















































































































































































































Author: Бутырская Е.В.
Tags: информационные технологии вычислительная техника обработка данных микропроцессоры химия программное обеспечение монография квантовая химия
ISBN: 978-5-91359-095-4
Year: 2011




Бутырс
он
Впервые в отечественой Н3С
литературе:
Работа в Gaussian и GaussView
-- примерами
Теооия и практика моделирования
.химических структур
• Насчет свойств атомно-молекулярных
гостем методами квантовой химии
Оценки «Отлично»!
Оцифровал ko'Chegara
kochegara@gmail.com
Серия «Библиотека студентов»
Бутырская Е. В.
Компьютерная химия: основы теории
и работа с программами Gaussian
и GaussView
Москва
СОЛОН-ПРЕСС
2011
УДК 004
ББК 32.97
Б 93
Е. В. Бутырская
Компьютерная химия: основы теории и работа с программами Gaussian и
GaussView. — М.: СОЛОН-ПРЕСС, 2011. — 224 с: ил. — (Серия «Библиотека
студентов»).
ISBN 978-5-91359-095-4
Монография является первым в отечественной литературе руководством по
работе с программными комплексами Gaussian и Gaussview. Рассмотрены
теоретические основы методов квантовой химии. Кратко описаны неэмпирические и
полуэмпирические методы решения электронного уравнения Шредингера, системы
базисных функций, методы расчета термодинамических свойств системы, модели
сольватации, теория ядерного магнитного резонанса, методы молекулярной
механики и молекулярной динамики. Приведено большое число примеров расчета
структуры и свойств молекул и анализа полученных результатов с использованием
указанных программ.
Предназначена для студентов, аспирантов и научных работников химических,
физических и биологических специальностей вузов.
КНИГА - ПОЧТОЙ
Книги издательс[ва «СОЛОН-ПРЕСС» можно заказать наложенным платежом (оплата при
получении) по фиксированной цене. Заказ оформляется одним из трех способов:
1. Послать открытку или письмо по адресу: 123001, Москва, а/я 82.
2. Оформить заказ можно на сайте www.solon-press.ru в разделе «Книга — почтой».
3. Заказать по гел. (499) 254-44-10, 252-36-96.
Бесплатно высылается кагалог издательства по почте. Для этого присылайте конверте маркой по
адресу, указанному в п. I.
При оформлении заказа следует правильно и полностью указать адрес, но которому должны быть
высланы книги, а также фамилию, имя и отчество получателя.
Желательно указать дополнительно свой телефон и адрес электронной почты.
Через Интернет 13ы можете в любое время получить свежий каталог издательства
«СОЛОН-ПРЕСС», считав его с адреса www.solon-press.ruAat.(loc.
Интернет-магазин размещен на сайте www.soIon-press.ru.
Сайт издательства «СОЛОН-ПРЕСС»:
Тел.: (499)254-44-10, (499)795-7326
www.solon-press.ru e-mail: avtor@coba.ru
ISBN 978-5-91359-095-4
© Е. В. Бутырская, 2011
© Обложка «СОЛОН-ПРЕСС», 2011
Предисловие
Прогресс в развитии вычислительной техники и программного
обеспечения сделал методы квантовой химии одним из наиболее важных инструментов
химических и физико-химических исследований. Компьютерное
моделирование структуры и свойств веществ не только дополняет экспериментальные
методы исследования, но и во многих случаях дает принципиально новую
информацию. Так с помощью компьютерного эксперимента стало возможным
исследовать переходные состояния и механизмы химических реакций, интермедиа-
ты, несуществующие вещества и др., что невозможно на основе имеющихся
экспериментальных методов.
Одним из самых распространенных комплексов для квантово-химических
расчетов в настоящее является пакет программ GAUSSIAN. Данный комплекс
программ позволяет рассчитывать структуры молекул и их энергии,
переходные состояния, электронные, колебательные и рамановские спектры,
поверхности потенциальной энергии, разнообразные свойства молекул в газовой фазе и
растворе в основном и в возбужденных состояниях и др.
Широкое использование пакета программ GAUSSIAN в практике
химических исследований тормозится практически полным отсутствием литературы,
обучающей практической работе с данным комплексом. Предлагаемая
монография частично восполняет этот пробел. Монография состоит из трех
разделов. Первый раздел посвящен краткому описанию методов квантовой химии.
Во втором разделе изложены основы практической работы с программой
GAUSSIAN под Windows. В третьем разделе излагаются основы практической
работы с программой GAUSVIEW - графическим приложением к комплексу,
облегчающим формирование стартовой структуры и входного файла
программы GAUSSIAN и позволяющим осуществить просмотр промежуточных и
конечных результатов.
Монография не претендует на полноту изложения методов квантовой
химии и всех возможностей программы GAUSSIAN, а представляет собой
начальный курс, ознакомление с которым позволит пользователю сделать первые
шаги практического использования программы в химических, физических,
физико-химических и биологических исследованиях.
Монография издана при поддержке ФЦП "Научные и научно-
педагогические кадры инновационной России" на 2009 - 2013 годы, ГК№ П846
от 25.05.2010
э
ГЛАВА I. Теоретические основы вычислительных методов
квантовой химии
1.1 Разделение электронного и ядерного движений в молекулах
Движение в молекулах является более сложным, чем движение в атомах.
В молекулах имеются 3 вида движения: электроны движутся вокруг ядер, ядра
колеблются около положения равновесия, молекула как целое может вращаться
вокруг некоторой оси. На первом этапе рассматривается движение молекулы в
системе координат, жестко связанной с молекулой, т.е. вращающейся вместе с
молекулой, начало этой системы координат находится в центре масс молекулы,
эта система координат называется молекулярной системой координат. В этой
системе координат отсутствует вращательное движение и движение центра
масс молекулы, имеются только электронное и колебательное движения.
Положение молекулярной системы координат относительно внешней неподвижной
системы координат задается шестью координатами, три из которых
характеризуют положение центра масс молекулы и еще три угла Эйлера характеризуют
вращение молекулы как целого относительно лабораторной системы. Поэтому
конфигурация нелинейной многоатомной молекулы, состоящей из М ядер, в
молекулярной системе координат определяется К = ЗМ-б независимыми
координатами, линейные и двухатомные молекулы имеют две вращательные
степени свободы, для них К = ЗМ-5 . Стационарное уравнение Шредингера,
определяющее состояние молекулы в молекулярной системе, имеет вид:
Н WfF.RJ^EWfr.R), (1.1.1)
г ={?], ?2,-- -гд.'}- координаты электронов, N- число электронов,
R-{R/ ^2>'"^К}~ координаты, определяющие конфигурацию ядер в
молекулярной системе, К - ЗМ - 6 для нелинейной многоатомной молекулы,
К=ЗМ-5 для двухатомных и линейных многоатомных молекул, М-число
ядер.
Оператор Гамильтона молекулы имеет вид
H = fR+fr + V(r,R)=fR+He, (1.1.1)
где Tr =— оператор кинетической энергии ядер,
Тг - оператор кинетической энергии электронов,
V(f,R) - оператор потенциальной энергии взаимодействия между всеми
частицами.
Оператор
He = H-TR=fr+V{r,R), (1.1.3)
содержащий дифференцирование только по электронным координатам,
называется электронным гамильтонианом.
He=ir+V{r,R)=ir+Vee+Vea + Vap> О-1-4)
Vee - описывает кулоновское взаимодействие электронов
Уеа~ описывает кулоновское взаимодействие ядер и электронов
4
Vap - описывает кулоновское взаимодействие ядер
Несмотря на то, что гамильтониан называется электронным, в него
включены все нерелятивистские виды взаимодействий, которые имеют место в
молекуле, это гамильтониан молекулы с фиксированной конфигурацией ядер.
Приближение Борна-Оппенгеймера решения уравнения (1. 1.1) основывается на
предположении, что оператором кинетической энергии ядер на первом этапе
можно пренебречь. На первом этапе решается уравнение Шредингера с
электронным Гамильтонианом
Не <pen(r,R)=en(R)<pen(r,R).
(1.1.5)
Функции (pen(r,R) называются электронными волновыми функциями, sn\R)
- электронной энергией, она зависит от координат ядер как от параметров, п -
электронное квантовое число, нумерующее электронные уровни энергии. Хотя
sn{R) называется электронной энергией, в нее входят все виды энергии
молекулы кроме кинетической энергии ядер. В случае двухатомной молекулы число
ядер М=2, электронная энергия зависит от одной переменной R — межъядерного
расстояния. Зависимость электронной энергии двухатомной молекулы от
межъядерного расстояния для устойчивого состояния представлена на
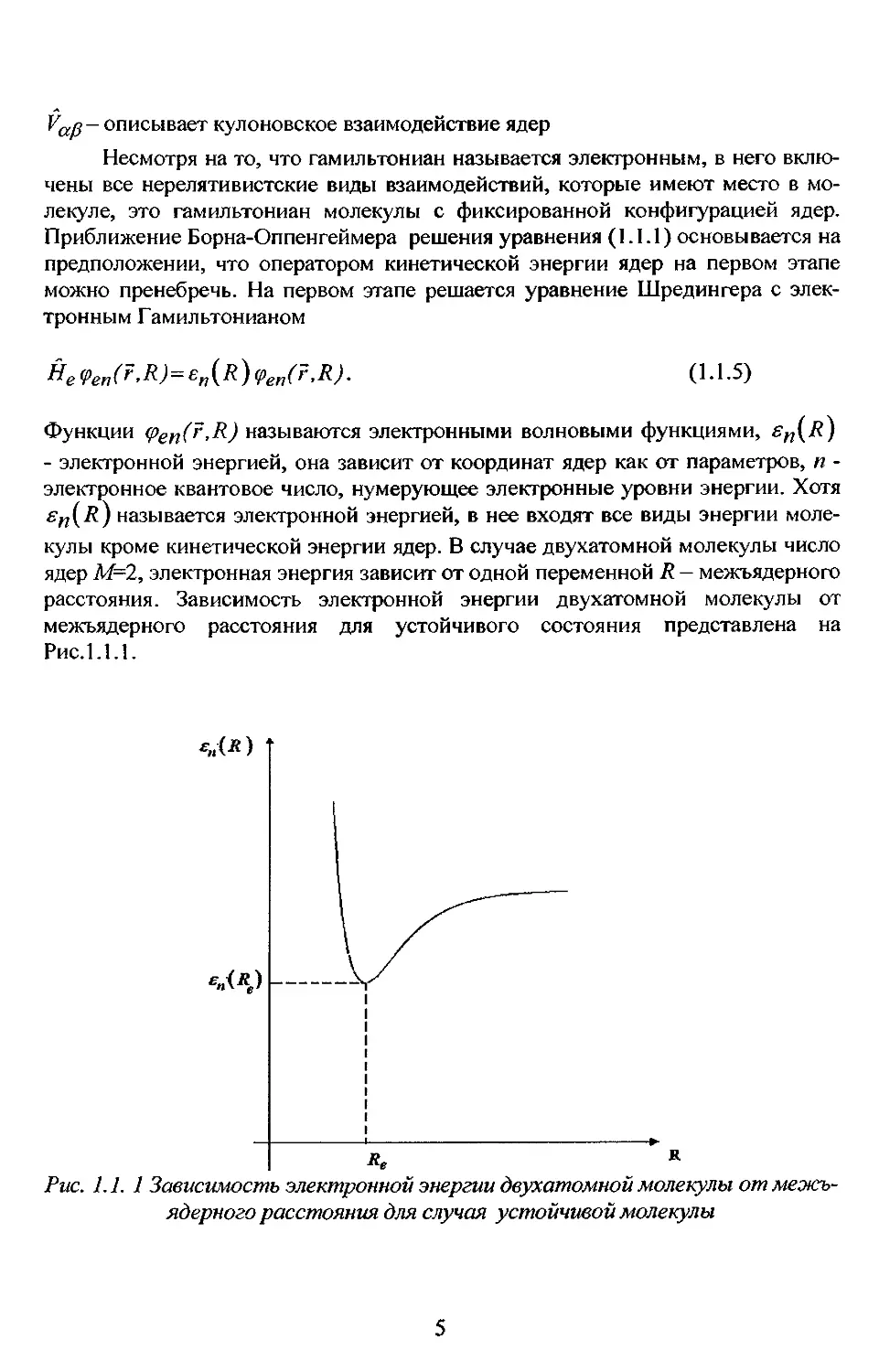
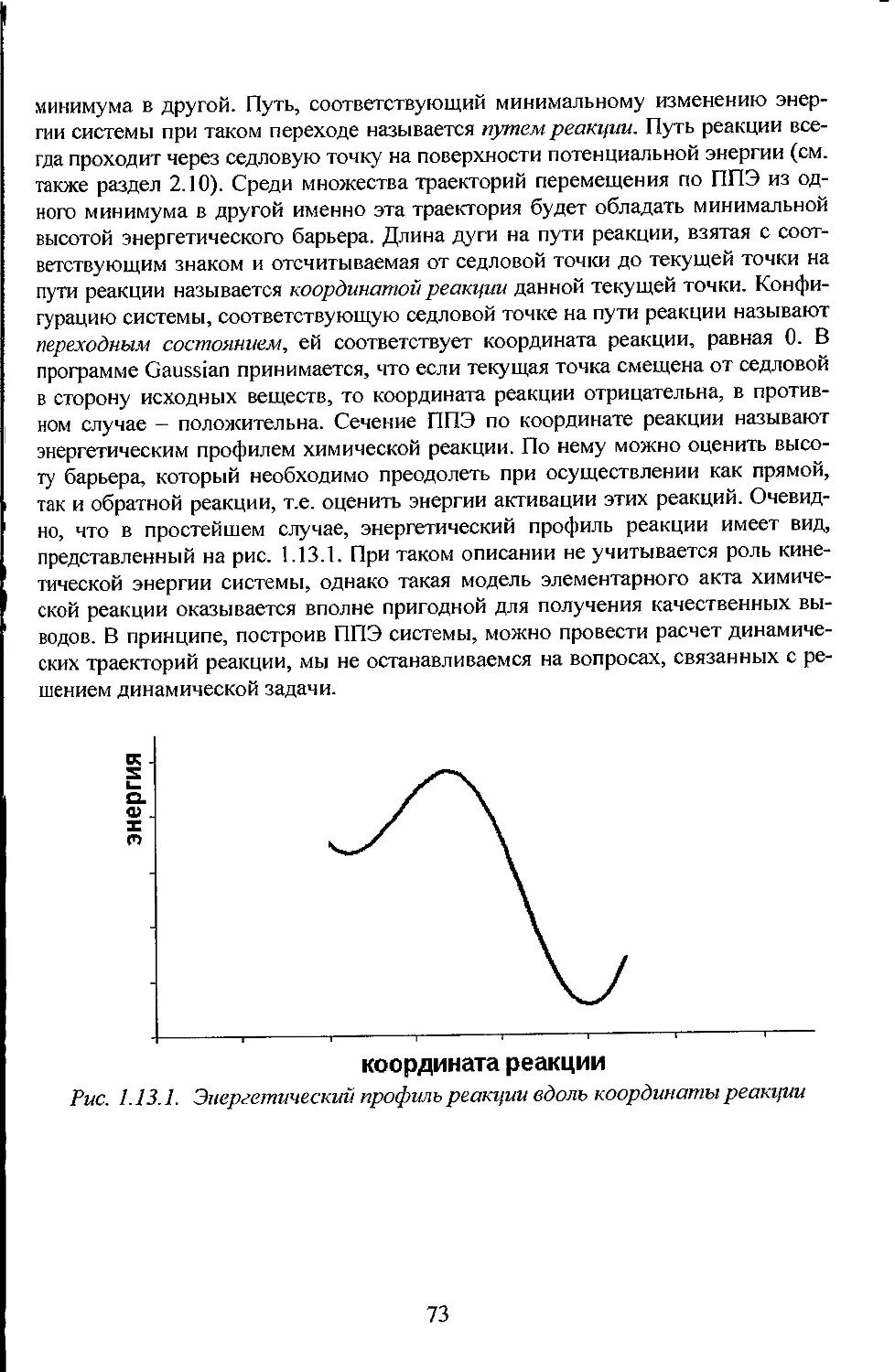
Рис.1.1.1.
*„(*)
*<*) -
Рис. 1.1. 1 Зависимость электронной энергии двухатомной молекулы от
межъядерного расстояния для случая устойчивой молекулы
5
Проанализируем зависимость sn(R) , представленную на Рис. 1.1.1. При
стремлении R к бесконечности молекула превращается в два атома, поэтому
при больших R значение sn(R)= const, равно сумме энергий атомов, на
которые распадается молекула. При стремлении R к нулю значение
еп\R )-> со вследствие роста энергии отталкивания ядер. Устойчивая молекула
будет существовать лишь в том случае, если имеется межъядерное расстояние
R-Re, при котором электронная энергия имеет минимум, это расстояние
является равновесным межъядерным расстоянием. Зависимость электронной
энергии от расстояния между ядрами различна для каждого электронного
состояния, т.е. вид функций Sfj(R) различен для разных п. Значение sn(Re)
называется чисто электронной энергией. Это название обусловлено тем, что при R=Re
колебания отсутствуют. Уровень энергии E3jin=sn(Re) называется чисто
электронным. Электронную энергию £и(/?)можно представить в виде суммы:
s„{R)=e„{Re)+U„rR) = E3„„+U„(R). (1.1.6)
Un (R) представляет собой разность
U„(R)=sn{R)-sn{Re) = sn{R)-E3Jin,
Что останется, если из энергии молекулы sn{R), включающей в себя все
виды энергии молекулы кроме кинетической энергии ядер, вычесть чисто
электронную энергию? Останется то, что относится к ядрам, а поскольку в
sn{R) не включается только кинетическая энергия ядер, то остаток,
относящийся к ядерной подсистеме, представляет собой энергию взаимодействия
ядер с потенциальным полем, в котором они движутся, это поле создается
всеми частицами системы. Поэтому Un(R) является потенциальной энергией для
подсистемы ядер. Для неустойчивых состояний зависимость sn(R) имеет вид
кривой без минимума, убывающей с ростом R. В таких состояниях с
увеличением межъядерного расстояния электронная энергия уменьшается, вследствие
чего происходит самопроизвольный распад молекулы.
Набор электронных волновых функций (pen(r,R) образует полную
систему функций. Молекулярную волновую функцию (1.1.1) можно разложить по
этому базису, коэффициенты разложения в общем случае, зависят от R:
у(г,л)=1фи(л)%иГг,л;.
п
Подстановкой этой функции в уравнение (1.1.1) можно показать, что если
переходами между электронными состояниями под влиянием оператора Где можно
пренебречь, то функции Фп (R) = <Pm (r) должны удовлетворять уравнению
6
(fR + U„{R)) Фт{к) = етФт(я),
(1.1.7)
которое с учетом смысла функции JJn(R) представляет собой ничто иное, как
уравнение Шредингера для колебаний ядер, при этом энергия V'п (R)
взаимодействия ядер с потенциальным полем, в котором они движутся, получается из
решения электронной задачи, v - колебательное квантовое число, нумерующее
колебательные уровни энергии в электронном состоянии п. Для двухатомных
молекул Un(R) называется потенциальной кривой, для многоатомных -
поверхностью потенциальной энергии.
Как следует из вида (1.1.7) Фпу (R) есть ничто иное, как колебательная
волновая функция.
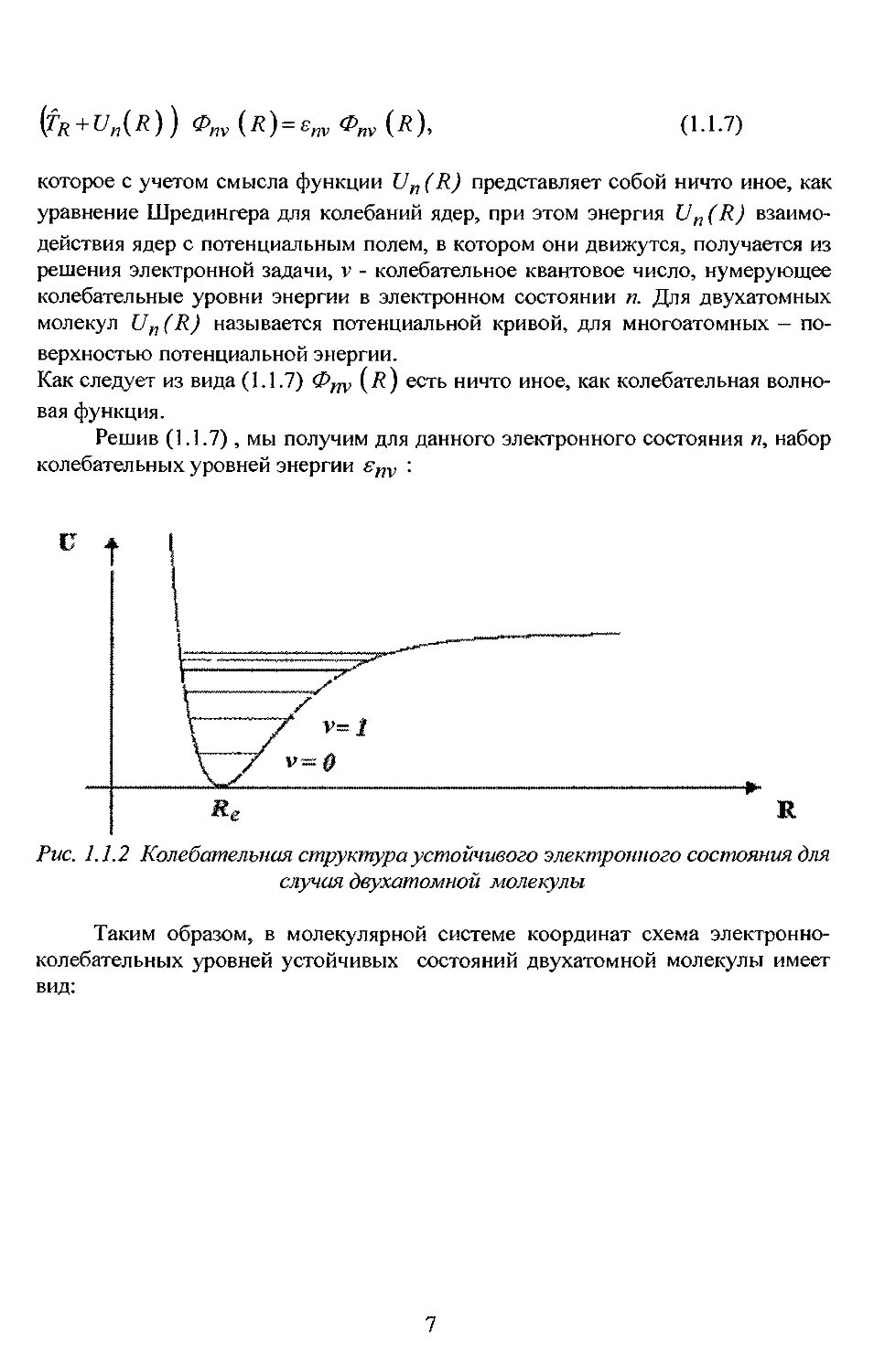
Решив (1.1.7), мы получим для данного электронного состояния п, набор
колебательных уровней энергии snv :
Рис. 1.1.2 Колебательная структура устойчивого электронного состояния для
случая двухатомной молекулы
Таким образом, в молекулярной системе координат схема электронно-
колебательных уровней устойчивых состояний двухатомной молекулы имеет
вид:
7
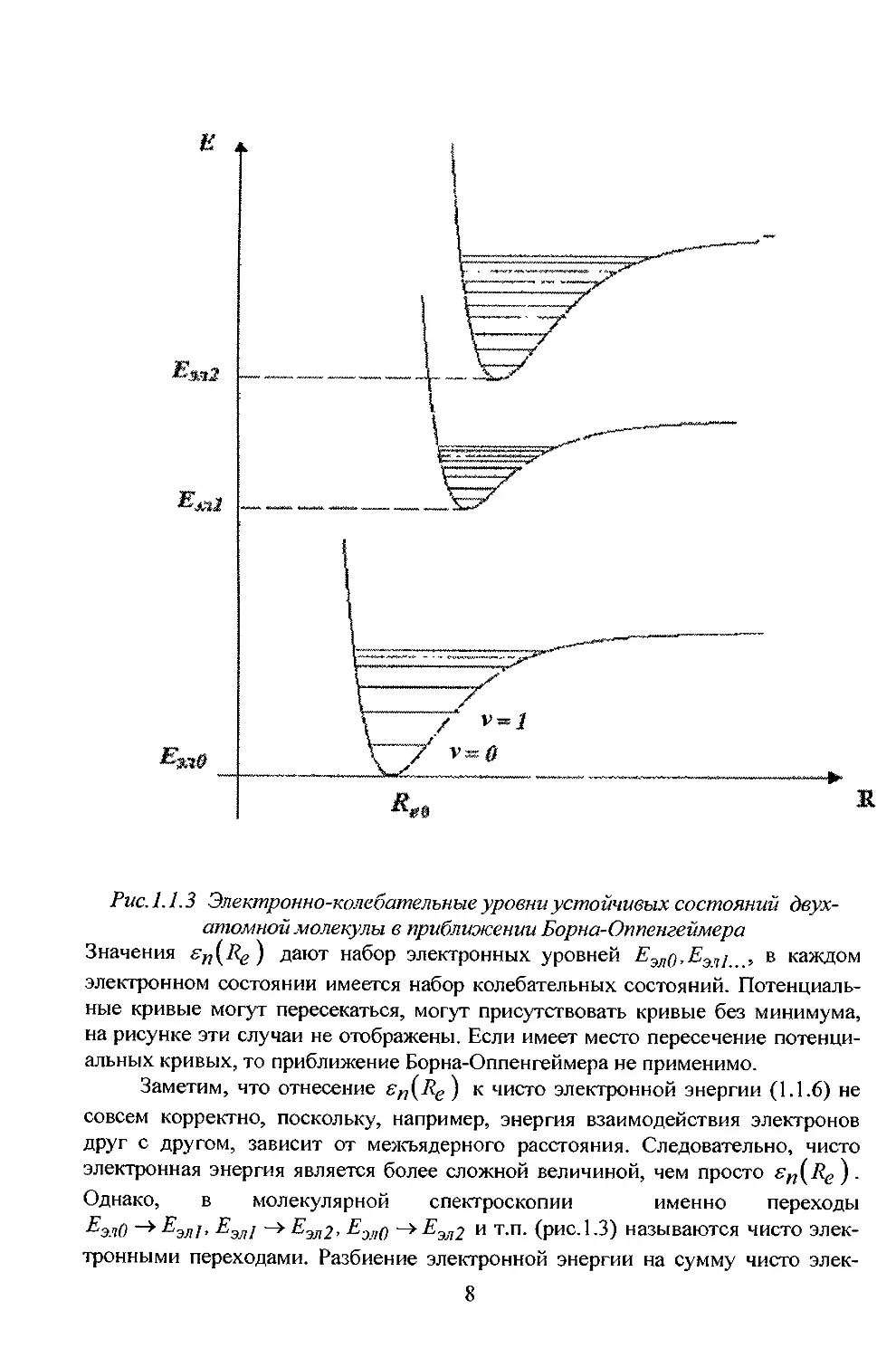
Рис. 1.1.3 Электронно-колебательные уровни устойчивых состояний
двухатомной молекулы в приближении Борна-Оппепгеймера
Значения sn{Re} дают набор электронных уровней Еэлд,ЕЭЛ{ , в каждом
электронном состоянии имеется набор колебательных состояний.
Потенциальные кривые могут пересекаться, могут присутствовать кривые без минимума,
на рисунке эти случаи не отображены. Если имеет место пересечение
потенциальных кривых, то приближение Борна-Оппенгеймера не применимо.
Заметим, что отнесение sn{Re ) к чисто электронной энергии (1.1.6) не
совсем корректно, поскольку, например, энергия взаимодействия электронов
друг с другом, зависит от межъядерного расстояния. Следовательно, чисто
электронная энергия является более сложной величиной, чем просто sn(Re).
Однако, в молекулярной спектроскопии именно переходы
ЕэлО ~^Еэл!> Еэл/ -» Еэл2, Еэ,,о ->£'M2 и т-п- (рис. 1.3) называются чисто
электронными переходами. Разбиение электронной энергии на сумму чисто элек-
8
тронной энергии в указанном смысле и потенциальной энергии ядер (1.1.6)
позволяет разделить электронное и колебательное движение в молекуле. Таким
образом, в молекулярной системе координат и приближении Борна-
Оппенгеймера, молекулярное уравнение Шредингера разбивается на два
уравнения: электронное, описывающее движение молекулы для фиксированных
межъядерных расстояний, и колебательное, описывающее колебания ядер в
определенных электронных состояниях.
1.2. Метод молекулярных орбиталей. Метод Хартри-Фока
Первым этапом решения молекулярного уравнения Шредингера (1.1.1)
является решение электронного уравнения (1.1.5). Электронный оператор
Гамильтона, описывающий атомно-молекулярную систему является
дифференциальным оператором в частных производных 3N переменных ( N - число
электронов). Один из наиболее распространённых приближённых методов решения
многоэлектронной задачи основан на введении самосогласованного поля,
позволяющего свести задачу многих частиц к задаче одной частицы, движущейся
в среднем самосогласованном поле, создаваемом всеми другими частицами.
При этом уравнение движения каждого электрона описывается волновой
функций, зависящей от координат только рассматриваемого электрона, которая
называется одноэлектронной волновой функцией. Такие одноэлектронные
функции называются орбиталями - атомными орбиталями (АО) в случае атома и
молекулярными орбиталями (МО) в случае молекулы. Молекулярная орбитапь
описывает описывает движение отдельного электрона в поле остальных
электронов и всех ядер молекулы и является многоцентровой функцией.
Геометрическим образом атомной (молекулярной) орбитали является область
пространства вокруг ядра атома (ядер молекулы), в которой высока вероятность
обнаружения электрона (обычно выбирают значение вероятности 90-95%). Контуры
атомной (молекулярной) орбитали - это графическое отображение поверхности
уровня волновой функции, полученной при решении волнового уравнения для
одного электрона.
Электронный гамильтониан имеет вид
Ле=1
( *2 л
U 2т J
IE^+IX-+IIZaZ/?'2
к а гка к j>krkj а р>а ra Р
к к j>krkj а р>а гаР
где т- масса электрона, Z0,Zn - заряды ядер а, /?соотвественно, ria -
расстояние от i-го электрона до ядра с номером а, гап - расстояние между ядрами
ti2 Z е2
а,/?, h(){k) = А^ ~Х~~—• Электронный гамильтониан не может быть
2т а гка
9
точно представлен в виде суммы одноэлектронных операторов вследствие на-
" _ е2
линия члена, описывающего отталкивание электронов ^ ее ~ 2~i 2~i .
к j>k rkj
Два первых слагаемых оператора /^зависят от координат только одного (/-го)
электрона, последнее слагаемое Vap не зависит от координат электронов, это -
постоянная величина при фиксированном межъядерном расстоянии. Van не
влияет на вид волновых функций, его наличие лишь сдвигает электронные
уровни энергии на постоянную величину, поэтому Vaa можно учесть на
последнем этапе.
Если бы Vee можно было представить в виде суммы операторов, зависящих от
координат только одного электрона Vee =Х'/А:(^А:)' то электронный гамильто-
к
ниан был бы представлен в виде суммы одноэлектронных операторов и
электронное уравнение можно было бы решить методом разделения переменных.
Наиболее совершенным методом представления электрон-электронного
взаимодействия в виде суммы одноэлектронных операторов (т.е. введения
самосогласованного поля) является метод Хартри-Фока [1,2]. Метод Хартри-
Фока применяется, в основном, для атомов. Атомная волновая функция
представляет собой детерминант Слэтера, построенный из атомных спин-орбиталей.
Развитием метода Хартри-Фока в применении к молекулам является метод
молекулярных орбиталей [3 - 6]. Основу этого метода составляет идея построения
молекулярной волновой функции в виде детерминанта Слэтера, элементами
которого являются не атомные, а молекулярные спин-орбитоли, описывающие
поведение отдельного электрона в поле, создаваемом всеми ядрами и
оставшимися электронами. Метод молекулярных орбиталей был развит в работах
Ф.Хунда, ДжЛеннард-Джонса и Р.С. Малликена [7-12], и заключается в
применении подхода Хартри-Фока для нахождения молекулярных орбиталей и
орбитальных энергий. В методе молекулярных орбиталей система уравнений для их
определения часто также называется системой уравнений Хартри-Фока. Для ее
получения нужно вычислить значение энергии молекулы, проварьировать
энергию, и получить систему уравнений для определения молекулярных орбиталей,
чтобы вычисленная с ними энергия еэч была минимальной.
Найдем эту систему для случая четырех электронов, находящихся на 2-х
заполненных молекулярных орбиталях (Рис. 1.2.1):
2 -4-t-
1 -\-\-
Рис. 1.2.1. Четыре электрона на 2-х молекулярных орбиталях
Это самый простой случай, когда получаемые уравнения имеют такой же вид,
как для систем с заполненными оболочками (четное число электронов). Описа-
10
ние молекулы водорода отличается от рассматриваемого случая тем, что, в
случае нахождения на нижней орбитали спаренных электронов, между ними
имеется только кулоновское взаимодействие, а обменное - отсутствует, поскольку
спины электронов антипараллельны, тождественными являются электроны с
параллельными спинами (см. ниже).
Однодетерминантная волновая функция молекулы имеет вид:
Ф =■
1
/4!
Ф]{г])а(\) ср1(г2)а(2) р,(г3)а(з) 9\(г4)а(4)
PlOi )/?0) Ф\{г2)Р{2) Фх(гъ)р{ъ) ^(г4)/?(4)
Р2(й)«(0 92 {г 2)«( 2 ) 02 (г з)«( 3 ) Ф2 {г 4)«( 4 )
Р2 0О/?(2) 9г{?2)Р{ 2 ) ф2$ъ)Р{ 3 ) ^2 (г 4 ) /?( 4 )
<?,-,- пространственные молекулярные орбитали, которые также называются
просто орбиташми, а и /? -спиновые функции с проекциями спина ms= — ,
Ф. -■
,(1.2.1)
/ws = —, соответственно. Запишем данную функцию в виде
Ф\а{?\У Ф\а{* г)* Ф\а{Т ъ) <Р\а (г 4)
#V(rl)* ^(«"г) ^(«"з)* #v(r4)
/4! <2>2or(rl) ^2or(^2) #>2ог (г 3 )* Ф2а {Т $)•
<Р2рМ Ф2/3 (г2)# ?>2/?0"з) ^2/?(г4)*
где г,- - совокупность пространственных и спиновых координат, функции
Ф1а (г/ )~ФI in )а(У) называются спин-орбитачями, каждая спин орбиталь-
одноэлектронная волновая функция всей молекулы, мы эти функции не знаем и
хотим получить уравнения для их определения. Пространственные орбитали и
спиновые функции ортонормированны. Символы • и * - см. ниже.
Вычислим среднее значение электронного Гамильтониана на однодетерми-
нантной функции q> .
4 4 4 J
Z*o(*)+SI —
k k j>krkj
k j>k'kj
|EM*)
k
(1.2.2)
11
Рассмотрим сначала один из одноэлектронных интегралов \Ф \ho(k) ш \.
Оператор ho(k) действует только на пространственные координаты к—го
электрона. Далее нам потребуется раскрыть определитель (1.2.1). Напомним, что
определитель квадратной матрицы п — го порядка - число, равное
алгебраической сумме и/ членов, каждый из которых является произведением и элементов
матрицы, взятых по одному из каждой строки и каждого столбца, причем, знак
каждого члена определяется как (-1)г(''\ где r(J) — число инверсий в
перестановке из номеров столбцов элементов матрицы, если при этом номера строк
записаны в порядке возрастания:
л=\А =
ац
а21
ап1
<*12 ■
а22 ■
ап2 ■
• а1п
• а2п
■■ апп
по всем перестановкам
номеров столбцов
(-1)г(»а1}1а2}2...а3}п
Возьмем какой либо член в разложении определителя, например, обозначенный
• в (1.2.1):
Х1=<Р1а(*1)-<Р2/]{т2)-<Р1/з(тз)(3У'Р2а{Т4)- О-23)
Этот член соответствует случаю, когда первый и третий электрон находятся на
орбитали 1, а второй и четвертый - на орбитали - 2 (Рис. 1.2.2).
2 4
-J-t-
-н-
3 1
Рис. 1.2.2. Расположение электронов на орбиталях, соответствующих члену
Х1=<Р1а{х1)-<Р2р{1[2)-<Р1р{*з)-<Р2а{ч)
Покажем, что матричный элемент ш \h()(k)\Xi ) будет отличен от нуля
только, если в разложении второго определителя (<р будет выбран член
(Xj |. Действительно, пусть k=I, тогда
{ <p3n\ho(1)\Xi) = { <P3J\ho{i)\<Pla{*l)-<P2p{*2)-<Pip{*3)-<P2a{4))
При выборе в разложении определителя ш члена {X} | получаем
12
( <PlaiTl)\hoU)\<Pla{Tl) )■
{<Р2/з(т2)-<Р1/з(тз)-<Р2а(и)\<Р2/з(т2)-<Р!/з(тз)-<Р2а{т4)} =
= ( <Pla(Tl)\hoU)\<Pla(Tl)) = ( <Pl(n)\hoO)\<Pl(n) } = £or
£„. - сумма кинетической и потенциальной энергии электрона на первой орби-
тали без учета электрон-электронного отталкивания. В последнем выражении
учтено, что гамильтониан действует только на пространственные орбитапи, а
спиновые функции нормированы.
Если в разложении определителя ш выбрать любой другой член, кроме
(Xi |, то хотя бы две из спин-орбиталей, входящих в выбранный член будут
отличаться от спин-орбиталей, входящих в1,-,и из-за ортогональности этот член
обратится в нуль.
В (1.2.2) имеются 4 оператора hg{k),k= 1,2,3,4.
Очевидно
(<Pl(n){ho{l)\<Pl(n)) = (<Pi{rk1{ho(k)\<Pl(Fk))
"{v2{b\ho{l)\<P2{ri)) = {(P2{rk\hQ(k)\<p2{rk)),
т.к. интегралы, стоящие в правой и левой частях этих выражений отличаются
только заменой переменной интегрирования. У нас в электронной функции 24
слагаемых. При раскрытии определителей в матричном элементе
ср \h(){k)\<p \ будут отличны от нуля 24 слагаемых, соответствующих
(
одинаковым членам разложения однодетерминантных функций q> эп и <р .Из
них 12 сводятся к вычислению матричного элемента ho(k) от
пространственной молекулярной орбитали ф ,у £}, и 12 от пространственной молекулярной
орбитали ср2 [г £ J. Таким образом
I iM*)
'А=/
2£(ЧЪ0{1)\Х;)
= 4Ы =
24
12(<pI{rl)lh0{l)\<pI{?I)) + 12(<p2{r}p0{l)\<p2{rI))_
24
2-{Pl{n}h0(l)\<Pl(n)) + 2{<P2(n]h0{l)\<p2(rl))= (1.2.4)
2- I (<Pm(nih0(l)\<pm(?}))
13
Здесь 1/24 - нормировочный коэффициент, множитель 4 появляется
вследствие суммирования четырех одинаковых вкладов от одноэлектронных операто-
4
ров1/?0(А;).
к
Смысл выражения (1.2.4) очевиден. Независимо оттого какие номера
присвоены электронам, это сумма энергий 4-х электронов (без учета межэлектронного
отталкивания), 2 из которых находятся на орбитали 1 , а 2 других - на орбитали
2, но подинтегральные выражения записаны через координаты первого
электрона.
Рассмотрим типичный двухэлектронный интеграл, например, который соответ-
ствует взаимодействию электронов 1 и 2, т.е. члену гамильтониана —.
П2
2
* е,
Вычислим интеграл ]срэл cp3Jldx .dx-dx,dx 4 . Рассмотрим в определителе
г12
(р любой член разложения, например, обозначенный в (1.2.1) символом •,
Рис. 1.2.2:
X-t ^Х; (1, 2, 3, 4)=(р[а (tj ) -<Р2/з(г2 )-<Р]/з(тз) {.$\<Р2а (т4 ) •
При вычислении матричного элемента
2
№эл Xi{l,2, 3, 4)dr ,dx dx?dx
rl2
из-за свойства ортогональности обратятся в нуль интегралы от большинства
членов разложения определителя <р Э1}, отличны от нуля будут два интеграла,
называемые кулоновскгш и обменным
2
1)J' =\Х*{1,2,3.4)~Х-,{1,2, 3,4)dx dx2dx3dx4, (1.2.5)
12 Г12
2
2) К' =\Х*{2,1,3,4)—X\l,2,3,4)dx.dT^dx2dx ,. (1.2.6)
12
Член Xj (2,1,3,4) разложения определителя <р в (1.2.6) соответствует
члену Xj(l,2,3,4) в разложении определителя (р , в котором переставлены
пространственные и спиновые координаты первого и второго электронов. Отличие
от нуля только этих интегралов является следствием ортогональности спин-
орбиталей третьего и четвертого электронов, поскольку на них не действует
14
оператор . Например, если в (р взять член разложения, равный произве-
Г12
дению функций, обозначенных в (1.2.1) символом * :
Xj=9l/3(Tl)-9la{'r2)P2a (тз) ' <Р2/з(т4)>
получим
\Х)—Х\ drjdz2dr3dT4
12
= j 91 {r,)cc{l)-<p2 (Ъ) ФУ? i (? з)Р(3)-<р2 {г4)а(4)
■<Pl in )P{1)-<Pl (b)a(2)-'P2 (r з)а{3)-<Р2 {r4)a{4)dTldT2dT3dT^
= 0.
например из-за fa j (? j) q>2 {r j ) dr^ -0.
Рассмотрим кулоновский интеграл:
У' ^\X*(l,2,3,4)^—Xi{l,2,3,4)dT.dr1dT^dT,.
12 Гп
Выполним в этом выражении интегрирование по пространственным и
спиновым переменным третьего и четвертого электронов и спиновым переменным
первого и второго электронов:
2
Jin = i<Pl{ri)a{l)-<p2{?2)P{2)<pI(r 3)p{3).<t>2{r4)a{4)Y —
Г12
<Pl{71)a{l)-(p2{r2)P{2)- (p i{r 3) p{i\(p2{r4)a{4)dT {dz 2dx 3dr 4 =
2
= j[<pI{r1)a{iy<p2{r2)fi{2)]* — <Pl{rl)a{l)-<p2{r2)P{2)dTIdT2
r12
{<Plir 3)p{3)-92{r4)a{4)\ 2dr3dr4 =
=/
= l\vi(n)\2\jtl)£]<P2(r'2)\2\j^2)\^f-dTldT2 =
j =/
J -1
12
l\viin)\ ■\(P2{r2)\2 ^— df[dr2
r12
15
- кулоновский интеграл для случая, когда электроны, для которых он
вычисляется, расположены на разных пространственных орбиталях. Аналогично, если
электроны находятся на одной орбитали и их спины антипараллельны, под ин-
2 2
тегралом будет стоять \<Pi(jj ) •р/(^) • Перед кулоновским инте-
I II I у
Г12
фалом будет знак +, поскольку перемножаются одинаковые члены в разложе-
нии определителей <р эл и (р .
г * е2
Вклад в матричный элемент шэя <p3Jldr,dt 2dr,dt 4дают 24 кулонов-
г12
ских интефала в соответствие с числом членов разложения определителя,
просуммируем их:
\<Рэл Рэл^т Idr2dr 3dr 4
г12
куп
24 , , ч,2 е2
l\\Xi{l2,3,4)2—dT}dT2
_''=' Г12
24 .
~ 12
_ i
41
24
Из этих 24 слагаемых 8 будут соответствовать случаю, когда первый электрон
находится на первой, а второй - на второй орбитали (Рис. 1.2.3):
2 3 24 32 42
2 _j_f_ -ц- -4-t- -4-t-
l ~\-\- -\-\~ -\~\- -\-\~
14 13 14 13
23 24 32 42
2 -М- -Ы- -\-\- S-\-
i -*-t- -И- -И- -M-
4 1 3 1 4 1 3 1
Pwc. 7.2. J Возможные заселенности первым электроном орбитали 1, авто-
рым электроном орбитали 2
16
Аналогично будут 8 возможностей расположения второго электрона на первой
орбитали, а первого на второй орбитали. Кроме того имеется 4 возможности
расположения обоих электронов на первой орбитали и 4 возможности
расположения обоих электронов на второй орбитали (Рис. 1.2.4):
43 34 43 34
2 _J_t_ H_t- -|-t- -M-
i -М- -М- -М- -М-
2 1 21 12 12
Рис. 1.2.4 Возможные заселенности двух орбиталей при расположении
первого и второго электронов на орбитали I
У нас 6 членов межэлектронного отталкивания:
2 2 2 2 2 2
е е е е е е
П2 ПЗ П4 г23 г24 г34
Очевидно, если электроны находятся па разных орбиталях, то
2 2
Jl2{l2))=\\q>,{r,)\ ■\<P2(r2)\2—dF1dr2 =
112
2 2
12 II I rl3
= Ji2{l,4) = Ji2(2,3) = Ji2(2,4) = Ji2(3,4)) = Jl2,
так как они отличаются только заменой под интегралом переменной
интегрирования, нижние индексы обозначают орбитали, индексы в скобках - номера
электронов. Аналогичный вывод можно сделать для случая, когда электроны
находятся на одной орбитали.
В соответствии со сказанным к Рис. 1.2.3, 1.2.4 вклад в энергию молекулы двух-
электронных кулоновских интегралов равен:
12е2_
Е =6 & +
*J|P/07)| ■\<P2{r2) \2 — dr,dr2
кул -' ~^4
2 е2
8\\<Р2{п)\ -\<Pl{r2)\2 d?ldr2
+ 6-
12
24
17
+ 6-
4\\<Pl{ri)\ \(P i{b)\2 ^—dr }dr
: \l2
24
2
4\\<P2{h)\ \(Р2{Ь) \2 ~ dr,dr2
+ 6 ^ ,т.е. (1.2.7)
24
Екул = 2J12 +2J21+Ju+ J22 ■ 0 -2-8)
Кулоновские интегралы в (1.2.8) определены на координатных функциях, по
спиновым функциям проведено интегрирование. Заметим, что J]2 =^2Ь т-к-
2 £2
J12=l\<Pl{n)\ ■\(P2(r2)\2 dr.dr7
Г12
отличается от
I \2 \ | ■) е^
J2l=l\<P2{ri )\ -|^/(б) | dr}dr2
Г12
только заменой переменной интегрирования. Смысл Екуп (1.2.8) очевиден. Это
кулоновская энергия отталкивания всех электронов, равная сумме энергий
отталкивания всевозможных пар электронов. Независимо от того, какие номера
имеют электроны на первой и на второй орбиталях, таких взаимодействий 6
(Рис. 1.2.5): 4 одинаковых взаимодействия для электронов находящихся на
разных орбиталях (сплошные линии), одно взаимодействие для случая, когда
электроны находятся на первой орбитали, и одно взаимодействие для случая,
когда электроны находятся на второй орбитали (пунктир).
1 ФФ
Рис. 1.2.5. Возможные парные взаимодействия 4-х электронов, находящихся на
двух заполненных молекулярных орбитачях
Рассмотрим обменный интеграл для слагаемого, соответствующего X. в
разложении определителя ср :
2
К' = \Х*{2,l,3,4)-^~Xi{l2,3,4)dTjdr2dr3dz4 . (1.2.9)
12 rl2
Проинтегрируем по пространственным и спиновым переменным третьего и
четвертого электронов и спиновым переменным первого и второго электронов,
учитывая, что оператор межэлектронного отталкивания действует только на
координаты первого и второго электронов:
18
KiI2 = $.Vl(}S2)<*(2)-q>2{n)p(l)-Vl{r3)0(3)'V>2fa)<*(-t)Y
e2 Г 1
[<?l(n )а{1)<Р2{г2)Р{2)-<рАТ з)р{з)<р2{г4)а(4) \ dTldr2dr3dt4 =
rl2
Г 1 2
= hl{?2)a{2).<p2{rI)p{l)-\*^<p,{n)a{l)-<p2{l:2)P{2)dTldT2
rl2
| <p i(r 3) P{3)-<p2{74)a{4)\2 dz 3dt 4 =
=/
&9>l{r2)-9i(n) J y~<P2in)-<P2 ih) drIdz2
\a{l)p{l)dal \a{2)p{2)da2.
Но спиновые функции ортогональны:
\a*{2)-p{2) dz2 =0.
(1.2.11)
Поэтому для членаХ> интеграл К' =0.
Интегралы К1 не будут равны нулю только в том случае, если вместо X,
выбрать слагаемое с параллельными спинами электронов 1 и 2, это возможно,
если они находятся на разных орбиталях, тогда интегралы по спиновым
функциям в выражениях, аналогичных (1.2.10), примут вид
(Ы/)|2- dr. =1,
Ji ' ' (1.2.12)
J|/?(i)|2-</*.=;.
Перед обменным интегралом всегда будет знак -, поскольку член в разложении
определителя <р эл отличается от члена в разложении определителя q> эп
переменой одного индекса, эти члены имеют противоположные знаки. Таким
образом, для каждого члена разложения однодетерминантной функции (р эл
е2
вклад оператора — в полную энергию молекулы равен либо кулоновскому
П2
интегралу, если спины электронов 1 и 2 антипараллельны, либо сумме кулонов-
ского и обменного интегралов, если спины электронов 1 и 2 параллельны. При
19
этом перед кулоновским интегралом будет знак «+», а перед обменным знак «
— » .
Просуммируем обменные интегралы, возникающие от всех членов
разложения определителей, при вычислении матричного элемента
е2
J <Рэл <p3J1dT}dt2dr3dT4.
г12
Их будет 4, если первый электрон занимает орбиталь 1, а второй — орбиталь 2:
2 3 2 4 32 42
-М- -4-t- -4-t- +Ч~
s-t- -*-t- -м- s-t-
14 13 41 31
и еще 4, если первый электрон занимает орбиталь 2, а второй - орбиталь 1.
Если оба электрона занимают одну и ту же орбиталь (1 или 2) обменный интеграл
будет равен нулю, вследствие того, что в этом случае спины первого и второго
электронов противоположны. В случае электронов с параллельными спинами
интегрирование по спиновым переменным дает I, для пространственных
координат получаем с учетом того, что у нас 6 операторов межэлектронного оттал-
2 2 2 2 2 2
е е ее е е
кивания —, —, —,—,—,—:
г12 ПЗ г14 г23 г24 Г34
2
* с
\<Рэл <Рэл^тjdr2dT3dT4
rI2
обм
■6
Г 1 * р
4\[<Pl{ri )-<Р2{г2 ) J ■ 9 2 {ri)-9 lib) drldr2
rl2
24
Г 1* е2
4 ][92(П )'<РАЪ ) J —-9i{r,)-<p2{r2) drldr2 (1.2.13)
-6 Г12 _
24
-к12-к2Г
20
*ЯЛ \±-> £—I
к j>k'kj
Здесь обменные интегралы определены на координатных функциях,
подынтегральное выражение - функция координат первого и второго электронов, г. и
г2 можно заменить на координаты любой другой пары электронов.
Таким образом вклад в электронную энергию двухэлектронных интегралов
равен
>2 \
~ Фчя J = 2J'2 + 2J2I + JH + J22 ~ Кп - K21 =
/ (1.2.14)
2 2
m*n m
где интегралы определены на координатных функциях, по спиновым функциям
произведено интегрирование.
