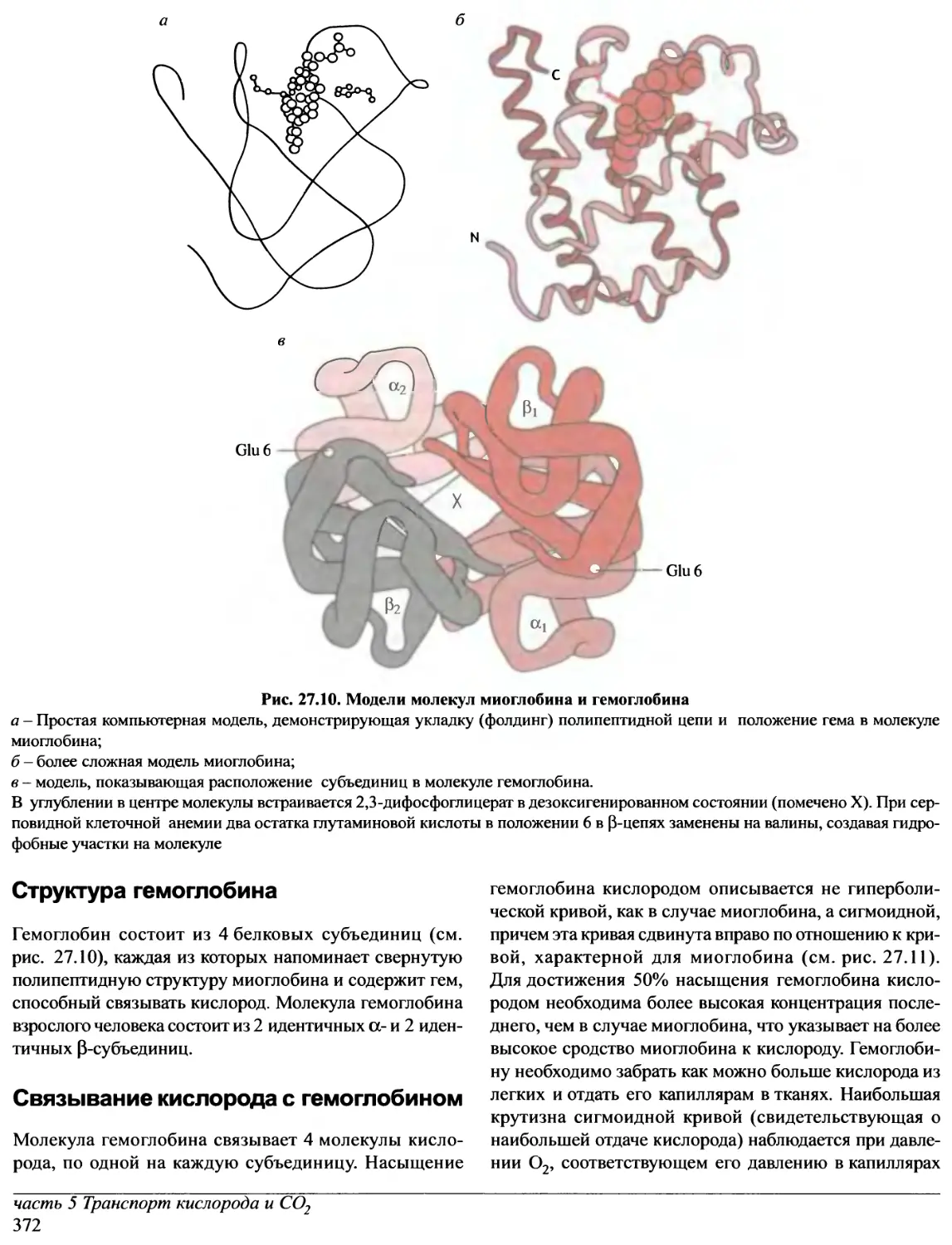
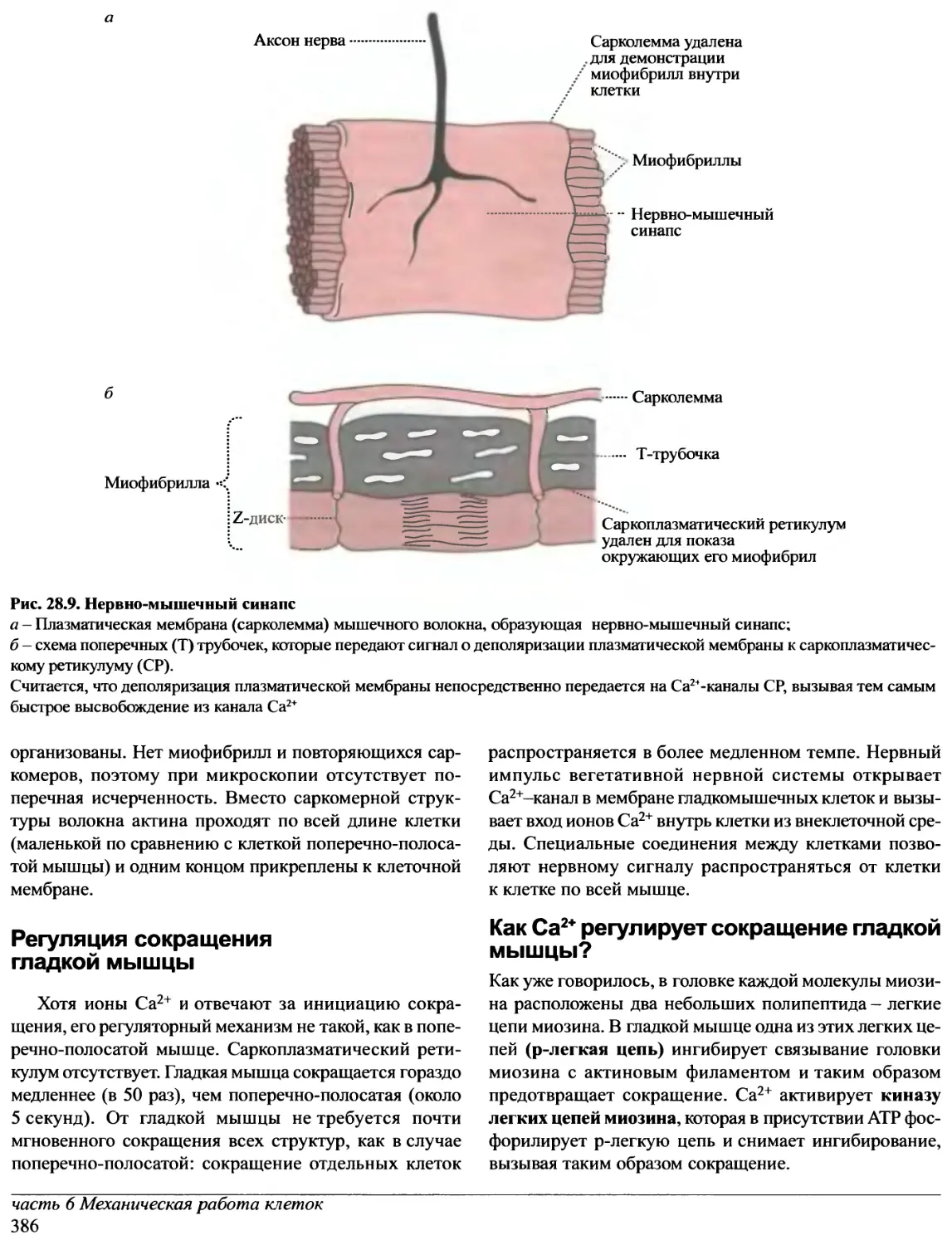
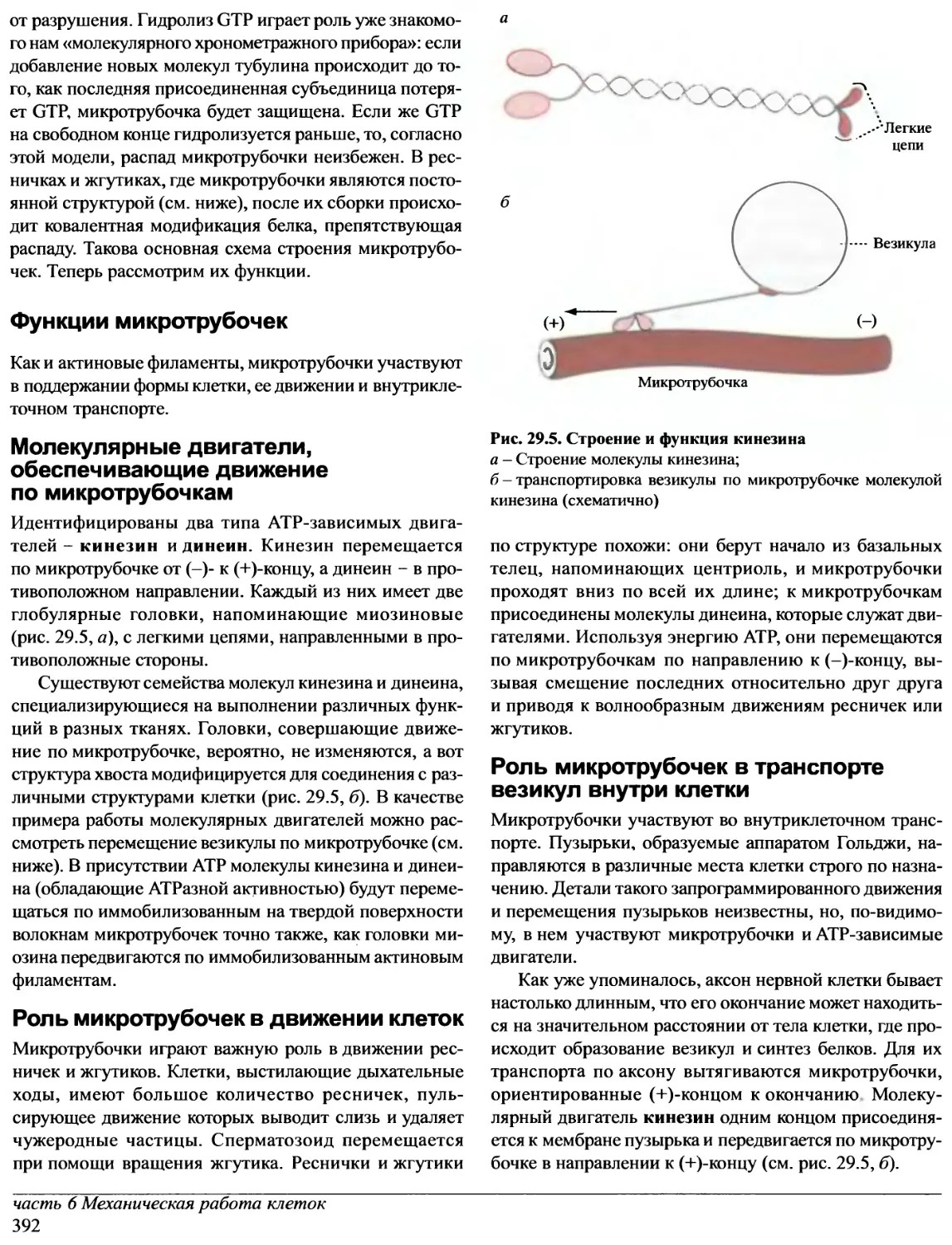
/






















































































































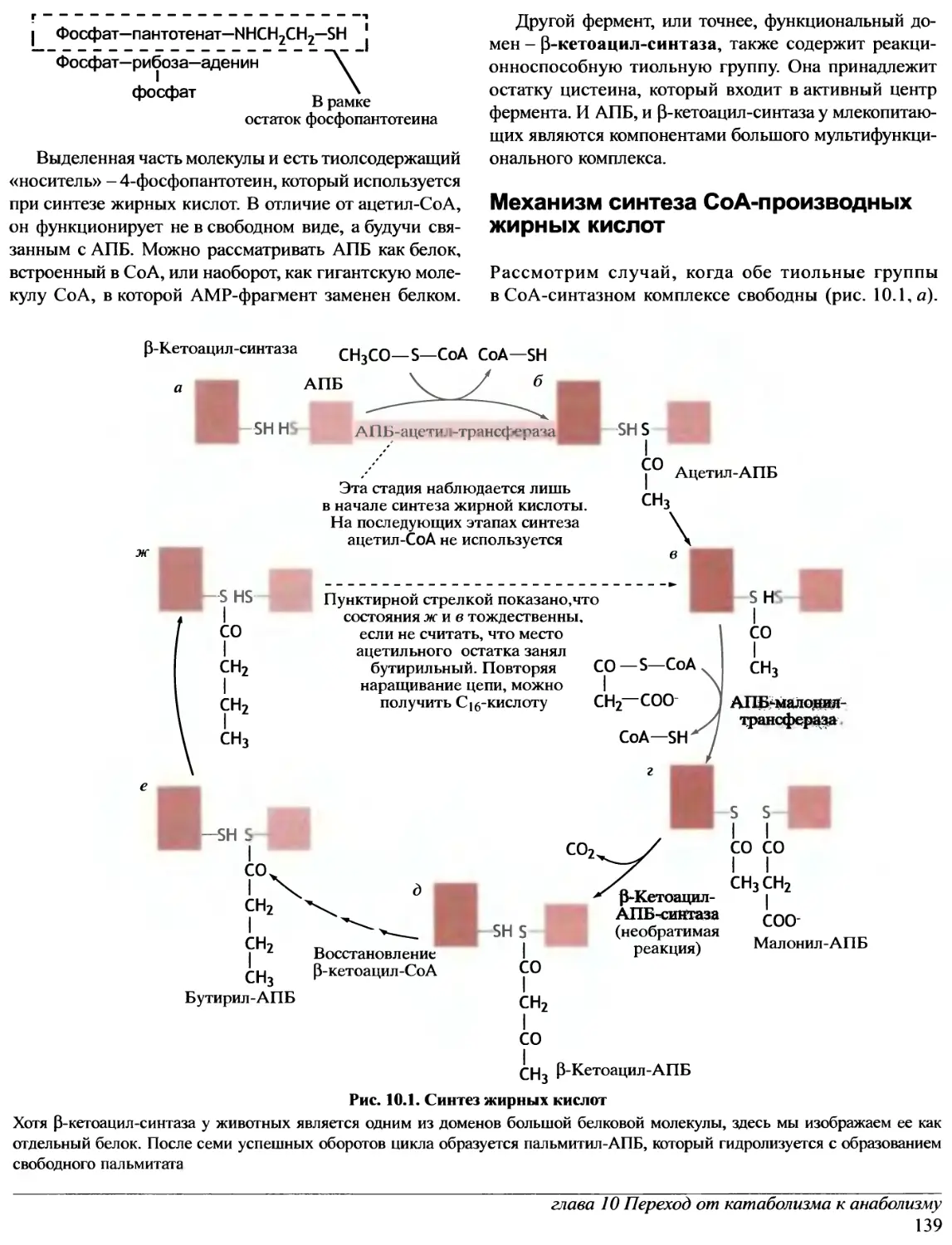


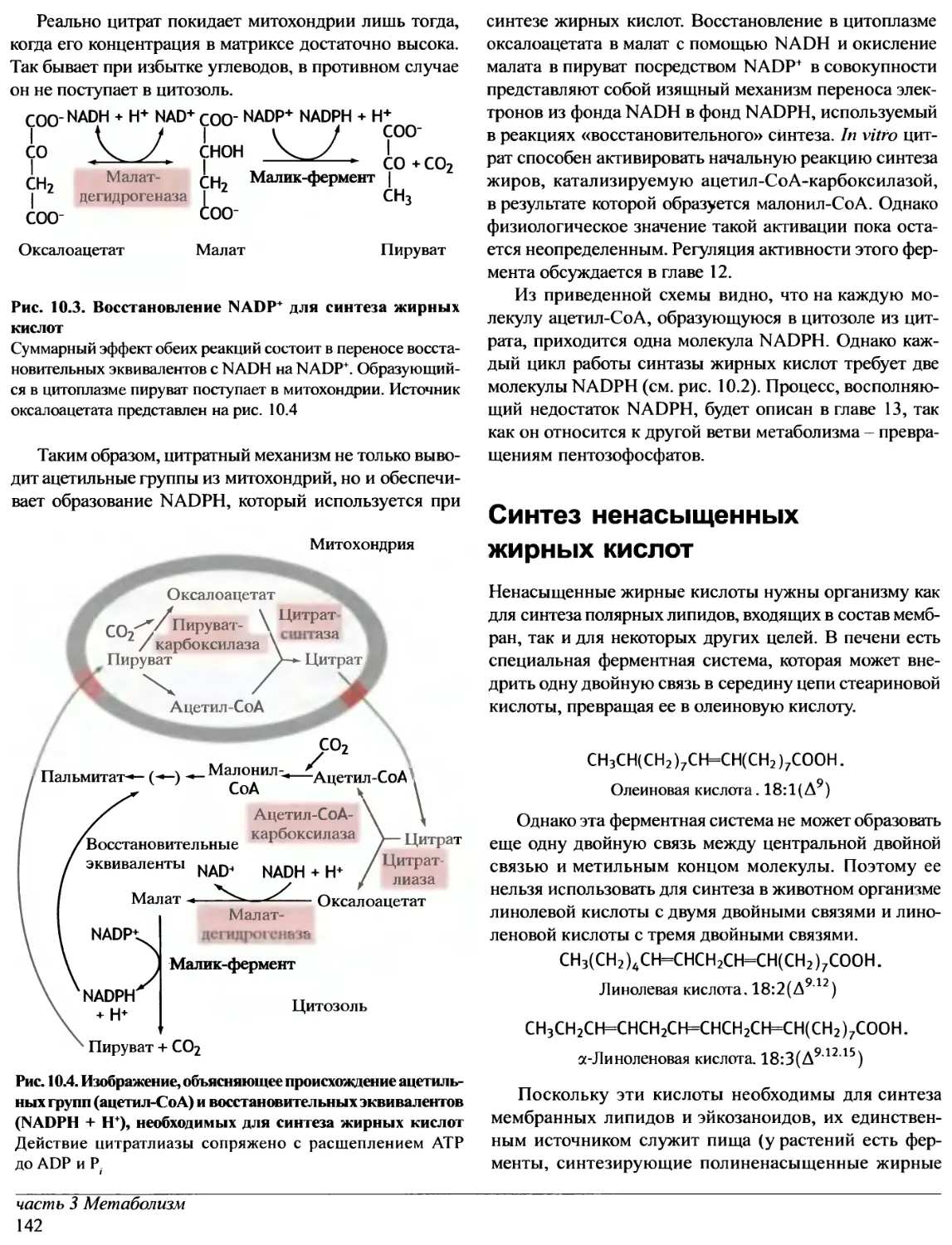


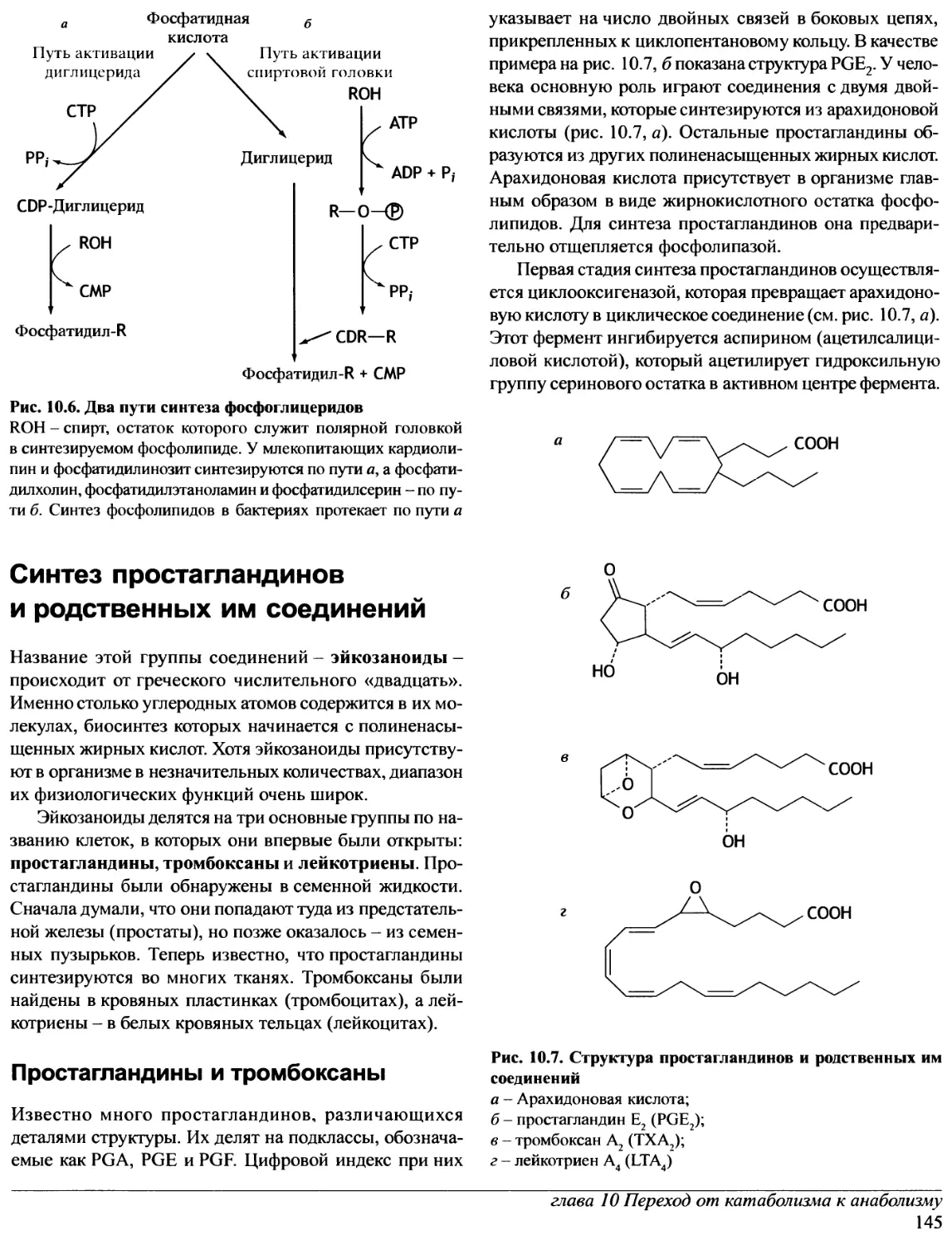





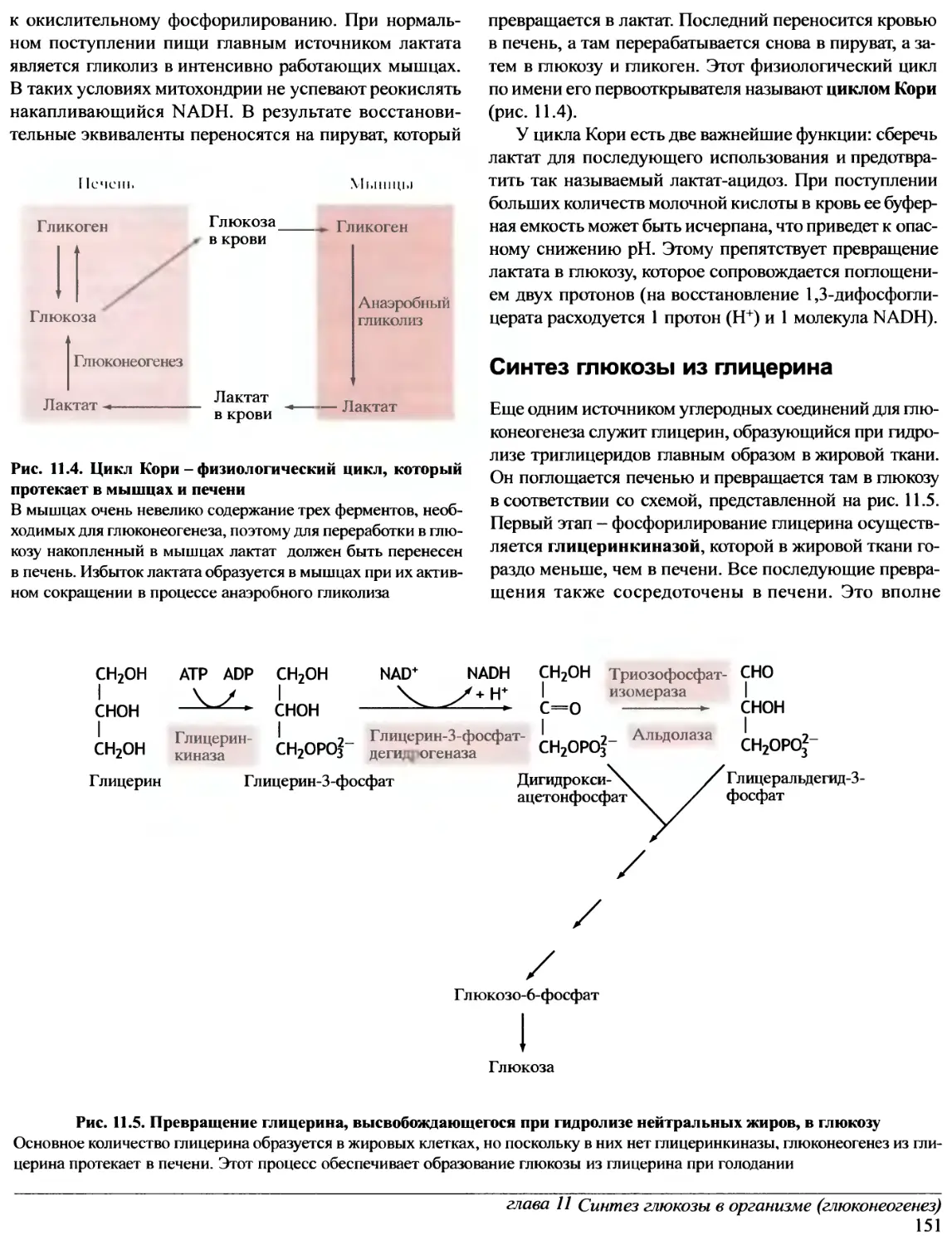
































































































































































































































































Author: Эллиот В. Эллиот Д.
Tags: материальные основы жизни биохимия молекулярная биология биофизика общая биофизика, общая биохимия и общая физиология
ISBN: 5-7846-0036-2
Year: 2002




Text
В. ЭЛЛИОТ
Д. ЭЛЛИОТ
БИОХИМИЯ И
МОЛЕКУЛЯРНАЯ
БИОЛОГИЯ
Рекомендовано Министерством образования Российской Федерации
для использования в учебном процессе студентами высших учебных
заведений, обучающихся по биологическим специальностям
Допущено Департаментом образовательных медицинских учреждений
и кадровой политики Министерства здравоохранения РФ в качестве
учебного пособия для студентов медицинских и фармацевтических
специальностей медицинских вузов, а также для интернов,
ординаторов и врачей системы последипломного образования
Москва
МАИК «Наука/Интерпериодика»
2002
Перевод с английского:
О.В. Добрыниной, И.С. Севериной, Е.Д. Скоцеляс, А.Е. Медведева,
A.M. Шкроба
Под редакцией:
А.И. Арчакова, М.П. Кирпичникова, А.Е. Медведева, В.П. Скулачева
Эллиот В.
Биохимия и молекулярная биология / В. Эллиот, Д. Эллиот; Под ред.
А.И. Арчакова, М.П. Кирпичникова, А.Е. Медведева, В.П. Скулачева;
Пер. с англ. О.В. Добрыниной, И.С. Севериной, Е.Д. Скоцеляс и др. —
М.: МАИК «Наука/Интерпериодика», 2002. —446 с: ил.
ISBN 5-7846-0036-2
Представлены новейшие данные по биохимии и молекулярной биологии клетки.
Учебный материал сгруппирован в пять основных разделов: структура белков и
мембран, метаболизм, хранение и переработка информации, транспорт кислорода и
углекислого газа, механическая работа в клетке. Отличительной особенностью этого издания
является его смысловой акцент на биологическом значении биохимических процессов,
которые рассматриваются в тесной взаимосвязи с физиологическими функциями.
Предназначена для студентов биологических и медицинских специальностей
высших учебных заведений, а также читателей, интересующихся молекулярными основами
процессов жизнедеятельности.
ISBN 5-7846-0036-2 О W.H. Elliot and D.C. Elliot, 1997
О НИИ биомедицинской химии РАМН,
(перевод на русский язык и издание),
1999
О МАИК «Наука/Интерпериодика»,
(издание, оформление), 2002
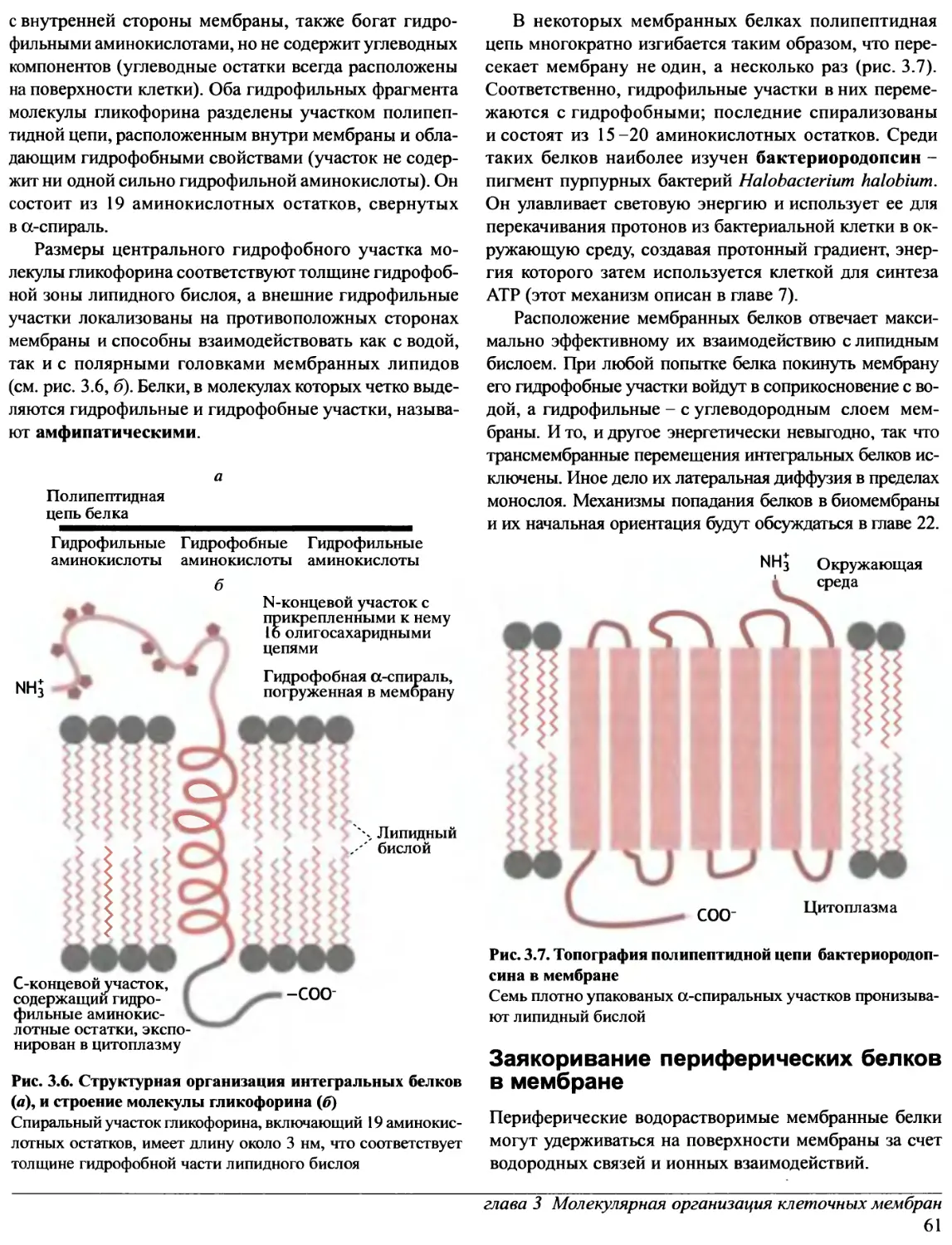
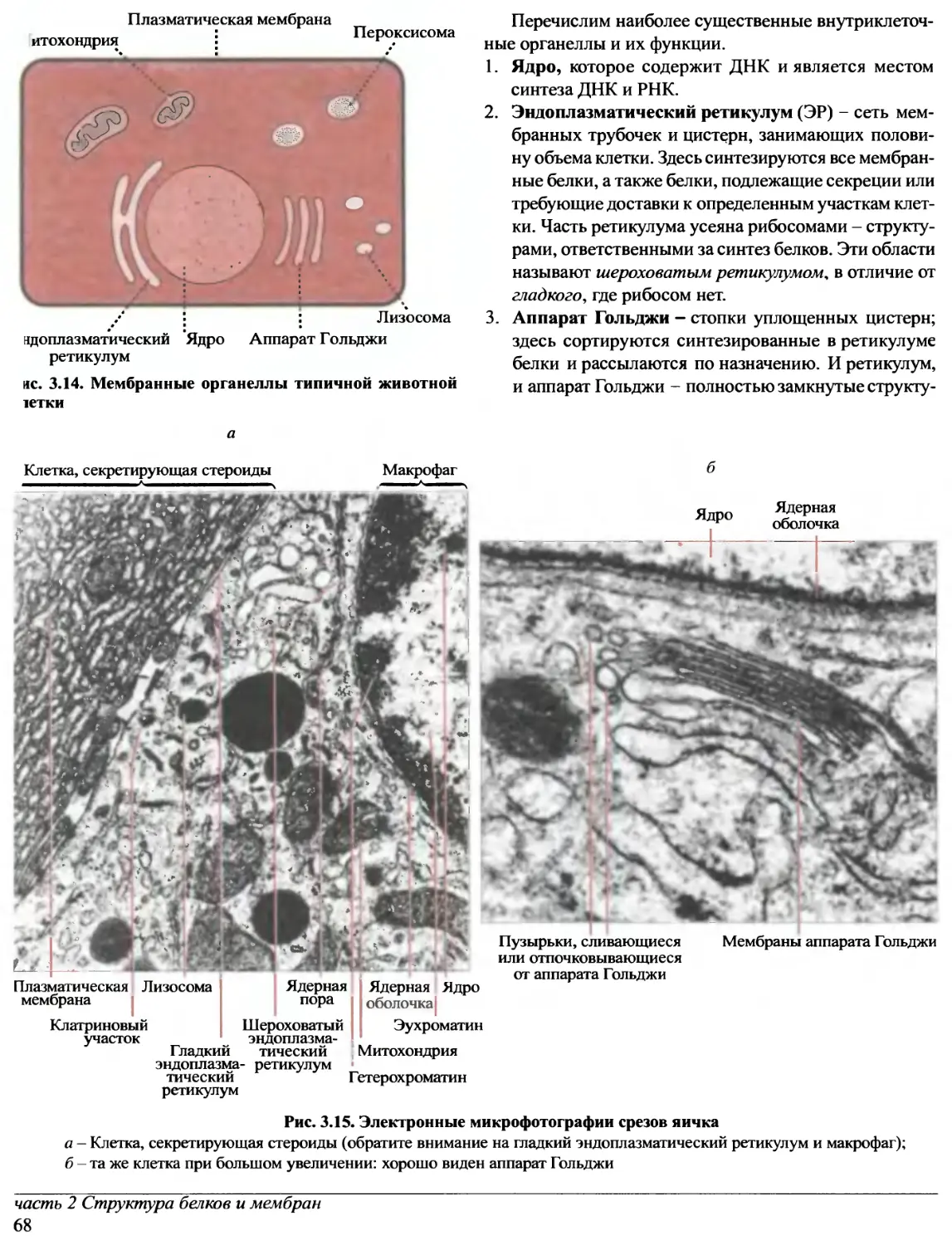
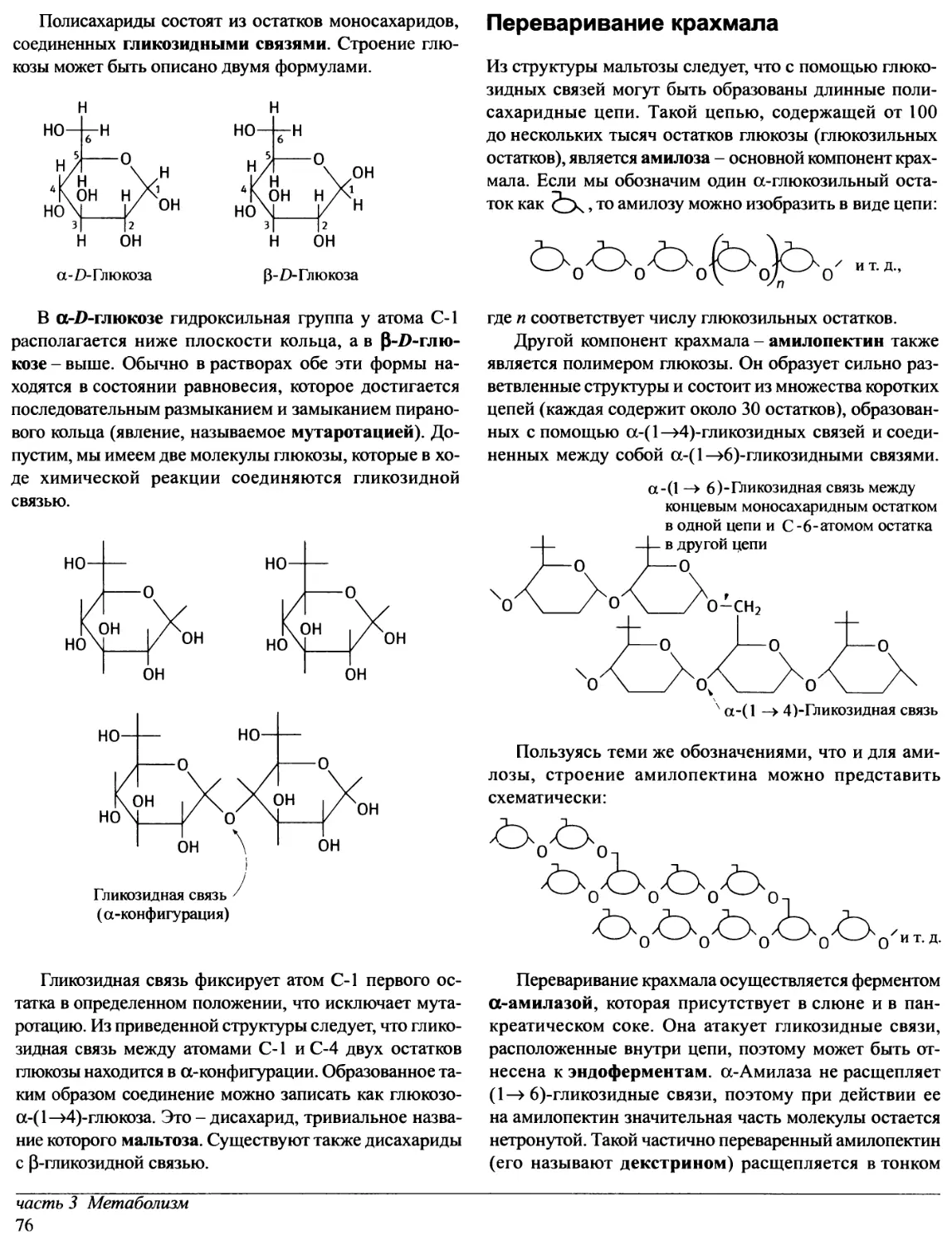
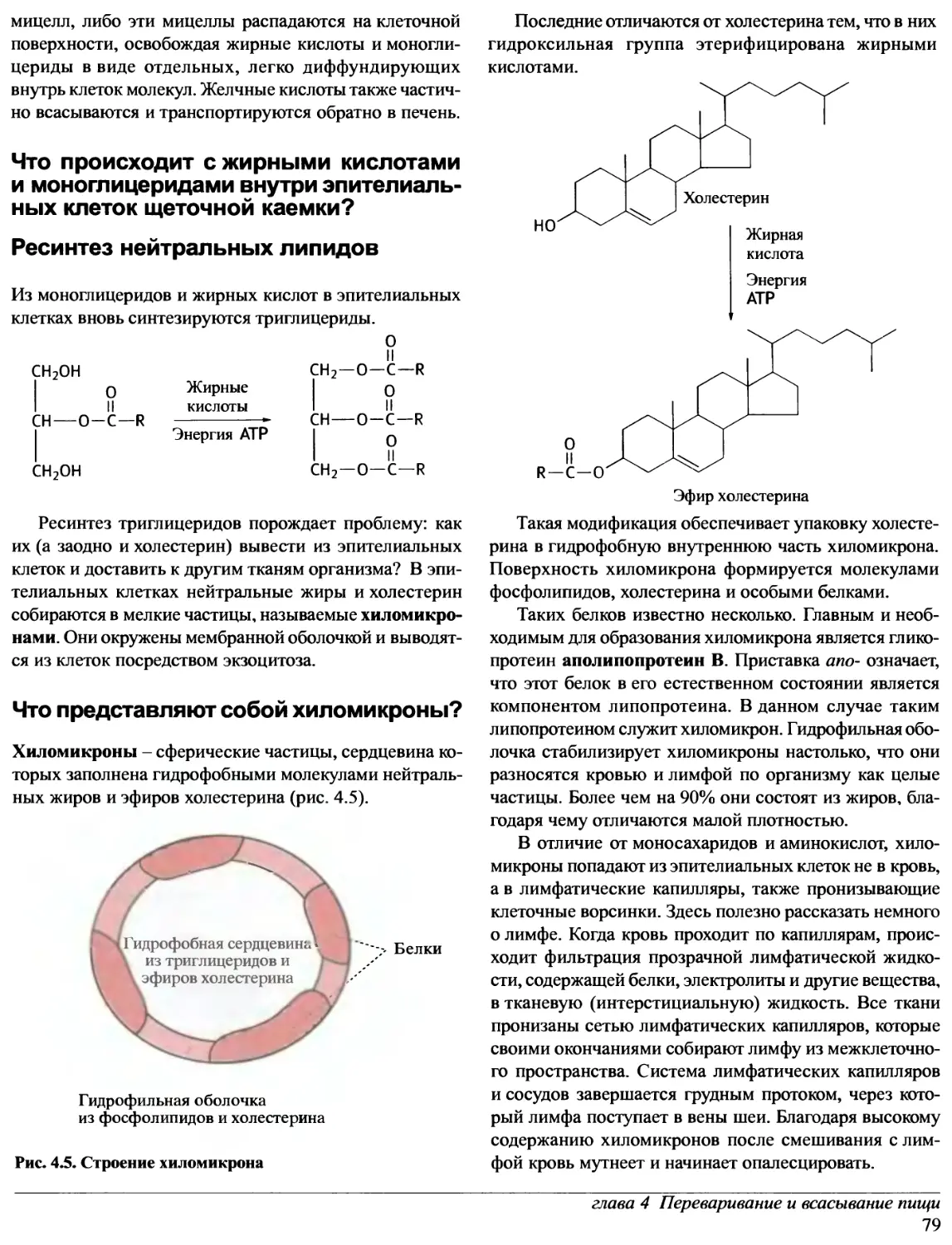
Предисловие редакторов перевода
Прошло уже около десяти лет с тех пор, как в нашей
стране были изданы переводы лучших в мировой практике
учебников по биохимии. За истекшее время в биохимии
и - особенно! - в молекулярной биологии очень многое
значительно изменилось. Сейчас в России потребность
в новых, современных учебных руководствах ощущается
так остро, как никогда. А между тем издательство «Oxford
University Press» в конце 1997 - начале 1998 гг. выпустило
ряд примечательных книг, предназначенных для
студентов, аспирантов и научных сотрудников,
специализирующихся в этих областях науки.
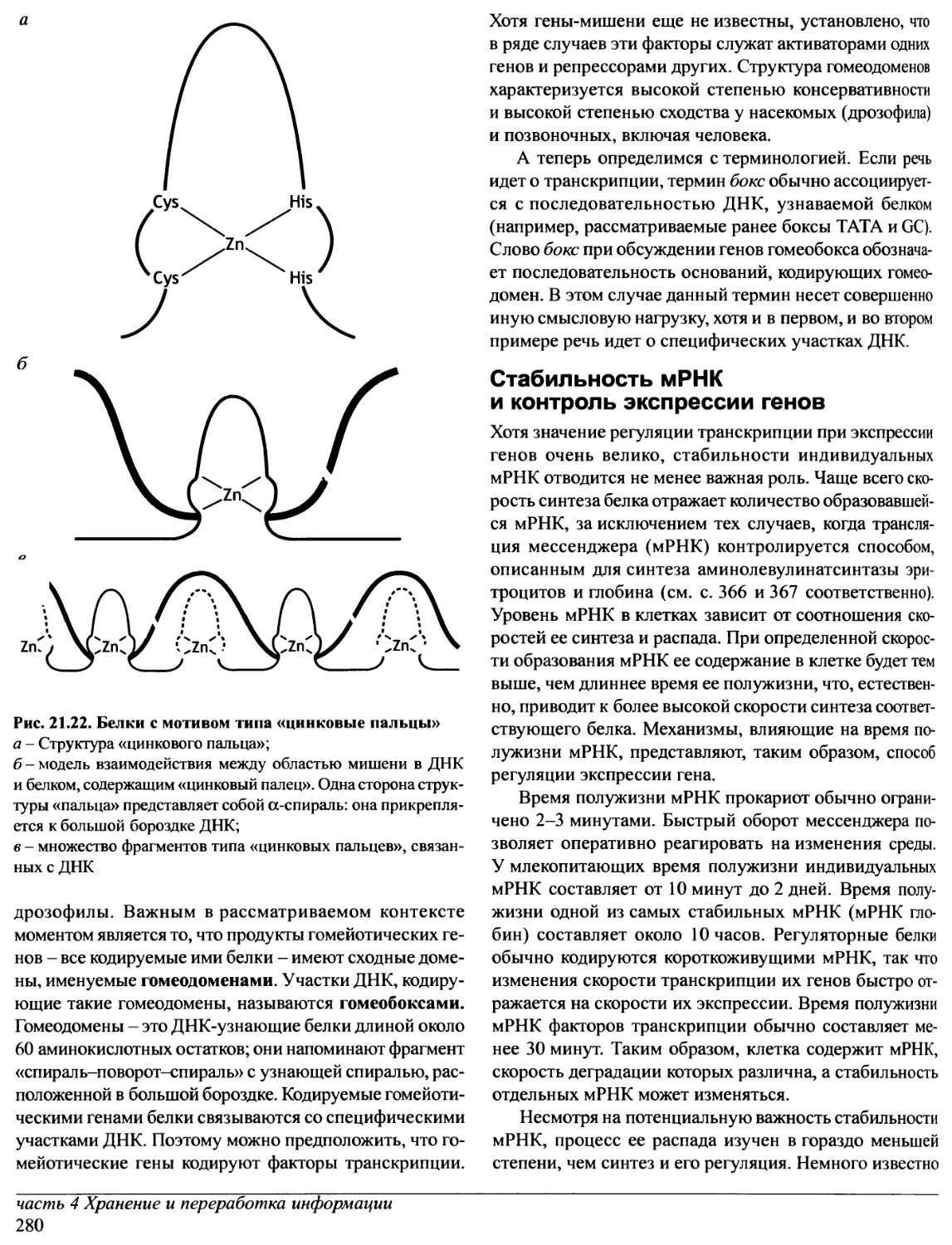
Переговоры с «Oxford University Press» о передаче
авторских прав на издание трехтомного руководства
по биохимии на русском языке, к нашему
удовлетворению, увенчались успехом. Редакторами перевода были
отобраны семь отличных книг, которые и легли в основу
этого русскоязычного издания.
В первом томе, который вы сейчас держите в руках,
представлен один из самых популярных в англоязычном
мире учебник Вильяма и Дафны Эллиот «Биохимия и
молекулярная биология». По сравнению с многими
другими учебниками его важной особенностью и главным
достоинством является, на наш взгляд, смысловой акцент
на биологическом значении биохимических процессов
в организме. Идея о неразрывной взаимосвязи
биохимических реакций с физиологическими функциями, о
развитии патологических процессов при аномальном
протекании этих реакций проходит через всю книгу красной
нитью. Пожалуй, больше ни в одном руководстве по
общей биохимии и молекулярной биологии не уделяется
столь пристального внимания проблемам возникновения
заболеваний на конкретной химической основе! Это,
конечно, особенно интересно и важно для студентов
медицинского и медико-биологического профиля.
Учебник Эллиотов дополнен сборником задач
и упражнений, который составлен Джоном Джеффер-
соном в качестве пособия для самостоятельной работы
студентов. Издание такого задачника в электронной
версии позволит вести занятия по биохимии в
компьютерных классах, что несомненно будет стимулировать
дальнейшее внедрение технических средств в преподавание
биохимии и молекулярной биологии.
В состав второго тома - «Биоинформатика» - входят
два учебных пособия: «Предсказание структуры белка»
(ред. М. Стернберг) и «Анализ последовательности ДНК
и белка» (ред. М. Бишоп, К. Роулингс). По сути,
впервые на русском языке представлено солидное
руководство по биоинформатике, которая ныне является
одним из наиболее динамично развивающихся
разделов современной биохимии и молекулярной
биологии. Ее методы позволяют на основе генетической
информации о первичной структуре макромолекул
выяснять их пространственные структуры и функции.
Полученные таким образом данные можно в
дальнейшем использовать (и они уже используются!) для
создания нового поколения лекарственных веществ,
точечными мишенями которых служат белки
(ферменты, рецепторы и др.) или кодирующие их гены.
Данное руководство будет полезно не только
студентам и аспирантам, специализирующимся в области
биохимии, молекулярной биологии и информатики,
но и всем специалистам
медико-фармакологического профиля (фармакологам, фармацевтам,
медицинским химикам и др.).
Второй том дополнен специальным разделом об
использовании в биоинформатике ресурсов Интернета,
который был написан специалистами Института
биомедицинской химии РАМН профессором В. В. Поройковым
и его сотрудниками.
В состав третьего тома - «Современные методы
исследования структуры и функции белка» - включены два
учебных пособия: «Структура белка» и «Функция
белка» (ред. Е. Крейгтон). Что особо примечательно, здесь
современные методы анализа структуры и функции
расписаны в виде протоколов (как это принято в настоящее
время в мировой литературе), которые легко
воспроизвести (разумеется, при наличии соответствующего
оборудования и реактивов). Пожалуй, это руководство также
впервые представляет на русском языке протоколы
широко используемых современных методов биохимии
и молекулярной биологии.
Несмотря на все сложности нашей сегодняшней
жизни, к идее создания такого учебного комплекса
с большим пониманием отнеслись и в Министерстве
здравоохранения, и в Министерстве науки РФ.
Итак, перед вами учебник «Биохимия и молекулярная
биология» Вильяма и Дафны Эллиот в переводе на
русский язык. Авторы поставили перед собой нелегкую
задачу: не только изложить в сжатой и понятной студентам
форме основы биохимии с элементами молекулярной
5
биологии (т. е. общий курс биохимии, как принято
говорить у нас), но еще и донести до читателя информацию
о новейших достижениях в этой области.
Студенты наверняка по достоинству оценят указания
и рекомендации авторов: с каким материалом следует
просто познакомиться, а какой - многократно и
вдумчиво проработать.
Биохимия и в особенности молекулярная биология
принадлежат к тем отраслям естественных наук, где
накопление новых научных данных, заставляющих подчас
пересмотреть сложившиеся за долгие годы
представления, происходит особенно быстро. Поэтому
оперативное издание новых учебных пособий по этим
дисциплинам имеет крайне важное значение. Подготовка
к печати этого учебника на русском языке осуществлена
в беспрецедентно сжатые сроки - всего за полтора года
с момента выхода англоязычной книги.
Следует специально подчеркнуть такие достоинства
учебника, как удачное расположение материала (в
порядке усложнения биологической функции) и подробное
описание тесной взаимосвязи обменов углеводов, жиров
и белков. При этом авторы ограничились минимальной
информацией по биологическим макромолекулам,
полагая, по-видимому, что предшествующий курс
биоорганической химии у студентов даром не пропал.
Некоторая конспективность отдельных разделов
компенсируется широтой охвата рассматриваемых вопросов:
от традиционных для курса биохимии разделов - до
проблем фолдинга белка и методов генной инженерии.
Мы уверены, что эта книга окажется весьма
полезной для студентов и биологических, и медицинских
вузов. К примеру, такие вопросы, как роль прионов в
развитии нейродегенеративных заболеваний или же механизмы
действия антилейкемических препаратов, имеют важное
значение не только для медиков, а полимеразная цепная
реакция (и ее диагностические возможности!)
заинтересует не только биологов.
Мы считаем также, что эта книга послужит ценным
учебным и справочным пособием для аспирантов,
преподавателей и научных сотрудников самых различных
биологических и медицинских специальностей.
А. И. Лрчаков
М. П. Кирпичников
A. Е. Медведев
B. П. Скулачев
6
Предисловие к английскому изданию
Эта книга предназначается для студентов, изучающих
биологию и медицину, которые еще только начинают
осваивать серьезный курс биохимии и молекулярной
биологии.
Приступая к изучению молекулярных основ жизни,
мы сталкиваемся с почти бесконечной
последовательностью химических превращений и механизмов. Все это,
на первый взгляд, выглядит настолько сложным и
запутанным, что может попросту испугать многих учащихся,
особенно если учебный курс для «всеобъемлющего»
изложения материала перегружен такими
подробностями, которые знают и помнят лишь узкие специалисты.
Мы считаем, что подобные детали не должны
заслонять восприятия основных принципов организации
молекулярных процессов в клетке, обусловливающих
существование жизни.
Назначение этой книги - стать «другом студента»: она
поможет вам выделить то главное, что вы должны
понять, что может пригодиться вам в самостоятельных
занятиях. Здесь вы найдете указания, с каким материалом
в принципе достаточно просто познакомиться,
специально его не запоминая (однако решающее слово в этом
вопросе принадлежит, конечно, вашим педагогам).
Книга построена таком образом, чтобы у студентов не
возникал постоянный и вполне понятный вопрос: «Что мы
должны выучить?», столь хорошо знакомый всем
преподавателям.
Расположив учебный материал в соответствии с
классификацией тем по биологическим функциям, мы
старались по возможности предлагать новую информацию
лишь в том случае, когда наглядно прослеживается
непосредственная взаимосвязь изучаемых структур и
процессов с их биологическим назначением. Такой подход,
как мы считаем, должен предотвратить
преждевременное знакомство с новым (и потому не всегда понятным)
материалом.
В разделах, посвященных метаболизму, мы
ограничились изложением данных, полученных на животных
(единственное исключение - фотосинтез), исходя из того, что
метаболические системы прокариот и других организмов
в общем аналогичны, хотя и могут отличаться
интересными деталями. Там же, где эти различия
принципиальны (например, в разделе, посвященном геному и
экспрессии гена), данные, полученные на про- и эукариотах,
приводятся раздельно. То же самое относится и к
некоторым другим разделам, где большая часть материала
получена при исследовании прокариот.
Мы постарались включить в наш учебник как можно
больше различных гипотез, которые так или иначе
объясняют общебиологическое значение рассмотренных
молекулярных систем. Ведь метаболические пути транспорта,
хранения и выделения энергии будут особенно
интересны и понятны, если взглянуть на них сквозь призму
физиологических потребностей клетки и организма в целом.
То же относится к элементам иммунной системы.
Там, где это было возможно, биохимические процессы
увязаны с биологическими функциями. Именно
поэтому процессы свертывания крови, реакции с участием
фермента супероксиддисмутазы, цитохрома Р450 и т. д.
собраны в главе, посвященной защитным системам
организма, а не рассеяны по всей книге в качестве отдельных
примеров специфических функций белков. По той же
самой причине читатель знакомится с гемоглобином в
главе, посвященной проблемам транспорта газов в крови.
Мы рассмотрели метаболизм в первой части книги,
постепенно увеличивая сложность излагаемого
материала. Так была создана база для изучения последующих
разделов по молекулярной биологии. В этой книге мы
широко применяли метод напоминания читателю, где он
может найти более подробное изложения того или иного
материала, что позволяет без затруднений в случае
необходимости обратиться к нужному разделу. Овладев
материалом, изложенным в нашем учебнике, студент сможет
без особых проблем перейти к более углубленному
изучению биологических, химических или медицинских
аспектов интересующего его предмета.
За последние годы биохимия и молекулярная
биология щедро пополнялись новой информацией, и это
привело к появлению множества небольших журналов,
специализирующихся на коротких, вполне понятных и легко
читаемых обзорах почти по каждой теме (такие
публикации практикуют и солидные научные журналы).
Подобные «мини-обзоры» могли бы составить прекрасное
продолжение нашей книги для тех, кто стремится более
глубоко вникнуть в отдельные аспекты интересующей их
проблемы. Вот почему в конце каждой главы мы
приводим список дополнительной литературы, где
представлены главным образом обзоры вышеупомянутого типа: они
7
распределены по отдельным подтемам и снабжены
лаконичными комментариями. Большинство из них нетрудно
найти в библиотеках. Кстати, в главах, посвященных
молекулярной биологии, списки дополнительной
литературы значительно длиннее, что отражает современный
уровень интенсивности исследований в данной области науки.
И даже простое знакомство с такими списками способно
расширить кругозор читателя по интересующему его
вопросу.
Мы надеемся, что издание фундаментального
учебника с таким подробным изложением базисного
материала предоставит преподавателям прекрасную
возможность сконцентрировать свои усилия на тех областях
предмета, которые особенно важны для читаемых ими
курсов.
Д. Эллиот, В. Эллиот
Аделаида, апрель 1996 г.
8
Благодарности
Мы глубоко признательны за ценные советы и
критические замечания по отдельным главам книги нашим
высокочтимым коллегам, чьи имена перечислены ниже.
Доктор М. Abbey (Эбби), Division of Human Nutrition,
CSIRO, Adelaide, South Australia (Австралия)
Доктор J. A. Berden (Берден), Ε. С. Slater Institute,
University of Amsterdam, The Netherlands (Нидерланды)
Доктор G. W. Booker (Букер), Department of Biochemistry,
University of Adelaide, South Australia (Австралия)
Профессор М. С. Clark (Кларк), Department of
Biochemistry, University of Tasmania (Австралия)
Доктор J. С. Coates (Коутс), Department of Chemistry,
University of Adelaide, South Australia (Австралия)
Профессор J. С. Crabbe (Краббе), University of Reading,
UK (Великобритания);
Профессор К. van Dam (ван Дам), University of
Amsterdam, The Netherlands (Нидерланды)
Доктор A. Daws (Доус), School of Biological Sciences,
University of East Anglia, UK (Великобритания)
Доктор J. В. Egan (Иген), Department of Biochemistry,
University of Adelaide, South Australia (Австралия)
Доктор I. W. Flynn (Флинн), University of Edinburgh, UK
(Великобритания)
Доктор Ch. Hahn (Хан), Department of Biochemistry,
University of Adelaide, South Australia (Австралия)
Доктор D. Hawcroft (Хоукрофт), De Montfort University,
Leicester, UK (Великобритания)
Доктор S. van Heyningen (ван Хейниген), University
of Edinburgh, UK (Великобритания)
Доктор Ε. Μ. J. Jaspars (Джаспере), The Leiden Institue
of Chemistry, University of Leiden, The Netherlands
(Нидерланды)
Профессор J. R. Jefferson (Джефферсон), Department
of Chemistry, Luther College, Decorah, Iowa, USA (США)
Профессор I. Kotlarski (Котларски), Department of
Microbiology and Immunology, University of Adelaide,
South Australia (Австралия)
Профессор I. de la Lande (де ла Ланде), Department
of Physiology, University of Adelaide, South Australia
(Австралия)
Профессор S. Lincoln (Линкольн), Department of
Chemistry, University of Adelaide, South Australia (Австралия)
Доктор В. К. May (Мэй), Department of Biochemistry,
University of Adelaide, South Australia (Австралия)
Доктор G. Mayrhofer (Мэйрхофер), Department of
Microbiology and Immunology, University of Adelaide, South
Australia (Австралия)
Профессор J. F. Morrison (Моррисон), John Curtin School
of Medical Research, Australian National University,
Canberra, Australian Capital Territory (Австралия)
Профессор D. L. Nelson (Нелсон), Department of
Biochemistry, University of Wisconsin, Madison, USA (США)
Профессор J. Μ. Palmer (Палмер), Department of Biology,
Imperial College of Science, Technology and Medicine,
London, UK (Великобритания)
Профессор Р. Rathjen (Расджен), Department of
Biochemistry, University of Adelaide, South Australia (Австралия)
Доктор A. Robins (Робине), Bresatec Limited, University
of Adelaide, South Australia (Австралия)
Доктор А. С. Robinson (Робинсон), Department of
Biological Sciences, Napier University, UK (Великобритания)
Профессор G. Ε. Rogers (Роджерс), Department of
Biochemistry, University of Adelaide, South Australia
(Австралия)
Миссис R. Rogers (Роджерс), Department of Biochemistry,
University of Adelaide, South Australia (Австралия)
Профессор D. Rowley (Роули), Queen Elizabeth Hospital,
Adelaide, South Australia (Австралия)
Профессор R. Saint (Сейнт), Department of Genetics,
University of Adelaide, South Australia (Австралия)
Доктор Т. Sadlon (Сэдлон), Department of Biochemistry,
University of Adelaide, South Australia (Австралия)
Доктор G. Scroop (Скруп), Department of Physiology,
University of Adelaide, South Australia (Австралия)
Профессор R. Η. Symons (Саймоне), Waite Agricultural
Research Institute, University of Adelaide, South
Australia (Австралия)
Профессор D. Thomas (Томас), Department of Paediatrics,
University of Adelaide, South Australia (Австралия)
Профессор, доктор P. Van de Putte (ван де Путте),
Department of Molecular Genetics, Leiden University, The
Netherlands (Нидерланды)
Профессор J. Veale (Вейл), Department of Physiology,
University of Adelaide, South Australia (Австралия)
Доктор Ε. W. de Vrind de Jong (де Вринд де Джонг),
The Leiden Institute of Chemistry, University of Leiden,
The Netherlands (Нидерланды)
Доктор J. С. Wallace (Уоллейс), Department of Biochemistry,
University of Adelaide, South Australia (Австралия)
9
Профессор J. M. Walker (Уолкер), Division of Biosciences,
University of Hertfordshire, UK (Великобритания)
Доктор Р. Wigley (Ригли), Department of Biochemistry,
University of Adelaide, South Australia (Австралия)
Доктор R. J. H. Williams (Уильяме), Cardiff Institute
of Higher Education, UK (Великобритания)
Доктор A. F. Wilkes (Уилкс), Ludwig Institute for Cancer
Research, Royal Melbourne Hospital, Melbourne,
Victoria (Австралия)
Доктор J. Wiskich (Уискич), Department of Botany,
University of Adelaide, South Australia (Австралия)
Авторы выражают особую благодарность доктору
L. A. Burgoyne (Бергойн), Biological Sciences, Flinders
University of South Australia, за терпеливое
вычитывание макета этой книги и ценные замечания. А
также Крису Мэтьюзу, подготовившему макеты
рисунков с использованием компьютерной графики,
и Дэвиду Керри за проведение компьютерного поиска
литературы.
Авторы благодарны персоналу издательства Oxford
University Press за подготовку этой книги.
И наконец - но не в последнюю очередь! - авторы
глубоко признательны миссис Рос Мьюррел за ее высокий
профессионализм и долготерпение при подготовке
машинописного варианта этой книги.
10
Некоторые традиционные сокращенные
обозначения соединений и единиц,
используемые в данной книге
Перечень не является полным и в большинстве
случаев в тексте при первом упоминании каждое
сокращенное обозначение приводится в скобках.
- аденин
- аденозиндифосфат
- 5-аминолевулиновая кислота
- аденозинмонофосфат
- антиген-представляющая клетка
- аполипопротеин
- аденозинтрифосфат
- азидотимидин
- белок-активатор катаболизма
- кофермент А
- цитидиндифосфат
- сАМР-чувствительный элемент
- CRE-связывающий белок
- колоний-стимулирующий фактор
- дезокси(нуклеозидфосфаты) (dNMP, dNTP
и т.д.)
- дидезокси-
- фактор элонгации
- фактор роста эпидермиса
- эукариотический фактор инициации
- флавинадениндинуклеотид
- восстановленный флавинадениндинуклеотид
- кристаллизуемый фрагмент
(иммуноглобулина)
- дигидрофолат
- тетрагидрофолат
- формилметионин
- флавинмононуклеотид
- гуанин
- γ-аминобутират
- восстановленный глутатион
- окисленный глутатион
- гемоглобин
- оксигемоглобин
- белок теплового шока
- спираль-поворот-спираль (ДНК-узнающий
мотив)
- фактор инициации
- иммуноглобулин (IgG, IgA и т. д.)
- инсулиноподобный фактор роста (IGFI и IGFII)
1Р3 - инозиттрифосфат
IRE - железочувствительный элемент
IS - инсерционная последовательность
ISRE - стимулируемые интерфероном
чувствительные элементы
JAK - Янус киназа (тип тирозинкиназы)
Км - константа Михаэлиса
LTR - длинный концевой повтор
МНС - главный комплекс гистосовместимости
NAD+ - никотинамидадениндинуклеотид (окисленная
форма)
NADH - никотинамидадениндинуклеотид
(восстановленная форма)
NADP^ - никотинамидадениндинуклеотидфосфат
(окисленная форма)
NADPH - никотинамидадениндинуклеотид
(восстановленная форма)
Р450
Р/
PAF
PDGF
Pol
PP..
PrP
Q
Rubisco
S
SAM
SH2
SRP
SSB
Τ
τ3
T4
TBP
TCR
TFIID
TPA
U
UDPG
UTR
YAC
-цитохромР450
- неорганический фосфат
- фактор активации тромбоцитов
- тромбоцитарный фактор роста
- ДНК-полимераза
- неорганическая пирофосфатаза
- прионовый белок
- убихинон
- рибулозо-1,5-дифосфат-карбоксилаза
- константа Сведберга
- S-аденозилметионин
- домен GRB
- сигнал-узнающая частица
- белки, связывающиеся с одноцепочечной ДНК
- тимин
- трииодтиронин
- тироксин
- ТАТА-связывающий белок
- рецептор Т-клетки
- транскрипционный фактор D для полиме-
разы II
- тканевой активатор плазминогена
- урацил
- уридиндифосфатглюкоза
- нетранслируемая область
- искусственные дрожжевые линейные
хромосомы
11
1 Химия, энергия и метаболизм
Жизнь - это химический процесс, включающий тысячи
упорядоченно протекающих реакций. Их называют
метаболическими реакциями или, обобщенно, метаболизмом.
Поэтому, изучая биохимию, вы встретитесь с
множеством (но все же не с тысячами!) химических уравнений,
описывающих наиболее существенные аспекты химии
клетки. Биохимические стратегии, отобранные и
отшлифованные за миллионы лет эволюции, элегантны и покоряю-
ще красивы, но чтобы оценить их по достоинству,
понимать и восхищаться ими, сначала нужно понять, с какими
проблемами связано само существование жизни.
Анализ любого биологического превращения
основан на представлении об энергии. Записать его в виде
химического уравнения недостаточно, если при этом
нет сведений о происходящих изменениях химической
энергии. Без этого нельзя понять, сможет ли это
превращение сколько-нибудь заметно протекать в растворе
и в какой мере оно будет уравновешиваться обратной
реакцией.
Насколько важнее такой подход, можно видеть на
следующем примере. При интенсивной работе мышц
полисахарид гликоген (полимер глюкозы) в серии
химических реакций превращается в молочную кислоту, а при
последующем отдыхе он вновь синтезируется из
молочной кислоты, причем не в результате простого
обращения реакции, а иным путем. Простые энергетические
соображения позволяют понять, почему клетка поступает
именно так.
Для понимания биохимических процессов
достаточно самых общих знаний термодинамики. Есть,
разумеется, области, где этого недостаточно, но в целом
большинству биохимиков хватает знаний основных
принципов, и они редко используют термодинамические
уравнения. Этот раздел призван обеспечить именно
такую степень понимания.
Что определяет возможность
протекания химических реакций?
Как уже отмечалось, только изменение энергии
определяет, какие химические реакции возможны, а какие нет.
Заметим, что в биохимии нас интересуют реакции,
протекающие с образованием большого количества
продукта. В принципе, термодинамические факторы
не препятствуют полному протеканию реакций (за
исключением случаев, когда субстраты и продукты
находятся в равновесных концентрациях; см.ниже). Но если
реакция останавливается после того, как произошло
минимальное превращение субстрата, то с точки зрения
жизни такая реакция не имеет значения.
Сначала определим, что имеется в виду под
изменением энергии химической системы, т. е. ансамбля
молекул, участвующих в химических реакциях. Это не так
легко понять, как, например, изменение гравитационной
энергии при падении тела. Химическая система
состоит из огромного числа отдельных молекул, в каждой из
которых заключено некоторое количество энергии,
определяемое ее структурой. Эта энергия может быть
представлена как теплосодержание, или энтальпия,
молекулы. Когда структура молекулы меняется в ходе
химической реакции, изменение её энергии
описывается как изменение энтальпии АН. Оно может быть
отрицательным (теплота теряется молекулами и
рассеивается, увеличивая температуру окружающей среды) или
положительным (теплота поглощается из окружающей
среды, которая при этом охлаждается).
На первый взгляд может показаться, что реакции с
положительным АН столь же нереальны, как тело,
самопроизвольно взлетающее к потолку. Но такая простая
физическая аналогия неприменима к химическим
реакциям: здесь знак изменения энтальпии не определяет
жестко направление процесса, а лишь показывает,
что этот фактор либо способствует ему, либо
препятствует. Куда будет падать тело, зависит только от
изменения гравитационной энергии, а какая пойдет реакция,
зависит не только от изменения энтальпии, но также
и от изменения энтропии (AS) химической системы.
Энтропия может быть определена как степень
неупорядоченности. В химической системе беспорядок
проявляется по-разному. Поскольку молекула обычно не
жесткая структура, она может вибрировать, вращаться как
единое целое и относительно отдельных своих связей. Чем
легче происходит любое молекулярное движение, тем
больше беспорядка, или энтропии. Кроме того, огромное
число молекул, образующих химическую систему, может
быть случайным образом рассеяно в пространстве или
существовать в нем в той или иной мере упорядочено,
как это имеет место в живых клетках. И наконец, число
отдельных молекул и ионов в системе может измениться
часть 1 Введение в химические реакции клетки
14
в результате химических реакций. Чем больше молекул,
тем больше их совокупная неупорядоченность, а
следовательно, и энтропия системы.
И АН, и AS имеют право голоса при решении
вопроса, пойдет ли химическая реакция. Отрицательное АН
и положительное AS вместе дают ответ «да»,
положительное АН и отрицательное AS - ответ «нет». Если же
изменения энтальпии и энтропии имеют одинаковые знаки,
их влияние противоположно, и вопрос решается
сравнением их величин.
Рост энтропии как движущей силы процесса можно
проиллюстрировать на примере таяния льда в теплой
воде. Плавление льда происходит с поглощением тепла
(АЯ- положительно), однако увеличение молекулярного
беспорядка при разрушении кристаллической решетки
столь велико, что именно оно является определяющим
фактором процесса. Гораздо сложнее представить, как
энтропия управляет химической реакцией. Общее
утверждение состоит в том, что при сопоставимых значениях
энергии предпочтительнее более неупорядоченное
состояние системы. Если вам трудно это понять - примите пока
на веру, далее мы к этому еще вернемся.
Сравнивать изменение энтальпии и энтропии, которые
могут быть союзниками или антагонистами, при оценке
возможности протекания реакции крайне неудобно; хотя
бы потому, что их величины имеют разную размерность.
Более того, в биологических системах прямое измерение
энтропии затруднительно или невозможно. Ситуация,
однако, значительно улучшается благодаря
предложенному Гиббсом понятию свободной энергии, которое
объединяет оба рассмотренных выше понятия - энтальпию и
энтропию. Изменение свободной энергии (следуя Гиббсу,
обозначим его AG) описывает знаменитое уравнение:
AG = AH-TAS,
в котором Т- абсолютная температура.
Определение свободная здесь означает не свободу
вообще, а свободу использовать эту энергию для
совершения полезной работы. AG представляет собой
максимальное значение энергии, которое доступно для совершения
полезной работы за счет химической реакции. Иначе
говоря, это та часть ваших денег, на которую вы можете
рассчитывать при покупках.
Применительно к биологии, полезная работа-это
мышечное сокращение, химический синтез в клетках,
преодоление осмотических или электрических сил.
Величину AG выражают в калориях (кал) или, что более принято,
в джоулях (Дж) на моль (1 кал = 4,19 Дж). Поскольку
значения AG обычно велики, чаще пользуются
килокалориями или килоджоулями.
Изменение свободной энергии в ходе любого
процесса - важнейший термодинамический параметр.
Применительно к химическим процессам можно
сформулировать общее правило: химическая реакция протекает
лишь в случае AG<0, т.е. есть в условиях, когда
свободная энергия продуктов реакции меньше, чем исходных
веществ.
Изменение свободной энергии
и обратимость реакций
Вероятно, вам уже говорили, что многие химические
реакции обратимы. Это может озадачить, ведь тогда
получается, что величины AG отрицательны и для
прямой, и для обратной реакции (вспомним, что это
критерий возможности спонтанного протекания любого
процесса). Разгадка кажущегося парадокса состоит в том,
что AG не является фиксированным параметром и
зависит от концентраций исходных веществ и продуктов
их превращения.
С уравнением, описывающим эту зависимость, мы
познакомимся позже. А пока заметим, что для
обратимой реакции А <-> В изменение свободной энергии AG
может быть отрицательным применительно к пре-
вращению А —>В и положительным применительно
к превращению В —> А, если только концентрация
вещества А больше концентрации вещества В. Приобратном
соотношении концентраций А и В отрицетельным
может стать изменение cвoбoдн^и_^н^rлw^_ΔGJΊЩJπpe-
вращении В —> А. Нетрудно сообразить, что существует
некоторое соотношение концентраций, при котором
изменения AG для прямой и обратной реакции равны нулю.
Такое соотношение будет сохраняться сколь угодно
долго - это точка химического равновесия.
Если величина AG для некоторой биохимической
реакции А <-> В невелика по модулю, то с большей ве^
роятностью^эта_реакция в клетке будет обратима, по-
скольку_изменение концентрации веществ А и В в ходе
метаболизма может привести к обращению знака AG.
Напротив, если абсолютное значение AG велико, ре-
акцию можно считать ^практически необратимой.
Диапазон^ изменений внутриклеточных концентраций
метаболитов]р5рш1н^збъ^д>шяющий всех участников
биохимических реакций) относительно узок: обычно
значения концентраций лежат в пределах^ 1 (Н-10~3 М.
6^^ртьт1те]5ёаЩ
Hbjj^3Ha4eHHflNiHJA(jj3ea^^ так как кон-
центраииошшх^13^нениЙ21едостаточно для обращения
знака AG. (Позже мы объясним,, почемукажется, что при
некоторых реакциях это положение нарушено; см. с. 110).
глава 1 Химия, энергия и метаболизм
15
Как правило, внутриклеточный гидролиз, т.е. реакции
расщепления связей с участием молекулы воды,
необратим в том смысле, что образование этих связей в клетках
не является результатом обращения гидролиза. О том,
насколько обратимы другие внутриклеточные реакции,
будет рассказано в главе о метаболизме.
Итак, внутриклеточные реакции, сопровождающиеся
незначительным изменением свободной энергии, могут
быть обратимы, причем их направление зависит от
небольших изменений концентраций участвующих
метаболитов. Напротив, при значительном изменении
свободной энергии метаболические реакции идут в одном
направлении и реально протекают до полного
завершения, поскольку именно в эту область концентраций
сдвинута точка равновесия. Иными словами, исходные
вещества полностью превращаются в продукты реакции.
Роль необратимых реакций
в стратегии метаболизма
В предыдущем разделе мы подчеркнули влияние
величины AG химической реакции на возможность ее
протекания в обратном направлении. Пора объяснить,
почему это так важно. Основные физиологически важные
внутриклеточные химические процессы обычно
включают не одну, а много последовательно протекающих
реакций, объединенных в так называемые
метаболические пути, где продукт первой реакции является
исходным веществом для второй и т.д. Например, в
упомянутом выше превращении гликогена в молочную кислоту
в мышцах вовлечено двенадцать последовательных
химических реакций.
Общим свойством метаболических путей является их
необратимость. Это не означает, что необратимы все
включенные в них реакции. Но по меньшей мере одна
реакция в физиологических условиях не протекает в
обратном направлении. Такие реакции, действуя подобно
клапанам на трубопроводе, и обеспечивают
термодинамическую основу завершенности суммарного
метаболического превращения.
Это, однако, не означает физиологической
необратимости результирующего превращения. Так, из
молочной кислоты в организме благополучно синтезируется
гликоген.
Некоторые из стадий биосинтеза гликогена
представляют собой обращенные реакции его расщепления.
Но есть другие реакции, которые не могут быть
обращены, и вместо них используются альтернативные пути.
Например, некоторые реакции протекают с использованием
энергии, что делает их необратимыми в обратном
направлении. В результате и прямой (гликоген—>молочная
часть 1 Введение в химические реакции клетки
16
кислота), и обратный (молочная кислота—>гликоген)
метаболические пути являются необратимыми. Типичная
метаболическая ситуация может быть описана
следующей схемой:
На этой схеме красными стрелками помечены
необратимые реакции с большими по модулю
отрицательными значениями AG. Чуть позже мы расскажем, как
достигается энергетическое обеспечение необратимости
в обоих направлениях.
Итак, стал ясен общий биохимический принцип.
Всякий раз, когда суммарный химический процесс,
характерный для данного метаболического пути, должен
быть физиологически обратимым, прямая и обратная
траектории не совпадают полностью и обязательно
содержат хотя бы пару различающихся по своей
природе необратимых реакций.
Почему клетки используют
такую стратегию метаболизма?
Тому есть две основные причины. Рассмотрим
альтернативную стратегию, при которой все метаболические
реакции обратимы.
Α ^ в Т=± С ^ D ^=Ζ Ε Ξ=Ξ F ^ Продукты
Главный недостаток при этом - подверженность
процесса в целом закону действующих масс. Если
концентрация вещества А возрастает (скажем, вы что-нибудь
съели), равновесие сдвигается вправо и возрастает
концентрация конечных продуктов. Напротив, при
уменьшении концентрации А некоторые продукты будут
превращаться в исходные вещества. Представьте теперь, что
речь идет о биосинтезе ДНК генов или жизненно
важных белков. И вы поймете, насколько неприемлем такой
сценарий.
Есть и вторая сходная причина, по которой прямое
и обратное направления метаболического пути должны
включать различные реакции: метаболизм обязан быть
контролируемым. Мы уже знаем, что при интенсивной
работе мышц гликоген превращается в молочную
кислоту, тогда как при отдыхе этот процесс выключается
и происходит ресинтез гликогена из молочной
кислоты. Чтобы независимо управлять обоими
процессами, т.е. включать один и выключать другой, они должны
различаться. Иначе их можно включать и выключать
только одновременно! Как правило, именно необратимые
стадии метаболизма являются местом приложения регуля-
торных механизмов. Подробнее об этом в главе 12.
Из чего складывается величина AG?
Хотя изменение свободной энергии при химической
реакции, как уже отмечалось, непостоянно, в некоторых
определенных условиях, принятых за стандартные,
величина AG является постоянной. Примем в качестве таких
стандартных условий концентрации всех
веществ-участников 1,0 М, температуру 25° С и значение рН 7,0.
Отвечающую этим условиям величину AG называют
стандартным изменением свободной энергии данной реакции
и обозначают AG0'. Заметим, что некоторые физики и
химики предпочитают в качестве стандартного условия
значение рН 1,0, вместо 7,0.
Для вычисления стандартного изменения свободной
энергии различных реакций часто используют
физико-химические таблицы, в которых приведены стандартные
свободные энергии образования большого числа
химических соединений. Если по отдельности сложить
значения для исходных веществ и продуктов реакции, то
разность между этими двумя суммами и будет искомой
величиной AG0. Альтернативой является
экспериментальное определение константы равновесия данной
реакции, из которой нетрудно вычислить AG0'.
При заданных концентрациях исходных веществ и
продуктов реакции величины AG и AG0' связывает простое
соотношение. Поэтому, зная внутриклеточные
концентрации метаболитов, можно вычислить изменение
свободной энергии, характеризующее соответствующую
биохимическую реакцию. Это было проделано для очень многих
биохимических реакций, и на полученные таким образом
значения часто ссылаются.
Однако совсем не просто определить внутри клеток
концентрации тысяч метаболитов, содержание которых
там мало и непостоянно. И потому для многих
биохимических реакций значения AG пока остаются
неизвестными. К счастью, оказалось, что во многих случаях
можно использовать величины AG0', хотя отвечающие
им условия в клетках никогда не реализуются.
Разумеется, это компромисс, но полезный.
Стандартная свободная энергия
и константы равновесия
Знать величины AG0' особенно полезно, потому что из
них легко вычисляется константа равновесия
соответствующей реакции. Константа равновесия К
представляет собой отношение произведений равновесных
концентраций продуктов реакции и исходных веществ
(штрих означает, что константа равновесия отвечает
рН 7,0). Так, например, для реакции А + В <-> С + D
константа К вычисляется из концентраций веществ
А, В, С и D, измеренных в условиях равновесия, т.е. когда
эти концентрации перестали изменяться.
~. ИМ
е" [А][В]
Связь между величинами AG0' и К описывается
уравнением:
AG0' = -RT - In К 'eq = -2,303Я Г - lg К 'eq,
где R - газовая постоянная (8,315 кДж ■ моль-1 · Кг1);
Τ - абсолютная температура в градусах Кельвина
(298 К = 25° С). При 25° С RT = 2,478 кДж · моль1.
Таким образом, измерив значение К'' , можно
вычислить величину AG0', и наоборот, располагая сведениями
о AG0', можно вычислить значение константы
равновесия К 'ед.
Таблица 1.1
Соотношение между константой равновесия реакции (/Г )
и изменением свободной энергии (AG0)
Таблица позволяет составить представление о соотношении
констант равновесия химических реакций со стандартными
изменениями свободной энергии
AG0' (кДж · моль-1)
+ 17,1
+ 11,4
+5,7
0
-5,7
-11,4
-17,1
К'еЧ
0,001
0,01
0,1
1,0
10,0
100,0
1000,0
Допустим, что AG отрицательно. От чего
зависит, будет ли протекать
соответствующая реакция в клетке с ощутимой
скоростью?
Д,ля протекания химической реакции необходимо, чтобы
ей отвечало уменьшение свободной энергии. Однако
из этого вовсе не следует, что она будет протекать с
ощутимой скоростью. Оценка с точки зрения термодинамики
реакции взаимодействия сахара с кислородом (т.е.
горения) с образованием Н20 и С02 показывает, что
энергетически это очень выгодный процесс. В то же время
сахар на воздухе вполне устойчив. Существует барьер
для протекания реакций даже с очень большим
уменьшением свободной энергии, иначе все горючие
материалы на Земле давно бы исчезли в огне. Однако сахар, столь
глава 1 Химия, энергия и метаболизм
17
устойчивый в чашке с чаем, быстро окисляется в
организме, как только мы выпиваем этот чай. Возникает
вопрос: почему реакции, протекающие в живых клетках, вне
их либо не идут вообще, либо идут с ничтожно малыми
скоростями? Потому что в клетках они осуществляются
с помощью ферментативного катализа.
Природа
ферментативного катализа
В простейшем случае фермент (или энзим) - это
катализатор, ускоряющий только одну химическую реакцию.
Известны тысячи биохимических реакций, и каждая из
них катализируется своим ферментом. Принцип «одна
реакция - один фермент» соблюдается и в случае муль-
тифункциональных ферментов, обладающих
различными каталитическими активностями, и в случае мульти-
ферментных комплексов.
Фермент является белком. И хотя строению белков
будет посвящена глава 2, краткий экскурс в эту область
здесь будет полезен. Белки построены из 20 аминокислот,
соединенных в длинные цепи. Это высокомолекулярные
соединения: даже у самых маленьких ферментов
молекулярная масса порядка 10 000 дальтон (Да),
а у многих она достигает сотен тысяч дальтон. Одна
из причин, по которой ферменты так велики, заключается
в том, что длинная цепь (или цепи), из которой они состоят,
должна свернуться с образованием некого кармана,
называемого активным центром. Попадая в такой
карман, молекула вещества с исключительной точностью
атакуется функциональными группами фермента. Под
«атакой» здесь следует понимать химическое
превращение вещества, которое принято называть субстратом,
при участии данного фермента.
В целом ферментативный процесс обычно
описывают моделью Михаэлиса-Ментен:
E + S<->ES<->EP->E + P,
где Ε - фермент; S - субстрат; ES и ЕР - соответственно
комплексы фермента с субстратом и продуктом
ферментативной реакции (Р).
Из уравнения, описывающего эту модель, следует, что
субстрат обратимо связывается с активным центром
фермента и далее в связанном виде превращается в новое
вещество, которое диффундирует из активного центра
в окружающий раствор. Мы еще вернемся к модели
Михаэлиса-Ментен при обсуждении регуляции
ферментативного катализа (см.с. 156), а пока рассмотрим основы
механизма катализа.
часть 1 Введение в химические реакции клетки
18
Как работает фермент?
Чтобы ответить на этот вопрос, нам следует понять
природу химических реакций. Они протекают в две стадии.
Рассмотрим превращение S —> Р.
На первой стадии исходная молекула претерпевает
определенные конформационные и электронные
изменения, которые служат предпосылкой ее последующего
легкого преобразования в конечный продукт.
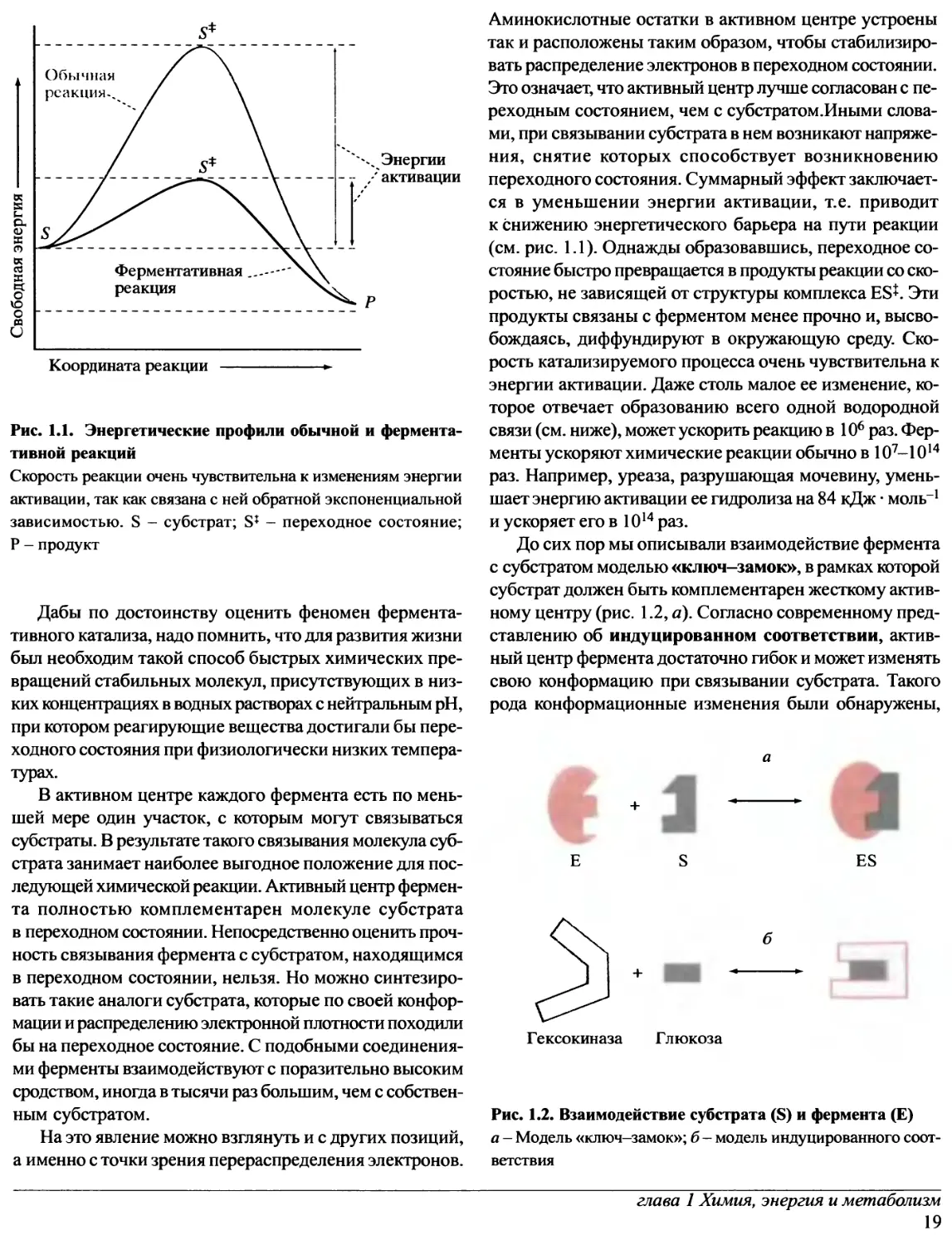
S-^-^P
В таком возбужденном состоянии S* (его называют
переходным) молекула существует очень недолго (10"14-
10~13 с). Ясно, что возникновение переходного состояния
обусловлено поступлением энергии извне.
Суммарная реакция S —> Ρ должна сопровождаться
уменьшением свободной энергии, иначе она вообще бы
не произошла. Этот термодинамический параметр
зависит только от строения исходного и конечного
соединений и инвариантен к характеру промежуточных
изменений свободной энергии, который обычно называют
энергетическим путем, или энергетическим профилем
реакции. Поэтому нет никакого противоречия в том, что в
переходном состоянии S* молекула обладает большей
свободной энергией, чем в исходном S. Положительное
изменение свободной энергии при превращении S —> S*
называют энергией активации для данной реакции (рис. 1.1).
Энергетический горб (см. рис.1.1) служит барьером
на пути химической реакции. Без него энергетический
профиль отражал бы непрерывное падение свободной
энергии на пути от S к Р, и все вещества, способные вступать
в реакции, сразу бы в них вступали.
Итак, чтобы реакция прошла, необходимо внести
в систему энергию активации. В некатализируемых
реакциях источником этой энергии служат столкновения
между молекулами в растворе. Если соударяющиеся
молекулы должным образом ориентированы, есть
шанс, что возникающие при этом искажения
молекулы S будут такого свойства, что ее
высокоэнергетическое состояние окажется переходным на пути S —> Р.
Следовательно, скорость протекания реакции
определяется вероятностью возникновения переходного
состояния исходных молекул. С ростом температуры
возрастает как скорость теплового движения, так и частота
соударений, и потому увеличивается вероятность
возникновения переходного состояния.
Химики-органики издавна используют нагревание для ускорения
реакций. Что же касается биохимических реакций, то
в физиологическом диапазоне температур и в
отсутствие катализаторов все они протекают с ничтожно
малыми скоростями.
-^Энергии
/'активации
Координата реакции
Рис. 1.1. Энергетические профили обычной и
ферментативной реакций
Скорость реакции очень чувствительна к изменениям энергии
активации, так как связана с ней обратной экспоненциальной
зависимостью. S - субстрат; S* - переходное состояние;
Ρ - продукт
Дабы по достоинству оценить феномен
ферментативного катализа, надо помнить, что для развития жизни
был необходим такой способ быстрых химических
превращений стабильных молекул, присутствующих в
низких концентрациях в водных растворах с нейтральным рН,
при котором реагирующие вещества достигали бы
переходного состояния при физиологически низких
температурах.
В активном центре каждого фермента есть по
меньшей мере один участок, с которым могут связываться
субстраты. В результате такого связывания молекула
субстрата занимает наиболее выгодное положение для
последующей химической реакции. Активный центр
фермента полностью комплементарен молекуле субстрата
в переходном состоянии. Непосредственно оценить
прочность связывания фермента с субстратом, находящимся
в переходном состоянии, нельзя. Но можно
синтезировать такие аналоги субстрата, которые по своей конфор-
мации и распределению электронной плотности походили
бы на переходное состояние. С подобными
соединениями ферменты взаимодействуют с поразительно высоким
сродством, иногда в тысячи раз большим, чем с
собственным субстратом.
На это явление можно взглянуть и с других позиций,
а именно с точки зрения перераспределения электронов.
Аминокислотные остатки в активном центре устроены
так и расположены таким образом, чтобы
стабилизировать распределение электронов в переходном состоянии.
Это означает, что активный центр лучше согласован с
переходным состоянием, чем с субстратом.Иными
словами, при связывании субстрата в нем возникают
напряжения, снятие которых способствует возникновению
переходного состояния. Суммарный эффект
заключается в уменьшении энергии активации, т.е. приводит
к снижению энергетического барьера на пути реакции
(см. рис. 1.1). Однажды образовавшись, переходное
состояние быстро превращается в продукты реакции со
скоростью, не зависящей от структуры комплекса ES*. Эти
продукты связаны с ферментом менее прочно и,
высвобождаясь, диффундируют в окружающую среду.
Скорость катализируемого процесса очень чувствительна к
энергии активации. Даже столь малое ее изменение,
которое отвечает образованию всего одной водородной
связи (см. ниже), может ускорить реакцию в 106 раз.
Ферменты ускоряют химические реакции обычно в 107-1014
раз. Например, уреаза, разрушающая мочевину,
уменьшает энергию активации ее гидролиза на 84 кДж · моль-1
и ускоряет его в 1014 раз.
До сих пор мы описывали взаимодействие фермента
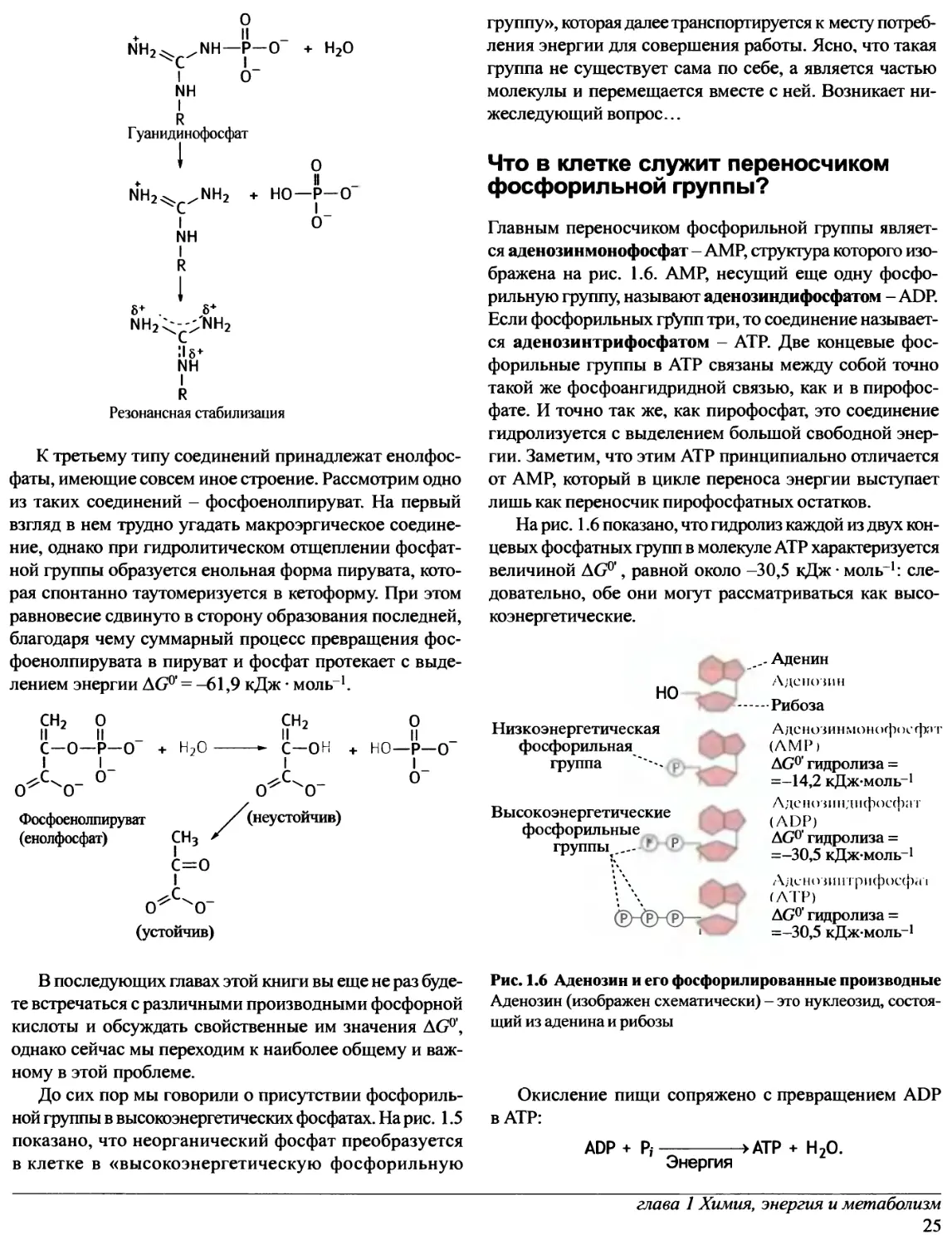
с субстратом моделью «ключ-замок», в рамках которой
субстрат должен быть комплементарен жесткому
активному центру (рис. 1.2, а). Согласно современному
представлению об индуцированном соответствии,
активный центр фермента достаточно гибок и может изменять
свою конформацию при связывании субстрата. Такого
рода конформационные изменения были обнаружены,
ES
Гексокиназа Глюкоза
Рис. 1.2. Взаимодействие субстрата (S) и фермента (Е)
а - Модель «ключ-замок»; б - модель индуцированного
соответствия
глава 1 Химия, энергия и метаболизм
19
например, при рентгеноструктурных исследованиях
гексокиназы, которая катализирует перенос фосфатного
остатка с АТР на глюкозу. В структуре этого фермента
можно выделить два «крыла», которые при связывании
глюкозы «складываются», образуя каталитический центр
(рис. 1.2, б). Этот конформационный переход определяет
специфичность гексокиназы к глюкозе. Структура,
которая связывает глюкозу, по-видимому, должна в какой-то
степени связывать и другие полиолы и даже молекулы
воды. В этом случае, исходя из модели «ключ-замок»,
можно ожидать от гексокиназы определенную активность
в отношении других молекул, отличных от глюкозы.
Применительно к воде это означало бы катализ гидролиза АТР,
каковой в действительности не имеет места.
Следовательно, каталитический центр возникает лишь при
связывании «правильного» субстрата - глюкозы. Другие
молекулы, если и связываются с ним, не способны вызвать
те конформационные изменения фермента, которые
обусловливают протекание ферментативной реакции.
Большинство молекул могут вступать в различные
химические реакции, каждой из которых отвечает свое
переходное состояние. В отсутствие катализа, когда
скорость превращения определяется лишь температурой,
молекулы соударяются непредсказуемым образом,
благодаря чему возникают самые разные переходные
состояния. Поэтому основная реакция обычно
сопровождается множеством побочных. Напротив, при
ферментативном катализе ускоряется только одна из возможных
реакций и образуются только определенные продукты
превращения.
Особую роль ферменты могут играть в ускорении
реакций посредством общего кислотно-основного
катализа, когда принадлежащие активному центру
протон-донорная или протон-акцепторная группы
расположены таким образом, что непосредственно участвуют
в переносе протона к переходному состоянию связанного
субстрата или от него. Точно так же (а иногда и
одновременно) ферменты ускоряют реакции благодаря тому, что
в их комплексах с субстратом «правильно»
относительно переходного состояния расположен каталитически
активный ион металла.
В ряде случаев ферменты выступают как участники
химической реакции, образуя промежуточное ковалент-
ное соединение с субстратом, которое, однако, далее
распадается, высвобождая исходную молекулу белка.
Примером здесь может служить важный класс
ферментов - сериновые протеиназы, названные так потому, что
их активные центры включают остаток аминокислоты
серина, ковалентно участвующий в протеолизе:
R-CO-NHR-R' + Н20 -> RCOCT + R'NH$ .
часть 1 Введение в химические реакции клетки
20
В ходе этой каталитической реакции ацильный
остаток, входящий в пептидную группу, сначала переносится
на реакционноспособный гидроксил серина - ОН, а
затем на молекулу воды
R-CO-NH-R" + Enz-OH + H+->Enz-0-CO-R + NH3+-R", (1)
Enz-O-CO-R + H20-> Enz-OH + R-COO" + H+. (2)
Разные сериновые протеиназы специфичны к разным
пептидам, но у всех каталитический центр содержит
реакционноспособный остаток серина. Механизм действия
этих ферментов детально изучен.
Скорость ферментативного катализа обычно
является «визитной карточкой» данного фермента. В целом, все
ферментативные реакции в клетке протекают
неизмеримо быстрее некатализируемых, вклад которых попросту
неощутим. Это ускорение, как уже отмечалось выше,
может составлять 1014 раз и даже больше. Реально в
ферментативные превращения вступает от нескольких до 107
молекул субстрата в секунду в расчете на одну молекулу
фермента. Как будет показано далее (см. с. 157), скорость
ферментативных реакций в определенном диапазоне
зависит от концентрации субстрата. Если принять ее
постоянной, то эта скорость будет определяться лишь
внутриклеточным содержанием соответствующего фермента.
Добавим, что многие ферменты обладают регуляторны-
ми механизмами, позволяющими изменять
каталитическую активность в соответствии с физиологическими
потребностями (см. главу 12, посвященную
метаболическому контролю).
Активность фермента зависит от многих факторов.
В их числе - степень ионизации его функциональных
групп, которая определяет устойчивость белковой
молекулы в целом и деятельность активного центра.
С другой стороны, имеет значение также ионизация
субстрата. Иными словами, скорость катализируемой
реакции обычно зависит от рН. Эта зависимость может
быть различной, но обычно она характеризуется
максимумом, расположенным в нейтральной области
значений рН (типичная кривая показана на рис. 1.3, а).
Случаются исключения. Так, пищеварительный фермент
пепсин работает в сильно кислой среде желудка; рН-оп-
тимум пепсина находится в области 2,0. Температура
тоже влияет на активность ферментов. С ее ростом
скорость катализируемых реакций сначала возрастает,
примерно вдвое на каждые 10° С, а далее начинает
падать из-за инактивации ферментов, поскольку они, как
и большинство белков, неустойчивы к нагреванию.
Типичная зависимость ферментативной активности
от температуры приведена на рис. 1.3, б, однако на
форму подобных кривых сильно влияют условий проведения
10 11
100
'К
20 30 40 50
Температура, °С
70
Рис. 1.3. Влияние рН (а) и температуры (б) на
относительную активность ферментов:
α -Кривая 1 с максимумом при физиологическом значении рН
характерна для большинства ферментов, кривая 2 отвечает
пепсину, функционирующему в сильно кислом содержимом
желудка;
б - резкое падение активности при высоких температурах
связано с денатурацией фермента (есть и термостойкие ферменты).
Положение максимума зависит от скорости нагревания
эксперимента: чем короче время пребывания
фермента при высокой температуре, тем меньше ее
повреждающее действие. Хотя некоторые ферменты выдерживают
нагревание, большинство из них инактивируется при
температурах выше 50° С, а некоторые и при более
низких. К этому следует добавить, что многие ферменты
активны лишь в присутствии определенных ионов
металлов, например, Mg2+, Zn2+, Fe2+ или Cu2+.
Активность ферментов изменяется также в
присутствии ингибиторов. Вещества, принадлежащие к клас-
су конкурентных ингибиторов, по строению подобны
субстрату и конкурируют с ним за активный центр
фермента. Неконкурентные ингибиторы уменьшают
активность фермента, вступая с ним в разного рода
специфические взаимодействия. Например, ион ртути
образует ковалентное соединение с тиоловой группой,
принимающей непосредственное участие в акте катализа.
В других случаях ингибитор необратимо ацилирует
фермент. Это происходит при действии аспирина на цикло-
оксигеназу, которая участвует в биосинтезе простаглан-
динов (подробности см. на с. 146).
СООН
Enz—ОН +
Циклооксигеназа Аспирин
О—С —Oh
СООН
Enz—О—С СН3 +
Ацилированный
фермент
2- Гидроксибензойная
кислота
Ацилируемая группа здесь принадлежит остатку сери-
на, который входит в состав активного центра циклоок-
сигеназы.
Как расщепление пищи в клетках
сопряжено с внутриклеточными
реакциями, потребляющими
энергию
Образование из низкомолекулярных предшественников
таких биополимеров, как белки и ДНК, не может быть
спонтанным, поскольку сопряжено со значительным
увеличением свободной энергии. В термодинамическом
контексте спонтанность означает отсутствие
потребности во внешнем источнике свободной энергии. Внешним
источником энергии является процесс окисления пищи.
Химические превращения, протекающие с
положительным изменением свободной энергии (синтетические, или
«строительные» процессы) в совокупности называют
анаболизмом, или анаболическими реакциями
(анаболические стероиды, которыми злоупотребляют
некоторые спортсмены, увеличивают массу тела - отсюда их
название). Остальная часть метаболических
превращений состоит из деструктивных процессов, протекающих
с отрицательным изменением свободной энергии. Их
называют катаболизмом, или катаболическими
реакциями. Таким образом, метаболизм включает в себя
анаболизм и катаболизм.
глава 1 Химия, энергия и метаболизм
21
В результате катаболизма высвобождается свободная
энергия, которая используется для приведения в действие
энергопотребляющих анаболических реакций.
Химическое превращение, происходящее с положительным
изменением свободной энергии, требует ее притока извне,
но значение AG, характеризующее процесс в целом,
должно всегда быть отрицательным. Катаболические
реакции, при которых энергия высвобождается, часто
называют экзоэргоническими, а энергопотребляющие
анаболические реакции - эндоэргоническими. Следует
четко различать такие понятия, как химическая реакция
и химическое превращение, т. е. процесс, происходящий
в ходе ряда химических реакций. Так, соединение А
в клетке реально может превратиться в соединение X,
даже если для последнего характерна большая
стандартная свободная энергия и его образование требует
притока энергии извне. Однако это превращение протекает
как последовательность отдельных химических реакций,
каждая из которых имеет отрицательное значение AG.
Обсудим кратко, как это может произойти.
Такую совокупность реакций можно представить
общей схемой, изображенной на рис. 1.4. В результате
окисления пищи высвобождается энергия, которая
используется для приведения в действие энергетически
невыгодных процессов. Чтобы придать этой схеме глобальное
значение, рассмотрим фотосинтез, при котором световая
энергия используется для синтеза пищевых молекул
(глюкозы) из С02 и Н20. Глюкоза превращается в организмах
в другие пищевые молекулы, например в жир. Хотя
сборка крупных клеточных структур связана с уменьшением
энтропии (т.е. невыгодна), окисление пищевых молекул
сопровождается значительным увеличением энтропии
(и потому выгодно). При этом для системы в целом (клетка
и окружающая среда) изменение энтропии
положительно, так что второй закон термодинамики выполняется
в полной мере (согласно этому закону, общая энтропия
Вселенной может только возрастать).
1 Пищевые Упорядоченные
I молекуль ма!фомолекулы
τ Ι υ2 в клетке
Шкала Энергия {имическа;
свободной —►
энергии света энергия
I комолекупярныс
I неупорядоченные
I молекулы в
I СОо+НоО окружающей среде
Фотосинтез Катаболизм Анаболизм
Рис. 1.4 . Энергетический цикл жизни
часть 1 Введение в химические реакции клетки
22
Здесь можно усмотреть аналогию с автомобилем,
который поднимается в гору за счет сгорания бензина.
Однако засунув пылающее ведро с горючим под капот,
нужного эффекта не получишь, ибо должен
существовать некий механизм для передачи выделяющейся
энергии горения от ведра к колесам. Точно так же и клетка
должна уметь сопрягать экзоэргонические и эндоэрго-
нические реакции, чтобы разумным образом
использовать энергию, которой иначе суждено бесполезно
рассеяться в виде тепла.
Как это происходит? Использовать
высвобождающееся тепло невозможно потому, что в физиологических
условиях оно рассеивается. Энергия,
высвобождающаяся при расщеплении пищевых веществ, используется
в химических реакциях для совершения механической
(мускульной) работы, в осмотических и
электрофизиологических процессах. С другой стороны, для каждого
потребляющего энергию процесса организму было бы
слишком сложно создавать специфический
преобразователь энергии. Вместо этого в клетках есть общий
энергетический интермедиат, который принимает энергию от
всех реакций окисления пищи и доставляет ее туда, где
совершается работа и требуется энергия. Это более
гибкий способ передачи энергии. Снова прибегнув к
аналогии, уподобим его энергетике, опирающейся на
электричество. Свободную энергию, выделяющуюся при
сгорании угля, нефти, дерева или газа, превращают
в электроэнергию, последняя же легко может быть
передана туда, где она нужна, и использована так, как это
требуется. (Впрочем, эта аналогия не вполне верна, ибо
производство электроэнергии здесь опосредовано
использованием тепла, чего нет в клетках.) Так мы
приходим к мысли, что универсальный переносчик энергии
должен иметь химическую природу. И потому
закономерен вопрос: что же это за вещество?
Высокоэнергетический фосфат
Ответ на вопрос замечательно прост. Такое вещество
универсально для всех форм земной жизни (строго
говоря, исключения есть, но они столь экзотичны, что
лишь подчеркивают универсальность).
Общую концепцию можно изложить предельно ясно:
при распаде пищевых молекул неорганические
фосфатные ионы превращаются в фосфорильные группы
молекул так называемых макроэргических фосфатных
органических соединений (что это значит, см. ниже).
Эти молекулы в клетке транспортируются туда, где
нужно совершить полезную работу. Такая работа
совершается за счет энергии, запасенной в химических связях и
выделяющейся при обратном превращении фосфорильных
остатков в неорганические фосфатные ионы.
Тут крайне важно отметить, что ни_в коем случае
нельзя рассматривать обратное превращение фосфо-
рильных остатков как обычный гидролиз, ибо при гид-
ролизе вся выделяющаяся энергия превращается в
бесполезное тепло. Рассмотрим упрощенный механизм,
который позволяет «обуздать» энергию химических
связей и использовать ее для совершения полезной работы.
Что такое «макроэргический фосфат»?
Внутриклеточные соединения, содержащие фосфориль-
ные остатки, можно разделить на две группы.
«Низкоэнергетические», при гидролизе которых до
неорганического фосфата (Pf.) отрицательное изменение свободной
энергии AG0' лежит в пределах 9-20 кДж · моль1. И
«высокоэнергетические», или макроэргические, у которых
AG0' превышает 30 кДж · моль1. Понятие
высокоэнергетический фосфат иногда используют для описания не
самого соединения, а фосфатной связи. Однако это
противоречит принятым в химии представлениям, согласно
которым высокоэнергетическая связь - такая связь,
расщепление которой требует больших затрат энергии. По-
этому мы будем для ясности говорить о
высокоэнергетической фосфорильнои группе, подразумевая, что
присутствие такой группы придает высокую энергетичность
молекуле в целом. С этой важной оговоркой именно
высокоэнергетическую фосфорильную группу можно
рассматривать как энергетическую «валюту» живой клетки.
Концепцию иллюстрирует рис. 1.5, уточняющий рис. 1.4.
1
i
Шкала
свободной
энергии
Пищевые
шекуль
со2+н2с
02
МИР* '
энергия
Перенос внутри
клеткикместу
ыполнения работы
ρ, ρ
Перенос внутри
клеткикместу
нлработки энергии
Биологическая
работа
(химическая,
^ектр^кхкая,
осмотическая,
механическая)
Рис. 1.5. Участие фосфатных групп в энергетическом
хозяйстве клетки
-©-Фосфатная группа в молекулах, гидролиз которых
приводит к отщеплению Р. и сопряжен с высвобождением более
30 кДж ■ моль '
В чем главные структурные
особенности высокоэнергетических фосфатов?
Фосфорная кислота Н3Р04 является гидроксипроизвод-
ным фосфора, содержащим в молекуле 3 протона.
О О
II II
- - =^но-р-о- + н+
Р*а1=2,2 ^
но-р-сг ^
он
Р*а2 = 7,2
но-р-сг+н+
II
?
Р*а3=12,3
-о-р-сг+н+
0"
При физиологических значениях рН эта кислота в
растворе представлена главным образом одно- и двухза-
рядными анионами, равновесную смесь которых
биохимики обозначают сокращенно Р/, где индекс ι (от англ.
inorganic) указывает на неорганическую природу
вещества. Смеси анионов (Pf.) соответствует самый низкий
уровень свободной энергии фосфата, который условно
можно принять за исходный, или нулевой.
Степень ионизации фосфорной кислоты можно
вычислить, исходя из значений трех констант ее
диссоциации. Они могут быть представлены в виде параметров
рКа (напомним, что под рК понимают такое значение
рН, при котором в диссоциированном состоянии
находится половина соответствующих групп). Более
подробные разъяснения рКа даны в приложении к этой главе.
Его следует изучить, если вы еще не знакомы с такими
понятиями, как диссоциация, буферные растворы и т. п.
При физиологическом значении рН (скажем 7,4) группа
с рКа 2,2 полностью диссоциирована, а группа с рКа 12,3
полностью находится в недиссоциированном состоянии.
Группа с рКа 7,2 диссоциирована частично. Для
вычисления степени диссоциации химических групп можно
воспользоваться уравнением Гендерсона - Хассельбаха:
[Соль]
pH = pA:fl2 + lg
[Кислота]
7,4 = 7,2 + lg
[HPOJ]
[Η2Ρθ4]
Таким образом,
lgiH^r0'2-
Откуда следует, что
нро*-
н2ро;
1,58
глава 1 Химия, энергия и метаболизм
23
Неорганический фосфат, этерифицированный
органическим спиртом, называют фосфоэфиром.
?
+ R-0-P-CT
0"
Фосфоэфир
-► R0. +
ч
-Го-
0"
Спирт Неорганический
фосфат (Pi)
При гидролизе такого фосфоэфира величина AG0'
составляет около -12,5 кДж · моль-1. Это означает, что
равновесие реакции сильно сдвинуто в сторону
образования продуктов гидролиза и обратная реакция внутри
клеток не происходит.
Точно так же, как фосфоэфиры образуются из
неорганического фосфата и спиртов, из 2 молекул фосфата
можно получить производное фосфорного ангидрида,
которое называют пирофосфатом и обозначают РР/
(пиро- означает огонь; пирофосфат может быть получен
отщеплением воды от Р, при высокой температуре).
Изменение свободной энергии при гидролизе этого
вещества значительно больше (AG0' = -33,5 кДж · моль1),
чем для фосфоэфиров.
? ?
но-р-о-р-сг
0" 0"
Неорганический
пирофосфат (РР/)
Н20
?
-**2Н0-Р-СГ+ Н+
0"
Неорганический
фосфат (Pi)
Столь значительное высвобождение свободной
энергии при гидролизе пирофосфата обусловлено
несколькими факторами.
1. Структура пирофосфата дестабилизирована
электростатическим отталкиванием двух отрицательно
заряженных фосфорильных групп. То же самое можно
представить иначе: при образовании пирофосфатной
связи нужно сблизить одноименно заряженные
остатки, преодолевая их отталкивание.
2. Продукты реакции гидролиза пирофосфатов (в
простейшем случае 2 Pf.) резонансно
стабилизированы, т.е. имеют большее число возможных
резонансных структур, чем структура пирофосфата.
Это происходит потому, что фосфорильные группы
в пирофосфатах конкурируют за электроны мости-
ковых атомов кислорода. Наличие таких электронов
необходимо для образования резонансных структур.
В результате конкуренции то одна, то другая фос-
форильная группа в РР, может резонировать, тогда
как два Pf. могут резонировать одновременно.
Кроме того, в неорганическом фосфате все Р-О связи
усреднены по свойствам, поэтому протон не связан
с каким-то определенным атомом кислорода, что
автоматически приводит к увеличению энтропии.
На приведенной ниже схеме d~ обозначает
частичный отрицательный заряд (d+ означал бы
частичный положительный заряд).
ч
О- р- 0"^
0"
δ-ο^— ρ-τ^οό
<ь-
Резонансная стабилизация фосфата
Все эти факторы способствуют протеканию реакции
гидролиза РР,, равновесие которой сдвинуто в сторону
образования Pf.. Эти рассуждения справедливы по
отношению не только к самому пирофосфату, но и к
любому соединению, содержащему фосфорноангидридную
группу, о которых речь пойдет далее. Отметим, что к
обычным сложным эфирам фосфата представленные
выше рассуждения неприложимы; именно поэтому при
их гидролизе вывобождается гораздо меньше энергии.
Пирофосфаты хотя и главные, но не единственные
биологические макроэргические производные
фосфорной кислоты. Известны еще три типа таких соединений.
К первому относятся смешанные ангидриды
фосфорной и карбоновых кислот, гидролиз которых
характеризуется величиной AG0' около -49 кДж · моль1. Столь
большое значение AG0' объясняется резонансной
стабилизацией обоих продуктов гидролиза: Pf. и карбоксилат-
ного аниона.
р
R-c' О
\ II _
О—Р-0
I
о
Ацетилфосфат
(ангидрид)
Н20
О О
// II _
— R—С +Н0—Р-0 +Н+
V А-
δ 0<->Όδ
I
R
Резонансная стабилизация
Ко второму типу соединений относятся гуанидино-
фосфаты, у которых величина AG0' благодаря
резонансной стабилизации обоих продуктов гидролиза
составляет около -АЪ кДж · моль1.
часть 1 Введение в химические реакции клетки
24
NH2^ /NH—P-0 + H20
I 0"
NH
Гуанидинофосфат
NH2^c/NH2
I
NH
I
R
+ HO—P-0
I
δ+ .
nh2;
δ+
:c>'nh2
:ΐδ+
NH
Резонансная стабилизация
К третьему типу соединений принадлежат енолфос-
фаты, имеющие совсем иное строение. Рассмотрим одно
из таких соединений - фосфоенолпируват На первый
взгляд в нем трудно угадать макроэргическое
соединение, однако при гидролитическом отщеплении
фосфатной группы образуется енольная форма пирувата,
которая спонтанно таутомеризуется в кетоформу При этом
равновесие сдвинуто в сторону образования последней,
благодаря чему суммарный процесс превращения фос-
фоенолпирувата в пируват и фосфат протекает с
выделением энергии AG0' = -61,9 кДж · моль1.
О'
СН2 О
С-
I
ч0"
-Р-
I
О"
Н20
Фосфоенолпируват
(енолфосфат)
СИ,
I
сн2
II
— с-он
I
o*Sr
'(неустойчив)
О
II
но—р—о
I
О"
(Г
(устойчив)
группу», которая далее транспортируется к месту
потребления энергии для совершения работы. Ясно, что такая
группа не существует сама по себе, а является частью
молекулы и перемещается вместе с ней. Возникает
нижеследующий вопрос...
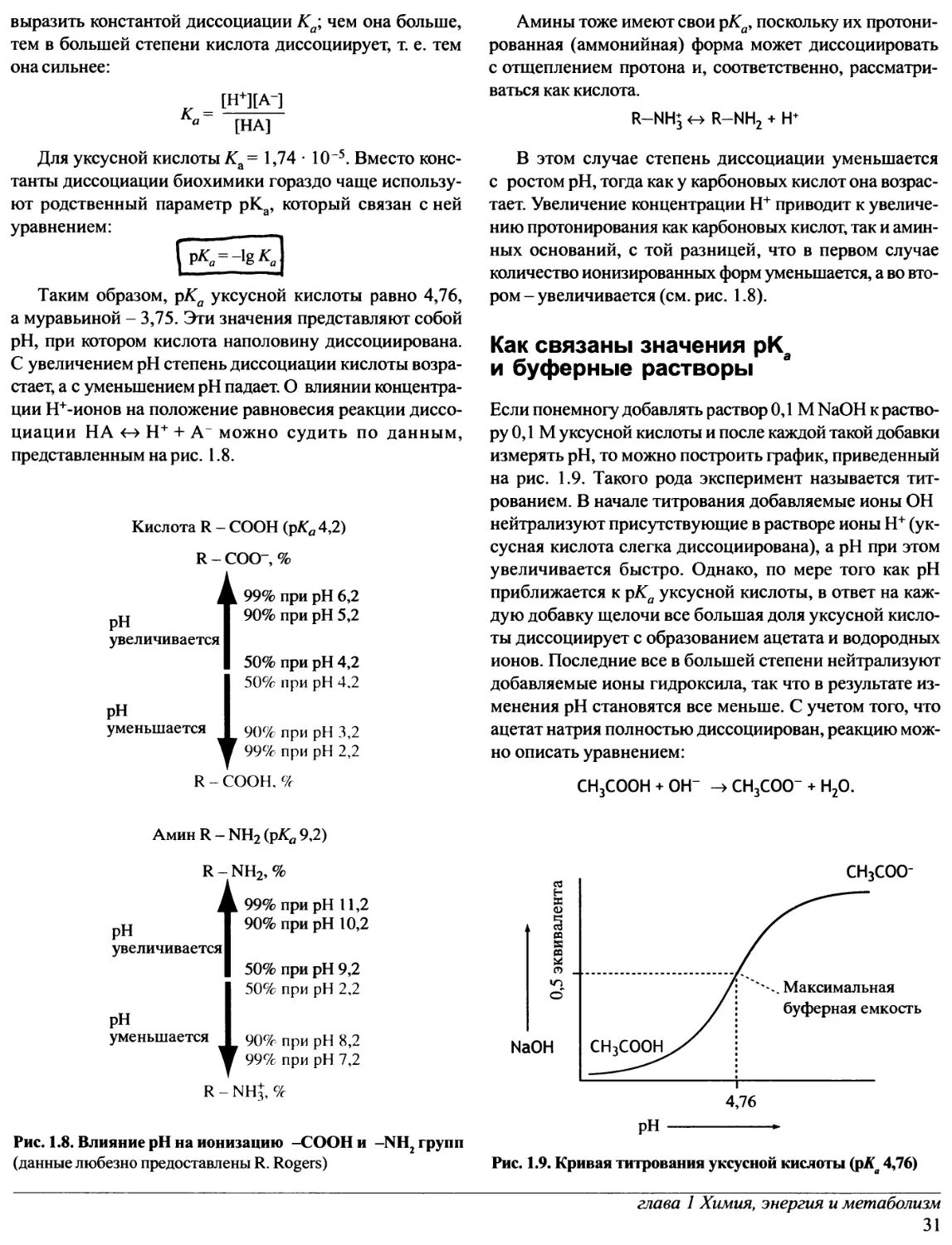
Что в клетке служит переносчиком
фосфорильной группы?
Главным переносчиком фосфорильной группы
является аденозинмонофосфат - AMP, структура которого
изображена на рис. 1.6. AMP, несущий еще одну фосфо-
рильную группу, называют аденозиндифосфатом - ADP.
Если фосфорильных групп три, то соединение
называется аденозинтрифосфатом - АТР. Две концевые фос-
форильные группы в АТР связаны между собой точно
такой же фосфоангидридной связью, как и в пирофос-
фате. И точно так же, как пирофосфат, это соединение
гидролизуется с выделением большой свободной
энергии. Заметим, что этим АТР принципиально отличается
от AMP, который в цикле переноса энергии выступает
лишь как переносчик пирофосфатных остатков.
На рис. 1.6 показано, что гидролиз каждой из двух
концевых фосфатных групп в молекуле АТР характеризуется
величиной AG0', равной около -30,5 кДж · моль1:
следовательно, обе они могут рассматриваться как
высокоэнергетические.
НО
,. Аденин
Λдспочин
Рибоза
Низкоэнергетическая
фосфорильная
группа
Высокоэнергетические
фосфорильные
группы ,,-
А д с н о ч и н м оно φ () с φί» τ
(AMP)
AG0' гидролиза =
=-14,2 кДж-моль-1
Аде ночи ндифосфат
(ADP)
AG0' гидролиза =
=-30,5 кДж-моль-1
АдсночинтрифосфсП
(АТР)
AG0' гидролиза =
=-30,5 кДж-моль-1
В последующих главах этой книги вы еще не раз
будете встречаться с различными производными фосфорной
кислоты и обсуждать свойственные им значения AG0',
однако сейчас мы переходим к наиболее общему и
важному в этой проблеме.
До сих пор мы говорили о присутствии
фосфорильной группы в высокоэнергетических фосфатах. На рис. 1.5
показано, что неорганический фосфат преобразуется
в клетке в «высокоэнергетическую фосфорильную
Рис. 1.6 Аденозин и его фосфорилированные производные
Аденозин (изображен схематически) - это нуклеозид,
состоящий из аденина и рибозы
Окисление пищи сопряжено с превращением ADP
в АТР:
ADP + Р/
Энергия
+ АТР + Н20.
глава 1 Химия, энергия и метаболизм
25
В каждый данный момент любая клетка содержит
небольшое количество АТР, которого ей хватает лишь на
короткое время. Ни АТР, ни ADP, ни AMP, молекулы
которых заряжены, не могут спонтанно диффундировать
через клеточную мембрану (см. главу 3). Поэтому
каждая клетка вынуждена сама вырабатывать АТР,
поддерживая его быстрый кругооборот, включающий гидролиз
АТР до ADP и фосфата и последующий ресинтез АТР из
этих соединений. С учетом изложенного, мы можем
модифицировать схему, представленную на рис. 1.5
(рис. 1.7). Яд цианид в основном блокирует
внутриклеточный ресинтез АТР, и смерть наступает почти
мгновенно из-за того, что энергопотребляющие процессы
останавливаются, как только резерв АТР будет исчерпан.
Шкала
свободной
энергии
1ищевые
молекул
^о2+н2о
о2
Химическая
энергия
Перенос внутри
клеткикместу
выполнения работы
ЛР АТР
Биологическая
работа
(химическая,
\dp+p, adp+p элеюрическая,
Переносвнутри осмотическая,
клеткикместу механическая)
вь ботки энергии
Рис. 1.7. Участие АТР в энергетическом хозяйстве клетки
На универсальности этого рисунка не сказываются отдельные
случаи, когда с выполнением работы сопряжен гидролиз АТР
до AMP
Как АТР подвергается
химическим превращениям?
Предположим, клетке требуется синтезировать
соединение X-Y из двух компонентов, Х-ОН и Y-H, причем для
этого процесса значение AG0' равно 12,5 кДж · моль1.
Прямая реакция Х-ОН + Y-H —» X-Y + Н20 не пойдет,
поскольку она эндоэргонична и равновесие сильно
смещено влево. Ниже показано, как решается эта проблема
путем сопряжения синтеза X-Y с гидролизом АТР.
Сопряженными будем называть те реакции, когда продукт одной
из них является исходным веществом для другой. При этом
речь идет только о системе реакций в чисто химическом
смысле. Все внутриклеточные реакции с участием АТР
катализируются ферментами. Тот факт, что гидролиз
каждой из двух фосфорноангидридных групп сильно экзоэр-
гоничен, не означает, что АТР является нестабильным или
высокореакционноспособным соединением (в виде
белого порошка его можно хранить неопределенно долго
в обычном холодильнике).
Реакция 1: ΧΟΗ + ΑΜΡ-(ΡΧΕ) —^ Χ"® + ΑΜΡ-®.
Реакция 2: X-(P) + YH —► X-Y + Ρ,-.
Сумма 1+2: ΧΟΗ + ΥΗ + ΑΜΡ-(Ρ}-® ►
► Χ-Υ + ΑΜΡ-(Ρ) + Ρ,·.
Результирующее значение AG0' здесь должно быть
арифметической суммой величин AG0',
соответствующих реакциям 1 и 2. Поскольку AG0' для реакции
Х-ОН + YH -^ X-Y + Н20 составляет 12,5 кДж · моль"1,
а для гидролиза АТР (АТР + Н20 -> ADP + Ρ.)
AG0' =-30,5 кДж · моль"1, результирующее значение
AG0'=-18,0 кДж · моль1. Это означает, что суммарный
процесс сильно экзоэргоничен и, следовательно,
протекает вплоть до практически полного завершения
(константа его равновесия ~103, т. е. в равновесных условиях
на 99,9% смещено в сторону X-Y; см. табл. 1.1).
Заметим, что здесь имеет место не прямой гидролиз АТР
до ADP и фосфата. Фосфатная группа сначала
переносится на молекулу одного из реагирующих компонентов
и лишь потом отщепляется в свободном виде.
В клетке реальное значение AG0 для любых
процессов гидролиза АТР до ADP и фосфата гораздо
больше стандартной величины AG0', поскольку
концентрации всех участвующих в реакции веществ
существенно меньше 1М. Если истинные
концентрации известны, свободную энергию можно вычислить
с помощью уравнения:
[Продукты реакции]
AG = AG° + /?77-2,3031g
[Исходные вещества]'
где R - газовая постоянная (8,315 Дж · моль1 · Кг1),
а Т - температура в градусах Кельвина (298 К = 25° С).
Хотя концентрации АТР, ADP и Pf. в различных
клетках неодинаковы, в среднем они близки к ΙΟ3 Μ для АТР
и Р1и 10^ Μ для ADP. Для простоты предположим, что
все эти концентрации равны 1 мМ. Подставляя это
значение в уравнение:
[ADP][Pf]
AG = AG°' + /?77-2,303lg
[АТР]
получаем:
AG = -30,5кДж · моль"1 +
+ (831,5-10-3)(298)2,303 lg [1°~ ][13°~ ] =
= -30,5 - 17,1= -47,6кДж · моль"1.
Описанная выше последовательность реакций
характерна для многих биосинтетических процессов,
сопряженных с расщеплением АТР. Однако при биосинтезе
таких макромолекул, как белки и нуклеиновые кислоты,
часть 1 Введение в химические реакции клетки
26
природа использует более эффективный энергетический
трюк: от АТР отщепляется не одна, а две фосфатные
группы. Благодаря чему отрицательное изменение AG столь
велико, что эти процессы протекают до полного
завершения и совершенно необратимы. Как это достигается,
мы покажем, опять-таки, на примере той же реакции
Χ-ΌΗ + ΥΗ->Χ-Υ + Η20.
Реакция 1: ΧΟΗ + АМР-®~® * Х-АМР + РР,.
Реакция 2: Х-АМР + ΥΗ ► Χ-Υ + AMP.
Сумма 1+2: ΧΟΗ + ΥΗ + АМР-<Р)-(Р) + Н2° ►
► Χ-Υ + AMP + 2Р,-.
Предположим, как и ранее, что для реакции
Х-ОН + ΥΗ -^ Χ-Υ + Н20 изменение AG0' составляет
12,5 кДжмоль1, AG0'для реакции АТР + Н20 -^
—> AMP + РР, равно -32,2 кДж · моль1, а для
гидролиза РР, AG0'равно -33,5 кДж · моль1.
Реакции АТР + Н20 —» AMP + 2Pf. соответствует
AG°=-65,7 кДж · моль1, а итоговому синтезу X-Y,
протекающему сопряженно с этой реакцией, будет отвечать
весьма значительная отрицательная величина
АСР=-65,7 - 12,5=-53,2 кДж · моль"1.
Эффективность суммарной реакции зависит от того,
насколько быстро в клетке расщепляется
неорганический пирофосфат РР,. Его гидролиз катализирует
специальный фермент пирофосфатаза, который можно
назвать движущей силой важнейших процессов
биосинтеза.
НО-Р-О-Р-0-0" ^—► ZHO-P-CT+H4.
I I I
О" О" 0"
Неорганический Неорганический
пирофосфат (РР/) фосфат (Pi)
Как с помощью АТР
совершаются другие виды работ?
Энергия, высвобождающаяся при расщеплении АТР,
помимо участия в химических реакциях обеспечивает
мышечное сокращение, генерацию электрических
сигналов, а также перекачку ионов и молекул против
градиентов их концентраций. Механизмы всех этих процессов,
которые подробнее будут рассмотрены позже,
принципиально сходны в том, что для их осуществления
используется свободная энергия, освобождающаяся при
расщеплении АТР.
Замечание о соотношении между AMP,
ADP и АТР
Выше было сказано о том, что во многих случаях
АТР-зависимый синтез химических соединений в клетке
приводит к образованию не ADP + Р/9 а АМР + РР,.
В то же время АМР, в отличие от ADP, не превращается
в АТР при окислении пищевых веществ (см. рис. 1.6).
Какова же судьба образующегося АМР? Ответ можно найти
в обратимой реакции переноса фосфатного остатка с АТР
на АМР: АМР + АТР <-> 2 ADP. Ее катализирует фермент
АМР-киназа. Киназами называют ферменты,
осуществляющие перенос фосфатного остатка с АТР на молекулу
акцептора. Термин АМР-киназа означает, что акцептором
фосфата является АМР (поскольку другое название АМР -
адениловая кислота; этот же фермент иногда называют
аденилаткиназой). Заметим, что фосфатный остаток при
этом переносится непосредственно от одной молекулы к
другой, а изменение свободной энергии относительно
невелико. Далее вы убедитесь, что такого рода «перетасовка»
энергетических ресурсов широко распространена.
Образующийся в данном случае ADP может быть вовлечен в
синтез АТР, сопряженный с окислением пищевых веществ.
Детальному рассмотрению механизмов, посредством
которых при окислении пищи из ADP и Р, образуется
АТР, будет уделено особое внимание в главах,
посвященных метаболизму. Здесь мы лишь в самом общем виде
затронули вопросы, касающиеся использования энергии
АТР для совершения полезной работы. Конкретные
механизмы утилизации энергии АТР будут подробно
рассмотрены в следующих главах.
А сейчас мы несколько изменим тему, хотя и
продолжим анализ изменений свободной энергии в химических
процессах.
Слабые связи
и изменения свободной энергии
До сих пор речь шла о явлениях, связанных с
образованием и расщеплением ковалентных связей. Теперь пора
перейти к рассмотрению нековалентных связей,
называемых также вторичными, или слабыми (два последних
эпитета не отражают их значимости для жизни).
В реакциях, катализируемых ферментами,
происходит перераспределение связей между атомами, однако
суммарное число связей при этом сохраняется. Когда два
атома соединяются посредством химической связи,
выделяется определенное количество энергии:
X + X -» X—X + энергия,
где X - свободный атом.
глава 1 Химия, энергия и метаболизм
27
Два таких атома соединяются связью, которая ранее
не существовала. Мы не рассматриваем здесь обычные
химические реакции, в ходе которых число ковалентных
связей не увеличивается.
Чтобы обратить рассмотренное превращение и
разорвать химическую связь, нужно затратить столько же
энергии, сколько выделилось при образовании связи:
X—X + энергия —> X + X.
Образование обычной ковалентной связи происходит
с выделением значительной энергии; следовательно,
значительная энергия должна быть затрачена и на ее
расщепление. Такого рода расщепления в биохимических
системах не происходят, что можно проиллюстрировать
на примере кислорода, который существует в виде
молекул 02. Их образованию из атомарного кислорода
отвечает большое значение AG0', около -460 кДж · моль1,
поэтому равновесное содержание атомарного кислорода
исчезающе мало.
В обычных химических реакциях расщепление
одной химической связи сопровождается
одновременным образованием другой ковалентной связи через
переходное состояние. При этом суммарное
изменение энергии оказывается гораздо меньше, чем
необходимо для расщепления ковалентных связей. Однако
нековалентные слабые связи между отдельными
атомами или группами атомов в растворе непрерывно
возникают и исчезают. Они достаточно прочны, чтобы
участвовать в формировании структуры вещества, но
не настолько устойчивы, чтобы исключить
разрушение этой структуры. Иными словами, такие связи
определяют гибкость структуры молекулы. Их
основополагающее значение выявится при рассмотрении
мембран, белков и ДНК. В действительности эти связи
слабы и вторичны не по их значению, а лишь по
энергии их образования (AG0' = -4-^30 кДж ■ моль1),
которая достаточно мала для того, чтобы они легко
разрывались. Известны три типа слабых связей: ионные,
водородные и ван-дер-ваальсово притяжение.
Ионные связи возникают как следствие
электростатического притяжения между достаточно близко
расположенными группами, несущими постоянные по
величине заряды. Типичным примером таких связей
может служить взаимодействие между отрицательно
заряженной карбоксилатной и положительно
заряженной аммонийной группами:
-СОСГ
+NH,
Связи также возникают вследствие
электростатического притяжения между атомами полярных
молекул, однако здесь речь идет о гораздо менее выраженном
разделении зарядов, чем в случае ионных связей. А
именно - об электрическом дипольном моменте. Рассмотрим
в качестве примера распределение зарядов в молекуле
воды.
о-р-сг
0"
^ΟτΡτ-Ό6"
4 К
н+.
Резонансная стабилизация фосфата
Атом кислорода в молекуле воды
электроотрицателен. Это означает, что он оттягивает на себя электроны,
образующие связь более эффективно, чем атомы
водорода. В результате в молекуле воды атом кислорода
приобретает частичный отрицательный заряд, а атомы
водорода - соответствующие частичные положительные
заряды. Это приводит к возникновению между атомами
О и Η соседних молекул воды слабых притяжений,
называемых водородными связями. Каждая молекула
воды может быть связана с 4 другими молекулами,
поскольку атом кислорода участвует в образовании 2
водородных связей, а каждый атом водорода - в одной.
Не только в воде, но и в других молекулах атомы
водорода участвуют в образовании водородных связей, если
они ковалентно присоединены к электроотрицательным
атомам. В биохимических структурах роль последних
чаще всего выполняют атомы кислорода и азота. Кроме
водорода в образовании водородной связи участвует
и его партнер - электроотрицательный акцепторный
атом, обычно также кислород или азот.
Здесь показаны наиболее часто встречающиеся типы
водородных связей.
-0-Н..-0-
-0-Η---Ν-
-Ν-Η---0-
-Ν-Η---Ν-
Водородная связь характеризуется определенной
направленностью. Ее прочность максимальна, когда все
вовлеченные в нее атомы расположены на одной прямой.
О ван-дер-ваальсовых силах говорят при описании
слабых взаимодействий между тесно сближенными
атомами, когда флуктуации электронной плотности в таких
атомах индуцируют образование диполей. Эти силы
неспецифичны и являются единственным типом слабых
связей, возникающих между неполярными молекулами,
которые неспособны образовать ионные и водородные
связи. Ван-дер-ваальсовы силы действуют между любыми
часть 1 Введение в химические реакции клетки
28
двумя атомами, если они сближены настолько, что их
электронные оболочки почти соприкасаются. При этом
притяжение обратно пропорционально расстоянию между
атомами в шестой степени. Отсюда понятна
необходимость тесного сближения атомов. Однако, если
атомы заставить сблизиться еще больше, их электронные
оболочки начнут перекрываться, что приведет к
возникновению сил отталкивания. Таким образом, ван-дер-вааль-
совы силы эффективны лишь в узком диапазоне
межатомных расстояний.
Все три типа слабых связей объединяет то, что они
возникают как короткодействующее притяжение.
Различаются же они своей длиной и энергией образования: AG0'
для ионных связей составляет около 20 кДж · моль1, для
водородных - 12ч-19 кДж · моль-1, а для ван-дер-вааль-
совых - 4^-8 кДжмоль-1. Для сравнения: энергия
образования ковалентной связи составляет сотни кДжмоль1.
Значение слабых связей состоит в том, что именно
они ответственны за внутри- и межмолекулярную
ассоциацию молекул. Здесь необходимо вспомнить о
факторе, который также способствует ассоциации, хотя его
и нельзя отнести к подлинному связыванию: речь идет
о так называемом гидрофобном связывании, или
гидрофобном взаимодействии.
Полярные вещества вроде сахара растворимы в воде,
потому что их молекулы хотя и нарушают водородные
связи между молекулами воды, но и сами образуют с
ними такие связи, что в целом оказывается энергетически
предпочтительным. Соли, например NaCl, растворимы
в воде потому, что вокруг ионов Na+ и С1~ возникают гид-
ратные оболочки, которые взаимодействуют с ионами
сильнее, чем эти ионы между собой. В результате ионы
расходятся, а это, в свою очередь, приводит к
значительному росту энтропии при переходе от кристалла к
раствору. Что произойдет, если попытаться растворить в воде
неполярное вещество, скажем оливковое масло? Его
молекулы не могут образовать водородные связи с
окружающими молекулами воды. Это означает, что у
пограничных с маслом молекул воды некоторая доля потенциально
способных к связыванию атомов останется без
партнеров. Чтобы уменьшить число подобных атомов,
молекулы воды перестраиваются таким образом, что
приобретают упорядоченность большую, чем в обычной
воде (энтропия уменьшается). Это препятствует
растворению в воде масла и других неполярных веществ -
и одновременно вызывает слипание отдельных
неполярных молекул, поскольку при этом уменьшается
совокупная площадь их контакта с молекулами воды. Внешне
это выглядит как подталкивание неполярных молекул друг
к другу. Силы, действующие при этом, принято называть
гидрофобными. Гидрофобные группы присутствуют
и в белках, и в ДНК, и в других внутриклеточных
молекулах и потому играют важную роль в формировании их
структуры. Просто удивительно, сколь многое в живых
клетках определяется необходимостью «спрятать»
гидрофобные группы от воды!
Что вызывает образование и разрыв
слабых связей?
Образование слабых связей происходит с уменьшением
свободной энергии системы и не требует преодоления
сколько-нибудь заметного активационного барьера.
Поэтому такие связи возникают между подходящими и
притом достаточно сближенными атомами совершенно
спонтанно, без участия ферментов. Мы уже упоминали
о соотношении, связывающем константу равновесия
реакции с величиной AG0' (результаты соответствующих
вычислений представлены в табл. 1.1). Из них следует,
что даже в том случае, когда при образовании слабой
связи AG0' составляет всего -5,7 кДж - моль1,
соответствующее равновесие реакций сильно смещено в
сторону связывания. Если же AG°= -11,4 кДж · моль1,
степень связывания достигает 99%. Отсюда видно,
насколько образование слабых связей чувствительно
к величине изменения свободной энергии.
Нельзя забывать, что равновесие - динамический
процесс: отдельные связи постоянно разрываются
и образуются вновь. Что вызывает разрыв связи?
Для этого достаточно передать связанным атомам
такое же количество энергии, какое ранее высвободилось
при образовании данной связи. Эта энергия черпается
из кинетической энергии соседних молекул,
соударяющихся со связанными атомами благодаря тепловому
движению. Даже при физиологических значениях
температуры кинетической энергии наиболее быстрых
молекул вполне хватает, чтобы разорвать слабую связь.
Если слабые связи так легко рвутся,
каково их значение в биохимических
системах?
Возьмем, например, ван-дер-ваальсово притяжение.
Отвечающая ему свободная энергия едва превышает
тепловую энергию молекул в растворе, и соответствующие
связи постоянно образуются и разрываются. Неужели
столь эфемерное взаимодействие может иметь какое-
либо значение? Хотя и в меньшей степени, но это
относится и к другим типам слабых связей.
Как ни парадоксально, именно слабость таких
связей определяет их центральную роль в явлениях жизни.
Их главная особенность состоит в исключительной
глава 1 Химия, энергия и метаболизм
29
специфичности. Взаимное узнавание биологических
молекул происходит с такой исключительной точностью, что
связываются лишь те из них, которые отобраны для этого
эволюцией.
Вот несколько примеров. Фермент избирательно
ускоряет только одну определенную реакцию: прежде всего
потому, что его активный центр связывает только один
или немногие из потенциально способных к
образованию такой связи субстратов. Гормон оказывает строго
определенное действие, поскольку он связывается не с
любым, а только с предназначенным для него белком-
рецептором. Функционирование генов зависит от
специальных белков, которые распознаются и связываются
с определенными участками ДНК. Антитело образует
комплекс только с соответствующим ему антигеном. Этот
список необъятен, и по мере изучения биохимии вы
будете встречать все новые и новые примеры
специфичности. Как же обеспечивается столь высокая
избирательность при межмолекулярных взаимодействиях?
Именно благодаря слабым связям. При этом важно
нижеследующее.
1. Объединение молекул в устойчивый нековалентный
комплекс возможно лишь при одновременном
образовании не одной, а многих слабых связей.
2. Слабые связи требуют тесного сближения
взаимодействующих атомов, а водородные связи, кроме того,
их определенной взаимной ориентации.
Поэтому связываются только те молекулы-партнеры,
которые по своей структуре комплементарны друг
другу. Это правило соблюдается настолько безукоризненно,
что иногда достаточно поменять всего один атом в
молекуле субстрата или активном центре фермента, чтобы они
перестали взаимодействовать.
Наряду со специфичностью важнейшей
особенностью взаимодействий в биологических системах
является их транзиторный характер. Связывание в
активном центре фермента не может быть очень прочным.
И функционирование гена, и контроль за ним также
предполагают легкую обратимость ответственных за это
взаимодействий. Это жестко ограничивает диапазон
допустимых для таких взаимодействий отрицательных
значений AG0' (т. е. констант равновесия): они должны
быть достаточно велики, чтобы слабые связи легко
образовывались, и вместе с тем достаточно малы, чтобы эти
связи легко нарушались, обеспечивая в целом
необходимую степень обратимости взаимодействия. В случае же
взаимодействия типа «антиген - антитело» большое
число слабых связей приводит к практически необратимому
связыванию. Итак, слабые связи позволяют точно
дозировать межмолекулярные взаимодействия и лежат в
основе биологической избирательности. Если таких связей
часть 1 Введение в химические реакции клетки
30
предусмотрено немного, то взаимодействие вполне
обратимо, в противном же случае образуются прочные
меж- или внутримолекулярные взаимодействия. Роль
слабых связей в биохимических системах станет выглядеть
еще более значимой при дальнейшем знакомстве
с мембранами, структурой белков, строением и
функционированием генов.
Таким образом, все изложенное выше можно
рассматривать как предисловие к двум последующим
главам о структуре белков и биологических мембранах. Обе
эти темы необходимы для понимания метаболизма,
которому посвящена первая половина книги.
Буферные растворы
и значения рКа
Биохимику совершенно необходимо понимать, что
такое буферные растворы и что такое рКа. Удобно
выделить эту тему в отдельный раздел, дабы она не
затерялась в основном тексте.
В живых клетках рН поддерживается в пределах
7,2 -7,4. Лишь в некоторых особых ситуациях рН
выходит за эти пределы, например в полости желудка,
где секретируется НС1, или в лизосомах, куда
специально закачиваются протоны. Постоянство рН
соблюдается несмотря на то, что при метаболических процессах
происходит, скажем, образование молочной и ацетоук-
сусной кислот или имеет место характерное для крови
превращение С02 в Н2СОэ.
В значительной мере постоянство рН обеспечивается
буферным действием слабых кислот. Термин слабая
означает, что в данных условиях такая кислота лишь
частично диссоциирована.
Карбоновая кислота диссоциирует, высвобождая
протон:
R-COOH <-> R-COCT+H+
Донор и акцептор протона в этом и подобных ему
уравнениях называют сопряженными кислотой и
основанием. В общем виде уравнение диссоциации кислот
можно записать так:
НА<->Н++А~
Кислоты различаются по склонности к диссоциации.
Говоря о том, что одна кислота более сильная, а другая -
более слабая, имеют в виду, что первая из них
диссоциирует легче. Так, в растворе 0,1 Μ муравьиной кислоты
(НСООН) рН меньше, чем в растворе уксусной кислоты
(СН3СООН): последняя является более слабой.
Способность кислоты к диссоциации можно количественно
выразить константой диссоциации Ка; чем она больше,
тем в большей степени кислота диссоциирует, т. е. тем
она сильнее:
К =
[Н+][А~]
[НА]
Для уксусной кислоты Ка= 1,74 · 10~5. Вместо
константы диссоциации биохимики гораздо чаще
используют родственный параметр рКа, который связан с ней
уравнением:
| рк„=-18л:а|
Таким образом, рКа уксусной кислоты равно 4,76,
а муравьиной - 3,75. Эти значения представляют собой
рН, при котором кислота наполовину диссоциирована.
С увеличением рН степень диссоциации кислоты
возрастает, а с уменьшением рН падает. О влиянии
концентрации Н+-ионов на положение равновесия реакции
диссоциации НА <-> Н+ + А~ можно судить по данным,
представленным на рис. 1.8.
Кислота R - СООН (рКа 4,2)
R - COO" %
рН
увеличивается
рН
уменьшается
99% при рН 6,2
90% при рН 5,2
50% при рН 4,2
50% при рН 4.2
90% при рН 3,2
99% при рН 2,2
Амины тоже имеют свои рКа, поскольку их протони-
рованная (аммонийная) форма может диссоциировать
с отщеплением протона и, соответственно,
рассматриваться как кислота.
R-NH^ <-> R-NH2 + Н+
В этом случае степень диссоциации уменьшается
с ростом рН, тогда как у карбоновых кислот она
возрастает. Увеличение концентрации Н+ приводит к
увеличению протонирования как карбоновых кислот, так и амин-
ных оснований, с той разницей, что в первом случае
количество ионизированных форм уменьшается, а во
втором - увеличивается (см. рис. 1.8).
Как связаны значения рКа
и буферные растворы
Если понемногу добавлять раствор 0,1 Μ NaOH к
раствору 0,1 Μ уксусной кислоты и после каждой такой добавки
измерять рН, то можно построить график, приведенный
на рис. 1.9. Такого рода эксперимент называется
титрованием. В начале титрования добавляемые ионы ОН
нейтрализуют присутствующие в растворе ионы Н+
(уксусная кислота слегка диссоциирована), а рН при этом
увеличивается быстро. Однако, по мере того как рН
приближается к рКа уксусной кислоты, в ответ на
каждую добавку щелочи все большая доля уксусной
кислоты диссоциирует с образованием ацетата и водородных
ионов. Последние все в большей степени нейтрализуют
добавляемые ионы гидроксила, так что в результате
изменения рН становятся все меньше. С учетом того, что
ацетат натрия полностью диссоциирован, реакцию
можно описать уравнением:
R-COOH, %
СН3СООН + ОН-
СН3СОСГ + Н20.
Амин R-NH2 (ρ/ς 9,2)
R - NH2, %
рН
увеличивается
рН
уменьшается
99% при рН 11,2
90% при рН 10,2
50% при рН 9,2
50% при рН 2,2
90% при рН 8,2
99% при рН 7,2
R - NH3, %
Рис. 1.8. Влияние рН на ионизацию -СООН и -NH2 групп
(данные любезно предоставлены R. Rogers)
NaOH
СН3СОО-
Максимальная
буферная емкость
4,76
рН
Рис. 1.9. Кривая титрования уксусной кислоты (рКа 4,76)
глава 1 Химия, энергия и метаболизм
31
Точно так же по другую сторону pKfl по шкале рН
добавление в среду ионов Н+ вызывает относительно
небольшие изменения рН благодаря следующей реакции:
СН3СОСГ + Н+ -> СН3СООН.
В этом и заключается буферный эффект уксусной
кислоты. Он максимален вблизи рКа, где уксусная
кислота и ацетат присутствуют в эквимолярных количествах.
Биохимики пользуются таким эмпирическим
правилом: буфер эффективен в интервале единицы рН, центр
которого приходится на величину рКа. Для вычисления
рН раствора, содержащего смесь сопряженной пары
кислота-основание, можно использовать уравнение Гендер-
сона-Хассельбаха:
pH = p*e + lg
[Акцептор протона]
[Донор протона]
которое обычно записывают в виде:
[Соль]
pH = pKa + \g
[Кислота]
Оно применимо для любой смеси уксусной кислоты
и ацетата, что позволяет подобрать ацетатный буферный
раствор с нужным значением рН. Для раствора,
содержащего 0,1 Μ уксусной кислоты и 0,1 Μ ацетата натрия,
рН = 4,76 + lg ψΐ = 4,76 + 0 = 4,76.
