/

























































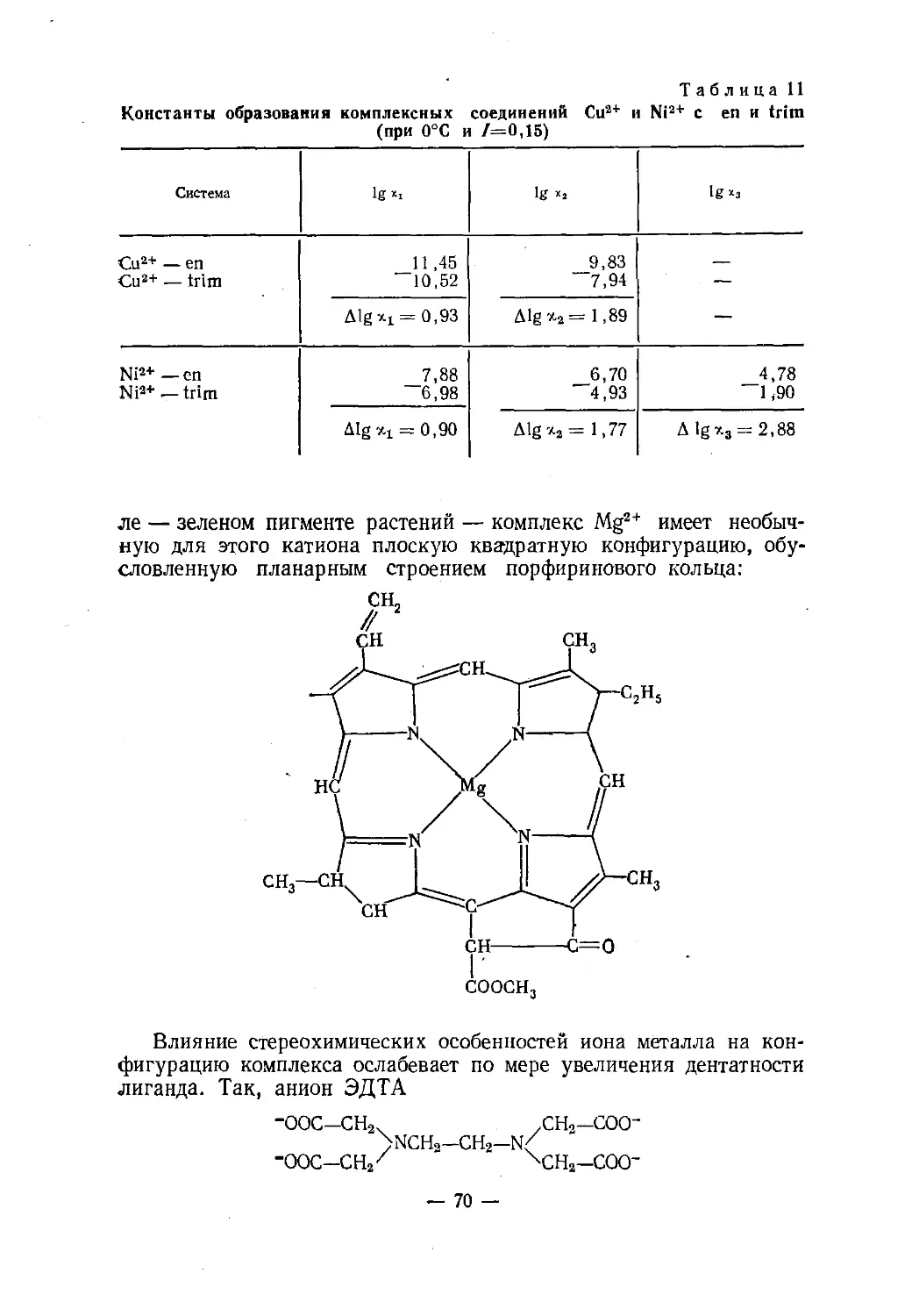

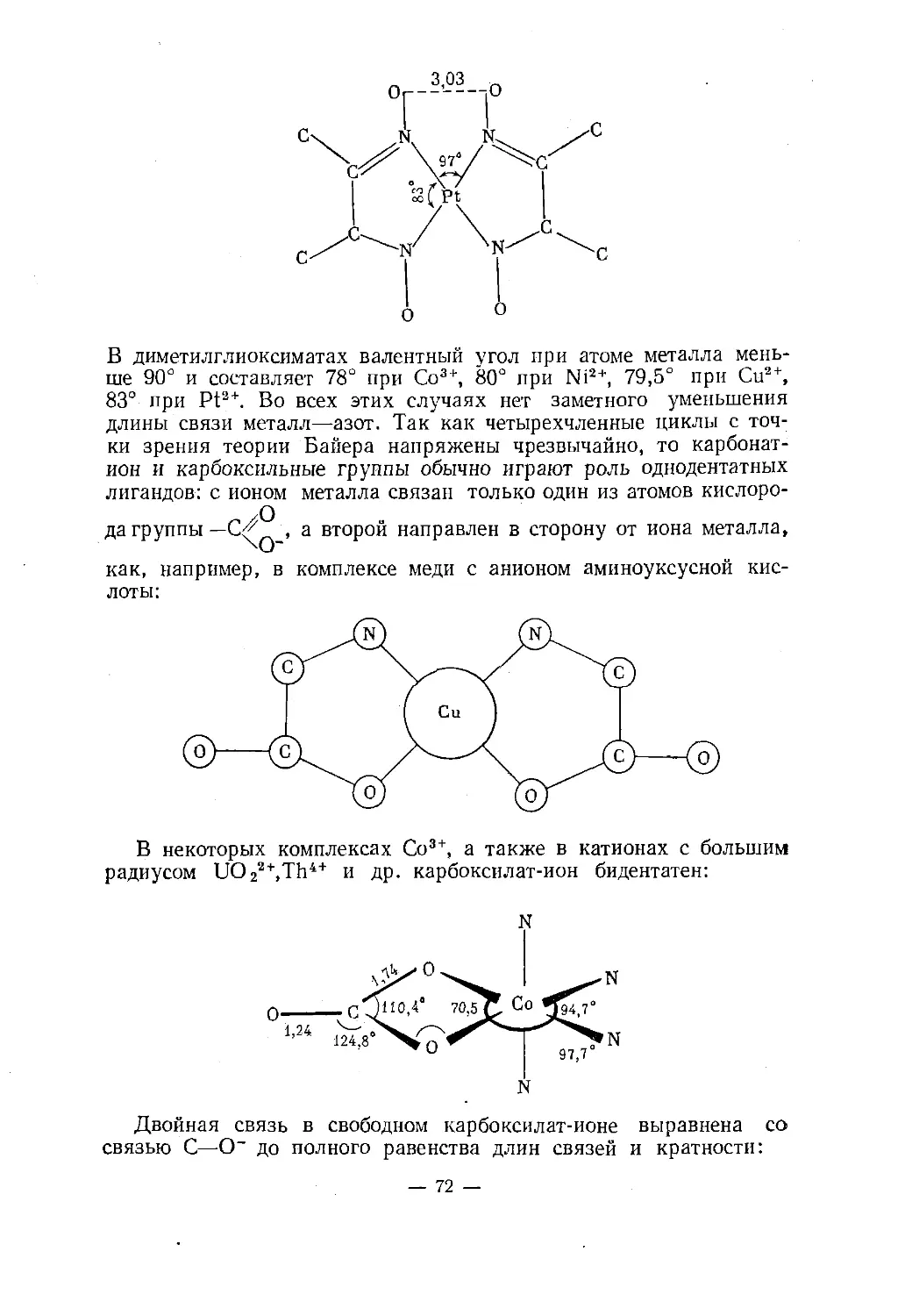


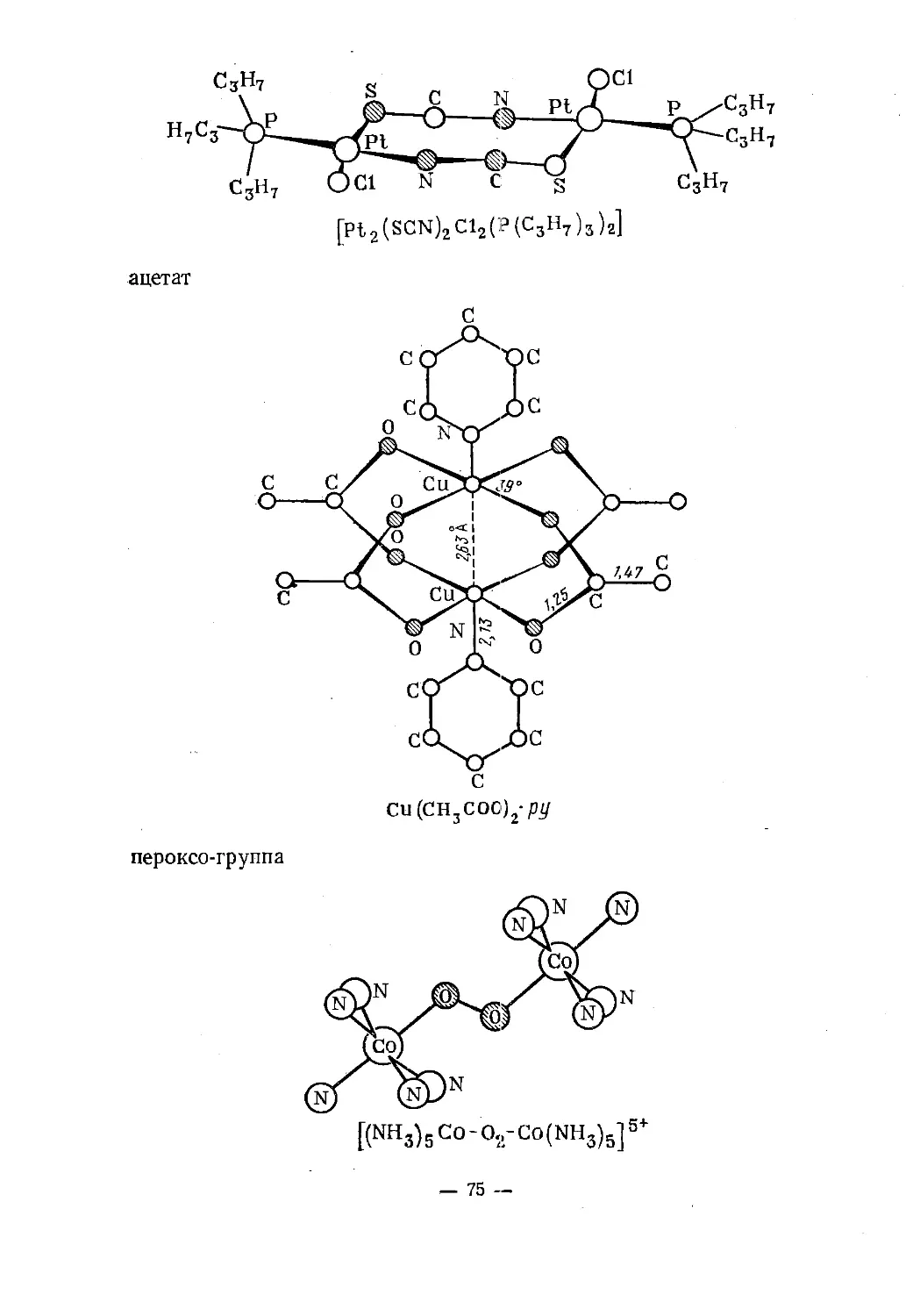
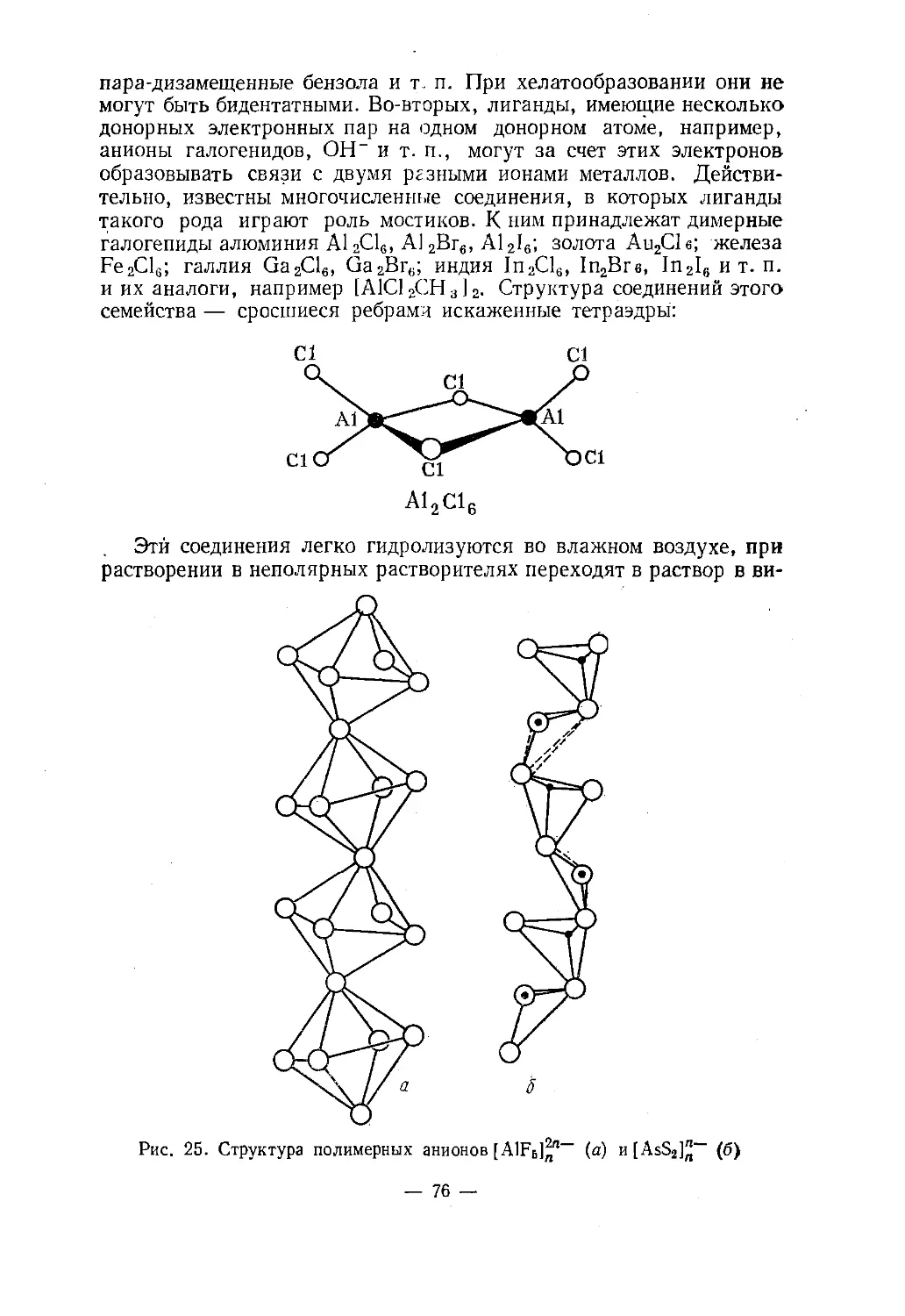

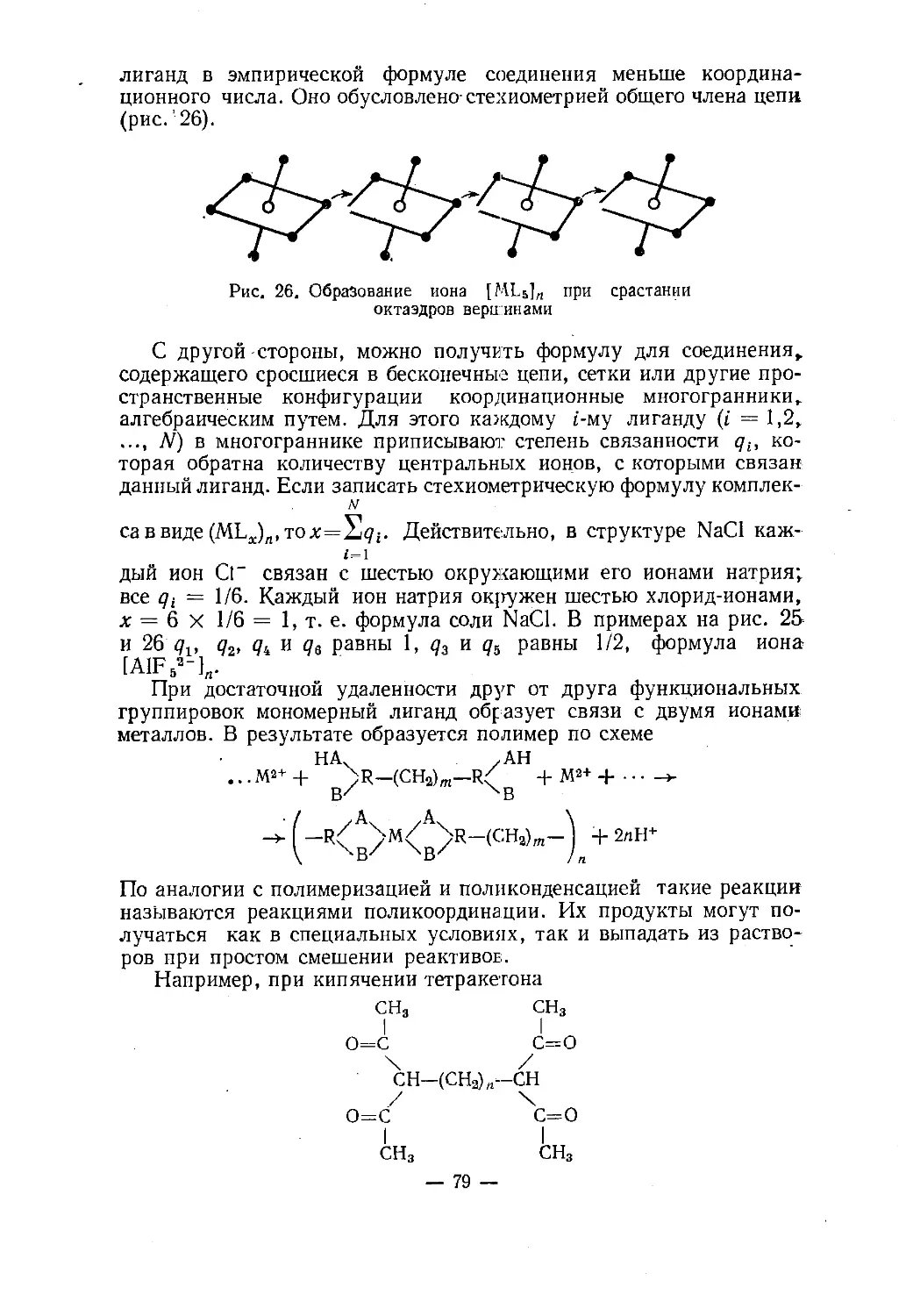
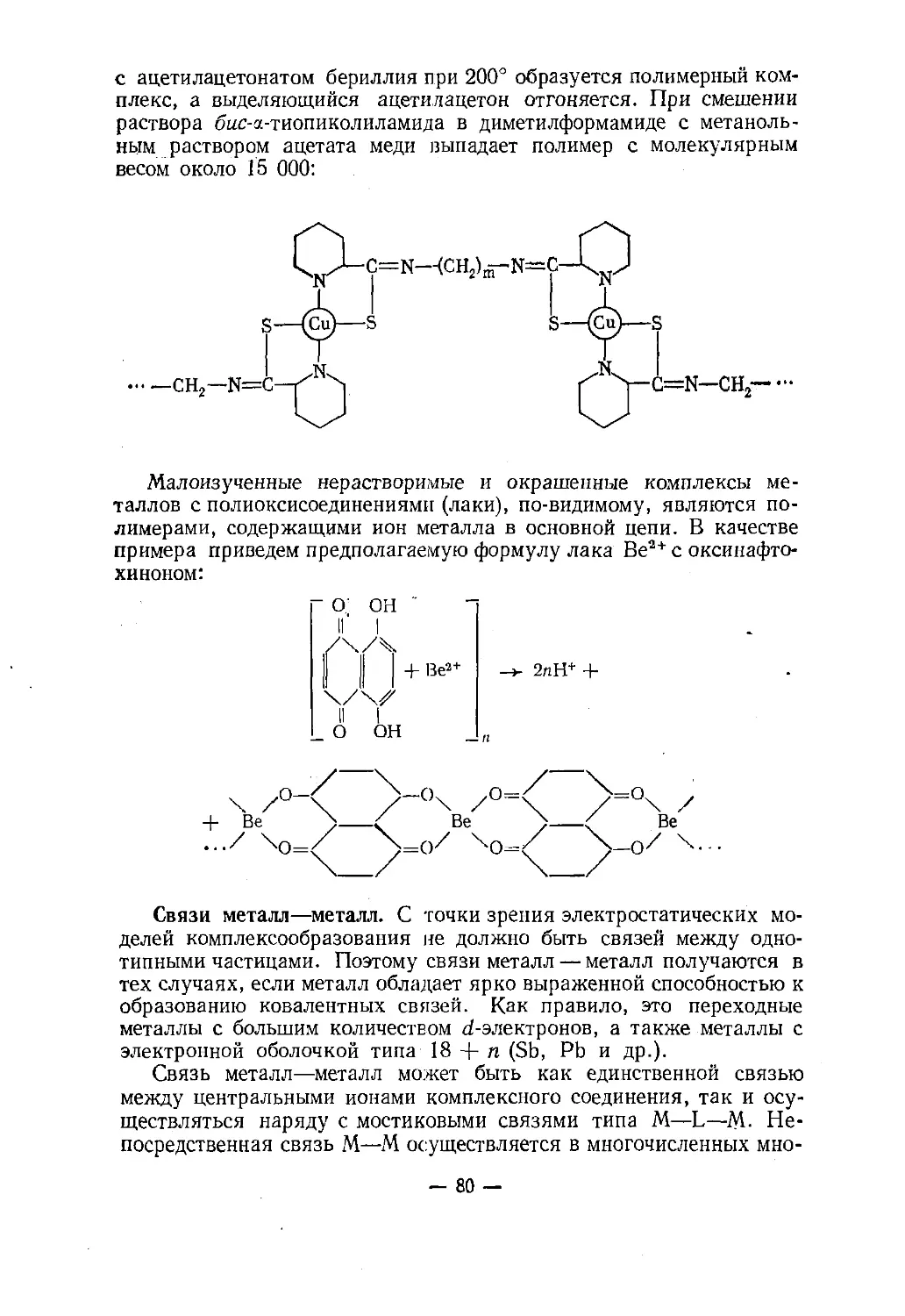
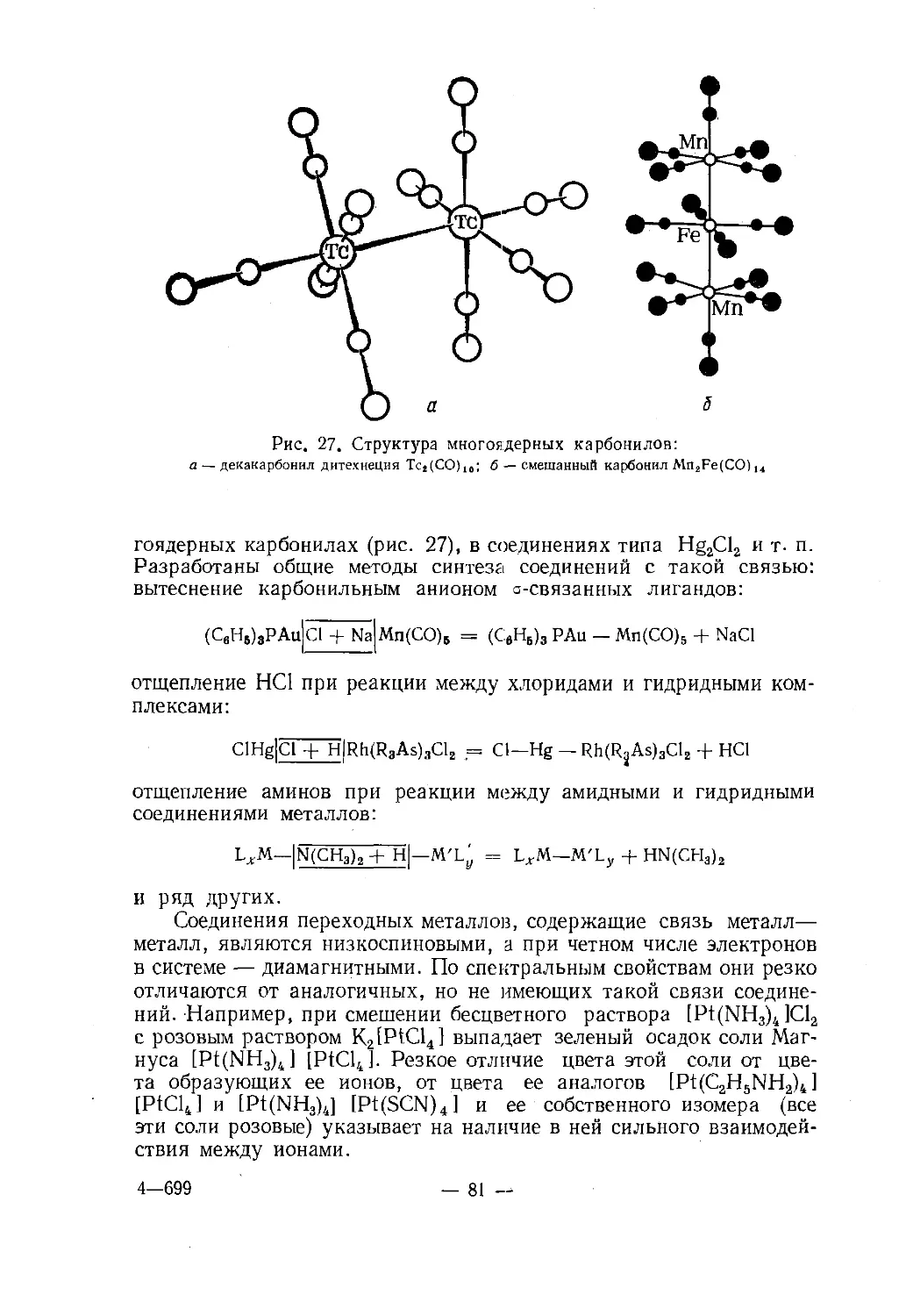


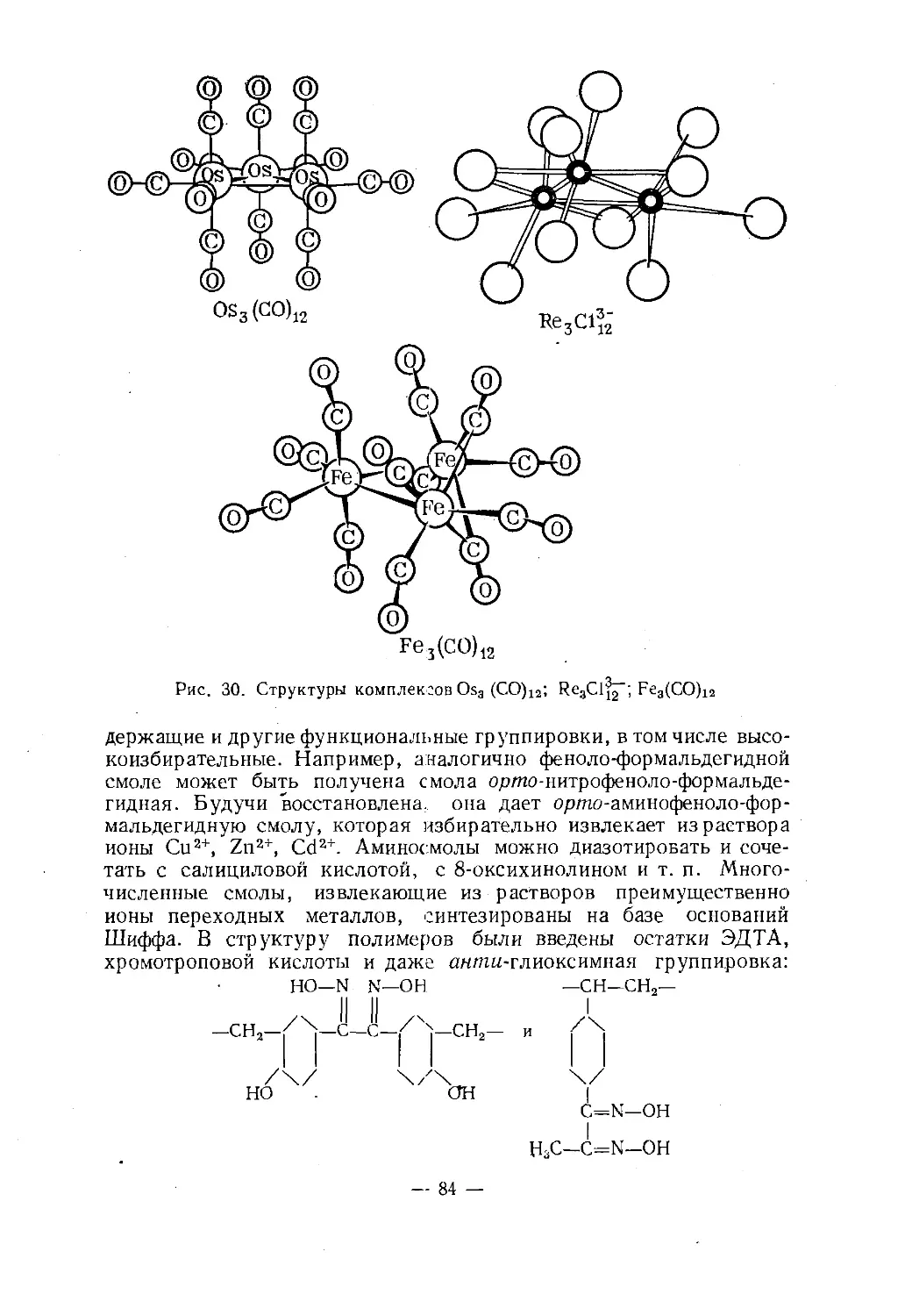
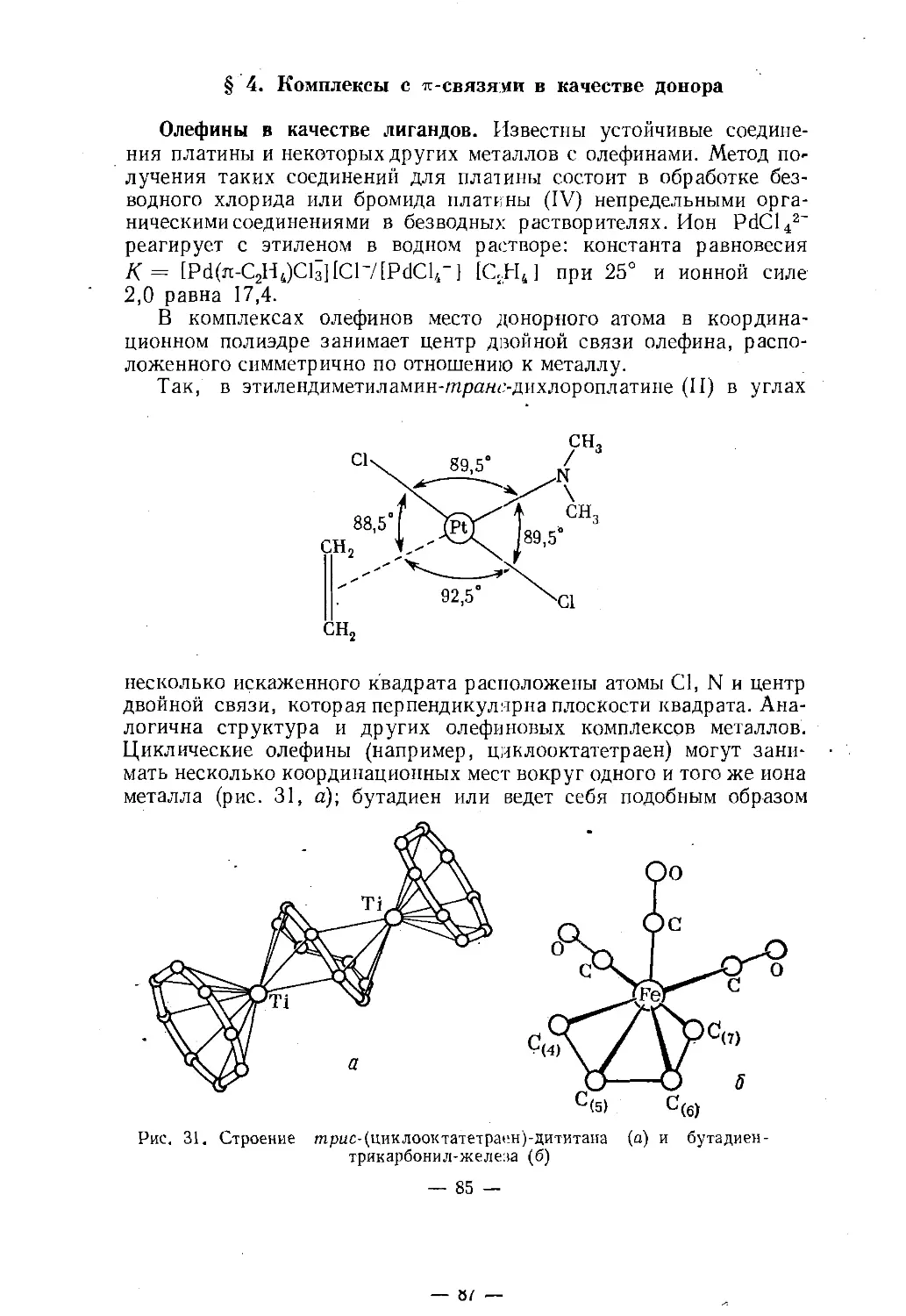




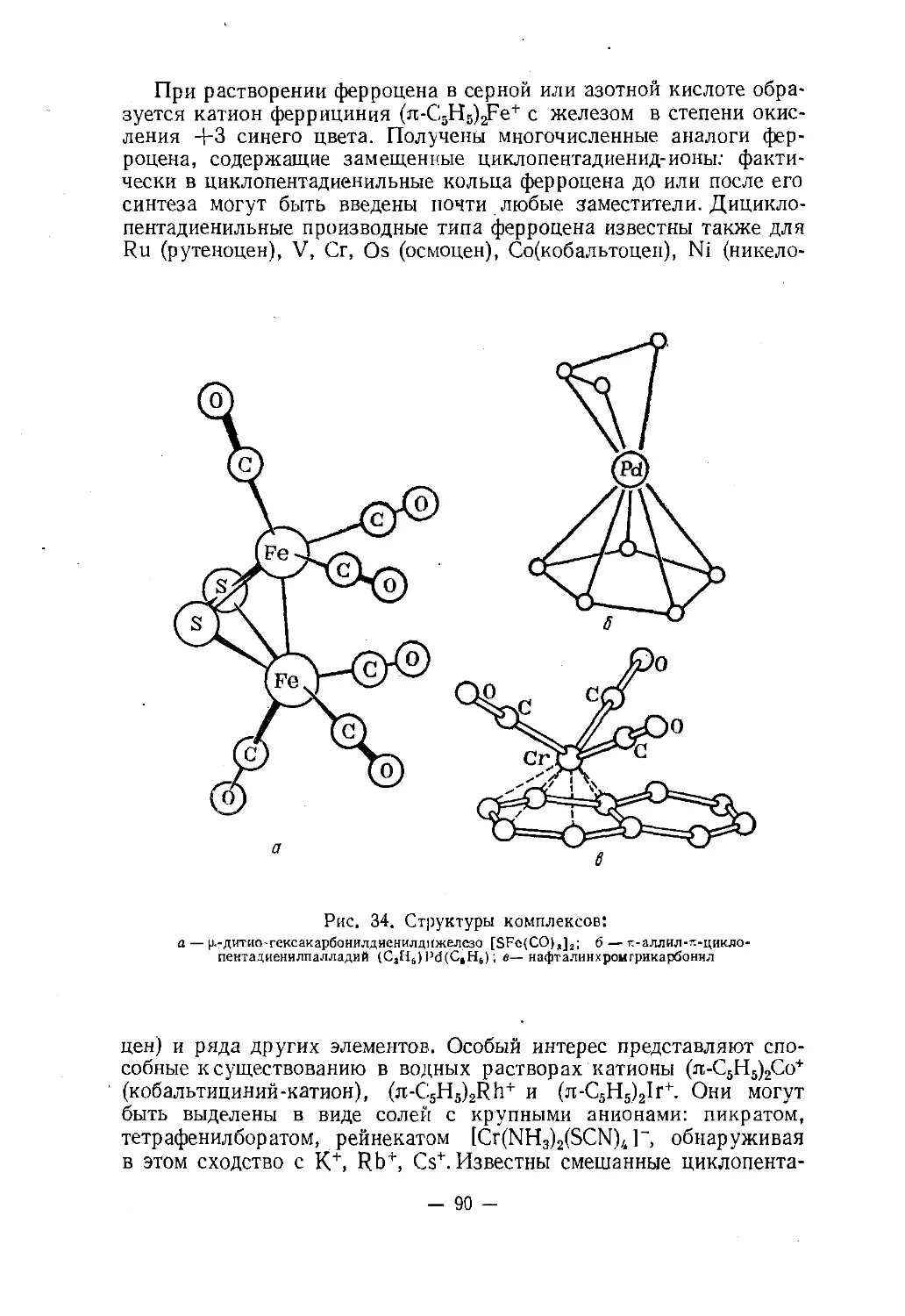



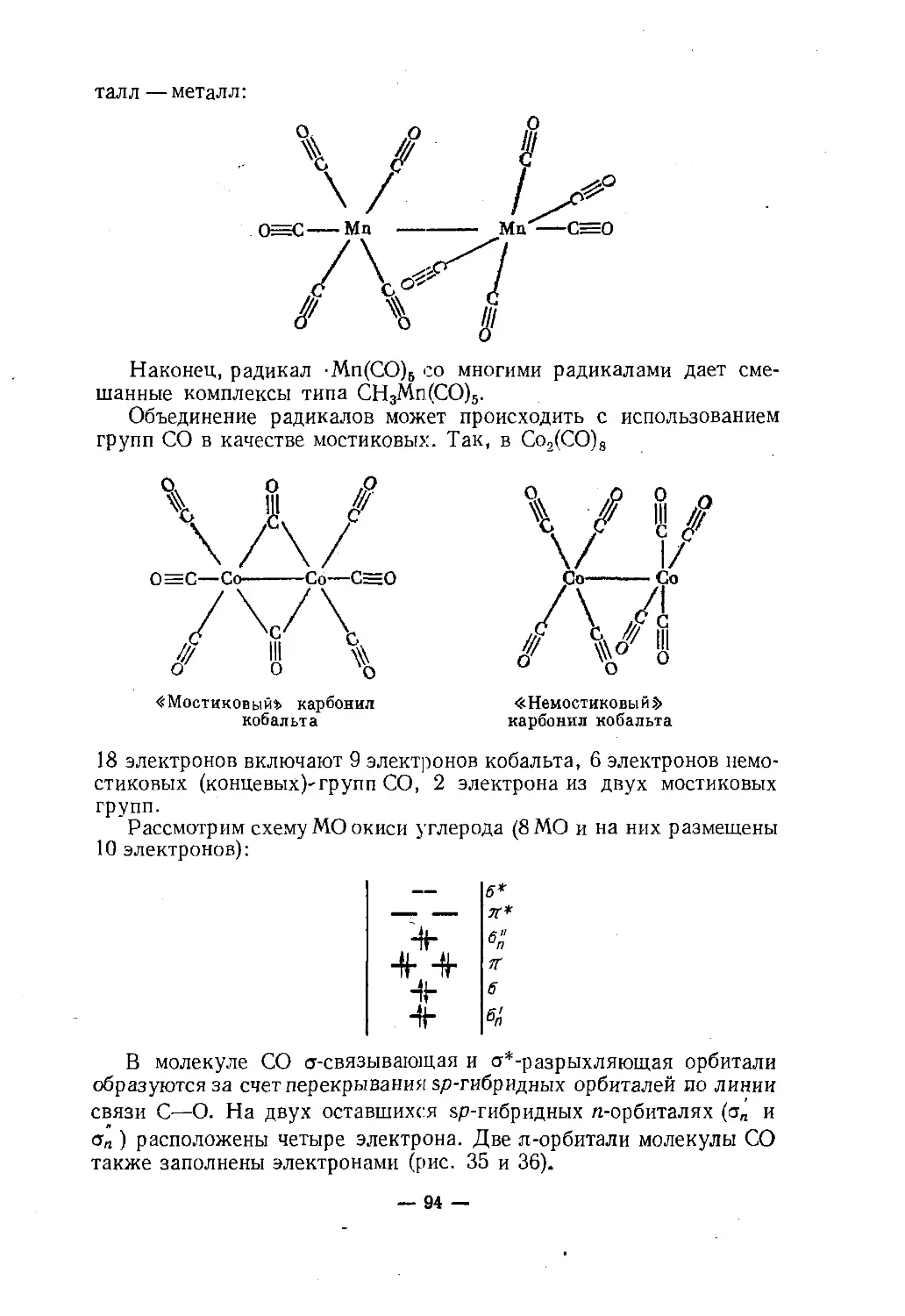
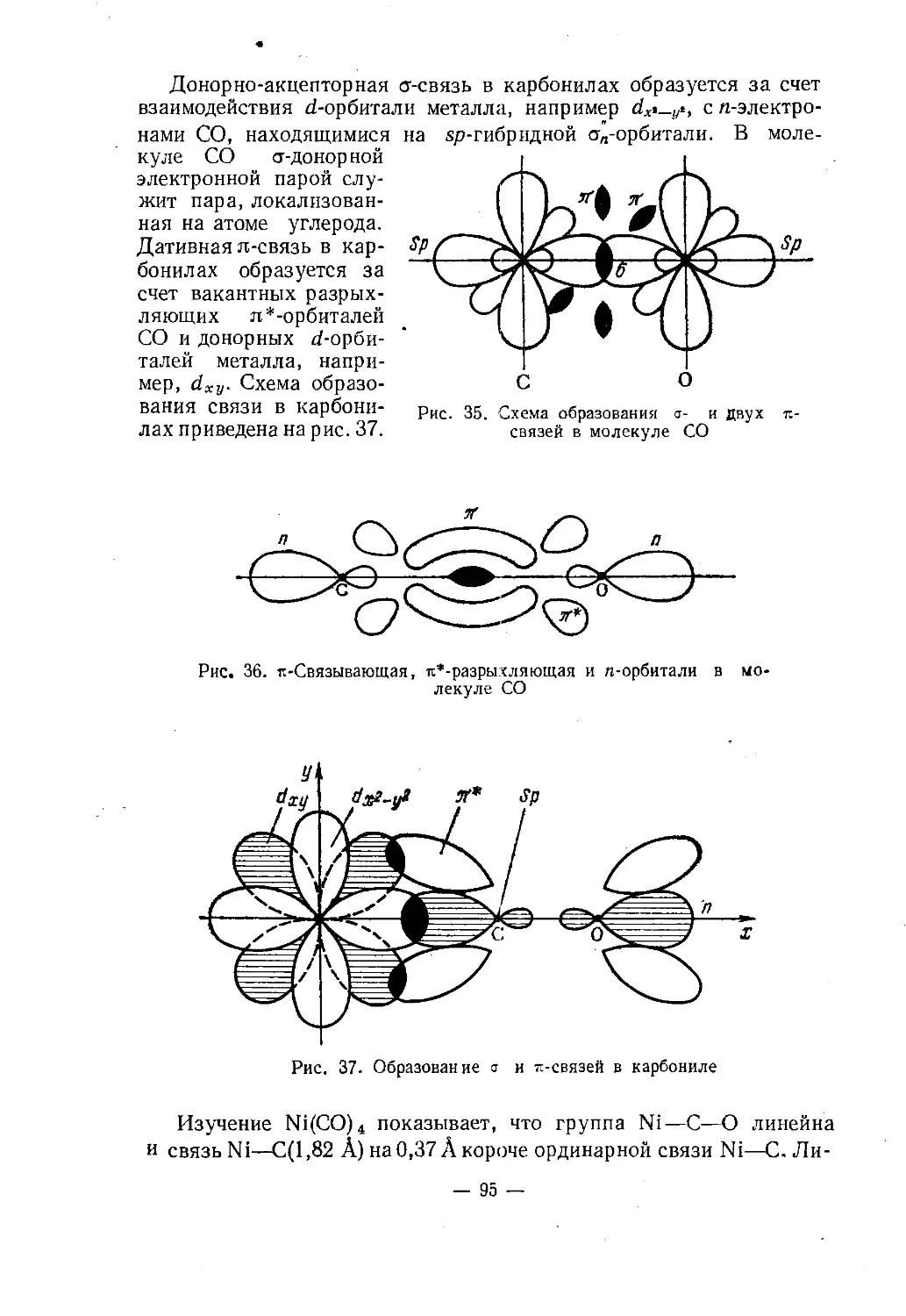








































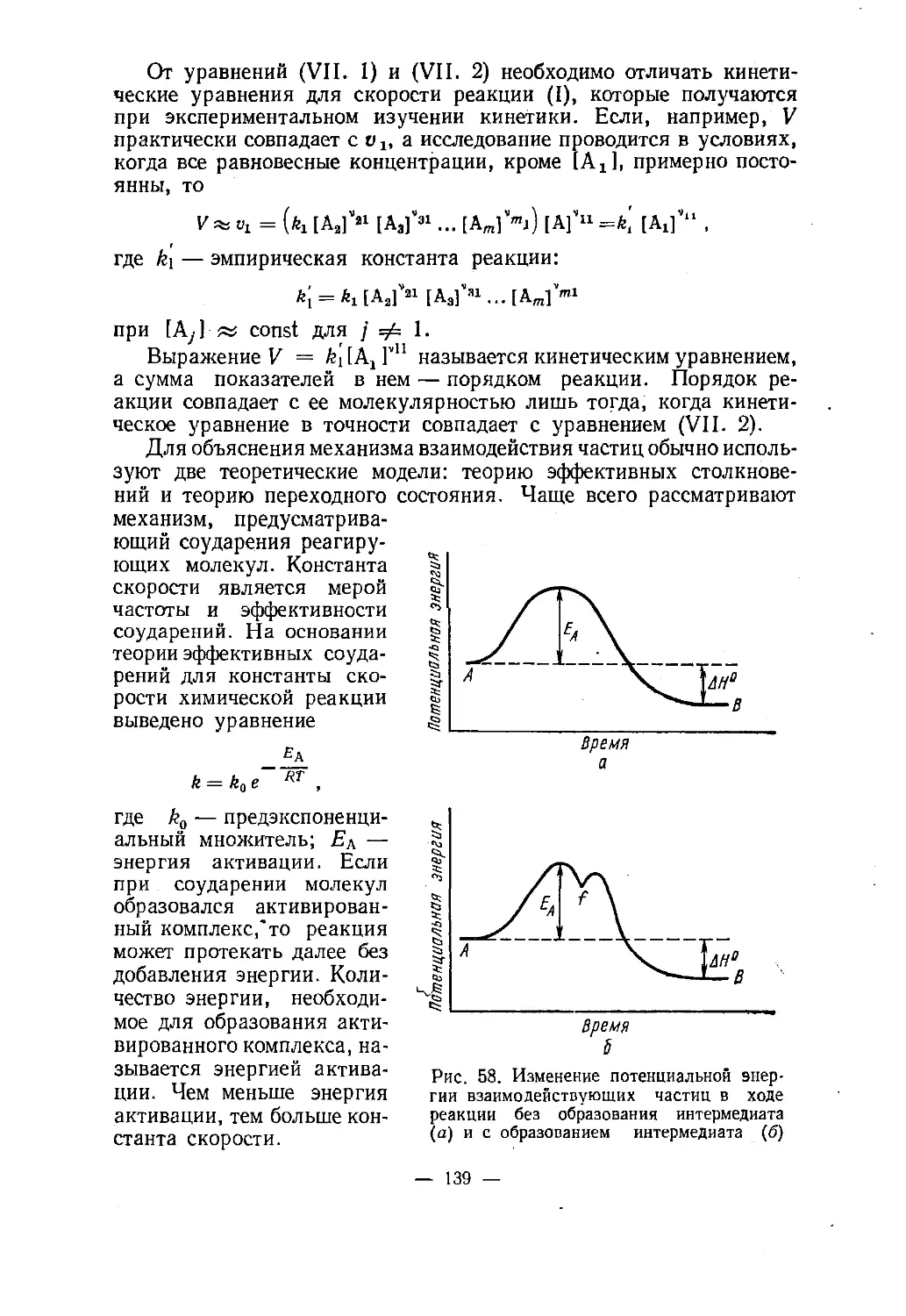




























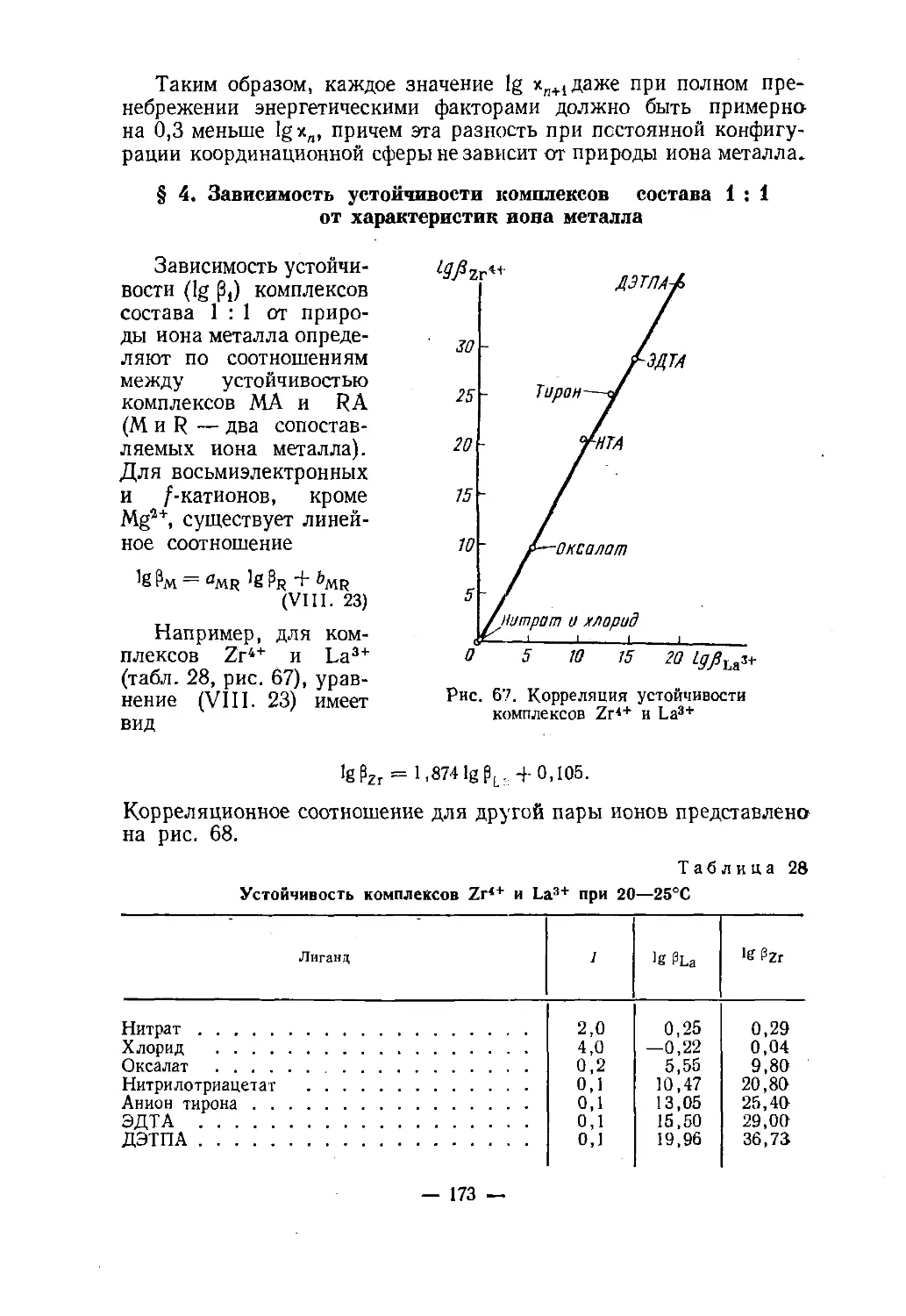





























Text
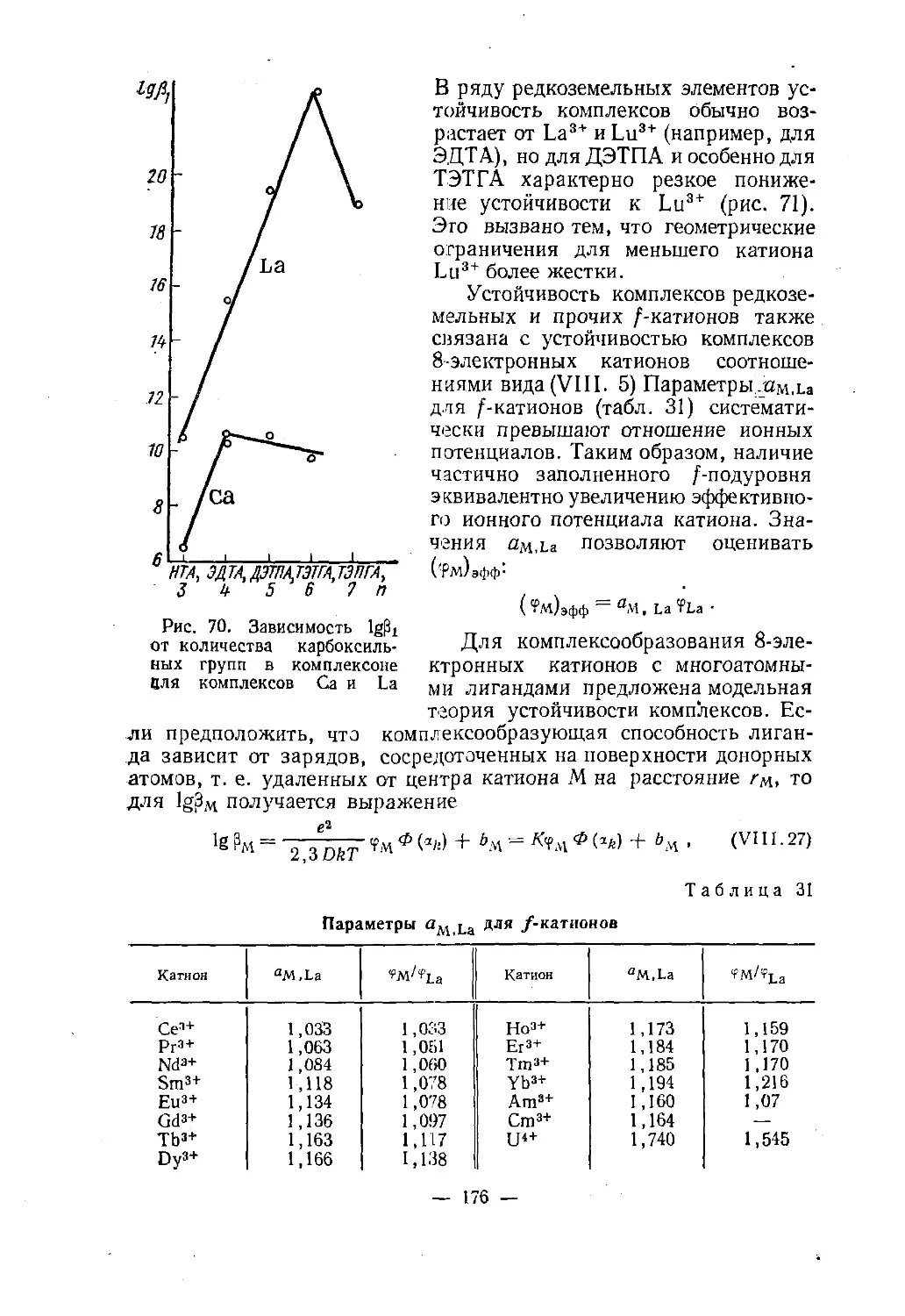
Н. А. СКОРИК, в. н. кумокхимияКООРДИНАЦИОННЫХ
СОЕДИНЕНИЙДопущено
Министерством высшего и среднего
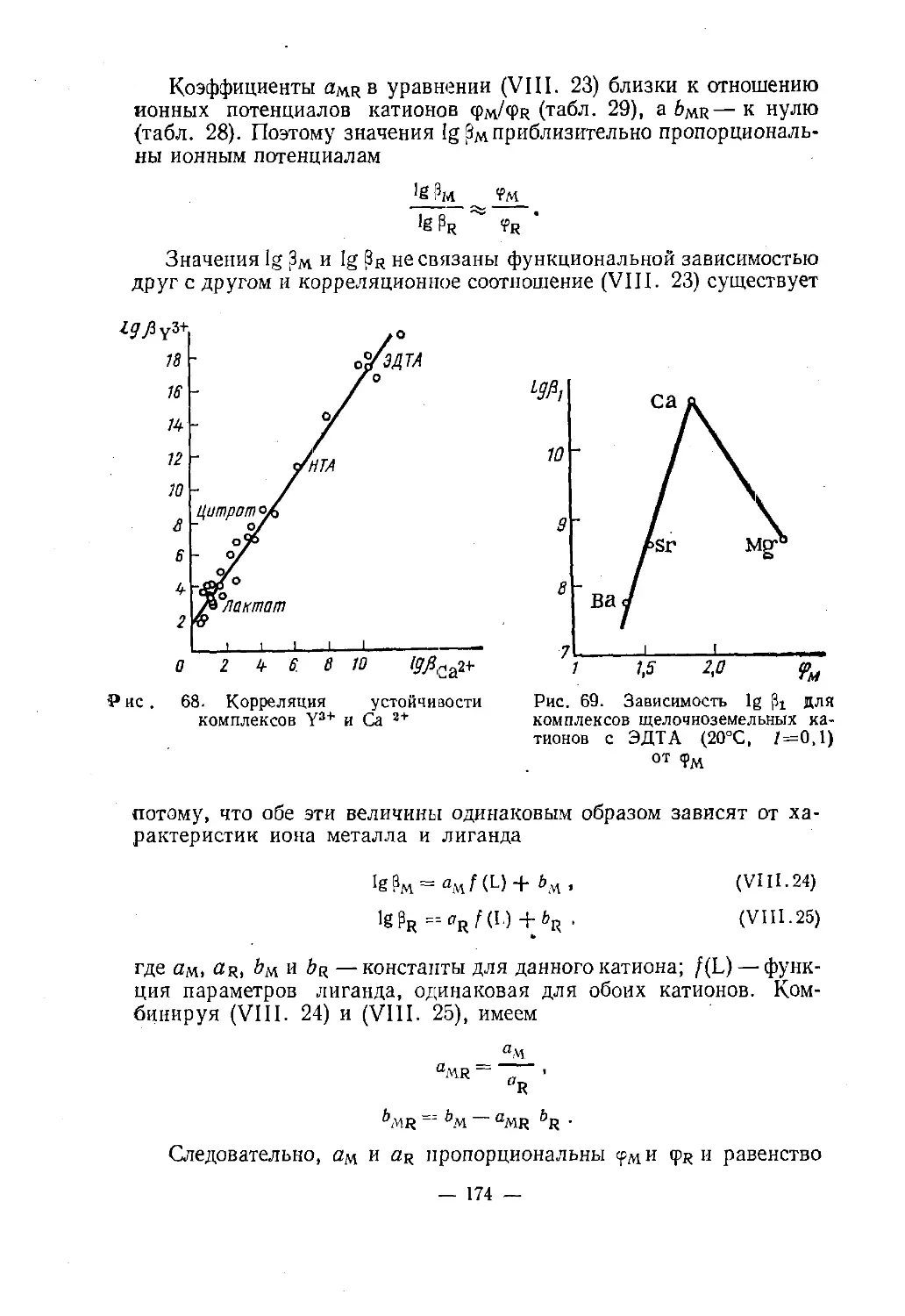
специального образования СССР
в качестве учебного пособия
для студентов химических специальностей
университетовМОСКВА «ВЫСШАЯ ШКОЛА» —1975
541С44УДК 541-49 (075>Рецензенты: доц. Э. Ю. Янсон (Латвийский ун-т) и кафедра
неорганической химии ЛТИ им. Ленсовета (зав. ка¬
федрой проф. Ю. И. Кукушкин)Скорик Н. А., Кумок В. Н.С 44 Химия координационных соединений. Учеб. пособие
для вузов. М., «Высш. школа», 1975.208 с. с ил.В книге даны основные понятия химии координационных соединений, рассмат¬
риваются номенклатура комплексных соединений, реакции, в которых участвуют
инертные н лабильные комплексные ионы. Приведен обзор отдельных классов коор¬
динационных соединений; классификация дана по структурному-принципу, отдель¬
но рассматривается класс комплексов с я-связью в качестве донора.Описаны основные принципы синтеза инертных и лабильных комплексов, ус¬
тойчивость комплексов в растворах, кинетика и механизм реакций замещения ли¬
гандов, виды изомерии комплексных ионов.Большое внимание уделено химии лабильных комплексных частиц, факторам,
влияющим на устойчивость этих частиц, основным способам определения констант
устойчивости комплексных соединений в растворе.20502—205 , 541С 109—75001(01)—75© Издательство «Высшая школа», 1975 г.
ОТ АВТОРОВОсновы химии координационных соединений, которая стала за
последнее время одним из ведущих направлений неорганической
химии, были заложены А. Вернером. Большой вклад в развитие
этой отрасли науки внесли советские научные школы И. И. Черня¬
ева и А. А. Гринберга. Советскими учеными создан и целый ряд
учебных пособий по химии координационных соединений.Предлагаемое пособие, в котором большее, чем обычно, внимание
уделено комплексам непереходных металлов и другим лабильным
комплексам, написано на основе лекций, прочитанных для студен¬
тов IV курса химического факультета Томского университета, спе¬
циализирующихся по неорганической химии. Содержание построе¬
но с учетом того, что дополнительные сведения по изучаемому-
предмету сообщаются в курсах «Строение вещества и спектры» и «Ме-
.тоды исследования неорганических соединений».Авторы приносят глубокую благодарность за ценные замеча¬
ния рецензентам пособия Ю. Н. Кукушкину, А. И. Стеценко и
Э. Ю. Янсону, а также В. В. Серебренникову, Л. А. Алексеенко,Н. Д. Стрельниковой, Г. А. Катаеву, А. А. Бугаевскому,
М. С. Новаковскому и Л. Н. Ушеренко за тщательный просмотр
рукописи, за помощь и поддержку в создании настоящего пособия.
1. ВВЕДЕНИЕ§ 1. Основные понятия химии координационных соединенийХимия координационных соединений изучает ионы и молекулы,
состоящие из центральной частицы и координированных вокруг нее
лигандов (аддендов). Центральную частицу называют также центром
или ядром координации. Многоядерные комплексные ионы содер¬
жат несколько центральных частиц*. Понятие «комплексные соеди¬
нения» более широко, чем понятие «координационные соединения».
Оно включает в себя также молекулярные комплексы, в которых
невозможно указать центр координации, а также соединения вклю¬
чения.Химия координационных соединений является частью неоргани¬
ческой химии, охватывающей как чисто неорганические соединения,
так и соединения, содержащие лиганды органической природы. Ли¬
ганды, как правило, не связаны друг с другом и между ними действу¬
ют силы отталкивания. Между лигандами могут возникать силы
межмолекулярного притяжения типа водородной связи. С централь¬
ным атомом лиганды могут быть связаны дву центровыми а-, тс- и 8-свя¬
зями и многоцентровыми связями. При двуцентровых связях ядро —
лиганд можно указать атомы лиганда, через которые связь осу¬
ществляется. Обычно эти атомы называют донорными.Внутренней координационной сферой называют совокупность
непосредственно связанных с ядром лигандов, а координационным
числом (к. ч.) — число атомов или групп их, координируемых цент¬
ральной частицей в данном соединении. Если связи ядро — лиганд
двуцентровые, то координационное число равно числу сг-связей, об¬
разуемых центральной частицей, т. е. числу непосредственно сосед¬
ствующих с ней донорных атомов. Дентатность (координационная
емкость) лиганда — это число атомов лиганда, образующих в дан¬
ном соединении координационные связи, т. е. число мест, которые
занимает лиганд в координационной сфере.Если связи ядро — лиганд многоцентровые, как, например, в
дибензолхроме Сг(С6Н6)2, то указать в лиганде донорные атомы,
определить дентатность лиганда и к. ч. ядра в комплексе становит¬* В данной книге в основном рассматриваются центральные частицы,
представляющие собой ионы металлов.
ся невозможным. В некоторых случаях, например для олефиновых
комплексов, можно рассматривать кратную связь в лиганде как экви¬
валент донорного атома, чтобы при помощи этого искусственного
приема сохранить понятия дентатности и координационного числа.Монодентатные лиганды используют в качестве донорного толь¬
ко один атом и занимают одно координационное место (F-, С1~, Вг, Г,
SCN", CN-, NO2, NO3, СО, Н20, NH3 и др.). Примерами полиден-п/0~татных лигандов могут служить оксалат-ион — ^0 ’этилендиамин Н 2N — СН 2 — СН 2 — NH 2, анион этилендиамин-
тетрауксусной кислоты-оос—сн2. ,сн2—соо->N—СН2—СН2—N<-ООС—СН/ хсн2—соо-Термин «валентность» применяется, с одной стороны, для указа¬
ния числа ковалентных связей между атомами, а с другой стороны,
как синоним степени окисления. Из-за многозначности этого тер¬
мина он, как правило, не употребляется в химии координационных
соединений. Вместо него используют более точные понятия: степень
окисления, ковалентность и координационное число.Степень окисления (электровалентность) центрального иона— это разность заряда комплексной частицы и суммы формальных
зарядов лигандов. При расчете степени окисления принимают, что
при координации лиганды не изменяют своей «нормальной» заряднос-
ти. Например, ион [Co(NH3)e]3+ относят к производным Со3+, при¬
писывая NH3 нулевой заряд как нейтральной молекуле, а ион
[Co(SCN)4]2_— к производным Со2+, приписывая группе SCN- за¬
ряд — 1, как в роданидах щелочных металлов.Понятие о степени окисления очень удобно при классификации
соединений, но условно, так как оно не указывает ни числа атомов,
с которыми связан ион металла, ни числа связей,, образуемых им.