Обменный интеграл для электронов, имеющих противоположные спины, равен
нулю за счет ортогональности спиновых функций, когда он определяется на
спин-орбиталях, в (1.2.14) он определяется на координатных функциях, в этом
случае, Jmm = Ктт, так как
2
Jmm=l\9m{n )\ '\ <Рт(Ь)
\2 е
12
dr.dr2, m = l,2.
* „2
Kmm= JlPmte )'9т(п ) I —Vmird-Vmih) drld?2.
r12
Поскольку Y.(Jmm- Kmm) = 0, выражение (1.2.14) можно переписать в более
т=1
симметричном виде:
\ k j>krkj
\ 2 2 2
*Рэп \ 2-S тп ~ упп) ^ 2-i^mm ^ 2-i'">mm ~ &•mm) ~
I m^n m т=1
2-i'^^mn *^mn/'
Учитывая выражение для одноэлектронных интегралов
(<?эл\ IM*) <РЭЛ) = 2- I {<?т(п)\Ь(){1)\<Рт(п)) = 2 ^т ,
\ к=1 I т=1 т=1
можно записать выражение полной электронной энергии системы с четырьмя
электронами, находящимися на двух нижних молекулярных орбиталях:
21
/ 4 4 4 е2
£эл= И%, Z*o(*)+ZZr-
\ * kj>k\j
2
I
т=1
<РЭЛ ) = 2- Z {<Pm(n)\ho(l)\<Pm(n)} +
2 2 2
2-i(^тп ~ К-mns ~2 2-i£om + Zj'^Jmn ~ Ктп/>
т,п т=1 т,п
причем все интегралы определены на координатных функциях.
Подавляющее большинство устойчивых молекул имеет закрытые
оболочки, т.е. их основное состояние является синглетным. Таким образом, занятые
спин-орбитали встречаются попарно. Тогда выражение электронной энергии
молекулы с закрытой оболочкой с N электронами (N -четное), имеет вид:
N/2 N/2
£эл ~ ■ь I* £Qm+ £*( ^Jmn~&~mn)>
е„ -сумма кинетической и потенциальной энергии электрона на т - ой орби-
тали без учета электрон-электронного отталкивания.
£эл=2- Z \<PmVi )ho{i)<pATi ) d?i
т
NJJ [r , ч , ч -,* „2
2-\\<рЛТ, УфЛ?, ) I
т,п [ 12
* 2 1
Г12 J
Такой же результат получается, если для вычисления одно- и двухэлектронных
матричных элементов использовать правила Слэтера[13].
Вывод уравнений Хартри-Фока. Дадим функции <р т[г. ) в (1.2.15) приращение
S (р т\г. ), тогда вариация энергии должна быть равна нулю
N/2 *( \ I \
5еэп=2- £ \S<PmVl)h0{l)<pm{rl)dr]
т
+ I \4Л5(Р\^1 \<P*m{fl )~ ^Л^иЫ dFldP2
-2\5<p*S?i У*%) г~ ^^ Ь^Ы^/^Н-
W2 J
22
В последнем выражении учтено, что при варьировании кулоновского интеграла
получились два равных слагаемых Sep m\f. )(р т\г. )+<р т\г. )S<p m\r. ), при
варьировании обменного интеграла учли, что т и п пробегают значения
независимо, поэтому в сумме £ для каждой пары тпп обменные интегралы, у ко-
т,л
е2
торых слева от стоит произведение <р (г/ )• срп (г/) встречаются два раза.
г12
Функции <рт\г.) нормированы
проварьируем это уравнение по <р т\г. )
умножим последнее уравнение на произвольное число
~2s т СО \5 <Р *„ fa)? т Yi)dT'i =0
и сложим с 8 8 , получим
N/2\ * ( \ I \
+ I \4-\s<p*m{f, )-^l(F/ )f" nfo^fo) dpi
m,n У 12
2
-2\5(P*S?1 )•*>% ) f- <РЛ?2 )^AP2)dFldj:2
12
~2E m W iS V w Ы? rn (?i)dr, =0.
Согласно основной лемме вариационного исчисления равенство нулю
последнего выражения означает равенство нулю выражения, на которое под
интегралом умножается 8 ср т\г. ). Отсюда для каждого т имеем
( N/2 ( \е2 /_ \ 1 ( \
ho0)+ E 2W„V2) <Ptl\r2)d72 рД'/Г
12
rN/2 (Ye2 ( \ Л ( \ ( \
2 \<P„V2) — <PmV2 rdT2 «M?/J ^„(^m^M1-2-16)
\ n 12 )
23
Здесь тип пробегают значения от 0 до N/2, г. и Я~ можно заменить на
координаты любой другой пары электронов, т.е. различных значений г. (г,)
может быть Л". (1.2.16) представляет собой систему уравнений, которая
называется системой уравнений Хартри-Фока для определения
пространственных орбиталей систем с закрытыми оболочками. Каждое уравнение этой
системы зависит от координат только одного электрона. Первое слагаемое в
круглых скобках в левой части (1.2.16) описывает одночастичный вклад,
второе - отвечает за кулоновское отталкивание, вторая круглая скобка левой
части (1.2.16) - за обмен.
Обычно (1.2.16) записывают через кулоновские и обменные операторы
J и К , которые определяются следующим образом:
2
J^rn (о )= \<Р „ [г2 )^—<Р „ (г2 ) dr2 ■ 9т (гj ),
г12
2
г12
т.е. оператор Кп действуя на функцию <pm [F. ) переводит ее в функцию
2
срп \г. J с коэффициентом \<р [fA—ср [f A dr. . Правая часть второго со-
г12
отношения (1.2.17) получается, если в правой части первого соотношения
поменять местами функцию, стоящую за оператором под знаком интеграла с
Г12
функцией, стоящей за интегралом с одновременной заменой их аргументов.
Операторы Кт называются обменными.
Тогда система уравнений Хартри-Фока для пространственных орбиталей
принимает вид:
Uo{i)+Ni:{2Jn -Кп)]<рт (г, ) = *„(/)?„ fc ),*.» = *. 2...N/2.
\ п )
(1.2.18)
(1.2.18) можно записать для каждого из электронов системы.
/ ч N/2l \
Оператор F = h 0 (/)+ Е [2Jn-Kj
п
называется оператором Фока или фокианом.
24
Собственные значения оператора Фока е (l) = £ называются орбитальными
энергиями. Из (1.2.16) следует, что они определяются выражениями:
£т=£0т+ £ 2 k*n,(rI)<P*n(r2)^r-<pm{r})<Pn{f2)df1d?2
п 42
-I \<Р*т{Ч)<р\ Ы— ^n{rI}pm{F2)drIdr2= (1.2.19)
12
N/2
'От ' ^ ' ■"' А/ °0т
*„_.+ in/,2j = fifl+£M,
где efl -сумма кинетической и потенциальной энергии электрона на т - ой
орбитали без учета электрон-электронного отталкивания (см.выше),
N/2
Ет = £ Етп( 1,2) - суммарное кулоновское и обменное взаимодействие
вз п вз
электрона, находящегося на орбитали т с другими электронами.
2
Еит(1,2) = 2 \<р*т (r} \p\ {r2¥-<pm{7} ]g,n {T2)dr,d72 -
k *т (F, \р % (г2 )z—'P n (Fl Vm (F2 ) d7l dr2 '
42
- сумма кулоновского и обменного взаимодействий двух электронов,
находящихся на орбиталях тип, причем т может быть равно п.
Вычислим сумму энергий электронов, находящихся на заполненных МО,
называемую в части монографий полной энергией системы в приближении Хартри-
Фока:
N/2 N/2
ЕХФ=2- Цет=2. ^е0т +
т т
2- 12 \ср*т (rl )<р\ {r2)^--(pm{rl )cpn [r2)drldr2 -
\ п 12
N/2 , . ( \е2 I \ ( \ ^
~ I \<Р*т (Ч k*n V2)—<Pn Vl)Vm V2 ) dndr2 .
n rl2 )
Сравним это значение со средним значением (1.2. J 5) электронного
Гамильтониана на однодетерминантной функции (1.2.1):
25
N/2
£эл =2' Ё /?и(?/ )h0{l)<Pm(r'i ) d?l
m
+ I \2-\[<pA71 )-<?m(f, )]*~ пЫ'^Ы d7id72-
m,n { '12
-jt <Pm{n)-9n{?l)]j- <Pm{f2 )-<pAP2)d?Id?2
12
Видно, что s ф E v H, а именно
эл ХФ
£эл=ЁХФ~ 2 W^fc/ \<Pm(fl Iff" ^Ы^лЫ d7ldT2
m,n [ 12
-j[ Pm0v )'<РЛп)] f- ^„(^ )-<Pn{r2)drldf2
rl2
Это различие обусловлено тем, что кулоновское и обменное
взаимодействия электронов 1 и 2 Етп( 1, 2) , находящихся на разных орбиталях тип, как
следует из (1.2.19) входят как в энергию орбитали т, так и в энергию орбитали
п, т.е. учитываются при вычислении полной энергии два раза. Вследствие этого
сумма орбитальных энергий не имеет смысла полной энергии. Придавать
орбитальным энергиям физический смысл величин одноэлектронных энергий по
мнению ряда авторов, на основании этого нельзя. На наш взгляд s Ф ЕХф не
может служить основанием для такого утверждения. Действительно,
Етп(1, 2) входит в одноэлектронную энергию и электрона, находящегося на
ез
орбитали т , и электрона на орбитали я, поскольку эти взаимодействия имеют
место для обеих электронов. Совпадения же s и £„. по причине двойного
учета Ет"(1, 2) при вычислении полной энергии и не должно быть. Чтобы не
вз
было двойного учета взаимодействия электронов, полная энергия системы в
приближении Хартри-Фока должна вычисляться по формуле
1
N/2
От ' 2
N/2 N/2 , , , ,J
^Ф=2-1^т+^,я =
2- I *ж + Z 2 W*m {г, )<Р*п (f2)~cpm [fl )cpn [r2)drld72 -
т п г12
~ t \<P*m (r, )q>*„ {г2)^<Рп [7l)(pm(r2 ) drldr2 =еэд.
12
26
Таким образом, при правильном суммировании, s и Е уф
совпадают, что позволяет интерпретировать орбитальные энергии как одноэлек-
тронные энергии. Теорема Купманса [14,15], согласно которой энергия орби-
тали Хартри-Фока s , взятая с обратным знаком, приближенно равна
потенциалу ионизации электрона с данной орбитали, позволяет установить
смысл орбитальных энергий е . Энергия перехода между молекулярными
орбиталями приближенно равна энергии одночастичных возбуждений [6].
Например, фотоэлектронный спектр молекулы ^[4, стр.145] хорошо
коррелируете рассчитанным энергетическим спектром молекулярных орбиталей.
1.3 Метод Хартри-Фока-Рутана
Уравнения Хартри-Фока представляют собой систему интегро-
дифференциальных уравнений, которым должны удовлетворять молекулярные
орбитали, чтобы вычисленная с ними энергия молекулы еэп была
минимальной. Решение этой системы в случае атомов частично упрощается вследствие
наличия центральной симметрии системы. Для расчета молекул уравнения
Хартри-Фока практически не применяются вследствие серьезных
математических трудностей. Большой успех в решении этих уравнений был достигнут в
1951 г., когда в Чикаго были написаны уравнения Рутана [15]. Минимум
функционала может находиться не только путем варьирования функций (орбиталей),
но и путем варьирования коэффициентов, от которых эти функции зависят.
Представим одноэлектронную волновую функцию (т.е. молекулярную орби-
таль) <р в виде линейной комбинации линейно-независимых функций:
М
<Р ( г, )= У С х (г, 1
m,n = l,2—N/2.
Функции х 1 > X? ■*"XM называются базисными функциями, М-число базисных
функций, индексы базисных функций, как правило, обозначаются греческими
буквами, индексы молекулярных орбиталей -латинскими.
Запишем систему уравнений Хартри-Фока:
Используя разложение молекулярной орбитали по базисным функциям,
получаем
FIC % (?,)=£ (/)1С х (?,)• (1-3.1)
V=l v=l
27
Мы рассмотрим вывод уравнений ХФР, отличный от вывода Рутана,
получившего свои уравнения варьированием коэффициентов С . Сначала
используем известный в квантовой механике прием для нахождения коэффициентов
разложения С , молекулярной орбитали по базисным функциям.
Умножим (1.3.1) слева на % v /) и проинтегрируем по d г., получим:
'=/ v=I
(1.3.2)
Будем рассматривать случай, когда система базисных функций не
ортонормирование, введем обозначения:
S/JV=iX*flif,)xv(r,)drr
FpV=IZ*pfa) F Zv(pl)d?r
Тогда (1.3.2) примет вид:
I С (F -e (l)S ) = 0. (1.3.3)
v=l г л
m = l,2—N/2.
Вычислим матричные элементы фокиана
F = h0(l)+ I \?J„-K„) d-3.4)
п
на базисных функциях.
Очевидно, что вклад первого слагаемого правой части (1.3.4) в матричный
элемент F равен:
*1Ы|М') КЫ)
Рассмотрим вклад в F кулоновского оператора. Выражения (1.2.17)
определяют действия кулоновского и обменного операторов J , К на молекулярные
28
орбитали. Выясним как ./ , К действуют на базисные функции х и
вычислим их матричные элементы от базисных функций. По определению J„ имеем
г,
J„(Pm{rI )=1<Р„ (r2)z-9>„ {r2)d72 ■(Pm{fl ) ИЛИ
Г12
J 1С x (r,)=\<P (r*Y—q> (r,)rfr, -1С X (r,\
n , m v л v \ / ' J Y и v 21 ~ v n \ 2' 2 , m v л v V / '
v=/ ' i2 v=I
Последнее равенство возможно при условии
2
42
тогда
2
\x\Ml)J„Xv^l)d7l =^(?/)^*и ^2)z-(Pn Ы ^2 *v(?/)rf?/ =
r/2
у=/ «7 У Г/2 S=l
= I С* Сп6 \\xAf)x\{r2)~x8{72)xv^l)drldr2
Y,S=1 "r r12
7.8=1 ny rn
В последнем выраэ/сении в интеграле, записанном через скобочные обозначения
Дирака, не указаны переменные интегрирования. Принято, что функция,
е2 .
стоящая на первом месте слева от является функцией г., а на втором
Г12
месте — функцией г_, то же самой относится и к произведению функций,
е2
стоящих справа от .
Г12
Рассмотрим вклад обменного оператора в F , по определению
2
Кп(Рт (?/ )='Р* (Я2 )т~^ m (F2 ) d72 ' 9п (?, )>
Г12
29
*,)-
м
К I С
п , т
v=l
М * * / \ о- М i \ М
/1С X [г-, ) 1С х К ) rfr, • I С у
^_у пу у r}7 v-} y-j ns 8
М
(г,>
I С I С* I С {j* (r)^X (r)dr -х (г).
v=I mv Y=i "У y=i n8 У r}7 vW/ J 8 J
Последнее равенство возможно при условии
К X (г,)=
Z С* 1С \% (гА—х \fAdr2 -х \гХ
'=/ "Г y=i п8 J ri2 s
-да
^(F/)Jfi,^(o)rfO =
у.
тогда
м
= I
у,8
М
}fCnrCns(^\~\Sy).
у, 8=1 "г гр
Порядок переменных в последнем выражении определяется также, как в
случае кулоновского интгерала. С учетом проведенных преобразований
матричный элемент фокиана принимает вид
N/2 М * . . е2 , N/2 М * , е2 ,
« Г,о=/ "У r}2 n yts=] "У г}7
м
I ■
у,8=1
>- 1 *
{Mr\—\v8)--{fiy\—\8v )
г12 2 r12
N/2
N/2 *
I 2C С
n "Y
nS
(1.3.5)
Величины Z 2 С С „ =Р ? называются порядками связей.
30
м
Уравнения 1С (F -e U)S ) = 0, (1.3.6)
v=l л л
где матричные элементы фокиана имеют вид
р*у={х*Л)\*о{1)\ х*Ь))+
Л/
I
Y,S=1
12
-i{x*»(PlK(F2)\f-\ Zv(Pl)*S(P2) ) Pys\
называются уравнениями Хартри-Фока-Рутана. В отличие от уравнений ХФ,
представляющих собой систему интегро-дифференциальных уравнений,
уравнения ХФР представляют собой алгебраическую систему для определения
коэффициентов разложения молекулярных орбиталей по базисным функциям.
Для решения данной системы нужно вычислить ~М одноэлектронных
интегралов и ~М двухэлектронных интегралов, поэтому размеры вычислений
быстро увеличиваются с увеличением размеров базиса. Система уравнений ХФР
не является линейной относительно С , поскольку эти коэффициенты через
величины порядков связей Р _ входят входят в F . Для их определения
прибегают к процедуре самосогласования. Используют некоторое нулевое
приближение для коэффициентов С *• ' и рассчитывают с ними порядки связей и
т v
F , подстановка этих величин в (1.3.6) приводит к однородной системе
уравнений для определения коэффициентов первого порядка С *■ . Чтобы система
т v
имела нетривиальные решения необходимо, чтобы ее определитель обращался
в нуль, что приводит к вековому уравнению для определения первого
приближения молекулярных орбиталей е^' :
det(F -s^S ) = 0 .
1 (.IV f.lV/
Каждому из N/2 корней этого уравнения соответствует свой набор
коэффициентов С ^ ' , определяющий молекулярные орбитали первого приближения
т v
Ф \р1 )• Процесс самосогласования продолжается до тех пор пока резуль-
т
тэты последующего шага с заданной точностью не совпадут с результатами
предыдущего шага.
31
При использовании в качестве базисных функций j _ атомных ор-
биталей, последние преобразуются по вырожденным представлениям группы
вращений конкретного атома. Это означает, что выбор направления осей
координат для каждого отдельного атома с их началом на этом атоме не должен
влиять на результаты расчета.
Из различных функций, которые можно выбрать в качестве базисных
наилучшими (хотя и не единственно возможными), являются атомные одноэлектрон-
ные функции, т.е. атомные орбитали. Если в качестве базисных функций
выбрать атомные орбитали, то мы получим наиболее распространенный вариант
метода Хартри-Фока-Рутана, называемый методом МО ЛКАО.
1.4 Наборы базисных функций
В приближении МО ЛКАО молекулярные орбитали ищутся в виде
разложения в ряд по атомным орбиталям:
м
<P-t = I cifiXfi ■
ц=1
Лучшими атомными орбиталями являются самосогласованные АО, полученные
путем решения уравнения ХФ для атомов. Однако такие функции получаются в
табличном виде и работать с ними неудобно. Удобнее работать с функциями,
которые заданы в аналитическом виде и с которыми интегралы, входящие в
уравнения ХФР также вычисляются в аналитическом виде. В качестве базисных
функций наиболее часто используют два типа функций: слэтеровский тип и га-
уссовский тип [4,5,18-21].
Слэтеровский тип АО . Нормированные слэтеровские АО в сферической
системе координат имеют вид [21,22]:
X ^Х nlm=Nf7r"-Iexp(-^nlf-)Ylm{e,<p), (1.4.1)
/'
£и/ называется орбитальной экспонентой.
Применение слэтеровского базиса оправдано тем, что именно такой вид имеет
решение уравнения Шредингера для атома водорода. Запись АО в виде (1.4.1)
предполагает, что центр системы координат для каждой АО находится на том
атоме, для которого эта АО записывается, как говорят, АО центрированы на
атомах. При использовании базисных функций данного типа возникают
сложности при вычислении многоцентровых двухэлектронных интегралов,
входящих в систему уравнений Хартри-Фока-Рутана. При их вычислении
необходимо все АО записать в одной и той же системе координат. Многоцентровые
интегралы намного легче вычислять, если в качестве базисных использовать гаус-
совский тип орбиталей.
Гаусовскш тип АО. Функцию Гауссовского типа обычно выражают в
декартовых координатах и обозначают буквой g [4, 5, 20,23]:
g^,r)=NGxiyJzk exp[-% r2), (1.4.2)
i,j, k— целые положительные или равные нулю числа.
Нормировочный коэффициент определяется из условия:
32
[g2(Z,r)dT = l.
Функции gig,г) называются примитивными гауссианами.
Нормированные примитивные гауссианы s, p и dxy типа имеют следующий вид:
&(£r)=(ii) exp{-$r2\
8Рх{£.г) =
gpy(Z>r) =
gPz{Z,r) =
(128%5
1/4
хехр
(-И.
f 128Z,5
1/4
уехр
' 128£5 '
М-
ехр(-£г2) gd (4,г):
(1.4.3)
1 2048 f '
хуехр
(-И
Декартовые координаты связаны со сферическими соотношениями:
х = rsinOcos (р ; у = rsin6sm<p ; z = rcos в ,
где
cos в = Yjq
' I—
ГУ
*2к'
Отсюда следует, что угловая часть АО заключена в предэкспонентах
примитивных гауссианов.
Поведение функций gig,г) при г—>0, и при г—><х> существенно отличается от
поведения АО. Чтобы улучшить аппроксимацию АО гауссовыми функциями
используют контрактированные (сгруппированные) базисные наборы.
Контрактацией называется объединение нескольких гауссовских функций в одну
функцию. Контрактированные базисные орбитали имеют вид:
2\ж
sin 6 sirup = i{Yj_i + Y//V
sin в cos (p = {Yj_i -Yjj)j л
N
Zls={r)= I kUsgiIs\gitIs,r\
i=l
N
X
I
Z2s=(r)= 2 ki.2sSi,2s\4i,2S'r)>
(1.4.4)
33
N
Z2p(r)= I k2Pgi,2p{uls-r)-
Ы1
кщ фиксированы в пределах данного базисного набора. Контрактированные
базисные функции нормированы.
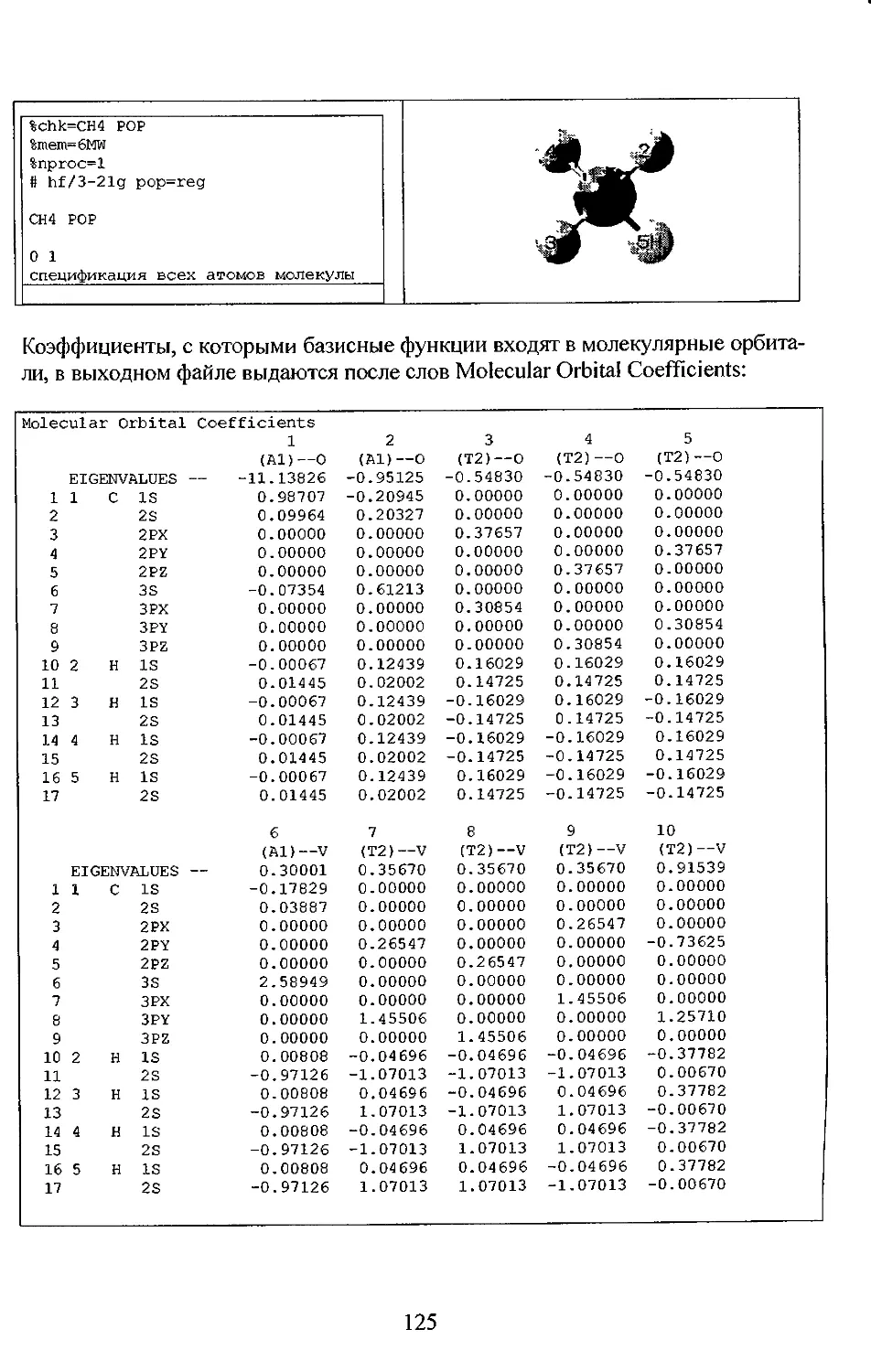
Индивидуальная молекулярная орбиталь в программе GAUSSIAN- линейная
комбинация атомных орбиталей, каждая из которых представляет
контрактацию функций Гаусса:
N N Ng i \
<Pi= I с-щхи= I c¥ I kii/Igii/i^ii/itr), (1.4.5)
ft=l /л=1 i=J
ci\x ~ Коэффициенты молекулярных орбиталей (Molecular Orbital Coefficients),
N- число базисных функций,
N - число примитивных гауссианов, входящих в базисную функцию.
Расчеты по методу Ругана можно разделить на два класса: расчеты с
минимальным базисным набором, который для построения МО использует только
АО, занятые электронами в основном состоянии атома, и с расширенным
базисным набором, который включает дополнительно АО, не занятые
электронами в основном состоянии атома. Перечислим некоторые базисные наборы.
1.Минимальный базисный набор состоит только из атомных орбиталей
заполненных электронами оболочек атомов. Наиболее простой тип базисных
наборов - наборы STO-NG (атомная орбиталь представляется орбиталью слэте-
ровского типа, она аппроксимируемая N функциями гауссова типа). N чаще
всего берут равным 3, поскольку при дальнейшем увеличении числа членов
точность расчета увеличивается медленно, при этом имеем минимальный
базисный набор STO-3G. Этот базис включает одну базисную функцию (атомную
орбиталь) для атома водорода (Is), 5 функций (Is, 2s, 2px , 2ру , 2pz) для атомов
второго периода от Li до Ne и 9 функций (Is, 2s, 2рх , 2ру, 2pz, 3s , Зрх , Зру ,
3pz) - для атомов третьего периода от Na до Аг. При этом каждая из
перечисленных базисных АО аппроксимируется линейной комбинацией трех функций
Гаусса. В рассмотрение включены только заполненные оболочки атомов,
входящих в состав молекулы.
2.Улучшение точности расчета достигается при использовании более
широких валентно расщепленных базисов (split-valence). В этих базисах АО
внешних электронов составлены из двух частей - внутренней более компактной и
внешней, более диффузной с разными значениями экспонент, например
Н :1s, Is
С:Is,2s, 2s ,2px,2py,2pz,2px ,2py ,2pz
или, обозначая внутреннюю часть волновой функции индексом / - inner , a
внешнюю индексом О — outher
H:ls rJsQ;
34
С; Is, 2s f,2s0,2px / ,2py / ,2pz } ,2px Q ,2py 2pZQ- (J -4-6)
При составлении МО в процедуре ССП коэффициенты каждой из орбиталей
этих двух типов можно варьировать независимо. В валентно расщепленных
базисных наборах на компактную и диффузную составляющие разделены только
валентные орбитали. Обозначение таких базисов М - NPG, (где М, N, Р- целые
числа, например 6-31G), здесь увеличение точности достигается благодаря
тому, что валентные орбитали представляются двумя наборами функций с
разными значениями экспонент, М - число гауссовых функций, входящих в каждую
АО внутренних электронов. Например, аббревиатура базиса 6-31G означает,
что каждая АО внутренних электронов является суперпозицией 6 гауссовых
функций, имеется два набора базисных функций валентных электронов, первый
набор состоит из 3 гауссовых функций, второй набор состоит из одной
гауссовой функции.
З.В валентно-расщепленных биэкспоненциальных (double zeta, DZ)
базисных наборах расщеплены АО как внутренних, так и валентных электронов.
4. Дальнейшее увеличение точности достигается включением в базисный
набор поляризационных функций, т.е. добавлением р орбиталей при описании
атома водорода и d орбиталей для элементов 2 периода, что обозначается 6-
31G(d,p) =6-3IG**. Аббревиатура 6-31(d) = 6-31G* означает, что добавляются
только d орбитали для элементов 2 периода.
5. Расчеты ионов в программе Gaussian целесообразно проводить в
базисах, дополненных диффузными функциями. Эти базисы включают для s ир АО
валентных электронов дополнительно гауссовы функции со значениями
орбитальных экспонент £ от 0,01 до 0,1, что позволяет лучше описать АО при
больших г . Включение в базис диффузных функций обозначается символом
«+» ( если они добавляются к s и р атомным орбиталям тяжелых атомов и нет
добавлений к s орбитали атома Н) или «++» ( тяжелые атомы+Н). Например 6-
31++G (дополнен диффузными функциями для тяжелых атомов и Н) или 6-
31++G* ( включает поляризационную d функцию для атомов второго периода и
дополнен диффузными функциями для тяжелых атомов и Н).
Все основные расчетные методы современной квантовой химии
используют приближение МО ЛКАО ХФР. Неэмпирические методы основаны на
точном решении уравнений ХФР, которые не включают никаких
экспериментальных параметров, кроме фундаментальных физических постоянных. Затраты
машинного времени быстро растут с увеличением размеров базиса.
Указанные выше и другие базисные наборы обсуждаются во многих
работах, например [24 - 27].
35
1.5. Электронная плотность, порядки связей, заряды на атомах,
поляризуемость
Плотность вероятности нахождения электрона, заселяющего молекуляр-
м
ную орбиталь (р = £ С % , в точке с радиус-вектором г определяется
17 y=i п У у
выражением:
■у М М
r=1 S=l (I.5.1)
S с cnS/ {f)xs{f).
n у ПО у О
М- число атомов в молекуле.
Результируюидую плотность вероятности нахождения электрона в точке г для
молекулы с закрытой оболочкой, где каждая молекулярная орбиталь занята
двумя электронами получим, просуммировав (1.5.1) по всем занятым орбита-
лям:
N/2. ,2 N/2 M „
D{r)=x_l_Il\^(r)\ =2E T.C Си£ХХб =
-> n=l n=l v ё—1
2енакаж- г'
дои МО
= I Ь*С СпбХ Хб= ZP\SX ir)xs(r)t 0-5.2)
у,8=1п=1 7 у у, 8=1 7
уд
N - число электронов в молекуле, Р ~ - по определению порядок связи (см.
1.3.5).
Результирующую плотность можно представить в виде суммы вкладов в
D (г)отдельных атомных орбиталей и попарных перекрываний различных АО,
а именно:
I I2
О J ,у = 1,2,---М совпадает с распределением плотности вероятности
нахождения электрона на атомной орбитали j
и
2) X Хх*У^8;у,д = 1,2,- ■ -М учитывает вклад в D (г ) перекрывания АО.
Из (1.5.2) следует, что Р ^характеризуют величины этих вкладов в
электронную плотность.
36
В квантовой химии часто используется понятие «заряд на атоме». Это
понятие неоднозначно и зависит от интерпретации члена
*
X Хх> У*o;y,S = J,2,---Mпопарного перекрывания атомных орбиталей.
Наиболее часто используется схема разложения этого члена, предложенная
Малликеном, называемая анализом заселенностей по Малликену [28,29].
Анализ заселенностей по Малликену. Рассмотрим двухатомную молекулу
с ядрами у и 5, запишем выражение ее молекулярной орбитали и, используя
метод МО ЛКАО:
Условие нормировки данной волновой функции в случае вещественных
коэффициентов
\\<р (r)\dr = C2 +2C С , \х*у(г) X я{г)<1г + С2 =1.
1"1 п у п У п ° ' ° п ё
Перепишем условие нормировки в виде:
\С2 +С С XS Х\+\С С rS г+С2 \ = 1 . (1.5.4)
\ пу п у п 5 уд) у п у п 6 уд „ g J
Пусть п-ая орбиталь заполнена, т.е. число электронов на ней равно 2. Умножим
обе части (1.5.4) на число электронов на данной орбитали и поменяем местами
правую и левую части:
2=2\С2 +С С -S Л + 2\С С -S г+С2 |. (1.5.5)
[ пу п у п ё уё) у п у п ё уё ns) х
Число электронов на молекулярной орбитали и представлено в виде суммы
двух слагаемых, первое из которых может быть проинтерпретировано как
число электронов на атоме у, а второе - как число электронов на атоме 5.
Просуммировав (1.5.5) по всем занятым МО для двухатомной молекулы с
заполненными оболочками получаем полное число электронов у атомов у и 5,
называемые полными заселенностями атомов у и 5:
Р„„=2 Ni2[c2ny+C„ .C.^.,1 (1.5.6)
УУ Л пу п у п 8 уё
N/2f ,
Рхх-2 Z С С XS Х+С2 I. (1.5.7)
год~* _.\ п У п ё уё п $ *
37
Заряды на атомах у (5) по Малликену в молекуле определяются как разность
между полным числом электронов п у{п g) в изолированном атоме и значе-
ниями р
ь
*s
у
= п8
ГУ
-р
-р
р
rr'
SS
SS
>
(1.5.8)
(1.5.9)
они представляют собой избыточное число электронов на атоме в молекуле по
сравнению с индивидуальным атомом. При вычислении зарядов на атомах по
Малликену заселенность перекрывания делится между атомами равным
образом. Существуют другие подходы ее разбиения на составляющие атомов,
например [30].
Величину 2-2С С 5 tXyi?) X fi)dr = 2-2 С С „S * ъ выражении
(1.5.5) называют заселенностью перекрывания. Разделив эту величину на 2,
получим характеристику числа пар электронов в области перекрывания АО.
Величину
N/2
р я=2 I С С . S я (1.5.10)
гу8 , п у п о yS v/
называют межорбитальной заселенностью связи, она является характеристикой
связывания двух атомных орбиталей. Для двухатомной молекулы эта величина
является также полной заселенностью связи. Для многоатомной молекулы с
числом атомов М выражения полных заселенностей атомов и связей,
учитывающие вклады всех атомов принимают вид:
(
.2
М М
Сг +1 1С С ,S .
«г y=is=i п у " д yS
м м
I I С С ~S Я+С2
{r=}S=i п У " s У6 "8
N/2
1г п=1
N/2
PSS-2 Z,
N/2 М М
Р я=2 Ъ Y. НС С , S . .
^уд , , ~ п у и 5 уд
п-1 у-1д=1 ' '
Заряды на атомах и межорбитальная заселенность связи многоатомных молекул
также определяются формулами 1.5.8 - 1.5.10.
Поляризуемость молекулы. Поляризуемость молекулы а определяет ее
способность поляризоваться, т.е. приобретать под действием электрического поля
индуцированный (наведенный) дипольный момент. В общем случае для молекул
составляющие дипольного момента /Lix,/uy,juz в молекулярной системе
координат являются линейными функциями составляющих Ex,Ey,Ez
напряженности поля:
38
j.lx — CCxxh,x + CC^ytLy + CCxziLz
My ~ CZyx^-'X (XyyE'y т" C^yz^z ''
Совокупность величин a^y (j3,y = x,y,z) образует тензор
поляризуемости. Можно показать, что этот тензор симметричный, т.е.
(Хху -ayx,ayz =cczy,azx — ccxz, и, следовательно, определяется шестью
составляющими ахх,ауу,а22,,ауХ,агу,агх.
Большое число расчетов тензора поляризуемости атомов и молекул в
современной литературе выполнено с применением методов квантовой химии.
При этом используются следующие основные методы расчета поляризуемости
атомов и молекул: Sum-Over-States, Response theor}', Polarization Propagator
model, Perturbed Electron Propagator method, Time Dependent Coupled Hartree-
Fock Theor}' [31]. Большинство неэмпирических расчетов тензора статической
поляризуемости атомов и молекул основано на теории отклика (Response
theor}') атомно-молекулярной системы на внешнее электромагнитное
воздействие [32]. В основе этой теории лежит разложение энергии атомно-
молекулярной системы или дипольного момента в ряд по напряженности
внешнего электрического поля F:
E(F) = E(0)-i:diFi —ZayFiFj -Up^F^ -... , (1.5.11)
i 2jj J 6iJk
di{F) = di(0)+I,aijFj+^l.J3ijkFjFk+... , (1.5.12)
где di - компоненты дипольного момента,
aij>Pijk ~ тензоры поляризуемости и первой гиперполяризуемости,
соответственно.
Современные вычислительные программные комплексы позволяют
решить уравнение Шредингера, описывающее поведение атомно-молекулярной
системы во внешнем электромагнитном поле с Гамильтонианом
H^Ho-Y.diFi-L Y.eyF0-..., (1.5.13)
/' i,j=x,y,z
где dt - оператор дипольного момента, 0у - оператор квадрупольного момента,
F-, - компоненты напряженности внешнего поля,/},- - градиент поля,
обусловленный внутренними зарядами, и найти энергию (1.5.11) или дипольный
момент атомно-молекулярной системы (1.5.12) как функцию напряженности
внешнего поля. Численное дифференцирование функций (1.5.11) и (1.5.12) по
напряженности внешнего поля позволяет найти дипольный момент,
поляризуемость, гиперполяризуемость рассматриваемой системы и т.д. Этот метод был
39
использован для расчета статических поляризуемостей двухатомных и малых
молекул во многих работах.
В работе [33] показано, что при выполнении дифференцирования в (1.5.11) и
(1.5.12) методом конечных разностей использование формулы для
индуцированного дипольного момента (1.5.12) приводит к более точным результатам для
поляризуемости, чем использование формулы (1.5.11).
1.6 Электронная корреляция и неограниченный метод ХФ(ХФР)
Рассчитанные по методу ХФ и ХФР энергии молекул всегда меньше
экспериментального значения [4,20]. Это обусловлено тем, что меяолектронное
взаимодействие входит в уравнения ХФ (1.2.16) в интегральном виде через
усредненное поле:
( N/2 ( \е2 ( \ 1 ( \
h()0)+ Z 2 \<Р „ V2) <Р „ \r2 ) dr2 <рт (г, )-
я ' 12 )
N/2 ( у е2 [ \ ~] ( \ I \
п 42 )
Потенциал взаимодействия электронов 1 и 2 определяется только
координатами электрона 1 (кулоновский и обменный интегралы после выполнения
интегрирования являются функциями г.). Он не учитывает того, что
электроны 1 и 2 стремятся находиться как можно дальше друг от друга. Поле, в
котором движется электрон 1 не зависит от координат электрона 2, поэтому с точки
зрения уравнения ХФ (ХФР) положения электронов а и б (Рис. 1.6.1)
эквивалентны
а
Рис. 1. б. 1. Кулоновская корреляция электронов в методе ХФР
На самом деле вследствие кулоновского отталкивания положение б
предпочтительнее, т.е. существует кулоновская корреляция в движении электронов,
не учтенная в уравнении ХФ(ХФР) .
Второй тип корреляции включает синхронизацию движений пар
электронов с параллельными спинами, расположенными на разных МО. Два таких
электрона слабее отталкиваются друг от друга, чем электроны с
антипараллельными спинами, поскольку для параллельных спинов имеет место обменная
поправка к отталкиванию, знак которой отрицателен (см. комментарии к
формуле (1.2.10)). Поэтому электроны стремятся так скоррелировать свое
движение, чтобы электроны с одинаковым спином в среднем были расположены
40
ближе друг к другу, чем электроны с антипараллельными спинами. Рассмотрим
систему с открытой оболочкой, в которой имеются МО с неспаренными
электронами, например, на верхней занятой МО (ВЗМО, или HOMO - highest
occupied МО) имеется 1 а электрон, а более низкие МО заполнены полностью,
Рис. 1.6.2а.
Для такой системы а электроны заполненных оболочек будут испытывать
меньшее электронное отталкивание со стороны неспаренного а электрона, чем
/? электроны, поскольку уравнение ХФ для пространственной волновой
функции а электрона орбиталей <р. и <р2 содержит отрицательную обменную
добавку, а для р электрона - не содержит. Поэтому пространственные волновые
функции а и р электронов заполненной МО будут различны. Метод ХФ для
систем с открытыми оболочками, учитывающий это различие, называется
неограниченным методом Хартри-Фока (UHF)[20]. Метод Хартри-Фока для систем
с заполненными оболочками, где такого различия нет называется
ограниченным методом ХФ (RHF) [20]. Различие между неограниченным и
ограниченным методом ХФ иллюстрируется схел
LUMO ф,
номо <Р 1-
v, -t-t-
я
RHF
Рис. 1.6.2 Молекулярные орбитали в ограниченном (RHF) и неограниченном
(UHF) методах ХФ (ХФР)
Вследствие наличия кулоновской электронной корреляции метод Хартри-Фока-
Рутана в принципе не может точно описать экспериментальные характеристики
молекулы. Разность между точной нерелятивистской энергией и энергией,
полученной методом ХФ называется энергией электронной корреляции [4,5,20]:
Е -Е - Е
корр точи Х—Ф'
Точная нерелятивистская энергия молекулы получается при решении
уравнения Шредингера с гамильтонианом, не учитывающим релятивистские члены (
спин-орбитальное, спин-спиновое взаимодействия и др.). Энергия электронной
корреляции составляет небольшую долю полной энергии молекулярной
системы (0,5% для молекулы воды [4]). Однако энергия связывания в молекулах
также имеет такой же порядок. Но можно заметить, что ошибки,
обусловленной:
<Р% —<
9>% = К
<Р%-\-—<
<ра2-\--\-К
6
UHF
41
ные приближением самосогласованного поля, во многих случаях
приблизительно постоянны [20], и при изменении молекулярной структуры можно
получать неплохие результаты для относительных характеристик. Сравнительная
характеристика энергии молекулы, рассчитанная различными методами
приведена на рисунке [4]:
■ ХФ (ХФР) расчет е минимальном базисе
_____ ХФ (ХФР) расчет в расширенном базисе
ХФ (ХФР) предел
почти полное КБ
точная нерелятивистская энергия
эксперимент
Наиболее часто применяемыми методами учета электронной корреляции
являются метод конфигурационного взаимодействия и метод теории возмущений
Меллера-Плессета (MP).
1.7 Метод взаимодействия конфигураций
Распределение электронов по МО молекулы называется электронной
конфигурацией молекулы. Определенной электронной конфигурации молекулы
соответствует однотерминантная волновая функция, построенная из спин-
орбиталей. Методы конфигурационного взаимодействия представляют
молекулярную волновую функцию в виде суперпозиции детерминантов Слэтера,
построение которых осуществляется следующим образом. Как правило, при
решении уравнений Хартри-Фока-Рутаана, число рассчитанных МО превышает
число занятых МО. Наименьшей энергии в однодетерминатном приближении
соответствует случай, когда электроны находятся на нижних орбиталях
(соответствующий детерминант (р п), а верхние орбитали свободны. При этом
незанятые орбитали называются виртуальными. Помещая на них электроны
различными способами, можно построить различные электронные конфигурации и
соответствующие им определители (р , (А=1,2...). В методе
конфигурационного взаимодействия полную волновую функцию записывают в виде линейной
комбинации слэтеровских определителей, соответствующих различным
электронным конфигурациям [34-36]:
42
к max
гэл " . . к ™ элк
к=0
Здесь <р - электронная волновая функция точного (истинного) гамильтониана
молекулы Н-НХф +V, <р , - однодетерминантная волновая функция
молекулы, полученная методом ХФР. Коэффициенты Ак находят методами
квантовой механики (вариационный, теория возмущений). Различают методы
взаимодействия конфигураций с одиночной, двойной, и т.д подстановкой, полное
конфигурационное взаимодействие [26,34-36]. При одиночной подстановке
одна виртуальная орбиталь заменяет занятую орбиталь в детерминанте, что
соответствует возбуждению электрона на данную виртуальную орбиталь. При
двойной подстановке две занятых орбитали замещаются виртуальными орбита-
лями и т.д.
1.8 Теория возмущений Меллера-Плессета (MP)
Среди методов учета электронной корреляции наибольшее распространение
получил метод Меллера-Плессета (MP) [37-48], согласно которому истинный
Гамильтониан молекулы представляется в виде суммы:
« = нхф+л(н-нхф)=нхф+у-
При X -0 имеем хартрифоковский предел, при X - 1 получаем точное значение
Гамильтониана. Искомая волновая функция и энергия разлагается в ряд по
степеням Я,
а
Y э.7 т эл(0) Y эл(1) Y эл(2)
Е — Е ,п. + X Е ,,. + X Е го.н—.
ал эл(0) Щ<) эл(2)
Для нахождения поправок к волновой функции и энергии применяют метод
теории возмущений. Обычно принимают X -1, поправки рассчитывают не
выше, чем до 5 порядка:
1ЕэлП) =№* /niF 9 dx.
™('J эл(0) эл(о)
Выражения для (р ,.., (р ,_. >ЕЭл(2)''' выражаются через характеристики ХФ
орбиталей по формулам теории возмущений. В зависимости от порядка теории
возмущений различают МР2, МРЗ, МР4, МР5 методы.
1.9 Молекула водорода
Электронный гамильтониан молекулы водорода, состоящей из двух протонов и
двух электронов имеет вид:
Яе= lh (k) + g{l,2), (I.9.1)
где
43
2т. гы гю
- одноэлектронный оператор, зависящий от координат только одного электрона,
к-1,2 - номера электронов, А, В - ядра.
g(l,2)= - двухэлектронный оператор, описывающий кулоновское оттал-
Г12
кивание электронов.
В одноэлектронном приближении поведение каждого электрона описывается
одноэлектронной волновой функцией - молекулярной орбиталью. Согласно
методу Рутаана молекулярную орбиталь будем искать в виде разложения по
атомным орбиталям:
<Р (F,hClXl(P,A)+C2%2(PlB)> (L9-3)
г. - расстояние от рассматриваемого электрона до центра масс молекулы,
Г1А '' IB ~ Расстояния рассматриваемого электрона (первого) до ядер А и В
соответственно,
Xi и Хо' базисные функции, в качестве которых можно выбрать атомные
орбитали атома водорода. Это могут быть, например, Is- орбитали (минимальный
базис):
1 е~г^; % =1е~г1В. (1.9.4)
/ ПГ ' л2
я ' у я
Заметим, что функции х j и Х2 (1-9.4) центрированы каждая на ядре А или В,
соответственно. Расстояния г. и г.„ легко могут быть выражены через г..
Уравнения Рутаана (1.3.6) с учетом того, что Бц = S22 =1 принимают вид:
IcAFu-e )*C2[F -e S12)=0
\CAF2l-e S2lYC2\F22-s )=° ' (L9'5)
Вследствие эрмитовости матриц и симметрии задачи выполняются условия
F12 = F21' Fll = F22' S12 ~S21~S-
Уравнения Рутаана решаются, как сказано выше, самосогласованно
итерационным путем, в данном случае вследствие простоты задачи, решение существенно
упрощается.
Система уравнений (1.9.5) имеет нетривиальное решение только в случае, если
детерминант системы равен 0:
44
Fn-s
12
Fn -s S
■s S F}[ -s
(1.9.6)
Отсюда, для определения е получаем квадратное уравнение
(Fn~£ Y -iF,2-£ S)2=°>
корни которого равны:
s = F11 +F12 . _
1 1 + S ' 2
11
12
1-S
(1.9.7)
Для нахождения коэффициентов С. и С_ подставим собственные значения
(1.9.7) в систему (1.9.5):
а) для собственного значения е получаем:
//
21
F + F
г11 + г 12
1 + S
+ С.
F12-
F +F
II .
1 + S
12
S\ = 0
F'I+Fl2s\+c7
1 + S I 2
F -
22
Fll+F12
1 + S
отсюда
\c1{fus-f12)-c2{f11s-f12)=o
\c](fJ2-f]1s)-c2{f12-fiis)^o-
Поскольку определитель системы равен нулю, уравнения линейно зависимы, из
любого уравнения получаем С, =С, =С, следовательно, <р } -С\х} +%2>'
Условие нормировки молекулярной орбитали :
j|p/ \2dr = C2( l\z, f dT + 2l%1Z2 dr + \\x2 \2dr) = l,
отсюда
J2+2S'
б) для собственного значения s 7 получаем:
(1.9.8)
45
С, \F„- il l2
7 /; 1-S
J
( F..-F..
С
+ C2\F,2 —IZ^8^0
F J n FnXC\F J11 F12
21 1-S 2{ 22 1-S
F +F F -F
e, = -^ ^-; s. =-U. 12- . (1.9.10)
отсюда
\Cl^2-FnShC2(FI2-FnSh0 c c
lCl(FI2-FnS)+C2(F12-FI/S)=0 ' 2~ '
<p2=c[Xi-%2).
Из условия нормировки С = , ,
'>-ТГйк--*'1 °-9'9)
Таким образом, молекулярные орбитали и орбитальные энергии молекулы
водорода в результате расчета получились равными:
+ F F -F
г/ f 12 . =2JL_J_
1+S ' 2 1-S
"' =W2S{l> +*>*'>'^=Ts(X' ''Л °'9H)
MO <p. симметрична относительно перестановки ядер, МО <з, -
антисимметрична. Расчет показывает, htoF}2 <0, поэтому s. <е2. Для любого значения
межъядерного расстояния в точке, соответствующей середине расстояния
между ядрами имеем %. =Х2> поэтому электронная плотность в середине связи
для связывающей орбитали отлична от нуля, а для разрыхляющей орбитали
равна нулю. Заметим, что выбор базисных функций %. и х2 влияет на
численные значения орбитальных энергий и МО, но не влияет на вид выражений
(1.9.10) и (1.9.11).
Получившееся число пространственных МО равно числу базисных функций М
( в данном случае 2), поскольку вековое уравнение является уравнением М-ой
степени и имеет М корней. Два электрона можно разместить на двух МО
шестью способами (Рис. 1.9.1). Молекулярные волновые функции этих
конфигураций можно представить в виде слейтеровских детерминантов второго
порядка, сконструированных из спин-орбиталей. При этом детерминанты
"2
А . , А 2 , А , , A f являются собственными функциями оператора 5" и
описывают чистые спиновые состояния, соответствующие полным спинам
системы 0, 1, 1 и 0, соответственно.
46
*i-h—^ 1 1 \— —
4 4 4 4( ^5 4
Рис. 1.9.1 Возможные электронные конфигурации при размещении двух
электронов на двух пространственных молекулярных орбиталях
Мультиплетность указывается надстрочным индексом слева у символа
детерминанта. Детерминанты А ,, А. не являются собственными функциями опера-
тора S , но из них можно сформировать линейные комбинации
Л3 =-т= \А, -А,) и Л4 =-j= [А, +А,), которые описывают
соответственно синглетное и триплетное чистые спиновые состояния [4, 13, 42].
Волновая функция основного состояния молекулы водорода. В однодетерми-
нантном приближении волновая функция основного состояния молекулы
водорода, когда оба электрона находятся на орбитали <р., один электрон имеет спин
а, а другой (3, выражается слэтеровским детерминантом:
tp,{l)a{l) <рЛ1Щ1)
<pI{2)a{2) <pi{2)(3{2)
~^<р1{1)<Р1{2){а{1)/3{2)-а(2)/3{1))
Электронная энергия молекулы в этом состоянии с учетом того, что
гамильтониан (1.9.1) не зависит от спиновых переменных имеет вид:
Z ft=/ rJ2
{а{1)(3{2)-а{2)р{1)\а{1)р{2Уа{2)р{1)).