Если же раствор содержит 0,1 Μ уксусной кислоты
и 0,2 Μ ацетата натрия, то
рН = 4,76 + lg ί^ = 4,76 + lg2 = 4,76 + 0,3 = 5,06.
Предположим, что к раствору 0,1 Μ уксусной
кислоты и 0,1Μ ацетата натрия добавлена щелочь до
концентрации 0,05 М. Тогда половина уксусной кислоты будет
превращена в ацетат. После этого величина рН составит:
рН = 4,76 + lg ^13 = 4,76 + lg3 = 4,76 + 0,48 = 5,24.
Смесь ацетат-уксусная кислота не обладает
буферным действием при физиологических значениях рН.
Однако в организмах есть вещества, рКа которых позволяет
им служить эффективными буферами при рН~7.
Важнейшими среди таких веществ являются фосфатный ион
и его производные. При диссоциации фосфорной
кислоты может отщепляться 3 протона, причем отщеплению
второго (Н2Р04 <-> НРО \+ Н+) соответствует рКа 6,86.
Поэтому фосфат служит превосходным
внутриклеточным буфером.
Буфером могут служить также имидазольные
радикалы в остатках аминокислоты гистидина, входящей
в состав белков. Протонированию этих радикалов
отвечает рА^~6.
H3N—CH-COO" H3N—CH-COO"
I I
сн2 ^ сн2
+ н+
ΝΗ^Ν+Η
ΝΗ^Ν
Вы можете наблюдать роль буфера, поставив
простейший опыт. Наполните пробирку дистиллированной водой,
доведя ее рН до 7 с помощью разбавленной щелочи
(дистиллированная вода обычно слегка кислая из-за
растворенного в ней С02). Если к воде прибавить несколько капель
разбавленной соляной кислоты, ее рН резко уменьшится.
Проделайте то же с децимолярным раствором фосфата
натрия: рН раствора почти не изменится. Вот это и есть
буферное действие фосфата.
Таким образом, буферное действие веществ с рКа~1
предохраняет клетки и межтканевые жидкости от
значительных изменений рН.
Дополнительная литература
Термодинамика
Newsholme Ε. Α., Leach A. R. II Biochemistry for medical
sciences. N. Y.: Wiley, 1983. P. 10-45.
(Изложение законов термодинамики применительно к
метаболизму.)
Nelsestuen G. L. IIA partial remedy for the non-relationship
between reversibility of a reaction in the cell and the value
of AG0'. Biochem. Education. 1989. Vol.17. P. 190-192.
(Обсуждается, почему реакция, катализируемая альдо-
лазой, обратима, хотя для нее характерно большое
значительное отрицательное значение AG0'.)
Ферментативный катализ
Steitz Т. A.t Shoham Μ., Bennett W. S. Jr. II Structural
dynamics of yeast hexokinase during catalysis. London: Phil.
Trans. Roy. Soc. Series B. 1981. Vol. 293. P. 43-52.
(Обсуждаются конформационные изменения в гексо-
киназе при связывании этим ферментом субстрата —
глюкозы.)
Srere P. А. II Why are enzymes so big? Trends Biochem. Sci.
1984. Vol. 9. P. 387-390.
(Вынужденно спекулятивная, но весьма интересная
статья.)
часть 1 Введение в химические реакции клетки
32
Koshland D. Ε. Jr. // Evolution of catalytic function. Cold
Spring Harbor Symp. Quant. Biol. 1987. Vol. 52.
P. 1-7.
(Четкое и лаконичное описание роли активных центров
и конформационных изменений фермента в
ферментативном катализе.)
Kraut J. II How do enzymes work? Science. 1988. Vol. 242,
P. 533-40.
(Ферментативный катализ описан исходя из принципа
стабилизации переходного состояния. В качестве
примеров использованы механизм действия цинк-содержащих
протеаз и получение каталитически активных антител,
специфичных к соединениям, являющимся
структурными аналогами истинных субстратов.)
Понятие макроэргического фосфата
Lipmann ΕII Metabolic generation and utilisation of
phosphate bond energy. Adv. Enzymol. 1941. Vol. 1. P. 99-162.
(Знаменитый обзор, в котором впервые изложена
концепция АТР-зависимых клеточных процессов. Обзор,
разумеется, устарел, но концепция эта жива и здравствует!)
Вопросы к главе 1
1. Представьте себе мужчину весом 70 кг, питание
которого позволяет удовлетворить его энергетические
затраты - 10 000 кДж в день. Допустим, что свободная
энергия, которую он получает с пищей, используется
для образования АТР из ADP и ?i с
эффективностью 50%. В клетке AG превращения ADP + Р,
приблизительно равно 55 кДж · моль1. Вычислите,
сколько весит АТР, синтезируемый этим человеком
за один день (количество выразить в граммах динат-
риевой соли АТР с молекулярной массой 551).
2. Стандартная величина изменения свободной энергии
гидролиза АТР до ADP и фосфата составляет
-0,5 кДж · моль1. Объясните, почему в предыдущем
вопросе значение 55 кДж · моль1 считается
количеством свободной энергии, необходимой для синтеза 1
моля АТР из ADP и Р, в клетке.
3. Почему AG0' гидролиза АТР в ADP + Р, и ADP
в AMP + Pf. составляет -30,5 кДж · моль1, тогда
как для гидролиза AMP до аденозина и Pf. величина
AG0' = -14,2 кДж · моль-1? В чем причина такого
большого различия?
4. Какую информацию можно извлечь из кривой
температурной зависимости ферментативной активности?
5. Объясните, почему:
а) бензол практически нерастворим в воде;
б) полярные молекулы глюкозы растворимы в воде;
в) NaCl растворим в воде.
6. Катаболизм пищи в клетке устроен так, что
обеспечивает превращение ADP в АТР, но не может
непосредственно превратить AMP в АТР. Между тем
многие ферментные системы превращают АТР
в AMP. Как организм справляется с накоплением
AMP?
7. Фермент катализирует синтез соединения X-Y из ΧΟΗ
и ΥΗ, сопряженный с расщеплением АТР до AMP и
неорганического пирофосфата. Стандартное
изменение свободной энергии при реакции
ΧΟΗ + ΥΗ -^Χ-Υ + Н20 составляет 10 кДж · моль1.
Вычислите AG0' ферментативной реакции в клетке, а
также при использовании очищенного фермента.
Объясните полученные результаты. (В расчетах
рекомендуем использовать для гидролиза АТР до AMP
и РР, величину AG0'= -33,2 кДж · моль1, а для
гидролиза РР/ AG0' = -33,4 кДж · моль-1.)
8. Какие типы слабых связей используются в
биологических системах и каковы их энергетические
характеристики? Почему для образования этих связей не
нужен ферментативный катализ? Каково значение
слабых связей?
9. Диссоциация фосфорной кислоты описывается
тремя рКа: 2,2; 7,2;12,3.
а) Какая из ионных форм доминирует при рН 0, рН 4,
рН9ирН 14?
б) Вычислите рН водного раствора эквимолярной
смеси NaH2P04 и ^НР04.
в) Какую аминокислоту из числа природных вы
выбрали бы в качестве буфера для поддержания
физиологического значения рН?
10. Окисление глюкозы до С02 и воды протекает с
выделением очень большого количества энергии. Как
объяснить тот факт, что глюкоза на воздухе вполне
устойчива к окислению?
11. Как ферменты катализируют химические реакции?
глава 1 Химия, энергия и метаболизм
33
глава
2
Структура белков
С полным правом можно утверждать, что нет другого
вещества с такими удивительными свойствами, как
белок. Ничего нельзя понять в биохимии, не оценив
всеобъемлющей роли белков в жизни. Если клетке нужно
совершить какую-либо работу, почти всегда ее
выполняет определенный белок. Жизнь зависит от тысяч
белков, которые устроены так, что их молекулы с
невероятной точностью распознают другие молекулы и
взаимодействуют с ними. Химические реакции в клетке
протекают благодаря связыванию ферментов с
субстратами, а скорость ферментативных реакций во многих
случаях определяется избирательным связыванием
аллостерических регуляторов со специфическими
участками фермента. Работа такой сложнейшей структуры,
как мышца, основана на белок-белковых
взаимодействиях. Действие генов управляется белками, которые
взаимодействуют с определенными участками ДНК. Точно
так же в основе гормонального контроля лежит
избирательное взаимодействие гормонов с белковыми
рецепторами. Распознавание молекул белками происходит при
трансмембранном транспорте, в том числе и при
генерации нервных импульсов. Белок-белковые
взаимодействия обеспечивают иммунитет путем образования
комплексов антиген-антитело. И таким избирательным
взаимодействиям нет конца.
Данная глава посвящена химическому строению
белков - гигантских молекул, которые выполняют
широкий спектр функций.
Первичная структура белков
Белки представляют собой полипептидные цепи. Их
молекулы могут состоять из нескольких цепей, но пока
ограничимся одноцепочечными белками. Полипептидная цепь
состоит из большого числа соединенных между собой
аминокислот. Природа использует в белках 20
аминокислот; если обозначить их буквами, то полипептидную
цепь можно представить как слово из сотен таких букв.
Самая длинная из известных полипептидных цепей
содержит около 5000 аминокислот, однако большинство
белковых цепей имеет менее 2000 аминокислотных
звеньев. В принципе аминокислотный «алфавит»
допускает существование практически бесконечного множества
различных белков - «слов».
Это означает, что эволюционный отбор не ограничен
числом белковых структур. Вероятно, именно поэтому
белки были избраны носителями столь большого числа
биологических функций. Будь число возможных цепей
ограниченным, из них нельзя было бы отобрать в ходе
эволюции оптимальные, поскольку шанс добиться
нужного результата при случайной сборке цепи крайне мал.
Ниже показана структура α-аминокислоты,
которая в нейтральных водных растворах находится в виде
цвиттер-иона.
H2N—СН—СООН
R
α-Аминокислота
+hUN-
-СН-
I
R
СОО"
Цвиттер-ион
Каждая из аминокислот, за исключением пролина,
содержит общий фрагмент H2N-CH-COOH, так что
различаются они только боковой группой R,
прикрепленной к α-углеродному атому. Все аминокислоты в белках
имеют L-конфигурацию (кроме глицина, в молекуле
которого нет асимметрического атома).
Две аминокислоты можно соединить вместе, если
отщепить от них молекулу воды (в клетке это достигается
не столь простым способом). При этом образуется ди-
пептид, его строение показано ниже (для простоты -
в неионизированном состоянии). Связь -СО-NH-
называют пептидной связью.
H2N—CH-COOH H2N—CH-COOH
R R'
Две аминокислоты
Пептидная связь
H2N—CH-C—NH-CH-COOH
R'
Дипептид
В дипептиде есть концевые амино- и карбоксильная
группы; поэтому к нему можно присоединять всё новые
аминокислоты, переходя таким образом к полипептиду.
О
О
HUN—CH-C—NH-CH—С-
R1
I
R2
I
R"
О
NH-CH-C4NH-CH-COOH
-1/7
I
R3
Пол и пептид
часть 2 Структура белков и мембран
36
Хотя четкого разграничения не существует,
короткие цепи принято называть пептидами, или олигопеп-
тидами (олиго- значит несколько), а под полипептидом
понимают обычно цепь из восьмидесяти и более
аминокислот. Белки могут состоять из нескольких
полипептидов, которые в этом случае называют субъединицами.
Субъединицы могут быть связаны в мультимерном
белке как исключительно слабыми связями, так и
посредством прочных ковалентных непептидных связей.
Немного терминологии. Участок цепи, на котором
находится концевая NH3+-rpynna, называют N-концевым,
а противоположный ему - С-концевым.
Собственно цепь без аминокислотных радикалов именуют
полипептидным скелетом.
(-NH-CH-CO-NH-CH-CO-NH-CH-CO-)
I I I
Аминокислоту, включенную в белок, именуют
аминокислотным остатком, а аминокислотные радикалы
R - боковыми цепями аминокислот, боковыми цепями
белка или просто боковыми цепями.
Порядок расположения аминокислот в полипептиде
называют аминокислотной последовательностью.
Процедуру установления этой последовательности
называют секвенированием. Секвенирование играет
очень важную роль в химии белка. Аминокислотная
последовательность составляет первичную структуру
белка. Сэнгер в Кембридже первым определил
аминокислотную последовательность белка (инсулина)
и был за это удостоен в 1958 г. Нобелевской премии.
Впоследствии Эдману, используя чисто химический
подход, удалось автоматизировать эту работу.
Современным автоматам для этого требуется всего 0,001 мкг
белка. Кроме того, есть приборы - аминокислотные
анализаторы для определения аминокислотного состава
белка. Последний при этом предварительно подвергают
полному кислотному гидролизу.
Что такое нативный.белок?
Изображение на бумаге первичной структуры белка
создает впечатление, что его молекула имеет вытянутую,
нитевидную структуру. Это заблуждение, что можно
доказать на примере яичного альбумина - бесцветного
водорастворимого белка из куриного яйца. Его можно
очистить и получить в виде острых кристаллов. Способность
к кристаллизации определяется компактной трехмерной
структурой молекул. Для процесса кристаллизации
необходимо, чтобы все молекулы имели одинаковую
форму. Полипептидная цепь в глобулярных белках в
естественном состоянии свернута в компактную сферическую
структуру - глобулу; в отличие от фибриллярных
белков, длинные цепи которых вытянуты вдоль одной оси,
они имеют небольшое осевое отношение. Нет четкого
определения, что такое глобулярные белки, но очевидно,
что их трехмерная структура - плотная укладка
полипептидной цепи - обычно нарушается при нагревании.
Белок с исходной, природной укладкой цепи называют
нативным, белок с развернутой, беспорядочной
укладкой цепи - денатурированным, а превращение натив-
ного белка в денатурированный - денатурацией.
Когда яйцо варят, прозрачный яичный белок
сначала мутнеет, а затем превращается в нерастворимую
белую массу. Что это такое? Отдельные, определенным
образом сложенные белковые цепи при нагревании
раскручиваются и беспорядочно и необратимо
переплетаются между собой (рис. 2.1). Это явление
свидетельствует о том, что укладка цепи в нативном белке
осуществляется в значительной мере благодаря слабым
нековалентным связям, поскольку ковалентные связи не
разорвались бы при умеренном нагревании. Сама же по-
липептидная цепь в этих условиях остается интактной.
Тепловая денатурация - общее свойство белков.
Биологическая активность белков, например
ферментов, почти всегда нарушается при нагревании (хотя
известны немногие термостабильные белки). По этой
причине нагревание смертельно для клетки.
Тепловая
денатурация
Рис. 2.1. Денатурация яичного альбумина (гипотетическая
схема)
глава 2 Структура белков
37
Теперь, получив представление о первичной
структуре белков, мы можем перейти к обсуждению
трехмерной укладки полипептидных цепей.
Каковы основные факторы,
определяющие трехмерную
структуру белка?
Как уже отмечалось, большинство белков свернуто
в глобулы. Такие глобулы устойчивы в водных
системах благодаря тому, что их полярные группы находятся
на поверхности в контакте с водой, а неполярные
обращены вглубь молекулы таким образом, что их
соприкосновение с водой сведено к минимуму. Если какие-
либо группы в глубине глобулы потенциально могут
образовать ионные и водородные связи, но реально
лишены партнеров, - это дестабилизирует упаковку.
Таким образом, ионизованная группа, которая находится
в гидрофобном окружении, будет дестабилизировать
молекулу белка.
На первый взгляд, проблемы образования
функционально-активного глобулярного белка могут
показаться неразрешимыми. Ведь сотни аминокислотных
остатков должны быть уложены в плотно упакованную
глобулярную структуру с полярными группами на
поверхности и гидрофобными внутри. Более того, в
случае ферментов снаружи должны еще располагаться
карманы, идеально соответствующие по форме молекулам
субстратов, а также аллостерическим регуляторам
активности белков (см. главу 12).
Теперь вы понимаете, почему так медленно - в
масштабе миллионов лет - действует эволюция, главной
задачей которой, если вдуматься, как раз и является
конструирование новых белков. Бесконечно мал шанс
случайно набрести на такую аминокислотную
последовательность, которая не только удовлетворила бы условиям
устойчивой упаковки цепи, но и обеспечила выполнение
функциональных задач. Поэтому не следует удивляться,
встретив белки с различной функцией, но сходные по
структуре настолько, что может навести на мысль об их
происхождении от общего предка или друг от друга.
Похоже, что эволюция, столкнувшись с необходимостью
решить определенную задачу, предпочитает не
конструировать для этого белки de novo, а приспособить уже хорошо
отлаженные структуры, адаптируя их для новых целей.
Прежде чем перейти к укладке цепей в белках, нам
следует побольше узнать о строении тех 20
аминокислот, которые являются их строительными блоками.
Структура 20 белковых аминокислот
Изучение структуры 20 различных аминокислот будет
более познавательным, если помнить, для чего
предназначены их боковые группы. Именно различия в форме,
размерах и полярности позволяют аминокислотам
служить теми строительными блоками, которые
использует эволюция, чтобы удовлетворить многочисленные
и жесткие требования к структуре белков. Нетрудно
представить, как это происходит: вот здесь нужна
маленькая гидрофобная группа, чтобы заполнить
полость возле соседней группы, тут нужна сильная
полярная группа, а там более слабая. Когда в игре участвуют
20 разных игроков, у великого тренера - эволюции-
есть возможность гибкого конструирования.
В разных организмах было обнаружено множество
аминокислот, не входящих в состав белков (так
называемые небелковые аминокислоты), однако все известные
организмы для строительства своих белков используют
одни и те же 20 аминокислот. Ф. Крик назвал их
«магической двадцаткой». Только они шифруются генетическим
кодом. (Если это вам ни о чем не говорит, подождите до
следующих глав.) Начнем с алифатических гидрофобных
цепей, представленных в порядке увеличения их размера.
+H3N— CH-COO"
I
Η
Глицин
В глицине роль боковой группы играет атом
водорода. Это самая маленькая аминокислота; ее остаток в белке
не имеет ярко выраженных гидрофобных или
гидрофильных свойств. Далее следуют алифатические неполярные
боковые цепи по мере увеличения их гидрофобности,
причем последние три цепи являются разветвленными.
+Η3Ν—СН—СОО"
I
сн,
Алании
*H3N — CH-COO"
I
сн.
сн, сн
Лейцин
+H3N—СН—СОО"
I
СН3 ^СН3
Валин
+H3N—CH-COO"
I
-™
СН; СН3
СН-з
Изолейцин
Особняком среди гидрофобных алифатических
аминокислот стоит метионин. Не потому, что в его боковую
часть 2 Структура белков и мембран
38
цепь входит атом серы, а из-за того, что его концевая
метильная группа играет важную роль в метаболизме.
+H3N—CH-COO"
I
сн2
сн2
5
I
СН3
Метионин
Далее мы переходим к действительно крупным
боковым цепям ароматических аминокислот:
H3N—СН—COO" +H3N— СН—COO" +H3N — СН—СОО
+H3N-
Фенилаланин
Триптофан
Заметим, что гидроксильная группа в тирозине
может участвовать в образовании водородных связей ,
поэтому тирозин нельзя однозначно классифицировать
как гидрофобную аминокислоту.
Гидрофильные аминокислоты
Все ионизированные группы гидрофильны, а потому
к гидрофильным аминокислотам, безусловно,
относятся и те, которые содержат в боковой цепи
карбоксильную или аминогруппы. Обе эти группы при
физиологических значениях рН ионизированы. Аспарагиновая
и глутаминовая кислоты - кислые аминокислоты, лизин
и аргинин - сильно основные, а гистидин - слабо
основная аминокислота. Кольцевую структуру в молекуле
гистидина называют имидазольным кольцом.
-сн-соо-
I
сн2
сн2
сн2
NH
I
NH2 NH2
Аргинин
+H3N—CH-COO'
I
CHp
II *CH
HC^NH
Гистидин
Обе кислые аминокислоты - аспарагиновая и
глутаминовая - в белках представлены и своими амидами -
аспарагином и глутамином.
+H3N—CH-COO"
I
сн2
Αχ
NH2 О
Аспарагин
+H3N— CH-COO"
I
сн2
сн2
NH2 О
Глутамин
К гидрофильным относятся также гидроксилсодер-
жащие аминокислоты: серии и треонин.
+H3N—CH-COO"
I
СН2ОН
Серии
+H3N — CH-COO"
I
снон
I
СН3
Треонин
Аминокислоты, используемые
для особых целей
Цистеин похож на серии, но содержит тиольную
группу -SH вместо гидроксильной -ОН. Его специфическая
роль в белках двояка: благодаря цистеину в активные
центры белков могут быть введены тиольные группы;
кроме того, два остатка цистеина в белках могут быть
соединены ковалентной связью -S-S-.
+H?N—CH-COO"
сн2
СОО"
Аспарагиновая
кислота
+H3N— CH-COO
I
сн2
сн2
СОО"
Глутаминовая
кислота
+H3N
—сн-
1
сн2
сн2
сн2
1
+NH3
Лизин
СОО"
Пролин примечателен тем, что его остаток вызывает
излом пептидной цепи. В отличие от других аминокислот,
свободный пролин содержит не амино-, а иминогруппу.
+H3N-CH-COO" . +H2N CH-COO"
ι II
сн2 сн2,сн2
sh СНз
Цистеин Пролин
глава 2 Структура белков
39
Ионизация аминокислот
Как уже упоминалось, свободные аминокислоты в
водных растворах находятся в виде цвиттер-ионов, в
которых α-амино- и α-карбоксильные группы
ионизированы, поскольку у первых рКа 8-10, а у вторых рКа 2.
В пептидной цепи все эти диссоциирующие группы
блокированы (кроме концевых), поскольку участвуют
в образовании пептидных связей. Поэтому ионный
статус белков практически полностью определяется
диссоциацией групп в боковых цепях аспарагиновой и глута-
миновой кислот, лизина, аргинина и гистидина.
Ниже показаны заряженные аминокислотные
остатки в составе пептидной цепи:
—СО—NH—СН—СО—NH—СН— Полипептидная цепь
сн2
соо
Боковая цепь
глутаминовой
кислоты
сн2
^н2
сн2
CH2-NH3+
Боковая цепь
лизина
Карбоксильные группы в боковых цепях
аспарагиновой и глутаминовой кислот имеют рКа~4 и при рН 7,4
практически полностью диссоциированы; при этом
их боковые группы заряжены отрицательно.
Аминогруппы в боковых цепях основных аминокислот лизина
(рКа 10,5) и аргинина (рКа 12,5) при физиологических
значениях рН также полностью ионизированы (NH3+).
Третья основная аминокислота- гистидин с имида-
зольным кольцом в качестве боковой группы имеет
рКа 6,04, достаточно близкое к физиологическим
значениям рН. Благодаря этому имидазол часто входит
в активный центр ферментов, которые катализируют
реакции, связанные с переносом ионов водорода. В ходе
этих реакций имидазольный остаток - в зависимости
от своего ионного состояния - может выступать как
донор или акцептор протона.
Распределение заряженных остатков аминокислот
в полипептидной цепи сильно влияет на ее конфор-
мацию, ибо близкорасположенные одноименные
заряды, как известно, отталкиваются, а разноименные -
притягиваются. Для регуляции ферментативной
активности в клетке часто используется фосфорилирование,
в ходе которого в молекулу белка вводится сильный
отрицательный заряд, вызывающий изменение ее кон-
формации.
Уровни структурной организации
белка: от первичной структуры
к четвертичной
Последовательность аминокислот, ковалентно связанных
в полипептидную цепь, как вы уже знаете, называется
первичной структурой белка (рис. 2.2, а). Сама по себе
первичная структура ничего не говорит о том, как
полипептидная цепь уложена в трехмерном пространстве
Т"
R1
R2
"Т"
R3
R4
Т"
R5
Τ
R6 R7 и др.
Эта структура ниже представлена линией
Отдельные участки полипептидной цепи
существуют в виде α-спирали, β-складчатого
слоя или беспорядочного клубка
\\\
α-Спираль β-Складчатые слои Беспорядочный
клубок или петля
Вторичные структуры уложены в компактный
глобулярный белок
Молекулу такого белка
можно представить
таким образом:
Отдельные белковые молекулы (субъединицы),
объединенные нековалентным взаимодействием
в мультимерный белок
> /
Рис. 2.2. Схематическое изображение первичной(а),
вторичной (б), третичной(в) и четвертичной (г) структуры белков
часть 2 Структура белков и мембран
40
Полипептидная цепь принимает специфическую кон-
формацию, известную как вторичная структура
белка (см. рис. 2.2, б), которая, в свою очередь,
укладывается в компактное образование, именуемое третичной
структурой (см. рис. 2.2, в). Такое образование,
сформированное первичной, вторичной и третичной
структурами, может представлять собой либо
самостоятельный белок, либо же в качестве мономера (субьединицы)
ассоциироваться с такими же или другими мономерами,
образуя сложный мультимерный белок. Под
четвертичной структурой понимают расположение в
пространстве взаимодействующих между собой
субъединиц, образованных отдельными полипептидными
цепями белка (см. рис. 2.2, г).
Приняв эту терминологию, мы далее более
подробно рассмотрим различные уровни организации белковой
структуры.
Вторичная структура белков
Здесь речь пойдет об организации не столько боковых
цепей, сколько собственно полипептидного скелета.
Мы уже говорили о том, что в водорастворимых
глобулярных белках полярные боковые группы должны быть
по возможности расположены снаружи глобулы, а
гидрофобные - внутри нее. Но как быть с полипептидным
скелетом? Как его ни скручивай в компактную форму, все
равно часть скелета окажется погруженной в
центральную, т. е. гидрофобную область. Проблема здесь в том,
что этот скелет, т. е. собственно пептидная цепь,
содержит множество С=0- и N-H- групп пептидных связей,
каждая из которых потенциально способна участвовать
в образовании водородных связей. До тех пор, пока это
потенциальное связывание не произойдет (с
выделением свободной энергии), структура будет неустойчивой.
Кто же может послужить партнерами этим
многочисленным группам? Ответ простой: аналогичные группы из
той же или соседней полипептидной цепи.
Еще раз подчеркнем, что речь пока не идет об
упаковке боковых аминокислотных групп, которая
приобретает значение главным образом на уровне третичной
структуры. Пока мы ограничиваемся обсуждением
проблемы, как удовлетворить потребность С=0- и N-H-
групп полипептидной цепи в партнерах для образования
водородных связей. Существуют два главных типа
структур, позволяющих это осуществить: α-спираль, в
которую цепь свертывается, как шнур от телефонной
трубки, и складчатая β-структура, в которой бок о бок
уложены вытянутые участки одной или нескольких цепей.
Обе эти структуры весьма стабильны; они встречаются
на периферии белковых глобул (где расположены
гидрофильные боковые группы аминокислот) и в их
внутренней области (где расположены гидрофобные
боковые группы).
α-Спираль
Термин α-спираль был введен Л. Полингом, открывшим
укладку пептидной цепи в виде правосторонней
спирали в белке α-кератине. Для L-аминокислот
правосторонняя спираль устойчивей левосторонней. Вы
можете составить представление о правосторонней
спирали, вообразив траекторию точки на краю головки
завертываемого шурупа; форма такой спирали
приведена на рис. 2.3, а.
Естественно предположить, что в подобной спирали
на один виток приходится целое число аминокислотных
остатков. От этой заманчивой идеи пришлось, однако,
отказаться, когда Полинг показал, что в действительности
Рис. 2.3. Схематическое изображение укладки
полипептидной цепи в виде α-спирали
а - Примерное расположение водородных связей (штриховые
линии) между С=0- и N-H-группами (боковые группы не
показаны); б - вид с торца α-спирали. Проекции боковых
групп R ориентированы произвольно, а сами они размещены
вдоль цепи на равных расстояниях друг от друга (3,6 остатка
на виток, или через 100° на проекции 360°/100° - 3,6)
глава 2 Структура белков
41
на один виток α-спирали приходится 3,6
аминокислотных остатков. Это означает, что группа С=0- одной
пептидной связи образует водородную связь с группой
N-H другой пептидной связи, отстоящей от первой на-
четыре аминокислотных остатка. И С=0-, и N-H- связи
направлены параллельно оси спирали и попарно
противостоят друг другу; такое расположение оптимально для
образования водородной связи и, следовательно, для
стабилизации α-спирали. В поперечном сечении α-спираль
выглядит диском, от которого наружу смотрят боковые
цепи аминокислот (рис. 2.3, б). Ван-дер-ваальсовы
радиусы атомов таковы, что внутри спирали нет пустого
пространства; это способствует устойчивости
α-спирали. Не вся полипептидная цепь в глобулярном белке
спирализована. В среднем отдельные спиральные
участки включают 10 аминокислотных остатков, но в
разных белках длина спиралей может сильно отличаться
от этой величины.
Аминокислоты различаются по частоте
встречаемости спиральных участксв. Особую роль здесь играет про-
лин, остаток которого служит своего рода терминатором
для α-спиралей. Во всех других случаях полипептидная
цепь может свободно вращаться вокруг двух простых
связей на каждый аминокислотный остаток. Заметим,
что свободное вращение относительно самой пептидной
связи CO-NH не происходит, так как благодаря
электронному сопряжению она по своим свойствам близка
к двойной, представляя собой гибрид двух структур:
а) где связь между атомами углерода и кислорода
двойная; б) где эта связь одинарная.
Η
//
О
С—N
С —
— С Η
\ /
C=N+
/ \
0~ С-
Пептидная группа с участием N-атома пролина не
содержит водорода, необходимого для образования
водородной связи, а главное, в этом месте поворотная
изомерия ограничена, и пептидная цепь не может принять
конформацию, соответствующую α-спирали.
R—СН-
0
II
сн2/
сн—с-
I
сн2
сн2
-NH—
Иминный атом
азота у пролина не способен
к образованию водородной связи
Прежде чем обсудить, какую роль α-спирали играют
в структуре белков, следует познакомиться с
альтернативным типом упорядочения полипептидных цепей -
β-складчатым слоем. Обычно структура белка
представляет собой комбинацию обоих типов укладки, которые
занимают разные участки полипептидной цепи.
β-Складчатый слой
Это тоже стабильная структура, в которой полярные
группы полипептидной связи попарно соединены
водородными связями, что придает ей стабильность даже
в гидрофобной центральной части белковой глобулы.
Принцип организации здесь предельно прост.
Полипептидная цепь находится в растянутом состоянии (или
β-форме - по названию белка β-кератина, в котором она
впервые была обнаружена), а ее С=0- и N-H- группы
связаны водородными связями с такими же группами
соседней, параллельно ориентированной
полипептидной цепи (рис. 2.4). Обе цепи могут быть независимыми
или представлять фрагменты одной, общей для них
цепи.
Когда такую структуру образуют несколько
параллельных цепей, термин слой (англ. sheet) становится
вполне оправданным. Складчатым же (англ. pleated)
Η О
и > и
■ ХК /Nk /C^ JLH
Η
Ν
Η
I
С СН
О "О
НО Η
ι
>ιι /"\ ^С. ХН ^NL
Ν" X СН ^Ν" "^
I
н
"С
II
о
tH
.CH^/NU
II
н о
Участок параллельного ρ-складчатого слоя
Η
ι
Η
>L СН X >L
СН ^Г "NT XH" ^C'
0
Η
Η
о
Ν" ^C CH ^hT
I II I
но н
о
Η
■сн Λ
ιι
о
Участок антипараллельного β-складчатого слоя
Рис. 2.4. Структура β-складчатого слоя
а - Водородные связи между однонаправленными
полипептидными цепями в параллельном β-складчатом слое. Боковые
группы R, прикрепленные к остаткам -СН-, расположены
выше и ниже плоскости бумаги; б - водородные связи между
противоположно направленными полипептидными цепями
в антипараллельном β-складчатом слое. Такой слой может
образоваться в пределах одной полипептидной цепи,
уложенной в складки
часть 2 Структура белков и мембран
42
его называют потому, что α-углеродные атомы
аминокислотных остатков расположены попеременно по обе
стороны центральной плоскости слоя. Образующие
складчатый слой полипептидные цепи могут быть
направлены в одном и том же или в противоположном
направлениях: в первом случае складчатый слой
называют параллельным, а во втором - антипараллельным.
Антипараллельная β-структура обычно возникает,
когда пептидная цепь поворачивает вспять, образуя так
называемую шпильку. Место поворота называют β-изги-
бом (англ. β-turn).
Беспорядочный клубок,
или петли полипептидной цепи
Беспорядочный клубок на самом деле вовсе не
беспорядочный и не клубок. Так стали называть участки
полипептидной цепи, которые по конформации нельзя
отнести ни к α-спирали, ни к β-складчатому слою. Более
правильный термин - соединительные петли. Их
структура в основном определяется взаимодействиями
боковых цепей входящих в них аминокислотных
остатков; в молекулах любого конкретного белка она
фиксирована, а не беспорядочна, как можно ожидать
из названия. Да и слово «клубок» здесь не слишком
уместно, поскольку речь идет не о произвольной
конформации. В соединительных петлях не все пептидные С=0-
и N-H- группы могут участвовать в образовании
водородных связей, поэтому участки полипептидной цепи
чаще находятся на поверхности белковой глобулы, в
области ее контакта с водой.
Третичная структура белков
Итак, эволюция располагает 20 аминокислотами для кон-
струирования белков. Последовательность
аминокислотных звеньев в полипептидной цепи характерна и
постоянна для каждого белка, определяя его первичную
структуру. Полипептидная цепь может свертываться в
виде α-спирали или β-складчатого слоя, либо находится
в менее упорядоченных петлях, причем только в
α-спирали и β-структуре пептидные связи участвуют в
образовании водородных связей. Все эти три типа укладки
распределены вдоль полипептидной цепи, занимая
отдельные ее участки и составляя вторичную структуру
белка. Дальнейшая укладка вторичных структур в
компактную структуру глобулярного белка носит название
третичной структуры.
Чтобы упростить изображения белковых молекул,
используют условные обозначения вторичных структур.
Так, α-спираль изображают в виде цилиндра (иногда
с вписанной в него спиралью) или в виде свернутой
в спираль ленты (рис. 2.5, а), а участки цепи, входящие
в β-складчатые слои, - в виде стрел, указывающих
направления хода пептидных цепей (рис. 2.5, б). Иногда
их изображают несколько скрученными, чтобы отразить
таким образом реальное слабое правостороннее
скручивание цепи в β-структуре. В обоих случаях условное
обозначение относится к некоторому фиксированному
участку полипептидной цепи. В категорию петель
попадают участки цепи, которые разделяют спирали и
складчатые слои: их обозначают простой линией.
Рис. 2.5. Символы, применяемые для изображения
участков α-спирали (а) и р-складчатых листков (б)
Справа показано их правостороннее искривление,
характерное для антипараллельных слоев. Промежуточные петли и
неупорядоченные участки цепи изображаются простой
линией
Как строятся белки из α-спиралей,
β-складчатых слоев и соединительных
петель?
В принципе для построения белков может быть
использована любая комбинация спиралей, складчатых слоев и
петель, однако при этом не должно возникать затруднений,
глава 2 Структура белков
43
χ Χ
■^
<fl
Χ Ν
>
^ _ 0
V.
ν
\Ν . ν
Ν *-
12 3 4 5 6 7 8
Рис. 2.6. Строение различных белков
а - Миоглобин, в центре которого расположен гем, выделенный красным цветом; б - стафиллококковая нуклеаза; в - триозо-
фосфатизомераза (под ней показано расположение в цепи α-спиралей и β-складчатых слоев); г - пируваткиназа
часть 2 Структура белков и мембран
44
связанных с упаковкой боковых цепей аминокислот
и обусловленных их притяжением или отталкиванием.
Кроме того, на упаковку белковой молекулы может
влиять расположение на ее поверхности боковых полярных
групп аминокислотных остатков, образующих
соединительную петлю. В целом должны выполняться
следующие требования: усилено образование водородных
связей и сведены к минимуму контакты гидрофобных групп
с полярными группами и водой. Этим требованиям,
однако, могут удовлетворить лишь немногие
аминокислотные последовательности из бесчисленного множества
возможных.
Трехмерное строение довольно большого числа
белков было установлено на основании данных о
дифракции рентгеновских лучей. При этом выяснилось,
что некоторые структурные мотивы предпочтительнее,
поэтому число основных конструкций белков может
быть ограниченным.
Трехмерные белковые структуры вряд ли интересны
для биохимика, ими не занимающегося, однако их
знание часто бывает необходимо, например, при
обсуждении бихимических механизмов какого-нибудь
процесса. Некоторые из этих структур представлены на рис. 2.6.
Миоглобин (см. рис. 2.6, а) содержит только
α-спирали, соединенные петлями, и гем, погруженный в
глубокую щель. В молекуле стафилококковой нуклеа-
зы - фермента, гидролизующего нуклеиновые кислоты
(см. рис. 2.6, б), видна комбинация антипараллельных
β-складчатых слоев (1-4) и трех α-спиралей. Еще один
структурный мотив-так называемая α/β-бочка,
центральная часть которой образована β-участками (1-4),
расположенными наподобие клепок в деревянной бочке
(за исключением того, что они наклонены), а
периферия заполнена α-спиралями. Этот мотив иллюстрирует
структура триозофосфатизомеразы (см. рис. 2.6, в). Еще
один мотив упаковки, образованный чередующимися
спиралями и участками β-структуры, прослеживается
в структуре пируваткиназы (см. рис. 2.6, г).
Какие силы поддерживают третичную
структуру?
Мы уже говорили о том, что элементы вторичной
структуры - α-спирали и β-складчатые слои -
стабилизируются водородными связями. Теперь обсудим, что же
скрепляет эти элементы в единую третичную структуру.
Разумеется, определенный вклад вносят ионные и
водородные связи между боковыми цепями аминокислотных
остатков, однако главную роль играют гидрофобные
силы, которые определяют локализацию гидрофобных
цепей в центральной части молекулы, уменьшая, таким
образом, площадь их контакта с водой и увеличивая
ван-дер-ваальсово взаимодействие. В большинстве
глобулярных белков одних слабых взаимодействий
достаточно для поддержания третичной структуры.
Какое отношение к третичной структуре
имеют ковалентные S—S- связи?
Ранее упоминалось об особых функциях остатков цис-
теина. Одна из них состоит в том, что тиольные группы
этих остатков участвуют в образовании активных
центров ферментов, например гликолитического фермента -
глицеральдегид-3-фосфатдегидрогеназы (см. с. 112).
Однако у цистеина есть и другая функция. Третичная
структура белков своим существованием обязана в
основном слабым взаимодействиям между боковыми цепями.
Этого достаточно для защиты белков от повреждающих
воздействий внутри клетки, однако внеклеточным
белкам - инсулину в крови, пищеварительным
ферментам и т. п. - нужна дополнительная защита от более
жесткого внеклеточного окружения. И это достигается
попарным связыванием пространственно сближенных
остатков цистеина с образованием ковалентных
дисульфидных связей, которые действуют подобно
стальным скобам, скрепляющим бревенчатые
конструкции. Даже небольшого числа таких связей достаточно
для того, чтобы весьма эффективно стабилизировать
белковую структуру; например, в инсулине их всего три.
Дисульфидные связи, или S-S-мостики, как их иначе
называют, не разрушаются при умеренном нагревании,
а содержащие их белки часто отличаются большей
термоустойчивостью.
Простой опыт помогает понять, как образуются
дисульфидные связи. Если оставить на воздухе
нейтральный раствор цистеина, то через несколько часов
поверхность жидкости покроется слоем белых нерастворимых
кристаллов. Протекающую при этом химическую
реакцию можно представить в виде уравнения:
СОО" СОО"
ι ι
+H3N —CH +H3N —CH
3 ι ι
сн2 сн2
| г ., |
s:h h:s
ι I
Цистеин Цистеин
|о2
f
СОО " СОО"
I I
+H3N—CH +H3N—CH
I I + H202
CH2 CH2
I I
s s
Цистин
глава 2 Структура белков
45
Точно так же происходит образование дисульфидных
связей между остатками цистеина в белках.
Предельно наглядно стабилизирующая роль S-S-moc-
тиков прослеживается на примере кератина - основного
белка волос. Пучки кератиновых полипептидов
прошиты множеством S-S-мостиков, придающих волосу
жесткость. При так называемом химическом
перманенте S-S-связи разрушают восстановителем, придают
волосам иную конфигурацию, а затем вновь замыкают
S-S-связи, используя подходящий окислитель.
Количество S-S-связей после этих процедур может остаться
таким же, как прежде, однако теперь они образуются
между теми остатками цистеина, которые оказались
сближенными при механическом изменении конфигурации
волоса.
Четвертичная структура белков
Третичная структура завершает описание строения
молекулы белка. Есть, однако, довольно много белков,
молекулы которых представляют собой комплексы,
образованные из нескольких белковых молекул, соединенных
нековалентными связями. Такие комплексы называют
олигомерными, мультимерными или
субъединичными белками. Их состав и стехиометрия постоянны.
Это доказывает, что объединяющиеся белковые
субъединицы «узнают» друг друга благодаря присутствию
на их поверхности комплементарных по форме участков.
К числу таких олигомерных белков относится, например,
гемоглобин (см. главу 27) или аллостерически
регулируемые ферменты (см. с. 158). Укладку субъединиц
в функционально активном белковом комплексе
называют четвертичной структурой белка (см. рис. 2.2, г).
Мембранные белки
До сих пор речь шла о глобулярных белках, у которых
структура глобулы обеспечивает водорастворимость
молекулы в целом. Многие же белки, пронизывающие
биологические мембраны, устроены иначе: они состоят из
двух экспонированных на поверхности мембраны
«водорастворимых» частей, которые соединены спиральным
тяжем, расположенным внутри мембраны (см. с. 61).
Недостаток такой конструкции очевиден: контакты
полярных групп белка с внутренним углеводородным
слоем мембраны делают молекулу нестабильной.
Белковые конъюгаты
Очень многие белки - не сложнее вышеописанных.
Для нормального функционирования им достаточно
правильно уложенной полипептидной цепи и, в крайнем
случае, таких кофакторов, как ионы металлов или не-
ковалентно связанные коферменты. Однако некоторые
дополнительные компоненты могут быть присоединены
к белкам ковалентно. Их называют простетическими
группами, их активный комплекс - холоферментом,
а его белковую часть - апоферментом. Примерами
таких структур могут служить цитохромы, содержащие
в качестве простетической группы гем, либо дегидроге-
назы, содержащие флавинадениндинуклеотид (FAD).
Белки, ковалентно связанные с углеводными
компонентами, относят к гликопротеинам. К ним, в
частности, принадлежат многие белки плазматических
мембран, к молекулам которых с внешней стороны
прикреплены олигосахариды (см. с. 61). Их остатки
замещают атом водорода в гидроксильной группе серина или
треонина (О-гликозиды), а также амидный протон в
боковой цепи аспарагина (N-гликозиды). Строение таких
N-гликозидов схематически показано ниже:
цепь
1
NH
1 >
сн—сн2
1
NH
1 _
к
Боковая цепь
аспарагина
О
II Η
— С—N
с
\
г
Остаток
"" сахара
Гликопротеинами являются секреторные белки,
многие белки крови. Назначение углеводных компонентов
в гликопротеинах не всегда понятно. Иногда они,
по-видимому, обеспечивают стабильность белка, в других
случаях - его долговечность. Так, разрушение углеводных
компонентов белков сыворотки крови или эритроцитар-
ных мембран служит сигналом для клеток печени к
поглощению и уничтожению соответствующих белков.
Напротив, гликозилирование белков защищает их от
действия протеиназ. В некоторых случаях углеводные
компоненты используются для распознавания, поскольку
разнообразные комбинации моносахаридов в
углеводных цепях позволяют создавать уникальные «метки».
Такие «метки» используются, например, аппаратом Голь-
джи при сортировке белков (см. главу 22).
В случае гликопротеинов муцина (слизи),
защищающих ткани от повреждения, О-гликозиды
способствуют формированию растянутой конформации
полипептидной цепи, которая необходима для поддержания
сеточной структуры муцинов даже в очень
разбавленных растворах. Поддержание белковой цепи в
растянутом виде, по-видимому, является функцией О-глико-
зидов и в других случаях. У рецепторов липопротеинов
часть 2 Структура белков и мембран
46
низкой плотности в молекулах можно различить
область, ответственную за связь с мембраной, и
собственно рецепторный домен, вынесенный за ее пределы. Обе
эти части рецептора соединены длинным пептидным тя-
жем, покрытым множеством О-гликозидных остатков
(рис. 2.7). Существует обширное семейство протеогли-
канов, в которых углеводные компоненты богаты ами-
носахарами, сульфосахарами и карбоксильными
производными Сахаров. Считается, что эти вещества
выполняют важную роль в формировании межклеточного
матрикса, однако эта проблема лежит вне рамок данной
книги
Глобулярная часть белка
.. Гликозилированная часть белка
Ю нм Гликокаликс
Плазмати* кая
ь брана
Рис. 2.7. Строение белка-рецептора липопротеинов низкой
плотности (ЛПНП)
Гликозилированный участок полипептидной цепи
представляет собой тяж, поддерживающий функционально активную
белковую глобулу на расстоянии более 10 нм от поверхности
плазматической мембраны
Белковые модули, или домены
У белков, образованных одной полипептидной цепью,
нативная структура обычно представляет собой
компактное образование, каждая часть которого не может
существовать сама по себе, сохраняя структуру, присущую
ей в исходной глобуле. Но так бывает далеко не всегда,
особенно если белок содержит более 200
аминокислотных остатков. В трехмерной структуре больших белков
иногда обнаруживается не одна, а несколько в той или
иной степени независимо образованных компактных
областей, соединенных малоструктурированными
полипептидными участками. Такое впечатление, что эти
компактные области, если бы их удалось выделить по
отдельности в нативном виде, сохранили бы свою упаковку
неизменной. Действительно, в некоторых случаях так
и получалось. На основании этого было введено
понятие белковых модулей, или доменов, под которыми
понимают фрагменты полипептидной цепи, сходные
по своим свойствам с самостоятельными глобулярными
белками. Считается, что домену всегда должен
соответствовать непрерывный отрезок полипептида, поэтому
исключается ситуация, когда цепь выходит из
компактного домена, а потом, как-то извернувшись,
возвращается в него обратно. Домен должен быть автономен.
Здесь уместна аналогия с отдельными частями
симфонии: они являются законченными произведениями
и могут исполняться по отдельности, но, тем не менее,
подчинены общему замыслу, и только в его рамках
проясняется их истинное значение и взаимосвязь. Что
реально представляют собой домены, видно на рис. 2.6, г, где
представлена структура фермента пируваткиназы, в
которой легко обнаруживаются три компактные области.
Чем интересны домены?
Доменная структура часто ассоциируется с
возможностью представить функциональную активность белка как
совокупность отдельных элементарных процессов.
Пожалуй, наиболее ярким примером здесь может служить
фермент млекопитающих - синтаза жирных кислот,
единственная полипептидная цепь которой содержит все
необходимое для катализа семи реакций. Поскольку у
бактерий каждая из этих реакций катализируется
отдельным ферментом, то вполне возможно, что домены син-
тазы некогда объединились в один белок в результате
слияния генов. Многие ферменты, выполняющие
сходные функции, связывают по меньшей мере два
субстрата. Типичным примером служат никотинамидаденинди-
нуклеотиддегидрогеназы, или (NAD+) -дегидрогеназы,
катализирующие реакцию:
AH2+NAD+<->A+NADH + H+
Все они связывают NAD+, но окисляемые
субстраты для них различны.
Оказалось, что в молекулах дегидрогеназ есть два
домена, один из них связывает NAD+ и сходен по строению
у всех ферментов этого семейства, тогда как другой
связывает окисляемые субстраты АН2 и отличается по
структуре у разных дегидрогеназ. Эти данные можно
истолковать двояко. Можно предположить, что сущес-
вует уникальная белковая структура, способная
эффективно связывать NAD+, и в ходе эволюции каждый
раз отыскивается единственно верное решение (это
называют конвергентной эволюцией). Альтернативная
гипотеза состоит в том, что однажды найденную удачную
глава 2 Структура белков
47
конструкцию NAD+-CBA3biBaioiijero модуля эволюция
использует вновь и вновь, занимаясь чем-то вроде
тасования готовых модулей.
Идея модульной конструкции ферментов и других
белков допускает частое появление и быструю
эволюцию новых функциональных белков. Это чем-то
напоминает сборку электронного устройства путем перебора
готовых универсальных микросхем разного назначения,
вставляемых в общую материнскую плату. Поскольку
речь идет о микросхемах, а не о триодах или
сопротивлениях, вполне возможно получить разумно действующую
схему. Точно так же самые разные функции могут
возникнуть у новых белков, которые образуются в
результате ассоциации разных доменов из уже существующих
молекул.
Все это может показаться чистой фантазией,
основанной на сходстве между родственными по функции
белками, тем более что существует иное толкование
конвергентной эволюции. В главе 21 будет рассказано
о том, что домены иногда кодируются отдельными
фрагментами генов, именуемыми экзонами.
Это создает некую материальную основу для
размышлений о «перетасовке» экзонов, а вместе с ними и
доменов, как о возможном механизме эволюции белков.
Во всяком случае, подобный механизм мог бы
действовать куда быстрее, чем случайные точечные мутации.
Белки волос
и соединительных тканей
Удивительно, что белки составляют основу таких
нерастворимых в воде и прочных материалов, как рога,
копыта, шерсть, кожа и сухожилия. К подобным белкам
предъявляются различные биологические требования.
Волос - это всего лишь длинное, достаточно прочное
и нерастворимое волокно. Его основой является
структурный белок - (Х-кератин. Сухожилия, связывающие
мышцы с костями, должны быть прочнее стали и
лишены эластичности. Эти свойства обеспечивает другой
структурный белок - коллаген. Совсем иные
механические свойства должны быть у кожи, стенок артерий
или у легочных альвеол. Здесь требуется не только
прочность, но также эластичность и упругость, т. е.
способность обратимо изменять форму под воздействием
нагрузки. Неудивительно, что ответственный за эти
свойства белок называется эластин.
Далее мы рассмотрим структуру кератина, коллагена
и эластина. Общей особенностью этих белков является
участие ковалентных непептидных связей в формировании
часть 2 Структура белков и мембран
48
их пространственной структуры. Вспомним, что у
обычных белков главный вклад в стабилизацию трехмерной
структуры вносят слабые взаимодействия. Однако их
явно недостаточно для образования свойственных этим
белкам уникальных структур.
α-Кератины волос, шерсти, рогов
и копыт
Кератины волос и шерсти образуют так называемые
промежуточные филаменты. Они состоят из длинных
полипептидных цепей с крупными доменами,
образованными α-спиралями и содержащими повторяющиеся
последовательности из семи аминокислотных остатков
(гептапептиды). Так выглядит центральная часть
полипептидной цепи, а домены на ее концах не обладают
какой-либо предпочтительной регулярной структурой,
и она может различаться у разных кератинов. Две
одинаково направленных цепи кератина образуют
суперспираль, в которой остатки неполярных аминокислот
защищены от воды, будучи обращенными внутрь. Такая
структура дополнительно стабилизируется
многочисленными дисульфидными связями, образованными
остатками цистеина соседних цепей. Суперспиральные
димеры, в свою очередь, объединяются с образованием
тетрамеров, подобных четырехжильному канату. Есть
данные, свидетельствующие о том, что в
промежуточных филаментах тетрамеры могут агрегировать,
образуя октамерные структуры.
Структура коллагенов
Коллаген образуется вне клеток из секретируемого
ими белка - проколлагена, который превращается
в собственно коллаген в результате воздействия
соответствующих ферментов. Молекула проколлагена
представляет собой тройную суперспираль, образованную тремя
скрученными вместе спирализованными полипептидами
(рис. 2.8).
Превращения проколлагена начинаются с
отщепления концевых пептидов. После этого белок, называемый
теперь уже тропоколлагеном, упаковывается в колла-
геновые волокна. Каждый из трех полипептидов в
молекуле тропоколлагена находится в виде левосторонней
спирали (напомним, что обычная для белков α-спираль -
правосторонняя). Примерно треть аминокислотных
остатков в тропоколлагене представлена пролином, а
каждый третий остаток - глицином.
В ходе образования коллагена многие остатки про-
лина и лизина гидроксилируются, превращаясь
соответственно в гидроксипролин и гидроксилизин. Заметим,
что этих аминокислот нет в «магической двадцатке»,
и они оказываются включенными в белок не в ходе его
матричного синтеза, а в результате химического
посттрансляционного превращения входящих в его состав
аминокислот. Гидроксилирование пролина требует в
качестве кофактора аскорбиновую кислоту (витамин С),
которая нужна для поддержания в восстановленном
состоянии иона Fe2+ в активном центре фермента про-
лил-гидроксилазы.Вот почему при недостатке
витамина С нарушается образование соединительных тканей,
это вызывает заболевание - цингу
-N
I I
СН2,СН2
СН2
О
п о2
С Η С Аскорбиновая кислота , "
О
II
-СН—С —
Остаток пролина
в полипептиде
.- .. ,,,: сн2,сн2
чсн
I
он
Остаток гидроксипролина
в полипептиде
(на самом деле гидроксилирование протекает сложнее
и в нем участвует а-кетоглуторат)
Конструкция коллагеновой суперспирали -
прекрасный пример того, как эффективно использует эволюция
возможности белковой инженерии. Три спирально
навитые друг на друга молекулы тропоколлагена ковалентно
связаны между собой, образуя очень прочную структуру.
Такая ассоциация невозможна в обычной белковой
спирали, поскольку ей препятствовали бы объемные
боковые цепи. В коллагене спирали более вытянуты - на один
виток приходится три остатка. Поскольку каждый
третий из них - глицин, у которого боковой цепи нет,
спирали в этих точках максимально приближены друг
к другу. Дополнительная стабилизация осуществляется
благодаря участию гидроксилированных остатков
лизина и пролина в образовании водородных связей.
Молекулы тропоколлагена содержат около 1000
аминокислотных остатков. Они собираются в коллагеновые
фибриллы, стыкуясь «голова к хвосту» (см. рис. 2.8, а).
Пустоты в этой структуре при необходимости могут
служить местом первоначального отложения кристаллов
гидроксиапатита Са5(ОН)(Р04)3, играющего важную
гУ
Молекула
тропоколлагена
**··· Поперечные
сшивки
Коллагеновая
фибрилла
Одна из левосторонних
спиралей, входящих в
правостороннюю тройную
спираль тропоколлагена
Часть молекулы Часть коллагеновой Строение
тропоколлагена фибриллы сухожилия
Полипептидные цепи
с=о с=о
ι ι
ΝΗ ΝΗ
I I
CH-(CH2)4-NH-(CH2)4-CH
I г I
С = О С = О
I / I
Сшивка между двумя боковыми
цепями лизиновых остатков
Рис. 2.8. Модели фибриллярных белков
а - Укладка фибрилл в коллагеновых волокнах
(цветом выделены сшивки между остатками лизина,
которые есть и в тройной спирали); б - структура
сшивок между близлежащими лизиновыми
остатками. Структура разных типов коллагена зависит
от выполняемых ими функций
глава 2 Структура белков
49
роль в минерализации костей. Для приобретения
необходимых механических свойств, например в
сухожилиях, коллаген подвергается ферментативной
модификации. При этом в концевых частях тропоколлаге-
новых цепей ковалентно .сшиваются остатки лизина
(см. рис. 2.8, б). Таким образом, сухожилия
представляют собой пучки параллельно ориентированных фибрилл
зрелого (т. е. прошедшего все этапы
посттрансляционной модификации) коллагена. В отличие от
сухожилий, в коже коллагеновые фибриллы образуют подобие
неупорядоченной двумерной сетки. Известно несколько
типов коллагенов, различающихся структурой
полипептидных цепей. Эти различия определяются как
содержанием гидроксипролина и гидроксилизина, так
и степенью их гликозилирования. Генетически
обусловленные нарушения созревания коллагена приводят к
тяжелым наследственным заболеваниям - коллагенозам.
Структура эластина
Эластин по своему строению совершенно отличен
от коллагена или α-кератина. Он содержит обычные
α-спирали, но благодаря некоторым особенностям
строения молекулы обладает упругостью большей, чем у
резины. Эластин образует поперечно-сшитую сеть,
которая своими необычными механическими свойствами
обязана уникальному способу ковалентного связывания
боковых цепей лизина: четыре сближенных лизиновых
остатка формируют так называемую десмозиновую
структуру (рис. 2.9), объединяющую в один узел четыре
участка пептидных цепей.
Тайна укладки белка
Полипептидные
цепи
Рис. 2.9. Десмозиновая сшивка между четырьмя
полипептидными цепями эластина. Такие сшивки возникают в
результате ферментативной модификации четырех
лизиновых остатков
некоторые из этих конформаций могут быть метаста-
бильными, и их переход в правильную конформацию
был бы сопряжен с преодолением значительного
энергетического барьера. Природа сумела решить эту
сложнейшую проблему.
Дополнительная литература
Как белки укладываются во вторичную и третичную
структуру? Это тема главы 21, однако здесь стоит
подчеркнуть, что аминокислотная последовательность лишь
определяет, какая трехмерная структура возможна
для полипептидной цепи, но отнюдь не диктует, каким
образом возникает эта структура. Последнее - одна из
величайших нерешенных проблем современной биологии.
Известно, что «плохая» последовательность
препятствует образованию «хорошей» структуры, но пока
неизвестно, как «хорошая» последовательность приводит к
образованию «хорошей» структуры.
В процессе биосинтеза белка в клетке укладка
полипептидных цепей в глобулы занимает время порядка
нескольких минут. Если бы при такой укладке
случайным образом перебирались все мыслимые конформаций
цепи, это потребовало бы миллионы лет. Более того,
Branden С, Tooze J. Introduction to protein structure.
Garland Publ., 1991.
(Иллюстрированное изложение всех аспектов
структуры белка, включающее рассмотрение основных
особенностей строения мембранных белков. Сверх того,
описаны ДНК-связывающие белки, антитела и рецепторы.)
Размер белка и структура
Chothia С. Principles that determine the structure of
proteins // Annu. Rev. Biochem. 1984. Vol. 53. P. 537-572.
(Превосходное введение в проблему структуры белка.)
Goodsell D. S. Inside a living cell // Trends Biochem. Sci.
1991. Vol. 16. P. 203-206.
(Предпринята попытка описать распределение
биологических молекул в клетках Е. coli)
часть 2 Структура белков и мембран
50
Goodsell D. S. Soluble proteins: size, shape and function //
Trends Biochem. Sci. 1993. Vol. 18. P. 65-68.
(Обзор, посвященный структуре белков - их размеру
и форме. Почему они такие большие? Почему они олиго-
мерны?)
Белковые модули или домены
Doolittle R. Ε The multiplicity of domains in proteins //
Annu. Rev. Biochem. 1995. Vol. 64. P. 287-314.
(Обсуждение доменной структуры белков и ее роли в
эволюции - перетасовка доменов, экзоны и интроны.)
Белки волос и соединительной ткани
Francis Μ. J. О., Duksin D. Heritable disorders of
collagen metabolism // Trends Biochem. Sci. 1983. Vol. 8.
P. 231-234.
(Сравнительно простое изложение очень сложной
проблемы, имеющей важное значение для медицины.)
Hulmes D. J. S. The collagen superfamily - diverse
structures and assemblies // Essays in Biochem. 1992. Vol. 27.
P. 49-67.
(Описание и классификация очень сложных структур,
обнаруженных у различных коллагенов. Медицинские
аспекты вопроса.)
Вопросы к главе 2
1. Что такое первичная структура белка?
2. Что такое денатурация белка?
3. Изобразите структуру аминокислоты, у которой
боковая цепь:
а) атом водорода;
б) алифатическая гидрофобная;
в) ароматическая гидрофобная;
г) кислая;
д) основная.
4. Приведите примерные значения рКа
функциональных групп боковых цепей:
а) кислых аминокислот;
б) основных аминокислот;
в) гистидина.
5. От каких аминокислотных остатков зависит
суммарный заряд белковой молекулы, содержащей все
20 аминокислот?
6. Опишите четыре уровня организации молекулы
белка.
7. Какую функциональную роль в структуре белка
играют α-спирали и β-слои?
8. Каким образом белкам, которые так чувствительны
к различного рода воздействиям, удается образовать
сухожилия, невероятно прочные при растяжении?
9. Какие свойства эластина определяют его эластичность?
глава 2 Структура белков
51
3 Молекулярная организация клеточных
мембран
В главе 1 мы вкратце рассмотрели роль трех типов
слабых нековалентных связей, а также гидрофобных
взаимодействий, возникающих при выталкивании
молекулами воды неполярных молекул, мешающих
водородному связыванию. Образование слабых связей
уменьшает свободную энергию структуры, поэтому чем их
больше, тем структура устойчивей. Это рассуждение
можно рассматривать как введение к данной главе,
посвященной структуре и функциям клеточной мембраны.
Нам еще предстоит познакомиться с клеточным
метаболизмом. Вспомним, что прежде чем вещества пищи
начнут усваиваться, они должны попасть в кровь,
разнестись по организму и добраться до всех его тканей.
И уже в начале своего пути им нужно пройти через
мембраны клеток пищеварительного тракта. Одного этого
достаточно, чтобы с биохимией мембран
познакомиться раньше, чем с процессом усвоения пищи. Кроме того,
компоненты клеточных мембран участвуют в
транспорте жиров в организме, так что заодно мы получим
некоторое представление и об этом
Зачем клетке нужна мембрана?
Ответ очевиден: содержимое клетки должно быть
отделено от окружающей среды. Мы пока не знаем, как
возникла жизнь, хотя предложено несколько гипотез,
пытающихся объяснить возможные механизмы этого
процесса. Одно из необходимых условий возникновения
жизни - наличие примитивной системы молекулярной
ауторепликации. Такая система должна
функционировать в небольшом замкнутом пространстве, иначе ее
компоненты рассеются благодаря диффузии.
Это требование на первый взгляд кажется
невыполнимым, потому что клеточная мембрана слишком
сложна по своей структуре, чтобы примитивная система
ауторепликации могла ее воспроизводить. Конечно, условия,
при которых возникла жизнь на Земле, существенно
отличаются от современных, однако есть вещества,
которые спонтанно образуют замкнутые мембраноподоб-
ные структуры при простом встряхивании их с водой.
Если бы при этом в воде присутствовали аутореплици-
рующиеся системы, то часть их оказалась бы
включенной в крохотные пузырьки, или везикулы,
ограниченные мембранами, которые очень похожи по своей
структуре на мембраны современных живых клеток.
Иными словами, сборка первых мембран могла
происходить спонтанно, без участия специализированных
механизмов ауторепликации.
Что же это за вещества, обладающие столь
необычными свойствами? Одно из них можно извлечь с
помощью органических растворителей из яичного желтка.
Такой экстракт содержит смесь полярных липидов.
Полярные липиды (рис. 3.1) называются амфипатичес-
кими (т. е. обладающими двумя противоположными
свойствами), поскольку их молекулы включают как
полярные (полярная головка), так и неполярные
гидрофобные (гидрофобный хвост) группы.
Полярные
-" " гидрофильные
головки
Неполярные
гидрофобные
хвосты
Рис. 3.1. Структурная организация амфипатических
молекул, входящих в состав биологических мембран
Молекулы, которые могли бы сформировать
примитивную мембрану в далеком прошлом, не обязательно
были похожи на современные полярные липиды.
Важно, что они обладали амфипатическими свойствами.
В присутствии воды такие молекулы образуют
структуры, в которых полярные головки обращены к воде,
а контакт гидрофобных хвостов с водой сводится к
минимуму. Взаимодействие между гидрофобными
хвостами до предела усиливается ван-дер-ваальсовыми
взаимодействиями, что существенно уменьшает свободную
энергию структуры и, следовательно, резко повышает
ее стабильность.
После встряхивания полярных липидов в водной
среде образуются липосомы - пузырьки, окруженные ли-
пидным бислоем. Последний образован двумя рядами
полярных липидов, гидрофобные концы которых
спрятаны внутрь, а гидрофильные головки обращены
наружу, контактируя с водой и друг с другом, как показано
на рис. 3.2, а.
часть 2 Структура белков и мембран
52
Полярная головка.
ΑΛΛΛΛ^νν
/4/VVN/4/W
Углеводородный слой
Если сделать срез липосомы и обработать его
тяжелым металлом, который взаимодействует с полярными
головками, в электронном микроскопе липидный бис-
лой будет выглядеть как темные рельсы
железнодорожных путей. Мембрана живой клетки после аналогичной
процедуры имеет сходный вид, так как липидный бис-
лой выступает в качестве ее основной структуры.
Другим возможным типом структур, образуемых ам-
фипатическими молекулами в воде, является плотная
цилиндрической формы мицелла (см. рис. 3.2, б).
Однако двойные гидрофобные хвосты полярных липидов
способствуют образованию бислойной, а не мицелляр-
ной структуры.
Липидный бислой
Каковы липидные компоненты
клеточных мембран?
Существует много полярных липидов, структуры
которых, если их записать в виде формулы, будут сильно
различаться. Однако схематически все они могут быть
представлены так, как это показано на рис. 3.1: с полярной
головкой и двумя гидрофобными хвостами. Рассмотрим
сначала для сравнения структуру неполярных липидов.
Рис. 3.2. Строене липосомы и мицеллы
а - Поперечный разрез синтетической липосомы,
образованной липидным бислоем;
б - поперечный разрез плотной цилиндрической формы
мицеллы.
Для образования бислоя предпочтительнее участие липидов
с двумя гидрофобными цепями, а для образования мицеллы -
с одной
Жиры - это неполярные липиды, которые можно
рассматривать как производные жирных кислот.
Свободные жирные кислоты никогда не встречаются в
мембранах. Жирная кислота имеет структуру RCOOH, где R -
длинная углеводородная цепь. Среди жирных кислот
наиболее часто встречаются гексадеканоевая (С16) и ок-
тадеканоевая (С18) кислоты, которые обычно называют
соответственно пальмитиновой и стеариновой
кислотой. Их также обозначают как 16:0 или 18:0, что
указывает на число углеродных атомов и количество
ненасыщенных двойных связей. Стеариновая кислота (С18)
имеет структуру СН3(СН2)16СООН, или
Нр Н2 Н2 Н2 Н2 Н2 Н2 П2 /О
h,c'VCYCYCYCYCVCYCY<oh
Н2 Н2 Н2 Н2 Н2 Н2 Н2 Н2
Для простоты углеводородную цепь жирных кислот
изображают зигзагообразной линией, не всегда заботясь
о том, чтобы число зигзагов отвечало числу углеродных
атомов.
глава 3 Молекулярная организация клеточных мембран
53
Соли жирных кислот называют мылами. Твердое
мыло, которым мы моемся, представляет собой смесь
натриевых солей длинноцепочечных жирных кислот.
О Na+
В состав пищи человека жирные кислоты входят в
виде жиров, которые составляют заметную долю нашей
диеты. В основном это нейтральные жиры -
производные трехатомного спирта глицерина, который этерифи-
цирован жирными кислотами по всем трем своим гидро-
ксильным группам. Такие сложные эфиры глицерина
называют триацилглицеринами, или триглицеридами.
сн2-он
сн—он
сн2-он
Глицерин
0
II
0—С —R
0
II
0—С—R
0
II
0—С—R
Три молекулы
жирной кислоты
/'"Эфирная
/ 0 связь
i II
СН2-0—С —R
1 о
1 и
СН—0—С —R
1 о
1 м
СН2—0—С —R
Одна молекула
нейтрального жира
(триаци л глицерин,
или триглицерид)
Если нейтральные жиры кипятить в водном
растворе щелочи (NaOH или КОН), сложноэфирные связи гид-
ролизуются и образуются глицерин и мыла. Люди
научились варить мыло гораздо раньше, чем узнали, что такое
сложные эфиры. Неудивительно, что термин омыление
часто используют, имея в виду гидролиз любых
сложных эфиров, а не только нейтральных жиров.
Нейтральные жиры съедобны и не являются детергентами. Они
не встречаются в биологических мембранах и не
способны образовывать бислойные структуры в воде. Здесь
мы рассматриваем их лишь для того, чтобы сравнить
с полярными липидами.
Наиболее распространенными мембранными
липидами являются производные глицерин-3-фосфата,
называемые глицерофосфолипидами (или фосфогли-
церидами).
НО-
КИ2~
-2с-н
он
о
3сн2-о-р-он
он
sw-Глицерин-З -фосфат
Центральный углеродный атом (С-2) в глице-
рин-3-фосфате асимметричен, поэтому он может
находиться в двух изомерных формах. В природных
фосфоглицеридах встречается только изомер,
относящийся к L-ряду; для указания на это используется
приставка sn. Две остальные гидроксильные группы
замещены обычно остатками жирных кислот. Простейшим
представителем фосфоглицеридов является фосфатид-
ная кислота:
О
II
СН2—О—С—R
I о
СН—0-С—R
I О
СН.-О-
-0"
Фосфатидная кислота
Стоит запомнить это название, чтобы потом легче
было усвоить названия более сложных соединений. Дело
в том, что в мембранах часто встречаются производные
фосфатидной кислоты, в которых к фосфорильному
остатку присоединены различные полярные радикалы.
Если, например, речь идет о радикале X, то
соответствующее соединение называется фосфатидил-Х:
О
II
СН2 —О—С—R
СН—0-
0
II
-с-
0
II
СН2—О—Р-О-Х
0~
Чем более полярен радикал X, тем полярнее будет
«головка» фосфолипидной молекулы. Распределение
полярности в ней станет более наглядным, если изобразить
молекулу иначе:
Полярная
головка
Неполярные
хвосты
Фосфоглицерид
часть 2 Структура белков и мембран
54
В таком виде в фосфоглицеридах можно узнать ам-
фипатический мембранный липид, способный
образовать бислои: он содержит полярную головку и два
гидрофобных хвоста.
Какие полярные группы связаны
в фосфолипидах с остатком
фосфатидной кислоты?
Бислои могут быть образованы одним типом полярных
липидов. Однако для строительства своих мембран
живые клетки обычно используют разные липиды, подчас
весьма сложные по структуре, но сходные по форме
и амфипатичности молекулы. У фосфоглицеридов это
проявляется в разнообразии полярных групп, связанных
с фосфатидной кислотой.
В качестве такой группы может выступать остаток
этаноламина HOCH2CH2NH3+, фосфатидильное
производное которого называют фосфатидилэтаноламином,
или кефалином:
О—CH2-CH2NH3
0=Р-СГ
I
о
I
сн2-сн—сн2
о о
Кефалин
Трижды метилированный по азоту этаноламин
HOCH2CH2N+(CH3)3 называют холином, а холинсо-
держащий фосфолипид - фосфатидилхолином, или
лецитином.
Карбоксилированный этаноламин есть не что иное,
как аминокислота серии HOCH2CHNH3+COO".
Соответствующий фосфолипид, фрагмент структуры
которого показан ниже, называют фосфатидилсерином.
о-сн2-сн;
о=р-сг
I
о
.NH3+
чсоо"
К другому типу полярных групп относится остаток
шестиатомного спирта инозита, который присутствует
в фосфатидилинозите (PI).
снон—снон
снон чснон
чснон—снон
Прикрепляется
так:
ΟΙ
0=Р-
I
о
о
снон—снон
сн хснон
чснон—снон
Инозит
Фосфатидилинозит
Особняком в этом ряду стоит дифосфатидил глицерин,
или кардиолипин, который представляет собой
глицерин, связанный с двумя остатками фосфатидной
кислоты. Кардиолипин также обладает амфипатическими
свойствами и присутствует во внутренней мембране
митохондрий и в мембранах бактериальных клеток.
-сн2-
V
0~
-снон—сн2—о
οι
-Р=0
I
о
I
сн2-сн—сн2
I I
о о
Ос
Центральный остаток
глицерина, связывающий
две молекулы
фосфатидной кислоты
Кардиолипин
Все перечисленные полярные липиды являются
производными глицерина. Однако эволюция с
непостижимым мастерством создавала полярные липиды с иной
структурой. Тем не менее их молекулы обладают сходной
формой и свойствами. Речь идет о производных
многоатомного аминоспирта сфингозина, который по
своему строению довольно сильно отличается от глицерина:
НО-СН—СН-СН2ОН
НЧн "н+
Н2С<
н2сч
н2сч
н2сч
Н2С<
Н2С<
сн2
сн2
сн2
сн2
:СН2
:СН2
Сфингозин
В отличие от глицерина, сфингозин содержит одну
из двух требующихся углеводородных цепей (как
правило, в ней 15 углеродных атомов). Вторая углеводородная
глава 3 Молекулярная организация клеточных мембран
55
цепь в полярных липидах этого типа связана со сфин-
гозином амидной связью, как это показано на примере
церамида, молекула которого по форме близка к ди-
ацилглицерину.
Фосфорилхолиновым производным церамида
является сфингомиелин - природный фосфолипид, главный
компонент миелиновой оболочки нервных клеток.
+/СНз
CH2-CH2-N-CH3
■О" ЧсНз
Церамид
Сфингомиелин
Эволюции свойственно, обнаружив удачное
решение, применять его снова и снова. Например, церамид
используется в различных полярных липидах в
сочетании с различными полярными заместителями по
свободной гидроксильной группе. В качестве таких
заместителей обычно выступают сахара.
Моносахариды глюкоза и галактоза типичны для
цереброзидов, которые присутствуют в мембранах
мозговых клеток. Напомним, что галактоза является стерео-
изомером глюкозы по С-4-атому
О— Пиактош
I
0=Р-0"
I
о
I
но—сн—сн—сн2
NH
Цереброзид
Известно много моносахаридов и их производных,
в том числе аминосахаров. Структура одного из них -
глюкозамина - изображена ниже:
СН2ОН
-О
Η ΝΗ2
Глюкозамин
Соединяя в разных комбинациях даже небольшой
набор моносахаридов, природа получает много разных
олигосахаридов (в них от 3 до 20 моносахаридных
остатков, тогда как в полисахаридах - сотни или даже
тысячи), в том числе с разветвленной структурой. Оли-
госахаридные производные церамида называются
ганглиозидами. В этих липидах полярные головки
особенно велики и разнообразны по строению.
Моносахарид
Олигосахарид
Схема строения ганглиозида
Трудно запомнить структуру Сахаров, однако
строение N-ацетилглюкозамина и сиаловой кислоты
необходимо знать.
СН2ОН
НзС-
СОО"
СН3
N-Ацетилглюкозамин
СНОН
Сиаловая кислота J-uqu
(N-Ацетилнейраминовая |
кислота) СН2ОН
часть 2 Структура белков и мембран
56
Оба эти соединения входят в состав углеводных
компонентов ганглиозидов и мембранных гликопротеинов.
Сиаловая кислота представляет особый интерес в связи
с тем, что участвует в инфицировании клеток вирусом
гриппа (см. с. 312). Ганглиозиды являются
производными сфингозина, они определяют группу крови у людей
(О, А, В). Углеводные части ганглиозидов обладают
антигенными свойствами и различаются концевыми
остатками в олигосахаридных цепях.
О номенклатуре мембранных липидов
Наряду с индивидуальными названиями полярных
липидов, часто используют их собирательные (групповые)
названия. Так, термины мембранные или полярные
относятся к липидам, встречающимся во всех
биологических мембранах. К фосфолипидам относят все
фосфорсодержащие липиды. Среди них выделяют фос-
фоглицериды, в основе структуры которых лежит
глицерин, и сфингомиелины - производные сфингозина.
Производные церамида, лишенные фосфорильных групп
и содержащие углеводные компоненты, называются гли-
колипидами, или гликосфинголипидами. Плазмалоген -
глицерофосфолипид, в котором один из углеводородных
радикалов связан с остатком глицерина не сложноэфир-
ной, а простой эфирной связью. Еще один важный
компонент мембран - холестерин, по своей структуре он
не имеет ничего общего с полярными липидами.
Почему существует так много типов
мембранных липидов?
Ответа на этот вопрос пока нет. Разные мембранные
липиды определяют различные свойства поверхности
мембран. Так, молекулы лецитина несут положительно
заряженные полярные группы, молекулы фосфатидил-
серина - отрицательно заряженные, а цереброзиды -
нейтральные. У разных клеток состав мембранных
липидов может существенно различаться. Мембраны клеток
мозга, например, богаты цереброзидами и ганглиози-
дами, а мембраны миелиновых оболочек нервных
клеток - гликосфинголипидами. Но липиды различаются
по составу не только в мембранах разных типов клеток.
Во всех изученных плазматических мембранах внешняя
и внутренняя поверхности бислоя сильно различаются
по липидному составу. Например, гликолипиды всегда
расположены во внешнем слое, и их углеводная часть
экспонирована в окружающую среду. Липидные
молекулы практически не могут самопроизвольно
перемещаться из одного слоя в другой (такой переход
называют флип-флоп), так как при этом полярные головки
должны пройти через гидрофобную центральную часть
мембраны, а это термодинамически невыгодно.
Известны ферменты, катализирующие этот процесс; вероятно,
они участвуют в создании и поддержании асимметрии
в биологических мембранах.
Напротив, липиды легко, свободно и быстро могут
перемещаться в пределах монослоя (латеральная
подвижность), так как практически не взаимодействуют
друг с другом.
Один из мембранных липидов - фосфатидилинозит,
служит переносчиком двух различных химических
сигналов, регулирующих функции клетки (см. главу 26).
Это, однако, особый случай. Пока нет разумных
объяснений, почему мембраны содержат так много разных
полярных липидов, чем обусловлено своеобразие липид-
ного состава у разных клеток и для чего нужна
асимметрия липидного состава внешнего и внутреннего
монослоев. Некоторые мембранные липиды, особенно
ганглиозиды, представляют интерес с медицинской
точки зрения. Так, при детских заболеваниях, относящихся
к группе гликосфинголипидозов (болезни Тея-Сакса
и Гоше), нарушается обмен гликосфинголипидов, и
накопление продуктов их частичного расщепления
вызывает тяжелое поражение нервной системы.
Подводя итог, можно сказать, что клетки используют
много разных полярных липидов, создавая свои
мембраны, однако причины такой вариабельности пока не ясны.
Каков жирнокислотный состав
мембранных липидов?
Остатки жирных кислот, образующих гидрофобный
хвост мембранных липидов, разнообразны по структуре.
Как правило, они имеют неразветвленную цепь и
содержат четное число атомов углерода - от С,4 до С24 (в
основном - от С14 до С18). (Причины четности числа
углеродных атомов обсуждаются в главе 9.) Два жирно-
кислотных остатка в одной молекуле фосфолипида
могут быть одинаковыми или разными, причем в фосфо-
глицеридах гидроксил в первом положении обычно
этерифицирован кислотой с насыщенной цепью,
а во втором - с ненасыщенной. Степень ненасыщенос-
ти жирнокислотных хвостов играет важную роль,
поскольку центральная гидрофобная область мембраны
должна быть по своим свойствам близка к жидкости,
а не к твердому телу. Чем выше ненасыщенность, тем
ниже температура соответствующего фазового перехода
у мембранных липидов.
Как правило, двойные связи в жирнокислотных
хвостах имеют ^мс-конфигурацию, благодаря чему
содержащие их хвосты (в отличие от насыщенных) имеют
глава 3 Молекулярная организация клеточных мембран
57
не прямую, а изломанную форму Такого рода изломы
препятствуют плотной упаковке молекул.
ХООН /СООН ХООН
\
Насыщенная Ненасыщенная Ненасыщеная
жирная кислота жирная кислота с жирная кислота
ipc-двойной связью с трянс...двойной
связью
Влияние степени ненасыщенности на фазовое
состояние липида наглядно видно при сравнении бараньего
жира с оливковым маслом. В обоих случаях мы имеем
дело с триглицеридами, но в липидах оливкового масла
много ненасыщенных жирнокислотных хвостов.
Однако в мембранах триглицериды отсутствуют, и
правильнее сравнивать жирнокислотный состав мембранных ли-
пидов теплолюбивых и холодоустойчивых организмов
близких видов: у первых преобладают полностью
насыщенные кислоты.
Важность поддержания «текучести» мембран
иллюстрируется тем, что бактерии регулируют
жирнокислотный состав своих мембран таким образом, чтобы
температура фазового перехода мембранных липидов была
близка к температуре среды культивации. У
теплокровных животных, впадающих в зимнюю спячку, степень
насыщенности жирных кислот мембранных липидов
также варьирует в зависимости от сезонных изменений
температуры тела.
Наиболее распространены мононенасыщенные
олеиновая (18:1) и пальмитолеиновая (16:1) кислоты,
достаточно часто встречаются линолевая (18:2) и арахи-
доновая (20:4) кислоты с двумя и четырьмя двойными
связями соответственно. О номенклатуре таких кислот
подробнее будет сказано в главе 10.
Какую роль играет холестерин
в мембранах?
При взгляде на структурную формулу холестерина
(рис. 3.3, а), возникает вопрос: почему это вещество
является компонентом биологических мембран? Однако
часть 2 Структура белков и мембран
58
Рис. 3.3. Молекула холестерина
а - Плоскостное изображение;
б - пространственное изображение
все становится на свои места, если ознакомиться с кон-
формацией его молекулы (рис. 3.3, б). Холестерин
представляет собой вытянутую молекулу с жестким
стероидным ядром и гибкой углеводородной цепью. Молекула
холестерина амфипатична, поскольку на одном из ее
концов находится полярная гидроксильная группа.
Встраиваясь между молекулами липидов (гидроксил
располагается в зоне полярных головок), холестерин
участвует в регуляции текучести мембраны, препятствуя
плотной упаковке углеводородных цепей и тем самым
снижая температуру плавления липидов. Внешне это
проявляется в «расплывании» области фазового
перехода в липидном бислое. Иными словами, в отсутствие
холестерина этот переход происходит в более узком
интервале температуры. Подобным образом на температуру
плавления кристаллических веществ влияет присутствие
в них примесей. Мембрана эритроцитов может
содержать до 25% холестерина, в бактериальных мембранах
его нет вообще, а в митохондриальных мембранах
животных холестерина очень мало. В растительных
клетках роль холестерина выполняют родственные ему
соединения - фитостерины.
Слияние липидных бислоев
Липидный бислой в некотором роде подобен двумерной
жидкости, в которой отдельные молекулы липидов
свободно перемещаются в пределах своего монослоя.
Липидные бислой обычно замыкаются сами на себя,
устраняя свободные края. По этой причине они
способны самопроизвольно восстанавливаться при
повреждениях, смыкая края разорванных участков; при тесном
контакте бислой сливаются. Такие свойства бислоев
придают клеткам гибкость и способствуют ликвидации
перетяжек, что особенно важно при клеточном делении
(рис. 3.4, а).
Подобные процессы происходят при эндоцитозе -
процессе поглощения клеткой частиц (или крупных
молекул), которые не могут пройти через мембранный бис-
лой. Поглощаемая частица постепенно окружается
небольшим участком плазматической мембраны, который
сначала впячивается, а затем отщепляется, образуя
внутриклеточный пузырек, или вакуоль (рис. 3.4, б).
Возможно и обратное явление. Так, перенос секре-
тируемых клеткой молекул белков осуществляется
путем упаковки их в окруженные мембраной пузырьки,
которые затем сливаются с плазматической мембраной,
высвобождая содержимое в окружающую клетку среду
(рис. 3.4, в).
Такой процесс называется экзоцитозом. Он имеет
место, например, при секреции пищеварительных
ферментов клетками поджелудочной железы.
а
б
Частица
^^ Плазматическая мембрана
— О -
Цитоплазма
в
Окружающая среда
ф. Плазматическая мембрана
Рис. 3.4. Деление клетки (а), эндоцитоз (б) и экзоцитоз (в)
Экзоцитоз используется для секреции пищеварительных
ферментов
Проницаемость липидных бислоев
Способность веществ диффундировать через липидный
бислой в основном определяется его растворимостью
в жирах. Жирорастворимые молекулы легко
преодолевают бислой, а для большинства полярных молекул
и ионов он почти непроницаем. Крупные незаряженные
полярные молекулы типа глюкозы проникают через
бислой хуже, чем более мелкие, такие как этанол и глицерин.
Заряженные группы полярных молекул и
неорганические ионы в водном растворе окружены сольватными
оболочками из определенным образом ориентированных
молекул воды. Для того чтобы пересечь гидрофобный
слой мембраны, им нужно избавиться от этих оболочек,
что энергетически крайне невыгодно. Поэтому
проницаемость липидных бислоев для таких молекул и ионов
ничтожно мала.
Большинство молекул, участвующих в
биохимических процессах, достаточно полярны и потому не могут
диффундировать через липидный бислой со скоростью,
соизмеримой с потребностями клетки. Отсюда следует,
что: во-первых, бислойная структура биологических
мембран идеально удовлетворяет требованию оградить
цитоплазму от окружающей среды; во-вторых, мембрана
должна обладать механизмами, позволяющими клетке
быстро и избирательно пропускать потоки молекул и
ионов в соответствии с метаболическими потребностями.
Удивительно, что молекулы воды, хотя они и
полярны, легко пересекают липидный бислой. Безусловно,
сказываются малые размеры молекулы воды и
отсутствие у нее заряда, но этого недостаточно; вероятно,
здесь действуют какие-то дополнительные факторы.
В мембранах клеток почечных канальцев и секреторных
эпителиальных клеток, где транспорт воды особенно
интенсивен, присутствует особый белок - аквапорин,
который обеспечивает свободное движение воды через
мембрану. Липидный бислой проницаем также для
растворенных газов, в частности для кислорода.
Суммируя сказанное, отметим, что клетки должны
быть окружены барьером, который препятствует утечке
их компонентов, представленных главным образом
полярными соединениями. Эта цель достигается благодаря
использованию липидного бислоя - структуры,
образование и стабильность которой определяются слабыми
взаимодействиями между различными амфипатичес-
кими полярными липидами.
Монослои, составляющие бислойную мембрану,
отличаются липидным составом. Это различие
появляется в ходе сборки мембраны и сохраняется вследствие
энергетической нецелесообразности обмена липидами
между монослоями. Латеральное движение липидов (в
глава 3 Молекулярная организация клеточных мембран
59
пределах каждого из монослоев) происходит вполне
свободно. Разные типы клеток различаются липидным
составом своих мембран. Непонятно, зачем природе
понадобился такой обширный набор липидов для
биомембран, тем более что только один из них - фосфа-
тидилинозит имеет специфическое предназначение,
выступая в качестве химического сигнала.
Углеводородный слой мембран находится в «жидком» состоянии.
В животных клетках это достигается поддержанием
баланса между насыщенными и ненасыщенными
остатками жирных кислот. Последние снижают температуру
фазового перехода в бислоях: их изломанные ^модвой-
ной связью цепи препятствуют плотной упаковке липид-
ных молекул. Для регуляции температуры фазового
перехода используется холестерин. В бактериальных
мембранах тот же эффект достигается с помощью
жирных кислот с разветвленными цепями. Липидный бис-
лой практически непроницаем для сильно полярных
молекул, однако через него относительно легко
диффундируют небольшие слабополярные молекулы. Эти
свойства позволяют бислою осуществлять свою главную
функцию - служить барьером, препятствующим утечке
компонентов цитоплазмы. Способность бислоев к
слиянию обеспечивает возможность протекания таких
процессов, как клеточное деление, эндоцитоз и экзоцитоз.
Липидный бислой является конструкционной
основой биологических мембран, однако сам по себе не спо-
способен осуществить все выполняемые ими функции,
в том числе избирательный трансмембранный перенос
молекул. За это ответственны мембранные белки.
Мембранные белки и структура
биологических мембран
Благодаря своей удивительной структуре биологическая
мембрана обладает разнообразными свойствами. Разные
клетки выполняют различные функции, и каждая из этих
функций может быть реализована с помощью
специализированного мембранного белка, который
синтезируется для выполнения определенной задачи, а затем
попадает в мембрану (о том, как это происходит, см. главу 22).
При этом необходимо, чтобы белок мог встроиться в эту
мембрану. Экспериментально доказано, что
функциональные белки, выделенные из биомембран, способны
встраиваться в искусственные липидные мембраны
(липосомы) и осуществлять там те же функции, что
и в исходной мембране.
Структуру биомембран часто называют жидкомоза-
ичной (рис. 3.5). Ее уподобляют двумерному морю
липидов, в котором плавают «айсберги» - интегральные
белки, частично погруженные в мембрану или
пронизывающие ее насквозь Кроме того, выделяют так
называемые периферические белки, молекулы которых
не встроены в мембрану, а удерживаются на ее
поверхности за счет слабых взаимодействий.
Периферический белок
t
интегральный белок
Интег-
альный
белок ^
Интегра
ный белок
Рис. 3.5. Жидкомозаичная модель строения биологических
мембран
Что удерживает интегральные белки
в липидном бислое?
Интегральные мембранные белки удерживаются в
липидном бислое благодаря особенностям своей
структуры, которые обеспечивают возможность слабых
взаимодействий между ними и липидным окружением.
Как известно, белки представляют собой
полипептиды, состоящие из ковалентно связанных аминокислот.
Если все аминокислотные остатки в полипептиде
содержат гидрофобные боковые цепи, то полипептид в целом
будет гидрофобен и т. д. У интегральных мембранных
белков концевые участки полипептидной цепи состоят
из остатков гидрофильных аминокислот, а центральная
часть цепи - из гидрофобных (рис. 3.6, а).
На рис. 3.6, б схематически показано строение гли-
кофорина - главного белкового компонента мембраны
эритроцитов. На внешней стороне мембраны
расположен N-концевой участок этого белка, богатый
гидрофильными аминокислотами. Гидрофильность этого
фрагмента усиливается благодаря наличию большого
числа остатков серина, треонина и аспарагина,
ковалентно связанных с углеводными радикалами. С-кон-
цевой участок полипептидной цепи, расположенный
часть 2 Структура белков и мембран
60
с внутренней стороны мембраны, также богат
гидрофильными аминокислотами, но не содержит углеводных
компонентов (углеводные остатки всегда расположены
на поверхности клетки). Оба гидрофильных фрагмента
молекулы гликофорина разделены участком
полипептидной цепи, расположенным внутри мембраны и
обладающим гидрофобными свойствами (участок не
содержит ни одной сильно гидрофильной аминокислоты). Он
состоит из 19 аминокислотных остатков, свернутых
в α-спираль.
Размеры центрального гидрофобного участка
молекулы гликофорина соответствуют толщине
гидрофобной зоны липидного бислоя, а внешние гидрофильные
участки локализованы на противоположных сторонах
мембраны и способны взаимодействовать как с водой,
так и с полярными головками мембранных липидов
(см. рис. 3.6, б). Белки, в молекулах которых четко
выделяются гидрофильные и гидрофобные участки,
называют амфипатическими.
Полипептидная
цепь белка
Гидрофильные Гидрофобные Гидрофильные
аминокислоты аминокислоты аминокислоты
N-концевой участок с
прикрепленными к нему
16 олигосахаридными
цепями
Гидрофобная α-спираль,
погруженная в мембрану
В некоторых мембранных белках полипептидная
цепь многократно изгибается таким образом, что
пересекает мембрану не один, а несколько раз (рис. 3.7).
Соответственно, гидрофильные участки в них
перемежаются с гидрофобными; последние спирализованы
и состоят из 15 -20 аминокислотных остатков. Среди
таких белков наиболее изучен бактериородопсин -
пигмент пурпурных бактерий Halobacterium halobium.
Он улавливает световую энергию и использует ее для
перекачивания протонов из бактериальной клетки в
окружающую среду, создавая протонный градиент,
энергия которого затем используется клеткой для синтеза
АТР (этот механизм описан в главе 7).
Расположение мембранных белков отвечает
максимально эффективному их взаимодействию с липидным
бислоем. При любой попытке белка покинуть мембрану
его гидрофобные участки войдут в соприкосновение с
водой, а гидрофильные - с углеводородным слоем
мембраны. И то, и другое энергетически невыгодно, так что
трансмембранные перемещения интегральных белков
исключены. Иное дело их латеральная диффузия в пределах
монослоя. Механизмы попадания белков в биомембраны
и их начальная ориентация будут обсуждаться в главе 22.
NH3 Окружающая
среда
ЫНз
, Липидный
бислой
соо-
Цитоплазма
С-концевой^часток, гпп-
содержащии гидро- —LUU
фильные
аминокислотные остатки,
экспонирован в цитоплазму
Рис. 3.6. Структурная организация интегральных белков
(а), и строение молекулы гликофорина (б)
Спиральный участок гликофорина, включающий 19
аминокислотных остатков, имеет длину около 3 нм, что соответствует
толщине гидрофобной части липидного бислоя
Рис. 3.7. Топография полипептидной цепи бактериородоп-
сина в мембране
Семь плотно упакованых α-спиральных участков
пронизывают липидный бислой
Заякоривание периферических белков
в мембране
Периферические водорастворимые мембранные белки
могут удерживаться на поверхности мембраны за счет
водородных связей и ионных взаимодействий.
глава 3 Молекулярная организация клеточных мембран
61
Однако существуют и другие способы. Так, белок
может содержать ковалентно связанный остаток длинно-
цепочечной жирной кислоты, который внедряется в ли-
пидный бислой и служит своего рода якорем (рис. 3.8).
В одних белках N-концевые остатки глицина связаны
с жирной миристиновой кислотой:
R-CO-NH-CH2-CO- полипептид f
в других - жирные кислоты С14, С16 и С18 этерифициру-
ют гидроксильные группы остатков серина и треонина
или тиольную группу цистеина. Используются кислоты
и более сложного строения, остатки которых
прикреплены к белку простыми эфирными связями.
Примером более усовершенствованного способа заякоривания
периферических белков в мембране является кова-
лентное связывание фосфатидилинозита с олигоса-
харидной частью мембранного гликопротеина. Такой
остаток фосфатидилинозита включается в мембрану
и ведет себя там как свободная липидная молекула.
Мембранные гликопротеины
Многие мембранные гликопротеины так же, как и глико-
липиды - цереброзиды и ганглиозиды, содержат
разветвленные олигосахариды с внешней стороны мембраны.
Эти олигосахариды, ковалентно связанные с боковыми
цепями остатков серина или аспарагина, состоят из фу-
козы, маннозы, галактозы, N-ацетилгалактозамина,
Белок, связанный
с жирной кислотой
с помощью ЫН2-группы
О HN
Якорный
" - гидрофобный
хвост
Плазматическая
мембрана
Рис. 3.8. Якорный остаток жирной кислоты,
прикрепленный амидной связью к молекуле белка
Иногда остатки жирных кислот прикреплены к белкам
сложноэфирной или тиоэфирной связями
часть 2 Структура белков и мембран
62
N-ацетилглюкозамина и сиаловой кислоты. В гликопроте-
ине эритроцитарной мембраны - гликофорине
углеводная часть составляет по массе половину молекулы.
Точно неизвестно, зачем нужны эти олигосахариды, хотя
в некоторых случаях они, по-видимому, играют роль
маркеров при распознавании клеточной поверхности.
Итак, разнообразие мембранных белков
определяется множественностью функций биологических мембран.
Интегральные белки погружены в мембраны, тогда как
периферические удерживаются на поверхности
благодаря нековалентным взаимодействиям с полярными
группами липидов. Интегральные белки амфипатичны:
в их структуре четко различаются центральная
гидрофобная область, соприкасающаяся с углеводородным
слоем липидной мембраны, и гидрофильные
фрагменты на С- и N-концевых участках цепи, экспонированные
в водное окружение по разные стороны мембраны.
Мембранные белки часто гликозилированы, т. е. содержат
ковалентно связанные остатки Сахаров, причем эти
сахара всегда находятся на внешней стороне клеточной
мембраны. В принципе в липидную мембрану может
быть встроено большое число типов белков. Это
придает ей функциональную гибкость.
Функции мембран
Мембраны выполняют множество разнообразных
функций. Мы ограничимся рассмотрением функций
плазматической мембраны, окружающей клетку, а о
внутриклеточных мембранах поговорим в следующих
главах. Кроме основной задачи - сохранить содержимое
клетки, плазматические мембраны выполняют и
множество других. В этом разделе будет рассмотрено их
участие в транспорте веществ в клетку и из клетки, передаче
сигналов, поддержании формы клеток, а также в
межклеточных взаимодействиях.
Транспорт веществ в клетку и из клетки
Уже говорилось о том, что если не большинство, то
многие из веществ, которые клетка должна получать из
окружающей среды, не способны проникать через
плазматическую мембрану со скоростью, соизмеримой с ее
метаболическими потребностями. (Заметим, что есть
внутриклеточные мембраны, например внешние
мембраны митохондрий и хлоропластов, у которых поры так
велики, что через них свободно проходят любые
молекулы с массой менее 600 Да.) Поэтому для переноса через
мембрану полярных структур - Сахаров, аминокислот,
неорганических ионов и т. п. - требуются специально
предназначенные для этой цели белки. Только они
могут обеспечить должную избирательность и управление
клеточными процессами извне. Требование
избирательности объясняет необходимость существования
множества специализированных транспортных систем и,
следовательно, транспортных белков.
Пассивный транспорт,
или облегченная диффузия
Транспортные системы подразделяют на пассивные
и активные. Первые осуществляют перенос веществ
через мембрану, не требуя притока энергии извне,
поскольку он совершается за счет концентрационного
градиента. Именно поэтому пассивный транспорт называют
также облегченной диффузией. Хорошим примером
здесь может служить белок, обеспечивающий
двунаправленное движение анионов (СГ и НСО 3) через эрит-
роцитарную мембрану (рис. 3.9). Физиологическое
значение этого процесса обсуждается в главе 27. Другим
примером облегченной диффузии может служить
перенос глюкозы через мембраны животных клеток -
необходимый и важный этап усвоения этого вещества,
разносимого кровью.
Важными элементами в системе пассивного
транспорта являются регулируемые поры, обычно
именуемые каналами. К их числу относятся структуры,
обеспечивающие трансмембранные ионные потоки,
пропускная способность которых зависит от внешнего
сигнала. Одни каналы открываются в ответ на
деполяризацию (см. главу 26), другие - под действием
определенных химических веществ, в частности ацетилхо-
лина. Структурные элементы каналов, непосредственно
отвечающие за их пропускную способность, называют
воротами, а структурные элементы, распознающие
сигнальную молекулу (химический сигнал), - рецепторами.
Каналы имеют важное физиологическое значение для
ионов Са2+, Na+ и К+.
Активный транспорт
Этот вид трансмембранного переноса веществ
совершается за счет внешнего источника энергии. Во всех
клетках (кроме бактериальных) таким источником
служит гидролиз АТР. При активном транспорте
перемещение вещества происходит против его
концентрационного градиента.Чтобы определить количество энергии,
необходимой для перемещения вещества в клетку
против концентрационного градиента, можно использовать
уравнение, приведенное в главе 1 для описания
химических реакций:
[Продукты]
Белок, ответственный
за транспорт анионов
СГ
л
НСО*
J
Мембрана
СГ
cr
ν
НСОз
НСО",
V
А
сг
НСО",
Рис. 3.9. Анионный канал в мембране эритроцита
Ионы СГ и НСО~ могут двигаться через канал в обоих, но
противоположных направлениях в соответствии с
концентрационными градиентами. Противоток этих анионов
обеспечивает сохранение электронейтральности
Для веществ, чья структура при переносе не
изменяется (AG0' = 0), это уравнение упрощается:
[С2]
AG = /?r-2,3031g-T-
где Cj - концентрация вещества вне клетки; С2 -
концентрация вещества внутри клетки. Если С2/С] = 10,
а Т= 298 К, то:
10
АО=8,315кДж · моль"1 ■ 1^X298К)-2,303 lg- =
AG = AG°' + /?r-2,303 1g
1
[Исходные вещества]
=5706кДж-моль-1.
глава 3 Молекулярная организация клеточных мембран
63
Итак, в принятых условиях перенос одного моля
вещества требует 5706 Дж (при 25° С). Неудивительно,
что нервные клетки, мембранный потенциал которых
в значительной мере определяется градиентом
концентрации ионов калия и натрия, расходуют на его
поддержание большую часть производимого в них АТР.
Примером системы активного транспорта может
служить так называемый Na~7K+-Hacoc в клеточных
мембранах животных клеток. В этих клетках поддерживается
высокая концентрация ионов калия (140 мМ) и низкая -
ионов натрия (12 мМ), тогда как в крови и
межклеточной жидкости соотношение этих концентраций
обратное: соответственно 4 и 145 мМ. Ионная проницаемость
плазматических мембран клеток такова, что
концентрационным градиентам калия и натрия соответствует
разность электрических потенциалов 50-70мВ
(положительный - снаружи мембраны, отрицательный -
внутри). За это приходится платить: на поддержание
ионных градиентов животные тратят около трети
расходуемой ими энергии. Но тратится она не впустую. Эти
градиенты необходимы для генерации и проведения
электрических сигналов в возбудимых мембранах, а
кроме того, запасенная в виде градиентов энергия
расходуется на транспорт веществ через мембрану.
Устройство Na7K+-Hacoca
Этот насос иначе называют NaVIC-АТРазой, поскольку
выкачивание из клетки натрия и закачивание в нее
калия сопряжено с гидролизом АТР до ADP + Pf..
Ыа+/К+-АТРаза представляет собой белок, состоящий
из двух пар субъединиц (αα и ββ). Некоторые белки
способны изменять свою форму при связывании
специфических лигандов (лигандами называют
соединения, которые связываются с другими соединениями
в строго определенных местах). Эти изменения
происходят в результате конформационных превращений в
отдельной белковой молекуле или вследствие упаковки
субъединиц в белковые комплексы (например в
гемоглобине). В Na+/K+-ATPa3e такие изменения могут быть
вызваны фосфорилированием белка, в ходе которого
может измениться его сродство к ионам натрия и калия.
Согласно модели, представленной на рис. 3.10,
Na+/K+ -АТРаза может существовать в двух конформа-
циях. Связывание Na+ и последующее фосфорилирова-
ние АТРазы со стороны цитоплазмы (см. рис. 3.10, а)
приводят к конформационным изменениям в белке,
в результате которых Na4" проходит через мембрану и
высвобождается в межклеточное пространство.
Одновременно с этим происходит связывание К+ на внешней
поверхности мембраны (см. рис. 3.10, б), а
последующее дефосфорилирование возвращает белок в перво-
часть 2 Структура белков и мембран
64
начальное состояние; при этом К+, пройдя через
мембрану, поступает в цитоплазму (см. рис. 3.10, в). Таким
образом, фосфорилирование фермента приводит к
выбросу иона натрия из клетки и к связыванию им иона
калия из окружающей среды. Если при дефосфорилиро-
вании фермент возвращается в первоначальную форму,
то процесс становится циклическим и выражается в
гидролизе АТР, сопряженном с трансмембранным
противотоком ионов натрия и калия. Действительно, один цикл
Ыа+/К+-АТРазы сопровождается встречным переносом
трех ионов натрия и двух ионов калия. Все
происходящее при этом можно описать уравнением:
ЗЫа+(внутри) + 2К+(снаружи) + АТР + Н20 ->
-> ЗЫа+(снаружи) + 2К+(внутри) + ADP + Р,- + Н\
Здесь интересен медицинский аспект. Существует
группа соединений, называемых сердечными гликози-
дами, которые представляют собой углеводные
производные стероидов (по структуре сходные с
холестерином). Они содержатся в наперстянке и близких к ней
видах растений.
Эти соединения ингибируют Na+/K+-ATPa3y,
препятствуя удалению фосфорильного остатка, и тем
самым останавливают транспорт ионов на стадии б
(см. рис. 3.10). В малых дозах они уже давно
используются в медицине при лечении сердечной
недостаточности, а в больших дозах - смертельно опасны.
Терапевтический эффект сердечных гликозидов обусловлен тем,
что они, ингибируя Ыа+/К+-АТРазу, увеличивают
концентрацию ионов натрия в клетках сердечной мышцы,
вследствие чего уменьшается натриевый градиент. Это
приводит к увеличению содержания в цитоплазме ионов
Са2+, поскольку существует другая система, которая
обеспечивает транспорт Na+ внутрь клетки, а Са2+
наружу, т. е. выход Са2+ из мышечных клеток происходит
путем его обмена на натрий и зависит от натриевого
градиента. Вызванное сердечными гликозидами снижение
градиента Na+ способствует снижению выхода Са2+
наружу и увеличению его внутриклеточного уровня. Это
в итоге стимулирует сокращение сердечной мышцы.
Сходным эффектом обладает уабаин - вещество
растительного происхождения, которым африканцы
отравляли наконечники своих стрел.
Совместный перенос молекул
через мембраны (симпорт)
Трансмембранный №+-градиент, создаваемый
Na+/K+-ATPa3oft, есть не что иное, как форма запасания
энергии. Эта потенциальная энергия рассеется в виде
тепла, если ионам натрия дать возможность просто так
Окружающая среда
+ АТР
Цитоплазма
3Na+
Окружающая среда
\ 2К+
3Na+
+ ADP
Цитоплазма
Окружающая среда
з
+ h
2К+
3Na+ \
Цитоплазма
Рис. 3.10. Гипотетический мех анизм работы Na7K+-Hacoca
а-в - Последовательные состояния №7К+-АТРазы
вернуться в клетку. Представим, однако, что они это
сделают вместе с другими молекулами. Такой совместный
перенос называется симпортом. Существует, например,
белок, ответственный за симпорт ионов натрия и
глюкозы, при котором переносятся их строго эквимолярные
количества. В результате глюкоза может двигаться
внутрь клетки против собственного концентрационного
градиента за счет градиента ионов натрия. А поскольку
последний возникает за счет гидролиза АТР, то АТР
косвенно является движущей силой транспорта глюкозы.
Подобный механизм функционирует при всасывании
аминокислот и глюкозы в кишечнике (рис. 3.11), причем
в переносе разных веществ участвуют разные
транспортные белки.
Симпорт №+/К+АТРаза
\
Глюкоза \
^.
Na+
(высокая
концентрация)
Глюкоза
S
\
Na+
(низкая
концентрация)
АТР
\
Na+
К+
ADP + Р/
/
-'
Na
К+
Рис. 3.11. Симпорт - система совместного транспорта
глюкозы и ионов натрия
Антипорт
В связи с действием сердечных гликозидов мы уже
упоминали о том, что вход ионов натрия в клетки
сердечной мышцы сопряжен с выкачиванием наружу ионов
кальция. Такой жестко связанный противоток называют
антипортом (рис. 3.12). И в этом случае кальций может
транспортироваться против собственного
концентрационного градиента за счет энергии натриевого
градиента, который, в свою очередь, возникает благодаря
гидролизу АТР. Когда мы подойдем к рассмотрению
мышечного сокращения, вы увидите, что встречаются
и Са2+-насосы (Са2+-АТРазы), которые
непосредственно используют гидролиз АТР как источник энергии для
трансмембранного переноса ионов кальция.
Антипорт Ыа+/К+-АТРаза
Са*
Na+
Са2+
Na4
АТР
Na+
К+
/
' -^Na
К+
ADP + Pj
Рис. 3.12. Антипорт - система жестко связанного
противотока ионов натрия и кальция
глава 3 Молекулярная организация клеточных мембран
65
Унипорт
Термином унипорт обозначают перенос любых
растворенных веществ или ионов с одной стороны мембраны
на другую с участием специализированных
транспортных белков, например Са2+-АТРазы из саркоплазматичес-
кого ретикулума.
Итак, большинство веществ переносится через
плазматическую мембрану с помощью специфического для
каждого вещества транспортного белка, что позволяет
понять, почему таких белков очень много.· Транспорт
называют пассивным, когда его движущей силой является
только градиент концентрации (перенос анионов в эритроцитах
или глюкозы в животных клетках). Однако так бывает
не всегда, и вещества могут двигаться через мембрану
против собственного градиента. Животные клетки обладают
Ыа7К+-АТРазой, которая создает на плазматической
мембране встречные градиенты концентраций ионов калия
и натрия. Ингибирование этого фермента сердечными
гликозидами используется в медицине при лечении
сердечной недостаточности. Натриевый градиент, возникающий
на плазматической мембране, может использоваться как
источник энергии для переноса других молекул (симпорт
натрия и аминокислот или глюкозы в кишечнике) или
выведения их в окружающую среду (антипорт ионов натрия
и кальция). И при симпорте, и при антипорте с участием
ионов натрия источником энергии для создания всех
градиентов является гидролиз АТР. Наряду с NaVIC-АТРазой
существуют и другие транспортные АТРазы.
Преобразование сигналов
Все клетки животного должны работать согласованно,
в соответствии с нуждами целого организма, о которых
они узнают из посылаемых им сигналов. Жизнь - это
химический процесс, и потому неудивительно, что
сигналы посылаются в виде химических соединений, будь
то гормоны, продуцируемые железами, или нейромеди-
аторы, выделяемые нервными окончаниями.
Среди сигнальных соединений есть неполярные
вещества, такие как стероидные гормоны, которые
проникают в клетки, диффундируя через липидный бислой, и
полярные, которые в клетку проникнуть не могут, а
связываются на ее поверхности со специализированными
белками-рецепторами, каковые и отвечают за передачу
сигнала. Связывание сигнальной молекулы с
мембранным белком-рецептором инициирует цепь
последовательно протекающих событий внутри клетки. Именно это
имеют в виду, когда говорят о плазматической мембране как
о преобразователе сигнала. Биохимические аспекты
преобразования сигналов будут рассмотрены в главах 12 и 26.
часть 2 Структура белков и мембран
66
Роль плазматической мембраны
в поддержании формы клеток
и их подвижности
Эукариотические клетки обладают своего рода каркасом,
который, с одной стороны, придает им определенную
форму, а с другой - допускает возможность ее
изменения, позволяя клеткам двигаться и перемещать свои ор-
ганеллы с одного места на другое. Этот каркас,
называемый цитоскелетом (см. главу 29), в отличие от костного
скелета животных, не жесткий и не имеет
фиксированной структуры. Он образован белковыми волокнами,
которые пронизывают цитоплазму и во множестве точек
связаны с белками плазматической мембраны.
Естественно, эти белки не способны к латеральной диффузии,
свойственной обычным мембранным белкам.
Наиболее наглядным примером, демонстрирующим
связь цитоскелета с мембранными белками, служит
эритроцит. Он имеет форму двояковогнутого диска, что
позволяет увеличить поверхность газообмена между
цитоплазмой и внешней средой. При движении сквозь тончайшие
капилляры эритроцит испытывает значительные
механические нагрузки и благодаря гибкости и упругости
претерпевает значительные, но обратимые изменения
формы. Эритроцит обладает этими свойствами из-за наличия
в нем плотного примембранного каркаса из волокон,
образованных белком спектрином. Название этого белка
происходит от английского слова spectre (тень, призрак),
которое является синонимом другого английского слова
ghost Так называют эритроциты, сохранившие форму,
но утратившие содержимое. Спектрин связан с белком
анкирином (англ. anchor - якорь), который, в свою
очередь, удерживается на внутренней поверхности
мембраны благодаря связыванию с белком, ответственным
за транспорт анионов. Другие якорные белки
прикрепляют спектрин к гликофорину (рис. 3.13).
Известны больные, у которых цитоскелет
эритроцитов нарушен из-за дефектов структуры спектрина или
анкирина. При этом эритроциты имеют необычную
форму и разрушаются в селезенке. Болезни, вызванные
такими нарушениями, называют наследственным сферо-
цитозом или наследственным эллиптоцитозом.
Межклеточные взаимодействия:
плотные соединения, контактные поры,
ответственные за клеточную адгезию
белки
Клетки эпителия, выстилающего пищеварительный тракт,
служат посредниками при переносе продуктов
расщепления пищи в кровь. Последние проникают в
эпителиальную клетку через обращенную в кишечный тракт
Окружающая среда Белок,
транспортирующий
... анионы -..
Гликофорин
··
Цитоплазма
Анкирин
Спектриновые Белки, связывающие
волокна гликофорин с цитоскелетом
Рис. 3.13. Взаимодействие с цитоскелетом белка, транспортирующего анионы, и гликофорина
плазматическую мембрану, переносятся к
противоположной стороне клетки и затем выходят через мембрану
наружу, попадая в кровеносный капилляр. Именно так
происходит всасывание, а не через промежутки между
клетками, что в принципе невозможно, ибо клетки
тесно прилегают друг к другу. Плотные соединения -
общее название непроницаемых белковых конструкций,
которые опоясывают прилегающие друг к другу клетки
и связывают их между собой. В области плотного
соединения не происходит даже латерального движения
мембранных белков. Это имеет важное функциональное
значение. Участки плазматической мембраны
эпителиальной клетки, обращенные в просвет кишки и в
кровеносный капилляр, содержат разные транспортные
белки, которые в отсутствие латеральной диффузии могут
переходить из одного участка в другой. В то же время
было бы разумно обеспечить перемещение молекул
между клетками для того, чтобы сделать их деятельность
синхронной в пределах всей ткани. Это и в самом деле
имеет место благодаря особым белкам, образующим
межклеточные туннели, или контактные поры,
способные пропустить такие молекулы, как АТР, но
задерживающие крупные молекулы, например белки. Важность
такого межклеточного контакта не вызывает сомнений.
Он необходим, например, для согласованного
сокращения клеток сердечной мышцы.
Специальные тканеспецифичные адгезионные белки
обеспечивают объединение однотипных клеток в ткань.
Если смешать эмбриональные клетки различных типов,
они образуют агрегаты из одинаковых клеток: печеночные
объединяются с печеночными, почечные с почечными
и т.д. Общее название белков, ответственных за адгезию, -
кадхерины. Среди них выделяют отдельные группы,
например NCAMS (от англ. Nerve Cell Adhesive MoleculeS -
адгезивные молекулы нервных клеток), играющие важную
роль в формировании нервной ткани.
Внутриклеточные мембраны
До сих пор в этой главе речь шла только о наружной,
или плазматической, клеточной мембране. Однако
внутри эукариотических клеток находится много разных
мембранных структур. Все они в основе имеют липидный
бислой, но различаются липидным составом и набором
входящих в мембраны функциональных белков.
Внутренние мембранные структуры разбивают клетку на
отдельные компартменты. Главные компоненты
животной клетки показаны на рис. 3.14.
глава 3 Молекулярная организация клеточных мембран
67
Плазматическая мембрана
итохондрия
Пероксисома
^
<&
Ядро
: Лизосома
Аппарат Гольджи
ядоплазматический
ретикулум
3.14. Мембранные органеллы типичной животной
не
1етки
Перечислим наиболее существенные
внутриклеточные органеллы и их функции.
1. Ядро, которое содержит ДНК и является местом
синтеза ДНК и РНК.
2. Эндоплазматический ретикулум (ЭР) - сеть
мембранных трубочек и цистерн, занимающих
половину объема клетки. Здесь синтезируются все
мембранные белки, а также белки, подлежащие секреции или
требующие доставки к определенным участкам
клетки. Часть ретикулума усеяна рибосомами -
структурами, ответственными за синтез белков. Эти области
называют шероховатым ретикулумом, в отличие от
гладкого, где рибосом нет.
3. Аппарат Гольджи - стопки уплощенных цистерн;
здесь сортируются синтезированные в ретикулуме
белки и рассылаются по назначению. И ретикулум,
и аппарат Гольджи - полностью замкнутые структу-
Клетка, секретирующая стероиды
Макрофаг
Ядро
Ядерная
оболочка
- t
4fi
4,
Плазматическая
мембрана
Клатриновый
участок
Лизосома Ядерная
пора ι
Шероховатый
эндоплазма- |
Гладкий тический
эндоплазма- ретикулум
тический
ретикулум
Ядерная Ядро
оболочка!
Эухроматин
Митохондрия
Гетерохроматин
Пузырьки, сливающиеся
или отпочковывающиеся
от аппарата Гольджи
Мембраны аппарата Гольджи
Рис. 3.15. Электронные микрофотографии срезов яичка
а - Клетка, секретирующая стероиды (обратите внимание на гладкий эндоплазматический ретикулум и макрофаг);
б - та же клетка при большом увеличении: хорошо виден аппарат Гольджи
часть 2 Структура белков и мембран
68
ры, все выходящее из них заключено в
отпочковывающиеся, окруженные мембранами пузырьки.
4. Митохондрии - место окислительного
метаболизма, где образуется большинство молекул АТР (во
внутренней митохондриальной мембране),
необходимых для различных энергетических нужд клетки.
5. Лизосомы содержат гидролитические ферменты,
разрушающие нежелательные для клетки вещества.
6. Пероксисомы - пузырьки с ферментами,
катализирующими различные окислительные реакции.
Разные клетки в разной мере нуждаются в тех или
иных органеллах. На электронной микрофотографии
среза яичка (рис. 3.15, а, б) видны участки гладкого рети-
кулума, где сосредоточены ферменты, участвующие
в синтезе стероидов. Особенно развит ретикулум в
клетках, секретирующих белки, например пищеварительные
ферменты или антитела. Клетки, которые нуждаются
в большом количестве АТР (например мышечные),
богаты митохондриями. Большой редкостью являются
клетки, которые вообще лишены органелл. Примером могут
служить зрелые эритроциты.
Дополнительная литература
Строение и подвижность мембранных
липидов
Higgins С. Ε Flip-flop: the transmembrane translocation of
lipids // Cell. 1994. Vol. 79. P. 393-395.
(Обсуждается, как молекулы липидов попадают в
отведенное для них место и как достигается асимметрия
распределения липидов между двумя слоями,
составляющими липидную мембрану.)
Rooney S. Α., Young S. L., MendelsonC. R. Molecular
and cellular processing of lung surfactant // FASEB J. 1994.
Vol. 8. P. 957-967.
(Рассматривается физиологическое значение липидов,
выстилающих альвеолы легких.)
Мембранные белки
Macdonald С. Gap junctions and cell-cell
communication//Essays Biochem. 1985. Vol. 21. P. 86-118.
(Представлен полный обзор данной проблемы.)
Neher E., Sakmann В. The patch clamp technique // Sci.
Amer. 1992. Vol. 266(3). P. 44-51.
(Современные методы изучения электрически и
химически управляемых ионных каналов.)
Von Heijne G. Membrane proteins: from sequence to
structure //Annu. Rev. Biophys. Biomolec. Struct. 1994. Vol. 23.
P. 167-192.
(Обзор, посвященный изучению структуры
интегральных мембранных белков.)
Von Heijne G Membrane protein assembly: rules of the
game // BioEssays. 1995. Vol. 17. P. 25-30.
(Рассуждения о том, как белки удерживаются в
мембранах за счет множественных трансмембранных
α-спиралей.)
Вопросы к главе 3
1. Какие структурные особенности амфипатических
соединений определяют их способность к образованию
в воде мицелл или липосом?
2. Входят ли триглицериды в состав биологических
мембран?
3. Могут ли белки пройти через липидный бислой?
4. Какова функция холестерина в мембранах эукарио-
тических клеток?
5. Изобразите строение фосфатидной кислоты.
6. Назовите три фосфолипида - производных
фосфатидной кислоты, и те группы, которыми они
различаются.
7. Опишите строение сфингомиелина.
8. Чем цереброзид и ганглиозид отличаются от
сфингомиелина?
9. Что такое сиаловая кислота? Какова ее структура?
10. В чем состоит функциональная роль ^моненасыщен-
ных жирнокислотных хвостов в фосфолипидах
мембран?
11. Почему липидные мембраны плохо проницаемы для
полярных молекул?
12. Чем облегченная диффузия через мембраны
отличается от обыкновенной? Приведите пример.
13. Объясните, как вычислить энергию, которая
требуется для переноса вещества в клетку. Сколько нужно
затратить энергии для переноса одного моля
вещества, если снаружи его концентрация в десять раз
меньше, чем внутри?
14. Как удается клеткам поддерживать внутреннюю
концентрацию ионов калия на высоком уровне, а
концентрацию ионов натрия на уровне гораздо
меньшем, чем в окружающей среде?
глава 3 Молекулярная организация клеточных мембран
69
__ Переваривание и
глава
4
В этой главе мы рассмотрим вопросы, связанные с
перевариванием и всасыванием пищи (именно с этого
начинается метаболизм), обсудим, что представляет собой
пища с химической точки зрения, как она
трансформируется, прежде чем попасть в кровь, и каким образом она
туда попадает.
Химический состав пищи
Пища содержит три главных компонента: белки,
углеводы и липиды.
Белки представляют собой высокомолекулярные
полимеры, образованные двадцатью видами аминокислот,
соединенных пептидными связями.
Углеводы - это сахара и их производные. Термин
своим происхождением обязан общей формуле обычных
Сахаров - Сп(Н20)п, из которой следует, что они
состоят из углерода и воды. Хотя пища и содержит
моносахариды, например глюкозу, но большая часть углеводов,
присутствующих в ней, встречается в виде дисахаридов,
в том числе сахарозы и полисахаридов типа крахмала.
Липиды представлены главным образом триглице-
ридами или нейтральными жирами. Их типичными
примерами могут служить сливочное и оливковое масло,
жировые прослойки говядины или баранины. Полярные
липиды также присутствуют в пище, но обычно их
гораздо меньше.
Переваривание и всасывание
За исключением моносахаридов типа глюкозы, вся пища
подвергается в кишечнике гидролитическому
расщеплению, распадаясь на элементарные строительные блоки,
из которых они состоят: белки - до аминокислот,
углеводы - до моносахаридов, нейтральные жиры - до
жирных кислот и моноглицеридов. Ни в каком ином виде
компоненты пищи не могут проникать в эпителиальные
клетки, выстилающие пищеварительный тракт.
Анатомия пищеварительного тракта
Рассмотрим отдельные участки пищеварительного тракта.
В ротовой полости пища разжевывается и
смачивается, что облегчает ее проглатывание. В незначительной
степени здесь происходит расщепление крахмала.
всасывание пищи
Желудок содержит соляную кислоту, которая
«стерилизует» пищу и денатурирует белки; здесь же
начинается расщепление белков.
Основной вклад в переваривание и усвоение всех
компонентов пищи вносит тонкий кишечник. Его
внутренняя поверхность образована крохотными
пальцеобразными выростами, или ворсинками, которые
покрыты эпителиальными клетками. На обращенной в
просвет кишки поверхности эпителиальных клеток имеется
множество микроворсинок, образующих вместе с
клетками так называемую щеточную каемку. Благодаря
микроворсинкам происходит существенное увеличение
площади контакта кишечника с его содержимым (рис. 4.1).
В толстом кишечнике из остатков пищи удаляется
избыток воды.
Что такое переваривание и всасывание
с энергетической точки зрения
Гидролитическое расщепление белков до аминокислот,
углеводов до моносахаридов, триглицеридов до
глицерина и жирных кислот представляет собой экзоэргони-
ческий процесс, при котором значительные
отрицательные величины AG полностью сдвигают равновесие
в сторону образования продуктов гидролиза.
Всасывание часто связано с перемещением веществ
против их концентрационных градиентов и потому
требует затрат энергии.
Почему организм сам себя
не переваривает?
По своему составу пища мало чем отличается от тканей
поедающих ее животных. Ферменты, расщепляющие
пищу, образуются внутри клеток, которые при этом сами
не подвергаются их разрушительному действию. Здесь
наблюдаются две линии защиты.
Образование проферментов зимогенов
Многие пищеварительные ферменты синтезируются
в виде неактивных белков-предшественников,
называемых зимогенами, или проферментами. Именно в
таком виде они секретируются клетками, и поэтому их
цитоплазма защищена от контакта с активной формой
фермента. Амилаза, разрушающая крахмал, секретируется
часть 3 Метаболизм
72
Просвет кишки
Ворсинки, покрытые
эпителиальными клетками
Лимфатический сосуд
Сеть капилляров
t f
Микроворсинки
Вена -
Артерия-
Лимфатический сосуд
Эпителиальные клетки
щеточной каемки
Рис. 4.1. Строение внутренней поверхности тонкого кишечника (схематично)
в активном состоянии, возможно потому, что внутри
клеток-продуцентов крахмал отсутствует. Вопросы
синтеза пищеварительных ферментов и их секреции не
имеют прямого отношения к пищеварению и будут
рассмотрены в главе 22.
Защита эпителиальных клеток
с помощью слизи
Слой слизи, покрывающий внутреннюю поверхность
кишечника, - основная защита эпителиальных клеток
от активных форм пищеварительных ферментов.
Основными компонентами слизи являются муцины. Это гли-
копротеины с большой молекулярной массой,
значительная доля которой приходится на олигосахариды,
состоящие из остатков фукозы, глюкозамина, сиаловой кислоты
и т. п. Благодаря межмолекулярному взаимодействию
муцины в воде создают трехмерные молекулярные сети;
вследствие этого образуется гель, защищающий
эпителиальные клетки. Белковая часть муцинов относительно
устойчива к перевариванию благодаря защитному
действию углеводов. Муцины синтезируются и секретиру-
ются особыми бокаловидными клетками кишечного
эпителия в соответствии с потребностями пищеварения.
Переваривание белков
В нативных белках полипептидная цепь плотно
уложена в компактную глобулу, так что значительная часть
пептидных связей не доступна для гидролитических
ферментов. Отсюда возникает необходимость в
предварительной денатурации белка. Она происходит
в желудке, содержимое которого благодаря секреции
НС1 имеет рН~2. В такой среде разрываются многие
слабые связи, стабилизирующие белковую глобулу,
и она разворачивается, открывая внутренние участки
полипептидной цепи для протеолиза -
ферментативного гидролиза белков. Кроме того, соляная кислота
убивает попадающие с пищей микроорганизмы.
Образование HCI в желудке
Соляную кислоту секретируют париетальные клетки
эпителия желудка. В основе этого процесса лежит
выкачивание из клеток ионов водорода против их
концентрационного градиента. Оно происходит в результате
трансмембранного Н+/К+-обмена, источником энергии для
которого служит гидролиз АТР (сравните Na"7K+- обмен,
глава 4 Переваривание и всасывание пищи
73
описанный на с. 65). Клетки, секретирующие кислоту,
содержат Н+/К+-АТРазу. Используя энергию гидролиза
АТР, они выкачивают Н+ в обмен на входящий внутрь
клетки К+, который впоследствии выводится из клетки
(рис. 4.2). Откуда берутся протоны? Двуокись углерода
(С02), попадающая в клетку из крови, под действием
фермента карбоангидразы превращается в угольную
кислоту, которая затем диссоциирует с образованием
бикарбонатиона.
С02
Н20
— н2со3
Карбоанг™ драза
Н+
HCOi
Бикарбонатные ионы при помощи анионтранспорт-
ного белка обмениваются на ионы С1~ в крови (см
рис. 4.2), затем выходят в полость желудка, образуя НС1.
Анионный канал
α
НСОз
Просвет желудка
Электронейтральный
совместный К+/С1~ -транспорт
С02
со2^М.н2со3
из крови Карбоангидраза
АТР
Н+ г^Н+
Н+/К+-АТРаза
Апикальная мембрана
Рис. 4.2. Механизм образования НС1 в желудке
Базолатеральная мембрана,
обращенная к крови
Пепсин - протеолитический фермент
желудка
В название ферментов обычно включается суффикс -аза,
однако пищеварительные ферменты традиционно
называют: пепсин, химотрипсин, трипсин.
Названия их неактивных предшественников получают
добавлением суффикса -оген: пепсиноген, химотрипсиноген,
трипсиноген.
Эпителиальные клетки желудка секретируют
пепсиноген. Эта секреция стимулируется гормоном гастри-
ном, который клетки желудка выделяют в кровь в ответ
на поступление в него пищи. Пепсиноген превращается
в пепсин после отщепления от его полипептидной цепи
фрагмента из 44 аминокислотных остатков, который
экранирует и тем самым блокирует активный центр. При
попадании в сильно кислую среду пепсиноген
претерпевает конформационные изменения, достаточные для
того, чтобы разблокировать активный центр и
«отщепить» дополнительный фрагмент цепи. Такая активация
фермента носит аутокаталитический характер,
поскольку, как только появляется небольшое количество
пепсина, он также вступает в процесс активации, превращая
пепсиноген в активный фермент.
Сильно кислая среда является оптимальной для
работы пепсина. Этим он отличается от подавляющего
большинства ферментов, которые активны в
нейтральных условиях (см. рис. 1.3, а). Пепсин гидролизует
пептидные связи, расположенные внутри полипептидной
цепи белка-мишени, поэтому его действие приводит
к образованию смеси пептидов. Такого рода ферменты
называют эндо пептид азам и, в отличие от экзопепти-
даз, расщепляющих только пептидные связи концевых
аминокислот.
Протеолитические ферменты (протеиназы, или про-
теазы) обычно обладают избирательностью, расщепляя
пептидные связи, образованные лишь определенными
аминокислотами. Это справедливо и для пепсина,
поэтому в желудке происходит только частичное
переваривание белков. Пепсин наиболее легко разрывает
пептидные связи, NH-группы которых принадлежат
ароматическим аминокислотам: тирозину, фенилаланину или
триптофану.
Детеныши млекопитающих большую часть белков
потребляют в виде материнского молока. В их
желудках оно створаживается под действием фермента рен-
нина. Образование сгустка задерживает продвижение
нерастворимого казеина в желудочно-кишечном
тракте, в результате чего он дольше подвергается действию
протеаз.
Переваривание белков
в тонком отделе кишечника
Частично переваренная в желудке пища (химус) далее
попадает в двенадцатиперстную кишку. Кислый химус
стимулирует выделение клетками кишечника в кровь
гормонов (секретина и холецистокинина), благодаря
которым поджелудочная железа начинает секретировать
панкреатический сок. Сок имеет щелочную реакцию (как
и желчь) и нейтрализует химус, останавливая тем самым
действие пепсина и способствуя деятельности
панкреатических ферментов, которые наиболее активны
в слабощелочной среде.
Клетки поджелудочной железы секретируют целый
ряд протеиназ. В составе панкреотического сока они
поступают в мелкие протоки, сливающиеся в один
часть 3 Метаболизм
74
главный, который соединен с двенадцатиперстной
кишкой. Среди панкреатических протеиназ имеются три
эндопептидазы: трипсин, химотрипсин и эластаза.
Они поступают в кишечник в виде неактивных
предшественников: трипсиногена, химотрипсиногена и про-
эластазы. В виде неактивного предшественника
выделяется и карбоксипептидаза - экзопептидаза,
последовательно отщепляющая аминокислоты с С-конца
полипептидной цепи.
H2N—СН—CO-NH-CH—CO-NH NH-CH—СООН
I I I
R R' 4 R"
N- концевая
аминокислота
Ка боксипептидаза
С- концевая
аминокислота
Активация
панкреатических проферментов
Как и пепсиноген, панкреатические проферменты
активируются благодаря их ограниченному протеолизу. Это
тоже аутокаталитический процесс, запускаемый особым
протеолитическим ферментом - энтеропептидазой,
которая в активной форме вырабатывается клетками
тонкого кишечника. Энтеропептидаза расщепляет в трип-
синогене единственную пептидную связь, а
образующийся при этом трипсин способен уже активировать
не только трипсиноген, но и другие проферменты, что
делает активацию лавинообразной (рис. 4.3). Вся эта
тонкая и вместе с тем достаточно сложная система
преследует единственную цель - избежать активации протеаз
до их попадания в просвет кишечника. Если это
происходит, то развивается болезнь - панкреатит. Ее причиной
Трипсиноген
Трипсин
Прокарбоксипептидаза
Химотрипсиноген
Химотрипсин
Проэластаза
Карбоксипептидаза
Эластаза
Рис. 4.3. Активация панкреатических протеиназ
Активные протеиназы выделены цветом
может быть закупорка протоков или воспалительное
поражение самой поджелудочной железы. Проферменты
после их синтеза в экзокринных клетках поджелудочной
железы оказываются включенными в окруженные
мембраной пузырьки. Под влиянием гормонов или нервной
стимуляции мембраны этих пузырьков сливаются
с плазматической мембраной, и их содержимое
выводится из клетки. В случае попадания проферментов из
пузырьков в цитоплазму начинает действовать
специальный клеточный белок - ингибитор трипсина. Он
образует с трипсином настолько прочный нековалентный
комплекс, что ингибирование становится практически
необратимым.
Кроме панкреатических протеиназ в тонком
кишечнике присутствует аминопептидаза, относящаяся к эк-
зопептидазам. Этот фермент осуществляет
последовательное отщепление N-концевых аминокислот
от пептидов, образовавшихся после частичного
гидролиза белков другими протеиназами. Таким образом,
в переваривании белков в кишечнике участвуют три
эндопептидазы, расщепляющие пептидные связи внутри
белковой молекулы, и две экзопептидазы, отщепляющие
от пептидов концевые аминокислоты. Совместное
действие всех этих ферментов приводит к полному
расщеплению белков пищи до свободных аминокислот.
Перенос аминокислот
из кишечника в кровяное русло
Чтобы попасть в кровь, аминокислоты, образующиеся
в кишечнике в результате гидролиза белков, должны
сначала пройти через мембрану щеточной каймы внутрь
эпителиальных клеток (см. рис. 4.1), а после этого
проникнуть в кровеносные капилляры ворсинок
На первой стадии этого процесса происходит
накопление аминокислот внутри клеток, которое
осуществляется как симпорт аминокислот и ионов натрия (см.
главу 3). На рис. 3.11 показан механизм симпорта ионов
натрия и Сахаров; он применим и к аминокислотам, хотя
в их переносе принимают участие совсем другие
транспортные белки.
Переваривание углеводов
Основные углеводы пищи - это крахмал и другие
полисахариды, а также дисахариды - сахароза и лактоза.
Из моносахаридов встречаются лишь глюкоза и
фруктоза. Установлено, что в кишечнике всасываются
только моносахариды, поэтому при переваривании пищи все
углеводные компоненты должны быть расщеплены
до свободных моносахаридов.
глава 4 Переваривание и всасывание пищи
75
Полисахариды состоят из остатков моносахаридов,
соединенных гликозидными связями. Строение
глюкозы может быть описано двумя формулами.
α-D-Глюкоза
Р-Л-Глюкоза
В a-D-глюкозе гидроксильная группа у атома С-1
располагается ниже плоскости кольца, а в β-Ζ)-πιο-
козе - выше. Обычно в растворах обе эти формы
находятся в состоянии равновесия, которое достигается
последовательным размыканием и замыканием пирано-
вого кольца (явление, называемое мутаротацией).
Допустим, мы имеем две молекулы глюкозы, которые в
ходе химической реакции соединяются гликозидной
связью.
Гликозидная связь
(α-конфигурация)
Гликозидная связь фиксирует атом С-1 первого
остатка в определенном положении, что исключает мута-
ротацию. Из приведенной структуры следует, что
гликозидная связь между атомами С-1 и С-4 двух остатков
глюкозы находится в α-конфигурации. Образованное
таким образом соединение можно записать как глюкозо-
сс-(1—>4)-глюкоза. Это - дисахарид, тривиальное
название которого мальтоза. Существуют также дисахариды
с β-гликозидной связью.
Переваривание крахмала
Из структуры мальтозы следует, что с помощью глюко-
зидных связей могут быть образованы длинные поли-
сахаридные цепи. Такой цепью, содержащей от 100
до нескольких тысяч остатков глюкозы (глюкозильных
остатков), является амилоза - основной компонент
крахмала. Если мы обозначим один a-глюкозильный
остаток как Оч, то амилозу можно изобразить в виде цепи:
f> Х^> ^>
^-^ о ч-^ о ^-^ о
о
и т. д.,
где η соответствует числу глюкозильных остатков.
Другой компонент крахмала - амилопектин также
является полимером глюкозы. Он образует сильно
разветвленные структуры и состоит из множества коротких
цепей (каждая содержит около 30 остатков),
образованных с помощью а-(1—>4)-гликозидных связей и
соединенных между собой а-(1—>6)-гликозидными связями.
а-(1 -> 6)-Гликозидная связь между
концевым моносахаридным остатком
в одной цепи и С -6-атомом остатка
в другой цепи
-О
4 а-( 1 -> 4)-Гликозидная связь
Пользуясь теми же обозначениями, что и для
амилозы, строение амилопектина можно представить
схематически:
Переваривание крахмала осуществляется ферментом
α-амилазой, которая присутствует в слюне и в
панкреатическом соке. Она атакует гликозидные связи,
расположенные внутри цепи, поэтому может быть
отнесена к эндоферментам. α-Амилаза не расщепляет
(1—> 6)-гликозидные связи, поэтому при действии ее
на амилопектин значительная часть молекулы остается
нетронутой. Такой частично переваренный амилопектин
(его называют декстрином) расщепляется в тонком
часть 3 Метаболизм
76
кишечнике ферментом амило-а-(1—»6)-глюкозидазой.
В результате гидролизуются (1—»6)-связи и образуются
ди- и трисахариды. В свою очередь, они атакуются
другим ферментом - α-глюкозидазой, или мальтазой,
с образованием свободной глюкозы. Амилаза слюны
действует на крахмал непродолжительное время, так как
после проглатывания пищи она инактивируется кислым
содержимым желудка. Поэтому основное переваривание
крахмала происходит в тонком кишечнике. Там же
расщепляются и дисахариды: лактоза и сахароза.
Лактоза-это галактозо-р-( 1 —>4)-глюкоза. Она
является основным углеводом молока. Напомним, что
галактоза отличается от глюкозы только оптической
конфигурацией С-4-атома.
На представленной ниже структурной формуле
лактозы изломы на связях не имеют определенного
смысла, а лишь упрощают понимание структуры.
HOMO,
Н0-
-0
ОН
ОН
ОН
он
Галактоза
* +
Глюкоза
Глюкоза поглощается эпителиальными клетками
вместе с ионами натрия (рис. 4.4). Движущей силой
переноса глюкозы при этом служит градиент
концентрации Ыа+-ионов, создаваемый №+/К+-АТРазой.
Дальнейшая судьба глюкозы такова: она покидает
эпителиальную клетку через мембрану, обращенную
к кровеносному капилляру, и переносится кровью через
воротную вену в печень. Выход глюкозы из клетки
происходит в сторону уменьшения ее концентрации и
представляет собой облегченную диффузию, которая
обеспечивается специальным транспортным белком (см.
рис. 4.4). Фруктоза, в отличие от глюкозы, поглощается
клетками пассивно, без участия ионов натрия.
Глюкоза и остальные моносахариды, а также
аминокислоты и другие молекулы (кроме липидов),
всасывающиеся в тонком кишечнике, с помощью воротной
системы кровообращения доставляются непосредственно
в печень. Тем самым достигается защита организма
не только от избытка первичных продуктов
переваривания, которые далее перерабатываются в печени, но
также и от всасываемых в кишечнике токсических
соединений, которые в печени обезвреживаются.
ОН
В кишечнике лактоза расщепляется до
моносахаридов ферментом лактазой, локализованной на
внешней мембране эпителиальных клеток. Так как лактоза
является β-галактозидом, лактазу называют также
β-галактозидазой. Многие взрослые люди, особенно
выходцы из Азии, с возрастом утрачивают способность
синтезировать лактазу. В результате поглощаемая ими
с молоком лактоза не усваивается в тонком кишечнике,
а попадая в толстую кишку, подвергается там распаду
в результате действия бактерий. Это сопровождается
такими неприятными явлениями, как неумеренная жажда
и понос. Другой важный пищевой дисахарид - сахароза
расщепляется в кишечнике на глюкозу и фруктозу с
помощью фермента сахаразы.
ОН ОН
Сахароза
СН20Н
б
Сахараза
Н20
но-
4
но
5
он
но-И-
ΌΗ
о
он
но
А
он
он
сн2он
Пассивный Совместный транспорт
транспорт глюкозы за счет Ма+-градиента
***- Глюкоза
АТР
*с .
к+ Na-
ADP + Ρ,
Глюкоза
Na+
Плотное
соединение
Просвет
кишечника
Глюкоза
Фруктоза
NaVK^-АТРаза Эпителиальные клетки
щеточной каемки
Капилляр
Рис. 4.4. Совместный транспорт глюкозы и ионов натрия
при всасывании глюкозы иэ просвета тонкого кишечника
глава 4 Переваривание и всасывание пищи
11
Переваривание и всасывание жиров
Пищевые липиды представляют собой в основном
нейтральные жиры, или триглицериды (см. главу 3). Они
нерастворимы в воде и образуют в ней более или менее
крупные капли. В таком виде они не могут быть
усвоены, поскольку недоступны для ферментов.
Переваривание жиров происходит главным образом
в тонком кишечнике благодаря действию
панкреатического фермента-липазы, которая гидролизует в тригли-
церидах сложноэфирные связи, преимущественно
образованные первичными гидроксильными группами
глицерина.
\ О
CH2-0-C-R
I О
I it
СН—0-С—R + 2Н20
I о
I и
СН2—О—С—R
/
Триглицерид
ИП '
СН2ОН
I О
I и
СН—0-С—R + 2R-C00 +2Н+.
Жирная
СН2ОН кислота
Моноглицерид
Скорость расщепления триглицеридов ограничена
их доступностью для липазы. Она увеличивается
по мере уменьшения размера капелек жира и,
соответственно, роста поверхности его соприкосновения
с водным окружением. Эмульгированию жиров
способствуют как моноглицериды и свободные жирные
кислоты, образующиеся в результате расщепления
триглицеридов липазой, так и соли желчных кислот.
Моноглицериды, кроме того, уже достаточно
растворимы в воде, чтобы свободно всасываться
эпителиальными клетками. Их растворимости также
способствуют желчные кислоты и их соли.
Желчные кислоты образуются в печени и
накапливаются в желчном пузыре, откуда затем поступают
в двенадцатиперстную кишку. Они образуются из
холестерина, в молекулу которого вводятся гидроксильные
и карбоксильная группы.
часть 3 Метаболизм
78
Холестерин
Холевая кислота
В желчи человека в основном преобладает холевая
кислота; другие кислоты отличаются от нее числом и
положением гидроксильных групп. Значительная доля хо-
левой кислоты в желчи представлена в виде ее амидов,
образованных глицином (гликохолевая кислота) или его
сульфоаналогом - таурином (таурохолевая кислота). Их
называют конъюгатами, или парными желчными
кислотами, так как они состоят из двух компонентов - холе-
вой кислоты и глицина или таурина.
Формула глицина в ионной форме NH+3CH2COO~,
а таурина - NH+CH2CH2S03.
Если остаток холевой кислоты представить как
RCOO", то строение гликохолевой и таурохолевой
кислот будет RCONHCH2COO" и RCONHCH2CH2S03
соответственно. Оба эти конъюгата обладают меньшими
рКа(3,1 и 1,5) по сравнению с холевой кислотой (5,5).
Весьма вероятно, что конъюгированию способствует
полная ионизация желчных кислот в кишечике.
В присутствии желчных кислот моноглицериды
и жирные кислоты образуют смешанные мицеллы -
дископодобные частицы, края которых заполнены
молекулами желчных кислот, а более гидрофобная
сердцевина образована продуктами расщепления жиров,
холестерином и фосфолипидами. Мицелла по размерам
гораздо меньше самой маленькой жировой капли.
Концентрация продуктов распада жиров в мицеллярном
растворе может быть весьма велика, но несмотря на это
они сохраняют гомогенность и даже прозрачность. Нет
единого мнения в отношении механизма всасывания
жировых мицелл Продукты переваривания жиров
могут проникать в эпителиальные клетки либо в составе
мицелл, либо эти мицеллы распадаются на клеточной
поверхности, освобождая жирные кислоты и моногли-
цериды в виде отдельных, легко диффундирующих
внутрь клеток молекул. Желчные кислоты также
частично всасываются и транспортируются обратно в печень.
Что происходит с жирными кислотами
и моноглицеридами внутри
эпителиальных клеток щеточной каемки?
Ресинтез нейтральных липидов
Из моноглицеридов и жирных кислот в эпителиальных
клетках вновь синтезируются триглицериды.
О
СН2ОН
О
СН—0-С—R
СН2ОН
Жирные
кислоты
»■
Энергия АТР
СН2—О—С—R
I О
I и
СН—0-С—R
I О
I и
СН2—О—С—R
Ресинтез триглицеридов порождает проблему: как
их (а заодно и холестерин) вывести из эпителиальных
клеток и доставить к другим тканям организма? В
эпителиальных клетках нейтральные жиры и холестерин
собираются в мелкие частицы, называемые хиломикро-
нами. Они окружены мембранной оболочкой и
выводятся из клеток посредством экзоцитоза.
Что представляют собой хиломикроны?
Хиломикроны - сферические частицы, сердцевина
которых заполнена гидрофобными молекулами
нейтральных жиров и эфиров холестерина (рис. 4.5).
Гидрофобная сердцевинь
из триглицеридов и
эфиров холестерина
"ν Белки
Гидрофильная оболочка
из фосфолипидов и холестерина
Рис. 4.5. Строение хиломикрона
Последние отличаются от холестерина тем, что в них
гидроксильная группа этерифицирована жирными
кислотами.
О
II
R—С—(Г
Эфир холестерина
Такая модификация обеспечивает упаковку
холестерина в гидрофобную внутреннюю часть хиломикрона.
Поверхность хиломикрона формируется молекулами
фосфолипидов, холестерина и особыми белками.
Таких белков известно несколько. Главным и
необходимым для образования хиломикрона является глико-
протеин аполипопротеин В. Приставка апо- означает,
что этот белок в его естественном состоянии является
компонентом липопротеина. В данном случае таким
липопротеином служит хиломикрон. Гидрофильная
оболочка стабилизирует хиломикроны настолько, что они
разносятся кровью и лимфой по организму как целые
частицы. Более чем на 90% они состоят из жиров,
благодаря чему отличаются малой плотностью.
В отличие от моносахаридов и аминокислот,
хиломикроны попадают из эпителиальных клеток не в кровь,
а в лимфатические капилляры, также пронизывающие
клеточные ворсинки. Здесь полезно рассказать немного
о лимфе. Когда кровь проходит по капиллярам,
происходит фильтрация прозрачной лимфатической
жидкости, содержащей белки, электролиты и другие вещества,
в тканевую (интерстициальную) жидкость. Все ткани
пронизаны сетью лимфатических капилляров, которые
своими окончаниями собирают лимфу из
межклеточного пространства. Система лимфатических капилляров
и сосудов завершается грудным протоком, через
который лимфа поступает в вены шеи. Благодаря высокому
содержанию хиломикронов после смешивания с
лимфой кровь мутнеет и начинает опалесцировать.
глава 4 Переваривание и всасывание пищи
79
Переваривание других компонентов
пищи
Пищеварение - это процесс ферментативного
гидролитического расщепления, и ему подвергаются почти все
компоненты пищи.Так, фосфолипиды пищи
расщепляются фосфолипазами, а нуклеиновые кислоты - нукле-
азами. Растительная пища содержит целлюлозу -
полисахарид, который не поддается пищеварительным
ферментам животных. Однако травоядные животные
все же могут использовать целлюлозу благодаря тому,
что ее расщепляют обитающие в их пищеварительном
тракте микроорганизмы. Для человека целлюлоза в
пище - всего лишь волокнистый наполнитель, полезный
для нормальной работы кишечника.
Дополнительная литература
Переваривание и всасывание жиров
Riddihough G. Picture of an enzyme at work //Nature. 1993.
Vol. 362. P. 793.
(Рассказ о том, как активируется при переваривании
жиров панкреатическая липаза.)
Вопросы к главе 4
1. Какие пищеварительные ферменты синтезируются
в виде неактивных предшественников?
2. Для чего это нужно? Почему амилаза синтезируется
сразу в активной форме? Почему активные
ферменты не действуют на стенки тонкого кишечника?
3. Как происходит активация проферментов в тонком
кишечнике?
4. Какими последствиями для организма чревата
преждевременная активация панкреатических
проферментов?
5. Почему нужно переваривать пищу?
6. Сахара и многие аминокислоты транспортируются
в эпителиальные клетки против своего
концентрационного градиента, однако при этом не используются
специальные транспортные системы, АТР или
другие макроэргические фосфаты. Как объяснить это
внешне парадоксальное явление?
7. Отчего многие люди, особенно выходцы из Азии,
плохо переносят молоко и молочные продукты?
8. Что представляют собой нейтральные жиры?
9. Пищевые жиры нерастворимы в воде. Как при этом
с ними справляются пищеварительные ферменты?
10. Аминокислоты и сахара, поглощенные в тонком
кишечнике, далее переносятся кровотоком в печень.
Какова судьба продуктов расщепления жиров?
часть 3 Метаболизм
80
5 Предварительные сведения
^^^^^ о распределении и утилизации источников
энергии в различных тканях организма
Эта глава посвящена собственно метаболизму. Прежде
всего будет рассмотрен обмен молекулами между
кровью и тканями, а также между различными тканями.
Затем мы обсудим механизмы регуляции этих
процессов, обеспечивающих физиологические потребности
организма, имея в виду распределение источников
энергии среди его потребителей.
Главная задача метаболизма - обеспечить организм
за счет окисления пищевых веществ энергией,
запасенной в виде АТР. Пищевые молекулы используются
как сырье для создания компонентов клеток и других
необходимых для организма соединений. Отходы, т. е.
ненужные организму вещества, преобразуются в
соединения, которые могут быть выведены с мочой.
Некоторые специализированные клетки новорожденных
используют окисление пищевых веществ для
образования тепла. Выделение тепла - это побочное явление
при метаболизме.
Запасание пищи
в теле животного
Как известно, животные принимают пищу через
определенные промежутки времени, а не постоянно. Многим
из них свойственны циклически повторяющиеся
периоды насыщения и голодания. У людей приемы пищи
могут быть разделены малыми интервалами времени (в
течение дня), большими перерывами (во время ночного
сна) или значительными сроками (при голодании).
Биохимические машины должны как-то приспосабливаться
к этим ситуациям.
Сразу после еды кровь насыщается пищевыми
продуктами, поступающими в нее из кишечника. Однако они
не сохраняются там долго, а быстро переходят в ткани,
так что их содержание в крови вскоре снижается до
прежнего низкого уровня. Например, после приема
жирной пищи развивается липемия (плазма крови
становится похожей на молоко), но это состояние проходит уже
через несколько часов. После еды концентрация глюкозы
в крови возрастает в течение часа от 5 до 7,8 мМ, но уже
через два часа (если человек не болен диабетом) она
возвращается к норме. Хотя ткани быстро поглощают
пищевые вещества из крови, используют они их не
сразу. Большая часть поглощенных тканями веществ
запасается впрок.
Как различные виды пищевых веществ
хранятся в клетках?
Глюкоза запасается в виде гликогена
Для клеток непрактично накапливать и сохранять
глюкозу в свободном виде, поскольку при этом
внутриклеточное осмотическое давление достигло бы
недопустимо высоких значений. Напомним, что осмотическое
давление пропорционально молярной концентрации
растворенного вещества. Но если множество отдельных
растворенных молекул объединяются в один полимер,
то накопление глюкозы не приведет к заметному росту
осмотического давления. Образующиеся при этом
полимеры не всегда остаются в растворе, они могут
выпадать из него в виде гранул. У животных глюкоза по-
лимеризуется в гликоген, или .-животный крахмал,
который по своему строению сходен с амилопектином, но
отличается большей разветвленностью цепей (см. с. 76).
Синтез гликогена требует расхода энергии. При
необходимости гликоген расщепляется в клетке с
высвобождением глюкозы. Следует подчеркнуть, что запасы
гликогена у животных крайне ограничены. Так, у человека
гликоген хранится в печени, из которой глюкоза,
образующаяся после расщепления гликогена доставляется
к другим органам и тканям. Весь запас гликогена
исчерпывается после суточного голодания. Это обстоятельство
имеет существенное значение для биохимии животных.
Что касается других пищевых моносахаридов -
галактозы и фруктозы, то в организме они превращаются
в глюкозу (либо гликоген) или в соединения,
являющиеся метаболитами глюкозы.
Хранение жиров
Основная часть жиров (триглицеридов) хранится в
жировой ткани или жировых клетках, рассредоточенных
по разным участкам тела. Эти клетки под микроскопом
часто выглядят как жировые капли, окаймленные
тонким слоем цитоплазмы и мембраной (рис. 5.1).
глава 5 Распределение и утилизация источников энергии в организме
81
Ядро —
Цитоплазма
Капли
/ жира
5 мкм
Рис. 5.1. Жировая клетка (схематично)
В отличие от гликогена, жиры могут накапливаться
в организме в неограниченном количестве. В норме
человек запасает в виде жиров в 50 раз больше энергии,
чем в виде гликогена. Чем же объясняется такое
соотношение, особенно если учесть важную метаболическую
роль глюкозы? Жиры представляют собой гораздо
более восстановленные соединения, чем глюкоза, а
следовательно, при окислении равных весовых количеств
жиров и глюкозы в первом случае выделится гораздо
больше энергии. Таким образом, жиры - наиболее
компактная форма запасания энергии. Парадоксально, но
мы выглядели бы куда более толстыми, если бы в нашем
теле накапливался не жир, а гликоген.