Например, в соединении Ni(CO)4 атом никеля имеет нулевую элект¬
ровалентность, хотя и связан с четырьмя молекулами СО и образует
минимум четыре связи.Понятие о степени окисления нужно проанализировать с трех
точек зрения: во-первых, насколько однозначно оно определяется;
во-вторых, соответствует ли оно реальному распределению зарядов
в комплексной частице и, в-третьих, насколько данные по распреде¬
лению зарядов характеризуют валентное состояние центрального
иона.Расчет формальной электровалентности затруднен, если свобод¬
ный лиганд может существовать в нескольких формах, различаю¬
щихся зарядом. Например, поскольку существуют ионы 1~и 13 и моле¬
кулы 12, то невозможно по формуле определить, какова электрова¬
лентность таллия в Т113: или Т1+ соединен с I3 (I- + 12), или Т13+
соединен с 1~.
В действительности существуют оба неразличимые по стехиометрии
соединения. Большие трудности возникают, когда лигандом явля¬
ется водород или углеводородный остаток: действительно, формаль¬
но гидриды можно считать производными Н+, Н° и Н~, а метал-
лорганические соединения — производными радикалов или ани¬
онов.В органической химии отказались от понятия электровалент¬
ности, причем одной из причин отказа были затруднения, возника¬
ющие при расчете формальной электровалентности атомов углеро¬
да, участвующих в связях —С—С—С—. Наличие связей металл—
металл и металл—углерод в комплексных соединениях также силь¬
но затрудняет расчет электровалентности иона металла. Однако в
большинстве координационных соединений электровалентность рас¬
считывается надежно.Идея электровалентности опирается на ионные представления
Берцелиуса. Представим себе, что образование комплексного иона,
например [СоС14 ]2~ из свободных ионов Со2+ и С1“, происходит в две
стадии: пусть первоначально образуется гипотетический ион [СоС14]2-
с чисто ионным характером связи без поляризации. Распределение
зарядов в таком ионе (2 + на Со и 1— на С1) соответствует фор¬
мальной электровалентности частиц. При переходе к реальному
распределению зарядов произойдет частичная передача донорных
электронов от лигандов к иону металла, что сопровождается умень¬
шением эффективного положительного заряда центрального иона,
эффективных зарядов лигандов и полярности связей. Этот процесс
иногда трактуют как внутримолекулярную реакцию окисления —
восстановления. Итак, эффективные заряды Со2+иС1~в [СоС14]2'
по модулю меньше формальных (двух и единицы соответственно).
Таким образом, электровалентность не отражает истинной карти¬
ны распределения зарядов в соединениях.Л. Полинг выдвинул «принцип электронейтральности», соглас¬
но которому эффективные заряды на атомах уменьшаются в такой
степени, что по модулю не превосходят единицы. Согласно этому
принципу такие ионы, как Fe3+, La3+, Th4+, должны нести эффектив¬
ные заряды существенно меньше формальных. Однако в последнее
время накапливаются данные о том, что в целом ряде соединений эф¬
фективные заряды катиона близки к формальным. Так, рентгено¬
спектральным методом в акво-ионе Zn2+ (водный раствор) был оп¬
ределен заряд на Zn около +2, в кристаллах ацетилацетоната цин¬
ка заряд на Zn + 1,8, в кристаллах CrS04-7H20 заряд на Сг +1,9,
в циклопентадиениде марганца Мп(С5Н5)2 заряд на Мп +1,5.
При квантовомеханическом расчете иона NiF|| Методом Рутана был
получен заряд на атоме Ni, равный +1,82. В то же время в согла¬
сии с принципом электронейтраЛьности заряд на Сг в КгСг04, оп¬
ределенный тем же рентгеноспектральным методом, равен +0,2
при формальном заряде +6.В комплексных ионах с формально отрицательным ионом метал¬
ла, например в Мп(СО)5, Со(СО)4 и др., отрицательный заряд рас-— 6 —
пределяется между лигандами. При этом эффективный заряд иона
металла, как правило, положителен.Характеристика распределения зарядов в комплексной частице
не исчерпывает вопроса о валентном состоянии центрального иона.
В принципе после образования соединения нужно рассматривать
не валентные орбитали атома, а возникшие молекулярные орбитали.
Однако ряд допустимых приближений иногда позволяет считать,
что атомные орбитали центрального иона металла сохраняются при
вхождении его в комплексную частицу, но испытывают возмуще¬
ние под действием лигандов, в связи с чем может измениться поря¬
док их заполнения электронами. В соответствии с этим ион Fe3+(3d5)
в комплексной частице может иметь все электроны неспареикыми(^ПЕБЖП 4pj. 1 1 I ) и общий спин 2,5 или принуди¬тельно спаренные электроны (^ШШЕГИ 4р\ ill)и общий спин 0,5. Первый вариант осуществляется в ионе FeFis ‘
а второй — в ионе Fe(CN)6 . Магнитные, спектральные, кинетичес¬
кие свойства этих комплексов настолько различны, что высоко¬
спиновые и низкоспиновые комплексы Fe3+ обсуждают обычно от-»
дельно, хотя не исключается возможность того, что в обоих случа¬
ях эффективный заряд на Fe одинаков.Величина спина определяет магнитные свойства комплекса, по¬
этому при сопоставлении комплексов наряду с электровалентностью
указывают также магнитное состояние иона металла.Координационное число надежно определяется методами рентгено¬
структурного анализа, устанавливающими структуру комплекса.
На основании только данных химического анализа и формулы сое¬
динений координационное число, как правило, определить невоз¬
можно. Надо учесть, что во многих кристаллосольватах, в том числе
в кристаллогидратах и аммиакатах, часть молекул растворителя
не связана с' центральным ионом металла*. Для многодентатных
лигандов зачастую неизвестно, какую именно дентатность они про¬
являют в данном соединении. В многоядерных комплексах некото¬
рые лиганды координируются нескэлькими ионами металлов од¬
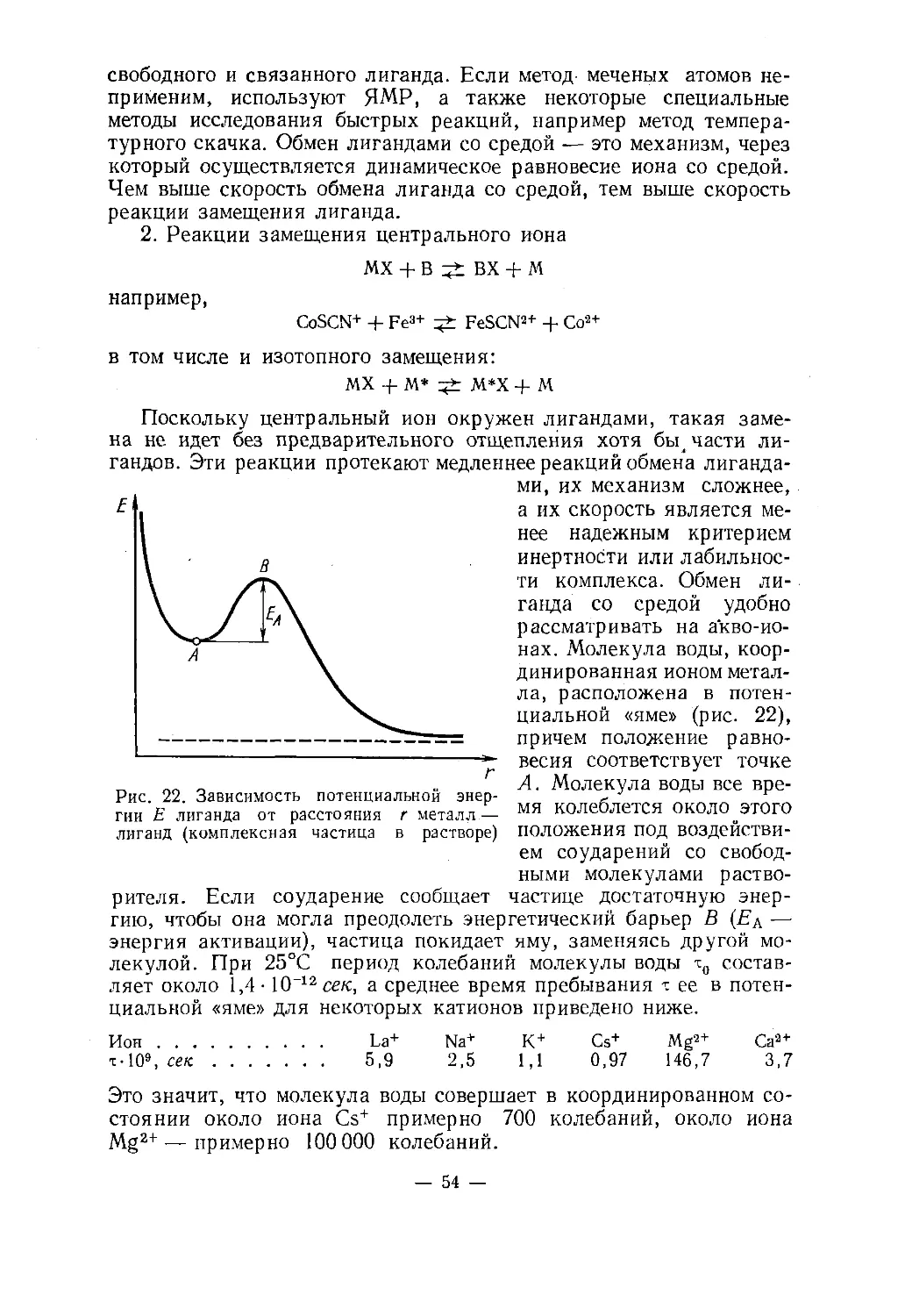
новременно.При исследовании структуры комплексов в растворах тоже иног¬
да удается применить рентгеноструктурный анализ, однако чаще* Так, в СиБСи-бНгО нон меди координирует четыре молекулы воды и
два иона сульфата, каждый из которых связан еще с одним ионом Си2+. Пятая
молекула воды расположена во внешней сфере и связана водородными связя¬
ми с внутрисферными частицами._ 7
используют методы спектроскопии: изучение инфракрасных (колеба¬
тельных) и электронных спектров соединения. При этом координа¬
ционное число определяется или при помощи теоретической трак¬
товки спектров, или экстраполяцией полученного для сходных
соединений соответствия между к. ч. и спектрами.Определение координационных чисел для комплексов в растворе
сильно осложняется. В частности, числа гидратации для некоторых
акво-ионов до сих пор не определены *.Ковалентностью центрального иона называют число, указываю¬
щее, сколько он образует неионных связей (связей с определенной
долей ковалентности). Минимальным значением ковалентности яв¬
ляется координационное число. В системах с многоцентровыми свя¬
зями применение понятия ковалентности затруднительно.Координационное число совпадает с числом образуемых атомом
а-связей. Ковалентность может быть больше к. ч. (больше числа
а-связей), если имеются еще я- или б-связи.Ковалентность практически всегда выше формальной электро¬
валентности. Так, Веа+всегда образует четыре о-связи с лигандами,
расположенными по тетраэдру, при этом обобществляется восемь
электронов. Ионы Се4+ и Th4+ имеют к. ч. 8, следовательно, мини¬
мальное значение ковалентности равно восьми.Ковалентность и координационное число — непостоянные ве¬
личины, зависящие от ряда факторов: типа связи, объемных со¬
отношений и т. п. Например, ион Со2+ в ярко-синем комплексе
[Co(SCN)4]2- имеет к. ч. 4, а в розовом акво-ионе [Со(Н20)в]2+ —
к. ч. 6. Карбонат-ион в комплексном ионе [Co(NH3)4C03]+ занимает
два места в координационной сфере, а в ионе [Co(NH 3) 5С0 3 ]+ одно;
ион Со3+ в обоих этих ионах имеет к. ч. 6. Вообще, к. ч. 6 харак¬
терно для Со3+и Сг3+; для иона Ag+ характерно к. ч. 2; для Hg2+ —
к. ч. 4. Один и тот же ион с одними и теми же лигандами может об¬
разовать ряд комплексных частиц, координационные числа в которых
различны: так, известны ионы [Си С14]2" в Cs2[CuCl4] и [СиС15]3_
в [Cr(NH3)в][CuCl5]. В соединениях Na3[TaF8], Кг[Тар7],
CstTaFe] содержатся комплексные ионы [TaF8]3~, [TaF7 ]2_, [TaFe]-.§ 2. Номенклатура координационных соединенийПри составлении названия комплексного иона пользуются сле¬
дующими правилами:1. К названиям лигандов-анионов добавляют суффикс «о» (суль¬
фате-, хлоро-, оксалато-); названия нейтральных лигандов, за исклю¬* Состав акво-ионов в водных растворах эффективно определяется ме¬
тодом ядерного магнитного резонанса (ЯМР). Так, при низких температурах
в растворах Al3+, Ga3+, In3+, Mg2+ и Ве2+ наблюдаются сигналы протонного
магнитного резонанса отдельно для свободной воды м отдельно для воды в
первой координационной сфере, что позволило установить для первых четы¬
рех катионов к. ч. 6, а для Ве2+ — к. ч. 4.
чением наименования координированной молекулы воды «акво-»,
не имеют суффикса (аммин-, пиридин-).2. Число координированных групп каждого рода указывают
греческими приставками моно-, ди-, три-, тетра-и т. д. Если лиганды
сложны, то применяют приставки бис-, трис-, тетракис-. Пристав¬
ку моно- часто опускают.3. Чтобы составить название комплексной частицы, первона¬
чально перечисляют лиганды-анионы, лиганды-молекулы, а затем
указывают центральный атом. Записывают формулу комплексного
иона в обратном порядке. Комплексную частицу принято выделять
квадратными скобками.Степень окисления центрального атома по системе ИЮПАК
(International Union of Pure and Applied Chemistry) обозначается
римской цифрой в скобках после названия частицы. Если комп¬
лексная частица является анионом, то ее название кончается суф¬
фиксом «ат». Таким образом, соединение [Co(NH 3) 5С1 ] С12 по системе
ИЮПАК называется хлоропентамминкобальт(Ш)хлорид. В рус¬
ском языке предпочитают ставить на первое место внешнесфер-
ный анион: хлорид хлоропентамминкобальта (III). Приставка мо¬
но- здесь опущена, буквы а в «пента» и в «аммин» срослись.Соединение NH4[Cr(NH3)2(SCN)4]— так называемая соль Рей-
неке — должно быть названо тетрароданодиамминхромат (III) аммо¬
ния. Комплексы аниона CNS- называют роданидными (тиоцианат-
ными), если координация идет через атомы серы (например, в
Hg(SCN)2_) или изороданидными (изотиоцианатными), если коорди¬
нация идет через атом азота (например, в FeNCS2+)*.Номенклатуру ИЮПАК можно сравнить с Женевской номенкла¬
турой в органической химии: кроме нее существуют многочислен¬
ные «практические» и «рациональные» названия комплексных сое¬
динений. Целая серия названий солей Со3+: лутео-соли, розео-соли,
празео-соли и т. д. — основана на их окраске. Немало солей наз¬
вано по имени химиков, их открывших (соль Рейнеке; от нее произош¬
ло название аниона [Cr(NH 3) 2(SCN) 4 ]- «рейнекат»).Система Вернера отличается от системы ИЮПАК отсутствием
цифры, указывающей на степень окисления. Эту роль выполняет
суффикс в наименовании металла:Валентность 1+ 2+ 3+ 4+ 5+ 6+ 7+ 84-Суффикс «а» «о» «и» «е» «ан» «он» «нн» «ен»Таким образом, ион Ag(NH3)2+ называют по этой системе диаммин-
аргента-ионом; CoSCN+ — монороданокобальто- ионом; FeSCN2+ —
монороданоферри-ионом, SbCl6 — гексахлоростибанат-ионом.Если в ионе содержатся моСтиковые группы, то они перечисля¬
ются после всех лигандов, перед ними ставится буква (j,; мостиковая
группа ОН- называется ол-группой.* В настоящем пособии используется название роданид, а формула иона
записывается в виде SCN- независимо от способа координации.
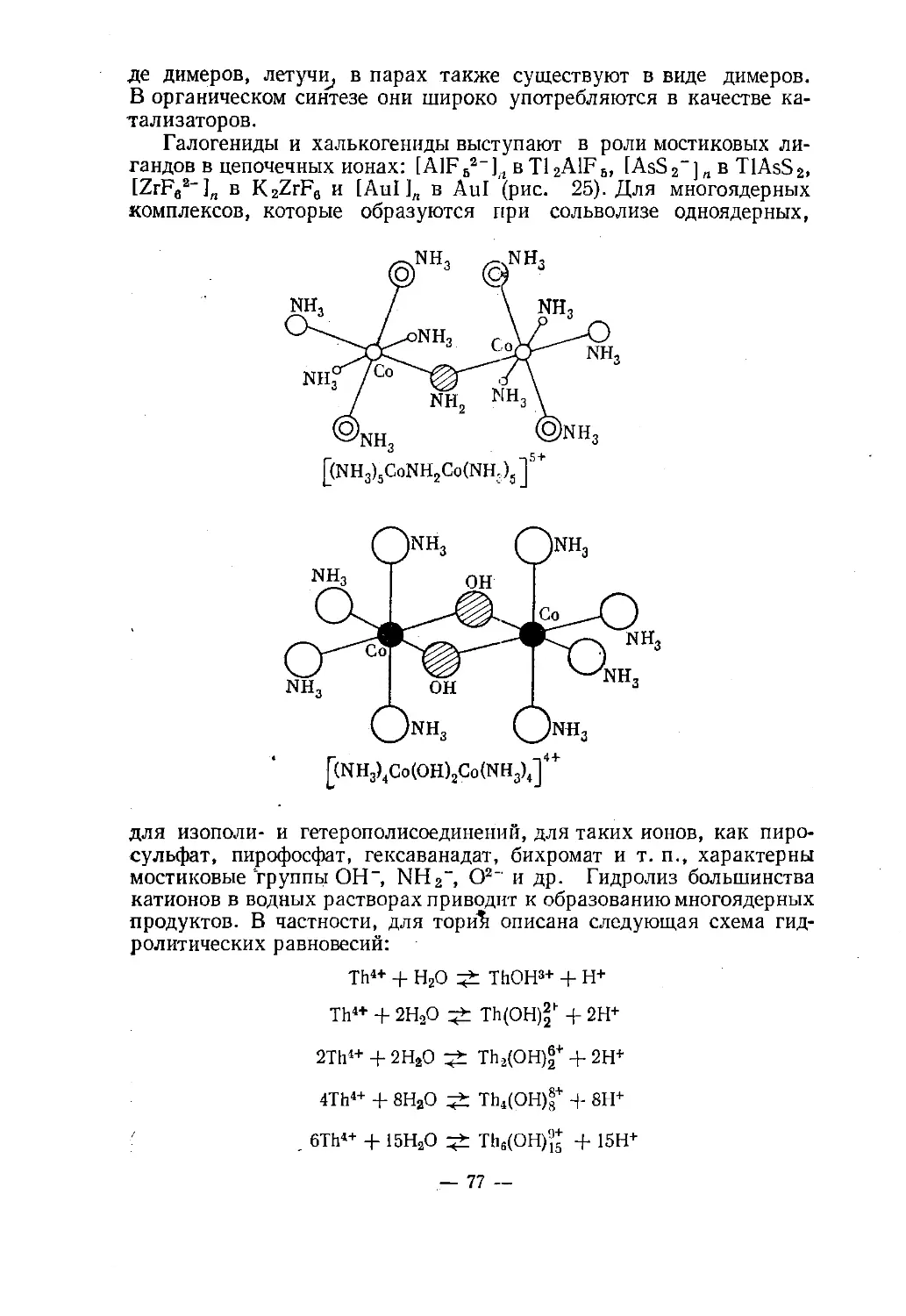
Ион/NH2x(nh,)*co< ;co(nh3)4
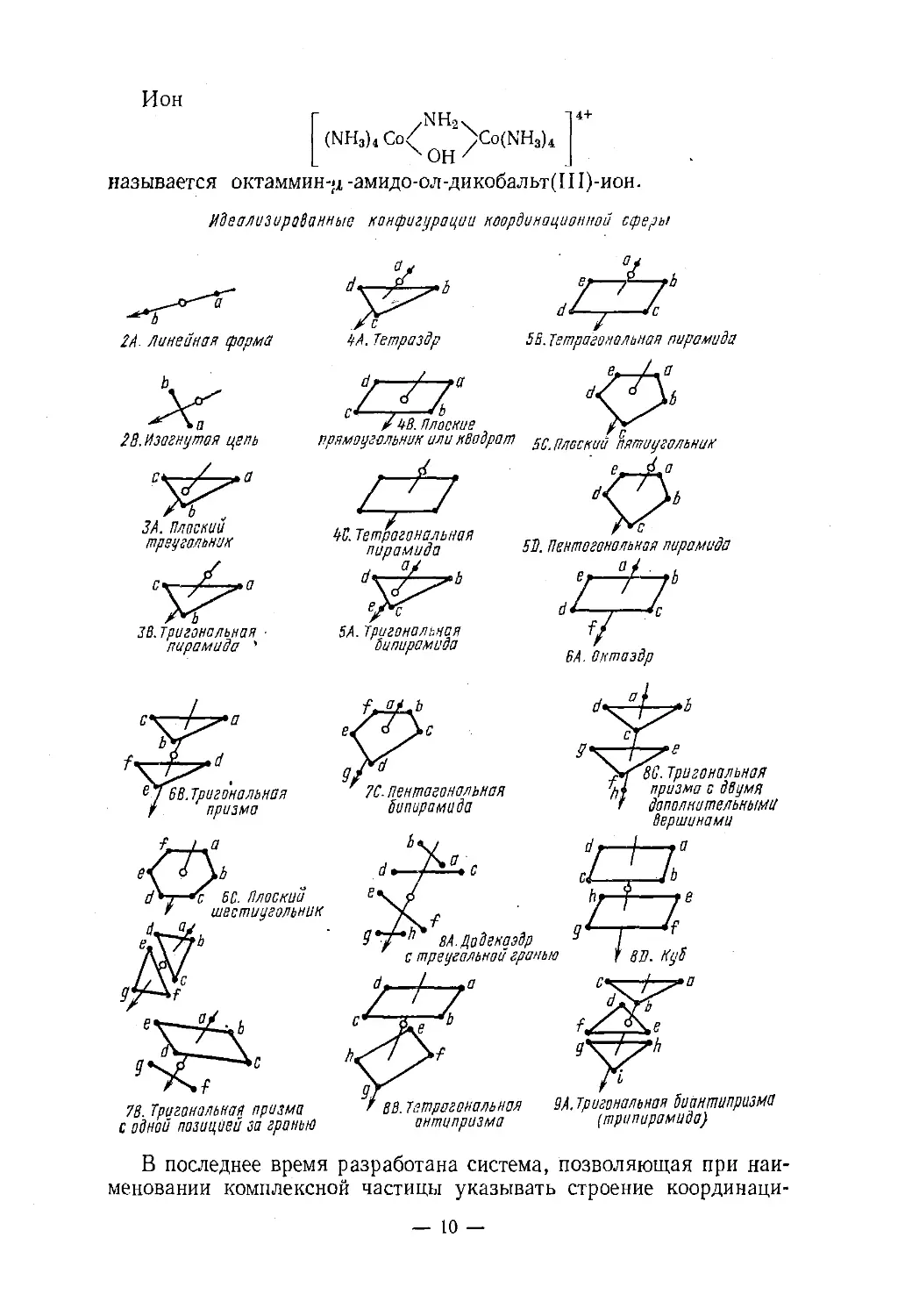
NOH /называется октаммин-<х -амидо-ол-дикобальт(Ш)-ион.Идеализированные конфигурации координационной сферы-L2А. Линейная формаАг28. Изогнутая цепьЗА. Плоек и и
треугольникЗВ.Тригональная ■
пирамида 'ZZ77SB. Тетрагональная пирамидаZXZ’/ 4В. Плоские гпрямоугольник или квадрат sc. Плоении пятиугольник№. Тетрагональная г- „пирамида № Пентагона, 'ая пирамидаd^L5А. Тригональная
бипирамида7С. пентагональная
Sun up амида™ 80. Тригональная
'U призма с двумя
дополнительнымиВС. Плоении
шестиугольник8А. Додекаэдрс треугольной гранью
& -
9'/4f78. Тригональная призма
с одной позицией за граньюВВ.Тетрагональная ЗА.Триганальная Ьионтпипразма
антипризма (трипирамида)В последнее время разработана система, позволяющая при наи¬
меновании комплексной частицы указывать строение координаци-— 10 —
онной сферы и способ присоединения лигандов и, таким образом,
различать геометрические изомеры комплексных- соединений. Эта
система состоит в том, что выделяются главнейшие идеализирован¬
ные конфигурации координационной сферы. Каждой конфигурации
присваивается индекс из цифры, равной координационному числу, и
буквы. Каждому донорному атому (или функциональной группе,
или всему лиганду в целом) приписывается индекс—малая буква ла¬
тинского алфавита, определяющая положение этой частицы в коор¬
динационной сфере. Идеализированные конфигурации и соответ¬
ствующие им индексы приведены выше.Чтобы избежать появления разных названий одной и той же ком¬
плексной частицы, необходимо ввести дополнительное условие о
значимости лигандов, например считать, что в рядуj 1~ Вг- Cl" !> F- NO2 NH3 пиридин(ру)* Н2Означимость убывает. При наименовании комплекса необходимо, что¬
бы среди аналогично расположенных лигандов индекс приписы¬
вался в первую очередь самому значительному из них. Тогда
[Co(NH3)5H20]3+ нужно назвать а, Ь, с, d, е-пентаммин-/- акво-6А-
кобальт(1П)-ион. Чтобы составить название изомерных октаэдри¬
ческих структурI Iследует в положение а поместить иодид, в положение Ъ — бромид.
Тогда первая из них — это а-иодо-б-бромо-с-аммин-й-хлоро-е-пири-
дин-/-нитро-6А-платина (IV), а вторая а-иодо-Ь-бромо-с-пиридин-d-
хлоро-е-аммин-/-нитро-6А-платина (IV).§ 3. Полные и сокращенные формулы координационных соединенииПолная формула комплексной частицы перечисляет все лиганды
внутренней координационной сферы и указывает способ их присое¬
динения.При невозможности записать полную формулу соединения, его
состав изображают сокращенной формулой или (для веществ,
полученных в результате реакций присоединения) применяют
термин «аддукты» и записывают их в виде формул исходных ве¬
ществ, соединенных точкой, например В13-Р13 или CuS04-5H20.
Слово «аддукты» означает «продукты присоединения». Как* Сокращенные названия некоторых лигандов приведены в табл. 1
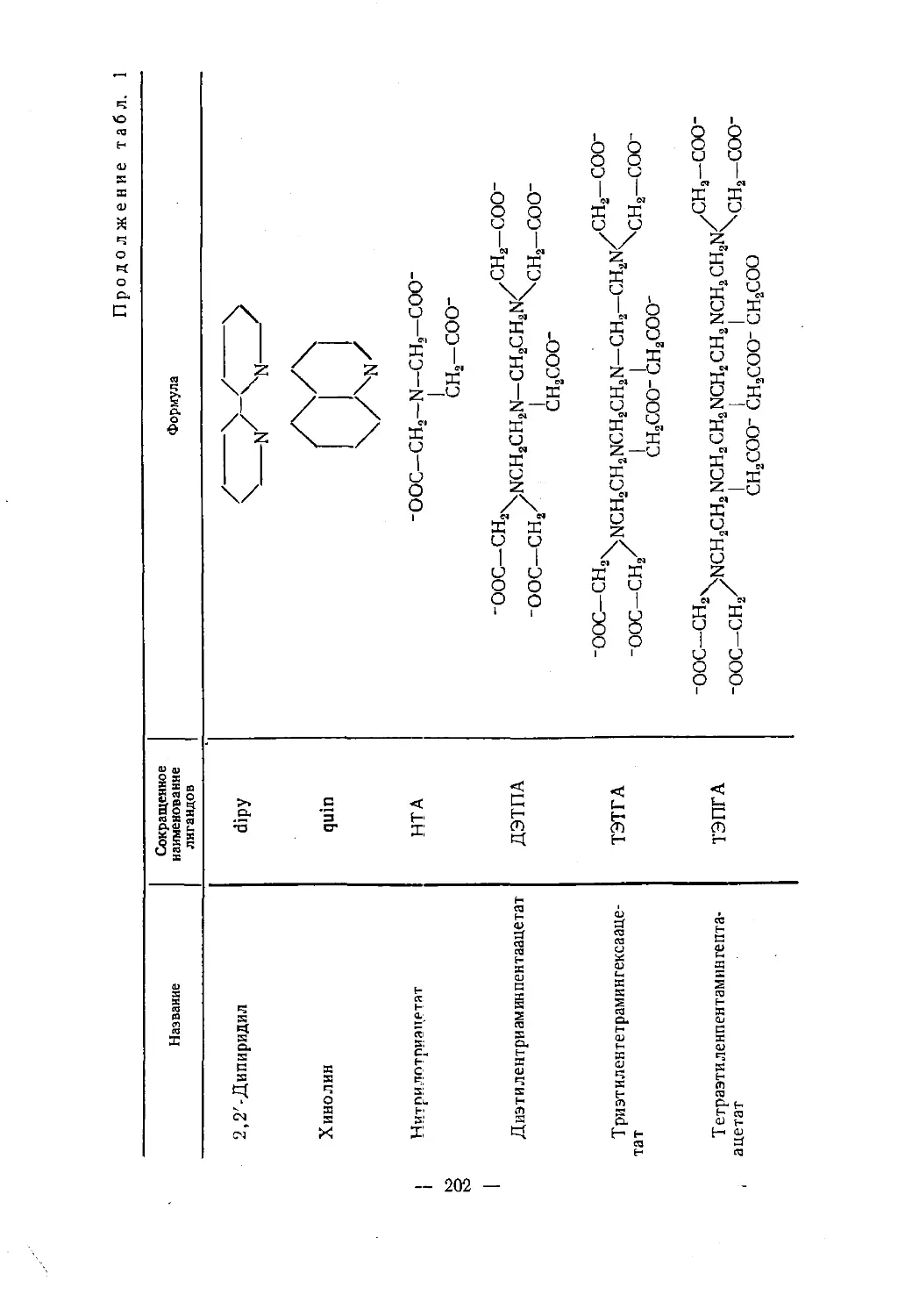
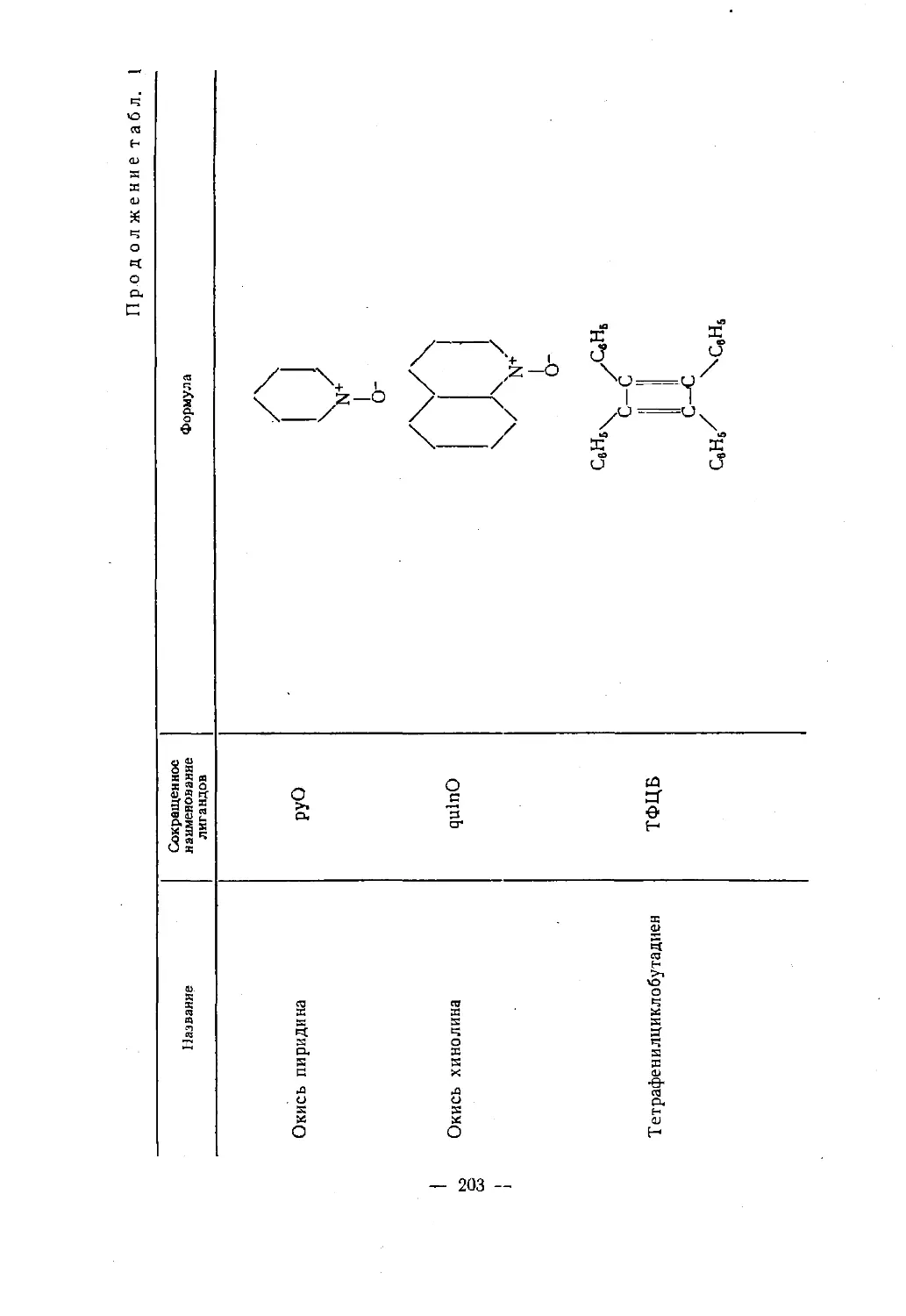
приложения.— 11 -
правило, его используют, если строение полученного соединения
неизвестно. Так, соединение 4ScCl3- С6Н12, полученное при длитель¬
ном выдерживании ScCl3 с циклогексаном в отсутствии влаги,
называют аддуктом, пока по имеющимся сведениям нельзя
определить его структуру и природу связей в нем. Аналогично
этому аддуктами названы вещества NbCl5• NOC1 и TaCl5-NOCl,
полученные взаимодействием NOC1 с хлоридами Та и Nb, или оса¬
док ТiCl 4 - PCI Б, выпадающий из дихлорэтановых растворов при
взаимодействии TiCl4 с РС15.Сокращенные формулы применяют по отношению к лабильным
комплексным ионам, существующим в растворах. Факт существо¬
вания таких ионов обычно определяется методами исследования
равновесий. Для определения их состава прослеживают зависимость
концентрации комплекса от концентраций компонентов. Если кон¬
центрации некоторых компонентов (обычно растворителя и индиффе¬
рентной соли) во время исследования не изменяются в достаточной
степени, невозможно установить, сколько частиц этих компонентов
входит в состав комплексного иона. Поэтому в формуле комплексно¬
го соединения предпочитают указывать только те лиганды, наличие
которых установлено достоверно. Остальные места в координацион¬
ной сфере могут быть заполнены по-разному. Сокращенная фор¬
мула, например дироданоферри-иона Fe^SCN)*, обнаруженного в
системе Fe3+ — SCN~ в присутствии 0,5 М НСЮ4, отвечает любому
из изображенных ниже соединений (для всех комплексов принято
к. ч. 6 и октаэдрическое строение координационной сферы):SCN SCN SCN SCNН20/—( уН20 Н2а—| 7^,0 н2а—| 7С104 Н20,—( 7СЮ+/ Fe / / Fe / / Fe / / Fe /H20z—-|—%0 Ciof 1—H20 Ciof 1—/H20 H207-' |^CIQ4SCN SCN SCN SCNпримеры транс - дироданоферри-ионовпримеры цис - дироданоферри - ионовт. е. приближенность сокращенной формулы доходит до того, что под
наименованием Fe(SCN)+ 2 выступают и ионы совершенно другого
заряда: [Fe(H 20) 3(C104)(SCN) 2 ]°, [Fe(H 20) 2(С104) 2(SCN)2 ]- и т. X-
Применение сокращенных формул характерно не только для хи¬
мии комплексных соединений.В сокращенной записи, например,— 12 —
уравнении диссоциации лимоннои кислоты
H3Cit H2Cit- + Н+ HCit2- + 2Н+С it3- + зн+не отражается существование гидроцитрат-и дигидроцитрат-ионов в
двух изомерных формах:СН,—СООНIнос—СООНсн2—СООНСОО-СОО”СООН —~СООНУСООН .уСОО-\СООНсоо-СОО- —COO"СООНСООНСОО-СОО“СОО'Аналогично этому формула аминоуксусной кислоты C2H502N
отвечает как кислоте, существующей в виде NH2CH2COOH, так и
бетаину +NH 3СН 2СОО_.Определение полной формулы комплексной частицы чаще всего
удается, если частица длительное время существует в растворе или
в кристаллическом соединении в виде стабильного образования.
Кроме полного химического анализа индивидуальных веществ при¬
меняются следующие методы установления формулы:1) синтетическое доказательство строения,2) проведение реакций на лиганды (у инертных комплексных
соединений при этом обычно обнаруживают только внешнесферные
частицы),3) рентгеноструктурный и электронографический анализ, изуче¬
ние электронных и инфракрасных спектров поглощения, ЯМР,
ЭПР, у -резонансная спектроскопия, криоскопия, изучение элек¬
тропроводности растворов и т. д.Простейший способ синтетического доказательства строения сос¬
тоит в том, чтобы обнаружить неизменность комплексной частицы
при реакциях двойного обмена. Так, возможность получения кри¬
сталлов [Co(NH3)5H20]C13 или [Co(NH3)5H20](N03)3 действием на
[Co(NH3)5H20]2(C204)3-4H20 холодной концентрированной со¬
ляной или азотной кислоты указывает на присутствие во всех этих
веществах комплексного катиона [Co(NH 3) 5Н 20 ]3+. Полные фор¬
мулы многих координационных соединений с большой вероятно¬
стью могут быть записаны при анализе схем их синтеза. Так,
соединение PtN5H14Cl3 получается при действии аммиака на рас¬
твор [Pt(NH3)5Cl ]С1 з- Предполагаемая схема синтеза
[Pt(NH3)5Cl]3+ + ОН" -> [Pt(NH2)(NH3)4Cl]2+ + НаОпозволяет считать, что продукт представляет собой амидохлоротет-
рамминплатехлорид [Pt(NH 3) 4(NH 2)С1 ]С12.При действии на раствор Co(NH3)5C13 нитратом серебра на хо¬
лоду обнаруживаются только два иона хлорида, что позволяет при¬
писать этому соединению формулу [Co(NH3)5C1 ]С12. При нагрева¬
нии осаждаются все три хлорид-иона.— 13 —
Из физико-химических методов исследования наиболее полные
и точные результаты, включая сведения о длинах связей и валент¬
ных углах, дает рентгеноструктурный анализ. Однако он чрезвы¬
чайно трудоемок и применим лишь к кристаллическим веществам.
Данные, получаемые другими методами, более ограничены. Поэтому
для окончательного решения вопроса о строении координационной
сферы применяют, как правило, несколько методов исследования.
Так, молекулярная электропроводность аддукта РС1Б-ReCls в
ацетонитриле (выбран неводный растворитель во избежание гид¬
ролиза) соответствует электропроводности двух однозарядных ионов.
На этом основании для данного соединения была предложена форму-
ла[РС14 ]+[ReCl6 ]“, а ИК-спектр соединения дал добавочные сведения,
говорящие в пользу этой формулы.Применение метода ИК-спектроскопии для изучения строения
частиц базируется или на теоретическом анализе спектра, или на
полуэмпирическом методе «характеристических частот». В самом
простом случае этот метод используется для идентификации частиц
сравнением спектра с литературными данными. Например, извест¬
но, что координация нитрит-иона через атом кислорода приводит к
появлению в ИК-спектре полос при 1460 и 1065 см-1, а через атом
азота — при 1430, 1315 и 825 см"1. Простое сопоставление ИК-спект-
ра исследуемого комплекса с этими данными позволяет установить
характер координации в нем NO2.Полосы поглощения, относящиеся к валентным колебаниям ме¬
талл—лиганд лежат в области 100—800 смг1 и очень мало характер¬
ны для различных типов связей. Поэтому основные сведения о струк¬
туре комплексов получают анализом положения полос, характер¬
ных для лигандов. Лигандные полосы поглощения подтверждают
присутствие лиганда в комплексе, а иногда позволяют указать ту его
таутомерную форму, которая участвует в комплексообразовании.
В результате смещения электронной плотности в лиганде под дейст¬
вием иона металла кратность связей в лиганде изменяется. Это ве¬
дет к сдвигу полос валентных колебаний (увеличение кратности
связи увеличивает частоту) и позволяет судить о способе присоедине¬
ния лиганда. Наконец, по расщеплению некоторых полос можно
судить о симметрии комплексной частицы и ее фрагментов или уста¬
новить присутствие неэквивалентно связанных и несвязанных ли¬
гандов или функциональных групп.Рассмотрим в качестве примера ожидаемые изменения в ИК-спек¬
тре координированной молекулы мочевины:В свободной мочевине донорные электронные пары на атомах азо¬
та можно считать занимающими рг-орбитали (эр2-гибридизация у
атома азота) и взаимодействующими с р2-орбиталями углерода и кис¬
лорода. Благодаря этому я-орбиталь делокализована и кратность— 14 —
связи С-0 меньше двух. При координации мочевины через один из
атомов азота его донорная электронная пара не сможет участвовать
в я-связи, делокализация я-орбитали уменьшится и кратность связи
С—О увеличится. Группы NH2 станут неэквивалентными.При координации мочевины через атом кислорода группы NH2 бу¬
дут эквивалентными, а кратность связи С—О уменьшится из-за
стремления лиганда к форме.NH+0'5-о—с/ " „ .При помощи таких соображений по ИК-спектрам установлено,
что в комплексе [Pt(NH2CONH2)2Cl2] мочевина координиро¬
вана через атом азота, а в соединениях [Fe(NH2CONH2) в ]С13,
[Cr(NH2CONH 2) в ]С1 з, в комплексах меди и цинка — через атом кис¬
лорода.§ 4. Комплексные частицы в раствореКомплексные ионы в растворе качественно можно обнаружить
одним из трех способов:1) по появлению у раствора свойств, принадлежащих образовав¬
шейся комплексной частице. Комплексообразование, например, наб¬
людается по изменению окраски раствора, когда спектр поглоще¬
ния комплексной частицы в видимой области резко отличается от
спектров поглощения лиганда и иона металла, а также протони-
рованных форм лиганда, присутствующих в растворе. В аналити¬
ческой химии комплексные соединения такого рода (роданидные ком¬
плексы Fe3+ и Со2+, сульфосалицилатный комплекс Fe3+ и др.) ис¬
пользуются для колориметрического определения иона металла;2) по исчезновению или ослаблению свойств раствора, которые
обусловливались несвязанным ионом металла или лигандом, их
протонированными или гидролизованными формами. Например,
прибавление трилона Б (динатриевой соли этилендиаминтетраук-
сусной кислоты Na2H2Y) к раствору LaCl3 приводит к тому, что
действием щелочи из такого раствора не осаждается La(OH) 3, так
как практически весь La3+ связан в прочный комплекс LaY";3) по такому изменению свойств раствора, которое можно счи¬
тать результатом распада конкурирующих соединений. Например,
при введении ионов F~ в раствор, содержащий Fe3+ и SCN-, исчеза¬
ет кроваво-красная окраска. Это— следствие разрушения FeSCN2+
и других комплексных роданидов Fe3+ за счет образования фторид-
ных комплексов железа. Аналогично этому растворение BaS04B
растворах полиметафосфатов натрия или калия указывает на сдвиг
равновесияBaS04 ^ Ва24 + SO2-— 15 -
вправо за счет связывания ионов Ва2+ в полиметафосфатный комп¬
лекс.Если образование устойчивых комплексов фиксируется легко,
то наличие малопрочных комплексов устанавливается при коли¬
чественном исследовании физико-химических свойств раствора: оп¬
тической плотности, электропроводности и т. п. Если между комп¬
лексами в растворе отсутствует динамическое равновесие, то их смесь
можно разделить и указать, из каких частиц она состоит. Например,
если смесь солей K3[Cr(SCN)6] и K[Cr(NH3) 2(SCN)4] (раствор 1) на¬
нести на бумажную ленту, смоченную смесью NH4OH+NH 4SCN, и к
концам ленты приложить разность потенциалов (метод электромиг¬
рации),™ через некоторое время пятно хромовых солей разделится
на два: каждый анион будет двигаться к аноду со своей собственной
скоростью. Или же, действуя на раствор 1 в присутствии NH4OH +
+ NH4SCN раствором соли Си2+, получим нерастворимый рейне-
кат [Cu(NH3)4 ] [Cr(NH „) 2(SCN)412, а ионы Cr(SCN)6 останутся в раст¬
воре. В обоих случаях разделение удается потому, что реакция[Cr(NH3)2(SCN)4]~ + 2SCN- -> [Cr(SCN)6]a- + 2NH3практически не протекает.Наоборот, если динамическое равновесие между комплексами
устанавливается, то разделить ионы нельзя. Так, в системе Fe3+ —— SCN~ равновесия комплексообразования устанавливаются быст¬
ро. При введении в раствор KSCN иона Fe3+ образуется равновес¬
ная смесь катионов Fe3+, FeSCN2+, Fe(SCN)2, нейтральных частиц
Fe(SCN)3 и анионов Fe(SCN)4, Fe(SCN)5 , Fe(SCN)! , концентрации
которых зависят от концентрации KSCN. Если при электромигра¬
ции какой-нибудь вид частиц этой смеси будет двигаться отдельно
от других, то возникнет область, где концентрации комплексов от¬
личаются от равновесных, но равновесие быстро восстановится за
счет расходования избыточных частиц. Поэтому смесь комплексов
такого рода может двигаться только как единое целое с некоторой
средней скоростью. Если такая смесь движется к катоду, то это сви¬
детельствует о доминировании катионных форм, но не об отсутствии
анионных.
11. ОБЩИЕ СВЕДЕНИЯ О ХИМИЧЕСКОЙ СВЯЗИ
В КОМПЛЕКСНЫХ СОЕДИНЕНИЯХВопрос о строении комплексных соединений можно обсуждать
с электростатической точки зрения, при помощи теории кристалли¬
ческого поля, используя модели донорно-акцепторных и дативных
связей в методе валентных схем или метод молекулярных орбиталей
(МО). Идеи и выводы каждого из методов с успехом применяются в
характерных для них сферах химии координационных соединений,
но приближенный характер методов ограничивает их применение.§ 1. Химическая связь с электростатической точки зренияЕсли считать, что связи центрального иона с лигандами чисто
электростатические, то должны наблюдаться следующие законо¬
мерности:а) при сферической симметрии центрального иона его взаимо¬
действие с лигандами в принципе ненаправленно. Направление си¬
ловых линий зависит от расположения лигандов;б) лиганд ориентируется в поле центрального иона так, чтобы
отрицательный конец его диполя или атом лиганда, несущий
отрицательный заряд, находились в непосредственном контакте
с центральным ионом. Ориентированные таким образом лиганды
электростатически расталкиваются. Если лиганды одинаковы, то
они должны при этом занять положения, максимально удаленные
друг от друга, а именно: при к. ч. 3 — в вершинах плоского треу¬
гольника, при к. ч. 4 — тетраэдра, при к. ч. 5 — тригональной
бипирамиды, при к. ч. 6 — октаэдра и т. д.;в) количество лигандов, которые могут поместиться вплотную
к центральному иону, зависит от соотношения между его радиусом
гм и радиусом лигандов rL • Малые лиганды разместятся вокруг кати¬
она, не касаясь друг друга. При большем размере лигандов они
касаются друг друга и центрального иона. Отвечающее этому значе¬
ние гь//"м называется критическим. При дальнейшем увеличении гь
получится конфигурация, в которой лиганды удалены от централь¬
ного иона, но касаются друг друга. Такая конфигурация неустойчи¬
ва и преобразуется в устойчивую с уменьшением координацион¬
ного числа путем удаления лишнего лиганда. Итак, по критическо¬
му отношению г^/гц (табл. 1) можно судить о максимальном к. ч., до¬
пускаемом пространственными факторами. Если, например, 6,5 >
> гl /гц> 4,4, то максимальное к. ч. равно 3. При этом лиганды ка¬
саются центрального иона, но не соприкасаются друг с другом. Уве¬
личение к. ч. до четырех невозможно, так как лиганды перестанут
касаться центрального иона;г) поляризующее действие центрального иона на лиганды пропор¬
ционально электростатическим характеристикам этого иона (кото¬
рые тем выше, чем больше его заряд и меньше радиус) и электрон¬
ной поляризуемости лигандов. .Учет происходящего при поляриза-
ции смещения электронной плотности к центральному иону с
уменьшением его эффективного заряда фактически описывает ча¬
стичную ковалентность связи;Таблица 1
Критические значения rL /гм и rM/rLК. ч.'L /ГМrM/rL36,50,1544,40,2252,40,4162,40,4171,70,5981,50,6591 ,40,73101 ,20,83111,10,90121,10,90д) устойчивость комплексных соединений должна расти по мере
увеличения электростатических характеристик центрального иона, а
также заряда или полярности (с учетом поляризуемости) лиганда.
У сложных лигандов необходимо учитывать локальную полярность
и поляризуемость.Основным недостатком чисто электростатического подхода яв¬
ляется невозможность описания с его помощью электронной структу¬
ры комплексной частицы и поэтому ее магнитных, спектральных, ки¬
нетических и других свойств. Он с трудом распространяется на ком¬
плексные частицы с ковалентными связями, а образования я-комп-
лексов он вообще не может предвидеть. Применение электростати¬
ческого подхода ограничено комплексами, центральный ион которых
имеет внешнюю электронную оболочку типа s2рв, а также некоторы¬
ми внешнесферными ассоциатами.§ 2. Теория кристаллического ноляРассмотрим электростатическое взаимодействие катиона метал¬
ла с ионными или полярными лигандами. Лиганды, ориентирован¬
ные в результате ион-ионного или ион-дипольного взаимодействия
отрицательными концами к иону металла, в первом приближении
будут рассматриваться как бесструктурные отрицательные заряды,
образующие так называемое «кристаллическое поле». При этом d- или
/-орбитали металла оказываются неравноценными относительно по¬
ля и их энергия становится неодинаковой: d-или /-подуровень рас¬
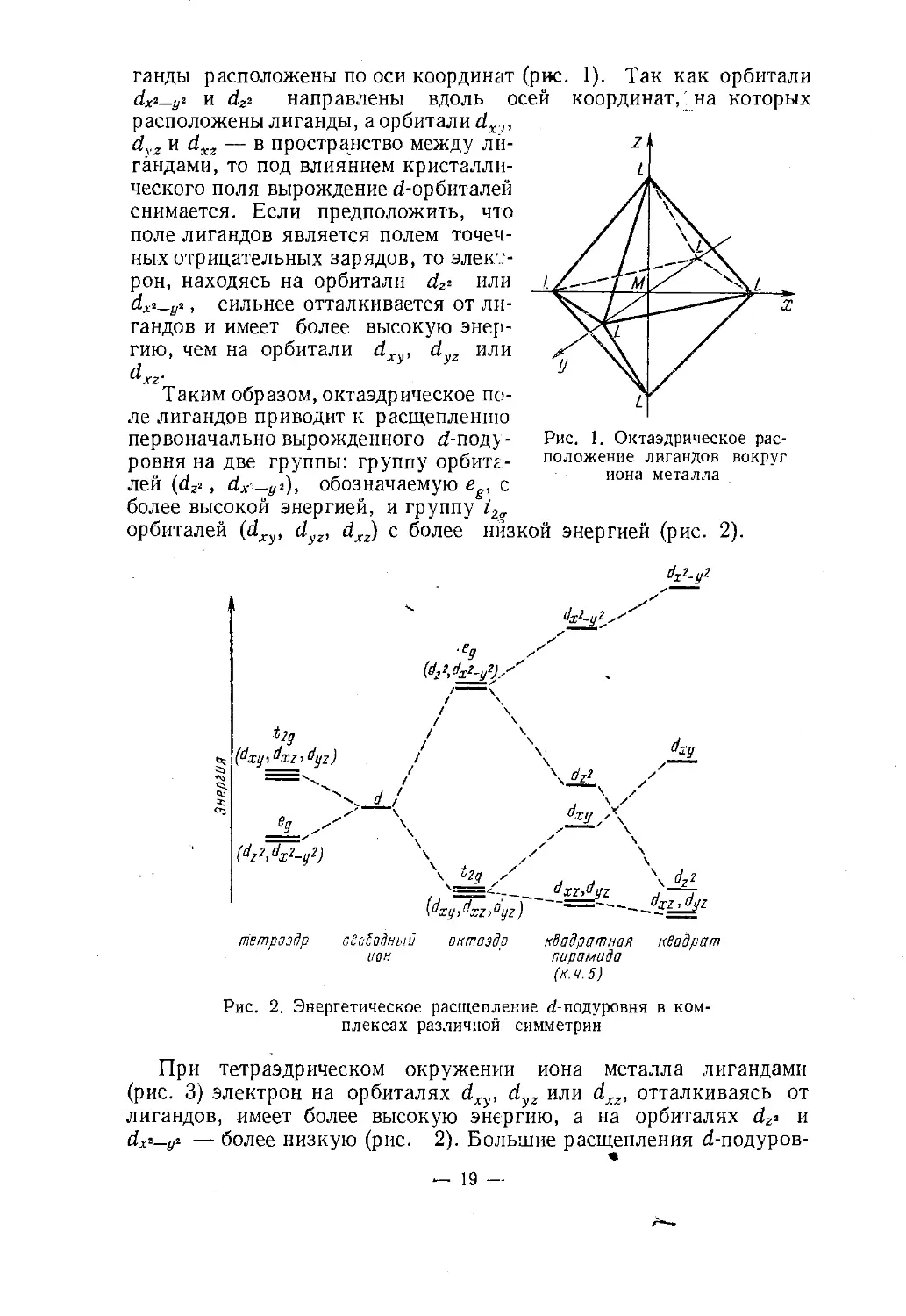
щепляется. Характер расщепления зависит от симметрии поля.Рассмотрим, например, октаэдрический комплекс. Пусть оси де¬
картовых координат совмещены с осями симметрии четвертого по¬
рядка, ядро иона металла служит началом системы координат, а ли¬— 18 —
ганды расположены по оси координат (рис. 1). Так как орбитали
dX‘~y2 и dzi направлены вдоль осей координат,' на которых
расположены лиганды, а орбитали йх,,,
dyz и dxz — в пространство между ли¬
гандами, то под влиянием кристалли¬
ческого поля вырождение d-орбиталей
снимается. Если предположить, что
поле лигандов является полем точеч¬
ных отрицательных зарядов, то элект¬
рон, находясь на орбитали d# или
dX2_yi, сильнее отталкивается от ли¬
гандов и имеет более высокую энер¬
гию, чем на орбитали dxy, dyz илиdxz'Таким образом, октаэдрическое по¬
ле лигандов приводит к расщеплению
первоначально вырожденного af-поду-
ровня на две группы: группу орбитг-
лей (d2г , dx'-y*), обозначаемую eg, с
более высокой энергией, и группу t2орбиталей (d , d , dxz) с более низкой энергией (рис. 2).Рис. 1. Октаэдрическое рас¬
положение лигандов вокруг
иона металлаdx2-y2dx2-y2s■Eg(°^2^x2-y2).-'i>2g(dxyi dxz ’ dyz)(dz?,dx2_y?)axy,jL>'\ 4-\ b2g ^X2ydjZ H~^7{dxy,dxz>°yz)тЕтраздр сСо£одньш октаэдр квадратная квадрат
ион 1 пирамида(к.ч. 5)Рис. 2. Энергетическое расщепление d-подуровня в ком¬
плексах различной симметрииПри тетраэдрическом окружении
(рис. 3) электрон на орбиталях dxy, dлигандов, имеет более высокую энергию, а на орбиталях dz* и
dX2-yi — более низкую (рис. 2). Большие расщепления d-подуров¬иона металла лигандами
у2 или йхг, отталкиваясь от— 19
ня вызываются так называемыми лигандами сильного поля (СО, CN“)
NO , NH3). При этом менее выгодные d-орбитали заполняются элект¬
ронами лишь после полного запол¬
нения более выгодных. Теория крис¬
таллического поля предсказывает до¬
полнительную стабилизацию некото¬
рых комплексных частиц полем ли¬
гандов, а также искажение высоко¬
симметричных конфигураций комп¬
лексов некоторых металлов (Си2+, Сг2+
и др.). Эта теория объясняет цвет сое¬
динений и магнитные свойства ком¬
плексов переходных металлов. Для
ионов с внешней электронной конфи¬
гурацией s2p® теория не дает каких-ли¬
бо интересных результатов. Для ком¬
плексных частиц с сильно выражен¬
ным ковалентным характером связей, особенно при наличии
я-взаимодействия, эта теория также мало пригодна. Теория кристал¬
лического поля наиболее эффективна для описания высокоспино¬
вых комплексных соединений переходных металлов и /-элементов.§ 3. Схема молекулярных орбиталей (МО)Орбитали, содержащие валентные электроны, а также ближай¬
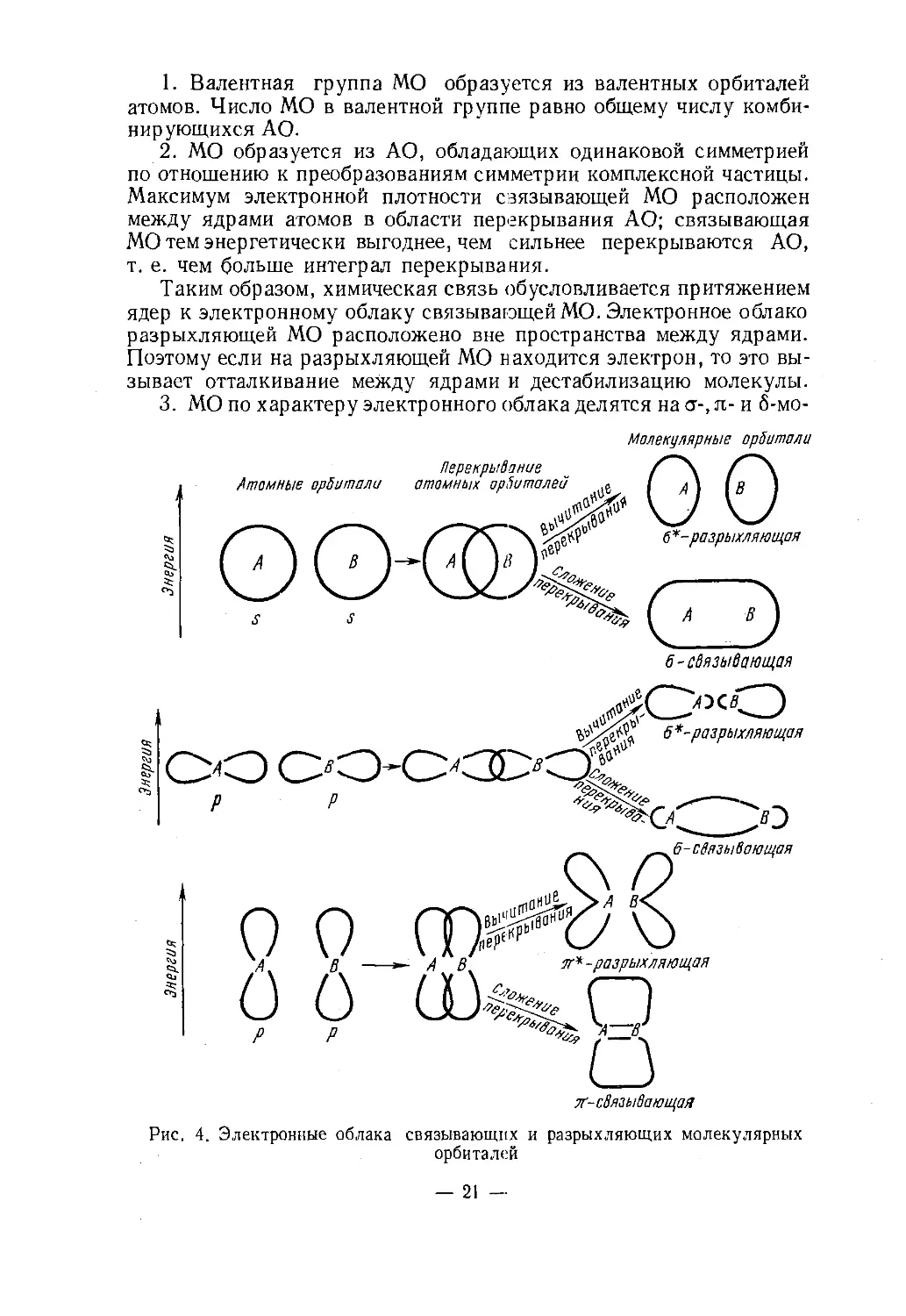
шие к ним по энергии вакантные орбитали, можно выделить в ва¬
лентную группу молекулярных орбиталей (МО). Различают связыва¬
ющие, несвязывающие и разрыхляющие МО. Связывающими
называются такие, переход электронов на которые с соответствую¬
щих атомных орбиталей (АО) энергетически выгоден, разрыхляющи¬
ми — такие, для которых этот переход невыгоден. Несвязывающие
МО как по энергии, так и по форме электронного облака мало от¬
личаются от соответствующих АО. Электронная плотность у связы¬
вающих орбиталей сосредоточена в пространстве между ядрами
связываемых атомов, у разрыхляющих — вне этого пространства.
При локализации электронной плотности МО между двумя ядрами
образуется двуцентровая МО, которая в обычной структурной фор¬
муле изображается чертой. Многоцентровые МО с трудом учитыва¬
ются в структурных формулах.Схему МО представляют в виде диаграммы,у которой вертикаль¬
ная ось соответствует энергии орбиталей (чем выше расположена ор¬
биталь, тем менее она выгодна энергетически), а каждая орбиталь
изображается горизонтальной чертой. При качественном построении
такой диаграммы обычно считают, что МО возникает при взаимодей¬
ствии нескольких АО. Соответствующий количественный метод из¬
вестен под названием «молекулярная орбиталь — линейная комбина¬
ция атомных орбиталей» (МО ЛКАО). При построении МО из АО ис¬
пользуются следующие правила:Рис. 3. Тетраэдрическое рас¬
положение лигандов вокруг
центрального иона— 20 —
1. Валентная группа МО образуется из валентных орбиталей
атомов. Число МО в валентной группе равно общему числу комби¬
нирующихся АО.2. МО образуется из АО, обладающих одинаковой симметрией
по отношению к преобразованиям симметрии комплексной частицы.
Максимум электронной плотности связывающей МО расположен
между ядрами атомов в области перекрывания АО; связывающая
МО тем энергетически выгоднее, чем сильнее перекрываются АО,
т. е. чем больше интеграл перекрывания.Таким образом, химическая связь обусловливается притяжением
ядер к электронному облаку связывающей МО. Электронное облако
разрыхляющей МО расположено вне пространства между ядрами.
Поэтому если на разрыхляющей МО находится электрон, то это вы¬
зывает отталкивание между ядрами и дестабилизацию молекулы.3. МО по характеру электронного облака делятся на <т-, я- и 6-мо-Молекупярные орбиталиАтомные орбиталиПерекрывание
атомных орбиталейООЧЗЭ■разрыхляющая6-связывающая„ЛОсОЪ' 6*-разрыхляющаяОО 00-0<3D<3! '8Рис. 4. Электронные облака связывающих и разрыхляющих молекулярныхорбиталей— 21 —
лекулярные орбитали. Электронные облака этих видов орбиталей
имеют соответственно один, два и четыре участка сгущения элект¬
ронной плотности. При взаимодействии АО с образованием а-,
я- и 6-связывающих орбиталей обычно получается такое же число
соответствующих разрыхляющих орбиталей, которые обозначают¬
ся ст*, я* и б*. Электронные облака связывающих и разрыхляю¬
щих ст- и я-молекулярных орбиталей представлены на рис. 4.4. МО заполняются электронами в порядке их энергетической
выгодности. При этом соблюдается правило Хунда о максималь¬
ности спина системы, т. е. в основном состоянии энергетически эк¬
вивалентные (вырожденные) орбитали сначала заполняются электро¬
нами по одному так, чтобы спины электронов были параллельны.Самые энергетически невыгодные из заполненных электрона¬
ми орбиталей легко становятся орбиталями-донорами электронов.
Наоборот, самые энергетически выгодные незаполненные электрона¬
ми (вакантные) орбитали становятся акцепторами электронов.Рассмотрим МО двухатомных молекул и ионов, образуемых
атомами второго периода: N2, 02, F2, СО, CN~, NO и т. д. Каждый
из этих атомов имеет в валентной группе одну s- и три р-орбитали.