Проинтегрируем по спиновым переменным, используя свойство ортонорми-
руемости спиновых функций (1.2.11, 1.2.12):
{а{1)/3(2)-а{2) /3{1)\а{1)/3{2)-а(2) /3{1)) =
{а{1)р{2)\а{1)р{2)) + {а{2) (3{1)\а{2) (3{1))
~{а{1)(3{2)\а{2) (3(1))-{а(2) (3{1)\а{1){3{2)) = 2.
Получаем
Л 1
1 JJ
47
E0=(<Pi{l)<Pi{2)\Y.h0{k)+^-\<p!{l)<p}{2)) =
ft=/ Г12
{<Pl(l)\h0{l){<P,{l)) + (<Pi{2)\h0{2\<pI{2))+ (1.9.12)
{9l{l)cpl{2)\~\cp}{l)<p}{2)).
Г12
Отметим, что в последнем выражении присутствует только кулоновский
интеграл, обменный интеграл отсутствует, поскольку электроны имеют разные
спины. Подставив в (1.9.12) выражение <pi из (1.9.11), значение энергии можно
выразить через матричные элементы от базисных функций. Уточнение
волновой функции и энергии основного состояния молекулы может быть
выполнено методом кофигурационного взаимодействия. С конфигурацией
основного состояния вследствие пространственной и спиновой симметрии
взаимодействует только конфигурация Л, (Рис. 1.9.1), поэтому уточненную функцию
Ч* основного состояния молекулы Нг можно искать в виде [49]:
Y = c}A}+c2A6,
используя для определения коэффициентов с . и с, , например, вариационный
принцип.
1.10 Теория функционала плотности
При численном решении уравнения Шредингера систем с большим
числом частиц возникают сложности, связанные с невозможностью
вычисления волновой функции с достаточной точностью и записью волновой
функции в цифровом виде в память компьютера (так называемая катастрофа
Ван Флека). Данные проблемы не могут быть решены посредством
увеличения точности расчёта или расширения памяти, так как в их основе заложен
экспоненциальный рост ошибок или экспоненциальный рост требуемого
объема памяти. Использование многочастичных волновых функций для
описания системы с числом электронов более 1000 неправомерно [51-53]. Путь
решения проблемы был найден в работах Вальтера Кона с сотрудниками, в
которых было показано, что электронная плотность основного состояния
атомно-молекулярной системы неявно определяет все ее свойства,
получаемые решением временного или стационарного уравнений Шредингера.
Иными словами, вся информация о многоэлектронной системе может быть
получена из электронной плотности основного состояния, зависящей всего от
трех переменных. Это достижение дало серьезный толчок для развития
квантовой химии и вычислительной физики сложных конденсированных систем.
Работы Кона были отмечены Нобелевской премией по химии в 1998г [52,53].
Первоначально теоремы Хоэнберга— Кона были сформулированы только
48
для основного состояния электронной подсистемы в отсутствие магнитного
поля. Они могут быть обобщены и для расчета возбуждённых состояний
электронов. Суть теории функционала плотности (ТФП) состоит в
использовании при описании атомно-молекулярных систем распределения
электронной плотности - свойства, которое измеряется в рентгеновской
кристаллографии. Электронная плотность - это функция, заданная в пространстве трех
измерений в отличие от волновой функции, заданной в Зи-мерном
пространстве (п -число электронов). Поэтому, если электронная плотность известна,
то методы функционала плотности более просты по сравнению с методами,
где описание идет на языке волновых функций.
В работах Хоэнберга— Кона оператор потенциальной энергии
рассматриваемой системы разделен на оператор электрон-электронного
взаимодействия U и оператор взаимодействия электронов с неподвижными ядрами
V, являющегося внешним для системы взаимодействующих электронов [52 -
55]. Тогда стационарное электронное уравнение Шредингера атомно-
молекулярной системы имеет вид
Н~.а =(T + V + U)<p =s со , (1.10.1)
->-" т Э.Ч ч /г ЭЛ ЭЛ ^ ЭЛ
здесь <р [г. ,г2,---г J - электронная волновая функция, Т- оператор
кинетической энергии. Для фиксированной конфигурации ядер (координаты
которых опущены) электронная плотность задается выражением
p{r)=n \<plJ,[r,r2,-i:n)(poj]\r,r2,-rn)dr2--drn . (1.10.2)
Энергия взаимодействия электронов с ядрами может быть выражена через
электронную плотность:
У(р)=( <РЭЛ\ У\срэд Y\V{r)p{?)d3r. (1.10.3)
Истоком ТФП явилась теория Томаса-Ферми, согласно которой
существует соответствие между потенциалом V(r), в котором движутся электроны и
распределением их электронной плотности. Это обстоятельство привело
авторов ТФП к гипотезе, что для любой электронной системы плотность электронов
основного состояния однозначно определяет эту систему и к построению
теории, в которой основной переменной является электронная плотность. В 1964г.
П. Хоэнбергем и В. Коном (ХК) была сформулирована основная лемма,
лежащая в основе методов функционала плотности.
Основная лемма ХК. Электронная плотность р~ (г) основного состояния
связанной системы взаимодействующих электронов, движущихся в некотором
внешнем потенциале V(r), однозначно определяет этот потенциал.
49
Доказательство.
Рассмотрим основное состояние системы и электронов с гамильтонианом
Я, =Т + U + V, V - внешний потенциал (состояние предполагается
невырожденным, в случае вырожденного основного состояния лемма верна для
каждого основного состояния). Пусть (р.- электронная волновая функция
основного состояния данной системы, £/- его энергия, P^i?) - электронная
плотность. Тогда из (1.10.1) - (1.10.3) следует
*/=(?/ |tf/M \<P}} = (<Pi \Т + У + и\<р,} =
{<Pi \VW,) + {<P, \T + U\9/} =
w " w " (1.10.4)
= \V{r)P(){r)d3r +{cpI \T + U\<p}).
Пусть существует внешний потенциал v(r)* V(r) + const, в котором движутся
и электронов, и эта система характеризуется такой же электронной плотностью
основного состояния />„(г). Следовательно, имеются два различных
электронных гамильтониана
Я, =T + U + V и Я, =T + U + v,
1эл 2эл
описывающих систему и электронов с одной и той же электронной плотностью
основного состояния pf (г). Пусть <р7 - волновая функция основного состоя-
ния гамильтониана Я_ , и <р2 &е <р ..
Поскольку состояние (р. невырождено, то принцип минимума Рэлея-Ритца для
Я. имеет вид
е1<(к<Р2 \Н,ЭЛ \<р2}, (1.10.5)
но Я. -Н2эл +V -v, с учетом этого правая часть (1.10.5) принимает вид:
{<Р2 \Н1ЭЯ \<Р2) = {<Р2 \Н2эл+У.-и\<р2} = ^
^г+^з \V-o\(p2} = e2+i(V-v)p0{f)d3r'
где е2 - энергия основного состояния системы с гамильтонианом Я_ .
50
Подставляя (1.10.6) в (1.10.5) получаем
ei<e2+!{V-v)p0(r)d3r. (1.10.7)
Состояние <р у не предполагается невырожденным, поэтому в записи принципа
минимума Рэлея-Ритца для Н2 , в выражении, аналогичном (1.10.5), знак
строгого неравенства нужно заменить на <, получаем
^Ц<Р, \Н2ЭЛ \<P}) = ei+l(v-V)p0(r)d3r . (1.10.8)
Складывая (1.10.7) и (1.10.8) приходим к противоречию
е1+е2<е1+е2.
Следовательно, предположение о существовании потенциала
v(r)^V(r) +const, дающего ту же самую плотность электронов основного
состояния р0 (г) является неверным.
Таким образом, плотность основного состояния р0 (г) определяет
полное число электронов п ( отсюда оператор кинетической энергиии электронов
и оператор электрон-электронного взаимодействия), и внешний потенциал
V(r). Следовательно, р» (г ) определяет гамильтониан системы Ни все
свойства системы, получаемые решением стационарного или временного уравнения
Шредингера. Если любая положительная функция р() (г) с регулярным
поведением, интеграл от которой равен целому положительному числу п ,
представляет электронную плотность основного состояния системы электронов,
движущихся в потенциале V(r), то такая плотность называется Р-представимой.
Вариационный принцип Хоэнберга-Кона. Энергия электронной
подсистемы, записанная как функционал электронной плотности, имеет минимум,
равный энергии основного состояния, достигающийся при равенстве пробной
электронной плотности р{г) плотности р() (?) основного состояния.
Действительно, рассмотрим систему п взаимодействующих электронов,
движущихся во внешнем потенциале V{r ). Электронная энергия основного
состояния атомно-молекулярной системы для фиксированной конфигурации ядер
может быть найдена из принципа минимума Рэлея — Ритца:
0 утэл эл^эл' л Ю9)
51
где <р - нормированная пробная электронная волновая функция. Но каждая
пробная функция в соответствии с (1.10.2) определяет некоторую пробную
плотность. Если <р равна волновой функции основного состояния (р( , то она
определяет плотность основного состояния р0(г), при этом энергия
основного состояния системы в соответствии с (1.10.4) равна
е0 =W{r)p0{r)d3r +[<р0 | T + U\<p0). (1.10.10)
Энергия как функционал электронной плотности на классе функций 7р имеет
вид
^v[p{r)]=W{r)p (r)d3r +F[p{r)l (1.10.11)
где
/>(')=» \fl„{r ,r2,-fn)<p M{f ,r2,-rn) dr2-dfn ,
F[p(r)] = (S I T + U\<p )- универсальный функционал, который
определяется внутренними свойствами системы: кинетической энергией
электронов и энергией их взаимодействия друг с другом, эта часть функционала
одинакова для всех систем, содержащих п электронов. Различие
функционалов £j/[p(^)] систем с и электронами друг от друга заключается в первом
слагаемом (1.10.11), описывающим взаимодействие электронов с внешним
полем. Очевидно
sv[p{r) \>£с>если р{г)*Р0{г)-
Это следует из соотношения между обычными функционалами энергии Рэлея
- Ритца от волновых функций
Ьэл ]'
что и доказывает вариационный принцип ХК.
Обычно из универсального функционала F[/)(F)] вычитают классическую
энергию кулоновского взаимодействия электронов
G[p{f)] = F[p(r)]-llp№p* 'drdr'.
2
г -г
Т.е. G [ р ] - часть универсального функционала, описывающего
гипотетическую систему, в которой электроны не взаимодействуют друг с другом.
52
Тогда энергия как функционал электронной плотности принимает вид
е [р ] = \V{f)p {r)d3r +LspV>py' 'drdr'+G[p ]. (1.10.12)
2 U -'
r -r
Минимизируя данный функционал можно определить энергию основного
состояния системы и плотность р Л?).
Уравнения Кона-Шэма. Представим функционал G [ р ] в виде суммы
двух слагаемых - кинетической энергии невзаимодействующих электронов
Г[/>] и остатка Е [р], который называется обменно-корреляционной
энергией и выражение (1.10.13) является ее определением
G[phTs[p]+Exc[p]. (1.10.13)
Запишем функционал энергии (1.10.12) с учетом выражения G [ р ] (1.10.13)
ev[p]4V(f)p(f)d3r^!^^drdr'+Ts[p]+Ejp]=
\г - г
= Ts[p] + IV(r)p{r)d3r +L^)PY ldrdr'+Exc[p]. (1.10.14)
F -F
Варьируя данный функционал энергии по р при условии сохранения числа
частиц приходим к уравнениям Кона-Шэма (КШ), описывающим движение
одного электрона в эффективном потенциале [55]:
где
0эфф№ = У^)+1, dr'+vxct?), (1.Ю.16)
53
5Е [р]
где v (г)-—— локальный обменно-корреляционный потенциал, зави-
хс Sp
сящий от полного распределения электронной плотности р (r)= £ Р/Ол •
Если в (1.10.16) пренебречь о (г), то уравнения КШ переходят в уравнения
Хартри. Как указывалось в 1.2, электронное уравнение Шредингера не может
быть решено методом разделения переменных вследствие наличия в
гамильтониане членов, описывающих взаимодействие электронов. Идея КШ
заключается в сведении многоэлектронной задачи к одноэлектронной путем введения
эффективного потенциалам . , (г), обеспечивающего электронную плотность
в гипотетической системе невзаимодействующих электронов, такую же как в
реальной системе взаимодействующих электронов. Таким образом, волновые
функции КШ обеспечивают истинную электронную плотность, которая
согласно основной лемме ХК определяет все свойства системы. Кроме этого, энергия
верхнего занятого уровня е ., отсчитанная от вакуумного нуля, совпадает с
потенциалом ионизации [56]. В отличие от метода Хартри-Фока, где наличие
электрон-электронного взаимодействия приводит к появлению кулоновского и
обменного операторов в одноэлектронных уравнениях, в уравнениях КШ
появляется обменно-корреляционный потенциал, учитывающий эффекты обмена и
корреляции. Обменно-корреляционная энергия и обменно-корреляционный
потенциал часто разбиваются на сумму обменной и корреляционной частей:
Ехс[р]=Ех[р} + Ес[р].
Орбитальная энергия в теории Хартри-Фока получается как частный случай
выражения энергии в теории функционала плотности с обменным интегралом
Е [ р ], равным обменному интегралу теории ХФ, и Е [ р ]= 0.
Схема расчета свойств атомно-молекулярной системы методом
функционала плотности состоит в следующем. Сначала задается некоторая
начальная плотность. По формуле (1.10.16) с учетом выражений обменно-
корреляционной энергии, некоторые из которых приведены ниже, вычисляется
потенциал о . . (г). Затем, путем решения уравнения (1.10.15) находятся од-
ноэлектронные волновые функции и энергии рассматриваемой системы.
Найденные волновые функции используются для вычисления уточненного
значения электронной плотности. Процедура повторяется, пока не выполнены
заданные критерии сходимости. На основе вычисленной плотности
рассчитываются свойства рассматриваемой системы.
Практическая пригодность теории функционала плотности заключается в
том, существуют ли достаточно простые приближения для универсального
функционала F[p] в формулировке ХК или обменно-корреляционной энергии
54
Е [ р ] в формулировке КШ, которые, в тоже время, обладали бы требуемой
точностью. Как оказалось, для обменно-корреляционной энергии можно найти
удачную аппроксимацию, которая и обуславливает успех теории Кона-Шэма в
практических применениях.
Приближения для обменно-корреляционной энергии Е [ р ]. Для
нахождения Е [р] применяют различные приближения: однородного
электронного газа; электронного газа почти постоянной плотности; электронного газа с
медленно меняющейся плотностью (градиентные и обобщенные градиентные
разложения) и гибридные методы.
Приближение локальной плотности. В приближении локальной
плотности (LDA - Local density appoximation) в качестве обменно-корреляционной
энергии на одну частицу выбирается таковая для однородного электронного
газа [52-54, 58]. При этом функционал обменно-корреляционной энергии
вычисляется как интеграл от обменно-корреляционной энергии е[ р{г)\ на одну
частицу однородного электронного газа в точке г (обычно величины, относящиеся
к одной частице, обозначаются малыми буквами, а ко всей системе -
большими):
^LDA[ph\exc[p(r)]p(r)dF,
хс
где е [/>(r)J в точке г зависит только от электронной плотности в данной
точке р{г). Обменная составляющая обменно-корреляционной энергии в
приближении LDA определяется формулой Дирака [57,58]:
ELxDA[p] = -Cxl P4/3(?)dr; eLD\[p{r)h-CxPI/3{r),CxJ-{£^/3.
Аналитическое выражение корреляционной составляющей обменно-
корреляционной энергии в приближении LDA может быть получено только в
предельных случаях. В пределе высоких плотностей получаем [26,58]:
£с = Aln{rs) + В + rs(C!n{rs)+ D).
A,B,C,D -константы, г - радиус эффективного объема приходящегося на один
4 з 1
электрон, определяется из условия — irrs = —.
3 р
В пределе низких плотностей
55
80 . Si .
3/2
Моделирование энергии однородного электронного газа методом Монте-
Карло, обеспечивающее точную плотность корреляционной энергии, было
выполнено в [59]. Для ее включения в ТФП необходимо аналитическое
выражение ес, интерполяционная формула для которого в локальном приближении
найдена в работе Vosko - Wilk - Nusair (VWN)[60]:
ec(rs,Q) = ec{rs.O)+ea{rs)
/(c)
./ ФУ
{lS4)+[ec(rSJhec(rs,0)]f(s)<;<
где
g = — - относительная спиновая поляризация. д = 0 соответствует pa-
Pa+Pp
венству плотностей а и /? электронов, д = 1, если р „ =0 и g = -l, если
р =0.
^ а
г - радиус эффективного объема приходящегося на один электрон, определя-
4 з 1
ется из условия —щ = — .
3 р
/(<
\{HrcY'3 + {l-cY'3-2]
■,4/3
2"/J-2
ес и еа - функционалы, параметризованные следующим образом
ФГ [ Х{х) Q {2х+е) Х{х0)
jc = a//7, Х(х) = х2 +£х+с, Q = ]4c-l2 .
(х-х0У
Х(х)
Q \2х + в
Параметры А,£,хд,с - константы, различные для ec(rs,0), ec(rs,l),
£a(rs)-
Модифицированная форма е / чпредложена Perdew и Wang [61]:
56
e%f=-2aplf + ax*)h,
1+-
\ 2 a \plX + fax2 + p3x3 + P4x3))'
где а, а,Р; - константы.
Отметим также работы Perdew-Zunger (PZ81) [62] и Cole-Perdew (CP) [63], в
которых вычислены выражения ес.
Ха метод представляет собой разновидность LDA метода, в котором
корреляционной составляющей обменно-корреляционной энергии пренебрегается, а ее
обменная составляющая вычисляется по формуле:
где первоначально полагалось а = 1, однако расчет показал, что а = — обеспе-
4
чивает лучшее согласование с экспериментом при расчете свойств атомно-
молекулярных систем.
Практика расчетов показала, что LDA дает энергии ионизации атомов, энергии
диссоциации молекул и энергии связи твердых тел с точностью 10 — 20%.
Точность расчета геометрической конфигурации молекул очень высока ~ 1 %.
Приближение LSDA (Local spin density approximation) - представляет
собой обобщение LDA для систем с неспаренными спинами. Если плотности
электронов со спинами а и /? не равны, то обменная энергия в приближении
LSDA вычисляется по формуле [64]:
Е™*[р]=-2"3Сх1[ра</3+Р,4/3)<1г;
Для систем с закрытыми оболочками приближения LDA и LSDA совпадают.
Квазилокальное приближение. В квазилокальном случае обменно-
корреляционная энергия е [г, р[г)J на одну частицу в точке г зависит от
плотности, близкой к точке г, то есть от плотности в точках г , близких к г:
Е [p]=\exc[>:,p\'r)\p{r)dr.
хс
Раскладывая р{ г ) в ряд вблизи г можно получить градиентное и обобщенное
градиентное приближение (generalized gradient approximation - GGA) обменно-
корреляционной энергии, где Е не зависят не только от электронной
хс
плотности, но и ее градиентов.
57
Perdew и Wang (PW86) добавили к обменной энергии приближения LSDA
градиентную поправку [65]
1/15 |Vp|
/xW86^eLDA{1 + ax2+bx4+cx6y
где х =1
J/3
Очень популярен градиентно коррелированный обменный функционал,
предложенный Веске в 1988 году [66]:
В88_ША_ Р1/Зх2
ьх ~ ьх У
где Л' =
„4/3
1 + бу sink х
, у - подгоночный параметр, обеспечивающий согласование
обменной энергии с известными данными по атомным системам.
Обменный функционал Perdew и Wang 91 [26] имеет вид
£PW91 _ £LDA
А
ионал rerdew и Wang У1 |/с
+ \a3+a4exp\-bx JJx
, где A = I + xaj sink
A + a^x
Широко используется градиентно коррелированный корреляционный
функционал Lee - Yang - Parr (LYP) [67]:
^LYP
Pa
(2t^+V2pa)+Pj3(
где А = -а-
a, l,d,c - параметры.
y = 2
2 , 2\
P2
,tw
vpi\
-vPff
Pa
у P = Pa+P/J>
В 1986 году Perdew была предложена градиентная поправка к LSDA
корреляционному функционалу [68,69]:
e™=e£DA+Ae™,
As.
P86_e<t>c(p)\Vp\2
/j . \5/3 / i \5/3
Ф = а4Ш> C(PhG]+ <h+G*+?4j
C{p)~7/6
1 + G5rs+G6r2+G7r3
58
где а и d■■ — постоянные.
Этот функционал был записан в другой форме Perdew и Wang в 1992 году, его
обозначение AePW92( ЛгРс92) [61].
Гибридные фунщионалы. Гибридный подход к конструированию обменно-
корреляционных функционалов был введен Веске [70]. В гибридных методах
обменная энергия рассчитывается с привлечением точного результата,
полученного методом Хартри — Фока (HF), и обменно-корреляционная энергия
имеет вид:
Ehybrid fhf+c EDFT
Из гибридных методов отметим метод B3LYP (Becke, Lee , Yang, Parr) [70,71],
согласно которому второе слагаемое в последней формуле представляет собой
трехпараметрический функционал:
EB3LYP=ELDA +Co(EHF_ELDA)+CxAEBS8+EmN3 ^LYP __ ^).п
осредством изменения коэффициентов «с» можно осуществлять различное
смешивание входящих в данную формулу функционалов. Заметим, что многие
описанные функционалы содержат подгоночные параметры, определяемые из
эксперимента. Однако, их «полуэмпиричность» существенно отличается от
таковой в полуэмпирических методах квантовой химии, где пренебрегается
многими двухэлектронными интегралами и часть интегралов заменяется
параметрами, оцененными из эксперимента. Оценить точность методов функционала
плотности практически невозможно, необходимо рассчитывать свойства
простых систем, сравнивать их с экспериментом и на основании этого
прогнозировать точность расчета свойств исследуемой системы.
1.11 Полуэмпирические методы квантовой химии
Орбитальные энергии и коэффициенты разложения МО по базисным
функциям в ограниченном методе Хартри-Фока-Рутаана вычисляются из
решения системы уравнений (см. раздел 1.3)
ЕС (F -e (l)S ) = 0. (1.11.1)
Матричные элементы фокиана метода ХФР имеют вид
59
м
z
у,s=i
(*;ы*;ы|^ы*,ы)- (,-п-2)
Решение системы ХФР сопряжено со значительными вычислительными
трудностями, связанными в большей степени с вычислением матричных элементов
оператора Фока. Методы квантовой химии решения этого уравнения делятся
на а)неэмпирические, б) полуэмпирические, в)метод функционала плотности,
основанные на приближении Борна-Оппенгеймера и методе МО ИКАО ХФР.
Неэмпирические методы основаны на точном решении уравнений ХФР,
которые не включают никаких экспериментальных параметров, кроме
фундаментальных физических постоянных. В полуэмпирических методах часть
интегралов, входящих в систему уравнений ХФР, заменяется параметрами, значения
которых вычисляются из эксперимента. Существующие полуэмпирические
методы можно разделить на две группы [72,73 ]. К первой относятся методы Мал-
ликена-Вольфсберга-Гельмгольца (МВГ) [73,74] и различные варианты метода
Хюккеля. В этих методах всеми двухэлектронными матричными элементами в
системе ХФР пренебрегается, а одноэлектронный оператор ho{l) заменяется
на некоторый эффективный оператор ИЭ(^, матричные элементы которого
вычисляются из эксперимента. Вторая группа методов базируется на
приближении нулевого дифференциального перекрывания, введенному в работах
[75,76].
Приближение нулевого дифференциального перекрывания (НДП). В основе
приближения нулевого дифференциального перекрывания лежит
предположение, что две различные атомные орбитали % их, нигде в пространстве не
перекрываются, т.е.
x\l(r)xv(r)dr^X\l(F)xv(F)dV = 0, M*v. (1.11.3)
Очевидно, что, если выполняется это условие, то интегралы перекрывания
между различными базисными АО обращаются в нуль
SMV=iXM{r)xv(f)dr=0, n*v. (1.1I.4)
Обратное утверждение, вообще говоря, неверно, из (1.11.14) не следует (1.1.13).
В некоторых случаях расчеты МО ЛКАО проводятся в приближении (1.11.4),
такое приближение называется пренебрежением перекрыванием в отличие от
приближения дифференциальным перекрыванием.
Запишем диагональный матричный элемент фокиана (1.11.2):
60
xl(?i)x*rif2)\j- х^,)х^2)
M
z
У,ё=1
-L2{ x\ hh], Ы I7- %g (f7k/y Ы
у 5
В приближении НДП второе слагаемое не равно нулю только при у = 8, а
третье при у = 6 = /и, таким образом, в приближении НДП
-!\^Ы*аЫ ^- *^Ц,Ы /V
Рассмотрим недиагональный матричный элемент фокиана (1.11.2)
+
*1 (Р/К if2) \v~\x„ ifi)хя if2))-
F =h +
M
z
y,S=l
" j( X\ ifjVy if2)\f~\ Xs {r,)xy (r2)
у8
, /u^v.
Первое слагаемое под знаком суммы в приближении НДП равно нулю,
поскольку /j*v. Второе слагаемое под знаком суммы не равно нулю, если S = fj и
y-v , т.е. от суммы в приближении НДП остается I член, для недиагонального
матричного элемента получаем:
/ 2
F =h --(/tv 1—1 fiv )P .
r12
Таким образом, используя приближение НДП для двухэлектронных интегралов,
приходим к системе уравнений ХФР:
М
1С (F -е Ш ) = 0,
v=l ^
где
Fllu=hLlA(w\—\w)Pu+hh,y\^-\,y)Pyy, (1.П.5)
ИИ
ИИ 2
ИИ
12
УУ
61
F =h -±(uv\—\fiv)P . (1.116)
12
Приближение НДП лежит в основе методов полного пренебрежения
дифференциальным перекрыванием - ППДП { или complete neglect of the differencial
overlap - CMX?)[77,78], полного пренебрежения двухцентровым
дифференциальным перекрыванием ПДДП (Neglecting of Diatomic Differential Overlap - NDDO)
[78-80], частичного пренебрежения дифференциальным перекрыванием ЧПДП
(intermediate neglecting of the differencial overlap - INDO)[80,8l],
модифицированного метода частичного пренебрежения дифференциальным перекрыванием
МЧПДП (modified intermediate neglecting of the differencial overlap - MINDO) [82
- 85], Modified Neglecting of diatomic overlap MNDO [86], AMI187-91], РМЗЩ-
91] и др.
Метод ППДП(СНИО) - метод полного пренебрежения дифференциальным
перекрыванием (ППДП) ( или complete neglect of the differencial overlap - CNDO).
В этом методе для всех пар атомных орбиталей, в том числе и для
принадлежащих одному и тому же атому вводится приближение НДП. Для записи
системы уравнений ХФР в этом приближении выделим в диагональном матричном
элементе (I.I1.5) члены, относящиеся к конкретному атому. При вычислении
диагонального матричного элемента фокиана (1.11.5)
^=(м')+уК-*„)}
/</'
обозначим м - номер атома, на котором центрирована функция х > матричный
элемент которой вычисляется. Рассмотрим оператор hAl) - первое слагаемое
оператора Фока (см.(1.2.18)).
ЪЖ>~*21-1
2те и . 1а
и разобьем потенциальную энергию взаимодействия электрона I с ядрами на
два слагаемых, выделив взаимодействие электрона 1 с ядром атома м, атомной
орбиталью которого является функция % , в отдельное слагаемое:
М'Ь-^-^-I^. (I.II.7)
В диагональном матричном элементе фокиана (1.11.5) в последней сумме по у,
также выделим слагаемые с АО, принадлежащей атому м ( напомним, что в
разложении МО по базисным АО, приведенным в 1.3, индексы /u,v,y,S незави-
62
симо пробегают значения от 1 до М, поэтому возможны ситуации, когда
атомная орбиталь х является атомной орбиталью атома м):
У ' 12 Уем Г12 У*м 12
(1.11.8)
Подставляя (1.11.7) и (1.11.8) в выражение диагонального матричного элемента
фокиана (1.11.5) получаем
РЧ*
2т г у г У 2
те 1м Уем 12
а*м > 1а
* / У** гi2 Z fi2
(1.11.11)
Первое слагаемое (1.11.11) представляет собой орбитальную энергию е
атома м, которая может быть найдена из атомных спектров. В приближении
ППДП/1 первое слагаемое заменяется потенциалом ионизации электрона с
атомной орбитали % с обратным знаком, что следует из теоремы Купманса.
Второе слагаемое (1.1
>рбитали х
него обозначение
I) описывает взаимодействие электрона на
атомной орбитали х со всеми со всеми ядрами кроме ядра атома м. Введем для
-г
аФм г 1а
X,
М
= I К.
эя-яд
а/л
(1.11.12)
Третье слагаемое (1.11.11) описывает электронное отталкивание
электрона на АО х от электронов, АО которых находятся на ядрах, отличных от м.
Этот член перегруппируем, выделив в нем члены, относящиеся к
определенному атому с индексом а ( а&м), идентичным индексу а (1.11.11), (1.11.12).
Тогда третье слагаемое (1.11.11) с учетом комментариев, следующих за
формулой (1.11.13), можно записать в виде
I (»v\f-\vv)ryy= S v™~™Pa,
2
meV™-9Jt={vy\—\vy),yea,pa= IP.
(1.1I.I3)
12
yea
63
Правая часть (1.1 I.I 3) равна левой при условии, что все двухэлектронные инте-
2
гралы (/иу I \fiy) одинаковы для каждой пары орбиталей у,/л уеа,/иел1,
г
Г12
вне зависимости от того, каким является состояние у атома а (например, 2s ,
2рх, 2ру или 2pz , если рассматривается валентный электрон атома второго
периода ). Только при этом для пар орбиталей у,/и yea, ftем имеем
(^2s\^r2s)Pr2sy2s +
\МГ2Р* \г12ГУ2рЧРУ2рхУ2рх +'"
= {МУ\ 1/'7)м° +Р +---\ = (МУ\ \му)Ра-
r12 I ¥2s r2s Г2рх Г2рх ) ^' ' rl2 V 1Уа
(1.11.14)
2
В методе ППДП при вычислении данного матричного элемента (fj у \ \fj у)
г12
р- атомные орбитали заменяются .у-орбиталями, что сохраняет инвариантность
интегралов кулоновского отталкивания для пар атомных орбиталей одного и
того же атома относительно поворота системы координат (более подробно см.
[6,4, 13]).В соответствии с данными раздела 1.5 величину р = Z Руу можно
yea
интерпретировать как величину, характеризующую электронную плотность на
атоме а.
Сумма второго и третьего слагаемого (1.11.11) определяют полное
взаимодействие выделенного электрона атома,и с электронами и ядрами других
атомов.
Последнее слагаемое (1.11.11) связано с наличием обменной поправки к
движению рассматриваемого электрона (см. комментарии к формуле (1.2.14)).
Объединяя второе и третье слагаемое (1.11.11) в один член с учетом
(1.11.12) и (I.I I.13) запишем диагональный матичный элемент фокиана в
методе ППДП
64
F ={X
-,2_
2 m,
Vl-^—+ Z UyV~Xydr2
1м уем
■l-{мм I—I мм )PPP + Z (C"Pe + *&-").
\ * "e 1m Г&М 12 j
1
P Vs
2 Ml1
Z ferVa+^r0)-
Недиагональный матричный элемент фокиана (1.11.6) в приближении
НДП для двухэлектронных интегралов
= /? —( и v I \ uv )Р =
f.1 V J.IV 7 Г ' fiV
где А = ( у *
^ -~У™~ЭЛР ,
pv 2 Pv
h2 о М7 е2
2те а г1а
уЭЛ-ЭЛ
энергия отталкивания электронов, находящихся на орбиталях // иг.
Причем, поскольку все двухэлектронные интегралы {{л v | \/и v) одинаковы
Г12
для каждой пары орбиталей v./л, ve«, /нем, то У^~эл = У^эл.
Выделим в сумме Z—-— слагаемые, относящиеся к центрам ммп орбиталей
а г1а
X, И *v :
//к \л li
м
Z^/v с
2т,
-vf-Z^
=U'
+U*,
a la
2 г, „2
й v2 Z„e Z^e
2m„ r, г
/jW
/«
*., +
W 7 J
a#M,n 1 la
65
Последним членом этого выражения пренебрегают, так как он отвечает
взаимодействию трех центров (м, и, а), которые обычно малы. Тогда
F =ix*
й2 ^2 2Me2 Zne2
-V
2те ' гы rln
)Л2(^\~\^К* =
J.IV 2 /<V
При расчете одноэлектронного матричного элемента h приближение НДП
не используется вследствие наличия в операторе А„ (/) оператора V/.
Матричный элемент h заменяется параметром, значение которого определяется
только природой атомов м и и. которым принадлежат АО % и X и не
зависит от вида конкретных АО:
где Р - эмпирический параметр,
S = \Х* | X )> - интеграл перекрывания, при вычислении которого НДП
не используется. Поскольку АО одного и того же атома ортогональны, то
h -0, если //, v - орбитали одного центра.
Таким образом, в приближении ППДП:
1. В системе уравнений ХФР (1.11.1) полагают & -S
2. При расчете двухэлектронных интегралов, входящих в диагональный
матричный элемент фокиана, НДП применяется для любой пары АО, как
принадлежащих различным атомам, так и одному и тому же атому.
3. При расчете диагонального матричного элемента фокиана разные двухцен-
2
тровые интегралы (/<у | |/'У) заменяются одним и тем же интегралом для
г12
каждой пары атомов, см. (1.11.14).
4. При расчете недиагонального матричного элемента фокиана приближение
НДП вводится только для двухэлектронных интегралов, одноэлектронный
матричный элемент заменяется выражением
Система уравнений ХФР в приближении ППДП имеет вид
м
X С (F -е Ш ) = 0,
где
66
F
h2 ? Z e2 * p2
e f Im У&Л1 f 12
% p
-Lp уэя-эл у (уэл-эл уэд-яд]
* И о.фм
F =Pu„S --У™~ЭЛР . (1.1 1.15)
Применяется два подхода к решению этой системы. В первом случае
величины V™~0JI, V™~ , fiMn и первое слагаемое F заменяются
параметрами, оцененными из экспериментальных данных. При этом используется
валентное приближение, и заряды ядер заменяются эффективными зарядами
остова. Обычно параметризация проводится таким образом, чтобы
рассчитываемые в рамках метода свойства молекул хорошо воспроизводили
соответствующие экспериментальные свойства для выбранного класса химических
соединений. Удачным подбором параметров удается отчасти скомпенсировать
погрешности метода. Поскольку число классов соединений и число рассчитываемых
свойств достаточно велико, существует большое число параметризаций [73],
каждая из которых применима только для расчета определенных свойств.
Существуют различные варианты метода ППДП - ППДП/1, ППДП/2,
различающиеся выбором параметров.
Второй подход связан с вычислением интегралов, входящих в (1.11.15),
отличие от неэмпирических методов заключается только в уменьшении числа
интегралов, входящих в уравнения Хартри-Фока-Рутаана. При этом расчет
усложняется, и часто приводит к результатам, более худшим, чем первый подход,
поскольку введение экспериментальных параметров может скоррелировать
погрешности, обусловленные приближением НДП.
Другие методы, основанные на приближении НДП.
ЧПДП и МЧПДП (INDO и MINDO) . В методе частичного пренебрежения
дифференциальным перекрыванием ЧПДП ( INDO — intermediate neglecting of
the differencial overlap) нулевое дифференциальное перекрывание
Xu {r)%v (r) dr = zfJ (#0Zv 00 dV = 0, ju*v
вводится только для атомных орбиталей, принадлежащих различным атомам,
т.е. условие р * v дополняется условием р, v - АО различных атомов, поэтому
формулы матричных элементов фокиана дополняются слагаемыми,
учитывающими двухэлектронные интегралы, построенными на АО, принадлежащих
одному атому. Методы ППДП и ЧПДП плохо воспроизводят теплоты атомизиции,
они неприменимы для построения поверхностей потенциальной энергии и
изучения механизмов реакций. Для замкнутых оболочек более предпочтителен
метод ППДП, а для открытых ЧПДП. Параметризация
модифицированного метода частичного пренебрежения дифференциальным перекрыванием
МЧПДП (MINDO -modified intermediate neglecting of the differencial overlap)
позволяет получить правильные значения теплот образования и геометрии моле-
67
кул для широкого класса стандартных соединений. Наиболее распространен
метод MINDO/3. Метод достаточно хорошо воспроизводит поверхность
потенциальной энергии вблизи минимумов, что позволяет рассчитывать силовые
постоянные и ИК спектры. Однако, он дает завышенные на 6 - 8° значения
валентных углов, переоценивает стабильность малых циклов, плохо работает для
описания свойств систем с водородной связью [4].
MNDO, AMI, РМЗ. Метод MNDO (Modified Neglecting of diatomic overlap)
, разработанный также как MINDO группой Дыоара основан на более строгом
приближении для расчета одноэлектронных и двухэлектронных интегралов. В
этом методе все интегралы рассчитываются с правильными АО, а не
аппроксимируются интегралами от s - орбиталей. Эти методы требуют больших
ресурсов ЭВМ и большего машинного времени. Метод MNDO является дальнейшим
развитием метода MINDO/3. Позволяет проводить качественные расчеты
электронной и атомной структур органических молекул, содержащих атомы I -й и 2-
й главных подгрупп (но не атомов переходных элементов). Этот метод
позволяет получать хорошие результаты для больших органических молекул при
расчетах электронных характеристик системы и теплот образования. Дальнейшее
усовершенствование метода MNDO отразилось в методе AM](Austm model l -
по названию Аустина - месторасположения группы Дыоара в Техасском
университете, автора метода). Это один из наиболее точных методов. Используется
для органических молекул, содержащих элементы из главных подгрупп 1 и 2
групп периодической системы. Возможно, этот метод позволяет получать более
качественные результаты, по сравнению с методом MNDO, для молекул,
содержащих как азот, так и кислород, особенно при описании водородных связей.
Вычисляет электронную структуру, оптимизирует геометрию, рассчитывает
полную энергию и теплоты образования.
Метод РМЗ является отличается от AMI только величинами параметров.
Параметры для РМЗ были получены сравнением большого числа
экспериментальных молекулярных характеристик с расчетными результатами.
Параметризация РМЗ позволяет рассчитывать свойства структур, содержащих переходные
металлы. Методы AMI и РМЗ считаются наиболее надежными из всех
полуэмпирических методов.
Полуэмпирические методы не всегда обеспечивают стабильность в
описании одних и тех же свойств даже в рядах родственных систем, причем, в
рамках самих этих схем затруднительно понять причину и предсказать те
случаи, где можно ожидать значительного расхождения с экспериментом.
68
L
Сравнительная характеристика полуэмпирических методов расчета по
данным монографии [4] определяется следующей таблицей.
Метод
Хорошо
воспроизводимые
свойства
Плохо
воспроизводимые
свойства
CNDO/2
Дипольные моменты,
длины связей, валентные
углы, силовые
константы, ЯМР
Теплоты образования,
потенциал ионизации,
электронное сродство,
спектр
CNDO/S
Спектр
Теплоты образования,
геометрия
INDO
Спиновые плотности,
константы сверхтонких
взаимодействий,
геометрия
Теплоты образования,
потенциал ионизации,
электронное сродство,
спектр
MINDO/3
Теплоты образования,
потенциал ионизации,
длины связей
Спектр,
связь
водородная
MNDO
Теплоты образования,
геометрия
Спектр,
связь
водородная
AMI
Теплоты образования,
геометрия
Спектр
Рекомендации об использовании полуэмпирических методов на
Вебстранице [92] немного отличаются от данных этой таблицы:
Метод CNDO используется для расчетов основного состояния электронных
характеристик систем с открытой и закрытой оболочками, оптимизации
геометрии и полной энергии.
Метод INDO позволяет проводить расчет основного состояния систем с
открытой и закрытой оболочками, оптимизации геометрии и полной энергии.
Метод MINDO/3 (Modified INDO, version 3, улучшенный метод INDO позволяет
получать хорошие результаты для больших органических молекул при расчетах
основного состояния систем с открытой и закрытой оболочками, оптимизации
геометрии и полной энергии.
Метод MNDO позволяет проводить качественные расчеты электронной и
атомной структур органических молекул, содержащих атомы 1-й и 2-й главных
подгрупп (но не атомов переходных элементов), дает хорошие результаты для
больших органических молекул при расчетах электронных характеристик
системы и теплот образования.
Метод AMI используется для органических молекул, содержащих элементы из
главных подгрупп 1 и 2 групп периодической системы. Возможно, позволяет
получать более качественные результаты, по сравнению с методом MNDO, для
молекул, содержащих как азот, так и кислород. Вычисляет электронную
структуру, оптимизирует геометрию, рассчитывает полную энергию и теплоты
образования.
69
РМЗ первоначально предназначался для расчета органических молекул, но
потом он был также параметризован и для ряда других групп элементов, в
частности - и для переходных металлов.
ZINDO/1 позволяет вычислять энергетику и геометрию молекул, содержащих
переходные металлы.
Метод ZINDO/S полезен для прогнозирования УФ и видимых спектров, но не
пригоден для оптимизации геометрии или молекулярной динамики.
Описанные в разделах 1.2 - 1.11 методы являются методами решения эле-
тронного уравнения Шредингера (1.1.5) и позволяют найти электронную
энергию и электронную волновую функцию молекулы. В соответствии с
приближением Борна-Оппенгеймера (раздел 1.1) электронная энергия молекулы в данном
состоянии как функция конфигурации ядер представляет собой потенциальную
энергию подсистемы ядер.
1.12 Поверхность потенциальной энергии многоатомной молекулы
Приближение Борна-Оппенгеймера позволяет разделить электронное и
ядерное движения в молекулах и лежит в основе понятия поверхности
потенциальной энергии многоатомной молекулы. Поверхностью потенциальной
энергии молекулы называется зависимость потенциального поля, в котором
движутся ядра, от всех независимых геометрических координат молекулы:
U(R) = U[ RrR2.-RK ), (1.12.1)
К = ЗМ-б для нелинейной многоатомной молекулы, К-ЗМ-5 для
двухатомных и линейных многоатомных молекул, М-число ядер.
Поверхность потенциальной энергии двухатомной молекулы U{r)
представляет функцию одной переменной - расстояния между ядрами R и называется
потенциальной кривой. Для трехатомной молекулы - выражению (1.12.1)
соответствует функция трех переменных U = Uy-j ,г2,ву.
При численном решении электронного уравнения Шредингера находится
конфигурация молекулы, соответствующая минимуму электронной энергии
молекулы, т.е. Еэд„ - значению электронной энергии в стационарной точке.
Затем изменением межъядерных расстояний и валентных углов вблизи
стационарной точки находится электронная энергия молекулы как функция
конфигурации ядер sn{R), и, наконец, Un{R) по формуле (1.1.6). Знание Un (r)
позволяет решить колебательное уравнение Шредингера (1.1.7), найти колебатель-
70
ные частоты, силовые постоянные, колебательные волновые функции,
рассчитать интенсивность ИК и рамановских спектров.
1.13. Классификация стационарных точек на поверхности потенциальной
энергии. Путь химической реакции
Каждое квантовое состояние системы (основное или возбужденное)
характеризуется своей поверхностью потенциальной энергии ППЭ
U(R., R„ ),K = 3M-6 для нелинейной многоатомной молекулы.
К-ЗМ-5 для двухатомных и линейных многоатомных молекул, М-число
ядер.
Особое значение имеют стационарные точки ППЭ, в которых значения
всех производных dU/dR, равны нулю. Стационарная точка может быть точкой
минимума (локального или глобального), точкой максимума (локального или
глобального) или седловой точкой.
Вблизи стационарной точки ППЭ с хорошей степенью точности предста-
вима многомерным параболоидом:
2
U(R,, RK) = -YI 8 U R.R.. (1.13.1)
• чJ
Чтобы определить вид поверхности потенциальной энергии в стационарной
точке необходимо исследовать матриц}' Гессе вторых частных производных
потенциальной функции (гессиан)
г.. -s s2(J .
'•J ^dR.dR.
' J
Путем эквивалентного преобразования координат
Q. =2>..Я. (1.13.2)
матриц}' Гессе можно привести к диагональному вид}' (r(i,j) = 0, при i* j).
При этом возможны следующие случаи:
1. Все диагональные элементы положительны, смещение из стационарной
точки вдоль любого направления приводит к увеличению значения
потенциальной энергии (стационарная точка — минимум). Наличие на ППЭ
нескольких минимумов говорит о существовании геометрических или
инверсионных изомеров рассматриваемой молекулы. Переход одной
изомерной формы в другую осуществляется вдоль пути реакции,
проходящего через соответствующую этому переходу седловую точку.
2. Все диагональные элементы отрицательны, стационарная точка -
максимум.
3. Наличие одного или нескольких различных знаков среди диагональных
элементов Гессиана свидетельствует о том, что при смещении из
стационарной точки существуют как направления возрастания
71
потенциальной энергии, так и направления ее убывания. Стационарная
точка, в которой Гессиан имеет только одно отрицательное собственное
значение, называется седловой точкой 1-го порядка (в дальнейшем просто
седловая точка). Эта точка является максимумом вдоль направления
отрицательного собственного значения Гессиана и минимумом вдоль
направлений остальных (положительных) собственных значений этой
матрицы. Стационарные точки, для которых среди диагональных элементов
Гессиана имеется 2 ( или более) отрицательных значения не имеют
химического смысла, система избегает попадания в эти области.
К какому из трех случаев принадлежит найденная стационарная точка
можно установить, проанализировав значения частот нормальных колебаний. Для
расчета частот нормальных колебаний используется теория малых колебаний.
Если преобразование (1.13.2) диагонализует выражение потенциальной и
кинетической энергии многоатомной молекулы:
U(QJ,-Qn) = ^k.QJ!,
2
T(Qr- e^ylm.e,,
z i
то координаты Qj называются нормальными координатами, энергия молекулы
вблизи стационарной точки в нормальных координатах равна сумме энергий
независимых гармонических осцилляторов
/ ( 2 )
H = T + U = ±T m.Qi+k.Q? (1.13.3)
* i
\ )
\m.
с частотами нормальных колебаний со. - 2nv. I—-.
V I
Очевидно, что
1. В случае 1 все &, положительны (все частоты действительны - локальный
минимум).
2. В случае 2 все k отрицательны (все частоты мнимые-локальный максимум).
3. В случае 3 имеются к, > 0 и к,, < 0, часть частот действительна, часть частот
мнимая.
Найденная в ходе оптимизации структура является переходным
состоянием (см.ниже), если среди частот ее нормальных колебаний имеется одна (и
только одна) мнимая частота, в этом случае через седловую точку можно
однозначно попасть к исходным реагентам и продуктам.
После завершения оптимизации геометрии, необходимо провести расчет частот
нормальных колебаний, чтобы проверить, действительно ли найденная в ходе
оптимизации структура является локальным минимумом или переходным
состоянием.
Путь химической реакции. В терминах поверхности потенциальной
энергии химическая реакция означает переход молекулярной системы из одного
72
минимума в другой. Путь, соответствующий минимальному изменению
энергии системы при таком переходе называется путем реакции. Путь реакции
всегда проходит через седловую точку на поверхности потенциальной энергии (см.
также раздел 2.10). Среди множества траекторий перемещения по ППЭ из
одного минимума в другой именно эта траектория будет обладать минимальной
высотой энергетического барьера. Длина дуги на пути реакции, взятая с
соответствующим знаком и отсчитываемая от седловои точки до текущей точки на
пути реакции называется координатой реакции данной текущей точки.
Конфигурацию системы, соответствующую седловои точке на пути реакции называют
переходным состоянием, ей соответствует координата реакции, равная 0. В
программе Gaussian принимается, что если текущая точка смещена от седловои
в сторону исходных веществ, то координата реакции отрицательна, в
противном случае - положительна. Сечение ППЭ по координате реакции называют
энергетическим профилем химической реакции. По нему можно оценить
высоту барьера, который необходимо преодолеть при осуществлении как прямой,
так и обратной реакции, т.е. оценить энергии активации этих реакций.
Очевидно, что в простейшем случае, энергетический профиль реакции имеет вид,
представленный на рис. 1.13.1. При таком описании не учитывается роль
кинетической энергии системы, однако такая модель элементарного акта
химической реакции оказывается вполне пригодной для получения качественных
выводов. В принципе, построив ППЭ системы, можно провести расчет
динамических траекторий реакции, мы не останавливаемся на вопросах, связанных с
решением динамической задачи.
к -
Q- / \
О) . / \
I / \
координата реакции
Рис. 1.13.1. Энергетический профиль реакции вдоль координаты реакг^ии
1Ъ
1.14 Энергия нулевых колебаний (ZPE)
В гармоническом приближении колебательная энергия молекулы
определяется по формуле
Е= I hv .( v,+- ),
где v .- гармонические частоты 1-го нормального колебания, v,-
колебательные квантовые числа, h - постоянная Планка, К - число нормальных
колебаний. Zero point energy (ZPE) - добавка к электронной энергии молекулы
энергии нулевых колебаний молекулы, не исчезающей при О К. Таким образом,
учет ZPE дает более точное значение полной энергии молекулы при О К.
Значение ZPE определяется по формуле:
ZPE = -hYvei, (1.14.1)
где ]>V . - сумма частот всех нормальных колебаний многоатомной молекулы.
Выражение (1.14.1) не учитывает ангармонические эффекты. Влияние
энгармонизма на ZPE для двухатомной молекулы, можно оценить из выражения
колебательного терма, имеющего в данном случае вид:
E{v)=hve[ v + !)-AveJte(v + !) , (1-14.2)
где ve - гармоническая частота колебаний, v х - постоянная
ангармоничности, v - колебательное квантовое число.
Отсюда
ZPE = ~hv --hv x . (1.14.3)
2 е ^ е е х '
Таким образом неучет энгармонизма приводит к ошибке вычислений в ZPE,
- К
равной —h v х .
к 4 e e
1.15 Сумма по состояниям системы
Решение электронного уравнения Шредингера с учетом ZPE позволяет
предсказать полную энергию молекулы Е при при О К. Чтобы предсказать
полную энергию молекулы при более высоких температурах к Е
необходимо добавить вклады поступательного, вращательного и колебательного
движений, сумма которых называется термической добавкой к Е . Вычисление
J v r эли
данных вкладов и расчет термодинамических свойств молекулярных газов,
проводят на основе их связи с суммой по состояниям. Сумма по состояниям
отдельной молекулы обычно обозначается Q, сумма по состояниям всей системы,
74
состоящей из большого числа молекул - Z. Сумма по состояниям отдельной
молекулы Q вычисляется по формуле:
е
и_
Q = Ze kT , (1.15.1)
и
где к — постоянная Больцмана, Т- абсолютная температура, п - совокупность
квантовых чисел, описывающих состояние молекулы, е - энергии молекулы в
этих состояниях (отдельные энергии могут быть одинаковы), которые в
адиабатическом приближении могут быть представлены в виде:
+ е +е , (1.15.2)
кол врспц v '
е - энергии поступательного, электронного, коле-
вращ г J r
бательного и вращательного движений молекулы в состоянии п. Три последних
слагаемых в (1.15.2)-характеристики внутренних движений молекулы.
Статистическая сумма обладает свойством мультипликативности. А
именно, если энергия системы может быть представлена в виде суммы
нескольких слагаемых Е =E]+E2+---Ejl, то статистическая сумма этой
системы равна произведению соответствующих статистических сумм:
Z =ZXZ2-Zk . (1.15.3)
В соответствии с (1.15.2) и (1.15.3) статистическая сумма молекулы может быть
представлена в виде:
е=еиос«а. o-is.4)
где
Qi^QwUKonQepaur (1-15.5)
- сумма по состояниям внутренних движений молекулы.
