. реакция катализируется солями кобаль-
та, которые участвуют в ней в виде НСо(СО)4 и Со2(СО)в, а реак-
ционная смесь состоит из ненасыщенного углеводорода или спир-
та, окиси углерода и водорода, находящихся под давлением, и ка-
кой-либо соли кобальта. В такой системе можно проводить многие
реакции, наиболее важными из которых являются следую-
щие [58]:
(1) Гидроформилирование:
НСо(СО)4
СН2=СН2 + СО + Н2 - > СН3СН2СНО
100—400 ат
(2) Гидрирование:
СН2=СН2+Н2 ---------------------------> СН3СН3
(3) Гомологенизация:
RCH2OH + СО + Н2 ---> RCH2CH2OH + Н2О
Активация малых молекул при координации с металлами 437
(4) Гидрогенолиз:
R2CHOH + Н2 ----> R2CH2 + Н2О
Основным процессом для многих систем подобного рода яв-
ляется реакция внедрения, в которой окись углерода или молекула
олефина внедряется между атомом кобальта и алкильной группой
или атомом водорода, связанными с ним:
HCo(CO)4-f-C2H4 ---> Н—С2Н4—Со(СО)4
СН3Со(СО)4 + СО ---> СН3ССо(СО)4
Важной особенностью этих реакций является способность кобаль-
та образовывать активный интермедиат, в котором он имеет бо-
лее низкое координационное число, и затем реагировать с окисью-
углерода или олефином. Карбонилы других металлов также могут
катализировать подобные реакции, преимущественно по сходному
механизму [59].
Во многих случаях плоские квадратные комплексы переходных
элементов типа d6 вступают в окислительно-восстановительные
реакции с малыми молекулами, причем последние расщепляются
на две части и активируются для последующих превращений [60].
Эти реакции обычно следуют схеме
где А—В может быть галогеном, псевдогалогеном, алкилгалоге-
нидом, ацилгалогенидом, Ог, Н2, ацетиленами, галогенводородами,
олефинами, галогенидами металла, неметаллическими гидридами
или протонными кислотами (см. также гл 20). Примерами таких
процессов могут служить следующие реакции:
СН3(СвН5)2Рч /СО СН3(С6НБ)2Р °^СО
Ir -f-CH3Br----> 1г
ос/ \р(СвНБ)2СН3 ОС'/111\р(СвНБ)2СН1
(C6HS)3PX ^СО
/1г\ * °2
С1 'Р(С6Н5)з
Р(С6Н5)з
ос-. I --о
Ir.-."'
Х^1 О
Р(С6Н5)3
Комплексы с конфигурацией d& могут быть весьма эффектив-
ными катализаторами гидрирования (например, RhCl[P(C6H5)3]3.
гидроформилирования (например, Ри(СО)3[Р(СбН5)3]2, декарбо-
нилирования и димеризации этилена [60].
438
Глава 13
Активация молекулярного водорода, особенно при гидрирова-
нии, достаточно эффективно осуществляется большим числом ком-
плексов переходных металлов. По Галперну [61], можно выде-
лить три механизма образования гидридных комплексов, служа-
щих активными восстанавливающими агентами:
Гетеролитическое расщепление:
[Ruincie]S- + Н2 <—>: [RuIHHCIJS- -f-H+ -f-СГ
Гомолитическое расщепление:
2[CoH(CN)6]3- + Н2 <==» 2[CoinH(CN)6]3~
Внедрение (образование дигидрида):
IriCl(CO) [Р(СвН5)3]а + Н2 1гШН2С1(СО)[Р(СвНь)312
Процессы катализа реакций гидрирования этими комплексами
включают образование координационной связи комплекса с оле-
фином, перенос на него водорода и отщепление продукта. Образо-
вание гидридного комплекса может происходить до или после об-
разования связи с олефином. Большое число низковалентных ком-
плексов железа, кобальта, никеля и металлов платиновой группы
могут катализировать гидрирование в гомогенных условиях; при
этом гидрируются такие органические субстраты, как алкены, ал-
кины, альдегиды и ароматические соединения. Некоторые из этих
комплексов весьма эффективны, например КЬС1[Р(С6Н5)з]з, ката-
лизирующий гидрирование олефинов при комнатной температуре
и давлении водорода порядка 1 ат. Эффективными катализатора-
ми также являются Р1С1[Р(С6Н5)з]25пС1з, КиС12[Р(СбН5)3]2,
НСо(СО)4, IrH(CO) [Р(СбНз)з]з и их аналоги. Многие из этих
комплексов сходны с комплексными соединениями, активирующи-
ми другие малые молекулы, и их общей чертой является способ-
ность координировать субстрат в процессе катализа.
Процессы активации молекулярных кислорода и азота обсуж-
даются в гл. 20 и 23 соответственно.
8. РАЗНЫЕ РЕАКЦИИ
К числу таких реакций можно отнести некоторые общие про-
цессы: 1) взаимодействие кислоты Льюиса с л-электронной аро-
матической системой и 2) фотохимические реакции комплексов.
Существует также большое число разнообразных процессов, пред-
ставляющих те особые случаи, когда уникальные или необычные
свойства одного или нескольких лигандов, сочетаясь с особыми
свойствами катиона (размер, окислительный потенциал, стерео-
химия), приводят к необычным реакциям.
Активация малых молекул при координации с металлами 439
Взаимодействие кислот Льюиса с ароматическими системами
хорошо известно; много довольно устойчивых аддуктов выделено
и охарактеризовано [62], например С6Н6-А12Вг6, (СН3)3С6Н3-
• GaBr3, C6H6-3TiC14, C6H6-2SbCl3 и (СН3)2СбН4-А1Вг3. Кислоты
Бренстеда образуют с ароматическими системами аналогичные
аддукты [62]. Структуры этих соединений в большинстве случаев
неизвестны. Для ряда известных структур трудно объяснить при-
роду связи. Так, оказывается, что в С6Н6А12Вг6 молекула бензола
связана с димером бромистого алюминия через атомы брома [63].
В CgHgCuAlCh бензол связан с атомом Сп(1), остальные коорди-
национные места которого заняты ионами хлора группы А1С1Г
[64]. В этом случае атом меди связан только с одним ароматиче-
ским кольцом в отличие от ионов Ag(I) в CeHe-AgClC^, связыва-
ющих два бензольных кольца [65]. Подобного типа соединения
участвуют в каталитических реакциях ароматических колец в не-
водных растворителях [62]. Образование этих соединений во мно-
гих случаях происходит довольно легко, без труда выделяются и
твердые аддукты. Исходные ароматические соединения, если они
не подверглись каким-либо изменениям, могут быть регенерирова-
ны путем обработки подобных аддуктов водой.
Фотохимические реакции комплексов обычно зависят от спо-
собности иона металла поглощать свет. Сюда относятся много-
численные реакции переноса электрона [66, 67], например:
[C^NH^I]2* + hv ----> [Co(NH3)6]2+ -f-1-
[Ce(H2O)x]4+ + hv --> Ce3+(aq) + H+ + -OH
[FeSCN]2+(aq) + hv -> Fe2+(aq) + -SCN
а также реакции с образованием гидратированного электрона
М*(Н£>)(,+Лу -—> М^(НаО)у+е(Н2О)х
который затем, реагируя с Н+, образует Н- [68].
Другим аспектом использования фотохимии может служить
фотохимическая изомеризация высших олефинов:
Нез(СО)12
октен-1 ——> изомеры [69]
Реакции, в которых особая комбинация лигандов и металла
приводит к необычным эффектам, открывают путь для выявления
колоссальных возможностей металлоорганического катализа. Од-
ним из примеров может служить цинковый комплекс пиридинкар-
440
Глава 13
боксальдоксима (I), катализирующий гидролиз 8-ацетоксихино-
лин-5-сульфоновой кислоты (II).
II
В этой реакции I и II образуют смешанный комплекс и ацетиль-
ная группа быстро переносится на кислород оксима, а затем гид-
ролитически отщепляется. Весь процесс, хотя он и протекает в
две стадии, идет значительно быстрее, чем некаталитический гид-
ролиз.
Другие процессы, более тесно связанные с ферментативными,
включают реакции, в которых комплекс металла образуется вбли-
зи гидрофобной «впадины» и ускоряет гидролитическое расщепле-
ние молекулы субстрата, находящейся в этой впадине [71]. Ис-
ходным соединением для такого рода процесса может служить
циклоамилоза — тороидальный полисахарид, содержащий в гид-
рофобном центре углеводородные группы. Гидроксильная группа
у открытой части тороида этерифицируется пиридин-2,5-дикарбо-
новой кислотой и продукт образует комплекс с Ni(II). Затем об-
разуется смешанный комплекс с пиридинкарбоксальдоксимом. При
добавлении к этому раствору n-нитрофенилацетата фенильная
группа располагается в гидрофобном центре, а металлокомплекс-
ная группа у открытой части тороида катализирует отщепление
ацетильной группировки. В этом примере ион металла использует-
ся только как катализатор и не участвует в связывании субстрата
и фермента. Возможно, эта модель ближе к некоторым фермен-
тативным реакциям, чем модель, допускающая использование
иона металла для обеих целей.
9. ЗАКЛЮЧЕНИЕ
Основные эффекты координации в реакциях лигандов базиру-
ются на том факте, что образование новых связей изменяет реак-
ционную способность исходных соединений. Если проследить, как
изменяются свойства координационных центров при переходе от
простых ионов, таких, как Mg(II) или Са(П), к ионам, свойства
Активация малых молекул при координации с металлами 441
которых резко изменены из-за присутствия лигандов в координа-
ционной сфере (например, железо в гемоглобине, кобальт в вита-
мине В12 и магний в хлорофилле), то становится ясно, что коорди-
национные центры могут взаимодействовать с разнообразными
орбиталями лигандов, от заполненных орбиталей с низкой энер-
гией до незаполненных орбиталей с высокой энергией. Практиче-
скими следствиями такого взаимодействия являются различные
эффекты, начиная от большей легкости нуклеофильной атаки,
когда лиганды координируются с катионом, до более сложных
взаимодействий, происходящих при ферментативной фиксации
азота с учатием комплексов железа и молибдена. Число типов
таких реакций ограничивается лишь изобретательностью человека
и природы в поиске новых типов координации для разнообразных
лигандов.
СПИСОК ЛИТЕРАТУРЫ
1. Pearson R. G., J. Am. Chem. Soc., 85, 3533 (1963); Science, 151, 172 (1966);-
Chem. Brit., 3, 103 (1967).
2. Meerwein H„ Ann. Chem. (Liebigs), 455, 227—253 (1927); Schriften Konigsber-
ger Geiehrten Ges. Naturiw. KI., 3, 129—166 (1927).
3. Werner A., New Ideas on Inorganic Chemistry, Longmans-Green, New York,
1911, p. 201.
4. Pedersen K- J., Kgl. Danske Vetensk. Akad. Math.-Phys. Medd., 20, (7), 20
(1943).
5. Bag S. P., Fernando Q., Freiser H., Inorg. Chem., 1, 887 (1962).
6. Hanania G. I. H., Irving D. H., J. Chem. Soc., 1960, 2745, 2750.
7. Smith E., in W. D. McElroy, B. Glass (eds.), The Mechanism of Enzyme Action,
The Johns Hopkins University Press, Baltimore, Md, 1954, p. 309.
8. Hellerman L., Perkins M. E., J. Biol. Chem., 112, 175 (1935); Hellerman L^,
Stock С. C., J. Biol. Chem., 125, 771 (1938); Hellerman L., Physiol. Rev., 17,
454 (1937).
9. Wicker W., Thilo E., Z. Anorg. Allgem. Chem., 306, 48 (1960); 313, 296 (1961);
Thilo E., Von Lampe F., Z. Anorg. Allgem. Chem., 349, 1 (1967).
10. Bender M., Adv. Chem. Ser., 37, 19 (1963).
11. Gibbs W., Genth F. M., Am. J. Sci., 23, (2), 241 (1857); 24, (2), 84 (1857).
12. Zsindely S., Pungor E., Mikrochim. Acta, 2, 209 (1963).
13. Beck M. T„ Kling O., Acta Chem. Scand., 15, 453 (1963).
14. Jones M. M., Johnston D. O., Barnett C. J., J. Inorg. Nucl. Chem., 28, 1927
(1966).
15. Laidler K. J-, Reaction Kinetics, Vol. 2, Pergamon Press, Oxford, 1962, p. 6.
16. Westheimer F. H., Trans. N. Y. Acad. Sci., 18, 15 (1955).
17. Conley H. L., Jr., Martin R. B., J. Phys. Chem., 69, 2914 (1965).
18. Kroll H., J. Am. Chem. Soc., 74, 2036 (1952).
19. Jones M. M., Ligand Reactivity and Catalysis, Academic Press, New York, 1968,
pp. 34 et seq.
20. Busch D. H., Alexander M. D., J. Am. Chem. Soc., 88, 1130 (1966).
21. Hix J. E., Jr., Jones M. M., Inorg. Chem., 5, 1863 (1966).
22. Regardh C. G., Acta Pharm. Suedica, 3, 101 (1966).
23. Hix J. E., Jr., Jones M. M., J. Am. Chem. Soc., 90, 1723 (1968).
24. Bamann E., Haas J. G., Trapmann H., Arch. Pharm., 294, 569 (1961).
25. Collman J. P., Buckingham D. A., J. Am. Chem. Soc., 85, 3039 (1963).
442
Глава 13
26. Ватапп Е„ Mutterlein W. P„ Chem. Ber., 91, 471, 1322 (1953).
27. Oestreich С. H., Jones M. M„ Biochemistry, 5, 2926 (1966).
28. Hofstetter R., Martell A. E„ J. Am. Chem. Soc., 81, 4461 (1959).
29. Clark H. T„ Dreger E. E., Org. Syntheses, 1, 87 (1941).
30. Hartman W. W., Dickey J. B., J. Am. Chem. Soc, 55, 1228 (1933).
31. Brockmann W., Smit P. J., Rec. trav. chim., 82, 757 (1963).
32. Butt V. S., Hallaway M., Arch. Biochem. Biophys. 92, 24 (1961).
33. Henry P. M„ Adv. Chem. Ser., 70, 127 (1968); also contains a discussion of
the previous literature.
34. Flynn J. H„ Hulburt H. M., J. Am. Chem. Soc., 76, 3393, 3396 (1954).
35. Boisonnas R. A., Adv. Org. Chem., 3, 159 (1963).
36. McOmie J. F„ Adv. Org. Chem., 3, 191 (1963).
37. Kurtz A. C., J. Biol. Chem., 122, 477 (1938); J. Biol. Chem., 140. 705 (1941).
38. Neuberger A., Sanger F„ Biochem. J., 37, 515 (1943).
39. Synge R. L. M., Biochem. J., 42, 99 (1948).
40. Kurtz A. C., J. Biol. Chem., 180, 1253 (1949).
41. Wolf D. E., Valliant J.. Peck R. L„ Folkers K-, J. Am. Chem. Soc., 74, 2002
(1952).
42. Smith L. H„ J. Am. Chem. Soc., 77, 6691 (1955).
43. Turba F„ Schuster K. H„ Z. Physiol. Chem., 283, 27 (1948).
44. Hanby W. E., Wales S. G., Watson J., J. Chem. Soc., 1950, 3241.
45. Ikutani Y., Okuda T., Sato M., Akabori S., Bull. Chem. Soc. Japan, 32, 203
(1959).
46. Ikutani Y., Okuda T„ Akabori S., Bull. Chem. Soc. Japan, 33, 582 (1960).
47. Okawa K., Sato M., англ. пат. 814063, 1959; Chem. Abstr., 54328g.
48. Gillard R. D., Mitchell P. R., Payne N. C., Chem. Commun., 1968, 1150.
49. Krause R. A., Goldby S. D., Adv. Chem. Ser., 37, 143 (1963).
50. Vaska L., Accounts Chem. Res., 1, 335 (1968).
51. Collman J. P., Accounts Chem. Res., 1, 136 (1968).
52. Allen A. D., Bottomley F., Accounts Chem. Res., 1, 360 (1968).
53. McGinnety J. A., Mays M. J., Rep. Progr. Chem., 65A, 353 (1969).
54. Homogeneous Catalysis, Adv. Chem. Ser., 70 (1968).
55. Homogeneous Catalysis with Special Reference to Hydrogenation and Oxidation,
Discuss. Faraday Soc., 1968, 46.
56. Otsuka S., Nakamura A., Tatsuno Y., J. Am. Chem. Soc., 91, 6994 (1969).
57. Otsuka S., Nakamura A., Yoshida T„ J. Am. Chem. Soc., 91, 7198 (1969).
58. Basolo F., Pearson R. G„ Mechanisms of Inorganic Reactions, John Wiley and
Sons, New York, 2nd edn., 1967, pp. 587—596.
59. Wender I., Pino P., Organic Syntheses via Metal Carbonyls, Vol. I, Interscience,
New York, 1968.
60. Collman J. P., Roper W. R., Adv. Organometall. Chem., 7, 53—94 (1968).
61. Halpern J.. Adv. Chem. Ser., 70, 7 (1968).
62. Olah G. A., Meyer M. W., in G. A. Olah (ed.), Friedel-Crafts and Related
Reactions, Vol. 1, John Wiley and Sons (Interscience, New York, 1963, p. 710.
63. Eley D. D., Taylor J. H., Wallwork S. C., J. Chem. Soc., 1961, 3867.
64. Turner R. W., Атта E. L„ J. Am. Chem. Soc., 85, 4046 (1963).
65. Smith H. G., Rundle R. E„ J. Am. Chem. Soc., 80, 5075 (1958).
66. Adamson A. W„ Coord. Chem. Rev., 3, 169 (1968).
67. Adamson A. W., Waltz W. L., Zinato E., Watts D. W., Fleischauer P. D., Lind-
holm R. D., Chem Rev., 68, 541 (1968).
68. Hart E. J., Solvated Electron Adv. Chem. Ser., 1965, 50.
69. Carr M. D., Капе V. V., Whiting M. C., Proc. Chem. Soc., 1964, 408.
70. Breslow R., Chipman D., J. Am. Chem. Soc., 87, 4196 (1965).
71. Breslow R., Overman L., J. Am. Chem. Soc., 92, 1076 (1970).
ГЛАВА 14
МЕТАЛЛОФЕРМЕНТЫ
М. С. Скраттон
Scrutton М. С., Department of Biochemistry, Rutgers Medical
School, Rutgers University, The State University of New
Jersey, New Brunswick, New Jersey 08903, USA
1. ВВЕДЕНИЕ
1.1. Роль ионов металлов в механизме
каталитического действия ферментов
Согласно Ленинджеру [1], примерно одна треть описанных к
1950 г. ферментов требовала для проявления своей максимальной
активности либо добавления иона металла, либо эти ферменты
содержали прочно связанный ион металла, по-видимому, прини-
мающий участие в каталитических процессах. С тех пор примене-
ние усовершенствованных методов анализа металлов в биологи-
ческих системах позволило выявить еще большое число фермен-
тов, относящихся к классу металлопротеинов. Кроме того, уста-
новлена ферментативная активность ряда металлопротеинов, для
которых ранее было показано наличие металла [2].
Целью настоящей главы является выяснение роли металлов
в ферментативном катализе, описание и оценка некоторых мето-
дов, применяемых для изучения роли металлов в ферментативных
реакциях, а также рассмотрение современных представлений об
участии ионов металлов в механизмах ферментативного катализа
дегидрогеназами, карбоксилазами и декарбоксилазами, изомераза-
ми, лиазами, альдолазами, трансферазами и синтетазами. После-
дующие главы этой части будут посвящены роли ионов металлов
в ферментативном катализе пептидазами (гл. 15), карбоангидра-
зами (гл. 16), киназами (гл. 17) и фосфатазами (гл. 18).
Мы не будем проводить здесь различий между металлофер-
ментами (ферментами, содержащими один или более прочно свя-
занных ионов металла) и металлактивируемыми ферментами (для
проявления максимальной каталитической активности которых
необходимо добавление ионов металла). Малмстрём и Розенберг
[3] подчеркивают, что во многих случаях не существует корреля-
ции между каталитической ролью иона металла и его сродством
к апоферменту. Из этого вытекает, что попытки разрешить проб-
лему, исходя из различия в сродстве ферментов к ионам метал-
лов, могут лишь сделать неясной сущность данного явления на
функциональном уровне [4, 5]. Например, фермент с резко выра-
444
Глава 14
женной специфичностью к активации ионом металла может вести
себя как металлопротеин в присутствии этого катиона, обладаю-
щего высоким сродством к апоферменту, но проявлять себя как
металлактивируемая система в присутствии более слабо связыва-
ющегося иона металла [6]. Изменения pH также могут вызывать
изменения в сродстве данного иона металла. Иногда, например
Рис. 14.1. Схемы взаимодействия фермента (Е) с ионом металла (Мх+) и лиган-
дом (L).
в случае р-метиласпартазы [7], изменения pH вызывают превра-
щение металлофермента в металлактивируемую систему. Однако
непохоже, что в этих условиях происходят существенные измене-
ния в механизме катализа. Было высказано предположение, что
константы устойчивости комплекса фермент—ион металла поряд-
ка 107—108 М-1 могут рассматриваться в качестве границы между
собственно металлоферментами и металлактивируемыми фермен-
тами [8].
Функции, которые выполняют ионы металлов в ферментатив-
ном катализе, проиллюстрированы на рис. 14.1, где L (лиганд) —
субстрат, активатор или ингибитор фермента, Мж+ — ион металла
и Е — фермент.
Металлоферменты
445
1.2 Комплексы с лигандом в качестве мостика*
На рис. 14.1,1 изображен комплекс с лигандом в качестве
мостика (мостиковый лиганд). В этом комплексе ион металла свя-
зан только с лигандом и участвует в катализе, не взаимодействуя
непосредственно с ферментом. Комплексы с мостиковыми лиган-
дами характерны для многих ферментов, взаимодействующих с
комплексами М—НДФ- и М—НТФ2-, например для креатин-
киназы [9, 10], аргининкиназы [11], аденилаткиназы [12]
и тетрагидрофолатсинтетазы [13]. Описанию этих и других
киназ посвящена гл. 18. В двойном комплексе М2+* с АТФ
и АДФ как фосфатные группы, так и атомы азота пуринового
кольца образуют координационные связи с ионом металла [14,
15] (ср. гл. 33). В большинстве случаев АДФ3-— (или АТФ4-)
имеет примерно такое же сродство к ферментам, как и активный
комплекс М2+ — нуклеотид, но действует как ингибитор реакции
[16]. Следовательно, роль иона металла в подобных комплексах
с мостиковым субстратом может заключаться в активации атома
фосфора, по которому протекает атака. Исключение, по-видимому,
составляет тетрагидрофолатсинтетаза [13] (разд. 12.2). Приме-
ров мостиковых комплексов с ингибитором (активатором) в каче-
стве мостика гораздо меньше, хотя влияние металлов на актив-
ность дТТФ (ингибитор) и дЦТФ (активатор) для дЦМФ-деами-
назы может быть объяснено образованием комплексов с мостико-
выми лигандами, так как эффекты активации и ингибирования
проявляются в присутствии Са2+, Mg34' и Мп2+ [17] (разд. 2.4).
1.3. Комплексы с металлом в качестве мостика
Еще одна возможность участия ионов металлов в ферментатив-
ном катализе заключается в образовании таких мостиковых ком-
плексов, в которых центром связывания лиганда является пол-
ностью (рис. 14.1, II, А), либо частично (рис. 14.1, II, Б) ион
металла. Существование мостикового комплекса фермент — ме-
талл— субстрат впервые предположил Хеллерман [18] для объ-
яснения данных, полученных при активации аргиназ ионами ме-
таллов [19], а также для описания медленной металлзависимой
активации некоторых пептидаз [20]. Впоследствии концепция
мостиковых металлов была применена для объяснения металлза-
висимой активации многих ферментов, но до последнего времени
* Применяемые сокращения: СРП — скорость релаксации протонов (скорость
продольной ядерной магнитной релаксации протонов воды); ФЕП — фосфоенол-
пируват; М2+ — ион двухвалентного металла; М+ — ион одновалентного металла;
НМФ, НДФ и НТФ — нуклеозидмоно-, нуклеозидди- и нуклеозидтрифосфат;
ТДФ — тиаминдифосфат; ФДФ — фруктозо-1,6-дифосфат, тРНК или транспорт-
ная РНК — РНК, которая несет антикодоны.
446
Глава 14
не было прямых доказательств существования комплексов с ме-
таллом в качестве мостика. Техника ядерного магнитного резо-
нанса (ЯМР), позволяющая проследить за внутренней координа-
ционной сферой некоторых парамагнитных ионов металлов, на-
пример Мп2+, применена и для изучения комплексов с мостиковым
металлом [21]. Первые исследования, в которых изучалась ско-
рость ЯМР-релаксации субстрата, однозначно доказали взаимо-
действие Мп2+ и фторфосфата с пируваткиназой из мышц кроли-
ка. Было показано, что при этом происходит образование мостико-
вого комплекса пируваткиназа — Мп2+ — фторфосфат. Свойства
этого комплекса согласуются с его предполагаемой ролью в ката-
лизе [22] (гл. 18). Впоследствии применение этого же метода
позволило установить существование комплексов фермент — ме-
талл — субстрат, фермент — металл — продукт или фермент — ме-
талл— ингибитор еще для семи ферментов [8]. Эти исследования
будут детально рассмотрены в разделах, посвященных отдельным
ферментам (разд. 3—10) и в гл. 18.
Существование комплексов с мостиковым металлом также про-
демонстрировано с помощью рентгеноструктурного анализа (кар-
боксипептидаза) [23] (гл. 15) и инфракрасной спектроскопии
(карбоангидраза) [24] (гл. 16). Однако данные, доказывающие
существование мостиковых комплексов, полученные с помощью
этих методов, не позволяют установить, участвуют ли такие ком-
плексы в катализе. Выяснить это можно сравнением кинетических
и термодинамических свойств мостикового комплекса с аналогич-
ными параметрами, полученными при изучении самих процессов
катализа. В связи с этим метод ЯМР имеет существенные преиму-
щества перед другими методами (разд. 2.3). Однако важность
этого критерия не может быть преувеличена, поскольку в одном
случае был обнаружен комплекс с мостиковым металлом, не при-
нимающий участия в катализе [25].
Во всех мостиковых комплексах фермент — металл — лиганд
ион металла благодаря своим уникальным координационным
свойствам играет важную роль во взаимодействии белок — ли-
ганд. Однако обычно предполагается, что ион металла в комплек-
сах фермент — металл — субстрат оказывает также и каталитиче-
ское влияние [8]. Предположение о каталитической роли часто
вытекает из рассмотрения катализа ионами металла в модельных
системах, которые лишь имеют сходство с биологическими реак-
циями [8, 26, 27]. Хотя предположение о каталитическом участии
ионов металла и весьма привлекательно, однако убедительные
доказательства их каталитической роли в биологических системах
были получены лишь в нескольких случаях. Итак, роль иона ме-
талла в связывании и в катализе в биологических системах не
легко разделить, а модельные исследования обладают лишь неко-
торой степенью приближения.
Металлофер менты
447
Часто ион металла, весьма эффективный при катализе в мо-
дельных системах, в биологической реакции либо неактивен, ли-
бо малоэффективен. Такая ситуация хорошо исследована для ре-
акций декарбоксилирования оксалоацетатов [28, 29] и гидролиза
пептидных связей [30]. Более того, хотя катализ в биологических
системах и должен подчиняться основным физическим и химиче-
ским законам, наличие третичной и четвертичной структур белков
может стабилизировать такие комплексы и способствовать таким
эффектам, например прямому переносу протона [31], которым
трудно найти аналогии в модельных системах. Эти положения
проиллюстрированы Вангом и сотрудниками на примере механиз-
мов, предложенных для химотрипсина [32] и карбоангидразы
[33]. Расхождение между Вангом и Каплоу во взглядах на воз-
можную роль белкового фермента отражено в дискуссии в гл. 16,
посвященной карбоангидразе [33]. Таким образом, хотя модель-
ные системы во многих случаях и дают полезную информацию,
необходимо соблюдать осторожность при экстраполяции этих
данных на каталитическую роль иона металла в комплексах с
мостиковым металлом.
Вейлли и Уильямс [34] предположили, что окружение иона ме-
талла в ферменте может создавать «энтатное» состояние в отсут-
ствие субстрата. «Энтатное» состояние определяется как «сущест-
вование в ферменте группировок с энергией, более близкой к энер-
гии мономолекулярного переходного состояния, чем к энергии
обычной стабильной молекулы, и, таким образом, образующих
энергетически богатую область» [34]. В случае металлоферментов
этот постулат основывается на изменении абсорбции, электронно-
го парамагнитного резонанса (ЭПР) и (или) спектров дисперсии
оптического вращения (ДОВ) и окислительно-восстановительных
потенциалов некоторых медь-, железо- и кобальтсодержащих фер-
ментов, для которых не найдено объяснения при сравнении их
свойств со свойствами обычных комплексов этих металлов с ма-
лыми молекулами или комплексов металл — белок, не обладаю-
щих каталитической активностью (см. библиографию в статье
Вейлли и Уильямса [34]). Однако «энтатные» спектры поглоще-
ния, полученные для ряда кобальтсодержащих ферментов, во мно-
гом походят на спектры поглощения комплексов, содержащих пя-
тикоординационный Со2+ [37а, б]. Нет также полной корреляции
между аномальными спектрами или окислительно-восстановитель-
ными свойствами, которые могут быть следствием асимметричной
или искаженной координации [34], и наблюдаемой каталитиче-
ской активностью. Так, например, Со2+-комплексы пируваткиназы
[35] и пируваткарбоксилазы [36] каталитически активны, но
имеют спектр, характерный для октаэдрических комплексов Со24".
Определение скоростей обмена лиганда на связанных с белком
ионах металлов также не дает убедительных доказательств нали-
448
Глава 14
чия необычных кинетических свойств, которые могли бы быть ха-
рактерны для активированного состояния [8, 37]. Однако до сих
пор не проводились кинетические исследования металлофермен-
тов, имеющих спектральные или окислительно-восстановительные
свойства, характерные для «энтатного» состояния.
Существуют по меньшей мере два пути образования комплек-
сов с мостиковым металлом в тех случаях, когда металл не обра-
зует предварительно прочной связи с ферментом (рис. 14.1):
Е + М2+ >- Е — М2+ + лиганд < > Е — М2+ — лиганд (1)
Е + лиганд ч. >. Е — лиганд + М2+ < > Е — М2+ — лиганд (2)
Возможен и третий путь, если несвязанный ион металла проявля-
ет значительное сродство к лиганду:
М2+ + лиганд < > М2+ — лиганд Е < > Е — М2+ — лиганд (3)
Чтобы определить путь, по которому протекает на самом деле об-
разование мостикового комплекса, необходимо исследовать на-
чальную скорость реакции и скорости связывания лиганда и ме-
талла, причем образование мостикового комплекса фермент — ме-
талл — лиганд должно протекать в кинетически контролируемой
стадии. Однако до сих пор лишь для немногих систем проведено
столь детальное кинетическое исследование. Для пируваткиназы
из мышц [35, 38, 39] данные, описывающие механизм образова-
ния комплексов мостиковых металлов с ФЕП и АДФ, хорошо
согласуются со всеми тремя возможными путями их образования
(ср. гл. 18). Эти данные противоречат предположению [16] о том,
что все комплексы нуклеотидов с ферментами, в которых прини-
мают участие ионы двухвалентных металлов, образуются в ре-
зультате взаимодействия фермента с комплексом металл — лиганд
[уравнение (3)].
1.4. Комплексы с ферментом в качестве мостика
Ион металла может также взаимодействовать с ферментом не
по тому месту, по которому связывается лиганд. Это взаимодей-
ствие может вызывать изменение в свойствах каталитического
центра (или центра связывания лиганда). В таких комплексах
фермент играет роль мостика между лигандом и металлом
(рис. 14.1, III). В противоположность комплексам с мостиковыми
металлами или мостиковыми лигандами (разд. 1.2 и 1.3) в случае
этих комплексов уделялось мало внимания механизмам с их уча-
стием, хотя многие эффекты иона металла могут быть объяснены
образованием комплексов с мостиковым ферментом. Например,
действие ионов одновалентных металлов на каталитические свой-
ства многих ферментов обусловлено стабилизацией каталитически
активного состояния путем образования комплекса с мостиковым
ферментом [40—42] (было предложено также и другое объясне-
Металлоферменты
449
ние [43]). Аналогичным образом восстановление активности глут-
аминсинтетазы из Escherichia coli при действии ионов некоторых
двухвалентных металлов, возможно, обусловлено стабилизацией
активной конформации этого фермента [44]. Образование ком-
плексов с мостиковым ферментом часто сопровождается измене-
ниями в параметрах, которые отражают конформацию данного
фермента [44 -46]. Однако эти эффекты не характерны для об-
разования только комплексов с мостиковым ферментом, поскольку
при образовании комплексов с мостиковым металлом взаимодей-
ствие металл — фермент также вызывает сходные изменения в па-
раметрах, отражающих конформацию белка [45].
Следует отметить, что для работы многих ферментов требуется
несколько ионов металлов, что может означать участие в меха-
низме катализа сразу нескольких типов мостиковых комплексов.
Так, например, для ацетил-КоА-синтетазы необходимы три иона
металла [47, 48].
2. ЭКСПЕРИМЕНТАЛЬНЫЕ ПОДХОДЫ К ИЗУЧЕНИЮ
МЕТАЛЛОФЕРМЕНТОВ
2.1. Общее рассмотрение
Многие из общих подходов к исследованию механизма дейст-
вия ферментов также применимы и к изучению роли ионов ме-
таллов в ферментативном катализе. Схемы координации, описы-
вающие взаимодействие фермента, металла и лиганда, могут быть
изучены методами, применяемыми при определении стехиометрии
и сродства связывания белками небольших молекул. Эти методы
включают гель-фильтрацию в присутствии или в отсутствие не-
больших молекул [49], метод скоростного диализа [50], ультра-
фильтрацию, метод ультрацентрифугирования по Хейесу — Вели-
ку [52], равновесный диализ [53], а также методы для измерения
только сродства взаимодействия [54—58]. Выбор схемы коорди-
нации ионов металлов и лигандов с ферментами с помощью этих
методов возможен только при отсутствии влияния других факто-
ров. Например, если образуется комплекс Е — лиганд — М2+, фер-
мент должен проявлять значительное сродство к иону металла
только в присутствии лиганда. И, наоборот, если образуется ком-
плекс Е — М2+ — лиганд, то не должно происходить значительно-
го связывания лиганда в отсутствие иона металла. Однако прак-
тически ферменты часто проявляют склонность к связыванию обо-
их компонентов комплекса, невзирая на выбранную схему коорди-
нации. Следовательно, важны данные, полученные с учетом сте-
хиометрических и кинетических критериев. Такие важные типы
комплексов, как Е — лиганд — М2+ и Е — М2+ — лиганд, обычно
содержат все три компонента в эквимолярных количествах. Более
29—2451
450
Глава 14
того, если данный комплекс важен при катализе, то константы
диссоциации, измеренные при изучении связывания, должны при-
ближаться к константам, определенным при изучении начальной
скорости реакции в целом, хотя различие в используемых концен-
трациях белка может вызвать в некоторых случаях трудности при
интерпретации. Необходимые константы могут быть получены из
изучения начальной скорости для иона металла (К л) и для лиган-
да, если он является ингибитором (Кг)- Однако константы диссо-
циации комплексов фермент — субстрат и фермент — продукт не
так легко получить, пока не сделаны допущения относительно
скоростьопределяющей стадии реакции и (или) порядка присоеди-
нения субстрата (или отщепления продукта). Следовательно,
утверждение о кинетической важности таких комплексов, наибо-
лее интересных с точки зрения изучения механизма действия ме-
таллоферментов, связано с большими сложностями.
Для выяснения роли ионов металлов в ферментативном ката-
лизе было изучено изменение начальных скоростей реакции как
функции концентрации субстрата и иона металла. Хотя Малмст-
рём и Розенберг [3] показали, что реакции образования комплек-
сов Е — М2+ — субстрат и Е — субстрат — М2+ для односубстрат-
ных систем имеют сходные кинетические уравнения, в случае мно-
госубстратных систем можно определить порядок при-
соединения иона металла и субстрата по методу Клеланда [59,
60] с введением соответствующих поправок на взаимодействие
металл — субстрат*. Некоторые примеры с использованием этого
подхода будут обсуждаться в следующих разделах (см. также
гл. 18). В этой связи необходимо заметить, что часто неудовлет-
ворительной является практика поддержания постоянной и насы-
щающей концентрации иона металла в ходе изучения кинетики
при выяснении влияния порядка присоединения субстрата и (или)
отщепления продуктов. Во многих системах незакомплексованный
М2+ действует как активатор или как ингибитор реакции. Более
того, в случае слабых комплексов, например А^АДФ-, относи-
тельная концентрация промежуточных соединений является функ-
цией общей концентрации обоих компонентов комплекса [16].
Наконец, схема координации при взаимодействии ферментов
с ионами металлов и лигандами может быть изучена с помощью
рентгеноструктурного анализа, хотя до сих пор этим методом бы-
ла исследована детально только карбоксипептидаза [23]. Однако
наряду с достоинствами этот метод обладает и рядом недостатков.
Во-первых, рентгеноструктурный анализ можно применять для
* В настоящее время нет хороших обзоров по всем аспектам ферментативной
кинетики. Простую обработку кинетических данных обычных субстратных систем,
куда входит определение Км (константы Михаэлиса) и Kt (константы ингибиро-
вания), можно найти в статье [60а]. Кинетика сложных субстратных систем
лучше всего описана в статьях [59, 60, 606].
Металлоферменты
451
изучения только статического связывания и с его помощью нельзя
оценить кинетическую значимость исследованного комплекса. Во-
вторых, в настоящее время рентгеноструктурные исследования
ограничены изучением комплексов ферментов с самыми простыми
субстратами или ингибиторами. Хотя структуры таких комплек-
сов достаточно интересны, выводы о нормальной каталитической
функции фермента, основанные на изучении комплексов с просты-
ми субстратами, могут оказаться неправильными, поскольку в ре-
зультате изменения характера комплексообразования может на-
блюдаться изменение в каталитической активности. Аналогичные
рассуждения справедливы и при изучении взаимодействия лиган-
дов с ферментсвязанным ионом металла методом инфракрасной
спектроскопии [24].
Все методы, которые мы обсудили, применимы для изучения
механизмов действия любых ферментов независимо от того, вклю-
чают они ион металла или нет. Однако имеются три метода, более
широкое применение которых в изучении металлоферментов осно-
вано на уникальных свойствах ионов металлов. Эти методы будут
рассмотрены более детально, а именно: 1) исследование спектров
ЭПР; 2) измерение парамагнитного вклада в скорости ядерной
магнитной релаксации магнитных ядер (например, протонов) в
лигандах; 3) изучение замены одного металла на другой.
2.2. Электронный парамагнитный резонанс
Теоретические основы применения ЭПР-спектроскопии для ис-
следования окружения парамагнитных ионов металла по характе-
ру влияния поля лиганда на неспаренные электроны были описа-
ны ранее [62]. В данном разделе обсуждается применение метода
ЭПР для изучения металлоферментов.
Во-первых, сверхтонкая структура ЭПР-спектра при высокой
степени разрешения, какая, например, была получена для ком-
плексов Fe3+ и Си2+, может экспериментально идентифицировать
природу и число связанных лигандов [63]. Эффективность метода
может быть повышена включением магнитных ядер в лигандные
группы белка. Возможности подобного изотопного замещения бы-
ли проиллюстрированы изучением ЭПР-спектров негемовых желе-
зосодержащих белков [64, 65], в которых атом 32S был заменен
на 33S и 77Se. Спин-спиновое взаимодействие дало сверхтонкое
расщепление, которое позволило довольно точно определить при-
роду лигандов, поскольку в данном случае тонкое спин-спиновое
взаимодействие могло протекать только через химические связи
[66].
Аналогичный подход может быть применен и для того, чтобы
установить существование комплекса Е — М2+ — лиганд, если
в лиганде присутствуют магнитные ядра. Так, этим методом бы-
ла показана непосредственная координация 19F~ с железом в мет-
29*
452
Глава 14
миоглобинфториде [67]. И, наоборот, отсутствие ожидаемого
сверхтонкого расщепления ЭПР-сигналов исключает металл-ли-
гандное взаимодействие в комплексе с ферментом [68]. Попытки
распространить этот метод на ферменты, содержащие другие ме-
таллы, например Мп2+, оказались неудачными из-за сложности
полученных ЭПР-спектров и уменьшения степени разрешения
спектра при комплексообразовании. Однако недавно для
Мп2+-комплекса конканавалина А [69] были получены хорошо
разрешенные ЭПР-спектры, что показывает возможность исполь-
зования ЭПР-спектров этого иона металла для исследования вы-
сокомолекулярных комплексов.
Во-вторых, для изучения схемы координации может быть ис-
пользована чувствительность амплитуд ЭПР-спектров Мп2+ к его
окружению. Так, например, если образуется комплекс Е — Мп2+ —
лиганд, то наблюдаемая амплитуда комплекса Мп2+ — лиганд
должна уменьшиться при добавлении фермента. Если же ампли-
туда ЭПР-спектра комплекса Мп2+ — лиганд не изменяется при
добавлении фермента, в то время как другие данные показывают
образование тройного комплекса, то это означает, что образуется
комплекс Е — лиганд — Мп2+. Такая ситуация наблюдалась при
взаимодействии МпАДФ- с креатинкиназой [9].
В-третьих, введение в биологические системы стабильных ор-
ганических молекул, содержащих свободные радикалы (спиновые
метки) [71], расширяет возможности использования ЭПР для
изучения таких спин-меченых систем. Спиновые метки могут быть
присоединены к остатку аминокислоты в активном центре фер-
мента или около него [72], а также могут быть включены в ана-
лог субстрата [73]. В любом из этих случаев может быть оцене-
на степень иммобилизации спиновой метки, связанной с белком,
путем сравнения ЭПР-спектров для свободного и связанного со-
стояний, а также может быть изучено действие различных аген-
тов, например диамагнитного иона металла, на окружение спино-
вой метки [72]. В таких экспериментах спиновая метка действует
как детекторная группа [74] и обнаруживается с помощью спек-
тров ЭПР. Однако в присутствии парамагнитных ионов, например
Мп2+ и Со2+, спин-спиновые взаимодействия преобладают над
процессами релаксации и вызывают заметное уменьшение ампли-
туды ЭПР-сигналов, обусловленных спиновой меткой, поскольку
спины ведут себя так, как будто бы они зафиксированы в жесткой
решетке [74а]. Этот эффект позволяет рассчитать расстояние меж-
ду спинами с учетом времени корреляции диполь-дипольного взаи-
модействия [72, 74а]. Таким образом, использование специфич-
ных спиновых меток для различных аминокислотных остатков или
для различных участков активного центра делает возможным соз-
дание карты активных центров металлоферментов [746].
Металлоферменты
453
2.3. Парамагнитные эффекты на скорости ядерной
магнитной релаксации ядер лигандов
2.3.1. Общие положения
Этот сложный метод не будет детально обсуждаться в данном
разделе, однако некоторые его аспекты требуют более подробного
рассмотрения. Наиболее подробно метод обсуждается в обзорах
[75, 76, 76а], а также в гл. 18.
В присутствии некоторых парамагнитных ионов, например
Мп2+, наблюдается парамагнитный вклад в продольную (Г/Лр) и
поперечную (1/Т2р) скорости ядерной магнитной релаксации, на-
пример протонов воды [77, 78]:
1 Р9 , 1
Т1Р = Лм + тм + 1™ (4)
1 pg 1
где р — отношение концентрации парамагнитного иона и концен-
трации лиганда; q—-координационное число лиганда; 1/тм—-ско-
рость обмена лиганда на парамагнитном ионе; 1/7’1Лг(1/7’2лг) —
скорости релаксации ядра связанного лиганда; 1/Г Js (l/7gs)—
вклад в скорость релаксации, обусловленный диполь-дипольным
взаимодействием парамагнитного иона с внешней координацион-
ной сферой ядра (внешнесферная релаксация). Следует заметить,
что уравнение (5) имеет силу для Мп2+, но применимо не для всех
парамагнитных ионов [77]. Уравнения (4) и (5) содержат не-
сколько важных для анализа каталитической роли ионов метал-
лов параметров при условии их применимости для высокомоле-
кулярных комплексов. Во-первых, сравнением q для протонов во-
ды в комплексах Е—Мх+ и Е — Мх+ — субстрат определяют схе-
му координации при этих взаимодействиях. Во-вторых, сравнени-
ем 1/тм обмена субстрата в комплексе Е — Мх+— субстрат с чис-
лом оборотов фермента в катализе можно выяснить, достаточно
ли быстро происходят образование и диссоциация этого комплек-
са, чтобы он мог участвовать в каталитических актах. В-третьих,
можно установить, образуется ли комплекс Е — Мх+— лиганд,
рассчитав расстояние г между протонами лиганда и парамагнит-
ным ионом. Этот расчет можно провести исходя из 1/Т\м в случае,
если известна величина времени корреляции (тс), описывающая
это диполь-дипольное взаимодействие [79, 80]. И, наконец, из
значений \JT2M и \!Т\М могут быть получены константы сверх-
тонких взаимодействий с учетом величины времени корреляции
(тс), описывающей эти сверхтонкие взаимодействия [79, 81]. Этот
последний параметр исключительно важен для доказательства не-
посредственной координации, поскольку сверхтонкие взаимодей-
454
Г лава 14
ствия могут происходить только через химические связи [66]. Та-
ким образом, измеряя одновременно 1/Т1р и i/T2p, можно полу-
чить в одном и том же эксперименте как кинетические, так и
структурные данные при условии, что они оказывают доминирую-
щее влияние на релаксационные процессы. Однако получение по-
добной информации часто затруднено в связи с тем, что в первую
очередь необходимо установить, какие процессы оказывают основ-
ное влияние на скорость релаксации. Например, 1/Т1р может опре-
деляться как 1/Т1м, так и 1/тм или 1/Т^ , а в свою очередь
1/Т1м может содержать вклад l/rs (время релаксации спина элек-
трона), 1/т„ (вращательное время корреляции) или 1/тм [81]. Ес-
ли имеется вклад только одного процесса в скорости релаксации,
то обычно можно идентифицировать этот процесс изучением час-
тотной и температурной зависимостей наблюдаемых скоростей ре-
лаксации, хотя малая температурная устойчивость большинства
биологических систем ограничивает возможности изучения темпе-
ратурной зависимости. Милдван и Кон [76] суммировали в таб-
лицы возможное в этом случае поведение для ряда примеров. Од-
нако, если скорости релаксации отражают вклад более чем одно-
го процесса, вышеописанный анализ становится невозможным.
В плане исследований, проводимых в настоящее время, важно
подчеркнуть три аспекта. Во-первых, чтобы установить, что вклад
хм доминирует в скорости процесса релаксации, необходимо нали-
чие как отрицательной температурной зависимости 1/7'1г,, так и
равенства i/Tip и \1Т2р*. Доминирование ts в 1/Г]Р или в 1/72р
также может привести к возрастанию отрицательной температур-
ной зависимости скорости релаксации, но в этом случае 1/Г2р>
>1/71р [76, 82]. Во-вторых, если для протонов воды в ряде ком-
плексов Е — Мх+ — лиганд вклад хм доминирует в значениях
1/Г1Р или 1/72у, то относительные числа гидратации могут быть
определены сравнением 1/Т1р—(1/Т2р) при данной температуре,
только если для хм наблюдаются сходные энергии активации [82].
В-третьих, в большинстве случаев должны быть установлены вре-
мена корреляции диполь-дипольных взаимодействий (тс) между
ионом металла и магнитным ядром лиганда в комплексе Е—Мх+—
лиганд. Если в качестве магнитных ядер используются протоны, то
часто величины, полученные для расстояния металл — лиганд, не
дают возможности идентифицировать лигандные группы, хотя и
согласуются с возможностью прямой координации. Эта проблема
может быть решена прямым определением тс из частотной зави-
симости l/7ip и (или) 1/Г2р, однако наилучшим решением проб-
* Поскольку доминирование Тм в 1/Г2р может сохраниться и после исчезно-
вения доминирования для 1/Tip, этот критерий может быть сформулирован более
точно либо как равенство 1/Г2р и 1/7iP, либо как достижение равенства при
низкой температуре. Надо заметить, что такой подход применим только тогда,
когда металл обладает большим т«, например в случае Мп2+.
Металлоферменты
455
лемы является изучение парамагнитных эффектов на скорости
релаксации магнитных ядер (13С, 17О), которые близко располо-
жены к иону металла. В случае 13С такой подход облегчается на-
личием больших химических сдвигов, наблюдаемых для этого
ядра.
2.3.2. Явление усиления и его применение
В исследованных к настоящему времени методом ядерного маг-
нитного резонанса (ЯМР) биологических системах много инфор-
мации дало изучение явления усиления, открытого Эйзингером
и др. [81] для комплексов ДНК с некоторыми парамагнитными
ионами, например Мп2+, Сг3+ и Си2+, Кон и Леем [9, 21] для ком-
плексов различных ферментов с Мп2+ в присутствии или в отсут-
ствие субстратов. Фактор усиления для 1/7\ (ei) определяется из
следующего уравнения [81]:
61 “ 1/Лр “ 1/Л-1/Л(0)
где l/T^o) — скорость релаксации, наблюдаемая в отсутствие па-
рамагнитного иона, а звездочкой отмечены скорости релаксации,
измеренные в присутствии макромолекулы. Фактор усиления для
1/7'2(62) определяется аналогично. В том случае если 1/7’[ измере-
но для протонов воды (СРП), то для ионов металла, например
Мп2+, наблюдается величина ei больше 1,0, причем тс зависит от
времени вращательной корреляции хг [81]:
В данной ситуации тс определяется временем полного поворота
молекулы комплекса. Поскольку высокомолекулярный комплекс
гораздо медленнее вращается, чем гидратированный ион металла
(акво-катион), образование такого комплекса может привести к
ei>l,0 при условии, что увеличение вклада времени корреляции
превышает уменьшение вклада в результате замены ионов воды
в ходе комплексообразования. И хотя в отдельных случаях тг мо-
жет определять величину тс как для акво-катионов, так и для вы-
сокомолекулярных комплексов, в большинстве случаев понижение
тг при образовании высокомолекулярного комплекса может быть
таким, что ts или тм становятся определяющими в значении тс.
В таком случае, следовательно, наблюдаемая для протонов воды
величина ei не будет иметь большого значения. Аналогичные рас-
суждения можно привести и для фактора усиления скоростей ре-
лаксации протонов лиганда. Однако этот параметр может прине-
456
Глава 14
сти пользу в качественной оценке, во-первых, доступности иона
металла для растворителя и, во-вторых, того, какую из трех воз-
можных ролей, описанных в разд. 1, выполняет ион металла в
ферментативной реакции. Как установлено Кон [21], фактор уси-
ления (ei) протонов воды для бинарного комплекса Е — М2+ (еь)
может быть больше, чем ei для тройного комплекса Е — М2+ —
лиганд (тип II) (ес). И наоборот, ферменты, образующие комплек-
сы Е —лиганд — М2+ (тип I), проявляют небольшое взаимодейст-
вие фермент — ион металла (либо вообще его не проявляют) и
имеют величину ес>£ь~ 1,0, в то время как в комплексах М2+ —
Е — лиганд (тип III) лиганд может оказывать небольшое влияние
на окружение иона металла и Еь~ес. Хотя эти закономерности
наблюдались для большинства комплексов типов I и II [21], из-
вестны исключения. Изучением скоростей релаксации протонов
субстрата в присутствии Мп2+ — фермента для ФДП-альдолазы
из дрожжей доказано существование мостиковых комплексов
Е — Мп2+ — субстрат (разд. 9), хотя и наблюдались небольшие
изменения для Е[ протонов воды при образовании этих комплек-
сов (т. е. еь~ес). Следовательно, хотя сравнение величины ei про-
тонов воды для бинарных и тройных комплексов фермента, метал-
ла и лиганда дает простой и быстрый метод определения типа
образующегося комплекса, однако эти результаты должны рассмат-
риваться как предварительные и подтверждаться с помощью дру-
гих методов, например определением г и A/h (константы сверхто-
ного взаимодействия) путем измерения скоростей релаксации маг-
нитного ядра лиганда. Быстрый метод определения констант дис-
социации комплексов дает также наблюдение за изменениями ej
протонов воды при взаимодействии фермента с Мп2+ и лигандом
[21].
2.4. Замена металла на другой металл
В большинстве исследований по металлактивированным фер-
ментам делались попытки оценить специфичность взаимодействия
либо изучением степени активации фермента различными ионами
металлов в стандартных условиях, либо в ряде случаев измере-
нием кажущихся констант активации и максимальных скоростей
для тех ионов металлов, которые вызывают существенную акти-
вацию. Из этих исследований можно сделать некоторые обобще-
ния. Например, большинство киназ и синтетаз активируются в
значительной степени только под действием Mg2+, Мп2+, Со2+ и
в некоторых случаях Са2+, в то время как другие ферменты, на-
пример дегидрогеназы, лиазы, в большинстве случаев проявляют
более широкую специфичность по отношению к активации под
действием М2+. Подобные высокие степени активации под дейст-
вием Mg2+ или Мп2+ наблюдались во многих случаях, хотя в об-
Металлоферменты
457
щем величина кажущейся КА Для Мп2+ на порядок выше, чем для
Mg24" (как и можно было бы ожидать из сравнения констант
устойчивости комплексов этих двух металлов с небольшими мо-
лекулами [3]).
По предположению Кон [21] наблюдение за поведением фер-
мента при добавлении Са2+ может служить критерием для выбора
схемы координации. Ферменты, образующие комплексы Е — суб-
страт — М2+, например креатинкиназы, активируются Са2+, в то
время как на ферменты, образующие комплексы Е — М2+ — суб-
страт, Са2+ обычно действует как ингибитор. Основой этого эмпи-
рического критерия является быстрая скорость обмена лигандов
в Са2+-комплексах [84], поскольку для Са2+ комплекс с мостико-
вым металлом неустойчив. Однако недавние исследования нукле-
азы стафилококка по связыванию Са2+, а также рентгенострук-
турные работы показали, что этот ион взаимодействует с фермен-
том, включаясь в центр связывания нуклеотидов или очень близко
к нему [85, 86]. Следовательно, в данном случае, по-видимому,
имеет место образование мостикового комплекса фермент —
Са2+ — нуклеотид, хотя не исключено и участие комплекса с фер-
ментом в качестве мостика (М2+ — Е — лиганд).
Аналогичные исследования по замещению металла были про-
ведены для таких металлоферментов, как карбоксипептидаза [6],
карбоангидраза [88], щелочная фосфатаза [89] и алкогольдегид-
рогеназа из печени [90]. Для всех этих ферментов замена иона
металла в природном ферменте другим металлом может быть до-
стигнута следующим образом: 1) получением апофермента и по-
следующей его рекомбинацией с ионом металла [4]; 2) прямой
заменой металла на металл в тщательно контролируемых усло-
виях [91] или 3) для ферментов микробного происхождения рос-
том микроорганизмов в среде, содержащей высокую концентрацию
металла, включение которого требуется в фермент [92]. Интерес-
но, что получение апофермента обычно сводится к обработке очи-
щенного природного фермента, например длительному диализу
против хелатирующих агентов [4], хотя апощелочная фосфатаза
была получена из Escherichia coli [93], а апогалактозооксидаза —
из D act у Hum dendroides [93а], когда эти микроорганизмы были
выращены в условиях, «свободных от металла».
Сравнительное изучение каталитических свойств различных
металлоферментов в начале было главным образом ограничено
оценкой их относительной каталитической активности. Однако
детальное изучение катализа и связывания лиганда для серии
металлоферментов может пролить свет на роль иона металла, ес-
ли при отсутствии каталитической активности сохраняется связы-
вание металла. Этот подход был недавно использован для объяс-
нения наблюдаемой каталитической неактивности Cd(II)-щелоч-
ной фосфатазы [94].
458
Глава 14
3. ДЕГИДРОГЕНАЗЫ
3.1. Содержание металла и некоторые общие подходы
при обнаружении и количественной оценке
ионов металла, связанных с белком
Общий вид реакций, характерных для этого класса ферментов,
изображен ниже:
X—Н + НАД+ (НАДФ+) <—fc X + НАДН (НАДФН) + Н+ (8)
Некоторые дегидрогеназы катализируют специфические виды этих
реакций и являются цинксодержащими металлоферментами; при-
мером могут служить алкогольдегидрогеназа из печени [95, 96,
96а] и из дрожжей [97], малатдегидрогеназа из сердца свиньи
[98] (см. [99]), глутаматдегидрогеназа из печени млекопитающих
[100] и лактатдегидрогеназа из скелетных мышц кролика [101].
Однако, за исключением алкогольдегидрогеназы, наличие связан-
ного цинка, в которой тщательно изучено [95—97], достоверность
остальных ранних сообщений вызывает сомнение. Так, при деталь-
ном изучении лактатдегидрогеназы из печени крысы [102] и серд-
ца свиньи [103], глицеральдегид-3-фосфатдегидрогеназы из мышц
кролика [104] и из дрожжей [105], глюкозо-6-фосфатдегидроге-
назы из дрожжей [Ю6] и глутаматдегидрогеназы из печени быка
[106а] было продемонстрировано отсутствие в значительных ко-
личествах иона этого металла. Последние исследования глюкозо-
6-фосфатдегидрогеназы [104] показали отсутствие в очищенных
препаратах магния, кальция, хрома, марганца, железа, кобальта,
меди и молибдена.
Значительные трудности встретились при определении соотно-
шения металл — белок в алкогольдегидрогеназе из печени. Име-
ются сообщения, что этот фермент содержит либо 2 г-атома [97],
либо 4 г-атома [107, 108] цинка на 1 моль фермента. Алкоголь-
дегидрогеназа из печени содержит два центра связывания НАДН
[109, ПО], в то время как фермент из дрожжей содержит четыре
центра связывания этого кофермента [111]. Недавно обширные
исследования [91] пролили свет на эту путаницу в отношении
стехиометрии соотношения цинк — белок и было однозначно по-
казано, что содержание цинка в алкогольдегидрогеназе из дрож-
жей составляет 4 г-атома/моль фермента, однако два из этих
атомов цинка играют в большей степени структурную, чем ката-
литическую роль [91, 107]. Однако при проверке этого вывода
изучением связи между содержанием цинка и каталитической
активностью были получены противоречивые данные [91, 108].
Эти разногласия не могут быть связаны с разными образцами
ферментов,, так как изоферменты алкогольдегидрогеназы дрожжей
содержат одинаковое количество цинка [ИЗ].
Металлоферменты
459
Множество проблем, описанных выше для алкогольдегидроге-
назы, не являются особенностью этого фермента. Аналогичные
трудности могут встретиться для любого металлофермента, если
не применять со всей строгостью критерии Вейлли [4], предло-
женные для идентификации таких ферментов, а именно: 1) проч-
ное связывание иона металла белком; 2) повышение соотношения
металл — белок и специфической активности фермента в ходе его
очистки; 3) постоянное число грамм-атомов металла на моль
очищенного фермента и 4) постоянное соотношение между содер-
жанием металла и содержанием кофактора (или его связывани-
ем). Хотя эти критерии вполне применимы для идентификации
простых металлоферментов, например алкогольдегидрогеназы из
дрожжей, в которой связанный металл и активные центры присут-
ствуют в эквимолярных количествах, они могут привести к ошиб-
кам в более сложных случаях, примером чего может служить
история исследований алкогольдегидрогеназы из печени. В по-
следнем случае получению ошибочных результатов также способ-
ствовала неопределенность в молекулярной массе и молярном
коэффициенте поглощения фермента [91], и надо заметить, что
неточность в определении этих параметров также приводит к оши-
бочному определению соотношения металл — белок.
Кроме ферментов, в которых весь связанный металл либо его
часть играют в большей степени структурную, чем каталитическую
роль, известны ферменты, содержащие два различных металла,
которые, по-видимому, играют одинаковую каталитическую роль,
например алкогольдегидрогеназа, выделенная из дрожжей, выра-
щенных в присутствии СоСЬ [92] (разд. 5). В таких случаях сме-
шанная стехиометрия может быть результатом либо наличия двух
металлоферментов в соответствующем соотношении, либо образо-
вания истинного гибридного металлофермента. Нет простых мето-
дов, чтобы установить разницу между этими двумя возможностя-
ми, кроме тех случаев, когда в результате замещения металла
происходит изменение свойств белка. И хотя включение несколь-
ких металлов в фермент является обычно результатом заранее
обдуманного введения этих металлов в пищу или в питательную
среду, однако такие же ситуации могут иметь место и в природе,
и, таким образом, могут возникать те же проблемы оценки числа
активных центров.
В том случае, если последней стадией очистки металлофермен-
та является его отделение от другого металлопротеина, содержа-
щегося в более высокой концентрации и содержащего тот же ме-
талл, описанный выше критерий (2) не может быть применен.
В данном случае повышение степени очистки фермента сопровож-
дается уменьшением соотношения металл — фермент, как и на-
блюдалось для цинка, связанного пируваткарбоксилазой из пе-
карских дрожжей [114]. Поскольку в таких ситуациях критерий
460
Глава 14
(2) более неприменим, сравнение соотношения каталитической
активности и содержания белка и металла может быть осущест-
влено фракционированием очищенного фермента по размеру или
заряду частиц. При фракционировании на колонке очищенного
фермента постоянство соотношения металл — ферментативная ак-
тивность должно сохраняться по всему пику белка. Кроме того,
если в качестве метода разделения применяется гель-фильтрация,
пик, содержащий металл, должен выходить в соответствии с мо-
лекулярной массой (или стоксовым радиусом) фермента и с уче-
том свободного объема колонки.
Трудно переоценить важность применения перечисленных выше
критериев для идентификации металлоферментов. Действительно,
правильная информация не может быть получена простым опре-
делением содержания металла в ферментах различной степени
очистки. Следует отметить, что данные, полученные при исследо-
вании ингибирования или активации ферментов металлхелатирую-
щими агентами, взаимодействующими со связанными металлами,
могут привести к ошибочной интерпретации. Например, причины
инактивации фермента при его инкубации с 1,10-фенантролином
могут быть следующими: взаимодействие хелатирующего агента
со связанным ионом металла или его удаление; окисление сульф-
гидрильных групп (как было продемонстрировано для ФДП-аль-
долазы [115]); включение в гидрофобную область белка плоской
молекулы 1,10-фенантролина. Было показано также, что актива-
ция фруктозо-1,6-дифосфатазы и замедление агрегации вируса
табачной мозаики, происходящие в присутствии ЭДТА, по-видимо-
му, не являются результатом взаимодействия ЭДТА со связанны-
ми ионами металла [116, 117]. В других случаях активация под
действием ЭДТА или других хелатирующих агентов может быть
обусловлена удалением ингибирующих ионов металла в ходе вы-
деления фермента [Н8]. Хотя эффекты ингибирования хелатирую-
щими агентами наблюдались для многих дегидрогеназ [119, 120],
только алкогольдегидрогеназа была достоверно классифицирована
как металлофермент.
3.2. Каталитическая роль связанного металла
Хотя каталитическая роль связанного цинка в алкогольдегид-
рогеназе изучалась в течение последних двух десятилетий, эта
проблема осталась неразрешенной, несмотря на то, что были вы-
двинуты многочисленные предположения [121]. Были предложе-
ны механизмы, в соответствии с которыми цинк алкогольдегидро-
геназы принимает участие в связывании субстратов [122, 123],
коферментов [121, 124] и субстратов и коферментов одновремен-
но [125, 128]. Более того, в этих различных гипотезах [124—128]
были предложены все возможные центры координации ионов ме-
Металлоферменты
461
талла с НАД+. До сих пор не была предположена всего лишь од-
на оставшаяся возможность (см., однако, работу Уильямса и Вейл-
ли [129]), когда цинк не участвует непосредственно во взаимо-
действии ни с субстратами, ни с коферментом. Так как подобная
путаница часто возникает при попытках интерпретации косвенных
доказательств, представляет интерес обсудить имеющиеся данные.
Изучение начальных скоростей для алкогольдегидрогеназы из
дрожжей [130] или из печени [131] показало, что обратимое ин-
гибирование 1,10-фенантролином является конкурентным для
НАД+ (НАДН) и неконкурентным по отношению к субстратам.
Эти данные по ингибированию были интерпретированы Вейлли
[121] как указание на то, что связанный цинк взаимодействует
только с НАД+ (НАДН) и не играет роли в связывании с субст-
ратом. Эта интерпретация была подвергнута сомнениям по двум
причинам. Во-первых, Дэлзиел [132] указал на то, что приведен-
ные примеры ингибирования согласуются также с предположени-
ем, что цинк связывается одновременно и с субстратом и с кофер-
ментом. Однако рассмотрение последовательности реакций, пред-
ложенной для алкогольдегидрогеназы (9) [133—135], показывает,
что данные по ингибированию не дают возможности однозначно
определить центры связывания субстрата или кофермента:
с2н5он
сн3сно
НАДН
I
(9)
В соответствии с последовательным механизмом (9) любой инги-
битор, реагирующий со свободным ферментом (Е), должен прояв-
лять себя как конкурентный по отношению к НАД+(НАДН) и не
являться конкурентным по отношению к субстратам [60]. Конку-
рирующее поведение по отношению к НАД+ или НАДН подразу-
мевает конкуренцию на тех же участках фермента, но не обяза-
тельно на тех же активных центрах. Во-вторых, аналоги 1,10-фе-
нантролина, например 1,5-фенантролин, 5,6-(или 7,8)-бензохино-
лин, не являющиеся хелатирующими агентами, тем не менее кон-
курентно ингибируют НАД+ и столь же или даже более активны,
чем 1,10-фенантролин [136]. Как 1,5-, так и 1,10-фенантролин да-
ют при взаимодействии с алкогольдегидрогеназой из дрожжей
одинаковые дифференциальные спектры, однако только 1,10-фе-
нантролин показывает такой же спектр при взаимодействии с
[Zn(H2O)4]2+ [136]. Центр связывания 1,10-фенантролина на ал-
когольдегидрогеназе определяют как место взаимодействия фер-
мента с никотинамидным кольцом НАД+. Это устанавливается
изучением взаимодействия между этим ингибитором и аналогами
462
Глава 14
части молекулы НАД+, которые сами по себе являются конкурент-
ными ингибиторами НАД+, например N'-метилникотинамидхлори-
дом [137] и АДФ-рибозой [138]. Однако, учитывая, что хелати-
рующие свойства не коррелируют с эффективностью ингибирова-
ния, возможно, что связывание в большей степени является
результатом гидрофобных взаимодействий, чем координации со
связанным цинком. Далее, некоторую ясность вносят исследования
связывания НАД+ и аналогов НАД+ со свободной от цинка апо-
алкогольдегидрогеназой. При выделении апофермента диализом
против ЭДТА при pH 6,0 удаление связанного цинка не сопровож-
дается изменениями в степени связывания НАД+ алкогольдегид-
рогеназой [139]. Однако другие исследования показали линейную
зависимость между содержанием цинка и связыванием НАД+
(или АДФ-рибозы) при удалении связанного цинка диализом при
pH 5,0 [108]. Известно, что алкогольдегидрогеназа неустойчива
при низких значениях PH [91] и наблюдаемое уменьшение спо-
собности к связыванию НАД+ может являться результатом вто-
ричных изменений в конформации фермента, происходящих при
удалении связанного цинка [140].
Хотя эти данные, по-видимому, свидетельствуют против широ-
ко признанной роли цинка в этой металлдегидрогеназе, т. е. про-
тив образования комплекса фермент — Zn2+— НАД(НАДН)
[121], они недостаточны для определения истинной каталитиче-
ской роли этого металла. Результаты, полученные Милдваном и
Винером [122, 141], могут быть интерпретированы в пользу об-
разования комплексов фермент — Zn2+ — субстрат [8], хотя, учи-
тывая имеющиеся данные, такая интерпретация является доволь-
но рискованной. В этих исследованиях при использовании спин-
меченого аналога АДФ-рибозы было определено расстояние между
неспаренным электроном спиновой метки и протонами субстрата
[121]. Если бы Zn2+ в центре связывания металла можно было за-
менить на парамагнитный ион металла, то можно было бы мето-
дом ЭПР измерить степень спин-спинового взаимодействия и, та-
ким образом, определить расстояние между спиновой меткой и
связанным металлом [72, 74а] (разд. 2.2). Опубликовано сообще-
ние о замене Zn2+ на Со2+ в алкогольдегидрогеназе из печени, и
при этом Со2+-фермент проявлял каталитическую активность [90].
Аналогичная замена Zn2+ на Мп2+ может непосредственно проде-
монстрировать наличие мостикового комплекса Е — М2+— субст-
рат изучением скоростей ядерной магнитной релаксации протонов
субстрата (разд. 2.3). Возможно использование ЯМР 35С1 для изу-
чения влияния субстратов и коферментов на свойства связанного
цинка в нативном ферменте [143]. Этот метод был использован
для изучения связанного Zn2+ в пируваткиназе [144], и он являет-
ся одним из немногих методов изучения окружения диамагнит-
ного атома цинка.
Металлоферменты
463
Недавно было сообщено о необычном влиянии Са2+, Мп2+ и
Mg2+ на кинетические свойства глутаматдегидрогеназы, выделен-
ной из Blastocladiella emersonii [146, 147]. Эти двухвалентные ка-
тионы являются сильными активаторами восстановления а-кето-
глутарата, но слабыми ингибиторами окисления глутамата. Такие
направленные эффекты, по-видимому, связаны с регуляторными
свойствами этого фермента и не доказывают, что ионы двухва-
лентных металлов принимают непосредственное участие в ката-
лизе. Регулярные эффекты одновалентных катионов также наблю-
дались для некоторых дегидрогеназ [148, 149], хотя подобные эф-
фекты более свойственны пиридиннуклеотидзависимым окисли-
тельным декарбоксилазам (разд. 4).
Хотя большинство дегидрогеназ полностью активно и без до-
бавления ионов двухвалентных металлов, были обнаружены неко-
торые полиолдегидрогеназы, для которых необходима активация
добавлением Mg2+ или Мп2+ [150—153]. Нет работ по выяснению
роли иона металла в каталитическом механизме этих металлакти-
вируемых дегидрогеназ.
4. ПИРИДИННУКЛЕОТИДЗАВИСИМЫЕ ОКИСЛИТЕЛЬНЫЕ
ДЕКАРБОКСИЛАЗЫ
Описан ряд ферментов, катализирующих окислительное декар-
боксилирование р-оксикислот и требующих активации добавлени-
ем М2+. Этот тип реакции проиллюстрирован ниже для фермента
декарбоксилирования яблочной кислоты (ь-малат-фермента)
[154—157]:
м2+
L-малат -|- НАД+ (НАДФ+) < пируват + СО2 + НАДН (НАДФН) + Н+ (10)
Аналогичные реакции катализируются р,|3-диметилмалатдегпдро-
геназой [158], 0-изопропилмалатдегидрогеиазой [159], НАД+- и
НАДФ+-зависимой изоцитратдегидрогеназой [160—164] и гомо-
изоцитратдегидрогеназой [165]. Сходные реакции, катализируе-
мые 6-фосфоглюконатдегидрогеназой [166] и оксалогликолят вос-
становительной декарбоксилазой [167], протекают без добавления
ионов двухвалентных металлов, однако неизвестно, содержат ли
сами ферменты связанные металлы.
Хотя прямые доказательства роли ионов двухвалентных метал-
лов в металлзависимых окислительных декарбоксилазах в настоя-
щее время отсутствуют, некоторые предварительные данные под-
тверждают, что ион металла участвует исключительно в образова-
нии мостикового комплекса фермент — М2+— субстрат. Во-пер-
вых, НАДФ+ и НАДФН образуют связь с ь-малат-ферментами и
в отсутствие М2+. Степень и сродство связывания НАДФ+ не за-
464
Глава 14
висят от добавления Мп2+, и наблюдаемая константа диссоциации
комплекса Е — НАДФН согласуется с константой, определенной
на основании изучения начальных скоростей [168, 169]. Во-вто-
рых, комплекс Мп2+— изоцитрат обеспечивает защиту НАД+-за-
висимой изоцитратдегидрогеназы от инактивации п-хлормеркур-
фенилсульфонатом, но ни изоцитрат, ни НАД+ ни сами по себе,
ни в присутствии Мп2+, ни в его отсутствие такую защиту не обес-
печивают [170]. В-третьих, некоторые из этих ферментов также
катализируют металлзависимое декарбоксилирование аналогич-
ных кетокислот, например оксалоацетата ( ь-малат-фермент) [155,
8 _ 8
с—о" . С—
| ^м2* НАДФ+
V- г
НАДФН СНг
СН3С0С00Н + м2
Рис. 14.2. Возможная роль ионов М2+ в реакциях, катализируемых пиридиннук-
леотидзависимыми окислительными декарбоксилазами. Приведенный механизм
предложен Бойером [176] для L-малат-фермента.
156], оксалосукцината (НАДФ+-зависимая изоцитратдегидрогена-
за) [171, 172]. Возможно, реакции протекают на том же активном
центре, что и окислительное декарбоксилирование 0-оксикислот
[164, 169], хотя кинетический анализ не доказывает в послед-
нем случае промежуточного образования ни свободной, ни фер-
ментсвязанной кетокислоты [16, 169]. Даже если декарбоксили-
рование кетокислот и протекает по другому пути, необходимость
иона металла указывает на участие во всех реакциях комплекса
Е — М2+ — субстрат по аналогии с металлзависимыми оксало-
ацетатдекарбоксилазами (разд. 5). В-четвертых, было установле-
но [169, 173] значительное связывание Мп2+ ь-малат-ферментом
и изучение НАД+-зависимой изоцитратдегидрогеназы показало,
что комплекс М2+ — изоцитрат действительно является субстра-
том этого фермента, в то время как центр активации специфичен
для свободного изоцитрата [173а]. Для НАДФ+-зависимой изо-
цитратдегидрогеназы наблюдалась заметная взаимосвязь между
кажущейся КА для М2+ и кажущейся Км для изоцитрата [174].
Интересно, что последний фермент характеризуется относительно
широкой специфичностью активации ионами металлов и что наи-
более эффективным активатором является Zn2+ [174].
Все эти факты согласуются с механизмом, предложенным Бойе-
Мёталлоферменты
465
ром [176] для малат-фермента, изображенным на рис. 14.2. В со-
ответствии с этим механизмом электрофильный характер иона
металла способствует снижению электронной плотности на
а-углеродном атоме и переносу гидридного иона на НАДФ+. Та-
ким образом, ион металла должен ускорять согласованные дегид-
рирование и декарбоксилирование. Роль, отведенная иону металла
на рис. 14.2, аналогична предложенной Штейнбергером и Вестгей-
мером [177] для каталитического декарбоксилирования 0-кетокис-
лот. Вытекающая из этого механизма структура комплекса М2+—
l -малат отличается, однако, от структуры, наблюдаемой для свя-
зывания [Мп(Н2О)б]2+ ь-малатом [178], и это означает, что при-
сутствие фермента может изменять структуру образующегося ком-
плекса М2+— малат.
Необходимо заметить, что анализ НАД+-связанной изоцитрат-
дегидрогеназы из сердца свиньи на содержание связанных ионов
металла показал отсутствие значительных количеств Mg, Са, Сг,
Мп, Fe, Со, Ni, Си, Zn, Мо и Cd в этом ферменте [178а]. Возмож-
но, что ингибирование этого фермента CN- происходит в резуль-
тате образования аддукта НАД-цианид [170]. Ситуация, анало-
гичная вышеописанной для декарбоксилирования 0-кетокислот не-
которыми окислительными декарбоксилазами, наблюдалась и для
металлзависимой изомероредуктазы, которая катализирует восста-
новительную конденсацию с алкильной миграцией как стадию в
ходе биосинтеза валина и изолейцина [179].
м2*
а-ацетолактатНАДФН < —>- а,р-диоксиизовалерат + НАДФ+ (П)
Этот фермент также катализирует НАДФН- и М2+-зависимую изо-
меризацию а-окси-|3-кетоизовалерата в а-ацетолактат, однако нет
доказательств участия а-ацетолактата в качестве промежуточного
соединения в реакции [179] и не определена также роль иона ме-
талла в этой реакции.
5. ФЕРМЕНТЫ МЕТАБОЛИЗМА СО,
5.1. Биотинсодержащие ферменты
Ферменты, содержащие биотин как кофактор, катализируют
три родственные друг другу реакции. Биотинкарбоксилазы ката-
лизируют карбоксилирование а-кетокислот и ацильных производ-
ных кофермента А [180, 181]:
м2
Е—биотин АТФ 4- НСО3 <=
Е—биотин~СО2 + АДФ 4- POf-
Е—биотин-~СО2
пируват
или
пропионил-КоА
Е—биотин -j-
оксалоацетат
или
метилмалонил-КоА
(12)
(13)
30—2451
466
Глава 14
Метилмалонил-КоА-оксалоацетаттранскарбоксилаза катализирует
транскарбоксилирование между кетокислотами и ацильными про-
изводными кофермента А [182].
Е—биотин 4- метилмалонил-КоА :< Е—биотин~СО2 + пропионил-КоА (14)
Е—биотин~СО2 пируват < > Е—биотин + оксалоацетат (15)
Биотиндекарбоксилазы также катализируют декарбоксилирование
кетокислот и ацильных производных кофермента А [183, 184]:
Е—биотин 4-
оксалоацетат пируват
или <— > Е—биотин~СО2 4* или
метилмалонил-КоА пропионил-КоА
Е—биотин~СО2 < >. Е—биотин 4~ СО2
(16)
(17)
Для участия биотинкарбоксилаз в реакции синтеза, когда из АТФ
и НСОз образуется комплекс Е — биотин~СО2 [уравнение (12)],.
необходимо добавление М2+. Роль ионов металла в реакции (12)
будет рассмотрена в разд. 12, посвященном синтетазам.
Все три типа катализируемого биотинзависимыми ферментами
транскарбоксилирования а-кетокислот [реакции (13), (15) и (16)]
или ацилпроизводных кофермента А [реакции (13), (14) и (16)]
протекают через образование комплекса Е — биотин ~СО2. Пер-
вое указание на участие связанного с белком иона металла в био-
тинзависимом транскарбоксилировании кетокислот было сделано^
на основании ЯМР-исследований пируваткарбоксилазы из пече-
ни цыпленка. Эти исследования пролили свет на роль ионов двух-
валентных металлов в отдельных стадиях реакций синтетаз [реак-
ция (12)] и показали присутствие связанной парамагнитной час-
тицы в ферменте, которая была идентифицирована как ион
марганца [185]. Дальнейшее изучение показало, что марганец яв-
ляется составным компонентом пируваткарбоксилазы и присутст-
вует в эквивалентных с биотином количествах [37, 185]. Следова-
тельно, пируваткарбоксилаза из печени цыпленка идентифициро-
вана и как первый металлофермент, содержащий марганец в
качестве природного иона металла, а также как первый металло-
биотиновый фермент [37, 185]. Впоследствии было показано, что
другие пируваткарбоксилазы, а также метилмалонил-КоА-окса-
лоацетаттранскарбоксилаза содержат связанные ионы металла
(табл. 14.1), а косвенные доказательства подтверждают, что свя-
занный ион металла может присутствовать в биотинзависимой
оксалатдекарбоксилазе [183]. Следовательно, ионы металлов, свя-
занные с белком, могут играть существенную роль в биотинзави-
симом транскарбоксилировании а-кетокислот. Хотя описаны окса-
латдекарбоксилазы, не содержащие биотина [28, 191], эти фер-
менты не проявляют каталитической активности без добавления
ионов двухвалентных металлов и, по-видимому, не катализируют
Металлоферменты
467
Таблица 14.1
Наличие связанных ионов металлов в различных биотинзависимых ферментах
Фермент Источник Связанный иои металла3 Литература
Пируваткарбокснлаза Печень цыпленка Мпб 185
Печень цыпленка (цыплята, лишенные Мп2+) Mg6 186
Печень индюка Мп 187
Печень крысы Мп 188
Печень теленка Mg-J-Mn 187
Пекарские дрожжи Zn 189
Метилмалонил-КоА-ок- салоацетаттранскарбо- ксилаза Propionibacterium shermanll Со+Zn 190
а Во всех случаях общее содержание связанного металла эквимолярно содержанию
биотина в ферменте, за исключением пируваткарбоксилазы из печени крысы.
® Пируваткарбокснлаза, выделенная из печени цыплят, выращенных при очень низкой
концентрации Мп, содержит Mn+Mg в том же соотношении, что и их содержание в корме,
стадию транскарбоксилирования, которая предшествует декар-
боксилированию [28].
Для объяснения роли ионов металла в декарбоксилировании и
биотинзависимом транскарбоксилировании оксалоацетата было
выдвинуто несколько предположений (см. рис. 14.3). На
основе данных по декарбоксилированию диметилоксалоацетата,
которое катализируется ионами М2+, Штейнбергер и Вестгеймер
[177] предложили механизм, применимый как для декарбокси-
лирования, так и для транскарбоксилирования кетокислот. В со-
ответствии с этим механизмом связывание иона металла карбо-
нилом и С-1 карбоксилом кетокислоты стабилизирует енольный
продукт реакции (рис. 14.3, Д). Предполагаемую структуру этих
комплексов подтверждает влияние добавок М2+ на ультрафиолето-
вые спектры некоторых кетокислот [193], однако другие доказа-
тельства [194] более согласуются с тем, что Мп2+-оксалоацетат-
ный комплекс является С-1,С-4-дикарбоксилхелатным, а Мп2+ —
пируват — монодентатным комплексом, включающим в качестве
лиганда карбоксильную группу. Хотя последние исследования ок-
салоацетатдекарбоксилазы из мышц трески [195] и были интер-
претированы в соответствии с механизмом Штейнбергера — Вест-
геймера [196, 197], ни стереоспецифическое восстановление пиру-
вата в d-лактат [197], ни повышенная скорость обмена метиль-
ных протонов пирувата [196], вызываемая присутствием фермента,
не исключает других механизмов.
30*
468
Глава 14
% —s, //
C-CHJ-C-----С
Г7 \ -
О OJ ,о
'..2+
м
о
CHj=C—с
z II \
°- Л>-
''М2+
Рис. 14.3. Возможная роль ионов металла в биотинзависимом транскарбоксили-
ровании Р-кетокислот.
Л—механизм Штейнбергера—Вестгеймера [177], модифицированный для декарбоксилирова-
ния оксалоацетата; Б — предложенный Стилсом [198] механизм, объясняющий роль М2+
в биотинзависимом транскарбоксилировании; В — механизм, предложенный Милдваном и др.
[201], объясняющий роль связанного марганца в реакциях, катализируемых пируваткарбоК-
силазой. Этот механизм объединяет особенности как механизма Штейнбергера—Вестгеймера»
так и механизма Стилса и показывает двойственную роль иона металла; Г — механизм, пред-
ложенный Брюсом и Хегарти [210] и основанный на предположении, что комплекс Е — био-
тин ~СО2 образует О-карбоксильное производное.
Металлоферменты
469
Второй механизм, применимый только для катализа ионами
металлов биотинзависимого транскарбоксилирования, был впер-
вые предложен Стилсом [198] (рис. 14.3,5). Возможность этого
механизма была продемонстрирована в исследованиях, которые
показали, что ионы двухвалентных металлов замедляют декар-
боксилирование Ь1-карбоксиимидазолона-2, аналога l'-N-карбокси-
биотина (связанного промежуточного продукта ферментативной
реакции) [199] и ускоряют реакцию эфиров N-карбоксиимидазо-
лона-2 с нуклеофилами [200]. Хотя эффекты, наблюдаемые в этих
модельных исследованиях, и должны приводить к транскарбокси-
лированию, нет доказательств координации остатка l'-N-карбокси-
биотина с ионом металла, связанным с белком [201]. Титриметри-
ческие и ЯМР-исследования взаимодействия М2+ с d-биотином и
некоторыми его аналогами указывают на роль атома серы в об-
разовании комплекса М2+ — биотин в растворе [202].
В настоящее время информация, полученная при прямом ис-
следовании такого рода ферментов, главным образом вытекает из
изучения ЯМР-спектров пируваткарбоксилазы из печени цыплен-
ка. Так как в активном центре этого фермента присутствует свя-
занный металл Мп2+, метод ЯМР является идеальным для изуче-
ния роли иона металла, как и было предсказано Кон [21]. Эти
исследования рассмотрены в работах [203, 204], здесь же приве-
дено только краткое их изложение. Изучение скоростей релакса-
ции для взаимодействия Мп2+ — протоны воды [76] (разд. 2.3)
показало, что в реакциях пируваткарбоксилазы [реакция (13)]
только субстраты и ингибиторы стадии транскарбоксилирования
заметно взаимодействуют со связанным марганцем. Этот харак-
тер участия марганца в реакции пируваткарбоксилазы подтвер-
ждается изучением влияния связанного иона металла на скорости
ядерной магнитной релаксации протонов пирувата и оксалоацета-
та. Расчеты расстояний Мп2+ — протоны (из 1/Ллг) и константы
сверхтонкого взаимодействия (из \]TzM и I/T^m) продемонстри-
ровали образование мостиковых комплексов фермент — Мп2+ —
пируват [205] и фермент — Мп2+ — оксалоацетат [194]. Кинети-
ческие свойства этих мостиковых комплексов согласуются с их
участием в общей реакции [194, 205]. Однако неопределенность
времени корреляции (тс) при взаимодействии комплекса Мп2+ —
субстрат с протонами не позволила приписать точную структуру
этим мостиковым комплексам. В результате предварительных ис-
следований сделано предположение, что при взаимодействии ме-
тилмалонил-КоА-оксалоацетаттранскарбоксилазы с пируватом об-
разуется мостиковый комплекс фермент — Со2+ — пируват [190].
Для образования мостиковых комплексов пируваткарбоксила-
за — Мп2+ — субстрат был предложен внешнесферный механизм
SnI [206], так как кажущаяся скорость образования этих ком-
плексов приблизительно равна кажущейся скорости отхода моле-
470
Глава 14
кул воды из координационной области связанного марганца [194,
205]. Это заключение было основано на предположении, что на-
блюдаемый отрицательный температурный коэффициент для \/Т2р
или ЦТ1Р означает, что тм доминирует в общей скорости релакса-
ции. Однако и Кон [82], и Шерд и Брэдбури [76а] указывают на
то, что отрицательный температурный коэффициент для \11\р
(Д/Т2р) может быть также результатом доминирования rs, что яв-
ляется наиболее вероятным объяснением для наблюдаемых в этом
случае низких энергий активации [37, 205]. Эти две возможности
можно различить (разд. 2.3), так как доминирование тм характе-
ризуется равенством между \jT\p и \1Т2р, в то время как при до-
минировании Ts i/T2p превышает 1/7\р- Последние СРП-исследова-
ния показали, что доминирование xs предпочтительнее, чем тм
(как предполагалось ранее [37]), в парамагнитном влиянии на
скорость релаксации протонов воды в присутствии пируваткар-
боксилазы, поскольку \]Т2р значительно превышает l/Tip при тем-
пературах от 0 до 40°С [207]. Таким образом, величина (1,0-
• 106±0,5-106 с-1), рассматривавшаяся ранее как 1/тм [37], яв-
ляется всего лишь нижним пределом этого параметра. Аналогично
значение \1Т2р метильных протонов пирувата в присутствии пиру-
ваткарбоксилазы повсеместно превышает \!Т\Р в том же темпера-
турном интервале, в котором rs (более предпочтительное, чем
Тм) доминирует в значениях \/Т2р [205]. Однако в комплексе пи-
руваткарбоксилаза — оксалоацетат при низких температурах и
высоких энергиях активации \1Т2р метиленовых протонов оксало-
ацетата приближается к l/Tip; в связи с этим выдвинуто предпо-
ложение, что в данном случае Тм доминирует в \1Т2р при темпера-
турах ниже 20 °C [194].