Поскольку глюкоза запасается в небольшом
количестве, а поступает ее в составе пищи довольно много,
избыток перерабатывается в жир. Схема создания
энергетических резервов в форме гликогена и жиров показана
на рис. 5.2.
Глюкоза в организме легко перерабатывается в
жиры, однако не существует метаболического пути
превращения жирных кислот в глюкозу Вклад глицерина
в клеточную энергетику несоизмеримо меньше, чем
от длинноцепочечных жирных кислот.
Другие сахара
из пищи „
Глюкоза ^^
из пищи
Гликоген
Нейтральные жиры
в жировых клетках
Запасаются ли аминокислоты
в организме?
Семена растений содержат белки, единственная
функция которых - служить источником аминокислот для
развивающегося зародыша. У животных только белки
яиц и молока можно рассматривать как форму
резервирования аминокислот. Пищевые аминокислоты,
поступающие в кровь животных, расходуются на синтез
белков, нейромедиаторов и т. п., а неиспользованные
аминокислоты подвергаются расщеплению. При этом
азот, входящий в состав аминокислот, оказывается
включенным в молекулу мочевины (нейтрального,
водорастворимого и нетоксичного соединения) и выводится
из организма животного с мочой. Лишенная азота
молекула в зависимости от ее строения перерабатывается
в жиры или в гликоген, либо окисляется в соответствии
с энергетическими нуждами организма (рис. 5.3).
Белки и другие
компоненты тканей
Аминокислоты
из пищи
Избыточные
аминокислоты
>'л ал с пни
Nib груши,ι
Мочевина
Моча
С—Η-остовы молекул
Жиры и их
производные
Глюкоза
Н20 + С02
> Л\НЛ \\ i :
Рис. 5.3. Судьба аминокислот, поступающих в организм
с пищей
Выбор одного из метаболических путей (я, б или в) зависит
от строения аминокислоты, физиологического состояния
организма и способа регуляции
В то же время все клеточные белки можно
рассматривать как форму хранения аминокислот. Особенно это
относится к сократительным белкам мышечных клеток,
которые расщепляются в условиях, когда организму
не хватает пищевых аминокислот.
Энергетический метаболизм
в различных тканях
Рис. 5.2. Запасание Сахаров и жиров после приема пищи
Гликогена запасается немного, а жиры накапливаются в
неограниченных количествах, главным образом в жировых клетках
Все ткани организма различаются по своим
биохимическим характеристикам. Мы остановимся на тех
тканях, которые имеют первостепенное значение при
часть 3 Метаболизм
82
распределении и усвоении пищевых веществ. Речь
пойдет о печени, скелетных мышцах, мозге, жировых
клетках и эритроцитах. Органы, выполняющие регуляторную
функцию, вроде поджелудочной железы и
надпочечников, здесь рассматриваться не будут.
1. Печень играет центральную роль в обеспечении
постоянства концентрации глюкозы в крови. Когда
уровень глюкозы в крови высок (после приема пищи), ее
избыток откладывается в печени в виде гликогена.
Когда появляется потребность в ней, гликоген в печени
распадается и глюкоза поступает в кровь.
Когда голодание длится более суток, запасы
гликогена в печени исчерпываются, и если ничего не
предпринять, содержание глюкозы в крови может упасть до
столь низкого уровня, что возникнет опасность для
жизни организма. В таком случае организм мобилизует
запасы жиров, которые выбрасываются в кровь клетками
жировой ткани. Однако с их помощью можно
поддержать мышечные и другие ткани, но не мозг и
эритроциты, которые нуждаются только в глюкозе и не
способны усваивать жирные кислоты. Печень справляется
с этой проблемой, превращая аминокислоты в глюкозу.
Этот процесс называют глюконеогенезом. Главным
источником аминокислот при этом служат
расщепляемые мышечные белки. Разумеется, мышцы страдают, но
все же это предпочтительней, чем смерть от нехватки
глюкозы.
Не менее важную роль печень играет и в
метаболизме жиров. Когда при длительном голодании в кровь
выбрасываются жирные кислоты, они усваиваются
всеми тканями (за исключением мозга и эритроцитов).
Если же мобилизация жиров достигает значительного
уровня, печень превращает жирные кислоты в
соединения, называемые кетоновыми телами. Они поступают
в кровь и также усваиваются тканями (исключая
эритроциты). Кетоновые тела могут обеспечить до
половины энергетических потребностей мозга, уменьшая тем
самым его зависимость от глюкозы. Печень способна
не только расщеплять, но и синтезировать жирные
кислоты, которые затем в виде триглицеридов
переносятся в другие ткани.
Таким образом, печень запасает глюкозу в виде
гликогена и освобождает ее, если в этом есть потребность
и не исчерпан запас гликогена. При голодании она
превращает аминокислоты в глюкозу, а жиры - в
кетоновые тела, снабжая ими другие ткани через кровоток
(рис. 5.4). Печень способна не только синтезировать
и экспортировать жирные кислоты, но и окислять их для
энергетических нужд. И это только те функции печени,
которые связаны непосредственно с энергоснабжением
организма.
2. Мозг не имеет собственных энергетических
резервов и должен непрерывно снабжаться глюкозой.
Транспорт глюкозы в нейроны носит пассивный
характер, а потому при уменьшении ее концентрации в крови
(гипогликемия) он прекращается, что приводит к
судорогам и коме. Тем не менее, хотя глюкоза абсолютно
необходима для работы мозга, до половины своих
энергетических потребностей он может удовлетворить за счет
кетоновых тел, экономя глюкозу для тех случаев, когда
она незаменима.
3. Скелетные мышцы расходуют много энергии
в ходе сокращения. Эта энергия черпается из различных
источников. Мышцы поглощают глюкозу из крови и
могут запасать ее в виде гликогена. Однако, в отличие от
печени, глюкоза, высвобождающаяся из гликогена мышц
не поступает в кровь. Помимо глюкозы, в мышцах
окисляются жирные кислоты, кетоновые тела и
аминокислоты. Более того, если уровень жирных кислот и
кетоновых тел в крови достаточно велик, они становятся
главными субстратами окисления. При голодании
мышцы «жертвуют» своими белками ради того, чтобы
снабдить печень аминокислотами для нужд глюконеогенеза.
4. Роль жировых клеток относительно проста.
После еды они поглощают из крови жирные кислоты
и глюкозу и преобразуют эти вещества в триглицериды.
V
Жирные
кислоты
мозгом не
используются
Печень
Мозг
Производство
энергии
Жирные
кислоты
Кетоновые
тела
Гликоген
(резерв на сутки)
лг
Аминокислоты
Мышца
г- щ
Производство
энергии
Мышечный
белок
Аминокислоты
L
Глицерин
Жирные
Триглице идь
Жировые клетки
Рис. 5.4. Потокои биологического топлива в голодающем
организме
глава 5 Распределение и утилизация источников энергии в организме
83
Напротив, при уменьшении уровня глюкозы в крови,
указывающем на голод, жировые клетки используют
запас триглицеридов для того, чтобы выделять в кровь
жирные кислоты с целью удовлетворения потребности
в них других тканей. Жировые клетки и печень -
основные производители триглицеридов. Жирные
кислоты либо поступают с пищей, либо синтезируются
в печени, а глицерин образуется из глюкозы.
5. Эритроциты способны усваивать только
глюкозу, превращая ее в молочную кислоту. У них
отсутствуют митохондрии, поэтому, в отличие от других клеток,
они не могут окислять пищевые вещества. Таким
образом, глюкоза обеспечивает все их энергетические
потребности, включая работу Na+/K+-ATPa3bi.
Роль гормонов в координации
распределения пищевых
веществ
После рассмотрения роли отдельных органов и тканей
в усвоении пищевых веществ целесообразно обсудить,
как осуществляется координация их деятельности,
позволяющая организму справиться с различными
физиологическими ситуациями. Вот наиболее типичные из них.
1. Избыток пищи после плотного обеда.
2. Легкий голод после некоторого перерыва между
приемами пищи, например, утром перед завтраком.
3. Голод - состояние, наступающее после отсутствия
пищи в течение одного-двух дней.
5. Аномальные условия (например, при диабете
глюкоза не усваивается из крови.)
6. Экстремальная ситуация (под воздействием стресса
в кровь выбрасывается огромное количество
адреналина).
После всасывания пищевых веществ
Все компоненты пищи в значительных концентрациях
присутствуют в крови. Глюкоза поглощается печенью,
мышцами и жировыми клетками и используется для
пополнения запасов гликогена. Наряду с этим часть
глюкозы в печени, жировых клетках и других тканях
превращается в нейтральные жиры.
Сама печень не накапливает синтезированные жиры
в значительных количествах, а отдает их другим тканям
(ожирение печени - патология).
Аминокислоты поглощаются всеми тканями и
используются для синтеза белков и других компонентов
клеток. Жиры поступают из хиломикронов и поглощаются
часть 3 Метаболизм
84
разными тканями, в том числе жировыми клетками, где
они откладываются для хранения, и клетками молочных
желез, которые используют их при образовании молока.
В период накопления гликогена и жиров расщепления
этих веществ в клетках не происходит.
Главным сигналом, побуждающим все ткани
организма перейти в режим накопления, служит гормон
поджелудочной железы инсулин, выброс которого в кровь
«извещает» все клетки о повышении в ней
концентрации глюкозы: ситуация благоприятна для синтеза
гликогена и жиров. В то же время высокое содержание
глюкозы в крови приводит к снижению уровня другого
панкреатического гормона - глюкагона, чье действие
противоположно инсулину. Глюкагон извещает клетки
о недостатке пищевых веществ и побуждает их
мобилизовать накопленные запасы: гликоген и нейтральные
жиры. Заметим, что мозг не чувствителен к инсулину; он
нуждается в непрерывном поступлении глюкозы.
Легкий голод
Стимулированное инсулином запасание приводит к
постепенному снижению содержания в крови глюкозы,
аминокислот и жиров в виде хиломикронов.
Параллельно с уменьшением концентрации глюкозы уменьшается
и секреция инсулина. Этот белковый гормон в крови
быстро расщепляется протеиназами, и потому торможение
секреции приводит к практически полному его
исчезновению в крови. С другой стороны, уменьшение
концентрации глюкозы побуждает поджелудочную железу сек-
ретировать глюкагон. Этот гормон служит сигналом для
печени и жировых клеток к мобилизации запасов
гликогена и жиров, чтобы обеспечить поступление в кровь
глюкозы и жирных кислот. Последние используются
в качестве топлива мышцами, печенью и другими
тканями, что позволяет сберечь глюкозу для нужд мозга.
Одновременно низкий уровень инсулина в сочетании с
высоким уровнем глюкагона подавляет синтез гликогена
и жиров из глюкозы.
Продолжительное голодание
Уже после суточного голодания запасы гликогена в
печени иссякают, и она далее не может поддерживать
на должном уровне концентрацию глюкозы в крови
(на большее время хватает гликогена в почках, но их
вклад в этот процесс незначителен) Уровень инсулина
низок, а глюкагона - высок. В этих условиях жировые
клетки начинают выделять в кровь жирные кислоты,
которые становятся главным источником энергии для
тканей. Запаса жиров в этих клетках хватает не на одну
неделю. В мышцах происходит распад белков до
аминокислот, из которых в печени под влиянием глюкагона
синтезируется глюкоза. Чем дальше, тем интенсивней
побуждает глюкагон жировые клетки увеличивать
содержание жирных кислот в крови, и в печени включается
механизм образования кетоновых тел. Под этим давно
используемым и неправильным термином понимают два
соединения - ацетоацетат (СН3СОСН2СОО~) и β-гид-
роксибутират (СН3СНОНСН2СОО ). Второе
вещество - не кетон, и оба не являются «телами». Раньше
полагали, что наличие кетоновых тел свидетельствует
о развитии патологических состояний, однако в
действительности их синтез происходит в процессе
нормального метаболизма.
Диабет
В отсутствие инсулина мышцы не могут использовать
глюкозу, как бы много ее ни было в крови. Именно так
обстоит дело у диабетиков, чью жизнь спасает только
то, что мозг и эритроциты усваивают глюкозу
независимо от действия инсулина. Состояние организма при
диабете напоминает длительное голодание, при котором
отношение концентраций инсулин/глюкагон крайне
мало, а жировые клетки продуцируют много жирных
кислот, часть которых перерабатывается печенью в
кетоновые тела. Содержание последних становится
значительно выше нормы, что легко обнаружить по характерному
сладковатому «органическому» запаху изо рта. Это
запах ацетона, который появляется путем спонтанного де-
карбоксилирования ацетоацетата:
сн3сосн2соо-
СН3СОСН3 + С02.
Экстремальная ситуация
Когда человеку грозит опасность, его надпочечники,
повинуясь нервному сигналу из мозга, выбрасывают в кровь
адреналин. Жировая ткань иннервирована, и ее
активность также стимулируется адреналином,
высвобождаемым нервными окончаниями. Адреналин
действует на организм подобно нажатию кнопки «Тревога!».
Он побуждает печень и жировые клетки выделять в кровь
глюкозу и жирные кислоты в таких количествах, чтобы
мышцы не испытывали недостатка в «топливе». Кроме
того, он вызывает распад гликогена в самих мышцах,
чтобы их клетки могли производить АТР с
максимальной скоростью. В совокупности все эти меры приводят
мышцы в рабочее состояние, позволяющее их хозяину
избежать опасности.
Вопросы к главе 5
1. Почему клетки запасают глюкозу в виде гликогена?
2. Почему триглицеридов запасается гораздо больше,
чем гликогена?
3. Может ли глюкоза перерабатываться в организме
в жиры и наоборот? Существуют ли какие-либо
специальные формы запасания аминокислот?
4. Опишите роль печени в преобразовании и
распределении питательных веществ.
5. Использует ли мозг в качестве источника энергии
жирные кислоты?
6. Перечислите основные метаболические свойства
жировых клеток.
7. Что служит источником энергии в эритроцитах?
8. Какие гормоны несут главную ответственность за
утилизацию пищевых веществ?
глава 5 Распределение и утилизация источников энергии в организме
85
глава
6 Биохимические механизмы транспорта,
хранения и мобилизации пищи
Превращение глюкозы
в организме
Механизм синтеза гликогена
Прежде всего отметим, что синтез гликогена из
глюкозы - процесс эндоэргонический, т. е. требующий затрат
энергии. AG0' гидролиза гликозидной связи составляет
—16 кДж · моль1, так что равновесному состоянию
отвечает практически полное гидролитическое
расщепление гликогена до глюкозы (гликоген + Н20 —> глюкоза).
Образование гликозидной связи протекает в организме
сопряженно с гидролизом макроэргического
фосфатного производного.
Синтез гликогена происходит путем увеличения
существующей затравки молекулы гликогена (именуемой
праймером) за счет последовательного присоединения
отдельных молекул глюкозы. Таким праймером служит
белок гликогенин, к одному из тирозиновых остатков
которого с помощью О-гликозидной связи
присоединена олигосахаридная цепочка из 8 остатков глюкозы.
Этот белок так и остается включенным в гранулу
гликогена. Молекулы глюкозы всегда присоединяются к не-
восстанавливающему (нередуцирующему) концу
полисахарида. Глюкоза принадлежит к классу Сахаров,
называемых альдозами. В водных растворах ее
циклическая (преобладающая) форма находится в равновесии
с открытой формой, которая содержит альдегидную
группу.
Альдегидная группа
Н0-
Н0-
НСУ
ч*ОН
ОН
но
Ч*0Н
-он
τ
iCHO
он
он
Невосстанавливающий Восстанавливающий
конец цепи конец цепи
Глюкоза Глюкоза
(циклическая форма) (открытая форма)
Альдегидная группа - это восстанавливающий агент,
так что С-1 -конец кольцевой формы называется
восстанавливающим, а С-4-конец - ^восстанавливающим.
Цепь гликогена поэтому всегда имеет
невосстанавливающий конец.
Итак, молекула гликогена схематически может быть
представлена в ел едущем виде:
Невосстанавливающий
конец цепи
й |£jX ХТ^Х ХГ^Х ХГУ- ,
t>
ОН
остаток глюкозы.
О
а ее биосинтез с участием АТР можно описать схемой:
он ^o^V^V™
Глюкоза ν/ Праймер гликогена
<— Энергия АТР
0>Ο0Ο0Ονитл·
Этот процесс повторяется многократно, что и
приводит к образованию длинной полисахаридной цепи.
Как синтез гликогена обеспечивается
необходимой энергией?
Когда глюкоза входит в клетку, она подвергается
фосфорилированию посредством АТР. Эту реакцию
в мозге и мышцах катализирует фермент гексокиназа.
+ АТР
ОН
Глюкоза
Гексокиназа
(глюкокиназа в печени)
Δ60 =-16,7кДж-моль1
ADP
ОН
Глюкозо-6-фосфат
часть 3 Метаболизм
86
Киназы - семейство ферментов, переносящих фосфо-
оильный остаток с АТР на различные молекулы, а глю-
ко3а - один из представителей моносахаридов - гексоз;
отсюда и название гексокиназа. В печени эту же
реакцию катализирует другой фермент - глюкокиназа.
фосфорилирование - экзоэргоническая и поэтому
практически необратимая реакция. Образующийся глю-
козо-6-фосфат в отличие от самой глюкозы является
сильно заряженной молекулой, из-за чего она не может
дйффундировать через плазматическую мембрану. В то
же время фосфорилирование сопровождается
уменьшением концентрации свободной глюкозы в
цитоплазме. Оба эти фактора способствуют диффузии глюкозы
в клетки из окружающей среды в соответствии с ее
концентрационным градиентом. Фосфорильный остаток
в молекуле глюкозофосфата под влиянием фермента
фосфоглюкомутазы может обратимо мигрировать
между гидроксильными группами при С-6- и С-1-атомах.
23ОРО-
нсг
.он
он
он
Глюкозо-6-фосфат (G-6-P)
Фосфоглюкомутаза
AG0' = -7,3 кДж· моль-1
Глюкозо-1-фосфат (G-1-P)
Величины свободной энергии гидролиза
глюкозо-1-фосфата и гидролиза а-(1—>4)-гли-
козидной связи гликогена одинаковы и равны
-А.0 кДж · моль-1. Свободная энергия
гидролиза щ 1 ->4)-гликозидной связи в дисахариде
мальтозе равна -15,5 кДж · моль-1. Поэтому вполне
естественно предположить, что наращивание
цепи гликогена происходит в результате реакции
между глюкозо-1-фосфатом и праймером
гликогена. Но это ошибочное мнение. Биохимические
реакции комбинируются таким образом, чтобы
суммарный процесс оказался термодинамически
необратимым. Между тем прямой синтез гликогена
из глюкозо-1-фосфата оказался бы реакцией обратимой
и, следовательно, не контролируемой.
Чтобы придать синтезу гликогена
термодинамическую необратимость, в него введена дополнительная
стадия. Чтобы разобраться в ней, вспомним структуру АТР
(см. рис. 1.6). Его аналогом является уридинтрифосфат
(UTP), в молекуле которого место аденина занимает ура-
цил. Клетка синтезирует UTP, используя АТР в качестве
источника энергии. Оказалось, что при синтезе
гликогена промежуточным продуктом является уридиндифос-
фоглюкоза (UDP-глюкоза), образующаяся из UTP и
глюкозо-1-фосфата (рис. 6.1)
По чисто формальной причине (так требуют правила
номенклатуры) фермент, катализирующий эту реакцию,
назван по обратной реакции, реально в клетке не
протекающей - UDP-глюкозопирофосфорилазой.
Необратимость этой реакции достигается тем, что
неорганический пирофосфат (продукт прямой реакции) в клетке
чрезвычайно быстро гидролизуется пирофосфатазой.
Именно UDP-глюкоза, а не глюкозо-1-фосфат,
выступает как донор остатка глюкозы при синтезе гликогена.
Ее можно рассматривать как активированный сахар,
и в таком качестве UDP-производные Сахаров
участвуют в биосинтетических реакциях у животных. Почему
Уридильный остаток
О
II
он о-
Гл юкозо-1 -фосфат
ОН
UDP-глюкоза
~0—Р—О—Р—О—Р—О—Урндин
I I I
О" О" О
UTP
UDP-глюкозопирофосфорилаза
О О
II II
-Уридии + "О— Р—О— Р—ОН
ι ι
о о-
Неорганический пирофосфат
/ (ΡΡί)
/ н2о
2Ό— Р— 0Н + Н+
I
0"
Неорганический фосфат (Р;)
Рис. 6.1. Образование UDP-глюкозы из UTP и глюкозо-1-фосфата
глава 6 Транспорт пищи, ее хранение и мобилизация
87
он
UDP-глюкоза
О О
I I т кон
О—Ρ—Ο—Ρ—О —Уридин НО
о- о-
ит.д.
ОН 'ОН ■ ОН
Праймер гликогена
(п звеньев глюкозы)
UDP +
и т.д.
Гликоген
(п + 1 звеньев глюкозы)
Рис. 6.2. Синтез гликогена путем удлинения затравки
именно они - загадка, вторую еще предстоит разгадать.
Во всяком случае, в растениях синтез крахмала
происходит с участием не урацильных, а адениновых
соединений. Схема синтеза гликогена с участием UDP-глюкозы
в качестве донора глюкозного остатка представлена на
рис. 6.2. Эту реакцию катализирует фермент гликоген-
синтаза (синтазами называют ферменты,
катализирующие синтетическую реакцию без использования АТР;
в противном случае используют термин синтетазы).
Еще раз вернемся к вопросу о необратимости реакции
Гексокиназа
Глюкоза + АТР ► Глюкозо-6-фосфат
(глюкокиназг
в печени
Фосфоглюкомутаза
UTP + Глюкозо-1 -фосфат
UDP-
глюкозопи ^ ol ^ илаза
UDPG + РР
ГликогеН(л+1ч
+ UDP
1 ликоген-
синтаза
Праймер гликогена
Рис. 63. Синтез гликогена из глюкозы
образования UDP-глюкозы из UTP и глюкозо-1-фосфата.
В ходе этой реакции (см. рис. 6.1) образуется
неорганический пирофосфат, гидролиз которого протекает
необратимо (AG°= -33,5 кДж · моль1). Именно это
обстоятельство придает необратимость всей совокупности
реакций, включенных в биосинтез гликогена (рис. 6.3).
Этим же объясняется невозможность распада гликогена
простым обращением реакций его синтеза.
Эта схема не отражает полностью процесса синтеза
гликогена, поскольку он должен представлять собой
неопределенно длинную олигосахаридную цепь, наподобие
той, которая встречается у амилозы - линейного
компонента растительного крахмала. Между тем гликоген,
также как и другой компонент крахмала - амилопектин,
имеет разветвленную структуру. Такую структуру ему
придает специальный фермент, который называют
разветвляющим ферментом. Как только синтаза
изготовила очередной линейный участок цепи длиной
примерно в 11 глюкозных остатков, разветвляющий фермент
переносит ее концевой фрагмент, содержащий в
среднем 7 остатков глюкозы, на гидроксильную группу
С-6-атома глюкозы этой или другой цепи (рис. 6.4).
Энергия образования а-(1—>6) связи примерно такая же,
что и связи а-(1—>4). Следовательно, это - реакция
простого переноса, не требующая затрат энергии. По мере
синтеза в гликогене лавинообразно возрастает число
концов цепей, являющихся как точками роста
молекулы, так и местами ее последующей фрагментации.
Итак, после еды глюкоза из крови поступает в тка- %
ни и превращается в ходе рассмотренных реакций
в гликоген.
часть 3 Метаболизм
88
но Ό ο ο ο 'ρΑΑ,ΑΑ^
гликогена
ос-(1—4)-связь
Разветвляющий фермент
ос-(1-* 6)-точка ветвления
1
НО
0 0
Рис. 6.4. Действие разветвляющего фермента при синтезе гликогена
Ядро
гликогена
Как в печени образуется глюкоза?
Печень запасает глюкозу в виде гликогена не столько для
своих собственных нужд, сколько для обеспечения
постоянного поступления глюкозы к другим тканям,
особенно к мозгу и эритроцитам. В перерывах между
приемами пищи печень расщепляет накопленный в ней
гликоген с такой скоростью, чтобы по возможности
удержать на постоянном уровне концентрацию глюкозы
в крови. Эта скорость обусловлена внешним сигналом,
который определяется соотношением глюкагон/инсулин.
Так же, как и синтез, расщепление гликогена
происходит с невосстанавливающего конца полисахарид-
ных цепей. При этом гликозидная связь атакуется не
водой, а фосфатным анионом; такого рода реакции по
аналогии с гидролизом называют фосфоролизом.
Соответственно и катализирующий эту реакцию фермент
носит название гликогенфосфорилазы.
О- Гликоген
(п-\ остатков)
Образующийся при фосфоролизе глюкозо-1-фосфат
далее изомеризуется фосфоглюкомутазой в глюко-
зо-6-фосфат (реакция, обратная описанной выше),
который гидролизуется до глюкозы, и последняя
выделяется в кровь (такой гидролиз, катализируемый глюкозо-
6-фосфатазой, происходит только в печени и почках):
Глюкозо-1 -фосфат ^ Гпюкозо-6-фосфат
Глюкозо-6-фосфат + Н20 -^ Глюкоза + Р,.
Общая схема образования глюкозы из гликогена
представлена на рис. 6.5.
Глюкозо-6-фосфатаза локализована на мембране эн-
доплазматического ретикулума. Этот фермент
примечателен тем, что его активный центр обращен не в
цитоплазму, а в просвет ретикулума. Чтобы связаться с ним,
глюкозо-6-фосфат с помощью особого транспортного
белка пересекает мембрану и только после этого
гидролизуется. В свою очередь, продукты гидролиза -
глюкоза и фосфат - с помощью специальных транспортных
систем возвращаются обратно в цитоплазму.
Цепь гликогена (п остатков глюкозы)
>] Гликогенфосфорилаза
Глюкозо-1-фосфат + цепь гликогена
(остатки глюкозы^ _ j
Фосфоглюкомутаза
Глюкозо-6-фосфат
Глюкозо-6-фосфатаза
Глюкоза + Ρ,
Рис. 6.5. Расщепление гликогена путем фосфоролиза,
завершающееся высвобождением глюкозы в кровь
глава 6 Транспорт пищи, ее хранение и мобилизация
89
Точки ветвления в гликогене затрудняют работу
фосфорилазы, видимо, из-за ее размера и
особенностей строения активного центра. Она не может
справиться со своей задачей, если конец цепи и точка
ветвления разделены четырьмя или меньшим числом гли-
козидных остатков. Эта проблема преодолевается в два
этапа. На первом этапе три концевых остатка
переносятся под действием деветвящего фермента на гид-
роксильную группу С-4-концевого остатка другой
короткой цепи, что удлиняет последнюю и тем самым
делает ее доступной для фосфорилазы. Второй этап
заключается в гидролитическом отщеплении
оставшегося бокового а-(1—>6)-гликозильного остатка
(рис. 6.6). Примечательно, что оба эти процесса
катализирует один и тот же белок - деветвящий фермент,
который, следовательно, обладает двумя разными
ферментативными активностями.
Общая схема поглощения, хранения и
высвобождения глюкозы из печени показана на рис. 6.7.
Перенос
Отметим некоторые принципиально важные
особенности процессов, отображенных на этой схеме.
1. Синтез и распад гликогена проходят разными
путями, а потому оба процесса могут регулироваться
независимо друг от друга.
2. Только в печени и почках глюкозо-6-фосфат,
образующийся из гликогена, превращается в глюкозу,
которая далее высвобождается в кровь, становясь
доступной другим тканям.
3. И в печени, и в других тканях глюкозо-6-фосфат
служит субстратом окисления (см. главу 8).
4. Инсулин побуждает клетки накапливать гликоген,
одновременно блокируя его распад. Глюкагон действует
наоборот.
Таким образом, инсулин вызывает синтез гликогена,
тогда как глюкагон стимулирует выход глюкозы из
печени. Механизм действия этих гормонов будет
рассматриваться в последующих главах, посвященных
молекулярным сигналам, которые управляют жизнью клеток.
НО
НО
гликогена
wo
-ex
Трансферазная активность деветвящего
фермента, удаляющего разветвления
<A<ix-
Новая ос-(1 — 4)-связь
^U j Гидролиз Ъ^-Х S~\
..^ χ)6 Ov
но
у\
гликогена
Рис. 6.6. Удаление разветвлений в молекуле гликогена
Инсулин активирует Глюкагон активирует
UDP ^ ^* ^^ Гликоген ^ \^ р.*
Гликоген- λ/^Глкжагон Инсулин^/
/ ингибирует ингибирует
ADP
UDP-глюкозо-
пирофосфорилаза
ι ликоген-
ос о илаза J фосфоглюкомутаза
АТР
Глюкозо-6-фосфат
Глюкокиназа Глюкозо-6-фосфатаза
Н70
UTP
Глюкозо-1 -фосфат>
Глюкоза + Р,-
Поглощение Г ) Выход
Глюкоза в крови
Рис. 6.7. Поглощение, хранение и высвобождение глюкозы в печени
Инсулин и глюкагон непосредственно не влияют на ферменты метаболизма гликогена
часть 3 Метаболизм
90
Почему в печени работает глюкокиназа,
а в других тканях - гексокиназа?
Что происходит с другими сахарами,
поглощенными в тонком кишечнике?
Как только глюкоза поступает в клетку, она немедленно
фосфорилируется в глюкозо-6-фосфат. Эту реакцию в
печени катализирует глюкокиназа, а в мозгу и других
тканях - гексокиназа. В условиях голода, когда
поступление глюкозы в кровь становится первоочередной
задачей, ее использование мышечными и другими
клетками ограничено уровнем инсулина, тогда как
потребление глюкозы мозгом, эритроцитами и печенью
не зависит от инсулина. Это кажется нелогичным.
Получается, что мышцы жертвуют своими белками ради
того, чтобы печень из аминокислот синтезировала
глюкозу для поддержания жизнеспособности клеток мозга, и в
то же время сама печень конкурирует с мозгом за эту
глюкозу. Этого, однако, не происходит! Дело в том, что
глюкокиназа печени обладает значительно меньшим
сродством к глюкозе по сравнению с гексокиназой
мозга (рис. 6.8). Это означает, что в условиях голода печень
поглощает гораздо меньше глюкозы, чем мозг,
поскольку пассивный транспорт глюкозы в клетки
определяется скоростью ее внутриклеточного фосфорилирования.
100 г-
&
о
О
Я
<
Концентрация глюкозы, мМ
Рис. 6.8. Влияние концентрации глюкозы на активность
гексокиназы (/) и глюкокиназы (2)
После еды, когда уровень глюкозы в крови высок,
а инсулин побуждает печень синтезировать гликоген,
глюкокиназа работает с максимально возможной
скоростью. Есть еще одно различие между гексокиназой
и глюкокиназой: последняя не ингибируется продуктом
реакции - глюкозо-6-фосфатом. Благодаря этому синтез
гликогена в печени может происходить при высоком
внутриклеточном содержании глюкозо-6-фосфата.
Кроме того, у транспортного белка, ответственного за
поступление глюкозы в клетки печени, константа Михаэ-
лиса (Км) выше, чем в других клетках. Аналогичная
ситуация наблюдается в тех клетках поджелудочной
железы, которые секретируют инсулин и координируют его
секрецию с уровнем глюкозы в крови.
При переваривании животными пищи в портальную
вену из кишечника наряду с глюкозой поступает
много других Сахаров. К их числу относится галактоза,
образующаяся из молочного сахара - лактозы, и
фруктоза - продукт гидролиза другого пищевого дисаха-
рида - сахарозы. Для энергообеспечения организма
необходимо превратить галактозу и фруктозу либо в
глюкозу, либо в продукты ее метаболизма в печени. Тогда
эти сахара вместе с глюкозой в конечном итоге будут
преобразованы в гликоген и жир либо же подвергнутся
окислению до С02 и Н20.
Чтобы превратить галактозу в глюкозу, необходимо
изменить оптическую конфигурацию С-4-атома
сахарного остатка (в химии этот процесс называют эпимери-
зацией).
ОН
Глюкоза
ОН
Галактоза
Эпимеризовать галактозу клетки не могут, но в них
есть фермент эпимераза, который превращает UDP-ra-
лактозу в UDP-глюкозу. Последняя является
промежуточным соединением при синтезе гликогена. Сначала
галактоза фосфорилируется галактокиназой в галакто-
зо-1 -фосфат:
НО-
-О
ОН
+ АТР
ОН
он
Галактоза
1 алактокиназа
HO-
HO,
ОН
+ ADP
ОРО^
он
Галактозо-1 -фосфат
глава 6 Транспорт пищи, ее хранение и мобилизация
91
Теперь, казалось бы, галактозо-1-фосфат должен,
по аналогии с глюкозо-1-фосфатом, вступить в реакцию
с UTP. Однако вместо этого галактозо-1 -фосфат
замещает глюкозо-1-фосфат в UDP-глюкозе, в результате чего
и образуется UDP-галактоза:
О
0-Р-0-Р-0-Уридин+ |\Он |/\)РОГ
UDP-глюкоза
ОН
Галактозо-1 -фосфат
У идил анс )е аза
О О
II II
Ό-Ρ-0-Ρ-0-
СГ
UDP-галактоза
СГ
- Уридин н£
ОН
Глюкозо-1 -фосфат
Поскольку эту реакцию можно рассматривать как
перенос уридинфосфатного (уридильного) остатка
с UDP-глюкозы, катализирующий ее фермент называют
уридилтрансферазой. Лишь после образования
UDP-галактозы происходит реакция эпимеризации.
Н0-
-0— Уридин
ОН
UDP-галактоза
UDP-галактозо-
эпимераза
НО-
НО
ОН
0 О
II II
О—Р—О—Р—О—Уридин
1 I
О" О"
1 ОН
UDP-глюкоза
Если все эти реакции собрать вместе, можно
представить следующую схему превращений галактозы, конечным
продуктом которых является глюкозо-1-фосфат (рис.6.9).
Известен генетический дефект, который
проявляется у детей в форме заболевания, называемого галак-
тоземией. У таких больных нет уридилтрансферазы,
вследствие чего в организме накапливается галактоза
Галактоза
АТР
N
А
ADP
Галактозо-1 -фосфат
UDP-глюкоза
Уридил-
т анссЬе аз
UDP-галактоза
Глюкозо-1 -фосфат
UDP-галактозо-
эпиме аза
Рис. 6.9. Вхождение галактозы в главные метаболические
пути посредством ее превращения в глюкозо-1-фосфат
и продукты ее метаболизма. Это приводит к
нарушениям развития мозга и слепоте. Чтобы избежать столь
тяжелых последствий, достаточно исключить галактозу
из пищевых продуктов. Потребности организма таких
больных в галактозе для синтеза собственных гликоли-
пидов и гликопротеинов удовлетворяются благодаря
тому, что эпимеризация UDP-галактозы в UDP-глюкозу
обратима, а указанные синтетические процессы
требуют как раз UDP-галактозу в качестве донора галакто-
зильных остатков.
Пути превращения аминокислот
в организме (в рамках проблемы
энергоснабжения)
Хотя для аминокислот не предусмотрено таких
специализированных способов запасания, как для глюкозы
и жира, тем не менее и они участвуют в
энергоснабжении. Особенно важна их роль при голодании: в этих
условиях расщепляются мышечные белки, а
образующиеся аминокислоты переносятся в печень и становятся там
субстратами для синтеза глюкозы.
Пути превращения жиров
и холестерина в организме
Следует пояснить, почему мы говорим о холестерине при
анализе энергоснабжения, хотя он не вносит в него
никакого вклада. Дело в том, что холестерин после пище-
частъ 3 Метаболизм
92
варения вместе с жирами транспортируется в составе
липопротеинов. Отсюда и целесообразность
совместного рассмотрения судьбы этих веществ.
Поглощение жира клетками
из хиломикронов
После приема жирной пищи кровь насыщается хило-
микронами - частицами, оболочка которых
образована фосфолипидами, холестерином и белками, а ядро
состоит из нейтральных жиров (триглицеридов) и эфиров
холестерина (см. рис. 4.5).
Триглицериды откладываются про запас в жировых
клетках, в молочных железах млекопитающих
(выделяются при секреции с молоком), а также в мышцах и
других тканях, где используются для производства энергии.
В отличие от триглицеридов свободные жирные
кислоты легко проходят через мембраны. Видимо,
именно поэтому триглицериды в хиломикронах гидро-
лизуются в кровотоке липазой, расположенной на
поверхности клеток, выстилающих капилляры.
Продуктами гидролиза являются жирные кислоты и глицерин,
которые тут же поглощаются близлежащими клетками.
О
II
СН2—О—С—Ra
I О
I II
СН—О—С—R2 + 3H20
I О
I и
СН2—О—С—R3
Триглицерид
СН2ОН
СНОН +
СН2ОН
Глицерин
V
+ зн*
Жирные
кислоты
Количество жирных кислот, поступающих при этом
из крови в ткань, определяется суммарной активностью
липазы в пронизывающих ткань капиллярах. Эта
активность особенно велика в жировой ткани и в молочных
Впячивание Захваченный остаток
мембраны в клетке печени
клетки печени (рецепторный белок
Аполипопротеин Ε возвращается в мембрану)
Клетка
печени
Рецепторный белок
Остаток
хиломикрона
Рис. 6.10. Поглощение остатка хиломикрона клеткой
печени путем эндоцитоза, управляемого рецептором, который
специфичен к аполипопротеину Ε в хиломикроне
железах в период лактации. Содержание липазы
зависит от физиологических потребностей организма. Оно,
например, возрастает с увеличением соотношения ин-
сулин/глюкагон. Поскольку хиломикрон можно
рассматривать как липопротеин (комплекс липида с белком),
липазу в капиллярах называют липопротеинлипазой,
чтобы отличать ее от других липаз. Видимо, эта липаза
может служить своего рода мостиком, временно
скрепляющим хиломикрон с поверхностью клетки.
По мере удаления триглицеридов из хиломикронов
последние уменьшаются в размерах и превращаются
в так называемые остатки, содержащие исходные
количества холестерина и его эфиров, а также около 10%
исходного количества триглицеридов. Эти остатки
поглощаются печенью в ходе рецептор-опосредованного
эндоцитоза (рис. 6.10) и заканчивают свое
существование внутри печеночных клеток, доставляя туда
триглицериды и холестерин. Полностью судьба хиломикронов
отражена на рис. 6.11.
Жиры и
холестерин
из пищи
_^ Хиломикрон
~*" в крови
Жиры в ткани
Остаток
хиломикрона
Доставка в печень
холестерина и некоторого
количества жира путем
эндоцитоза
Хиломикрон
в лимфе
Липиды]
в пище
Жирные Клетки (кроме
кислоты клеток печени)
t
Клетка печени
Холестерин
жирные
кислоты
t
Ж!
Эндоцитоз
Рецептор
аполипопротеина Ε
Липопротеинлипаза
на стенках капилляров
Остаток хиломикрона,
содержащий 10%
исходных триглицеридов,
а также холестерин и его
эфиры
Рис. 6.11. Транспорт жира и холестерина с помощью
хиломикронов (ХМ)
а - Общее изображение;
б - детальное изображение
глава 6 Транспорт пищи, ее хранение и мобилизация
93
Судьба жиров и холестерина
Кроме хиломикронов, переносящих жир и холестерин
непосредственно из кишечника, существенный вклад
в транспорт этих веществ вносят липопротеины, в виде
которых жир и холестерин поступают в кровь из печени.
Печень сама способна синтезировать триглицериды
из глюкозы и других метаболитов, а некоторое
количество триглицеридов она получает извне вместе с
остатками хиломикронов. Тем не менее печень никак нельзя
рассматривать как место хранения липидов (ожирение
печени - патология). Триглицериды экспортируются
из печени в другие ткани в виде так называемых липо-
протеинов очень низкой плотности (ЛПОНП), которые
по своему строению напоминают хиломикроны. В
результате именно печень оказывается главным
поставщиком жиров, снабжающим основных потребителей:
мышцы, где жиры окисляются, и жировые клетки,
где происходит их запасание.
Печень не только собирает холестерин пищи,
поступающий в нее в составе остатков хиломикронов, но
является основным местом синтеза холестерина в
организме. Из печени холестерин вместе с триглицеридами
разносится по всему телу в составе ЛПОНП. Вместе с тем
существует и встречный поток холестерина из
периферических тканей в печень (рис. 6.12). Смысл такого
встречного движения пока не вполне ясен, тем более что
большинство клеток сами способны синтезировать
холестерин. Одно из возможных объяснений заключается
в том, что таким образом достигается наиболее гибкая
регуляция общего содержания холестерина в организме,
поскольку печень способна удалить его избыток в виде
желчных кислот. Другая гипотеза предполагает, что
выход холестерина из печени - необходимое следствие
Глюкоза и др.
Остатки
хиломикронов
\ Выходящий поток
Три_ триглицеридов
(глицериды и холестерина
[холестерин Входящий поток
холестерина
Печень Периферические
w клетки
Желчные кислоты
Рис. 6.12. Движение триглицеридов и холестерина в печень
и из печени (схематично)
В печени эти вещества синтезируются из метаболитов и
включаются в составе остатков хиломикронов. Там же холестерин
превращается в желчные кислоты
часть 3 Метаболизм
94
«секреции» триглицеридов, поскольку холестерин
является непременным компонентом переносящих их
ЛПОНП. Согласно этой гипотезе, встречный поток
холестерина в печень есть не что иное, как способ
замкнуть транспортный цикл, вернув переносчик
(холестерин) назад. Решение этой проблемы имеет важное
значение для медицины, ибо оно тесно связано с
пониманием природы атеросклероза.
Использование холестерина
в организме
Холестерин - важный компонент плазматической
мембраны животных клеток. В то же время его избыток в
организме служит причиной сердечно-сосудистых
заболеваний. Вот почему важное значение имеют механизмы
выведения холестерина из организма.
Главным из этих механизмов является переработка
холестерина в желчные кислоты в печени. Таким способом
у человека выделяется примерно около 0,5 г холестерина
за сутки. Из печени желчные кислоты в составе желчи
поступают в тонкий кишечник, затем всасываются его
стенками вместе с жиром. Поэтому одна из терапевтических
задач - снижение содержания холестерина -
заключается в подавлении обратного всасывания желчных кислот.
Это достигается введением в пищу веществ, образующих
в кишечнике прочные комплексы с желчными кислотами,
которые не могут быть поглощены эпителиальными
клетками. Другой, более современный подход состоит в
регуляции синтеза желчных кислот. В надпочечниках и в
половых органах из холестерина синтезируются не только
желчные кислоты, но и стероидные гормоны. В форме
эфиров холестерин хранится в клетках и в составе липоп-
ротеинов транспортируется в организме.
Липопротеины, участвующие
в транспорте жира и холестерина
Печень производит липопротеины двух типов: ЛПОНП
и ЛПВП (липопротеины высокой плотности). В этом
процессе принимают участие эндоплазматический рети-
кулум и аппарат Гольджи, где липопротеины
упаковываются в покрытые мембраной пузырьки, которые
выводятся с помощью экзоцитоза. У крыс около 80% липо-
протеинов (не хиломикронов) образуется в печени,
а остальная часть - в клетках кишечника. Далее мы
коснемся превращения ЛПОНП в ЛППП (липопротеины
промежуточной плотности) и ЛПНП (липопротеины
низкой плотности). Так что вместе с хил омикронами речь
идет о пяти типах липопротеинов. Электронные
микрофотографии некоторых из них представлены на рис. 6.13.
a
б
v. -x. ** ~'Γ'
* 7»
.41
V"
Рис. 6.13. Электронные микрофотографии хиломикронов и липопротеинов
а - Хиломикроны; б - липопротеины очень низкой плотности ; в - липопротеины низкой плотности; г - липопротеины
высокой плотности. Масштаб, указанный жирной чертой на каждой фотографии, соответствует 100 нм. Фотографии предоставлены
доктором Т. Forte (Lawrence Berkeley Laboratory, University of California)
глава 6 Транспорт пищи, ее хранение и мобилизация
95
Аполипопротеины
Каждому из пяти липопротеинов отвечает свой набор
аполипопротеинов, т. е. входящих в их состав чисто
белковых компонентов. Известно более двенадцати
таких белков. Функция каждого из них точно не
определена, но в целом роль этих белков сводится к следующему.
1. Некоторые аполипопротеины необходимы для
образования липопротеинов. Таковы белки ароВ48 в хи-
ломикронах и ароВЮО в ЛПОНП.
2. Другие апобелки отвечают за распознавание липо-
протеина рецепторами на поверхности клеток-мишеней.
Такое распознавание, в свою очередь, является сигналом
к запуску эндоцитоза (см. рис. 6.10). Избирательное
распознавание позволяет отдельным липопротеинам
связываться только с предназначенными для них
клетками. Примерами адресных аполипопротеинов могут
служить ароВ 100 в ЛПНП, который связывается с ЛПНП-
рецепторами, и ароЕ, позволяющий клеткам печени
распознавать остатки хиломикронов.
3.Часть аполипопротеинов служит активаторами
ферментов. Так, ароСП в хиломикронах активирует ли-
попротеинлипазу, которая отщепляет жирные кислоты
от триглицеридов. Не все апобелки изначально
присутствуют в липопротеинах. Некоторые включаются в них
в процессе внеклеточного транспорта. Тот же ароСП
синтезируется в печени, но встраивается в хиломикроны
уже после их образования.
Механизм транспорта триглицеридов
и холестерина из печени к другим
тканям и обратный перенос
холестерина в печень
Начнем с выхода из печени триглицеридов и
холестерина в составе ЛПОНП. У больных с нарушениями
синтеза ЛПОНП происходит накопление триглицеридов
в печени.
Как только ЛПОНП попадают в кровоток, они
постепенно теряют триглицериды благодаря действию липо-
протеинлипазы. При этом в них возрастает
относительное содержание холестерина и его эфиров (табл. 6.1),
Таблица 6.1
Содержание (в % от сухой массы) триглицеридов и
холестерина в различных липопротеинах
Показатель
Триглицериды
Холестерин
и его эфиры
Хиламикроны
80
8
ЛПОНП
50
20
ЛППП
30
30
ЛПНП
10
50
ЛПВП
8
30
часть 3 Метаболизм
96
увеличивается их плотность и уменьшаются линейные
размеры. В результате ЛПОНП превращаются сначала
в ЛППП, а затем в ЛПНП. Именно ЛПНП распознаются
рецепторами клеток периферических тканей и далее
поглощаются ими путем эндоцитоза, снабжая эти клетки
остатками триглицеридов и холестерином.
Интенсивность такого поглощения регулируется изменением
числа рецепторов на поверхности клеток-мишеней.
Выход холестерина из печени компенсируется его
встречным потоком из периферических тканей. Это
происходит благодаря ЛПВП. Они образуются в основном
в печени и представляют собой дископодобную
структуру, состоящую из апобелков, фосфолипидов и
относительно малого количества триглицеридов и
холестерина. В периферических тканях ЛПВП обогащаются
холестерином, предположительно благодаря диффузии
последнего из плазматических мембран в
прикрепившийся к ним липопротеин. Исходно гидроксильные
группы молекул холестерина экспонированы в водное
окружение ЛПВП и доступны для этерификации.
Преобразование амфипатического холестерина в
полностью гидрофобные эфиры сопровождается изменением
формы ЛПВП: из диска он превращается в сферу.
Внутри клеток для этерификации холестерина
(CHOL) требуется энергия АТР, в отличие от
внеклеточной этерификации; поэтому для этерификации
холестерина в ЛПВП используется другая стратегия.
Особый фермент лецитин-холестерин-ацилтрансфераза
(ЛХАТ) катализирует перенос остатков жирных кислот
с лецитина, присутствующего в ЛПВП, на холестерин
О
II
СН2-0—С—Ra
I °
СН—0-C-R2 + CHOL—ОН
I О
I и
СН-, — О—Р—О—Холин
1 ι
0"
Лецитин + Холестерин
(фосфатидилхолин)
I
О
II
СН2—О—С—Ra
I О
I и
СНОН + R2 —С-О—CHOL
I о
I и
СН-,—О—Р— Холин
I
0"
Лизолецитин + Эфир
холестерина
Эта реакция не требует внешнего источника энергии,
поскольку исходный и образующийся сложные эфиры
энергетически почти эквивалентны.
Эфиры холестерина, заключенные в ЛПВП,
переносятся на хиломикроны, ЛПОНП, ЛППП и ЛПНП с
помощью особого белка-переносчика (белок-переносчик
эфиров холестерина). Хотя главная функция ЛППП
и ЛПНП состоит в транспорте их содержимого в
периферические ткани, некоторая доля возвращается в
клетки печени. Таким образом, механизм встречного
потока холестерина может быть описан в виде нескольких
стадий.
1. Холестерин переносится из мембран
периферических клеток в ЛПВП.
2. Холестерин ЛПВП этерифицируется;
образующиеся при этом эфиры мигрируют с периферии липоп-
ротеина к центру.
3. Эфиры холестерина переносятся из ЛПВП в
хиломикроны, ЛПОНП, ЛППП и ЛПНП.
4. Некоторая доля ЛППП и ЛПНП (а после еды - и хи-
ломикронов) передает эфиры холестерина клеткам
печени, обусловливая перенос холестерина из
периферических клеток в печень (рис. 6.14).
В действительности все обстоит гораздо сложнее,
поскольку параллельно с описанными здесь процессами
осуществляется обмен между разными липопротеина-
ми, триглицеридами и эфирами холестерина, обмен
триглицеридов на эфиры холестерина и т.п. Все эти
явления в деталях пока не изучены.
Медики рассматривают ЛПНП как «плохой
холестерин», потому что избыток холестерина в организме,
повышающий вероятность атеросклероза, коррелирует
с долей липопротеинов этого типа. Атеросклероз прояв-
Псрснос эфиров лпвп Перенос
холестерина холестерина
лппп —-· лпнп
I i \ I
Печень Удаление триглице- Периферичес
ридов в виде жирных Жирные кие ткани
кислот липопротеин- кислоты
липазой
Рис. 6.14. Роль липопротеинов в переносе холестерина в
печень и из печени
Жирными линиями показано перемещение эфиров холестерина
между липопротеинами. Все его этапы обратимы, а сам
рисунок упрощен
ляется в образовании специфических бляшек внутри
кровеносных сосудов, в значительной степени
состоящих из холестерина. Такие бляшки закупоривают
сосуды. В коронарных сосудах это приводит к сердечным
приступам. Известен генетический дефект, при котором
в клетках мало рецепторов ЛПНП. В результате
ослабляется «откачка» содержащих холестерин ЛПНП из
крови, вследствие чего в ней увеличивается уровень
ЛПНП-связанного холестерина. Это заболевание,
называемое семейной гиперхолестеринемией,
сопровождается поражениями сердечно-сосудистой системы.
Напротив, ЛПВП врачи называют «хорошим
холестерином», поскольку высокое содержание ЛПВП связано
с уменьшением риска заболеть атеросклерозом.
Благодаря ЛПВП ускоряется поток холестерина в печень,
однако механизм их защитного действия до конца еще не
ясен. Возможно (хотя это и не доказано), что ЛПВП
способны остановить развитие атеросклероза, извлекая
холестерин из атеросклеротических бляшек в
кровеносных сосудах.
Как жиры покидают жировые клетки?
Липаза высвобождает свободные жирные кислоты,
хранящиеся в виде нейтральных жиров. Однако липаза,
функционирующая внутри жировых клеток,
принципиально отличается от липазы кровяных капилляров. Это
отличие состоит в том, что внутриклеточная липаза
регулируется гормонами: ее активирует глюкагон и инги-
бирует инсулин (см. главу 12). После еды, когда
инсулина много, а глюкагона мало, жировые клетки поглощают
глюкозу из плазмы крови и жирные кислоты из хиломик-
ронов, превращая их в триглицериды; гидролиз
последних заторможен. При голоде ситуация
противоположная: образуются жирные кислоты, которые затем выходят
из жировых клеток в кровь и доставляются к другим
тканям. При юношеском диабете, когда у больных очень
низкое соотношение инсулин/глюкагон, гормончувстви-
тельная липаза обеспечивает поступление в кровь
большого количества жирных кислот, которые улавливаются
печенью и перерабатываются ею в кетоновые тела, что
приводит к сильной кетонемии (избыток кетоновых тел
в крови) - дополнительному тяжелому осложнению
сахарного диабета
Гормончувствительную липазу активирует не
только глюкагон, но и адреналин. После «адреналинового»
сигнала жировые клетки выбрасывают жирные
кислоты, которые доставляются мышцам и позволяют им
развить максимальную активность (см. главу 12). Жировые
клетки иннервированы симпатическими нервными
глава 6 Транспорт пищи, ее хранение и мобилизация
97
волокнами, окончания которых при возбуждении секре-
тируют норадреналин. Этот нейромедиатор также
активирует гормончувствительную липазу.
Как жирные кислоты переносятся
кровью?
Если бы жирные кислоты присутствовали в крови
в свободном виде, она была бы похожа на мыльный
раствор, потому что в нейтральной среде'эти кислоты
полностью диссоциированы. В действительности жирные
кислоты в крови адсорбированы на поверхности белка -
сывороточного альбумина (СА). На его молекуле
имеются гидрофобные участки, к которым прилипают
различные гидрофобные молекулы. Образующиеся некова-
лентные комплексы не слишком прочны, что делает
связывание обратимым.
Дополнительная литература
Синтез гликогена
Alonso М. D., LomakoJ., Lomako W. M., Whelan W. J.
A new look at the biogenesis of glycogen // FASEB J. 1995.
Vol. 9. P. 1126-1137.
(Превосходная статья о роли гликогена, в которой
обобщены новые концепции.)
Van Schaftingen E., Detheux M, Da Cunha Μ. V. Short-term
control of glucokinase activity: role of a regulatory protein //
FASEB J. 1994. Vol. 8. P. 414-419.
(Описана локализация, свойства и регуляция глюко-
киназы.)
Транспорт липидов и липопротеины
Nilsson-Ehle P., Garfinkel A. S., Schotz M. С. Lipolytic
enzymes and plasma lipoprotein metabolism // Annu. Rev.
Biochem. 1980. Vol. 49. P. 667-693.
(Общее резюме о липопротеинах, в том числе об их
физиологических функциях.)
Cannon В., NedergaardJ. The biochemistry of a inefficient
tissue // Essays Biochem. 1985. Vol. 20. P. 110-164.
(Обзор знакомит с проблемой генерации тепла
клетками бурого жира.)
часть 3 Метаболизм
98
Angelin В., Gibbons G. Lipid metabolism // Curr. Opin. Li-
pidology. 1993. Vol. 4. P. 171-176.
(Прекрасный обзор о липопротеинах.)
Flier J. S. The adipocyte: storage deposit or node on the
energy information superhighway?// Cell. 1995. Vol. 80.
P. 15-18.
(В обзоре приведены данные о том, что адипоциты
выполняют эндокринную функцию и являются
элементами регуляторной системы, защищающей организм
от ожирения.)
Tall A. Plasma lipid transfer proteins // Annu. Rev. Biochem.
1995. Vol.64. P. 235-257.
(Обзор посвящен транспорту липидов и холестерина.
Особое внимание уделено белку, который обеспечивает
перенос эфиров холестерина.)
Вопросы к главе 6
1. Объясните, как достигается термодинамическая
необратимость синтеза гликогена из
глюкозо-1-фосфата.
2. Как вы истолкуете название UDP-глюкозопирофос-
форилаза?
3. Какие ткани способны выделять в кровь глюкозу,
образующуюся при расщеплении гликогена, и
почему?
4. Какую реакцию катализируют ферменты глюкокина-
за и гексокиназа? Почему печень содержит глюкоки-
назу, а мозг и другие ткани - гексокиназу?
5. Детскую галактоземию предотвращают диетой,
лишенной галактозы. Это возможно благодаря UDP-ra-
лактозоэпимеразе. Почему?
6. Как триглицериды покидают хил омикроны и
усваиваются тканями?
7. Что такое ЛПОНП и для чего они нужны?
8. Что имеется в виду под встречным переносом
холестерина?
9. Каким образом большая часть холестерина
выводится из организма?
10. Этерификация холестерина - эндоэргоническая
реакция. Каким образом тогда в ЛПВП холестерин эте-
рифицируется без участия АТР?
11. Как и при каких условиях жировые клетки
освобождаются от жира?
глава
7
Получение энергии из пищи. Введение
Генерация энергии в форме АТР осуществляется
сложными и переплетенными путями, рассмотрение которых
лучше всего начать с превращения глюкозы.
Образование энергии
из глюкозы
Основные этапы окисления глюкозы
Окисление глюкозы можно описать суммарным
уравнением:
С6Н1206 + 602 -> 6С02 + 6Н20.
Изменение свободной энергии AG0' этой реакции
равно-2820 кДж ■ моль1. В клетках окисление
молекулы глюкозы сопряжено с синтезом более 30 молекул
АТР из ADP и Pf.. Окисление глюкозы до С02 и Н20
можно разделить на три этапа.
1. Гликолиз - процесс расщепления глюкозы на два
трехуглеродных фрагмента (молекулы пировиног-
радной кислоты), сопряженный с восстановлением
переносчика электронов; протекает в цитоплазме
клетки.
2. Цикл Кребса (синонимы: цикл лимонной кислоты,
цикл трикарбоновых кислот) - совокупность
реакций, в результате которых второй и третий атомы
углерода пировиноградной кислоты превращаются
в С02, с восстановлением переносчиков электронов.
В этом процессе, проходящем внутри митохондрий,
молекулярный кислород не участвует.
3. Электронтранспортная цепь- это цепь переноса
электронов на 02, после чего он, забирая из
окружающей среды водород в виде протонов, превращается
в Н20. У эукариот эта стадия происходит во
внутренней мембране митохондрий, и именно она
сопровождается образованием наибольшего количества АТР.
Прежде чем подойти к более детальному
рассмотрению окисления глюкозы, необходимо поговорить о
природе биологического окисления.
Биологическое окисление и системы
переноса водорода
Окисление совсем не обязательно должно быть связано
с участием кислорода. Этот термин в общем случае
отражает не что иное, как потерю электронов. Окисление
может реализоваться либо в виде переноса электронов,
как при превращении феррикатиона в феррокатион:
ре2+ _> рез+ + е 9 либо в виде переноса электронов,
сопровождаемого отщеплением водорода (протона) от
окисляемой молекулы: АН2 —> А + 2е~ + 2Н+.
В химических системах окисление можно
рассматривать как перенос электронов от молекулы-донора
к молекуле-акцептору. Протоны могут высвобождаться
в окружающий раствор или переноситься вместе с
электронами на акцептор, что эквивалентно переносу
атомов водорода.
В аэробных клетках конечным окислителем служит
кислород. Кислород электрофилен, т. е. может
приобретать электроны; при этом образуется вода, причем
требующиеся для этого протоны забираются из раствора:
02 +4е" +4Н+ ^2Н20.
Между кислородом (конечным окислителем) и
окисляемым метаболитом расположены промежуточные
акцепторы электронов. Связывая электроны, они
передают их следующему акцептору, образуя цепь, по которой
электроны переносятся с исходного метаболита на
кислород. Каждый из таких переносчиков попеременно
выступает то акцептором электронов, то донором. Именно
это позволяет рассматривать каждого из участников
цепи в качестве переносчика электронов. С цепью
переноса электронов связан синтез основного количества
АТР. Общая схема устройства такой цепи изображена
на рис. 7.1. Два участника этой цепи играют особенно
важную роль, и их свойства нужно очень хорошо знать.
NAD+ - важный переносчик электронов
Первым переносчиком электронов, который
непосредственно встречается с большинством окисляемых
метаболитов, является никотинамидадениндинуклеотид,
или NAD+. На примере AMP (см. рис. 1.6) мы уже
встречались с нуклеотидами - соединениями,
имеющими строение типа азотистое основание-сахар-фосфат.
глава 7 Получение энергии из пищи
99
Η ► Электроны + Протоны (Н+) (могут
(из пищи) (далее перемещаются связываться с
по цепи переносчиков) переносчиками или
высвобождаться в
раствор)
Выделение
свободной энергии
ADP + Р,·
Рис. 7.1. Общая концепция переноса электронов на
кислород и генерации АТР
В молекуле AMP нуклеиновым основанием служит аде-
нин (строение аденина и других оснований см. в
главе 18). В случае NAD+ мы имеем дело с двумя нуклео-
тидами, которые связаны друг с другом своими
фосфатными группами. Поэтому данное соединение
и называют динуклеотидом. По своей структуре NAD+
отличается от динуклеотидных элементов нуклеиновых
кислот, в которых мононуклеотиды также соединены
фосфоэфирной связью. Строение NAD+ можно
представить следующим образом: основание-сахар-фосфат-
фосфат-сахар-основание. Одно из этих оснований -
аденин, как и в АТР, а другое - никотинамид (амид
никотиновой кислоты); сахара одинаковые - рибоза. Итак,
строение NAD+ можно изобразить следующим образом:
Аденин
I
Рибоза
ι
Фосфат
Никотинамид
Рибоза
Фосфат
NAD+ является коферментом: так называют
низкомолекулярные органические соединения, которые
принимают участие в ферментативных реакциях. От обычного
субстрата NAD+ отличается тем, что, будучи
восстановленным, он покидает один фермент и тут же
окисляется другим ферментом. Оба фермента - «всего лишь»
катализаторы, так что в действительности NAD+
восстанавливается субстратом первого фермента, а окисляется
субстратом второго, выполняя функцию переносчика
электронов, постоянно окисляясь и восстанавливаясь.
«Рабочим участком» этого кофермента служит
остаток никотинамида; никотиновая кислота (другое
название - ниацин) - витамин. Для некоторых животных он
является необходимым компонентом пищи, однако
организм человека способен синтезировать его
самостоятельно из аминокислоты триптофана. Остальная часть
молекулы NAD+ служит для связывания с ферментами
и не подвергается химическим превращениям в ходе
каталитического цикла.
Никотинамид имеет следующее строение:
.У
Строение NAD+ можно представить так:
Н CONH,
где R - остальная часть молекулы NAD+
Восстановление NAD+ можно рассматривать как
присоединение двух электронов и протона или, что
эквивалентно, так называемого гидрид-иона (Н~).
Восстановленный продукт (NADH) имеет следующее строение:
Η Η
(J -Η-
I
R
Восстановленный NADH + H4
NAD+ служит коферментом различных дегидрогеназ,
осуществляющих реакции типа:
АН2 + NAD+ ~ А 4- NADH + Н+
Восстановленный NAD+ затем диффундирует к
другому ферменту, где подвергается окислению:
В + NADH + Н+ *-+ ВН2 + NAD+
Таким образом, NAD+ действует в качестве
переносчика двух атомов водорода от субстрата А к субстрату В:
АН2 + В -* А + ВН2.
часть 3 Метаболизм
100
В биохимии такие реакции часто изображают в виде
карусели:
АН2Х х NAD+X rBH2
NADH
+ Н+
Хотя формально происходящее можно представить
как перенос двух атомов водорода, в действительности
один из них «путешествует» с восстановленным NAD+,
тогда как движение другого реализуется в виде двух
последовательно протекающих и разнесенных в
пространстве событий: отщепления протона с его выходом
в окружающую среду и связывания протона из
окружающей среды. Поэтому при использовании аббревиатуры
NADH всегда подразумевается комбинация NADH + Н+.
FAD - важный переносчик электронов
FAD - флавинадениндинуклеотид - служит важным
переносчиком электрона (водорода). Он является
производным витамина В2 (рибофлавина). Восстанавливаясь,
FAD присоединяет два атома водорода и превращается
в FADH2. В отличие от NAD+, который перемещается
от одной дегидрогеназы к другой, FAD прикреплен
к апоферменту, являясь его простетической группой.
FAD имеет следующее строение: циклическая
система изоаллоксазина-рибит-фосфат-фосфат-рибоза-
аденин. Изоаллоксазин - это химическое название
системы циклов в рибофлавине; рибит - пятиатомный
спирт, остаток которого также входит в структуру
рибофлавина. Термин «динуклеотид» еще менее применим
к FAD, чем к NAD, поскольку рибит не является
сахаром. Структурные изменения при восстановлении FAD,
точнее изоаллоксазинового остатка, приведены ниже:
О
й н
/С. ^NL /С. /СН3
1 1 II 1
* н
FAD
(окисленная форма)
Η
II
Η
FADH2
Vвосстановленная форма)
Еще одним переносчиком электронов является фла-
винмононуклеотид, или FMN. Он имеет следующее
строение: циклическая система изоаллоксазина-рибит-
фосфат и отличается от витамина В2 (рибофлавина)
только наличием фосфата.
И окисленные, и полностью восстановленные
формы FMN и FAD имеют тождественные структуры
изоаллоксазинового остатка. Оба флавиновых кофермента
могут существовать и в форме так называемых семихи-
нонов. На полное восстановление флавина расходуется
два электрона, а на частичное - один. Это позволяет
флавинам участвовать в реакциях как одноэлектронно-
го, так и двухэлектронного переноса.
Гликолиз - первый этап окисления
глюкозы
Окисление глюкозы происходит в три стадии, первой
из которых является гликолиз. Это - бескислородный
процесс, в ходе которого синтезируется всего две
молекулы АТР на молекулу глюкозы. Как показано на рис. 7.2,
конечными продуктами гликолиза являются пируват (т.е.
анион пировиноградной кислоты) и NADH. Если
гликолиз протекает в аэробных условиях (в присутствии
кислорода), оба эти вещества поступают в митохондрии,
где пируват окисляется до С02 и Н20, a NADH - в NAD+.
2NAD+ 2(NADH + Н+)
1 Глюкоза (С$)
■ -en,- -
2ADP 2АТР
+2Р,
2 Пируват (2 х Сз)
сн3сосоо-
Рис. 7.2. Аэробный гликолиз глюкозы (общая схема)
Однако обеспечение организма кислородом не
всегда соответствует потребности в нем. Например, на
начальных фазах реакции тревоги, когда частота
сердечных сокращений возросла еще не очень сильно, резко
повышается скорость гликолиза, чтобы обеспечить
мышечное сокращение необходимым количеством АТР.
Поскольку NAD+ - это своего рода катализатор,
присутствующий в клетках в малых количествах, очень важно,
чтобы NADH успевал окисляться обратно в NAD+: ведь
если нет NAD+, то нет и гликолиза. Никак нельзя
допустить торможения гликолиза, который остается
единственным источником энергообеспечения мышц, если
митохондрии не справляются с этой задачей. На этот
случай предусмотрена запасная система регенерации
NAD+ из NADH, которая основана на окислении NADH
пируватом. Эту реакцию катализирует лактатдегидро-
геназа, которая в достаточном количестве присутствует
в мышечных клетках.
СН3СОСОО"
Пируват
NADH + Н+ -СН СНОНСОО" + NAD+
Лактат- Лактат
цегидрогеназа
глава 7 Получение энергии из пищи
101
Образование лактата (т.е. аниона молочной кислоты)
из глюкозы называют анаэробным гликолизом, чтобы
отличить его от аэробного гликолиза, при котором
образуется пиру ват и окисляемый в митохондриях NADH.
Подчеркнем, что смысл анаэробного гликолиза вовсе
не в образовании лактата. Это лишь способ поддержать
синтез АТР в условиях, когда блокирован нормальный
путь реокисления NADH (рис. 7.3). Анаэробный
гликолиз - экономически крайне невыгодный способ
производства АТР, поскольку на каждую израсходованную
молекулу глюкозы синтезируются всего лишь две молекулы
АТР. Когда сердце переключится на ускоренный ритм
и снабжение мышц кислородом станет адекватным их
нагрузке, возобновляется реокисление NADH, и
гликолиз становится аэробным. Накопившийся в мышцах
лактат не является бесполезным шлаком, он выводится
в кровь и утилизируется печенью (см. с. 151).
Заметим, что, используя аналогичный подход,
дрожжи могут существовать в отсутствие кислорода сколь
угодно долго только за счет анаэробного гликолиза. В их
Глюкоза(С^)
2ADP
+2Р,
2АТР
3
2NAD+
Лактат рогеназа
2 Пируват
2 Лактат
Рис. 7.3. Анаэробный гликолиз глюкозы (общая схема)
клетках пируват декарбоксилируется (т.е. теряет С02)
с образованием ацетальдегида. Последний же
расходуется на окисление NADH, катализируемое алкогольде-
гидрогеназой. Из-за низкого выхода АТР огромное
количество глюкозы расщепляется с образованием спирта
иС00.
сн3сосоо
+ Н+->СН3СНО + С02.
ι ируватдекарооксилаза
СН3СНО + NADH + Н+ — СН СН2ОН + NAD".
А к голь ги рогеназа
Приведенное описание гликолиза упрощено, чтобы
выделить его главные особенности. Так, основное
количество глюкозы в мышцах сосредоточено в гликогене.
Если продукцию АТР относить не к свободной глюкозе,
а к одному из ее остатков в гликогене, то гликолиз
приводит к образованию не двух, а трех молекул АТР на
молекулу глюкозы.
Цикл лимонной кислоты - второй этап
окисления глюкозы
Митохондрии - небольшие внутриклеточные
мембранные органеллы (см. рис. 3.15). Их можно считать
главными электростанциями клеток, так как в них
синтезируется основное количество АТР. Митохондрии окружены
двумя мембранами. Внешняя мембрана проницаема для
многих соединений и особой роли в производстве
энергии не играет. Внутренняя мембрана обладает крайне
низкой проницаемостью для большинства веществ,
кроме тех, для которых в ней предусмотрены
специализированные транспортные системы. Внутренняя
мембрана образует складки, или кристы,
увеличивающие ее площадь. Чем больше энергии образуется в
ткани, тем больше крист в митохондриях и тем плотнее они
упакованы (рис. 7.4). Внутри митохондрия заполнена
Межмембранное
пространство
Митохондриальный
матрикс
Внешняя мембрана
Внутренняя мембрана
"" Кристы
Рис. 7.4. Митохондрии печени (а) и сердечной мышцы (б)
Число крист отражает потребности клеток в АТР
"*" Кристы
часть 3 Метаболизм
102
концентрированным раствором ферментов: его
называют матриксом. Вторая стадия метаболизма
глюкозы - цикл лимонной кислоты - протекает в матриксе,
и лишь одна из реакций цикла происходит во
внутренней мембране.
Как уже отмечалось, аэробный гликолиз,
протекающий в цитоплазме, приводит к образованию пирувата
и NADH. Пируват попадает в митохондриальный мат-
рикс с помощью специальной транспортной системы,
a NADH туда проникнуть не может. Вместо него
посредством особого челночного механизма через внутреннюю
мембрану переносится его «восстановительный
эквивалент», который внутри митохондрий передается NAD+-
и FAD-ферментам. В конечном итоге NADH,
образовавшийся в ходе гликолиза, реокисляется митохондриями.
NAD+ остается в цитоплазме для дальнейшего участия
в гликолизе. Внутри митохондрий NADH и FADH2
окисляются электронтранспортной системой.
Необходимо отметить, что в некоторых клетках,
например эритроцитах, нет митохондрий, и у них
гликолиз является единственным источником АТР.
Естественно, что существование таких клеток полностью зависит
от поступления глюкозы.
Рассмотрим ферментативную реакцию, посредством
которой поступивший в митохондрии пируват
превращается в соединение, играющее ключевую роль во
многих метаболических превращениях - ацетил-кофер-
мент А, или ацетил-СоА. Ацетил-кофермент А в
биохимических реакциях часто обозначают как CoA-SH,
потому что тиольная группа представляет собой
реакционный центр его молекулы. В отличие от NAD+ и FAD,
кофермент А служит для переноса не электронов, а
ацильных остатков. Он является динуклеотидом, в
состав которого входит остаток водорастворимого
витамина - пантотеновой кислоты.
СН3 Η О
I I II //
но—сн2—с—с—c-nh-ch2—сн2—с
II ii.
сн3 он
Пантотеновая кислота
О
ОН
Любопытно, что пантотеновая кислота в кофермен-
те А играет иную роль по сравнению с другими
витаминами - ниацином в NAD+ и рибофлавином в FAD,
которые принимают непосредственное участие в
химических реакциях. Остаток пантотеновой кислоты,
по-видимому, нужен для распознавания кофермента А
белками, хотя совершенно непонятно, почему эволюция для
этого избрала столь уникальную в биохимии структуру.
Строение кофермента А можно описать следующей
схемой:
Аденин β-Меркаптоэтиламин
I I
Фосфат Рибоза Пантеноновая кислота
I I
Фосфат Фосфат
β-Меркаптоэтиламин - главная «рабочая» группа
молекулы:
RCO—NHCH2CH2-SH.
Остаток
β-меркаптоэтиламина
Молекула кофермента А переносит ацильные
остатки в виде тиоэфиров, например ацетил-СоА, который
имеет строение: CH3CO-S-CoA. В отличие от обычных
сложных эфиров, тиоэфиры являются макроэргичес-
кими соединениями. Величина AG0' их гидролиза равна
~ -31 кДж · моль1, тогда как при гидролизе эфиров кар-
боновых кислот AG0'—20 кДж · моль-1. Разницу в
значении AG0' можно объяснить тем, что эфиры карбоно-
вых кислот резонансно стабилизированы и,
следовательно, характеризуются более низким содержанием
свободной энергии, чем тиоловые эфиры, резонансно
не стабилизированные. Эта особенность тиоэфиров
используется в разных биохимических системах.
Пируват в митохондриях подвергается
окислительному декарбоксилированию, которое включает
отщепление С02 (декарбоксилирование), перенос двух
электронов на NAD+ (окисление) и перенос
образовавшейся ацетильной группы на кофермент А. В
дрожжах тоже происходит декарбоксилирование пирувата,
катализируемое пируватдекарбоксилазой, однако оно
не сопровождается окислением (NAD+ не участвует
в реакции) и приводит к образованию не ацетильного
остатка, а ацетальдегида. Большое отрицательное
изменение свободной энергии свидетельствует о том, что
окислительное декарбоксилирование пирувата
необратимо:
Пируват + СоА—SH + NAD+ ->
-> Ацетил—S—СоА + NADH + Н+ + С02
AG°' = - 33,5 кДж · моль"1.
Ацетильный остаток в ацетил-СоА - это та форма,
в которой пируват вводится в цикл лимонной кислоты.
Иначе этот цикл называют циклом Кребса (по имени
первооткрывателя), или циклом трикарбоновых
кислот (поскольку ряд участвующих в нем метаболитов
глава 7 Получение энергии из пищи
103
содержит три карбоксильные группы). Ацетильные
углеродные атомы в ацетил-СоА, которые ранее
принадлежали пирувату, превращаются в С02, и параллельно
с этим 3 молекулы NAD+ восстанавливаются в NADH,
а I молекула FAD - в FADH2. Кроме того, следствием
всех этих превращений является синтез одной
«высокоэнергетической» фосфорильной группы из Р, на
каждую исчезнувшую ацетильную группу (рис. 7.5).
Итак, молекула пирувата, поступившая из
цитоплазмы в митохондрию, превратилась в 3 молекулы С02;
при этом восстановились 3 молекулы NAD+ и I
молекула FAD, а также образовалась одна молекула АТР.
Учитывая, что 1 молекула глюкозы расщепляется на 2
молекулы пирувата, гликолиз вместе с циклом лимонной
кислоты позволяет получить 4 моля АТР на моль
глюкозы. Но еще 30 молекул АТР будет образовано потом.
Перенос электронов на кислород -
третий этап окисления глюкозы
Речь пойдет об окислении NADH и FADH2, которое
происходит во внутренней мембране митохондрий, где
осуществляется последовательная передача электронов
специальными переносчиками.
Иерархия переносчиков электронов
в электронтранспортной цепи
Напомним, что задача состоит в переносе электронов от
NADH и FADH2 к кислороду с образованием воды:
NADH + Н+ + У202 -> NAD+ + Н20.
Изменение свободной энергии этого процесса AG0'
составляет -220 кДж · моль1. Чтобы разобраться в том,
как он протекает, нужно воспользоваться данными об
окислительно-восстановительных потенциалах
участвующих в нем соединений (их часто называют
редокс-потенциалами). В любой
окислительно-восстановительной реакции участвуют акцептор электронов
(окислитель) и донор электронов (восстановитель).
Суммарную реакцию:
X" + Υ <-> Χ + Υ
можно условно разделить на две полуреакции:
Х"<->Х + е (1)
Υ + е <-> Υ (2)
Внутренняя
митохондриальная
мембрана
Цитоплазма
СН3СОСОО
Пируват
Перенос
восстановите *ых
эквиваленте:
\ NAD+
\ пи FAD
CH3COCOO
_NAD+
CoA-SH
NsJ "NADH
CH3CO-S-CoA + C02
NADH
NAD+
1---4
NADH
ли FADH9
FADH2
CoA SH
Цикл \
?i [лимонной ] *-2C02
FAD
3NAD+
3NADH
Рис. 7.5. Упрощенное изображение, показывающее, что
происходит с NADH, образовавшимся при гликолизе, и как
осуществляется генерация NADH и FADH2 в митохондриях
В каждой из них участвуют окисленная и
восстановленная формы одного соединения; их называют
сопряженной парой, окислительно-восстановительной
парой, или редокс-парой. Здесь такими парами
являются X и Х~, а также Υ и Υ". Ясно, что в
действительности в реакции участвуют обе редокс-пары, одна из
которых отдает электрон, а другая его принимает.
Разные редокс-пары обладают различным сродством
к электрону. Те, у которых это сродство меньше, отдают
электрон тем, у кого оно больше. Мерой сродства
редокс-пары к электрону служит так называемый
окислительно-восстановительный потенциал (или ре-
докс-потенциал) EQ. Значение этого параметра позволяет
предугадать направление переноса электрона между ре-
актантами. Не менее важно и то, что величина Е^
непосредственно связана с изменениями свободной энергии.
Величину Eq выражают в вольтах.Чем она меньше
(отрицательнее), тем меньше сродство вещества к
электронам, тем выше способность их отдавать, тем
значительнее восстановительный потенциал и тем выше энергия
электронов.
Причина, по которой, казалось бы, чисто химическое
свойство веществ выражается в вольтах, объясняется
способом измерения редокс-потенциала. В
окислительно-восстановительной реакции должны участвовать две
редокс-пары, между которыми осуществляется перенос
часть 3 Метаболизм
104
электронов. Они могут быть разнесены по разным
сосудам (полуэлементам). Если соединить такие
полуэлементы медным проводом (проводником электронов),
можно направлять электронный поток. При измерении
Е'0 в качестве одной из редокс-пар используют реакцию
2Н++2е~ «-» Н2, катализируемую платиновой чернью (так
называемый водородный электрод), тогда как другой
сопряженной парой служит смесь окисленной и
восстановленной форм интересующего нас соединения.
Помимо положительных ионов в каждом полуэлементе
присутствуют еще и анионы. Электроны перетекают
по проводу в направлении, зависящем от величины их
потенциалов (от сродства двух систем к электрону).
Допустим, что водородный электрод соединен с
полуэлементом, содержащим ионы Fe2+ и Fe3+.
Перераспределение электронов приведет в этом случае к изменению
количества катионов в обоих сосудах. Чтобы ток
продолжал идти по проводу, нужно сохранить
электронейтральность каждого раствора, обеспечив поток анионов
для выравнивания заряда. С этой целью сосуды
соединяют трубкой, заполненной агар-агаровым гелем (чтобы
совместить ионную проводимость с отсутствием потока
жидкости). Такое устройство называют агаро-солевым
или агаровым мостиком (рис. 7.6). Разность
электрических потенциалов полуэлементов измеряют
вольтметром, врезанным в соединительный провод. Потенциал
водородного электрода принято считать равным нулю.
Таким образом, все экспериментальные значения Е'0
рассчитываются исходя из этого стандарта, и потому они
являются относительными величинами. У физиков
принято считать, что электрический ток направлен
навстречу движению электронов. Поэтому полуэлемент,
отдающий электроны, находится под более отрицательным
напряжением.
Величина Е0 (без штриха) - стандартная величина
окислительно-восстановительного потенциала, которая
определяется в стандартных условиях, когда
концентрации всех веществ равны 1 М, а давление водорода
составляет 1 атмосферу. В биохимии обычно используют
значения потенциала Е^, отвечающее рН 7 (вместо рН 0).
При этом потенциал электрода сравнения (т. е.
водородного электрода) равен -0,42 В. Для реакции
NAD++2H++2e--> NADH + Н+ величина Ё0= -0,32 В,
а для 1/Ю2+2Н++2е~-> Н20 значение Е'0= +0,82 В. Столь
большая разница значений редокс-потенциалов
указывает на то, что NADH способен восстановить кислород
до воды, а обратная реакция невозможна.
Редокс-потенциалы ^связаны с изменением
свободной энергии AG0' уравнением Нернста:
AG°' = -nFAE^
Вольтметр
-О
Солевой
мостик
с агар-
агаровым I
Полуэлемент А- Полуэлемент Б-
водородный исследуемая
электрод редокс-пара
Рис. 7.6. Схема прибора для измерения редокс-потенциалов
Водородный электрод сравнения (А) содержит редокс-пару
Н2/2Н+, окислительно-восстановительная реакция которой
катализируется платиновой чернью, нанесенной на электрод.
Второй полуэлемент (Б) содержит редокс-пару, потенциал
которой должен быть измерен. Если Б более восстановлен (Е'0 имеет
более отрицательное значение, чем Е'0 водородного
электрода), чем А, электроны будут двигаться от Б к А и
восстанавливать 2Н+ до Η (если полуэлементы соединены медным
проводом), а анионы будут перемещаться от А к Б по агар-солевому
мостику, чтобы нейтрализовать заряд. Если Б менее
восстановлен, чем А (величина Е'0 более положительна, чем Е'0
водородного электрода), весь процесс будет протекать в
противоположном направлении
где η - число перенесенных в реакции электронов; F -
постоянная Фарадея (96,5 кДж · В-1 · моль-1); ΔΖ^-
разность редокс-потенциалов электронодонорной и элект-
роноакцепторной пар.
Реакцию окисления NADH:
NADH + Н+ + УгОг -> NAD+ + Н20
можно представить как комбинацию двух полуреакций:
NADH + Н+ -> NAD+ + 2Н+ + 2е Е'п= -0,320 В;
Уг07 + 2Н+ + 2е- -> Н20 Е'0 = -0,816 В.
Для суммарной реакции:
Щ= - 0,320 - 0,816 = 1,136 В,
поэтому:
AG0' = -2(96,5 кДж · В1 · моль »)(—1,136 В) =
= -219,25 кДж · моль"1.
глава 7 Получение энергии из пищи
105
-0,32-
Редокс-
потенциал
(Eb)B
Уровень
свободной
энергии
+0,82-^
NADH
[Переносчик а
[Переносчик б
[Переносчик в
АТР
/i
|Переносчик г
Кислород
^-Н+в
Н+ в растворе
2с
ADP + Р/
Рис. 7.7. Цепь переноса электронов (принципиальная схема)
Некоторые переносчики принимают только электроны, а протоны высвобождаются в воду. Другие переносчики передают и
электроны, и протоны. Конечная стадия - перенос электронов на молекулу кислорода. FADH9 расположен в цепи переноса ниже,
чем NADH. (Число переносчиков выбрано произвольно)
Стадия 1. Стадия 3. Цепь переноса Стадия 2. Цикл лимонной
Гликолиз электронов в митохондриальной кислоты в матриксе и внутрен-
в цитоплазме мембране
ней мембране митохондрий
Глюкоза
-NADH
Транспорт электронов к
кислороду происходит не в один этап.
Система транспорта электронов в
митохондриях представляет собой
цепочку из переносчиков электронов,
у которых по мере приближения
к кислороду возрастает редокс-потен-
циал (и соответственно уменьшается
восстановительный потенциал).
Движение электронов от NADH и FADH2
к кислороду можно уподобить
скатыванию с лестницы, ступеньками
которой служат переносчики электронов.
При каждом прыжке со ступеньки на
ступеньку высвобождается порция
свободной энергии (рис. 7.7),
которая не пропадает впустую,
рассеиваясь в виде тепла, как при горении
глюкозы, а с помощью особых
механизмов используется для синтеза
АТР из ADP и Pf.. Вот почему этот
сложный процесс называют
окислительным фосфорилированием
При окислении 1 моля глюкозы
образуется более 30 молей АТР. Схемы
всех трех рассмотренных выше
этапов окисления глюкозы объединены
на рис. 7.8.
Челночный перенос
восстановительных эквивалентов
NADH
Источники электронов ^_
для дыхательной цепи ^_
2 Пируват-
Транспорт
электронов
переносчиками
Окислительное
фосфорилирование
НПГируват
С02 -— I -
Ацетил- СоА
N
NADH +
FADH2
Цикл
лимонной
кислоты
со2
о2 н+
к
Н20
Обмен
Рис. 7.8. Окисление глюкозы
Для простоты показаны только продукты превращений (очевидно, что
восстановленный NADH образуется из NAD+). При окислении гликогена на каждый остаток
глюкозы при гликолизе образуются три молекулы АТР. В цикле лимонной кислоты
в качестве макроэргического соединения образуется не АТР, a GTP. АТР из
митохондрий поступает в цитоплазму в обмен на ADP. NADH и FADH2
вырабатываются не только при окислении глюкозы, но также при окислении жирных кислот и др.
NADH и FADH2 передают электроны дыхательной цепи в разных ее участках
часть 3 Метаболизм
106
Генерация энергии
при окислении жиров
и аминокислот
Для обеспечения себя энергией в виде АТР организм
окисляет не только глюкозу и гликоген, но также жиры
и избыточные количества аминокислот. В случае три-
глицеридов с точки зрения энергообеспечения главное
значение имеет окисление не глицеринового остатка,
а жирной кислоты. Химическое строение жирных
кислот ничего общего не имеет со строением глюкозы, и
естественно ожидать, что механизм их окисления
совершенно иной. Именно поэтому так поражает магическая
простота, с которой увязаны воедино метаболические
пути самых разных веществ. В ходе превращений
глюкозы образуется ацетил-СоА, который вступает в цикл
лимонной кислоты. Жирные кислоты подвергаются
последовательному расщеплению, в результате
каждого из них пара углеродных атомов также превращается
в ацетильную группу ацетил-СоА, поступающего в цикл
лимонной кислоты. Поэтому метаболизм жирных
кислот и глюкозы различается только до момента
образования ацетил-СоА. Более того, на первых стадиях
превращения глюкозы и жирных кислот образуются
восстановленные формы NAD (NADH) и FAD (FADH2),
которые далее окисляются в митохондриальной цепи
переноса электронов (рис. 7.9).
Окисление аминокислот сходно с окислением
глюкозы и жирных кислот, хотя и сложней в деталях. Все
20 аминокислот могут использоваться для генерации
энергии, если их количество превышает сиюминутные
потребности организма в синтезе белков. Схема их
метаболизма, несмотря на различия в строении,
универсальна: они дезаминируются, а лишенная азота
углеводородная часть молекулы используется в конечном итоге на
образование пирувата и/или ацетил-СоА,
вовлекающихся в цикл лимонной кислоты (рис. 7.10). Таким образом,
этот цикл играет центральную роль в метаболизме.
Взаимозаменяемость разных
видов «топлива»
Избыток глюкозы может перерабатываться в жиры. Это
происходит благодаря тому, что жирные кислоты
синтезируются из ацетил-СоА:
Глюкоза —» пируват —» ацетил-СоА —» жирные кислоты.
i ЛЮКО ίίΐ
Гликолиз
Пируват
е
/KlIpHiMiJ
KllLVliM "Ы
Ацетил-СоА
I
Цикл
лимонной
кислоты
ADP + Pi
Д ι л\ател ι
ним нет»
АТР^|
2 .Н+
Н20
Рис. 7.9. Окисление жиров и глюкозы
На рисунке показано, как формируется поток электронов,
поступающий в дыхательную цепь
Гликолиз
Аминокислоты
Αΐΐρϊϊϊ.Κ1 а
KIICIU'I Ы
Амино- ^
кислоты
Амино- <*
кислоты
Пируват
е
Ацетил-СоА
L
Цикл
лимонной
кислоты
ADP + Р,·
АТР
,Ί.Ι.ΐν.ΊΊΓ'Ί..
пая п.сп:.
?2 .Н+
Н20
Рис. 7.10. Окисление глюкозы, жиров и аминокислот для
генерации энергии
глава 7 Получение энергии из пищи
107
Однако у животных жирные кислоты не могут быть
переработаны в глюкозу, поскольку ее синтез протекает
с непременным участием пирувата, а его нельзя
получить из ацетил-СоА из-за необратимости реакции,
катализируемой пируватдегидрогеназой. Поэтому жирные
кислоты не могут превращаться в глюкозу (рис. 7.11),
но глюкоза может превращаться в триглицериды.
Глюкоза ^
Гликолиз
Пируват>;
Расщепление
Жирные
Глюконеогенез
Реакция
невозможна
кислоты
Ацетил-СоА-
Синтез
I
Цикл
лимонной
кислоты
Рис. 7.11. Упрощенное изображение, показывающее,
почему у животных глюкоза превращается в жиры, но жиры
не могут быть превращены в глюкозу
Обратные пути превращений не полностью совпадают с
прямыми. Растения и бактерии могут превращать жиры в
глюкозу, не прибегая к обращению пируватдегидрогеназной реакции
Те аминокислоты, которые при распаде поставляют
пируват или карбоновые кислоты для цикла лимонной
кислоты, могут перерабатываться животными в
глюкозу (их называют глюкогенными). В условиях голода
мышечные белки расщепляются, а образующиеся при-
этом аминокислоты утилизируются печенью для
образования глюкозы. Растения и бактерии умеют
превращать в глюкозу жир и двухуглеродные соединения вроде
ацетата, однако они используют для этого особый, так
называемый глиоксилатный цикл (разновидность
цикла лимонной кислоты), которого нет у животных.
Вопросы к главе 7
1. Что представляют собой три этапа окисления
глюкозы и где они протекают?
2. Опишите строение NAD+. Изобразите его электро-
ноакцепторную группу в окисленной и
восстановленной форме. Каким образом NAD+ переносит
водород между субстратами?
3. Расскажите о строении и функции FAD.
4. В чем разница между аэробным и анаэробным
гликолизом в мышцах? Когда используется анаэробный
гликолиз?
5. Опишите строение кофермента А. Какая часть его
молекулы используется в качестве акцептора ациль-
ных остатков? Каково AG0' гидролиза тиолового
эфира? Сравните эту величину с AG0' гидролиза
карбоксильного эфира.
6. Опишите реакцию, катализируемую
пируватдегидрогеназой, и приведите значения AG0'.
7. Какова в норме судьба образующегося в
пируватдегидрогеназной реакции ацетил-СоА?
8. В результате гликолиза и цикла лимонной кислоты
образуются NADH и FADH2. Какова дальнейшая
судьба этих соединений?
9. Редокс-пара FAD+2H++2e~-> FADH2
характеризуется Eq= -0,219 В, а редокс-пара V2 02+2H+ + 2е~->
-> Н20 имеет Е^= +0,816 В. Вычислите AG0'
при окислении FADH2 кислородом до воды.
10. Как помимо пируватдегидрогеназной реакции в
организме может синтезироваться ацетил-СоА?
1 \.А. Может ли глюкоза превращаться в жиры?
Объясните ваш ответ
Б. Могут ли жирные кислоты у животных
превращаться в глюкозу? Объясните ваш ответ.
В. Могут ли жирные кислоты у Е. coli превращаться
в глюкозу? Объясните ваш ответ.
часть 3 Метаболизм
108
глава
О Гликолиз, цикл лимонной кислоты,
система транспорта электронов:
реакции этих метаболических путей
Стадия 1. Гликолиз
Гликолизом называют расщепление молекулы глюкозы
или глюкозильного остатка гликогена на две молекулы
пирувата.
Глюкоза или гликоген?
До сих пор мы обсуждали главным образом метаболизм
глюкозы. Однако в обычных условиях большинство
тканей обладает запасом гликогена, и он подвергается
гликолизу в большей степени, чем глюкоза. Между этими
процессами есть разница. В случае расщепления гликогена
из него образуется глюкозо-1 -фосфат, который далее
превращается в глюкозо-6-фосфат (см. с. 86). Последний
в печени гидролизуется с образованием глюкозы,
которая поступает в кровь. Однако глюкозо-6-фосфат также
НОСН2
подвергается гликолитическому превращению и во всех
тканях расщепляется до пирувата. Заметим, что глюко-
зо-6-фосфат образуется не только из гликогена, но
также и из глюкозы в результате ее фосфорилирования АТР.
Эта реакция является одной из стадий синтеза
гликогена (см. с. 86). Будет ли использован глюкозо-6-фосфат
для получения гликогена, пирувата или для насыщения
крови глюкозой (в случае печени), зависит от
физиологических потребностей организма и систем регуляции.
Взаимосвязь между гликолизом, глюкозой и гликогеном
показана на рис. 8.1. На фосфорилирование молекулы
глюкозы с образованием глюкозо-6-фосфата
затрачивается одна молекула АТР. В то же время АТР не
расходуется при образовании глюкозо-6-фосфата из гликогена,
поскольку энергия глюкозильной группы в гликогене
(AG0' гидролиза гликозильной группы) эквивалентна
энергии фосфоэфирной связи.
НОСН2
■ /+ р' т
О Гликоген-
ОН
Невосстанавливающий
конец молекулы
гликогена (п остатков)
фосФо илаза
ОН
Глюкозо-1 -фосфат
ОРОз + Гликоген
(/2-1 остатков)
Фосфоглюком таза
НОСН2
АТР ADP
ОН Гексокиназа НО
гчюкокиназа в печени)
зОРОСН2
ОН
Глюкозо-6-фосфат
Глюкозо-6
фосфатаза
в π чени
(все ткани)
Гликолитический
путь
ОН
Глюкоза в крови
Рис. 8.1. Образование глюкозо-6-фосфата из гликогена или глюкозы и его превращения
О регуляторных механизмах этих процессов см. в главе 12
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
109
Почему на начальном этапе гликолиза
расходуется АТР?
Может показаться странным, что метаболический путь,
рассчитанный на производство АТР, начинается с его
потребления. На что расходуется АТР? Дело в том, что
в гликолизе участвуют фосфорилированные соединения,
а чтобы профосфорилировать глюкозу, необходимо
пожертвовать макроэргическим фосфатом. Однако в
дальнейшем эта затрата компенсируется, так как в ходе
гликолиза образуются две молекулы АТР.
Зачем глюкозо-6-фосфат превращается
во фруктозо-6-фосфат?
Следующий этап гликолиза состоит в изомеризации
производного альдозы - глюкозо-6-фосфата, в производное
кетозы - фруктозо-6-фосфат. Конечной целью
гликолиза является расщепление глюкозы на два С3-фрагмента.
Реакция альдольной конденсации протекает между
альдегидом и кетоном (или между двумя альдегидами),
которые конденсируются в ос-кетоспирт, называемый
альдолем, или β-гидроксикарбонильным соединением
(рис. 8.2). Образование альдолей обратимо, и поэтому
обратная реакция может быть использована для их
расщепления. Вернемся к гликолизу. В отличие от глюко-
зо-6-фосфата, фруктозо-6-фосфат обладает альдольной
структурой; это хорошо видно на линейных формулах
данных соединений, представленных на рис. 8.3.
Глюкозо-6-фосфат изомеризуется во фрукто-
зо-6-фосфат в реакции, катализируемой глюкозофос-
фатизомеразой. Затем другой фермент - фосфофрук-
токиназа с помощью АТР превращает фруктозо-6-фос-
фат во фруктозо-1,6-дифосфат. После этого альдолаза
R
С О
С R"
Η
+
Кетон
R
С -О
R" -C-R"
Альдегид
н^о
н-с-он
I
R
Альдоль
(β-гидроксикарбонильное
соединение)
Рис. 8.2. Альдольная конденсация: реакция альдегида с
кетоном или альдегидом
сно
снон
снон
снон
снон
СН2ОРОз
Глюкозо-6-фосфат
(альдоза - не альдоль)
СН2ОН
г°
снон
снон
снон
СН2ОРОз
Фруктозо-6-фосфат
(кетоза - альдоль)
Рис. 8.3. Линейные формулы изомеров моносахаридов:
альдозы и кетозы
Глюкозо-6-фосфат в растворе находится в равновесии с
циклической пиранозной (шестичленной) формой, а фрук-
тозо-6-фосфат - с фуранозной (пятичленной)
катализирует расщепление гексозодифосфата на два
триозофосфата: глицеральдегид-3-фосфат и дигидрок-
сиацетонфосфат (рис. 8.4).
На рис. 8.5 представлена та же реакция, но
приведены не линейные, а циклические структуры
Сахаров; фруктозо-6-фосфат показан в виде пятичленно-
го кольца (фуранозная форма). Изменение свободной
энергии при альдолазном расщеплении составляет
AG0'= +24,3 кДж · моль1. Казалось бы, такая реакция
не должна протекать; возможна лишь обратная реакция.
Но здесь есть тонкость: одна молекула фруктозо-1,6-ди-
фосфата превращается в две, и потому не стандартное,
а реальное изменение свободной энергии (AG) сильно
зависит от концентраций участников реакции В
условиях клетки величина AG мала, так что реакция вполне
обратима.
Изменения свободной энергии AG0' и AG
при альдолазном расщеплении
Несмотря на то, что изменение стандартной свободной
энергии при альдолазном расщеплении составляет
AG0' = +24,3 кДж · моль ], эта реакция легко протекает,
хотя гораздо менее эндоэргонические реакции в клетке
невозможны. Мы знаем, что величина AG0' соответствует
стандартным условиям, когда концентрации исходных
веществ и продуктов равны I M. Так как
внутриклеточная концентрация метаболитов обычно находится в
пределах 10~4-10"3 М, величина AG всегда отличается
от значений AG0'. Знание величины AG0 позволяет
оценить возможность протекания той или иной реакции.
В случае альдолазы это не так, поскольку величины AG
прямой и обратной реакции в клетке отличаются мало.
Это и является причиной необычного поведения альдо-
лазной реакции.
часть 3 Метаболизм
ПО
оч н
ν
I
снон
I
снон
I
снон
I
снон
AG0' = + 1,7 кДж · моль-1
Глюкозофосфат-
изомераза
СН20
Глюкозо-6-фосфат
(фосфат альдозы)
СН2ОН
С = 0
I
СНОН
I
снон
I
снон
I
сн2о ^;
Фруктозо-6-фосфат
(фосфат кетозы)
СН2Ог
I
с=о +
I
СН2ОН
Дигидроксиацетон-
фосфат
AT
ADP
N
Л
Фосфофруктокиназа
AG0' = -14.2 кДж · моль"1
сно
| AG0' = +24,3 кДж · моль-
снон ■
CH2Oi:
I
с=о
I
снон
I
сн2о
Глицеральдегид-
3-фосфат
(триозофосфат)
СНОН
Альдолаза |
СНОН
I
сн2о
Фруктозо-1,6-дифосфат
Рис. 8.4. Превращение глюкозо-6-фосфата в два С3-соединения
Сахара для ясности представлены в виде цепочек
3ОРОСН2
ОН
Глюкозо-6-фосфат
Глюкозофосфат-
изомераза
3ОРОСН2 о СН2ОН
НО/
он
он
Фруктозо-6-фосфат
АТР\|
ADP-^I
Фосфофруктокиназа
СН2ОРОз
С=0
I
СН2ОН
Дигидроксиацетон-
фосфат
СНО
I
снон
I
Альдолаза
сн2оро;
Глицеральдегид-
3-фосфат
;оросн2 о сн2орОз
но/]
он
он
Фруктозо-1,6-дифосфат
Рис. 8.5. Превращение глюкозо-6-фосфата в два С3-соединения
Приведены наиболее часто встречающиеся циклические структуры Сахаров
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
111
Дело в том, что в ходе прямой реакции число
молекул удваивается. Реальное изменение свободной энергии
AG связано со стандартным значением AG0' следующим
уравнением (см. главу 1):
[Продукт]
AG° + RT\g-
AG-
или в данном случае:
' [Исходные вещества]'
+iW-lg
AG = AG°' +
[глицеральдегидфосфат][гидроксиацетонфосфат]
[фруктозо-6-фосфат]
Поскольку концентрация продуктов невелика,
величина RT ■ lg {[продукты]/[реактанты]} имеет большое
отрицательное значение, за счет чего реакция в клетке
становится обратимой. Предположим, что концентрации
всех реактантов в клетке равна 10-4 М, тогда
AG = + 24,3 кДж · моль"1 + (8,315 · 10"3)(298)χ
(1(Н)(1(Н)
χ 2,30 ■ lg-
(1(H)
= + 24,3 - 22,8 = 1,5 кДж ■ моль"1.
Значение AG при выбранных концентрациях
реактантов допускает свободное обращение реакции.
Взаимопревращение
дигидроксиацетонфосфата
и глицеральдегид-3-фосфата
Глицеральдегид-З-фосфат и дигидроксиацетонфосфат
являются молекулами-изомерами. Их
взаимопревращение катализирует фермент триозофосфатизомераза.
сно
ι
снон
I
СН20Н
I
со
СН2ОРОз СН2ОР05
AG0' = +7,6 кДж-моль1
Глицеральдегид- Дигидроксиацетон-
3-фосфат 3-фосфат
Два вещества находятся в равновесии, но поскольку
глицеральдегид-3-фосфат постоянно используется
в последующей стадии гликолиза, весь
дигидроксиацетонфосфат постепенно превращается в глицераль-
дегид-3 -фосфат.
Глицеральдегид-3-фосфатдегидро-
геназа. Образование макроэргического
фосфата
Альдегидная группа в глицеральдегид-3-фосфате
подвергается окислению с помощью NAD+. Для обычной
реакции окисления группы -СНО в группу -СОО
характерно большое отрицательное значение AG, вполне
соизмеримое со свободной энергией образования макро-
эргической фосфорильной группы. Поэтому в данном
случае главная задача катализирующего реакцию фермента
заключается в том, чтобы свободную энергию окисления
использовать для получения макроэргического фосфата.
В активном центре глицеральдегид-3-фосфатде-
гидрогеназы расположен остаток аминокислоты цисте-
ина, содержащий в боковой цепи тиольную (сульф-
гидрильную) группу, которая реагирует с альдегидной
группой субстрата, образуя тиополуацеталь:
Е—SH +
Фермент с
тиольной
группой
Η
/^
О
Глицеральдегид-
3-фосфат
R
I
E-S-Cx
Η ОН
Тиополуацеталь
Это соединение подвергается окислению, отдавая
электроны молекуле NAD+. При этом тиополуацеталь
превращается в тиоэфир:
R
I
E-S-C4
Η ОН
+ NAD+
Ε—S—С
'*,
NADH
О
Ε—S—С + НО
чЧо
Как и ацетил-СоА, тиоэфир (R-CO-S-) является мак-
роэргическим соединением, которое при фосфоролизе
превращается в смешанный ангидрид фосфоглицерино-
вой и фосфорной кислот:
D О 0 0
II II II
Р-0 τ—^ R—С-0—Р-0 + Е—SH
I I
0" 0"
Высокоэнергетическая группа R-CO-PO|" может
переносить фосфорильный остаток на ADP с
образованием АТР. По чисто номенклатурным соображениям
фермент, катализирующий такой перенос, назван не по
прямой, а по обратной реакции фосфоглицераткина-
зой. Киназы всегда получают свое название по
соединению, находящемуся по одну сторону с АТР в уравнении
реакции (рис. 8.6.).
Образовавшаяся в ходе этого процесса фосфориль-
ная группа остается прикрепленной к истинному
субстрату фермента. Поэтому процесс получил название
субстратного фосфорилирования. З-Фосфоглицерат -
низкоэнергетическое соединение фосфата, которое не
может фосфорилировать ADP. Однако на следующих
этапах гликолиза внутримолекулярные перестановки
приводят к тому, что низкоэнергический фосфоэфир
становится высокоэнергетической фосфорильной
группой, переносимой на ADP с образованием АТР.
часть 3 Метаболизм
112
AG0' = +6,3 кДж · моль"1 I ЧОРОз"
= СНОН + NADH + Н+
Глицеральдегид- I 2.
3-фосфатдегидрогеназа ^r^uruj
1,3-Дифосфоглицерат
(13-ДФГ)
1ЧН
СНОН + NAD+ + Ρ,
I 2
СН2ОРОз
Глицеральдегид-
3-фосфат
З-Фосфоглицерат-
киназа //. ^о' ι ο η ττ -ι
"AGU = -18,9 кДж· моль 1
росной
I 2
СН2ОРОз
З-Фосфоглицерат
(3-ФГА)
Рис. 8.6. Превращение глицеральдегид-3-фосфата в 3-фосфоглицерат
Заключительные стадии гликолиза
Сначала фосфорильная группа 3-фосфоглицерата
переносится из положения 3 в положение 2 фосфоглицерат-
мутазой.
С00"
I
СНОН
I 7
СН2ОРОз
С00"
СНОРО^
СН2ОН
AG0' = +4,4 кДж · моль'1
З-Фосфоглицерат
2-Фосфоглицерат
В таком виде реакция внутримолекулярного
переноса фосфатной группы протекает лишь в растениях.
В мышцах кролика, например, все происходит сложнее.
Фермент содержит фосфатный остаток, который
переносится на 2-ОН-группу 3-фосфоглицерата,
превращающегося при этом в 2,3-дифосфоглицерат. После
этого с 2,3-дифосфоглицерата 3-фосфорильная группа
переносится на фермент, чтобы восполнить отданный
фосфат, и в результате из 3-фосфоглицерата образуется
2-фосфоглицерат. Следующая стадия гликолиза
заключается в отщеплении молекулы воды от 2-фосфоглице-
рата. Обычно дегидратирующие ферменты так и
называют дегидратазами, однако в данном случае
используется историческое название енолаза, которое отражает
химический смысл реакции - образование
замещенного енола.
С00"
1 2"
СНОРОз
I
СН2ОН
2-Фосфоглицерат
Енолаза
-Н20
С00"
1 2
С-0—Р0§
II
СН2
Пируват-
киназа
ADP
С00
I
с
ОН
Фосфоенолпируват
СН2
+ АТР
Енолпируват
С00"
I
с=о
I
СН3
Пируват
Хотя изменение свободной энергии реакции,
катализируемой енолазой, составляет всего AG®= +1,8 кДж · моль-1,
но фосфоенолпируват, в отличие от фосфоглицерата,
является макроэргическим соединением; его гидролизу
соответствует AG0' = -62,2 кДж · моль-1. Причина такой
разницы величин AG0' кроется в том, что продукт
реакции - енольная форма пирувата - спонтанно
превращается в кетоформу в реакции, которой соответствует
большое отрицательное значение AG0'. Кето-енольная
изомерия хорошо изучена. Кетоформа (в данном
случае - пируват) обычно термодинамически выгоднее
енольной. Поэтому значительное по величине
отрицательное изменение свободной энергии суммарной
реакции есть следствие вклада изомеризации. Фосфатная
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
ИЗ
группа с фосфоенолпирувата переносится на ADP
ферментом пируваткиназой (опять-таки по номенклатуре
киназы называют по обратной реакции фосфорилирова-
ния субстрата, даже если сама реакция в таком виде
не имеет места). Необратимость превращения
фосфоенолпирувата в пируват имеет важное значение для глю-
конеогенеза. Интересно, что в растениях и
микроорганизмах пируват превращается в фосфоенолпируват,
но происходит это под действием другого фермента,
а главное, при этом расходуется не эквимолярное, а
вдвое большее количество АТР.
Полностью метаболические превращения при
гликолизе представлены на рис. 8.7.
Гексокиназа
Глюкоза ДО0'. кДж · моль-1
L^ATP
Ρ ADP
Глюкозо-6-фосфат
-16,7
Глюкозофосфат-
изоме аза
+ 1,7
Фруктозо-6-фосфат
tATP
-14,2
ADP
Глицеральдегид
3-фосфат
Глицеральдегид-3-
фогфатдег др геназа
Фруктозо-1,6-дифосфат
+24,3
+7,5
Гриозофосфат-
изомераза
Дигидрокси-
ацетонфосфат
NAD+
+6,3
-NADH + Н+
1,3-Дифосфоглицерат
З-Фосфоглицерат- ][ -18.5
1ГАТР
З-Фосфоглицерат
Фосфоглицерат-
м таза
+4,4
2-Фосфоглицерат
Енолаза
U
Пируваткиназа
Н20
Фосфоенолпируват
Uadp
f^ATP
Пируват
1,8
-31,4
Рис. 8.7. Гликолитический путь
Красной стрелкой помечены необратимые реакции
Баланс АТР при гликолизе
Если гликолиз начинается с глюкозы, то на образование
глюкозо-1-фосфата и фруктозо-1,6-дифосфата
расходуются 2 молекулы АТР. Однако в результате фосфогли-
цераткиназной и пируваткиназной реакций образуются
2 молекулы АТР (см. рис. 8.7). А если учесть, что 1
молекула фруктозо-1,6-дифосфата расщепляется на 2
молекулы глицеральдегид-3-фосфата, то их последующее
превращение в пируват сопровождается образованием
4 молекул АТР. Следовательно, суммарный выход мак-
роэргических соединений в результате гликолитическо-
го расщепления 1 молекулы глюкозы равен 2
молекулам АТР. Если же гликолиз начинается с гликогена, то
на образование фпуктозо-1,6-цифосфата расходуется
всего 1 молекула АТР, а синтезируются те же 4
молекулы, и, следовательно, в этом случае образуется 3
молекулы АТР.
Реокисление цитоплазматического
NADH челночными системами
переноса электронов
В аэробных условиях NADH, образующийся при
гликолизе, подвергается реокислению путем переноса
электронов внутрь митохондрий. Сам NADH не может
проникнуть внутрь митохондрий. Для переноса электронов
предусмотрены две системы.
Первая из них использует дигидроксиацетонфос-
фат (ДГАФ), образующийся при расщеплении
фруктозо-1,6-дифосфата альдолазой. В цитоплазме
электроны от NADH передаются на ДГАФ с образованием
глицерин-3-фосфата. Это происходит при участии
фермента глицерин-3-фосфатдегидрогеназы (здесь
фермент также назван по обратной реакции). Схема
глицеро-фосфатной челночной системы представлена
на рис. 8.8.
Глицерин-3-фосфат достигает внутренней мембраны
митохондрий (вспомним, что внешняя мембрана
для большинства низкомолекулярных веществ не
является преградой), где глицерин-3-фосфатдегидрогена-
за, встроенная в мембрану (простетической группой
фермента служит FAD), передает электроны с глице-
рин-3-фосфата в митохондриальную электронтранс-
портную цепь. Образующийся при этом ДГАФ
возвращается в цитоплазму, замыкая таким образом
челночный цикл. Необходимо отметить, что переносчик
электронов глицерин-3-фосфат не проникает в матрикс
митохондрий. Его задача - перенести электроны с
цитоплазматического NADH на митохондриальную элек-
тронтранспортную цепь.
часть 3 Метаболизм
114
Цитоплазма
Проницаемая внешняя
митохондриальная
мембрана "*--.._
Дигидроксиацетонфосфат
СН2ОН
С=0
I 2-
СН2ОР03
NADH + Н+
Цитоплазматическая
глицерин-3-
фосфатдегидрогеназа
NAD+
СН2ОН
НОСН
2-
СН2ОР03
Глицерин-3-фосфат
Внутренняя
митохондриальная
мембрана
Дыхательная
цепь переноса
электронов
Митохондриальная
глицерин-3-фосфат-
дегидрогеназа
Рис. 8.8. Схема глицерофосфатной челночной системы, передающей электроны от цитоплазматического NADH
на митохондриальную дыхательную цепь
Другая челночная система - малат-аспартатная -
переносит электроны от цитоплазматического NADH
к митохондриальному NAD+. Это приводит к
образованию митохондриального NADH, который далее
окисляется в электронтранспортной цепи. В цитоплазме NADH
восстанавливает оксалоацетат до малата. Последний
с помощью специального переносчика попадает в
митохондрии, где реокисляется в оксалоацетат с
восстановлением NAD+. Сам оксалоацетат выйти из митохондрий
не может, поэтому он сначала превращается в аспартат,
который и транспортируется переносчиком в
цитоплазму. В цитоплазме аспартат дезаминируется, превращаясь
в оксалоацетат и замыкая тем самым челночный цикл
(рис. 8.9). Результатом этого цикла является окисление
цитоплазматического NADH митохондриальным NAD+.
В различных тканях глицерофосфатный и малат-ас-
партатный челночные механизмы, по-видимому,
работают с разной интенсивностью. Они существенно
отличаются. Первый с помощью цитоплазматического NADH
обеспечивает восстановление митохондриального FAD,
являющегося простетической группой флавопротеина -
глицерин-3-фосфатдегидрогеназы. Редокс-потенциал
у FADH2 выше, чем у NADH. FADH2 передает
электроны далее по цепи переноса. Чем дальше в этой цепи
кислород расположен от переносчика, тем больше АТР
будет синтезировано при движении электронов к
кислороду. При окислении молекулы цитоплазматического
NADH и реокислении митохондриального FADH2
образуется 1,5 молекулы АТР. Начинаясь с молекулы
цитоплазматического NADH, малат-аспартатный челночный
цикл завершается восстановлением молекулы
митохондриального NAD+, при реокислении которой
образуется 2,5 молекулы АТР.
Транспорт пирувата в митохондрии
Наряду с NADH продуктом гликолиза является пиру ват.
Если он не восстанавливается до лактата, то попадает
внутрь митохондрий благодаря транспортной системе,
обеспечивающей его антипорт с ионами ОН .
Стадия 2.
Цикл лимонной кислоты
Продукт гликолиза пируват вступает в метаболические
превращения, относящиеся к циклу лимонной кислоты,
не сам по себе, а будучи предварительно
переработанным в ацетил-СоА. Поэтому мы и начнем этот раздел
с рассмотрения механизмов образования ацетил-СоА
из пирувата.
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
115
Проницаемая внешняя
митохондриальная
мембрана "*---..
Цитоплазма
ДХГ
CHNH3 Аспартат
СН2
СОО"
сосг
I Оксалоацетат
ι 2
СОСГ
Внутренняя
митохондриальная
-"' мембрана
NADH
Η
NAD+
Малат-
дегидрогеназа
СОО
I
онсн
I
сн2
СОО"
Малат
L_ J
Митохондриальный матрикс
СОО
Аспартат CHNH3
сн2
СОО"
Оксалоацетат
СОО"
I
с=о
I
сн2
СОО"
Малат-
дегидрогеназа
NADH
Н+
NAD+
Малат
СОО"
I
онсн
I
сн2
СОСГ
Реакция превращения пирувата в ацетил-СоА
необратима (AG°'= -33,5 кДж · моль1). Это означает, что
жирные кислоты в организме животных не могут
превращаться прямо в глюкозу, хотя бактерии и растения
делают это с помощью специального механизма.
Пируватдегидрогеназа устроена довольно сложно.
Она состоит из множества полипептидных цепей и
объединяет в одной молекуле три различные
ферментативные активности, каждая из них ответственна за
протекание одной из стадий реакции. Первая стадия - де-
карбоксилирование, в результате которого от пирувата
отщепляется С02, а остаток молекулы превращается
в гидроксиэтильную группу
Η
сн3с-,
он
которая присоединяется к кофактору тиаминпирофос-
фату. Этот кофактор образуется в организме из
витамина В j (тиамина), недостаток которого приводит к
нарушениям метаболизма углеводов.
CH2V/Hf о О
\ I и и
Х=С-СН2-СН2-0—Р-0—Р-0"
3 СН3 О О"
Тиаминпирофосфат
Рис. 8.9. Малат-аспартатная челночная система для
переноса электронов от цитоплазматического NADH к митохон-
дриальному NAD*
Механизм взаимопревращения оксалоацетата и аспартата, в
отличие от глицерофосфатного, обратим и поставляет NADH
в митохондрии лишь при условии, что отношение NADH/
NAD+ в цитоплазме выше,чем в митохондриальном матриксе
Превращение пирувата вацетил-СоА-
предварительный этап цикла лимонной
кислоты
В матриксе митохондрий пируват превращается в аце-
тил-СоА, после чего ацетильная группа пирувата
включается в цикл лимонной кислоты. Суммарно реакцию,
катализируемую пируватдегидрогеназой, можно описать
следующим уравнением:
Пируват + NAD+ + CoA-SH ->
-> Ацетил-S-CoA + NADH + Н+ + С02.
Гидроксиэтильная группа пирувата в несколько
стадий преобразуется в ацетильный остаток ацетил-СоА,
причем такое превращение сопровождается
восстановлением NAD+. По понятным причинам этот процесс
называется окислительным декарбоксилированием.
В окислительно-восстановительных реакциях здесь
участвует еще один важный кофактор - липоевая
кислота, которая может существовать в двух формах:
ациклической (восстановленной) и циклической (окисленной).
5Н
SH
I
СН2 ^СН-СНз-СНз-СНз-СНз-СОО"
чсн2
Липоевая кислота
(восстановленная)
S S
ί !
СН2 СН-СН2-СН2-СН2-СН2-СОО"
сн2
Липоевая кислота
(окисленная)
часть 3 Метаболизм
116
Остаток липоевой кислоты входит в состав
фермента в качестве простетической группы: он прикреплен
к боковой цепи одного из лизиновых остатков
посредством амидной связи -CO-NH-. На рис. 8.10, который
иллюстрирует участие липоевой кислоты в синтезе аце-
тил-СоА, липоиллизиновые остатки изображены в виде
дисульфидного фрагмента, а символами Е,, Е2 и Е3
обозначены три фермента, входящих в состав единого
ферментного комплекса.
В чем истинная магия цикла?
Цикл лимонной кислоты, на первый взгляд
представляющий серию обычных химических реакций, является
примером удивительной изобретательности природы,
придумавшей, как получить универсальное топливо
в форме восстановительных эквивалентов NADH
и FADH2, часть которых образуется при расщеплении
воды. Это топливо далее используется митохондриаль-
ной цепью переносчиков электронов для генерации АТР
из ADP и Pf.. Расщепление воды не является здесь
стартовым процессом накопления энергии, как при
фотосинтезе (см. главу 14), а кислород высвобождается не
в свободном виде, а в составе С02. Для управления
процессом используется энергия превращения
ацетильной группы ацетил-СоА. Необходимо
последовательно разобраться в деталях цикла, чтобы понять его
суть и значение.
Ацетил-СоА вступает в цикл, взаимодействуя с окса-
лоацетатом, в результате чего образуется цитрат (анион
лимонной кислоты). В ходе этой реакции поглощается
молекула воды. Когда цикл замыкается, вновь
образуется оксалоацетат, а ацетильная группа ацетил-СоА
исчезает. В результате одного оборота цикла (рис. 8.11)
образуются 2 молекулы С02; 3 молекулы NAD+
восстанавливаются до NADH; 1 молекула FAD
восстанавливается до FADH2; синтезируется 1 молекула GTP из
GDP и Pf.; ацетил-СоА теряет ацетильную группу и
превращается в CoA-SH.
Если сложить все образующиеся восстановительные
эквиваленты в трех NADH и одном FADH2, то всего
их будет восемь, поскольку в обоих случаях
восстановление является двухэлектронным. К этому числу
надо прибавить единицу, поскольку еще один эквивалент
выделяется в виде образовавшейся тиольной группы
в СоА—SH. Итак, на один оборот цикла выделяется
9 восстановительных эквивалентов, что равносильно
9 атомам водорода.
ТРР
ТРР
ТРР
NADH + Н+
NAD4
О
СН^С-
Липоевая кислота
FAD
1 СН^СОСОО"
он X
FADH7
СН3С
н ι
ТРР
FAD
S-CoA
О SH'
jppCH3C—S~
CoA-SH
FAD
Ei
E2 E3
FAD
_l
Рис. 8.10. Механизм пируватдегидрогеназной реакции
ТРР - тиаминпирофосфат; Е,, Е2, Е3 - ферменты в составе комплекса
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
117
сн3со-
S-CoA CoA-SH
Оксалоацетат
Цикл
Н20
\
Цитрат
GDP + Р/
3NADH + Н+
1FADH2
2С02
Упрощенное описание
цикла лимонной кислоты
Изучая одну за другой множество химических реакций,
составляющих цикл, можно потерять представление
о его «архитектуре» в целом. Поэтому имеет смысл
сначала рассмотреть упрощенную версию цикла.
Оксалоацетат - вещество, с которого все
начинается и которым все завершается. Ацетат - это СН3СОО~,
оксалильная группа (ацильный остаток щавелевой
кислоты) - это "ООС-СО- , а оксалоацетат имеет
структуру*
Рис. 8.11. Включение в цикл и вывод веществ из цикла
лимонной кислоты
Отдельные реакции цикла не показаны
Три атома водорода и один атом кислорода
поставляются ацетильной группой CH3CO-S-CoA. Остается
понять, откуда берутся остальные шесть атомов
водорода и три атома кислорода. Кислород не участвует
в цикле, поэтому нужно выявить источник трех атомов
кислорода, необходимых для образования двух молекул
С02 (четвёртый - из ацетильной группы). Их, как,
впрочем, и атомы водорода, могут поставлять молекулы
воды. Одна из них используется при синтезе цитрата,
другая - при образовании оксалоацетата. Остается
объяснить происхождение еще двух атомов водорода
и одного атома кислорода, однако это удобнее будет
сделать несколько позже (см. с. 120).
Получается удивительный результат. Внешне все
выглядит так, будто электроны воды сами собой
поднимаются по энергетической шкале, с
достигая уровня
восстановительных эквивалентов NADH и FADH2
(это же справедливо для
электронов ацетильной группы аце-
тил-СоА). Однако речь идет
не о орямом переносе атомов
водорода воды на NADH и FADH2,
а лишь о химическом балансе,
в соответствии с которым водород
из воды восстанавливает NAD+
и FAD, а кислород воды
расходуется на образование С02.
Разумеется, это эндоэргонические
процессы, которые требуют затрат
свободной энергии. Источником
этой энергии является
превращение ацетильной группы пирува-
та в ацетил-СоА и последующие
продукты.
V
I
н2с-
СОО"
-сект
Ацетильная группа ацетил-СоА присоединяется к окса-
лоацетату, который при этом превращается в цитрат.
Глядя на формулу последнего, можно понять, где идет
присоединение:
СН2С00"
ι
но -с
Н;С-
соо~
-соо
Цитрат - анион оптически симметричной трикарбо-
новой С6-кислоты - изомеризуется в анион другой три-
карбоновой кислоты - изоцитрат, в молекуле которого
есть один центр асимметрии. Изоцитрат далее
последовательно превращается в ос-кетоглутарат (С5), сукцинат
(С4), фумарат (С4), малат (С4) и, наконец, в исходный
оксалоацетат (рис. 8.12).
СН2-СОО"
I
но-с-сосг
I
сн2-сосг
с2
Ацетильная,
группа ^
ацетил-СоА
к Цитрат
С6
сн2-сосг
НС-СОО"
I
НС*-СОО"
он
Изоцитрат
со2
^.
-сосг
-сосг
н2с-
Оксалоацетат
С4
ОН
I
нс-сосг
I
Н2С-СОО"
Малат
Η СОО
V
оос н
Фумарат
СН2-СОО"
I
сн2
I
С-СОО"
а-Кетоглутарат
со7
СН2-СОО"
I
СН2-СОО"
Сукцинат
Рис. 8.12. Реакции цикла лимонной кислоты
Рисунок знакомит со структурой кислот, участвующих в цикле, и их взаимосвязью
часть 3 Метаболизм
118
Цикл завершился, а ацетильный остаток исчез.
Теперь перейдем к более подробному рассмотрению
химических превращений всех веществ, участвующих
в цикле.
Механизмы реакций
цикла лимонной кислоты
Для удобства эти реакции можно разбить на три группы:
1) синтез цитрата- реакция, благодаря которой в цикл
вводится ацетильная группа; 2) превращение цитрата
(С6) в α-кетоглутарат (С5); 3) превращения
(^-соединений - сукцината в оксалоацетат.
Синтез цитрата
Фермент, ответственный за синтез цитрата, называют
цитратсинтазой (заметим, не синтетазой, поскольку
в реакции не участвует АТР). Он катализирует
реакцию конденсации ацетил-СоА с оксалоацетатом,
приводящую к образованию цитрил-СоА, который
неустойчив и быстро гидролизуется до цитрата и
свободного СоА. Суммарная реакция необратима, поскольку
AG0' = -32 кДж ■ моль1.
О 0=С—С00"
II I
СН3 —С—S—СоА Н2С—С00"
Ацетил-СоА
Оксалоацетат
Цитратсинтаза
СН2-С—S—СоА
НО-С —С00"
I
СН2-СОО"
Цитрил-СоА
СН2-СОО
Н20
I
НО-С—COO" + CoA-SH
I
СН2-СОО"
Цитрат
Превращение цитрата в а-кетоглутарат
Суть реакции Цитрат—> Изоцитрат состоит в
перемещении гидроксильной группы симметричной молекулы
цитрата из положения 3 в положение 2 с образованием
асимметричной молекулы изоцитрата. Механизм
реакции заключается в одновременной обратимой
дегидратации цитрата и изоцитрата в общий для них продукт -
г/мс-аконитат (аконитовую кислоту впервые нашли в
растениях рода Aconitum).
1 но I
Н—С-ОН н2и Н—С
I
н—с—н
Н20
н—с
Обе реакции катализирует фермент аконитаза. Это
название - дань традиции, поскольку ферменты этого
класса принято называть дегидратазами.
СН2-СОО"
НО—С—С00"
I
СН2-СОО"
Цитрат
-н2о сн2-соо- Нг0 сн2-соо
н2о ин_
С—С00"
I
1С—С00"
-н2о ι
-С00" 2 СНОН-СОО"
г/м>Аконитат
Изоцитрат
В клетке изоцитрат легко превращается в
а-кетоглутарат, поэтому равновесие реакции сдвинуто в сторону
образования изоцитрата:
Цитрат -> цис-Аконитат -^ Изоцитрат.
На примере лактатдегидрогеназы мы уже познакомились
с ЫАО+-зависимыми дегидрогеназами. Реакция
окисления изоцитрата и лактата принадлежит к одному типу
реакций:
Н—С—Η
I
NAD+
Η—С-
I
с=
NADH + Н+
Н—С-ОН
I I
В цикле изоцитратдегидрогеназа катализирует
реакцию образования ос-кетоглутарата:
СН2—С00"
I
сн-соо-
I
СНОН-СОО"
Изоцитрат
NAD
СН2—COO"
I
аСН—С00~
I
ВС—С00"
II
о
Оксалосукцинат
+ NADH + Н+
СН2—С00"
I
сн2
С—С00"
+ со?
а- Кетоглутарат
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
119
Промежуточным продуктом окисления изоцитрата
является оксалосукцинат, который, как и все β-кетокис-
лоты (кетогруппа здесь в β-положении по отношению
к центральному карбоксилу), нестабилен и легко теряет
карбоксильную группу в виде С02. Декарбоксилирова-
ние происходит на поверхности изоцитратдегидрогена-
зы с образованием α-кетоглутарата (С5).
Четырехуглеродные кислоты
α-Кетоглутарат является аналогом пирувата, и оба эти
соединения имеют общую формулу:
R
C-COO"
II
о
в которой пирувату соответствует R= -CH3, а а-кетоглу-
тарату - R= -CH2CH2COO~. Ранее мы встречались
с превращением пирувата в ацетил-СоА и С02, которое
катализирует пируватдегидрогеназа:
С—С00~ + NAD+ + CoA—SH
II
о
R
I
C-S-
-СоА + С02 + NADH + Н+
Подобно пируватдегидрогеназному комплексу,
α-кетоглутаратдегидрогеназа (также ферментный
комплекс) катализирует образование сукцинил-СоА
из а-кетоглутарата:
СН2—С00"
I
снг
С—S—СоА
II
о
Сукцинил-СоА
Однако, если ацетил-СоА в цикле используется
для синтеза цитрата, то сукцинил-СоА гидролизуется
до сукцината и свободного CoA-SH. Изменение
свободной энергии гидролиза этого тиоэфира составляет
AG0' = -35,5 кДж · моль-1. Этого достаточно для
синтеза макроэргического фосфата, что и происходит на
следующем этапе.
Сопряжение гидролиза сукцинил-СоД
с синтезом GTP
Суммарное уравнение для сопряженных реакций
выглядит так:
Сукцинил-СоА + GDP + Р, <-> Сукцинат + GTP + CoA-SH
д£°'=-2,9кДж моль"1.
Фермент, катализирующий эту реакцию, называется
сукцинил-СоА-синтетаза (и здесь по номенклатурным
соображениям фермент назван по обратной реакции,
которая в клетке не протекает). У растений, в отличие от
животных, аналогичный фермент синтезирует не GTP,
аАТР.
Сукцинил-СоА-синтетазная реакция начинается
с фосфоролиза тиоэфирной группы в сукцинил-СоА:
сн2—соо о
ι и
сн2 + но—р-о"
I
C-S—СоА
II
о
Сукцинил-СоА
О"
СН2 — СОО"
СН2 О + СоА—SH.
С-0—Р-0"
II I
о о"
Сукцинил-фосфат
Далее фосфатный остаток переносится на GDP с
образованием GTP и сукцината:
СН2—СОО"
ι о
сн2 о _ и
I 2 и + О—P-0-GMP
С-0—Р-0" !_
II I О
О О"
Сукцинил-фосфат
GDP
СН2—СОО"
СН2—СОО"
О О
II II
"О—Р-0—P-0-GMP,
1_ 1_
о о
Ранее (см. с. 118) мы подводили баланс атомов
водорода и кислорода в цикле, и для сведения этого баланса
не хватило двух атомов водорода и одного - кислорода.
Теперь можно указать причину этого дисбаланса:
участие неорганического фосфата в расщеплении сукцинил-
СоА. Для ясности представим истинную реакцию в
виде эквивалентной комбинации двух реакций:
GDP + Р,-> GTP + Н20,
Сукцинил-СоА + Н20 -> Сукцинат + CoA-SH.
часть 3 Метаболизм
120
Подчеркнем, что такая комбинация двух реакций
не отражает истинного механизма процесса, но зато
позволяет разобраться, откуда берется недостающая
молекула воды.
Превращение сукцината в оксалоацетат
На первом этапе этого превращения FAD-содежащий
фермент сукцинатдегидрогеназа окисляет сукцинат
до фумарата. Это окисление сопровождается
отщеплением двух атомов водорода:
I
н—с—н
I
н—с—н
I
I
2H
сн
II
сн
Почему для окисления сукцината здесь
используется FAD, а не NAD+? Дело в величине редокс-потенци-
ала пары сукцинат-фумарат. По термодинамическим
причинам электроны перетекают от пары с меньшим ре-
докс-потенциалом к паре с большим редокс-потенци-
алом. Сукцинат оказывается слишком слабым
восстановительным агентом для NAD+, однако он может
восстановить FAD. Эта реакция описывается уравнением:
СН2-СОО"
I + Enz-FAD-
СН2-СОСГ _
Сукцинат-
К .COO
+ Enz-FADH2.
Сукцинат
дегидргеназа
"ООС"" ^Н
Фумарат
Последний этап, который замыкает цикл, состоит
в превращении фумарата в оксалоацетат. Сначала из
фумарата в результате присоединения молекулы воды
образуется малат (малат - анион яблочной кислоты).
Фермент, осуществляющий эту реакцию, логично было бы
назвать фумаратгидратазой, однако за ним
закрепилось традиционное название - фумараза.
К .COO
ОН
I
"ООС
Фумарат
Фумараза Н-С—С00
+ Н20 . - I
\н Н2С-СОО"
Малат
Малат далее окисляется в оксалоацетат с помощью
ЫАО+-зависимого фермента малатдегидрогеназы:
ОН
Малат-
н_(2 С00— дегидрогеназа
+NAD =^ Г "~ + NADH+H+
Н2С—COO Н2С—СОСГ
0
II
С-СОО"
Малат
Оксалоацетат
Это эндоэргоничная реакция (AG°= +29,7 кДж · моль1),
и сама по себе она бы не протекала Однако
образование оксалоацетата замыкает цикл, и дальше следует
первая из рассмотренных нами реакций - превращение
оксалоацетата в цитрат. Это очень экзоэргоничный
процесс, вследствие чего концентрация оксалоацетата мала.
Равновесие реакции Малат «-> Оксалоацетат смещается
в сторону образования оксалоацетата. Иными словами,
реальная величина AG гораздо меньше, чем
стандартная AG0', благодаря чему малат окисляется.
Полностью цикл лимонной кислоты представлен на
рис. 8.13.
Что определяет общее направление
реакций в цикле?
Из рис. 8.13 следует, что все реакции в цикле протекают
согласованно в одном направлении. Это обусловлено
тем, что трем из всех реакций отвечают столь большие
по абсолютной величине отрицательные значения AG0',
что они являются практически необратимыми. Одна
из таких реакций - это синтез цитрата из ацетил-СоА
и оксалоацетата (AG°'=-32,2 кДж · моль-1), другая -
декарбоксилирование изоцитрата в а-кетоглутарат
(AG0'= -20,9 кДж · моль1), третья - образование сукци-
нил-СоА из α-кетоглутарата (AG°'= -33,5 кДж · моль1).
Это приводит к тому, что цикл работает в одном
направлении, несмотря даже на то, что равновесие малат-
дегидрогеназной реакции сдвинуто в противоположную
сторону (AG°'= +29,7 кДж · моль1). Суммарная
величина изменений свободной энергии всех реакций цикла
отрицательна, что и обеспечивает его протекание в
одном направлении.
Стехиометрия цикла
Суммарное уравнение, подводящее итоги работы
цикла, имеет следующий вид:
CH3CO-S-CoA + 2Н20 + 3NAD+ + FAD + GDP + Р,->
-> 2C02 + 3NADH + 3Η+ + FADH2 + CoA-SH + GTP
ЛО0' = ^ЮкДж· моль"1.
Если попытаться подвести в этом уравнении баланс
атомов водорода и кислорода, обнаружится нехватка
одного моля Н20. Причина этого была рассмотрена
нас. 120.
Субстратное обеспечение
цикла лимонной кислоты
Цикл начинается с реакции конденсации оксалоацетата
и ацетил-СоА и заканчивается образованием
оксалоацетата. Цикл существует не изолированно от других
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
121
Аконитаза
СН2-СОО"
I
но-с-coo-
I
сн2-соо
Цитрат
сн2-соо
I
с-соо
СН2-С00"
1_
Аконитаза НС СОО
НС-СОО"
Изоцитрат-
дегид огеназа
а-Кетоглутарат
+ Н20
НС-СОО"
«{мс-Аконитат
CoA-SH
CH^-C-S-CoA
Н70
^
С-СОО"
I
н2с-соо~
Оксалоацетат
NAD+
Малат-
егидрогеназг
ОН
Изоцитрат
CoA-SH
3(NADH + Н+)
1 FADH2
α-Кетоглутарат-
цегидрогеназа
со2
Малат
^укцинат-
цегидрогеназа
Сукцинил-СоА
/^ GDP + Р,
У? Сукцинил-
сн2-соо- [ \тр
сн2-соо- CoA_SH
Сукцинат
Фумарат
Рис. 8.13. Полный цикл лимонной кислоты
Цветом помечено образование восстановительных эквивалентов. FAD - простетическая группа сукцинатдегидрогеназы
процессов метаболизма, и некоторые из участвующих
в нем кислот используются для других целей. Однако они
не поступают в организм с пищей в достаточных
количествах. Метаболизм пищевых углеводов сопровождается
образованием большого количества С3-соединений
в форме пирувата, превращением липидов - появлением
С2-соединений в виде ацетильных групп. Характерные
для цикла С4-, С5- и С6-кислоты могут образовываться
из аминокислот, но они и расходуются для синтеза
некоторых из них (см. главу 15), да и других метаболитов,
в силу чего их содержание в клетке ограничено. В
действительности между разными метаболическими
системами устанавливаются сложные стационарные
состояния. Главной задачей цикла является бесперебойное
обеспечение «топливом» процесса генерации энергии
в митохондриях, поэтому особое значение имеет так
называемая анаплеротическая реакция,
заключающаяся в синтезе оксалоацетата из пирувата и С02:
СОО"
I
О
АТР + С=0 + HCOf — С-СОО" + ADP + Р; + Н+.
II
СН3 Н2С—СОО"
Реакцию катализирует фермент пируваткарбокси-
лаза (не надо путать с пируватдекарбоксилазой
дрожжей!). Особенность анаплеротической реакции
заключается в том, что она является уникальным процессом
превращения С3-кислоты в С4-кислоту. Рабочим
элементом пируваткарбоксилазы служит биотин. Он является
кофактором синтетических реакций карбоксилирования,
в которых используется так называемый активный С02.
Биотин представляет собой водорастворимый витамин
группы В. Будучи ковалентно связанным с ферментом,
он реагирует с бикарбонатом, образуя карбоксибиотин.
Эта реакция энергетически сопряжена с гидролизом
АТР. Карбоксибиотин является реакционноспособным,
но достаточно устойчивым соединением; он может кар-
боксилировать другие вещества, перенося на них
карбоксильный остаток. Движущей силой таких реакций
служит значительная отрицательная величина
изменения свободной энергии при отщеплении С02 от карбок-
сибиотина (AG°'=-19,7 кДж· моль1). В данном случае
субстратом фермента служит пируват, но и другие кар-
боксилазные ферментные системы используют биотин
в качестве кофактора.
часть 3 Метаболизм
122
о
ϊ
ΗΝ ΝΗ
\S^^(CH2)4-C—Enz
Биотин
♦ \-o-
но
ATP
О f^ADP
- if Л
О—С—N ΝΗ
W- if
^sy^{CH2)A-C—Enz
Карбоксибиотин
Пируваткарбоксилаза содержит два каталитических
центра: один для карбоксилирования биотина, а
другой - для переноса карбоксильной группы с биотина
на пиру ват (у некоторых бактерий эти функции
выполняют два разных фермента). Биотин образует амидную
связь с одним из белковых остатков лизина. Таким
образом, он оказывается связанным с белком как бы
длинной гибкой «ножкой». Полагают, что изгиб этой ножки
позволяет биотину перемещаться из одного
каталитического центра фермента в другой (рис. 8.14).
Биотин у Фрагмент
''' пируваткарбоксилазы
■ Центр
карбоксилирования
пирувата
Центр /
карбоксилирования
биотина
Рис. 8.14. Биотин в активном центре пируваткарбоксилазы
Рабочий элемент биотина прикреплен к белку с помощью
длинной «ножки». Она позволяет биотину перемещаться от одного
активного центра к другому
Стадия 3. Цепь переноса
электронов от NADH и FADH2
на кислород
Окисление глюкозы (или гликогена) включает три
основных этапа. Первым^ является гликолиз, вторым - цикл
лимонной кислоты. Теперь мы подошли к рассмотрению
конечного этапа. С точки зрения генерации энергии,
на первых двух этапах результат весьма скромен: всего
2 молекулы АТР на 1 молекулу глюкозы образуется
в процессе гликолиза и столько же - в цикле лимонной
кислоты (с учетом энергетической эквивалентности GTP
и АТР); еще одна молекула АТР образуется, если
источником глюкозы служит гликоген. Весьма важно, что
большая часть энергии после первых двух этапов запасается
в виде 10 молекул NADH (две из гликолиза, две из пи-
руватдегидрогеназной реакции и шесть из цикла
лимонной кислоты) и 2 молекул FADH2 (из цикла лимонной
кислоты). Следует помнить, что из глюкозы образуется
2 молекулы пирувата, которые обеспечивают 2 оборота
цикла.
Итак, основное количество АТР, синтезируемого
из ADP и Pf. в ходе окисления глюкозы, образуется в
результате окисления NADH и FADH2.
Цепь переноса электронов
Переносчики электронов размещены на поверхности или
в глубине внутренней митохондриальной мембраны. Она
уложена в кристы (см. рис. 7.4), число и плотность
упаковки которых коррелирует с энергетическими
потребностями клетки.
Что представляют собой
переносчики электронов?
Многие переносчики электронов - это белки,
содержащие в качестве простетической группы гем.
Переносчики электронов называют цитохромами, так как они
окрашены в красный цвет. Разные цитохромы
обозначаются буквенными индексами: сх, с, а и аъ - в порядке
их расположения в цепи (роль двух цитохромов Ъ
обсудим позже). Главная часть структуры гема показана на-
рис. 8.15, а полностью она изображена на рис. 27.3.
Основное, что нужно знать о геме как о
простетической группе цитохромов-переносчиков, это изменение
валентности его атома железа при их
функционировании: Fe2+ при получении электрона от предыдущего
\ : / -е- \ : /
N Fe2+ N , N Fe3+ Ν
/ ! \ +р- / ! \
Восстановленный гем Окисленный гем
Рис. 8.15. Схематическое изображение структуры гема
\ /
N
1
1
1
Fe2+—
1
1
1
/\
/
-N _
\
-е~
+е~
\
"* N-
/
\ /
N
1
1
— Fe3+
1
1
1
/N\
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
123
переносчика цепи и Fe3+ - после передачи электрона
последующему. Свойства самой молекулы гема зависят
от белка, к которому он присоединен. Кроме того, гемы
в разных цитохромах могут отличаться строением
боковых групп и способом прикрепления к апобелку.
Поэтому нет никакого противоречия в том, что цитохромы
отличаются редокс-потенциалами, хотя у них всех про-
стетические группы почти одинаковы.
К другому типу негемовых железосодержащих
переносчиков электронов относятся белки, в которых атомы
железа связаны с сульфгидрильными группами остатков
цистеина белка, а также с сульфидными анионами,
образуя железо-серные комплексы, или центры.
Простейший из них показан на рис. 8.16.
Как и в цитохромах, атомы железа в таких центрах
могут принимать и отдавать электроны, поочередно
переходя в ферро(Ре2+)- и ферри(Ре3+)-состояния.
Железо-серные центры функционируют совместно с фла-
винсодержащими ферментами, принимая электроны
от сукцинатдегидрогеназы и дегидрогеназ,
участвующих в окислении жиров (см. главу 9). Еще одним типом
переносчиков является FMN-содержащий белок. FMN
(флавинаденин-мононуклеотид) - соединение, которое
представляет собой флавиновую половину молекулы
FAD (см. с. 101). FMN переносит электроны otNADH
на железо-серные центры.
Единственный небелковый переносчик электронов -
убихинон (рис. 8.17), названный так потому, что, с
одной стороны, он - хинон, а с другой - встречается
повсеместно (англ. ubiquitous — вездесущий). Сокращенно его
обозначают CoQ, UQ или просто Q. Все железо-серные
центры отдают электроны убихинону.
Убихинон при восстановлении приобретает не
только электроны, но и протоны. При одноэлектронном
восстановлении он превращается в семихинон
(органический свободный радикал), а при двухэлектронном -
в гидрохинон. Именно промежуточное образование
свободного радикала позволяет убихинону служить
переносчиком не двух, а одного электрона. Очень длинный
гидрофобный «хвост» (40 углеродных атомов в десяти
последовательно соединенных изопреноидных
остатках) придает убихинону способность легко внедряться
и свободно перемещаться в неполярном слое
внутренней митохондриальной мембраны.
О
Cys
\
С s
Fe-
Cys"
Полипептидная
цепь белка
Атом железа
координирован
. атомами серы,
принадлежащими
четырем остаткам
цистеина
Рис. 8.16. Строение простейшего железо-серного центра
В более сложных центрах больше атомов железа и серы
б
СНз
СНз
(сн2-сн=с-сн2)-сн2-сн=с-сн3
(л=5*9)
Убихинон (кофермент Q)
Итак, митохондриальная электронтранспортная цепь
содержит:
• FMN-белок;
• белки, имеющие негемовые железо-серные центры;
• убихинон, не связанный с белком и свободно
перемещающийся в мембране;
• цитохромы - белки, содержащие гем.
R3
R2
Длинный
гидрофобный
остаток
Окисленная форма
QH" QH2
Семихинон Восстановленная форма
(форма свободного радикала)
Рис. 8.17. Убихинон - кофермент Q (а) и его окислительно-восстановительные превращения (б)
R, -длинный гидрофобный остаток; R2=CH3; R3=OCH3. Семихинон может находиться в форме аниона (Q~)
часть 3 Метаболизм
124
Важный момент заключается в том, что один из ци-
тохромов - цитохром с (небольшой водорастворимый
белок с молекулярной массой —12,5 кДа, содержащий
чуть больше 100 аминокислотных остатков), непрочно
связан с внешней поверхностью внутренней митохон-
дриальной мембраны и легко покидает ее. Все другие
белковые переносчики - интегральные белки,
занимающие в мембране строго фиксированное положение
и ориентированные определенным образом.
Расположение
переносчиков электронов
В главе 7 говорилось о редокс-потенциалах акцепторов
электронов, в частности о том, что поток электронов
между переносчиками направлен от переносчика с
более высоким восстановительным потенциалом (т.е.
меньшим редокс-потенциалом) к переносчику с более низким
восстановительным потенциалом (т.е. более
окисленному, с большим редокс-потенциалом). В митохондриаль-
ной цепи переносчики обладают разными редокс-потен-
циалами.
Значения редокс-потенциалов непосредственно
связаны с изменениями свободной энергии (AG0).
Переносчики электронов расположены в цепи так, что AG0'
постепенно уменьшается, а редокс-потенциал
соответственно возрастает. Таким образом, на каждом этапе
передачи электрона соседнему по цепи переносчику
высвобождается свободная энергия (рис. 8.18).
Применительно к окислению глюкозы, задача,
стоящая перед электронтранспортной цепью, заключается
в переносе электронов от NADH и FADH2 на кислород.
В этом процессе участвует очень много переносчиков,
-0,4
Редокс
потенциал
да», в
Уровень
свободной \
энергии
ОН
+0,4 Η
+0,8 4
однако их можно сгруппировать в четыре комплекса,
которые встроены во внутреннюю митохондриальную
мембрану (рис. 8.19). Между комплексами электроны
перемещаются вместе с подвижными переносчиками:
убихиноном и цитохромом с. Убихинон получает
электроны от комплексов 1 и II и передает их комплексу III.
Цитохром с служит посредником между комплексами III
и IV. Комплекс I переносит электроны от NADH на Q;
комплекс II - от сукцината через FADH2 на Q;
комплекс III использует QH2 для восстановления цитохро-
ма с, а комплекс IV передает электроны с цитохрома с
на кислород. Комплексы I, III и IV называют
соответственно NADH-CoQ-редуктазой, СоС>Н2-цитохром с-ре-
дуктазой и цитохромоксидазой.
Цитохром с
^ ^ *- \е
Цитозоль
Внутренняя ]
митохондриальн
мембрата
-k
PQ
I
t
III
IV
Матрикс
NADH FADH2
ч
о2
Рис. 8.19. Четыре комплекса электронных переносчиков
в митохондриальной дыхательной цепи
Комплекс I - NADH-CoQ-редуктаза; комплекс II - сукцинат-
CoQ-редуктаза; комплекс III - Со(ЗН2-цитохром с-редуктаза;
комплекс IV - цитохромоксидаза. Q - убихинон, или кофер-
мент Q. FADH2 образуется в цикле лимонной кислоты из
сукцината в сукцинатдегидрогеназной реакции. Все комплексы
встроены во внутреннюю митохондриальную мембрану.
Кофермент Q и цитохром с - подвижные переносчики
электронов. FADH2 входит в состав флавопротеинов - дегидрогеназ,
окисляющих сукцинат и жирнокислотные производные СоА
Цитохром с >ч е-
| Цитохром а ~^
Цитохром д3
f H+ в растворе
[Кислород—*- НзО
Рис. 8.18. Редокс-потенциалы некоторых главных компонентов цепи переноса электронов в митохондриях
(дыхательной цепи)
Стрелки указывают направление переноса электронов
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
125
Комплекс IV - цитохромоксидаза - состоит из
нескольких белков. Он получает электроны от цитохрома с
с внешней стороны внутренней митохондриальной
мембраны. На пути к кислороду эти электроны проходят
через цитохромы а и av содержащие атомы меди,
которые поочередно переходят в состояния Си+ и Си2+.
Цитохромоксидаза осуществляет восстановление
свободного кислорода:
02 + 4е"+4Н+ -^2Н20.
Позже (см. главу 17) мы объясним, почему так
важно, чтобы цитохромоксидаза передала кислороду все
четыре электрона, необходимые для его восстановления
именно в воду, а не в какие-либо частично
восстановленные продукты: перекись водорода или реакцион-
носпособные свободные радикалы.
Как свободная энергия,
высвобождающаяся при переносе электронов,
используется для синтеза АТР?
Механизм синтеза АТР из ADP и Р|5 сопряженный
с транспортом электронов, принципиально отличается
от субстратного фосфорилирования в процессе
гликолиза (см. с. 112). В последнем случае синтез АТР
неотделим от собственно гликолитических реакций. Реакция
не протекает без образования АТР, который является ее
неотъемлемым компонентом. Точно так же обстоит дело
с синтезом GTP в цикле лимонной кислоты.
При рассмотрении движения электронов по цепи
переносчиков мы даже не упоминали о синтезе АТР.
Хотя именно ради синтеза АТР функционирует цепь,
транспорт электронов происходит и в поврежденных
митохондриях, которые вообще не способны
синтезировать АТР. Эта ситуация десятилетиями ставила
биохимиков в тупик. Если в клетках или интактных
митохондриях транспорт электронов приводит к генерации
АТР, то почему же в поврежденных митохондриях, где
успешно осуществляется передача электронов, АТР
не образуется? Ответить на этот вопрос с точки зрения
хорошо известного по гликолизу субстратного
фосфорилирования не представлялось возможным. Решение
этой проблемы нашел английский биохимик Питер
Митчелл, который в 1961 г., работая в своей домашней
лаборатории, придумал принципиально новую концепцию
механизма сопряжения транспорта электронов с
синтезом АТР. Эта концепция оказалась столь оригинальной,
что поначалу ее никто не принял всерьез, и прошло
немало времени, прежде чем она была признана всеми
(только в 1978 г. Митчеллу присудили Нобелевскую
премию). Идея Митчелла предельно проста: источником
часть 3 Метаболизм
126
энергии для совершения работы служат градиенты. Так,
перепад уровня воды приводит в действие
турбогенераторы гидростанций, разница атмосферного
давления порождает воздушный поток, заставляющий
работать ветряные мельницы, и т. д. Химические
градиенты не могут быть исключением. Молекулы движутся
по концентрационному градиенту (от высокой
концентрации к низкой), и если на их пути разместить
подходящее устройство, этот поток можно использовать для
выполнения полезной работы.
Исходя из своей концепции, Митчелл пришел к
выводу, что для сопряжения транспорта электронов с
синтезом АТР необходимо выполнение трех условий.
Во-первых, транспорт электронов должен создавать
некий градиент; во-вторых, обратный поток против
градиента может осуществляться лишь через устройство,
использующее энергию градиента для синтеза АТР
из ADP и Pf.. Третье условие концепции Митчелла
заключается в том, что биохимическая машина, работающая
на этих принципах, должна представлять собой
замкнутый пузырек, целостная мембрана которого позволяет
создать градиент. Однако градиент может существовать
лишь в том случае, если мембрана пузырька
непроницаема для создающего его вещества. Так просто и
изящно был получен ответ на вопрос: почему при
повреждении митохондриальной мембраны, когда все градиенты
исчезают, синтез АТР прекращается - но ничто не
препятствует транспорту электронов?
Митчелл обнаружил, что поток электронов
вызывает выкачивание протонов из митохондрий в
окружающую среду, создавая градиент протонов через мембрану
(рН внешнего раствора уменьшается). Поскольку
протоны являются положительно заряженными частицами,
вследствие их выкачивания из митохондрий на
мембране возникает разность электрического потенциала
(минус - внутри) и разность рН (выше - внутри). В
совокупности электрический и концентрационный градиенты
составляют то, что Митчелл назвал протондвижущей
силой, которая и является источником энергии для
синтеза АТР. Ясно, что нативная мембрана при этом
должна быть непроницаема для протонов. С другой стороны,
в такой мембране должны существовать специальные
каналы: проходя через них из окружающей среды
обратно в матрикс, протоны отдают свою энергию на синтез
АТР.
Такими специализированными протонными
каналами являются грибовидные выросты, которыми покрыта
внутренняя поверхность крист (рис. 8.20). Эти выросты
представляют собой АТР-синтазные комплексы,
использующие поток протонов для синтеза АТР из ADP и Pf..
Такой комплекс представлен двумя связанными между
Матрикс
митохондрий
Внутренняя
митохондриальная
мембрана
Цитозоль
АТР
АТР-синтаза
Меньше [Н+]
чПротонный *ч Мембраннь
/ градиент /* потенциал
Больше [Н+]
Н+
ADP + Р/
Н+
Дыхательные
/ комплексы
+ + + + + +
Протонный канал
1_1+ го н+ Протоны, транспортируемые
за счет потока электронов через
дыхательные комплексы
Рис. 8.20. Система синтеза АТР во внутренней митохондриальной мембране
F, состоит из девяти субъединиц. F0 также включает несколько субъединиц
собой компонентами F0 и F,, каждый из которых
состоит из нескольких белковых молекул. F0 утоплен в
мембране, a Fj расположен на ее поверхности. Именно в ¥{
синтезируется АТР, тогда как F0 выполняет функцию
собственно протонного канала. Под словом канал здесь
следует понимать упорядоченную в пространстве
последовательность заряженных боковых групп
аминокислотных остатков. Протоны, перемещаясь вдоль этой
последовательности, «перескакивают» с одной группы
на другую.