При рассмотрении идеального молекулярного газа, состоящего из N молекул
одного сорта, значение уровня энергии всей системы равно сумме энергий
отдельных молекул е ,е ,---е :
nl п2 nN
£„=£ +£ Н + е ,
" п\ П2 ид'
здесь одинаковые значения уровней энергии не объединены в одно слагаемое.
Статистическая сумма всей системы
75
+£■ +■•£■
"1 «2 "N
Z =— Те АГ
«L "2 "/У
■ и/ «2 "/V
=—Q,-Qo—Qb,=—QN. (I.15.6)
где gm- - кратность вырождения уровня еп}.
Учтено, что газ состоит из одинаковых молекул Q. = 6? ~"'Q\i и чт0 со~
стояния, полученные перестановкой частиц неразличимы. Число различных
состояний будет в N! раз меньше, N - число молекул газа.
Знание суммы по состояниям системы позволяет вычислить все основные
термодинамические функции состояния системы.
1.16. Термодинамические функции состояния
К основным термодинамическим функциям состояния относятся
внутренняя энергия, свободная энергия Гельмгольца, энтропия, энтальпия,
свободная энергия Гиббса. Все эти величины, кроме энтропии имеют размерность
энергии, энтропия имеет размерность энергия/ температура.
Внутренняя энергия системы - это разность между полной энергией
системы и суммой энергии движения системы как целого и потенциальной энергии
системы в поле внешних сил ( т.е. это энергия поступательного и
вращательного движения молекул, энергия внутримолекулярных колебаний атомов и
атомных групп, энергия движения электронов в атомах и молекулах, внутриядерная
энергия, энергия межмолекулярных колебаний и др.).
Согласно первому началу термодинамики, выражающему закон
сохранении энергии при тепловых процессах, изменение внутренней энергии системы
dE равно количеству сообщаемой системе теплоты 8Q минус количество
работы 8W, совершаемой системой:
dE=8Q-8WvjweQ = dE+SW, (I.16.1)
Свободная энергия Гельмгольца. Внутренняя энергия условно делится на
свободную и связанную:
E^F + T-S. (1.16.2)
Свободная составляющая F внутренней энергии называется свободной
энергией Гелъмгольца. При изотермических процессах работа совершается системой
не за счет убыли внутренней энергии (как при адиабатных процессах), а за счет
76
убыли свободной энергии F. Величина Т ■ S называется связанной энергией, Т
- абсолютная температура, S - энтропия. В изотермическом процессе все
изменение внутренней энергии нельзя превратить в работу, часть этого изменения
TdS теряется в виде теплоты (см. ниже (1.16.4)).
Энтропия. Статистический смысл энтропии заключается в том, что S
является мерой вероятности состояния системы
S = k-lnw, (1.16.3)
где w - термодинамическая вероятность состояния, определяемая числом
микросостояний, реализующих данное макросостояние, поэтому энтропия является
мерой хаотичности и неупорядоченности системы, к - постоянная Больцмана.
Замкнутая система самопроизвольно переходит от менее вероятных состояний
к более вероятным, что находит отражение в законе возрастания энтропии.
Равновесие характеризуется максимумом энтропии. По определению,
элементарное изменение энтропии равно элементарной приведенной теплоте:
dS = ^-. (1-16.4)
Приведенной теплотой называется отношение количества теплоты,
полученного системой в изотермическом процессе, к температуре этого процесса.
Приведенная теплота более показательна для характеристики изменений в системе,
чем просто подведенное количество теплоты, т.к. одно и то же количество
теплоты, сообщенное системе при различных температурах, вызывают различные
изменения в системе.
Энтальпия. Для изобарных процессов, в которых работа расширения
системы равна SW' = pdV', первое начало термодинамики (1.16.1) можно записать
в виде
SQ = dE + pdV = d(E + pV).
Отсюда в изобарных процессах количество теплоты, подведенной к системе
равно изменению функции
H = E + pV, (1.16.5)
называемой энтальпией системы.
Энергия Гиббса. Энтальпия системы, также как и внутренняя энергия
системы, условно делится на свободную и связанную:
H = G + TS.
Свободная составляющая энтальпии называется свободной энтаньпией, или
свободной энергией Гиббса, или просто энергией Гиббса. При изобарно-
изотермических процессах работа совершается системой за счет убыли энергии
Гиббса. Энергия Гиббса играет в термодинамике важнейшую роль, поскольку
многие процессы приближенно можно считать изобарно-изотермическими.
Изменение энергии Гиббса системы
dG=dH-TdS
11
характеризует возможность самопроизвольности процесса: при dG<0 процесс
может протекать самопроизвольно, при dG>0 не может. При равновесии
dG = 0.
Для идеальных систем зависимость свободной энергии GA компонента А
от его концентрации [А ] имеет вид
GA=G°A+nRTln[A], (1.16.6)
где GA - энергия Гиббса в стандартном состоянии, которое выбирается
следующим образом: для твердых веществ - чистый кристалл в наиболее
устойчивой модификации при давлении 1 атм, для жидкостей - чистая жидкость при
давлении 1 атм, для газов - идеальный газ при давлении 1 атм, для
растворенных веществ - гипотетический идеальный раствор с концентрацией /Мпри
давлении 1 атм. В определение стандартного состояния ИЮПАК не входит
стандартная температура, хотя часто говорят о стандартной температуре,
которая равна 25°С (298,15 К).
Изменение энергии Гиббса в химической реакции a A +b£=cC+dD
идеальной системы согласно (1.16.6) равно
(Cf[Df
AG=AG +RTln-
[Af[Bf
(1.16.7)
где AG -Gq + Gq -GA-G{.
В равновесии A G = 0, следовательно,
AG =-RTln
[cY[Df
[Af[Bf
(1.16.8)
' равн
Индексравн означает, что все концентрации в правой части (1.16.8) берутся для
равновесного состояния.
Величина, стоящая под знаком логарифма
KJcf[D]d
[Af[Bf
называется константой равновесия (напомним, что последнее выражение
записано для идеальной системы, для реальной надо концентрации заменить на
активности). Поскольку при постоянной температуре значение AG постоянно,
то в этом случае постоянно и значение К. Значение К зависит от температуры, а
также природы растворителя. Значение К изменяется при изменении
температуры по закону:
о
AG
K = e'RT
78
Выражая в (1.16.7) стандартное изменение энергии Гиббса AG с помощью
(1.16.8) изменение энергии Гиббса при протекании химической реакции можно
записать в виде
AG = RT
In
[cY[Df
In
[cY[Df
= RT
In
[Cf[D]
InK
[Af[Bf {[Af[Bf )рши) I [Af[Bf
В состоянии равновесия первое и второе слагаемые в круглых скобках равны и
AG = 0, при отсутствии равновесия - все концентрации в первом слагаемом
неравновесные и AG характеризует отклонение системы от состояния
равновесия.
1.17 Вычисление суммы по состояниям для поступательного движения
Вычислим сумму по состояниям частицы, свободно лижущейся в ящике,
Р2
энергия которой равна е--—, р - импульс, т - масса. В классическом при-
2т
ближении число состояний частицы, движущейся в ящике, с энергией, лежащей
в интервале от е до s+de равно отношению объема фазового пространства,
отвечающего данному интервалу энергий к h . Объем фазового пространства,
отвечающий энергии е, равен
Г = jdxdydz \d pxd pyd pz,
где интегрирование осуществляется по всем координатам и импульсам,
удовлетворяющим соотношению 0 < р < -yj2 m e . Вычислим интеграл в
сферических координатах:
2 те
Г = 4яУ J p2dp =
4nV p*
2ms
4n(2ms)3/2V
V - объем газа. Отсюда объем фазового пространства, отвечающего интервалу
энергий от е до s+ds, равен
йГ ds=4nmVJ2mede.
де v
Тогда функция состояния поступательного движения отдельной молекулы
79
3/2 rr<x>
V f^e~bTd£J2™kT
3/2
V.
(1.17.1)
1.18 Формулы, выражающие термодинамические функции через сумму по
состояниям
Все основные функции состояния внутренняя энергия, энтальпия,
энтропия, свободная энергия Гиббса могут быть найдены, если известна сумма по
состояниям системы Z, ее первая и вторая производные по температуре.
Статистическая физика приводит к следующим формулам, выражающим
термодинамические функции системы через сумму по состояниям системы Z [93,94]:
Свободная энергия Гельмгольца:
F = -0lnZ.
Свободная энергия Гиббса
dlnZs
G = 0
dlnV
-InZ
Внутренняя энергия
Е = в2
( dlnZ\
Энтальпия
Н = 0
(dlnZ\ (dlnZ
[dlnV)T+[dlnT
Энтропия
S = k
InZ+T
f dlnZ)
(1.18.1)
(1.18.2)
(1.18.3)
(1.18.4)
(1.18.5)
где @ = кТ - статистическая температура.
Статистическая сумма идеального газа, состоящего из N молекул одного
сорта согласно (1.15.6) равна
Z = ±- QN,
N!
80
i
тогда из (1.18.1) - (1.18.5) следуют следующие выражения термодинамических
функций идеального газа
/. Свободная энергия Гелъмголъца F:
F = -&NInQ+&lnN!.
Используя формулу Стирлинга
1nN!=NlnN-N,
получим
(1.18.6)
Представим N = vNA, где v - число молей, N^-число Авогадро, тогда
F = -@NlnQ--&N.
N
F = -kTvNAn-
Q
-kTvNA=- vTRln-
Q
vRT.
vNA vNA
Из уравнения Клайперона-Менделеева pV = vRT, тогда
Q
F = - vTRln-
pV.
2. Энергия Гиббса G
Поскольку изобарно-изотермический потенциал G = F + p V, то
G = - vTRln-^.
vNA
3. Внутренняя энергия.
Согласно (1.18.3) внутренняя энергия равна
(1.18.7)
Е = &2
din— QN
N!
д©
{ ет L { от
(1.18.8)
4. Энтропия.
Энтропию вычислим по формуле (1.18.5) с учетом выражения статистической
суммы идеального газа, состоящего из N молекул одного сорта (1.15.6)
S = k
In-*-QN+T
N!
din— QN
N!
dT
Jv
О Е
vRln—+vR+—,
N T
(1.18.9)
5. Энтальпия.
Энтальпию определим из выражения (1.16.5) с учетом (1.18.8)
81
{ дТ )v { дТ }p
(1.18.10)
(din q) (din q) i
поскольку — = — + —.
Ч дТ )р { дТ )v T
Выражение суммы по состояниям (1.15.4) сучетом (1.17.1) имеет
е-а—a=(^)J/%a.
Подставляя это выражение в формулы 1.18.6 - 1.18.10 можно получить
выражения термодинамических функций через сумму по состояниям внутренних
движений отдельной молекулы.
1.19 Сумма по состояниям внутренних движений молекулы
При расчете суммы по состояниям молекулы в программе Gaussian
используются следующие приближения: молекулярный газ рассматривается как
идеальный, колебания описываются набором невзаимодействующих
осцилляторов, для вращений используется приближение жесткого ротатора.
Сумма по электронным состояниям. При расчете суммы по состояниям
для электронного движения используется, что, как правило, при вычислении
Q3n достаточно учитывать только низший электронный уровень, энергия
которого равна нулю, поэтому
о_ _ff
Q9J,-slJ'e~kr^gTe~kT +•••**?, (1.19.1)
те- бэл приближенно равно кратности вырождения основного электронного
уровня gQ4. Для многих молекул основное состояние невырождено, поэтому
для них бал = 1.
Сумма по колебательным состояниям. Колебательная энергия молекулы
в гармоническом приближении равна сумме энергий отдельных нормальных
колебаний:
KJ , ( Г
екоя= I hvekUk+-\, (1.19.2)
где К/ - число нормальных колебаний, v* — колебательное квантовое число к-го
нормального колебания.
Вклад в молекулярную сумму по состояниям к-го нормального колебания
рассчитывается по формуле бесконечно убывающей геометрической прогрессии:
82
I
00 —
кТ -„ 2кТ
КШ ч=о
hvek
~ 2kT
( hvek 2hvek nhvek
1 + e kT + e kT + — + e kT +•
hvek '
1-е ЬТ
Вследствие свойства мультипликативности суммы по состояниям (1.15.3), для
молекулярной колебательной суммы по состояниям получаем:
hvek
N е 2кТ
QK0*= П j—. (1.19.3)
k=I nvek
1-е М
Сумма по вращательным состояниям.
1. Для линейной молекулы, в системе координат с осью z, проходящей через
ядра молекулы, для главных моментов инерции имеем Ix=Iy = I, Iz=0 и
вращательная энергия определяется по формуле
h2
J(j+l), (1.19.4)
У 8я21
где J- вращательное квантовое число.
Сумма по состояниям бесспиновой линейной молекулы с учетом кратности
вырождения 2J + 1 уровня J имеет вид:
h2JJJ+l)
— VI? 7х/Ь
1вр
Qep= I(2J + l)e 8я2Ш' (1 195)
J=0
При малых температурах значение выражения под знаком суммы быстро
убывает с ростом J и сумма вычисляется непосредственным суммированием.
При больших температурах суммирование приближенно можно заменить
интегрированием, интеграл легко вычисляется, при этом
h2
Существуют более точные численные методы вычисления вращательной
суммы (1.19.5).
2. Для симметричного волчка главные моменты инерции 1х=1уФ Iz, где ось z
- ось симметрии волчка, выражение вращательной энергии имеет вид:
83
"6/7
№1
j{j + i) + k*
У
^
(1.19.7)
У J
где К - квантовое число проекции момента количества движения на ось
симметрии волчка.
В этом случае при вычислении вращательной статистической суммы
молекулы необходимо проводить суммирование по J от 0 до со и по К от -/до
J:
' ' 'у,
ъ1
1я'1
Qep=l i (2J + l)e~
J=OK=-J
kT
(1.19.8)
При высоких температурах, заменяя суммирование интегрированием, для
(1.19.8) можно получить:
060=8"*
(2якТ1у\(2пкТ1.
h'
1/2
3. Для сферического волчка, Ix=Iy- Iz=l, имеем:
jep
1пЧ
-J(j + j)
(1.19.9)
(1.19.10)
Таким образом, вращательные уровни молекулы типа сферического волчка
определяются такой же формулой, что и уровни линейной молекулы. Однако,
степень вырождения уровня J для линейной молекулы равна 2J + 1, а для
сферического волчка (2J + 1) [95]. Для жесткого сферического волчка, при
высоких температурах приближенно получаем:
п _ 2(2л1кТ\3/2
Значения энергетических уровней молекулы е , определяющие согласно
(1.15.1) сумму по состояниям молекулы, зависят от внешних условий, в
частности от природы растворителя. От природы растворителя зависят и большинство
свойств атомно-молекулярных систем.
1.20 Модели сольватации
Большинство физико-химических процессов протекает в растворах, где
существенны эффекты сольватации. Существуют два типа методов оценки
эффектов сольватации. Методы первого типа (континуальные) рассматривают
растворитель как непрерывную среду, методы второго типа (дискретные) явно
учитывают молекулярную структуру растворителя [96,97]. В дискретных
моделях сольватации молекула растворенного вещества окружается достаточным
84
числом молекул растворителя. Имеются также комбинированные подходы,
например, явное введение молекул растворителя в первую координационную
сферу, и рассмотрение оставшейся части растворителя как непрерывной среды.
В свою очередь, континуальные и дискретные методы подразделяются в
соответствии с тем, как они рассматривают растворенное вещество и растворитель:
классически, квантовым образом или используют гибридные схемы (т.е.
сочетающие уравнения квантовой и классической механики). Широко используются
гибридные подходы, в которых раствор подразделяется на две подсистемы:
молекулу сольвата, описываемую квантовомеханически и растворитель,
описываемый классически (молекулярная механика, молекулярная динамика). Из
континтуальных моделей наиболее широко используется модель реактивного
поля.
Модель реактивного поля. В этой модели сольвент рассматривается как
однородная поляризуемая среда с диэлектрической проницаемостью е,
растворенное вещество помещается в полость, находящуюся в сольвенте. Любое
распределение зарядов, погруженное в среду с диэлектрической
проницаемостью е, порождает электрическое поле в растворителе. Это поле,
взаимодействуя с растворенным веществом, изменяет его характеристики. Состояние и
структура молекулы растворенного вещества в растворителе вследствие этого
взаимодействия изменяются по сравнению с таковыми в вакууме, молекула
сольвата ориентируется определенным образом в полости.
Свободная энергия сольватации растворенной молекулы представляет
собой сумму следующих вкладов:
AG , = AG +AG , + AG .,
solv cav disp el
где AG - кавитационная энергия, т.е. энергия реорганизации растворителя,
необходимая для образования полости, в которой помещается растворенная
молекула.
AG ,. - дисперсионная составляющая энергии взаимодействия, основу
которой составляет ван-дер-ваальсово взаимодействие сольвент - сольват,
AG . - энергия электростатического взаимодействия между собственными
зарядами растворенного соединения и индуцированными зарядами в
растворителе, поскольку распределение зарядов растворенной молекулы поляризует
среду, создавая в ней наведенные заряды.
Образование полости требует затрат энергии, наличие дисперсионных и
электростатических взаимодействий ведет к стабилизации растворенной молекулы
в растворителе, т.е. понижает энергию, поэтому
AG > О, AG ,. <0, AG , < 0. Расчет показывает, что в ряду близких по
cav ' disp ' е/ r -
строению структур для оценки AG . достаточно оценить электростатическую
составляющую AG ., поскольку AG,, и AG во многих случаях
компенсируют друг друга или малы по величине [97].
85
Модели реактивного поля отличаются друг от друга тем, как они
определяют полость и поле реакции. При кпассическом рассмотрении
электростатическую энергию AG , можно вычислить, заменив распределение зарядов в соль-
вате на точечное (или представив его в виде разложения по мультиполям) и,
рассчитав по формулам электростатики, энергию взаимодействия системы
зарядов растворенной молекулы с наведенными зарядами в растворителе. При
таком способе расчета предполагается неизменность электронной структуры
растворенной молекулы в растворителе и в газовой фазе, что не всегда оправдано.
Наиболее точными являются гибридные теории самосогласованного
реактивного поля, в которых растворитель описывается классически, а молекула
растворенного вещества квантовомеханически (гамильтониан сольвата без
учета растворителя Н' , влияние растворителя на молекулу сольвата
учитывается введением оператора взаимодействия сольвата со средой U). Таким
образом модельный гамильтониан молекулы растворенного вещества имеет вид:
H = HQ+U . (1.20.1)
Соответствующее уравнение Шредингера решается самосогласованно:
плотность растворенного вещества стартует с зарядовой плотности молекулы
растворенного вещества в газовой фазе. Распределение зарядов сольвата на каждой
итерации определяется по формуле:
р(г)=Ъ2.8('г-К.)+ре(г\ (1.20.2)
где первый член описывает вклад в полную плотность растворенной молекулы
в точке г ядер сольвата, второй - электронной плотности сольвата, Z., R{ -
заряды ядер сольвата и их координаты.
Электростатическое поле, порождаемое данным распределением зарядов,
определяется уравнением:
VD = 4?rp[r) или V [f(rj£:(r)J = 4яр[г ),
где D=£\r JE [r J - индукция, а Е (г ) -напряженность наведенного поля, syrj
- диэлектрическая проницаемость, зависящая в общем случае от
пространственных координат.
С учетом соотношения Е [г J= -grady/\rj получаем уравнении Пуассона,
определяющее потенциал у/\г) наведенного поля:
v[e(r)gra<tfy/(r )] =-4icp(r\ (1.20.3)
86
I
Диэлектрическая проницаемость часто принимается равной £\г ]=1 внутри
полости, е\г )=£, т.е. диэлектрической проницаемости растворителя, вне
полости. В этом предположении для сферической полости радиуса а решение
уравнения (1.20.3) можно записать в виде разложения по сферическим функциям
Y»{0.p)[96]:
2/ + 1
ю=/
оо . т=1
/=0 г т=-1
где В, - коэффициенты разложения, у/ . - потенциал, создаваемый зарядовым
распределением растворенной молекулы в вакууме (г < а) , у/ - потенциал
поля реакции, создаваемый в области г < а индуцированными зарядами на
поверхности сферы.
Случай 1-0 соответствует модели Борна, 1 = 1 - модели Онзагера,
учитывающей только отраженный диполь, создаваемый сольватом в сольвенте.
Более высокие члены разложения соотвутствуют учету высших мультиполей в
разложении у/. С помощью у/(г < а) вычисляется потенциальная энергия U
(1.20.1), которая включается в матрицу Фока, процесс самосогласования
продолжается до нахождения решения с заданной точностью.
Одной из популярных моделей реактивного поля является модель РСМ
(Polarized Continuum Model). Согласно этой модели молекула растворенного
вещества помещается в полость, поверхность которой задается набором сфер,
центры которых находятся на атомах молекулы растворенного вещества, а
радиусы определяются атомными радиусами Ван-дер-Ваальса [98]. Свободная
энергия сольватированной молекулы определяется по формуле [98]
G{W)^\H0\F) + ^\U\^),
где *F - волновая функция молекулы, находящейся в растворителе,
"' • I- - -Г'
-?,Яр \ГГГ{\ .
Ra-ri
v=Y,zaq]P
a.j
Za, Ra заряды и координаты ядер,
q-L - заряды, индуцированные полем на поверхности полости, формулы для
расчета которых приведены в [98],
87
r Js - координаты зарядов q'p ,
Fj - эффективные координаты электронов растворенного вещества.
У и U вычисляются самосогласованно, при этом начальная электронная
плотность растворенного вещества принимается равной электронной плотности
газовой фазы, а начальная волновая функция ^тч - собственной функции
гамильтониана Hq.
Упрощенной модификацией метода РСМ является модель COSMO
(Conductor - like Screening Model), согласно которой сильно полярные
жидкости с большими значениями е можно рассматривать как проводники (е = <х>).
Считается, что такая модель сольватации подходит для описания сольватации
больших биомакромолекул.
Если полость, в которую помещается сольват, определяется как
поверхность равной электронной плотности растворенной молекулы (типичные
значения 0,001 - 0,0004), то соответствующая модель называется моделью IPCM
(Isodensity Polarizable Continuum Model ). Если при этом еще самосогласованно
учитывается взаимовлияние формы полости и электронной плотности, такая
модель называется моделью SCI-PCM(учитывает, что форма полости
определяется электронной плотностью, которая сама зависит от формы полости).
1.21 Ядерный магнитный резонанс Метод GIAO
Ядерный магнитный резонанс (ЯМР) - резонансное поглощение энергии
переменного электромагнитного поля веществом, которое находится в
постоянном магнитном поле [99 - 102]. Ядерный магнитный резонанс связан с
наличием у атомных ядер момента количества движения
/- спиновое квантовое число ядра (ядерный спин),
и магнитного момента
м = уР,
у - коэффициент пропорциональности (гиромагнитное отношение),
постоянный для определенного типа ядер. Спектры ядерного магнитного резонанса
можно получить только для молекул, в состав которых входят ядра,
обладающие ядерным угловым моментом и магнитным моментом. Наиболее важными
для химиков являются ядра Н и С, поскольку резонанс этих ядер наиболее
важен для определения структуры органических молекул. Для получения
спектров ЯМР вещество помещается в постоянное магнитное поле напряженности
Но (обычно его выбирают направленным по оси г). При этом угловой момент
ядра ориентируется вдоль направления поля так, что его проекция на
направление магнитного поля может принимать значения:
88
P =mft,
m = l,I~l,— -I,
m - магнитное квантовое число ядра.
Соответственно, проекция магнитного момента ядра на направление поля
принимает значения
ju^ -ymti.
При включении магнитного поля с индукцией В0 ядро приобретает
дополнительную зеемановскую энергию, зависящую от магнитного квантового числа
ядра
Е =-и В~ = -утЛВп.
т 'г 0 ' О
Поэтому в магнитном поле каждый энергетический уровень ядра расщепляется
на 21 + 1 подуровней. Расщепление энергетического уровня ядра со спином
1 = 1/2, помещенного в постоянное магнитное поле, схематически представлен
на Рис. 1.21.1. Переменное электромагнитное поле может индуцировать
переходы, если его частота соответствует энергетической разности между этими
подуровнями. Расщепление уровней мало, соответствующая частота лежит в
радиодиапазоне (1-10 МГц).
l = V2 {*Н,13с)
т = -1/2
О
т = 1/2
Рис. 1.21.1. Схема энергетических уровней ядра (I -1/2), помещенного в
постоянное магнитное поле
В молекуле ядра окружены электронами и другими атомами. Поэтому ядра в
определенной степени экранированы и эффективная индукция магнитного поля
на ядре зависит от окружения рассматриваемого ядра.
Е
*гВэфф =-Mz{l-o-)BQ=-ymh(l-o-)B0,
о - постоянная экранирования.
Частота перехода между подуровнями:
89
F — F
h u
где к -уAmu.
Поскольку резонансная частота определяется индукцией эффективного
поля, она зависит от постоянной экранирования. Поэтому молекулы,
содержащие однотипные ядра (например, протоны) с различным окружением и с
различными <т, будут иметь различные частоты поглощения. Сдвиг частоты
поглощения для однотипных ядер, обусловленный различием в постоянных
экранирования называют химическим сдвигом. По частоте линии в спектре ЯМР
можно сделать вывод об окружении ядра, вызывающего эту линию.
Интерпретация спектров ЯМР является относительно однозначной. Главной областью
применения спектроскопии ЯМР является определение структуры молекул.
При получении спектров ЯМР выбирают эталон - некоторую молекулу,
содержащую исследуемые ядра. Химический сдвиг для линий других молекул,
обусловленный этими ядрами, рассматривают относительно частоты эталона.
Относительный химический сдвиг очень мал, поэтому его умножают на 10 и
определяют по формуле:
v.-vn , k(l-<j.)- k(l-an) * <jn-a. ,
S = ~^ 1-106=- '- u±w6=-° '-М^а -а. (м.д.),
vQ k(l-a0) J-a0 О Л
v. - резонансная частота в спектре ЯМР исследуемой молекулы,
у» - резонансная частота в спектре ЯМР молекулы-эталона,
в последнем выражении этих равенств множитель 10 заменен обозначением
м.д. - миллионная доля.
Характеристика Sявляется безразмерным числом и обычно выражается в
миллионных долях (м.д.) (ppm [part per million] - промиль (миллионная часть)).
Для ядер Я и С за эталон обычно принимают спектр ЯМР тетраметилси-
лана (ТМС, Si\f!Hi J , Рис. 1.21.2). Примеры химических сдвигов в спектрах
ЯМР (l H) представлены в Табл. 1.21.1
Рис. 1.21.2. Молекула тетраметилсгтапа, принимаемая за эталон, при расчете
спектров ЯМР
90
Табл.1. 21.1 Химические сдвиги ( Н) для различного окружения протона
Функциональная группа
сн3-сн2 -
Т
сн3-с^с-
т
CH^-N
сн3-сн2-
т
Интервал,м.д.
0,5-1,6
1,6-2,6
2,2 -3,2
0,9-2,1
Общая теория химических сдвигов была дана Рэмси [103 - 105]. Согласно
этой теории химический сдвиг обусловлен взаимодействием ядерных спинов с
движением электронов атома (молекулы), находящихся во внешнем магнитном
поле. Природа этого взаимодействия состоит в следующем. Если электрон
атома обладает орбитальным (1^0), а следовательно, и магнитным моментом, то
при включении магнитного поля, магнитный момент электрона прецессирует
вокруг вектора напряженности магнитного поля (Рис. 1.21.3.)
i
J
0
Но
' ДН
Рис. 1.213.Ларморовская прецессия магнитного момента р. в магнитном
поле.
Вследствие прецессии магнитного момента электрона в атоме (молекуле)
возникает ток J, создающий в месте расположения ядра (т.О на Рис. 1.21.3)
дополнительное поле АН, направленное противоположно исходному Hq
(диамагнитное), или сонаправленное с исходным (парамагнитное). Поэтому
результирующее магнитное поле в месте расположения ядра Hq+AH отличается от
приложенного магнитного поля. Это отличие зависит от состояния
окружающих ядро электронов, что и является причиной химического сдвига.
Для расчета величины химического сдвига необходимо исследовать
поведение электронов, входящих в состав исследуемой системы, в магнитном
поле. Системе в магнитном поле Hq отвечает гамильтониан, получаемый путем
91
замены оператора импульса р в гамильтониане без поля на выражение
в —
р — Aq (знак е отрицателен), где А0 - векторный потенциал, определяемый
с
уравнениями
<HvAq =0, rotAg = Hq.
Хорошо известно, что одно и тоже магнитное поле можно описать при помощи
различных векторных потенциалов. Так векторные потенциалы А и
A + V<p, (1.21.1)
где (р - произвольная скалярная функция определяют одно и то же магнитное
поле. Инвариантность поля по отношению к этому классу преобразований
называется калибровочной инвариантностью (gauge invariance) [100- 105].
Помимо поля Hq на электрон действует магнитное поле, создаваемое ядром
На, возникающее в месте расположения электрона вследствие прецессии
магнитного момента ядра в поле Hq . Нерелятивистский гамильтониан атома,
результирующий спин которого равен 0, взаимодействующего с двумя
магнитными полями Hq и На равен [101, 103,105]
H = ^-Z\pk--AQk--Aak) +V-Hflp,
2т k\ с с )
Aq/c - значение векторного потенциала, создаваемого внешним магнитным
полем в месте /^ расположения к электрона, Аак - значение векторного
потенциала, создаваемого ядерным моментом ядра в месте расположения к электрона,
Г- оператор потенциальной энергии, /2 - магнитный момент ядра.
Векторные потенциалы Aq^ и Аак выберем в виде [ 103]:
-л 1 Гг - ~л Г1Х?к
А0к=^Н0хгк> Лх*=—Г-
Проекции результирующего векторного потенциала Аь = Aq^ + Аак на оси
координат для случая направленности магнитного поля Hq вдоль оси 0z имеют
вид:
Axk=~H0y-^ft Аук=±Н0х + 2£, Az=0.
'"к гк
Тогда гамильтониан Н запишется в виде
# = -/-£
2m k
Рк — ~;но хгк г"
с 2 с ,->
92
Раскрывая квадрат, получим с точностью до членов квадратичных по //иЯ^с
учетом рк =-i'»VA и \рк --Ак I = рк2 --(ркАк +Акрк\ следующее
выражение гамильтониана
*2 _
к 2т ft 2mc k
( \ 7
#0+^г
2 тс г,
2р.
J
К 'к
Ham+WI+W2+W3,
(1.21.2)
где Lk -Ркхрк - оператор орбитального момента количества движения в
отсутствие поля,
Л 2т
П2 2
Ham=-Y-—Vk+V - гамильтониан атома в отсутствие поля,
Щ-^-zZ
2тс
( \
2
h; ^2=^Н0хгк^; W3=-H0m
2tnc
3
V 'к J ",,L- rk
- операторы возмущения, возникающие вследствие включения магнитного
поля.
Дополнительное поле АН в месте расположения ядра согласно определению
постоянной экранирования о равно
АН = -аН0,
Это поле приводит к дополнительной зеемановской энергии
AE=-JiAH = раН(),
поэтому тензор магнитного экранирования может быть рассчитан по формуле
д2АЕ д2 Е
<Ф dfladHofi дцадНор'
где Е = Eq + АЕ, Eq - энергия системы в отсутствие поля, a. /? = х, у, z.
Таким образом, оап есть коэффициент при члене /^Ядяпри разложении
энергии системы с гамильтонианом (1.21.2) в ряд по степеням jua и Hqq.
Энергия Жданной системы может быть вычислена по теории возмущений,
поскольку электростатическая энергия V значительно больше энергии
взаимодействия электрона с внешним магнитным полем и магнитным моментом ядра.
При этом, члены, содержащие в выражении энергии слагаемые,
пропорциональные цаНор> появляются в первом порядке теории возмущений вследствие
2 - -
наличия в гамильтониане возмущения Щ -Ндхгк——, и во втором
2 тс гк
порядке теории возмущений вследствие наличия в гамильтониане возмущения
93
2mc /(
Hn +
2p
т.к. произведение матричных
элементов
Wj"w" =(0 \Wj |«){и \Wj |f) дает члены, пропорциональные naHGp.
Используя при расчете вклада в постоянную экранирования возмущения И^
формулу преобразования скалярного произведения двух векторных произведений
для Одф во втором порядке теории возмущений получаем [100 - 105]:
+ комл. сопр.
(1.21.3)
где | 0 ), | п ) - волновые функции основного и возбужденного состояний
невозмущенного гамильтониана Нат.
rak'rpk=xk-)>k>zk-
Первое слагаемое в правой части (1.21.3), обусловленное W2 (токами ларморов-
ской прецессии), дает диамагнитный вклад в компоненту тензора магнитного
экранирования, второе, обусловленное W3 (поляризацией электронных
оболочек)- парамагнитный [100 - 105]. Можно показать, что выражение (1.21.3)
инвариантно по отношению к градиентному преобразованию векторных
потенциалов, хотя каждое из слагаемых таким свойством не обладает.
Указанный подход можно использовать, когда необходимо вычислить
химический сдвиг одного из ядер, входящих в состав многоатомной молекулы, выбрав
начало координат на этом ядре. При расчете химических сдвигов нескольких
ядер многоатомной молекулы возникает сложность, связанная с тем, что АО
центрированы каждая на своем ядре. Константа экранирования не должна
зависеть от выбора начала системы отсчета. Значение напряженности магнитного
поля в точке /^ конечно не зависит от такого выбора. Однако в уравнение Шре-
дингера системы в постоянном магнитном поле входит векторный потенциал,
зависящий от начала отсчета. Действительно, при выборе нового начала
координат радиус-векторы электрона в новой (гд) и старой (гд.) системах связаны
соотношением
h =rk+R,
где R - вектор, идущий из старого начала в новое. При этом значение
векторного потенциала А в новой системе координат равно
94
Оба векторных потенциала А и А определяют одно и то же поле и отличаются
калибровкой. Скалярная функция (р(г) (1.21.1) имеет вид
<?{?) = — H0xR-r , (т.к. при этом V<p = --H0y.R ). (1.21.4)
Известно, что при замене A—>A + W<p волновая функция (атомная орбиталь)
умножается на фазовый множитель [17]:
Ф у-^фувХр] <Р
\пс
при этом наблюдаемые физические величины (в определение которых не
входят в явном виде векторные потенциалы) не меняются. Учитывая значение (р\г)
(1.21.4) получаем
( ie - -
фу-^фуехр\ -—-H0xRr
\ 2Ьс
Таким образом, калибровочная зависимость может быть устранена введением
зависящих от поля базисных функций. Такие АО называются калибровочно
независимыми атомными орбиталями (GIAO - Gauge-Independant Atomic Orbital).
Метод расчета спектров ЯМР с использованием такого подхода называется
методом GIAO.
Мы кратко рассмотрели спектры ЯМР без учета тонкой структуры линий
спектра. При учете спин-спиновых взаимодействий линии в спектре расщепляются,
т.е. имеют мультиплетную структуру.
1.22 Методы молекулярной механики
Методы расчета свойств молекулярных систем можно разбить на 3
класса:
1. Методы молекулярной механики.
2. Методы квантовой химии, рассмотренные выше.
3. Методы молекулярной динамики.
Методы молекулярной механики позволяют определить геометрическое
строение и энергию молекул.
Молекулярная механика [106] рассматривает молекулу как набор атомов,
движущихся в потенциальном поле, которое подбирается эмпирически из
спектральных, калориметрических, кристаллографических и других экспериментов.
В этом отличие молекулярной механики от методов квантовой химии, в
которых поверхность потенциальной энергии молекулы находится путем решения
уравнения Шредингера. В молекулярной механике электроны системы явно не
рассматриваются. Потенциальное поле, в котором движутся ядра, в
молекулярной механике аппроксимируется определенными эмпирическими функциями
разной степени сложности, представляющими собой, например, суммы парных
95
потенциалов взаимодействия атомов. Существуют различные наборы
параметров для потенциалов взаимодействий. Эти потенциальные функции,
определяющие так называемое силовое поле молекулы, содержат некоторые
параметры, численное значение которых выбирается оптимальным образом так, чтобы
получить согласие рассчитанных и экспериментальных характеристик
молекулы. В простейшем случае параметрами являются равновесные межьядерные
расстояния (длины связей) и валентные углы, а также силовые постоянные.
Метод основан на допущении возможности переноса этих параметров из одной
молекулы в другую, так что численные значения параметров, подобранные для
некоторых простых молекул, используются далее при прогнозировании свойств
других более сложных соединений. Потенциальная энергия молекулы обычно
задается в виде суммы вкладов отдельных взаимодействий:
V ~ U раст + Uдеф + ^торс + U ВДВ + ^ эл.стат. + U Нее. >
где Uраст - описывает изменение потенциальной энергии молекулы при
изменении длин химических связей; идеф- при изменении валентных углов; Umopc-
торсионных углов; V'щв, Иэп стат , Uj-jce - описывают вклад в потенциальную
энергию молекулы ван-дер-ваальсовых, электростатических взаимодействий и
энергии водородной связи, соответственно.
Эти вклады имеют различный функциональный вид:
Rj - длина, R . - равновесная длина, Ау - силовая постоянная i - ой химической
связи.
Аналогично
i
Зависимость потенциальной энергии молекулы от угла внутреннего
вращения Ф (потенциал торсионных взаимодействий):
и„юрс = 1кф(<Х№ (пФ - S)+J),
Ф
где п - кратность торсионного барьера, S- сдвиг фазы, константы кф -
константы, зависящие от величин барьеров внутреннего вращения.
Ван-дер-ваальсовы взаимодействия атомов, разделенных тремя и более
валентными связями (невалентные взаимодействия), описываются
потенциалами Леннард-Джонса:
U вдв = £
i.j
\
_А В_
'и 'и J
96
Параметры потенциала А и В зависят от типов атомов / и j, участвующих во
взаимодействии; г- - расстояние между этими атомами.
Невалентное электростатическое взаимодействие описывается законом
Кулона:
U - kr ' 2
^ эл-Стат. Л^
£Г
q ., q~- заряды взаимодействующих частиц, г - расстояние между ними, е -
эффективная диэлектрическая проницаемость.
Функциональный вид потенциала водородной связи похож на потенциал
ван-дер-ваальсовых взаимодействий, но с более короткодействующими силами
притяжения:
Aj__B2_
'и ru )
и т.д. Сумма всех перечисленных вкладов определяет энергию V молекулы как
функцию геометрической конфигурации ядер. Оределяя минимум V с
помощью алгоритма поиска стационарных точек на многомерных потенциальных
поверхностях нахождят равновесную конфигурацию исследуемой молекулы.
Для многих задач, например для конформационного анализа, уровень
моделирования методами молекулярной механики оказывается вполне достаточным
для качественных и даже количественных заключений. В каждом конкретном
случае необходимо анализировать, для каких классов соединений
параметризована версия программы, которую предполагается применять при
моделировании свойств нового соединения. Особенно осторожно следует относиться к
оценкам энергий, хотя и для геометрических конфигураций возможны грубые
ошибки. При расчетах молекулярной структуры на более высоком уровне
методами квантовой химии полезно использовать координаты ядер молекулы,
найденные с помощью молекулярной механики, в качестве начального
приближения.
1.23 Метод молекулярной динамики
Движение атомно-молекулярной системы между химическими актами
хорошо может быть описано уравнениями классической механики при
температурах выше 1 ОК. Исключение составляют атомы водорода и протон, для
которых существенны квантовые поправки даже при 1000К. В классической
механике состояние системы из N частиц (атомов) в момент времени /
однозначно определяется заданием набора векторов координат и скоростей частиц.
Основная задача механики состоит в том, чтобы найти состояние системы в
произвольный момент времени /, если известно состояние системы в начальный
момент времени (при / = 0). В классической механике эта задача решается пу-
97
тем вычисления траектории движения, определяемой при решении уравнений
движения. В классической механике это хорошо известные уравнения Ньютона,,
для атомно-молекулярной системы из N частиц имеем: \
d2n(t) * !
щ—■yL=Fr
dt
т, - масса атома /, Fj - результирующая сила, действующая на / атом со сторо- \
ны остальных частиц: '
^(f/,Ъ"'^м) ' потенциальная энергия, зависящая от взаимного расположения
всех атомов.
В методе молекулярной динамики принципиальный вопрос состоит в
способе задания u{rj ^'"^м)- При моделировании систем, состоящих из
большого числа частиц, в качестве t/(r/ гг*--/^) используют потенциальные
функции метода молекулярной механики, в принципе С/(г/ ?2 • • -гд/ ) может быть
рассчитана методами квантовой химии. Скорости атомов - первые
производные координат по времени
_ _d?i
V'l~~dl
Уравнения движения решаются численно. Выбор начальных условий зависит
от цели исследования. Пусть необходимо изучить траекторию теплового
движения частиц системы. Например, для ряда систем характерны флуктуации
длин валентных связей около положений равновесия, величин валентных углов,
повороты вокруг одинарных химических связей. С этими флуктуациями
связаны конформационные степени свободы, которые ответственны за многие
функции системы. Начальные значения координат атомов выбираются
соответствующими конфигурации с минимумом потенциальной энергии, а начальные
значения скоростей задаются случайным образом. Если необходимо изучить
механизм химической реакции, то начальные значения координат могут
соответствовать минимуму потенциальной энергии реагентов, а скорости -
направлению движения вдоль координаты реакции.
Первое, с чем нужно определиться при численном решении уравнений
движения атомно-молекулярной системы - с шагом интегрирования, т.е. с
минимальным отрезком времени, на котором мы будем считать приращение
координаты частицы пропорциональным мгновенному значению скорости, а
приращение скорости пропорциональным мгновенному значению ускорения:
Г; (t + At) = 7t (t) + Vj (I) At,
0; (/ + At) = 0,- (t) + —At.
98
Шаг интегрирования должен быть много меньше характерного времени
самых быстрых движений в молекуле - валентных колебаний. Обычно шаг
составляет 1 фемтосекунду (/0 с). Расчет обычно проводят при постоянной
температуре. В качестве последней понимают среднюю кинетическую энергию
атомов :
T(i) = —!— I (m, v? ); ЕКШ1 =-NkBT .
w 3NkB / ' ' ' ™" 2 b
Применяют различные приемы для поддержания постоянной температуры
(различные способы задания термостата).
В результате расчета методом молекулярной динамики получается траек-
торный файл, содержащий координаты всех атомов в определенные моменты
времени (обычно запись координат в траекторный файл осуществляют с шагом
0,1 пс). Траекторный файл может быть использован для просмотра
молекулярного кино, т.е. пространственных движений молекулы в процессе расчета.
Такая визуальная информация хотя и полезна, но не может быть использована для
количественного анализа. Количественное изучение динамических свойств
молекул проводят с использованием функций распределения и средних значений
от различных величин.
99
Глава II. Основы работы с программой GAUSSIAN под WINDOUS
2.1 Возможности программы Gaussian и ее структура
Программный пакет Gaussian [107] один из самых первых программных
пакетов квантовой химии (первая версия вышла в 1970 г.), его основные
алгоритмы хорошо отлажены и надежны; он активно развивается, и в каждой новой
версии этого программного пакета отражены многие новейшие достижения в
области квантовой химии. Руководитель группы разработчиков данного
программного комплекса Джон Попл за развитие методов квантовой химии
удостоен Нобелевской премии по химии 1998 г [108-109].
Пакет программ Gaussian позволяет рассчитывать целый ряд свойств
молекул и характеристик химических реакций, в том числе [41]:
• структуры молекул и переходных состояний, их энергии;
• колебательные частоты, ИК- и рамановские спектры;
• термохимичекские свойства, энергии связей;
• энергии реакций и пути реакций;
• молекулярные орбитали и заряды атомов;
• мультипольные моменты, поляризуемости и гиперполяризуемости;
• электростатический потенциал и электронную плотность;
• сродство к электрону и потенциал ионизации;
• тензоры экранирования ЯМР и магнитную восприимчивость и др.
Могут быть рассчитаны не только свойства систем в газовой фазе, но и в
растворах. Расчеты могут быть проведены для основных и возбужденных
состояний.
Основной программной единицей Gaussian'a является линк - программа,
выполняющая определенную задачу [раздел Help программы Gaussian, 20].
Линки обмениваются друг с другом информацией посредством файлов,
расположенных на внешних носителях. Линки, выполняющие близкие функции,
объединяются в оверлеи. Каждый линк имеет трех или четырехзначный номер,
последние две цифры в котором обозначают номер линка в оверлее, а первые две
- номер оверлея. Например, линк 105 обозначает линк 5 в оверлее 1, линк 1002
- линк 2 в оверлее 10.
Оверлей 0 определяет набор дисковых файлов и машинных параметров,
необходимых для решения задачи и задает последовательность выполнения
отдельных линков, исходя из значений ключевых слов входного файла. Этот
оверлей состоит из 2-х линков L0 - инициализирует программу и управляет
оверлеями, L1 - обрабатывает ключевые слова в route section, формирует
список линков для выполнения.
Оверлей 1 выполняет считывание исходных данных и осуществляет
контроль оптимизации структуры. Оверлей I включает следующие линки: L101 -
считывает задание, L102, LI03, L105 осуществляют контроль процедуры
оптимизации с использованием методов Флетчера - Пауэлла (LI02), Берни (LI03),
Мэтага - Сержента (LI05), LI06 выполняет численное дифференцирование для
вычисления поляризуемостей и гиперполяризуемостей. Линк L107 ищет пере-
100
ходное состояние методом линейного синхронного транзита, L108
выполняет сканирование поверхности потенциальной энергии, L109 - оптимизацию
Ньютона-Рафсона. L110 численно вычисляет вторые производные
потенциальной энергии для расчета частот колебаний, LI 11 выполняет численное
дифференцирование зависящей от напряженности внешнего поля энергии для
определения поляризуемостей и гиперполяризуемостей, L113 для оптимизации
применяет алгоритм следования собственным векторам (EF), используя
аналитические градиенты, L114 проводит численную оптимизацию с алгоритмом
EF, L1I5 вычисляет путь реакции, L116, LI17 - численно вычисляет
самосогласованное реактивное поле, L118 - выполняет вычисления траектории
химической реакции, L120 контролирует ONIOM вычисления.
Оверлей 2 состоит из одного линка L202, который на основе заданных
координат атомов определяет центр масс молекулы, вводит систему центра
масс, определяет координаты атомов молекулы в этой системе, определяет
симметрию молекулы.
Оверлей 3 управляет выбором базисных функций и вычислением
интегралов. L301 генерирует базисные функции для каждого атома, L302 вычисляет
одноцентровые интегралы, L303 - мультипольные интегралы, L308 -,
L309 - ЕСР (effective core potential) интегралы, линки L310, L311, L314
вычисляют двухэлектронные интегралы, L316 печатает двухэлектронные интегралы,
L319 - вычисляет одноцентровые интегралы при наличии спин-орбитальной
связи.
Оверлей 4 состоит из 3-х линков. L401 формирует начальный набор
молекулярных орбиталей, выполняющий роль начального приближения для
процедуры самосогласованного поля, L402 выполняет расчеты с использованием
полуэмпирических методов и методов молекулярной механики. L405
инициализирует вычисления методом многоконфигурационного самосогласованного
поля.
Оверлей 5 содержит линки, осуществляющие процедуры
самосогласованного поля. L502 выполняет процедуру ССП ограниченным и неограниченным
методами Хартри-Фока (ОХФ и НХФ), как для систем в газовой фазе, так и с
учетом сольватационных эффектов в различных растворителях. При этом
используются все прямые методы, заключающиеся в перевычислении по
необходимости промежуточных результатов, особенно двухцентровых интегралов, а
не сохранении их на диске. L503 осуществляет расчет ОХФ и НХФ методами,
используя прямую минимизацию (ключевое слово вызова процедуры SCFDM).
Этот метод позволяет почти всегда достичь сходимости, но затраты машинного
времени при этом также возрастают. L506 выполняет вычисления НХФ или
методом GVB-PP. L508 осуществляет процедуру ССП с использованием
методов квадратичной сходимости, L510 - осуществляет процедуры
многоконфигурационного самосогласованного поля.
Оверлей 6 осуществляет анализ свойств атомно-молекулярных систем на
основе волновой функции, полученной в оверлее 5. L601 осуществляет анализ
орбитальных заселенностей, заселенностей перекрывания, дипольных и муль-
типольных моментов, L602 вычисляет одноэлектронные свойства (электро-
101
статический потенциал, электрическое поле и его градиент). L604 производит
оценку молекулярных орбиталей и электронной плотности, L607 осуществляет
анализ натуральных связевых орбиталей в терминах локализованных
электронных пар, L608 безитерационно рассчитывает энергии в теории
функционала плотности, L609 рассчитывает свойства атомов в молекуле.
Оверлей 7 рассчитывает первые и вторые производные и силы на атомах.
L701 рассчитывает первые и вторые производные одноэлектронных
интегралов, L702 рассчитывает первые и вторые производные двухэлектронных ин- i
тегралов (sp), L703 рассчитывает первые и вторые производные
двухэлектронных интегралов (spdf), L709 формирует вклад производных интег ралов эффек- i
тивного потенциала остова в градиенты, L716 обрабатывает информацию для
процесса оптимизации и расчета частот колебаний.
Оверлеи 8 и 9 предназначены для расчета электронной корреляции и
возбужденных состояний. L801, L802, L804 выполняют преобразование атомных
интегралов в молекулярные. L803 выполняет экстраполяцию матрицы Фока
при использовании сложных базисных наборов при расчете энергии методом I
МР2. L811 преобразует производные интегралов и вычисляет их вклады во
вторые производные МР2. L901 антисимметризует двухэлектронные
интегралы. L902 определяет стабильность ХФ волновых функций. L903, L905,
L906 выполняют вычисления с использованием модификаций метода МР2.
L908, L909 - выполняют вычисления методом внешней валентной функции
Грина OVGF. L913 вычисляет post-SCF энергии и градиентные члены, L914 -
управляет расчетом возбужденных состояний методом конфигурационного
взаимодействия (CI-Singles, RPA и Zindo). L915 - выполняет расчеты по
теории возмущений 5 порядка (для МР5, QCISD(TQ) и BD(TQ)). L918-
выполняет переоптимизацию волновых функций.
Оверлей 10 предназначен для выполнения процедур самосогласования при
использовании сложных методов. L1002 решает самосогласованно CPHF
уравнения (Coupled-Perturbed Hartree-Fock Equation); вычисляет различные
свойства, включая ЯМР. L1003 Самосогласованно решает CP-MCSCF уравнения,
L1014 вычисляет аналитически вторые производные при использовании метода
конфигурационного взаимодействия с одиночной подстановкой.
Оверлей 11 предназначен для вычисления производных различных интегралов.
L1I01 вычисляет производные одноэлектронных интегралов. L1102
вычисляет матричные элементы от производных дипольного момента, L1110
вычисляет вклад производных двухэлектронных интегралов в силы, действующие
на атомы. L11112 вычисляет PDM и post-SCF производные, LI 112 - МР2 вто- '
рые производные.
L9999 выполняет окончание вычислений и формирует output файл. '
I
1
102
2.2. Запуск программы GAUSSIAN для операционной системы WINDOWS
Начало работы с программой начинается со щелчка мышкой по иконе
программы GAUSSIAN c^ow, после чего на экране монитора появляется
главное окно программы ( Рис. 2.2.1 ):
Рис. 2.2.1 Главное окно программы
Первая строка File, Process, Utilities, View, Help - зона меню.
Во второй строке представлены иконы, контролирующие процесс работы.
Для введения исходных данных нажимаем File, после чего открывается File-
меню, состоящее из 5 строк (Рис.2.2.1):
New -создает новый input файл и открывает окно ввода работы.
Open - открывает меню существующих файлов.
Modify - редактирует текущий загруженный input файл.