В результате этих и других исследований марганца, связанно-
го пируваткарбоксилазой, был предложен механизм катализа этим
металлом (рис. 14.3, В) [201], в котором соединяются особенности
механизмов Штейнбергера — Вестгеймера и Стилса (рис. 14.3, А
и В). В соответствии с этим механизмом как карбонильная груп-
па пирувата, так и l'-N-карбоксильная группа остатка карбокси-
биотина являются лигандами связанного марганца, что позволяет
иону металла в результате его электрофильных свойств облегчить
и уход протона от метильной группы пирувата, и образование кар-
бониевого иона нз l'-N-карбоксибиотинильного остатка. Так как
при инкубации 3Н-пирувата с пируваткарбоксилазой в отсутствие
других компонентов реакции [201] не освобождаются значитель-
ные количества трития, возможно, что уход протона и перенос
карбоксила протекают по согласованному механизму, изображен-
ному на рис. 14.3, В. Недавно были продемонстрированы два фак-
та, предсказанных на основании этого согласованного механизма.
Во-первых, наблюдался значительный изотопный кинетический
эффект /гн/Ат=4,2, во-вторых, изучение карбоксилирования хи-
Металлоферменты
471
ральных пируватов показало, что карбоксилирование протекает
с обращением конфигурации, т. е. карбоксильная группа находит-
ся в ^uc-положении к отщепляющимся протонам [208]. Для окон-
чательной оценки механизма, приведенного на рис. 14.3, В, необ-
ходимо, однако, выяснить структуры мостиковых комплексов фер-
мент — Мп2+ — пируват и фермент — Мп2+ —- оксалоацетат.
Согласованный механизм (рис. 14.3, В) также предполагает,
что промежуточное карбоксилированное производное в реакциях
биотинкарбоксилазы имеет структуру l'-N-карбоксибиотина, как
и было установлено на основании экспериментов по улавливанию
с использованием диазометана [209]. Однако недавно это заклю-
чение было подвергнуто сомнению Брюсом и Хегарти [210] на
основе приведенных ими модельных исследований, которые пока-
зали, что уреидный кислород является лучшим нуклеофилом, чем
уреидный азот. Механизм, изображенный на рис. 14.3, Г, предло-
женный на основе этих модельных исследований [210], должен
решить многие проблемы, с которыми столкнулись в предыдущих
механизмах биотинзависимого транскарбоксилирования. Кроме
того, можно ожидать, что промежуточное О-карбоксильное произ-
водное при метилировании перегруппировывается в N-карбоксиль-
ное [210].
Таким образом, было однозначно продемонстрировано участие
связанного с белком иона металла в биотинзависимом транскар-
боксилировании кетокислот. Поэтому можно ожидать, что сходную
роль ион металла будет играть в аналогичных реакциях ацильных
производных кофермента А. Однако до сих пор не было получено
доказательств присутствия связанного с белком иона металла в
какой-либо из изученных ацетил-КоА-карбоксилаз [201], хотя в
этой области до сих пор не было проведено сколько-нибудь де-
тальных исследований. К тому же метилмалонил-КоА-оксалоаце-
таттранскарбоксилаза содержит, скорее всего, только 1 г-атом
связанного металла на остаток биотина [190], а не 2 г-атома, как
следовало бы ожидать в том случае, если ионы металла присутст-
вуют как в центре связывания ацил-КоА, так и в центре связыва-
ния кетокислоты [реакции (13) и (14)] [210а]. Эти исследования
содержат явный парадокс, так как дейтерообмен протекает более
медленно в метильных группах ацетил-КоА [211] п обычных аце-
тилтиоэфирах [212], чем в метильной группе пирувата в тех же
условиях [212], что указывает на меньшую степень активации тио-
эфиров. Однако возможно, что для реакций, катализируемых
ацил-КоА-карбоксилазами, степень активации метильных (пли
метиленовых) групп, по которым протекает связывание СО2, мо-
жет и не играть роли, поскольку для некоторых ферментов не
наблюдалось первичного кинетического изотопного эффекта [213,
214], хотя карбоксилирование и протекает с обращением конфи-
гурации [213].
472
Глава 14
5.2. Ферменты карбоксилирования
фосфоенолпирувата
Описаны три фермента, катализирующих гомологичные реак-
ции карбоксилирования ФЕП в присутствии иона двухвалентного
металла, например Мп2+:
м2+
ФЕП -f-НДФ -f-СОа ч—оксалоацетат-f-НТФ (18)
м2+
ФЕП + POf" + СО2 < • оксалоацетат + РаО|“ (19)
м2+
ФЕП-f-HCOa ---► оксалоацетат + POJ- (20)
Все эти реакции заключаются в нуклеофильной атаке по фосфо-
рильной группе ФЕП, в таутомерном сдвиге двойной связи и
в присоединении электрофила к атому С-3 ФЕП [8]. Различие
состоит в природе используемого акцептора фосфорила, которым
может быть НДФ (ФЕП-карбоксикиназа) [реакция (18)] [215],
РО1- (ФЕП-карбоксифосфотрансфераза) [реакция (19)] [216]
или НгО (ФЕП-карбоксилаза) [реакция (20)] [217]; могут также
быть различными карбоксилирующие агенты (СО2 или НСОз)
[218, 219]. ФЕП-карбоксилирующие ферменты также гомологич-
ны с реакцией, катализируемой пируваткиназой [220]:
м2+
ФЕП 4-АДФ 4-Н+ < > пируват 4-АТФ (21)
Эту гомологию подчеркивает и тот факт, что в отсутствие СОг
ФЕП-карбоксифосфотрансфераза катализирует фосфоролиз ФЕП
[221]:
м2+
ФЕП -f- POf- 4- Н+ < пируват 4- РаО|_ (22)
Степень гомологичности проиллюстрирована изучением механиз-
ма действия этих ферментов [222]. Во-первых, результаты изуче-
ния начальных скоростей пируваткиназы [35, 38, 223] и ФЕП-кар-
боксикиназы [222, 224] согласуются с тем фактом, что для этих
двух ферментов безразличен порядок добавления субстратов и
М2+. Этот вывод подтверждается и изучением связывания, в ре-
зультате которого показано образование кинетически важных
комплексов ФЕП, М2+ и НДФ для пируваткиназы и ФЕП-карбо-
ксикиназы [37, 55, 222], а также комплексов Мп2+ с ФЕП-карбо-
ксилазой [222]. И пируваткиназа [225], и ФЕП-карбоксикиназа
[222] также образуют комплексы более высокого порядка типа
Е — МНДФ-— М2+, которые проявляют каталитические свойст-
ва, аналогичные свойствам комплекса Е — МНДФ-. Во-вторых,
комплексы Е — Мп2+, образованные ФЕП-карбоксикиназой, пиру-
ваткиназой или ФЕП-карбоксилазой, заметно увеличивают влия-
Металлоферменты
473
ние на СРП воды (еь=14—33), которое снижается при добавле-
нии или ФЕП, или НДФ (ФЕП-карбоксикиназа, ппруваткиназа)
[35, 38, 222]. В случае ФЕП-карбоксилазы это снижение еь являет-
ся временным, так как в этих же условиях протекает вся реакция
[222]. Эти последние данные предполагают, но не доказывают, что
общей чертой этих механизмов реакции является образование мо-
стикового комплекса фермент —Мп2+—ФЕП, как показано на
Рис. 14.4. Возможный механизм действия ФЕП-карбоксилазы, описывающий
роль М2+ в ферментах карбоксилирования ФЕП и в пируваткиназе [222].
рис. 14.4 для ФЕП-карбоксилазы. Исследования, продемонстри-
ровавшие образование мостикового комплекса пируваткиназа —
Мп2+— субстрат, обсуждаются в гл. 18. СРП-исследования
ФЕП-карбоксикиназы [222] показали еще один подвох при интер-
претации таких данных. Отрицательный температурный коэффи-
циент наблюдался для \!Т1Р протонов воды в присутствии как
Е — Мп2+, так и тройных комплексов с ФЕП и НДФ. Следователь-
но, предполагая доминирующее влияние хм на 1/7’1р (разд. 2.3),
соотношение ес/вь (при 25 °C) было использовано для вывода, что
НДФ заменяет одну, а ФЕП — две молекулы воды в координа-
ционной сфере Мп2+, связанного с ферментом. Однако для этих
трех комплексов наблюдается большая разница в энергиях акти-
вации для IJTip протонов воды [222]. В этой ситуации вычислен-
ное для любой данной температуры соотношение ет/еь является
ошибочным и по отношению к схеме координации [82], и, таким
образом, механизм на основе этих данных для ФЕП-карбоксики-
назы [222] является неверным.
Недавно проведенные исследования также дают повод для сом-
нений относительно места, которое занимает ФЕП-карбоксифосфо-
трансфераза в этой гомологичной группе ферментов. Данный фер-
мент очень чувствителен к ингибированию широким набором хела-
тирующих агентов, например ЭДТА, 1,10-фенантролином, CN-
[226]. Изучение ингибирования ЭДТА различных стадий и учет
влияния других хелатирующих агентов показывают, что образо-
474
Глава 14
вание комплекса фермент — ФЕП требует связанного иона метал-
ла [227]. Однако прямые попытки продемонстрировать присутст-
вие связанного металла в этих ферментах до сих пор давали лишь
двусмысленные и неопределенные результаты [228].
5.3. Рибул озо-1,5-д ифосф аткарбокси л аз а
Карбоксилирование рибулозо-1,5-дифосфата, в результате ко-
торого образуются 2 моля 3-фосфоглицерата [реакция (23)], ка-
тализируется ферментом, который, как принято считать, требует
активации ионом двухвалентного металла [229, 230] (однако см.
Андерсон и др. [231]):
м2+
Рибулозо-1,5-дифосфат + СОа -> 2,3-фосфоглицерат +Н+ (23)
Потенциальный ингибитор, 2-карбоксирибит-1,5-дифосфат (I), ко-
торый является аналогом предполагаемого промежуточного про-
дукта в реакции рибулозо-1,5-дифосфаткарбоксилазы (II) [232],
прочно связывается с ферментом только в присутствии М2+ [233]:
СН2ОР СН2ОР СН2ОР
С(ОН)СОО~ 1 1 С(ОН)СОО“ C(OH)CN
с=о н—с—он Н—С—ОН Н—с—он 1 1 Н—С—ОН н—с—он
СН2ОР II 1 СН2ОР СН2ОР I III
Однако прочное связывание рибулозо-1,5-дифосфата в присутствии
CN- [возможно, в виде аддукта рибулозо-1,5-дифосфатцианида
(Ш)] не требует присутствия М2+ [234]. Эти данные позволяют
предположить, что М2+ играет роль в стабилизации промежуточ-
ного соединения II в реакции рибулозо-1,5-дифосфаткарбоксилазы.
Более ранние сообщения об образовании стабильного комплекса
фермент — Mg2+—СО2 не подтвердились [233].
Кроме того, было показано, что связанный Си2+ является
структурным компонентом рибулозодифосфаткарбоксилазы из
шпината [236]. Этот связанный ион металла не играет непосред-
ственной роли в механизме катализа, так как его удаление не вы-
зывает уменьшения каталитической активности. Однако наблюда-
лись некоторые тонкие изменения при связывании природным
ферментом рибулозодифосфата плюс цианид или 2-карбоксирибит-
дифосфата по сравнению с ферментом, свободным от Си2+ [233].
Следовательно, связанный Си2+ участвует в поддержании конфор-
мации рибулозо-1,5-дифосфаткарбоксилазы.
Металлоферменты
475
5.4. Декарбоксилазы
Помимо металлактивируемых оксалатдекарбоксилаз (разд.
5.1), имеется ряд других декарбоксилаз, которым необходима ак-
тивация М2+, например оротидин-5-фосфатдекарбоксилаза [237],
пиридоксиндекарбоксилаза [238]. Роль ионов двухвалентных ме-
таллов в механизме действия этих декарбоксилаз детально не ис-
следована. Однако следует заметить, что необходимость иона ме-
талла является скорее исключением, чем правилом среди фермен-
тов этого типа [239].
6. ТИАМИНПИРОФОСФАТЗАВИСИМЫЕ ФЕРМЕНТЫ
Ферменты, использующие ТДФ как кофактор, катализируют ре-
акции, в которых расщепление углерод-углеродной связи (или ее
синтез) сопровождается образованием аддукта между частью рас-
щепленного субстрата и ТДФ; примером могут служить ферменты
декарбоксилирования а-кетокислот, транскетолазы, фосфокетола-
зы, глиоксалаткарболигазы [240]. Для многих ТДФ-зависимых
ферментов требуется активация ионом М2+ [240], однако имеется
ряд примеров, где для работы ферментов этого типа либо вообще
не требуется присутствия ионов М2+, как, например, для транске-
толазы из печени крысы [241], бензоилформиатдекарбоксилазы
[242], либо проявляется только частичная зависимость активно-
сти от добавления М2+, как, например, для пируватдекарбоксила-
зы из ростков пшеницы [243], оксидазы а-кетокислот с разветвлен-
ной цепью [243а, 244]. Наблюдалось широкое разнообразие в ха-
рактере взаимодействия этих апоферментов с ТДФ и (или) М2+.
Эти кофакторы связываются очень прочно с некоторыми ТДФ-за-
висимыми ферментами, например с пируватдекарбоксилазой, и
удалить их можно только .в специальных условиях [245]. В противо-
положность другим ферментам оксалил-КоА-декарбоксилаза и
глиоксалаткарболигаза, не проявляющие активности в отсутствие
ТДФ и М2+, связываются с кофакторами обратимо [246, 247а]. Ес-
ли фермент после отделения ТДФ оказывается лишенным связан-
ного М2+, как в случае с пируватдекарбоксилазой из ростков
пшеницы [243], нельзя сделать вывода, нужен ли М2+ наряду с
ТДФ для активации полученного апофермента.
Изучение пируватдекарбоксилазы [248] подтвердило предпо-
ложение о включении М2+ в образование комплекса фермент —
ТДФ [249]. После отделения природного фермента от ТДФ и Mg2+
холофермент может снова образоваться лишь при инкубации апо-
фермента с избытком ТДФ и М2+ [250]. Как скорость образова-
ния, так и стабильность полученного холофермента зависит от
природы иона двухвалентного металла, хотя каталитическая ак-
476
Глава 14
тивность одинакова для всех изученных М2+ (Са2+, Mg2+, Мп2+ и
/Са2+
Со2+). Например, комплекс и образуется, и диссоциирует
ХТДФ
быстрее, чем соответствующий М22+-комплекс [250], как и было
предсказано из изучения свойств простых комплексов этих ионов
металлов [84]. Исследование концентрационных зависимостей про-
цессов рекомбинации показало, что ТДФ и Mg2+ взаимодействуют
независимо с апоферментом и первоначально образуется диссоци-
ирующий тройной комплекс (IV), который затем в результате мед-
ленной и квазинеобратимой реакции превращается в другой трой-
ной комплекс (V), который и является каталитически активным
холоферментом [251]:
% /
^Е-ТДФ^
(24)
IV
Превращение IV в V можно блокировать действием ЭДТА [244].
Однако еще не определены ни природа медленной реакции прев-
ращения IV в V, ни какие-либо группы ТДФ, которые являются
лигандами по отношению к иону металла. Кроме того, неясно, при-
меним ли механизм реакции, предложенный в уравнении (24), к
другим ТДФ-зависимым ферментам, особенно к тем, в которых
этот кофактор легко диссоциирует, например к глиоксалаткарболи-
газе [246, 247], или же в этом случае ион металла не только облег-
чает связывание с ТДФ, но и играет какую-то дополнительную
роль в реакции.
7. ИЗОМЕРАЗЫ
7.1. Пентозизомеразы
Ферменты, катализирующие изомеризацию таких сахаров, как
ь-и d-арабиноза [252, 253], D-ксилоза [254], d-ликсоза [255] и
ь-рамноза [256], согласно уравнению (25), проявляют максималь-
ную активность только в присутствии иона двухвалентного метал-
ла, например Мп2+, и в большинстве случаев неактивны в отсутст-
вие М2+:
м2*
альдопентоза •< кетопентоза
(25)
Металлоферменты
477
Многие эти ферменты действуют менее эффективно в качестве гек-
созизомераз. Изотопные исследования ряда пентозизомераз ука-
зывают на промежуточное образование 4{ыс-ендиолов в ходе реак-
ции [257]:
н>.^н
Vz
II
3 /Сх
-С ОН
,он
с1
,?с о
(26)
В реакции (26) электрофильный агент, например М2+, должен об-
легчать реакцию путем поляризации карбонильной группы альдо-
зы или кетозы в ходе образования промежуточного ^ис-ендиольно-
го производного [257].
Механизм активации этой группы ферментов под действием М2+
наиболее тщательно был изучен для D-ксилозизомеразы, которая
активируется Мп2+, но практически неактивна в присутствии Mg2+
[258]. Изучение начальных скоростей позволило предположить, что
мостиковый комплекс фермент — Мп2+—D-ксилоза образуется
главным образом при добавлении d-ксилозы вслед за Мп2+
[259] (рис. 14.1, II, А). Это предположение об образовании комп-
лекса с мостиковым металлом было подтверждено ЯМР-исследова-
ниями [260]. Мп2+-ксилозизомераза оказывает повышенное влия-
ние на l/Tip протонов воды (еь=4), которое уменьшается при
добавлении как субстратов (D-ксилоза, D-глюкоза), так и инги-
биторов ( d-ксилит, D-сорбит) в диапазоне концентраций, соответ-
ствующем константам диссоциации, которые получены при изуче-
нии начальных скоростей [259]. Фермент также усиливает влияние
Мп2+ на скорости релаксации протона при С-1 ксилозы. На ос-
новании изменений l/Tip этого протона можно определить расстоя-
ние от Мп2+ до протона при С-1, равное 5,3±2,4 А,
что соответствует непосредственному связыванию d-ксило-
зы через С-1 с образованием мостикового комплекса фермент —
Мп2+ — субстрат [260]. Возможно, что в связывании участвует гид-
роксильная группа и (или) кислород гликозидного кольца, хотя
детальная структура этого комплекса не определена. Механизм,
описывающий роль М2+ в этой реакции, был предложен, основы-
ваясь на такой структуре мостикового комплекса, в которой обе
эти группы связаны как лиганды с ионом металла [8]. Интересно,
что в этих исследованиях изучался реакционноспособный комплекс,
что было возможно благодаря равновесному соотношению альдо-
за : кетоза, равному примерно 3: 1 (pH 7,0) [258]. Всего лишь еще
один реакционноспособный объект был изучен так же тщательно
ЯМР-методом— это мостиковый комплекс пируваткарбоксилаза—
Мп2+ — оксалоацетат [194] (разд. 5.1).
478
Глава 14
Необходимо заметить, что D-ксилозизомераза обратимо дезак-
тивируется при добавлении хелатирующих агентов, например
ЭДТА или 1,10-фенантролина [261], хотя и не доказано наличие
связанного иона металла в этом ферменте.
7.2. Гексозо- и пентозофосфатизомеразы
В противоположность пентозизомеразам большинство гексозо-
или пентозофосфатизомераз полностью активны в отсутствие ионов
двухвалентных металлов, хотя в нескольких случаях изотопные
исследования указывают на промежуточное образование цпс-ен-
диольного производного [261а]. Недавно фосфоманнозизомераза
из дрожжей была строго идентифицирована как цинксодержащий
металлофермент, в котором ферментативная активность зависит от
присутствия связанного иона металла [262]. И хотя факт непосред-
ственного участия Zn2+ в механизме катализа не установлен, за-
манчивым является предположение, согласно которому этот ион
металла играет ту же роль, которая предложена для Мп2+ в реак-
циях, катализируемых D-ксилозизомеразой [8]. В противополож-
ность ферменту, выделенному из дрожжей, фосфоманнозизомера-
за из красных кровяных телец является металлактивируемым фер-
ментом [262а]. Это наблюдение, а также тот факт, что Zn2+ уско-
ряет неферментативную изомеризацию маннозо-6-фосфата во
фруктозо-6-фосфат [2626], должны помочь разъяснить роль М2+
в ферментативной реакции.
Другие гексозо- и пентозофосфатизомеразы, например фосфо-
глюкозизомер аза ['262, 263] и фосфорибозизомер аза [264], не со-
держат связанного иона металла, хотя для фосфоглюкозизомеразы
[261а] было показано промежуточное образование цыс-ендиольно-
го производного. Возможно, что в этих ферментах вместо М2+ име-
ется другой электрофильный центр.
7.3. Другие изомеразы
Описан ряд изомераз, действующих на другие субстраты и про-
являющих частичную необходимость в активации ионами двухва-
лентных металлов. Примером могут служить изопентилпирофосфа-
тизомераза [265], цис, ццс-муконатлактоназа [266], оксалоаиетат-
кетоенолтаутомераза [267] и манделатрацемаза [267а]. До сих
пор не проводилось детальных исследований по выяснению роли
ионов металлов в этих реакциях.
8. МУТАЗЫ И ТРАНСФЕРАЗЫ
8.1. Фосфоглюкомутаза и другие мутазы
Ферментам, которые катализируют реакции переноса между
двумя атомами углерода в гексозо- или пентозофосфатах (мута-
зы), необходим для активации ион двухвалентного металла, как
Металлоферменты
479
показано в реакции (27) [268—270] для фосфоглюкомутазы, хотя
другие мутазы полностью активны и в отсутствие М2+ (например,
фосфоглицератмутаза [271]):
глюкозо-1,6-дифосфат, М2+
глюкозо-1-фосфат-----------------*• глюкозо-6-фосфат (27)
Ход реакции, катализируемой фосфоглюкозомутазой и установ-
ленный на основании изучения начальных скоростей, изображен на
£+глюкозо-1,в-дифосфат
Е- глюкозо-1,Б-дифосфат
,ф глюкозо-1-фосфат\\ ф ^ф
£Z / <- — 'V « *- Е
глюкозо-1-фосфат Ф глюкозо-Б-фосфат елюкозо-6-фосфат
Рис. 14.5. Ход реакции, катализируемой фосфоглюкомутазой (Ф—фосфат) [272].
рис. 14.5 [272]. Эти же исследования показали, что Mg2+ может
быть добавлен как к фосфо- или дифосфоферментам, так и к любо-
му из промежуточных комплексов. При этом оказалось безразлич-
ным, добавлять ли Mg2+ и глюкозо-1-фосфат (или глюкозо-6-ди-
фосфат) к фосфоферменту или Mg2+ и глюкозо-1,6-дифосфат к не
содержащему фосфора ферменту [273]. Однако скорость отщеп-
ления Mg2+ от любой из этих форм фермента примерно на три
порядка меньше скорости диссоциации Mg-комплекса любого из
гексозофосфатов [274]. Таким образом, обычные выводы, следую-
щие из отсутствия влияния порядка добавления реагентов на ско-
рости реакций, становятся несостоятельными, поскольку фосфоглю-
комутаза в данных условиях ведет себя как комплекс фермент—
Mg2+.
Ранее предполагалось, что фосфоглюкомутаза проявляет час-
тичную активность и в отсутствие М2+ [275], а также что предва-
рительная инкубация с Mg2+ плюс хелатирующий агент приводит
к зависящей от времени активации этого фермента [275]. Теперь
известно, что эти эффекты являются результатом присутствия в
виде примесей прочно связанных ионов металлов [274]. После уда-
ления ионов металлов образовавшийся каталитически неактивный
апофермент проявляет широкую специфичность к активации М2+,
причем наиболее эффективным являются Mg2+ и Ni2+ [277]. Эф-
фективность активации коррелирует со скоростью диссоциации
комплекса фермент — М2+. Так, Mg2+, который является наиболее
эффективным активатором, и отщепляется от комплекса с наиболь-
480
Глава 14
шей скоростью [277]. Некоторые менее эффективные ионы, напри-
мер Mn2+, Cd2+, Zn2+, взаимодействуют с фосфоглюкомутазой с
образованием двойных комплексов, которые проявляют себя как
металлоферменты, в противоположность двойному комплексу
Mg2+— фосфоглюкомутаза. Имеются данные, указывающие на не-
большое различие в механизме катализа между металлактивируе-
мыми ферментами и металлоферментами в случае фосфоглюкому-
тазы [277].
Попытки определить роль М2+ в реакциях, катализируемых
фосфоглюкомутазой, путем сравнения свойств различных металло-
фосфоглюкомутаз не имели успеха. Все изученные ионы металлов
давали одинаковые дифференциальные спектры при взаимодейст-
вии с апоферментом, и это взаимодействие сопровождалось неболь-
шими изменениями конформации фермента, что было показано
путем замены растворителя [278]. Дифференциальные спектры,
отвечающие образованию промежуточного комплекса (рис. 14.5),
изменяются при добавлении ионов металлов, причем степень изме-
нения этих спектров в районе тирозина обратна эффективности
активации различными М2+ [279].
Недавно были измерены скорости ядерной магнитной релакса-
ции протонов воды в присутствии как Мп (II)-фосфоглюкомутазы,
так и тройных комплексов этого фермента с различными субстра-
тами [279а]. Эти исследования показали, что при образовании
тройных комплексов координационные молекулы воды при Мп(II)
замещаются на прочно связывающиеся субстраты, например глю-
козофосфат или ксилозофосф ат. Однако замены координационной
воды при Мп2+ не происходит, если образуется тройной комплекс
или с довольно прочно связанными субстратами, например с фрук-
тозофосфатом, галактозофосфатом или с неорганическим фосфа-
том, который является конкурентным ингибитором фосфоглюкому-
тазы [2796].
Эти исследования показали, что ион металла может играть как
непосредственную, так и непрямую роль в ориентации субстратов
в каталитическом центре, а также что активатор не принимает
прямого участия в каталитическом процессе. Однако, поскольку
фосфоглюкомутаза не проявляет активности в отсутствие М2+[277],
очевидно, что для проявления каталитической активности необхо-
димо сохранение конформации активного центра, которая зависит
от присутствия М2+ [279].
8.2. Трансферазы
Описаны многие нуклеотидил- и пентозилтрансферазы, которым
необходима активация ионом двухвалентного металла [обычно
Мп2+ и (или) Mg2+] [280], однако нет исследований, проливающих
свет на роль иона металла в реакциях этого типа.
Металлоферменты
481
9. АЛЬДОЛАЗЫ
ФДФ-альдолазы, которые катализируют реакцию (28), могут
быть разделены .на два основных класса.
ФДФ < > диоксиацетонфосфат + глицеральдегид-3-фосфат (28)
Класс II ФДФ-альдолаз, обнаруженный в грибах, бактериях,
дрожжах и сине-зеленых водорослях, в значительной степени ин-
гибируется в присутствии таких хелатирующих агентов, как ЭДТА
и 1,10-фенантролин [280, 281]. Подобное действие хелатирующих
агентов было объяснено участием связанных ионов металлов в ка-
тализе альдолазами класса II [281]. Это предположение подтверж-
дается тем фактом, что ФДФ-альдолаза из дрожжей содержит
связанный цинк в соотношении примерно 2 г-атома на 1 моль фер-
мента [282, 283]. Хотя большинство ФДФ-альдолаз класса II ведет
себя как металлоферменты, некоторые ферменты этого класса об-
разуют менее стабильные комплексы с М2+ и могут быть выделены
в виде апоферментов. Так, для ФДФ-альдолаз, выделенных из Clo-
stridium perfringens [284] и Anacystis nidulans [285], необходима
активация добавлением М2+, например Fe2+, Со2+.
В отличие от этого класса ФДФ-альдолазы класса I, характер-
ные для высших животных и растений, простейших и кишечнопо-
лостных, нечувствительны к ингибированию хелатирующими аген-
тами и не содержат связанного иона металла [280, 281]. ФДФ-аль-
долазы класса I проявляют типичную субстратзависимую
инактивацию при выдерживании комплекса фермент — диоксиаце-
тонфосфат в боргидриде [286]. Инактивация происходит в резуль-
тате восстановления основания Шиффа, которое образуется при
конденсации диоксиацетонфосфата с е-МН2-группой остатка лизина
в активном центре фермента [287]. В соответствии с этим механиз-
мом действия ФДФ-альдолаз класса I основания Шиффа играют
роль электрофильных центров и заменяют ион металла, который
играет эту роль в случае ФДФ-альдолаз класса II [281,
288]. Это предположительное сходство механизмов действия про-
иллюстрировано на рис. 14.6 и подтверждается тем, что ФДФ-аль-
долазы класса I и II катализируют сходные реакции обмена SH и
18О в субстратах [288]. Сходство механизмов действия оснований
Шиффа и связанных с ферментом ионов металла, изображенное
на рис. 14.6, распространяется и на другие типы реакций, особен-
но на декарбоксилирование а-кетокислот [28, 289, 289а]. Для та-
кого сходства механизмов действия ФДФ-альдолаз необходимо,
чтобы ферменты класса II образовывали мостиковый комплекс
фермент — М2+ — субстрат.
При удалении из ФДФ-альдолазы из дрожжей связанного цин-
ка с помощью диализа против ЭДТА получен каталитически не-
активный апофермент, молекулярные свойства которого сходны со
31—2451
482
Глава 14
свойствами холофермента [290]. Апоферменту может быть возвра-
щена активность путем инкубации как с ионами Со2+, Fe2+, Мп2+,
Ni2+, так и с Zn2+, причем образуется серия металлоальдолаз. За-
мена природного Zn2+ на другие М2+ приводит к заметным измене-
ниям в максимальных скоростях катализа. Однако такое замеще-
ние не изменяет существенным образом ни значения Км для ФДФ,
А. Класс I
Рис. 14.6. Сравнение стадий механизмов реакций, предложенных для ФДФ-аль-
долаз классов I и II [281, 288, 289а].
ни степени активации под действием К+ [291]. Эти данные показы-
вают, что ион металла участвует в скоростьлимитирующей стадии
реакции, но мало, либо вообще не играет никакой роли в поддер-
жании активной конформации фермента. Мп2+- и Со2+-альдолазы
были изучены методом ЯМР; в результате этих исследований од-
нозначно показано существование мостикового комплекса фер-
мент— М2+ — субстрат, участвующего в механизме, изображенном
на рис. 14.6. Хотя СРП-исследования не показали взаимосвязи
между эффектом усиления и связыванием Мп2+ в двойных и трой-
ных комплексах, которую можно было бы ожидать для комплек-
сов с мостиковым металлом (ср. разд. 2.3) [83], образование та-
ких комплексов однозначно доказано измерением скоростей релак-
сации протонов ФДФ и диоксиацетонфосфата. Найденное расстоя-
ние Мп2+— протоны субстрата (из 1/Т1р) и константа сверхтонкого
взаимодействия (из 1/Т2р и \{Т\Р) согласуются с предполагаемым
образованием тройного комплекса [292]. Более того, сравнение
Металлоферменты
483
данных для комплексов Мп2+ — диоксиацетонфосфат и фермент —
Мп2+ — диоксиацетонфосфат указывает на разницу в их структу-
рах, которая соответствует связыванию карбонила субстрата [292]
в присутствии фермента в соответствии с предложенным механиз-
мом (рис. 14.6). Однако следует с осторожностью относиться к
экстраполяции этих данных на механизм действия природного
7п2+-содержащего фермента, поскольку замена Zn2+ на Мп2+ на
порядок понижает каталитическую активность фермента [291].
Описаны также и другие альдолазы, относящиеся к классу II.
Некоторые из этих ферментов являются аналогами ФДФ-альдола-
зы из дрожжей, так как они нуждаются в добавлении ионов ме-
талла только после продолжительного диализа против ЭДТА.
Примерами могут служить фукулозо-1-фосфатальдолаза [292а] и
рамнулозо-4-фосфатальдолаза [293]. Природный ион металла,
присутствующий в этих ферментах, до сих пор не определен. Одна-
ко некоторые другие альдолазы класса II, которые образуют менее
устойчивые двойные комплексы Е—М2+, как, например, в случае
ФДФ-альдолазы из С. perfringens, выделены в виде апоферментов.
К ним относятся также 2-кето-З-дезоксиглутаратальдолаза [293а]
и кетопантоатальдолаза [294]. Этим ферментом необходима акти-
вация добавлением М2+, и, хотя обычно принято считать, что к ним
применим механизм, изображенный на рис. 14.6 [289а], в настоя-
щее время нет доказательств правильности такого допущения.
Кроме того, некоторые альдолазы, например 2-кето-4-окси-4-метил-
глутаратальдолаза [294а], были классифицированы как ферменты
класса II [289а] лишь на основании того, что ингибирование в
присутствии ЭДТА уменьшается при добавлении М2+. Однако этот
критерий неоднозначно демонстрирует роль М2+ в катализе, так
как ослабление ингибирования может протекать в первую очередь
благодаря удалению ЭДТА в виде комплекса с металлом, а не в
результате восстановления активности при комплексообразовании
М2+ с апоферментом.
Несколько лиаз кетокислот, близкородственные альдолазам,
также требуют активации добавлением М2+, например изоцитрат-
лиаза [295], р-окси-р-метилглутарил-КоА-лиаза [296] и цитрат-
лиаза [297]. Было высказано предположение, что в катализе лиа-
зами кетокислот [295] участвуют мостиковые комплексы фер-
мент— М2+— субстрат, однако имеется мало данных, подтвержда-
ющих это предположение. Было показано образование кинетически
важного комплекса Мп2+ — цитратлиаза, в котором Мп2+ повыша-
ет СРП. Однако не наблюдалось снижение этого эффекта ни при
добавлении субстрата (цитрата), ни при добавлении продуктов
этой реакции (оксалоацетата, ацетата) [298]. Эти данные, однако,
недостаточны, чтобы исключить образование комплекса с метал-
лом в качестве мостика. Сходные результаты были получены при
СРП-исследованиях Мп2+-ФДФ-альдолазы из дрожжей, однако в
31*
484
Глава 14
этом случае последующее изучение влияния этого фермента на
скорости релаксации протонов дало однозначные доказательства
существования комплекса фермент —Мп2+ —субстрат [83, 292].
Цитратлиаза из Escherichia coli и Aerobacter aerogenes [299]
подвергается необычной инактивации в результате собственной ре-
акции («самоубийство»), которая может вызываться взаимодейст-
вием фермента с енолоксалоацетатом в присутствии М2+ [300].
Специфичность активации под действием М2+ для этой реакции
отличается от специфичности, наблюдаемой для реакции расщеп-
ления [300, 301]. Причины такого «самоубийства» до сих пор не
объяснены; важно, однако, отметить, что этот эффект не является
результатом образования стабильного комплекса фермент — М2+—
енолоксалоацетат [302].
Ю. ГИДРОЛИАЗЫ И АММОНИЙЛИАЗЫ*
Поскольку эти ферменты, по-видимому, не имеют единого меха-
низма действия (однако см. Милдвана [8]), каждый класс будет
рассмотрен отдельно.
10.1. Гидролиазы
10.1.1. Енолаза
Изучение начальных скоростей при активации енолазы из
дрожжей ионами двухвалентных металлов [303, 304] дало осно-
вания для предположения, что мостиковый комплекс фермент —
М2+ — субстрат участвует в реакции, катализируемой этим фер-
ментом [3].
м2+
3-фосфоглицерат < > фосфоенолпируват + Н8О
(28а)
Изучение магнитного резонанса дало еще одно подтверждение об-
разования такого мостикового комплекса. Енолаза взаимодейству-
ет с Мп2+ с образованием двойного комплекса, который усиливает
СРП воды (еь=13,8). Этот эффект ослабляется при добавлении
субстратов [9, 21]. Как этот эффект, так и ингибирование под дей-
ствием Са2+ [305] согласуются с участием в реакции комплекса
с мостиковым металлом (разд. 2.3 и 2.4). Было выдвинуто несколь-
ко умозрительных механизмов, основанных на участии этого комп-
лекса в механизме катализа [8]. Однако эта интерпретация кине-
тических данных и данных ядерного магнитного резонанса пред-
* Лиазы — это ферменты, которые катализируют негидролитическое расщеп-
ление связей углерод — азот, углерод — кислород и углерод — углерод.
Металлоферменты
485
ставляется не очень надежной в свете последних данных,
полученных при исследовании прямого связывания М2+ и дальней-
шего изучения начальных скоростей [306]. Последние данные бо-
лее согласуются с участием в реакции комплекса с мостиковым
ферментом (разд. 1.4). Эти эксперименты показывают, что енолаза
из дрожжей имеет много центров связывания М2+. В отсутствие
субстрата найдены два центра связывания Mg2+ или Мп2+, отли-
чающихся на порядок по сродству к металлу. Однако в этих же
условиях появляется до шести центров взаимодействия с Са2+.
В результате возникает сомнение, можно ли интерпретировать сам
факт ингибирования под действием Са2+ как доказательство уча-
стия в реакции комплекса с мостиковым металлом.
В присутствии субстрата появляются два дополнительных цент-
ра связывания Mg2+ (или Мп2+) с гораздо меньшим сродством к
этим металлам. В то же время присутствие субстрата в 3—4 раза
повышает сродство того из двух прежде существовавших центров,
который проявлял меньшее сродство к этим ионам в отсутствие
субстрата. Найденное для Mn2+ Kd этого последнего центра хоро-
шо согласуется с определенным в результате СРП-исследований
[9, 21]. Однако необъяснимым остается то, что при этих исследо-
ваниях не удалось обнаружить второго более прочного места свя-
зывания Мп2+, особенно учитывая, что более прочный центр связы-
вания Mg2+ наблюдался при изучении изменений ультрафиолето-
вых спектров и спектров флуоресценции енолазы, вызываемых
этим ионом металла [308]. Различие в предположениях о роли
ионов двухвалентных металлов в реакциях енолазы является пря-
мым результатом противоречий в имеющихся экспериментальных
данных. Эти противоречия должны быть разрешены до того, как
будут сделаны какие-либо заключения о механизме этих реакций.
10.1.1. Аконитаза
Поскольку в случае аконитазы несомненно требуется активация
ионами Fe2+ и цистеином [309, 310], неоднократно предполагалось,
что существование мостикового комплекса фермент— Fe2+— суб-
страт является особенностью механизма катализа этого фермента
[310—313], катализирующего реакцию
±н2о ±н2о
цитрат < > циг-аконитат изоцитрат (286)
Для этой реакции был предложен элегантный механизм «железно-
го колеса», который является наиболее современным из всех пред-
ложенных механизмов. Он основан на кристаллографических ис-
следованиях конформаций цитрата и изоцитрата и исходит из до-
пущения, что ^ыс-аконитат может существовать в конформациях,
отвечающих соответственно цитрату или изоцитрату [313]. Меха-
486
Глава 14
низм «железного келеса» позволяет удовлетворительно объяснить
ряд необычных свойств реакции аконитазы, таких, как стереохимия
внутримолекулярного переноса протонов [314, 315], обязательный
обмен гидроксильной группы субстрата с растворителем [315],
специфичность ингибирования и инактивация различными изоме-
рами фторцитрата [316—318].
Хотя Мп2+ и не активирует аконитазу [310], этот ион металла
снижает скорость и степень активации ионами Fe2+, что позволяет
предположить конкуренцию этих двух металлов за один и тот же
центр (или центры) связывания [319]. Доказательства существо-
вания мостиковых комплексов фермент — М2+ — субстрат, предпо-
лагаемых как в механизме «железного колеса», так и в других
предложенных механизмах [310—313], были получены изучением
влияния комплекса Мп2+ — аконитаза на СРП в присутствии и в
отсутствие субстратов, а также изучением его влияния на скорости
релаксации метиленовых протонов цитрата. Эти исследования
[319] показали, что 1) образуется комплекс фермент — Мп2+ —
цитрат, свойства которого соответствуют его участию в катализе;
2) Fe2+ может заменять Мп2+ в этом мостиковом комплексе;
3) цитрат и изоцитрат конкурируют за один и тот же центр (или
центры) на ферменте. Это сродство цитрата и изоцитрата к Мп2+-
аконитазе близко к константам Михаэлиса взаимодействия этих
субстратов с природным Ре2+-ферментом. Однако цис-аконитат
взаимодействует с Мп2+-ферментом в гораздо меньшей степени.
Это последнее обстоятельство может, в частности, объяснить низ-
кую каталитическую активность Мп2+-аконитазы [319]. Хотя эти
данные и согласуются с механизмом «железного колеса», нужны
более определенные доказательства структуры мостиковых комп-
лексов фермент — М2+ — цитрат и фермент — М2+— изоцитрат.
Также интересно определить причины строгой специфичности ако-
нитазы к активации ионами М2+. По-видимому, Fe2+ является
единственным ионом металла, способным активировать этот фер-
мент [310, 319].
ь-Цитромалатгидролаза и (+)-тартратдегидраза, которые ка-
тализируют аналогичные реакции, также нуждаются в активации
Fe2+ и тиолом, например меркаптоэтанолом [320, 320а]. Однако
для реакции, катализируемой цитромалатгидролазой, необходимы
еще два белковых компонента [320].
10.1.3. Другие гидролиазы
Для некоторых других гидролиаз (дегидратаз) также необхо-
дима активация ионами М2+; примерами могут служить альтронат-
дегидратаза [321], дегидратаза диоксикислот [322], 6-фосфоглюко-
натдегидратаза [323], имидазолглицерофосфатдегидратаза [324]
и, по предварительным данным, 6-аминолевулинатдегидратаза из
Металлоферменты
487
Ustilago sphaerogena, возможно являющаяся медьсодержащим
металлоферментом [325] (однако см. Уилсон и др. [326]). Мало
что известно о роли ионов металла в реакциях, катализируемых
этими ферментами. Однако многие другие гидролиазы не являют-
ся металлоферментами, например дегидратазы аминокислот, ко-
торым в качестве кофактора необходим пиридоксальфосфат [327],
а также такие ферменты, как фумараза и кротоназа, которые не
нуждаются в кофакторе [328, 329].
10.2. Аммонийлиазы
Несколько аммонийлиаз, катализирующих реакцию (29), клас-
сифицированы как металлоферменты:
м2+
R—СН2—CH(NH2)—СООН <—te R—СН=СН —СООН-f-NH3 (29)
Имеются две наиболее изученные металлзависимые аммонийлиазы:
0-метиласпартаза и гистидиндезаминаза. Для обоих ферментов
имеются доказательства участия в реакциях мостиковых комплек-
сов типа фермент — М2+— субстрат [7, 330, 331], причем было
выдвинуто предположение, что электрофильный характер иона
двухвалентного металла облегчает депротонирование субстрата
[8, 330]. Однако имеются некоторые различия между р-метилас-
партазой и гистидиндезаминазой. Во-первых, p-метиласпартаза не-
активна в отсутствие М2+ [332—334], в то время как скорость ка-
тализа гистидиндезаминазой при добавлении М2+ повышается
только на 30—40% [335]. Возможно, что остаточная каталитиче-
ская активность, наблюдаемая в отсутствие М2+, объясняется на-
личием связанного иона металла, поскольку ЭДТА в этом случае
является сильнодействующим ингибитором каталитической актив-
ности. Кроме того, степень активации при добавлении М2+ варь-
ирует в зависимости от условий реакции и от методов выделения
фермента [335].
Во-вторых, p-метиласпартаза катализирует М2+-зависимый об-
мен протонов при p-углероде субстрата. Такой обмен позволяет
предположить промежуточное образование карбаниона, который
может быть стабилизирован ионом двухвалентного металла [336].
Отщепление аммиака от этого промежуточного продукта является
скоростьлимитирующей стадией реакции р-метиласпартазы [330,
336]. Наоборот, гистидиндезаминаза не катализирует обмен про-
тонов на p-углероде субстрата ни в присутствии, ни в отсутствие
М2+, а дезактивируется инкубацией с боргидридом, что указывает
на наличие «электрофильного центра», участвующего в дезамини-
ровании [335, 337]. В реакции гистидиндезаминазы стадией, лими-
тирующей скорость, является диссоциация комплекса фермент—
аммоний [338].
488
Глава 14
Фенилаланиндезаминаза напоминает в некоторых аспектах гис-
тидиндезаминазу, например природой реакции, которую она ката-
лизирует, наличием «электрофильного центра» и отсутствием ката-
лиза реакции p-протонного обмена. Однако этот фермент обладает
полной активностью и в отсутствие М2+ при всех до сих пор изу-
ченных условиях [339, 340], причем нет доказательств присутствия
связанного металла в ферменте, хотя такая возможность и не иск-
лючена. Эти исследования фенилаланиндезаминазы надо иметь в
виду, когда делается предположение о включении М2+ в реакцию
гистидиндезаминазы, поскольку в соответствии с последними дан-
ными [340а] возникают сомнения в участии М2+ в этой реакции, а
также в интерпретации эффекта ингибирования ЭДТА [335].
11. ГИДРОЛАЗЫ
Две большие группы гидролаз (пептидазы и фосфатазы) обсуж-
даются в гл. 15 и 18 соответственно. Эти две группы включают не-
которые хорошо изученные металлоферменты, например карбокси-
пептидазы и щелочную фосфатазу. Описан и ряд других металло-
гидролаз.
11.1. Гидролазы, требующие или содержащие Са2+
Ряд гидролаз либо содержит связанный Са2+, как, например,
а-амилазы [341—343], либо требует активации этим ионом метал-
ла, например нуклеаза стафилококка [85], некоторые фосфолипа-
зы [345, 346], у-лактоназа [347]. Хотя амилазы дезактивируются
при удалении связанного металла [341], роль Са2+ в этих фер-
ментах (возможно, за исключением нуклеазы стафилококка [85,
86]) является скорее структурной, чем каталитической [8, 341],
поскольку соотношение Са2+: фермент широко меняется в а-ами-
лазах, выделенных из разных источников [347]. Прямым доказа-
тельством структурной (матричной) роли Са2+ является потреб-
ность в этом ионе на некоторых стадиях действия така-амплазы
[341а], в результате чего каталитическая активность восстанавли-
вается. Однако ни эти данные, ни теоретические расчеты, основан-
ные на скорости обмена лигандов [76, 84], не достаточны, чтобы
исключить непосредственное участие Са+2 в катализе этими гид-
ролазами. В этом отношении будет уместно заметить, что физиоло-
гическими субстратами многих из этих ферментов являются макро-
молекулы и в ходе реакции могут образовываться мостиковые ком-
плексы; таким образом, могут наблюдаться отличия от действия
этих металлоферментов на небольшие молекулы субстратов [4].
Например, в образовании мостикового комплекса между гидрола-
зой и ее макромолекулярным субстратом могут участвовать не
один, а несколько ионов металла.
Металлоферменты
489
11.2. Аргиназа
На основе исследований по активации аргиназы под действием
М2+ можно было сделать вывод об образовании комплекса с мо-
стиковым металлом [18] (ср. разд. 1.3). После этого аргиназе, а
также родственным ей металлактивируемым пептидазам (гл. 15)
уделялось довольно мало внимания. Недавно было показано, что
аргиназа из дрожжей активируется ионами металла только после
обработки хелатирующими агентами [349]. Однако попытки иден-
тифицировать природный ион металла в этом ферменте путем срав-
нения его свойств со свойствами рекомбинированных металлоарги-
наз дали неоднозначные результаты [350]. Ни одно из этих иссле-
дований не дает возможности разобраться в механизме активации
аргиназ М2+.
12. СИНТЕТАЗЫ
Все синтетазы можно разделить на два класса на основе места
расщепления молекулы НТФ, которое происходит в ходе реакции
[351].
12.1 Синтетазы, катализирующие отщепление
пирофосфата
Эти ферменты, например ацил-КоА-синтетаза жиров, амино-
ацилпереносящие РНК-синтетазы, катализируют реакции, в кото-
рых активация карбоксильной группы сопровождается расщепле-
нием НТФ до НМФ+Р2О7 [351]. Во многих из этих реакций мо-
гут промежуточно образовываться ациладенилаты, связанные с
ферментом [352—354]. Для всех до сих пор изученных синтетаз
этого типа показана необходимость М2+ для их активации. Однако
только в немногих случаях исследована роль иона металла в ката-
лизе.
12.1.1. Ацетилкофермент А-синтетаза
Необходимость добавления ионов металлов для различных ре-
акций, катализируемых ацетил-КоА-синтетазой, дала основание
для предположения о механизме действия этого фермента
(рис. 14.7), который предусматривает наличие центров взаимодей-
ствия с тремя ионами металлов, необходимых для работы этого
фермента [48]. Участие М2+ наблюдали только для стадии (а)
(рис. 14.7), и тут могут быть использованы такие ионы, как Mg2+,
М.п2+, Са2+ и Fe2+, в концентрациях, превышающих 1 ммоль [47].
490
Глава 14
Такая же специфичность наблюдается при активации аналогичной
реакции, катализируемой бутирил-КоА-синтетазой [357]. Тот факт,
что реакция активируется Са2+, позволяет предположить участие
в стадии (а) мостикового комплекса фермент — АТФ—М2+ и так-
же может объяснить специфическую потребность этой реакции в
Е + ацетат + АТФ
м|+, Мд , М
(О) 4_
Р207*
Е + ацетил-АМФ
(в)|мд
Е (ацетил-АМФ)
Е+ацетил-КоА +АМФ
Рис. 14.7. Предложенный механизм реакции ацетил-КоА-синтетазы, отражающий
стадии активации фермента одновалентными и двухвалентными катионами [359].
KoASH— кофермент А (—SH-форма); ацетил-КоА — кофермент А (—SCOCHs-форма).
добавлении Mi+ [21]. После обработки фермента и реагентов хе-
латирующими смолами для катализа, как всей реакции в целом
[47], так и ее отдельных стадий [48], необходимо уже добавление
в стехиометрическом соотношении к ферменту второго иона двух-
валентного металла (Mii"), которым может быть Fe2+, Cd2+, Cu2+
или Ni2+ (рис. 14.7). Комплекс фермент — Мп+ медленно диссоци-
ирует и содержит 2 г-атома Мц+ на 1 моль фермента [48]. Однако
недавно была выделена ацетил-КоА-синтетаза, которой после обра-
ботки хелатирующими агентами не нужен М2ц. Исследование син-
тетазы, обработанной таким образом, не дает оснований думать о
присутствии в ней связанного металла [350]. Следовательно, аце-
тил-КоА-синтетаза может существовать в двух формах, только од-
ной из которых необходим М1Г. Не было сообщений о том, что
другим ацил-КоА-синтетазам жиров также необходима аналогич-
ная активация вторым ионом двухвалентного металла.
Кроме того, в случае ацетил-КоА-синтетазы для активации, как
реакции в целом, так и стадий (а) и (б) (рис. 14.7), необходимы
одновалентные катионы [359]. Однако связывание ацетиладенила-
та ферментом [стадия (в), рис. 14.7] протекает и в отсутствие
ионов одновалентных металлов. Многие аминоацил-тРНК-синтета-
зы также требуют одновалентных катионов для своей активации
[360].
Металлоферменты
491
12.1.2. Алшноацил-тРНК-синтетазы
Эти ферменты катализируют реакции, аналогичные изображен-
ным на рис. 14.7 для ацетил-КоА-синтетазы.
м2+
Е -f-аминокислота -f- АТФ < Е (аминоацил-АМФ) + Р2О|_ (31)
Е (аминоацил-АМФ) -f-тРНК ч Е -f- АМФ -f- аминоацил-тРНК (32)
Во всех случаях для реакции (31) необходим М2+ [361—363], од-
нако специфичность активации варьирует для различных синтетаз.
В большинстве случаев наблюдается активация под действием
Mg2+ и Мп2+ [360]. Однако Са2+ также активирует некоторые из
этих ферментов, например тирозил-тРНК-синтетазу [364], пролил-
тРНК-синтетазу [365], а другие — ингибирует, например глицил-
тРНК-синтетазу [366]. Последние исследования изолейцил-тРНК-
синтетазы показали^ что в реакции участвуют комплексы
[МАТФ]2- и [МР2О7 ]. Однако необходимы дальнейшие исследо-
вания, которые пролили бы свет на разную специфичность актива-
ции различными М2+, а также на возможное отражение этих раз-
личий в механизме реакции.
Хотя для каталитического действия некоторых аминоацил-
тРНК-синтетаз в реакции (32) [367, 368] необходимы ионы М2+,
эта необходимость проявляется только после диализа тРНК против
ЭДТА [367] и не является общей чертой, характерной для всех
ферментов этого типа [353, 363]. В данном случае потребность в
М2+ может явиться результатом обратимой денатурации некоторых
тРНК, протекающей при удалении связанного М2+ диализом про-
тив ЭДТА [3686], хотя нельзя исключить и возможности непосред-
ственного участия М2+ в реакции (32), если учесть некоторые дру-
гие данные [368а]. В тех случаях, когда было непосредственно
показано, что для протекания реакции (32) требуется М2+, специ-
фичность активации и требуемая концентрация иона металла сход-
ны с теми, которые определены для реакции (31) [367]. Следова-
тельно, в этом случае наблюдаются отличия от ацетил-КоА-синте-
таз и их активации под действием Mu' (см. выше).
12.2 . Синтетазы, катализирующие отщепление
ортофосфата
Хотя для активации всех ферментов этого типа необходимы
ионы М2+, для большинства из них к настоящему моменту этот
факт либо недостаточно исследован, либо, что встречается реже,
плохо изучена специфичность активации под действием М2+. Иск-
лючение составляют ферменты, рассматриваемые ниже.
492
Глава 14
12.2.1. Формилтетрагидрофолатсинтетаза
Для специфического связывания Мп2+ формилтетрагидрофолат-
синтетазой, как показали СРП-исследования [13], необходимо до-
бавление АТФ и АДФ. Так, влияние Мп2+ на увеличение СРП про-
является только при добавлении АТФ или АДФ к системам, содер-
жащим фермент 4-Мп2+. Эти данные совместно с данными о том,
что Са2+ является эффективным активатором фермента [369], ука-
зывают, что ион металла, необходимый для активации формилтет-
рагидрофолатсинтетазы, участвует в образовании комплекса фер-
мент— АТФ (АДФ)—М2+ (разд. 2.3). Добавление тетрагидрофо-
лата (или формилтетрагидрофолата) к комплексу фермент —
АТФ — Мп2+ вызывает снижение эффекта связанного Мп2+ вслед-
ствие образования кинетически важных четвертичных комплексов.
Формиаты оказывают сходный, хотя и более слабый, эффект [13].
Эти данные свидетельствуют о том, что субстраты изменяют окру-
жение иона металла в комплексе фермент — нуклеотид — М2+, хо-
тя прямого взаимодействия этих субстратов со связанным марган-
цем не происходит.
12.2.2. Биотинкарбоксилазы
Участие и роль связанных ионов металла в процессе переноса
СО2 от промежуточного комплекса фермент — биотин — СО2 на
карбоксильный акцептор [реакция (12)] обсуждались в разд. 5.1.
Однако биотинкарбоксилазам также необходима активация легко
диссоциируемыми М2+, который специфически включается в обра-
зование промежуточного комплекса фермент — биотин — СО2 из
АТФ-|-НСОз [реакция (12)] [370]. К настоящему времени роль
М2+ в реакции (12) однозначно не определена. Было показано об-
разование диссоциируемого комплекса Мп2+ — пируваткарбоксила-
за. Этот комплекс проявляет эффект усиления СРП, однако значе-
ние его в кинетике реакции не определено, поскольку он содержит
2—3 Мп2+ на остаток биотина. Более того, добавление субстратов
реакции (12) (АТФ и НСО3) вызывает либо небольшое, либо во-
обще не вызывает снижение эффекта усиления СРП комплексом
Е—Мп2+. Учитывая то, что Са2+ является конкурирующим ингиби-
тором пируваткарбоксилазы по отношению к Мп2+ [372], нельзя,
по-видимому, объяснить роль М2+ в реакции (12) участием его в
комплексах Е—АТФ—М2+ или Е—М2+—АТФ. Однако некоторые
последние кинетические данные указывают на то, что в случае пи-
руваткарбоксилазы из надпочечников овец как Mg2+, так и
А^АТФ2^, могут принимать участие в реакции (12) [373, 374].
В этой ситуации интерпретация данных магнитного резонанса и
данных ингибирования Са2+ представляет собой нелегкую задачу.
Металлоферменты
493
12.2.3. Глутаминсинтетаза
Интенсивное изучение Мейстером [375] механизма реакции глу-
таминсинтетазы из мозга овец указывает на то, что ион двухва-
лентного металла, необходимый для активации фермента, участ-
вует в образовании комплекса фермент—-АТФ. Этот вывод осно-
ван на изучении различных реакций, катализируемых ферментом,-
а также того, какие М2+ требуются для связывания субстратов и
защиты фермента от инактивации. Однако, поскольку эти реакции
отличаются между собой по специфичности активации различными
М2+ [375], может оказаться, что ионы металлов принимают уча-
стие на различных стадиях реакций [376]. Поэтому для дальней-
шего понимания механизма активации под действием М2+ необхо-
димы исследования глутаминсинтетазы с использованием методов,
которые бы дали информацию об окружении иона металла. Сле-
дует заметить, что введение остатка адениловой кислоты в глут-
аминсинтетазу из Е. coli вызывает заметные изменения специфич-
ности активации ионами М2+ [376а].
Недавно были получены новые данные о роли М2+ в активно-
сти глутаминсинтетазы из Е. coli [367]. При выделении фермента
в присутствии буфера, содержащего ион Мп2+, этот фермент со-
держит связанный Мп2+ [367]. При диализе такого фермента про-
тив ЭДТА наблюдается потеря каталитической активности. Однако
эта инактивация обратима, и при инкубации свободного от метал-
ла фермента с Mg2+, Мп2+ или Са2+ [377] происходит восстановле-
ние его активности. Эта реактивация фермента сопровождается
включением 1 г-атома связанного М2+ на субъединицу [378]. Об-
разовавшийся в результате холофермент весьма сходен с природ-
ным ферментом, но не идентичен ему [379]. Следует отметить, что
свободная от металла глутаминсинтетаза не только теряет катали-
тическую активность, но и имеет менее компактную четвертичную
структуру по сравнению с природным ферментом [380]. Это ука-
зывает на возможное участие связанного М2+ в поддержании ка-
талитически активной конформации фермента.
13.. ЗАКЛЮЧЕНИЕ
В этой главе суммированы имеющиеся в настоящее время дан-
ные о роли ионов металлов в механизме катализа некоторыми
классами сравнительно малоизученных металлоферментов. В боль-
шинстве случаев наши знания ограничены данными о специфично-
сти активации ионами М2+, и лишь для немногих ферментов мы
понимаем характер участия этого кофактора в механизмах реак-
ции. Для небольшого числа подробно исследованных металлофер-
ментов понять роль ионов металлов удалось в первую очередь в
494
Глава 14
результате широкого применения чувствительных биофизических
методов, таких, как ЯМР, ЭПР и рентгеноструктурный анализ.
Вклад этих и других биофизических методов является наибольшим
для ферментов, описанных в гл. 15, 16 и 19. В случае ферментов,
рассмотренных в настоящей главе, в основном был применен опи-
сательный подход, и поэтому для большинства из них имеющаяся
информация не дает возможности удовлетворительно проанализи-
ровать каталитическую роль иона металла с точки зрения коор-
динационной химии. Более аналитический подход к проблеме име-
ет место в гл. 18, а также в последнем обзоре Милдвана [8] (см.
также имеющиеся там ссылки).
В заключение следует упомянуть о нескольких интересных, но
менее хорошо изученных эффектах ионов металла в ферментатив-
ных реакциях. Описано несколько случаев, когда необходимость в
активации ионом М2+ является особенностью источника, из кото-
рого выделен фермент. Это явление наилучшим образом охарак-
теризовано для трех ферментов (цитратсинтетазы [381], фосфо-
трансацетилазы [382] и сериндегидратазы [383]), которым необ-
ходима активация ионами М2+ только в том случае, если они вы-
делены из Clostridium acidiurici. Эта потребность в активации
ионом металла может быть связана с конформационными особен-
ностями ферментов, выделенных из этого микроорганизма. Ту же
природу может иметь нетипичное поведение аденилатдеаминазы
из Porphyra crispata [384] при ее активации ионами металлов.
Наконец, описаны ферменты, которые используют ионы метал-
ла в качестве субстратов. Хорошо известны гемсинтетазы, которые
включают ионы металла, например Fe2+, в протопорфириновую си-
стему [385], хотя механизм этой интересной реакции и не изучен
во всех деталях. Имеются косвенные доказательства предпочти-
тельного (и, возможно, ферментативного) включения in vivo спе-
цифичного иона металла в металлоферменты, например включения
Мп2+ в пируваткарбоксилазу из печени цыпленка [186]. Наконец,
ионы металлов могут влиять на регуляторные эффекты ферментов,
примером чего может служить влияние М2+ на ингибирование цит-
ратсинтетазы под действием АТФ [386], однако подобные эффекты
в настоящее время еще недостаточно изучены.
В заключение я выражаю благодарность доктору Альберту
Милдвану за предоставление мне ряда еще неопубликованных ра-
бот и мисс Джоан Ковач за ценную редакторскую помощь.
СПИСОК ЛИТЕРАТУРЫ
1. Lehninger A. L., Physiol. Rev., 30, 393 (1950).
2. McCord 1. G., Fridovich I., J. Biol. Chem., 244, 6049 (1969).
3. Malmstrom B. G., Rosenberg A., Adv. Enzymol., 21, 131 (1959).
4. Vallee B. L., Adv. Protein Chem., 10, 317 (1955).
М еталлоферменты
495
5. Smith Е. L., Discuss. Faraday Soc., 20, 264 (1955).
6. Coleman J. E„ Vallee B. L„ J. Biol. Chem., 235, 390 (1960); 236, 2244
(1961).
7. Fields G. F., Bright H. J., Biochemistry, 9, 3801 (1970).
8. Mildvan A. S„ in P. D. Boyer (ed.), The Enzymes, Vol. 1, 3rd edn., Academic
Press, New York, p. 445.
9. Cohn M„ Leigh J. S., Nature, 193, 1037 (1962).
10. O’Sullivan W. J., Cohn M., J. Biol. Chem., 241, 3104 (1966).
11. O’Sullivan W. J., Virden R., Blethen S., Eur. J. Biochem., 8, 562 (1969).
12. O’Sullivan W. J., Noda L., J. Biol. Chem., 243, 1424 (1968).
13. Himes R. H., Cohn AL, J. Biol. Chem., 242, 3628 (1967).
14. Sternlicht H„ Shulman R. G., Anderson E. W„ J. Chem. Phys., 43, 3123
(1965).
15. Cohn M., Hughes T. R., J. Biol. Chem., 235, 176 (1962).
16. Cleland W. W., A. Rev. Biochem., 36, 77 (1967).
17. Scarano E., Geraci G., Rossi M., Biochemistry, 6, 192 (1962).
18. Heller man L., Physiol. Rev., 17, 454 (1937).
19. Hellerman L., Stock C. C„ J. Biol. Chem., 125, 771 (1938).
20. Smith E. L., Adv. Enzymol., 12, 191 (1951).
21. Cohn M„ Biochemistry, 2, 632 (1963).
22. Mildvan A. S., Leigh J. S., Cohn M., Biochemistry, 6, 1805 (1967).
23. Lipscomb W. N., Hartsuck J. A., Rieke G. N., Quiocho F. A. et al., Brookhaven.
Symp. Biol., 21, 24 (1968).
24. Riepe M„ Wang J. H., J. Biol. Chem., 243, 2779 (1968).
25. Mildvan A. S., Scrutton M. C., unpublished observations.
26. Busch D. H., in «Reactions of Coordinated Ligands», Adv. Chem. Ser., 37, 1
(1963).
27. Jones M. M., Ligand Reactivity and Catalysis, Academic Press, New York,
1968.
28. Herbert D., Discuss. Symp. Soc. Exptl. Biol., 5, 52 (1951).
29. Williams R. J. P„ Biol. Revs., 28, 381 (1953).
30. Bamann E., Rother H., Trapmann H., Naturwissenschaften, 93, 326 (1956).
31. Wang J. H., Science, 161, 328 (1968).
32. Wang J. H., Parker L., Proc. Natn. Acad. Sci. U. S., 58, 2451 (1967).
33. Wang J. H., in R. E. Forster, J. T. Edsall, A. B. Otis, F. J. W. Roughton
(eds.), CO2: Chemical, Biochemical and Physiological Aspects, NASA SP-188,
Gov’t Printing Office, Washington D. C., 1969, p. 101.
34. Vallee B. L., Williams R. J. P., Proc. Natn. Acad. Sci. U. S., 59, 498
(1968).
35. Mildvan A. S., Cohn M., J. Biol. Chem., 240, 238 (1965).
36. Scrutton M. C., unpublished observations (1968).
37. Scrutton M. C., Mildvan A. S., Biochemistry, 7, 1490 (1968).
37a. Dorey Z., Gray H. B., J. Am. Chem. Soc., 88, 1394 (1966).
376. Ciampolini M., Nardi N., Inorganic. Chem., 6, 445 (1967).
38. Mildvan A. S., Cohn M., J. Biol. Chem., 241, 1178 (1966).
39. Mildvan A. S., Hunsley J., Suelter С. H., in B. Chance, T. Yonetani, M. Cohn
(eds.). Probes for Macromolecular Structure and Function, Vol. 2, 1970,
p. 131.
40. Kachmar J. F., Boyer P. D., J. Biol. Chem., 200, 669 (1953).
41. Happold F. C., Beechey R. B., Biochem. Soc. Symp., 15, 52 (1958).
42. Evans H. T., Sorger G. I., A. Rev. Plant. Physiol., 17, 47 (1966).
43. Suelter С. H., Abstr. 158th Meeting ACS, Biol-56 (1969).
44. Stadtman E. R., Shapiro В. M., Kingdon H. S., Woolfolk C. A., Hubbard J. S.,
AAn. Enzymol. Regulation, 6, 257 (1968).
45. Suelter С. H., Singleton R., Kayne F. J., Arrington S., Glass J., Mildvan A. S.,
Biochemistry, 5, 131 (1966).
46. Kayne F. J., Suelter С. H., Biochemistry, 7, 1678 (1968)..
496
Глава 14
47. Webster L. T„ J. Biol. Chem., 240, 4010 (1965).
48. Webster L .T., J. Biol. Chem., 242, 1232 (1967).
49. Hummel J. P., Dryer W. J., Biochim. Biophys. Acta, 63, 530 (1962).
50. Colowick S. P„ Womack F. C., J. Biol. Chem., 244, 774 (1969).
52. Velick S. F„ Hayes J., Harting J., J. Biol. Chem., 203, 527 (1953).
53. Klotz I. M„ Walker F. M., Pivan R. B., Biochem. J., 68, 1486 (1946).
54. Labeyrie F., Stachiewicz E., Biophys. Acta, 52, 136 (1953).
55. Mildvan A. S., Leigh R. A., Biochim. Biophys. Acta, 89, 393 (1964).
56. Scrutton M. C„ Utter M. F„ J. Biol. Chem., 240, 3714 (1965).
57. Suelter С. H„ Melander W„ J. Biol. Chem., 238, PC 4108 (1963).
58. Boyer P. D„ Theorell H., Acta. Chem. Scand., 10, 447 (1956).
59. Cleland W. W., Biochim. Biophys. Acta, 67, 104 (1965).
60. Cleland W. W., Biochim. Biophys. Acta, 67, 188 (1963).
60a. Christensen H. N., Palmer G. A., Enzyme Kinetics, W. B. Saunders, Philadel-
phia, 1967.
606. Dalziel K, Acta. Chem. Scand., 11, 1706 (1957).
62. Beinert FL, Palmer G. A., Adv. Enzymol., 27, 105 (1965).
63. Peisach I., Levine W. G„ Blumberg W. E., J. Biol. Chem., 242, 2847 (1967).
64. Tsibris J. С. M., Tsai R. C., Gunsalus I. C., Orme-Johnson W. H., Han-
sen R. E„ Beinert H., Proc. Natn. Acad. Sci. U. S., 59, 959 (1968).
65. Orme-Johnson W. H., Hansen R. E., Beinert H., Tsibris J. С. M., Bartholo-
maus R. C., Gunsalus I. C., Proc. Natn. Acad. Sci. U. S., 60, 368 (1968).
66. Barfield M., Karplus M., J. Am. Chem. Soc., 91, 1 (1969).
67. Kotani M., Morimoto H., in A. Ehrenberg, B. G. Malmstrom, T. Vanngard
(eds.), Magnetic Resonance in Biological Systems, Pergamon Press, New
York, 1967, p. 135.
68. Blumberg W. E., Goldstein M., Lauber E., Peisach J., Biochim. Biophys. Acta,
99, 187 (1965).
69. Reed G. H., Cohn M., J. Biol. Chem., 245, 662 (1970).
71. Hamilton C. L., McConnell H. M„ in A. Rich, N. Davidson (eds.). Structural
Chemistry and Molecular Biology, W. Freeman, San Francisco, 1968, p. 115.
72. Taylor J. S., Leigh J. S., Cohn M., Proc. Natn. Acad. Sci. U. S., 64, 219
(1969).
73. Weiner H., Biochemistry, 8, 526 (1969).
74. Burr M., Koshland D. E., Proc. Natn. Acad. Sci. U. S., 52, 1017 (1964).
74a. Leigh J. S., J. Chem. Phys., 52, 2608 (1970).
746. Cohn M., Q., Rev. Biophys., 3, 61 (1970).
75. Kowalsky A., Cohn M„ A. Rev. Biochem., 33, 481 (1964).
76. Mildvan A. S., Cohn M., Adv. Enzymol., 33, 1 (1970).
76a. Sheard B., Bradbury E. M., in J. A. V. Butler, D. Noble (eds.), Progress in
Biophysics and Molecular Biology, Pergamon Press, New York, 1970,
p. 189.
77. Swift T. J., Connick R. E., J. Chem. Phys., 37, 307 (1962).
78. Luz Z., Meiboom S., J. Chem. Phys., 40, 2686 (1964).
79. Solomon I., Phys. Rev., 99, 559 (1955).
80. Bloembergen N., J. Chem. Phys., 27, 572 (1957).
81. Eisinger J., Shulman R. G„ Szymanski В. M., J. Chem. Phys., 36, 1712
(1962).
82. Cohn M., in A. Ehrenberg, B. G. Malmstrom, T. Vanngard (eds.), Magnetic
Resonance in Biological Systems, Pergamon Press, New York, 1967, p. 101.
83. Kobes R. D„ Mildvan A. S., Rutter W. J., Abstr. 158th ACS Meeting, Biol-58,
(1969).
84. Eigen M., Hammes G. G., Adv. Enzymol., 25, 1 (1963).
85. Cuatrecasas P., Fuchs S., Anfinsen С. B., J. Biol. Chem., 242, 3063 (1967).
86. Arnone A.. Bier C. J., Cotton F. A., Hazen E. E., Richardson D. C., Richard-
son J. C., Proc. Natn. Acad. Sci. U. S., 64, 420 (1969).
88. Coleman J. E., Biochemistry, 4, 2644 (1965).
Металлоферменты
497
89. Applebury М. L., Coleman J. E., J. Biol. Chem., 244, 709 (1969).
90. Drum D. E., Federation Proc., 29, 608 (1970).
91. Drum D. E., Li T. K-, Vallee B. L., Biochemistry, 8, 3792 (1969).
92. Curdel A., Iwatsubo M., FEBS Lett., 1, 133 (1968).
93. Harris M. L, Coleman J. E., J. Biol. Chem., 243, 5063 (1968).
93a. Markus Z., Miller G., Avigad G„ Appl. Microbiol., 13, 686 (1965).
94. Applebury M. L., Johnson B. L., Coleman J. E., J. Biol. Chem., 245, 4968
(1970).
95. Vallee B. L., Hoch F. L., Proc. Natn. Acad. Sci. U. S., 41, 327 (1955).
96. Theorell H., Nygaard A. P., Bonnichsen R., Acta Chem. Scand., 9, 1148
(1955).
96a. von Wartburg J. P., Bethune J. C., Vallee B. L., Biochemistry, 3, 1175
(1964).
97. Vallee B. L.. Hoch F. L., J. Biol. Chem., 225, 245 (1959).
98. Harrison J. H., Federation Proc., 22, 493 (1963).
99. Pfleider G., Hohnholz E., Biochem. Z., 331, 245 (1959).
100. Adelstein S. J., Vallee B. L„ J. Biol. Chem., 233, 589 (1958).
101. Vallee B. L., Wacker W. E. C„ J. Am. Chem. Soc., 78, 1771 (1956).
102. Terayama H., Vestling C. S., Biochim. Biophys. Acta, 20, 586 (1956).
103. Pfleiderer G., Jacket D., Wieland T., Biochem. Z., 330, 296 (1958).
104. Burkman D. E., Sandstead H. H., Park J. H., J. Biol. Chem., 245, 1036
(1970).
105. Vanderheiden B. S., Meinhaet J. O., Dodson R. G., Krebs E. G., J. Biol. Chem.,
237, 2095 (1962).
106. Yue R. H., NoItmann E. A., Kuby S. A., J. Biol. Chem., 244, 1353 (1969).
106a. Foster D. S., Colman R. F., J. Biol. Chem., 245, 6190 (1970).
107. Akeson A., Biochem. Biophys. Res. Commun., 17, 552 (1967).
108. Oppenheimer H. L., Green R. W., McKay R. H., Arch. Biochem. Biophys., 119,
552 (1967).
109. Theorell H., Bonnichsen R., Acta. Chem. Scand., 5, 1105 (1951).
110. Ehrenberg A., Dalziel K., Acta Chem. Scand., 12, 465 (1958).
111. Hayes J. E„ Velick S. F„ J. Biol. Chem., 207, 225 (1954).
113. Sandler N., McKay R. H., Biochem. Biophys. Res. Commun., 35, 151 (1969).
114. Scrutton M. C., Young M. R„ Utter M. F., J. Biol. Chem., 245, 6227 (1970).
115. Kobashi K-, Horecker B. L., Arch. Biochem. Biophys., 121, 178 (1969).
116. Pogell В. M., Scrutton M. C., unpublished observations (1969).
117. Shalaby R. F., Lauffer M. A., Biochemistry, 6, 2465 (1967).
118. Vallee B. L., Proc. 4th Int. Congr. Biochem., Vienna, 8, 138 (1969).
119. Stoppani А. О. M., Schwarez M. N., Freda C. A., Arch. Biochem. Biophys.,
113, 464 (1966).
120. Lin E. E. C., Magasanik B„ J. Biol. Chem., 235, 1820 (1960).
121. Vallee B. L., in P. D. Boyer, H. A. Lardy, K. Myrback (eds.), The Enzymes,
Vol. 3, 2nd edn, Academic Press, New York, 1960, p. 225.
122. Mildvan A. S., Weiner H., J. Biol. Chem., 244, 2465 (1969).
123. Theorell H., Yonetani T., Biochem. Z., 338, 537 (1963).
124. Kosower E. AL, Biochim. Biophys. Acta, 56, 474 (1962).
125. Mahler H. R„ Douglas J., J. Am. Chem. Soc., 79, 1159 (1957).
126. Evans N„ Rabin B. R., Eur. J. Biochem., 4, 548 (1968).
.127. Wallenfels K., Sund H., Biochem. Z., 329, 59 (1957).
128. Palmer G., Brintzinger H., in M. Klingenberg, T. E. King (eds.), A Treatise on
Electron and Coupled Energy Transfer in Biological Systems, 1970, in press.
129 Williams R. J. P-, Vallee B. L., Discsuss. Faraday Soc., 20, 262 (1955).
130 Hoch F. L., Williams R. J. P., Vallee B. L„ J. Biol. Chem., 232, 453 (1968).
131 Vallee B. L., Williams R. J. P., Hoch F. L., J. Biol. Chem., 234, 2621 (1959).
132. Dalziel K. Nature, 197, 462 (1963).
133. Theorell H., Chance B., Acta. Chem. Scand., 5, 1127 (1951).
134. Wratten С. C., Cleland W. W., Biochemistry, 2, 935 (1963).
32—2451
498
Глава 14
135. Silverstein Е„ Boyer Р. D., J. Biol. Chem., 239, 3908 (1964).
136. Anderson В. M., Reynolds M. L., Anderson C. D., Biochim. Biophys. Acta., 113,
235 (1966).
137. Anderson В. M., Reynolds M. L., Arch. Biochem. Biophys., Ill, 1 (1965).
138. Yonetani T., Theorell H„ Arch. Biochem. Biophys., 106, 243 (1964).
139. Hoagstrom C. W., Iweibo I., Weiner H„ J. Biol. Chem., 244, 5967 (1969).
140. Coleman P. L., Weiner H., Federation Proc., 29, 868 (1970).
141. Mildvan A. S„ Weiner H., Biochemistry, 8, 552 (1969).
143. Stengle T. R., Baldeschweiler J. D., Proc. Natn. Acad. Sci. U. S., 55, 1020
(1966).
144. Cottam G. L., Ward R. L., Arch. Biochem. Biophys., 132, 308 (1969).
146. Lelohn H. B., J. Biol. Chem., 243, 5126 (1968).
147. Lelohn H. B., Jackson S. G., Klassen G. R., Sawula R. G., J. Biol. Chem.,
244, 5346 (1969).
148. Powell G., Rajagopalan К. V., Handler P., J. Biol. Chem., 244, 4793 (1969).
149. Black S., Arch. Biochem. Biophys., 34, 86 (1951).
150. Arsenis C., Touster O., J. Biol. Chem., 244, 3895 (1965).
151. Kersters K., Wood W. A., DeLey J., J. Biol. Chem., 240, 965 (1965).
152. Jakoby W. B„ Fredericks J., Biochim. Biophys. Acta, 48, 26 (1961).
153. Mortlock R. P., Fossitt D. D., Wood W. A., Proc. Natn. Acad. Sci. U. S., 54,
572 (1965).
154. Mehler A. H„ Kornberg A., Grisolia S., Ochoa S., J. Biol. Chem., 174, 961
(1948).
155. Rutter W. J., Lardy H. A., J. Biol. Chem., 233, 374 (1958).
156. Korkes S., del Catnpillo A., Ochoa S., J. Biol. Chem., 187, 891 (1950).
157. Hayaishi M„ Hayaishi M„ Unemoto T., Biochim. Biophys. Acta, 122, 374
(1966).
158. Magee P. T., Snell E. E., Biochemistry, 5, 409 (1966).
159. Burns R. O., Umbarger H. E., Gross S. R., Biochemistry, 2, 1053 (1963).
160. Adler E., Von Euler J., Gunther G„ Plass M„ Biochem. J., 33, 1028 (1939).
161. Ochoa S., J. Biol. Chem., 174, 133 (1948).
162. Kornberg A., Pricer W. E., J. Biol. Chem., 189, 123 (1951).
163. Plaut G. W. E„ Sung S. C„ J. Biol. Chem., 207, 305 (1954).
164. Plaut G. W. E., in P. D. Boyer, H. A., Lardy, K. Myrback (eds.), The Enzy-
mes, Vol. 3, 2nd edn., Academic Press, New York, 1963, p. 105.
165. Rowley B„ Tucci A. F., Federation Proc., 29, 922 (1970).
166. Pontromeli S., DeFlora A., Grazi E., Mangiarotti G., Bonsignore A., Ho-
recker B. L., J. Biol. Chem., 236, 2975 (1961).
167. Kohn L. D„ Jakoby W. B., J. Biol. Chem., 243, 2486 (1968).
168. Hsu R. Y., Lardy H. A., J. Biol. Chem., 242, 527 (1967).
169. Hsu R. Y„ Lardy H. A., Cleland W. W„ J. Biol. Chem., 242, 5315 (1967).
170. Chen R. F., Brown D. M., Plaut G. W. E„ Biochemistry, 3, 552 (1963).
171. Ocha S., Weisz-Tabori E., J. Biol. Chem., 174, 123 (1948).
172. Siebert G., Carsiotis M., Plaut G. W. E., J. Biol. Chem., 226, 977 (1957).
173. Spina J., Bright J. J., Biochemistry, 9, 3794 (1970).
173a. Duggleby R. G., Dennis D. T., J. Biol. Chem., 245, 3745 (1970).
174. Northrop D. B., Cleland W. W., Federation Proc., 29, 408 (1970).
175. Parvin R., Pande S. V., Venditasubramanian T., Biochim. Biophys. Acta. 92,
260 (1964).
176. Boyer P. D., A. Rev. Biochem., 29, 17 (1960).
177. Steinberger R., Westheimer F. H., J. Am. Chem. Soc., 73, 429 (1961).
178. Scrutton M. C., Mildvan A. S., unpublished observations (1969).
178a. Scrutton M. C., Fleming J., Plaut G. W. E., unpublished observations (1970).
179. Arfin S. M., Umbarger H. E., J. Biol. Chem., 244, 1118 (1969).
180. Scrutton M. C„ Keech D. B„ Utter M. F„ J. Biol. Chem., 240, 574 (1965).
181. Kaziro Y., Leone E„ Ochoa S., Proc. Natn. Acad. Sci. U. S., 46, 1319 (1950).
Металлоферменты
499
182. Wood Н. G„ Lochmuller H., Rie per ting er C., Lunen F., Biochem. Z., 337, 247
(1963).
183. Stern J. R., Biochemistry, 6, 3545 (1967).
184. Galivan J. H., Allen S. H. G., J. Biol. Chem., 243, 1253 (1968).
185. Scrutton M. C., Utter M. F., Mildvan A. S„ J. Biol. Chem., 241, 3480 (1966).
186. Griminger P., Scrutton M. C., Federation Proc., 29, 765 (1970).
187. Scrutton M. C„ Griminger P., Wallace J. C., J. Biol. Chem., 247, 3305 (1972).
188. McClure W. R.. Federation Proc., 28, 728 (1969).
189. Scrutton M. C., Young M. R„ Federation Proc., 29, 597 (1970).
190. Northrop D. B., Wood H. G., J. Biol. Chem., 244, 5801 (1969).
191. Plaut G. W. E„ Lardy H. A., J. Biol. Chem., 180, 13 (1949).
193. Kornberg A., Ochoa S., Mehler A. H., J. Biol. Chem., 174, 159 (1948).
194. Scrutton M. C., Mildvan A. S„ Arch. Biochem. Biophys., 140, 131 (1970).
195. Kosicki G. W., Biochemistry, 7, 4299 (1968).
196. Kosicki G. W„ Biochemistry, 7, 4310 (1968).
197. Kosicki G. W„ Westheimer F. H., Biochemistry, 7, 4303 (1968).
198. Stiles M„ Ann. N. Y. Acad. Sci., 88, 332 (1960).
199. Caplow M., Yager M., J. Am. Chem. Soc., 89, 4513 (1967).
200. Caplow M., J. Am. Chem. Soc., 87, 5774 (1965).
201. Mildvan A. S., Scrutton M. C., Utter M. F., J. Biol. Chem., 241, 3488 (1966).
202. Sigel FL, McCormick D. B„ Griesser R., Priiys B., Wright L. D., Biochemistry,
8, 2687 (1969).
203. Scrutton M. C., Mildvan A. S., in R. E. Forster, J. T. Edsall, A. B. Otis,
F. J. W. Roughton (eds.), CO2: Chemical Biochemical and Physiological
Aspects NASA SP-188, Cov’t Printing Office, Washington, D. C., 1969,
p. 207.
204. Utter M. F., Scrutton M. C., in B. L. Horecker, E. R. Stadtman (eds.), Cur-
rent Topics in Cellular Regulation, Vol. 1, Academic Press, New York,
1969, p. 253.
205. Mildvan A. S., Scrutton M. C., Biochemistry, 6, 2978 (1967).
206. Eigen M., Tamm K., Z. Elektrochem., 66, 107 (1962).
207. Mildvan A. S., Scrutton M. C., unpublished observations (1970).
208. Rose I. A., J. Biol. Chem., 245, 6052 (1970).
209. Knappe 7., Wenger B., Wiegand U., Biochem. Z., 337, 232 (1963).
210. Bruice T. C„ Hegarty A. F., Proc. Natn. Acad. Sci. U. S., 65, 805 (1970).
210a. Northrop D. B., J. Biol. Chem., 244, 5808 (1969).
211. Srere P. A., Biochem. Biophys. Res. Commun., 26, 609 (1967).
212. Graham R. G., Scrutton M. C., unpublished observations (1969).
213. Prescott D. J., Rabinowitz J. L., J. Biol. Chem., 243, 1551 (1968).
214. Stoll E., Lane M. D., personal communication.
215. Utter M. F„ Kurahashi K-, J. Biol. Chem., 207, 787 (1954).
216. Lochmuller H„ Wood H. G., Davis J. J., J. Biol. Chem., 241, 5678 (1966).
217. Bandurski R. S., Greiner С. M., J. Biol. Chem., 204, 781 (1953).
218 Cooper T. G„ Tchen T. T„ Wood H. G„ Benedict C. R., J. Biol. Chem., 243,
3857 (1968).
219 Maruyama H., Easterday R. L., Chang H. C„ Lane M. D., J. Biol. Chem., 241,
2405 (1966).
220. Tietz A., Ochoa S., Arch. Biochem. Biophys., 78, 477 (1958).
221. Davis J. 7., Willard J. M., Wood H. G., Biochemistry, 8, 3127 (1969).
222 Miller R. S.. Mildvan A. S., Chang H. C., Easterday R. L„ Maruyama H„ La-
ne M. D., J. Biol. Chem., 243, 6030 (1968).
223. Reynard A. M„ Hass L. F., Jacobson D. D., Boyer P. D„ J. Biol. Chem., 236,
2277 (1961).
224. Miller R. S., Lane M. D„ J. Biol. Chem., 241, 6041 (1968).
225 Kirsch K., Mildvan A. S., Kowalsky A., Abstr. 158th ACS Mtg. BioL-52
(1969).
226. Willard J. M„ Davis J. J., Wood H. G„ Biochemsitry, 8, 3137 (1969).
32*
500
Глава 14
227. Wood Н. G., Davis J. J., Willard J. M., Biochemistry, 8, 3145 (1969).
228. Scrutton M. C., Willard J. M., Wood H. G„ unpublished observations (1970).
229. Weissbach A., Horecker B. L., Hurwitz J. J., J. Biol. Chem., 218, 795 (1956).
230. Kuehn G. D., McFadden B. A., Biochemistry, 8, 2394 (1969).
231. Anderson L. E., Price G. B., Fuller R. C., Science, 161, 182 (1968).
232. Calvin M., Federation Proc. 13, 697 (1954).
233. Wishnick M., Lane M. D., Scrutton M. C., J. Biol. Chem., 245, 4939 (1970).
234. Wishnick M., Lane M. D., J. Biol. Chem., 244, 55 (1969).
235. Akoyunoglou G„ Calvin M., Biochem. Z., 338, 20 (1963).
236. Wishnick M., Lane M. D., Scrutton M. C., Mildvan A. S., J. Biol. Chem., 244,
5761 (1969).
237. Lieberman L, Kornberg A., Sims E. S., J. Biol. Chem., 215, 403 (1955).
238. Snell E. E., Smucker A. A., Ringlemann E„ Lynen F„ Biochem. Z., 341, 109
(1964).