Общая схема, иллюстрирующая хемиосмотический
механизм (так называют концепцию Митчелла),
представлена на рис. 8.21.
Возникают два вопроса.
1. Каким образом восстановление кислорода
посредством NADH и FADH2 вызывает выкачивание
протонов из матрикса митохондрий в окружающую среду?
2. Как встречный поток протонов из окружающей
среды в матрикс становится движущей силой для
синтеза АТР из ADP и Р/?
Как выкачиваются протоны?
Механизм транслокации протонов в результате потока
электронов через комплексы I и IV пока не вполне ясен.
Иная ситуация складывается с комплексом III. Идея,
выдвинутая в свое время Митчеллом для сопряжения
потоков протонов и электронов, столь же проста, сколь
остроумна. Она состоит в том, что на поверхности
мембраны, обращенной к матриксу, атомы водорода
формируются из протонов, забираемых из матрикса, и
электронов, поставляемых цепью. Эти атомы образуются
Внутренняя митохондриальная мембрана,
непроницаемая для протонов
Больше [Н+] -
Протонный
градиент
Дыхательные
комплексы,
транспортирующие
протоны «%в
NADH Меньше [Н+]
FADH2
Матрикс митохондрий
— н+
АТР I ADP + Р,
Ч I / '
.АТР-синтаза
+ + + + --Мембранный
потенциал
Рис. 8.21. Синтез АТР в митохондриях по хемиосмотичес-
кому механизму
FADH2, получаемый при окислении сукцината и жирных
кислот, используется одинаково
на молекуле убихинона Q, который превращается в
восстановленную форму QH2. Последняя диффундирует
к противоположной стороне мембраны, где события
протекают в обратном направлении: электроны
отщепляются от атомов водорода, а образующиеся протоны уходят
наружу. Иными словами, Q восстанавливается на одной
стороне мембраны, движется в виде QH2 к другой, там
окисляется до Q и возвращается обратно. Дело в том, что
Q участвует в двух редокс-парах, разнесенных по обе
стороны мембраны и способных передавать атомы
водорода через мембрану с одной поверхности на другую.
Как восстановление Q, так и окисление QH2
катализируют специализированные белки. Вся эта система
получила название Q-цикла (рис. 8.22).
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
127
Цитозоль
2£"из
комплекса
I или II
4Н+ 2е~
QH2
Q
2Н+
Матрикс
2QH2 2Q· —Г— 2Q
2е~\
U,UTbL
QH2
Цит^//
К мил kcIIL
Цитохром с
ι
Комплекс 1У
2Н+
Рис. 8.22. Сопряжение переноса электронов через
дыхательный комплекс III с транспортом протонов через мембрану
Цветом помечены траектории подвижных переносчиков
электронов убихинона и его производных; черным -
химические превращения; прерывистой черной линией -транспорт
электронов
Его схема, на первый взгляд, кажется сложной, но
в действительности она очень проста.
Полная схема Q-цикла включает два разных
циклических процесса, каждый из которых приводит к
выбросу из мембраны 2 протонов. В мембране существует
стационарный общий фонд Q и QH2. В левой части
рис. 8.22 показано, что восстановление Q до QH2
комплексами I и II сопровождается поглощением 2 протонов
из матрикса митохондрий. Образовавшийся QH2
мигрирует к внешней поверхности мембраны, где отдает
1 электрон на восстановление цитохрома с,
переносящего его к комплексу IV (цитохром с, подобно убихи-
нону, подвижен, но не в мембране, а в воде и
располагается с наружной стороны мембраны, как и участок
комплекса IV, акцептирующий электрон).
Одновременно с потерей электрона QH2 отдает во внешнюю среду
2 протона и превращается в анион-радикал семихино-
на СГ- Последний далее снова отдает 1 электрон, но уже
не цитохрому с, а цитохрому Ъ. Этими событиями
заканчивается первая половина рассказа о Q-цикле. Молекула
QH2, покинувшая комплекс II, была окислена, 2
электрона (по одному) были переданы от нее цитохромам с
и Ь, а 2 атома водорода в виде протонов вышли наружу
во внемитохондриальное пространство. Убихинон
поступает в общий фонд этого вещества в мембране.
Другая половина истории начинается с того, что вторая
молекула QH2 подвергается точно такому же
окислению, что и первая, отдавая 2 протона в окружающую
среду. Таким образом, после окисления 2 молекул QH2
2 электрона передаются комплексу IV через цитохром с,
а еще 2 электрона - цитохрому Ъ. Далее электроны
с одного гема, входящего в состав цитохрома Ь,
переходят на другой гем, связанный с тем же белком, но
обладающий большим редокс-потенциалом (или, что то же
самое, меньшей энергией). Этот перенос эквивалентен
переходу электронов на другую, обращенную к матрик-
су сторону мембраны. Здесь они расходуются на
восстановление Q в QH2, при этом атомы водорода в виде
протонов извлекаются из матрикса. Далее QH2
возвращается к внешней стороне мембраны, цикл замыкается, и эта
«карусель» готова к очередному обороту. (Для простоты
и ясности на рис. 8.22 показано, будто каждая из
изображенных молекул последовательно обегает цикл,
подобно детали, движущейся по автоматической линии
станков. В действительности же каждая из
окислительно-восстановительных реакций протекает с молекулой,
произвольно выбранной из общего фонда.) Если
рассматривать только процессы, протекающие в пределах
комплекса III, то их можно описать уравнением,
согласно которому окисление одного QH2 сопровождается
выбросом из мембраны не двух, а четырех протонов:
QH2 + 2Н+(матрикс) + 2 Цит с (Fe3+) ->
-> Q + 4Н+(цитозоль) + 2 Цит с (Fe2+).
Анализ Q-цикла наглядно демонстрирует, что на
работу, совершаемую при выкачивании протонов,
расходуется свободная энергия, которая освобождается при
переносе электронов по градиенту редокс-потенциала
Перенос протонов через внутреннюю
NADH митохондриальную мембрану
-0,4 Ч
И
1*3
К
Я"
Я
о
о
с
I
υ
О
С£
О
ОН
+0,4 Η
+0,8 Η
II е~ Q^
111 е Цитс\
е Цит с
IV
е~ 4Н+
о2-
2Н70
200
о
Moog
ι-Ο
Рис. 8.23. Примерное расположение главных переносчиков
электронов относительно шкалы редокс-потенциала
Справа - шкала, позволяющая оценить, сколько энергии
высвобождается при переносе пары электронов от данного
переносчика на кислород. Q - убихинон
часть 3 Метаболизм
128
Как поток протонов может привести
к синтезу АТР?
Грибовидные выросты F, в пробирке обладают АТРаз-
ной активностью (гидролизуют АТР до АДР и Pf.),
но в митохондриях они осуществляют обратную
реакцию - синтезируют АТР.
Вопрос заключается в том, как поток протонов
делает возможной реакцию ADP + Р/—> АТР + Н20?
Наиболее примечательная особенность синтеза АТР ми-
тоходриальной синтазой ¥{ состоит в том, что не
удалось обнаружить никаких признаков промежуточных
продуктов, в которых ADP или фосфат были бы кова-
лентно связаны с белком. Создается впечатление, что
происходит прямая конденсация этих двух молекул. По-
видимому, когда ADP и Pf. связаны с каталитическим
центром, для образования АТР (тоже связанного с
каталитическим центром) достаточно небольшого
изменения свободной энергии; а вот для высвобождения АТР
из каталитического центра необходимы существенные
затраты энергии. Полагают, что свободная энергия,
высвобождаемая при переносе электронов по дыхательной
цепи, вызывает конформационные изменения в
молекуле белка. Конформационные переходы, вероятно,
требуют затрат энергии того же порядка, что и синтез АТР
в растворе.
До сих пор мы встречались с транспортом веществ
через мембрану, движущей силой которого является
гидролиз АТР. АТР-синтазу можно рассматривать как
протонный насос, движимый АТР, но работающий в
обратном направлении.
Есть нечто магическое в концепции Митчелла.
Миллионы лет все аэробные формы жизни на Земле
существуют благодаря созданию градиента рН и
заряда через липидный бислой мембраны. Воистину, это
одна из самых великих и замечательных концепций
в биологии!
Транспорт ADP в митохондрии и АТР
из митохондрий
В большинстве эукариотических клеток синтез
основного количества АТР происходит внутри
митохондрий, но основные потребители АТР расположены
вне ее. Следовательно, должны существовать
механизмы, обеспечивающие поступление ADP и фосфата
в матрикс митохондрий и выведение АТР из него
наружу. Эти заряженные молекулы пересечь липидный
бислой сами никак не могут. Существует так называемая
ATP-ADP-транслоказа, которая обменивает внутри-
митохондриальный АТР HaADP, находящийся вне
митохондрии (рис. 8.24).
Откуда берется энергия для ADP-ATP обмена? Мы
уже упоминали о том, что выкачивание протонов из
митохондрий создает на внутренней мембране не только
градиент рН, но и разность электрических потенциалов
(минус - внутри). Молекула АТР несет четыре
отрицательных заряда, a ADP - только три. Нетрудно видеть, что
ADP-ATP обмен эквивалентен переносу одного
отрицательного заряда из митохондрии наружу, т. е. по
градиенту потенциала. Другими словами, движущей силой
такого обмена является мембранный потенциал,
возникающий в результате переноса электронов. Расчет
показывает, что на ADP-ATP-обмен расходуется около
четверти свободной энергии, высвобождающейся при
транспорте электронов по цепи. Другие транспортные системы
также могут приводиться в действие электрохимическим
Н+
Н+
АТР-синтаза
ATP-ADP-
транслоказа
Транслоказа
фосфата
Матрикс
митохондрий
Внутренняя
митохондриальная
мембрана
Цитозоль
\ АТР4" ADP3
Р/
Дыхательные
/ комплексы
АТР4" ADP3" P/ Н+
Антипорт Симпорт
Н+
Рис. 8.24. Трансмембранный транспорт в митохондриях, связанный с синтезом АТР
Все потоки проходят через специализированные белковые транслоказы
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
129
градиентом. К их числу, например, относится транслоказа,
обеспечивающая поступление в митохондрии фосфата,
необходимого для синтеза АТР (см. рис. 8.24).
Баланс между синтезом АТР
и транспортом электронов
Если исходить из допущения, что ADP и фосфат уже
находятся в матриксе, то каждой синтезированной
молекуле АТР соответствует прохождение через синтазу
3 протонов. ADP-ATP-обмен в чистом виде создает
на мембране электрический потенциал, поскольку
заряды у этих молекул отличаются на единицу Чтобы такой
обмен стал электронейтральным, в митохондрию
должно поступать эквимолярное количество протонов,
которые затем предстоит выкачать обратно. Поэтому, с
учетом «транспортных расходов», синтез 1 молекулы АТР
(доступной для использования в цитоплазме) сопряжен
с переносом 4 протонов из матрикса наружу.
Каждой паре электронов, перенесенных от NADH
на кислород, соответствует 10 протонов, перекачанных
из митохондриального матрикса. Таким образом,
окисление 1 молекулы NADH, уже находящейся внутри
митохондрии, должно привести к синтезу 2,5 молекул АТР,
а окисление 1 молекулы FADH2 (что эквивалентно
молекуле сукцината) - к синтезу 1,5 молекул АТР. Раньше
полагали, что синтезируются соответственно, три и две
молекулы АТР. Эти величины принято называть
отношениями Р/О, поскольку перенос 2 электронов
эквивалентен восстановлению 1 атома кислорода.
Молекулы NADH, образовавшиеся в ходе гликолиза,
находятся в цитозоле. Они отдают свои пары
электронов либо внутримитохондриальному NAD+, либо мито-
хондриальному FAD, в зависимости от того, какой
челночный механизм преобладает в данной клетке (см.
с. 114). Благодаря этой неопределенности одной цитоп-
лазматической молекуле NADH отвечает синтез от 1,5
до 2,5 молекул АТР.
Выход АТР при окислении
молекулы глюкозы до С02 и Н20
Если в процесс окисления вступает молекула глюкозы,
а не гликогена, валовый выход АТР из расчета на
полностью окисленную молекулу субстрата составляет от 30
до 32 молекул в зависимости от того, какой тип челнока
используется для цитоплазматического NADH.
Откуда берутся эти значения?
Некоторое количество АТР синтезируется в ходе
субстратного фосфорилирования: 2 молекулы АТР
поставляет гликолиз (продуцируются 4, но 2 расходуются)
и еще 2 молекулы АТР (у млекопитающих это GTP)
образуются в цикле лимонной кислоты (из 1 молекулы
часть 3 Метаболизм
130
глюкозы образуются 2 молекулы ацетил-СоА,
запускающие два оборота цикла). Итак, в процессе
субстратного фосфорилирования синтезируются всего 4
молекулы АТР, а все остальные образуются в митохондриях
с участием цепи переноса электронов.
При гликолизе в цитоплазме образуется также 2
молекулы NADH на 1 молекулу глюкозы. Их окисление
увеличит выход АТР на 3-5 молекул в зависимости от
типа используемого челнока. Кроме того, в расчете на
1 молекулу глюкозы пируватдегидрогеназа производит
2 молекулы NADH, а цикл лимонной кислоты - 6
молекул NADH. Их окисление приводит к синтезу 20
молекул АТР. Еще 3 молекулы АТР образуются за счет
окисления FADH2 при превращении сукцината в фумарат.
Суммируя все эти числа (в скобках - значения для
глицерин-3-фосфатного челнока), получаем:
2 + 5(3) + 2 + 20 + 3 = 32 (30).
Заметим, что эта величина приблизительна. Точно
оценить выход АТР можно только при субстратном фос-
форилировании, а соотношение между выбросом
протонов и синтезом АТР не является фиксированным.
Клетка кишечной палочки Escherichia coli в
известном смысле подобна митохондрии. Ее плазматическая
мембрана выполняет функцию внутренней мембраны
митохондрии, а цитоплазма - матрикса. В такой клетке
не нужно прибегать к челнокам для доставки NADH
к дыхательной цепи. Она не нуждается в транспортных
системах для ADP и АТР, поэтому выход АТР на
молекулу окисленной глюкозы у Е. coli заметно выше.
Используется ли потенциальная
энергия в виде π ротон движу щей силы
для выполнения других задач,
не связанных с синтезом АТР?
У новорожденных детей поддержание температуры тела
на постоянном уровне требует усиленного производства
тепла, которое обеспечивают клетки бурого жира.
Своим цветом они обязаны обилию митохондрий,
содержащих окрашенные цитохромы. Уровень синтеза АТР
в митохондриях зависит от проницаемости внутренней
мембраны для протонов; если она ничтожна, обратный
поток протонов вынужден проходить через АТР-синтазу.
При увеличении проницаемости мембраны происходит
«короткое замыкание»: АТР не вырабатывается и вся
энергия рассеивается в виде тепла. В митохондриях
клеток бурого жира есть специальный белок термогенин,
благодаря которому во внутренней мембране появляются
протонные каналы. Такой же эффект можно получить
и на обычных митохондриях, используя соединения,
увеличивающие протонную проницаемость липидных
бислоев (самое популярное из них - 2,4-динитро-
фенол). Все вещества, действующие подобным
образом, называют разобщителями окислительного
фосфорилирования, ибо они нарушают передачу
энергии от окислительных систем к фосфорилирующим.
Бактерии также используют протонные насосы для
создания протонного градиента на плазматической
мембране и протонные АТР-синтазы для синтеза АТР (мы
уже отмечали, что бактериальная клетка во многом
напоминает митохондрию). Однако у бактериальных
клеток есть своя специфика: они используют мембранные
градиенты для закачивания в цитоплазму различных
веществ из окружающей среды. Примером может служить
симпорт протонов и лактозы, протекающий подобно
симпорту Na+-HOHOB и метаболитов в животных клетках.
Удивительно, что протонный градиент служит
движущей силой для вращения бактериальных жгутиков.
Дополнительная литература
Гликолиз
Boiteux Α., Hess В. Design of glycolysis // Phil. Trans. Roy.
Soc. Series B. 1981. Vol. 293. P. 5-22.
(Обширный обзор о гликолизе и его регуляции.)
Цепь переноса электронов
BoyerR D., Chance В., ErnsterL, Mitchell P., Racker Ε.,
Slater E. С. Oxidative phosphorylation and photophospho-
rylation // Annu. Rev. Biochem. 1977. Vol. 46. P. 955-1026.
(Каждый раздел обзора автономен и написан корифеем
в своей области. Устарев в деталях, он сохраняет
научную ценность, поскольку в нем изложены исторические
аспекты и основные принципы изучения механизмов
окислительного фосфорилирования.)
Slater Ε. С. The Q cycle, an ubiquitous mechanism of
electron transport.// Trends Biochem. Sci. 1983. Vol. 8.
P. 239-242.
(Описание протонных насосов в митохондриях и хло-
ропластах.)
Mitchell P. Chemiosmosis: A term of abuse // Trends
Biochem Sci. 1984. Vol. 9. P. 205.
(Создатель хемиосмотической теории объясняет,
почему неудачен термин «хемиосмос».)
ВаЪсоск G. Т., Wikstrom M. Oxygen activation and the
conservation of energy in cell respiration// Nature. 1992.
Vol.356. P. 301-309.
(Детальный обзор данных о цитохромоксидазах и их
роли в перекачивании протонов.)
Capaldi R. Α., Aggeler R., Turina P., Wilkens S. Coupling
between catalytic sites and the proton channel in F,F0-type
ATPases// Trends Biochem. Sci. 1994. Vol.19.
P. 284-289.
(Обзор, посвященный исследованию комплекса, который
синтезирует АТР из АДР и Pf. и управляется потоком
протонов.)
Вопросы к главе 8
1. Дайте химическое обоснование глюкозофосфатизо-
меразной реакции.
2. Что подразумевается под субстратным фосфорили-
рованием? Приведите примеры.
3. Реакция, давшая название пируваткиназе, в
действительности никогда не протекает. Почему ферменту
дали такое название и почему не протекает эта реакция?
4. Сколько молекул АТР образуется при расщеплении
в гликолитическом пути 1 молекулы глюкозы
и 1 глюкозильного остатка гликогена?
5. Почему нельзя однозначно ответить на вопрос:
сколько молекул АТР образуется при окислении
молекулы цитоплазматического NADH митохондриями
эукариотических клеток?
6. В цикле лимонной кислоты превращению каждой
ацетильной группы соответствует образование 9
восстановительных эквивалентов (или, что то же самое,
9 атомов водорода) и 2 молекул С02 (4 атомов
кислорода). Откуда они взялись? 3 атома водорода и 1 атом
кислорода - из ацетильной группы, еще 4 водорода
и 2 кислорода принадлежат 2 молекулам воды,
вступающим в цикл. Итак, всего 7 атомов водорода
и 3 атома кислорода. Для баланса элементов не
хватает 1 молекулы Н20. Откуда она берется?
7. Почему продукт окисления изоцитрата декарбокси-
лируется, а сам он - нет?
8. Объясните, какое значение для цикла лимонной
кислоты имеет анаплеротическая реакция.
9. Какой кофактор участвует в реакции декарбоксили-
рования? Объясните, как он работает.
10. Как дыхательные комплексы складываются в единую
цепь переноса электронов?
11. Что общего у убихинона и цитохрома с и чем они
различаются как переносчики электронов? Где они
локализованы в клетке?
12. Для чего нужна митохондриям цепь переноса
электронов?
13. Выход АТР при окислении глюкозы в
эукариотических клетках составляет 30 или 32 молекулы. В
случае бактериальных клеток выход больше. Чем это
вызвано?
глава 8 Гликолиз, цикл лимонной кислоты, транспорт электронов
131
9 Энергетическая
Жиры - основной источник энергии для синтеза
АТР. Они обеспечивают образование примерно
половины энергии, потребляемой сердцем и покоящимися
скелетными мышцами. Количество энергии, запасенной
организмом в виде нейтральных жиров, хранящихся
в жировых клетках, практически ничем не
ограничивается, в отличие от ее лимитированных запасов в виде
гликогена. Кроме того, жиры - более компактная форма
хранения энергии, поскольку они менее окислены и гид-
ратированы по сравнению с гликогеном.
АТР образуется как при окислении жиров, так и при
окислении глюкозы. В обоих процессах участвуют цикл
лимонной кислоты и электронтранспортная цепь.
Сходство этих двух метаболических путей объясняется тем,
что как при окислении глюкозы, так и при окислении
жиров образуется ацетил-СоА. Кроме того, распад
жиров отличает универсальный механизм окисления
жирных кислот путем последовательного отщепления от
углеводородной цепи двух углеродных атомов в виде
ацетил-СоА. При этом образуются NADH и FADH2,
которые затем окисляются. Нельзя не подивиться
эффективности использования одной и той же биохимической
машины для получения энергии из самых разных
пищевых веществ. Пути их окисления различаются лишь
начальными стадиями, а в цикле лимонной кислоты они
сливаются в общий поток.
В первую очередь необходимо обратить внимание
на следующие замечания.
1. Прежде чем начнется окисление, жиры должны
подвергнуться гидролизу для высвобождения жирных
кислот. При этом из триглицеридов образуется
также и глицерин, метаболическая судьба которого иная,
чем у жирных кислот: он фосфорилируется
окисляется в дигидроксиацетонфосфат. Таким образом,
при голодании глицериновый остаток триглицеридов
может использоваться печенью при глюконеогенезе
(см. главу 11).
2. Периферические ткани извлекают жирные кислоты
из крови, куда они экскретируются жировыми
клетками в ответ на повышение уровня глюкагона. Эти
кислоты попадают в клетки также и из хиломик-
ронов и ЛПОНП, атакуемых липопротеинлипазой
(см. с. 93).
3. Свободные жирные кислоты разносятся кровью в
виде анионов, обратимо связанных с сывороточным
частьЗ Метаболизм
132
ценность жиров
альбумином. Они легко проникают в клетки,
поэтому их концентрация там находится в прямой
зависимости от уровня в крови. По мере того как свободные
жирные кислоты поглощаются из крови, их
постоянная концентрация поддерживается благодаря
диссоциации альбуминовых комплексов.
4. Жирные кислоты расщепляются путем
последовательного отщепления двух углеродных атомов в
виде ацетил-СоА.
5. В ходе превращения в ацетил-СоА жирные кислоты
постоянно находятся в форме ацил-СоА. Поэтому
первая стадия их метаболизма, известная как
активация жирных кислот, заключается в синтезе ацил-
СоА-производного жирной кислоты.
Механизм образования
ацетил-СоА из жирных кислот
Активация жирных кислот путем синтеза
их ацил-СоА-производных
Термин активация отражает тот факт, что тиоэфиры
жирных кислот являются макроэргическими реакцион-
носпособными соединениями. Реакция активации
выглядит следующим образом:
RCOCT + АТР + CoA-SH -> RCO-S-CoA + AMP + РР;
AG°'= -0,9 кДж ■ моль1.
Изменение свободной энергии при этом невелико
(из-за высокой энергии тиоэфира), однако действие
пирофосфатазы приводит к тому, что суммарный
процесс оказывается сильно экзоэргоничным и
необратимым (AG°'=-32,5 кДж ■ моль ').
Реакцию активации жирных кислот катализируют
три родственных фермента - ацил-СоА-синтетазы
жирных кислот, специализирующиеся на кислотах
с короткими, средними и длинными цепями.
Транспорт СоА-производных жирных
кислот в митохондрии
Жирные кислоты активируются на внешней мембране
митохондрий, которую топологически можно отождествить
с цитоплазмой, однако их превращение в ацетил-СоА
происходит только в митохондриальном матриксе.
Ацильная группа переносится в митохондрии без СоА
при помощи специальной транспортной системы и уже
внутри митохондрий вновь взаимодействует с CoA-SH
с образованием ацил-СоА. В ходе транспорта
сохраняется макроэргическая природа ацильного остатка, так
что по другую сторону мембраны его не надо повторно
активировать. Это возможно потому, что перенос жир-
нокислотного остатка в митохондрии осуществляет
молекула карнитина:
СИ,
Η
+1 I //
Н3С—Ν—СН2-С—СН2-С
1 ' О"
СН3 ОН и
Карнитин
Хотя сложные эфиры обычно не являются
макроэргами, структура карнитина такова, что связь ацил-кар-
нитин является макроэргической. Возможно, именно
поэтому эволюция остановила свой выбор на карнитине
как на ацилтранспортирующем соединении. Ацильные
производные карнитина проникают в матрикс, где
протекает обратная реакция: ацильный остаток
переносится с карнитина на тиольную группу свободного СоА,
а карнитин возвращается назад за очередным остатком
жирной кислоты.
СН3
Η
ι '/
Н3С—Ν—СН2—С—СН2-С
О
СН3
О
I
с=о
Ацилкарнитин
Схема трансмембранного переноса жирных кислот
в митохондриях представлена на рис. 9.1. Две реакции
переноса ацильных групп - на карнитин и на митохон-
дриальный СоА - катализируют два фермента: первую,
протекающую на внешней стороне митохондрии, -
карнитин-ацилтрансфераза I, а вторую, имеющую
место в матриксе, - карнитин-ацилтрансфераза И.
При некоторых генетических дефектах ощущается
нехватка карнитина или карнитин-ацилтрансферазы, что
приводит к мышечным болям и к ожирению мышц
Превращение ацил-СоА в ацетил-СоА
внутри митохондрии
Этот процесс включает четыре последовательно
протекающие реакции, три из которых похожи на уже
встречавшиеся нам в цикле лимонной кислоты стадии
Связана с внешней стороны
митохондриальной мембраны
RCH7CO-S-CoA Карнитин-ацил- CoA-SH
трансфераза I
Цитоплазма
Внутренняя
щтохондриальная
мембрана
Матрикс
Карнитин
RCH2C0-KapHHTHH
КСНгСО-Карнитин
RCH2CO - S - СоА Карнитин-ацил £0д _ $н
трансфераза II
Расположена на внутренней стороне
митохондриальной мембраны
(со стороны матрикса)
Рис. 9.1. Механизм транспорта длинноцепочечных жирно-
кислотных остатков в митохондрии: там они окисляются
Перенос ацильных остатков с ацилкарнитина на СоА не
требует затрат энергии
превращения сукцината (Сп) в оксалоацетат:
дегидрогенизация с помощью FAD-зависимого фермента,
гидратация и ЫАО+-зависимая дегидрогенизация. Вспомните
последовательные превращения Сукцинат—> Фумарат—>
—>Малат—Юксалоацетат в цикле лимонной кислоты.
Соответствующие реакции с производными ацил-СоА
представлены на рис. 9.2.
Ключевой реакцией в метаболизме жиров служит
реакция взаимодействия CoA-SH с β-кетоацил-СоА,
в ходе которой разрывается углеводородная цепь остатка
жирной кислоты. Ее двухуглеродный фрагмент
отщепляется в виде ацетил-СоА, и остается укороченный на два
атома углерода остаток жирной кислоты (ацил-СоА).
Фермент, осуществляющий эту реакцию, называют
тиолазой, поскольку молекула исходного ацил-СоА
атакуется тиольной группой свободного СоА.
Природные длинноцепочечные жирные кислоты содержат
четное число атомов углерода, поэтому последовательное
отщепление двухуглеродных фрагментов рано или
поздно приводит к образованию бутирил-СоА, который
превращается затем в β-ацетоацетил-СоА. Расщепление
этого соединения на два ацетил-СоА завершает процесс
укорачивания жирнокислотной цепи:
CH3COCH2CO-S-CoA + CoA-SH — 2CH3CO-S-CoA .
Ацетоацетил-СоА Ацетил-СоА
Итак, на каждом этапе расщепления жирных кислот
образуется β-кетоацил-СоА со все более укороченной
глава 9 Энергетическая ценность жиров
133
Η Η О
I I II
R—СН2—С—С —С—S—СоА (Ацил-СоА)
I I
н н
L-FAD
|^FADH2
Η О
\цил-СоА-дегидрогеназа
R—СН2—С=С—С—S—СоА (гарднс-Л2-еноил-
| СоА)
Η
Н.,0 Ьноил-Соа-
гидратаза
Г1
он н о ,_ ГлАЧ
ι ι и (Гидроксиацил-СоА)
R—СН2—С—С—С—S—СоА
Η Η
L-NAD+
Гидроксиацил-LoA-
дегид огеназа
j^NADH + Н+
ОНО
I I II
R—СН2—С—С—С—S—СоА (β-Кетоацил-СоА)
Η
СоА - SH Кетоацил-СоА-тиолаза
О
II
R—СН2—С—S—СоА + СН3—С—S—СоА
Cn-Ацил-СоА Ацетил-СоА
Рис. 9.2. Четыре последовательные реакции, посредством
которых укорачивается углеводородная цепь жирн окислот-
ных производных СоА
Цепь укорачивается на два атома углерода,
высвобождающихся в виде ацетильной группы ацетил-СоА
углеводородной цепью. Это дает основание весь
рассмотренный выше процесс в целом назвать
β-окислением жирных кислот. NADH и FADH2,
образующиеся при β-окислении, отдают затем свои
электроны митохондриальной электронтранспорт-
ной цепи (см. рис. 8.19).
Энергетический выход при окислении
жирных кислот
Молекула пальмитиновой кислоты (С16) превращается
в 8 молекул ацетил-СоА, при этом дополнительно
образуются 7 молекул NADH и столько же FADH2. Ацетил-
СоА далее окисляется в цикле лимонной кислоты,
a NADH и FADH2 - в митохондриальной электрон-
транспортной цепи (см. главу 8). При окислении NADH
синтезируются 2,5 молекулы АТР, а при окислении
FADH2- 1,5 молекулы (поскольку NADH образуется
в матриксе, челночный механизм не задействован в его
переносе внутрь митохондрий).
Таким образом, окисление 1 молекулы
пальмитиновой кислоты приводит к синтезу 106 моль АТР из ADP
и РДс учетом затраты двух молекул АТР на образование
пальмитоил-СоА и синтеза одной молекулы GTP на
каждый ацетил-СоА в цикле лимонной кислоты). Это
означает, что КПД процесса равен 33%; такая доля
свободной энергии преобразуется в полезную работу в виде
синтеза АТР. Результаты нашего расчета можно
представить и по-другому: окисление 256 г пальмитиновой
кислоты дает 45 кг АТР. Разумеется, такого количества АТР
никогда не бывает, он быстро гидролизуется,
обеспечивая энергетические потребности организма.
Окисление
ненасыщенных жиров
Оливковое масло - жидкое, потому что в нем
высокое содержание мононенасыщенных жиров - тригли-
церидов и фосфолипидов, в состав которых входят
ненасыщенные жирные кислоты с одной и более двойными
связями в углеводородной цепи. Примером может
служить пальмитоолеиновая кислота с двойной связью
между девятым и десятым атомами углерода. Вплоть до
двойной связи цепь такой кислоты укорачивается
обычным окислением с образованием г/м>А3-еноил-СоА:
Η Η м
R—С=С—СН2—С—S-CoA
(4) (3) (2) (1)
Двойная связь в этом соединении не позволяет
ацил-СоА-дегидрогеназе ввести еще одну, соседнюю
двойную связь между вторым и третим С-атомами. На
этом β-окисление могло бы закончиться, однако
фермент изомераза сдвигает двойную связь в нужное
положение и образует транс-изомер, который становится
субстратом еноил-СоА-гидратазы (см. рис. 9.2).
Η Η м
R—С=С—СН2—С—S—СоА г/г/с-Д3-еноил-СоА
Μ (3)
Изомераза
н 8
R—СН2—С=С—С—S—СоА транс-А2-еноил-СоА
Η
частъЗ Метаболизм
134
Это ухищрение решает проблему биологического
окисления мононенасыщенных жирных кислот.
Полиненасыщенные жирные кислоты создают
дополнительные трудности. Возьмем, например, линолевую кислоту
с двумя двойными связями (Δ9 и Δ12). Судьба одной из
них (Δ9) при окислении такова, как описано выше.
Другая связь (Δ12) восстанавливается в нужный момент
особым ферментом, благодаря чему гидролиз линолевой
кислоты осуществляется по механизму β-окисления (на
самом деле все обстоит несколько сложнее).
Всегда ли ацетил-СоА,
образующийся при β-окислении,
поступает в цикл лимонной
кислоты?
До сих пор рассматривалась ситуация, когда весь
синтезированный ацетил-СоА поступает в цикл лимонной
кислоты. Так обычно и бывает во всех тканях, однако
случаются чрезвычайные ситуации, когда в печени этого
не происходит.
Иногда метаболизм жиров становится для
организма основным источником энергии (см. главу 5). Это
состояние наступает, например, при голоде, когда
исчерпаны все запасы гликогена. У диабетиков глюкозный голод
возникает не из-за недостатка глюкозы, а вследствие
невозможности ее превращения. Как бы то ни было,
торможение гликолиза побуждает жировые клетки
высвобождать жирные кислоты, а печень - использовать их
для синтеза избыточного количества ацетил-Со А.
Избыточного в том смысле, что оно превышает потребности
цикла лимонной кислоты. Ситуация усугубляется еще
и тем, что прекращение углеводного метаболизма
приводит к недостатку оксалоацетата, необходимого для
производства цитрата из ацетил-СоА (см. с. 121).
В этой ситуации в клетках печени два ацетильных
остатка соединяются в ацетоацетат (СН3СОСН2СОО~),
который частично восстанавливается в β-гидрокси-
бутират (СН3СНОНСН2СОО ). Эти два вещества -
кетоновые тела - поступают в кровь.
Как ацетоацетат образуется
из ацетил-СоА?
Ацетоацетил-СоА образуется из ацетил-СоА в
результате обращения реакции, катализируемой кето-
ацил-Со А-тиол азой:
2CH3CO-S-CoA <- CH3COCH2CO-S-CoA + CoA-SH
Казалось бы, свободный ацетоацетат должен был
образоваться в результате гидролиза ацетоацетил-СоА (что
сдвигало бы равновесие приведенной реакции в сторону
образования продукта). Вместо этого ацетоацетил-СоА
взаимодействует с третьей молекулой ацетил-СоА
с образованием 3-гидрокси-З-метилглутарил-СоА
(ГМГ-СоА), который расщепляется до ацетоацетата
и ацетил-СоА (рис. 9.3).
Почему синтез ацетоацетата происходит таким
образом, непонятно. Гидроксиметилглутарил-СоА,
синтезируемый многими животными клетками, является
предшественником холестерина - важного компонента ци-
топлазматической мембраны (см. с. 58). Образование
кетоновых тел происходит в митохондриях (именно там
жиры превращаются в ацетил-СоА). Более того,
образование гидроксиметилглутарил-СоА, необходимого
для синтеза холестерина, сосредоточено в цитоплазме,
где находится специальный фермент - гидроксиметил-
глутарил-СоА-синтаза, прикрепленная к мембране
эндоплазматического ретикулума.
О
II
с-
-S—СоА
СН2
I
о=с—сн3
Ацетоацетил-СоА
СН3
I
с=о
I
S—СоА
Ацетил-СоА
ГМГ-СоА-
синтаза
О
II
С—S—СоА
I
сн2
I
но—с—сн3
ГМГ-СоА
NADH
+ Н
СН2
I
соо-
З-Гидрокси-3-метил-
глутарил - СоА
+ CoA-SH
О
II
СН3—С—S—СоА
Ацетил-СоА
+
о=с—сн3
ГМГ-СоА
лиаза
Η
I
но— с— сн3
? У
сн2
сн2
I
соо-
β-Гидроксибутират
NAD+ J/ COO
^^^~/ Ацетоацетат
β-Гидроксибутират-
дегидрогеназа
(Спонтанно)
СН3
I
с=о + со:
I
сн3
Ацетон
Рис. 9.3. Образование кетоновых тел в митохондриях
печени при избыточном окислении жиров (голодание,
диабет)
3-Гидрокси-З-метилглутарил-СоА (ГМГ-СоА) служит
предшественником холестерина и в этом случае синтезируется в
цитоплазме в результате реакции, катализируемой гидроксиме-
тилглутарил-СоА-синтазой
глава 9 Энергетическая ценность жиров
135
Периферические ткани могут использовать ацетоа-
цетат для генерации энергии. В митохондриях в
результате реакции обмена ацильным остатком с сукцинил-
СоА он превращается в ацетоацетил-СоА:
Ацетояцетят
Ацетоацетил-СоА
+ сукцинил-СоА
CoA-SH
Ацетоацетил-СоА —-^
+ сукцинат
—- 2-Ацетил-СоА
Тиолаза
Ацетоацетил-СоА далее расщепляется тиолазой (с
участием свободного СоА) на две молекулы ацетил-
СоА β-Гидроксибутират после предварительной
дегидрогенизации также превращается в ацетоацетат.
Окисление жирных кислот
с нечетным числом углеродных
атомов
Небольшая доля жирных кислот в пищевых продуктах
(например, растительного происхождения) имеет
нечетное число углеродных атомов. При β-окислении таких
кислот в качестве предпоследнего продукта образуется
не ацетоацетил-СоА, а пятиуглеродный β-кетоацил-СоА.
Расщепление такого остатка тиолазой приводит к
появлению ацетил-СоА и трехуглеродного пропионил-СоА:
CH3-CH2-CO-CH2-CO-S-CoA + CoA-SH —
-> CH3-CH2-CO-S-CoA + CH3-CO-S-C0A.
Пропионил-СоА Ацетил-СоА
Пропионил-СоА превращается в сукцинил-СоА и
вовлекается в цикл лимонной кислоты. Соответствующие
реакции приведены ниже. Эпимераза катализирует
превращение D-конфигурации метилмалонил-СоА в L-koh-
фигурацию:
11ропионил-Со-А-
ка боксилаза
СН3—СН2—CO-S—СоА + НС03~ +А Р-
Пропионил-СоА
Метилмалонил-СоА-
7 эпиме аза ,
Последняя реакция (изомеризация метилмалонил-
СоА) интересна тем, что в ней участвует наиболее
сложно устроенный кофермент - дезоксиаденозилкобала-
мин, производное витамина В12 (рис. 9.4).
Пропионат также образуется при расщеплении
четырех аминокислот (валина, изолейцина, метионина и
треонина), а еще из боковой цепи холестерина.
Если не хватает метилмалонил-СоА-мутазы или
нарушен синтез кофермента из витамина В12, развивается
смертельно опасный ацидоз.
ОН
СН
СН2
ОН
I I
с —с
^н н^
/
СН
СН
/N\C
/
СН
NH2—СО—СН2-
Н3С СН3 СН2—С0-
I
NH2
-NH2
СН2
nh2 - со- сн2 cVrAhr/4
^c If ι сн-сн2-сн2-со-
< CH3>Nn l^\
CH3 Co+ CH
CH—μ . n—c CH3
CH CH3
νη2-<:ο—сн2-
V ΐ
со-сн2-сн2сн3/£н
-СН
NH
I
сн2
сн—сн3
I
CH2— CH2— CO—NH2
>
но-
СН II
Η
ли*
но
с-
/н
СН
ιν-
сн2
-с
н\
'^Чн,
.сн
AMP + РР/ Рис. 9.4. Структура дезоксиаденозилкобаламина
►СН3—С—CO-S—СоА -СН3—С—CO-S—Со—
I I
Η COO"
D-Метилмалонил-СоА L-Метилмалонил-СоА
Метилмалонил-Co-A-
— " OOC—CH2—CH2—CO-S—СоА
Сукцинил-СоА
Окисление жирных кислот
в пероксисомах
Пероксисомы - небольшие, окруженные мембраной
пузырьки, обнаруженные во многих животных клетках.
Подробнее о них рассказано в главе 16. Заметим лишь,
частьЗ Метаболизм
136
что они также окисляют жирные кислоты, хотя основная
масса последних подвергается окислению в
митохондриях. При пероксисомальном β-окислении жирных
кислот также образуется FADH2, который затем окисляется
не в митохондриях, а в самих пероксисомах, при
непосредственном участии кислорода и с образованием
перекиси водорода и тепла. Функции пероксисом менее
изучены, чем функции других внутриклеточных органелл.
Можно предположить, что одной из них является
метаболизм жирных кислот, содержащих более 18
углеродных атомов, которые не окисляются митохондриальной
системой.
До сих пор мы имели дело с образованием энергии
из глюкозы и жиров. Остается третий главный
компонент пищи - аминокислоты, и логично было бы теперь
рассмотреть их превращения. Однако целесообразнее от
метаболизма глюкозы и жиров перейти
непосредственно к их синтезу и его регуляции в организме, тем более
что при генерации энергии из аминокислот уже на
начальных стадиях метаболизма образуются вещества,
участвующие либо в гликолизе, либо в цикле лимонной
кислоты, так что особым своеобразием эти
превращения не отличаются. Наиболее важные аспекты
метаболизма аминокислот не имеют отношения к производству
энергии.
Дополнительная литература
McGarryJ. D., Foster D. W. Regulation of hepatic fatty
acid oxidation and ketone body production // Annu. Rev.
fiiochem. 1980. Vol. 49. P. 395-420.
(Обзор данных об образовании кетоновых тел.)
Lardy Η., Shrago E. Biochemical aspects of obesity // Annu.
Rev. Biochem. 1990. Vol. 59. P. 689-710.
(Сведения об ожирении, гормонах и метаболизме.)
Вопросы к главе 9
1. Периферические ткани получают жирные кислоты
из крови. Расскажите о трех возможных способах их
доставки.
2. Какие клетки организма не используют жирные
кислоты как источник энергии?
3. Что является первой стадией расщепления жирных
кислот? Где она происходит? Где в клетках эукариот
жирные кислоты превращаются в ацетил-СоА? Как
жирные кислоты туда попадают?
4. Что общего между реакциями окисления жирных
кислот до ацетил-СоА и некоторыми
превращениями в цикле лимонной кислоты?
5. Каков выход АТР при окислении пальмитиновой
кислоты? Поясните ответ.
6. Объясните, как мононенасыщенный жир (Δ9)
окисляется в ацетил-СоА.
7. Всегда ли ацетил-СоА, образовавшийся при
расщеплении жирных кислот, попадает в цикл лимонной
кислоты?
8. Гидроксиметилглутарил-СоА - промежуточный
продукт, образующийся при синтезе ацетоацетата и
холестерина. Где протекают эти процессы?
глава 9 Энергетическая ценность жиров
137
глава
10
Переход от катаболизма к анаболизму.
Синтез в организме жиров
и родственных им соединений
В предыдущих главах, посвященных метаболизму, речь
шла о катаболических реакциях, в ходе которых
происходит расщепление жиров и углеводов. Другая задача
метаболизма - синтез молекул, или анаболизм. Начать
изучение анаболизма лучше всего с синтеза жиров.
Синтез жиров осуществляется главным образом из
углеводов, поступающих в избыточном количестве и
не используемых для пополнения запаса гликогена.
Кроме того, в синтезе участвуют и некоторые
аминокислоты. Накоплению жиров способствует и избыток пищи.
Жир - наиболее компактная форма запасания энергии
организмом. Нам пришлось бы сильно увеличить свои
размеры, если бы мы запасали энергию исключительно
в виде гликогена.
Механизм синтеза жиров
Общие принципы
В процессе метаболизма жирные кислоты
превращаются в ацетил-СоА. В свою очередь, и ацетил-СоА
превращается снова в жирные кислоты (тот факт, что жирные
кислоты синтезируются из ацетил-СоА - двухуглеродного
соединения, приводит к тому, что число углеродных
атомов в природных жирных кислотах почти всегда четное).
Избыточное поглощение углеводов сопровождается
отложением жиров, так как глюкоза расщепляется до пиру-
вата, который превращается в ацетил-СоА. Эта последняя
реакция, катализируемая пируватдегидрогеназой,
необратима. Поэтому в животных клетках ацетил-СоА не может
превращаться в пируват и, соответственно, жиры не
могут превращаться в глюкозу, в отличие от бактерий и
растений, осуществляющих подобные превращения.
Любой метаболический путь включает по меньшей
мере одну стадию, которая в прямом и обратном
направлениях реализуется разными биохимическими
реакциями. Это справедливо для расщепления и синтеза жиров.
Соответствующие им метаболические пути местами
совпадают, а местами - нет. Благодаря этому оба
процесса термодинамически выгодны, необратимы и
регулируются по-разному.
Чтобы синтез жирных кислот из ацетил-СоА стал
термодинамически выгодным, он должен включать
необратимую стадию, для которой требуется дополнительная
энергия. Уже на первом этапе синтеза жирных кислот -
образовании малонил-СоА из ацетил-СоА и С02 -
используется энергия гидролиза АТР. Позже молекула
жирной кислоты теряет С02.
Это выглядит, на первый взгляд, бессмысленно,
однако таким путем превращению придается
термодинамическая необратимость; решается не столько химическая,
сколько энергетическая проблема. Малоновая кислота
имеет структуру НООС-СН2-СООН, а малонил-СоА -
HOOC-CH2-CO-S-CoA. Приведенную ниже реакцию
катализирует ацетил-СоА-карбоксилаза:
О
II
СН3—С—S—СоА + АТР + НС03"
Ацетил-СоА
О
О
С—СН2 —С—S—СоА + ADP + Р/ + Н+
Малонил-СоА
Простетической группой этого фермента является
биотин. Как вы уже знаете, все карбоксил азы
используют энергию гидролиза АТР для введения в субстраты
С02. В качестве промежуточного продукта в этой
реакции образуется «активированный С02» - карбоксибио-
тин (см. с. 123).
Таким образом, чтобы синтезировать жирные
кислоты, нужно последовательно присоединять к ацетил-СоА
двухуглеродные единицы. Активным донором таких
единиц является малонил-СоА, хотя малонильный
остаток сам по себе содержит не два, а три С-атома. Но
прежде чем остановиться на синтезе жирных кислот
подробнее, нужно пояснить, что такое ацилпереносящий
белок, или АПБ.
Ацилпереносящий белок
и β-кетоацил-синтаза
Все катаболические превращения происходят не с
самими жирными кислотами, а с тиоэфирами, которые они
образуют с СоА. Во всех реакциях синтеза жирных
кислот также участвуют тиоэфиры, но не СоА, а более
простой молекулы, которую можно рассматривать как
половину СоА. Напоминаем, как выглядит структура СоА.
часть 3 Метаболизм
138
I Фосфат—пантотенат—NHCH2CH2—SH !
-аденин \
фосфат
Фосфат—рибоза—аденин
В рамке
остаток фосфопантотеина
Выделенная часть молекулы и есть тиолсодержащий
«носитель» - 4-фосфопантотеин, который используется
при синтезе жирных кислот. В отличие от ацетил-СоА,
он функционирует не в свободном виде, а будучи
связанным с АПБ. Можно рассматривать АПБ как белок,
встроенный в СоА, или наоборот, как гигантскую
молекулу СоА, в которой АМР-фрагмент заменен белком.
Другой фермент, или точнее, функциональный
домен - β-кетоацил-синтаза, также содержит реакци-
онноспособную тиольную группу. Она принадлежит
остатку цистеина, который входит в активный центр
фермента. И АПБ, и β-кетоацил-синтаза у
млекопитающих являются компонентами большого мультифункци-
онального комплекса.
Механизм синтеза СоА-производных
жирных кислот
Рассмотрим случай, когда обе тиольные группы
в СоА-синтазном комплексе свободны (рис. 10.1, а).
β-Кетоацил-синтаза СН3СО—S—СоА СоА—SH
а АПБ б
SHH
АПБ-ацети -тр^нсс^^ра^а
Эта стадия наблюдается лишь
в начале синтеза жирной кислоты.
На последующих этапах синтеза
ацетил-СоА не используется
SHS
I
со
I
Ацетил-АПБ
СН3
\
SHS
I
со
I
сн2
I
сн2
I
сн3
Пунктирной стрелкой показано,что
состояния жив тождественны,
если не считать, что место
ацетильного остатка занял
бутирильный. Повторяя
наращивание цепи, можно
получить С ^-кислоту
S—СоА
СО
I
сн2—соо
СоА—SH
N
SH
I
со
I
сн3
АПБ-малонил-
7
—SH
со
L.X
сн2
I
сн2
I
сн3
Бутирил-АПБ
Восстановление
β-кетоацил-СоА
SHS
I
со
I
сн2
I
со
I
со2
β-Кетоацил-
АПБ-синтаза
(необратимая
реакция)
S S
I I
со со
I I
сн3сн2
соо-
Малонил-АПБ
СН β-Кетоацил-АПБ
Рис. 10.1. Синтез жирных кислот
Хотя β-кетоацил-синтаза у животных является одним из доменов большой белковой молекулы, здесь мы изображаем ее как
отдельный белок. После семи успешных оборотов цикла образуется пальмитил-АПБ, который гидролизуется с образованием
свободного пальмитата
глава 10 Переход от катаболизма к анаболизму
139
Ацетильная группа (СН3СО-) переносится с аце-
тил-СоА на тиольную группу АПБ специальной транс-
феразой (рис. 10.1, б). Далее она передается на
тиольную группу β-кетоацил-синтазы, в результате чего
место ее посадки в АПБ снова становится вакантным
(рис. 10.1, в). Его занимает малонильный остаток,
перенесенный другой трансферазой с малонил-СоА;
образуется малонил-АПБ (рис. 10.1, г). После этого синтаза
катализирует реакцию переноса ацетильной группы
на малонильную группу, что приводит к образованию
β-кетоацил-АПБ и С02 В данном случае речь идет
о β-кетобутирил-АПБ (рис. 10.1, д). Последний,
оставаясь в составе белкового комплекса, восстанавливается
в бутирил-АПБ (рис. 10.1, е). В конечном итоге система
переходит в состояние (рис. 10.1, ж\ аналогичное
изображенному на рис. 10.1, в: тиольная группа АПБ
свободна, а в синтазе она ацилирована остатком насыщенной
жирной кислоты. Единственное различие состоит в том,
что место ацетильного остатка (см. рис. 10.1, в) теперь
занимает бутирильный (см. рис. 10.1, ж). Если все
описанные выше реакции пройдут еще раз, бутирильный
остаток превратится в гексаноильный. Еще пять оборотов
цикла приведут к образованию пальмитоил-АПБ; затем
под действием фермента гидролазы происходит
высвобождение в раствор пальмитиновой кислоты (С16):
CH3(CH2)14CO-S-AnB + Н20 -^
-^ СН3(СН2)14СОСГ + АПБ-SH.
Реакция, катализируемая β-кетоацил-АПБ-синтазой,
необратима, так как имеет место процесс декарбокси-
лирования. Благодаря этой стадии весь путь синтеза
жирных кислот становится подобен дороге с
односторонним движением.
Если требуется синтезировать жирную кислоту,
содержащую более 16 углеродных атомов, например
стеариновую (С18), удлинением цепи пальмитиновой
кислоты будут заниматься другие ферментные системы.
Организация процесса
синтеза жирных кислот
Описанная последовательность реакций кому-то
покажется простой, а кому-то сложной, но несомненно одно:
этот свойственный животным ансамбль ферментов
организован удивительно. В клетках Е. coli ту же
последовательность реакций катализируют отдельные
ферменты, так что после каждой реакции продукты
диссоциируют в раствор и диффундируют к следующему ферменту.
У животных все стадии синтеза жирных кислот,
следующие за образованием малонил-СоА, реализуются
часть 3 Метаболизм
140
внутри одного гигантского белкового комплекса,
состоящего из нескольких субъединиц. Каждая из них
обладает ферментативной активностью, но функционирует
обычно только в составе комплекса (на самом деле два
таких мультифермента объединены в димер). Растущий
жирнокислотный остаток поочередно связывается
с тиольными группами АПБ и β-кетоацил-синтазы,
ни разу не покидая комплекс, пока синтез не
завершится образованием пальмитата. И удлинение цепи, и
восстановление кетогрупп происходит, когда субстраты
связаны с АПБ. Длинная гибкая ножка 4-фосфопантотеина,
вероятно, нужна, чтобы позволить различным
промежуточным соединениям взаимодействовать с нужными
каталитическими центрами мультиферментного
комплекса. Такая организация ускоряет синтез, так как
позволяет избежать многократной диссоциации и
диффузии продукта и способствует его связыванию с другим
ферментом.
Стадии восстановления в ходе
синтеза жирных кислот
В последовательности событий, приводящих к
удлинению жирнокислотной цепи, присоединенной к АПБ,
важное место занимает восстановление β-кетоацильных
остатков, которое проходит в три стадии (рис. 10.2).
Восстановителем здесь служит NADPH (не NADH!) -
переносчик электронов.
0 О
II II
R—С—СН2—С—S—АПБ β-Кетоацил-АПБ
\^ NADPH + Н+
К β-Кетоацил-АПБ
|Ч NADP+ ^дуктаза
Η О
1 II
R—С—СН2 —С—S—АПБ β-Гидроксиацил-АПБ
ОН
к β-Гидроксиацил-АПБ-
t^ Н20 цегидратаза
Η О
I II
R—С=С—С—S—АПБ
А
\^ NADPH + Н+
К Еноил-АПБ-
N NADP+ Редуктаза
О
II
R—СН2—СН2 —С—S—АПБ Ацил-АПБ
Рис. 10.2. Восстановительные стадии синтеза жирных
кислот
Что такое NADP+?
NAD+ имеет следующую структуру:
Р—рибоза—никотина!
Р—рибоза—аденин
А структура NADP+ выглядит так:
Р—рибоза—никотинамид
Р—рибоза—аденин
Ρ
Дополнительная фосфатная группа в молекуле
NADPH присоединена к 2'-гидроксильной группе рибо-
зы, связанной с аденином:
ΧΟΝΗ2
О—Ρ
I
о
I
О—Ρ
I
о
=0
ζ0 . ОН ОН
^0
Аденин
ОН
ОРОз"
Эта фосфатная группа практически не
сказывается на редокс-потенциале молекулы и фактически
используется в качестве сигнала при идентификации
и распознавании этой молекулы белками. ΝΑΟ+-φερ-
менты не реагируют с NADP+ и наоборот (за редкими
исключениями).
Долгое время думали, что использование NADPH
в процессе синтеза жирных кислот - не более чем
прихоть природы, своего рода курьез. Однако постепенно
стало ясно, что параллельное существование двух
пиридиновых коферментов играет важную роль при
разделении путей метаболизма. Чтобы в этом разобраться,
необходимо вспомнить, что производство энергии можно
представить как окисление восстановительных
эквивалентов, а синтез жира - как потребление
восстановительных эквивалентов. Цели диаметрально
противоположные! Клетка разделяет эти задачи, не разводя их
в пространстве: окислительные ветви метаболизма
используют NAD+ в качестве переносчика электронов,
а восстановительные ветви метаболизма для этих целей
используют NADP+. По-разному протекает
восстановление этих химически сходных соединений. NAD+
восстанавливается в ходе гликолиза, в пируватдегидро-
геназной реакции, в цикле лимонной кислоты, при
окислении жира. А как восстанавливается NADP+? Об этом -
в следующем разделе.
Где синтезируются жирные кислоты?
Основным местом синтеза жирных кислот является
печень и в меньшей степени жировые клетки, хотя жиры
образуют и многие другие органы, например молочные
железы в период лактации.
Внутри клетки синтез пальмитата из ацетил-СоА
сосредоточен в цитозоле, в то время как окисляются
жирные кислоты в митохондриях. Главным источником
ацетил-СоА для синтеза жирных кислот служит пиру-
ватдегидрогеназная реакция, протекающая в митохонд-
риальном матриксе (см. с. 116). Ацетил-СоА не может
самостоятельно пройти через митохондриальную
мембрану, поэтому должен существовать механизм переноса
ацетильных остатков из митохондрий в цитозоль, к
месту синтеза жирных кислот. В митохондриях ацетил-СоА
превращается в цитрат (см. с. 119), который
переносится из митохондрий в цитозоль особой транспортной
системой. В цитозоле цитрат расщепляется ферментом
цитратлиазой на ацетил-СоА и оксалоацетат. Эта
реакция сопряжена с гидролизом АТР, благодаря чему
практически необратима
Цитрат + ATP + CoA-SH + Н20 ->
—> Ацетил-СоА + Оксалоацетат +ADΡ +Pf.
Оксалоацетат не может вернуться в митохондрию
сам, так как митохондриальная мембрана для него
непроницаема, а переносчиков не существует. Он
восстанавливается в малат посредством NADH (заметим,
не NADPH!). Малат же подвергается окислительному
декарбоксилированию в пируват с помощью особого
«малик»-фермента, использующего в качестве кофер-
мента NADP+ (не NAD+). Образующийся NADPH
участвует в синтезе жирных кислот (рис. 10.3).
Пируват может теперь транспортироваться назад
в митохондрии, где он снова превращается в
оксалоацетат с помощью пируваткарбоксилазы (см. с. 120):
Пируват + НС03" + АТР
Пируват-
карбоксилаза
Оксалоацетат + ADP + Р,- + Н+
глава 10 Переход от катаболизма к анаболизму
141
Реально цитрат покидает митохондрии лишь тогда,
когда его концентрация в матриксе достаточно высока.
Так бывает при избытке углеводов, в противном случае
он не поступает в цитозоль.
COO-NADH + Н+ NAD+ rnn- NADP+ NADPH + Н+
\ /IV / С0°"
V У СНОН W, I ,
* ^ ' > ι —^~*-—·* СО + (
СО
сн7
Малат-
| " дегидрогеназа [
coo- Cl
Оксалоацетат
ςχχ Малик-фермент
Малат
со+со2
I
сн3
Пируват
Рис. 10.3. Восстановление NADP* для синтеза жирных
кислот
Суммарный эффект обеих реакций состоит в переносе
восстановительных эквивалентов с NADH на NADP+.
Образующийся в цитоплазме пируват поступает в митохондрии. Источник
оксалоацетата представлен на рис. 10.4
Таким образом, цитратный механизм не только
выводит ацетильные группы из митохондрий, но и
обеспечивает образование NADPH, который используется при
Митохондрия
Оксалоацетат
Цитрат
аза
С02^/Пируват- \
/карбоксилаза \
Пируват У-+~ Цитрат
X /
Ацетил СоА
С02
/ Пальмитат^ (*-) — Ма^онил--^Ацетил-СоА\
СоА w \
Ацетил-СоА- \ ^
Восстановительные / ЦитРат
эквиваленты
Малат -
NAD* NADH +
Малат-
"· П
Цитрат-
лиаза
Оксалоацетат
Малик-фермент
Цитозоль
Пируват + С02
Рис. 10.4. Изображение, объясняющее происхождение
ацетильных групп (ацетил-СоА) и восстановительных эквивалентов
(NADPH + Н+), необходимых для синтеза жирных кислот
Действие цитратлиазы сопряжено с расщеплением АТР
до ADP и Ρ
синтезе жирных кислот. Восстановление в цитоплазме
оксалоацетата в малат с помощью NADH и окисление
малата в пируват посредством NADP+ в совокупности
представляют собой изящный механизм переноса
электронов из фонда NADH в фонд NADPH, используемый
в реакциях «восстановительного» синтеза. In vitro
цитрат способен активировать начальную реакцию синтеза
жиров, катализируемую ацетил-СоА-карбоксилазой,
в результате которой образуется малонил-СоА. Однако
физиологическое значение такой активации пока
остается неопределенным. Регуляция активности этого
фермента обсуждается в главе 12.
Из приведенной схемы видно, что на каждую
молекулу ацетил-СоА, образующуюся в цитозоле из
цитрата, приходится одна молекула NADPH. Однако
каждый цикл работы синтазы жирных кислот требует две
молекулы NADPH (см. рис. 10.2). Процесс,
восполняющий недостаток NADPH, будет описан в главе 13, так
как он относится к другой ветви метаболизма -
превращениям пентозофосфатов.
Синтез ненасыщенных
жирных кислот
Ненасыщенные жирные кислоты нужны организму как
для синтеза полярных липидов, входящих в состав
мембран, так и для некоторых других целей. В печени есть
специальная ферментная система, которая может
внедрить одну двойную связь в середину цепи стеариновой
кислоты, превращая ее в олеиновую кислоту.
СН3СН(СН2)7СН=СН(СН2)7СООН.
Олеиновая кислота. 18:1(Δ9)
Однако эта ферментная система не может образовать
еще одну двойную связь между центральной двойной
связью и метильным концом молекулы. Поэтому ее
нельзя использовать для синтеза в животном организме
линолевой кислоты с двумя двойными связями и лино-
леновой кислоты с тремя двойными связями.
СН3(СН2)4СН=СНСН2СН=СН(СН2)7СООН.
Линолевая кислота, 18:2(А912)
СН3СН2СН=СНСН2СН=СНСН2СН=СН(СН2)7СООН.
α-Линоленовая кислота. 18:3(Δ9"12"15)
Поскольку эти кислоты необходимы для синтеза
мембранных липидов и эйкозаноидов, их
единственным источником служит пища (у растений есть
ферменты, синтезирующие полиненасыщенные жирные
часть 3 Метаболизм
142
кислоты с двойной связью в концевой части молекулы).
В печени же могут происходить дальнейшее удлинение
линолевой кислоты и образование в ней
дополнительных двойных связей. Таким способом синтезируется
арахидоновая С20-кислота (20:4, Δ5-8-11'14) с четырьмя
двойными связями, а также С22- и С24-кислоты,
входящие в состав липидов нервных тканей. Все эти
превращения совершаются с ацильными производными Со А.
Синтез триглицеридов
и мембранных липидов
из жирных кислот
Организм синтезирует нейтральные жиры с целью
пополнения запасов. Для образования эфирной связи
между глицерином и жирными кислотами последние нужно
активировать путем присоединения ацил-СоА; эту
реакцию катализирует ацил-СоА-синтетаза:
RCOOH + CoA-SH-ATP -^ RCO-S-CoA + AMP + РР,.
У обычных эфиров карбоновых кислот свободная
энергия гидролиза меньше, чем у соответствующих тио-
эфиров, поэтому такая активация жирных кислот делает
синтез триглицеридов экзэргоническим процессом.
Обратите внимание, что акцептором ацильных групп
выступает не глицерин, а глицерин-3-фосфат, который
образуется в результате восстановления дигидроксиаце-
тонфосфата - промежуточного продукта гликолиза.
СН20Н
I
NADH
+ Н+
NAD+
S^L
СН20Н
I
снон
СН2ОРО3 |ίιη *лрофг>сфа'гпегицрогеназа СН2ОРОз
Дигидрокси-
ацетонфосфат
Глицерин-3-фосфш
В печени (но не в жировых клетках) глицерин-3-фос-
фат образуется также в реакции прямого фосфорилиро-
вания глицерина. Все этапы синтеза триглицеридов
представлены на рис. 10.5.
Синтез глицерофосфолипидов
В состав мембраны входят фосфолипиды, основу
структуры которых составляет остаток глицерина.
2R-
0
II
-с-
сн2он
I
снон о
I II
сн2—о—р—о-
о-
Глицерин-3-фосфат
S—СоА 2СоА-
-SH CH2-0-C-R
сн
-о—с-
0
СН2—О—С—R
I о
СоА—SH R-
СН2—О—Р— 0-
Фосфатидная '
кислота
Н20^|
р А
о
JLc-rnA CH2-0^C-R
-S—СоА
СН
-0—С—R
0
СН
0
II
-О—С—R
СН2—О—Р—R
Триглицерид
СН20Н
Диглицерид
Рис. 10.5. Синтез триглицеридов (нейтральных жиров)
из глицерин-3-фосфата
Фосфоглицериды различаются лишь природой
спиртового остатка, связанного фосфоэфирной связью с фос-
фатидной кислотой.
СН2—0-
СН—0—С—R2
I 0
I II
СН2—0—Ρ—ΟΙ
0"
Фосфатидная
кислота
СН2—О—С-
I °
I м
сн—о—с-
I о
R2
СН2—О—Р-0—R3
I
О"
Фосфолипид
В роли R3 может выступать остаток серина, этанол-
амина, холина, инозита или диацилглицерина. В
мембранах эукариот преобладают фосфатидилэтаноламин
и фосфатидилхолин, поэтому рассмотрим сначала их
синтез. Этаноламин и холин имеют следующее строение:
NH2CH2CH2OH
Этаноламин
СНзч
СН3—N+CH2CH2OH
СН,
Холин
Оба эти соединения - спирты и могут быть
обозначены как R-OH. Для участия в синтезе фосфолипидов
эти спирты предварительно «активируются» в ходе
двухстадийного процесса. На первой стадии они фос-
форилируются с помощью АТР:
глава 10 Переход от катаболизма к анаболизму
143
R-OH + ATP
R-O—P-O"
I
0"
ADP
На второй стадии фосфорилированные спирты
реагируют с цитидинтрифосфатом (СТР), который
является аналогом АТР. В нем адениновый остаток заменен
на цитозиновый (цитидином называют цитозилрибозу).
СТР присутствует во всех клетках, поскольку он
необходим для синтеза РНК и ДНК (см. главу 21). Почему
при фосфорилировании спиртов используется именно
СТР, а не АТР, остается только гадать. То ли это
«причуда» эволюции, то ли нам пока не известен ее замысел.
Во всяком случае, реакция СТР с фосфоэфирами очень
похожа на образование UDP-глюкозы из глюкозо-1
-фосфата и UTP (см. с. 87):
О
II
о о
II II
О—Р—О—Р—О—Р—О— Цитидин
I I I
О" О" О"
R—О—Р—О—Р—О— Цитидин + PP.·
I I
О" О"
CDP-холин или CDP-этаноламин
Н20
2Р/
Гидролиз пирофосфата делает эту реакцию
необратимой и сильно экзоэргоничной. В конечной реакции
синтеза фосфатидилэтаноламина и фосфатидилхолина
участвует 1,2-диглицерид, образующийся под
действием фосфатазы на фосфатидную кислоту (см. с. 54).
СН2-
СН—0-C-R2
-O-C-Ri
О
СН20Н
1,2-Диглицерил
0 О
II II
О—Р—О—Р—О—Цитидин
1 I
О" 0"
+ СМР
-Р-0—R3
I
0"
Фосфатидилхолин или фосфатидилэтаноламин
Таким образом, описанный путь синтеза фосфо-
глицеридов сводится к активации спиртовой полярной
головки СТР с последующим переносом фосфорилиро-
ванного спирта на другой спирт - диглицерид.
Энергетически возможен и другой путь - активация дигли-
церида и перенос остатка диглицеридфосфата на холин
или этаноламин. Так в клетках эукариот синтезируются
фосфатидилинозит и кардиолипин (см. с. 55). В
последнем случае роль спирта играет вторая молекула дигли-
церида.
0
II
СН2-0-С—Rx
1 о
1 и
СН—0-С—R2+CTP—
1 о
1 м
СН2-0-Р-СГ
0"
Фосфат идная
кислота
СН
-СН
СН
0
II
2-0-С—
Ri
0
II
—0-С—R2 + РР,-
0 0
II II
j—0—Ρ—0—Ρ—О-Цитидин
ι ι
Ι Ι
0" 0"
'
Инозит
1
0
II
СН2—0—С—Rx
Ι о
1 и
СН—0—С—R2 + CMP
1 и
CH2—0—Ρ—0— Инозит
ι
0"
Фосфатидилинозит
Схема обоих путей синтеза глицерофосфолипидов
представлена на рис. 10.6. В действительности все
обстоит значительно сложнее из-за различных
взаимопревращений фосфолипидов. Например, они могут
обмениваться остатками серина и этаноламина, фос-
фатидилсерин декарбоксилируется с образованием
фосфатидилэтаноламина, а последний метилируется
до фосфатидилхолина. Все эти процессы в разной
степени представлены у различных организмов.
Место синтеза мембранных липидов
В животных клетках синтез мембранных липидов
осуществляется на поверхности гладкого эндоплазмати-
ческого ретикулума. Синтезированные липиды
транспортируются по назначению либо в виде мембранных
пузырьков, которые отпочковываются от мембран
аппарата Гольджи, либо с помощью белков-переносчиков
(механизм такого переноса до конца не выяснен).
часть 3 Метаболизм
144
а Фосфатидная б
кислота
Путь активации у ч Путь активации
диглицерида / \^ спиртовой головки
ROH
СТР
рр,
CDP-Диглицерид
ROH
t
CMP
Фосфатидил-R
Диглицерид
ATP
К
ADP + Ρ,
R— 0-®
t
СТР
РР/
CDR—R
Фосфатидил-R + CMP
Рис. 10.6. Два пути синтеза фосфоглицеридов
ROH - спирт, остаток которого служит полярной головкой
в синтезируемом фосфолипиде. У млекопитающих кардиоли-
пин и фосфатидилинозит синтезируются по пути я, а фосфати-
дилхолин, фосфатидилэтаноламин и фосфатидилсерин - по
пути б. Синтез фосфолипидов в бактериях протекает по пути а
указывает на число двойных связей в боковых цепях,
прикрепленных к циклопентановому кольцу. В качестве
примера на рис. 10.7, б показана структура PGE2. У
человека основную роль играют соединения с двумя
двойными связями, которые синтезируются из арахидоновой
кислоты (рис. 10.7, а). Остальные простагландины
образуются из других полиненасыщенных жирных кислот.
Арахидоновая кислота присутствует в организме
главным образом в виде жирнокислотного остатка
фосфолипидов. Для синтеза простагландинов она
предварительно отщепляется фосфолипазой.
Первая стадия синтеза простагландинов
осуществляется циклооксигеназой, которая превращает арахидоно-
вую кислоту в циклическое соединение (см. рис. 10.7, а).
Этот фермент ингибируется аспирином
(ацетилсалициловой кислотой), который ацетилирует гидроксильную
группу серинового остатка в активном центре фермента.
соон
Синтез простагландинов
и родственных им соединений
Название этой группы соединений - эйкозаноиды -
происходит от греческого числительного «двадцать».
Именно столько углеродных атомов содержится в их
молекулах, биосинтез которых начинается с
полиненасыщенных жирных кислот. Хотя эйкозаноиды
присутствуют в организме в незначительных количествах, диапазон
их физиологических функций очень широк.
Эйкозаноиды делятся на три основные группы по
названию клеток, в которых они впервые были открыты:
простагландины, тромбоксаны и лейкотриены.
Простагландины были обнаружены в семенной жидкости.
Сначала думали, что они попадают туда из
предстательной железы (простаты), но позже оказалось - из
семенных пузырьков. Теперь известно, что простагландины
синтезируются во многих тканях. Тромбоксаны были
найдены в кровяных пластинках (тромбоцитах), а
лейкотриены - в белых кровяных тельцах (лейкоцитах).
Простагландины и тромбоксаны
Известно много простагландинов, различающихся
деталями структуры. Их делят на подклассы,
обозначаемые как PGA, PGE и PGF. Цифровой индекс при них
СООН
НО
ОН
СООН
ОН
СООН
Рис. 10.7. Структура простагландинов и родственных им
соединений
а - Арахидоновая кислота;
б - простагландин Е2 (PGE2);
в - тромбоксан А2 (ТХА2);
г - лейкотриен A (LTA )
глава 10 Переход от катаболизма к анаболизму
145
Поскольку тромбоксаны (рис. 10.7, в) образуются путем
модификации некоторых простагландинов, аспирин
блокирует и их синтез.
Простагландины выполняют множество
физиологических функций. Сразу после синтеза они
выбрасываются из клеток и служат в качестве местных гормонов,
воздействуя на рецепторы соседних клеток. Они
вызывают боль, воспаление и жар, стимулируют сокращение
гладкой мускулатуры, участвуют в регуляции давления
крови, подавляют секрецию соляной кислоты в
желудке. Тромбоксаны вызывают агрегацию тромбоцитов, тем
самым стимулируя свертывание крови.
Ингибируя циклооксигеназу, аспирин подавляет
многие из перечисленных эффектов. В частности, в малых
дозах (100 мг в день) он блокирует синтез тромбоксанов,
уменьшая агрегацию тромбоцитов. Такая
профилактическая терапия предотвращает инфаркт, поскольку
снижает вероятность образования сгустков в
коронарных артериях.
Лейкотриены
Строение одного лейкотриена показано на рис. 10.7, г.
Лейкотриены также синтезируются из арахидоновой
кислоты, но другим ферментом - липоксигеназой.
Вызывая продолжительное сокращение гладкой мускулатуры,
они сужают дыхательные пути, участвуя тем самым в
развитии астматических приступов. Кроме того,
лейкотриены играют роль в регуляции деятельности лейкоцитов.
Синтез холестерина и его регуляция
Считается, что большинство клеток нашего тела может
синтезировать холестерин. Он является необходимым
компонентом плазматических мембран и служит пред-
0
шественником желчных кислот и стероидных гормонов.
Печень и в меньшей степени тонкий кишечник -
наиболее активные продуценты холестерина. В главе 6
рассказывалось о двух механизмах, посредством которых
холестерин в печени и других тканях образует единый
сбалансированный фонд.
Удивительно, что при синтезе большой сложной
молекулы холестерина единственным исходным
материалом служит ацетил-СоА. Путь этого синтеза очень
сложен. Наиболее интересны первые этапы синтеза,
поскольку именно на этом уровне определяется количество
продуцируемого холестерина. Речь идет о биосинтезе
мевалоновой кислоты - первого метаболита,
предназначенного исключительно для синтеза холестерина.
Соответствующие реакции, показанные на рис. 10.8,
протекают в цитозоле. Гидроксиметилглутарил-СоА служит
также предшественником ацетоацетата при образовании
кетоновых тел, однако для этой цели он синтезируется
внутри митохондрий.
На уровне гидроксиметилглутарил-СоА-редуктазы
синтез холестерина регулируется посредством трех
механизмов. По механизму обратной связи холестерин:
1) подавляет синтез редуктазы на уровне гена; 2)
вызывает деструкцию фермента; 3) инактивирует фермент
с помощью фосфорилирования (о последнем см. с. 161).
Для снижения в терапевтических целях уровня
холестерина в крови разработаны специальные лекарственные
препараты. Напоминая по структуре мевалоновую
кислоту, они при взаимодействии с гидроксиметилглута-
рил-СоА-редуктазой выступают как аналоги
переходного состояния, что обеспечивает их прочную ассоциацию
с активным центром фермента. Два таких лекарства
известны под названиями ловастатин и симвастатин.
2CH3-C-S-CoA
Ацетил-СоА
СН3 Η
1 1
2СН3—С—С
Н2—С—S—СоА+С
Тиолаза Ацетоацетил-СоА
Ацетил-СоА \
+ Н20 \
)
ГМГ-СоА-
редуктаза
Г-С А-синтаза
СН3 0
1 II
00С-СН2-С-СН2-С-0Н s V
I I ' \
ОН Η 2NADP+ 2NADPH
Мевалонат + 2Н+
00С-СН2-С—СН2—С—S-CoA
I
он
Гидроксиметилглутарил-СоА
+ СоА—SH
Рис. 10.8. Синтез мевалоната из ацетил-СоА
часть 3 Метаболизм
146
Дополнительная литература
Вопросы к главе 10
Синтаза жирных кислот
Smith S. The animal fatty acid synthase: one gene,
one polypeptide, seven enzymes // FASEB J. 1994. Vol. 8.
P. 1248-1259.
(Статья посвящена описанию структуры и функции
замечательного белка - синтазы жирных кислот.
Обсуждается роль каждого компонента и димерная
организация комплекса.)
Синтез мембранных липидов
Dawidowicz Ε. A. Dynamics of membrane lipid metabolism
and turnover //Annu. Rev. Biochem. 1987. Vol. 56. P. 43-61.
(Хорошо написанный обзор данных о мембранных липи-
дах, их синтезе, транспорте и формировании мембраны.)
Rooney S. Α., Young S. L., Mendelson C. R. Molecular
and cellular processing of lung surfactant // FASEB J. 1994.
Vol. 8. P. 957-967.
(Обзор данных о физиологической функции фосфоли-
пида, покрывающего изнутри легочные альвеолы.)
Kent С. Eukaryotic phospholipid biosynthesis // Annu. Rev.
Biochem. 1995. Vol. 64. P. 315-343.
(Описаны пути биосинтеза различных мембранных фос-
фолипидов.)
1. На ранних этапах синтеза жирных кислот аце-
тил-СоА сначала карбоксилируется, затем почти
сразу декарбоксилируется. Для чего это нужно?
2. Изобразите с помощью диаграммы основные этапы
цикла удлинения жирнокислотной цепи, опуская
при этом восстановительные реакции.
3. Как устроена синтаза жирных кислот у эукариот?
Чем она отличается от фермента Е. colP. Где более
эффективно протекает синтез жирных кислот -
у бактерий или у животных?
4. Изобразите структуру NAD+ и NADP+. В чем причина
одновременного существования обоих коферментов?
5. В каких тканях сосредоточен синтез жирных кислот
у эукариот?
6. Синтез пальмитата из ацетил-СоА сосредоточен в
цитоплазме, а главный производитель ацетил-СоА -
пируватдекарбоксилаза - в митохондриях. Ацетил-
СоА не проникает через мембрану. Как он попадает
из митохондрий в цитоплазму?
7. Откуда берется NADPH, необходимый для синтеза
жирных кислот?
8. Опишите, как из жирных кислот синтезируются
триглицериды.
9. Какую роль в метаболизме липидов играет СТР?
10. Что такое эйкозаноиды? Из чего они образуются?
Каково их физиологическое значение? Какое
отношение к ним имеет аспирин?
11. Как действуют лекарственные препараты,
подавляющие синтез холестерина?
глава 10 Переход от катаболизма к анаболизму
147
глава
11
Синтез глюкозы в организме
(глюконеогенез)
Организм может синтезировать глюкозу из соединений,
способных предварительно превратиться в пируват, т. е.
из большинства аминокислот и лактата,
поступающего в кровь из работающих мышц. Совокупность таких
превращений называют глюконеогенезом. Глюкоза
не может быть синтезирована из ацетил-СоА и жирных
кислот. Глюконеогенез позволяет как бы сохранить
энергию превращений в виде гликогена. Однако
помимо этого глюконеогенез в ряде случаев спасает
организм от гибели.
Мозг требует непрерывного обеспечения глюкозой.
Это означает, что во избежание комы или смерти
уровень глюкозы в крови должен поддерживаться в
пределах нормы. Между тем запасы гликогена в печени
невелики и полностью исчерпываются после суточного
голодания.
Люди, однако, остаются живы и при более
продолжительном отсутствии пищи. В этом случае поступление
глюкозы в кровь обеспечивается печенью. Ни один
другой орган (кроме почек, вклад которых невелик) не
способен выполнить эту задачу. После суточного голодания
печень должна удовлетворить потребность человека
в глюкозе (около 100 г в день).
На удовлетворение основных энергетических
потребностей организма запаса жиров хватает на
недели, но жирные кислоты не проникают через ге-
матоэнцефалический барьер и потому не могут
использоваться мозгом. Мобилизация жиров приводит
к образованию кетоновых тел, чье присутствие в
крови в какой-то мере уменьшает потребность в глюкозе
(см. с. 84), однако необходимость ее синтеза при этом
не исчезает. Потребность мозга в глюкозе при
голодании остается прежней. К тому же мозг лишь один
из основных ее потребителей. В глюкозе нуждаются
клетки сетчатки, мозгового слоя почек, эритроциты,
т. е. все ткани и клетки, жизнедеятельность которых
в значительной мере или полностью (эритроциты
вообще лишены митохондрий!) поддерживается
анаэробным метаболизмом.
Глюконеогенез в печени начинается с пирувата,
который служит исходным соединением и при синтезе
жирных кислот. Казалось бы, он может связать две ветви
метаболизма. Однако превращение пирувата в ацетил-СоА
у животных необратимо, поэтому они не могут
переработать жирные кислоты в глюкозу.
Механизм синтеза глюкозы из пирувата
Среди реакций гликолиза три термодинамически
необратимы (см. рис. 8.7):
1) АТР-зависимое фосфорилирование глюкозы гексо-
киназой (или глюкокиназой);
2) фосфорилирование фруктозо-6-фосфата фосфоф-
руктокиназой;
3) превращение фосфоенолпирувата в пируват.
Глюкоза синтезируется из пирувата с образованием
тех же промежуточных соединений, что и при
гликолизе, но для того, чтобы обойти необратимые реакции,
приходится избирать иные пути.
Первый термодинамический барьер нужно
преодолеть в ходе превращения пирувата в фосфоенолпируват.
Поскольку спонтанное превращение енольной формы
пирувата в кетоформу сильно экзоэргонично (большая
отрицательная величина AG0), реакция
Фосфоенолпируват—» Пируват необратима (см. с. ИЗ). Поэтому
у животных превращение пирувата в фосфоенолпируват
происходит обходным путем, в две стадии с
использованием двух макроэргических фосфатов, делающих это
превращение термодинамически выгодным:
Пируват-
Пируват + ATP + HCOf
(1)
► Оксалоацетат + ADP + Рг + Н+ ;
Фоссроенолпируват-
карбоксикиназа
Оксалоацетат + GTP ~* ►
Фосфоенолируват 4- GDP + С02
(2)
Суммируя реакции (1) и (2), получаем:
Пируват + ATP + GTP + Н20 ->
—» Фосфоенолпируват + ADP + GDP + р. + 2Н+.
Схема этих превращений представлена на рис. 11.1.
Почему на второй стадии используется GTP, а не АТР,
непонятно. Энергетически оба эти вещества
совершенно эквивалентны. Заметим, что реакция (1) - синтез ок-
салоацетата, катализируемый пируваткарбоксилазой,
играет важную роль в регуляции цикла лимонной
кислоты. Однако это не имеет отношения к глюконеогенезу.
глава 10 Метаболизм
148
Оксалоацетат
ФЕП-к оксикиназа
атка оксилазг
GTP ADP
+ Р/ ] \^~ С02
GDP ATP
ФЕП |-J ► Пируват
(Фосфоенолпируват) Ингибирование в печени
| Г„„Ко„ео„„„
Глюкоза
Рис. 11.1. Глюконеогенез в печени: образование фосфо-
енолпирувата из пирувата (ФЕП - фосфоенолпируват)
Обратите внимание, что эти реакции образуют холостой цикл,
если не блокировать обратное превращение ФЕП в пируват.
О том, как это достигается, см. в главе 12
Вторая реакция осуществляется фосфоенолпиру-
ват-карбоксикиназой. Такое название фермента
объясняется тем, что обратную реакцию можно
рассматривать как карбоксилирование фосфоенолпирувата,
сопряженное с переносом фосфатной группы.
После образования фосфоенолпирувата все гликоли-
тические реакции обратимы вплоть до реакции
образования фруктозо-1,6-дифосфата, превращение которого
из фруктозо-6-фосфата в ходе гликолиза необратимо.
Это препятствие легко преодолевается, поскольку
удаление фосфатной группы путем обычного гидролиза
не требует затрат энергии.
Аналогичная реакция гидролиза в процессе глюко-
неогенеза имеет место при превращении глюко-
зо-6-фосфата в глюкозу (при гликолизе образование глю-
козо-6-фосфата, катализируемое гексокиназой или глю-
кокиназой, необратимо).
В печени присутствует глюкозо-6-фосфатаза,
которая гидролизует глюкозо-6-фосфат, после чего
свободная глюкоза выходит из клетки. Совокупность
реакций глюконеогенеза представлена на рис. 11.2. О том,
как регулируется уровень глюкозы крови, рассказано
в главе 12.
Итак, существуют четыре фермента, ответственные
за глюконеогенез и не принимающие участия в
гликолизе: пируваткарбоксилаза, фосфоенолпируват-кар-
боксикиназа, фруктозо-1,6-дифосфатаза и глюко-
зо-6-фосфатаза. Естественно, они сосредоточены
преимущественно в печени. У крыс концентрация этих
ферментов в печени в 20-50 раз больше, чем в
скелетных мышцах.
О О
II II
о—р—о—сн2 ^о сн2—о—р—о"
он
он
Фруктозо-1,6-дифосфат
|н2о
Фруктозо-1,6-
дифосфотаза
О
II
"О—Р—О—СН2 О СН20Н
О" \Г .>J + Р/·
ч
но
он
он
Фруктозо-6-фосфат
Откуда печень получает пируват
для глюконеогенеза?
Когда при голодании истощается запас гликогена,
главным источником пирувата становится гидролитическое
расщепление мышечных белков, в ходе которого
образуются все 20 аминокислот. Хотя аланин - всего лишь
одна из них, более 30% всех аминокислот, поступающих
в печень, приходится на него. Аланин - одна из глюко-
генных аминокислот; его углеводородный скелет
используется печенью для строительства молекулы
глюкозы (метаболизм аминокислот будет рассмотрен
в главе 15).
Почему при расщеплении мышечных белков
образуется так много аланина? В результате метаболизма
многих аминокислот и цикла лимонной кислоты
накапливается оксалоацетат, который превращается в пируват
(см. рис. 11.1). Последний преобразуется в аланин;
аминогруппа достается ему от других аминокислот.
Заметим, что при голодании для производства
энергии мышцы используют в основном жирные кислоты
и кетоновые тела; поэтому они располагают
достаточным количеством ацетил-СоА, и для его получения им
не нужно окисление пирувата. Как будет показано в
главе 12, высокое соотношение концентраций ацетил-СоА/
СоА приводит к инактивации пируватдегидрогеназы.
Благодаря этому пируват, образующийся из аминокислот,
используется для синтеза аланина. При расщеплении
мышечных белков образуются различные аминокислоты,
глава Π Синтез глюкозы в организме (глюконеогенез)
149
11счсмь
Глюкозо-6-фосфат r·
Гексозофосфатизомераза
Фруктозо-1,6-дифосфатаза
Альдолаза
Дигидрок-
сиацетон-
фосфат
^ Глицеральдегид-3-
фосфат
р.-*^ NAD+ Глицеральдегид-3-
Т4 NADH + Н+ ФосФатДегиДрогеназа
(2х) 1,3-Дифосфоглицерат
γ ADP
(2х) З-Фосфоглицерат
З-Фосфоглицераткиназа
Фосфоглицератмутаза
(2х) 2-Фосфоглицерат
Ь-НгО
(2х)
Фосфоенолпируват
соАг GDP
S- GTP
(2х) Оксалоацетат
t^ADP+Pj
HCOj-^KaTP
(2х)
ируват
Енолаза
Фосфоенолпируват-
карбоксикиназа
Пируваткарбоксилаза
Рис. 11.2. Глюконеогенез в печени: синтез глюкозы из
пирувата
Реакции, отличающиеся от соответствующих гликолитичес-
ких превращений, выделены цветом
многие из которых превращаются в аланин; он
переносится кровью в печень. (Кроме аланина в мышцах
синтезируется и доставляется в печень для синтеза
глюкозы также и глутамин; принцип его превращения тот же.)
В печени аланин преобразуется снова в пируват, а
затем - в глюкозу Схема участия аланина в глюконеоге-
незе представлена на рис. 11.3.
По мере того как при голодании в крови
увеличивается содержание кетоновых тел, мозг более активно
начинает использовать их вместо глюкозы для
производства энергии. Это снижает (хотя и не устраняет)
потребность в глюконеогенезе. Поскольку на производство 1 г
Мышца
Мышечные белки
ι
20 аминокислот
Глюкоза
Некоторые
аминокислоты
Кислоты цикла лимонной кислоты
Оксалоацетат
Ит попрованпс
при голодании
Ацетил-СоА
Пируват
Аланин ·♦
Рис. 11.3. Механизм, посредством которого при голодании
мышечные белки обеспечивают печень пируватом для глю-
конеогенеза
Схема предполагает, что пируват не превращается в
ацетил-СоА в ходе пируватдегидрогеназной реакции. Наряду
с аланином из мышц поступает другой субстрат глкжонеоге-
неза — глутамин
глюкозы расходуется 2 г мышечных белков, замедление
процесса их расщепления благотворно отражается на
выживании голодающего организма.
В главе 15 подробно обсуждается схема глюкозо-ала-
нинового цикла (см. рис. 15.9). На ней показано, как
аланин, синтезированный в мышцах и доставленный в
печень, перерабатывается там в глюкозу. Однако в рамках
этой схемы пируват, необходимый для синтеза аланина,
образуется в результате гликолиза. При этом имеет
место следующая последовательность событий:
глюкоза в печени —> глюкоза в мышцах —> аланин в
мышцах —> аланин в печени —» глюкоза в печени.
Понятно, что такой цикл не приводит к увеличению
количества глюкозы и не решает проблемы снабжения
ею тканей, а является лишь формой транспорта амин-
ного азота из мышц в печень.
Другой источник пирувата для глюконеогенеза важен
не столько при голодании, сколько при нормальной
жизнедеятельности организма. Речь идет о лактате,
образующемся при анаэробном гликолитическом
расщеплении глюкозы или гликогена (см. с. 101). Некоторые
клетки, в частности мозгового вещества почек и
сетчатки, фактически анаэробы, а в зрелых эритроцитах
вообще нет митохондрий, и они не способны, следовательно,
глава 10 Метаболизм
150
к окислительному фосфорилированию. При
нормальном поступлении пищи главным источником лактата
является гликолиз в интенсивно работающих мышцах.
В таких условиях митохондрии не успевают реокислять
накапливающийся NADH. В результате
восстановительные эквиваленты переносятся на пируват, который
Печень
Гликоген
Глюкоза
Глюкоза _
в крови
Мышцы
Гликоген
Анаэробный
гликолиз
Глюконеогенез
Лактат -*
Лактат
в крови
— Лактат
Рис. 11.4. Цикл Кори - физиологический цикл, который
протекает в мышцах и печени
В мышцах очень невелико содержание трех ферментов,
необходимых для глюконеогенеза, поэтому для переработки в
глюкозу накопленный в мышцах лактат должен быть перенесен
в печень. Избыток лактата образуется в мышцах при их
активном сокращении в процессе анаэробного гликолиза
превращается в лактат. Последний переносится кровью
в печень, а там перерабатывается снова в пируват, а
затем в глюкозу и гликоген. Этот физиологический цикл
по имени его первооткрывателя называют циклом Кори
(рис. 11.4).
У цикла Кори есть две важнейшие функции: сберечь
лактат для последующего использования и
предотвратить так называемый лактат-ацидоз. При поступлении
больших количеств молочной кислоты в кровь ее
буферная емкость может быть исчерпана, что приведет к
опасному снижению рН. Этому препятствует превращение
лактата в глюкозу, которое сопровождается
поглощением двух протонов (на восстановление 1,3-дифосфогли-
церата расходуется 1 протон (Н+) и 1 молекула NADH).
Синтез глюкозы из глицерина
Еще одним источником углеродных соединений для
глюконеогенеза служит глицерин, образующийся при
гидролизе триглицеридов главным образом в жировой ткани.
Он поглощается печенью и превращается там в глюкозу
в соответствии со схемой, представленной на рис. 11.5.
Первый этап - фосфорилирование глицерина
осуществляется глицеринкиназой, которой в жировой ткани
гораздо меньше, чем в печени. Все последующие
превращения также сосредоточены в печени. Это вполне
СН2ОН ATP ADP СН2ОН
NAD+
СНОН
I
Х^
I
снон
NADH СН2ОН Триозофосфат- СНО
^ ^ ^ с=о
изомераза
Глицерин
киназа
Глицерин Глицерин-3-фосфат
ι
снон
I
СНгОН киназа СН2ОРОз деги огеназа
^ Глицерин-3-фосфат- ^^2- Альдолаза ^^г-
Дигидрокси-
ацетонфосфат
/
/
Глюкозо-6-фосфат
Глицеральдегид-3-
фосфат
Глюкоза
Рис. 11.5. Превращение глицерина, высвобождающегося при гидролизе нейтральных жиров, в глюкозу
Основное количество глицерина образуется в жировых клетках, но поскольку в них нет глицеринкиназы, глюконеогенез из
глицерина протекает в печени. Этот процесс обеспечивает образование глюкозы из глицерина при голодании
глава И Синтез глюкозы в организме (глюконеогенез)
151
логично, поскольку в ситуации, когда образование
глюкозы становится жизненно необходимым, эту задачу
решает печень. Поэтому жировые клетки сами не
используют глицерин.
При продолжительном голодании после небольшого
начального снижения уровень глюкозы в крови
поддерживается неизменным в течение нескольких недель
благодаря постоянному поступлению жирных кислот
из жировых клеток. Удивительно, однако, что животным,
в отличие от растений и бактерий, не был дарован
природой более простой способ, не связанный с
«поеданием» собственных мышц. Видимо, у эволюции на этот счет
были какие-то неведомые нам соображения.
Синтез глюкозы посредством
глиоксилатного цикла
Е. coli прекрасно существует, используя в качестве
единственного источника углерода ацетат. В отличие от
животных, бактерии способны превращать ацетил-СоА
в С4-кислоты, задействованные в цикле лимонной
кислоты, и использовать их для синтеза глюкозы и других
необходимых компонентов клетки. В прорастающих
семенах растений для синтеза глюкозы также
используются запасенные триглицериды. Как же это удается
бактериям и растениям?
Эти организмы обладают обычным циклом
лимонной кислоты, но часть образующихся в нем
соединений они могут использовать в других превращениях,
не свойственных животным. Так, в цикле лимонной
кислоты 2 углеродных атома ацетил-СоА вводятся
сначала в молекулу оксалоацетата (С4) с образованием
цитрата (С6), а затем 2 углеродных атома выводятся в виде
2 молекул С02 (переход от С6- к С4-кислотам) с
образованием сукцината. Этой безвозвратной потери удается
избежать благодаря так называемому глиоксилатному
пути, который позволяет вывести из цикла лимонной
кислоты 2 атома углерода не в виде С02, а в виде глиок-.
силата, который образуется непосредственно при
расщеплении изоцитрата на сукцинат и глиоксилат:
С00"
I
снон
I
СНСОО"
I
СН2СОО~
Изоцитрат
Изоцитра -
\ а
СН2СОО" СОСГ
I +1
СН2СОО" СНО
Сукцинат Глиоксилат
Глиоксилат (С2) далее реагирует с ацетил-СоА,
превращаясь в малат - обычный компонент цикла
лимонной кислоты.
СН-СОО"
II
0
Глиоксилат
+
сн3—c-s-
Ацетил-СоА
СоА
Н20
Малатсинтаза
ОН
I
СН—С00 + СоА—SH
I
сн2-соо~
Малат
Общая схема, иллюстрирующая взаимосвязь всех
рассмотренных процессов, представлена на рис. 11.6.
Их суммарный эффект сводится к тому, что ацетил-
СоА плюс оксалоацетат превращаются в малат плюс
сукцинат. И малат, и сукцинат могут быть
преобразованы в оксалоацетат, одна молекула которого без ущерба
для цикла может быть направлена на синтез глюкозы.
В растениях эти реакции протекают в мембранах орга-
нелл, называемых глиоксисомами.
Ацетил-СоА
Оксалоацетат
1(мс-Аконитат
Глиоксилат
/
Ацетил-СоА
ФЕП \ СоА-SH
/
^ г >п Малат
/
Глюкозо-6-фосфат ^>умарат"
Изоцитрат
\^С02
а-Кетоглутарат
I
Ы С02
г
Сукцинил-СоА
Сукцинат
Рис. 11.6. Глиоксилатный цикл, с помощью которого
растения и бактерии (у животных он отсутствует)
осуществляют синтез углеводов из ацетил-СоА
Специфичные для этого цикла реакции выделены цветом.
Штриховой линией отмечены реакции цикла лимонной
кислоты, которые не задействованы в глиоксилатном цикле. Таким
образом удается сохранить 2 молекулы С02, необходимые для
превращения цитрата в 2 молекулы оксалоацетата, одна из
которых опять используется для синтеза цитрата, а другая
превращается в фосфоенолпируват
глава 10 Метаболизм
152
В заключение можно напомнить, что подавляющая
доля углеводов на Земле образуется благодаря
фотосинтезу, в ходе которого энергия солнечного света
используется для фиксации С02 в виде дифосфоглицерата -
соединения, знакомого нам по гликолизу. Механизмы
этого процесса и дальнейшего превращения
дифосфоглицерата в глюкозу будут рассмотрены в главе 14
как составные части фотосинтеза.
Итак, мы познакомились с использованием жиров
и глюкозы в качестве источников энергии, а также с
механизмами синтеза этих веществ. Следует напомнить,
что эти метаболические процессы не существуют
изолированно, а образуют интегрированную
метаболическую систему, все части которой взаимозависимы и
нуждаются в регуляции.
Дополнительная литература
Felig P. Amino acid metabolism in man // Annu. Rev. Bio-
chem. 1975. Vol. 44. P. 933-955.
(Очень полезный обзор, в котором обсуждается
метаболизм аминокислот на уровне органов и
рассматриваются проблемы, связанные с голоданием, диабетом и
ожирением с точки зрения глюконеогенеза.)
Snell К. Alanine as a gluconeogenic carrier // Trends Bio-
chem. Sci. 1979. Vol. 4. P. 124-128.
(Обзор, в котором анализируется роль мышц в снабжении
печени веществами, необходимыми для глюконеогенеза.)
Pilkis S. J., El-Maghrabi Μ. R.. Claus Τ. Η. Hormonal
regulation of hepatic gluconeogenesis and glycogen // Annu.
Rev. Biochem. 1988. Vol. 57. P. 755-783.
(Исчерпывающий обзор.)
Вопросы к главе 11
1. После суточного голодания запасы гликогена в
печени истощаются, но в организме имеются довольно
большие запасы жиров. Зачем при голодании
протекает процесс глюконеогенеза, когда в организме есть
практически безграничные запасы ацетил-СоА (из
жирных кислот), которых вполне хватает для
производства энергии?
2. Почему фосфоенолпируват, необходимый для
протекания глюконеогенеза, не может быть получен путем
фосфорилирования пирувата с помощью пируват-
киназы?
3. Начиная от фосфоенолпирувата, глюконеогенез
в печени осуществляется путем обращения реакций
гликолиза. Какие два гликолитических фермента
катализируют необратимые реакции? Как
существование этих реакций сказывается на механизме
глюконеогенеза?
4. Есть ли в мышцах глюкозо-6-фосфатаза? Поясните
ответ.
5. Что такое цикл Кори и какова его физиологическая
роль?
6. В жировых клетках практически нет глицеринки-
назы - фермента, катализирующего превращение
глицерина в глицерин-3-фосфат, хотя там образуется
глицерин (путем гидролиза триглицеридов). В
печени же этот фермент есть. Логично ли это? А если да,
то почему?
7. Как бактериям и растениям, в отличие от животных,
удается превратить ацетил-СоА в углеводы?
глава 11 Синтез глюкозы в организме (глюконеогенез)
153
А О Регуляция метаболизма
глава I £л углеводов и жиров
Эта глава логически завершает все то, что было ранее
изложено о метаболизме, в ней будет показано как
отдельные метаболические пути пересекаются и
функционируют вместе. До сих пор мы в основном изучали
каждый их них отдельно. Первоначальное рассмотрение
индивидуальных метаболических путей, на наш взгляд,
оправданно, поскольку: 1) каждый из метаболических
путей достаточно сложен, чтобы отягощать знакомство
с ним еще и проблемами регуляции; 2) гораздо
эффективнее изучать вопросы регуляции применительно ко
всему ансамблю метаболических процессов; 3) всегда
можно вернуться к уже прочитанным, изученным главам.
Хотя в регуляции нуждаются все звенья
метаболизма, интеграция и управление превращениями углеводов
и жиров имеют особое значение, поскольку по этим
метаболическим путям идут особенно мощные потоки
веществ, интенсивность и направление которых часто
изменяются. Это связано с тем, что у животных периоды
насыщения сменяются голодом, периоды отдыха -
различными видами деятельности. Все это требует
изменения направления потоков веществ, участвующих в
метаболических превращениях. В то же время на примере
обмена углеводов и жиров можно рассмотреть все
основные принципы регуляции.
Поскольку все биохимические реакции
катализируются ферментами, регуляция метаболизма в конечном
итоге представляет собой регуляцию ферментативной
активности. Поэтому основное внимание уделяется
именно способам регуляции активности ферментов.
После этого мы покажем, как регуляторные ферменты,
активность которых зависит от уровня различных
метаболитов, поддерживают баланс между
метаболическими превращениями. Это далеко не все аспекты
регуляции, ибо метаболическая активность отдельных клеток
зависит от гормональных и нервных сигналов,
поступающих в соответствии с нуждами всего организма.
Поэтому в заключение мы разберем, как внешний по
отношению к клетке сигнал регулирует метаболизм.
Зачем необходима регуляция?
Совершенно ясно, что все метаболические процессы,
с которыми мы познакомились - синтез и распад
гликогена, гликолиз и глюконеогенез, синтез и расщепление
жиров, цикл лимонной кислоты, транспорт электронов
и т. п. не могут одновременно протекать с максимальной
для каждого из них скоростью. Если метаболические
превращения протекают в одном направлении, то
обратные превращения не должны иметь места. Скорость
реакций каждого метаболического пути должна
изменяться в широких пределах, в зависимости от текущих
потребностей организма в энергии. Так, скорость
основных реакций метаболизма у человека, играющего
в теннис, увеличивается шестикратно, а при еще более
тяжелой физической нагрузке - в пятнадцать раз.
Укажем на два наиболее важных момента, связанных
с регуляцией реакций, ответственных за производство
энергии:
• производство энергии должно согласовываться с
постоянно меняющимися в ней потребностями;
• скорость и направление превращений должны
подчиняться нуждам и жизненному ритму всего
организма, который периодически принимает пищу
и голодает, временами кормит детенышей молоком,
а иногда болеет.
Еще одна, не столь очевидная необходимость
регуляции - потенциальная опасность возникновения
«холостых» (или субстратных) циклов.
Потенциальная опасность «холостых»
циклов в метаболизме
Данную проблему обсудим на примере глюконеогенеза
и гликолиза. При гликолизе фруктозо-6-фосфат фосфо-
рилируется фосфофруктокиназой во фруктозо-1,6-ди-
фосфат, тогда как при глюконеогенезе дифосфатаза
гидролизует фруктозо-1,6-дифосфат до исходного фрук-
тозо-6-фосфата (рис. 12.1). Такая неуправляемая
последовательность событий не приведет ни к чему, кроме
бессмысленного перевода энергии АТР в тепло,
подобно короткому электрическому замыканию (этим приемом
пользуются шмели, чтобы холодным утром перед
вылетом разогреть летательные мышцы).
Аналогичный гигантский «холостой» цикл, занятый
уничтожением АТР, мог бы объединить весь гликолиз
и глюконеогенез, замыкая их друг на друга (рис. 12.2).
В такие же циклы потенциально могут объединиться
реакции, связанные с образованием и распадом гликогена
и жиров, т. е. любые парные комбинации процессов
синтеза и распада отдельных метаболитов.
часть 3 Метаболизм
154
Глюконеогенез
Фруктозо-6-фосфат
Фруктозо-1,6-
дифосфатаза
Гликолиз
АТР
Фосфофруктокиназа
ADP
Фруктозо-1,6-дифосфат
Рис. 12.1. «Холостой» цикл на уровне гликолитичес-
кой фосфофруктокиназы, который может возникнуть
в отсутствие регуляторных механизмов
Понятно, что расщепление и синтез метаболитов
должны регулироваться противоположным образом:
когда активируется один из этих процессов, другой
должен ингибироваться. Такая регуляция возможна лишь
в том случае, если синтез и расщепление протекают
по разным метаболическим путям. Как мы уже
отмечали, для этого оба пути должны включать по меньшей
мере по одной необратимой стадии, которые
катализируются разными ферментами и, следовательно, могут
по-разному регулироваться. В случае же обратимых
процессов прямые и обратные реакции катализируются
одними и теми же ферментами, так что их раздельная
регуляция невозможна.
«Холостые» циклы называют также субстратными
циклами, что отражает их сущность: «объединение
реакций» синтеза и распада субстрата. Хотя
возникновение таких больших циклов в принципе неразумно, они,
тем не менее, могут функционировать как элементы
Глюкозо-6-фосфат
Глюконеогенез [ ) Гликолиз
(потребляет АТР) V J (производит АТР)
^"^ Пируват
Гликоген ■
_ , λ Расщепление
Синтез гликогена ( J гликогеНа
* Гл юкозо-1 -фосфат
Жирная
s^ кислота **\
Синтез жира
к.
) Расщепление жира
Ацетил-СоА
^ -ле"^
Рис. 12.2. Крупномасштабные «холостые» циклы,
потенциально возможные в отсутствие регуляции
регуляторных систем. Предположим, например, что
вещество А превращается в вещество С через
промежуточное вещество В, причем возможна реакция В <-> А.
Фермент 1
Фермент 2
Скорость образования С можно снизить, либо ин-
гибируя фермент 1, либо активируя фермент 2.
Наибольший эффект будет достигнут, если одновременно
произойдет и то, и другое. При этом для полной
остановки реакции В —» С понадобится значительно меньше
ингибитора фермента 1. Как будет показано ниже,
именно такой тип регуляции характерен для реакции фрукто-
зо-6-фосфат <-> фруктозо-1,6-дифосфат.
Как регулируется активность
ферментов?
Метаболическая регуляция есть не что иное, как
регуляция скорости по меньшей мере одной из
последовательных каталитических реакций, образующих в
совокупности метаболический путь. При этом реакция
необязательно должна быть химической, например,
транспортные белки являются катализаторами
переноса метаболитов через мембраны. Существуют два
главных способа обратимо влиять на скорость
ферментативных процессов в клетке.
1. Изменить количество фермента.
2. Изменить его каталитическую активность.
Разумеется, существуют способы необратимой
активации фермента. К ним можно отнести протеолити-
ческое превращение трипсиногена в активный трипсин
(см. с. 75). Однако в этой главе мы будем касаться
только таких регуляторных механизмов, которые полностью
обратимы, поскольку только они имеют существенное
значение для эффективного управления метаболизмом.
Управление метаболизмом путем
изменения количества ферментов
Уровень белка в клетке можно изменить, воздействуя
либо на скорость его синтеза, либо на скорость его
распада. Белки - короткоживущие компоненты клетки;
время полураспада ферментов печени - от часа до
нескольких дней.
глава 12 Регуляция метаболизма углеводов и жиров
155
У животных изменение количества ферментов
вносит заметный вклад в регуляцию метаболизма. Такая
долгосрочная регуляция реализуется в шкале часов и
дней, но никак не секунд. Она используется при
адаптации к новым физиологическим нуждам. Здесь можно
привести множество примеров. Вспомним липопроте-
инлипазу кровеносных капилляров (см. с. 93). Ее
количество коррелирует с потребностью ткани в липидах,
увеличиваясь, к примеру, в молочных железах на
период лактации. В ответ на смену диеты содержание
ферментов в печени меняется в течение часов (если надо
справиться с непривычной, очень жирной или
изобилующей углеводами пищей). При богатом рационе в
печени возрастает количество ферментов, участвующих
в синтезе жиров, тогда как при голодании уже через
несколько часов этот процесс обращается. При
попадании в организм чужеродных химических веществ,
например лекарств, в печени быстро нарастает количество
окисляющих их ферментов (см. с. 210). Еще раз
подчеркнем, что у животных уровень ферментов в клетке
меняется в течение нескольких часов, а то и дней, а для ко-
роткоживущих ферментов этот срок бывает меньшим.
В общем случае такой способ регуляции (изменение
количества фермента) используется животными лишь
в тех случаях, когда возможное время ответа системы
на внешнее воздействие достаточно велико.
У бактерий уровень ферментов изменяется гораздо
быстрее. Если клетки Е. coli поместить в среду, где
единственным источником углерода служит лактоза, то
синтез фермента β-галактозидазы, гидролизующего этот
сахар, начинается немедленно. Его количество
становится ощутимым уже через несколько минут, а через
несколько часов возрастает тысячекратно. Тем не менее
это не самый быстрый ответ.
После того как потребность в ферменте пропадает, его
концентрация в клетке уменьшается. Время ответа
системы при этом определяется скоростью распада белков (у
бактерий также разбавлением при делении), коррелируя
со временем их полураспада в организме. Для
оперативной автоматической регуляции метаболизма изменение
количества ферментов - неподходящий способ,
поскольку этот тип регуляции имеет дело только с теми
ферментами, которые в данный момент присутствуют в клетке.
Управление метаболическими
процессами путем изменения
активности ферментов
При регуляции активности фермента изменяется не его
количество, а скорость его работы. Главное в этом
способе управления - его быстрота. Чтобы понять основные
часть 3 Метаболизм
156
принципы такой регуляции, необходимо разобраться,
какие факторы влияют на каталитические свойства
фермента.
Основные сведения
о ферментативной кинетике
Гиперболическая кинетика
«классического» фермента
При ферментативном катализе субстрат (S) обратимо
связывается в активном центре фермента (Е) с
образованием фермент-субстратного комплекса (ES). После того
как в пределах этого комплекса произошла химическая
реакция, образовавшийся продукт (продукты, Р)
высвобождается из активного центра фермента, оставляя его
свободным для нового взаимодействия и реакционного
цикла. Такое представление о работе фермента лежит
в основе модели, предложенной Михаэлисом и Ментен
для описания кинетики ферментативного катализа:
E + S<->ES->E + P.
При низкой концентрации субстрата [S] скорость
ферментативной реакции определяется лишь частотой
столкновений молекул субстрата S и свободного
фермента Е. В этой ситуации концентрация
фермент-субстратных комплексов [ES] и скорость ферментативной
реакции малы. Однако с ростом [S] количество молекул
фермент-субстратного комплекса возрастает, а вместе
с этим увеличивается и скорость реакции. Наступит
момент, когда увеличение концентрации субстрата
приведет к тому, что практически все молекулы фермента
будут насыщены субстратом и превратятся в
комплексы ES, после чего дальнейший рост [S] уже не будет
сказываться на скорости ферментативной реакции. В
этом случае говорят о насыщении фермента, а
максимальную скорость реакции обозначают Vmax.
Графически такая зависимость описывается гиперболической
кривой (рис. 12.3). Такого рода кинетику называют
кинетикой Михаэлиса-Ментен, а подчиняющиеся этой
модели ферменты - ферментами Михаэлиса-Ментен.
Долгое время они были единственными известными
науке и потому получили название классических ферментов.
До сих пор мы использовали понятие низкая
концентрация субстрата, что не вполне корректно,
поскольку при одном и том же значении [S] один фермент
будет насыщен субстратом, а другой - нет. Это зависит
от прочности связывания субстрата ферментом или,
как говорят, от степени сродства фермента к субстрату.
Сродство может быть количественно выражено
константой диссоциации фермент-субстратного комплекса или
изменением свободной энергии при его образовании
и зависит от природы реагирующих веществ и числа
слабых связей, образующихся между ними.
Относительное сродство фермента к субстрату в
большинстве случаев можно оценить, зная константу
Михаэлиса Км. Этот параметр определяется как
концентрация субстрата, при которой скорость
ферментативной реакции составляет половину максимально
возможной (см. рис. 12.3) и, как нетрудно понять,
не зависит от количества фермента. (Заметим, однако,
что истинное значение Км можно определить, когда
скорость диссоциации комплекса ES на Ε + S
значительно выше, чем скорость распада ES на Ε 4- Р.)
Известно много графических методов преобразования
модели Михаэлиса-Ментен, позволяющих просто и точно
оценить величину Км, однако здесь мы ограничимся
самым простым изложением основных принципов
ферментативной кинетики, необходимых для понимания ре-
гуляторных механизмов.
Чем больше величина Км, тем ниже сродство
фермента к своему субстрату. Следовательно, значения Км
могут быть использованы для сравнения сродства
различных ферментов к их субстратам. Величины Км могут
о §■
8 еЗ
U Он
У Область насыщения
фермента субстратом
У * * * * Некатал изируемая
реакция
Км Концентрация субстрата [S]
(для некатализируемой реакции -
исходного вещества)
Рис. 12.3. Влияние концентрации субстрата на скорость
реакции (V). катализируемой ферментом, кинетика
которого описывается моделью Михаэлиса-Ментен
Км - константа Михаэлиса. Пунктирная линия отражает
свойства некатализируемой реакции, однако следует обратить
внимание только на форму этой зависимости, но не на абсолютные
значения скорости. Скорости реакций, протекающих в клетке,
были бы ничтожно малыми, если бы их не катализировали
ферменты
варьировать в диапазоне от наномолярных до миллимо-
лярных концентраций, но в целом они лежат в пределах
ΙΟ^-ΙΟ-4 М. Внутриклеточная концентрация субстрата
обычно соизмерима по порядку величины с Км, т. е.
фермент полностью субстратом в клетке не насыщен.
Какие ферменты метаболического
пути нужно регулировать?
Прямой ответ на данный вопрос таков: те ферменты,
которым эволюция придала это свойство. Любой
метаболический путь включает реакции, катализируемые
специфическими регулируемыми ферментами. Обычно они
расположены в стратегически важных местах, например,
на необратимой стадии в целом обратимого процесса.
Как правило, стратегическим пунктом контроля
является первый фермент метаболического пути. Рассмотрим
такую последовательность реакций: А—> В—> С—> D—> Е,
в которой метаболический путь заканчивается
образованием нужного клетке вещества Е. Важно, чтобы Ε
не производился в количестве, превышающем текущие
потребности в нем. Простейшей схемой автоматической
регуляции этого каскада реакций будет торможение
первого фермента, катализирующего превращение А—> В,
конечным продуктом (Е):
Ингибированис
Такой эффект достигается двумя путями: ингиби-
рованием фермента и/или уменьшением его количества.
Торможение именно первой стадии превращений
позволяет избежать накопления промежуточных продуктов В,
С и D, образующихся на последующих стадиях. Когда
метаболит Ε исчезает или его концентрация снижается,
ингибирование прекращается, в результате чего
синтез Ε возобновится с новой силой. Такой тип регуляции
в биохимии называется регуляцией по принципу
обратной связи, или ингибированием конечным продуктом. Он
часто встречается у бактерий (например, при синтезе
аминокислот) и осуществляется у них с
необыкновенной точностью. Однако этот тип регуляции слишком
примитивен для таких сложных процессов, как
метаболизм жиров или углеводов, где далеко не просто даже
назвать конечный продукт. Тем не менее он позволяет
регулировать метаболически разнесенные
ферментативные реакции ключевыми интермедиатами (продуктами
промежуточных реакций).
глава 12 Регуляция метаболизма углеводов и жиров
157
В ряде случаев регуляция осуществляется не
конечным продуктом (по принципу отрицательной обратной
связи), а исходным соединением (положительная прямая
связь), которое активирует ферменты, участвующие в его
превращении в данном метаболическом пути.
Природа регуляторных ферментов
Известны два основных способа регуляции
каталитической активности ферментов (изменение их количества
не рассматривается).
1. Аллостерическая регуляция.
2. Ковалентная модификация белка - обычно фос-
форилирование и дефосфорилирование.
Рассмотрим оба эти способа.
Аллостерическая регуляция
ферментов
Аллостерическая регуляция ферментов особенно важна
при управлении метаболизмом. Приставка алло-
означает другой. Она подразумевает, что на ферменте кроме
участка связывания субстрата есть по меньшей мере еще
один (их может быть несколько) участок связывания
другого соединения (или соединений).
Низкомолекулярные соединения, связываемые белком, принято называть
лигандами; этот термин часто распространяют и на
небольшие белки. Лиганды, связывающиеся с аллостери-
ческими участками, называют аллостерическими
эффекторами, или аллостерическими регуляторами.
Они обычно не похожи на субстрат. Действие
эффекторов, как правило, наблюдают при определенной
концентрации субстрата. Положительные эффекторы при
связывании с аллостерическим участком фермента
увеличивают его активность, отрицательные - ее
понижают. Иными словами, аллостерические эффекторы
можно подразделить на активаторы и ингибиторы.
В некоторых случаях аллостерические ингибиторы
снижают скорость ферментативной реакции, уменьшая
Vmax. Чаще, однако, их действие проявляется в
уменьшении сродства фермента к субстрату. Выше
отмечалось, что ферменты в клетке обычно работают, когда
концентрация субстратов ниже насыщающей. Поэтому
уменьшение сродства субстрата к ферменту приводит
к снижению активности последнего, а увеличение
сродства, наоборот, к его активации. При насыщающих
фермент концентрациях субстрата аллостерические
эффекторы не изменяют Утах, даже если сродство фермента
к субстрату изменяется. Однако в реальных условиях,
часть 3 Метаболизм
158
когда концентрации субстратов в клетке по большей
части далеки от насыщающих, эффекторы могут влиять
на величину скорости реакции.
Механизм аллостерической регуляции
ферментов
Мы обсудим сейчас природу аллостерических
ферментов, для которых связывание эффектора отражается
на сродстве фермента к субстрату (это основной тип
аллостерической регуляции ферментов). Такие
аллостерические ферменты состоят из нескольких каталитически
активных белков, которые посредством нековалентных
связей объединены в единый ферментный комплекс.
Компоненты такого комплекса называют белковыми
субъединицами, протомерами или мономерами. Хотя
каждая из субъединиц обладает каталитической
активностью, общая активность фермента зависит от их
взаимодействия друг с другом. Типичные кривые
зависимости скорости ферментативной реакции от
концентрации субстрата представлены на рис. 12.4. Аллостери-
ческий фермент отличается от «классического» тем, что
его кривая имеет не гиперболическую, а сигмоидную
форму (ее также называют S-образной). Что из этого
следует и почему так получается?
Ответим на первый вопрос. Когда аллостерический
активатор связывается ферментом, сигмоидная кривая
сдвигается влево, тогда как аллостерический ингибитор
сдвигает ее вправо (рис. 12.5). Сдвиг кривой влево
V l
v max χ- .
t* я
о< 5 /ι
^ α /ι
/fM K{) ^ Концентрация субстрата [S]
Рис. 12.4. Влияние концентрации субстрата на скорость
реакции (К), катализируемой типичным аллостерическим
ферментом
Пунктирной линией для сравнения показана соответствующая
кривая для «классического» фермента, подчиняющегося
кинетике Михаэлиса-Ментен. Поскольку понятие Км не имеет
физического смысла вне модели Михаэлиса-Ментен, здесь
используется обозначение К{) 5
свидетельствует об увеличении сродства фермента к
субстрату, а сдвиг вправо - о его уменьшении. Сигмоидная
форма кривой, отражающей зависимость скорости
ферментативной реакции от концентрации субстрата,
указывает на то, что скорость реакции более чувствительна
к изменению концентрации субстрата, чем при
гиперболической зависимости, особенно в области скоростей,
близких к 0,5 Vmax.
Почему зависимость скорости реакции от
концентрации субстрата приобретает сигмоидную форму?
Взаимодействие одной из субъединиц аллостерического
фермента с субстратом способствует лучшему
связыванию молекул субстрата другими субьединицами.
Такой эффект называют гомотропным кооперативным
связыванием субстрата. Термин гомотропный
отражает то обстоятельство, что все субьединицы связывают
один и тот же лиганд - субстрат. Поскольку в данном
случае сигмоидная форма кривой обусловлена только
связыванием субстрата, такую регуляцию не совсем
верно интерпретировать как результат аллостерического
конформационного изменения (предполагающего
взаимодействие эффектора с участком, отличным от
каталитического). Однако кооперативность часто относят
к аллостерическим эффектам несмотря на то, что
участки связывания лиганда и субстрата идентичны.
По-видимому, кооперативное взаимодействие можно считать
аллостерическим, так как связывание субстрата каждой
из субъединиц зависит от присутствия лиганда на
соседней субъединице.
0,5VWi
Аллостерическии
активатор
Аллостерическим
ингибитор
^0,5" ^0,5' К из"
Концентрация субстрата [S]
Рис. 12.5. Влияние концентрации субстрата на скорость
реакции (V), катализируемой типичным аллостерическим
ферментом
Vv V2 и Уъ- скорости реакции соответственно без лиганда,
в присутствии аллостерического активатора и
аллостерического ингибитора при фиксированной концентрации субстра-
xaSitfJ
Каков механизм взаимного влияния участков
связывания различных субъединиц? Предложены две
теоретические модели, согласно которым участок связывания
субстрата может находиться в двух состояниях,
различающихся по сродству к субстрату. Назовем их
состояниями с высоким и низким сродством. Предполагается,
что связывание субстрата увеличивает долю субъединиц
с высоким сродством к субстрату. Обе модели
различаются деталями распространения взаимовлияния
субъединиц в пределах единого белкового комплекса.
В рамках согласованной модели, предложенной
Моно, Уайменом и Шанже (рис. 12.6), все субъединицы
могут одновременно находится либо в состоянии с
высоким сродством, либо с низким. Обе формы белкового
комплекса при этом находятся в динамическом
равновесии, которое сдвинуто в сторону белка с низким
сродством субъединиц. Связывание субстрата с любой
субъединицей сдвигает это равновесие в противоположную
сторону - к образованию белка с высоким сродством.
Таким образом, с ростом концентрации субстрата все
большее число молекул фермента переходит в
состояние с высоким сродством.
Заметим, что все сказанное относится к
кооперативному связыванию субстрата, а не к аллостерическим
модификаторам или эффекторам, регулирующим
фермент. Это особый вопрос. Согласно данной модели,
аллостерические регуляторы изменяют положение
равновесия между состояниями молекулы с низким
сродством (его обозначают буквой Т, от англ. tense -
напряжение, т. е. напряженное состояние) и высоким
сродством (обозначаемым буквой R, от англ. relaxed, т. е.
расслабленное состояние). Если аллостерическии
регулятор более прочно связывается с ферментом в
состоянии R, он будет смещать равновесие T<->R вправо.
Спонтанное
равновесие
сильно
сдвинуто влево
Рис. 12.6. Модель «согласованного» кооперативного
связывания субстрата
Согласно этой модели, фермент существует в двух
состояниях - Τ и R, находящихся в равновесии, которое, однако, в
отсутствие субстрата сдвинуто в сторону формы Т. В случае
связывания молекул субстрата с формой R равновесие
сдвигается вправо, что увеличивает сродство к субстрату всех
субъединиц в этой молекуле. Обозначения Η (High) и L (Low)
указывают на сродство фермента к субстрату
глава 12 Регуляция метаболизма углеводов и жиров
159
Это приведет к активации фермента, так как большее
число его молекул при данной концентрации субстрата
будет в форме R. По мере увеличения концентрации
положительного аллостерического регулятора все больше
молекул фермента перейдет в состояние R (с высоким
сродством), а кривая зависимости скорости реакции
от концентрации субстрата станет гиперболической.
Обратная ситуация возникает, когда регулятор
отрицательного типа более прочно связывается с молекулами
фермента в состоянии Τ и переводит другие молекулы
в это же состояние, уменьшая скорость ферментативной
реакции в целом.
Последовательную модель разработали Кошланд,
Немети и Филмер. Они предположили, что в отсутствие
субстрата все молекулы фермента находятся в
состоянии Τ с низким сродством. Равновесия между
состояниями Τ и R нет (поскольку нет состояния R).
Связывание молекулы субстрата с одной субьединицей
вызывает изменение ее конформации (от Τ к R), а также
конформации соседней субъединицы, увеличивая ее
сродство к молекуле субстрата, которому теперь легче с
ней связаться. Связывание второй молекулы субстрата
будет способствовать переходу третьей субъединицы
в состояние R, и этот процесс будет повторяться до тех
пор, пока все субъединицы фермента не перейдут в
состояние R с высоким сродством (рис. 12.7).
Обе модели удовлетворительно объясняют
наблюдаемые эффекты, их нельзя рассматривать как
взаимоисключающие. Истинный механизм аллостерической
регуляции вполне может оказаться промежуточным.
Некоторые ферменты, например глицеральдегид-
3-фосфатдегидрогеназа (см. с. 112), проявляют
^оперативность при связывании субстрата, однако для них
неизвестны аллостерические регуляторы. Такие
ферменты более чувствительны к изменениям концентрации
субстратов, чем классические, и, возможно, именно это
служит причиной кооперативного связывания. В
дальнейшем мы встретимся с более сложным вариантом
аллостерической регуляции, когда участок связывания
регулятора расположен на отдельной регуляторнои
субъединице, лишенной каталитических свойств.
Связывание аллостерического регулятора с этим участком
приводит к тому, что регуляторная субъединица покидает
комплекс, а каталитические субъединицы изменяют
сродство к субстрату. Такое управление ферментативной
реакцией может показаться чрезмерно громоздким,
но эволюция прежде всего заботится о надежности и
работоспособности системы.
Обратимость
аллостерической регуляции
Наиболее важной особенностью аллостерической
регуляции является ее мгновенное действие. Аллостеричес-
кий лиганд практически мгновенно связывается с регуля-
торным центром за счет нековалентных взаимодействий.
Регулятор диссоциирует из комплекса с ферментом,
когда его концентрация в среде уменьшается; при этом
фермент возвращается в исходное состояние.
Аллостерическая регуляция -
метаболическая концепция
огромной мощности
Существенным в аллостерической регуляции
является тот факт, что эффектор по своему строению может
не иметь ничего общего ни с субстратом данного
фермента, ни с любым другим веществом, образующимся
в процессе метаболических превращений. Это
означает, что любой метаболический путь может быть регуля-
торно связан с любым другим. Метаболит(ы) одного
из каскадов биохимических превращений может(гут)
быть регулятором(ами) другого. Более того, фермент
может управляться не одним, а несколькими
регуляторами, каждому из которых отведен специфический
участок связывания, и получать таким образом регулятор-
ные сигналы от нескольких метаболических путей, что
увеличивает гибкость регуляции.
+S
S S S S
+S +S S +s
S S
S S
Рис. 12.7. Модель «последовательного» кооперативного связывания субстрата аллостерическим ферментом
Связывание одной молекулы субстрата с субъединицей вызывает конформационное изменение последней. Это облегчает кон-
формационную перестройку второй субъединицы при взаимодействии её со следующей молекулой субстрата и т. д. Суммарный
эффект заключается в изменении общей конформации белка
часть 3 Метаболизм
160
Мы уже познакомились с тем, как сложны и
взаимозависимы метаболические пути, так или иначе связанные
с производством и хранением энергии. В такой сложной
системе каждый отдел должен «знать», как дела у
«соседей», оценивая уровень ключевых метаболитов.
Достаточно АТР или нет? Поставляется ли циклу лимонной
кислоты нужное количество ацетил-СоА? Протекает ли
гликолиз слишком быстро или недопустимо медленно?
Представьте, какой химический хаос воцарится, если
каждый метаболический путь не будет ежесекундно
подстраиваться к текущей ситуации, исходя из информации,
поступающей от других метаболических путей или
различных участков собственного пути! Принцип аллостери-
ческой регуляции открыл для эволюции возможность
создавать множество информационных каналов, которые
позволяют ферментам получать сигналы из любой точки
на метаболической карте. Как заметил Моно, один из
первооткрывателей аллостерической регуляции, без нее
просто невозможно существование столь сложной
системы, как клетка. Он называл аллостерическую регуляцию
«вторым секретом жизни» (первый - ДНК).
Роль фосфорилирования в регуляции
ферментативной активности
Теперь приступим к рассмотрению второго метода
регуляции ферментов - фосфорилирования. Строго
говоря, следовало бы посвятить этот раздел более общей
проблеме регуляции за счет ковалентной модификации
ферментов. Однако фосфорилирование имеет
настолько важное значение, что обсуждение этого процесса
отдельно вполне оправданно.
Принцип предельно прост. Существуют ферменты,
называемые протеинкиназами, которые переносят
остаток фосфата с АТР на соответствующий белок. При
этом изменяется конформация фосфорилированного
белка и, как следствие, его каталитические свойства.
Мишенью для фосфорилирования может быть не
только фермент, но и регуляторный белок - ингибитор или
активатор фермента. В этом случае фосфорилирование
влияет на его регуляторные свойства. Нет ничего
удивительного в том, что введение сильно заряженной
группы отражается на структуре белковой молекулы. С
другой стороны, фосфатные остатки легко отщепляются
протеинфосфатазами (рис. 12.8).
Фосфорилированию подвергаются гидроксильные
группы отдельных остатков серина и треонина в
полипептидной цепи фермента, которые киназа различает
по их аминокислотному окружению (в главе 26 описано
фосфорилирование тирозина, в данном разделе мы
этого вопроса касаться не будем).
е ос рилированный
dbe мент
Фо
-о-®
АТР
Про инкиназ'
ADP
Фосфорилированный
фермент
Рис. 12.8. Регуляция активности фермента посредством его
фосфорилирования
В разных случаях фосфорилированный фермент может быть
либо более, либо менее активным, чем нефосфорилированный.
Возможна также регуляция фермента с помощью ингиби-
торного белка, активность которого меняется при его фосфо-
рилировании
На схеме показана гидроксильная группа серина,
подвергающаяся фосфорилированию:
ι ι Я
СН—СН2ОН + АТР СН—СН2—О—Р-СГ + ADP.
I II
со со ο-
Ι I
Остаток серина
в полипептиде
Итак, мы познакомились с двумя основными
механизмами, регулирующими активность ферментов -
аллостерической регуляцией и обратимым фосфорилиро-
ванием. Теперь следует рассмотреть, как эти механизмы
используются для регуляции метаболизма.
Регуляция отдельных
метаболических путей
Два типа управления - внутреннее
и внешнее, или внеклеточное
Аллостерическая регуляция ферментов осуществляется
при непосредственном участии фермента и субстрата,
тогда как при регуляции с помощью фосфорилирования
активность фермента-мишени зависит от соотношения
киназ и фосфатаз. Чем же определяется это
соотношение? В большинстве случаев (хотя и не всегда) оно
определяется гормональным сигналом, поступающим с
наружной стороны клетки.
глава 12 Регуляция метаболизма углеводов и жиров
161
Управление метаболизмом - сложный комплексный
процесс. Чтобы сделать его рассмотрение более
доступным, разделим материал на два обширных раздела.
Сначала мы расскажем о внутриклеточной регуляции,
которая координирует взаимодействие метаболических
путей и отдельных их участков внутри отдельной
клетки, чтобы избежать дефицита или перепроизводства
необходимых веществ. На этом уровне регуляторными
сигналами служат сами метаболиты, а управление
обычно (хотя далеко не всегда) осуществляется посредством
аллостерической регуляции. В одних случаях при этом
используется принцип обратной связи, который,
например, не позволяет гликолизу производить больше пиру-
вата, чем его способен переработать цикл лимонной
кислоты. В других случаях, напротив, регуляция по
принципу положительной связи позволяет удаленным
по цепи превращений метаболическим звеньям
справиться с нарастающим потоком субстратов.
Полностью изолированная клетка могла бы
довольствоваться внутренним управлением метаболическими
процессами. Однако в организме функционируют
системы внешнего управления жизнедеятельностью клеток
(разумеется, внешнего в отношении клеток, но не
организма). Сигналы поступают к клеткам от гормонов или
нейромедиаторов, заставляя их подчинять основные
метаболические превращения интересам всего организма,
например запасать или сжигать топливо. Такая регуляция
особенно необходима, если работа клетки важна для
организма в целом. Клетки не могут сами принять решение
о том, что нужно организму. Они должны получить
сигнал извне, и лишь после этого внутриклеточная
регуляция включится в поддержание необходимых процессов.
Основные моменты
внутренней регуляции метаболизма
углеводов и липидов
Внутриклеточная регуляция
метаболизма гликогена
Метаболизм гликогена включает синтез этого полимера
гликогенсинтазой и его расщепление гликогенфосфори-
лазой. Гликоген синтезируется в период хорошего
питания, и этот синтез регулируется внешними сигналами
(см. с. 169). Расщепление гликогена преследует две
физиологические цели. Общей для всех тканей задачей
является поставка гликолизу исходного материала - глю-
козо-6-фосфата. Именно в него с помощью фосфоглю-
комутазы превращается первичный продукт распада
гликогена - глюкозо-1-фосфат, а гликогенфосфорилаза
часть 3 Метаболизм
162
(фермент, расщепляющий гликоген) занимает ключевое
место в метаболизме. Энергия, образующаяся в процессе
гликолиза в виде АТР, необходима для всех тканей,
но особое значение она имеет для мышц, сокращение
которых сопровождается интенсивным потреблением
АТР (см. главу 28). В печени распад гликогена
происходит с целью поддержать на должном уровне
концентрацию глюкозы в крови.
Механизмы регуляции гликогенфосфорилазы,
которая является примером «классического» регуляторного
фермента, подробно изучены (это особенно касается
фермента из мышц). В покоящейся мышце она
находится в виде неактивной фосфорилазы Ь, которая
частично активируется AMP, действующим в качестве ал-
лостерического эффектора (максимальную активацию
вызывают внешние сигналы). Почему именно AMP
выступает в данном случае в роли аллостерического
сигнала? Гликогенфосфорилаза должна быть
активирована, когда резерв энергии в клетке достаточно мал, т. е.
в клетке мало АТР. Поскольку аденилаткиназа
катализирует реакцию:
2ADP<->AMP + ATP,
AMP служит индикатором накопления ADP, который
образуется при гидролизе АТР и, следовательно,
отражает уменьшение концентрации последнего. Расчеты
показывают, что даже незначительный расход АТР
приводит к относительно большому увеличению
концентрации AMP, активирующего гликогенфосфорилазу,
чтобы обеспечить производство дополнительной энергии.
С другой стороны, АТР и глюкозо-6-фосфат являются ал-
лостерическими ингибиторами гликогенфосфорилазы
(рис. 12.9). Если их достаточно, зачем производить еще?
Простая внутриклеточная регуляция контролируется
извне и зависит от физиологического режима всего
организма (см. с. 169).
В печени ситуация та же: AMP активирует фосфо-
рилазу Ъ до -20% от максимального уровня, а внешние
сигналы имеют приоритет над внутренними, хотя здесь
они преследуют достижение иных физиологических
целей (см. с. 169).
Итак, и в мышцах, и в печени в отсутствие
внеклеточных регуляторов (о них мы поговорим чуть позже)
фосфорилаза находится в неактивной форме Ъ. AMP
акивирует фермент, а АТР и глюкозо-6-фосфат ингиби-
руют активность фосфорилазы.
Гликолиз и глюконеогенез
Из общей схемы регуляторных путей, изображенных
на рис. 12.9, следует, что:
• рост концентрации AMP указывает на увеличение
отношения ADP/ATP;
• AMP активирует гликогенфосфорилазу и фосфоф-
руктокиназу;
• AMP ингибирует фруктозо-1,6-дифосфатазу;
• активация распада гликогена сопровождается
увеличением уровня фруктозо-6-фосфата;
• фруктозо-6-фосфат активирует фосфофруктокиназу;
• активация фосфофруктокиназы приводит к
увеличению концентрации фруктозо-1,6-дифосфата;
• фруктозо-1,6-дифосфат активирует пируваткиназу.
Из перечисленного видно, что система аллостеричес-
кой регуляции гликолиза включает как отрицательные
и положительные обратные связи, так и положительные
прямые связи. Эволюция редко ограничивается каким-то
одним способом регуляции. Так, в данной системе фос-
фофруктокиназа активируется AMP, но ингибируется
АТР. Это позволяет гликолизу точно реагировать на
изменения соотношения ATP/ADP (через концентрацию
AMP).
С ростом уровня АТР замедляется цикл лимонной
кислоты и происходит накопление цитрата. Последний
выходит из митохондрий в цитоплазму и аллостери-
чески ингибирует фосфофруктокиназу. Таким способом
процесс гликолиза приостанавливается. Та же цель
достигается и другим путем: ацетил-СоА аллостерически
ингибирует пируваткиназу.
Особо следует остановиться на активации пируват-
карбоксилазы посредством ацетил-СоА, в результате
чего образуется оксалоацетат (см. рис. 12.9). Накопление
ацетил-СоА происходит при торможении цикла
лимонной кислоты (вследствие дефицита оксалоацетата).
Вполне естественно, что при этом автоматически
запускается анаплеротическая реакция (см. с. 122). Эта регу-
ляторная связь существенна также для глюконеогенеза.
Внутриклеточная регуляция
пируватдегидрогеназы,
цикла лимонной кислоты
и окислительного фосфорилирования
Пируватдегидрогеназа (см. с. 116) занимает
стратегически важное положение в метаболизме, катализируя
необратимую реакцию превращения пирувата в аце-
тил-СоА, который поступает в цикл лимонной кислоты,
а также используется при синтезе жиров. Разные
способы регуляции этого фермента показаны на рис. 12.10.
Оба его продукта - ацетил-СоА и NADH, являются ал-
лостерическими ингибиторами фермента, а субстраты -
CoA-SH и NAD+ - активаторами. Таким образом,
активность пируватдегидрогеназы определяется
соотношениями ацетил-СоА/СоА и NADH/NAD+. Логика
Гликоген
LJDPG
Глюкозо-1 -фосфат *
1 ликогенфосфс илаз
+ AMP
- АТР
Глюкозо-6-фосфат^
Фруктозо-6-фосфат
Фруктозо-1 6
дифосфатаза
AMP -
) ос о τ
+ ^
+ AMP
- - АТР
- -« Цитрат
Фруктозо-1,6-дифосфатч
Оксалоацетат
г Фосфоенолпируват
Пи ваткиназа
- - АТР
- -· Ацетил-СоА
Шруваткар- \ Пируват
юксилаза |
Ацетил-СоА
Рис. 12.9. Главные пути аллостерической регуляции
метаболизма гликогена, гликолиза и глюконеогенеза
Цветом выделены активирующие звенья регуляции, а
пунктиром - ингибирующие. UDPG - уридиндифосфатглюкоза
очевидна. Если существует значительный избыток аце-
тил-СоА и NADH, значит их образование с помощью
пируватдегидрогеназы должно быть приостановлено.
И наоборот, если высока концентрация CoA-SH и NAD+,
то фермент должен активно работать.
Особое значение имеет отрицательная регуляция
фермента высокими концентрациями АТР. Накопление АТР
свидетельствует о необходимости замедлить его
производство. Регуляция активности фермента с помощью
АТР - не прямой процесс. При увеличении молярного
отношения ATP/ADP активируется протеинкиназа
пируватдегидрогеназы, которая фосфорилирует фермент, тем
самым ингибируя его (напомним, что фосфорилированию
всегда противостоит дефосфорилирование,
катализируемое фосфатазами) (см. рис. 12.10). Киназа входит в
состав пируватдегидрогеназного комплекса. Помимо АТР
она активируется также ацетил-СоА и NADH. Все эти ре-
гуляторные эффекты сводятся к одному: прикрыть
«топливный кран», если вырабатывается избыток энергии.
глава 12 Регуляция метаболизма углеводов и жиров
163
Прямая аллостерическая регуляция ПДГ:
ингибирование продуктами (ацетил-СоА, NADH)
Регуляция
фосфатазы
(Са2+-зависимая)
Н70
Активная]
пдг У_он
Пируват Ацетил -СоАл
+ C0A-SH +NADH
+ NAD+
Неактивная ПДГ" _0-Гр)
АТР
иротеинкиназа
Регуляция протеинкиназы:
актвация продуктами ПДГ
(ацетил-СоА, NADH, АТР),
ингибирование субстратами
ПДГ (пируват. CoA-SH, NAD+)
ADP
Рис. 12.10. Аллостерическая регуляция и обратимое фосфорилирование как способы управления активностью пируват-
дегидрогеназного ферментного комплекса млекопитающих (ПДГ)
Множественность способов контроля обусловлена стратегическим положением этого комплекса в общем метаболизме, так
как ацетил-СоА далее поступает в цикл лимонной кислоты и используется для синтеза жиров. Все способы регуляции
фактически призваны обеспечить ингибирование ПДГ продуктами его активности и активацию субстратами (АТР можно
рассматривать как отдаленный продукт превращения ацетил-СоА в цикле лимонной кислоты). Регуляция посредством обратимого фос-
форилирования обычно сопряжена с гормональной регуляцией, а ионы кальция, стимулирующие активность фосфатазы,
накапливаются под действием катехоламинов
Аллостерическая регуляция ферментов
осуществляется также и в цикле лимонной кислоты, и в цепи
переноса электронов. Однако в этих системах,
расположенных в митохондриях, основные регуляторные
механизмы связаны с изменениями концентрации таких
ключевых субстратов, как NAD+ и ADP. Если большая часть
NAD представлена восстановленной формой NADH,
активность дегидрогеназ в цикле лимонной кислоты
снижается. Поскольку NADH накапливается в том
случае, когда не успевает окислиться в цепи переноса
электронов (например, из-за недостатка кислорода),
ингибирование некоторых реакций цикла лимонной кислоты
можно рассматривать как регуляторный ответ. Точно
также при малом молярном соотношении ADP/ATP
тормозится перенос электронов по митохондриальной
цепи, поскольку процессы фосфорилирования и
окисления в митохондриях тесно взаимосвязаны. Эта
взаимосвязь носит специальное название - дыхательный
контроль - и имеет огромное значение.
Наряду с NAD+ и ADP цикл лимонной кислоты
регулируется на уровне цитратсинтазы (ингибируется АТР),
изоцитратдегидрогеназы (ингибируется АТР,
активируется ADP) и сх-кетоглутаратдегидрогеназы
(ингибируется NADH и сукцинил-СоА).
Все эти регуляторные звенья вполне логичны и
помогают поддерживать баланс метаболитов в цикле
лимонной кислоты, чтобы избежать их избытка или
дефицита.
Внутриклеточная регуляция процессов
окисления и синтеза жирных кислот
Схема регуляции этих процессов представлена на
рис. 12.11. Смысл ее в том, чтобы избежать
одновременного протекания в одной клетке окисления и синтеза
жирных кислот.
Эти два метаболических пути всячески подавляют
друг друга. Исходные вещества окисления - СоА-про-
изводные жирных кислот (см. с. 132) - аллостерически
ингибируют первый фермент синтеза жирных кислот -
ацетил-СоА-карбоксилазу. Напротив, ключевой
метаболит системы синтеза жиров - малонил-СоА (см. с. 138) —
аллостерически ингибирует перенос ацильных остатков
на карнитин (см. с. 133). Из-за этого жирнокислотные
остатки не могут попасть внутрь митохондрий и
подвергнуться там окислению: если вы синтезируете жиры,
вряд ли стоит одновременно сжигать их в
митохондриальной топке. Ацетил-СоА-карбоксилаза in vitro
активируется цитратом, превращение которого приводит
к образованию ацетил-СоА - субстрата этого же
фермента (см. с. 141). Цитрат выходит из митохондрий лишь
в том случае, если его там много, т. е. при изобилии
пищи, когда самое время накапливать жиры. Нет
сомнений, что в пробирке цитрат активирует ацетил-СоА-кар-
боксилазу, однако пока не установлено, имеет ли место
такая активация в клетке. В то же время бесспорно, что
часть 3 Метаболизм
164
Нейтральные жиры Нейтральные жиры
Синтез Τ J Окисление
Свободная жирная кислота
I
Малонил-СоА -..
АМР-
-V --- Ацил-СоА
Ацетил-СоА
Ацилкарнитин
Цитрат в цитоплазме
Цитоплазма
Цитрат в цикле цил~ ° Матрикс
лимонной кислоты
* Митохондриаль-
Окисление ная мембрана
Рис. 12.11. Основные места внутриклеточной регуляции
при окислении и синтезе жиров
Поскольку при окислении жиров образуется ацетил-СоА,
скорость его последующих превращений зависит от регуляции
реакций цикла лимонной кислоты и т. д. Пунктиром показаны
аллостерические воздействия. Цитрат активирует аце-
тил-СоА-карбоксилазу in vitro, однако пока не ясно, имеет ли
это какое-то физиологическое значение
ацетил-СоА-карбоксилаза ингибируется при фосфори-
лировании. Оно осуществляется протеинкиназой,
активность которой зависит от присутствия AMP, тогда как
обратная фосфатазная реакция контролируется на
гормональном уровне (см. с. 176).
Все, что было сказано выше о внутриклеточной
регуляции метаболизма углеводов и жиров, разумеется,
не претендует на исчерпывающее изложение
накопленных сведений. Но этих данных более чем достаточно
для понимания того, что метаболизм не является
суммой последовательно протекающих реакций. Это
сложный, вполне упорядоченный процесс, в котором все
метаболические пути тесно взаимосвязаны между собой.
Чтобы понять, как клеточный метаболизм
согласуется с физиологическими потребностями организма
в целом, необходимо уяснить внеклеточную регуляцию
гормонами и другими соединениями. Они часто
контролируют те же реакции, что и внутриклеточные
соединения, поэтому некоторые реакции подвергаются как
внутри-, так и внеклеточной регуляции.
Регуляция метаболизма углеводов
и жиров внеклеточными агентами
Особую роль среди внеклеточных агентов, участвующих
в регуляции углеводного и жирового обмена, играют
гормоны глюкагон, инсулин, адреналин и норадрена-
лин. Гормоны действуют мгновенно, вызывая подчас
сильный эффект. Они помогают организму
приспособиться к периодическим чередованиям состояний
насыщения и голода (см. с. 84).
В целом гормональная регуляция чрезвычайно
сложна. Гормоны гипофиза, надпочечников, щитовидной
железы и др. (см. табл. 26.1) воздействуют на
метаболизм далеко не всегда понятным образом. Обычно их
действие опосредовано множеством событий и
растянуто во времени. Например, при избытке тироксина люди
постепенно худеют и становятся легко возбудимыми,
а его недостаток вызывает обратные и тоже медленно
развивающиеся эффекты. Однако есть гормоны,
которые проявляют себя направленно и быстро.
Как вы помните, глюкагон - это «гормон голода»,
который вырабатывается поджелудочной железой в
ответ на снижение уровня глюкозы в крови. И наоборот,
при повышении содержания глюкозы выделяется
инсулин (см. с. 84). Адреналин и норадреналин образуются
в мозговом слое надпочечников и вызывают активацию
процессов, связанных с превращением пищевых
веществ. Норадреналин выделяется также
симпатическими нервными окончаниями. Симпатическая нервная
система управляет непроизвольными сокращениями
гладких мышц внутренних органов, а также иннер-
вирует жировую ткань. Ее можно рассматривать как
способ быстрой доставки дозированного количества
гормона непосредственно к регулируемым клеткам
и органам.
Как регулируется содержание в крови
инсулина, глюкагона и адреналина?
Инсулин - небольшой белок, синтезируемый и секрети-
руемый в кровь β-клетками островков Лангерганса
поджелудочной железы. Время жизни инсулина в крови
невелико, поэтому его действие затухает, как только
прекращается секреция. β-Клетки очень чувствительны
к изменениям концентрации глюкозы и начинают секре-
тировать инсулин не позже чем через минуту после
увеличения содержания глюкозы в крови в результате
приема пищи. Важно отметить, что сродство глюкозы
к транспортному белку, обеспечивающему ее
поступление в клетки, очень мало, поэтому белок начинает
функционировать лишь после того, как содержание
глюкозы в крови превысит нормальный уровень - 90 мг
глава 12 Регуляция метаболизма углеводов и жиров
165
в 100 мл, или 5 мМ. Зависимость скорости секреции
инсулина от концентрации глюкозы описывается сигмо-
идной кривой; отсюда высокая чувствительность
ответа на изменения ее содержания.
Глюкагон, напротив, секретируется поджелудочной
железой при снижении концентрации глюкозы в
крови. Адреналин же выделяется надпочечниками в ответ
на нервный сигнал.
Как работают гормоны
глюкагон, адреналин и инсулин?
Гормоны представлены веществами различной
химической природы, поступающими в кровь. Они
передают химические сигналы и доступны всем тканям.
Избирательность их действия достигается тем, что не все
клетки, а лишь клетки-мишени воспринимают эти
сигналы.
Жирорастворимые гормоны (стероиды, тироксин)
беспрепятственно проникают через плазматическую
мембрану, тогда как водорастворимые гормоны
(глюкагон, адреналин, инсулин) через нее пройти не могут. Их
молекулы связываются рецепторными белками,
специфичными для каждого гормона и расположенными
на поверхности клеток. Избирательность связывания
гормона с рецепторным белком имеет ту же природу, что
и специфичность взаимодействия фермента с
субстратами. Только клетки, предназначенные быть мишенью
для гормона, имеют «настроенные» на него рецепторы.
Остальные клетки присутствия гормона не ощущают,
они как бы слепы по отношению к нему.
Гормоны быстро выводятся из крови, поэтому как
только железа перестает выделять гормон, его
концентрация падает, гормон-рецепторные комплексы
диссоциируют и сигнал исчезает. Однако, пока гормон связан
с рецептором, его присутствие вызывает определенные
химические изменения внутри клетки, которые и
составляют гормональный ответ (рис. 12.12).
Клеточный рецептор
Гормон
Рис. 12.12. Гормон, инициирующий химические реакции
в цитоплазме, связывается рецептором на поверхности
клетки
В основе современных представлений о природе
гормонального ответа лежит концепция вторичного
мессенджера (англ. messenger - гонец, посредник,
посланец).
Что такое вторичный мессенджер?
Гормон можно рассматривать как первичный
мессенджер: связываясь со своим клеточным рецептором, он
передает гуморальный сигнал клетке. При этом
изменяется уровень вторичного мессенджера -
внутриклеточной молекулы, запускающей в клетке те или иные
химические превращения в ответ на связывание
гормона с рецептором.
Что служит вторичным мессенджером
для глюкагона, адреналина
и норадреналина?
В случае глюкагона и адреналина вторичным
мессенджером служит циклический аденозин-3',5'-монофосфат,
или циклический AMP (cAMP), который синтезируется
из АТР ферментом аденилатциклазой (рис. 12.13).
В свою очередь, с AMP аллостерически активирует
сАМР-зависимую протеинкиназу, которая фосфори-
лирует гидроксильные группы серина или треонина
белков-мишеней, изменяя активность последних. Каскад
регуляторных реакций, передающих гормональный
сигнал, представлен на рис. 12.14.
Механизм, посредством которого сАМР
активирует протеинкиназу, показан на рис. 12.15. В отсутствие
сАМР киназа представляет собой тетрамер,
образованный двумя каталитическими и двумя регуляторными
субъединицами. В составе тетрамера каталитические
субъединицы неактивны. Когда сАМР связывается с
регуляторными субъединицами, тетрамер распадается
на мономеры с высвобождением каталитически
активных субъединиц.
Фермент сАМР-фосфодиэстераза гидролизует
сАМР до AMP (рис. 12.16). Поэтому активация проте-
инкиназы зависит от постоянного образования сАМР,
которое прекращается, как только гормон покидает
рецептор. Как уже упоминалось, гормоны в крови тоже
постоянно разрушаются или удаляются, так что их
присутствие в ней зависит от секреции. Таким образом,
каждый этап передачи гормонального сигнала
включает механизм его торможения. Когда уровень гормона
падает, образование сАМР прекращается, фосфорили-
рованные белки дефосфорилируются протеинфосфата-
зами и все возвращается в исходное состояние.
В этой главе мы не обсуждаем, как гормон запускает
синтез с AMP и как этот синтез прекращается, когда
гормон покидает рецептор. Вопросы передачи сигналов
часть 3 Метаболизм
166
NH7
NH2
А енила иклаза
0 0 0
II II II
ο — ρ—ο—ρ—ο—ρ—о—сн2
ill н н
οο-ο- . Η Η
Η Η
он он
Τ"
ΡΡ, О—СН2
Ι η η ι
й 3. η
ο=ρ — о он
ο
Аденозинтрифосфат (АТР)
Рис. 12.13. Синтез циклического AMP из АТР, катализируемый аденилатциклазой
Циклический аденозин-3\5'-монофосфат
(с AMP)
Гормон
(адреналин, глюкагон)
Связывание с
рецептором клетки-мишени
ι
Увеличение концентрации
сАМР внутри клетки
г
R
R
С
Неактивная
протеинкиназа А
R
R
сАМР
Регуляторные субъединицы,
связавшие сАМР
с
сАМР активирует
протеинкиназу
Протеинкиназа фосфорилирует
специфические белки
\\\
Фосфорилирование белков приводит к
изменению их ферментативной активности
и метаболическому ответу
Рис. 12.14. Этапы гормональной регуляции метаболизма
Здесь не показано, что с AM Ρ постоянно разрушается и дефос-
форилирование белков осуществляется фосфатазами.
Метаболический ответ поддерживается до тех пор, пока гормон
связан с рецептором
через клеточные мембраны детально рассматриваются
в главе 26. Пока достаточно понять, что связывание глю-
кагона и адреналина клеточными рецепторами приводит
к увеличению внутриклеточной концентрации сАМР
Управление числом рецепторов
Число специфических рецепторов на поверхности
клетки может регулироваться. Продолжительный контакт
клетки с агонистом (так называют любое соединение,
которое связывается с рецептором и активизирует его) может
привести к обратимому уменьшению числа рецепторов
Активные каталитические
субъединицы протеинкиназы А
Рис. 12.15. Активация сАМР-зависимой протеинкиназы
под действием сАМР
R и С - соответственно регуляторные и каталитические
субъединицы протеинкиназы
и к снижению чувствительности клетки к данному аго-
нисту, а следовательно, к более слабому ответу. Это
явление называют даунрегуляцией (англ. downregulation).