Поэтому общее число МО равно восьми.Пусть ядра атомов расположены вдоль оси г. Тогда орбитали
рхи pz обоих атомов взаимодействуют, давая две я-связывающие и
две я-разрыхляющие МО. Из-за эквивалентности орбиталей рх и ру
связывающие орбитали эквивалентны друг другу, и также равно¬
ценны я*-разрыхляющие орбитали:ад—- 'Рх’РуОрбитали s и р2обоих атомов взаимодействуют, давая четыре ор¬
битали ст-типа. В первом приближении можно считать, что орбитали
s и р2 одного атома взаимодействуют только с одноименными ор¬
биталями другого атома. Тогда должны появиться орбитали cr(2s)
и cr*(2s), которые по энергии ниже л-орбиталей; орбиталь ст(2рг)
по энергии ниже, чем орбитали я, так как взаимодействие р-орби¬
талей по tr-типу сильнее, чем по я-типу, и, наконец, орбиталь ст*(2рг),
которая по энергии выше, чем л*. Схема МО приобретает вид:Орбитали
■ первого атомаPx,Py,PzОрбитали
Второго атомаРх Ру Pz— 22 —
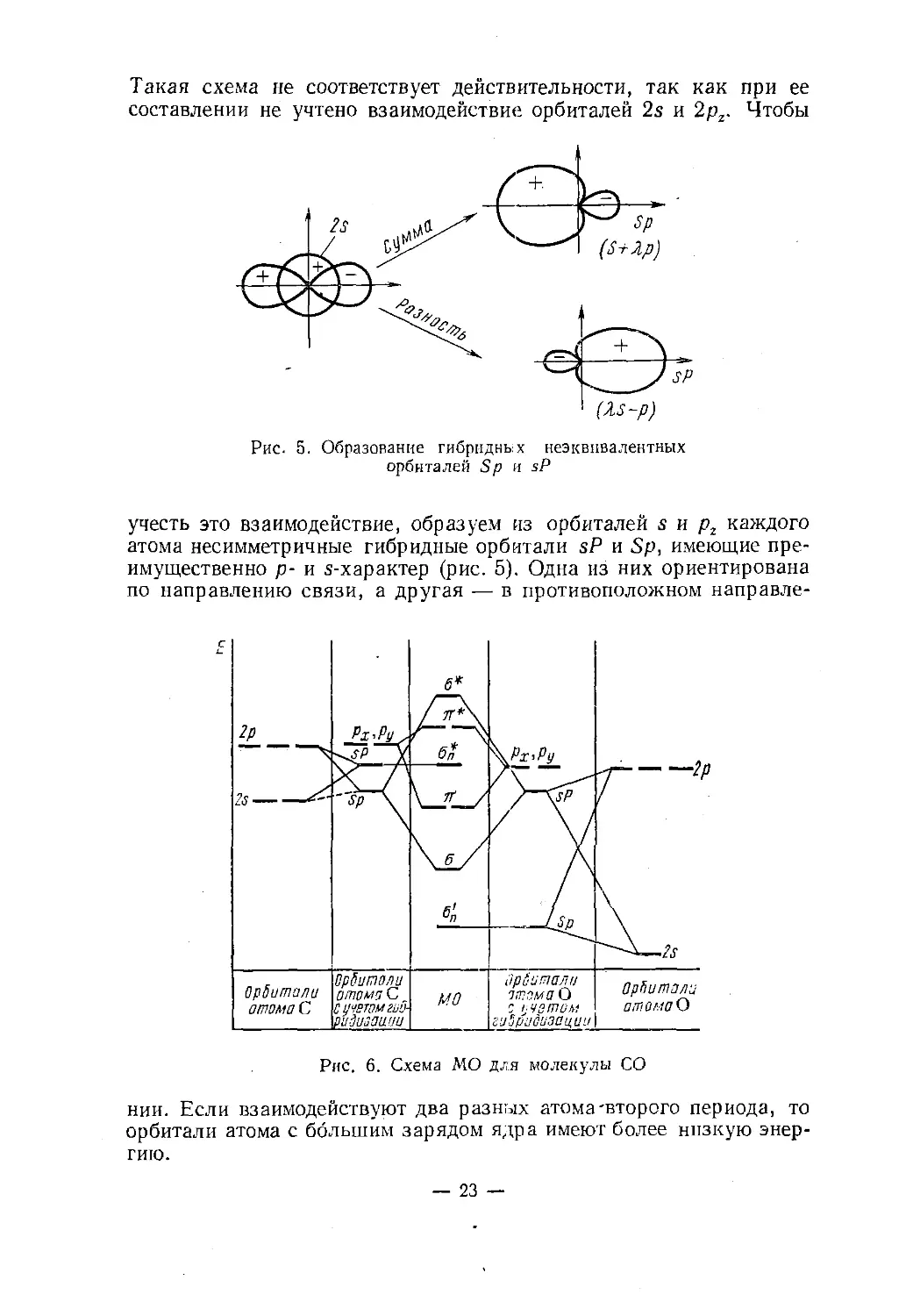
Такая схема не соответствует действительности, так как при ее
составлении не учтено взаимодействие орбиталей 2s и 2pz. ЧтобыРис. 5. Образование гибридньх неэквивалентных
орбиталей Sp н sPучесть это взаимодействие, образуем из орбиталей s и р2 каждого
атома несимметричные гибридные орбитали sP и Sp, имеющие пре¬
имущественно р- и s-характер (рис. 5). Одна из них ориентирована
по направлению связи, а другая — в противоположном направле¬нии. Если взаимодействуют два разных атома'второго периода, то
орбитали атома с большим зарядом ядра имеют более низкую энер¬
гию.— 23 —
Рассмотрим построение схемы МО для СО (рис. 6.). Орбитали
Sp атома кислорода и sP атома углерода, электронные облака ко¬
торых расположены, в основ¬
ном, в пространстве вне ядер
(рис.7, а), практически не
взаимодействуют, т. е. прев¬
ращаются в <т-орбитали «-ти¬
па. Взаимодействие орбиталей
sP атома кислорода и Sp ато¬
ма углерода (рис. 7, б) при¬
водит к возникновению ст-свя-
зывающей и сг- р аз р ы х л я юще й
орбиталей. Такая схема ка¬
чественно отличается от ра¬
нее приведенной тем, что од¬
на из стп-орбиталей по энер¬
гии расположена между я- и
д*-орбиталями. Аналогичный
характер схема МО имеет так¬
же в гомоядерных молекулах
второго периода. Упрощенно эту схему можно представить сле¬
дующим образом:'б*
п*а"ап
ST
6
%Рассмотрим теперь заполнение МО электронами. У молекул
СО, N2 и аниона CN- имеется по 10 валентных электронов. Они
заполняют все связывающие и я-орбитали. Таким образом, в этих
молекулах тройная связь (одна ст- и две я-связи), а л-электроны,
занимающие орбиталь сг/, являются донорами. Орбиталь оп" прак¬
тически совпадает с sP-орбиталью более легкого атома; следова¬
тельно, в молекуле СО и в анионе CN- можно считать ст-донорной
электронную пару, локализованную на атоме углерода.В молекуле NO одиннадцатый валентный электрон расположит¬
ся на одной из я-разрыхляющих МО. Молекула N0 представляет
собой молекулу-радикал, т. е. молекулу, имеющую неспарен¬
ный электрон. Она парамагнитна.В молекуле Оа 12 валентных электронов: на двух я-разрыхляю¬
щих орбиталях по правилу Хунда расположатся два неспаренных
электрона. Молекула кислорода сильно парамагнитна и являет¬
ся молекулой-бирадикалом. Наличие в NO и 02 электронов на
я-разрыхляющих орбиталях дестабилизирует эти молекулы. Энер¬
гия связи в них ближе к энергии двойной, чем тройной связи.С ОРис. 7. Образование а-орбиталей
в молекуле СО— 24 —
Схемы МО для комплексов металлов более сложны. Рассмотрим
МО в октаэдрическом комплексе [CoF6 ]3~, учитывая в качестве
валентных шесть р-орбиталей лигандов (от каждого по одной,
имеющей подходящую направленность для участия в ст-связыва¬
нии) и орбитали 3d, 4s, и Ар иона Со3+. Энергия орбиталей лиган¬
дов ниже энергии орбиталей иона металла, поэтому связывающие
МО по энергии ближе к энергии АО лигандов. Вдоль осей х, у и2, на которых расположены лиганды, ориентированы орбитали
Со3+ Ар, 3d2* и Мх^у1. Шесть d2sp:i -гибридных орбиталей Со3+
перекрываются с шестью АО лигандов и образуют шесть свя¬
зывающих и шесть разрыхляющих МО: За2ad 2?d* as* За• *.При использовании только ра-орбиталей лиганда орбитали
dXy, dXi и dyz не имеют соответствующих по симметрии орбиталей
лиганда и поэтому дают несвязывающие МО, близкие по энергии к
исходным АО. При рассмотрении всех орбиталей лигандов среди
них найдутся такие, с которыми орбитали dxlJ, dxz и dyz взаимодей¬
ствуют, образуя МО я-типа. Заполнение МО электронами происхо¬
дит следующим образом: сначала заполняются шесть связываю¬
щих МО, обладающих низкими энергиями, а остальные электроны
распределяются между несвязывающими и ad*-разрыхляющими
МО. Так, например, схема МО иона [CoF6]3~ имеет видАО41-«-+■¥++3dСО3+МО+ 4-
■W- + +dxy dyz dXz■ff Bd
-ff -If 6p
Hf 6s[C0F6]3'AO6F'Квантовомеханическое рассмотрение молекул сводится к ре¬
шению уравнения Шрёдингера:Щ = Е^,в котором гамильтониан Н учитывает взаимодействие электронов
со всеми ядрами молекулы, а также межэлектронные и межъядер-
ные взаимодействия. Задачи для молекулы решаются приближен¬
но.Обозначим атомы в молекуле индексами k, а валентные орби¬
тали каждого атома — индексом т. Тогда ^mh обозначает т-ю ва¬— 25 —
лентную АО &-го атома. Представим МО, т. е. волновую функцию
ф электрона в молекуле в виде линейкой комбинации фтй с коэффи¬
циентами стк\4 = (II.1)k тПри этом мерой участия фтЬв характеристиках ф, не зависящих от
координат, является c„k. Например, если орбиталь ф занята N
электронами (N — нуль, единица или два), то заселенность ф рав¬
на N, а вклад фтй в заселенность ф равен chkN.Из выражения (II. 1) следует, что электрон, описываемый функ¬
цией ф, с какой-то вероятностью можно найти на любом из ато¬
мов, т. е. электрон делокализован по всей молекуле.Гибридные орбитали. Сгруппируем в выражении (II. 1) члены,
содержащие орбитали k-vo атома, и обозначим сумму через с/гук:ckLh = 2 cnik^mk • (И-2)тгде ук — гибридная орбиталь; будем считать ее нормированной,
т.е. \yjidv = I. Перепишем (II. 2) в виде•/.a = 2c«*'W (н.з)тгдеCnih Clnk/C и •Ортонормированность функций фтй приводит при этом к соот¬
ношениям= Sc^ = i.т тВеличина Cfnk = cllklck2 является мерой участия tymh в характе¬
ристиках yk, не зависящих от пространственных координат. Если
заселенность орбитали yk электронами равна nk, то доля еептк, вне¬
сенная орбиталью фтй, составляет,2„2 mk .nmk— mknb~~ r2 nk'
ckПри помощи гибридных орбиталей выражение (II. 1) преобразуется
к виду-Ф = 2С*7** (п-5>kв котором каждый атом представлен единственной гибридной ор¬
биталью. ВеличиныSkr = j1 't-kf-fdv— 26 —
называются интегралами перекрывания гибридных орбиталей.
При этомSkr = Srk. (II.6)Заселенность и эффективный заряд по Малликену. Пронуме¬
руем МО в молекуле индексом i. Для каждой из введем засе¬
ленность Nt и запишем уравнения, аналогичные уравнениям (II. 2)
и (II. 5):cih^-ik — 2 citntt'\'mh > ’ii — 2 cili^'ik-
m kРассчитаем iV* как среднее значение физической величины с
учетом независимости Nt от пространственных координат:= Ni 2 cili lcik + 2 cirSil!r\ ■ (I I-7)k 1 r 4 k jПредставим Nt в виде суммы вкладов атомов. Выпишем члены,
содержащие индексы k\ Nffk, Nt cihcirSikr и Nt cir cih Slrk (при
разных г). Две последние величины согласно (II. 6) равны между
собой. Логично считать Nдолей Nь принадлежащей &-му ато¬
му, а 2N, cihcirSihr — принадлежащей совместно k- му и му ато¬
мам. Малликен предлагает делить величину 2 Л^с;й cir Sihr между
k-м и г-м атомами поровну. Тогда величина Nih, рассчитанная
по уравнениюNik = NiCik | Сц; 2 cir^ikr j •определяет долю Nсвязанную с k-м атомом, а точнее — с его
орбиталью yih, и соответствует nh в уравнении (II. 4). Сумма зна¬
чений Nih должна составлять полную заселенность i-й орбитали2 Nik = Nt.ftСуммируя Nih по i, получим общее количество электронов Nk на
k-м атоме:2^ = л*.
iРазность заряда ядра zh и Nh равна эффективному заряду qh на
k-м атоме:Qk — Zk—Из уравнения (II. 4) вытекает, что с орбиталью i>mk k-ro атома свя¬
зана часть Nit выражаемая формулой,2»j imknimk — С imk Nik — 2 JVjj,.cik— 27 —
Суммирование по всем МО дает заселенность nmk для АО фтА:ftmk = ^ ftimk •
iВалентное состояние. При помощи коэффициентов cik и cihm
можно оценить заселенность отдельных АО в соединении. Напри¬
мер, при расчете схемы МО для Мп04“ обнаружено, что суммар¬
ная заселенность З^-орбиталей атома Мп равна 5,82 электрона;
45-орбиталей—0,18 электрона, 4р-орбиталей—0,34 электрона.
Запись 3d5'82 4s')'18p0'34 можно рассматривать как часть электронной
формулы валентного состояния атома Мп.Для оценки эффективного заряда атома Мп учтем, что ниже¬
лежащие орбитали Мп полностью заполнены и содержат в сумме
18 электронов (Is2 2s2p63s2pe), a Ad и вышележащие — пустые.
Тогда общее число электронов на марганце jVMn = 18 + 5,82 -f-
+ 0,18 + 0,34 = 24,34. Так как 2Мп= 25, то <7мп= 25 — 24,34 =
= +0,66. Это мало по сравнению с формальной электровалент¬
ностью марганца в Мп04“, равной +7. Малое дмп указывает на
значительную ковалентность связей в системе.§ 4. Метод валентных связейЭтот метод описывает образование комплексных соединений
при помощи ковалентных двухэлектронных связей. Обобществлен¬
ная электронная пара сосредотачивается в
том месте, где перекрываются орбитали
связываемых частиц. Для образования свя¬
зи, кроме того, необходимо отличие интег¬
рала перекрывания от нуля, поэтому, нап¬
ример, если ядра атомов расположены вдоль
оси х, то при перекрывании s-орбитали од¬
ного атома и ру-орбитали другого связь не
образуется (рис. 8).Важную роль при рассмотрении ком-
плексообразования методом валентных свя¬
зей играет понятие о донорно-акцепторной
и дативной связях.Донорно-акцепторная связь образуется
при перекрывании занятой орбитали, при¬
надлежащей донору, и свободной орбитали,
принадлежащей акцептору, и после образования ничем не отли¬
чается от обычной ковалентной.Образование донорно-акцепторных ст-связей происходит за счет
перекрывания вакантной орбитали иона металла с заполненными
электронами орбиталями донорной группы лиганда. При этом
электронные пары лиганда «поступают в общее пользование» ли¬Рис. 8 Перекрывание
s- и р-орбиталей. Ин¬
теграл перекрывания
равен нулю— 28 —
ганда и центрального иона. Донорами электронов могут служить
Н20:, :NH3, :СО, :CN“, :С1;- ЮН',CH3C;f , СНэ-С-СНэ и др.хО ||6Донорно-акцепторные связи я-типа (дативные) образуются толь¬
ко наряду с ст-связями. При этом донором служит ион металла,
представляющий в общее пользование свои спаренные d-электроны.
Поэтому дативные связи образуются ионами переходных метал¬
лов, имеющими большое число d-электронов. Акцепторами явля¬
ются лиганды, которые имеют достаточно энергетически выгодные
вакантные орбитали.Дативные связи образуются: 1) за счет перекрывания d-орбитали
иона металла, на которой находятся электроны, не вступившие в
ст-связь, с вакантными р-орбиталями или я*-МО лиганда (взаимо¬
действие dn — р„, рис. 9); 2) за счет перекрывания заполненныхРис. 9. Образование п-
связи при перекрывании
атомных орбиталей dXz
и pzОРис. 10. Схема взаимодействия
d_ — d.d-орбиталей металла и пустых d-орбиталей лигандов (взаимодей¬
ствие dn—dn, рис. 10).Взаимодействие dn — ря происходит, если лиганд координи¬
руется через атомы элементов второгэ периода (С, N, О), а взаимо¬
действие d„ — dn — через атомы третьего и последующих пе¬
риодов, имеющие вакантные d-орбитали (Р, S и др.).При образовании дативных я-связей осуществляется перенос
электронов от металла к лиганду. В результате увеличивается по¬
ложительный заряд и акцепторные свойства иона металла, поэтому
становится более прочной ст-связь металл—лиганд. Наоборот, чем
прочнее ст-связь металл-лиганд, тем выше электронная плотность
на ионе металла и его донорные свойства, которые ведут к образо-— 29 —
ванию я-связи. Таким образом, донорно-акцепторные ст-связи и да¬
тивные связи взаимно усиливают друг друга. Например, ион
Ni^+(ds) в донорно-акцепторных ст-связях служит акцептором за
счет вакантных орбиталей 4s, 4р, Ad3d 4s ty bd и одной орбитали 3d, если З^-электроны предварительно спарены.Но если донорный атом лиганда имеет вакантные d-орбитали,
то ион Ni2+ может служить и донором. Так, между ионом Ni^+ и ато¬
мом серы в серусодержащем лиганде (диэтилдитиокарбамате, тио-
мочевине и др.) наряду с донорно-акцепторой ст-связью возникает
и дативная, в которой Ni2+ является донором d-электронов, а атом
серы — акцептором (рис. 10). Большее сродство никеля к сере,
чем к кислороду, может быть объяснено тем, что атом кислорода
не может принимать электроны никеля в общее пользование, так
как у него нет энергетически выгодных вакантных d-орбиталей
(второй период).При помощи метода валентных связей объяснено строение и
многие свойства, особенно магнитные, большого числа комплек¬
сных соединений.§ 5. Гибридизация орбиталей. Пространственная
структура комплексного ионаПри использовании метода МО ЛКАО полагают, что МО возни¬
кает как линейная комбинация атомных орбиталей, причем в ком¬
бинации могут участвовать по нескольку орбиталей каждого атома.
Для применения метода валентных связей удобно представить,
что АО металла перед образованием связи переходят в гибриди-
зованное состояние, причем гибридным орбиталям соответствуют
линейные комбинации атомных орбиталей, «смешанных» в тех же со¬
отношениях, в которых они входят в образующиеся затем МО ком¬
плекса.Представление о гибридизации позволяет наглядно описать
геометрию образующейся молекулы, исходя из направленности
гибридных орбиталей. Некоторые типы гибридизаций являются
очень характерными и повторяются в многочисленных соединени¬
ях. Так, гибридизация sp приводит к возникновению из двух орби¬
талей вир (например, рх) двух «гибридных» орбиталей sp, электрон¬
ные облака которых имеют вид асимметричных гантелей и вытяну¬
ты вдоль оси х, образуя угол 180° друг, с другом. Доля участия
АО в гибридной орбитали (ГО) пропорциональна C2mk—квадрату
коэффициента в выражении (II. 3).— 30 —
Коэффициенты Cmh вычисляются с учетом требования симмет¬
ричности и ортонормированности Г’0. Так, при гибридизации sp3
гибридные орбитали имеют вид:Vi(s + Рх + Ру + Pz)¥2 = — (S + p, — Ру— Pz)
1Ъ = — (S — Рх —Р у + Pz)= — (S — Р.г + Ру — Pz)Рассмотрим электронное облако гибридной орбитали cp^.1+ (Рх + Ру + Pz)}¥1 =Так как волновые функции орбиталей рх, р
нальны векторам х, у и z, то их комбинации
шинам тетраэдра (рис. 11)
по биссектрисам октантов.Суммирование рх, ру и pz
дает орбиталь p-типа, нап¬
равленную вдоль биссект¬
рисы первого октанта. От¬
рицательная часть этой
гантели при сложении с
положительной s-функцией
уменьшится, а положитель¬
ная увеличится; гантель
станет ассиметричной (рис.12). Можно показать, что
электронные облака осталь¬
ных типов ГО также име¬
ют вид асимметричных ган¬
телей, иногда несколько ус¬
ложненных. При гибридизации spsу и pz пропорцио-
направлены к вер-Рис. 11.Вершины, к которым направлены
комбинации р-функцийи ds3 валентный угол равен 109,5°.
При гибридизации sp3d2 или d2sp3 шесть эквивалентных гибридныхGXРт+Р„+Р*spJРис. 12. Образование гибридной зр3-орбиталн— 31 —
орбиталей направлены к вершинам октаэдра, валентный угол со¬
ставляет 90°. При тригональной гибридизации sр2 одна из р-ор¬
биталей (например, рг) остается неизменной, а смешиваются ор¬
битали s, рх и ру. Получаются три гибридные АО, которые располо¬
жены в плоскости под углом 120° друг
относительно друга (рис. 13).При sp-гибридизации остаются не¬
изменными, например, орбитали ру и
рг, а смешиваются рх и s-орбитали.
В результате получаются две эквива¬
лентные орбитали, направленные под
углом 180° по отношению друг к дру¬
гу (рис. 14). Если в гибридизации
участвуют АО, ориентированные в од¬
ной плоскости, гибридные орбитали не
могут выйти из этой плоскости. По¬
этому при гибридизации dsp2, если d-
и обе р-орбитали лежат в одной плос¬
кости (например, dxy< рх и ру,) то че¬
тыре гибридные орбитали направле¬
ны к вершинам плоского квадрата.
Валентный угол равен 90°. В неко¬
торых случаях можно определить на¬
бор гибридизующихся орбиталей пос¬
ле исключения орбиталей, используе¬
мых на образование я-связи. Напри¬
мер, в молекуле СаН4 каждый атом
углерода отдает одну р-орбиталь на
образование я-связи, остальные ва¬
лентные орбитали углерода вступа¬
ют в sp2-гибридизацию. В ацетилене
два атома углерода соединены a-связью за счет гибридных sp-ор¬
биталей и двумя я-связями.У ионов металлов в комплексах рассматривают гибридизацию
вакантных орбиталей с образованием группу равноценных акцеп¬
торных орбиталей.Вид гибридизации и структура комплексного иона зависят
как от электронной структуры иона металла, так и от природы
лиганда. Например, в основном состоянии свободного иона никеля
распределение электронов следующее:JdПри взаимодействии ионов Ni2+ с С1 донорные Зр-орбитали
ионов хлора перекрываются с акцепторными зр3-гибридными— 32 —Рис. 13. Образование гиб¬
ридных $р2-орбиталейУГ~~Ъ-Рис. 14. Две вр-гибридные
орбитали
орбиталями иона Ni2+:3d Us Up[NiCl4]2'-ff “H" -ff ® (B©®sp3- гибридизация,
тетраэдрическое
строение комплекса(кружками обозначены гибридные орбитали, участвующие в обра¬
зовании связей)). У переходных металлов четвертого, пятого и
шестого периодов внутренними называют орбитали 3d, 4d и 5d,
внешними — 4d, 5d и 6d соответственно. В зависимости от исполь¬
зуемых орбиталей различают внутреннюю или внешнюю гибриди¬
зацию и внутриорбитальные или внешнеорбитальные комплексы.Если на всех внутренних орбиталях имеются электроны, то необ¬
ходимо ожидать внешней гибридизации. Так, для комплекса
[№(1МНз)6]2+ осуществляется гибридизация sp3d2. Комплекс име¬
ет октаэдрическое строение и парамагнитен:3d us Up ud[Nl(NH3)6]2Mf-ff-H-f-|- ® ®@) V'" ' /Внутренняя гибридизация может осуществляться, если часть
внутренних d-орбиталей центрального иона или свободна, или
может освободиться за счет принудительного спаривания электро¬
нов. Так, для комплекса [Cr(CN)6]3_3d Us Up
Cr3+ -f-f-f — И™)в]3--(- + +Ф® ®Vгибридные вакантные орбитали хрома d2sp3 взаимодействуют с
неподеленными электронными парами цианид-иона, образуя МО
химической связи. Для комплекса [Ni(CN)4]2_ за счет принудитель¬
ного спаривания d-электронов осуществляется внутренняя гибри- *
дизация dsp2. Комплекс имеет плоское строение и диамагнитен:3d us up[Ni(CN)4]2_4f-ff -И- -Н-@) @ —> v *Можно считать, что при образовании комплекса Ni(CO)4 сна¬
чала 45-электроны атома никеля переходят на Зс^-орбитали и спа¬
риваются с имеющимися там неспаренными Зс?-электронами, за¬
тем происходит гибридизация sp3 и образуются донорно-акцептор-2—699— 33 —
ные связи за счет донорных электронных пар молекулы СО:3d ks k-pNKCO),-Ц--fj--f*-#-fr ф фф®Для образования двух или более эквивалентных связей могут
быть использованы только определенные комбинации орбиталей
центрального атома, которые зависят от пространственного рас¬
положения связей, т. е. от симметрии молекулы, и могут бытьопре-CXJ-dsp2, л'вадрат1iк/sd3} тетраэдрdsp3, тригональная dsp3: кдадратная
бипирамида пирамидаРис. 15. Гибридизация с использованием d-орбиталейТаблица 2Виды гибридизации орбиталей центрального ионаК. Ч.Гибридные орбита¬
ли центрального
ионаПространственная конфигу¬
рация гибридных связейПримеры2spПрямая линия[Ag(CN)2]-, [Ag(NH3)2]+3sp°-Равносторонний тре¬
угольникNOJT4sp3, d3sТетраэдрnh+,bf4-, Сг024-4dsp2Квадрат[Ni(CN)4r, '[Cu(NH3)4r,
[Pl(NH,)4]2+6d2sp3Октаэдр[Co(NH3)e]3+, [Fe(CN)6]3_,
[Cd(NH3)6]3+, SF65dsp3Тригональная бипи¬
рамида; квадратная пи¬
рамидаFe(CO)6, [CuCl6]3~— 34 —
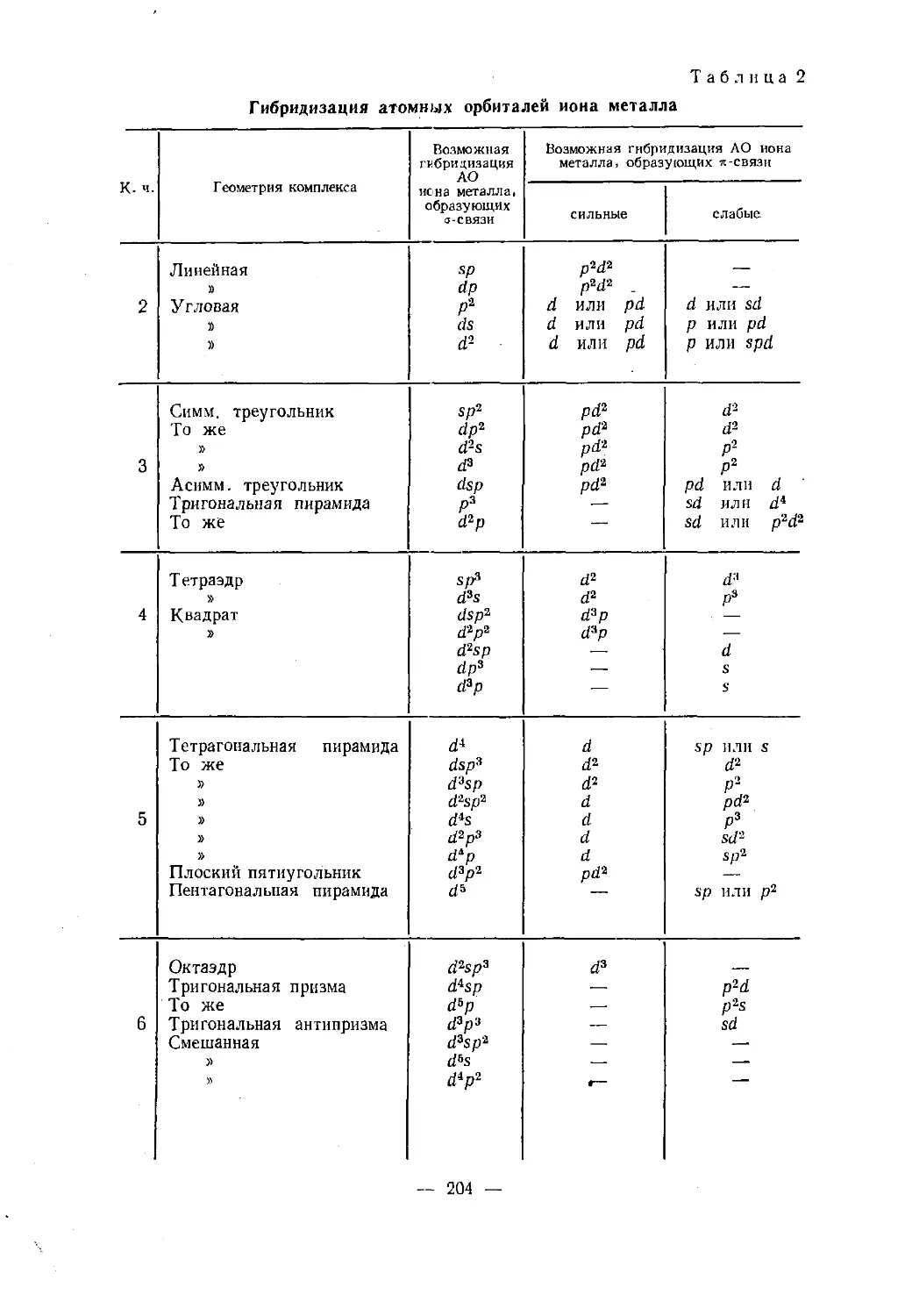
делены методами теории групп. Возможные способы гибридизации
при образовании сг- ия-связей для различных типов симметрии при¬
ведены в табл. 2 приложения. Наиболее часто встречающиеся ви¬
ды гибридных орбиталей и конфигурации образующихся комплек¬
сов приведены в табл. 2 и на рис. 15. Стрелки показывают направ¬
ления, по которым ориентируется большая часть электронного
облака гибридных орбиталей.Метод валентных связей позволяет предвидеть магнитные свой¬
ства комплексов. Так, он указывает на парамагнетизм комплек¬
сов [NiCl 4 ]2_ и [Ni(NH з) 6 ]2+ и диамагнетизм комплекса
[Ni(CN) 412-, что подтверждается экспериментом. Этот метод по¬
зволяет предсказать, что реакции замещения лигандов прохо¬
дят быстро у внешнеорбитальных комплексов. Расчет электронного
строения комплексов, а также анализ и предсказание их спектров
при помощи метода валентных связей затруднены.§ 6. Основные типы конфигураций внутренней координационнойсферыРаботы по синтезу комплексных соединений повлекли за со¬
бой многочисленные структурные исследования, в результате ко¬
торых определяются координационное число иона металла, форма
полиэдра (координационного многогранника), межатомные рас¬
стояния и валентные углы.Конфигурация комплекса зависит от электронного строения
иона металла и лиганда, от взаимодействий металл—лиганд и ли¬
ганд—лиганд и от геометрических свойств лигандов.В методах приблизительного предсказания конфигурации ком¬
плексов обычно рассматривают только один из факторов: или схему
гибридизации АО центрального иона, или схему расталкивания
электронов связывающих МО. При использовании метода валент¬
ных связей рассматривают гибридизацию АО иона металла, при¬
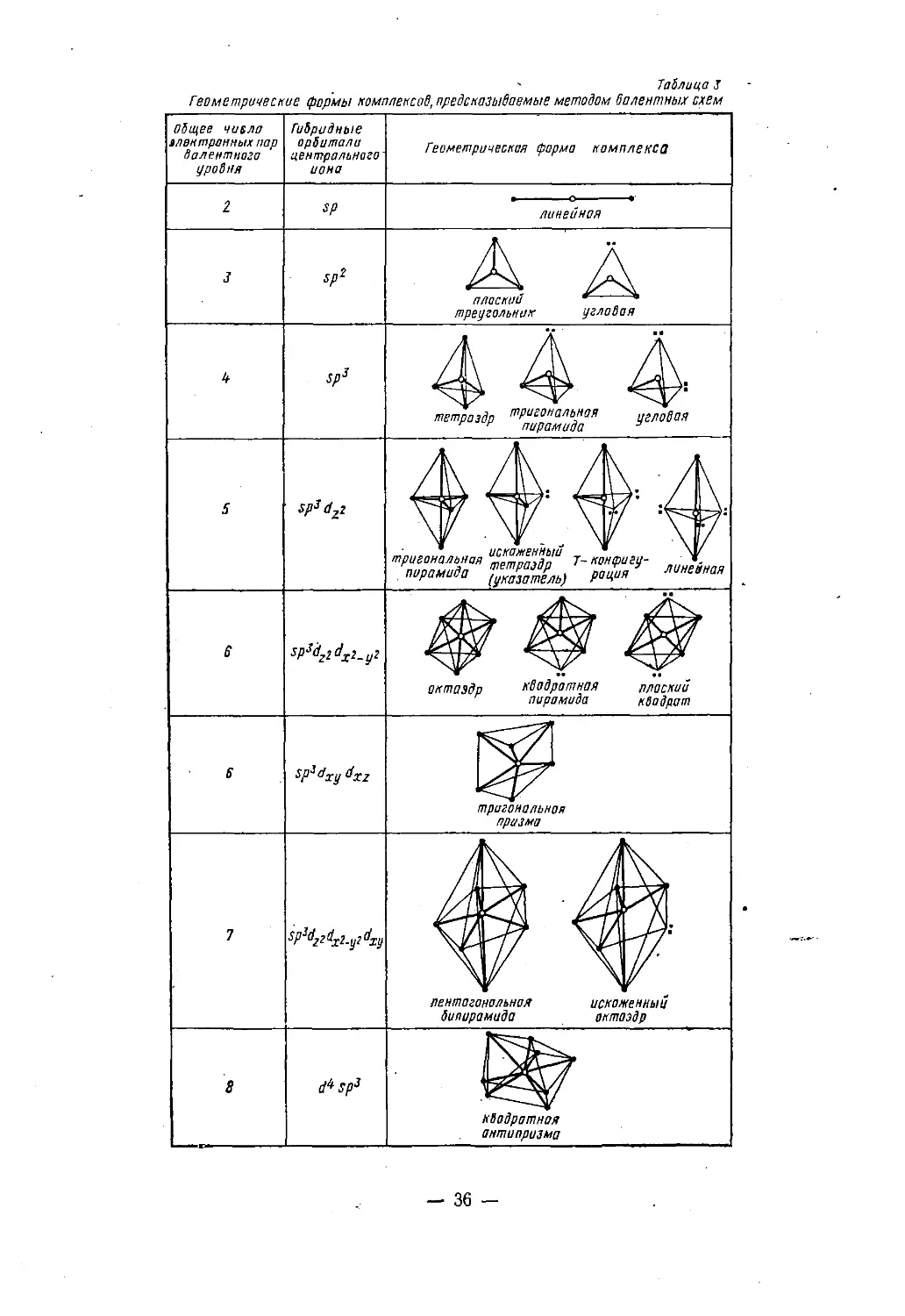
чем учитывают неэквивалентность d-орбиталей (табл. 3).Например, при пяти парах валентных электронов и гибриди¬
зации sp3dX!-yi должна реализоваться конфигурация квадратной
пирамиды с ионом металла в центре, а при гибридизации sp3dzг
конфигурация тригональной бипирамиды. Для шести электронных
пар гибридизация sp3dx2_у2 dZ2 соответствует октаэдру, а гибриди¬
зация sp3dxydyz — тригональной призме.Конфигурации некоторых молекул объясняют при помощи бо¬
лее простых моделей.Стереохимия соединений непереходных элементов. Согласно
концепции Джиллеспи форма комплекса зависит от общего числа
валентных электронных пар центрального атома, числа неподе-
ленных электронных пар и электроотрицательности элементов,
образующих координационный многогранник. Основные положе¬
ния этой концепции: 1) валентные электронные пары находятся в
среднем на одинаковом расстоянии от ядра; 2) поведение электрон-2*— 35 -
Таблица J— 36 -
ных пар моделируется при помощи системы частиц, передвигаю¬
щихся по поверхности сферы. Каждая частица соответствует элек¬
тронной паре. Частицы взаимно отталкиваются; сила отталкивания F
пропорциональна 1 /гп(п ^ 2). Наиболее выгодное энергетически *
взаимное расположение частиц, соответствующее обычно макси¬
мальному взаимному удалению, отвечает геометрии комплекса
(табл. 4). •Таблица 4Равновесное распределение одинаковых частиц на
сфере при силе взаимодействия, пропорциональной 1/лп(я > 2)Число частицГеометрическая форма22А.Линейная*3ЗА. Равносторонний треугольник44А.Тетраэдр55А. Тригональная билирамида66А.Октаэдр77С.Пентагональная бипирамида (п = 2)88В.Квадратная антипризма99А.Трипирамида* См. стр. 10.Как прип =2 (чисто кулоновское отталкивание, F ~ Mr2), так
и при п > 2 получаются одни и те же конфигурации, за исклю¬
чением многогранника с семью электронными парами. Для него
при п = 2 получается пентагональная бипирамида (осуществляется
у IF7, UF7 , U02F5), при п >- 6 — неправильный октаэдр с рас¬
положением седьмой электронной пары против середины одной
из граней октаэдра (реализуется у окислов La, Се, Рг, Nd), а при
2 < п < 6 — тригональная призма с положением седьмой пары
электронов против одной из прямоугольных граней (у TaFf ,NbFT).Приведенные в табл. 3 и 4 геометрические конфигурации иска¬
жаются в двух случаях: во-первых, при неравноценности лигандов;
во-вторых, при наличии стереохимически активной неподеленной
электронной пары. Если центральный ион имеет в валентном слое
свободную электронную пару, то у переходных элементов такая
пара, обычно принадлежащая d-подуровню, не участвует в гибри¬
дизации и не влияет на геометрию комплекса. Так, и Ti3+ и Со3+
дают октаэдрические комплексы, хотя Со3+ имеет на пять электро¬
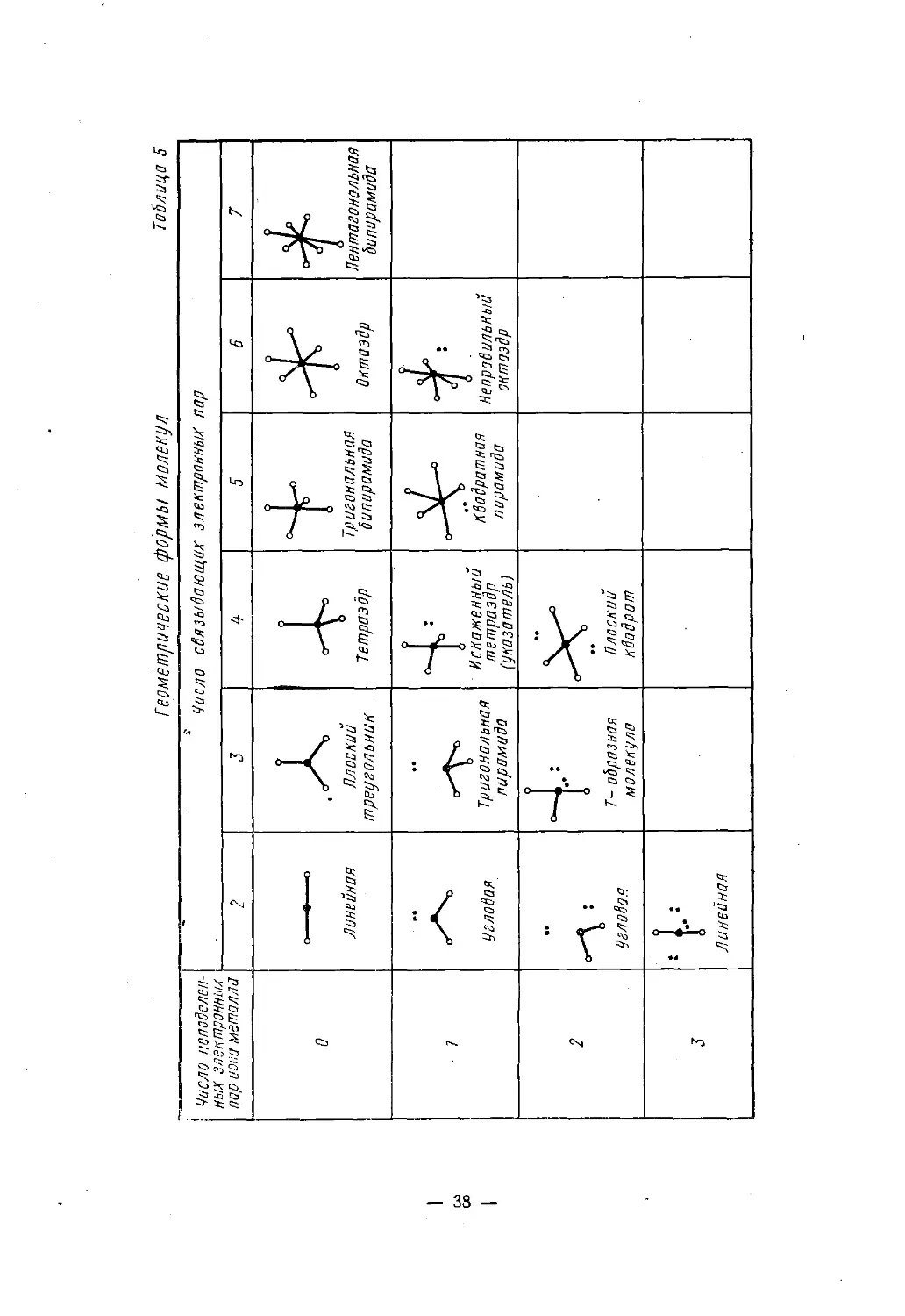
нов больше.У непереходных элементов неподеленная пара участвует в гибри¬
дизации и занимает место в вершине фигуры, описанной в табл. 3— 37 —
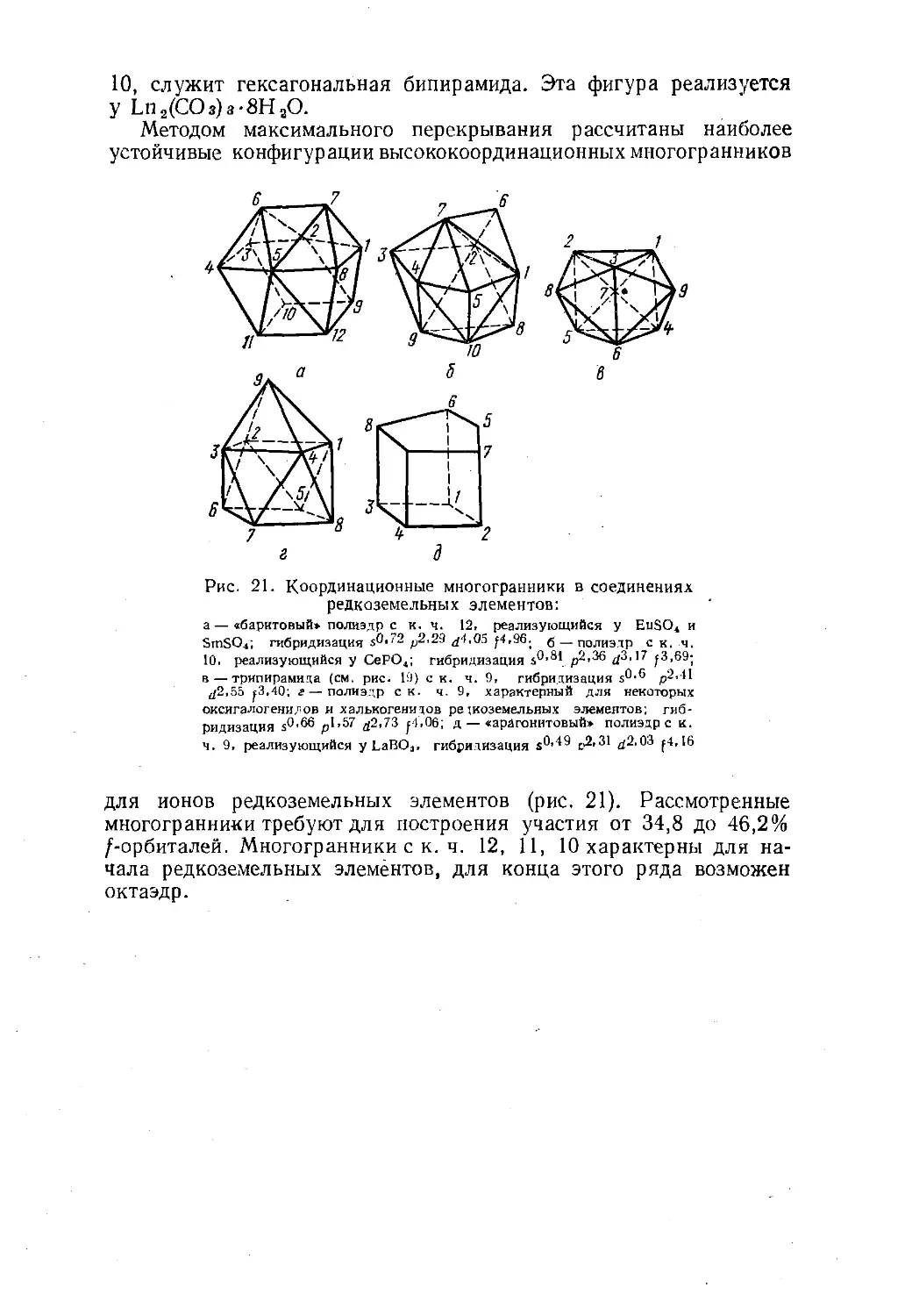
Геом'етричвские формы молекул Таблица 5— 38 —
или 4, т. е. является стереохимически активной*. Поскольку против
такой пары не расположен лиганд, то симметричная фигура иска¬
жается (табл. 5).Так, увеличивая при общем числе электронных пар, равном
пяти, число неподеленных пар от нуля до трех, можно перейти от
тригональной бипирамиды к искаженному тетраэдру («указателю»),
затем к Т-образной и, наконец, к линейной молекуле. Центральный
атом с двумя электронными парами в валентном слое образует
линейные молекулы или ионы. При трех электронных парах, если
все они дают a-связи, реализуется треугольник с углами 120° меж¬
ду связями, а если одна из электронных пар не поделена, то обра¬
зуется угловая молекула.Свободные электронные пары приводят к искажению конфигу¬
раций. Как правило, максимально отталкиваются друг от друга
неподеленные (донорные) электронные пары: слабее — неподелен-
ная и связывающая и еще слабее — связывающие пары. У квадрат¬
ной пирамидыцентральный атом выводится из плоскости квадрата вниз', а непо-
деленная пара располагается под основанием пирамиды. Отталки¬
вание неподеленной и связывающей пар приводит к излому «ука¬
зателя»Ои Т-образной формыКроме того, у указателя сближаются экваториальные лиганды.
Например, HOHSb2F72- в Cs2Sb2F7 состоит из двух таких изло¬
манных указателей SbF4, сросшихся вершиной. Угол FaKCSbFaKc
у них 150° вместо 180°, а угол F3Kl)SbF3KB 90,5° вместо 120°.* Если в комплексе шесть лигандов, то это правило нарушается: так,
ионы SeBre2-, SbBre3_ и BiCle3~ являются октаэдрическими, несмотря на на¬
личие у Se4+, Sb3+ и Bi3+ донорной электронной пары.— 39 -координируются атомом ртути, а атомы азота •— атомами меди/<-. 1 О £Г\ ТТ ТТ -.С' / n г J\ 1,110
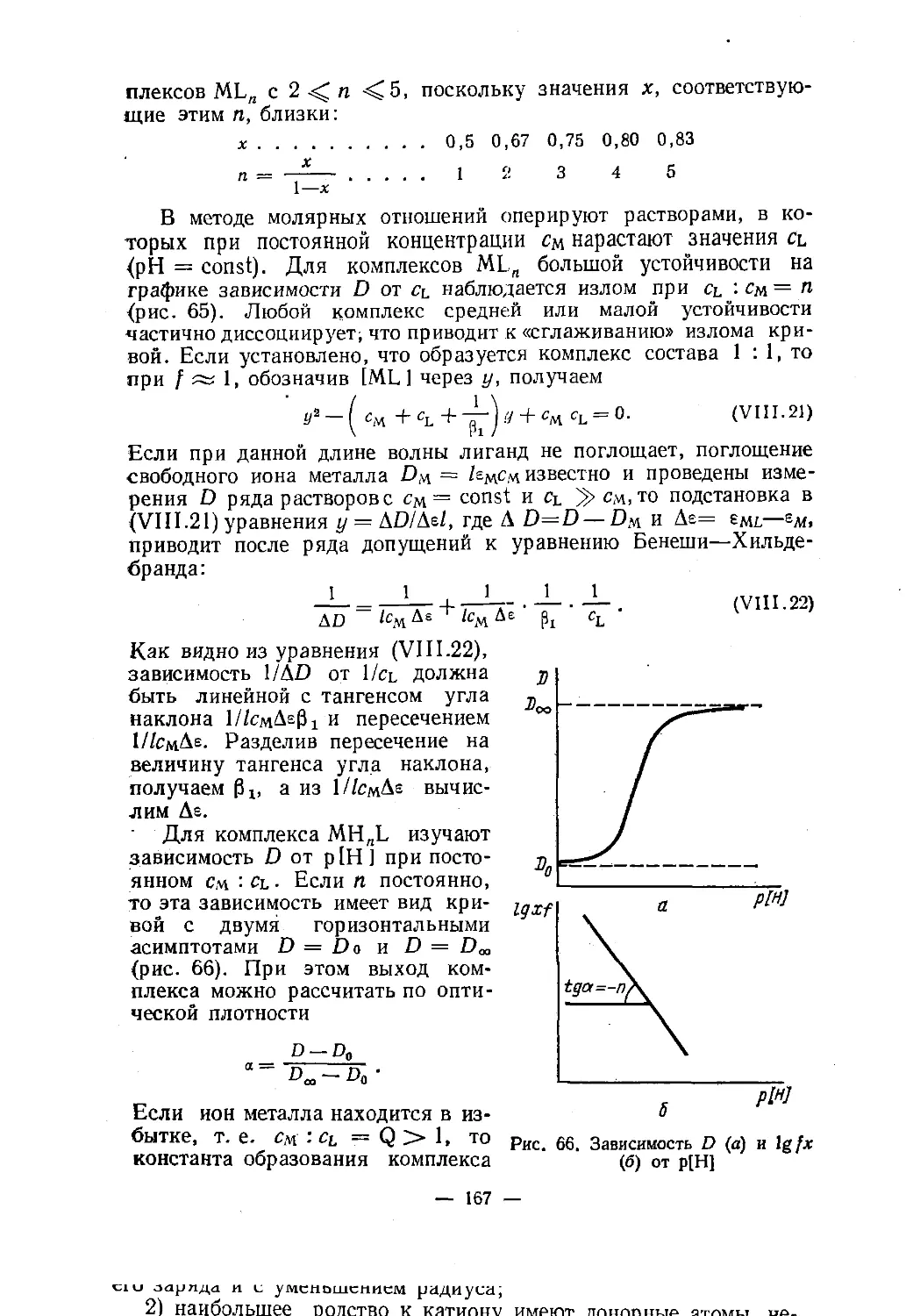
У тригональной бипирамиды связи, как правило, неравноцен¬
ны. В РС15 и SbCl 5 аксиальные (ориентированные вдоль оси 2,
рис. 16) связи длиннее экваториальных. На¬оборот, в CuCl53_, CdCl53-, Ni(CN)53- и Fe(CO) 5Рис. 16. Аксиальные
и.экваториальные связи
в тригональной бипи¬
рамидеаксиальные связи короче.Стереохимия переходных элементов. Кон¬
цепция Джиллеспи неудовлетворительна для
переходных элементов, так как не учитыва¬
ет разнообразия дативных я-связей, связей
металл — металл и многоцентровых связей, в
комплексах этих элементов. У переходных
элементов пятого и шестого периодов наблю¬
дается увеличение к. ч. сверху вниз в каждой
подгруппе и от Pd—Pt к У—La:ЭлементыК. и.Pd, Pt6,4Rh, Ir6(4)*Ru, Os6(4,5)Те, Re6(7, 8, 9)Mo, W6,9Nb, Та7,6(8, 9)Zr, Hf8,7(6)Y, La8,9(7, 6, 10,12)В этом направлении возрастает также степень ионности свя¬
зей в комплексных ионах, увеличивается радиус иона металла
и число вакантных мест на d-орбиталях. Для элементов четверто¬
го периода от хрома до цинка характерны к. ч. 6 (октаэдр) и к. ч. 4:
тетраэдр реализуется у высокоспиновых комплексов и у низкоспи¬
новых с кратными связями, квадрат — у низкоспиновых комплек¬
сов с конфигурацией центрального иона металла d* (Ni2+, Pd2+,
Pt2+).У ионов с конфигурацией d10(Zn2+, Cd2+, Cu+, Ag+, Hg2+) наблю¬
даются различные к. ч. Для цинка характерны как тетраэдричес¬
кие (Csa[ZnCl4], KtZn(H20)Cl3], Znpy2Cl2), так и октаэдрические
(ZnCl2-2A, где А — бидентатный лиганд) комплексы. В ZnCl2-2A
четыре связиZnдополняются двумя связями Zn—Cl, которые длиннее связей Zn—Cl
в тетраэдрических комплексах, и по-видимому, отличаются боль¬
шей ионностью.Ион Cd2+ в большинстве исследованных структур имеет окта¬
эдрическое окружение. Октаэдрические комплексы цинка и кад¬
мия, например Zn(N2H4)2Cl2 и Cd(N2H4)2Cl2, часто изоструктурны.В скобках приведены реже встречающиеся к. ч.— 40 —
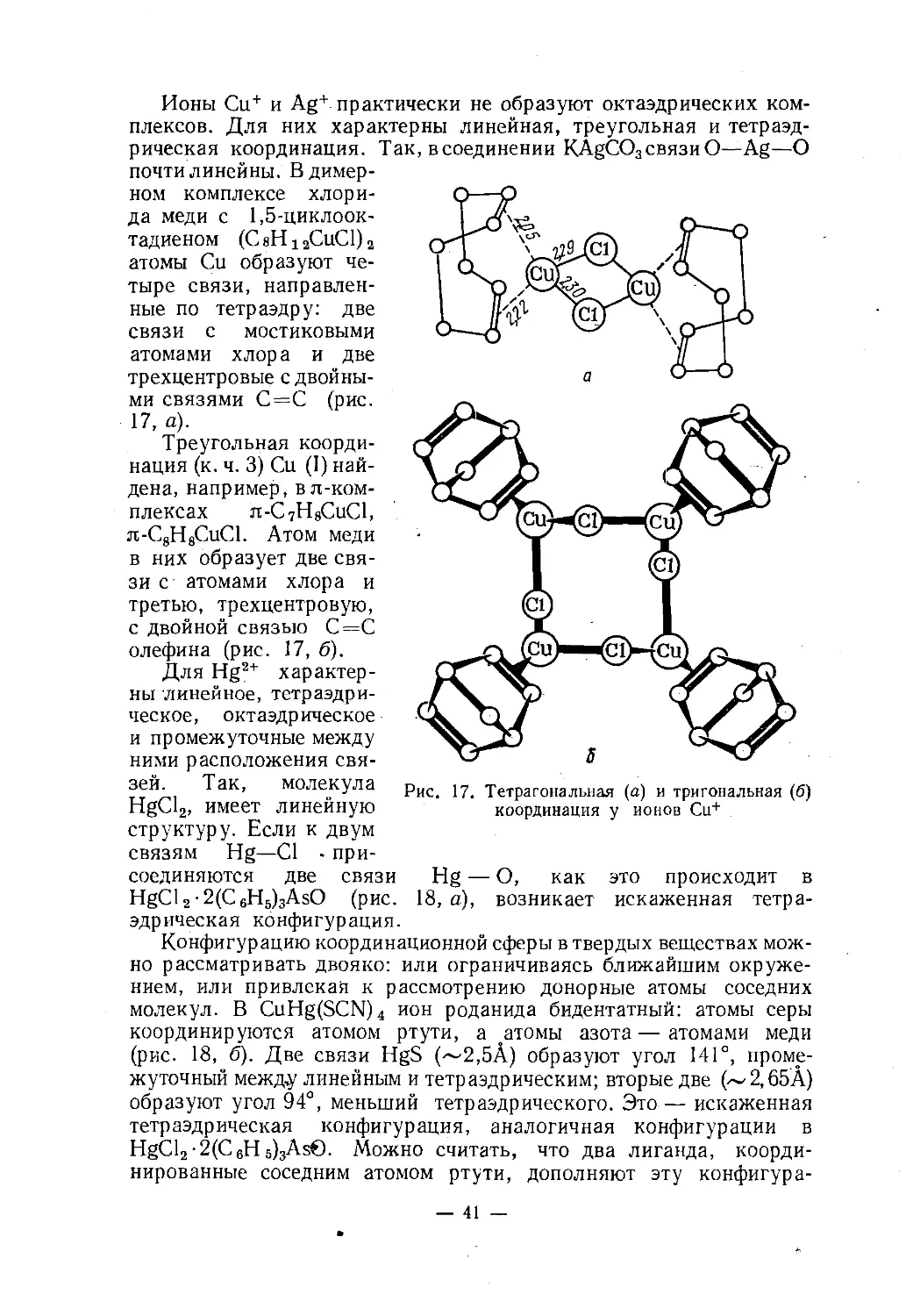
Ионы Cu+ и Ag+ практически не образуют октаэдрических ком¬
плексов. Для них характерны линейная, треугольная и тетраэд¬
рическая координация. Так, в соединении KAgC03 связи О—Ag—О
почти линейны. В димер¬
ном комплексе хлори¬
да меди с 1,5-циклоок¬
тадиеном (С8Н12СиС1)2
атомы Си образуют че¬
тыре связи, направлен¬
ные по тетраэдру: две
связи с мостиковыми
атомами хлора и две
трехцентровые с двойны¬
ми связями С=С (рис.17, а).Треугольная коорди¬
нация (к. ч. 3) Си (I) най¬
дена, например, вл-ком-
плексах я-С7Н8СиС1,
я-С8Н8СиС1. Атом меди
в них образует две свя¬
зи с атомами хлора и
третью, трехцентровую,
с двойной связью С=С
олефина (рис. 17, б).Для Hg2+ характер¬
ны линейное, тетраэдри¬
ческое, октаэдрическое
и промежуточные между
ними расположения свя-тт^Ач Так> молекула pHCi Тетрагональная (а) и тригональная (6)
HgCl2, имеет линеиную координация у ионов Си+структуру. Если к двум
связям Hg—Cl - при¬
соединяются две связи Hg — О, как это происходит в
HgCl2-2(C6H5)3AsO (рис. 18, а), возникает искаженная тетра¬
эдрическая конфигурация.Конфигурацию координационной сферы в твердых веществах мож¬
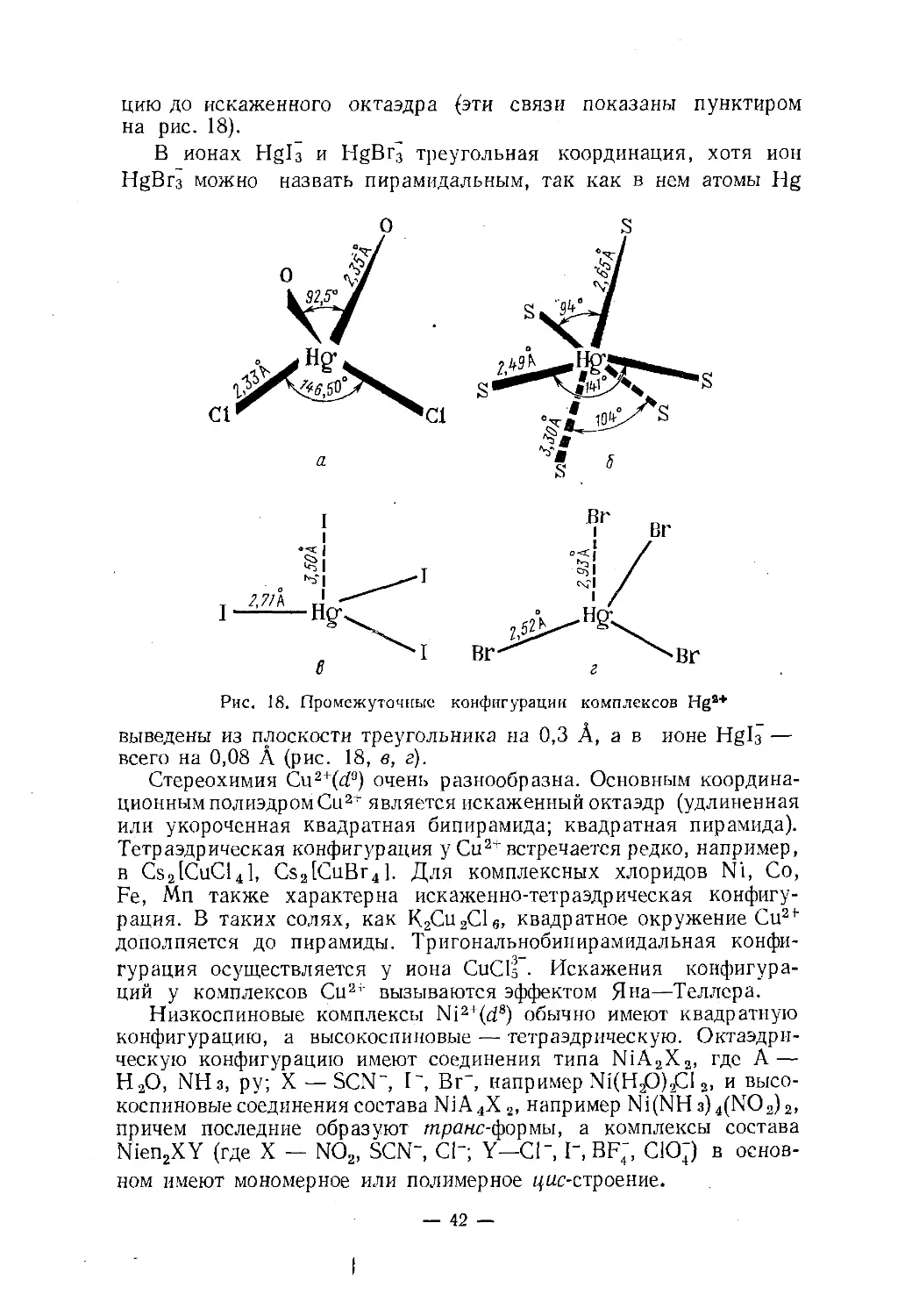
но рассматривать двояко: или ограничиваясь ближайшим окруже¬
нием, или привлекая к рассмотрению донорные атомы соседних
молекул. В CuHg(SCN)4 ион роданида бидентатный: атомы серы
координируются атомом ртути, а атомы азота — атомами меди
(рис. 18, б). Две связи HgS (~2,5А) образуют угол 141°, проме¬
жуточный между линейным и тетраэдрическим; вторые две (~ 2,65А)
образуют угол 94°, меньший тетраэдрического. Это — искаженная
тетраэдрическая конфигурация, аналогичная конфигурации в
HgCl2-2(C6H5)3As0. Можно считать, что два лиганда, коорди¬
нированные соседним атомом ртути, дополняют эту конфигура¬— 41 —
цию до искаженного октаэдра (эти связи показаны пунктиром
на рис. 18).В ионах Hgl3 и HgBr3 треугольная координация, хотя ион
HgBr3 можно назвать пирамидальным, так как в нем атомы Hgвыведены из плоскости треугольника на 0,3 А, а в ионе Hgb —
всего на 0,08 А (рис. 18, в, г).Стереохимия Си2+(d9) очень разнообразна. Основным координа¬
ционным полиэдром Си2+ является искаженный октаэдр (удлиненная
или укороченная квадратная бипирамида; квадратная пирамида).