239. Scrutton M. C. in J. R. Norris, D. W. Ribbons (eds.), Methods in Microbiology,
Vol. 6A, Academic Press, New York, 1971, p. 479.
240. Krampitz L. O., A. Rev. Biochem., 38, 213 (1969).
241. Horecker B. L., Smyrniotis P. Z., Klenow H., J. Biol. Chem., 205, 661 (1963).
242. Gunsalus C. F., Stanier I. Y., Gunsalus I. C., J. Bact., 66, 548 (1953).
243. Singer T. P„ Penksy J., J. Biol. Chem., 196, 375 (1962).
243a. Namba Y., Yoshigawa K-> Ejima A., Hayaishi T., Kaneda T., J. Biol. Chem.,
244, 4437 (1969).
244. Morey A. V., Juni E., J. Biol. Chem., 243, 3009 (1968).
245. Hussain Qadri S. M„ Hoare D. S., Biochim. Biophys. Acta, 148, 304 (1967).
246. Krakow G., Barkulis S. S., Hayaishi J., J. Bact., 81, 509 (1961).
247. Kornberg H. L., Gotto A. M., Biochem. J., 78, 69 (1961).
247a. Quayle J. R., Biochem. J., 89, 492 (1963).
248. Schellenberger A., Angew. Chem. (Int. Edn.), 6, 1024 (1967).
249. Green D. E., Herbert D., Subrahmanyan V., J. Biol. Chem., 138, 327 (1941).
250. Schellenberger A., Winter K-, Hubner G., Schwaiberger R. et al., Hoppe-
Seylers Z. Physiol. Chem., 346, 123 (1966).
251. Schellenberger A., Hubner G., Hoppe-Seylers Z. Physiol. Chem., 348, 491
(1967).
252. Mortlock R. P., Wood W. A., J. Bact., 88, 835 (1964).
253. Mortlock R. P., Wood W. A., J. Bact., 88, 838 (1964).
254. Yamanaka K-, Agric. Biol. Chem. (Japan), 27, 271 (1963).
255. Anderson R. L., Allison D. P., J. Biol. Chem., 240, 2367 (1965).
256. Domagk G. F„ Zech R., Biochem. Z„ 339, 145 (1963).
257. Rose I. A., O’Connell E. L., Mortlock R. P., Biochim. Biophys. Acta, 178, 376
(1969).
258. Yamanaka K-, Biochim. Biophys. Acta, 151, 670 (1968).
259. Yamanaka K-, Arch. Biochem. Biophys., 131, 502 (1969).
260. Mildvan A. S., Rose I. A., Federation Proc., 28, 534 (1969).
261. Yamanaka K., in S. P. Colowick, N. O. Kaplan (eds.), Methods in Enzymolcr-
gy, Vol. 9, Academic Press, New York, 1966, p. 588.
261a. Rose I. A., Btrookhaven Symp. Biol., 15, 293 (1962).
262. Gracy R. W., NoItmann E. A., J. Biol. Chem., 243, 4109 (1968).
262a. Bruns F. H., NoItmann E. A., Nature, 181, 1467 (1958).
2626. Gracy R. W., NoItmann E. A., Federation Proc., 28, 520 (1968).
263. Scrutton M. C., unpublished observations (1969).
264. Rutner A., Scrutton M. C., unpublished observations (1969).
265. Shah D. H„ Cleland W. W., Porter J. W., J. Biol. Chem., 240, 1946 (1965).
266. Sistrom W. R., Stanier R. Y„ J. Biol. Chem., 210, 821 (1954).
267. Annett R. G., Kosicki G. W., J. Biol. Chem., 244, 2059 (1969).
267a. Weil-Malherbe H., Biochem. J., 101, 169 (1966).
268. Cori C. F., Colowick S. P., Cori G. T., J. Biol. Chem., 121, 465 (1937).
269. Ressig J. L., J. Biol. Chem., 219, 753 (1956).
Металлоферменты
501
270. Lang К., Hartmann К. V., Experientia, 14, 130 (1958).
271. Rodwell К. V., Towne J. C., Grisolia S., J. Biol. Chem., 228, 875 (1957).
272. Ray W. J., Roscelli G. A., J. Biol. Chem., 239, 1228 (1964).
273. Ray W. J., Roscelli G. A., Kirkpatrick D. S., J. Biol. Chem., 241, 2603 (1966).
274. Ray W. J., Roscelli G. A., J. Biol. Chem., 241, 3499 (1966).
275. Najjar V. A., J. Biol. Chem., 175, 281 (1948).
276. Robinson J. P., Najjar V. A., Biochem. Biophys. Res. Commun., 3, 62 (1960).
277. Ray W. J., J. Biol. Chem., 244, 3740 (1969).
278. Peck E. J., Ray W. J., J. Biol. Chem., 244, 3748 (1969).
279. Peck E. J.. Ray W. J., J. Biol. Chem., 244, 3754 (1969).
279a. Ray W. J., Mildvan A. S., Biochemistry, 9, 3886 (1970).
2796. Peck E. J., Kirkpatrick D. S., Ray W. J., Biochemistry, 7, 152 (1968).
280. Imsande J., Handler P., in P. D. Boyer, H. A. Lardy, K. Myrback (eds.), The
Enzymes, Vol. 5, 2nd edn., Academic Press, New York, 1961, p. 281.
280a. Warburg 0., Christian W., Biochem. Z., 314, 419 (1943).
281. Rutter W. J., Federation Proc., 23, 1248 (1964).
282. Jagannathan V., Singh K., Daniodavan M., Biochem. J., 63, 94 (1956).
283. Richards О. C„ Rutter W. J., J. Biol. Chem., 236, 3177 (1961).
284. Bard R. C., Gunsalus I. C„ J. Bact., 59, 387 (1950).
285. Fewson C. A., Al-Hafidh M„ Gibbs M., Pl. Physiol., 37, 402 (1962).
286. Grazi E., Cheng T., Horecker B. L., Biochem. Biophys. Res. Commun., 7, 250
(1962).
287. Grazi E., Rowley P. T., Cheng T„ Tchola O., Horecker B. L., Biochem. Bio-
phys. Res. Commun., 9, 38 (1962).
288. Morse D. E., Horecker B. L., Adv. Enzymol., 31, 125 (1968).
289. Warren S., Zerner B., Westheimer F. H., Biochemistry, 5, 817 (1966).
289a. Rutter W. J., in V. Bryson, H. J. Vogel (eds.), Evolving Genes and Proteins,
Academic Press, New York, 1965, p. 279.
290. Harris С. E., Kobes R. D„ Teller D. C., Rutter W. J., Biochemistry, 8, 2442
(1969).
291. Kobes R. D., Simpson R. T., Vallee B. L., Rutter W. J., Biochemistry, 8, 508
(1969).
292. Mildvan A. S., Kobes R. D., Rutter W. J., Biochemistry, 10, 1191 (1971).
292a. Heath E. C., Ghalambor M. A., J. Biol. Chem., 237, 2427 (1962).
293. Chiu T. H., Smith C. J., Feingold D. S„ Federation Proc., 27, 520 (1968).
293a. Fish D. C., Blumenthal H. J., Bact. Proc., 1963, 110.
294. McIntosh E. N., Purko M„ Wood W. A., J. Biol. Chem., 228, 499 (1957).
294a. Shannon L. M., Marcus A., J. Biol. Chem., 237, 3342 (1962).
295. Olson J. A., J. Biol. Chem., 234, 5 (1959).
296. Stegink L. D., Coon M. J., J. Biol. Chem., 243, 5272 (1968).
297. Dagley S., Dawes E. A., Biochim. Biophys. Acta, 17, 177 (1955).
298. Ward R. L., Srere F. A., Biochim. Biophys. Acta, 99, 270 (1965).
299. Bowen T. J., Rogers L. J., Biochim. Biophys. Acta, 77, 685 (1963).
300. Eisenthal R., Tate S. S., Datta S. P„ Biochim. Biophys. Acta, 128, 155
(1966).
301. Blair J. McD., Datta S. P., Tate S. S., Eur. J. Biochem., 1, 26 (1967).
302. Singh M., Srere P. A., Federation Proc., 29, 929 (1970).
303. Malmstrom B. G., Vanngard T., Larsson M., Biochim. Biochim. Biophys. Acta,
30, 1 (1958).
304. Wold F„ Ballou C. E„ J. Biol. Chem., 227, 313 (1957).
305. Malmstrom B. G„ Arch. Biochem. Biophys., 58, 398 (1955).
306. Hanlon D. P., Westhead E. W., Biochemistry, 8, 4247, 4255 (1969).
308. Brewer J. M., Weber G„ J. Biol. Chem., 241, 2550 (1966).
309. Dickman S. R., Cloutier A. A., J. Biol. Chem., 188, 379 (1951).
310. Morrison J. F„ Biochem. J., 58, 685 (1954).
311. Speyer J. F„ Dickman S. R„ J. Biol. Chem., 220, 193 (1956).
502
Глава 14
312. Sichhorn G. L., in Reactions of Coordinated Ligands, Am. Chem. Soc. Adv.
Chem. Ser., 37, 37 (1963).
313. Glusker J. P„ J. Mol. Biol., 38, 149 (1968).
314. Gawron O., Glaid A. J., Fondy T. P„ J. Am. Chem. Soc., 83, 3634 (1961).
315. Rose I. A., O’Connell E. L„ J. Biol. Chem., 242, 1870 (1967).
316. Fanshier D. W., Gottwald L. К., Kun E„ J. Biol. Chem., 239, 425 (1964).
317. Kun E., in J. M. Lowenstein (ed.), The Citric Acid Cycle: Control and Com-
partmentation, Marcel Dekker, New York, 1969, p. 318.
318. Carrell H. L., Glusker J. P„ Villafranca J. J., Mildvan A. S., Duminel R. J.,
Kun E., Science, 170, 1412 (1970).
319. Villafranca J. J., Mildvan A. S., J. Biol. Chem., 246, 772 (1971).
320. Wang С. C„ Barker H. A., J. Biol. Chem., 244, 2516 (1969).
320a. Hurlbert R. E„ Jakoby W. B„ J. Biol. Chem., 240, 2774 (1965).
321. Smiley J. D., Ashwell G., J. Biol. Chem., 235, 1571 (1960).
322. Wixom R. L., Shatton J. B., Strassman M., J. Biol. Chem., 235, 128 (1960).
323. Palleroni N. J., Doudoroff M„ J. Biol. Chem., 223, 499 (1956).
324. Ames B. N„ J. Biol. Chem., 228, 131 (1957).
325. Komai H., Neilands J. P., Biochim. Biophys. Acta, 171, 13 (1969).
326. Wilson M. L„ lodice A. /., Shulman M. P„ Richert D. A., Federation Proc.,
18, 352 (1959).
327. Greenberg D. M., in P. D. Boyer, H. A. Lardy, K. Myrback (eds.), The Enzy-
mes, Vol. 5, 2nd edn.. Academic Press, New York, 1961, p. 563.
328. Alberty R. A., in P. D. Boyer, H. A. Lardy, K. Myrback (eds.), The Enzymes,
Vol. 5, 2nd edn.. Academic Press, New York, 1961, p. 531.
329. Stern J. R., in P. D. Boyer, H. A. Lardy, K. Myrback (eds.), The Enzymes,
Vol. 5, 2nd edn., Academic Press, New York, 1961, p. 511.
330. Bright H. J.. J. Biol. Chem., 240, 1198 (1965).
331. Givot I. L., Mildvan A. S., Abeles R. H„ Federation Proc., 29, 531 (1970).
332. Barker H. A., Smyth R. D., Wilson R. M., Weissbach H., J. Biol. Chem., 234,
320 (1959).
333. Bright H. J., Ingraham L. L., Biochim. Biophys. Acta, 44, 586 (1964).
334. Williams V. R„ Selbin J., J. Biol. Chem., 239, 1635 (1964).
335. Givot I. L., Smith T. A., Abeles R. H., J. Biol. Chem., 244, 6341 (1969).
336. Bright H. J., J. Biol. Chem., 239, 2307 (1964).
337. Rechler M. M., J. Biol. Chem., 244, 551 (1969).
338. Peterkofsky A., J. Biol. Chem., 237, 787 (1962).
339. Havir E. A., Hanson K. R., Biochemistry, 7, 1904 (1968).
340. Hodgins D. S„ Biochem. Biophys. Res. Commun., 32, 246 (1968).
340a. Frankfort A., Fridovich I., Biochim. Biophys. Acta, 206, 457 (1970).
341. Hsiu I., Fischer E. H„ Stein E. A., Biochemistry, 3, 61 (1964).
341a. Takagi T„ Isemura T., J. Biochem. (Tokyo), 57, 89 (1965).
342. Friedmann T., Epstein C. J., J. Biol. Chem., 242, 5131 (1967).
343. Pfueller S. L„ Elliot W. H., J. Biol. Chem., 244, 48 (1969).
345. Neumann W., Habermann E., Z. Physiol. Chem., 296, 166 (1954).
346. Davidson F. M„ Long C„ Biochem. J., 69, 458 (1958).
347. Vallee B. L„ Stein E. A., Sumerwell W. N., Fischer E. H., J. Biol. Chem., 234,
2901 (1959).
348. Fishbein W. N., Bessman S. P., J. Biol. Chem., 241, 4842 (1966).
349. van der Drift C., Vogels G. D„ Biochim. Biophys. Acta, 198, 339 (1970).
350. Middlehoven W. J., Biochim. Biophys. Acta, 191, 110, 122 (1969).
351. Jencks W. P., in P. D. Boyer, H. A. Lardy, K. Myrback (eds.), The Enzymes,
Vol. 6, 2nd edn., Academic Press, New York, 1962, p. 373.
352. Webster L. T„ J. Biol. Chem., 238, 4010 (1963).
353. Norris A. T., Berg P., Proc. Natn. Acad. Sci. U. S., 52, 330 (1964).
354. McElroy W. D„ Green A. F., Arch. Biochem. Biophys., 64, 257 (1956).
357. Webster L. T., Gerowin L. D„ Rakita L., J. Biol. Chem., 240, 29 (1965).
Металлоферменты
503
358. Londesborough J. C., Webster L. T., Scrutton, M. C., unpublished observations
(1969).
359. Webster L. T., J. Biol. Chem., 241, 5504 (1966).
360. Svensson I., Biochim. Biophys. Acta, 146, 239 (1967).
361. Kingdon H. S., Webster L. T„ Davie E. W., Proc. Natn. Acad. Sci. U. S., 44,
757 (1958).
362. Webster L. T., Davie E. W„ Biochim. Biophys. Acta, 35, 559 (1959).
363. Lagerkvist U., Rymo L., Waldenstrom J., J. Biol. Chem., 241, 5391 (1966).
364. Clark J. M., Eyzaguirre J. P., J. Biol. Chem., 237, 3698 (1962).
365. Bublitz C., Biochim. Biophys. Acta, 128, 165 (1966).
366. Bublitz C., Biochim. Biophys. Acta, 133, 158 (1966).
366a. Cole F. X., Schimmel P. R., Biochemistry, 9, 3143 (1970).
367. Allende J. E., Mora G., Gatica M., Allende С. C., J. Biol. Chem., 241, 2245
(1966).
368. Grosjean G., Charlier J., Vanhumbeeck J., Biochem. Biophys. Res. Commun.,
32, 935 (1968).
368a. Loftfield R. B., Eigner E. A., J. Biol. Chem., 242, 5355 (1967).
3686. Lindahl T., Adams A., Fresco J. R., Proc. Natn. Acad. Sci. U. S., 55, 944
(1961).
369. Himes R. H., Rabinowitz J. C„ J. Biol. Chem., 237, 2903 (1962).
370. Kaziro Y., Ochoa S., Adv. Enzymol., 26, 283 (1964).
371. Scrutton M. C., Mildvan A. S„ unpublished observations (1963).
372. Scrutton M. C., Olmsted M. R., Utter M. F., in S. P. Colowick, N. O. Kaplan
(eds.), Methods in Enzymology, Vol. 13, Academic Press, New York, 1969,
p. 235.
373. Keech D. B„ Barritt G. J., J. Biol. Chem., 242, 1983 (1968).
374. Blair J. McD., FEBS Lett., 2, 245 (1968).
375. Meister A., in P. D. Boyer, H. A. Lardy, K. Myrback (eds.), The Enzymes,
Vol. 6, 2nd edn., Academic Press, New York, 1962, p. 443.
376. Woolfolk C. A., Shapiro В. M., Stadman E. R., Arch. Biochem. Biophys., 116,
177 (1966).
376a. Kingdon H. S., Shapiro В. M., Stadtman E. R., Proc. Natn. Acad. Sci. U. S„
58, 1703 (1967).
377. Kingdon H. S., Hubbard I. S., Stadtman E. R., Biochemistry, 7, 2136 (1968).
378. Denton M. D., Ginsburg A., Biochemistry, 8, 1714 (1969).
379. Valentine R. C., Shapiro В. M., Stadtman E. R., Biochemistry, 7, 2143
(1968).
380. Shapiro В. M., Ginsburg A., Biochemistry, 7, 2153 (1968).
381. Gottschalk A., Eur. J. Biochem., 7, 301 (1969).
382. Robinson J. R., Sagers R. D., Bact. Proc., 1970, 124.
383. Benziman M., Sagers R„ Gunsalus I. C., J. Bact., 79, 474 (1960).
384. Su J. C., Li С. C., Ting С. C., Biochemistry, 5, 536 (1966).
385. Margoliash E., A. Rev. Biochem., 30, 549 (1961).
386. Kosicki G. W„ Lee L. P. K., J. Biol. Chem., 241, 3571 (1966).
ГЛАВА 15
КАРБОКСИПЕПТИДАЗА А
И ДРУГИЕ ПЕПТИДАЗЫ*
Марта Л. Людвиг**, В. Н. Липскомб
Ludwig Martha L„ Biophysics Research Division, Institute
of Science and Technology, University of Michigan and
Biological Chemistry Department, University of Michigan
Medical School, Ann Arbor, Michigan, USA
Lipscomb W. H., Chemistry Department, Harvard University,
Cambridge, Massachusetts, USA
1. КАРБОКСИПЕПТИДАЗА А БЫКА: ВЫДЕЛЕНИЕ,
СВОЙСТВА И СПЕЦИФИЧНОСТЬ
Для карбоксипептидазы А (КПА) из поджелудочной железы
быка известны и трехмерная структура [1—3], и полная амино-
кислотная последовательность [4]. Роль существенно важного ме-
талла установлена менее определенно, но она доступна изучению
с помощью разнообразных спектроскопических [5, 6] и кинетиче-
ских [7, 8] методов. Следовательно, этот фермент можно включить
во все увеличивающийся список белков, для которых возможно
провести корреляции структуры и функции. Любой предлагаемый
механизм катализа под действием КПА должен теперь учитывать
как накопленные химические данные, так и взаимодействия, на-
блюдавшиеся при структурном исследовании кристаллов комплек-
са карбоксипептидазы с модельным субстратом глицил-ь-тирози-
ном [3, 9].
Карбоксипептидаза А вырабатывается ацинарными клетками
поджелудочной железы в виде неактивного профермента. Молеку-
ла прокарбоксипептидазы быка имеет молекулярную массу при-
мерно 87 000 [10] и состоит из трех субъединиц, одна из которых
с молекулярной массой 40 000—42 000 и является непосредствен-
ным предшественником КПА [11]. Активную КПА можно полу-
чить любым из следующих трех методов: 1) фракционированием
автолизата замороженных поджелудочных желез (Энсон [12]);
2) фракционированием водного экстракта ацетонового порошка
поджелудочной железы после активации трипсином (Аллан [13]);
3) очисткой про-КПА хроматографией на ДЭАЭ-ионообменни-
ках с последующим расщеплением трипсином (Кокс [14]). Про-
* Сокращения: КПА — карбоксипептидаза А, КПВ — карбоксипептидаза В,
про-КПА — прокарбоксипептидаза А, ЛАП — лейцинаминопептидаза, КБЗ —
карбобензокси, Е — фермент, S — субстрат, I — ингибитор, Ак;1Т — каталитическая
константа, Км — константа Михаэлиса.
** Обладатель поощрительной премии (1 KO4GMO6611) службы здравоохра-
нения США.
Карбоксипептидаза А и другие пептидазы
505
5 10 15 20 25
ALA-ARC-SER-THR-ASN-THR-PHE-ASN-TYR-ALA-TKR-TYR-HIS-THR-LEU-ASP-GLU-ILE-TYR-ASP-PHE-MET-ASP-LEU-LEU-
Г f t
X ₽ Y
30 35 40 45 50
VAL-ALA-GLN-H1S-PRO-GLU-LEU-VAL-SER-LYS-LEU-GLN-ILE-GLY-ARG-SER-TYR-GLU-CLY-ARG-PRO-ILE-TYR-VAL-LEU-
X.YS-EHE-SER-THR-GLY-CLY-SER-ASN-ARG-PR0-A1A-ILE-TRP-ILE-ASP-LEU-CLY-ILE-HIS-SER-ARG-GLU-TRP-ILE-THR-
80 85 90 95 100
GLN-ALA-THR-GLY-VAL-IRP-PHE-ALA-LYS-LYS-PHE-THR-GLU-ASN-TYR-GLY-GLN-ASN-PRO-SER-PHE-THR-ALA-ILE-LEU-
ASP-SER-MET-ASP-ILE-PHE-LEU-GLU-ILE-VAL-THR-ASN-PRO-ASN-GLY-PHE-ALA-PHE-THR-HIS-SER-GLU-ASN-ARG-LEU;
130 135 140 145 150
TRP-ARG-LYS-THR-ARG-SER-VAL-THR-SER-SER-SER-LEU-CYS-VAL-GLY-VAL-ASP-ALA-ASN-ARG-ASN-TRP-ASP-ALA-CLY-
PHE-GLY-LYS-ALA-CLY-ALA-SER-SER-SER-PRO-CYS-SER-GLtJ-THR-IYR-HIS-CLY-LYS-TYR-ALA-ASN-SER-GLU-VAL-GLU-
VAL-LYS-SER-^^-tl£-ASP-PHE-VAL-LYS-l§^-HIS-CLY-ASH-PHE-LYj-ALA-PHE-LEU-SER-ILE-HIS-SER-TYR-SER-GLH-
205 210 215 220 225
IE U-LEU-LEU-TYR-PRO-TYR-GLY-TYR-THR-THR-GLN-SER-ILE-PRO-ASP-LYS-THR-GLI1-LEH-ASN-GLN-VAL-A1.A-LYS-SER-
ala-val-^^-aia-£3B-lys-ser-leu-tyr-cl?-thr-ser-tyr-lys-tyS-gly-ser- ILE-ILE-THR-THR-ILE-TYR-CLN-аЙ-
ser-gly-cly-ser-iS-asp-trp.ser-tyr-asn-cln-cly-ile-lys-tyr-ser-phe-thr-phe-glu-leu-arc-asp-thr-gly-
ARC-TYR-CLY-PHE-£88-LEU-PRO-ALA-SER-CT.N-TLE-TLE-PRn-THR-A?A-CT.N-GU1-THR-TRP-rF:n-CLY-VAI.-T.EII-THR-T1>ik
305
MET-GLU-HIS-THR-^-ASN-ASN
Рис. 15.1. Аминокислотная последовательность КПА из поджелудочной железы
быка [4].
N-концы форм а. р и у (или 6) соответствуют остаткам 1, 3 и 8, как показано стрелками.
В KnALeU в положениях 179, 228 и 305 находятся аминокислоты, приведенные над чертой,
Vai
тогда как в КПА их место занимают аминокислоты, указанные под чертой. Сегменты
цели, которые, судя по трехмерным моделям комплекса КПА с ацнлтрипептндом, образуют
активный центр, подчеркнуты.
дукт, полученный любым из этих трех методов, содержит пример-
но один атом цинка на молекулу [15] и состоит из одной полипеп-
тидной пепи [16].
Последующее хроматографическое разделение в присутствии
p-фенилпропионата обнаружило в этих препаратах два типа ге-
терогенности [17, 18]. Поскольку в процессе активации не проис-
ходит количественного расщепления единственной пептидной связи
506
Глава 15
профермента, то в зависимости от условий активации (см. работу
[19]) могут образовываться формы карбоксипептидазы, обознача-
емые буквами а, р или у (или б) [20, 21]. Различия в длине цепи
показаны на рис. 15.1. Препаративные методы Энсона (у-форма)
[12] и Аллана (б-форма) [13] дают главным образом цепь дли-
ной 300 остатков, в то время как при активации очищенной про-
КПА получается в основном a-форма с молекулярной массой
35472 [4, 22], состоящая из 307 остатков [1] (рис. 15.1). Кроме
того, КПА может существовать в виде двух аллотипных форм [23].
В одной из них, КПАУа1, остаток 179 является изолейцином, оста-
ток 228 — аланином, а остаток 305 — валином. Остатки, находя-
щиеся на их месте во втором аллотипе, КПАЬеи, приведены на
рис. 15.1.
Кристаллы КПАб и КПАО изоморфны. Неодинаковое распреде-
ление электронной плотности в районе N-конца приводит к неболь-
шим различиям в интенсивностях дифракционных пятен. Кристал-
лы препарата Энсона, т. е. КПАу, принадлежат к той же прост-
ранственной группе, что и кристаллы двух других карбоксипепти-
даз, однако по своим характеристикам и дифракционной картине
они очень сильно отличаются от них. Хотя и КПА и КПА? состоят
главным образом из цепей, имеющих на N-конце Asn, различия в
кристаллографических свойствах и стабильности апоферментов
[24] указывают на то, что они химически неодинаковы. При ана-
лизе кристаллической структуры была использована КПАа, в то
время как аминокислотная последовательность была определена
для КПА?.
Специфичность карбоксипептидазы А такова, что она отщепля-
ет l-аминокислоты от С-конца белков и модельных пептидных
субстратов*. При работе с чистыми препаратами эндонуклеазная
активность не обнаруживается. КПА не гидролизует простые ами-
ды с образованием аммиака, а также не отщепляет от С-конца
пептидной цепи а-амиды аминокислот. Гидролиз протекает быст-
рее всего в том случае, если боковая цепь остатка на С-конце име-
ет гидрофобный характер [26. 27]. Величина /г11ат для классиче-
ских модельных пептидных субстратов — КБЗ-глицил-ь-фенилала-
нина (КГФ) (рис. 15.2) и бензоилглицил-l-фенилаланина
(БГФ)—составляет примерно 104 мин-1. Эфирный аналог БГФ,
бензоилгилицил-ь-фениллактат (БГФЛ), также быстро гидролизу-
ется с каталитической константой примерно 3-104 мин-1 [28, 29].
В противоположность этому N-ациламинокислоты и дипептиды реа-
гируют значительно медленнее (табл. 15.4). Замещение у любого
из атомов азота пептидной связи в ацилдипептидных субстратах
* Стереоспецифичность не является абсолютной: в некоторых модельных пеп-
тидах наблюдалось очень медленное отщепление от D-конца остатка D-аланина
[25]. Освобождение концевых D-аминокислот большего размера не наблюдалось.
Карбоксипептидаза А и другие пептидазы
507
заметно уменьшает скорость гидролиза [26, 27]. Остатки дикарбо-
новых кислот, а также глицина и аланина отщепляются от пепти-
дов тоже сравнительно медленно в диапазоне pH 7,5—8,0, т. е. в
условиях, оптимальных для гидролиза таких модельных субстра-
тов, как БГФ. Появление в ходе гидролиза аргинина или пролина
на С-конце практически прекращает дальнейший процесс [30]. На
величины кинетических параметров реакции влияет природа не
только двух аминокислотных остатков, образующих расщепляе-
мую связь, но также, хотя и в меньшей степени, трех ближайших
Рис. 15.2. Карбобензоксиглицилфенилаланнн — модельный пептидный субстрат
карбоксипептидазы А. Гидролизуемая связь указана штриховой линией.
к ним остатков [31]. Следовательно, размеры участка связывания
пептидов таковы, что обеспечивают взаимодействие с пятью остат-
ками, причем предпоследний остаток цепи оказывает заметное вли-
яние на кинетику реакции. Присутствие остатков некоторых D-ами-
нокислот в положениях, отличных от С-концевого, не препятствует
взаимодействию с ферментом, однако если объемистая d-амино-
кислота является предпоследним остатком от С-конца, то гидролиз
идет только в случае дипептидных субстратов [32, 33].
Карбоксипептидазы, выделенные из млекопитающих и из бо-
лее примитивных видов, таких, как обыкновенная колючая акула,
требуют для активности ион металла. Предположение о присутст-
вии в КПА иона металла было сделано на основании анализа с
помощью ингибиторов [34]. Впоследствии было показано, что он
является ионом цинка и существен для активности [35]. Неактив-
ный апофермент может быть получен диализом против растворов
разнообразных хелатирующих агентов, в том числе о-фенантроли-
на и окснхинолинсульфоната [36, 37]. Апоферменты, полученные
из КПА а и КПА б. достаточно стабильны для использования в изу-
чении связывания металлов и субстратов, в то время как апофер-
мент КПА? по неизвестным причинам быстро денатурируется [24].
Реконструкцией из апо-КПА можно приготовить целый набор ме-
таллокарбоксипептидаз [38, 39]. Их каталитическая эффектив-
ность зависит от природы металла и наиболее высока в случае
Со2+, Мп2+ и Ni2+ (табл. 15.5). Выдерживание Zn-КПА в присут-
ствии другого подходящего иона в соответствующей концентрации
приводит к замещению им цинка [40].
Кажущиеся константы стабильности Со (И)-КПА были опреде-
лены измерением количества связанного 60Со в равновесных сме-
508
Глава 15
сях металла и апофермента. Для более прочно связывающихся
металлов (табл. 15.5) эти константы были получены из экспери-
ментов, в которых с белком уравновешивались пары конкурирую-
щих металлов [41]. Кроме ионов, приведенных в табл. 15.5, актив-
ность апо-КПА восстанавливают Сг3+ и Fe2+ [38]. Если предполо-
жить, что реакция цинка с КПА в диапазоне pH 7—9 протекает
стехиометрически (что подтверждается данными, приведенными в
табл. 15.5), то выделение протонов при присоединении цинка к
КПА контролируется двумя группами с рКа 7,7 и 9,1. Предполага-
лось, что эти группы участвуют в связывании цинка [41]*.
2. СТРУКТУРА КАРБОКСИПЕПТИДАЗЫ А
И ЕЕ КОМПЛЕКСА С ГЛИЦИЛТИРОЗИНОМ
2.1. Общее описание конформации белка
Атомные модели карбоксипептидазы А получены совмещением
последовательности 307 остатков с картой электронной плотности
при номинальном разрешении 2 А [1—3]. Фазы отраженных рент-
геновских лучей были определены методом изоморфного замеще-
ния с помощью четырех производных тяжелых атомов при разре-
шении 2,8 А и двух производных с разрешением, промежуточным
между 2,0 и 2,8 А. В двух ртутных производных ртуть замещала
цинк в активном центре (табл. 15.1). При совмещении полипептид-
ной цепи с картой электронной плотности не возникло чрезмерных
трудностей, хотя для некоторых остатков, например 133—137,
электронная плотность была существенно меньше, чем в среднем
по цепи. Оказалось, что остатки 138 и 161 соединены дисульфид-
ным мостиком [1]. Как и ожидалось, наибольшая электронная
плотность соответствовала иону цинка. При анализе карты элект-
ронной плотности было обнаружено, что цинк имеет три лиганда,
соответствующие остаткам 69, 72 и 196. Остатки 69 и 72 были пра-
вильно идентифицированы как His и Glx [1]. Отождествление ос-
татка 196 с Lys или Glx оказалось ошибочным [22].
При определении аминокислотной последовательности, приве-
денной на рис. 15.1, начальная фрагментация проводилась дейст-
вием BrCN по остаткам метионина 22, 103 и 301 [42—44]. Взаим-
ное расположение этих фрагментов было определено изучением
пептидов, содержащих метионин [21], и было подтверждено срав-
нением с картой электронной плотности. Последующее расщепле-
ние самого большого фрагмента цепи, 104—301, потребовало ис-
пользования пяти различных протеолитических ферментов [4].
* Теперь, когда известно, что лигандами цинка служат остатки гистидина и
глутаминовой кислоты .интерпретация этих значений р/Са 'Представляется более
сложной.
Карбоксипептидаза А и другие пептидазы
509
Таблица 15.1
Присоединение тяжелых металлов к карбоксипептидазе
Атом Заполнение (число элек- тронов на молекулу) Координаты тяжелого атома Остаток
Pbt 58 —0,094 0,500 —0,089 Glu-270
Pb2 53 —0,089 0,540 -0,147 Небелковый цитрат
Hg,ga 50 —0,071 0,455 —0,115 His-69, Glu-72, His-196
HgA 47 —0,071 0,452 —0,115 His-69, Glu-72, His-196
Hg,s2 46 —0,506 0,069 —0,257 His-29
Hg,s3 48 —0,475 0,109 —0,136 His-29, Lys-84
Pti 74 0,341 0,430 0,034 Cys-161
Pt2 45 —0,438 0,305 —0,568 Met-103
Pt3 68 —0,292 0,082 0,141 Ala-1 (N-конец)
Pti 27 —0,484 0,485 —0,500 His-303
Agj 0,238 0,498 —0,278 His-166, Ser-158
Aga —0,082 0,220 0,193 His-120
Ag3 —0,457 0,090 —0,143 His-29 (Lys-84)
Ag4 —0,483 0,477 —0,516 His-303
Coj —0,500 0,500 —0,500 His-303
Co2 —0,500 0,070 —0,130 His-29 (Lys-84)
Znd —0,087 0,443 —0,115 His-69, Glu-72, His-196
аДля получения КПА с Hg.g 5-10-4 М фермент диализовали против 1 мМ HgCi,. 1 М
LiCl, 0,2 М трис-буфера (pH 8) и кристаллизовали диализом против 0,18 М LIC1, 0,02 М
трнс-буфера (pH 8). Это производное, содержащее один атом металла, практически не дава-
ло информации о фазовых углах при высоком разрешении. Центры Hg.si, Hg.sj и Hg.Sa
заполняются при суспендировании кристаллов КПА в 0,8 мМ Hg^.
В согласии с данными рентгеноструктурного анализа остатки ци-
стеина были обнаружены в положениях 138 и 161. При определе-
нии последовательности удалось установить, что остаток 196, тре-
тий лиганд цинка, является гистидином. Природа двух других
лигандов, остатков 69 (His) и 72 (Glu), была при этом подтвер-
ждена.
Для сравнения с результатами определения аминокислотной
последовательности была предпринята попытка идентифицировать
боковые цепи методом рентгеноструктурного анализа [2, 3]. В за-
висимости от участка молекулы правильность определения остат-
ков варьировала от 60 до 85% [22]. Остатки, участвующие в свя-
зывании субстрата и в катализе (Arg-145, Туг-248 и Glu-270), были
510
Глава 15
идентифицированы правильно. При окончательном рассмотрении
некоторых участков структуры, в которых обнаруживались проти-
воречия между картой электронной плотности и последовательно-
стью, были рассчитаны дополнительные карты, причем для опре-
деления фазовых углов использовались координаты атомов. (Для
учета уменьшения электронной плотности на месте остатков 133—
137 были введены необходимые температурные параметры [3]).
Как это принято в обычных кристаллографических исследованиях,
Рис. 15.3. Стереоизображение молекулы карбокснпептидазы А.
Представлен вид молекулы вдоль оси В. Модельный субстрат КБЗ-А!а-А1а-Туг занимает вы-
емку связывания субстрата и полость, примыкающую к атому цинка. Слева видны несколь-
ко больших спиральных участков. В центре молекулы находится участок с P-структурой. Ди-
сульфидная связь расположена в нижней правой части рисунка.
атомы, расположение которых вызывало сомнения, не использова-
лись при расчете фаз. В результате этого были исключены атомы
растворителя и около 5% атомов белка. Появление этих атомов
впоследствии на результирующих картах служило подтверждением
их важности. После повторной обработки на вычислительной ма-
шине все же остались некоторые противоречия в последовательно-
стях, определенных двумя способами [22], причем они не затра-
гивали остатки, различающиеся в двух аллоформах. Действитель-
но, из плотностей, соответствующих остаткам 179, 228 и 305,
можно было заключить, что если сок поджелудочной железы для
приготовления кристаллов взят от одной коровы, то они отвечают
КПА^а|. Тем не менее остатки 151 и 93, по данным химического
анализа являющиеся соответственно Phe и Asn, занимают на кар-
тах больший объем и похожи скорее на Тгр и Glx. Кроме того, два
остатка кислых аминокислот, Glu-108 и Asp-256, находятся в поло-
жениях, в которых присутствие амидов вероятнее присутствия сво-
бодных кислот [22].
Общая конформация полипептидной цепи видна на рис. 15.3,
на котором помечены все а-углеродные атомы. Молекула размером
Карбоксипептидаза А и другие пептидазы
511
Таблица 15.2
Вторичная структура карбоксипептидазы
Остатки Структура Остатки Структура
14—28 Спираль 173—187 Спираль
32—36 0-Слой 190—196 0-Слой
49—53 200—204
60—66 215—231 Спираль
72—80, 82—88 Спираль 239—241 0-Слой
94—103 254—262 Спираль
104—109 0-Слой 265—271 0-Слой
112—122 Спираль 285—306 Спираль
122—174 «Неупорядоченная»
50X42X38 А имеет почти сферическую форму и, следовательно,
компактнее, чем было предсказано на основании величины f/f0, оп-
ределенной из коэффициентов диффузии [45]. В непосредственной
близости от атома цинка находится полость, которая оказалась
участком связывания ароматической боковой цепи глицилтирозина
(ср. рис. 15.6 и 15.8). Это углубление на поверхности молекулы хо-
рошо просматривается даже на картах КПА с низким разрешени-
ем [46]. При высоком разрешении в этой полости видно несколь-
ко молекул воды. От атома цинка в сторону, противоположную ей,
проходит выемка или канал, в который можно поместить ацилиро-
ванные трипептиды (разд. 2.4.1). Из общего числа 307 остатков
115 входят в спиральные структуры*, 45 — в протяженные р-струк-
туры, а оставшиеся 147 — в структуры, которые можно назвать
«нерегулярными» в том смысле, что они отличаются от типичных.
В табл. 15.2 приведены типы вторичной структуры, которые на-
блюдаются при рассмотрении пептидной цепи. Все спиральные уча-
стки, за исключением трех сравнительно коротких (остатки 112—
122, 173—187 и 254—262), расположены в левой части молекулы,
изображенной на рис. 15.3. Фактически ни один из них нельзя
считать находящимся внутри молекулы. Центр КПА и одна из
сторон активного центра образованы участком с искаженной
p-структурой, состоящим из восьми сегментов, которые расположе-
ны таким образом, что четыре пары цепей параллельны, а три ан-
типараллельны (рис. 15.4). На рис. 15.3 участок с p-структурой
расположен перпендикулярно плоскости листа, в результате чего
* Считается, что остатки входят в а-спиральный участок, если они образуют
одну водородную связь, характерную для этой структуры.
512
Глава 15
хорошо видны искривления в слое: самый верхний сегмент (остат-
ки 238—243) повернут по отношению к самому нижнему (остатки
32—37) почти на 120°. Правая часть молекулы (рис. 15.3) состоит
главным образом из «неупорядоченных» участков и содержит ди-
сульфидный мостик, соединяющий остатки 138 и 161 и находящий-
ся на расстоянии около 20 А от атома цинка. Эта «неупорядочен-
9
-C-241N - C-240-N - С-239- «—
О \ О
' » i
I О I
-2O4-NC-203-N-C-202-N-С-201- N-C-2OO-N- «—
6 ! 6 i ;
О ; О ; О 6
-N-265-C-N-266-C-N-267-C-N-268-C- N-269-C-N-27O-C-N-271-<5- —»
I 1 /1 t ' t
i О I О , О I
t ' i I .1 XI
О О 1 О 'О
-190-C-N-191-C- N -192-C-N-193-C-N-194 -С - N-195-C-N-196-C- —♦
х / 1
О / о
° \ р 9
-60 - С -N - 61 - <5-^4- 6 2-С- N- 63-С- N-64-9- N- 65- С -N- 66- —*
О / О / О /
1 О \ О б
' < х i .1
-N-1O4-C-N-105-9- N-K)6-C-N-l07-^-N-1Oe-C -N-1O9- —>
! 9 : 9
9 : 9 :
-53-N-C-52- N-C-51-N-C-5O-N-C-49-N- «—
: ? : ? :
9 ! 9 19
•32-C-N-33-C-N-34-C-N-35-C-N-36-C-►
6 6
Рис. 15.4. Диаграмма расположения 0-слоев в молекуле карбоксипептидазы А.
Предполагаемые водородные связи показаны штриховыми линиями. Стрелками обозначены
параллельное и антипараллельное расположения сегментов.
пая» часть молекулы сравнительно подвижна, и в ней происходит
большая часть конформационных изменений, вызываемых связы-
ванием субстрата. Окончательная структура КПА, изображенная
на рис. 15.3, не может получиться сворачиванием полипептидной
цепи субъединицы про-КПА в ходе ее синтеза, идущего от N-koh-
ца. Так, например, цепь 249—254 должна разместиться между дву-
мя сегментами около остатков 150 и 208, разделенными расстояни-
ем всего в 5—6 А. Аналогичным образом участок 265—271 в конеч-
ной структуре расположен между сегментами 200—204 и 190—196.
Координаты атомов были уточнены с помощью программы, ко-
торая, варьируя углы поворота относительно отдельных связей,
оптимизирует согласие с известной последовательностью на осно-
вании некоторых контрольных координат [47]. Уточненное значе-
ние угла т, образованного связями Скарбонил—Ce~N, составило
Карбоксипептидаза А и другие пептидазы.
513
112°, а не 109,5°, как в правильном тетраэдре. Это указывает на то,
что структура пептидных единиц несколько искажена. По извест-
ным координатам атомов для каждого остатка были рассчитаны
величины диэдральных углов ср и яр [3, 48, 49]. Описание структу-
ры посредством диэдральных углов дано на рис. 15.5, представля-
ющем собой конформационную карту молекулы карбоксипептпда-
Рис. 15.5. Карта диэдральных углов КПА.
Границы размещения областей обозначены сплошными линиями для Т=П0° и штриховыми
линиями для т=115°. Светлые кружки соответствуют остаткам глицина. Величины углов
<р и ip в а-спирали составляет 132 и 123°, а в антипараллельиом Р-слое — 40 и 315" соот-
ветственно. Отсчет углов Ф и ф приводился так же, как в работе [49].
зы. Хотя сгущение точек примерно соответствует углам, характер-
ным для модельной а-спирали, разброс означает, что ни один из
спиральных участков не является абсолютно правильным. Напри-
мер, часть цепи, содержащая остатки с 72 по 88, изогнута посере-
дине таким образом, что образование водородной связи с остатком
триптофана 81 оказывается невозможным. Точно так же обнаружи-
ваются отклонения от модельной структуры на участке с р-формой
[50].
Несколько остатков, по-видимому, находятся в конформациях,
которые по теоретическим соображениям запрещены. Аналогичные
результаты были получены для лизоцима [51], однако значение
этих высокоэнергетических конформаций пока непонятно. При
33—2451
514
Глава 15
встраивании остатка гистидина 196, являющегося лигандом цинка,
в карту электронной плотности оказалось необходимым ввести ис-
каженную цис-пептидную связь между серином 197 и тирозином
198. Поскольку это первый пример образования цис-пептидной свя-
зи у остатка, отличающегося от пролина, к нему следует отнестись
с некоторой осторожностью. С другой стороны, возможно, что та-
кой способ образования связи имеет значение для активности фер-
мента.
Распределение остатков внутри и снаружи молекулы согласу-
ется с данными для других глобулярных белков. Гидрофобные
остатки предпочтительнее располагаются внутри молекулы, а заря-
женные группы — снаружи [52]. Поскольку участок в p-форме на-
ходится главным образом внутри глобулы, в нем обнаружено мно-
го гидрофобных аминокислот, в том числе лейцина и фенилалани-
на. Всего в контакте с водой не принимают участия 78 остатков.
Из них 22 могут образовывать водородную связь с атомами пептид-
ной связи или близлежащих остатков, и, по-видимому, эта возмож-
ность почти во всех случаях реализуется [3, 52]. Два остатка трип-
тофана (63 и 147) и один остаток тирозина (238) спрятаны внутри
молекулы КПА. Остальные остатки этих аминокислот находятся в
частичном контакте с растворителем. Существование водородной
связи между ОН-группой Туг-238 и карбонильной группой Glu-270,
вероятно, имеет некоторое значение для конформационного изме-
нения с участием Glu-270 при связывании субстрата, как описано
ниже. Четыре из десяти остатков пролина расположены у N-концов
спиральных участков, а три — у концов наиболее длинных цепей в
слое с p-структурой. Во внутренней части молекулы находятся три
карбоксильные группы, принадлежащие остаткам 104, 108 и 292.
Конечно, справедливость этого утверждения зависит от того, на-
сколько правильно установлен тот факт, что они являются свобод-
ными и не участвуют в образовании амидных связей. Карбоксиль-
ная группа Glu-292 образует солевой мостик с Arg-272, так что ее
заряд локально нейтрализован. Детальное изучение карт электрон-
ной плотности обнаружило неизвестный ранее факт внедрения в
молекулу карбоксипептидазы десяти молекул воды [52].
2.2. Активный центр
2.2.1. Конформация белка
Структура активного центра образована теми атомами фермен-
та, расстояния которых от природных субстратов не превышают
расстояния, на котором действуют силы Ван-дер-Ваальса. Если
учесть все остатки вблизи молекулы глицилтирозина, а также те
остатки, близость которых к ацилтрипептиду, находящемуся в «вы-
емке» поверхности, постулируется из модельных опытов (рис. 15.3),
Карбоксипептидаза А и другие пептидазы
515
то можно заключить, что в образовании активного центра участву-
ют следующие сегменты цепи: 69—72, 125—127, 142—145, 155, 163,
164, 194—203, 243, 247—256, 268—270 и 275—279. Боковые цепи
остатков 70, 126, 143, 195, 197, 200, 202, 249, 252—253 и 275—276
направлены в сторону, противоположную месту присоединения суб-
страта, и, по-видимому, не контактируют с ним непосредственно.
Остатки 194—203 и 268—270 входят в слой с p-структурой, кото-
рый образует одну из стенок участка связывания глицилтирозина.
Остальные остатки, за исключением лигандов цинка 69 и 72, нахо-
дятся в извилистой правой части молекулы, показанной на
рис. 15.3. Верхняя часть полости, в которой помещается тирозиль-
ная боковая цепь глицилтирозина, образована остатками 243 и
246—250, а остальная ее часть — остатками 155 и 253—256. Левая
часть выемки, являющейся участком связывания, образована сег-
ментом 275—279 (рис. 15.3), а правая — сегментами 125—127,
142—145 и 163—164. На рис. 15.6 карта электронной плотности в
районе связывания субстрата совмещена с изображениями глицил-
тирозина и части контактирующих остатков.
Большая часть центра присоединения субстрата не имеет регу-
лярной вторичной структуры. За исключением нескольких сегмен-
тов в p-форме и нескольких остатков в начале одной из спираль-
ных частей (255—256), подавляющее большинство остатков нахо-
дятся в «неупорядоченных» участках. Конформационная карта
остатков активного центра подтверждает то, что только незначи-
тельная их часть имеет значения <р и ф, близкие к наблюдаемым в
а-спиральной структуре.
2.2.2. Цинк
В присоединении цинка к карбоксипептидазе участвуют три
белковых лиганда: His-69, Glu-72 и His-196. Связи гистидин—цинк
осуществляются через атом азота в положении Ni кольца. Кроме
того, лиганды принимают участие и в других взаимодействиях.
Атом азота N3 в His-69 образует водородную связь с остатком
Asp-142, а атом азота N3 в His-196 соединен такой же связью с мо-
лекулой воды. На предварительных картах вблизи атома цинка
была обнаружена повышенная электронная плотность, не относя-
щаяся к белку. С помощью карт, построенных по рассчитанным
значениям фаз, удалось установить, что она принадлежит един-
ственному небелковому лиганду металла. Поскольку величина
электронной плотности соответствовала атому кислорода, было
предположено, что он является молекулой воды или ОН-ионом.
Понятно, что некоторые молекулы КПА вместо воды могут связы-
вать СН, однако малая величина электронной плотности свидетель-
ствует о том, что их число невелико. Расположение лигандов отно-
сительно атома цинка показано на рис. 15.7, а значения углов
яя*
516
Глава 15
Рис. 15.6. Центр связывания глицил-l-тирозина. Модель глицил-L-тирозина в
ориентации, определенной по разностным картам, совмещена с несколькими се-
чениями карты электронной плотности КПА.
Близлежащие остатки аминокислот приведены в той конформации, в которой они находятся
в свободной КПА. Справа от субстрата расположен кластер из трех остатков аргинина.
Плотность в районе фенольного кольца субстрата происходит, как полагают, от четырех
молекул. воды, вытесняемых субстратом {Dickerson R. Е.» Geis I., The Structure and Action
of Proteins with Stereo Supplement, Harper and Row Publishers, Inc., 1969). Рисунок приве-
ден с разрешения авторов и издателей.
Карбоксипептидаза А и другие пептидазы
517
между связями приведены в табл. 15.3. Даже если допустить, что
ошибка при определении углов составляет ±10°, отклонения от
тетраэдрической конфигурации значительны главным образом из-
за близости атома цинка к |3-СН2-группам гистидина вследствие
того, что связь с ним осуществляется через атом азота.
Рис. 15.7. Стереоизображение лигандов цинка и части (3-слоя, построенное с по-
мощью ЭВМ по программе OR-ТЕР д-ра К. Джонсона.
Искажения в геометрическом расположении лигандов цинка не
являются исключительным свойством белков, так как они встреча-
ются и в хелатах, образуемых цинком с аминокислотами и пепти-
дами (гл. 4). Из них можно упомянуть Zn(Gly-Gly-Gly) (SO4)i/2*
• 2Н2О, Zn(L-His)2-2H2O и Zn(D, l-H1s)2-5H2O. Комплекс с тригли-
цином представляет собой деформированную бипирамиду, углы
Таблица 15-3
Углы между связями, образуемыми лигандами металла
в Zn-карбоксипептидазе
Lj—Zn—Lg Угол, град
N(69)—Zn—N(196) N(69)—Zn—0(72) 0(72)—Zn—N( 196) 0(72)—Zn—0(H20) N(69)— Zn—O(H2O) N(196)—Zn—O(H2O) 86 99 143 99 120 111
518
Глава 15
между связями в которой отличаются от идеальных на 20° [53].
В соединениях с гистидином четыре азотных лиганда образуют
тетраэдр с угловыми искажениями около 10°. Кроме того, с цин-
ком связаны два атома кислорода [54, 55].
2.3. Комплекс КПА с глицилтирозином
Фермент-субстратный комплекс глицилтирозина и КПА был
изучен кристаллографически при атомном разрешении с помощью
разностного фурье-метода [1—3]. Это оказалось возможным бла-
годаря чрезвычайно низкой скорости превращения глицилтирози-
на. В растворе АКат имеет порядок 1 мин-1, тогда как в кристалли-
ческом состоянии величина периода полупревращения соизмерима
со временем съемки дифракционной картины. В кристаллическом
комплексе обнаружен только один тип связывания глицилтирозина
с ферментом. Взаимодействия, наблюдаемые в этом комплексе, лег-
ли в основу предполагаемого механизма действия КПА (разд.
3.5.1) [2].
Обычно в кристаллографии белков для расчетов по
разностному методу Фурье используются коэффициенты
co(|/*esI — |АЕ|)ехр(гаЕ)*- Поскольку вклад, соответствующий
структуре нативного фермента, вычитается, то суммирование ря-
дов Фурье позволяет получить изображение различий между струк-
турами Е и ES. Если некоторые атомы белка смещаются в резуль-
тате комплексообразования, то на их месте появляются участки
с отрицательной электронной плотностью. На предварительных
разностных картах комплекса с глицилтирозином при разрешении
2,8 А хорошо наблюдалось расположение атомов тирозильной
группы. Однако глицильный остаток субстрата, который на
рис. 15.6 и 15.8 находится над атомом цинка, особенно его карбо-
нильная группа, был виден плохо. Причиной этого могло быть то,
что при связывании субстрата электронная плотность небелкового
происхождения вблизи атома цинка замещалась на эквивалент-
ную ей. Были рассмотрены две возможные ориентации глицильной
части субстрата. Более вероятная из них предполагает образова-
ние связи между атомом металла и кислородом карбонильной
группы [2].
Для того чтобы более надежно определить ориентацию моле-
кулы глицилтирозина, был проведен новый обсчет дифференциаль-
ной карты. В общем случае можно ожидать, что карта, рассчитан-
* |^es|—амплитуда рассеивания для кристаллов, содержащих субстрат,
Ив| —амплитуда рассеивания для кристаллов «свободного» фермента, Ие—фа-
зовый угол, рассчитанный методом изоморфного замещания. Множитель о харак-
теризует вес отдельного члена.
Рис. 15.8. Стереоизображение конформационных изменений, сопровождающих
присоединение субстрата
а — свободная КПА; б — комплекс КПА с Gly-Tyr. Остатки Arg-145 Glu-270 и Туг-248, из-
меняющие свое положение при связывании субстрата, и молекула Gly-Tyr обозначены утол-
щенными линиями. Тирозильный остаток Gly-Tyr занимает «полость специфичности», а атом
кислорода карбонильной группы расщепляемой пептидной связи становится лигандом цинка.
520
Глава 15
ная по коэффициентам (|/*es| — |Крассч|)ехр(1арассч)*, обнаружит
расположение всех атомов комплекса с субстратом, для которых
оно будет иным, чем в нативной структуре. К сожалению, из-за
того, что только часть молекул (х) в кристалле связана с субстра-
том, карта, основанная на этих коэффициентах, содержала бы
плотности, соответствующие всем атомам той доли молекул (1—
х), которые не образовали комплекс, за исключением опущенных
при расчете структуры, а именно, молекулы воды в полости, и
плотность небелкового происхождения вблизи атома цинка. По-
этому коэффициенты, соответствующие доле свободного фермента,
были также вычтены. Таким образом, в новом модифицированном
варианте расчета [3] коэффициенты были равны
(| Tes | X | -Ррассч | — (1—х) | Те |) ехр (Юрассч).
Контакты фермента с субстратом, обнаруженные на окончатель-
ной карте, представлены на рис. 15.6 и 15.8. Существенные взаимо-
действия между глицилтирозином и белком можно разбить на пять
групп.
1. Фенольная боковая цепь остатка тирозина занимает полость
на поверхности фермента, вытесняя оттуда несколько молекул во-
ды. Соответствие фенольной группы и этой полости неидеально, что
согласуется с наличием довольно широкой специфичности в отно-
шении этой части субстрата, которая может быть ароматической
или разветвленной алифатической. Вблизи ароматического цикла
находятся остатки Пе-243, Пе-247, Пе-255 и Asp-256 (рис. 15.6).
2. Карбоксильная группа субстрата присоединена к гуанидино-
вым группам Arg-145 посредством двух водородных связей.
3. Фенольный атом кислорода в Туг-248 находится на расстоя-
нии, меньшем длины водородной связи, от NH-группы пептидной
связи субстрата. Расстояние это равно 2,7 А при точности ±1,0 А.
4. Атом кислорода карбонильной группы субстрата занимает
по отношению к атому цинка примерно то же положение, что и мо-
лекула воды или ион ОН~ в свободном ферменте. Хотя атом кис-
лорода карбонильной группы субстрата виден очень плохо, его
локализация подтверждается расположением плотностей, соответ-
ствующих атомам азота аминогруппы и Са в субстрате. Кроме то-
го, на картах нет других сгущений электронной плотности, которые
можно было бы приписать карбонильной группе. Атом углерода
этой группы расположен на расстоянии 3,5 А от атома кислорода,
принадлежащего остатку Glu-270.
5. Аминогруппа субстрата связана с остатком Glu-270 через мо-
лекулу воды. Это взаимодействие считается «непродуктивным»
(разд. 3.5.1). По-видимому, оно не является электростатическим,
* l^paccnl и ссрассч — амплитуда рассеивания и фазовый угол, рассчитанные
по положению атомов.
Карбоксипептидаза А и другие пептидазы
521
поскольку из экспериментов по ингибированию вытекает, что с
КПА связывается только анионная форма глицилтирозина [56].
Образование фермент-субстратного комплекса происходит в ре-
зультате ряда конформационных изменений в структуре белка. Са-
мые значительные из них затрагивают остатки 248, 270 и 145 и
видны из сравнения рис. 15.8, а и б. Гуанидиновая группа остатка
Arg-145 перемещается примерно на 2 А в результате поворота вок-
руг связи Ср—Су. Кислородные атомы карбоксильной группы ос-
татка Glu-270 смещаются примерно на 2 А вдоль оси у вследствие
поворотов вокруг связей Са—Ср и Ср—CY (на рис. 15.8, б карбо-
ксильная группа переворачивается и сдвигается вверх по отноше-
нию к наблюдателю). Более других изменяет свое положение оста-
ток Туг-248. Кислородный атом боковой цепи этой аминокислоты
сдвигается на 12 А в результате поворота приблизительно на 120°
вокруг связи С а—Ср и небольшого смещения пептидного скелета.
Фенольное кольцо остатка Туг-248 поворачивается по направлению
к субстрату, заключая С-концевой участок в полость. В своем но-
вом положении этот остаток близок к расщепляемой пептидной
связи. Движение остатков Arg-145 и Туг-248, вероятно, взаимосо-
гласовано посредством нескольких менее значительных перемеще-
ний. В свободном ферменте остатки Туг-248 и Arg-145 соединены
системой водородных связей, в которой принимают участие остат-
ки 155, 154 и 249. После присоединения глицилтирозина взаимо-
действия в парах Arg-145—Glu-155 и Gln-249—Туг-248 нарушают-
ся и пептидная цепь на участке 247—249 несколько изменяет свое
положение.
Присоединение субстрата превращает заполненную молекулами
воды полость в сравнительно гидрофобный участок. Остаток
Leu-203 выступает внутрь полости, тогда как в свободном фермен-
те он взаимодействует с другими аминокислотами, образующими
p-структуру. Однако вода удаляется из полости лишь частично, и
три или четыре ее молекулы остаются внутри после присоединения
тирозильной группы субстрата. Поскольку в этом участке происхо-
дит связывание боковых групп гидрофобных С-концевых остатков
и субстраты с такими остатками гидролизуются быстрее всего
(рис. 15.2), то эту полость можно назвать участком специфичности.
При этом надо иметь в виду, что другие участки связывания также
влияют на специфичность.
Константа диссоциации комплекса глицилтирозина с КПА при
25 °C равна ЫО-3 М (табл. 15.4). Соответствующая ей величина
AG составляет —4,1 ккал/моль, что меньше суммы вкладов всех
наблюдаемых взаимодействий [57, 58]. Часть энергии связывания
предположительно используется для перевода субстрата и (или)
фермента в напряженные, т. е. энергетически невыгодные конфор-
мации [59—62] (разд. 3.5.1). Таким образом, благоприятные фер-
мент-субстратные взаимодействия служат источником энергии для
522
Глава 15
конформационных изменений белка. Так как остаток Туг-248, по-
видимому, существен для пептидазной активности (разд. 3.3), а его
правильное расположение по отношению к субстрату достигается в
результате структурной перестройки, вызываемой присоединением
субстрата, то взаимодействие КПА — глицилтирозин служит хоро-
шим примером «индуцированного соответствия» фермента субстра-
ту [63, 64].
2.4. Дополнительные структурные исследования*
2.4.1. Моделирование
Для увеличения взаимодействия с ферментом можно увеличить
размер пептидного субстрата, исходя из известной специфичности
КПА в отношении связывания [2, 3]. Одна из моделей фермент-
субстратного комплекса, в котором субстратом является КБЗ-А1а-
Ala-Туг, показана на рис. 15.3. После того как остаток Туг-248 из-
менил свое положение, пептидная связь Ala-Ala находится по
отношению к нему на расстоянии, при котором возможно образо-
вание водородной связи. Ее существование объясняет большую ре-
акционную способность пептидов с NH-группой в ближайшем от
конца положении по сравнению с пептидами, у которых в положе-
нии Si (рис. 15.9) находится Ы-метильный [66] или р-аланильный
остаток [67]. Остальная часть субстрата располагается в «выем-
ке» на поверхности КПА, что согласуется с тем, что в контакте с
белком могут находиться до пяти аминокислотных звеньев субстра-
та [31]. Положение бензильного остатка КБЗ-группы вблизи аро-
матического остатка Phe-279 и атома кислорода карбоксильной
группы третьей от конца аминокислоты субстрата вблизи гуани-
диновой группы остатка Arg-71 согласуется с известным влиянием
заместителей на величину /См [31].
Можно отметить, что моделирование подтверждает и некоторые
известные особенности стереоспецифичности КПА [3, 52], если до-
пустить, что молекула субстрата в кинетически важном комплексе
расположена так же, как молекула глицилтирозина. Так, если суб-
страты, замещенные в области С-конца (КБЗ-С1у-тиазолидинкар-
боновая кислота [68], КБЗ-Gly-Pro [69]), поместить таким обра-
зом, что группа R оказывается в полости, СОО-—вблизи Arg-145,
а С=О — вблизи атома цинка, то возникают стерические помехи
со стороны остатков Пе-247 и Туг-248. Пептиды, оканчивающиеся
* В настоящее время проводится кристаллографическое изучение комплекса
КПА и Phe-Gly-Phe-Gly [65]. Карты электронной плотности отчетливо указывают
на существование связи между карбонильной группой субстрата и атомом цинка.
Кроме того, ион цинка заметно смещается (примерно на 0,6 А) при связывании
этого субстрата.
Карбоксипептидаза А и другие пептидазы
523
объемистыми d-аминокислотами, тоже не могут образовать все те
три типа связей, которые наблюдаются в фермент-субстратном
комплексе.
Рис. 15.9. Диаграмма предпо-
лагаемых фермент-субстрат-
ных взаимодействий для пен-
тапептидного субстрата.
Отщепляемый остаток Si ориенти-
рован так же, как в случае глицил-
тирозина (рис. 15.6 и 15.8), ио в
образовании связи с остатком
Туг-248 участвует группа NH пеп-
тидной связи, соединяющей Si и Sa-
2.4.2. Комплексы с ингибиторами
Рентгеноструктурному анализу при разрешении 6 А были под-
вергнуты комплексы фермента с несколькими ингибиторами [70].
В результате не было получено детального описания взаимодейст-
вий, возникающих при связывании. Тем не менее совмещение раз-
ностных карт с картами белка, построенными при высоком разре-
шении, дало некоторую положительную информацию. Три
ингибитора — n-иод-р-фенилпропионат, l-фенилаланин и ь-лизил-
ь-тирозинамид — при pH 7,5 связываются вблизи атома цинка,
причем в их поведении существуют интересные различия. Проще
всего обстоит дело с ь-фенилаланином. Положительная плотность
обнаруживается на разностных картах в том месте полости, кото-
рое на разностных картах с низким разрешением соответствует
молекуле глицилтирозина. Эта плотность происходит от молекулы
ь-фенилаланина, причем свободная группа СОО- остатка взаимо-
действует с остатком Arg-145. Кроме того, обнаруживаются участ-
ки с положительной и отрицательной плотностью, возникающие
524
Глава 15
при перемещении Туг-248. Лизилтирозинамид, напротив, располага-
ется иначе, чем глицилтирозин, и на картах нет признаков конфор-
мационного изменения, затрагивающего остаток Туг-248. Иодфе-
нилпропионат связывается кристаллической КПА на четырех цент-
рах. Если предположить, что центр пиков электронной плотности
при низком разрешении совпадает с положением атомов иода, то
два из них оказываются в районе активного центра (рис. 15.10).
Рис. 15.10. Стереоизображение активного центра, показывающее расположение
атомов иода иодфенилпропионата. В отличие от других рисунков здесь представ-
лен вид вдоль кристаллографической оси а (на рис. 15.8. справа вдоль плоско-
сти листа). Атом иода ,11 находится в субстратсвязывающей полости.
Атомы иода К и 12 находятся в положениях, при которых возмож-
но взаимодействие между группой СОО- ингибитора и атомом
цинка. Ряд других данных подтверждает предположение о том, что
фенилпропионат присоединен к иону металла [71, 72]. Таким об-
разом, иодфенилпропионат не может служить хорошим аналогом
субстрата, глицилтирозина. Несмотря на это, его присоединение,
по-видимому, также вызывает изменение конформации в районе
остатка Туг-248. Множественность центров связывания фенилпро-
пионата не является неожиданной, так как кинетика ингибирова-
ния им довольно сложна (разд. 3.2.1).
2.4.3. Апофермент и металлы, отличные от цинка
Для удаления и замещения цинка в КПА, находящейся в крис-
таллическом состоянии, можно использовать ту же методику, что
и для КПА в растворе, несколько изменив условия [73]. Кристал-
лическая Hg-КПА была получена двумя способами: кристаллиза-
цией после замены Zn на Hg и диализом кристаллов против рас-
твора, содержащего ионы Hg2+ [46]. В последнем случае ртуть не
только замещала цинк, но и присоединялась к другим участкам
молекулы (табл. 15.1). Кристаллическая апо-КПА может быть по-
лучена обработкой кристаллов Zn-фермента оксихинолинсульфона-
том. Cd- и Cu-ферменты были приготовлены из кристаллов апо-
Карбоксипептидаза А и другие пептидазы
525
Карты электронной плотности для нативной КПА и апофермен-
та при разрешении 6 А практически совпадают, за исключением
большого отрицательного пика в положении атома цинка [46]. На
первый взгляд можно было предположить, что в апо-КПА остаток
His-196, несмотря на жесткость p-структуры, изменит свое поло'
жение таким образом, что между ним и остатками Asn-144 и
Gly-270 образуются водородные связи. Однако на разностных
картах при низком разрешении отсутствуют участки с положитель-
ной и отрицательной плотностью, которые согласовались бы с этим
предположением. Если допустить, что в кристалле, так же как в
растворе, потеря активности коррелирует с удалением цинка, то из
анализа карт апо-КПА можно заключить, что металл участвует
непосредственно в катализе, а не в возможной структурной пере-
стройке апофермента. Этот вывод строго подтверждается взаим-
ным расположением субстрата и металла в кристаллических фер-
мент-субстратных комплексах. Можно отметить, что результаты
химических исследований в растворе не позволили выяснить роль
цинка.
Карты электронной плотности активного центра Hg-КПА были
рассчитаны с высоким разрешением. Центр иона металла смещен
на 1,0 А главным образом вдоль осей х и у по сравнению с поло-
жением иона цинка. На рис. 15.7 атом ртути был бы выше и бли-
же к остатку His-69. Связь этого металла с белком тоже осущест-
вляется через атомы Ni двух остатков гистидина, 69 и 196, кото-
рые расположены так же, как в комплексе с Zn. Возможность
поворота имидазольного кольца в His-196, в результате чего в свя-
зи с металлом мог бы вместо атома Ni участвовать атом N3, сле-
дует исключить. Причиной этого является обнаружение на картах
молекулы воды, которая, как и в случае цинка, соединена с ато-
мом N3 этой аминокислоты водородной связью. Электронная плот-
ность, соответствующая карбоксильной группе остатка Glu-72, не-
сколько понижена. Тем не менее и эта группа, по-видимому, взаи-
модействует с атомом ртути. В остальном карты электронной плот-
ности для комплексов белка с Zn и Hg совпадают. Возможность
использовать Hg-КПА при расчете фаз подтверждает изоморфизм
двух структур. В растворе ртутное производное карбоксипептидазы
проявляет высокую эстеразную активность, но не катализирует
гидролиз пептидов [41]. Однако в кристаллическом виде Hg-КПА
в отличие от Zn-КПА обладает и высокой пептидазной активно-
стью, которая составляет примерно 1/1000 активности Zn-фермента
в растворе [73].
Для изучения структур Mn-КПА и Cu-КПА использовали ме-
тоды магнитного резонанса. Геометрия лигандного окружения этих
ионов в комплексе с белком отличается от существующей в
Zn-КПА. Анализ резонанса иона F~ и протонов воды в присутствии
Mn-КПА дает основания предположить, что в растворах, содержа-
526
Глава 15
щих F~, во внутренней координационной сфере иона металла при-
сутствуют по меньшей мере два небелковых лиганда: один ион
галогена и не менее одной молекулы воды. Если, кроме того, ион
металла связан с тремя белковыми лигандами, то его координаци-
онное число должно быть не менее пяти [6, 72]. Спектры электрон-
ного парамагнитного резонанса кристаллов Cu-КПА согласуются
с плоской геометрией лигандов, а не с искаженной тетраэдриче-
ской. Анализ сверхтонкой составляющей спектра указывает на то,
что среди лигандов есть два атома азота. Кристаллы КПА7, ис-
пользованные в измерениях электронного парамагнитного резонан-
са, изоморфны соответствующим кристаллам Zn-КПА [74]. Уча-
стие остатков His-196 и His-69 в связывании атома меди не вызыва-
ет сомнения, в то время как природа остальных лигандов и
расположение плоскости, в которой находятся лиганды, по отноше-
нию к молекуле КПА точно не установлены.
3. ФЕРМЕНТАТИВНАЯ АКТИВНОСТЬ
карбоксипептидазы а
3.1. Кинетика (обзор)
Для описания кинетики реакций, катализируемых КПА, в со-
ответствии с современным уровнем знаний требуется использовать
сложную схему, показанную на рис. 15.11. Она суммирует различ-
ные кинетические явления, которые в какой-то степени подтверж-
дены экспериментально: непродуктивное связывание (путь 2);
конкурентное ингибирование продуктами или аналогами субстра-
тов (путь 3); взаимодействие с модификаторами М (которые мо-
гут включать как продукты, так и субстраты), ведущее к смешан-
ному и неконкурентному ингибированию или активации (путь 4).
Ингибирование избытком субстрата можно считать вариантом пу-
ти 4. Существование тех или иных путей зависит от природы суб-
страта. Например, при гидролизе ГФЛ* ингибирование субстратом
проявляется даже при его концентрации 2-10~4 М [28, 75], в то
время как при использовании похожего пептидного субстрата, гип-
пурилфенилаланина, вместо ингибирования наблюдается актива-
ция [76] (см. также [78]). Активация субстратами обнаружена
также при гидролизе эфира — гиппурилгликолата [77] и пептида—
КБЗ-Gly-Phe [78]. Продукт расщепления последнего субстрата,
КБЗ-Gly, активирует гидролиз некоторых пептидов и в то же вре-
мя конкурентно ингибирует гидролиз циннамоилфениллактата [79]
и ацилтрипептидов [76]. К модификаторам относятся также бенз-
амид, циклогексанон и ряд замещенных спиртов [80].
Формулы субстратов приведены в табл. 15.4.
Карбоксипептидаза А и другие пептидазы
527
1’ Основная реакция (ковалентные промежуточные соединения опущены)
/ *1 Z *2 /₽1 *3 ,
Ex +S =?==* Ех +Н2о =f==* Ex +Ра Е+Рх
\ к—1 \ К—2 \ R 3
2. Образование непродуктивного комплекса
3. Конкурентное ингибирование
/
Е^ + 1 <==* Е^
4. Реакция с модификаторами
/ ЛМ / А1М *2М /Р1
Еч+Мч=*Ех +S Ех +Н2О=?=^Ех +Р2
\ \м *-1М \м -гм ХМ
Рис. 15.11. Кинетическая схема реакций, катализируемых КПА. Приведенные ме-
ханизмы являются упрощенными и включают только нековалентные промежу-
точные соединения. Для объяснения ингибирования продуктом реакции показана
стадия его диссоциации.
Непродуктивное связывание, проявляющееся в конкурентном
ингибировании субстратом, довольно трудно обнаружить с по-
мощью кинетических измерений [81]. Однако сравнение величин
/См и Ум в ряду субстратов (Gly)n-Tyr, где и=1—6, позволило од-
нозначно установить его существование в случае дипептида. Ве-
личина /См для Gly-Tyr в 5 раз меньше, чем для более длинных
субстратов, в то время как Ум для него составляет всего 1/1000
соответствующей величины для (С1у)г-Туг [82]. Аналогичная за-
кономерность при удлинении олигосахаридной цепи субстрата на-
блюдается для лизоцима. В случае этого фермента о существова-
нии непродуктивного связывания, определяющего величину /См,
хорошо известно [83, 84]. Анализ кристаллического комплекса
глицилтирозин—КПА показывает, что сравнительно прочное при-
соединение субстрата, которое, возможно, мешает образованию пе-
реходного состояния, объясняется взаимодействием а-аминогруппы
глицилтирозина через молекулу воды с остатком Glu-270 (разд.
3.5). Для более длинных субстратов такое взаимодействие невоз-
можно.
Некоторые кинетические параметры для ряда субстратов и мо-
дификаторов приведены в табл. 15.4. Незнание полного механизма,
Кинетические параметры для некоторых субстратов
Соединение
Структурная формула
Пептиды
1. Карбобензокси-
глицил-L -фенил-
аланин (КГФ)
II II
—сн2—ос—nhch2c—nh—сн—соо-
2. Бензоилглицил-
L-фенилаланин
(БГФ)
3. Бензоилглицил-
глицилфенил-
аланин (БГГФ)
4- Тетра-Ь-аланин
5. Глицил-ь-тиро-
зин
ООО
+ II II II
NH,—СН—С—NH—СН—С—NH—СН—С—NH—СН—СОО-
1111
СН3 сн3 сн8 сн8
о
+ II
NH8CH2—с-мн—со—соо-
Ациламинокисло-
ты
1. Ацетил-ь-феиил-
аланин
2. транс-Цннна-
моил-Ь-феиил-
аланин
Таблица 15.4
и модификаторов КПА (pH 7,5, 25 °C)
хм, М Ks или к., м *кат, МИН-1 Важные побочные процессы Литература
2-Ю-3— 37-Ю-3 4,2- 10"за 5,5-103— 12-Ю3 Активация субстратом, ингибирование избыт- ком субстрата 26, 78, 85, 89
0,8-10~3— 11 • ю-3 11.10~за 5,6- 10s— 11 • 103 Активация субстратом 78, 89
bio-» 1.2-103 76
-0,08 6-Ю3 31
0,7-10~3 1. ю-’а (^)ЫО-* 0,9 76, 82
0,155 26
6- ю-4 0,2 90
34—2451
Соединение
Структурная формула
Сложные эфиры
1- Бензоилглицил-
фениллактат
(гиппурил-ь-фе-
ниллактат,
ГФЛ)
2. Циннамонл-L-
феннллактат
3. Гиппурнлгли-
колат
4. Ацетилманделат
Ингибиторы
а) Продукты
1. L-Фенилаланин
2. L -Фениллактат
3. Циннамат
б) Аналоги
1. Р-Фенилпропио-
нат
2. D-Фенилаланмн
Модификаторы
1. Карбобензокси-
глицин
NH3—CH—СОО"
НО—СН—СОСГ
СН.—СОО"
а Присоединение к КПА (метод гель-фильтрации).
Продолжение табл. 15.4
кк.м Ks или Kt, М *кат- мнн"1 Важные побочные процессы Литература
5,1- IO-5— 8,8-10“5 28- Юз- 35-Ю3 Ингибирование избытком субстрата 28, 78
1,5-Ю-з— 1,9-10-* 4,6-10» 90, 91
1,2-Ю-з 4-10» Активация субстратом 77
6 - IO-* 30 92
33, 78
5,8-10-3 91
0,005 Связывается не так, как другие продукты? 90, 93
6,2-10-3— 19- Ю-з Конкурентное, смешан- ное неконкурентное 26, 76, 94, 95
0,002 26, 96
0,016—0,029 Активатор для ацил- дипептидов, ингибитор для эфиров и более длинных пептидов 76 , 79, 80
532
Глава 15
конечно, не позволяет выразить &KaT и Км через индивидуальные
константы скорости. Для большого числа субстратов, кинетика ре-
акций с которыми не подчиняется уравнению Михаэлиса—Ментен,
побочные пути учитывались введением соответствующих поправок.
В таблице приведены некоторые константы связывания, использо-
ванные ниже при обсуждении влияния модификации и замещения
металла. Для субстратов приведены значения Км, определенные в
стационарной фазе реакции. Понятно, что предположение о равен-
стве константы Михаэлиса равновесной константе связывания не
всегда справедливо для быстро превращающихся субстратов. Как
видно, константы Км и kKM не связаны простой пропорциональной
зависимостью [76], однако из рассмотрения параметров активации
гидролиза КГФ было заключено, что йКат вносит вклад в Км [85].
Присоединение к апоферменту изучалось методом, основанном на
анализе влияния субстратов на скорость рекомбинации цинка и
апо-КПА [86]. Если скорость этого процесса в присутствии субст-
рата существенно уменьшается, то при добавлении цинка к смеси
фермента и субстрата и последующем быстром отделении белка
образование Zn-КПА пропорционально количеству КПА, не свя-
занной в комплекс с субстратом. Любые взаимодействия, не влия-
ющие на скорость рекомбинации с металлом, оказываются при
этом незамеченными. Более того, любое изменение равновесной
константы связывания металла под действием субстрата должно
вызывать обратное влияние металла на сродство к субстрату [87].
Качественные оценки связывания были получены также по зави-
симости скорости обмена металлов от природы субстратов и инги-
биторов [88].
Выведение общих правил на основании данных, приведенных в
табл. 15.4, представляется несколько опасным. Частично это объ-
ясняется заметной величиной поправок на существование множе-
ственных путей реакции, а кроме того, известным фактом влияния
таких переменных, как ионная сила, на кинетические параметры
для пептидов. Тем не менее можно отметить некоторые закономер-
ности. Увеличение субстрата в определенных пределах уменьшает
величину Км, что указывает на усиление взаимодействия с фер-
ментом (ср. ацетилфенилаланин, БГФ и БГГФ). Изучение эффек-
та заместителей для ряда пептидов на основе аланина подтверж-
дает это общее заключение [31]. Аномальная кинетика чаще на-
блюдается для малых субстратов. Например, ацилирование
трипептидов упрощает кинетические закономерности [31, 76]. Кро-
ме того, аномалии обычно связаны с присутствием ароматических
остатков в защитных группах [2]. Из сравнения изостерических
соединений ГФ Л и БГФ ранее был сделан вывод о том, что эфиры
связываются ферментом значительно прочнее соответствующих
пептидов. Однако для других субстратов это правило не всегда
сохраняется. Если малая величина Км для эфиров типа ГФЛ и
Карбоксипептидаза А и другие пептидазы 533
циннамоилфениллактата является следствием непродуктивного
связывания, то эффективность гидролиза этих субстратов должна
быть очень велика.
3.2. Кинетико-структурные корреляции
3.2.1. Присоединение субстратов и ингибиторов
Существование дополнительных кинетических путей подразуме-
вает определенные особенности структуры КПА. Субстраты, кото-
рые способны активировать или ингибировать свой гидролиз, долж-
ны связываться более чем на одном центре. Модификаторы, по-ви-
димому, тоже присоединяются одновременно с субстратами.
Комплексы с модификаторами или субстратами, связанными на не-
скольких центрах, не подвергались прямому кристаллографическо-
му изучению. Однако моделирование позволило найти способ свя-
зывания, объясняющий кинетический эффект модификатора,
КБЗ-Gly [2, 3] (рис. 15.12). Если КБЗ-группа этого соединения на-
ходится вблизи остатка Туг-198, а группа СОО- взаимодействует
с остатком Arg-71, то возможны помехи ориентации субстрата в
продуктивном комплексе. Для объяснения ингибирования субст-
ратом при гидролизе КГФ было предположено, что вторая молеку-
ла КГФ образует с ферментом те же связи, что и КБЗ-Gly [2, 3]
(рис. 15.12). Однако эта модель не может полностью объяснить ки-
нетику субстратного ингибирования. В предполагаемой области
связывания субстрата трудно разместить более двух молекул ацил-
пептидов, в то время как кинетический анализ указывает на при-
соединение четырех или пяти молекул субстрата в растворе [85]
и более чем двух в кристалле [97]. Высокий кинетический порядок
реакции может объясняться связыванием на других участках мо-
лекулы фермента или более сложным образом [28].
По-видимому, при непродуктивном или конкурентном связыва-
нии используются некоторые из взаимодействий, возникающих в
продуктивном фермент-субстратном комплексе. Так, из пяти свя-
зей, образуемых молекулой Gly-Tyr, только одна — между амино-
группой субстрата и остатком Glu-270 — считается непродуктивной.
Другие неактивные комплексы могут образовываться в результате
смещения субстрата вдоль «связывающей выемки», приводящего
к присоединению карбоксильной группы к атому цинка, или в ре-
зультате «неправильного» связывания, при котором N-ацильный
заместитель ацилдипептида занимает полость, определяющую спе-
цифичность, а концевая группа ООО- вступает в контакт с остат-
ком Arg-71 [2,3].
Структурные и кинетические данные показывают, что присоеди-
нение некоторых ингибиторов общего вида RCOO~ осложняется
S34
Глава 15
наличием множественных типов взаимодействий. Ранее считали,
что 0-фенилпропионат, прототип таких ингибиторов, строго конку-
рентен по отношению к субстрату [26]. Однако более поздние ра-
боты показали, что в ингибировании можно выделить конкурент-
ную и неконкурентную компоненты для одного и того же пептидно-
го субстрата, что подразумевает сложный механизм связывания
Рис. 15.12. Модели связывания, объясняющие активирующий эффект КБЗ-Gly
при гидролизе КБЗ-Gl-Phe (а) и ингибирование избытком субстрата (б).
[8, 95]. Тем не менее данные по ингибированию и связыванию, по-
видимому, согласуются с образованием комплекса типа 1:1с Ki=
= 10-4 М [76, 98]. Способ связывания, соответствующий этой кон-
станте, требует присутствия цинка. Дополнительные молекулы фе-
нилпропионата присоединяются при концентрациях, больших
10~3 М. К сожалению, эти центры нельзя прямо сравнить с обнару-
живаемыми при кристаллографическом исследовании, так как в
кристаллическом состоянии сродство к фенилпропионату на два
порядка ниже и величина Ki составляет примерно 10-2 М [97].
Аналог этого соединения, содержащий тяжелый атом, иодфенил-
пропионат, частично занимает два центра при концентрации выше
10~3 М. Один из них расположен вблизи остатка Туг-198, а дру-
гой— в полости специфичности (рис. 15.10). В обоих случаях, как
показано методом ЯМР для Mn-КПА, молекула лиганда ориенти-
рована таким образом, что возможно взаимодействие его карбо-
Карбоксипептидаза А и другие пептидазы 535
ксильной группы с атомом металла активного центра [71]. Более
того, в отсутствие цинка сродство центра, расположенного в поло-
сти, уменьшается, а центр вблизи остатка Туг-198 вообще переста-
ет связывать иодфенилпропионат при его концентрации 0,10 М.
При большей концентрации ингибитора кристаллографическим ме-
тодом обнаруживаются два дополнительных места присоединения
иодфенилпропионата, однако они удалены от атома цинка.
Влияние центров, изображенных на рис. 15.10, на кинетику ре-
акции не вполне ясно. Связывание в полости, по-видимому, предпо-
лагает конкурентное ингибирование по отношению к любому суб-
страту, располагающемуся подобно Gly-Tyr. С другой стороны,
возможно, что ингибитор, находящийся вблизи остатка Туг-198,
присоединяется к атому цинка, не создавая стерических препятст-
вий связыванию длинных пептидов, рассмотренному в разд. 2.4.1.
Координационное число атома цинка при таком неконкурентном
взаимодействии, вероятно, становится равным пяти. Данные по
связыванию, из которых вытекает стехиометрия 1:1, казалось бы,
свидетельствуют в пользу того, что ингибитор не может одновре-
менно заполнять оба центра, обнаруживаемые кристаллографиче-
ским методом [70]. Однако факт защиты от модификации двух
тирозиновых остатков при высокой концентрации фенилпропионата
[99] проще всего объясняется одновременным присоединением
двух (или более) молекул ингибитора. Оглядываясь назад, прихо-
дится сожалеть, что ингибитор, ведущий себя столь сложным об-
разом, так часто использовался для изучения активного центра
КПА.
Обсуждение моделей присоединения эфирных субстратов наме-
ренно до сих пор откладывалось. Прямые данные о структуре от-
сутствуют, так как попытки получить кристаллические комплексы
КПА с эфирными субстратами пока не увенчались успехом. Неко-
торые, но не все эфирные субстраты ведут себя иначе, чем пепти-
ды. Например, активаторы гидролиза пептидов ингибируют гидро-
лиз циннамоилфениллактата [79], а ингибирование 0-фенилпро-
пионатом неконкурентно по отношению к пептидам и конкурентно
по отношению к ГФЛ [76]. Исходя из этих различий, было вы-
сказано предположение о том, что продуктивное связывание эфи-
ров и пептидов может происходить различным образом [100].
Полное описание взаимодействия с эфирами потребует эксперимен-
тов, позволяющих установить взаимное расположение важных
групп на ферменте и субстрате, т. е. использования флуоресцент-
ных и резонансных методов или проведения дополнительного рент-
геноструктурного анализа*.
* Недавно реитгеноструктуриым методом с разрешением 6 А было показано,
что продукт гидролиза эфира L-P-фениллактат присоединен к КПА так же, как
Ь-фенилаланин [65].
536
Глава 15
3.2.2. Зависимость активности от pH
Влияние pH на ферментативную активность иногда можно объ-
яснить в рамках модели, предполагающей, что катализ зависит от
состояния ионизации некоторых аминокислот. Тогда, если зависи-
мость от pH описывается колоколообразной кривой, по ней
можно определить константы ионизации К[е и К2е каталитически
важных групп в свободном ферменте. Константы ионизации, отно-
сящиеся к комплексу ES, можно получить из зависимости kKaT от
pH [101]. В случае КПА определение величин К\е и Kse было бы
полезным для установления роли тирозина и других остатков, рас-
положенных вблизи субстрата. Однако имеющиеся данные доволь-
но скудны. Пептидный субстрат КБЗ-Gly-Gly-Phe был исследован
недавно в условиях, при которых интерпретация результатов не
осложняется ингибированием или активацией [7]. Кривые зависи-
мости Акат/Км от pH оказались колоколообразными, что предпола-
гает участие в катализе как кислой, так и основной групп. Кривые,
описывающие влияние pH на kKaT, для этого субстрата имеют точ-
ку перегиба около pH 6. В щелочной области (до pH 10,5) /?Кат
практически постоянна* **. Отсутствие данных, указывающих на тит-
рование второй группы в комплексе ES, не исключает возможности
участия в реакции кислотного катализа, если предположить, на-
пример, что соответствующая стадия не лимитирует скорость реак-
ции (ср., однако, работу [103]). Зависимость гидролиза КГФ от
pH представлена в нескольких ранних работах. Данные, получен-
ные при высокой концентрации субстрата, указывают на сложный
характер влияния pH на kKaT и Км [26]. По зависимости началь-
ной скорости от pH [104] были определены величины pKes, равные
6,5 и 8,6, однако сравнение с ацилтрипептидами показывает, что эти
данные нуждаются в уточнении. При более низкой концентрации
КГФ влияние pH на /гКат менее выражено [85].