Кроме того, рецептор может быть инактивирован в
результате его фосфорилирования цитоплазматической
киназой, после чего связывание его с гормоном не
будет приводить к синтезу сАМР. Поскольку такого рода
фосфорилирование стимулируется сАМР, интенсивность
клеточного ответа регулируется им по принципу
отрицательной обратной связи.
Все эти виды регуляции имеют место в случае
рецепторов адреналина, инсулина и глюкагона.
Как инсулин управляет
метаболизмом?
Инсулин влияет на энергетический метаболизм
диаметрально противоположно глюкагону (см. главу 5).
Инсулин - белковый гормон, который известен довольно
давно и изучен, пожалуй, тщательнее, чем какой-либо
глава 12 Регуляция метаболизма углеводов и жиров
167
NH2
NH2
сАМР-фосфодиэстераза
0-СН2
+ H20
0 = Р
I
0"
Н Η η
Η |3. Η
— о он
4 Гидролизуемая связь
О
II
о-—р—о—сн2
I ! н н
Η Ι Η
+ н+
он он
Аденозинмонофосфат (AMP)
Циклический аденозин-3\5'-
монофосфат (сАМР)
Рис. 12.16. Реакция, катализируемая сАМР-фосфодиэстеразой
Это название фермент получил за то, что он гидролизует фосфодиэфирную связь в молекуле сАМР
другой белок или гормон. Причиной тому является
не только его ключевая роль в углеводном и жировом
обмене, но и терапевтическое применение при лечении
диабета. К сожалению, несмотря на все усилия, механизм
действия инсулина еще во многом неясен. Установлено
присутствие инсулиновых рецепторов на внешней
поверхности клеток. Совсем недавно стало известно, что
его рецепторы пронизывают плазматическую
мембрану, а цитоплазматический домен является тирозиновой
протеинкиназой. Она отличается от ранее описанных
протеинкиназ и стимулируется при связывании с
инсулином (см. главу 26). Эту киназу характеризует то, что
она фосфорилируеттирозиновые остатки. Более
подробно механизм действия инсулина обсуждается в главе 26.
Регуляция поглощения клетками
глюкозы и жиров
Глюкоза не может пройти через липидный бислой,
поэтому в клеточных мембранах присутствует специальный
мембранный транспортный белок, ответственный
за ее перенос. В пищеварительном тракте глюкоза
всасывается за счет активного транспорта, во всех других
тканях она поступает в клетки путем пассивного
переноса. Такой тип трансмембранного переноса
сводится к тому, что молекулы преодолевают мембранный
барьер благодаря наличию градиента концентраций,
и называется облегченной диффузией (см. с. 63).
В мозге и в печени поглощение глюкозы клетками
не зависит от инсулина, но в мышечных и жировых
клетках оно ускоряется этим гормоном. Судя по всему,
в цитоплазме таких клеток имеются неактивные
транспортные белки. Инсулин стимулирует их переход
в активную форму и встраивание в мембрану, что
увеличивает пропускную способность мембраны для
глюкозы (рис. 12.17). На жировых клетках было показано,
.. Рецептор инсулина
Жировая клетка
Внутриклеточные мембранные
пузырьки со встроенным в
них переносчиками глюкозы
Инсулин
Пузырек сливается с
клеточной мембраной,
и переносчики
глюкозы начинают
функционировать
Глюкоза
Пузырек, готовый
к слиянию с клеточной
мембраной
Рис. 12.17. Мобилизация переносчиков глюкозы под
действием инсулина в жировых и некоторых других клетках
В клетках печени и мозга этого не происходит. Транспорт
глюкозы через плазматическую мембрану осуществляется по типу
облегченной диффузии
часть 3 Метаболизм
168
что встраивание транспортного белка завершается уже
через 7 минут после связывания инсулина с рецептором.
После удаления инсулина процесс обращается, и через
20-30 минут транспортные белки выходят из мембраны
в цитоплазму.
Механизм этого удивительного явления пока
неизвестен. Возможно, инсулин стимулирует движение
транспортного белка и встраивание его в мембрану или
вызывает смещение динамического равновесия между
мембранным и цитоплазматическим фондами этого белка.
Облегченная диффузия может лишь выровнять
концентрации глюкозы в крови и клетках. Однако внутри
клеток глюкоза быстро исчезает, либо превращаясь
в гликоген, либо расходуясь на метаболические нужды.
Это препятствует выравниванию концентрационного
градиента глюкозы и служит движущей силой ее
транспорта внутрь клетки.
Первый этап внутриклеточных превращений
глюкозы - ее фосфорилирование. Он осуществляется в
печени глюкокиназой, а в других тканях - гексокиназой:
Глюкоза + АТР -» глюкозо-6-фосфат + ADP.
Величина Км (для глюкозы) глюкокиназы по
сравнению с гексокиназой гораздо выше, т. е. сродство
первого фермента к субстрату меньше (см. с. 91). Глубокий
смысл этого различия становится понятным, если
вспомнить о глюкостатической функции печени. Ей
не нужно поглощать глюкозу, если последняя в крови
не присутствует в избытке. Если глюкозы в крови мало,
печень ее производит и направляет в кровоток. Было бы
нелогично одновременно выводить глюкозу в кровоток
и поглощать ее оттуда. Этого и не происходит,
поскольку глюкокиназа не фосфорилирует глюкозу, если
концентрация последней мала по сравнению с Км.
Для глюкокиназы предусмотрен еще один регуля-
торный механизм. Этот фермент индуцируется
инсулином. Под термином индукция понимают ускорение
биосинтеза под влиянием индуктора, в данном случае
инсулина. Механизм, посредством которого инсулин это
делает, пока неизвестен.
Метаболизм жиров начинается с поступления в
клетки не самих триглицеридов, а жирных кислот.
Последние легко проходят через мембраны без участия
каких бы то ни было транспортных систем, поскольку
липидный бислой не служит для них барьером.
Поэтому необходимо контролировать поступление жирных
кислот в клетку. При этом следует регулировать их
содержание в крови. Такая регуляция осуществляется
глюкагоном, адреналином, норадреналином и
инсулином, действующими на жировые клетки.
Получив общие представления о внеклеточной
регуляции, мы можем перейти к рассмотрению действия
гормонов на отдельные метаболические пути.
Регуляция синтеза и расщепления
гликогена внеклеточными агентами
Регуляция расщепления гликогена
Мы уже познакомились с регуляцией гликогенфосфори-
лазы Ъ в покоящихся мышцах и в печени, в основе
которой лежит активация фермента под действием AMP. Эта
регуляция обеспечивает постоянный контроль уровня
глюкозы на фоне стабильного режима ее расходования.
Существует, однако, совершенно иная регуляторная
система, основанная на превращении фосфорилазы Ъ
в фосфорилазу а, чья активность не зависит от AMP.
Такое превращение происходит в результате фосфори-
лирования фермента протеинкиназой, которая
переносит фосфатный остаток АТР на гидроксильную группу
одного из сериновых остатков, вызывая тем самым кон-
формационные изменения молекулы фермента и его
активацию. Важно помнить о разнице между фосфори-
лированием, когда на гидроксильную группу серина
протеинкиназой переносится фосфатный остаток АТР,
и фосфоролизом - катализируемым фосфорилазой
процессом расщепления гликогена с участием
неорганического фосфата. В мышечных клетках протеинкиназа
активируется адреналином. Этот гормон, циркулирующий
в крови, распознается и связывается рецепторами на
поверхности мышечной клетки, запуская при этом
внутриклеточный синтез сАМР. Мы еще вернемся к
рассмотрению механизма, посредством которого сАМР
стимулирует фосфорилирование фосфорилазы Ь, а
пока обсудим физиологические аспекты всего процесса.
Адреналин образуется в надпочечниках в ответ на
нервный сигнал, идущий из мозга при возникновении
экстремальной ситуации, требующей мгновенной и
активной мышечной деятельности (в англоязычной
литературе такую ситуацию называют fight or flight-
бегство или борьба). Адреналин как бы нажимает кнопку
«тревога». Мышечной клетке некогда дожидаться,
когда в ней накопится индикатор сниженной
концентрации АТР-АМР - и сработает внутриклеточная
положительная обратная связь. Клетка должна мгновенно
обеспечить неограниченный поток топлива для
генерации АТР, чтобы организм успел справиться с грозящей
опасностью. Далее мы поясним, почему в такой
ситуации оптимальным топливом является глюкозо-6-фос-
фат, образующийся из продукта фосфоролиза
гликогена- глюкозо-1-фосфата. Здесь отметим только, что
из-за низкого выхода АТР для продолжения гликолиза
глава 12 Регуляция метаболизма углеводов и жиров
169
нужно иметь про запас большое количество топлива.
Эту задачу решает мгновенная активация гликогенфос-
форилазы.
Адреналин также стимулирует высвобождение
глюкозы из печени в кровь. Его цель - быстро снабдить
мышцы топливом в экстремальной ситуации. И здесь
эффект адреналина обусловлен быстрым фосфорилиро-
ванием гликогенфосфорилазы.
Любопытно, что в картофельных клубнях тоже есть
фосфорилаза, расщепляющая крахмал. Степень
гомологии растительной и мышечной фоефорилазы очень
высока: их аминокислотные последовательности
наполовину идентичны. Тем не менее картофельная
фосфорилаза не активируется фосфорилированием,
возможно потому, что картофелю не нужен ответ типа
«бегство или борьба» и не нужно контролировать
уровень глюкозы в крови.
Добавим, что печень выбрасывает в кровь глюкозу
также в ответ на связывание глюкагона рецепторами.
Этот гормон выделяется поджелудочной железой при
снижении уровня глюкозы в крови и, подобно
адреналину, вызывает увеличение содержания
внутриклеточного сАМР и активацию гликогенфосфорилазы.
Мышечные клетки не реагируют на присутствие глюкагона,
поскольку на их поверхности нет специфичных к нему
рецепторов.
Механизм регуляции синтеза и
расщепления гликогена с помощью сАМР
Гликогенфосфорилаза существует в двух формах: в
форме а сериновый гидроксил фосфорилирован, а в форме Ъ
нет. Схема взаимопревращений обеих форм
представлена на рис. 12.18.
Гликогенфосфорилаза Ъ
(неактивная)
Фосфопротеин
фо таза
сАМР
АТР
Киназа
фоефорилазы
сАМР
_0-(р) ADP
Гликогенфосфорилаза а
(активная)
Рис. 12.18. Взаимопревращения гликогенфосфорилаз а и Ъ
под действием киназы и фосфатазы
сАМР влияет на эти превращения лишь опосредованно. Гид-
роксильная группа принадлежит сериновому остатку белка
Активация под действием сАМР киназы
фоефорилазы, осуществляющей фосфорилирование
гликогенфосфорилазы, не является прямой. сАМР сначала активирует
протеинкиназу А (ПКА; А - от сАМР), которая, в свою
очередь, фосфорилирует киназу фоефорилазы, переводя
ее в активное состояние. А та уже фосфорилирует глико-
генфосфорилазу Ъ. Полная последовательность событий,
разворачивающихся после контакта клетки с гормоном,
представлена на схеме:
Гормон
(глюкагон и адреналин в печени, адреналин - в мышцах)
ι
Связывание гормона с рецептором
ι
Активация аденилатциклазы
ι
Образование сАМР из АТР
Аллостерическая активация протеинкиназы А (ПКА)
ΙΙΚΛ
N1/
(неактивная)
-> ПКА
(активная)
АТР
ADP
Киназа
фоефорилазы в
(неактивная)
Фосфорилаза б'
(неактивная в
отсутствие сАМР)
^ν] Фосфорилирование
λ киназы фоефорилазы в
Ψ
-> Киназа
фоефорилазы в
(активная)
АТР I
^^ Фосфорилирование
] фоефорилазы в
ADP^|
^ \ Фосфорилаза а
(активная в отсутствие сАМР)
ι
Расщепление гликогена
ι
Глюкозо-1 -фосфат
ι
Глюкозо-6-фосфат
Печень Мышцы
Глюкоза крови Гликолиз
К чему такой сложный механизм регуляции? Каждая
клетка связывает небольшое число молекул гормона.
Между тем ее ответ, особенно в экстремальной
ситуации, должен быть быстрым и мощным. Напомним, что
клетка печени содержит примерно 1 мкмоль остатков
глюкозы (180 мкг), т. е. ~ 6,02 ■ 1017 молекул.
Связывание нескольких молекул гормона с рецепторами
должно заставить клетку переработать астрономическое
число молекул глюкозы, при этом очень быстро. Так же
часть 3 Метаболизм
170
обстоит дело и с образованием глюкозо-6-фосфата в
ходе генерации энергии в мышцах.
Следовательно, слабый сигнал - связывание
гормона с рецептором - должен быть усилен до такой
степени, чтобы вовлечь в ответ максимально возможное
число исполнителей команды - молекул фермента. Таким
образом, рассмотренная выше схема есть не что иное,
как мощный регуляторный каскад. Предположим,
1 молекула гормона активирует 1 молекулу аденилат-
циклазы, которая образует 100 молекул сАМР в минуту.
Это уже стократное усиление. Если каждая молекула
сАМР активирует 1 молекулу протеинкиназы А, которая
с такой же производительностью фосфорилирует кина-
зу фосфорилазы Ь, то усиление составит 100 · 100 и т. д.
В действительности гормональная активация гликоген-
фосфорилазы включает четыре ступеньки усиления.
Каскадное усиление сигнала - общий принцип действия
гормонов.
Каждая молекула гликогенфосфорилазы атакует
конец одной из олигосахаридных цепей гликогена.
Если бы гликоген, подобно амилозе, представлял собой
длинные линейные цепи, гормональная стимуляция
стала бы бессмысленной, поскольку множеству молекул
активированной гликогенфосфорилазы не хватило бы
мишеней. Видимо, именно в этом причина крайней раз-
ветвленности гликогена (по сравнению с крахмалом),
позволяющей в критической ситуации подвергнуться
быстрому расщеплению. Неторопливые растения в этом
не нуждаются и могут позволить себе хранить глюкозу
в виде амилозы.
Обращение активации фосфорилазы
Все регуляторные метаболические процессы должны
быть обратимыми. «Включение» и «выключение»
открывает дополнительную возможность управления
регулируемым процессом. В случае фосфорилазы форма а
превращается обратно в форму Ъ с помощью особой
фосфатазы (так называемой протеинфосфатазы I),
которая отщепляет фосфат от фосфорилированного
остатка серина:
Е-О-Р + Н20-> Е-ОН + Ру + Н+.
Форма а Форма b
В печени эта реакция стимулируется свободной
глюкозой благодаря ее аллостерическому действию,
оказываемому на форму а.
На время развития гормонального ответа -
превращения фосфорилазы Ъ в фосфорилазу а -
целесообразно подавить деятельность фосфатазы, чтобы она не
препятствовала быстрому нарастанию числа активных
молекул гликогенфосфорилазы. Оказывается, во многих
клетках присутствует белок, который называют
ингибитором фосфатазы I. Он фосфорилируется той же самой
протеинкиназой А, что и киназа фосфорилазы, и инги-
бирует фосфатазу. Таким образом, в конечном итоге
сАМР не только «включает рубильник», но и не дает его
сразу «выключить».
При нормальном мышечном сокращении (т. е. в
ситуации, не требующей участия сАМР в регуляции)
киназа фосфорилазы аллостерически активируется
ионами Са2+, концентрация которых при сокращении мышц
в ответ на сигнал от двигательного нерва резко
возрастает (см. главу 28). Поскольку такая активация киназы
фосфорилазы никак не связана с фосфорилированием,
она исчезает, как только поступает первый сигнал к
расслаблению. Активация фермента с помощью ионов Са2+
опосредована регуляторным белком кальмодулином,
который присутствует почти во всех эукариотических
клетках. Этот небольшой растворимый белок в случае
киназы фосфорилазы является прочносвязанной
субъединицей фермента. Кальмодулин действует как детектор
концентрации Са2+: связывание кальмодулином этих
ионов вызывает в нем конформационные изменения,
благодаря которым он взаимодействует с большим числом
белков-мишеней, в том числе и с ферментами, активируя их.
Подведем итог основным фактам, изложенным
выше, чтобы закрепить в памяти главные элементы этой
сложной регуляторной системы (рис. 12.19).
1. Фосфорилаза- фермент, катализирующий
расщепление гликогена посредством фосфоролиза. Эта
реакция не имеет ничего общего с фосфорилированием -
переносом фосфата с АТР на гидроксильную группу
серина или треонина, которое катализирует фермент про-
теинкиназа. Фосфатаза осуществляет гидролитическое
отщепление фосфата от фосфорилированных белков.
2. В печени и в покоящихся мышцах фосфорилаза
находится в виде неактивной и нефосфорилированной
формы Ь, которую частично активирует сАМР. Эта
активация не сопровождается фосфорилированием фермента.
3. Адреналин, воздействуя на клетки мышц и
печени, а также глюкагон, воздействуя на клетки печени (но
не мышц), вызывают увеличение внутриклеточной
концентрации сАМР.
4. с AMP аллостерически активирует протеинкина-
зу, которая в свою очередь активирует киназу
фосфорилазы, специфически активирующую фосфорилазу Ъ.
Последняя при этом превращается в активную форму а.
Процесс в целом можно рассматривать как
усилительный каскад.
5. При нормальном мышечном сокращении под
влиянием нервного импульса в мышечных клетках
увеличивается концентрация Са2+. Эти ионы аллостерически
глава 12 Регуляция метаболизма углеводов и жиров
171
активируют (частично) киназу фосфорилазы Ъ. В
результате происходит частичная активация фосфорилазы.
В отличие от сАМР-индуцированной активации киназы
фосфорилазы, активация Са2+ не сопряжена с ее фосфо-
рилированием.
6. Превращение активной фосфорилазы а обратно
в неактивную форму Ъ катализирует фермент протеин-
фосфатаза I. Его активность регулирует ингибитор про-
теинфосфатазы I - белок, который проявляет свои инги-
бирующие свойства только после того, как будет фос-
форилирован. Это фосфорилирование осуществляется
той же самой сАМР-стимулируемой протеинкиназой,
которая фосфорилирует киназу фосфорилазы Ъ. Таким
образом, активация фосфорилазы сопровождается ин-
гибированием фосфатазы. В печени превращение
формы а фосфорилазы в форму Ъ активируется глюкозой.
Адреналин
I Активация
сАМР-зависимая
Частично
активированная Неактивная
киназа ·*—— киназа
фосфорилазы Са2+ фосфорилазы
Активация
фосфорилазы
ATP ADP
Полностью
активированная
киназа
фосфорилазы
Высвобождение ионов Са2+
в мышечной клетке под
влиянием нервного импульса
Максимальная
активация
фосфорилазы
Расщепление гликогена
для умеренного (нормального)
сокращения мышц
Ускоренное расщепление
гликогена для энергичного
сокращения мышц
Рис. 12.19. Регуляция киназы фосфорилазы мышц
Представлен также механизм активации киназы ионами
кальция, который не зависит от сАМР. Ионы кальция не только
инициируют мышечное сокращение, но также обеспечивают
его энергией. Для каталитической активности фосфорилиро-
ванной (активной) киназы необходимы ионы Са2\ Они
связываются с кальмодулином, который является субъединицей этого
фермента, вызывая активацию последнего
Как регулируется гликогенсинтаза?
Синтез и распад гликогена должны управляться таким
образом, чтобы эти два процесса не могли протекать
одновременно, ибо в противном случае возникает
холостой цикл. В предыдущем разделе говорилось о том,
что активация гликогенфосфорилазы является
следствием активации протеинкиназы А под влиянием сАМР.
Гликогенсинтаза непосредственно фосфорилируется
протеинкиназой А, но в этом случае фосфорилирование
инактивирует фермент. Регуляторный каскад активации
гликогенсинтазы короче, что вполне понятно,
поскольку синтез гликогена проходит в более спокойных для
организма условиях, чем его расщепление в
экстремальных ситуациях.
Гормон
(глюкагон или адреналин в печени; адреналин в мышцах)
ι
Гормон-рецепторный комплекс
ι
Активация аденилатциклазы
4
Образование сАМР из АТР
Аллостерическая активация протеинкиназы А (ПКА)
I
ΙΙΚΛ
(неактивная)
-^ ПКА
(активная)
ΑΤΡ~Ν
ADP^I
Фосфорилирование
гликогенсинтазы
-> Гликогенсинтаза
(неактивная)
Гликогенсинтаза —
(активная)
Мы уже видели, как происходит инактивация
фосфорилазы а. Ее осуществляет фосфатаза, которую в свою
очередь ингибирует белок (ингибитор протеинфосфат-
азы), фосфорилируемый протеинкиназой А. Нечто
похожее происходит при регуляции гликогенсинтазы.
Однако есть существенные отличия двух регуляторных
путей: 1) дефосфорилирование синтазы приводит к ее
активации; 2) протеинкиназа не активирует, а напротив,
инактивирует белковый ингибитор фосфатазы. Эта
протеинкиназа не идентична протеинкиназе А и, в отличие
от последней, активируется не сАМР, а инсулином.
Таким образом, инсулин активирует фосфатазу 1, которая
в свою очередь активирует гликогенсинтазу.
Удивительно, что один и тот же белковый ингибитор
участвует в регуляции и синтеза, и расщепления
гликогена; однако протеинкиназа А его активирует, а инсу-
линзависимая киназа - инактивирует.
Механизмы регуляции гликогенсинтазы
представлены на рис. 12.20, а общая схема регуляции метаболизма
гликогена - на рис. 12.21.
Регуляция гликолиза и глюконеогенеза
внеклеточными агентами
Эти два метаболических пути логично рассматривать
вместе, так как в них вовлечены, по большей части,
одни и те же ферменты. Глюкагон сигнализирует печени
часть 3 Метаболизм
172
Гликогенфосфорилаза b
(неактивная)
p.·,
Фосфопротеин- \ /
Фс .^ γ
сАМР----- - 4
сАМР-стимулируемая Λ
протеинкиназа Ат / \
активирующая / ^
белок-ингибитор .
t·
Λ
ATP
£Синаза
илазы
Ι + ^..-cAMP
фосфатазы
Н70
ADP
Р/.
Гликогенфосфорилаза а
(активная)
Гликогенсинтаза (активная)
АТР
Фосфопротеин
фосфатаза I
Инсулин ► + у
Инсулин-стимулируе
мая протеинкиназа,
инактивирующая
беЛОК-ИНГИбиТОр |_| q
фосфатазы
Гликогенсинтаза (неактивная)
;λ
теинкиназа А
сАМР
0-®
ADP
Рис. 12.20. Схема взаимной регуляции гл и ко ген фосфор и-
лазы и гликогенсинтазы
Цветными кружками обозначены молекулы ферментов:
активные - темнее, неактивные - светлее
Глюкоза в крови -
Печень
(глюкагон, адреналин)
Гликоген
1Гликогенолиз
{ включен
Глюкозо-6-фосфат
Глюконеогенез включен ♦ [
Гликолиз
выключен
Пируват
Мышцы (адреналин)
Гликоген
Гликогенолиз
Не происходит \ т включен
Глюкоза -*—Η— Глюкозо-6-фосфат
μ
Глюконеогенез
(в мышцах очень
незначительный)
Гликолиз
включен
Пируват
Г ликогенсинтаза
Инсулин
сАМР
f Гликоген >
L ликогенфосфорилаза
сАМР
UDPG
\
Гл юкозо-1 -фосфат
Рис. 12.21. Регуляция метаболизма гликогена
Действие инсулина и сАМР на активность ферментов
является опосредованным. UDPG - UDP-глюкоза
о необходимости выделения в кровь глюкозы либо за
счет гликогенолиза (расщепления гликогена), либо -
глюконеогенеза. Поэтому логично, что в печени сАМР
включает расщепление гликогена и глюконеогенез
(рис. 12.22, а). Было бы странно, если сАМР
включал бы при этом в печени гликолиз - процесс,
обратный глюконеогенезу.
В мышцах ситуация совершенно иная. Там
стимуляция синтеза сАМР адреналином является командой
Рис. 12.22. Различия в регуляции ответа на сигнал
глюкагон и /или адреналин в печени (а) и мышце (б)
Оба гормона используют сАМР в качестве вторичного мес-
сенджера. Термин гликогенолиз здесь означает расщепление
гликогена
к увеличению генерации энергии. Поэтому логично, что
сАМР в мышцах включает расщепление гликогена и
активирует гликолиз (в отличие от печени), поскольку
именно в ходе этого процесса образуется глюкозо-6-фос-
фат (рис. 12.22,6), дальнейшее превращение которого
приводит к синтезу АТР.
Таким образом, в печени сАМР должен ингибиро-
вать, а в мышцах активировать гликолиз. Наиболее
важной регуляторной стадией является реакция,
катализируемая фосфофруктокиназой (см. рис. 8.7). В печени
сАМР (в ответ на действие глюкагона) вызывает инги-
бирование фосфофруктокиназы, подавляя тем самым
гликолиз, тогда как в мышцах сАМР (в ответ на действие
адреналина) ускоряет гликолиз.
Влияние сАМР на фосфофруктокиназу в печени
заслуживает отдельного обсуждения.
глава 12 Регуляция метаболизма углеводов и жиров
173
Как сАМР регулирует активность
фосфофруктокиназы?
Этот регуляторный механизм хорошо известен для
печени, и в нем не так уж трудно разобраться. Если
уровень глюкозы в крови низок, то высоко содержание
глюкагона, в результате в печени увеличивается
внутриклеточная концентрация сАМР. Благодаря этому в
клетке уменьшается содержание фруктозо-2,6-дифосфата.
(Нет, здесь не опечатка!) Речь идет именно об этом,
ранее не встречавшемся нам соединении, а не о хорошо
знакомом фру ктозо-1,6-д и фосфате, который
образуется в процессе гликолиза при участии
фосфофруктокиназы. Фруктозо-2,6-дифосфат - чрезвычайно мощный
аллостерический активатор фосфофруктокиназы, а
вместе с ней и всего гликолиза. Это настоящая регулятор-
ная молекула. Стоит увеличить его концентрацию -
гликолиз ускоряется, если ее уменьшить - замедляется.
Существенно, что фруктозо-2,6-дифосфат не только
активирует фосфофруктокиназу, но также ингибирует
фруктозо-1,6-дифосфатазу (рис. 12.23)-один из
ключевых ферментов глюконеогенеза.
Фруктозо-6-фосфат
Нетрудно догадаться, что в печени сАМР должен
снижать уровень 2,6-дифосфата: при этом блокируется
гликолиз и активируется глюконеогенез. Как это
достигается?
Образование фруктозо-2,6-дифосфата
катализируется особой фосфофруктокиназой - ФФК7. После ее
открытия ранее описанную фосфофруктокиназу стали
сокращенно называть ФФК,.
сАМР вызывает ингибирование ФФК2 (рис. 12.24).
Происходит это так: сАМР активирует протеинкиназу,
которая фосфорилирует ФФК2 (рис. 12.25). Фосфори-
лированная ФФК2 не только перестает синтезировать
фруктозо-2,6-дифосфат, но напротив, активно его гид-
ролизует (нефосфорилированная ФФК2 не гидролизует
фруктозо-2,6-дифосфат). Таким образом, фосфорилиро-
вание меняет не просто скорость, а направление
катализируемой реакции.
ФФК!
Фруктозо-6-фосфат ► Фруктозо-1,6-дифосфат '.
Фруктозо-1,6-
и<Ьосфатаза
Фо" _ >ктокиназа
Фруктозо-2,6-дифосфат
Ингибируется
фруктозо-2,6-
дифосфатом
1 Активируется фрук-
тозо-2,6-дифосфатом
Фруктозо-1,6-дифосфат
Рис. 12.23. Регуляция гликолиза фруктозо-2,6-дифосфатом
Рис. 12.24. Синтез фруктозо-2,6-лифосфата второй формой
фосфофруктокиназы (ФФК2) и ингибирование фермента в
печени при участии сАМР
Это ингибирование опосредованное: сАМР активирует
протеинкиназу, которая фосфорилирует ФФК2, подавляя тем
самым синтез фруктозо-2,6-дифосфата. Фосфорилированная
ФФК, гидролизует фруктозо-2,6-дифосфат. Таким образом
с AMP снижает уровнь фруктозо-2,6-дифосфата в печени
ФФК2-активнйсть
дифосфатазная активность ингибирована
Образование
*" фруктозо-2,6-дифосфата
ФФК2—ОН
сАМР-зависимая
протеинкиназа
Фосфопротеин-
фосфатаза
ФФК2-0-®— Г=из
фруктозо-2,6-дифосфата
Дифосфатазная активность,
ФФК-активность ингибирована
Активация
ФФК,
Ингибирование
ФФК,
Рис. 12.25. Регуляция активности ФФК2 в печени посредством фосфорилирования сАМР-зависимой протеинкиназой
ФФК2 - бифункциональный фермент: в нефосфорилированной форме он синтезирует фруктозо-2,6-дифосфат, а после
фосфорилирования гидролизует его
часть 3 Метаболизм
174
Что хорошо для печени, то плохо для мышц,
поскольку там нельзя подавлять гликолиз, поставляющий
топливо при экстренных нагрузках; и следовательно, фос-
фофруктокиназа не может быть ингибирована.
Известно, что под влиянием адреналина в мышцах возрастает
уровень фруктозо-2,6-дифосфата. Возможно, это
объясняется тем, что сАМР ускоряет расщепление гликогена,
благодаря чему увеличивается содержание фрук-
тозо-6-фосфата - субстрата и активатора ФФК2. Однако
этот механизм регуляции требует дальнейшего изучения.
Глюкагон регулирует активность не только фосфо-
фруктокиназы, но также и пируваткиназы,
катализирующей заключительную стадию гликолиза.
Индуцированный глюкагоном рост внутриклеточной
концентрации сАМР приводит в клетках печени (но не мышц)
к фосфорилированию пируваткиназы, которое
сопровождается ее инактивацией.
Смысл этого совершенно ясен. Если требуется
насытить кровь глюкозой, в печени нужно стимулировать
глюконеогенез и блокировать гликолиз. Ингибирование
пируваткиназы, так же как и фосфофруктокиназы,
решает вторую из этих задач.
Мышцы, как уже не раз отмечалось, заняты не
синтезом, а исключительно расщеплением глюкозы, поэтому
с AMP здесь не должен подавлять гликолиз.
Действительно, увеличение уровня сАМР не приводит к
фосфорилированию пируваткиназы.
Стимулируемое глюкагоном ингибирование
пируваткиназы печени важно также и с точки зрения регуляции
глюконеогенеза. Синтез глюкозы из пирувата включает
две промежуточные реакции:
Пируват + АТР + НСО^ -> Оксалоацетат + ADP + Р, + Н~, (1)
Оксалоцетат + GTP <-> Фосфоенолпируват + GDP + С02 (2)
Нетрудно понять, что существует потенциальная
опасность возникновения холостого цикла, поскольку
пируваткиназа катализирует реакцию:
Фосфоенолпируват + ADP -> Пируват + АТР.
Такой потенциальный холостой цикл можно было бы
описать следующей схемой:
Фосфоенолпируват
Оксалоацетил
Пируват
Во избежание холостого цикла необходимо, чтобы
в процессе глюконеогенеза пируваткиназа была инакти-
вирована. Только тогда весь фосфоенолпируват будет
участвовать в реакциях глюконеогенеза. В печени
инактивация фермента происходит при участии сАМР
(рис. 12.26). Таким образом, глюкагон вызывает
ингибирование фосфофруктокиназы и активацию фрукто-
зо-1,6-дифосфатазы, тем самым стимулируя процесс
глюконеогенеза, что приводит к увеличению уровня
глюкозы в крови.
Гликоген
Гликогенсинтаза
Инсулин -► + ι
сАМР -
1 ликогенфосфо-
ил аза
1+ -—- сАМР
Глюкозо-1 -фосфат
Глюкозо-6-фосфат
Глюкоза
в крови
Не происходит
в мышцах Фруктозо-6-фосфат
сАМР -----^ +
МО··· К ИН
ι САМР
Фруктозо-1,6-дифосфат
Оксалоацетат
Фосфоенолпируват
Пируваткина-
- - сАМР
Пируват
Рис. 12.26. Внешняя регуляция участка метаболизма
углеводов в печени
Здесь под сАМР подразумевается воздействие гормонов: глю-
кагона и адреналина. Влияние сАМР на активность
ферментов везде опосредовано
Регуляция метаболизма жиров
внеклеточными агентами
У жировых клеток есть выбор: запасать триглицериды
или расщеплять их запас до жирных кислот и
выбрасывать последние в кровь. Инсулин служит сигналом для
реализации первого варианта, а глюкагон - второго. Но
у некоторых животных, в том числе и у человека,
подобно глюкагону действуют адреналин и норадреналин.
Жировые ткани иннервированы симпатическими
нервами, окончания которых выделяют норадреналин;
адреналин же секретируется надпочечниками.
Действие на метаболизм жировых клеток всех трех
гормонов - глюкагона, адреналина и норадреналина -
опосредовано участием общего для них вторичного
глава 12 Регуляция метаболизма углеводов и жиров
175
мессенджера - сАМР. Главной мишенью реакции
служит чувствительная к гормонам липаза, которая
катализирует реакцию:
триглицерид + Н20 ->
-^ диглицерид + свободная жирная кислота.
Диглицериды далее расщепляются до глицерина
и жирных кислот. Эта гормончувствительная липаза
активируется в результате фосфорилирования про-
теинкиназой и инактивируется протеинфосфатазой
(рис. 12.27). Связывание инсулина клеточными
рецепторами блокирует эти эффекты.
Стимулируемое инсулином поглощение глюкозы
способствует превращению ее избытка жировыми
клетками в жиры. Ключевым ферментом синтеза жиров
служит ацетил-СоА-карбоксилаза, катализирующая
необратимую реакцию. Она инактивируется при фосфорили-
ровании протеинкиназой, аллостерическим активатором
которой служит AMP (не сАМР), и реактивируется фос-
фатазой, которая отщепляет фосфатную группу. В свою
очередь, эта фосфатаза ингибируется сАМР -
вторичным мессенджером глюкагона и адреналина.
Таким образом, если уровень АТР в клетке низок,
AMP вызывает инактивацию карбоксилазы. При низком
уровне глюкозы в крови карбоксилазу в неактивном
состоянии поддерживает глюкагон. Инсулин вызывает
обратный эффект: он несколько стимулирует активность
карбоксилазы, тем самым способствуя синтезу жиров.
Гормончувствительная
липаза(неактивная)
Фосфопротеин-
фосфатаза
АТР
Протеинкиназа
ь- сАМР
0-®
ADP
Гормончувствительная
липаза(активная)
Рис. 12.27. Активация гормоночувствительной липазы
в жировых клетках путем ее сАМР-зависимого
фосфорилирования
Инсулин служит антагонистом катехоламинам и глкжагону,
которые способствуют росту концентрации сАМР. Глюкагон
и адреналин ингибируют синтез жира соответственно в
печени и в жировых клетках, предотвращая дефосфорилирование
ацетил-СоА-карбоксилазы. Инсулин активирует карбоксилазу
Дополнительная литература
Субстратные циклы
Newsholme Ε. Α., Challiss R. A. J., Crabtree B. Substrate
cycles: their role in improving sensitivity in metabolic
control // Trends Biochem. Sci. 1984. Vol. 9. P. 277-280.
(Обсуждается регуляторная роль субстратных циклов.)
Регуляция метаболизма гликогена
Johnson L. N. Glycogen phosphorylase: control by
phosphorylation and allosteric effectors// FASEB J. 1992. Vol. 6.
P. 2274-2282.
(Исчерпывающий обзор, посвященный аллостерической
и гормональной регуляции расщепления гликогена.)
Walsh D. Α., Van Patten S. Μ. Multiple pathway signal
transduction by the cAMP-dependent proteinkinase // FASEB J.
1994. Vol. 8. P. 1227-1236.
(Серьезный, но доступный источник сведений о
структуре и механизме действия протеинкиназ.)
Регуляция синтеза кетоновых тел
McGarry J. D., Foster D. W. Regulation of hepatic fatty acid
oxidation and ketone body production // Annu. Rev.
Biochem. 1980. Vol. 49. P. 395-420.
(Несмотря на заголовок, эта статья представляет собой
общий обзор биосинтеза кетоновых тел.)
Quant P. A. The role of mitochondrial HMG-CoA synthase
in regulation of ketogenesis // Essays Biochem. 1994.
Vol. 28. P. 13-25.
(Суммируются данные о регуляции процесса
образования кетоновых тел и обсуждается ее физиологическое
значение.)
Вопросы к главе 12
1. Каковы два главных способа обратимого изменения
активности ферментов?
2. Как сказываются изменения концентрации
субстрата на скорости реакций, катализируемых неаллосте-
рическими и аллостерическими ферментами?
3. Почему зависимость скорости реакции от
концентрации субстрата у аллостерических ферментов в
области Утах12 круче, чем у обычных?
4. Как влияют аллостерические эффекторы на скорость
ферментативной реакции при насыщающих
концентрациях субстрата?
5. Опишите две модели, объясняющие гомотропное
кооперативное связывание субстрата ферментом.
часть 3 Метаболизм
176
6. Почему в клетках так широко используется аллосте-
рическая регуляция?
7. Каковы основные принципы внутриклеточной и
внеклеточной регуляции?
8. Пользуясь схемой, разъясните смысл аллостеричес-
кой регуляции метаболизма гликогена, гликолиза
и глюконеогенеза.
9. Пируватдегидрогеназа - ключевой регулируемый
фермент. Как правило, продукты реакции ингибиру-
ют ее. Какие три механизма используются для
регуляции активности пируватдегидрогеназы?
10. Как происходит внутриклеточная регуляция синтеза
и окисления жиров?
11. Чем определяется секреция инсулина и глюкагона
поджелудочной железой?
12. Что такое вторичный мессенджер? Назовите
вторичный мессенджер для адреналина и глюкагона. Как
вторичному мессенджеру удается влиять на
клеточный метаболизм?
13 Как инсулин регулирует скорость поступления
глюкозы в жировые клетки?
14. Как сАМР активирует процесс расщепления
гликогена?
15.Глюкагон посредством сАМР активирует фосфори-
лазу печени. Адреналин точно так же активирует
фосфорилазу в мышцах. Объясните, почему сАМР
по-разному влияет на процесс гликолиза в печени
и мышцах.
16. Некоторые гормоны, вызывающие в клетках разные
эффекты, используют в качестве вторичного мессен-
джера сАМР. Как одно вещество может регулировать
разные процессы?
17. Фосфофруктокиназа - ключевой регуляторный
фермент. Назовите главный аллостерический регулятор
этого фермента. Как осуществляется контроль за его
содержанием в печени?
18. Для синтеза глюкозы в печени необходим фосфо-
енолпируват. Однако его образование в печени
окажется очень неэффективным, если фосфоенолпи-
руват будет дефосфорилироваться под действием
пируваткиназы с образованием пирувата. Как
избежать этого холостого цикла? Почему этот механизм
не подходит для мышц?
19. Как глюкагон вызывает высвобождение жирных
кислот из жировых клеток?
глава 12 Регуляция метаболизма углеводов и жиров
177
глава
13
Зачем нужен альтернативный путь
окисления глюкозы -
пентозофосфатный?
Существует совершенно отличный от гликолиза путь
окисления глюкозы - пентозофосфатный, который иногда
называют прямым окислением, или гексозомонофос-
фатным шунтом. Зачем он нужен, если основной путь
окисления глюкозы (гликолиз —» цикл лимонной
кислоты —> цепь переноса электронов) вполне справляется
с производством энергии из глюкозы? Парадоксально, но
этот альтернативный путь имеет целью не окисление
глюкозы или производство энергии. В его задачу входят:
• образование рибозо-5-фосфата для синтеза нуклео-
тидов и нуклеиновых кислот;
• производство NADPH для синтеза липидов;
• вовлечение избытка пентоз, усваиваемых из пищи,
в общее русло метаболизма глюкозы.
Окислительная стадия
Пентозофосфатный путь включает в себя две основные
стадии. На первой (окислительной) глюкозо-6-фосфат
превращается в рибозо-5-фосфат и С02, одновременно
NADP+ восстанавливается до NADPH. 6-Фосфоглюко-
натдегидрогеназа катализирует реакцию образования
β-кетокислоты, которая затем подвергается декарбокси-
лированию до производного кетопентозы - рибуло-
зо-5-фосфата. Последний изомеризуется в альдозный
изомер - рибозо-5-фосфат. Полная схема окислительных
реакций представлена на рис. 13.1. Таким образом,
окислительная стадия завершается образованием рибо-
зо-5-фосфата и NADPH.
Неокислительная стадия и ее значение
Потребности различных тканей в рибозо-5-фосфате
и NADPH разные. Например, много NADPH
потребляется при синтезе жиров. Поэтому жировые клетки и
клетки печени содержат гораздо больше ферментов,
участвующих в пентозофосфатном пути, чем, скажем, клетки
мышц. Используя этот путь для синтеза NADPH, клетка
вырабатывает одновременно много рибозо-5-фосфата,
гораздо больше, чем может ей потребоваться, например,
для синтеза нуклеотидов. С другой стороны, часто
делящимся клеткам, не синтезирующим жиры, требуется
много рибозо-5-фосфата для синтеза нуклеотидов,
но сравнительно мало NADPH. Это крайние случаи,
но они наглядно демонстрируют суть проблемы:
потребности в рибозо-5-фосфате и NADPH у разных клеток
неодинаковы. Как же используются их излишки?
В неокислительной стадии пентозофосфатного пути
участвуют два фермента: трансальдолаза и транске-
толаза. Они отщепляют от фосфокетосахаров
соответственно С3- и С2-фрагменты и переносят их на фос-
фальдосахара (рис. 13.2). В результате
взаимопревращения фосфосахаров их количество приводится в
соответствие с потребностями клеточного метаболизма.
Транскетолаза, подобно пируватдегидрогеназе,
использует в качестве кофермента тиаминпирофосфат
(см. с. 116). Напомним, что пируватдегидрогеназа
также отщепляет С2-фрагмент (от пирувата) и переносит
его на акцептор (СоА). Трансальдолаза и транскетолаза
способны осуществлять столь большое число
взаимопревращений моносахаридов, что найдется немного
биохимиков, способных удержать все это в памяти.
Суммарный баланс окислительного этапа
пентозофосфатного пути описывается уравнением:
Глюкозо-6-фосфат + 2NADP+ + Н20 ->
-» Рибозо-5-фосфат + 2NADPH + 2Н+ + С02.
I
снон
снон
снон
I U
СНОН ι
I
сн '
СН2ОРОз"
Глюкозо-6-
фосфатдегидрогеназа
Т^
NADP4
NADPH
+ Н+
I
с=о
снон
снон
I U
СНОН I
I
сн '
СН2ОРОз
Н20
соон
снон
снон
снон
I
снон
СН2ОРОз
NADP+ NADPH + Н+ СН2ОН
^z
6-Фосфоглюконат-
дегидрогеназа
ι
С=0
СНОН
снон
I
сн2оро5
Рибулозо-5-фосфат
сно
снон
► снон
снон
I 2-
СН2ОРОз
Рибозо-5-фосфат
Глюкозо-6-фосфат
6-Фосфоглюконолактон 6-Фосфатглюконат
С02
Рис. 13.1. Окислительные реакции пентозофосфатного пути
часть 3 Метаболизм
178
Η
CH2OH
c=o с
I I
CHOH + CHOH
CHOH R7
Кетоза Альдоза
(донор) ( акцептор)
(Ч /н ίΗ2°Η
С С=0
— снон + снон
Τ анскетолаза r CHOH
1 ι
R2
с=о с
снон + снон
снон L
I
снон
I
Ri
Кетоза
(донор)
0^ /Н СН2ОН
с с=о
— снон + снон
1рансальдолаза η СНОН
СНОН
ι
R2
Альдоза
(акцептор)
Рис. 13.2. Реакции, катализируемые транскетолазой
и трансальдолазой
В ситуациях, когда потребности в рибозо-5-фосфате
и NADPH сбалансированы, неокислительного этапа
не нужно. Но как быть, если неделящимся жировым
клеткам нужно NADPH гораздо больше, чем рибо-
зо-5-фосфата? В этом случае благодаря трансальдолазе
и транскетолазе избыток рибозо-5-фосфата
превращается в глюкозо-6-фосфат:
6 Рибозо-5-фосфат -> 5 Глюкозо-6-фосфат + Pf.
В общих чертах механизм данного превращения
сводится к следующему (кто хочет познакомится с ним
подробнее, может обратиться к приложению в конце этого
раздела). Сначала часть общего количества альдозы -
рибозо-5-фосфата (R-5-P) - изомеризуется в кетозу -
ксилулозо-5-фосфат (Х-5-Р). Это необходимый шаг,
поскольку и трансальдолаза, и транскетолаза используют
в качестве доноров переносимых фрагментов только
кетозы. Оставшаяся часть R-5-P будет служить их
акцептором. Далее происходит серия превращений,
которая открывается реакцией (1) между Х-5-Р и R-5-P:
2С5 <г-> С3 + С7
С7+ С3<-+С4 + С6
(1)
Г ансаль лазг (2)
(3)
С5 + С4 <-> С3 + С6 Гранскетолаза
Конечное С3-соединение - глицеральдегид-3-фосфат,
две молекулы которого могут быть превращены в глю-
козо-6-фосфат при обращении реакции гликолиза. С
учетом этого обстоятельства, суммарный результат
приведенных выше трех реакций сводится к превращению
3 молекул С5-сахаров в 2,5 молекулы С6-сахаров.
Заметим, что совокупность этих реакций позволяет
переработать рибозу, потребляемую с пищей, в глюкозу при
условии, что сначала рибоза будет фосфорилирована
соответствующей АТР-зависимой киназой.
Пентозофосфатный путь отличается крайней
гибкостью. Рассмотрим другую ситуацию, когда клетка для
синтеза нуклеотидов нуждается в рибозо-5-фосфате,
но ей совершенно не нужен NADPH. В таком случае
имеет место превращение:
5 Глюкозо-6-фосфат + АТР ->
-^ 6 Рибозо-5-фосфат + ADP + Н+.
Схема этого превращения приведена на рис. 13.3.
Оно начинается с того, что в ходе гликолиза часть глю-
козо-6-фосфата превращается во фруктозо-6-фосфат,
а часть - в глицеральдегид-3-фосфат. Иными словами,
мы располагаем теми же С3- и С6-соединениями,
которые были продуктами приведенной выше реакции (3).
Если все три реакции обратить, получится смесь рибо-
зо-5-фосфата и ксилулозо-5-фосфата, а последнее
соединение можно изомеризовать в рибозо-5-фосфат. При
этом окислительные реакции пентозофосфатного пути
вообще не используются.
Глюкозо-6-фосфат
Глюкозо * ос атизоме аза
(4 молекулы)
Фруктозо-6-фосфат
АТР ч|
J
ADP
Фруктозо-1,6-дифосфат
Аль ^лаз*
Дигидроксиацетонфосфат
Фосфо- \ Гранскетолаза/
φ ктокиназД г ансальдолаза
II
^ Рибозо-5-фосфат
(6 молекул)
(2 молекулы)
Глицеральдегид-3-фосфат
Рис. 13.3. Превращение глюкозо-6-фосфата в рибозо-5-фос-
фат без образования NADPH
Окислительная часть пентозофосфатного пути здесь не показана
Как полностью окислить глюкозу?
Окислительные реакции пентозофосфатного пути
(см. рис. 13.1) иногда называют прямым окислением
глюкозы до С02.
глава 13 Зачем нужен пентазофосфатный путь ?
179
Формально такое окисление можно описать
уравнением:
6Глюкозо-6-фосфат + 12NADP+ + 7Н20 ->
-> 5Глюкозо-6-фосфат + 12NADPH + 12Н+ + 6С02 .
Однако это всего лишь уравнение суммарного
баланса сложной последовательности реакций. Речь идет
вовсе не о том, что 1 молекула глюкозо-6-фосфата
превратилась в 6 молекул С02; на самом деле из 6
молекул глюкозо-6-фосфата образовалось 6 молекул рибо-
зо-5-фосфата и 6 молекул С02. После этого 6 молекул
рибозо-5-фосфата были переработаны трансальдолазой
и транскетолазой в 5 молекул глюкозо-6-фосфата.
Последовательное повторение этих реакций позволяет
клеткам производить NADPH, необходимый для синтеза
жиров, не накапливая рибозо-5-фосфат.
Зачем эритроцитам
пентозофосфатный путь?
Эритроциты не делятся и не синтезируют жиры.
Поскольку в этих клетках нет митохондрий, единственным
источником энергии для них служит анаэробный
гликолиз. Зачем же эритроцитам нужны ферменты пентозо-
фосфатного пути?
У людей, в эритроцитах которых отсутствует первый
фермент пентозофосфатного пути - глюкозо-6-фосфат-
дегидрогеназа, противомалярийное лекарство памахин
вызывает тяжелую гемолитическую анемию. Конечной
причиной этого является дефицит NADPH, который
нужен для поддержания гемоглобина в восстановленном
состоянии (подробнее см. на с. 213).
Любопытно, что генетический дефект,
определяющий низкий уровень эритроцитарной глюкозо-6-фос-
фатдегидрогеназы, распространен на территориях, где
люди традиционно страдают от смертельно опасной
разновидности малярии. Он дает больным некоторые
преимущества в борьбе с паразитом, живущим в их
эритроцитах. Возможно, малярийный плазмодий нуждается
в каких-то веществах, образующихся в реакциях
пентозофосфатного пути, или же дефектные эритроциты
настолько ослаблены, что лопаются раньше, чем у
гнездящегося в них паразита заканчивается цикл
размножения. Это не единственный случай, когда наследственный
генетический дефект «дарит» его носителям
дополнительный шанс в поединке с малярией. Примером может
служить так называемая серповидноклеточная анемия -
наследственное заболевание, связанное с нарушением
формы эритроцитов (см. главу 27).
часть 3 Метаболизм
180
Дополнительная литература
Beutler E. Glucose-6-phosphate dehydrogenase
deficiency // The metabolic basis of inherited disease / Ed. J. B. Stan-
bury, J. B. Wyngaarten, D. S. Fredrickson, M. S. Brown.
5th edn. McGraw-Hill, 1983. P. 1629-1653.
(В доступной форме рассмотрены медицинские
аспекты дефицита глюкозо-6-фосфатдегидрогеназы.)
Вопросы к главе 13
1. Какие функции выполняет пентозофосфатный путь?
2. Какие реакции образуют окислительную стадию
этого пути?
3. Какие ферменты осуществляют превращения
пентозофосфатного пути, не связанные с окислением?
4. Жировым клеткам для синтеза жира нужно много
NADPH и очень мало рибозо-5-фосфата. На первой
стадии пентозофосфатного пути образуется равное
количество этих веществ. Как клетки решают эту
проблему?
5. Пентозофосфатный путь иногда называют прямым
окислением глюкозы. Почему? Насколько корректен
этот термин?
6. Зрелые эритроциты не синтезируют ни жиров, ни
нуклеотидов. Зачем им нужна глюкозо-6-фосфатде-
гидрогеназа?
Приложение: Реакции, участвующие в превращении
рибозо-5-фосфата в глюкозо-6-фосфат
о
II
с-н
снон
I
н—с-он
I
н-с-он
сн2-о—роГ
Рибозо-5-фосфат
(R-5-P)
СН2ОН СН2ОН
С = 0 Фосфопемтозо^п^ 4ераза^ = 0
Н —С-ОН
I
н—с-он
СН2-0—POf
Рибулозо-5-фосфат
н—с-он
I
н—с-он
сн2-о—ро*~
Ксилулозо-5-фосфат
(Х-5-Р)
Этим превращениям подвергается только часть молекул R-5-P.
Оставшаяся часть участвует в реакциях 1-3.
сн2он
I
с=о
I
снон
I
снон
I
сн2—о-
Х-5-Р
-роГ
Реакция 1
СНО +
I
снон
сн2—о—роГ
Глицеральдегил -
3-фосфат
(G-3-P)
Реакция 2
СНО
I
СНОН +
I
СНОН
сн2—о—роГ
Эритрозо-
4-фосфат
М-Р)
СНО
I
снон
I
снон
I
снон
сн2—о—роГ
R-5-P
СН20Н
С=0
I
снон
I
снон
I
снон
I
снон
сн2-о—ро^
Седогептулозо-
7-фосфат
(S-7-P)
Грансал олаза
сн2он
с=о
I
снон
I
снон
I
снон
I
сн2
о-
Фруктозо-
6-фосфат
(F-6-P)
сн>он
I ~
с=о
I
снон
I
снон
I
сн2-о-
Х-5-Р
-Р01
Реакция 3
сн2он
с=о
I
снон
I
снон +
I
снон
сн2—о—роГ
F-6-P
СНО
I
СНОН
I
снон
сн2-о—ро§"
Е-4-Р
Τ ран к-ет ча^а
СНО
I
снон
сн2—о—ро§
G-3-P
Два цикла реакций 1-3 приведут к образованию двух молекул G-3-P
снон
I
2 СНОН
сн2—о—ро§"
G-3-P
1 F-6-P + Ρ,-
(Реакции
глюконеогенеза)
Глюкозофосфатизомераза превращает
фруктозо-6-фосфат в глюкозо-6-фосфат
В совокупности эти превращения могут
быть представлены так:
2С5 + 2С5 —
2С7 + 2С3 ^=
2С5 + 2С4 =
2С3 —
2С3 + 2С7
2С4 + 2С6
2С3 + 2С6
1С6.
Или суммарно:
6Рибозо-5-фосфат -
5Глюкозо-6-фосфат + Pf-.
глава 13 Зачем нужен пентазофосфатный путь ?
181
глава
А А Фотосинтез - способ передачи энергии
I *т электронам воды
Образование АТР в аэробных клетках зависит от
переноса электронов с веществ, обладающих высоким
энергетическим потенциалом, вниз по энергетической шкале
на кислород. Поскольку количество пищи на Земле
ограничено, для продолжения жизни должен существовать
какой-то способ «подъема» электронов на самый верх
энергетической шкалы. Небольшая оговорка может быть
сделана в отношении недавно открытых глубоководных
морских организмов. Они обитают в районе разломов
земной коры, где бьют горячие источники, богатые
сероводородом (H2S). Сероводород- мощный
восстановитель, поэтому его электроны могут транспортироваться
по энергетическому градиенту с выделением энергии,
которая может быть использована для генерации АТР
в клетках при условии, что они снабжены
соответствующим биохимическим аппаратом. Однако эти формы
жизни могут существовать лишь до тех пор, пока
сероводород и другие подобные ему вещества образуются в
разломах земной коры. Для существования же громадного
большинства других организмов необходим круговорот
электронов. Это утверждение верно и в отношении
любых ранних форм жизни в первичном океане. Какие бы
виды готовых к употреблению пищевых веществ в нем
ни содержались, экспоненциально размножающиеся
организмы давно бы их использовали. По этой причине
резервуары для «стока» электронов могли бы
истощиться еще в анаэробной атмосфере, которая, как полагают,
существовала до появления фотосинтеза.
Общие сведения
Фотосинтез - биологический процесс,
осуществляющий кругооборот электронов. Он имеет несколько
явных преимуществ:
• неисчерпаемость источника энергии - солнца;
• неисчерпаемость донора электронов - воды;
• выделяющийся кислород является неисчерпаемым
резервуаром для стока электронов, позволяющим
осуществлять высвобождение энергии из
высокоэнергетических электронов пищевых молекул.
Весь этот перечень свидетельствует о том, что
появление фотосинтеза было самым важным событием с
момента возникновения жизни. Суть поддерживаемого
фотосинтезом глобального энергетического цикла
иллюстрирует рис. 14.1.
Шкала
свободной
энергии
Молекулы
пищевых
веществ
р»
Энергия
света
-ι Η О
Окислительное
фосфорилирование
Фотосинтез
Рис. 14.1. Цикл переноса электронов, связывающего
процессы фотосинтеза и окислительного фосфорилирования
в живой природе (изображено схематично)
Ассимиляция С02 представляет собой вторичный процесс по
отношению к подъему электронов воды на высокий
энергетический уровень
Вероятно, вы знаете, что при фотосинтезе С02 и Н20
преобразуются в углеводы (обычно в моносахариды или
крахмал). Общее уравнение, описывающее этот
процесс, можно представить в виде:
6С02 + 6Н20 -> С6Н1206 + 602
AG0' = 2872 кДж · моль1 .
Чтобы синтезировать глюкозу из С02 и Н20,
необходимо выполнение двух условий. Во-первых, нужно
иметь восстанавливающий агент с достаточно низким
редокс-потенциалом (с высокой энергией). При
фотосинтезе таким восстановителем служит NADPH; при
глюконеогенезе у животных восстановителем служит
HeNADPH, aNADH (см. главу И). Во-вторых, должен
быть доступен АТР как движущая сила синтеза. Именно
на выполнение этих двух условий - переноса
электронов с воды на NADP+ и синтеза АТР - расходуется
поглощенная при фотосинтезе энергия света
Место фотосинтеза - хлоропласт
Реакции фотосинтеза протекают в хлоропластах клеток
зеленых растений. Подобно митохондриям, хлоропласта
являются внутриклеточными органеллами, внешняя
часть 3 Метаболизм
182
мембрана которых проницаема для протонов, а
внутренняя - нет. Как и митохондрии, хлоропласты имеют
собственную ДНК. Она кодирует часть их белков, и
собственный аппарат биосинтеза белков, который по
своим свойствам подобен прокариотическому (см. с. 296).
Считается, что хлоропласты - далекие потомки прока-
риотических фотосинтезирующих одноклеточных
организмов, некогда вступивших в симбиоз с эукариотичес-
кими клетками.
В отличие от митохондрий, внутренние мембранные
структуры хлоропластов - тилакоиды представлены
замкнутыми уплощенными мембранными мешочками,
уложенными в стопки - граны — и соединенными
между собой мембранными выростами (ламеллами).
Пространство внутри хлоропласта (без гран) называют стро-
мой (рис. 14.2).
Внешняя
Внутренняя мембрана
мембрана
Грана
Строма
Тилакоиды
Ламеллы Внутритилакоидное
в строме пространство
Рис. 14.2. Строение хлоропласта
Граны образованы стопками тилакоидов
Хлорофилл (пигмент, улавливающий свет) и все
переносчики электронов находятся в мембране
тилакоидов, тогда как синтез углеводов из С02 и Н20
происходит в строме. Синтетические процессы в хлоропласте
называют темновыми, имея в виду, что они не
приводятся в действие светом, хотя и происходят на свету,
а являются следствием генерации N ADPH и АТР в
мембране тилакоидов. Распределение ролей между тилако-
идами и стромой иллюстрирует рис. 14.3.
Фотосинтетический аппарат
и его организация
в тилакоидной мембране
Стоит вспомнить, как устроена цепь переноса электронов
в митохондриях, потому что у нее много общего с
фотосинтетическим аппаратом. Во внутренней мембране
митохондрий находятся белковые комплексы четырех
«Световые реакции»
Мембрана тилакоида
гЬО
«Темновые реакции»
Строма хлоропласта
NADPH + Η
NADP+
Углево!
Рис. 14.3. Фотосинтетический процесс (изображено
схематично)
типов, между которыми перемещаются электроны,
используя для этого подвижные переносчики: убихинон -
низкомолекулярное жирорастворимое вещество -
и цитохром с — небольшой водорастворимый белок.
В тилакоидной мембране есть три типа комплексов
(рис. 14.4). Первые два связываются диффундирующим
переносчиком электронов - пластохиноном, похожим
по структуре на убихинон, а второй и третий -
небольшим водорастворимым белком пластоцианином,
также участвующим в переносе электронов. Он содержит
атом меди, который служит то донором, то акцептором
электронов, и поэтому находится поочередно в
состоянии Си+ или Си2+.
1М/\1 11-»+
Свет Свет
Комплекс цит bf \
NADP+
А
Ферредоксин
-NADPH
ФСИ
Qe
ФС1
2Н20 02 + 4Н+ Н+
Рее·
е~ Строма хлоропласта
Мембрана тилакоида
Внутренняя часть
тилакоида
Рис. 14.4. Световая фаза фотосинтеза (схематично)
Главная задача системы - передать электроны из воды на
NADP+ и создать на тилакоидной мембране протонный
градиент, достаточный для хемиосмотического синтеза АТР. Этот
градиент создается благодаря расщеплению воды
фотосистемой II и активному транспорту протонов комплексом ци-
тохромов bf. Электроны доставляются от фотосистемы II
к цитохромам bfc помощью пластохинона (Q). Все это
похоже на цепь передачи электронов в митохондриях (где Q -
убихинон). Пластоцианин (Рс), подобно цитохрому с в
митохондриях, служит белковым переносчиком электронов, а
ферредоксин расположен на противоположной стороне мембраны
глава 14 Фотосинтез
способ передачи энергии электронам воды
183
Эти три комплекса называются соответственно
фотосистемой II (ФСИ), комплексом цитохромов bfu
фотосистемой I (ФС1). Пусть вас не смущает, что
фотосистема II стоит в цепи раньше фотосистемы I - нумерация
отражает всего лишь очередность их открытия.
Функция всего этого аппарата заключается в осуществлении
реакции:
2NADP+ + 2Н20 -> 2NADPH + 2Н+ + 02.
Реакция сопровождается очень большим
увеличением свободной энергии. До сих пор мы рассматривали
только такие эндоэргонические реакции, которые реали-
зовывались за счет энергии, накопленной в виде
химических связей. При фотосинтезе используется энергия
света: на образование каждой молекулы NADPH
расходуется энергия двух поглощенных фотонов. Этой
энергии хватает на то, чтобы восстановить молекулы NADP+
водой, а излишек использовать на синтез АТР. Такой
расклад позволяет полностью удовлетворить
сформулированные выше два основных условия синтеза
углеводов из С02 и Н20: наличие восстанавливающего агента
и АТР. Теперь перейдем к рассмотрению механизмов
улавливания энергии света фотосистемами II и I.
Что такое хлорофилл?
В зеленых растениях рецептором, воспринимающим
свет, служит хлорофилл. В бактериях и водорослях есть
другие светоулавливающие пигменты, но здесь мы
ограничимся рассмотрением высших растений.
СНз
СН2 СН3
Н,С
Н2С=СН
связей, что определяет его интенсивную окраску. В
растениях присутствуют два хлорофилла (а и 6),
различающиеся боковыми группами: в хлорофилле Ъ вместо
одной из боковых метальных групп присутствует
альдегидная. Оба хлорофилла поглощают свет в голубой
и красной областях видимого спектра (хотя максимумы
поглощения слегка различаются) и отражают зеленый
свет, что и придает этому пигменту и листьям растений
соответствующую окраску.
Молекула хлорофилла, поглотив фотон, переходит
в возбужденное состояние, сопровождающееся
перемещением одного из электронов на более высокий
энергетический уровень (он перепрыгивает на новую атомную
орбиту). Однако возбужденная молекула нестабильна
и стремится вернуться к исходному состоянию. В
случае, когда фотон поглощается отдельной молекулой
хлорофилла, эта энергия затем рассеется в виде тепла и
света (флуоресценция), и ничего не произойдет. Если же
молекулы хлорофилла упакованы плотно и
упорядочение, как в тилакоидной мембране, возбуждение
передается соседним молекулам при помощи так называемого
резонансного переноса энергии. В зеленых растениях
молекулы хлорофилла упакованы в функциональные
комплексы, называемые фотосистемами, поэтому
область распространения энергии ограничена размерами
этих комплексов (рис. 14.5).
В каждой фотосистеме наряду с большим числом
молекул «обычного» хлорофилла есть так называемые
реакционные центры, которые содержат предположительно
Антенные
молекулы
хлорофилла
Реакционный центр
К цепи переноса
электрона
СН2—СН2—С—О— Фитил
Структура хлорофилла а
Хлорофилл представляет собой тетрапиррол,
напоминающий по строению гем (см. главу 27). От гема
его отличает природа центрального атома (магний
вместо железа), а также строение боковых групп. Одна из
этих групп в хлорофилле содержит длинную
гидрофобную углеводородную цепь, которая служит якорем,
удерживающим хлорофилл в липидном бислое. Как и гем,
хлорофилл обладает системой сопряженных двойных
Фотон
Рис. 14.5. Активация молекулы хлорофилла в
реакционном центре
Реакционный центр представлен светлым кружком. Молекула
хлорофилла в этом центре возбуждается благодаря
резонансному переходу энергии от улавливающих квант света
антенных молекул хлорофилла (темные кружки)
часть 3 Метаболизм
184
две молекулы хлорофилла в комплексе с белками. В этих
центрах у хлорофилла энергия возбужденного
состояния невелика, так что ее резонансного переноса на
свободные молекулы хлорофилла не происходит. Таким
образом, реакционный центр представляет собой нечто
вроде ловушки, в которую рано или поздно попадает
энергия любого фотона, поглощенного любой
«обычной» молекулой хлорофилла в пределах данной
фотосистемы. Как только это происходит, электрон,
находящийся на верхнем энергетическом уровне, передается
акцептору, обладающему соответствующим редокс-по-
тенциалом. Этот акцептор стоит в начале
фотосинтетической цепи переноса электронов.
Итак, фотосистемы представляют собой сложные
комплексы, состоящие из молекул хлорофилла,
улавливающих свет, реакционного центра и цепи переноса
электронов. Хлорофилл в составе реакционного
центра фотосистемы II обозначают как Р680, а в составе
фотосистемы I - Р700 (от англ. Pigment - пигмент; числа
соответствуют длине волны максимума поглощения
света в нм). Молекулы хлорофилла, закачивающие
уловленную энергию в такие центры, называют антенными.
Механизм светозависимого
восстановления NADP+
На рис. 14.6 изображена так называемая Z-схема двух
фотосистем, демонстрирующая не только организацию
цепи переноса электронов, но также редокс-потенциа-
лы отдельных компонентов. Почему фотосистем две?
Ответить на этот вопрос поможет следующая аналогия:
чтобы питать трехвольтовую лампочку, нужны две
последовательно включенные полуторавольтовые
батарейки. При фотосинтезе восстановление NADP+ играет роль
«нагрузки», а «батарейками» служат фотосистемы II и I.
-1,2
-0,8 А
-0,4 -|
Шкала
энергии
или о J
Редокс-
потенциал
+0,4 -\
+0,8 А
+1,2 А
+1,6
Фотосистема I
Фотосистема 11
Место
расщепления
воды
2Н70
Возбужденное
состояние
Р680
Μη
4е~ е'
Ph
щЪ
Возбужденное
состояние
Р700
Ι-
\сс
^е~
Р700
основное
состояние
А
NADP+ NADP + Н+
4Н+ + 02
Возникают
внутри
тилакоида
ι
Синтез АТР
Р680
основное
состояние
Закачивание
протонов
внутрь тилакоида
Протон
Синтез АТР
Протон
Рис. 14.6. Фотосинтез: перенос электронов от воды к NADP+ (Z-схема)
Анализ этого рисунка следует начать с переноса электрона от возбужденной молекулы Р680 к Р700. Р680 затем приобретает
утраченный электрон от воды и ожидает нового акта возбуждения. Электрон, поступивший на Р700, после возбуждения молекулы
передается на NADP+. Ph - феофитин; Q - пластохинон; Рс - пластоцианин; Fd - ферредоксин; Fp - ферредоксин-ЫАОР-редуктаза
глава 14 Фотосинтез -
способ передачи энергии электронам воды
185
В действительности эти «батарейки» создают большую
разность редокс-потенциалов, чем нужно для
восстановления NADP+, и излишек расходуется на синтез АТР.
Начнем с хлорофилла Р680 в реакционных центрах
фотосистемы II. В темноте он находится в основном
(невозбужденном) состоянии, не проявляя никаких
восстановительных свойств. Когда Р680 получает энергию
фотона от антенного хлорофилла, он переходит в
возбужденное состояние и стремится отдать
возбужденный электрон, оказавшийся на верхнем энергетическом
уровне. В результате этот электрон приобретает
переносчик электронов фотосистемы II - феофитин. Это -
пигмент; по своему строению он похож на хлорофилл,
но не содержит Mg2+.
Две восстановленные молекулы феофитина
последовательно отдают полученные электроны на
восстановление пластохинона - растворимого в липидах
переносчика электронов от фотосистемы II к комплексу
цитохромов bf.
Н,С
Н3С
2е~
2Н+
R = длинная
углеводородная
гидрофобная
цепь
Последний состоит из двух цитохромов и
железо-серного центра. Он осуществляет перенос электронов
от восстановленного пластохинона к пластоцианину.
Что же происходит с фотосистемой I? На хлорофилл
Р700 в реакционном центре этой фотосистемы также
стекает энергия фотона, уловленного антенным
хлорофиллом, так что он становится мощным
восстанавливающим агентом. Электрон с возбужденного
хлорофилла Р700 передается по короткой цепочке переносчиков
(на рис. 14.6 она для простоты не показана) на ферре-
доксин - водорастворимый белок стромы, содержащий
электроноакцепторный кластер атомов железа.
Именно ферредоксин с помощью FAD-зависимого
фермента ферредоксин^АВР+-редуктазы восстанавливает
NADP+ в NADPH.
2Ферредоксинвосст + NADP+ + 2Н+ ->
► 2Ферредоксин0
NADPH + Н+
Ясно, что обе фотосистемы, отдав по электрону,
должны получить их обратно, чтобы вернуться в исходное
состояние.
Иными словами, из восстановителей они переходят
в ранг окислителей. Р700+ приобретает электрон у
восстановленного пластоцианина:
Р700+ + РсСи+ -> Р700 + РсСи2+ .
Что же касается фотосистемы II, то Р680+
возвращается в исходное состояние необычным образом:
электрон он получает от воды!
Расщепление воды фотосистемой II
Р680+ очень сильный окислитель; его сродство к
электрону даже выше, чем у кислорода, так что он вполне
может извлечь электрон из воды. Чтобы превратить
2 молекулы воды в 4 протона (Н+) и 02, требуется отнять
у них 4 электрона. При этом, как и при восстановлении
02 в митохондриях, необходимо избежать образования
реакционноспособных промежуточных продуктов
вроде перекиси водорода, крайне опасных для
биологических систем. Эту задачу в фотосистеме II
выполняет Мп2+-содержащий белковый комплекс (рис. 14.8, а),
известный как водорасщепляющий центр, который
отбирает электроны у воды, расщепляя ее на кислород
и протоны, и передает их на Р680+, после чего последний
готов к дальнейшим реакциям.
Как генерируется АТР?
Комплекс цитохромов bf который использует пластогид-
рохинон для восстановления пластоцианина, похож на-
митохондриальный комплекс III (см. рис. 8.19) тем, что
перенос им электронов сопряжен с трансмембранным
транспортом протонов с внешней стороны тилакоидной
мембраны вовнутрь. По аналогии с митохондриальным
Q-циклом можно ожидать, что при этом внутрь тилако-
ида попадут четыре протона на каждую окисленную
молекулу пластогидрохинона (см. с. 127). Однако такая
стехиометрия не подтверждается экспериментальными
данными. При расщеплении воды образуются также
протоны внутри тилакоидов. Оба эти процесса (перенос
протонов через мембрану и их образование внутри тилако-
ида) способствуют уменьшению рН внутри тилакоида
до 4,5; на мембране тилакоида создается протонный
градиент. Захват протона при восстановлении NADP+ фер-
редоксином в строме способствует поддержанию
трансмембранного градиента.
Таким образом, фото синтетический аппарат
включает в себя протонный насос. Этот насос создает на ти-
лакоидальной мембране Н+-градиент, достаточный для
хемиосмотического синтеза АТР из ADP и Pf.. Работа
протонного насоса не связана жестко с восстановлением
часть 3 Метаболизм
186
NADP+. Если весь NADP+ восстановился в NADPH, фер-
редоксин отдает электроны цитохромному комплексу bf
а тот переносит их на пластоцианин. Такой перенос
сопряжен с закачиванием протонов. Электроны при этом
движутся по кругу: фотосистема I —> ферредоксин —>
комплекс цитохромов bf —> пластоцианин —>
фотосистема I (рис. 14.7). Поэтому такой вариант синтеза АТР
называют циклическим фотофосфорилированием,
управляемым циклическим током электронов.
Н+
λ
н+ /ЕслиИАОР"
ι I недоступен
NADP . -
Ферредоксин
- NADPH
е
\
Рее
Строма хлоропласта
\
ФС1 Мембрана тилакоида
Внутреннее пространство
тилакоида
Н+
Рис. 14.7. Циклический перенос электронов
Когда все молекулы NADP4 восстановлены, ферредоксин
переносит электроны на комплекс цитохромов bf. Это приводит
к интенсификации транспорта протонов и, следовательно,
способствует синтезу АТР
_,, Λ Мп-центр
2Н20 i-^ 4Н+ + 4е" + 02
Высвобождаются _ Выделяется в
Переносятся к четырем ,
втилакоид г тл^пги. атмосферу
молекулам Р680+ от которых Ύ rj
передаются фотосистеме II
Свет NADP*-J—NADPH
Н+ \ / е"
I Ферредоксин
Комплекс цит bf ·.
С" вот
ФСП
Μη
2Н20 02 + 4Н+ Н+
V I
Φα
о2
АТР-синтаза
Внутреннее пространство
тилакоида
1ембрана тилакоида
Строма хлоропласта
Полностью схема электронных и протонных
потоков при фотосинтезе в хлоропластах представлена на
рис. 14.8.
Почему протонный насос в митохондриях
выкачивает протоны из матрикса в окружающую среду, а насос
втилакоидах закачивает их внутрь? Согласно одной
из гипотез, как митохондрии, так и хлоропласты
являются потомками прокариотических организмов,
обитавших внутри эукариотических клеток. Поэтому
направление активного транспорта протонов и ориентация
протонной АТР-синтазы в мембране хлоропластов и
митохондрий должны совпадать. Однако мембрана тила-
коидов образуется отшнуровыванием впячиваний
внутренней мембраны хлоропласта; следовательно, ее
полярность противоположна, что объясняет, почему
протонные градиенты и АТР-синтазы митохондрий и ти-
лакоидов имеют противоположную ориентацию.
Как С02 превращается в углеводы?
Среди всех рассмотренных биохимических процессов
фотосинтез отличается тем, что восстановление N ADP+
и синтез АТР происходят за счет энергии света.
ADP + Р,-
Н+
АТР
Рис. 14.8. Электронные и протонные потоки,
происходящие внутри тилакоида (схематично)
а - Суммарная реакция в Мп2+-содержащем центре;
б - движение протонов и электронов
Все дальнейшие химические превращения, в ходе
которых образуется глюкоза и другие углеводы,
свойственны лишь растениям и ничем принципиально не
отличаются от обычных ферментативных реакций.
Ключевым метаболитом здесь является 3-фосфогли-
церат, из которого затем углеводы синтезируются так же,
как и в печени (см. рис. 11.5), с той лишь разницей, что
восстановителем в этих процессах служит NADPH,
aHeNADH (рис. 14.9). Таким образом, проблема
сводится к выяснению механизма образования 3-фосфогли-
церата при фотосинтезе.
Как образуется 3-фосфоглицерат?
Из всех ферментов самым распространенным на Земле
является рибулозо-1,5-дифосфат-карбоксилаза, которую
глава 14 Фотосинтез - способ передачи энергии электронам воды
187
Запасные углеводы (крахмал)
t
Глюкозо-1 -фосфат
t
Глюкозо-6-фосфат
t
Фруктозо-6-фосфат
н7о
л
Фруктозо-1,6-дифосфат
Дигидроксиацетонфосфат , *~ Глицеральдегид-3-фосфат
'N
к
NADP+
NADPH +Н+
1,3-Дифосфоглицерат
ADP
Г
К
АТР
З-Фосфоглицерат
Рис. 14.9. Образование крахмала из 3-фосфоглицерата при
фотосинтезе
Этот процесс сходен с глюконеогенезом, отличаясь лишь тем,
что восстанавливающим агентом здесь служит NADPH, а не
NADH. В отличие от синтеза гликогена, активированной
формой глюкозы при получении крахмала служит ADP-глюкоза,
а не UDP-глюкоза
часто называют кратко «Rubisco». Именно этот фермент
использует С02 для синтеза 3-фосфоглицерата.
СН2ОРОГ
I
снон
со2 + н2о
СН2ОРО§
снон
I
С00"
2Н+
СНОН и,Р!1буЛ030;1'5' С0°"
упип дифосфаткарбоксилаза |
СН2ОРОГ
Рибулозо-1,5-дифосфат
СНОН
СН2ОРО|
Две молекулы 3-фосфоглицерата
Напомним, что рибоза является альдопентозой, а ри-
булоза (ее оптический изомер) - кетопентозой. Карбок-
силаза расщепляет рибулозо-1,5-дифосфат на 2
молекулы 3-фосфоглицерата, фиксируя при этом 1 молекулу
С02. Затем 3-фосфоглицерат превращается в
разнообразные моносахариды.
Из чего синтезируется
рибулозо 1,5-дифосфат?
Ответ, в принципе, очень прост. Из 6 молекул рибуло-
зодифосфата (всего 30 С-атомов) и 6 молекул С02
(6 С-атомов) образуются 12 молекул 3-фосфоглицерата
(36 С-атомов). Две из них (2С3) направляются на
синтез углеводов (С6), который протекает согласно схеме,
показанной на рис. 14.9.
Оставшиеся 10 молекул фосфоглицерата (всего 30
С-атомов) преобразуются в 6 молекул рибулозодифос-
фата (всего 30 С-атомов). Этот процесс достаточно
сложен и сводится к различным превращениям С3-, С4-,
С5-, С6- и С7-сахаров с помощью альдолазы и транс-
кетолазы (см. с. 181; напомним, что транскетолаза
переносит С2-блоки). В упрощенном виде происходящее
можно описать следующей схемой:
сз+ сз->сб>
С6 + С3
> С4 + С5,
С4 + С3 -> С7,
ь ол
Τ анскетолаза
Альдолаза
► С5 + С5.
С7+ С3
Суммарный результат: 5 С3 —> 3 С5.
Итак, 6 молекул рибулозодифосфата, 6 молекул С02
и 6 молекул Н20 преобразуются в 12 молекул
фосфоглицерата; они превращаются в 6 молекул
рибулозодифосфата и 1 молекулу фруктозо-6-фосфата, из которой
затем могут быть образованы запасные углеводы,
например крахмал. Весь процесс в целом известен как цикл
Кальвина (рис. 14.10). Суммарную реакцию описывает
довольно громоздкое уравнение:
6С02 + 18АТР + 12NADPH + 12Н+ + 12Н20 ->
^C6H1206+18ADP+18P,.+12NADP+ + 6Н+ .
На чем споткнулась эволюция?
Химические процессы, протекающие в организме,
настолько совершенны и эффективны, что кажется
невероятным, чтобы эволюция споткнулась на едва ли
не самом главном из ферментов - фиксирующей С02 ри-
булозо-1,5-дифосфат-карбоксилазе.
часть 3 Метаболизм
188
6 Рибулозо-1,5-дифосфат
6ADP
6 ATP
Ν
6 C02 + 6 Η20
12 З-Фосфоглицерат
V
12 ATP
12NADPH+H+
[Ч^ 12ADP+ 12 Pf
12 NADP+
\
2 молекулы
12 Глицеральдегид-3-фосфат
К) молекул /\
(это не прямое J
превращение) У
^ 1 Фруктозо-1,6-дифосфат
6 Рибулозо-5-фосфат I
t
Запасные углеводы
Рис. 14.10. Реакции, входящие в цикл Кальвина (упрощенная схема)
Здесь показана лишь стехиометрия превращений, но не их механизм
На заре фотосинтеза в первичной атмосфере совсем
не было кислорода, а содержание С02 намного
превышало нынешнее. Однако, благодаря фотосинтезу,
содержание кислорода постепенно увеличивалось. Кар-
боксилаза реагирует не только с С02, но и с кислородом.
Эти две реакции конкурентны, так что фермент
правильнее было бы называть рибулозо-1,5-дифосфаткар-
боксилазой/оксигеназой. Реакция карбоксилазы с
кислородом совершенно бесполезна, она приводит к
бессмысленному расходу АТР (этот процесс называют
фотодыханием).
На ярком свету, при достаточно высокой температуре
воздуха фотосинтез в зеленом листе настолько
интенсивен, что в его непосредственном окружении можно
зарегистрировать падение уровня С02 и увеличение
содержания кислорода. В таких условиях и протекают
реакции оксигенирования, которые могут на треть
уменьшить усвоение С02. В давние времена, когда уровень
С02 в атмосфере был существенно выше, чем уровень
02, оксигенирование не могло иметь места, но в
настоящее время это не так.
Странно, что в ходе эволюции не происходило
усовершенствования карбоксилазы. Возможно, тому есть
неведомые нам причины, ибо трудно предположить
небрежность в столь важном для судеб всего живого
процессе. Известно, правда, что у некоторых растений
существуют специальные приспособления, позволяющие
поддерживать высокий уровень С02 вокруг тех клеток,
где функционирует карбоксилаза. Вследствие этого
потери от оксигенирования сведены у них до
минимума. К числу таких растений, произрастающих на ярком
солнце и в теплом климате, относятся кукуруза и
сахарный тростник.
С4-путь
Название С3-растения означает, что первым продуктом
фотосинтеза, который удается обнаружить, используя
меченый 14С02, является С3-соединение - 3-фосфо-
глицерат, образующийся при участии рибулозо-1,5-ди-
фосфаткарбоксилазы. Однако у некоторых растений
первичным продуктом фиксации атмосферного С02
является оксалоацетат (рис. 14.11). Оксалоацетат
содержит четыре атома углерода; отсюда другое название -
С4-растения.
С3- и С4-растения различаются анатомическим
строением своих листьев. У последних в мезофильных
клетках на поверхности листа нет рибулозо-1,5-дифос-
фат-карбоксилазы, а фиксация С02 у них происходит
путем карбоксилирования фосфоенолпирувата. Эту
реакцию катализирует фосфоенолпируват-карбоксила-
за - фермент, не встречающийся у животных:
С02 + Н20 + Фосфоенолпируват —> Оксалоацетат + Р/.
У этого фермента высокое сродство к С02, так что
никаких проблем с оксигенированием не возникает.
Оксалоацетат затем восстанавливается в малат и
транспортируется в клетки, где протекает фотосинтез. Там же
глава 14 Фотосинтез — способ передачи энергии электронам воды
189
NADPH NADP+
V У*
Оксалоцетат ^^ ^ ► Малат
NADP+ -
малатдегидрогеназа
г/
-► Малат
' Фосфоенилпируваткарбоксилаза NADP - I
«малик-фермент>|\^
СО,
Пируват-Рг-дикиназа
ФЕП^ —^—^ Пируват -*
AMP + PPf- ATP + Ρ,
NADP+
NADPH +H+
+ C02
- Пируват
I
Углеводы
икл
Кальви
Рис. 14.11. С4-Путь, обеспечивающий повышенное содержание С02 в фотосинтезирующих клетках
Механизм образования оксалоацетата из пирувата здесь коренным образом отличается от аналогичных процессов в животных
клетках, где пируват никогда не превращается в фосфоенолпируват, а последний никогда не карбоксилируется. Кроме того,
здесь для восстановления оксалоацетата используется не NADH, a NADPH
малат декарбоксилируется так называемым
малик-ферментом (см. с. 141), благодаря чему в клетках в 10-60 раз
возрастает концентрация С02:
Малат + NADP+ -> Пируват + С02 + NADPH + Н\
Ясно, что таким образом создаются условия для
эффективной работы рибулозо-1,5-дифосфат-карбок-
силазы. Образующийся пируват возвращается в ме-
зофильные клетки, где перерабатывается пируватфос-
фат-дикиназой снова в фосфоенолпируват. Эта реакция
любопытна тем, что участвующий в ней АТР
превращается не в ADP, а в AMP:
СН3-СО-СОО" + АТР + Р,
СН2=С—СОО"
I 2_
ΟΡΟ?
+ AMP + РР, + Н\
У животных превращение пирувата в
фосфоенолпируват происходит совершенно иначе - через образование
оксалоацетата (см. с. 148). Как только фосфоглицерат
образуется в фотосинтезирующих клетках, он сразу
вовлекается в цикл Кальвина.
С4-путь можно рассматривать как плату за
полноценный фотосинтез, поскольку на синтез фосфоенолпиру-
вата и транспорт кислот расходуется драгоценный АТР.
Тем не менее в жарком климате он сулит значительные
преимущества. Во всяком случае, кукуруза и сахарный
тростник - одни из главных поставщиков углеводов.
Отдельные биохимические реакции у С4-растений
могут сильно различаться. Например, у некоторых
видов первой С4-кислотой оказывается не малат, а аспар-
тат. В таких растениях много аспартатаминотрансфе-
разы вместо ЫАОР+-зависимой малатдегидрогеназы
(малик-фермента; см. рис. 14.11). В
фотосинтезирующих клетках С4-растений есть дополнительные декар-
боксилазы С4-кислот. Две такие декарбоксилазы
расположены не в цитоплазме, а в хлоропластах. Однако,
какие бы механизмы ни использовались, цель одна -
увеличить содержание С02 в клетках, где работает
рибулозо-1,5-дифосфат-карбоксилаза.
Дополнительная литература
Использование энергии света
Slater Ε. С. The Q cycle, an ubiquitous mechanism of
electron transport // Trends Biochem. Sci. 1983. Vol. 8.
P. 239-242.
(Описаны протонные насосы, существующие как в
митохондриях, так и в хлоропластах.)
часть 3 Метаболизм
190
С4-путь
Rawsthorne S. Towards an understanding of C3-C4
photosynthesis // Essays Biochem. 1992. Vol. 27. P. 135-146.
(Обсуждается распределение С3 и С4 биохимических
систем в растительном царстве и их функционирование.)
Вопросы к главе 14
1. Объясните, чем различаются «темновые» и
«световые» реакции фотосинтеза.
2. Что означает выражение «антенный хлорофилл»?
3. Что означают термины «фотофосфорилирование»
и «циклическое фотофосфорилирование»?
4. Для окисления воды требуются очень сильные
окисляющие агенты. Что окисляет воду при фотосинтезе?
5. Протонный насос обусловливает транспорт
электронов в фотосистеме II (ФСП) и обеспечивает
движение протонов с внешней стороны тилакоидов внутрь.
В митохондриях протонный насос работает в
обратном направлении. Объясните почему.
6. Если растение на свету на очень короткое время
поместить в атмосферу с радиоактивным 14С02, метка
сначала появляется в 3-фосфоглицерате. Почему?
7. Объясните, что такое цикл Кальвина.
8. Опишите биосинтетические реакции на пути от
3-фосфоглицерата до крахмала.
9. Фермент рибулозо-15-дифосфат-карбоксилаза (Ru-
bisco) может реагировать и с кислородом, и с С02.
При низких концентрациях С02 (при интенсивном
солнечном свете) реакция оксигенации
интенсифицируется. Как растения избегают оксигенирования под
действием рибулозо-1,5-дифосфат-карбоксилазы?
10. В С4-растениях пируват перерабатывается в фосфо-
енолпируват с использованием АТР. Как это можно
прокомментировать?
глава 14 Фотосинтез
способ передачи энергии электронам воды
191
глава
15
Метаболизм аминокислот
При нормальном пищевом рационе переваривание
белков в тонком кишечнике приводит к всасыванию
довольно большого количества аминокислот. Эти
аминокислоты поступают в воротную вену и доставляются
кровотоком прямо в печень. Все клетки, за исключением
некоторых высокоспециализированных (таких,
например, как эритроциты), используют аминокислоты для
синтеза белков и множества других веществ:
компонентов мембран, нейромедиаторов, гема и т. п. Поэтому все
клетки обладают специальными системами транспорта
аминокислот, поскольку последние в ионизированном
состоянии не могут преодолеть липидный бислой
плазматических мембран.
Какой-либо специальной формы хранения
аминокислот, подобно высокомолекулярным полисахаридам,
которые при необходимости расщепляются до мономеров,
не существует. Поэтому резервными для аминокислот
веществами служат все функциональные белки, но
основными здесь являются белки мышц, поскольку их
больше всего. Однако эти белки участвуют в
сократительной деятельности мышц, и при их интенсивном
использовании для удовлетворения потребностей в
аминокислотах, например, при глюконеогенезе в печени,
наблюдается мышечная атрофия. Такая ситуация чревата
для человека неприятностями, поскольку в ходе
эволюции мы утратили способность синтезировать половину
(10 из 20) аминокислот (их называют незаменимыми).
Любопытно, что к их числу относятся и те
аминокислоты, синтез которых включает особенно много стадий
и требует большого числа специализированных
ферментов, кодируемых многими генами. Иными словами, речь
идет об аминокислотах, которые «дороги» в производстве.
Почему так получилось, можно только гадать.
Возможно, что это - чисто экономическая мера, так как
выгоднее получать самые сложные аминокислоты с
пищей, самостоятельно производя лишь самые простые.
В то же время, если бы человек получал все
необходимые продукты от дикой природы, его пищевой
рацион, вероятно, содержал бы все необходимые
аминокислоты. В подобном случае неспособность синтезировать
отдельные аминокислоты не так уж страшна по
сравнению с отсутствием пищи как таковой.
Развитие сельского хозяйства предоставило
быстро размножающемуся человеческому роду источник
химической энергии в виде растительных углеводов.
Растительная пища позволяет людям выжить, но далеко
не всегда удовлетворяет их потребности в незаменимых
аминокислотах, поскольку белки некоторых
культивируемых растений, например кукурузы, бедны лизином
и триптофаном. Однако даже при длительном
употреблении пищи, богатой полноценными белками, человек
не может отложить про запас незаменимые
аминокислоты. Лишь незначительная их доля используется по
назначению, а невостребованная часть просто окисляется
или перерабатывается в жиры или гликоген. В
некоторых ситуациях это равносильно сжиганию бриллиантов
ради обогрева.
Результатом неадекватного пищевого рациона,
недостаточного даже по одной незаменимой
аминокислоте, является патологическое состояние, называемое
квашиоркор. Так как практически все белки содержат
полный набор аминокислот, недостаток любой из них
приведет к синтезу неполноценных белковых молекул.
Это вызывает истощение, апатию, недостаточный рост,
а также снижение уровня сывороточных белков в
крови. Последнее обуславливает уменьшение онкотическо-
го давления крови и, по-видимому, является
непосредственной причиной отеков.
Возникает порочный круг. Выстилающие тонкий
кишечник клетки должны постоянно обновляться и
производить пищеварительные ферменты. При квашиор-
коре их способности к этому существенно снижены.
В результате даже та пища, которая доступна, плохо
переваривается и всасывается. От квашиоркора особенно
страдают дети, поскольку растущему организму нужно
синтезировать много белков.
Получается, что утрата в ходе эволюции способности
к синтезу некоторых аминокислот и отказ от запасания их
излишков оборачиваются для человека крупными
неприятностями. Нельзя, правда, исключить, что у эволюции
были на то серьезные причины. Например,
промежуточные продукты биосинтеза незаменимых аминокислот
могут быть токсичными для высших организмов. В
частности, они могут нарушать развитие или работу мозга.
Баланс азота в организме
Аминокислоты участвуют в поддержании азотистого
баланса в организме Если организм потребляет азота
столько же (главным образом в виде аминокислот),
часть 3 Метаболизм
192
сколько выделяет, то такое стационарное состояние
называют сбалансированным по азоту. В период роста или
при заживлении ран азота потребляется больше, чем
выделяется - баланс считается положительным. Баланс
азота становится отрицательным, если азота
потребляется меньше, чем выделяется; это наблюдается,
например, при голодании, когда мышечные белки
перерабатываются в глюкозу, а азот выводится с мочой. У животных
белки находятся в состоянии непрерывного
кругооборота: они постоянно расщепляются и ресинтезируются.
Хотя большая часть аминокислот, высвобождающихся
при расщеплении белков, вовлекается в последующий
синтез белка, мы ежедневно теряем в виде мочевины
около 0,3% азота нашего организма.
Незаменимыми считаются аминокислоты, удаление
которых из диеты приводит к отрицательному азотному
балансу и, как результат, к останавке роста
экспериментальных животных. Некоторые из таких аминокислот
являются абсолютно незаменимыми: для человека это
лизин, фенилаланин и триптофан. Ниже приводится
список заменимых и незаменимых аминокислот для
человека.
Заменимые
аминокислоты
Алании
Аспаргин
Аспаргиновая кислота
Цистеин**
Глутаминовая кислота
Глутамин
Глицин
Пролин
Серии
Тирозин***
Незаменимые
аминокислоты
Аргинин*
Гистидин
Изолейцин
Лейцин
Лизин
Метионин
Фенилаланин
Треонин
Триптофан
Валин
* Требуется лишь во время роста.
** Может синтезироваться из фенилаланина.
*** Может синтезироваться из метионина.
Тирозин не является абсолютно незаменимой
аминокислотой, поскольку он может быть получен из
фенилаланина, если тот поступает в организм в
достаточном количестве. Точно так же млекопитающие могут
синтезировать цистеин из другой незаменимой
аминокислоты - метионина. В целом нельзя сформулировать
четкие правила, которым подчинялась бы потребность
организма в аминокислотах. «Первоклассные» белки
богаты незаменимыми аминокислотами, а в
растительных белках некоторых незаменимых аминокислот мало.
Поскольку аминокислотный состав разных белков
сильно различается, вегетарианская диета заведомо должна
быть более разнообразной.
Общий метаболизм аминокислот
Общая схема метаболизма аминокислот представлена
на рис. 15.1.
NH3+
Белки
пищи
— R-CH-COO-
Аминокислоты
Белки
организма
Дезаминирование
избытка
аминокислот
А минны и а ют
Специфические компоненты клеток:
нейромедиаторы, катехоламины,
компоненты мембран, гем,
нуклеиновые основания и т.п.
R-CO-COO-
Кетокислоты
Мочевина, Пируват или
выделяемая компоненты цикла
с мочой лимонной кислоты
Кетогепные
аминокислоты
Ацетил-СоА
/\
СОг + НгО Глюкоза
(гликоген)
Жир С02 + Н20
Рис. 15.1. Катаболизм аминокислот
Некоторые аминокислоты отчасти кетогенны, а отчасти глю-
когенны
Аспекты метаболизма аминокислот
Избыток аминокислот (не использованных в синтезе
белка и на другие специфические нужды) расщепляется для
производства энергии или создания энергетических
запасов (жиров или гликогена). Возникает ряд существенных
вопросов, которые необходимо рассмотреть отдельно:
как организм синтезирует заменимые аминокислоты?
Как некоторые аминокислоты превращаются в другие
физиологически важные низкомолекулярные
соединения? В этой главе мы коснемся следующих проблем.
1. Как из аминокислот удаляются аминогруппы
(иными словами, как происходит дезаминирование). Хотя
все аминокислоты отличаются по строению,
большинство из них дезаминируется сходным образом,
что значительно упрощает изучение этой проблемы.
2. Что происходит с кетокислотами (углеродными
скелетами), образующимися при дезаминировании
аминокислот. У каждой из них своя собственная
глава 15 Метаболизм аминокислот
193
метаболическая судьба, и здесь мы рассмотрим лишь
отдельные примеры, особо интересные или важные
для понимания метаболизма в целом.
3. Как удаленный из аминокислот азот
перерабатывается в мочевину.
4. Как синтезируются аминокислоты. У животных это
касается, естественно, только заменимых
аминокислот. Если есть соответствующая кетокислота (т. е.
углеродный скелет аминокислоты), синтез часто
сводится к обращению дезаминирования. Мы
рассмотрим лишь несколько таких примеров. В
бактериях и растениях все аминокислоты синтезируются
по индивидуальным планам, а исходные вещества
поступают из реакций общего метаболизма.
Синтетические возможности животных ограничены
существованием незаменимых аминокислот. Изучение
путей биосинтеза всех аминокислот - это гигантский
объем информации. Мы ограничимся здесь лишь
наиболее интересными примерами.
5. Некоторые превращения аминокислот весьма
любопытны и важны для создания целостной картины
аминокислотного метаболизма. Часть из них также
будет рассмотрена в этой главе.
Дезаминирование аминокислот
Сначала немного химии. Шиффовы основания
образуются в результате обратимой реакции между
карбонильной группой альдегида или кетона со свободной
аминогруппой:
I I
С=0 + H2N—R =^ C=N—R + Н20
I I
Эта реакция обратима, следовательно, Шиффово
основание легко гидролизуется. Если R=H, то в
результате гидролиза высвободится аммиак (NH3). Таким
образом, отщепление двух атомов водорода от
аминокислоты приведет к дезаминированию, а связывание пары
атомов водорода - к синтезу аминокислоты из кетокис-
лоты и аммиака (для простоты приведены неионизиро-
ванные формы соединений):
ι ι Н2° ι
I -2Н I V I
CHNH2 » C=N—Η ^— С=0 + ΝΗ3.
I I I
Теперь вернемся к биологическому
дезаминированию. Одной из важнейших в метаболизме
аминокислот является глутаминовая кислота, дезаминирование
часть 3 Метаболизм
194
которой катализирует глутаматдегидрогеназа. Она
использует в качестве окислителя либо NAD+, либо NADP4
(при физиологических значениях рН NH3 протонирован
и находится в ионизированной форме - NH4+):
COO" COO"
I I
CH2 CH2
CH2 + NAD(P)+ + H20 — NAD(P)H + CH2 + NH4+.
CHNH3+ C=0
I I
COO" COO"
Глутамат а-Кетоглутарат
Глутаматдегидрогеназа - ключевой фермент
дезаминирования, участвует в окислении многих аминокислот.
Она аллостерически ингибируется АТР и GTP (это
индикаторы высокого уровня энергии: запасы энергии
большие - топлива не нужно) и активируется ADP и GDP.
Увеличение содержания последних свидетельствует
о необходимости окислительного фосфорилирования
(энергетические запасы малы - нужно топливо).
α-Кетоглутарат участвует в цикле лимонной
кислоты, что делает возможным окисление глутаминовой
кислоты (после дезаминирования) до Н20 и С02
Поскольку α-кетоглутарат может превращаться в окса-
лоацетат (см. рис. 8.12), глутаминовая кислота при
определенных условиях может участвовать в синтезе
глюкозы (см. главу И, рис. 11.3). Таким образом,
глутаминовая кислота принадлежит к числу глюкогенных
аминокислот.
Для других аминокислот не существует
соответствующих дегидрогеназ. Как же они дезаминируются?
Для этого есть несколько специальных механизмов,
но большинство из них основано на переносе
аминогруппы с аминокислот на α-кетоглутарат, в результате
чего образуется соответствующая кетокислота и
глутамат. Последний дезаминируется глутаматдегидроге-
назой. Иными словами, дезаминирование других
аминокислот протекает в две стадии. Первая называется
трансаминированием (или переаминированием),
а вторая - дезаминированием.
Стадия трансаминирования может быть
представлена следующим образом:
С00" С00"
I I
R CH2 R СН2
II II
CHNH3+ + СН2 С=0 + СН2
I I I I
С00" С=0 COO" CHNH3+
I I
COO" COO"
Предположим, что R=CH3, следовательно,
аминокислота- аланин. Дезаминирование аланина происходит
в соответствии со следующей схемой:
Аланин + α-Кетоглутарат -^ Пируват + Глутамат, (1)
Глутамат + NAD+ + Н20 -> α-Кетоглутарат + NADH + NHJ .(2)
Итого:
Аланин + NAD+ + Н20 -> Пируват + NADH + NHJ .
В совокупности обе стадии этого процесса - транс-
аминирование и дезаминирование глутамата - по
понятной причине называют трансдезаминированием.
Ферменты, катализирующие трансаминирование, называют
трансаминазами, или аминотрансферазами.
Известно много трансаминаз, специфичных к различным
аминокислотам, и большинство из них может быть дезами-
нировано с их участием.
Обратимость трансаминазной реакции означает, что
аминокислоту можно синтезировать из
соответствующей кетокислоты (однако кетокислоты
соответствующих незаменимых аминокислот не синтезируются в
организме). Примером трансаминирования может служить
малат-аспартатный челнок (см. с. 116), в котором
задействована обратимая реакция:
С00" С00" С00" С00"
СН2 С=0 Аспартатамино- ^н £HNH +
I I трансфераза \ I
СН2 + СН2 , СН2 + СН2
I I II
CHNH? С00" С=0 С00"
СН7ОН
сно
I
С00"
Глутамат Оксалоацетат
С00"
α-Кетоглутарат Аспартат
Механизм реакций трансаминирования
(переаминирования)
В активных центрах всех трансаминаз имеется прочно
связанный кофермент пиридоксаль-5'-фосфат (ПФ).
Он участвует во многих ферментативных
превращениях аминокислот, выступая в качестве электрофильного
интермедиата. Его главные функции заключаются в том,
чтобы сначала в качестве акцептора принять
аминогруппу от аминокислоты, а затем в качестве донора передать
ее кетокислоте. Все это происходит при участии одного
фермента. Как это часто бывает, кофермент оказывается
производным витамина (в данном случае В6). В
действительности группа витамина В6 включает три
родственных вещества: пиридоксаль, пиридоксин и пиридокса-
мин. Все они в организме превращаются в пиридоксаль-
фосфат (рис. 15.2).
XX XX
Η
ЧГ СН3
Η
Пиридоксин
CH2NH2
Пиридоксаль
СНО
.он
HOH2C\^k/OH 23OP-0-H2C.l^(
Х*Х XX
ϊΓ СН3 ™ СН3
Η Η
Пиридоксамин
Пиридоксальфосфат
Рис. 15.2. Структура соединений группы витамина В6 и
кофактора трансаминазы - пиридоксальфосфата
«Рабочей группой» пиридоксальфосфата служит
альдегидная группа —СНО. Реакция
трансаминирования в общем виде может быть представлена следующим
образом:
Е-ПФ + аминокислота 1 ^-> Ε-ΠΦ-ΝΗ2 + кетокислота 1
Е-ПФ—ΝΗ2 + кетокислота 2 <-> Е-ПФ + аминокислота 2
Здесь ПФ - пиридоксальфосфат, а ΠΦ-ΝΗ2 - пиридок-
саминфосфат.
Механизм этой реакции показан на рис. 15.3. Обе
реакции протекают в активном центре трансаминазы,
а пиридоксальфосфат и пиридоксаминфосфат прочно
связаны с этим ферментом.
Рассмотренный путь трансдезаминирования
является наиболее общим для аминокислот, однако некоторые
из них теряют свои аминогруппы по-другому.
Особые механизмы дезаминирования
Серии (гидроксилсодержащая аминокислота) дезамини-
руется в ходе реакции дегидратации, катализируемой
специфической дегидратазой. Цистеин, отличающийся
от серина тем, что содержит тиольную группу вместо
гидроксильной, дезаминируется после отщепления H2S.
В обоих случаях продуктом реакции является пируват
(рис. 15.4).
Судьба кетокислоты,
или углеродных скелетов
дезаминированных аминокислот
Учитывая роль аминокислот в общем метаболизме,
некоторые из них называют глюкогенными, а другие -
кетогенными. Термин кетогенная аминокислота
означает, что в ходе ее метаболизма образуется ацетил-СоА,
глава 15 Метаболизм аминокислот
195
Рис. 15.3. Механизм трансаминирования (упрощенное изображение)
P-CH=NH- - комплекс пиридоксальфосфата с аминогруппой остатка лизина белка. P-CH2-NH3+ - пиридоксальфосфат.
Прямая реакция (красные стрелки) приводит к превращению аминокислоты в кетокислоту, а пиридоксальфосфата - в пи-
ридоксаминфосфат. Обратная реакция (черные стрелки) представляет собой переаминирование между кетокислотой и
аминокислотой
СН2ОН СН2
| Сериндегидратаза ||
CHNH3+
С—ΝΗί
СОСГ
Аминоакриловая
кислота
СН3
I
coo-
τ
н2о
СН3
С=0 + ΝΗΙ
I
соо-
Пируват
соо-
Цистеин
Цистеиндесуль^^р
(в бактериях)
Рис. 15.4. Превращение серина и цистеина в пируват
который далее превращается в кетоновые тела. Такое
определение внешне противоречит утверждению, что
кетоновые тела возникают лишь в условиях
чрезмерного метаболизма жиров (см. с. 85), а в нормальных
условиях ацетил-СоА не расходуется на их синтез.
В действительности термин кетогенные возник очень
давно, когда на голодающих животных в качестве
тест-системы изучали судьбу полученных ими с
пищей аминокислот. Именно в этих условиях любое
повышение уровня ацетил-СоА сопровождалось
нарастанием в крови легко обнаруживаемых кетоновых тел.
В нормальных условиях кетоновые тела из кетогенных
аминокислот не образуются, а истинный смысл
термина в том, что эти аминокислоты превращаются не в
пируват, а в ацетил-СоА. Глюкогенные аминокислоты
стимулируют повышение уровня глюкозы в крови (у
диабетиков - в моче).
Аспарагиновая кислота дезаминируется в оксалоаце-
тат - один из метаболитов цикла лимонной кислоты,
который может превращаться в глюкозу. Следовательно,
аспарагиновая кислота- глюкогенная аминокислота.
Сюда же относятся глутаминовая кислота и аланин,
которые превращаются в ос-кетоглутарат и пируват, а
также серии, цистеин и ряд других аминокислот.
часть 3 Метаболизм
196
Некоторые аминокислоты являются одновременно
и кетогенными, и глюкогенными. Так, при метаболизме
фенилаланина образуется и ацетил-СоА, и фумарат,
участвующий в цикле лимонной кислоты. Из 20
аминокислот только две - лейцин и лизин - исключительно
кетогенные. О расщеплении изолейцина, метионина,
треонина и валина см. на стр. 136.
Метаболизм фенилаланина
Фенилаланин - ароматическая аминокислота, избыток
которой в норме превращается в тирозин ферментом
фенилаланингидроксилазой (рис. 15.5). Этот фермент
интересен тем, что два атома водорода в реакции
поставляются донором электронов - коферментом тетра-
гидробиоптерином.
Аномальныи метаболи зм
фенилаланина при фенилкетонурии
СОСГ Транс-
/=v | аминирование
(J-ch2-ch ■
RH4
RH2
А
NHJ
02
Фенил ал анингидроксилаз;
Н20
ζ^>—сн2—со—соо~
Фенилпируват
соо-
Нормальныи метаболизм
фенилаланина
НО—С )—СН2—СН —-—-—- Фумарат + ацетоацетат
Тирозин NHj
Рис. 15.5. Нормальный и аномальный метаболизмы
фенилаланина
RH4 - тетрагидробиоптерин; RH2 - дигидробиоптерин
H2N
СН—СН—СН3
н н ι
о он он
Тетрагидробиоптерин
HoN
N?
Η Η
Ν
Ν" "СН—СН—СН3
О ОН ОН
Дигидробиоптерин
Может показаться странным, что для данной
реакции необходимы как кислород, так и
восстанавливающий агент, но такое явление достаточно
распространено, и мы еще будем с ним встречаться. Дело
в том, что один атом кислорода используется здесь для
образования гидроксильной группы в ароматическом
кольце. Второй атом кислорода восстанавливается
в молекулу Н20, причем два атома водорода
поставляются ему тетрагидробиоптерином. Специальная
ферментная система, использующая N ADH в качестве
восстановителя, превращает дигидробиоптерин в
исходный тетрагидробиоптерин, так что последний
действует как катализатор.
Фенилаланингидроксилаза принадлежит к классу
ферментов монооксигеназ (они вводят в субстрат один
атом кислорода) или к классу оксигеназ смешанного
действия, так как оксигенируются два соединения -
аминокислота и пара атомов водорода (см. с. 210, где
обсуждаются различия между окислением - удалением
электронов, и оксигенированием - введением в
молекулу атома кислорода).
Сам фенилаланин в норме не подвергается дезами-
нированию; предварительно он должен превратиться
в тирозин. Известен сравнительно часто
встречающийся генетический дефект, при котором превращение
фенилаланина в тирозин затруднено или полностью
блокировано из-за недостатка фермента или, реже,
тетрагидробиоптерина. Накапливающийся при этом
фенилаланин вынужденно подвергается дезаминирова-
нию в аномальный метаболит - фенилпируват, который
сразу выводится из организма с мочой (см. рис. 15.5).
Это заболевание называют фенилкетонурией (ФКУ).
Оно особенно опасно для младенцев, поскольку
фенилпируват необратимо повреждает их мозг и вызывает
раннюю смерть. Если болезнь выявлена сразу после
рождения (по анализу мочи, а еще лучше - по анализу
крови на наличие фенилпирувата), ребенку назначают
диету, бедную фенилаланином, но содержащую
достаточное количество тирозина. Это позволяет
предотвратить заболевание и нормализовать развитие. Почему
фенилпируват так разрушительно действует на мозг,
пока неизвестно.
Довольно любопытно еще одно генетическое
нарушение, которое называют болезнью кленового сиропа.
При нем в организме накапливаются кетокислоты,
соответствующие трем алифатическим аминокислотам:
лейцину, изолейцину и валину. Это заболевание,
получившее столь странное название по характерному
запаху мочи, также сопряжено с нарушениями
биохимических превращений в мозге. Оно встречается гораздо
реже, чем фенилкетонурия.
глава 15 Метаболизм аминокислот
197
Еще одно генетическое заболевание - алкаптонурия
внешне проявляется в том, что моча, оставленная на
воздухе, быстро чернеет. Причиной является нарушение
расщепления тирозина, при котором в организме
накапливается один из промежуточных продуктов - гомо-
гентизиновая кислота. Она представляет собой дифенол,
который, окисляясь кислородом воздуха, превращается
в черный пигмент.
Метионин и перенос метильных групп
Метионин - одна из незаменимых аминокислот.
сн3—s—сн2—сн2—сн-сосг
Самая важная часть метионина - метильная группа.
Эти группы играют огромную роль в жизни клетки.
Многие соединения подвергаются метилированию,
причем именно метионин служит донором метильной
группы, которая переносится на другие вещества. Сам
метионин представляет собой устойчивое соединение и не
может служить метилирующим агентом. Однако его
метильная группа активируется, когда метионин,
реагируя с АТР, превращается в сульфониевый катион
S-аденозилметионин, или SAM (рис. 15.6).
Наличие группы, обладающей повышенной
реакционной способностью, делает термодинамически
благоприятным ее перенос с этого вещества на другие
соединения. Три фосфатные группы АТР, обеспечивающего
синтез S-аденозилметионина энергией, превращаются
в пирофосфат РР/ и неорганический Р/5 а затем РР,
расщепляется до двух молекул Р/ (см. рис. 15.6). Перенос
метильной группы с S-аденозилметионина на другие
соединения катализируют трансметил азы. При этом
S-аденозилметионин превращается в S-аденозилгомо-
цистеин. Последний гидролизуется в гомоцистеин -
метионин, у которого вместо S-CH3-rpynnbi
появляется - SH-группа. Далее тиольная группа с гомоцис-
теина переносится на серии с образованием цистеина
(см. рис. 15.6). Промежуточное соединение в этом
процессе (цистатионин) при определенном генетическом
дефекте накапливается в организме и выводится из
него с мочой. Это заболевание называют цистатио-
нурией.
Куда переносятся метильные группы?
Продуктами метилирования S-аденозилметионином
являются креатин (см. с. 380), фосфатидилхолин (см. с. 55),
адреналин (см. с. 200), азотистые основания
нуклеиновых кислот (кроме тимина).
часть 3 Метаболизм
198
Синтез аминокислот
В организме синтезируются только заменимые
аминокислоты. Мы не будем здесь касаться всех деталей этих
процессов, химия которых столь же разнообразна, сколь
и специфична. Наша задача - рассказать о наиболее
интересных и важных аспектах этой проблемы и
продемонстрировать, как аминокислоты синтезируются из
промежуточных соединений гликолиза и цикла лимонной
кислоты.
На самом деле всего пять таких соединений - 3-фос-
фоглицерат, фосфоенолпируват, пируват, оксалоацетат
и α-кетоглутарат, вместе с двумя моносахаридами пен-
тозофосфатного пути, служат предшественниками всех
20 аминокислот, которые синтезируются в бактериях
и растениях.
Синтез глутаминовой кислоты
Дезаминирование глутаминовой кислоты
осуществляется глутаматдегидрогеназой, кофакторами которой
служат NAD+ или NADP+. Реакция обратима, но,
по-видимому, для животных образование глутамата из
аминокислоты - процесс менее существенный, чем транса-
минирование α-кетоглутарата. Донорами аминогруппы
при этом служат другие аминокислоты, например ала-
нин или аспарагиновая кислота. У прокариот
существует и другой путь аминирования α-кетоглутарата,
который реализуется при малых концентрациях NH4+
и требует притока энергии.
Синтез аспарагиновой кислоты
и аланина
Эти аминокислоты образуются в клетках при транс-
аминировании оксалоацетата (R = -СН2СОО~) и пиру-
вата (R = -СН3):
R R
I I
С=0 + Глутамат CHNH3 + α-Кетоглутарат.
С00" С00"
Синтез серина
Серии синтезируется в три стадии из промежуточного
вещества гликолиза - 3-фосфоглицерата. Сначала 3-фос-
фоглицерат окисляется в кетокислоту - 3-фосфогидрок-
сипируват:
COO" NAD+ NADH C00"
I \ у I
снон —^—^-—- с=о
СН2ОР023" СН2ОР023"
CH3
I
s
I
CH2 + ATP —
I
CH2
CHNH3
I
COO Метионин
Аденозин
SH
I
CH2
CH2
I +
CHNH3
I
COO"
Гомоцистеин
- Серии
H20
COO"
I +
CHNH3
I
сн2
I
s
I
CH2
CH2
CHNH3+
COO-
I
CHNH3+
I
CH2OH
H20
аденин
+ PP/ + P,·
CHNH3+ OH OH H2°S
I 1
coo- I
S-Апснозилметионин
2P/
Неорганическая
пирофосфатаза
H70
Перенос СНз-группы на другие
соединения
аденин
CHNH3+ он 0Н
I
СОО"
S-Аденозилгомоцистеин
СОО"
I +
СНЫНз
I
сн2
I
SH
Цисте и н
+
сн3
сн2
I
со
СОО сс-Кетобутират
+
νη;
СОО Цистатионин
Рис. 15.6. Синтез S-аденозилметионина из метионина (я), а цистеина - из гомоцистеина (б)
Затем эта кетокислота подвергается трансаминиро-
ванию глутаминовой кислотой и превращается в 3-фос-
фосерин, который далее гидролизуется до серина и Р,..
I Трансаминирование |
С=0 CHNH3+
I
Гидролиз
СН2ОРОз
СН2ОРОз"
СОО"
I
- CHNH3+ + Ρ/.
СН2ОН
Синтез глицина
Глицин (1ЧНз-СН2-СОСГ) - простейшая из
α-аминокислот. Синтез его осуществляется путем удаления
концевой гидроксиметиленовой группы серина. Реакция
протекает с участием кофермента -тетрагидрофолиевой
кислоты, которая служит переносчиком одноуглеродных
групп. Такого рода перенос играет важную роль в
синтезе нуклеотидов (см. главу 18).
глава 15 Метаболизм аминокислот
199
Синтез других соединений
из аминокислот
Широкий круг низкомолекулярных физиологически
активных соединений синтезируется в организме из
аминокислот. Амины образуются в результате декарбокси-
лирования аминокислот:
RCH2NH*COCT -> RCH2NH* + C02 .
Катехоламины, к числу которых принадлежат
гормоны дофамин, адреналин и норадреналин, свое
название получили за структурное сходство с катехолом -
1,2-дигидроксибензолом.
НСк /^ + H0V
-СНОН—СН2—NH2—CH3
НО" ^ НО"
Адреналин Катехол
Среди соединений, образующихся из аминокислот,
встречаются нейромедиаторы: γ-аминобутират (GAB А;
от англ. Gamma AminoButyric Acid), 5-гидрокситрипта-
мин и те же катехоламины. Гормон тироксин
синтезируется из тирозина. Этот далеко не полный перечень
показывает, что аминокислоты служат предшественниками
многих биологически важных веществ.
Что происходит
с аминогруппами после
их удаления из аминокислот?
Цикл мочевины
Аминный азот катаболизируемых аминокислот
выводится из организма млекопитающих с мочой в виде
мочевины - инертного водорастворимого
нетоксичного вещества. Мочевина образуется в печени при
отщеплении гуанидиновой группы от аргинина с помощью
гидролитического фермента аргиназы. Попутно образуется
орнитин - аминокислота, не входящая в состав белков.
- -с
ι
ΝΗ
I
сн
I 2 аргиназа Η^Ν ΝΗ2
сн2 + н2о - с
I II
сн2 о
' KIU+ Мочевина
LHNn3
сосг
Аргинин
ΝΗ5
I
сн2
сн2
сн2
CHNH^
I
СОСГ
Орнитин
Для превращения орнитина обратно в аргинин
используется атом углерода из С02 и аминный азот,
высвобождающийся в ходе превращения всех 20
катаболизируемых аминокислот. Аргинин-орнитиновый цикл
стал первым из описанных метаболических циклов.
Кребс (который позже открыл цикл лимонной кислоты)
и Хенселейт обнаружили, что если к клеткам печени
добавить аргинин, то количество образовавшейся после
этого мочевины существенно превышает количество
использованной аминокислоты. То же самое происходит
при добавлении орнитина. Ученые заключили, что
превращение обеих аминокислот - процесс циклический,
и в него вовлечены как аргинин, так и орнитин.
Образование аргинина из орнитина протекает в
несколько стадий. В качестве промежуточного продукта
образуется еще одна аминокислота - цитруллин (как
и орнитин, она не входит в состав белков).
H2N^O
I
ΝΗ
\и2
сн2
сн
CHNH^
СОСГ
Цитруллин
Подобно аргинину и орнитину, цитруллин
стимулирует синтез мочевины клетками печени. Так был
окончательно расшифрован знаменитый цикл мочевины
(рис. 15.7).
, Аргинин N / 2
Мочевина
Орнитин
Цитруллин
Рис. 15.7. Цикл образования мочевины из аргинина
Включение в цикл азота и С02 обсуждается в тексте
Механизм синтеза аргинина
Первый этап синтеза аргинина - образование цитрул-
лина - протекает в митохондриальном матриксе. Он
начинается с того, что из ионов аммония и С02
образуется карбамоилфосфат, который представляет собой
смешанный ангидрид карбаминовой (NH2COOH)
и фосфорной кислот. Карбамоилфосфат - макроэрги-
ческое соединение, мощный ацилирующий агент.
часть 3 Метаболизм
200
Его синтез катализирует карбамоилфосфатсинте-
таза в следующей реакции:
NHJ + НСОз- + 2АТР
+ 2ADP + Р/ + 2Н+
О О
II II
ΝΗ2—С-О—Р—О"
0"
На синтез каждой его молекулы расходуются 2
молекулы АТР. Одна из них, расщепляясь до ADP и Р|5
обеспечивает образование из аммония и С02 карбама-
та, а вторая участвует в его фосфорилировании. Далее
карбамоильная группа карбамоилфосфата с помощью
фермента орнитинтранскарбамоилазы переносится
на орнитин, превращая его в цитруллин (орнитин
представлен в виде R—ΝΗ3+):
0 0 О
II II
RNH^ + NH2—С—О—Ρ—О- ΗΝ-
II
-с-
-ΝΗ7
Ρ/.
Орнитин
Карбамоил фосфат
R
Цитруллин
Превращение цитруллина в аргинин
Второй этап синтеза аргинина протекает в цитозоле
и состоит в том, что карбонильная группа (С=0) в цит-
руллине превращается в имидную (C=NH) группу
аргинина. Карбонил цитруллина значительно менее реакци-
онноспособен, чем кетонный карбонил в кетокислотах,
поэтому такое превращение не может быть
произведено как трансаминирование. В действительности оно
происходит в две стадии. На первой - цитруллин с
участием АТР конденсируется с аспартатом, превращаясь
в аргининосукцинат:
С=0
-NH 4NH2
Цитруллин
Η,Ν-
соо
i
-сн
сн2
I
соо
Аспартат
АТР
^АМР + РР/
СОО
ι
С—NH-CH
/ W I
R—NH NHJ СН2
сосг
Аргининосукцинат
Название продукта этой реакции обусловлено тем,
что его молекула по структуре напоминает аргининовое
производное сукцината, хотя при его расщеплении ар-
гининосукцинатлиазой образуются аргинин и фума-
рат, а не сукцинат:
СОО
I
С — NH-CH
COO^-CHNH;—(СН2)3—NH \н*2 сн,
Аргининосукцинат
I
СОО
NH2
COO^CHNH;—(СН2)3—NH—С
NH2
Аргинин
СОО
!
сн
II
сн
СОСГ
Фумарат
Полностью цикл мочевины представлен на рис. 15.8.
В основном он регулируется путем изменения
содержания ферментов. Их количество возрастает как при
значительном поступлении аминокислот из пищи, так
и при голодании, когда расщепляются мышечные белки.
Фумарат Аргинин
Аргинино-
сукцинатлиаза
Аргининосукцинат
AMP + PPf
Аргинино-
Сукцинатсинтетаза
Орнитинтранс
арб λ ила г
Цитруллин
Н20
Аргиназг
Мочевина
Орнитин
Карбамоилфосфат
ρ
Аспартат г»
+АТР КарОамоилфосфат К
синтетаза I ^
ι λ 2ADP
2АТР
ΝΗ4 + НС03
Рис. 15.8. Ферменты цикла мочевины
Уровень ферментов коррелирует с количеством белка,
потребляемого в виде пищи. Цикл аллостерически
регулируется на стадии карбамоилфосфатсинтетазы (по-видимому,
N-ацетилглутаматом). Орнитин преобразуется в цитруллин
внутри митохондрий, тогда как другие стадии цикла
протекают в цитоплазме
глава 15 Метаболизм аминокислот
201
Кроме того, карбамоилфосфатсинтетаза аллостери-
чески регулируется N-ацетилглутаматом, уровень
которого коррелирует с концентрацией аминокислот.
Как аминный азот
транспортируется в печень
для переработки в мочевину
Глутамин - транспортная форма
аммиака в крови
Аммиак, образующийся при дезаминировании
аминокислот, токсичен. И потому его содержание в крови
должно быть предельно мало, иначе возможно нарушение
функции мозга и развитие комы. Поэтому в свободной
форме в печень он не транспортируется. Аммиак
взаимодействует с глутаминовой кислотой, и с помощью
фермента глутаминсинтетазы образуется глутамин (амид
глутаминовой кислоты). В качестве промежуточного
соединения при этом появляется γ-глутам ил фосфат -
смешанный ангидрид глутаминовой и фосфорной
кислот. Это макроэргическое соединение, способное
взаимодействовать с ионами аммония (обе реакции
протекают в активном центре синтетазы).
СОО"
I
сн,
СН2 + NH* + ATP
CHNH^
I
coo-
Глутамат
I лутамин- q η
синтетаза
CH2
CO-NH2
12
+ ADP + Ρ/ + H+
CHNHt
I
coo-
Глутамин
Источником глутамата при этом служит а-кетоглу-
тарат из цикла лимонной кислоты, который
подвергается трансаминированию другими аминокислотами.
Глутамин переносится кровью в печень, где гидролизуется
глутаминазой, а высвободившийся аммиак
используется для синтеза мочевины.
CO-NH2
I
сн2
сн2 + н2о
I
CHNH^ ·
I
соо-
Глутамин
Глутаминаза
СОСГ
!*
СН2 + NHj-
chnh;
I
сосг
Глутамат
Глутаминаза есть и в почках Там ее функция
состоит в том, чтобы нейтрализовать аммиаком кислоты,
выводимые из крови. Помимо того, что глутамин
служит переносчиком аммиака и является одной из 20
аминокислот, входящих в состав белков, он участвует в
синтезе некоторых других метаболитов. В этом случае он
используется в форме глутаминамида в качестве
источника азота.
Алании - транспортная форма
аминного азота в крови
Около 30% аминного азота, который поступает в печень
после расщепления мышечных белков, сосредоточено
в аланине. Разумеется, доля аланина в белках гораздо
ниже; в основном аланин образуется при трансамини-
ровании пирувата другими аминокислотами. Попадая
в печень, аланин подвергается там дезаминированию
Аммиак используется на производство мочевины, а пи-
руват - глюкозы. Последняя возвращается с кровью
в мышцы, замыкая тем самым глюкозо-аланиновый
цикл переноса аммиака (рис. 15.9).
"N
Аминокислоты в мышце
Пируват в мышце -
I
Глюкоза в мышце Аланин в мышце —►Алании в крови
ι
Аланин в печени
Тра^аш ров ие
-NH2
^** Мочевина
Глюкоза в крови -·-
Пируват в печени
Глюкон ог н I
Глюкоза в печени
Рис. 15.9. Глюкозо-аланиновый цикл поставляет азот (в
виде аланина) из мышц в печень, а из печени возвращает
в мышцы глюкозу
При голодании этот цикл приобретает особенно
важное значение. После исчерпания запасов гликогена
печень должна снабжать глюкозой мозг и другие
нуждающиеся в ней ткани. Главным источником метаболитов,
используемых печенью для глюконеогенеза, являются
аминокислоты, образующиеся при расщеплении
мышечных белков. О регуляции такого расщепления будет
рассказано в главе 22. Многие аминокислоты еще
часть 3 Метаболизм
202
в мышцах превращаются в пируват, который
транспортируется в печень в форме аланина. Еще раз
подчеркнем, что глюкозо-аланиновый цикл не увеличивает
количество глюкозы, а лишь является способом
разделения метаболизма аминокислот между мышцами
и печенью, а также безвредным механизмом
транспорта в печень аминного азота.
Дополнительная литература
Felig P. Amino acid metabolism in man // Annu. Rev. Bio-
chem. 1975. Vol. 44. P. 933-955.
(Очень полезный обзор, посвященный метаболизму
аминокислот на уровне органов. Рассматриваются
проблемы, связанные с голоданием, диабетом, ожирением. На
этом же уровне обсуждается процесс глюконеогенеза.)
Snell К. Alanine as a gluconeogenic carrier // Trends Bio-
chem. Sci. 1979. Vol. 4. P. 124-128.
(Обзор, посвященный роли мышц в снабжении печени
метаболитами для синтеза глюкозы.)
News holme Ε. Α., Leach A. R. Biochemistry for medical
sciences. Wiley, 1983. P. 417^29.
(Очень полезное описание метаболизма и
физиологического значения аминокислот в различных тканях
организма.)
Вопросы к главе 15
1. Объясните, как окисление аминокислот может
привести к их дезаминированию.
2. Какие аминокислоты дезаминируются посредством
окисления?
3. Опишите подробно, как дезаминируется аланин.
4. Какой кофермент участвует в переаминировании?
Каково его строение и как происходит переамини-
рование?
5. Как дезаминируются серии и цистеин?
6. Что означают термины «кетогенные» и «глюкоген-
ные» по отношению к аминокислотам? Назовите
чисто кетогенные аминокислоты.
7. Что представляет собой заболевание фенилкето-
нурия?
8. Какова роль тетрагидробиоптерина при гидроксили-
ровании фенилаланина?
9. Метионин - источник метильных групп в некоторых
биохимических процессах. Объясните, как
активируется метионин в реакциях биологического
метилирования.
10. Опишите реакции, входящие в цикл мочевины.
11. Почему содержание ферментов цикла мочевины
возрастает как при обильном белковом питании, так
и при голодании?
12. Как аммиак и аминный азот попадают из
периферических тканей в печень для образования мочевины?
глава 15 Метаболизм аминокислот
203
глава
14* Внутриклеточные органеллы,
О удаляющие отходы
Глава посвящена лизосомам, задача которых состоит
в разрушении нежелательных для клетки молекул,
образовавшихся в процессе метаболизма или попавших в нее
в ходе эндоцитоза.
Такая роль кажется неприглядной, однако о ее
важности можно судить хотя бы по тому, что более сорока
генетических заболеваний связаны с дефектами лизосо-
мальных ферментов.
Кроме того, мы расскажем еще о внешне очень
похожих на лизосомы пероксисомах, функция которых
несколько отличается от функции лизосом: они заняты
химической переработкой того, с чем не сумела справиться
вся метаболическая машина клетки.
АТР + Н20 ADP + Pf
Протонный насос
Мембрана
Н+
(рН 4,5-5)
Гидролитические
ферменты
Лизосомы
Лизосомы - окруженные мембраной органеллы,
присутствующие в цитоплазме всех эукариотических
клеток (см. рис. 3.15, а).
В клетках печени их насчитывается несколько
сотен. Образно говоря, лизосомы можно назвать
«мешочками с оружием массового поражения»!
Действительно, внутри них находится целый набор
гидролитических ферментов, способных разрушить практически
любой компонент клетки. Клетку спасает от
разрушения не только лизосомальная мембрана. Лизосо-
мальные ферменты работают в кислой среде (рН 4,5),
которая внутри лизосомы поддерживается АТР-зави-
симым протонным насосом (рис. 16.1). Если мембрану
лизосомы повредить, ее ферменты окажутся в среде
с рН -7,3, где они практически неактивны (буферная
емкость цитоплазмы несоизмеримо больше, чем у
содержимого лизосом).
Первичные лизосомы отпочковываются от аппарата
Гольджи в виде пузырьков, начиненных ферментами.
Вновь синтезируемые ферменты упаковываются в
лизосомы в процессе их формирования (подробнее об этом
в главе 22). Среди них - протеиназы, фосфатазы, эсте-
разы, ДНКазы, РНКазы, ферменты, разрушающие
полисахариды и мукополисахариды. Всего этого достаточно,
чтобы гидролизовать любые биологические молекулы,
а гидролиз в водной среде необратим.
Рис. 16.1. Первичная лизосома, отпочковавшаяся от
аппарата Гольджи (схематическое изображение)
Первичная лизосома - это транспортный пузырек,
доставляющий гидролитические ферменты в эндосомы
Как предназначенные для разрушения
вещества попадают в лизосомы?
Поскольку лизосомальные ферменты не могут
работать в цитоплазме, их субстраты должны проникать
в лизосомы. Объекты, подлежащие разрушению,
исходно могут находиться как внутри, так и вне клетки. К
первым относятся митохондрии. Время жизни митохондрии
в клетке печени составляет около десяти дней, после чего
состарившиеся органеллы каким-то образом
распознаются и заключаются в пузырек, который образуется
из мембраны эндоплазматического ретикулума. Такого
рода пузырьки называют аутофагосомами (рис. 16.2),
чтобы отличить их от фагосом. Последние образуются
в фагоцитах, обволакивающих бактерии или другие
посторонние частицы.
Фагоцитоз бактерий осуществляют белые кровяные
тельца, но большинство клеток нашего организма
могут также поглощать внешние частицы посредством
эндоцитоза, управляемого рецепторами. Хорошим
примером может служить поглощение ЛПНП и остатков
хиломикронов (см. с. 96). Механизм этого процесса
иллюстрирует рис. 16.2. Рецепторы на поверхности
плазматических мембран размещены на участках,
часть 3 Метаболизм
204
Захватываемые частицы
Эндоцитоз,
управляемый
рецепторами
и
I
Клеточная мембрана
Участок мембраны,
покрытый клатрином
Покрытый
клатрином "
пузырек
Первичная
Клатрин возвращается
к клеточной мембране
О
О
Непокрытая
эндосома
Первичная
лизосома
\
Слияние с
эндосомой
СО
Вторичная ξ ]
лизосома ^^^/
Аутофагосома,
содержащая
митохондрию
$R
Переваривание содержимого
о
Рис. 16.2. Переваривание содержимого пузырьков,
покрытых клатрином (эндосом и аутофагосом)
Сходным образом перевариваются бактерии, захваченные при
фагоцитозе
содержащих белок клатрин. После связывания на
рецепторе, к примеру, ЛПНП эти участки впячиваются
внутрь клетки и отпочковываются, так что ЛПНП
оказываются заключенными в мембранный пузырек,
покрытый клатрином. Затем клатрин возвращается в
плазматическую мембрану. Мембранные пузырьки, содержащие
захваченные извне частицы, называют эндосомами.
С помощью эндоцитоза происходит уничтожение
состарившихся эритроцитов в печени и селезенке.
Углеводные цепи на поверхности эритроцитов
заканчиваются остатками сиаловой кислоты. Эти концевые группы
со временем теряются, обнажая остатки галактозы, что
служит сигналом к уничтожению эритроцита (средняя
продолжительность его жизни в крови - 120 дней). Ли-
зосомы также играют важную роль в лизисе липопроте-
инов, таких как ЛПНП, поглощаемых при эндоцитозе.
Аутофагосомы, фагосомы и эндосомы сливаются
с первичными лизосомами (последние
отпочковываются от аппарата Гольджи), так что ферменты и
вещества, на которые направлено их действие, оказываются
в одном пузырьке, называемом вторичной лизосомой
(терминология здесь еще не устоялась; иногда под
лизосомой понимают вторичную лизосому). Считается,
что на поверхности первичных лизосом есть
специальные белки, которые связываются с
комплементарными белками на мембране частицы-мишени (с ней
лизосома должна слиться). Слияние лизосомы с мишенью -
энергозависимый процесс, требующий гидролиза GTP.
Лизосомы участвуют не только в разрушении орга-
нелл и захваченных при эндоцитозе структур, но также
в расщеплении некоторых белков, нуклеиновых кислот,
липидов и даже гликогена. Как все эти соединения
попадают внутрь пузырьков с ферментами, пока неясно,
как и способ, посредством которого возвращаются в
цитоплазму продукты распада. По всей видимости, для
этого существуют специальные мембранные транспортные
системы - специфические мембранные белки.
Некоторые компоненты мембран также
разрушаются в лизосомах. Известно, например, целое семейство
генетических заболеваний, называемых сфинголипи-
дозами (классический пример - болезнь Тея-Сакса).
Их причиной является неспособность лизосомальных
ферментов расщеплять ганглиозиды (см. с. 56).
О важности лизосомального
переваривания
Отсутствие одного или нескольких лизосомальных
ферментов чревато тяжелыми заболеваниями.
Лизосомы при этом оказываются перегруженными
непереваренными веществами.
Загадочна роль лизосом в расщеплении гликогена.
Из главы 6 вы знаете, что гликоген расщепляется глико-
генфосфорилазой. Между тем известна так называемая
болезнь Помпе, которая обусловлена недостаточностью
лизосомального фермента, гидролизующего в гликогене
а-( 1 —>4)-гликозидные связи. Это приводит к
накоплению в клетках чрезмерного количества гликогена,
вызывая смерть в младенчестве. Лизосомы никак не
вписываются в хорошо известную схему расщепления
гликогена. Возможно, молекулы гликогена захватываются
лизосомами на последнем этапе расщепления. Однако
о значении такого расщепления известно очень мало.
Пероксисомы
Эти мембранные пузырьки, присутствующие в клетках
млекопитающих, содержат ферменты класса оксидаз.
Оксидазы используют кислород для окисления различных
глава 16 Внутриклеточные органеллы, удаляющие отходы
205
субстратов, причем продуктом восстановления
кислорода при этом является не вода, а перекись водорода:
R'H2 + 02 -> R' + Н202.
Перекись водорода, в свою очередь, сама окисляет
другие субстраты (в том числе и часть вводимого в
организм алкоголя):
RH2 + Н202 -> R + 2Н20.
В пероксисомах окисляются некоторые фенолы,
D-аминокислоты, а также жирные кислоты с очень
длинными цепями, которые не могут быть (до
укорачивания) окислены в митохондриях. Пероксисомы
участвуют также в синтезе некоторых сложных липи-
дов. Есть основания полагать, что они причастны к
переработке холестерина в желчные кислоты.
Любопытно, что клофибрат - лекарство, снижающее уровень
холестерина, вызывает значительное увеличение
числа пероксисом.
Как и в случае л изо сом, биологические функции
пероксисом до конца не изучены, однако об их значении
можно судить по последствиям генетических дефектов.
Известно, например, редкое смертельное генетическое
заболевание, вызываемое накоплением С24- и С26-жир-
ных кислот, а также предшественников желчных кислот.
Дополнительная литература
Neufeld Ε. F. Lysosomal storage diseases // Annu. Rev. Bio-
chem. 1991. Vol. 60. P. 257-280.
(В обзоре приведены общие сведения об отдельных ли-
зосомальных болезнях.)
Mallabiabarrena Α., Malhotra V. Vesicle biogenesis: the coat
connection // Cell. 1995. Vol. 83. P. 667-669.
(В этом обзоре, где вопросов больше, чем ответов,
обсуждается роль клатрина в эндоцитозе.)
Вопросы к главе 16
1. Зачем нужны л изо сомы?
2. Что такое первичные л изо сомы и где они образуются?
3. Как лизосомальные ферменты вступают в контакт со
своими субстратами?
4. Какие доводы можно привести в пользу участия
лизосом в расщеплении отдельных компонентов
клетки?
5. Чем обусловлено возникновение болезни Помпе?
6. Что такое пероксисомы и каковы их функции?
часть 3 Метаболизм
206
17 Защитные
I организма
Иммунная система - главный механизм, защищающий нас
от воздействия болезнетворных патогенов (см. главу 25).
Однако организму приходится защищаться и от многого
другого. В этой главе речь пойдет о защитных
механизмах, которые связаны со своеобразными
ферментативными реакциями. У них мало общего между собой, и при
сопоставлении они выглядят случайным набором
превращений. Но все это жизненно важные реакции, и
выполняют они общую задачу - защиту организма.
Свертывание крови
Свертывание (или коагуляция) крови имеет целью
образование сгустков, которые предотвращают кровотечение,
затягивая раны в поврежденных сосудах. По понятным
причинам такой физиологический ответ должен быть
быстрым и массированным, хотя первоначальный
сигнал, с химической точки зрения, очень слаб. Отсюда
возникает необходимость эффективного усиления этого
сигнала.
Мы уже видели, как биохимическое усиление
сигнала достигается с помощью каскада реакций
(см. с. 170). Суть такого усиления в том, что фермент,
являющийся продуктом предыдущей ферментативной
реакции, активирует другой фермент, а тот, в свою
очередь, следующий и т. д. В результате лавинообразно
нарастает число вовлеченных в работу ферментов и
продуктов очередной реакции. Свертывание крови удобно
рассматривать как совокупность двух процессов: каскад-
но развивающейся активации фермента, образующего
сгусток, и образование самого сгустка. Каскадная
активация основана на ограниченном протеолизе:
неактивные предшественники протеиназ активируются
после того, как в их молекулах произойдет разрыв
определенных пептидных связей (ср. образование трипсина
из трипсиногена; см. с. 75). Все необходимые
белки-предшественники протеиназ исходно присутствуют
в крови в ожидании сигнала о повреждении стенок
сосудов. До этого момента они неактивны.
Что служит сигналом
к образованию сгустка?
Свертывание крови осуществляется с помощью двух
механизмов.
механизмы
1. В нормальных условиях кровеносные сосуды
изнутри выстланы слоем эндотелиальных клеток. Если
этот слой будет нарушен, обнажатся лежащие под
ним структуры, в том числе коллагеновые волокна.
Когда свойства внутренней поверхности сосуда
изменятся, первой реакцией на это будет «наложение
временного пластыря» на поверхностный участок
благодаря агрегации близлежащих кровяных
пластинок (тромбоцитов). Пусковым механизмом для
образования сгустка является адсорбция на
поврежденной поверхности ряда белков крови, что
приводит к взаимной аутокаталитической активации двух
протеиназ. Одну из них называют фактором XII
(здесь непросто разобраться в номенклатуре: фактор
может оказаться либо ферментом, либо кофактором
фермента, а нумерация вовсе не соответствует
очередности участия в процессе). Фактор XII
запускает трехэтапный каскад реакций, результатом
которого является активация еще одной протеи-
назы - фактора X. А она, в свою очередь,
активирует протромбин, превращая его в главный фермент
свертывания крови - протеиназу тромбин. Именно
тромбин непосредственно участвует в образовании
сгустков крови, или тромбов. Тем, кто уже знает, что
названия ферментов заканчиваются на -аза,
напомним, что за давно известными протеиназами
сохранены традиционные названия: пепсин, трипсин,
химотрипсин и тромбин.
2. Все компоненты описанного выше внутреннего
механизма запуска свертывания крови находятся
в ней самой. Поэтому кровь свертывается даже вне
организма: достаточно налить ее в стеклянную
пробирку, поверхность которой для крови будет как бы
поврежденной стенкой сосуда.
Существует, однако, и другой, внешний
механизм свертывания крови, при котором сигнал
приходит от поврежденных тканей. Таким сигналом
служит белковый комплекс, называемый тканевым
фактором. Суть его действия заключается в том, что
он активирует протеиназу, которая затем активирует
фактор X, а дальше все развивается по уже
описанному сценарию. Оба способа инициации
свертывания показаны на рис. 17.1.
В лабораторных условиях (in vitro) кровь
свертывается быстрее по второму из этих механизмов. Известно,
глава 17 Защитные ферментативные механизмы
207
Внутренний механизм
При повреждении
кровеносного сосуда
обнажается
субэндотелиальный слой
Это приводит к
активации фактора
XII (протеиназы)
Внешний механизм
Тканевый фактор
из поврежденных
кровеносных сосудов
Фибринопептиды
Неактивная
протеиназа"""
Здесь нужен_
Активная
протеиназа
фактор VIII"
Неактивный
фактор X
Активная ^
протеиназа
Неактивная
"протеиназа
Активный ^*-
фактор X
(протеиназа)
Неактивный
фактор X
Протромбин
-Тромбин
Тромб (кровяной сгусток)
Рис. 17.1. Внутренний и внешний механизм свертывания
крови
Для простоты не приводятся названия многочисленных проте-
иназ и других факторов, вовлеченных в этот процесс (их всего
тринадцать). Все эти протеиназы строго специфичны.
Недостаток фактора VIII приводит к заболеванию - гемофилии А
однако, заболевание гемофилия А, при котором
внутренний механизм свертывания крови нарушается из-за
отсутствия фактора VIII, необходимого для протеоли-
тической активации фактора X. Казалось бы, этот
дефект может быть компенсирован внешним механизмом,
в котором фактор VIII не принимает участия. Но в
физиологических условиях оба механизма работают
слаженно и взаимозависимо, как единое целое.
Как тромбин вызывает образование
тромбов?
В крови присутствует белок фибриноген. Его молекула
состоит из двух палочкообразных субъединиц, каждая
из которых образована тремя полипептидными цепями.
В области N-концевых участков цепей субъединицы
соединены тремя дисульфидными мостиками.
Поблизости от них в двух цепях из трех содержатся отрицательно
заряженные пептидные фрагменты - фибринопептиды,
препятствующие ассоциации мономеров фибриногена
(рис. 17.2).Тромбин превращает фибриноген в фибрин,
отщепляя фибринопептиды. Благодаря этому
открывается возможность для спонтанной агрегации фибрина
С - конец
Мономер фибриногена
S-S>
S-S
^S-S
ТР
^ ^ife";: Отщепленные
/ I \г фибринопептиды
Мономер фиб ина
S-S
S-S
S-S
Благодаря удалению зарядов
мономеры фибрина приобретаю!
способность к полимеризации
Рис. 17.2. Превращение мономеров фибриногена в
мономеры фибрина
Каждая половинка мономерного фибриногена состоит из трех
полипептидных цепей, две из которых содержат
отрицательно заряженные фибринопептиды
за счет межмолекулярных нековалентных
взаимодействий. Участки на концах мономера фибрина
связываются с комплементарными участками в середине
соседнего мономера, так что в ходе полимеризации возникает
неустойчивая структура, называемая мягким сгустком
(рис. 17.3). После того как боковые группы соседних
мономерных молекул фибрина скрепятся ковалентными
сшивками, мягкий сгусток превращается в плотный
нерастворимый твердый сгусток.
Сшивками служат амидные связи, образующиеся
в ходе реакции трансамидирования между остатком глу-
тамина в одной полипептидной цепи и лизина в другой:
-CO-NH- + NH;.
Сшивка
-CONH2 +
Боковая
группа
глутамина
H3N+-
Боковая
группа
лизина
Образовавшийся сгусток объединяет в единое целое
фибриновые тяжи и запутывающиеся в них клетки крови.
Регуляция свертывания крови
Если свертывание крови не ограничится областью
повреждения сосуда, оно может нанести непоправимый
ущерб организму, поскольку носит аутокаталитический
характер и может распространиться на все кровяное
русло. Однако существует целый ряд препятствующих
этому защитных приспособлений. Во-первых, в крови
часть 3 Метаболизм
208
***... Вновь образованные
/мономеры фибрина
\
Спонтанная полимеризация
за счет нековалентного связывания
Мягкий
сгусток
Ферментативная сшивка
Плотный
фибриновый
тяж
Фибриновые тяжи
оплетают эритроциты
Кровяной
""" сгусток
.в Фибриновые
/ тяжи
Рис. 17.3. Образование фибриновых тяжей
Тяжи образуются благодаря спонтанной полимеризации
фибриновых мономеров и их последующей ферментативной
сшивке (места и число сшивок в значительной мере произвольны).
Мономеры фибриногена полимеризоваться не могут, поскольку
несут значительный отрицательный заряд, а места
потенциальной ассоциации экранированы фибринопептидами
присутствуют ингибиторы протеиназ (в том числе
антитромбин), которые тормозят процесс
свертывания. Во-вторых, стенки сосудов содержат сульфополи-
сахарид гепарин, усиливающий действие ингибиторов.
В-третьих, имеется специальная протеиназа плазмин,
растворяющая сгустки. Плазмин сам образуется из
неактивного плазминогена под воздействием белка -
тканевого активатора плазминогена ТРА (от англ. Tissue
Plasminogen Activator), который выделяется из
поврежденных тканей. В последнее время его используют
в терапевтических целях. Хотя ТРА присутствует в
тканях в ничтожном количестве, он стал вполне доступен,
поскольку его научились получать с помощью методов
генетической инженерии (см. главу 24). Все эти
защитные механизмы препятствуют распространению
свертывания крови, ограничивая его областью,
непосредственно примыкающей к месту повреждения сосуда,
послужившему инициатором образования сгустка.
Регуляция свертывания крови очень сложна.
Неудивительно, что нарушения в системе свертывания крови
являются причиной гибели многих людей.
Крысиный яд, свертывание крови
и витамин К
Широко распространенный крысиный яд варфарин
предотвращает свертывание крови, благодаря чему
грызуны погибают от внутренних кровотечений, вызванных
ничтожными и в нормальных условиях неизбежными
повреждениями кровеносных сосудов. В медицине
варфарин используют для профилактики образования
тромбов после инсульта. По своей структуре он похож
на витамин К (от нем. Koagulation - коагуляция,
свертывание) и действует как его конкурентный ингибитор
(рис. 17.4).
СНз СНз СНз
СН2-СН=С-СН2-(СН2-СН2-СН-СН2)2-СН2-СН2-СН - СН3
Рис. 17.4. Структура витамина К (а) и варфарина (б)
Витамин К необходим для свертывания крови, а варфарин является его антагонистом
глава 17 Защитные ферментативные механизмы
209
Витамин К необходим для превращения
протромбина в тромбин. Он служит кофактором в необычной
ферментативной реакции карбоксилирования остатка глу-
таминовой кислоты в протромбине.
Смысл такой модификации в том, что остаток кар-
боксиглутамина хорошо связывает ионы кальция,
которые необходимы для активации тромбина. Аналогичным
образом модифицируются и другие факторы,
участвующие в каскаде.
I I
γ^2 СН2 Эта группировка
СН2 +С02 ~ НС-СОО-ЗГаехСа-
С00" СОСГ
Боковая цепь Карбоксиглутамат
остатка глутаминовой
кислоты в протромбине
Защита от попавших в организм
чужеродных веществ
С крупными чужеродными молекулами имеет дело
иммунная система (см. главу 25). Здесь же речь пойдет
о низкомолекулярных соединениях, которые не
являются продуктами жизнедеятельности патогенных
организмов.
В организм современного человека попадает
огромное число самых разных посторонних химических
соединений, которые называют ксенобиотиками (от греч.
ксено - чужой). Среди них лекарства, пестициды,
гербициды и другие продукты химической
промышленности, а также терпены, алкалоиды и дубильные вещества
растительного происхождения. Многие из них плохо
растворяются в воде, зато хорошо^ растворимы в жирах.
Они накапливаются в углеводородном слое мембран,
в вакуолях жировых клеток и не выводятся из организма
с мочой. Чтобы избежать вредных последствий
накопления таких веществ, необходимо сделать их
растворимыми в воде. Существует несколько способов
модификации ксенобиотиков, способствующих их выведению
из организма, однако главную роль в этом процессе
играют ферменты семейства цитохрома Р450 в печени.
Наиболее типичной реакцией, которую катализируют
эти ферменты, является гидроксилирование как
алифатических, так и ароматических группировок в молекулах
ксенобиотиков. Затем уже другие ферменты замещают
водород в гидроксильных группах на высокополярные
остатки (обычно глюкуроновой кислоты), после чего
вещество становится достаточно растворимым, чтобы
быть удаленным с мочой.
часть 3 Метаболизм
210
Перейдем теперь к более детальному рассмотрению
этих процессов, начиная с системы цитохрома Р450.
Цитохром Р450
Как и цитохромы дыхательной цепи (см. с. 123),
цитохром Р450 (для краткости просто Р450) представляет
собой гемопротеин. Его название указывает на то, что он
окрашен (от англ. Pigment) и что максимум поглощения
комплекса Р450 с окисью углерода лежит в области 450
нм. Окись углерода не имеет никакого отношения к
функции Р450; она добавляется лишь для того, чтобы
облегчить определение содержания Р450 по
интенсивности спектра поглощения. Р450 находится на внутренней
стороне мембраны эндоплазматического ретикулума.
Катализируемые им реакции обычно можно описать
уравнением:
АН + 02 + NADPH + Н+ -> А-ОН + Н20 + NADP+ .
Такие реакции называют монооксигеназными,
потому что в модифицируемое вещество внедряется
только один атом из молекулы кислорода (02), второй же
восстанавливается до воды с помощью NADPH. P450
называют также оксигеназой смешанного действия,
поскольку он не только гидроксилирует субстрат, но
и восстанавливает 02 до Н20. Перенос электронов
от NADPH к Fe3+ в геме цитохрома осуществляется
Р450-редуктазой, также связанной с мембраной
эндоплазматического ретикулума.
Мы уже встречались с ферментом подобного типа
при гидроксилировании фенилаланина (см. с. 197).
Обратите внимание на разницу между окислением и ок-
сигенированием. Окисление подразумевает удаление
электронов, а оксигенирование - внедрение в молекулу
субстрата атома кислорода из молекулы 02 (не путать
с оксигенированием гемоглобина, когда молекула 02
обратимо связывается с атомом железа в геме).
Поразительно, что система Р450 справляется с
окислением огромного числа соединений, с которыми живые
организмы стали вступать в контакт только с развитием
современной химической промышленности. Об
эволюционной целесообразности этой системы можно только
гадать, но она позволяет нам выжить в созданных нами
новых условиях. Не исключено, что действие
изначально было направлено на терпены, алкалоиды и другие
вещества растений, которые защищают их от поедания
животными. Разносторонность системы Р450
определяется двумя факторами: во-первых, каждый из
ферментов этой системы обладает достаточно широкой
субстратной специфичностью в пределах определенного
класса соединений; во-вторых, спектры субстратной
специфичности различных ферментов хотя и разные,
но перекрывают друг друга.
Вторичная модификация:
присоединение полярных остатков
к продуктам реакций,
катализируемых Р450
На рис. 17.5 представлена схема, показывающая, как
продукты гидроксилирования гидрофобных
ксенобиотиков приобретают способность к растворению в воде
путем присоединения к ним остатка глюкуроновой
кислоты. Этот остаток переносится с глюкуронил-UDP,
который образуется при окислении в мембранах эндо-
плазматического ретикулума UDP-глюкозы (см. с. 87).
Ферменты, осуществляющие вторичную модификацию
ксенобиотиков, расположены там же.
Глюкозо-1 -фосфат
UTP
К
ГЧ РР,
UDP-глюкоза
Н20
2NAD+
Τ
Ν< 2 NADH + H+
UDP-глюкуронат
R-OH
К
UDP - глюкуронозилтрансфераза
ОН
Глюкуронид
Рис. 17.5 Система глюкуронилирования (а) и структура глю-
куронида (б)
Реакция печени на ксенобиотики
При попадании ксенобиотика (например,
фенобарбитала) в клетки печени начинается быстрое увеличение
поверхности эндоплазматического ретикулума.
Одновременно индуцируются системы глюкуронилирования
и Р450: включаются соответствующие гены и синтез
компонентов этих систем многократно усиливается.