Preferences
Exit
Input фат программы Gaussian. После выбора в зоне меню File -> New,
открывается окно вводы работы, имеющее вид ( Рис. 2.2.2), в которое
записываются входные параметры задачи. Примеры входных файлов представлены в
разделе 2.15. Окно ввода работы состоит из 4-х разделов и окна молекулярной
спецификации. До ввода информации - все поля пустые.
1. Раздел % Section содержит директивы по использованию ресурсов сис-
103
темы и хранению промежуточной информации. Наиболее часто используются в
этом разделе являются директивы:
% спк=имя файла.chk - указывает на создание в процессе работы
checkpoint файла, который хранится в каталоге с расширением chk. В этом файле
содержится информация о промежуточных вычислениях в процессе оптимизации,
молекулярных орбиталях и т.д. Этот файл используется, например, для
возобновления расчетов после прерывания работы, для графического изображения
молекулярных орбиталей, электронной плотности и электростатического
потенциала.
%mem=nMB - задает максимальное количество оперативной памяти, ко- |
торую может использовать программа ( п мегабайт). При ее отсутствии исполь- I
зуется значение по умолчанию (2000 мегабайт). I
%nproc=m — указывает число используемых процессоров. i
Заполнение этого раздела не является обязательным. I
Рис. 2.2.2. Окно ввода работы
2. Раздел Route section - секция выбора маршрута решения задачи,
начинается с символа "#", после которого ставится пробел, затем указываются все
необходимые директивы (см. примеры входных файлов, п. 2.15). Порядок их
следования значения не имеет, для их записи можно использовать любое число
строк, несколько директив можно указывать в одной строке. Обычно в начале
104
раздела директив указывается метод расчета (полуэмпирический, Хартри-
Фока, функционала плотности и др.), базисный набор (см. раздел 1.4) и тип
вычислений. Тип вычислений задается ключевым словом (см. п.2.14),
например, Opt - означает, что необходимо провести оптимизацию конфигурации
молекулы, Freq - означает требование расчета инфракрасных и рамановских
спектров и т.п. Каждое ключевое слово, как правило, имеет несколько опций
(вариантов использования). Выбор требуемых опций задается с помощью
следующего формата
ключевое слово = ( опция!, опция2,...)
Если при этом используется только одна опция, то скобки можно
опустить, если используется опция по умолчанию, то можно указать только
ключевое слово.
Для задания ключевых слов и опций используются только установленные
программой обозначения, которые описаны ниже (раздел 2.14). Если строка
секции маршрута начинается с символа #Т , то означает, что output файл
выводится в сжатом виде, в нем отражены только существенные результаты (terse -
сжатый), #Р - означает, что в output файле выводится максимальное число
компьютерных вычислений, # - означает, что output файл имеет традиционный вид.
3. Раздел Title section - раздел комментариев, в который программист
записывает необходимую для него информацию о выполняемой работе
(используется только английский язык). Здесь можно использовать любое число строк,
запись в этом раздела никак не отражается на ходе вычислений. Заполнение
этого раздела не является обязательным.
4. В разделе Charge & multiplicity указываются заряд и мультиплетность
рассчитываемой системы, их значения отделяются друг от друга пробелом .
В окне молекулярной спецификации указываются координаты всех
атомов начальной структуры исследуемой системы и при необходимости
некоторые дополнительные директивы. При выборе начальной конфигурации
руководствуются, обычно, экспериментальной информацией о строении молекул, для
ее задания можно использовать графический редактор GaussView (см. главу
III). На рис. 2.2.2 начальная конфигурация молекулы в окне молекулярной
спецификации задается декартовыми кординатами. Задание начальной
конфигурации можно осуществить различными способами, описанными в разделе 2.3.
При необходимости в окне молекулярной спецификации можно указать
дополнительные разделы, отделяемые от раздела спецификации и между собой
пустой строкой. Их назначение описано ниже.
Назначение кнопок окна ввода работы и главного окна программы.
Кнопки в правой части окна ввода работы (Рис.2.2.2) обозначают следующее [раздел
Help программы]:
тш
\iijss - кнопка запуска программы на решение.
105
.ИШ - кнопка возврата в главное меню, при необходимости затем можно
вернуться в окно ввода работы посредством File -> Modify.
кнопка сохранения input файла, записанного в окне ввода работы.
у™* - кнопка закрытия окна ввода работы.
Иконки во второй строке главного меню программы (Рис. 2.2.1)
*=йШ| обозначают следующее:
запуск процесса решения сначала (Begin Processing).
ШШ - пауза в работе.
пауза после текущего линка.
•тает» - продолжение решения после паузы, перед продолжением решения
появится информация о линке, исполняемом программой перед паузой
ЭДя^йЯ^оЬ Processing resumed. C:\G03W\t703.exe ,_.
я»?жК? t-i,*',%.3s^^^v%t«»!WJ№rf:>'.>iiJssjifiis«.:+..^«-.^i»». Перед паузой программа
выполняла линк 703.
I - остановка вычислений (Kill Job).
Запуск программы на решение. Запуск программы на решение осуществляется
после заполнения input файла (главного окна программы, Рис. 2.2.2) с помощью
После запуска программы на решение программа запросит под
каким именем сохранить output файл вычислений, появится каталог output файлов
программы. Внимание: при выборе имени input и output файла запрещается
использовать русский шрифт.
В правом верхнем углу окна ввода работы имеется кнопка
«дополнительные шаги» ( показана стрелкой на Рис.2.2.2).
Если, после того как окно ввода работы заполнено, нажать эту кнопку, то
появится меню ввода дополнительных шагов.
■АМШт$ЩЩШШ§§
«КЯЗШ ю я,
чюиючлгъ-'-г
При выборе в этом меню step -> add step появится окно ввода
дополнительного шага, которое имеет такой же вид как и окно ввода работы:
106
Это окно заполняется также как окно ввода работы. Верхние две кнопки
справа
кнопки перехода к предыдущему и последующему шагам.
Кнопка 13Щ выводит список всех имеющихся шагов. Двойной щелчок левой
кнопкой мыши по имени любого шага в этом списке открывает окно ввода
этого шага. Кнопка Шш1 закрывает окна всех шагов, кроме начального. Запуск
программы на решение осуществляв гея кнопкой ISS2U начального шага. После
окончания решения предыдущего шага, программа переходит к выполнению
задания следующего шага и не закончит работу, пока все шаги не будут
выполнены.
При выполнении решения в строке Run Progress главного окна (Рис. 2.2.1)
и в нижней строке главного окна печатается информация о текущем процессе
вычислений. При нормальном завершении вычислений в строке Run Progress
печатается Processing Complete, при наличии ошибки - Link died! После
завершения вычислений можно открыть output файл посредством выбора в зоне
меню главного окна (Рис.2.2.1) View -> Editor output file, или с помощью кнопки
главного окна ШШ , или открыв обычным образом соответствующий файл в
WordPad, куда программа записывает input и output файлы по умолчанию.
107
Output файл заканчивается указанием времени вычислений, информацией
об использованных ресурсах при выполнении работы и причиной окончания
расчетов ( нормальное завершение или ошибка):
Job cpu time: 0 days 0 hours 0 minutes 3.0 seconds.
File lengths (MBytes): RWF= 12 Int= 0 D2E= 0 Chk= 7 Scr= 1
Normal termination of Gaussian 03 at Sun Feb 03 18:55:22 2008
Формирование стартовой структуры и input файла для программы
Gaussian, просмотр промежуточных и конечных результатов расчета можно ,
также осуществить, используя программу «Gaussview»(GV), см. Гл.Ш.
2.3 Задание исходной конфигурации молекулы '
Начальная структура молекулы в окне молекулярной спецификации (Рис.2.2.2)
может быть задана четырьмя способами: с помощью внутренних координат
атомов, декартовых координат, комбинированным способом и с помощью
псевдоатомов. Единицами измерения, принятыми по умолчанию, являются
ангстремы (10"10 м) для расстояний и градусы для углов.
Пример молекулярной спецификаг/ии в декартовых координатах. При задании
конфигурации молекулы в декартовых координатах в окне молекулярной
спецификации (Рис.2.2.2) на первом месте указывается химический символ атома,
затем разделитель (пробел или несколько пробелов, или запятая), затем через
разделитель декартовы координаты атома х, yv\z. При записи десятичных чисел
используется точка, а не запятая. Если координаты атома - целые числа, то
наличие десятичной точки обязательно. Например, задание стартовой
конфигурации молекулы воды с помощью декартовых координат имеет вид
о
в
И
0.
0.96
- 0.32
0.
0.
0.90
0.
0.
0.
Выбор начала координат является произвольным (это может быть любой
атом молекулы или совершенно произвольная точка пространства).
Использование внутренних координат атомов. Взаимное расположение атомов
в молекуле можно описать с помощью внутренних координат атомов,
которыми являеотся межатомные расстояния (длины связей), валентные и диэдральные
углы. Для этой цели принято использовать так называемую Z-матрицу, которая
располагается в окне молекулярной спецификации. Перед ее построением всем
атомам структуры присваиваются номера, например, для молекулы метана:
108
Рис. 2.3.1 Молекула метана (атомы пронумерованы)
В первой строке Z-матрицы пишется только символ элемента, которому
присвоен номер 1. Вторая строка начинается с символа химического элемента,
которому присвоен номер 2, затем идет разделитель (пробел или запятая),
затем цифра 1 (номер элемента, стоящего в первой строке), затем — разделитель,
после которого указывается длина связи атом 2 - атом 1. Например, для
молекулы метана (длина С-Н связи равна 1.093 А ) имеем
с
н,
н,
н.
н,
1,
1,
1,
1,
1.093
1.093, 2, 109.47
1.093, 2, 109.47 ,3,
1.093, 2, 109.47 ,3,
120.,0
-120.,0
Третья строка кроме длины связи между атомом водорода Нр) и атомом
углерода C(i) (1.093 А0) содержит угол 3-1-2, равный 109.47°. Структура всех строк,
начиная с четвертой, одинакова. Например, последняя строка Z — матрицы
метана специфицирует атом Н (5) (№ 5 на рис.2.3.1) , после символа элемента и
разделителя идет номер любого предшедствующего элемента ( I в данном
примере), 1.093 А0 - расстояние от атома Н(5) до атома 1; 109.47° - угол Н(5) - атом I
- атом 2; - 120 - двугранный угол Н[5) - атом 1 - атом 2 - атом 3;
Внимание: При записи длин связей и углов обязательно использование
десятичной точки (даже если значение целое), при записи номеров атомов
использование точки недопустимо.
Наряду с приведенной здесь формой записи Z-матрицы можно
использовать переменные для обозначения некоторых внутренних (или всех) координат
молекулы. Такая форма будет иметь следующий вид :
с
Н, 1,
Н, 1,
Н, 1,
Н, 1,
R
R, 2,
R, 2,
R, 2,
Variables:
R=1.093
109
109
109
47
47
47
,3,
,3,
120.
-120.
,0
,0
109
В этой записи длины связи С-Н обозначены одной переменной. Переменными
можно обозначить обычные и двугранные углы. Использование переменных
позволяет уменьшить число оптимизируемых параметров, в данной структуре
длина связи С-Н рассматривается как один параметр, в предыдущем примере -
как различные.
При конструировании Z -матрицы можно присваивать номера атомам, напри-
мер:
С1
С2
СЗ
HI
Н2
нз
Н4
Н5
Нб
С1
С2
С1
С1
С2
СЗ
СЗ
СЗ
1.34
1.52
1.0S
1.09
1.09
1.09
1.09
1.09
С1
С2
С2
С1
С2
С2
С2
120
120
120
120
109
109
109
5
5
5
СЗ
СЗ
HI
C1
С1
С1
0.
180.
180.
180.
60.
-60.
Запись этой структуры с использованием переменных в Z матрице
C1
C2
СЗ
HI
Н2
нз
Н4
Н5
Н6
CI
C2
CI
CI
C2
C3
C3
C3
Variables:
Rl=
R2=
R3=
Al=
A2=
Dl=
=1.34
=1.52
=1.09
120.
=109.5
=60.
Rl
R2
R3
R3
R3
R3
R3
R3
CI
C2
C2
CI
C2
C2
C2
Al
Al
Al
Al
A2
A2
A2
C3
C3
HI
CI
CI
CI
0.
180.
180.
180.
Dl
-60.
Смешанное использование внутренних и декартовых координат. При
проведении квантово-химических расчетов иногда возникает необходимость
при составлении Z-матрицы использовать для описания положения атомов как
внутренние, так и декартовы координаты атомов. Ниже приведена Z-матрица
для оптимизации молекулы перекиси водорода в смешанных координатах
(атомы H(i) и 0(2) - в декартовых координатах, Ор) и Н(4) - во внутренних):
Н 0.95
0 0.
0 2 R2
Н 3 R1
Variables:
Rl=0.95
R2=1.48
А=105.0
D=120.0
0.
0.
1
2
0.
0.
А
AID
Использование псевдоатомов. Многие молекулы обладают высокой
симметрией, вследствие которой отдельные внутренние координаты молекулы имеют
НО
одинаковые значения. Например, в молекуле бензола С^Н^ соседние С -С и
С — Н связи имеют одинаковую длину, одинаковое значение имеют и
валентные углы. Чтобы при описании структуры молекулы с помощью Z-матрицы
учесть симметрию системы удобно использовать так называемые псевдоатомы.
Псевдоатомы обозначаются символами X или XX, их может быть любое
количество, а располагаться они могут в любой строке Z-матрицы. Псевдоатомы
используются программой только для описания структуры, на выполнение
расчетов они не влияют. Например, Z-матрицу молекулы бензола (Рис. 2.3.2 )
удобно записать, введя два псевдоатома Х(/) и X^j, один из которых
расположен в центре молекулы, а второй - в произвольной точке оси симметрии Qj
вне плоскости молекулы. Тогда расстояния Хп) ~ С равны длинам соседних
связей С -С, равны приближенно R-1.39A0, угол X(j) -X(2) ~С равен 90 ,
углы С - Х(2) - С для соседних атомов углерода равны 60 .
Рис. 2.3.2. Структура молекулы бензола с псевдоатомами Xpj и Х^)
Z-матрица молекулы имеет следующий вид:
XI
Х2
СЗ
С4
С5
С6
С7
С8
Н9
НЮ
НИ
Н12
Н13
Н14
г
R
1
2
2
2
2
2
2
2
2
2
2
2
2
1.39
2.46
1.0
г
г
г
г
г
г
R
R
R
R
R
R
1
1
1
1
1
1
1
1
1
1
1
1
90.
90.
90.
90.
90.
90.
90.
90.
90.
90.
90.
90.
3
4
5
6
7
3
4
5
6
7
8
60.
60.
60.
60.
60.
180.
180.
180.
180.
180.
180.
Введение в Z-матрицу псевдоатома позволило вследствие учета
симметрии молекулы бензола уменьшить число оптимизируемых параметров до двух.
111
Иногда бывает необходимо разные атомы рассчитывать с различными
базисными наборами. В этом случае вместо встроенного базисного набора в
секции маршрута указывается директива Gen, которая указывает, что информацию
о базисных наборах атомов необходимо прочитать из дополнительного раздела
входного файла, который размещается после задания молекулярной
спецификации и отделяется от нее пустой строкой. В этом случае формат ввода,
определяющий базисные наборы атомов имеет вид:
Эл1 Эл2ЭлЗ...ЭлЫ0либоп1 n2n3...nN0
базисный набор
****
где Эл1 Эл2 ЭлЗ ...ЭлЫ - символы химических элементов, nl n2 n3 ...nN-
их порядковые номера, 0 - символ окончания перечисления, **** - означает,
что определение базисного набора для перечисленных атомов закончено.
Пример.
%Chk=waterl
#Т RHF/Gen Opt Freq
water freq
0 1
О
HI 0.97
H 1 0.97 2 109.
О 0
3-21G
2 3 0
6-31G(d,p)
В этом для атома кислорода будет использован базис 3-21G, а для атомов
водорода - базис 6-31G(d,p). Данный и приводимые ниже примеры входных
файлов записаны не в окне вводы работы (Рис.2.2.2), а в формате, принятой в
операционной системе LINUX (см.[41]), где input файл записывается в виде
текста, разделы % Section, Route section, Title section, Charge Multipl.
отделяются друг от друга пустой строкой.
2.4. Расчет энергии молекулы для фиксированной конфигурации
Самый быстрый тип работ программы Гауссиан - расчет энергии молекулы
для фиксированной конфигурации многоатомной системы или Single point
Energy Calculation. Он позволяет рассчитать полную энергию молекулы для той
конфигурации молекулы, которая задана в окне молекулярной спецификации (
Рис.2.2.2 ). Это не обязательно структура, соответствующая минимуму энергии.
Рассчитанная энергия включает в себя кинетическую энергию движения
электронов для заданных межъядерных расстояний, и все виды потенциальной
энергии (взаимодействия электронов друг с другом, взаимодействия электронов
с ядрами, взаимодействия ядер друг с другом). Это собственное значение элек-
112
тронного гамильтониана для фиксированных межъядерных расстояний,
заданных исходной структурой. При расчете энергии в фиксированной точке для
заданной конфигурации будут вычислены дипольный и высшие мультипольные
моменты, поляризуемость, гиперполяризуемость, заряды на атомах,
молекулярные орбитали и некоторые другие характеристики. Вычисление Single point
Energy бывает необходимо для многих целей, из которых отметим следующие:
- Получение базисной информации о структуре.
- Для исследования данной структуры как стартовой для расчета
оптимальной структуры.
- Для получения информации о структуре, полученной грубыми методами.
- Когда нет возможности провести оптимизацию.
- Для выбора метода и базиса расчета, если имеется экспериментальная
информация о характеристиках данной конфигурации.
Single point Energy Calculation может быть выполнена на различных уровнях
теории: можно выбрать любой метод решения электронного уравнения Шре-
дингера, минимальный или расширенный базис для волновой функции.
Указание выполнить этот тип работ задается ключевым словом SP , оно может быть
опущено, это директива, выполняемая по умолчанию.
Например, входной файла расчета энергии молекулы воды для
конфигурации, заданной декартовыми координатами имеет вид:
Schk
=water
tt RHF/6-31G(d)
wate
0 1
0
Н
н -
tr Single
0.
0.96
0.32
Point
0
0
0
90
0.
0.
0.
Ограниченным методом Хартри-Фока в базисе 6-31G(d) программа выполнит
расчет полной энергии молекулы воды, если ее атомы расположены в точках с
координатами 0{ 0,0,0}; Н{0.96,0,0}; Н{-0.32,0.90, О}.
Открытие output файла и просмотр результатов. После окончания
расчета открываем output файл с помощью кнопки ШИ, или пути View -> Editor
Output File (Рис. 2.4.1). Можно также открыть обычным образом
соответствующий файл в WordPad, куда программа по умолчанию записывает input и
output файлы. Энергия исследуемой молекулы для конфигурации заданной в
input файле печатается в output файле после слов «SCF Done»:
|SCF Done:E(RHF) = -76.0099630509А.V. after 5 cycles |
113
Ниже значения полной энергии печатается значение сходимости по матрице
плотности и первый вириальныи коэффициент (отношение потенциальной
энергии к кинетической, равный 2 согласно теореме вириала):
| Convg = 0.4603D-04 -У/Т = 2.0026 "]
Рис. 2.4.1. Открытие output файла
Секция output файла Standart Orientation Geometry показывает положения
атомов в молекуле в декартовых координатах в системе центра масс молекулы:
Standard
Center
Number
1
2
3
orientation:
Atomic
Number
8
1
1
Atomic
Type
0
0
0
0
0
-0
Coordinates (Angstroms)
X Y 2
000000
782357
782357
0.110436
-0.445907
-0.437577
0.000000
0.000000
0.000000
Большинство молекулярных свойств, вычисляемых на каждом шаге,
соответствует стандартной ориентации. Standart Orientation Geometry обычно не
совпадаете Input Molecule specification:
114
Input
Center
Number
1
2
3
orientation:
Atomic
Number
8
1
1
Atomic
Type
0
0
0
Coordinates
X
0.000000
0.960000
-0.320000
Angstroms)
Y
0.000000
0.000000
0.900000
2
0
0
0
000000
000000
000000
Наряду с энергией системы, при расчете SP программа выдает электрические
характеристики системы: заряды на атомах по Малликену, дипольный и
высшие мультипольные моменты.
Mulli
1
2
3
О
н
н
ken
-0
0
0
atomic charges:
876631
438158
438473
Dipole moment (field-independent basis, Debye):
X= -0.0036 Y= -2.1352 Z= 0.0000 Tot=
Quadrupole moment (field-independent basis, Debye-Ang):
XX= -3.8964 YY= -6.1136 22= -7.2273
XY= -0.0144 XZ= 0.0000 YZ= 0.0000
Traceless Quadrupole moment (field-independent basis, Debye-
XX= 1.8494 YY= -0.3679 Z2= -1.4816
XY= -0.0144 XZ= 0.0000 YZ= 0.0000
Octapole moment (field-independent basis, Debye-Ang**2):
XXX= -0.0084 YYY= -1.3415 ZZZ= 0.0000 XYY=
XXY= -1.3953 XXZ= 0.0000 XZZ= -0.0034 Y2Z=
YYZ= 0.0000 XYZ= 0.0000
Hexadecapole moment (field-independent basis, Debye-Ang**3)
XXXX= -5.2761 YYYY= -5.9940 ZZZZ= -5.2092 XXXY=
XXXZ= 0.0000 YYYX= 0.0003 YYYZ= 0.0000 ZZZX=
ZZZY= 0.0000 XXYY= -1.6045 XX22= -2.0532 YY2Z=
XXYZ= 0.0000 YYXZ= 0.0000 ZZXY= 0.0034
2.1352
-Ang):
0.0088
-0.3680
-0.0002
0.0000
-1.9073
2.5 Управление процессом самосогласования
Для SP и всех других типов работ, которые рассмотрены ниже,
управление процессом самосогласования осуществляется с помощью ключевого слова
SCF (самосогласованное поле). Имеется несколько вариантов управления
процессом самосогласования, каждая опция определяется своим параметром. При
SP вычислениях в интересах скорости расчета в процедуре самосогласования
используются скромные критериями сходимости: по умолчанию энергия SCF
вычисляется с точностью 0.1 ккал/моль, а матрица плотности имеет 3 верных
десятичных знака. Это достаточно для анализа заселенности,
электростатического потенциала, производных и т.п.
Некоторые опции ключевого слова SCF.
Tight - означает, что в процедуре самосогласования будут использованы
улучшенные критерии сходимости.
Опции, опредеяяюгцие способы хранения интегралов.
Direct — указывает программе, что необходимые процедуре самосогласования
двухэлектронные интегралы вычисляются заново каждый раз. Это используе-
115
мая по умолчанию процедура. Она возможна для всех методов, за
исключением вычисления вторых производных MCSCF (см. раздел аббревиатура методов
квантовой химии) и некоторых величин с использованием сложных орбита-
лей.
InCore - указывает, что всю необходимую информацию следует хранить в
оперативной памяти, что значительно ускоряет расчеты. Это делается
автоматически в Direct вычислении, если достаточно памяти. NoInCore запрещает
использование Incore, как для SCF так и CPHF. Если памяти не хватает, то программа
автоматически переключается на Direct.
Conventional - означает, что двухэлектронные интегралы загружены на диск и
считываются в каждой итерации самосогласованного поля. NoDirect - синоним
Conventional.
Опции выбора алгоритма.
DIIS (Direct Inversion in the Iterative Subspace) требует и NoDIIS запрещает
использование прямой инверсии в методе экстраполяции итеративного
подпространства Pulay's [561].
QC - включает метод квадратичной сходимости вместо стандартного DIIS.
Этот метод((}С) более надежен, но и значительно медленнее стандартного.
Возможно его использование только для методов RHF, UHF, DFT.
Conver=N - Установить критерий SCF сходимости 10"N. По умолчанию N=8.
См. пример в п. 2.6.
Опции, относягциеся к перезапуску
Save - сохраняет волновые функции в checkpoint файле на каждой итерации,
так что процедура самосогласования может быть перезапущена, используется
по умолчанию.
NoSave - подавляет Save.
Restart - продолжить расчет SCF, пользуясь информацией из chk файла.
2.6 Оптимизация структуры атомно-молекулярной системы
Требование выполнить оптимизацию структуры задается ключевым
словом Opt в строке Route section, осуществляющим поиск стационарных точек на
поверхности потенциальной энергии. В процессе оптимизации в соответствии с
используемым методом поиска экстремума изменяются длины связей и
валентные углы исследуемой структуры, пока не будет найдена стационарная точка.
Здесь надо помнить, что поверхность потенциальной энергии многоатомной
молекулы кроме глобального мимнимума может иметь большое число
локальных минимумов и седловых точек (см. раздел 1.13). К сожалению,
используемые процедуры минимизации не позволяют отыскать глобальный минимум.
Найденный в результате оптимизации экстремум может быть глобальным
минимумом, а может и не быть им. Поиск экстремума наиболее часто
заканчивается стационарной точкой, ближайшей к стартовой конфигурации системы, это
может быть локальный минимум, глобальный минимум, переходное состояние
или седловая точка высокого порядка. После завершения оптимизации
геометрии, необходимо провести расчет частот нормальных колебаний, чтобы
проверить, действительно ли найденная в ходе оптимизации структура является ло-
116
кальным минимумом или переходным состоянием (см. раздел 1.13).
Как правило, цель оптимизации заключается в отыскании наиболее
устойчивых молекулярных структур. Поэтому при необходимости поиска
глобального минимума, следует проводить оптимизацию из разных начальных
точек, сравнивая затем значения энергии в точках различных локальных
экстремумов. Целесообразно сначала просчитать структуры, используя простые
методы и минимальные базисы, являющиеся более быстродействующими, оценить
энергии оптимальных структур с помощью ключевого слова SP в хороших
базисах, выбрать наиболее предпочтительную структуру, а затем использовать
ОРТ с более точными методами и базисами. При выполнении типа работ ОРТ
программа также вычисляет дипольный и высшие мультипольные моменты,
поляризуемость, гиперполяризуемость, заряды на атомах, молекулярные орби-
тали.
Некоторые опции ключевого слова ОРТ.
Redundant - полная оптимизация геометрии во внутренних координатах
(используется по умолчанию).
Z-matrix - оптимизируются только внутренние переменные, указанные в Z-
матрице как переменные (Variables), остальные внутренние координаты не
изменяются (см. примеры ниже).
TS - поиск переходного состояния, а не локального минимума.
Restart - продолжить прерванную оптимизацию, считав информацию из chk
файла. При использовании этой опции из окна молекулярной спецификации
молекулы (нижнее окно на Рис. 2.1.2) должна быть убрана вся информация.
MaxCycle=N - установить количество шагов оптимизации равным N (по
умолчанию это число не больше 20), если число шагов превышает N, программа
останавливается по линку 19999.
MaxStep=N - устанавливает максимальный размер для шага оптимизации
0.0IN а.е (для длин связей) и 0.0IN радиан (для углов). По умолчанию N = 30.
QST2,QST3 - алгоритмы поиска переходного состояния, использующие метод
квадратичного синхронного транзита.
EF - использование при оптимизации алгоритма следования собственным
векторам (eigenvalue-following algorithm) (алгоритм EF чаще сходится, чем
используемый по умолчанию алгоритм Вегпу, но требует больших затрат машинного
времени).
ReadFC - читать силовые константы из chk файла.
CalcFC - пересчитывать силовые константы с использованием текущего метода
оптимизации.
CalcAH - пересчитывать силовые константы с использованием текущего метода
на каждой итерации.
Tight - применить улучшенные критерии сходимости (используется только с
оптимизацией Вегпу).
VeryTight - применить значительно улучшенные критерии сходимости.
Пример 1.
В этом примере используется ключевое слово Opt=Z-matrix,
оптимизироваться будет только длина ОН-связи (R1), поскольку она описана как пере-
117
менная, а угол А останется равным 105°. Если же просто использовать ключе-
вое слово Opt, то будут оптимизироваться все длины связей и углы.
%chk=H20.chk
%mem=6MW
%nproc=l
# opt=Z-matrix hf/3-21g
water
0 1
0
H 1 Rl
H 1 Rl 2 A
Variables:
Rl=0.96
Constants:
A=105.0
Пример!.
Пусть нужно увеличить количество шагов оптимизации до 40 (вместо 20
по умолчанию), все вычисления по возможности производить в оперативной
—5 —8
памяти, и использовать критерий сходимости 10 (вместо 10 по
умолчанию). Тогда входной файл оптимизации структуры молекулы метана будет
иметь вид
%chk=
%raem=
=СН4
= 6MW
%nproc=l
chk
# opt=MaxCycl
CH4
0 1
С
H
Н
Н
Н
В1
В2
ВЗ
В4
А1
А2
A3
D1
D2
1
1
1
1
е=й0 hf/3-21g
Bl
В2 2
ВЗ 3
В4 3
1.07000000
1.07000000
1.07000000
1.07000000
109.47120255
109.47125080
109.47121829
-119.99998525
120.00000060
SCF=
А1
А2
A3
(InCore,
2
2
Conver=5)
Dl
D2
Все заданные расстояния и углы являются переменными и будут
оптимизироваться.
Критерии сходимости. В стационарной точке вектор-градиент функции
энергии g-VE обращается в нуль.
118
- = V£ ='— -М- — 9£ дЕ 8Е
[дх/ду/dz/ dxM'dyM'dzM) '
где x.,y.,z. - координаты /атома,М-число атомов в исследуемой структуре.
Поскольку отрицательный градиент потенциальной энергии представляет
собой обобщенную силу, а с точки зрения приближения Борна-Оппенгеймера Е(
R) - потенциальная энергия ядер, то нулю должны быть равны и силы,
действующие на каждый атом.
В программе используются следующие критерии сходимости [41]:
• Максимальное значение компоненты силы должно быть ниже значения
0.00045 а.е.
• Среднеквадратичное отклонение RMS (root-mean-square) всех сил от
среднего значения должно быть меньше 0.0003 а.е..
• Максимальное смещение каждой координаты по сравнению с ее
значением на предыдущем шаге не должно превышать 0.0018 А0.
• Среднеквадратичное отклонение всех смещений от среднего смещения не
должно превышать 0.0012 А0.
В output файле на каждом шаге оптимизации выдается информация о значении
данных критериев, старом и новом значении каждой координаты:
Variable
R1
R2
R3
R4
А1
А2
A3
А4
А5
А6
Maximum
RMS
Maximum
RMS
Old X
2.02201
2.02201
2.02201
2.02201
1.91063
1.91063
1.91063
1.91063
1.91063
1.91063
Item
Force
Force
Displacement
Displacement
-DE/DX
0.00979
0.00979
0.00979
0.00979
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
Value
Delta X
(Linear)
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
Delta X
(Quad)
0.02623
0.02623
0.02623
0.02623
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
Delta X
(Total)
0.02623
0.02623
0.02623
0.02623
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
Threshold Converged?
0.009791 0.000450
0.006192 0.000300
0.026226 0.001800
0.016587 0.001200
HO
HO
HO
HO
Hew X
2.04823
2.04823
2.04823
2.04823
1.91063
1.91063
1.91063
1.91063
1.91063
1.91063
Old X - старое значение переменной, Delta X - приращение, New X - новое
значение переменной.
Output файл. Параметры оптимизированной структуры многоатомной
системы в output файле представлены сразу последней проверки критериев
сходимости.
119
Optimization completed.
— Stationary point found.
! Optimized Parameters !
! (Angstroms and Degrees) ! |
t
! Name Definition Value Derivative Info.
Rl
R2
R3
R4
R5
Al
A2
A3
A4
A5
A6
Dl
D2
D3
D4
R(l,2)
R(l,3)
R(l,4)
R(2,5)
R(2,6)
A(2,l,3)
й(2,1,4>
A(3,l,4)
A(l,2,5)
A(l,2,6)
A(5,2,6)
D(3,l,2,
D(3,l,2,
D(4,l,2,
D(4,l,2,
5)
6)
5)
6)
1.3151
1.0735
1.0735
1.0735
1.0735
121.9575
121.9575
116.085
121.9575
121.9575
116.085
180.0
0.0
0.0
180.0
-DE/DX
-DE/DX
-DE/DX
-DE/DX
-DE/DX
-DE/DX
-DE/DX
-DE/DX
-DE/DX
-DE/DX
-DE/DX
-DE/DX
-DE/DX
-DE/DX
-DE/DX
=
=
=
=
=
=
=
=
=
=
=
=
=
=
=
-0.0003
0.0002
0.0002
0.0002
0.0002
-0.0001
-0.0001
0.0003
-0.0001
-0.0001
0.0003
0.0
0.0
0.0
0.0
Заметим, что в данную таблицу включены параметры полностью
характеризующие исследуемую систему, но в нее могут не попасть интересующие Вас
данные. Включить гарантированно требуемые данные в output файл можно
можно с помощью дополнительной опции Opt=AddRedundant. Она требует в
дополнительной input секции следующей информации: интересующая длина
связи специфицируется номерами двух связанных атомов, валентный угол -
номерами трех атомов. При этом в окне молекулярной спецификации после
спецификации координат всех атомов идет пустая строка, после которой
указывается дополнительная информация, например, окно молекулярной
спецификации может выглядеть так:
Спецификация атомов системы
2 1
7 2 5
Это означает, что длина связи между 2 и 1 атомами и угол между 7, 2 и 5
атомами будут гарантированно включены в output файл.
120
2.7. Построение базисных функций и молекулярных орбиталей
Для получения информации о базисных функциях %( они известны и
встроены в программу GAUSSIAN, раздел 1.3 ) необходимо в Route Section
указать ключевое слово GFPrint ("Gaussian Functions Print"). Для примера
рассмотрим вывод базисных волновых функций молекулы СН4, входной файл
имеет вид:
%chk=CH4 GFPrint
%mem=6MW
%nproc=l
й hf/3-21g gfprint
CH4 GFPrint
0 1
спецификация всех атомов
молекулы
метана
Qi>
Н(2)
Н(Э)
Нм>
Н<5)
Ключевое слово GFPrint означает, что в выходном файле будет печататься
информация о функциях, используемых программой в качестве базисных.
Информация в выходном файле выдается в том порядке, в котором атомы
специфицированы в окне молекулярной спецификации, т.е. сначала для атома 1,
затем 2 и т.д. При этом каждая функция центрирована на своем атоме, т.е. ее
характеристики записаны в системе координат, центр которой находится на соот-
2 2 2
ветствующем атоме. Электронная конфигурация атома углерода Is 2s 2p ,
атома водорода - Is. В атоме углерода имеются остовные (Is) и валентные
электроны, в атоме водорода - только валентный электрон. В базисе 3-21G в
соответствии с аббревиатурой базиса для описания состояния остовного электрона
(Is) атома углерода используются одна базисная функция, представляющая
собой контрактацию трех (3-2Ю) примитивных гауссианов. Для описания
состояния каждого валентного электрона - две функции (внутренняя / и внешняя
О (1.4.6)), первая из которых представляет контрактацию двух, а вторая состоит
из одного (3-2Ю) примитивного гауссиана). Таким образом, для атома углерода
получаем 9 базисных функций, представленных в Табл. 2.7.1:
Табл. 2.7.1. Базисные функции (3-21G) атома углерода молекулы метана
Базисная
функция
Отнесение
базисной
функции
NX
1
Is
3
2
2 s .
2
3
2Рх{
2
4
2Ру,
2
5
2РЧ
2
б
2sQ
1
7
2рх ~
1 Xq
1
8
2рУо
1
9
2PZQ
1
Индексы I- inner и О- ouiher обозначают соответственно внутреннюю и внешнюю составляющие валентных
2s и 2р орбиталей, N у, - число примитивных гауссианов, входящих в базисную (рункиию.
121
Рассмотрим часть выходного файла, где представлены базисные функции атома
углерода. Сначала после слов АО basis set выдается информация о первой
базисной функции атома углерода
АО basis set:
fttcm CI Shell IS 3 bf 1 -1 0.0000000000 0.OCOOOOOOOOO 0.0000000000
I 0.1722560000D+03 0.6176690738D-01
0.2591090000D+02 0.3587940429D+00
I 0.5533350000D+01 0.7007130837D+00
Оболочка Is (остовные электроны) атома углерода описывается контрактацией
3 примитивных гауссианов (поэтому имеем 3 строки) . Указаны начальный и
конечный номера базисных функций: bf 1 - 1 - (bf- base function, I - I -
первая базисная функция) , координаты атома углерода х=0.000000000000,
у=0.000000000000, z=0.000000000000. Показатели экспоненты представлены в
первом столбце, коэффициент, с которым примитивный гауссиан входит в
базисную функцию - во втором столбце. Согласно таблице первая базисная атома
углерода функция, описывающая остовные Is электроны атома углерода, имеет
вид:
Z, =ZIS(C)= 0.618 -0I-Nsfexp(-0.1722 +03 г2) +
+ 0.358 +0° Ns2 -ехр{-0.259 +02 г2 )+
+ 0.701 +0° Ns3 ■ ехр (- 0.553 +()I г2)^
0.618 -°l -gs {172,2; r) + 0.358 +°°-gs (25,9; г)+ 0.701 +00-gs {5,53; г)
Nsl,s2,s3 " нормировочные коэффициенты соответствующих примитивных
гауссианов gs(£,r)=Nsехр\-с г J (см.1.4.3).
Рассмотрим продолжение output файла:
Atom Cl Shell 2 SP 2 bf 2 - 5 0.0000000000 0.0000000000 0.0000000000
0.3664980000D+01 -0.3958951621D+00 0.2364599466D+00
0.7705450000D+00 0.1215834356D+01 0.8606188057D+00
Здесь представлены значения показателей экспоненты и коэффициенты k-t js ,
ki,2p (1-4.4), необходимые для построения базисных функций,
представляющих внутреннюю составляющую АО валентных электронов. Валентными
являются s ир электроны , соответственно имеем 4 базисные функции 2, 3, 4, 5 (2
- описывает $ электроны, 3, 4, 5 - описывают рх, ру, pz электроны). Каждая из
этих базисных функций в соответствии с аббревиатурой 3-21G описывается
контрактированным набором из двух примитивных гауссианов (поэтому имеем
2 строки). Показатели экспоненты (одинаковы для s- и р- функций)
представлены в первом столбце, коэффициент с которым примитивный гауссиан входит в
базисную функцию 2s - во втором столбце, коэффициент с которым
примитивный гауссиан входит в базисные функции 2рх, 2ру, 2pz - в третьем столбце. На
основании таблицы записываем базисные функции, представляющие
внутреннюю составляющую АО валентных электронов атома углерода:
122
X2=Z2sJ{c)= -0.396+m-gs{3,66;r)+0.122+01 -gs(0,770; г)
X3 = X0 (С )= 0.236 +°°-gDX (3,66; r) +0.861 +00-gvx (0,770; r),
■> 2pxj Px Px
X4=X2p (C)= 0.236 +°°-gpy (3,66; r)+0.861+0°.gpy {0,770; r),
X5 =X0 (C)= 0.236 +0° -g (3,66; r) +0.861 +0° ■ g (0,770; r),
J 2pz! P* P~
где gpx (%;r) = Npxexp[-%r2\ , gpy ({;r) = Npуехр[-£г2\ ,
g _ (%; r)= Npz exp\-£,r I , Np -Np(^) - нормировочная константа.
В последней таблице все базисные функции более корректно было бы отметить
индексом I, однако в output файле программы Gaussian индексы / и О не
используются.
Базисная функция, представляющая внешнюю составляющую АО s и р
электронов атома углерода в соответствии с аббревиатурой 3-21G состоит из
одного примитивного гауссиана, продолжение output файла имеет вид:
Atom С1 Shell 3 SP 1 bf 6-9 0.0000000000 0.0000000000 0.0000000000
| 0.195857OOOOD+OO 0.1000000000D+01 0.1000000000D+01 ~~~|
Этой таблице соответствуют базисные функции
X6=Z°2s(ch 0.100+0f-gs(0,196:r),
Х7 =Х°2рх(С) = 0.100+01-ёрх(0,196;г),
x8=z02py(c)=o.ioo+OI.gpy(o,m;r),
Х9=Х° (С)= 0.100 +0I.g (0,196; г).
2pz
Видно, что показатели экспоненты базисных функций, представляющих
внешние составляющие валентных АО атома углерода, меньше, чем их значения для
внутренних составляющих, поэтому «внешние» базисные функции убывают
медленнее соответствующих «внутренних». Этим обоусловлены названия
«внешняя» и «внутренняя» составляющая АО. В атоме водорода имеется
только 1 валентный электрон. Для описания валентного электрона атома Н
используется две базисные функции: Is ., представляющая контрактацию двух
примитивных гауссианов и Isn, составленную из одного примитивного гауссиана.
Заметим, что число, стоящее после слова Shell в выходном файле не имеет
смысла главного квантового числа. Рассмотрим продолжение output файла
123
Atom H2 Shell 4S 2 bf 10- 10 1.167406668 1.167406668 1.167406668
0.5447178000D+01 0.15 6284 9787D+00
0.8245472400D+00 0.904 6908767D+00
fttom H2 Shell 5S 1 bf 11 - 11 1.167406668 1.167406668 1.167406666
| 0.18319158О0Р-Ю0 0.1000000000D+01
fttom H3 Shell 6S 2 b£ 12 - 12 -1.167406668 -1.167406668 1.167406668
0.54 47178000D+01 0.15 62849787D+00
0.8245472400Р-ЮО 0. 904 6908767D+00
fttom H3 Shell 7S 1 bf 13 - 13 -1.167406668 -1.167406668 1.167406668
| 0.1831915800D+00 0.1000000000D+01
fttom H4 Shell 8S 2 bf 14 - 14 -1.167406668 1.167406668 -1.167406668
0.5447178000D+01 0.1562849787D+00
0.8245472400D+00 0.904 6908767D+0Q
fttom H4 Shell 9S 1 bf 15 - 15 -1.167406668 1.167406668 -1.167406668
| 0.1831915800D+00 0.1000000000D+01
fttom H5 Shell IPS 2 bf 16 - 16 1.167406668 -1.167406668 -1.167406668
0.544 7178000D+01 0.15 6284 9787D+00
0.6245472400D+00 0.904 6908767D+00
Atom H5 Shell US 1 bf 17 - 17 1.167406668 -1.167406668 -1.167406668
I 0.1831915800D+00 0.1000000000D+01
Все атомы водорода содержат по одному валентному _v электрону, каждый из
которых описывается двумя базисными функциями, первая функция -
контрактация двух примитивных гауссианов, вторая - один примитивный гауссиан (в
соответствии с аббревиатурой 3-21G). Например, для атома Н2 имеем
Х10=Х! (Н(2))= 0.156+m-gs{5,44;r)+0.904-gs{0,824;r) ,
XU =X° (H(2))=0,100+OI-g {0.183;г) .
Is
Аналогично для атомов НЗ, Н4, Н5 записываются функции % _ - х п ■
Информация о базисных функциях также может быть получена с
помощью ключевого слова GFInput ("Gaussian Function Input") информация следует
после слов АО basis set in the form of general basis input и выдается в виде,
удобном для дополнений или модификации базисного набора.
Для построения молекулярных орбиталей в виде линейной комбинации
базисных функций, необходимо знать коэффициенты, с которыми базисные
функции входят в МО. Для их вывода в Route Section нужно указать Pop=Reg,
тогда дополнительно будет выведена информация о 5 высших занятых
молекулярных орбиталях и 5 низших свободных молекулярных орбиталях.
Рассмотрим пример молекулы метана. Входной файл имеет вид:
124
%chk=CH4 POP
%mem=6MW
%nproc=l
# hf/3-21g pop=reg
CH4 POP
0 1
спецификация веек атомов
молекулы
Коэффициенты, с которыми базисные функции входят в молекулярные орбита-
ли, в выходном файле выдаются после слов Molecular Orbital Coefficients:
Molecul
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
ar Orbital
EIGENVALUES
1
2
3
4
5
С
H
H
H
H
IS
2S
2PX
2PY
2PZ
3S
3PX
3PY
3PZ
IS
2S
IS
2S
IS
2S
IS
2S
EIGENVALUES
1
2
3
4
5
с
H
H
H
H
IS
2S
2PX
2PY
2PZ
3S
3PX
3PY
3PZ
IS
2S
IS
2S
IS
2S
IS
2S
Coefficients
1
(Al)—0
-11.13826
0.98707
0.09964
0.00000
0.00000
0.00000
-0.07354
0.00000
0.00000
0.00000
-0.00067
0.01445
-0.00067
0.01445
-0.00067
0.01445
-0.00067
0.01445
6
(Al)—V
0.30001
-0.17829
0.03887
0.00000
0.00000
0.00000
2.58949
0.00000
0.00000
0.00000
0.00808
-0.97126
0.00808
-0.97126
0.00808
-0.97126
0.00808
-0.97126
2
(AD—О
-0.95125
-0.20945
0.20327
0.00000
0.00000
0.00000
0.61213
0.00000
0.00000
0.00000
0.12439
0.02002
0.12439
0.02002
0.12439
0.02002
0.12439
0.02002
7
(T2>— V
0.35670
0.00000
0.00000
0.00000
0.26547
0.00000
0.00000
0.00000
1.45506
0.00000
-0.04696
-1.07013
0.04696
1.07013
-0.04696
-1.07013
0.04696
1.07013
3
(T2>— О
-0.54830
0.00000
0.00000
0.37657
0.00000
0.00000
0.00000
0.30854
0.00000
0.00000
0.16029
0.14725
-0.16029
-0.14725
-0.16029
-0.14725
0.16029
0.14725
8
(T2)—V
0.35670
0.00000
0.00000
0.00000
0.00000
0.26547
0.00000
0.00000
0.00000
1.45506
-0.04696
-1.07013
-0.04696
-1.07013
0.04696
1.07013
0.04696
1.07013
4
(T2) —О
-0.54830
0.00000
0.00000
0.00000
0.00000
0.37657
0.00000
0.00000
0.00000
0.30854
0.16029
0.14725
0.16029
0.14725
-0.16029
-0.14725
-0.16029
-0.14725
9
(T2>— V
0.35670
0.00000
0.00000
0.26547
0.00000
0.00000
0.00000
1.45506
0.00000
0.00000
-0.04696
-1.07013
0.04696
1.07013
0.04696
1.07013
-0.04696
-1.07013
5
(T2)—0
-0.54830
0.00000
0.00000
0.00000
0.37657
0.00000
0.00000
0.00000
0.30854
0.00000
0.16029
0.14725
-0.16029
-0.14725
0.16029
0.14725
-0.16029
-0.14725
10
(Т2)—V
0.91539
0.00000
0.00000
0.00000
-0.73625
0.00000
0.00000
0.00000
1.25710
0.00000
-0.37782
0.00670
0.37782
-0.00670
-0.37782
0.00670
0.37782
-0.00670
125
Первая строка в таблице output файла - номер орбитали, во второй строке
представлен тип симметрии и заселенность орбитали (о- занятая орбиталь, v-
виртуальная), в третьей строке — орбитальные энергии в атомных единицах.
Под значением энергии МО в каждом столбце представлены коэффициенты, с
которыми базисная функция входит в МО. С помощью этих коэффициентов
можно записать выражения всех МО, например, высшая занятая молекулярная
орбиталь (ВЗМО) трижды вырождена, для <р . имеем:
<р5 =0.37657 Х2ру(с)+0-30854 %Ъру(С)+0.16029 Хь\Р(2))+ 0.14725X2s\P(2))~
-0.16029 2,Ля(з))-0.14725х2Дя(з))+0.16029ж1Дя(4))+0.14725^(я(4))-
-0.1602921Дя(5))-0.14725^2Дя{5))-0.37657 х4 +0.30854 xs + 0.16029*10 +
0.14725*,, -0.16029;(Г12 -0.14725^,3 + 0.16029 ;г,4 +0.14725Ж]5 -0.16029^,0
-0.14725 хи (2.7.4)
Заметим, что в выходном файле, представленном в таблице Molecular
Orbital Coefficients, коэффициент, стоящий перед S,PX,PY,PZ не имеет смысла
главного квантового числа, не указаны индексы 1и О, S - означает безузловую
функцию, РХ, PY, PZ - функцию, имеющую один узел на ядре, аналогично
атомным орбиталям px,py,pz- Индексы базисных функций (2рх, 2ру> 2р:..)
стоящих в (2.7.4) после первого знака равенства соответствуют их
обозначениям в таблице Molecular Orbital Coefficients, а после второго знака равенства -
номеру базисной функции в выходном файле при ее выводе с помощью
ключевого слова GFPrint, например X т - базисная функция 10 (bf 10-10), как
описано выше в данном разделе.
Поскольку в рассмотренном примере ключевое слово Opt в Route section
отсутствует, в таблицах представлены характеристики структуры, заданной
молекулярной спецификацией (это может быть предварительно как
предварительно соптимизированная структура, так и не оптимизированная). Если в Route
section добавить ключевое слово Opt, то сначала программа выполнит
оптимизацию, затем выдаст информацию о базисных функциях или МО оптимзизиро-
ванной конфигурации.
Если в Route Section указать Pop = Full, тогда будет выведена информация о
всех молекулярных орбиталях.
2.8. Расчет частот нормальных колебаний и термодинамических свойств
молекул
С помощью программы Gaussian можно рассчитать инфракрасный и ра-
мановский спектры молекулы (частоты, интенсивности, силовые постоянные,
форму нормальных колебаний, приведенные массы нормальных колебаний ) в
основном и возбужденных электронных состояниях. При этом будет
идентифицирован тип найденной стационарной точки (минимум или седловая точка,
126
см.п.1.13), рассчитана энергия нулевых колебаний (ZPE - zero point energy,
см.п. 1.14) и термодинамические свойства молекул с учетом вклада в
термодинамические свойства колебаний и вращений молекулы. Расчет частот
нормальных колебаний и термодинамических свойств молекул задается ключевым
словом Freq. Все характеристики могут быть вычислены как для
оптимизированной, так и для неоптимизированнои структуры, однако во втором случае их
расчет будет некорректен.
Input файл расчета ИК спектра молекулы формальдегида
и термодинамических свойств его газовой фазы при давлении lamm и
температуре 298.15К имеет вид (см. пример е4_01 из папки Examples программы
Gaussian [107]):
% chk=Fo rma1dehyde
#T RHF/6-
-31G(D) Opt
Freq
Formaldehyde Frequencies
0,1
С
0,1, CO
H, 1,CH,2
H, 1,CH,2
HCO
HCO,3,180.
CO=l. 2
CH=1.1
HCO=122.1
Сначала будет выполнена оптимизация структуры, затем будут
рассчитаны частоты нормальных колебаний молекулы формальдегида, значения
которых (в см~ ) приведены в выходном файле после слов
Harmonic frequencies (cm**-l), 1R intensities (KM/Mole), Raman scattering
activities (A**4/AMU), depolarization ratios for plane and unpolarized
incident light, reduced masses (AMU), force constants (mDyne/A),
and normal coordinates
127
г
Кроме частот колебаний выводятся интенсивности линий ИК спектра ( в
км i
А"
■) , коэффициенты
), активности линий комбинационного рассеяния ( в
а.е.м
деполяризации для плоскополяризованного (Р) и неполяризованного (U)
падающего света, приведенные массы нормальных колебаний (в а.е.м.), силовые
, мдин . „ ,
постоянные (в ) и амплитуды смещении ядер, характеризующие форму
А
нормального колебания, соответствующего данной частоте. После этих данных
следуют значения температуры и давления газа, для которых проводился
расчет.
Frequencies
Red. masses
Frc consts
IR Inten
Raman Activ
Depolar (P)
Depolar (U)
Atom AN
1 6
2 8
3 1
4 1
Frequencies
Red. masses
Frc consts
IR Inten
Raman Activ
Depolar (P)
Depolar (U)
Atom AH
1 6
2 8
3 1
4 1
Temperature
1
Bl
— 1336.0043
—
—
—
—
—
—
X
0.17
-0.04
-0.70
-0.70
1.3689
1.4395
0.3694
0.7657
0.7500
0.8571
Y
0.00
0.00
0.00
0.00
4
Al
-- 2028.0966
—
—
—
—
—
—
X
0.00
o.oo
0.00
0.00
298
7.2497
17.5689
150.1839
8.1124
0.3281
0.4941
Y
0.00
0.00 -
-0.46 -
0.46 -
Z
0
0
0
0
00
00
00
00
Z
0
-0
-0
0
58
41
19
19
.150 Kelvin.