Зависимость &кат/Км от pH описывается колоколообразной кри-
вой в случае двух эфирных субстратов — ацетилманделата [92] и
циннамоилфениллактата [91]. Константы ионизации свободного
фермента оказались равными 10-6’9 и 10-7-5, и 10-6-5 и 10-9-4 М со-
ответственно. Не предложено никаких объяснений расхождения
кривых в щелочной области. Предварительное сравнение рН-зави-
симостей начальных скоростей гидролиза ГФЛ и КГФ обнаружи-
* Kje— константа ионизации свободного фермента, определяющая более
«кислую» ветвь колоколообразной кривой, аналогичная константа для вто-
рой ветви кривой, Kps — кажущаяся константа ионизации комплекса ES, опреде-
ленная по зависимости Акат от pH.
** Опубликована полная сводка данных по влиянию pH на гидролиз трипеп-
тида [102]. Как Км, так и Акат/Км зависят от состояния ионизации группы
с рК около 9. Этой группой может быть остаток Туг-248, что означало бы, что
ои принимает участие в связывании субстрата. Существует несколько предполо-
жений о природе группы с меньшим значением рК, влияющей на £Кат и Акат/Км.
Карбоксипептидаза А и другие пептидазы
537
ло, как казалось, принципиальное отличие эфиров от пептидов, по-
скольку кривая pH-зависимости для эфиров была сигмоидальная,
а для пептидов — колоколообразная. Однако работы с КБЗ-Gly-
Gly-Phe, ацетилманделатом и модифицированными карбоксипепти-
дазами [Ю5] не подтвердили универсальности этой закономерно-
сти.
Влияние природы субстрата на характер pH-зависимости мож-
но объяснить изменением стадии, лимитирующей скорость [106].
Кроме того, следует иметь в виду, что простейшая модель рН-эф-
фектов не включает конформационные переходы и изменения при-
роды стадии, лимитирующей скорость, при изменении pH [58].
3.3. Химическое изучение роли различных
остатков в активном центре
3.3.1. Модификация остатков в центре связывания
субстратов
Химическая модификация тирозина убедительно доказывает,
что эта аминокислота играет некоторую роль в активном центре
КПА*. Особенно удачной оказалась «дифференциальная модифи-
кация» в присутствии и в отсутствие субстратов или ингибиторов.
Впоследствии она была дополнена определением расположения
центра модификации в аминокислотной цепи. Модификация тиро-
зина включала ацетилирование уксусным ангидридом [Ю4] или
ацетилимидазолом [99], иодирование [107], нитрование тетранит-
рометаном [Ю5] и диазотирование диазоний-1Н-тетразолом [108]
или диазо-п-бензоларсонатом [109]. При реакции с ацетилимида-
золом в КПА модифицируются пять тирозиновых остатков, однако
влияние ацетилирования на активность связывается с модификаци-
ей только двух остатков, которые можно защитить 0-фенилпропиона-
том. В результате ацетилирования kK3JI для пептидов (включая
ацилтрипептиды) понижается до 3—6% первоначальной величины
[76, 89], в то время как гидролиз ГФЛ (но не ацетилманделата
[ПО]) происходит все еще достаточно быстро. Основной эффект
ацетилирования на гидролиз ГФЛ заключается в уменьшении суб-
стратного ингибирования, в результате чего оно появляется при
большей концентрации субстрата [78]. Величины &кат и Км для
ГФЛ увеличиваются при ацетилировании [78, 89].
Реагенты, отличающиеся от ацетилимидазола, селективно моди-
фицируют тот или иной из двух остатков тирозина, что оказывает
разное влияние на пептидазную активность. Нитрование одной ти-
розильной группы приводит к пропорциональному уменьшению ак-
тивности [105], тогда как присоединение 1 моля диазобензоларсо-
* В механизме, рассматриваемом в разд. 3.5.1., тирозину отводится роль
донора протонов.
538
Глава 15
ната к этому же, по-видимому, остатку вызывает уменьшение ак-
тивности лишь на 50%- Модификация же диазо-1Н-тетразолом
примерно одного остатка тирозина, происходящая при умеренном
избытке реагента, почти не сказывается на пептидазной активно-
сти. Нитро-, арсанилазо- и тетразолиазо-КПА способны гидроли-
зовать эфирный субстрат, ГФЛ, причем они менее чувствительны к
субстратному ингибированию. Дополнительная модификация тет-
разолилазо-КПА тетранитрометаном приводит к включению одной
нитрогруппы и значительной потере пептидазной активности. От-
сюда был сделан вывод, что при такой двойной модификации КПА
нитро- и тетразолилазогруппы присоединяются к разным остаткам
тирозина [105]. Если это так, то только один из них существен для
гидролиза пептидов, тогда как ингибирование избытком ГФЛ конт-
ролируется обоими. Однако для окончательного установления мест
модификации необходимо определить первичную структуру получа-
ющихся производных.
Отождествление одного из каталитически важных остатков ти-
розина с Туг-248 основывается на пространственном расположении
последнего и аминокислотной последовательности пептида, полу-
ченного иодированием КПА действием 1311 в присутствии фенилпро-
пионата и затем 12SI в его отсутствие. После гидролиза иодирован-
ной КПА пепсином был выделен пептид с наибольшим отношени-
ем 1251/1311. Его аминокислотная последовательность оказалась Пе-
Туг-Gln-Ala [111], что совпадает только с участком полипептидной
цепи 247—250 [4]. Уменьшение реакционной способности остатка
Туг-248 при связывании фенилпропионата можно объяснить изме-
нением конформации белковой молекулы.
Хотя остаток Туг-198 и удален от расщепляемой пептидной свя-
зи, он может участвовать в вандерваальсовых взаимодействиях с
субстратами, большими, чем глицилтирозин (рис. 15.3 и 15.4). Бо-
ковые цепи других остатков тирозина, расположенных в районе ак-
тивного центра, направлены в сторону от участков присоединения
субстрата. Таким образом, второй остаток этой аминокислоты, ко-
торый, судя по результатам модификации, участвует в образовании
активного центра, вероятно, является остатком 198. Иодфенилпро-
пионат присоединяется в кристаллах КПА к центру, расположен-
ному вблизи фенольной группы Туг-198 (рис. 15.10). Если фенил-
пропионат связывается аналогичным образом, то становится понят-
ным влияние этого ингибитора на модификацию. Высокую реак-
ционную способность остатков Туг-198 и Туг-248 можно частично
объяснить тем, что они находятся в контакте с окружающим рас-
творителем. Кроме того, рК нитруемого остатка тирозина меньше,
чем рК остальных [112].
Расположение остатка Туг-248 в комплексе КПА с Gly-Tyr
вполне согласуется с предположением о том, что модификация
именно этой аминокислоты оказывает существенное влияние на
Карбоксипептидаза А и другие пептидазы 539
гидролиз пептидов. Так, судя по структуре, в нитро- и арсанилазо-
карбоксипептидазах заместители присоединены к остатку Туг-248.
Изменения спектров, наблюдаемые при прибавлении р-фенилпро-
пионата [109, 112, 113] или глицилтирозина [109] к нативной или
модифицированной КПА, хотя и не могут быть однозначно интер-
претерированы, однако хотя бы отчасти обусловлены переориента-
цией остатка Туг-248 [2, 3]. (Сообщение [114] о том, что спектры
тетразолилазо-КПА, в которой модифицирован не остаток 248
[105], тоже изменяются в присутствии глицилтирозина, подчерки-
вает необходимость определения аминокислотной последователь-
ности соответствующих пептидов в различным образом модифи-
цированной КПА.)
Влияние ацетилирования на гидролиз пептидов объясняется
главным образом уменьшением kKaT, хотя Км тоже может изме-
няться при этом. Для ацетил-КПА величина Км Для КГФ в каче-
стве субстрата [78] и К< для этого же пептида в качестве ингиби-
тора гидролиза ГФЛ [100] на порядок выше значения Км для
превращения КГФ нативной КПА. Ранее сделанный вывод о том,
что влияние ацетилирования на активность КПА связано с нару-
шением способности связывать ГФЛ и дипептиды [115], не согла-
суется с результатами экспериментов по конкуренции и рентгено-
структурными данными об образовании комплекса между Gly-Tyr
и ацетил-КПА. Действительно, присоединение гиппурилфенилала-
нина и КБЗ-Gly-Gly-Phe почти не изменяется при ацетилировании
КПА [76, 78]. Тем не менее из этих данных по модификации не
следует, что тирозин непосредственно участвует в катализе*. Для
проверки постулата об участии остатка Туг-248 в качестве донора
протона [2, 3] было бы полезно исследовать кинетические пара-
метры и pH-зависимость гидролиза пептидов арсанилазо-КПА, в
которой рК этой группы понижено по сравнению с нативной КПА
[П6].
Обнаружение взаимодействий субстрата с остатками Glu-270 и
Arg-145 в кристаллическом комплексе стимулировало попытки мо-
дифицировать эти остатки [117]. Реакция примерно трех остатков
аргинина в КПА с бутандионом приводит к значительному (85%)
уменьшению пептидазной активности [118]. При удалении моди-
фицирующей группы с одного остатка аргинина активность восста-
навливается. Так же как при реакциях с тирозином, эстеразная ак-
тивность уменьшается не пропорционально пептидазной. Место мо-
дификации и то, каким образом измеряется kKaT и Км для гидро-
лиза пептидов, не определены. Модификация карбоксильной груп-
пы М-этил-5-фенилизоксазолий-3'-сульфонатом (К-реагент Вудвор-
* Возможно, что величина Км, измеренная в кинетических экспериментах,
относится лишь к небольшой доле фермента [78]. Несмотря на это, вызывает ин-
терес то, что ацетил-КПА сохраняет значительную часть пептидазной активности,
хотя модифицирующий реагент находится в 500-кратном избытке [76].
540
Глава 15
да) сопровождается потерей и пептидазной и эстеразной активно-
сти, причем скорость инактивации уменьшается в присутствии 0-
фенилпропионата или глицилтирозина. Еще одна группа СОО~
быстро реагирует с этим соединением, что, однако, не влияет на ак-
тивность. Пептид, выделенный после замещения реагента К на
ОСНз, по-видимому, содержит остаток Glu-88 [93]. Для химиче-
ского подтверждения роли, приписываемой остатку Glu-270, было
бы очень важно получить монопроизводное белка по этой амино-
кислоте (разд. 3.5).
Химическая модификация дает основания предположить, что,
кроме тирозильной, карбоксильной и аргинильной групп, для фер-
ментативной активности КПА важен гистидин. Фотоокисление ги-
стидина в присутствии метиленового синего [П9] или бенгальского
розового [120] или реакция одного остатка этой аминокислоты с
диазо-1Н-тетразолом [108] вызывает потерю пептидазной актив-
ности. Положение реагирующих остатков не установлено, хотя
единственными остатками гистидина, расположенными в активном
центре, являются His-69 и His-196. Исчезновение активности в этих
экспериментах не было просто результатом удаления цинка из фер-
мента. Хотя при окислении метиленовым синим и происходила ча-
стичная потеря цинка, в тетразолилазо-His-KHA металл сохраня-
ется полностью. Последнее производное фермента обладало эсте-
разной активностью, причем величина kKBLT для ГФЛ составляла
примерно половину /гКат для немодифицированной КПА [108].
3.3.2. Химическая природа лигандов, связанных
с металлом, и дисульфидная связь
Значительные усилия были потрачены на установление природы
лигандов атома цинка до того, как стали известны трехмерная
структура и аминокислотная последовательность КПА. Как и при
изучении связывания субстрата и поиске каталитических групп, в
основном сравнивали реакционные способности различных остат-
ков в присутствии и в отсутствие присоединяемого компонента, в
данном случае иона металла. В результате был сделан вывод, что
атом цинка связан с цистеином и а-аминогруппой. На самом деле
лигандами цинка являются остаток глутамата и два остатка гисти-
дина. Было бы полезно для будущего проанализировать некото-
рые опасности, подстерегающие при химическом определении при-
роды лигандов.
Результаты изучения реакции КПА с ПХМБ методом разност-
ной спектроскопии согласуются с предположением о появлении в
молекуле фермента при удалении металла одной реакционноспо-
собной сульфгидрильной группы. Вывод о том, что цистеин служит
лигандом цинка, казалось бы, подтверждался влиянием на апо-
фермент феррицианида и конкуренцией за апофермент ионов се-
Карбоксипептидаза А и другие пептидазы
541
ребра и цинка [24]. Поскольку константы устойчивости для ряда
металлов находились в согласии с моделью бидентатного лиганда,
в котором донорами служат атомы азота и серы [121], было изу-
чено влияние реагентов, специфически взаимодействующих с ами-
но- и имидазольными группами. Фотоокисление гистидина вызыва-
ло постепенное уменьшение активности и содержания цинка [119],
однако прибавление цинка не влияло на скорость инактивации, что
послужило основанием для вывода о том, что гистидин не связан
с металлом. Модификация а-аминогруппы, напротив, затруднялась
при образовании металлофермента [119]. Поскольку величину
рК 7,7, определенную комплексометрическим титрованием апофер-
мента, вполне можно было отнести к а-аминогруппе, было предпо-
ложено, что концевой остаток NH2 является лигандом цинка, не-
смотря на то, что его ацетилирование не влияло на ферментатив-
ную активность [122].
Пересмотр этих данных на основании известной структуры КПА
заставляет сделать вывод о том, что при спектрофотометрическом
титровании ПХМБ реагировал с лигандом, связанным с цинком.
Отсутствие SH-групп и высокое сродство центра присоединения
цинка к Hg2+ предполагает, что спектральные изменения вызыва-
ются взаимодействием ПХМБ с имидазольной и (или) карбоксиль-
ной группой. Появление изменений в спектре при 250 нм было ра-
нее отмечено при взаимодействии ПХМБ и ЭДТА [123]. Опыт
изучения КПА должен служить указанием на то, что некоторые
специфические центры связывания металла, не содержащие сульф-
гидрильные группы, могут реагировать с производными ртути, вы-
зывая спектральные изменения. Корреляции с модельными
соединениями следует признать ненадежными, поскольку считается
[124], что трудно подыскать модели, похожие по свойствам на ме-
талл сфер менты. Единственным методом строгого установления
взаимодействий металл—фермент в трехмерном пространстве сей-
час является трехмерный рентгеноструктурный анализ. Действи-
тельно, кристаллография представляет собой наиболее точный ме-
тод установления природы лигандов, связанных с металлом. Одна-
ко из-за трудностей в идентификации боковых цепей аминокислот
[22] для полного описания центра присоединения металла необхо-
димо знать первичную структуру белка.
Вывод [121] об участии сульфгидрильной группы в связывании
цинка привел к тому, что существование дисульфидной связи [1]
оставалось незамеченным. Аминокислотный анализ обнаружил
2 моля цистеина в расчете на 1 моль белка. Поэтому из результа-
тов титрования ПХМБ вытекало присутствие в КПА второй, не-
реакционноспособной SH-группы. Ни одну из этих SH-групп нель-
зя было легко алкилировать без предварительной обработки бел-
ка восстанавливающими агентами, такими, как меркаптоэтанол.
После восстановления и алкилирования в присутствии фенилпро-
542
Глава 15
пионата фермент сохранял свою активность и в нем обнаруживал-
ся 1 моль/моль карбоксиметилцистеина. Последующее удаление
фенилпропионата и повторение реакции приводило к появлению
второй карбоксиметильной группы и инактивации фермента. Уча-
сток аминокислотной последовательности, включающий второй
остаток цистеина, был отождествлен с активным центром [125].
После определения первичной структуры алкилирование «цис-
теина» было изучено повторно. С помощью методик выделения
пептидов с высоким выходом было показано, что при восстановле-
нии и алкилировании в присутствии фенилпропионата реагирует
тот или иной из остатков 138 и 161, но не оба сразу. Таким обра-
зом было подтверждено существование дисульфидного мостика,
связывающего эти аминокислоты [126].
3.4. Зачем нужен металл?
Функцию металла в карбоксипептидазе можно рассматривать
в двух аспектах: участие в связывании субстрата и участие в ка-
тализе. Данные свидетельствуют в пользу того, что в активной ме-
талло-КПА ионы металла участвуют как в связывании субстрата,
так и в катализе, однако логически удобно разделить влияние ме-
талла на ориентацию субстрата на поверхности молекулы фермен-
та и его участие в понижении энергии переходного комплекса.
Надежные данные о типах прямых связей, образуемых атомом
металла активного центра с субстратами и ингибиторами, можно
получить с помощью рентгеноструктурного анализа, а также мето-
дом ЯМР. Уширение резонансных линий протонов нескольких ин-
гибиторов общего вида RCOO- при добавлении Мп-КПА можно
объяснить тем, что группа СОО- находится во внутренней коорди-
национной сфере иона Мп2+ [66, 71]. Влияние фенилпропионата
на резонанс ядер I9F~, присоединяемых к Мп в Мп-КПА, подтвер-
ждает этот вывод [67, 72]. Однако полное описание влияния ме-
талла на геометрию связывания субстрата или ингибитора требу-
ет точного сравнения геометрии апо-КПА и различных металло-
КПА в присутствии и в отсутствие субстратов.
Оценка прочности взаимодействий субстрат—металл представ-
ляется довольно трудной. Один из методов заключается в сравне-
нии констант прочности в присутствии и в отсутствие цинка. По
различию между этими константами можно определить изменение
свободной энергии, соответствующее взаимодействию металл—суб-
страт, если допустить, что удаление металла не изменяет структу-
ру Е и ES. На практике в присутствии цинка вместо значений кон-
стант равновесия приходится использовать значения Км. Некото-
рые пептиды присоединяются к апо-КПА почти с той же констан-
той равновесия, что и к Zn-КПА [86] (табл. 15.4). Прочность свя-
зи пептидов с активными формами КПА, получающимися заменой
Карбоксипептидаза Д и другие пептидазы.
543
Таблица 15.5
Термодинамические и кинетические константы для различных
металлокарбоксипептидаз [41, 76, 89]
Ион металла igKa Субстрат
БГГФ БГФ КГФ ГФЛ
к6 ЛМ лкат ^кат *М *кат *м *кат
Zn 10,5 8,0 1,2 8,1 5,6 19,5 5,5 0,76 28,6
Ni 8,2 7,4 1,1 2,1 27,6
Со 7,0 6,0 5,9 4,8 7,4 Н,7 12,3 0,98 37,7
Мп 5,6 2,9 0,23 1,1 0,45 22,9 2,3 3,2 56,8
Cd 10,8 5,5 61,5
Hg 21,0
Си 10,6 Неактивна со всеми субстратами
а Константы устойчивости комплекса с металлом с поправкой на влияние 1 М CI— и
> М трис-буфера (pH 8).
б Величины К доданы в 10* М, а *кат— в I0-3 мин-1.
цинка на другой металл, почти не зависит от его природы
(табл. 15.5). Так как известно, что атом металла взаимодействует
с субстратом, и предварительные расчеты показывают, что в фер-
мент-субстратном комплексе происходит частичный перенос заряда
от карбонильной группы пептида к иону металла [127], то отсут-
ствие его влияния на присоединение пептида не может не озада-
чить. Частичное объяснение этого экспериментального факта мож-
но найти в том, что ион металла, кроме карбонильной группы суб-
страта, прочно связывает молекулу воды, в результате чего
изменение свободной энергии для реакции М- • -ОНг^М- • -О=С^
может быть малым. Кроме того, предположение о сохранении
структуры фермент-субстратного комплекса при удалении металла,
скорее всего, неправильно. Рентгеноструктурный анализ показыва-
ет, что, хотя молекула Gly-Tyr в комплексах с апо-КПА и Zn-КПА
расположена одинаково, в случае комплекса с апо-КПА отсутству-
ет участок электронной плотности, соответствующий перемещению
остатка Glu-270 [70]. Другие изменения конформации тоже могут
зависеть от природы металла в ферменте.
Константы связывания ГФЛ, ациламинокислот и ингибиторов,
содержащих карбоксильные группы, существенно изменяются при
удалении цинка. В отсутствие посторонних металлов эти соедине-
ния в концентрациях 0,01—0,05 М не влияют на реконструкцию
544
Глава 15
Zn-КПА из апофермента [88]. Константы Км (или Кг), описываю-
щие взаимодействие этих соединений с Zn-КПА, изменяются от
10“5 до 10-2 М (табл. 15.5). Отсюда можно заключить, что удале-
ние металла изменяет константу равновесия для ГФЛ более чем
в 100 раз. Сродство КПА к фенилпропионату изменяется в зави-
симости от природы металла в следующем ряду: Zn>Cd>Co
[98], а константы Кг в случае ингибиторов, содержащих карбок-
сильные группы, для Mn-КПА примерно на порядок ниже, чем для
Zn-КПА [72]. Можно отметить, что структурная интерпретация
такого влияния металлов затруднена.
Металло-КПА, не катализирующие гидролиз, могут тем не ме-
нее присоединять субстраты. При образовании комплексов дппеп-
тидов или ацилпептидов с Cd-, Си- и Hg-КПА происходит измене-
ние скорости обмена металлов. В качестве примера можно
привести присоединение ГФЛ к неактивной Си-КПА [88]. Количе-
ственные данные по константам связывания отсутствуют, однако
само сохранение связывания означает, что утрата ферментатив-
ной активности не является следствием потери способности присо-
единять субстрат в активном центре.
Роль металла в катализе, по всей вероятности, заключается в
том, что он в качестве кислоты Льюиса оттягивает электроны от
углеродного атома карбонильной группы. Эта точка зрения нашла
отражение в разнообразных предполагаемых механизмах действия
КПА [128, 129]. Ее прямым подтверждением служит обнаружение
связи Zn—О в кристаллическом состоянии. Кроме того, изменение
природы металла сказывается прежде всего на величине /гкат. Тем
не менее активности нескольких металло-КПА не укладываются в
ряд Ирвинга—Уильямса, в котором кислоты Льюиса располагают-
ся в порядке изменения их силы (Mn<jFe<Co<?Ni<Cu>Zn)
[5]. Для пептидных субстратов эффективность металлов изменяет-
ся в ряду Co>Zn=Ni>Mn>Cu=0, а для эфирных — в ряду
Cd>Mn>Co>Zn=Ni>Hg>Cu=0 (табл. 15.5). Выяснение спо-
соба, которым белок изменяет «естественный» ряд каталитической
эффективности металлов, необходимо для понимания функцио-
нальных свойств этого металлофермента. Нельзя сказать, что сей-
час в этой области достигнуты значительные успехи. Особенно
большую роль в ферментативном гидролизе могут иметь простран-
ственные и геометрические факторы. Например, выпадение Си-
КПА из ряда Ирвинга—Уильямса может быть результатом того,
что из-за ограничений, накладываемых ориентацией белковых ли-
гандов и геометрией иона Си2+, атом кислорода карбонильной
группы субстрата не может занять положение, при котором возмо-
жен перенос части заряда на ион металла. Действительно, на кар-
тах электронной плотности комплекса Cu-КПА и глицилтирозина
с низким разрешением [70] не наблюдается участка с положи-
тельной плотностью около остатка Glu-270, что предполагает отсут-
Карбоксипептидаза А и другие пептидазы 545
ствие его взаимодействия с а-аминогруппой. К сожалению, низкое
разрешение не позволяет сделать заключение о состоянии связи
металл—кислород. В модельных системах Cd2+ не уступает Со2+
как кислота Льюиса, тем не менее Cd-КПА не катализирует гидро-
лиз пептидов. Отсутствие активности у Cd-КПА трудно объяснить
только на основании геометрии лигандов у металла [5]. По-види-
мому, следует учитывать и другие факторы, такие, как размер
иона. Существенными могут быть также ограничения, накладыва-
емые складчатой структурой белка. Примером служит смещение
атома Hg2+ на ~1 Апо сравнению с Zn2+ (разд. 2.4.3). Если мо-
лекула присоединенного субстрата соответственно изменяет свое
положение, то в результате водородная связь между ним и остат-
ком Туг-248 [2, 3] может исчезнуть. Аналогичная ситуация может
существовать для Cd-КПА, поскольку ион Cd2+ не уступает по раз-
меру иону Hg2+. В качестве общего объяснения уникальной реак-
ционной способности белка в присутствии иона активного металла
предложены изменения в количестве лигандов и их освобождение
или обмен в ходе реакции [5].
Известны модельные системы с Си2+ и Со34", способные ката-
лизировать синтез и гидролиз пептидных и эфирных связей [130—
133]. В комплексах Со111, рассматриваемых в гл. 1, активируется
атака по карбонильной группе, которая присоединена к атому ме-
талла. Однако эффективность катализа ферментом значительно
выше, чем в случае моделей. Это может частично объясняться эк-
ранирующим эффектом белковой молекулы по отношению к иону
металла [22]. После присоединения субстрата атом цинка, фор-
мально несущий заряд +1, перестает контактировать с массой рас-
творителя и оказывается в среде с низкой диэлектрической прони-
цаемостью. В результате этого происходит усиление электрического
поля вокруг атома металла, что вызывает увеличение поляризации
карбонильной группы субстрата.
3.5. Механизм действия
3.5.1. Предположение, основанное на анализе
кристаллической структуры
Предполагаемый механизм гидролиза пептидов, основную роль
в котором играют атом цинка и остатки Glu-270 и Туг-248, основан
на предположении о большом сходстве комплекса с глицилтирози-
ном и кинетически важного ферментативного комплекса ES [2, 3].
Рентгеноструктурное исследование показывает, что боковые груп-
пы только этих аминокислот белка находятся вблизи атомов пеп-
тидной связи. Следовательно, в интересующем диапазоне pH доно-
ром протона и нуклеофильным агентом могут быть только остатки
Туг-248 и Glu-270. Кроме них, обе эти функции могут выполняться
35—2451
546 Глава 15
молекулами воды. Поскольку глицилтирозин является конкурент-
ным ингибитором гидролиза более длинных пептидов, а другие пеп-
тиды присоединены в кристаллах нативной и модифицированной
КПА сходным образом, то этот механизм считается применимым к
пептидам вообще. Гидролиз некоторых эфиров, таких, как ГФЛ,
обнаруживает столько особенностей, что его надо обсуждать от-
дельно. Расщепление пептидов протекает в несколько стадий, по-
следовательность которых и относительные скорости не вполне из-
вестны.
1) Связывание. Субстрат присоединяется таким образом, что
его С-концевая часть оказывается в полости специфичности, а
СООН-группа — вблизи остатка Arg-145. Пептидная группа NH
образует водородную связь с остатком Туг-248, который изменяет
свое положение. Атом кислорода карбонильной группы присоеди-
няется к атому цинка. Эти взаимодействия приводят к некоторому
искажению плоскостного расположения атомов пептидной связи,
в результате чего геометрия образующейся аминогруппы становит-
ся в какой-то степени тетраэдрической.
2) Катализ металлом. Атом цинка поляризует связь С=О, уве-
личивая электрофильность атома углерода, в результате чего ус-
коряется атака его водой или нуклеофильной группой белка.
3) Атака атома углерода карбонильной группы. Карбоксильная
группа остатка Glu-270 смещается, однако не вступает во взаимо-
действие с а-аминогруппой субстрата, как в комплексе с дипепти-
дом. Скорее всего, она либо образует ангидрид, в котором субст-
рат ковалентно присоединен к белку, либо действует в качестве
обобщенного основания, катализируя атаку водой по атому угле-
рода карбонильной группы.
4) Расщепление пептидной связи. При разрушении пептидной
связи, сопровождающемся передачей протона от остатка Туг-248
уходящей аминогруппе, образуются продукты реакции или ацил-
фермент.
5) Выброс продуктов реакции. Если в реакции участвует кова-
лентный интермедиат, то он атакуется молекулой воды. Возмож-
но, что на этой стадии происходит основной катализ комплексом
Zn2+—Н2О или Zn2+OH~. После завершения гидролиза состояние
ионизации вновь образовавшихся кислотной и основной групп из-
меняется в соответствии с pH среды, а фенольная группа Туг-248
присоединяет протон и возвращается в исходное состояние (рис.
15.13).
Для объяснения высокой скорости гидролиза длинных пептидов,
в которых предпоследняя NH-группа не замещена, предполагается
образование водородной связи [66, 67] между этой группой и ос-
татком Туг-248 [2]. Для этого NH-группа смещается в результате
поворота вокруг простых связей по отношению к положению, зани-
маемому ею в комплексе с Gly-Tyr. Циклическая структура, возни-
Карбоксипептидаза А и другие пептидазы
547
кающая при образовании водородной связи, фиксирует конформа-
цию субстрата и, возможно, вызывает в нем напряжение. Кроме
того, она облегчает передачу протона уходящей аминогруппе
(рис. 15.13). Взаимодействие а-аминогруппы с остатком Glu-270,
обнаруженное в комплексе с Gly-Tyr и невозможное в случае
длинных пептидов, по-видимому, затрудняет стадию (3).
Рис. 15.13. Две возможные роли остатка Glu-270 при гидролизе: основной катализ
атаки водой (а) и образование промежуточного ангидрида (б).
Этот механизм гидролиза пептидов использует некоторые об-
щие представления о природе высокой эффективности фермента-
тивного катализа. Так, в результате формирования циклической
структуры с участием Туг-248, воздействия металла на субстрат и
образования солевой связи между остатком Arg-145 и СОО~-груп-
пой субстрата может возникнуть напряжение [59—62]. Геометрия
лигандов у цинка в фермент-субстратном комплексе такова, что
переходное состояние энергетически выгоднее исходного. В этом
отношении интерес представляет смещение атома цинка в комп-
лексах с Phe-Gly-Phe-Gly [65] и Gly-Tyr. Кроме того, локализация
заряда на атоме металла [22] может влиять независимо от гео-
метрического напряжения [132], поскольку она дает фермент-суб-
стратный комплекс с относительно высоким содержанием энергии.
Рассмотренный выше механизм имеет ряд общих черт с неко-
торыми из механизмов, предложенных до определения структуры
[95, 128, 129]. Поляризационный эффект металла, действующего
как кислотный катализатор, предполагался ранее. Следует ожи-
дать участия в катализе и основания и кислоты. Рентгеноструктур-
ный анализ дал возможность идентифицировать боковые цепи ами-
нокислот, находящиеся вблизи субстрата, и определить детальную
стереохимию, необходимую для создания схемы механизма дейст-
вия. Хотя кинетика реакций с такими белками, как КПА [97], в
35*
548
Глава 15
кристаллическом состоянии часто количественно отличается от ки-
нетики в растворе, рассмотрение имеющихся структур комплексов
фермент—субстрат и фермент — ингибитор позволяет сделать не-
которые строгие выводы. Так, близость остатков Glu-270 и Туг-248
к субстрату делает их участие в реакции очень возможным, тогда
как отсутствие связи между атомом цинка и азота пептида пред-
полагает, что стабилизация уходящей группы металлом [5] играет
лишь незначительную роль в катализе. Однако кристаллографиче-
ские методы пока нельзя использовать для изучения быстрых реак-
ций со специфическими субстратами. Поэтому кинетические гипо-
тезы, основанные на знании структуры, необходимо проверять дру-
гими методами. Некоторые проблемы, касающиеся механизма
действия, рассмотрены в заключительных разделах этой главы.
3.5.2. Некоторые альтернативные механизмы
Рассмотренный выше механизм можно назвать «минимальным»
в том смысле, что он предполагает использование на каталитиче-
ских стадиях всего двух групп белка (Туг-248 и Glu-270), молеку-
лы воды и иона цинка. Изучение химической модификации указало
также на участие в катализе остатка гистидина. Хотя из-за уда-
ленности остатков His-69 и His-196 от субстрата представляется
маловероятным, что они служат нуклеофилами, вполне возможно,
что один из них перестает быть лигандом у атома цинка [5] и в
качестве основания принимает участие в переносе протона. Эта
стадия может не лимитировать скорость реакции. Однако при свя-
зывании не обнаруживается удаления какого-либо из остатков ги-
стидина от атома цинка. По аналогии с карбоангидразой рас-
сматривался также механизм, включающий атаку по карбониль-
ному атому углерода субстрата ОН~-группой, присоединенной к
иону цинка [2]. Из-за стерических препятствий такой механизм
менее вероятен, чем тот, в котором роль цинка заключается в по-
ляризации кислородного атома карбонильной группы субстрата.
3.5.3. Специфичность и изменение конформации
Способность КПА отщеплять только С-концевые аминокислоты
и отсутствие у нее эндопептидазной активности можно объяснить
стерическими эффектами. Внутренние пептидные связи субстрата
нельзя поместить около атома цинка таким образом, чтобы оста-
ток 145 и, возможно, 248 не мешали бы связыванию пептида. От-
бор субстратов, у которых на С-конце находятся ароматические
аминокислоты, а не основные, по-видимому, также происходит на
стадии связывания [86]. Труднее объяснить, почему не расщепля-
ется связь в глицилтирозинамиде, поскольку он присоединяется
к КПА [88]. По-видимому, необходимость присутствия свободной
Карбоксипептидаза А и другие пептидазы 549
группы С00“ как-то связана с движением остатка Туг-248. При
отсутствии заряда на конце пептида перемещение этой аминокис-
лоты не происходит, как, например, в случае лизилтирозинамида,
и активный центр остается недостроенным, поскольку в нем нет не-
обходимой для гидролиза фенольной группы. Изменение конфор-
мации остатка Туг-248, по-видимому, можно наблюдать в раство-
ре* [109, 112, 113], что облегчает дальнейшие эксперименты по
выяснению необходимости этого превращения.
3.5.4. Промежуточные соединения и стадия,
лимитирующая скорость
Природа стадии, лимитирующей скорость, и соответствующего
переходного комплекса пока не установлена. По аналогии с боль-
шим числом реакций карбонильной группы, при которых в ней про-
исходит разрыв связи углерод—кислород, предполагается образо-
вание нестойкого тетраэдрического промежуточного соединения
[58, 134]. Возникает интересный вопрос: включение какой группы
приводит к его образованию — молекулы воды или остатка
Glu-270? Вторая возможность подразумевает ковалентное присо-
единение субстрата к белку и требует протекания второй реакции
замещения, в которой происходит атака ацилфермента водой. Ис-
ходя из особенностей структуры, наиболее вероятен в качестве ко-
валентного промежуточного соединения ангидрид с участием ос-
татка Glu-270. Однако нуклеофильная способность карбоксильной
группы обычно невелика, а амин является плохой уходящей груп-
пой [58]. С другой стороны, есть основания полагать, что остаток
Glu-270 как основание катализирует атаку воды по атому углерода
карбонильной группы.
Для того чтобы различить нуклеофильное замещение и прямую
атаку водой, предпринимались попытки обнаружить ковалентные
промежуточные соединения в гидролитических реакциях, катализи-
руемых КПА. Однако пока они не увенчались успехом. При ис-
пользовании в качестве субстратов циннамоил- и индолакрилоил-
фенилаланина не происходит начального выброса продукта и из-
менений оптического поглощения, которые указывали бы на
существование интермедиатов [90]. Конечно, невозможность обна-
ружить их может объясняться тем, что стадия деацилирования не
является скоростьлимитирующей для этих субстратов.
Указания на существование промежуточных соединений можно
получить, изучая соответствующие реакции обмена и переноса
[58]. Однако транспептидазная или трансэстеразная активности
* По спектральным изменениям трудно различить перемещение ароматическо-
го остатка из гидрофильного в гидрофобное окружение и прямое взаимодействие
с модификатором, а также установить, что глубина изменения коррелирует со
степенью образования комплекса.
550
Глава 15
типа А—Вч^А—X или А——В у КПА не обнаружены [135,
136]. Отчасти это объясняется тем, что равновесие реакции сильно
смещено в сторону гидролиза, поэтому сопряжение обратной реак-
ции с каким-либо термодинамически выгодным процессом, как,
например, при синтезе карбоксипептидазой В ингибитора трипсина
[137], могло бы облегчить выяснение механизма. Перенос ациль-
ного фрагмента на гидроксиламин оказался очень информатив-
ным при изучении механизма действия химотрипсина, однако в
реакциях, катализируемых КПА, аналогичное образование гидро-
ксамата не обнаружено. КПА катализирует обмен атомов 18О кар-
боксильной группы ацетилфенилаланина. Эта реакция ингибирует-
ся о-фенантролином, и ее зависимость от pH примерно такая же,
как для гидролиза [135]. В случае химотрипсина похожие обмен-
ные реакции в «псевдосубстратах» объясняют образование кова-
лентных промежуточных соединений. Однако сам факт существо-
вания обмена кислорода не служит строгим доказательством про-
исходящей атаки нуклеофильной группой белка; обмен может
происходить и путем прямого замещения. Предполагают, что суб-
страты, обменивающие атомы кислорода, связываются таким об-
разом, что карбоксильная группа оказывается на месте карбониль-
ной группы пептидной связи. Связь обмена 18О в ацетилфенилала-
нине с механизмом действия карбоксипептидазы можно было бы
подтвердить, определив ориентацию ациламинокислот по отноше-
нию к иону металла.
Чтобы узнать, происходит ли перенос протона на стадии, лими-
тирующей скорость, были измерены изотопные эффекты с дейте-
рием. Если скорость определяется общим основным катализом ата-
ки водой по субстрату или ацилферменту, то отношение £h2o/^d2o
может быть равно 2 или больше. По-видимому, перенос протонов
при гидролизе пептидов происходит не на самой медленной ста-
дии, поскольку отношение скоростей для КГФ [85] и БГФ [138]
меньше 1,2. При гидролизе эфирных субстратов, ГФЛ и циннамо-
илфениллактата дейтериевый изотопный эффект равен 2 [22,
138]. Однако нельзя с уверенностью сказать, что гидролиз этих
эфиров происходит посредством общего основного катализа.
Так как изучение изотопных эффектов не позволило охаракте-
ризовать важные промежуточные соединения, то можно было ожи-
дать, что для определения механизма будет полезным измерение
индивидуальных констант скорости. Использование таких субст-
ратов, как дансил-Gly-Tyr, для которого можно применить флуо-
риметрию в соответствии с методом остановленной струи, дает
возможность количественно охарактеризовать некоторые стадии в
катализе [8]. Скорость всех последовательных стадий реакции
должна быть по крайней мере не меньше общей скорости гидро-
лиза субстрата в стационарной фазе. Например, если постулирует-
ся, что остаток Туг-248 служит донором протона, то он должен
Карбоксипептидаза А и другие пептидазы
551
изменить свое положение при каждом обороте реакции и, следо-
вательно, спектральные изменения, наблюдаемые при добавлении
субстратов или ингибиторов к нитро-КПА или арсанилазо-КПА
при условии, что они служат правильной мерой переориентации
этого остатка, должны протекать по крайней мере так же быстро,
как превращение субстрата в целом. По предварительным данным,
константа скорости изменения спектра при взаимодействии Gly-Tyr
и арсанилазо-КПА при 25 °C не меньше 200 с-1. Таким образом,
скорость этого процесса равна скорости гидролиза лучших пептид-
ных субстратов КПА, что свидетельствует о том, что конформаци-
онный переход может быть одной из стадий реакции [139].
Можно надеяться, что другие детали механизма гидролиза пеп-
тидов также будут вскоре установлены. Гидролиз эфиров, таких,
как ГФЛ, обнаруживает ряд особых черт. Ряд различий в гидро-
лизе эфиров и пептидов сказывается на влиянии модификации ти-
розина, замещения металла и прибавления D2O, ингибиторов и
активаторов. Было высказано предположение о том, что механиз-
мы гидролиза эфиров и пептидов различны [128] и что центры
присоединения двух типов субстратов полностью или частично не
совпадают [100, 117]. Из известного несходства двух реакций дей-
ствительно вытекает существование количественных различий в
соответствующих им наборах констант скорости, из-за которых
стадии, лимитирующие скорость, для гидролиза пептидов и эфиров
могут не совпадать. Однако это можно объяснить не только с по-
мощью гипотезы «двух механизмов». Например, активность нитро-
КПА, Cd-КПА и Hg-КПА при гидролизе ГФЛ можно объяснить,
предположив, что некоторые эфиры могут превращаться без пере-
дачи протона от остатка тирозина [2, 3]. Узловой вопрос о воз-
можности превращения некоторых субстратов по различным меха-
низмам с участием различных каталитических групп заслуживает
более тщательного изучения.
4. ДРУГИЕ ПЕПТИДАЗЫ
4.1. Карбоксипептидаза В
Карбоксипептидазы, отщепляющие с большой скоростью от
С-конца пептидов остатки Lys и Arg, выделены из свиньи [140,
141] и быка [142, 143]. Наилучшими субстратами для них служат
пептиды, имеющие на концах основные аминокислоты, и эти фер-
менты называются карбоксипептидазами В (КПВ). По ряду
свойств КПВ очень похожа на КПА: ее молекулярная масса равна
34 000 [142, 144], содержание цинка составляет 1 г-атом/моль
[144] и его удаление или замена сопровождается изменением ка-
талитической активности [145]. Из аминокислотного состава и
552
Глава 15
величины молекулярной массы следует, что длина цепи КПВ та-
кая же, как у КПА; для КПВ свиньи она составляет 304 остатка
[144], а для КПВ быка — 300—301 остаток [142, 143, 146]. Пер-
вичная структура КПВ быка определена лишь частично [146—
148], а о рентгеноструктурных исследованиях сообщений не было.
Активная форма КПВ образуется при гидролизе профермента с
молекулярной массой 57 000 трипсином [142, 149].
Для описания специфичности КПВ использовано несколько суб-
стратов и ингибиторов [141]. По отношению к ним этот фермент
напоминает КПА. С наибольшей скоростью гидролизуются такие
модельные пептиды, как гиппуриллизин [150] (йКаТ=1,32-
• 104 мин-1, Км=7,7-10~4 М), гиппуриларгинин (Акат = 6,3-
• 10-3 мин-1) и его эфирный аналог гиппуриларгининовая кислота
(^кат = 1,43-Ю4 мин-1, Км=0,4-10-4 М). Сравнение с данными, при-
веденными в табл. 15.4, показывает, что эти величины такие же,
как для соответствующих субстратов КПА. Катализируемый КПВ
гидролиз эфиров ингибируется избытком субстрата, а образующие-
ся при реакции амино- и оксикислоты ингибируют гидролиз и эфи-
ров и пептидов [151]. Отсутствие активности КПВ по отношению
к гиппурил-с-лизину и гиппурил- ь-лизинамиду напоминает КПА.
Это же можно сказать относительно способности использовать в
качестве субстратов пептиды, содержащие некоторые необычные
аминокислоты, такие, как гиппурил-ь-гомоаргинин и бензоил-0-ала-
нил-ь-лизин. Присутствие пролина в предпоследнем от конца по-
ложении уменьшает скорость реакции, а дипептид глицил-ь -лизин
является очень плохим субстратом [141]. КПВ быка гидролизует
типичные пептидные субстраты КПА, но с меньшей эффективно-
стью. Гиппурил-0-фениллактат, по-видимому, одинаково хорош как
для КПА, так и для КПВ быка [142]. Гидролиз субстратов КПА
под действием КПВ не является результатом наличия примеси
КПА, так как КПВ ингибируется аналогами своих субстратов, на-
пример е-аминокапроновой кислотой, которая не влияет на соот-
ветствующие реакции КПА [142]. Самыми эффективными конку-
рентными ингибиторами служат соединения, содержащие группу
СОО- и радикалы, похожие на боковые цепи Arg и Lis [150]. Как
и в случае КПА, картина ингибирования фенилпропионатом слож-
на [142].
Сравнение аналогичных последовательностей КПВ и КПА быка
дает основание предположить, что эти ферменты представляют со-
бой структурные гомологи [152, 153]. Предполагается, что струк-
турные гены двух карбоксипептидаз происходят от общего пред-
шественника [152—154]. По аминокислотному составу два белка
довольно близки. Поразительные различия наблюдаются только в
содержании полуцистина, которое составляет семь для КПВ [142]
и два для КПА. КПВ быка содержит три дисульфидных мостика
и одну сульфгидрильную группу. Одна из петель цепи КПВ раз-
Карбоксипептидаза А и другие пептидазы 553
мером в 14 остатков, образуемая дисульфидной связью, по-види-
мому, гомологична участку 65—79 в КПА [148, 152]. Если это так,
то лиганды у цинка — His и Giu, занимающие в КПА положения
69 и 72,— сохраняются и в КПВ. Более того, остатки 66 и 79 в
КПА, которые гомологичны остаткам цистеина, образующим упо-
мянутую петлю в КПВ, расположены поблизости друг от друга.
В активном центре этих белков есть еще одна гомологичная после-
250 255
КПА: Thr-Ile-Tyr-Gln-Ala-Ser-Gly-Gly-Ser-Ile-Asp-Trp
КПБ: Thr-I le-Tyr-Pro-Ala-Ser-Gly-Gly-Ser-Asp-Asp-Trp
Рис. 15.14. Гомологичные аминокислотные последовательности в активных цент-
рах карбоксипептидаз А и В быка.
довательность. Под действием афинного реагента 4-бромацетил-
амидобутилгуанидина, в активном центре КПВ модифицируется
остаток тирозина [155]. Сравнение обнаруживает высокую степень
гомологичности последовательностей около этой аминокислоты и
Туг-248 в КПА, как показано на рис. 15.14 [153]. С другой сторо-
ны, гомологичность последовательностей у С-конца (14 остатков)
выражена слабее.
Известна последовательность еще одного участка КПВ, содер-
жащего сульфгидрильную группу [156]. Единственный остаток ци-
стеина в КПВ алкилируется иодацетамидом в отсутствие восста-
навливающих агентов при условии, если удален цинк. Таким об-
разом, для КПВ химические данные более определенно указывают
на то, что цистеин служит лигандом цинка. Однако последователь-
ность [157] вокруг этого остатка цистеина гомологична последова-
тельности КПА не в районе третьего лиганда цинка (His-196), а
в начале спирального участка у С-конца. Остаток цистеина соот-
ветствует остатку А1а-290, а-углеродный атом которого удален от
атома цинка на 11 А. Если считать, что химические данные до-
казывают участие цистеина в связывании металла, то возникает
следующая дилемма: или две высокогомологичные последователь-
ности расположены в двух белках в разных положениях, или их
третичные структуры не одинаковы. Объяснение противоречия мо-
жет заключаться в том, что остаток цистеина, как и А1а-290,
спрятан внутри молекулы и вступает в реакцию только после не-
которого ее разворачивания, вызываемого удалением цинка. Ско-
рее всего, цистеин не является лигандом цинка, однако обращение
к истории установления природы лигандов металла в КПА (разд.
3.3.2) показывает, что разумнее отложить этот вопрос до установ-
ления полной первичной структуры КПВ и сравнения третичных
структур прямыми методами.
554
Глава 15
4.2. Другие карбоксипептидазы
и прокарбоксипептидазы
Ферменты с карбоксипептидазной активностью выделены из ря-
да источников [21, 158]. Некоторые из них, как, например, фер-
менты свиньи [19, 141] и колючей акулы, обитающей в Тихом оке-
ане [159, 160], по специфичности очень похожи на карбоксипепти-
дазы быка [21], тогда как другие отличаются от них. Фермент из
Pseudomonas, названный карбоксипептидазой G, быстро отщепля-
ет глутамат от С-конца пептидов [161, 162]. Все вышеупомянутые
карбоксипептидазы содержат цинк [21] или по крайней мере нуж-
даются в нем для проявления активности [161]. Модификация ти-
розина в некоторых из них влияет на активность [21]. Молеку-
лярные массы в тех случаях, когда они определены, близки к
35000 [21]. Размер же молекул проферментов очень зависит от
источника. Так, про-КПА свиньи и акулы представляют собой мо-
номеры молекулярной массы чуть больше 40 000 [19, 159], что
соответствует размеру субъединицы I про-КПА быка [11]. Другие
карбоксипептидазы как бактериального [163], так и растительно-
го [164, 165] происхождения также частично охарактеризованы.
Некоторые из них не ингибируются хелатирующими агентами
(165], тогда как остальные предположительно являются металло-
ферментами [163].
Центры связывания металла и субстрата обнаружены в
про-КПА быка [114, 166]. При выделении этот белок сохраняет
около 1 моля цинка. Однако комплексометрическое титрование
про-КПА обнаруживает существенные различия между центрами
присоединения металла в КПА и про-КПА. Стерические требова-
ния для связывания субстрата двумя белками также неодинаковы
[166]. Активация прокарбоксипептидаз интенсивно изучалась, и
библиографию по этому вопросу можно найти в нескольких недав-
но опубликованных работах [21, 167, 168].
4.3. Лейцинаминопептидаза
Лейцинаминопептидаза (ЛАП) является экзопептидазой, от-
щепляющей аминокислоты от N-конца пептидной цепи. Субстра-
тами могут служить только пептиды, оканчивающиеся ь-аминокис-
лотами со свободной NH2-rpynnofi. Следующая аминокислота может
иметь d-конфигурацию [32, 164]. N-Ацетилпептиды не гидролизу-
ются. Название фермента связано с тем, что пептиды, имеющие на
N-конце лейцин, и их аналоги, удобные для измерения активности,
такие, как лейцил-п-нитроанилид, гидролизуются быстрее всего.
Однако специфичность ЛАП очень низка. Сообщалось, что субст-
ратами могут быть даже ь-лизинамид и ь-пролинамид, причем ак-
тивность по последнему соединению составляет 1 % активности по
Карбоксипептидаза А и другие пептидазы 555
лейцинамиду [169, 170]. Эфирные аналоги, такие, как н-бутил-ь-
лейцинат [169], гидролизуются медленно [171]. р-Аланинамид в
отличие от лейцил-р-аланинамида не является субстратом [170].
Систематических определений Км и ^кат не производилось, поэто-
му неизвестно, в какой степени специфичность определяется проч-
ностью фермент-субстратных комплексов. Исследование серии пеп-
тидов с d- и l-аланином на конце показало, что активный центр
ЛАП, по-видимому, может взаимодействовать по крайней мере с
четырьмя остатками субстрата [32].
Самым доступным источником ЛАП служат почки свиньи [172].
Молекулярная масса этого фермента больше молекулярной массы
карбоксипептидазы и составляет 200 000 [172, 173]. Описан также
препарат из хрусталика быка [174, 175]. Детальное изучение ЛАП
затруднено неустойчивостью фермента при выделении, присутстви-
ем сопутствующих эндопептидаз [176, 177] и существованием мно-
жественных молекулярных форм [173, 178]. Ионы Мп2+ и Mg2+
стабилизируют и активируют белок, вследствие чего выделение
обычно проводят в присутствии одного из этих металлов,'чаще все-
го Mg2+. Зависимость степени активации ЛАП от концентрации
Мп2+ [170] хорошо описывается кривой связывания при п— 1 (чис-
ло катионов на молекулу) и /fa=3,3-104 М-1. На основании этих
данных и ингибирующего действия цитрата и ЭДТА ЛАП была
недавно отнесена к Mg (Мп)-активируемым или Mg (Мп)-содержа-
щим металлоферментам, хотя данные о наличии в ней этих метал-
лов отсутствуют.
Модификация [173] метода выделения ЛАП из почек свиньи
позволяет получить активный фермент, содержащий 4—6 молей
цинка в расчете на молекулярную массу 300 000, и очень малые
количества других металлов [180]. ЛАП, выделенная этим мето-
дом, устойчива в отсутствие Mg2+, однако этот ион необходим во
время очистки. Таким образом, ЛАП, по-видимому, представляет
собой металлофермент. Ингибирование о-фенантролином и бипи-
ридилом и отсутствие ингибирования фторидом и ЭДТА подтверж-
дают это предположение [180]. Пока не удалось получить свобод-
ную от металла форму фермента, способную к реактивации,
однако некоторые ионы, например Са2+, замещают цинк с одно-
временным изменением активности. Интересно, что замена немно-
гим более половины цинка на Мп2+ приводит к видимому увели-
чению активности по отношению к лейцил-п-нитроанилиду. Дан-
ные [170] по влиянию на активность ионов некоторых других
металлов, включая Cd2+, Со2+ и Ni2+, нуждаются в пересмотре.
Согласно предложенным схемам гидролиза, незаряженная ами-
ногруппа субстрата связывает ион металла (Mg2+ или Мп2+) [129,
170, 181]. Химическая информация об активном центре почти от-
сутствует. Известно только, что гидрофобные соединения являются
очень сильными ингибиторами [170]. Судя по этому, а также по
556
Глава 15
субстратной специфичности, активный центр имеет гидрофобную
природу. Число активных центров в молекуле неизвестно [175].
По-видимому, ЛАП может быть хорошим объектом для изучения
взаимодействий металла с субстратом методами ЯМР. Транспеп-
тидазная активность у ЛАП отсутствует [182], что следует учиты-
вать при создании гипотетических механизмов ее действия.
4.4. Некоторые другие металлсодержащие протеиназы
Наряду с сериновыми, сульфгидрильными и кислыми протеина-
зами металлопептидазы составляют отдельный большой класс про-
теолитических ферментов [183]. Существуют списки пептидаз, тре-
бующих для своей активности присутствия цинка, кобальта или
марганца [181, 184]. Хотя многие из известных металлопептпдаз
являются экзопептидазами или дипептидазами, из этого правила
есть исключения. Недавно было показано, что две эндопептида-
зы—термолизин [185] и нейтральная протеаза В. subtilis [186] —
содержат цинк. Ионы Са2+ стабилизируют оба эти фермента. Их
молекулярная масса составляет 35 000—40 000 [186, 187]. Термо-
лизин гидролизует лучше всего пептидные связи, образованные
аминогруппами гидрофобных аминокислот [44, 188—190]. Похо-
жей специфичностью обладает протеаза В. subtilis [186], хотя для
этого фермента трудно получить удобные низкомолекулярные суб-
страты [191]. Нет сомнений в том, что другие бактериальные про-
теиназы окажутся металлоферментами [185]. По мере увеличения
числа хорошо охарактеризованных металлопептидаз будет получен
ответ на интересный вопрос о том, в какой степени механизмы ка-
тализа являются для них общими.
СПИСОК ЛИТЕРАТУРЫ
1. Reeke G. N., Hartsuck J. A., Ludwig М. L., Quiocho F. A., Steltz Т. А.,
Lipscomb W. N., Proc. Natn. Acad. Sci. U. S., 58, 2220 (1967).
2. Lipscomb W. N., Hartsuck J. A., Reeke G. N., Quiocho F. A. et al., Brookhaven
Symp. BioL, 21, 24 (1968).
3. Lipscomb W. N., Reeke G. N„ Hartsuck J. A., Quiocho F. A., Bethge P. H„
Phil. Trans. R. Soc., B257, 177 (1970).
4. Bradshaw R. A., Ericsson L. H., Walsh K. A., Neurath H., Proc. Natn. Acad.
Sci. U. S„ 63, 1389 (1969).
5. Dennard A. E., Williams R. J. P., Transition Metal Chemistry. A Series of
Advances, Vol. 2, Marcel Dekker, New York, 1966, p. 115.
6. Shulman R. G., Navon G„ Wyluda B. J., Douglass D. C., Yamane T„ Proc.
Natn. Acad. Sci. U. S., 56, 39 (1966).
7. Auld D. S., Federation Proc., 28, 346 (1969).
8. Latt S. A., Auld D. S., Vallee B. L„ Federation Proc., 29, 666 (1970).
9. Ludwig M. L., Hartsuck J. A., Steitz T. A., Muirhead H., et al., Proc. Natn.
Acad. Sci. U. S., 57, 511 (1967).
10. Yamasaki M., Brown L R., Cox D. J., Greenshields R. N., Wade R. D„ Neu-
rath H., Biochemistry, 2, 859 (1963).
Карбоксипептидаза А и другие пептидазы
557
II. Freisheim L Н., Walsh К- A., Neurath Н., Biochemistry, 6, ЗОЮ (1967).
12. Anson М. L„ J. Gen. Phisiol., 20, 663 (1937).
13. Allan В. J., Keller P. J., Neurath H., Biochemsitry, 3, 40 (1964).
14. Cox D. J., Bovard F. C., Bargetzi J. P., Walsh K. A., Neurath H., Biochemistry,
3, 44 (1964).
15. Vallee B. L., Neurath H„ J. Biol. Chem., 217, 253 (1955).
16. Thompson E. О. P„ Biochim. Biophys. Acta, 10, 633 (1953).
17. Petra P. H., Neurath H„ Biochemistry, 8, 2466 (1969).
18. Petra P. H., Neurath H„ Biochemistry, 8, 5029 (1969).
19. Folk J. E„ Schirmer E. W., J. Biol. Chem., 238, 3884 (1963).
20. Sampath Kumar K. S. V., Clegg J. B., Walsh K. A., Biochemistry, 3, 1728
(1964).
21. Neurath H., Bradshaw R. A., Petra P. H„ Walsh K- A., Phil. Trans. R. Soc.,
B257, 159 (1970).
22. Lipscomb W. N„ Hartsuck J. A., Quiocho P. A., Peeke G. N., Proc. Natn. Acad.
Sci. U. S., 64, 28 (1969).
23. Petra P. H., Bradshaw R. A., Walsh K. A., Neurath H., Biochemistry, 8, 2762
(1969).
24. Vallee B. L., Coombs T. L., Hoch F. L., J. Biol. Chem., 235, PC45 (1960).
25. Schechter L, Eur. J. Biochem., 14, 516 (1970).
26. Neurath H„ Schwert G. W., Chem. Rev., 46, 69 (1950).
27. Smith E. L„ Adv. Enzymol., 12, 191 (1951).
28. McClure W. O., Neurath H., Walsh K. A., Biochemistry, 3, 1897 (1964).
29. Bender M. L., Whitaker J. R., Menger F., Proc. Natn. Acad. Sci. LJ. S., 53, 711
(1965).
30. Ambler P. P., in S. P. Colowick, N. O. Kaplan (eds.), Methods in Enzymolo-
gy, Vol. 11, Academic Press, New York, 1967, p. 436.
31. Abramowitz N„ Schechter L, Berger A., Biochem. Biophys. Res. Commun., 29,
862 (1967).
32. Schechter L, Berger A., Biochemistry, 5, 3371 (1966).
33. Vanari S., Mitz M. A., J. Am. Chem. Soc., 79, 1150 (1957).
34. Smith E. L., Hanson H. T., J. Biol. Chem., 179, 802 (1949).
35. Vallee B. L., Neurath H., J. Am. Chem. Soc., 76, 5006 (1954).
36. Coombs T. L., Felber J. P„ Vallee B. L., Biochemistry, 1, 899 (1962).
37. Felber J. P., Coombs T. L., Vallee B. L„ Biochemistry, 1, 231 (1962).
38. Vallee B. L., Rupley J. A., Coombs T. L., Neurath H., J. Am. Chem. Soc., 80,
4750 (1958).
39. Vallee B. L., Pupley J. A., Coombs T. L., Neurath H., J. Biol. Chem., 235, 64
(1960).
40. Coleman J. E., Vallee B. L., J. Biol. Chem., 235, 390 (1960).
41. Colleman J. E., Vallee B. L„ J. Biol. Chem., 236, 2244 (1961).
42. Nomoto M., Srinivasan N. G., Bradshaw R. A., Wade P. D., Neurath H.. Bio-
chemistry, 8, 2755 (1969).
43. Bradshaw R. A., Babin D. R., Nomoto M„ Srinivasin N. G., et al., Biochemistry,
8, 3859 (1969).
44. Bradshaw R. A., Biochemistry, 8, 3871 (1969).
45. Putnam F. W., Neurath H., J. Biol. Chem., 166, 603 (1946).
46. Lipscomb W. N., Coppola J. C., Hartsuch J. A., Ludwig M. L. et al., J. Mol.
BioL, 19, 423 (1966).
47. Diamond R., Acta Cryst., 21, 253 (1966).
48. Ramachandran G. N., Ramakrishnan C„ Sasisekharan V., J. Mol. BioL, 7, 95
(1963).
49. Edsall J. T., Flory P. J., Kendrew J. C., Liquori A. M. et al., J. Biol. Chem.,
241, 1004 (1966).
50. Pauling L., Corey R. B., Proc. Natn. Acad. Sci. U. S„ 37, 251, 729 (1951).
51. Blake С. C. F., Mair G. A., North A. С. T., Phillips D. C., Sarma V. R„
Proc. R. Soc., B167, 365 (1967).
558
Глава 15
52. Hartsuck J. A., Lipscomb W. N., in Boyer P. D. (ed.), The Enzymes, 3rd edn.,
Vol. Ill, Academic Press, New York, 1971, p. 1.
53. Van der Helm D., Nicolas H., Acta Cryst., 26 B, 1858 (1970).
54. Kretsinger R. H., Cotton F. A., Bryan R. F., Acta Cryst., 16, 651 (1963).
55. Harding M. M., Cole S. J., Acta Cryst., 16, 643 (1963).
56. Yanari S., Mitz M. A., J. Am. Chem. Soc., 79, 1154 (1957).
57. Kauzmann W., Adv. Protein Chem., 14, 1 (1959).
58. Дженкс В., Катализ в химии и энзимологии, «Мир», М., 1972.
59. Eyring Н„ Lumry R„ Spikes J. D., in W. D. McElroy, B. Glass, (eds.), The
lechanism of Enzyme Action, Johns Hopkins, Baltimore, 1954, p. 123.
60. Lumry R., in P. D. Boyer, H. Lardy, K. Myrback (eds.), The Enzymes, 2nd
edn., Vol. 1, Academic Press, New York, 1959, p. 157.
61. Pauling L., Am. Scient., 36, 51 (1948).
62. Jencks W. P., in N. O. Kaplan, E. P. Kennedy (eds.), Current Aspects of Bio-
chemical Energetics, Academic Press, New York 1966, p. 273.
63. Koshland D. E., Proc. Natn. Acad. Sci. U. S., 44, 98 (1958).
64. Koshland D. E., Cold Spring Harbor Symp. Quant. Biol., 28, 473 (1963).
65. Quiocho F. A., Bethge P. H., Lipscomb W. N., Studebaker J. F. et al., Cold
Spring Harbor Symp. Quant. Biol., 36, 561 (1971).
66. Snoke J. £., Neurath H„ J. Biol. Chem., 181, 789 (1949).
67. Hanson H. T., Smith E. L., J. Biol. Chem., 175, 833 (1948).
68. Smith E. L., J. Biol. Chem., 175, 39 (1948).
69. Stahmann M. A., Fruton L S., Bergman J., J. Biol. Chem., 164, 753 (1946).
70. Steitz T. A., Ludwig M. L., Quiocho F. A., Lipscomb W. N., J. Biol., Chem., 242,
4662 (1967).
71. Navon G., Shulman R. G., Wyluda B. L, Yamane T., Proc. Natn. Acad. Sci.
U. S„ 60, 86 (1968).
72. Navon G., Shulman R. G., Wyluda B. J., Yamane T., J. Mol. BioL, 51, 15
(1970).
73. Bishop W. H., Quiocho F. A., Richards F. M., Biochemistry, 5, 4077 (1966).
74. Brill A. S., Kirkpatrick P. R., Scholes С. P„ Venable J. H., Jr., Proc. Fourth
Johnson Foundation Colloq., University of Pennsylvania, 1969, Academic Press,
New York, 1970.
75. Snoke J. E., Schwert G. W., Neurath H., J. Biol. Chem., 175, 7 (1948).
76. Auld D. S., Vallee B. L., Biochemistry, 9, 602 (1970).
77. Kaiser E. T„ Awazu S., Carson F. W., Biochem. Biophys. Res. Commun., 21,
444 (1964).
78. Whitaker J. R., Menger F., Bender M. L., Biochemistry, 5, 386 (1966).
79. Awazu S., Carson F. W„ Hall P. L„ Kaiser E. T., J. Am. Chem. Soc., 89, 3627
(1967).
80. Davies R. C., Auld D. S., Vallee B. L., Biochem. Biophys. Res. Commun., 31,
628 (1968).
81. Thoma J. A., Koshland D. E., J. Am. Chem. Soc., 82, 3329 (1960).
82. Izumiya N., Uchio H., J. Biochem. (Japan), 46, 235 (1959).
83. Blake С. C. F., Johnson L. N., Mair G. A., North A. С. T., Phillips D. C., Sar-
ma V. R„ Proc. R. Soc., Bl 67, 378 (1967).
84. Rupley J. A., Gates V., Proc. Natn. Acad. Sci. U. S., 57, 496 (1967).
85. Lumry R., Smith E. L., Glantz R. R., J. Am. Chem. Soc., 73, 4330 (1951).
86. Coleman J. E., Vallee B. L., J. Biol. Chem., 237, 3430 (1962).
87. Wyman J., Adv. Protein Chem., 19, 223 (1964).
88. Coleman J. E., Vallee B. L., Biochemistry, 1, 1083 (1962).
89. Davies R. C., Riordan J. F„ Auld D. S., Vallee B. L., Biochemistry, 7, 1090
(1968).
90. McClure W. €)., Neurath H., Biochemistry, 5, 1425 (1966).
91. Hall P. L., Kaiser B. L., Kaiser E. T., J. Am. Chem. Soc., 91, 485 (1969).
92. Carson F. W., Kaiser E. T., J. Am. Chem. Soc., 88, 1212 (1966).
93. Petra P. H., Neurath H„ Federation Proc., 29, 666 (1970).
Карбоксипептидаза А и другие пептидазы
559
94. Kaiser Е. Т., Carson F. Ж, Biochem. Biophys. Res. Common., 18, 457 (1965).
95. Lumry R., Smith E. L„ Discuss. Faraday Soc., 20, 105 (1955).
96. Neurath H., DeMaria G., J. Biol. Chem., 186, 653 (1950).
97. Quiocho F. A., Richards F. M., Biochemistry, 5, 4062 (1966).
98. Coleman J. E., Vallee B. L., Biochemistry, 3, 1874 (1964).
99. Simpson R. T., Riordan J. F., Vallee B. L., Biochemistry, 2, 616 (1963).
100. Vallee B. L„ Riordan J. F., Bethune J. L., Coombs T. L„ Auld D. S., Sokolov-
sky M., Biochemistry, 7, 3547 (1968).
101. Alberty R. A., Massey V., Biochim. Biophys. Acta, 13, 347 (1954).
102. Auld D. S., Vallee B. L„ Biochemistry, 9, 4352 (1970).
103. Bruice T. C., Schmir G. L., J. Am. Chem. Soc., 81, 4552 (1959).
104. Riordan J. F., Vallee B. L., Biochemistry, 2, 1460 (1963).
105. Riordan J. F., Sokolovsky M„ Vallee B. L., Biochemistry, 6, 3609 (1967).
106. Bender M. L., Clement С. E., Biochem. Biophys. Res. Commun., 12, 339
(1963).
107. Simpson R. T., Vallee B. L., Biochemistry, 5, 1760 (1966).
108. Sokolovsky M„ Vallee B. L., Biochemistry, 6, 700 (1967).
109. Kagan И. M., Vallee B. L., Biochemistry, 8, 4223 (1969).
110. Quiocho F. A., unpublished results.
111. Roholt O. A., Pressman D., Proc. Natn. Acad. Sci. U. S., 58, 280 (1967).
112. Riordan J. F., Sokolovsky M., Vallee B. L., Biochemistry, 6, 358 (1967).
113. Fujuoka H„ Imahori K-, J. Biol. Chem., 237, 2804 (1962).
114. Behnke W. D., Federation Proc., 29, 462 (1970).
115. Coleman J. E., Pulido P., Vallee B. L„ Biochemistry, 5, 2019 (1966).
116. Sokolovsky M., Riordan J. F., Vallee B. L., Biochem. Biophys. Res. Commun.,
27, 20 (1967).
117. Vallee R. L., Riordan J. F., Brookhaven Symp. Biol., 21, 91 (1968).
118. Riordan J. F., Federation Proc., 29, 462 (1970).
119. Coombs T. L„ Omote Y„ Vallee B. L„ Biochemistry, 3, 653 (1964).
120. Freude K- A., Biochim. Biophys. Acta, 167, 485 (1968).
121. Vallee B. L„ Williams R. J. P., Coleman J. E., Nature, 190, 633 (1961).
122. Ando T„ Fujioka H., J. Biochem. (Japan), 52, 363 (1962).
123. Boyer P. D„ in Boyer P. D., Lardy H., Myrback K. (eds.), The Enzymes, 2nd
edn., Vol. 1, Academic Press, New York, 1959.
124. Vallee B. L., Williams R. J. P., Proc. Natn. Acad. Sci. U. S., 59, 498 (1968).
125. Walsh K- A., Sampath Kumar K. S. V., Bargetzi J. P., Neurath H., Proc. Natn.
Acad. Sci. U. S„ 48, 1443 (1962).
126. Walsh K. A.. Ericsson L. H., Bradshaw R. A., Neurath H„ Biochemistry, 9, 219
(1970).
127. Epstein 1. R., Lipscomb W. N., personal communication.
128. Vallee B. L., Riordan J. F., Coleman J. E., Proc. Nant. Acad. Sci. U. S., 49, 109
(1963).
129. Smith E. L., Proc. Natn. Acad. Sci. U. S., 35, 80 (1949).
130. Meriwether L„ Westheimer F. H., J. Am. Chem. Soc., 78, 5119 (1956).
131. Buckingham D. A., Davis С. E., Foster D. M., Sargeson A. M., J. Am. Chem.
Soc., 92, 5571 (1970).
132. Blow D. M„ Steitz T. A., A. Rev. Biochem., 39, 63 (1970).
133. Angelici R. J., Leach В. E., J. Am. Chem. Soc., 90, 2499 (1968).
134. Robinson D. R., J. Am. Chem. Soc., 92, 3138 (1970).
135. Гинодман Л. M., Мальцев H. И., Орехович В. H., Биохимия, 31, 1073 (1966).
136. Hall Р. L., Kaiser E. T., Biochem. Biophys. Res. Commun., 29, 205 (1967).
137. Sealock R. W., Laskowski M., Jr., Biochemistry, 8, 3703 (1969).
138. Kaiser B. L., Kaiser E. T„ Proc. Natn. Acad. Sci. U. S., 64, 36 (1969).
139. Ballou D., Ludwig M. L„ Palmer G., unpublished observation.
140 Folk J. E., J. Am. Chem. Soc., 78, 3541 (1956).
141. Folk J. E., Gladner I. A., J. Biol. Chem., 231, 379 (1958).
142. Wintersberger E., Cox D. J., Neurath H., Biochemistry, 6, 1069 (1962).
560
Глава 15
143. Kycia J. H., Elzinga M., Alonzo N., Hirs С. H. W„ Arch. Biochem. Biophys.,
123, 336 (1968).
144. Folk J. E., Piez K. A., Carroll W. R., Gladner J. A., J. Biol. Chem., 235, 2272
(1960).
145. Folk J. E., Wolff E. C„ Schirmer E. W„ J. Biol. Chem., 237, 3100 (1962).
146. Elzinga M., Hirs С. H. W„ Arch. Biochem. Biophys., 123, 343 (1968).
147. Elzinga M., Lai C. Y., Hirs С. H. W., Arch. Biochem. Biophys., 123, 353
(1968).
148. Elzinga M., Hirs С. H. W„ Arch. Biochem. Biophys., 123, 361 (1968).
149. Cox D. J., Wintersberger E., Neurath H., Biochemistry, 6, 1078 (1962).
150. Wolff E. C., Schirmer E. W., Folk J. E., J. Biol. Chem., 237, 3094 (1962).
151. Folk J. E., Gladner 1. A., Biochim. Biophys. Acta, 33, 570 (19591.
152. Bradshaw R. A., Neurath H., Walsh K. A., Proc. Natn. Acad. Sci. U. S., 63,
406 (1969).
153. Plummer T. H., Jr., J. Biol. Chem., 244, 5246 (1969).
154. Lipscomb W. N., Accounts Chem. Res., 3, 81 (1970).
155. Plummer T. H„ Jr., Lawson W. B., J. Biol., Chem., 241, 1648 (1966).
156. Wintersberger E„ Neurath H„ Coombs T. L., Vallee B. L., Biochemistry, 4,
1526 (1965).
157. Wintersberger E., Biochemistry, 4, 1533 (1965).
158. Reeck G. R., Winter W. P., Neurath H., Biochemistry, 9, 1398 (1970).
159. Lacko A. G„ Neurath H., Biochem. Biophys. Res. Common., 26, 272 (1967).
160. Prahl J. W., Neurath H., Biochemistry, 5, 4137 (1966).
161. Goldman P., Levy С. C., Proc. Natn. Acad. Sci. U. S., 58, 1299 (1967).
162. Levy С. C., Goldman P., J. Biol. Chem., 243, 3507 (1968).
163. Izahi K., Strominger J. L„ J. Biol. Chem., 243, 3193 (1968).
164. Zuber H., Nature, 201, 613 (1964).
165. Wells J. R„ Biochem. J., 97, 228 (1965).
166. Piras R., Vallee B. L., Biochemistry, 6, 348 (1967).
167. Freisheim J. H., Walsh K. A., Neurath H., Biochemistry, 6, 3020 (1967).
168. Brown J. R., Greenshields R. N. et al., Biochemistry, 2, 867 (1963).
169. Smith E. L., Hill R. L„ in P. D. Boyer, H. Lardy, K- Myrback (eds.), The Enzy-
mes, Vol. 4, Academic Press, New York, 1960, p. 37.
170. Smith E. L.. Spackman D. H„ J. Biol. Chem., 212, 271 (1955).
171. Bryce G. F„ Rabin B. R., Biochem. J., 90, 509 (1964).
172. Spackman D. H., Smith E. L., Brown D. M., J. Biol. Chem., 212, 255 (1955).
173. Himmelhoch S. R., Peterson E. A., Biochemistry, 7, 2085 (1968).
174. Hanson H., Glaeser D., Kirschke H., Z. Physiol. Chem., 340, 107 (1965).
175. Kretschmer K., Hanson H., Z. Physiol. Chem., 249, 831 (1968).
176. Folk J. E., Gladner J. A., Viswanatha T„ Biochim. Biophys. Acta, 36, 356
(1959).
177. Frater R., Light A., Smith E. L., J. Biol. Chem., 240, 253 (1956).
178. Beckman L„ Bjorling G., Christodoulou C., Acta. Genet., 16, 223 (1966).
179. Johnson M. J. et al., J. Biol. Chem., 116, 515 (1936).
180. Himmelhoch S. R., Arch. Biochem. Biophys., 134, 597 (1969).
181. Bryce G. F„ Rabin B. R., Biochem. J., 90, 513 (1964).
182. Hill R. L„ Smith E. L„ J. Biol. Chem., 224, 209 (1957).
183. Hartley B. S., A. Rev. Biochem., 29, 45 (1960).
184. Vallee B. L., Coleman J. E., in M. Florkin, E. H. Stotz (eds.), Comprehensive
Biochemistry, Vol. 12, Elsevier, Amsterdam, 1964, p. 194.
185. Latt S. A. et al., Biochem. Biophys. Res. Commun., 37, 333 (1969).
186. Keay L., Biochem. Biophys. Res. Commun., 36, 257 (1969).
187. Ohta Y., Ogura Y., Wada A., J. Biol. Chem., 241, 5919 (1966).
188. Matsubara H., Singer A., Sasaki R., Jukes T. A., Biochem. Biophys. Res. Com-
mun., 21, 242 (1965).
189. Matsubara H., Biochem. Biophys. Res. Commun., 24, 427 (1966).
190. Matsubara H. et al., Biochem. Biophys. Res. Commun., 34, 719 (1969).
191. McConn J. D., Tsuru D., Yasunobu K-, J. Biol. Chem., 239, 3706 (1964).
ГЛАВА 16
КАРБОАНГИДРАЗА
Дж. Э. Коулмен
Coleman J. Е., Department of Molecular Biophysics
and Biochemistry, Yale University, New Haven,
Connecticut, USA
1. ВВЕДЕНИЕ
Карбоангидраза — первый металлофермент, в котором был об-
наружен цинк. Данные о том, что в эритроцитах имеется белок,
способный катализировать гидратацию — дегидратацию СОг, впер-
вые были получены в лаборатории Рафтона [1—4]. Кейлин и
Манн [5, 6] очистили фермент из эритроцитов быка и обнаружили
в его препаратах 0,33% цинка. Молекулярная масса этого фермен-
та около 30 000 [7—9]. В расчете на это значение, содержание цин-
ка в белке колеблется от 0,92 до 1,52 г-атом/моль [5, 10, 11]. Было
показано, что агенты, связывающие в комплекс металл (особенно
анионы — цианид, сульфид, азид [4] и тиоцианат [12] и 2,3-димер-
каптопропанол-1 [13]), являются сильными ингибиторами фермен-
та. Эти данные указывали на важную роль ионов цинка для функ-
ционирования фермента из эритроцитов.
Ранние исследования растительной карбоангидразы также сви-
детельствовали о наличии цинка и об ингибировании фермента
анионами [14—18], хотя и не так убедительно, как для фермента
из эритроцитов. Данные последних лет о растительной и бактери-
альной карбоангидразах обсуждаются в разд. 2.
Манн и Кейлин [19] обнаружили сильное ингибирование фер-
мента из эритроцитов сульфамидами. Это свойство — особенность
карбоангидразы, так как больше ни один из известных ферментов
не ингибируется этими соединениями так же специфично. Посколь-
ку эти ингибиторы сыграли очень важную роль в физико-химиче-
ских и энзимологических исследованиях фермента, разд. 4 посвящен
детальному обсуждению свойств сульфамидных комплексов кар-
боангидразы. Хотя по своей природе сульфамиды не являются
агентами, связывающими металлы в комплекс, последние данные
свидетельствуют о том, что в молекуле фермента центр связыва-
ния сульфамидов содержит ион цинка в качестве одной из коорди-
нирующих групп. Металл взаимодействует с группой —SO2NH2
[20—23].
36—2451
562
Глава 16
О карбоангидразе часто упоминали как о самом специфичном
из всех известных ферментов, так как считалось, что единственной
ее функцией является катализ обратимой гидратации СО2:
а) Н2О СО2 *. - > HgCOg < — Н"*" НСОз
Ъ (1)
б) ОН" + СО2 НСОз
*-2
Реакция может быть написана с водой или с гидроксилом в ка-
честве активных частиц. Как это происходит с ферментом, окон-
чательно пока не известно, однако имеется вполне убедительные
данные в пользу того, что атаку на СО2 осуществляет гидроксил,
координированный у цинка (разд. 7).
В последние годы было показано, что, кроме реакций (1), кар-
боангидраза катализирует некоторые процессы гидролиза и гидра-
тации, затрагивающие карбонильную группу (гидролиз п-нитро-
фенилацетата [24—27] и сультона 2-окси-5-нитро-а-толуолсульфо-
кислоты [28—30] и гидратацию ацетальдегида [31—33]). Эти
реакции детально обсуждаются в разд. 7.
2. ИЗОФЕРМЕНТЫ И ВИДОВЫЕ ВАРИАНТЫ —
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
В последние 10 лет в нескольких лабораториях были разрабо-
таны методы получения высокоочищенной карбоангидразы из эрит-
роцитов [34—42]. Они заключаются в осаждении гемоглобина
смесью хлороформ—этанол [37, 39, 42] с последующей хромато-
графией на колонке, заполненной ДЭАЭ-целлюлозой [34, 37, 42],
ДЭАЭ-сефадексом [43] или гидроксилапатитом [37, 39, 42]. Белок
хорошо переносит лиофилизацию, и в большинстве методов она
используется на той или иной стадии выделения. Накопленный
опыт препаративной работы выявил некоторые особенности мето-
дов очистки фермента. Обработка смесью хлороформ—этанол су-
щественно не меняет большинство физико-химических свойств. Но,
как показал рентгеноструктурный анализ, воздействие этой смеси
на фермент С человека снижает качество кристаллов [22, 23]. Для
удаления гемоглобина лучше использовать гель-фильтрацию [37]
или хроматографию на ДЭАЭ-сефадексе [43]. Качество кристал-
лов повышается, если проводится электрофоретическая очистка
фермента [23] и если не применяется лиофилизация [22, 23].
С помощью этих методов удалось выделить в чистом виде
много различных типов карбоангидразы из эритроцитов животных.
Некоторые из них получены в кристаллическом виде [39, 44, 45]
(рис. 16.1). Для фермента В человека кристаллы легче получают-
Карбоангидраза
563
Рис. 16.1. Кристаллы карбоангидразы из эритроцитов приматов и ее сульфамид-
ных комплексов.
а — карбоангидраза С обезьяны; б — комплекс карбоангидразы В обезьяны с азосульфами-
дом; в — карбоангидраза В обезьяны; г— комплекс карбоангидразы В человека с ацетазол-
амидом.
36'
Таблица 16.1
Физико-химические свойства различных типов карбоангидраз (КА) эритроцитов
Тил Молекулярная масса ’ао, а> Электрофоретическая подвиж- ность , 105 см2. в-1 • с-1 Молярное поглощение, 10-4 И 1-см-1 Парциальный удельный объем, мл-г-1 Изоэлектри- ческая точка Литература
КАВ человека 29 600 2,95-3,1 —1,72(рН 8,6) 4,90 0,737 5,85±0,2 35, 37, 38
28 000а —1,06(рН 7) 0,736а 36
КАС человека 31600 2,95-3,1 —1,01(рН 8,6) 5,34 0,723 7,25 35, 37, 38
—0,5 (pH 7) 0,736а 36
КАВ обезьяны 29 800 2,94 —2,28 (pH 8,6) 4,88 39
28 300а
КАС обезьяны 29 800 2,94 —1,29 (pH 8,6) 5,35 39
28 600а
КАЛ быка 31000 3,06 -2,8 (pH 7) 5,7 34
КАВ быка 31000 3,06 —2,3 (pH 7) 5,7 0,742 5,65 34
КАВ лошади 29 000 2,74 3,95 0,725а ~6 41
КАС лошади 28 000 2,71 3,73 0,732а ~11
КА собаки 29 000 2,82 0,734а 40
КА акул 42
шестижаберной 40 000 3,5
тигровой 40 000 4,65
а Рассчитано по аминокислотному составу.
Карбоангидраза
565
ся в форме сульфамидных комплексов [22, 30]. Все эти типы кар-
боангидразы являются мономерными формами с молекулярной
массой около 30 000, содержащими в молекуле 1 г-атом Zn(II)
[34—41]. Некоторые физико-химические свойства этих ферментов
приведены в табл. 16.1.
2.1. Аминокислотный состав
Аминокислотный состав карбоангидраз эритроцитов представ-
лен в табл. 16.2. Характерно наличие только одного остатка цис-
теина у большинства изоферментов и отсутствие этой аминокис-
лоты в ферменте быка. Имеется один или два остатка метионина
(см. ниже о расщеплении бромцианом) и небольшое число остат-
ков тирозина и триптофана. С эволюционной точки зрения самой
старой является карбоангидраза из эритроцитов акул. Фермент
шестижаберной акулы имеет, вероятно, самую низкую для своей
группы изоэлектрическую точку, равную 4,7, что согласуется с от-
носительно высоким содержанием аспарагиновой и глутаминовой
кислот (табл. 16.2). Единственный остаток цистеина карбоангидра-
зы С человека, по-видимому, не участвует непосредственно в ка-
тализе. По данным рентгеноструктурного анализа, он расположен
на расстоянии примерно 14 А от иона цинка [22, 23].
При выделении карбоангидразы из эритроцитов млекопитаю-
щих стало очевидным, что у каждого биологического вида можно
обнаружить несколько электрофоретически различных белков
с карбоангидразной активностью [34—40, 46]. У быка это фермен-
ты А и В [34], у человека — карбоангидразы В и С [35—38].
Наиболее существенные отличия физико-химических свойств таких
изоферментов определяются различием их аминокислотного соста-
ва (табл. 16.2) и последовательности аминокислот. А это в свою
очередь проявляется в функциональных и структурных особенно-
стях (см. ниже). Например, для изоферментов человека наблюда-
ется значительная разница в уровне активности. При гидратации
СО2 карбоангидраза В проявляет удельную активность около
40 000 ед./мг белка, а тип С — примерно 1 200 000 ед./мг белка
[47] (подробнее см. разд. 7). С физиологической точки зрения ин-
тересно, что содержание этих изоферментов в организме неодина-
ково— количество высокоактивной карбоангидразы С в 5—10 раз
меньше, чем фермента типа В. Результатом этого является при-
мерно равное распределение суммарной карбоангидразной актив-
ности между двумя изоферментами.
Наличие в эритроцитах двух изоферментов карбоангидразы с
различной первичной структурой является, по-видимому, отличи-
тельной особенностью приматов. Различные белковые фракции с
карбоангидразной активностью, выделяемые из гемолизатов низ-
ших видов млекопитающих, имеют одинаковый аминокислотный
Таблица 16.2
Аминокислотный состав карбоангидраз из эритроцитов человека и животных
Аминокислота Человек (37, 77] Обезьяна 139] Собака 140] Бык [48] Лошадь [41] Акулыа [42]
В С В с В С тигро- вая шести- жабер- иая
Лизин 18 25 18 24 22 19 19 19 22 21
Гистидин 11 12 9 12 12 И 12 10 11 7
Аргииин 7 7 7 8 7 9 9 5 10 8
Аспарагиновая кислота 31 29 31 30 34 32 27 32 30 27
Треонин 14 13 13 11 11 15 12 12 10 9
Серин 30 19 30 18 25 16 18 28 23 24
Глутаминовая кислота 22 24 22 26 26 24 26 25 30 33
Пролин 17 28 17 16 19 20 16 18 16 20
Глицин 16 22 15 22 24 20 23 23 24 24
Аланин 19 13 16 12 18 17 17 15 17 16
Полуцистин 1 1 1 1 1 0 1 2 9 21
Валин 17 17 16 14 15 20 19 20 10 8
Метионин 2 1 1 2 3 1 2 2 1
Изолейцин 9 9 10 10 13 5 7 9 12 10
Лейцин 20 27 19 24 25 26 22 21 26 27
Тирозин 8 9 9 7 8 8 7 8 7 7
Фенилаланин 11 13 10 11 12 11 11 11 13 10
Триптофан 6 7 7 7 6 7 5 5 5 4
Zn 1 1 1 1 1 1 1 1 а а
Амидный NHa (26) (21) — — — (24) (32) (22) — —
Общее число ос- татков амино- кислот 259 266 256 255 278 264 252 265
® Для удобства сравнения аминокислотный состав карбоангидраз акул в этой таблице
дается в расчете на молекулярную массу 30 000, хотя истинная ее величина — 40 000, а содер-
жание цинка —1 г-атом/39 000 [42].
Карбоангидраза
567
состав и удельную активность [34, 40, 48]. (Единственное исключе-
ние— фермент лошади [41]). Эти «изоферменты» представляют
собой слабо различающиеся конформации одного и того же белка.
Отличия в заряде у таких макромолекул могут быть следствием
различного распределения водородных связей между остатками
аминокислот. Индивидуальные изоферменты приматов могут на-
блюдаться в виде отдельных полос при электрофорезе в геле [39,
49].
Среди приматов детально изучена также карбоангидраза эрит-
роцитов макак-резус, которая состоит из двух изоферментов — В и
С, почти идентичных изоферментам человека (табл. 16.1 и 16.2).
Подобные результаты получены и при исследовании гемолизатов
некоторых других приматов [46]. Известны также мутанты [46,
49].
2.2. Карбоангидразы растений
Вследствие простоты выделения и большого физиологического
значения карбоангидразы эритроцитов животных привлекали вни-
мание исследователей. Однако после появления первых сообщений
о том, что растительные карбоангидразы не содержат цинка [51]
и не ингибируются сульфамидами [51], возрос интерес и к этим
ферментам. Опубликованы результаты исследований высокоочи-
щенных растительных карбоангидраз.