Когда ксенобиотик будет полностью удален, все
возвращается к норме. Такая быстрая реакция позволяет избежать
потенциальной опасности, связанной с попаданием
в организм посторонних веществ. Однако она создает
трудности для химиотерапии. Эффективность многих
лекарств ограничена скоростью их метаболического
разрушения, тем более что мощность детоксицирующих
ферментных систем тем больше, чем дольше эти
лекарства присутствуют в организме. К сожалению, у столь
мощной системы детоксикации есть обратная сторона.
Те же лекарства обычно химически инертны и
связываются с молекулами, на которые направлено их действие,
благодаря нековалентным взаимодействиям. Однако при
окислении цитохромами Р450 они часто превращаются
в реакционноспособные соединения, причиняющие
массу неприятностей. И не только лекарства! Совершенно
безвредный бензпирен, который присутствует в
табачном дыме, попадая в печень и легкие курильщика,
окисляется там, превращаясь в химически активные
канцерогенные соединения, реагирующие с ДНК.
Мишенями для цитохромов Р450 служат не только
ксенобиотики. Они участвуют также в разрушении гема
и стероидных гормонов. В митохондриях
надпочечников цитохромы Р450 катализируют отдельные стадии
синтеза стероидных гормонов из холестерина.
Множественная защита
Защитить организм от ксенобиотиков можно,
воспрепятствовав их накоплению в клетках. В плазматической
мембране многих клеток, в том числе и разных тканей
человека, присутствует гликопротеин, ответственный
за выведение разнообразных веществ из клетки за счет
энергии гидролиза АТР. Круг таких соединений весьма
широк, и в него, в частности, попадает много
лекарственных препаратов и цитотоксических веществ. О
биологической важности этой транспортной системы можно
судить по тому, что с ее помощью осуществляется
транспорт стероидных гормонов из клеток надпочечников.
Транспортируемые таким образом вещества могут
различаться по строению, но все они обладают амфипати-
ческими свойствами (содержат полярные и
гидрофобные группы) и растворимы в жирах.
глава 17 Защитные ферментативные механизмы
211
Защита организма
от воздействия
собственных протеиназ
В главе 4 рассказывалось, как пищеварительная
система избегает разрушительного действия собственных
протеолитических ферментов. Однако протеиназы в
организме присутствуют повсюду, так что решение этой
проблемы актуально и вне желудочно-кишечного тракта.
Здесь мы расскажем лишь об одном из ее аспектов,
связанном с ферментом эластазой из нейтрофилов.
Нейтрофилы - фагоцитирующие белые кровяные
тельца, которые концентрируются вокруг инфицированных
и воспаленных участков тканей. Здесь нейтрофилы
начинают секретировать эластазу, которая разрушает
прилежащие участки соединительной ткани. Действие эла-
стазы направлено на один из ее главных компонентов -
белок эластин.
В легких воздух попадает в пузырьковидные
образования - альвеолы; с их помощью увеличивается
поверхность газообмена между воздухом и кровью.
Нейтрофилы присутствуют в альвеолах и выделяют
эластазу, однако ее разрушительное действие на легочные
структуры предотвращается белковым ингибитором
а,-антитрипсином (α,-антипротеиназой), который
секретируется в кровь печенью. Этот ингибитор
подавляет активность многих протеиназ, в том числе и
трипсина, но особенно он эффективен по отношению к эла-
стазе. Антитрипсин прочно связывается с ферментом,
блокируя его каталитический центр.
а,-Антитрипсина в крови вполне достаточно, чтобы
защитить легочные альвеолы от эластазы. Известен,
однако, генетический дефект, при котором содержание
антитрипсина в крови существенно уменьшается. В
результате эластаза может частично разрушать легочную
ткань, что проявится в увеличении размера альвеол
и уменьшении поверхности газообмена. У людей
развивается эмфизема легких.
Эмфиземе подвержены и курильщики табака. Это
происходит по двум причинам. Во-первых,
раздражение, вызываемое табачным дымом, привлекает в легкие
нейтрофилы, что способствует увеличению там
содержания эластазы. Во-вторых, компоненты дыма инакти-
вируют а,-антитрипсин, окисляя в нем функционально
важный остаток метионина всульфоксид (S —» S=0).
Этого достаточно, чтобы предотвратить инактивацию
эластазы ах-антитрипсином и способствовать протеоли-
зу легочной ткани, что ведет к эмфиземе. Существуют
и другие антипротеиназы, но α{-антитрипсин имеет,
пожалуй, самое важное значение.
часть 3 Метаболизм
212
Защита от активных
форм кислорода
Молекулярный кислород служит конечным акцептором
электронов в митохондриальной дыхательной цепи (см.
главу 7). Его восстановление описывается уравнением:
02 + 4е- + 4Н+ -> 2Н20 .
На первый взгляд, нет причин думать о какой-либо
защите, поскольку и кислород, и вода вполне безвредны.
Однако восстановление кислорода может привести к
образованию не только воды, но и других продуктов,
крайне опасных для организма. Переход от анаэробного
метаболизма к использованию кислорода как акцептора
электронов был важнейшим достижением эволюции.
Опасность возникает при одноэлектронном
восстановлении кислорода, которое приводит к появлению ре-
акционноспособного анион-радикала - супероксида
(радикалами называют молекулы с неспаренным
электроном):
02 + е- -> 0^ .
Неспаренный электрон находит партнера, атакуя
ковал ентную связь другой молекулы.
Супероксид в организме образуется по нескольким
причинам. В норме восстановление молекулярного
кислорода в дыхательной цепи происходит только с
помощью цитохромоксидазы, которая отдает ему все четыре
электрона, необходимые для образования воды. Однако
неизбежная утечка электронов с промежуточных
переносчиков на кислород приводит к генерации некоторого
количества 02. Кроме того, мутации митохондриальной
ДНК могут сопровождаться блокированием электрон-
транспортных путей, и электроны будут использоваться
на образование супероксида (например, прямое
окисление убихинона кислородом). Такого рода мутации
особенно опасны, потому что митохондрии лишены
системы репарации ДНК (см. с. 259).
Другим источником супероксида может служить
спонтанное окисление гемоглобина. В норме гемоглобин (НЬ)
обратимо связывает кислород: НЬ + 02—» О^ + НЬ02.
Однако оксигемоглобин (НЬ02) с некоторой
вероятностью превращается в метгемоглобин (содержащий
не Fe2+, a Fe3+) и супероксид О^.
Известно также, что образование супероксида
вызывает ионизирующая радиация.
Кроме супероксида, в организме образуется и такое
опасное вещество, как перекись водорода (Н202). Это
происходит при окислении некоторых метаболитов ок-
сидазами (цитохромоксидаза дыхательной цепи к ним
не относится).
Фагоциты, захватывая бактериальные клетки,
потребляют много кислорода. Он восстанавливается с
помощью NADPH с образованием супероксида.
Последний, попав в вакуоль, превращается в перекись
водорода, которая способствует разрушению бактериальной
клетки. Нейтрофилы, в избытке собирающиеся в
воспаленных суставах, также способны выделять
супероксид, внося тем самым существенный вклад в развитие
артритов.
Количество образующегося в организме
супероксида ничтожно мало. Однако и этого достаточно, чтобы
вовлечь в нежелательные превращения большое число
других молекул. Дело в том, что супероксид - радикал,
а это означает, что один из продуктов его реакций
с обычными молекулами непременно должен быть
тоже радикалом. А поскольку все радикалы, за редкими
исключениями, крайне реакционноспособны, этот
продукт, в свою очередь, вступит в какую-нибудь другую
реакцию, порождая очередной радикал. Таким
образом, процесс носит цепной характер и потенциально
может продолжаться бесконечно. Остановиться он
может либо вследствие рекомбинации радикалов с
образованием обычных молекул (это маловероятно из-за
низкой концентрации радикалов), либо из-за аномально
низкой реакционной способности дочерних радикалов.
Какие дефекты в организме обусловлены
протеканием таких цепных реакций, пока точно не известно,
но полагают, что супероксид причастен к старению,
образованию катаракты хрусталика, инфаркту
миокарда и т. д. Существуют три основных способа защиты
от супероксидов - один химический и два
ферментативных.
Витамины С и Ε
как ловушки кислородных радикалов
Чисто химический подход к обезвреживанию
супероксида и блокированию цепных реакций заключается
в применении антиоксидантов. Так называют вещества,
которые, вступая в реакции со свободными радикалами,
превращаются в радикалы с малой реакционной
способностью. На них цепной процесс прерывается.
Главными природными тушителями цепных реакций являются
аскорбиновая кислота (витамин С) и а-токоферол
(витамин Е). Первая растворима в воде, а второй в
жирах, так что в тандеме они способны защитить и
компоненты цитозоля, и мембранные липиды. Эти два
вещества не единственные антиоксиданты; аналогичной
активностью обладают, например, β-каротин и мочевая
кислота. Эффективным антиоксидантом служит также
билирубин - продукт расщепления гема гемоксигеназой
(см. с. 370). Интересно, что гемоксигеназа активируется
продуктами частичного восстановления кислорода (их
накопление называют окислительным стрессом; он
может быть вызван, например, ионизирующим
излучением). Пока неясно, однако, может ли образование
билирубина рассматриваться как защитная реакция организма.
Ферментативное уничтожение
супероксида супероксиддисмутазой
По-видимому, все ткани животных содержат фермент
супероксиддисмутазу. Больше всего ее в
митохондриях, есть она и в лизосомах, и в пероксисомах (см.
главу 16). Супероксиддисмутаза присутствует не только
в клетках, но и в плазме крови, лимфе, синовиальной
жидкости. Этот фермент катализирует реакцию:
202 + 2Н+ -> Н202 + 02 .
Перекись же водорода разлагает другой фермент -
каталаза:
2Н202 -> 2Н20 + 02 .
Перекись водорода появляется не только в
результате дисмутации супероксида, но также благодаря
действию оксидаз. FAD-содержащие оксидазы
катализируют реакции, которые в обобщенном виде выглядят так:
АН2 + 02 -> А + Н202 .
К их числу принадлежит, в частности, ксантинок-
сидаза, участвующая в метаболизме пуринов (см. гла-
ву 18).
Перекись водорода опасна тем, что в присутствии
ионов тяжелых металлов (например Fe2+) она
распадается с образованием высокореакционноспособных
гидроксильных радикалов (не путать с безобидными
гидроксильными анионами), атакующих ДНК и другие
биомолекулы:
н2°2 + Fe2+ -> Fe3+ + он"+ он~·
Оба фермента - и супероксиддисмутаза, и каталаза -
защищают ткани организма от атак радикалов. Однако
есть еще один фермент, разрушающий перекись
водорода, - глутатионпероксидаза. Его защитная функция
важна для мозга, который содержит мало каталазы.
Стратегия защиты
с помощью глутатиона
Глутатионом называется тиолсодержащий трипептид
γ-глутамил-цистеинил-глицин (GSH; рис. 17.6). Он
присутствует в большинстве клеток и используется главным
глава 17 Защитные ферментативные механизмы
213
Glu-Cys-Gly
I
SH
Восстановленный глутатдон (GSH)
Glu-Cys-Gly
S
I
s
Glu-Cys-Gly
Окисленный глутатион (GSSG)
Рис. 17.6 Структура восстановленной (GSH) и окисленной
(GSSG) форм глутатиона
Glu, Cys, Gly - трехбуквенное обозначение аминокислот глу-
тамата, цистеина и глицина соответственно. Однобуквенная
система используется для записи протяженных
аминокислотных последовательностей
образом как восстанавливающий агент, например,
для поддержания в восстановленном состоянии цис-
теиновых остатков в белках. Эту защитную функцию
глутатион выполняет без участия ферментов. Другая
защитная функция глутатиона связана с восстанавле-
нием перекиси водорода (а также органических
гидроперекисей R-O-OH). Эта реакция катализируется
глутатионпероксидазой:
Н202 + 2GSH -> GSSG + 2Н20 .
Окисленный глутатион (GSSG) затем
восстанавливается NADPH при участии глутатионредуктазы:
GSSG + NADPH + Н+ -> 2GSH + NADP+.
Целостность эритроцитов зависит от глутатиона,
который восстанавливает ферригемоглобин (метгемогло-
бин) в феррогемоглобин и разрушает перекиси. Это
объясняет целесообразность существования в
эритроцитах пентозофосфатного метаболического пути, который
поставляет NADPH (см. рис. 13.1), необходимый для
восстановления глутатиона.
Людям с дефектной глюкозо-6-фосфатдегидрогена-
зой - первым ферментом пентозофосфатного пути -
вполне хватает этой активности в обычных условиях. Однако
при накоплении в их клетках перекисей (например, после
приема антималярийного лекарства примахина)
возникает дефицит NADPH, необходимого для восстановления
GSSG. Это приводит к повреждению плазматической
мембраны эритроцитов и гемолизу последних.
В этой главе мы уделили внимание защитным
механизмам у животных. Однако они существуют и у
растений, причем более разнообразные, возможно потому,
что у них нет иммунной системы и они не могут ни убе-
часть 3 Метаболизм
214
жать, ни прогнать хищника. Вот пример, взятый наугад:
некоторые растения обладают ферментом хитиназой,
который разрушает хитиновый покров насекомых.
Дополнительная литература
Свертывание крови
Scully Μ. Ε The biochemistry of blood clotting: the
digestion of a liquid to form a solid// Essays Biochem. 1992.
Vol.27. P. 17-36.
(Хорошо написанный обзор, посвященый механизму
свертывания крови и медицинским аспектам этого
процесса.)
Цитохром Р450
Сооп М. J., DingX., Pernecky S., VazA. D. N. Cytochrome
Р450; progress and predictions// FASEB J. 1992. Vol. 6.
P. 669-673.
(Четкий и ясный обзор, посвященный системе цитохро-
ма Р450.)
EstabrookR. W. The remarkable P450s: a historical
review of these versatile hemoprotein catalysts// FASEB J.
1996. Vol. 10. P. 202-204.
(Анализ состояния и перспектив исследований
метаболической роли цитохромов Р450; содержит более 2000
библиографических ссылок.)
Свободные радикалы
Babior В. М. The respiratory burst oxidase // Trends
Biochem. Sci. 1987. Vol. 12. P. 241-243.
(Обзор, посвященный изучению механизма образования
супероксида фагоцитами.)
Rusting R. L Why do we age? // Sci. Amer. 1992. Vol. 267(6).
P. 130-141.
(Одна из глав просвящена роли свободных радикалов
в старении.)
Harris Ε. Regulation of antioxidant enzymes // FASEB J.
1992. Vol. 6. P. 2675-2683.
(Обзор, посвященный регуляции количества ферментов,
разрушающих свободные радикалы.)
Rose R. С, Bode A. M. Biology of free radical
scavengers: an evaluation of ascorbate // FASEB J. 1993. Vol. 7.
P. 1135-1142.
(Обсуждается вопрос о том, какими свойствами
должны обладать ловушки свободных радикалов. Главное
внимание уделено аскорбиновой кислоте.)
Fridovich I. Superoxide radical and superoxide dismutase //
Annu. Rev. Biochem. 1994. Vol. 64. P. 97-112.
(Лаконичный, но полный обзор, посвященный
супероксид-радикалам и супероксиддисмутазе.)
Uddin S., Ahmad S. Dietary antioxidants protection against
oxidative stress // Biochem. Education. 1995. Vol. 23. P. 2-7.
(Полезный обзор, в котором изложено, как ионы меди
и железа катализируют образование ОН "-радикалов
и почему эти ионы всегда прочно связаны с белками.)
Вопросы к главе 17
1. Свертывание крови - каскадный процесс. В чем его
смысл?
2. Объясните, каким образом тромбин участвует в
образовании кровяного сгустка.
3. Спонтанная полимеризация фибриновых мономеров
приводит к образованию мягкого сгустка. Как он
превращается в более стабильную структуру?
4. Какова роль витамина К в свертывании крови?
5. Какова функция цитохрома Р450?
6. Зачем нужен NADPH в реакциях оксигенирования?
7. Какова функция глюкуронил-UDP при выведении
из организма нерастворимых в воде соединений?
8. Что такое множественная защита?
9. Почему курение вызывает эмфизему легких?
10. Что такое супероксид?
11. Какие механизмы защищают организм от вредного
действия супероксида?
глава 17 Защитные ферментативные механизмы
215
глава
18
Метаболизм нуклеотидов
В ряде предыдущих глав основное внимание уделялось
образованию энергии из питательных веществ. При этом
центральное место занимает АТР, хотя GTP, СТР и UTP
также участвуют в различных реакциях метаболизма.
Однако до сих пор мы в основном рассматривали
фосфатные группы этих молекул. Природа оснований, будь
то A, G, С или U, была важна только для их узнавания
соответствующими ферментами и не имела прямого
отношения к метаболическим процессам. По этой причине мы
и не обсуждали раньше строение самих оснований.
В этой главе мы переходим к новой важной области
биохимии - изучению механизма передачи информации;
мы обратимся к нуклеиновым кислотам и процессу
синтеза белка, а для этого необходимо знать структуру
основных нуклеотидов. Эта глава посвящена структуре,
синтезу и метаболизму нуклеотидов и является необходимой
для понимания последующих глав.
Структура и номенклатура
нуклеотидов
Термин нуклеотид происходит от названия
нуклеиновых кислот, впервые обнаруженных в ядре (лат. nucleus),
и представляющих собой полимеры нуклеотидов.
Нуклеотиды имеют общую структуру: фосфат-сахар-
основание
Структуру нуклеозидов можно записать как сахар -
основание.
Так, AMP и соответствующий нуклеозид, аденозин,
имеют следующее строение:
О
II
"О—Р-0-
I
О"
4'
Аденин
ОН
ОН
Основание = аденин
V
Нуклеозид = аденозин
J
к_
V
Нуклеотид = AMP
J
Строго говоря, AMP записана здесь как 5'-AMP. Штрих (')
означает, что речь идет о нумерации атомов в цикле
сахара рибозы, а не в адениновом цикле. Однако 5'-положе-
ние фосфата, как наиболее распространенное, обычно
не отмечается. Поэтому 5'-АМР часто называют просто
AMP, в то же время положение фосфата у третьего
углеродного атома рибозы обязательно обозначается З'-АМР.
Углеводный компонент нуклеотидов
Углеводный компонент нуклеотидов всегда представлен
пентозой - чаще рибозой или 2'-дезоксирибозой,
которые всегда имеют D-конфигурацию и никогда не
встречаются в L-форме.
Н0-
Н0-
ОН
ОН ОН
D-Рибоза
ОН
I'
Η
ОН Η
D -2!-Дезоксирибоза
В РНК углевод всегда представлен рибозой (отсюда
и название - рибонуклеиновая кислота), а в ДНК -
дезоксирибозой (отсюда - дезоксирибонуклеиновая
кислота). Нуклеотид, содержащий рибозу, является
рибонуклеотидом, но это обычно не отмечается
при написании; если не имеется специальных указаний,
то нуклеотид, обозначенный как AMP, относится к ри-
бонуклеотидам. При обозначении дезоксирибонуклео-
тидов либо используют приставку дезокси- (например,
дезоксиаденозинмонофосфат), либо добавляют «d»
к символу нуклеотида - dAMP (за единственным
исключением, о котором ниже).
Основания нуклеотидов
Номенклатура
Прежде всего нас интересуют пять главных
оснований - аденин, гуанин, цитозин, урацил и тимин,
кратко обозначаемые первыми буквами своих английских
названий. A, G, С и U обнаружены в РНК; A, G, С и Τ -
в ДНК.
часть 3 Метаболизм
216
Рибонуклеотиды обозначаются как AMP, GMP, CMP
и UMP. Их старые, но еще употребляемые названия -
соответственно адениловая, гуаниловая, цитидиловая
и уридиловая кислоты (или аденилат, гуанилат, цити-
дилат иуридилат - для их ионизированных при
физиологических значениях рН форм). К дезоксирибо-
нуклеотидам относятся dAMP, dGMP, dCMP и dTMP.
Последний чаще обозначается ТМР (или тимидилат),
без приставки d, поскольку Τ обнаружен только в де-
зоксирибонуклеотидах (редких исключений мы
касаться не будем).
Если необходимо обозначить нуклеотид без
указания конкретного основания, то часто используют
аббревиатура ΝΜΡ или 5'-ΝΜΡ для рибонуклеотидов
и dNMP или 5'-dNMP для дезоксирибонуклеотидов.
Дезокси- UMP встречается только в виде
промежуточного соединения при синтезе dTMP; он не
присутствует в ДНК (за исключением случаев ее химического
повреждения).
Соответствующими рибонуклеозидами
(основание-сахар) для дезоксирибонуклеозидов являются аде-
нозин, гуанозин, цитидин, уридин и d-аденозин и т. д.
Названия цитидин и уридин созвучны с названиями
свободных пуриновых оснований (сравните аденин
и гуанин), в то же время название свободного
основания цитозина похоже на название нуклеозида (сравните
с аденозином), так что здесь необходимо быть
внимательным. В транспортной РНК присутствуют и «минорные»
основания (см. с. 287). Одним из них является гипоксан-
тин. На пути биосинтеза пуриновых оснований еще
образуются гипоксантинриботид и инозинмонофосфат;
гипоксантинриботид называется инозином.
Структура оснований
Прежде всего необходимо указать, что А и G -
относятся к пуринам; а С, U и Τ - к пиримидинам.
Эти термины произошли от соответствующих
химических структур пурина и пиримидина, которые в
свободном виде в природе не встречаются.
Структура оснований, входящих в состав нуклеоти-
дов, представлена на рис. 18.1.
НС
Η
NfC6-C'
I1 5II
Пурин
-Ν
•и
Η
Η
Nf^CH
I 3 5 и
НС
;сн
Пиримидин
В дальнейшем эти циклы будут изображаться в
упрощенном виде.
Η2Ν Ν
Гуанин;
2-амино-6-оксипурин
N 12
и N
Цитозин;
2-окси-4-аминопиримидин
N
Аденин;
6-аминопурин
О
Урацил;
2,4-диоксипиримидин
Пурин
Пиримидин
СН3
N
Тимин;
2,4-диокси-5-метилпиримидин
Рис. 18.1. Схематичное изображение структуры пуриновых
и пиримидиновых оснований, обнаруженных в
нуклеиновых кислотах
Присоединение оснований
в нуклеотидах
Основания присоединяются к углеводному компоненту
нуклеотида через атом азота в 9 положении пурина
и в 1 положении пиримидина. Гликозидная связь имеет
β-конфигурацию; она расположена над плоскостью
сахарного кольца. Структуры AMP и СМР приведены
на рис. 18.2.
Синтез пуриновых
и пиримидиновых нуклеотидов
Пуриновые нуклеотиды
Большинство клеток может синтезировать пуриновые
основания de novo из предшественников меньшего
молекулярного веса. При этом основания не синтезируются
в свободном виде, а пуриновый цикл собирается
постепенно путем присоединения необходимых фрагментов
к рибозо-5-фосфату, так что к моменту завершения
построения цикла образуется готовый нуклеотид.
Это касается только синтеза пуринов de novo, поскольку
свободные пуриновые основания, образовавшиеся в
результате деградации нуклеотидов, используются для
их же синтеза (см. с. 222). Механизм присоединения
глава 18 Метаболизм нукеотидов
217
NH2
Ο—Ρ—0—CH2 η
I °
Q- Η H*\
Η Η
он он
Аденозинмонофосфат (AMP)
NH2
? °
О—Ρ—О—СН2 п
I °
о- н н и,
Η Η
он он
Цитидинмонофосфат (СМР)
Рис. 18.2. Структура пуринового и пиримидинового нук-
леотидов
к рибозо-5-фосфату (риботилирование) в обоих случаях
одинаков. Он же используется и в ходе синтеза пири-
мидиновых нуклеотидов (но соединения, которые
подвергаются риботилированию, различны). Это подводит нас
к ФРПФ, метаболиту, участвующему в риботилировании.
5-Фосфорибозил-1 -пирофосфат (ФРПФ)
ФРПФ образуется из рибозо-5-фосфата, который в
свою очередь, образуется в пентозофосфатном пути
(см. главу 13) в результате переноса ферментом
ФРПФ-синтетазой пирофосфатной группы с АТР.
О
О—Ρ—ΟΙ
0"
Н
+ АТР
ОН
О
II
о—р-о-
I
0"
он он
Рибозо-5-фосфат
I
о
он он
ФРПФ
н о о
II II
О—Р-0—Р-0"
I I
о- о-
АМР.
ФРПФ является «активированной» формой рибозо-
5-фосфата; соответствующие ферменты могут,
достраивая его, образовать основания нуклеотидов и отщепить
пирофосфат. Реакция термодинамически направляется
гидролизом последнего до 2Pf.
О
II
-о—ρ—οι
О"
N [/ О—Р-0—Р-0"
1 Г ι _ ι _
ОН ОН О" О
- основание
-о—р-о-
I
о-
основание
ОН ОН
2Р/
РР
Н20
В этой реакции конфигурация С-1 -углеродного
атома меняется так, что основание занимает нужное
β-положение.
Обратите внимание,что при синтезе de novo (см.
реакцию 1 на рис. 18.4) в качестве основания выступает
просто NH2-rpynna, полученная из глутамина; продуктом
реакции является 5-фосфорибозиламин.При реутилизации
пурина в качестве основания используется уже готовый
пурин. Обычно термин нуклеотид ассоциируется с пури-
новыми или пиримидиновыми основаниями, но он
может быть применен и к другим основаниям, связанным
с сахарофосфатом подобным образом.
Вернемся от общей схемы к синтезу пуринов de novo.
После образования 5-фосфорибозиламина
осуществляется 9 последовательных реакций, завершающихся
сборкой первого пуринового нуклеотида с гипоксантином
в качестве основания (рис. 18.3). Этим нуклеотидом
является IMP, или инозиновая кислота.
IMP служит своеобразным метаболическим
перекрестком, так как его основание гипоксантин
превращается или в аденин, или в гуанин, образуя
соответственно AMP или GMP. Этот путь кратко представлен
на рис. 18.3; и все реакции этого процесса приведены
на рис. 18.4 и 18.5. Обсудим наиболее интересные
реакции. Для синтеза 1 молекулы пуринового нуклеотида
используется 6 молекул АТР. При этом от АТР отщепляются
только фосфатные группы, а адениновый нуклеотид
не претерпевает никаких изменений, так что все сводится
к синтезу AMP. Разберем реакции (3) и (9) на рис. 18.4,
которые имеют особое значение, поскольку они
относятся к реакциям переноса одноуглеродных фрагментов.
Реакции одноуглеродного переноса
в синтезе пуриновых нуклеотидов
Реакции синтеза (3) и (9) включают введение фор-
мильной (НСО-) группы в промежуточные продукты
синтеза. Донором этих групп в обоих случаях служит
часть 3 Метаболизм
218
Рибозо-5-фосфат + ATP
ФРПФ + AMP
COO"
Ιορ-
NH2 /
I
-рибоза
(я)
зОР —О—рибоза
IMP
ν
AMP
9 XMP
ϊ
3ΟΡ—О—рибоза
,1
Η2Ν
9 GMP
Ν
Ι
зОР —Ο—рибоза
Рис. 18.3. Схематичное изображение синтеза пуринов GMP,
AMP и XMP de novo
Основание в IMP, или инозинмонофосфате, - гипоксантин.
Основанием в ХМР, или ксантозинмонофосфате, является
ксантин
Ы10-формилтетрагидрофолат. В клетке тетрагидрофолат
(FH4) является переносчиком формильных групп. Это
кофермент, образующийся из витамина - фолиевой
кислоты (F), или птероилглутаминовой кислоты.
С00"
" χ)—C—Ν—CH
Η '
C00"
Фолиевая кислота (птероилглутаминовая кислота)
Витамин F восстанавливается до FH4 с участием
NADPH в две стадии.
NADPH
+ н+1
NADP+
Фолат—^—*
(Θ V^
NADPH
+ H+l
Дигидрофолат —
(FH2)
NADP*
- Тетрагидрофолат
(FH4)
иги ро олат уктаза
Структуры FH2 и FH4 приведены ниже.
Дигидрофолат (FH2)
—N—(7 х)—C-N—
Η \=/ Η
Тетрагидрофолат (FH J
COO"
соо-
I
сн2
сн2
сн
I
С00"
Заметим, в структуре FH4 атомы N-5 и N-10
расположены таким образом, что только один углеродный атом
может заполнить промежуток между ними. Для наших
целей можно представить FH4 в таком виде:
HN
/
Тетрагидрофолат (FH4)
А N10-φopмилτeτpaгидpoφocφaτ в таком виде:
Ьк
NH ^СН2
O^C-N1-^'
Ып I
Η
Ν10- формилтетрагидрофолат
(формил-РНд)
Это донор формильной группы в реакциях (3) и (9)
биосинтеза пуринов; катализирует эти реакции
формилтрансфераза
Откуда поступает формильная группа
в формил-РН4?
Формильная группа поступает из аминокислоты серина
(который легко синтезируется из промежуточного
продукта гликолиза 3-фосфоглицерата).
СН2ОН
CHNHi
I
COO"
Серии
глава 18 Метаболизм нукеотидов
219
ΦΡΠΦ
R'CONH2 -Λ®
\Ч _ Глицин
R'COOH И
?)Р-0-СН2 о
рр,
NH7
+
ATP
NH7 М10-формил-РН4
ΉΗ.
H2V^ V Н2С^ ^СНО
он он
ί Η
Θ
R'CONH2^bATP
R"COOH
H^ADP + Ρ/
ООС м
IN
>
H2N N"F
2.
©
H2N
N
>
■N-R
ADP + P,· ATP
CH2 CHO
Аспарагиновая I
кислота ~~\~ ©
coo- ГADP + p'·
CH2H О
I I II
HC— N — С
I
coo-
coo-
I
H20 HN^ ^lj|-R
© Η
©
^°-формил-РН4
0
II
X
coo- A
(R = Рибозо-5'-фосфат)
(R'CONH2 = Глутамин)
Рибозо-5'-фосфат
IMP
Рис. 18.4. Последовательность реакций синтеза de novo пуринового цикла из ФРПФ до инозиновой кислоты
Реакции, отмеченные цифрами в кружках, обсуждаются в тексте. Выделены структурные изменения, произошедшие в результате
очередной реакции цикла
Фермент серингидроксиметилаза переносит гидроксиметильную группу (-СН2ОН) на FH4, в результате
образуются глицин и N5,N10-MemneH-FH4.
СН2ОН
CHNH^
I
coo
'ΗΝ
Η
CH2
Ι..Μ.
Μ
10
'Ν "СН2
I I
CH2—Ν—
CH-,ΝΗί
I
COO"
2,ΝΠ3 + H70.
Серии
FHL
N5, N10-MemneH-FH4 Глицин
часть 3 Метаболизм
220
ΗΝ'
О
II
.с.
Аспартат
Η
NAD
NADH + H+
ΗΝ'
0^-4N/<>y
I || CH + OOC—CH2—C —COO"
"SHY U
IMP Рибозо-5-фосфат
GDP + P.- x H
ooc— сн2—с—coo-
I
NH
I
Аденилосукцинат | || CH
CH
*H'
Рибозо-5-фосфат
ATP
AMP + PP,·
r
Рибозо-5-фосфат
coo-
H
II
CH
I
coo-
Фумарат
О
II
HN^C^C
NH7
HzN^^N^^1^7
CH
II ch
^
HC
N
■N/CvV
Рибозо-5-фосфат
ι
Рибозо-5-фосфат
GMP
AMP
Рис. 18.5. Синтез GMP и AMP из инозиновой кислоты (IMP)
Выделены структурные изменения, произошедшие в результате очередной реакции
Образовавшийся метилен-РН4 еще не готов для учас- Метиленовая группа окисляется NADP+-3aBHCHMbiM
тия в реакции формилирования, поскольку СН2-группа ферментом до метинильного производного, которое пос-
более восстановлена, чем формильная группа. ле гидролиза превращается в 1Ч10-формил-РН4- донор
формильной группы.
CH,-lU
NADP* NADPH
ΙιΜ.
ν; "ch2
11 Ίο
CH Ν10
H20
Ч^СН,
'Ν
Ο.Η
Η
Поставляет формильные
_^ группы для биосинтеза
пуриновых нуклеотидов
Ν5,Ν10- метилен -FHL
N^N^-MeTHHRn-FHL
Ы10-формил-РН4
глава 18 Метаболизм нукеотидов
221
Как образуются ATP и GTP из AMP
и GMP?
Для протекания большинства синтетических реакций
в клетке необходимы нуклеозидтрифосфаты. Прежде
всего, они нужны для синтеза нуклеиновых кислот.
Существует простое, но очень интересное решение
этого вопроса: в клетке есть ферменты киназы, которые на
высокоэнергетическом уровне осуществляют перенос
фосфатных групп между нуклеотидами. Поскольку
изменение свободной энергии в этой реакции
незначительно, то перенос фосфатной группы от нуклеотида к нук-
леотиду происходит легко. Основным источником
фосфатных групп служит АТР. Необходимо помнить,
что энергия, образующаяся в результате метаболических
процессов, постоянно используется для регенерации АТР
из ADP и Р#. Синтезированные de novo AMP и GMP фос-
форилируются киназами, как показано ниже.
AMP + АТР —> 2ADP, V енилаткиназа
GMP + АТР —> GDP + ADP, 1 уанилаткиназа
уклеозиддифосфат ·- г
GDP + АТР
или
ADP
GTP + ADP.
или
АТР
Реутилизация пуриновых оснований
при синтезе нуклеотидов
Мы уже подчеркивали, что синтез пуринов de novo
не дает свободных пуриновых оснований - сразу
образуются пуриновые нуклеотиды. Однако существует путь
синтеза пуриновых нуклеотидов, в котором свободные
Гуанин
или + ФРПФ
Гипоксантин
—>
GMP
или
IMP
основания превращаются в нуклеотиды после
взаимодействия с ФРПФ. Свободные основания образуются
в процессе деградации нуклеотидов; они не
подвергаются дальнейшему расщеплению, а сберегаются. В этом
процессе участвуют два фермента - две фосфорибозил-
трансферазы. Один из них образует нуклеотиды из аде-
нина, а другой из гипоксантина и гуанина. Второй
фермент, известный как ГГФРТ (гипоксантин-гуанин-
фосфорибозилтрансфераза), катализирует следующую
реакцию.
+ РР,-
По всей видимости, у человека сбережение аденина
имеет меньшее значение по сравнению с гуанином и ги-
поксантином, поскольку основные пути распада этих
нуклеотидов приводят к образованию свободного
гипоксантина (из AMP) и гуанина (из GMP) (рис. 18.6).
Какова физиологическая роль
реутилизации пуриновых оснований?
Так как синтез пуринов требует весьма большого
количества энергии механизм реутилизации свободных
пуриновых оснований выгоден, так как позволяет
клетке ограничить синтез de novo. Кроме того, эритроциты,
например, не способны синтезировать пурины de novo
и используют только имеющиеся готовые основания.
Физиологическое значение реутилизации пуринов
особенно наглядно демонстрирует генетическое
заболевание у детей - синдром Леша-Нихана, связанный
AMP
Н20 Ρ,· Η20 ΝΗ3 Ρ;
—^ * —► Аденозин ^ S ►Инозин ^*-
4
5'-Нуклеотидаза \денозиндезамина
удаляет фосфатную группу удаляет Nh^-rpynny
_► Гипоксантин +
рибозо-1 -фосфат
дф фор
GMP
ΗιΟ Ρ,
♦-Гуанозин
р»
Гуанин +
~*~ рибозо-1-фосфат
Рис. 18.6. Схема синтеза свободных пуриновых оснований из гипоксантина и гуанина, образующихся при деградации
нуклеотидов
Больные, у которых в лимфоцитах отсутствует аденозиндезаминаза, страдают иммуннодефицитом. Лечение этого заболевания
стало первым успешным применением метода генной терапии - введение in vitro нормального гена аденозиндезаминазы в
стволовые клетки костного мозга с последующей пересадкой их пациенту
часть 3 Метаболизм
222
с отсутствием фермента ГГФРТ. Он приводит к
неврологическим патологиям и проявляется в умственной
отсталости, нарушении координации движений
и агрессивности, часто направленной против самого
себя и приводящей к различным травмам. В мозге
синтез de novo выражен слабо, и потому этот орган так
чувствителен к нарушениям в процессе реутилизации
оснований. Отсутствие реакций реутилизации пурино-
вых оснований у таких больных сопровождается
избыточным синтезом пуриновых нуклеотидов de novo
в печени и возрастанием уровня ФРПФ. Этим
объясняется тот факт, что у больных резко увеличивается
образование мочевой кислоты, как и при подагре, что
может привести к повреждению почек кристаллами
уратов. Связь между биохимическим дефектом и
неврологическими симптомами при заболевании
Леша-Нихана не совсем ясна. Хотя гиперпродукцию
мочевой кислоты можно снизить аллопуринолом, это
не облегчает неврологическую симптоматику. Следует
отметить, что у больных подагрой неврологическая
симптоматика не выражена.
Источников свободных пуриновых оснований,
используемых для ресинтеза пуринов, скорее всего,
несколько. Хотя пищевые продукты содержат пурины
в виде нуклеиновых кислот, есть данные,
свидетельствующие о том, что большая их часть разрушается
эпителиальными клетками кишечника и не поступает в кровь.
Напротив, пурины, введенные в кровоток,
утилизируются клетками. Место синтеза большинства пуринов -
печень. Доказано, что она высвобождает их в кровь для
использования другими клетками, например ретикуло-
цитами, которые не обеспечены полной системой синтеза
de novo. Возможно также, что клетки используют пури-
новые основания, образовавшиеся при распаде
нуклеиновых кислот. При лизосомальном разрушении
компонентов клеток, содержащих нуклеиновые кислоты, по-
видимому, освобождаются преимущественно свободные
основания. И хотя значение синтеза пуриновых
оснований из фрагментов не вызывает сомнений, мы не
располагаем полной информацией о перемещении
свободных оснований в организме.
Повторное использование образованных ранее
пуриновых оснований, конечно, имеет явное
преимущество по энергетическим затратам, что ведет к
соответствующему снижению уровня их синтеза de novo. Это
достигается двумя путями: во-первых, реутилизация
снижает уровень ФРПФ и, следовательно, вклад всего
пути в образование пуринов; во-вторых,
образовавшиеся в процессе реутилизации пуриновых оснований
AMP и GMP осуществляют ингибирование по типу
обратной связи.
Образование мочевой кислоты
из пуринов
Распад нуклеотидов приводит к образованию свободных
гипоксантина и гуанина. Часть из них снова
преобразуется в нуклеотиды, а часть окисляется с образованием
мочевой кислоты (рис. 18.7).
V
Ксантиноксидаза
При лечении больных
п аллопуринолом ингибируется
N \ / °
Гипоксантин ^s^
о
N
0 У ν Ксантиноксидаза
Гуанозиндезаминаза / КсантинХ При лечении больных
\^ аллопуринолом
"С о ингибируется
9 90
Ν ^ Ν
H2N
Гуанин
Мочевая кислота
Рис. 18.7. Превращение гипоксантина и гуанина в мочевую
кислоту
Лекарственный препарат аллопуринол близок по структуре
к гипоксантину. Под действием ксантиноксидазы аллопуринол
превращается в аллоксантин, который, в свою очередь, инги-
бирует ксантиноксидазу
Фермент ксантиноксидаза, образующий мочевую
кислоту, присутствует в основном в печени и клетках
слизистой кишечника. Подагра обусловлена повышением
содержания уратов в крови, что сопровождается
отложением их кристаллов в тканях. По-видимому,
основным источником мочевой кислоты скорее всего
является избыточное образование de novo пуриновых
нуклеотидов, вызванное у некоторых пациентов повышенной
активностью ФРПФ-синтетазы. К избыточному
синтезу пуриновых нуклеотидов приводит также дефицит
фермента ГГФРТ. Лекарственный препарат аллопуринол,
применяемый для лечения подагры, по структуре
сходен с гипоксантином.
N
I
НС
он
г н
с /
Η
ΝΗ
Ν
Аллопуринол
г н
1 и /
O^N/CY
Η н
Аллоксантин
ОН
I
N С ^
I II СН
Енольная форма
гипоксантина
глава 18 Метаболизм нукеотидов
223
В организме под действием самой ксантиноксидазы ал-
лопуринол превращается в аллоксантин - ингибитор
ксантиноксидазы. Этот процесс известен как суицидное
ингибирование, или самоинактивация. Он приводит
к преимущественному накоплению ксантина и гипоксан-
тина, а не мочевой кислоты (см. рис. 18.7). Эти продукты
более водорастворимы, чем мочевая кислота, и быстрее
выводятся из организма, предотвращая таким образом
отложение нерастворимых кристаллов мочевой
кислоты в тканях и снимая клинические симптомы подагры.
Регуляция синтеза пуриновых
нуклеотидов
Как и во всех метаболических путях, во избежание
химического хаоса необходима четкая регуляция синтеза
пуриновых нуклеотидов. Путь синтеза de novo является
классическим примером аллостерического ингибирования
по принципу обратной связи (англ. feedback). «Местом
контроля» является первая (обратимая) реакция синтеза
пуринов. De novo ее катализирует ФРПФ-синтетаза,
ингибируемая AMP, ADP, GMP и GDP. Следующий
фермент, катализирующий первую необратимую реакцию
синтеза пуриновых нуклеотидов (см. рис. 18.4, реакция 2),
ингибируется так же, как и предыдущий (рис. 18.8).
Однако этим дело не заканчивается, поскольку в
процессе синтеза de novo образуется IMP, который затем
расходуется на образование AMP и GMP. Эта реакция
регулируется продуктами по принципу обратной связи
(см. рис. 18.8). Как положительные, так и
отрицательные регуляторные механизмы служат для
сбалансированного образования АТР и GTP, поскольку оба нук-
леотида требуются для биосинтеза нуклеиновых кислот
(см. главу 19).
Синтез пиримидиновых нуклеотидов
Большинство клеток организма синтезирует пиримиди-
новые нуклеотиды de novo. В отличие от бактерий,
у млекопитающих путь реутилизации свободных
пиримидиновых оснований не выражен так ярко, как для
пуринов. Однако нуклеозид тимидин легко фосфорилиру-
ется до ТМР под действием тимидинкиназы; таким
образом, ресинтез этого нуклеотида имеет место.
Путь синтеза пиримидинов схематично
представлен на рис. 18.9 и полностью приведен на рис. 18.10. Он
начинается с аспарагиновой кислоты и приводит к
образованию вещества с циклической структурой - орото-
вой кислоты. Оротовая кислота в ходе реакции ФРПФ
Рибозо-5-фосфат + АТР
ФРПФ-синтетаза
ш@
AMP
ФРПФ -4ir. Ингибирование
GMP
ФРПФ-аминотрансфераза
АТР
GTP
Рис. 18.8. Упрощенное изображение регуляции пути
биосинтеза пуриновых нуклеотидов
Положительная обратная регуляция АТР и GTP
осуществляется на уровне энергетического обеспечения реакций
превращается в соответствующий нуклеотид, а тот
в свою очередь превращается в UMP. Подобно пуринам,
UTP образуется под действием ферментов киназ, а СТР -
при аминировании UTP.
Как образуются
дезоксирибонуклеотиды?
Для синтеза ДНК необходимы dATP, dGTP, dCTP и dTTP
(см. главу 19). Восстановление рибонуклеотидов
до дезокси-производных происходит на уровне дифос-
фатов при использовании NADPH в качестве
восстановителя (процесс переноса электронов на редуктазу не
приведен).
ADP
GDP
CDP
UDP
Рибонуклетидредуктазг.
NADPH
+ Н+
NADP+
dADP
dGDP
dCDP + H20
dUDP
часть 3 Метаболизм
224
Аспарагиновая кислота
О
Карбамоилфосфат
Оротовая кислота
) х| ООН
I / ФРПФ
l·^ PP/
Оротидилат
UMP
ι
UDP dUDP
UTP
ι
СТР
ι г ииг
I I
dUMP
ι
dTMP
dTTP
dCDP
t
CDP
dCTP
Рис. 18.9. Синтез пиримидиновых нуклеотидов включает
превращение СТР в CDP
Полностью путь синтеза пиримидинов представлен на рис. 18.10
Образовавшиеся dADP, dGDP, dCDP и dUDP
превращаются в трифосфаты в результате их фосфорилирова-
ния соответствующими киназами с использованием АТР.
Однако dUTP не участвует в синтезе ДНК, поскольку
ДНК в качестве одного из своих оснований содержит
тимин (метилированный урацил), но никак не урацил.
dUTP превращается в dTTP. Это происходит в три
этапа. На первом dUTP гидролизуется до dUMP:
dUTP + H20->dUMP + PP,. .
Затем dUMP превращается в dTMP, который после
фосфорилирования преобразуется в dTTP. Для поддер-
О
II
ρ—ο-
Ι
ΝΗ2
СН2 о^ ^0—Ο" р.
+H3N^C^COO ^ ^ '
Аспартат
ООС
NH7
\
сн2
с—соо-
Η
Карбамоиласпартат
к
Н20
О
II
HN^C^CH
Η
Оротат
О
NADH + Н+
NAD+
HhK ^CH2
соо-
~^С\ /С-СОО-
Η
Дигидрооротат
Г4" РР,
ФРПФ
ΗΝ-
Ύ
со2
о
II
.с.
^_Τ"Ύ
Рибо ю-5-фосфат
Оротидилмонофосфат
о^^сн
Рибозо-5-фосфат
UMP
Рис. 18.10. Путь синтеза пиримидиновых нуклеотидов
de novo
Цветом выделены структурные изменения, которые
произошли в предшествующей реакции
жания баланса образования четырех дезоксинуклеотид-
трифосфатов существует система аллостерической
регуляции по принципу обратной связи.
Превращение dUMP в dTMP
Метилирование dUMP представляет особый интерес.
Участвующий в этом процессе фермент тимидилатсин-
тетаза использует в качестве кофермента метилентет-
рагидрофолат (М5,М10-метилен FH4).
глава 18 Метаболизм нукеотидов
225
На приведенной ниже схеме представлена только
наиболее важная (для рассматриваемых проблем) часть
Ы5,Ы10-метилен-РН4.
l,J<"
^ГГ "СН2
I I ю
CH2—N—
2- 0 Ρ—О— дезоксирибоза
dUMP N5, Ы10-метилен-РН
2ς Ο Ρ— О —дезоксирибоза
dTMP
Дигидрофолат CFH2)
Напомним, что в синтезе пуринов метиленовая
группа FH4 окислялась с образованием формильной группы.
При синтезе тимидилата метиленовая группа
переносится и одновременно восстанавливается до метильной
группы тимина; реакцию катализирует тимидилатсин-
тетаза. Восстановителем служит FH4, который в ходе
реакции превращается в FH2.
Образованный в этой реакции FH2 восстанавливается
до FH4 под действием дигидрофолатредуктазы. В свою
очередь, FH4 может быть превращен в метилен-FH4
в реакции с серином:
NADPH
+ Н+\
NADP+
Серии Глицин
FHo
^ ^ » FHA ^ S ■ N5,N10-метилен-FHA.
Для продолжения синтеза тимидилата под действием
дигидрофолатредуктазы FH2 должен быть восстановлен
до FH4. Антилейкемические препараты метотрексат
(аметоптерин) и аминоптерин (их называют
антифолатами), являясь структурными аналогами
фолата, ингибируют РН2-редуктазу.
Приведенную ниже структуру метотрексата (аметоп-
терина) запоминать не нужно; аминоптерин имеет
похожую структуру, но без 1Ч10-метильной группы.
СО—NH-
Метотрексат (аметоптерин)
С00
I
-сн
I
сн2
сн2
С00"
Раковые клетки, например лейкозные, для синтеза
ДНК требуют быстрого образования dTMP, а с помощью
этих лекарственных препаратов оно может быть
избирательно заторможено. Схема представлена на рис. 18.11.
Fh^-pe ктазг
FH2-
4
FH4
Ингибируется
метотрексатом
иаминоптерином
-еринпщрс симетипаза
► Метилен-РКЦ
dUMP
К'
dTMP
+
■ FH2
Рис. 18.11. Действия противоопухолевого препарата
метотрексата (схематично)
Сравните его структуру со структурой фолиевой кислоты,
изображенной на с. 219
Тетрагидрофолат, витамин В12
и пернициозная анемия
Помимо участия в приведенной выше реакции,
1Ч5,М10-метилен-РН4 может восстанавливаться до
1Ч5-метил-РН4, поставляющего метильные группы
для превращения гомоцистеина в метионин (см. с. 199)
в реакции, катализируемой метионинсинтазой.
Последней в качестве кофактора необходим витамин В12 (см.
с. 136). При его отсутствии, например, при заболевании
пернициозной анемией, имеющиеся запасы FH4
превращаются в метил-РН4 и становятся недоступными
для участия в биосинтезе пуриновых нуклеотидов
и других реакциях. Гипотеза «метильной ловушки»
объясняет тот факт, что недостаток витамина В12
вызывает мегалобластическую анемию, идентичную
наблюдаемой при дефиците фолата. Неврологические
симптомы пернициозной анемии могут быть обусловлены
метилмалоновым ацидозом (см. с. 136).
Хотя у бактерий существует несколько реакций,
зависимых от витамина В, у человека их известно
только две. Эти реакции катализируются метилмало-
нил-СоА-мутазой (см. с. 136) и метионинсинтазой.
Недостаток витамина В12 при пернициозной анемии
связан в большей степени с отсутствием внутреннего
фактора - желудочного гликопротеина, необходимого для
всасывания витамина в кишечнике, чем с
недостаточным его поступлением с пищей.
часть 3 Метаболизм
226
Дополнительная литература
Антифолатные лекарства
Huennekens F. Μ. The methotrexate story: a paradigm
development of cancer therapeutic agents. // Adv. Enzyme
Regulation. 1994. Vol. 34. P. 392^119.
(Наряду с историей антифолатных лекарственных
препаратов рассмотрены вопросы, связанные со структурой
дигидрофолатредуктазы и развитием устойчивости к
метатре^ату в клетках.)
Вопросы к главе 18
1. Опишите механизм риботилирования.
2. Какой кофактор участвует в двух реакциях синтеза
пуриновых нуклеотидов? Приведите структурные
формулы его формильного производного.
3. Каково происхождение формильной группы?
Используя структурные формулы кофактора, объясните, как
она образуется.
4. Что является функцией гипоксантин-гуанин-фосфо-
рибозилтрансферазы (ГГФРТ)?
5. Синдром Леша-Нихана включает несколько
генетических заболеваний. Объясните их биохимическую
природу.
6. Как лекарственный препарат аллопуринол снижает
образование мочевой кислоты?
7. Нарисуйте схему, иллюстрирующую основные пути
аллостерической регуляции синтеза пуриновых
нуклеотидов.
8. Некоторые соединения в организме метилируются"
с использованием в качестве источника метальных
групп метионина. Справедливо ли это в случае
синтеза тимидинмонофосфата? Обоснуйте ответ.
9. Как антилейкемическое лекарство метотрексат инги-
бирует репродукцию раковых клеток?
10. Симптомы пернициозной анемии в некоторой степени
напоминают симптомы, связанные с недостатком фо-
лата. В чем заключается возможная причина этого?
глава 18 Метаболизм нукеотидов
227
глава
19
ДНК: структура и локализация в клетках
В этой главе мы познакомимся со структурой ДНК
и понятием гена, а в двух последующих будет
рассказано о синтезе ДНК и о ее роли в образовании белка.
Главная функция ДНК связана с ее участием в синтезе
белков, аминокислотная последовательность которых
определяет все множество процессов, составляющих
основу жизни в течение миллионов лет эволюции.
Что такое
нуклеиновые кислоты?
Дезоксирибонуклеиновая кислота, или ДНК, впервые
была выделена из клеточных ядер. Поэтому она была
названа нуклеиновой (от лат. nucleus - ядро). Ее
кислотные свойства обусловлены присутствием в составе
молекулы остатков фосфорной кислоты. В качестве
углеводного компонента выступает 2-дезокси-/)-рибоза.
В совокупности все это и определило название -
дезоксирибонуклеиновая кислота. В клетках присутствуетм
и другой тип нуклеиновых кислот - рибонуклеиновые,
в которых углевод представлен D-рибозой. Поэтому и
называются они рибонуклеиновыми, или РНК (см.
главу 21). В отличие от D-рибозы 2-дезокси-£>-рибоза не
содержит кислорода при втором углеродном атоме; ее
обычно называют просто дезоксирибозой.
Н0-
Н0-
ОН
ОН
ι
3| |2
ОН ОН
D-Рибоза
ОН Η
2-Дезокси-Л-рибоза
В эукариотических клетках основная масса ДНК
заключена в ядре (незначительные количества
присутствуют в митохондриях и хлоропластах), в то время как
большая часть РНК находится в цитоплазме, хотя
синтезируется она в ядре (кроме РНК митохондрий и хло-
ропластов). Хотя синтез нуклеотидов был уже
рассмотрен в главе 18, для удобства изложения мы вновь
вернемся к этому классу соединений. Но сначала о ДНК.
Первичная структура ДНК
ДНК - полинуклеотид. Строение нуклеотида можно
представить следующим образом:
фосфат-сахар-основание.
А так выглядит структура дезоксирибонуклеотида,
для простоты приведенная в неионизированной форме.
основание
Сахар представлен, как уже отмечалось выше,
2-дезокси-/>-рибозой (дезоксирибозой) и находится
в форме фуранозы, образуя пятичленный цикл. При
нумерации атомов дезоксирибозного цикла добавляется
штрих ('), чтобы отличить ее от нумерации атомов в
основаниях, поэтому атомы углерода в углеводном
остатке обозначаются как Г, 21, З1, 41 и 51 (произносится «пять
штрих» и т. д.) и номера записываются с внешней
стороны цикла. Названия нуклеотидов приведены на с. 216.
Какие основания входят в ДНК?
В отличие от белков, образованных 20 аминокислотами,
в составе ДНК встречается только 4 различных
основания, структура которых ненамного сложнее , чем
структура аминокислот. Основания представлены аденином.
гуанином, цитозином и τ и ми ном, или, сокращенно, А,
G, С и Τ соответственно. А и G - пурины, С и Τ - пири-
мидины. Нумерация атомов в основаниях записывается
внутри цикла.
N
I
НС
Обратите внимание на метильную группу в тимине!
В дальнейшем названия оснований будут
приводиться в сокращенном варианте.
ΝΗ2
^\ N
1 6 SC 7\\
Ι 5П 8СН
2 о 4Г 9 /
^|Г N
Η
Аденин (А)
Η2Ν
0
II
HN1 6 С 7\\
Г у5М 8СН
С2 з АС 9/
-" Ч\' N
Η
Гуанин (G)
часть 4 Хранение и переработка информации
230
NH?
«Лен
Η
Цитозин (С)
О
II
HNf 4^С
1 Iй
o-cVCH
Η
Гимин (Т)
■СН,
Основания могут существовать в таутомерных
формах (кето-енол и амино-имино), и при нейтральных
значениях рН основания в ДНК находятся
преимущественно в форме кето- и амино-таутомеров:
(—С=0 и —ΝΗ7)
I I 2 '
а не в енольной и иминной формах:
II I
(С=ОН и C=NH)
I I
Связь оснований с дезоксирибозой
Первый атом углерода сахара связывается гликозидной
связью с атомом азота в положении 9 или 1 пуринового
либо пиримидинового основания соответственно. Эта
связь называется β-гликозидной, что определяется
структурой углеводного кольца.
Соединение, состоящее из основания и углевода,
называется нуклеозидом; если углевод представлен
дезоксирибозой -дезоксирибонуклеозидом. Все дезокси-
рибонуклеозиды имеют свои названия.
NH2 О
НОСН2 η
ОН Η
Дезоксиаденозин
ΝΗ2
О
НОСН2 η
I
ОН Η
Дезоксицитозин
Η2Ν
НОСН2 η
ОН Η
Дезоксигуанозин
О
О
НОСН2 η
ι
ОН Η
Дезокситимидин
СНЯ
Нуклеозид, содержащий в качестве основания аде-
нин, называется дезоксиаденозин, производное
гуанина - дезоксигуанозин. Дезоксицитидин и
дезокситимидин являются дезоксинуклеозидами цитозина и тимина
соответственно.
Каковы физические свойства
компонентов полинуклеотидов?
В ДНК нуклеотиды присутствуют в виде 5'-фосфатных
производных - dAMP, dGMP, dCMP и dTMP. Ранее
указывалось, что последний нуклеотид можно
обозначать просто как ТМР, так как его рибозный аналог
практически не встречается.
Обе гидроксильные группы фосфорной кислоты при
физиологических значения рН ионизированы, так что
рКа одной ~2, а другой ~7. Это свидетельствует об их
гидрофильности. Благодаря фосфатным остаткам
молекула ДНК несет сильный отрицательный заряд. Сахар
с его гидроксильными группами также проявляет
гидрофильные свойства. Основания, наоборот, почти
нерастворимы в воде и имеют ярко выраженный гидрофобный
характер, хотя ряд атомов при этом сохраняет
способность к образованию водородных связей.
Структура дезоксирибополинуклеотида
Мысленно отнимем от двух нуклеотидов молекулу воды.
В этом случае получившаяся структура будет
представлять собой динуклеотид (приведена неионизированная
форма):
ОН
ι
0=Р-0—СН2 Λ
он
основание
ОН;
га;
L|--J 5·
0=Р-0—СН7
он
основание
ОН
Образование динуклеотида сопровождается
большим положительным изменением свободной энергии
AG0', следовательно, синтез не может идти путем
прямой конденсации двух нуклеотидов.
глава 19 ДНК: структура и локализация в клетках
231
он
I
0=Р-0-СН2
он
XL основание
Η
О
0=Р-0-СН2
I I
он
основание
ОН
динуклеотид
В мононуклеотиде фосфатная группа соединена
с дезоксирибозой простой эфирной связью. В динукле-
отиде образуется фосфодиэфирная связь. Она
называется диэфирной потому, что фосфат соединен с двумя
остатками углеводов:
I
о
I
0=Р-СГ
I
о
I
В приведенном выше динуклеотиде это 3', 5'
-фосфодиэфирная связь, где фосфат связывает З'-ОН-группу
одного нуклеотида с 5'-ОН-группой другого нуклеотида.
Если далее подобным образом присоединять нуклеотиды,
то получим структуру, называемую полинуклеотидом.
Первичная структура ДНК - полинуклеотид огромной
длины. Говоря о белках, мы отмечали наличие
полипептидного остова с выступающими из него
остатками аминокислот. Полинуклеотид также имеет свой
остов, состоящий из чередующихся групп сахар-
фосфат-сахар, а основания присоединяются к
каждому остатку сахара в положении Г. Кодированная
информация записана в последовательности оснований.
Цепь ДНК можно схематически представить
следующим образом.
Фрагмент Информационная или кодирующая
остова часть структуры
2'-дезоксирибоза - основание
фосфат
2'-дезоксирибоза - основание
фосфат
2'-дезоксирибоза - основание
Структуру цепи можно изобразить в следующем виде
I
-о—р=о
I
о
СН2 о
/и\ основание
3', 5'-фосфодиэфирная
связь
О
"О—Р=0
I
О
IS'
сн2 ,
основание
О
I
О—Р=0
I
о
15-
сн2
основание
Почему дезоксирибоза?
Почему не рибоза?
В клетке присутствует ряд нуклеотидов (например AMP),
а также рибонуклеиновые кислоты, содержащие рибозу
в качестве углеводного компонента. Предполагается, что
рибонуклеотиды появились в ходе эволюции раньше
дезоксирибонуклеотидов, а следовательно, РНК
появилась раньше ДНК. В то же время клетки идут на
значительные энергетические затраты, чтобы превратить
рибонуклеотиды в дезоксирибонуклеотиды,
необходимые для синтеза ДНК. Почему?
Одна из весьма убедительных причин хранения
генетической информации, миллионы лет
накапливавшейся в виде молекул ДНК, состоит в следующем:
химические молекулы всегда характеризуются определенной
степенью нестабильности и способностью к
спонтанному распаду. Молекулы же ДНК остаются
неизменными и, в основном, интактными в течение многих
поколений. Кроме того, ДНК более стабильна, чем РНК.
Это связано с тем, что 2'-ОН-группа рибозы легко
подвергается нуклеофильной атаке в присутствии гидро-
ксильных ионов фосфата. В результате происходит
расщепление фосфодиэфирной связи с образованием
2\ 3'-циклических нуклеотидов, а в ДНК, где 2'-ОН-груп-
пы отсутствуют, этого быть не может.
часть 4 Хранение и переработка информации
232
Ион ОН" способствует превращению 2'-ОН-группы
в группу 2'-0 , которую атакует атом фосфора. Это
приводит к разрыву полинуклеотидной цепи и образованию
2',3'-циклического нуклеотида. Последующий гидролиз
циклического нуклеотида дает смесь 21- и З'-нуклеоти-
дов, в зависимости от места гидролиза.
основание
О
I
сн2
ОН"
основание
О
\ /
0=Р
I
°^
но—сн2
Разрыв
полинуклеотидной
цепи
основание
Ч
о он
I I
Различие в стабильности РНК и ДНК
подтверждается еще и тем, что разбавленный раствор NaOH
полностью разрушает РНК при комнатной температуре
и не действует на ДНК. Получается, ДНК более
стабильный и надежный хранитель информации, чем РНК.
Заметим, что факт использования некоторыми
вирусами РНК в качестве носителя информации не
противоречит высказанному утверждению.
Двойная спираль ДНК
Все ДНК почти всегда существует в виде двойной цепи,
за исключением ДНК некоторых вирусов. Другими
словами, ДНК - это две полинуклеотидные молекулы,
составляющие пару. Что же удерживает их вместе?
Комплементарность пар оснований. Это означает, что
когда А и Τ в двух цепях ДНК расположены друг против
друга, между ними спонтанно образуются 2 водородные
связи. Напомним, что водородные связи имеют малый
радиус действия, поэтому для их образования требуется
точное расположение пары атомов. G и С также
представляют комплементарную пару, правда, между ними
возникает 3 водородных связи. Другие комбинации
оснований не могут быть комплементарными, поэтому
G не образует пару с А или с Τ и т. д. В ДНК встречаются
только пары А-Т и G-C, они известны как пары
оснований Уотсона-Крика. Поражает то, что образование
водородных связей - полностью спонтанный процесс, не
требующий ферментативного катализа. Ведь водородные
связи - слабы и легко рвутся, например при повышении
температуры. Незначительное нагревание будет
сопровождаться нарушением спаривания оснований, а
охлаждение - обратной ассоциацией молекул.
Геометрия пар оснований приведена на рис. 19.1.
Н.
\
Г8
N
7\
Ь *%
Аденин l 9
Сахар фосфат \ 32/
N—С
\
Тимин
Ν-Η-Ο^ _ 7СН3
H-N3 6C-H
\2 1 /
/С=\
Η О Сахар фосфат
Η
Гуанин
>й! ',Vh
Сахар фосфат \ 3 2 /
А
Η
\ _ /
//*~А 5С\ Цитозин
Ыз 6С-Н
\2 1 /
с=н
\ // \
Ν—Η- -О Сахар фосфат
/
Η
Рис. 19.1. Водородные связи в парах оснований Уотсона-
Крика
Заметим, что пара оснований всегда содержит один
пурин (большая молекула) и один пиримидин (меньшая
молекула); следовательно, пара оснований всегда имеет
одинаковый размер. Спонтанный процесс образования
пар оснований называют гибридизацией. Если
молекулу ДНК разрезать на тысячи коротких двухцепочеч-
ных фрагментов длиной от 20 до 100 нуклеотидов, а
затем нагреть до температуры около 95° С, спаренные
цепи каждого фрагмента ДНК отделятся друг от друга:
наступит плавление ДНК. Оно происходит из-за
разрыва водородных связей под действием тепла и приводит
к образованию одноцепочечных фрагментов ДНК с
различной последовательностью оснований. Впрочем, если
раствор вновь охладить, то фрагменты начнут
медленно спариваться со своими прежними
комплементарными цепями (рис. 19.2). Такая техника гибридизации,
иногда называемая отжигом, лежит в основе
молекулярной биологии гена (см. главу 24).
Термодинамической движущей силой гибридизации становится
высвобождение энергии, происходящее при образовании
водородных связей и других слабых взаимодействиях.
В наиболее стабильном состоянии величина свободной
энергии минимальна. Это соответствует такому
спариванию оснований, при котором появляется
максимальное количество водородных связей. Экспериментальное
доказательство гибридизации свидетельствует: G-C
и А-Т пары оснований образуются спонтанно, и это
реально существующий феномен.
глава 19 ДНК: структура и локализация в клетках
233
Рис. 19.2. Спонтанная гибридизация комплементарных
фрагментов ДНК
Для демонстрирации принципов гибридизации
последовательность оснований приведена только на одном фрагменте ДНК
Вследствие комплементарности оснований
количество G в молекуле ДНК совпадет с количеством С, а
количество А - с количеством Т. Разные ДНК различаются
по содержанию пар [А+Т] и [G+C] ввиду несходства их
нуклеотидного состава. Это и объясняет их различия как
носителей генетической информации.
В первом приближении цепь ДНК можно
представить в виде длинных сплошных линий,
соответствующих сахарофосфатному остову; удерживаемые
водородными связями основания всегда ориентированы внутрь.
Сахаро-фосфатный
остов
-G=C—
-А=Т—|
-Т=А-
-C=G—I
-А=Т—I
-Т=А-
-C=G-
-C=G-
Заметим, что на участках ДНК, богатых [G+C], две
цепи будут удерживаться вместе сильнее, чем на
участке, где больше пар [А+Т].
Однако слабые взаимодействия оказывают
существенное влияние на конформацию крупных молекул,
и ДНК здесь - не исключение. Поэтому приведенная
выше структура с прямыми цепями в нормальных
условиях не встречается.
Дезоксирибоза —
I
0.6 nm Фосфат
Ι ι
_!_ Дезоксирибоза -
Фосфат
Дезоксирибоза -
Основание
Основание
>
Основание
t
0.33 nm
ί
0.27 nm
I
Гидрофобная поверхость
при взаимодействии
с молекулами Н20
Длина фосфодиэфирной связи составляет 0,6 нм
(1 нанометр = 10~9 м), а толщина оснований - около
0,33 нм, так что в прямой, похожей на лестницу
структуре между ними должны оставаться промежутки.
Плоскости оснований, как уже говорилось, гидрофобны.
В «прямой» структуре они вынуждены находиться в
водном окружении, что придает молекуле нестабильность.
Что же можно сделать для связи между собой
гидрофобных оснований и исключения их контакта с Н20?
Нужно уложить их друг на друга, изогнув при этом
фосфодиэфирную связь; тогда гидрофобные
взаимодействия между плоскостями оснований будут
способствовать их укладке (рис. 19.3, а).
Каждая из лежащих в одной плоскости пар
оснований параллельна соседним близлежащим парам, и все
они образуют так называемую стопку оснований.
Причем основания ориентированы таким образом, что
между противоположными цепями могут образоваться
водородные связи. Впрочем, такая «лестничная» структура
не встречается в ДНК, поскольку невозможна стереохи-
мически. На самом деле две цепи ДНК образуют
спираль, в которой основания расположены внутри, а
гидрофильные сахарные и фосфатные группы снаружи
(рис. 19.3, б, в).
Благодаря такой структуре обеспечивается тесное
взаимодействие пар оснований и исключается их
контакт с водой, но стопка оснований уже не абсолютно
вертикальна. Пары оснований слегка смещены
относительно друг друга; как показано на рис. 19.3, г, на один виток
приходится около 10 пар.
Спираль правозакрученная; если смотреть вдоль ее
оси, то повороты следуют по часовой стрелке
(представьте, что вы закручиваете винт, держа отвертку в правой
руке: направление вращения дает представление о
направлении поворота спирали). Структура двойной спирали
часть 4 Хранение и переработка информации
234
Сахаро-фосфатный S В В S
остов ДНК Р' >'
У' в в D-s'
S BBS
Соединенные вместе пары оснований,
минимально взаимодействующие
своими гидрофобными поверхностями с водой
б большая малая
бороздка бороздка
в
* ' <
г
-<32°
Рис. 19.3. Модель укладки нуклеотидов в ДНК
а - Тип связи пар оснований в структуре «наклонной
лестницы»;
б - контур остова в двойной спирали ДНК;
в - то же, что и б, изображены пары оснований в центре
спирали (каждый цилиндр - пара оснований);
г - расположение двух соседних пар оснований в двойной
спирали. Показано смещение, определяемое структурой
двойной спирали. S - сахар, Ρ - фосфат, В - основание. На рис. 19.4
приведена более подробная модель, соответствующая е
такова, что она имеет две бороздки - большую и малую
(см. рис. 19.3, б). Хотя бороздки показаны на плоскости,
их трудно изобразить вне трехмерного пространства.
Если у вас есть возможность ознакомиться с объемной
моделью ДНК, это поможет понять направление
спирали и обнаружить на ней две бороздки, что чрезвычайно
важно. Часть каждого основания видна как из большой,
так и из малой бороздок: это делает их доступными для
взаимодействия с другими молекулами. Большая
бороздка обеспечивает более легкое взаимодействие с
белками, которые «узнают» соответствующие основания.
Пары оснований совершенно не похожи, если их
рассматривать со стороны двух разных бороздок (рис. 19.4);
значение этого факта будет разъяснено в главе 21, где
обсуждается генетическая регуляция.
Описанная выше конформация ДНК известна как
В-форма (рис. 19.5): именно в такой форме ДНК
обычно находится в клетке. Однако ДНК может изменять
свою конфигурацию в зависимости от условий. При
дегидратации двойная спираль приобретает более
сплющенную форму с большим углом наклона оснований, и
в этом случае ее называют Α-формой. В такой форме
ДНК встречается в спорах. Известна и другая
конфигурация - Z-форма, когда сахарофосфатный остов
образует зигзагообразную (англ. zigzag) линию вдоль
спирали. В Z-форме двойная спираль закручена влево, а
в В-форме - вправо. Есть сведения, что Z-форма
встречается в условиях высокой ионной силы раствора в
коротких молекулах синтетической ДНК с
чередующимися попеременно пуриновыми и пиримидиновыми
основаниями. Биологическое значение А- и Z-формы пока
неизвестно, однако не исключена возможность
приспособительного изменения конфигурации ДНК на
коротких участках хромосом.
Важным свойством двойной спирали является ее
способность изгибаться. Молекула ДНК может быть
в миллион раз длиннее, чем клетки или ядра самых
больших размеров; чтобы поместиться в них, молекула
должна быть гибкой.
Следует отметить, что хотя ДНК почти всегда
находится в форме двойной спирали, существуют и одно-
цепочечные ДНК, например в некоторых вирусах
бактерий. Тогда для обеспечения термодинамической
стабильности молекулы образуется сложная скрученная
структура. Главное, что и в этом случае жизненный цикл
вируса включает стадию, когда ДНК находится в форме
двойной спирали (см. главу 23); следовательно,
основные принципы передачи генетической информации,
предусматривающие образование комплементарных пар
оснований, остаются в силе.
Цепи ДНК антипараллельны: что это
значит?
Под антипараллельной мы понимаем структуру, в
которой две цепи в двойной спирали ДНК имеют
противоположную направленность. Две антипараллельные цепи
ДНК приведены на рис. 19.6.
Что же означает полярность, или направление цепи
ДНК? Каждая линейная цепь имеет два конца, на одном
из них расположена 5'-ОН-группа дезоксирибозы (не свя-
заная с другим нуклеотидом). Иногда она бывает фос-
форилирована. Это - 5'-конец. На другом расположена
свободная З'-ОН-группа (она также не связана с другим
нуклеотидом и может быть этерифицирована
фосфатной группой). Это - З'-конец. Поэтому на каждом конце
глава 19 ДНК: структура и локализация в клетках
235
Малая бороздка
Гуанин
Большая бороздка
н-
Малая (
Тимин
N
-С
СНз
г
\
N-
гъ
-н
С
• н-
эорозд*
Η
V
С
-г/
1
1
Η
са
Аден к
N
С 2
N
ΙΗ
N
Ч
Большая бороздка
Рис. 19.4. Схематичное изображение, показывающее, что со стороны большой и малой бороздок одна и та же пара
оснований в ДНК выглядит по-разному
ДНК-связывающие белки, предназначенные для узнавания специфических последовательностей в ДНК, могут присоединяться
к соответствующим участкам без раскручивания спирали
двойной спирали всегда есть один 5'-конец и один З'-ко-
нец. Вы никогда не увидите два 5'-конца или два З'-конца
вместе! Это и означает антипараллельность. Если
фрагмент ДНК имеет кольцевую форму (свободные концы
отсутствуют), полярность отдельных нитей все равно
сохраняется. Это можно проиллюстрировать на примере
расположения в цепях углеводных компонентов.
5'—-3'
I Направления
- 3' - 5' фосфоди )фирная
связь
Вы можете узнать направление, ориентируясь на
положения 5'и 3'. Так, на приведенной слева структуре цепь
идет в направлении 5'^3' сверху вниз, а справа - 5'—>3*
соответствует направлению снизу вверх (условимся
обозначать направление последовательности нуклеотидов
в ДНК от 5'->3').
Обратите внимание на кажущееся противоречие!
Когда вы движетесь по цепи ДНК в направлении 5'—>3'
(от 5'-конца к З'-концу), каждая фосфодиэфирная
связь, которую вы «проходите», образована между
Рис. 19.5. Модель В-формы ДНК
Представлена трехмерная модель фрагмента ДНК с двумя
большими и одной малой бороздками
З'-ОН-группой дезоксирибозы предыдущего и 5'-ОН-груп-
пой дезоксирибозы последующего нуклеотида. Это
означает, что - исходя из положения фосфата - вы
следуете от З'-конца к 5'-концу цепи. Следует помнить, что
направление 51—>3' для нити линейной молекулы ДНК
означает, что вы двигаетесь от концевой 5'-ОН-группы
часть 4 Хранение и переработка информации
236
/ν
5--31
направление
5-конец
Рис. 19.6. Две антипараллельные цепи ДНК
В - основания
к концевой З'-ОН-группе, или от 5'-группы одного
остатка дезоксирибозы к 3'-группе соседнего. Это
необходимо запомнить как в силу важности самого вопроса,
так и для понимания материала последующих глав.
Антипараллельное расположение цепей
предъявляет определенные стереохимические требования к
структуре двойной спирали.
Существуют правила написания
последовательности расположения нуклеотидов в ДНК. Простую поли-
нуклеотидную структуру принято изображать в виде
последовательности букв, обозначающих
соответствующие основания нуклеотидов. А иногда в виде буквы
«р», располагаемой между основаниями, указывается
фосфодиэфирная связь (например, CpApTpGp, и т. д.).
Предположим, у нас имеется фрагмент двухцепочеч-
ной ДНК со следующей последовательностью оснований:
5' CATGTA 3'
3' GTACAT 5'
Иногда записывают последовательность для каждой
цепи, но в большинстве случаев нет необходимости
изображать обе цепи, так как вторая комплементарна
первой. Поэтому, несмотря на наличие в гене двух
цепей, его структуру часто представляют в виде простой
последовательности оснований. При условии, что она
записывается с 5'-конца слева, специального
обозначения 5'- и З'-концов не требуется. Таким образом,
приведенная выше структура может быть обозначена как
CATGTA.
Каковы размеры молекулы ДНК?
Размеры молекул ДНК, если ориентироваться по числу
входящих в их состав оснований, сильно различаются.
Но даже самые короткие из них представляют собой
очень крупные молекулы, а молекулы ДНК с наиболее
длинными цепями просто гигантские. Небольшие ДНК
вирусов состоят из нескольких тысяч пар оснований;
ДНК Е. coli содержит около 4 млн пар оснований.
Геном человека (полная сумма ДНК хромосом) достигает
длины 1-2 м и включает 6 млрд пар оснований. Если
учесть, что человек состоит из 1013 клеток, и подсчитать
суммарную длину его ДНК, получится астрономическая
величина, сравнимая с диаметром солнечной системы.
Ясно, что чем больше информации необходимо
сохранить, тем большее количество ДНК требуется.
Следовательно, чем сложнее организм, тем большее
количество ДНК содержат его клетки. Впрочем, это
соблюдается не всегда. Например, крупные клетки бобов
содержат больше ДНК, чем клетки человека, а у
земноводных ее больше, чем у других позвоночных.
Появление аномалий (ошибок) в структуре ДНК приводит к
потере некоторых функциональных свойств.
Каким образом ДНК упакована
в ядре?
В биохимии часто приходится сталкиваться с
поразительными решениями эволюцией тех или иных проблем!
Упаковка ДНК - одна из них.
В клетках человека содержится 46 хромосом, а
общая длина заключенной в клетке ДНК составляет 1-2 м,
причем она упакована в ядре, диаметр которого в
миллионы раз меньше. Значит, требуется тщательно
продуманная система укладки. В эукариотических клетках
ДНК находится в составе хроматина - комплекса ДНК
с белками. Белки представлены преимущественно
гистонами - основными белками, богатыми аргинином
и лизином: благодаря своему положительному заряду
они образуют ионные связи с расположенными на
внешней стороне двойной спирали ДНК отрицательно
заряженными фосфатными группами. Аминокислотная
глава 19 ДНК: структура и локализация в клетках
237
последовательность эукариотических гистонов высоко
консервативна. Например, первичная структура
гистонов, выделенных из растения (горох) и
млекопитающего (корова), отличается только на две аминокислоты, да
и эти замены не очень существенны (валин на изолей-
цин, лизин на аргинин). Столь выраженное постоянство
структуры позволяет предположить, что такой белок
должен точно взаимодействовать с настолько же
консервативной структурой или структурами.
Начнем с рассмотрения четырех гистонов - Н2А,
Н2В, НЗ и Н4. Они образуют октамерный белковый
комплекс, называемый нуклеосомной сердцевиной (англ.
nucleosome core), вокруг которого молекула ДНК
совершает два оборота (около 146 пар оснований). Такой
комплекс октамера гистоновых белков с ДНК является
основной структурной единицей хроматина и называется
нуклеосомой. Отдельные нуклеосомы соединяются
линейными участками ДНК - линкерами (рис. 19.7, а, б).
Если хроматин в пробирке обработать ферментом,
выделенным из микроорганизмов и способным ^идроли-
зовать ДНК на нуклеотиды, линейные участки ДНК
действительно подвергаются гидролизу, а около 146 пар
оснований, «намотанных» на нуклеосомную
сердцевину, оказываются защищенными от действия фермента
(он не в состоянии гидролизоватьТэту часть ДНК).
Экстрагированная после такой обработки ДНК содержит
неповрежденные фрагменты. Это наблюдение легло в
основу представлений о нуклеосомной структуре.
Нуклеосомы имеют примерно ΪΟημ в диаметре
и образуют нити толщиной 10 нм (см. рис. 19.7, б).
И все же такой упаковки ДНК недостаточно, в ее,
дальнейшей организации участвует пятый гистон: он не
входит в состав нуклеосомы и роль его не совсем ясна.
Далее нить 10 нм толщиной конденсируется с
образованием нити толщиной 30 нм (рис. 19.7, в). Такая
упаковка нуклеосом зачастую описывается как
соленоидная; впрочем, это лишь одна из моделей. Согласно
другой модели, упаковка имеет зигзагообразную
форму. Поскольку точная структура соленоида еще не
установлена, схема 30-нанометровой нити приводится
без деталей упаковки. Электронная микрофотография
такой нити представлена на рис. 19.8. При этом ДНК
конденсируется в 100 раз, т. е. происходит стократное
уменьшение ее длины. Нити толщиной 30 нм
образуют длинные петли, прикрепленные к центральному
поддерживающему белку хромосом (рис 19.7, г). За
счет формирования петель структура становится еще
более компактной, но пока не выяснено, включает ли
такое скручивание дополнительную спирализацию,
обеспечивающую 10 000-кратную конденсацию
исходной молекулы ДНК.
Линкерная ДНК, соединяющая
соседние нуклеосомы
Гистон HI
Линкерная ДНК
. Нуклеосома
Октамер, содержащий
по две копии гистонов
Н2А, Н2В, НЗ, Н4
Нуклеосомная сердцевина,
около 140 пар оснований.
ДНК обвивают гистоновый
октамер по принципу
левозакрученной сверхспирали
Линкерная ДНК -около
30-40 пар оснований
200 пар
, оснований ,
'50 нм
30 нм
30-нм нить нуклеосом, *
- расположение которых
окончательно
не установлено
Петли из 30-нм нити,
. лрикрепленные к центральному
поддерживающему белку
Центральный
поддерживающий белок
Рис. 19.7 Модель упаковки хроматина у эукариот
а- Нуклеосома;
б - нуклеосомы с линкерными участками ДНК;
в — нить хроматина 30 нм толщиной;
г - петли этой нити, прикрепленные к центральному
поддерживающему белку. Петли далее конденсируются путем сверх-
спирализации, образуя чрезвычайно компактную метафазную
хромосому. Последняя стадия конденсации на рисунке не.пред-
.ставлена
Таким образом, есть несколько уровней организации
ДНК: 1) закручивание вокруг нуклеосом; 2) упаковка
нуклеосом в 30-нанометровую нить, объединяющую
3-5 нуклеосом; 3) образование петли длиной в тысячи
нуклеосом, прикрепленной к центральной
поддерживающей структуре; 4) формирование из петли спирали
и/или складки (см. рис. 19.7, а, г).
Для выполнения своей функции упакованная ДНК
должна быть доступна ферментам, участвующим в
считывании генетической информации и репликации.
В клетках Е. coli двойная спираль ДНК имеет
кольцевую форму. Она длиннее клетки примерно в 1000 раз.
У прокариот нет ядерной мембраны и нет
упорядоченных структур наподобие эукариотических
нуклеосом. Тем не менее, в Е. coli найдены гистоноподобные
белки, которые, предположительно, выполняют сходную
часть 4 Хранение и переработка информации
238
Две одинаковых хромосомы
V
■ Центромера
Микротрубочки,
образующие
часть веретена
Кинетохор
Рис. 19.9. Хромосома в метафазе состоит из двух дочерних
хроматид
Рис. 19.8. Электронная микрофотография нити хроматина
30 нм толщины
функцию. Поскольку в бактериальных клетках белки
связаны с ДНК менее прочно, чем эукариотические гис-
тоны, нуклеосомоподобные комплексы в них не
обнаруживаются. Петельная структура ДНК в Е. coli
стабилизируется белками, но детали этого процесса до конца
не ясны.
Как структура ДНК соотносится
с компактными хромосомами эукариот,
видимыми в световой микроскоп?
Хромосомы в окрашенных делящихся эукариотических
клетках видны в световом микроскопе как компактные
плотные образования, подобные тем, что изображенны
на рис. 19.9.
Компактные структуры в делящихся клетках
представляют так называемые метафазные хромосомы.
Там ДНК находится в наиболее конденсированном
состоянии, что помогает перемещению хромосом после
их деления в каждую из дочерних клеток.
Наиболее плотно упакованная область ДНК,
состоящая из повторяющихся последовательностей,
приходится на центромеру. Она удерживает вместе пару
хроматид, а на их внешних поверхностях располагаются
комплексы белков кинетохора. Для разделения
дочерних хромосом к кинетохору прикрепляются
микротрубочки митотического веретена (см. главу 29). После
деления клетки хромосомы становятся менее
упакованными; их называют интерфазными хромосомами.
ДНК метафазных хромосом функционально инертна,
а интерфазных функционально активна (по сравнению
с простой нитью ДНК интерфазные хромосомы более
упакованы). Степень конденсации ДНК в
эукариотических клетках очень важна. Прежде чем использовать
содержащуюся в молекулах информацию, ДНК должна
стать доступна ферментам и другим белкам и не иметь
плотно упакованной структуры. Определенные участки
эукариотических хромосом содержат ДНК, которая,
предположительно, не входит в структуру гена, т. е.
не несет информации, необходимой для синтеза белка.
глава 19 ДНК: структура и локализация в клетках
239
ДНК в этих местах даже в интерфазных хромосомах
остается конденсированной в виде так называемого
гетерохроматина, в то время как функционирующие
области - эухроматин - менее конденсированы.
Функция гетерохроматина до конца не ясна.
В хроматине помимо гистонов присутствуют и
другие, негистоновые белки. Они участвуют в образовании
длинных петель ДНК, прикрепленных к осевым
поддерживающим белковым структурам. Таким образом,
хроматин является сложной структурой. В зависимости
от экспрессии гена меняются и степень его упаковки
и степень упаковки самой ДНК в хроматине. Термин
экспрессия гена относится к процессу синтеза белка,
контролируемого геном, и также используется в
случаях кодирования генами специфических молекул РНК
(см. с. 283).
Что такое ген
в молекулярных терминах?
Ген - единица наследственности. Он представляет
собой часть гигантской молекулы ДНК и содержит
закодированную информацию о последовательности
аминокислот одной полипептидной цепи (см. с. 264, где дается
детальное описание гена). Информация хранится в виде
последовательности оснований ДНК, и один ген
отличается от другого только закодированной информацией.
Для большей наглядности можно провести аналогию
между хромосомой и магнитной лентой: тогда ген
сравним с фрагментом музыкальной записи, а
последовательность оснований - с магнитными сигналами. Выше
сказанное относится к «стандартным» (типичным)
кодирующим белок генам. Их подавляющее большинство.
Некоторые вариации
«стандартного» гена
Иногда существуют множественные копии одного и того
же гена. Например, гены некоторых гистоновых белков:
сотни тандемно повторяющихся копий обнаружены у
морских ежей. Это может быть результатом дупликации гена
в процессе эволюции. Множественные копии гена
способны обеспечивать ускоренный синтез белка. В
эволюционном плане это позволяет приспособить одну из
копий к выполнению какой-нибудь иной (альтернативной)
функции. Существуют также псевдогены - копии
функциональных генов, способные к экспрессии,
синтезирующие нефункциональный белок или белковый фрагмент.
часть 4 Хранение и переработка информации
240
Есть группа генов, которые не кодируют белков: это
гены рибосомной и транспортной РНК. Они кодируют
молекулы РНК, необходимые для синтеза белков
(см. главу 22).
Обычный «стандартный» ген представляет собой
фрагмент ДНК, расположенный в определенном месте
хромосомы. Последовательности в ДНК не всегда
жестко фиксированы; иногда в них имеются фрагменты,
способные перемещаться из одного места ДНК в другое.
Они называются подвижными, или прыгающими
генами (см. с. 259). Хромосомы также могут включать
новые гены из ретровирусов (см. с. 313).
Структура гена не всегда постоянна. При
определенных условиях отрезок ДНК, соответствующий
какому-либо гену, может амплифицироваться, так что
получится огромное количество копий гена и соответственно
резко увеличится (примерно в 1000 раз) синтез
закодированного в нем белка. Такое явление наблюдается
в экспериментах на культуре клеток рака мышей,
обработанных метотрексатом - лекарственным препаратом
«антифолатного» действия, используемым для лечения
лейкемий (см. с. 226).
Повторяющаяся ДНК
В клетках человека не вся ДНК находится в виде генов.
Например, в наших хромосомах существуют довольно
протяженные (длиной в несколько сотен оснований)
участки ДНК, повторяющиеся сотни и тысячи раз,
разбросанные по всей длине хромосомы. Примером
могут служить Alu-последовательности (Alu -
аббревиатура полного названия фермента, осуществляющего
их гидролиз). Функция Alu-последовательностей
не установлена.
Еще один вид повторяющихся последовательностей
ДНК - сателлитная ДНК: она часто встречается в
области центромеры хромосом и является частью
гетерохроматина. Термин сателлитная возник потому, что
гигантская молекула эукариотической ДНК при выделении
обычным способом расщеплялась на фрагменты. При
их анализе в градиенте плотности большая часть ДНК
оседала одной полосой, в то время как повторяющиеся
участки ДНК, имеющие другую плотность, отделялись
от предыдущей полосы (т. е. выявлялись как сателлиты).
Сателлитная ДНК состоит из коротких
последовательностей, повторяющихся в тандемном порядке (друг за
другом). Длина каждого фрагмента примерно 105
оснований. Другие повторяющиеся последовательности ДНК
рассеяны по хромосоме.
Где мы сейчас находимся?
Дабы не потерять основной нити рассуждения среди
обилия деталей, напомним, что мы сначала имели дело со
структурой белка, после чего перешли к вопросам,
связанным с процессом его биосинтеза. Теперь мы
обратимся к биосинтезу ДНК. Во-первых, это логичное
продолжение изучения структуры ДНК; во-вторых, поможет
пониманию последующих этапов синтеза белка.
Поэтому в следующей главе мы обсудим синтез ДНК, а
после перейдем к изучению путей передачи генетической
информации, непосредственно связанной с биосинтезом
белка.
Дополнительная литература
Ptashne Μ. A genetic switch: gene control and phage λ //
Cell Press and Blackwell Scientific Publications, 1987.
(Классическая книга о структуре и функции ДНК, а
также молекулярной биологии фага.)
Callandine С. R., Drew Я. R. Understanding DNA:
the molecule and how it works. Acad, press, 1992.
(Очень четкое обсуждение структуры ДНК,
суперскручивания и организации ДНК в клетке. Оригинальный
стиль изложения материала.)
RennieJ. DNAs new twists //Sci. Amer. 1993. Vol. 266 (3).
P. 88-96.
(Интересная оценка прыгающих генов, хрупких
хромосом, протяженных генов, редактированных мРНК,
нестандартного генетического кода в митохондриях
и хлоропластах.)
Cambers D. Α., ReidK. В. Μ, Cohen R. L DNA: the double
helix and biomedical revolution at 40 years // FASEB J. 1994.
Vol. 8. P. 1219-1226.
(Материалы конференции, посвященной 40-летию
открытия двойной спирали ДНК. Биохимические
воспоминания, обобщение достижений в этой области и взгляд
в будущее. Настоятельно рекомендуется всем студентам
и научным сотрудникам.)
Вопросы к главе 19
1. Напишите структуру динуклеотида.
2. Рибонуклеиновая кислота (РНК) появилась раньше
дезоксирибонуклеиновой кислоты. Почему
появилась ДНК?
3. Основания в ДНК гидрофобны. Объясните, как это
влияет на структуру двуцепочечной ДНК.
4. Назовите основные характеристики двойной
спирали ДНК. Это лево- или правозакрученная спираль?
Сколько пар оснований формируют один виток
спирали ДНК?
5. Объясните, что означает антипараллельность цепей
ДНК в двойной спирали.
6. Объясните, что означает направление 51—»3' в
линейной молекуле ДНК.
7. Если вы видите структуру ДНК, записанную как
CATAGCCG, что она означает, исходя из структуры
двойной спирали и полярности ее цепей? Объясните
свой ответ.
8. Что такое нуклеосома? Что подтверждает ее
существование?
глава 19 ДНК: структура и локализация в клетках
241
глава
20
Синтез и репарация ДНК
Синтез ДНК - сложный процесс, механизмы которого
лучше изучены на бактериальных клетках Е. coli, чем
на эукариотических. Впрочем, в обоих случах
процессы в основном сходны.
Каждый раз во время деления клетки содержание
ДНК должно удвоиться или, как обычно говорят, хромо-
сома(ы) должна(ы) реплицироваться.Тогда каждая
дочерняя клетка получает полный набор ДНК. Нет
необходимости подчеркивать важность точной репликации,
поскольку наличие в гене одного неправильного
основания может привести к синтезу белка с нарушенной
функцией.
N15 N15
Ν14 Ν15 Ν15 Ν14
Общие принципы
репликации ДНК
Основу хромосомы составляет одна непрерывная двух-
цепочечная молекула ДНК. Ее репликация происходит
полуконсервативным способом. Две исходные цепи,
называемые родительскими, отделяются друг от
друга: каждая служит матрицей для синтеза новой цепи,
каждая новая двойная спираль содержит одну старую
и одну новую цепь. Это было доказано в классическом
эксперименте (рис. 20.1).
В основе образования новой цепи лежит принцип
комплементарности оснований (G с С и А с Т), так что
последовательность оснований в родительской цепи
однозначно определяет последовательность оснований
в новой - дочерней - цепи.
Поскольку в ДНК пары оснований Уотсона-Крика
образуют водородные связи, синтез новых молекул
может произойти только при расхождении цепей, что
делает основания доступными для новых взаимодействий
с комплементарными основаниями. Хромосома E.coli
имеет кольцевую форму и содержит около четырех
миллионов пар оснований. В определенной точке,
называемой точкой начала репликации, цепи расходятся
и образуются две репликативные вилки, которые
движутся в противоположных направлениях. ДНК
синтезируется со скоростью примерно 500 пар оснований
в секунду, причем разделение родительских ДНК и
синтез новых цепей происходит одновременно (рис. 20.2).
Две вилки встречаются на противоположной стороне
кольца. В результате репликации два новых кольца
Клетки,
выращенные
на среде,
содержащей Ν15,
Клетки
перенесены
в среду,
содержащую Ν14
►
Репликация ДНК
Тяжелая Тяжелая
ДНК, которая должна
реплицироваться,
содержит обе нити с Ν11
Тяжелая Легкая Легкая Тяжелая
Дочерние цепи
Дочерние Ν15/Ν14- молекулы ДНК
Рис. 20.1. Доказательство полу консервативного способа
репликации ДНК по Мезельсону и Сталю
ДНК клеток метили путем выращивания на среде,
содержащей в качестве источника азота тяжелый изотоп Ν15; он
включался в состав ДНК, и обе родительских нити ДНК
получались «тяжелыми». Затем клетки переносили на среду,
содержащую обычный природный изотоп Ν14. Анализ в градиенте
плотности показал, что в ходе первого деления после
пересева каждая молекула ДНК содержала одну «тяжелую» и одну
«легкую» цепь. Появление ДНК с «легкими» и «тяжелыми»
цепями указывало на полу консервативный способ репликации.
Продолжение эксперимента в последующих делениях
подтвердило этот результат. Цветом выделены вновь
синтезированные цепи
Точка начала репликации
Двунаправленные
репликативные вилки
Направление /
репликации /.
Хромосома Е. coli
Частично
реплицированная
хромосома
Рис. 20.2. Двунаправленная репликация хромосомы Е. coli
Вновь синтезированные нити выделены цветом
оказываются сцепленными друг с другом. Этот факт
представляли доводом против описанной выше модели
репликации ДНК. Остается лишь удивляться проница-
часть 4 Хранение и переработка информации
242
тельности Крика, утверждавшего, что эволюция
позаботилась о механизме, разделяющем кольца.
Впоследствии выяснилось, что эту функцию выполняет фермент
топоизомераза II (см. с. 245).
У эукариот синтез ДНК в репликативной вилке идет
со скоростью примерно 50 пар оснований в секунду -
слишком медленно для того, чтобы в хромосоме за
время, отведенное на клеточное деление, синтезировалась
ДНК огромных размеров. На самом деле репликация
эукариотической ДНК начинается одновременно во
многих точках (их, вероятно, более тысячи). От каждой
такой точки в обоих направлениях движутся две репли-
кативные вилки (рис. 20.3); в остальном процесс
протекает аналогично репликации у Е. coli.
Точка начала репликации имеет специфическую
последовательность оснований, богатую парами А-Т, что,
вероятно, облегчает разделение нитей (напомним, пары
А-Т имеют две водородные связи, а пары G-C три,
поэтому первые слабее связаны друг с другом). Во
время инициации к этой области присоединяется
множество молекул белка, названного DnaA, после чего
происходит отделение нитей. Это, в свою очередь, помогает
основному расплетающему цепи ферменту хеликазе,
работающему в каждой репликативной вилке, начинать
и продолжать последовательное расплетание цепей
в обоих направлениях. О хеликазе (называемой также
DnaB, т. е. белком, кодируемым геном dnaB) будет
рассказано при описании механизма синтеза ДНК.
Регуляция инициации
репликации ДНК в Е. coli
Перед делением клетки Е. coli должно произойти
полное удвоение хромосомы; само клеточное деление
протекает позже, минут через 20. Точно не известно, каким
образом скоординировано клеточное деление и
репликация ДНК. Для осуществления этих процессов
необходимы синтез белка и значительное увеличение размера
клетки. Ранее было показано (см. рис. 20.2), что у E.coli
существует единственная точка начала репликации ДНК
(oriC), откуда в двух направлениях идет репликация.
Целую хромосому E.coli называют репликоном; у
эукариот реп л икон - отрезок ДНК, репликация которого
протекает под контролем единственной точки начала
репликации.
Линейная эукариотическая хромосома
Направление репликации
Репликация начинается
во множестве точек
начала репликации
Репликативные вилки
\s—V.J—κμ~
Реиликативный глазок
Завершение репликации
Две дочерние хромосомы
Инициация репликации ДНК
у эукариот
Эукариотические клетки имеют более сложный
клеточный цикл, чем клетки Е. coli, и в них синтез ДНК
приурочен к определенному периоду, который
называется S-фазой (от англ. synthesis phase - фаза синтеза)
(рис. 20.4). Продолжительность клеточных циклов
различных клеток сильно различается; в культуре клеток
животных из общего 24-часового клеточного цикла
S-фаза занимает около 8 часов. S-фазе предшествует
Gj-фаза (от англ. gap in DNA synthesis - промежуток
в синтезе ДНК). Чтобы приступить к делению, клетки
млекопитающих должны получить извне
соответствующий сигнал. Таким сигналом служат белковые
молекулы - факторы роста.
С2-фаза. Клетка готовится
к митозу. Синтеза
ДНК нет (5-6 часов4
Митоз и клеточное
деление (1-2 часа)
Gj-фаза (6-9 часов).
Синтеза ДНК нет,
но сигнал к репликации
ДНК клеткой уже
получен. Если клетка не
получила внешнего
сигнала к делению, она
переходит в фазу
покоя G0
S-фаза.
Происходит
репликация ДНК (8-9 часов)
Рис. 20.3. Схематичное изображение, демонстрирующее
образование множества двунаправленных репликативных
вилок в эукариотической хромосоме
Рис. 20.4. Цикл эукариотической клетки
Продолжительность клеточного цикла разная у различных
типов клеток. Здесь приведены фазы цикла для быстроделя-
щейся культуры клеток млекопитающих (полный цикл 24 часа)
глава 20 Синтез и репарация ДНК
243
На поверхности клеток они связываются с рецепторами,
которые посылают сигнал к делению внутрь клетки
(см. главу 26). Как уже отмечалось, в хромосоме
животных имеется множество точек начала репликации,
от которых репликация идет в двух направлениях. Пока
неизвестно, как регулируется инициация репликации.
Существенно, что каждый репликон в ходе одного
клеточного деления активируется только один раз.
Расплетание двойной спирали
ДНК и сверхспирализация
Разделение цепей ДНК с помощью хеликазы
представляет собой топологическую (физическую) проблему.
Для объяснения этого явления мы должны
познакомиться с понятием сверхспирализации ДНК.
Две цепи ДНК (дуплекс ДНК) образуют правозак-
рученную спираль, на один виток которой приходится
10 пар оснований. Изолированный короткий фрагмент
линейной ДНК, который может легко вращаться вокруг
своей длинной оси, спонтанно принимает свободную
конфигурацию, известную как релаксированная форма.
Представим, что один конец дуплекса не может
вращаться, а на противоположном конце образуются
дополнительные витки. В этом случае спираль начинает
сжиматься, а количество оборотов на этом отрезке ДНК
возрастает, в то время как количество пар оснований,
приходящихся на один оборот, уменьшается. Это
явление называется положительной сверхспирализацией,
или сверхскрученностью. Если же поворачивать
дуплекс в противоположном направлении, спираль
раскручивается: количество оборотов на единицу длины
начинает уменьшаться, а количество оснований в одном
витке будет возрастать. ДНК перейдет в слабоскрученное
состояние, или состояние отрицательной
сверхспирализации. Обе формы - и сверхскрученная, и слабо-
скрученная - представляют напряженное состояние
молекулы, для выхода из которого необходимо
закручивание двойной спирали ДНК самой на себя с
образованием спирализованной спирали, или сверхспирали
(рис. 20.5). Сверхспирализацию легко представить, взяв
кусочек дважды закрученного каната. Пусть кто-нибудь
держит один конец каната или закрепит его так, чтобы
он не мог свободно вращаться, и крутит канат вдоль оси.
Образуется спираль, закрученная в соответствии с
напряжением вращения. Если отпустить конец сверхспи-
рализованного каната, он немедленно примет исходную
(релаксированную) форму.
Что происходит с ДНК во время репликации?
. Зажим
ι-
Релаксированный дуплекс ДНК
Конец освобожден
и разрешено
свободное вращение
\
вращение при
зажиме
Сверхспирализованная ДНК
и
. Зажим
\
Рис. 20.5. Схематичное изображение, поясняющее, как
закручивание фрагмента ДНК, не имеющего свободы
вращения, приводит к сверхспирализации, устраняющей
напряжение вращения
Клеточная ДНК прочно фиксирована и не обладает свободой
вращения. Впрочем, если свободное вращение станет
возможным, сверхспираль перейдет в релаксированное
состояние. Если вращение происходит в направлении
раскручивания, сверхспираль будет отрицательной; если вращение
производить в противоположном направлении, сверхспираль
будет положительной (различия между этими двумя формами
не приведены)
Ведь в клетке ее нить не может свободно вращаться
относительно своей оси. У Е. coli замкнутое кольцо
хромосомы прочно «фиксирует» ДНК. У эукариот
огромная молекула ДНК может образовывать петли (см.
рис. 19.7, г), закрепленные белковыми структурами; при
этом возникает подобие кольцевых структур, и в
результате участки становятся неспособными к свободному
вращению. Но при разделении нитей ДНК обязательно
происходит вращение дуплекса. Это вызывает
сверхспирализацию, которая приводит к появлению
положительных сверхвитков в начале репликационной вилки;
и так как спираль сжимается, дальнейшее расплетание
становится затруднительным. Если напряжение не
исчезнет, то оно будет препятствовать расплетанию нитей,
и репликация ДНК остановится.
В этом вас убедит очень простой эксперимент.
Возьмем короткий кусок двунитевого каната и
потянем его концы в разные стороны: канат будет быстро
раскручиваться без накопления положительных
сверхвитков. При этом нити каната легко и полностью
отделятся друг от друга. Теперь свернем длинный кусок
такого же каната в кольцо или закрепим один его
конец таким образом, чтобы канат не мог свободно
вращаться, и попытаемся развести его нити.
Положительные супервитки быстро «заклинят» образовавшуюся
часть 4 Хранение и переработка информации
244
вилку и воспрепятствуют последующему разделению.
То же самое будет происходить при репликации ДНК
в клетке, если что-либо не помешает этому.
Отсюда следует, что для продолжения синтеза ДНК
положительные сверхвитки в начале репликационной
вилки должны ослабляться. Это по мере необходимости
происходит за счет кратковременных расщеплений по-
линуклеотидной цепи.
Как устраняются
положительные сверхвитки
перед репликативной вилкой?
Этот процесс катализирует группа ферментов - топо-
изомераз. Они действуют на ДНК, изомеризуя или
меняя ее топологию. Существует два класса топоизомераз
(тип I и II). Сначала мы рассмотрим принцип их
действия, а затем объясним их роль в репликации ДНК.
Ферменты типа I расщепляют одну из двух цепей
сверхспирализованной двойной спирали, что позволяет
всему дуплексу вращаться вокруг единственной фосфо-
диэфирной связи противоположной цепи, которая в
молекуле ДНК играет роль эффективного шарнира. После
окончания вращения фермент восстанавливает дуплекс
(рис 20.6). Очень важно, что фермент не гидролизует
^Й ^Ь Топоизомераза I
Дуплекс ДНК
I Фермент расщепляет одну нить и
^^^ I ковалентно присоединяется к фосфату ДНК
Туг-0 Н
ι ι
Ρ О
I Дуплекс вращается вокруг единственной
I фосфодиэфирной связи
^^Пуг-0 Н
^^^^ Восстановление фосфодиэфирной связи
Рис. 20.6. Реакция, катализируемая топоизомеразой I
Туг-ОН - остаток тирозина в молекуле фермента
фосфодиэфирную связь, которую атакует. Он просто
переносит ее от З'-ОН группы дезоксирибозы к ОН-г-
руппе одного из тирозинов, входящих в состав
молекулы фермента. Процесс легко обратим, поскольку при
разрезании не происходит потери энергии. Отметим,
что топоизомераза I не использует энергию АТР. Сверх-
спирализованная молекула ДНК находится в состоянии
напряжения, и уровень энергии будет выше, чем в ре-
лаксированной форме, вне зависимости от вида сверх-
спирализации - положительной или отрицательной.
Топоизомераза I способна только релаксировать
сверхспирализованную ДНК, но не способна ввести
новые витки. В то же время в Е. coli имеется особая
топоизомераза I, которая может релаксировать только
отрицательную сверхспираль ДНК. Впрочем, это не решает
проблему раскручивания молекулы, хотя и играет
важную роль.
Релаксация положительной сверхспирали,
образующейся впереди репликативной вилки Е. coli, происходит
путем активного введения отрицательных сверхвитков,
которые «нейтрализуют» положительные сверхвитки.
Это происходит под действием топоизомеразы II.
Топоизомераза II разрывает обе нити ДНК в
двойном дуплексе, перенося связи на себя, что делает
процесс разрыва полинуклеотидных цепей легко
обратимым. Кольцевой дуплекс ДНК фермент физически
переносит между концами разрыва без вращения цепей.
Для этого требуется энергия АТР; возможно, именно
благодаря его гидролизу происходят конформационные
изменения в белке. У Е. coli топоизомераза II,
называемая гиразой, вводит в ДНК отрицательные сверхвитки
(рис. 20.7). Для простоты на рисунке изображено
образование отрицательных сверхвитков в релаксированной
циклической ДНК (см. рис. 20.7, а). Но этот же принцип
применим к ДНК в начале репликативной вилки у Е. coli,
где возникает положительная сверхспирализация в
результате расплетания нитей ДНК. Установлено, что
включение отрицательных сверхвитков в этой области
соответствует релаксации положительной сверхспирали.
Гираза взаимодействует с ДНК таким образом, что
последняя наматывается вокруг белка. При этом возникает
сверхскручивание (положительная сверхспирализация)
в тех местах молекулы, которые связаны с белком (см.
рис. 20.7, б). Однако до тех пор, пока не происходит
разрывов ковалентных связей, не нарушается и целостность
структуры, а следовательно, локальная положительная
сверхспираль, образовавшаяся в ответ на включение
белка, может уравновешиваться отрицательной сверхспи-
рализацией где-нибудь в другом месте молекулы ДНК:
в результате сверхспирализация сводится к нулю. Затем
фермент разрывает обе нити ДНК (см. рис. 20.7, в)
глава 20 Синтез и репарация ДНК
245
и переносит двойную нить с внутренней стороны
на внешнюю, после чего скрепляет оба разрыва,
превращая положительную петлю в отрицательную (см.
рис. 20.7, г). Иными словами, происходит вставка
отрицательного сверхвитка. Физическое перемещение ДНК
совершается за счет гидролиза АТР. Состояние
отрицательной сверхспирали также является напряженным;
следовательно, введение отрицательных сверхвитков
относится к энергозависимым процессам. Установлено, что
расщепление связей в ДНК происходит не путем
гидролиза, а в результате временного переноса однонитевых
Релаксированная кольцевая ДНК
Стабилизированная положительная петля
^
Разрыв в заднем сегменте дуплекса
<<&
Разрез
Вновь соединенные цепи
дуплекса
с внешней стороны
Рис. 20.7. Реакция включения отрицательных сверхвитков
в кольцевую ДНК, катализируемая гиразой
Этот процесс может превращать положительно сверхскручен-
ную ДНК в отрицательно сверхскрученную ДНК
концевых фрагментов ДНК на фермент без потери
свободной энергии; поэтому процесс разрыва связей легко
обратим.
Какова биологическая роль
топоизомераз?
С эволюционной точки зрения для выживания клеток
чрезвычайно важно решение топологических проблем,
и оно происходит с помощью двух типов топоизомераз
(I и 11), обладающих различными механизмами действия.
У многих организмов свойства этих ферментов
несколько отличаются. Описано по меньшей мере 11
разновидностей топоизомераз.
У Е. coli топоизомераза 1 релаксирует только
отрицательные сверхспирали. В то же время гираза
(топоизомераза II) активно включает отрицательные сверхвитки,
и поскольку это приводит к релаксации положительной
сверхспирали, тем самым обеспечивается продолжение
синтеза ДНК.
У эукариот топоизомераза I может релаксировать оба
типа сверхспиралей - и положительную, и
отрицательную, а топоизомераза II - только положительную, но
не может вводить отрицательные сверхвитки (табл. 20.1).
Таким образом, при репликации ДНК у про- и эукариот
спутывание цепей при их расплетании
предотвращается введением положительных сверхвитков.
Таблица 20.1
Топоизомеразы Е. coli и эукариот: типы и механизм
действия
Тип*
Действие на ДНК
Эффект на сверх-
спирализацию
Топоизомераза I
E.coli Разрывает
одну нить ДНК
Эукариоты Разрывает
одну нить ДНК
Топоизомераза II (гираза)
E.coli Разрывает
обе нити ДНК;
АТР-зависимая
Эукариоты
Разрывает
обе нити ДНК;
АТР-зависимая
Релаксирует
отрицательную сверхспира-
лизацию
Релаксирует
положительную и отрица-
тельнуюсверхспира-
лизацию
Релаксирует
положительные сверхвитки;
включает
отрицательные сверхвитки;
разделяет сцепленные
кольцевые молекулы
ДНК
Релаксирует
положительные сверхвитки,
но не может вводить
отрицательные
сверхвитки
* Топоизомеразы обоих типов встречаются и в других
организмах
часть 4 Хранение и переработка информации
246
Однако сверхспирализация ДНК имеет огромное
значение не только для процессов репликации.
Аккуратно выделенная из клеток ДНК находится в виде
отрицательной сверхспирали. В релаксированной ДНК на
один оборот двойной спирали приходится 10,5 пар
оснований, а в клеточной - 12, т. е. она слабо
скручена. Для ДНК, выделенной из различных клеток,
степень сверхспирализации сравнима, что
свидетельствует о важности и контролируемости этого явления.
Известно, что у Е. coli отрицательная
сверхспирализация необходима как для репликации, так и для
транскрипции (см. главу 21), из чего следует, что в сверхспи-
рализованном состоянии легче происходит отделение
нитей ДНК. Также предполагают, что взаимодействие
белка с отрицательно сверхспирализованными
участками ДНК способствует уменьшению конформацион-
ного напряжения и увеличивает выход энергии,
необходимой для связывания, иначе говоря, облегчает
присоединение белков к ДНК.
Подводя итог, следует еще раз подчеркнуть, что
в клетках Е. coli необходимая отрицательная
сверхспирализация ДНК создается гиразой (топоизомеразой II),
а топоизомераза I релаксирует отрицательные
сверхвитки, но не действует на положительные. Равновесие
между двумя противоположными активностями (ги-
разы и топоизомеразы I) поддерживает необходимую
степень сверхспирализации (рис. 20.8); правильное
соотношение активностей двух ферментов необходимо
для жизнедеятельности клетки. Для контроля за этим
специально синтезируются два фермента; степень и знак
сверхспирализации регулируется фактором, синтез
которого контролируется соответствующим геном.
Необходимость гиразы для жизнедеятельности бактерий
подтверждается тем, что антибиотик налидиксиновая
кислота угнетает их размножение путем инактивации
гиразы. У эукариот этот фермент отсутствует, но нали-
диксиновую кислоту можно использовать для лечения
определенных инфекций у человека.
Интересно, что у термофильных бактерий, живущих
при очень высоких температурах, обнаружена обратная
гираза, механизм действия которой прямо
противоположен механизму действия гиразы Е. coli. Такое действие
фермента можно объяснить тем, что положительно сверх-
спирализованная ДНК лучше защищена от разделения
цепей, которое происходит при высоких температурах.
Эукариотическая ДНК в клетке, подобно прокарио-
тической, недокручена или отрицательно сверхспира-
лизована. Но, в отличие от прокариот, эукариотическая
топоизомераза, которая могла бы активно вставлять
отрицательные сверхвитки в ДНК, неизвестна. Что же
происходит в эукариотических клетках?
Гираза активно вводит
отрицательные сверхвитки
и превращает положительно
сверхспирализованную ДНК
в отрицательно
сверхспирализованную ДНК
g ^^^^^^" Топоизомераза I пассивно
^&Й^^^^ уменьшает отрицательную
f ^^^^^ сверхспирализацию
^^ ^^^k (не удаляет положительные
^^^^^^ Jf сверхвитки)
Рис. 20.8. Сбалансированное действие гиразы и
топоизомеразы I у Е. coli
В результате совместной работы этих ферментов достигается
необходимая для репликации ДНК и транскрипции степень
отрицательной сверхспирализации. Гираза- АТР-зависимый
фермент. Считается, что количество и соотношение
синтезированных ферментов контролируется степенью
сверхспирализации промоторов генов
При образовании хроматина ДНК наматывается на
нуклеосомную сердцевину, и в области контакта с
белком молекула ДНК недокручена. Такие нуклеосомные
витки не имеют разрывов, а ДНК в хромосоме не
может свободно вращаться. Поэтому локальная
отрицательная сверхспирализация на нуклеосоме
компенсируется положительной сверхспирализацией в другом
месте, так что суммарные изменения структуры
сводятся к нулю (рис. 20.9, б). Эукариотические
топоизомеразы релаксируют положительный сверхспиральный
фрагмент за счет включения отрицательного
супервитка (рис. 20.9, в). Прокариотический тип гиразы, таким
образом, здесь не нужен.
Итак, мы имеем хеликазы, раскручивающие нити,
и топоизомеразы, позволяющие разделить их
благодаря уменьшению положительной сверхспирализации,
вызываемой вращением двойной спирали по типу
шарнирного механизма во время расплетания цепей. Но это
еще не все: для раскручивания спирали нужны также
белки SSB.
Что такое SSB?
Это белки, связывающиеся с одноцепочечной ДНК
(от англ. Single Strand Binding Proteins). Они
обладают большим сродством к одноцепочечной ДНК -
глава 20 Синтез и репарация ДНК
247
ДНК хромосомы (нулевая суперспираль);
обратите внимание на то, что эта
цепь свободно не вращается
Когда ДНК наматывается вокруг нуклеосомы,
образуются локальные отрицательные сверхвитки.
Так как связи не разрываются, невозможно ввести
отдельную отрицательную сверхспираль,
следовательно, она должна компенсироваться
локальной положительной сверхспирализацией
Расположение
отрицательного
сверхвитка
ДНК имеет
суммарную нулевую
сверхспирализацию
Эукариотическая топоизомераза I уменьшает
локальную положительную сверхспирализацию,
что приводит к суммарной отрицательной
сверхспирализации
ДНК с суммарной
отрицательной
сверхспиралью
Рис. 20.9 Механизм, с помощью которого эукариотическая
ДНК приобретает отрицательные сверхвитки
В эукариотических клетках отсутствует фермент, способный
активно включать отрицательные сверхвитки подобно прока-
риотической гиразе
вне зависимости от последовательности основании -
и взаимодействуют с ней по всей длине разделившихся
нитей. Освобождающаяся при связывании энергия не
позволяет расплетенным нитям снова соединяться; таким
образом одноцепочечная форма защищается от
обратимого отжига. Когда нити используются в качестве
матрицы для синтеза новой ДНК, SSB-белки удаляются.
Положение на данный момент
Пока мы имели дело с общими аспектами процесса
репликации ДНК - полуконсервативным механизмом,
основанном на комплементарном взаимодействии пар
оснований Уотсона-Крика, биологическими аспектами
клеточного цикла, инициацией репликации и
механизмом раскручивания ДНК. Вышеизложенное
свидетельствует о сложности процесса репликации ДНК. Мы
получили представление о разделение нитей ДНК, но пока
ничего не узнали о механизме синтеза ДНК в реплика-
тивной вилке. Вернемся к этой проблеме. Прежде всего
надо понять: как нуклеотиды связываются между собой
при синтезе новой цепи ДНК? Ферменты,
катализирующие этот процесс, называются ДНК-полимеразами,
так как они полимеризуют нуклеотиды с образованием
молекулы ДНК.
Основные ферментативные
реакции, катализируемые
ДНК-полимеразами
Мы рассмотрим основную химическую реакцию
образования ДНК, но прежде остановимся на некоторых
аспектах.
• У Е. coli существует три ДНК-полимеразы,
называемые Pol I, Pol II и Pol III.
• Синтез ДНК, происходящий в репликативной вилке,
катализируется Pol III или ее эукариотическими
аналогами; Pol I играет важную роль как в репликации,
так и в репарации ДНК. Pol II изучена меньше;
предполагается, что она связана с определенным типом
репарации ДНК.
• Субстратами ДНК-полимеразной реакции служат
четыре дезоксинуклеозидтрифосфата dATP, dCTP,
dGTP и dTTP. В ходе репликации образуются
дочерние цепи - копии с материнских цепей.
• Для копирования полимеразе нужна одноцепочечная
матрица ДНК. Термин копирование используется
в смысле комплементарности: G на матрице
«копируется» (переписывается) в С дочерней цепи;
соответственно А «копируется» в Т, С - в G, Τ - в А.
• Очень важно запомнить: ДНК-полимераза может
только элонгировать (удлинять) уже существующую
нить, называемую затравкой (праймером). Праймер
комплементарен матрице и состоит из 2-10 нук-
леотидов; без него синтез начаться не может.
ДНК-полимераза не инициирует образование
новой цепи, так как она не может соединить два
свободных нуклеотида.
Полимераза присоединяет нуклеотиды к свободной
концевой З'-ОН-группе праймера, высвобождая при
этом неорганический пирофосфат (рис. 20.10).
Гидролиз фосфата увеличивает отрицательное значение AG0
синтеза полинуклеотидной цепи, способствуя таким
образом протеканию реакции полимеризации. Включение
нуклеотида в новую цепь ДНК сопровождается
образованием водородных связей с матрицей. При этом
происходит высвобождение энергии, что делает процесс
термодинамически выгодным.
Порядок взаимодействия четырех дезоксинуклеозид-
трифосфатов с ДНК-полимеразой определяют
основания копируемой родительской цепи.
Синтез ДНК всегда протекает в направлении 5'->3'
растущей цепи. Это означает, что растущая цепь элон-
гируется (наращивается) в направлении 5'—» 3'; нуклео-
тид добавляется к свободному 3 -ОН-концу
предшествующего нуклеотида. Напомним, что когда мы говорим
часть 4 Хранение и переработка информации
248
Праймер ДНК
0 0 0
II II II 5'
О—Р—О — Р— О — Р— О — СН2
1_ 1_ 1_
О О О j
/
Дезоксирибонуклеозидтрифосфат qu
(dATP, dGTP, dCTP, dTTP,
но в данном случае dATP)
ДНК-полимераза
G = C
0 О
II II
О—Ρ—О —Ρ—ОН +
1 I
О О
Неорганический пирофосфат
/н2о
2Р,-
А = Т-
Рис. 20.10. Реакция, катализируемая ДНК-полимеразой.
На схеме показано присоединение аденинового дезоксинук-
еотида (от АТР) к З'-концу затравки (праймеру); выбор
присоединяемого основания определяется основанием в матрич-
юй цепи. Синтез протекает в направлении 51—»3', т. е. цепь
удлиняется в этом направлении
) синтезе в направлении 51—» З1, мы всегда имеем в виду
тправление элонгации или полярности новой нити.
Матричная цепь имеет противоположную полярность.
3 полярности нитей ДНК мы говорили в предыдущей
лаве (см. с. 235).
Проблемы и еще раз проблемы
при синтезе ДНК
Как инициируется новая нить?
ДНК-полимераза не может инициировать образование
новой цепи, поэтому в любой начальной точке синтез
должен быть инициирован.
Решение этого вопроса довольно необычно и
заключается в том, что цепи ДНК инициируются при помощи
РНК. РНК имеет структуру, аналогичную структуре
одноцепочечной ДНК, только в качестве углеводного
компонента в ней выступает рибоза, а тимин (Т)
заменен на урацил (U). РНК синтезируется РНК-полиме-
разой практически при помощи такого же химического
механизма, как и ДНК, за исключением того, что для
синтеза используются рибонуклеозидтрифосфаты АТР,
СТР, GTP и UTP. Но в данном случае
принципиальное различие состоит в том, что РНК-полимераза
способна инициировать новые цепи. Она может
соединить два нуклеотида (при этом также требуется
матрица), а ДНК-полимераза - нет.
После того, как специальной РНК-полимеразой,
называемой праймазой, будет синтезирован небольшой
участок РНК-праймера (например, из 5 нуклеотидов),
подключается ДНК-полимераза и продолжает
наращивать цепь.
Теперь рассмотрим антипараллельное расположение
цепей в молекуле ДНК.
Как решается проблема полярности
цепей при репликации ДНК?
Вспомним предыдущую главу, где обсуждается
полярность цепей ДНК. ДНК реплицируется в каждой
репликативной вилке, которая постепенно смещается
вдоль хромосомы. У Е. coli в каждой репликативной
вилке работают две молекулы ДНК-полимеразы III (Pol III),
по одной на цепь. Эти две молекулы объединяются в
единый асимметричный димер и образуют холофермент.
Молекулы полимеразы должны двигаться в одном
направлении (рис. 20.11). Однако направление 51—»З1
одной родительской цепи противоположно направлению
движения вилки, а в другой цепи совпадает с ним.
Поскольку ДНК-полимераза может осуществлять
синтез только в направлении 5'—> 3', матричная цепь
должна идти в направлении З1—> 51, которое противоположно
направлению синтеза. Это вполне подходит для синтеза
одной новой нити (на рис. 20.11 - левой), а что же с
другой? Она идет в направлении перемещения вилки
(51—»З1), следовательно, синтез дочерней цепи должен
глава 20 Синтез и репарация ДНК
249
5'3'
Реплицируемая
ДНК
Репликативный аппарат после
раскручивания цепей ДНК
должен двигаться
в направлении, указанном
черной стрелкой
Синтез отстающей цепи
ДНК должен протекать
в направлении, указанном
пунктирной стрелкой: 5'->
3'
Вновь
синтезированная ДНК
3' 5'
3' 5'
Обратите внимание:
данная схема представляет
только одну репликативную
вилку двунаправленного
репликативного «глазка»,
показанного на рис. 20.3
Рис. 20.11. Распределение полярности цепей при
репликации ДНК
идти в противоположном направлении. Рис. 20.11
отражает на первый взгляд неразрешимую проблему: поли-
мераза на правой нити должна двигаться вверх по
странице, что она и делает, а ДНК должна синтезироваться в
направлении вниз по странице. Левая нить называется
лидирующей цепью, другая - запаздывающей.
Но как же инициируется синтез запаздывающей
цепи? С лидирующей цепью все ясно: праймаза
закладывает простой праймер в точке начала репликации,
а ДНК-полимераза наращивает цепь. Но это не
подходит для запаздывающей цепи. Проблема решается
следующим образом: пока цепи ДНК разъединены,
на правой цепи происходит неоднократно
инициированный праймазой (РНК-полимеразой) синтез
множества коротких цепей ДНК (рис. 20.12). На первый
взгляд это кажется невероятным!
5'3'
Синтез ДНК
Короткие цепи ДНК, связанные с РНК-праймерами
на запаздывающей цепи, называются фрагментами
Оказаки - по имени открывшего их исследователя.
Однако из схемы все еще неясно, как ДНК-полимераза
может синтезировать ДНК в направлении,
противоположном ее движению. Кроме того, запаздывающая цепь
состоит из серии разрозненных коротких фрагментов,
соединенных с праймером. Для начала разберем, как
работает репликативный аппарат в репликативной вилке
Ферментативный комплекс
репликативной вилки Е. coli
Функциональный комплекс белков репликативной
вилки изображен на рис. 20.13. Ключевыми ферментами
являются хеликаза, раскручивающая двойную спираль,
праймаза, синтезирующая праймеры на участках
запаздывающей цепи, и конечно комплекс Pol III.
Раскручивающая активность хеликазы зависит от АТР; фермент
(вероятно, благодаря конформационным изменениям)
перемещается по цепи ДНК и разделяет две цепи
двойной спирали. После присоединения SSB-белков
происходит стабилизация одиночных цепей. У Е. coli в
репликативной вилке присутствуют две связанные между
собой молекулы Pol III, одна из которых синтезирует
лидирующую цепь, а другая запаздывающую. Это по
сути один и тот же фермент, но димер холофермента
асимметричен в связи с наличием дополнительной
субъединицы со стороны запаздывающей нити. Pol III
является высокопроцессивным ферментом: однажды
присоединившись к цепи ДНК, он удерживается на ней
на отрезке в тысячи оснований, что удобно для
лидирующей цепи. Его фиксации на матричной цепи помогают
так называемые кольцевые скользящие «зажимы»
(β-субъединицы), состоящие из двух половинок. Они
образуют позади Pol III скользящее кольцо, связанное
с ферментом, что предотвращает отделение последнего
35"
РНК - праймеры
Фрагменты Оказаки
%
35'
• Лидирующая
цепь
Запаздывающая
цепь
3'5'
Рис. 20.12. Репликативная вилка
Лидирующая цепь синтезируется постоянно, в то время как
запаздывающая - отдельными короткими фрагментами
(фрагменты Оказаки)
SSB - белок
(белок, связывающийся
с одиночной цепью)
Pol III
Реплицируемая ДНК
Хеликаза
Праймаза
Pol III
Poll
Poll
/
\
Праймер для
фрагмента
Оказаки
• Лидирующая
цепь
Запаздывающая
цепь
Рис. 20.13. Расположение ферментов в репликативной вилке
Е. coli (упрощенная схема)
часть 4 Хранение и переработка информации
250
от матричной нити ДНК. Круговой зажим имеет
отверстие для свободного протягивания ДНК. Структуру
зажима иллюстрирует ленточная модель (рис. 20. 14).
ч^
ι
\
На запаздывающей цепи фермент (вместе с
кольцевым зажимом), встречая праймер, синтезированный прай-
мазой для предыдущего фрагмента, останавливается.
Пока неизвестно, отделяется ли комплекс Pol III,
чтобы присоединиться вновь после встречи с новым прай-
мером, который будет использован для синтеза
очередного фрагмента Оказаки, или все происходит как-то
иначе.
Теперь перейдем к топологической проблеме,
возникающей при синтезе фрагментов Оказаки в
направлении 5'—> 3', так как репликативная машина не имеет
обратного хода. Основной принцип предельно понятен.
Если запаздывающая цепь образует петлю, то на
коротком участке она будет ориентирована в том же
направлении, что и лидирующая. Таким образом, репликатив-
ный аппарат может перемещаться в направлении вилки
и одновременно синтезировать обе новые цепи.
Хотя принцип прост, проблема, связанная с
перемещением петли вдоль родительской цепи при
синтезе фрагментов Оказаки, гораздо сложнее. Для ее
решения Корнбергом была предложена модель, названная
моделью петли (рис. 20.15).
По мере распространения петли и перемещения
репликативного аппарата новые нити ДНК будут
встречать 5'-концы РНК от предыдущего фрагмента Оказаки.
В этой точке полимераза должна освобождаться, петля
разрушиться, а затем образоваться новая. А теперь
разберемся: каким образом фрагменты Оказаки
соединяются в непрерывную цепь ДНК?
5' 3'
КЗ
Репликативная вилка
Рис. 20.14. Ленточная модель скользящих «зажимов»
дрожжей и Е. coli
а - «Зажим» дрожжевых клеток, обеспечивающий процессив-
ность ДНК-полимеразы. является тримером;
б- «зажим» Е. coli, присоединяющийся к ДНК-полиме-
разе III, - димер.
Отдельные субъединицы внутри каждого кольца показаны
разными оттенками. Цепи β-слоя изображены плоской лентой,
aot-спираль- спирализованной. В центре каждой структуры
помещена модель В-формы ДНК, так как кольцо может
охватывать дуплекс
Участок
синтеза ДНК
3'5'
Расширяющаяся
| „СЕ,Я 5.
ч5' /
Праймер ДНК
Завершенный
фрагмент Оказаки
\
35'
Рис. 20.15. «Петельная» модель синтеза фрагментов Оказаки
Более темные стрелки указывают направление синтеза ДНК
Согласно модели, при встрече нового фрагмента Оказаки с
предыдущим необходимо разрушение петли. Затем должна
образоваться новая петля. В этой модели обе нити могут
синтезироваться в направлении 5'—> 3', так как репликативный
аппарат перемещается в направлении вилки. Точно описать детали
образования петли невозможно
глава 20 Синтез и репарация ДНК
251
Что происходит
с фрагментами Оказаки?
У Е. coli Pol III отделяется от ДНК в момент
достижения РНК-праймера предшествующего фрагмента
Оказаки, оставляя разрыв в месте соединения ДНК и РНК.
Здесь подключается ДНК-полимераза I (Pol I). Это
удивительный фермент, объединяющий в одной молекуле
три различных ферментативных активности.
Если мы рассмотрим схему на рис. 20.12, то увидим,
что отдельные фрагменты ДНК (фрагменты Оказаки)
должны быть превращены в непрерывную цепь ДНК.
Каждый фрагмент начинается с РНК, которую нужно
удалить, затем заделать бреши, а отдельные фрагменты
ДНК соединить между собой. Pol I присоединяется
к разрывам между завершенными фрагментами
Оказаки, добавляет нуклеотиды к З'-ОН-концу предыдущего
фрагмента, двигаясь в направлении 51—» З1, как и Pol [II
(при синтезе ДНК добавление нуклеотидов всегда
происходит в направлении 5'—» З1). По мере перемещения
передняя часть Pol I сталкивается с РНК предыдущего
фрагмента Оказаки и отщепляет его рибонуклеотиды,
а расположенный позади участок фермента
присоединяет дезоксирибонуклеотиды, заполняя бреши.
«Передняя» активность называется 5'—> З'-экзонуклеазной:
экзо- потому, что фермент удаляет нуклеотиды с конца
молекулы, нуклеазная - потому, что фермент гидролизу -
ет нуклеиновую кислоту, а 5'—» З1 указывает направление
движения фермента от 5'-конца РНК в направлении
З'-конца. Обратите внимание на то, что Pol III, в отличие
от Pol I, не обладает 5'—> З'-экзонуклеазной
активностью и при встрече с фрагментом Оказаки не может
отщеплять праймер РНК. Поэтому она отделяется в этой
точке от ДНК, и всю остальную работу проделывает Pol I.
Но это еще не все. «Тыльная» сторона молекулы Pol I
наряду с полимеразной проявляет 3'—> 5'-экзонуклеазную
активность. Эта экзонуклеаза гидролизует нуклеотиды
с З'-конца цепи ДНК, которую только что синтезировала
Pol I, и отрезает их в направлении 5'-конца, т. е. в
противоположном действию 5'—» З'-экзонуклеазы.
Парадоксально, но факт: фермент, который
одновременно разрывает ДНК, в тоже время синтезирует ее.
Но это можно объяснить. 3'^ 5' экзонуклеаза
гидролизует концевой нуклеотид вновь образованной нити ДНК
только тогда, когда его основание не спаривается с
соответствующим основанием матричной цепи; а
ДНК-полимераза добавляет нуклеотиды только в том случае,
если предыдущее основание дочерней цепи
надлежащим образом спарено с соответствующим основанием
матричной цепи. Так фермент проверяет правильность
включения в цепь предыдущего основания, вырезает
часть 4 Хранение и переработка информации
252
его, если оно не соответствует матрице и заменяет
другим. Этот процесс называется коррекцией
неправильного спаривания; с его помощью контролируется
правильность синтеза.
ДНК-полимераза I (в отличие от Pol III) низкопро-
цессивна. Она непрочно удерживается на матричной
цепи и довольно быстро отделяется от нее после
удаления РНК, не имея кругового зажима, удерживающего
ее на ДНК. Это понятно, иначе она продолжала бы
замещение длинных цепей новосинтезированной ДНК.
После ее отделения в цепи остаются разрывы, для
соединения которых используется фермент, называемый
ДНК-лигазой.
Этот фермент синтезирует фосфодиэфирную связь
между З'-ОН одного фрагмента ДНК и 5'-фосфатом
следующего. Процесс требует затраты энергии. У
некоторых прокариот и эукариот ее источником служит АТР.
Механизм действия фермента (Е) состоит в том, что он
принимает AMP от АТР, освобождая пирофосфат, и
переносит AMP на 5'-фосфат ДНК. Затем ДНК-AMP
реагирует с ДНК-З'-ОН, высвобождая AMP и заделывая
разрыв:
Ε + АТР -> Е-АМР + РР,.;
Е-АМР + Р-5'-ДНК -> Ε + АМР-Р-5'-ДНК;
ДНК-З'-ОН + АМР-Р-5'-ДНК ->
-> ДНК-З'-О-Р-б'-ДНК + АМР.
AMP соединяется с ферментом и ДНК через 5'-фос-
фатную группу.
У Е. coli вместо АТР используется NAD+. Он может
служить источником AMP точно также, как и АТР, и
у Ε coli происходит именно так. Потом Pol I
связывается с ДНК в определенной точке (рис. 20.16). Поли-
мераза добавляет нуклеотиды к З'-концу фрагмента
ДНК, проверяя каждое присоединенное основание, и
перемещается в направлении 5'—» 3'. РНК и некоторая
часть ДНК отщепляются - и промежуток заполняется
ДНК. Затем Pol 1 отделяется, и лигаза соединяет вместе
два фрагмента ДНК. Таким образом, из серии
фрагментов Оказаки образуется непрерывная новая цепь ДНК.
Коррекция неправильного спаривания
оснований полимеразой III
Последовательность оснований во вновь образованной
цепи ДНК должна быть воспроизведена точно,
поскольку включение даже одного неправильного основания
может привести к летальному исходу. Но при огромном
количестве оснований ошибки неизбежны.
Когда Pol III встретит на матричной цепи, например,
G, ее высокая специфичность проявится в том, что
Здесь присоединяется Pol I
Матричная нить
3 s^-v2«2»«ebipiai-^^i.irst<i^^
5'dB-dB- dB
Направление движения Pol I
►
гдеар*=«*я№р^^у?| .тяти*
Pol I имеет низкую ироцессивность
и после удаления праймера
быстро отделяется от ДНК;
затем лигаза соединяет разрывы,
образуя непрерывную нить ДНК
ГштПГмшТГятШштт
5' гВ - гВ - гВ - гВ - гВ - dB - dB - dB - dB -
3'- конец фрагмента
Оказаки
dB 3' ОН
Если последнее
основание ошибочно
и не может
спариваться
с основанием
матричной цепи,
оно удаляется
и замещается другим,
при участии Pol I
ДНК-праймер Нить ДНК, присоединенная
на 5'-конце фрагмента к ДНК-праймеру
Оказаки. Он должен
быть замещен ДНК
Рис. 20.16. Схема действия полимеразы I по преобразованию фрагментов Оказаки
dB - Дезоксирибонуклеотид; гВ - рибонуклеотид
в абсолютном большинстве случаев только dCTP будет
присоединен к активному центру фермента и перенесен
на растущую цепь ДНК. Аналогичное утверждение
справедливо для других оснований. Механизм, с
помощью которого Pol III подбирает правильный нуклеотид
к основанию матрицы, основан на модели Уотсона-Кри-
ка, когда все пары комплементарных оснований имеют
одинаковую геометрическую форму, отличную от любой
другой пары оснований; это строго распознается
ферментом (рис. 20.17). Образование водородных связей
между нужными парами и между основаниями одной
цепи (стэкинг-взаимодействия) также способствуют
правильному отбору, поскольку это сопряжено с
высвобождением энергии.
Впрочем, этот механизм имеет предел точности. Хотя
образование пар по Уотсону-Крику является
определяющим моментом, в любом наборе нуклеотидов
несколько молекул, например dATP, будут находиться в ими-
но-форме (=NH вместо -NH2), и при этом они могут
спариваться с С. Следовательно, Pol [II может
совершать ошибки при копировании ДНК с частотой
примерно одну на 105-106 оснований. Это дало бы слишком
высокий уровень мутаций. Однако у Е. coli реальная
конечная частота ошибок при репликации ДНК
значительно ниже, около 10~10 за один раунд репликации.
Оказалось, что Pol III обладает также 3'—»5'-экзо-
нуклеазной активностью, благодаря которой последнее
из включенных в синтезируемую цепь оснований
может быть удалено, если оно неправильно спаривается.
В результате такого устранения ошибок считывания их
уровень сводится к минимуму. Большинство событий,
происходящих при репликации ДНК у Е. coli и эука-
риот, показано на рис. 20.18.
7Н
СН3 О -H-N
Ν-Η
11.1
Η
M*-H-NH M
Ю.З
10.8
Η
N.
У \ A .Ν
c'r V-Vh c1
chm°
(tn-h-o
hV^n 4c1
Η
Ю.З
10.7
Рис. 20.17. Геометрические характеристики пар оснований
Уотсона-Крика и «ошибочных» пар
Данные получены на основе рентгенографии кристаллов дву-
нитевых олигодезоксирибонуклеотидов (В-форма ДНК).
Наблюдается строгое геометрическое соответствие пар
Уотсона-Крика и неустойчивое состояние «качающихся» пар А-С
и G-T или пары G-A (в конформации анти- и син-
соответственно). Термин «качающиеся» пары будет объяснен на с. 287.
Указаны расстояния (в нм) между С-1-атомами дезоксири-
бозы и углы вращения гликозидной связи между основанием
и дезоксирибозой
глава 20 Синтез и репарация ДНК
253
Эукариоты
Топоизомераза II
Релаксирует положительные
сверхвитки, но не может
вводить отрицательные витки
ТопоизомеразаI
Релаксирует как
положительные, так
и отрицательные сверхвитки
Хеликаза Ρ как и у Е. coli
SSB-белки как и у Е. coli
δ-Полимераза на ·.«::
лидирующей цепи.
а-Полимераза на
запаздывающей цепи.
Обладает
праймазной
активностью
γ-Полимераза при
репликации
митохондриальной
ДНК
Е. coli
Гираза
.. (Топоизомераза II)
Релаксирует
положительные
сверхвитки при помощи
введения
отрицательных витков
Хеликаза раскручивает
двойную спираль
в репликативной вилке,
вызывая образование
положительных
,. сверхвитков
/ впереди нее
SSB-белки,
связывающиеся
, с одноцепочечной ДНК
/ Праймаза Ρ -
часть праймосомы.
\ Синтезирует праймеры
\ для фрагментов
: Оказаки
\ Полимераза III Ρ
'·..· по одной молекуле
" на каждую нить
\ Полимераза I вырезает
РНК-праймер
из фрагмента Оказаки
и достраивает ДНК
й**%. ДНК-лигаза
1 * соединяет
1 разрывы в ДНК
1 после удаления Pol I
Рис. 20.18. Репликации ДНК Е. coli и эукариот
Метил-зависимое
исправление (репарация) ошибок
Несмотря на использование описанных выше
механизмов устранения ошибок считывания, обеспечивающих
высокую точность в системе репликации ДНК, ошибки
при работе с таким огромным числом присоединяемых
нуклеотидов, неизбежны. У Е. coli имеется другой
механизм; он используется для исправления ошибок в
последовательности оснований вновь синтезированной
ДНК. Если ошибка не была устранена полимеразой,
она вызовет деформацию дуплекса.
Новая нить
Родительская нить
Ошибка появляется в новой цепи, и репарирующая
система обязана распознать именно ее. Важно, что при
ошибках такого рода основание родительской цепи
является правильным по определению, а неправильным
является комплементарное основание в новой цепи.
Репарирующая система должна четко различать обе цепи
(родительскую и дочернюю), чтобы удалить
мутированное основание из новосинтезированной цепи и
исправить ошибку. Как же происходит распознавание цепей?
В ДНК Е. coli в последовательности GATC аденин
всегда метилирован ферментом Dam-метилазой.
Метилирование основания не нарушает спаривания оснований
или структуры ДНК, так как оно происходит спустя
некоторое время после синтеза новой цепи. Таким
образом, в течение какого-то времени родительская нить
будет метилирована, а вновь синтезированная - нет.
Репарирующая система включает меньше компонентов
по сравнению с системой, участвующей в репликации
ДНК. Один белок Mut S узнает нарушение в спирали;
Mut Η, связанный с Mut S при участии Mut L, находит
неметилированную последовательность GATC на одной
из цепей дуплекса (идентифицируя таким образом вновь
синтезированную цепь) и разрывает ее в этом месте
(рис. 20.19). Место разрыва может быть удалено от
места ошибки на тысячи оснований. Затем, при
совместном действии хеликазы, SSB-белков и экзонуклеазы,
удаляется участок цепи от разрыва до места ошибки,
а полимераза III застраивает образовавшуюся брешь
в ДНК. Завершает репарацию ДНК-лигаза,
соединяющая нуклеотиды в месте разрыва. Для исправления
одного основания бывает необходимой замена тысячи
оснований (см. рис. 20.19). Эта система исправления
ошибок тысячекратно увеличивает точность
репликации. Подобная система репарации ошибок
существует и у эукариот. У человека известны белки,
аналогичные белкам Mut S и Mut L Ε. coli. Мутации в их генах
сопровождаются увеличением риска возникновения
раковых опухолей.
Репарация повреждений ДНК
у Е. coli
Описанный выше механизм обеспечивает репликацию
ДНК с очень высокой степенью точности,
гарантирующей продолжение жизни. Однако все еще остается
большая проблема. Последовательность оснований в ДНК
обязательно должна быть неизменной, поскольку
информация, которую она несет, необходима организмам
в течение всей их жизни. Но ДНК - с точки зрения
химической стабильности - обычная молекула, и с ней
могут происходить химические изменения, которые
способны вызывать большое число мутаций, если бы
не процессы постоянной репарации. Некоторые
повреждения носят спонтанный характер. Гликозидная связь,
при помощи которой пуриновые и (в меньшей степени)
пиримидиновые основания соединяются с остатками
часть 4 Хранение и переработка информации
254
Шйаяаагаавдиа
GATC
CTAG
ι
эдд»йг^«':*^р^-.4т:мил^з^>*1*>й'''1 «ь*«**.-<ч>4*»».
С
G
ι
GATC
CTAG
- mUi
Mut S находит
ошибку
Mut L связывает Mut S
с Mut H, который находит
последовательность GATC
Mut Η идентифицирует
вновь синтезированную
нить по отсутствию
метилирования
в последовательности GATC
и разрывает
полинуклеотидный остов
Экзонуклеаза, хеликаза
SSB-белки удаляют
участок новой цепи
Полимераза III и
лигаза восполняют
удаленный участок
Рис. 20.19. Метил-зависимый путь репарации ошибок
дезоксирибозы, нестабильна, и нередко происходит
апуринизация и апиримидинизация, когда большое
количество пуринов и пиримидинов отщепляется от ДНК.
Кроме этого, цитозин и аденин подвергаются дезамини-
рованию, превращаясь в урацил и гипоксантин
соответственно (см. формулы на с. 217 и рис. 18.7). ДНК также
является объектом атаки множества соединений.
Ионизирующая радиация, свободные радикалы
кислорода (см. с. 212), канцерогены, УФ-излучение могут
повреждать ДНК и, следовательно, вызывать мутации.
В общем, повреждения, затрагивающие обе нити
двуспиральной молекулы ДНК и происходящие точно в
одном и том же месте обеих цепей, относительно редки,
хотя и случаются. При повреждении какого-то участка
одной из цепей вторая цепь всегда может служить
матрицей и управлять репарацией поврежденной части.
В клетке существует множество систем, репарирующих
практически любые повреждения ДНК, поскольку
поддержание целостности этой молекулы имеет
исключительное биологическое значение. ДНК нужно репа-
рировать любой ценой.
Существуют различные типы репарирующих систем.
• Прямая репарация. Облучение ДНК
ультрафиолетовыми лучами может вызывать ковалентное
связывание двух расположенных рядом (в одной и той же
цепи) тиминовых оснований с образованием Т-ди-
мера.
Т-димер
У Е. coli нехарактерные связи разрываются при
помощи активируемого светом механизма, благодаря чему
остатки тимина возвращаются в исходное состояние.
Система широко распространена и имеет большое
значение для растений. Алкильные группы оснований (они-
образуются под действием некоторых мутагенов) могут
напрямую удаляться под действием так называемого
«фермента-самоубийцы»: он служит их акцептором,
после чего сам подвергается разрушению. Строго
говоря, это скорее специфичный белковый реагент, чем
фермент.
• Репарация с вырезанием (эксцизией) нуклеотида.
Повреждения, деформирующие двойную спираль,
например Т-димеры, могут также репарироваться
путем вырезания включающих повреждение коротких
последовательностей нуклеотидов с последующим
замещением их на правильную последовательность.
При этом в качестве матрицы используется
противоположная цепь ДНК. У Е. coli есть необычная эндо-
нуклеаза: эксцинуклеаза, или комплекс uvrABC -
по названию трех генов, кодирующих фермент. Она
разрезает ДНК по обе стороны от повреждения и
удаляет однонитевой фрагмент из 12-13 нуклеотидов
(рис. 20.20). К разрыву присоединяется ДНК-по-
лимераза I и добавляет нуклеотиды к З'-концу
разорванной цепи; лигаза соединяет разрыв. Работа этой
системы зависит от правильного распознавания
поврежденной нити ДНК.
глава 20 Синтез и репарация ДНК
255
ДНК, содержащая повреждение,
например, тиминовый димер Повреждение
ксцин клеаза
Эксцинуклеаза разрезает цепь
с двух сторон от повреждения
Фрагмент ДНК длиной 12-13 оснований,
h" содержащий повреждение, удаляется
- ^ Μ
5'
3'
Образовавшаяся брешь
заполняется при участии
ДНК-полимеразы I и лигазы I
Рис. 20.20. Путь эксцизионной репарации у Е. coli
• Репарация с эксцизией основания и репарация
АР-сайта. Дезаминирование превращает цитозин
в урацил и аденин - в гипоксантин. ДНК-гликозида-
за узнает обычное основание и отщепляет его,
оставляя АР-сайт (от англ. apurinic-apyrimidinic) -
дезоксирибозу, лишенную основания (рис. 20.21).
АР-сайты могут также образовываться спонтанно,
поскольку связь пурин-дезоксирибоза достаточно
нестабильна. Репарация АР-сайтов включает
разрезание полинуклеотидной цепи, прилежащей к месту
повреждения, с последующим замещением этого
фрагмента при участии ДНК-полимеразы I и лигазы.
Образованный из цитозина урацил также необходимо
удалять (вероятно, это и объясняет, почему в состав ДНК
вместо U входит Т). Напомним, что Τ отличается от U
наличием дополнительной метильной группы (см.
с. 217). Если бы нормальная ДНК содержала U, то было
бы невозможно отличить обычный U от «ложного»,
образованного при дезаминировании С.
ΝΗ2 О
Цитозин
Урацил
Использование в составе ДНК основания Τ вместо
U решает эту проблему. Как будет рассказано в
следующей главе, U используется в составе РНК, но поскольку
она имеет относительно более короткое время жизни
и гораздо меньшие размеры, ошибки в ней не так
критичны. Вероятно, поэтому РНК и не репарируется.
Замечена довольно четкая корреляция: в клетках
Е. coli может присутствовать множество разрывов ДНК,
U Μ .5 & Урацил, образованный в
результате дезаминирования
цитозина
G В
ι ι
S S
I I
в в
ι ι
S S
1 ■
1ДНК-гл икозил аза
удаляет урацил
I I I I I I I
S S S S S S S
I I I I I I I
В В В АР-сайт В В В
В
S
S
в
в
S
я1ят
Т"
S
в
в
S
в
S
J
S
в
в
S
шя1т
—г
S
в
в
S
в
S
Разрыв
1
S
в
в
S
G
S
\
τ
S
G
S
\
В
S
В
S
в
S
Эндонуклеаза
разрывает цепь
—г—
S
В
в
S
-г-
S
в
в
S
-J
S
в
в
S
ι
Полимераза I и лигаза
восстанавливают цепь
S
в
в
S
S
с
G
S
-L
S
В
В
S
-JL.
S
в
в
S
S
в
в
S
Рис. 20.21 Образование и репарация АР-сайта: S - сахар;
В - основание
В приведенном примере АР-сайт образуется после удаления
урацила под действием гликозилазы. Но такие сайты могут
возникать и в ходе спонтанного гидролиза пуриновых
оснований (в меньшей степени - пиримидиновых оснований)
требующих присоединения Pol I и проведения ею
репарации, но имеется только две репликативных вилки,
требующие участия Pol III. Поэтому в клетках Е. coli
присутствует всего 10-20 молекул Pol III и значительно
больше молекул Pol I.
Устройство
репликативной вилки эукариот
Общие принципы репликации ДНК у Е. coli применимы
и к эукариотам, правда, с некоторыми поправками.
Главная заключается в том, что вместо димера Pol 111 у Е. coli
часть 4 Хранение и переработка информации
256
в синтезе лидирующей и запаздывающей цепей
участвуют разные полимеразы.
У эукариот описано пять ДНК-полимераз, которые
обозначаются греческими буквами. Полимеразы α и δ
участвуют в репликации ядерной ДНК (Pol 111 у £. coli).
Pol ос находится в комплексе с праймазой, но не
проявляет активности З1—>5'-экзонуклеазы. Полимераза δ
обладает З1—>5' экзонуклеазной активностью. Похоже,
что полимеразы α и δ формируют у эукариот реплика-
тивный комплекс для синтеза лидирующей цепи. δ-Πο-
лимераза дрожжей соединена с круговым скользящим
зажимом, известным как PCNA (от англ. Proliferating
Cell Nuclear Antigen - ядерный антиген пролиферирую-
щих клеток) и обеспечивающим ей процессивность, как
и у фермента Е. coli. На рис. 20.14 на примере ленточной
модели показана структура зажимов дрожжей и Е. coli.
Несмотря на внешнее сходство, их аминокислотные
последовательности сильно различаются; кроме того, зажим
Е. coli является димером, а дрожжевой белок - тример.
Это свидетельствует о независимом формировании этих
белков в ходе эволюции.
Две другие ДНК-полимеразы эукариот - β и ε -
выполняют репарирующую функцию, а γ-полимераза
участвует в репликации митохондриальной ДНК.
Проблема репликации
концов хромосом эукариот
Хромосомы эукариот линейны, что вызывает проблемы,
не возникающая при репликации кольцевых хромосом.
Рассмотрим репликацию хромосомы,
представленной на рис. 20.22. Она реплицируется в двух
направлениях от одного инициирующего сайта, расположенного
в центре (на самом деле хромосома эукариот имеет
много таких сайтов). 5'-концевая область лидирующей цепи
реплицируется полностью. Но этого не происходит при
синтезе запаздывающей цепи, потому что участвующий
в образовании фрагментов Оказаки РНК-праймер
расположен у З'-конца матричной цепи (см. рис. 20.22).
После удаления праймера эти концы оказываются нере-
плицированными, и нет механизма, позволяющего
восполнить их, используя ДНК-синтезирующий комплекс,
описанный выше. Для восполнения теряемых частей
требуются РНК-праймеры, но матриц, с которых их
можно было бы образовать, тоже нет. Значит, с каждым
клеточным делением хромосомы будут последовательно
укорачиваться; дальнейшие предположения
затруднительны, так как механизм синтеза ДНК подразумевает
полную репликацию двуцепочечной ДНК. Решение
проблемы состоит в том, что на концах хромосом эукариот
находятся специальные повторяющиеся
последовательности ДНК - теломерная ДНК (содержащие ее концы
хромосом - тел ом еры). Концы все еще не могут быть
реплицированы при помощи описанного ранее
синтезирующего ДНК комплекса, но полнота репликации
«настоящей» хромосомы осуществляется при помощи
праймера на 3'-конце матричной цепи, расположенной
против теломерной ДНК. Более того, в быстроделя-
щихся клетках теломера добавляется, чтобы хромосома
никогда не подвергалась риску укорочения.
Как синтезируется теломерная ДНК?
Теломера состоит из коротких повторяющихся
фрагментов (последовательность расположения оснований в этих
фрагментах варьирует у разных видов). У человека
имеются сотни тандемных повторов последовательности
TTAGGG. Фермент теломераза добавляет эти
последовательности (одну за другой) к З'-концу предсуществу-
ющей теломерной ДНК и таким образом удлиняет ее.
Теломераза имеет две особенности: 1) использует РНК
РНК-праймер Нереплицированный
фрагмента Оказаки участок
Синтез 1
ДНК 1
► 1
* N
И
ι Ч
1 i
1 и
Ι 11
ι f I
1 τ 1
ч
III
3' 5' 3' 5' 3' 5'
Реплицируемая
хромосома
Рис. 20.22. Схематичное изображение укорочения вновь
образованных линейных хромосом при репликации
Представлена двунаправленная репликация очень короткого
фрагмента ДНК. Следует отметить, что удаление праймеров
из фрагментов Оказаки происходит постоянно (здесь оно
представлено как отдельное событие) У обычной хромосомы
имеется множество точек начала репликации. Светлым выделены
синтезируемая ДНК и РНК-праймеры фрагментов Оказаки
глава 20 Синтез и репарация ДНК
257
в качестве матрицы для синтеза ДНК - это обратная
транскриптаза (см. главу 23); 2) эта матрица включена
в структуру фермента. У человека РНК имеет
последовательность, включающую 1,5 повтора
комплементарных TTAGGG. Матрица РНК гибридизуется с концом
теломерной ДНК (рис. 20.23), затем постепенно (по
одному нуклеотиду) добавляется еще один фрагмент.