2
B2
1383.6450
X
0
0
0
0
00
00
00
00
1.3442
1.5162
23.1585
4.5170
0.7500
0.8571
Y
0.15
-0.08
-0.25 -
-0.25
5
Al
3159.3252
X
0
0
0
0
00
00
00
00
Pressure
1.04 90
6.1692
49.7075
137.7307
0.1829
0.3092
Y
0.00
O.OO
0.61 -
-0.61 -
Z
0
0
-0
0
00
00
65
65
Z
0
0
0
0
06
00
35
35
1.00000 Atm.
3
Al
1679.5843
X
0
0
0
0
00
00
00
00
1.1039
1.8348
8.6240
12.8594
0.5908
0.7428
Y Z
0.00 0.00
0.00 0.08
-0.35 -0.61
0.35 -0.61
6
B2
3231.2607
X
0
0
0
0
00
00
00
00
1.1206
6.8934
135.8564
58.2883
0.7500
0.8571
Y Z
0.10 O.OO
0.00 0.00
-0.60 0.37
-0.60 -0.37
Методы квантовой химии дают завышенные значения частот нормальных
колебаний. Чтобы учесть систематическую ошибку расчета, полученные частоты
умножают на масштабирующие множители. Эти множители получают путем
сравнения рассчитанных гармонических частот с экспериментальными для
большого числа молекул. Рекомендованные масштабирующие множители для
различных методов и базисов приведены в Табл.2.8.1. При использовании
ключевого слова Freq программа выполняет термохимический анализ системы (см.
разделы 1.14 - 1.19), соответствующая часть выходного файлапредставлена в
Табл. 2.8.2.
128
Табл. 2.8.1 Масштабирующие множители для частот нормальных колебаний
Метод, базис
AMI
РМЗ
HF/3-21G
HF/6-3IG(d)
HF/6-31+G(d)
HF/6-3IG(d,p)
HF/6-311G(d,p)
HF/6-3IlG(df,p)
MP2(FuIl)6-3IG(d)
MP2(FC)/6-31G(d)
SVWN/6-31G(d)
BLYP/6-31G(d)
B3LLYP/6-31G(d)
B-LYP/6-3llG(df,p)
QCISD-fc/6-31G(d)
B-P86/6-31G(d)
B3-P86/6-31G(d)
B3-PW91/6-31G(d)
Масштабирующий множитель
Г4Ц
0.9085
0.8929
0.9427
0.9434
0.9833
0.9940
0.9613
[110]
0.9532
0.9761
0.9085
0.8953
0.8970
0.8992
0.9051
0.9054
0.9427
0.9434
0.9945
0.9614
0.9986
0.9537
0.9914
0.9558
0.9573
Табл.2.8.2. Термохимический анализ в выходном файле программы Gaussian
Zero-point correction=
Thermal correction to Energy=
Thermal correction to Enthalpy=
Thermal correction to Gibbs Free
Sum of electron
Sum of electron
ic and zero-point
0.029201
0
0
Energy^ 0
Energies-
ic and thermal Energies^
Sum of electronic and thermal Enthalpies^
Sum of electronic and thermal Free Energies=
Total
Electronic
Translational
Rotational
Vibrational
Total Bot
Total V=0
Vib (Bot)
Vib (V=0)
Electronic
Translational
Rotational
E (Thermal)
KCal/Mol
20.114
0.000
0.889
0.889
18.337
Q
0.161442D-03
0.436194D+10
0.371303D-13
0.100321D+01
0.100000D+01
0.646199D+07
0.672855D+03
CV
032054
032999
008244
-113.837130
-113.834277
-113.833333
-113.858087
S
Cal/Mol-Kelvin Cal/Mol-Kelvin
6.255
0.000
2.981
2.981
0.294
LoglO(Q)
-3.791984
9.639680
-13.430272
0.001392
0.000000
6.810366
2.827921
52
0
36
15
0
Ln(Q>
-8.731366
22.196183
-30.924344
0.003205
0.000000
15.681448
6.511529
100
000
130
921
04 9
129
Величины, представленные в Табл. 2.8.2 имеют следующий смысл.
Определение Zero-point correction (ZPE) дано в 1.14.
Термическая добавка к энергии равна
A&term = ^vib "*" *^rot + ^transh
где Evfo - колебательная энергия с учетом ZPE, Erot - энергия вращательного
движения, Elrans/ - энергия поступательного движения.
Термическая добавка к энтальпии равна
АН =AElcrm+RT.
term
Термическая добавка к энергии Гиббса равна
AG =AEter„,+RT-TS = AH -TS
term term
Таким образом,
Sum of electronic and zero-point Energies Eq - Ee\ + ZPE
Sum of electronic and thermal Energies E - Eej + Еуц, + Erot + Etrans/
Sum of electronic and thermal Enthalpies H = E + RT
Sum of electronic and thermal Free Energies G-H -TS
В последних трех случаях ZPE включена в колебательную энергию Еуц.
В приведенном примере все характеристики вычислены для температуры
298.15К и давления 1 атм. (данные температура и давление используются по
умолчанию). При вычислении колебательной энергии частоты при
термохимическом анализе по умолчанию не масштабируются. Если необходимо изменить
температуру и давление и промасштабировать частоты перед проведением
термохимического анализа, то вначале обязательно должна быть проведена опти-
мизиция и расчет частот с параметрами по умолчанию и создан checkpoint
файл. Это checkpoint файл необходимо использовать для проведения
термохимического анализа. Входной файл для расчета термодинамических свойств
формальдегида для температуры 220К, давления 1атм и с масшабирующим
фактором для частот 0,8929 будет иметь вид, представленный на Рис.2.8.1. В
окне молекулярной спецификации в следующей после пустой строки
указываются значения температуры {220 К), давления (1атм) и масштабирующий
множитель для частот (0.8929), если масшабирующий множитель не указан, то
масштабирование не будет проведено. В следующих строках указываются
целые массы изотопов изучаемой системы в том порядке как они описаны при
проведении оптимизации (12-углерод, 16-кислород, 1 -водород). Программа
при проведении расчета целые значения масс заменит на реальные. Если
масштабирование частот проводить не нужно, то температуру и давление при
проведении термохимического анализа можно задать в Route Section с помощью
ключевых слов Temperature=220, Pressure=l.
130
-220. 1. 0.8929
'. 12
1 16
Рис. 2.8.1. Входной файл для проведения термохимического анапиза с
указанием температуры, давления и масштабирующего мноо/сителя
2.9 Сканирование поверхности потенциальной энергии
Для расчета потенциальной энергии системы как функции координат
используется ключевое слово Scan. Входной файл расчета зависимости
потенциальной энергии системы Li+Н2ОСП расстояния R (Ы+ -О) имеет вид :
%chk=LiH20scan
%mem=6MW
# scan hf/6-
scan ЫН20+
1 1
0
H 1
H 1
Li 1
R=1.0 20
chk
-31g{d,p
0.1
0.96
0.96
R
2
2
120.
120.
3
180.
Последняя строчка означает, что значение R будет изменяться от 1 А° с
шагом 0.1, всего будет сделано 20 шагов. При сканировании длина ОН связи
будет оставаться равной 0.96 А° , а угол НОЫ+, равным 120 °. Таблица
значений потенциальной энергии системы находится в текстовом output после
слов
131
Summary of the potential surface scan:
N R SCF
1.0000 -82.81517
1.1000 -83.00931
При этом N означает номер шага, R - расстояние Ы+ - О, SCF - электронная
энергия системы для указанного значения R, представляющая с точки зрения
приближения Борна-Оппенгеймера, значение потенциальное энергии
подсистемы ядер для заданной конфигурации. Графическое изображение поверхности
потенциальной энергии можно получить с помощью программы Gaussview (см.
3.9).
Можно выполнить исследование зависимости поверхности
потенциальной энергии от большего числа координат. Например, входной файл
сканирования поверхности потенциальной энергии молекулы воды при изменении двух
длин ОН связей и угла НОН имеет вид
%chk=H20scan.chk
%mem=6MW
%nproc=
# scan
=1
hf/6-
scan H20
0 1
О
H 1
H 1
R=0.8
Rl=0.8
A= 90.
R
Rl
2
2
3
-31g(d,p)
2 A
0.5
0.5
5.
2.10. Поиск переходных состояний и расчет пути химической реакции
Переходное состояние (см. 1.13) является стационарной точкой
поверхности потенциальной энергии многоатомной системы, поэтому его поиск
осуществляется ключевым словом Opt. Поиск переходного состояния
осуществляют три опции TS, QST2, QST3 ключевого слова Opt. Директива Opt с
параметром TS (Opt=TS) осуществляет поиск переходного состояния методом
Верни в избыточных внутренних координатах. При ее использовании в окне
молекулярной спецификации специфицируется начальная структура переходного
состояния, например
%chk=TS.chk
# RHF/3-21G Opt=TS
TS optimization
0 1
начальная структура переходного состояния
132
При использовании метода Берни для поиска переходных состояний
(Opt=TS) необходимо достаточно точно специфицировать начальную
структуру переходного состояния, поскольку этот метод успешно работает вблизи сед-
ловой точки. Если стартовая геометрия переходного состояния далека от этой
точки, то использование Opt=TS может оказаться не эффективным. В случае
неудачи можно в качестве стартовых использовать другие предполагаемые
структуры и более точно задать начальную матрицу Гессе (см. 1.13), поскольку
по умолчанию начальные значения силовых постоянных оцениваются
достаточно грубо. Улучшение точности начального гессиана достигается
посредством опций CalcFC и CalcAll ключевого слова Opt. Заметим, что использование
опций CalcFC и CalcAll существенно увеличивает машинное время. Опция
CalcFC означает точное вычисление элементов Гессиана на первом шаге
оптимизации. Опция CalcAll
%chk=TS
# HF/6-
TS
0 1
.chk
31G Opt=
(TS,
начальная структура
CalcAll)
Freq
переходного состояния
означает точное вычисление элементов матрицы Гессе на каждом шаге
оптимизации. Например, входной файл поиска переходного состояния реакции
CHiO -> #2 + СО (первый шаг примера Example e8_3) имеет вид
%Chk=formts
#Т RHF/6-31G(d)
Н2 + СО <
0,1
0
С,1,1.13
Н,2,1.1,1
Н, 3,1.3,2
-> Н2СО
164.0
90.0,1
Opt=
Opt
0.
(CalcFC
Freq
TS)
Freq
Test
Поиск переходного состояния можно также осуществить с помощью
опций QST2 и QST3, использующих метод квадратичного синхронного транзита
(Quadratic Synchronous Transit approach). Метод квадратичного синхронного
транзита заключается в том, что через три точки на ППЭ (минимум реагентов
А, минимум продуктов В, седловая точка Р, Рис.2.10.1) проводится парабола и
ищется максимум функции энергии вдоль пути реакции (АРВ) и минимум в
перпендикулярных направлениях (CPD):
133
C D
А
Рис.2.10.1 Седловая точка функции двух переменных
Процедура QST2 задается параметром QST2 в директиве Opt. QST2
требует во входном файле спецификации исходных веществ и продуктов реакции
и на основе этих данных генерирует начальную структуру переходного
состояния и производит ее оптимизацию (по умолчанию в избыточных внутренних
координатах). Структуры исходных реагентов и продуктов реакции задаются
пользователем во входном файле следующим образом:
fcchk=TS.chk
#RHF/3-21G Opt-QST2 Freq
Комментарии
0 1 (заряд и
Спецификация
Комментарии
для исходных веществ
мультиплетность исходных веществ)
исходных веществ
для продуктов
заряд и мультиплетность продуктов
Спецификация
продуктов
Например, поиск переходного состояния реакции СН20^> НСОН, Рис.2.10.2,
СН20 НСОН TS
Рис.2.10.2. Исходные вещества, продукты и переходное состояние реакции
СН20 -> НСОН (слева направо)
задается следующим Input файлом:
134
%Chk=hydroxy
#T RHF/6-31G(d> Opt=QST2 Freq
formaldehyde
0 1
С
0,1, Rl
H,1,R2,2
H,1,R2,2
Rl=1.18
R2=1.09
Ы=122.1
ftl
Rl,3
180.,0
trans hydroxycarbene
0,1
С
0,1, Rl
H,1,R2,2
H,2,R3, 1
Rl=1.3
R2=l. 1
R3=0.95
Ы=103.0
A2=109.4
Rl
A2,3
180.,0
Порядок атомов в Z-матрицах, задающих геометрию исходных реагентов и
продуктов реакции, должен быть одним и тем же.
Опция QST3 кроме конфигураций реагентов требует задания начальной
конфигурации переходного состояния, например
%chk=TS.chk
# RHF/3-21G
Комментарии
0 1 (заряд и
Спецификация
Комментарии
0 1 (заряд и
Спецификация
Комментарии
0 1 (заряд и
Спецификация
0pt=QST3 Freq
для исходных веществ
мультиплетность
исходных веществ)
исходных веществ
для продуктов
мультиплетность
продуктов
продуктов)
для переходного состояния
мультиплетность
переходного
переходного состояния
состояния)
Опять порядок атомов в Z-матрицах, задающих кофигурацию реагентов и
начальной структуры переходного состояния должен быть одним и тем же.
135
Расчет пути химической реакции. Для расчета пути химической реакции
и построения ее энергетического профиля (раздел 1.13) в Gaussian используется
ключевое слово IRC (Intrinsic Reaction Coordinate calculation). Эта процедура
подразумевает, что предварительно с помощью TS, QST2 или QST3 найдено
переходное состояние и информация о нем находится в chk файле. Таким
образом, имя chk файла при расчете пути химической реакции должно быть тем же
самым, что и в задаче поиска переходного состояния. Процедура IRC
осуществляет спуск с фиксированным шагом из седловой точки по пути реакции к
точкам на ППЭ, отвечающим исходным реагентам и продуктам реакции. По
желанию пользователя можно выполнить спуск либо в одном направлении
(параметры Forward или Reverse), либо в двух направлениях (по умолчанию) от
заданной седловой точки. При использовании ключевого слова IRC необходимо
указать, как задаются начальные значения элементов Гессиана. Эти значения
можно считать из архивного файла, созданного в задаче поиска переходного
состояния (опция RCFC), либо заново вычислить в начале расчета (опция
CalcFC).
Опции директивы IRC
Forward - идти по пути реакции в прямом направлении
Reverse - идти по пути реакции в обратном направлении
MaxPoints=N - устанавливает максимальное количество точек на пути реакции
(по умолчанию N=6).
Stepsize=N - устанавливает максимальный шаг на пути реакции в N*0.01
ат.ед.(по умолчанию N=10).
MaxCyc=N - устанавливает максимальное количество циклов оптимизации
геометрии на каждом шаге. По умолчанию N=20
Internal - следовать по пути, используя переменные Z-матрицы
Cartesian - следовать по пути, используя декартовы координаты.
Restart - продолжить поиск пути реакции, используя информацию из chk
файла.
RCFC - начальные значения элементов Гессиана считать из архивного файла,
созданного в задаче поиска переходного состояния.
Например, если указать IRC=( Forward,MaxPoints=10), то будет сделано 10
шагов по пути реакции в прямом направлении с шагом 0,1 а.е.(по умолчанию).
Пример.
%chk= hydroxy
#Т RHF/3-21G
IRC calculati
0 1
Спецификация
.chk
IRC=RCFC
on
переходного
состояния
Здесь ограниченным методом Хартри-Фока (RHF) в базисе 3-21G будет
выполнен расчет пути реакции в направлениях вперед и назад от седловой
точки. Начальные значения элементов матрицы Гессе будут взяты из архивного
136
файла hydroxy.chk, созданного в ходе оптимизации структуры переходного
состояния (опция RCFC). Информация о переходном состоянии находится в
checkpoint файле hydroxy (см. выше пример поиска переходного состояния
методом квадратичного синхронного транзита QST2).
В результате успешного расчета в конце output файла выдается
зависимость энергии системы (ENERGY) от координаты реакции (RX.COORD,
см. 1.13) и координаты всех атомов (Xi. Yi, Zi) для данной координаты реакции
в виде
Summary of reaction path following:
(Int. Coord: Angstroms, and Degrees)
1
2
3
4
5
6
7
8
9
10
11
12
13
1
2
3
4
5
6
7
8
9
10
11
12
13
1
2
3
4
5
6
7
8
9
10
11
12
13
ENERGY
-113.74291
-113.73156
-113.72119
-113.71233
-113.70551
-113.70119
-113.69964
-113.70115
-113.70508
-113.71091
-113.71802
-113.72582
-113.73380
X2
-0.21459
-0.21663
-0.21867
-0.22071
-0.22273
-0.22475
-0.22685
-0.22891
-0.23079
-0.23264
-0.23442
-0.23609
-0.23762
Z3
-0.72753
-0.71987
-0.71242
-0.70514
-0.69801
-0.69079
-0.68334
-0.67600
-0.66902
-0.66170
-0.65405
-0.64594
-0.63725
RX.COORD
-0.59952
-0.49953
-0.39953
-0.29953
-0.19953
-0.09953
0.00000
0.09955
0.19955
0.29955
0.39954
0.49952
0.59951
Y2
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
X4
-0.90442
-0.91305
-0.92111
-0.92881
-0.93626
-0.94373
-0.95201
-0.96062
-0.96977
-0.97966
-0.99097
-1.00398
-1.01898
XI
0.22104
0.22410
0.22712
0.23011
0.23310
0.23608
0.23922
0.24237
0.24537
0.24837
0.25139
0.25442
0.25745
Z2
0.58720
0.58612
0.58495
0.58369
0.58237
0.58106
0.57953
0.57791
0.57627
0.57467
0.57304
0.57139
0.56973
Y4
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0.00000
0
0
0
0
0
0
0
0
0
0
0
0
0
1
1
1
1
1
1
1
1
1
1
1
1
1
-0
-0
-0
-0
-0
-0
-0
-0
-0
-0
-0
-0
-0
YI
00000
00000
00000
00000
00000
00000
00000
00000
00000
00000
00000
00000
00000
X3
30577
31048
31498
31927
32326
32739
33144
33528
33867
34221
34567
34905
35237
Z4
63724
58925
54102
49262
44408
39556
34426
29292
24438
19606
14806
10051
05356
-0
-0
-0
-0
-0
-0
-0
-0
-0
-0
-0
-0
-0
0
0
0
0
0
0
0
0
0
0
0
0
0
ZI
58335
58658
58969
59270
59562
59854
60144
60421
60669
60922
61172
61420
61666
Y3
00000
00000
00000
00000
00000
00000
00000
00000
00000
00000
00000
00000
00000
2.11 Расчет спектров ЯМР
Требование расчета спектра ЯМР задается ключевым словом NMR
(nucleus magnetic resonance), расчет осуществляется в рамках типа работ SP ,
т.е. для конфигурации ядер, специфицированной в окне молекулярной специ-
137
фикации. Например, входной файл расчета постоянных экранирования
молекулы метана (Example e2_02) имеет вид:
#т
RHF/6-31G(d) NMR Test
Methane
0,1
С
Н, 1
Н, 1
Н,1
Н,1
R=l
R
R,2
R,2
R,2,
NMR
109
109
109
Variabl
@ B3LYP/6-31G(d)
471221
471221,
471221,
es:
09349799
3,120.
3,-120
0
,0
Geomet
ry
В данном примере раздел % Section отсутствует. Из раздела Route section
следует, что ограниченным методом Хартри-Фока в базисе 6-31G(d) с
использованием метода GIAO (по умолчанию), будут вычислены постоянные экранирования
молекулы метана. Из раздела Title section следует, что структура молекулы
метана, специфицированная в окне молекулярной спецификации, рассчитана
методом B3LYP в базисе 6-31G(d). В выходном файле выдаются значения
компонент тензора экранирования (см. раздел 1.21) для каждого атома(в ррт):
Calculating GIRO nuclear magnetic shielding tensors.
SCF GIAO Magnetic shielding tensor (ppm):
1 С Isotropic = 199.0521 Anisotropy = 0.0000
XX= 199.0521 YX= 0.0000 ZX= 0.0000
XY= 0.0000 YY= 199.0521 ZY= 0.0000
XZ= 0.0000 YZ= 0.0000 ZZ= 199.0521
Eigenvalues: 199.0521 199.0521 199.0521
2 H Isotropic = 32.0502 Anisotropy = 9.0696
XX= 32.0502 YX= 3.0232 ZX= 3.0232
XY= 3.0232 YY= 32.0502 ZY= 3.0232
XZ= 3.0232 YZ= 3.0232 ZZ= 32.0502
Eigenvalues: 29.0270 29.0270 38.0966
3 H Isotropic = 32.0502 Anisotropy = 9.0696
XX= 32.0502 YX= 3.0232 ZX= -3.0232
XY= 3.0232 YY= 32.0502 ZY= -3.0232
XZ= -3.0232 YZ= -3.0232 ZZ= 32.0502
Eigenvalues: 29.0270 29.0270 38.0966
4 H Isotropic = 32.0502 Anisotropy = 9.0696
XX= 32.0502 YX= -3.0232 ZX= 3.0232
XY= -3.0232 YY= 32.0502 ZY= -3.0232
XZ= 3.0232 YZ= -3.0232 ZZ= 32.0502
Eigenvalues: 29.0270 29.0270 38.0966
5 H Isotropic = 32.0502 Anisotropy = 9.0696
XX= 32.0502 YX= -3.0232 ZX= -3.0232
XY= -3.0232 YY= 32.0502 ZY= 3.0232
XZ= -3.0232 YZ= 3.0232 ZZ= 32.0502
Eigenvalues: 29.0270 29.0270 38.0966
Для расчета химического сдвига необходимо рассчитать тензор ядерного
магнитного экранирования для молекулы, принятой за эталон. В спектроскопии
.ЯМР на ядрах Я и С в качестве точки отсчета принимают сигнал тетраме-
138
тилсилана (ТМС) 5,/(СЯз)4- Расчет тензора магнитного экранирования
молекулы тетраметилсилана методом B3LYP в базисе 6-31G(d) дает:
Calculating GIAO nuclear magnetic shielding tensors.
SCF GIRO Magnetic shielding tensor (ppra):
1 Si Isotropic = 410.4333 Anisotropy =
XX= 410.4 333 YX= 0.0000 ZX= 0.0000
XY= 0.0000 YY= 410.4333 ZY= 0.0000
XZ= 0.0000 YZ= 0.0000 ZZ= 410.4333
Eigenvalues: 410.4333 410.4333 410.4333
2 С Isotropic = 189.0916 Anisotropy =
XX= 189.0916 YX= 1.7849 ZX= 1.7849
XY= 1.7849 YY= 189.0916 ZY= 1.7849
XZ= 1.7849 YZ= 1.7849 ZZ= 189.0916
Eigenvalues: 187.3067 187.3067 192.6613
3 H Isotropic = 32.1736 Anisotropy =
XX= 32.8261 YX= -1.8416 ZX= 4.6257
XY= -1.7463 YY= 30.8686 ZY= -1.7463
XZ= 4.6257 YZ= -1.8416 ZZ= 32.8261
Eigenvalues: 28.2004 30.0044 38.3161
Опции директивы NMR
SpinSpin - Вычисляет константы спин-спинового взаимодействия.
CSGT -Вычисляет свойства спектра NMR используя метод CSGT (Continuous
Set of Gauge Transformations).
GIAO -Вычисляет свойства спектра NMR используя метод GIAO.FIo умолча-
нию.( Gauge-Independant Atomic Orbital).
IGAIM - Использует для масштабирования центры атомов.
SingleOrigin - Использует для масштабирования одно начало координат (не
рекомендуется).
АН - Вычисляет свойства ЯМР, используя SingleOrigin, IGAIM, и CSGT
методы .
PrintEigenvectors - Выводит собственные векторы тензора экранирования для
каждого атома.
2.12. Расчет эффектов сольватации
Моделирование свойств систем в растворах задается посредством
ключевого слова SCRF (self-consistent reaction field, самосогласованное реактивное
поле, см. 1.20). Данное ключевое слово используется вместе с опцией,
определяющей метод расчета и растворитель.
Опции, определяющие метод расчета [107]:
Dipole - модель Онсагера.
РСМ- модель поляризационного континуума Томаси.
139
IPCM - модель поляризационного континуума с формой полости,
определяемой поверхностью уровня электронной плотности (Isodensity Polarizable
Continuum Model).
SCI-PCM - самосогласованная модель IPCM.
COSMORS - модель COSMO (см. раздел 1.20).
Опции, определяющие растворитель:
Water или Н20: £=78.39
Acetonitrile или CH3CN: e=36.64
DiMethylSulfoxide или DMSO: е=46.7
Methanol или СН3ОН: е=32.63
Ethanol или СН3СН2ОН: е=24.55
Isoquinoline: £=10.43
Quinoline: £=9.03
Chloroform или СНС13: е=4.9
Ether или DiEthylEther или СН3СН2ОСН2СН3: е=4.335
DiChloroMethane или MethyleneChloride или СНЬСЬ: £=8.93
DiChloroEthane или СН2С1СН2С1: е= 10.36
CarbonTetrachloride или ССЦ: 8=2.228
Benzene или СбНб: е=2.247
Toluene или С6Н5СН3: £=2.379
ChloroBenzene или С6Н4С1: £=5.621
NitroMethane или CH3N02: £=38.2
Heptane или С7Н16: £=1.92
CycloHexane или С6Н12: £=2.023
Aniline или C5H5NH2: £=6.89
Acetone или СН3СОСН3: £=20.7
TetraHydroFuran или THF: £=7.58
DiMethylSulfoxide или DMSO или CH3SOCH3: £=46.7
Argon или Ar: e=l .43
Krypton или Кг: £=1.519
Xenon или Хе: £=1.706
2.13 Расчет возбужденных состояний
В программе Gaussian расчет возбужденных состояний осуществляется
методом взаимодействия конфигураций. Электронной конфигурацией
молекулы называется размещение электронов молекулы по молекулярным орбиталям
с указанием чисел их заполнения. В приближении Хартри-Фока волновая
функция, отвечающая электронной конфигурации, представляет собой слэте-
ровский определитель, составленный для основного состояния молекулы из
занятых МО. В методах конфигурационного взаимодействия из этого
детерминанта конструируются другие определители путем замены занятых орбиталей
на виртуальные. При этом возможны одиночные, двойные и тройные подста-
140
новки. В одиночных подстановках одна виртуальная орбиталь заменяет одну
занятую орбиталь в детерминанте, составленном из занятых МО. В двойных -
две виртуальные орбитали заменяют две занятые, в тройных — три.
Сконструированные однотерминантные функции в методе кофигурационного
взаимодействия являются базисом, в виде разложения по которому, ищется полная
волновая функция молекулы. Коэффициенты разложения находятся
вариационным методом.
Для расчета возбужденных состояний используется ключевое слово CIS
(CI-Singlet), в котором используется одиночная подстановка и без указания
опций рассчитывается одно синглетное состояние. При этом возможен расчет
систем с закрытыми (RCIS) и открытыми (UCIS, см. 1.6) оболочками.
Для указания номера рассчитываемого возбужденного состояния
используется опция CIS=(Root=n). Состояния располагаются в ряд по возрастанию
энергии, п — номер состояния в этом ряду. По умолчанию п=1, т.е. будет
рассчитано только первое возбужденное состояние.
Для расчета нескольких возбужденных состояний используется опция
CIS=(NStates=n), n - число рассчитываемых состояний. По умолчанию п=3.
CIS=50-50 означает требование расчета синглетных и триплетных
состояний, опция 50-50 вместе с опцией NStates=n, указывает, что будет
рассчитано п синглетных и п триплетных состояний.
2.14 Ключевые слова программы
В настоящем разделе представлено краткое описание ключевых слов
программы Gaussian [107], подробное описание их использования представлено в
разделе Help программы.
Archive — указывает программе, чтобы в случае нормального завершения
вычислений результаты расчета были отправлены в архив ( базу данных
результатов работ).
FormCheck - данное ключевое слово указывает, чтобы форматированная
версия checkpoint файла была записана в конце успешного завершения программы.
Применяется в случае необходимости использования выходных данных
завершенной программы в последующих вычислениях.
Charge — данное ключевое слово указывает, чтобы программа при расчете
свойств атомно-молекулярной системы учитывала наличие внешних зарядов,
расположенных в фиксированных точках пространства.
CheckPointBasis (синонимы ReadBasis, RdBasis ) - указывает программе, чтобы
базисный набор был прочитан с checkpoint файла. Удобно для типов работ,
включающих несколько шагов, каждый из которых использует один и тот же
базисный набор, поскольку позволяет указать базисный набор только для
первого шага. CheckPointBasis также позволяет извлечь базисный набор из
checkpoint файла, независимо оттого, как он первоначально был определен.
CID, CISD — указывает, чтобы при расчете учитывалось конфигурационное
взаимодействие.
Constants - специфицирует, какой набор физических констант будет
использован в расчетах. Используется в виде Constants=rofl (например, Constants=1979).
141
При этом будут использованы физические постоянные, заложенные в версию
программы 1979 года. Используется для изучения влияния на результаты
расчета уточнения физических постоянных.
Counterpoise — осуществляет уравновешивающую корректировку поверхности
потенциальной энергии системы, состоящей из фрагментов, имеет цель
устранить ошибку BSSE.
CPHF - выбирает алгоритм, использующий решение CPHF (связанных
возмущенных уравнений Хартри-Фока).
Density - осуществляет анализ заселенности и других свойств, используя
электронную плотность, полученную методом самосогласованного поля.
External - производит вычисления с использованием внешних программ.
ExtraBasis, ExtraDensityBasis - указывает, что к основному базисному набору
будут добавлены дополнительные базисные функции, специфицированные в
route section. Эти базисные функции описываются отдельно в специальном
формате, что более подробно обсуждается при описании ключевого слова Gen.
Frozen Core Options - определяют какие внутренние орбитали будут
заморожены в вычислениях, выполняемых после процедуры самосогласованного поля.
Field - выполняет расчеты системы, находящейся во внешнем
электростатическом поле.
FMM — включает вычисления с использованием быстрого мультипольного
метода. Обеспечивает ускорение вычислений для несимметричных молекул с чи-
лом атомов до 60, для высокосимметричных молекул - с числом атомов до 240
( гибридные методы функционала плотности) и до 360 ( чистые методы
функционала плотности).
Force - вычисляет силы на атомах, т.е. градиент энергии. При этом также будет
вычислен дипольный момент для МР2, СС, QCI и CI.
Freq - осуществляет вычисление силовых постоянных, частот ИК и раманов-
ских спектров и интенсивностей, форму нормальных колебаний, коэффициенты
деполяризации, термохимический анализ.
G Keywords - включает методы G1,G2,G2MP2, G3, G3MP2, G3B3, G3MP2B3
высокоточного расчета энергии системы.
Gen - позволяет для каждого атома системы использовать базисный набор,
определенный пользователем.
Geom - указывает программе откуда считывается информация о начальной
структуре молекулы. По умолчанию считывание осуществляется из окна
молекулярной спецификации молекулы (см.2.1). Geom может указать
альтернативный источник начальной структуры. Geom также управляет относящейся к
геометрии молекулы информацией.
Geom=Connectivity определяет связанные валентной связью атомы,
посредством дополнительного ввода, записанного после пустой строки в окне
молекулярной спецификации, идущей после спецификации атомов системы.
GFInput (Gaussian Function Input) - в output файле выдает информацию об
используемом базисном наборе в форме удобной для дополнений или
модификации стандартных базисных наборов.
142
GFPrint - в output файле выдает информацию об используемом базисном
наборе в табличной форме. GFOldPrint выдает информацию в формате Gaussian.
Guess - управляет выбором стартовых хартри-фоковских волновых функций
для процедуры самосогласования. По умолчанию используются волновые
функции, диагонализующие функционал Harris'a. Guess применяется только с
указанием опции, идентифицирующей метод, лежащий в основе выбора
начальных волновых функций.
GVB - указывает на то, чтобы был использован метод улучшенного спаривания
электронов для валентных связей.
Integral - модифицирует метод вычисления, используя двумерные интегралы и
их производные.
Юр — указывает на использование набора внутренних опций, описание
которых прилагается к программе.
IRC - означает требование вычисления пути реакции.
IRCMax - выполняет вычисление максимальной энергии вдоль указанного
пути реакции.
MaxDisk - в 8- байтовых словах определяет сумму дисковой памяти, доступной
для временных данных.
Name - определяет имя пользователя, которое сохранено на архивном входе.
Желаемое имя пользователя используется как параметр (напр, Name=RChavez).
Это ключевое слово наиболее часто используется для пользователей Gaussian,
которые также используют Базу данных Системы Квантовой Химии.
NMR - предсказывает характеристики спектра ЯМР, тензоры экранирования и
магнитные восприимчивости, расчеты возможны при использовании метода
Хартри-Фока, методов функционала плотности и метода МР2.
ONIOM - позволяет провести расчет больших молекулярных структур,
разбив все атомы молекулы на 3 области, которые рассматриваются с разной
степенью точности. Возможны три уровня точности: низкий, средний и
высокий.
Output - требует вывода неформатированного файла на фортране.
Используется с указанием опции, отражающей содержание файла. Например, output=wfn
выводит файл расчета волновых функций.
OVGF - требует при вычислении сродства к электрону и потенциала ионица-
ции использования внешней валентной функции Грина.
РВС - позволяет специфицировать опции для выбора периодических
граничных условий.
Polar — означает требование расчета поляризуемостей и гиперполяризуемостей.
Population — управляет выводом молекулярных орбиталей, мультипольных
моментов, анализом заселенностей. По умолчанию выводятся полные атомные
заряды по Малликену и орбитальные энергии.
Pressure - указывает величину давления при проведении термохимического
анализа, по умолчанию давление принимается равным I атмосфере. Значение
давления указывается с помощью опции # ... Pressure=1.5.
Prop - означает требование расчета электростатических свойств. По
умолчанию вычисляются потенциал, электрическое поле и его градиенты.
143
Pseudo - использует в расчетах метод псевдопотенциала.
Punch - позволяет вывести необходимую информацию в формате, удобном для
пользователя.
QCISD - указывает на использование квадратичного метода
многоконфигурационного взаимодействия.
ReArchive - использует checkpoint файл для генерации входа в архив. При
наличии этого ключевого слова новые расчеты не проводятся.
SAC-CI - требует использования метода Symmetry Adapted Cluster /
Configuration Interaction.
Scale - указывает на использование масштабирующего множителя для частот
колебаний при проведении термохимического анализа. По умолчанию
масштабирующий множитель принимается равным 1.
Scan - сканирует поверхность потенциальной энергии.
SCF - управляет функционированием процедуры самосогласованного поля, т.е.
используемыми методами и критериями сходимости. SCF=QC -указывает на
использование квадратичной сходимости в методе самосогласованного поля,
основанной на методе Bacskay. Он представляет комбинацию линейной
минимизации и метода Ньютона-Рафсона. Обычно данный метод требует почти в
два раза большего машинного времени по сравнению со стандартной
процедурой самосогласованного поля. Метод достаточно надежен и рекомендуется в
первую очередь при наличии проблем сходимотсти процедуры
самосогласования.
SCRF - указывает, чтобы расчеты были выполнены в присутствии
растворителя.
SP - указывает на вычисление энергии для фиксированной конфигурации ядер.
Sparse - используется при вычислении больших систем (более 400 атомов) ,
указывает программе заменить матричные элементы, значение которых меньше
указанного пользователем значения нулем.
Stable - указывает, чтобы программа протестировала устойчивость волновых
функций, полученных методом Хартри-Фока или методами теории
функционала плотности.
Symmetry - требует использования в вычислениях молекулярной симметрии.
TD - требует расчета возбужденных состояний с использованием зависящего
от времени метода Хартри-Фока или метода теории функционала плотности.
При этом также будет вычислен электронный круговой дихроизм.
Temperature - позволяет пользователю указать температуру системы при
проведении термохимического анализа. Температура задается в градусах Кельвина
виде опции
#... Temperature=300
По умолчанию принимается Т = 298,15 К.
Test - подавляет автоматическое создание архивного входа, антоним Archive.
TestMO - используется по умолчанию и прерывает расчет, если любой из МО
коэффициентов больше тысячи. NoTestMO отменяет этот останов.
Transformation - управляет алгоритмом, использованным для преобразования
интегралов и типами используемых преобразований.
144
Units -контролирует единицы для расстояний и углов в Z-матрице. По
умолчанию длины выражаются в ангсремах, а углы - в градусах.
Volume - указывает на вычисление молекулярного объема, ограничеснного
контуром с электронной плотностью 0.001 ——, а^ - боровский радиус. Интег-
а0
рирование производится методом Монте-Карло, точность - два знака после
запятой, но этого достаточно для оценки радиуса сферы Онзагера при учете
растворителя.
W1 - указывает на использование в вычислениях W1U или W1BD методов,
представляющих собой модифицированный метод связанных кластеров.
Obsolete Keywords
таблицей [107]
устаревшие ключевые слова представлены следующей
[Устаревшее ключевое слово |3амеряющее ключевое слово с опцией
|Alter
|вр-т
[BeckeHalfandHaff
|Camp-King
|CCSD-T
|CubeDensity
|Cube=Divergence
|diis
|Direct
|GridDensity
|Guess=Restart
[MP2=Stingy and VeryStingy
|NoDIIS
|NoExtrap
|NoRaff
|OIdConstants
[Opt^AddRedundant
|OptCyc=«
[oss
|PlotDensity
|Prop=Grid
^CID
QCISD-T
J|Guess=AIter
|BD(T)
]BHandH
lsCF=Camp-King
J|CCSD(T)
Jcubegen
Jcubegen
||SCF=DIIS
J|SCF=Direct
Jcubegen
JsCF=Restart
\none (options are a no-op)
||SCF=NoDIIS
JSCF=NoExtrap
J^nt=NoRaff
JConstants=1979
|Opt=ModRedundant
J|Op_t(MaxCyc=n)
JGVB(OSS)
Jcubegen
fcubegen
Ice i) |
|OCISD(T)
145
Iqcscf
|Raff
|Save
^CFCon=n
(SCFCyc=«
^CFDM
[SCFQC
[sCRF=Checkpoint
|VShift[=i7]
SCF=QC
|[lnf=NoRaff
\none (Save is a no-op)
|SCF(Conver=»)
|SCF(MaxCyc=«)
|SCF=DM
|SCF=QC
^|Field=EChk
JSCF(VShift[=w])
2.15 Примеры входных файлов программы GAUSSIAN
В данном параграфе приведены примеры некоторых входных файлов папки
Example программы GAUSSIAN под Windous. Входной файл состоит из
четырех разделов и окна молекулярной спецификации (Рис.2.1.2), которые ниже
представлены не в окне ввода работы, а в виде текстовых строк, в которых один
раздел отделен от другого пустой строкой. Тексты входных файлов
воспроизведены с разрешения Gaussian Inc.[107].
Example e2_01. Расчет энергии и свойств формальдегида для
фиксированной конфигурации ядер.
Входной файл расчета энергии, зарядов на атомах, дипольных, высших
мультипольных моментов и молекулярных орбиталей молекулы формальдегида
для конфигурации ядер, указанной в окне молекулярной спецификации имеет
вид:
# RHF/6-31G(d) Pop=Full Test
Formaldehyde Single Point
0 1
С 0. 0. 0.
О 0. 1.22 0.
H .94 -.54 0.
H -.94 -.54 0.
В данном примере раздел % Section отсутствует, входной файл начинается с
раздела Route section. По умолчанию ограниченным методом Хартри-Фока в
базисе 6-31G(d) программа выполнит вычисление электронной энергии
системы для фиксированной конфигурации ядер ( SP, см. раздел 2.4) молекулы
формальдегида. Директива Pop=Full указывает, что для данной конфигурации
будет выведена информация о всех рассчитанных молекулярных орбиталях.
После раздела комментариев Formaldehyde Single Point идет строка,
показывающая, что заряд молекулы равен нулю, мультиплетность рассчитывае-
146
мого состояния равна 1. В данном примере разделителем между числами,
указывающими заряд и мультиплетность, символы атомов и их координаты,
является пробел. Обращаем внимание, что целые числа, идентифицирующие
координаты, представляются обязательно с десятичной точкой
Молекула формальдегида
Example e2_02. Расчет спектра ЯМР молекулы метана
Входной файл имеет вид:
#Т RHF/6-31G(d) HMR Test
Methane NMR @ B3LYP/6-31G(d) Geometry
0,1
С
H,1,R
H, 1,R,2, 109.471221
H, 1,R, 2,109.471221, 3,120., 0
H, 1,R, 2,109.471221,3,-120., 0
Variables:
R=l.09349799
В данном входном файле раздел % Section отсутствует. Раздел Route section
(первая строка) указывает, что методом GIAO (по умолчанию) с
использованием ограниченного метода Хартри-Фока в базисе 6-31G(d) будет выполнен
расчет спектра ЯМР ( ключевое слово NMR) молекулы метана для указанной
конфигурации ядер. Символ Т после знака # показывает, что текстовая
информация в выходном файле будет выводиться в сжатом виде. Раздел комментариев
(пишется пользователем) подсказывает, что данная конфигурация молекулы
метана получена методом B3LYP в базисе 6-31G(d). Заряд рассчитываемого
состояния равен 0, мультиплетность 1. Структура молекулы метана задана во
внутренних переменных, разделителем данных является запятая (можно
использовать пробел как в предыдущем примере).
Example еЗ_02. Расчет оптимальной конфигурации молекулы этилена
Требование нахождения оптимальной конфигурации задается ключевым
словом ОРТ (см. раздел 2.6). При этом для оптимальной структуры будут также
147
вычислены полная энергия, заряды на атомах и мультипольные моменты
системы.Входной файл имеет вид:
RHF/6-31G(d) Opt
#Т RHF/6-31G(d) Opt Test
Ethylene Geometry Optimization
0 1
С
С 1 CC
H 1 CH 2 HCC
H 1 CH 2 HCC 3 180.
H 2 CH 1 HCC 3 180.
H 2 CH 1 HCC 4 180.
Variables:
CC=1.31
CH=1.07
HCC=121.5
Оптимизация структуры молекулы этилена будет выполнена ограниченным
методом Хартри-Фока в базисе 6-31G(d).
Example еЗОЗ. Расчет переходного состояния реакции НзСО => НгСОН
В данном примере методом квадратичного синхронного транзита QST2
(см.раздел 2.10) с использованием неограниченного методом Хартри-Фока в
базисе 6-31G(d) производится поиск переходного состояния реакции
изомеризации Н3СО => Н2СОН.
Входной файл имеет вид:
#Т UHF/6-31G(d) Opt=QST2
НЗСО —> H2COH Reactants
0,2
С
О 1 1.48
Н 1 R 2 А
Н 1 1.08 2 110. 3 120.
Н 1 1.08 2 110. 3 -120.
R=1.08
А=110.
НЗСО —> H2COH Reactants
0,2
С
О 1 1.48
Н 1 R 2 А
Н 1 1.08 2 110. 3 120.
Н 1 1.08 2 110. 3 -120.
R=1.9
А=30.
Первая спецификация идентифицирует исходную молекулу НзСО, вторая —
конечную Н2СОН. Программа методом квадратичного синхронного транзита на-
148
ходит переходное состояние. Начальное, конечное, и рассчитанное переходное
состояние представлены на рисунке.
ЩСО Н2СОН TS
Example e4_0l. Расчет частот колебаний формальдегида
Требование расчета ИК и КР спектров атомно-молекулярной системы задается
ключевым словом Freq ( см. раздел 2.8), входной файл имеет вид:
#Т RHF/6-31G(D) Freq Test
Formaldehyde Frequencies
0,1
С
0,1,AB
H, 1,AH, 2,HAB
H, 1,AH,2,HAB, 3,180.
№=1.18429
AH=1.09169
HAB=122.13658
Программа в соответствии с input файлом рассчитает ИК и рамановский
спектры молекулы формальдегда, выполнит термохимический анализ системы (см.
раздел 2.8). В качестве стартовой использована предварительно соптимизиро-
ванная структура. Программа рассчитает спектры и в случае, если заданная
структура не является оптимальной, но в этом случае результаты будут
неверны.
Example e4_02a, b, с, d. Оптимизация структуры и расчет ИК спектров
изомеров 1-фторпропена.
Примеры е4_02а, Ь, с, d представляют собой входные файлы оптимизации
структур изомеров trans (0е), trans (180°), cis (0°) и переходного состояния trans
- cis молекулы 1-фторпропена. Оптимизированные структуры изомеров trans
(0е), trans (180е), cis (0°) и переходного состояния trans - cis молекулы 1-
фторпропена приведены на рисунке с указанием их полных энергий
Ea,E[,,Ec,Ecj.Входной файл оптимизации структуры trans (0°) изомера (пример
е4_02а) имеет вид:
149
#T RHF/6-31G(d) Opt Freq Test
trans 1-fluoropropene HCCC=0 Optimization
0 1
С
H, 1,R2
C,1,R3,2,A3
H,1,R4,2,A4,3,D4,0
H, 1,R4,2,A4,3,-D4,0
H,3,R6,1,A6,2,180.,0
C,3,R7,1,A7,2,0.,0
H,7,R8,3,A8,1,0.,0
F,7,R9,3,A9, 1,180.,0
Variables:
R2=1.091
R3=1.52
R4=1.092
R6=1.081
R7=1.34
R8=1.082
R9=1.33
A3=109.471
A4=109.472
A6=120.00
A7=120.01
A8=120.02
A9=120.03
D4=120.04
Наиболее энергетически выгодной является cis (0°) структура. Анализ
рассчитанных ИК спектров исследуемых молекул показывает, что все
частоты изомеров trans (0°) и trans (180°) действительны, и структуры
соответствуют минимуму на поверхности потенциальной энергии. В ИК
спектрах trans (180 ) и ts структур имеется по одной мнимой частоте, что
подтверждает, что структура ts является переходным состоянием, и
указывает на то, что переходным состоянием является также и изомер trans
(180 ). Значения модулей мнимых частот данных структур соответственно
равны 1515.3 см и 226 Л см , что указывает на то, что в первом случае
активационный барьер является более узким и высоким. Небольшое значение
модуля мнимой частоты для trans (180°) изомера указывает на пологость
активационного барьера и незначительное искажение конфигурации
Example e5_01. Влияние базиса на результаты расчета молекулы
метанола СН3ОН и аниона метоксида СНзО .
Входной файл оптимизации молекул СНзОН и СН30~на уровнях теории
RHF/6-31G(D), 6-31+G(D) и 6-311 ++G(3df,2pd) имеет вид:
150
tST RHF/6-31G(D> Opt Test
Methanol 6-31G{D) Structure
I ° 1
С
0,1, R2
H,1,R3,2,A3
H,1,R4,2,A4,3,D4
H,1,R4,2,A4,3,-D4
H,2,R6,1,A6,3,180.
Variables:
R2=l. 399645
R3=l.081060
M=l.087345
R6=0.946290
R3=107.159
114=112.008
Йб=109.406
D4=118.774
—Linkl—
ttT RHF/6-31+G{D> Opt Test
Methanol 6-31+G(D> Structure
0 1
С
0,1, R2
H,1,R3,2,A3
H,1,R4,2,A4,3,D4
H,1,R4,2,A4,3,-D4
H,2,R6, 1,A6,3,180.
Variables:
R2=l.401858
R3=l.080467
R4=l. 086479
R6=0. 94 6353
R3=106.959
R4=111.691
Йб=110.346
D4=118.764
—Linkl—
#T RHF/6-31G(D) Opt Test
Methoxide Anion 6-31G(D> Structure
-1,1
С
0,1, AB
H, 1,AH, 2,HAB
H,1,AH,2,HAB,3,120.
H,1,AH,2,HAB,3,-120.
AB=1.310651
№=1.133220
HftB=116.537
151
—Linkl—
#T RHF/6-31+G{D) Opt Test
Methoxide Anion 6-31+G(D) Structure
-1Д
С
0,1,AB
H, 1,AH, 2,HAB
H,1,AH,2,HAB,3,120.
H, 1,AH, 2, HAB, 3,-120.
AB=1.330449
AH-1.121031
HAB=114.991868
—Linkl —
#T RHF/6-311++G(3df,2pd> Opt Test
Methoxide Anion HF Basis Set Limit
-1,1
С
О, 1,АВ
H, 1,AH, 2,HAB
H, 1,AH, 2,HAB, 3,120.
H,1,AH,2,HAB,3,-120.
AB=1.33044 9
AH=1.121031
HAB=114.991868
Входной файл, описанный после разделителя —Linkl—, является работой
дополнительного шага (см.2.2) и идентифицируется в окне ввода работы
дополнительного шага. Все шаги input файла, представленного выше, при их
выдаче в текстовом формате, располагаются друг за другом и отделяются один от
другого пустой строкой и разделителем —Linkl— .
Результаты расчета представлены в таблице, из которой видно, что учет
диффузных функций наиболее сильно изменяет углы в структуре аниона и
незначительно изменяет структурные характеристики метанола:
теристика
гСО
гсн
гон
ZHCH
ZOCH
ZCOH
Система
СН3ОН
6-31G(d)
1.3966
1.0873
0.9463
108.4127
112.008
-
6-31+G(d)
1.4019
1.0865
0.9464
108.6555
111.691
-
Experiment
1.427 ±0.007
1.096 + 0.01
0.956 ±0.015
109.3 + 0.75
-
-
СНзО"
6-31G(d)
1.3107
1.1332
-
101.5713
116.537
-
6-31+G(d)
1.3304
1.121
-
103.4298
114.9919
-
6-311++G
(3df,2pd)
1.32.23
1.1209
-
103.2409
115.1097
-
152
Example e5_02. Учет в базисных функциях высших угловых
моментов и его влияние на длину связи молекулы РО.
Представлен входной файл оптимизации структуры молекулы РО с
использованием метода B3LYP в базисах 6-31G(d), 6-311G(d), 6-311 (2d), 6-
31 lG(2df), 6-31 lG(3df). В результате оптимизации длины связи молекулы РО в
данных базисах получились равными 1.4986, 1.4914, 1.4818, 1.4796 , 1.4758 А0
соответственно. Экспериментальное значение длины связи 1.476 А [41].
Example e6_01. Анализ молекулярных орбиталей молекулы тетрафе-
нилпрофина.
Входной файл имеет вид:
%Chk=tpp
#Т AMI Test Cube=Orbital Pop=Reg
TetraPhenylProphine AMI
0 1
спецификация молекулы тетрафенилпрофина
пустая строка
tpp_homo.cub
HOMO
—Linkl—
&Chk=tpp
ffT AMI Test Guess= (Read, Only) Geom=AllCheck Cube=Orbital
tpp_lumo.cub
LUMO
—Linkl—
%Chk=tpp
#T AMI Test Guess=(Read,Only) Geom=AllCheck Cube=Orbital
tpp_occ.cub
112
—Linkl—
&Chk=tpp
SNoSave
#T AMI Test Guess=(Read,Only) Geom=AllCheck Cube=Orbital
tpp_virt.cub
115
Ключевое слово Cube с опцией Orbital используется для расчета одной
или нескольких молекулярных орбиталей в точках, расположенных с
определенным шагом в объеме трехмерного куба. Входной файл, описанный после
разделителя —Linkl--, является работой дополнительного шага (см.2.2) и
идентифицируется в окне ввода работы дополнительного шага. Рассчитанная
информация хранится в checkpoint файле, поэтому, при выборе данного типа
работ, указание имени checkpoint файла обязательно. GAUSSIAN в зависимости
от размеров молекулы устанавливает по умолчанию разумный размер куба.