Таблица 16.3
Физико-химические свойства растительных карбоангидраз
Источник S20, W Молекулярная масса Содержание цинка Литература
Шпинат 6,8 140 000 145 000 (равновес- мое центрифуги- рование) 0,0013 мкг/мг 52
Петрушка 8,8 170 000 180 000 (равновес- ное центрифуги- рование) 1 г-атом/29 000 г белка 53, 54
Neisseria sica 28 000 1 г-атом/30 000 г белка 56
Физико-химические свойства ферментов из листьев шпината
[52] и петрушки [53, 54] суммированы в табл. 16.3. Их аминокис-
лотный состав приведен в табл. 16.4 Наиболее заметная особен-
ность этих ферментов — большая кажущаяся молекулярная мас-
са-— около 140 000 для фермента из шпината (определена несколь-
568
Глава 16
Таблица 16.4
Аминокислотный состав карбоангидраз из листьев петрушки и шпината
[52, 53]
Аминокислота Шпинат, среднее содержание, мол. % Петрушка, моли/28 150 г белка
Аспарагиновая кислота 8,85 24
Треонин — 10
Сернн 11,85® 18
Глутаминовая кислота 9,50 21
Пролин 4,70 22
Глицин 11,60 22
Аланин 8,25 17
В алии 8,10б 24
Полуцистин 4,0 7
Изолейцин 5,80 8
Лейцин 5,90 21
Тирозин 2,20 8
Фенилаланин 4,20 17
Лизин 5.90 21
Г петидин 2,90 5
Аргинин 4,05 5
Триптофан <1 3
Метионин 4
® Значение, полученное экстраполяцией к нулевому времени гидролиза.
® Значение, полученное после 72 ч гидролиза.
кими методами) [52] и около 170 000 для фермента из петрушки
[53] (табл. 16.3). Первоначально полученные препараты карбоан-
гидразы из шпината содержали только 1,3 мкг Zn на 1 г белка
[52], а из петрушки— 1 г-атом Zn на 51 000 г белка [53]. В даль-
нейшем были получены более активные образцы фермента, содер-
жавшие 1 г-атом Zn на 29 000 г белка [53]. Согласно последним со-
общениям, в растительной карбоангидразе имеется металлсодер-
жащая субъединица, во многом похожая на фермент животных.
Однако функциональная роль цинка в растительных карбоангидра-
зах пока еще не исследована. Можно только сказать, что между
растительными и животными ферментами имеются функциональ-
ные различия. Карбоангидраза из растений не проявляет эстераз-
ной активности [53], хотя этот фермент ингибируется ацетазол-
Глава 16
569
амидом и азидом [53]. Интересно, что в некоторых случаях при
определении молекулярной массы карбоангидразы акулы также
получены значения больше 100 000 [42]. Необходимо отметить, что
это единственная карбоангидраза животного происхождения, кото-
рая содержит значительное количество цистеина (табл. 16.2).
2.3. Прочие карбоангидразы
Проведена частичная очистка карбоангидразы на куколок ба-
бочки Heleothis zea [55]. При гель-фильтрации на сефадексе G-100
получены активные фракции, отвечающие молекулярной массе
80 000—90 000. Однако эти препараты могли содержать комплексы
фермента с посторонним клеточным материалом [55]. По некото-
рым свойствам (ингибирование азидом и ацетазоламидом, K.i=
= 0,03 мкМ) этот фермент имеет сходство с карбоангидразой эрит-
роцитов. Наймен и сотр. [56] получили гомогенный препарат бак-
териальной (из Neisseria sica) карбоангидразы с молекулярной
массой 28 000. Фермент содержит на каждую субъедини-
цу молекулярной массы ~ 30 000 один атом цинка. Количество
триптофана и тирозина значительно меньше, чем у ферментов эри-
троцитов млекопитающих, но в остальном их свойства одинаковы
[56].
2.4. Последовательность аминокислот
Ведутся активные исследования первичной структуры изофер-
ментов карбоангидразы быка и карбоангидраз В и С человека [49,
57—61]. Опубликованы предварительные результаты этой работы
[49, 60, 61], однако к настоящему времени определены последова-
тельности аминокислот только для некоторых фрагментов поли-
пептидной цепи. Они представлены в конце данного раздела.
Относительно ограниченные исследования N- и С-концевых
фрагментов макромолекулы тем не менее привели к довольно ин-
тересным результатам. Риккли [37] и Маррик [62] и их сотруд-
ники показали, что все формы карбоангидразы человека не содер-
жат свободной аминогруппы на N-конце полипептидной цепи. Ни
при каких условиях с этими изоферментами не удалось получить
положительной реакции с динитрофторбензолом [49]. Лоран с
сотр. [63], анализируя обе формы (В и С) по методу Филлипса
[64], обнаружил только по одной N-ацетильной группе на 1 моль
каждого изофермента.
Лоран и Дерриан [49] после обработки белка гидразином в
течение 17 ч при 100 °C смогли выделить примерно 1 моль
ДНФ-ацетилгидразида. Эти результаты свидетельствуют о том,
что у большинства (если не у всех) форм карбоангидразы N-koh-
цевая аминокислота ацетилирована. В дальнейшем тем же авто-
570
Глава 16
Таблица 16.5
N-Концевые аминокислоты, изолированные из карбоангидраз человека
и быка [49]
1 2 3 4 5 6
КАА человека N-aiieiwi-Ala-Ser-Pro-Asp-Trp-Gly-Tyr-Asp-Asp-Lys
... Lys- Ser-Trp-Glu-Pro-GIn-Gly-Asn
КАВ человека N-auemn-Ala-Ser-Pro-Asp-Trp-Gly ...
КАС человека Ы-ацетил-5ег . ..
КАВ быка Ы-ацетил-5ег ...
рам удалось изолировать N-ацетилированные пептиды [49]
(табл. 16.5). На N-концах карбоангидразы С (КАС) человека и В
(КАВ) быка оказалось возможным идентифицировать только одну
аминокислоту (N-ацетилсерин), так как от этих форм отщепление
большего пептидного фрагмента осуществляется труднее, чем от
изофермента В [49]. Карбоангидраза А — электрофоретически от-
деляемый изофермент эритроцитов человека, по активности и ами-
нокислотному составу идентичный форме В [35, 38]. Это сходство
дополняется одинаковой последовательностью N-концевых амино-
кислот (табл. 16.5).
Определение С-концевой структуры молекулы основано на спо-
собности бромциана отщеплять от С-конца изоферментов карбоан-
гидразы быка и человека пептиды, содержащие 19 или 20 остатков
аминокислот [57—59]. Строение этих пептидов показано в
табл. 16.6. При нумерации остатков предполагается, что 21-й ами-
нокислотой во всех случаях является метионин. Это позволяет во
всех трех последовательностях выделить наибольшее число гомо-
логичных участков (отмеченных рамками в табл. 16.6). Карбоан-
гидраза В человека при этом оказывается короче на одну С-конце-
вую аминокислоту (лизин). Наблюдается также большое структур-
ное сходство карбоангидразы С человека с изоферментами низших
видов млекопитающих. Последнее согласуется с результатами фи-
зико-химических и кинетических исследований (разд. 2.3).
При третьем способе определения первичной структуры анали-
зировался пептидный фрагмент, содержащий остаток гистидина,
способный реагировать с иодацетатом в активном центре карбо-
ангидразы В человека (подробнее об этой реакции см. в разд. 7).
После взаимодействия фермента с иодацетатом получается единст-
венный карбоксиметилированный остаток гистидина, входящий в
пентапептид ТЬг-3-карбоксиметил-Н1з-Рго-Рго-Ьеи, выделенный
Брэдбери [65, 66]. Андерссон и сотр. [58] определили, что амино-
кислоты этого фрагмента занимают места 59—64, считая от
С-конца.
С другой стороны, показано, что остаток гистидина реагирует
с N-хлорацетилхлортиазидом, когда последний замещает сульф-
Карбоангидраза
571
Таблица 16.6
C-Концевые аминокислотные последовательности карбоангидраз человека
и быка
21 20 19 18 17 16 15
Met-Glx-His|Asn-Asn^Arg-Pro
I l *
i । ;
Met-Val -Asp Asn-TrpJArg-Pro
Met-Leu-Al a I Asn Trp-Arg-Pro^-
МВ человека
КАС человека
КАВ быка
14 13 12 11 10 9 8___7 6 5 4 3 2 1
?-Thr jGln-Pro-Leu-LysjGlyjArg-'Thr Vai -Arg-Ala-Ser-Phe-
i I I । I
।-Ala-Gin-Pro-Leu-LySj-Asn-Arg ^Gln-Ile-Lys-Ala-Ser-Phe-Lys
i i III
AlajGln-Pro-Leu-Lys*AsnArgjGin-Vai-Arg-Gly-Phe Pro-Lys
амид, связанный с активным центром [60, 67]. При кислотном гид-
ролизе хлортиазид отщепляется и образуется карбоксиметилгисти-
дин. Этот гистидин отличается от упоминавшегося выше остатка
модифицируемого иод- или бромацетатом [60, 68]. Гистидин, взаи-
модействующий с сульфамидами, в первичной структуре карбоан-
гидразы В человека занимает место в области от 65-й до 76-й ами-
нокислоты (считая от N-конца).
Хотя полностью первичная структура не определена ни для од-
ной формы карбоангидразы, продолжаются интенсивные исследо-
вания фрагментов, получаемых при гидролизе трипсином [60, 61].
Результатом всех этих работ является картина частичной последо-
вательности аминокислот двух изоферментов человека, представ-
ленная в табл. 16.7.
2.5. Кислотно-основные свойства
Методами потенциометрического и спектрофотометрического
титрования обеих форм карбоангидразы человека [69, 70] и изо-
фермента В быка [71] были получены детальные кривые титрова-
ния в области ионизации гидроксильной группы тирозина. При кис-
лотном титровании карбоангидразы В человека наблюдается ска-
чок при pH 4,0—4,2, соответствующий поглощению 7 протонов на
1 моль [69]. Эти семь групп, по-видимому, являются скрытыми в
глубине молекулы остатками гистидина. Четыре остальных гисти-
дина в нативном белке титруются нормально. При обратном тит-
ровании щелочью от pH <2 число групп, титруемых при pH 5—8,
возрастает до 11. По-видимому, скрытые в глубине нативной струк-
туры остатки гистидина освобождаются при кислотной денатура-
ции [69].
При спектрофотометрическом титровании карбоангидразы В
человека обнаружены три типа тирозильных групп [69]. При рН<
<11,5 теплоты и энтропии ионизации первых четырех остатков
тирозина имеют нормальные значения, однако одна или две из
этих групп характеризуются повышенной величиной р/Са [69].
572
Глава 16
Ионизация оставшихся трех или четырех тирозинов протекает
очень медленно и в более щелочной области. При этом наблюдает-
ся одновременное медленное снижение ферментативной активнос-
ти [69].
Главное отличие кислотно-основных свойств изофермента С от
описанной выше формы В заключается в положении изоэлектриче-
ской точки (при pH 7,3 и 5,8 соответственно) [70]. Оба изофермен-
та имеют похожие кривые титрования (скачок при pH 4; 7 скры-
тых остатков гистидина [70]). Три тирозильных остатка титруются
нормально, остальные шесть находятся в глубине молекулы. Мед-
ленная ионизация последних сопровождается необратимыми изме-
нениями конформации. Форма С, по-видимому, более чувствитель-
на к щелочной денатурации, чем форма В [70].
Карбоангидраза В быка имеет изоэлектрическую точку при
pH 5,65 [71]. При титровании этого фермента кислотой также об-
наруживаются 7 остатков гистидина, протонирование которых про-
исходит в узком интервале около pH 4 и сопровождается значи-
тельной перестройкой конформации. При обратном титровании все
11 гистидинов титруются нормально [71]. Только один тирозин
титруется свободно с повышенным значением рКа (10,8). Осталь-
ные семь медленно ионизируются только при рН>12 — и тоже с
потерей ферментативной активности [71]. Таким образом, во всех
исследованных до сих пор препаратах карбоангидразы из эритро-
цитов млекопитающих обнаружены остатки гистидина и тирозина,
заключенные в глубине молекулы. Тем самым подтверждается
предположение о существовании гистидил-тирозильных взаимодей-
ствий (возможно, водородных связей), являющихся характерной
структурной особенностью карбоангидразы [69—71]. Во всяком
случае, наличие внутри глобулы фенольных гидроксилов и имида-
зольных групп представляется существенным для каталитических
свойств этого фермента.
2.6. Спектры поглощения и оптическая активность
Спектры поглощения в ближней ультрафиолетовой области у
всех разновидностей карбоангидразы эритроцитов имеют вид, ти-
пичный для белков. При 280 нм наблюдается максимум и четко
выраженное плечо при 290 нм. Величины молярного поглощения
при 280 нм для некоторых изоферментов приведены в табл. 16.1.
Риккли и сотр. [37] исследовали абсорбционные спектры кар-
боангидразы человека в области 185—230 нм. Полученные резуль-
таты свидетельствуют о том, что структура макромолекулы, по-
видимому, не содержит (или содержит очень мало) а-спиральных
участков, так как наличие последних обычно проявляется в виде
значительной гипохромии, не характерной для изученных образцов
Таблица 16.7
Предварительная схема последовательности аминокислот карбоангидразы В и С
человека [58, 59, 63, 154] (подчеркнуты участки наибольшей гемологичности)
। Фрагмент В IV _______
Фермент В'- Лцетил-Ala-Ser-I’ro Asp-Trp Gly-Tyr-Asp-Asp-Iys-Asn Gly-Glu Pro-GIu-Trp-Ser-I.ys-
Фермент Ct Acetyl-Ser-Uis-His(Gly ,Trp,Tyr)Gly-l.ys (Gly,Pro,Glx,Asx,Trp,His,His ,Lys)Asp-Phe-Pro-1le Ala l.ys(Gln Gly-Arg)
I Фрагмент С 1____________ _____________________I
-Leu-Tyr-Pro- He-Ala-Asp* Gly-Asn-Asn-Gin - Ser-Pro-Vai-Asp-1le-Lys-Thr-Ser-Clu-Thr-Lys-lUs-Asp-Thr-Ser-Leu-Lys-Pro-1le-Ser-Vai-Ser-
(Leu,Tyr,Pro,lle,Ala,Asx,Gly,Asx,Asx,Glx,Ser,Pro,Val ,Asx lie,Lys)Thr-Ser-Glu-Thr-l.ys (His-Asp*Thr-Ser*Leu) (l.ys,Pro,He,Ser,Val,Ser,
| Фрагмент C 2
______________________Фрагмент Bill
Тут*Asn-Pro-Ala-Thr-Ala-Lys-GIu-1le-1le-Asn-Vai Gly His-Ser-Phe-aUsj-Vai-Asn-Phe-Glu-Asp-Asn-Asn-Asp-Arg--Ser-Vai Leu-Lys-Gly-Gly-
Tyr;Asx,Pro,Ala,Thr)Ala-Lys( неизолированные пептиды )(Ser,Vai ,Glx,Phe,Glx,Asx,Asx,Asx,Asx,Lys)(Ala,Val ,Leu,Lys) (неизо-
।Фрогмен/п В I
• Pro-Phe*Ser-Asp-Ser-Tyr-Arg-Leu-Phe( 79 остатков )!le-Lys-Thr-Lys-Gly-Lys-Arg-Ala-Pro-Phe-Thr-Asn-Phe-Asp-Pro-Ser-Thr-Leu-Leu-
Лированные пептиды )Arg( 87 остатков )( неизолированные пептиды )(Clx,Ser,Phe-Gly)Ile-Val-Leu*Lys-
Фрагмент С 3| ___Фрагменте 4 ________
• Pro*Ser-Ser-Leu-Asp-Phe-Trp-Thr-Tyr-Pro--Gly-Ser-!.eu-Thr OliJ Pro-Pro-Leu-Tyr-Glu-Ser-Val-Thr-Trp-- Не-He-Cys-Lys-
(Pro,Glx,Ser,Leu,Asx,Leu,Leu, Tyr,Trp)(Gly,Thr,Leu,Thr,Gcu,Pro,Pro,I.cu,Tyr,Glx,Ser ,Val ,Thr,Trp) (He,Ile,Cys,1-ys)
___________________।___________Фрагмент В V
- Glu-Ser- Ile-Ser-Val-Ser-Gln-Ser-Clu-Leu-Ala*Gin-Phe-Arg ....Ser-Leu-Leu-Ser-Asn-Val-Glu-Asp-Asn-Gly--Ala-Val-Pro-Met-Glx--
(Glx-Pro-He-lie (Vai ,Ser ,Glx,Ser ,Glx)Leu-Val-Lys JPhc-Arg (Lys .Lcu.Asn.Phe ,Glx ,Cly) (Glx.Clx ,Clx ,Gly,Pro,Glx) (Asp ,Leu,Trp).Met-Val *
________________________________________________________।________________Фрагмент C S
His-Asn-Asn-Arg-Pro-Thr-Gln-Pro-Leu-Lys-Gly-Arg-Thr-Val-Arg-Ala-Ser-PheO(l
«Asp-Asn-Trp-Arg-Pro-Ala-Gln-Pro-Leu-Lys-Asn-Arg-Cln- Ile-Lys-Ala-Scr-Phe -l.ysOH
модифицируемый йром-или иодацетатом остаток фермента В,
модифицируемый N-хлорацетилхлортиазидом остаток фермента В.
574
Глава 16
фермента [37]. Интересно, что при подкислении растворов белка
до pH 1,67 эта гипохромия возрастает. Независимыми измерениями
дисперсии оптического вращения (ДОВ) и кругового дихроизма
(КД) при низких значениях pH выявлены резкие изменения кон-
формации глобулы, сопровождаемые потерей цинка [72—74]. Как
следует из результатов определения ДОВ и КД. при pH 1,5—1,6
белок содержит довольно много а-спиральных участков, отсутству-
ющих в нативной структуре при нейтральных pH [72—74]. Однако
спектры ДОВ и КД нативного фермента сложны для интерпрета-
ции (см. ниже).
2.6.1. Оптическое вращение
Рис. 16.2 иллюстрирует необычные особенности оптического
вращения карбоангидразы [30]. Хорошо заметны сложные эффек-
ты Коттона, связанные с полосами поглощения в ближней ультра-
фиолетовой области. В расчете на 1 моль эти эффекты по меньшей
мере на порядок превосходят величины эффектов Коттона, наблю-
давшиеся для большинства модельных соединений ароматических
хромофоров в асимметричном окружении. Большая эллиптичность
этих хромофоров при их введении в структуру белка должна инду-
цироваться окружающими группами.
Ароматические эффекты Коттона исчезают при кислотной дена-
турации и в растворах гидрохлорида гуанидина [72, 76, 77]. По
данным работы [73], после удаления денатурированного агента
(или после возвращения к нейтральному pH) наблюдавшиеся ра-
нее хорошо выраженные полосы эллиптичности в ближней ультра-
фиолетовой области полностью уже не восстанавливаются. Однако
в одном из последующих сообщений говорится о возможности вос-
становления исходной конформации при правильном выборе усло-
вий и при использовании гидрохлорида гуанидина [77]. Причины
возникновения этих полос эллиптичности подробно обсуждаются в
работах [72—76].
Анализ данных ДОВ для определения конформации полппеп-
тидной цепи карбоангидразы обычно проводится по классическому
методу (построение графиков Янга—Доти, определение Ьо) [37].
Карбоангидраза В человека имеет значение 6о~О, что говорит о
низком содержании или отсутствии а-спиралей в нативном белке
[37]. При кислотной денатурации Ьо становится более отрицатель-
ным (—97°). Следовательно, в денатурированном белке увеличи-
вается количество спиральных участков [37] (см. выше обсужде-
ние спектров поглощения). Однако слишком большие эффекты
Коттона в ближней ультрафиолетовой области и необычное поло-
жение эффектов Коттона в дальней ультрафиолетовой области вы-
зывают сомнение в применимости классических методов обработки
данных дов.
Карбоангидраза
575
Спектры КД и ДОВ в дальней ультрафиолетовой области так-
же имеют характер, необычный для белков. Значительную полосу
эллиптичности, наблюдаемую для обоих рассмотренных изофер-
ментов при 222 нм (рис. 16.2), можно с некоторой вероятностью
Рис. 16.2. Спектры КД (вверху) и ДОВ (внизу) карбоангидразы В человека
(---------) и карбоангидразы С обезьяны (------------) (0,025 М трис, pH 7,5,
25 °C) [30].
соотнести с а-спиралью. Большие отрицательные полосы находят-
ся около 211 нм (форма В человека) и 217 нм (форма С обезь-
яны). В спектре ДОВ это проявляется в необычном положении
первой отрицательной впадины — при 222 и 225 нм соответственно.
Поскольку для карбоангидразы характерны оптически активные
ароматические полосы поглощения в ближней ультрафиолетовой
области, необходимо учитывать возможность того, что некоторые
ароматические полосы поглощения тирозина или триптофана в
дальней ультрафиолетовой области также могут проявлять опти-
ческую активность и вносить свой вклад в спектры КД и ДОВ в
коротковолновой области. Этот вклад существенно осложняет ана-
лиз конформации по спектральным данным. Неблагоприятным об-
стоятельством является также необычно высокая оптическая плот-
ность карбоангидразы млекопитающих (около 200 нм), затрудняю-
щая определение спектров КД в этой области.
576
Глава 16
Исключительную сложность интерпретации спектров КД и
ДОВ в ультрафиолетовой области можно проиллюстрировать сле-
дующим примером. При рентгеноструктурном анализе с разреше-
нием 2 А оказалось, что молекула карбоангидразы С человека со-
держит только несколько спиральных витков, причем лишь неболь-
шая часть этих витков соответствует параметрам истинной
Рис. 16.3. Спектры КД карбоангидразы тигровой акулы в ближней (а) и дальней
(б) ультрафиолетовой области (0,025 М трис, pH 7,5, 25 °C) [42J.
а-спирали [23]. Преобладающую роль в структуре молекулы иг-
рает p-конфигурация, которая образует центральную часть глобу-
лы (разд. 6). Похоже, что большая отрицательная полоса эллип-
тичности в спектрах КД (около 211—217 нм) определяется именно
P-структурой. Последнее согласуется и с исследованиями модель-
ных систем, демонстрирующих аналогичные полосы при 215—
218 нм [79]. Сильное отрицательное вращение при Х<200 нм, на-
блюдаемое для спектров ДОВ, пока не нашло объяснения.
На рис. 16.3 показаны спектры КД в дальней ультрафиолето-
вой области, полученные для образцов чистой карбоангидразы из
эритроцитов тигровой акулы [42]. Они интересны в двух отноше-
Карбоангидраза
577
ниях. Во-первых, большая эллиптичность в ближней ультрафиоле-
товой области фермента эритроцитов является структурной особен-
ностью, характерной на ранней стадии эволюции. Во-вторых,
спектры КД в дальней ультрафиолетовой области имеют вид, бо-
лее типичный для белка. Малая оптическая плотность около
200 нм позволяет работать с ферментом вплоть до 190 нм. Значи-
тельная полоса при 222 нм свидетельствует о наличии в нативном
белке а-спиральных участков. Однако наблюдается сдвиг другой
а-спиральной полосы (обычно расположенной около 190 нм) к
196 нм, а глубина впадины при 222 нм почти равна высоте пика
при 195 нм. Обе последние особенности, по-видимому, объясняются
наличием больших областей с ^-конфигурацией [79, 80].
3. МЕТАЛЛОКАРБОАНГИДРАЗЫ: АПОФЕРМЕНТЫ,
СВЯЗЫВАНИЕ МЕТАЛЛА, СПЕКТРЫ ПОГЛОЩЕНИЯ
И СВЯЗЫВАНИЕ ИНГИБИТОРОВ
При нейтральных значениях pH не наблюдается обмена иона-
ми цинка между металлокарбоангидразой и окружающим раство-
ром [73, 81, 82]. Однако при подкислении до pH<5 этот обмен
происходит, хотя и в небольшой степени [82]. По-видимому, он со-
провождается изменениями геометрии координационного центра,
как следует из спектра поглощения в видимой области Со(II)-кар-
боангидразы (см. ниже).
3.1. Апофермент
Обработка фермента 1,10-фенантролином или 8-оксихинолин-
сульфонатом [20, 83, 84] в течение 15 дней при pH 5,5 привела к
удалению цинка (II). Полученный апофермент совершенно неакти-
вен, хотя и довольно стабилен, так как после прибавления цинка
он полностью восстанавливает свою активность [20, 73, 83]. Уда-
ление металла приводит к небольшому изменению спектров ДОВ,
но основные физико-химические свойства апо- и металлофермента
одинаковы. Существенны в этом отношении данные о том, что
единственной особенностью разностных карт электронной плотно-
сти между кристаллами апо- и Zn-карбоангидразы является мак-
симум плотности, отвечающий ионам цинка (рис. 16.4) [85]. Кри-
сталлы металле- и апофермента изоморфны. Последние могут быть
получены при длительной (16 суток) обработке кристаллов натив-
ной карбоангидразы 0,01 М раствором 2,3-димеркаптопропанола-1,
содержащим 2,45 М (NH^SC^, в атмосфере водорода [85].
Кроме Zn(II)-карбоангидразы, приготовлены металлоферменты,
содержащие Mn(II), Co(II), Ni(II), Cu(II), Cd(II) и Hg(II).
37—2451
578
Глава 16
Имеется много данных о том, что все эти ионы занимают в фер-
менте одно и то же место [20, 23, 84, 86]. Ниже излагаются ре-
зультаты исследований этих препаратов.
Рис. 16.4. Разностная карта электронной плотности. Карбоангидраза С—апокар-
боангидраза С человека [45].
3.2. Ферментативная активность и устойчивость
комплексов металл — белок
Несмотря на то что все указанные выше ионы входят в актив-
ный центр фермента, только Zn(II)- и Со(II)-карбоангидразы об-
ладают свойствами, необходимыми как для катализа гидратации
СОг, так и для проявления эстеразной активности (табл. 16.8)
[20, 21]. Отсутствие каталитических свойств у остальных металло-
ферментов может объясняться небольшими изменениями геомет-
рии координационной сферы или неспособностью к присоединению
или быстрому замещению лигандов [21]. Связывание сульфамидов
также зависит от природы металла [20, 21], и неактивные метал-
локарбоангидразы не взаимодействуют с этими ингибиторами
(табл. 16.8; подробнее см. разд. 4 и 7).
Линдског и Наймен [84] определили константы устойчивости
(К) различных комплексов фермента с металлами, используя ме-
тод конкурентного комплексообразования с ЭДТА, 1,10-фенантро-
лином и 8-оксихинолин-5-сульфонатом. В табл. 16.9 приведены
значения К, измеренные при pH 5,5, где ионы металлов удаляются
Карбоангидраза
579
Таблица 16.8
Ферментативная активность и связывание ацетазоламида
металлокарбоаигидразами человека [21]
Металл3 Гидратация COj , ед. Гидролиз п -нитрофен илацетата® Число молей ацетазоламида, связанного с 1 молем фермента г
Апофермент 400 0,09 0,04
Мп(П) В 400 0,38 0,40
Со(П) В 5 700 8,70 1,00
Ni(II) В 500 0,32 0,02
Cu(II) В 127 0,50 0,14
Zn(II) В 10 200 2,70 1,00
Zn(II) С 30 000 7,30 1,00
Cd(II) В 430 0,20 0,06
Hg(II) В 5 0,09 0,05
а В и С обозначают соответствующие изоферменты карбоангидразы эритроцитов чело-
века.
б Определено по методу Уилбура и Андерсона.
в Условия: 0,025 М трнс, 5%-ный ацетонитрил, pH 7,5, 23 °C.
г Определено в присутствии 1-10—e М свободного ацетазоламида.
легче всего. Для цинка получена зависимость этой константы от
pH. Величина lg/С линейно возрастает с увеличением pH от 5 до
10 и в более щелочной области выходит на плато, которому соот-
ветствует К.= Ю15. Это означает, что ионы водорода взаимодейст-
вуют с активным центром при pH<10. Не исключена также воз-
можность небольших изменений структуры белка, обусловленных
изменением pH и влияющих на стабильность комплекса.
Хенкенс и Стюртевант [87] детально изучили кинетику взаимо-
действия цинка с апоферментом. Реактивация белка цинком сопро-
вождается небольшими изменениями ультрафиолетовых спектров
[87]. Измерения скорости комплексообразования проводились ме-
тодом остановленной струи. Степень взаимодействия определялась
по изменению оптической плотности и по увеличению ферментатив-
ной активности. Реакция имеет первый порядок как по апофермен-
ту, так и по цинку. Значение константы скорости — около 104 л-
моль-1-с-1. Эта величина примерно на два порядка меньше, чем
при взаимодействии цинка с небольшими хелатирующими агента-
ми, для которых получены значения 106—108 л-моль-1-с-1 [87—
89]. В последнем случае считают, что скорость процесса лимитиру-
ется вытеснением воды из координационной сферы [87]. Скорость
37*
580
Глава 16
Таблица 16.9
Устойчивость металлокарбоангидраз [84]
Металл IgK (pH 5,5) Независящая от pH величина IgK
Мп(П) 3,8 —
Со (II) 7,2 —
Cu(II) 11,6 —
Ni(II) 9,5 —
Zn(II) 10,5 15,0
Cd(II) 9,2 —
Hg(II) 21,5 —
взаимодействия цинка с апокарбоангидразой возрастает при изме-
нении pH от 5,5 до 7,5 (табл. 16.10), но все же не достигает того
же уровня, как для модельных систем. Температурная зависимость
этого процесса удовлетворительно выражается кривой Аррениуса.
Рассчитанные значения энергии и энтропии активации приведены
в табл. 16.10. Наблюдаемые величины энергии активации выше
энергий активации модельных систем, но частично компенсируют-
ся большим выигрышем в энтропии активации. Значение послед-
ней при взаимодействии цинка с малыми комплексонами состав-
ляет 7—8 кал-моль-1-град-1 [88, 89].
Константа скорости второго порядка, полученная для модель-
ных систем, представляет собой произведение константы присоеди-
нения к внешней сфере (Ко) и константы скорости первого поряд-
ка (kj) для замещения воды. Возможны различные предположе-
ния о причинах низкой скорости взаимодействия цинка с
апоферментом. По-видимому, взаимодействие с металлом не сопро-
вождается значительной перестройкой конформации белка. Необ-
ходимо учитывать, что в металлоферменте ион цинка находится на
дне глубокой полости с несколько необычной геометрией (рис.
16.19). Вероятно, свой вклад вносят и процессы вытеснения воды
из координационной сферы.
Используя определенное ранее значение константы равновесия,
Хенкенс и Стюртевант [87] рассчитали константу скорости диссо-
циации комплекса Zn(II) с апоферментом (1,5-10—9 с-1). Интерес-
ные результаты получены при сравнении с аналогичными данными
для карбоксипептидазы А, которая легче обменивает свой ион цин-
ка на ионы из раствора [90]. Время, необходимое для замещения
половины цинка на кадмий для этого фермента при pH 8,0 и 4 °C,
составляет около 20 ч [90]. Отсюда следует, что константа скоро-
сти диссоциации равна 10-6 с-1 [87]. При обмене с изотопом 65Zn
Карбоангидраза
581
Таблица 16.10
Скорость рекомбинации Zn(II) с апокарбоангидразой при 25 °C. Теплота и
энтропия активации [87]
pH Ионная сила, М Ионы Н+ выделяю- щиеся при реакции Эффективная константа скорости второго порядка, 10—4 л -моль—1.с—1 Е, ккал/моль AS, кал-моль— •град-1
по диффе- ренциаль- ным УФ-спект- рам восстано- вление активности в реакции гидратации восста- новление эстеразной активности
5,5 0,051 — — — — 21,2 27,2
5,90 0,001 0,18 0,27 — —
5,90 0,021 0,42 0,54 — —
6,27 0,056 0,85 — — 0,81
7,0 0,056 1,5 —- 1,36 —
7,40 0,020 1,6 1,33 — —
7,56 0,056 2,2 —- — 2,0
7,5 0,051 — — — 20,8 30,0
эта величина имеет значение около 2,5-10~5 с-1 [91], т. е. на 4—
5 порядков больше, чем для карбоангидразы. По-видимому, раз-
личие в скоростях ассоциации для обоих ферментов не превышает
одного порядка. Поэтому неудачи при попытках обнаружить обмен
ионами цинка между металлокарбоангидразой и раствором при
pH 8,0 можно объяснить исключительно медленной диссоциацией
металлофермента. Расчеты показывают, что период полураспада
этого комплекса при pH 8,0, ионной силе 0,1 М. и 25 °C составляет
5—6 лет [87].
3.3. Спектры поглощения и оптическая активность
Наиболее существенным изменением физико-химических
свойств карбоангидразы при замене цинка ионами переходных ме-
таллов Зй-ряда является возникновение полос поглощения в види-
мой области спектра. Незаполненные Зй-орбитали этих ионов де-
лают возможным d—d-переходы, которые и проявляются в спект-
рах поглощения. Как и следует из теоретических соображений, эти
полосы лучше всего выражены у комплексов фермента с Со(II) и
Си (II) [92] (рис. 16.5). Со (И)-Карбоангидраза отличается наи-
большим поглощением, е~300. По интенсивности эта полоса ско-
рее соответствует тетраэдрическим, чем октаэдрическим, комплек-
сам Со(II), так как последним отвечает е~10 [92]. Но по струк-
туре (расщепление на четыре побочных максимума при 520, 555.
582
Глава 16
615 и 645 нм) она не похожа ни на один из известных тетра- или
октаэдрических кобальтовых комплексов, для которых наблюда-
ются простые полосы без расщепления. Это указывает на своеб-
разную геометрию координационной сферы металла в активном
центре.
Рис. 16.5. Спектры поглоще-
ния в видимой области
Co(ll)-, Ni(II)- и Cu(II)-
карбоангидраз В человека
и их комплексов с ингиби-
торами [73, 102, 156].
Наиболее близким к Со(II)-карбоангидразе по спектральным
свойствам оказался недавно полученный комплекс Со(II) с коор-
динационным числом 5 [93—96]. В результате этого появились
мнения о том, что и в белке для кобальта характерно координа-
ционное число пять. Однако по данным рентгеноструктурного ана-
лиза с разрешением 2 А координационная сфера Со(П) напоми-
нает сильно искаженную тетраэдрическую конфигурацию
(рис. 16.20, Б) [98]. Подобная структура наблюдалась и у недав-
но исследованных методом дифракции рентгеновских лучей кри-
сталлов быс-ацето-быс-(этилентиокарбамид)кобальта(П) [97]. При
комнатной температуре спектр этого соединения очень похож на
спектры Со(II)-карбоангидразы.
Координационная сфера этого комплекса имеет псевдотетра-
эдрическое строение. Ближайшими соседями атома Со являются
два атома кислорода и два атома серы, удаленные от него на 1,957
и 2,328 А соответственно. Наибольшее искажение в тетраэдриче-
Карбоангидраза
583
скую конфигурацию вносят два других атома кислорода, распо-
ложенные от центра на расстоянии 2,928 А. Имеются данные о том,
что их влияние и является причиной изменения спектров [97].
Спектр поглощения в видимой области Си(II)-карбоангидра-
зы, на котором можно различить три полосы поглощения, также
X, нм
Рис. 16.6. Спектры поглощения в видимой области Со(II)-карбоангидразы В че-
ловека при разных pH (а) и зависимость коэффициента молярного поглощения
при 640 нм от pH (б) [84].
свидетельствует о нарушенной геометрии. Спектр Ni (II)-карбо-
ангидразы имеет вид, более типичный для октаэдрических комп-
лексов Ni(II). По-видимому, различные металлоферменты имеют
неодинаковую геометрию координационных сфер. Результат
этого—отсутствие активности у большинства комплексов карбо-
ангидразы с металлами.
Кроме сведений о геометрии координации с помощью спект-
ров поглощения в видимой области было получено много других
полезных данных об активном центре. Это особенно относится к
Со(II)-карбоангидразе, поскольку она обладает ферментативной
активностью. Добавление сульфамидов или анионов, подобных
цианиду, приводит к резкому увеличению .интенсивности и умень-
шению ширины полосы поглощения [20, 39, 73, 84] (рис. 16.5).
Эти результаты были первым серьезным доказательством внед-
рения ингибиторов обоих типов в первую координационную сферу
иона металла.
На рис. 16.6 показано изменение спектра Со(II)-карбоангид-
разы, впервые обнаруженное Линдскогом [20, 84] при изменении
pH. Интенсивность полосы уменьшается, а два длинноволновых
584
Глава 16
пика при рН<6 вообще не наблюдаются. Величина р/Са, отве-
чающая этому сдвигу, в зависимости от условий лежит в интер-
вале pH 7—8 [20, 73, 84, 86] и, по-1видимому, идентична той кон-
станте, которая определяет pH-зависимость активности фермен-
та [26, 27, 29, 86, 99—101]. По всей вероятности, геометрия коор-
Рис. 16.7. Спектры КД в видимой области для трех изоферментов и видовых
вариантов Со(П)-карбоангидразы [102, 119].
I — карбоангидраза С человека; 2 — карбоангидраза В быка; 3 — карбоангидраза В человека;
4 — карбоангидраза В человека ± 0,2 — Де.
динационной сферы металла и каталитические свойства фермента
зависят от одного и того же процесса ионизации. Возможно, эти
эффекты связаны с диссоциацией координированной молекулы
воды (разд. 5 и 7).
Еще более чувствительными к изменениям в активном центре
оказались спектры КД [102] (рис. 16.7). Полосы поглощения у
всех трех представленных на этом рисунке разновидностей
Со(II)-карбоангидразы почти одинаковы — как по поглощению,
так и по интенсивности. На основании этого можно было бысчи-
Карбоангидраза
585
тать одинаковой и структуру координационной сферы. Но вели-
чины эллиптичности указывают на существенные различия. Изо-
ферменты С человека и В быка весьма оптически активны
(Де^З—5), а у формы В человека во всей области Де<0,2.
При 10-кратном увеличении чувствительности и у этого фер-
мента можно обнаружить небольшие положительные полосы эл-
липтичности, соответствующие всем полосам поглощения
Рис. 16.8. Спектры КД Со (.11) -карбоангидразы В человека в D2O при pD 6.81
(-------------------------------) и 9,21 (---------).
(рис. 16.8). С понижением pH они одновременно возрастают и су-
жаются, следуя за изменением спектров поглощения (см. выше).
Результаты измерений КД У различных видовых форм Со (II)-
карбоангидразы говорят о том, что оптическая активность ко-
бальтового хромофора более чувствительна к локальным особен-
ностям структуры изофермента, чем к симметрии самого коорди-
национного комплекса. Решающим фактором здесь может быть
асимметричность потенциального поля, создаваемая окружающи-
ми заряженными и полярными остатками аминокислот. Поэтому
становятся понятными трудности использования данных КД для
получения структурной информации. Очень низкая оптическая
активность Со (II)-карбоангидразы В человека, по-видимому, сви-
детельствует о наличии в строении комплекса металла элементов
симметрии, маскирующих большую оптическую активность, при-
сущую самому кобальтовому хромофору. Такими структурными
факторами могут быть плоские тетраэдрические или пятикоорди-
национные комплексы.
При добавлении комплексообразующих анионов наблюдается
увеличение значения рКа, определяемого по изменению спектра
586
Глава 16
при изменении pH. Увеличение этого параметра тем больше, чем
значительнее у добавляемых ионов сродство к металлу [101].
Такой же сдвиг рКа в присутствии тех же анионов заметен и на
кривых зависимости скорости реакции от pH [101]. Одно из воз-
можных объяснений этого — конкуренция между гидроксилом и
анионами за свободное место в координационной сфере иона ме-
талла. Такое объяснение согласуется с многочисленными данны-
ми о наличии в активном центре связей фермент — Zn—ОН
(см. ниже).
3.4. Связывание ингибиторов
Исследование функциональных и физико-химических аспектов
взаимодействия ингибиторов с активным центром карбоангидразы
способствовало разработке новых методов изучения фермента.
Кинетические, спектральные и рентгеноструктурные данные о
взаимодействии карбоангидразы с ингибиторами двух главных
типов (сульфамидами и небольшими анионами) свидетельствуют
о том, что их ферментативное связывание осуществляется одним
и тем же центром белка [21, 101, 103]. Имеется достаточно осно-
ваний считать, что главную роль в этом взаимодействии играет
обычная координация ионами металлов, хотя реальная картина
явлений ингибирования может включать и другие типы взаимо-
действий [23].
Изменение спектральных свойств под воздействием сульфами-
дов будет обсуждено в разд. 4. С помощью 3Н-ацетазоламида
можно непосредственно показать, что для связывания этого суль-
фамида необходим ион металла (рис. 16.9, а) [21]. При концент-
рациях, меньших 10-4 М, ацетазоламид не связывается апофер-
ментом, тогда как Zn (II)-карбоангидраза присоединяет 1 моль
ингибитора уже при концентрации последнего 5-Ю-7 М.
(рис. 16.9, а). Характерно, что подобный тип взаимодействия об-
наружен только с цинковыми и кобальтовыми комплексами кар-
боангидразы—-единственными активными разновидностями ме-
таллофермента (табл. 16.8). По-видимому, только реакционноспо-
собная конфигурация активного центра допускает возможность
связывания сульфамидов [21].
Азид-, цианат-, сульфид- и цианид-анионы способны вытеснять
сульфамид из активного центра (рис. 16.9, б). Концентрация
3Н-ацетазоламида, необходимая для связывания с ферментом в
присутствии равных и постоянных концентраций этих анионов,
оказывается прямо пропорциональной величинам их сродства к
активному центру, рассчитанным из кинетики ингибирования
(CN-^ SH- >OCN->N з) [21].
При исследовании инфракрасных спектров поглощения Рипе и
Уонг [105] также смогли показать непосредственное взаимодейст-
Карбоангидраза
587
вие одного из этих анионов (N3) с ионом металла в активном
центре. Азид-анион имеет полосу поглощения при 2049 см
обусловленную асимметричными валентными колебаниями этой
линейной молекулы. Эта полоса обычно смещается в сторону
Рис. 16.9 Связывание 3Н-ацета-
золамида в зависимости от его
концентрации (а) с металлокар-
боангидразой и (б) с Zn(II)-кар-
боангидразой В в присутствии
0,2 мМ металлсвязывающих ани-
онов и в их отсутствие (кривая
слева) [21]. Условия: 0,025 М
трис, pH 8,0, 4 °C.
больших частот при связывании азида с металлами [102]. В при-
сутствии фермента максимум поглощения Na находится при
2081 см-1 [Со(II)-карбоангидраза] и 2094 см-1 [Zn(II)-карбоан-
гидраза] (рис. 16.10). Хотя такое смещение и меньше, чем в мо-
дельных смесях азида с Со2+ и Zn2+, оно свидетельствует о при-
соединении аниона к координационной сфере металла в активном
центре. Один из сульфамидов, этоксзоламид (6-этоксибензотиа-
зол-2-сульфамид), устраняет этот сдвиг полосы в ИК-спектре.
Взаимодействие с субстратом (СО2) происходит иначе. В при-
сутствии металлофермента полоса поглощения свободного раство-
ренного СО2 (2343 см-1) смещается лишь очень мало (до
2341 см-1). Это означает, что в ферменте молекула СО2 почти
не деформируется._Связанная двуокись углерода легко вытесня-
ется анионами N3, NO3 и НСОз Поэтому считают, что СО2
не входит в координационную сферу металла, а каким-то обра-
588
Глава 16
зом (возможно, гидрофобно) связывается неподалеку от него
[105]. Это согласуется с современными представлениями о меха-
низме действия карбоангидразы (разд. 7).
Вообще этот фермент ингибируется, хотя бы и в малой степе-
ни, многими анионами. Такое действие оказывает даже ацетат,
продукт эстеразной реакции [86]. Более интересный тип ингиби-
рования был открыт при исследовании влияния иодацетата на
карбоангидразу В человека [65, 66, 68]. Ингибирование этим
Рис. 16.10. Дифференциальные ИК-спектры карбоангидразы быка в присутствии
азида [105].
I — фермент (0,01 М) с избытком азида (0,05 М) при pH 5,56; пик при 2094 см-' типичен для
азидного комплекса Zn(II); 2 —фермент (0,01 М) с азидом (0.05 М) и с избытком этоксзол-
амида (0,01 М) [105].
соединением необратимо, но при этом наблюдаются некоторые
особенности. Фермент никогда не теряет свою активность пол-
ностью. Даже при эквимолярном присоединении ингибитора со-
храняется 5—10% активности. Эта остаточная активность харак-
теризуется сигмоидальной pH-зависимостью с точкой перегиба
при pH 8,5 [68]. Следовательно, и после модифицирования в
ферменте присутствует та же ионогенная группа, которая опре-
деляет pH-профиль нативной карбоангидразы [68].
Иодацетат реагирует с гистидином, входящим в пептидный
фрагмент, упоминавшийся в разд. 2. Имеется ряд доказательств
[65, 66] того, что эта реакция происходит в активном центре:
а) параллельно с образованием 3-карбоксиметилгистидина теря-
ется эстеразная активность; б) иодацетат инактивирует карбоан-
гидразу необратимо только тогда, когда присоединяется к ней
как обратимый ингибитор; в) сульфиды предотвращают необра-
тимое ингибирование; г) для быстрой дегидратации необходима
нативная конформация фермента; в присутствии 8 М мочевины
быстрая реакция с гистидином уже не наблюдается.
Карбоангидраза
589
Спектры поглощения и КД Со (II)-карбоангидразы в присут-
ствии ацетата и иодацетата указывают на одинаковый характер
их присоединения к ферменту — координации к иону Со (II) [107]
(рис. 16.11). Затем иодацетат с относительной малой скоростью
необратимо реагирует с ферментом [65, 66, 107]. Для завершения
Рис. 16.11. Влияние иодацетата на спектр Со (II)-карбоангидразы В человека при
pH 9,0 (кривые 1—б). Влияние цианида на спектр карбоксиметилированной
Со (II)-карбоангидразы В человека (кривые 7—9).
Кривые 1—6 соответствуют спектрам, полученным при концентрациях иодацетата от 0 до
2 10"2 М, как показано на вставке (Ф Dass; О645). Ks для иодацетата лежит в пределах от
4-Ю-3 до 8-I0-3. После 24 ч необратимо прореагировавший с иодацетатом фермент титро-
вался цианидом (избыток CN- 7,1, 8,2 и 9 экв) [156].
процесса при взаимодействии карбоангидразы В обезьяны с
3Н-иодацетатом в концентрации 10-2 М потребовалось около 12 ч
[107]. Характер концентрационной зависимости свидетельствует
о необходимости предварительного насыщения иодацетатом
центра его обратимого связывания. С кинетической точки зрения
причины этого не вполне ясны.
Инактивированный иодацетатом фермент сохраняет способ-
ность присоединять к активному центру цианид и крупные мо-
590
Глава 16
лекулы сульфамидов [107]. При этом цианид так же изменяет
спектры карбоангидразы, как и в случае нативного фермента
(рис. 16.11). Следовательно, структура координационного комп-
лекса не нарушается при модифицировании гистидина. Эти дан-
ные соответствуют схеме на рис. 16.12, согласно которой метили-
руемый азот гистидина удален от атома Zn на расстояние не бо-
лее чем три длины связи, т. е. достаточно близко для переноса
Рис. 16.12. Схема взаимодействия иодацетата с активным центром карбоангид-
разы В человека.
протона. Установлено, что этот гистидин имеет р/Со=5,8 и реа-
гирует с иодацетатом только в виде основания [65, 66]. Для фер-
ментативной активности это слишком низкое значение. С другой
стороны, независимые измерения показали, что обратимое присое-
динение иодацетата зависит от группы с р/Со~7 (координирован-
ная молекула воды) [65]. Активность полностью карбоксимети-
лированного фермента составляет 10%, причем сохраняется сиг-
моидальный вид pH-зависимости с рКа=8,5. Эту величину также
можно объяснить ионизацией воды в модифицированном фер-
менте. По данным спектров КД, в модифицированном ферменте
сульфамиды попадают в более полярное окружение, чем в натив-
ной карбоангидразе [108].
Разные видовые варианты фермента не одинаково чувстви-
тельны к иодацетату [107]. Используя 3Н-иодацетат, удалось
установить, что при концентрации 10-2 М реакция необратимого
ингибирования карбоангидразы В человека завершается через
24 ч поглощением 1 моля иодацетата. Примерно так же взаимо-
действует и изофермент В обезьян. Однако с карбоангидразой С
человека реакция при этих условиях лишь слегка заметна, иона
Карбоангидраза
591
вообще не происходит в случае карбоангидразы В быка [107].
Отсюда вытекает, что если гистидин есть во всех разновидностях
фермента, то в некоторых из них он обладает пониженной реак-
ционной способностью.
Для выяснения механизма катализа оказались полезными ис-
следования смещения кислотно-основных равновесий в фермента-
тивной системе вследствие присоединения лигандов к свободным
местам координационной сферы металла активного центра. При
ингибировании ионами, подобными CN- или HS~, в области зна-
чений pH, меньших величины pH для диссоциации HCN или
H2S (при 23 °C — 9,3 или 6,9 соответственно [108]), должно на-
блюдаться выделение протонов. Общее количество последних
будет зависеть уже от нового положения кислотно-основных рав-
новесий системы. Для карбоангидразы исследование этих явле-
ний было проведено с помощью дифференциального титрования
в интервале pH от 6 до 10 [86], т. е. в области устойчивости циа-
нидного комплекса фермента. Метод заключался в фиксировании
(на pH-стате с точностью 1 ±0,02 мкмоль Н+) выделения или по-
глощения протонов после прибавления к раствору фермента экви-
молярного количества ингибитора, содержащегося в небольшом
объеме его концентрированного раствора.
Для решения поставленной задачи полезно провести сравне-
ние экспериментальных результатов с теоретическими зависимо-
стями, основанными на использовании какой-нибудь гипотетиче-
ской модели исследуемого процесса. Одна из таких схем, пред-
полагающая замещение цианидом координированной молекулы
воды, может иметь вид:
(a) EZn -H2O ± HCN <—> EZn-CN_±HaO ±Н+
pKQ=8 ||-Н+ рЛо=9.зЦ-Н+ (2)
(б) EZn-OH" + С№ EZn CN- + OH"
Реакции (2а) и (26) должны иметь место в очень кислых и
в очень щелочных растворах соответственно, если, конечно, зна-
чения рЛа координированной молекулы Н2О не выходят за пре-
делы экспериментального интервала pH. Как показано на
рис. 16.13, кривая титрования для реакции цианида с Zn(ll)-Kap-
боангидразой пересекает ось абсцисс; 1 моль Н+ освобождается
при pH 6,0 и 0,7 моля НО- освобождается при pH 10,1. Подоб-
ное изменение кислотно-основных равновесий является функцией
металла, так как при добавлении ионов CN- к апоферменту pH
системы не смещается. Аналогичная кривая наблюдается и для
случая кобальта (рис. 16.13). Эти результаты указывают на су-
ществование еще одного равновесия с рКо 8 (пунктирная кривая
на рис. 16.13). Экспериментальная картина согласуется со схе-
592
Глава 16
мой (2), если последнее значение относится к диссоциации моле-
кулы Н^О. При увеличении pH возрастает доля фермента в
ОН-форме. Ион ОН-, вытесняемый цианидом, постепенно нейтра-
лизует HCN в соответствии с теоретической кривой на рисунке.
В конце концов CN~ вытесняет только ОН-. Дополнительной осо-
бенностью, предсказываемой схемой (2), является зависимость
Рис. 16.13. Замещение Н+ и ОН— в карбоангидразе В человека цианидом и суль-
фидом.
1 и 3 — кривые титрования HCN и H2S, приведенные в виде доли неднссоциированного ин-
гибитора (ордината слева); 2 — теоретическая кривая титрования для группы с рКа=8,1,
приведенная в виде отношения количества диссоциировавшего Н+ в молях на моль белка
(ордината справа). Д эстеразная активность КАВ (мкмоль гидролизованного субстрата
в I мни на I мкмоль фермента). 4 и 5 — теоретические [по уравнению (2)] кривые диффе-
ренциального титрования для замещения Н+ и ОН— цианидом и сульфидом соответственно
как функции pH: ф 2п(П)-карбоангидраза; О апофермент: □ Со(П)-карбоангидраза+С1М—
(максимальное количество Н+ и ОН- освобождается лишь тогда, когда добавлено ~1,5 экв
цианнд-аннона); 2п(П)-карбоангидраза+сульфид [86].
дифференциального титрования от рКв ингибитора. Это можно
проверить, используя сульфид, так как H2S имеет р/Са=6,3. Как
и ожидалось, титрование происходит на 1,5 единицы pH в более
кислой области. И опять для совпадения теоретических и экспе-
риментальных данных необходимо учитывать равновесие с
рКа«8.
Эти результаты согласуются с присутствием координированной
молекулы воды в активном центре карбоангидразы [уравнение
(2) ], но не доказывают его.
Карбоангидраза
593
Возможны и другие объяснения этих явлений. Одно из них
предполагает, что анионом замещается какой-то белковый ли-
ганд, диссоциации которого отвечает очень большая величина
pKQ. Последний затем присоединяет протон. А взаимодействие
самой этой группы с металлом также зависит от pH со значе-
нием рКа«8. Однако данная гипотеза не очень убедительна.
Результаты, полученные методом ЯМР, дают все основания при-
писывать значение р/Со~8 координированной молекуле воды
(разд. 5).
4. КОМПЛЕКСЫ КАРБОАНГИДРАЗЫ
С СУЛЬФАМИДАМИ
После того как Манн и Кейлин [21] обнаружили ингибирова-
ние карбоангидразы сульфамидами, было получено еще много
подобных соединений. Установлено, что для эффективного инги-
бирования структура их молекул должна удовлетворять двум
общим требованиям. Во-первых, необходима незамещенная суль-
фамидная группа (I, А).
NH2
R А
сульфаниламид (I)
Обычно модифицирование этой группы приводит к утрате инги-
бирующей способности. Правда, недавно появились сообщения о
том, что некоторые модификации хотя и сильно снижают эффект
ингибирования, но еще оставляют возможности для связывания
nh2
хлортиазид(й')
CHjCONH C^ ^-SO2-NH2
N----------N
з/поксзолслх/йПУ)
ацетсзалслшй(Ш)
4-okcu-3 - нигпро^ензолсульфамид (V)
38—2451
594
Глава 16
с ферментом [67]. Второе требование к структуре молекулы — на-
личие объемистого ароматического или гетероциклического ради-
кала (R), присоединенного к свободной сульфамидной группе.
Как видно из приведенных схем (II—V), детали строения этого
радикала могут быть самыми различными. (Подробный список
этих соединений дан Мареном [109] и Баром [ПО].) Сульфамиды
интересны также тем, что они оказывают фармакологический и
терапевтический эффекты на различные обменные реакции орга-
низма, завершающиеся превращением карбонатов [109]. В по-
следнее время выяснилось, что сульфамиды являются очень цен-
ными для исследования физико-химии и механизма действия фер-
мента. Ниже эти сведения изложены в специальных разделах.
4.1. Образование комплексов, константы диссоциации
Величины констант диссоциации, рассчитанные по кинетиче-
ским данным для некоторых комплексов карбоангидразы с суль-
фамидами, изменяются от 2-10-9 для этоксзоламида до 2,6-10—5
для сульфаниламида и представлены в табл. 16.11. Исключи-
тельно низкие значения констант говорят о весьма высокой проч-
ности связей в этих нековалентных комплексах. Эта особенность
была использована для разработки удобного метода определения
концентрации карбоангидразы титрованием ее растворов этокс-
золамидом [103, 105, 112]. Как отмечалось выше, спектральные
изменения при связывании сульфамидов и исследования с по-
мощью 3Н-ацетазоламида свидетельствуют о непосредственном
взаимодействии сульфамидов с ионами металла в активном
центре [20, 21], причем координация, очевидно, происходит за
счет группы —SO2—NH2 [21, 111]. Это согласуется с результатами
рентгеноструктурного анализа [22, 23] кристаллического комплек-
са карбоангидразы С человека с сульфамидом. Они показывают,
что во внутреннюю координационную сферу цинка включена амин-
ная группа сульфамида (разд. 6). По-видимому, в связывании
участвуют анионная группировка (—SOa—NH-) с одним прото-
ном, замещенным на металл [30, 103]. В отсутствие конкуренции
последнего величина рКа для этой группировки колеблется от 7
до 10 (в зависимости от структуры R) [109, 115]. Некоторыми
исследователями было обнаружено максимальное связывание
при pH —7. В кислых и щелочных растворах прочность взаимо-
действия быстро падает [86, 115, 116]. Причиной распада комп-
лекса в кислой области может являться уменьшение количества
анионной формы ингибитора.
В щелочных растворах сульфамиды, вероятно, вытесняются из
координационной сферы конкурирующими гидроксильными иона-
ми. Точно так же зависит от pH и константа скорости второго
порядка для образования комплекса фермент — сульфаниламид.
Карбоангидраза
595
Таблица 16.11
«Константы диссоциации» для комплексов карбоангидразы с сульфамидами»
Соединение Константа диссоциации, М Литература
Сульфаниламид 2,8-10-» 111
N4-Ацети лсу льф анил ам ид 5,0-10-7 111
Дихлорфеиамид 2,5-10-8 111
Ацетазоламид 8,0-10-s 111
1,5-10-76 21
Метазоламид 9,0-10-» 111
Этоксзоламид 1,0-10-9 111
Хлоротиазид 2,2-10-8 111
Циклотиазид 6,0-10-7 67
Хлорацетилциклотиазид 6,5-10-5 * * 67
5-Диметиламиионафталин-1 -сульфамид (ДНСА) 2,5-10-7» 103
4'-Сульфамилфенил-2-азо-7-ацетамидо-1-ок- 1,5-10-»г 30
синафталин-3,6-дисульфонат (неопронтозил)
а Если не указано иначе, считается, что величина К£- равна константе диссоциации,
определенная непосредственно по связыванию 3Н-ацетазоламида.
определенная флуоресцентным методом.
ГЛ5, определенная спектрополярн.метрическим методом.
значение которой составляет примерно 7-104 л-моль-1-с-1
(pH 7,9, 25 °C, измерена методом остановленной струи) [115].
Поэтому величины рКа должны представлять ионизацию как сво-
бодного фермента, так и свободного ингибитора [115]. Однако
при интерпретации этих данных надо соблюдать осторожность.
Высокая прочность связей карбоангидразы с сульфамидами явля-
ется результатом не только координационного взаимодействия,
но и других эффектов (гидрофобных связей, энтропийных и про-
чих вкладов). Поэтому любые изменения в структуре белка
вследствие изменения кислотности среды могут также сказы-
ваться на величине сродства к ингибиторам.
При действии сульфамидов на карбоангидразу наблюдается
кинетика ингибирования как конкурентного, так и неконкурент-
ного типа в зависимости от времени наблюдения и порядка сме-
шивания реагентов [116—118]. Причина подобных расхождений,
вероятно, кроется в чрезвычайно низкой скорости диссоциации
ингибиторных комплексов, следствием чего является и малая
скорость установления равновесия с конкурирующей молекулой
субстрата [115, 117].
38*
596
Глава 16
4.2. Спектры поглощения и круговой дихроизм
Особенно ценным оказалось использование сульфамидов в ка-
честве молекулярных зондов, чему способствует наличие хромо-
форов в молекулах сульфамидов и белка. Лучший пример белко-
вого хромофора — ион Со(II). Выше уже отмечалось (рис. 16.5),
Рис. 16.14. Аппроксимация полос в спектрах поглощения и КД Со(II)-карбоан-
гидразы В человека и ее комплексов с этоксзоламидом и цианидом (-----------)
сериями перекрывающихся гауссовских линий (-----------).
Сплошные линии на всех частях рисунка представляют собой огибающие кривые для сумм
составляющих гауссовских линий, которые показаны штриховыми линиями и пронумерованы
в порядке возрастания энергии. Все огибающие согласуются в пределах ошибки измерения
с экспериментальными данными. А—спектр поглощения Со(II)-фермента; Б — спектр погло-
щения комплекса с этоксзоламидом; В — спектр КД комплекса с этоксзоламидом; Г —
спектр поглощения цианидного комплекса; Д — спектр КД цианидного комплекса. Аппрок-
симация выполнена с помощью аппаратуры Du Pont. 310 (119].
что при взаимодействии этого хромофора с сульфамидами меня-
ются и интенсивность, и энергетическая структура полос поглоще-
ния в видимой области. Эти сдвиги, как и в случае ингибирова-
ния анионами, ясно свидетельствуют о вхождении лиганда в
координационную сферу иона металла. Главные особенности
спектра поглощения обсуждались в разд. 3.
Изменения энергетических характеристик и оптической актив-
ности полос поглощения кобальта, проявляющиеся после присое-
динения анионных и сульфамидных ингибиторов к активному
Карбоангидраза
597
центру карбоангидразы, являлись объектом многих исследований.
Основные результаты суммированы в примерах, приведенных на
рис. 16.14, где показаны спектры поглощения и КД этоксзоламид-
ных и цианидных производных Со (II)-карбоангидразы В челове-
ка. Эти спектры разложены на соответствующие гауссовские
полосы поглощения и эллиптичности для демонстрации (хотя бы
приблизительной) их тонкой структуры [119].
Из проведенного анализа становятся ясными два обстоятель-
ства. Во-первых, все спектры характеризуются расщеплением ви-
димых полос, что говорит о заметном искажении координацион-
ной геометрии даже в ингибированном ферменте. Во-вторых,
для аппроксимации спектров этоксзоламидного комплекса
(рис. 16.14, Б и В) требуется такое же количество гауссовских
полос, что и для спектра немодифицированной Со(II)-карбоан-
гидразы (рис. 16.14,Л). Результатом введения сильного лиганда
сульфамида, по-видимому, является также смещение в коротко-
волновую область двух слабых полос. Однако общая мульти-
плетность спектра при этом не снижается. Последнее можно объ-
яснить строгими структурными условиями замещения лиганда
(см. рентгеноструктурные данные, разд. 6). Одновременно изме-
няются спектры КД в ближней ультрафиолетовой области хро-
мофоров, что особенно четко проявляется для цианидного комп-
лекса (рис. 16.14, Г). Эти полосы также легко обнаруживаются в
спектре КД немодифицированного Со(II)-фермента (рис. 16.7 и
16.8). Специфическая геометрия координации активного центра,
свойственная ей необычная серия d—d-переходов (рис. 16.14,Л),
а также сродство к сульфамидам являются хорошо известными
особенностями белковой химии карбоангидразы. Такие же спек-
тральные полосы имеются и у Со (II)-производных фермента,
выделенного из акул, который, по-видимому, развивался по обо-
собленному эволюционному пути за несколько сотен миллионов
лет до наступления эры млекопитающих [42].
4.3. Взаимодействие с сульфамидами,
содержащими хромофоры
4.3.1. Оптические спектры
Введением хромофоров в молекулу сульфамида можно выз-
вать появление серии спектральных линий, соответствующих
электронным переходам в области активного центра. Многие
сульфамиды обладают хромофорными группировками, оптиче-
ские свойства которых меняются после связывания с карбоангид-
разой .[30, 102, 115, 120]. Но наиболее ярко эти эффекты прояв-
ляются при взаимодействии с оксинафталиновым производным
598
Глава 16
азосульфаниламида — 2- (4-сульфанилфенилазо)-7-ацетамидо-1-
оксинафталин-3,6-дисульфонатом (VI)
Для этого сульфамида характерны интенсивные полосы поглоще-
ния в видимой области около 500 нм и ряд полос в ультрафиоле-
товой области. При связывании с карбоангидразой наблюдается
Рис. 16.15. Спектры КД карбоангидразы В (------) и ее эквимолярного ком-
плекса с азосульфамидом (----------------------) [120].
сильная гипохромия в полосах в видимой области и смещение в
длинноволновую область на несколько нанометров. Количествен-
ные характеристики этих явлений специфичны для каждого изо-
фермента и видового варианта карбоангидразы [30].
Очень похожие изменения спектральных свойств у этого хро-
мофора наблюдаются и в неполярных растворителях [30]. Можно
заключить, что ферментативный центр связывания сульфамидов
имеет гидрофобную природу [30]. Такой вывод подтверждается и
рядом других спектральных исследований [103, 105].
Дополнительная большая оптическая активность появляется у
азосульфаниламида .вследствие связывания этой симметричной
молекулы хромофора с асимметричным белковым окружением
[30, 120]. Спектры КД в видимой области для комплексов этого
Карбоангидраза
599
сульфамида с карбоангидразой В обезьяны приведены на
рис. 16.15, где для сравнения также показан спектр нативного
фермента [120]. Наряду с большими полосами эллиптичности в
видимой области имеются дополнительные интенсивные полосы
эллиптичности в ультрафиолетовой области, которые полностью
затемняют КД-спектр нативного белка. Сила вращения некото-
рых этих полос (/Д«2-10-38 ед. CGS) по своей величине больше
соответствует собственно несимметричным хромофорам, чем сим-
метричным, оптическая активность которых возбуждена асиммет-
ричным окружением [121]. Следовательно, структуре белка свой-
ственно особенно эффективное асимметрическое потенциальное
поле [30].
У разных типов карбоангидразы величина и знак полос эллип-
тичности для комплексов с этим сульфамидом сильно различа-
ются [30]. Очевидно, это является следствием различной молеку-
лярной структуры областей, окружающих активный центр.
К такому же заключению приводят и результаты, полученные ме-
тодом ЯМР с 35С1 [122, 123] и комплексометрическим титрова-
нием [86]. Подобные структурные расхождения могут служить
причиной неодинаковой удельной активности у изоферментов и
видовых вариантов карбоангидразы [77].
4.3.2. Флуоресценция
Карбоангидраза быка образует сильно флуоресцирующий ком-
плекс с 5-диметиламинонафталин-1-сульфамидом (ДНСА) [103].
Исследуя усиление флуоресценции лиганда и тушение ультрафио-
летовой белковой флуоресценции, Чен и Кернохан [103] показа-
ли, что с одной молекулой фермента связывается одна молекула
флуоресцирующего сульфамида. Константа диссоциации этого
комплекса равна 2,5-10—7 М. 'Флуоресценция свободного сульфа-
мида имеет пик испускания при 580 нм и квантовый выход 0,055.
Соответствующие значения для связанного ингибитора — 468 нм
и 0,84. Большое смещение максимума испускания в коротковол-
новую область можно объяснить сильной гидрофобной природой
центра связывания, а также отрывом одного протона от сульфа-
мидной /группы лиганда при присоединении к ферменту [103].
Расчеты эффективности переноса энергии привели к неожидан-
ным результатам: 85% фотонов, поглощенных семью остатками
триптофана, переносятся к единственной связанной молекуле
ДНСА. Такая эффективность намного больше когда-либо ранее
наблюдавшейся у белков, содержащих только одну 5-диметила-
минонафталин-1-сульфанильную группировку. Хотя диаметр гло-
булы равен 51 А, связанная молекула ДНСА, очевидно, удалена
от каждого из семи остатков триптофана не больше, чем на кри-
тическое расстояние переноса, составляющее для триптофана
600
Глава 16
21,3 А. Согласно этим данным, остатки триптофана и центр свя-
зывания сульфамида находятся в глубине молекулы. Рентгено-
структурный анализ (разд. 6), выполненный, правда, на другом
видовом варианте (карбоангидраза С человека), привел к такому
же заключению. Тем самым доказывается аналогичность струк-
тур этих двух форм фермента.
4.3.3. Фосфоресценция
Гэлли и Страйер [124] использовали явление фосфоресценции,
возникающей при триплет-триплетном переносе энергии, для
изучения взаимодействия еще одного связанного сульфамида с
остатками триптофана в белке. Между синглетными состояниями
хромофоров перенос электронной энергии возбуждения возможен
на расстояния порядка 30 А, тогда как триплет-триплетный
перенос энергии требует значительно меньших расстояний-—
около 12 А. Если синглетное состояние триплетного акцептора
обладает большей энергией, чем синглетное состояние триплетно-
го донора, последний может возбуждаться светом с длиной вол-
ны, не поглощаемым триплетным акцептором. В этих условиях
фосфоресценция триплетного акцептора наблюдается только при
триплет-триплетном переносе энергии. Гэлли и Страйер [124]
обнаружили подобные явления у комплекса карбоангидразы быка
с .w-ацетилбензолсульфамидом (МАБС). Фосфоресценция трипто-
фана карбоангидразы вызывается светом с Л='28О нм. Фосфорес-
ценция сульфамида возбуждается светом с длиной волны 330 нм,
которая не вызывает свечения фермента. Однако в комплексе,
фосфоресцирующем при воздействии Х=330 нм, свечение, веро-
ятно, обусловлено скорее триптофаном, чем МАБС [124]. Следо-
вательно, остатки триптофана расположены очень близко от
центра связывания, образуя, видимо, поверхность его полости.
5. ЯДЕРНЫЙ МАГНИТНЫЙ РЕЗОНАНС
И ЭЛЕКТРОННЫЙ ПАРАМАГНИТНЫЙ РЕЗОНАНС
До недавнего времени методы магнитного резонанса не ис-
пользовались для исследования карбоангидразы. Однако теперь
методы протонного магнитного резонанса и электронного спино-
вого резонанса применяются и для этого фермента благодаря хи-
мическим особенностям его активного центра, содержащего ион
металла. Используя хорошо известный эффект увеличения скоро-
сти релаксации протонов воды, индуцированный парамагнитными
ионами металлов, Фабри и сотр. [125] доказали присутствие ко-
ординированных НгО или ОН- в первой координационной сфере
Со (II) в активном центре карбоангидразы. Парамагнитный ион
Карбоангидраза
60i
металла имеет 'магнитный момент, в несколько тысяч раз боль-
ший, чем другие ядра. Поэтому беспорядочное движение таких
ионов оказывает гораздо больший эффект на релаксирующие
соседние протоны растворителя, особенно если парамагнитный
центр включен в макромолекулу с относительно слабым тепловым
Рис. 16.16. pH-Зависимость удельной скорости релаксации при малых частотах;
Со(П)-карбоангидраз В быка (7) и человека (2).
Сплошные линии получены при обработке методом наименьших квадратов. Стрелки указы»»
вают величины pKfl. Условия: 0,1 М трис, 0,1 М SO|“, 25 °C, ~10 масс. % [125].
движением. Поскольку замена Zn(II) на Со(II) в активном цент-
ре карбоангидразы сохраняет активность фермента, Со (II)-произ-
водные, содержащие парамагнитный центр, являются идеальным
объектом для таких исследований.
Со(II)-Карбоангидраза вызывает заметное увеличение скоро-
сти релаксации (Т71) протонов воды, особенно при больших pH
[125]. Это увеличение в значительной степени исчезает при добав-
лении азида или этоксзоламида; это несомненно подтверждает,
что наблюдаемый эффект обусловлен активным центром. Усиле-
ние релаксации в зависимости от pH представлено на рис. 16.16 в.
виде удельной скорости релаксации. Ингибируемая составляю-
щая общей Т~' зависит от pH с рК=7,0±0,2 Для Со(П)-карбоан-
602
Глава 16
гидразы быка и с рК=8,2 + 0,2 для Со (II)-карбоангидразы В чело-
века. Из зависимости величины ингибируемой .компоненты общей
скорости релаксации от напряженности магнитного поля рассчи-
таны значения времен корреляции для дипольного взаимодейст-
вия протона с электронным моментом кобальта (Ю-11 с), расстоя-
ния между протоном и кобальтом (2,2—2,5 А) и времена пребы-
вания протона тм (10-5 с) [,125]. Лучше всего эти данные можно
объяснить тем, что протон, определяющий ингибируемую часть
l/Ti, находится либо в гидроксильном ионе, связанном с Со (II)
в активном центре, либо в молекуле воды, которая одним ато-
мом водорода связана с соседним остатком. Характерно, что зна-
чения р/<а, описывающие изменения увеличения скорости релак-
сации, очень похожи на величины, полученные для координиро-
ванной молекулы воды при комплексометрическом титровании
(разд. 7) —-8,1 для карбоангидразы В человека и 7,4 для изофер-
мента быка [86].
Трудно объяснимой особенностью этого явления остается отно-
сительно слабое усиление релаксации при 'низких значениях pH,
где координированной частицей, скорее всего, является молекула
Н2О. Можно предположить, что в кислой области затруднен (или
происходит по измененному механизму) обмен с протонами воды
из раствора [125]. Напомним, что титрование при pH от 9 до
6 сопровождается явным изменением геометрии координации
(разд. 3.3). Однако пока не установлена четкая взаимозависи-
мость между возможными изменениями строения координацион-
ной сферы и уменьшением усиления релаксации.
Несколько отличным методом для изучения тех же особенно-
стей активного центра воспользовался Уард [122, 123]. В качест-
ве JIMP-зонда в этих работах применялся 35С1, ядро которого
имеет спин 3/2 и электрический квадрупольный момент Q. Для
сред, в которых ядра хлора находятся в условиях градиента на-
пряженности электрического поля, ширина линии для резонанса
35С1 определяется уравнением
2л
Av = —g- (e2qQ)2x
О
где Av — полная ширина при половинной амплитуде; q — градиент
напряженности электрического поля у ядра; т — .время корреля-
ции, описывающее неупорядоченные молекулярные движения,
ответственные за изменения во времени градиента q [122]. Со-
гласно этому соотношению, связывание 35С1 с Zn(II) приводит к
ускорению релаксации ядра и уширению линии (рис. 16.17).
Zn (II)-Карбоангидраза тоже вызывает это уширение, а апофер-
мент нет (рис. 16.17). Это свидетельствует о том, что хлорид мо-
жет занимать одно координационное место у иона Zn(II) в актив-
ном центре. Уширение линии устраняется цианидом, что также
Карбоангидраза
603
позволяет отнести это взаимодействие к активному центру. Проч-
ность связи Zn—Cl сильно зависит от pH. Она максимальна при
pH 6 и резко падает при pH 9,0. Величина рЛо, описывающая
этот процесс, может быть найдена экстраполяцией данных ЯМР,
полученных при нескольких концентрациях хлорида, к нулевой
его концентрации [122, 123]. Эти значения рЛо составляют
Рис. 16.17. Спектр ЯМР 35С1 в 0,5 М растворе
NaCl.
а — без фермента, Avq—=12,5 Гц; б— 3,5-10-5 М
апокарбоангидразы в 0,01 М ацетатном буфере
(pH 4,8), Av—Av q—=7,0 Гц; в — 4,1-10-5 М карбоаН'
гидразы в 0,01 М трис-буфере (pH 8,2), Av—=
=49,4 Гц [122].
7,0±0,1 для фермента быка и 8,1 ±0,1 для изофермента В челове-
ка, что соответствует величинам константы ионизации координи-
рованной молекулы воды, найденным при изучении протонного
резонанса Со (II)-фермента [125]. Как отмечалось выше, то же
обнаруживается и при комплексометрическом титровании [86].
Эти результаты свидетельствуют о конкуренции между С1_ и
ОН- за место в координационной сфере металла при высоких
значениях pH. Еще раньше Линдског [101] в своих исследованиях
различного влияния анионов на спектры Со(II)-карбоангидразы
при различных pH объяснил такие же соотношения (см. выше).
Как и в случае протонного резонанса, при рН<6 наблюдается
значительное уменьшение уширения линии 35С1, по-видимому,
вследствие каких-то структурных изменений в белке.
5.1. Карбоангидраза со спиновыми метками
Введение парамагнитных групп в белок посредством привязы-
вания к нему стабильных свободных радикалов является ценным
методом спектроскопического исследования [126, 127]. Получае-
мые от введенного свободного радикала сигналы ЭПР можно ис-
пользовать для контроля за изменениями в белке. При этом
можно сделать определенные выводы и об окружении свободного
радикала. Первые примеры применения этой техники представили
МакКоннел с сотр. [128—130]. Удобнее всего оказался стабиль-
ный свободный радикал иминоксильной группы (нитроксид)
[131, 132], поскольку сам он .мало реакционноспособен, но может
604
Глава 16
быть привязан к различным соединениям, содержащим опреде-
ленные активные группы. Последние могут присоединяться к ак-
тивному центру или к специфическим аминокислотным остаткам
белка [127—130].
Рис. 16.18. Спектр ЭПР соединения
V1.1 (Л) в растворе (магнитное поля
3401 Гс, аплитуда модуляции 0,5 Гс)
и его эквивалентных комплексов с
различными изоферментами карбо-
ангидраз (Б, В и Г) (магнитное поле
3401 Гс, амплитуда модуляции 6,3
Гс) [133] (Б — Zn(iII)-карбоангид-
раза В быка; В — Zn(II)-карбоан-
гидраза В человека; Г — Со(П)-кар-
боангидраза В человека).
При исследовании иминоксильного радикала установили, что
свободный электрон в основном локализован на 2рл-атомной
орбитали азота. Сигнал от свободного радикала вследствие сверх-
тонкого взаимодействия с ядром азота 'расщепляется на три ли-
нии [126, 127] (рис. 16.18). Это сверхтонкое расщепление зависит
от ориентации иминоксила относительно приложенного поля.
Поэтому три различные линии наблюдаются вдоль трех осей
строго ориентированного монокристалла иминоксила [127].
Однако в разбавленных растворах небольших молекул, содержа-
щих свободный радикал, вращение молекул происходит значи-
тельно быстрее, чем обращение спина. Поэтому различные поло-
жения свободного радикала относительно внешнего поля усред-
няются. С другой стороны, если радикал привязан к макромоле-
Карбоангидраза
605
куле с гораздо меньшей скоростью вращения, так что обращение
-спина происходит прежде, чем -молекула примет .все возможные
ориентации, то спектр будет комбинацией спектров для различных
ориентаций свободного радикала, например -комбинацией спект-
ров, отвечающих трем осям монокристалла. .Подобная картина
наблюдается при дроблении кристалла, когда ориентация полу-
чаемого порошка является комбинацией всех ориентаций кри-
сталлов [1-29]. Поэтому спектры таких заторможенных свободных
радикалов относят к спектрам порошкового типа [127, 128].
Поскольку свободный электрон парамагнитного радикала при-
вязан к группе +N—-О-, он вообще не взаимодействует с элек-
тронными облаками и ядрами окружения, как это имеет место у
свободных электронов ионов переходных металлов. Поэтому из-
менения спектров ЭПР говорят главным образом о степени затор-
моженности. Этот метод только начинает применяться к металло-
ферментам, но некоторые результаты уже получены.
Сульфамиды связываются с активным центром, координируясь
у иона металла (разд. 4). Их способность к связыванию мало
зависит от структуры группировок, присоединенных к сульфа-
миду. Поэтому удается довольно просто ввести в карбоангидразу
иминоксильные метки, привязывая их к сульфамиду. Одно из та-
ких соединений (VII) синтезировали и использовали Мушак и
Коулмен '[133].
VII
Спектр ЭПР этого соединения в растворе показан на
рис. 16.18, А, а спектры его эквимолярных комплексов с различ-
ными изоферментами карбоангидразы—на рис. 16.18, Б, В и Г.
Связывание со всеми тремя формами фермента затормаживает сво-
бодный радикал. Заторможенность несколько больше у фермента
быка, что согласуется и с большей прочностью связей сульфамидов.
Сравнение этого спектра с расчетными данными Ицковича '[126,
134] для заторможенных свободных радикалов приводит к вели-
чине времени корреляции (т) для связанного с ферментом быка
иминоксила в 2-10-8—4-10~8 с. Интересно, что для времени вра-
щательной релаксации, рассчитанного для комплекса той же
формы карбоангидразы с флуоресцирующим сульфамидом, Чен и
Кернохан [103] получили значения 2,-89-10~8 с.
Со(II) и Zn(II) вызывают почти одинаковую заторможенность
иминоксильной метки, а так как связывание определяется метал-
лом, то и центры связывания в обеих активных металлокарбоан-
606
Глава 16
гидразах должны быть почти одинаковыми. Ни Hg(II)-, ни
Cd(II)-производные не затормаживают иминоксильный сульфа-
мид, что также согласуется с данными об отсутствии связывания
сульфамидов с этими металлоферментами.
Другой характерной особенностью является практически пол-
ное отсутствие спин-спинового взаимодействия между парамаг-
нитным Со(II) и иминоксилом. Это, видимо, не является неожи-
данным, так как в соответствии с представлениями о способе
связывания сульфамидов свободный радикал должен находиться
от Со(II) на расстоянии 15—20 А.
5.2. Магнитная восприимчивость
Линдског и Эренберг [.135] детально исследовали магнитную
восприимчивость Со(II)-карбоангидразы В человека и некоторых
ее комплексов с ингибиторами. Все комплексы фермента содер-
жат высокоспиновый Со(II) с магнитными моментами от 4,2 до
4,7 магнетонов Бора. Поскольку чисто спиновый момент предпо-
лагается равным 3,9 магнетона Бора, все комплексы показывают
ожидаемое увеличение магнитного момента вследствие спин-орби-
тальных связываний [92]. Согласно многочисленным данным о
магнитной восприимчивости высокоспиновых комплексов Со (II),
при октаэдрической координации магнитные моменты составляют
4,7—5,2 магнетона Бора, а при тетраэдрической — 4,1—4,9 маг-
нетона Бора. Все описанные квадратно-плоскостные комплексы
Со (II) являются низкоспиновыми и имеют магнитные моменты от
1,8 до 2,0 магнетона Бора. Последнее, вероятно, не слишком
удивительно, так как низкоспиновая конфигурация б/7-иона может
стабилизироваться благодаря эффекту Яна—Теллера [92]. Ве-
личина магнитной восприимчивости Со (II)-карбоангидразы согла-
суется с несколькими вариантами тетраэдрической геометрии.
К сожалению, интерпретация тонких различий восприимчивости
невозможна. Независимо от силы поля лигандов тетраэдрические
комплексы Со(II) являются высокоспиновыми вследствие распре-
деления орбитальных энергетических уровней. Со (II)-карбоанги-
драза остается высокоспиновой и при относительно высоком поле
лигандов, особенно в сульфамидном комплексе, который к
тому же имеет наибольшую восприимчивость.
6. РЕНТГЕНОСТРУКТУРНЫЙ АНАЛИЗ КАРБОАНГИДРАЗЫ С
ЧЕЛОВЕКА
Рентгеноструктурные исследования кристаллических препара-
тов карбоангидразы С человека Страндберг с сотр. [22, 23, 44,
45, 85] довели до степени разрешения 2 А. Наряду с более ран-
Рис. 16.19. Модель карбоангидразы человека (А) и ее комплекса с ацетоксимер-
курсульфаниламидом (Б), выполненная по картам электронной плотности с раз-
решением 5,5 А [22].
608
Глава 16
Карбоангидраза
609
Рис. 16.20. Схемы электронной плотности с разрешением 2А в области атома
Zn(II)-карбоангидразы С человека [23].
А—в плоскости атома цинка; Б — в плоскости, расположенной над атомом цинка [23]:
В — карта электронной плотности активного центра, предварительная расшифровка. Цинк
координирован с тремя остатками гистидина — с двумя остатками посредством Ns и с одним
при помощи Nj. Молекула воды занимает четвертое координационное место. Четвертый
гистидин располагается при входе в полость активного центра и может быть тем остатком,
который, как сообщалось, модифицировался бромпируватом в изоферменте С [23]. Элек-
тронные плотности между этим гистидином и ионом цинка обусловлены растворителем,
вероятно молекулами воды’.
ними результатами, полученными при разрешении 5 А, эти дан-
ные позволяют теперь подойти к более обоснованному обсужде-
нию механизма действия с точки зрения структурных характери-
стик фермента. Д-р Б. Страндберг и д-р А. Лильяс любезно
предоставили нам свои данные с разрешением 2 А еще до публи-
кации [23]. 1
Общая структура молекулы, видимая при разрешении б А,
показана на рис. 16.19,А [22]. Ион цинка(П) находится в глубо-
кой щели вблизи центра глобулы. N- и C-Концевые области по-
липептидной цепи предварительно отнесены на позиции 1 и
33 соответственно. Такое отнесение азотного конца подтвердилось
при разрешении 2 А [23].
Положение единственной свободной SH-группы определено по
разнице в картах электронной плотности нативного фермента и
фермента, модифицированного ацетоксимеркурсульфаниламидом.
Две молекулы последнего реагируют с карбоангидразой — одна с
39-2451
610
Глава 16
активным центром, а другая со свободной сульфгидрильной
группой [22]. Модель этого комплекса показана на рис. 16.19,5.
SH-группа удалена от иона Zn(II) на 14 А.
Карта электронной плотности с разрешением 2 А показывает,
что центр тяжести сульфамида находится от иона цинка на рас-
стоянии 3,2 А. Это в пределах длины связи и согласуется с пред-
ставлением о координации сульфамидного атома азота у иона
цинка (разд. 4).
Атом ртути ацетоксимеркурсульфаниламида находится на зна-
чительно большем расстоянии от цинка, т. е. бензольная часть
сульфамида лежит в щели, ведущей к иону цинка [22]. Низкие
константы диссоциации сульфамидных комплексов карбоангидра-
зы в растворах (10~5—10-9 М) [111—114] свидетельствуют о том,
что кроме координационных имеются и другие типы связей между
белком и циклической частью молекулы сульфамида. Изменения
в спектрах поглощения и флуоресценции связанных сульфамидов
свидетельствуют о гидрофобном характере связывающей полости
[30, 103, 105]. Однако основание этой полости, расположенное
вокруг атома цинка, может быть достаточно гидрофильным.
На рис. 16.20, А и Б приведены две карты электронной плот-
ности с разрешением 2 А. Одна из них соответствует плоскости,
в которой находится ион цинка, а вторая — плоскости, располо-
женной сразу над ним. Наибольшей плотности на картах отве-
чает ион металла. Вокруг него заметны три координированные
группы белка, электронные плотности которых хорошо совмеща-
ются со структурой остатков гистидина (рис. 16.20,5). Так как
аминокислотная последовательность пока не известна, идентифи-
кация должна считаться лишь предварительной. Интересно, что
два из этих трех лигандов (на рис. 16.20,5 они расположены ле-
вее) относятся к одному и тому же участку пептидной цепи и их
разделяет только один остаток.
Кроме описанных трех лигандов на схеме заметно еще одно
скопление над атомом цинка (на рис. 16.20,Л — немного правее).
Оно несколько слабее, чем у белковых групп, и, видимо, отно-
сится к молекуле воды из раствора. По направлению это скопле-
ние соответствует местам связывания сульфамида, которые на
рис. 16.20,5 обозначено символами Аз (сульфамидная группа) и
X. Шп п бензольной части). При связывании сульфамида слабое
< oi:.-'i'!i:i ;;:м' '.зстся более плотным. Имеется значительное ко-
личеств-) пых о -ом, что в растворе фермента это четвертое
место снимают координированные Н2О или ОН- (см. ниже).
Точная структура вокруг иона Zn(II) не может быть опреде-
лена и с помощью карт при разрешении 2 А. Для этого необхо-
дима еще большая степень разрешения. Строение этой области
белка приближается к тетраэдрической геометрии, хотя спектры
Со (II)-карбоангидразы указывают на значительное искажение.
Карбоангидраза
611
Некоторые общие особенности структуры глобулы карбоан-
гидразы заметны и на этих картах. На пространственной модели
полипептидной цепи, построенной в соответствии с ними [23],
обнаруживаются четыре фрагмента правой спирали, по два витка
в каждом. Только один из этих участков соответствует парамет-
рам а-спирали, остальные значительно искажены и скорее похо-
дят на Зю-спираль [23]. Как отмечалось в разд. 2, спектры ДОВ
и КД различных карбоангидраз также указывают на малую сте-
пень спирализации.