Тетраэдрическая конфигурация у Си2+ встречается редко, например,
в Cs2[GuCl4], Cs2[CuBr4], Для комплексных хлоридов Ni, Со,
Fe, Мп также характерна искаженно-тетраэдрическая конфигу¬
рация. В таких солях, как К2Си2С1в, квадратное окружение Си2+
дополняется до пирамиды. Тригональнобипирамидальная конфи¬
гурация осуществляется у иона CuCls . Искажения конфигура¬
ций у комплексов Си2+ вызываются эффектом Яна—Теллера.Низкоспиновые комплексы Ni2+(d8) обычно имеют квадратную
конфигурацию, а высокоспиновые — тетраэдрическую. Октаэдри¬
ческую конфигурацию имеют соединения типа №А2Х2, где А —
Н20, NHa, ру; X — SCN", Г, Вг~, например №(Н20),С12, и высо¬
коспиновые соединения состава №А4Х 2, например Ni(NH3)4(N02)2,
причем последние образуют транс-формы, а комплексы составаОSаЯS5IвгI6вггРис. 18. Промежуточные конфигурации комплексов Hga+Nien2XY (где X — N02, SCN~, Cl"; Y—СГ, I“, BF", СЮ') в основ¬ном имеют мономерное или полимерное ^ис-строение.— 42 —
В комплексах Со2+(сР) состава СоХ"-2, где X — CN-, N02_,
SCN-, F-, С1_, Вг-, полярность связи повышается при переходе от
иона CN~ к иону F-. Лиганды, помещен¬
ные в этом ряду правее нитрит-иона, обра¬
зуют высокоспиновые, комплексы. В от¬
личие от Ni2+ у низкоспиновых соедине¬
ний Со2+ преобладает не квадратная, а ок¬
таэдрическая конфигурация, среди высо¬
коспиновых — не октаэдрическая, а тет¬
раэдрическая.Стереохимия редкоземельных элемен¬
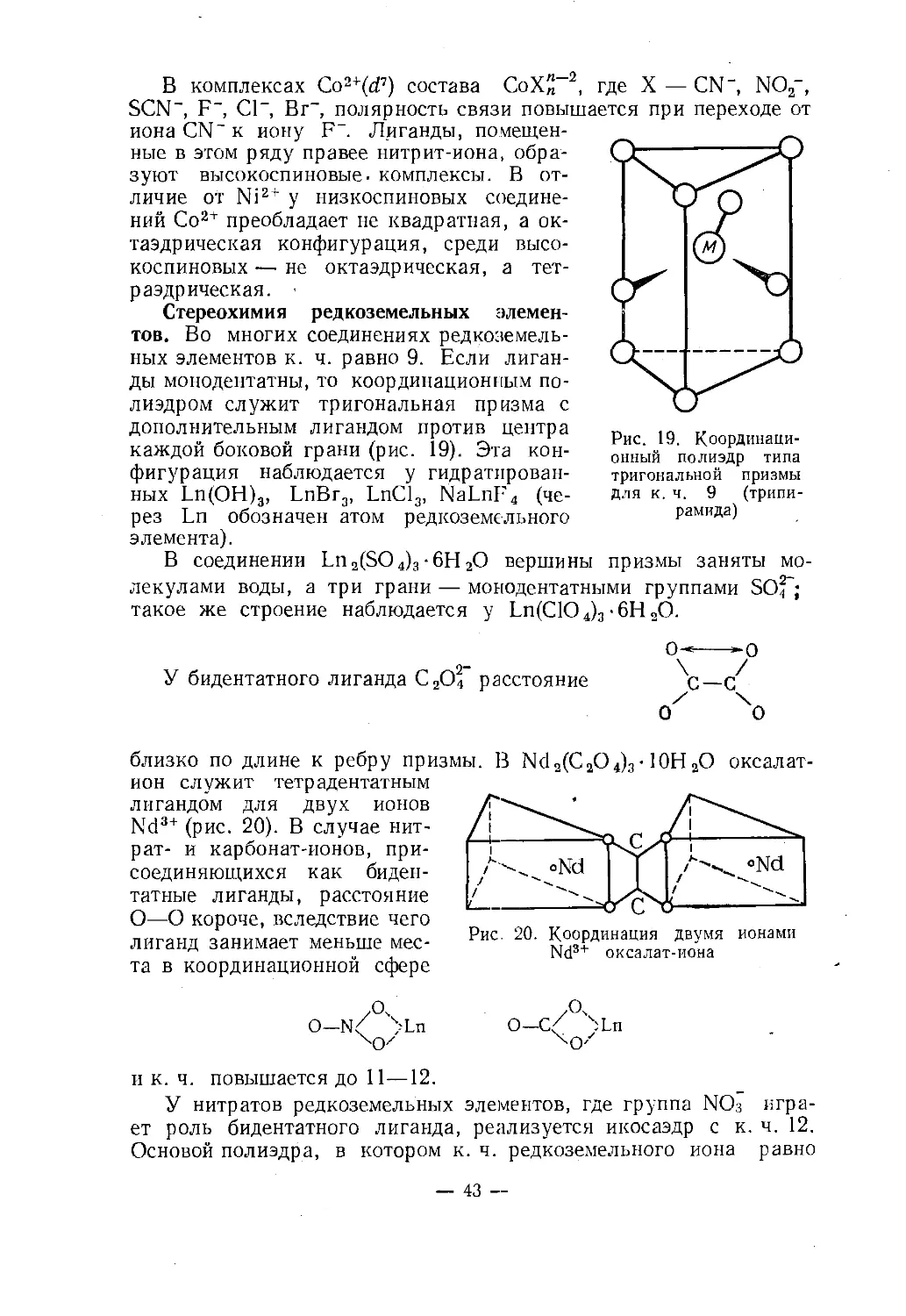
тов. Во многих соединениях редкоземель¬
ных элементов к. ч. равно 9. Если лиган¬
ды монодентатны, то координационным по¬
лиэдром служит тригональная призма с
дополнительным лигандом против центра
каждой боковой грани (рис. 19). Эта кон¬
фигурация наблюдается у гидратирован¬
ных Ln(OH)3, LnBr3, LnCl3, NaLnF4 (че¬
рез Ln обозначен атом редкоземельного
элемента).В соединении Ln2(S04)3-6H20 вершины призмы заняты мо¬
лекулами воды, а три грани — монодентатными группами SOf;
такое же строение наблюдается у Ln(C104)3-6H20.Рис. 19. Координаци¬
онный полиэдр типа
тригональной призмы
для к. ч. 9 (трипи-
рамида)0-У бидентатного лиганда С2С>4 расстояние—ОW(/ чоблизко по длине к ребру призмы. В Nd2(C204)3-ЮН20 оксалат-
ион служит тетрадентатным
лигандом для двух ионов
Nd3+ (рис. 20). В случае нит¬
рат- и карбонат-ионов, при¬
соединяющихся как биден-
татные лиганды, расстояние
О—О короче, вследствие чего
лиганд занимает меньше мес¬
та в координационной сфере£—1 КС А^ 1 ^1Г 1/\ oNd°Nd/ 4V.// ^гсЧРис. 20. Координация двумя ионами
Nd3+ оксалат-иона/°-О—N< >Ln
ХСК0—С( \'Ln
хОи к. ч. повышается до 11—12.У нитратов редкоземельных элементов, где группа ЫОз игра¬
ет роль бидентатного лиганда, реализуется икосаэдр с к. ч. 12.
Основой полиэдра, в котором к. ч. редкоземельного иона равно— 43 —
10, служит гексагональная бипирамида. Эта фигура реализуется
у Ьп2(СОз)з-8НаО.Методом максимального перекрывания рассчитаны наиболее
устойчивые конфигурации высококоординационных многогранниковРис. 21. Координационные многогранники в соединениях
редкоземельных элементов:
а—«баритовый* полиэдре к. ч. 12, реализующийся у EuS04 и
SmS04; гибридизация р2,29 ^4,05 f4,96. g — полиэдр с к. ч.10, реализующийся у СеР04; гибридизация &0.S1 р2,36 ^3,17 ^3,69;
в — трипирамица (см. рис. 19) с к. ч. 9, гибридизация s^.6 р2,41
^2,55 ^3,40; г — полиэдр с к. ч. 9, характерный для некоторых
оксигалогени^ов и халькогеницов ре поземельных элементов; гиб¬
ридизация pi.57 д2,73 ^4,06; д — «арагонитовый» полиэдре к.ч. 9, реализующийся yLaBOa. гибридизация d^I ^2.0^ f4.16для ионов редкоземельных элементов (рис. 21). Рассмотренные
многогранники требуют для построения участия от 34,8 до 46,2%
/-орбиталей. Многогранники с к. ч. 12, 11, 10 характерны для на¬
чала редкоземельных элементов, для конца этого ряда возможен
октаэдр.
111. РЕАКЦИИ КОМПЛЕКСНЫХ ЧАСТИЦ§ 1. Основные типы реакций комплексных частицРеакции комплексных частиц можно разделить на четыре ос¬
новные группы:1. Реакции присоединения, замещения или отщепления ли¬
ганда.2. Реакции связанного лиганда.3. Реакции изомеризации (в том числе рацемизации).4. Окислительно-восстановительные реакции.,К первой группе относятся реакции:1) присоединения лиганда к изолированному атому или ионуМ + nL = ML„напримерNi + 4СО == Ni(CO)i
и присоединения лиганда к комплексной частицеMLm + nA = MA„Lmнапример реакции плоского фосфинного комплекса иридия (0)
с СОсо(Сснр,р^сн—с^Р(анл. . W2p^CH3~^p(c6H5)a/ Ir / + СО / Ir /(C6H5)2PNch_Ch/P(C6H5)2 (c6h,,)2pNch_cH/P(c6h5)2в результате которой образуется пирамидальный комплекс;2) замещения лиганда с разрывом связи ядро—донорный атом.
К ним относятся:а) реакции непосредственного замещенияML„ + тк = ML„_mAm + mLб) реакции обмена лигандами между двумя комплексамиML„ + M'Lm = ML„_ гА; + M'LjAm_; (I)илиML„ + M'Am = МЬ„_гАг + M'Am_; + /L (II)Примером могут служить реакции обмена ионом металла между
комплексамиМА„ + M'Lm = MLm + М'А„в) реакции полимеризации комплекса с возникновением лй-
гандного мостикаML„ + M'Am = L„.1M-A-M'Am_1 + L- 45 -
г) рэахции лигандного диспропорционированияz ML„ = a ML; + 6 MLy
причем а + Ь = г и ai +■ bj = /гг;д) реакции замещения, сопровождаемые окислительно-восста¬
новительной реакцией, не затрагивающей ст-связей лиганда;е) реакции замещения, сопровождаемые реакциями между ли¬
гандами, в результате чего получаются новые лиганды, отличающие¬
ся от исходных. Примером реакции непосредственного замещения
является акватация[Co(NH3)6Cl]2+ + Н20 [Co(NH3)6H20]3+ + Cl-Утверждать, что реакция идет по механизму обмена лигандами
(I) или (II), можно только в том случае, если соединение МА не
диссоциирует, т. е. лиганды А в виде самостоятельных частиц в
растворе не существует. Так, при кипячении раствора дибромо-
(тетрафенилциклобутадиен)палладия (ТФЦБ) PdBr2 с Fe(CO)5
в ксилоле в атмосфере азота образуется (ТФЦБ) Ре(СОз), хотя
ТФЦБ в свободном состоянии в растворе не существует. Сдругой
стороны, несмотря на то что реакция[Fe(H20)6]3+ +[ Co(H20)BSCN] + [Fe(H20)5SCNp+ + [Co(H20)ep+ (III)записана в форме (I), она относится к реакциям непосредственного
замещения. Ионы [Со(Н 20) 5SCN ]+ в растворе сильно диссоцииро¬
ваны и фактическое протекание реакции (III) совпадает с кинети¬
кой процесса[Fe(H20)ela+ + SCN' ^ [Fe(H20),SCN]2+ + Н20Примером димеризации комплексов и лигандного диспропор¬
ционирования могут служить следующие реакции:2Fe(CO)B = Fe2(CO)9 + СО
3Fe2(CO)fl = Fe3(CO)12 + 3Fe(CO)5При взаимодействии дифенилацетилена с некоторыми низко¬
спиновыми комплексами образуются металлорганические соедине¬
ния, лигандом в которых являются дифенилэтилен, ТФЦБ, или
гексафенилбензол. Примером реакции замещения, которая ослож¬
нена дальнейшими превращениями лигандов, служит реакция/\
Образование молекул ТФЦБ и гексафенилбензола облегчается
благоприятной ориентацией, которую занимают во внутренней
координационной сфере относительно друг друга первоначально
координируемые молекулы дифенилацетилена.Частицей — «носителем» лиганда ;з реакциях замещения может
быть не только комплекс (например, МАт), но и некомплексная
частица, при расщеплении которой возникает лиганд. Так, при
реакции(R3P)2PdC!2 + 2CH3Li = (R3P)2Pd(CH3)2 + 2LiClгруппаCH3образуется изСНДл, а при восстановлении Мп2 (СО)10
водородом получается НМп(СО)5.Ко второй группе реакций (т. е. к реакциям связанного лиган¬
да) относятся:1) реакции замещения или присоединения у донорного атома
лигандаМ—D—X + Y = М—D—Y + X (IV)I IR R2) реакции отщепления частицы лиганда, содержащей донорный
атом, причем остаток лиганда связываетсяМ—D—Х=М—R + DX (V)IRилиМ—D—R + D'R' = М—D'—R + DR'а также обратные им реакции внедрения;3) реакции, на первых стадиях которых донорный атом не за¬
трагивается.Примерами реакций связанного лиганда могут служить конден¬
сация дихлородигидроксиламинпалладия (II) с диацетилом:сн3 он сн3
СК /NIZZo]=C С1Ч /N=G^Pd4 + | = | + 2H,0Cl^ N|H2 Ol=r: СК ^N=0 • 2L i •' 1он сн3 он сн3При этой реакции, которая относится к типу (IV), получается
комплекс диметилглиоксима.Уравнением типа (V) описывается обратимая реакция разло¬
жения ацетилпентакарбонила марганца:СН3—С—Mn(CO)5 СН3—Mn(CO) j -f- соIIо— 47 —
Если при нагревании удалять СО, то равновесие сдвигается
вправо,а при давлении окиси углерода 35 атм равновесие сдвига¬
ется влево — протекает реакция внедрения.Одной из реакций координированных неорганических лигандов,
не затрагивающей донорный атом, является отщепление протона.
Так, кислотные свойства аммиачных комплексов Pt(IV), которые
ведут себя как слабые кислоты, близкие по силе к борной, обус¬
ловлены протеканием реакций типа[Pt(NH,)e]‘* [Pt(NH3)6NH2r+FPа гидролиз инертных акво-ионов — отщеплением протона от коорди¬
нированной молекулы воды:[Сг(Н20)6]3' [Сг (Н20)л0Н]2+ + Н+Реакции органических координированных лигандов, не затра¬
гивающие донорного атома, например сульфирование циклопен-
тадиенила в ферроцене или ацетилирование ацетилацетона в его
комплексах, протекают как обычные реакции в органической хи¬
мии. Так как координированные лиганды отличаются от свободных
распределением электронной плотности и конфигурацией, то ста¬
новится возможным протекание реакций, не характерных для сво¬
бодных лигандов.§ 2. Лабильные и инертные комплексные ионыПонятие о лабильности и инертности. Комплексные ионы де¬
лятся на лабильные и инертные. У лабильных ионов реакции заме¬
щения проходят быстро, т. е. лабильные ионы находятся со средой
в динамическом равновесии. У инертных комплексных ионов реак¬
ции замещения лигандов протекают медленно и поэтому они медленно
реагируют на изменение условий в системе. Инертные ионы могут
участвовать в быстрых реакциях отщепления частей лигандов, оса¬
ждения, ассоциации и окисления—восстановления, которые будут
рассмотрены отдельно.Различие между инертными и лабильными комплексами на¬
ложило глубокий отпечаток на хиМию координационных соедине¬
ний в растворах, поскольку методы изучения обоих классов ионов
различны.Понятия «инертный» и «лабильный» относятся к области кине*
тики и их нельзя смешивать с выражениями «устойчивый» и «не¬
устойчивый», которые определяют термодинамическую устойчи¬
вость. Так, инертный комплексный ион [Со(ЫНз)6]3+ в кислой
среде термодинамически неустойчив: константа равновесия[Co(NH3)e]3f +6Н30' ^ [Co(H,0),]»+ + 6NHj
равна 1025, а лабильный комплексный ион [Hgl4]2- очень устой-- 48 —
чив: для равновесия[Hgl4p- Hg;e+ + 41-константа равна ~ 10~30.Наиболее примечательными группами инертных комплексов
являются комплексы Со3+ и Сг3+, а также цианидные комплексы
многих катионов. Так, например, инертны [Fe(CN)6]4-, iFe(CN)6]3“,
хотя почти все остальные комплексы Fe2+ и Fe3+ лабильны. Инер¬
тность комплексов свойственна, главным образом, ионам с неза¬
конченной d-оболочкой (табл. 6).Таблица 6Распределение элементов, образующих инертные комплексные ионы
(заштрихованы) в Периодической системе химических элементов
Д. И. МенделееванHeLiBeШ’СN0FNeNaMgA1SimIIClArКCaScTi11W///////.mVS///IIЩ/////pS\w\CuZnGaGe'Ш/VksA'/////IIBrKrньSrYZrNbW'.-Mo;/////TcШ'///ХУ///Л;RW/////.PdAgCdInSn'/////,;Sb^'/////////;Te|////AIXeCsBaLaHfТаУУУУ/,'////,''Ш;Re^mW///Ш$}//AuHgTlPbВPoAtRnFrRaAcCePrNdLuThPaUNpPuCfКомплексные соединения катионов с внешней электронной обо¬
лочкой s2p6, а также лантаноидов и актиноидов лабильны.Рассмотрим более подробно кинетические свойства комплексов,
образованных ионами переходных металлов. Лабильны все внешне¬
орбитальные комплексы этих ионов. Использование внешних d-орби-
талей для гибридизации характерно для конфигураций d9(Cu2+)
и d10(Ag+, Cu+, Zn2+, Cd2+, Hg2+, Qa3 ~, In3+, Tl3+) и для высокоспи¬
новых комплексов ионов с конфигурациями от d5 (а для октаэдри¬
ческих комплексов, когда нужны две акцепторные d-орбитали, и
от d4) до ds (Mn2+, Fe2+ и Fe3+, Со2+, №2+ и др.).При рассмотрении кинетических свойств внутриорбитальных
комплексов видно, что так называемым диссоциативным механиз¬
мам реакций замещения, связанным с временным удалением за¬
мещаемого лиганда или с переходом его в слабо связанное состоя¬
ние, должны соответствовать очень большие энергии активации и— 49 —
малые скорости процесса. Другой путь для реакций замещения
открывается ассоциативными механизмами, для которых харак¬
терно временное присутствие обоих (уходящего и входящего) ли¬
гандов в связанном состоянии. При этом для связывания входящего
лиганда в комплексе должна иметься акцепторная орбиталь.Предложен критерий, согласно которому внутриорбитальный
комплекс лабилен, если в нем не занята электронами и может быть
использована в качестве акцепторной одна из нижележащих (в
приближении теории кристаллического поля) «внутренних» d-op-
биталей, т. е. для реакции замещения возможен ассоциативный
механизм.При октаэдрическом строении комплексов энергетически более
выгодны три орбитали i2g■ Поэтому комплексы ионов с одним или
двумя d-электронами (Ti3+, V3+) должны быть лабильными, а с
тремя (Сг3+)— инертными, что и наблюдается на практике. Окта¬
эдрические высокоспиновые комплексы • ионов Cr2+, Mn2+, Fe3+,
Fe2+, Со3+, например [Сг(Н 20) „ ]2+, [Mn(H 20) 6]2+, [FeF6]3_,
[Fe(H20)6]2+, [CoF6]3_ и т.д., являются внешнеорбитальными и
лабильными, а низкоспиновые, например, [Fe(CN) 6 ]4_, [CoNH3)b]3+,—
внутриорбитальными и инертными. При этом высокоспиновые
комплексы для Со3+(d6) нехарактерны. В частности, не получены
соединения акво-иона [Со(Н 20) 6 ]3+: Со3+ разлагает воду с вы¬
делением кислорода.Для инертных комплексных ионов в растворе некоторое вре¬
мя может существовать неравновесная смесь изомеров и инертные
комплексы могут быть переведены в твердую фазу без изменения
строения. Так, соединение [Co(NH3)5С1 ]С12и в кристалле, и.в рас¬
творе состоит из ионов [Co(NH3) 5С1 ]2+ и С1_. При растворении же,
например, алюмокалиевых квасцов, в кристаллах которых ионы
SO ^координированы алюминием, оказывается, что лишь очень
малая доля ионов А13+ в растворе связана в сульфатные комплексы.
По мере разбавления раствора комплексного соединения степень
диссоциации лабильных комплексов увеличивается, так как ли¬
ганды из них вытесняются растворителем, инертные же комплексные
ионы не изменяются.Разграничение между инертными и лабильными комплексами
условно. В'табл. 7 приведены данные по скоростям обмена ли¬
ганда для некоторых комплексов, по которым можно судить об их
инертности или лабильности. Поскольку скорости реакций при на¬
гревании увеличиваются, то при повышении температуры все боль¬
шее число комплексов становится лабильным.В комплексных ионах с разнородными лигандами часть лиган¬
дов может вести себя инертным образом, а часть — лабильным
Центральный ион с инертными лигандами часто рассматривается
как неизменная структурная единица в реакциях замещения и
присоединения. Так ведут себя металлорганические фрагменты типа
C2H5Hg+ или (С2Н5)зРЬ+, оксокатионы типа U0 22+ и др. Присут¬
ствие таких фрагментов в комплексе иногда отражают в его назва-— 50 —
Таблица 7Скорость обмена лиганда у некоторых комплексных ионов
в водных растворах при 20—30°мML„Время протекания реакции zaL*на 5%на 50%на 95%Сг3+Cr(H20)fCr(CN)g~Cr(NH3)fH20**CN~*NH317-58 ч
24—30 суш
25 чW3+W2Clf*ci-6,5 чFe3+Fe(CN)g~*CN~64 чFea+Fe(CN)g~*CN-33 чPf2+Ptlf*Г~4 минPta+Ptlf*1-30 минCu2+Cu(en)|+*en3 сек (0° С)Zn2+Zn(H20)fH20*2 минAl3+A1(H20)63+А1(Са04)Гh2o**c2of3 мин
20 секЦЗ +tici;*C1-1 минTh*+Th(H20)fH20*3 минBi3+Bil~*1-5 миннии. Например, оксоацетатные комплексы U(VI) называют аце¬
татными комплексами уранил-иона.Оксокатионы и оксоанионы (см. "абл. 8) можно рассматривать
как комплексные ионы с лигандами О2-. В водной среде лиганд
О2- в свободном состоянии не существует, тем не менее обмен кис¬
лородом со средой у этих ионов идет — у некоторых очень медлен¬
но, а у феррат-иена, например, довольно быстро: при 25° С •;%% =
= 3 мин. Относительно некоторых оксокатионов, например Zr02+,
существует мнение, что в водных растворах они превращаются в
гидроксокомплексы, например в Zr(OH)22f, т. е. в (ZrO • Н20)2+.Взаимодействие комплексной частицы с аналитическими реакти¬
вами. Пусть реактив Z быстро и обратимо реагирует в растворе
с лигандом L или ионом металла М. Рассмотрим реакцию этого реак¬
тива в растворе, в который М или L введены только в составе ком-— 51 —
Таблица 8Распространенность оксо-катионов (у заштрихованных элементов)
в Периодической системе химических элементов Д. И. МенделееванLiшШШ.il0FNaMg;////,mЩifif]'/////ШлКCaЩ'////11II'///ЛW'':Mn<////A'////ACoNiCuШ,////A■/////,IPlS'mRbSr-Y[il'/////■Mo''////,W//M,ЩRhPdAgCdЩW/У/m7///<VSbt'4У/,IICsBaLaHf'/////?Тай'////яЩ'/////'////,ГгPtW//,;Au'HgT1'////уP Щ'////,у///.mvmшFrRaAcСеРгNdPmj • ■ ■LuThУ"//;РКтшцУ//У%У/У/.Cfплексной частицы. Например, реактив AgN03 добавлен к раство¬
ру Н2[PtCl в ] или металлическое железо введено в раствор
[Cu(NH3)4]C12.Уравнение, описывающее взаимодействие комплекса с реакти¬
вом на лигандML„ + mZ MLn_! + LZm (I)представляет собой разность уравненийL -f- tfiZ : LZmиMLn-1 -f- L MLnс константами равновесия Kl и *n(xn — ступенчатая константа
образования комплекса MLJ. Следовательно, лиганд можно обна¬
ружить, если константа равновесия (I), равная Kl /*п> много боль¬
ше единицы, т. е. Kl ^ *п-Уравнение взаимодействия комплекса с реактивом на ион ме¬
таллаML„ + ml MZm -f- nL
является разностью уравненийМ + mZ MZmиМ + nL ML„с константами равновесия Км и Рп (рп — общая константа образо-- 52 -
вания комплекса MLn). Следовательно, ион металла можно обна¬
ружить, если Км. »Рп-Если комплекс MLn лабильный, то равновесие будет достигать¬
ся быстро, если же инертный — медленно.Таким образом, качественная реакция не откроет присутствия
иона металла или лиганда в растворе очень прочных комплексов
(лабильного или инертного). В растворах малопрочных комплек¬
сов ион металла или лиганд будет открыт немедленно, если комп¬
лекс лабилен. Если же он инертен, то положительный результат
реакции будет развиваться во времени. Нагревание ускорит этот
процесс.Некоторые реакции свободного и связанного в комплексе ли¬
ганда могут протекать аналогично, отличаясь лишь скоростями и
условиями. Например, бензол в дибензолхроме сульфируется так
же, как и свободный. Вместе с тем для связанных лигандов харак¬
терен ряд реакций, не наблюдающихся у свободных лигандов.Итак, при отрицательной качественной реакции открываемая
частица может содержаться в растворе, но быть замаскированной,
т. е. связанной в комплекс. Возможность «маскировки» часто ис¬
пользуется как в качественном, так .и в количественном анализе.Связывание в комплексные ионы служит средством сдвига рав¬
новесия реакций. Очень характерны трансформации в ряду актив¬
ности металлов, если раствор содержит какой-либо мощный комплек¬
сообразующий лиганд. Так, железо не вытесняет меди из аммиачных
растворов медного купороса. Цинк не восстанавливает платины
из растворов H2fPt(CN)4], а растворяется в них с выделением
водорода. Наоборот, в растворах, содержащих комплексообразую¬
щие агенты, легко растворяются даже благородные металлы: так,
общеизвестно окисление Аи и Та азотной кислотой в присутствии
НС1 и HF соответственно, растворение золота в цианидных ваннах
цод действием кислорода воздуха.Обмен комплексного иона со средой лигандами или ионом металла.
Реакции обмена позволяют судить о том, лабильна или инертна
комплексная частица. К ним относятся:1. Реакции замещения лигандаMX + Y ^ MY + XВажными частными случаями этой реакции являются, во-пер¬
вых, реакция образования комплексной частицы из акво-иона, на¬
примерСо2+ + SCN- C0SCN+ + Н20
во-вторых, реакция обмена лигандами со средойМХ + Х* MX* + Xкоторую можно раблюдать, если свободные первоначально лиган¬
ды каким-либо образом (например, изотопно) помечены. Скорость
такой реакции определяют по выравниванию изотопного состава— 53 -
свободного и связанного лиганда. Если метод меченых атомов не¬
применим, используют ЯМР, а также некоторые специальные
методы исследования быстрых реакций, например метод темпера¬
турного скачка. Обмен лигандами со средой — это механизм, через
который осуществляется динамическое равновесие иона со средой.
Чем выше скорость обмена лиганда со средой, тем выше скорость
реакции замещения лиганда.2. Реакции замещения центрального ионаMX + В ВХ + Мнапример,CoSCN+ -f Fe3+ FeSCN2+ + Со2+в том числе и изотопного замещения:мх + м* м*х + мПоскольку центральный ион окружен лигандами, такая заме¬
на не идет без предварительного отщепления хотя бы_ части ли¬
гандов. Эти реакции протекают медленнее реакций обмена лиганда¬
ми, их механизм сложнее,
а их скорость является ме¬
нее надежным критерием
инертности или лабильнос¬
ти комплекса. Обмен ли¬
ганда со средой удобно
рассматривать на акво-ио-
нах. Молекула воды, коор¬
динированная ионом метал¬
ла, расположена в потен¬
циальной «яме» (рис. 22),
причем положение равно¬
весия соответствует точке
А. Молекула воды все вре¬
мя колеблется около этого
положения под воздействи¬
ем соударений со свобод¬
ными молекулами раство¬
рителя. Если соударение сообщает частице достаточную энер¬
гию, чтобы она могла преодолеть энергетический барьер В (Ед —
энергия активации), частица покидает яму, заменяясь другой мо¬
лекулой. При 25°С период колебаний молекулы воды z0 состав¬
ляет около 1,4-10~12 сек, а среднее время пребывания : ее в потен¬
циальной «яме» для некоторых катионов приведено ниже.Ион La+ Na+ К+ Cs+ Mg2+ Са2+т-109, сек 5,9 2,5 1,1 0,97 146,7 3,7Это значит, что молекула воды совершает в координированном со¬
стоянии около иона Cs+ примерно 700 колебаний, около иона
Mg2+ — примерно 100 000 колебаний.Рис. 22. Зависимость потенциальной энер¬
гии Е лиганда от расстояния г металл —
лиганд (комплексная частица в растворе)— 54 —
Значения т, оцененные для ряда ионов переходных металлов,
приведены ниже.Ион Си2+ Мп2+ Со2+ №2+ Fe3+1, сек 3-10-7 4,5- 10-з 3,2-10-3 3,1Ю~8 4,2-10“3и=1,-/т0 (число колеба¬
ний около точки А) 2-105 3-104 2-103 2■ 107 3-10®Они показывают, что внутренняя координационная сфера акво-
комплексов этих ионов длительное время существует в виде ста¬
бильного образования. Если продолжительность активированного
скачка примерно равна периоду колебания молекулы воды
(1,4-10-12 сек), то доля акво-иенов, в гидратной оболочке которых
в данный момент времени происходит по крайней мере один ска¬
чок при координационном числе N ~ 6, равна N~0Дг, т. е. для
Cs+ ~ 0,9%, а для Ni2+ всего 3-10“5%.Быстрые реакции инертных комплексных ионов. С участием
инертных комплексных ионов быстро протекают реакции, при
которых не затрагиваются связи металл—лиганд. Рассмотрим че¬
тыре типа таких реакций:1. Реакции осаждения. В реакциях этого типа комплексный
ион участвует как единое целое. Они протекают как типичные быс¬
трые ионные реакции. Растворимость полученных при этом соеди¬
нений отличается некоторыми особенностями. Например, для ве¬
сового определения пикрат-иона (Pic-) был предложен в ка¬
честве осадителя катион [Co(NH3)5C1 ]2+. Осадок [Co(NH3)5C1 ]Pic2
малорастворим. При 20° общая концентрация кобальта в растворе
(сСо) заметно возрастает даже через 48 ч после начала насыщения.
Дело в том, что кроме реакциибыстро[Co(NH3)8C1J Pica [Co(NH3)5Ci]2+ + 2Pic"
на ссо влияет медленная акватация иона [Co(NH3)3Cl ]2+:медленно[Co(NH3)5CIF + Н20 [Со(ЫНз)5Н2Ор + CI-и отождествлять ссо с равновесной концентрацией [Co(NH3)5C1 ]2+
нельзя. Растворимость осадков, содержащих инертные комплекс¬
ные ионы, может сильно увеличиваться со временем за счет мед¬
ленных реакций этих ионов в растворе. Накопление продуктов
таких реакций может привести к выпадению осадков. Общим эффек¬
том тогда будет медленно проявляющаяся инконгруэнтность рас¬
творения.2. Реакции ассоциации. Данные реакции часто называют также
реакциями внешнесферного комплексообразования, образования
сверхкомплексных соединений и др. Наиболее обычно обра¬
зование комплексным ионом так называемой ионной пары, т. е.
ассоциата, в котором частица, находящаяся во внешней сфере,- 55 -
связана с комплексным ионом только электростатическими силами:
[Co(NH3)6Cl]2+ + Cl" = ([Co(NH3)6Cl] CI) +La3+ + [Fe(CN)„p- = {La [Fe(CN)6])Ассодиаты могут образовываться инертными и лабильными ком¬
плексными ионами. В случае инертных ионов ассоциацию, не ос¬
ложненную процессами замещения, которые заторможены, можно
изучать в более чистом виде. Когда процесс замещения внешнесфе-
рным лигандом внутрисферного выгоден термодинамически, ион¬
ную пару можно рассматривать как промежуточный продукт реак¬
ции замещения. Так, при взаимодействии Fe3+ с Fe (CN)63- на пер¬
вой стадии образуется ионная пара ([Fe(H20)6 ] [Fe(CN) e ]}, а
на второй происходит вытеснение молекулы воды из внутренней
координационной сферы и образование мостикового двуядерного
комплекса [(H20)5Fe(CN)Fe(CN)5].Для лабильных комплексов быстро устанавливается равнове¬
сие{[M(H,0)^A} ^ [М(Н20)у А] + (х — у) НаОНапример, комплекс NiSCN+cocTOHT из 25% {[Ni(Н 20)жlSCN}
и 75% [Ni(H20)ySCN ]+. При stqm частицы {[М(Н20)Л ]А} и
[М(Н20)уА] (ионная пара и комплекс), отличающиеся по составу
только числом молекул растворителя, различимы лишь метода¬
ми, вскрывающими структуру частиц, например методом ИК-
спектроскопии и др.Внешнесферная частица может быть связана неэлектростати¬
ческими силами. Например, катион [Co(NH3)5SCN ]2+ ассоцииру¬
ет с катионом Ag+. Ассоциация осуществляется за счет свободного
донорного атома S у роданид-иона, и связь в значительной мере
ковалентна. При внешнесферной координации катиона образуется
двуядерная комплексная частица. Реакции ассоциации инертных
комплексных ионов приводят часто к образованию двуядерных
частиц, у которых одна из половин ведет себя как инертная, дру¬
гая — как лабильная. Вся двуядерная частица при этом быстро и
обратимо диссоциирует на одноядерные.3. Быстрые реакции координированных лигандов. К данным
реакциям относятся прежде всего протолитические реакции. Гидро¬
лиз инертного комплексного иона на первой стадии обычно является
реакцией отщепления протона от одного из лигандов, например
от молекулы воды[Co(NH3)5H2OP+ ^ [Co(NH3)6OH]2+ + Н+
или аминокислоты'НООС—CH2NH2nh3,NH3ГООС—CH„NHo. ,NH.
Если протон в молекуле лиганда связан лабильно, то вся реакция
идет быстро и обратимо. Лабильно связанный в лиганде протон
имеет высокую скорость обмена со средой. Поэтому исследования
обмена комплексной частицы со средой мечеными атомами водорода,
с одной стороны, и мечеными донорными атомами — с другой,
дают разные результаты. Критерий лабильности всей частицы
получается только при исследованиях второго типа.Отщепление протона от координированного лиганда происхо¬
дит гораздо легче, чем от свободного. Так, при ионной силе 0,1 р/С
диссоциации гидроэтилендиаминтетраацетат-иона HY3- составля¬
ет 10,3, а комплекса LaHY — около 2,2. С электростатической точки
зрения это объясняется контраполяризацией — выталкиванием по¬
ложительно заряженного иона Н+ положительно заряженным цен¬
тральным ионом. Этот эффект может повести к диссоциации ли¬
гандов, которые в водных растворах не отщепляют протона. Так,
в аммиачных комплексах Pt4+ и Hg2+ протон отщепляется от ам¬
миака, превращающегося в координированный амид-ион NH“,
а в комплексах Ga3+ и 1п3+ с оксикислотами (винной, лимонной
и т. п. ), глицерином и гликолем отщепляется протон спиртовых
групп.Явление контраполяризации равносильно смещению электрон¬
ной плотности к центральному иону едоль a-связей. ^-Электронная
плотность в лиганде часто смещается при комплексообразовании
в противоположном направлении. Это объясняется отвлечением
донорного атома от участия в сопряжении с ^-связями лиганда при
его координации (см. строение комплекса мочевины на стр. 14).4. Окислительно-восстановительные реакции. Окислительно-вос¬
становительная реакция в растворе может быть представлена как
сумма двух сопряженных электрохимических реакций. Такие
реакции на внесенном в раствор индифферентном электроде про¬
текают быстро, если они не сопровождаются изменением конфигура¬
ции комплексного иона, оксоаниона или оксокатиона и связаны
только с принятием или отдачей электрона:[Fe(CN)e]3_ + «- ^ [Fe(CN),]«“UOf + е~ UO2
Mn04 + е~ МпО^—Быстрое и обратимое протекание на электроде электрохими¬
ческих реакций приводит к возникновению на нем равновесного
окислительно-восстановительного потенциала. Медленно проте¬
кающие реакции, напримерMnOJ- + 8Н+ + 5е~ = Мп2+ + 4Н20
UC>2+ + 4Н+ 4- 2е~ = U‘+ + 2Н20
связаны с изменением состава и конфигурации реагирующих частиц.— 57 —
Потенциал индифферентного электрода в системе с такими реакция¬
ми обычно далек от равновесного.Окислительно-восстановительные процессы в растворе в от¬
сутствие электродов протекают по такому механизму, гори котором
частицы окислителя и восстановителя объединяются в активи¬
рованный комплекс. Если активированный комплекс образуется
легко (например, при помощи реакции ассоциации) и его строение
способствует внутримолекулярному переносу электрона, то реакция
окисления — восстановления протекает быстро.§ 3. Возможность существования изомеровПусть изомерия двух веществ вызвана изомерией комплексной
частицы, входящей в их состав. Обозначим комплексную частицу
R, все вещество в целом RA, тогда через А обозначены все частицы,
не входящие во внутреннюю координационную сферу. Рассмотрим
возможные случаи существования и сосуществования изомеров
с точки зрения термодинамики, считая, что все реакции прихо¬
дят к состоянию равновесия.I. Изомеризация в твердой фазе. Запишем реакцию в видеRA R'A (I)Соответствующее ей изменение стандартного изобарно-изотерми-
ческого потенциала (свободной энергии Гиббса) обозначим через
AG°. Возможны два варианта:1) если RA и RA' образуют отдельные фазы, то AG° равно раз¬
ности свободных энергий образования R'A и RA соответственно:AG° = AG^r,A) — AG^ (RA) . (Ill -1)Если AG°<0, т. e. AGf(R'A)< AG/(RA),to вещество R'A энерге¬
тически более выгодно и реакция идет до конца вправо. Если же
AG° > 0, то реакция идет до конца влево. Если при изменении
внешних условий (например, температуры) AG° сохраняет знак, то
устойчив один и тот же изомер. Если AG0 изменяет знак, то на фа-
зввой диаграмме точка или линия, соответствующая равенству
AG° = 0, является границей областей устойчивости обоих изо¬
меров. Изомеры могут находиться в равновесии только при внеш¬
них условиях, соответствующих этой границе (линии перехода);2) если RA и R'A. дают друг с другом твердые растворы, то
AG° = —RT\nK, где К = ац<a/ora — константа равновесия реак¬
ции (I). Пусть активности веществ пропорциональны их мольным
долям (AVa иА'кд = 1—AVa соответственно). Тогда из уравненияNR'AAG° = —RT\nI ^R'Aвидно, что при AG° = 0 концентрации изомеров равны (N' = 0,5),
а при увеличении AG° концентрация энергетически менее выгодно-— 58 —
го изомера резко уменьшается: при 25° и ДО0 = 1,3 ккал/моль
N' = 0,1, при Д(3° ж 2,7 ккал/моль N’ = 0,01 и далее уменьшается
в 10 раз при увеличении AG° на 1,3 ккал/моль.Таким образом, изомер практически исчезает из равновесной
смеси, если на его образование требуется затрата работы на
7—8 ккал/моль больше, чем на образование более устойчивого
изомера.2. Изомеризация в растворе. Поскольку частицы внешней ко¬
ординационной сферы в растворе можно не учитывать, ре¬
акция (I) упрощается доR ^ R'Тогда AG° — AG/(r'> — &Gf(R) — A(AG^), т. е. стандартное изме¬
нение свободной энергии в ходе реакции равно разности свободных
энергий образования частиц R и R' в данном растворе. С другой
стороны,д<5° = — ЯТ\пКъ— ЯГ In — • (П1.2)[R]Итак, отношение равновесных концентраций зависит от разности
свободных энергий образования изомеров. Как и в случае твердых
растворов, изомер практически исчезает из равновесной смеси, если’
на его образование тратится работы на 7—8 ккал/моль больше,
чем на образование более устойчивого изомера. При малых раз¬
личиях в энергии (как, например, у двух конфигураций катиона
[Си(Н20)6 ]2+ — удлиненного и укороченного октаэдра ДG° в водном
растворе при комнатных температурах составляет 1 ккал/моль)
концентрации обеих изомерных форм сопоставимы.3. Кинетические возможности существования изомеров. Со¬
гласно термодинамике сосуществовать с наиболее устойчивым
изомером могут только изомеры, близкие к нему по энергии обра¬
зования.На самом же деле возможно сосуществование соединений-изо¬
меров, свободные энергии образования которых ДG; отличаются
на очень большие величины. Так, AGf диметилового эфира
СН3ОСН3 на 13 ккал/моль выше, чем у изомерного ему этилового
спирта. Равновесная смесь этих веществ практически не должна
содержать диметилового эфира, а смеси с другим составом должны
быть термодинамически неустойчивы. Тем не менее устойчивые
во времени смеси диметилового эфира и этилового спирта могут быть
составлены с каким угодно соотношением концентраций. Это объ¬
ясняется тем, что термодинамическое равновесие реакции R R'
не достигается, т. е.-реакции R -> R' и R ч- R' протекают с неиз¬
меримо малой скоростью. При заметной скорости взаимопревраще¬
ния термодинамическое равновесие достигается с течением времени,
но неустойчивый изомер довольно долго сосуществует с устойчи¬
вым. При больших скоростях реакций изомеризации существова¬
ние изомеров определяется только термодинамическими критериями.— 59 -
Таким образом, область существования термодинамически не¬
устойчивых изомеров существенно расширяется кинетическими
факторами, действие которых в свою очередь зависит от механизма
реакции. Механизмы эти можно условно разбить на три группы:1. Изомеризация изменением валентных углов внутри частицы
без разрыва существующих химических связей для поворотных
изомеров, конформационных изомеров и т. п.2. Изомеризация внутримолекулярной перегруппировкой.3. Изомеризация, протекающая с разрывом связей в частице
и с временным образованием промежуточных продуктов.В кристаллическом состоянии, когда подвижность атомов в мо¬
лекулах и комплексных ионах сильно ограничена, осуществление
любого из этих механизмов изомеризации сильно затруднено, а
третьего — практически исключено. Поэтому легче всего неустой¬
чивые изомеры могут быть стабилизированы в твердом виде. Пере¬
ход их в раствор облегчает изомеризацию, однако даже в жидком
и в газообразном состояниях известны (особенно для органических
соединений) изомеры с ничтожной скоростью взаимопревращения,
если оно идет с разрывом связей в частице.Верхней границей области кинетической устойчивости изомера
является условная температура так называемой термо-изомериза¬
ции, при которой метастабильный комплекс быстро изомеризуется.
Например, для цис- [Pt(NH3)2Br2 ] эта температура 205°, а для
цис- [Pt(C2H5NH2)Br2 ] 120°.
IV. ОСНОВНЫЕ ТИПЫ КОМПЛЕКСНЫХ СОЕДИНЕНИЙКомплексные соединения в этой главе классифицированы по
структурному принципу. Выделены группы одноядерных комплек¬
сов с однодентатными лигандами, комплексов с полидентатными
лигандами и многоядерных комплексов и, наконец, группа коорди¬
национных соединений, при образовании которых существенным
образом используются тг*-орбитали лигандов (олефиновые комплек¬
сы, сэндвичевые соединения, карбонилы, цианиды и нитрозилы).§ 1. Одноядерные комплексы с однодентатными лигандамиДонорными в комплексных соединениях могут быть атомыС N О F
р s С1As Se Вг
Sb Те IПри этом атомы галогенов служат донорами почти исключитель¬
но в виде галогенид- или полигалогенид-ионов (F-, С1“, Вг-, I-,
13- и т. д.). Остальные атомы выступают в качестве донорных в
разнообразных по характеру молекулах. В табл. 9 приведены при¬
меры проявления донорных свойств атомом кислорода.Таблица 9Некоторые типы лигандов, содержащие атом кислородаТип лигащовЛигандКомплексная частицаВода и ее ионын2оAi (н2о);+он-FeOH2+02-wofАнионы кислородныхso;_, сог[Со (NH3)5 С03]+неорганических кислотАнионы карбоновыхCHgCOO-.QAf[CuCH3COO]+кислотСоединения с ионизи¬свн5о-[LaC0H5OP+рованной или неионизи-рованной спиртовой груп¬пойОкиси аминов, фос-C5H5NO (руО)[La(pyO)e]3+финов; сульфоны, суль-фоксиды и т. д.Эфиры, в том числеС4Н802 (диоксан)циклическиеСоединения с кето-СН3СОСН3 (ацетон)группой— 61 -
Лигандами являются обычно анионы или нейтральные молеку¬
лы, поскольку приближение положительно заряженного лиганда к
иону металла энергетически невыгодно. Тем не менее известны при¬
меры комплексных соединений с лигандами-катионами; как пра¬
вило, при этом положительный заряд удален от донорного атома.
Так, в кристалле (N2Hs)2Zn(S04)2 катион Zn2+ координирует
гидразиний-ионН НI I
н—N+—N:I IН Нчерез незаряженный атом азота.Рассмотрим следующие группы ионов металлов:1) восьмиэлектронные катионы (с внешней электронной обо¬
лочкой типа s2р6): Na+, К+, Rb+, Cs\ Fr+, Mg2+, Ca2+, Sr2+, Ba2+,
Ra2+, Al3+, Sc3+, Y3+, La3+, Ac3+, Th'4+, Ce4+, Hf4+, Zr4+ и ряд других;2) двухэлектронные катионы: Li+, Ве2+;3) /-катионы: Се3+, Pr3+,..., Lu3+, U3+, U4+, Am3+,...;4) переходные d-катионы: Tia+, Va+, Cr2+, Cr3+, Mn2+, Fe3+, Fe2+,
Co2+, Ni2+, Cu2+, Pda+, Pt2+,...;5) восемнадцатиэлектронные (s2p6d10) катионы: Cu+, Zna+,
Ga3+, Ag+, Cd2+, In3+, Sn4+, Hg2+, Tl3+, Pb4+,...;6) катионы с надстроенной восемнадцатнэлектронной оболоч¬
кой: Ge2+, Sn2+, Pb2+, Tl+, Sb3+ и т. д.Восьми- и двухэлектронные катионы образуют комплексы элек¬
тростатического типа, которые лабильны. Устойчивость этих ком¬
плексов зависит от электростатических характеристик центрального
иона металла и лиганда. Чем больше заряд катиона и чем меньше его
радиус, тем комплекс устойчивее. Поэтому более устойчивые ком¬
плексы дают те лиганды, на донорных атомах которых имеется наи¬
больший эффективный заряд при минимальном радиусе: фторид-
ион и лиганды с заряженными донорными атомами кислорода. Срод¬
ство донорных атомов к катионам этого типа можно изобразить
схемой:в V группе N>P>As
в VI группе 0>S>Se
в VII группе F“ > Cl" > Br-> I-Комплексные фториды известны для большинства высокозаряд¬
ных ионов этой группы, а в некоторых случаях (при разделении
ниобия и тантала в виде соединений K2TaF7 и K2NbOF5 и т. д.)
играют важную роль в технологии. Известно, что такие металлы, как
титан, ниобий, тантал, хорошо сопротивляются действию кислот.
Однако их можно растворить в смеси азотной и плавиковой кислот,— 62 —
причем первая играет роль окислителя, а вторая—комплексо-
образователя.Катионы с частично заполненной /-электронной оболочкой ве¬
дут себя аналогично, т. е. образуют электростатические лабильные
комплексы; различия в структуре / оболочки не вызывают значи¬
тельных качественных изменений. Однако наличие частично за¬
полненной /-оболочки увеличивает устойчивость комплексов за
счет энергии стабилизации кристаллическим полем (ЭСКП).
Этот эффект должен отсутствовать у катионов с конфигурацией
/7(Gd3+, Eu2+) и /14(Lu3+). Двухэлектронные, восьмиэлектронные и
/-катионы объединяют в «класс А».Низкозарядные 18-электронные катионы и некоторые d-катио¬
ны со значительным числом d-электронных пар склонны к обра-'
зованию дативных связей с акцепторными d-орбиталями донорных
атомов S, Se и Те (но не О), Р и As (но не N) и анионами С1~, Вг-,
1“ (но не F~). Эти катионы (Pd2+, Pt2\ Ag+, Au3+, Ir3+, Hg2+ и т. д.),
а также некоторые катионы с надстроенной 18-электронной обо¬
лочкой относятся к «классу Б».Элементы, проявляющие в некоторых степенях окисления (Rh,
Ir, Pd, Pt, Au, Ag и Hg проявляют эти свойства во всех степенях
окисления) свойства катионов класса Б, выделены в табл. 10 штри¬
ховкой.Таблица 10Расположение элементов класса Б в Периодической системе элементовД. И. МенделееваwШilN0FLiMgA1SiPsClNaCaScTiVCrж7/Mi'llv///y///M7//XS////
;СоЖ№#Сий
. . 1.. JZnGaGeAsSeBrКSrYZrNbЩ7////V////'////mv/M§1111111ЩV/MInSnSbW/У/IRbBaLaHfТаУ'///mm'млm7/MW///яч|y///,;Pb;7/s/s.WM,mШ,AtCsRaAcThPaОбразование дативных связей обычно проявляется в увеличе¬
нии инертности комплекса, а у переходных катионов — в прину¬
дительном спаривании их d-электронов с соответственным умень¬
шением магнитного момента комплекса (низкоспиновые комп¬
лексы).Для катионов класса Б чрезвычайно характерны прочные хло-
ридные (а не фторидные, как у катионов класса А) комплексы. Бла¬
городные металлы (Au, Pt) не растворяются в азотной кислоте, но
растворимы в смеси соляной и азотной кислот (царской водке)
благодаря образованию хлоридных комплексов АиС1( и PtCl6 .— 63 -
В отличие от катионов класса А здесь устойчивости комплексов
благоприятствуют низкий заряд и большая поляризуемость цен¬
трального иона. Низкий заряд увеличивает число электронов у
центрального атома и менее препятствует образованию дативной
связи. Поэтому, например, ион Си+ относится к классу Б, а Си2+ нет.
Схема сродства донорных атомов к катионам класса Бв V группе N < Р > As > Sbв VI группе 0<CS%Se^Teв VII группе F- < С1~ < Вг" <; I-Для этих катионов чрезвычайно характерны цианидные ком¬
плексы.18-Электронные и d-катионы обладают рядом особенностей в
комплексообразовании. Прежде всего это высокое сродство к до-
норным атомам азота и серы, причем сродство к донорному атому
азота обычно выше, чем к донорному атому кислорода. Особенно
это видно при взаимодействии этих катионов с аммиаком. В раство¬
ре аммиака содержатся «кислородные» лиганды ОН” и «азотные»
лиганды NH3. Если катионы класса А образуют при действии ам¬
миака гидроокиси или комплексные гидроксо-ионы (бериллий), то
катионы рассматриваемой группы, как правило, дают аммиачные
комплексы. Эта группа катионов велика и по свойствам очень раз¬
нообразна, являясь как бы переходной между классами А и Б.
Некоторые факторы (увеличение заряда у Fe3+, появление d5-обо¬
лочки у Мп2+ и т. д.) способствуют повышенной электростатич¬
ности комплексов. Так, ион Fe3+ образует прочные фторидные ком¬
плексы, а под действием NH4OH этот ион осаждается в виде гид¬
роокиси, т. е. ведет себя как катион класса А.В электронной теории кислот и оснований Льюиса акцепторы
играют роль кислот, а доноры — оснований. Согласно этой теории
кислоты и основания делятся на «жесткие» (слабо поляризующиеся
частицы с высокими электростатическими характеристиками) и
«мягкие» (легко поляризующиеся частицы с низким зарядом и
большими размерами). Мягкие кислоты эффективно взаимодей¬
ствуют с мягкими основаниями, а жесткие кислоты — с жесткими
основаниями. Таким образом, катионы класса А являются жесткими
кислотами Льюиса, а катионы класса Б — мягкими. Соответствен¬
но к мягким основаниям Льюиса относятся сульфиды, олефины и
другие лиганды, обладающие повышенным сродством к катионам
класса Б, а к жестким — кислородсодержащие лиганды и фторид-
ион.Существенные отличия в устойчивости комплексных соедине¬
ний ионов металлов различных классов позволяют создавать груп¬
повые аналитические реактивы и применять метод «маскировки».
Этот метод, используемый в технологии и аналитической химии,
состоит в том, что раствор, содержащий смесь ионов металлов, об-— 64 —
рабатывается двумя реактивами, один из которых — групповой —
связывает ряд катионов в комплексы, маскируя их. Благодаря
этому второй реактив связывает в комплексы или осаждает только
незамаскированные ионы и его действие становится более специфич¬
ным. Катионы класса А обычно маскируют фторидом, с которым они
дают очень прочные комплексы или осадки; хорошо они маскиру¬
ются также многими кислородсодержащими реактивами. Переход¬
ные ионы чаще всего маскируют аминами. Для катионов класса
Б и некоторых переходных катионов, не входящих в этот класс,
превосходным маскирующим агентом служит цианид. Эти катио¬
ны также весьма чувствительны к действию серусодержащих ли¬
гандов, с которыми катионы класса А практически не реагируют.
Фторид и цианид успешно применяются для маскировки в комплек-
сонометрии, где второй реактив — этилендиаминтетрауксусная кис¬
лота или ее динатриевая соль, трилсн Б.Группа одноядерных комплексов очень многочисленна. Сюда
относится большая группа координационных соединений общей фор¬
мулы [M(NH3)e]X„, где М — центральный ион, X — однова¬
лентный кислотный остаток; п — степень окисления металла.
В аммиакатах молекулы аммиака могут быть частично заме¬
щены ионами или нейтральными молекулами: [Co(NH3)8C1]C12,
[Соеп3]Х з, [Co(NH3)2en2)X3, [Co(NH3)en2py]X3, [Cr(NH3)5H20]X3,
[Co(NH3)4(H20)2]X3. Соединения гексамминового типа известны
для Сг3+, Ir3+, Pt4+, Ni2+, Со3+, Fe2* и др. Соединения тетраммино-
вого типа чаще всего образуются у
двухвалентных металлов (Pt2+, Pd2+,Zn2+, Cd2+, Cu2+, Hg2+, Cos+):[Pd(NH3)4]X2, [Cu(NH3)4]X 2и др.Разнообразна группа галоидных
ацидо-комплексов [МХв]ге-й. В та¬
ких соединениях центральными ио¬
нами чаще всего служат трех- или
четырехзарядные катионы, иногда и
неметаллов: [SiF в ]а~, [AIFg]3',[PF в ]-, [FeFe]3-, [PtCleP-,[SnCle]2-, [PtCle]2-. Известны гало-,
генидные комплексы с высоким к. ч.:[NbF,]2-, [TaF7]2-, [ZrF9 ]s- Со
многими металлами устойчивые аци-
докомплексы образуют также циа¬
нид- и роданид-ионы: K[Ag(CN)2],Na3[Mo(CN)8], Co[Hg(SCN)4).Одноядерные комплексные ионы играют роль изолированных
структурных единиц в кристаллической решетке и занимают ее
узлы. Так, при кристаллизации K2PtCle ионы К+ и [PtCle]2- за¬
нимают соответственно положения F" и Са2+ в структуре флюорита
(рис. 23).Рис. 23.
ионы PtCLОктаэдрические
и катионы К+в кристаллической решетке
КгР1С1в (структура флюо¬
рита)3—699— 65 —
§ 2. Комплексные соединения с полидентатными лигандамиПри образовании комплексов полидентатными лигандами полу¬
чаются циклы, содержащие ион металла*.Очень часто при комплексообразовании замыкают циклы орга¬
нические аналитические реактивы: так, 2,2'-дихинолил (I) или
бицинхониновая кислота (II)образуют с ионами Си+ комплекс, имеющий ярко-красную окраску
и содержащий циклыИзвестный красный осадок диметилглиоксимата Ni2+ содержит
два цикла:Комплекс Ti4+с хромотроповой кислотой (III) содержит шести¬
членный цикл (IV):Первоначально были известны лишь циклообразующие аналитические
реактивы (а-нитрозо-p-нафтол, 8-оксихинолин и др.), дающие с определяе¬
мыми ионами осадки. Поэтому исторически сложилось представление о «внут-
рикомплексных соединениях», как о нейтральных молекулах, в которых анион
связан силами как «главной», так и «побочной» валентности. В настоящее вре¬
мя термин «внутрикомплексные соединения» устарел, а комплексные соедине¬* Циклы получаются также при образовании многоядерных комплексных
соединений с несколькими мостиковыми группами.III"СиСН3\ уОН—/СН3
С=М'СНз^ \>~нс/ \ж3но онH0,sIIIIV— 66 —
ния с полидентатными лигандами часто называют хелатами или хелатными
соединениями — от английского слова «chelate» (клешневидный).Строение и устойчивость циклических соединений зависят от
величины цикла и его сопряженности. Несопряженные циклы в
комплексах в отличие от сопряженных обычно неплоские. Конфи¬
гурацию их можно предсказать, используя теорию напряжения
Байера. Согласно этой теории наиболее выгодны энергетически те
соединения, в которых сохраняются нормальные валентные углы
и нормальные длины связей.Сумма внутренних углов плоского выпуклого я-угольника, рав¬
ная 180°(п — 2), больше, чем у неплоского. Поэтому цикл должен
быть неплоским, если сумма нормальных валентных углов между
образующими его связями больше 180° (п — 2). Рассмотрим, на¬
пример, строение циклопарафинов. Нормальный валентный угол
а в полиметиленовой цепочке—СН2—СН2—СН2—равен 109,5°
(тетраэдрический угол при гибридизации sp3).По формулерассчитаем разности между валентным углом а в гипотетических
плоских циклопарафинах и нормальным значением этого угла:Из значений Д следует, что циклогексан и циклогептан должны
быть неплоскими, а циклобутан и циклопентан — плоскими, при¬
чем циклопентан практически ненапряжен. Термин «напряжение»
применяется для конфигураций, в которых валентные углы или дли¬
ны связей сильно отличаются от нормальных. Этот термин вызы¬
вает представление о связях как о пружинах, исходящих из атома
под определенным углом друг к другу и способных с некоторым
напряжением изгибаться или растягиваться.Неплоские ненапряженные конфигурации (изомеры) циклогекса-
на с валентными углами 109,5° имеют видНормальные длины некоторых связей (А)С—С 1,54 С—О 1,43С=С 1,34 С=() 1,22С=С 1,20 С—Н 1,09С—N 1,47 С—С1 1,76C==N 1,16 С—Вг 1,91л4567п а, град . ... .
Д=а—109,5° . .(циклобутан) (циклопентан) (циклогексан) (циклогептан)
90 108 120 128,3—19,5 —1,5 +10,5 +18,8VКреслаВанна3*— 67 —
Расчеты конфигураций циклических комплексных соедине¬
ний осложняются разнообразием валентных углов и длин связей
в них. Тетраэдрический угол 109,5° является нормальным дляI Iатомов лиганда, в том числе донорных, с к. ч 4: —С—, —N—I Iи т. д. (гибридизация sp3). При атомах лиганда с к. ч. 3 (гибри¬
дизация sp2): —С= , —N=, —О— и —S— нормальныйI I I Iвалентный угол должен быть 120°, поэтому плоская конфигурация
бензола в отличие от гипотетической плоской конфигурации цик-
логексана не напряжена. Что касается нормальных валентных
углов у атома металла, то они разнообразны и зависят от строения
координационной сферы; часты углы 120, 109,5 и 90°.Рассмотрим результаты расчета конфигурации пятичленного
цикла, образуемого Со3+ с этилендиамином NH 2—СН 2—СН 2—NH 2:Если задать все длины связей и четыре валентных угла, то пятый
угол определится из требования замкнутости цикла. Если он
близок к нормальному, то цикл не напряжен. Пусть валентные углы
при атомах С и N равны 109,5° (атом азота имеет в данном случае
к. ч.4) и длины связей составляют С—С 1,54 А; С—N 1,47 А; Со—N2,00 А.При расчете получаются следующие координаты атомов (А):XУ2Со . . . . -1,4600Nr . . . . 01,370N2 . . . . 0—1,370Ci .... 1,280,740,30С2 . . . . 1,28—0,74—0,30при этом угол NCoN равен 86,2°. Нормальный валентный угол при
атоме Со с октаэдрической конфигурацией комплексного соедине¬
ния равен 90°. Таким образом, искажение угла равно 3,8°, к тому
же оно может быть перераспределено между всеми углами, поэ¬
тому цикл почти ненапряженный. Однако цикл не лежит-в плос¬
кости ху: атом Сх расположен выше нее, а атом С2 ниже. Результа¬
ты расчета удовлетворительно совпадают с экспериментом: рент¬— 68 —
генографическое определение структуры Со(еп) 33+ показало, что при
тех же длинах связей угол CoNC равен 109,5°, угол NCC— 109,69
и угол NCoC — 87,4°.Пятичленные циклы не напряжены и являются неплоскими так¬
же и в других комплексах переходных металлов.Аналогично этому можно построить также неплоские ненапря¬
женные шестичленные циклы, однако они будут более изогнуты,
чем циклогексан. Это ведет к возникновению добавочного фактора
нестабильности комплексных соединений с такими циклами. Акси¬
альные лиганды, расположенные перпендикулярно к той части цик¬
ла, которая содержит донорные атомы, препятствуют выходу цикла
из экваториальной плоскости, т. е. его изгибанию. Из-за этого эффек¬
та, который называют /'-напряжением, шестичленные несопряженные
циКлы менее устойчивы, чем пятичленные. Это можно показать,
сравнив константы образования комплексов Си2+ и Ni2+ с этилендиа-
мином (еп) и триметилендиамином (trim), в которых реализуются
соответственно пяти- и шестичленный циклы:Комплекс с еп комплекс с trimКонстанты у.п для реакцииMLn_x + L MLnопределены при 0° и ионной силе 0,15 (табл. 11). Во всех случаях
значения ^„дляеп выше, чем для trim, и этот эффект проявляется
тем отчетливее, чем больше п, т. е. чем больше циклов в комплек¬
сной частице.Стереохимические свойства иона металла играют менее важную
роль в циклообразовании, чем стереохимические свойства лиганда.
Различные типы гибридизации орбиталей иона металла сравни¬
тельно мало отличаются по энергии. Поэтому может наблюдать¬
ся невыгодный способ гибридизации, если при этом возрастает
устойчивость комплекса, например, за счет увеличения числа свя¬
зей между металлом и лигандом. Возникающие при этом конфигу¬
рации комплексов называют вынужденными. Например, в ком¬
плексе Pt2+ с р, р', р"-триаминотриэтиламином четыре донорных
атома азота занимают вершины тетраэдра, а не квадрата, явля¬
ющегося обычной координационной фигурой для Pt2+. В хлорофил-69
Таблица 11Константы образования комплексных соединений Си2+ и Ni2+ с еп и trim(при 0°С и /=0,15)Системаlg *ilg *2lg *3Са'2+ — еп11,459,83 Cu2+ — trim10,527,94—Alg = 0,93Alg *2 = 1,89—Ni2+ —еп7,886,704,78Ni2+ — trim6,98""4,93—1,90Alg xx = 0,90Alg*,= 1,77Д lg r.3 = 2,88ле — зеленом пигменте растений — комплекс Mg2+ имеет необыч¬
ную для этого катиона плоскую квадратную конфигурацию, обу¬
словленную планарным строением порфиринового кольца:соосн3Влияние стереохимических особенностей иона металла на кон¬
фигурацию комплекса ослабевает по мере увеличения дентатности
лиганда. Так, анион ЭДТА“ООС-СН2ч -СНо-СОСГ>NCH2-CH2-N/“ООС-СН/ хСНа-СОО-— 70 —
образует комплексы с Zn4+, Hg2+, Cd2+, Со2+, Cu2+ и т. п., как пента-
или гексадентатный лиганд, т. е. к. ч. этих катионов не меньше
5, в то же время в галогенидных комплексах координационное число
Zn2+ и Hg2+ равно 4.Однако в комплексах с Ga3+, Ве2+ и некоторыми другими ка¬
тионами даже этилендиаминтетраацетат-ион проявляет дентат-
ность 4 из-за малого объема центрального иона.Плоские шестичленные циклы содержатся практически во всех
ацетилацетонатах металлов. Ацетилацетон СН3—С—СН2—С—СН3о Ореагирует с ионами металлов в енольной формеРентгенографические исследования показывают, что циклпостроен симметрично и является плоским. При этом связи С—СН,
соответствуют по длине ординарным, а связи С—О и С—С имеют
кратность 1 С п < 2. Ацетилацетонаты устойчивы; многие из
них перегоняются без разложения. Хелатный цикл под воздей¬
ствием ряда агентов ведет себя как ароматический-, при централь¬
ном атоме углерода в цикле проходят реакции галогенирования,
нитрования, ацилирования и т. п.В ацетилацетонатах лиганд в результате сопряжения двойных
связей становится полностью симметричным:Сопряжение иона металла с лигандом не наблюдается: связи
М—О имеют длину, соответствующую ординарным.Плоские пятичленные циклы с двумя сопряженными двойными
связями образует с ионами переходных металлов диметилглиок-сн3—С—СН=С—СН3II Iо онсн/чН3С—С С—СНзо о
ч/
мСН3—С—С—СН3в анти-форме*.СИМIIно—N N—ОН* R—С С—R R—С—С—RR—С-■С—RN—OHNOH НО—N N—ОНNOH HONамфи- формаawmw-формаfwtt-форма
В диметилглиоксиматах валентный угол при атоме металла мень¬
ше 90° и составляет 78° при Со3+, 80° при Ni2+, 79,5° при Си2+,
83° при Pt2+. Во всех этих случаях нет заметного уменьшения
длины связи металл—азот. Так как четырехчленные циклы с точ¬
ки зрения теории Байера напряжены чрезвычайно, то карбонат-
ион и карбоксильные группы обычно играют роль однодентатных
лигандов: с ионом металла связан только один из атомов кислоро¬
да группы—С<^ , а второй направлен в сторону от иона металла,как, например, в комплексе меди с анионом аминоуксусной кис¬
лоты:В некоторых комплексах Со3+, а также в катионах с большим
радиусом U022+,Th4+ и др. карбоксилат-ион бидентатен:Двойная связь в свободном карбоксилат-ионе выравнена со
связью С—О- до полного равенства длин связей и кратности:— 72 —
-с1°оРавноценность связей несколько нарушается при мо-нодентатном присоединении группы СОО-. При образовании же че¬
тырехчленного цикла она сохраняется.Известно образование четырех членных циклов сульфат-ионом,
иодатом в Zr(I03)4, перйодатом в производном Си3+ [Си(Юб)2]7-и т. д. Дитиокарбоксильная группа —координируется ионамипереходных металлов через два атома серы с
образованием четырехчленного цикла3J3to свидетельствует о чрезвычайной эне¬
ргетической выгодности образования связи
ионов переходных металлов с атомом серы. 'Как правило, многодентатный лиганд за- ?
нимает во внутренней координационной сфе- [
ре соседние места. В октаэдрических и ква- \
дратных конфигурациях бидентатные лиган- ^ v-/
ды занимают два цис-положения* (рис. 24, а).Согласно А. А. Гринбергу, это обстоятель- бство можно использовать для определения рИс. 24. Расположе-геометрической конфигурации комплексов, ние бидентатного ли-для которых установлена генетическая связь ганда в октаэдричес-
м г j ком комплексе', до-с циклически построенным соединением. норнЫе атомы в цис-Например, при взаимодействии нитрита нат- положениях (а) и врия с [Co(en)2C03]X (X — галоген) обра- транс-положениях (б)зуются соли, которым можно приписатьцис-строение, т. е.7NCL2NO,"+ со.NO,* Чтобы донорные атомы заняли транс-положения (3 и 5, см. рис. 24,6)
необходима цепь с большим количеством звеньев, так как нужно обогнуть
лиганд, находящийся в положении 1 или в эквивалентном ему. Предполагают,
что в фиолетовых изомерах комплексов Си2+ с бис-а-аминокислотами НООС——CRR'—NH—(СНг)л—NH—CRR'—СООН при п > 5 (когда в цикле восемь
или более членов) наблюдается транс-%,оординация аминогрупп лиганда.