После пустой строки, следующей за спецификацией молекулы, пользователем
указывается имя выходного файла, а в следующей строке указывается номер
молекулярной орбитали (см пример входного файла). По умолчанию каждая
153
сторона куба содержит 80 точек. Численные значения молекулярной орбитали
содержатся в checkpoint файле и в выходном файле в виде матрицы. При этом
координаты точек трехмерной сетки куба не указываются. При идентификации
элементов матрицы, нужно иметь ввиду, что сначала с определенным шагом
меняется координата х от начального до конечного значения, определяемого
размерами куба, координаты у иг фиксированы, затем это повторяется для
увеличенной на величину шага координаты у, пока все значения у не
исчерпаны и затем то же самое повторяется для координаты z. Графическое
изображение молекулярной орбитали можно просмотреть с помощью программы
GAUSVIEW (см. 3.11).
Example e6_02. Оптимизация структуры димера фтористого водорода
методами AMI, PM3 и Хартри-Фока.
Входной файл расчета системы HF...HF имеет вид:
%Chk=Dimer
#Т AMI Opt=AddRedundemt Test
HF Dimer (Planar)
0,1
F 0.013733 1.510707 0.000000
F 0.013733 -1.364162 0.000000
H -0.473347 -2.030563 0.000000
H 0.226146 0.711660 0.000000
1 2
2 3
2 4
1 4
12 3
—Linkl—
%Chk=Dimer
#T PM3 Opt Geom=AllCheck Guess=Read Test
—Linkl —
%Chk=Dimer
SPIoSave
#T HF/6-31+G{d) Opt(MaxCycle=30) Geom=AllCheck Guess=Read Test
Данный пример позволяет сравнить результаты расчета длин ковалентных
связей, водородных связей и углов, входящих в состав водородного мостика,
димера фтористого водорода полуэмпирическими методами и методом Хартри-
Фока. Входной файл, описанный после разделителя —Linkl—, является работой
дополнительного шага (см.2.2) и идентифицируется в окне ввода работы
дополнительного шага. Известно, что полуэмпирические методы плохо работают
для систем с водородными связями и при расчете переходных структур. Output
файл данного примера подтверждает вывод о плохой применимости
полуэмпирических методов для расчета систем с водородными связями.
154
Во входном файле использована опция AddRedundant (добавить
избыточные) ключевого слова Opt, означающая что к набору 3N-6 независимых
внутренних координат системы при оптимизации будут добавлены координаты,
указанные после пустой строки, следующей за спецификацией атомов (т.е.
длины связей между атомами F1F2, F2H3, F2H4, F1H4 и угол F1F2H3), если их
нет в этом наборе. Результирующий набор координат будет в общем случае
избыточным (т.е. число координат более 3N - б, необходимых для однозначного
определения мимнимизируемой функции - энергии, зависящей от
конфигурации молекулы). Существуют специальные методы оперирования такими
наборами, приводящие к тем же самым результатам, что при использовании
однозначных наборов. При использовании этой опции в output файл обязательно
будут включены значения избыточных координат оптимизированной структуры.
Опция Geom=AHCheck второго шага данного примера,
специфицированного после разделителя — Linki-означает, что спецификация молекулы
(включая переменные, заряд, мультиплетность и информацию строки Title) на
втором шаге будет считана из checkpoint файла (один и тот же в данном
примере для всех шагов). Опция Guess=Read означает, что начальная оценка
волновой функции системы таюке будет считана из checkpoint файла. Опция
Opt(MaxCycle=30) означает, что максимальное число шагов оптимизации
равно 30 ( по умолчанию 20).
Example ебОЗ. Расчет энергии связи молекулы HF.
В данном примере методом QCISD(T,E4T) /6-311++G(3df, 3pd)
рассчитывается энергия двухатомной молекулы HF, атома водорода и атома фтора.
Энергию связи можно рассчитать как разность между суммой энергий атомов
водорода и фтора и энергией молекулы. Расчет показывает, что рассчитанная
энергия связи HF (140.6 ккал/моль) удовлетворительно согласуется с
экспериментальной (141.2 ккал/моль).
Example e6_04. Оптимизация молекулы озона.
Example e6_05 (a - g). Расчет оптимальной структруры и энергии
атомизации молекулы СОг различными методами и базисами.
Example e6_06. Расчет оптимальной структуры и ИК спектра аниона
FJ методами теории функционала плотности в базисе d95v+.
155
Example e7_01. Расчет энергии атомизации молекулы РН2 с
использованием улучшенных критериев сходимости процедуры
самосогласования.
Энергия атомизации молекулы РН2 рассчитывается как разность между
суммой энергий составляющих молекулу атомов и энергией молекулы
Еат=Е{Р) + 2Е(нуЕ{РН2)
Входной файл данного примера требует, чтобы программа вычислила данные
энергии, используя улучшенный критерий сходимости процедуры
самосогласования, что задается с помощью опции SCF=Tight:
#Т Test UB3LYP/6-31+G(d,p> SCF=Tight
Phosphorous atom
0 4
P
—Linkl—
#T Test UB3LYP/6-31+G(d,p) SCF=Tight
Hydrogen atom
0 2
H
—Linkl—
%Chk=ph2
#T UB3LYP/6-31G<d) Opt Freq Test
PH2
0 2
P
H,1,R2
H,1,R2,2,A3
Variables:
R2=l.43382202
A3=90.35361918
—Linkl—
%Chk=ph2
SNoSave
#T UB3LYP/6-31+G(d,p> SCF=Tight Test Geom=AllCheck Guess=Read
Входной файл, описанный после разделителя —Linkl—, является работой
дополнительного шага (см.2.2) и идентифицируется в окне ввода работы
дополнительного шага. При расчете Е{РН2)дяя улучшения точности расчета
необходимо учесть ZPE коррекцию электронной энергии (см.2.14), которая
вычисляется на основе рассчитанных частот нормальных колебаний, и выдается в а.е.
в output файле при термохимическом анализе системы ( см.2.8) . Для молекулы
(РЯ?) имеем
Sum of electronic and zero-point Energies^ -342.496027,
Sum of electronic and thermal Energies=-342.493172 a.e.
156
Первая сумма представляет собой внутреннюю энергию молекулы РН2 при
о
О С. Вторая сумма представляет собой внутреннюю энергию молекулы РН2
при 25 С (по умолчанию). При вычислении энергии атомизации для О К
используем первую сумму, для температуры 25 С- вторую сумму.
Расчет энергий атомов фосфора и хлора дает
Е{Р) = - 341.25930 а.е.,
£(#)= - 0.5002728 а.е.,
Еат =\Р°К) = 0.2361814 а.е.= 148.2 ккал/моль,
Еат =[25°С ) = -0,2333264 а.е.= 146.4 ккал/моль,
Elm" = 144.7 ккал/моль [41].
Example e7_02. Расчет сродства к электрону молекулы PHj •
Сродство к электрону атомно-молекулярной системы определяется изменением
энергии системы при присоединении к ней электрона. Сродство к электрону
(electron affinity - ЕА) молекулы Р#2 представляет собой разность между
энергией нейтральной молекулы и энергией аниона РН~. ЕА-Е^РН^)-ЕуРН~).
Значение энергии молекулы РН2 рассчитано в предыдущем примере. Входной
файл для расчета энергии РН~ методами B3LYP/6-31G(d) и B3LYP/6-
31+G(d,p)HMeer вид
$Chk=electron
#Т B3LYP/6-31G<d) Opt Freq Test
PH2 (-1)
-1 1
P
H, 1,R2
H,1,R2,2,A3
Variables:
R2=l.44164194
A3=90.87779967
—Linkl—
%Chk=electron
%NoSave
#T B3LYP/6-31+G(d,p) SCF=Tight Test Geom=AllCheck Guess=Read
При использовании базиса 6-31+G(d,p) для аниона получаем (в)
Sum of electronic and zero-point Energies=-342.541467 а.е..
Sum of electronic and thermal Energies=-342.538609 a.e.
Используя результаты предыдущего примера, получаем
157
ЕА (о° к) = -342.490753 - (-342.541467) = 0.050714 а. е. = 31.82 ккап / моль
Еа[25°с)= -342.493172 - (-342.538609) = 0.047856 а.е. = 28.51 ккал/молъ,
ЕАжсп =29.06 ккал/молъ[41].
Example e7_03. Расчет энергии ионизации молекулы />#2* Потенциал
ионизации молекулы определятся как отношение наименьшей энергии,
необходимой для удаления из молекулы электрона, к абсолютному значению заряда
электрона. В результате удаления валентного электрона получается катион в
основном состоянии. Энергия, необходимая для ионизации молекулы />#2>
вычисляется по формуле EI = Е\РН2')- Е(РН2). Энергия молекулы РН2
рассчитана в примере 7.01. Входной файл для расчета внутренней энергии катиона
РН2 методами B3LYP/6-31+G(d,p) и B3LYP/6-31 G(d) имеет вид:
%Chk=ionpot
#Т B3LYP/6-31G(d> Test Opt Freq
PH2 (+1)
1 1
P
H,1,R2
H, 1,R2,2,A3
Variables:
R2=l.44426244
A3=90.45083822
—Linkl—
%Chk=ionpot
SNoSave
#T B3LYP/6-31+G(d,p) SCF=Tight Guess=Read Geom=AllCheck
В результате расчета катиона получаем
Sum of electronic and zero-point Energies--342.130623,
Sum of electronic and thermal Energies=-342.127770 a.e.
Энергия ионизации молекулы РН2 с учетом результатов примера 7.01 равна
ЕЩк)== -342.130623- (-342.490753) = 0.36013 а.е. = 225.98 ккал/молъ = 9.80 ev
Е/[25°с)=-342.127770-(-342.493172)= 0.365402 а.е. = 229.29 ккап/моль = 9.94ev
Е1жсп =982ev = 226.45 ккач/молъ [41].
Example e7_04, е7_05, е7_06. Расчет изменения энергии молекулы
PHj в реакции протонирования. Изменение энергии молекулы при
присоединении к ней протона может служить мерой сродства молекулы к протону.
Изменение энергии молекулы РН$ при ее протонировании рассчитывается по
формуле
ае = е(рн3)-е[рн$).
158
В примере е7_04 расчет энергий РН% и PHj проводится методом B3LYP/6-
31+G(d,p), SCF=Tight, в е7_05 - сверхточным методом расчета энергии G2,
в е7_06 - методом CBS-Q (complete basis set) . Входные файлы расчета систем
Р#3 и РН4 имеют вид:
Пример е7_04.
%Chk=proton
#Т B3LYP/6-31G(d,p) Opt Freq Test
PH4 (+1)
1 1
P
H, 1,R2
H,1,R2,2,109.47122063
H, 1,R2,2, 109. 47122063, 3,D4,C
H,1,R2, 2,109. 47122063, 3,-D4,0
Variables:
R2=l.41205824
D4=120.
—Linkl—
%Chk=proton
#T B3LYP/6-31+G(d,p) Test SCF-Tight Guess=Read Geom=AllCheck
—Linkl—
%Chk=proton
#T B3LYP/6-31G(d) Opt Freq Test
PH3
0 1
P
H, 1,R2
H,1,R2,2,A3
H,1,R2,2,A3,3,D4,0
Variables:
R2=l.42928344
A3=91.84753714
D4=-91.90910853
—Linkl—
%Chk=proton
%NoSave
#T B3LYP/6-31+G(d,p) Test SCF=Tight Guess=Read Geom=AllCheck
Пример е7_05.
#T G2 Test
PH4 (+1)
1 1
P
H,1,R2
H, 1,R2,2, 109.47122063
H,1,R2,2,109. 47122063, 3,D4,0
H,1,R2,2,109. 47122063, 3,-D4,0
Variables:
R2=l.41205824
D4=120.
159
—Linkl—
#T G2 Test
РНЗ
0 1
Р
H,1,R2
H,1,R2,2,A3
H,1,R2,2,A3,3,D4,0
Variables:
R2=l.42928344
A3=91.84753714
D4=-91.90910853
Пример е7_06.
#T CBS-4 Test
PH4 (+1)
1 1
P
H,1,R2
H,1,R2,2,109.47122063
H,1,R2, 2,109. 47122063, 3,D4,0
H,1,R2, 2,109.47122063, 3,-D4,0
Variables:
R2=l.41205824
D4=120.
—Linkl—
#T CBS-4 Test
PH3
0 1
P
H,1,R2
H,1,R2,2,A3
H,1,R2,2,A3,3,D4,0
Variables:
R2=l.42928344
R3=91.84753714
D4=-91.90910853
—Linkl—
#T CBS-Q Test
PH4 (+1)
1 1
P
H,1,R2
H,1,R2,2,109.47122063
H,1,R2, 2,109. 47122063, 3,D4,0
H,1,R2, 2,109. 47122063, 3,-D4,0
Variables:
R2=l.41205824
D4=120.
—Linkl—
160
#T CBS-Q Test
РНЗ
H,1,R2
H,1,R2,2,A3
H,1,R2,2,A3,3,D4,0
Variables:
R2=l.42928344
A3=91.84753714
M=-91. 90910853
Результаты расчета АЕ((РК) примеров е7_04 - e7_06 [41] приведены в таблице
B3LYP
G2
G2 (MP2)
CBS-Q
эксперимент
ЛЕ, ккал/моль
185.9
186.14
186.80
186.24
187.1
Example e8_01a. Расчет электронной плотности промежуточного
комплекса в реакции нитрования хлорбензола.
Известно, что продуктами реакции нитрования хлорбензола являются изомеры
нитрохлорбензола в мета (1%), пара (70%) и орто (29%) положениях.
Объяснение состава смеси заключается в анализе распределения электронной плотности
промежуточного комплекса реакции.
Рис. Промежуточный комплекс реакции нитрования хлорбензола (атакующая
электрофильная частица - нитроич-катион N(?2 в мета (а) и пара (Ъ)
положениях).
Входной файл для расчета распределения электронной плотности имеет вид
161
%chk=eB_01a.chk
%mem=6MW
%nproc=l
# hf/6-31g(d) nosymm geom=connectivity cube=(55,density) test
Nitrated Chlorobenzene Cation Meta Position
1 1
спецификация промежуточного метакомплекса
cln_meta.cube
—Linkl —
#T HF/6-31g* Cube=(55,Density) NoSymm Test
Nitrated Chlorobenzene Cation Para Position
1 1
спецификация промежуточного паракомплекса
cln_para.cube
#T HF/6-31g* Cube=(55,Density) NoSyrara Test
Nitrated Chlorobenzene Cation Para Position
1 1
спецификация промежуточного паракомплекса
cln_para.cube
Здесь ключевое слово cube=(55,density) означает, что для расчета плотности
будет формироваться куб с числом точек 55 . Вычисление электронной
плотности проводятся методом hf/6-31g(d) для предварительно
оптимизированной структуры, заданной в окне молекулярной спецификации. Для анализа
электронной плотности можно исследовать значения электронной плотности в
точках плоскости бензольного кольца и параллельной ей плоскостях. Gaussian
такие построения не выполняет. Их необходимо сделать самостоятельно по
данным, хранящимся в checkpoint файле или файле, описанном после
спецификации системы. Графическое изображение электронной плотности
промежуточных (мета и пара) комплексов реакции нитрования хлорбензола в точках
плоскостей, параллельных плоскости бензольного кольца представлено в [41].
Анализ распределения электронной плотности показал, что для мета структуры
при удалении от центральной плоскости сохраняется резонансная структура
кольца. Для пара структуры по мере удаления от плоскости кольца электронная
плотность сосредотачивается вблизи заместителей, что делает их более
реакционно способными.
Example e8_01b. Расчет электронной плотности промежуточного
комплекса в реакции нитрования нитробензола.
Example e8_02. Расчет энтальпии реакции гидратации иона водорода
(газовая фаза).
162
Уравнение реакции гиратации иона водорода одной молекулой воды имеет вид
н++н2о=н3о+.
Изменение энтальпии системы в процессе реакции равно разности энтальпий
продуктов и реагентов. Входной файл для расчета энтельпий иона #}0+и
молекулы воды имеет вид:
SChk=hydrate
#Т B3LYP/6-31G(D) Opt Freq^Readlso
НЗО+
1,1
О
X, 1,1.
H,1,R,2, A
H,1,R,2,A,3,120.
H,1,R,2,A,3,-120.
R 0.96876
A 105.50217
298.15 1.0 0.9804
16
1
1
1
—Linkl—
SChk=hydrate
#T B3LYP/6-311+G(2df,2p) Geom=Allcheck Guess=Read
—Linkl—
%Chk=hydrate
#T B3LYP/6-31G(D) Opt Freq=ReadIso
H20
0,1
0
H,1,R
H,1,R,2,A
R 0.94734
A 105.49542
298.15 1.0 0.9804
16
1
1
—Linkl—
%Chk=hydrate
%HoSave
ftT B3LYP/6-311+G(2df,2p) Geom=AUcheck Guess=Read
Расчет дает следующие значения энтальпий (см.2.8) иона Н^О+и молекулы
воды:
163
\)Н^0+ (первая строка - расчет в базисе 6-31G(D), вторая - в базисе 6-
311+G(2df,2p)):
Sum of electronic and thermal Enthalpies= -76.651613,
Sum of electronic and thermal Enthalpies= -76,696908 a.e.
2).H20:
Sura of electronic and thermal Enthalpies= -76.384426,
Sum of electronic and thermal Enthalpies= -76.437850 a.e.
Электронная энергия Н+ равна нулю, поскольку в данном катионе нет электро-
. ? ккст
нов, энтальния Н равна термической добавке, т.е. —RT-0.889 . Таким
2 моль
образом, энтальпия гидратации иона водорода равна:
(-76.651613+ 76.384426)-627.5095-0.889 = -168,551^-, B3LYP/6-3IG(D),
моль
(-76.696908+ 76.437850)-627.5095-0.889 = -163,45^^-, B3LYP/6-311+G(2df,2p)
моль
Экспериментальное значение энтальпии гидратации -165.5±1.8 [41].
моль
Множитель 627.5095 переводит атомные единицы энергии в .
моль
Example e8_03. Расчет переходного состояния и пути химической
реакции н2 +со<^н2со.
Входной файл расчета пути химической реакции состоит их двух шагов:
%Chk=formts
#Т RHF/6-31G(d) Opt(CalcFC,TS) Freq Test
H2 + CO <-> H2CO Opt Freq
0,1
О
С,1,1.13
H,2,1.1,1,164.0
Н,3,1.3,2,90.0,1,0.
—Linkl—
%Chk=formts
%NoSave
#Т RHF/6-31G(d) IRC=RCFC Guess=Read Geom=AllCheck Test
На первом шаге вычисляется переходное состояние реакции, начальная
конфигурация которого задана в окне молекулярной спецификации (ключевое слово
TS), на втором шаге — путь химической реакции (ключевое слово IRC),
комментарии см. в разделе 2.10.
Example e8_04. Расчет переходного состояния (QST2) и пути
химической реакции СН20 -» НСОН.
164
При расчете переходного состояния методом QST2 в окне молекулярной
спецификации входного файла указываются конфигурации исходных веществ и
продуктов реакции (Комментарии см. в разделе 2.10).
%Chk=hydroxy
#Т RHF/6-31G(d) Opt=QST2 Freq Test
formaldehyde
0,1
С
0,1,AB
H, 1,AH,2,HAB
H,1,AH,2,HAB,3,180.,0
ЙВ=1.1842 9
№=1.09169
HAB=122.13658
trans hydroxycarbene
0,1
С
0,1, AB
H, 1,AH0,2,BAH0
H,2,BH6,1,ABH6,3, 180., 0
№=1.29994
№0=1.09897
BH6-0.95075
BAH0=103.00645
№H6=109.43666
—Linkl—
%Chk=hydroxy
%NoSave
#T RHF/6-31G(d) IRC=RCFC Guess=Read Geom=AllCheck Test
Example e9_01. Расчет возбужденных состояний молекулы этилена.
Расчет возбужденных состояний в программе Gaussian задается с помощью
ключевого слова CIS:
ST RCIS(NStates=2,50-50)/6-31+G(d) Test
Ethylene Excited States
0,1
С
C,1,R{C-C)
H,1,R(C-H) ,2,A(CCH)
H,1,R(C-H) , 2,A(CCH),3, 180.
H,2,R(C-H) , 1,A(CCH),3,0.
H,2,R(C-H),1,A(CCH),3, 180.
R(C-C)=1.31696
R(C-H)=1.07601
ft(CCH)=121.81335
165
В данном примере будет рассчитано 2 синглетных и 2 триплетных
возбужденных состояния (onm™NStates=2,50-50). В базис включены диффузные функции,
которые позволяют улучшить результат. В выходном файле будут выданы
энергии возбуждения (в электронвольтах eV и нанометрах nm) и силы
осцилляторов (f), соответствующие переходам из основного состояния в данные
возбужденные состояния. После этой информации будет указано на какие номера
орбиталей возбужденного состояния проведены замены в детерминанте
основного состояния и коэффициенты, с которыми соответствующие детерминанты
входят в волновую функцию возбужденного состояния.
Excitation energies and oscillator strengths:
Excited State 1: Triplet-BlU 3.7768 eV 328.27 nm f=0.0000
8 -> 11 0.52952
8 -> 17 -0.45942
This state for optimization and/or second-order correction.
Copying the excited state density for this state as the 1-particle RhoCI
density.
Excited State 2: Triplet-B3U 7.4297 eV 166.88 nm f=0.0000
8 -> 10 0.60741
8 -> 15 -0.34842
Excited State 3: Singlet-B3U 7.8294 eV 158.36 run f=0.1884
8 -> 10 0.64288
8 -> 15 -0.27205
8 -> 18 -0.10534
Excited State 4: Singlet-BlU 7.9753 eV 155.46 nm f=0.5532
8 -> 11 0.66068
8 -> 17 -0.20317
Example e9_02. Оптимизация и расчет частот колебаний первого
возбужденного состояния молекулы формальдегида.
Входной файл состоит из двух шагов:
%Chk=c2ho_es
#Т RCIS/6-31+G(D) Test
Formaldehyde (C2V) Vertical Excited States
0,1
С
0,1,АВ
H,1/AH,2,HRB
H,1/AH,2,HAB,3,180.
AB=1.18429
AH=1.09169
HAB=122.13658
—Linkl—
%Chk=c2ho_es
fcNoSave
# RCIS(Root=l,Read)/6-31+G(D) Guess=Read Opt Freq Test Geom=AllCheck
На первом шаге в окне молекулярной спецификации задается
конфигурация основного состояния молекулы. Для данной кофигурации будут рассчитаны
энергии трех (по умолчанию) наиболее низких возбужденных состояний
молекулы этилена, что задается ключевым словом RCIS. Результаты первого шага
используются на втором шаге в качестве стартовых для конфигурации
166
(Geom=AHCheck) и волновых функций (Guess=Read) первого возбужденного
состояния. Требование расчета частот в первом возбужденном состоянии
задается ключевым словом Freq.
Example elOOla. Расчет энергий конформеров дихлорэтана в
растворе циклогексана. В данном примере рассчитываются энергии транс- и гош-
конформеров молекулы дихлорэтана в растворе циклогексана. Входной файл
состоит из двух шагов:
#Т B3LYP/6-31+G(d) SCRF(IPCM) SCF=Tight Test 6D
TRAHS-1,2-DICHLOROETHANE
0,1
С
С, 1,RCC
CL, 1,RCL,2,A1
CL,2,RCL,1,A1,3,180.
X,1,1.,2,A2,3,180.
H,1,RH,5,A3,3,90.
H,l,RH,5,A3,3,-90.
X,2,l., 1,A2,4,180.
H,2,RH,8,A3,4,90.
H,2,RH,8,A3,4,-90.
RCC=1.51610
RCL=1.79175
RH=1.07764
Al=109.40364
A2=129.36050
A3=54.80917
2.0
—Linkl—
%Chk=gauche
#T B3LYP/6-31+G(D) SCRF(IPCM) SCF=Tight Test 6D
GAUCHE-1,2-DICHLOROETHANE IN CYCLOHEXANE
0,1
С
C,1,RCC
CL, 1,RCL,2,A1
CL,2,RCL,1,A1,3,B1
H,1,RCH1,2,A2,4,B2
H,1,RCH2,2,A3,4,B3
H,2,RCH1,1,A2,3,B2
H,2,RCH2,1,A3,3,B3
RCC=1.51490
RCL=1.78594
RCH1=1.08019
RCH2=1.07802
Al=112.88944
A2=108. 96431
ft3=lll.38813
Bl=70.51710
52=188.89555
B3=-50.42251
2.0
167
На первом шаге рассчитывается энергия транс-конформера молекулы 1,2
дихлорэтана. Начальная структура (предварительно соптимизированная)
задается с помощью двух фиктивных атомов X (см.раздел 2.3). После пустой
строки, следующей за заданием значений переменных, указывается величина
диэлектрической проницаемости растворителя е = 2. Учет растворителя
осуществляется методом IPCM (см. раздел 1.20). Выбор растворителя и метода расчета
во входном файле можно также задать с помощью ключевого слова
scrf=(IPCM,soIvent=cycIohexane) и не указывать значение е.
На втором шаге аналогично рассчитывается энергия гош-конформера.
Расчет дает:
Етранс=-999.024746 а.е.
Егош = -999.0224846 а.е.
Из расчета следует, что в растворе циклогексана более энергетически
выгодным является транс-конформер
^гош ~ Етрас = 0.0022614а.е. = 1.42 ккая/моль
Example el0_01b. Расчет энергий конформеров дихлорэтана в растворе
циклогексана и в газовой фазе.
Входной файл состоит из 4-х шагов. На первом шаге данного примера
методом МР2/6-31+G(d) рассчитывается энергия предварительно соптимизиро-
ванной структуры транс-формы 1,2 дихлорэтана в газовой фазе, checkpoint
файл на первом шаге отсутствует. Второй, третий и четвертый шаги имеют
общий checkpoint файл dichlor. На втором шаге методом HF/6-31+G(D) с
помощью модели Онзагера (опция Dipole) рассчитывается энергия гош-формы 1,2
дихлорэтана в циклогексане. На третьем шаге эта же энергия рассчитывается
методом МР2/6-31+G(D). На четвертом шаге методом MP2/6-31+G(D)
рассчитывается энергия гош-формы 1,2 дихлорэтана в газовой фазе. Необходимая для
168
расчета начальная информация на третьем и четвертом шаге считывается с
checkpoint файла. Входной файл имеет вид:
#Т MP2/6-31+G(d) SCF=Tight Test
TRANS-1,2-DICHL0R0ETHANE
0,1
С
С, 1, RCC
CL, 1,RCL,2,A1
CL,2,RCL,1,A1,3,180.
X,1,1.,2,A2,3,180.
H,1,RH,5,A3,3,90.
H,1,RH,5,A3,3,-90.
X,2,1.,1,A2,4,180.
H,2,RH,8,A3,4,90.
H,2,RH,8,A3,4,-90.
RCC=1.51610
RCL=1.79175
RH=1.07764
Al=109.40364
A2=129.36050
A3-54.80917
—Linkl—
%Chk=dichlor
#T HF/6-31+G(D) SCRF(Dipole) SCF^Tight Test
GAUCHE-l,2-DICHLOROETHANE IN CYCLOHEXANE
0,1
С
С, 1, RCC
CL, 1,RCL,2,A1
CL,2,RCL,1,A1,3,B1
H,1,RCH1,2,A2,4,B2
H,1,RCH2,2,A3,4,B3
H,2,RCH1,1,A2,3,B2
H,2,RCH2,1,A3,3,B3
RCC=1.51490
RCL=1.78594
RCH1=1.08019
RCH2=1.07802
Al=112.88944
A2=108.96431
A3=lll.38813
Bl=70.51710
B2=188.89555
B3=-50.42251
3. 65 2.0
—Linkl—
%Chk=dichlor
#T MP2/6-31+G(D) SCRF(Dipole) SCF=Tight Test Guess=Read Geom=AllCheck
3.65 2.0
—Linkl—
£Chk=dichlor
%HoSave
#T MP2/6-31+G(D) SCF=Tight Test Guess=Read Geom=AllCheck
169
Строка 3.65 2.0, следующая за пустой строкой после идентификации
структуры, обозначает радиус сферы Онзагера (3.65 А0) и диэлектрическую
постоянную циклогексана. Рассчитав данные энергии можно выяснить как изменяется
энергия молекулы 1,2 дихлорэтана в реакции изомеризации и при ее переходе
из газовой фазы в раствор циклогексана.
Example el0_01c. Расчет частот колебаний молекулы формальдегида
в растворе ацетонитрила.
Входной файл состоит из двух шагов:
%Chk=formsolv
# RHF/6-31+G(D) SCRF Opt Test
Formaldehyde in ftcetonitrile
0,1
С
0,l,ftB
H, l,ftH,2,HAB
H, 1,AH, 2,HAB, 3,180.
AB=1.18587246
AH=1.09071133
HAB=121.85573823
2.82 35.9
—Linkl —
%Chk=formsolv
%NoSave
#T RHF/6-31+G(D) SCRF Freq Guess=Read Geom=AllCheck Test
2.82 35.9
На первом шаге производится оптимизация стартовой структуры
молекулы формальдегида в ацетонитриле, на втором шаге рассчитываются частоты ее
колебаний. Числа 2.82 35.9 определяют радиус сферы полости в
растворителе, в которую помещается молекула, и диэлектрическую проницаемость
ацетонитрила, соответственно.
Кроме папки Example в программе имеется еще папка Exercise, которая в
данной монографии не представлена.
170
ГЛАВА ИГ. ОСНОВЫ РАБОТЫ С ПРОГРАММОЙ «GAUSSVIEW»
В настоящей главе использована информация, изложенная в разделе Help
программы GV [107]. Формирование стартовой структуры и input файла для
программы Gaussian, просмотр промежуточных и конечных результатов
расчета можно осуществить, используя программу «Gaussview»(GV). Эта программа
представляет собой набор подпрограмм, применяемых в сочетании с основной
программой. Она позволяет получить графическое изображение молекулярных
структур, осуществляет визуализацию вращения молекулы, облегчает
формирование сложных input файлов, с ее помощью можно осуществлять запуск
программы на решение и анализировать результаты. Данная программа позволяет
визуализировать следующие свойства рассчитанных систем:
• Структуру оптимизированной системы.
• Молекулярные орбитали, электронную плотность и электростатический
потенциал.
• Магнитные свойства.
• Заряды на атомах.
• Форму нормальных колебаний с помощью их анимации.
• Спектры ИК, КР, ЯМР, колебательного кругового дихроизма и др.
• Пути реакций.
• Результаты сканирования поверхности потенциальной энергии.
• Траектории движения, рассчитанные методом молекулярной динамики.
• Графики полной энергии и других свойств атомно-молекулярных систем.
3.1. Старт программы Gaussview
Рис..3.1.1 Контрольная панель программы GV
171
Рис..3.1.2 Активное (рабочее) окно программы GV
В верхней части контрольной панели расположена зона меню Gaussview. Для
графического изображения требуемой структуры имеется библиотека
элементов и фрагментов.
3.2. Кнопки элементов и молекулярных фрагментов на контрольной
панели
Ж- двойной щелчок левой кнопкой мыши открывает панель атомов (Рис.3.2.1
). Внизу панели, имеются указатели, позволяющие выбрать атом с нужным
направлением связей.
^ - двойной щелчок левой кнопкой мыши открывает панель циклов, (Рис.3.2.2
к - двойной щелчок левой кнопкой мыши открывает панель молекулярных
групп, ( Рис.3.2.3 ).
й*8 - двойной щелчок левой кнопкой мыши открывает библиотеку
биологических фрагментов, которая состоит из панелей 2-х типов - аминокислот и нук-
леозидов, соответствующую панель можно выбрать в поле Туре (Рис.3.2.4).
По умолчанию можно открыть только одну из перечисленных панелей,
если одна панель открыта, то при открытии следующей панели текущая панель
автоматически закрывается. В левом верхнем углу этих панелей изображена
шпилька острием вверх, если 1 раз щелкнуть по ней левой кнопкой мыши, то
шпилька повернется острием вниз и текущая панель не будет закрыта при от-
172
крытии следующей. С помощью шпильки можно открыть любое число
перечисленных панелей.
Рис..3.2.1. Панель библиотеки элементов
Рис..3.2.2. Панель библиотеки циклов
173
После открытия соответствующей панели и щелчка левой кнопкой мыши
по требуемому фрагменту (элементу) этот фрагмент появляется в окне
контрольной панели. Для расчета требуемой структуры с помощью программы
Gaussian необходимо нарисовать ее в активном окне. Чтобы фрагмент
структуры появился в активном окне нужно щелкнуть левой кнопкой мыши по той
части активного окна, где должен быть фрагмент, изображенный на контрольной
панели. Можно отправить в активное окно последовательно несколько
фрагментов и соединить их в одну структуру, можно изменять длины связей и углы
в структуре активного окна. Перемещение фрагментов в активном окне осуще-
ствяется посредством использования мыши.
3.3. Использование мыши. Просмотр и регулировка параметров
структуры
С помощью левой кнопки осуществляется вращение фрагмента в
текущем окне. Shift+левая кнопка мыши - перемещение молекулы в плоскости
экрана. Правая кнопка мыши (или cntrH- левая кнопка мыши) - перемещение
молекулы перпендикулярно плоскости экрана. Если в активном окне имеются 2 не
174
связанных фрагмента, ими можно манипулировать как с отдельными
фрагментами и как с целой структурой. При необходимости манипулирования с целой
структурой используется описанное выше применение мыши. Для
манипуляции с отдельным фрагментом дополнительно используется клавиша alt.
Устанавливаем курсор на фрагмент которым мы хотим манипулированть: alt+левая
кнопка мыши - вращение фрагмента, alt+ Shift +левая кнопка мыши -
перемещение фрагмента в плоскости экрана. С помощью GV можно посмотреть или
изменить параметры структуры, расположенной в активном окне.
Просмотр параметров структуры осуществляется посредством клавиши
запроса моды *«:. Делаем щелчок левой кнопкой мыши по этому значку. Затем
для просмотра длины связи левой кнопкой мыши выделяем два атома в
молекуле активного окна, для просмотра угла - три атома и для просмотра значения
двугранного угла - 4 атома. Значения длин связей (в А ) и углов (в градусах)
появляются в левом нижнем углу активного окна, в котором расположена
молекула, параметры которой просмотриваются. Для изменения длины или
кратности связи используется клавиша **£* - модифицировать связь. Делаем
щелчок левой кнопкой мыши по этому значку. Выделяем два атома в молекуле
активного окна, после чего появляется окно модификации связи (Рис.3.3.1).
{! "~™.^, ft
I ОМШ** >*-S5^i. Г afgglpB йГ ||§§||§ К |р§ШЩ ] *
Рис. 3.3.1. Окно модификации связи
Ползунком в средней части окна можно установить требуемую длину,
тип связи можно изменить, установив флажок в нужном месте части окна Bond
Туре. Если выбрать кнопку None, то связь между выделенными атомами будет
разорвана. В окне Displacement (atom l, atom 2) можно выбрать тип смещения
групп, присоединенных к выделенным атомам, при этом для каждого атома
можно выбрать: translate group - переместить всю группу, связанную с данным
атомом, translate atom - переместить только атом, а связанную с ним группу
оставить на месте, fixed - атом, отмеченный этим значком перемещаться не будет,
перемещаться будет только второй из выделенных атомов вместе с
присоединенной к нему группой или без в соответствии с выбранным типом смещения
175
для атома 2. Если мы хотим изменить значение угла, выбираем кнопку ?»-
модифицировать угол. Выделяем три атома в молекуле активного окна, после
чего появляется окно модификации угла, аналогичное окну модификации связи
(Рис.3.3.1). Ползунком в средней части окна можно установить требуемое
значение угла. В средней части окна Displacement можно выбрать тип смещения
групп, присоединенных к выделенным атомам, при этом для каждого атома
можно выбрать: rotate group - вращать всю группу, связанную с данным
атомом, translate group - переместить всю группу, связанную с данным атомом,
rotate atom - вращать только атом, а связанную с ним группу оставить на месте,
fixed - атом, отмеченный этим значком вращаться не будет, вращаться будут
только оставшиеся из выделенных атомов в соответствии с выбранным типом
вращения, связанных с ними групп. Аналогично кнопкой ' ш можно изменить
двухгранный угол, выделив 4 атома. Следующая кнопка на контрольной панели
1-" г- добавить валентность. При нажатии этой кнопки после щелчка по
любому атому в активном окне у этого атома появляется дополнительная
валентность с атомом водорода. Кнопка удаляет атомы и висячие валентности
в активном окне. Кнопка Тп.. удаляет все содержание активного окна. CntrZ
или undo ^: - возвращение на 1 шаг назад. Кнопка """ (подчистить) -
подчистит построенную структуру в соответствии с характерными значениями
длин связей и углов между связями. Кнопка (rebonding strucrure) - разорвет
связи, длины которых больше характерной длины связи между атомами.
Кнопка Ш Л ( центрировать) располагает молекулу в центре окна.
Кнопка "^ (симметризовать) симметризует молекулу в соответствии с
точечной группой симметрии. При необходимости симметризовать молекулу в
соответствии с определенной группой симметрии лучше использовать путь Edit -^
Point group, при этом появляется окно установления точечной группы
симметрии (Рис.3.3.2).
176
.rfePg;
D2d
C3v
S4
D2
C2v
3 :C2
Pnc.3.3.2. Окно установления точечной группы симметрии
В этом окне при наличии флажка в строке Enable Point Group Symmetry
(допустимая точечная группа симметрии) в меню Constrain to subgroup можно
выбрать требуемую группу симметрии.
Кнопка Ш- служит для открытия текстового файла, соответствующего
структуре, находящейся в активном окне, если этот файл (input, output и т.д.)
был ранее сохранен.
3.4 Расположение активных окон на рабочем столе
На рабочем столе может быть одновременно открыто несколько активных
окон. Открыть новое активное окно можно с помощью кнопки . Если
открыто хотя бы одно окно, то еще одно окно с помощью этой кнопки можно
открыть двумя способами 1. Create to Molgroup 2. Add to Molgroup, выбор
которых осуществляется посредством полосы прокрутки справа от иконки "" .В
первом случае появится новое окно, во втором случае 2 окна будут соединены в
одно. Выберем первый вариант, а вас появилось еще одно окно. Выберем
второй вариант, у вас появилось 2 совмещенных окна в одном, с каждым из этих
окон можно работать независимо. В обоих режимах можно открыть
необходимое число окон. Если используется первый способ и открыто несколько
активных окон их можно упорядочить с помощью кнопки ""•"-а,: - расположить
каскадом, или с помощью кнопки S~t ;- упорядочить на рабочем столе. Кнопка -
■ВЩ :*
- - - добавить вид служит для целей одновременного наблюдения за
молекулой под разными ракурсами. При наличии молекулы в активном окне и
нажатии на эту кнопку появляется еше одно окно с той же самой молекулой. Оба
окна независимы и можно одновременно наблюдать требуемые виды.
3.5 Стандартные кнопки ввода - вьшодад^дактор^писка^толюв.
Стандартные кнопки ввода - вывода "-' " '• "" -:-°"
означают соответственно открыть, сохранить, напечатать, сохранить как рисунок,
177
вырезать, копировать. Нельзя выделять и копировать часть молекулы, всегда
выделяется полная структура в активном окне. Существует несколько способов
вставить структуру, скопированную в буфер. Выбор нужного способа
осуществляется с помощью полосы прокрутки справа от иконки «вставить» IsSifj,
которая активна, если в буфере находится скопированный фрагмент. Replace
molecule - удаляет структуру в текущем окне и вставляет вместо нее другую.
Appended molecule - добавляет молекулу к имеющейся в активном окне
структуре, как отдельный фрагмент. Add to moleculare groop - создает новую модель
в пределах текущей модели и вставляет структуру внутрь, если так вставить
нельзя, то формирует совмещенное с активным окно, в которое вставляет
структуру. Кнопка IS - сохранить как рисунок используется для сохранения
структуры активного окна как рисунка, который может быть вставлен в
текстовый файл.
Редактор списка атомов (The Atom List Editor). Это окно отображается, если
выбирать пункт меню Edit=>Atom или щелкнуть по кнопке зШт - the Atom List
Editor. Этот редактор показывает координаты и другие параметры молекулы в
редактируемом формате электронной таблицы. С помощью этого редактора
можно изменить координаты требуемых атомов молекулы активного окна,
добавлять новые атомы к имеющейся структуре, управлять отнесением слоев
ОМОМ(см. 3.8) и другими данными.
3.6. Регулировка качества структуры молекулы в активном окне
Качество структуры в активном окне может быть отрегулировано с помощью
кнопки '~: - Display Format на контрольной панели, при нажатии на которую
открывается диалоговое окно, где представлены General, Text, Molecule панели
и панель Surface (Рис. 3.6.1 - 3.6.4).
Обгцая Панель (General Panel) (Рис.3.6.1). Пункты на General контролируют
дисплейное качество молекулы, книзу отображены дополнительные пункты,
поставим в них флажки show tags, show symbols, show casterian axes - на
элементах молекулы в активном окне появятся метки, символы и оси координат.
Оси координат проходят через центр масс молекулы только после выполнения
операции симметризации^^ .
Опция Use Fog to improve depth perception (использовать туман для
улучшения глубины восприятия) позволяет более наглядно изображать трехмерные
молекулярные структуры. Поставим флажок в соответствующем окне и
установим ниже видимость 20А. Введем в активное окно, например, антрацен
(откроем библиотеку циклов, выберем 4 молекулу в верхнем ряду). Вид активной
молекулы изменился; более удаленные атомы видны хуже, ближние атомы более
четкие. На глубине 20А туман делает видимость нулевой.
Опции в верхней части общей панели управляют видом молекулы в
активном окне в стационарном режиме (Stationary Object Display) и в режиме
анимации (Moving Object Display). В медленных и неподвижных системах часто
178
полезно устанавливать движок ближе к реалистичному (Realistic) концу шкалы,
для быстро движущегося объекта ближе к быстрому (Fast) концу шкалы.
Опция плавный (Smooth ) выводит молекулу на дисплей более ровно.
Прожектор (spotlight) запускает/отключает освещение в активном окне.
Молекулярная (Molecule Panel) панель (Рис.3.6.2). Опции на этой панели
управляют видом атомов и связей в активном окне. Три меню (High layer, Medium
layer, Low layer) определяют вид атомов для этих трех уровней методики
расчета ОМОМ ( см.3.8 ). Если средство ОМОМ не используется, то вид атомов и
связей молекулы в активном окне будет таким как задано в строке High Layer
(высокий уровень).
.^йИЙШШё^р^-'-^Й*1
if:., тш | kmd
1 ^^ШЫ^-Д1>1#ЙК .-.3
Рис.3, б. 1. Диалоговое окно Display Format. Общая панель.
179
fficwtlsSSK | Wireframe]>§}
1 ШШШШШЩ
Puc.3. 6. 2. Диалоговое окно Display Format. Молекулярная панель.
Предусмотрены следующие виды атомов и связей, которые можно выбрать с
помощью полосы прокрутки справа от кнопки соответствующего уровня:
1. Ball&Stick (Шар и Палка). 2. Ball&Bond Type (Шар и тип связи). 3. Tube
(Трубка). 4.Wireframe: молекула появляется как серия тонких цветных строк,
тип связи указывается через количество строк. 4.None: Атомы невидимы
(полезен только для многоуровневых моделей).
Установив флажок Use Vanderwaals Radii и масштабирование, можно
отобразить атомы шарами с радиусами, равными радиусам Ван дер Ваальса.
Движок Z-Clip используется для удаления части изображения.
Текстовая Панель (Text Panel) (Рис.3.6.3). Текстовая панель регулирует шрифт
и цвет номеров атома и символов, позволяет исключить маркировку
определенных атомов (Exclude View Labels, Exclude View Symbols). Шрифтовое меню
(Font File) позволяет выбирать шрифт меток из предусмотренных GaussView.
Поле Size определяет относительный размер символов и меток: Область Render
определяет тип шрифта и его цвет.
180
Puc.3. 6. 3. Диалоговое окно Display Format. Текстовая панель.
Панель поверхностей (Surface Panel) (Рис.3.6.4). Позволяет выбрать вид
отображаемых поверхностей (Solid, Mesh, Transparent - твердый, прозрачный,
сетчатый, см. примеры в разделе 3.11).
Заметим, что большинство описанных в разделах 3.3 - 3.б действий может
быть выполнено не только с помогцью указанных кнопок, но и выбрав путь
File -> соответствующая опция, Edit -> соответствующая опция, View ->
соответствующая опция.
-!«^|.
Рис.3. 6. 4. Диалоговое окно Display Format. Панель поверхностей.
181
3.7 Запуск программы Gaussian и просмотр результатов с помощью
Gaussview
Программа Gaussview позволяет сформировать input файл расчета
свойств системы, находящейся в активном окне. Для его формирования и
запуска программы Gaussian с помощью программы Gaussview в командной
строке контрольного окна выбираем Calculate -> Gaussian:
После этого открывается диалоговое окно (Рис.3.7.1) задания входных
параметров задачи, которое включает ряд панелей:
1. Job Туре (Тип работы) - здесь указаны ключевые слова, с помощью
которых программе задается тип необходимых вычислений. Например, для расчета
энергии молекулы, изображенной в активном окне (т.е. SP, см.2.4), выберем
energy ( Рис.3.7.1). Ключевые слова программы Gaussian описаны в разделе 2.14.
Если необходимо выполнить работы, отсутствующие в меню Job Type, их
можно указать в строке Additional Keywords.
2. Для указания метода и базиса расчета открываем панель Method
(Рис.3.7.2.). На Рис.3.7.2 в качестве расчетного метода основного состояния
выбран метод Хартри-Фока, базис 6-311G, в строке Charge указывается заряд
рассчитываемой системы. Мультиплетность основного состояния программа
задает автоматически, при необходимости расчета возбужденного состояния
мультиплетность задается пользователем. С помощью полос прокрутки (отмечены
Мл ) желаемые характеристики расчетной процедуры можно выбрать из
соответствующего меню.
3. Панель Title - панель комментариев (Рис.3.7.3.). Здесь можно записать
любые необходимые комментарии, объем неограничен, но возможно
использование только латинского шрифта.
4. LinkO панель (Рис.3.7.4) служит для указания имени chk - файла, в
который будут записываться промежуточные результаты, максимального объема
182
памяти, который необходим для вычислений и числа процессоров,
используемых в расчете.
5Й1
$шдощзЩ?яШйШШШШ''
| Energy j§ t
Optimization "*j ■.
Frequency \ j
Opt+Fieq
IRC
Scan
Stability
NMR
BOMD
ADMP
Рис. З.7.1. Панель выбора типа работы
■ЩЩтжттШШтШМ.
Ground State~]|§|j Hartiee^Fock jf§ \ Default Spin |Ц
;Sljag||, ||C
3-21G Щ
i-31G -^S"** 1
cc-pVQZ
LanU2DZ j f
LanL2MB
SDD
DGDZVP
DGDZVP2 Л!
DGTZVP
АййопШЙЩк
Puc.3.7.2. Панель задания метода и базиса расчета
183
% йхм}тммШ0шштштш
\ -'ШГ0 | ffiffibtt | Щ | Ж* |)%*я# f 1ш |йЖШ РйР I ШФШё
£chk=ENERGY HF
%mem=6MW
%пргос-1
Tjxf
^^^g^fejl
P4 ашшда» к,.,, -;
t,Jfggj,,,,,| t^Hilb „... J „ fe_ I j^jjjjljl ,,ъ ]|Ш
Рис. 3.7.4. LinkO панель
184
5. The General Panel. Здесь можно выбрать некоторые обычно используемые
опции. В таблице представлены соответствия между ключевыми словами и
этими опциями
Опция
Use Quadratically Convergent SCF
Use Modified Redundant Coordinates
Ignore Symmetry
Additional Print
Write Connectivity
По умолчанию
выключено
включено
выключено
выключено
включено
Ключевое слово
SCF=QC
Geom=Mod Redund ant
NoSymm
#P
Geom=Connectivity
6. The Guess Panel. Здесь указываются характеристики стартовых
молекулярных орбиталей. Чем «лучше» выбраны стартовые орбитали, тем меньше число
итераций в процедуре самосогласования. Необходимость вычисления на
каждой итерации ~М одноэлектронных интегралов и ~М двухэлектронных
интегралов (см. 1.3) является причиной выбора стартовых молекулярных орбита-
лей наилучшим образом. По умолчанию в Gaussian03 используются стартовые
орбитали, получаемые диагонализацией функционала Harris'a. Указав (в
additional keywords) одну из опций INDO, Huckel, AMI, Core и другие, описанные в
разделе Help к программе Gaussian, в качестве стартовых можно выбрать МО,
рассчитанные данными методами (Core - получены диагонализацией матрицы
Фока, состоящей из одноэлектронных вкладов). Можно также выбрать одну из
опций, поставив флажок в соответствующей клетке (Рис. 3.7.5).
7. NBO панель используется при необходимости проведения анализа
заселенности с использованием натуральных связевых орбиталей.
8. Панель Solvation (Рис.3.7.6) служит для формирования input файла задачи
моделирования систем в растворах. Сначала в меню моделей выбрается
желаемый метод расчета, по умолчанию используется метод РСМ (см. 1.20). После
выбора модели сольватации открывается меню выбора растворителя и можно
выбрать любой из описанных в 2.12 растворителей.
После описания расчетной процедуры посредством заполнения
перечисленных панелей с помощью Gaussview можно осуществить запуск программы
Gaussian на решение. Для запуска необходимо задать имена входного и
выходного файла. Выбираем submit - представить на рассмотрение (Рис.3.7.7) и в
диалоговом режиме отвечаем на задаваемые вопросы (имя входного и
выходного файлов, их местонахождение), после чего Gaussview запускает программу
Gaussian.
185
,%даоМ^ Plillp^ewHa-egMieggfe
Штш I даш § »щ,к|шйьап |шш |!j||| | ыаа faggi] &!«1Бн |i
T уш&тшщшщщщтщт
Pwc.J?.7.5. Окно выбора стартовых МО
Рис.3.7.6. Панель учета эффектов сопъвитации
186
^^^Ш'ШЧш^^тш: $ш$ II ШЬ&Щ 1 Щщ ЩШ £ш ! ?#*"
sll " * ,.„„„I!...„.^,!
!»ШаЩИ fDefault ' j£| ж«
5о$й$* ij Water
f3 Readj^h^bstrajj *Ы^' :^^ysuii!etsSrf«aoinp5ie'6ciw?
:UV$$$,;
LjJ^fe„J .-. Jll^Lt4biai^§^ £;;M -.. -I.?; f## J,- .0..--.№fe„„ Л
Рис. 3.7.7. Запись входного файла с помощью программы Gaussview
Информацию о работе программы Gaussian или ее завершении с помощью
Gaussview можно получить, выбрав на контрольной панели Calculate
->Current Jobs.
После окончания работы программы Gaussian для просмотра
результатов расчета с помощью Gaussview необходимо сначала открыть выходной
файл. На контрольной панели Gaussview нажимаем ".', с помощью
прокрутки выбираем тип файлов output, в меню файлов output находим свой
файл и открываем его, при этом появится окно с графическим изображением
рассчитанной структуры. Для анализа результатов при использовании типа
работ Energy выбираем сначала Rezalts -> Summary, затем Rezalts -> Charge
(Рис.3.7.8). При выборе Rezalts -> Summary появляется окно вывода
основных результатов расчета, включающее основные характеристики расчетной
процедуры, полную энергию, дипольный момент и точечную группу
симметрии рассчитываемой системы (Рис.3.7.9). При выборе Rezalts -> Charge
открывается окно вывода зарядов на атомах рассчитанной структуры. При
выборе Rezalts -> View File открывается текстовой output файл задачи.
Текстовый файл задачи, соответствующий структуре, находящейся в активном
окне также можно открыть также с помощью кнопки 83 на контрольной
панели. При выполнении сложных типов работ в меню Rezalts активны и
другие опции: Surface, Vibration, NMR, Scan, IRC, Trqjectory, Optimization (см. в
следующих разделах).