Основной особенностью вторичной структуры белка, как сле-
дует из карт электронной плотности с разрешением 2 А, является
большая область p-конфигурации, занимающей центральную часть
молекулы. По меньшей мере восемь таких фрагментов, направ-
ленных антипараллельно друг к другу, образуют изогнутую
складчатую структуру. Еще три или четыре участка цепи также
расположены параллельно или антипараллельно к этой структу-
ре. В результате в центре глобулы образуется большая гидро-
фобная область. В этом отношении структура карбоангидразы
очень похожа на строение карбоксипептидазы А [136, 137]
(гл. 15). Три гистидиновых лиганда, связанных с ионом Zn(II),—
тоже из этой р-конфигурации.
7. МЕХАНИЗМ ДЕЙСТВИЯ
Первоначально считали, что карбоангидраза является высоко-
специфичным ферментом, катализирующим только одну реак-
цию— обратимую гидратацию СОг [уравнение (1)]. В последние
годы были найдены и другие реакции, катализируемые этим фер-
ментом. к ним относятся гидролиз п-нитрофенилацетата [24—27]
NO2 + Н* (3)
Н* <4)
о
'I
сн3-с-н + н2о
он
I
сн3—с—н
он
(5)
612
Глава 16
{уравнение (3)], гидролиз сультона 2-окси-5-нитро-а-толуолсуль-
фокислоты [28—30] [уравнение (4)] и гидратация ацетальдегида
|[31—33] [уравнение (5)], а также некоторых других альдегидов
[138, 139].
Гидролиз сультона катализируется ферментом исключительно
эффективно [28—30]. Число оборотов меняется от 600 до
20 000 молей субстрата/мин на 1 моль фермента в зависимости от
природы изофермента, pH, присутствия или отсутствия ингиби-
рующих анионов и органического растворителя [30, 41]. Такая
большая скорость реакции (4), особенно если сравнить с относи-
тельно медленным гидролизом л-нитрофенилацетата (от 6 до
20 молей/мин на 1 моль фермента), по-видимому, объясняется
специфическим характером взаимодействия с активным центром,
так как структура сультона похожа на структуры сильных суль-
фамидных ингибиторов (разд. 4).
Физиологическое значение реакций (3) —(5) в настоящее вре-
мя неизвестно. Важность же обратимой гидратации СОг оче-
видна. В водных растворах эта реакция протекает относительно
быстро и без помощи ферментативного катализа. Кинетическая
схема этого процесса, предложенная Эйгеном и сотр. [140]
[уравнение (6)], имеет то преимущество, что она учитывает непо-
средственное равновесие СО2 с НСО3 и Н2СО3.
н* + нсо3_ < н2со3
fe2l
Кинетические .константы уравнения (1) следующим образом вы-
ражаются через параметры этой схемы:
= ^31 “Ь ^32 И k—1 = ^13^0 “Ь ^23 (7)
где Ка — истинная константа диссоциации угольной кислоты,
рК=3,6. Константа первого порядка для скорости гидратации
—d [СО2]
dT
К
(8)
определена в некоторых работах. Сообщались значения: 0,03 с-1
[141], 0,0358 с-1 ,[142], 0,0375 с"1 [143] для 25°С и 0,0434 с-1
[114] для 37 °C. Лучшее значение для константы скорости деги-
дратации k_b по-видимому, составляет 15 с-1 [142, 143] при 25 °C.
В присутствии фермента гидратация СО2 осуществляется в
Ю7 раз быстрее, чем без катализатора (см. ниже). При рН>10
Карбоангидраза
613
преобладает, однако, реакция между ОН- и СОг, и константа
скорости этой реакции второго порядка
d [СО,]
= dF ~/[ОН] [СО2] (9)
составляет примерно 8500 л •моль-1-с-1, а&_2=2-10-4 с-1 [141].
Это может быть важным для механизма действия фермента
(см. ниже).
Расчеты, основанные на учете физиологических факторов, та-
ких, как время циркуляции крови по капиллярам легочных аль-
веол, первоначально подтверждали полную недостаточность ско-
рости самопроизвольного (без катализатора) установления рав-
новесия между НСОз и СОг для необходимого освобождения эри-
троцитов от углекислого газа за один проход капилляров [,144,
145]. Те же расчеты предсказывали быструю гибель организма
при полном ингибировании карбоангидразы [146]. Данные, полу-
ченные в последнее время, говорят о менее катастрофических по-
следствиях [147], однако ясно, что карбоангидраза играет наи-
более значительную роль в быстром установлении равновесия
НСОз *^СО2 (обзор по этой проблеме см. [109]).
Карбоангидраза играет важную роль и в других тканях — в
почках, глазах, поджелудочной железе, желудке, где она косвен-
но участвует в транспорте ионов. Ее роль в этих процессах заклю-
чается в том, что она катализирует реакции с освобождением или
поглощением ионов Н+ или ОН-. Эти ионы участвуют в различ-
ных клеточных транспортных процессах, которые могут заканчи-
ваться секрецией бикарбоната (поджелудочная железа) или кис-
лоты (желудок). Физиологические, фармакологические и химиче-
ские аспекты карбоангидразы рассмотрены в обстоятельном и эле-
гантном обзоре Марена [109]. Целью же этого раздела является
суммирование физико-химических данных, непосредственно отно-
сящихся к механизму действия чистых препаратов фермента.
Изофермент С (разд. 2) присутствует в эритроцитах человека
и обезьяны в гораздо меньших количествах, чем изофермент В,
однако первый намного активнее [47]. Удельная активность, вы-
раженная в единицах, равных количеству фермента, необходимо-
му для гидратации 1 мкмоля СО2 в 1 мин при 25 °C, составляет
44 000 ед./мг белка для В-формы и 1300 000 ед./мг белка для
С-формы [47]. Последняя цифра соответствует числу оборотов
6-105с-1 или 3,6* 107 мин-1. Это позволяет считать гидратацию
СОг карбоангидразой самой быстрой из всех известных фермен-
тативных реакций — факт, который нужно обязательно учитывать
при сопоставлении гипотез о механизме действия (см. ниже).
Кривая pH-зависимости скорости карбоангидразной реакции
имеет простую сигмоидальную форму, соответствующую иониза-
ции группы с рКа, величина которой в зависимости от ионного
614
Глава 16
окружения, буфера, субстрата и природы изоферментов состав-
ляет 6,9—8,4 [26, 29, 31, 42, 86, 100, 101]. Эта величина р/Са
очень чувствительна к анионному окружению [101], что может
объяснить некоторые из наблюдавшихся расхождений. Данные,
полученные методом ЯМР на активном кобальтовом препарате
Е125], а также результаты комплексометрического титрования
86] позволяют отнести это значение к координированной у ме-
талла молекуле воды (разд. 5). Правда, эта величина несколько
меньше, чем свойственно аквокомплексам металлов, однако это
можно объяснить воздействием других лигандов.
Привлекательной гипотезой о действии фермента является
механизм, показанный на схеме (10). Он основан на представле-
нии об атаке координированного гидроксила на молекулу СОг
[86, 105, 148—150].
(Ю)
Этот механизм объясняет отсутствие ингибирования слабо связы-
вающимися анионами при высоких значениях pH [101], возвра-
щение спектральной картины комплексов Со (II)-карбоангидразы
с анионами или сульфамидами к типичным спектрам щелочной
формы неингибированного фермента, наблюдаемое также при вы-
соких значениях pH [101], и смещение pH-зависимости в щелоч-
ную область в присутствии анионов [101]. Все эти явления
вызваны конкуренцией с ОН-, который при достаточно больших
концентрациях вытесняет анионы и реактивирует фермент. Этот
механизм согласуется и с кинетическими данными, свидетельст-
вующими о том, что в месте связывания анионов находится груп-
па, основная форма которой существенна для гидратации СОг,
а кислая форма необходима для дегидратации бикарбоната
[99, ПО]. Инфракрасные спектры комплекса карбоангидразы с
Карбоангидраза
615
СОг при pH 5,0 указывают на то, что последний связывается с
гидрофобной полостью в неизменном (линейном) виде [105].
Спектральные данные также свидетельствуют об образовании
комплекса Со(II)-карбоангидразы с бикарбонатом при больших
равновесных концентрациях последнего при pH 8,2 [73, 119].
При pH 7 и 25 °C константа первого порядка для стадии фер-
ментативной гидратации равна 4-105—6-105 с-1 [47]. Как уже
отмечалось, это на семь порядков выше, чем для неферментатив-
ной гидратации СОг, но только на два порядка больше, чем для
самопроизвольной реакции между ОН- и СО2 при рН>10, где
^2=8,5-103 л-моль-1 •с-1. Уонг [150] ввел константу скорости
реакции псевдопервого порядка для гипотетической системы, в ко-
торой ОН- помещается рядом с молекулой СОг. Значение Этой
константы равно 4-10-5с-1, что по порядку совпадает с величи-
ной, свойственной карбоангидразной реакции. Снижение значе-
ния рКа для диссоциации Н2О с 15,7 до 7—8, конечно, удовлет-
воряло бы требованию о преобладании реакции между ОН- и
СО2 в области нейтральных pH. Уонг сделал также и другой вы-
вод, основанный на определенных нуклеофильных свойствах сво-
бодного и координированного гидроксильного иона. Хотя отноше-
ние нуклеофильности двух групп может и не совпадать с отно-
шением их констант диссоциации, однако различие в этих
константах не может составлять несколько порядков. Если посту-
лированный механизм соответствует действительности, то, соглас-
но этим выводам, нуклеофильность иона ОН- не должна в ре-
зультате координирования уменьшаться до степени, соответствую-
щей указанному снижению р/Са. В противном случае реакция
ускоряется благодаря каким-то другим особенностям активного
центра.
На первый взгляд описанная выше схема (10) кажется
вполне естественной. Однако при более глубоком анализе выяв-
ляются некоторые ее «узкие места». К ним прежде всего отно-
сится вопрос о скорости переноса протонов. Необходимо, чтобы
она соответствовала очень высокой скорости ферментативной реак-
ции. Для одного из протонов, переносимых на стадии образова-
ния бикарбонатного интермедиата (fe2), переход происходит между
соседними атомами кислорода. В этих условиях транспорт прото-
на действительно может быть исключительно быстрым, хотя о
точном его механизме, последовательности и природе промежу-
точных состояний можно говорить только предположитель-
но [150].
Более серьезные осложнения возникают при рассмотрении
второго протона, переносимого к растворителю. На схеме (10) —
это регенерация активной формы Zn—ОН-. Согласно модельным
системам, предельные значения скорости транспорта протона,
контролируемой диффузией, приближаются к 1010—10п лХ
П6
Глава 16
Кмоль-1-с-1 [151]. Поскольку Н3О+— сильная кислота, а ОН-—
сильное основание, то выигрыш свободной энергии будет наблю-
даться, если переход осуществляется при условии рЛн3о+ С рК на
или рЛн2о 3>рЛна, где А — акцептор. Только в этом случае ско-
рость переноса протона приближается к диффузионной.
Эйген [151] представил зависимость 1g fe (константа скорости
транспорта протона) от ДрК (разница в значениях р/С для донора
и акцептора). По этим данным, скорость процесса только тогда
лимитируется диффузией, когда рК акцептора на 2—3 единицы
больше, чем у донора. А если рК донора выше, чем у акцептора,
lg fe линейно уменьшается с увеличением ДрК. Используя наи-
меньшее значение для рКо (~7), можно заключить, что переход
протона к молекуле Н2О из раствора требует значения p/С акцеп-
тора для Н3О+ около —1,7. Следовательно, для прямого перехода
протона от координированной у цинка молекулы воды в раствор
необходимо, чтобы ДрК«—9. Поэтому даже при самых благо-
приятных обстоятельствах А не может быть больше 102—103 с-1.
Тем не менее наблюдаемая на опыте константа скорости для фер-
ментативной реакции псевдопервого порядка составляет 105 с~1.
Некоторые исследователи предполагают транспорт протона от
соседнего имидазольного ядра с образованием иона имидазолия
[26, 27, 31—33]. Однако и в этом случае вопрос не снимается,
так как передача имидазольного протона к воде из раствора
также происходит с константой скорости порядка 103 с-1 [151].
На основании изучения гидролиза эфиров сульфокислот Кай-
зер и Лоу [29] предложили гипотезу о циклическом механизме, в
котором отсутствует перенос протона к растворителю:
Гидролиз этих субстратов описывается сигмоидальными кривыми
с рКа, равным 7,28—7,8 (в зависимости от природы изофермента)
[29, 42]. В своей схеме Кайзер и Лоу приписывают это значение
координированной воде. Для завершения цикла включена допол-
нительная молекула Н2О из раствора.
Подобная схема позволяет обойти вопрос о транспорте протона
и в реакции гидратации. При этом надо только предположить
промежуточное образование не бикарбоната, а угольной кислоты:
Карбоангидраза
617
-Zn2+— ОН + Н2СО3
НСО3“+Н+ (12)
Поскольку для Н2СО3 рК=3,6, то .последующий переход протона
от угольной кислоты к растворителю вряд ли затруднен. Коор-
динация возле иона цинка, естественно, способствует усилению
кислотности протонов воды.
Однако и эта гипотеза .встречает возражения, основанные на
том, что при обратной реакции (дегидратации) субстратом долж-
на быть угольная кислота. Следовательно, необходимо учитывать
скорость протонирования НСОз. Некоторые данные указывают на
то, что скорость этого протонирования недостаточна для протека-
ния реакции по первому порядку по ферменту при больших его
концентрациях [152]. Кроме того, расчеты показывают, что даже
предельные (диффузионные) значения скорости взаимодействия
угольной кислоты с ферментом оказываются слишком малыми по
сравнению с реально наблюдаемыми [100, 152] (.скорости гидрата-
ции и дегидратации при pH 7,05 примерно одинаковы [47, 100]).
Де Во и Кистяковский [153] еще раньше представили кинетиче-
ские данные против участия в реакции Н2СО3 в качестве суб-
страта.
Кэплоу [152] предложил согласованную реакцию с разрывом
связи С—О для механизма дегидратации:
—Zn—ОН + со2 + Н2О
(13)
Разрыв связи С—О наблюдается в модельных реакциях декарбо-
ксилирования с участием пентааминовых соединений кобаль-
та (III) [154] и уже обсуждался в связи с возможным механиз-
мом действия карбоангидразы [73]. При всей своей привлека-
тельности эта схема все же не разрешает вопроса о транспорте
протона на стадии гидратации.
Деннард и Уилльямс [98] предполагают участие белкового
азота в атаке на углерод СО2. При этом в качестве интермедиа-
та образуется нестабильный карбамат VIII
618
Глава 16
N-" | VN
VIII
Однако, согласно известным механизмам распада карбаматов,
реакция может не пойти в направлении образования нужных про-
дуктов [152].
Итак ясно, что центральной проблемой при составлении гипо-
тез о механизме реакции гидратации — дегидратации в настоящее
время является вопрос о переносе протона. Его скорость должна
быть лимитирующей. Эстеразные реакции значительно медлен-
нее. Хотя они и могут протекать по тому же механизму, однако
заранее нельзя быть уверенным в абсолютном сходстве всех
деталей.
В проведенном выше анализе допускалось, что в ферментатив-
ном процессе механизм переноса протона такой же, как и в мо-
дельных системах. Однако нельзя забывать и о том, что свое
влияние на величину скорости протонного перехода может ока-
зывать и специфический (например, льдообразный) характер мо-
лекулярной структуры водной фазы вблизи поверхности белка.
В этом случае скорость передачи протона может отличаться от
величины, наблюдавшейся в простых системах [151, 155]. В связи
с этим весьма интересен тот факт, что предварительное рассмот-
рение карт электронной плотности, по-видимому, указывает на
присутствие рядом с активным центром высоко структурированной
группы молекул растворителя (рис. 16.20,В).
ДОПОЛНЕНИЕ, ВНЕСЕННОЕ В КОРРЕКТУРУ
Пока рукопись данной книги готовилась к печати, были опуб-
ликованы новые работы, посвященные исследованию карбоанги-
дразы; они относятся к структуре фермента в растворе и в кри-
сталлическом состоянии, субстратной специфичности и кинетике.
Ниже приведен список этих публикаций с названиями работ.
Taylor J. S„ Coleman J. Е., «Electron spin resonance of metallo-carbonic an-
hydrases», J. Biol. Chem., 246, 7058 (1971).
Kannan К. K- et al., «Crystal structure of human erythrocyte carbonic anhydra-
se С. VI. The three-dimensional structure at high resolution in relation to other mam-
malian carbonic anhydrases», Cold Spring Harbor Symp. Quant. Biol., 36, 221 (1972)-
Карбоангидраза
619
Lindskog S., Henderson L. E., Kannan K. K-, Liljas A., Nyman P. O., Strand-
berg B., «Carbonic anhydrase», in «The Enzymes», Vol. V, 1971, p. 587.
Khalifah R. G„ «The carbon dioxide hydration activity of carbonic anhydra-
se. I. Stop-flow kinetic studies on the native human isoenzymes В and С», J. Biol.
Chem., 246, 2561 (1971).
Khalifah R. G., Edsall J. T., «Carbon dioxide activity of carbonic anhydrase:
kinetics of alkylated anhydrases В and C from humans», Proc. Natn. Acad. Sci.
U. S„ 69, 172 (1972).
Taylor P. W., King R. W., Burgen A. S. V., «Kinetics of complex formation bet-
ween human carbonic anhydrases and aromatic sulfonamides», Biochemistry, 9, 2638
(1970).
Taylor P. W., King R- W., Burgen A. S. V., «Influence of pH on the kinetics
of complex formation between aromatic sulfonamides and human carbonic anhydra-
se», Biochemistry, 9, 3894 (1970).
Taylor P. W., Burgen A. S. V., «Kinetics of carbonic anhydrase-inhibitor com-
plex formation. A comparison of anion- and sulfonamide-binding mechanisms», Bio-
chemistry, 10, 3859 (1971).
Mushak P., Coleman J. E., «Electron spin resonanse studies of spin-labeled car-
bonic anhydrase», J. Biol. Chem., 247, 373 (1972).
Hower I. F., Henkens R. W., Chestnut D. В., «А spin label investigation of the
active site of an enzyme. Bovine carbonic anhydrase», J. Am. Chem. Soc., 93, 6665
(1971).
Packer Y., Beug M. W., Ainardi V. R„ «Coprecipitation of carbonic anhydrase
by 1,1 -bis (p-chlorophenyl) -2,2,2-trichloroethane, 1,1-bis (p-chlorophenyl) -2,2-dichlo-
roethylene, and dieldrin», Biochemistry, 10, 1390 (1971).
Lanir A., Navon G., «Nuclear magnetic resonance studies of bovine carbonic
anhydrase. Binding of sulfonamides to the zinc enzyme», Biochemistry, 10, 1024
(1971).
Taylor P. W., Feeney J., Burgen A. S. V., «Investigation of the mechanism of
ligand binding with cobalt(II) human carbonic anhydrase by *H and 19F nuclear
magnetic resonance spectroscopy», Biochemistry, 10, 3866 (1971).
Pocker Y., Watamori N., «Catalytic versatility of erythrocyte carbonic anhydra-
se. IX. Kinetic studies of the enzyme-catalyzed hydrolysis of 3-pyridyl and nitro-3-
pyridyl acetates», Biochemistry, 10, 4843 (1971).
Tanis R. I., Tashian R. E., «Purification and properties of carbonic anhydrase
from sheep erythrocytes», Biochemistry, 10, 4852 (1971).
Pocker Y., Guilbert L. I., «Catalytic versatility of erythrocyte carbonic anhydra-
se. Kinetic studies of the enzyme-catalysed hydrolysis», Biochemistry, 11, 180
(1972).
Henkens R. W.. Sturtevant J. M., «Extrinsic Cotton effects in a metal chelator —
bovine carbonic anhydrase complex», Biochemistry, 11, 206 (1972).
Cohen J. S„ Yim С. T., Kandel M., Gornall A. G., Kandell S. I., Freedman M. H.,
«Studies of the histidine residues of carbonic anhydrases using high-field proton
magnetic resonance», Biochemistry, 11, 327 (1972).
Pocker Y., Beug M. W., «Kinetic studies of bovine carbonic anhydrase-catalyzed
hydrolyses of para-substituted phenyl esters», Biochemistry, 11, 698 (1972).
Yazgan A., Henkens R. W., «Role of zinc(II) in the refolding of guanidine
hydrochloride denatured bovine carbonic anhydrase». Biochemistry. 11, 1314 (1972).
Coleman J. E„ Coleman R. V., «Magnetic circular dichroism of Co(II) carbonic
anhydrase», J. Biol. Chem., 247, 4718 (1972).
A group of papers on carbonic anhydrase is published in «Oxvgen affinity of
haemoglobin and red cell acid-base status», Alfred Benzon 'Symposium IV,
Munksgaard. Copenhagen, 1972.
Whitney P. L. «Inhibition and modification of human carbonic anhydrase В with
bromoacetate and iodoacetamide», Eur. J. Biochem., 16, 126 (1970).
Henkart P., Dorner F., «The dinitrophenylation of human carbonic anhydra-
se В», J- Biol. Chem., 246, 2714 (1971).
Глава 16
Funakoshi S., Deutsch H. F., «Human carbonic anhydrases. VI. Levels of isozy-
mes in old and young erythrocytes and in various tissues», J. Biol. Chem., 246,
1088 (1971).
Dorner F., «Human carbonic anhydrase B. Location of tyrosine residues that
react with tetranitromethane», J. Biol. Chem., 246, 5896 (1971).
СПИСОК ЛИТЕРАТУРЫ
1. Brinkman R., Margaria R., Meldrum N. V., Roughton F. J. W„ J. Physiol. (Lon-
don), 75, 3 (1932).
2. Meldrum N. V., Roughton F. J. W., J. Physiol. (London), 75, 4 (1932).
3. Meldrum N. V., Roughton F. J. W., J. Physiol. (London), 75, 15 (1932).
4. Meldrum N. V., Roughton F. J. W., J. Physiol. (London), 80, 113 (1933).
5. Keilin D., Mann T., Biochem., J., 34, 1163 (1940).
6. Keilin D., Mann T., Nature, 153, 107 (1944).
7. Eirich F. R., Rideal E. K, Nature, 146, 107 (1944).
8. Petermann M. N., Hakala N. V., J. Biol. Chem., 145, 701 (1942).
9. Smith E. С. B., Biochem. J., 34, 1176 (1940).
10. Scott D. A., Mendive J. R., J. Biol. Chem., 140, 445 (1941).
11. Hove E., Elvehjem C. A., Hart E. B., J. Biol. Chem., 136, 425 (1940).
12. Davenport H. W., J. Physiol. (London), 97, 32 (1939).
13. Webb E. C„ van Heyingen R., Biochem. J., 41, 74 (1946).
14. Bradfield J. R. G„ Nature, 159, 467 (1947).
15. Day R., Franklin I., Science, 104, 363 (1946).
16. Sibly P. M„ Wood J. G., Aust. J. Sci. Res., B4, 500 (1951).
17. Neish A. C., Biochem. J., 33, 300 (1939).
18. Wood J. G„ Sibly P. M„ Aust. J. Sci. Res., 5, 244 (1952).
19. Mann T., Keilin D., Nature, 146, 164 (1940).
20. Lindskog S., J. Biol. Chem., 238, 945 (1962).
21. Coleman J. E., Nature, 214, 193 (1967).
22. Fridborg K-, Kannan К. K-, Liljas A., Lundin J. et al., J. Mol. Biol., 25, 505-
(1967).
23. Liljas A., Kannan К. K-, Bergsten P. C., Waara L, Fridborg K. et al., Nature,.
235, 131 (1972).
24. Schneider F„ Lieflander M., Z. Physiol. Chem., 334, 279 (1963).
25. Tashian R. E., Plato С. C., Shows T. B., Science, 140, 53 (1963).
26. Packer Y., Stone I. T., J. Am. Chem. Soc., 87, 5497 (1965).
27. Pocker Y., Stone J. T., Biochemistry, 7, 2936 (1968).
28. Lo К-W., Kaiser E. T., Chem. Commun., 1966, 834.
29. Kaiser E. T„ Lo K.-W., J. Am. Chem. Soc., 91, 4912 (1969).
30. Coleman J. E., J. Biol. Chem., 243, 4574 (1968).
31. Pocker Y„ Meany J. E., Biochemistry, 4, 2535 (1965).
32. Pocker Y., Meany J. E., Biochemistry, 6, 239 (1967).
33. Pocker Y„ Dickerson D. G„ Biochemistry, 7, 1995 (1968).
34. Lindskog S., Biochim. Biophys. Acta, 39, 218 (1960).
35. Laurent G., Marriq C., Nahon D., Charrel M., Derrien Y., Compt. Rend. Soc.
Biol., 156, 1456 (1962).
36. Nyman P. O., Biochim. Biophys. Acta, 52, 1 (1961).
37. Rickli E. E., Chazanfar S. A. S., Gibbons В. H., Edsall J. T., J. Biol. Chem.,.
239, 1065 (1964).
38. Laurent G., Charrel M., Luccioni F., Autran M. F., Derrien Y., Bull. Soc. Chim.
Biol., 47, 1101 (1965).
39. Duff T. A., Coleman J. E., Biochemistry, 5, 2009 (1966).
40. Byvoet P., Gotti A., Mol. Pharmacol., 3, 142 (1967).
41. Furth A. J. Biol. Chem., 243, 4832 (1968).
42. Maynard J., Coleman J. E„ J. Biol. Chem., 246, 4455 (1971).
Карбоангидраза
621
43. Armstrong J. McD., Myers D. V., Verpoorte J. A., Edsall J. T., J. Biol. Chem.,
241, 5137 (1966).
44. Strandberg B., Tilander B., Fridborg K-, Lindskog S., Nyman P. O., J. Mol.
Biol., 5, 583 (1962).
45. Tilander B., Strandberg B., Fridborg K, J. Mol. Biol., 12, 740 (1965).
46. Tashian R. E„ Am. J. Human Genet., 17, 257 (1965).
47. Gibbons В. H„ Edsall J. T„ J. Biol. Chem., 239, 2539 (1964).
48. Nyman P. O., Lindskog S„ Biochim. Biophys. Acta, 85, 141 (1964).
49. Laurent G., Derrien Y., in R. E. Forster, J. T. Edsall, A. B. Otis, F. J. W. Rough-
ton (eds.), CO?: Chemical, Biochemical and Physiological Aspects, NASA
Symp,. SP-188, Washington, D. C., 1969, p. 115.
50. Shows T. B„ Biochem. Genet., 1, 171 (1967).
51. Fellner S. K, Biochim. Biophys. Acta, 77, 155 (1963).
52. Rossi C., Chersi A., Cortivo M., in R. E. Forster, J. T. Edsall, A. B. Otis,
F. J. W. Roughton (eds.), CO2: Chemical, Biochemical and Physiological
Aspects, NASA Symp., SP-188, Washington, D. C., 1969, p. 131.
53. Tobin A. J., in R. E. Forster, J. T. Edsal, A. B. Otis, F. J. W. Roughton (eds.),
CO2: Chemical, Biochemical and Physiological Aspects, NASA Symp., SP-188,
Washington, D. C„ 1969, p. 131.
54. Tobin A. J., J. Biol. Chem., 245, 2656 (1970).
55. Whitney P. L., in R. E. Forster, J. T. Edsall, A. B. Otis, F. J. W. Roughton (eds.),
CO2: Chemical, Biochemical and Physiological Aspects, NASA Symp., SP-188,
Washington, D. C., 1969, p. 109.
56. Nyman P. 0., in R. E. Forster, J. T. Edsall, A. B. Otis, F. J. W. Roughton
(eds.), CO2: Chemical, Biochemical and Physiological Aspects, NASA Symp.,
SP-188, Washington, D. C., 1969, p. 109.
57. Nyman P. O„ Strid L., Westermark G., Biochim. Biophys. Acta, 122, 554
(1966).
58. Andersson B„ Gothe P. O., Nilsson T„ Nyman P. 0., Strid L., Eur. J. Biochem.,
6, 190 (1968).
59. Nyman P. 0., Strid L., Westermark G., Eur. J. Biochem., 6, 72 (1968).
60. Andersson B., Nyman P. O., Strid L., in R. E. Forster, J. T. Edsall, A. B. Otis,
F. J. W. Roughton (eds.), CO2: Chemical, Biochemical and Physiological
Aspects, NASA Symp., SP-188, Washington, D. C., 1969, p. 109.
61. Henderson L. E., in R. E. Forster, J. T. Edsall, A. B. Otis and F. J. W. Rough-
ton (eds.), CO2: Chemical, Biochemical and Physiological Aspects, NASA
Symp., SP-188, Washington, D. C., 1969, p. 109.
62. Marriq C., Gignoux D., Laurent G„ Compt. Rend. Acad. Sci., Paris, 260, 1810
(1965).
63. Laurent G., Marriq C., Garcon D„ Luccioni F., Derrien Y„ Bull. Soc. Chim.
Biol., 49, 1035 (1967).
64. Phillips D. M. P„ Biochem. J., 86, 397 (1963).
65. Bradbury S. L., J. Biol. Chem., 244, 2002 (1969).
66. Bradbury S. L., J. Biol. Chem., 244, 2010 (1969).
67. Whitney P. L„ Folsch G., Nyman P. O., Malmstrom B. G„ J. Biol. Chem., 242,
4206 (1967).
68. Whitney P. L., Nyman P. O., Malmstrom B. G., J. Biol. Chem., 242, 4212
(1967).
69. Riddiford L. M., J. Biol. Chem., 239, 1079 (1964).
70 Riddiford L. M., Stellwagen R. H., Mehta S., Edsall J. T„ J. Biol. Chem.,
240, 3305 (1965).
71. Nilsson A., Lindskog S., Eur. J. Biochem., 2, 309 (1967).
72. Myers D. B., Edsall J. T., Proc. Natn. Acad. Sci. U. S., 53, 169 (1965).
73. Coleman J. E., Biochemistry, 4, 2644 (1965).
74 Beychok S., Armstrong J. M„ Lindblow C., Edsall J. T., J. Biol. Chem., 241,
5150 (1966).
75. Rosenberg A., J. Biol. Chem., 241, 5126 (1966).
622
Глава 16
76. Edsall J. T„ Harvey Leet Ser., 62, 191 (1968).
77. Edsall J. T„ Ann. N. Y. Acad. Sci., 151, 41 (1968).
78. Wong K. P-, Tanford C., Federation Proc., 29, 335 Abstr. (1970).
79. Greenfield N., Fasman G. D., Biochemistry, 8, 4108 (1969).
80. Myer Y. P., Biophys. J., 9, A-215 (1969); J. Chem. Path. Pharm., 1, 607
(1970).
81. Tupper R., Watts R. W. E„ Wormall A., Biochem. J., 50, 429 (1952).
82. Lindskog S., Malmstrom B. G„ J. Biol. Chem., 237, 1129 (1962).
83. Rickli E. E„ Edsall J. T„ J. Biol. Chem., 237, PC 258 (1961).
84. Lindskog S., Nyman P. O., Biochim. Biophys. Acta, 85, 462 (1964).
85. Tilander B., Strandberg B., Fridborg K, J. Mol. Biol., 12, 740 (1965).
86. Coleman J. E., J. Biol. Chem., 242, 5212 (1967).
87. Henkens R. W., Sturtevant J. M., J. Am. Chem. Soc., 90, 2669 (1968).
88. Holyer R. H., Hubbard C. D., Kettle S. F. A., Wilkins R. G., Inorg. Chem., 4,
929 (1965).
89. Holyer R. H„ Hubbard C. D„ Kettle S. F. A., Wilkins R. G., Inorg. Chem., 5,
622 (1966).
90. Vallee B. L., Coleman J. E., Comprehensive Biochemistry, Vol. 12, 1964,
p. 165.
91. Coleman J. E., Ph. D. Dissertation, Massachusetts Institute of Technology,
1963.
92. Коттон Ф., Уилкинсон Дэю., Современная неорганическая химия, тт. 1—3,
«Мир», М., 1969.
93. Ciampolini М., Structure and Bonding, 6, 52 (1969).
94. Sacconi L„ Orioli P. L., DiVaira M., J. Am. Chem. Soc., 87, 2059 (1965).
95. Sacconi L., Nannelli P., Nardi N., Campigll U., Inorg. Chem., 4, 943 (1965).
96. Dori Z., Gray H. B., J. Am. Chem. Soc., 88, 1394 (1966).
97. Holt E. M„ Holt S. L„ Watson K. L, J. Am. Chem. Soc., 92, 2721 (1970).
98. Dennard A. C., Williams R. I., Trans. Metal Chem., 2, 115 (1966).
99. Kemohan J. C„ Biochim. Biophys. Acta, 81, 346 (1964).
100. Kemohan J. C., Biochim. Biophys. Acta, 96, 304 (1965).
101. Lindskog S., Biochemistry, 5, 2641 (1966).
102. Coleman J. E., Proc. Natn. Acad. Sci. U. S., 59, 123 (1968).
103. Chen R. F., Kemohan J. C„ J. Biol. Chem., 242, 5813 (1967).
104. Wilbur К. M„ Anderson N. G„ J. Biol. Chem., 176, 147 (1948).
105. Riepe M. E., Wang L H„ J. Biol. Chem., 243, 2779 (1968).
106. Verpoorte J. A., Mehta S„ Edsall J. T., J. Biol. Chem., 242, 4221 (1967).
107. Coleman J. E„ in E. T. Kaiser, Kedzy (eds.), Progress in Bioorganic Che-
mistry, Vol. 1, Interscience, New York, 1970, pp. 159—344.
108. Sillen L. G., Martell A. E., Stability Constants of Metal-Ion Complexes, The
Chemical Society, London, Special Publication, No. 17, 1964.
109. Maren T. H., Physiol. Rev., 47, 595 (1967).
110. Bar D., Actualities Pharmacol., 15, 1 (1964).
111. Maren T. H., Robinson B., Palmer R. F., Griffith M. E., Biochem. Pharmacol.,
6, 21 (1960).
112. Maren T. H., Parcell A. L., Malik M. N., J. Pharm. Exp. Therapeut., 130, 389
(1960).
113. Maren T. H., J. Pharm. Exp. Therapeut., 139, 129 (1963).
114. Maren T. H., J. Pharm. Exp. Therapeut., 139, 140 (1963).
115. Lindskog S., Thorslund A., Eur. J. Biochem., 3, 453 (1968).
116. Kemohan J. C., Biochim. Biophys. Acta, 118, 405 (1966).
117. Kemohan J. C„ Biochem. J., 98, 31 (1966).
118. Maren T. H., personal communication.
119. Coleman J. E., in R. E. Forster, J. T. Edsall, A. B. Otis, F. J. W. Roughton
(eds.), CO2: Chemical, Biochemical and Physiological Aspects, NASA Symp.
SP-188, Washington, D. C., 1969, p. 141.
120. Coleman J. E., J. Am. Chem. Soc., 89, 6757 (1967)
Карбоангидраза
623
121. Moskowitz A., Proc. R. Soc., A297, 16 (1967).
122. Ward R. L„ Biochemistry, 8, 1879 (1969).
123. Ward R. L., Biochemistry, 9, 2447 (1970).
124. Galley W. C., Stryer L., Proc. Natn. Acad. Sci. U. S., 60, 108 (1968).
125. Fabry M. E., Koenig S. H., Schillinger W. E., J. Biol. Chem., 245, 4256
(1970).
126. Hamilton C. L., McConnell H. M., in A. Rich, N. Davidson (eds.), Structural
Chemistry and Molecular Biology, Freeman, San Francisco, 1968, p. 115.
127. Griffith О. H., Waggoner A. S., Accounts Chem. Res., 2, 17 (1969).
128. Stone T. J., Buckman T., Nordio P. L., McConnell H. M., Proc. Natn. Acad.
Sci. U. S„ 54, 1010 (1965).
129. McConnell H. M., Hamilton C. L., Proc. Natn. Acad. Sci. U. S., 60, 776
<1968).
130. Hubbell W. L., McConnell H. M., Proc. Natn. Acad. Sci. U. S., 63, 16
(1969).
131. Rozantzev E. G., Neiman M. B., Tetrahedron, 20, 131 (1964).
132. Rozantzev E. G., Krinitzkaya L. A., Tetrahedron, 21, 491 (1965).
133. Mushak P„ Coleman J. E„ unpublished data.
134. Itzkowitz M. S., Ph. D. Thesis, California Institute of Technology, 1966.
135. Lindskog S., Ehrenberg A., J. Mol. Biol., 24, 133 (1967).
136. Lipscomb W. N„ Hartsuck J. A., Reeke G. N„ Quiocho F. A. et al., in «Structu-
re Function and Evolution of Proteins», Brookhaven Symp. Biol., 21, 24
(1969).
137. Lipscomb W. H., Hartsuck J. A., Quiocho F. A., Reeke G. N., Proc. Natn. Acad.
Sci. U. S„ 64, 28 (1969).
138. Pocker Y., Storm D. R., Biochemistry, 7, 1202 (1968).
139. Pocker Y., Stone J. T., Biochemistry, 7, 3021 (1968).
140. Eigen M., Kustin K., Moss G., Z. Phys. Chem., 30, 130 (1961).
141. Kern D. M„ J. Chem. Educ., 37, 14 (1960).
142. Ho C„ Sturtevant J. M„ J. Biol. Chem., 238, 3499 (1963).
143. Gibbons В. H., Edsall J. T., J. Biol. Chem., 238, 3502 (1963).
144. Roughton F. I. W., Physiol. Rev., 15, 241 (1935).
145. Roughton E. I. W., Harvey Leet. Ser., 39, 96 (1943).
146. Davies R. E., Biol. Rev. Cambridge Phil. Soc., 26, 87 (1951).
147. Cain S. M., Otis A. B„ J. Appl. Physiol., 16, 1023 (1961).
148. Davis R. P., J. Am. Chem. Soc., 60, 5209 (1958).
149. Davis R. P., in P. Boyer, H. Lardy, K. Myrback (eds.), The Enzymes, Vol. 5,
Academic Press, New York, 1961, p. 545.
150. Wang L H., Science, 161, 328 (1968).
151. Eigen M„ Discuss. Faraday Soc., 39, 7 (1965).
152. Caplow M„ J. Am. Chem. Soc., 93, 230 (1971).
153. DeVoe H„ Kistiakowsky G. B., J. Am. Chem. Soc., 83, 274 (1961).
154. Sargeson A. M„ in F. P. Dwyer, D. P. Mellor (eds.), Chelating Agents and
Metal Chelates, Academic Press, New York, 1964, p. 183.
155. Eigen M., Wilkins R. G., Mechanisms of Inorganic Reactions, Adv. Chem. Ser.,
No. 49, American Chemical Society, Washington, D. C., 1965, p. 55.
156. Coleman J. E., unpublished data.
157. Malmstrom B. G., personal communication, 1971.
ГЛАВА 17
ФОСФАТНЫЙ ПЕРЕНОС
И ЕГО АКТИВАЦИЯ ИОНАМИ МЕТАЛЛОВ;
ЩЕЛОЧНАЯ ФОСФАТАЗА
Т. Г. Спиро
Spiro Т. G., Department of Chemistry, Princeton University,
Princeton, New Jersey, USA
1. ФОСФАТЫ и биоэнергетика
Можно утверждать, что фосфор является пятым из важнейших
для биологии элементов вслед за углеродом, водородом, кислоро-
дом и азотом. Значительный интерес для химиков-неоргаников
представляет тот факт, что, в то время как четыре основных эле-
мента входят в огромное множество сложных органических моле-
кул, биохимия фосфора ограничивается главным образом произ-
водными иона ортофосфата. Подавляющее большинство этих
производных являются просто эфирами и ангидридами фосфорной
кислоты, хотя существуют и такие, в которых есть связи между
фосфором и азотом или серой. Интересно также то, что биохимия
фосфора в основном тесно связана с ионами двухвалентных ме-
таллов. Можно полагать, что фосфаты и их производные, а также
их взаимодействия с ионами металлов составляют раздел неор-
ганической химии, однако вклад химиков-неоргаников в эту
область в действительности довольно скромен. Хотя им и принад-
лежит несколько превосходных работ по химии фосфатов, их ин-
терес к этим соединениям невелик. В биохимическом же мире,
напротив, по крайней мере три десятилетия после того, как Лип-
ман :[;1—3] указал на их важность, они находятся в центре
внимания.
Фосфаты играют две ключевые роли в биологии. Во-первых,
они служат структурными элементами ряда биологических ком-
понентов: например, сахарофосфатный остов нуклеиновых кислот
или отложения фосфата кальция костей и зубов. Вторая, более
интересная <роль связана спереносом энергии. По-видимому, фос-
фат представляет собой универсальную энергетическую «размен-
ную монету» в живых организмах. Сокращение мышц, перенос
ионов через мембраны против градиента концентрации и очень
большое число реакций 'биосинтеза — эти потребляющие энергию
процессы осуществляются благодаря переносу фосфорильных
групп (РОз) от высокоэнергетических акцепторов к низкоэнерге-
тическим.
Фосфатный перенос и его активация ионами металлов 625
1.1. Потенциалы переноса фосфорильной группы
Свободная энергия, запасаемая в 'фосфатных связях в природ-
ных соединениях, может составлять 13 ккал/моль. На основе сво-
бодной энергии гидролиза фосфатных производных, называемой
также потенциалом переноса фосфорильной группы [1], построена
удобная энергетическая шкала. Некоторые характерные величины
:[4, 5] приведены в табл. 17.1. Эти свободные энергии сильно
зависят от концентраций протонов и ионов металлов, которые по-
разному взаимодействуют с реагирующими соединениями и про-
дуктами гидролитических реакций. Данные, приведенные в
табл. 17.1, относятся к стандартным условиям (pH 7,0, концентра-
Таблица 17.1
Стандартная свободная энергия гидролиза фосфатных
производных [5]
Соединение8 -bF', ккал/моль0
Енолпируват-Ра 12,8
3 - Ф осф оглицерн л- Р Н.8
Креатин-Р 10,5
Ацетил-Р 10,1
Аргинин-Р 9,0
АТФ (а-ангидрид) 8,0
АТФ (₽-ангидрид) 6,9
Пирофосфат 6,6
АДФ 6,4
Глюкозо-1-Р 5,0
Глицеро-2-Р 4,2
Глюкозо-6-Р 3,3
Глицеро-1-Р 2,3
aR—Р обозначает R—РОз .
6 Стандартные условия: pH 7,0, 25 °C, 0,01 М Mg4-.
ция iMg2+ 0,01 М, 25°C), которые .близки к обычным физиологи-
ческим. Кроме того, стандартное состояние, как обычно, основы-
вается на шкале молярных концентраций для реагирующих
веществ и продуктов реакции, но не для растворителя. Актив-
ность растворителя принята равной единице, хотя в рассматри-
ваемом случае он тоже участвует в реакции. Если бы учитыва-
40—2451
626
Глава 17
лась молярная концентрация воды (55,5 М), то все изменения
свободной энергии уменьшились бы на 2,4 ккал/моль и тогда
стало бы очевидным, что простые фосфорные эфиры, такие, как
глицеро-1-фосфат, по стабильности не намного уступают ортофос-
фату. Тем не менее в водном растворе из-за большого избытка
воды по отношению к глицерину реакция гидролиза протекает
почти до конца.
Известно, однако, несколько фосфатных производных, таких,
как фосфоенолпируват, креатинфосфат и ацетилфосфат, при гид-
ролизе которых освобождается 10—13 ккал/моль свободной энер-
гии. Неустойчивость фосфатной связи в этих молекулах объясня-
ется их структурными особенностями [6]. В фосфоенолпирувате
существует значительное отталкивание зарядов анионных фос-
фатной и карбоксильной групп, энергия которого оценивается в
3—4 ккал/моль, однако основной движущей силой реакции гидро-
лиза служит переход образующейся пировиноградной кислоты из
енольной в более устойчивую кето-форму, свободная энергия кото-
рой на 6—9 ккал/моль меньше:
ОРО|- ОН
I I
СН2=С—СОО~ + НОН ——» СН2=С—СОО"+НОРОГ (1)
фосфоенолпируват пировиноградная кислота
(енольная форма)
о
II
сн3—с—соо-
пировиноградная кислота
(кето-форма)
В ацетилфосфате существует аналогичное отталкивание атомов
кислорода фосфатной и находящейся в непосредственной близо-
сти карбоксильной групп, но и здесь основной энергетический
вклад вносит последующее превращение продукта, заключающее-
ся в этом случае в ионизации уксусной кислоты при нейтральном
pH благодаря резонансной стабилизации ацетата-иона.
О О
II II
СН3—С—О—РО|-+НОН --------> СН3—С—ОН + Н0РО3_
СН3—СОО~ + Н+ (2)
Гидролиз креатинфосфата
Н3С +NH2
I II
~ООС—СН2—N—С—NH—РО|-
приводит к продуктам — креатину и ортофосфату — с повышенной
резонансной стабилизацией, но основным фактором здесь явля-
ется меньшая прочность связи Р—N по сравнению с Р—О.
Фосфатный перенос и его активация ионами металлов 627
Пирофосфат занимает промежуточное положение на шкале
потенциалов групп (6,6 ккал/моль). В нем также есть электро-
статическое отталкивание двух фосфатных групп, а образование
ортофосфат-иона, по-видимому, приводит к некоторому увеличе-
нию делокализации электронов кислорода. Похожая ситуация
обнаружена и для нуклеозидполифосфатов, которые все харак-
теризуются близкими значениями свободной энергии гидролиза.
Свободные энергии гидролиза полифосфатов особенно чувстви-
тельны к pH, поскольку и продукты реакции и реагирующие ве-
щества ионизуются в нейтральной области. Соответствующие зна-
чения р/(а таковы Г7]: Н2РО4 — 6,9; Н2Р20у —6,1; НР2О‘? —8,4;
Н2АТФ2- —4,1; НАТФ3- — 6,5; Н2 АДФ-—4,0; НАДФ2-—6,4;
Н2АМФ°—3,8; НАМФ-—6,0. Для других нуклеотидов эти вели-
чины примерно такие же. Кроме того, большое значение имеет
связывание двухвалентных катионов как продуктами' реакции, так
и реагирующими соединениями. Логарифмы констант устойчиво-
сти [7] для комплексов типа 1:1 с Mg2+ следующие: 1,9 для
НРО4, 6,4 для Р2О7-, 4,0 для АТФ4-, 3,1 для АДФ3- и 2,0 для
АМФ2- (т. 2, гл. 33, табл. 33.1).
Изменение свободной энергии при переносе фосфорильной
группы от одного акцептора к другому можно определить, вычи-
тая потенциал переноса группы второго фосфатного производ-
ного из аналогичной величины для первого. Почему эта свобод-
ная энергия используется организмом, а не рассеивается в окру-
жающей воде в результате гидролиза? Причина заключается в
том, что гидролиз фосфатов при нейтральном pH представляет
собой медленный процесс, несмотря на большое изменение свобод-
ной энергии. По-видимому, такие нуклеофилы, как вода или гид-
роксил, отталкиваются отрицательно заряженным кислородным
атомом фосфата. Поэтому перенос фосфорильных групп может
контролироваться ферментами. Сравнительно широкий диапазон
потенциалов переноса групп в сочетании со значительным кине-
тическим барьером являются отличительными особенностями фос-
фатных производных, позволяющими им служить посредниками
в биологическом переносе энергии.
1.2. Фосфорилирование и фосфоролиз
Оказывается, что существует единственный посредник в дви-
жении энергии при переносе фосфорильной группы. Это молекула
аденозинтрифосфата (АТФ). В высших организмах энергия, вы-
деляющаяся при восстановлении кислорода, запасается в виде
АТФ в результате прямого синтеза из аденозиндифосфата (АДФ)
и ортофосфата. Во всех организмах гликолиз сахаров приводит
к образованию высокоэнергетических 1,3-дифосфоглицерата и
фосфоенолпирувата, которые отдают свои фосфорильные группы
40*
628
Глава 17
АДФ [8, 9]. Последующее использование энергии происходит при
передаче фосфорильной группы АТФ акцепторам, которые обес-
печивают сокращение мышц, транспорт ионов и реакции биосин-
теза [8]. В конце концов эта фосфорильная группа гидролизу-
ется до ортофосфата и может опять участвовать в синтезе АТФ.
Существует, конечно, много поступлений в этот фосфатный поток.
Например, клетки мышц образуют большое количество высоко-
энергетического креатинфосфата, который может быстро фосфо-
рилировать АДФ, если в течение короткого интервала времени
для сокращения мышцы необходимо много АТФ.
Еще один процесс, который имеет отношение к превращению
энергии, — это фосфоролиз, названный так по аналогии с гидро-
лизом. В этом процессе расщепление связей в субстрате происхо-
дит под действием фосфата, а не воды. Так, например, гликоген,
запасной полимер глюкозы, распадается не путем гидролиза до
глюкозы, а в результате фосфоролиза до глюкозо-1-фосфата:
гликоген + НРОд~ -> глюкозе-1-фосфат (3)
Из энергетических соображений преимущество фосфоролиза над
гидролизом состоит в сохранении примерно 5 ккал на моль глю-
козы (табл. 17.1.), тем более что глюкозо-1-фосфат может быть
непосредственно использован в дальнейшем метаболизме.
Изменение свободной энергии для реакции (3), как и для дру-
гих реакций фосфоролиза, невелико. Несмотря на это, биосинтез
гликогена не протекает в результате обращения реакции (3).
Глюкозо-1-фосфат предпочтительнее соединяется с уридинтрифос-
фатом (УТФ) с образованием УДФ-глюкозы и освобождением
пирофосфата:
глюкозе-1-фосфат + УТФ -> УДФ-глюкоза НР3О?“ (4)
Затем УДФ-глюкоза присоединяет свой остаток глюкозы к
растущей цепи гликогена:
УДФ-глюкоза гликогенп --> гликогенп+1 УДФ (5)
и УДФ опять фосфорилируется под действием АТФ
УДФ + АТФ ----> УТФ + АДФ (6)
Реакция (4) представляет собой обращение процесса, аналогич-
ного фосфоролизу, но с участием пирофосфата, т. е. гшрофосфо-
ролиз. Как и для фосфоролиза, изменение свободной энергии для
этой реакции мало. Однако она сдвигается в сторону завершения
в результате последующего гидролиза пирофосфата:
НР2О|- + Н2О --> HPOf- + Н2РО; (7)
с освобождением 6,6 ккал/моль. В целом процесс расходует мно-
го энергии, но ясно, что он надежнее, чем обращение реак-
ции (3), которое зависело бы от неожиданных изменений кон-
Фосфатный перенос и его активация ионами металлов 629
центрации свободного фосфата в организме. По-видимому, общей
закономерностью распада биологических макромолекул — нуклеи-
новых кислот, белков и липидов, а также полисахаридов, явля-
ется то, что он протекает посредством фосфоролиза, в то время
как их синтез включает стадию активации, энергию для которой
дает гидролиз выделившегося пирофосфата [10].
2.. МЕХАНИЗМЫ ПЕРЕНОСА ФОСФАТА
2.1. Расщепление связей Р—О и X—О
Возможны два основных пути протекания реакций, объединяе-
мых названием «перенос фосфата». Их можно представить сле-
дующим образом:
O(Ri)
ох
° O(R2)
zO(Ri)
Y—Р—О+ ХО:
XO(R2)
(I)
O(R,)
Y:^X-^-P^o
O(R2)
zO(Ri)
Y_x + o-Pro
O(R2)
(II>
Для простоты здесь опущены протоны и заряды. R] и R2 могут
присутствовать или отсутствовать. Тризамещенные фосфаты, по-
видимому, редко встречаются в биохимических системах.
Путь (I) представляет перенос фосфорильной группы от одно-
го акцептора (ХО) к другому (Y) или обратный процесс. Он
включает разрыв связей Р—О или Р—Y. Важнейшим классом
реакций этого типа являются реакции переноса фосфорильной
группы к АДФ или от нее. Ферменты, которые их катализируют,
называются киназами. Они рассмотрены в гл. 18. Путь (II)
представляет перенос электрофила X от фосфата к Y или наобо-
рот. В этом случае разрываются связи X—О или X—Y. При от-
сутствии Ri и R2 реакцией, обратной (II), будет процесс, извест-
ный как фосфоролиз [,см. уравнение (3)], а осуществляющие его
ферменты называются фосфорилазами [11]. Если Ri — фосфо-
рильная группа, a R2 отсутствует, то путь (II) —это реакция, об-
ратная пирофосфоролизу. Например, в реакции (4) кислородный
атом фосфата в глюкозо-1-фосфате представляет собой нуклео-
фил Y, а а-фосфорный атом УТФ— электрофил X. Ферменты,
катализирующие подобные реакции, раньше назывались пирофос-
<630
Глава 17
форилазами, однако теперь их называют синтетазами, чтобы под-
черкнуть тот факт, что они осуществляют стадии активации в
процессах биосинтеза. Общим правилом оказывается то, что ак-
'глюкоза—О—Р О
/
О
уридин
.J /ОРО3
Р-О-Р.
/ \ | О
о О о
уридиндифосфатглюкоза + Р20*
(4)
тивирующей группой, как и в реакции (4), служит нуклеотид.
'Следовательно, синтетазные реакции можно также рассматривать
как путь (I), если Ri — это нуклеозид [уридин в реакции (4)], а
ОХ — пирофосфат. Важно то, что разрывается связь Р—О. Даже
фосфоролиз может приводить к расщеплению связи Р—О, если
•атаке фосфатом под действием полинуклеотидфосфорилазы под-
вергается нуклеиновая кислота [12].
2.2. Гидролиз
На вопрос о том, протекает ли рассматриваемая реакция пе-
реноса фосфата посредством разрыва связи Р—О или X—О,
часто можно ответить, анализируя продукты реакции, хотя фер-
ментативные реакции могут быть сложнее, чем представляются
при поверхностном рассмотрении, и нередко включают образова-
ние промежуточных соединений. Иногда их можно обнаружить,
используя метку 18О, по методу, предложенному Кон [13].Если
нуклеофилом служит вода, то расщепление связей Р—О и X—О
приводит к одним и тем же продуктам реакции и использование
воды, меченной 18О, является единственным способом сделать
выбор. В случае пути (I) метка оказывается в фосфате, а в слу-
чае пути (П)—в НО—X:
O(R,)
\ г*
н2о‘^рох
° O(R2)
O(Ri)
---> НО* Р._ + нох
\ О
O(R2)
/OfRi)
H2O*^iXZO-P^f.
O(R2)
^O(Ri)
но*х + но—Р^_
\ °
O(R2)
(Г)
(If)
Этим методом изучено большое число реакций ферментативного
гидролиза, и, как оказалось, все они протекают исключительно
через расщепление связи Р—О (путь I')-
Фосфатный перенос и его активация ионами металлов
631
В неферментативных гидролитических реакциях, напротив, в
зависимости от условий происходит расщепление как Р—О-, так
и С—О-связей [14]. В случае монозамещенных фосфатов дианион
ХОРОГ и моноанион ХОРО3Н~ гидролизуются с расщеплением
связи Р—О, в то время как нейтральная молекула ХОРО3Н2
гидролизуется с расщеплением связи С—О, причем X может быть
алкильной, арильной, ацильной или гликозильной группой.
Известен также катализируемый кислотой процесс, при кото-
ром расщепляются обе связи, Р—О и С—О, но в разной степени
в зависимости от эффективности X в качестве уходящей группы.
В диэфирах фосфорной кислоты одновременное расщепление
связей Р—О и С—О происходит как в нейтральной, так и в мо-
ноанионной формах. Триэфиры фосфорной кислоты сравнительно
быстро атакуются ионом гидроксила с разрывом связи Р—О.
Для установления детального механизма реакций интенсивно
изучался неферментативный гидролиз производных фосфата [14].
Естественно, что внимание обычно фокусируется на пути, сопро-
вождающемся разрывом связи Р—О, поскольку именно он ис-
пользуется гидролитическими ферментами. Этот путь включает
нуклеофильную атаку на фосфор и разрыв связи Р—О, и инте-
ресно установить порядок, в котором происходят эти две стадии.
Две крайние возможности таковы: 1) диссоциативный механизм,
в котором расщепление Р—О приводит к производному трехко-
ординационного .метафосфата (РОз), реагирующему затем с нук-
леофилом, и 2) ассоциативный механизм, в котором при нуклео-
фильной атаке образуется пятикоординационное производное,
отщепляющее затем уходящую группу ОХ. Нет необходимости
пятикоординационное
более вероятно,
стабильных
в том
пяти
промежуточное соединение,
чем трехкоординационное,
пятикоордин анионных сое-
кото-
на-
говорить о возможности механизма, занимающего промежуточное
положение между двумя крайними случаями, такого, в котором
появление и разрушение связей происходит одновременно, без
образования промежуточного соединения.
A priori
по-видимому,
Существует много
динений фосфора, в том числе оксифосфораны, у
рых во всех пяти координационных положениях
ходится атомы кислорода. Стабильные же трехкоординационные
соединения фосфора(V) неизвестны. [Соединения фосфора(III),
конечно, являются трехкоординационными, однако нет указаний
на то, что гидролиз фосфатов включает окислительно-восстанови-
RO
НО: Р-ОХ
//\
° OR
OR
I
но-р-о:
О XOR
Z°R
HOP,.
I O
OR
+ ХОН + ОН ‘
•632
Глава 17
тельные реакции фосфора.] Пятикоординационное промежуточное
соединение, по-видимому, участвует в гидролизе триэфиров фос-
форной кислоты, энергия активации для которого меньше, чем
для гидролиза диэфиров и моноэфиров [3] (см. также гл. 34).
Несомненно, быстрый гидролиз этиленметилфосфата трудно по-
нять без механизма псевдовращения Вестхаймера, который вклю-
чает такой интермедиат [16] (разд. 4.5).
С другой стороны, есть основания полагать, что гидролиз мо-
нозамещенных фосфатов происходит по диссоциативному меха-
низму. Имеются разнообразные доказательства участия мета-
фосфатного интермедиата в гидролизе ацил- [17] и арилфосфатов
[18]. Их можно обобщить следующим образом:
1. Скорость гидролиза дианионов ХОРОз строго коррелирует
с константой кислотности уходящей группы ХО-, что указывает
на значительное разрушение связи Р—О в переходном состоянии.
В случае очень кислых уходящих групп, таких, как 2,4-дини-
трофенолят и n-нитробензоат, дианион гидролизуется быстрее
моноаниона ХОРОзН-, хотя можно было бы ожидать, что боль-
ший отрицательный заряд дианиона затруднит нуклеофильную
атаку на фосфорный центр.
2. Скорость гидролиза моноанионов ХОРОзН- значительно
хуже коррелирует с кислотностью уходящей группы. При слабо-
кислых уходящих группах моноанион гидролизуется быстрее диа-
ниона. Считают, что этот особый эффект связан с протонирова-
нием уходящей группы, после которого удаляется нейтральная
молекула ХОН и образуется метафосфатный ион [14]:
Q4 /О-гН*э
о /О н н
X
+ хон
Если протон заменен на алкильную или арильную группу, этот
эффект исчезает и моноанионы диэфиров XO(RO)PO гидроли-
зуются быстрее моноанионов моноэфиров.
3. При сольволизе n-нитрофенилфосфата как в виде моно-,
так и дианиона в системе метанол — вода образование метил-
фосфата и ортофосфата происходит в том же отношении, в каком
присутствуют метанол и вода, хотя метанол является лучшим
нуклеофилом. Это служит доказательством того, что реакционно-
способное промежуточное соединение, метафосфат, реагирует с
водой и метанолом в одинаковой степени. В других смешанных
системах растворителей можно обнаружить отклонения от стати-
стического распределения продуктов, но эти отклонения легко
объяснить эффектами селективной сольватации.
Фосфатный перенос и его активация ионами металлов 633.
4. Энтропии активации гидролиза моно- и дианионов близки
к нулю, что указывает на мономолекулярный механизм. Бимоле-
кулярные реакции сольволиза обычно имеют значительную отри-
цательную энтропию активации (около 20 э. е.) из-за необходимо-
сти ориентации атакующей молекулы растворителя.
2.3. Фосфатазы и фосфотрансферазы
Ферменты, которые катализируют гидролиз производных фос-
фата, называются фосфатазами [5]. Существует несколько фер-
ментов, роль которых, по-®идимому, заключается в образовании
ортофосфата из моноэфиров фосфорной кислоты. Они малочувст-
вительны к природе заместителя в фосфате и называются неспе-
цифическими фосфомоноэстеразами. Существуют, однако, замет-
ные различия в диапазонах pH, в которых они работают, и по-
этому они делятся на кислые [19] и щелочные [20] фосфатазы,
хотя бывают и промежуточные случаи. Другие фосфатазы дейст-
вуют на определенные субстраты, такие, как глюкозо-6-фосфат
[21], фосфосерин [21] и 5'-нуклеотиды [22]. Важным ферментом
является неорганическая пирофосфатаза [23], поскольку гидро-
лиз пирофосфата сопряжен со всеми синтетазными реакциями.
Некоторые киназы обнаруживают небольшую АТФ-азную актив-
ность. Активную АТФ-азу можно выделить из разрушенных ми-
тохондрий [9], и она без сомнения участвует в окислительном
фосфорилировании, посредством которого обращается гидролити-
ческая реакция.
Дизамещенные фосфаты гидролизуются ферментами фосфо-
диэстеразами. Некоторые из них неспецифичны, но большинство
участвует в гидролизе нуклеиновых кислот и называется нуклеаза-
ми. Существуют рибонуклеазы и дезоксирибонуклеазы (гл. 34), и
по действию они совершенно различны. Экзонуклеазы атакуют
фосфодиэфирные связи последовательно, начиная с одного конца
цепи нуклеиновой кислоты, в то время как эндонуклеазы атакуют
также и связи внутри цепи.
Часто предполагают, что при действии фосфатаз образуется
промежуточное фосфорилферментное соединение, которое затем
гидролизуется, и в некоторых случаях эта особенность механизма
вполне установлена. Некоторые фосфатазы, кроме гидролиза
фосфатов, катализируют также перенос фосфорильной группы к
нуклеофилам, отличным от воды, т. е. обнаруживают фосфотранс-
феразную активность. Так как водные растворы содержат 55,5 М
воды, то эти реакции иногда трудно заметить, если не использо-
вать смешанные растворители. В некоторых случаях, однако,
найдены такие нуклеофилы, которые даже в малой концентрации
эффективно конкурируют с водой. Неизвестно, имеют ли такие
реакции биологическое значение.
634
Глава 17
Важность катализа определенных стадий фосфотрансферазных
реакций обычно очевидна. Киназы и синтетазы уже упомянуты
выше. Некоторые ферменты этого класса переносят фосфатную
группу из одной части молекулы в другую. Примером может слу-
жить фосфоглюкомутаза [24], которая превращает глюкозо-6-фос-
фат, продукт фосфорилирования глюкозы под действием АТФ, в
глюкозо-1-фосфат, служащий субстратом в реакции (4). Интерес-
но, что это превращение протекает через два фосфорилированных
промежуточных соединения — фосфорилированный фермент и
глюкозо-1,6-дифосфат. Фосфатная группа первого из них перено-
сится на ОН-группу глюкозо-6-фосфата, находящуюся в положе-
нии 1, а затем образовавшийся глюкозо-1,6-дифосфат отдает фос-
фатную группу из положения 6 ферменту.
Ферменты, относящиеся к классу фосфотрансфераз, не фосфо-
рилируют воду, т. е. не способны катализировать гидролиз. Не-
известно, каким образом это оказывается возможным в водном
растворе, и этот факт представляет собой один из самых интерес-
ных аспектов механизма переноса фосфата. В случае некоторых
рибонуклеаз гидролизу предшествует стадия переноса фосфата,
приводящая к образованию лабильного циклического 2',3'-нуклео-
зидфосфата. Наиболее изучена из них панкреатическая рибону-
клеаза [25].
2.4. Необходимость ионов металлов
Среди всех ферментов, переносящих фосфат, по-видимому,
единственным хорошо охарактеризованным металлоферментом,
т. е. ферментом, который сохраняет существенно важный металл
при выделении, является щелочная фосфатаза, рассматриваемая
в следующем разделе. Согласно имеющимся данным, возможно,
что фосфатаза из селезенки быка, гидролизующая фосфобелки
и другие монозамещенные фосфаты, представляет собой железо-
содержащий металлофермент [26].
Большинство ферментов, переносящих фосфат, нуждаются для
активности в ионах двухвалентных металлов. Эти ионы сравни-
тельно легко удаляются из ферментов, что приводит к потере ак-
тивности. При добавлении металлов она восстанавливается.
Поскольку единственными двухзарядными ионами, которые в фи-
зиологических условиях присутствуют в достаточном количестве,
являются Mg2+ и Сае+, то считают, что именно они выполняют
биологическую функцию. Однако влияние ионов металлов на фер-
менты может быть разным. Чаще всего наибольшей эффектив-
ностью обладает M.g2+. Некоторые металлы первого переходного
ряда, особенно Mns+, могут заменять его, но активность фермента
при этом несколько понижается. Это же относится к Са2+, однако
иногда он ингибирует реакцию, тогда как в некоторых случаях
Сае+ служит активатором, a Mg2+ — ингибитором.
Фосфатный перенос и его активация ионами металлов
635
Установление факта необходимости ионов металлов представ-
ляет иногда сложную задачу. Их влияние на активность может
маскироваться взаимодействиями с субстратом, примесями в пре-
парате фермента или дополнительными центрами связывания на
самом ферменте. Обычно существует оптимальная концентрация,
при превышении которой наблюдается эффект ингибирования, и
величина этой оптимальной концентрации зависит от природы ме-
талла. Иногда хелатирующие агенты, например ЭДТА, увеличи-
вают активность, что может объясняться селективным связыва-
нием следов таких ионов, как Си2+, которые сильно ингибируют
реакцию.
Несмотря на эти трудности, имеющиеся надежные данные
позволяют с уверенностью сделать несколько общих выводов о
потребности в ионах металлов. Двухзарядные ионы необходимы
для всех киназ и синтетаз. Фосфатазы обычно также нуждаются
в катионах, хотя известны и исключения, например .неспецифиче-
ская кислая фосфатаза. Для фосфоглюкомутазы ион Mgt2+ необ-
ходим, а для похожей на нее фосфоглицератмутазы нет .[24].
Потребность в двухзарядном катионе характерна для нуклеаз,
механизм действия которых не включает образование на первой
стадии циклического нуклеотида [25]. Фосфорилазы, расщепляю-
щие связь С—О, не зависят от двухзарядных ионов, а расщеп-
ляющие связь Р—О зависят, примером чему служит фосфоролиз
полинуклеотидов. Кроме того, ионы двухвалентных металлов
обычно, ио не всегда необходимы для ферментов, переносящих
фосфат в том случае, если катализируется расщепление связи
Р—О. Эти выводы суммированы в табл. 17.2.
Таблица 17.2
Необходимость ионов металлов для ферментов, переносящих фосфат
Нуждаются в двухзарядном катионе Не нуждаются в двухзарядном катионе
Неспецифические щелочные фосфатазы Большинство других фосфомоиоэстераз Пирофосфатаза Ф осф ог люкомутаза Нуклеазы, действующие как фосфоди- эстеразы Киназы Синтетазы Полинуклеотидфосфорилаза (расщепле- ние связи Р—О) Неспецифические кислые фосфатазы Фосфоглицератмутаза Рибонуклеазы, образующие цикличе- ский интермедиат, 2',3'-нуклеозид- ф осф ат Другие фосфорилазы (расщепление связи С—О)
636
Глава 17
3 .- ЩЕЛОЧНАЯ ФОСФАТАЗА
Щелочная фосфатаза из бактерии Escherichia coli заслуживает
особого внимания, поскольку это единственный хорошо охарак-
теризованный металлофермент из числа переносящих фосфат.
В настоящее время он изучается в нескольких лабораториях, и
описание его структуры и механизма функционирования еще
далеко от завершения. Проводимый [27] рентгеноструктурный
анализ кристаллов фермента в значительной мере прояснит су-
ществующую картину. Тем не менее уже сейчас известны неко-
торые удивительные особенности этого фермента. Их подробное
обсуждение с акцентом на ферментативной кинетике дано Рей-
дом и Вильсоном [28].
Используя обедненную фосфатом среду, можно вызвать обра-
зование в Е. call большого количества фосфатазы [29—31], кото-
рую затем сравнительно нетрудно очистить и охарактеризовать.
Она является неспецифической фосфомоноэстеразой [32] с опти-
мумом pH в диапазоне 8,5—10,0. Фосфатаза катализирует также
гидролиз соединений с фосфоангидридной связью (например, пи-
рофосфата и АТФ) и фторфосфата, но неактивна по отношению к
связям Р—N и Р—С [33, 54]. Интересно, что S-алкилтиофосфаты
(RS—РО3) быстро расщепляются [34, 35], тогда как О-алкилтио-
фосфаты (RO—PO2S) или не гидролизуются вообще [35], или
гидролизуются значительно медленнее [36] обычных эфиров фос-
форной кислоты. Кислая фосфатаза, напротив, расщепляет
€)-замещенные, но не S-замещенные эфиры тиофосфорной кис-
лоты [35]. Известно также, что щелочная фосфатаза переносит
•фосфат на нуклеофилы, отличные от воды [37].
3.1. Присоединение металлов
Очищенный фермент содержит прочно связанные ионы цинка
и похож в этом отношении на два других гидролитических фер-
мента— карбоксипептидазу (гл. 15) и карбоангидразу (гл. 16).
Молекулярная масса нативной фосфатазы равна 80000 [38—40].
В кислом растворе молекула белка диссоциирует на две иден-
тичные субъединицы [40—43]. При высокой концентрации цинка
образуется тетрамерная форма, причем положение равновесия
димер^тетрамер зависит от pH [44].
Число атомов цинка в составе нативного фермента составляет,
по данным разных авторов [34, 45—50], от двух до четырех на
димерную молекулу. Можно считать установленным, что только
два атома цинка определяют каталитическую активность фер-
мента. Из экспериментов Коэна и Вильсона [45] по равновесному
связыванию, в которых фосфатазная активность измерялась в
присутствии буферных растворов цинка, следует, что активность
Фосфатный перенос и его активация ионами металлов
637
молекулы фермента, содержащей один ион цинка, составляет
12% активности комплекса с двумя (или более) ионами этого
металла. Константы комплексообразования, полученные в этой
работе при pH 8,5 и 25 °C в присутствии 1 М NaCl, оказались
различными для двух ионов цинка: pKi=7,66 и рКг= 10,22. По
данным Чопак [46], использовавшей метод равновесного диали-
за, обе эти константы при pH 8,5 и 25 °C в присутствии 0,1 М
KNO3 значительно меньше и различаются только на статистиче-
ский множитель, равный четырем: p/G = 11,16 и рК2=11>76.
Уменьшение этих величин при pH меньше 8,5 описывается кривой
с наклоном, примерно равным единице. Третий и четвертый ионы
цинка диссоциируют значительно легче двух первых, определяю-
щих активность [48].
Ионы цинка можно удалить обработкой хелатирующими аген-
тами. Образующийся апофермент может присоединять затем раз-
нообразные двухзарядные ионы [49, 51]: Мп124-, Со2-*-, Ni2+, Cu2+,
CdE+, Hg2+. Прочность комплексов для центра с наименьшим
сродством уменьшается в ряду Cd2+=Mn2+>Zn2+>Col2+>Ni2+
.[49, 50]. Положение Си2+ и Hg2+ в этом ряду неизвестно, однако
Си2+, по-видимому, присоединяется прочнее ZnB+ [50]. Каталити-
ческая активность обнаружена лишь у Со2+-фосфатазы и состав-
ляет только 12% активности нативного фермента, содержащего
ZnB+ [52—54]. Со2+-форма фермента не обнаруживает трансфе-
разную активность с нуклеофилами, которые эффективны с
гп2+-ферментом.
Со2+-Фосфатаза представляет особый интерес, так как элек-
тронный спектр Со(П) дает возможность судить о геометрии ко-
ординации в активном центре. Видимый спектр Со2+-фермента
имеет необычный вид [48, 55], и для него характерны максимумы
при 640, 610, 555 и 510 нм. Молярные коэффициенты поглощения
при этих длинах волн равны соответственно 250, 210, 350 и 280.
Эта картина похожа на наблюдаемую для Со (II)-карбоангидразы
(гл. 16). Она не отвечает ни октаэдрической, ни тетраэдрической
координации лигандов Со (II). В некоторой степени похожие
спектры получены для пятикоординационных комплексов ко-
бальта с расположением лигандов в вершинах псевдотригональ-
ной бипирамиды [56, 57], хотя в случае фермента расщепление
пол ос более выражено, что свидетельствует о пониженной симмет-
рии [48]. Спектры кругового дихроизма в видимой области слож-
ны [48, 55]. Спектры ЭПР присоединенного к фосфатазе иона
Си2+ указывают на изменение его окружения при связывании вто-
рого иона этого металла [50].
Информация о центрах присоединения металлов на ферменте
неполна. Тейт и Вейлли [58] предположили, что лигандами цинка
служат три остатка гистидина, поскольку фотоокисленный апо-
фермент, степень разрушения гистидина в котором выше, чем в
638
Глава 17
нативном ферменте, не способен к присоединению обычных коли-
честв пинка и неактивен. Рейнольдс и Шлезингер [47] из сравне-
ния кривых титрования нативного и апофермента также заклю-
чили, что в связывании цинка участвуют два или три остатка
гистидина. Характерный спектр и активность Со2+-фосфатазы ис-
чезают при добавлении кислоты [48]. Точка перехода находится
при pH 7,0, что согласуется с ионизацией гистидина [47]. Спек-
трофотометрическим титрованием установлено, что при удалении
из фермента цинка шесть остатков тирозина приходят в контакт
с растворителем [59]. Это согласуется с участием тирозина в
связывании цинка, хотя другой причиной такого эффекта может
быть изменение конформации.
3.2. Фосфорилфермент
Тот факт, что перенос фосфата протекает через ковалентное
промежуточное соединение, фосфорилфермент, служит основой
предполагаемого механизма катализа щелочной фосфатазой. Фос-
форилированный белок можно осадить из смеси фосфата с фер-
ментом прибавлением трихлоруксусной кислоты [60]. Анализ про-
дуктов расщепления белка показывает, что фосфат присоединен
к остатку серина [61]. В щелочной среде обнаружить ковалентно
присоединенный фосфат не удается, однако Вильсон и сотр.
[62—67] с помощью кинетических и изотопных методов показали,
что фосфорилфермент образуется при всех значениях pH и что он
идентичен фосфорилированному белку, выделяемому из кислого
раствора. Свободная энергия гидролиза этого промежуточного
соединения удивительно мала, и по величине соответствующей ей
константы равновесия он в 106 раз устойчивее обычных фосфор-
ных эфиров [62—64]. Причина того, что фосфатаза не оказыва-
ется в термодинамической ловушке, заключается в образовании
нековалеитного комплекса фермент — фосфат, который при pH 8
в 100 раз устойчивее фосфорилфермента [62]. В результате этого
равновесие
Е—РО3 ч- Е РО4 (8>
в щелочной среде сильно смещено вправо и свободный субстрат
может вытеснить нековалентно связанный фосфат и регенериро-
вать катализатор. Как и следовало ожидать, сам фосфат сильно
ингибирует фосфатазную реакцию.
В кислых растворах, напротив, равновесие (8) смещено влево
и фермент неактивен [63, 64]. Однако положение этого равновесия
не является определяющим фактором, так как рКа группы, кото-
рая контролирует инактивацию фермента, близок к 7 как для
Zn2+-, так и для Со2+-фосфатаз [53, 54]. При pH 7 основная часть
фосфата связана нековалентно [63]. Предполагается, что для ак-
Фосфатный перенос и его активация ионами металлов 639
тивации фермента необходимо депротонирование остатка гисти-
дина [53]. Фосфорилирование Zn2+- и Со2+-ферментов протекает
очень быстро, причем Со^-форма реагирует с фосфатом быстрее.
Стадия дефосфорилирования лимитирует скорость гидролиза
Со2+-фосфатазой [53]. С 2п2+-ферментом отщепление фосфата
протекает значительно быстрее, и в щелочной среде эта стадия,
по-видимому, не определяет скорости гидролиза [66, 67]. Сущест-
вует предположение, что скорость ферментативной реакции ли-
митируется стадией изменения конформации [68, 69]. Оно под-
тверждается кинетическими данными по связыванию ферментом
обратимого ингибитора [70]. Этот вопрос детально рассмотрен
Рейдом и Вильсоном [28]. Максимальная скорость гидролитиче-
ской реакции не зависит от природы субстрата, несмотря на зна-
чительные различия в эффективности уходящей группы (напри-
мер, в нитрофенил- и динитрофенилфосфатах [52]). При высоких
значениях pH константа Михаэлиса увеличивается, что означает
ослабление связи с субстратом. Величина рКо этого перехода для
2п2+-фосфатазы составляет 8,6 [54], а для Со2+-фосфатазы она
несколько больше (8,9 [54] или 9,6 [52] по данным разных ав-
торов). Этот эффект объясняют ионизацией молекулы воды, свя-
занной с ионом металла [52]. Аналогичным образом можно объ-
яснить pH-зависимость константы диссоциации цинка с точкой
перехода при pH 8,5 [46].
Коулмен и сотр. [51] обнаружили, что Cd2+- и Мп124--фосфата-
зы, у которых каталитическая активность совершенно отсутству-
ет, очень прочно присоединяют фосфат с образованием значитель-
ного количества фосфорилфермента. В то время как для Zn2+-
фосфатазы степень образования ковалентного соединения с фос-
фатом максимальна при pH 5 и очень мала при pH выше 6, для
Cd2+- и Мпе+-фосфатаз оптимальное значение pH этой реакции
равно 7. В случае Сб2+-фермента при этом значении pH большая
часть присоединенного фосфата связана ковалентно. Следователь-
но, Cd2+- и Мп2+-производные так же эффективны на стадии фос-
форилирования белка, как и фермент, содержащий цинк. Причи-
на отсутствия у них ферментативной активности заключается,
вероятно, в неспособности к дефосфорилированию в щелочной
среде. Степень ковалентного присоединения фосфата к Сое+-фос-
фатазе, как сообщается в этой работе, очень мала при всех значе-
ниях pH и достигает максимума (всего 0,2 моля на 1 моль фер-
мента) при pH 6 [51].
Последний результат противоречит сообщению Лаздунски и
др. [71] о том, что при 0 °C (группа Коулмена проводила иссле-
дования при 4 °C) можно выделить до 1 моля ковалентно при-
соединенного фосфата иа 1 моль Со^-фосфатазы после ее инку-
бации с фосфатом в кислой среде. В щелочных растворах кова-
лентное соединение не было обнаружено, причем pH точки пере-
640
Глава 17
хода было равно 5,6. Для 7п2+-фермента эта величина была почти
такой же (5,1), но максимальная величина включения в кислом
растворе была равна двум молям на 1 моль белка. Более того,
максимальная величина ковалентного присоединения фосфата при
инкубации с субстратами (АМФ и АТФ) в кислом растворе при
0°C и для Zn2+-, и для Со^+'фосфатаз составляла 2 моля/моль,
а минимальная величина включения в щелочной среде была равна
одному молю/моль. Однако 'величины pH перехода были для них
различными: 6,6 для цинкпроизводного и 4,6 для кобальтпроиз-
водного.
3.3. Присоединение фосфата
Интересные результаты, полученные при определении числа
фосфатсвязывающих центров, привлекли в последнее время зна-
чительное внимание. Несмотря на димерную природу фермента,
кинетические данные указывают на существование одного актив-
ного центра [62—64]. Экспериментальное подтверждение этого
было получено методом остановленной струи [66, 68, 72], при
помощи которого было обнаружено, что величина предстационар-
ного выброса продукта равна примерно 1 молю на моль фермен-
та. Однако при высокой концентрации субстрата (2-10-3 М) этот
метод дал величину 2,7 [73]. Включение фосфата в большинстве
работ, за исключением вышеупомянутой работы Лаздунски и др.
[71], составляло 0,6—1,3 моля/моль [64, 74, 75]. Нейман [35]
обнаружил связывание 1 моля ингибитора. О-п-нитрофенилтио-
фосфата, на 1 моль белка.
В экспериментах по изучению равновесного связывания обна-
ружено присоединение одного или двух ионов фосфата на моле-
кулу. По последним данным, один ион фосфата связывается
прочно и одни или несколько слабо [51, 68, 69, 76, 77]. Кроме
того, Симпсон и Вейлли [76] получили кинетическое подтвержде-
ние подобной антикооперативности, обнаружив в соответствующих
условиях активацию субстратом, выражающуюся в двухфазном
характере зависимости активности от концентрации субстрата.
Отрицательная кооперативность подробно обсуждена для другой
системы Левицки и Кошландом [77]. Симпсон и Вейлли предпо-
лагают, что функциональная роль антикооперативности заключа-
ется в сохранении определенного уровня активности фермента в
широком диапазоне концентрации субстрата. Их вывод о суще-
ствовании в фосфатазе аллостерических взаимодействий отчасти
подтверждается изменением конформации фермента при связыва-
нии ингибитора [70] [см. «Дополнения, внесенные в корректуру»,
пункт 1].
Присоединение фосфата к Со2+-фосфатазе вызывает значи-
тельное изменение спектра белка в видимой области [48, 55].
Фосфатный перенос и его активация ионами металлов 641
Оказывается, что две высокоэнергетические полосы — при 510 и
555 нм — сдвигаются соответственно к 475 и 550 нм, интенсив-
ность низкоэнергетической полосы при 640 нм сильно уменьша-
ется, а плечо при 610 нм исчезает совсем. Аналогичные измене-
ния происходят при связывании арсената [51]. Оба аниона силь-
но влияют на спектр кругового дихроизма, однако если фосфат
уменьшает [48, 55], то арсенат увеличивает эллиптичность основ-
ной полосы при 550 н.м [51]. Следовательно, хотя фосфат и арсе-
нат вызывают примерно одинаковые изменения энергетических
уровней d-электронов кобальта (II) в Со2+-фосфатазе, их влияние
на диссимметрию координационной сферы металла оказывается
поразительно различным. Существует противоречие в вопросе о
том, сколько ионов фосфата, один [55] или два [48], должны
присоединиться к молекуле фермента для достижения макси-
мального спектрального эффекта. Связывание фосфата Сп2+-фос-
фатазой вызывает значительное изменение спектра ЭПР иона
меди [50, 128].
3.4. Модели активного центра
Как следует из изложенного выше, в описании щелочной фос-
фатазы и ее действия остаются неясными некоторые ключевые
моменты, которые подчас трактуются взаимоисключающими спо-
собами. Не приходится сомневаться в том, что дальнейшие иссле-
дования, в том числе рентгеноструктурный анализ кристаллов
фермента, прояснят положение. Однако сейчас остается только
гадать о деталях процессов, происходящих в активном центре.
По-видимому, можно без опасений считать, что молекула суб-
страта в активном центре прямо взаимодействует с ионом цинка.
Ярко выраженная зависимость ферментативной реакции от приро-
ды металла, чувствительность электронного спектра и спектра
кругового дихроизма Со2+-фосфатазы и спектра ЭПР Си2+-фос-
фатазы к фосфату и арсенату — все эти факты делают маловеро-
ятным косвенное участие ионов цинка в катализе. Кроме того,
прямое присоединение субстрата к иону цинка было уже надеж-
но устаиовлеио кристаллографически [78] для другого фермен-
та— карбоксипептидазы (гл. 15).
Электронный спектр Со2+-фосфатазы обнаруживает некоторую
асимметрию в координационном окружении иона кобальта. При-
соединение фосфата или арсената сильно влияет на спектр, одна-
ко форма полос и их расщепление остаются почти неизменными.
Из этого можно заключить, что в целом координационная гео-
метрия сохраняется и что фосфат или арсенат просто замещают
лиганд атома металла. Самым удобным для замещения лигандом
могла бы быть вода. Это предположение также напрашивается из
данных по кристаллической структуре карбоксипептидазы,
41—2451
642
Глава 17
согласно которым вода служит четвертым лигандом цинка в силь-
но искаженной координационной сфере. С этой гипотезой согла-
суется также предположение Готтесмана и сотр. [52] о том, что
увеличение константы Михаэлиса для Zne+- и Со2+-фосфатаз
связано с ионизацией молекулы воды, служащей лигандом ме-
талла. Вытеснение субстратом воды из комплекса с металлом
должно происходить легче, чем вытеснение иона гидроксила.
Прочность связи фосфата с металлоферментом удивительно
велика. Константа устойчивости комплекса, равная около 106 М-1
[51, 63, 76], выше, чем можно было бы ожидать для взаимодей-
ствия НРОГ и иона двухвалентного металла. Так, константа
устойчивости комплекса типа М(НРС>4) равна 370 М-1 для Мп2+
и 77 М.-1 для Mg124- [7]. Вероятно, вблизи иона цинка находится
еще один катионный центр связывания, например протон амино-
группы боковой цепи аминокислоты, взаимодействующий с кис-
лородными атомами фосфата. Образование фосфорилфермента
при реакции с субстратом и НРО4 можно считать твердо уста-
новленным. После разрушения белка фосфат оказывается присое-
диненным к серину. Отсюда делается вывод о том, что в актив-
ном центре вблизи иона цинка находится остаток серина, ориен-
тация которого делает возможной его атаку по фосфорильному
атому присоединенного производного фосфата. Высокая термоди-
намическая устойчивость получающегося фосфорилфермента при
гидролизе предполагает сохранение в нем значительной части
взаимодействий комплекса фермент — фосфат [62]. Вполне ве-
роятно, что фосфорильная группа, присоединенная к серину, со-
храняет связь с ионом цинка.
Бреслоу и Кац [36] пришли к интересному выводу о механиз-
ме действия из сравнения относительных скоростей гидролиза
О-алкилфосфатов и тиофосфатов. При ферментативном гидролизе
моноэфиры тиофосфата реагируют быстрее эфиров фосфата, тогда
как при неферментативном гидролизе наблюдается обратная за-
кономерность. С другой стороны, при неферментативном гидро-
лизе триэфиров производные тиофосфата реагируют медленнее.
Считают, что триэфиры гидролизуются по «ассоциативному меха-
низму», который предусматривает уменьшение кратности связи
Р=О (или P=S) и увеличение заряда атома кислорода (серы)
в переходном состоянии, тогда как гидролиз моноэфиров проте-
кает по механизму элиминирования с образованием метафосфата,
при котором кратность связи Р=О (или P=S) увеличивается,
а заряд атома кислорода в переходном состоянии уменьшается
(разд. 2.1). Соотношение скоростей неферментативных реакций
можно объяснить меиыпей электроотрицательностью серы по
сравнению с кислородом. Следовательно, изменение соотношения
скоростей распада моноэфиров в ферментативной реакции пред-
полагает переход от одного механизма к другому. Эта гипотеза
Фосфатный перенос и его активация ионами металлов
643
согласуется с существованием сильных взаимодействий с кис-
лородными атомами фосфата в активном центре.
После фосфорилирования фермента происходит нуклеофильная
атака на образовавшееся промежуточное соединение. Нуклео-
филом может быть не только вода. Вильсон и др. [37] показали,,
что ряд акцепторов фосфата, таких, как этаноламин и этилен-
гликоль, эффективно конкурирует с водой, даже если их кон-
центрация 1 М или меньше. /Молекулы этих акцепторов содержат
Рис. 17.1. Модель активного центра щелочной фосфатазы.
а — фосфорилирование фермента; б—перенос фосфата на воду и другие нуклеофилы.