Однако эта гипотеза не проверена рентгеноструктурным анализом.— 73 —
Генетическое доказательство строения возможно, если равно¬
весия диссоциации и изомеризации комплекса сильно заторможе¬
ны, т.е.внутренняя координационная сфера существует как ста¬
бильное образование длительные промежутки времени. Метод ге¬
нетического доказательства строения является общим для химии
комплексных соединений и органической химии. В химии комплек¬
сных соединений его можно применять к инертным комплексам,
если реакция замещения не сопровождается внутримолекулярной
перегруппировкой.Генетические связи между начальным и конечным продуктами
реакции осуществляется через активированный комплекс, кото¬
рый может быть исходной частицей, потерявшей лиганд А—А или
присоединившей два лиганда В. В обоих случаях изменяется коор¬
динационное число, а значит, и вся координационная фигура. Гене¬
тические связи при изменении координационного числа нарушаются:и появляется возможность образования /пранс-изомера.Образование цикла многодентатным лигандом затрудняется,
если число атомов в цикле больше 6—7, при дальнейшем же уве¬
личении числа звеньев, разделяющих функциональные группи¬
ровки, начинает сказываться стремление к полимеризации. Дей¬
ствительно, гексаметилендиаминтетраацетатв отличие от ЭДТА обладает высокой способностью к образованию
двуядерных комплексов.К многоядерным комплексным частицам, которые содержат не¬
сколько ионов металлов, относятся комплексы с лигандами в
качестве мостика, комплексы с непосредственной связью металл—ме¬
талл и комплексы, в которых роль каркаса молекулы играет поли¬
мерный лиганд, присоединяющий большое число ионов металла.Комплексы с мостиковыми лигандами. Во-первых, мостиковым
может быть лиганд, имеющий две функциональные группы (или
два их набора), которые выступают в качестве доноров по отноше¬
нию к двум различным ионам металлов. При этом бидентатными ста¬
новятся такие лиганды, как роданид+А - А—ООССН2ч
—ооссн./К /^ШзСНаСНаСНаСНгСНйСНгШа/^СНаСОО—^СНаСОО—§ 3. Многоядерные комплексные соединения— 74 —
[Pt2(SCN)2Cl2(P (C3H7)3)2]ацетатСCU (СН3СОО)2-рупероксо-группа— 75 —
пара-дизамещенные бензола и т. п. При хелатообразовании они не
могут быть бидентатными. Во-вторых, лиганды, имеющие несколько
донорных электронных пар на одном донорном атоме, например,
анионы галогенидов, ОН- и т. п., могут за счет этих электронов
образовывать связи с двумя разными ионами металлов. Действи¬
тельно, известны многочисленные соединения, в которых лиганды
такого рода играют роль мостиков. К ним принадлежат димерные
галогениды алюминия А12С16, А] 2Вг6, А1216; золота Аи2С1 в; железа
Fe2С16; галлия Ga2Cl6, Ga2Brs; индия In2Cl6, 1п2Вгв, In2I6 и т. п.
и их аналоги, например [А1С12СН3]2. Структура соединений этого
семейства — сросшиеся ребрами искаженные тетраэдры:Эти соединения легко гидролизуются во влажном воздухе, при
растворении в неполярных растворителях переходят в раствор в ви-Рис. 25. Структура полимерных анионов [AlFe]^7 (а) и [AsS2]” (б)С1а— 76 —
де димеров, летучи, в парах также существуют в виде димеров.
В органическом синтезе они широко употребляются в качестве ка¬
тализаторов.Галогениды и халькогениды выступают в роли мостиковых ли¬
гандов в цепочечных ионах: [A1Fа2_bT12A1F5, [AsS2"]„ в T1AsS2,
[ZrFe2"]„ в K2ZrFe и [Aul ]я в Aul (рис. 25). Для многоядерных
комплексов, которые образуются при сольволизе одноядерных,[(NH3)5CoNH2Co(NH,)5]5+0NH3 0Шз[(NH3)4Go(OH)2Co(NH3)4]4+для изополи- и гетерополисоединений, для таких ионов, как пиро¬
сульфат, пирофосфат, гексаванадат, бихромат и т. п., характерны
мостиковые‘группы ОН-, NH2-, О2' и др. Гидролиз большинства
катионов в водных растворах приводит к образованию многоядерных
продуктов. В частности, для ториЪ описана следующая схема гид¬
ролитических равновесий:Th4+ + Н20 ThOH3+ + Н+Th4+ + 2Н20 Tb(OH)f + 2Н+2Th4+ + 2Н20 Thj(OH)®+ + 2Н+4Th4+ + 8H20 Th4(OH)f + 8H+. 6Th4+ + 15HaO Th6(OH)j5 + 15H+— 77 —
Описан многоядерный гидролиз и для лантана:La3+ + Н20 LaOH2+ + Н+2La3+ + Н20 -g: La2OH5+ + Н+5La3+ + 9Н20 La5(OH)f + 9Н+Донорному атому необходимо несколько донорных электронных
пар только в том случае, если он связан с каждым ионом металла
двуцентровой донорно-акцептОрной a-связью. Если же он способен
образовывать многоцентровые* связи с несколькими ионами ме¬
таллов одновременно, то достаточно одной донорной пары электро¬
нов. Так, в соединении [(С2Нft)] [НСг2(СО)10] линейный ани¬
он [(СО)5Сг—Н—Cr(CO 5) V состоит из двух фрагментов Сг(СО)6,
связанных, гидрид-ионом :Н“, при этом образуется трехцентровая
связь Сг—Н—Сг.По-видимому, трехцентровые связи образуются и мостиковыми
атомами, имеющими несколько донорных электронных пар, на¬
пример атомами хлора в димерах М2С16. Структуры этих димеров
совпадают со структурой диборана В2Не, в котором атомы Н за¬
ведомо не являются донорами для обоих атомов бора. Как дибо-
ран, так идимерыМ2С1в стабилизируются двумя трехцентровыми
связями В—Н—В и М—Cl—М соответственно.Цепочечные ионы типа [AsS 2~ ]п (рис. 25, б) образуются при
кристаллизации соли, если при этом достигается наибольшая
энергетическая выгодность кристаллической решетки как целого.
Так, взаимодействием K2S2 с платиной и серой при 950° С в токе
сухого азота получается соединение КгР^г. содержащее цепочеч¬
ный ИОН; ’— s—S—S—S —/pt /Pt / pt /—s—s—s—-s—При растворении соли они разрушаются до мономеров. Существо¬
вание же димерных конфигураций вызывается или энергети¬
ческой выгодностью этих конфигураций, или малой скоростью их
распада. Димерные конфигурации склонны сохраняться в раство¬
рах, а иногда даже в парах вещества. Однако бывают исключения,
например, ион [(CN)5CoN202Co(CN)6]e_ с гипонитритным мо-о остиком || у в водных растворах распадается на два иона
—N—N—[Co(CN)5NO]3-.Существование многоядерных соединений, в особенности це¬
почечных ионов, делает невозможным определение координацион¬
ного числа центрального иона по эмпирической формуле молекулы.
Так, многоядерный ион [AlFf2-]n (рис. 25, а) построен из окта¬
эдров [AlFe], сросшихся вершинами, т. е. А1 имеет к. ч. 6, а не5. При сращивании координационных полиэдров отношение металл:— 78 —
лиганд в эмпирической формуле соединения меньше координа¬
ционного числа. Оно обусловлено-стехиометрией общего члена цепи
(рис. ’26).Рис. 26. Образование иона [ML5]„ при срастании
октаэдров вершинамиС другой стороны, можно получить формулу для соединения,,
содержащего сросшиеся в бесконечные цепи, сетки или другие про¬
странственные конфигурации координационные многогранники^
алгебраическим путем. Для этого каждому i-му лиганду (/ = 1,2,
..., N) в многограннике приписывают степень связанности qit ко¬
торая обратна количеству центральных ионов, с которыми связан
данный лиганд. Если записать стехиометрическую формулу комплек¬
сусаввиде(МЬЛ.)я,то^=2^;. Действительно, в структуре NaCl каж-
i= 1дый ион Сг связан с шестью окружающими его ионами натрия*
все qt = 1/6. Каждый ион натрия окружен шестью хлорид-ионами,
х = 6 х 1/6 = 1, т. е. формула соли NaCl. В примерах на рис. 25
и 26 <71( q2, qk и qe равны 1, q3 и qb равны 1/2, формула иона
[AlF5a_]n.При достаточной удаленности друг от друга функциональных
группировок мономерный лиганд образует связи с двумя ионами
металлов. В результате образуется полимер по схеме
НАЧ ,АН... М»+ + >R-(CH2)m-R<; + М2+ + в/ ХВ^ —R\ g/ M\g/R—(С^а) m—^ + 2nH+По аналогии с полимеризацией и поликонденсацией такие реакции
называются реакциями поликоординации. Их продукты могут по¬
лучаться как в специальных условиях, так и выпадать из раство¬
ров при простом смешении реактивов.Например, при кипячении тетракетонаСН3 СНз
с ацетилацетонатом бериллия при 200° образуется полимерный ком¬
плекс, а выделяющийся ацетилацетон отгоняется. При смешении
раствора бис-а-тиопиколиламида в диметилформамиде с метаноль-
ным раствором ацетата меди выпадает полимер с молекулярным
весом около 15 ООО:Малоизученные нерастворимые и окрашенные комплексы ме¬
таллов с полиоксисоединениями (лаки), по-видимому, являются по¬
лимерами, содержащими ион металла в основной цепи. В качестве
примера приведем предполагаемую формулу лака Веа+ с оксинафто-
хиноном:Связи металл—металл. С точки зрения электростатических мо¬
делей комплексообразования не должно быть связей между одно¬
типными частицами. Поэтому связи металл — металл получаются в
тех случаях, если металл обладает ярко выраженной способностью к
образованию ковалентных связей. Как правило, это переходные
металлы с большим количеством d-электронов, а также металлы с
электронной оболочкой типа 18 + п (Sb, Pb и др.).Связь металл—металл может быть как единственной связью
между центральными ионами комплексного соединения, так и осу¬
ществляться наряду с мостиковыми связями типа М—L—М- Не¬
посредственная связь М—М осуществляется в многочисленных мно-- О' он
If I
/\/ч,-f Ве2+ 2пН+ +\/Ч^О он п■о-/~V- 80 —
MnРис. 27. Структура многоядерных карбонилов:
а — декакарбонил дитехнеция Тс2(СО)10; 6 — смешанный карбонил Mn2Fe(CO)i4гоядерных карбонилах (рис. 27), в соединениях типа Hg2Cl2 и т. п.
Разработаны общие методы синтеза соединений с такой связью:
вытеснение карбонильным анионом a-связанных лигандов:(C6He)sPAu Cl + Na Мп(СО)5 = (С:вН6)3 PAu — Мп(СО)5 + NaClотщепление НС1 при реакции между хлоридами и гидридными ком¬
плексами:ClHglCl + H[Rh(R3As)3Cl2 = Cl-Hg - Rh(R2As)3Cl2 + HC1отщепление аминов при реакции между амидными и гидридными
соединениями металлов:L^M—|ЩЩ)Г+Н|—МТ' = L^M—M'Ly + HN(CH3)2и ряд других.Соединения переходных металлов, содержащие связь металл—
металл, являются низкоспиновыми, а при четном числе электронов
в системе — диамагнитными. По спектральным свойствам они резко
отличаются от аналогичных, но не имеющих такой связи соедине¬
ний. Например, при смешении бесцветного раствора [Pt(NH3)4 ]С12
с розовым раствором [PtCl4 ] выпадает зеленый осадок соли Маг¬
нуса [Pt(NH3)4] [PtCl4]. Резкое отличие цвета этой соли от цве¬
та образующих ее ионов, от цвета ее аналогов [Pt(C2H5NH2)4 ]
[PtCl4] и [Pt(NH3)4] [Pt(SCN)4] и ее собственного изомера (все
эти соли розовые) указывает на наличие в ней сильного взаимодей¬
ствия между ионами.4—699 — 81 —
Соль Магнуса диамагнит¬
на. Квадратные катионы
[Pt(NH3)4 ]2+ и анионы
[PtCl 4 ]2_ упакованы в ней
друг под другом (рис. 28).
Расстояние между атомами Pt
составляет 3,31 А. Все это,
наряду с малой растворимо¬
стью соли Магнуса, т. е.
большой энергией ее крис¬
таллической решетки, согла¬
сованно указывает на нали¬
чие в ней связей Pt—Pt.Связи металл — металл
сильно изменяют свою длину
и кратность при переходе от соединения к соединению. При 90° вос¬
становлением Re04 в водных растворах НС1 при помощи Н3Р02 об¬
разуются соли иона[^е2С18 Р~. Методами рентгеноструктурного ана¬
лиза для этого иона установлено строение с расстоянием Re—Re2,24 А, тогда как в металлическом Re эти расстояния составляют
2,74—2,76 А. В этом анионе между атомами рения осуществляется
максимально возможная — четырехкратная — связь, а именно:
если считать, что ось г направлена вдоль линии Re—Re, то на связи
с хлорид-ионами используются орбитали рх, ру, dxi^ и гибрид¬
ная орбиталь dz2 s, причем степень использования орбитали йгг в этой
гибридизации мала: атомы С1 образуют вокруг атомов Re почти плос¬
кие квадраты. Тогда перекрывание р2- и (^-орбиталей атомов Re
дает две ст-МО, одна из которых несвязывающая; за счет dxz и dyz
образуются две л-связи, а за счет орбиталей dxtJ—-б-связь. В карбо¬
ниле Re2(CO)10 орбитали рения в большей мере используются на связи
с лигандами, связь Re—Re близка к однократной, длина ее 3,02 А.В принципе взаимодействие между ионами металла может при¬
вести к возникновению только несвязывающих или разрыхляю¬
щих МО. При этом молекула при наличии мостиковых связей между
этими ионами металла будет устойчива. Такое взаимодействие, ко¬
торое может привести к спариванию спинов, но вносит отрицатель¬
ный или близкий к нулевому вклад в энергию связи, избегают на¬Рис. 28. Структура соли Магнуса— 82 —
зывать связью. Поэтому при наличии мостиковых лигандов во¬
прос о том, есть ли в соединении связь металл—металл, становится
довольно сложным. В частности, хорошо известно о наличии взаи¬
модействия между атомами Си в структуре монопиридинкупроаце-
тата (см. стр. 75). Атомы Си удалены в нем друг от друга на 2,63 А,
что всего на 0,1 А больше, чем в металле, однако сколько-нибудь
прочной связи Си—Си не образуется.Критерием связи М—М при на¬
личии мостиковых лигандов слу¬
жит спаривание спинов у атомов
переходных металлов и уменьше¬
ние расстояния М—М до значений,
меньших ковалентного диаметра
металла. Сравним, например,два
аниона: W2C193- и Сг2С193~. Оба
они имеют одинаковую структуру
(рис. 29). Однако при ковалент¬
ных радиусах атомов W 1,40 А и Рис' 29- СтРУктУРа комп-п , ос a J/VW \i7\ лексных анионов W2ClaСг 1,25 А расстояние d(W — W) = з-= 2,41 A, a d (Сг—Сг) = 3,12 А, и Сг2С1дпричем анион вольфрама диамаг¬
нитен. Таким образом, в W2C193- помимо трех мостиковых связей
М—Cl—М имеется непосредственная связь М—М.Структуру, аналогичную монопиридинкупроацетату, имеют
Си(СН3СОО)2 • Н20, Сг(СН3С00)2-Н20 и Rh(CH3COO)2 • Н20.
Связь металл—металл имеется в ацетатах хрома (II) и родия (II).
Так, красный ацетат хрома (II) резко отличается по цвету от синих
растворов Сг2+, а расстояние Сг—Сг в нем составляет 2,385 А.Некоторые группировки со связью металл—металл (с допол¬
нительными мостиковыми связями или без них) очень прочны и
характерны для целых серий соединений. Так, ReCl3 состоит из
частиц Re3Cle, которые сохраняются даже в газовой фазе. Почти
во всех веществах, которые удается получить изЯеСЦ, сохраняется
группировка атомов Re3. Такие группировки получили название
кластеров от английского слова cluster — гроздь, группа. Неко¬
торые характерные кластеры приведены на рис. 30. Группу мно¬
гоядерных комплексов составляют полимеры, содержащие ионы
металлов. Основная цепь полимера построена по типу обычных
органических полимеров или поликонденсатов и на нее «навеша¬
ны» функциональные группировки, способные образовывать ком¬
плексы с ионами металлов, либо же металл непосредственно содер¬
жится в главной цепи полимера и участвует в поликонденсации.Полимерные лиганды — это, во-первых, полиэлектролиты и
другие растворимые полимеры: поливиниловый спирт, полиакри¬
ловая кислота, белковые вещества и т. д. Во-вторых, это смолы —
иониты. Кроме катионитов типа сульфоуглей, которые редко рас¬
сматриваются как полимерные лиганды, получены катиониты, со-4*— 83 —
Рис. 30. Структуры комплексов Os3 (СО)12; ReaClf2 ; Fea(CO)i2держащие и другие функциональные группировки, в том числе высо¬
коизбирательные. Например, аналогично феноло-формальдегидной
смоле может быть получена смола ор/ло-нитрофеноло-формальде-
гидная. Будучи восстановлена, она дает орто-аминофеноло-фор-
мальдегидную смолу, которая избирательно извлекает из раствора
ионы Cu2+, Zn2+, Cd2+. Аминосмолы можно диазотировать и соче¬
тать с салициловой кислотой, с 8-оксихинолином и т. п. Много¬
численные смолы, извлекающие из растворов преимущественно
ионы переходных металлов, синтезированы на базе оснований
Шиффа. В структуру полимеров были введены остатки ЭДТА,
хромотроповой кислоты и даже анти-глиоксимная группировка:
НО—N N—ОН —СН—СН2—I—сн,—с—с—,/ч/чно\/\он\/C=N—ОНIНзС—C=N—ОН— 84 —
§ 4. Комплексы с тс-связяии в качестве донораОлефины в качестве лигандов. Известны устойчивые соедине¬
ния платины и некоторых других металлов с олефинами. Метод по-
лучения таких соединений для платины состоит в обработке без¬
водного хлорида или бромида платины (IV) непредельными орга¬
ническими соединениями в безводных растворителях. Ион PdCl42'
реагирует с этиленом в водном растворе: константа равновесия
К = [Р(3(л-С2Н4)С1з][С1~/[Рс1С14_] [СН4] при 25° и ионной силе
2,0 равна 17,4.В комплексах олефинов место донорного атома в координа¬
ционном полиэдре занимает центр дюйной связи олефина, распо¬
ложенного симметрично по отношению к металлу.Так, в этилендиметиламин-транодихлороплатине (II) в углахнесколько искаженного квадрата расположены атомы Cl, N и центр
двойной связи, которая перпендикулярна плоскости квадрата. Ана¬
логична структура и других олефиновых комплексов металлов.
Циклические олефины (например, циклооктатетраен) могут зани¬
мать несколько координационных мест вокруг одного и того же иона
металла (рис. 31, а); бутадиен или ведет себя подобным образомРис. 31. Строение трис-(циклооктатетраен)-дититана (а) и бутадиен -
трикарбонил-желе:)а (б)— 85 —— В/ —
(рис. 31, 6), или занимает по одному координационному месту у
двух центральных атомов металлов. Неплоский 1,5,9-циклодо-
декатриен, образуя комплексы как тридентатный лиганд, целиком
окружает атом металла:СН,СН;"Чпз
/сн2 \
сн//Чм.Дн
/Н \1 9яг\Н2 >СН—СН2
сн-снКоординационную связь в металл-олефиновых соединениях
можно представить в виде донорно-ащепторной, причем роль до¬
нора играет я-связь между атомами углерода. Таким образом, два
я-электрона образуют связь в пространстве между тремя атомами;
это является одним из примеров многоцентровой (в данном случае
трехцентровой) связи. Рассмотрим образование связи между Pt2+
и этиленом в комплексе [Pt(C2H4)(CH3NHCH3)Cl2]. Схема МО
этилена имеет вид-fr•fr6*(С-С)7Г*яб (С-С)Электронную структуру HOHaPt2+(d8) после принудительного спари¬
вания d-электронов можно представить в видеSd 6s Sp■tHHfft— —Донорно-акцепторная ст-связь между ионом Pt2+ и молекулой С2Н4
образуется электронами связывающей л-орбитали С2Н4 и пустой
^5/о2-гибридной орбиталью платины; дативная я-связь между Pt2+
и G2H4 осуществляется за счет пустой разрыхляющей я-орбитали
этилена и двух d-электронов платины. На рис. 32 показано пере¬
крывание электронных облаков при образовании в- и я-связей в
олефиновом комплексе платины. Олефиновые я-комплексы обра¬— 86 —
зуются атомами металлов, у которых больше пяти d-электро-
нов.Координироваться таким же образом может пероксо-группа
022- в Сг05 • ру*, в ионе Сг3083~, в ионе [(NH3)5Co—02—Co(NH3)5]5+\//\Рис. 32. Образование а-связи ("сгН4; dsPpt) (а) и образование^■СВЯЗИ (dpf, 71*0, Hl) (б)(см. стр. 75) и др. В нитрозо-диметилдитиокарбамате Со3+ таким
же образом координирована нитрозо-группа, причем это единствен¬
ный пока пример несимметричного по отношению к связи располо¬
жения координируемой группы.Сэндвичевые соединения. Представления о многоцентровой свя¬
зи позволили объяснить строение «сэндвич»-соединений, примером
которых может служить ферроцен. Ферроценом называют кристал¬
лическое соединение (C5H5)2Fe, где С6Н6~т анион циклопента-диена \ / ' Ферроцен (т. кип. 249 ') оранжевого цвета, устойчив
снна воздухе, не разлагается при нагревании до 470°С, перегоняется
с водяным паром и выдерживает нагревание с концентрирован¬
ной НС1 или со щелочью. Эти свойства показывают, что ферро¬
цен не является диарильным производным железа с ст-связями
Fe—С:Связь Fe—С в алкильных и арильных производных крайне непроч¬
на и легко разрушается.Рентгенографические исследования показали, что в кристалли¬
ческом состоянии средняя по времени конфигурация ферроцена* Перекись хрома СгОб относится к группе синих надхромовых кислот,
образование которых используется для открытия хрома.— 87 —
Рис. 33. Строение молекулы ферроценаявляется пентагональной антипризмой (рис. 33). Не¬
возможно указать, с каким именно атомом углеро¬
да в циклопентадиенильном кольце связан атом же¬
леза, поскольку относительно него кольца занима¬
ют симметричное положение.Циклопентадиенид-анион •сн-имеет характерную особенность — систему из шести я-электронов,
аналогичную ароматическому секстету бензола, а еще точнее, его
гетероциклических аналогов: пиррола, тиофена и фурана:В таких системах происходит делокализация я-электронов, и их
систему можно рассматривать как единое целое. Отрицательный
заряд циклопентадиенид-иона также делокализуется до такой
степени, что эффективные заряды всех пяти атомов углерода ста¬
новятся одинаковыми, а центром тяжести заряда становится центр
кольцаСНСНСвязь между атомами железа и циклопентадиенидом в ферро¬
цене неионная. Во-первых, ион Fe2+, обладающий шестью d-элек¬
тронами, по правилу Хунда имел бы четыре неспаренных электрона,
общий спин 2,0 и большой магнитный момент. На самом деле фер¬
роцен диамагнитен. Во-вторых, большая устойчивость ферроцена
в химических реакциях резко отличает его от ионных соединений с
углеводородными анионами, которые крайне легко гидролизуют¬
ся, например, от циклопентадиенида калия.В ферроцене ароматические секстеты я-электронов являются
донорами, используя в качестве акцепторных две 3d-, одну 4s- и
три 4(о-орбиталиРе2+, гибридизованные по схеме d2sp3. Образуются
шесть многоцентровых связей. Шесть d-электронов Fe2+ при этом
принудительно спариваются (диамагнетизм) и в свою очередь участ¬
вуют в дативных связях с кольцом, что приводит к тому, что на— 88 —
атоме железа в общем остается небольшой положительный эффек¬
тивный заряд, а молекула дополнительно стабилизируется.Для ферроцена при помощи метода МО были развиты и коли¬
чественные теории связи.Циклопентадиенид-анион и другие рассматриваемые лиганды
являются амбидентатными и могут координироваться тремя спо¬
собами. Присоединение лиганда- с использованием в качестве до¬
норных его я-орбиталей называется я-координацией. Кроме того,
возможна координация через один из атомов углерода при помо¬
щи ст-связи, т. е. ст-координация, а если лиганд является анионом,
то он может и координироваться при помощи электростатических
сил. Способ координации указывают при построении названия ком¬
плекса: так, ферроцен называют бш>(я-циклопентадиенил)-желе-
зом. Ковалентная ст-связь осуществляется в бис-(ст-циклопентадие-
нил)-олове и в аналогичных соединениях сЬннца, висмута, индия,
ртути — в общем, у ионов с электронной оболочкой dla или d10 s2.
Ионные соединения, образуемые щелочными металлами, Mg2+,
Мп2+, называют циклопентадиенидами.Сэндвичевое строение молекулы не обязательно указывает на
наличие в ней л-связанных лигандов. Если лиганд связан электро¬
статически, а отрицательный заряд на лиганде делокализован,
то при отсутствии геометрических затруднений наиболее близкой к
катиону должна быть точка, соответствующая центру тяжести от¬
рицательного заряда. У ароматических анионов типа циклопента-
диенида она близка к центру кольца, поэтому в ионных цикло-
пентадиенидах могут осуществляться конфигурации, сходные с
сэндвичевыми. Такие конфигурации получаются также, если цикли¬
ческий лиганд имеет два симметрично расположенных донорных
атома, образующих с катионом ст-связи. Так, дикетопиперазин,СН2—NH—С=оI | в* водном растворе в присутствии КОН взаимо-
0=С NH—СН2действует со свежеосажденной Си(ОН)2, образуя K2tCuL2]— пур¬
пурные призматические кристаллы. Анион этой соли имеет
сэндвичевое строение— 89 —
При растворении ферроцена в серной или азотной кислоте обра¬
зуется катион феррициния (n-C5H5)2Fe+ с железом в степени окис¬
ления +3 синего цвета. Получены многочисленные аналоги фер¬
роцена, содержащие замещенные циклопентадиенид-ионы: факти¬
чески в циклопентадиенильные кольца ферроцена до или после его
синтеза могут быть введены почти любые заместители. Дицикло-
пентадиенильные производные типа ферроцена известны также для
Ru (рутеноцен), V, Сг, Os (осмоцен), Со(кобальтоцен), Ni (никело-Рис. 34. Структуры комплексов:а — ^-дитио-гексакарбонилдиенилдижелезо [SFe(CO),]2; б — г-аллил-тс-цикдо-
пентадиенилпалладий (С3Н6) 1М(С§Н6); а— нафталинхромгрикарбонилцен) и ряда других элементов. Особый интерес представляют спо¬
собные к существованию в водных растворах катионы (я-С5Н5)2Со+
(кобальтициний-катион), (n-C5H3)2Rh+ и (л-С5Н5)21г+. Они могут
быть выделены в виде солей с крупными анионами: пикратом,
тетрафенилборатом, рейнекатом [Cr(NH3)2(SCN)4 Г, обнаруживая
в этом сходство с К+, Rb+, Cs4". Известны смешанные циклопента-- 90 -
диенильные соединения: гидриды, карбонилы, нитрозилы
(рис. 34)Другие ароматические молекулы также способны быть ли¬
гандами в соединениях сэндвичевого типа. В первую очередь — бен¬
зол. В настоящее время синтезированы дибензолхром (л-С6Н4)2Сг и
его аналоги, например, дитолуолхром; соли катиона (я-С6Нв)2Сг+;
дибензольные производные ванадия, молибдена и др. В соедине¬
ниях типа (л-С6Н6)2М металл находится в нулевой степени окис¬
ления, катионы типа (л-СвН6)2М+ содержат однозарядный металл
Mo, Re, V и т. д.Остальные ароматические карбоцяклы также с успехом исполь¬
зовались в качестве лигандов. Эту роль играли инденил-анион,
дифенил, тетралин и др.Длительное время не были синтезированы л-комплексы арома¬
тических гетероциклических систем. Поскольку в их состав, вхо¬
дит гетероциклический донорный атом, они склонны к образованию
«т-связи с атомом металла. В настоящее время известны трикарбо-
нилы тс-тиофенхрома и ^-2,4,6-трифенилфосфоринхрома, я-тио-
фенжелезо-дикарбонил, иодид л-1-метилпиридиниймолибдентри-
карбонила.Нагреванием Мп2(СО)10 с пирролом в петролейном эфире полу¬
чены оранжевые кристаллы я-пирролилтрикарбонила марганцаВ ферроцене, дибензолхроме и подобных соединениях при обра¬
зовании химической связи участвует система делокализованных
я-орбиталей лиганда. При этом формулы комплексов изображают в
видеСНинденил-анион дифенилтетралинЯ - (С4Н 4N)Mn (СО)3.СгFeВ сэндвичевых комплексах хинонов, циклооктадиена и некото¬
рых других лигандов я-связи лиганда несопряжены и лиганд
имеет конфигурацию ванны:МПолучены я-комплексы металлов с производными циклобута¬
диена. Циклобутадиен, гораздо более напряженный, чем цикло¬
бутан, неустойчив и не выделен, но он стабилизируется, входя
в качестве я-лиганда в комплекс. Так, реакцией дихлороциклобу-
тена с Fe2(CO)9 в пентане при 30° получено трикарбонил (я-циклобу¬
тадиен) железо:НСНСCHCI Fe,(CO)g 9^CHCI"С/П'сиHCzz ^снFeос7со СОВсе металлы, способные образовывать комплексы с я-связью в
качестве донора, являются переходными. Для образования сэн-
двичевых соединений в d-оболочке иона металла должно отсутство¬
вать не менее двух электронов и присутствовать хотя бы один. Не¬
известны сэндвичевые я-комплексы для элементов подгрупп цинка
и меди, а также в подгруппах никеля и кобальта со степенью окисле¬
ния металлов 0 и +1.§ 5. Карбонилы в цианиды переходных металловКарбонилы могут быть получены прямым взаимодействием окиси
углерода с некоторыми переходными металлами, например,I атм, 25°Ni + 4СО >- Ni(CO)iПолучены карбонилы для переходных металлов V—VIII групп
периодической системы:V V(CO)e
Сг, Mo, W Сг(СО)в, Мо(СО)в, W(CO),Mn, Re Mn2(CO)10, [Re(CO)6]„Fe.Ru, Os Fc.(CO)5, Ru(CO)5, Os(CO)6Fi;2(CO)9, Ru(CO)9 , Os2(CO)9,Fe3(CO)12, Ru3(CO)12
Co, Rh, Ir Co2(CO)8, [Rh(CO)4]„, [lr(CO)4]n,Co4(CO)i2, [Rh(CO)3]„Ni Ni(CO)4— 92 —
Карбонилы Ni(CO)4, Fe(CO)5, Ru(CO)5, Os(CO)5, Co2(CO)8
представляют собой жидкости, последние три разлагаются ниже
100° с образованием устойчивых многоядерных карбонилов. Соеди¬
нения металлов VI группы сублимируются при 100—200° без раз¬
ложения.При более сильном нагревании все карбонилы разлагаются
на металл и СО. Карбонилы не смешиваются с водой, но раство¬
римы в органических растворителях. Легко летучие карбонилы
Ni и Fe сильно ядовиты.Если подсчитать внешние электроны атома металла и донорные
электроны лигандов, то их сумма ei устойчивых карбонилах равна
18 (правило 18 электронов). При таком подсчете для металла п-го
периода учитываются d-электроны (я—1)-го уровня и s-электроны
п-го, а для молекулы СО — два электрона, если она не мостиковая.
Атомы V, Сг, Мп, Fe, Со и Ni представляют 5, б, 7,8, 9 и 10 электро¬
нов соответственно, так как имеют конфигурации от 3d3 4s2 до 3d8 4s2.
Поэтому для Сг, Fe и Ni возможно образование одноядерных кар¬
бонилов с присоединением шести, пяти и четырех молекул СО соот¬
ветственно: Сг(СО) 6, Fe(CO) s и Ni(CO) 4.Металлы с нечетным числом электронов (V, Мп и Со и их
аналоги) не могут образовать незаряженные одноядерные карбо¬
нилы. Существование V(CO)6 противоречит данному правилу и
его приходится рассматривать как исключение. Для этих метал¬
лов возможны следующие варианты образования простейших
карбонилов:а) комплекс, имеющий избыточное число электронов, стабилизи¬
руется, теряя электрон: так, у-Мп(СО)6 было бы 19 электронов, но
образуется катион [Мп(СО)6]+ с 18 электронами и степенью окис¬
ления марганца +1;б) комплекс с недостаточным числом электронов, например,
■Мп(СО)Б с 17 электронами, может стабилизироваться несколькими
способами: приобретение электрона ведет к образованию анионно¬
го комплекса [:Мп(СО)БГ со степенью окисления марганца — 1*;
объединение двух частиц по схеме(СО)БМп • + • Мп(СС% —Мп2(СО)10
приводит к образованию двуядерного карбонила со связью ме-* Некоторые соли этого аниона, например Hg[Mn(CO) &] 2, Zn[Mn(CO) 5] 2,
Cd[Mn(CO) 5] 2, аналогично иону [НСг2(СО)10]_ имеют линейную структуру
(СО)5Мп — М — Мп(СО)б со связью металл — металл. Cd[Mn(CO) в] 2 полу¬
чают при взаимодействии порошка кадмия с Mn2(CO)io при 120° С в инертной
атмосфере.— 93 —
тал л — металл:о=с—мп мп—с=оМП (J=(JНаконец, радикал -Мп(СО)Б со многими радикалами дает сме¬
шанные комплексы типа СН3Мп(СО)5.Объединение радикалов может происходить с использованием
групп СО в качестве мостиковых. Так, в Со2(СО)818 электронов включают 9 электронов кобальта, 6 электронов немо-
стиковых (концевых)-групп СО, 2 электрона из двух мостиковых
групп.Рассмотрим схему МО окиси углерода (8 МО и на них размещены
10 электронов):В молекуле СО ст-связывающая и а*-разрыхляющая орбитали
образуются за счет перекрывания s/5-гибридных орбиталей по линии
связи С—О. На двух оставшихся sp-гибридных /г-орбиталях (ст„ и
ап ) расположены четыре электрона. Две я-орбитали молекулы СО
также заполнены электронами (рис. 35 и 36).0=С—Со-Со—С=0Со-СоОоои<<Немостиковый
карбонил кобальта«Мостиковый» карбонил
кобальта6*—- Я*
Донорно-акцепторная a-связь в карбонилах образуется за счет
взаимодействия d-орбитали металла, например d*»—,,», с /г-электро¬
нами СО, находящимися на sp-гибридной <т„-орбитали. В моле¬
куле СО сг-донорной
электронной парой слу¬
жит пара, локализован¬
ная на атоме углерода.Дативная я-связь в кар¬
бонилах образуется за
счет вакантных разрых¬
ляющих я*-орбиталей
СО и донорных d-орби-
талей металла, напри¬
мер, dxy. Схема образо¬
вания связи в карбони¬
лах приведена на рис. 37.Sp( п(QbSf: с)Рис. 35. Схема образования aи Двух тс-связей в молекуле СО7Г*ЛРис. 36. ^-Связывающая, ^’-разрыхляющая и n-орбитали в мо¬
лекуле СОИзучение Ni(CO)4 показывает, что группа Ni—С—О линейна
и связь Ni—С(1,82 А) на 0,37 А короче ординарной связи Ni—С. Ли¬— 95 —
нейность подтверждает участиеп-электронов СО в образовании свя-Сзи, так как участиел-электронов привело бык структуре М || .ОУменьшение расстояния Ni—С подтверждает существование да¬
тивной связи.Цианид-ион изоэлектронен с молекулой СО. В изоэлектронном
рядуМ=0+, С=0, C=N~ цианид-ион имеет наибольшую склонность
к образованию ст-связей. Акцепторные свойства цианид-иона выра¬
жены слабее, чем у молекулы СО, но доказательством способности
образовывать я-связи является стабилизация цианидами низких
степеней окисления центрального иона металла (см. табл. 12).Таблица 12Цианидные комплексы с низкими степенями окисления
иона металлаКоличест*
во внеш¬
них d-и s-
электро-
нов в ионе
металлаСостав комплексаК. ч. ме¬
таллаСтруктура комплексаИзоэлектрон-
ный карбонил1099[Pt° (CN)4]4-
[Pd°(CN)4]4_,
[Ni°(CN)4]4_
[Ni1 (CN)4p-[ Ni' (CN)*]*-444Тетраэдр»Планарная со
связью М—М[Ni°(CO)4]987[Co° (CN)*]»-,
[Pd° (CN)e]«-
-[Co1 (CN)4]3~
[Co”(CN)10]e-546Квадратная пи¬
рамида со связью
М—М
Плоский квадрат
Октаэдр со
связью М—М[Со°(СО)е][Fe°(CO)4][Мпа(СО)10)67 1[Mn1 (CN).]»",
[Re1 (CN).]»",
[Co°(CN)6]e_
[Mn«(CN)6]«-66Октаэдр»[Сг°(СО)0]V»(CO)eК наиболее прочным цианидным комплексам относятся комплексы
Mn3+, Fe3+, Fe2+, Со3+, Ni2+, Cu+, Мо4+, Ru2+, Rh3+, Pd2+, Ag+, W4+,
Ree+, Re5+, Os2+, Ir3+, Pt2+, Au3h, Au+. Цианиды Ta3+, Ti3+, V2+,
V3+, Сг2+, Сг3+, Mn2+ и Co2+ имеют заметно меньшую устойчивость.
Для таких металлов, как Zr4+, Hf4+ и Nb5+, цианидные комплексы
неизвестны.Группа NO+ изоэлектронна с CN- и СО, и известны многочислен¬
ные смешанные нитрозилкарбонилы и нитрозилцианиды. Формаль¬
но они могут быть произведены или от NO+, или от N0. Так, про¬
дуктом взаимодействия K.4[Fe(CN)e] с НЫОз является нитропрус-— 96 —
сид калия K2lFe(CN) 5N0 ], который можно рассматривать или как
производное Fe3+ и N0, или Fe2+ и NO+.Согласно правилу 18 электронов в этих соединениях группа
N0 вносит три электрона (два донорных и п*-разрыхляющий),
а группа N0+ —два. Смешанные нитрозилкарбонилы Fe и Со, обра¬
зующиеся при взаимодействии N0 с карбонилами Fe и Со, имеют
формулы Fe(CO)2(NO)2 и Co(CO)3NO, подчиняющиеся этому пра¬
вилу. Атомы М, N и О в них расположены линейно и расстояние
М—N существенно короче ординарной связи, т. е. в этих соедине¬
ниях имеются дативные связи.Правилу 18 электронов подчиняются также смешанные цикло-
пентадиенилкарбонилы металлов (анион С 5Н 5" является донором
шести электронов). В соответствии с этим известны следующие ней¬
тральные соединения:(CaH.),Ti<CO)*С5Н6М(СО)4, где M==V, Nb, Та;[С6Н6М(СО)3]2, где M = Cr, Mo, W;С5Н6М(СО)3, где М =;Мп, Тс, Re;[С6Н6М(СО)2]2, где M==Fe, Ru, Os;С5Н6М(СО)а, где М == Со, Rh, Ir;[С5Н6М(СО)]2, где M = Ni, PL* Два d-электрона дает ион Ti2*, 12 электронов дают ионы С6Н6 и 4 элект¬
рона — молекулы окиси углерода.
V. ВИДЫ ИЗОМЕРИИ КОМПЛЕКСНЫХ ионовНекоторые виды изомерии комплексных соединений не связаны
с изомерией самого комплексного иона. Их классифицируют следу¬
ющим образом:1. Координационная изомерия соединений, состоящих по
крайней мере из двух комплексных ионов, обусловлена неодинако¬
вым распределением лигандов в составе комплексных ионов:
[Co(NH3) 6] [Cr(CN)6 ] и [Cr(NH3)e][Co(CN)6].2. Координационная полимерия — явление, сходное с предыду¬
щим видом изомерии, но соединения одинакового эмпирического
состава обладают разным молекулярным весом.Так, формулы [Pt(NH3)4][PtCl4] и [Pt(NH3) 4 ] [Pt(NH3)Cl3 ] 2
можно записать в виде (Pt(NH-;)2Cl2}2 и (Pt(NH3) 2С12}3. По резуль¬
татам элементарного химического анализа эти вещества невозможно
отличить друг от друга и отдихлородиамминплатины [Pt(NH3) 2С12 ]3. Г ид ратная изомерия: изомеры различаются по функции
воды, входящей в состав соединения. Для хлорного хрома, напри¬
мер, известны три модификации:[Сг(Н20)в]С13 сине-серая (растворы фиолетовые)[Сг(Н20)5С1]С1:, • Н20 светло-зеленая
[Сг(Н20)4С12]С ■ 2НаО темно-зеленая4. Ионизационная метамерия: изомеры различаются по распре¬
делению анионов между внешней и внутренней сферами комплекс¬
ного иона: [Pt(NH3)4Cl2] Вг2 и [Pt(NH3)4Br2]Cl2.Перечисленные виды изомерии наблюдаются для соединений с
инертными комплексными ионами.Рассмотрим более подробно изомерию собственно комплексной
частицы.§ 1. Структурная изомерияСтруктурными изомерами называются такие комплексные час¬
тицы, в которых при одинаковых лигандах наблюдается разная кон¬
фигурация координационной сферы. В качестве примера можно при-Со(СН3—С=СН—С—СН3)2.
вести комплекс | ц В твердом состоянииО- NHего молекула имеет плоское строение, а в безводных раст¬
ворах существует равновесие между плоской и тетраэдрической
конфигурацией. Обе конфигурации сильно отличаются по спектраль¬
ным и даже магнитным свойствам: плоские комплексы низкоспи¬
новые, а тетраэдрические — Еысокоспиновые. Это связано с мень¬
шей симметричностью плоской конфигурации. Аналогичные явления
наблюдаются у хелатных комплексов никеля, например у бис -N-
метилсалицилальдимината никеля. Многие димерные карбонилы— 98 —
существуют в виде мостикового и немастикового (со связью металл-
металл) изомеров. К ним относятся Со2(СО)в (см. стр. 94),
[(СН3)аР ]2Со2(СО)6, [(n-C5H5)Fe(CO)2]2 и др.Некоторые полиморфные превращения комплексных соединений
представляют собой реакции структурной изомеризации. Так, при
переходе a-модификации Cs2MnCl4, содержащей бесконечную цепь
октаэдров {МпС16}, срос¬
шихся ребрами, в р-модифи¬
кацию образуются изолиро¬
ванные тетраэдры МпС1^~.Структурная изомерия
часто связана с искажением
правильной координационной
сферы за счет эффекта
Яна — Теллера. Так, при
искажении октаэдрической
структуры высокоспиновых
комплексов Си2+, Сг2+ и
Mn3+(d9 и d4) и низкоспино-
вых комплексовCo2+(d7) коор¬
динационная сфера может иметь конфигурацию удлиненного и
укороченного октаэдра (рис. 38). Удлиненная конфигурация при¬
мерно на 1 ктл/моль энергетически выгоднее, т. е. в равновесных
растворах ее концентрация при 25° С примерно на порядок выше.Близки по смыслу к структурной изомерии конфигурационные
равновесия в растворах, например равновесия между голубыми тет¬
раэдрическими и розовыми октаэдрическими комплексами Со2+,
которые связаны с изменением координационного числа и явля¬
ются реакциями присоединения, а не структурной изомеризации.§ 2. Изомерия связиНекоторые лиганды, например ионы CN“, SCN~ N02 и др.W:C=№H:N=C = S:
н:0 -^N= Osимеют несколько донорных атомов и могут координироваться раз¬
ными способами.Координация NO2 возможна как через атом азота, так и через
кислород. Комплексы первого типа называются нитро-; а второго —
нитритокомплексами. Координация роданида возможна через азот
и серу, наконец, координация цианида — через азот и углерод.
Лиганды такого типа иногда называются амбидентатными.Рис. 38. Изомерные конфигурации ком¬
плексен Си2+ в виде удлиненного (а) и
укороченного (б) октаэдров— 99 —
В оранжевом кристаллическом комплексе PdL(SCN) 2, где L—это
(C6H5)2PCH2CH2CH2N(CH3)2, роданид-ион, находящийся в транс-
положении к атому фосфора, координирован через атом азота, а вто¬
рой— через атом серы. В решетку KAg(CN)2 входят линейные
ионы:Присоединение CN через углерод наблюдается также BFe(CN)e ,
Co(CN);T, Ru(CN)r, Мо(СЫ)б и др. В решетках Zn(CN)2,AuCN, AgCN,
KCu(CN)3 наряду со связями Me—С имеются связи Me—N.Комплексы, отличающиеся только способом присоединения ам-
бидентатного лиганда, называются связевыми изомерами. Наибо¬
лее известна связевая нитрито-нитро изомерия: при реакциях обмена
комплексов Со3+ с нитрит-ионами вначале получаются розовые нит-
рито-изомеры, например, [Co(NH3)5ONO ]2+, транс-[Co(NH3)2
(py)2(ONO)2]+ или цис- [Co(NH3)4(ONO)21+. Как в растворах, так
и в составе твердых веществ они переходят со временем в желто-ко¬
ричневые нитро-изомеры [Co(NH з) 5NO 2 ]2+, транс-[Со(NH3) 2(ру) 2
(N02)2]+ и t}uc-[Co(NH3)4(N02)2 ]+. Изомеризация идет довольно
быстро, поскольку возможен механизм внутримолекулярной пере¬
группировки. Аналогично ведут себя нитритные комплексы Rh3+,
Ir3+, Pt4+; нитритный комплекс Сг3+ не изомеризуется. Описаны свя-
зевые изомеры с амбидентатными лигандами SCN-. В комплексах
Fe3+ роданид координирован через азот, а в комплексах Hg2+
и других катионов класса Б — через серу. В некоторых комплек¬
сах Со3+, Pt2+, Ni2+ и т. д. эта координация различается по энер¬
гетике незначительно и равновесие между связевыми изомерами
может быть смещено изменением внешних условий. Так, равнове¬
сна связевой изомеризации(здесь L — 4-трет-бутилпиридин, Н 2R — диметилглиоксим) в ап-
ротонных растворителях с диэлектрической постоянной D < 10
сдвинуто влево, при D > 30 — вправо, а при 10 < D < 30 оба
комплекса присутствуют в растворе в соизмеримых количествах.Некоторые лиганды проявляют амбидентатность за счет присо¬
единения или поя-, или по сг-типу. Так, для бис-(инденил) ртутив растворах существует равновесие между я- и ст-изомерами причал
скорости взаимопревращения довольно высоки.N—С—Ag—С—N[CoL(HR)2SCN] [CoL(HR)2NCS]- 100 —
В названии комплексного соединения, содержащего амбидентат-
ный лиганд, указывается донорный атом, через который осуществля¬
ется координация. Например, комплекс [Zn(H 20)4(C5H4NCOO)21
называют бис- (N-никотинато) - тетрааквоцинком. Способ коорди¬
нации позволяет определить ИК-спектроскопия. Рассмотрим ИК-
спектр координированного роданид-нона, строение которого явля¬
ется промежуточным между структурами<—>N=C=S N = C—S(->I IIПри координации через азот строение роданида приближается к
структуре I. При этом кратность связи С—N уменьшается, а свя¬
зи С—S увеличивается. При координации через атом серы увеличи¬
вается кратность связи С—N, а связь С—S приближается к орди¬
нарной. С ростом кратности связи должна расти частота ее валент¬
ных колебаний v. При координации роданида через азот v(CS) по¬
вышается от 750 до 800 см'1, а при координации через серу понижа¬
ется до 700 слг1, однако v (CN) в обоих случаях повышается. Из¬
менение способа координации амбидентатного лиганда может про¬
исходить при отщеплении от него иона водорода. Так, ион Си2+ при
pH 5 координирует H2L~— однозарядный анион 3,4-диоксифенил-
глицинаNH—СН2—('ООНI/\через атомы азота и группу — СОО". При pH 9 анион L3" коорди¬
нируется через ионизированные фенольные группы. Это значит,
что реакция[Cu(H2L)2] [CuL2]4-+4H+сопровождается существенным изменением способа координации
лиганда в комплексе.§ 3. Геометрическая и оптическая изомерияРассмотрим наиболее часто встречающиеся конфигурации коор¬
динационной сферьь начиная с к. ч. 3. Все возможные построения
координационной сферы можно разбить на два типа: открытые и за¬
крытые, при этом под открытыми понимаются такие структуры, в ко¬
торых центральный ион металла не экранирован полностью лиганда¬
ми и доступен извне. Укажем на два ряда открытых конфигураций:
плоские правильные многоугольники с центральным атомом в цент¬
ре и правильные пирамиды с центральным атомом в вершине. Вид— 101 —
сверху у них одинаков:а = 120* 90” 72* 60“В плоских многоугольниках и пирамидах при к. ч. больше 3 ли¬
ганды сближены сильнее, чем в правильных многогранниках с тем
же к. ч. Координационные сферы, представляющие собой плоские
многоугольники с к. ч. больше 6, не способны к существованию:
во-первых, из-за того, что однотипные лиганды с одинаковым эф¬
фективным зарядом и не связанные друг с другом химической связью,
расталкиваются; и, во-вторых, из-за расталкивания электронных
облаков валентных орбиталей центрального атома. Это же справед¬
ливо и для пирамид. Угол а в них меньше, чем в соответствующем
плоском многоугольнике, поэтому расталкивание начинает прояв¬
ляться здесь еще раньше.Практически пирамиды и плоские многоугольники с к. ч. 5 и б
неизвестны. Они встречаются лишь изредка как составные части
'более сложных конфигураций: в соединении Na[U02(CH3C00)3]
атомы карбоксильного кислорода расположены по искаженному
шестиугольнику вокруг урана, атомы кислорода из группы U0£ —
на прямой, перпендикулярной плоскости шестиугольника, т. е.
шестиугольник присутствует здесь как составная часть бипирамиды.Изомерия открытых форм. Будем рассматривать лиганд А как
основной, а*Б, В, Г — как монодентатные заместители. Тогда од-
нозамещенные конфигурации не имеют изомеров,— все положения
заместителей равноценны.Двузамещенные конфигурации у треугольника не имеют изо¬
меров. У квадрата 1,2-замещенная конфигурация — г^ис-изомер;
1,3-замещенная ■—транс-изомер. Цис-транс-юомерш является
частным случаем геометрической изомерии. При геометрической
изомерии одни и те же лиганды одним и тем же способом присоеди¬
няются к центральному атому. При этом в основном сохраняется
конфигурация координационной сферы, но изменяется порядок при¬
соединения лигандов, в результате чего изменяются ближайшие со-А Вседи лиганда. Так, в цис-изомере группа А имеет сосе-А Б— 102 —
А Вдями группы А и Б, а в транс-изомере | —■ две группыБ АБ. Геометрическая правильность координационной сферы после
введения в нее заместителей не сохраняется.При наличии в комплексном соединении двух или более одина¬
ковых лигандов линии поглощения в инфракрасном спектре долж¬
ны расщепляться из-за появления нескольких типов колебаний
лигандов относительно центрального атома. Однако у транс-изо¬
меров, обладающих центром симметрии, расщепления полос не про¬
исходит, что позволяет отличить их от ^ыс-изомеров. Так, в области
1100 см"1 транс-изомер [Coen2(SCN)2] SCN имеет одну полосу
(1126 см'1), а г{«с-изомер—две (1123 и 1144 см'1). Комплекс с
центром симметрии (транс-изомер) можно отличить также по ну¬
левому дипольному моменту, однако измерение дипольного момента,
как правило, удается провести только для нейтральных комплекс¬
ных частиц с крупными органическими лигандами.У треугольной и четырехугольной пирамид (рис. 39) зеркально¬
отраженные конфигурации с двумя разными заместителями относят¬
ся к разным ионам. Соответствующие плоские фигуры совмещают¬
ся поворотом на 180° вокруг осиС2. У пирамид при таком вра¬
щении основания совместятся, но вершина окажется с другой
стороны плоскости основа¬
ния. Изомерия такого рода
называется оптической. Она
наблюдается, если каждый
атом в обоих изомерах име¬
ет одинаковое окружение,
но комплексный ион не обла¬
дает ни одним элементом сим¬
метрии. Изомеры, являющие¬
ся зеркальным отображением
друг друга, называются опти¬
ческими антиподами. Физи¬
ческие свойства их крайне
близки, а энергии образова¬
ния одинаковы, т. е. равно¬
весная смесь должна быть ра¬
цематом — состоять из 50%
одного и 50% другого анти¬
пода. Название «оптический»
происходит от способности оп¬
тических изомеров вращать
плоскость поляризации поля¬
ризованного луча, быть «оп¬
тически деятельным». Враще¬
ние плоскости поляризации уРис. 39. Оптическая изомерия у.
пирамидальных комплексов (вид
сверху)- 103 -
антиподов происходит в одинаковой степени и во встречных напра¬
влениях; в результате рацемат является оптически недеятельным.При синтезах комплексных соединений, имеющих оптические
изомеры-антиподы, образуется, как правило, рацемат. Для разде¬
ления рацемата на изомеры или для синтеза только одного из изо¬
меров необходимы специальные меры. Выделенный изомер рацеми-
зуется: быстро в случае лабильных комплексов и тех инертных комп¬
лексов, рацемизация которых возможна по механизму внутримо¬
лекулярной перегруппировки; медленно — в случае остальных
инертных комплексов*. Поскольку рацемизация лабильных комп¬
лексов идет чрезвычайно быстро, они фактически не могут быть рас¬
щеплены на оптические антиподы, но это не значит, что оптическая »
изомерия у лабильных комплексов не существует.Плоские правильные многоугольники и пирамиды наиболее важ¬
ны из открытых конфигураций координационной сферы. Однако их
существование ограничивается, по-видимому, кристаллическим сос¬
тоянием и, может быть, растворами в растворителях с большим
объемом молекул. Действительно, трудно представить, как может
сохраниться открытая конфигурация, например, в водном растворе.Изомерия закрытых форм. Е1ажнейшие закрытые конфигурации
с полностью равноценными лигандами — это или правильные много¬
гранники (тетраэдр при к. ч. 4; октаэдр при к. ч. 6; куб при к. ч. 8;
додекаэдр, икосаэдр), или призмы и антипризмы для четных коорди¬
национных чисел (6, 8, 10
и 12), соответственно три-,
тетра-, пента- и гексаго¬
нальные. Пример триго-
нальной антипризмы —ок¬
таэдр, тетрагональной при¬
змы — куб.Рассмотрим изомерию
замещенных комплексов
только для тетраэдра и
октаэдра. У тетраэдра и
октаэдра из-за равноцен¬
ности мест в координа¬
ционной сфере однозам'е-
щенные производные изо¬
меров не имеют. Двузамещенные производные у тетраэдра изомеров
не имеют; трехзамещенные производные типа МАБ3 и МАБ2В
можно представить как соответственно одно- и двузамещенные тет¬
раэдра МБ4, т. е. они тоже не ймеют изомеров. У тетраэдров с че¬
тырьмя различными лигандами МАБВГ (рис. 40) каждый лиганд
в обеих конфигурациях имеет тех же соседей, но конфигурация I не
может быть переведена в конфигурацию II иначе, как зеркальным* Например, правовращающий комплекс [Со(еп)з]С18 не теряет оптичес¬
кой активности при нагревании до 127° в течение 85 ч.— 104 ——РБРис. 40. Зеркальная изомерия у тетраэ¬
дрических комплексов МАБВГ
отображением, т. е. здесь наблюдаются оптические антиподы. Дей¬
ствительно, тетраэдр МАБВГ не имеет уже ни одного элемента сим¬
метрии. При к. ч. 4 для квадрата характерна цис-транс-изомерия и
отсутствует оптическая. Наоборот, у тетраэдра полностью отсут¬
ствует геометрическая
изомерия, но возможна
оптическая. Тетраго¬
нальная пирамида до¬
пускает существование
изомеров обоих типов.Такое резкое различие
в характере изомерии
позволяет определить
конфигурацию у комп¬
лексов с к. ч. 4.Валентные углы ок¬
таэдра (рис. 41) состав¬
ляют 90°, длины связей
равны. Это высокосим¬
метричная фигура, сим¬
метрия ее отвечает формуле 3L44L36L29PC. После введения замести¬
теля (в положение 1) образуется конфигурация, не имеющая изо¬
меров, но обладающая резко пониженной симметрией: L44P. Вве¬
дение нового заместителя возможно з 2-м и в б-м положениях; при
этом могут возникнуть if «с-транс-геометрические изомеры. Сим¬
метрия транс-МА4Б2 и /лранс-МА4БВ — L44L25PC и L44P; симмет¬
рия цис-МА4Б2 и г^ис-МА4БВ — L22P и Р. Это значит, что двуза-
мещенные производные октаэдрического строения не имеют опти¬
ческих изомеров, однако уже трехзгмещенные будут их иметь.Интересен вопрос о закрытых конфигурациях с неравноценным
положением лигандов: здесь уточняется постулат о полной равно¬
ценности всех связей в комплексном ионе с одинаковыми лигандами.
Ранее комплексные соединения рассматривали как возникшие в
результате объединения нескольких «валентно-насыщенных» моле¬
кул и записывали, например, в виде 2КС1 -PtCl4 вместо современг
ной записи Kz[PtCl6]. После того как выяснилось, что все шесть
атомов хлора координируются платиной, встала задача определить,
отличаются ли в комплексном ионе «свои» четыре атома хлора от
«чужих». Исследование показало, что ион представляет собой ок¬
таэдр, в котором все лиганды равноценны. Возникло и распростра¬
нилось представление о том, что все связи (в конфигурации с оди¬
наковыми лигандами, по крайней мере) в комплексных ионах равно¬
ценны. И действительно, связи «металл—лиганд» совершенно не зави¬
сят от происхождения лиганда. Что же касается их равноценности в
статическом (длина, направленность, полярность, энергия и т. д.)
и динамическом (реакционная способность) смысле, то этот вопрос
требует уточнения с двух точек зрения. Во-первых, некоторые кван¬
товомеханические эффекты ведут к более или менее сильному иска¬Рис. 41. Октаэдрическая конфигурация коор¬
динационной сферы (а) и валентные углы в ок¬
таэдре (б)— 105 —
жению симметричных конфигураций (эффект Яна—Теллера). Во-вто¬
рых, лиганды принципиально неравноценны в некоторых бипира¬
мидах и пирамидах с центральным расположением иона металла.
При одинаковых лигандах конфигурация тригональной бипирами¬
ды осуществляется в пентакарбониле железа Fe(CO)6, в ионе CuCls
и т. п. Три связи в горизонтальной плоскости расположены здесь
под углом' 120° друг к другу; с остальными двумя связями каждая из
них составляет угол 90°. При этом даже если длины всех связей оди¬
наковы, положения 1 и 5 и 2, 3, 4 неравноценны; если при реакци¬
ях замещения конфигурация бипирамиды сохранится, то можно ожи¬
дать появления двух однозамещенных геометрических изомеров —
экваториального и аксиального.Тетрагональную бипирамиду можно рассматривать как искажен¬
ный октаэдр. При этом также появляются новые виды изомерии,
происходящие от неравноценности координационных позиций, од¬
нако этот вопрос связан с природой искажения правильной конфигу¬
рации.§ 4. Конформационвая изомерияСуществует несколько типов изомерии, связанных с образовани¬
ем цикла многодентатным лигандом. Прежде всего изменяются
возможности для геометрической изомерии. При определении числа
геометрических изомеров лиганды можно рассматривать бесструк-
турно: бидентатные симметричные'лиганды в виде А ^ А, а несим¬
метричные в виде А В (А и В—донорные атомы). Например,
СН3—СН-СН21,2-пропилендиамин | | должен изображаться в видеNH3 NH2А ^ В. Вернером были выделены а- и (3-изомеры цис-
[Co(en)(pn)(N02)2]+ со строениемh,c-nh2
Iн2с
/H,Nна\ nh2НС/Iсн3;.ли в упрощенном видеH2N,— 7no2 h2n,/ С° / /W—T-m HjNf-A/~r~7N0*
В L—H,C—NH,
/H2C
/H2N/?NO,CoNO,HCV NH3HjO^hVГ~7L 'N(no2NO,— 106 —
За счет возникновения асимметрии донорного атома при коор¬
динации могут появиться оптические изомеры у квадратных ком¬
плексов, например, у комплексаП'-'о ^с асимметричным атомом азота.Конформационная изомерия специфична для хелатных комплек¬
сов. Конформационные изомеры отличаются другот друга прост¬
ранственным расположением атомов в циклах и переходят друг
в друга без разрыва связей. Конформационную изомерию вызывает,
например, шестичленный несопряженный цикл, присутствуя в одной
комплексной частице в форме ванны, а в другой — в форме кресла.Так, ион [Co(NH2CH2CH2CH2NH2)3]3+ в бромидных раство¬
рах присутствует в виде двух изомеров: в одном все шестичлен¬
ные циклы имеют конфигурацию кресла, а во втором — скошен¬
ной (во избежание F-напряжения) ванны. Первый изомер примерно
на 0,5 ккал/моль более выгоден энергетически. Конформационные
изомеры появляются также у комплексного иона, содержащего не¬
сколько пятичленных несопряженных неплоских циклов. Напри¬
мер, в квадратных комплексах возможны конформациипричем в конформации I над горизонтальной плоскостью, в кото¬
рой лежат атомы азота, расположены атомыCj и С3, а в конформа¬
ции II — атомы С2 и С3.III
VI. ХИМИЧЕСКАЯ СВЯЗЬ В КОМПЛЕКСНЫХ СОЕДИНЕНИЯХ
ПЕРЕХОДНЫХ МЕТАЛЛОВ§ 1. Применение теории групп к молекулярным структурамПри помощи теории групп для молекулярных структур осущест¬
вляют классификацию атомных орбиталей по симметрии, что поз¬
воляет указать орбитали, взаимодействующие между собой с обра¬
зованием МО. Это сильно облегчает качественное рассмотрение
электронного строения молекул, а при количественном его анализе
существенно упрощает вычисления.Операцией симметрии для молекулы является такая операция,
которая переводит ядра молекулы в положение, не отличимое от ис¬
ходного. Обычные для молекул операции симметрии приведены в
табл. 13. Операциям симметрии ставят в соответствие операторы
Е, Сп, и, i и т. д. Действие оператора А на объект X, в резуль¬
тате чего получается объект Y, записывают в виде У — АХ. Про¬
изведением В А операторов В и А называется оператор, действие
которого на объект равносильно действию на этот же объект операто¬
ра Л, а на результат — оператора В. В общем случае В А не равно
АВ.Таблица 13Наиболее обычные операции симметрииОбозначениеОперацияЕс*Тождественное преобразование
fe-Кратный поворот нюкруг оси на угол IninОтражение в плоскости: ад—отражение в плоскости, пер¬
пендикулярной главной оси; — отражение в плоскости, со¬
держащей главную ось; ad — отражение в диагональной пло¬
скости; h — горизонтальная плоскость;
v — вертикальная плоскостьfe-Кратный поворот вокруг оси на угол Ъz/n с последую¬
щим отражением в плоскости, перпендикулярной оси поворота
Инверсия в центре симметрииСовокупность операций симметрии для молекулы образует
точечную группу симметрии молекулы. Точечной группой порядка
h называется множество из h элементов, которое имеет следующие
свойства:1) произведение АВ любых двух элементов множества принад¬
лежит тому же множеству;2) соблюдается ассоциативный закон умножения, т. е. (АВ) С =
= Л(ВС)-— 108 —
3) в группе существует единичный элемент Е и для всех элементов
группы справедливо равенство ЕА = АЕ = А;4) каждому элементу А соответствует элемент А_1, называемый
обратным, и справедливы равенстваА~1А = АА~ 1 = Е.При поисках элементов симметрии молекулу обычно изобража¬
ют так, чтобы ось высшего порядка совпадала с осью г; если имеетсяРис. 42. Элементы сим¬
метрии для молекулы
формальдегида/1Fr'13by'’srУуУ-+F2б*Рис. 43. Элементы еим
метрии молекулы SF4точка, в которой оси и плоскости симметрии пересекаются, то ее выби¬
рают в качестве начала координат. Элементами симметрии для мо-/Нлекулы формальдегида 0=сС (рис. 42) являются вращениеХНвокруг оси z(C2) на угол я, отражения в плоскостях симметрии
xz(<tb) и yz(aB') и операция идентичности (£). Действие каждой из
этих операций приводит к расположению ядер атомов, которое экви¬
валентно первоначальному. Совокупность элементов Е,С2, и а0'
образует точечную группу симметрии С 2р.Для построения таблицы умножения элементов группы рас¬
смотрим последовательное проведение: двух операций симметрии.