После этого фермент перемещается и связывается с концом
нового, только что образовавшегося фрагмента. Таким
образом, теломера непрерывно достраивается. На
удлиненной теломере одной цепи затем формируется
комплементарная цепь, так что теломера имеет двуцепочеч-
ную структуру. Однако механизм образования такой
структуры пока не выяснен. По-видимому, в этом
процессе участвует ДНК-полимераза.
Необходимость теломер доказана на примере YACs,
или искусственных дрожжевых линейных хромосом.
Они включают три участка ДНК, необходимых для
репликации хромосомы: центромеры, точки начала
репликации и теломеры. Показано, что после встраивания
в дрожжевые клетки YACs могут сохраняться в течение
нескольких поколений, но по мере потери теломерных
концов они со временем исчезают.
Этого не происходит у прокариот, поскольку в
кольцевой хромосоме всегда есть матрица, доступная для
образования праймера. Существование теломер у эукариот
очень важно. Теломераза обнаружена в эмбриональных
клетках, которые постоянно и быстро делятся, и
непрерывное удлинение хромосом необходимо для
компенсации их постоянного укорочения.
Однако в соматических клетках, которые делятся
не часто, добавление теломер не столь важно. И в
самом деле, теломераза в них не обнаружена. Если это так,
то напрашивается следующий вывод: соматические
клетки получают свою «порцию» теломерной ДНК,
необходимую на время жизни клетки и ее потомства.
Теломеры в большинстве клеток укорачиваются с
возрастом, и это может являться важным фактором,
определяющим продолжительность жизни. Если
соматические клетки трансформируются в канцерогенные, они
переходят в состояние неограниченного деления, в
результате чего становятся бессмертными - нет предела их
способности к делению в культуре. Интересно, что в
раковых клетках доказано присутствие теломеразы.
Репарация повреждений ДНК
у эукариот
Деалкилирующий «суицидный» фермент, описанный
для Е. coli, обнаружен и в клетках человека. У эукариот
Дезоксинуклеозид +
трифосфаты
Основания матрицы РНК спариваются
с концом теломерной ДНК (матрица
представляет собой короткий отрсчок
длинной молекулы РНК. но
для наглядности здесь приведена
только ее масть)
Теломераза
Теломераза синтезирует фрагмент
теломерной ДНК, используя
включенную в нее РНК-матрицу
Новый повторяющийся фрагмент
5· С
3Έ
I
Τ
РНК-ма и а3
—■ ' Теломераза -+~
ч. >
Затем теломераза перемещается вправо
и повторяет процесс синтеза теломерной
ДНК фрагмент за фрагментом.
В результате образуются
сотни таких фрагментов
(я)
)тот промежуток заполняется
в процессе синтеза ДНК
Рис. 20.23. Механизм синтеза теломеразой теломерной ДНК
Темными линиями обозначена хромосомная ДНК
(«настоящая» хромосома), а светлыми - предсуществующая на одном
конце хромосомы теломерная ДНК. В теломеразу встроена
короткая молекула РНК, содержащая последовательность,
комплементарную повторяющемуся видоспецифичному
фрагменту. Фермент прикрепляется за счет взаимодействия РНК
с расположенными на конце основаниями предсуществующей
теломеры и добавляет (последовательно по одному основанию)
один повторяющийся фрагмент. Синтез всегда идет в
направлении 5'—> 3'. Фермент перемещается таким образом, что
матрица РНК постоянно спарена с концевыми основаниями
нового повторяющегося фрагмента, затем добавляется
следующий фрагмент и так далее. Заново синтезированная тело-
мерная ДНК служит матрицей для построения
противоположной цепи, так что теломера присутствует в обеих цепях
также имеет место эксцизионная репарация. Например,
у людей с генетическим заболеванием - ксеродермальным
пигментозом - эксцизионная репарация пиримиди-
новых димеров обычно не встречается. При УФ-воз-
действии на кожу в результате ковалентного связывания
двух соседних тиминовых оснований (одной цепи)
образуется тиминовый димер. Пребывание пациентов с
ксеродермальным пигментозом на солнце вызывает рак
кожи. Таким образом, эксцизионная репарация имеет
большое значение для человека. Описаны гены белков,
соответствующих Mut S и Mut L Ε. coli (см. с. 254);
установлено, что мутации в этих генах связаны с развитием
рака.
часть 4 Хранение и переработка информации
258
Системы репарации у эукариот играют большую
роль в сохранении целостности генома. Сегодня многие
исследователи выясняют связь старения человека и
снижения способности к репарации ДНК, в том числе с
увеличением частоты мутаций.
Транспозоны,
или прыгающие гены
Оказывается, некоторые фрагменты ДНК могут
перемещаться с одного места на другое в пределах одной
хромосомы или встраиваться в другую хромосому.
Существование прыгающих генов впервые было показано
Б. Мак-Клинток при изучении генетики кукурузы. Она
выяснила, что элементы регуляторного гена
перемещаются в геноме с одного места на другое и влияют
на экспрессию генов, приводя к появлению фенотипи-
ческих вариаций. Эта работа не привлекала внимания
в течение 30 лет, пока исследования на Е. coli не
подтвердили способность генов к передвижению и
внедрению в другое место генома, и Мак-Клинток была
присуждена Нобелевская премия. Способные к
перемещению последовательности ДНК получили название
транспозонов, или прыгающих генов.
Простейший тип транспозона Е. coli называется
инсерционной последовательностью (англ. insertion
sequence, IS). Он представляет собой отрезок ДНК,
включающий ген транспозазы - фермента,
ответственного за перемещение. На первый взгляд, присутствие
IS вызывает недоумение: ген, не кодирующий ничего,
кроме возможности перемещения с места на место!
IS может встроиться в любой участок хромосомы,
поскольку для него не существует специфичности
встраивания в определенные районы хромосомы. Когда транс-
позон встраивается в какой-то ген, то последний инак-
тивируется, так как в нем нарушается нуклеотидная
последовательность; однако частота перемещений
довольно низка. Кроме гена транспозазы инсерционная
последовательность содержит на концах инвертированные
концевые повторы. Они состоят из коротких
фрагментов, ориентированных как показано на рис. 20.24.
Возможны варианты механизмов транспозиций.
Один из них - предварительная репликация
транспозона, а не его перенос. Копия транспозона, покинув свое
исходное положение, встраивается в какое-то другое
место. Появление множества повторяющихся
последовательностей в геноме - это путь создания
благоприятных возможностей для гомологичной рекомбинации.
Возможно, что перегруппировки в хромосомах,
осуществляемые при участии транспозонов, чрезвычайно
важны для эволюционного процесса.
Является ли единственным
описанный механизм,
при помощи которого
синтезируется ДНК?
Известно, что для достижения одних и тех же целей
эволюция использовала различные пути. Например, для
синтеза цепей ДНК некоторые вирусы используют в качестве
ДНК хромосомы
Транспозон, имеющий
инвертированные
концевые повторы из
9 пар оснований. Номера
с 1 по 9 обозначают
поеледовател ьность
расположения оснований
Сайт-мишень (место встраивания IS)
■ATGCA·
■TACGT-
t
123456789
123456789
■ATGCA 123456789
■TACGT 123456789
987654321
987654321
Встраивание транспозона в сайт-мишень ДНК
вызывает дупликацию его нуклеотидной
последовательности
987654321ATGCA
987654321 TACGT
Прямые повторы в
сайте-мишени
Рис. 20.24. Встраивание транспозона (типа IS) в хромосому
Точки разрывов в последовательности ДНК-реципиента показаны стрелками
глава 20 Синтез и репарация ДНК
259
промотора белки вместо РНК. Однако ретровирусы
имеют принципиально другой механизм синтеза - у них
РНК генома копируется в ДНК (см. главу 23).
Интересно, что митохондрии имеют собственную ДНК, которая
должна реплицироваться при увеличении их
численности. В то же время в митохондриях нет систем
репарации ДНК. Предположительно это связано с тем, что их
хромосома значительно короче, и поэтому снижается
вероятность появления ошибок при репликации; а так как
в клетке содержится большое число митохондрий,
незначительное количество дефектов в них не столь
существенно. Однако отсутствие репарации означает, что
мутации в митохондриальной ДНК могут со временем
накапливаться и, возможно, обусловливать развитие
процессов старения. Известно большое количество
генетических заболеваний, связанных с мутациями митохонд-
риальных ДНК.
Дополнительная литература
Книги
Kornberg Α., Baker Т. Л. DNA replication. 2nd edn.,
W. Η. Freeman., 1992
Lewin B. Genes V. Oxford University Press, 1994
Singer M., Berg Ρ Genes and genomes: a changing
perspective. University Science Books, 1991
ДНК-полимераза
Kelman Ζ, Ο 'Donnel M. DNA polymerase III holoenzyme:
structure and function of chromosomal replicating
machine // Annu. Rev. Biochem.1995. Vol. 64. Ρ 171-200.
(Прекрасный обзор, посвященный сложному ферменту,
структуре его субъединиц, скользящим зажимам и их
роли в синтезе лидирующей и запаздывающей цепей.
Очень компактный обзор.)
Репликация
Coverley D., Las key R. A. Regulation of eukaryotic DNA
replication // Annu. Rev. Biochem. 1994. Vol. 63. P. 745-776.
(Описываются точки начала репликации и механизм
регуляции репликации в связи с клеточным циклом.
Обзор сложен по содержанию, но читается неплохо.)
LiJ.J. Once and once only// Curr. Biol. 1995. Vol. 5.
P. 472-475.
(Обсуждается важность взаимосвязи между клеточным
циклом эукариотической клетки и инициацией сайтов
начала репликации ДНК.)
Wyman С, Botcham M A familiar ring to DNA polymerase
processivity // Curr. Biol. 1995. Vol. 5. P. 334-337.
часть 4 Хранение и переработка информации
260
(Обзор, посвященный роли кольцевых скользящих
зажимов в репликации ДНК. Прекрасно иллюстрирован.)
Теломеры
Greider С. W., Blackburn Ε. Η. Telomeres, telomerase and
cancer// Sci. Amer. 1996. Vol. 274 (2). P. 80-85.
(Обсуждаются проблемы, связанные с репликацией
концов ДНК, укорочения ДНК и с защитой теломеразой
концевых сегментов хромосом. Обсуждается рольтело-
мераз в связи со старением и канцерогенезом.)
Репарация
Echols Η., Goodman Μ. Ε Fidelity mechanisms in DNA
replication // Annu. Rev. Biochem. 1991. Vol. 60. P. 477-511.
(Статья является интересным современным обзором,
посвященным вопросам избирательности встраивания
оснований ДНК-полимеразами, контроля правильности
считывания ДНК-полимеразой и мутаций, возникающих
при повреждении ДНК.)
Demple В., Karran P. Death of an enzyme: suicide repair
of DNA// Trends Biochem. Sci. 1983. Vol 8. P. 137-139.
(Обзор, посвященный ферментам, участвующим в деал-
килирующей репарации ДНК.)
Hammans S. R. Mitochondrial DNA and disease // Essays
Biochem. 1994. Vol. 28. P. 99-112.
(Обсуждение заболеваний, возникающих при
аномалиях митохондриальной ДНК.)
Вопросы к главе 20
1. Что такое репликон?
2. Какие топологические проблемы возникают при
разделении нитей родительской ДНК в ходе
репликации?
3. Как разрешаются проблемы, обозначенные в
предыдущем вопросе, у Е. coli и у эукариот?
4. Объясните при помощи схем действие топоизоме-
раз I и II.
5. У эукариот нет топоизомеразы, способной вводить
отрицательные сверхвитки в ДНК. В то же время у
эукариотической ДНК такие сверхвитки есть.
Объясните, как они образуются.
6. Что является субстратом для синтеза ДНК?
7. Почему при синтезе ДНК используется ТТР,
а не UTP, как в РНК?
8. А Может ли цепь ДНК синтезироваться с
использованием только четырех нуклеозидтрифосфатов?
Объясните свой ответ.
Б. В каком направлении происходит синтез ДНК?
Объясните свой ответ.
9. Какие термодинамические силы движут синтезом
ДНК?
10 Полимераза III E. coli является высокопроцессивной.
Объясните, что это значит, за счет чего достигается
высокая процессивность и причины ее возникновения.
11. Полимераза I E. coli является сложным ферментом.
Какой активностью обладают его субъединицы и
какова их роль в синтезе ДНК?
12 При помощи какого механизма ДНК-полимераза III
Е. coli достигает высокой степени точности при
синтезе ДНК?
13. Для достижения требуемого уровня точности
считывания активности полимеразы III E. coli
недостаточно. При встраивании неправильного нуклеотида его
необходимо заменить. Следовательно, репарирую-
щая система должна распознавать ошибочное
основание. Как это происходит? Существует ли такой
механизм в клетках человека?
14. Объясните, что такое тиминовый димер, как он
образуется и как репарируется.
15. Объясните, как укорачиваются хромосомы эукариот
при каждой последующей репликации.
16. Объясните, почему предотвращается укорочение
ДНК.
глава 20 Синтез и репарация ДНК
261
глава
21
Транскрипция гена - первый этап
в механизме контроля за биосинтезом
белка
В молекуле ДНК в виде последовательности из 4
азотистых оснований закодирована информация о
структуре белков. Одну аминокислоту кодирует
последовательность из 3 оснований. При комбинации 4 азотистых
оснований возможны 4-4-4 вариантов триплетов,
поэтому ограничения кодирующих возможностей нет. Ген
в биосинтезе белка непосредственно не участвует.
У эукариот ДНК окружена ядерной мембраной, а белок-
синтезирующий аппарат находится в цитоплазме. Каким
образом тогда ген управляет синтезом белка? Он
посылает из ядра в цитоплазму копии, в которых
закодирована информация (например, у Е. coli такая копия
поступает в цитоплазму непосредственно). Поскольку
скопированная информация представлена в виде
последовательности азотистых оснований, копия тоже
должна быть нуклеиновой кислотой. Она носит название
рибонуклеиновой кислоты (РНК), о которой речь шла
выше; с учетом функции ее называют матричной (или
информационной, или мессенджерной) РНК (для
краткости - мРНК).
Матричная РНК
Структура РНК
РНК - полинуклеотид, похожий на ДНК, но имеющий
свои особенности.
• Сахар в РНК представлен рибозой, имеющей во
втором положении ОН-группу, а не дезоксирибозой.
НОЛИ
D-Рибоза
ОН Η
2'-Дезокси-£-Рибоза
мРНК - одноцепочечная молекула, а не дуплекс двух
молекул, из чего можно заключить, что мРНК -
копия только одной из двух цепей ДНК гена.
В РНК 4 азотистых основания: А, С, G и U. Τ -
отсутствует. И U, и Τ образуют пару с А. Можно
рассматривать Τ как U, к которому присоединена
метальная группа (объяснение возможной причины ее
происхождения приведено на с. 256).
Если бы не эти различия, структуру одноцепочеч-
ной ДНК (см. с. 235) вполне можно было бы принять
за РНК: те же З1—» 5' фосфодиэфирные связи между
соседними нуклеотидами, на 5'-конце нет нуклеотид-
ного заместителя у 5'-ОН, а 3'-конец имеет свободную
З'-ОН-группу. Гидроксильная группа в 2'-положении
делает молекулу РНК химически более нестабильной
по сравнению с ДНК (см. с. 235). В разведенном
растворе щелочи РНК разрушается при комнатной
температуре, а молекула ДНК устойчива.
Как синтезируется мРНК?
«Строительными блоками» для синтеза мРНК служат
ATP, CTP, GTP и UTP, синтезируемые во всех клетках
(см. главу 18). В Е. coli синтез мРНК катализирует
ДНК-зависимая РНК-полимераза (для краткости-
РНК-полимераза). В клетках эукариот существуют три
таких полимеразы.
Чтобы происходил катализируемый РНК-полимера-
зой синтез РНК, т. е. на одной цепи-матрице нуклеоти-
ды собирались в определенной последовательности,
необходимо разделение цепей ДНК. На участке синтеза
РНК две цепи ДНК временно расходятся, а после
прохождения полимеразы они соединяются вновь.
Процесс синтеза РНК во многом схож с синтезом
ДНК: азотистое основание входящего рибонуклеотида
должно быть комплементарно основанию на матрице
ДНК (рис. 21.1).
РНК-полимераза, продвигаясь вдоль матрицы,
соединяет нуклеотиды в том порядке, который определен
матрицей ДНК. Как и в случае ДНК, синтез РНК всегда
А
"G РНК полимераза
С GTP\
Τ АТРч\
5'
CTP- Входящие
нуклеозид
|~G 3* трифосфаты
с э-1
Матричная
цепь ДНК
Нуклеозид
трифосфаты
из раствора
3'
Начало новой
цепи мРНК
Рис. 21.1. Копирование мРНК на матричной цепи ДНК
Вторая цепь ДНК не указана. Обратите внимание, что
разделение цепей ДНК - временное
часть 4 Хранение и переработка информации
262
осуществляется в направлении 5'—» 3\ Нуклеотиды
присоединяются к З'-ОН-группам, и поэтому элонгация
цепи происходит в направлении 5'—» 3\ Химическая
реакция, катализируемая полимеразой, заключается в
переносе ос-фосфорильной группы нуклеозидтрифосфатов
(прямо соединенной с рибозой) на З'-ОН-группу
предшествующего нуклеотида с высвобождением
неорганического пирофосфата. Последний гидролизуется с
образованием двух молекул Р/5 что делает эту реакцию
(рис. 21.2) сильно экзоэргонической.
РНК-полимераза способна инициировать новые
цепи, и ей не нужен праймер. В присутствии матрицы
РНК-полимераза может сразу синтезировать всю
молекулу мРНК. В этом заключается еще одно важное
отличие процесса транскрипции от синтеза ДНК, поскольку
ДНК-полимеразе всегда необходим праймер (см. с. 248).
Некоторые общие свойства мРНК
Обычная хромосома содержит тысячи различных
генов. Каждую молекулу мРНК кодирует один ген (или
небольшая группа генов у прокариот), поэтому в клетке
постоянно образуется множество различных мРНК
в соответствии с числом активных в данный момент
генов. По сравнению с длинной молекулой ДНК молекула
мРНК невелика. В цитоплазме многочисленные мРНК
управляют синтезом множества белков, закодированных
соответствующими генами.
Для данной живой клетки молекула ДНК
бессмертна. Время полужизни мРНК у эукариот - от 20 минут
до нескольких часов, а у бактерий - около двух минут.
Таким образом, для экспрессии гена (термин экспрессия
означает, что ген работает и белок синтезируется)
необходима постоянная транскрипция молекул мРНК.
Можно сказать, что ген «штампует» копию за копией, а РНК-
полимераза - его «штамповочная машина». Как только
ген прекращает образование мРНК, деградация
последней служит главным «выключателем» синтеза белка.
Клетки Е. coli ядерной мембраны не имеют, и гены
контактируют с цитоплазмой. У эукариот одна мРНК
всегда соответствует одному гену, а у прокариот одна
мРНК может нести информацию сразу о нескольких
белках. Ее называют полицистронной (генетический
термин цистрон обозначает ген, контролирующий одну
функцию). Кластеры генов, транскрибируемых в одну
полицистронную мРНК, обеспечивают
координированный синтез белков, функционирующих как единое
целое (например, ферментов одного метаболического
пути). Существуют и другие значительные различия
в образовании мРНК про- и эукариот, которые будут
рассмотрены ниже.
Некоторая важная терминология
Поток информации при экспресии гена происходит в
направлении:
(транскрипция) (трансляция)
ДНК ► мРНК ► белок .
Стрелки обозначают направление переноса информации.
цепьРНК
-о—р—о—р—о—р—о
I I I
о- о- о-
Входящие
нуклеотидг
основание
ОН ОН
\Точка присоединения
следующего нуклеотида
Рис. 21.2. Реакция, катализируемая РНК-полимеразой
глава 21 Транскрипция гена
263
При копировании ДНК в РНК происходит
переписывание (транскрипция) информации, поэтому
образование мРНК называют транскрипцией гена, или чаще
просто транскрипцией. О ДНК же говорят, что она
транскрибируется. Образовавшиеся молекулы РНК
называют транскриптами. Синтез белка на матричной
РНК называют трансляцией.
До сих пор мы обсуждали синтез мРНК как
копирование ДНК. Матрица цепи ДНК «копируется» только
комплементарным способом: путем спаривания по Уот-
сону-Крику азотистых оснований входящих рибонукле-
отидов с основаниями матрицы. Против А в матрице
становится U в копии и т. д. Ту цепь ДНК, которая
служит матрицей для синтеза мРНК, называют матричной
цепью, а другую - нематричной цепью.
Кроме того, широко используются термины:
кодирующая и некодирующая цепи, смысловая и несмысловая
цепи, хотя нередко различные авторы понимают их
по-разному. Единой согласованной терминологии не
существует, однако важно знать, что имеется в виду при
использовании того или иного термина. В этой книге
подразумевается, что мРНК несет информацию об
аминокислотной последовательности белка и таким образом
передает послание гена на трансляционную машину.
Последовательность азотистых оснований в мРНК
та же, что и в нематричной цепи ДНК (за исключением
замены Т—» U), поэтому мы будем называть нематричную
цепь кодирующей, или смысловой. Матричная же цепь
здесь определяется как несмысловая, или некодирующая
(рис. 21.3). У вирусов матричную цепь часто
называют минус-цепь, или (-)-цепь, а некодирующую -
плюс-цепь, или (+).
О чем пойдет речь дальше
До сих пор мы знакомились с экспрессией гена,
рассматривая ген только как матрицу ДНК. Однако есть
много других важных и интересных вопросов,
например: каким образом, в клетке происходит отбор генов,
подлежащих экспресии? Как РНК-полимераза «узнает»,
Нематричная цепь
... 5' С G AT G С AT 3' (кодирующая,
ДНК ч или смысловая)
* 3' G С ТА С G ТА 5' Матричная цепь
(некодирующая,
или несмысловая)
5'CGAUGCAU3' Цепь мРНК
Рис. 21.3. Схематичное изображение, показывающее на
примере мРНК термины «матричная» и «нематричная» для
цепи ДНК как переносчика информации
У вирусов матричную цепь часто называют (-)-цепью,
а некодирующую (+)-цепью
часть 4 Хранение и переработка информации
264
с какого места в транскрибируемом гене нужно начинать
копирование ДНК и где остановиться? Каким образом
контролируется скорость экспрессии гена? Один белок,
кодируемый своим геном, может образовываться в
больших количествах, а другой - в очень малых или не
образовываться совсем. Некоторые гены могут быть эксп-
рессированы только в определенное время. Как же это
достигается? Для ответа на поставленные вопросы мы
должны рассмотреть структуры реальных генов, точнее,
последовательности их оснований.
До сих пор мы рассматривали биохимию животной
клетки. Впрочем, различия многих метаболических
процессов, протекающих у прокариот и эукариот, невелики
и не затрагивают фундаментальных принципов. Однако
при переходе к экспрессии генов они становятся столь
значительными, что требуют самостоятельного
рассмотрения. Начнем с Е. coli.
Транскрипция гена Е. coli
Что определяется термином «ген»
у прокариот?
В главе 19 (см. с. 240) мы отмечали, что ген - это
фрагмент большой молекулы ДНК. Теперь нам предстоит
уточнить это определение.
Итак, ген функционирует только в качестве матрицы
для транскрипции молекул РНК, у которых
последовательность азотистых оснований соответствует
нематричной цепи ДНК. Ген - это фрагмент ДНК,
транскрибируемый в РНК. Большая группа генов кодирует
рибосомные и транспортные РНК.
Остальные гены ответственны за образование
молекул мРНК, которые и кодируют аминокислотные
последовательности соответствующих белков. На каждом
конце молекулы мРНК есть области, которые не
транслируются в белок. На 5'-конце находится нетрансли-
руемая область (НТО; в англоязычной литературе -
untranslated region, UTR), которая содержит
закодированный сигнал, необходимый для инициации
трансляции. Участок на З'-конце сигнализирует о терминации
трансляции. Таким образом, ген, помимо
закодированной информации о структуре белка, включает также
участки - сайты - ДНК, которые транскрибируются, но не
используются для кодирования данного белка.
Этим перечень участков ДНК, связанных с геном,
не исчерпывается. Примыкающий к 5'-концу гена
участок ДНК называют промотором. Он важен для
транскрипции гена, но сам в РНК не транскрибируется.
Противоположный З'-конец - терминаторная
область - необходим для терминации транскрипции.
В РНК он также не транскрибируется. Схематическая
структура гена приведена на рис. 21.4. Транскрипция
начинается со стартовой точки, которая служит матрицей
для первого нуклеотида мРНК. Матрица первого нукле-
отида обозначена как +1, а соседний нуклеотид с 5'-кон-
ца как -Ь Стартовая точка обозначена стрелкой,
указывающей направление транскрипции. Нуклеотиды,
расположенные по отношению к стартовой точке в сторону
5'-конца, обозначаются термином против хода, а в
сторону З'-конца - по ходу транскрипции. На рис. 21.4 один
конец гена помечен 5\ Это подводит нас к следующему
вопросу.
Промоторная Стартовая точка Участок терминации
область транскрипции транскрипции
-1
и.
ДНК
Это 5'-конец
гена
J,.
мРНК
5'-UTR содержит..-"" '
сигналы З'-UTR содержит
инициации Кодирующая белок сигналы терминации
трансляции область мРНК трансляции
Рис. 21.4. Структура прокариотного гена и его мРНК
5'-Конец гена относится к нематричной, или смысловой цепи.
В соседнем гене нематричной может быть другая цепь, а
транскрипция будет происходить справа налево
Что мы имеем в виду,
говоря о 5а-конце гена?
5'-Конец гена- это конец, содержащий промотор.
Поскольку синтез РНК всегда происходит в направлении
5'—» 3\ то 5'-конец гена всегда соответствует З'-концу
матричной цепи (см. рис. 21.3). Следует отметить, что
в физическом смысле концов нет: цепь ДНК
продолжается и в следующем гене.
5'-Конец гена на схеме хромосомы обычно
располагают слева. Цепь ДНК, являющаяся для одного гена
матричной, для другого может быть и нематричной
(смысловой). Использование цепи в качестве матрицы может
переключаться от одного гена к другому. Если
противоположная цепь используется таким образом на втором,
соседнем гене, транскрипция происходит в
противоположном направлении, так как синтез идет в
направлении 51—» 3'.
Фазы транскрипции гена
Существуют три фазы транскрипции гена: инициация,
элонгация и терминация. Наиболее сложной из них
является инициация.
Инициация транскрипции в Е. coli
В промоторах есть короткие последовательности
азотистых оснований, называемые боксами или элементами.
Это принятые термины, но последовательности
оснований в любом смысле не являются «боксами» и не
имеют ничего общего с элементами атома. В типичном
промоторе Е. coli есть два бокса: бокс Прибнова (названный
так по имени открывшего его автора), который
расположен около участка -10, и бокс, находящийся левее
на участке -35 по отношению к стартовой точке
транскрипции. Нуклеотидные последовательности боксов
приведены на рис. 21.5.
5'— ТАТААТ—3'
3' — АТАТТА —5',
г гт « Стартовая точка
Бокс Прибнова у
-35
TTGACA
15
транскрипции
«—16-19п.о.— ТАТААТ^ 5-9 п.о.^ГьТ
3'*
зг-сг-гаг'-зях
*-- * **Ь&Ъ.1У** .^Гай
Рис. 21.5. Консенсусные последовательности элементов
промотора Е. coli
Эти последовательности получили название
консенсусы ых - из-за частой встречаемости у различных генов
и малой вариабельности (хотя известны случаи
отсутствия некоторых из этих последовательностей).
Одиночные последовательности всегда приводятся в
направлении 51—» З1, но в гене есть вторая цепь ДНК. Таким
образом, хотя мы и говорим, что бокс Прибнова имеет
последовательность ТАТААТ, в действительности дело
обстоит так:
5" - ТАТААТ - 3'
3' - АТАТТА - 5'
Т. е. это двойная цепь, которая и распознается белками,
регулирующими процесс транскрипции.
Правильная инициация транскрипции чрезвычайно
важна, поскольку синтез мРНК должен начаться с точно
определенного нуклеотида на матрице. Каким же
образом РНК-полимераза оказывается в нужном месте?
Важная роль в этом процессе принадлежит боксу
Прибнова и элементу -35. ДНК-зависимая РНК-полимераза
Е. coli - большой белковый комплекс, сердцевину
которого формируют 4 субъединицы. Обладая сродством к
любому отрезку ДНК, к которому он может произвольно
глава 21 Транскрипция гена
265
присоединиться, фермент не может «самостоятельно»
узнать сайт инициации. При соединении с другим ци-
топлазматическим белком - сигма-белком, или сигма-
фактором - образующийся холофермент теряет
сродство к случайным последовательностям ДНК, но прочно
связывается с элементом -35 и боксом Прибнова
(несмотря на то, что азотистые основания ДНК внутри
дуплекса спарены по Уотсону-Крику, они могут быть
узнаны белками в бороздках ДНК; см. рис. 19.4). Это
должным образом ориентирует фермент. РНК-поли-
мераза может теперь разделить цепи ДНК и
инициировать синтез РНК. В месте разделения цепей ДНК
формируется «глазок» (длиной примерно 1,5 витка
спирали), благодаря которому азотистые основания
матричной цепи становятся доступны для соединения с нуклео-
зидтрифосфатами. Из них фермент образует несколько
первых фосфодиэфирных связей - происходит
инициация. В этот момент сигма-белок отходит, а полимераза
движется вдоль гена, синтезируя мРНК со скоростью
около 40 нуклеотидов в секунду. Так продолжается до
тех пор, пока фермент не достигнет терминатора. Что
вызывает отделение сигма-белка от фермента, до сих
пор неизвестно.
Расплетание ДНК
Для транскрипции гена необходимо временное
разделение двойной спирали ДНК. Бокс Прибнова,
характеризуемый высоким содержанием АйТи потому более
слабыми водородными связями, является местом
первоначального разделения цепей. ДНК расплетается
перед полимеразой и закручивается позади нее.
Отрицательная сверхспирализация ДНК облегчает этот процесс
(см. с. 244). Таким образом, временно образовавшийся
«глазок» движется вдоль гена вместе с полимеразой.
На коротком участке вновь синтезированная мРНК
спаривается с матричной цепью, но затем отделяется от нее.
На рис. 21.6 показано продвижение РНК-полимеразы
вдоль двойной спирали ДНК.
Терминация транскрипции
В конце некоторых генов прокариот находятся
последовательности, которые в транскрибируемой РНК
формируют структуру шпильки. Хотя молекула мРНК и
является одноцепочечной, ее термодинамические особенности
допускают спаривание оснований в пределах одной
цепи. Вновь синтезированная мРНК связана с
матричной цепью ДНК за счет спаривания оснований. Недалеко
от конца гена последовательность оснований в
транскрибированной мРНК способствует образованию
шпилечной структуры (рис. 21.7). Последовательность
оснований в этом месте содействует образованию
часть 4 Хранение и переработка информации
266
ДНК
закручивается РНК-полимераза F ^
"Zt Транскрипционный глазок
Хромосома § Движение
/ ч полимеразы
спираль РНК-ДНК Участок
5" мРНК длиной 12 п.о. синтеза РНК
Рис. 21.6. Схематичное изображение транскрипции ДНК
РНК-полимеразой Е. coli
Полимераза расплетает участок ДНК длиной около 17 пар
оснований, образуя транскрипционный глазок, который
продвигается вдоль ДНК. ДНК расплетается впереди полимеразы
и закручивается позади нее. Синтезированная РНК образует
с ДНК двойную спираль РНК-ДНК длиной около 12 пар
оснований
стабильных пар G-C за счет 3 водородных связей
в каждой. Сразу же вслед за шпилечной структурой
в транскрипте мРНК находится несколько остатков U,
слабо связывающих РНК с ДНК. Это, по-видимому,
облегчает отделение мРНК и терминацию транскрипции.
Шпилечная структура нарушает связывание мРНК
с матрицей: ведь если основания преимущественно
спариваются внутри цепи мРНК, они не могут
образовывать пар с матрицей ДНК. Серия остатков U облегчает
окончательное отделение мРНК из-за слабости
водородных связей между А и U. Каков бы ни был точный
механизм, эта шпилечная структура с последующими
остатками U приводит к терминации транскрипции.
У многих генов прокариот есть и второй способ
терминации транскрипции. В его осуществлении
участвует так называемый белок Rho, или фактор Rho,
необходимый для отделения мРНК от гибрида ДНК-РНК.
Фактор Rho присоединяется к транскрибированной
мРНК и двигается вдоль нее позади РНК-полимеразы.
G-C ...
g-c ;
G«C Область, богатая G-C
G-C
sG-C
мРНК „-ОС
--—·■< -■ - - —- —он
ииии
Рис. 21.7. Структура шпильки транскрипта РНК, который
обусловливает Rho-независимую терминацию
транскрипции гена
В участке терминации полимераза останавливается,
возможно, из-за трудности разделения богатого парами
G-C участка ДНК, что позволяет Rho-фактору догнать
полимеразу. Этот белок обладает расплетающей (хели-
казной) активностью, которая специфична в отношении
ДНК-РНК дуплекса, образующегося в ходе
транскрипции. Поэтому, догнав полимеразу, Rho-фактор отделяет
РНК-транскрипт, тем самым заканчивая транскрипцию.
мРНК управляет синтезом белка. У Е. coli она начинает
делать это еще до завершения образования полной
молекулы мРНК, поскольку транскрипция происходит при
непосредственном контакте с цитоплазмой.
Однако остается открытым вопрос о контроле за
транскрипцией генов в Е. coli.
Скорость инициации транскрипции гена
у прокариот
К конститутивно экспрессируемым генам относятся
гены, которые все время находятся во «включенном»
состоянии, и скорость их экспрессии избирательно
не модулируется. Такие гены кодируют ферменты и
другие белки, постоянно необходимые для
жизнедеятельности клетки. Однако иногда потребность в некоторых
из таких конститутивных белков может сильно
возрастать. Основное влияние на скорость экспрессии генов
у бактерий оказывает скорость образования мРНК, а это
в значительной степени определяется частотой
инициации транскрипции данного гена. У разных генов она
может сильно варьировать, так как они имеют
промоторы разной «силы». «Сильный» промотор будет часто
инициировать транскрипцию, что приведет к образованию
многочисленных мРНК транскриптов и большого
количества соответствующего белка. «Слабый» промотор
оказывает противоположный эффект. Сила промотора
является функцией последовательности оснований в
боксах Прибнова и -35, расстояния между ними и
основаниями в участке от +1 до +10. Более высокое сродство
полимеразы к этим областям и сила промотора могут
коррелировать и не так строго.
Контроль транскрипции различными
сигма-факторами
Сигма-факторы являются одним из эффективных
«инструментов» контроля целых блоков генов у
прокариот. При определенных условиях обычный
сигма-фактор заменяется иным, который побуждает полимеразу
инициировать транскрипцию другого набора генов. К
таким условиям относятся: 1) споруляция бацилл, когда
после получения сигнала о неблагоприятных условиях
окружающей среды образуется новый сигма-фактор; это
вызывает экспрессию ряда генов, ведущих к споруляции;
2) азотное голодание, при котором образуется особый
фактор; 3) тепловой шок (внезапный подъем
температуры), после которого у Е. coli временно увеличивается
синтез, стабильность и активность другого
сигма-фактора, чей уровень в норме очень низок. Этот фактор
управляет транскрипцией генов, кодирующих «белки
теплового шока», которые защищают клетку от его
последствий.
Lac-оперон
Каким образом осуществляется
регуляция
отдельных генов £. coli?
Бактерии вынуждены приспосабливаться к постоянно
меняющимся условиям окружающей среды. К тому же
среди бактериальных популяций существует жестокая
конкуренция. Типичная клетка Е. coli в оптимальных
условиях делится каждые 20 минут. Если штамм способен
сократить это время на 1 минуту, он очень быстро
станет преобладать над другими штаммами.
Неэффективность биохимических процессов в условиях такой
конкуренции недопустима.
На образование ферментов затрачиваются ресурсы
и энергия, и если в данный момент эти ферменты не
нужны, Е. coli не будет их синтезировать. Как уже
отмечалось, конститутивные ферменты образуются
постоянно при любых обстоятельствах. Поскольку глюкоза
является основным сахаром, а другие сахара могут пойти
по пути метаболизма глюкозы, ферменты, метаболизи-
рующие глюкозу, - конститутивны. Клетка
«предполагает», что они будут нужны всегда, поэтому промоторы
кодирующих их генов не имеют «выключателя», они
постоянно «включены».
Однако Е. coli может столкнуться с другими саха-
рами, например, с присутствующим в молоке дисаха-
ридом лактозой. Если это единственный доступный
источник углерода, его утилизация позволит клетке
выжить. Для этого необходим фермент β-галактозидаза,
названный так потому, что лактоза - это β-галактозид,
который при гидролизе распадается на галактозу
и глюкозу (рис. 21.8). Для транспорта лактозы в
клетку необходим дополнительный транспортный белок
β-галактозидпермеаза. Третий белок - галактозид-
трансацетилаза - защищает клетку от неметаболизи-
рующихся, потенциально токсичных β-галактозидов.
В норме эти белки синтезируются в течение нескольких
глава 21 Транскрипция гена
267
минут. Они не нужны клетке до момента встречи с
лактозой, но когда она оказывается единственным
источником энергии, происходит практически мгновенная
активизация синтеза этих трех белков - и клетка может
использовать лактозу в качестве источника углерода
и энергии. Однако, если помимо лактозы присутствует
и глюкоза, продукция этих ферментов невыгодна: зачем
образовывать глюкозу внутри клетки, когда ее и так
много в окружающей среде? В этом случае клетка
«игнорирует» лактозный сигнал и не продуцирует указанные
выше ферменты. Эта регуляция, к которой мы сейчас
переходим, осуществляется на уровне инициации
транскрипции гена.
Структура /ас-оперона £. coli
Для начала несколько терминов: синтез белка в ответ
на химический сигнал называется индукцией, а само
химическое вещество - индуктором. Подавление
синтеза белка называется репрессией.
Как уже отмечалось ранее, гены эукариот
представляют собой одиночные структуры. Многие из них
сгруппированы, и такая группа может находиться под
транскрипционным контролем одного промотора. РНК-поли-
мераза транскрибирует всю группу, создавая молекулу
полицистронной мРНК, которая кодирует несколько
белков. Такая группа генов, контролируемая одним
промотором, называется опероном, а промотор - часть этого
оперона. Гены β-галактозидазы, лактозопермеазы
и трансацетилазы принадлежат к такому оперону.
Их часто обозначают как ζ, у и а соответственно.
Существует также /-ген (i обозначает индуцибелъ-
ность), кодирующий белок /ас-репрессор. Репрессор
может связываться с участком ДНК, который
называется оператором. Наконец, есть фрагмент ДНК, с
которым способен связываться белок: рецептор с AMP.
Он назван белком-активатором катаболизма (САР),
потому что участвует в индукции и других ферментов,
осуществляющих катаболизм субстратов. Примерная
организация этих элементов приведена на рис. 21.9.
Теперь рассмотрим собственно механизм контроля.
Следует заметить, что сам по себе /яопромотор - слабый
промотор, и РНК-полимераза для инициации
транскрипции практически не связывается с ним. Без посторонней
помощи транскрипция с /яооперона осуществляется
только на низком уровне. Помощь приходит в виде
САР-белка, который связывается с матрицей на
фрагменте, соседствующем с местом связывания полимеразы.
Связывание САР-белка вызывает изгиб двойной спирали
на участке присоединения, что усиливает свойства
промотора. Однако САР не присоединяется до тех пор, пока
не вступит в контакт с сАМР, который является аллосте-
рическим регулятором этого белка. сАМР образуется
в клетке только при низком уровне глюкозы. Когда
содержание глюкозы велико, сАМР в клетке Е. coli мало,
САР-белок не связывается с опероном, так же как
и РНК-полимераза, и поэтому транскрипция /яооперона
минимальна. Это показано на рис. 21.10, а.
но—сн2
НО
но—сн2
ОН
N
он
он
Лактоза
(β-галактозид)
ОН
Н70
но—сн2
НО
ОН
ОН
но—сн2
г VH
ОН ' ОН
Галактоза
Минорная реакция
\ необходимая для
\ индукции /ас-оперона
о—сн2
ОН
Глюкоза
ОН НО
Аллолактоза
Рис. 21.8. Реакции, катализируемые β-галактозидазой
Фермент, участвующий в метаболизме лактозы, часто называют лактазой
часть 4 Хранение и переработка информации
268
Участок lac
связывания САР Промотор Оператор
| /-ген [ JJ р| | z-ген [ у-ген | д-ген [
I Транскрипция I
мРНК Полицистронная мРНК
I Трансляция 111
/ас-белок- β-Галакто- Пермеаза Ацетилаза
репрессор зидаза
Рис. 21.9. Структура /ас-оперона
/-ген кодирует /яс-белок-репрессор
Всегда ли /яс-оперон транскрибируется при низком
уровне глюкозы? Нет, для транскрипции необходимо
присутствие лактозы. В отсутствие лактозы /яс-белок-ре-
прессор присоединен к оператору, блокирует РНК-поли-
меразу, и транскрипции не происходит (рис. 21.10, б).
Ltfopenpeccop - аллостерический белок. В отсутствие
лактозы в окружающей среде он имеет высокое сродство
к оператору. Если в среде есть лактоза, низкий уровень
активности пермеазы обеспечивает попадание
небольших количеств сахара в клетку. В ходе гидролиза
лактозы небольшое количество этого дисахарида
превращается в изомер- аллолактозу (см. рис. 21.8). Она
связывается с белком-репрессором, и в результате алло-
стерических изменений последний диссоциирует от
оператора, тем самым разблокируя оперон.
Почему индуктором является аллолактоза, а не
лактоза? Возможно потому, что аллолактоза, будучи
побочным продуктом β-галактозидазной реакции, образуется
в достаточном для индукции количестве только при
высоких концентрациях лактозы. Это предотвращает
индукцию в присутствии следовых количеств лактозы.
Использование аллолактозы (а не лактозы) в
качестве индуктора крайне целесообразно. Если бы
индуктором являлась лактоза с низким сродством к бел-
ку-репрессору, то индукция была бы невозможна, так
как индуцированная β-галактозидаза поддерживала бы
низкий уровень лактозы независимо от концентрации
последней вне клетки. Когда вся лактоза будет
исчерпана, β-галактозидаза станет использовать и аллолактозу,
которая также является субстратом этого фермента,
обеспечивая тем самым механизм терминации
индукции. Система будет, таким образом, постоянно
тестировать скорость поступления лактозы, решая, стоит
продолжать индукцию или нет.
После освобождения оператора РНК-полимераза
может свободно двигаться по оперону, образуя полицист-
ронную мРНК (рис. 21.10, в), трансляция которой в
конечном итоге и приводит к синтезу трех ферментов.
а Высокая концентрация глюкозы, нет сАМР, нет
лактозы, нет транскрипции /ас-оперона
Участок связывания САР ► Старт транскрипции
Промотор
у-ген
в отсутствие индуктора
белок-рецептор связан с
опероном
САР-белок в отсутствие Не взаимодействует эффективно если
сАМР не связывается с ДНК сАМР не связался с САР-белком
б Низкая концентрация глюкозы, высокий уровень сАМР
комплекс САР—сАМР связывается с САР-связывающим
участком ДНК
Участок связывания САР
| Промотор I Оператор! z-ген | y-i
САР [рНК-пошшсраза!
сАМР
в Низкая концентрация глюкозы, высокий уровень с AMP,
присутствует лактоза, комплекс белок—репрессор—аллолактоза
отсоединяется от оператора, происходит транскрипция оперона
Участок связывания САР ι ·*
Промотор Оператор z-ген
САР РНК-полиме аза
у-ген
Транскрипция
сАМР
_..в присутствии индуктора аллоктозы
/ас-репрессор отсоединяется
Аллолактоза
Рис. 21.10. Экспрессия /ас-оперона
а - При высоких концентрациях глюкозы отсутствует сАМР,
который способствует связыванию САР-белка, что
необходимо для присоединения РНК-полимеразы к промотору;
б - при низких концентрациях глюкозы, но в отсутствие
лактозы, происходит связывание САР-белка, что способствует
присоединению РНК-полимеразы к промотору. Однако
транскрипция все еще не происходит, поскольку /яс-репрессор связан
с оператором, блокируя движение полимеразы;
в - аллолактоза связывается с репрессором, вызывая
отделение последнего от оператора. Начинается транскрипция (в
присутствии лактозы образуется небольшое количество ее
изомера - аллолактозы, которая, собственно, и является индуктором).
САР-белок - активатор катаболизма
На примере лактозного оперона был впервые
расшифрован механизм регуляции генов прокариот,
который, как оказалось, применим ко многим
метаболическим путям. Триптофановый оперон (trp-оперон) клеток
Е. coli, содержащий 5 структурных генов, необходим
для образования трех ферментов, участвующих в
синтезе триптофана. Он контролируется Гф-белком-репрессо-
ром. Будучи связанным с триптофаном, этот белок
блокирует оператор, препятствуя транскрипции мРНК.
глава 21 Транскрипция гена
269
Второй механизм регуляции ίφ-оперона вступает
в действие в отношении тех молекул РНК-полимеразы,
которым удается преодолеть репрессорный блок при
высокой концентрации триптофана. Этот механизм
называется аттенуацией.
У прокариот трансляция мРНК начинается задолго
до завершения образования полной молекулы (после
синтеза небольшого участка), и поэтому полимеразу
сопровождает рибосома, осуществляющая трансляцию
(механизм трансляции подробно изложен в следующей
главе). Рибосома - это нуклеопротеиновая частица,
которая движется вдоль мРНК, добавляя остатки
аминокислот к растущей полипептидной цепи. Оперон
первоначально кодирует так называемый лидерный пептид
длиной 14 аминокислот. Он отличается от лидерного
пептида, присоединяемого к определенным белкам (см.
главу 22). Лидерный пептид аттенуации разрушается сразу
же после образования. Он содержит 2 следующих один
за другим остатка триптофана. Если молекул триптофана
много, рибосома присоединяет их и движется дальше
вдоль мРНК; если же триптофана недостаточно, то
рибосома останавливается на триплетах триптофана в
области аттенуации. Когда уровень триптофана позволяет
рибосоме перейти этот участок при трансляции, пары нуклео-
тидов, образующиеся внутри частично синтезированной
молекулы мРНК, формируют терминаторную шпильку -
и транскрипция преждевременно заканчивается. Если
же рибосома находится в области аттенуации (в
отсутствие триптофана), она взаимодействует со
спаривающимися основаниями, что позволяет полимеразе
завершить транскрипцию оперона. Таким образом, уровень
транскрипции регулируется содержанием триптофана.
Чем триптофана меньше, тем больше молекул полимера-
зы способны провести полный синтез мРНК.
Известно, что аттенуация контролирует работу
шести оперонов, ответственных за биосинтез
специфических аминокислот. Среди них только ίφ-оперон
обладает репрессорным контролем. У всех шести оперонов
есть один и тот же чувствительный метод определения
уровня своей аминокислоты в клетке, который
заключается в присутствии множественных остатков данной
аминокислоты в лидерном пептиде. В гистидиновом
опероне этот пептид содержит 7 остатков гистидина.
Механизм контроля /яс-оперона (и других оперонов)
у бактерий подробно изучен. Он работает по-военному
четко: САР связывается и способствует присоединению
полимеразы, белок-репрессор связывается и блокирует
транскрипцию, а индуктор ее разблокирует. Контроль
транскрипции генов эукариот более сложен. Однако
сначала нам необходимо рассмотреть процесс
транскрипции у эукариотических генов.
часть 4 Хранение и переработка информации
270
Транскрипция генов
и ее контроль у эукариот
Основные процессы,
обусловливающие образование мРНК
у эукариот
У эукариот составные части для синтеза РНК те же, что
и у прокариот. Поэтому рис. 21.1, иллюстрирующий
синтез РНК из 4 рибонуклеотидов на основе матрицы ДНК,
полностью применим и для РНК эукариот.
ДНК-зависимая РНК-полимераза, которая транскрибирует гены,
кодирующие эукариотические белки, называется
РНК-полимеразой II. (РНК-полимеразы I и III будут
рассмотрены в главе 22, где описана транскрипция
генов молекул РНК.)
Процесс инициации транскрипции генов эукариот
сложен и будет рассмотрен в разделе, посвященном
регуляции. Допустим, что инициация уже произошла.
Это позволит нам познакомиться с образованием РНК
у эукариот, которое отличается от разобранного нами
выше процесса у прокариот.
Терминация транскрипции у эукариот
До сих пор непонятно: каким образом РНК-полимераза II
осуществляет терминацию транскрипции у генов
эукариот? Ведь в эукариотических генах нет ничего
похожего на то, что соответствовало бы прокариотическим
сигналам терминации. Когда РНК-полимераза II достигает
З'-ОН-конца гена, она синтезирует последовательность
AAUAAA, кодируемую матричной цепью; после этого
транскрипция продолжается, и только потом фермент
каким-то образом завершает свою работу. Особый
фермент отщепляет транскрипт РНК (за пределами уже
упоминавшейся последовательности), а затем другой
фермент, независимый от матрицы, осуществляет поли-
аденилирование, присоединяя (с использованием АТР)
примерно 200 адениновых нуклеотидов, образующих
поли-А-«хвост». У мРНК гистоновых белков такого
хвоста нет, поэтому, возможно, он не имеет существенного
значения для трансляции. Некоторые
экспериментальные данные позволяют преположить, что полиадени-
лирование обеспечивает стабильность мРНК в клетке.
РНК-полимераза II кэпирует
транскрибируемую РНК
Сразу же после инициации синтеза РНК первичный
транскрипт РНК гена эукариот претерпевает
модификацию с 5'-конца, или кэпирование. На 5'-конце РНК есть
трифосфатная группа: первый нуклеотидтрифосфат,
включенный в РНК, просто принимает нуклеотид на
свою З'-ОН-группу, причем исходная трифосфатная
группа остается неизменной. Концевой фосфат этой группы
удаляется, его место занимает остаток GMP,
переносимый otGTP (рис. 21.11). 5'-Трифосфатная связь
встречается очень редко. Затем происходит метилирование G
в положении N-7, а также 2'-ОН второго и иногда
третьего нуклеотида. По причине формирования кэпа на
первом нуклеотиде РНК стартовая точка гена (нуклеотид +1)
часто обозначается как кэп-сайт. Считается, что кэп
защищает конец мРНК от атаки экзонуклеазы и
участвует в инициации трансляции.
Это различие в образовании мРНК у про- и эукари-
от не единственное, так как большинство генов эукариот
относятся к прерывистым генам.
Ы7-метилгуаниновая
группа
СНз
5 '-5 '-трифосфатная
связь
основание
Оставшаяся цепь РНК
Рис. 21.11. Структура 5'-кэпа у мРНК эукариот
[Сонцевой нуклеозидтрифосфат первичного транскрипта РНК
5 ходе реакции с GTP превращается в дифосфат, при этом
выделяется пирофосфат. Затем происходят реакции
метилирования (третья метильная группа может быть присоединена
к 2'-ОН следующего нуклеотида первичного транскрипта).
После чего кэпированный первичный транскрипт
достраивается до мРНК
Что такое прерывистые гены?
За исключением коротких нетранслируемых 51- и
3'-областей, протяженность мРНК прокариот и эукариот
пропорциональна размеру кодируемых ими белков (или,
в случае полицистронных мРНК, - нескольких
кодируемых белков). Это утверждение справедливо для
первичного транскрипта мРНК бактерий, однако для
большинства генов эукариот первичные транскрипты намного
длиннее (иногда в 10 раз), чем можно было бы ожидать.
Последнее обусловлено тем, что транслируемая в белок
последовательность оснований РНК в молекуле мРНК
разделена на несколько частей, связанных
промежуточными участками, не кодирующими аминокислотные
последовательности. Последовательности ДНК, с
которых транскрибируются такие фрагменты, не несущие
информации о структуре белков, получили название инт-
ронов, а кодирующие белок последовательности -
экзонов. В гене человека может быть от 2 до 50
нитронов (рис. 21.12, а), длина которых варьирует от 50
до 20 000 пар оснований. Длина экзонов обычно не
превышает 1000 пар оснований. Процесс удаления
нитронов из первичного транскрипта и объединения экзонов
с образованием зрелой молекулы мРНК называется
сплайсингом (рис. 21.12,6). Хотя и принято считать, что
интроны обычно не несут никакой информации,
известно, что некоторые из них, например энхансеры,
содержат регуляторные последовательности (см. ниже).
„ „ Интроны
Первичный транскрипт..--- ч
5'-кэп Экзон
Экзон
Экзон АААААА(А)и
Сплайсинг
мРНК
it Удаленные интроны
5'-кэп I Экзон | Экзон | Экзон 1 АААААА(А)и
Трансляция
Белок
Рис. 21.12. Первичный транскрипт РНК-полимеразой II
гена эукариот
а - Интроны после кэпирования и добавления «хвоста» поли-А;
б - удаление интронов с образованием зрелой мРНК
называется сплайсингом
Механизм сплайсинга
Удаление интронов из первичного транскрипта РНК
и соединение экзонов в молекулу мРНК - непростая
задача. Ключом для ее решения является реакция
трансэтерификации, в ходе которой фосфодиэфирная
связь переносится на другую —ОН-группу. При этом
глава 21 Транскрипция гена
271
не происходит ни гидролиза, ни значительной потери
энергии:
X R
О
I
0=Р-СГ
I
о
I
Υ
+ R-OH
X
I
он
о
I
0=Р-СГ
I
о
I
Υ
Если Χ-Υ - цепь РНК, то она будет разорвана.
Теперь мы подошли к вопросу созревания (процес-
сингу) РНК - сплайсингу. Сочленения экзона с
нитроном «помечены» консенсусными последовательностями.
Все интроны начинаются с GU, а заканчиваются AG,
хотя сами консенсусные последовательности длиннее.
В приведенной реакции ROH в действительности
является 2'-ОН-группой адениннуклеотида в интрон-
ной цепи (рис. 21.13). Эта группа атакует 5'-фосфат
G-нуклеотида в месте соединения, образуя лассоподоб-
ную структуру. В результате происходит разрыв цепи с
З'-конца экзона 1 и освобождение З'-ОН, который
атакует 5'-конец экзона 2, соединяя оба экзона.
У большинства эукариот сплайсинг в ядре
катализируют сложные рибонуклеопротеиновые частицы -
сплайсосомы. Молекулы РНК этих частиц обозначают
мяРНК - малая ядерная РНК.
По-видимому, мяРНК образуют гибридные
молекулы путем спаривания с основаниями консенсусных
последовательностей в местах сплайсинга на первичном
транскрипте РНК и определяют местоположение
подлежащих удалению интронов. Мутация в консенсусной
последовательности может привести к неправильному
5'
| он
Экзон 1 pGU д
Первичный 1
транскрипт РНК ♦
fG*p^
'l· i~
OH U^A -
AGp Экзон 2
*,7,^,т<^.->-«*- ;. _w
AGp
A
J\^ *# Qm ρ ^
Экзон 1 I Экзон 2 I I
I \ ^ AG
I ^U" A^*й&*'!'й?чк,й*
мРНК Удаленный интрон
Рис. 21.13. Два этапа механизма сплайсинга мРНК
Поскольку сплайсинг осуществляется путем переноса фосфо-
эфирных связей, он не сопровождается потреблением энергии
сплайсингу или к невозможности удаления интрона.
При генетическом заболевании β-талассемии не
происходит образования нормальных количеств β-цепи
гемоглобина по причине замены с 5'-конца интрона G на А,
и поэтому первичный транскрипт не преобразуется
в зрелую мРНК должным образом.
У простейших Tetrahymena точный сплайсинг
осуществляется без помощи белков самим РНК-транскрип-
том. На биохимиков этот факт произвел впечатление
разорвавшейся бомбы: молекула РНК сама катализирует
специфические химические реакции?! Ведь до этого
подобные функции приписывали только белкам.
Поэтому данное открытие ознаменовало начало новой эры
в биохимии РНК и, кроме того, дало дополнительные
аргументы в пользу некоторых эволюционных теорий,
согласно которым первые формы жизни основывалась
на РНК, а белки появились позднее.
Таким образом, первичный транскрипт
кодирующего белок гена эукариот, образованный с участием РНК-
полимеразы II, кэпируется, «подрезается», снабжается
хвостом, подвергается сплайсингу - и только после
этого выходит из ядра в виде зрелой мРНК.
Каков биологический статус интронов?
Биологический смысл существования интронов
заключается в том, что они облегчают эволюцию. Подобно тому
как исследования структуры белка выявили домены,
генетические исследования выявили наличие экзонов: было
обнаружено, что последние часто кодируют дискретные
домены белка, хотя это соответствие и не всегда полное.
Как уже отмечалось ранее (см. с. 47), считается, что
новые белки появлялись в процессе эволюции в результате
«тасования доменов». Согласно этой концепции, один
и тот же домен может использоваться повторно,
объединяясь с другими, так что новое семейство белков
составляется из уже существующих доменов. Разделение
частей гена на экзоны, кодирующие дискретные белковые
домены, облегчает их тасование. Последнее
обеспечивает более высокую скорость образования новых белков
за счет рекомбинации, а не точечных мутаций ДНК,
ведущих к заменам отдельных аминокислот.
Роль интронов в тасовании экзонов нуждается
в разъяснении. Тасование экзонов может происходить
в результате перегруппировки хромосомы.
Последовательность ДНК, кодирующая белковый домен, должна
оставаться интактной, и существование интронов с
каждой стороны экзона может облегчить успешную
транспозицию. Перестановка внутри области, кодирующей
белковый домен, вероятно, нарушит его структуру и будет
нежизнеспособной, поскольку каждый белковый домен
(см. с. 47) представляет собой автономную структуру,
часть 4 Хранение и переработка информации
272
которая может существовать в качестве дискретного
объекта Так как интроны не несут кодирующих белок
последовательностей, миграция генов сопровождается,
вероятно, меньшим риском повреждения доменов и,
следовательно, более успешным тасованием экзонов.
Каково происхождение
прерывистых генов?
Существует две гипотезы. Согласно первой,
непрерывный ген прокариот примитивен, а интроны появились
в ходе эволюции позднее (модель «поздних интронов»).
Но это не объясняет происхождение интронов.
Вторая гипотеза представлена в экзонной теории
генов — моделью «ранних интронов». Она постулирует
примитивность интронов, которые, возможно,
представляли собой слившиеся, нетранскрибируемые
фланкирующие области древних минигенов - «строительного
материала» современных генов. Тот факт, что молекулы
РНК могут обладать способностью к самосплайсингу,
снимает вопрос о необходимости существования слож-
ног механизма сплайсинга на ранних этапах эволюции.
В соответствии с этой гипотезой, непрерывные гены
прокариот рассматриваются как более поздний
результат эволюции, облегчающий быстрое деление клеток.
Однако, если интроны были встроены в древние
непрерывные гены беспорядочно, некоторые части генов,
кодирующие белковые домены, могли бы приобрести
интроны случайно, и они, удачно помещенные с каждой
стороны этих генов, обеспечили бы успешное тасование
экзонов. Если допустить, что подобное тасование дало
эволюционное преимущество, то, вероятно, многие из
современных генов, образовавшихся в результате
тасования экзонов, должны автоматически демонстрировать
соответствие между белковыми доменами и экзонами.
В свете этого именно тасование, а не что-либо иное,
привело к соответствию экзонов и доменов. Впрочем,
это соответствие проясняет происхождение интронов.
Исследование нескольких древних генов,
кодирующих белки, сохраненные в течение длительного
эволюционного периода, не выявило соответствия между эк-
зон-интронной структурой и структурными свойствами
белков. Вероятно, эти факты дают аргументы против
модели «ранних интронов» и указывают на
примитивность непрерывных генов. Заметим, что тасование
экзонов не оспаривается ни одной из гипотез. Это предмет
другого обсуждения, не связанного с происхождением
инттюнсв.
Прерывистые гены у прокариот могли бы создать
сложные проблемы со сплайсингом, поскольку мРНК
транслируется рибосомами с 5'-конца еще до того, как
завершен синтез З'-конца.
Альтернативный сплайсинг
Существует одно хорошо известное преимущество,
которое дают прерывистые гены - альтернативный
сплайсинг. При типичном сплайсинге все экзоны
первичного транскрипта соединяются вместе с
образованием зрелой мРНК. Однако известно много случаев,
когда сплайсинг происходит по различным схемам: одна
группа экзонов образует одну мРНК, а другая группа того
же транскрипта гена - другую, что приводит в конечном
счете к появлению различных белков. Такой механизм
может быть применен для образования белков,
необходимых в различных тканях или в одной ткани, но в
разное время.
Механизм транскрипции
генов эукариот и ее контроль
Хотя инициация транскрипции генов эукариот
предшествует транскрипции, процессингу и т. д., мы
отложили обсуждение этой темы до настоящего момента,
поскольку она сложна и неотделима от проблемы контроля
за активностью гена.
Распаковывание ДНК для транскрипции
Напомним, что для размещения в ядре громадная молек-
ла эукариотической ДНК должна быть хорошо
упакована (см. с. 237). Перед транскрипцией (или во время нее)
структура ДНК избирательно распаковывается в
транскрибируемых участках. Известны два факта,
свидетельствующие об избирательном распаковывании ДНК. Во-
первых, в политенных хромосомах слюнных желез
насекомых «распаковывание» ДНК можно увидеть в
световом микроскопе. Поскольку эти уникальные
хромосомы использовались в классических исследованиях,
рассмотрим их в первую очередь.
Клетки слюнной железы личинок фруктовой мушки
Drosophila велики и имеют необычную структуру.
Хромосомы реплицируются около 1000 раз без деления
клетки и лежат выстроившись в линию, удлиненные
и плотно упакованные- Такая упорядоченная структура
под микроскопом имеет вид полос. В отдельной
хромосоме их увидеть нельзя, однако при увеличении в
тысячи раз они становятся различимыми. В процессе
развития личинки экспрессируются последовательные
группы генов, и можно наблюдать разуплотнение
специфических полос (так называемых хромосомных пуфов)
в ходе транскрипции ДНК (рис. 21.14).
глава 21 Транскрипция гена
273
Полосы на хромосоме,
видимые в световой
микроскоп
t—
Хромосомный пуф
Рис.21.14. Схематичное изображение хромосомных пуфов транскрипционно-активного хроматина политенных
хромосом слюнных желез насекомых
Во-вторых, в экспериментах in vitro показано, что
транскрипция генов эукариот связана с разрыхлением
упаковки ДНК. Оказалось, что транскрипционноак-
тивные области хроматина становятся более
восприимчивыми к добавлению ДНКазы - фермента, гидролизу-
ющего ДНК. Известно, например, что гены глобина
становятся особенно чувствительными к действию ДНКазы
только в хроматине клеток, синтезирующих глобин,
и только в момент активного транскрибирования.
Необходимость генетического контроля
у дифференцированных эукариот
В клетке Е. coli имеется около 4000-5000 генов. В
человеческой - 50 000-100 000. В клетке Е. coli
большинство генов или все они экспрессируются во время
деления. Несмотря на то, что /яооперон, например, активен
лишь при низкой концентрации глюкозы и в присутствии
лактозы, каждой клетке Е. coli, в отличие от эукариот,
заведомо необходима транскрипция всех генов.
У эукариот индукция определенных генов
происходит под действием химических индукторов. В печени,
например, фенобарбитал вызывает увеличение
количества метаболизирующих его ферментов. Встречается
и репрессия генов: холестерин, например,
репрессирует первый фермент метаболического пути своего
синтеза (см. с. 146). В большинстве своем подобные
эффекты не столь критичны для жизнедеятельности
клетки как, скажем, в случае /яс-оперона, но все равно
очень важны.
У животных имеется целый ряд гормонов и других
регулирующих агентов, которые вызывают селективную
модуляцию синтеза специфических белков, контролируя
транскрипцию их генов. Сложность таких регуляторных
эффектов очень велика, и действительно, активность
гена может контролироваться различными факторами
(см. главу 26). Столь сложной регуляции у Е. coli не
найдено.
У эукариот различные гены экспрессируются в
разные моменты времени. Некоторые из них, так
называемые гены домашнего хозяйства, экспрессируются
постоянно и во всех тканях. Ферменты гликолиза,
белки, необходимые для синтеза ДНК и белка, и т. д.
нужны любым клеткам, так что гены «домашнего хозяйства»
экспрессируются практически в каждой, кроме высоко
специализированных клеток, например зрелых
эритроцитов. Другие белки необходимы только в
специализированных тканях: незрелые красные кровяные клетки
образуют гемоглобин; есть белки, специфичные для
печени, мышц, почек и т. д. Каждый тип клеток одного
организма содержат одинаковый набор генов. Это
означает, что в мышечной клетке, например,
экспрессируются только определенные гены. Однако проблема
экспрессии тканеспецифичностью не исчерпывается: ведь
в ходе эмбрионального развития на ранней стадии
процесса дифференциации требуются одни гены, другие
начинают работать позднее, в ходе развития отдельных
тканей. Из этого следует, что гены эукариот, с одной
стороны, могут экспрессироваться с постоянной
скоростью конститутивно, с другой - подвержены
множественному контролю различными гормонами и другими
факторами.
Проблема заключается в том, что у прокариот можно
«выключить» ген репрессором либо, в случае /яс-оперо-
на, - сАМР или лактозой. Но как осуществить
множественную регуляцию гена эукариот?
часть 4 Хранение и переработка информации
274
Структура генов эукариот II типа
В этом разделе мы обсудим транскрипцию кодирующих
белок генов, которые используют РНК-полимеразу II.
У эукариотических и прокариотических генов с
5'-конца стартового фрагмента существует промотор
с короткой последовательностью между нуклеотида-
ми -3 и +5, в так называемой области инициации
(рис. 21.15). У 80 % генов есть ТАТА-бокс с центром
в положении -25. Его последовательность
напоминает бокс Прибнова в промоторе прокариот.
СААТ-бокс
Ζ ΑΑ
Хромосома
GC;60KC
ТАТА-бокс
■ г
Комплекс
TFID-полимераза II
Рис. 21.15. Некоторые активирующие последовательности
в области промотора гена эукариот, расположенные перед
стартовой точкой
На расстоянии 100-200 пар оснований перед
стартовой точкой располагаются короткие специфические
последовательности ДНК, распознаваемые особыми
белками -транскрипционными факторами: СААТ-боксом,
GC-боксом и октамерным боксом (включающим 8 пар
оснований). В различных генах они расположены в
области между -100 и -200 нуклеотидами. Впрочем, есть
и другие боксы, или элементы, ДНК - энхансеры. Они
связывают транскрипционные факторы, ускоряющие
транскрипцию гена. Энхансеры могут располагаться
на расстоянии тысяч пар оснований от гена, поскольку
их функционирование не зависит от ориентации в ДНК.
Все это делает определение промотора гена
эукариот менее точным по сравнению с геном прокариот.
Является ли удаленный энхансер частью промотора? Одно
полезное рабочее определение дано в учебном пособии
Льюина «Гены»: «Промотор эукариот включает
последовательности ДНК, которые должны находиться в
относительно фиксированных положениях по отношению
к стартовой точке». Следовательно, промотор включает
область инициации, ТАТА-бокс и боксы GC и С A AT,
расположенные выше точки инициации, но исключает
энхансеры.
Одним из наиболее трудных для понимания
аспектов является как бы сделанная наугад природа
механизма генетического контроля у эукариот. Это подводит
нас к замечательному и несколько смущающему
заключению о том, что ни один из элементов не является
обязательным для транскрипции и что различные гены
имеют разные комбинации элементов. У 20% генов,
включая многие гены «домашнего хозяйства», нет
ТАТА-боксов, некоторые лишены и других элементов,
или же у них существуют множественные копии
одного и того же элемента. Вариации, найденные в трех
образцах генов, приведены на рис. 21.16.
SV40
ранний
Тимидин-
киназа
Гистон 2В
J_
_L
J_
J_
J_
_1_
_L
_L
J
-140 -120 -100 -80 -60 -40
-20 |
Стартовая точка
Типы модулей ψ ^
Октамер СААТ
GC
ТАГА
Рис. 21.16 Элементы промоторов трех генов эукариот
Промоторы содержат различные комбинации ТАТА-боксов, СААТ-боксов, GC-боксов и других элементов
глава 21 Транскрипция гена
275
Механизм инициации
транскрипции генов эукариот
Стабильный комплекс инициации
У бактерий РНК-полимераза узнает правильный сайт
на промоторе и непосредственно связывается с ДНК,
чему в ряде случаев способствует, например, САР.
У эукариот для образования основного инициаторного
комплекса на ДНК, необходимого для всех генов II типа,
к которому присоединяется РНК-полимераза, требуется
ряд белков - общих факторов транскрипции,
присутствующих во всех клетках. К ним относятся: ТВР-белок
(англ. TATA binding protein) присоединяющийся
к ТАТА-боксу; с ТАТА-боксом связаны восемь и более
TAF-белков (TBP-associated factors), образующих
TFIID-комплекс (transcriptional factor D for
polymerase II - фактор транскрипции D для полимеразы И);
с TFIID связывается РНК-полимераза II и другие
белки, завершая образование основного инициаторного
комплекса. Все это происходит у генов, имеющих
ТАТА-бокс. У генов без ТАТА-бокса комплекс
собирается в инициаторной области; при этом, возможно, для
достижения правильного положения полимеразы
используется другой фактор. Если гены транскрибируются
полимеразами I и III, то собираются различные другие
стабильные комплексы, которые мобилизуют
соответствующую полимеразу.
Однако, как уже отмечалось, существуют различные
гены. Некоторые из них, например гены «домашнего
хозяйства», экспрессируются постоянно, другие - ткане-
специфичные - экспрессируются только в
определенных клетках, третьи - индуцибельны (например,
специфические гены, транскрипция которых активируется
стероидными гормонами). Наконец, существуют гены,
которые, наоборот, являются объектами негативного
контроля, а некоторые - объектами для различных
комбинаций указанных выше типов регуляции. Как же
осуществляются все эти типы регуляции? Здесь уместно
рассмотреть специфические факторы транскрипции,
которые комбинируются с энхансером и элементами,
расположенными перед стартовой точкой.
Какова роль
транскрипционных факторов?
Все гены в хроматине (это ДНК, связанная с белками;
см. с. 237) в отсутствие факторов транскрипции
находятся в «выключенном», или репрессированном,
состоянии, так как нуклеосома блокирует область инициации
часть 4 Хранение и переработка информации
276
каждого промотора. До тех пор, пока нуклеосома не
будет вытеснена (это позволит осуществить сборку
инициаторного комплекса), ген не транскрибируется.
В активации одного и того же гена могут участвовать
различные факторы транскрипции. Прежде всего к ним
относятся факторы, которые связываются с боксами GC
и СААТ, расположенными перед стартовой точкой. Они
есть во всех клетках, и конститутивно экспрессирующи-
еся гены могут нуждаться только в них. Каким образом
факторы транскрипции, связывающиеся с удаленными
элементами, участвуют в формировании комплекса? Это
может происходить в результате образования петли
ДНК. Каждый фактор имеет по меньшей мере два
участка связывания - один с ДНК, а другой с белками,
участвующими в формировании транскрипционного
комплекса, как показано на рис. 21.18.
Каким образом осуществляется избирательный
контроль экспрессии генов? Это функция элементов,
расположенных в разных участках ДНК. Даже на расстоянии
тысяч пар оснований они могут влиять на
транскрипцию гена. Например, присутствие в ДНК удаленного
энхансера увеличивает в сотни раз транскрипцию гена
β-глобина.
Ген β-глобина экспрессируется только в незрелых
эритроцитах; его транскрипция зависит от связывания
транскрипционного фактора с GATA-боксом. Данный
фактор присутствует лишь в этих клетках, что и
обеспечивает специфичность экспрессии.
Как регулируется скорость транскрипции индуци-
бельного гена? В качестве примера приведем активацию
ряда генов стероидными гормонами.
В клетках-мишенях присутствуют неактивные
факторы транскрипции. Они не способны
самостоятельно связываться с соответствующими элементами ДНК.
Гормон, поступающий в клетку, активирует фактор
транскрипции, который затем связывается с ДНК,
Комплекс
TFIID
TAFs \
РНК-полимераза
ТВР
TATA
ДНК
Рис. 21.17. Модель основного инициаторного комплекса для
РНК-полимеразы Π
Комплекс TFIID включает ТАТА-связывающий белок (ТВР)
и многочисленные TAF-белки. Формы, размеры и положения
компонентов указаны произвольно
вызывая активацию транскрипции. Механизм активации
этого фактора описан в главе 26, где мы познакомимся
с клеточной сигнализацией, основным механизмом
которой является активация факторов транскрипции.
Отметим, что существует и негативный контроль
транскрипции гена.
Основной вопрос - каким образом взаимодействие
факторов транскрипции и инициаторного комплекса
регулирует инициацию - остается пока без ответа.
Важное свойство регуляции экспрессии генов эукариот - ее
высокая гибкость; т. е. многие факторы могут внести
свой вклад в регуляцию транскрипции при условии
наличия для них соответствующего участка связывания
на молекуле ДНК (рис. 21.19).
Структура ДНК-связывающих белков
Из изложенного в этой главе становится понятной
важная роль белков, связывающихся со специфическими
участками ДНК. Существуют многочисленные репрес-
соры и факторы транскрипции, участвующие в
процессе дифференцировки, эмбрионального развития и т. д.
Эффективность их действия зависит от способности этих
соединений узнавать ДНК и связываться с ней.
Для выяснения структуры белков,
взаимодействующих с соответствующими участками ДНК, были
предприняты широкомасштабные исследования.
Оказалось, что на основании структурных характеристик
Энхансерный элемент
Комплекс/ TAFs >
TFIID-:;■/—" ,
|. РНК-полимераза|
ТВР
TATA '. I **
ДНК
Рис. 21.18. Гипотетическая модель, показывающая, каким
образом белок, связывающийся с сайтом ДНК, удаленным
от стартовой точки, может участвовать в инициации
транскрипции РНК-полимеразой II
Участки взаимодействия между транскрипционным фактором,
основным инициаторным комплексом и РНК-полимеразой
нарисованы произвольно. Энхансерные элементы,
расположенные за тысячи пар оснований, могут участвовать в образовании
комплекса за счет механизма образования петли (см. рис. 21.19)
большинство ДНК-связывающих белков может быть
сгруппировано в несколько семейств, среди которых
наиболее важными являются белки с мотивами
«спираль-поворот-спираль», гомеодоменные белки, белки
со структурами типа «лейциновой молнии» и, наконец,
белки с мотивами «цинковые пальцы».
Структуры, или мотивы, давшие эти названия,
представляют собой лишь небольшие участки белков,
характеризующие все семейство. Например, на рис. 21.20
показан только характерный для всего семейства
фрагмент «спираль-поворот-спираль» мономера ДНК-свя-
зывающего белка.
ДНК-связывающие белки присоединяются к двухце-
почечной ДНК; сайт-специфическое связывание
происходит за счет взаимодействия между боковыми цепями
аминокислотных остатков и основаниями ДНК.
Дополнительная стабилизация может достигаться
связыванием с сахарофосфатным скелетом. Контактные
взаимодействия узнающих мотивов белка с азотистыми
основаниями происходят преимущественно в большой
бороздке двойной спирали, куда экспонированы
некоторые атомы азотистых оснований. Во многих случаях
такое связывание обусловлено узнаванием α-спирали
белка, которая укладывается в большую бороздку.
Важно отметить, что для специфического
присоединения белка на молекуле ДНК необходимо наличие
двух соседних участков. Они могут быть
идентичными, образованными палиндромным расположением
оснований (см. ниже) или различаться. С палиндром-
ными участками способны связываться, например,
сАМР-чувстви- Тканеспецифичный
тельный элемент / элемент
Стероид-чувстви- / / Энхансерный
тельный элемент / /' / элемент
Комплекс TAFs ж
TFIID - — \,
I РНК-полимераза
GC-
бокс/ . днк
Фактор СААТ-
транскрипции Spl бокс
Рис. 21.19. Множественный контроль инициации
транскрипции гена эукариот (схематично)
Размеры, положение и взаимодействие белков, а так же
комбинация регуляторных элементов - условны
глава 21 Транскрипция гена
277
->
V J
Поворот
Взаимодействие между
\ двумя α-спиралями
стабилизирует
ДНК- структуру фрагмента
узнающая спираль
Фрагмент
«спираль-поворот-спираль»
:П
ДНК-связывающие
.· домены
•\&
Двойная ось
Рис. 21.20 Фрагмент одного из ДНК-связывающих белков
и его взаимодействие с ДНК
а - Фрагмент «спираль-поворот-спираль»;
б - связывание этого белка с ДНК в больших бороздках
белки-гомодимеры, состоящие из двух одинаковых
субъединиц, а с другими - белки-гетеродимеры,
состоящие из различных субъединиц. На белках с
фрагментами «цинковые пальцы» могут располагаться
множественные ДНК-узнающие мотивы.
Сущность вариабельного контроля гена состоит
в том, что ДНК-связывающие белки, например факторы
транскрипции и репрессоры, во многих случаях
взаимодействуют с ДНК только после соответствующего
«инструктажа». Мы уже познакомились с ситуациями, при
которых активация транскрипционного фактора
достигается аллостерической модификацией регуляторного
белка при связывании лиганда (вспомним регуляцию
/яс-оперона). У эукариот такая «инструкция» обычно
приходит в виде внешнего внеклеточного сигнала,
скажем, гормона или фактора роста. Подобные
механизмы активации факторов транскрипции рассмотрены
в главе 26.
Теперь рассмотрим семейства ДНК-связывающих
белков.
Белки с мотивом
«спираль-поворот-спираль» (НТН)
Это первое идентифицированное семейство
ДНК-связывающих белков. У прокариот обнаружено много
белков такого типа, например, лямбда-репрессор.
Бактериофаг лямбда Е. coli описан на с. 314. Мы не будем
разбирать здесь роль белка-репрессора - это довольно
сложно; скажем только, что он связывается с
определенной областью ДНК фага лямбда и участвует в
«принятии решения» о вхождении инфицирующего вируса
в литический или лизогенный циклы (разъяснения
см. на с. 314-315). Сейчас гораздо важнее, что механизм
его связывания с ДНК изучен очень подробно.
Как мы уже объясняли, мотив «спираль-поворот-
спираль» (англ. helix-turn-helix, НТН) представляет
собой небольшой участок белка, с помощью которого
происходит связывание с операторным участком ДНК. (Это
не домен белка, и он не может существовать сам по
себе при выделении; мотив означает узнаваемое
структурное свойство). Мотив включает две α-спирали,
соединенные β-изгибом. Одна из спиралей (узнающая)
располагается в большой бороздке ДНК (см. рис. 21.20, а).
Оператор или ДНК-узнающий участок представляет
собой неполный палиндром. Палиндромом называют
фразу или слово, которые можно одинаково прочесть
слева направо или справа налево. Например, фраза
«Madam I'm Adam» симметрична относительно буквы
«I» (в русском языке в качестве примера можно
привести фразу «ум его Бог ему»; прим. переводчика.). Палин-
дромные ДНК-связывающие участки имеют парную
симметрию (когда две единицы действуют как одна или
как группа из двух). Ниже показана структура оператора.
5' TATCACCGCCAGTGGTA У
У ATAGTGGCGGTCACCAT 5'
Две половины палиндрома представляют собой
идентичные участки связывания, хотя нуклеотиды
показывают, что палиндром не абсолютен. Белок-репрес-
сор - это димер, узнающие спирали двух
фрагментов НТН которого подходят к данным нуклеотидам
(см. рис. 21.20, б).
часть 4 Хранение и переработка информации
278
Белки, содержащие
«лейциновую молнию»
Обнаружены среди многочисленных факторов
транскрипции эукариот. В данном случае название
обозначает не фрагмент узнавания (как у НТН-белков), а
характерную структуру, которая вызывает димеризацию двух
субъединиц (последние могут и не иметь идентичных
участков узнавания). Во фрагменте «лейциновой
молнии» каждая седьмая аминокислота представлена
лейцином. Поскольку виток образуют 3,6 аминокислотных
остатка, все остатки лейцина появляются на одной
стороне α-спирали, формируя гидрофобную поверхность.
Две таких субъединицы соединяются за счет
гидрофобных взаимодействий между боковыми цепями лейцинов
(рис. 21.21, а). Кстати говоря, термин молния - неверен.
Он был предложен, когда предполагали, что остатки
лейцина переплетаются подобно зубцам застежки-молнии.
В действительности ДНК-связывающим участком
каждого мономера является область, богатая остатками
положительно заряженных аминокислот - аргинина и
лизина. Образно говоря, димер присоединяется к ДНК
у рукоятки «ножниц», располагаясь между двумя
«лезвиями» в соседних сегментах большой бороздки
дуплекса (рис. 21.21, б).
Белки с мотивом «цинковые пальцы»
В этом ДНК-связывающем мотиве атом цинка,
соединенный с 2 остатками гистидина и 2 остатками цистеина,
стабилизирует пальцеподобную структуру (рис. 21.22, а),
одна часть которой является узнающей α-спиралью,
располагающейся в большой бороздке ДНК (рис. 21.22, б).
Факторы транскрипции могут иметь несколько таких
пальцев, обусловливающих множественные контакты
с ДНК (рис. 21.22, в). Один такой белок-регулятор гена
имеет около 30 цинковых пальцев. Не все цинковые
пальцы специфически связываются с ДНК. Некоторые из -
них неспецифичны, но они помогают стабилизировать
специфическое связывание. Мотив «цинковые пальцы»
характерен для многих регуляторных белков различных
эукариотических генов, например для внутриклеточных
рецепторов стероидных гормонов.
Гомеодоменные белки
и эмбриональное развитие
Расшифровка механизма процесса дифференцировки
и эмбрионального развития - одна из важнейших задач
биологии. Оплодотворенное яйцо многоклеточного
организма делится и дифференцируется в различные
ткани: эпидермис, нервы, мышцы, печень и т. д. Поскольку
каждая клетка имеет один и тот же набор генов, диффе-
ренцировка включает селективную экспрессию генов
в различных тканях. О том, как это происходит, до сих
пор известно немного. Однако несколько лет назад было
идентифицировано несколько гомейотических генов,
ответственных за правильное эмбриональное развитие.
Их важность была установлена в ходе изучения влияния
мутаций в этих генах на развитие плодовой мушки
* Два мономера
белка
L
L L
L I
L
α-Спиральный полипептид,
_ каждая седьмая аминокислота
которого представлена
лейцином (L)
Фрагмент
«лейциновой
молнии»
Ось ДНК— Основной участок
узнавания ДНК
Рис. 21.21. Белки, содержащие «лейциновую молнию»
а - Строение фрагмента «лейциновой молнии». Гидрофобные остатки лейцина располагаются друг против друга, но не
переплетаются подобно зубцам на молнии;
б - прикрепление к ДНК белка, содержащего «лейциновую молнию» (вид вниз по оси ДНК)
глава 21 Транскрипция гена
279
Рис. 21.22. Белки с мотивом типа «цинковые пальцы»
а - Структура «цинкового пальца»;
б - модель взаимодействия между областью мишени в ДНК
и белком, содержащим «цинковый палец». Одна сторона
структуры «пальца» представляет собой α-спираль: она
прикрепляется к большой бороздке ДНК;
в - множество фрагментов типа «цинковых пальцев»,
связанных с ДНК
дрозофилы. Важным в рассматриваемом контексте
моментом является то, что продукты гомейотических
генов - все кодируемые ими белки - имеют сходные
домены, именуемые гомеодоменами. Участки ДНК,
кодирующие такие гомеодомены, называются гомеобоксами.
Гомеодомены - это ДНК-узнающие белки длиной около
60 аминокислотных остатков; они напоминают фрагмент
«спираль-поворот-спираль» с узнающей спиралью,
расположенной в большой бороздке. Кодируемые гомейоти-
ческими генами белки связываются со специфическими
участками ДНК. Поэтому можно предположить, что го-
мейотические гены кодируют факторы транскрипции.
часть 4 Хранение и переработка информации
280
Хотя гены-мишени еще не известны, установлено, что
в ряде случаев эти факторы служат активаторами одних
генов и репрессорами других. Структура гомеодоменов
характеризуется высокой степенью консервативности
и высокой степенью сходства у насекомых (дрозофила)
и позвоночных, включая человека.
А теперь определимся с терминологией. Если речь
идет о транскрипции, термин бокс обычно
ассоциируется с последовательностью ДНК, узнаваемой белком
(например, рассматриваемые ранее боксы TATA и GC).
Слово бокс при обсуждении генов гомеобокса
обозначает последовательность оснований, кодирующих гомео-
домен. В этом случае данный термин несет совершенно
иную смысловую нагрузку, хотя и в первом, и во втором
примере речь идет о специфических участках ДНК.
Стабильность мРНК
и контроль экспрессии генов
Хотя значение регуляции транскрипции при экспрессии
генов очень велико, стабильности индивидуальных
мРНК отводится не менее важная роль. Чаще всего
скорость синтеза белка отражает количество
образовавшейся мРНК, за исключением тех случаев, когда
трансляция мессенджера (мРНК) контролируется способом,
описанным для синтеза аминолевулинатсинтазы
эритроцитов и глобина (см. с. 366 и 367 соответственно).
Уровень мРНК в клетках зависит от соотношения
скоростей ее синтеза и распада. При определенной
скорости образования мРНК ее содержание в клетке будет тем
выше, чем длиннее время ее полужизни, что,
естественно, приводит к более высокой скорости синтеза
соответствующего белка. Механизмы, влияющие на время
полужизни мРНК, представляют, таким образом, способ
регуляции экспрессии гена.
Время полужизни мРНК прокариот обычно
ограничено 2-3 минутами. Быстрый оборот мессенджера
позволяет оперативно реагировать на изменения среды.
У млекопитающих время полужизни индивидуальных
мРНК составляет от 10 минут до 2 дней. Время
полужизни одной из самых стабильных мРНК (мРНК
глобин) составляет около 10 часов. Регуляторные белки
обычно кодируются короткоживущими мРНК, так что
изменения скорости транскрипции их генов быстро
отражается на скорости их экспрессии. Время полужизни
мРНК факторов транскрипции обычно составляет
менее 30 минут. Таким образом, клетка содержит мРНК,
скорость деградации которых различна, а стабильность
отдельных мРНК может изменяться.
Несмотря на потенциальную важность стабильности
мРНК, процесс ее распада изучен в гораздо меньшей
степени, чем синтез и его регуляция. Немного известно
и о ферментах, участвующих в процессе распада,
однако некоторые механизмы, определяющие
продолжительность жизни мРНК, уже установлены.
Структуры, определяющие
стабильность мРНК и их роль
в регуляции экспрессии
Почти все мРНК имеют поли-А-«хвост»,
присоединяющийся к З'-концу перед выходом молекулы РНК из ядра
(рис. 21.23, я); исключение составляют мРНК гистонов.
Считается, что поли-А-«хвост» защищает мРНК эука-
риот от быстрой деградации. Деаденилирование часто
предшествует деструкции мРНК. Уже обнаружен белок,
который связывается с «хвостом» поли-А, тормозя тем
самым распад мРНК с З'-конца молекулы, но механизм,
посредством которого поли-А защищает мРНК,
детально не выяснен. Предполагают, что поли-А влияет на т-
ранспорт, трансляцию и распад мРНК.
У мРНК гистонов поли-А-«хвост» отсутствует, и
стабильность этих молекул определяется крайней
шпилечной петлей наЗ'-конце (рис. 21.23,6). Синтез
гистонов необходим только во время S-фазы клеточного
цикла эукариот, когда происходит синтез ДНК и сборка
нуклеосом. Гены гистонов транскрибируются во время
S-фазы клеточного цикла, но в С2-фазе этот процесс
а
Кодирующая
5' UTR область
3' UTR Хвост поли-А
κ&ν^Λ^κ ААААААААААиАА
• Ίί
3' Шпилечная петля
Железочувствительный элемент
Богатый AU-элемент (AURE)
'ААААААиАА
Рис. 21.23. Организация структур, присутствующих в не-
транслируемой области З'-конца (untranslated region, UTR)
мРНК млекопитающих, которые влияют на время
полужизни молекул в клетке
а - «Хвост» поли-А найден в большинстве мРНК эукариот;
б - шпилечная петля, обнаруженная у гистоновых мРНК;
в - железо-чувствительный элемент (iron-responsive element)
мРНК трансферрина;
г - AU-элемент (A\J-rich element; AURE) найден у большого
числа нестабильных мРНК эукариот
прекращается - и уровень мРНК гистонов быстро
падает. Последнее частично обусловлено остановкой
транскрипции, но кроме того происходит сокращение
времени полужизни мРНК с 40 до 10 минут, которое зависит
от присутствия шпилечной петли на 3'-конце. Перенос
последней на мРНК глобина приводит к образованию
гибридного мессенджера, который после введения
в культивируемые клетки дестабилизируется в конце
S-фазы, подобно мРНК гистона. Для дестабилизации
мРНК гистонов необходимо присутствие свободных
мономеров гистонов, которые накапливаются вскоре
после окончания синтеза ДНК (и следовательно, сборки
нуклеосом) в конце S-фазы. Быстрое выключение
синтеза гистонов необходимо по причине цитотоксичности
их мономеров. Четырехкратное уменьшение времени
полужизни гистоновых мессенджеров означает, что
после прекращения транскрипции гена гистона процесс
деградации мРНК длится в течение 2 часов, а без
дестабилизации он занимает около 9 часов (рис. 21.24).
Синтез β-тубулина регулируется при помощи
механизма обратной связи, модулирующего стабильность
мРНК. В главе 29 описана роль агрегации этого белка
и ее влияние на формирование микротрубочек.
Присутствие мономеров свободного тубулина дестабилизирует
его мРНК. Для того, чтобы происходила деградация,
мРНК должна участвовать в процессе трансляции,
поскольку частично синтезированный пептид каким-то
образом способствует процессу дестабилизации.
Механизм вызываемого посредством тубулина распада мРНК
пока непонятен, но регуляторный смысл этой системы
очевиден.
Синтез рецепторного белка трансферрина
представляет еще один пример того, как стабильность мРНК
регулирует синтез белка. Рецептор ответственен за
транспорт железа в клетки (см. с. 368). В З'-нетранслируемой
области мРНК есть группа из пяти шпилечных петель,
10000
Τι η = 40 мин
Время, мин
Рис. 21.24. Влияние четырехкратного уменьшения времени
полужизни мРНК на ее количество в клетке после
прекращения синтеза
глава 21 Транскрипция гена
281
называемая железочувствительным элементом (англ.
iron-responsive element', [RE) (см. рис. 21.23, в). В
отсутствие железа IRE-связывающий белок присоединяется
к IRE и стабилизирует мРНК, увеличивая тем самым
синтез рецептора и повышая поступление железа в
клетку. При избытке железа происходит образование
комплекса железа с этим белком, в результате последний
несвязывается с IRE, что, как полагают, делает эту область
доступной для эндонуклеазы.
Короткоживущие мРНК могут содержать так
называемые последовательности нестабильности, которые
служат сигналом для быстрой деградации мРНК в
клетке. Структурной особенностью многих нестабильных
мРНК является существование богатых аденином и ура-
цилом элементов (англ. adenine/uracil-rich elements',
AURE) в З'-нетранслируемых областях молекул
(см. рис. 21.23, г). Эти элементы в разных мРНК
различны, однако у каждого из них есть
последовательности AU длиной по меньшей мере в 9 оснований. Они
вызывают дестабилизацию мРНК, возможно, при помощи
AURE-связывающих белков. До сих пор неизвестно,
как AURE функционируют. Их влияние на время
полужизни мРНК может быть непрямым, обусловленным
воздействием на трансляцию; впрочем, присутствие
AURE всегда ассоциируется с быстрым деаденилиро-
ванием мРНК. Особенно интересный пример
представляет мРНК - продукт гена c-fos, который кодирует
факторы транскрипции, содержащих лейциновую молнию.
Эта мРНК имеет в нетранслируемой области AURE
(термины c-fos, v-fos и онкогенностъ объяснены на с. 356).
Ген v-fos является сильным онкогеном, вызывающим
развитие рака; у мРНК этого гена нет AURE,
найденного у мРНК c-fos, и время полужизни в клетке дольше,
чем у мРНК c-fos. При делеции области AURE
нормальный ген c-fos превращается в онкоген. (О взаимосвязи
регуляторных белков с онкогенностью см. главу 26).
Механизм, определяющий стабильность мРНК
в клетке, сложен. В дополнение к изложенному выше,
отметим, что ряд гормонов модулирует стабильность
специфических мессенджеров. В заключение добавим,
что регуляция распада мРНК представляет собой один
из важных методов контроля за экспрессией гена.
Транскрипция гена
в митохондриях
В предыдущих главах уже рассматривалась центральная
роль митохондрий в выработке энергии в ходе окисления
поступающих с пищей субстратов. Митохондрии -
реплицирующиеся органеллы клеток эукариот. У них
часть 4 Хранение и переработка информации
282
есть собственная ДНК и белок-синтезирующий аппарат
(так же, как и у хлоропластов). Митохондрии делятся;
поэтому в разных клетках поддерживается
определенное число этих органелл: около 1000 в гепатоците
крысы и 107 в овоците лягушки. Митохондриальная ДНК
у млекопитающих обычно представляет собой
кольцевой дуплекс, содержащий менее 20 000 нуклеотидов;
в каждой митохондрии может быть представлено 5 или
10 копий такого дуплекса.
Митохондриальная ДНК кодирует лишь небольшую
часть белков этих органелл; остальные белки
кодируются ядерной ДНК. Белки синтезируются в цитоплазме
и транспортируются в митохондрии (см. главу 22). В
митохондриях обе цепи ДНК полностью
транскрибируются в линейные длинные транскрипты (молекулы поли-
меразы движутся в противоположных направлениях,
так как синтез всегда происходит в направлении 51—»3').
Эти первичные транскрипты (непонятным пока
образом) подвергаются процессингу с образованием мРНК,
тРНК и рРНК. Чтобы образовалось больше рРНК,
производится много более коротких транскриптов,
окружающих гены рРНК.
В отличие от синтеза белка в цитоплазме (цитоплаз-
матическая трансляция), трансляция в митохондриях
обладает рядом особенностей, свойственных
прокариотам. Отсюда возникло предположение о том, что
митохондрии появились в результате поглощения эука-
риотическими клетками прокариотических, ставших
симбионтами (аналогичная теория объясняет
происхождение хлоропластов в растительных клетках).
Свойства митохондриальной системы транскрипции и в самом
деле представляют собой смесь прокариотических и эу-
кариотических типов. В митохондриях млекопитающих
мРНК полиаденилированы (что характерно для
эукариот), но не кэпированы (свойство прокариот), и (как
у прокариот) в митохондриальных генах нет интронов.
В митохондриях трипаносом образующиеся мРНК
подвергаются последующему редактированию.
Транскрипты РНК обладают дополнительными U,
расположенными в особых местах для образования
правильной последовательности, кодирующей белки. Эти U
не были предусмотрены в ДНК. Позже было
обнаружено, что редактирование мРНК может происходить
и в ядерных транскриптах. В результате транскрипции
гена аполипопротеина В млекопитающих (см. с. 96)
из одного транскрипта образуется две мРНК. «Нере-
дактированная» мРНК кодирует апопротеин В100,
а меньший аполипопротеин В48 кодируется
«отредактированной» версией, где соответствующий С из ДНК
конвертируется в U с образованием трансляционного
стоп-кодона (см. главу 22).
Вирус обезьян (гены которого обладают свойствами
генов эукариот) образует мРНК, которые
редактируются путем инсерции (встраивания) двух остатков G, не
закодированных в ДНК. Пока неясно, имеют ли эти
вариации биологический смысл или же представляют собой
пример эволюционного «ремесленничества».
Гены, не кодирующие белки
Помимо генов, ответственных за синтез белков,
имеются гены, кодирующие особые молекулы РНК, которые
не являются мессенджерами. В следующем разделе вы
узнаете, что белки синтезируются нуклеопротеиновыми
частицами - рибосомами и что для построения
полипептидных цепей из аминокислот необходимы небольшие
многочисленные транспортные РНК (тРНК).
Рибосомы содержат молекулы рибосомных РНК (рРНК); у
прокариот они содержат 3 различных молекулы рРНК,
а у эукариот - 4 молекулы. При транскрипции около
половины от общего количества РНК приходится на долю
рРНК. Поскольку тРНК и рРНК по сравнению с мРНК
живут долго, большая часть РНК в клетке представлена
именно этими формами. В клетках Е. coli три рРНК
и несколько тРНК транскрибируются вместе в виде
большой молекулы предшественника, которая затем
разрезается на соответствующие более мелкие фрагменты.
В отличие от прокариот, у эукариот существует три
РНК-пол имеразы.
РНК-полимераза II, которая транскрибирует гены,
кодирующие белки, уже была описана.
РНК-полимераза I транскрибирует большую часть рРНК, а
РНК-полимераза III - тРНК и более мелкие рРНК. Поскольку
клетке нужно много тРНК и рРНК, существуют
множественные копии их генов. У эукариот могут быть
сотни и тысячи генов рРНК, организованных в виде
тандема - голова к хвосту; они находятся в нуклеолярных
областях ядра. Гены, транскрибируемые полимеразой III,
необычны тем, что регуляторные элементы, или боксы,
встречаются не только с 5'-конца стартовой точки ДНК,
но также в транскрибируемой области, а именно после
стартовой точки.
Дополнительная литература
Lewin В. Genes V. Oxford University Press, 1994
Экспрессия генов
Lewin В. Chromatin and gene expression: constant questions,
but changing answers // Cell. 1994. Vol. 79. P. 397-406.
(Легко читаемая общая дискуссия о нуклеосомах и об
их роли в экспрессии генов эукариот.)
Интроны и экзоны
Gilbert W. The exon theory of genes // Cold Spring Harbor.
Symp. Quant. Biol. 1987. Vol. LII. P. 901-905.
(Статья описывает теорию сборки генов из минигенов.)
Go Μ, Nosaka M. Protein architecture and the origin of In-
trones.// Cold Spring Harbor. Symp. Quant. Biol. 1987.
Vol. LII. P. 915-924.
(В работе рассмотрены две теории происхождения
нитронов.
MattickJ. S. Introns-evolution and function// Curr. Opin.
Genet. Develop. 1994. Vol. 4. P. 823-831.
(Легко читаемый обзор, в котором очень доходчиво
суммированы последние дискуссии, посвященные теориям
«ранних и поздних интронов» и затронуты
эволюционные аспекты этой темы.)
Белковые модули и домены
Doolittle R. F. The multiplicity of domains in proteins //
Annu. Rev. Biochem. 1995. Vol. 64. P. 287-314.
(Интересный обзор, посвященный доменам, экзонам
и интронам.)
Сплайсинг
Breibart R. Е., Andreadis Α., Nadal-Ginard В. Alternative
splicing: a ubiquitous mechanism for the generation of
multiple protein isoforms from single genes // Annu. Rev.
Biochem. 1987. Vol. 56. P. 467-495.
(Хороший обзор о биологической роли
альтернативного сплайсинга.)
Sharp P. A. Splicing of messenger РНК precursors //
Science. 1987. Vol. 235. P. 766-771.
(Краткое резюме по данной проблеме.)
OrgelL. E. The origin of life on earth// Sci. Amer. 1994.
Vol. 271(4). P. 52-61.
(Все больше данных свидетельствуют в пользу того, что
происхождение каталитической РНК было решающим
шагом для возникновения жизни.)
Саморасщепление РНК
Symons R. Η. Cell cleavage of РНК In the replication
of small pathogens of plants and animals //Trends Biochem.
Sci. 1989. Vol. 14. P. 445^50.
(В статье рассмотрены вироиды и классическая
саморасщепляющаяся структура РНК - «головка молотка».)
Факторы транскрипции эукариот
Sheldon Μ, Reinberg D. Tuning up transcription // Curr.
Biology. 1995. Vol. 5. P. 43-46.
(Транскрипция у эукариот - сложный процесс;
приведенные данные описывают множественные этапы,
глава 21 Транскрипция гена
283
необходимые для активации транскрипции; например,
конформационное изменение транскрипционного
комплекса TFIID.)
Tjian R. Molecular machines that control genes // Sci. Amer.
1995. Vol. 272 (2). P. 38-45.
(Активность генов, кодирующих белки, регулируется
многочисленными факторами транскрипции, каждый
из которых может включать несколько белков.)
Zawel L., Reinberg D. Common themes in assembly and
function of eukaryotic transcription complexes //Annu. Rev.
Biochem. 1995. Vol. 64. 533-561.
(Подробный обзор, посвященный сборке
транскрипционных комплексов эукариот.)
Surridge С. The core curriculum // Nature. 1996. Vol. 380.
P. 287-288.
(В статье обсуждается состав TFIID инициаторного
комплекса при транскрипции генов эукариот.)
ДНК-связывающие белки
Brennan R. G., MatthewsB. W. Structural basis of DNA-
protein recognition//Trends Biochem. Sci. 1989. Vol. 14.
P. 287-290.
(Обзор структур белков, участвующих в контроле
транскрипции генов, и способы их взаимодействия с ДНК.)
ДНК-связывающие белки - «цинковые
пальцы»
Klevit R. Ε. Recognition of DNA by Cys2, His2 zinc
fingers. // Science. 1991. Vol. 253. P. 1367, 1393
(Краткое резюме по данной проблеме.)
Rhodes D., KlugA. Zinc fingers // Sci. Amer. 1993.
Vol 263 (2). P. 56-65.
(В статье обсуждаются структура, функция и
распределение этих факторов транскрипции.)
KlugA., SchwabeJ. W. R. Zinc fingers// FASEB J. 1995.
Vol. 9. P. 597-604.
(Статья, посвященная структуре этого фрагмента белка
и его роли в связывании ДНК.)
ДНК-связывающие белки - «лейциновые
молнии»
McKnightS.L. Molecular zippers// Sci. Amer. 1991.
Vol. 264 (4). P. 32-39.
(Доступно изложенное сообщение об этих факторах
транскрипции.)
Ellenberger Т. Е., Brandil С. J., Struhl K.t Harrison S. С.
The GCN4 basis region leucine zipper binds DNA as a di-
mer of uninterrupted α-helices: crystal structure of the pro-
tein-DNA complexes // Cell. 1992. Vol. 71. P. 1223-1237.
(Экспериментальная, но легко читаемая статья, в
которой приведены заслуживающие внимания иллюстрации.)
часть 4 Хранение и переработка информации
284
ДНК-связывающие белки - гистоны
Grunstein Μ. Hi stones as regulators of genes // Sci. Amer.
1992. Vol. 267(4). P. 40-47.
(В статье обсуждается роль гистонов в репрессии и
активации генов.)
Lu Q., Wallrath L. L., Elgin S. R. Nucleosome positioning
and gene regulation// J. Cell. Biochem. 1994. Vol.55.
P. 83-92.
(В статье обсуждается, каким образом нуклеосомы
могут выступать в качестве репрессоров транскрипции
у эукариот.)
Редактирование мРНК
Hodges R., Scott J. Apolipoprotein B mRNA editing: anew
tier for the control of gene expression // Trends Biochem.
Sci. 1992. Vol. 17. P. 77-81.
(Интересное сообщение о том, каким образом
редактирование мРНК ведет к образованию двух форм аполи-
поротеина В из одного транскрипта мРНК.)
Транскрипция в митохондриях
Clayton D. A. Transcription of the mammalian mitochondrial
genome // Annu. Rev. Biochem. 1984. Vol. 53. P. 573-594.
(Обзор, посвященный процессу транскрипции в
митохондриях, отличному от ядерной транскрипции.)
Вопросы к главе 21
1. Каковы отличия синтеза РНК от синтеза ДНК?
2. С помощью схем опишите компоненты гена Е. coli
и фланкирующих областей.
3. Опишите процесс инициации транскрипции гена
Е. coli.
4. Опишите два способа терминации транскрипции
гена Е. coli.
5. Какие факторы определяют силу промотора у
конститутивного гена Е. colil
6. Опишите регуляцию /яооперона.
7. Опишите основные отличия между образованием
РНК у эукариот и прокариот.
8. Пользуясь схемами, объясните механизм сплайсинга.
Какова возможная биологическая роль
существования интронов?
9. Каковы отличия инициации транскрипции у
эукариот сравнительно с прокариотами?
10. Какие семейства факторов транскрипции выделены
на основе сходства структурных мотивов?
QQ Синтез, внутриклеточный транспорт
глава ^^ и деградация белков
В предыдущей главе мы познакомились с процессом
транскрипции гена, который приводит к образованию
мРНК. В этой главе мы обсудим процессы синтеза,
адресной доставки и избирательной деградации белков. Вы
узнаете, каким образом происходит процесс трансляции
мРНК в белок. Большинство белков эукариотической
клетки синтезируется в цитоплазме (за исключением белков
митохондрий и хлоропластов), и для их адресной доставки
в ядро, плазматическую мембрану, мембраны
митохондрий и другие клеточные органеллы существуют
специальные механизмы. Наконец, разные белки
программируются для короткого или продолжительного времени жизни.
Основные принципы процесса
биосинтеза белка
Сначала рассмотрим проблему в общих чертах. мРНК -
достаточно длинная молекула, в ее состав входят 4 вида
азотистых оснований. Белки синтезируются из 20
различных аминокислот, и последовательность оснований
в мессенджере (мРНК) определяет последовательность
аминокислот в белке. Код, состоящий из 1 азотистого
основания, может кодировать только 4 аминокислоты;
код, включающий 2 азотистых основания - 16
аминокислот, что по-прежнему явно недостаточно для 20.
Таким образом, для кодирования 20 аминокислот
необходимо минимум 3 азотистых основания. Каждая
аминокислота в мРНК «записывается» в виде
триплета азотистых оснований, называемых код оном.
4 различных основания могут образовать 64 триплета,
или кодона (4-4-4).
На первый взгляд, эволюция вполне могла
ограничиться использованием 20 кодонов, игнорируя
оставшиеся. Однако тогда в результате случайных мутаций
неизбежно появлялись бы триплеты, не кодирующие
аминокислоты. Это привело бы к бездействию генов, так
как при встрече с триплетом мРНК, не кодирующим
аминокислоту, синтез белка прекращается.
Эволюция выбрала альтернативную стратегию. Три
кодона были зарезервированы в качестве стоп-сигналов,
указывающих белок-синтезирующему аппарату, что
синтез белка завершен. Эти 3 триплета (один из них - UAA)
не кодируют аминокислот.
Если в результате мутации стоп-сигнал появится
в кодирующей области мРНК, белок-продукт данного
гена образовываться не будет. Если бы таких
стоп-триплетов было больше (44), то мутации, приводящие к
остановке синтеза белков, встречались бы гораздо чаще
(впрочем, у бактерий стоп-мутаций можно преодолевать
путем мутаций супрессора, но на этом мы
останавливаться не будем).
Все оставшиеся (числом 61) триплеты кодируют
аминокислоты, значит, одну аминокислоту могут
кодировать несколько различных триплетов. Это явление
называется вырожденностью генетического кода.
Как показано в табл. 22.1, только две аминокислоты -
метионин и триптофан - имеют по одному кодону
(для метионина - AUG).
Таблица 22.1
Генетический код
Основание
5'-конца
и
С
А
G
Среднее основание
и
UUU Phe
UUCPhe
UUA Leu
UUGLeu
CUU Phe
CUC Phe
CUA Leu
CUG Leu
AUU He
AUC He
AUA He
AUG Met*
GUU Phe
GUC Phe
GUA Leu
GUG Leu
С
UCU Ser
UCC Ser
UCA Ser
UCG Ser
CCU Pro
CCC Pro
CCA Pro
CCG Pro
ACU Thr
ACC Thr
АСА Thr
ACG Thr
GCU Ala
GCC Ala
GCA Ala
GCG Ala
A
UAU Туг
UAC Туг
UAA Стоп*
UAG Стоп*
CAU His
CAC His
CAA Gin
CAG Gin
AAU Asn
AACAsn
AAA Lys
AAGLys
GAU Asp
GAC Asp
GAA Glu
GAG Glu
G
UGU Cys
UGC Cys
UGA Стоп*
UGGTrp
CGUArg
CGC Arg
CGA Arg
CGG Arg
AGU Ser
AGC Ser
AGA Arg
AGG Arg
GGU Gly
GGC Gly
GGA Gly
GGG Gly
Основание
З'-конца
и
С
А
G
и
С
А
G
и
С
А
G
и
С
А
G
* Стоп-триплет не кодирует аминокислот
+ Триплет AUG является также кодоном, инициирующим
трансляцию
Остальные аминокислоты имеют 2 и более кодонов;
у лейцина их даже 6. Распределение кодонов не было
случайным. В тех случаях, когда одну аминокислоту
кодирует несколько кодонов, они похожи друг на друга.
Например, кодоны изолейцина - AUU, AUC и AUA -
различаются только по третьему основанию. Кроме того,
глава 22 Синтез, внутриклеточный транспорт и деградация белков
285
сходные триплеты кодируют аминокислоты,
обладающие структурным сходством: например, алифатические
аминокислоты изолейцин и лейцин (см. с. 38) имеют ко-
доны CUU (лейцин) и AUU (изолейцин). Благодаря этим
особенностям генетического кода многие мутации,
приводящие к изменению лишь одного основания,
часто не влияют на структуру синтезируемого белка
(изменение CUU на CUC не влияет на кодируемую
аминокислоту - оба кодона соответствуют лейцину)
или сопровождаются заменой на аминокислоты со
сходной структурой (изменение CUU на AUU приводит к -
замещению лейцина на изолейцин). Изолейцин и
лейцин настолько схожи по размеру и гидрофобным
свойствам, что такая замена может и не сказаться на-
функции белка. Таким образом, организация
генетического кода создает своеобразный генетический «буфер»,
при котором влияние многих точечных мутаций на
синтезируемый белок сводится к минимуму.
Как транслируются код он ы
Синтез белка, называемый трансляцией, происходит
на рибосомах. Их структуру и функции мы обсудим
позже. Сейчас же мы познакомимся с трансляцией кодонов
в аминокислоты.
В структуре аминокислоты и ее кодона нет
физического или химического сходства, которое могло бы
приводить к их непосредственной ассоциации. Поэтому
было высказано предположение о существовании адап-
торных молекул, обеспечивающих соединение
аминокислот с соответствующими кодонами. В дальнейшем
оказалось, что в роли таких адапторов выступают
небольшие молекулы РНК. Их назвали транспортными
РНК, или тРНК.
Транспортные РНК, или тРНК
Это небольшие молекулы РНК. Схематически их
структуру изображают в виде кленового листа (рис. 22.1, а).
Благодаря спариванию оснований образуется двуспи-
ральный стебель с петлей из неспаренных оснований.
Наиболее важными частями считаются 3 неспаренных
основания, которые формируют антикодон, и гибкое
плечо - З'-ССА, к которому присоединяется
аминокислота.
В процессе биосинтеза 2 молекулы тРНК должны
одновременно рядом поместиться на рибосоме, а их ан-
тикодоны - спариться с соседними кодонами на мРНК.
В реальной жизни молекулы тРНК компактно свернуты
(рис. 22.1, б). На рис. 22.2 представлена
пространственная модель тРНК.
/
А—ОН
Гибкое плечо ССА.
Аминокислоты
присоединяются к
З'-ОН концевого
адениннуклеотида
Стебли внутренних
спаренных оснований
Петли, сформированные
неспаренными основаниями
Триплет антикодона
неспаренных нуклеотидов
Антикодон
Рис. 22.1. Транспортная тРНК
а - Структура транспортной тРНК (кленовый лист)
б - схематическое изображение свернутой молекулы тРНК
Структура молекул тРНК является как бы «вещью в
себе». Хотя в тРНК есть модифицированные, необычные
основания, основными частями являются антикодон
и участок связывания аминокислоты. Антикодон -
триплет оснований, комплементарный кодону (рис. 22.3).
Таким образом, если кодоном на мРНК является UUU
(кодирующий фенилаланин), то соответствующим анти-
кодоном на молекуле тРНК будет AAA. Важно то, что
данная молекула тРНК будет связывать только
фенилаланин. Вскоре мы рассмотрим процесс присоединения
аминокислоты. Как уже отмечалось, аминокислоты
часть 4 Хранение и переработка информации
286
τ7 l· J! (.' VScV * Λ^
W^v
7Ч>^
так что U в этом положении будет
взаимодействовать с А или G на кодоне,
a G - с С или U. Это и есть
неоднозначное образование пар (см. рис. 20.17).
Понятие первое основание антико-
дона нуждается в разъяснении. Когда
последовательность оснований антико-
дона приводится сама по себе, как и
в случае любой другой нуклеотидной
последовательности, она записывается
в направлении 5'—> 3'. Однако при
взаимодействии кодон-антикодон
расположение триплетов антипараллельно,
поэтому, если мы хотим показать тот же
антикодон взаимодействующим с кодо-
ном, последний всегда приводится в
направлении 5'—> 3'; а «первое» основание
антикодона в этом случае должно быть
записано в противоположном
направлении - справа. Вот почему на схемах
молекула тРНК сама по себе
приводится (на рис. 22.1) с 5'-концом,
расположенным слева, но когда показывают ее
Рис. 22.2. Структурная пространственная модель дрожжевой тРНКРЬе
Цветом выделены конец ССА и петля антикодона
кодирует 61 кодон, поэтому можно предположить, что
для трансляции необходимо столько же молекул
различных тРНК. В действительности разновидностей тРНК
меньше. Понятно, что их должно быть хотя бы по одной
на 20 аминокислот. Некоторые молекулы тРНК могут
узнавать несколько кодонов, которые, конечно, должны
представлять одну и ту же аминокислоту. Это
достигается за счет механизма неоднозначного соответствия.
Механизм неоднозначного соответствия,
или «качания»
До сих пор, рассматривая процессы репликации и
транскрипции, мы видели незыблемость принципа Уотсона-
Крика - комплементарного спаривания азотистых
оснований. Поэтому небольшое отклонение от этого правила
при спаривании оснований кодон-антикодон может
внести некоторую неразбериху. Однако упомянутое
отклонение относится только к первому основанию антикодона
(за одним исключением, о котором позже).
«Неправильное» спаривание этого основания есть результат
гибкости близлежащей структуры тРНК (см. рис. 22.3),
*РНК 5'
кодон
Основание,
демонстрирующее
Рис. 22.3. Схематичное изображение спаривания
оснований антикодона молекулы тРНК с кодоном мРНК
Для достижения антипараллельного спаривания молекула
тРНК переворачивается. Поэтому структура тРНК,
приведенная на рис. 22.1 (с 5'-концом слева), в спаренной форме
находится в перевернутом виде (слева З'-конец); мРНК всегда
изображается 5'-концом слева
глава 22 Синтез, внутриклеточный транспорт и деградация белков
287
спаривание с основаниями кодона, то 5'-конец
расположен справа (на рис. 22.3). Таким образом, антикодон
GGC будет взаимодействовать с кодоном как показано
ниже:
3'CGG 5' антикодон
5'GCC 3' кодон
Механизм «качания» допускает неправильное
спаривание того же антикодона:
3'CGG 5' антикодон
5'GCU 3' кодон
Поскольку и GCC, и GCU кодируют одну и ту же
аминокислоту - аланин, такое спаривание не изменяет
аминокислотной последовательности синтезируемого
белка, но способствует трансляции одной тРНК
нескольких кодонов. Это позволяет клетке синтезировать
меньшее число молекул тРНК.
Эволюция позаботилась и о других путях,
допускающих более гибкое взаимодействие кодон-антикодон
без уменьшения точности трансляции. Один из них
заключается в использовании в антикодоне
нестандартного основания гипоксантина (см. рис. 18.7) для его
спаривания с С, U или А в кодонах.
Как аминокислоты присоединяются
к молекулам тРНК
Молекула тРНК присоединяет аминокислоту в
соответствие с кодоном мРНК, комплементарному антикодону.
Таким образом, молекула тРНК с антикодоном AAA
«отвечает» только за фенилаланин, так как UUU -
комплементарный ему кодон мРНК. Присоединение любой
другой аминокислоты к этой тРНК привело бы к ошибке
в структуре белковой молекулы: ведь в таком случае
фенилаланин будет замещен другой аминокислотой.
Специфичная для фенилаланина тРНК обозначается как
тРНКРИе. тРНК остальных аминокислот записываются
аналогичным образом, с использованием трехбуквенной
аббревиатуры аминокислот (заметим, что тРНКРЬе
уточняет только вид тРНК, а не то, что Phe присоединен
к ней: для этого используется обозначение Рпе-тРНКРЬе).
Присоединение аминокислот к тРНК катализируют
ферменты аминоацил-тРНК-синтетазы. В каждой клетке
должно быть по меньшей мере 20 различных видов этих
ферментов, каждый из которых присоединяет
определенную аминокислоту к соответствующей тРНК. Это
означает, что каждый фермент узнает одну или
несколько специфических тРНК и соответствующую
аминокислоту, соединяя их вместе. Узнавание синтетазой
часть 4 Хранение и переработка информации
288
соответствующей тРНК может происходить двумя
путями: в одних случаях узнается антикодон, в других -
несколько оснований в разных частях молекулы. Полная
реакция, включающая АТР в качестве источника
энергии, выглядит так:
Аминокислота + тРНК + АТР <->
<-> Аминоацил-тРНК + РР, + AMP
Этот процесс иногда называют активацией
аминокислот.
Неорганический пирофосфат гидролизуется до Р,,
что обеспечивает протекание реакции слева направо.
От правильного выбора синтетазами определенной
аминокислоты зависит точность трансляции. После
связывания тРНК со своей аминокислотой аминоацил-тРНК
поступает в белок-синтезирующий аппарат, который уже
не проверяет правильность выбора тРНК
присоединенной аминокислоты. Поэтому очень важно, чтобы
фермент, присоединяющий аминокислоты, при работе
допускал мало ошибок. Активный центр фермента обычно
высокоспецифичен относительно своего субстрата,
однако пределы точности все же существуют. Фермент
довольно легко отличает аминокислоты с сильно
различающимися свойствами, но ему намного труднее
распознать похожие аминокислоты, такие, например, как
валин и изолейцин. Напомним их структуру.
СНз
сн3 сн3 сн3 сн3
V / J \3/ 3
сн сн
CH-NH7 CH-NH7
соон соон
Валин Изолейцин
Различие в энергии связывания, обусловленное
одной СН2-группой, не может обеспечить высокой
селективности для изолейцина и валина. Без дополнительных
мер предосторожности ошибка была бы слишком
серьезной, и специфичная для лейцина аминоацил-тРНК-
синтетаза присоединяла бы валин к тРНК11е, приводя
к неприемлемым ошибкам в трансляции мРНК.
Впрочем, этого не происходит из-за наличия механизма
коррекции, который можно описать в виде суммарной двух-
стадийной реакции.
Аминокислота + АТР <-> Аминоацил-АМР + РР, (1)
Аминоацил-АМР + тРНК ^ Аминоацил-тРНК + AMP (2)
Аминоацил-АМР не покидает фермента.
Присоединение тРНК вызывает такое конформационное
изменение молекулы фермента, при котором формируется
дополнительный каталитический участок, способный
гидролизовать ошибочный аминоациладенилат. В
случае ошибочного аденилирования изолейцин-специфич-
ный фермент гидролизует валил-АМР, в то время как
изолейцил-АМР (возможно, из-за гораздо больших
размеров) в этот центр не входит. Такой механизм
уменьшает ошибку присоединения изолейцина до 1 на 60 000
молекул. Другие аминоацилсинтетазы «вычитывают
свою корректуру» аналогичным образом, гидролизуя
ошибочные аминоацил-тРНК. Гидролитический центр
а О R
3 II I
А—О—С—С—ЫНз
/ I
с н
Эфирная связь с
3 '-ОН-концевым
аденозином
Аденин
тРНК—О —Р—О—С—Η
Рис. 22.4. Присоединение аминокислоты к тРНК
а - Молекула тРНК с основаниями ССА на З'-конце, к которой
с помошью эфирной связи присоединена аминокислота;
б - структура концевого нуклеотида с присоединенной
аминокислотой. Аминоацильная группа в растворе быстро
мигрирует между 21- и З'-ОН-группами цикла рибозы. В тексте
мы рассматриваем ее в положении 3
соответствующей аминоацилсинтетазы узнает тРНК,
несущие аминокислоты меньшего размера (по
сравнению с правильным субстратом) или похожего размера,
но содержащие другие боковые группы. Например,
размеры треонина и валина схожи, однако
гидролитический центр специфичного для валина фермента
преимущественно связывает гидрофильную ОН-группу
треонина, а не гидрофобную СН3-группу валина.
Двойная избирательность - преимущественное связывание
правильной аминокислоты с тРНК и
преимущественный гидролиз аминоацил-тРНК, содержащих
неправильную аминокисоту, - уменьшает ошибку до 1 молекулы
на несколько тысяч. Не все аминоацил-тРНК-синтетазы
имеют такие механизмы коррекции. Они нужны только
для распознавания структурно похожих аминокислот.
Случайная ошибка в биосинтезе белка не дает серьезных
последствий, так как образуется множество молекул
белка, и все они обязательно подвергаются деградации
(дефектные молекулы, по-видимому, быстрее).
Подобные ошибки не так критичны, как ошибки в синтезе
ДНК; впрочем, высокая степень точности все-таки
необходима.
Молекула тРНК имеет концевую З'-тринуклеотидную
последовательность - ССА. Аминокислота соединяется
эфирной связью со свободной З'-ОН группой рибозы
последнего нуклеотида (А) (рис. 22.4). Этот тринуклео-
тид образует гибкое плечо, которое может поместить
аминоацильную группу на соответствующий
реакционный центр рибосомы (см. ниже). Эфир, образованный
аминоацильной группой, имеет тот же энергетический
уровень, что и пептид. Таким образом,
термодинамической проблемы переноса аминоацил-эфирной группы
на -ΝΗ2 другой аминоацильной группы при
образовании пептидной связи не существует. Другими словами,
энергия, необходимая для образования пептидной связи,
обеспечивается аминоацил-тРНК-синтетазой,
использующей АТР. Теперь познакомимся с организацией
аминокислот на молекулах тРНК в полипептидную цепь.
Рибосомы
Рибосомы - маленькие частицы, присутствующие
в больших количествах в клетках (за исключением
высоко дифференцированных клеток, например
эритроцитов, которые белков не синтезируют). Свое
название они получили от рибонуклеиновой кислоты (РНК),
на долю которой приходится около 60% их сухого веса.
Рибосома состоит из двух частиц: у Е. coli большая
субчастица содержит 2 молекулы РНК, а малая
субчастица - одну. Мы уже познакомились с мРНК и тРНК.
Рибосомная РНК (рРНК) также имеет свои особенности.
глава 22 Синтез, внутриклеточный транспорт и деградация белков
289
Из-за внутреннего спаривания оснований рРНК
представляет собой сильно скрученные компактные
структуры. Схема на рис. 22.5 поможет представить сложную
форму одной из молекул РНК рибосомы Е. coli. Это -
16S рРНК малой субчастицы. Большая субчастица
имеет две различные рРНК, оседающие при 23S и 5S.
С рРНК ассоциированы многочисленные белки, в
результате чего образуются плотные частицы; 34 белка входят
в состав большой субчастицы и 21 - в состав малой.
Когда речь идет об очень больших структурах, таких
как рибосомы, их размеры оценивают по скорости
седиментации (оседания) при ультрацентрифугировании,
выраженной в единицах Сведберга (Svedberg), которые
обозначаются S. Чем больше молекулы, тем быстрее они
оседают. Размер рибосомы Е. coli - 70S, а ее субчастиц -
50S и 30S (суммарная величина не является результатом
арифметического сложения субчастиц, так как
величина S зависит не только от размера, но и от формы).
Общий принцип синтеза белка в сильно упрощенном
виде выглядит так. Рибосома присоединяется около
5'-конца мРНК и затем движется вдоль мРНК по
направлению к З'-концу, соединяя аминоацильные группы
нагруженных молекул тРНК (т. е. молекул тРНК с
присоединенными молекулами аминокислот) в
полипептидную цепь в соответствии с последовательностью
Центральный домен
^J L-_,r/N rx
-\N г ! 3'-Главный
Ι,ι ) домен
( ) 3'-Конец
В 3'-Минорный домен
Рис. 22.5. Структура 16S рРНК (схематично)
кодонов. В конце мРНК рибосома встречает стоп-кодон:
в этой точке белок высвобождается, рибосома
отсоединяется и диссоциирует на субчастицы. Обратите
внимание, что «особых» рибосом нет. В данной клетке любая
рибосома может использовать любую мРНК, как плейер
может использовать любую записанную ленту;
ограничения существуют у рибосом митохондрий и хлоропла-
стов, которые отличаются от цитоплазматических
рибосом (см. с. 296).
Но это только принцип. Для понимания процесса
нам необходимо остановиться на деталях. В ходе
синтеза белковой молекулы можно выделить три стадии:
инициацию, элонгацию и терминацию.
Инициация трансляции
Трансляция начинается со строго фиксированной точки
в цепи мРНК, которая имеет 51- и З1- нетранслируемые
области и расположенную между ними кодирующую
область. Рибосома должна узнать первый триплет
кодирующей последовательности и начать трансляцию с него.
Необходима абсолютно точная инициация, поскольку
правильная трансляция мРНК рибосомой зависит от
точной установки рамки считывания. Предположим, что
кодирующая область мРНК начинается с
последовательности:
5' AUGUUUAAACCCCUG 3'.
Первые 5 аминокислот определены кодонами AUG,
UUU, AAA и т.д. Нет никаких указаний на то, какие же
основания составляют кодон; исключение составляют
три первых основания кодирующей части мРНК,
формирующие конститутивный кодон 1, следующая тройка -
кодон 2 и т.д. Между ними нет ни запятых, ни точек.
Следовательно, правильное считывание информации
определяется лишь началом считывания. Предположим,
произошла ошибка в одном основании и мы начали
считывание с основания 2. В этом случае кодоны
читались бы так:
(A) UGU, UUA, AAC, ССС, UG ...
Иными словами, транслируемые кодоны, как и
соответствующие им аминокислоты, были бы
совершенно другими. Такая ошибка получила название сдвига
рамки считывания. Мутации, вызванные делецией или
добавлением одного или двух оснований в мРНК,
вызывают сдвиг рамки считывания. Это приводит к тому, что
аминокислотная последовательность пептида,
синтезированного после сдвига рамки считывания,
оказывается ошибочной, а образовавшийся продукт не способен
часть 4 Хранение и переработка информации
290
выполнять функции белка, отобранного миллионами лет
эволюции и закодированного в данном гене. Общая
схема синтеза белка и участвующих в нем компонентов
в основном применима к про- и эукариотам, однако
существуют значительные различия в реальных
механизмах, поэтому мы рассмотрим их отдельно. Начнем
с прокариот.
Инициация трансляции у Е. coli
Как уже отмечалось, перед стартовой точкой в мРНК есть
последовательность, расположенная с 5'-конца цепи,
которую определяет рибосома. Раз инициация не
начинается с 5'-конца мРНК, то первый кодон должен быть
каким-то образом идентифицирован.
Стартовой точкой является кодон AUG (реже GUG);
однако, как ни странно, триплет AUG может
располагаться в любой части мРНК, так как он кодирует
аминокислоту метионин. Долгое время оставалось
непонятным: каким же образом рибосомы инициируют
трансляцию именно с первого AUG, а не с любого другого
AUG в составе мРНК? Почему бы для инициации не
использовать особый кодон? Оказывается, существует две
различные тРНК, специфичные для метионина. Обе
тРНК обладают одним и тем же кодоном, но одна тРНК
используется только для инициации, а другая - только
для добавления метионина в процессе элонгации.
Инициирующая трансляцию тРНК может
спариваться с кодонами AUG или GUG за счет «качания»
(см. с. 287). Обычно качание касается 5'-основания анти-
кодона. В используемой для инициации трансляции
тРНК это З'-концевое основание антикодона (рис. 22.6).
а Аминокислота
3'
\
тРНК, используемая при
элонгации трансляции
Что же определяет различные функции двух
специфичных в отношение метионина тРНК? Инициатор-
ная аминоацил-тРНК имеет структурные особенности,
которые распознаются инициаторным белком, или
фактором инициации IF2, доставляющим инициатор-
ную аминоацил-тРНК к формирующемуся инициатор-
ному комплексу. Аминоацил-тРНК, участвующая в
элонгации, распознает другой цитоплазматический фактор,
который и доставляет их к рибосоме; он не связывается
с инициаторной тРНК. У Е. coli метионин,
присоединяемый к инициаторной тРНК, подвергается формили-
рованию по NH2-rpynne. Этот процесс катализирует
трансформилаза, использующая 1Ф°-формилтетрагид-
рофолат (см. с. 219) в качестве донора формильной
группы, так что синтез белков прокариот, по непонятным
пока причинам, начинается с N-формилметионина. Фор-
мильная группа (а часто и метионин) удаляются до
завершения синтеза. Инициаторная тРНК обычно
называется TPHKfMet («f» обозначает формил), а нагруженная
аминокислотой тРНК - fMet-TPHKfMet (часто
сокращается до fMet-TPHKf). Вышеизложенное проясняет,
каким образом происходит правильное использование
метионин-специфичных тРНК для инициации и
элонгации: особые белковые факторы, участвующие в
инициации и элонгации, узнают только Met-TPHKfMet
и fMet-TPHKfMet соответственно.
В цитоплазме существует фонд свободных 30 и 50S
субчастиц рибосом, находящихся в равновесии с 70S
рибосомами. Фактор инициации (IF3) связывается
с 30S субчастицей и предупреждает на этой стадии ее
преждевременную реассоциацию с 50S субчастицей
(рис. 22.7, а). Кроме того, необходимы и другие факторы,
6 fMet
3'
\
тРНКМег, используемая
только при инициации
^трансляции
Положение
качания
мРНК 5'
Рис. 22.6. Участок антикодона, демонстрирующий положение «качания»
а - Обычные тРНК, участвующие в элонгации;
б - инициаторная TPHKfMet.
Непостоянное основание (англ. wobble base) определяется близлежащей структурой тРНК. Значение тРНКгМе' объяснено в
тексте
глава 22 Синтез, внутриклеточный транспорт и деградация белков
291
IF1 и IF2. Эти белковые факторы связываются с 30S
субчастицами, участвуют в процессе инициации и
освобождаются в цитоплазму, чтобы функционировать вновь
в новом раунде инициации. Функция IF1 неизвестна,
a IF2 необходим для связывания fMet-TPHKfMet.
мРНК, fMet-TPHKfMet и GTP образуют комплекс
с 30S субчастицей, a IF3 высвобождается в цитоплазму.
Как показано на рис. 22.7, б, в мРНК существует
последовательность оснований, известная как
последовательность Шайна-Дальгарно, которая
комплементарна участку 16S рРНК. При их связывании мРНК
должным образом располагается на малой субчастице
рибосомы, тРНК подходит к Р-участку субчастицы, а ее
антикодон спаривается с кодоном AUG (рис. 22.8).
50S
30S
50S
+
30S
[F3
30S
if3
70S рибосома, которая :
только что завершила цитопла3матический фактор
синтез оелка и инициации предупреждает
отсоединилась от мРНК реассоциаци|о р'ибосомальных
субчастиц
б Последовательность Инициаторный Кодон 2
Шайна-Дальгарно кодон AUG
5'
AUG
Ύ
мРНК
30S
Фрагмент 16S рРНК из 30S субчастицы,
содержащий последовательность,
комплементарную последовательности
Шайна-Дальгарно; это связывание
и определяет точное положение
для инициаторного кодона AUG
Рис. 22.7. Влияние фактора инициации трансляции IF3
и последовательности Шайна-Дальгарно на субчастицы
70S рибосом
а -Диссоциация 70S рибосомы на 50S и 30S субчастицы.
Фактор инициации IF3 предупреждает реассоциацию 50S и 30S
субчастиц. При инициации трансляции IF3 должен быть
высвобожден присоединения 50S;
б - рисунок, показывающий роль последовательности Шайна-
Дальгарно в расположении 30S рибосомы Е. coli на мРНК
при инициации. Ρ - пептидильный участок, А - аминоациль-
ный центр. Участки Ρ и А окончательно формируются только
при присоединении 50S субчастицы
После высвобождения IF3 этот комплекс соединяется
с 50S субчастицей, что сопровождается гидролизом GTP
и высвобождением GDP и Pf, а также IF 1 и IF2 (см.
рис. 22.8). Теперь мы имеем полную 70S рибосому
с мРНК, расположенной в Р-участке с fMet-тРШ^
(антикодон которой спарен с инициаторным кодоном AUG)
и свободным Α-участком, ожидающим доставки второй
аминокислоты соответствующей тРНК. Инициация
завершена.
Частично сформированные участки
Ρ ι
ι
30S
IF1,2h3
GTP n
Последовательность
Шайна-Дальгарно в
мРНК, основания
которой спариваются с
поеледовател ьностью
16S рРНК
τ
fMet К
fMet-TPHKMet
мРНК
F3
fMet-TPHKMet
, Р. , Α
мРНК 4 AUG N
30S
50S субчастица
Ν
k
GDP + P;
IF1, IF2
Сформированные
участки v
50S
fMet] Г
.. ι
*>- ι
ι >«
Ι ι
I I
, Ρ ι ι Α
мРНК
5'
AUG
30S
Рис. 22.8. Инициация трансляции у Е. coli
Инициаторная тРНК - TPHKfMet показана серой линией;
антикодон - короткой горизонтальной линией. Фактор IF2
доставляет fMet-TPHKfMet к 30S субчастице. NNN - любой кодон (N -
любой нуклеотид). Рибосома имеет также центр выхода, не
показанный на диаграмме
часть 4 Хранение и переработка информации
292
Некоторые бактериальные мРНК - полицистронны;
например lac мРНК. На одной молекуле мРНК находятся три
области, кодирующие 3 различных белка. В этой ситуации
каждая кодирующая область соседствует с последова-
тельностю Шайна-Дальгарно, и в каждом случае
инициация трансляции происходит независимо (рис. 22.9).
Инициаторные кодоны AUG
AUG
AUG
AUG
Последовательности Шайна-Дальгарно
Рис. 22.9. Структура полицистронной мРНК прокариот
Кодирующие области lac мРНК обозначают ζ, у, и а (см. рис. 21.9)
Элонгация - следующий
за инициацией этап трансляции
Механизм элонгации
Начнем с инициаторного комплекса, приведенного на
рис. 22.10, а. В этом комплексе есть fMet-TPHKfMet
в Р-участке и свободный Α-участок. Только в процессе
инициации Р-участок принимает тРНК, нагруженную
аминокислотой - N-формилметионином; все
последующие аминоацил-тРНК поступают в Α-участок. Амино-
ацил-тРНК (отличные от инициаторных) образуют
комплекс с фактором элонгации EF-Tu, связанным с
молекулой GTP. Комплекс EF-Tu-GTP-аминоацил-тРНК
связывается с рибосомой таким образом, что аминоацил-
тРНК занимает Α-участок, а ее антикодон располагается
у кодона мРНК (рис. 22.10, б). Важно отметить, что
Ρ
fMet
мРНК
5'
АА2
Доставка
АА2-тРНК
Аминоацил-тРНК,
связанная
с EF-Tu-GTP
fMet AA2
Цитоплазматические факторы
элонгации
В цитоплазме есть два растворимых белковых фактора
элонгации. Они являются G-белками, которые в
комплексе с молекулой GTP могут связываться с
рибосомами. Оба белка представляют собой латентные вТРазы.
На рибосоме они гидролизуют связанную молекулу GTP
до GDP и Pf.; освобождение последнего вызывает кон-
формационные изменения в данных белках. В
комплексе с GDP они отсоединяются от рибосомы. В цитоплазме
GDP обменивается на GTP, и факторы готовы
участвовать в следующем этапе элонгации.
Этими двумя факторами являются EF-Tu - нетермо-
стабильный фактор элонгации (англ. elongation factor
temperature unstable) и EF-G. Задача EF-Tu состоит в
доставке аминоацил-тРНК к рибосоме. Как только ами-
ноацильная группа добавится к растущему пептиду,
EF-G способствует продвижению рибосомы по мРНК
в направлении 51—» З1 к следующему кодону.
Эти два фактора попеременно двигаются по
рибосоме в связанном с GTP состоянии, выполняют свои
задачи и отсоединяются в связанном с GDP состоянии,
чтобы затем повторно участвовать в последующих раундах
элонгации. А теперь мы можем перейти к детальному
рассмотрению синтеза пептидной связи.
tMet
AA2
Доставка
АА9-тРНК
GTP
Пауза для «чтения ч
корректуры» \
(гидролиз GTP) \
Пептидилтрансферазная»
Транслокация реакция не происходит N
/ ^ \ до тех пор. пока не I
высвободится EF-Tu
ρ,
+
GDP
EF-G-GTP
fMet AA2
OH
fMet
AA9
Дипептидил-тРНК
Синтез пептидной
связи
Теперь может произойти
пептидилтрансферазная реакция
Рис. 22.10. Процесс элонгации
тРНК указаны в виде серых линий; АА - аминоацильная
группа. Положение EF-Tu-GTP на тРНК и на рибосоме показано
условно
глава 22 Синтез, внутриклеточный транспорт и деградация белков
293
EF-Tu-GTP не связывается с fMet-TPHKf (инициаторной
тРНК), а взаимодействует только с аминоацил-тРНК,
участвующими в элонгации. Таким образом, инициация
и элонгация осуществляются раздельно.
Концентрации EF-Tu в клетках Е. coli вполне достаточна для
связывания всех аминоацил-тРНК. На рибосоме EF-Tu,
обладающий вТРазной активностью, гидролизует
связанную молекулу GTP до GDP и Р/5 высвобождая
последний. Это служит сигналом для отделения EF-Tu от
аминоацил-тРНК, по-видимому, вследствие аллостери-
ческих изменений, a EF-Tu-GDP покидает рибосому
для регенерации в ходе обмена GDP-GTP в цитоплазме
(рис. 22.10, в). Аминоацильные группы на двух
молекулах тРНК, расположенных в Р- и Α-участках,
находятся вблизи рибосомной пептидилтрансферазы:
последняя катализирует перенос fMet от тРНК,
расположенной в Р-участке, на свободную аминогруппу
аминоацил-тРНК в Α-участке, образуя дипептид,
присоединенный ктРНК (рис. 22.10, г). Мы описали пепти-
дилтрансферазу как фермент, но есть данные,
свидетельствующие о том, что эта активность сохраняется
в рибосомах даже после экстракции 95% рибосомного
белка. Это убедительный довод в пользу того, что
данная активность является свойством рибосомной РНК.
Схема пептидилтрансферазной реакции выглядит так:
тРНК—О—СО—fMet +
(Р-участок)
NH2—СН—СО-О—тРНК-
ή· (А-участок)
-тРНК— ОН + fMet—CO-NH-CH—СО-О—тРНК
(Р-участок) ρ· (А-участок)
После синтеза первой пептидной связи А-участок
заполнен пептидил-тРНК, а Р-участок содержит ненагру-
женную тРНК. Рибосома передвигается на один кодон
вдоль мРНК: этот процесс называется транслокацией.
Освободившаяся тРНК двигается к «выходу» - центру,
расположенному на 50S субчастице, из которого она
высвобождается для последующего использования (рис.
22.10, д). Для движения рибосомы по мРНК необходим
фактор EF-G, который известен также как транслоказа.
Для процесса транслокации необходим гидролиз GTP.
Связывание EF-Tu-GTP и EF-G на рибосоме
происходит таким образом, что в данный момент времени
только один из факторов может быть связан с рибосомой.
Таким образом, синтез пептида и транслокация происходят
поочередно (друг за другом).
На рис. 22.10 не уточнен механизм, при участии
которого пептидилтрансферазная реакция и транслокация
проходят от стадии (в) до (г). Недавние исследования
показали, что в ходе этого процесса каждая тРНК занимает
на рибосоме два участка (рис. 22.11). После освобождения
от пептидильной группы тРНК занимает участки Ρ и Ε
(от англ. exit - выход); антикодоновый конец молекулы
по-прежнему находится в Р-участке, но другой конец -
уже в Ε-участке. Аналогично этому тРНК, расположенная
в Α-участке (и несущая пептид), занимает А и Р-участки.
Обе модели основаны на наблюдении, что тРНК
занимают участок Р/Е и А/Е на рибосоме. Согласно
модели I, АА-тРНК раскачивается для того, чтобы сделать
шаг; модель II предусматривает формирование на
рибосоме гибридных участков. Предполагается, что синтез
происходит таким образом, что пептид всегда остается
в одном положении относительно большой субчастицы.
Для объяснения механизма этого процесса
предложены две альтернативные модели. Модель I (см.
рис. 22.11) предусматривает, что один конец аминоацил-
тРНК раскачивается так, как показано на рисунке;
происходит перенос пептида, а освободившаяся тРНК
поворачивается одним из концов к Ε-участку. Следующая
затем транслокация вновь возвращает нас к ситуации,
показанной на рис. 22.10, г, когда все готово для
следующего раунда элонгации.
ЕРА
ОН
Перенос пептида
ЕРА
ОН ^ ' Модель 1
Транслокация
I
Кодон
Модель 2
Ε/Ρ Ρ/Α AJ-
ОН
3
1
Движение 50S
субчастицы
и перенос пептида
Движение 30S
субчастицы
Рис. 22.11. Альтернативные модели транслокации и
синтеза пептида на рибосоме
На этом рисунке для примера выбран синтез первой
пептидной связи, так что серый кружок обозначает fMet,
присоединенный к тРНК в Р-участке, который располагается у первого
кодона мРНК; в конце этого раунда Р-участок располагается
у второго кодона. Прямоугольники обозначают участки
связывания, Ε - центр выхода, Ρ - пептидильный участок, А -
акцепторный участок. Е/Р и А/Р - гибридные центры,
образованные движением большой субчастицы относительно малой
часть 4 Хранение и переработка информации
294
Согласно альтернативной модели II, движется
большая субчастица рибосомы, а малая остается
неподвижной. Это приводит к образованию гибридных центров
связывания Р/Е и А/Р. После пептидилтрансферазной
реакции гибридные участки оказываются занятыми
(см. рис. 22.11). Транслокация малой субчастицы затем
приводит к ситуации, предусмотренной моделью I,
когда все готово к приему следующей аминокислоты.
Согласно обеим моделям, синтезирующийся пептид
остается в фиксированном положении относительно
большой субчастицы. Это позволяет преодолеть
проблему физического передвижения тРНК, нагруженной
достаточно большим пептидом. С помощью модели II
можно также объяснить, почему рибосомы всегда состоят
из двух субчастиц. Рибосома имеет очень сложную тун-
нелеподобную структуру (рис. 22.12).
Итак, рибосома теперь находится в состоянии,
показанном на рис. 22.10, д. Р-участок занят дипепти-
дил-тРНК. Α-участок, расположенный у третьего кодо-
на, свободен. Элонгация полипептидной цепи включает
повторные циклы одного и того же процесса. Амино-
ацил-тРНК, образующая комплекс с EF-Tu-GTP,
доставляется в Α-участок, на нее переносится пептидильная
группа с Р-участка; освободившаяся тРНК движется
к «выходу» (Ε-участок), пептидил-тРНК перемещается
из Α-участка в Р-участок, а рибосома перемещает один
кодон - и весь процесс начинается вновь. Если
синтезируется белок, насчитывающий 200 аминокислот,
конечный раунд включает перенос пептида длиной 199
аминокислот на последнюю аминокислоту и ее тРНК, и это
Рис. 22.12. Рибосома с вероятным расположением рибо-
сом-связанных тРНК в А/А-, Р/Р- и Ε-центрах (схематично)
Серая область справа - примерный участок взаимодействия
EF-Tu. На рисунке показана полярность фрагмента мРНК,
содержащей кодоны в А- и Р-участках. Стрелки - вероятный
путь тРНК на рибосоме
глава 22
приводит к образованию комплекса белок-тРНК.
Образно говоря, собака добавляется к хвосту, а не наоборот.
Вначале синтезируется N-конец полипептидной цепи,
а последняя присоединяемая аминокислота формирует
С-конец.
Точность трансляции достигается взаимодействием
кодон-антикодон, которое и определяет выбор
соответствующей тРНК; однако пока неясно, каким образом
происходит подобная селекция. Дело в том, что EF-Tu
«не узнает», какая аминоацил-тРНК должна быть
следующей, и похоже, что этот фактор доставляет их
в Α-участок беспорядочно. Существует гипотеза,
согласно которой «неузнаваемый» комплекс амино-
ацил-тРНК-EF-Tu-GTP диффундирует еще до реакции,
поскольку он не будет удерживаться в Α-центре так же
прочно, как это происходит при формировании
водородных связей в ходе правильного взаимодействия кодона
с антикодоном. Синтез пептида не происходит до тех пор,
пока не будет высвобожден EF-Tu-GDP, а это может
случиться только после гидролиза GTP. Предполагается, что
небольшая задержка нужна для «вычитывания
корректуры», т. е. для обеспечения эффективности процесса.
Кроме того, рибосомы, как полагают, могут каким-то
(пока невыясненным) образом участвовать в узнавании
правильной аминоацил-тРНК. Но пока это только
предположение, для объяснения которого еще не предложен
механизм.
Терминация биосинтеза белка
На конце мРНК находится по крайней мере один из трех
стоп-кодонов, называемых также терминаторными,
для которых не существует tRNA: это триплеты -
UAG, UAA и UGA. Когда рибосома достигает одного
из стоп-кодонов, особый цитоплазматический фактор
высвобождения белка (фактор терминации)
способствует отделению готового полипептида от тРНК,
вызывая изменения пептидилтрансферазы, в результате
которых последняя гидролизует эфирную связь между
СООН-группой белка и ОН-группой 3'-концевого нук-
леотида. Рибосома отсоединяется от мРНК и
диссоциирует на субчастицы, готовые к следующей инициации.
Известны два фактора, узнающих различные стоп-кодоны.
Что такое полисома?
Синтез одного белка Е. coli на рибосоме,
присоединяющей около 15 аминокислот в секунду, занимает около
20 секунд. Однако, как только инициированная рибосома
тез, внутриклеточный транспорт и деградация белков
295
продвинулась примерно на 30 кодонов, может
произойти еще одна инициация трансляции, так что рибосомы
одна за другой будут двигаться по одной мРНК, и
каждая независимо друг от друга будет синтезировать
белковую молекулу. Обычно на 1 молекулу мРНК
приходится около 5 рибосом; впрочем, их количество
изменяется в зависимости от длины мРНК. Такая
структура из нескольких рибосом называется полисомой.
Чем отличается синтез белка у
эукариот?
У эукариот процесс трансляции, в сущности, протекает
так же, как и у прокариот, правда, с некоторыми
отличиями. Рибосомы эукариот больше (80S, с
субчастицами 40S и 60S) и содержат больше молекул рРНК и
белка. Первой аминокислотой в синтезируемом белке всегда
является метионин, однако метионил-тРНК,
используемая для инициации, не формилируется (значение фор-
милирования у прокариот непонятно). У эукариот, как
и у прокариот, для инициации трансляции существует
особая метионил-тРНК, которая отличается от тРНК,
используемой в ходе элонгации. Это не означает, что все
белки эукариот начинаются с метионина, так как часто
эта аминокислота удаляется впоследствии из
полипептидной цепи. Сборка 40S инициаторного комплекса
аналогична образованию 30S инициаторного комплекса
прокариот. Образовавшийся комплекс затем
взаимодействует с большой 60S субчастицей рибосом,
окончательно формируя инициаторный комплекс. Процесс
биосинтеза белка также сопровождается гидролизом
GTP. Однако у эукариот правильное расположение
мРНК - строгое соответствие Р-участка инициаторному
кодону AUG - обеспечивает совершенно иной механизм.
Напомним, что с 5'-конца мРНК эукариот кэпированы
метилированным гуаниновым нуклеотидом (с. 270).
В мРНК эукариот нет последовательности Шайна-Даль-
гарно; вместо нее к кэпу присоединяется группа
белковых факторов, которая объединяется с 40S рибосомной
субчастицей (рис. 22.13). Таким образом, с каждого
5'-конца мРНК на некотором расстоянии от кодона AUG
мы имеем 40S преинициаторный комплекс. При помощи
АТР-зависимого механизма комплекс 40S субчастицы
движется вдоль мРНК до тех пор, пока не встретится
первый триплет AUG: тогда происходит присоединение
60S субчастицы и завершение инициации. Этот процесс
сопровождается гидролизом GTP. В отличие от мРНК
прокариот, которые часто бывают полицистронными
и содержат несколько последовательностей Шайна-
Дальгарно, обеспечивающих инициацию трансляции
часть 4 Хранение и переработка информации
296
Met
+ elF2 + GTP + кэп-белки и другие факторы инициации
Met-TPHKMet /
(инициаторная аминоацил-тРНК) /
Met χ „
X + 40S субчастица рибосом
/ + мРНК
σπ
1F2
AUG мРНК
5' 3'
иле Комплекс движется по мРНК
: до тех пор, пока не достигнет
Комплекс кэп-белков первого AUG кодона
и других факторов I
У5-концамРНК Теперь присоединяется
субчастица 60S, а Р|Ч GDP
и факторы инициации I
высвобождаются *
60S _
Met '
Инициация трансляции
завершена
I I
I I
Ι ι ■
мРНК ΝΝΓ
5' 3'
40S
Рис. 22.13. Упрощенное изображение инициации у эукариот
Помимо elF2 в этом процессе участвует несколько факторов
инициации. тРНКМе' - инициаторная РНК; elF2 - фактор
инициации эукариот, соответствующий фактору IF2 прокариот
каждого цистрона, мРНК эукариот - моноцистронны
(кодируют один полипептид) и содержат только один
инициаторный участок на молекулу. Функции прокари-
отических EF-Tu и EF-G у эукариот выполняют EFla
и EF2 соответственно.
Как происходит синтез белка
в митохондриях?
Митохондрии содержат ДНК и имеют собственный бе-
лок-синтезирующий аппарат. Широко распространена
гипотеза о том, что митохондрии будто бы произошли
из прокариотических клеток, которые внедрились в
клетки эукариот (с. 282).
Рибосомы митохондрий, подобно прокариотам, для
инициации используют fMet-TPHKfMet. Для них
характерны другие интересные особенности: несколько
отличающийся генетический код и более простые
взаимодействия кодона и антикодона, вследствие чего
митохондрии млекопитающих могут обходиться 22 видами
тРНК. Такие упрощения возможны, по-видимому,
потому, что митохондрии синтезируют ограниченное
количество различных белков. Митохондрия - не
автономное образование. Большинство митохондриальных
белков кодируются генами клеточного ядра и
транспортируются в эти органеллы. Считается, что и хлоропласты
также произошли из внедрившихся в эукариотические
клетки фотосинтетических прокариот.
Влияние антибиотиков
и токсинов на синтез белка
Антибиотики - химические «ракеты» микроорганизмов:
последние выпускают их друг в друга в борьбе за
выживание. Антибиотики атакуют наиболее важных
участников клеточного метаболизма, некоторые из них
задействованы и в процессе трансляции.
У прокариот стрептомицин нарушает инициацию,
кирромицин предупреждает высвобождение EF-Tu,
а эритромицин и хлорамфеникол ингибируют пепти-
дил-трансферазу (последнее также справедливо и для
митохондриальных рибосом). Фусидовая кислота
тормозит транслокацию, блокируя высвобождение EF-G-GDP.
Кроме того, дифтерийный токсин ингибирует
ЕР2-транслоказу эукариот (которая соответствует EF-G
у бактерий). Рицин - токсин клещевины
обыкновенной - это N-гликозидаза, удаляющая адениновое
основание в одной из рибосомных РНК эукариот и инактиви-
рующая большую субчастицу. Одна молекула рицина
может разрушить клетку, содержащую десятки тысяч
рибосом.
Как сворачивается
полипептидная цепь,
синтезированная на рибосоме?