ОН- или NH2-rpynny, находящуюся на расстоянии, не превышаю-
щем длины цепи из четырех углеродных атомов от акцептирую-
щей гидроксильной группы. Это структурное требование указы-
вает на существование у фермента центра связывания, несущего
положительный заряд и способствующего реакции с нуклеофилом.
Молекулы акцепторов, которые не удовлетворяют этому требова-
нию, оказываются неэффективными в экспериментах по переносу
[37], однако нет оснований полагать, что при достаточно высо-
кой концентрации они не смогли бы конкурировать с водой.
По-видимому, фермент не имеет специального центра для связы-
вания воды. Эти выводы из экспериментальных данных иллюст-
рируются схемой на рис. 17.1. Об участии металлов в придании
активному центру фермента стеричеекой селективности свиде-
тельствует тот факт, что Cd2+ и Мпе+ облегчают фосфорилиро-
вание белка, но не последующий гидролиз промежуточного сое-
динения, тогда как Со24-, активность с которым составляет 12%
активности с Zn124-, делает трансферазные реакции невозможными.
Обсуждение механизма действия фосфатазы до сих пор не
касалось осложнения, связанного с существованием лишь одного
центра прочного присоединения фосфата при участии в катализе
двух ионов металла. Согласно аллостерической модели [76, 77],
связывание одной молекулы фосфата затрудняет связывание дру-
гой из-за возникающих дальнодействующих взаимодействий меж-
ду субъединицами. Альтернативный механизм [51] объясняет это
41*
644
Глава 17
тем, что основное место присоединения фосфата находится в
области контакта двух субъединиц. Предварительные данные
рентгенографического анализа [27, 28] свидетельствуют о том,
что кристаллы нативного фермента принадлежат к моноклинной
пространственной группе Р3121 с размерами ячейки а=70,5 А,
6=70,5 А, с—155,6 А, р=120°. Элементарная ячейка содержит
три димерные молекулы, однако асимметрической единицей явля-
ется молекула мономера. Пары идентичных мономеров связаны
посредством оси вращения второго порядка. Если эти пары пред-
ставляют собой функциональные молекулы димеров, то единст-
венный центр связывания фосфата может находиться на оси сим-
метрии второго порядка. В результате появляется интересная
возможность того, что фосфат одновременно взаимодействует с
двумя ионами цинка и что эта неизвестная ранее структурная
особенность объясняет активацию производных фосфата при ну-
клеофильной атаке.
4 . РОЛЬ ИОНОВ МЕТАЛЛА
Ввиду большого разнообразия ферментов, переносящих фос-
фат при участии ионов металлов, следует учитывать возможность
такого же разнообразия функций металла. Действительно, если
обратиться к сравнению эффективности различных металлов,
природы ингибиторов и концентрационных зависимостей актива-
ции и ингибирования, то можно увидеть, что специфические тре-
бования к металлам для различных ферментов неодинаковы.
Вполне возможно, что для каждого фермента характерен свой
механизм участия ионов металла в реакции. Тем не менее можно
попытаться найти некоторую объединяющую основу, базирую-
щуюся на фундаментальных законах химии.
4.1. Тройные комплексы ферментов с металлами
и субстратами
Активация фермента ионами металлов предполагает образова-
ние тройного комплекса фермент — ион металла — субстрат.
Можно представить несколько вариантов организации этого
комплекса, и все они встречаются в реальных системах. Схемы
координации, в которых связывающим мостиком служат лиганд
(Е—S—М), металл (Е—М—S) или фермент (S—Е—М), рас-
смотрены в гл. 14 и в исчерпывающем обзоре Милдвана [79].
4.2. Нейтрализация заряда
Наиболее очевидным свойством ионов металлов, которое
могло бы помочь в объяснении их роли при активации фосфат-
переносящих ферментов, является положительный заряд. Катио-
Фосфатный перенос и его активация ионами металлов
645
ны двухвалентных металлов сохраняют свой заряд в нейтральном
растворе и могут снимать отрицательный заряд кислородных
атомов фосфата, облегчая тем самым присоединение субстрата
к ферменту. Хорошо известно, что двухзарядные катионы обра-
зуют с производными фосфата сравнительно прочные комплексы
(хотя этот эффект часто не виден из-за образования осадка), осо-
бенно если они содержат ди- и трифосфатные группировки
[7, 87]. Кроме того, эти катионы сильно стабилизируют третич-
Рис. 17.2. Предполагаемый механизм реакции 2пг+-ПКА+1тРО3—>-Zn2+-
(ПКА)Р0з+1т.
ную структуру транспортной РНК [88], вероятно, вследствии
взаимодействия с рибозофосфатным скелетом.
Модельная система, в которой нейтрализация заряда, по-види-
мому, играет важную роль, обнаружена Куперманом и Ллойдом
[89]. Они нашли, что Zna+ катализирует перенос фосфорильной
группы от фосфорилимидазола к кислородному атому 2-пиридин-
карбальдоксима (ПКА), о комплексе которого с Zn2+ было из-
вестно, что он является эффективным нуклеофилом [90]. Реакция
описывается кинетикой с насыщением, что свидетельствует об
образовании тройного комплекса ПКА — Zn — фосфорилимида-
зол. Кривая pH-зависимости скорости реакции имеет колоколооб-
разную форму с максимумом при pH 6. В этих условиях комп-
лекс Zn2+ — КПА и фосфорилимидазол ионизованы. Полагают, что
роль иона цинка заключается в экранировании фосфатной группы,
в результате чего облегчается атака отрицательно заряженным
нуклеофилом (рис. 17.2). Величина эффекта очень большая, и в
оптимуме pH скорость этой реакции примерно в 104 раз превы-
шает скорость атаки фосфорилимидазола водой.
По похожему механизму, вероятно, протекает неферментатив-
ное фосфорилирование ортофосфата до пирофосфата под дейст-
вием АТФ, которое, как показал Ловенштейн [91], требует обяза-
тельного присутствия ионов двухвалентных металлов, предпочти-
тельнее всего Mri2+, Cd2+ и Са2+. В этом случае не наблюдается
эффекта насыщения при высокой концентрации реагентов, однако,
имея в виду низкую концентрацию фосфата в эксперименте,
можно заключить, что реакция в этих условиях протекает значи-
тельно быстрее гидролиза АТФ [92]. Необычные характеристики
этого процесса, а именно рН-оптимум 9, оптимальное соотношение
42—2451
646
Глава 17
концентраций АТФ и Мв+ около 0,6, показывают, что ее меха-
низм непрост.
Хотя в участии металлов в нейтрализации зарядов при фер-
ментативных реакциях переноса фосфата не приходится сомне-
ваться, можно отметить, что эта функция не обязательно выпол-
няется металлом. Белки содержат и другие заряженные центры,
не содержащие металл, — аминные и имидазольные группы, ко-
торые с успехом могут участвовать в связывании отрицательно
заряженных фосфатных групп. Многие ферменты, в том числе
некоторые из переносящих фосфат, работают в отсутствие метал-
лов, хотя их субстраты заряжены отрицательно.
4.3. Поляризация
Можно ожидать, что, кроме нейтрализации заряда, ион ме-
талла, присоединенный производным фосфата через атом кисло-
рода, вызывает поляризацию соответствующих связей Р—О. Рань-
ше при обсуждении роли металла в переносе фосфата функция
поляризации рассматривалась чаще всего. Ее идея заключается
в том, что, оттягивая электроны от атома кислорода, ион металла
ослабляет связь Р—О и увеличивает положительный заряд на
атоме фосфора. Это должно облегчать и нуклеофильную атаку,
и разрыв связи Р—О. Иллюстрацией служит рис. 17.3 (см. также
т. 2, гл. 34).
Если бы поляризация играла важную роль в активации фос-
фата, то можно было бы ожидать, что ионы металлов сами по
себе ускоряют гидролиз производных фосфата, поскольку ком-
плексы с металлами образуются и в отсутствие ферментов. Одна-
ко это предположение не подтверждается. Хотя каталити-
ческая роль металлов в модельных системах и обнару-
живается [93], эффекты вовсе не велики, если реагент не
имеет необычных структурных особенностей. Например, ионы
магния ускоряют гидролиз дианиона ацетилфосфата СН3СООРО3~
[94], однако всего ib 3 раза при концентрации металла 0,1 М.
Если предположить, что константа диссоциации комплекса
Mg(CH3COOPO3) не больше, чем комплекса Mg(HPO4) [7] (она
может быть меньше ввиду возможного участия кислородного
атома карбонильной группы в связывании), то оказывается, что
СН3СООРО23” гидролизуется в комплексе с Mg2+ всего в 4—5 раз
быстрее, чем в свободном виде.
Влияние M.g'2+ на гидролиз АТФ еще меньше [92], несмотря
на то что этот металл служит активатором большинства киназ.
При pH 9, т. е. когда молекула АТФ депротонирована, магний
ускоряет гидролиз в 2 раза, тогда как при pH 5, когда молекула
свободного АТФ присоединяет один протон, магний даже несколь-
ко ингибирует гидролиз. В случае Са2+ и Мп2+ ускорение не-
Фосфатный перенос и его активация ионами металлов
647
сколько больше и составляет 2—10. Существенное увеличение
скорости (в бОраз) дает только Си24-, однако реакция с этим
катионом протекает исключительно через димерный хелат
[ (АТФСиОН)2]6-, в котором ионы меди, вероятно, соединены
двумя гидроксильными ионами [95]. Неясно, почему этот необыч-
ный комплекс оказывается таким эффективным при гидролизе
АТФ. Скорее всего, дело здесь не в простой поляризации.
Ускорение гидролиза салицилфосфата ионами Cu2+, Fe3+,
VO2+ и VO2 также объясняется структурной особенностью суб-
/6Я1
М2+Ъ :0—Р—О
или
МОН+ +
RjO. м—о
0 = Р— Y- или 0=Р—Y- + ROH
RzO^ RjO
Рис. 17.3. Схема активации посредством поляризации.
страта, заключающейся в присутствии рядом карбоксильной
группы [96]. Эфиры фосфорной кислоты быстро гидролизуются
гидроокисями La (III), Ce(III), Ce(IV), Th(IV) и Pb(II) [97, 98].
В присутствии гидроксигеля La (III) при pH 8,5, 78 °C скорость
распада оксиэтилфосфата увеличивается в 1000 раз [99]. Следует
отметить, что детали этих гетерогенных реакций практически не-
известны.
Можно предположить, что хотя в водных растворах бинарных
комплексов выраженный эффект поляризации отсутствует, он
может существовать в тройных ферментных комплексах. Эффек-
тивная диэлектрическая проницаемость в активном центре, веро-
ятно, значительно больше. С другой стороны, есть основания счи-
тать, что отсутствие активации фосфата в простых комплексах
объясняется не отсутствием поляризационного влияния на связь
Р—О. Скорее всего, поляризация компенсируется оттягиванием
42*
648
Глава И
атомом фосфора заряда от остальной части молекулы. Это свой-
ство атома фосфора строго вытекает из анализа колебательных
спектров производных фосфата и их комплексов с металлами,
проведенного Бринцингером и Хестором [100]. Оказывается, что
силовая константа для растяжения связи Р—О уменьшается
только для атомов кислорода, связанных с ионом металла, а для
свободных атомов кислорода она увеличивается, в результате
чего средняя силовая константа связи Р—О остается неизменной.
Похожим образом ведут себя другие оксианионы: сульфат, карбо-
нат и нитрат. Предполагается, что поляризация компенсируется
О3
I 1,517
О -Н—o4|.56q_p1.608 О
| 1.497 'R
Рис. 17.4. Структура серилфосфата [19J.
Значительным образованием зт-связей между атомами кислорода
и центральным атомом с использованием вакантных 2р-орбита-
лей атомов углерода и азота и Зй-орбиталей атомов фосфора и
серы. Электроны легко передаются от одного атома кислорода к
другому без большого изменения заряда центрального атома.
Структурные данные подтверждают такую точку зрения. Кру-
икшанк [101] обнаружил, что средняя длина связи Р—О в произ-
водных фосфата почти постоянна. Присоединение заместителя к
одному из кислородных атомов ортофосфата может существенно,
на 0,15 А, увеличить длину связи Р—О, однако остальные связи
Р—О укорачиваются таким образом, что средняя длина сохраня-
ется равной 1,54 А. Структура серил фосфата [102], показанная
на рис. 17.4, иллюстрирует этот эффект. Видно также, что влия-
ние протона на длину связи Р—О вдвое меньше влияния остатка
аминокислоты. Эта закономерность является общей и, вероятно,
объясняется образованием в кристалле прочных межмолекуляр-
ных водородных связей [101]- Можно полагать, что она справед-
лива и в водных растворах, и на протонных центрах белков.
В солях, образуемых металлами с фосфатом и пирофосфатом
[105], значительное удлинение связей Р—О под действием ме-
таллов не происходит, однако все исследованные кристаллы этих
соединений представляют собой трехмерные ионные решетки, в
которых все кислородные атомы присоединены к ионам металла
и наоборот, в результате чего поляризационный эффект уничто-
Фосфатный перенос и его активация ионами металлов 649
жается. По-видимому, до сих пор неизвестна структура ни одного
отдельного комплекса металл — фосфат.
Хотя образование комплекса с ионом металла обычно не уско-
ряет значительно гидролиз простых производных фосфата, с поли-
рибонуклеотидами дело обстоит иначе. В этих полимерах фосфо-
диэфирные связи быстро гидролизуются в присутствии некоторых
металлов, особенно Zn2+, с образованием монорибонуклеотидов,
которые не распадаются дальше до фосфата [106]. Атом фос-
фора фосфодиэфирной группы уже поляризован под действием
двух эфирных связей, и это может служить объяснением того, что
дополнительная поляризация при связывании иона металла не
может быть компенсирована оттягиванием электронов от кисло-
родных атомов (гл. 34).
4.4. Активация воды
Усиление нуклеофильных свойств воды представляется более
вероятной функцией ионов металлов по сравнению с активацией
фосфата посредством поляризации. У кислородного атома воды
нет богатых электронами заместителей, которые могли бы ком-
пенсировать поляризующее влияние металла, поэтому прочность
связей О—Н координированной молекулы воды существенно
Рис. 17.5. Предполагаемый механизм гидролиза зарина (Ri=изопропил, Р2=ме-
тил), ускоряемого [(АА)СиОН] + (АА=хелатирующий диамин).
уменьшается и оказывается возможной ионизация молекулы.
Таким образом, ион металла способен образовать гидроксильный
ион или близкое к нему состояние при физиологических значе-
ниях pH. Хотя нуклеофильные свойства связанного иона гидро-
ксила, скорее всего, хуже, чем в свободном состоянии, они тем
не менее могут быть лучше, чем у воды. Более того, активность
связанного иона ОН- может существенно увеличиваться благода-
ря его благоприятной для реакции ориентации. Этот эффект объ-
ясняет способность комплекса [Сц(АА)ОН]+ (АА=хелатирую-
щий диамин) ускорять гидролиз метилизопропилфторфосфата
(зарина) [108]. Вероятно, ион меди взаимодействует с атомом
фгора во время атаки иона гидроксила на атом фосфора
(рис. 17.5). В некоторых хелатах связанный ион ОН- активнее
свободного [108]. Быстрый гидролиз производных фосфата гид-
роксидами лантанидов также, вне всякого сомнения, объясняется
650
Глава 17
участием связанного гидроксильного иона. Об эффективности
этого катализа известно давно, однако образование осадков за-
трудняет изучение его механизма [109—111]. 'Судя по данным
Селвина [112], в случае АТФ гидролизуется растворимый комп-
НзМ
H3N
МНз
NH3
ин2-снг
с=о
НН3
I
0С2Н5
+ HjO
быстро
/rlt быстро
МНз OCjHs
H3N
H3N
Н;0
быстро
медленно \ К2
HOCjHj 4- “ОН
Рис. 17.6. Предполагаемый механизм образования хелатированного имида гли-
цина из этилового эфира глипина, связанного с пентаампнокобальтом(Ш) [113].
леке АТФ(М.3+)г и pH-зависимость скорости реакции согласуется
с участием координированного иона гидроксила.
Пока нет прямых данных в пользу того, что определенная мо-
лекула воды или гидроксил-ион, связанные с катионом металла,
оказываются в конце концов присоединенными к атому фосфора.
Однако Бакингем и сотр. [113] показали прямым методом нали-
Фосфатный перенос и его активация ионами металлов
651
чие нуклеофильных свойств у координированной молекулы ам-
миака. Если этилглицинат присоединен своим аминным концом к
комплексу [Со(МНз)5]3+, то продуктом его гидролиза в NaOH
оказывается хелатированный имид NH2CH2CH2CONH. Очевидно,
что для этого необходима атака одной из координированных мо-
лекул аммиака по карбонильному атому углерода. Предполагае-
мый механизм изображен на рис. 17.6. Трудно сомневаться в том,
что молекула воды может выполнять в гидролитических реакциях
похожую роль.
В ферментативных реакциях самые убедительные доказатель-
ства участия механизма с координированным ионом гидроксила
получены для карбоангидразы. По данным инфракрасной спек-
троскопии, молекула субстрата (СОг) не связана с атомом цин-
ка, но расположена сравнительно близко от него, чтобы быть
атакованной координированным гидроксильным ионом [82]
(гл. 16, разд. 7). С этим механизмом согласуется зависимость
активности от pH и природы разнообразных ингибиторов [114,
115]. Селвин [112] предложил похожий механизм для АТФ-азы
митохондрий, участвующей в окислительном фосфорилировании.
Он основывался на сходстве pH-зависимостей ферментативной
реакции и упомянутого выше ускорения гидролиза АТФ ионами
лантанидов. Однако величина рКа для АТФ-азной реакции (при-
мерно 7) была намного выше рКа воды в комплексе с активирую-
щими двухзарядными ионами. Для наилучших активаторов
Mg2+, Со24- и ZnE+ эта величина примерно равна соответственно
~ 12, ~9 и ~9 [87]. Селвин [112] предполагает, что кислот-
ность комплексов может увеличиваться под влиянием положи-
тельно заряженных групп фермента, расположенных поблизости.
Надо признать, что расхождение в 5 единиц pH для M.g2+ слиш-
ком велико, чтобы его можно было объяснить таким образом
(необходимая для этого энергия электростатического взаимодей-
ствия должна составлять примерно 7 ккал/моль). Выше говори-
лось о том, что ионизация свободной молекулы воды предложена
в качестве объяснения перехода при pH 8,5, наблюдаемого для
2пи+-содержащей щелочной фосфатазы. При этом увеличение pH
приводило к уменьшению активности фермента. В силу этого
можно считать, что механизм с координированным гидроксиль-
ным ионом вряд ли играет существенную роль при ферментатив-
ном гидролизе фосфатов.
4.5. Стереохимические модели
Тот факт, что в бинарных комплексах с производными фосфа-
та ионы металлов лишь незначительно ускоряют гидролиз, пред-
полагает качественное различие во взаимном расположении ионов
металла и активируемой фосфатной группы в бинарном комплек-
652
Глава 17
се и в тройном комплексе с участием фермента, переносящего
фосфат. Возможно, что ион металла играет лишь косвенную роль,
создавая надлежащую ориентацию фермента и субстрата. С дру-
гой стороны, может быть, что присоединение бинарного комплек-
са к ферменту сопровождается такими стереохимическими изме-
нениями, которые оказываются благоприятными для реакции. Эту
дилемму можно разрешить, изучая модельные системы фермент—
н+ + CL Л 4- СН3ОН
. P'S
(Г хон
Рис. 17.7. Предполагаемый механизм псевдовращения, объясняющий быстрый
гидролиз этиленметилфосфата с образованием этиленфосфата н метанола fl6].
свободный металл — фосфат, выбранные с таким расчетом, чтобы
проверить конкретные стереохимические гипотезы.
Интересная гипотеза была недавно выдвинута Куперманом
[116], однако она, по-видимому, опровергается его же экспери-
ментальными данными. Исходя из того, что несимметричные ди-
эфиры пирофосфорной кислоты гидролизуются в 10® раз быстрее
соответствующих моноэфиров [117], Куперман предположил, что
металл, присоединяясь к замещенной фосфатной группе моно-
эфира пирофосфата, дает похожий эффект. Обычно ионы метал-
лов образуют с полифосфатами хелатные комплексы, но в ак-
тивном центре фермента металл может вовлекаться в связь толь-
ко со второй от конца фосфатной группой полифосфата, например
АТФ, и активировать таким образом концевую фосфатную груп-
пу. Для того чтобы обеспечить связывание металла только со
второй от конца, но не с концевой фосфатной группой, было
получено несколько замещенных фосфонилфосфатов и фосфонил-
пирофосфатов. В качестве заместителей были использованы хе-
латообразующие полиаминные и фенолдиаминные группы. Кон-
станты скорости гидролиза комплексов с катионами различных
двухвалентных металлов существенна не превышали константы
Фосфатный перенос и его активация ионами металлов
653
скорости гидролиза свободных лигандов. Методом ЯМР с 31Р
было показано, что по крайней мере в комплексе с Zn124- реали-
зуется желаемая структура. Отсюда следует, что механизм при-
соединения металла ко второй от конца фосфатной группе можно
исключить из рассмотрения.
Другим возможным механизмом влияния металлов является
механизм бидентатной активации, предложенный Фаррелом и др.
[118]. Имеются данные в его пользу, однако строго он не доказан.
Этот механизм также основывается на аналогии с быстрогидро-
лизующимся фосфатным эфиром, а именно пятичленным цикли-
ческим эфиром, этиленфосфатом. Давно известно, что этиленфос-
фат гидролизуется намного быстрее нециклических диэфиров и
что циклическая пятичленная структура объясняет нестойкость
циклического промежуточного соединения, образующегося под
действием панкреатической рибонуклеазы [14]. Кроме того,
этиленфосфат быстро обменивается кислородными атомами с во-
дой [119] и гидролиз этиленметилфосфата до метанола и этилен-
фосфата с сохранением циклической структуры происходит в не-
сколько миллионов раз быстрее гидролиза триметилфосфата
[120, 121]. Вестхаймер [16] предложил для этих необычных зако-
номерностей красивое объяснение, которое сводится к тому, что
реакции гидролиза и обмена протекают через пятикоординацион-
ное соединение со структурой тригональной бипирамиды
(рис. 17.7). Этиленовая группа соединяет аксиальный и эквато-
риальный атомы кислорода тригональной бипирамиды, поскольку
угол у атома фосфора, равный 90°, легко встраивается в пяти-
членное кольцо, чего нельзя сказать об угле в 120° между двумя
экваториальными атомами кислорода. Известно, что цикл этилен-
фосфата напряжен, так как угол О—Р—О, образуемый этилено-
вой группой, на 10° меньше, чем в правильном тетраэдре [122].
Энергия активации образования постулированного промежуточ-
ного соединения в форме тригональной бипирамиды понижается
благодаря энергии, освобождающейся при снятии этого напря-
жения. Можно считать, что аксиальные положения промежуточ-
ного соединения будут более реакционноспособными по сравне-
нию с экваториальными, и легкий разрыв связи Р—О снаружи и
внутри кольца объясняется быстрым внутримолекулярным взаи-
мопревращением аксиального и экваториального положения
(рис. 17.7). Этот переход, являющийся по сути дела продолже-
нием одного из нормальных колебательных движений промежу-
точного соединения, назван «псевдовращением», поскольку его
результатом является поворот экваториальной плоскости триго-
нальной бипирамиды на 90°.
Фаррелл и сотр. [36] предположили, что ион металла может
вызывать аналогичную активацию, если он соединяет два кисло-
родных атома, принадлежащих одной фосфатной группе, т. е. если
654
Глава 17
активируемая фосфатная группа служит бидентатным лигандом.
В результате должны получиться четырехчленная циклическая
структура, и хотя связи металл — кислород менее направленны и
более гибки по сравнению со связями этилен — кислород, угол
О—Р—-О в ней будет заметно напряжен. Если это так, то и в
этом случае может образоваться промежуточное соединение в
форме тригональной бипирамиды, при псевдовращении которого
может происходить быстрый обмен акцепторов фосфорильной
группы.
Возможно, что бидентатная координация не является пред-
почтительным способом взаимодействия ионов металлов с произ-
водным фосфатов в бинарных комплексах. Действительно, если
напряжение в кольцевой структуре бидентатного комплекса ве-
лико, то можно ожидать, что он будет неустойчив по сравнению
с монодентатным комплексом. Активация в тройном комплексе
фермент—металл — субстрат может быть результатом сдвига в
сторону бидентатной координации фосфатной группы вследствие
стерического влияния активного центра. Для АТФ известно, что
связываемые ионы металлов хелатируются атомами кислорода,
принадлежащими разным фосфатным группам, с образованием
циклических структур [423, 124]. Для бидентатной активации
переноса фосфата от АТФ необходимо присоединение металла к
двум кислородным атомам концевой фосфатной группы. Анало-
гично этому для бидентатной активации переноса нуклеотидной
группы необходима координация с двумя кислородными атомами
а-фосфатной группы. Хотя любая из этих структур гораздо менее
выгодна по сравнению с шестичленными кольцевыми структура-
ми, под влиянием подходящего фермента они могут стабилизиро-
ваться.
Пытаясь определить, действительно ли бидентатная координа-
ция активирует производные фосфата, Фаррелл и др. [118] про-
анализировали скорость гидролиза метилфосфата в составе
комплексов с Со (III), имеющих одну или две вакантные связи.
Кобальт (III) был выбран в качестве центра координации из тех
соображений, что инертность его координационных связей должна
была облегчить распознавание моно- и бидентатных структур.
Экспериментально было обнаружено, что гидролиз СН3РО^ уско-
ряется примерно в 130 раз при связывании с [Co(trien)]3+
(trien = триэтилентетрамин; в этом комплексе с тетрадентатным
лигандом свободны два координационных места) по сравнению
с гидролизом СН3РО4Н". Последний анион можно рассматривать
как продукт соединения СН3РОГ с Н+, и он кинетически важен
при гидролизе метилфосфата в воде [125]. Скорость гидролиза
СН3РО4 в составе комплекса с [Co(NH3)5]3+ (свободно одно'
место координации), напротив, по меньшей мере на два порядка
ниже. Значительная устойчивость метилфосфата к гидролизу в-
Фосфатный перенос а его активация ионами металлов 655
том случае, если он присоединен через один атом кислорода к
имеющему тройной положительный заряд иону [Со(ЫНз)5]3+,
служит еще одним доказательством против важности эффекта
поляризации.
К сожалению, трудно объяснить сравнительно быстрый гидро-
лиз в случае триэтилентетрааминокобальта, поскольку вопреки
ожиданиям метилфосфат соединен с ним главным образом через
один атом кислорода, а не через два, причем шестое координа-
ционное положение занимает вода. Линкольн и Штранкс [126]
показали, что для ортофосфата, присоединенного к аналогично-
ОНг
/°\Ь°
|tn«n)Co
V Ъоснз
о О
(trien)Co ^-Р-^ОСНз
^0 о
«2
Рис. 17.8. Альтернативный механизм гидролиза метилфосфата, ускоряемого три-
этилентетрааминокобальтом (III).
а — бидентатная активация фосфата; б — активация связанной воды [118].
му комплексу [Со(еп)г]3+( еп=этилендиамин; свободны два ко-
ординационных положения), существует равновесие между моно-
и бидентатными структурами, которое устанавливается при ком-
натной температуре за несколько минут. При этом бидентатная
координация преобладает для РО|', а монодентатная — для
HPOV - Ясно, что СНзРО?- похож на НРОГ . Следовательно,
если ускорение гидролиза СН3РО?г, присоединенного к
[Со (trien) ]3+, объясняется бидентатной активацией, то на самом
деле эффект значительно больше, чем представляется, поскольку
бидентатная координация существует только в небольшой доле
комплекса. Нельзя, однако, не учитывать возможность того, что
наблюдаемое ускорение связано с нуклеофильной атакой связан-
ной молекулы воды в монодентатном комплексе. В этом случае
можно было бы говорить о механизме с активацией воды, рас-
смотренном в предыдущем разделе. Оба возможных объяснения
представлены на рис. 17.8.
Пока нет прямых данных за или против гипотезы бидентатной
активации в ферментативных реакциях, однако Милдван [79]
указывает на возможность объяснить сравнительно низкую ско-
рость присоединения фторфосфата к комплексу пируваткиназа —
Мп|2+ [127] образованием напряженного комплекса с бидентатной
координацией (см. «Дополнения, внесенные в корректуру»).
656
Глава 17
ДОПОЛНЕНИЯ, ВНЕСЕННЫЕ В КОРРЕКТУРУ
1. Лаздунски и др. [128, 129] обнаружили, что фосфорилиро-
вание щелочной фосфатазы в кислой среде также характеризу-
ется антикооперативностью. На кривой зависимости отношения
количества ковалентно связанного фосфата к количеству фермен-
та от концентрации фосфата отчетливо виден излом в точке, от-
вечающей присоединению 1 моля фосфата на 1 моль белка.
Степень антикооперативности, определяемая величиной излома,
зависит от pH, температуры и природы металла в активном
центре. Во всех случаях димерная молекула фермента прочно
присоединяет ковалентным образом одну молекулу фосфата. Свя-
зывания второй молекулы фосфата можно достигнуть достаточ-
ным увеличением его концентрации. В щелочной среде фосфорил-
фермент менее стабилен, чем нековалентный комплекс с фосфатом,
за исключением неактивного Сб2+-фермента. В случае активных
Zn2+ и Со2+-фосфатаз фосфорилированное промежуточное соеди-
нение можно обнаружить после реакции с субстратами при низ-
кой температуре. В этих условиях ковалентно присоединяется не
более 1 моля фосфата на 1 моль фермента. При реакции моно-
фосфорилфермента с конкурентным ингибитором, п-хлоранилидо-
фосфонатом, связанный фосфат быстро освобождается. Лаздунски
и др. [129] предложили для этого фермента механизм типа
«качелей». В его основе лежит предположение о том, что в обыч-
ных условиях в каждый момент времени в фосфорилированном
состоянии может находиться только одна из субъединиц фермен-
та. Вторая субъединица может связать молекулу субстрата. Когда
это происходит, она фосфорилируется, а ее партнер одновремен-
но освобождает ковалентно присоединенный фосфат и может в
свою очередь быть фосфорилирован другой молекулой субстрата.
Таким образом, две половины фермента осциллируют между фос-
форилированным и нефосфорилированным состояниями, которые
предположительно связаны конформационным переходом.
2. Кон и сотр. [130, 131] объяснили данные для креатинкина-
зы по релаксации линии спектра ЯМР взаимодействием иона
металла-активатора с двумя фосфатными группами АДФ, а не с
фосфорильной группой, переносимой от креатина к АДФ. Это со-
гласуется с механизмом элиминирования метафосфата, тем более
что неферментативный гидролиз креатинфосфата также протека-
ет с отщеплением метафосфата [132], поскольку креатин пред-
ставляет собой весьма благоприятную уходящую группу. Следо-
вательно, для этого фермента механизм бидентатной активации
можно исключить. Он может все же существовать для ферментов,
которые катализируют перенос фосфорильной группы между худ-
шими уходящими группами и для которых ассоциативный меха-
низм может быть более вероятен [36].
Фосфатный перенос и его активация ионами металлов 657
СПИСОК ЛИТЕРАТУРЫ
I. Lippman F., Adv. Enzymol., 1, 99 (1941).
2. Kaplan N. О., Kennedy E. P. (eds.), Current Aspects of Biochemical Energe-
tics, Academic Press, New York, 1966.
3. Kalckar H. M„ Biological Phosphorylations, Prentice-Hall, Englewood Cliffs,
4. Atkinson M. R., Morton R. K., in M. Florkin, H. S. Mason (eds.), Comparative
Biochemistry, Vol. II, Academic Press, New York, 1960, p. 1.
5. Morton R. K. in M. Florkin, E. H. Stotz (eds.), Comprehensive Biochemistry,
Vol. 16, Elsevier, New York, 1965.
6. Segel I. H., in J. Williams, E. M. Lansford, Jr. (eds.), The Encyclopedia of
Biochemistry, Reinhold, New York, 1967, p. 642.
7. Bock R. M., in P. Boyer, H. Lardy, K. Myrback (eds.), The Enzymes, Vol. 2,
Academic Press, New York, 1960, pp. 15, 16.
8. Lehninger A., Bioenergetics, W. A. Benjamin, New York, 1965.
9. Racker E., Mechanisms in Bioenergetics, Academic Press, New York, 1965.
10. Kornberg A., in M. Kasha, B. Pullman (eds.), Horizons in Biochemistry, Aca-
demic Press, New York, 1962, p. 251.
11. Cohn M„ in P. Boyer, H. Lardy, K. Myrback (eds.), The Enzymes, Vol. 5, Aca-
demic Press, New York, 1961, p. 179.
12. Grunberg-Manago M., in P. Boyer, H. Lardy, K. Myrback (eds.), The Enzymes,
Vol. 5, Academic Press, New York, 1961, p. 257.
13. Cohn M., J. Biol. Chem., 180, 771 (1949).
14. Bruice T. C., Benkovic S.. Bioorganic Mechanisms, Vol. II, W. A. Benjamin,
New York, 1966, Ch. 5.
15. Quin L. D., in J. Hamer (ed.), 1,4-Cycloaddition Reactions, Academic Press,
New York, 1967, pp 47—96.
16. Westheimer F. H., Accounts Chem. Res., 1, 70 (1968).
17. DiSabato G„ Jencks W. P., J. Am. Chem. Soc., 83, 4400 (1961).
18. Kirby A. J., Varvoglis A. G., J. Am. Chem. Soc., 89, 415 (1967).
19. Schmidt G., in P. Boyer, H. Lardy, K. Myrback (eds.), The Enzymes, Vol. 5,
Academic Press, New York, 1961, p. 37.
20. Stadtman T. C., in P. Boyer, H. Lardy, K. Myrback (eds.), The Enzymes, Vol. 5,
Academic Press, New York, 1961, p. 55.
21. Byrne W. L., in P. Boyer, H. Lardy, K. Myrback (eds.), The Enzymes, Vol. 5,
Academic Press, New York, 1961, p. 73.
22. Heppel L. A., in P. Boyer, H. Lardy, K- Myrback (eds.), The Enzymes, Vol. 5,
Academic Press, New York, 1961, p. 49.
23. Kunitz M., Robbins P. W., in P. Boyer, H. Lardy, K- Myrback (eds.), The En-
zymes, Vol. 5, Academic Press, New York, 1961, p. 169.
24. Cori C. F„ Brown D. H„ in M. Florkin, E. H. Stotz (eds.). Comprehensive Bio-
chemistry, Vol. 15, Elsevier, New York, 1964, p. 212.
25. Barnard E. A., A. Rev. Biochem., 38, 677 (1969).
26. Revel H. R., Racker E.. Biochim. Biophys. Acta, 43, 465 (1960).
27. Hanson A. W., Applebury M. L., Coleman J. E„ Wyckoff H. W., Richards F. M„
J. Biol. Chem., 245, 4975 (1970).
28. Reid T. W., Wilson I. B., in P. D. Boyer (ed.), The Enzymes, 3rd edn., Vol. 4,
Academic Press, New York, p. 373.
29. Horiuchi T., Horiuchi S., Mizuno D„ Nature, 183, 1528 (1959).
30. Torriani A., Biochim. Biophys. Acta, 38, 460 (1960).
31. Garen A., Levinthal C, Biochim. Biophys. Acta, 38, 470 (1960).
32. Stadtman T. C., in P. D. Boyer et al. (eds.), The Enzymes, Vol. 5, Academic
Press, New York, 1961, p. 55.
33. Heppel L. A., Harkness D. R„ Hilmoe R. J., J. Biol. Chem., 237, 841 (1962),
34. Neumann H„ Boross L„ Katchalski E„ J. Biol. Chem., 242, 3142 (1967).
35. Neumann H., J. Biol. Chem., 243, 4671 (1968).
f>58
Глава 17
36. Breslow R., Katz J., J. Am., Chem. Soc., 90, 7376 (1968).
37. Wilson I. B„ Dayan I., Cyr K., J. Biol. Chem., 239, 4182 (1964).
38. Garen A., Levinthal C., Biochim. Biophys. Acta, 38, 470 (1960).
39. Simpson R. T., Vallee B. L., Tait G. H., Biochemistry, 7, 4336 (1968).
40. Applebury M. L., Coleman I. E., J. Biol. Chem., 244, 308 (1969).
41. Levinthal C., Signer E. R., Fetherolf K-, Proc. Natn. Acad. Sci. U. S., 48, 1230
(1962).
42. Rothman F., Byrne R., J. Mol. Biol., 6, 330 (1963).
43. Schlesinger M. I., Barret K., J. Biol. Chem., 240, 4284 (1964).
44. Reynolds I. A., Schlesinger M. J., Biochemistry, 8, 4287 (1969).
45. Cohen S. R., Wilson I. B., Biochemistry, 5, 904 (1966).
46. Csopak H„ Eur. J. Biochem., 7, 186 (1969).
47. Reynolds J. A., Schlesinger M. L, Biochemistry, 6, 3552 (1967); 7, 2080
(1968).
48. Simpson R. T., Vallee B. L., Biochemistry, 7, 4343 (1968).
49. Lazdunski C., Petitclerc C., Lazdunski M., Eur. J. Biochem., 8, 510 (1969).
50. Csopak H., Falks K- E„ FEBS Lett., 7, 147 (1970).
51. Applebury M. L., Johnson В. P., Coleman J. E., J. Biol. Chem., 245, 4968
(1970).
52. Gottesman M., Simpson R. T., Vallee B. L., Biochemistry, 8, 3776 (1969).
53. Tait G. H., Vallee B. L., Proc. Natn. Acad. Sci. U. S., 56, 1247 (1966).
54. Lazdunski C., Lazdunski M., Eur. J. Biochem., 7, 294 (1969).
55. Applebury M. L., Coleman J. E., J. Biol. Chem., 244, 709 (1969).
56. Dori Z., Gray H. B., J. Am. Chem. Soc., 88, 1394 (1966).
57. Ciampolini M., Nardi N., Inorg. Chem., 6, 445 (1967).
58. Tait G. H., Vallee B. L., Proc. Natn. Acad. Sci. U. S., 56, 1247 (1966).
59. Reynolds J. A., Schlesinger M. J., Biochemistry, 8, 589 (1969).
60. Engstrom L., Agren G„ Acta Chem. Scand., 12, 357 (1958).
61. Schwartz J. H., Lippman F., Proc. Natn. Acad. Sci. U. S., 47, 1996 (1962).
62. Wilson I. B., Dayan I., Biochemistry, 4, 645 (1965).
63. Levine D., Reid T. W., Wilson I. B., Biochemistry, 8, 2374 (1969).
64. Reid T. W., Pavlic M., Sullivan D. J., Wilson I. B., Biochemistry, 8, 3184
(1969).
65. Aldridge W. N., Barman T. E„ Gutfreund H., Biochem. J., 92, 23c (1964).
66. Fernley H. N„ Walker P. G.. Biochem. J., 10, lip (1968).
67. Fernley H. N„ Walker P. G., Nature, 212, 1435 (1966).
68. Trentham D. R„ Gutfreund H„ Biochem. J., 106, 455 (1966).
69. Gutfreund FL, Biochem. J., 110, lip (1968).
70. Halford S. E., Bennett N. G„ Trentham D. R., Gutfreund H., Biochem. J., 114,
243 (1969).
71. Lazdunski C., Petitclerc C„ Chappelet D., Lazdunski M„ Biochem. Biophys.
Res. Commun., 37, 744 (1969).
72. Ko S. H. D., Kezdy F. J., J. Am. Chem. Soc., 89, 7139 (1967).
73. Fife W. K., Biochem. Biophys. Res. Commun., 28, 309 (1967).
74. Engstrom L., Biochim. Biophys. Acta, 56, 606 (1962), Arkiv. Kemi, 19, 129
(1962).
75. Pigretti M. M., Milstein C., Biochem. J., 94, 106 (1968).
76. Simpson R. T„ Vallee B. L., Biochemistry, 9, 953 (1970).
77. Levitzki A., Koshland D. E., Jr., Proc. Natn. Acad. Sci. U. S., 62, 1121
(1969).
78. Lipscomb W. N., Hartsuck J. A., Quiocho F. A., Reeke G. N., Jr., Proc. Natn.
Acad. Sci. U. S„ 64, 28 (1969).
79. Mildvan A. S., in P. D. Boyer (ed.), The Enzymes, 3rd edn., Vol. I, Academic
Press, New York, 1970.
80. Hellerman L., Physiol. Rev., 17, 454 (1937).
81. Mildvan A. S.. Cohn M., Leigh J. S., Jr., Biochemistry, 6, 1805 (1967).
82. Riepe M. C„ Wang J. H., J. Biol. Chem., 243, 2779 (1968).
Фосфатный перенос и его активация ионами металлов
659
83. Cohn М„ Leigh J. S., Jr.. Nature, 193, 1037 (1962).
84. Cuatrecasas P., Fuchs Anfinsen С. B., J. Biol. Chem., 242, 3063 (1967).
85. Cuatrecasas P., Tanuichi H., Anfinsen С. B., Brookhaven Symp. Biol., 21, 172
(1968).
86. Arnone A., Bier C. J., Cotton F. A., Hazen E. E., Jr., Richardson D. C.,
Richardson J. S., Proc. Natn. Acad. Sci. U. S., 64, 420 (1969).
37. Sillen L. G., Martell A. E., Stability Constants of Metal Complexes, The Che-
mical Society, London, 1964.
88. Lindahl T., Adams A., Fresco J. R„ Proc. Natn. Acad. Sci. U. S., 55, 941
(1966).
89. Cooperman B., Lloyd G. J., J. Am. Chem. Soc., 93, 4883 (1971).
90. Breslow R., Chipman D., J. Am. Chem. Soc., 87, 4195 (1965).
91. Lowenstein J. M., Biochem. J., 70, 222 (1958).
92. Tetas M„ Lowenstein J., Biochemistry, 2, 350 (1963).
93. Bender M. L„ in «Reactions of Coordinated Ligands and Homogeneous Cata-
lysis», Adv. Chem. Ser., 37, 19—35 (1963).
94. Koshland D. E., J. Am. Chem. Soc., 74, 2286 (1952).
95. Spiro T. G., Kjellstrom W. A., Zydel M. C., Butow R. A., Biochemistry, 7, 859
(1968).
96. Hofstetter R., Murakami Y., Mont G., Martell A. E., J. Am. Chem. Soc., 84,
3041 (1962).
97. Bamann E., Mutterlein W. D., Chem. Ber., 91, 471, 1322 (1958), and oter re-
ferences cited therein to the work of Bamann and his associates.
98. Dimroth K., Witzel H., Hillsen W., Mirbach H., Ann., 620, 94 (1959).
99. Butcher W. W., Westheimer F., J. Am. Chem. Soc., 77, 2420 (1955).
100. Brintziner H., Hester R. E., Inorg. Chem., 5, 980 (1966).
101. Cruichshank D. W. J., J. Chem. Soc., 1961, 5486.
102. McCallum G. H., Robertson J. M„ Sim G. A., Nature, 184, 1863 (1959).
103. Tables of Interatomic Distances and Configuration in Molecules and Ions, Spe-
cial Publication No. 11, The Chemical Society, London 1958.
101. Geller S., Durand J. L., Acta Cryst., 13, 325 (1960).
105. Calvo C., lonrg. Chem., 7, 1345 (1968).
106. Eichhorn G. L., in S. Kirschner (ed.), J. C. Bailar Symposium on Coordination
Chemistry, Plenum Press, New York, 1969, p. 92.
107. Butzow J. J., Eichhorn G. L., Biopolymers, 3, 95 (1965).
108. Gustafson R. L., Martell A. E., J. Am. Chem. Soc., 84, 2309 (1962).
109. Bamann E., Fischer F., Trapmann H., Biochem. Z., 325, 413 (1954).
110. Bamann E., Trapmann H., Adv. Enzymol., 21, 169 (1959).
111. Butcher W. W., Westheimer F. H., J. Am. Chem. Soc., 77, 2420 (1955).
112. Selwyn M. J., Nature, 219, 490 (1968).
113. Buckingham D. A., Foster D. M., Sargeson A. M., J. Am. Chem., Soc., 91, 3451
(1969).
114. Davis R. P„ in P. D. Boyer, H. Lardy, K. Myrback (eds.), The Enzymes, Vol. 5,
2nd edn., Academic Press, New York, 1961, p. 545.
115. Edsall J. T„ Harvey Leet. Ser., 62, 191 (1967).
116. Cooperman B„ Biochemistry, 8, 5005 (1969).
117. Miller D. L., Ukena T., J. Am. Chem. Soc., 91, 3050 (1969).
118. Farrell F. J., Kjellstrom W., Spiro T. G., Science, 164, 320 (1969).
119. Haake P. C„ Westheimer F. H., J. Am. Chem. Soc., 83, 1102 (1961).
120. Covitz F„ Westheimer F. H„ J. Am. Chem. Soc., 85, 1773 (1963).
121. Dennis E. A., Westheimer F. H., J. Am. Chem. Soc., 88, 3432 (1966).
122. Steitz T. A., Lipscomb W. H„ J. Am. Chem. Soc., 87, 2488 (1965).
123. Cohn M„ Hughes T. R., Jr., J. Biol. Chem., 235, 3250 (1960); 237, 176
(1962).
124 Sternlicht H., Shulman R. G., Anderson E. W., J. Chem. Phys. 43, 3123
(1965).
БбО
Глава 17
’25. Bunion С. A., Llewellyn D. Oldham К.. G., Vernon С. A., J. Chem. Soc.,
3574 (1958).
126. Lincoln F„ Stranks D. R., Aust. J. Chem., 21, 57 (1968).
127. Mildvan A. S., Leigh J, S„ Jr., Cohn M., Biochemistry, 6, 1805 (1967).
128. Lazdunski C., Chappelet D., Petitclerc C., Leterrier F„ Douzou P„ Lazdun-
ski M., Eur. J. Biochem., 17, 239 (1970).
129. Lazdunski M„ Petitclerc C„ Chappelet D., Lazdunski C., Eur. J. Biochem., 20,
124 (1971).
130. Cohn M„ Reuben J., Accounts Chem. Res., 4, 215 (1971).
131. Leigh J. S., Jr., Ph. D. Dissertation, University of Pennsylvania, 1971.
132. Haake P., Allen G. W., Proc. Natn. Acad. Sci. U. S, 68, 2691 (1971).
ГЛАВА 18
КИНАЗЫ
У. Дж. О’Сулливан
O’Sullivan W. Department of Medicine, University
of Sydney, Sydney, N. S. W, Australia
1. ВВЕДЕНИЕ
Киназы составляют большую группу ферментов, катализирую-
щих перенос концевой фосфорильной группы от АТФ к молекуле
акцептора по общему уравнению:
м2+
АТФ4- + НХ АДФ3- + РХ2- + Н+
(1)
Читателю можно порекомендовать обзор Нордли и Ларди [1], в
котором рассматриваются различные типы молекул акцепторов,
другие обзорные статьи шестого тома «Ферменты» [2], посвящен-
ные многим отдельным киназам, а также рекомендации Комис-
сии по ферментам о включении индивидуальных ферментов в эту
группу [3]. В данной главе мы попытались сделать некоторые
обобщения и проиллюстрировать их на примерах нескольких
более подробно изученных ферментов.
С точки зрения основной концепции этой книги наиболее ин-
тересным представляется обязательное участие катионов двухва-
лентных металлов в киназных реакциях. Чаще всего это Mg2+,
который является, вероятно, самым типичным физиологическим
активатором. Обычно магний можно заменить марганцем. Воз-
можно также участие и других ионов двухвалентных металлов —
Со2+, Zn2+ [2]. Многие киназные реакции активируются каль-
цием, однако для некоторых он является ингибитором. Такие раз-
личия, по-видимому, обусловлены разными типами взаимодейст-
вий с ионами Са2+. По классификации Кон [4, 5] (основанной на
изучении спектров ЯМР), ферменты, активируемые кальцием,
относятся к типу I. В этом случае металл связывается с фермен-
том не непосредственно, а через молекулу субстрата (Е—S—М).
При ингибировании кальцием он, скорее всего, присоединяется
прямо к белку (тип II, Е—М—S, см. гл. 14).
Исследование характера взаимодействия ферментов, субстра-
тов и активаторов в киназных реакциях потребовало привлечения
многих физико-химических методов. Наряду с традиционными
43—2451
662
Глава 18
способами изучения равновесий к ним относятся методы коорди-
национной химии и тонкие химические эксперименты (темпера-
турный скачок и т.п.). Но наиболее полезным для выяснения
природы ферментативной реакции оказалось использование мето-
дов магнитного резонанса, применение которых основано на па-
рамагнитных свойствах ионов Мп2+ [4, 5].
Основное внимание в данной главе уделено роли ионов метал-
ла в киназных реакциях. При этом мы старались выделить общие
положения, применимые, по возможности, ко всем киназам, хотя
некоторые ферменты описаны несколько более подробно.
На данной стадии изложения можно указать следующие об-
щие аспекты:
1. Ионы металлов-активаторов образуют умеренно прочные
комплексы с нуклеотидными субстратами. При pH 7—9, где ле-
жат оптимумы pH большинства киназных реакций, главными
компонентами реакционных систем являются МАТФ2-, МАДФ-
и т. д. Во многих случаях кинетический анализ свидетельствует о
том, что эти комплексы — «истинные» субстраты реакций, хотя
из этого не всегда можно сделать какие-то выводы о природе
промежуточных комплексов между ферментом, металлом и суб-
стратом [5, 6].
.2. Наиболее общими нуклеотидными субстратами являются
адениновые нуклеотиды, АТФ и АДФ. Однако многие киназы,
например креатинкиназа [7, 8] и пируваткиназа [9], обладают
более широкой специфичностью, а для некоторых ферментов
естественными субстратами могут быть и другие нуклеотиды: ГТФ
для ГТФ-аденилаткиназы и ГТФ или ИТФ для фосфоенолпиру-
ваткарбоксикиназы (фосфопируваткарбоксилазы, КФ 4.1.1.32)
[10].
3. Реакции киназ обычно обратимы, хотя для некоторых (на-
пример, для гексоииназы) реакция в направлении образования
АТФ может быть очень медленной (ср. сноски [11, 12] в гл. 14).
4. Значения Км для металлонуклеотидных субстратов обычно
имеют порядок Ю-5—10~4 М. Такого же порядка и константы
диссоциации (не обязательно равные величинам Км).
5. Молекулярные массы и аминокислотный состав киназ при-
мерно такие же, как и ферментов других групп. Заметно только
относительно большое количество киназ с цистеиновыми остатка-
ми в активных центрах.
6. Реакция в прямом направлении (т. е. перенос фосфориль-
ной группы от АТФ) обычно сопровождается небольшим отрица-
тельным изменением свободной энергии. Аткинсон и Мортон [12]
провели обстоятельный анализ термодинамики реакции переноса
фосфорильной группы. В этой области еще надо многое сделать,
поскольку лишь в немногих работах надлежащим образом учи-
тывалось присутствие различных ионных форм.
Киназы
663
2. ОБЩИЕ СВОЙСТВА КИНАЗ
2.1. Природа киназных реакций
Киназы осуществляют 'Перенос фосфорильной группы —РОз-
от АТФ. При этом образуется фосфорилированный продукт и
АДФ. Реакция, по-видимому, заключается в нуклеофильной атаке
на концевой атом фосфора в АТФ молекулой субстрата-акцепто-
ра [13]. Доказательства того, что разрывается именно концевая
связь О—Р, получены Кон [14] и Харрисоном с сотр. [15] при
изучении превращения соединений, содержащих 18О, различными
киназами. По мнению Милдвана [16], это можно объяснить сле-
дующим образом. Если фосфатная группировка АТФ имеет те же
размеры, что и неорганический трифосфат, то концевая связь
О—Р должна быть несколько длиннее (1,68 А), чем неконцевые
связи (1,61 А). Поэтому она легче разрывается. Проведенный не-
давно рентгеноструктурный анализ АТФ подтверждает это [17],
однако все подробности анализа ко времени завершения работы
над рукописью данной главы еще не были опубликованы.
Как правило, при киназной реакции осуществляется непо-
средственный перенос фосфорильной группы от донора к акцеп-
тору. Крейн [11] собрал доказательства того, что у многих киназ
не происходит промежуточное фосфорилирование фермента.
Исключение составляет реакция нуклеозиддифосфаткиназы, для
которой показано образование фосфофермента [18, 19].
2.2. Равновесие металл — субстрат
Как упоминалось выше, ноны металлов-активаторов образуют
комплексы с нуклеотидными субстратами. В некоторых случаях
комплексы образуются и с ненуклеотидными субстратами, но они
гораздо менее устойчивы и не очень существенны. Ниже рассмат-
риваются только адениновые нуклеотиды, АТФ и АДФ, поскольку
они лучше всего изучены. Однако все это относится и к другим
нуклеотидам, так как природа основания очень мало влияет на
величину рКа фосфатных групп и на их способность связывать
ионы металлов [20, 21].
Значения рКа концевых фосфатных групп у АТФ, АДФ и род-
ственных соединений лежат в интервале 6,7—7,0 [22—25]. Резуль-
таты отдельных измерений могут отличаться из-за различий
температуры, ионной силы, состава среды и т. д. [24, 25]. Так, при
pH значительно выше 7 в растворе находятся главным образом
АТФ4- и АДФ3-, имеющие 4 или 3 окси-аниона. В начале
50-х годов было установлено, что эти группы могут образовывать
комплексы с Mg2+ и другими катионами (включая одновалентные
43*
664
Глава 18
ионы Na+ и К+ [26]), хотя величины соответствующих констант
устойчивости были определены позже.
Критический обзор работ в этой области, выполненных до
1960 г., сделал Бок [27], который отметил неадекватность многих
экспериментов по определению констант устойчивости комплексов
металлов с нуклеотидами. Он также выдвинул положение о том,
что «исследования должны проводиться в растворах известной
ионной силы с нехелатирующими буферами и при таких концен-
трациях ионов металлов и фосфатов, которые позволяют добиться
наибольшей точности измерений и расчетов». В настоящее время
это положение в большинстве случаев выполняется и теперь на-
блюдается меньше расхождений в определяемых величинах.
Например, при pH 8,0 в нехелатирующем буфере и в отсутствие
мешающих ионов экспериментальные константы устойчивости для
MgATO2- лежат в интервале 5-Ю4—7-Ю4 М-1 (lgK=4,70—4,85),
а для MgAAO- — от 2-103 до 4-103М-1 (lgK=3,30—3,60).
Некоторое представление о сложности анализа подобных
систем с помощью классической потенциометрической техники
можно получить при рассмотрении системы равновесий между
АТФ и Mg2+:
Р*О1 РКаг
н2атф2- +=* НАТФ3- атф4-
К111
MgH.AT® <—> MgHAT®~ <—> MgAT®2~
Даже при разумных упрощениях полный анализ с определением
всех констант остается очень сложным. Во многих случаях эту
проблему можно решить только при помощи ЭВМ [28].
Интересующимся читателям можно рекомендовать обзорную
статью Филлипса [25] с критическим рассмотрением этой обла-
сти, особенно определений термодинамических параметров комп-
лексообразования, а также другие статьи той же группы [29] и
работу Такуи Хана и Мартелла [30]. Однако здесь следует под-
черкнуть, что не все биохимики при работе с ферментами и, в
частности, при исследовании этих комплексов подходят к ним
со строгих физико-химических позиций. Это проявляется в недо-
статочно правильном выборе ионной силы, интервалов pH и
температуры и особенно буфера, который почти никогда не бы-
вает инертным по отношению к ионам металлов, хотя его взаи-
модействие с ними иногда незначительно. Таким образом полу-
чаются «кажущиеся» (или «эффективные») значения констант
устойчивости, которые зависят от выбора экспериментальных
условий:
_ [МАТФ2~]
лЭфф— [М2+] [АТФ]Г,
Киназы
665
где [АТФ]т' — суммарная концентрация всех форм незакомп-
лексованного АТФ.
В идеальном случае измерения следует проводить точно в тех
условиях, в которых будет изучаться фермент. Кроме того, обыч-
но можно экстраполировать имеющиеся данные по комплексооб-
разованию к условиям ферментативной реакции или выбрать эти
условия таким образом, чтобы свести различия к минимуму.
Обширные сводки данных по константам устойчивости комп-
лексов металлов с нуклеотидами содержатся в обзоре Филлипса
[25], в публикациях Химического общества [24] (в которых
много типографских ошибок), а также в «Data for Biochemical
Research» [31].
2.2.1. Скорости образования МАДФ~ и МАТФ2~
При измерении скоростей образования комплексов металлов с
АДФ и АТФ Хамме и сотр. [32, 33] пришли к заключению, что-
они возникают по внешнесферному механизму SnI с промежу-
точным образованием ионной пары. По-видимому, самым инте-
ресным в их результатах является то, что Са2+ взаимодействует
на два порядка быстрее, чем другие применявшиеся ионы двух-
валентных металлов, включая Mg2+ и Мп2+. Остается невыяс-
ненным, в какой степени этот факт связан с активацией кальцием
ферментов типа I (Е—S—М) и ингибированием им же ферментов
типа II (Е—М—S).
2.3. Структура комплексов металл — нуклеотид
2.3.1. В растворе
Обнаружив образование в растворе достаточно прочных комп-
лексов металл — нуклеотид, естественно попытаться установить и
их структуру. Ясно, что связывание щелочноземельных металлов
должно осуществляться фосфатными окси-анионами. С металла-
ми, подобными марганцу, возможно также присоединение к азоту
по типу донорно-акцепторной связи. В случае ионов Си2+ послед-
ний вид комплексов может быть даже преобладающим.
Вызывает большой интерес вопрос о том, в какой степени ион
металла, связанный с окси-анионами фосфата, может взаимодей-
ствовать еще и с атомами азота. Хотя для определения структуры
таких комплексов применялись разнообразные физико-химические
методы, эта проблема пока еще не решена. Она детально рас-
сматривается в гл. 33.
2.3.2. На ферменте
В этом случае ситуация еще менее ясная, чем для комплексов
в растворе. Почти все факты, относящиеся к этому вопросу, по-
лучены косвенным путем.
666
Глава 18
Некоторая информация была получена методом ЭПР. Спектры
ЭПР очень чувствительны к напряженности поля около иона
металла и, следовательно, к любым изменениям его лигандов.
При связывании МпАДФ- с креатинкиназой [34, 35] и МпАТФ2-
с аденилаткнназой [36] не обнаружено никаких изменений в
•спектрах ЭПР. Эти данные подтверждают (хотя и не доказы-
вают) гипотезу о том, что в растворе, как и на ферменте, струк-
тура комплексов одинакова. В противоположность этому в
спектре МпАДФ- при связывании с пируваткиназой наблюдались
определенные изменения, вызванные, вероятно, появлением
дополнительных лигандов со стороны фермента [37].
Косвенные данные получены и из кинетических экспериментов
с ферментами; для креатинкиназы установлено, что М2+ более
прочно связан с Е—АДФ, чем со свободным нуклеотидом [35,
38]. Однако такие выводы несколько искусственны, поскольку они
зависят от выбора модели, которая может не отражать реальной
ситуации (разд. 2.4).
2.4. Кинетические исследования киназ
Главные особенности любого фермента состоят в том, что он
намного (в 107—1012 раз [39]) ускоряет реакцию, которая в его
отсутствие идет очень медленно или вообще не идет. Поэтому
было бы естественным, чтобы рациональной основой для нашего
понимания действия ферментов служило изучение кинетики фер-
ментативных реакций. К сожалению, будучи важным подспорьем
другим методам исследования, кинетические эксперименты редко
ведут к точному установлению механизма действия ферментов.
Изучение киназ осложняется образованием большого числа
возможных промежуточных соединений, которые необходимо учи-
тывать. Кроме двух субстратов и фермента, в системе всегда
присутствуют ионы металла-активатора, которые образуют комп-
лексы с нуклеотидным субстратом, а иногда также и с фермен-
том и со вторым субстратом. Вообще говоря, кинетическое иссле-
дование всех киназ, по-видимому, можно^ проводить, полагая, что
истинными активными субстратами являются МАДФ- и МАТФ2-
[6]. Однако нельзя быть полностью уверенным в том, что данная
гипотеза правильно отражает действительный механизм фермен-
тативной реакции. Причиной ошибочных заключений в таких
случаях может являться неспособность модели различать отдель-
ные пути реакции.
Хорошим примером развития представлений о роли ионов ме-
талла-активатора в кинетике киназных реакций может служить
исследование креатинкиназы. Результаты ранних работ (Эннор и
Розенберг [40], с одной стороны, и Куби с сотр. [41]—с дру-
гой) противоречили друг другу. Первая группа исследователей на
Киназы
667
основании полученных ими результатов полагала, что образуется
комплекс металла с ферментом, который затем реагирует со сво-
бодным нуклеотидом. Вторая же группа считала реальным суб-
стратом металлону.клеотидный комплекс, даже несмотря на то, что
опубликованные данные о значении констант устойчивости таких
комплексов были слишком малы для количественной обработки
экспериментальных результатов в соответствии с их моделью.
Затем работу продолжили еще две группы. В лаборатории Нода
[42, 43] были получены данные, определенно говорившие о том,
что если принять большие значения соответствующих констант
устойчивости, то подлинными субстратами могут являться
МёАДф- и MgATO2-. Моррисон с сотр. [44] попытались решить,
проблему. Предполагая постоянство концентрации второго суб-
страта (фосфокреатина), они постулировали, что реакция прохо-
дит через тройной комплекс фермент — металл — субстрат, кото-
рый может возникать тремя способами:
М
S
MS
11*6
*i к2 к4 к3
Е + S Е S EMS ЕМ 5=t Е + М
---------------. 1----------------------
I продукты реакции II
Все они эквивалентны, поэтому KiK^—К?,К^=Если извест-
на величина константы диссоциации МёАДФ“ (As), то на опыте
можно определить и все другие константы.
Таким образом, по' этой схеме ион металла действительно мог
присоединяться к ферменту. Но такой вывод противоречил ре-
зультатам термодинамических [45] и ЯМР [34] измерений. Это
потребовало внесения изменений в дальнейшие исследования [38].
Самым важным из них было использование более достоверных
значений констант устойчивости MgAДф-, отвечающих экспери-
ментальным условиям, и проведение опытов по выяснению приро-
ды ингибирования избытком АДФ и Mg2+. Было обнаружено,
что по отношению к MgAДФ- избыточный магний является не-
конкурентным ингибитором, а АДФ3- — конкурентным (рис. 18.1).
Иными словами, участие реакций II в образовании комплекса
EMS оказалось маловероятным, хотя по этим данным еще нель-
зя сделать выбор и между реакциями I и III. Дальнейшие иссле-
668
Глава 18
дования подтвердили предположение (подразумевавшееся во всех
предыдущих работах) о том, что кинетический механизм може
быть описан как быстрое статистическое равновесие [46] и что
действие Са2+ и Мп2+ такое же, как и Mg2+ [47].
4 - ю 4м
[Мд ддф")
Рис. 18.1. Ингибирование креатинкиназы.
а — АДФ3— конкурентен по отношению к МрАДф—; б — Mg2+ неконкурентен по отношению
к М®АДФ-. (Данные заимствованы из работы [101]. Полное описание этих экспериментов
см. в работе [38]).
Кинетические исследования других ферментов будут рассмот-
рены в разд. 3. Здесь же необходимо еще раз подчеркнуть, что
несмотря на некоторую неоднозначность результатов кинетиче-
ских исследований, они являются предпосылкой для других мето-
дов изучения промежуточных комплексов. Уместно также отме-
тить относительно большое количество информации, которое мож-
но получить в кинетических экспериментах с очень малым коли-
Киназы
669
чеством фермента, когда многие другие методы оказываются
недостаточно чувствительными. Для обычного энзимолога послед-
нее обстоятельство весьма существенно.
2.5. Методы исследования фермент-субстратных комплексов
в равновесии
Основы для понимания механизма функционирования фермен-
тов могут быть получены только при сочетании кинетических и
других физико-химических методов исследования. В принципе
современному биохимику доступны многие такие средства, вклю-
чая равновесный диализ, ультрацентрифугирование, различные
оптические методы, ДОВ и др. Можно указать и на общие руко-
водства по применению физических методов в биохимии [48].
Эти методы пока еще недостаточно широко использовались
для изучения киназ. Ниже приведены результаты некоторых ра-
бот, показывающие ту информацию, которую можно получить
таким путем.
1. Обширное исследование связывания субстратов и т.п. с
креатинкиназой и аденилаткиназой выполнили Куби и сотр. [45].
Методами равновесного диализа и ультрацентрифугирования
были получены доказательства слабого связывания Mg2+ и отно-
сительно сильного связывания свободного нуклеотида и его комп-
лекса с металлом.
2. Кинетический анализ защитного действия лиганда приме-
няли Милдван и Лей [49] для пируваткиназы, а для креатинки-
назы— О’Сулливан с сотр. [50] и Каннингам с сотр. [51].
3. При обстоятельном исследовании пируваткиназы, которое
выполнили Сьюлтер с сотр. [52], с помощью ультрафиолетовой и
флуоресцентной спектроскопии были получены ясные доказатель-
ства изменения конформации фермента при присоединении раз-
личных субстратов и кофакторов.
К этой группе методов следует отнести и различные методы,
использующие явления магнитного резонанса. Но вследствие их
особой важности для исследования киназ и других ферментов они
более подробно рассмотрены ниже.
2.6. Применение методов магнитного резонанса
В последние годы для изучения белков широко используются
различные методы магнитного резонанса. Мы рассмотрим два из
них: электронный парамагнитный резонанс (ЭПР) и ядерный
магнитный резонанс (ЯМР). При помощи этих методов получают
данные о химических сдвигах и скоростях релаксации. Данные
с химических сдвигах пока еще мало используются в биохимии.
670
Глава 18
но с развитием техники высокого разрешения они, по-видимому,
приобретут большее значение. Измерения скоростей релаксации
могут проводиться импульсным или сканирующим методом.
Ко времени написания этой книги результаты, наиболее полезные
для понимания киназных реакций, были получены при помощи
импульсной техники, особенно в случае измерения скоростей ре-
лаксации протонов воды. Можно ожидать, что при комплексном
использовании различных методов будут получены еще более
ценные данные.
2.6.1. Электронный парамагнитный резонанс
Спектры ЭПР дают группировки с неспаренными электронами.
К ним относятся парамагнитные ионы металлов, что, например,
оказалось очень полезным при исследовании металлоферментов,
содержащих Си и Мо [53—55]. Впервые для изучения киназных
реакций этот метод использовали Кон и Таунсенд [56], которые
таким способом измерили константы устойчивости некоторых
комплексов субстратов с марганцем. Последнее оказалось воз-
можным благодаря тому, что амплитуда сигнала от комплекса
гораздо меньше, чем от свободного металла. Кроме этого случая,
ЭПР-спектроскопия марганца успешно использовалась совместно
с измерениями протонного резонанса для определения концентра-
ции свободных ионов Мп2+ [35, 57]. Другие попытки использова-
ния этого метода, основанные на парамагнитных свойствах мар-
ганца, оказались неудачными. Было очень трудно получить сами
спектры комплексов и еще труднее их интерпретировать. Однако
благодаря этим измерениям были получены доказательства,
позволившие Кон разделить киназы на две группы: тип I (напри-
мер, креатинкиназа) и тип II (например, пируваткиназа) в со-
ответствии с тем, как ион металла реагирует с ферментом — через
субстратный мостик (Е—S—М) или непосредственно (Е—М—S)
(см. разд. 2.6.2. и гл. 14).
Использование ЭПР оказалось особенно ценным в одном спе-
циальном случае — при связывании иминоксильной метки с реак-
ционноспособной сульфгидрильной группой активного центра
креатинкиназы. Этот радикал имеет только один неспаренный
электрон (спин Va). и поэтому его спектры ЭПР значительно
проще, чем у ионов марганца. Другое преимущество метода ими-
ноксильной метки состоит в том, что хотя модифицирование
SH-группы инактивирует фермент, однако адсорбционный центр
нуклеотида остается ненарушенным. А это позволяет изучать
изменения спектров при введении субстратов или диамагнитных
солей. Ясно, что синтез новых имипоксильных меток откроет перед
этим методом дополнительные возможности.
Киназы
671
2.6.2. Ядерный магнитный ргзонанс
Измерение химического сдвига. Это наиболее известный тип
измерений магнитного резонанса. В основном используется ска-
нирующий метод, который позволяет определить спектры иссле-
дуемых ядер. Чаще всего получают спектры протонов, хотя для
изучения металлонуклеотидных комплексов оказались полезными
и спектры 31Р и 15N (гл. 33).
В принципе сканирующим методом можно получить больше-
всего сведений, так как он дает информацию об электронной
структуре активного центра и о конформации фермент-субстрат-
ных комплексов. Однако его практическое использование часто
затрудняется сложностями при разрешении широких перекрываю-
щихся линий в спектрах больших белковых молекул, а также еще,
и необходимостью выполнения измерений протонного резонанса
в D2O, для чего обычно требуется неоднократная лиофилизация
белка. Первое осложнение преодолевается применением новейшей
техники с большой разрешающей способностью [59]. О ее воз-
можностях можно судить по работе Ярдецкого с сотр. [60], по-
священной механизму действия рибонуклеазы. Предварительные
эксперименты такого рода были выполнены и с аденилаткиназой
(разд. 3.3).
Релаксационные методы уже обсуждались в гл. 14. Можно
указать также на подробный обзор Милдвана и Кон [5].
В данной главе имеет смысл лишь суммировать те сведения,
которые можно получить при измерениях скоростей релаксации
протонов и которые наиболее важны при изучении киназ.
1. На этом методе основана полезная классификация фермен-
тов, активируемых марганцем, на два типа — I (субстрат в каче-
стве мостика) и II (металл в качестве мостика). Желательно,
чтобы эта классификация была бы подтверждена измерениями
ЭПР (разд. 2.6.1) и скоростей релаксации ядер субстрата (для
определения расстояний от марганца).
2. При изучении парамагнитных частиц эти методы имеют
преимущество перед остальными термодинамическими методами-
определения констант диссоциации различных комплексов фер-
ментов. Можно ожидать, что точность получаемой при этом ин-
формации будет повышена при применении ЭВМ [61]. Такие-
методы использовались для определения прочности связывания
субстратов со специфически инактивированными формами фер-.
ментов [62].
3. Методом протонного резонанса были получены доказатель-
ства того, что субстрат вызвает конформационные изменения в.
белке. Данные такого рода относятся прежде всего к креатинки-.
назе (они обсуждаются ниже, см. разд. 3.1).
4. Исследования температурной и частотной зависимости мо-.
гут способствовать определению механизма релаксации, В ре-.
€72
Глава 18
зультате можно получить сведения о микроокружении иона мар-
ганца, а в благоприятном случае — и о количестве молекул воды
в первой координационной сфере.
5. Введение спиновых меток, первоначально применявшихся
для изучения изменений в спектрах ЭПР [63], влияет также и на
скорости релаксации соседних с ними протонов. Благодаря этому
значительно расширяются возможности применения и методов
протонного резонанса. Примером таких работ могут служить ис-
следования алкогольдегидрогеназы, которые выполнили Вейнер и
Милдван [64], а также введение спиновых меток в креатинки-
назу [58].
3. ОБСУЖДЕНИЕ ОТДЕЛЬНЫХ ФЕРМЕНТОВ
Хотя известно большое количество киназ, только немногие из
них изучены подробно с физико-химической точки зрения. По-ви-
димому, наибольшее внимание было уделено креатинкиназе и
пируваткиназе, которые послужили прототипами ферментов
типа I (Е—S—М) и II (Е—М—S) соответственно (табл. 18.1).
Таблица 18.1
Классификация ферментов в соответствии со структурой комплексов, содержащих
фермент, металл н субстрат
Тип I (Е—S—М)
Тип II (Е—м—S)
Креатинкиназа (из мышц и мозга)
Адеиилаткиназа
Аргининкиназа
Г ексокиназа
Фосфоглицераткиназа
Пируваткиназа (из мышц и мозга)
Фосфоенолпируваткарбоксикиназа
(Енолаза)8
(Пируваткарбоксилаза)8
а * 1
Не относится к классу киназ.
Ниже они рассмотрены более детально. Несколько менее подроб-
но обсуждаются и некоторые другие киназы, включая и такие,
которые не соответствуют «нормальным» моделям.
3.1. Креатинкиназа
Креатинкиназа (АТФ: креатинфосфотрансфераза, КФ 2.7.3.2)
катализирует обратимый перенос фосфорильной группы от АТФ к
креатину. Реакцию можно представить в следующем виде:
М АТФ2-+ креатин < » МАДФ' + фосфокреатин2-+ Н+
Киназы
673
где обязательным ионом металла может быть Mg, Мп, Са или
Со. Хотя раньше это и оспаривалось, однако теперь в результате
кинетических, термодинамических и ЯМР-исследований оконча-
тельно установлено, что металл связывается с ферментом не не-
посредственно, а через нуклеотидный субстрат (тип I).
Больше всего работ выполнено с ферментом из мышц кро-
лика, который с хорошим выходом получается в кристаллическом
виде [65]. Он является димером с молекулярной массой 82 600 и,
по-видимому, образован из идентичных мономеров [66]. Диссо-
циация димера, наблюдаемая только в жестких денатурирующих
условиях, приводит к утрате активности и деструкции центра
связывания нуклеотида [62, 67]. Если же принять меры для за-
щиты сульфгидрильных групп, освобождающихся при денатура-
ции, то впоследствии удается восстановить в активном виде и
димерную форму. Считается, что глобула содержит два активных
центра (по одному на мономер), в которых имеется по одному
реакционноспособному остатку цистеина.
Выше уже рассматривались результаты кинетических исследо-
ваний фермента (разд. 2.2). Мы не будем обсуждать работы
группы Куби [66] и других лабораторий [68, 69], посвященные
белковой химии креатинкиназы. Основное внимание будет уделено
лишь тем сведениям, которые были получены методом магнит-
ного резонанса, а также при параллельных исследованиях.
В одной из первых работ Кон и Лей [34] использовали изме-
рения ЯМР и ЭПР для демонстрации образования комплекса
МпАДФ — фермент. Эти результаты были дополнены О’Суллива-
ном и Кон [35], которые, используя графический метод анализа
данных ЯМР, определили значения констант ассоциации фермен-
та с МпАДФ- и МпАТФ2-, хорошо согласующиеся с кинетиче-
скими результатами. Впоследствии благодаря использованию вы-
числительной техники в эти величины были внесены некоторые
поправки, не изменившие представления о наличии у фермента
двух активных центров [61, 70].
В работе [7] в качестве субстратов изучались различные
нуклеозиддифосфаты. Одновременно исследовалась способность
их марганцевых комплексов воздействовать на скорости релакса-
ции протонов воды. Были обнаружена строгая корреляция между
наблюдаемым усилением для соответствующего тройного комп-
лекса и способностью дифосфата участвовать в реакции в качест-
ве субстрата. Третья из исследованных в этой работе характери-
стик— способность металл-нуклеотидных комплексов влиять на
реакцию иодацетата с каталитической сульфгидрильной груп-
пой— подчиняется той же закономерности (рис. 18.2). Был сде-
лан вывод о том, что наблюдаемые явления — следствие конфор-
мационных изменений в активном центре фермента, индуцируе-
мых субстратами. Обнаруженные различия в усилении
674
Глава 18
релаксации, скорости реакции и доступности SH-группы можно
объяснить различием в степени этих конформационных измене-
ний при воздействии разных нуклеотидов. Аналогичные соотно-
шения получены и для нуклеозидтрифосфатов, хотя в этом случае
результаты менее убедительны, чем для дифосфатов.
Скорость
реакции
Увеличение
реакционной
способности
SH-группы
Усиление
релаксации
Рис. 18.2. Сравнение относительных значений максимальной скорости реакции,
усиления протонной релаксации и возрастания реакционной способности
SH-групп для комплексов металлов с различными нуклезиддифосфатами.
А — МАДФ—; Б — М-З'-дДДф—; в — М-2'-дАДФ~; Г — МИДФ—; Д — МГДф—. Максимальные
скорости и реакционная способность SH-групп определялись с Mg2+, кроме случая ИДФ.
где использовался Мп;+; усиление релаксации измерено с Mn2+ [J. Biol. Chem., 241, 3116
(1966)].
Наблюдались и значительные различия в характере влияния
свободных нуклеотидов и их комплексов с металлами на реак-
цию иодацетата с SH-группой. Например, АДФ3" защищает фер-
мент от инактивации, хотя эта защита никогда не бывает полной
даже при насыщающих концентрациях, тогда как комплекс фер-
мента с MgAflO- реагирует с алкилирующим агентом в большей
степени, чем нативная креатинкиназа. Поскольку ни АДФ3-, ни
А^АДФ- не влияют на скорость взаимодействия сульфгидриль-
ной группы с электронейтральным иодацетамидом [50], можно
заключить, что при конформационных изменениях активного
центра происходит перераспределение зарядов.
Доказательства наличия таких изменений конформации ак-
тивного центра были получены и при исследовании креатинкина-
зы методом температурного скачка [71]. Согласно этим данным,
при добавлении АДФ3- или МАДФ- (M=Mg2+, Са2+ или Мп2+)
происходит изомеризация фермента. Константы связывания ме-
талл-нуклеотидных комплексов лишь в незначительной степени за-
висят от природы металла, что согласуется с изложенным выше
представлением о характере взаимодействия (ESM). (Величины
констант скоростей образования самих металл-нуклеотидных
Киназы
675
комплексов отличаются у разных металлов на несколько по-
рядков.)
Образование непродуктивного четверного комплекса (присое-
динение креатина к МпАДФ—Е) приводит к резкому изменению
скорости протонной релаксации. Причина этих изменений до не-
которой степени объясняется результатами измерений при разных
Рис. 18.3. Зависимость 1/Tip от температуры для тройного и четверного ком-
плексов креатинкиназы [J. Biol. Chem., 243, 2737 (1968)].
Условия: 4 мг/мл (0,05 мМ) креатинкиназы, 0,1 мМ MnCk, 0,1 мМ АДФ, 0,05 М N-этилмор-
фолина. pH 8,0. ф нативный фермент; нативный фермент в присутствии 30 мМ креатина;
А неактивный карбоксиметилирсванный фермент; Q неактивный карбоксиметнлированный
фермент в присутствии 30 мМ креатина
температурах (рис. 18.3). Скорость релаксации для тройного ком-
плекса МпАДФ—Е имеет нулевой или отрицательный температур-
ный коэффициент, а у четверного комплекса МпАДФ—Е —креа-
тин этот коэффициент положителен. Раньше считалось, что по-
следнее является следствием процесса, определяемого тм [62].
Однако, согласно недавно полученным данным (зависимость этого
явления от частоты), величина Tip, по-видимому, определяется
значением rs [5].
На рис. 18.3 показаны также результаты, полученные скреа-
тинкиназой, обработанной иодацетатом (замещены две активные
SH-группы). Модифицированный фермент связывает МпАДФ~
так же хорошо, как и нативная креатинкиназа, однако при этом
уже не наблюдается «эффект креатина».
Свидетельства наличия конформационных изменений при об-
разовании четверного комплекса получены в исследованиях инак-
тивации доступных сульфгидрильных групп иодацетатом и иода-
676
Глава 18
цетамндом [50] и чувствительности фермента к трипсину [51].
Некоторые сведения о природе этих изменений получены в на-
чальных работах со спин-меченой креатинкиназой.
Использование в качестве спиновых меток иминоксильных про-
изводных иодацетамида, стехиометрически реагирующих с сульф-
гидрилом, способствовало значительному увеличению количества
информации, получаемой резонансными методами [58]. Подобно
другим модификаторам SH-группы, иминоксильные производные
полностью инактивируют фермент из мышц кролика. Однако при
этом он еще сохраняет способность связывать нуклеотидные суб-
страты, хотя уже и не показывает «эффект креатина». Благода-
ря этому оказалось возможным использовать методы ЭПР и про-
тонного резонанса для определения констант диссоциации раз-
личных диамагнитных металлов (например, Mg2+, Са2+) из их
тройных комплексов с ферментом. Выяснилось, что добавление
МАДФ- приводит к еще большей степени заторможенности ими-
ноксильного радикала, и так уже значительно заторможенного
вследствие присоединения к ферменту.
Два следующих примера демонстрируют возможности метода
парамагнитных зондов [70]. По уширению сигнала ЭПР от ими-
нокснльного радикала, наблюдавшемуся после добавления
МпАДФ-, удалось определить расстояние между неспаренным
электроном метки и связанным ионом марганца. Оно составило
8—12 А (для МпАТФ на поверхности белка это расстояние равно
13—18 А). Аналогичные исследования были проведены с фермен-
том из сердца цыпленка, который после связывания иминоксиль-
ной метки еще сохраняет около 25% активности. Из наблюдений
за уширением линий протонов Н-8 и Н-2 в кольце связанного
аденина, происходящем при переходе от АДФ—Е к МАДФ—Е,
а затем к МАДФ—Е—креатин, было усгановно что адениновое
кольцо удаляется от сульфгидрильной группы.
Эти эксперименты пока еще имеют характер предварительных.
Тем не менее уже и по их результатам видно, какие широкие
возможности предоставляют для изучения активных центров
киназ методы магнитного резонанса.
3.2. Аргининкиназа
Аргининкиназа катализирует обратимый перенос фосфориль-
ной группы от АТФ к ь-аргинину аналогично реакции креатин-
киназы [1]. Она широко распространена среди беспозвоночных,
особенно у членистоногих. Ферменты, выделенные из некоторых
ракообразных, почти не отличаются друг от друга по физическим
свойствам. Аргининкиназа состоит из одной полипептидной цепи с
молекулярной массой около 40 000 (половина молекулярной массы
креатинкиназы) с одним каталитическим центром.
Киназы
677
Подобно креатинкиназе аргининкиназа, по-видимому, подчи-
няется кинетике быстрого статистического равновесия. (Сообще-
ние [72] о том, что у фермента из Jasus verrauxi реакция имеет
механизм типа «пинг-понг», не содержало достаточно убеди-
тельных доказательств.) Это согласуется с ЯМР-данными, полу-
ченными с аргининкиназами из трех источников: Homarus vul-
garis, Homarus americanus и Panullrus longipes [73]. Все они
являются ферментами типа I с относительно высокими значе-
ниями е* для комплексов МпАДФ—Е и МпАТФ—Е. Хотя и
имеются некоторые количественные различия, однако характер
усиления релаксации одинаков во всех случаях, а константы
диссоциации комплексов марганца с нуклеотидами, определен-
ные по скоростям релаксации протонов, хорошо согласуются со
значениями, полученными из кинетических данных.
Эксперименты с различными Мп-нуклеотидами (АДФ,
2'-дАДФ и ИДФ) обнаружили такую же корреляцию между et
и максимальной скоростью, как и для креатинкиназы. Между
этими ферментами наблюдается аналогия и в изменениях ско-
рости релаксации при образовании четверных комплексов
(МпАДФ—Е—креатин и МпАДФ—Е—ь-аргинин). С о'-аргини-
ном, который является либо слабым ингибитором, либо вообще
инертным по отношению к аргининкиназе, подобный эффект
не обнаружен.
На данной стадии исследований представляется, что арги-
нинкиназа и, вероятно, другие гуанидинкиназы [74, 75] имеют
механизм, подобный креатинкиназе. Гораздо меньшая молеку-
лярная масса аргининкиназы делает ее более удобной для изу-
чения методом сканирующей ЯМР-юпектроскопии.
3.3. Пируваткиназа
Пируваткиназа катализирует реакцию
м2+. м+
АТФ4- -|- пируват t > АДФ3- + фосфоенолпируват (1)
а также две нефизиологичеокие реакции — «фторкиназную» и
«гидроксиламинкиназную»:
нс°“
AT®4--f-F“ > АДфз-+РРО|- (2)
нсо~
АТФ4- + NHaOH -< АДФ3- + гидроксиламинфосфат (3)
Все эти реакции, по-видимому, происходят в одном и том же
центре фермента. Для всех них необходимо присутствие ионов
как двух-, так и одновалентных металлов [76]. Ионами двух-
Здесь et —то же, что и ес в гл. 14.
678
Глава 18
валентных металлов для реакций (1) и (2) могут быть Mg2*
или Мп2+, для реакции (3) — Zn2+. а одновалентных —
K+>Rb+>NHt>Cs+>Na+ [76, 77].
Наиболее подробные исследования проведены с ферментом
из мышц кролика — тетрамером с молекулярной массой 237 000,
состоящим, по-видимому, из одинаковых субъединиц [78]. Име-
ются некоторые расхождения в мнениях по поводу кинетической
схемы [6, 79], однако представляется вероятным (главным
образом на основании экспериментов со слабо диссоциирующим
ионом Ni2+ [80]), что тройной комплекс металл — фермент —
АДФ может образовываться как при взаимодействии АДФ3- с
ЕМ, так и непосредственно из фермента и МАДФ- (пути 11 и
III на схеме в разд. 2.4). Было также обнаружено, что только
предварительная инкубация фермента с фосфоенолпируватом
(ФЕП), но не с МАДФ~ приводит к увеличению скорости реак-
ции. Последнее свидетельствует об определенной упорядоченно-
сти в последовательности связывания субстратов, фермент из
дрожжей подчиняется тому же механизму, однако с существен-
ным отличием — фруктозодифосфат является аллостерическим
активатором [80].
Обе пируваткиназы (из мышц кролика и из дрожжей) ведут
себя как ферменты типа II. Наблюдается значительное усиление ре-
лаксации у комплексов фермента с марганцем при малых значе-
ниях для тройных комплексов. Результаты кинетических экспери-
ментов свидетельствуют о существовании структуры с металлом
(в качестве мостика. О том же говорят данные резонансных ис-
следований. Лучше всего в этом убеждают результаты изучения
«фторкиназной» реакции, катализируемой ферментом [81], так
как ядра фтора дают удобный для наблюдения спектр. Исследо-
вание влияние марганца, связанного с ферментом, на скорости
релаксации 1/Tj и 1/Т2 ядер I9F позволило рассчитать расстояние
между атомами Мп и F. Эти расчеты хорошо согласуются с не-
давним рентгеноструктурным анализом кристаллов СаЕРОз [82]
и соответствуют мостиковым структурам
0 0 о
Е—Мп—О—Р—F или Е—Мп; ,Р^
Скорость обмена (1/тм) аниона FPO1- в координационную сферу
иона марганца по порядку величины согласуется с кинетическими
данными.
Различия в значениях Et для Е—МпАДФ и Е—МпАТФ соот-
ветствуют различиям самих этих комплексов (потеря одной моле-
кулы воды в последнем случае) [83]. По-видимому, Мп образует
Киназы
679
связи с у-фосфатом АТФ (т. е. с фосфорильной группой, перено-
симой <к ФЭП), а возможно, и с 0-фосфатом АТФ и АДФ.Однако
добавление ФЕП к Е—Мп приводит к гораздо большим измене-
ниям в усилении релаксации, чем в тех случаях, когда это можно
объяснить простым замещением молекулы воды. Вероятно, свой
вклад в это явление вносят и конформационные изменения фер-
мента. Независимые доказательства наличия структурных преоб-
разований глобулы при добавлении различных субстратов и одно-
и двухвалентных активаторов получены также при спектроскопи-
ческих исследованиях в лаборатории Сьюлтера [52].