Так, при операциях симметрии точечной группы С2о в молекуле
SF4 (рис. 43) атомы фтора изменяют положения согласно табл. 14 в
отличие от атома серы.Из табл. 14 видно, что последовательное проведение опе¬
раций С2 и ст0 дает такой же результат, как операция сг/,т. e.C2cjv=
= сг1/. Аналогичным образом можно получить всю таблицу умно-,
жения группы С2о (см. табл. 15).Рассмотрим преобразование орбиталей s, рх, ру, рг под действи¬
ем операций симметрии группы С а„. Пусть орбитали принадлежат— 109 —— 111 -
Таблица 14Действие операций симметрии группы C2v на молекулу SFtНачальное положение атома фтора1234Положение атома фтора после действияЕ1234оператора с2214312432134Таблица 15
Таблица умножения для группы симметрии С2г,Первый множительВторой множительЕ2Сгa'v°VЕЕс2a'vavс2СЕavaiavЕС2avс2Еатому, ядро которого имеет декартовы координаты (0, 0, 0). Тогда
уравнения для функций s,px,pyup2 в декартовых и сферическихРис. 44. Связь декартовых
и сферических координатРис. 45. Элементы симмет¬
рии группы С2о— 110 —
координатах (рис. 44) будут/2 Мs = /х (г), ру = /2 (г) sin a sin <Р = —-— у,, . . . „ /2 (Г) г , , с f sW
P.r =/ 2 М Sln ^ C0S 9 — *» Рг — / 2 (Г) C0S ^ = 2 ■Если ось С 2 совпадает с осью z, а плоскости а^, и ст0 с плоскостями
xz и уг соответственно (рис. 45), то координаты х, у и z любой точки
преобразуются согласно табл. 16.Таблица 16Преобразование декартовых координат элементами группы
симметрии C^vНачальные значения координатх у ZРезультат Ех У 2действия С2—X —у 2оператора avх —У 2—X у 2Ех = х; С2х = — х;avx = х; а0 х — — х;Ьй1IIи£II(ЦavU = —у> г/ = У,Ez = z; С22 = г;°vy = г; 2 = 2.Величина радиус-вектора точки г = V х2 + у2 + z2 и функцияs, зависящая только от г, при действии рассматриваемых операций
симметрии не изменяются, а функции рх, ру и pz преобразуются так
же, как координаты х, у и г.Из табл. 17 преобразований" p-функций видно, что каждая
из них преобразуется через себя (со знаком «+» или «—»), но не
смешивается с другими функциями.Таблица 17
Преобразования /7-функций элементами группы C%vИсходные функцииРхРУpzРезультатыЕРхPvPzдействияС2—Рх-РуPzоператоровРх-РуPz%—РхРуPzТеория представлений. Если декартовы координаты (хъ уъ zx)
некоторой точки в результате преобразования симметрии перехо-— 111 —
дят b(jc2,y2,z2),TO эти новые координаты являются линеиными
функциями xlt уъ zx:*2 = auxi + аиУ1 + а1зг1>Уг = °21*1 + °22!/l + а232Ъ
г2 = a3i*i + ааг!/1 ~Ь азз21-(VI.1)Квадратная таблицааП Д12 Л13
Д22 й23
^31 й32 fl33составленная из коэффициентов aijt называется матрицей преобра¬
зования. Матрица размерности ту,п (сначала указано число строк,
а затем — число столбцов) может быть умножена на матрицу раз¬
мерностью п хN. Результатом будет матрица размерностью mxN,
элемент которой cVj является скалярным произведением i-й строки
первой матрицы на j-й столбец второй. Если N = 1, то вторая мат¬
рица — это вектор из п элементов, а результат — вектор из т эле¬
ментов. В таком случае система уравнений (VI. 1) равносильна мат¬
ричному уравнениюИтак, вектор [хг, у2, z2) получается из (л^, уъ или применени¬
ем к нему некоторой операции симметрии, или умножением на мат¬
рицу преобразования. Эту матрицу называют представлением опе¬
рации симметрии в данном базисе, понимая подбазисом преобразуе¬
мый вектор [хи уъ zх}. Матрицы-представления квадратны и
имеют размерность, равную числу элементов базиса. Из табл.
17 преобразования p-функций видно, что^2 [рх 1 Ру 1 Рг] “ ( Pxi Ру 1 Рг] •Вектор-результат можно записать в форме— Рх = — 1 • Рх + 0 • ру + 0 ■ Рг.— Ру = 0 • Рх + (— 1) • Ру + 0 • рг
Pz = 0 • Рх + 0 • Ру + 1 • PzОтсюда видно, что матрица-представление операции Са в базисе
p-функций будетДля разных преобразований группы С2о базис p-функций нуж-— 112 —
но умножить слева на следующие матрицы:Группа матриц, действие которых на базис из данных функций
(например, p-функций) совпадает с действием элементов симметрии
на этот же базис, называется представлением точечной группы сим¬
метрии в данном базисе. Табл. 15 умножения элементов симметрии
группы C2v справедлива для элементов симметрии и их представле¬
ний-матриц. Набор четырех матриц Е, С2, ov, а0 образует представ¬
ление группы С2v в базисе p-функций. Можно получить представле¬
ние группы С2о в базисе пяти d-функций. В табл. 18 показано
преобразование d-функций поддействием операций симметрии груп¬пы С2d-Матрицы преобразований группы С2о в базисе d-функций имеютвидСг&/ ш000о'/По 001/ЕВ о о оdй]000°\010000-1 00°\01 0 000-1000001000 0-70*\00-1 000010000010ООО100 0 1000010\ 0000\о 0 00\оООО7/\ 0о.00чРазмерность и вид матриц-представлений зависят от выбора ба¬
зиса. Совокупность элементов базиса, члены которой преобразуются
в функции элементов только этой совокупности может сама быть
базисом представления. Процесс разложения базиса на базисы мень¬
шей размерности называется приведением. Приведение заканчива¬
ется, если полученные базисы не поддаются дальнейшему приведе¬
нию; тогда они называются неприводимыми. Этим неприводимым
базисам соответствуют неприводимые представления (НП) груп¬
пы симметрии.Таблица 18
Преобразование d-функций элементами группы С2оИсходные функции*XZУ2Згг—г2ХарактерППРезультат Еху*хгУ*X2—у2322—Г25действия Счху—хг—угх"—I/2323—Л21операторов av—хухг-yzX2—у2322—Г21av—ху—хгУгX2—у2Зг2—г21* Символ d у функции опущен.5-699— 113 -
При разложении приводимого представления (ПП) на неприво¬
димые, как правило, приходится предварительно подвергнуть базис
такому линейному преобразованию, чтобы матрицы-представления
для нового базиса имели блочную (квазидиагональную) структуру.PiР 2Такую матрицу приводят, выделяя отдельные блоки, например
матрицы Dx и Г)2, соответствующие НП р1 и р2- Если все матрицы-
представления диагональны, та их разбивают на блоки единичной
размерности, т. е. каждый элемент базиса является базисом НП.Рассмотрим представления группы С2с в базисе р- и d-функций.
Соответствующие матрицы диагональны. Одинаково расположенные
диагональные элементы, например, четыре единичные матрицы, отме¬
ченные квадратами, образуют НП. Приводимое представление груп¬
пы С2в в базисе р-функций распадается на три НП: {1, —1, 1, —1);
{1/—1, —1, 1}; (1, 1, 1, 1}, а в базисе d-функций на четыре НП:
{1, 1,-1,-1}; {1,-1, 1,-1}; {1, —1, —1, 1}; {1, 1, 1, 1}. Последнее
из них повторяется дважды.Для группы С2в существует только четыре разных набора мат¬
риц размерности 1x1, т. е. четыре НП типа Аъ А 2, Въ В2
(табл. 19).Таблица 191Матрицы, являющиеся неприводимыми представлениями
точечной группы С2эПредставлениеЕЭлементыс.симметрии"avаОБазисы соответствующих
представлений-^1(О0)(1)(1)Pz 1 dxi_yt i dztЛ2(ОО)(-1)(-1)d.xyВх(О(-о(1)(-1)Pz > d,XZв2-(1)(-D(-1)(1)Py, dy2В сложных случая-х приведение представлений сильно облег¬
чается использованием таблиц характеров. Характером матрицыНулевыеэлементыНулевыеэлементыD%- 114 —
называется сумма ее диагональных элементов, поэтому матрицам-
представлениям группы С 2„ в базисе p-функций соответствуют сле¬
дующие характеры:*£=3; = хв =1: х ' = 1>е *-t av Соа в базисе d-функцийХр = 5; = — 1; Х„ =1 иХ, = 1.£ с« °v аVХарактер матрицы приводимого представления должен быть
равен сумме характеров матриц НП. Обозначим через kt, k2, k3 и
it4 коэффициенты, указывающие, сколько раз в данном приводимом
представлении содержатся НП Аъ А2, В х и В2 соответственно.
После этого, составив и решив систему линейных алгебраических
уравнений, найдем значения ^ит. д. Для d-базиса характеры ПП
приведены в табл. 18. Используя характеры НП из табл. 19, полу¬
чим уравнениядля Е 5 = ki -f- k2 -f- k3 -(- (1)для Ca 1 = ki + fe2 — k3 — kt (2)для av 1 = — ,k2 + ks — ki (3)для o' 1 = kx — кг — k3 + kt (4)Суммируя уравнения (1) — (4), получаем 8 = 4klt откуда кг = 2.
Суммируя уравнения (1) и (2), получаем б = 2(k1 + k2), откуда
k2 = 1. Суммируя уравнения (1) и (3), получаем 6 = 2(кх + kз),
откуда kz = 1. Суммируя уравнения (1) и (4), получаем 6 =
= 2(kx + &4), откуда А4 = 1. Итак, d-базис распадается на
■2Aj + А2 + В, + В2.Характеры НП точечных групп 0Л, C4v, Dih, Dtd и Td приве¬
дены в табл. 20 и табл. 3 приложения. Буквами х, у, и z указано,
по какому представлению преобразуются сами координаты.Таблица 20Классы элементов симметрии и характеры НП точечной группы Од°АЕ8Сзбс26 с*ЗС2=ЗС,4S2=i6 st8S,ъ°п6odAig+1+1+1+1+1+1+1+1+1+1Аы+1+1+1+1+1—1—1—1—1—1fyg4"!+1—1—1+1+1—1+1+1—1А 2 и+ 1+1—1—1+1—1+1—1—1+1Es+2—100+2+20—1+20El+ 2—100+2—20+1—20Tlg+30—1+1—1+3+10—1—1Ты+30—1+1—1—3—10+ 1+1T2g+30+1—1—1+3—10—1+1Т2а+30+1—1—1—3+10+ 1—15* - 115 -
Классификация НП и буквенные обозначения для них вводят¬
ся следующим образом: если для какого-либо элемента симметрии
характер НП равен 1, будем называть его симметричным относитель¬
но этого элемента; у антисимметричных НП характер равен — 1. Не¬
приводимое представление, базис которого состоит из т элементов,
имеет размерность т, т. е. является m-кратно вырожденным. НП с
размерностью 1 называют невырожденными.Символ А обозначает невырожденное НП, симметричное
относительно главной оси, а символ В—антисимметричное. Символы
Е и Т обозначают дважды и трижды вырожденные НП. Если мо¬
лекула обладает центром инверсии, то нижний индекс g исполь¬
зуется для обозначения симметрии (+1), а индекс и — антисиммет¬
рии (—1) относительно него. Индексы 1 и 2 указывают на симметрич¬
ное или антисимметричное расположение относительно других осей
вращения (в частности относительно оси 2-го порядка, перпендику¬
лярной главной оси). Если других осей
нет, то индексы относятся к симметрии
относительно плоскости av. Симмет¬
рия и антисимметрия относительно го¬
ризонтальной плоскости симметрии сгА
обозначается одним или двумя штри¬
хами. Малыми буквами а, Ь, е, t обоз¬
начают совокупность функций, пре¬
образующихся по представлениям типа
А, В, Е, Т.Элементы симметрии с одинако¬
выми характерами можно объединить
в классы. Рассмотрим, например,
преобразование p-функции атома азо¬
та в молекуле NH3 под влиянием
элементов симметрии группы С3„
(рис. 46). Элементы симметрии груп¬
пы C3v запишем в виде Е, 2С3, 3а„,
т. е. два поворота вокруг оси третьего порядка Сз и С\ (на 120 и
240°) объединим в один класс 2С 3, а отражения в трех вертикаль¬
ных плоскостях симметрии аг — в класс Зз0.Действие операции Сз на базис (х, у, г\ представим при по¬
мощи общего выражения для вектора {х'} у', z'j, возникающего
при вращении базиса вокруг оси z на угол ср:х’ = a:c:os<p—у sin <р,у' = X МП ср + у cos <р,= г.Подставив ф = 120° и ф — 240°, получим первые три матри-Рис. 46. Расположение мо¬
лекулы аммиака относитель¬
но осей координат— 116 —
цы ПП группы C3v в базисе р-функций:/7 000 10\о 01Для полученияплоскости хъ (а»)сз(1 а\г г0Уз 1г г00 01г = г, т. е. к матрицесз(1 Узп\г го \Уз 1пг ги\ 0 0иЧ■ а00-10007а сг0 = Сзсго иаа = С|сг0. Матрицы этих элементов имеют виддля бi! 1
2Уз2ЛдляпУз2Уз10•Уз12222001\ °0\Характеры матриц одного класса действительно одинаковы: для
Е X = 3; для Сз и Сз X = 0; для всех ov X = 1. Способ приведе¬
ния базиса координат указан клетками в матрицах ПП. Базис ко¬
ординат распадается на базис [х, у], преобразующийся по двукрат¬
но-вырожденному НП Е, и базис [г], преобразующийся по НП А.Базис p-функций приводится так же, как базис координат: он
распадается на базис [рх, ру) и базис [рг), т. е. трехкратно вырож¬
денный р-подуровень в поле симметрии С3v расщепляется на орби¬
таль ах и две вырожденные орбитали е\РхI Ру < Pz Рх-PzРу01свободный атомХарактеры НП группы C3vатом в поле
с симметрией С„ЕА111а211— 1Е2—10— 117 —
позволяют подтвердить правильность проведенного разложения р-
базиса:X (Е) = 2 —10+X = 1 1 1X {Рх< Ру> Pz} 0 1Найдем характеры приводимого представления группы С3о в
базисе d-функций. Для этого достаточно провести по одному преоб¬
разованию d-функций в каждом классе (для удобства символ d опус¬
тим), напримерс\(ху)= cUx)c\{y) = (- *--Т“ х~\У) =1 ут
= ~-~y ху ~ “Т- — ^ ■Результат действия оператораПехотныефункцииЕсзаV322 - Г2
хгЗг2 - г2
хгЗг2 - г3
1_ хг2Зг2 -— г2
хгчг1/2/3“г- *z1 UZ2— У*хуХУ12 А !/-(дг* —»•)4— хух*-у*х2 — у2V" 3 ху —4- (*2 — у2)X2 (/2X5•—11Коэффициент — -^-появляющийся перед ху при преобразованииэтой величины, является диагональным элементом и при расчете
характера суммируется с коэффициентами, стоящими перед другими
функциями в выражении для их преобразования. Поэтому характер
ПП для Сз равен2 2 2 2Аналогично этому в базисе d-функций для оператора Е характер
ПП равен 5, а для операторов класса av + 1.
Для приведения базиса d-функций используем еще один метод
разложения ПП на НП. Введем'понятие вектора характеров, у кото¬
рого столько элементов, сколько классов в группе. Каждый элемент
этого вектора равен произведению характера класса на корень квад¬
ратный из отношения числа элементов в этом классе к порядку груп¬
пы h, т. е. вектор характеров ПП d-базиса1 хг={5х л[т!-1х/та векторы характеров НПг-Н,х У т;1хУт^.-{1х Vт;1х Ут-' *я = {ах -у/--lxiУт}‘
Ут}--л/~Скалярное произведение X на вектор характеров НП указывает,
сколько раз данное НП входит в Г1П:т--3-— = о,6Отсюда следует, что в поле симметрии С 3v базис d-функций разлага¬
ется на три НП: одно типа Аг и два типа Е. При этом функция d2*
является базисом одномерного представления A lt а остальные
d-функции — базисами двумерных представлений:—► ■—►5 2X =6 ~ з'5 2XXAt =6 _ 6■ ► ■—V10 2xxE =: 6 + (3Свободный ион— _ е (dXz’dyz)
~■ е О, (dZ2) •Ион в поле симметрии C3v.§ 2. Теория кристаллического поляВ октаэдрическом поле представление A lg индуцируется s-ор¬
биталью; Т1и — орбиталями рх, ру и рг\ Т 2g — орбиталями dxv
dyz, dxl и представление Eg — орбиталями dz« и dx*-y* цент¬— 119 —
рального атома. Базис из пяти d-орбиталей распадается на два не¬
приводимых представления Eg и Т Zg. По этим двум однократно вхо¬
дящим НП орбитали классифицируются следующим образом:fgg Idjry , dyz, dX2),
eg [ > dxi_yt },В поле лигандов cf-подуровеиь расщепляется (рис. 47). Величи¬
на этого расщепления измеряется разностью энергий А между ор¬
биталями eg и 12g и
определяется спектро¬
скоп ическим'и метода¬
ми. Величина А может
быть определена по
энергии, необходи¬
мой для перехода
электрона с одного
уровня на другой*.
Так, для комплек¬
са [Ti(H20)6]3+ пере¬
носу единственного
d-электрона с уровня
12g на eg соответствует полоса вблизи 5 ООО А (А а* 20 ООО смг1). Обыч¬
но значения А заключены между 10000 и 30 000 см'1. По возраста¬
нию величин А лиганды можно расположить в спектрохимический
рядI" < Вг~ < SCN- < Cl" < NOJ- < F" < ОН" < С20‘~ < НаО << ру < NH3 < en < NOJ < CN-который почти не зависит от иона металла и геометрии комплекса.-
В спектрохимическом ряду лиганды находятся в порядке возраста¬
ния силы их кристаллического поля. Если энергия пяти вырож¬
денных d-орбиталей равна 5E(d), то после расщепления в октаэ¬
дрическом поле5Е (d) = 2Е (eg) + ЗЕ (t2g) (VI.2)A = E(eg)-E(f2g). (VI.3)Умножив (VI. 3) на три и сложив (VI. 2) и (VI. 3), получимE(eg)—E{&) = -^~ Д = 0,6Д;Оумножив (VI.3) на два и вычтя (VI.3) из (VI.2), получим* Поскольку энергия кванта'пропорциональна частоте или волновому
числу, выраженному в см'1, то энергию в спектроскопии часто выражают не¬
посредственно в см'1.е9а 5Рис. 47. Расщепление d-подуровня в полях
симметрии Ой (<з) и Td (б)- 120 —
E (tig)-Е (d) = - Д = — 0,4Д,Огде 0,6Д и —0,4Д — приращения к энергии E(d) после расщепле¬
ния.Шкала энергии выбирается так, что за начало отсчета берется
E(d) — положение центра тяжести всех пяти d-орбиталей, которое
остается неизменным под действием поля лигандов. При октаэдри¬
ческой симметрии орбитали eg подняты на 0,6Д, а орбитали 12g опу¬
щены на 0,4Д. Поэтому, если d-электрон попадает на орбиталь 12г.то
комплекс становится на 0,4Д более стабильным, чем следует из про¬
стой электростатической модели. Эта дополнительная энергия назы¬
вается энергией стабилизации кристаллическим полем (ЭСКП). Ее
можно вычислить, прибавляя 0,4Д на каждый электрон, занимаю¬
щий 12г-орбиталь, и вычитая 0,6Д на каждый электрон, занимающий
е^-орбиталь, т. е. если заселенность d-подуровня t\g epg, тоПри небольших значениях Д (слабое поле) электроны в основ¬
ном состоянии занимают орбитали по таким же правилам, как
будто Д = 0. Образуются высокоспиновые комплексы, например
пять d-электронов располагаются по одному с параллельными спи¬
нами на каждой из орбиталейи ЭСКП = 3 • 0,4Д — 2 • 0,6Д = 0. ЭСКП для слабых полей
приведена в табл. 21.При больших значениях Д (сильное поле) пять электронов в ос¬
новном состоянии дают низкоспиновый комплекс, конфигурациюи ЭСКП = 5 -0,4Д — 0 = 2Д.У одного и того же центрального иона в зависимости от рас¬
положения лиганда в спектрохимическом ряду могут образовываться
как высокоспиновые, так и низкоспиновые комплексы. Так, комп¬
лекс CoFe высокоспиновый (4 неспаренных электрона), а комплекс
Co(NH 3)б+низкоспиновый и диамагнитный, т. е. в соответствии со
спектрохимическим рядом расщепление, вызываемое NH3, пре¬
вышает вызываемое фторидом.ЭСКП = —4- (2q — Зр).
5g— 121 —сы возбуждения центрального атома, связанные с изменением глав-
Таблица 21Энергия стабилизации октаэдрических комплексов
слабым кристаллическим полемЧисло d-электроновЭлектровная конфигурацияЭСКП1е°
l2g eg—2/52t2 е¥—4/53t е
l2geg—6/54l2g g—3/55el2g eg06t4 p2
2g eg—2/57/5 e2
{2geg—4/58e2—6/59t eIs *—3/510t6 e4
eg0У высокоспиновых комплексов Сг2+ (d4) и у комплексов
Cu»+ (d9) наблюдаются тетрагональные искажения октаэдра.
В основном электронном состоянии этих ионов (tig elg и )
орбитали 12g заполнены электронами одинаково, а из орбиталей eg
занята электроном (у Сг2+) или дыркой (у Си2+) лишь одна:
dxt_yt или dz« ■ Если электрон находится на орбитали
4t*-u* (рис. 48), то лиганды L2, L3, L4, L6 испытываютбольшее отталкивание со стороны
d-электрона и располагаются на
большем расстоянии от централь¬
ного иона, чем лиганды Ll и Le.
Вместо правильного октаэдра обра¬
зуется укороченный. Наоборот, ес¬
ли орбиталь занимает дырка, то об¬
разуется удлиненный октаэдр. Та¬
ким образом, искажение комплек¬
са снимает вырождение d-орбита¬
лей и понижает энергию одной из
них (dz 1). Увеличение Д при эф¬
фекте Яна— Теллера в системах d4
и d9 обусловлено приближением
части лигандов к центральному ио¬
ну. Дополнительная устойчивость
этих систем называется энергией стабилизации Яна — Теллера.Появление внутренней асимметрии возможно в октаэдрическихРис. 48. Возникновение
внутренней асимметрии в ок¬
таэдрическом комплексе— 122 —
высокоспиновых комплексах при электронной конфигурации иона
d1, d2, di, da, d1, d9 ив октаэдрических низкоспиновых комплекс
сах при конфигурации d1, d2, dk, d5, d7, d9.Теория кристаллического поля хорошо объясняет поведение ио¬
нов редкоземельных элементов, так как химическая связь в комплек¬
сах этих ионов преимущественно ионная (внешние 5s2pe оболочкиРис. 49. Зависимость энергии стабилизации в октаэдрическом поле
от атомного номера редкоземельного элементаэкранируют 4/-орбитали, которые становятся малодоступными для
образования а-и тс-связей). Под влиянием электростатического поля
лигандов семикратное вырождение/-подуровня частично или пол¬
ностью снимается; расщепление подуровня зависит от симметрии
поля. На рис. 49 представлено изменение ЭСКП в ряду редкозе¬
мельных элементов при октаэдрическом расположении лигандов.
Максимум ЭСКП приходится на элементы с 4 и 11 электронами
(Pm, Ег). Для La, Gd и Lu энергия стабилизации равна нулю.Большим достижением теории кристаллического поля является
объяснение окраски и магнитных свойств комплексных соединений.
При качественном рассмотрении принято различать в спектрах ком-
лексных соединений следующие группы полос поглощения:1) полосы интенсивного поглощения, соответствующие разре¬
шенным электронным переходам (коэффициент молярного погашения
е « 103 — 105);2) полосы слабого поглощения (е « 10—100), наблюдаю¬
щиеся у комплексов d- и /-катионов. Они относятся к интраконфи-
гурационным (d — d или / — /) переходам, которые в первом приб¬
лижении запрещены по Лапорту и могут наблюдаться лишь как
смешанные электронно-колебательные переходы или быть так на¬
зываемыми квадрупольными переходами;3) полосы очень слабого поглощения (е л* 0,01 — 1), относя¬
щиеся к запрещенным переходам между термами различной муль-
типлетности, т. е. между состояниями комплексного иона, которые
различаются спином. Нарушение запрета вызывается сочетанием
чисто электронного перехода с различными второстепенными про¬
цессами.Среди полос интенсивного поглощения выделяют Ридберговы поло¬
сы возбуждения центрального атома, связанные с изменением глав¬
ного или побочного квантового числа, например 3dn->- 2>dn~l 4s1. Они— 123 —
аналогичны линиям возбуждения изолированного иона или атома с
той разницей, что при комплексообразовании изменяется схема энер¬
гетических уровней. Энергии таких переходов довольно велики, по¬
этому связанное с ними поглощение наблюдается в дальней ультра¬
фиолетовой области, а влияние лигандов на частоту поглощения щ
обычно мало.Связанные с изменением электронной структуры лиганда и
близкие к полосам поглощения свободного лиганда интралигандные
полосы также интенсивны. В спектрах комплексов, образованных
катионами с внешней электронной оболочкой типа s2р6 (а отчасти
и s2p*d10), обычно имеются только интралигандные и Ридберго-
вы полосы, поэтому цвет таких комплексов совпадает с цветом ли¬
ганда. Так, ксиленоловый оранжевый (H6L), применяемый в качестве
металлохромного индикатора* при комплексонометрическом титрова¬
нии, дает фиолетовые комплексы ScH 2L-, происходящие от аниона
H2L4-, имеющего фиолетовую окраску. Наблюдать образование та¬
ких комплексов на фоне реактива удается потому, что свободные
лиганды H2L4_ при pH 6 протонируются до желтых анионов
H3L3-, H4L2~h т. д., тогда как комплекс разрушается лишь при
pH < 2.Третьим типом интенсивных полос поглощения являются поло¬
сы переноса заряда, которые отвечают переходу электрона с орбита¬
ли, локализованной преимущественно на лиганде, на орбиталь, ло¬
кализованную, в основном, на центральном атоме или наоборот.
Переходы такого рода можно рассматривать как внутримолеку¬
лярные процессы окисления — восстановления. Они особенно ха¬
рактерны для комплексов центральных атомов, имеющих несколь¬
ко степеней окисления.К лигандам, отдающим электроны, относятся, например, гало-
генид-ионы. Интенсивность и длина волны полос переноса заряда
растут в ряду С1~ < Вг-С I-. К переходам этого типа принадлежат
интенсивные полосы 36 500 и 43 960 см_1 для комплекса
lCo(NH3)5Cl]2+, 31 800 и 39 450 смг1 для [Co(NH3)5Br]2+ и 26 110
и 34 930 смг1 для [Co(NH3)5I ]2+. Легко протекающий перенос* Металлохромными индикаторами называются органические красители,
образующие с ионами металлов комплексы, резко отличающиеся по цвету от
свободного индикатора. Так, пиридил-азо-резорцин (ПАР)НО<I^>-n=n-\Z>-ohпри рН5 дает с ионами Си2+ и РЬ2+ ор анжево-красные комплексы. При титро¬
вании трилоном Б (динатриевой солью ЭДТА) раствора, содержащего Си2* и
индикатор, вначале связываются в этилендиаминтетраацетатный комплекс
свободные ионы меди, а затем разрушаются комплексы меди с ПАР. Переход
окраски раствора в зеленую (это цвет свободного ПАР при pH 5) служит при¬
знаком конца титрования.— 124 —
заряда у иодидных комплексов объясняет их нестабильность. Пере¬
нос электронов с лиганда на центральный атом вызывает появле¬
ние полос в спектре оксоанионов VO3,-, СгОГ» М0О4 , WOf, МпО-р
ReO^, FeOf и др.Полосы переноса заряда с центрального иона металла наблю¬
даются у 1,10-фенантролиновых комплексов Fe2+, у пиридиновых
комплексов 1г3+ и т. п. Максимумы поглощения у полос переноса
заряда лежат обычно в
ультрафиолетовой области 600
спектра, а в видимую об- £
ласть (760—400 нм, т. е.13000 — 25 000 смг1) могут
попасть их «хвосты», вызы¬
вая желтый, бурый или
красный цвет комплекса.Полосы переноса заряда и
Ридберговы полосы назы¬
вают интермолекулярными.И нтраконфигурацион¬
ные переходы d — d или
/—/ чрезвычайно характер¬
ны для комплексов пере¬
ходных металлов, лантано¬
идов и актиноидов. По энергии они захватывают видимую,. близ¬
кую ультрафиолетовую и близкую инфракрасную область спектра
(рис. 50) и поэтому сильно влияют на окраску комплексов. Прибли¬
женный анализ возможных переходов может быть проведен при по¬
мощи теории кристаллического поля.Рассмотрим, например, октаэдрические высокоспиновые комп¬
лексы. При конфигурации dl (Ti3+) основным состоянием комплекса
является t\g e°g, а возбужденным— t]g е\. Поэтому возможен единст¬
венный d — d переход t2g-> eg, который и обуславливает, например,200002500030000Рис. 50. Кривая поглощения акво-ком-
плекса Мп2+ в видимой и близкой уль¬
трафиолетовой области-4-_1 Ti3+Свет
5 ООО к^<н2о,бГ *[Ti(H20)6FРис. 51. Интраконфигурационный переход d—d у Tis+фиолетовую окраску комплекса [Ti(H20)e]3+ (рис. 51). При кон¬
фигурации d2(V3+) основное состояние t\g el с неспаренными спинами.— 125 —
Переходы в состояния■н~++запрещены по спину (по мультиплетности). Разрешены по спину
переходыпричем вероятность первого из них (двухэлектронного) очень мала.
Второй из них приводит к конфигурации t\g е\, которой при учете
межэлектронного отталкивания соответствуют два терма, обозначае¬
мые 3Tlg и 3T2g, и, следовательно, у рассматриваемых комплексов
возможны два электронных d — d -перехода. Действительно, ион
[V(H20)fi]3+ имеет две полосы поглощения: при 17 700 (565 нм)
и 25 ООО слс1 (400 нм).Конфигурации de и da ведут себя аналогично конфигурациям d1
и d2. Электронные конфигурации d10~n рассматривают как дырочные
конфигурации dn, Так, электронная конфигурация d9(^ej) экви¬
валентна конфигурации дырокd1(t2gelg) т. е. возможный переход
дырки eg ->- t2g строго соответствует (противоположен по направле¬
нию) электронному переходу в электронной конфигурации d1.У высокоспиновых конфигураций d5(Mn2+,. Fe3+) и /7 (Gd3+)
невозможны интраконфигурационные переходы с сохранением спи¬
на. Поэтому в спектре соответствующих ионов интраконфигураци¬
онные переходы наблюдаются лишь в виде полос очень слабого по¬
глощения. В спектрах ионов с конфигурацией d°, d10, f° и /14
интраконфигурационные переходы вообще невозможны. Комплексы
перечисленных ионов или практически бесцветны, или окрашены за
счет полос переноса заряда (например, комплексы Fe3+ с феноль¬
ными лигандами, роданидом и т. д.) и интралигандных полос.Диамагнитные свойства присущи всем веществам без исключе¬
ния. Они вызывают возникновение силы, выталкивающей вещество
из неоднородного магнитного поля. Если все электроны в веществе
спарены, оно обладает только диамагнитными свойствами. При на¬
личии неспаренных электронов вещество обладает также парамаг¬
нетизмом, который вызывает втягивание вещества в магнитное поле;
диамагнетизм проявляется в этом случае в виде малой поправки
и вещество ведет себя как парамагнитное. Наиболее выражен пара¬
магнетизм у свободных радикалов (*СН3, *С6Н5 и др.), молекул-§ 3. Магнитные свойства комплексов— 126 —
радикалов (N0, 02 и др.), у ионов переходных элементов, лантано¬
идов и актиноидов.Некомпенсированные спиновые и орбитальные моменты электро¬
нов превращают атомную систему в микромагнитный диполь с мо¬
ментом ц = (L + 25)jiB> где L — полный спиновый угловой мо¬
мент набора электронов в атоме (в атомных единицах), S = 2Sf,—"►где S; — спиновый угловой момент/-го электрона. Единица магнит¬
ного момента— магнетон Бора^в — 0,927 ■ 10-20 эрг/гс. Наличие
магнитных диполей приводит к парамагнетизму.Вследствие взаимодействия микромагнитных диполей соседних
атомов могут возникать ферромагнетизм и антиферромагнетизм.
Этими явлениями можно пренебречь для большинства комплексов,
так как ионы металлов, являющиеся источниками парамагнетизма,
изолированы друг от друга диамагнитными атомами лигандов.Напряженность магнитного поля Н связана с магнитной индук¬
цией В внутри образца соотношением~в = Я + 42x7*,—+где / — намагниченность.Объемная магнитная восприимчивость х и удельная восприим¬
чивость х связаны с / и Н соотношениями / Н ' X~ Hd ~ d 'где d — плотность вещества.Чтобы оценить магнитную восприимчивость иона металла, необ¬
ходимо в наблюдаемую магнитную восприимчивость вещества внес¬
ти поправку на диамагнетизм атомов лиганда. При введении диамаг¬
нитных поправок удобно пользоваться молярными (%м) или атомны¬
ми (ха) восприимчивостями:1м = гм и '(.a = ia<где М — молекулярный вес; А — атомный вес.Все методы измерения магнитной восприимчивости основаны
на том, что сила, действующая на элементарный объем вещества
dv с массой dm, в неоднородном магнитном поле напряженности Н
направлена вдоль градиента поля и пропорциональна ему:dF = (•/. Н grad Н) da = (7 Н grad Н) dm.Если поле по направлениям осей координат у и z однородно, то
сила направлена вдоль оси х и равнаdH dH
dF = ч.Н dv - чН dm.dx dxДля определения магнитной восприимчивости часто использу¬
ют метод Фарадея. Согласно этому методу образец помещают в об¬— 127 —
ласть максимального градиента магнитного поля, которая прихо¬
дится на верхний конец зазора между полюсами электромагнита.Размер образца выбира¬
ется малым, чтобы в его
объеме градиент поля
йШйх можно было счи¬
тать постоянным. Сила,
действующая на обра¬
зец, измеряется кварце¬
вой пружиной, микроа¬
налитическими весами
или специальными мик¬
ровесами (рис. 52). Ус¬
тановку калибруют по
веществам с известной во¬
сприимчивостью (NiCl2;
(NH4)2Fe(S04)2-6H20;
CuS04 • 5Н20;
CoHg(SCN)4).Для парамагнитных веществ магнитная восприимчивость имеет
положительное значение и зависит от температуры, а для диамаг¬
нитных веществ — отрицательное и от температуры не зависит.
Закон Кюри выражает связь магнитной восприимчивости вещества
с его магнитным моментом^эфф = 2,84 "|/" Т ,где 2,84= Y^RlNk |iB; Na —число Авогадро.Рассмотрим вычисление магнитного момента иона металла по
экспериментально определенной магнитной восприимчивости для
диметилглиоксимового комплекса меди C4HeN202Cl2Cu/ОНH3C-C=N/ у ClI '.'Си/Н3С—C=N< ХС1Ч)НПри 30° х = 5,47 ■ 10-6; молярная магнитная восприимчи¬
вость хм = 1370-10'® (мол. вес = 250,5). Для вычисления
полной диамагнитной поправки используем значения Д из табл. 22:С 4 • 6,0 • 10-е = 24,0 • 10-«Н 8-2,93 • 10-« = 23,4- 10-«N 2 • 5,6 • 10-е = 11,2 • 10-«О 2 • 4,6 • 10-» = 9,2 • 10-*С1 2-20,1 • 10-е = 40,2- 10"вДвойные связи 2 ■ (— 8,2 • 10~<;) = — 16,4 • 10_вПолная диамагнитная поправка = 91,6 • 10_6— 128 —Рис. 52. Установка для определения: магнит¬
ной восприимчивости методом Фарадея:/ — образец; 2 — коромысло весов; 3 — уравновешиваю¬
щий груз; 4 — диск торзиоииых весов; 5 — кварцевая
нить
Следовательно, xjn = 1370 • 10-8 + 91,6 ■ 10-e = 1462 • 10'*
и(^эфф = 2,84 V 303,0 • 1462 ■ 10~* = 1,89 .Состояние свободного атома описывается термом, который
представляет собой совокупность уровней энергии с данными L и S,
характеризующими полный спиновый и орбитальный угловые мо¬
менты электронов.Таблица 22Диамагнитные поправки Л для некоторых частицЧастицаА-10е, г-атомNa+6,8Hg*+40Со2+12,8Cl20,1Н2,930(в спиртах и эфирах)4,61N (в открытой цепи)5,6С6,0<х\2IIи-8,2Полный механический момент количества движения всех элект¬
ронов в атоме характеризуется квантовым числом J. Для данного
набора L и 5 оно может принимать все целые значения от | L + S|,
| L + S — 11 до | L — S |, так что имеется 2L + 1 или 2S + 1
значений, смотря по тому, какая из этих величин меньше. Если
подуровень заполнен электронами менее чем наполовину, J в ос¬
новном состоянии равно | L — S}, а при заполнении подуровня
более чем наполовину J равно | L + S|.В магнитном поле магнитный момент системы связан с полным
механическим моментом, пропорциональным у j (У + 1) , для
которого могут наблюдаться различные ориентации. Проекции ме¬
ханического момента Jг на направление поля z принимают целые
значения от —■ J до J, всего 2J + 1 значений. Каждому из них от¬
вечает соответствующая ориентация магнитного момента. Из-за
различия в энергиях взаимодействия с полем энергия каждой ори¬
ентации будет разной, т. е. терм с данным J расщепляется на
2J + 1 компонентов. Расстояние между двумя уровнями рав¬
но £1-1 в Н, где g — фактор спектроскопического расщепления
(g-фактор). Он указывает долю орбитального момента в полном
моменте количества движения.Общий магнитный момент, выраженный в магнетонах Бора,
при учете спин-орбитального взаимодействия электронов определя-— 129 —Alg И I ла/пдиш ПО ftUIWpUIA vjnpj/nvu «IV 1 1 р J f-> '-'«‘А’-" ■ «*«Ч “и,,ит“. vl“роданид-ионы имеют мостиковый характер и обращены атомом азота к иону
ется выражениемгдеJ(J+\)+S(S+\)-L(L + l)2 J (J + 1)(VI .4)Так, для электронной конфигурации d2 основным термом яв¬
ляется aF, где индекс 3 указывает на мультиплетность, равную 25 +
+ 1, следовательно, S = 1, L = 3, 7 = 2. Зная J, можно найти
g и рассчитать для данного атома р.Эфф- Если L = 0 и J = S, то
g = 2 и является чисто спиновым фактором; спин-орбитальное
взаимодействие отсутствует и при образовании элементарных магнит¬
ных диполей существенны только спиновые моменты электронов.
Тогдагде п — число неспаренных электронов в системе.Вычисленные по уравнению (VI. 6) магнитные моменты для 1,
2, 3, 4 и 5 неспаренных электронов равны соответственно 1,73;
2,83; 3,87; 4,90 и 5,9 цв .Сходимость вычисленных по формуле (VI. 6) и эксперименталь¬
ных величин |л,Эфф (табл. 23) хорошая для ионов, имеющих до пяти
З^-электронов. Гораздо хуже она у ионов 4-го периода, имеющих
от 6до9 d-электронов и у переходных ионов 5-го и 6-го периодов.
У редкоземельных элементов парамагнетизм обусловлен частично
заполненным 4/-подуровнем и участие орбитальных моментов явля¬
ется значительным. Уравнение (VI. 6), учитывающее только спино¬
вые эффекты, непригодно для них,так как велика роль орбитальных
моментов. Например, у иона Рг3+ электронная конфигурация 4/2; S=
= 1, L = 5, J = \ L — S| = 4. По уравнению (VI. 6) для
Рг3+[хЭфф= 2,83 [лв , тогда как экспериментальное значение [хЭфф =3.47(хв. При использовании уравнения (VI.4), учитывающего спин-
орбитальное взаимодействие, получается близкое к эксперимен¬
тальному значение р,эфф = 3,58[хв .По результатам измерений магнитной восприимчивости можно
судить о степени окисления центрального иона в комплексе, об эле¬
ктронной, а иногда и о геометрической структуре, комплекса.
Так, соединение Hg[Co(SCN)4] имеет |хэфф = 4,5|хв. Формально его
можно представить соединением или Hg+ и Со3+, или Hg2+H Со2+.
У ионов Со3+ высокоспиновые комплексы имеют 4 неспаренных элект¬
рона и (j, Эфф > 5; низкоспиновые — диамагнитны. У ионов Со2+
высокоспиновые комплексы имеют [i Эфф = 4,30 — 5,20, а низкоспи¬
новые—р эфф = 1,8. Ионы Hg2+ и Hg22 диамагнитны. Сопоставле-^эфф = 2 V"S (S + 1).
Выражение (VI. 5) можно упростить до(VI. 5)Иэфф У П(п + 2) ,(VI.6)— 130 —
Таблица 23Магнитные моменты ионов переходных элементов 4-го периодаИовЧисло d-электроновВысокоспиновые комплексыНизкоспиновые комплексычисло неспа¬
ренных элект¬
роновМагнитныерасчет
по (VI. 6)моменты, р.DОПЫТ.числонеспаренныхэлектроновопытные
магнитные
моменты, itдTi3+i11 ,731,73V<i+i11,731,68—1,78——V3+222,832,75—2,85——V2+333,883,80—3,90——Сг3+333,883,70—3,90——Mn4+333,883,8—4,0——Cr2+444,904,75—4,9023,20—3,30Mn3+444,904,90—5,023,18Мп2+555,925,65—6,1011,80—2,10Fe3+555,925,70—6,012,0—2,5Fe2+644,905,10—5,700—Co3+64 -4,90>50—Co2+733,884,30—5,2011,8Ni3+733,88>41001юоNi2+822,832,80—3,500—Cu2+911,731,70-2,200—ние данных указывает единственно возможный вариант:
Hg[Co(SCN)4] является высокоспиновым комплексом Со2+*.Низкоспиновые и высокоспиновые комплексы одного и того же
катиона очень четко разделяются по магнитной восприимчивости.
Так, наличие ц эфф = 2,8 у [NiCl4]2' показывает, что в комплексе
есть два неспаренных электрона, а в связи с лигандом участвуют
sp3-гибридные орбитали, комплекс имеет тетраэдрическую конфигу¬
рацию. В диамагнитном комплексе [Ni(CN)4]2~ два d-электрона
никеля спарились и образовался плоский комплекс с dsp2-гибри¬
дизацией:3d ksNi2+ “44- — мз<рф=м[NiCl4]2“-ff--ff (§) Мзф<р=2,8[Ni(CN)4]2iff -ff -ff -ftr (D @ Лз<р<р=0* Рентгеноструктурные исследования показали, что в узлах кристалли¬
ческой решетки этого ярко-синего соединения расположены поочередно ионы
Hg2+ и Соа+, каждый из которых окружен по тетраэдру роданид-ионами. Все
роданид-ионы имеют мостиковый характер и обращены атомом азота к иону
Со2+, а атомом серы — к иону Hg2 . Поэтому формулы Hg[Co(SCN)4] и
Co[Hg(SCN)4] лишь приблизительно отражают реальное строение вещества.— 131 —
Одно время применяли так называемый магнитный критерий типа
связи,согласно которому связи в низкоспиновых комплексах ковалентны, а
высокоспиновые комплексы имеют ионную связь. Если низкоспиновость обус¬
ловлена принудительным спариванием электронов с образованием дативных
связей, например, в цианидных комплексах переходных металлов, то дейст¬
вительно очень велика роль ковалентности. Но в некоторых случаях переход
от высокоспиновых комплексов к низкоспиновым происходит при одном и
том же характере связи за счет понижения симметрии. Иногда монодентат-
ные лиганды образуют высокоспиновые комплексы, а аналогичные хелаты
являются низкоспиновыми. Более того, некоторые комплексы Ni2+
имеют изомерные конфигурации (тетраэдрическую и квадратную) с разными
магнитными свойствами. Природа снязи в них одинакова. Поэтому в настоя¬
щее время магнитный критерий для характеристики типа связи почти не при¬
меняется.§ 4. Применение метода МО к координационным соединениямРасчет схемы молекулярных орбиталей в методе МО ЛК.АО
сводится к составлению и решению системы линейных уравнений
относительно коэффициентов разложения МО на АО. Если базисный
набор АО составлен из N орбиталей, то система уравнений имеет
N-й порядок. Решение таких систем затруднено даже для относите¬
льно простых молекул: так, для мо¬
лекулы SFe учет только внешних s и
р-орбиталей атомов серы и фтора при¬
водит к системе из 28 уравнений.
Классификация орбиталей по типам
симметрии позволяет разбить такую
систему уравнений на несколько под¬
систем, каждая из которых решается
отдельно; порядок каждой подсис¬
темы равен порядку соответствую¬
щего неприводимого представления
(НП).Рассмотрим применение теории
групп к октаэдрической частице. У
группы октаэдра Oh (рис. 53) имеет¬
ся 48 операций симметрии, образую¬
щих 10 классов. Четыре оси третьего порядка проходят через
центры противоположных граней и генерируют восемь поворо¬
тов С\ и Сз, три оси четвертого порядка проходят через противопо¬
ложные вершины и генерируют 6 поворотов С\ и Cl и три оси второго
порядка С2 = С\. Шесть осей второго порядка С\ проходят через
середины противоположных ребер. Звездочка * отличает эти оси
от осей С2 = С\.Поскольку октаэдр имеет центр симметрии, то к операциям сим¬
метрии группы октаэдра также относятся операция инверсии i и ее— 132 —Рис. 53. Оси координат в
октаэдреновые—(х Эфф = 1.8- Ионы Ugi+ и Hg"2 диамагнитны. Сопоставле-
произведения на все остальные операции симметрии:8i С3 = 8Se; 6iCi = 6Si, 3('C2 = 3a„; 6iC*2=6ad,где Se и S4 — зеркально-поворотные оси шестого и четвертого по¬
рядка; три плоскости ап являются плоскостями ху, yz и xz\ плос¬
костная делят пополам противоположные ребра.Группа симметрии Oh имеет десять НП: Alg, A2g, Eg, Тlg,
T2g, Alu, A 2Ц, Ea, Tla, T2a. Характеры для этих НП приведены
в табл. 20. Как и ранее, для анализа поведения орбиталей в окта¬
эдрическом поле необходимо пользоваться таблицей действия опе¬
раций симметрии на координаты точки (табл. 24).Таблица 24Преобразование координат при операциях группы Oh
(центр инверсии в начале координат)E8 C33C26 Ctее;I8S,3*h6 s.6’dXXz—X—yУ—X—2XX—yУУX—УXX—y—XУX—XzzУ—2zz—z—y—2—2zX30 111—301— 11 jИз табл. 24 видно, что s-функция преобразуется по типу alg,
р-орбитали принадлежат к типу 11и и в поле симметрии октаэдра не
расщепляются, а остаются вырожденными, как в свободном атоме.
Базис из пяти d-орбиталей распадается на два НП типа Eg и Т 2g,
которые входят в ПП однократно. Орбитали называют соответствен¬
но неприводимым представлениям i2g (dxy, dyz, dxz) и eg(d7*, dX‘-y‘).Найдем уровни энергии МО в ионе [Ti(H20)6 ]3+. Шесть лиган¬
дов расположены по октаэдру. Выберем на центральном ионе и на
каждом лиганде свою систему координат, чтобы ось г на каждом
лиганде была направлена в сторону иона металла. На рис. 54 стрел¬
ками показаны положительные направления осей координат. Для
простоты примем во внимание только a-орбитали лигандов, т. е. ор¬
битали pz. о-Валентными орбиталями центрального иона являются
орбитали 4s, 4р.,., 4ру) 4pz, 3dzt, 3dxt_yi. По симметрии они
классифицируются следующим образом:4s*Рх, 4Ру, 4pz3d~-> . 3d 2 2z • x—у— 133 —algha
Рис. 54. Локальные системы координат в октаэ¬
дрическом комплексеПреобразования о-орбиталей шести лигандов под действием опе¬
раций симметрии группы 0Н приведены в табл. 25.Таблица 25Действие элементов симметрии группы Oh на базис с-орбиталей
лигандов (рис. 55)Классы элементов
симметрииРезультаты действия одного из операторов классаЕ8Сзбс'23 с216S,SS,Зод6ad6Ct«1°1°4в1ов«2с„Ol°а°3°з°4=4°з°5°236°3Элементы°з°2а6=1°334°4базиса°4“бЧ2°6°3°4с3°б°В°1°4°3°3°2°В®2°2ав°в°2°1“в°1°4°1“вХарактеры Х{а}6002000422Представление {а} в базисе о-орбиталей распадается следующим
образом:{<*} = Alg + Т1и + Eg.Соответствующие представлениям A lg, Т 1а и Eg а-орбитали в
октаэдрическом комплексе обозначают a lg, tla и eg. Если построить
линейные комбинации о-орбиталей лигандов (рис. 55), принадлежа¬
щие НП A lg, Т1ии Es, то получатся те волновые функции, которые— 134 —
способны "взаимодействовать с ор¬
биталями центрального иона (табл.26) alg, tlu и eg. Например, орби¬
таль о2 — а4 соответствует по сим¬
метрии орбитали ру и такое же со¬
ответствие наблюдается между ор¬
биталями рг и Cj —<г«; Рх и
Оз —05, s И + Ст2 “Ь °3 “Ь °4 "Ь
+ ог5 + <т6 и т. д. Для орбиталей
иона металла dXy, dxz, dj,2 нет со¬
ответствующих по симметрии ком¬
бинаций о-орбиталей лигандов,
поэтому при использовании о-приб¬
лижения они остаются несвязыва¬
ющими МО. Для образования я-свя¬
зей с лигандами используются орбитали иона металла dxy, dxz,
dyz, Рх. Ру. Рг- По симметрии они разбиваются на типы:dxy > , dyZ t$gPx j Ру I Рг ^litТаблица 26Распределение орбиталей иона металла
и а-орбиталей лигандов по НП группы симметрии ОлРис. 55. Система а-орбиталей ли¬
гандов в октаэдрическом комплексеТипсимметрииОрбитали иона
металлаЛКАО лигандов
(ненормированные)°l gS°1 + + а3 + а4 + аБ + аб*lgРх> Ру> Рга1 — °2 —а4* а3 — °6eg2^1 + 2тв—о2—а3—а6;°2 °3 + °4 аБЛ0ММОи!д— “Н?■ff "If ез
"H- ■fH■ff" °fgРис. 56. Схема МО [Ti(H20)]e3+ в а-приближении— 135 —АО,
Таблица 27Линейные комбинации орбиталей иона металла и лигандов
для октаэдрических комплексовТип орбиталиОрбитали
иона металлаНормированные комбинации орбиталей лигандов<hg («)
eg (а)*1и (о. *)*«*(*)tig («)dx,_t,,Р*РуPzdxzЛугdxy1Кб1— (Sj + а2 + а3 + а4 + °5 + ав)(2ах + 2а„ — о6 — а2 — а3 — о*)2 К?12 (а5 — а2 + а3 — а*)-j^=r к - о.); 7 (*„ + )1 1(=* - =и); — (*,, + )Vi1Vi— К — »•); — (*,. + **, - )*« У<.1Т(я«* +1~г- (п, + я + я„ + я_ )
2 v *« i'l 1 </* 1 *« '1— К. + ”„, + %. + *,4)Т v-**.+*«.)1Т" ("// ——”, + )2 х* у*' far, )2 у* Xl х% у*(7t TZ + те 7С )2 б'в * б'в *i'—— (и_ —Я + 7С„ —ТС. )
2 4 *• V» У» ^47136 —
Это значит, что орбитали типа tlu могут принимать участие и в
а-, и я-взаимодействии. Линейные комбинации орбиталей лигандов
типа t2g(n) и tlu (я) составляют так же, как и для a-связей. При этом
затрачивается шесть из 12 я-орбиталей лигандов. Кроме того, вид¬
но, что эти орбитали дают
линейные комбинации типа
Zlg.(rc) и t2u(Tz). Классифика¬
ция валентных АО октаэд¬
рического комплекса при¬
ведена в табл. 27.Рассмотрим, например,
схему МО в [Ti(H20)6 ]3+.На рис. 56 эта схема при¬
ведена с учетом только
с-взаимодействия и указано
распределение 13 валент¬
ных электронов в основном
состоянии иона. Неспарен¬
ный электрон располагается
на орбитали t2g. Если учи¬
тывать а- и и-взаимодейст-
вия, то получаются 27 ва¬
лентных МО (рис. 57):
шесть ст-МО alg, a* lg, eg и
е\, шесть л -МО 12g и tig,
шесть несвязывающих ор¬
биталей 12и(к) и tlg (тг) и де¬
вять МО типа tla, которые
образуются при взаимо¬
действии орбиталей, ионаметалла рх, ру и рг с орбиталями лиганда tu{o) и tu(b). У иона
металла нет орбиталей, соответствующих линейным комбинациям
орбиталей лигандов типа t1 (к) и ^„(гс). поэтому такие орбитали
лигандов остаются в комплексе несвязывающими МО.В этом приближении приходится учитывать 37 валентных элект¬
ронов. В основном состоянии они распределяются по МОтаким об¬
разом, что неспаренный электрон, как и в упрощенной схеме, зани¬
мает орбиталь t2g.Рис. 57. Схема МО в [Ti (H20)e]s* при
учете a-и п-связываний
VII. КИНЕТИКА И МЕХАНИЗМ РЕАКЦИЙ КОМПЛЕКСНЫХЧАСТИЦ§ 1. Основные понятия. Теоретические моделиКинетические методы исследования комплексных соединений
позволяют оценить реакционную способность, взаимное влияние
лигандов, зафиксировать необычные координационные состояния
центрального иона металла, решить вопрос о природе связи ме¬
талл — лиганд.Скорость V химического процесса в конденсированной фазе оп¬
ределяется как изменение концентрации одного из реагирующих
веществ в единицу времени. В реальных условиях химический про¬
цесс обычно реализуется при помощи нескольких параллельно или
последовательно протекающих химических реакций, которые назы¬
вают элементарными актами процесса.Экспериментально определяется скорость химического процес¬
са в целом. Она зависит от скоростей v элементарных актов, вид
этой зависимости соответствует рассматриваемой системе элемен¬
тарных актов. Например, если протекает несколько (/ = 1, ..., п)
параллельных реакций, в ходе которых расходуется вещество Аг,
то' ПV — 2 ^j [моль!л - сек]./= 1Если протекают обратные реакции, то их скорости с учетом знака
также должны быть внесены в сумму. Если процесс многоступенча¬
тый и нет параллельных реакций, то общая скорость реакции оп¬
ределяется скоростью самой медленной стадии.Если уравнение j-го элементарного акта реакциичд Ai + '<]2 Аа + ... + ijm Ат -> продукты (I)где Ах, А2, ..., Ат — реагирующие вещества; v;-2, ..., vjm —— стехиометрические коэффициенты, то скорость его выражается
уравнениемV,- = kj П а ^== k, а У а 2у ... a , (VI1.1)J J t ‘'12 тгде at — активность вещестЕ.а Аг; kj — константа скорости,
1 —(моль/л) ‘ сек'1. Сумма показателей в уравнении (VII. 1)
называется молекулярностью реакции (I). Она равна общему числу
молекул, участвующих в реакции. При некоторых допущениях зна¬
чения at в уравнении (VII. 1) можно заменить концентрациями
[AJ, изменив соответственно значение константы скорости:vj=kj[kl\1i...[ Affl]V. (VII.2)— 138 —
От уравнений (VII. 1) и (VII. 2) необходимо отличать кинети¬
ческие уравнения для скорости реакции (I), которые получаются
при экспериментальном изучении кинетики. Если, например, V
практически совпадает с vlt а исследование проводится в условиях,
когда все равновесные концентрации, кроме [Ах ], примерно посто¬
янны, тоV^vt = [h [A/*1 [А,]’» ... [Am]4) [А]’“ =k[ [А1]',“ ,
где k\ — эмпирическая константа реакции:A; = MA2]v2i 1А3Га1... [A^i4"*1
при [Ay] « const для j Ф 1.Выражение V = /г! [А* Г11 называется кинетическим уравнением,
а сумма показателей в нем — порядком реакции. Порядок ре¬
акции совпадает с ее молекулярностью лишь тогда, когда кинети¬
ческое уравнение в точности совпадает с уравнением (VII. 2).Для объяснения механизма взаимодействия частиц обычно исполь¬
зуют две теоретические модели: теорию эффективных столкнове¬
ний и теорию переходного состояния. Чаще всего рассматривают
механизм, предусматрива¬
ющий соударения реагиру¬
ющих молекул. Константа
скорости является мерой
частоты и эффективности
соударений. На основании
теории эффективных соуда¬
рений для константы ско¬
рости химической реакции
выведено уравнениеk AIq 6£антгде k0 — предэкспоненци-
альный множитель; Е.\ —
энергия активации. Если
при соударении молекул
образовался активирован¬
ный комплекс,'то реакция
может протекать далее без
добавления энергии. Коли¬
чество энергии, необходи¬
мое для образования акти¬
вированного комплекса, на¬
зывается энергией актива¬
ции. Чем меньше энергия
активации, тем больше кон¬
станта скорости.6Рис. 58. Изменение потенциальной энер¬
гии взаимодействующих частиц в ходе
реакции без образования интермедиата
(а) и с образованием интермедиата (б)— 139 —
Согласно теории переходного состояния при взаимодействии
двух молекул, обладающих необходимой энергией активации, вна¬
чале образуется активированный комплекс (переходное состояние),
который затем разлагается с образованием конечных продуктов. Пе¬
реход от исходного состояния А реагирующих веществ к конечно¬
му состоянию В полученных продуктов может осуществляться как
непосредственно после получения системой энергии (рис. 58, а),
так и через интермедиат /, который является стабильной частицей
(рис. 58, б). Чтобы из состояния f перейти в состояние В, нужно
получить дополнительную энергию активации. Скорость реакции
определяется скоростью прохождения через потенциальный барьер.
Высота этого барьера по отношению к энергии исходного состоя¬
ния равна энергии активации £А.а разность между энергиями на¬
чального и конечного состояния равна теплоте реакции —АН°.§ 2. Механизм реакций замещения лигандовДля реакций замещения с разрывом'связи металл—лиганд раз¬
личают диссоциативный механизм D, ассоциативный механизм А,
а также механизмы диссоциативной активации Id и ассоциативной
активации 1а.На первой и лимитирующей стадии диссоциативного механизма
из исходной комплексной частицы MXnL образуется активированный
комплекс с уменьшенной связанностью «уходящего» лиганда L, а
затем —интермедиат МХ„ с пониженным координационным числом*МХ„ L -* МХ„ + L (а)Oi = —MMX„L], (VII.3)В отличие от активированного комплекса интермедиат устой¬
чив к малым возмущениям, обладает конечным временем жизни,
на кривой энергия — путь реакции (рис. 58) ему соответствует
не максимум, а минимум, а концентрация его в состоянии равнове¬
сия рассчитывается согласно закону действия масс. Образующийся
интермедиат расходуется, вступая в реакцию со «входящим» лиган¬
дом Y* Одним из наиболее стабильных и изученных интермедиатов такого типа
является ион Co(CN)b2~, имеющий форму тетрагональной пирамиды.Равнове¬
сиеСо (CN)3- Со (CN)§_ + CN"можно сильно сдвинуть вправо связыванием выделяющегося цианида. Ион
Co(CN)^ активно вступает во многие реакции присоединения, в частности,
он реагирует с молекулярным водородом, при этом образуется октаэдричес"
кий гидридный комплекс2 Со (CN)?- + На 2 НСо (CN)§“— 140 —
MX„ + Y —МХЛ Y (6).2=-ft2[MX„][Y] (VII. 4>
и с отщепленным лигандомМХ„ + L МХ„ L (в)«3 = — *3 [МХ„] [L] (VI 1.5)причем реакция (в) является обратной по отношению к реакции (а).
Если реакция (б) достаточно быстрая, то в ходе ее, начиная с некото¬
рого времени, установится стационарная концентрация интермеди¬
ата МХП, т. е. скорости образования и исчезновения интермедиата
станут приблизительно равными. Чтобы рассчитать стационарную
концентрацию, в выраженииd[ МХ„]dz■ = -v1 + va + vi = k1 [MX.,L] - (ft2 [Y] + k3 [L]) [MX„]пренебрегают производной d[M.Xn]/dт, так как остальные величины
намного больше ее, и получаютrMX1 feitMX„L11 л1стац k2 [Y] + k3 [L] *Экспериментально измеряемая скорость разрушения комплекса
MX„L (v) должна быть равна ь1 — v3, что соответствует кинетичес¬
кому уравнениюd[MX„L]di■ = k± [МХП L] + k3 [MX„] [L] == -^[MX„L] - - = ~^"L] ■ ■ (VII. 6)^2 1*1 + ^3 [L] I _l. *3 [L]MY]Если входящий лиганд является' молекулой растворителя, значе¬
ние [Y] не меняется сколько-нибудь существенно в ходе реакции.
Допустив, что [Y] a; const и объединяя константы в уравнении
(VII. 6), получаемki [MX„L1р=~ ' ■ 1ь гч ’ <VIL7>1 + й4 [L]где k4 = k3/k2[Y]. Если &JL ] < I, тоv = —kx [MX„ L],По окончании реакции концентрация интермедиата уменьша¬
ется от стационарной до равновесной. Доказательствами участия
интермедиата в процессе могут служить или непосредственное на¬
блюдение его в стационарном периоде реакции по каким-либо харак¬
терным свойствам, или уменьшение скорости с ростом [L] согласно
(VII. 6) или (VII. 7).На первой стадии остальных механизмов происходит образова¬
ние группировки (MX„L)Y. В ассоциативном механизме (А) первой и— 141 —v-, = — k, IML1.fVIT 17V
лимитирующей стадией является образование интермедиата с по¬
вышенным координационным числомМХЛ L + Y МХ„ LYо^-ММХяЬЦУ] (VII.8)а второй стадией — распад его или на продукты реакцииМХЛ LY -у МХ„ Y + L
vt = — kt[MXnLY] (VI 1.9)или на исходные веществаМХЛ LY -v МХ„ L + Y
va = — k3 [МХ„ LY] (VII.10)При расчете стационарной концентрации интермедиата полагают
—V-! + v2 + v3 = 0, откуда[МХП LY] = - fei - [MX„L] [Y].«2 + 3Поэтому экспериментальная скорость v ~ vt — vs должна быть
равнаv = -kl [MX„L] [Y] + ft, ^ [MX„L] [Y] = -ft4 [MX„L] [Y], (VII. 11)“Г *3где &4 = k^kzlikz + ka). Таким образом, уравнение (VII. 11) —
это кинетическое уравнение второго порядка и не отличается по
форме от уравнения скорости для реакции (VII. 8), т. е. интерме¬
диат в механизме А нельзя обнаружить по форме кинетического
уравнения.Если входящий лиганд — растворитель и [Y ] л? const, уравне¬
ние (VII. 11) преобразуется в кинетическое уравнение для реакции
псевдопервого порядкаV=— ft6[MX„L], (VII. 12)где k5 = &4[Y]. При этом по форме кинетического уравнения ме¬
ханизм А может совпасть с механизмом D.При использовании терминологии Ингольда механизмы D и А называют
5дг1 и Sv2 соответственно, причем S v обозначает нуклеофильное замещение
{nucleophilic substitution), т. е. замещение у положительно заряженного
атома, а цифры — мономолекулярную и бимолекулярную реакции. Иногда
механизмы D и А обозначают S vl(lim) и 5л/2( 1 im). Добавление (lim) указыва¬
ет, что они. являются предельными вариантами механизмов Sa/1 и S,v2.В механизмах 1 а и 1 d первой стадией является обратимая и быст¬
рая (нелимитирующая) реакция образования группировки (MXnL)Y.
Эта группировка может быть или ионной парой [MXnL]Y, или
практически несвязанной парой молекул, заключенных в «сетку»
молекул растворителя. В обоих случаях можно считать концентра¬— 142 —
цию такой группировки в ходе реакции равновесной и подчиняю¬
щейся закону действия масс:[(MX„L) Y] = iC[MX„L][Y]. (VII. 13>На второй (лимитирующей) стадии группировка (MX„L)Y перест¬
раивается плавным согласованным перемещением лигандов L и Y:
по мере того как входящий лиганд внедряется во внутреннюю сферу,
лиганд L из нее уходит. Процесс перестройки идет в одну стадию,
это значит, что интермедиатов не образуется. Механизмы различа¬
ются между собой конфигурацией активированного комплекса.