187
Suminaiji
Рис.3.7.8. Меню Results
l-aczHiOT.taicMtotinftSu^D^^aisiiMBS-?
•Ш&Щт
" ICafculaj^tfifjjje
„ ;Са1сц1аиоп'ЙЁЙгоа;
•Basis $5ёК
С ;ЩЙ£+НЩШ:
ШШРШШ-Нрш
Jjna$inarj>;to<i;
ЙфЩЩпшй:
ШКШЭф
:dcb)q^uf»№:2td^ С
^.-■tK-Hlf-.ij.CrMli-.Jf.W
СОД;
1
зЙШ I
mm г
ятаж я
гЩШЩЙ! |
яшшшее щ ii
ВЛЙВШйВ ж 11
S V,
mm ъёцм а
.т !•
*е№Л;«ШйШ;Р1Шй№ •:
Рис.3.7.9. Окно вывода основных результатов расчета
3.8 ONIOM расчет молекулярных структур
В последних версиях Gaussian (G98 и далее) были развиты методы
расчета сверхбольших молекулярных систем. Это вызвано интересом к расчетам
больших биомолекул, и в том числе потребностями задач конструирования
лекарств. Среди таких методов можно отметить методику ONIOM, развитую
профессором Морокума с сотрудниками, в которой вся молекула разбивается
на 3 области, которые рассматриваются с разной степенью точности. Такой
подход эффективен, в частности, для биомолекул. Например, "активное место"
188
лекарства можно рассчитать с максимально высокой степенью точности,
непосредственное окружение активной части можно считать можно посчитать менее
точно, а дальние атомы - учесть максимально приближенно. Разбиение
молекулы на области различной расчетной точности осуществляется с помощью
программы GV с использованием кнопки -№ (трехуровневая модель) на
контрольной панели (Рис.3.8.1).
Рис. 3.8. 1. Кнопка трехуровневой модели на контрольной панели
Предварительно молекула должна быть построена в активном окне. После
нажатия кнопки открывается инструмент выбора уровня расчетной точности
атомов исследуемой системы (Рис.3.8.2).
Для задания уровня выполняем следующие действия:
1. Нажимаем Select Layer . Открывается 3 возможных уровня точности:
низкий, средний и высокий. Выбираем нужный уровень.
2. В активном окне щелкаем по атому, расчет которого хотим вести с
выбранной точностью. Нажимаем Apply.
189
3. Процедуры 1-2 выполняем для каждого атома. (По умолчанию атом
рассчитывается с высокой степенью точности, поэтому для атомов,
рассчитываемых с Higt Layer процедуру можно не выполнять).
Вид атомов и связей между ними для структуры, рассчитываемой с ONIOM,
задается в редакторе Displey format (кнопка " -, см. раздел 3.6, Рис.3.6.2). На
Рис.3.8.3. справа представлена вид структуры молекулы пропана,
рассчитываемой с ONIOM, используемый по умочанию (концевые атомы водорода 1 - 6
рассчитываются с низкой степенью точности, средние атомы водорода 7,8 - со
средней степенью точности, а атомы углерода 9 - 11 - с высокой степенью
точности).
Рис. 3.8.3. Вид молекулярной структуры в трехуровневой модели
слева : атомы рассчитываются на высоком уровне точности,
справа: трехуровневая модель(атомы 1-6- низкий уровень точности, 7-8-
средний, 9—11 - высокий)
3.9 Анализ результатов сканирования поверхности потенциальной энергии
с помощью программы Gaussview
Сканирование поверхности потенциальной энергии задается с помощью
ключевого слова Scan. При выборе этого типа работы на панели Job Type
(Рис.3.7.1) программы Gaussview будет указано требование дополнительного
ввода. Это означает, что осуществить запуск сканирования поверхности
потенциальной энергии системы только с помощью программы Gaussview нельзя,
необходимо вводить дополнительную информацию в окно ввода работы
(Рис.2.2.2). Дополнительная информация вводится в соответствии с примерами,
представленными в разделе 2.9. после чего программа запускается на решение.
Для анализа зависимости поверхности потенциальной энергии от координат с
помощью программы Gaussview после окончания расчета необходимо в
Gaussview открыть output файл. При этом откроется совмещенное активное окно
190
(Рис. 3.9.1). число окон в котором равно числу шагов сканирования. Каждое из
этих окон отображает конфигурацию системы для определенного шага.
..аваамяэдт; яатнвгаип-':
Рис. 3.9.1. Совмещенное активное окно, включающее 21 окно. Открыта
конфигурация первого шага сканирования поверхности потенциальной энергии
системы Ы+ -Н2О. С помощью полосы прокрутки можно осуществить
просмотр конфигурации произвольного шага
Путь Results -> Scan (Рис. 3.9.2) открывает графическое изображение
поверхности потенциальной энергии.
Рис. 3.9.2. Путь Results -9 Scan
191
Программа выводит зависимость полной энергии системы как функцию
номера шага, т.е. как U\N) независимо от числа варьируемых переменных
(Рис.3.9.3, см. также примеры раздела 2.9):
ш
^й^^^шзМшш^ш\&цЗ
sillштщщШшжж
ц^**
Рис. 3.9.3. Зависимость поверхности потенциальной энергии системы
Ы Н2О как функции номера шага, определяющего расстояние Ы+ — О
3.10 Просмотр орбитальных энергий в программе GAUSSVIEW
Графическое изображение орбитальных энергий можно открыть в программе
GAUSSVIEW при выполнении любого типа работ. Просмотр орбитальных
энергий в программе GAUSSVIEW осуществляется после открытия output
файла с помощью кнопки -ML, нажатие на которую открывает окно редактора МО.
В этом окне слева представлена молекула, справа - ее одноэлектронные уровни
энергии (Рис.3.10.1).
Рис. 3.10.1. Орбитальные энергии молекулы метана
192
3.11 Изображение молекулярных орбиталей в программе GAUSSVIEW
Изображение молекулярных орбиталей в программе GAUSSVIEW можно
открыть при выполнении любого типа работ. Для этого сначала нужно выполнить
следующие действия:
а) В активном окне открыть checkpoint файл исследуемой структуры, поскольку
информация о молекулярных орбиталях содержится в checkpoint файле, после
открытия chk станет активным пункт Surface в меню Rezalts (Рис.3.11.1).
S^GaussViewaus
.Ж&Щ#!2ЙШ1 ЩЭШШ|Ще5ШяШ1Й»В1Ёь!й(
Рис.3.11.1. Путь Rezalts -> Surface на контрольной панели GV
б) Выбрать Rezalts -> Surface. При этом станет активным окно Surfaces and
Cubes (Рис.3.П.2), которое позволяет отображать различные химические
данные в 3-х измерениях. Это окно включает 2 строки Cubes Available (доступные
кубы) и Surface Available (доступные поверхности). Сначала для генерации
трехмерного куба заполняется Cubes Available посредством выбора
соответствующего пункта (создать новый куб или загрузить куб) из меню Cube Actions
(Рис.3. П.2).
ЩагттШШтшЖ^Шш
Cubes iAyailafile:
SurfacesiSvailefcflfe
|ооои
Рис. 3.11.2. Окно Surfaces and Cubes
Для генерации нового куба выполяем следующие действия:
193
1. В меню Cube Actions выбираем New Cube — сгенерировать новую
трехмерную сетку для построения требуемой поверхности. Здесь также имеются
пункты Load Cube - загрузить куб из внешнего файла (Рис.3.11.2). Save Cube —
сохранить текущий куб во внешний файл. Remove Cube - удалить указанное в
строке Cubes Available. Последние два пункта становятся активными после
появления записи в строке Cubes Available.
После выбора New Cube появится окно Generate Cubes с активными опциями
Kind (тип), Orbitais и Grid (сетка), Рис.3.11.3.
слева меню Kind, справа — меню Orbitais
2. В меню Kind (Рис.3.11.3) выбираем свойство, которое нужно отобразить (МО
- молекулярную орбиталь, ESP - электростатический потенциал - потенциал,
создаваемый в пространстве точечными зарядами ядер и непрерывно
распределенными зарядами электронов молекулы и др.). В меню Orbitais выбираем
орбиталь, для которого это свойство отображается (HOMO, LUMO, Occupied,
Virtual, All, Other). Для выбора орбитали можно в соседней пустой строке указать
номер орбитали, которую необходимо вывести. Рис.3.11.3 показывает, что
будет генерироваться сетка для отображения седьмой МО (HOMO).
тт.Щ Molecular Orbital jg|
I HOMO jjpl i{ Alpha j*j$
%ВШЩ Coarse jy| tjsJT:
^ Ха31к1к<С1;-;" ТЩ *■:■ -IS, -Jl
Puc.3.11.4 Заполненное окно Generate Cubes для построения а-орбитали
HOMO (№1)
После выбора орбитали становится активным меню выбора проекции спина
электрона [а, /?) ( Рис. 3.11.4, справа). В ограниченном методе Хартри-Фока
пространственные МО а, (3 электронов одинаковы. Пункты меню Grid (сетка)
194
определяют плотность трехмерной сетки. Для целей визуального наблюдения
достаточна установка Coarse (грубая). Сетка Medium (средняя) требуется для
целей печати. При генерации кубов в случае выбора в меню Kind (тип)
требования отобразить МО, под полем Orbitals высвечивается число МО,
информация о которых имеется в сЫс^йлеПзанятых и 11 виртуальных на Рис.3.11.5).
&Шй1 Molecular Oibtal Яр '
Рис. 3.11.5. В chk файле имеется информация о 7 занятых и 11
виртуальных МО, будет генерироваться МО №11
После заполнения окна Generate Cubes щелчок OK сгенерирует куб с
требуемыми свойствами и выход из диалога Cubes Available. Новый куб появится в
списке Cubes Available (доступные кубы, рис.3.11.6).
jAiphaMOJmo = 1 ; npts -16,18,18; res = 0.333Шаз!зЗЗЗЗДЗЗЗЗЗЗ] gj.
Рис. 3.11.6. Сформированный куб для построения первой МО
Можно сформировать любое число трехмерных кубов для изображения
интересующих поверхностей. Поэтому в списке доступных кубов, который
открывается при щелчке мышью по знаку полосы прокрутки Cubes Available может
быть любое количество записей. Поверхность будет строится для случая,
видимого на экране в строке Cubes Available (первая МО для случая,
представленного на Рис.3.11.6).
Кубы, сгенерированные посредством New Cube автоматически не
сохраняются и будут потеряны после завершения работы GV.
Следующим этапом является построение изображения требуемой поверхности.
GAUSSVIEW отображает изоповерхности - поверхность постоянной плотности
вероятности (для орбитали), постоянной электронной плотности (для электрон-
195
ной плотности), эквипотенциальную поверхность (для потенциала). Для
построения изображения нужно выполнить действия:
1. В строке isovalue for new surface (Рис.3.11.6) указываем требуемое изозначе-
ние (или оставляем указанное программой по умолчанию).
2. GAUSSVIEW может отображать несколько поверхностей в одном активном
окне, и может каждую новую поверхность строить в новом окне. В первом
случае в поле Add views for new surfaces (Рис. 3.11.6) не ставится флажок, во
втором-ставится.
3. Открываем пункты меню Surface action (Рис.3.11.7). Меню Surface Actions
содержит пункты New surface - создать новую поверхность, New Mapped
surface-показать новое свойство отображаемой поверхности (см.ниже), Show
surface (показать поверхность), Hide surface ( спрятать поверхность), Remove
surface (удалить поверхность). Последние 3 пункта становятся активными после
появления хотя бы одной записи в строке Surface АуаПаЫе.Выбираем New
surface. В активном окне появляется заданная при генерации кубов изоповерх-
ность, в строке Surface Available появляется запись, характеризующая
созданную поверхность.
Р Alpha МО (то = 1 ; npts = 18,18.18;res = 0.333333,0.333333,0.333333]
^J5iliig..,.....ia
Рис. 3.11.7 Меню Surface action
На Рис.3.11.8 показаны 2р - орбитали атома водорода.
1Пг ', Р
Рис. 3.11.8. 2р^.2р ,2 р атомные орбитали атома водорода
Поверхности могут быть отображены в твердом, прозрачном и сетчатом виде
(Рис.3.11.9).
196
Рис.3.11.9. Вид изображаемых поверхностей(слева направо): тведый,
сетчатый, прозрачный для твердого вида поверхности использован ползунок
Z-Clip (Рис.3.11.10) , позволяющий «срезать» часть поверхности
Вид изображаемых поверхностей задается в окне Display Format на
панели Surface (Рис.3.11.10).
Рис.3.11.10. Выбор вида изображаемой поверхности
Ползунок Z-Clip используется, чтобы удалять части переднего плана отбра-
женной поверхности, при этом видна часть внутренней поверхности
(Рис.3.11.9).
Пример. Определение симметрии и построение молекулярных орбиталей
молекулы Л^2- Если МО обладают симметрией и программа ее определяет, то
информацию о типе симметрии можно получить из текстового output файла,
который можно открыть из GV щелкнув по кнопке Шт после того как в активном
окне изображена соответствующая output структура.
Часть output файла, отражающего симметрию МО начинается после слов
Population analysis using the SCF density.
Orbital symmetries:
197
Occupied (SGU) (SGG) (SGG) (SGU) (РШ) (PIU) (SGG)
Номер МО 1 2 3 4 5 6 7
Virtual (PIG) (PIG) (SGU) (SGG) (PIU) (PIU) (PIG) (PIG) (SGU) (SGG) (SGU).
Номер МО 8 9 10 11 12 13 14 15 16 17 18
Здесь, SG =a, PI =я, например SGU = a , PIG = n . Список занятых и
виртуальных орбиталей приведен для молекулы N2- Из списка следует, что HOMO
орбиталь ( номер 7) имеет симметрию а , a LUMO орбиталь (номер 8) -
симметрию ж . Построенная с помощью программы GAUSSVIEW HOMO
орбиталь молекулы N' (т.е. поверхность, в каждой точке которой, плотность
вероятности равна определенной величине, в рассматриваемом примере 0,02)
представлена на Рис.3.11.11.
Рис. 3.11.11. HOMO орбиталь молекулы N' ( с ),
объяснения в тексте
Качественно вид изображенной поверхности определяется видом
комбинирующих атомных орбиталей. Так вид HOMO орбитали (Рис. 3.11.11) позволяет
сделать вывод, что основной вклад в HOMO дает линейная комбинация двух р
атомных орбиталей, направленных вдоль линии связи. Действительно, элек-
9 9 4
тронная конфигурация атома N Is 2s 2p , тройная связь молекулы N =
Nобразуется тремя р - электронами первого и тремя р -электронами второго атомов
азота ( 2р ,2р ,2р_ ). При этом одна из трех «восьмерок» каждого атома
направлена вдоль линии связи и приводит к образованию с-связи (Рис. 3.11.12а,
HOMO, орбиталь №7). Образование HOMO происходит таким образом, что
вклады в нее от 2pz атомных орбиталей атомов азота (ось z направлена вдоль
линии связи) противоположны по знаку (Рис.3.11.12а). Это приводит к умень-
198
шению электронной плотности в пространстве между ядрами. Однако
электронная плотность в центре не равна нулю (Рис.3.11.11), и орбиталь является
связывающей, о чем свидетельствует достаточно высокая энергия диссоциации
молекулы Nj, находящейся в основном состоянии.
С>С0Ез*3-
a be
Рис. 3.11.12 Образование HOMO а-орбитали (а), связывающих я (Ъ)и
разрыхляющих ж (с) орбитшей молекулы Л^.
Две оставшиеся «восьмерки» {2р ,2р^) перпендикулярны линии связи, их
суперпозиция приводит к образованию двух п -связей. В орбитали л (№5 и
№6) основной вклад дают 2р и 2р АО азота, сближающиеся «лопастями» с
■*■ У
одинаковыми знаками (Рис.3.12 Ь)
<P5{N2)~c{x2pxN{}) +X2PXfN(2)) ,
п орбитали №5 и №6 вырождены и по энергии лежат ниже HOMO, орбитали
являются связывающими и имеют вид, представленный на Рис. 3.11.13.
199
Рис.3.11.13. Связывающая п орбиталъ молекулы N2
к орбитали №8 (LUMO) и №9 выролодены и по энергии лежат выше HOMO,
при их образовании «восьмерки» волновых функций в областях перекрывания
имеют разный знак (Рис. 3.11.12с):
1>8Ш = С1 \X2pXtN(,)-X2pXiN(2)) '
<Р9Ш = С1[Х2руЛ}) -X2pytN(2)) >
орбитали являются разрыхляющими и имеют вид, представленный на Рис.
3.11.14.
Рис.3.11.14. Изображение LUMO орбитали молекулы 7V2 ( ж ) в GV.
Орбиталъ является разрыхляющей: вероятность нахождения электрона в
пространстве между ядрами равна 0.
200
Создание новых свойств отображаемых поверхностей. При выборе
опции New Mapped surface (Рис.3.11.7) откроется окно диалога создания новых
свойств отображаемых поверхностей.
й GI:M1:V1 - Suifaee Mappmg
рсдщ! ш
Рис.3.11.15. Окно создания новых свойств отображаемых поверхностей
Выберем ESP - электростатический потенциал, программа раскрасит
построенную изоповерхность МО в соответствии со значениями электростатического
потенциала (Рис.3.11.16).
Ш а»м5Л&*ш:
Рис.3.11.16. Изображение значения электростатического потенциала (справа)
на изоповерхности МО (слева)
3.12 Изображение ИК спектров молекул с помощью программы GAUSS-
VIEW
Для запуска на решение задачи расчета ИК спектров молекул с помощью
программы GAUSSVIEW необходимо:
1. Построить в активном окне исследуемую молекулу.
201
2. Запустить Gaussian (тип работ OPT+FREQ, метод, базис по усмотрению,
раздел 3.7).
Для просмотра результатов расчета с помощью программы GAUSSVIEW
необходимо:
1. После окончания расчета с помощью программы GAUSSVIEW открыть
output файл как описано в 3.7.
2. На контрольной панели программы GAUSSVIEW выбрать Rezalts ->
Vibration (Рис.3.12.1), после чего откроется окно Display Vibration (Рис.3.12.2).
WGaussVieWjaq^j:
Ifl-y^ffe
5-йЩик"'Г':'-"
Рис.3.12.1. Открытие окна Display Vibration
При наличии у молекулы ИК и КР спектров в окне Display Vibration будут
отображены (Рис.3.12.2):
В столбце Freq - немасштабированные частоты колебаний, в столбце Infrared -
интенсивности линий ИК спектра (в км/моль), в столбце Raman - активности
А4
линий КР ( в ), в двух последних столбцах - коэффициенты деполяриза-
а.е.т
ции плоскополяризованного (Р) и неполяризованного (U) света. Для получения
информации о конкретном нормальном колебании необходимо выделить
обычным образом соответствующую строку в окне Display Vibration. При наличии
флажка в окне Show Displacement Vectors на молекулярной output структуре в
активном окне будут показаны векторы амплитуд смещений всех атомов
молекулы, соответствующие выделенному нормальному колебанию (Рис.3.12.3),
позволяющие сделать вывод о форме данного нормального колебания.
202
Рис.3.12.2. Окно Display Vibration
Так из Рис.3.12.3 следует, что анализируемая частота соответствует колеба-
Рис.3.12.3. Форма нормального колебания молекулы тназина,
соответствующая колебанию N- Н связи.
При нажатии кнопки Start окна Display Vibration (Рис.3.12.2) на
молекулярной output структуре в активном окне будет показана анимация колебаний
атомов, соответствующая выделенному нормальному колебанию, что
позволяет исследовать форму колебаний. При нажатии кнопки Spectrum окна
Display Vibration (Рис.3.12.2) на экране монитора появится изображение рассчи-
203
-= о ЭД1 шШГ"""""шт. _ аш" Mir ШШ Пий! J»y
Рис.3.12.4. ИК, КР спектры и коэффициенты деполяризации молекулы тиа-
зина
3.13 Изображение спектров ЯМР в программе Gaussview
Для запуска на решение задачи расчета спектров ЯМР молекул с помощью
программы GAUSSVIEW необходимо:
1. Выполнить оптимизацию структуры исследуемой молекулы.
2. Открыть оптимизированную структуру в активном окне.
204
3. Запустить Gaussian на решение (тип работ NMR). При этом в меню
методов ЯМР (Рис.3.13.1) можно выбрать интересующий метод расчета
спектра ЯМР (GIAO, CGST, 1GAIM, All NMR methods). Остальные панели
окна Gaussian Calculation заполняем обычным образом, как описано в
разделе 3.7.
: fj СК!МШ116ШШ|1|Ш,Ж^ЖС,
ЗММ№ ^V
ЩйркЖ
CGST Method
IGAIM Method
All NMR Methods
^ScSi%nafR%woi3K: |
Рис. 3.13.1 Выбор метода расчета спектра ЯМР
Для просмотра результатов расчета с помощью программы GAUSSVIEW
необходимо:
1 . После окончания расчета задачи NMR открыть ее output файл.
2. На контрольной панели программы GAUSSVIEW выбрать Rezalts ->
NMR (Рис.3.13.2), после чего откроется окно графического изображения
рассчитанных постоянных экранирования (Рис.3.13.3).
205
Рис.3.13.2. Открытие окна NMR Spectra
Рис.3.13.3. Окно NMR Spectra с изображением постоянных экранирования ядер
атома водорода молекулы бутана
В окне NMR Spectra внизу слева в клетке Element можно указать химический
элемент, постоянная экранирования которого интересует, при этом будет
показана область спектра, соответствующая данному элементу. При наличии в
строке Reference (эталон) указателя None (Рис.3.13.3) в окне NMR Spectra по шкале
абсцисс откладываются значения рассчитанных постоянных экранирования. На
Рис.3.13.3 графически изображены рассчитанные постоянные экранирования
протонов молекулы бутана, нумерация атомов представлена на Рис.3.13.4. Если
курсор мыши подвести к интересующей вертикальной линии (Рис.3.13.3) и
206
щелкнуть по ней левой кнопкой мыши, то внизу окна NMR Spectra появится
численное значение указанной постоянной экранирования (не показано на
Рис.3.13.3). Для химически эквивалентных атомов (например, 5 и 6, 7-10, 11-
14), постоянные экранирования одинаковы, для химически неэквивалентных
атомов - различны. Химически эквивалентными ядрами называют ядра одной
индивидуальности (например, протоны), имеющие одинаковую постоянную
экранирования, ядра одной индивидуальности, имеющие различные постоянные
экранирования называют химически неэквивалентными.
Рис.3.13.4. Молекула бутана
Программа GAUSSVIEW позволяет вывести графическое изображение
химического сдвига интересующего ядра относительно ядра эталона, указанного в
строке Reference. Для указания эталона открываем меню эталонов (Рис.3.13.5).
После выбора интересующего эталона на мониторе появляется окно NMR
Spectra с изображением химических сдвигов ядер рассчитываемой молекулы
относительно выбранного эталона (Рис.3.13.6). Если в этом окне щелкнуть по
интересующей линии, что внизу окна появится значение химического сдвига
ядер выбранной линии относительно соотвествующих ядер эталона. Внизу окна
(Рис. 3.13.6) представлен химический сдвиг для протонов 11-14 СИ 2 ГРУПП
молекулы бутана, его значение 1,29 хорошо согласуется с экспериментальным
значением 0,9 - 2,1 (Табл. 1.21.1).
207
G2;Ml;VI -V*.* . Mr
f^^^*l^^^1?]nM^^^^Vift*i*-^ff{fmumin
w
■ ш~,ллл^аш&х1:££&
тшшшшшшшшшш
Д1|ВсяЬ?|Н j|j ШШЙпЖ INone
t;lj""*)":"l-r"~l'""'l-iJ'!'u'"'jhi^'^"»j-''
6*«Ш11ШЙ»
Рыс. 3.13.5. Выбор эталона для расчета химического сдвига (TMS'- тетраме-
тилсилап)
s^immmmmm.
Рис.3.13.6. Окно NMR Spectra с изображением химических сдвигов протонов
молекулы бутана (эталон — тетраметичсжан, расчет ctms методом ХФ в
базисе б-ЗЮ(ф)
208
Аббревиатура методов компьютерной химии
ACM - Adiabatic connection model
ANO - Atomic Naturfl Orbitals
APT - Atomic Polar Tensor
AIM - Atoms in Molecules
AMI - Austin Model 1
ACPF - Averaged Coupled Pair Functional
АО - атомная орбиталь
ВЗМО - высшая занятая молекулярная орбиталь
BD - Brueckner Doubles calculation
BSSE - basis set superposition error (ошибка суперпозиции базисных наборов)
ВО - Born Oppenheimer approximation
CASSCF - Complete Active Space Self- Consistent Field method
CBS - Complete basis set ( сложные базисные наборы)
CC - Coupled cluster methods
CCD - Coupled cluster, using double substitutions
CCSD - Coupled cluster, using single and double substitutions
СЕРА - Coupled Electron Pair Approximation
CGTO - Contracted Gaussian Type Orbitals
CI - configuration interaction ( взаимодействие конфигураций)
CID - configuration interaction with all double substitutions (взаимодействие
конфигураций с двойной подстановкой)
CISD - configuration interaction with all single and double substitutions
(взаимодействие конфигураций с одиночной и двойной подстановкой)
CIDS и CI синонимы CISD
CNDO - complete neglect of the differencial overlap
COSMO - Conductor - like Screening Model
CPHF - Coupled-Perturbed Hartree-Fock Equation (связанные возмущенные
уравнения Хартри-Фока)
СР - Counterprise
CSF - Configurational State Function
DFT - Density Functional Theory (теория функционала плотности)
DSO - Diamagnetic Spin-Orbit
DODS - Different Orbitals for Different Spin
DMA - Distributed Multipole Analysis
DZ (TZ, QZ) - Double (Triple, Quadrupole) Zeta basic set
ECP- Effective Core Potential
EF - Eigenvector Following method
EC - Electronic Correlation
ESP - Electrostatic Potential
FMM - fast multipole method (быстрый мультипольный метод)
FORS - Full Optimazed Reaction Space
GAPT - Generalized Atomic Polar Tensor
GGA - Generalized gradient approximation (обобщенное градиентное
приближение)
209
GTO -Gaussian Type Orbitals
Gl, G2, G2(MP2), G2(MP2SVP), G2Q - Gaussian-1, Gaussian-2 models
GVB - Generalized Valence Bond method
GVB-PP - perfect-pairing Generalized Valence Bond method
HE - Half-electron method
HF - Hartree-Fock method
HL - Heitler-London Function
IGLO - Individual Gauge for Localized Orbitals
SCRF - self-consistent reaction field (самосогласованное реактивное поле)
МО - молекулярная орбиталь
SCF - самосогласованное поле
ЕСР - effective core potential (эффективный остовный потенциал)
MCSCF - Multiconfiguration SCF ( многоконфигурационное самосогласованное
поле)
NBO - Natural Bond Orbital ( натуральные (естественные) связевые орбитали )
OVGF - Outer Valence Green's Function
QCISD - Quadratic CI calculation, including single and double substitutions
PES - potential energy Surface (ППЭ - поверхность потенциальной энергии)
Юр - internal options ( внутренние опции)
IRC - intrinsic reaction coordinate ( существенная координата реакции)
PBC - Periodic Boundary Conditions ( периодические граничные условия)
SAC-CI - Symmetry Adapted Cluster/Configuration Interaction
ECD - Electronic circular dichroism (электронный круговой дихроизм)
QC - quadratic convergence (квадратичная сходимость)
EHT - extended Huckel theory (расширенный метод Хкжкеля)
VCD - vibrational circular dichroism (колебательный круговой дихроизм)
LDA - Local density appoximation (Приближение локальной плотности)
LSDA - Local spin density approximation (обобщение LDA для систем с неспа-
ренными спинами)
В88 - приближение Веске. 1988 год
LYP - C.Lee, W.Yang, R. G Parr
PW -J. P. Perdew , Y. Wang
ХК-П.Хоэнберг,В. Кон
КШ - В. Кон, Л. Шэм
НДП - Приближение нулевого дифференциального перекрывания
ППДП - полное пренебрежение дифференциальным перекрыванием
ПДДП - пренебрежение двухцентровым дифференциальным перекрыванием
ЧПДП - частичное пренебрежение дифференциальным перекрыванием
INDO - intermediate neglecting of the differencial overlap
МЧПДП - модифицированный метод частичного пренебрежения
дифференциальным перекрыванием
MINDO - modified intermediate neglecting of the differencial overlap
MNDO - Modified Neglecting of diatomic overlap
NDDO - Neglecting of Diatomic Differential Overlap
210
Литература
1. V. Fock. Naherungsmethoden zur Losung des Quantenmechanischen Mehrkor-
perproblems. Z. Phys. 1930. vol. 61. p. 126.
2. V. Fock. Naherungsmethoden zur Losung des Quantenmechanischen Mehrkor-
perproblems. Z. Phys. 1930. vol. 62. p. 795.
3. Ч. Коулсон. Валентность. М.: Мир. 1965. 428 с.
4. В.И.Минкин, Б.Я. Симкин, P.M. Миняев. Теория строения молекул. Ростов-
на-Дону, 1997. 558с.
5. С.Фудзинага. Метод молекулярных орбиталей. М.:, Наука. 1983. 464 с.
6. М.Дьюар. Теория молекулярных орбиталей в органической химии. М.:, Мир,
1972. 590с.
7. F. Hund. Zur Deutung einiger Erscheinungen in den Molekelspektren [On the
interpretation of some phenomena in molecular spectra]. Zeitschrift fur Physik,
1926. vol. 36, p. 657-674.
8. F. Hund. Zur Deutung der Molekelspektren. Zeitschrift fur Physik. Part I. 1927.
vol. 40, p. 742-764; Part II. 1927.vol. 42, p. 93-120; Part III. 1927. vol. 43, p. 805-
826; Part IV. 1928. vol. 51, p. 759-795 ; 1930. Part V, vol. 63, p. 719-751.
9. R. S. Mulliken. Electronic states. IV. Hund's tlieory; second positive nitrogen and
Swan bands; alternate intensities. Physical Review. 1927. vol. 29, p. 637 - 649.
10. R. S. Mulliken.The assignment of quantum numbers for electrons in molecules.
Physical Review. 1928. vol. 32. p 186 - 222.
11.R.S. Mulliken's Nobel Lecture. Science. 1967. 157. №. 3785. p. 13-24.
12. J. E. Lennard-Jones. The electronic structure of some diatomic molecules.
Transactions of the Faraday Society. 1929. vol. 25, p. 668-686.
13.Н.Ф. Степанов. Квантовая механика и квантовая химия. М.: Мир. 2001. 519
с.
14. Т. Koopmans. Uber die Zuordnung von Wellenfunktionen und Eigenwerten zu
den Einzelnen Elektronen Eines Atoms. Physica. 1934. vol.1, p. 104-113.
15.4. Киттель, Квантовая теория твердых тел. М.:Наука. 1967.492 с.
16. C.C.J. Roothaan. New Developments in Molecular Orbital Theory. 1951. Rev.
Mod. Phys. Vol. 23. p. 69-89.
17.Л.Д. Ландау, Е.М. Лифшиц. Квантовая механика.М.:, Наука, 1974. 752 с.
18. E.R. Davidson, D. Feller. Basis set selection for molecular calculations. Chem.
Rev. 1986. vol.86, p.681-696.
19. M.E. Соловьев, М.М. Соловьев. Компьютерная химия. М.: Солон-Пресс.
2005. 536 с.
20. Кларк. Компьютерная химия. М.: Мир, 1990. 383 с.
21.D. Young. Computational Chemistry: A Practical Guide for Applying Techniques
to Real World Problems. New York: John Wiley & Sons. 2001. pp. 322-359.
22. J.C. Slater. Atomic Shielding Constants. Phys. Rev. 1930. vol. 36, p. 57 .
23. S.F. Boys. Electronic wafe functions. I. A general method of calculation for the
stationary states of any molecular system. Rev. Mod. Phys. 1960. vol. 32, p.296 -
299.
24.1. N. Levine. Quantum Chemistry. Englewood Cliffs, New jersey: Prentice Hall.
1991. p. 461^66.
211
25. С. J. Cramer. Essentials of Computational Chemistry. Chichester: John Wiley &
Sons, Ltd.. 2002. p. 154-168.
26. F. Jensen. Introduction to Computational Chemistry. Chichester, England: John
Wiley and Sons. 1999. p. 150-176.
27. A. R. Leach. Molecular Modelling: Principles and Applications. Singapore:
Longman. 1996.pp. 68-77.
28. R. S. Mulliken. Electronic Population Analysis on LCAO-MO Molecular Wave
Functions. I. J. Chem. Phys. 1955. vol. 23. p. 1833-1840.
29. G. Csizmadia, Theory and Practice of MO Calculations on Organic Molecules,
Elsevier, Amsterdam, 1976. 378 p. .
30. F. M. Bickelhaupt, N. J. R. van Eikema Homines, C. Fonseca Guerra, E. J.
Baerends. The Carbon-Lithium Electron Pair Bond in (CFbLi),, (n = 1, 2, 4).
Organometallics 1996. 15. p. 2923-2931.
31. S.P.A. Sauer. The Ab Initio Calculations of Molecular Properties .Textbook for
the 3rd MERCOSUR Institute on Molecular Physics, Universidad National del
Nordeste, Corrientes. 2001.
32. O.Jeppe. Linear and nonlinear response functions for an exact state and for an
MCSCF . J. Chem. Phys. 1985. vol.82, p.3235 - 3264.
33. V.Kello. Quasi-relativistic coupled cluster calculations of electric dipole moments
and dipole polarizabilities of GeO, SnO, PbO . J. Сотр. Meth. Sci. Eng. 4. 2004.
special issues 3 and 4. p.753 - 764.
34. R.Krishnan, H.B.Schlegel, J.A.Pople. Derivative studies in configuration
interaction theory. J. Chem.Phys. 1980. vol.72, p.4654-4655.
35.M.Head-Gordon, R.J.Reco, M.Oumi, T.J. Lee. A doubles correction to electronic
excited state from configuration interaction in the space of single substituon.
Chem.Phys.Lett. 1994. vol.219, p.21-29.
36. С J. Cramer. Essentials of Computational Chemistry. Chichester: John Wiley &
Sons, Ltd.. 2002. p. 191-232.
37.C.M0ller, M.S. Plesset. Note on an Approximation Treatment for Many-Electron
Systems . Phys. Rev. 1934. vol.46. 618-622.
38. M.L. Leininger, W.D. Allen, H.F.Schaefer, CD. Sherrill. Is Moller-Plesset
perturbation theory a convergent ab initio method? J. Chem. Phys. 2000. vol. 112
(21). p. 9213-9222.
39. T. Helgaker, P. Jorgensen , J. Olsen. Molecular Electronic Structure Theory.
Wiley. 2000.
40. C. J. Cramer. Essentials of Computational Chemistry. Chichester: John Wiley &
Sons, Ltd.. 2002. p. 207-211.
41. J. B. Foresman, Л51ееп Frisch. Exploring Chemistry with Electronic Structure
Methods. Pittsburgh, PA: Gaussian Inc.. 1996, p. 267-271.
42. A. Szabo, S.O. Neil. Modern Quantum Chemistry. Mineola, New York: Dover
Publications, Inc. 1996. p. 350-353.
43. M.Head-Gordon, J.A.Pople, M.J. Frisch. MP2 energy evaluation by direct
methods. Chem.Phys.Lett. 1988. vol.153, p.503-506.
44. M.J. Frisch, M.Head-Gordon, J.A.Pople. A direct MP2 gradient method.
Chem.Phys.Lett. 1990. vol.166, p.275-280.
212
45. M.J. Frisch, M.Head-Gordon, J.A.Pople. Semi- direct algoritms for the MP2
energy and gradient. Chem.Phys.Lett. 1990. vol.166, p.281-289.
46. G.W. Trucks, E.A.Salter, C. Sosa, R.J.Bartlet. Theory and implementation of the
MBPT density matrix. An aplication to one-electron properties. Chem.Phys.Lett.
1988. vol.147, p.359-366.
47. G.W. Trucks, J.D. Watts, E.A.Salter, R.J.Bartlet. Analytical MBPT (4) gradients.
Chem.Phys.Lett. 1988. vol.147, p.490-495.
48.R.Krishnan, J.A.Pople. Approximate fourth-order perturbation theory of the
electron correlation energy. Int. J. Quant.Chem. 1978. vol.14, p.91-100.
49. P. Заградник, Р. Полак. Основы квантовой химии. М.: Мир. 1979. 504 с.
50. O.K. Давтян. Квантовая химия. М.: ВШ. 1962. 783с.
51. J. H.Van Vleck. Nonorthgonality and Ferromagnetism. Phys. Rev. 1936. vol.49.
p.232 - 240.
52. В.Кон. Нобелевская лекция. Электронная структура вещества - волновые
функции и функционалы плотности. УФН., 2002, т.172, №3, с.336 - 344.
53. W. Kohn. Nobel Lecture. Electronic structure of matter — wave functions and
density functionals. Reviews of Modern Physics. 1999. vol. 71. N 5, p. 1253-
1266.
54. R. Hochenberg, W. Kohn. Inhomogeneous Electron gas. Phys. Rev. 1964. vol.
136. B864-B871.
55. W. Kohn, L. J. Sham. Self-Consistent Equations Including Exchange and
Correlation Effects. Physical Review. 1965. vol. 140. N 4A. p. Al 133-A1138.
56. C-0 Almbladh, U. von Barth. Exact results for the charge and spin densities,
exchange-correlation potentials, and density functional eigenvalues. Phys.Rev. 1985.
B31. p. 3231 - 3247.; O.V. Gritsenko, E.J. Baerends. The analog of Koopmans'
theorem in spin-density functional theory. J. Chem. Phys. 2002. vol.117. p. 9154 -
9160.
57. P. A. M. Dirac. Note on exchange phenomena in the Thomas-Fermi atom. Proc.
Cambridge Phil. Roy. Soc. 1930. vol. 26 p. 376-385.
58.R. G Parr,W.Yang . Density-Functional Theory of Atoms and Molecules. Oxford:
Oxford University Press. 1994.
59.D. M. Ceperley , B. J. Alder . Ground State of the Electron Gas by a Stochastic
Method. Phys. Rev. Lett. 1980. vol.45, p.566-569.
60. S. H. Vosko, L. Wilk, M. Nusair . Accurate spin-dependent electron liquid
correlation energies for local spin density calculations: a critical analysis. 1980.
Can. J. Phys. vol. 58. p. 1200-1211.
61. J. P. Perdew, Y. Wang . Accurate and simple analytic representation of the
electron-gas correlation energy. Phys. Rev. 1992. В 45. p. 13244-13249.
62. J. P. Perdew, A. Zunger . Self-interaction correction to density-functional
approximations for many-electron systems. 1981. Phys. Rev. В 23. p. 5048-5079.
63. L. A. Cole, J. P. Perdew . Calculated electron affinities of the elements. Phys.
Rev. 1982. A 25. p. 1265- 1272.
64.LJ.von Barth, L. Hedin. A local exchange-correlation potential for the spin
polarized case. J. Phys. C: Solid State Phys. 1972. vol.5, p. 1629-1642.
213
65.J. P. Perdew, Y. Wang . Accurate and simple density functional for the electronic
exchange energy: Generalized gradient approximation Phys. Rev. 1986. В 33. p.
8800-8802.
66. A.D. Becke. Density-functional exchange - energy approximation with correct
asymptotic bihavior. Phys. Rev. 1988. В 38. p.3098-3100.
67. C.Lee, W.Yang, R. G Parr. Development of the Colle - Salvetti correlation-
energy formula into a functionalof the electron density. Phys. Rev. 1988. В 37.
p.785 - 789.
68. J. P. Perdew. Density-functional approximation for the correlation energy of the
inhomogeneous electron gas. Phys. Rev. 1986.B 33. p.8822 - 8824.
69.J. P. Perdew. Phys. Rev. Erratum: Density-functional approximation for the
correlation energy of the inhomogeneous electron gas. 1986. В 34. p.7406- 7406.
70. A.D. Becke. A new mixing of Hartree-Fock and local density-functional theories.
J. Chem. Phys. 1993. vol.98, p.1372-1377.
71. C. Fiolhais, F. Nogueira, M.Marques. A Primer in Density Functional Theory.
Springer. 2003. p. 60.
72.B.А.Губанов, А.Л. Ивановский, М.В. Рыжков. Квантовая химия в
материаловедении. М: Наука. 1987. 335с.
73.Губанов В.А., Жуков В.П., Литинский А.О. Полуэмпирические методы
молекулярных орбиталей в квантовой химии. М: Наука. 1976. 217 с.
74. C.J.Ballhausen, H.B.Gray. Molecular orbital theory. N.Y.:Benjamin. 1964 . 317p.
75. J. A. Pople. Electron Interaction in Unsaturated Hydrocarbons. Trans. Faraday
Soc. 1953. vol. 49. p. 1375- 1386.
76.1. Fisher-Hjalmors. Deduction of zero differential overlap Approximation from an
Orthogonal Atomic Orbital Basis. J. Chem. Phys.1965. vol.42, p.1962-1973.
77. J. A. Pople , D. L. Beveridge, Approximate Molecular Orbital Theory, N. Y.
Series in Advanced Chemistry. 1970. 236 p.
78. J. A. Pople , A. Segal. Approximate Self-consistent Molecular Orbital Theory II.
Calculations with Complete Neglect of Differential Overlap. J. Chem. Phys.1965.
vol.43, 5136-5151.
79. J. A. Pople, D. Santry, G. A. Segal. Approximate Self-consistent Molecular
Orbital Theory I. Invariant Procedures. J. Chem. Phys. 1965, vol. 43, 5129 - 5135.
80.J. A. Pople, D. Beveridge, P. Dobosh. Approximate Self-consistent Molecular
Orbital Theory V. Intermediate Neglect of Differential Overlap. J. Chem. Phys.
1967. vol.47. 2026-2034.
81. R.N. Dixon. Approximate self-consistent field molecular orbital calculations for
valence shell electronic states. Mol. Phys. 1967. vol.12, p.83-90.
82. M.J.S. Devvar, R.C.Haddon. MINDO [modified intermediate neglect of
differential overlap]/3 study of the multiplicity of cyclopentadienate cations. J.Amer.
Chem. Soc. 1974. vol.96, p.253 -256.
83. Полуэмпирические методы расчета электронной структуры / Под ред. Дж.
Сигала. М.: Мир.1980. Т.1. 327с.
84.R. С. Bingham. M. J. S. Dewar, D. H. Lo. Ground States of Molecules. XXV.
MINDO/3. An Improved Version of the MINDO Semiempirical SCF-MO
Method. J. Am. Chem. Soc. 1975. vol. 97. p. 1285-1293.
214
85.М. J. S. Dewar , G. P. Ford. Ground States of Molecules. 44 MINDO/3
Calculations of Absolute Heat Capacities and Entropies of Molecules without Internal
Rotations. J. Am. Chem. Soc. 1977. vol.99. 7822-7829.
86. M. J. S. Dewar, W. Thiel. Ground States of Molecules, 38. The MNDO Method.
Approximations and Parameters. J. Am. Chem. Soc. 1977. vol. 99. 4899-4907.
87. M. J. S. Dewar, E. G. Zoebisch, E. F. Healy, J. J. P. Stewart. AMI: A New
General Purpose Quantum Mechanical Model. J. Am. Chem. Soc. 1985. vol.107, p.
3902-3909.
88. J. J. P. Stewart. Optimization of Parameters for Semi-Empirical Methods 1-
Method. J. Сотр. Chem. 1989. vol.10, p.209-220.
89. J. J. P. Stewart. Optimization of Parameters for Semiempirical Methods II.
Applications. J. Сотр. Chem. 1989. vol. 10. p.221-264.
90. Stewart, J. J. P. Optimization of parameters for semiempirical methods. HI
Extension of PM3 to Be, Mg, Zn, Ga, Ge, As, Se, Cd, In, Sn, Sb, Те, Hg, Tl, Pb,
and Bi. J. Comput. Chem. 1991. vol. 12. p. 320-341.
91.Stewart, J. J. P. Optimization of parameters for semiempirical methods IV:
extension of MNDO, AMI, and PM3 to more main group elements. Journal of
Molecular Modeling 2004. vol. 10. p. 155-164.
92. http://physics-of-molecules.odessit.org/library/books/romanova/ Program_ Hy-
perChem/chapter_06_2.htm
93.И.Н. Годнев. Вычисление термодинамических функций по молекулярным
данным. М.: ГИТТЛ. 1956. 419с.
94.Ландау, Л. Д., Лифшиц, Е. М. Статистическая физика. Часть 1. Издание 5-е.
М.: Физматлит. 2005. 616 с.
95. М.А. Ельяшевич, Атомная и молекулярная спектроскопия. М.: ФМ. 1962.
892 с.
96.Г.Н. Чуев, Базилевский М.В. Молекулярные модели сольватации в полярных
жидкостях. Успехи химии. 2003. т. 72 № 9. с.827-851.
97.Б.Я.Симкин, И.И.Шейхет. ЬСвантовохимическая и статистическая теория
растворов. Вычислительные методы и их применение. М.: Химия. 1989. 256с.
98. J. В. Foresman, Т. A. Keith, К. В. Wiberg, J. Snoonian, M.J. Frisch. Solvent
Effects. 5. Influence of Cavity Shape, Truncation of Electrostatics, and Electron.
Correlation on ab Initio Reaction Field Calculations. J. Phys. Chem. 1996. 100. p.
16098-16104.
99. Б.И. Ионин, Б.А. Ершов, А.И.Кольцов. ЯМР спектроскопия в органической
химии. Л.: Химия. 1983. 267с.
100. И.В. Александров. Теория ядерного магнитного резонанса. М.: Наука.
1964. 208 с.
101. А. Абрагам. Ядерный магнетизм. М.: ИЛ. 1963. 551с.
102. Гюнтер X. Введение в курс спектроскопии ЯМР: Пер. с англ. М.: Мир.
1984. 478 с.
103. N.F. Ramsey. Chemical effects in nuclear magnetic resonance and in diamag-
netic susceptibility . Phys. Rev. 1952. v 86. p.243 - 248.
104. 4. Сликтер. Основы теории магнитного резонанса. М.: Мир. 1981.448с.
215
105. N.F. Ramsey. Magnetic shielding in molecules . Phys. Rev. 1950. vol. 78.
p.699-703.
106. У.Буркерт, Н. Л. Эллинджер. Молекулярная механика, пер. с англ., М. :
ИЛ. 1986.364 с.
107. М. J.Frisch, G. W.Trucks, H. B.Schlegel, P. M. W.Gill, В. G.Johnson, M.
A.Robb, J. R.Cheeseman, Т. A.Keith, G. A.Petersson, J. A.Montgomery,
K.Raghavachari, M. A.Al-Laham, V. G.Zakrzewski, J. V.Ortiz, J. B.Foresman,
J.Cioslowski, B. B.Stefanov, A.Nanayakkara, M.Challacombe, C. Y. Peng, P. Y.
Ayala, W.Chen, M. W. Wong, J. L. Andres, E. S. Replogle, R. Gomperts, R. L.
Martin, D. J. Fox, J. S. Binkley, D. J. Defrees, J. Baker, J. J. P. Stewart, M. Head-
Gordon, C. Gonzalez, J. A. Pople. Gaussian 03, Revision C.2, Gaussian Inc.:
Pittsburgh PA. 2003
108. Зеленин К.Н., Ноздрачев А.Д., Поляков Е.Л. Нобелевские премии по
химии за 100 лет. СПб, "Гуманистика". 2003. 873 с.
109. J.A. Pople. Nobel lecture: quantum chemical models. Rev. Mod. Phys. 1999.
vol. 71. p.1267- 1274; Дж. Попл. Квантово-химические модели. УФН. 2002.
Т. 172. №3. С. 349-356.
110. А. Р. Scott, L. Radom. Harmonic Vibrational Frequencies: An Evaluation of
Hartree-Fock, Meller-Plesset, Quadratic Configuration Interaction, Density
Functional Theory, and Semiempirical Scale Factors. J. Phys. Chem. 1996. vol. 100. p.
16502-16513.
216
Содержание
Предисловие 3
Глава I. Теоретические основы вычислительных методов
квантовой химии 4
. 1 Разделение электронного и ядерного движений в молекулах 4
.2. Метод молекулярных орбиталей. Метод Хартри-Фока 9
.3 Метод Хартри-Фока-Рутана 27
.4 Наборы базисных функций 32
.5. Электронная плотность, порядки связей, заряды на атомах,
поляризуемость 36
.6 Электронная корреляция и неограниченный метод ХФ(ХФР) 40
.7 Метод взаимодействия конфигураций 42
.8 Теория возмущений Меллера-Плессета (MP) 43
.9 Молекула водорода 43
. 10 Теория функционала плотности 48
.11 Полуэмпирические методы квантовой химии 59
.12 Поверхность потенциальной энергии многоатомной молекулы 70
.13. Классификация стационарных точек на поверхности
потенциальной энергии. Путь химической реакции 71
. 14 Энергия нулевых колебаний (ZPE) 74
. 15 Сумма по состояниям системы 74
.16. Термодинамические функции состояния 76
.17 Вычисление суммы по состояниям для поступательного
движения 79
18 Формулы, выражающие термодинамические функции
через сумму по состояниям 80
.19 Сумма по состояниям внутренних движений молекулы 82
.20 Модели сольватации 84
.21 Ядерный магнитный резонанс. Метод GIAO 88
.22 Методы молекулярной механики 95
.23 Метод молекулярной динамики 98
Глава П. Основы работы с программой Gaussian под WINDOUS
2.1 Возможности программы Gaussian и ее структура 100
2.2. Запуск программы Gaussian для операционной
системы WINDOWS 103
2.3 Задание исходной конфигурации молекулы 108
2.4. Расчет энергии молекулы для фиксированной конфигурации 112
2.5 Управление процессом самосогласования 115
2.7. Построение базисных функций и молекулярных орбиталей 121
2.8. Расчет частот нормальных колебаний и термодинамических
свойств молекул 126
2.9 Сканирование поверхности потенциальной энергии 131
2.10. Поиск переходных состояний и расчет пути
химической реакции 132
217
2.11 Расчет спектров ЯМР 137
2.12. Расчет эффектов сольватации 139
2.13 Расчет возбужденных состояний 140
2.14 Ключевые слова программы 141
2.15 Примеры входных файлов программы Gaussian
Глава III. Основы работы с программой Gaussview 171
3.1. Старт программы Gaussview 171
3.2. Кнопки элементов и молекулярных фрагментов
на контрольной панели 172
3.3. Использование мыши. Просмотр и регулировка параметров
структуры 174
3.4 Расположение активных окон на рабочем столе 177
3.5 Стандартные кнопки ввода - вывода. Редактор списка атомов 177
3.6. Регулировка качества структуры молекулы в активном окне 178
3.7 Запуск программы Gaussian и просмотр
результатов с помощью программы Gaussview 182
3.8 ONIOM расчет молекулярных структур 188
3.9 Анализ результатов сканирования поверхности потенциальной
энергии с помощью программы Gaussview 190
3.10 Просмотр орбитальных энергий в программе Gaussview 192
3.11 Изображение молекулярных орбиталей
в программе Gaussview 193
3.12 Изображение ИК спектров молекул с помощью
программы Gaussview 201
3.13 Изображение спектров ЯМР в программе Gaussview 204
Аббревиатура методов компьютерной химии 209
Литература 211
Содержание 217
218
Серия «Библиотека студента»
Бутырская Е. В.
Компьютерная химия: основы теории
и работа с программами Gaussian и GaussView
Обложка
СОЛОН-ПРЕСС
ООО «СОЛОН-ПРЕСС»
123242, Москва, а/я 20
Телефоны:
(499) 254-44-10. 795-73-26, 252-25-21
E-mail: avtor@coba.ru
Формат 70х 100/16. Объем 14 п. л. Тираж 500