Кроме АДФ и АТФ пируваткиназа может превращать и дру-
гие нуклеотидные субстраты, но эта способность зависит от при-
роды нуклеотида и от величины pH [9]. Пока неизвестно, сущест-
вует ли корреляция между максимальными скоростями и усиле-
нием релаксации для соответствующих комплексов, подобно
наблюдавшейся у креатинкиназы. Интересно, что предварительные
эксперименты свидетельствуют об обратной зависимости между
усилением релаксации комплекса Е—М—ФЕП и природой при-
сутствующего иона одновалентного металла [77].Чем слабее ион
М+ как активатор, тем значительнее усиление релаксации трой-
ного комплекса. Пока еще не ясно, в какой степени это является
следствием изменений в значениях Et и констант диссоциации
ФЭП из комплекса [112]. Тем не менее подобные данные свиде-
тельствуют о важной роли одновалентных ионов, которые, воз-
можно, образуют центр связывания для карбоксильной группы
ФЕП [77].
Дополнительные доказательства присутствия одновалентных
катионов в каталитическом центре пируваткиназы получены в
предварительных экспериментах с таллием [85]. Наиболее рас-
пространенный изотоп 205Т1 имеет спин ‘/г. что позволяет исполь-
зовать его для резонансных измерений. Результаты этих экспери-
ментов говорят о наличии в ферменте взаимодействия между Т1
и Мп. Следовательно, их центры связывания достаточно близки
друг к другу.
3.4. Фосфоенолпируваткарбоксикиназа
(фосфопируваткарбоксилаза)
Подобно пируваткиназе и другим ферментам, превращающим
ФЕП [5], фосфоенолпируваткарбоксикиназа относится к типу II.
Ее реакция очень похожа на реакцию пируваткиназы и пируват-
карбоксилазы. Она подробно рассмотрена в гл. 14.
3.5. Аденилаткиназа
Реакция, катализируемая аденилаткиназой, может быть пред-
ставлена в виде [86]
МАТФ2- -f- АМФ2- х—МАДФ- 4- АДФ3-
680
Глава 18
где роль металла могут играть Mg, Мп, Са или Со [36]. Специ-
фичность этого фермента к ионам металлов требует некоторых
пояснений. Первоначально сообщалось, что для обратной реак-
ции, катализируемой ферментом из мышц кролика, ион Са2+
не является активатором. Однако тот факт, что по данным ЯМР
аденилаткиназа относится к типу I, заставил более внимательно
изучить явление активации. При этом было обнаружено, что хотя
Са+2— действительно слабый активатор при протекании реакции
в направлении образования АТФ (его эффективность в 10 раз
меньше, чем у Mg2+), он почти так же хорошо, как и Mg2+, акти-
вирует прямую реакцию [36]. Такое аномальное поведение каль-
ция пока остается не понятым, хотя и представляется заманчи-
вым объяснить его тем, что Са2+ вступает в комплексы с АТФ и
АДФ и выходит из них примерно на два порядка быстрее, чем
Mg2+ или Мп2+. К сожалению, в литературе отсутствуют убеди-
тельные данные о подобных свойствах и других киназ.
Наиболее подробно изучена аденилаткиназа, выделенная из
мышц кролика. Она построена из единственной полипептидной
цепи с молекулярной массой 21000 и имеет два активных центра.
Низкая молекулярная масса делает ее удобной для измерений
методом сканирующей ЯМР-спектроскопии. Предварительные
эксперименты [87] продемонстрировали наличие значительных
сдвигов протонов Н-2 и Н-8 АТФ при связывании с ферментом.
Как уже указывалось, этот фермент относится к типу I, т. е.
Мп связывается с аденилаткиназой через АТФ. Первоначально
графический анализ результатов измерений протонного резонанса
привел к таким значениям усиления релаксации комплекса
МпАТФ—Е, которые превосходили даже теоретически возможный
максимум (особенно при температурах 30—40 °C) [5, 36]. Впо-
следствии при обработке этих данных на ЭВМ [61] были полу-
чены более реальные значения, аналогичные наблюдавшимся у
других ферментов (табл. 18.2). Согласно результатам машинного
анализа, аденилаткиназа, скорее всего, имеет два центра связы-
вания для МпАТФ2-.
Было найдено, что различные нуклеозидтрифосфаты действуют
как дополнительные субстраты для аденилаткиназы. Обнаружена
и соответствующая корреляция между максимальными скоро-
стями и усилением релаксации марганцевого комплекса [36].
Другими словами, и для этого фермента наблюдаются индуци-
руемые субстратами изменения конформации, степень которых
зависит от природы субстрата. Комплекс МпАДФ—Е исследовать
не удалось.
Аденилаткиназа имеет два реакционноспособных остатка ци-
стина, и хотя они необходимы при проявлении каталитической
активности, в некоторых случаях оказалось возможным провести
их модифицирование без полной инактивации фермента. Так, ре-
Киназы
681
Таблица 18.2
Усиление релаксации у различных комплексов марганца
еа относится к усилению релаксации бинарного комплекса М—S; 8 соответствует бинар-
ному комплексу Е—М, а — тройному комплексу, содержащему фермент, металл и суб-
страт. Усиление релаксации для ионов Мп!+ в отсутствие комплексующего агента принято
за 1,0. Данные заимствованы из работ [35, 36, 57, 59, 72. 100]. В некоторых случаях они бы-
ли пересчитаны на ЭВМ [61].
Комплекс Усиление релаксации
Еп Еь *7
МпАДФ ~ 1,6
МпАТФ2- Мп-пируваткиназа АДФ—Мп-пируваткиназа 1,7 33 20
АТФ—Мп-пируваткиназа 13
Фосфоенолпируват—Мп-пируваткиназа Мп-креатинкиназа 1,5 23
Мп-АДФ—креатинкиназа 20
Мп-АТФ—аденилаткиназа 13
Мп -дАТФ—аденилаткиназа 8
Мп-АДФ—аргининкиназа Мп—фосфоенолпируваткарбоксикиназа 14 45
акция SH-группы с дитио-быс-нитробензойной кислотой (ДТНБ)
приводит к полной потере активности [88], но после реакции с
некоторыми органическими производными ртути, также взаимо-
действующими с сульфгидрилом, фермент еще сохраняет значи-
тельную часть каталитической активности [89]. По данным про-
тонной релаксации, МпАТФ2- связывается с аденилаткиназой,
модифицированной одним из таких соединений (с производным
меркурбензоата) [36], и не связывается при использовании про-
изводных ДТНБ [88].
Милдван и Кон [5] считают, что роль металл а-активатор а со-
стоит в сближении лигандов для облегчения реакции. Можно
предположить, что использование сканирующей ЯМР-спектроско-
пии и спиновых меток приведет к более подробным сведениям об
активном центре.
3.6. Гексокиназа
Гексокиназа катализирует перенос фосфорильной группы от
АТФ к глюкозе. Наиболее подробно изучен дрожжевой фермент,
хотя некоторое внимание исследователи уделили и гексокиназе
44—2451
682
Глава 18
мышц и мозга. Фермент обладает широкой 'специфичностью как к
нуклеотидным, так и к углеводным субстратам [90].
Гексокиназа, особенно дрожжевая, была объектом широких
кинетических исследований. При этом были получены как свиде-
тельства того, что имеет место упорядоченный механизм [91] с
определенной последовательностью связывания глюкозы и
MgATO2-, так и противоположные аргументы в пользу независи-
мого присоединения субстратов к свободному ферменту по типу
быстрого статистического равновесия [92]. Подобное противоре-
чие еще раз подчеркивает, что на основании чисто кинетических
экспериментов очень трудно получить однозначный ответ о меха-
низме действия фермента.
Группа французских исследователей впоследствии представи-
ла (результаты физико-химического изучения гексокиназы, кото-
рые в некоторой степени подтверждают их гипотезу об упорядо-
ченном механизме. Им удалось показать, что фермент из дрож-
жей связывает глюкозу прочнее, чем MgATO2- [93]. Подробное
исследование ингибирования фермента позволило Косоу и Роузу
[94] заключить, что при некоторых условиях (малые или уме-
ренные ингибирующие концентрации MgAflO-) продукты реак-
ции выделяются в определенной последовательности — сначала
глюкозо-6-фосфат, а затем АДФ.
Первые результаты исследования протонной релаксации, вы-
полненные Кон [4], свидетельствуют о том, что гексокиназа от-
носится к типу I. Тем самым в какой-то степени поддержи-
вается представление об упорядоченном механиме. Оказалось, что
усиление релаксации МпАДФ- или МпАТФ2- наблюдается только
в присутствии глюкозы. Однако последнее можно объяснить и
тем, что комплекс Мп—нуклеотид может ферментом связываться
двояко: без глюкозы — со слабым усилением, в присутствии глю-
козы — с большим усилением [5]. Очевидно, для выяснения меха-
низма необходимы дальнейшие исследования.
3.7. З-Фосфоглицераткиназа
Этот фермент катализирует обратимый перенос фосфорильной
группы от АТФ к 3-фосфоглицерату. По данным широких кине-
тических исследований фермента из дрожжей его свойства пред-
ставляются очень похожими на свойства креатинкиназы [95].
Результаты измерений протонной релаксации свидетельствуют
о том, что 3-фосфоглицераткиназа относится к типу I [4]. Срав-
нительно небольшая молекулярная масса (45 000) и простота
выделения из многих тканей позволяют надеяться, что этот фер-
мент будет удобным объектом в дальнейших физико-химических
исследованиях.
Киназы
683
3.8. Фосфофруктокиназа
Фосфофруктокиназа занимает ключевое место в гликолитиче-
ском цикле и участвует во многих контрольных механизмах.
Большинство работ было направлено на выяснение именно этой
роли фермента, например на изучение его аллостерических
свойств [96]. Последнее не относится к теме данной главы.
3.9. Нуклеозиддифосфаткиназа
Этот фермент катализирует фосфорилирование нуклеозидди-
фосфатов нуклеозидтрифосфатами. Он проявляет относительно
широкую специфичность к субстратам обоих классов. Кинетиче-
ские исследования свидетельствуют о том, что реакция протекает
по механизму типа «пинг-понг» [97]. Первый продукт отделяется
от фермента еще до присоединения второго субстрата, т. е. про-
текают реакции
АТФ-+-Е АДФ-+-Р—Е
Р—Е + ВДФ Е +ИТФ
АТФ + ИДФ ч~=^ АДФ + ИТФ
Доказательства такого необычного для киназ протекания реак-
ции (аналогичной фосфоглюкомутазному процессу [97]) первыми
получили Гарсис и Клеланд [18], которые обнаружили образова-
ние 32Р-меченого фермента после инкубации с АТФ, меченным в
у-положении. При щелочном гидролизе из такого препарата фер-
мента удалось выделить 4 экв фосфорилированного гисти-
дина [19].
4. ЗАКЛЮЧЕНИЕ
Во введении основное внимание было уделено свойствам, об-
щим для всех киназных реакций. По этим данным можно
было бы заключить, что все эти ферменты функционируют при-
мерно одинаково [6, 11]. Однако даже на примерах немногих
более подробно изученных ферментов мы убедились в существо-
вании значительных различий — неодинаковые способы связыва-
ния ферментами ионов двухвалентных металлов, необходимость
присутствия в отдельных случаях одновалентных катионов, раз-
личные способы переноса фосфорильной группы (как это пока-
зано на примере нуклеозиддифосфаткиназы), неодинаковые ки-
нетические механизмы.
Для большинства ферментов пока еще остается невыясненной
роль ионов двухвалентных металлов. В этом отношении наиболее
44*
«84
Глава 18
ясная картина вырисовывается для пируваткипазы, для которой
получены надежные экспериментальные доказательства образо-
вания комплекса с металлом в качестве мостика между фермен-
том и переносимой фосфорильной группой. Трудно оценить воз-
можное значение других свойств ионов металлов в тех или иных
киназных реакциях [98], например их электрофильности или
влияния нейтрализации заряда и приведения молекулы нуклео-
тида в специальную конфигурацию на образование металл-нук-
леотидного комплекса. Конечно, и без ферментов ионы металлов
увеличивают скорость гидролиза АТФ [99], хотя этот эффект
намного меньше того, что наблюдается в присутствии фермента,
а его значение для киназ точно не известно. Во многих случаях
комплексы металлов с нуклеотидами связываются с ферментами
прочнее, чем свободные нуклеотиды, — но и этот эффект незна-
чителен [35, 38]. Можно упомянуть и вторичный «мостиковый»
эффект ионов металла: в обратной киназной реакции при пере-
ходе от а- и р-фосфатов АДФ к р- и у-фосфатам АТФ ион ме-
талла может образовывать промежуточное соединение между
АДФ и переносимой фосфорильной группой, даже не входя в
контакт с поверхностью фермента.
Работа с киназами продолжается во многих лабораториях.
Делаются попытки приготовления кристаллических препаратов,
удобных для рентгеноструктурного анализа. Конечно, эти иссле-
дования важны для понимания механизма реакций, катализируе-
мых ферментами этой группы. Однако наибольший вклад в ре-
шение этой проблемы внесли (и, по-видимому, еще внесут) раз_-
личные методы магнитного резонанса.
СПИСОК ЛИТЕРАТУРЫ
1. Nordlie R., Lardy И. A., in «The Enzymes», Vol. 6, Academic Press, New York,
1962, p. 1.
2. Boyer P. D., Lardy H. A., Myrback K. (eds.), The Enzymes, Vol. 6, Academic
Press, New York, 1962.
3. Номенклатура ферментов, ВИНИТИ, M., 1966.
4. Cohn М., Biochemistry, 1963, 623.
5. Mildvan A. S., Cohn M„ Adv. Enzymol., 33, 1 (1970).
6. Cleland W. W., A. Rev. Biochem., 36, 77 (1967).
7. O’Sullivan W. J., Cohn M., J. Biol. Chem., 241, 3116 (1966).
8. James E„ Morrison J. F., J. Biol. Chem., 241, 4758 (1966).
9. Plowman К- M., Krall A. R., Biochemistry, 4, 2809 (1965).
10. Chang H. C„ Lane M. D., J. Biol. Chem., 241, 2413 (I960).
11. Crane R. K-, in M. Florkin, E. H. Stotz (eds.), Comprehensive Biochemistry,
Vol. 15, Elsevier, Amsterdam, 1964, p. 200.
12. Mahler FL R., Cordes E. H., Biological Chemistry, Harper and Row, New York,
1966, pp. 411, 422; Atkinson M. R., Morton R. K„ in M. Florkin, H. S. Mason
(eds.), Comparative Biochemistry, Vol. 2, Academic Press, New York, 1960,
Ch. 1.
13. Cohn M., J. Cell. Comp. Physiol., Suppl. 1, 54, 17 (1959).
Киназы
685
14. Cohn М., Biochim. Biophys. Acta, 20, 92 (1956).
15. Harrison IL. H., Boyer P. D., Falcone A. B„ J. Biol. Chem., 215, 303 (1955).
16. Mildvan .4. S., in «The Enzymes», 3rd edn., Academic Press, New York, Vol. II,
1970, p. 445.
17. Kennard O., Isaacs N. It7., Coppola J. C., Kirby A. J. et al.. Nature, 225, 333
(1970).
18. Garces E., Cleland ll7. №.. Biochemistry, 8, 633 (1969).
19. Edlund B., Rask L., Olsson P., Walinder 0., Zetterquist O., Engstrom L., Eur.
J. Biochem., 9, 451 (1969).
20. Phillips R., Eisenberg P., George P., Rutman R. I., J. Biol. Chem., 240, 4393
(1965).
21. Woos £., Acta Chem. Scand., 12, 528 (1958); 11, 1082 (1957).
22. O’Sullivan W. J., Perrin D. D., Biochemistry, 3, 18 (1964).
23. Phillips R. C., George P., Rutman R. J., Biochemistry, 2, 501 (1963).
24. Sillen L. G., Martell A. £., Stability Constants, Special Publication No. 17,
The Chemical Society, London, 1964.
25. Phillips R. C., Chem. Rev., 66, 501 (1966).
26. Melchior N. C., Melchior J. B., J. Biol. Chem., 231, 609 (1958).
27. Bock R. M„ in P. D. Boyer, H. Lardy, K. Myrback (eds.), The Enzymes, Vol. 2,
Academic Press, New York, 1960, p. 3.
28. Perrin D. D., Sharma V. S., Biochim. Biophys. Acta, 127, 35 (1966).
29. Phillips R. C., George P„ Rutman R. J., J. Am. Chem. Soc., 88, 2631 (1966);
J. Biol. Chem., 244, 3330 (1969).
30. Taqui Khan M. M„ Martell A. E., J. Am. Chem. Soc., 88, 668 (1966).
31. O'Sullivan W. J., in R. M. C. Dawson, D. C. Elliott, W. H. Elliot, К- M. Jones
(eds.), Data for Biochemical Research, Oxford University Press, London, 1969,
p. 423.
32. Hammes G. G., Levison S. A., Biochemistry, 3, 1504 (1964).
33. Diebier H., Eigen M., Hammes G. G„ Z. Naturforsch., 156, 554 (1960).
34. Cohn M„ Leigh J. S., Jr., Nature, 193, 1037 (1962).
35. O'Sullivan It7 J., Cohn M., J. Biol. Chem., 241, 3104 (1966).
36. O'Sullivan W. J., Noda L„ J. Biol. Chem., 243, 1424 (1968).
37. Mildvan A. S., unpublished observations.
38. Morrison J. F., O’Sullivan W. J., Biochem. J., 97, 37 (1965).
39. Koskland D. E., Jr., Neet К. E., A. Rev. Biochem., 37, 359 (1968).
40. Ennor A. H., Rosenberg H., Biochem. J., 57, 203 (1954).
41. Kuby S. A., Noda L., Lardy H„ J. Biol. Chem., 210, 65 (1954).
42. Noda L., Nihei T., Morales M. F., J. Biol. Chem., 235, 2830 (1960).
43. Nihei T., Noda L., Morales M., J. Biol. Chem., 236, 3203 (1961).
44. Morrison J. F., O’Sullivan W. J., Ogston A. G., Biochim. Biophys. Acta 52, 82
(1961).
45. Kuby S. A., Mahowald T. A., NoItmann E. A., Biochemistry, 1, 748 (1962).
46. Morrison J. F., James E., Biochem. J., 97, 37 (1965).
47. Morrison J. F., Uhr M. L., Biochim. Biophys. Acta, 122, 57 (1966).
48. Hughes T. R., Klotz I. M., in D. Glick (ed.), Methods of Biochemical Analy-
sis, Vol. 3, Interscience, New York, 1956, p. 265; Hayes J. E., Velick S. F.,
J. Biol. Chem., 207, 225 (1954); Schachman H. K-, Biochemistry, 2, 887 (1963).
49. Mildvan A. S., Leigh R. A., Biochim. Biophys. Acta, 89, 393 (1964).
50. O'Sullivan W. J., Diefenbach H., Cohn M., Biochemistry, 5, 2666 (1966).
51. Jacobs G., Cunningham L. It7, Biochemistry, 7, 143 (1968); Lui N. S., Cun-
ningham L„ Biochemistry, 5, 144 (1966).
52. Suetter C. H„ Singleton R., Kayne F. J., Arrington S., Glass J., Mildvan A. S„
Biochemistry, 5, 131 (1966); Suelter С. H., Biochemistry, 6, 418 (1967)-
Kayne F. J., Suelter С. H„ Biochemistry, 7, 1678 (1968).
53. Beinert H„ Palmer G., Adv. Enzymol., 27, 105 (1965).
54. Ehrenberg A., Malmstrom B. G., Vanngard T., Magnetic Resonance in Biolo-
gical Systems, Pergamon Press, New York, 1967.
686
Глава 18
55. Malkin R., Malmstrom В. G., in G. Eichhorn (ed.), Inorganic Biochemistry,
Elsevier, Amsterdam, 1970.
56. Cohn M„ Townsend J., Nature, 173, 1090 (1954).
57. Mildvan A. S., Cohn M., J. Biol. Chem., 240, 238 (1965).
58. Taylor I. S., Leigh J. S., Jr., Cohn M., Proc. Natn. Acad. Sci. U. S., 64, 219
(1969).
59. Bradbury E. M„ Crane-Robinson C., Nature, 220, 1079 (1968); McDonald С. C.,
Phillips IF. D., in S. N. Timasheff, G. Fasinon (eds.), Biological Macromole-
cules, Vol. Ill, Marcel Dekker, New York, 1970.
60. Roberts G. С. K-, Dennis E. A., Meadows D. H„ Cohen J., Jardetzky O., Proc.
Natn. Acad. Sci. U. S., 62, 1151 (1969).
61. Reed G., Cohn M., O’Sullivan W. J., J. Biol. Chem., 245, 6547 (1970).
62. O’Sullivan W. J., Cohn M., J. Biol. Chem., 243, 2737 (1968).
63. Hamilton C. L., McConnell H. M., in A. Rich, N. Davidson (eds.), Structural
Chemistry and Molecular Biology, W. H. Freeman and Co., San Francisco,
1968, p. 115.
64. Weiner H., Biochemistry, 8, 526 (1969); Mildvan A. S., Weiner H„ Bioche-
mictry, 8, 552 (1969); Mildvan A. S., Weiner H., J. Biol. Chem., 244, 2465
(1969).
65. Ruby S. A., Noda L., Lardy H. A., J. Biol. Chem., 209, 191 (1954).
66. Yue R. H., Palmieri R. H„ Olson О. E., Kuby S. A... Biochemistry, 6, 3204
(1967).
67. O’Sullivan W. J., Int. J. Protein Res., 3, 131 (1971).
68. Thomson A. R., Eveleigh J. W., Miles B. J., Nature, 202, 267 (1964).
69. Mahowald T. A., Biochemistry, 4, 732 (1965); Mahowald T. A., Agodoa L.,
Biochem. Biophys. Res. Commun., 37, 576 (1969).
70. Leigh J., Cohn M., unpublished results, cited in Cohn M„ Q. Rev. Biophys.,
3, 61 (1970).
71. Hammes G. G., Hurst J. K-, Biochemistry, 8, 1083 (1969).
72. Uhr M. L., Marcus F., Morrison J. F., J. Biol. Chem., 241, 5428 (1966).
73. O’Sullivan W. J., Virden R., Blethen S., Eur. J. Biochem., 8, 562 (1968); O’Sul-
livan W. J., Smith E., Marsden K, Proc. Aust. Biochem. Soc., 1970.
74. Gaffney T. J., O’Sullivan W. J., Biochem. J., 90, 177 (1964).
75. Kassab R., Pradel L. A., der Terrossian E., Thoai N. V., Biochim. Biophys. Acta,
132, 347 (1967).
76. Boyer P. D„ in ref. [2], p. 95.
77. Mildvan A. S., Cohn M., unpublished results.
78. Cottam G. L., Hollenberg P. F., Coon M. J., J. Biol. Chem., 244, 1481 (1969).
79. Melchior J. B., Biochemsitry, 4, 1518 (1965); Reynard A. M„ Haas L. F.,
Jacobsen D. O., Boyer P. D., J. Biol. Chem., 236, 2277 (1961).
80. Mildvan A. S„ Hunsley J. S., Suelter С. H., submitted for publication.
81. Mildvan A. S., Leigh J. S„ Cohn M., Biochemistry, 6, 1805 (1967).
82. Perloff A., Abstr. Am. Cryst. Ass., 1970, 74.
83. Mildvan A. S„ Cohn M., J. Biol. Chem., 241, 1178 (1966).
84. Reuben J., Cohn M„ J. Biol. Chem., 245, 6539 (1970).
85. Kayne F. J., Reuben J., J. Am. Chem. Soc., 92, 220 (1970).
86. Noda L., J. Biol. Chem., 232, 551 (1957); p. 139 in ref. [2].
87. Cohn M., unpublished experiments.
88. Kress L. F., Noda L„ J. Biol. Chem., 242, 558 (1967).
89. Kress L. F„ Bono V. H., Noda L„ J. Biol. Chem., 241, 2293 (1968).
90. Crane R. K-, in ref. [2], p. 47.
91. Noat G., Ricard J., Borel M., Got C., Eur. J. Biochem., 5, 55 (1968).
92. Fromm H., Eur. J. Biochem., 7, 385 (1959).
93. Noat G., Ricard J., Borel M., Got C., Eur. J. Biochem., 11, 106 (1969).
94. Kosow D. P., Rose I. A., J. Biol. Chem., 245, 198 (1970).
95. Malmstrom B. G„ Larsson-Raznikiewicz M„ ref. [2], p. 85; Larsson-Razni-
kiewicz M., Biochim. Bophys. Acta, 85, 60 (1964).
Киназы
687
96. Lindell Т. L, Stellwagen E., J. Biol. Chem., 243, 907 (1968).
97. Ray W. J., Roscelli G. A., J. Biol. Chem., 239, 1228 (1964).
98. Scrutton Л1. C., in G. Eichhorn (ed.), Inorganic Biochemistry, Elsevier, Amster-
dam, 1970.
99. Tetas Л1., Lowenstein J. M., Biochemistry, 2, 350 (1963).
100. Miller R. S., Mildvan A. S„ Chang H.-С., Easterday R. L., Maruytima H„ La-
ne M. D., J. Biol. Chem., 243, 6030 (1968).
101. O’Sullivan W. J., Ph. D. Thesis, Australian National University, 1963.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аденилатдезамипаза 494
Аденилаткиназа 445, 662, 666, 669,
672, 679—681
Аденозиндифосфат (АДФ) 127, 445,
492, 656, 662,663, 664,665,666 и сл.,
668, 673, 674, 675, 676, 677, 678,
679, 680, 681, 682, 683
Аденозинмонофосфат, комплексы 127
Аденозинтрифосфат (АТФ)
гидролиз 270, 625, 627, 646 и сл.,
650, 683
комплексы 126, 127. 445, 492,
493, 627, 661, 662, 663, 664, 665,
666, 667, 668, 676, 677, 678, 679,
680, 681, 682, 683
в креатинкиназной реакции 672,
673
Аквокомплсксы 90
Аконитаза 485 и сл.
Актинонин 241
Актины 249—253, 260, 269
Аламетицин 259, 269
Аланин, комплексы 151, 158, 188
Алкогольдегидрогеназа 457, 458, 459,
460 и сл., 462
Альбомицин 213, 216, 218, 220, 228,
езо
61 228
62 228
е '228
Альбумин сывороточный бычий 127
комплекс с медью 292—294
реакция с ионами цинка 280—282
Альдолазы 481—484
Амилазы 488
Аминоацетил-тРНК-синтетазы 491
Аминокислоты
в качестве лигандов 113, 121—
125, 151 и сл.
связывание металлом 152 и сл.
центры связывания металлов
154 и сл.
Аммонийлиазы 487—488
Антаманид 258 и сл.
Апокарбоангидраза 577 и сл., 580,
581. 586
Апока1рбоксипептидаза А 524, 525 и
сл., 543
Апотрансферрин 355
Апофермент, получение 457
Апоферритин 303, 304—305, 319
биосинтез 322
дисперсия оптического вращения
320
четвертичная структура 305 и сл.
.Апоцерулоплазмин 363, 368 и сл., 374
Аргиназа 445, 489
Аргинин 433
Аргининкиназа 445, 672, 676 и сл.
Ароматические системы, реакция с
ионами металлов 439
Асиалоцерулоплазмин 371. 372, 373
Асимметрии 240
Аспергилловая кислота 209, 239
АТФ-аза 251
3Н-Ацетазоламид 593, 595
связывание с карбоангидразой
586, 594, 595
N-Ацетиллизин 433
Ацетил-КоА-синтетаза 449, 489—490
Ацетилфосфат, гидролиз 625, 626, 646
Ацетоксимеркурсульфаинламид, комп-
лекс с карбоангидразой 607, 609,
610
Аэробактин 208, 218, 233
Белки, комплексы с металлами 152—
162, 274—298
методы исследования 275—276
pH и другие факторы, влияющие
на сродство иона металла к
лиганду 276—279
число связывающих центров, ме-
тоды определения 275 и сл.
Предметный указатель
689
Белки, переносящие кислород см.
Гемоглобин, Гемэритрин и Гемо-
цианин
Бензоилформилдекарбоксилаза 475
Биотин, комплексы 492
Биотинзависимые ферменты 465—471
Биотинкарбоксилазы 465 и сл., 492
и сл.
Биурет, комплексы с металлами 151
и сл., 163—165, 197
Боварицин 253
Бора эффект 380
Вазопрессин, комплекс с медью 285
L - ВалинГ4-моноацетатмедь(П) 120,
121
Валиномицин 247, 248, 250, 254—256,
260, 261, 264, 269
комплекс с калием 255 и сл.
Ванадий
в гидролизе фосфатов 647
координационные числа и сте-
реохимия комплексов 35 и сл.
Васки комплекс 401
Внедрения реакции посредством ко-
ординации 437
Вольфрам, координационные числа
и стереохимия комплексов 44 и сл.
Галлин, комплексы
с микобактином 236
с трансферрином 343, 351
Гексозофосфатизомеразы 478
Гексокиназа 662, 672, 681 и сл.
Гем 100
Гемин 143
Гемоглобин 300, 378, 379, 380
Гемопексин 383
Гемосидерины 300, 325
Гемоцианины 149, 377, 378, 404—418
каталазная активность 414
круговой дихроизм 409
молекулярная масса 416 и сл.
распространение в животном
мире 404 и сл.
связывание кислорода 409, 410
и сл.
— лигандов, иных, чем кислород
412 и сл.
спектры поглощения 408—410
старение и регенерация 413
центры связывания меди 416 и
сл.
четвертичная структура 406 и сл.
Гемсинтетаза 494
Гемэритрин 145—149, 377—402
активный центр, свойства 384—
395
--------строение 395 и сл.
взаимопревращение химических
форм 379
координация железа 382 и сл.
магнитная восприимчивость 387
и сл.
Мессбауэра спектры 390—394
модельные соединения 385 и сл.
окисление и реакции с малыми
молекулами 380 и сл.
первичная структура 382
равновесие взаимодействия с
кислородом 379 и сл.
распространение и выделение
378—379
связывание молекулярного кис-
лорода 145—140
спектры поглощения 146, 387
— — и круговой дихроизм 387
строение и функции 400—402
ЭПР 395
Ген операторный 206
Ген-репрессор 206
Гидролиазы (дегидратазы) 484—487
Гистамин, комплекс с медью (II) 187
Гистидин, комплексы с металлами
120, 122, 123, 124, 158, 177 и сл.,
183—184, 191, 197, 515, 516, 610
Гистилиндезаминаза 487
Гликоген, фосфоролиз 628
Глицеральдетид-З-фосфатдегпдроге-
наза 458
Глицерофосфаты, гидролиз 625
Глицилглицилглицилглицин, комплек-
сы с металлами 160, 161, 163, 164,
165, 166, 175, 197, 286
Глицилглицин 125, 152, 165
комплексы с металлами 155, 169,
170, .176, 177, 178
Глицилтирозин, комплекс с карбо-
ксипептидазой А 518—522
Глицин 102, 103, 122, 124, 152
комплексы с металлами 155, 156,
157, 159, 165, 166, 178, 190, 191,
434 и сл.
Глутаматдегидротеназа 458, 463
Глутаминовая кислота, комплексы с
металлами 122, 123, 160
Глутаминсинтетаза 449, 493
Глюкозо-6-фосфатдегидрогеназа 458
Глюкозофосфаты 634
Гомоизоцитратдегидрогеназа 463
Грамицидин 248, 256, 260
Грамицидины 256
690
Предметный указатель
Гризеип 218, 228
Призорексин 262, 264
Даномицин 239
Дегидрогеназы 458
активация ионами металлов 465
каталитическая роль металла
460—463
Дезоксигемэритрин
мессбауэровские спектры 390,
391, 392
структура 400
ЭПР 395
Дезоксирибонуклеиновая кислота,
взаимодействие с ионами метал-
лов 128
Дезоксиродоторуловая кислота 231
Дейтеропорфирин IX, диметиловый
эфир, комплексы 129, 143
Декарбоксилазы 466, 475
окислительные 463—465
Деферр,ней дер о хромы 208, 209, 215,
216
Деферриферриоксамин В 221
Деферриферрихромы 215, 218
Деферришизокинин 208
Деферроферровердин 242
Дианмицин 247, 261, 264, 271
Диастереомеры 63—70
Дибензо- 18-корона-6, комплексы с
металлами 255, 257, 258, 264
Дигидроксомостиковые димеры 142
и сл.
Диглицилглицин, комплекс с медью
167—169
Димерумовая кислота 232, 233
Р Д - Дим ети л м а л атдеги д роген аза 463
Динактин 249, 252
Диоксибензоилглицин см. Итоевая
кислота
6-М-Ди-(2,3-диоксибензоил)-1.-лизин
225
2,3-Д'иокси-М-бепзоил-1-серин 224,
225 и сл.
2,3-Диоксибензойиая кислота 213,
220, 222, 226
Донорные атомы 18, 98
классификация 54 и сл.
Енолаза 484—485, 672
Железо
в аконитазе 485, 486
внедрение .в гем 494
в гемэритрине 378
гидрат оксида 312, 316
в гидролизе фосфатов 647
депо 300
в карбоксипептидазе А 508
комплекс с альдолазой 481, 482
— с аминокислотами 122, 123
— с аэробактииом 233
— гидроксаматные 207, 212, 222,
226—239
— с микобактином 234, 235, 236
— с пиримином 241, 242
— с порфириновыми димерами
143
— с родоторулатом 230, 231
— фенолятные 207, 212, 222,
223—226
— с фосфатазой 634
— с цистеином 190, 191
— с энтеробактином 223, 224
комплексы биядерные 140—143,
385—386, 396
— Fe(II) 134—136
— Fe(lII) 136—139
— электронное строение 133, 135
координационные числа 39
координация в гемэритрине 382
и сл.
связывание с трансферрином 338,
346
стереохимия 38 и сл.
транспорт трансферрином 354 и
сл.
транспортные соединения 205,
206
удаление патологическое 221
в ферритине 300, 302. 318 и сл.
в цитратгидролазе и тартратгид-
ролазе 486
Железо(Ш)гемоглобин 144, 145
Железо (III) гемопротеины, ги дрокси-
ды 144 и сл.
Железо(Ш)порфирины 144 и сл.
Железо(Ш)родоторулат 230, 231
Железо(III)энтеробактин 223, 224
Желеэосвязывающие белки 331. См.
также Трансферрины
Железосерусодержащие белки 100
Изомеразы 476—478
Изомерия
геометрическая 63
конфигурационная 62, 63—79
конформационная 62, 79—86
лигандов 68, 69
оптическая 20 и сл.
связей 68
Предметный указатель
691
Изомеры
конфигурационные 62
конформационные 62, 79—86
лигандов 68, 69
оптические 70—79
связей 68
структурные 68—70
транс 63, 64
цис 63, 64
Изопентенилпирофосфатизом е р а з а
478
Р-Изопропилмалатдегидрогеназа 463
Изоцитратдегидрогеназа 463, 464
Изоцитратлиаза 483
Имидазол
комплексы с металлами 178, 180,
197, 278, 280, 281, 283 и сл„
289, 290, 291
связывание в трансферрине 350
Индий, комплекс с трансферрином
343
Инсулин, реакция с цинком 282—285
Иодацетат, реакция с карбоангидра-
зой 589, 590
Ионофоры
как антибиотики 269
динамика образования комплек-
сов 265—268
нейтральные 249—262
карбоксилатные 262—265
катализ транспортных равнове-
сий 266—268
катионная активность 259—262
применение 269—270
Ирвинга — Уильямса последователь-
ность 100, 101, 102, 103, 117, 125,
127, 129, 544
Иридиндифосфатглюкоза 630
Иридий, комплексы 438
Итоевая кислота 172, 211, 212, 213,
215, 219, 226
Кадмий, комплексы
с биуретом 164
с карбоангидразой 577, 579, 580,
587, 606
с карбоксипептидазой А 524, 543
с трансферрином 343
с фосфоглюкомутазой 480
со щелочной фосфатазой 457,
637, 656
Калий
комплексы с валиномицином 255
и сл.
— монамицинами 260
— с нонактином 251, 252
транспорт 247
Кальций
и аденилаткиназа 680
в аминоацил-тРНК-сннтетазах 491
в амилазах 488
влияние иа глутаматдегидроге-
назу 463
— на окисление гемоцианина 411
в гидролиазах 488
в глутаматсинтетазе 493
в киназах 661, 668
комплексы с АДФ 674
— АТФ 665
— с ГЛИШ1ЛГЛИЦИЛГЛИЦИНОМ 161,
163
в креатинкиназе 673
Карбоангидразы 446, 447, 457, 561—
618
аминокислотный состав 565—567
апофермент 577
взаимодействие с сульфамидами
586
----содержащими хромофоры
597—600
видовые варианты 562—565
гидратация — дегидратация СО2
561, 562
кинетика 613
кислотно-основные свойства 571
и сл.
комплексы с кобальтом 577, 579,
580, 581, 582, 583, 585, 587, 589
— с медью 577, 579, 580, 582,
583, 587
— с металлами, устойчивость
580 и сл.
— с никелем 577, 579, 580, 582,
583, 587
— спектры поглощения и опти-
ческая активность 581—586
— с сульфамидами 593—600
магнитная восприимчивость 606
методы получения 562
механизм действия 611—618
оптические свойства 574—577
последовательность аминокислот
'569—571, 573
растений 567—569
реакция с циан- и сульфид-иона-
ми 591, 592
рентгеноструктурный анализ
606—611
связывание ангибиторов 586—593
со спиновыми метками 603—606
спектры поглощения 564, 572
581—584
— кругового дихроизма 584—
586, 596
692
Предметный указатель
удаление цинка 577
устойчивость 578, 580
ферментативная активность 578—
581
физико-химические свойства 564,
567
флуоресценция 599 и сл.
фосфоресценция 600
цинк в 577
-— места связывания 610
ЭПР-спектр 604 и сл.
ЯМР-спектр 601—603
Карбоксилазы 465 и сл.
Карбоксикиназа 472
Карбоксипептидаза А 446, 457, 504—
556
активность как функция pH 535
и сл.
активный центр 514—518
-----роль различных остатков
537—542
аминокислотная последователь-
ность 505, 508, 509, 552, 553
взаимодействие фермент—суб-
страт 523
вторичная структура 511 и сл.
выделение 504, 507
гидролиз 506 и <сл.
— пептидов, механизм 545—551
кинетика катализируемых реак-
ций 526—533
кинетико-структурные корреля-
ции 533—537
комплекс с глицилтирозином
518—522
комплексы, с ингибиторами 523
и сл.
— моделирование 522 и сл.
механизм действия 545—551
присоединение субстратов и ин-
гибиторов 533—535
специфичность 506, 548 и сл.
стереоизображение 510
структура 508—516
углы между связями цинк — ли-
ганд 517
устойчивость 507 и сл.
формы 506
функция металла 542—545
центры связывания цинка 200
Карбоксипептидаза В 551—553
Карбоксипептидаза С 554
Кариозии, комплекс с медью 178, 188
Катоптрометры 63. См. также
Энантиомеры
Кетокислоты
биотинзависимое декарбоксили-
рование 466, 467—471
транскарбоксилирование ацил-
коферментом 466, 467—171
Киназы 629, 633, 634, 661—684
активация ионами металлов 165
кинетика 666—669
классификация 670, 672
природа реакций 663
равнове ие металл - субстрат
663—655
ЭПР 670
ЯМР 671 и сл.
Кислоты
жесткие 54, 95
мягкие 54, 55
Кобальт
и аденилаткиназа 680
в алкогольдегидротеназе 459
в карбоксипептидазе А 507, 509
в киназе 661
комплексы, гидролиз триметил-
фосфата 654, 655
— с гистидином 120, 158, 181—
184
— имидазолом 180
— с карбоангидразой 577, 579,
580, 581, 582, 583, 587, 589, 601,
605, 606
— с карбоксипептидазой А 509,
543
— с лейцинаминопептидазой 555
— с пептидами 167—174, 176,
177
— с пируваткиназой 447
— с трансферрином 338, 343
— с фосфатазой 638, 639
— со щелочной фосфатазой 637,
641, 642, 656
координационные числа и сте-
реохимия комплексов 39—40
в креатинкиназе 673
накопление сидерохромов 217
Колицин В 225
Комплексы с металлами
1высокоопиновые 134
квадратио-плоскостнь е 72
конфигурации 18
мостиковые, с металлом в каче-
стве мостика 445—448, 463,
483, 484, 486, 487, 670, 672
— с ферментом в качестве мо-
стика 448 и сл., 452, 670, 672
ниэкоспиновые 134
октаэдрические 30, 70, 72
смешанные (тройные) 115—118,
644
стереоизомерия 62—86
Предметный указатель
693
тетраэдрические 72
•устойчивость, константа 89—93
— последовательность 100—104
фотохимические реакции 439
Кональбумин 331, 332, 337
аминокислотный состав 334, 335
выделение и очистка 333
комплекс с железом 339 и сл.,
343
•молекулярная масса 337
и лактоферрин, биологические
функции 357
оптические свойства 347
углеводный состав 336
из яичного белка 332
Конканавалин А, комплекс с марган-
цем 452
Координационное место 20
Координационные соединения
стереохимия 21 и сл.
устойчивость 89—98
Координационные числа 18, 19—33
вычисление 46
необычные 23. 61
н стереохимия 19—33
Координация с металлами, основные
эффекты 423—424
Копроген 232 и сл.
Короны 248, 257 и сл.
Креатинкиназа 445, 452, 656, 662, 666,
669, 670, 672—676
ингибирование 669
ЭПР- и ЯМР-методы исследова-
ния 673 и сл.
Креатинфосфат, гидролиз 625, 626
Криптанды 258, 264
D-Ксилозизомераза 477, 478
Лактатдегидрогеназа 458
у-Лактоназа 488
Лактоферрин 332, 333 и сл., 340
биологическая функция 357
коровий 334, 335, 336, 337
человека 334, 335, 336, 337
Лейцинаминопептидаза 554—556
Лиазы
активация ионами металлов 465
кетокислот 483
Лиганды 18
изомеры 68
конформации 18
в качестве мостика 445
кислотность 423
координация с металлами, ак-
тивность 422—444
маскирование реакций 424
окислительно-восстановительн ы е
свойства 430—432
•оптически активные 118 и сл.
основность 104—108
размеры 56 и сл.
стереохимия 57—60
хелатный эффект 108—113
Льюиса
кислота 54, 104
основание 98, 104
L.L-491 259
Магний
в аминоацил-тРН'К-синтетазе 491
влияние на глутаматдегидроге-
назу 463
в гидролизе АТФ 646
— фосфатов 625
в глутаматсинтетазе 493
енолазные реакции 485
в киназах 661, 667, 668
комплекс с АДФ 674
— с АТР 655
— с лейцинаминопептидазой 555
в креатинкиназе 673
и пируваткиназа 678
в фосфоглюкомутазе 479, 480,
634
Магнитный ядерный резонанс 669—
672
Малатдегидрогеназа 458
L -Малат-фер мент 463, 464
Малоновая кислота, комплексы с ме-
таллами 103, 109, НО, 111
Манделатрацемаза 478
Марганец
и аденилаткиназа 680, 681
активация пентозизомераз 476,
477
активирование ферментов 671
в аминоацил-тРНК-синтетазах
491
влияние на глутаматдегидрогена-
зу 463
гидролиз фосфатов 646
в глутамипсиптетазе 493
енолазные реакции 485
в карбоксипептидазе А 507
в киназах 661, 662
комплексы с АДФ 665, 666, 674,
675
— с аконитазой 486
— с альдолазой 482
— с АТФ 594
— с карбоангидразой 577, 578,
579, 587
694
Предметный указатель
— с карбоксипептидазой А 526,
542, 543
— с конканавалином А 452
— с креатинкиназой 452
— с лейцинаминопептидазой 555
— с трансферрином 338, 343, 347,
355
— с цитратом 486
— с цитратлиазой 483
— ЯМР-иоследование 680 и сл.
в креатинкиназе 673
координационные числа 37
в ксилозизомеразе 477
в пируваткарбоксилазе 455, 466,
467, 468, 469, 470
реакция ic пируваткиназой 446
парамагнитные эффекты 453
реакция с пируваткиназой 446
скорость ядериой магнитной ре-
лаксации 453 и сл.
стереохимия 37
в фосфоглюкомутазе 480
Медь
в гемоцианине 149, 404, 405
гидролиз АТФ 647
— метилизопропилфосфата 649
— • фосфатов 647, 648
комплексы с аминокислотами
121, 122
— с биуретом 164
— с вазопрессином 285
— с гистамином 187
— с гистидином 186 и сл.
— с глицилглицилглицилглици-
ном 286
— с глицилглицилглицином 164,
170
— с глицином 434 и сл.
— с имидазолам 178, 278, 289
— с карбоангидразой 577, 579,
580, 582, 583, 587
— с карбоксипептидаэой А 525,
543
— с карнозином 178
— с кональбумином 339
— с миоглобином 177, 287 и сл.
— с окситоцином 285 и сл.
— с пептидами 165 и сл., 167 и
сл., 292 и сл.
— с рибонуклеазой 294—298
— стереохимия 42 и сл.
— с сывороточным альбумином
292—294
— с трансферрином 349, 351,
352, 355
— с фосфатазой 637
координационное преимущество
280
координационные числа 43
недостаток 361
реакция с цистином 195
в рибулозодифосфаткарбоксила-
зе 474
связывание пептидного азота 285
и сл.
в церулоплазмине 366—368
Мезопорфирин, диметиловый эфир,
комплекс с цинком 114, 128
Мембраны биологические 265
Меркаптоэтиламин 123
Метаквогемэритрин, магнитные мо-
менты 148
Металлоферменты 443—494
замена металла .на металл 456
и сл.
метаболизм СО2 465—475
методы исследования 449—451
мостиковые 445—448
парамагнитные эффекты 453—
457
пиридиннуклеотидзави с и м ы е
463—465
тиаминпирофосфатзавис и м ы е
475—476
ЭПР 451—452
Металлы, ионы
взаимодействие с карбоксильным
кислородом, типы 158—162
жесткие [класс(а)] 97, 98, 100
мягкие [класс(б)] 97, 98, 190,
193
нежелательные, выведение из
организма 15
переходные, влияние на коорди-
национные числа и стереохи-
мию комплексов 49—54
связывание белком, зависимость
от pH 277 и сл.
щелочноземельные, влияние иа
координационные числа и сте-
реохимию комплексов 45—48
щелочные, влияние на координа-
национные числа и стереохи-
мию комплексов 45—48
Метгемоцианин 414
Метгемэритрин 379, 380, 381
мессбауэровские спектры 391 и
сл.
----комплексов 393, 394
структура 396—В98
ЭПР 395
Р-Метиласпартаза 444, 487
Метилизопропилфторфосфат (зарин),
гидролиз 649
Предметный указатель
695
Метилмалонил-КоА-оксалоа ц е т а т-
траискарбоксилаза 466, 467, 469,
471
Метилфосфат, гидролиз 654, 655
Метионин 122, 123
комплексы с металлами 193—195
Метхлорогемэритрин, электронные
спектры 145, 146, 147
Микобактин 208, 209, 214, 218, 234,
'235
М 235
Р 235, 236
Микобактины 234—236
Миоглобин, комплексы с металлами
126, 177, 287—292
Мицелианамид 239, 240
Модельные соединения 196
Молибден
комплекс с цистеином 192 и сл.
координационные числа 45
стереохимия комплексов 44 и
сл.
Моназомицин 259, 260
Мон актин 249, 252
Монамицины 259
Моненсин 250, 261, 262, 263, 264, 269
цис, quc-Муконатлактоназа 478
Мутазы 478—480
Нигерицин 247 и сл., 261, 262, 263,
264, 269
Никель
в карбоксипептидазе А 507
комплексы с альдолазой 482
— с аминокислотами 122
— с биуретом 165
— с гистидином 120, 182
— с глицилглицином 176
— с карбоангидразой 577, 579,
580, 582, 583, 587
— с карбоксипептидазой А 543
— с лейцинаминопептидазой 555
— с пептидазами 167, 174—176
— с пептидами 167 и сл.
— стереохимия 40—42
— с трансферрином 343
— со щелочной фосфатазой 637
координационные числа 41
и пируваткиназа 678
Ниодим, комплекс с глицином 159
Нонактин, комплексы 248, 249, 251
и сл., 264
Норлейцин, комплекс с серебром 156
Нуклеаза стафилококка 488
Нуклеазы 633
Нуклеиновые кислоты, комплексы с
металлами 126—128
Нуклеозиддифосфаты 100, 673, 683
Нуклеотидтрифосфаты 100
Нуклеотиды, комплексы с металла-
ми 126—128, 492, 663—666
Нуклеофильная атака, катализ вслед-
ствие координации 424—426
Овотрансферрин 331
Оксалоацетат, декарбоксилирование
447, 467, 470
Оксалоацетатдекарбоксилаза 467
Оксалоацетаткетоенолтаутоме р аза
478
Оксигемо цианин 149
Оксигемэритрин 380, 381
магнитный .момент 148
мессбауэровские спектры 391,
392, 393
спектр поглощения 146, 147
структура 398—400
ЭПР 395
Р-Окси-Р-метилглутарил-КоА-ли аза
483
Окси.мочевина 221
З-Окоинитробензолсульфамид 593
Окситоцин, комплекс с медью 285—
287
Оксомостиковые димеры 140—142
Оляция 142
Орнитин 433
Ортоклаз, электронный спектр 139
Основания
жесткие 54, 95
мягкие 54
Палладий, комплексы
с гистидином 185
с метионином 193, 194
с пептидами 167, 176
Пацифарин 220
Пеницилламин 15, 123
Пентозизомеразы 476 и сл.
Пентозофосфатизомеразы 478
Пептидазы 445
Пептиды 40, 125 и сл.
азот, связывание медью 285, 291
гидролиз связи 40
— КПА 545-551
группы как доноры 162 и сл.
комплексы с металлами 164, 165,
167, 168—176
связывание металла 152—154
— через N 166—177
696
Предметный указатель
---------О 163—166, 543
хелатообразование 154—162
Пиримин, комплекс с железом 241,
242
Пирофосфатаза 633, 635
Пирофосфорилазы 629 и сл. см.
Синтетазы
Пируватдекарбоксилаза, комплексы
с металлами 447, 466, 475
Пируваткарбокснлаза 466, 467, 475,
477, 494, 672
Пируваткиназа 446, 448, 462, 472 и
сл., 655, 662, 669, 670, 672, 677—679
комплексы с металлами 446, 447
Платина, комплексы 15, 438
с гистидином 184, 185
с глицином 157
с карбоксипептидазой А 509
с метионином 193, 194
с пептидами 167, 176 и сл.
Полинуклеотидфосфорилаза 630
Полифосфаты, гидролиз 627
Полициклические эфиры 248, 257—
258
Порфирины 28, 114, 128 и сл.
Протеаза В. subtilis 556
Пулчерри минован кислота 240
Ретрородоторуловая кислота 230,
231, 232
Рибонуклеаза, введение иона метал-
ла 294—298
Рибонуклеиновая кислота, взаимо-
действие с ионами металлов 128
Рибулозо-1,5-дифосфат, карбсжсили-
рование 474
Рибулозо-1,5-дифосфаткарбоксила з а
474
Родий, комплексы 437, 438
Родоторуловая кислота 215, 218,
230, 231, 232
Рутений, комплексы 437, 438
Ртуть комплексы
с биуретом 164
с карбоангидразой 577, 579, 580,
587, 606
с карбоксипептидазой А 508, 524,
525, 543
с цистеином 190
со щелочной фосфатазой 637
Салицилфосфат, гидролиз 647
Свинец
в гидролизе фосфатов 647
в карбоксипептидазе 509
Сейка краситель 228
Серебро 155, 190
в карбоксипептидазе А 509
комплексы с актинами и поли-
эфирами 262
— с глицином 156, 159
— с карбоксипептидазой А 509
— с метионином 194
— с моненоином 262
— с нигерицином 262
— норлейцином 156
— с цистеином 190
Серин, комплексы 122, 124, 188 197,
642
Сериндегидратаза 494
Сиаловая кислота 333, 336, 371
Сидерамины 206
Сидеромицины 206
Сидерофилины 331. См. также
Трансферрины
Сидерохромы 206—243
биологическое действие 211—214
биосинтез 208—210
гидроксаматные 215, 222, 227—
239
классификация 207 и сл.
константы устойчивости 215
структуры и свойства 222 и сл.
фенолятные 222, 223—226, 242
Синтетазы 489—493, 630, 635
активация ионами металлов 465
Спектры переноса заряда 139 и сл.
Стереоизомерия 62—86
Стереоселективность 118 и сл., 182,
429
Сукцинимицин 239
Сульфамиды, связывание с карбоан-
гидразой 586
Сульфаниламид 583, 595
«Суперкислоты» 426
Талий, комплексы
с актинами и полиэфирами 262
с пируваткиназой 679
(+)-Тартратдегидраза 486
Теория поля лигандов 133—139
расщепление в поле лигандов
134, 136
Термолизин 556
Тетрагидрофолатсинтетаза 445
Тирозин 188, 189
в карбоксипептидазе А, роль в
активном центре 537 и сл.
.места связывания в трансферри-
не 346 и сл.
связывание металла в трансфер-
рине 353
Предметный указатель
697
Тироцидин 256
Титан, координационные числа и сте-
реохимия комплексов 34
Торий в гидролизе фосфатов 647
Транскарбоксилнрование 466, 667
Транскетолаза 475
Трансферазы 480
Трансферрин 334, 335, 336
биологические функции 354—357
как буфер для иоиов железа 356
и сл.
взаимодействие с ретикулоцита-
ми 355 и сл.
выделение и очистка 333—334
дисперсия оптического вращения
и круговой дихроизм 347 н сл.
кинетика связывания железа 345
комплексы с галлием 343, 351
— с индием 343
— с кадмием 343
— с медью 351, 352, 355
— с металлами 338, 343, 349
— с хромом 338, 343, 349, 355
— ЭПР 348—352
константы связывания железа
342
металлсвязываюшие центры
339—348, 353 .и сл.
молярный коэффициент погло-
щения 337, 338, 346
перенос железа к ферритину 325
роль бикарбоната 344 и сл., 349
и сл.
спектроскопия магнитного резо-
нанса 348—352
субъед 1ничная структура 339
сывороточный 332, 333
химические модификации 352 и
сл.
Трансферрины 331—358
аминокислотный состав 334 и сл.
гидродинамические свойства 338
иммунохимия 337
молекулярная масса 337
оптические свойства 337 н сл.
пептидные карты 355 и сл.
углеводный состав 336
Триметилфосфат, гидролиз 653
Тринактин 249, 252
Фенилаланиндезаминаза 488
Ферменты
активные центры 422
биотинсодержащие 465—474
металлактивируемые 422 и сл.,
443
в качестве мостиков 448 и сл.
роль ионов металлов в механиз-
ме каталитического действия
443—444
схема взаимодействия с метал-
лом и лигандом 443, 444
Ферредоксин 192
Феррибактин 218, 228, 239
Феррикроцин 227
Ферримицин Й19, 220, 237
А, 238
Ферриоксамин 219, 237—239
Ферриредуктаза 328
Ферриродин 227, 228
Феррирубин 227, 228, 232
Ферритин 220, 300—328
аминокислотный состав 305
аналоги железосодержащего яд-
ра 312, 313
биосинтез 320—325
влияние железосодержащей ком-
поненты на свойства и струк-
туру 319—320
дисперсия оптического вращения
320
магнитная восприимчивость 313
и сл.
Мессбауэра спектры 314 и сл.
мобилизация железа 327
образование из апоферритина
325 и сл.
получение 301 и сл.
рентгеноструктуриый анализ 320
роль в организме 301
удаление железа 303—305
четвертичная структура 305—307
ядра, атомная структура 307,
316—319
— дифракция рентгеновских лу-
чей и электронов 312 и сл.
— расположение 318
— рассеяние рентгеновских лу-
чей 311
— спектры Мессбауэра 314—316
— структура 305—307
— электронная микроскопия 310
Феррихризин 227, 228
Феррихром 207, 209, 211, 213, 214,
215, 216, 217, 227, 228, 230
А 215, 227, 228, 229, 230
С 228, 230, 231
Фцрровердин 241, 242
Ферроксидаза 369
Феррохелатаза 217
Флавопротеины 217
Формнлтетрагидрофолатсинтетаза 492
45—2451
698
П редметный указатель
Фосфата перенос 634—635
активирование ионами металлов
624 и сл., 644—651
механизм 629—635
стереохимические модели 651—
655
Фосфатазы 457, 633, 635
щелочная 633, 634, 636—644
— активация 636 и сл.
— механизм катализа 638—640
— модель активного центра
641—644
— прочность связи с металлом
641
— спектры 640, 641
— фосфорилирование 656
кислые 633, 634
Фосфаты
гидролиз, механизм 625 и сл.,
642, 647
— неферментативный механизм
631 и сл.
— ферментативный механизм
629 и сл.
роль в биологии 624
Фосфоглицераткиназа 672, 682
Фосфоглицератмутаза 479, 635
Фосфоглюкозизомераза 478
Фосфоглюкомутаза 478, 479, 480,
634, 635
Фосфодиэстеразы 633
Фосфоенолпируват 484
гидролиз 625, 626
карбоксилирование ферментами
472 и сл.
комплексы 472 и сл.
Фосфоенолпируваткарбоксики н а з а
672, 679
Фосфолипазы 488
Фосфоманноизомераза 478
Фоофомоноэстеразы 633
Фосфор, биохимия 624 и сл.
Фосфорибозизомер аза 478
Фосфорилазы 629, 635. См. также
Синтетазы
Фосфорилирование 627 и сл., 645
Фосфорильная группа, перенос 624,
625 и сл., 633 и сл., 661 и сл.,
663—684
Фосфоролиз 628
Фосфотрансацетилаза 494
Фосфотрансферразы 633, 634
Фосфофруктокииаза 683
Фузаринины 236, 237
Фузиген 237
Хадацидин 209, 240
Хелатные
кольца 60—62
соединения 108—113
Хелатный
комплекс 153
эффект 153
Хелатообразование 20
Химотрипсин, прямой перенос прото-
на 447
Хлоротиазид 593
Хром
комплекс с трансферрином 349
координационные числа 36 и сл.
• стереохимия комплексов 36 и сл.
Х-206 264, 271
Х-537 250, 262, 263, 264
Церий в гидролизе фосфатов 647
Церулоплазмин 346, 361—374
аминокислотный состав 363, 365
биосинтез 373 и сл.
генетический полиморфизм 370
как гликопротеид 370—373
десиалированный 371, 372 и сл.
медь, внедрение 373
— обмен 368
— степень окисления 367
оксидазное действие 369 и сл.
оптические свойства 364
физические свойства 362, 364
электрофоретические свойства
364
Циклоамилоза 440
Цинк
как активатор изоцитратдегидро-
геназы 465
в алкогольдегидрогеназе 458
ионы, реакция с сывороточным
альбумином 280—282
в карбоангидразе 561, 577
в карбоксипептидазе А 125, 196,
504, 507, 509, 542, 543
в киназах 661
комплексы 197
— с альдолазой 482
— с аминокислотами 122
— с биуретом 165, 197
— с гистидином 182, 197, 515,610
— с глицилглицилглицином 160.
197
— с глицилглицином 155
— с глицином 155
— с глутаминовой кислотой 160
— с имидазолом 178 и сл., 197,
280 и сл., 283 и сл., 289
Предметный указатель
699
— с инсулином 282—285
— с карбоангидразой 586
— с лейцинаминопептидазой 555
— с мезопорфирином, диметило-
вым эфиром 114
— с металлоферментами 458
— с миоглобином 287—289
— с серином 188, 197, 642
— с трансферрином 343, 3'55
— со щелочной фосфатазой 636,
637, 656
координационная предпочтитель-
ность 280
лиганды, длина связи 198
— в карбоксипептидазе А 515—
518
— углы связей 515, 517
пептидная связь 542 и сл.
' и пируваткиназа 678
связывающие центры в карбоан-
гидразе 610
стереохимия комплексов 44
в термолнзине 556
в фосфоглюкомутазе 480
в фосфоманнозоизомеразе 478
Цистеин, комплексы с металлами
122, 123. 190, 191 и сл., 192, 195 и
сл.
Цистин 192, 193
Цитратлиаза 483, 484
Цитратсинтетаза 494
3'-Цитидинмонофосфат, комплексы
294, 297, 298
Цитохром с 194, 276
L-Цитромалатгидролаза 486
Цитр ул чин 433
Шизокинин 211, 226, 233
Щавелевая кислота, комплексы 109,
НО, 111
Экзонуклеазы 633
Эккрииосидерофилин 332
Электронный парамагнитный резо-
нанс 451 и сл., 670
Энантиомеры 63. См. также Изоме-
ры оптические
Эндонуклеазы 633
Энергия стабилизации кристалличе-
ским полем 49, 50, 53, 157, 158, 175,
176, 185
Энниатин 248, 250, 253 и сл., 260
В 253, 264
Энниатины 253 и сл.
Энтатное состояние 447
Энтеробактин 207, 209, 210, 211, 212,
214, 218, 219, 222, 223, 224, 225
Энтерохелин 223
Этилендиамин, комплексы 102, 103,
108 и сл., 111
Этилендиаминтетрауксуоная кислота
104, 345, 388, 391, 460, 462, 473, 476,
478, 481, 483, 487, 488, 491, 493,
541, 555, 578, 635
Этиленфосфат, гидролиз 653
Этоксзоламид 593, 595, 596
Эффекты
вицинальный 70, 74—79
конфигурационный 70—74
конформационный 70
«креатина» 675, 676
макроциклический 113—115
хелатный 153
Ядерная магнитная релаксация ядер
лигандов, парамагнитные эффекты
453—457, 671 и сл.
ЯМР-изучение биологических систем
455 и сл.
явление усиления 455—456
Янтарная кислота, комплексы 109,
НО, 111
45’
СОДЕРЖАНИЕ
Предисловие редакторов перевода ................................. 5
Предисловие редактора американского издания ....................... 7
Введение............................................................ 13
Список литературы....................................................16
ЧАСТЬ I. КООРДИНАЦИОННАЯ ХИМИЯ
Глава 1. Структура и стереохимия координационных соединений. Д. А. Бу-
кингем (перевод Н. К. Давиденко) .... 17
1. Введение.................................................17
2. Координационное число н стереохимия..................... 19
2.1. Координационное число 2.............................23
2.2. Координационное число 3.............................23
2.3. Координационное число 4.............................24
2.3.1. Тетраэдр.......................................24
2.3.2. Плоский квадрат................................26
2.4. Координационное число 5.............................28
2.5. Координационное число 6.............................29
2.6. Координационное число 7.............................31
2.7. Координационное число 8.............................32
2.8. Координационное число 9.............................32
2.9. Координационные числа больше 9...................33
3. Координационные числа и стереохимия комплексов обычных
переходных элементов....................................34
3.1. Титаи................................................34
3.2. Ванадий..............................................35
3.3. Хром.................................................36
3.4. Марганец.............................................37
3.5. Железо...............................................38
3.6. Кобальт............................................. 39
3.7. Никель...............................................41
3.8. Медь.................................................42
3.9. Цинк.................................................44
3.10. Молибден и вольфрам.............................44
Содержание 701
4. Факторы, влияющие на координационное число н стереохимию 45
4.1. Эффекты ионов металлов ................45
4.1.1. Ионы щелочных и щелочноземельных металлов 45
4.1.2. Ионы переходных металлов ..... 49
4.2. Свойства лиганда................................54
4.2.1. Донорные атомы 54
4.2.2. Размер лиганда............. 56
4.2.3. Стереохимия лиганда.......................57
4.2.4. Хелатные кольца . 50
5. Стереоизомерия.....................................52
5.1. Конфигурационные изомеры ... 63
5.1.1. Диастереомеры . . ... 63
5.1.2. Оптические изомеры . 70
5.2. Конформационная изомерия . 79
5.2.1. Общие положения . 79
5.2.2. Конформационная изомерия в хелатах . 80
Список литературы ........................................................ 86
Глава 2. Устойчивость координационных соединений. Р. Дж. Ангеличи (пе-
ревод Н. К. Давиденко) . . 89
1. Введение......................................................89'
2. Факторы, влияющие на устойчивость комплексов ионов ме-
таллов ...........................................................93
2.1. Природа иона металла.......................................93
2.1.1. Заряд и размер иона металла . 93
2.1.2. Жесткая и мягкая природа ионов металлов и до-
норных атомов лигандов ...... 96
2.1.3. Последовательность Ирвинга — Уильямса устойчи-
востей комплексов ионов металлов первого переход-
ного ряда................................100
2.2. Природа лиганда....................................104
2.2.1. Основность лиганда ... 104
2.2.2. Хелатный эффект .... ... 108
2.2.3. Макроциклический эффект . . ... 113
2.2.4. Смешанные (или тройные) комплексы . . . . 115
2.2.5. Оптически активные лиганды и стереоселективность 119
3. Некоторые биохимические лиганды..............................121
3.1. Аминокислоты.................................121
3.2. Пептиды......................................125
3.3. Нуклеотиды и нуклеиновые кислоты ..... 126
3.4. Порфирины .............................................. 128
Список литературы.........................................................129
Глава 3. Электронное строение комплексов железа. Г. Б. Грей, Г. Дж. Шу-
гар (перевод Н. К. Давиденко) .... ... 133
1 . Введение....................................................133
2 . Теория поля лигандов для октаэдрических комплексов . . 133
702
Содержание
2.1. Комплексы Fe2+....................................134
2.2. Комплексы Fe3+....................................136
3 Спектры переноса заряда...............................139
4 Димерные комплексы Fe3+ . . 140
4.1. Оксомостпковые димеры.............................140
4.2. Дигидроксомостиковые димеры.......................142
5 Электронное строение модельных хелатов железа и родствен-
ных соединений, представляющих биологический интерес . 143
5.1. Димеры железопорфирина . . 143
5.2. Гидроксиды железо(1Т1)гемопротеинов . 144
5.3. Гемэритрин и проблема связывания кислорода 145
Список литературы................................................149
ЧАСТЬ II. ВЗАИМОДЕЙСТВИЕ ИОНОВ МЕТАЛЛОВ
С АМИНОКИСЛОТАМИ И ПЕПТИДАМИ И РОДСТВЕННЫМИ
ИМ ПРИРОДНЫМИ ХЕЛАТИРУЮЩИМИ АГЕНТАМИ И БЕЛКАМИ
Глава 4. Комплексы металлов с аминокислотами и пептидами. Ганс К. Фри-
ман (перевод В. А. Бидзпля).......................................151
1. Функциональные группы аминокислот и пептидов как металл-
связывающие центры........................................154
1.1. Концевые аминогруппы.............................154
1.2. Концевые и находящиеся в боковых цепях карбоксиль-
ные группы.............................................157
1.3. Пептидные группы..................................162
1.4. Атомы кислорода пептидных групп...................163
1.5. Пептидные атомы азота.............................166
1 6. Имидазольные атомы азота........................177
1.6.1. Взаимодействия металл—имидазол без образования
хелатов.......................................177
1.6.2 Хелатообразование с гистидином .... 181
1.7. Гидроксильные атомы кислорода боковых цепей . . 188
1.8. Сульфгидрильные атомы серы........................189
1.9. Тиоэфирные атомы серы.............................193
1.10. Мостиковые (дисульфидные) атомы серы . 195
2. Ценность информации, получаемой при исследовании «мо-
дельных соединений»...................................196
Список литературы................................................ 200
Глава 5. Транспорт соединений железа микробами (сидерохромы). Дж.
Б. Нейлендс (перевод В. А. Бищзиля) ... 205
1. Общий обзор...........................................205
1.1. Введение....................................... . 205
1.2. Терминология......................................206
1.3. Классификация.....................................207
1.4. Биосинтез........................................ 208
1.5. Биологическое действие............................210
Содержание 703-
1.6. Назначение структуры сидерохромов . 214
1.7. Распространение 21&
1.8. Заключение . . . . 220
2. Структура и свойства.....................................222
2 .1. Методы исследования . 222
2 2 Феноляты . 223
2.2.1. Энтеробактин 223
2.2.2. 2-М-6-К-Ди(2,3-диокеибензоил)-1_-лизин(П) 225-
2.2.3. 2,3-Диокси-М-бензоил-ь -серии (III) . . 225-
2.2.4. 2,3-Диоксибензоилглицин (итоевая кислота) 226
2 3. Гидроксаматы 227
2.3.1. Семейство феррихрома 227
2.3.2. Семейство родоторулиевой кислоты . ... 230
2.3.3. Аэробактип ... ... 233
2.3.4. Семейство микобактина . . 234
2.3.5. Семейство фузаринина 236
2.3.6. Семейство ферриоксамина . 237
2 .4. Другие соединения..................................239
3. Заключение . 242
Список литературы.....................................................243
Глава 6. Ионофоры — хелатирующие агенты для щелочных металлов.
В. К Прессман (перевод В. А. Бидзиля) . . . 246
1. Историческая справка.....................................247
2. Основные структурные особенности ионофоров . . 248
3. Специфические группы ионофоров . 249
3 1 Макротетралидные актины............................ 249'
3.2. Энниатин............................................ 253
3.3. Валиномицин..........................................254
3.4. Грамицидин.......................................... 256
3 5. Полициклические эфиры . ... 257
3.6. Другие соединения....................................258
3.7. Катионная селективность..............................259
4. Карбоксилатные ионофоры............................262
5. Динамика комплексообразования с ионофорами .... 265
6. Транспортные равновесия, катализируемые ионофорами . . 266
7. Применение ионофоров............................. 2691
Дополнения, внесенные в корректуру . ... 271
Список литературы.....................................................271
Глава 7. Комплексы металлов с белками. Е. Бреслоу (перевод В. А. Бидзиля) 274
1. Введение.................................................274
2. Рассмотрение комплексов металлов с белками и методы их
исследования .............................................. 274
3. pH и другие факторы, влияющие на сродство иона металла
к лиганду...................................................276
704
Содержание
4. Выбранные системы................................. • 279
4.1. Ионы цинка и сывороточный альбумин 280
4.2. Взаимодействие цинка с инсулином....................282
4.3. Комплексы иона меди с окситоцином и вазопрессином 285
4.4. Взаимодействие метмиоглобина кашалота с ионами меди
и цинка ...................................287
4.5. Комплексы ионов меди с сывороточным альбумином . 292
4.6 Взаимодействие ионов металлов с рибонуклеазой 294
5. Заключение . 298
Список литературы.............................................. . 298
ЧАСТЬ III. РОЛЬ МЕТАЛЛОПРОТЕИНОВ В НАКОПЛЕНИИ
И ТРАНСПОРТЕ
Глава 8. Ферритин. 77. Л1 Харрисон, Т. Хой (перевод В А Бидзиля) 300
1. Ферритин как соединение, накапливающее железо . . 300
2. Получение и характеристика ферритина ... 301
3. Удаление железа из ферритина: получение и свойства не со-
держащего железа белка апоферритина . ... 303
4. Четвертичная структура апоферритина....................305
5. Морфология и атомная структура ферритиновых ядер . 307
5.1. Электронная микроскопия............................307
5.2. Рассеяние рентгеновских лучей под малыми углами . . 311
5.3. Исследование дифракции рентгеновских лучей с больши-
ми углами рассеяния и дифракции электронов на железо-
содержащих ядрах ферритина и их аналогах .... 312
5.4. Магнитная восприимчивость и спектры Мессбауэра фер-
ритина и гидролизатов Fe (III) . . 313
6. Атомная структура ядер ферритина.......................316
7. Связь белковой оболочки с ядром, состоящим из гидрата
оксида железа............................................ 318
8. Влияние железосодержащего компонента на свойства и струк-
туру белка.................................................319
9. Биосинтез ферритина и его синтез in vitro..............320
10. Механизм образования ферритина из апоферритина . 325
11. Мобилизация железа из ферритина . . 327
Дополнения, внесенные в корректуру . . 327
Список литературы.................................................. 328
Глава 9. Трансферрины (сидерофилины). П. Айсен (перевод В А. Бидзиля) 331
1. История и номенклатура ............................... 331
2. Выделение и очистка ....... 333
2.1. Сывороточный трансферрин............................333
2.2. Кональбумин........................................ 333
2.3. Лактоферрин.........................................333
Содержание 705.
3. Химический состав.............. . .. 334
3.1. Аминокислотный состав.............................. 334
3.2. Пептидные карты . . . . 335
3.3. Углеводный состав и структура . 336
3.4. Иммунохимия...................... . . 337
4. Основные физические свойства............. . . 337
4.1. Молекулярная масса..................................337
4.2. Оптические свойства.................................337
4.3. Гидродинамические свойства 338
4.4. Субъединичная структура.............................339
5. Металлсвязывающие центры................................339
5.1. Существует ли взаимодействие между связывающими
центрами?.................................................339
5.2. Значение констант связывания .... 342
5.3. Типы и состояния ионов металлов, специфически связан-
ных с трансферрином ................................... 343
5.4. Роль бикарбоната....................................344
5.5. Кинетика связывания железа..........................345
5.6. Спектроскопические исследования.....................346
5.6.1. Оптические исследования ......................346
5.6.2. Исследование дисперсии оптического вращения и
кругового дихроизма..................................347
6. Спектроскопия магнитного резонанса ...... 348
7. Изучение химической модификации....................352
8. Обзор современного состояния вопроса о металлсвязывающих
центрах сидерофилинов................................353
9. Биологические функции сидерофилинов................354
9.1. Потребность в физиологическом переносчике железа 354
9.2. Взаимодействие трансферрин—ретикулоцит ... 355
9.3. Трансферрин как буфер для ионов железа . . . 356
9.4. Биологическая функция кональбумина н лактоферрина 357
9.5. Генетическая разнородность трансферринов . 357
10. Заключение.............................................357
Дополнения, внесенные в корректуру .... 357
Список литературы....................................................358
Глава 10. Церулоплазмин И. Г. Шейнберг, А. Дж. Морелл (перевод
А. П. Филиппова) 361
1. Введение................................................361
2. Физические и химические свойства церулоплазмина . 362-
3. Состояние меди в церулоплазмине . . . 366
3.1. Число атомов меди в молекуле церулоплазмина . . 366
3.2. Свойства церулоплазмина, обусловленные присутствием в
нем меди..................................................366
3.3. Степень окисления меди в церулоплазмине .... 367'
706
Содержание
3.4. Обмен меди церулоплазмина на медь из растворов ее
солей.....................................................368
4. Апоцерулоплазмип........................................368
5. Оксидазные свойства церулоплазмина . . . . 369
6. Неоднородность и генетический полиморфизм церулоплазмина 370
7. Характеристика церулоплазмина как глпкопротеида . 370
8. Биосинтез церулоплазмина................................373
Список литературы...................................................374
Глава 11. Гемэритрин. М. И. Окамура и И. М. Клотц (перевод А. П. Фи-
липпова) .............................................................377
1. Введение................................................377
2. Распространение белка среди живых организмов и его выде-
ление .................................................. 378
3. Химические формы гемэритрина............................379
4. Равновесие взаимодействия с кислородом ... 379
5. Процессы окисления и реакции с малыми молекулами . . 380
6. Образование субъединиц..................................382
7. Координация железа в белке..................... 382
8. Магнитные и спектральные свойства активного центра . 384
8.1. Модельные соединения................................385
8.2. Гемэритрин..........................................387
8.2.1. Спектры поглощения и кругового дихроизма (КД) 387
8.2.2. Магнитная восприимчивость.....................387
8.2.3. Спектры Мессбауэра . .............390
8.2.4. Электронный парамагнитный резонанс ... 395
9. Строение активного центра...............................395
9.1. Метгемэритрин.......................................396
9.2. Оксигемэритрин.................................... 398
9.3. Деоксогемэритрин....................................400
9.4. Строение и функции гемэритрина ... 400
Дополнения, внесенные в корректуру........................402
"Список литературы..................................................402
Глава 12. Гемоцианин. Р. Лонтье, Р. Виттерс (перевод А. П. Филиппова) 404
1. Введение..........................................................404
2. Распространение в животном мире.........................404
3. Молекулярная масса и четвертичная структура ... 405
4. Спектры поглощения......................................408
5. Связывание кислорода.................................. 410
6. Связывание других лигандов..............................412
7. Старение и регенерация..................................413
8. Апогемоцианин и реставрация гемоцианина . 415
9. Центры связывания меди ................................ 416
10. Выводы.................................................417
Дополнения, внесенные в корректуру..................... 417
Список литературы...................................................418
Содержание
707
ЧАСТЬ IV. РЕАКЦИИ ЛИГАНДОВ, ОБУСЛОВЛЕННЫЕ
КООРДИНАЦИЕЙ С МЕТАЛЛОМ, И МЕТАЛЛОФЕРМЕНТЫ
Глава 13. Активация малых молекул при координации с металлами.
М. М- Джонс, Дж. Э. Хикс мл. (перевод Е. А. Миронова) . . 422
1. Введение............................................422
2. Основные эффекты координации........................423
3. Катализ нуклеофильной атаки.........................424
4. Некоторые важные гидролитические процессы .... 426
4.1. Эфиры аминокислот...............................427
4.2. Пептидные свизи.................................429
4.3. Фосфатные связи.................................429
5. Окислительно-восстановительные свойства лигандов . . . 430
6. Маскирование реакционной способности лигандов в комплек-
сах .......................................................432
7. Активация малых молекул посредством координации . . 436
8. Разные реакции......................................438
9. Заключение..........................................440
Список литературы...................................................44|
Глава 14. Металлоферменты. М. С. Скраттон (перевод А. А. Филиппова) 443
1. Введение................................................443
1.1. Роль ионов металлов в механизме каталитического дей-
ствия ферментов.........................................443
1.2. Комплексы с лигандом в качестве мостика ... 445
1.3. Комплексы с металлом в качестве мостика ... 445
1.4. Комплексы с ферментом в качестве мостика ... 448
2. Экспериментальные подходы к изучению металлоферментов 449
2.1. Общее рассмотрение..................................449
2.2. Электронный парамагнитный резоиаис..................451
2.3. Парамагнитные эффекты на скорости ядерной магнитной
релаксации ядер лигандов .............................. 453
2.3.1. Общие положения...............................453
2.3.2. Явление усиления и его применение .... 455
2.3.3. Замена металла на другой металл...............456
3. Дегидрогеназы...........................................458
3.1. Содержание металла и некоторые общие подходы при
обнаружении и количественной оценке иоиов металла,
связанных с белком . 458
3.2. Каталитическая роль связанного металла .... 460
4. Пиридиннуклеотидзависимые окислительные декарбоксилазы 463
5. Ферменты метаболизма СО2 ... 465
5.1. Биотинсодержащие ферменты ..........................465
5.2. Ферменты карбоксилирования фосфоенолпирувата . 472
5.3. Рибулозо-1,5-дифосфаткарбоксилаза ... 474
5.4. Декарбоксилазы . ...................................475
708
Содержание
6. Тиаминпирофосфатзависимые ферменты.....................475
7. Изомеразы...............................................476
7.1. Пентозизомеразы......................................476
7.2. Гексозо- и пентозофосфатизомеразы . . . 478
7.3. Другие изомеразы.....................................478
8. Мутазы и трансферазы ...................................478
8.1. Фосфоглюкомутаза и другие мутазы 478
8.2. Трансферазы..........................................480
9. Альдолазы...............................................481
10. Гидролиазы и аммоннйлиазы ...... 484
10.1. Гидролиазы..........................................484
10.1.1. Енолаза......................................484
10.1.2. Аконитаза • 485
10.1.3. Другие гидролазы ... . . • 486
10.2. Аммоннйлиазы...............................487
11. Гидролазы......................................488
11.1. Гидролазы, требующие или содержащие Са2+ . . 488
11.2. Аргиназа...................................489
12. Синтетазы......................................489
12.1. Синтетазы, катализирующие отщепление пирофосфата 489
12.1.1. Ацетилкофермеит А-сиитетаза.........489
12.1.2. Аминоацил-тРНК-синтетазы............491
12.2. Синтетазы, катализирующие отщепление ортофосфата 491
12.2.1. Формилтетрагидрофолатсинтетаза ............. 492
12.2.2. Биотиикарбоксилазы...........................492
12.2.3. Глутаминсинтетаза........................... 493
13. Заключение.....................................493
Список литературы....................................................494
Глава 15. Карбоксипептидаза А и другие пептидазы. Марта Л. Людвиг,
В. Н. Липскомб (перевод А. А. Байкова) . ... 504
1. Карбоксипептидаза А быка: выделение, свойства и специфич-
ность .....................................................504
2. Структура карбоксипептидазы А и ее комплекса с глицилти-
розином.....................................................508
2.1. Общее описание конформации белка . . 508
2.2. Активный центр.......................................514
2.2.1. Конформация белка..............................514
2.2.2. .Цинк..........................................515
2.3. Комплекс КПА с глицнлтирозииом.......................518
2.4. Дополнительные структурные исследования . 522
2.4.1. Моделирование..................................522
2.4.2. Комплексы с ингибиторами...................... 523
2.4.3. Апофермент и металлы, отличные от цинка 524
3. Ферментативная активность карбоксипептидазы А . 526
3.1. Кинетика (обзор).....................................526
3.2. Кинетико-структурные корреляции......................533
Содержание
709
3.2.1. Присоединение субстратов и ингибиторов 533
3.2.2. Зависимость активности от pH................. 536
3.3. Химическое изучение роли различных остатков в активном
центре...................................................537
3.3.1. Модификация остатков в центре связывания суб-
стратов .............................................537
3.3.2. Химическая природа лигандов, связанных с метал-
лом, и дисульфидная связь . . . . 540
3.4. Зачем нужен металл? ..... 542
3.5. Механизм действия...................................545
3.5.1. Предположение, основанное на анализе кристалли-
ческой структуры.....................................545
3.5.2. Некоторые альтернативные механизмы .... 548
3.5.3. Специфичность и изменение конформации . 548
3.5.4. Промежуточные соединения и стадия, лимитирую-
щая скорость.......................................549
4. Другие пептидазы.....................................551
4.1. Карбоксппептидаза В...............................551
4.2. Другие карбоксипептидазы и прокарбоксипептпдазы . . 554
4.3. Лейцинаминопептидаза................................554
4.4. Некоторые другие металлсодержащие протеиназы . . 556
Список литературы...................................................556
Глава 16. Карбоангидраза. Цж. Э. Коулмен (перевод А. А. Байкова) 561
1. Введение...............................................561
2. Изоферменты и видовые варианты — физико-химические свой-
ства . ...........................................562
2.1. Аминокислотный состав...............................565
2.2. Карбоангидразы растений.............................567
2.3. Прочие карбоангидразы...............................569
2.4. Последовательность аминокислот..................... 569
2.5. Кислотно-основные свойства..........................571
2.6. Спектры поглощения и оптическая активность . . , 572
2.6.1. Оптическое вращение...........................574
3. Металлокарбоангидразы: апоферменты, связывание металла,
спектры поглощения и связывание ингибиторов ... 577
3.1. Апофермент..............................................
3.2. Ферментативная активность и устойчивость комплексов
металл — белок...........................................578
3.3. Спектры поглощения и оптическая активность . 581
3.4. Связывание ингибиторов . ................586
4. Комплексы карбоангидразы с сульфамидами................593
4.1. Образование комплексов, константы диссоциации . . 594
4.2. Спектры поглощения и круговой дихроизм .... 596
4.3. Взаимодействие с сульфамидами, содержащими хромо-
*°РЫ ....................................................597
710
Содержание
4.3.1. Оптические спектры.............................597
4.3.2. Флуоресценция..................................599
4.3.3. Фосфоресценция . . 600
5. Ядерный магнитный резонанс и электронный парамагнитный
резонанс................................................ • 600
5.1. Карбоангидраза со спиновыми метками.................603
5.2. Магнитная восприимчивость ... . . 60&
6. Рентгеноструктурный анализ карбоангидразы С человека 606.
7. Механизм действия........................................611
Дополнение, внесенное в корректуру...................... . 61&
Список литературы...................................................620
Глава 17. Фосфатный перенос и его активация ионами металлов; щелочная
фосфатаза. Т. Г. Спиро (перевод А. А. Байкова) 624
1. Фосфаты и биоэнергетика..................................624
1.1. Потенциалы переноса фосфорильной группы . . 625
1.2. Фосфорилирование и фосфоролиз ... 627
2. Механизмы переноса фосфата...............................620
2.1. Расщепление связей Р—О и X—О.................... . 629
2.2. Гидролиз............................................630
2.3. Фосфатазы и фосфотрансферазы 633
2.4. Необходимость ионов металлов 634
3. Щелочная фосфатаза.......................................636
3.1. Присоединение металлов ...... 636
3.2. Фосфорилфермент.....................................638
3.3. Присоединение фосфата...............................640
3.4. Модели активного центра........................ . 641
4. Роль ионов металлов......................................644
4.1. Тройные комплексы ферментов с металлами и субстра-
тами .....................................................644
4.2. Нейтрализация заряда............................... 644
4.3. Поляризация.........................................646
4.4. Активация воды......................................649
4.5. Стереохимические модели ..... 651
Дополнения, внесенные в корректуру..........................656
Список литературы....................................................657
Глава 18. Киназы. У. Дж. О’Сулливан (перевод А. А. Байкова) 661
1. Введение.................................................661
2. Общие свойства киназ........................ 663
2.1. Природа киназных реакций....................... . 663
2.2. Равновесие металл — субстрат ..... 663
2.2.1. Скорости образования МАДФ- и МАТФ2~ . 665
2.3. Структура комплексов металл — нуклеотид . 665
2.3.1. В растворе.................................... 665
2.3.2. На ферменте................................... 665
2.4. Кинетические исследования киназ.....................666
Содержание
711
2.5. Методы исследования фермент-субстратных комплексов в
равновесии...............................................669
2.6. Применение методов магнитного резонанса .... 669
2.6.1. Электронный парамагнитный резонанс . . 670
2.6.2. Ядерный магнитный резонанс....................671
3. Обсуждение отдельных ферментов 672
3.1. Креатинкиназа......................................672
3.2. Аргпнпнкпназа ... 676
3.3. Пируваткиназа..................................... 677
3.4. Фосфоенолпируваткарбоксикиназа (фосфопируваткарбок-
силаза)..................................................679
3.5. Аденилаткиназа.....................................679
3.6. Гексокиназа........................................681
3.7. З-Фосфоглицераткиназа..............................682
3.8. Фосфофруктокиназа..................................683
3.9. Нуклеозиддифосфаткиназа............................683
4. Заключение.............................................683
Список литературы...................................................684
Предметный указатель ...............................................688
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании книги, ее оформлении,
качестве перевода и другие просим присылать по адре-
су: 129820, Москва, И-110, ГСП, 1-й Рижский пер. д. 2.
ИВ № 1117
неорганическая
БИОХИМИЯ
том 1
Редактор Г. Б. Шкляева
Художник Н. Г. Блинов
Технический редактор Е. С. Потапенкова
Корректор К. Л. Водяницкая
Сдано в набор 05.01.78. Подписано к печати 04.05.78. Формат
60X90'/ib. Бумага типографская Кг 1. Гарнитура латинская.
Печать высокая. Объем 22,25 бум. л., 44,50 печ. л.,
уч.-изд. л. 48,56. Тираж 6.000 экз. Зак. 2451. Цеиа 4 р. 60 к.
ИЗДАТЕЛЬСТВО «МИР»
Москва, 1-й Рижский пер., 2
Московская типография № 11 Союзполиграфпрома при Госу-
дарственном комитете Совета Министров СССР по делам^ изда-
тельств, полиграфии н книжной торговли. Москва, 113105, На-
гатинская ул., д. 1.