При механизме I d лиганды L и Y находятся в слабо связанном сос¬
тоянии и активированный комплекс является нестабильным анало¬
гом интермедиата МХ„, а при механизме 1а лиганды L и Y сильно
связаны и активированный комплекс аналогичен интермедиату
MXnLY. Таким образом, для-механизмов D и Id энергия актива¬
ции должна зависеть, в основном, от свойств уходящего лиганда, а
для механизмов А и Id от свойств обоих лигандов.На третьей стадии механизмов /а или Id должен происходить
быстрый обратимый распад группировки (MXnY)L.Будем считать стадию(MX„L) Y -*■ (МХ„) Lt>i = -M(MXnL) Y] (VII. 14)необратимой и мономолекулярной. Комбинируя уравнение скорости
для нее с уравнением (VII. 13), получаем уравнение скорости для
реакции второго порядка:»! = — kiK [MX„L] [ Y]. (VII .15)Однако интерпретация уравнения (VII. 15) существенно зависит от
того, каково относительное содержание частиц MXnL и (MX„L)Y
в растворе и можно ли отличить их друг от друга.При определении концентраций MXnL и (MXnL)Y можно исполь¬
зовать одновременно уравнения (VII. 13) и (IV. 14) и вычислитьи К- Если можно определить только суммарную концентрацию[ML] = [MX„L] + [(MX„L)Y]fто наблюдаются два крайних случая. Если доля частиц (MXnL)Y
мала, то [ML] « [MX„L] и, согласно (VII. 15),v1 = —klK[ML][Y\. ■ (VII. 16)Когда доля частиц (MX„L)Y близка к единице, например если
лиганд Y является растворителем и практически все частицы MXnL
окружены им, то [ML] ж [(MXrtL)Y] и, согласно (VII. 14),0l = —ftj[ML], (VII. 17)Таким образом, экспериментальное кинетическое уравнение с
использованием условной частицы ML имеет в пределе второй или
первый порядок.— 143 —
Существуют дополнительные факторы, влияющие на форму ки¬
нетического уравнения. Параллельно вхождению лиганда всегда
идет вхождение молекул растворителя. Например, при проведении
в водной среде реакций замещения в октаэдрических комплексах
Со3+, Сг3+и Rh3+ акватация настолько преобладает, что лигандыY (за исключением иона гидроксила ) в первых стадиях процесса
практически не участвуют. Замещение лиганда L на Y в этих комп¬
лексах происходит через образование акво-комплекса в роли интер¬
медиата, напримерСо (NH3)6L2+ + Н20 Со (NH3)5 Н203+ + L"Со (NH3)6 Н203+ + Y- Со (NH3)6 Y*+ + Н20Обмен лигандом со средой (например, реакция Co(NH3)6Cl2+ +*+ Cl") также протекает через активированный комплекс:Со (NH3)6 С1П + Н2С> ^ Со (NH3)6 Н203+ + С1-Со (NH3)6 Н203+ + Cl- _>■ Со (NH3)5 С12+ + Н20В водных растворах плоских квадратных комплексов Pt2+
замещение происходит по механизму А. На лимитирующей стадии
образуются два конкурирующих между собой интермедиата с к. ч. 5:
один с присоединением входящего лиганда Y, а второй — с присое¬
динением молекулы воды. При этома = - (kY [Y] + kHiQ [Н20]) [MX„L]и, полагая k та knto Ш20], получаемv = — (fe + йу IY]) [MX„L], (VII. 18)Уравнение (VII. 18) является модификацией (VII. 11) для парал¬
лельных реакций.Кроме того, вблизи от состояния равновесия на скорость процес¬
са должна влиять обратная реакция, т. е. реакция между MXnY и
L. Действительно, состояние равновесия определяется равновесны¬
ми концентрациями всех четырех частиц: MX„L, MXnY, L и Y,
тогда как в уравнения вида (VII. 6) или (VII. 11) входят равновес¬
ные концентрации только двух или трех. Поэтому вблизи от состоя¬
ния равновесия при любом механизме замещения, а не только при
механизме D, скорость реакции уменьшается с ростом [L]. Этот эф¬
фект может быть пренебрежимо малым в стационарной стадии про¬
цесса, однако утверждать это заранее практически невозможно.Таким образом, иногда механизм процесса очень трудно устано¬
вить по кинетическому уравнению. Так, кинетические уравнения
для механизмов А, 1а и Id практически идентичны. Если входящий
лиганд является растворителем, то этим механизмам соответствуют
уравнения псевдопервого порядка (VII. 12) или (VII. 17), которые— 144 —
вблизи состояния равновесия (когда появляется тормозящее дейст¬
вие уходящего лиганда) становятся аналогичными уравнению
(VII. 7) для механизма D. Поэтому для установления истинного
механизма реакции часто приходится пользоваться косвенными до¬
казательствами.Рассмотрим, например, механизм реакций замещения в окта¬
эдрических комплексах Со3+, Сг3+ и Rh3+. Поскольку эти катионы
практически не способны повышать координационное число, их ре¬
акции идут по механизмам D или Id.Для доказательства того, что реакция [Co(NH3)5H20]3+ с ани¬
онами Ym~ (так называемая реакция анации) идет по механизму Id,
рассмотрим вначале форму кинетического уравнения для нее. Ско¬
рость этой реакции увеличивается с ростом [Y ] согласно уравне¬
нию (VII. 15). Установлено, что образуются ионные пары
[Co(NH 3) ВН 20 ]SCN2+, [Co(NH 3) 5Н 20 ]S04+, [Co(NH 3) 5Н аО ]H2POf
и др., определены константы их образования, показано, что ско¬
рость пропорциональна равновесной концентрации ионных пар со¬
гласно (VII. 14) и вычислены значения кг. Перечисленные факторы
указывают на то, что осуществляется один из двух механизмов: Iа
или 1 й. Различить их позволяет зависимость энергии активации и
скорости реакции от характера входящей частицы. Поскольку зна¬
чения /гх близки между собой, то реализуется механизм 1й.Механизм D доказан, например, для реакции анации комплек¬
са [Co(CN)5Н20]2~. Этот отрицательно заряженный ион практичес¬
ки не образует ионных пар с анионами. Поскольку концентрация
уходящего лиганда постоянна (L == Н20), из (VII. 6) следует,
что скорость реакции должна выражаться уравнениемo = -*1[MX„L]/(L+ft4/[Y]), (VII. 19)где k4 = &3[Н20]/&2. Уравнение (VII. 19) получено эксперимен¬
тально для реакции [Co(CN) 5Н 20 ]2_ с азидом, роданидом, иодидом и
бромидом, причем значение константы скорости для реакции[Со (CN)6 НаО]2- [Со (CN)6p- + НаО(к х) оказалось во всех случаях одним и тем же и равным(при 40°,
ионной силе 1,0 и pH 6,4) 1,6 • 10-3 сект1.Однако механизм так называемого кислотного гидролиза, т. е. ак-
ватации катионных октаэдрических комплексов Со3+, неемотря на
множество проведенных исследований, не установлен. Скорость ак-
ватации в водных растворах зависит только от концентрации комп¬
лекса, т. е. является реакцией первого или псевдопервого порядка.
Это может соответствовать кинетическим уравнениям всех четырех ме-.
ханизмов (VII. 12), (VII. 17) или (VII. 7) при &4[L] < 1. Константа
скорости k сильно зависит от природы уходящего лиганда: так, при
25° k равны для [Co(NH 3) 5N02 ]2+ 2,7 • 10-® секЛ для [Co(NH3)5Cl ]2+
1,7* 10“® сек-1, для [Co(NH3) 5SCN ]2 + 5 ■ 10“10 сект1. Из механизмов
D и Id можно было бы выбрать D, или установив тормозящее дей¬6—699— 145 —
ствие вытесняемой группы согласно (VII. 7), или обнаружив ин¬
термедиат [Co(NH3)6]3+.Механизм А установлен для плоских квадратных комплексов.
Кинетика их реакций подчиняется уравнению (VII.18), причем ky
сильно зависит от природы как входящего, так и уходящего лиган¬
да. Например, значения ky (л!моль ■ сек) для реакций замещения в
транс- [Pt(py)2Cl 2 ] составляют при 30° С для аммиака 470, для
иодида 1,07 - 10s, для селеноцианата 5,15 ■ 10е. Для реакцийPt (dien) Х+ + ру Pt (dien) ру2+ + Х~илиPt (dien) Х2+ + ру _> Pt (dien) ру2+ + Xгде dien — диэтилентриамин NH 2СН 2СН 2NHCH 2СН 2NH 2,
константа скорости при 25°С изменяется от 1,9 • 109 сек'1 для
X = Н20 до 3,5 • 106 се/с"1 при X = С1_ и 5 • 104 сек'1 при
X = N02.Ассоциативное замещение в квадратных комплексах происхо¬
дит через промежуточные конфигурации тетрагональных пирамид
и тригональной бипирамиды:После перемещения в квадратной пирамиде атомов, расположен¬
ных на оси СМЕ, в направлении, указанном стрелками, треуголь¬
ник ABD образует экваториальную плоскость тригональной бип.и-
рамиды:Например, при структурной изомеризации красного сое¬
динения [CoL 2С1 ]SnCl з, в катионе которого лиганды L =
= (С6Н 5) 2РСН 2СН 2Р(С0Н 5) 2 занимают по два места в основании
квадратной пирамиды, а С1~ расположен в вершине, получается
зеленый изомер с тригонально-бипирамидальным катионом, и атом— 146 —
хлора становится экваториальным:Таким образом, аксиальное направление при такой изомеризации
(так называемом псевдовращении) поворачивается на 90° .Разница в энергиях между конфигурациями тригональной бипи¬
рамиды и квадратной пирамиды у комплексных ионов обычно не¬
велика. Поэтому, например,в структуре [Cr(en)3][Ni(CN)5]-1,5Н20
часть анионов имеет одну из этих конфигураций, а часть — другую.
Энергетический барьер псевдовращения также мал. Поэтому в от¬
личие от октаэдра эти конфигурации называют стереохимически
нежесткими.Механизмы A, D, /а и 1й могут применяться и для интерпретации
реакций лабильных комплексных ионов. У катионов с внешней
электронной оболочкой s2(Be2+) и s2pa (катионы подгруппы скандия,
редкоземельных элементов и актиноидов, щелочных и щелочнозе¬
мельных металлов) скорость реакций образования комплексов в
водных растворах тем меньше, чем выше электростатические харак¬
теристики иона металла. Расположение 8-электронных катионов в
порядке убывания их ионных потенциалов приведено ниже.Катион, Мп+А13+Zr4+Се4+Th4+Sc3+уз+Mg2+La3+¥м5,0854,4943,8463,5713,5292,7772,5002,419Катион, Мп+Са2+Sr2+Ва2+!Ra2+Na+K+Rb+Cs+Ы1,8521 ,5501,3791,2991,0000,7410,6620,599У катионов с минимальными ионными потенциалами срм от Cs+
до Са2+ константы скорости k (для уравнений псевдопервого порядка)
выше 108 сек-1, у ионов с 2,0 срм <; 3,5 т. е. у Mg2+, Sc3+, Y3+
и редкоземельных элементов константы скорости меньше 108 сек~1
и почти не зависят от природы входящего лиганда. Еще меньшие зна¬
чения наблюдаются у А13+(~1), Ве2н(~100). В аналогичном порядке
изменяются и скорости обмена молекулами воды между акво-ионами
и внешней средой. Это указывает на то, что лимитирующая стадия
реакции замещения является мономолекулярной реакцией плавно¬
го обмена молекулы воды на внешнесферный лиганд (механизм* Фм ~ г^г!Л’ где гм~ заРяД иона; гм — радиус иона.6*— 147 —
Id). Аналогичные выводы справедливы для катионов с внешней элект¬
ронной оболочкой s2p®d10.Ионные потенциалы 18-электронных катионов приведены ниже.Катион, Мя+Ga3+1п3+ЦЗ+Zn2+Cd2+Hg2+Ag+<Рм4,6883,1912,8042,3531,9051,7540,870<PM/<Pzn1,9921,3561,1921,0000,8100,7450,370Здесь также ионы с 2,0 < срм< 3,5 (Zn2+, In3+) имеют k = 105—
—108 сек-1, a Ga3+ с <рм ~ 4,7 — гораздо меньшее. Образование
лабильных комплексов переходных металлов из акво-ионов харак¬
теризуется, как правило, значениями k = 105 — 108 сек'1.Большой положительный заряд центрального иона и комплекса
и малый радиус центрального иона уменьшают скорость и увеличи¬
вают энергию активации при протекании реакции замещения по ме¬
ханизму D или 1 d. Действительно, большая напряженность поля
центрального иона должна затруднять уход отрицательно заряжен¬
ного лиганда.Реакции замещения хелатных лигандов имеют ряд характерных
особенностей. Отщепление или присоединение такого лиганда про¬
исходит ступенчато. Например, при вытеснении из внутренней сфе¬
ры донорные атомы лиганда отщепляются поочередно, заменяясь
входящим лигандом или молекулой растворителя. Пока лиганд не
отщепился полностью, ушедшие донорные атомы находятся вбли¬
зи узлов координационной сферы и вероятность обратного внедре¬
ния их остается высокой. Протонизация донорного атома (например,
связывание —СОО~ в —СООН или —NH2 в —NH3+) подавляет об¬
ратную реакцию, поэтому ион водорода катализирует замещение по-
лидентатного лиганда.Реакции хелатов из-за многоступенчатости сравнительно медлен¬
ны даже у катионов, образующих лабильные комплексы. Интерме¬
диаты в реакциях хелатов — это, как правило, протонированные
комплексы. Такие интермедиаты могут отличаться местом протони¬
рования от стабильной формы протонированного комплекса, и поэ¬
тому при резком подкислении растворов хелатных комплексов мо¬
гут возникать необычные для данного лиганда окраски. Для ре¬
акций замещения лигандов,которые не могут самостоятельно сущест¬
вовать в растворе, предложен так называемый четырехцентровой
механизм, в котором интермедиатом служит двуядерный комплекс:ДчMX + M'Y ->■ М' >М' MY + М'ХY /— 148 —
По такому механизму проходит, например реакция(CH3)2Hg + HgCl2 _> 2CH3HgClВ некоторых случаях реакция замещения идет без разрыва свя¬
зи металл—донорный атом. Так, при помощи изотопа О18 было дока¬
зано, что реакция акватации иона [Co(NH3)БС03]+в кислой среде
протекает без разрыва связи Со—О. ИонСОз не уходит из внутрен¬
ней сферы, а расщепляется по схеме[(NH3)6 Со—О—С02]2+ +*2Н+н(NH3)6 Со—о/
ХН+ соа§ 3. транс-Влияние в квадратных и октаэдрических комплексахтранс-Влияние, открытое и изученное И. И. Черняевым, харак¬
терно для реакций замещения в плоских квадратных, а иногда и в
октаэдрических комплексах. Оно представляет собой одну из форм
взаимного влияния лигандов. трснс-Влияние лиганда состоит в
том, что он изменяет лабильность связи иона металла с транс-рас¬
положенным лигандом. Так, если действовать аммиаком на квад¬
ратный комплекс [Pt(NH3)Cl3 ]“, то в первую очередь замещается
хлорид-ион, находящийся в транс-положении к другому хлорид-
иону:Cl NH3NH3Pt+ NH3 —>Pt+ ClCl ClClПолучается i{«c-[Pt(NH3)2Cl2]. В этой реакции хлорид-ион про¬
являет большее транс-влияние, чем молекула NH3. Такое же со¬
отношение между этими лигандами наблюдается во многих реакциях
комплексов Pt2+. Для комплексных соединений Pt2+, Pt4+, Pd2+,
Ir3+, Rh3+, Co3+ и некоторых других катионов довольно устойчив
ряд транс-влиянияСО % CN- % С2Н4 > R2S > NO' > Г > В г- > Cl" > F" > ОН- > RNH2 >> NH3 > Н20в котором каждый из лигандов обладает более высоким транс-вли¬
янием, чем последующие. Наиболее активны в этом ряду СО, CN”,
олефины, лиганды, содержащие серу: тиоэфиры (например, СН3—
—S—СН3), роданид-ион SCN“, тиосульфат-ион S20, , тиомочевина
(NH2)2CS и др. Менее транс-активны гидроксил-ион, амины, амми¬
ак и вода.Внутрисферные лиганды в инертных, как правило, комплексах
Pt2+, подвергаясь сильному транс-влиянию, вступают в характер¬
ные для них качественные реакции.— 149 —
Так, среди изомерных соединенийIIIIIс AgN03 реагируют комплексы II и III, образуя AgCl и AgBr со¬
ответственно. Соединение I практически не взаимодействует с AgN.03,
но реагирует с НС1, причем во внутренней сфере замещается ам¬
миак:С2Н4С1ВгPt+ НС1NHLС2Н4ClPtBrCl+ NH/Использование эффекта транс-влияния помогает проводить нап¬
равленные синтезы. Например, цис-[Pt(NH3)2Cl2] получается при
действии аммиака на [Pt(NH3)Cl3]_, а транс-изомер — при дейст¬
вии хлорида на [Pt(NH3)3Cl ]+: действительно, более сильное транс-
влияние хлорид-иона приведет к замещению транс-расположенной
молекулы аммиакаNH3NH,NH3ClPt+ Cl"ClNH,PtNH,+ NH3ClА. А. Гринберг и Б. В. Некрасов рассматривали транс-влия¬
ние с точки зрения поляризационных представлений. Б. В. Нек¬
расов объясняет транс-влшпт различной прочностью связей меж¬
ду центральным ионом и лигандом. Более реакционноспособным
оказывается тот лиганд, связь которого с центральным ионом сла¬
бее. Если лиганды имеют одинаковый заряд, а суммарный (постоян¬
ный и индуцированный) диполь лиганда Y больше, чем X, то ион
металла становится диполем, положительный конец которого обра¬
щен к Y. Поэтому лиганд X, находящийся в транс-положенш к Y,
будет испытывать повышенное отталкивание, связь его с М станет
слабее, и этот лиганд будет легче замещаться.Согласно теории Чатта — Оргела реакционная способность ли¬
ганда зависит от способности транс-лиганда к я-взаимодействию
с центральным ионом, а увеличение реакционной способности объяс¬
няется не ослаблением связи М — L, а уменьшением энергии акти¬вации промежуточного активированного комплекса в реакции транс¬
замещения. При рассмотрении транс-влияния Чатт и Оргел
предполагают, что реакция замещения идет по ассоциативному меха¬
низму, где скорость определяется образованием промежуточного
соединения, включающего старый и новый «атакующий» лиганды.
Скорость образования промежуточного соединения зависит от несим¬
метричности распределения зарядов в исходном комплексе; замеще¬
ние по координате с меньшей электронной плотностью требует мень¬
шей энергии активации.При замене лиганда L2 на L (рис. 59) электроны на орбитали L
должны взаимодействовать с орбиталью металла d**-,,» , образую¬
щей а-связи М—L2 и М—L. Этому мешают гантели орбиталей dyz.
Если у L! есть незаполненная орбиталь, способная к тс-взаимодейст-
вию с dj^-орбиталью металла, то электронное облако dyz частично
смещается в сторону Lx, тогда связь М—L2 легко атакуется L.
Чем сильнее я-взаимодействие М—L, тем легче замена L2 на L.Недостатком теории Чатта —Оргела является то, что она не объ¬
ясняет те случаи, когда транс-активный лиганд не способен обра¬
зовывать тс-связи с ионом металла, и то, что эту теорию трудно при¬
ложить к реакциям замещения в октаэдрических комплексах.Таким образом, по А. А. Гринбергу и Б. В. Некрасову транс¬
влияние связано с термодинамическими свойствами комплексной
частицы, а по Чатту — Оргелу — эго кинетический эффект. Послед¬
няя точка зрения в настоящее время более распространена. Дейст¬
вительно, в кинетических исследованиях обнаруживается большое
влияние транс-заместителей на подвижность уходящего лиганда.— 151 —
Так, для комплекса ^M£-[Pt(NH£;)LCl2]'1'' скорости реакции замеще¬
ния хлорид-иона, находящегося в транс-положении к лиганду L,
на молекулу пиридина резко уменьшаются в ряду L = С2Н4, N02,
Вг“, С1~, который совпадает с рядом транс-влияния. Измеренная при
25° и постоянной концентрации пиридина в этаноле константа
скорости псевдопервого порядка k для реакциитранс-[Pt {Р (СаН5)3}а LC1] + ру +4 транс-[Pt {Р (С2Н6)а’а Lpy]+ + С1~имеет значения (сек-1): 6,0 • 10-4; 1,2 * 10-4 и 3,5 « 10-в для L =
= СНз, СвНз и С1~, а при L = Н- она даже при 0° равна 4,7 • ЮЛ
И здесь скорость замещения хлорид-иона тем больше, чем больше
транс-влияние лиганда L.Тот факт, что Н“, СНз, С2Н; и аналогичные анионы обладают
большим транс-влиянием, указывает на значимость не только я-,
но и а -электронных эффектов. тракс-Расположенные лиганды кон-
курируют за р3 -орбиталь Pt2+, например, за рх. Поскольку ст-ор¬
битали Н ", фосфинов, СНз и некоторых других лигандов перекрыва¬
ются с ней очень сильно, то транс-расположенный лиганд становится
относительно слабо связанным* и легко замещается по механиз¬
му А. Действительно, в интермедиате MXnLY входящий лиганд Y
также относительно слабо связан орбиталью pz, перпендикулярной
плоскости комплекса. Так как обмен слабо связанных групп тре¬
бует низкой энергии активации, то скорость его будет высока. Та¬
ким образом, за счет а-транс-влияния должна уменьшаться и проч¬
ность, и инертность связи уходящего лиганда. Экспериментально
наблюдаемый эффект, по-видимому, может быть или чистым а-транс-
влиянием ( в случае Н“, СНз) или чистым я -транс-влиянием (в слу¬
чае олефинов), или суммой двух эффектов (в случае SCN", СО, га-
логенид-ионов и т. п.).* Например, в [ЯЬ(МНз)б(СгН5)]Вг2 расстояние Rh — N для транс-_ Орасположенного по отношению к СгКь лиганда NH3 равно 2,26 А, а для ос-Отальных — 2,07 А.
VIII. УСТОЙЧИВОСТЬ КОМПЛЕКСНЫХ СОЕДИНЕНИЙВ РАСТВОРАХ§ 1. Определение констант устойчивости комплексных соединенийв растворахВ среде с постоянной ионной силой 7=const можно с удовлетво¬
рительной точностью считать коэффициенты активности постоян¬
ными и пользоваться концентрационными (т. е. Еыраженными в
виде произведения равновесных концентраций) константами равно¬
весия. Константы равновесия реакций, записанных без указания
зарядов веществL + iH H;L
М + iL MLj
MLг_! + L ^ MLj
называются константой протонизации лигандаd IHjL]* [L][H]' *общей константой устойчивости комплексаВ [MLi]Р‘ [М] [L]‘и ступенчатой константой устойчивости комплекса[ML;] '[ML,_]] [L] 'При этом рг = Pj = Ион металла называют ну¬левым комплексом [ML0] и вводят понятие р0:_[MLo]Р° [М] [L]°Равновесный состав раствора при 7=const описывается системой,
состоящей из: а) уравнений закона действия масс (з. д. м ), ко¬
личество которых равно количеству независимых равновесий в
системе; б) уравнениями материального баланса. Например, если
ион металла превращается в комплексы ML, MLZ, ML3...,cm = [M] + [ML] + [ML2] + [ML3]+ ...в) уравнения электронейтральности, требующего, чтобы сумма
зарядов ионов в системе была равна нулю. Например, для раствора
пирокатехина в воде уравнение электронейтр-альности имеет вид[Н*] - [ОН-] - [ С„Н607] - 2 [ СвН40|-] =0.— 153 —
Для удобства записи уравнений материального баланса вво¬
дится несколько вспомогательных функций:1. При протонизации аниона слабой кислоты и отсутствии побоч¬
ных реакций (димернзации, комплексообразования и др.) матери¬
альный баланс по аниону записывается в видеCL = [L] + [HL] + ... + [H„L] + ... + [Ндг L] ,а материальный баланс по ионам водорода в кислой среде ([Н]>
>[ОН]) в видеch=[H] + [HL] + ... + tn [HmL] + ... + N [Hv L] .Используя уравнение закона действия масс [HjL] = BjLHH]',
получимcL=[4(i + 2МН]г) и cH = [L]'£iBi[н]г.NВведем функцию протонизации /= 1 -\-51в£[Н]1 и функцию/=1N =<р = ]г и перепишем при: помощи их уравнения материаль-г=1ного баланса в видеcL=[L)f, (VIII. 1)cH-[H] = [L]y. (VIII.2)Разделив (VIII. 2) на (VIII. 1), получимсн —'[Н] <е ^iBt [И]/ 1+2в»1н1' 'Функция N имеет смысл среднего
числа протонов, присоединенных к
каждому аниону, который не участ¬
вует в побочных реакциях. Графи¬
ческая зависимость функций f, <р и
N от [Н ] представлена на рис. 60.2. При образовании ионом ме¬
талла системы одноядерных ком¬
плексов с лигандом-анионом силь¬
ной кислоты уравнения материаль¬
ного баланса по иону металла и
лиганду имеют видРис. 60. Зависимость функций N,
•у и / от равновесной концентра¬
ции ионов водорода [Н+]см =[М] +[ML] + ... +[МЦЯ,(VIII.3)
CL = [L] + [ML] + 2 [ML,] + ... + n [ML„]. (VIII.4)Преобразуем их, используя уравнение закона действия масс
(MLJ = [М ] [LcM=[M](l + 2Pi[L]‘') и cL = [L] +• ППри помощи функции закомплексованности F = 1 + [L ]* =1=1Л П= 2lML]J и ФунВДии Ф = [L 1г запишем уравнения (VIII. 3)
1=0 £—1
и (VIII. 4) в видеcM = [M]F,СЬ - [L] = [М] Ф.Определим функцию комплексообразования (функцию образования
Бьеррума)_ *-14 фП = =-Г= п . (VIII. 5)М 1 + 2 Pi [L1*i=lкоторая имеет смысл среднего числа лигандов, связанных с ионом
металла. Между функциями /, <р и N с одной стороны, и Р,Ф и п —
с другой, существует аналогия. Зависимости F, Ф и п от [L] аналогич*
ны графической зависимости f, ф, N от [Н ] (рис. 60)При комплексообразовании с лигандом-анионом слабой кислоты
уравнения материального баланса записываются в виде
cyi = [M]F и cL = [L] / + сМ/Г.В кислой среде можно считать, чтоСН= [H] + [L]<p. (VIII. 6)Рассмотрим систему одноядерных комплексов М, ML, ML2.-.-
Выход комплекса ML; (считая М «нулевым комплексом») есть от¬
ношение его равновесной концентрации к сумме концентраций
комплексов:[ML;] = _[М_у = р, [L]*
см [М] F FЕстественно, что 0 аг 1 и сумма выходов всех комплексов
равна 1. Так как аг являются функциями только [L], то возможно
построение диаграммы выходов в координатах at — ]g [L ]. На этих
диаграммах (рис. 61) <*о — выход свободного иона металла—при
росте [L ] падает от 1 до 0, выход ап — комплекса с максимальным
числом лигандов — при [L ] -> оо растет от 0 до I. При реально-- 155 —
достижимых концентрациях лиганда величина ап может далеко не
достигать единицы. Выходы остальных комплексов по мере роста
(L ] проходят через максимум: комплекс [МЬг ] уступает место ком¬
плексу [ML/+1 ] и т. д. При этом комплексы могут иметь областидоминирования, в ко¬
торых их выход бли¬
зок к 1. В области до¬
минирования реакцию
образования комплек¬
са можно считать про¬
ходящей количествен¬
но и пригодной поэто¬
му для аналитических
целей.Некоторые свойст¬
ва растворов линейно
зависят от равновес¬
ных концентраций
компонентов. Для сис¬
темы одноядерных комплексов такое аддитивное свойство W запи¬
сывается в видеW = W0 [М] + Гд [ML] + [MLa] + ... Wn [ML„].К аддитивным свойствам относятся оптическая плотность D, пре¬
дельный ток в полярографии J, подвижность системы лабильных ком¬
плексов U нт. п. Если отнести свойство W к молю вещества, то это
характеризует среднее молярное свойство: W= Wlc^. Тогда W =
= 2а;Ц7г, а величины Wt приобретают смысл молярных величин для
отдельных сортов частиц. Так, если определить средний молярный
коэффициент погашения е как D/см то по закону Бугера—Ламбер -
та—Бера он будет равен1='%чч<где £; — коэффициенты молярного погашения отдельных комплек¬
сов. Функцию п также можно рассматривать как аддитивную, при¬
чем Wt — i.Увеличение равновесной концентрации ионов водорода [Н+]
в системе усиливает протонизацию лиганда-аниона слабой кислоты,
уменьшает его равновесную концентрацию и приводит к смещению
на диаграмме а* — lg [L ] по абсциссе влево вплоть до полного раз¬
рушения комплексов. Например, в системе Fe3+ — сульфосалици-
ловая кислота в присутствии избытка лиганда при pH 5—6 наблю¬
дается желтая окраска комплекса ML3, при подкислении она пере¬
ходит в оранжевую окраску комплекса ML2, и затем — в крас¬
ную комплекса ML. Красная окраска достигает максимума при pH
2,9 и при дальнейшем подкислении разрушается: доминироватьРис. 61. Зависимость выходов для системы
одноядерных комплексов от lg[L]— 156 —
начинают свободные ионы Fe3+, Подкисление системы не влияет на
выход комплексов с анионами сильных кислот. Поэтому в доста¬
точно кислой среде можно, например, определять Fe3+роданидом в
присутствии ацетата, цитрата и других органических лигандов. Под-
щелачивание часто разрушает комплексы в результате связыва¬
ния иона металла в гидроксокомплексы или- гидроокиси.В некоторых случаях возможно комплексообразование не только
с лигандом L, но и с его протонированными формами HL, Н2Ь и
т. д. В частности, это происходит при комплексообразовании La3+
с ЭДТА или тартратом. Такие комплексы называются кислыми или
протонированными. Для них вводятся константы равновесия
k= [MHL]/[ML] [Н 1 и (3хх = [MHL]/[M][HL]. При этом 0Х1 —
обычная константа образования комплекса MHL, a k — константа
протонизации комплекса ML; \gk равен р [Н ], при котором [MHL ] =
= [ML]. Константы й и рп связаны соотношением Рц =
Аналогичным образом возможно образование комплексов, в состав
которых входят гидроксильные ионы ОН-.Определение констант равновесия связано с применением ста¬
тистических методов. Пусть необходимо определить N констант
равновесия и для этого измеряется какая-то функция этих констант
у, вид которой известен. Проводят К измерений, причем К > N и
разность К — N = р называется числом статистических степеней сво¬
боды.Принцип наименьших квадратов состоит в том, что наилучшим
набором констант считается такой, который приводит к минимальнойКдисперсии s2. Дисперсией называется величина 1/р^]б|, причемЙ=1~ Ун — Ук>тДе У к — функция, рассчитанная с использованием
выбранных констант; при этом необходимо, чтоб была минималь¬
на сумма квадратов отклонений функции измеренной от рассчи¬
танной.Расчет констант в уравнениях по экспериментальным данным
осуществляется несколькими способами:а) при многократном определении константы вычисляется сред¬
нее арифметическое;б) при измерениях функции одной переменной у = а + Ьх
строится и решается относительно а и Ь система нормальных урав¬
нений2 Ук = па + Ь ^ xk,2 ^ = а 2 ^ + 6 2 *1 •При измерении линейной функции многих переменныхУ — о О + а1х1 + а2х2 + + aN— I XN— 1строят и решают относительно параметров систему нормальных
уравнений— 157 —
^Vk — nao + Ol 2 *1* + a2 2 X‘k + ••• + aN— 1 s X(N— 1)A2 xik = до 2 -ья* 2*?* "I- дз 2 *2* ■Ci* + •■• H" ajv—12 x(N—\)kXi/t
2 x*b УЬ = ao 2 *2ft + ai 2 *1* *2fc + “2 2 *2fc + ••• + aN-1 2 -*( V—l)je *a*2 x(N—l)k Vk — a0 2 *(ДГ—])A + al 2 *1* *(ЛГ—1)A + a2 2 *** *(JV—1)* + ••• ++ %—12 xf.v—илРасчет коэффициентов решением системы нормальных уравне¬
ний называется методом наименьших квадратов (МНК);в) при линеаризации уравнений более сложного вида часто исполь¬
зуют также МНК- В частности, степенные и показательные функции
линеаризуются при логарифмировании с последующей подстанов¬
кой. Например, из уравнения Аррениуса k = kQe—EalRT получает¬
ся \gk = \gk0 — Ea/2,3Q3RT. Подстановка у = lgfe; х = 1 IT, а =
lg&0 и b = —EJ2,303RT позволяет получить уравнение вида у —
= а + Ьх. Полином у = а0 + Haj V приводится к виду у = а0 + HajXj,
если положить х3 = V. Многие функции, не сводимые к линейно¬
му виду, позволяют после приблизительного определения их пара¬
метров (констант) провести уточнение этих констант. Рассмотрим,
например, функциюу = Л е+ А2 e~b,t.Если известны приближенные значения А°и А°, Ьи и b\ то, раз¬
лагая у в ряд Тейлора по параметрам в точке (Aj0, А2°, Ьг° 62°)
и пренебрегая членами разложения с порядком выше первого,
получим функцию, линейную относительно поправок к параметрам:г = у — у0 = е~~blt ДА\ + ё~‘ДУ12 — А° 1ё~ь\* Д61 — A^te-'Ь2 * ДЬ2.Обозначив*! = e~b°it,xz = e~b02t, х3 = —Л? tx1 и xt = —Л2 tx2,получим линейное уравнение,
позволяющее, определить АА lf
ДЛг, ДЪг и А62 по МНК. Введя
эти поправки и получив Л^ =
= А?+АЛ! и т. д., можно
вновь провести уточнение и пос¬
тупать так до достижения необ¬
ходимой точности:г) методом прямого поиска
такого набора констант, кото¬
рый обеспечивает минимум дис¬
персии. Этот самый универсаль¬
ный метод является в то же вре-
Рис. 62. Определение коэффици- мя наиболее трудоемким и поэто-
ентов полинома му обычно реализуется на ЭВМ.158
Часто применяются графические методы расчета констант равно¬
весия как параметров полиномиальных уравнений. Коэффициенты
полинома у = а0+ а^х + а2х2+ ... с положительными зна¬
чениями х и аг, например функции закомплексованностиможно найти, если построить график у от х и провести по экспери¬
ментальным точкам кривую (рис. 62). Отрезок, отсекаемый этой
кривой на ординате, равен а0, а наклон касательной в точке пере¬
сечения— аг. Затем рассчитывается и строится вспомогательная
функцияТаким же образом определяется для нее пересечение а х и наклон
а2 (рис. 62). Расчеты продолжают до тех пор, пока на графике оче¬
редной вспомогательной функции не будет наблюдаться совокуп¬
ности беспорядочно разбросанных точек. Общее уравнение для
построения вспомогательных функций имеет видпричем фо = у. Для функции закомплексованности, где а о =1, можно непосредственно перейти к построениюПотенциометрические методы определения констант равновесия.
При образовании комплексов с лигандом-анионом слабой кислоты
вытесняются ионы водорода за счет конкуренции за лиганд между
ионами металла и водорода. Пусть vo мл раствора с концентра¬
циями соли М(С104)„, кислоты НСЮ4, кислоты HmL, анион ко¬
торой является лигандом, см. снсю, и с?, соответственно и с ион¬
ной силой /о (создается добавлением NaC104) титруется ще¬
лочью NaOH концентрацией сщ. Измеряют объем добавленной
щелочи и р[Н J^ = —lglH]/ в каждой i-й точке кривой ти¬
трования. Если щелочь имеет ионную силу /о, изменениями / в
ходе титрования можно пренебречь. Считая h = [Н+] > [ОН-],
получим Vi = v0 + vmi) hi = 10-?™', cLi = d\yalvt\ сщ=с°р°lvi'
сИ[ = (mc0Lv0 + chcio^o — Cmvi)/vf В методе Бьеррума учиты¬
вают существование только непротонированных комплексов.
Если в изучаемом интервале pH кислота HmL заметно диссоцииро¬
вана, то необходимо знание констант протонизации аниона Lm~.
Используя уравнение материального баланса по ионам водорода
(см. VIII. 6), можно рассчитать [Ь]г=(снг — Лг)Лр4, а затем
щ = (ci. — [L ]г^)/сМг. После линеаризации уравненияF= l+pi [L] + 3,[L]»+ ...(VIII.7)= (У — ай) / * = + а2х + аах* -f ...(VIII.8)получаем выражение(1 — 7Г) [L]П2 — п . 3 — п= Pl + P»T^I:L1 + p,TZj [Lli+ - ■— 159 —
которое может служить для определения констант равновесия по
МНК. Если кислота HmL, например глицерин или гликоль, прак¬
тически недиссоциирована в изучаемом интервале pH, то перехо¬
дят к константам обмена:[ML,] [НГ'js ' 13~ [HmL]'[M] *Значение л; вычисляют по формуле_ I Н]г — сн.
п> = — ,"“-м,причем сн. рассчитывают, не учитывая ионы водорода, содержа¬
щиеся в HmL. Обозначив t-= [HmL}/km, получают [ML,-] =
=KjlM.W и записывают п в виде- Kit + 2K,P+ ... +nKntn Q41 + Kyt + KJ* + ... + Knin 'Линеаризация выражения (VII 1.9) даетп 2 — п .. / — п- = Кх + Ка ~ t + + Kj ~ i1 1 + ...(l— n)t 1—n I— nМетодом наименьших квадратов вычисляют К)- Если константы
протонизации лиганда известны, то = KjBm.Равновесные концентрации некоторых катионов могут быть из¬
мерены при помощи металлических (для Ag+, Au2+, Hg2+), стацио¬
нарных амальгамных (для Bi3+, Pb2+, Cd2+, Zn2+) или капельных
амальгамных (для Cu2+, Ga3+, Li", Na+ К+, Rb+, Cs+) электродов 1-го
рода. При постоянной ионной силе можно считать, что потенциал
tcVthx электродов (для Мл+)Гвыражается уравнением0я = я0 + — Ig[Mn+],пгде 0 = 2,3RT/F (59,16 мв при 25° С); тг0 — стандартный элек¬
тродный потенциал. Чаще всего составляют цепи с переносом: или
с применением стандартного полуэлемента с известным потенциа¬
лом, например насыщенного каломельного, или двойные концентра¬
ционные. В первом случае составляется цепьКС1HgHg2Cl2насыш.растворИсследуемый раствор с
известными см и cLМДанную цепь часто приходится усложнять, вводя промежуточные
мостики, чтобы избежать образования осадков КС104, AgCl и т. п.
Э. д. с. такой цепи Е равна п1—я2+я;-, причем л;- — диффу¬
зионный потенциал, который стремятся по возможности учесть— 160 —
или уменьшить (обычно его считают равным нулю), а п1 и л2 —
потенциалы первого и второго электродов. Обозначив л? —л2 +
+ я;- через Е0, получают0Е = Е° + —lg [Мя+1.
пОпределяют Е° при помощи растворов, в которых лиганда нет к
см = [Мя+]. Зная Е°, а также Е и см для исследуемого раствора,
вычисляют = См/[М].Двойную концентрационную цепь составляют из тождественных
половинММ (С104)„ раствор
при 1 = 1 о, см =М (С104) растворПри 1 = /о, Сдо =МЭ. д. с. и £° такой цепи равны нулю. При титровании правого-
раствора раствором лиганда концентрацией сь и / = /о, имеем:си1'» 0 смг 0 , vo + vLiMi Ко + »ц * 11 ' П tMb n v0
Следовательно,„(Ег—я n)n v0lg Fi = * _±_ + ,g _____ . (VI, 1. 9>Если пренебречь диффузионным потенциалом (я;*» 0) в (VIII.9),
то вычисление F упростится.Расчет констант равновесия при помощи функции F осложнен
тем, что в уравнение (VIII. 7) входят равновесные концентрации
лиганда, которые неизвестны. Для вычисления равновесных кон¬
центраций применяют метод последовательных приближений Леде-
на. Для этого используют уравнения (VIII. 6) и (VIII. 8). Задают
начальные приближения для значений п (в простейшем случае пола¬
гают все п равными нулю) и рассчитывают [L ] по уравнению (VIII. 6)_
Полученные значения [L] подставляют в уравнение (VIII. 7).
и графическим методом (или по МНК) рассчитывают приближенные
значения констант |3j. Подставляя эти значения в (VIII. 8), опре¬
деляют новое приближение для значений п, снова вычисляют зна¬
чения [L ] и Pj и продолжают расчет до постоянства значений flj.Обычно метод Ледена применяют только при / » 1, когда ли¬
гандом служит анион сильной кислоты или анион слабой кислоты
при высоких значениях pH. При этом измерение pH исследуемых
растворов становится излишним.Электродом второго рода называют электрод, обратимый по от¬
ношению к ионам какого-либо металла и находящийся в контакте
с труднорастворимой солью этого же металла. Электрод второго рода
обратим по отношению к аниону этой соли L"1- и потенциал его вы¬— 161 —
ражается уравнением0 .л = я0 — — lg[L],Из электродов второго рода наиболее известны галоидсереб-
ряные и галоидртутные, особенно каломельный и хлорсеребря-
ный, а кроме того Ag/AgSCN, Ag/AgN3, Ag/Ag2C204, Ag/Ag3P04,
Hg/HgaS04 и др. Используя цепи с переносом, можно определить
в изучаемых растворах [L ].., рассчитать по уравнению (VIII. 5) п и
после линеаризации выражения для п вычислить константы равно¬
весия. Поскольку для расчета п, если f Ф 1, необходимы измерения
pH, то этот метод применяют лишь для определения устойчивости
галогенидных, роданидных и прочих комплексоц с лигандами-анио¬
нами сильных кислот, а для анионов слабых кислот применяют
метод Бьеррума.Расширить возможности потенциометрии можно с использованием ,
конкурентных методик. Фактически уже метод Бьеррума является
конкурентным: ионы водорода вытесняются в результате конкурен¬
ции за лиганд'между ионами водорода и металла. Аналогично мо¬
жет происходить конкуренция за лиганд между ионами металла и
ртути с вытеснением последних.Пусть раствор, содержащий Hg2f, Ми+ и лиганд с концентраци¬
ями CHg, См и cl соответственно, выдерживается с металлической рту¬
тью в атмэсфгре азота. При этом часть ртути растворяется:Hg + Hg2+ 5»: Hgf+ (а)Если ион Hg22+_ не образует комплексов а ионы Hga+ и Мя+ обра¬
зуют только комплексы состава 1 : 1, то уравнения материального
баланса приобретают видcHg = I н§2+1 + 2 [Hg22+] + [HgL] = c°Hg + [Hg2+] , (VIII. 10)
cL=[L]/ + [HgL]+[ML], (VIII.11)cm=[M] + [ML]. (VIII.12)Измерив при помощи ртутного электрода [Hg2+], вычислим
CHgl+] > используя закон действия масс для равновесия (а),
<7= [Hgf]/[Hg2+], и рассчитаем [HgL 1 согласно (VIII. 10):[HgL] = с°д - [Hg*4 (9 + 1).Если известна константа устойчивости комплекса HgL, то можно
последовательно вычислить[L] = Д г ; [ML] = cL —[L] / — [HgLli[ML][M] = cM — [ML] и pML =[M] [L]— 162 —
При этом величина [L ]/ часто становится пренебрежимо малой и
измерения pH — излишними.Методы, связанные с изучением гетерогенных равновесий.Пусть соль NixLy конгруэнтно растворяется в растворах, содержа¬
щих различные концентрации аниона-осадителя L, причем ионная
сила насыщенных растворов примерно постоянна. Мерой раствори¬
мости соли M^Ly служит аналитически определяемая концентра¬
ция металла см> a fL= ycs\/x, где х и у— стехиометрические коэф¬
фициенты в формуле соли. Подставляя в выражение для функции
закомлексованности F = см/[М ] произведение растворимости ПР =
= [М И [L ]у, получаемПРF= см [Ц»'* = ПР + (ПР Э,) [L] + (ПР р2) [L]* + ... (VIII. 13)Как и в методе Ледена, задав начальные приближения для п, вы¬
числяют [L ] и произведение ПР/1, оценивают приближенно коэффи¬
циенты уравнения (VIII.13): ПР, ПР^, ПР02 и т.д., вычисляют
приближенно @j и при помощи их уточняют п. Расчет продолжают
до самосогласования.Рассмотрим зависимость растворимости соли от избытка осадите-
ля, т. е. см от [L]. Из соотношения (VIII.13) следует, что если в
системе доминируют положи¬
тельно заряженные (катион¬
ные) комплексы ML; с i < у/х,
значение см с ростом [L]
уменьшается. Концентрация
нейтрального’комплекса МЬг
(при i = у/х), если он сущест¬
вует, равна ПРрг, постоянна
и определяет тот минимум,
ниже которого величина см
опуститься не может. Далее
по мере роста (L ] можно дос¬
тичь области, где доминиру¬
ют отрицательно заряжен¬
ные (анионные) комплексы сi > у!х. При этом растворимость увеличивается с ростом ILJ
(рис. 63). Так, например, запишем выражение для растворимости
РЬС12:срь= [Pb*+] + [РЬС1+] + [РЬС1°] + [PbClr] +[PbClJ“] + ... == ПР ([Cl]-* + р! [Cl]-» + р2 + За [С1] + р4 [С1Р +...),откуда видно, что срьуменьшается сростом [С1 ], когда доминируют
комплексы РЬ2+ и РЬС1+, концентрация комплекса РЬС12° постоян¬
на, а комплексы РЬС13~ и РЬС142- повышают растворимость с рос¬
том [С1]. Растворение в избытке аниона-осадителя характерно для
амфотерных гидроокисей (в избытке ОН-), для сульфидов мышь¬Рис. 63. Зависимость растворимости
соли от lg [L] при образовании ани¬
онных комплексов— 163 —
яка, сурьмы и т.д. (в избытке S2'), для AgCl и РЬС1а (в избытке
СП.Рассмотрим экстракцию в системах с комплексными соединени¬
ями. Пусть ML„° — единственный сорт металлосодержащих частиц,
экстрагирующихся в органическую фазу, т. е. (см)0рг = 1МЬя]орг.При постоянстве среды в водной фазе и незначительных изме¬
нениях в составе органического растворителя закон распределения
можно представить в виде[МЬфр^ (VHI 14)[ л]воднгде q — константа распределения. Определим аналитически (см)0рг
и (см)водн в равновесных фазах, их отношение называется коэффи¬
циентом извлечения Q:Q = l_C^0gL 1 1ML"]o^ . (VIII.15)( См)вОДН [М]ВОдн FИз (VIII. 14) следует, что[ML„]0pr = п [М]СОдн[Ь]”0дН
и уравнение (VIII. 15) преобразуется*:Л ?P«[L]"Q = J: = ?<*л.т. е. коэффициент извлечения пропорционален выходу нейтраль¬
ного комплекса. Если при росте [L ] анионных комплексов не обра¬
зуется, то а„ стремится к единице, a Q — к q, если же анионные
комплексы образуются, то значение Q, достигнув максиму¬
ма, начинает затем убывать. Обозначив l/q$n через S, получаем£F= = 5 + (6 РО IL] + (6P,)[L]*+ ... (VIII.16)Способ расчета констант равновесия рг в уравнении (VIII. 16)
аналогичен способу расчета в методе растворимости.Для определения констант равновесия изучают также раствори¬
мость солей слабых кислот в сильных кислотах. Растворимость
таких солей с ростом кислотности увеличивается. Если поддержи¬
вать в насыщенных растворах постоянную ионную силу и вычислять
ПРдля солиМжЬупо формулеnp=(CM;,(f См)У/р,не учитывающей комплексообразования, то эта величина при ма¬
лых значениях pH, когда комплексы разрушены, будет близка кВ дальнейшем индекс «водн» в уравнениях опускается.
истинному значению ПР, а с ростом pH будет увеличиваться.
В общем случае константы равновесия по зависимости раствори¬
мости от pH находят методом минимизации дисперсии на ЭВМ.
Если состав соли ML и не нужно учитывать образования комплек¬
сов, кроме [ML0], то расчет упрощается: поскольку см = сь\
[М] = см— [ML], [L] = (cL— [ML])// и [ML] = х= ПР|31, то( см — %)2// == ПР. (VIII.17)Линеаризация уравнения (VIII.17) даетсм = -|/ПР. у7+*.т. е. по зависимости см от j/у можно графически определить У и?
и х и вычислить ПР и (3! = х/ПР.Методы, связанные с изучением аддитивных функций соста¬
ва раствора. Поскольку интерпретация измерений всех аддитив¬
ных функций состава аналогична, рассмотрим только спектрофо¬
тометрические методы.Электронный спектр поглощения в координатах D — X обычно
имеет вид суммы искаженных гауссиан. При любой длине волны
соблюдается закон Бугера—Ламберта—БераD=*f2«i[Ati, (VIII. 18)где [А;]—равновесные концентрации веществ А;, Для системы
одноядерных комплексов уравнение (VIII. 18) преобразуется к ви-
ДУD _ ХЧ ео + ei Pl [L] + е2р2 [L]2 + .• ■ 1+ЫЧ+ЫЧ-+... • (VII,'IS>)Расчет констант устойчивости и коэффициентов е,- чаще всего про¬
водят на ЭВМ методом минимизации дисперсии. Из графических
или расчетно-графических методов наиболее известен метод Яцимир-
ского-Бударина, в котором уравнение (VIII. 19) представляют в
виде0 = (7-e0) + p1[L](7-e1) + Р2 [L]2 (7— е2) + ... (VIII.20)При условии cl см Для анионов сильных кислот принимают
[L] л} cL. Считая, что при малых [L] члены с [L]2, [L]3 и т. д.
несущественны, их отбрасывают и получают- е — е0 1 е — 1е = ег — • —— = eL — • .CL Pi CL **Обозначают (e— s0)/cl через 11 и экстраполируют линейный учас¬
ток графика зависимости е от t ъ соответствующий малым cl (боль¬
шим ?j) на t! = 0. Получают пересечение ег и наклон— 1/хг. Урав¬
нение (VIII.20) с учетом квадратичного члена приводится к виду— 165 —
где = — (— + е — s1) может быть рассчитано с исполь-CL \*1 /зованием полученных значений у.х и ег. По графику зависимости
еот t2 могут быть вычислены х2 и е2 и построена новая вспомога¬
тельная функция tz = — + s — s2) и т. д.СЬ \ <3 )Если образуется или доминирует один комплекс, можно исполь¬
зовать ряд упрощенных ме¬
тодик. Для определения соста¬
ва доминирующего комплек¬
са применяется метод изомо-
лярных серий (метод Остро-
мысленского—Жоба) или ме¬
тод молярных отношений. Ме¬
тод изомолярных серий сос¬
тоит в том, что составляют¬
ся смеси с переменным См/сь
при см+ cL = const. При
исследовании комплексов сла¬
бых кислот нужно поддержи¬
вать также постоянство pH.
Если Dm и Dh — оптические
плотности растворов иона ме¬
талла и лиганда соответст¬
венно, то оптическая плот¬
ность их смеси с составомРис. 64. Определение состава комп¬
лекса методом изомолярных серийcL/cM = */(1 —х)* при от¬
сутствии комплексообразова-
ния (£>адд) была бы равна
xDj_ + (1 — x)Djvf. Разность
реально измеренной оптиче¬
ской плотности D и £>адд на¬
зывается отклонением от ад*
дитивности: AD = D — DaM.
Значение х/(\ — х), соответ¬
ствующее максимуму кривой
AD — состав, равно отноше¬
нию коэффициентов р и и в
доминирующем комплексе
Ма LB (рис. 64). Если этот
комплекс одноядерный, то
х/(\ — х) равно п в формуле
MLn. Метод мало пригоден
для определения состава ком¬* Через х обозначено отношение cL/(смфс£), тогда cM/(cM-|>cL) = 1 —х.Рис. 65. Определение состава комп¬
лекса методом молярных отношений,
если ион металла и лиганд неоира-
шены— 166 —
плексов ML„ с 2<n <5, поскольку значения х, соответствую¬
щие этим п, близки:* 0,5 0,67 0,75 0,80 0,83* .\—х4В методе молярных отношений оперируют растворами, в ко¬
торых при постоянной концентрации См нарастают значения сь
{pH = const). Для комплексов ML,„ большой устойчивости на
графике зависимости D от cl наблюдается излом при cl См = п
{рис. 65). Любой комплекс средней или малой устойчивости
частично диссоциирует, что приводит к «сглаживанию» излома кри¬
вой. Если установлено, что образуется комплекс состава 1 : 1, то
при / « 1, обозначив [ML] через у, получаем(VIII. 21)1/2 —LMЕсли при данной длине волны лиганд не поглощает, поглощение
свободного иона металла Дч = /емсм известно и проведены изме¬
рения D ряда растворов с см = const и Cl ^>см,то подстановка в
(VI 11.21) уравнения у = AD/Azl, где A D=D — Dm и Де= £ml—
приводит после ряда допущений к уравнению Бенеши—Хильде¬
бранда:1 1.1'ДО /смД£ + /смДеКак видно из уравнения (VIII -22),
зависимость 1/AD от 1/cl должна
быть линейной с тангенсом угла
наклона 1//cmAs|3i и пересечением
1//смАе. Разделив пересечение на
величину тангенса угла наклона,
получаем а из 1//смАе вычис¬
лим As.■ Для комплекса MH„L изучают
зависимость D от р[Н] при посто¬
янном см :cl■ Если п постоянно,
то эта зависимость имеет вид кри¬
вой с двумя горизонтальными
асимптотами D = Do и D =(рис. 66). При этом выход ком¬
плекса можно рассчитать по опти¬
ческой плотностиD-A,(VIII.22)Da~D0Если ион металла находится в из¬
бытке, т. е. см ■ Ci = Q > 1, то
константа образования комплексаРис. 66. Зависимость D (а) и Igfx
(б) от р[Н1— 167 —ciu ларлда и и умсношснисм радиуса;2) наибольшее COOTICTRO К KflTITfmv ИИРШТ лпнппнир атгаш оо.
равнаacLf[MHnL]Pln [M][H„L] (1 a) (Q a) cl Bn [H]nОбозначив a/(l —a) (Q — a) через x, получаем
lg xf = —лр [H] + Ig pln cL Bn.Построив график зависимости lg xf от p[H], по наклону прямой
определяют п, а из пересечения ее с осью ординат вычисляют§ 2. Понятие об устойчивости комплексного соединенияТермодинамическая устойчивость характеризуется следующими
величинами:1) стандартным изменением изобарного изотермического потен¬
циала Аб°для процесса образования комплекса ML„ из иона металла
и лигандов в газовой фазе (заряды ионов в приводимых уравнениях
реакций опущены):2) значением AG° для реакции, протекающей в водном растворе»Реакцию (б) можно представить в виде суммы более простых
реакций:где реакция (в) совпадает с реакцией (а), а остальные — это реак¬
ции гидратации иона металла, лиганда и комплекса соответственно;3) выходом комплексного иона при стандартных условиях. В ана¬
литической практике принято сравнивать устойчивость комплексов
одного и того же катиоца с разными лигандами по выходу или по по¬
ведению этих комплексов при разрушении их каким-либо реаген¬
том в одинаковых условиях. Устойчивость такого рода зависит от
выходов ап. Если выбраны pH и стандартные концентрации ли¬
ганда cl и иона металла См, причем <С сь. тогде р„= [ML„] /[М] [L]л — константа равновесия реакции (б).
Если сравнивается выход комплексов 1: 1, и в системе образуются
только они, то IL ] / иМ (г) + nL (г) = ML„ (г)М (Н20)* + nL (Н20)г = М (Н20)у Ln + (х - nz - у) Н20 - (б>М (г) + nL (г) = ML„ (г)(в)(Г)(Д>(е)М(Н2ОЬ (р) = М(г)-МН20 (р)
nL (Н20)г (р) == nL (г) + nz Н20 (р)
ML„ (г) + у Н20 (р) = М (Н20)у Ln (р)Pi [L] Pi cL168 —
Если с а = с^, то отношение выходов для лигандов А и L равноa‘A:a‘L /д+PlA^A : fL+PlLcL 'Если выход комплексов мал и а /a3>PiaCa, отноше¬ние выхфов равноПри таком способе оценки устойчивость определяется величиной
Pi— lo/).т- е- она зависит не только от константы равновесия
реакции (б), но и от констант протонизации лиганда и pH.Значение lgpj для комплекса катиона М с /г-дентат’ным лигандом,
как правило, больше значения lgp„ для комплекса с п аналогич¬
ными однэдентатными лигандами. Так, для комплекса Co(NH2CH2CH2NH3)21' lgpj = 5,93, а для Co(NH3)2f lgp3 = 3,74. Одна¬ко делать' из этого вывод о каком-то особом влиянии циклообра¬
зования на константы устойчивости неверно, так как j34 и (Зп имеют
разную размерность.Особенностью хелатных комплексов является то, что при умень¬
шении концентрации лиганда или при разбавлении раствора вы¬
ход комплекса с n-дентатным лигандом убывает медленнее, чем вы¬
ход аналогичного комплекса с п однодентатными лигандами, по¬
тому что выход ML зависит от [L], а выход ML„ — от [LK§ 3. Факторы, влияющие на устойчивость комплексных частицЭлектростатические факторы. Если комплекс образуется в зна¬
чительной мере за счет электростатических сил, то проявляются
следующие закономерности:1) устойчивость комплексной частицы увеличивается с ростом
напряженности поля центрального катиона, в частности, с ростом
его заряда и с уменьшением радиуса;2) наибольшее сродство к катиону имеют донорные атомы, не¬
сущие наибольший отрицательный заряд. Наличие у лиганда отри¬
цательно заряженных функциональных групп, не участвующих в
связи, увеличивает устойчивость комплекса, если воздействие этих
групп не уменьшает заряда донорного атома.Так, сравнение комплексов лантана с анионами пирокатехина
и тиронаalА : alL —/А fhо-0-/\/,о-\/анион пирокатехинаанион тирона— 169 —
показывает, что сульфогруппы тирона, не участвующие в комплек-
сообразовании, тем не менее вносят вклад в устойчивость комплекса:
значения lg^ равны 9,46 для пирокатехина и 12,93 для тирона.Накопление донорных групп в молекуле лиганда увеличивает
устойчивость комплексного соединения;3) после присоединения к иону металла первого лиганда-анио¬
на положительный заряд комплекса уменьшается, поэтому второй
лиганд должен притягиваться слабее, третий — еще слабее и т. д.,
и ступенчатые константы устойчивостидля лигандов-анионов уменьшаются с ростом я;4) когда незаряженная функциональная группа, например
—ОН или— СООН, теряет ион водорода за счет диссоциации, ее
склонность к комплексообразованию возрастает. Поэтому анион
кислоты образует более прочные комплексы, чем его протониро-
ванные формы (HL, Н2Ь и т. д.).Эти закономерности не распространяются на комплексы, обра¬
зованные преимущественно по ковалентной схеме. Однако практи¬
чески всегда при комплексообразовании действуют силы, которые
удобно считать электростатическими. Их значение растет с увели¬
чением формального заряда центрального иона металла; так у Си2+
оно выше, чем у Си+, а у Fe3+, выше, чем у Fe2+,Геометрические факторы. .'Если отрезок прямой, соединяющий
два донорных атома лиганда, пересекает какие-либо связи в мо¬
лекуле, или длина его больше 4 А, то эти донорные атомы не могут
замкнуть цикл на одном ионе металла. Так, среди производных
бензола бидентатны лишь орто-замещенные, а среди производных
этилена — соединения с цис-расположенными (как у малеиновой
кислоты) функциональными группами.Дентатность «жестких» лигандов вообще легко поддается ана¬
лизу: ниже приведены лиганды, которые обычно бидентатны, и ли¬
ганды, которые в этой роли выступать не могут.[ML„][ML*.!] [L]Бидентатные лигандыМонодентатные лиганды/ \
\—к/ \N_ //Ч /1,7-фенантролин'Y’^N'7он8-оксихннолин7-оксихинолин170 —
но онАЛ ноу\/у°нHOaS^^/^/^SOaH H0aS'/\/N''/N4S03Hхромотроповая кислота 3,7-диокси-2,8-нафталнн-дисульфокислотаДля «гибких» лигандов часть конформаций может способство¬
вать полидентатности, а часть препятствовать. Закрепление вы¬
годной конформации увеличивает устойчивость комплексов. При¬
мером могут служить комплексы этилендиаминтетраацетата
(ЭДТА) — лиганда со сравнительно свободным вращением, — и
аналогичного ему транс- 1,2-циклогександиаминтетраацетата
(ДЦТА), у которого выгодная конфигурация закреплена образо¬
ванием цикла:.сн2—соо-Жги /СН2—соо-2 N'Wcoo- «4 У ХсН!“со°'ХН2—СОО- Н-Сч Я* хна—соо-хсн2—соо- ЬПз хсн2—соо-ЭДТА ДЦТАУстойчивость комплексов ДЦТА выше, чем ЭДТА: так, например,
при / = 0,1 для ЬаДЦТА"^ ^ = 16,26, а для ЬаЭДТА- lg^= 15,13.Стерические препятствия создаются крупными заместителями
и мешают образованию выгодных для комплексообразования кон¬
фигураций. Во-первых, заместитель может мешать некоордини¬
рованному лиганду принимать выгодную конфигурацию. Так,
молекула 2,2'-дипиридила/ \_/ \\_N/ \N_/2,2'-дипириднлнаходится в выгодной для комплексообразования конформации и
ведет себя аналогично молекуле 1,10-фенантролина, когда является
плоской. Крупные заместители, введенные в 3- и З'-положенияN03 NO,/ \_/ \\—п/ \N—/3,3' динитро*2,2'-днпиридилотталкиваясь друг от друга, ведут к повороту пиридиновых колец
вокруг линии связи и удалению донорных атомов азота друг от— 171 -
друга, что ослабляет склонность молекулы к комплексообразо-
ванию.Во-вторых, наличие в молекуле реагента свсеобразного «во¬
ротника», окружающего клешнеобразующую группировку, напри¬
мер метальных групп в молекуле 2,7-диметил-8-оксихинолина/\/\Н3с/\N'/N'f/N'CHa
ОН2 • 7 * диметил- 8 - оксихинолинили бензольного кольца в молекуле N-окиси хинолинаI°-N-окись хинолинаувеличивает, место, занимаемое лигандом во внутренней коор¬
динационной сфере. Поэтому образование комплекса 1 : 1 должно
сопровождаться дегидратацией и быть энергетически менее еыгод-
ным, т. е. уменьшается lg [3j. Лиганды такого типа меша-ют друг
другу накапливаться во внутренней сфере, что резко уменьшает
значения lg*2, lg*3H т.д. и координационное число иона металла
(если оно у него переменно). Так, синтезированы комплексы лан¬
тана с N-окисью пиридина (р>0) состава [La(pyOg][Cr(SCN)„};
а с N-окисью хинолина (guin О) состава [La(quinO)7][Cr(SCN)e].Кроме того, геометрическими факторами, влияющими на устой¬
чивость комплексной частицы, являются отношение размеров ли¬
ганда и иона металла (см. главу II, § 1) и число членов цикла в
хелатных комплексах (см. главу IV, § 2).Статистические факторы. Во-первых, статистическими факто¬
рами определяется доля выгодной для комплексообразования кон¬
фигурации лиганда среди прочих конфигураций. Во-вторых, ста¬
тистические факторы влияют на соотношение между ступенчатыми
константами устойчивости. Пусть конфигурация координацион¬
ной сферы, например октаэдр, задана. Вероятность внедрения
однодентатного лиганда в координационную сферу пропорцио¬
нальна количеству молекул воды в комплексе MLK_X, подвергаю¬
щемся атаке, а вероятность отщепления этого лиганда от образо¬
вавшегося комплекса ML„ — числу лигандов в ML„, т. е. п. Сту¬
пенчатая константа устойчивости хл пропорциональна отнсшению
этих двух чисел. Для октаэдра хп пропорциональна (7 — л)/л,
т. е.х1:х2:хз'х4;хб:хв==:б: 2,5 : 1,3 : 0,75 : 0,4 : 0,17.— 172 —
Таким образом, каждое значение lg хя+1даже при полном пре¬
небрежении энергетическими факторами должно быть примерно
на 0,3 меньше lgx„, причем эта разность при постоянной конфигу¬
рации координационной сферы не зависит от природы иона металла.§ 4. Зависимость устойчивости комплексов состава 1 : 1
от характеристик иона металлаЗависимость устойчи¬
вости (lg Р() комплексов
состава 1 : 1 от приро¬
ды иона металла опреде¬
ляют по соотношениям
между устойчивостью
комплексов МА и RA
(Ми R — два сопостав¬
ляемых иона металла).Для восьмиэлектронных
и /-катионов, кроме
Mg2+, существует линей¬
ное соотношениеlg Рм = амR 'б ?R + ^MR(VIII. 23)Например, для ком¬
плексов Zr4+ и La3+(табл. 28, рис. 67), урав¬
нение (VIII. 23) имеет
видlg PZr = 1 tOf* ‘ё Р[_ < Т" U.1UO.Корреляционное соотношение для другой пары ионов представлено
на рис. 68.Таблица 28
Устойчивость комплексов Zr4+ и La3+ при 20—25°СЛиганд1lg PLa>g PzrНитрат 2,00,250,29Хлорид 4,0—0,220,04Оксалат 0,25,559,80Нитрилотриацетат 0,110,4720,80Анион тирона 0,113,0525,40ЭДТА 0,115,5029,00ДЭТПА 0,119,9636,73Рис. 67. Корреляция устойчивости
комплексов Zr4+ и La3+— 173 —
Коэффициенты «mr в уравнении (VIII. 23) близки к отношению
ионных потенциалов катионов фм/фя (табл. 29), a 6mr—к нулю
(табл. 28). Поэтому значения Ig Змприблизительно пропорциональ¬
ны ионным потенциалам]еРм ы_
lg Pr 88 ?r *Значения lg и Ig {3r не связаны функциональной зависимостью
друг с другом и корреляционное соотношение (VIII. 23) существуетРис. 68. Корреляция устойчивости Рис. 69. Зависимость lg Pi для
комплексов Y3+ и Са 2+ комплексов щелочноземельных ка¬тионов с ЭДТА (20°С, /=0,1)
от Фмпотому, что обе эти величины одинаковым образом зависят от ха¬
рактеристик иона металла и лигандаIg рм=амНЧ+Лм, (VIII.24)1gPR==tfR/(b)+> • (VIII.25)где ам> <2r, и Ьц — константы для данного катиона; /(L) — функ¬
ция параметров лиганда, одинаковая для обоих катионов. Ком¬
бинируя (VIII. 24) и (VIII. 25), имеемам~= ЙМ ~ aMR 6R •Следовательно, ам и а% пропорциональны срл1 и фя и равенство— 174 —
(VIII. 24) переписывается в видеIgpM=K<?M/(L) + ftM. (VIII.26)Если лиганд присоединяется к катионам разным способом, та
/(L) =j= const и соотношение (VIII. 23) не выполняется.Пропорциональность значений lg(3M ионным потенциалам 8-
электронных катионов (см. стр. 147) нарушается при неполном
использовании дентатности лигандаТаблица 29Коэффициенты ам^ в уравнении (VIИ.23)мп+°M,LaV¥ LaM"+°M,LaV'fLaM"+°M,BaV'fBaуз+1,141,15Th<+1,461,48Sr2+1,121,12Lu3+1,201,22Ce*+1,551,59Ca2+1,351,34Sc3+1,481,46Ba2+0,580,57Zr4+1,871,86Таблица 30
Коэффициенты &MR в уравнении (VIII.23)M Л +4M.LaM«+^M.Laуз+—0,23Ce4++ 0,19Lu3+—0,02Zr4++0,10Sc3++0,63Ba2+—1.0Th< ++0,31вследствие малых размеров катиона. По этой причине у много-
дентатных лигандов уменьшается сродство к ионам Mg2+ и А13+
по сравнению с Са2+ и Sc3+ соответственно (рис. 69). У комплек¬
сов катионов с большим радиусом (Са2+) или зарядом (La3+) гео¬
метрические ограничения сказываются только для лигандов с очень
высокой дентатностью. Проследим, например, за устойчивостью
комплексов Са2+ и La3+ в ряду аминополикарбоновых кислот (ком¬
плексонов), начиная с нитрилотриуксусной (НТА). У ионов НТА,
ЭДТА, диэтилентриаминпентаацетата (ДЭТПА), триэтилентетра-
мингексаацетата (ТЭТГА) и тетразтиленпентамингептаацетата
(ТЭПГА) при переходе от лиганда к лиганду число функциональных
групп увеличивается на две: на атом азота и карбоксильную груп¬
пу. При этом устойчивость комплексов первоначально почти ли¬
нейно возрастает (рис. 70). Начиная с ДЭТПА у Са2+ и с ТЭПГА
у La3+, рост lg прекращается, что указывает на невозможность
использования этими лигандами всех своих функциональных групп.— 175 —
В ряду редкоземельных элементов ус¬
тойчивость комплексов обычно воз¬
растает от La3+ и Lu3+ (например, для
ЭДТА), нодляДЭТПА и особенно для
ТЭТГА характерно резкое пониже¬
ние устойчивости к Lu3+ (рис. 71).
Это вызвано тем, что геометрические
ограничения для меньшего катиона
Lu3+ более жестки.Устойчивость комплексов редкозе¬
мельных и прочих /-катионов также
связана с устойчивостью комплексов
8-электронных катионов соотноше¬
ниями вида (VIII. 5) Параметры jaM,La
для /-катионов (табл. 31) системати¬
чески превышают отношение ионных
потенциалов. Таким образом, наличие
частично заполненного /-подуровня
эквивалентно увеличению эффективно¬
го ионного потенциала катиона. Зна¬
чения ам.ьа позволяют оцениватьv НТА, ЭД ТА, ДЭТПА. ТЭТГА, ТЭПГА," ('Рм) эфф:J 4 5 6 7 п(<?м)эфф — аМ, LafLa •Рис. 70. Зависимость lgpt аот количества карбоксиль- Для комплексообразования о-эле-
ных групп в комплексом ктронных катионов с многоатомны-
вля комплексов Са и La ми лигандами предложена модельнаятеория устойчивости комплексов. Ес¬
ли предположить, что комплексообразующая способность лиган¬
да зависит от зарядов, сосредоточенных на поверхности донорных
атомов, т. е. удаленных от центра катиона М на расстояние гм, то
для lgj3M получается выражениеlg = 2 3 DkT Тм ф ^ + ьл = к<*л ф ^ + ьл ' (VI 11'27)Таблица 31Параметры aMiLa для /-катионовКатионaM,LaКатионaM,La•fm/vLaСе3+1,0331,033Ho3+1,1731,159ргз+1,0631,051Er3+1,1841,170Nd3+1,0841,000Tm3+1,1851,170Sm3+1,1181,078Yb3+1,1941,216Eu3+1,1341,078Am3+1,1601,07Gd3+1,1361,097Cm3+1,164 Tb3+1,1631,117U4+1,7401,545Dy3+1,1661,138176
Порядковый номер элементаРис. 71. Зависимость lg^ для комплексов лантанои¬
дов с комплексонами (/=0,1) от порядкового номера
лантаноидагде е — заряд электрона; k—постоянная Больцмана; 6м—-коэф¬
фициент, учитывающий влияние второстепенных эффектов; Ф^) —
функция параметров л-дентатного лиганда;П® (а/г) = 2 Haft aw 'k=\ak и aw — заряды донорного атома лиганда и молекулы воды;
8„ — изменение к. ч. при комплексообразовании.Параметр К = е2/2,3DkT при 25° С для водных растворов
равен 3,1 -10-8 см. Выражение (VIII. 27) качественно согласуется
с выражением (VIII. 26), причем /(L) = Ф(ак).Уравнение (VIII. 27) верно описывает линейную зависимость
lgpM от ионного потенциала <рмиот обратной величины диэлектри¬
ческой проницаемости (1 ID), а также аддитивную зависимость
lgp м от лА— количества донорных групп каждого k-ro сорта в ли¬
гандеlgpM = 1fo (VII 1.28)где f0 и -[k — константы. Зависимость (VIII. 28) наблюдается при
малом взаимовлиянии донорных групп. Коэффициенты функций
вида (VIII. 28) для редкоземельных элементов приведены в табл. 32.
Для La3+, Y3+, Lu2+ и I = 0 уравнение (VIII. 28) преобразуется
к видуРьа’+ = —2,53 + 4,89/Iqqq-+ 1 ,14rtQfj -f- 0,60я^ , (VIII.29)
lgP°.+ = —3,13 + 5,51ясоэ_ + ЬЗбЛон + 0,67ядг , (VIII.30)
1е^+ = -3-01 +5,70псоэ_ + 1,31яон + 0,89лд, , (VIII.31)7-699— .177 —
Сопоставление (VIII. 28) и (VIII. 27) показывает, что име¬
ет смысл /Сфм(«* — О- При делении*^ из уравнений (VIII. 29) —
(VIII. 31) на /Сфм (табл. 33)получаются значения (ак — aw), ко¬
торые практически не зависят от того, какое уравнение использо¬
вано.Таблица 32Коэффициенты f0 и 7* в уравнении (VIM.28) при 7=0,1 и 20—30°СКатнонToTCOO-ЮН7/VКатионTo7COO-lOHVLa—2,534,251,140,6Dy—3,144,911,340,9Се—2,824,481,390,6Ho—3,134,931,360,9Рг—2,684,451,310,8Er—3,174,951,370,9Nd—2,734,501,350,9Tm—3,395,171,500,8Sm—2,904,641,410,9Yb—3,075,041,330,9Eu—2,894,691,370,9Lu—3,015,061,310,9Gd—3,084,781,370,8Y—3,134,871,350,7Tb—3,134,841,360,9Таблица 33Оценка разности зарядов (а*.— аш) при помощи уравнений (VIII.29) —(VII1.31)Катион“coo-“oh “i*aNLa**7,520,650,150,08уз*8,640,€40,160,08Lu3+9,230,620,140,10Устойчивость комплексов 18-электронных катионов. Донор¬
ные атомы азота и серы имеют гораздо большее сродство к 18-элек-
тронным катионам, чем к8-электронным. Растворы аммиака, которые
осаждают большинство 8-электронных и /-катионов в виде гидро¬
окисей, переводят 18-электронные катионы в устойчивые раство¬
римые аммиачные комплексы. Сродство 18-электронных катионов к
атому азота [см. уравнение (VIII. 29) ] сопоставимо со срод¬
ством их к заряженным функциональным группам, содержащим атом
кислорода. Так, для Zn уравнение (VIII. 29) приобретает видр2п ~ — 1,522, 90/i^qq - 3,38/2дг ■—0,36/2ру,где лсоо — число карбоксильных групп в лиганде; nN — общее
число атомов азота в лиганде; пру — число «пиридиновых» атомов- 178 —
азота. Отношение ' Чсоо составляет 3,38 : 2,90=1,17 тогда как
для La3* оно равно 0,14.При сравнении 8- и 18-электронных катионов, например Са2+
и Zn2+, можно выделить группу лигандов, содержащих в качестве
донорных только атомы кислорода. Корреляционная диаграмма
для этой группы приведена на рис. 72. Уравнение (VIII.27) при¬
обретает видIgpZn = 1.25 lgpc, + 0,38*3Величина 1,25 близка к отношению ионных потенциалов Zn2+ и
Са2+, равному 1,27.Введение в лиганд донорных атомов азота, серы или фенольных
атомов кислорода увеличивает
lgfW Это значит, что при пе-
реходе от 8-электронных к
18-электронным катионам
возрастают величины ~{k для
некоторых функциональных
групп. Если значения / (L)
для лигандов постоянны для
всей группы 18-электронных
катионов, то внутри этой
группы также будут справед¬
ливы уравнение (VIII.25) и
следующее из него соотноше¬
ние (VIII.23).Уравнение (VIII.25) для
разных групп лигандов может
иметь различные коэффициен¬
ты • Тогда и на корреляци¬
онной диаграмме, например
Cd2+ — Zn2+ или Ag+ — Zn2+,
точки расположатся вдоль па¬
раллельных прямых, каждаяиз которых соответствует группе родственных лигандов. На кор¬
реляционных диаграммах In3+ — Zn2+ и Ga3+ — Zn2+ (рис. 73)
точки для большинства лигандов сосредоточены вокруг одной пря¬
мой, описываемой уравнениемРис. 72. Корреляция устойчивости
комплексов Zn2+ и Са2+ для «кис¬
лородных» лигандовlg р,п= 1,365 lg pZn +1,485(VI11.32)илиlgPQ,= 1,965 lg?Zn+0,14.(VI11.33)При этом у Ga3+ точки для лигандов с дентатностью 4 и более
(нитрилотриацетат, ЭДТА и др.) лежат ниже прямой (VIII.33),
так как часть функциональных групп лигандов не используется.7*— 179 —
а 5Рис. 73. Корреляционные диаграммы для комплексов индия и
цинка (а), галлия и цинка (б)Отношения ионных потенциалов ф1п/ф2п= 1,356 и ф0а/ф2п =
= 1,992 (см. стр. 148) близки к коэффициентам aM,zn в уравнениях
(VIII.32) и (VIII.33).Линейным соотношением связаны значения lg рх для комплек¬
сов Ni2+ и Zn2+, однако распространить соотношения вида (VI 11.23)
на остальные катионы с частично заполненным d-подуровнем уда¬
ется только для «кислородных» лигандов. Согласно Ирвингу и
Уильямсу, значения lg рп при п 4 для ионов Mn2+, Сг2+, Fe2+,
Со2+, Ni2+, Cu2+, Zna+ увеличиваются с ростом ионизационного по¬
тенциала М -*■ М2+, т. е. увеличиваются от Мп2+ до Си2+ и уменьша¬
ются у Zn2+.Параметр &mr сильно влияет на избирательность аналитических
реагентов. Чем больше AlgP1( тем больше при равных условиях
разница в закомплексованности катионов М и R. Если они подчи¬
няются соотношению AlgPM = aMRlgPR~f 6MR( тоA lg Pi = lg Рм — 'g Pr = ( “mr 0 ^ Pr ^mr •Рассмотрим два случая: когда r »0и когда &mr >0- Пусть
lg Pr > 0- Если 6MR fa 0, то знак Alg p i определяется знаком (aMR—
—1), а сама величина увеличивается с ростом lg или/(Ь),т. е. с
ростом дентатности лиганда. Повышение дентатности лиганда де¬
лает реакцию избирательной по отношению к катиону с высокими
электростатическими характеристиками.Если &mr > 0, a aMR <С 1, то при малых lg Pr (левее точки с
на рис. 74) более устойчивы комплексы катиона со слабыми электро¬
статическими характеристиками. При увеличении lg Pr , величины
/(L) или дентатности лиганда более устойчивыми становятся ком-— 180 —
плексы катиона с высокими элект¬
ростатическими характеристиками.
Так, при сравнении Zn2+ и Ag+
(табл. 33) устойчивость комплексов
однодентатных аминов выше для
серебра, а полидентатных — для
цинка.Диссоциация протонированных
комплексов. Из соотношений вида
(VIII.23) следует, что протониро-
ванные комплексы ионов металла
отщепляют протон тем легче, чем
выше фм- Качественно это вытека¬
ет из представления о контрапо-
ляризации лиганда HL катионом М.
Рассмотрим равновесие в системе,
содержащей катион М, лиганд L и
протоны:Рис. 74.ЗависимостьPRAlgpi отРавновесияКонстантыM+L=MLPiM+HL=MHLhiH+L=HLSiMHL=ML+HКТаблица 34Зависимость относительной устойчивости комплексов цинка и серебра отдентатности аминаЛигандДентат-ность$znl8?AgA lg?,Пиридин 10,952,01+ 1,06Аммиак 12,183,32+ 1 ,14Этилендиамин 25,564,70—0,861,10-фенантролин 26,835,02—1,812,2'-диаминодиэтиламин 38,906,10—2,80Триэтилентетрамин 412,107,70—4,40Четыре приведенные константы связаны соотношением
Pi _ ' [ML] 1 HL]Pit= кв±.(VIII.34)[MHL] (L]Уравнение (VIII.34) в применении к катионам М и R имеет видР,■м= *м fii;л1
м— 181 -(VIII. 35)
= KR В!. (VIII.36)Рц RПреобразуя (VIII.35) и (VIII.36) с использованием (VIII.34),
получаемIfi = aMR 'g ^R•g = ]g + ( aMR — ]) Kr B1ИЛИpKM = pKjj ( aA1R 1) lg Si-Итак, соотношение между р/(м и р/Си должно зависеть от зна¬
ка (aMR— 1), так как Ig(PR/pnR) = lgКЯВ{> 0, поскольку про-
тонированный лиганд имеет меньший заряд и меньшую дентат-
ность. Таким образом, р/См<Р ^к, если амr> 1. В середине инди¬
каторного перехода, т. е. при [ML] = IMHL], pH = p/f. Поэтому
pH этого перехода сдвигается н кислую область при увеличении
Фм-Зависимость устойчивости комплексов от а-констант Тафта.Для констант устойчивости комплексов с родственными лигандами
справедливо уравнение ТафтаlgpM = 1fipM + PM°*. (VIII.37)где or* — константа Тафта для лиганда; рД —коэффициент,
постоянный для данной группы лигандов; lg?„— логарифм кон¬
станты устойчивости комплек¬
са эталонного лиганда, для
которого а* = 0. Например,
существует линейная зависи¬
мость lg Зх для комплексов
Th4+ с монокарбоксилат-ио-
нами от рК диссоциации со¬
ответствующих кислот (рис.
75) и от линейно связанных
с ними а*-констант Тафта.
Константы Тафта связаны с
эффективным зарядом донор-
ных атомов лиганда.Из уравнений (VIII.23) и
(VIII.37) следует, что. --и*- — • (VIH-38)Pr ^rт. е. пропорциональна <рми lg(3M зависит от эффективного заря¬
да донорного атома тем сильнее, чем выше ионный потенциал
катиона М.Рис. 75. Зависимость lgpj комплек
сов Th'*+ с моно-, ди-и трихлороаце
тат-ионами (/=0,5) от рК диссо^ла
ции соответствующих кислотРм
XI. СИНТЕЗ КОМПЛЕКСНЫХ СОЕДИНЕНИИ§ 1. Основные принципы синтезаПри оценке пригодности какой-либо реакции для синтеза ком¬
плексной частицы необходимо, чтобы было отрицательным изменение
изобарного изотермического потенциалаД0 = — ЯГ In K + ai’ 0х-1)где К — константа равновесия; v£ — стехиометрический коэффи¬
циент; аг — активность i-ro вещества. Из уравнения (IX. 1) сле¬
дует, что условия синтеза можно подбирать, изменяя концентрации
веществ, а также давление и температуру, от которых зависит кон¬
станта равновесия. Согласно принципу смещения равновесий
Ле-Шателье повышение температуры увеличивает выход продук¬
тов эндотермических реакций, повышение давления увеличивает
выход продуктов реакций, идущих с уменьшением объема. Выход
комплекса увеличивается с ростом концентрации исходных ве¬
ществ и уменьшением концентрации продуктов синтеза.Чтобы уменьшить концентрации продуктов, применяют или
удаление их в гетерофазу (например, растворенную двуокись се¬
ры удаляют, продувая раствор азотом), или связывают их в нерас¬
творимые, легколетучие или малодиссоциирующие вещества. Та¬
кое связывание означает, что в системе протекает еще по крайней
мере одна реакция. Например, для проведения реакцииС2Н4/ 7NH3 С2Н4/ 7NH3/ Pt / + Н20 / Pt / + сг (I)Вг ЧД Вг %0в систему добавляют ионы Ag+, осаждающие хлорид-ионы;Ag+ + Cl- AgCl i (II)Оценить направление процесса синтеза при этом можно, получив
уравнение суммарной реакциии константу равновесия для него. Поскольку равновесие (III) яв¬
ляется суммой равновесий (I) и (II), то/Сш — К\ Ки, и, чтобы
синтез осуществлялся, необходимы условия Кт >1, VKn,т. е. произведение растворимости AgCl должно быть существенно
меньше, чем Кг .Если связываются одновременно и исходные вещества, и про¬
дукты реакции (например, в комплекс), то необходимо, чтобы про¬
дукты реакции связывались в более устойчивый комплекс. Рассмо-— 183 —
трим, например, уксуснокислый раствор ацетата Со2+ в равновесии
с кислородом воздуха:Со2+ + Оа + 4Н1 z£. Со3+ + 2Н20 (IV)Равновесие (JV), для которого K\v <С I, сдвинуто влево, т. е. окис¬
ление Со2+ практически не происходит. При добавлении в систему
нитрит-иона устанавливаются равновесия:Со3+ + 6NO^~ [Со (N02)e]®- (V)Со2+ + п N0^ [Со (Ша)я]»-Я (VI)Константы равновесий Kv и Kv^ больше I, равновесия реакций сдви¬
нуты вправо. Суммарная реакция[Со (N02)„p-" + (6 - п) NC£- + 02 + 4Н+ [Со (NO,),]»- + 2Н20 (VII)с константой равновесия Kvn используется для синтеза гексани-
трокобальтиата натрия. Реакция (VII) протекает с хорошим вы¬
ходом, т. е. Со3+ стабилизируется в виде нитритного комплекса.
Поскольку Kvn = (Kv/Kvi) Kiv I» то необходимо, чтобы
Kv Kvi/Kiv, т. е. Kv l/Kiv и Kv^Kvi- Условие Kv Kvi озна¬
чает, что нитритные комплексы Со3+ должны быть гораздо прочнее
комплексов Со2+.Чтобы при синтезе стабилизировать неустойчивую частицу,
нужно ввести ее в такую реакцию, у которой константа равновесия
велика, а стабилизируемая частица является самостоятельным струк¬
турным фрагментом продукта реакции. На практике в систему
вводят добавочный реагент, осаждающий стабилизируемый ион
или связывающий его в растворимый комплекс. С заметным вы¬
ходом идут те реакции синтеза, для которых AG отрицательно,
т. е. константа равновесия больше единицы. Термодинамические
условия синтеза важны как для лабильных, так и для инертных
комплексов. Но при работе с инертными комплексами необходимо
учитывать и кинетические факторы, в особенности наличие под¬
ходящего механизма реакции.Нельзя указать универсальные методы, пригодные для синтеза
любого комплексного соединения. Но имеется ряд частных зако¬
номерностей, позволяющих осуществлять целенаправленные
синтезы.§ 2. Выделение комплексного иона из системы
лабильных комплексовДля получения максимального выхода требуемого комплек¬
сного иона в системе создают необходимые условия и выделяют его,
подбирая подходящий противоион или растворитель. Чтобы
обеспечить большую скорость выделения и большой выход осадка,
следует добиться доминирования выделяемого иона. Если не удает¬— 184 —
ся достичь доминирования, то стремятся сделать выход максималь¬
ным. Максимальный выход комплексного иона не обязательно вле¬
чет за собой доминирование его в системе. Так, например, про¬
межуточные комплексы часто не имеют области доминирования во
всем интервале концентраций компонентов, а для преобладания
комплекса с максимальным числом лигандов или многоядерных
комплексов может потребоваться слишком высокая концентрация
лиганда или ионов металла.Иногда удается выделить из раствора ион, присутствующий в
системе в очень малых количествах, если он успешно конкурирует
с другими ионами за ион-осадитель (противоион). Тогда будет про¬
исходить разрушение доминирующих комплексов с образованием
выделяемого иона и связывание его в осадок. Противоионы подби¬
рают с учетом полуэмпирических правил К- Б. Яцимирского:1. Отношение радиусов ионов rjru, при котором достигается
минимум растворимости, для солей типа MX составляет 0,7, а для
солей типа МХ2— 1,1. В качестве ссадителей комплексных ионов,
размеры которых обычно велики, можно использовать анионы пи-
крат, гексацианоферроат, тетрароданодиамминхромиат, пслииодид
и т. п.; катионы: аммиакаты Со3+ и Сг3+, катионы четЕертичных
аммониевых оснований NR4" и др.2. Растворимость солей при прочих равных условиях тем мень¬
ше, чем выше заряды комплексных катионов.3. Входящие в состав осадка группы, способные к образованию
водородной связи с молекулами воды (—ОН, —ССОН, —NH2 и
т. д.), увеличивают растворимость ссли.4. Наличие полярности внутри иона при прочих равных усло¬
виях уменьшает растворимость соли.Ионы-осадители часто применяются для выделения в виде осад¬
ков чрезвычайно неустойчивых в водном растворе продуктов. Из-за
медленного протекания реакций в твердой фазе метастабильнсе
состояние кристаллических веществ устойчиво кинетически. Так,
выделены соединения ионов [СгОС14]~ и [СтОС15]2-, содержащих
хром в степени окисления +5. При этом противоионами для [СгОС14]_
служили катионы изохинолиния, хинальдиния, акридиния и дру¬
гих гетероциклических аминов, для [СгОС15]2- — катионы
1,10-фенантролиния и 2,2/-дипиридилияВ табл. 35 приведены ионы-осадители, применяемые в ряде
синтезов. Для выделения комплексных катионов [LnA6]3+, гдеЬп3+—
ион редкоземельного элемента, а лиганд А — пиридин, хинслин,
пиперидин, никотиновая кислота в бетаинсвсй форме и другиеNH HN. / \ / \\ / \ /NH :HN++катион I .Ю-фенантро/шния катион 2,2'-дипиридилня— 185 —
органические амины, были применены анионы гексароданохро-
миат [Cr(SCN)e]3- и тетрароданомеркуроат [Hg(SCN)4]2_.Приведенные правила применимы и для осаждения инертных
комплексных ионов. Так, катион [Re(CO)6]+ осаждается перхло¬
ратом, рейнекатом и тетрафенилборатом [В(С6Н5)4]_. Катион
транс-IPtA4Cl2 ]2+, где А — этаноламин, легко осаждается ионом
lPtCl4]2-.Свойство крупных катионов и анионов взаимно осаждать друг
друга широко используется в анализе. Например, обычные реак¬
тивы, осаждающие К\ Rb+nCs+: гексанитрокобальтиат [Co^O^g]3-,
тетрахлороплатоат [PtCl4]2' тетрафенилборат [В(СвН5)4]~, в мень¬
шей степени — перхлорат. Для весового определения [Co(NH3)e]3+
предложен ион [Т1С1„]3~. Осаждение [Ag(NH3)2]+, [Cd(NH3)4]2+,
[Ni(NH3)4 ]2+, [Zn(NH3)J2+, [Cu(NH3)4]2+, [Co(NH3)4]2+ рейне-
кат-ионом позволило разработать методики количественного ана¬
лиза катионов цветных металлов.Таблица 35Условия осаждения некоторых комплексных ионовЦентраль¬
ный ионКомплексныйнояСреда и условия
осажденияИоны-осадителиGe4+[GeCle]2-Смесь этанола и
коиц. НС1Cs+, Rb+, N(CH3)+Мо5+[МоОСЦр-M0CI5 в конц.
НС1 при 0°СCs+, Rb+, CH3NH^,
(CH3)2NH+, (CH3)3NH+W5+[WOClB]2~WC15 в конц. НС1C5H6NH+ (пиридиний-ион)и»+[U(C204)4]*-при 0°С[Cr(NH2CONH2)e]3+и<+[U(N03)e]2—Азотнокислые ра¬
створы, -,HNOj > ЗМCs+, Rb+, NH^", ионы пи-
ридиния, ХИНОЛИНИЯ, a-
аминопиридиния и а,а'-
дипиридилияTi<+[Ti Fe]2_Раствор TiF4 + HF
в ацетонеПиридиний, морфолиний,
пиперазиний-ионы
(C2H5)3NH+, (C2H5)2NH+Со3+[Co(SeCN)4]2-Этанольный раст¬
вор СоС12 + KSeCN[(C2H5)4N1+, (CeH6)4As+1п3+[InCl5]2-NH+, Rb+, Cs+Th4+[Th(C03)5]<>-Из раствора
Th(N03)4 +(NH4)2C03[Co(NH3)e]3+При проведении реакций замещения с инертными комплексными
ионами в растворе иногда существуют как катион, так и анион,—
производные от одного и того же иона металла. Они могут взаим¬
но осаждаться. Например, при взаимодействии [Co(NH3)e]Cl 3
с NaCN в водном растворе при соотношении компонентов 1 : 3
образуется осадок [Co(NH3)0] [Co(CN)6]. При взаимодействии— 186 —
ионов lNiA3P+, где А — молекула 1,10-фенантролина или 2,2'-
дипиридила, с цианат-ионом в атмосфере азота из водных растворов
выпадает осадок [NiAa][Ni(CN0)4]-2H20. То же самое может быть
при диспропорционировании комплексного иона в какой-либо
среде.Синтез комплексных соединений часто ведут в неводных рас¬
творителях или в водно-органических смесях. Поскольку диэлек¬
трическая проницаемость D у них меньше, чем у воды (исключени¬
ями являются Н202, жидкая HCN, формамид HCONH2), работа
образования ионов в этих средах повышена. Работа образования
г-го иона в растворе примерно пропорциональна z^./r^ и линейно
зависит от 1 ID, т. е. растет с уменьшением D.Поэтому в неводных растворах усиливается комплексообразо-
вание с лигандами-анионами. Если растворитель не оказывает
серьезной конкуренции лиганду, могут образоваться комплексы
МАП с таким большим п, которого не удается достичь в водном
растворе. Так, из метанольных растворов можно выделить
K3[In(SCN)„]-CH30H, тогда как из диметилформамидных из-за
большого сродства диметилформамида HCON(CH3)2 к иону 1п3+—
только [In(SCN)3Dmfs ].Повышенная работа образования ионов в неводных средах и
водно-органических смесях уменьшает растворимость осадков.
Чтобы выделить соли из водных растворов, к ним часто прибав¬
ляют метиловый или этиловый спирты, эфирно-спиртовую смесь
или ацетон. Например, добавление ацетона к 17М HF, содержащей
Ра5+ и NaF, вызывает выпадение осадка Na3PaF8. Выделенные
осадки отмывают от маточника чаще всего не водой, а спиртом и за¬
тем эфиром. Это позволяет уменьшить потери при промывании и
быстро высушить осадок.Органические растворители часто оказывают на систему ком¬
плексов дифференцирующее действие, т. е. расширяют интервалы
устойчивости отдельных комплексов. При этом для промежуточных
комплексов может проявиться область доминирования и создадутся
условия для их выделения.Если молекула воды является слишком сильным. конкурентом
для лиганда, то он с трудом вытесняет ее из акво-ионов. Проведение
реакции в растворителе, молекулы которого обладают меньшим
сродством к иону металла, позволяет получить комплексы с вво¬
димым лигандом. Например, при сливании спиртовых растворов
NiBr2, СиВг2 или Со(С104)2 и N-окиси пиридина (C5H5N+0~)*
происходит немедленное выделение осадков [Ni(pyO)6 ]Вг6,
[Cu(pyO)2Br2] и [Со(руО)„](СЮ4)2. Из водных растворов выделить
комплексы Ni2+, Cuz+ и Со2+ с N-окисью пиридина не удается. При
взаимодействии [N(CH3)4]NOs с Co(N03)2-2H20 в соотношении
2:1 в нитрометане получено соединение [N(CH3)4 ]2 [Co(N03)4 ]* N-Окись пиридина сокращенно обозначается руО.— 187 —
фиолетового цвета. Из водных растворов нитратные комплексы
Со2+, тем более с координационным числом 4, не выделены.
Если перед синтезом вода удалялась из-за ее высокой донорной
активности, получающиеся соединения гигроскопичны.Воду необходимо удалять из-за возможного участия ее в окис¬
лительно-восстановительных реакциях с продуктом реакции или -
с одним из реагентов.Реакции с сильными восстановителями проводят обычно в тетра-
гидрофуране, диэтиловом эфире, жидком аммиаке или в жидких
алифатических аминах и т. д. Так, при взаимодействии LiAlH4
с (CH3)2Zn в эфире выделяются не растворимые в этом растворите¬
ле LiZnH3, Li2ZnH4 или Li3ZnH5. Обычно применяемые сильные
восстановители: щелочные металлы, их амальгамы, гидриды или
борогидриды — разлагают воду с выделением водорода.Обычные окислители, за исключением фтора, фторидов гало¬
генов и ряда высших фторидов металлов, более или менее устой¬
чивы в водных растворах. Поскольку фторсодержащие окислите¬
ли — это газы или легколетучие жидкости, окисление ими часто
производится без растворителя. Рассмотрим пример синтеза, со¬
провождающегося окислением иона металла. При нагревании экви¬
молекулярной смеси LiF и MnF2 в токе F2 при 350° образуется
LiMnF5 кирпично-красного цвета. Примером синтеза, сопровождаю¬
щегося окислением и замещением лиганда, является получение
голубых соединений M3[CoFe] (М — Li, К, Rb или Cs) при фто¬
рировании M3[Co(CN)e ] смесью СОа и F2- При действии HaM3[CoF6]
воды образуется черный осадок и выделяется кислород;
при действии НС104 также выделяется кислород и образуется
раствор соли Со2+.Аналогичная методика применяется для_синтеза комплексных
фторидов церия (IV) и празеодима (IV). Соединения M2LnF6 и
M3LnF7, где М—Na, К, Rb или Cs, получают действием на смесь
Се02 или Рг6Ои с MCI фтором в присутствии С02 при 400—500°.
Продукты реакции быстро охлаждают до —78°.§ 3. Окисление и восстановление доминирующего комплекса
в системе комплексных ионовВ системе лабильных комплексных ионов равновесие может быть
сдвинуто довольно легко. Например, ионы Со2+ и Сг2+ образуют
лабильные комплексы, а ионы Со3+ и Сг3+— инертные, поэтому
комплекс Со3+ или Сг3+ иногда легче получить окислением соот¬
ветствующего комплекса Со2+ или Сг2+, чем проводить реакции
замещения, исходя из других комплексов Со3+ или Сг3+. Так осу¬
ществляют, например, синтез гексамминхроми-иона. Приготовлен¬
ный раствор соли Сг2+ сливают в атмосфере инертного газа со смесью
525 г NH4C1 и 540 г 25%-ного раствора аммиака и оставляют в со¬
суде с газоотводной трубкой, снабженной гидрозатвором, до пре-— 188 —
кращения выделения водорода (сосуд снаружи охлаждают до 0°).
Красный раствор сливают и обрабатывают равным объемом 95 %-ного
спирта. Через несколько часов выделяются оранжево-желтые кри¬
сталлы [Cr(NH3)6 ]С13. Их промывают спиртом и сушат на воздухе.Методика такого типа может быть применена и тогда, когда
исходный комплекс инертен, но легко синтезируется. Метод син¬
теза комплексных соединений с нехарактерными степенями окисле¬
ния центрального иона состоит в восстановлении комплекса амаль¬
гамами металлов, борогидридами, гидридами и т. д. Некоторые
лиганды: цианиды, ' фосфины, карбонилы, некоторые серусо-
держащие лиганды, 2,2'-дипиридил и 1,10-фенантролин— обла¬
дают явно выраженной способностью стабилизировать низшие
состояния окисления переходных элементов. В табл. 36 приво¬
дятся некоторые примеры синтезов комплексов металлов.Таблица 36Некоторые синтезы комплексов металловСтепень окисления
в синтезируемом
комплексеСпособ синтезаСг(0)Со(1+)Re(l+)Re(3+)Mn(l—)При восстановлении Кз [Cr(CN)e] металлическим
калием в жидком аммиаке получают KetCr(CN)e]
(зеленого цвета), окисляющийся и гидролизую¬
щийся на воздухе, но устойчивый в атмосфере
инертного газа
При восстановлении [Co(dipy)2(H20)2](C104)3 бо-
рогидридом натрия NaBH4 в водно-спиртовом ра¬
створе получают мелкие темно-синие кристаллы
Co(dipy)2C104
К водному раствору K2[ReCle] и KCN прибав¬
ляют 0,4%-ную амальгаму калия, 2-3 мин пока¬
чивают реакционный сосуд при охлаждении. От¬
деляют сине-аеленый осадок K5[Re(CN)e]-3H20 и
промывают спиртом и эфиромАнион [Re(CN)e]3- образуется при восстановле¬
нии водного раствора K3[Re(CN)s] борогидридом
калия. Он выделяется в виде зеленого осадка
[Co(NH3)6][Re(CN)e] при добавлении к восстанов¬
ленному раствору [Co(NH3)e]Cl3
При восстановлении раствора Мп2(СО)10 в тетра-
гидрофуране амальгамой натрия получается
Na[Mn(CO)5]. Эту соль можно выделить испарени¬
ем раствора в вакуумеПри синтезе Ke[Cr(CN)6] (см. табл. 36) восстановление про¬
текало без изменения координационого числа центрального иона
и фактически без изменения структуры комплекса, т. е. протека¬
ла окислительно-восстановительная реакция типа[МАд,]и+ + ke~ [МАд,]"-*— 189 —
Такие реакции легко осуществляются с применением «чисто
электронных» окислителей или восстановителей, напримерСе4* + «Г CeSfFes* + (Г Fe2+Na Na+ + e~Быстрое протекание окислительно-восстановительных реак¬
ций инертных комплексных ионов позволяет использовать окислен¬
ную или восстановленную форму комплекса в качестве катализа¬
торов реакций замещения или изомеризации. Рассмотрим, например,
действие лабильного и поэтому рацемического иона [Соеп3]2+ на
смесь рацемического [Соеп3]3+ и d-тартрата. Диастереомерный
jd-[Coen3])2 (d-C4H604)3 плохо растворим. При добавлении
[Соеп3]2+ осаждается почти весь рацемат [Соеп3]3+ в виде диасте¬
реомера, так как в результате электронного обмена возникают новые
порции d-изомера:Z-[Coen3]3+ + d., 4Coen3]2*=d, /-[Coen3]3+ + /-[Coen3]2+Эта окислительно-восстановительная реакция, не сопровождаю¬
щаяся изменением внутренней координационной сферы, протека¬
ет быстро, а образующийся лабильный ион /-[Соеп3Р+ быстро
превращается в рацемат.При применении некоторых окислителей или восстановителей
очень часто во внутренней сфере комплекса «застревают» продук¬
ты распада реактива или ионы растворителя. Например, при окис¬
лении оксалатных растворов Со2+ церием (IV) получаются анионы
[G^QOJg]3', а при окислении кислородсодержащими окислите¬
лями — гипохлоритом или перекисью водорода — двуядерный ани¬
он [(Q.04)2C0(0H)2C0(C204)2]4-.Если при окислении координационное число центрального иона
увеличивается, реакция часто идет с присоединением окислителя
и представляет собой реакцию присоединения. Так, соль Гро
(транс-дихлоротетрамминплатехлорид) получают, пропуская хлор
через солянокислый раствор [Pi(NH3)4 ]С12. При этом Pt2+ с к. ч.4 переходит в Pt4+ с к. ч. 6:[Pt (NH,)*] Cl, + Cl, . [Pt (NH3)4 C!,] Cl,Схему данной реакции можно представить следующим образом:С1CIПлоским комплексам Pt2+ строго соответствуют октаэдрические
транс-замещенные комплексы Pt4+, что служит методом установ¬
ления строения комплексов Pta+ и Pt4+.— 190 —
Если окислителем является кислород, его восстановленная фор¬
ма может координироваться в виде пероксо-группы 0%~. Пусть
реакция окисления обратима; тогда при большом парциальном
давлении кислорода образуется комплекс, содержащий пероксо-
группу [МАд, -02), а после уменьшения парциального давления ком¬
плекс распадается:MAW • Од -f- ОдИменно такие соединения — гемоглобины и гемоцианины —
обеспечивают перенос кислорода по организмам животных. Они
представляют собой комплексные соединения Fe2+ и Си+, способные
присоединять кислород, превращаясь в производные Fe3+ и Си2+.
В настоящее время изучаются более простые комплексы, способ¬
ные эффективно переносить кислород и вновь регенерироваться.
В качестве переносчиков кислорода было предложено соединение
Со2+ с Шиффовым основанием, полученным из салицилового альде¬
гида и этилендиамина,1 /СН2ш^м СН2а также аминокислотные и дипептидные комплексы Со2+; системы
Fe2+ — диметилглиоксим и Ni2+ — днметилглиоксим (сильно щелоч¬
ные растворы диметилглиоксимата Ni более или менее обратимо
поглощакУг кислород) и, наконец, система Мп2+—фталоцианин в пи¬
ридиновом растворе. Большинство этих комплексов планарны,
причем планарность обусловлена конфигурацией лиганда. Присое¬
динение пероксо-группы происходит по свободным вершинам окта¬
эдра; часто одновременно присоединяется еще какой-нибудь
лиганд. Исключение представляют собой комплексы Со2+ с амино¬
кислотами, которые имеют октаэдрическое строение: пероксо-груп-
па здесь вытесняет функциональную группу лиганда и становится
на ее место. Комплексы с (Щ~ разрушают продуванием азота.В последнее время усиленно изучались также комплексные сое¬
динения, способные связывать атмосферный азот. Так, с азотом
быстро и обратимо реагирует при низких температурах [(С5Н5)2Тi ]х—
один из продуктов восстановления (я-СбН5)2Т1С12 металлическим
натрием. При этом получается голубой комплекс C6H5(N2)TiC5H5.
Комплекс t|«c-[Ru(en)2(H20)2]2+ реагирует с азотом при ком¬
натной температуре в водном растворе с образованием цис-
[Ru(en)2(H20)(N2)]2+.— 191 —
Молекула азота является электронным аналогом окиси углерода
и координируется аналогичным образом: получается линейная
группировкаМ ч- : N=NКомплексы, содержащие молекулу азота, образуются также при
некоторых реакциях координированного гидразина и азосоедине¬
ний. Подобные комплексы могут быть* получены при взаимодей¬
ствии координированного нитрит-иона с ионом NH+. Так, при вза¬
имодействии K2[Pt(N02)4] с (NH4)2S04 при 60—70° и pH 1—2 по¬
лучаются комплексы, содержащие линейную группировкуPt — N=N — Pt.§ 4. Использование реакций замещения лиганда
во внутренней сфере инертного комплексного ионаМетоды, используемые при синтезе одного инертного комплекса
из другого замещением лиганда в растворе, очень сходны с анало¬
гичными методами в органической химии.Как для лабильных, так и для инертных комплексов идут толь¬
ко термодинамически возможные процессы. Изменение изобарного
изотермического потенциала ДG связано с константой равновесия
реакции К выражениемДО= — ДГ In K + RT >£чг1паг, (IX.2)где at — активность i-то участника реакции; —стехиометрический
коэффициент в уравнении■'iAi + ч2А2 + ... -f "v*Aj + ... +^ЛА„ = 0,в котором V; для конечных продуктов взяты со знаком «+», а для
исходных веществ — со знаком «—». Если взять все реагенты в
стандартном состоянии (at = 1), то ДG = AG° = —RTlnK- Рас¬
положим лиганды в порядке увеличения AG0 для реакции вытесне¬
ния ими какого-либо стандартного лиганда из комплекса. Это будет
ряд активности лигандов, в котором предыдущий вытесняет каждый
последующий, если все участники реакции находятся в стандартных
состояниях. Для каждого иона металла должен быть построен свой
ряд активности лигандов. Так, для Pt2+ он имеет видS,Of- > Г > Thio > SCN“> NOf > Вг" > NHS> py > Cl" > OH">
>sqj-, NOJ", CI07Согласно принципу Ле-Шателье и уравнению (IX. 2) можно
заставить и последующий лиганд в ряду активности вытеснять
предыдущий: для этого надо создать большой избыток его по срав¬
нению с вытесняемым лигандом или же связывать или удалять этот
последний по мере вытеснения. Так, анион [Re(CN)e]4_ устойчив— 192 —
в водных растворах в присутствии избытка цианида; в кислых
растворах он акватируется до [Re(CN)5H20 ]3_. Для полу¬
чения KjRe(CN)6 ]-ЗН20 концентрированный водный раствор
K3[Re(CN)6H20 ] кипятят с избытком KCN до перехода окраски
раствора из коричневой в оранжево-красную и затем упаривают в
вакуум-эксикаторе.Для синтеза методом замещения удобно использовать карбонат¬
ные комплексы, так как СО^- по мере вытеснения можно легко уда¬
лить в виде С02. Например, сначала синтезируют трикарбонато-
кобальтиат натрия Na3[Co(C03)3]-ЗН20: раствор Co(N03)2 с из¬
бытком Н202 по каплям добавляют ко взвеси NaHC03 в малом
количестве воды. Выпавшие оливковые кристаллы промывают по¬
следовательно водой, спиртом и эфиром. Прибавив к взвеси
Na3[Co(C03)3 ]-ЗН20 избыток дигидрохлорида этилендиамина
NH2CH2CH2NH2-2НС1 и нагрев на водяной бане, получают рас¬
твор, из которого спиртом осаждается [Со(еп)3 ]С13.Если замещению подвергается бидентатный лиганд, то в коор¬
динационной сфере освобождаются цис-положепия и при после¬
дующем присоединении монодентатных лигандов образуются
цис- изомеры. Особую ценность для синтеза представляют собой
комплексы, в которых в качестве бидентатного лиганда высту¬
пает карбонат-ион. Так, ц«с-диаквотетрамминкобальтисульфат
[Co(NH3)4(H20)2 ]2 (S04)3-3H20 получают акватацией карбонато-
тетрамминкобальтисульфата [Co(NH3)4C03]2S04-ЗН20: 5 г кар¬
бонатного комплекса растворяют в 100 мл холодной воды, в кото-
руйУ*-предварительно добавляют 10 мл разбавленной H2S04. После
выделения С02 раствор фильтруют и, добавив 50—60 мл спирта,
осаждают из фильтрата карминово-красные кристаллы продукта.
Промывают эти кристаллы на фильтре 5%-ным спиртом и высуши¬
вают на воздухе.В целенаправленном синтезе комплексных соединений платины
и некоторых других металлов может быть использована закономер¬
ность mpawc-влияния. Так, из ряда активности лигандов (см.
стр. 192) следует, что дихлоротетрамминплате-ионы можно получить
из трихлоротриамминплате-ионоввытесняя хлорид-ион аммиаком. Поскольку транс-влияние хло¬
рида больше транс-вшяння аммиака (стр. 149), то в комплексе II
все ионы хлорида одинаково нелабильны и замещение их аммиа¬
ком идет трудно. В комплексе I легко замещается один из транс¬С1■ С1II- 193
расположенных ионов хлорида, и в результате получается практи¬
чески чистый цис-изомер [Pt(NH3)4Cl2]2+. Синтез [Pt(NH3)4Cl2]2+
был впервые осуществлен И. И. Черняевым именно через комплекс I,
который предварительно был получен окислением [Pt(NH3)3Cl ] +
хлором.Синтез комплекса существен но зависит от наличия подходящего
механизма реакции. Для смеси реагирующих компонентов возможны
как правило, несколько направлений реакции. Могут образовы¬
ваться разные конечные продукты. Протекает та из реакций, энер¬
гия активации которой минимальна по сравнению с остальными
реакциями и достаточно мала для того, чтобы реакция могла про¬
текать при данной температуре. Если энергии активации всех воз¬
можных реакций слишком высоки, ни одна из них не будет осу¬
ществляться, а если энергии активации нескольких реакций
сопоставимы, то они будут протекать приблизительно в равной
степени. ,Для увеличения скорости синтеза применяют катализаторы и
повышенные температуры. Однако эти факторы могут полностью
изменить результаты синтеза, так как могут измениться направле¬
ние реакции, термодинамическая или кинетическая устойчивость-
продукта реакции при повышенной температуре и, наконец, уси¬
литься побочные процессы, например, гидролиз. Так, окисление
растворов Сг2+ в смеси NH4OH + NH4C1 при 0° дает [Cr(NH3)e]3+,
при 20° в продуктах реакции преобладают [Cr(NH 3) 5ОН ]2+ и
[Cr(NH3)4(OH)2]+, при нагревании же выпадает [Cr(NH3)6Cl ]Cf2.Типичным примером реакции замещения во внутренней сфере
инертного комплекса может служить акватация: замещение ли¬
ганда молекулой воды. Эта реакция протекает уже при растворе¬
нии многих комплексов в воде: ионы Н+ и ОН” ускоряют ее.Так, при
синтезе [Co(NH3)5H20 ]2 (QOJj^F^O 10 г тонкоизмельченно-
го [Co(NH 3) 5С1 ]С12 помещают в коническую колбу и раство¬
ряют при перемешивании в 75 мл воды, добавляя при этом 50 мл
10%-ного водного раствора аммиака. Хлоропентаммин в слабоще¬
лочных растворах превращается в аквопентаммин[Со (NH3)6JCI]2+ + Н20 [Со (NH,)5 Н2Ор+ + CI"Раствор отфильтровывают от окислов кобальта и нейтрализуют
щавелевой кислотой. Выпадает осадок [Co(NH3)5H20]2)C204)3-4H20.
Данная реакция протекает при комнатной температуре. В качестве
осаждающего противоиона использован оксалат.Аналогично в [Co(NH3)s С1 ]С12 идет замещение С1“ на S042'.
Для этого [Co(NH3)6 Cl ]С12 обрабатывают концентрированной H2S04,
после выделения НС1 смесь 4 часа нагревают на водяной бане, раз¬
бавляют двойным объемом воды, фильтруют и фильтрат оставляют
кристаллизоваться. Выделяется [Co(NH3)5S04 ]HS04 -2Н20. При
таких довольно жестких условиях синтеза замещению во внутрен¬
ней сфере подвергся только хлорид-ион.194 —
Из рассмотренных примеров можно составить ряд вытеснения
лигандов из внутренней сферы комплексов Со3+: NH3 > Н20 >
>501“ >£1-.§ 5. Синтез комплексных соединений при помощи
хроматографии и электрофорезаДля выделения комплексного иона из смеси инертных комплексов
пригодны методы, применяемые для лабильных комплексов. Одна¬
ко требования к избирательности выделяющего агента, противо-
иона-осадителя, растворителя и т. п. здесь гораздо выше. При обра¬
ботке избытком осадителя смеси лабильных комплексов получает¬
ся осадок, образованный только одним из них. Если же инертные
комплексные ионы близки по свойствам, то при осаждении избыт¬
ком осадителя выпадает смесь продуктов.В смеси инертных комплексов миграция их под действием диф¬
фузионных электрофоретических или каких-либо других сил про¬
исходит для каждого сорта ионов с характерной скоростью. На этом
основано применение хроматографии при синтезе комплексных со¬
единений. Хроматография и электрофорез могут быть использова¬
ны при синтезе, очистке и разделении координационных соединений.
Например, очистка [Cr(NH3)„]3+ от [Cr(NH3)6 (Н20) ]3+ и
[Cr(NH3)4 (Н20)2]3+ на ионообменной колонке с А1203; использо¬
вание для синтеза хлороаквокомплексов Rh3+, Ir3+ и Pt4+ и т. д.
высоковольтного электрофореза на бумаге.При окислении окисью азота азотнокислого раствора Сг(СЮ4)а
получается смесь катионов [Cr(H20)6N0]2+, [Cr(H20)e]3+ и
1(Н20)5Сг0Сг(Н20)5]4+. Эту смесь сорбируют катионом в Н^фор¬
ме, затем элюируют раствором НС104. Элюирование сильно за¬
висит от заряда иона: 0,2 М НС104 вымывает [Cr(H20)5N0]2+,
0,5 М НС104 — [Сг(Н20)в]8+, а 2,0 М НСЮ4—димерный четырех¬
зарядный катион.При электрофорезе на бумаге в 1 М растворе гликолевой кисло¬
ты наблюдается следующий порядок скорости движения ионов ме¬
таллов к катоду:La > Се > ... > Yb > Lu > Sc ^ Th > Zrа в 3M растворе гликолята натрия частицы, содержащие ионы
металла, передвигались к аноду. При электромиграции комплексов
Со(Ш) в присутствии NH4C1 за счет различной подвижности за 6 ч
катионы [Co(NH 3) в ]3+продвинулись к катоду на 12, 95 см, катионы
[Co(NH3)5C1 ]2+—на 9,30 см, а катионы транс-(Co(NH3)4(N02)2]+—
на 4,55 см.У большинства цыс-изомеров подвижность в условиях хрома¬
тографии ниже, чем у транс-изомеров. У незаряженных или одно¬
зарядных частиц различие в скорости достаточно велико, что обес¬
печивает надежное разделение. У высокозарядных изомерных ио¬
нов подвижности близки и разделение их затруднено. Для разде¬— 195 -
ления могут быть использованы любые виды хроматографии. Чаще
всего применяют колонки с А1203 или ионообменными смолами.
Так, при помощи хроматографии были разделены цис- и транс-
изомеры [Pt(NH3)2Br2] и [Pt(NH3)2I2]. Для разделения опти¬
ческих изомеров применяют наполнитель из оптически активного
вещества. Применение неводных растворов позволяет использовать
D-винную кислоту! D-лактозу и др., для водных растворов упот¬
ребляют D- или L-кварц или ионообменную целлюлозу.§ 6. Синтез комплексных соединений в отсутствие
растворителяВ синтезах при высоких температурах обычно участвуют газо¬
вые фазы, твердые вещества и их расплавы. Направление этих син¬
тезов зависит от возможности удаления какого-либо из продуктов
реакции в виде газовой фазы и от энергий кристаллических реше¬
ток конечных веществ. Высокая температура способствует уста¬
новлению равновесия в системе, поэтому значение механизма умень¬
шается.Высокотемпературная изомеризация. Нагревание может вы¬
звать 1) переход комплекса из метастабильной модификации в ста¬
бильную; 2) переход комплекса в высокотемпературную модифи¬
кацию, если такая имеется. При
подходящем режиме охлаждения
можно «заморозить» эту модифика¬
цию и получить ее в низкотемпера¬
турной области.Высокотемпературные реакции
соединения. Типичным примером та¬
ких реакций может служить обра¬
зование NaCoF3 при нагревании
смеси безводных NaF и CoF2. В не¬
которых случаях такие реакции
идут только после плавления одно¬
го из компонентов, но возможно и
протекание их между твердыми фа¬
зами. Комплексные соединения мо¬
гут быть получены при участии
легколетучих компонентов. Так, для
получения оранжевого комплекса
[C5H5NH ]3 [ТiCl6 ] смешивают без¬
водный TiCl3 с большим избытком
хлористого пиридина и нагревают
смесь в ампулах при 150е. Непро¬
реагировавший [CjH5NH ]С1 экстра¬
гируют хлороформом.Образование нового комплекс¬
ного соединения из исходных про¬ТемператураРис. 76. Термогравиметрическая
кривая разложения вещества:/ — область устойчивости начального ве¬
щества; 2 — область перехода начального
вещества в промежуточное; 3 — область
устойчивости промежуточного вещества;
4 — область перехода промежуточного ве¬
щества в конечное; 5 — область устойчи¬
вости конечного вещества— 196 —
дуктов при проведении реакции в твердой фазе подтверждается
термографическими и рентгеноструктурными исследованиями. Воз¬
никновение нового соединения может быть выявлено также по¬
строением диаграмм состав—свойство, в частности диаграмм плав¬
кости.Реакции разложения комплексных соединений. Эти реакции про¬
ходят особенно легко при удалении летучего лиганда или про¬
дуктов его разложения. Если лиганд удаляется ступенчато, то в
некотором интервале температур устойчиво промежуточное ком¬
плексное соединение. Этот температурный интервал можно уста¬
новить, исследуя зависимость веса вещества от температуры: ему
соответствует горизонтальный участок термогравиметрической кри¬
вой (рис. 76). Примерами синтезируемых таким образом комплек¬
сных соединений могут служить светло-желтый NiCl2-py, который
получается при нагревании синего NiCl2-4py в течение часа при
115°; соединения [Co(NH3)sCl ]С12 и транс- [Pt(NH3)2Cl2], полу¬
чающиеся при разложении [Co(Nby5H20]Cl3 (100 — 125°С) и
[Pt(NH3)4]Cl2 (195—200°С) соответственно и др.Известны координационные соединения, над которыми давление
паров вещества, играющего роль лиганда, значительно при комнат¬
ной температуре. Так, при хранении на воздухе выветриваются
некоторые аммиакаты, кристаллогидраты и др. Такие вещества
нужно хранить в атмосфере паров веществ, входящих в состав
комплекса в качестве лиганда.§ 7. Использование реакций координированных лигандовИспользование обычных методов органического синтеза в ус¬
ловиях, в которых лиганд не может отщепиться от центрального
иона металла, является мощным средством получения разнообраз¬
ных координационных соединений. Реакции такого типа объединяют
под названием «реакции комбинирования». Так, реакция полу¬
чения биферроценила из иодоферроцена (60—150°) аналогична реак¬
ции конденсации иодзамещенных углеводородов при помощи меди:О1 ’ОFe + 2Cu + Fe —*" Cu2I2 + Fe FeО О О ОЕсли свободный лиганд представляет собой совокупность изо¬
мерных форм или конформаций, то. при координации он закрепляет¬
ся в одной определенной форме, которая может быть и нехарактер¬— 197 -
ной для свободного лиганда*. Эффективные заряды на его атомах
изменяются под воздействием иона металла и остальных лигандов.Влияние координации на реакции лигандов может быть в пер¬
вом приближении представлено в виде двух независимых эффектов:
электронного, изменяющего относительную активность реакционных
центров, и геометрического, который проявляется в том, что а) часть
реакционных центров при координации может оказаться экрани¬
рованной от воздействия атакующих реагентов; б) некоторые функ¬
циональные группы лиганда могут оказаться сближенными, что бла¬
гоприятствует реакциям внутримолекулярной перегруппировки или
замыкания цикла; в) жесткая взаимная ориентация соседних ли¬
гандов позволяет проводить их конденсацию, так называемый
«матричный синтез». Так, квадратный комплекс никеля (II) с мерка-
птоэтиламином конденсируется с ацетилацетономсн3\с=оСНс=о/H,N'\/CHjCH^
JNk /S'SСН3\CHjCHjC=ISkIC=N-
/ Nr/СНзСН^;NiC + 2H,0
\s ■а продукт синтеза — с дибром ксилолом, давая соединение с лиган¬
дом, содержащим 14-членный цикл:СН3\ /СН;С.НаЧ
C=N\ ^-SC=N^CH3/ NCHjCH2/Электронный эффект наиболее ярко проявляется в комплексах
ацетилацетона и других |3-дикетонов, анионы которых при коор¬
динации стабилизируются в енольной форме. Благодаря поляри¬
зующему действию катиона на центральном атоме углерода ока¬
зывается повышенный заряд и атом водорода на нем легко заме¬
щается группами I, Вг, Cl, SCN, N02 и т. д.Реакции координированных лигандов чаще используются для
проведения органического синтеза, а не для получения новых ком¬
плексных соединений. Ион металла играет при этом роль катали¬
затора, а координационное соединение — роль интермедиата.* Так, молекула глицина входит в комплексы цис- и транс-
[Pt(NH2CH2СООН)2CI2], координируясь через азот, хотя в растворе она
существует в таутомерной форме бетаина +ЙНзСН2СОСГ, в которой азот
лишен донорных свойств.— 198 —
Так, 2,2'-дипиридил синтезируют, нагревая пиридин с безводным
FeCl3. Образование пиридинового комплекса железа является про¬
межуточной стадией процесса.Наиболее широко известно применение соединений металлов
в качестве катализаторов реакций полимеризации олефинов.ЛитератураБабко А. К. Физико-химический анализ комплексных соединений в
растворах. Изд-во АН УССР, Киев, 1955.Б а с о л о Ф., Джонсон Р. Химия координационных соединений.
«Мир», 1968.Б а с о л о Ф., Пирсон Р. Механизмы неорганических реакций.
«Мир», 1971.Берсукер И. Б., А б л о в А. Б. Химическая связь в комплексных
соединениях. Изд-во АН МССР. Кишинев, 1962.Гринберг А. А. Введение в химию комплексных соединений. «Хи¬
мия», М.—Л., 1966. .Гельман А. Д. и др. Комплексные соединения трансурановых эле¬
ментов. Изд-во АН СССР, М., 1961.Дей К., С е л б и н Д. Теоретическая неорганическая химия. «Химия»,
М.— Л., 1969.Джаффе Г., Орчин М. Симметрия в химии. «Мир», 1968.Желиговская Н. Н., Черняев И. И. Химия комплексных
соединений. «Высшая школа», 1966.Журнал Всесоюзного химического общества им. Д. И. Менделеева,
1972, 17, № 3.Карапетьянц М. X., Д р а к и н С. И. Строение вещества.
«Высшая школа», 1967.К е н д л и н Дж., Тейлор К., Томпсон Д. Реакции координа¬
ционных соединений переходных металлов. «Мир», М., 1970.Коттон Ф., Уилкинсон Дж. Современная неорганическая
химия, ч. 1,2, 3. «Мир», 1969.Ко у л сон Ч. Валентность. «Мир», 1965.Ключников Н. F. Руководство по неорганическому синтезу. Гос-
химиздат, М. —Л., 1964.Кребс Г. Основы кристаллохимии неорганических соединений. «Мир»,
1971.Лэнгфорд К., Грей Г. Процессы замещения лигандов, «Мир»,
1966.Маррел Дж, Кеттл С, Теддер Дж. Теории валентности.
«Мир», 1968.Некрасов Б. В. Основы общей химии, т. I, 2, 3. «Химия», М., 1965—
1970.Новаковский М.С. Лабораторные работы по химии комплексных
соединений. Изд-во Харьковского гос. ун-та, 1964.О р г е л Л. Введению в химию переходных металлов. «Мир», 1964.Орчин М., Джаффе Г. Разрыхляющие орбитали. «Мир», 1969.Паулинг Л. Природа химической связи. ИЛ., М. — Л., 1947.Порай -Кошиц М. А., Гали некая Э. А. Кристаллохимия,
ВИНИТИ, М., 1966.Р о с с о т и Ф., Россоти X. Определение констант устойчивости и
других констант равновесия в растворах. «Мир», 1965.— 199 —
Сб. статей «Колебательные спектры в неорганической химии» под ред.
Ю. Я. Харитонова. «Наука», М. — Л., 1971.Современная химия координационных соединений» под ред. Дж. Льюи¬
са и Р. Уилкинса. ИЛ, 1963.«Синтезы неорганических соединений», под ред. У. Джолли, т. I, «Мир»,
1966.«Синтез комплексных соединений металлов платиновой группы», под ред.
И. И. Черняева, «Наука», М. — Л., 1964.«Спектроскопические методы в химии комплексных соединений» под ред.
М. В. Вдовенко, «Химия», М. — Л., 1964.Фишер Э., Вернер Г. я-Комплексы металлов. «Мир», 1968.
«Химия координационных соединений» под ред. Дж. Бейлара и Д. Буша.
ИЛ, 1960.Ш л е ф е р Г. Л. КомплексооСразование в растворах. «Химия», М.—Л.,
1964.Я неон Э. Ю. Комплексные соединения. «Высшая школа», 1968.
Яцимирский К- Б. Термохимия комплексных соединений. Изд-во
АН СССР, М. — Л., 1951.Яцимирский К. Б., Васильев В. П. Константы нестой¬
кости комплексных соединений. Из£-во АН СССР, М., 1959.Яцимирский К. Б. и др. Химия комплексных соединений редко¬
земельных элементов. «Наукова думка», Киев, 1966.
ПРИЛОЖЕНИЕЯх«=;юиое*.Xcdс.S3ххалО,*ОCJООUООиX2С5XиСЧх0CJ12XVXи/2\хияхи/2\XоIиооXиIиоо§хX л
Чи
X I
и л
хЧ-
Ч ■-
X хZ 2X с<-О о
z —0Z/\ XXX
х х z и—ич- I оCJ « 'w*\/-Zg и-о^1Iо.!>К<Н<ПЦц
£ £
^ нXXS«аяttкО)р;Sн<Т)CUКсЯЕ<яСПXX3яXXS=(соXиSt=tXЧЯXфXh-чнCDЯеаесиXо*сисиSНса•9-оа.г=СSН Н— 201
СокращенноеНазвание наименованне Формула
лигандовп\оXSО)XпсиЕ/\\/ \ /а < >z \ /\/ооиX Iч «Xz-uхииООо о0 оо о1 IМ С*X Xо о\/2■мХ !е» 1хиIeqа:иС9IUZ/\М ЯК X
о иооX-ииооио010 о
о о
и о1 IО! МX I
о о
\/
ZоINXиIо
о
иegх
z—иа II о
и от?1 и
и I
Z —ОС4XО01XиZ/\I
иXUIиоо8 §и\ Iя о*Ж I
о и
\/z„х о
о оКГ? ОО XZ—ОXиNXоZ-ооопX-и1 о
Ч. О
гг U« ми хz-0NXиD)XиZ/\N 91X Xо оI Iи оо оо о202
Продолжение табл.£а/-ч—/I-О/\+ к\Z —о//~'\\/X<3\XвоV|—ч/и °\•о «вX X«О вU ио>1О*ОСсаЕГенsсЛ. о
К
*
О33К4
о
£
Я
XЛо5
£
Ое;Wяа-пкхо>*0"<Яа.— 203 —
Таблица 2Гибридизация атомных орбиталей иона металлак. Ч.Геометрия комплексаВозможнаягибридизацияАОиена металла,
образующихо-СВЯЗИВозможная гнбри
металла, образсильныедизация АО иона
ующих «-связислабыеЛинейнаяspp2d2 »dpp2d2 _—2УгловаяР2d или pdd или sd»dsd или pdр или pd»d2d или pdр или spdСимм. треугольникsp2pd2d2То жеdp2pd2d2»d2spd2Р°-3»d3pd2р2Асимм. треугольникdsppd2pd или dТригональная пирамидаP3—sd или d4То жеd2p sd или p2d2Т етраэдрsp3d2d-1»d3sd2p34Квадратdsp2dap»d2p2d3p—d2sp—ddp3—sd?p—sТетрагональная пирамидаd*dsp или sТо жеdsp3d2d2»d3spd2p2»d2sp2dpd25d4sdp3d2p3dsd2»dipdsp2Плоский пятиугольникd3p2pd2—Пентагональная пирамидаd5sp или p2Октаэдрd2sp3d3_Тригональная призмаdlsp—p2dТо жеdsp—p2s6Тригональная антипризмаd3p3—sdСмешаннаяd3sp2——»d6s——»dip2— 204 —
Продолжение табл. 2ВозможнаягибридизацияВозможная гибридизация АО иона
металла, образующих г.-связик. ч.Геометрия комплексаметалла.образующиеа-связисильныеслабыеZrFf~d3sp3—л2dbsp—р°-7TaF^-d*sp2—dpdip3—dsdtp*—ps8ДодекаэдрАнтипризмаГранецентрированная призмаdisp3
dbp3
dbsp2dРsТаблица 3Характеры неприводимых представлений точечных группЕс22 С,2®0 ^Группас*гл2Вгв2Е\ х, у111121111—211— 1— 1
01—11—101— 1
— 1
-1
0Е2 СчС22 С22 С2i2S4аЛ2'02‘dГруппа®lhR2gRgE2gAgАщBiaBlaE„:—2—1—1—1•к. У—2—2— 1
1— 1
0
— 1
1— 1
1
0— 1
— 1
1
0
—1
1
1—10— 205 —
Е23,2 С,2Sgс.4С24<vAy11111II11111—1—1Г руппаBi1—11—111—1Didв2; z1—11—11— 11Ей х, у2У~т-0-V2—200Еъ20—20200Е32-ГГ0V~2—200 'Е8С„6 сг6*а6 S,Ау11111ГруппаА 2111—1—1ТаЕ2— 1200Т2; *, «, 230—11—IТу30—1—11
СОДЕРЖАНИЕОт авторов . 3I. Введение 4§ 1. Основные понятия химии координационных соединений (4). § 2. Номенклатура
координационных соединений (8). § 3. Полные и сокращенные формулы координа¬
ционных соединений (И). § 4. Комплексные частицы в растворе (15).II. Общие сведения о химической связи б комплексных соединениях . . 17§ 1. Химическая связь с электростатической точки зрения (17). § 2. Теория кристал¬
лического поля (18). § 3. Схема молекулярных орбиталей (МО) (20). § 4. Метод
валентных связей (28). § 5. Гибридизация орбиталей. Пространственная структура
комплексного иона (30). § 6. Основные типы конфигураций внутренней координа¬
ционной сферы (35).III. Реакции комплексных частиц 45§ 1. Основные типы реакций комплексных частиц (45). § 2. Лабильные и инертные
комплексные ионы (48). § 3. Возможность существования изомеров (58).IV. Основные типы комплексных соединений .61§ 1. Одноядерные комплексы с однодентатными лигандами (61). § 2. Комплексные
соединения с полидентатными лигандами (66). § 3. Многоядерные комплексные
соединения (74). § 4. Комплексы с я-связями в качестве донора (85). § 5. Карбони¬
лы и цианиды переходных металлов (92).V. Виды изомерии комплексных ионов 98§ 1. Структурная изомерия (98). § 2. Изомерия связи (99). § 3. Геометрическая
и оптическая изомерия (101). § 4. Конформацнонная изомерия (106).VI. Химическая связь в комплексных соединениях переходных металлов 108§ 1. Применение теории групп к молекулярным структурам (108). § 2. Теория кри¬
сталлического поля (119). § 3. Магнитные свойства комплексов (126). § 4. Примене¬
ние метода МО к координационным соединениям (132).VII. Кинетика и механизм реакций комплексных частиц 138§ 1. Основные понятия. Теоретические модели (138). § 2. Механизмы реакций заме¬
щения лигандов (140). § 3. гране-Влияние в квадратных и октаэдрических комплек¬
сах (149).VIII. Устойчивость комплексных соединений в растворах 153§ 1. Определение констант устойчивости комплексных соединений в растворах (153).§ 2. Понятие об устойчивости комплексного соединения (168). § 3. Факторы, влияю¬
щие на устойчивость комплексных частиц (169). § 4. Зависимость устойчивости ком¬
плексов состава 1 : 1 от характеристик иона металла (173).IX. Синтез комплексных соединений 183§ 1. Основные принципы синтеза (183). § 2. Выделение комплексного иона из сис¬
темы лабильных комплексов (184). § 3. Окисление и восстановление доминирую¬
щего комплекса в системе комплексных ионов (188). § 4. Использование реакций
замещения лиганда во внутренней сфере инертного комплексного иона (192). § 5. Син¬
тез комплексных соединений при помощи хроматографии и электрофореза (195).§ 6. Синтез комплексных соединений в отсутствие растворителя (196). § 7. Исполь¬
зование реакций координированных лигандов (197).Литература 199Приложение 201— 207 -
Нина Алексеевна Скорик, Виктор Нафтулович КумокХимия координационных соединенийРедактор Т. П. Федорова, художественный редактор Т. М. Скворцова.
Технический редактор Н. Н. Баранова, художник Ю. Д. Федичкин.
Корректор С. К. МарченкоСдано в набор 9/Х—74 г. Подп. к печати 7/IV—75 г. Формат 60x90Vle,
Объем 13 печ. л. Бум. тип. Ка 3. (Уел. п. л. 13). Уч.-изд. л. 11,72
Изд. № ХИМ—486. Тираж 11 ООО экз. Зак. 699. Цена 42 коп.План выпуска литературы изд-ва
«Высшая школа» (вузы и техникумы) на 1975 г. Позиция № 109
Москва, К-51, Неглинная ул., д. 29/14,Издательство «Высшая школа»Ярославский полиграфкомбинат Союзполиграфпрома при Государственном
комитете Совета Министров СССР по делам издательств, полиграфии и
книжной торговли. 150014, Ярославль, ул. Свободы, 97.