Не так давно было принято считать, что после
образования полипептидной цепи на рибосоме белок
автоматически сворачивается в трехмерную структуру за счет
взаимодействия аминокислотных остатков. Конечно,
последнее очень важно, поскольку мы знаем, что структуру
белка определяет только один тип информации -
последовательность оснований в гене, которая в ходе
трансляции преобразуется в аминокислотную
последовательность полипептида. Более того, существует
экспериментальное подтверждение этой концепции. Фермент
рибонуклеазу денатурировали, обрабатывая мочевиной,
нарушающей водородные связи, и восстанавливая
дисульфидные связи. После удаления мочевины при
диализе и реокислении часть фермента вновь
правильно сворачивалась и даже проявляла каталитическую
активность. Таким образом, аминокислотная
последовательность белка, вне всякого сомнения, определяет его
нативную конфигурацию. Однако в полипептидной
цепи есть практически безграничные возможности
ассоциации аминокислотных остатков друг с другом.
Фрагмент из гидрофобных аминокислот в одной части
цепи может - по мере синтеза белка - вступать во
взаимодействие с другим гидрофобным фрагментом;
однако этот контакт может быть совершенно «непригодным»
и отсутствовать в нативном белке. Теоретически
можно предположить, что полипептид «пробует» любой
мыслимый вариант внутренней ассоциации, пока
не достигнет свободной энергии нативного белка. Это
беспорядочный метод фолдинга. Для такого
процесса, как было подсчитано, необходимы многие
миллионы лет, в то время как в клетке все происходит за
считанные минуты.
После повторного сворачивания (рефолдинга) рибо-
нуклеазы подобные эксперименты были проведены
in vitro с другими небольшими белками, состоящими
из одного домена. Попытки рефолдинга более крупных
(особенно мультидоменных) белков были менее
успешными. Поскольку большинство белков принадлежит
к последней категории, очевидно, что сворачивание
крупных молекул представляет серьезную проблему
Механизм фолдинга белков in vivo, под которым
мы понимаем пути приобретения вновь
синтезированными пептидами правильной трехмерной
конфигурации, окончательно не выяснен. Считается, что
определенные фрагменты полипептида могут быстро принять
вторичную структуру, и это каким-то образом облегчает
правильный фолдинг всей молекулы. Последнее
предположение лежит в основе концепции модульного
фолдинга, при котором быстро образуются свернутые
модули, способствующие фолдингу остальной
молекулы. Таким образом, фолдинг белка, по-видимому,
происходит в несколько этапов, однако детали этого процесса
пока остаются невыясненными.
До сих пор мы обсуждали фолдинг белка как
автономный процесс, осуществляющийся без
посторонней помощи. Однако существуют два класса белков,
глава 22 Синтез, внутриклеточный транспорт и деградация белков
297
участвующих в фолдинге. К первому относятся
традиционные ферменты, ко второму - молекулярные шапе-
роны. В таком порядке мы их и рассмотрим.
Ферменты, участвующие в фолдинге
белка
Первый фермент - протеин-дисульфидизомераза
(ПДИ), которая «перемещает» S-S- связи в
полипептидной цепи. Если образовалась ошибочная S-S-связь, то
в силу своей ковалентной природы она не может
разорваться спонтанно, и поэтому полипептид будет
зафиксирован в неправильной конфигурации. Разрушая и вновь
образуя S-S-связи между различными остатками цисте-
ина, ПДИ способствует коррекции фолдинга. Высокие
концентрации ПДИ обнаружены в эндоплазматическом
ретикулуме, где происходит сворачивание белков,
доставляемых для секреции, причем, многие из них имеют
дисульфидные связи.
Другой фермент - пептидилпролин-изомераза
(ППИ) - катализирует цис-, гяряноизомеризацию
пептидных связей пролина, что помогает белку
принять правильную конфигурацию. Обнаружен целый
класс совершенно удивительных белков, названных
циклофилинами, которые обладают пролинизоме-
разной активностью. Они связывают антибиотик-им-
мунодепрессант циклоспорин (используемый при
трансплантации органов). Связь между пролин-изо-
меразой и биологической ролью циклофилинов до
сих пор непонятна.
Молекулярные шапероны и фолдинг
Шапероны (из англ.; букв. - пожилая дама,
сопровождающая молодую девушку на балах) - семейство
специализированных внутриклеточных белков, обеспечивающих
быстрое нахождение правильной пространственной
структуры. Они могут узнавать и связываться с частично
свернутыми (или развернутыми) белками. При выходе из
рибосомы в белке могут происходить «ошибочные»
взаимодействия, например, между гидрофобными фрагментами; это
будет препятствовать правильному сворачиванию
молекулы. Связывание шаперонов с подобными фрагментами
стабилизирует частично свернутую молекулу до того
момента, пока не произойдет правильный фолдинг белка.
Это означает, что шапероны должны «отойти» от
полипептида: после диссоциации возникает благоприятная
возможность завершения правильного фолдинга. Хотя
детали этого механизма остаются неясными, очевидна важная
роль шаперонов в фолдинге полипептидных цепей. О
сложности системы свидетельствует и необходимость АТР для
часть 4 Хранение и переработка информации
298
отделения шаперонов от полипептидной цепи. Обратите
внимание, что шапероны имеют отношение к кинетике
процесса фолдинга, а не к третичной структуре белка,
определяемой аминокислотной последовательностью.
Болезни, вызываемые прионами,
и фолдинг белка
Существует группа смертельно опасных
неврологических болезней, поражающих человека и животных.
Она включает болезнь Крюцфел ьда-Якоба и куру у
человека. Последняя известна как «смеющаяся болезнь»
из-за вызываемых ею гримас на лице. Предполагается,
что в некоторых племенах Новой Гвинеи куру
заражаются при каннибализме. У овец эту болезнь называют
почесухой, потому что животные счесывают шерсть
о столбы изгороди. У коров ее именуют губчатой
энцефалопатией (англ. bovine spongiform
encephalopathy, BSE), или болезнью коровьего бешенства. Эти
болезни могут передаваться при употреблении в пищу
инфицированной ткани. Реже встречаются
наследственные заболевания. Раньше считали, что все
перечисленные заболевания вызываются «медленными вирусами»,
потому что они заразны и развитие их происходит
исподволь в течение ряда лет. Однако любые попытки
обнаружить нуклеиновые кислоты в инфекционном
материале, выделенном из мозга, оказались
безуспешными, что позволило исключить возможность участия
какого-либо инфекционного агента (например вируса)
в развитии заболевания. Впрочем, инфекционный агент,
по-видимому, реплицируется.
Оказалось, что инфекция обусловлена устойчивой
к протеазам формой нормального белка - прионовым
белком, (англ. prion protein; PrP), обнаруженным в
мозге. Вызывающий болезнь белок называется прионом
по аббревиатуре от англ. proteinaceous infectious particle
(белковая инфекционная частица).
Белок, заражающий мышей почесухой, называется
PrPsc, а его нормальный «двойник» - РгРс («с» - от англ.
constitutive, конститутивный). Оба представляют собой
один и тот же полипептид и кодируются одним геном,
однако их конформации различны: PrPsc
характеризуется высоким содержанием β-слоев, которые
практически отсутствуют у РгРс. В отличие от РгРс, белок PrPsc
легко образует агрегаты, приводящие к образованию
амилоидных бляшек, свойственных болезням,
вызываемым прионами. Возникает вопрос: каким образом
неправильно свернутый белок может стать инфекционным
и воспроизводить себя? Ведь ни один из рассмотренных
нами биохимических механизмов образования белка
непозволяет ему управлять своей собственной
репликацией. Тем не менее, существуют веские доказательства
того, что PrPsc каким-то образом переводит РгРс
(нормальный белок) в аномальную форму. Это было
подтверждено после совместной инкубации двух белков.
Модели механизма данного превращения
подразумевают участие шаперона и источника энергии в
раскручивании белка РгРс и его рефолдинг под влиянием
молекулы PrPsc. Согласно альтернативной модели
«образования ядра», молекулы РгРс захватываются
агрегатом PrPsc, после чего и происходит конформационная
реорганизация РгРс. Из-за ограниченных
экспериментальных возможностей до сих пор нет доказательств
приобретения белком in vitro инфекционных свойств
в ходе описанных выше событий.
В отсутствие инфекции превращение РгРс в PrPsc
происходит нечасто, поэтому спонтанные случаи
заболевания встречаются чрезвычайно редко. Считается,
будто мутации в гене, кодирующим нормальный РгР,
могут увеличить вероятность этого посттрансляционного
превращения, что приводит к появлению
наследственных случаев болезни. Таким образом, однажды
возникнув, PrPsc способен запустить свое дальнейшее аутока-
талитическое образование.
Другие функции
молекулярных шаперонов
Мы рассмотрели роль шаперонов в фолдинге белка, но
это лишь часть выполняемых ими функций. Как уже
отмечалось, шапероны способны присоединяться к
несвернутым или частично свернутым белкам: полипептид,
сошедший с рибосом, - один из них.
Однако белки могут частично денатурировать.
Например, до определенной степени разворачиваться под
действием различных факторов: тепла, чрезмерного
окисления, ионизирующей радиации,
ультрафиолетового света и т.д. Денатурацию белка также могут вызвать
различные клеточные стрессы. Длительное время
считалось, что тепловой шок индуцирует синтез особых
белков, роль которых, как полагали, заключается в
защите клетки. Если температуру Е. coli поднять до 42° С,
произойдет вспышка синтеза белков теплового шока
(с. 267). Оказалось, что это шапероны. Поэтому иногда
их называют белками стресса.
Кроме участия в фолдинге вновь синтезированных
белков, шапероны играют важную роль в разворачивании
и стабилизации денатурированных белков, а также в ме-
чении состарившихся белков с целью дальнейшей их
деструкции лизосомами. Шапероны также необходимы для
переноса белков через митохондриальные мембраны, для
реорганизации субъединиц в комплексах и для сборки
олигомерных белковых комплексов. Особенно удачное
определение этого класса белков дали Хендрик и Хортл
(см. раздел «Дополнительная литература»): «В настоящее
время мы определяем молекулярный шаперон как белок,
который связывается и стабилизирует неустойчивую
форму другого белка. Контролируя связывание и
высвобождение этого белкового субстрата, шаперон способствует
образованию его правильной формы in vivo: будь это фол-
динг, олигомерная сборка, транспорт в определенный
компартмент клетки или контролируемое переключение
активной/неактивной конформаций».
Последнее положение, касающееся переключения
конформаций, относится к внутриклеточным
рецепторам глюкокортикоидных гормонов (см. с. 346).
Для выполнения столь разнообразных функций
нужны различные шапероны, и в настоящее время уже
известны многие из них.
Как синтезированные белки
доставляются по назначению?
В клетке существует только один тип рибосом, и все они
локализованы в цитоплазме клетки (за исключением
рибосом прокариотического типа, локализованных
в митохондриях и хлоропластах). Какой белок будет
в данный момент времени синтезироваться на рибосоме,
зависит только от мРНК, которая в данный момент
времени транслируется. У вновь синтезированных белков
различные конечные пункты назначения. Многие из них
являются цитоплазматическими, после синтеза
высвобождаются из рибосом и остаются в цитоплазме;
вместе с тем многие белки локализованы в мембране клетки.
Как они туда попадают? Рассмотрим клетку печени: она
продуцирует белки плазмы крови. Это крупные белки,
а клеточные мембраны устроены так, что не допускают
«утечку» белков. Каким же образом происходит
избирательное высвобождение именно белков крови?
Аналогичный вопрос возникает и при высвобождении любых
внеклеточных белков, например, пищеварительных
ферментов или инсулина из поджелудочной железы. Есть
и другие проблемы. Большинство митохондриальных
белков синтезируется в цитоплазме. Каким образом
происходит их избирательная транспортировка в митохондрии?
Лизосомы и пероксисомы (см. главу 16) - окруженные
мембраной пузырьки, наполненные ферментами; но они
не могут синтезировать белки. Каким образом последние
транспортируются в эти пузырьки?
Сейчас мы познакомимся с различными типами
перемещения (транслокации) белков. Начнем с тех,
которые сначала транспортируются в эндоплазматический
ретикулум (ЭР).
глава 22 Синтез, внутриклеточный транспорт и деградация белков
299
Что такое
эндоплазматический ретикулум (ЭР)?
Если посмотреть на срез печени или поджелудочной
железы в электронный микроскоп, внутри клетки
можно увидеть большое количество мембран,
ограничивающих огромное количество сплющенных полостей. Эта
сложная сеть взаимосвязанных полостей называется
эндоплазматическим ретикулумом (ЭР). Цитоплазма
находится снаружи ЭР, а просвет ЭР представляет
собой отдельный компартмент, ограниченный мембраной
ЭР (см. электронную микрофотографию на рис. 3.15).
Часть ЭР усеяна рибосомами, обусловливающими
шероховатость ЭР в электронном микроскопе: это
шероховатый эндоплазматический ретикулум. У
части ЭР рибосомы отсутствуют, поэтому такой ЭР
называют гладким эндоплазматическим ретикулумом
(рис. 22.14). Белки, предназначенные, например, для ядра
или митохондрий, сначала транспортируются в просвет
шероховатого эндоплазматического ретикулума; это
подводит нас к следующему вопросу.
Как белки проходят через мембрану ЭР?
Белок, который должен проникнуть через плазматическую
мембрану, внутрь лизосом или в просвет ЭР, имеет на
N-конце лидерную последовательность, обычно
состоящую из 25±11 аминокислот. Лидерная аминокислотная
последовательность определяет общее структурное
свойство многих белков, транспортируемых в различные
компартменты (рис. 22.15). Зрелый белок высвобождается
после протеолитического расщепления в участке
со специфической последовательностью.
Обратите внимание, что в цитоплазме свободная
рибосома сначала синтезирует лидерную
последовательность белка, которая сразу же распознается
сигнал-узнающей частицей (англ. signal recognition
particle; SRP). Последняя представляет собой цито-
плазматический комплекс РНК-белок. Эта частица
связывается с комплексом рибосома-синтезирующийся
пептид, препятствуя дальнейшей элонгации
полипептидной цепи. Механизм, обеспечивающий перенос
«арестованной» рибосомой растущего пептида через
мембрану ЭР, довольно сложен и до конца не изучен;
впрочем, ряд важных моментов уже известен (рис. 22.16).
В мембране ЭР есть рецепторы SRP, или так
называемые докинг-белки, к которым присоединяется цитоп-
лазматический комплекс рибосома-SRP. В результате
серии последовательных этапов, в ходе которых
происходит гидролиз одной молекулы GTP до GDP,
рибосома присоединяется к мембранному белку, который
образует полипептид-транслоцирующую пору, иногда
называемую транслоконом (см. рис. 22.16), a SRP
освобождается в цитоплазму для повторного использования.
Растущий полипептид переносится через белковую пору
в мембране в виде петли с сигнальным пептидом,
который прикрепляется к поре (см. рис. 22.16).
Расположенная рядом с порой сигнальная пептидаза отщепляет
лидерную последовательность, а полипептид поступает
в просвет ЭР.
Гладкий эндоплазматический
ретикулум
Просвет
:ι^^> ^""""""П Цитоплазма
»ГО U U U U U U 7тч /С UU U U U IT"
ρ Г. Ω J I П ПОП ПП nq Ui ЪПГ>Г
/'\°oouu—uo—^ (υ υ υ и υ цц и иц οσ
Шероховатый эндоплазматический
ретикулум
Рибосомы
Рис. 22.14. Строение эндоплазматического ретикулума
Более короткие области Зрелый белок
содержат полярные аминокислоты
+ΝΗ3
Гидрофобная область,
состоящая из 10-15
аминокислот
Участок
расщепления
Рис. 22.15. Присоединение типичной лидерной
последовательности к N-концу белка, транспортируемого через
мембрану ЭР
Подобные лидерные последовательности образованы
определенным типом аминокислот, но не обладают специфической
аминокислотной последовательностью; кислые аминокислоты
в ней отсутствуют
мРНК
Цитоплазма
Мембрана ЭР
Образовавшийся
пептид
Просвет ЭР
'чЗдесь сигнальная пептидаза
отщепляет лидерную
последовательность
Рис. 22.16. Перенос полипептида через мембрану ЭР при
участии мембранного транслокона
часть 4 Хранение и переработка информации
300
Пока неизвестно, что же управляет транслокацией
растущего пептида. В фолдинге полипептида,
по-видимому, участвуют находящиеся в просвете ЭР шапероны.
Гликозилирование белков
в просвете ЭР
В главе 3 (см. с. 62) мы упоминали, что некоторые белки
(особенно мембранные и секреторные) соединены с оли-
госахаридами. Местами присоединения являются либо
МН2-боковые группы аспарагина (N-гликозилирование)
или ОН-группы остатков серина и треонина (О-гликози-
лирование). Присоединение осуществляется в
несколько этапов. Внутри ЭР происходит N-гликозилирование.
Первый этап этого процесса особенно интересен тем,
что «ядро» олигосахарида, состоящее из 14 мономеров
сахара, образуется в цитоплазме и транспортируется
через мембрану будучи присоединенным к длинной
гидрофобной цепи. Последняя включает более 100
атомов углерода и называется долихолфосфат.
Расположенный на внутренней стороне мембраны ЭР фермент
трансфераза переносит олигосахаридную группу
на образующийся пептид, как только тот появляется
в просвете ЭР. О-гликозилирование происходит в
аппарате Гольджи (см. следующий раздел).
Что происходит с полипептидом,
перенесенным в просвет
шероховатого ЭР?
ЭР представляет собой «мешок», из которого нет выхода.
В просвете шероховатого ЭР белок окружается
мембраной, и образующиеся пузырьки отделяются от не
содержащих рибосомы областей ЭР, доставляя содержимое
просвета ЭР в другую органеллу - комплекс Гольджи
(аппарат Гольджи) путем слияния с ее мембраной.
Структура аппарата Гольджи проста: около
полудюжины вытянутых мембранных мешочков, не имеющих
ни входа, ни выхода. При помощи рассмотренных выше
транспортных пузырьков он получает белки из ЭР и
посылает их по назначению, отпочковывая пузырьки, как
показано на рис. 22.17. Электронная микрофотография
аппарата Гольджи приведена на рис. 3.15.
Не все белки, поступающие в аппарат Гольджи,
предназначены для секреции. Конечным пунктом назначения
одних являются лизосомы или пероксисомы. Другие,
например протеин-дисульфид-изомераза (с. 298) или
ферменты гликозилирования, должны вернуться в
шероховатый ЭР, потому что они оказались «унесенными»
в аппарат Гольджи случайно, вместе с остальными
белками. Аппарат Гольджи сортирует эти белки,
«упаковывает» их в пузырьки, которые и доставляют содержимое
в нужное место. Поэтому аппарат Гольджи, пожалуй,
Эндоплазматический
ретикулум
Транспортные
пузырьки из ЭР
цыс-область,
обращенная
к эндоплазма-
тическому
ретикулуму
Стеллаж
вытянутых
пузырьков
аппарата
Гольджи
транс-область, / © ®
обращенная к мембране ι ι Секреторные
♦ t пузырьки
Другие конечные пункты
назначения (лизосомы,
пероксисомы, новая
цитоплазматическая
мембрана, возвращение
в ЭР)
Рис. 22.17. Схематическое представление центральной роли
аппарата Гольджи в посттрансляционной сортировке и
мечен и и белков
Вновь синтезированные мембранные липиды метятся для
доставки транспортных пузырьков точно по назначению.
Образование пузырьков для транспорта между цистернами
аппарата Гольджи происходит при участии удивительного
механизма: комплекс GTP-белок взаимодействуют с мембраной, что
приводит к связыванию «покровных» белков. Отпочковывание
пузырьков от участков связывания ведет к образованию
покрытых везикул. «Раскрываются» они при контакте с мембраной-
мишенью в результате гидролиза GTP до GDP; «раскрытие»
приводит к слиянию везикулы и мембраны-мишени.
Молекулы-мишени на везикуле направляют последнюю точно по
назначению. Механизм «укрывания» пузырьков, по-видимому,
широко распространен
наиболее удивительное функциональное
«подразделение» клетки, способное к сортировке белков по их
конечному назначению и отправлению последних в
«посылках» по точно указанным адресам.
Предназначенные для секреции белки аппарат
Гольджи упаковывает в пузырьки, мигрирующие по
направлению к плазматической мембране. Есть два типа
секреции. Некоторые белки высвобождаются постоянно
по мере образования: к ним относятся белки крови,
выделяемые из клеток печени без дополнительных
сигналов. Для этого пузырьки сливаются с плазматической
мембраной клетки по мере поступления, а освобождение
глава 22 Синтез, внутриклеточный транспорт и деградация белков
301
их содержимого осуществляется посредством экзоцито-
за. Высвобождение пищеварительных ферментов,
например из поджелудочной железы, необходимо только
при поступлении пищи в кишечник. В этом случае
пузырьки имеют больший размер. Они хранят ферменты
до тех пор, пока нейрональный или гормональный
стимул не вызовет их массового освобождения в ходе экзо-
цитоза секреторных везикул (сигнал и механизм этого
процесса описаны на с. 358). Отпочковывание
транспортных пузырьков из мешочков аппарата Гольджи
происходит после того, как мембраны будут окружены
покровным белком в комплексе с G-белком и присоединенным
к нему GTP. Гидролиз GTP служит сигналом для
раскрытия и слияния везикул с мембраной-мишенью. В
процессе узнавания важную роль играют особые белки,
расположенные на везикулах и мембранах-мишенях.
Что такое «адресные сигналы»
на белках, участвующих в сортировке
в аппарате Гольджи?
Как описано ниже, в случае ферментов, направляемых
для включения в лизосомы, сигнал несет углеводная
часть гликопротеина. Однако в большинстве случаев
механизм сортировки должен включать рецепторы,
узнающие определенные структурные свойства или
специфические последовательности белков, предназначенных
для сортировки. Они еще окончательно не установлены,
но одна категория рецепторов уже известна. У тех
белков (например, у протеин-дисульфид-изомеразы),
которые должны быть возвращены в ЭР, «меткой» служит
последовательность, состоящая из четырех
аминокислот: Lys-Asp-Glu-Leu. В системе однобуквенного
обозначения аминокислот (табл. 22.2) эта
последовательность записывается как KDEL.
Таблица 22.2
Однобуквенные символы аминокислот
Алании
Аргинин
Аспарагин
Аспарагиновая кислота
Цистеин
Глутамин
Глутаминовая кислота
Глицин
Гистидин
Изолейцин
А
R
N
D
С
Q
Ε
G
Η
I
Лейцин
Лизин
Метионин
Фенилаланин
Пролин
Серии
Треонин
Триптофан
Тирозин
Валин
L
К
Μ
F
Ρ
S
Τ
w
Υ
ν
Упаковка лизосомных белков
До сих пор мы знакомились с белками,
предназначенными для секреции. Внутриклеточный путь секреторных
белков похож на путь лизосомальных белков. Как
описано в главе 16, лизосомы - небольшие окруженные
часть 4 Хранение и переработка информации
302
мембраной внутриклеточные органеллы, содержащие
группу высокоактивных гидролитических ферментов.
Ферменты образуются на ЭР тем же способом, что
и секреторные белки, и в просвете ЭР они
подвергаются N-гликозилированию.
В аппарате Гольджи ферментная система узнает ли-
зосомные ферменты и фосфорилирует маннозу оли-
госахаридной части последних. В мембране аппарата
Гольджи есть внутренние рецепторы, к которым
прикрепляется фосфорилированная манноза; после этого
происходит отпочковывание пузырьков, содержащих ли-
зосомные ферменты. Пузырьки сливаются с другими
«сортировочными пузырьками», внутри которых
поддерживается низкое значение рН (за счет протонных
насосов в мембранах). Кислый рН пузырьков способствует
диссоциации рецепторов маннозофосфата от гликопро-
теинов, и они возвращаются (путем отпочковывания
пузырьков) назад, в аппарат Гольджи; в это время
ферменты доставляются к лизосоме. При генетическом
заболевании, называемом болезнью 1-клеток («I»
обозначает включение мукополисахаридов), фосфорилиру-
ющая маннозу система дефектна, что приводит к
нарушению поступления группы лизосомных ферментов
в лизосомы. Последнее, в свою очередь, приводит к
нарушению деградации мукополисахаридов, накопление
которых в лизосомах сопровождается развитием
серьезной клинической симптоматики.
Когда было обнаружено, что фосфорилирование
остатков маннозы является своеобразным «ярлыком» лизо-
сомного фермента, появилось предположение, что
углеводное «мечение» должно быть широко распространено.
Однако в большинстве случаев сортирующий механизм
аппарата Гольджи узнает определенные структурные
особенности белков.
Каким образом метятся интегральные
белки цитоплазматических мембран?
Интегральные белки мембран также синтезируются
на шероховатом ЭР, включаются в мембрану ЭР,
транспортируются в аппарат Гольджи в составе везикул и
оттуда (также в составе пузырьков) направляются к
плазматическим мембранам. Синтез мембранных липидов
также происходит в ЭР, и новые мембраны образуются
в этой органелле.
Интересен, конечно, вопрос: почему в случае
секреторного белка полипептид идет прямо через мембрану
шероховатого ЭР, а интегральный белок заякоривается
в ней? Вся необходимая информация, которая
определяет, будет или нет (и каким образом) данный пептид
интегрироваться в структуру мембраны, заложена в
аминокислотной последовательности белка.
Наиболее проста ситуация, когда в полипептиде есть
якорная последовательность (называемая также
останавливающей перенос) (рис. 22.18). Она фиксируется
в гидрофобной части бислоя и останавливает
последующий перенос. Затем рибосома завершает синтез С-кон-
цевой части полипептида, приводя к образованию белка,
N-конец которого находится снаружи ЭР, а С-конец
выступает в просвет ЭР. Когда участок ЭР становится
фрагментом клеточной мембраны, этот белок имеет ту же
ориентацию, а N-конец выступает снаружи клетки.
Мембранные белки могут также иметь и противоположную
ориентацию, «прошивая» мембрану несколько раз.
Механизм встраивания таких белков предусматривает
участие несколько лидерных и останавливающих перенос
сигналов.
мРНК
Цитоплазма
Якорная
последовательность
Ό
Ό
СОО-
Мембрана ЭР
Просвет ЭР
Рис. 22.18. Иллюстрация роли якорных сигналов во
встраивании интегральных мембранных белков в мембрану ЭР
N-Конец полипептида ориентирован наружу, а С-конец -
внутрь. Мембранные белки, участвующие в транслокации, не
приведены. Другая организация якорных сигналов может
способствовать расположению белков в противоположной
ориентации
Является ли перенос всех белков
ко-трансляционным?
До сих пор мы рассматривали перенос (транслокацию)
белков в ходе их синтеза через или в мембраны. Однако
в клетках эукариот существует и другой тип транспорта
белка - в митохондрии или хлоропласты у
растительных клеток. Как уже отмечалось, у митохондрий есть
хромосома и собственная белок-синтезирующая
машина, но они ответственны лишь за синтез небольшой
части белков этих органелл. Несколько сотен различных
белков или субъединиц транспортируются в митохондрии,
составляя более 95% присутствующих там белков. Они
синтезируются в цитоплазме. По аналогии с ЭР можно
было бы предположить, что синтез белка происходит
на связанных с митохондриями рибосомах, однако дело
обстоит не так. Митохондриальные белки
синтезируются на свободных рибосомах. Кодируемые в ядре белки
митохондрий (т.е. белки, кодируемые генами
клеточного ядра) синтезируются и освобождаются в цитоплазму
в виде π ре-бел ков, после чего к ним присоединяются
шапероны, поддерживая их в развернутой форме.
Высвободившиеся (с рибосомы) белки пересекают мембрану
в вытянутой (несвернутой) форме, и этот процесс
управляется мембранным потенциалом. После того как белок
пересечет мембрану, другие шапероны внутри митохон-
дриального матрикса осуществляют его сворачивание.
Лидерная последовательность митохондриального
белка насчитывает примерно 12-70 аминокислот. Сами
последовательности варьируют от одного пре-белка
к другому, но общим структурным свойством является
амфипатическая α-спираль, в которой одна сторона
заряжена положительно, а другая в значительной
степени гидрофобна. Она присоединяется к рецепторам
внешней митохондриальной мембраны.
В простейшем случае N-концевая лидерная
последовательность направляет белок в матрикс митохондрий
через пору, расположенную на внешней и внутренней
мембранах в точках контакта. Затем лидерная
последовательность отщепляется (рис. 22.19). В том случае,
когда пункт назначения находится в межмембранном
пространстве, пре-белок имеет две лидерных
последовательности. Как уже было описано, первая направляет
его в матрикс. Но ее удаление приводит к
экспонированию второй, которая направляет полипептид из
матрикса через внутреннюю мембрану в межмембранное
пространство. Здесь происходит удаление второй лидер-
ной последовательности.
Ядро клетки окружено двойной ядерной мембраной.
Белки, синтезируемые в цитоплазме, переносятся в
ядро через специальные поры. Во многих случаях для
прохождения белков через эти поры необходима сигнальная
последовательность, содержащая короткий
аминокислотный фрагмент; для транспорта небольших белков-ги-
стонов он не требуется. В случае доставляемых в ядро
факторов транскрипции сигнальная последовательность
находится внутри полипептидной цепи.
Не нужно путать пре-белок с προ-белком! Инсулин
секретируется в просвет ЭР в виде одиночной
полипептидной цепи - проинсулина. В ходе удаления среднего
фрагмента проинсулина он превращается в 2
полипептида, соединенных дисульфидной связью. мРНК
инсулина кодирует лидерную последовательность
проинсулина, что привело к появлению термина пре-проинсулин,
хотя - поскольку пре-последовательность (лидер)
удаляется при пересечении мембраны ЭР - пре-проинсулина
как такового в клетке не существует.
глава 22 Синтез, внутриклеточный транспорт и деградация белков
303
а Сигнальный пептид
матрикса
грикса ^^щ^
Шапероны поддерживают
белок, направляющийся
в митохондриальный
матрикс в частично
развернутом состоянии
Митохондриальные
мембраны
Белок, свернутый
шаперонами
Митохондриальный
матрикс
Рис. 22.19 Перенос митохондриальных пре-белков
а - Схема строения митохондриального пре-белка,
синтезированного в цитоплазме; конечный пункт назначения - матрикс;
б - полипептид пересекает митохондриальную мембану через
белковую пору, а лидерная последовательность удаляется
сигнальной пептидазой матрикса.
Начальное прикрепление полипептида к митохондрии
происходит за счет рецепторного белка в мембране (не показано). Если
пунктом назначения является межмембранное пространство,
вторая лидерная последовательность управляет движением
белка из матрикса через внутреннюю мембрану митохондрий
Деградация белков
Мы познакомились с синтезом белка и его транслокацией.
Теперь рассмотрим деградацию белка.
Важно отметить следущее: 1) деградация белка
происходит во всех клетках, поскольку старые молекулы
белка замещаются новыми; 2) деструкция белковых
молекул высокоселективна. Некоторые белки имеют
время полужизни более 20 часов (белки печени -
несколько дней), другие - 10 и даже 2 минуты.
Есть несколько причин, обусловливающих
деградацию белков. Поскольку абсолютная точность в трансляции,
по-видимому, невозможна, неизбежно образуются
белковые молекулы, содержащие ошибочную аминокислоту, что
может привести к неправильному фолдингу молекулы.
Белки, пострадавшие от химического повреждения,
например окисления, также должны быть уничтожены.
Однако «удалением отходов» эта тема не
исчерпывается. Некоторые белки, похоже, намеренно созданы
для быстрого разложения: об этом свидетельствует
присутствие определенных аминокислотных
последовательностей. Почему клетка должна заботиться о
быстрой деградации очень хороших ферментов? Во
многих случаях в интересах метаболического контроля.
Если фермент разрушается быстро, то его уровень
можно тонко и постоянно контролировать скоростью его
синтеза. Такие ферменты характеризуются
относительно коротким временем жизни. Структурные белки
и гемоглобин, наоборот, относятся к «долгожителям».
В одном из механизмов селективной деструкции
белков участвует убиквитин - небольшой белок,
найденный у всех эукариот, но не бактерий; так что его
название (от англ. ubiquitous - вездесущий) явно
преувеличено. В ходе АТР-зависимой реакции его концевая
карбоксильная группа связывается с ε-аминогруппой
боковой цепи остатков лизина белка-мишени, и последний
таким образом оказывается помеченным для деструкции
(рис. 22.20). Выбор убиквитином белков,
предназначенных для разрушения, по-видимому, определяется их
N-концевой аминокислотой. Шапероны тоже могут
участвовать в мечении белков, подлежащих деструкции.
В деградации долгоживущих структурных белков
определенную роль играют лизосомы (см. рис. 16.2):
они ответственны за аутолитическую деструкцию
клеток в ходе развития (например, за исчезновение хвоста
у головастиков).
В целом о деградации белка нам известно намного
меньше, чем о гораздо более сложном процессе его
биосинтеза. А эта область жизненно важна. Вспомним
(см. с. 149), что при голодании распад мышечных
V
Предназначенный
ε- ΓΙ3—LyS дЛЯ деГрадацИИ белок
γ
к
АТР
AMP + РР(
*-\ Предназначенные
N —Lys для деградации белок
Цитозольные ферменты
Гидролиз
белка-мишени
Рис. 22.20. Убиквитин-зависимое мечение белков,
подлежащих деградации
Механизм выбора белков для деструкции неизвестен, хотя
отмечено, что концевая аминокислота влияет на время
полужизни белка
часть 4 Хранение и переработка информации
304
белков обеспечивает глюконеогенез аминокислотами:
без этого человек не смог бы прожить дольше 24 часов
из-за полного истощения запасов гликогена. Деградация
белка в клетке сейчас активно изучается.
Дополнительная литература
Транспортная РНК
Schimmel P., de Pouplana L. R. Transfer RNA: from
minihelix to genetic code // Cell. 1995. Vol. 81. P. 983-986.
(В обзоре рассматривается вопрос о соответствии
определенных триплетов нуклеотидов специфическим
аминокислотам. Предполагается, что тРНК имеют два
информационных домена: акцепторную Т\|/С - мини-
спираль, кодирующую операционный код РНК для
аминокислот, и домен, содержащий антикодон с тринукле-
отидами генетического кода.)
Рибосомная РНК
Noller К F. Ribosomal RNA and translation // Annu. Rev.
Biochem. 1991.Vol. 60. P. 191-227.
(В доступной форме обзор знакомит со структурой
и функцией рРНК и содержит полезный раздел о
взаимодействии с ними антибиотиков. Опровергает мнение,
будто рРНК - лишь инертные «строительные блоки»
рибосомы.)
Молекулы РНК и ДНК
как терапевтические агенты
Cohen J.S., Hogan Μ. Ε. The new genetic medicines // Sci.
Amer. 1994. Vol. 271 (6). P. 50-55.
(В качестве лекарственных препаратов используются
последовательности ДНК. Эти синтетические цепи
называются антисмысловыми, или триплексными, агентами;
они могут атаковать вирусы и раковые клетки без
поражения здоровой ткани.)
Факторы элонгации и точность
транскрипции
Thompson R. С. EF-Tu provides an internal kinetic standard
for translational accuracy // Trends Biochem. Sci. 1989.
Vol. 13. P. 91-93.
(Описывается механизм «паузы» и его значение для
точности трансляции.)
WeylandA., Parmeggiani A. Towards a model for the
interaction between elongation factor Tu and the ribosome //
Science. 1993. Vol. 259. P. 1311-1314.
(Обсуждается возможность гидролиза двух молекул
GTP в ассоциированных с фактором EF-Tu реакциях
вместо одной, как предполагали ранее (см. следующую
ссылку).
WeylandA., Parmeggiani A. Why do two EF-Tu molecules
act in the elongation cycle of protein biosynthesis? // Trends
Biochem Sci. 1994. Vol. 19. P. 188-193.
(Обсуждаются данные об участии двух молекул EF-Tu-
GTP в присоединении каждой аминокислоты в ходе
синтеза белка.)
Powers Т., Noller Η. Ε. The 530 loop of 16S rRNA - a
signal to EF-Tu? // Trends Genet. 1994. Vol. 10. P. 27-31.
(Дискуссия о том, как достигается точность в синтезе
белка.)
Трансляционный контроль
VassalliJ. D., StutzA. Awakening dormant mRNAs // Curr.
Biology. 1995. Vol. 5. P. 476-479.
(Обсуждается роль «хвоста» поли-А в активации мРНК
ооцитов для трансляции.)
Chen J.-J., London 1. Μ. Regulation of protein synthesis by
heme-regulated eIF-2oc kinase // Trends Biochem. Sci. 1995.
Vol.20. P. 105-108.
(Подробный обзор, посвященный регуляции синтеза
гемоглобина при участии киназы eIF-2oc.)
Циклофилины, пептидилпролинизоме-
раза и фолдинг белка
SchmidEX. Prolyl isomerases join the fold // Curr. Biology.
1995. Vol. 5. P. 993-994.
(Обсуждается участие циклофилинов, обладающих про-
линизомеразной активностью, в фолдинге митохондри-
ального белка; связь с иммуносупрессией
свидетельствует о существовании у циклофилинов и других функций.)
Молекулярные шапероны и фолдинг
белка
Ellis R. J., van der Vies S. M. Molecular chaperones // Annu.
Rev. Biochem. 1991. Vol. 60. P. 321-347.
(В обзоре приводятся определение и примеры действия
шаперонов.)
АН N, Вапи N. Heat shock proteins: molecular chaperones. //
Biochem. Educ. 1991. Vol. 19. P. 166-172.
(Подробное представление семейства белков теплового
шока и их роли.)
глава 22 Синтез, внутриклеточный транспорт и деградация белков
305
Craig E. A. Chaperones: helpers along the pathway to protein
folding// Science. 1993. Vol. 260. P. 1902-1903.
(Краткое резюме о роли шаперонов в фолдинге белка.)
AgardD. A. To fold or not to fold // Science. 1993. Vol. 260.
P. 1903-1904.
(Резюме стратегий, которые клетки могли бы
использовать для предупреждения агрегации полипептидов.)
HendrikJ. P., Hortl E.-U. Molecular chaperone functions
of heat-shock proteins //Annu. Rev. Biochem. 1993. Vol. 62.
P. 349-384.
(Подробный обзор многих функций шаперонов.)
Welch W. J. How cells respond to stress // Sci. Amer. 1993.
Vol. 268(5). P. 34-41.
(Обзор, посвященный белкам теплового шока, их роли
в качестве шаперонов и других функций.)
Hortl E-U., Hlodon R., hanger T. Molecular chaperones
in protein folding: the art of avoiding sticky situation // Trends
Biochem. Sci. 1994. Vol. 19. P. 20-25.
(Прекрасный обзор по фолдингу белка.)
Horwich A. L. Resurrection or destruction? // Curr. Biology.
1995. Vol. 5. P. 455^*58.
(Обсуждается, каким образом шапероны «избавляют» или
«обрекают» белки на протеолитическую деградацию.)
Glick В. S. Can hsp70 proteins act as force-generating
motors? // Cell. 1995. Vol. 80. P. 11-14.
(Миниобзор по нуклеотид-зависимым конформацион-
ным изменениям одного класса шаперонов - семейства
hsp70. Обсуждается путь, посредством которого они
могут действовать в качестве двигателей транслокации.)
HendrickJ. P.t Hortl E-U. The role of molecular chaperones
in protein folding// FASEB J. 1995. Vol. 9. P. 1559-1569.
(Хорошо иллюстрированное сообщение о роли и
значении шаперонов в транспорте белка через мембрану.)
BuchnerJ. Supervising the fold: functional principles of
molecular chaperones // FASEB J. 1996. Vol 10. P. 10-19.
(Хорошо иллюстрированный обзор о роли шаперонов
в фолдинге.)
Болезни, вызываемые прионами
Weissmann С. Yielding under the strain// Nature. 1995.
Vol. 375. P. 628-629.
(Краткое резюме, посвященное молекулярным основам
болезней, вызываемых прионами.)
Prusiner S. В. The prion diseases// Sci. Amer. 1995.
Vol. 272(1). P. 30-37.
(Прекрасный обзор, посвященный болезням,
вызываемым прионами.)
Thomas P. J., Qu В.-Η., Pedersen P L. Defective protein
folding as a basis of human disease // Trends Biochem. Sci.
1995. Vol. 20. P. 456-459.
част 4 Хранение и переработка информации
30b
(Предполагается, что помимо болезней, вызываемых
прионами, большое число других заболеваний может
быть также обусловлено нарушениями фолдинга белка.)
Taubes G. Misfolding the way to disease // Science. 1996.
Vol.271. P. 1493-1495.
(На основании экспериментальных данных
формулируется гипотеза, согласно которой неправильный фол-
динг белка может стать причиной амилоидных
болезней, например болезни Альцгеймера, а также болезней,
вызываемых прионами. Высказывается мнение, что
агрегация белка в нерастворимые комплексы может быть
более распространена и более важна, чем это считалось
ранее.)
Мечение белка
Von Hijne G Membrane protein assembly: rules of the game //
BioEssays. 1995. Vol. 17. P. 25-30.
(Интересное обсуждение способа «зашивки» белков
в мембраны с помощью множественных
трансмембранных спиралей.)
McNewJ. Α., Goodman J. M. The targeting and assembly of
peroxisomal proteins: some old rules do not apply // Trends
Biochem. Sci. 1995. Vol. 21. P. 54-58.
(Обсуждается доказательство импорта свернутых
белков в пероксисомы.)
SchmidS. L, DamkeH Coated vesicles: a diversity of form
and function // FASEB J. 1995. Vol. 9. P. 1445-1453.
(Рассматриваются различные пути отпочкования
покрытых пузырьков от аппарата Гольджи и других мембран.)
Schekman R., Orel L. Coat proteins and vesicle budding//
Science. 1996. Vol. 271. P. 1526-1533.
(Прекрасный обзор, посвященный транспорту
пузырьков и их роли в сортировке белков.)
Schatz G., Dobberstein В. Common principles of protein
translocation across membranes // Science. 1996. Vol. 271.
P. 1519-1525.
(В обзоре представлены общие принципы транспорта
всех белков через мембраны.)
Gorlich D., Mattaz L W. Nucleocytoplasmic transport//
Science. 1996. Vol. 271. P. 1513-1518.
(Подробное, легко читаемое резюме о движении белка
и РНК между ядром и цитоплазмой через ядерные поры
и о механизмах, участвующих в реализации этого
движения. Рассмотрена роль GTP в этом процессе.)
Гликозилирование белка
Abeijon С, Hirschberg С. В. Topography of glycosylate
reactions in the endoplasmic reticulum // Trends Biochem.
Sci. 1992. Vol. 17. P. 32-36.
(Обсуждаются различные реакции гликозилирования
белка, происходящие в эндоплазматическом ретикулуме.)
Деградация белков
Mayer R. J., DohertyF. Ubiquitin // Essays in Biochemistry.
1992. Vol. 27. P. 37^8.
(Структура, механизм действия и функции убиквитина.)
Ciechanover Л. The ubiquitin-proteasome proteolytic
pathway//Cell. 1994. Vol. 79. P. 13-21.
(Освещается роль убиквитин-протеосомной системы
в качестве главного нелизосомного протеолитического
и регуляторного пути для онкобелков и регуляторов
транскрипции, циклинов, рецепторных молекул
поверхности клетки, I-антигенов главного комплекса гисто-
совместимости.)
JentschS., SchlenkerS. Selective protein degradation: a
journey's end within the proteasome// Cell. 1995. Vol. 82.
P. 881-884.
(Репарация ДНК, прогрессия клеточного цикла,
передача сигнала, транскрипция и представление антигена -
все эти процессы регулируются опосредуемой убикви-
тином деградацией.)
Jentsch S. When proteins receive deadly messages at birth //
Science. 1996. Vol. 271. P. 955-956.
(Описывается необычный механизм, недавно
обнаруженный у бактерий. Если у мРНК отсутствует стоп-ко-
дон, синтезированный пептид может заблокировать
рибосому. С помощью метода мечения получены
доказательства того, что полипептид гидролизуется, а
рибосома освобождается.)
Вопросы к главе 22
1. Почему для кодирования 20 аминокислот
используется 61 кодон, а не 64?
2. Типов тРНК меньше, чем 61. Почему?
3. Почему на схемах в спаренном с кодоном состоянии
молекулу тРНК располагают в перевернутом виде
по сравнению с той же РНК, показанной отдельно?
4. Какие механизмы обеспечивают точность
трансляции?
5. Опишите участие и (там, где это известно) роль GTP
в синтезе белка.
6. Исследования показали, что у Е. coli молекулы тРНК
(с прикрепленными аминоацильными или пепти-
дильными фрагментами) занимают А, Р и Е-участки
на рибосоме. Объясните причину и механизм этого
явления.
7. Механизм инициации трансляции у эукариот не
совместим с полицистронной мРНК. Объясните почему.
8. Объясните роль шаперонов в синтезе белка.
9. Какое заболевание может быть вызвано
неправильным фолдингом белка?
10. Объясните, каким образом белки транспортируются
через эндоплазматический ретикулум.
глава 22 Синтез, внутриклеточный транспорт и деградация белков
307
QO Вирусы и вироиды
глава ^О
Помимо того, что вирусы имеют огромное
биологическое и медицинское значение, они нашли широкое
применение в биохимических исследованиях. Вирусы
используются в качестве простых моделей для изучения
сложных процессов функционирования гена и служат
полезным инструментом, предназначенным для
достижения самых разных целей, включая создание рекомбинан-
тных ДНК (см. главу 24).
За исключением особых стратегических путей
выживания, например, образования спор, в которых
химические превращения сведены к минимуму, живые
клетки всегда метаболически активны: синтезируют АТР,
выполняют осмотическую работу, синтезируют
клеточные компоненты и т.д. Еще клетка увеличивается в
размерах и делится. Для этого требуются тысячи генов,
кодирующих необходимые белки.
Вирусы отличаются от клеток. Они намного меньше,
и чтобы их увидеть, необходим электронный
микроскоп.
У вирусов отсутствует метаболизм. Вирусная
частица - вирион - сама по себе не может ровным счетом
ничего. Она не генерирует энергию, в ней не протекают
химические реакции: это инертный организованный
комплекс молекул.
Вирусы размножаются только после инфицирования
живых клеток. Различные вирусы инфицируют
животные и растительные клетки, а также бактерии (вирусы
бактерий называются бактериофагами). Задача
вирусов заключается в доставке генетического материала
в клетки и в использовании аппарата клетки для своей
собственной репликации.
Вирусная частица представляет собой нуклеиновую
кислоту, окруженную одной или несколькими защитными
оболочками. Хотя защитные оболочки содержат много
копий белковых молекул, у большинства вирусов они
образованы всего лишь несколькими видами белков.
Общее число вирусных генов может изменяться от сотен
(например, у вируса коровьей оспы) до трех-четырех.
Белковая оболочка, окружающая геном, называется
капсидом, а геном вируса, покрытый этой оболочкой, -
нуклеокапсидом. У некоторых вирусов есть
дополнительная липидная бислойная оболочка, в которую
встроены белковые молекулы, экспонированные на
поверхность и участвующие в начальных стадиях
инфекционного процесса.
Жизненный цикл вируса
Вирус должен войти в клетку, которую он инфицирует,
внедрив в нее свой генетический материал (РНК
или ДНК). Вирусные гены управляют синтезом
виру сспецифических ферментов, нужных для репродукции
вируса, а также белков, необходимых для сборки новых
вирусных частиц. Вирус обязан произвести множество
копий своего генома и из синтезированных
компонентов собрать потомство вирионов, которое должно затем
покинуть клетку.
Опишем стадии жизненного цикла вирусов, а затем
для иллюстрации общих принципов более детально
разберем ряд специфических вирусов.
Живая клетка окружена липидной бислойной
мембраной, которая препятствует проникновению вируса.
Однако существуют пути для преодоления этого
барьера, например рецептор-опосредуемый эндоцитоз у
животных клеток (с. 204). Некоторые вирусы используют
этот клеточный механизм импорта для проникновения
в клетку. На первом этапе вирион связывается с ее
поверхностью при помощи покровного белка,
комплементарного специфическому рецептору,
расположенному снаружи клетки-хозяина. Наличие такого рецептора -
главное условие для инфицирования вирусом данной
клетки. После прикрепления комплекс
вирус-рецептор движется в мембране к углублению, называемому
окаймленной ямкой, внутренняя поверхность которой
выстлана белком клатрином (рис. 23.1). Как и в случае
нормального рецептор-опосредуемого эндоцитоза, ямка
впячивается до поглощения вириона, заключенного в
окаймленный пузырек (везикулу), внутрь клетки. Затем
клатрин возвращается к клеточной мембране, а
содержащий вирус пузырек сливается с цитоплазматическим
пузырьком - эндосомой, оставляя вирус внутри
последней. При нормальном (невирусном) эндоцитозе эн-
досомы доставляют содержимое к пузырькам,
образованным аппаратом Гольджи (лизосомам), которые
содержат набор гидролитических ферментов, разрушающих
захваченную частицу (см. рис. 16.2). Работа
протонного насоса поддерживает кислое значение рН внутри
эндосомы. Кислая среда вызывает диссоциацию вируса
от мембранного рецептора клетки-хозяина. В случае,
если вирус имеет мембранную оболочку, она сливается
часть 4 Хранение и переработка информации
308
Белок мембраны вируса
прикрепляется к
специфическому рецептору
на поверхности
клеточной мембраны
Цитоплазма
Вирус подвергается эндоцитозу
Пузырек теряет белковую ι
оболочку и клатрин возвраща- I
ется к клеточной мембране
Содержащий вирус эндоцитоз
ный пузырек (без белкового
покрытия), склеивается с кис>
лой эндосомой
Кислое
Кислое значение рН внутри
эндосомы вызывает слияние
ее мембраны с мембраной вируса!
Нуклеокапсид попадает I
в цитоплазму '
Цитоплазма
Липидный бислой
вирусной мембраны
Нуклеокапсид
Нуклеиновая
кислота вируса
'-- Клеточная
мембрана
Клатрин покрывает
инвагинированную
мембрану
Покрытая клатрином
везикула в цитоплазме
результатом взаимодействия вирусного и
специфического клеточного белков. При этом происходит слияние
двух мембран (вирусной и клеточной) с образованием
сквозного отверстия, через которое содержимое вируса
входит в клетку. Это путь использует вирус СПИДа.
Бактериальные вирусы - бактериофаги - проникают
в клетку другим способом. Бактерии имеют прочную
клеточную стенку. Фаг лямбда (λ) похож на головастика
(рис. 23.2), его головка представляет собой белковую
капсулу, внутри которой находится молекула ДНК. Фаг
прикрепляется к клеточной стенке нитевидным хвостом
и с его помощью, словно шприцом для подкожных
инъекций, вводит ДНК в бактериальную клетку.
Типы генетического материала
у различных вирусов
Генетический материал вирусов может быть
представлен двуцепочечной или одноцепочечной ДНК, двуцепо-
чечной или одноцепочечной РНК. До сих пор в этой
книге в качестве генетического материала рассматривалась
ДНК
Головка
Рис. 23.1. Процесс инвазии животной клетки вирусом,
содержащим мембранную оболочку, с использованием
рецептор-опосредованного эндоцитоза
Хвост
с мембраной эндосомы - и содержимое вируса
оказывается в цитоплазме. Здесь вирус теряет белковую
оболочку, высвобождая в цитоплазму генетический материал
Механизм проникновения вируса, лишенного мембраны,
из эндосомы в цитоплазму еще не выяснен до конца.
Второй путь, доступный только для вирусов,
имеющих липидную мембрану, заключается в прямом
слиянии с внешней мембраной клетки-хозяина. В этом
случае прикрепление вируса к поверхности клетки является
Рис. 23.2 Фаг лямбда
Головка фага образована хромосомой, содержащий около
50 000 пар оснований ДНК. Хромосома окружена белковой
оболочкой
глава 23 Вирусы и вироиды
309
двуцепочечная ДНК. Ранее мы отмечали (см. с. 232), что
ДНК более стабильна, чем РНК: наличие у нее двух
цепей означает, что повреждение в одной цепи можно
исправить, используя для репарации другую цепь в
качестве матрицы. Реплицирующий аппарат ядра включает
исправление ошибок, что сильно уменьшает число
последних. Для механизма синтеза РНК такой аппарат
не предусмотрен, тем не менее у многих вирусов
генетическим материалом служит РНК. Почему? Среди
вероятных причин можно назвать следующие: 1) геномы
вирусов крайне малы по сравнению с размером генома
клетки, следовательно, вероятность вредных мутаций
в ходе репликации вирусной РНК меньше; 2) хотя
ошибок при репликации РНК больше, чем при репликации
ДНК, и некоторые из них могут быть летальными, этот
недостаток компенсируется высокой скоростью
размножения. (В условиях жесткого селекционного отбора она
становится даже преимуществом, поскольку быстрая
мутация помогает вирусу избежать иммунологической
атаки со стороны организма-хозяина.)
После проникновения в клетку хозяина судьба
различных типов генетического материала вирусов
складывается по-разному. Двуцепочечная ДНК вируса
транскрибируется РНК-полимеразой клетки-хозяина
с образованием мРНК. (В этом классе вирусов вирус
коровьей оспы необычен тем, что у него есть собственная
РНК-полимераза.) Минус (матричная) цепь двухцепо-
чечного генома копируется для образования мРНК, а
последняя управляет синтезом вирусспецифических
белков. В случае вирусов, содержащих одноцепочечную
ДНК - в виде (+)- или (-)-цепи - в ходе репликации
с использованием ферментов клетки-хозяина
образуется двухцепочечная ДНК, с которой транскрибируется
мРНК (определение цепей см. ниже).
У клеток нет аппарата для репликации вирусной
РНК. Вирусы, обладающие двуцепочечной РНК, имеют
в составе вирусных частиц молекулы РНК-зависимой
РНК-полимеразы. В клетке хозяина этот фермент
транскрибирует мРНК с матрицы вирусной РНК. мРНК
транслируется аппаратом клетки хозяина с образованием
вирусспецифических белков, необходимых для
репликации вируса. Геномы вирусов с одноцепочечной РНК
существуют в двух формах. Вспомним (см. с. 263), что
в клетке одна из двух цепей ДНК называется смысловой
{нематричной) цепью, последовательность
оснований которой идентична (за исключением замены Τ
на U) мРНК транскрибируемой с другой, несмысловой
{матричной) цепи. У различных вирусов, содержащих
одноцепочечную РНК, цепь РНК может быть
эквивалентна как смысловой, так и несмысловой цепи. Они
называются «плюс» (+)- или «минус» (-)-цепи соответственно.
часть 4 Хранение и переработка информации
310
Иными словами, РНК вируса, содержащего (+)-цепь
РНК, является матричной (информационной) РНК, в то
время как у вируса с (-)-цепью - нет. Если (+)-РНК
попадает в клетку, то ее белоксинтезирующий аппарат
может немедленно транслировать РНК, производя
вирусные белки, в том числе ферменты, необходимые
для репродукции вирусов, - ведь это мРНК. Такой вирус
нуждается только в РНК, кодирующей все белки,
необходимые для репродукции после инфицирования.
Сложнее обстоит дело с вирусом, содержащим (-)-цепь РНК:
это не мРНК, клетка не может ни транслировать ее,
ни реплицировать. Для копирования (-)-РНК в (+)-РНК
(мРНК) требуется дополнительный РНК-реплицирую-
щий фермент, имеющийся у вируса.
Другой класс вирусов, содержащих (+)-цепь РНК -
ретровирусы, среди которых наибольший интерес
представляет вирус иммунодефицита человека (ВИЧ),
вызывающий синдром приобретенного иммунодефицита
(СПИД). Его вирион несет фермент, превращающий
одноцепочечную РНК в двуцепочечную ДНК, которая
может встраиваться в хромосому клетки-хозяина.
Как вирусы высвобождаются из клеток?
Некоторые вирусы, например вирус полиомиелита,
высвобождаются в ходе простого лизиса клетки. В случае
бактериофага λ прочная клеточная стенка бактерий могла
бы препятствовать высвобождению новых фаговых
частиц. Однако один из фаговых генов кодирует фермент
лизоцим, который разрушает клеточную стенку,
вызывая лизис клетки. Этот ген транскрибируется только
на поздней стадии заражения, так что клетка не
разрушается до тех пор, пока не образуются новые фаговые
частицы. Несмотря на то, что бактериофаг λ - всего
лишь небольшой «кусочек» ДНК длиной в 48 502
нуклеотида с 63 генами, последние являются объектом
сложной системы контроля.
Механизм высвобождения животных вирусов,
которые окружены бислойной липидной мембраной, более
сложен. Вирус и в этом случае использует
инфицированную им клетку. Напомним, что в обычной клетке новая
клеточная мембрана, содержащая необходимое
количество белков, образуется в шероховатом эндоплазмати-
ческом ретикулуме (см. с. 301). Белки гликозилируются
и при помощи пузырьков аппарата Гольджи
транспортируются к клеточной мембране; пузырьки сливаются
с ней, образуя новую клеточную мембрану, содержащую
нужные мембранные белки.
Некоторые вирусы, окруженные липидной
оболочкой, адаптируют этот процесс для своих
собственных целей. мРНК белков оболочки кодирует л и дерную
и якорную последовательности (см. с. 303), поэтому
положение белков в шероховатом ЭР, откуда они
доставляются, такое же, как было описано выше для клеточной
мембраны. Там мембранные белки вируса
накапливаются. Новый нуклеокапсид вируса (геном + белки
оболочки капсида) отпочковывается от мембраны, собирая
свою липидную оболочку с включенными в нее
мембранными белками. Вирусы гриппа и СПИДа
используют именно этот метод формирования частиц (рис. 23.3).
Мембрана
эндоплазматического
ретикулума Чч
Мембрана ЭР с
вирусными белками
Вирус управляет синтезом белков,
предназначенных для липидного
бислоя. Они встраиваются в
I мембрану ЭР
<χχχχχχΧίθΐΧΧ*
Плазматическая мембра
на, содержащая теперь
участок, с включенными в|
Участок мембраны ЭР, содержащий
белки оболочки вируса, переносится
посредством везикул Гольджи, для
того, чтобы стать частью
него вирусными белками τ плазматической мембраны клетки
Цитоплазма
Тем временем вирусный геном
управляет синтезом нуклеокапсидов.
Каждый из них отпочковывается от
плазматической мембраны,
осуществляя сборку
собственной мембраны,
со встроенными в нее белками
Цитоплазма
Высвободившаяся
вирусная частица
Плазматическая
мембрана клетки
Цитоплазма
Рис. 23.3. Высвобождение вирусных частиц из клетки
путем отпочкования от клеточной поверхности
Механизмы репликации
некоторых вирусов
Вирусы успешно преодолевают трудности, связанные
с различными путями репликации. Их обобщение
заняло бы слишком много места, поэтому мы познакомимся
лишь с несколькими вирусами, размножение которых
иллюстрирует основные способы репликации. Кроме
того, эти вирусы представляют самостоятельный интерес.
Вирус коровьей оспы
Вирус, вызывающий оспу у коров, использовался для
создания вакцины против оспы. Он является одним из
самых крупных и наиболее сложных вирусов, геном
которого представлен двуцепочечной ДНК. Последняя
транскрибируется в мРНК, обеспечивающую синтез
рибосомами клетки-хозяина белковых компонентов,
необходимых для образования новых вирионов (включая
ферменты репликации ДНК вируса).
Геном большинства вирусов, представленный
двуцепочечной ДНК, находит свой путь в ядро, где РНК-поли-
мераза хозяина транскрибирует вирусные гены в мРНК.
Вирус коровьей оспы необычен тем, что полная
репликация происходит в цитоплазме, где нет РНК-полимера-
зы хозяина. Поэтому вирус должен приносить в своих
частицах молекулы собственной РНК-полимеразы,
обеспечивая тем самым образование мРНК и синтез всех
необходимых белков, включая РНК-полимеразу.
Полиовирус
Это «голый» вирион, содержащий лишь оболочку
из покровного белка. Он связывается со* специфическим
рецептором, найденным только в эпителиальных клетках
человека и других приматов. Поскольку его одноцепочеч-
ная (+)-РНК является матричной (информационной), она
транслируется при попадании в цитоплазму. При этом
образуется РНК-репликаза, которая синтезирует РНК,
используя РНК вируса в качестве матрицы. Репликаза
копирует исходные (+)-цепи в (-)-цепи, последние же
служат матрицами для синтеза большего числа (+)-цепей.
Среди РНК-синтезирующих ферментов РНК-репликаза
полиовируса уникальна тем, что не может сама по себе
инициировать синтез цепей РНК. В этом случае прайме-
ром является не олигонуклеотид, а белок, к которому
фермент добавляет первый нуклеотид и затем удлиняет цепь.
При трансляции мРНК образуется большой
«полипротеин», который затем расщепляется на отдельные
белки. Цель образования полипротеина не совсем
понятна: ведь в ходе трансляции образуется равное число
молекул покровного белка и репликазы, хотя для
образования вирусных частиц требуется гораздо больше
молекул покровного белка, чем нужно репликазы для
репликации.
глава 23 Вирусы и вироиды
311
Вирус гриппа
Этот вирус обладает (-)-цепью РНК. Его геном
разделен на восемь сегментов, каждый из которых заключен
в спиральный нуклеокапсид, окруженный липидной
мембраной, куда встроены молекулы двух различных
гликопротеинов (рис. 23.4). Одним из них является
поверхностный белок гемагглютинин. Это название
белок получил из-за того, что при смешивании с
красными кровяными клетками он вызывает их агглютинацию.
Углеводный компонент на поверхности эритроцитов,
присоединенный к молекулам трансмембранного белка -
гликофорина, оканчивается нейраминовой (сиаловой)
кислотой (см. с. 56). При смешивании с красными
кровяными клетками in vitro гемагглютинин
присоединяется к нейраминовой кислоте и сшивает клетки в
агрегаты. Вирус находит вход в клетку-хозяина, присоединяясь
к ее рецептору (который также несет концевой остаток
нейраминовой кислоты), а затем проникает в нее
посредством эндоцитоза.
На поверхности вириона находится 700 молекул ге-
магглютинина. Кроме того, есть еще один
поверхностный белок - фермент нейраминидаза, который гид-
ролизует концевые группы нейраминовых кислот
гликопротеинов. Функциональное значении этого
фермента до конца не выяснено. На первый взгляд кажется
странным наличие у вируса поверхностного фермента
для разрушения клеточных рецепторов, от которых
зависит его проникновение в клетку. Возможно, данный
фермент разжижает муцин, содержащий сиаловую
кислоту (см. с. 56). Это облегчает доступ к клеточной
поверхности для инфицирования и выход новых вирусных
частиц. Разжижение муцинов может также
способствовать более эффективному распространению вируса при
чихании. Наконец нельзя исключить, что вновь
образовавшиеся вирионы фиксированы на клетке-хозяине
., Гликопротеин гемаглютинин
__. Гликопротеин нейраминидаза
Липидный бислой мембраны
РНК-репликаза-ферментный
... -комплекс, переносимый
вирусом
- Спиральные нуклеокапсиды;
они состоят из сегментов од-
ноцепочечной РНК, связаных
с белком. Геном образован
восемью такими сегментами
Рис. 23.4. Схематическая структура вируса гриппа
Слой матриксного белка внешней мембраны не показан
часть 4 Хранение и переработка информации
312
рецепторами нейраминовой кислоты. Нейраминидаза
противодействует этому, разрушая рецепторы и
высвобождая вирусы. Есть данные о важности этого фермента
для эффективного размножения вируса в ходе инфекции.
Как уже отмечалось, геном вируса в виде
спирального нуклеокапсида сосредоточен в 8 отдельных
сегментах (-)-РНК (см. рис. 23.4). Поскольку (-)-цепь РНК
не является матричной, для ее репликации необходима
РНК-репликаза.
Вирус несет в своих нуклеокапсидах молекулы
РНК-репликазы, которая копирует (-)-цепи в (+)-цепи
РНК (эквивалент мРНК), а в ходе трансляции
образуются все необходимые вирусные белки, включая большое
число молекул РНК-репликазы.
Действие иммунной системы направлено главным
образом против гемагглютинина вируса гриппа. Он
нейтрализуется таким образом, что переболевший этой
болезнью человек становится невосприимчивым к
повторной инфекции идентичным штаммом вируса. Впрочем,
в результате мутаций (ведь это РНК-вирус)
аминокислотный состав вирусного гемагглютинина постоянно
меняется, и защитные свойства иммунитета постепенно
снижаются. Это явление известно как антигенная
изменчивость. Замена отдельных аминокислот не
сопровождается большими эпидемиями, потому что участки
связывания антигена на поверхности белка
сохраняются при постепенных изменениях в структуре.
Происходит только уменьшение их числа, так что у населения
наступает лишь частичная потеря иммунитета, и
многие инфекции протекают достаточно мягко. Однако,
если у вируса присутствует совершенно новый
гемагглютинин, то в силу антигенной изменчивости может
возникнуть пандемия, поскольку остаточная иммунная
защита против нее отсутствует. Такая ситуация
возможна в ходе инфицирования клетки двумя различными
штаммами вируса. Из-за разделенного генома при
сборке новых вирусных нуклеокапсидов способна
происходить пересортировка РНК. Пандемия 1918 года, которая
как полагают, произошла в результате такой
пересортировки между штаммами вируса человека и птиц, унесла
20 млн жизней.
Новые подходы к лечению гриппа связывают с
воздействием на структуру белка. С помощью метода рент-
гено-структурного анализа была установлена точная
структура центра связывания нейраминовой кислоты
кристаллической нейраминидазой вируса гриппа. Это
позволило получить синтетический аналог
нейраминовой кислоты, который с высоким сродством
присоединяется к ферменту, блокируя его действие. В настоящее
время проводятся клинические испытания этого
субстратного аналога в качестве лекарственного средства
против вируса гриппа. Данное соединение не
блокирует участок гемагглютинина, ответственный за
связывание с рецептором клетки-хозяина, поэтому
инфицирование клетки вирусом гриппа все-таки происходит.
Торможение активности нейраминидазы, по-видимому,
предупреждает дальнейшее распространение инфекции,
так как для высвобождения новых вирусов из клетки
необходим активный фермент.
Ретровирусы
Как уже отмечалось, есть четвертый класс одноцепочеч-
ных РНК-вирусов, вызывающих в настоящее время
большой интерес. Это ретровирусы, к которым
принадлежит и вирус СПИДа. Они относятся к числу вирусов,
содержащих (+)-цепь РНК. Однако при инфицировании
клетки ретровирусы не следуют по пути полиовируса.
Частица ретровируса несет несколько молекул особого
фермента. Его находка сначала вызвала недоверие,
а завершилась присуждением Нобелевской премии
первооткрывателям - Говарду Темину и Дэвиду Балтимору.
Этот фермент называется обратной транскриптазой.
До его обнаружения, конечно, знали, что ДНК может
управлять синтезом РНК, но чтобы наоборот?!
Обратная транскриптаза - удивительный
полифункциональный фермент. Когда ретровирус инфицирует
клетку, обратная транскриптаза вируса копирует
(+)-цепь РНК в ДНК. Как и во всех случаях репликации,
для синтеза ДНК необходим праймер. У ретровирусов
эту функцию выполняет тРНК, попадающая в вирион
из клетки-хозяина и взаимодействующая с вирусной
ДНК по принципу комплементарности.
Образовавшийся гибрид РНК/ДНК после гидролиза РНК
превращается в одноцепочечную ДНК. Эту РНКазную реакцию
также катализирует обратная транскриптаза. Затем
одноцепочечная ДНК копируется тем же ферментом
с образованием двуцепочечной ДНК. Вирусный геном
теперь находится в форме нормального дуплекса ДНК
(так называемой провирусной ДНК) и встраивается
в хромосому клетки-хозяина (рис. 23.5). В этом
процессе важную роль играет еще один вирусный фермент -
интеграза, которая и интегрирует провирусную ДНК
в хромосому хозяина. У двуцепочечной ДНК вирусного
генома (провирусной ДНК) на каждом конце есть
последовательность оснований, называемая длинным
концевым повтором (англ. long terminal repeats;
LTR) (рис. 23.6), которая участвуют в образовании
«стыков» с клеточной ДНК. В результате двуцепочечная
ДНК вируса представляет собой, в сущности,
дополнительный набор генов клетки, который реплицируется
вместе с ДНК хозяина при делении. Для образования
Цитоплазма
Нуклеокапсид, содержащий
капсидные белки и РНК генома
Липидный бислой с
погруженными в него белками
Ретровирус прикрепляется к рецептору
клетки, и его мембрана сливается с
клеточной мембраной, высвобождая
РНК в цитоплазму
Вирусная оболочка
Клеточная мембрана
Ядро
Обратная транскриптаза
Обратная транскриптаза вируса
копирует вирусную РНК в ДНК
РНК разрушается, а одноцепочечная
ДНК при участии обратной
транскриптазы превращается
в двухцепочечную ДНК
ДНК встраивается в
ДНК клетки-хозяина
Вирусная ДНК транскрибируется
в РНК аппаратом клетки-хозяина
Множественные копии РНК-вируса
Геном вируса Вирусные белки
Новые вирусные частицы
Рис. 23.5. Репликация гипотетического ретровируса
^Транскрипция
1 .г^т
: LTR
ДНК
клетки-хозяина
г
Гены, необходимые
для репликации вируса
Онкоген
-,—, ζ
LTR j
ДНК
клетки-хозяина
Рис. 23.6. Провирусный геном онкогенного ретровируса,
встроенный в геном клетки хозяина
LTR - длинный концевой повтор (long terminal repeat).
Онкоген может кодировать аномальный регуляторный белок или
способствовать образованию чрезмерных количеств такого
белка
новых ретровирусных частиц провирусные гены (гены
вируса в хромосомах хозяина) транскрибируются в
(+)-РНК транскрипты. Одни из них становятся геномом
глава 23 Вирусы и вироиды
313
нового «потомства» ретровирусов, а другие
подвергаются процессингу в мРНК и используются для трансляции
белков, необходимых для сборки вирусных частиц.
Препарат AZT - азотимидин, структура которого
приведена ниже, используется для лечения СПИДа:
он ингибирует обратную транскриптазу. Для этого AZT
должен превратиться в трифосфат, являющийся
аналогом dTTP. AZT-трифосфат ингибирует обратную
транскриптазу, а также обрывает элонгацию цепи ДНК из-за
отсутствия З'-ОН группы. ДНК-полимераза тормозится
в гораздо меньшей степени.
НОСН
N3
Азотимидин
Ретровирусы могут быть использованы в генотерапии
для доставки к клеткам больного, у которого
присутствует дефектный ген, «хорошего» гена, способного
исправить этот дефект. Принцип заключается в
нарушении репродуктивной способности ретровируса путем
делеции гена и замещения участка его РНК фрагментом
в виде ДНК, который эквивалентен гену,
предназначенному для интеграции в хромосому хозяина.
Недостаток метода заключается в беспорядочном течении
интеграции, а это таит в себе потенциальную опасность.
В настоящее время развиваются методы
сайт-направленного встраивания генов.
Онкогенные ретровирусы
Интеграция в хромосому генетического материала онко-
генного ретровируса (в форме своей ДНК, см. рис. 23.6)
может привести к образованию опухоли. Первым среди
описанных онкогенных вирусов был вирус саркомы
Рауса, вызывающий опухоли мышечной ткани цыплят.
Развитие опухоли обуславливал один ген ретровируса -
онкоген.
С тех пор у млекопитающих было открыто 18 или
20 онкогенов. Ранее мы говорили о гибридизации ДНК.
Согласно этому методу, небольшой фрагмент ДНК,
ставший одноцепочечным под действием нагревания,
безошибочно найдет среди тысяч оснований
комплементарный ему фрагмент и соединится с ним при помощи
водородных связей. Разработаны методы определения
гибридизационных комплексов (см. с. 233).
Удивительно, что в каждом случае ретровирусный онкоген,
вызывающий опухоль, имеет своего «двойника» в
нормальных клетках. Это относится только к экзонам
нормального гена, поскольку из-за сплайсинга форма вирусной
РНК гена не имеет интронов. В ходе эволюции
вирусный онкоген, по-видимому, произошел от своего
клеточного двойника. Единственным различием между
ними может быть расхождение всего в одном основании.
Вирусный ген обозначают приставкой «ν», а
соответствующий клеточный ген - приставкой «с» (англ.
cellular). Генам дают любопытные сокращения (см.
объяснение названий на с. 356): например, с-тус и
v-myc, c-ras и ν-гas и т. д. Клеточный ген называется
протоонкогеном. Оказалось, что протоонкогены
кодируют белки, участвующие в важных регуляторных
клеточных механизмах (см. главу 26), нарушения которых
могут привести к неконтролируемому делению клеток.
Некоторые из них кодируют аномальные факторы
транскрипции (см. с. 355). Эволюционно ретровирусные
онкогены могли произойти от ретровирусов, случайно
«подобравших» молекулу мРНК, транскрибированную с ре-
гуляторного гена клетки. Онкогенная вирусная форма
такого регуляторного гена могла возникнуть в ходе
мутации нормального клеточного двойника на стадии
«подбора» или после него. При повторном встраивании
этого гена (при ретровирусной инфекции) в хромосому
клетки-хозяина, с помощью уже описанного механизма,
присутствие онкогена приведет либо к синтезу
аномального регуляторного белка, либо к синтезу чрезмерного
количества такого белка с последующим нарушением
клеточной регуляции (подробнее см. главу 26).
Бактериофаг лямбда
Этот бактериальный вирус интенсивно изучается и
используется в молекулярной биологии и при
клонировании генов (см. главу 24 и рис. 23.2). Фаг λ содержит
двуцепочечную ДНК, и его жизненный цикл
характеризуется рядом особенностей. При попадании в клетку
у вирусной ДНК есть два пути «на выбор». Она может
немедленно реплицироваться и образовать новые
вирусные частицы, высвобождаемые из клетки при участии
кодируемого фагом фермента лизоцима, который
разрушает клеточную стенку, приводя к лизису клетки. Это -
вирулентный, или литический путь (рис. 23.7).
Альтернативным является лизогенный путь, при
котором геном фага интегрируется в хромосому Е. coli, где
он может оставаться инертным неопределенно долго,
реплицируясь вместе с ДНК клетки-хозяина. Таким
часть 4 Хранение и переработка информации
314
Хромосома
о
Клетка с фагом лямбда
инъецирующим свою
ДНК; внутри клетки
она приобретает
кольцевую форму
о
о
Лизогенный путь;
ДНК фага встраивается
в хромосому клетки и
репродуцируется при
каждом ее делении
Литический путь;
ДНК фага ответственна
за образование новых
фаговых частиц
О
о
Популяция клеток, каждая из
которых несет провирус лямбда.
Такие агенты, как УФ, повреждают
клетку и индуцируют экцизию
провируса из хромосомы хозяина и
его вхождение в литическую фазу
Клетка разрушается,
и новые фаговые
частицы
высвобождаются
ttt
Рис. 23.7. Инъекция ДНК бакатериофага λ в
бактериальную клетку
Литический и лизогенный пути репликации
образом, большая популяция нормальных в других
отношениях клеток Е. coli может нести ДНК фага. Клетка,
содержащая в своей хромосоме ДНК лямбда,
называется лизогенной клеткой, а встроенная ДНК фага -
профагом. Если ДНК клеток Е. coli повреждается
ультрафиолетом, химическими реагентами или
ионизирующей радиацией, происходит активация так называемых
SOS-функций, предназначенных для устранения
нарушений. В лизогенной клетке подобные повреждения при-
водият к выщеплению (рксцизии) профага из ДНК по
сложному механизму, который мы не будем
рассматривать. Отщепившийся профаг вступает в литический цикл
репликации. Бактериофаг лямбда представляет нам
красивый пример процесса самосборки. Если в пробирке
белковые компоненты вируса смешать с молекулами
вирусной ДНК, произойдет полная сборка
функционально активного вируса с упакованной в головке ДНК. Фаг
лямбда - мощный инструмент клонирования генов.
Что такое вироиды?
Вироиды - самые маленькие из всех известных
инфекционных частиц, которые меньше и проще, чем
вирусы. Они представляют собой голые молекулы РНК,
не покрытые ни белком, ни какой-либо иной оболочкой
(рис. 23.8). Для инфицирования вироидами
растительных клеток необходимо механическое повреждение
последних (в условиях эксперимента инфицирование
растений вироидной РНК проводят путем втирания ее
в листья с помощью абразивного материала). Действие
вироидной инфекции может проявляться по-разному,
!
с
с
i
J
с
с
с
с
с
С
С
с
1
1
1
)
)
)
)
)
J
ь
г
J
Рис. 23.8. Структура вироида кокосового ореха каданг-каданг
Петли представляют собой неспаренные области, а
поперечные линии - спаренные основания. Это наименьший из всех
известных вироидов (246 нуклеотидов). Пальмы погибают
через 5-10 лет после инфицирования. Любезно предоставлена
профессором R. Symons, Department of Plant Science, Waite
Institute, University of Adelaide
глава 23 Вирусы и вироиды
315
в виде некоторого ухудшения еостояния растения. Но
может также привести его к гибели. Так происходит,
например, у пальмовых деревьев. До сих пор неизвестно,
каким образом вироиды вызываю! болезни. Остается
загадкой и способ репликация вироидной РНК.
Удивительно и го, что у вироидов не найдены гены,
кодирующие белки. Для го го, чтобы определить ген, кодирующий
белок, надо найти открытую рамку считывания, т. е.
последовательность триплетов, кодирующих длинный
участок аминокислотной последовательности. Хотя у
вироидов и были найдены последовательности,
ответственные за инициацию трансляции, последующие
рамки считывания столь коротки (за счет стоп-колонов),
что образование белка исключено.
Дополнительная литература
Watson ./. D. The molecular biology of the gene. 4th edn.
Benjamin/Cummings. 1987.
Ptashne M. A genetic switch: gene control and phage 1 //
Cell Press and Blackwell Scientific Publications. 1987.
Вирус гриппа
LaverC, Gorman E. Flu neuraminidase: the virus4 Achilles
heel? /7 Today's Life Sciences. 1994. Vol. 6 (9). P. 40-47.
(Дается прекрасное объяснение новому подходу
рациональной терапии гриппа.)
Hughson F. Μ. Structural characterisation of viral fusion
proteins // Curr. Biology. 1995. Vol. 5. P. 265-274.
(Знакомите инфицированием вирусом гриппа и
слиянием липидных бислоев. Описывает выход вируса из эн-
досомы.)
Ретровирусы
Katz R. A. Skalka А. М. The retroviral enzymes // Annu. Rev.
Biochem. 1994. Vol. 63. P. 133 173.
(Детальный об'юр, посвященный ретровирусным
ферментам протсазе, обра гной транскриптазс и интегразе,
в котором также описаны ингибиторы ВИЧ и факторы,
определяющие устойчивость к последнему.)
Xowak Μ. Α., McMichael A. J. How HIV defeats the
immune system /7 Sci. Amer. 1995. Vol. 273 (2). P. 42-49.
(Описывается, как продолжительная эволюция вируса
иммунодефицита человека в организме ведет к
иммунному истощению, определяющему развитие СПИДа.)
Вироиды
Symons R. //. Self-cleavage of RNA in the replication
of small pathogens of plants and animals //Trends Biochem.
Sci. 1989. Vol· 14. P. 445-450.
(Приводится описание саморасщепляющейся
структуры «молотка» РНК.)
Вопросы к главе 23
1. Объясните, каким обраюм вирус может получить
доступ внутрь клетки эукариот.
2. Чем должен отличаться вирус (но не ретровирус),
содержащий одноцепочечную (+)-РНК, от вируса,
содержащего одноцепочечную (-)-РНК?
3. Почему вирус коровьей оспы в своем вирионс несет
собственную ДНК-зависимую РНК-1 юл им сразу, хотя
у клетки-хозяина сеть свой фермент?
4. Ретровирус это вирус, содержащий (+)-цепь РНК.
Чем он отличается от перетровируса, содержащею
одноцепочечную (-)-РНК?
5. Объясните, как вирус, обладающий липидной
мембраной, приобретает ее со всем набором
интегральных белков.
6. Эпидемии гриппа регулярно охватывают земной
шар. Большинспю из них протекает достаточно
мягко, но случаются, как в 1918-м, эпидемии с большим
числом летальных исходов. Почему?
7. Почему вирус гриппа вызывает агглютинацию
эритроцитов, если вирусы и эритроциты смешать вместе?
8. Поверхность вируса гриппа обладает нейраминидаз-
ной активностью. Чем необычен этот факт? Какова
функция нейраминидазной активности?
9. Опишите «выбор», который бактериофа! λ должен
сделать при инфицировании клетки Е. eoli.
\0.А. Что такое вироид?
Б. Известно, что вироиды инфицируют растения
и размножаются там; следовательно, они должны
иметь гены для образования бедка. Так ли это?
часть 4 Храпение и переработка информации
316
*У А Клонирование гена, метод
глава £τΎ рекомбинантной ДНК, генная инженерия
Практическое использование достижений в области
биохимии в данной книге специально не рассматривается,
однако методы, в основе которых лежат манипуляции
с ДНК, произвели поистине революционные изменения
в биологии и особенно в биохимии. Поэтому мы
посчитали целесообразным посвятить современным методам
молекулярной биологии отдельную главу.
Благодаря им стало возможным выделение
отдельных генов, определение в них последовательности
оснований, а также перенос генов от одного вида организмов
к другому и т. д. Зная последовательность оснований
кодирующей области, можно определить
аминокислотную последовательность белка, соответствующего
данному гену.
Выяснение принципов функционирования и
механизмов регуляции гена невозможно без данных о его
структуре. Большая часть информации о регуляции
активности гена, приведенной в главе 21, была
получена в экспериментах с использованием
рекомбинантной ДНК.
Изолированный ген может быть включен во
множество различных типов клеток для продуцирования
любых количеств кодируемого этим геном белка.
Рассматриваемые в данной главе методы находят свое
применение и в медицинской диагностике генетических
заболеваний.
В чем заключались проблемы
выделения генов?
Прежде всего - в гигантском размере молекул ДНК
хромосом. В диплоидной эукариотической клетке
каждый гомозиготный ген представлен двумя копиями - по
одной на каждую из пары гомологичных хромосом.
Среди тысяч других генов они образуют фракцию общей
ДНК клетки.
Единственное, что отличает каждый конкретный ген
от остальной ДНК, это закодированная в
последовательности его оснований информация. Задача выделения
гена сопоставима даже не с поиском иголки в стоге сена,
а с поиском определенного пучка сена в этом стоге!
Не нужно быть слишком большим пессимистом, чтобы
оценить «тупиковость» ситуации. Все изменила
технология рекомбинантной ДНК.
Первый шаг: разрезание ДНК
эндонуклеазами рестрикции
Первый шаг при выделении гена заключается в
разрезании клеточной ДНК на определенные, достаточно
малые фрагменты, с которыми можно дальше работать. Эта
цель сама по себе казалась недостижимой.
Единственным ферментом, гидролизующим фосфодиэфирные
связи в ДНК (см. с. 231), была ДНКаза, обнаруженная
среди пищеварительных ферментов панкреатического сока.
Если ДНК инкубировать с таким ферментом,
произойдет фрагментация молекулы, однако разрыв связей
происходит беспорядочно.
Это объясняет, почему открытие другого класса
эндонуклеаз, или ДНКаз, у бактерий имело
революционное значение: эндонуклеаза атакует внутренние связи
в молекуле, а не концевые, как экзонуклеаза. Новый
класс ферментов называют ферментами рестрикции,
а вызываемые ими разрывы - рестрикцией. О
молекуле ДНК, обработанной такими ферментами, говорят, что
она была подвергнута рестрикции. Эндонуклеазы
рестрикции гидролизу ют ДНК не беспорядочно: каждый
узнает свою короткую специфическую
последовательность оснований и производит разрез в точно
установленном месте. У различных бактерий имеются
ферменты рестрикции, узнающие разные последовательности
в ДНК и, следовательно, осуществляющие разрезы в
разных участках (сайтахрестрикции). Например, фермент
из Е. coli разрезает двуцепочечную ДНК у
последовательности:
GAATTC
CTTAAG
А фермент из Bacillus amyloliquefaciens специфичен в
отношении другого сайта рестрикции:
GGATCC
CCTAGG
Запоминать эти последовательности не стоит, но
обратите внимание, что они имеют двойную симметрию,
т. е. при чтении обеих цепей в направлении 5'—» 3!
последовательности оснований идентичны. Ферменты
получают свое название по имени бактерий (или
бактериальных штаммов), из которых они выделены,
глава 24 Клонирование гена, метод рекомбинантной ДНК, генная инженерия
317
а римская нумерация используется в тех случаях, когда
штаммы бактерий имеют несколько таких ферментов.
Вышеупомянутые ферменты названы соответственно
EcoRl и ВатШ. EcoRl был первым ферментом,
выделенным из R-штамма Е. coli. Другие ферменты рестрикции
узнают последовательности из 4, 5 и 8 оснований.
Ферменты рестрикции делают возможным разрезание ДНК
в строго установленных местах, определяемых
последовательностями оснований, с хирургической
точностью, образуя заданные фрагменты.
Какова биологическая функция
ферментов рестрикции?
Ферменты рестрикции в бактериальных клетках
предназначены для разрушения чужеродных ДНК. Например,
бактериофаг, или фаг лямбда (λ), инъецирует свою ДНК
в клетку Е. coli (см. с. 314). Фермент рестрикции
разрезает эту ДНК в сайтах рестрикции, чем предупреждает
успешное размножение фага и не дает возможности
чужеродной ДНК инфицировать клетку. Почему фермент
не разрушает собственную ДНК клетки? Гексамерная
последовательность оснований, подобная
распознаваемой EcoRl, должна встречаться в хромосоме Е. coli
много раз; статистически - каждые 46 (4096) пар
оснований. Клетка защищает от разрезания собственную
ДНК, добавляя сразу же после ее синтеза метильную
группу во все последовательности обеих цепей,
узнаваемые ферментом рестрикции. Это не мешает
спариванию оснований и экспрессии гена, но фермент
рестрикции не узнает метилированную последовательность,
а следовательно, собственная ДНК клетки
нечувствительна к атаке этого фермента.
Биологическая роль ферментов рестрикции может
быть проиллюстрирована на примере фага λ,
инфицирующего клетки Е. coli. Разные штаммы Е. coli
различаются по ферментам рестрикции, поэтому фаг λ, который
уже реплицировался в одном штамме Е. coli, будет
инфицировать его с более высокой эффективностью,
поскольку собственная ДНК фага защищена
метилированием также, как и ДНК хозяина - Е. coli. Однако, если
фаг пытается инфицировать другой штамм Е. coli,
содержащий другой фермент рестрикции, последний будет
быстро атаковать ДНК фага. Насколько успешным
окажется инфицирование в этом случае - определит
«соперничество» между системой метилирования клетки и
ферментом рестрикции. Степень защиты от инфекции
очень высока, а ДНК фага λ достаточно длинна и может
включать несколько участков, на которые способны
воздействовать ферменты рестрикции.
часть 4 Хранение и переработка информации
318
Когда неметилированная в этих участках ДНК фага
входит в клетку, для успешного ее инфицирования
процесс метилирования должен выиграть все «раунды
соревнования» у фермента рестрикции: единственный
разрез в ДНК фага будет сопровождаться потерей его
способности к репродукции в клетке.
Теперь обратимся к технологии выделения генов.
Клонирование гена,
или как выделяют гены
В этом разделе мы опишем два метода, используемые
при получении рекомбинантной ДНК. Первый из них
основан на выделении гена из ДНК.
Возьмем в качестве примера ДНК человека, хотя
методы применимы к ДНК из любого источника. Клоны
генов представляют собой фрагменты ДНК, идентичные
по последовательности оснований участку клеточной
ДНК, содержащему нужный ген. Второй метод
заключается в выделении кДНК человека («к» обозначает
комплементарный). кДНК представляет собой двух-
цепочечную копию мРНК. Геномная ДНК и кДНК
отличаются друг от друга тем, что первая содержит интроны,
а вторая - нет. Это различие имеет важное
практическое значение. Дело в том, что клетки бактерий
(подобных Е. coli) часто используют для получения инсулина
или гормона роста человека. Поскольку первичный
РНК-транскрипт генов Е. coli не подвергается
сплайсингу (см. с. 271), клонированные гены эукариот
(содержащие интроны) не могут управлять синтезом белка в этих
клетках. В то же время Е. coli транскрибирует кДНК
в мРНК-подобный транскрипт (при условии, что
соответствующие транскрипционные сигналы помещены
на эту к ДНК). Транскрипты будут управлять синтезом
белка в Е. coli; для этого необходимо также, чтобы
с кДНК были внесены соответствующие
трансляционные сигналы. Иногда выделение искомого гена
начинают с получения соответствующей этому гену кДНК,
которая затем может быть использована в качестве гиб-
ридизационного зонда (см. ниже). Такой подход
оправдан тогда, когда можно получить препараты,
обогащенные одним видом мРНК, например индуцибельного
фермента. Такой препарат мРНК можно использовать
в качестве гибридизационного зонда для выделения
кДНК.
Таким образом, клоны генов нужны для изучения
структуры гена, а клоны кДНК используются при на-
работкке инсулина человека или других белков в
клетках Е. coli.
Мы выбрали два метода - геномное клонирование
и клонирование кДНК, потому что они хорошо
иллюстрируют общие принципы клонирования гена.
Что такое клонирование?
Клоном называют множество идентичных копий,
образованных из одного предшественника. Клонирование
гена или кДНК позволяет получить большое число
копий фрагмента ДНК.
Сущность клонирования заключается во введении
участка ДНК, полученного, например, в результате
рестрикции общей ДНК, в бактериальную клетку, где он
будет реплицироваться. Одиночная клетка даст начало
колонии, содержащей большое число идентичных клеток:
например, при выращивании на агаре в течение ночи
одиночная клетка Е. coli произведет ~107 клеток в
одной колонии. Кроме того, в каждой клетке может
существовать множество копий вектора, содержащих свой
фрагмент встроенной ДНК. Поскольку это произойдет
с каждым фрагментом ДНК рестрикционной смеси,
клонирование обеспечивает как амплификацию
(увеличение числа копий), так и разделение каждого фрагмента
ДНК. Последнее объясняется тем, что клетки Е. coli,
содержащие различные векторы, легко отделить друг
от друга, выращивая их в колониях на питательных
чашках. Если вам удастся выбрать из тысяч различных
колоний ту, которая содержит интересующий вас фрагмент
ДНК, вы сможете получить фактически неограниченное
число копий такого фрагмента, выращивая чистую
культуру выбранной колонии.
Выделение клона гена человека
Создание библиотеки генов человека
Прежде всего нужно выбрать клонирующий вектор,
который может принять фрагмент ДНК человека, а затем
реплицироваться в клетках Е. coli. Для этого есть
несколько возможностей, и мы выберем в качестве вектора
фаг λ (см. с. 314). Центральная часть молекулы ДНК
фага может быть заменена фрагментом ДНК человека
без ухудшения возможности репликации такого
рекомбинантного фага в клетках Е. coli. Важные для
собственной репликации гены фага будут располагаться
по обе стороны от встроенного фрагмента. Фаг λ может
принять фрагмент чужеродной ДНК длиной 15-20
тысяч пар нуклеотидов (т. п. н.) без ущерба для его
репликации. Такой размер фрагмента удобен для большинства
случаев геномного клонирования.
глава 24 Клонирова
Как конструируются рекомбинантные
молекулы?
В основе этого метода лежит использование «липких
концов» ДНК. Многие ферменты рестрикции, например
EcoRl, осуществляют не прямой разрез через обе цепи
двухцепочечной ДНК, а «ступенчатый».
—X-X-G \ А-А—Т—Т—С-Х-Х—
— Х-Х-С-Т—Т-А-А t G-X-X—
EcoR\
— X-X-G А-А—Τ—Τ—С-Х-Х—
— Χ-Χ—С—Τ—Τ-Α^Α / G—Χ-Χ—
Липкие концы
Это приводит к образованию липких концов, причем
«выступающие» комплементарные последовательности
могут автоматически спариваться друг с другом (в
данном случае - А с Т). Если смешать два фрагмента ДНК,
имеющих идентичные липкие концы, они соединятся
(рис. 24.1). Если же смешать предварительно
подвергнутые рестрикции ДНК человека и фага λ, липкие
концы будут способствовать образованию рекомбинантных
молекул ДНК фага, как описано выше. Для ковалентного
связывания разрывов используется фермент лигаза,
катализирующий образование связи между З'-ОН- и
5'-фосфорильной группой.
При добавлении рекомбинантных молекул к
белковым компонентам фага λ происходит самосборка фага,
и рекомбинантная ДНК автоматически пакуется в его
головку с образованием зрелого фага.
Упаковка рекомбинантной ДНК в инфекционные
фаговые частицы произойдет только в том случае, если
фрагмент ДНК человека не более 15-20 т. п. н. будет иметь
на своих концах ДНК фага λ. Таким образом проходит
отбор рекомбинантных молекул (при получении
фрагментов ДНК человека предпринимаются определенные меры,
чтобы их размеры соответствовали требованиям, но мы
их рассматривать не будем). Фаг λ несущий молекулу
рекомбинантной ДНК, используется для заражения клеток
Е. coli посредством процедуры, в ходе которой
инфицируется лишь небольшая порция клеток. Это необходимо
для увеличения вероятности проникновения одной
фаговой частицы в данную клетку. Важно также, чтобы штамм
Е. coli был мутантным по гену фермента рестрикции.
В конечном итоге полностью фрагментированный геном
человека будет находиться в культуре клеток Е. coli (по
одному фрагменту на клетку). Такое «полное собрание»
генома человека, распределенное по «отдельным томам»
\е гена, метод рекомбинантной ДНК, генная инженерия
319
Рестрикционные фрагменты Рестрикционные фрагменты
ДНК фага λ ДНК человека
Липкие-"-"—
концы
Спонтанная
гибридизация
ДНК-лигаза
Белки головки и
хвоста фага λ
"1
Упаковка ДНК in vitro
Рис. 24.1. Основные этапы получения рекомбинантных
ДНК, используемые при создании библиотеки ДНК на
основе фага λ
Следует отметить, что благодаря липким концам могут иметь
место другие виды ассоциации фрагментов ДНК; но в головку
фага будут упаковываться только те рекомбинантные
молекулы, в центре которых расположен фрагмент ДНК человека
(15-20 т. п. н.), а по бокам - фрагменты фаговой ДНК
рекомбинантных молекул фаговых частиц, заключенных
в «суперобложку» бактериальной клетки, называют
геномной библиотекой. Культура клеток затем
наносится на твердую питательную среду и инкубируется. Не-
инфицированные клетки растут, образуя непрозрачный
«газон» по всей поверхности чашки. Среди них
находятся и инфицированные фагом клетки, колонии которых
проявляются в виде пятна, или бляшки. Происхождение
последней обусловлено тем, что после размножения фага
в клетке происходит ее лизис, и фаговые частицы
инфицируют соседние клетки, разрушая «газон»,
образованный исходно неинфицированными бактериями. Каждая
бляшка содержит множественные копии фага,
размножившиеся из одиночной инфекционной частицы, и несет
одну и ту же копию фрагмента ДНК человека (рис. 24.2).
Если мы сможем идентифицировать бляшку,
которая содержит фаг, несущий требуемый ген, то это
позволит вырастить неограниченное число фага в Е. coli
и выделить его ДНК Как же идентифицировать
нужную бляшку?
Скрининг бляшек на выбранный ген
человека
Для идентификации фага, несущего искомый ген
человека, наиболее широко используется
ДНК-ДНК-гибридизация. Для этого необходим зонд гибридизации,
состоящий из короткого фрагмента ДНК (длиной около
20 нуклеотидов), последовательность оснований
которого комплементарна известной последовательности в
гене. Метод предусматривает перенос фага с каждой
бляшки на мембрану с ДНК-адсорбентом. Если наложить
мембранный диск на поверхность культивируемой
чашки (не забывая его пометить, чтобы потом можно было
идентифицировать положение каждой бляшки), то фаг с
каждой бляшки прилипнет к мембране. Затем мембрану
обрабатывают щелочью для разрушения комплекса бе-
лок-ДНК (головки фага), высвобождения и разделения
цепей ДНК. Одноцепочечная ДНК фиксируется на
мембране, которую затем обрабатывают неспецифической
ДНК, чтобы предотвратить фиксацию на мембране
зонда. Для гибридизации с любой комплементарной ДНК
гибридизационный зонд инкубируется с мембраной
(предварительно, с помощью ферментативного фосфо-
рилирования с использованием меченого АТР, в него
вводится радиоактивная метка). При этом к зонду
«прилипает» только комплементарная ДНК - ген-мишень.
Затем при помощи рентгеновской пленки определяют
положение гибридизованного зонда (см. рис. 24.2).
Идентифицировав бляшку, содержащую нужный ген,
можно вырастить неограниченное количество фага с
этой бляшки, инфицируя Е. coli, а после этого извлечь
из ДНК фага вставки ДНК человека, используя
соответствующую рестриктазу.
Как получить соответствующий зонд - с нуклеотид-
ной последовательностью, комплементарной
последовательности гена, если структура последнего
неизвестна? В этом случае можно работать в обратном
направлении, опираясь на генетический код (см. табл. 22.1). Если
полностью или частично известна последовательность
аминокислот в белке, кодируемом интересующим нас
геном, мы можем сделать вывод о последовательности
оснований в этом гене. Конечно, из-за вырожденности
генетического кода такой обратный подход не всегда
приемлем. Там, где аминокислота имеет несколько кодо-
нов, мы не можем дедуктивно установить, какой из
часть 4 Хранение и переработка информации
320
Перенос фаговых бляшек
на мембрану, фиксация ДНК"
на мембране и блокада
оставшихся на мембране
мест связывания
неспецифической ДНК
Фаг, содержащий
рекомбинантную ДНК
Инфицирование Е. coli
Посев инфицированной культуры клеток
на чашки. Неинфицированные клетки
образуют непрозрачный клеточный «газон»
Рентгеновская
пленка
Гибридизация с радиоактивнь
зондом. Экспонирование
рентгеновской пленкой
и проявление
Отбор позитивной бляшки
и разведение Е. coli
Пятно соответствует
искомой бляшке
Выделение ДНК фага
Вырезание вставки при
помощи рестриктаз
Клонирование фрагмента ДНК
(в неограниченном количестве)
Рис. 24.2. Этапы клонирования гена с использованием бактериофага λ в качестве вектора
них действительно использовался клеткой для синтеза
белка (ученым, клонирующим гены, доставляет
удовольствие найти в секвенируемых фрагментах метионин или
триптофан, кодируемые только одним триплетом). В тех
случаях, когда необходимо учитывать вырожденность
генетического кода, используют методы,
предусматривающие синтез смеси зондов. Специальные приборы
легко синтезируют короткие зонды ДНК с
определенной последовательностью оснований. Зонд завершает
добавленная ферментативным способом радиоактивная
глава 24 Клонирование гена, метод рекомбинантной ДНК, генная инженерия
321
фосфатная группа. Если такой зонд недоступен,
используют альтернативные возможности скрининга, которые
в этой книге не рассматриваются.
Рассмотрим выделение клона кДНК.
Клонирование кДНК человека
Создание библиотеки кДНК человека
Принцип метода заключается в том, что из клеток
человека, в которых экспрессируется интересующий нас
ген, выделяется мРНК; затем она копируется in vitro
в двуцепочечную ДНК, а последняя клонируется.
Значительное преимущество этого подхода состоит в выборе
ткани, в которой искомый ген высоко активен,
следовательно, соответствующая мРНК находится в изобилии.
Среди всей РНК клетки доля мРНК невелика, однако
в клетках человека все мРНК (за редким исключением)
имеют поли-А-«хвосты» (см. с. 270). Если препарат РНК
пропустить через колонку с инертной матрицей,
несущей синтетический, лиганд олиго-dT (короткая одноце-
почечная «ДНК», составленная только из оснований Т),
рибосомная и транспортная РНК проходят через
колонку не задерживаясь, а мРНК связывается с ней за счет
образования водородных связей А=Т между хвостом
и лигандом. Затем мРНК можно элюировать, используя
растворы низкой ионной силы.
Изолированная смесь мРНК копируется in vitro в ДНК
с использованием полимеразного домена обратной
транскриптазы вируса (см. с. 313). Образуется дуплекс
РНК-ДНК. РНК разрушается при обработке NaOH или
РНКазой, а одноцепочечная ДНК превращается в
двуцепочечную под действием свободной от экзонуклеазы
ДНК-полимеразы I, выделенной из бактериофага.
Для клонирования молекул кДНК в качестве
клонирующего вектора можно использовать фаг λ, как это уже
было описано для геномных библиотек. Молекулы
кДНК не имеют липких концов, однако есть методы
ферментативного присоединения липких концов к так
называемым молекулам с тупыми концами.
Используются и другие методы лигирования «тупых концов».
Что можно сделать
с клонированной ДНК?
Итак, мы имеем фаг λ, содержащий нужную геномную
вставку (ее называют также инсертом) ДНК или инсерт
кДНК. Мы можем получить его в любых количествах,
часть 4 Хранение и переработка информации
322
инфицируя клетки Е. coli, затем выделить ДНК и
вырезать инсерты путем рестрикции.
Дальнейшее использование последних для
различных целей почти всегда включает встраивание
интересующей нас ДНК в другой клонирующий вектор -
наиболее часто в бактериальную плазмиду. Фаг λ удобен для
создания библиотек, поскольку он может принять
большие фрагменты ДНК (около 15 т. п. н.) и эффективно
инфицировать клетки, что позволяет получить полную
геномную библиотеку или комплексную библиотеку
кДНК. Однако, когда дело доходит до изучения
клонированного фрагмента геномной ДНК или кДНК,
недостатком фага становится его собственная большая ДНК по
обе стороны от инсерта (около 30 т. п. н.), которая
необходима для его репликации, но не представляет
никакого интереса для решения поставленных задач. Плазми-
ды намного меньше и более удобны в обращении. Они
не могут принять больших фрагментов чужеродной
ДНК подобно фагу λ, но если, например, цель работы
заключается в секвенировании гена, выделенного с
помощью фага λ, то этот ген разрезается на фрагменты,
которые потом встраивают в плазмиды (см. ниже), и
каждый фрагмент секвенируют. Аналогичным образом
можно обработать клоны кДНК. Они достаточно малы для
встраивания в экспрессирующие векторы, в том
числе плазмиды Е. coli, но требуют добавления
соответствующих транскрипционных и трансляционных сигналов.
При инфицировании ими клеток Е. coli белок,
кодируемый кДНК, может быть получен в нужных
количествах. Теперь мы рассмотрим бактериальные плазмиды.
Бактериальные плазмиды
Клетка Е. coli имеет одну главную кольцевую хромосому,
несущую тысячи генов, которые составляют большую
часть генетического материала клетки. Однако в
цитоплазме есть также и отдельные мелкие минихромосомы,
или плазмиды: кольцевые молекулы ДНК, несущие
небольшое количество генов и играющие защитную роль
в клетке. Обычно они несут гены, придающие клетке
устойчивость к антибиотикам, кодируя, например,
разрушающий их фермент. Каждая плазмида способна
к репликации, что обеспечивает ее удвоение в клетке.
Плазмиды неинфекционны в том смысле, в каком ин-
фекционен бактериофаг. Однако, если клетки Е. coli
обработать, например, СаС12 при 0° С, а затем повысить
температуру до 42° С то они становятся компетентными -
способными к принятию плазмиды.
Предположим, что мы имеем дело с клонированной
молекулой кДНК, выделенной по вышеописанному
методу. Сначала выбранная нами плазмида разрезается
ферментом рестрикции, а к молекулам кДНК
добавляются соответствующие липкие концы (мы не будем
описывать, как это делается). Когда разрезанные плазмиды и
модифицированная кДНК смешиваются и лигируются
(т. е. их концы соединяются ферментативным путем) -
образуются рекомбинантные плазмиды (рис. 24.3).
Во избежание излишнего усложнения
рассматриваемого процесса мы не описываем здесь меры,
предотвращающие восстановление исходных кольцевых плазмид
и способствующие образованию рекомбинантных
плазмид. Клетки Е. coli инфицируют рекомбинантными плаз-
мидами и выращивают на агаровых чашках таким
образом, что каждая колония вырастает из одной клетки.
В результате плазмидного заражения инфицируется
только небольшая часть клеток, и возможность захвата
двух плазмид одной клеткой минимальна. Поскольку
Изолированная
рекомбинантная ДНК фага λ
Плазмида, подготовленная
для секвенирования
или экспрессии
Клонированный фрагмент
ДНК вырезается при
помощи фермента
рестрикции
**fc, "^та- •mn»~i~ ^.f
*^&&&*ж&&в&ъг!£&#
Рестрикционный
разрез
Липкие концы инсерта и
разрезанной плазмиды
спонтанно гибридизуются
Отжиг и лигирование
Плазмида выращивается в Е. coli и
используется для нужных целей
Рис. 24.3. Перенос инсерта (вставки) ДНК из фага λ в
плазм иду
Фрагмент ДНК, который клонировали в фаге или в
предназначенной для экспрессии кДНК, с целью наработки нужного
белка переносят в Е. coli в составе специально
сконструированной плазмиды
^трансформированные клетки численно намного
превосходят трансформированные, нужен быстрый метод
отбора последних.
Одна из сконструированных плазмид, используемых
для клонирования, - pBR322; она несет встроенные
в ее ДНК гены устойчивости к ампициллину и
тетрациклину. Каждый из этих генов имеет различный участок
рестрикции. Предположим, что чужеродный фрагмент
ДНК встроен в ген тетрациклина; последний уже не
может управлять синтезом белка, придающего
устойчивость к этому антибиотику. Клетка, несущая такую
плазмиду, будет устойчива к ампициллину, но
чувствительна к тетрациклину Клетка, содержащая плазмиду
без вставки ДНК, будет устойчива к обоим
антибиотикам, в то время как клетка, не содержащая плазмиду,
будет чувствительна к ним. Это служит основой для
отбора только тех клеток, которые содержат
интересующую вас плазмиду.
Определение последовательности
оснований клонированного фрагмента
ДНК
Вся информация в гене заключена в последовательности
его оснований, определение которой имеет
первостепенное значение. Существует два метода секвенирования
ДНК (так называется этот процесс). Один из них -
прямой химический метод Максама-Гилберта. Другой,
используемый наиболее часто, основывается на
репликации ДНК и называется методом Сэнгера, или
дидезокси-методом.
Схема дидезокси-методд
секвенирования ДНК
Предположим, что у вас есть клонированный фрагмент
ДНК, который вы хотите секвенировать. Он встраивается
в специально сконструированную плазмиду,
размножающуюся вместе с инфицированной ею клеткой Е. coli.
Плазмиды выделены, так что у вас в распоряжении
имеется большое число рекомбинантных плазмид,
содержащих выбранный фрагмент ДНК. Процедура
секвенирования (см. ниже) требует, чтобы
предназначенный для секвенирования фрагмент копировался in vitro
ДНК-полимеразой. Помимо 4 дезоксинуклеозидтрифос-
фатов (dNTPs) для этого необходимо: 1) чтобы
копируемая ДНК была одноцепочечной; 2) чтобы праймер гиб-
ридизовался со стартовым участком (напомним, что
ДНК-полимераза не может инициировать новую цепь).
Предназначенные для секвенирования плазмиды
конструируют таким образом, что у З'-конца встроенного
клонированного фрагмента всегда располагается одна
глава 24 Клонирование гена, метод рекомбинантной ДНК, генная инженерия
323
и та же нуклеотидная последовательность (плазмидная
ДНК). Зная ее, можно синтезировать комплементарный
(олиго)нуклеотид, который и будет служить праймером.
Инкубация предназначенной для секвенирования
одноцепочечной ДНК с праймером, полимеразой I
(свободной от экзонуклеазы), dATP, dGTP, dCTP и dTTP
обеспечивает копирование фрагмента ДНК. Один из
трифосфатов (или праймер) должен быть
радиоактивным, так что все новые ДНК оказываются мечеными.
При проведении секвенирования решающим^
являются два момента. Первый связан с использованием
электрофоретического метода разделения молекул ДНК,
которые в электрическом поле двигаются вдоль
пластины акриламидного геля со скоростью, определяемой
длиной цепи: чем короче цепь ДНК, тем быстрее она
движется. Каждое присоединение нуклеотида изменяет
подвижность цепи. На геле цепям с различной
подвижностью соответствуют полосы, причем эти полосы
соответствуют цепям, которые отличаются друг от друга
на один нуклеотид. Их идентифицируют при помощи
радиоавтографии, которая включает экспонирование
геля с рентгеновской пленкой, чувствительной к
радиоактивности. Второй момент касается дидезокси-произ-
водных нуклеозидтрифосфатов (ddNTPs). ДНК-полиме-
раза добавляет З'-нуклеозид к З'-ОН-концу растущей
цепи ДНК. УдидезоксиЫТРБ З'-ОН-группа отсутствует
(рис. 24.4); они могут быть добавлены к цепи
посредством их 5'-Р-группы, но после этого цепь
наращиваться не может.
Важно подчеркнуть, что когда мы говорим о секве-
нируемом «фрагменте» ДНК, в эксперименте
участвуют множество копий этого «фрагмента». Даже
ничтожное количество ДНК содержит большое число молекул
0 0 0
II II II 5-
"0—Р-0—Р-0—Р-0—СН2 Λ
ι ι ι ι ^о
0~ О" О"
Аденин
dATP
ОН Η
О
О
"О—Р—О—Р—О—Ρ—ΟΙ I I
О" О" О"
ddATP
-сн2
Аденин
Η
Η
Рис. 24.4. Структуры дезоксиАТР (dATP) и дидезоксиАТР
(ddATP)
Отсутствие З'-ОН-группы означает, что в момент
присоединения ddNTP к растущей цепи последняя обрывается
(помните, что 1 моль любого химического вещества
содержит астрономическое число 6,03· 1023 молекул).
Предположим, что для копирования мы располагаем
всеми четырьмя dNTPs и небольшим количеством
одного дидезоксиЫТР; для примера возьмем
дидезоксиАТР (А). Количества завершенных цепей достаточно
для того, чтобы с помощью электрофореза выявить
их на геле в виде отдельных полос. Рост цепей будет
продолжаться до тех пор, пока к ним не будет добавлен
А; отсутствие З'-ОН-группы делает невозможным
присоединение следующего нуклеотида.
Как все это интерпретировать в виде
последовательности оснований?
Предположим, что секвенируемый фрагмент ДНК
имеет показанную ниже последовательность, которая
включает основания Т:
3'-Х-Х-Т-Х-Х-Х-Х-Т-Х-Х-Т-5'.
Если копирование происходит в присутствии
дидезоксиАТР (вместе с избытком дезоксиАТР), то при
добавлении каждого А рост части цепей будет
заканчиваться, и в конечном итоге образуются следующие цепи
(связанные с праймером).
а) Б'-Х-Х-сЮА-З',
б) Б'-Х-Х-А-Х-Х-Х-Х-сЮА-З',
в) Б'-Х-Х-А-Х-Х-Х-Х-А-Х-Х-сЮА-З'.
На геле (называемом секвенирующим) эти цепи
будут видны в виде полос (рис. 24.5, столбик слева).
N
/
Направление
электрофореза
Включение
дидезоксинуклеозидтрифосфата
А
(в) —
(б)—
(а) —
G С Τ
_ —
— _
■—
шш
Последовательность
оснований на:
вновь синте- исходной
зированнои цепи
цепи
A3' Τ 5'
С G
G С
Τ А
А Т
G С
С G
С G
Τ А
А Т
G С
G 5' С 3'
Рис. 24.5. Радиоавтография секвенирующего геля
Последовательность полинуклеотида читается снизу вверх,
я, б, в - Полосы, соответствующие полинуклеотидам,
образованным в присутствии дидезоксиАТР
часть 4 Хранение и переработка информации
324
Если вторая инкубационная смесь содержит дидезок-
сиТТР вместо дидезоксиАТР, то образуется набор цепей,
оканчивающихся на Т. Если же в среде присутствуют
дидезоксиСТР или GTP, цепи будут оканчиваться на С
или G соответственно. При проведении всех четырех
инкубации продукты реакций располагаются на геле
друг за другом в виде так называемой секвенирующей
лестницы (см. рис. 24.5). С ее помощью легко
определить последовательность оснований фрагмента ДНК,
которая читается с нижней части геля вверх: это
последовательность цепи-копии (т. е. партнера по отношению
к матрице), а не матрицы. В результате
последовательность располагается в направлении 5'—» 3', поскольку
именно в этом направлении всегда происходит синтез.
В каждой секвенирующей лестнице можно
определить последовательность около 200-300 оснований.
Если при использовании различных ферментов
рестрикции получаются перекрывающиеся фрагменты
исходной кДНК, можно определить ее полную
последовательность. Фотография полученной в ходе эксперимента
секвенирующей лестницы приведена на рис. 24.6.
Если же кДНК секвенируется с целью определения
аминокислотной последовательности белка, то,
используя генетический код, определяют правильную рамку
считывания (из 6 возможных на 2 цепях).
Описанная выше методика применяется во многих
лабораториях. Однако технология секвенирования
продвинулась вперед настолько, что позволила сделать этот
процесс автоматизированным. Принцип нового метода
не отличается от использованного Сэнгером, но
предусматривает применение флуоресцентно-меченых нукле-
озидтрифосфатов, причем каждый из 4 типов имеет свой
максимум флуоресценции. Для инкубации ДНК
используется среда, в которой присутствуют все 4 дидезокси-
соединения и, конечно, 4 дезоксинуклеозидтрифосфата;
продукты анализируются на одной электрофоретичес-
кой дорожке. Гель автоматически сканируется, и
компьютер печатает последовательность оснований.
Использование полимеразной цепной
реакции (ПЦР) для амплификации
сегментов ДНК
Полимеразная цепная реакция (ПЦР) становится в
последние годы одним из наиболее популярных методов
работы с ДНК. Она позволяет амплифицировать сегмент
молекулы ДНК путем синтеза огромного числа его
копий. Для проведения ПЦР достаточно ничтожно малых
количеств ДНК (всего из нескольких клеток).
Принцип метода прост и изящен. Для проведения ПЦР
необходимо знать нуклеотидную последовательность
Рис. 24.6. Фотография секвенирующей лестницы
А, С, G и Τ обозначают дидезоксинуклеозидтрифосфаты,
присутствовавшие в инкубационной смеси. Любезно
предоставлена Dr. Cris Hahn, Departament of Biochemistry, University
of Adelaide
интересующей нас ДНК и синтезировать два олиго-
нуклеотида, каждый из которых комплементарен
участку одной из двух цепей молекулы ДНК, примыкающему
к выбранному для амплификации сегменту. Эти олиго-
нуклеотиды будут праймерами при синтезе ДНК in vitro.
Цепи дуплекса ДНК разделяют нагреванием и
копируют выбранный сегмент с использованием
синтетических праймеров (рис. 24.7). В конце каждого цикла
синтеза дуплексы ДНК, образованные в процессе
копирования, разделяются нагреванием для осуществления
следующего раунда праймирования и копирования.
глава 24 Клонирование гена, метод рекомбинантной ДНК, генная инженерия
325
Двухцепочечная ДНК
Фрагмент, выбранный
/ для амплификации
Нагрев для разделения цепей
(для простоты приведена
только одна цепь)
Праймирование
Копирование
Нагрев для разделения цепей
(для простоты приведена
только одна цепь)
Праймирование
Копирование
Нагрев для разделения цепей
(для простоты приведена
только одна цепь)
Рис.24.7. Схематичное изображение, иллюстрирующее
основной принцип амплификации участка ДНК в ходе по-
лимеразной цепной реакции (ПЦР)
а - В центре - участок, выбранный для амплификации с
использованием соответствующих праймеров (—><—); каждый из
них комплементарен З'-концу участка амплифицированной
цепи;
б - нагревание приводит к разделению цепей (с этого
момента для простоты схемы показана амплификация только одной
цепи, хотя в действительности амплифицируются обе);
в - инкубационные смеси содержат 4 dNTPs,
термостабильную ДНК-полимеразу и соответствующие праймеры;
г - ДНК реплицируется;
д - в ходе нагревания цепи разделяются;
е - праймируется новая цепь;
ж - новая цепь реплицируется;
з - нужный фрагмент отделяется.
За 25 циклов репликации, нагревания, праймирования и
синтеза выбранный участок может быть амплифицирован в
миллионы раз. Обратите внимание, что каждый раз происходит
праймирование и копирование
Использование термостабильных ДНК-полимераз (из
термофильных бактерий) исключает необходимость
добавления фермента после каждого цикла копирования и
разделения цепей в ходе нагревания.
До последнего времени можно было амплифициро-
вать участок ДНК, состоящий не более, чем из 2 т. п. н.,
однако новейшие технологии позволяют амплифициро-
вать фрагменты, включающие до 35 т. п. н. Полученные
таким образом копии ДНК могут использоваться для
самых разных целей. Этот метод важен для судебной
медицины и в диагностике генетических аномалий плода
in utero (см. ниже). Он используется также и в
биохимических исследованиях.
Применение технологии амплификации
рекомбинантной ДНК
Как уже отмечалось, главная роль клонирования гена
заключается в определении последовательности
оснований. Это необходимое условие для изучения функции
гена и его регуляции. Последовательность оснований
ДНК - это прямое «окно» в эволюционные взаимосвязи
организмов. Амплификация рекомбинантных ДНК
нашла практическое применение и в ряде других случаев,
которые мы здесь вкратце разберем.
Экспрессия гена в клетках бактерий
и эукариот
Выделенный клон кДНК или бактериальный ген могут
быть встроены в специально сконструированный вектор.
Например, в плазмиду, в которую включены
соответствующие промоторы бактериальной ДНК и сигналы
инициации трансляции. Встроенная в каждую плазмиду
молекула кДНК экспрессируется: клетка синтезирует
из кДНК мРНК и транслирует ее в белок. Таким
образом, встроенные гены покариот или кДНК эукариот
(помните, что она не имеет интронов) могут быть
эффективно транскрибированы, а соответствующие мРНК
транслированы, в результате чего можно получить
нужный белок.
При помощи описанных методов бактерии (Е. coli
и др.) способны производить неограниченное
количество белков человека, применяемых в качестве
лекарственных средств. Кроме наработки белков,
присутствующих в организме человека в ничтожно малых
количествах, использование Е. coli исключает
возможность присутствия в белке примесей инфекционных
агентов. Так продуцируется человеческий инсулин и
некоторые другие белки, например гормон роста. В
качестве вакцины, без риска занесения инфекции,
используется иммуногенный белок вируса гепатита В,
образующийся в дрожжах. В некоторых случаях Е. coli может
продуцировать чужеродный белок в таких количествах,
что он преципитирует в виде телец включений и не
может принимать нужную конформацию. В ряде случаев
часть 4 Хранение и переработка информации
326
лишь in vitro происходит правильное сворачивание
белка. Бактерии не могут гликозилировать белки, но если
использовать в качестве экспрессионных хозяев
животные клетки, например клетки насекомых, глико-
зилированные белки могут образовываться в больших
количествах.
Направленный мутагенез
Это очень мощный метод анализа роли отдельных
аминокислотных остатков в белке. Рассмотрим случай,
когда у нас есть фермент, для которого известна
аминокислотная последовательность, установленная либо
в результате прямого секвенирования, либо на
основании последовательности оснований его гена или к ДНК.
Теперь мы хотим исследовать роль боковой цепи
отдельной аминокислоты в функционировании данного белка.
Если бактериальный ген или кДНК эукариот
клонировать в экспрессирующемся векторе, например плазми-
де, клетки Е. coli будут продуцировать белок в
количествах, достаточных для исследования. Например,
в случае фермента нас может интересовать его
каталитическая активность или субстратная специфичность.
Направленный мутагенез позволяет осуществить
специфическое замещение одной или нескольких
выбранных аминокислот в белке. Это можно сделать,
замещая соответствующий фрагмент выделенного гена
синтетическим олигонуклеотидом, последовательность
оснований которого такова, что кодирует измененную
аминокислоту. Получив мутантный белок, можно затем
определить его каталитическую активность.
Трансгенез
Трансгенезом называется встраивание генов животным
или растениям. Встраивание нормальных генов
пациентам для коррекции дефектных генов (генотерапия)
может быть использовано для лечения генетических
заболеваний. В качестве векторов для введения генов
в хромосомы человека можно было бы использовать
ретровирусы, «покалеченные» генетически для
предупреждения их репликации. Однако этот способ не
позволяет определить инсерционную точку, а беспорядочная
инсерция потенциально опасна. В настоящее время
разрабатываются методы инсерции гена в специфических
точках. Инсерция гена аденозиндезаминазы человека
в лейкоциты больных лечит иммунодефицит у детей
(см. рис. 18.6). Для инсерции чужеродных генов в
хромосомы растений в качестве клонирующего вектора
используются природные (но соответствующим образом
модифицированные) Ti-плазмиды (от англ.
tumor-inducing - опухоль-индуцирующая), содержащиеся в
патогенной почвенной бактерии Agrobacterium tumefaciens.
Метод дробовика предусматривает «стрельбу» генами
по клеткам растений при помощи специального «ружья».
Таким способом конструируются растения, устойчивые
к гербицидам; это позволяет уничтожать гербицидами
сорняки, не влияя на рост. Трансгенные животные
могут также использоваться в качестве «экспрессирую-
щихся векторов». Например, белок человека может
нарабатываться после присоединения его гена к
сигнальной последовательности гена, ответственного за
синтез белка молока, что в конечном итоге приводит
к секреции такого белка вместе с молоком. Этот подход
был использован для получения белка человека в
молоке овец.
Определение генетических аномалий
анализом рестрикции или блоттингом
по Саузерну
Когда известна структура генов, можно сделать гиб-
ридизационные зонды для определения их аномалий.
Для этого анализа требуется минимум ДНК, что
позволяет проводить пренатальную диагностику плода
(при использовании полимеразной цепной реакции для
таких анализов достаточно ничтожно малых количеств
ДНК). Наличие информации о потенциальных
заболеваниях позволяет принять решение о возможном
прерывании беременности. Например, при мышечной
дистрофии аномалия гена обычно обусловлена делецией
части кодирующей ДНК, и это можно определить
Саузерн-блот-анализом. Этот анализ, названный по
имени автора, включает разрезание ДНК одним или
несколькими ферментами рестрикции, разделение
фрагментов при помощи гель-электрофореза, перенос
разделенных фрагментов на мембрану и зондирование
мембраны ген-специфичным радиоактивным зондом
гибридизации. Все это позволяет выявить участки ДНК в виде
полос. Аномалии гена могут давать различные картины
полос, потому что в результате мутации могут
появляться новые участки для атаки фермента рестрикции либо
разрушаться или удаляться ранее существовавшие. Даже
если на геле будет присутствовать много различных
фрагментов ДНК, выявляться будут лишь те немногие из них,
что гибридизуются со специфическим зондом.
Если локализация гена, обусловливающего
возникновение генетической болезни, точно не известна, или
он не выделен, можно применить RFLP-анализ -
анализ полиморфизма длины рестрикционных
фрагментов (англ. restriction fragment length polymorphism).
Метод здесь не обсуждается. Его можно использовать
для идентификации родственников больного -
носителей аномального гена.
глава 24 Клонирование гена, метод рекомбинантной ДНК, генная инженерия
327
Понять принцип применения RFLP-анализа легче
всего на примере метода «отпечатков пальцев» ДНК,
или фингерпринтинга (англ. fingerprinting) ДНК.
Повторяющиеся последовательности в ДНК, разбросанные
по всему геному, свидетельствуют о большом
полиморфизме сайтов рестрикции: рестрикционные образцы
уникальны у каждого человека. Этот метод до сих пор
используется в судебной медицине, однако новая
технология, базирующаяся на ПЦР-реакции, постепенно его
вытесняет. В геноме человека идентифицировано
большое число повторяющихся последовательностей.
Число повторов в последовательности сильно варьирует
у разных индивидуумов. Этот метод заключается в
выборе праймерных сайтов с каждой стороны
повторяющейся последовательности и ее амплификации при
помощи ПЦР.
Подвижность анализируемых образцов ДНК при
электрофорезе зависит от числа повторов в амплифици-
рованном сегменте. Выбирая число таких локусов для
амплификации, получают картину полос, характерную
для каждого индивидуума.
Дополнительная литература
Клонирование гена
Jordan Ε., Collins К S. Human genome project: a march of
genetic maps // Nature. 1996. Vol. 380. P. 111-112.
(Краткое резюме проекта «Геном человека», которое
подчеркивает роль генетических карт в анализе
генетических болезней.)
Полимеразная цепная реакция
SaikiR. К., Gelfand D. К, Stoffel S., ScharfS. J., Higuchi
R., Horn G. Т., Mullis К. В., Erlich H. A.Primer-directed
enzymatic amplification of DNA with a thermostable DNA
polymerase // Science. 1988. Vol. 239. P. 487-491.
(Прекрасное объяснение технологии ПЦР-реакции.)
Arnheim N., Erhlich H. Polymerase chain reaction
strategy // Annu. Rev. Biochem. 1992. Vol. 61. P. 131-156.
(Хорошо написанный обзор, посвященный всем
аспектам ПЦР-реакции.)
часть 4 Хранение и переработка информации
328
Генотерапия
Morgan R. Α., Anderson W. F. Human gene therapy //
Annu. Rev. Biochem. 1993. Vol. 62. P. 191-217.
(Обзор знакомит с болезнями, которые можно лечить
с помощью генотерапии, в том числе лечением дефицита
аденозиндезаминазы, и методами переноса генов;
также обсуждаются вопросы безопасности и этики.)
CapechiM. R. Targeted gene replacement // Sci. Amer. 1994.
Vol. 270(3). P. 34-41.
(Описывает замещение гена методами гомологичной
рекомбинации и трансгенеза.)
Anderson W. Ε Gene therapy // Sci. Amer. 1995. Vol. 273 (3).
P. 96-98.
(Описание успешной инсерции гена
аденозиндезаминазы в белые кровяные клетки и их возврат пациентам
для лечения сочетанного синдрома иммунодефицита.
Включает список из 14 болезней, лечение которых
методами генотерапии прошло клиническое испытание.)
Вопросы к главе 24
1. Чем фермент рестрикции отличается от
панкреатической ДНКазы?
2. Фермент рестрикции Е. соli RI разрезает гексамер-
ную последовательность оснований. Такая
последовательность должна многократно встречаться в ДНК
Е. coli. Почему же фермент не разрушает свою
собственную ДНК?
3. Что обозначает термин «липкие концы»
применительно к молекулам ДНК?
4. Что такое клон генов? Что такое клон кДНК? Чем они
отличаются у эукариот?
5. Каковы этапы подготовки геномной библиотеки в
бактериофаге λ?
6. Каковы этапы выделения определенного клона из
геномной библиотеки, помещенной в фаг λ?
7. Что такое дидезоксинуклеозидтрифосфат?
Какова его функция в секвенировании ДНК по методу
Сэнгера?
8. Кратко изложите суть полимеразной цепной реакции
и условия ее проведения.
9. Что такое бактериальный экспрессирующий плаз-
мидный вектор?
10. Что такое рестрикционный анализ?
ОС Иммунная система
глава ^%J
Организм постоянно находится под угрозой заражения
различными патогенными агентами, например
бактериями и вирусами. Им противостоит иммунная система.
Смерть больных с генетическими (дефицит аденозиндез-
аминазы, см. рис. 18.6) или приобретенными (СПИД)
нарушениями иммунной системы красноречиво
свидетельствует о жизненно важном значении иммунитета.
Иммунная система защищает организм от
макромолекул нескольких типов, наиболее важными среди
которых являются белки. Особенна важна защита от
посторонних белков. Вместе с тем иммунная система
не реагирует на небольшие чужеродные молекулы,
например на лекарства, которые, попадая в организм,
становятся «добычей» других систем химической защиты
(см. главу 17). Поступление чужеродной
макромолекулы в организм служит для него предупреждением о
необходимости защиты против «оккупанта».
Впрочем, у больных возможны аллергические
реакции и на небольшие молекулы, например на
пенициллин. Однако в этом случае организм реагирует не на сам
пенициллин, а на комбинацию его с другой
макромолекулой, например с белком. В результате этой
комбинации белок организма начинает «выглядеть» чужеродным
для иммунной системы.
Из главы 4 вы, наверное, помните, что одной из
главных задач пищеварительной системы является
предотвращение самопереваривания, т. е. разрушения
организмом самого себя. Аналогичная проблема стоит и перед
иммунной защитой.
В отличие от распознавания липополисахаридных
компонентов бактериальной клетки, которые
отличаются от соответствующих компонентов любых бактерий,
присутствующих в нашем организме, и могут быть,
следовательно, легко отнесены к чужеродным, проблема
распознавания посторонних белков гораздо сложнее.
В самом организме есть тысячи белков, отличающихся
от чужеродных только отдельными аминокислотными
последовательностями, так что сортировка «своих»
и «чужих» белков должна проводиться с большой
осторожностью.
Когда иммунная система совершает ошибку и
атакует один из собственных белков, может возникнуть
аутоиммунное заболевание. Например, миастения -
аутоиммунная реакция, в результате которой
разрушаются ацетилхолиновые рецепторы мышцы (см. с. 384)
и нервная стимуляция сокращения становится
невозможной. При ревматической лихорадке иммунный
ответ против белка, продуцируемого некоторыми
штаммами Streptococcus, сопровождается образованием
антител, перекрестно реагирующих с сердечными
белками и вызывающих повреждение сердечных клапанов.
Инсулинзависимый диабет возникает при
аутоиммунной атаке панкреатических клеток.
У иммунной системы существуют
два защитных механизма
Первый - образование антител. Антитела -
растворимые белки, секретируемые клетками иммунной системы,
которые специфически связываются с чужеродными
веществами - антигенами (от англ. antibody generation).
Антигеном называют любую молекулу, способную
вызвать специфический иммунный ответ, в данном случае -
с образованием антител. Образование комплекса
антиген-антитело приводит к развитию защитных реакций,
о которых будет рассказано позже. Такой тип иммунитета
называется гуморальным иммунитетом (от лат. humor -
жидкость); этот механизм защиты обусловлен
присутствием растворимых антител в жидких средах организма.
Второй тип иммунитета, в ходе реализации которого
особые цитотоксические, или киллерные клетки
узнают аномальные клетки в организме, известен как
клеточный иммунитет. Он работает в клетке,
например, при вирусной инфекции. Киллерная клетка
прикрепляется к инфицированной вирусом клетке и разрушает
ее. Это эффективный путь защиты от дальнейшего
распространения вирусной инфекции, поскольку
разрушение клеток прерывает репликацию вируса. Основным
моментом данного механизма является прямой контакт
между киллерной клеткой и клеткой-мишенью -
отсюда и термин клеточный иммунитет. Гуморальный и
клеточный иммунитеты дополняют друг друга. В
рассмотренном нами примере вирусной инфекции гуморальная
система обеспечивает защиту до инфицирования
клетки вирусом, а клеточная система разрушает
клетку-хозяина после ее инфицирования, что позволяет прервать
репликацию вируса в этой клетке.
Антитела вырабатываются особой группой
лимфоцитов, называемых плазматическими клетками
(разновидность лейкоцитов), которые образуются при
глава 25 Иммунная система
329
соответствующей стимуляции В-клеток (В-лимфоцитов).
Цитотоксическими (киллерными клетками) являются
Т-лимфоциты (буква «Т» указывает на место их
образования -тимус, т. е. вилочковую железу, которая у
человека располагается за грудиной). Все лимфоциты
постоянно образуются в костном мозге (у плода - в печени).
В-клетки созревают в костном мозге.
Есть очень важное условие в «разделении труда»
между В- и Т-лимфоцитами. Для того чтобы В-клетка
стала вырабатывать антитела, она должна, во-первых,
встретить антиген, а во-вторых, вступить в контакт
с особым типом Т-клетки, которая уже «познакомилась»
с тем же антигеном. Так как эта Т-клетка «помогает»
В-клетке выполнять свои функции, ее называют хелперной
Т-клеткой (от англ. help - помогать). Хелперные
Т-клетки - самостоятельная группа клеток, отличающаяся
от цитотоксических Т-клеток.
Итак, в организме есть зрелые В-клетки, которые
вырабатывают антитела, хелперные Т-клетки,
помогающие В-клеткам образовывать антитела, и цитотокси-
ческие (киллерные) Т-клетки, ответственные за
клеточный иммунитет.
Где в организме локализована
иммунная система?
Клетки иммунной системы не сгруппированы в один
орган, а распределены по организму: 30% находятся в
селезенке, 20% - в лимфатических узлах, 40% - в
кишечной и слизистой лимфоидной ткани и 10% - в
кровеносных и лимфатических сосудах. Для гуморального
и клеточного иммунитета главными клетками являются
лимфоциты, но в иммунном ответе участвуют и другие
лейкоциты, например макрофаги. Как уже отмечалось
выше, и В-, и Т-клетки являются лимфоцитами. Они
циркулируют в лимфе (в организме есть две
циркулирующих жидкости - кровь и лимфа). В тканях плазма
крови из капилляров поступает в тканевую
(межклеточную) жидкость, которая омывает все клетки. Она отдает
им принесенные с кровью питательные вещества и
забирает продукты жизнедеятельности. Затем эта жидкость
поступает в венозные и лимфатические капилляры, а из
них в более крупные лимфатические сосуды (они
в конечном итоге открываются в две центральные вены
шеи). Лимфатические сосуды в определенных местах
расширяются, образуя лимфатические узлы, в которых
содержание В- и Т-клеток особенно высоко. Узлы
расположены в подмышечной ямке, паховой области,
миндалинах, аденоидах и кишечнике: находящиеся в них
лимфоциты могут встретить любой чужеродный антиген,
попавший в ткани, из которых вытекает лимфа. Поэтому
можно сказать, что узлы постоянно «отслеживают»
тканевую инфекцию, действуя в качестве фильтра, а также
создают определенную среду для размножения и диф-
ференцировки лимфоцитов при их контакте с
антигенами. Лимфоциты образуются в костном мозге. Начало
всем клеткам крови дают стволовые клетки; по мере диф-
ференцировки некоторые из них становятся В-клетками,
другие - Т-клетками, третьи - красными кровяными
клетками и т. д. (рис. 25.1). Как уже отмечалось,
В-клетки развиваются в костном мозге, а Т-клетки мигрируют
в вилочковую железу (тимус), где происходит их
размножение и первичное созревание. Миграция Т-клеток
из костного мозга и высвобождение зрелых Т-клеток
из тимуса происходит, главным образом, на ранних
стадиях развития организма, так что удаление тимуса
у взрослых почти не влияет на иммунные реакции.
Образование В-клеток и их высвобождение из костного
мозга у взрослых людей не прекращается.
Костный мозг и тимус называют первичными
лимфоидными органами, а лимфатические узлы -
вторичными лимфоидными органами. Созревание
лимфоцитов в первичных лимфоидных органах не
зависит от антигена, а все последующие дифференцировки
высвобожденных зрелых лимфоцитов во вторичной
лимфоидной ткани - зависят.
Стволовая клетка может Клетки детерминированы для
дифференцироваться в формирования одного
различные типы клеток специфического типа клеток
ι 1 ι 1
Ч Стволовая
Ч клетка _
Ч О"
Т-лимфоциты
В-лимфоциты
Ч
О
U
о-
о-
о-
Ό-
о-
Эритропоэтин
~** *" ·- О Эритроциты
-► *■ ► О Тромбоциты
► —о
-—о
►—о
—о
Различные типы
фагоцитов и
воспалительных
лейкоцитов
Дифференциация
и размножение
Размножение
Рис. 25.1. Этапы гемопоэза (упрощенно)
Каждая стрелка указывает на возможное размножение клетки,
которое специфически регулируется различными белками -
цитокинами. К ним относятся колоний-стимулирующие
факторы, интерлейкины и эритропоэтины. Последние
стимулируют образование эритроцитов
часть 4 Хранение и переработка информации
330
В этой главе мы знакомимся с иммунным процессом;
сначала вкратце рассмотрим стратегию иммунной
защиты, используемую организмом.
Основная стратегия иммунной защиты
Иммунная система должна решить две задачи: 1)
организацию продукции антител В-клетками в ответ на
чужеродные антигены, но не на собственные компоненты
организма; 2) обеспечение избирательной атаки цитоток-
сическими Т-клетками только аномальных клеток
организма. Основной принцип иммунной защиты
состоит в том, что данная В-клетка может образовывать
только одно специфическое антитело (для связывания
со своим антигеном) и данная Т-клетка отвечает только
на один антиген. В организме образуется громадное
количество различных антител. Им должно
соответствовать эквивалентное число В-клеток, каждая из которых
содержит ген, кодирующий антитело, образуемое этой
клеткой. Возникает естественный вопрос: как может
существовать такое большое число генов? Мы обсудим
это позднее.
В организме также есть множество Т-клеток с
различными рецепторами, каждый из которых специфичен
в отношении своего антигена.
Незрелые В-клетки способны образовывать
антитела, атакующие практически любые макромолекулы в
организме. Для предупреждения аутоиммунных реакций
любая клетка, продуцирующая антитело против
компонентов собственного организма, должна удаляться в
первую очередь. Сначала это происходит в процессе
первичного созревания В-клеток в костном мозге; по мере
созревания В-лимфоциты, по-видимому, адаптируются
к большинству аутоантигенов, с которыми они когда-
либо могут повстречаться. Предполагается
существование и других механизмов подавления аутоиммунных
реакций, но они пока неизвестны.
В организме незрелая В-клетка образует около 105
молекул своего антитела и выставляет их на
поверхность (фиксированными в мембране); при этом В-лим-
фоцит не секретирует антитело, которое играет роль
рецептора. В ходе первичного созревания В-клетки
в костном мозге с антителом, фиксированным на ее
поверхности, может взаимодействовать только антиген,
являющийся составной частью организма. Иммунная
система не должна реагировать на него, в противном
случае на этой стадии развития В-клетки гибнут (или
функционально элиминируются другим способом).
После такого избирательного «умерщвления» выжившие
клетки должны отвечать только на чужеродные антиге-
- ны. Теперь они высвобождаются в кровоток.
Высвобожденная популяция виргильных В-клеток -
так их теперь называют - при встрече с
соответствующими антигенами активируется, и теперь это вполне
оправдано, поскольку встретившийся антиген - чужероден.
Активированная В-клетка не созревает в секретиру-
ющую антитело плазматическую клетку до тех пор, пока
ей не встретится уже активированная при встрече с тем
же антигеном зрелая Т-клетка. Т-клетки реагируют
на специфический антиген при помощи своего
рецептора; при созревании в тимусе они продуцируют
антиген-специфический рецептор, реакция которого на
компоненты собственного организма ведет к элиминации
незрелых клеток. Отбор Т-клеток проиходит на ранних
стадиях развития в ходе их созревания в тимусе, где они,
подобно В-клеткам, встречают большую часть
аутоантигенов. Выжившие в результате селекции Т-клетки
высвобождаются в виде неактивных хелперных
Т-клеток (или предшественников цитотоксичных Т-клеток,
см. ниже), которые при встрече со специфическими
антигенами активируются. Роль активированных
хелперных Т-клеток заключается в доставке сигнала к
специфически активированным В-клеткам - для
размножения и созревания последних в секретирующую
антитело плазматическую клетку (рис. 25.2). Все сказанное
отражает основные принципы теории клональной
селекции. Антиген осуществляет отбор
соответствующих клеток для размножения. Из описанного выше
механизма есть исключение: некоторые компоненты
клеточной стенки грам-отрицательных бактерий -
липополисахариды - могут стимулировать образование
антител соответствующими виргильными В-клетками
и без сигнала хелперных Т-клеток. Контакта антигена
Виргильная
В-клетка
Активация тем
Хелперная же антигеном
Т-клетка *■
Антиген связывается с
антителом
Активированная
*~ В-клетка
Активированная
хелперная Т-клетка
Размножение клеток
Секретирующие антитело
плазматические В-клетки
Циркулирующее антитело
Рис. 25.2. Активация В-клеток для секреции антитела
(основные этапы)
Механизм активации антигеном хелперных Т-клеток несколько
отличается от активации В-клеток тем же антигеном
глава 25 Иммунная система
331
этого типа с В-клеткой вполне достаточно для
созревания последней в секретирующую антитело
плазматическую клетку.
Плазматическая клетка представляет конечный
продукт дифференциации В-клетки. Она специализируется
на синтезе и секреции антитела. Если вспомнить роль
эндоплазматического ретикулума в секреции белка, то
неудивительно, что у плазматических клеток эти ор-
ганеллы очень хорошо развиты. (Регуляция деления
В-клеток и хелперных Т-клеток будет описана позже.)
В тимусе некоторые из незрелых клеток
развиваются в предшественника цитотоксических, или киллерных
Т-клеток, которые играют важную роль в уничтожении
зараженных вирусом клеток. При этом происходит
аналогичный отбор: любая потенциально цитотоксическая
клетка, рецептор которой реагирует на компонент
собственного организма, инактивируется или
элиминируется. Прошедшие селекцию клетки высвобождаются
из тимуса в кровоток. При узнавании чужеродного
антигена на поверхности зараженной клетки-хозяина они
становятся функционально активными цитотоксически-
ми клетками. Когда активированной цитотоксической
Т-клетке встречается клетка, несущая чужеродный
антиген, с ним взаимодействует ее рецептор. В результате
Т-клетка выделяет перфорин - белок, убивающий
клетку-мишень путем увеличения проницаемости ее
мембраны или вызывающий апоптоз- программируемое
саморазрушение клетки.
Механизмы иммунитета:
В-клетки, хелперные Т-клетки
и образование антител
Структура антител
Антитела являются иммуноглобулинами. Термин
глобулин относится к ранней системе классификации: так
обозначали белок, растворимый в солевом растворе.
Иммуноглобулин обычно пишется сокращенно как Ig.
Существует несколько классов иммуноглобулинов,
одним из них является IgG, вырабатываемый клетками
в больших количествах при длительном контакте с
антигеном. Поэтому рассмотрим сначала именно его
структуру. Молекула IgG имеет Y-образную форму. Она
состоит из 4 полипептидных цепей: 2 одинаковых
легких L (от англ. light) и 2 одинаковых тяжелых Н-цепей
(от англ. heavy), соединенных дисульфидными связями
(рис. 25.3). Концы лучей «Y» имеют вариабельные
области как на тяжелых, так и на легких цепях. Именно эти
фрагменты формируют антиген-связывающий участок
на каждом луче. Связывание с антигеном происходит
за счет нековалентных связей. Таким образом, антитело
имеет два идентичных антиген-связывающих центра,
следовательно, оно может образовать с молекулами
антигена поперечную связь. У развилки «Y» находятся гибкие
«шарнирные» области, способствующие формированию
поперечных молекулярных связей. Аналогичная (но
не идентичная) структура свойственна и другим
иммуноглобулинам. Некоторые из них (например IgM),
представляют собой полимеры, состоящие из 5 субъединиц,
каждая из которых очень похожа на молекулы IgG.
а Участки,
N .... взаимодействующие ...
Вариабельные
области
обеих цепей
Fc - фрагмент
тяжелой цепи
Константная
область
Антиген
Рис. 25.3. Структура молекулы IgG
а - Молекула IgG. Молекулы IgM и IgA отличаются Fc-фраг-
ментами, но схожи в вариабельной части. Η - тяжелая цепь; L -
легкая цепь. N и С соответственно обозначают амино- и кар-
бокси-концы полипептида. Фрагмент Fc - С-концевая часть
Η-цепей, ковалентно соединенных в один домен S-S-связями;
б - поперечная связь антигена с отдельными
специфическими эпитопами;
в - антигены с множественными антигенными
детерминантами образуют с антителом поперечносшитый нерастворимый
кластер
часть 4 Хранение и переработка информации
332
Несмотря на большие размеры антигена,
вызывающего иммунный ответ, данное антитело связывается
только с небольшой его частью (в случае белкового
антигена - всего с несколькими аминокислотами).
Специфическая часть антигена, узнаваемая антителом,
называется эпитопом. Большой белковый антиген будет
провоцировать образование ряда различных антител,
каждое из которых связывается со специфическим
эпитопом и вырабатывается отдельным клоном В-клеток.
Организм может контактировать с громадным
числом различных антигенов и потенциально способен
вырабатывать различные антитела, взаимодействующие
с ними. Специфические антитела различаются по
аминокислотной последовательности антиген-связываю-
щих участков. У антител, вырабатываемых различными
клетками, и L-, и Η-цепи содержат вариабельные
аминокислотные последовательности (V-домены),
определяющие особенности структуры антиген-связывающих
участков и обусловливающие тем самым большое
разнообразие антител. Как уже отмечалось, принцип
организации иммунной системы заключается в произвольном
образовании В-клеток, отличающихся генами,
кодирующими антитела. При этом каждая вновь
развившаяся В-клетка может вырабатывать антитела только
одинаковой антигенной специфичности, молекулы которых
на этой стадии зафиксированы в плазматической
мембране, а взаимодействующие с антигеном участки
располагаются снаружи. Образно говоря, клетка выставляет
антитела на поверхность, словно товар на витрину
магазина. Когда подойдет антиген, одно или несколько
зафиксированных на мембране антител свяжутся с ним,
инициируя активацию В-клетки.
Каковы функции антител?
Молекула антитела IgG бивалентна: она содержит 2
идентичных участка связывания, каждый из которых может
связываться с одним и тем же эпитопом молекулы
антигена. Если антиген имеет более двух идентичных эпито-
пов, одно антитело будет образовывать целую сеть в
комплексе антиген-антитело. Большинство
вырабатываемых у животных антител содержит множественные
участки узнавания различных эпитопов данного
антигена, так что взаимодействие антител с антигенами
приведет к образованию целой сети молекул, которая
активирует систему комплемента. Название последней
указывает на то, что она «дополняет» (англ. complement)
действие антител при лизисе бактериальной или других
инвазивных клеток. Комплемент - название комплекса
примерно из 20 белков крови. После взаимодействия
антигена с антителом на бактериальной клетке происходит
фиксация некоторых из этих компонентов. Белки
комплемента окружают комплекс антиген-антитело,
приводя к гибели бактериальной или другой клетки
вследствие перфорации клеточной мембраны. Кроме
того, комплекс покрытой антителом бактерии может
поглощаться фагоцитирующими лейкоцитами
(например макрофагами и нейтрофилами), которые
убивают и переваривают микроорганизмы.
Фагоцитирующие клетки имеют на своей поверхности рецепторы
(Fc-рецепторы). Некоторые из них связываются с
константной Fc-областью антител (ствол «Y», см. рис. 25.3, а),
которая выступает из комплекса антиген-антитело, в то
время как другие (рецепторы комплемента) связывают
прикрепившиеся компоненты комплемента. Эти
события вызывают адгезию фагоцита и его мишени и
запускают механизм поглощения частицы. Процесс
активации комплемента сопровождается выделением
биологически активных веществ, расширяющих кровеносные
сосуды, что способствует поступлению фагоцитов к
инфицированному участку.
Классы антител
Существует 5 основных классов иммуноглобулинов,
отличающихся один от другого константными областями
Η-цепей. Эти различия не имеют ничего общего с анти-
ген-связывающими участками, но ответственны за
физиологическую функцию антитела.
Когда организм отвечает на антиген, первым из
антител образуется IgM. Это мультисубъединичная форма
антитела с 10 антиген-связывающими участками,
которая из-за наличия множественных участков связывания
особенно эффективна при обезвреживании вирусов
и бактерий. Этот иммуноглобулин участвует в активации
комплемента и обеспечении фагоцитоза.
Повторные введения того же антигена приводят
к массированному образованию IgG. Он также
активирует комплемент и эффективно переносится к плоду
через плаценту.
Иммуноглобулин A (IgA) практически незаменим
на первой линии защиты. Он транспортируется через
мембрану эпителиальных клеток (в сочетании с особым
секреторным пептидом), в слизистый слой кишечника,
дыхательной системы и т. д. и секретируется в молоко.
IgA играет важную роль, например, в выработке
иммунитета к холерному вибриону, который в ходе
инфекционного процесса должен прикрепиться к эпителиальной
выстилке кишечника в участках Пейеровских бляшек
и других местах. Бактерии, покрытые специфическим
IgA, не могут взаимодействовать с эпителиальными
клетками кишечника и поэтому не участвуют в заражении.
глава 25 Иммунная система
333
IgA обладает свойствами, определяющими его роль
в мембранах слизистых оболочек. Он не сразу
разрушается протеолитическими ферментами кишечника
и не может эффективно активировать комплемент.
Иммуноглобулин Ε (IgE) участвует в выделении
гистамина и способствует развитию симптомов
аллергии в присутствии специфического антигена или
аллергена. У людей с повышенной чувствительностью
он может приводить к синтезу фактора активации
тромбоцитов (англ. platelet activating factor; PAF). PAF
имеет сходство с лецитином, только его центральная
жирно-ацильная группа замещена ацетильной. Он
способствует воспалению дыхательных путей и вызывает
агрегацию тромбоцитов, снижение артериального
давления и др. Функции IgD неизвестны.
Хотя каждая В-клетка может продуцировать только
антитела одинаковой антигенной специфичности, она
способна, сохраняя свою антигенную специфичность,
переключаться с выработки одного класса антител на
другой. Это переключение позволяет клону В-клетки
увеличивать диапазон физиологических функций,
осуществляемых ее антителами. Переключение выработки
одного класса антител на другой происходит за счет
рекомбинации ДНК.
Как достигается разнообразие антител?
Клетка человека содержит примерно 50 000-100 000
генов, кодирующих все белки организма. Для
кодирования, скажем, миллиона антител требуется миллион
различных генов. Ясно, что для синтеза такого громадного
разнообразия антител необходимо предпринять нечто
особенное.
Чтобы понять, как достигается такое разнообразие,
подробно рассмотрим L-цепь. У нее есть два участка:
концевая вариабельная область для связывания
антигена и константная область, идентичная для всех
молекул Ig. Процесс организации функционально
активного гена, кодирующего L-цепь иммуноглобулина,
включает соединение сегментов, кодирующих константную
область, и одного из многочисленных сегментов,
кодирующих вариабельные области (рис. 25.4).
Рассмотрим стволовую клетку в костном мозге до
того, как она дифференцировалась в В-клетку. ДНК,
кодирующая L-цепь, состоит из нескольких участков. Есть
сегмент, кодирующий константную (С) область; есть
4 отдельных сегмента, каждый из которых кодирует
различные короткие пептидные последовательности. Они
могут быть использованы для объединения константной
и вариабельной областей. Эти последовательности
называются J-участками (от англ. joining - соединение).
Примерно 300 генов
V-сегментов
Сегменты Одиночный
J-гена сегмент С-гена
ДНК
24
Номер 24
V-сегмента и |
номер 3 J-сегмента
выбраны для
иллюстрации условно
* 300
Этот фрагмент
элиминируется в
ходе рекомбинации
Расположение
сегментов гена
б ДНК
24
Рекомбинация Расположение сегментов
гена в В-клетке. Оно будет
I— различаться у разных
клеток в зависимости от
того, какие сегменты выбраны
Транскрипция
РНК
24
Сплайсинг
мРНК 3
24
Первичный транскрипт РНК
мРНК легкой цепи
иммуноглобулина
Рис. 25.4. Процесс рекомбинации ДНК, ведущий к
образованию функционального гена, кодирующего L-цепь
иммуноглобулина
а - Расположение сегментов гена в стволовой клетке, где гены
иммуноглобулина не экспрессируются;
б - случайно выбранный V-ген (в этом примере V24)
двигается к одному из J-генов (здесь J3), а находящийся между ними
сегмент ДНК вырезается;
в - транскрипция начинается в сегменте гена V24,
транскрибируются также J-гены (J3 и J4);
г - после сплайсинга РНК, удаляющего транскрипты J4 и
нитронов, транскрипты, соответствующие V24, J3 и С,
образуют мРНК.
Благодаря аллельной эксклюзии только один из пары аллель-
ных генов становится функциональным геном
иммуноглобулина (см. текст), так что данная клетка продуцирует только
один иммуноглобулин, а не два
И наконец, есть около 300 сегментов (V), любой из
которых кодирует альтернативные вариабельные концы
L-цепи. Все 300 V-сегментов отличаются друг от друга,
также как и 4 J-сегмента. В каждом конкретном случае
V-сегменты будут кодировать различные пептидные
последовательности. Когда стволовая клетка костного
мозга дифференцируется в В-лимфоцит, происходит
реорганизация, вызванная рекомбинацией и эксцизией
ДНК и приводящая к соединению одного из 300
V-сегментов к одному из сегментов J-участков. Это приводит
к образованию нового сложного гена у каждого из В-
лимфоцитов. Ген иммуноглобулина может содержать
более одного сегмента J-участка, но в ходе сплайсинга
первичного транскрипта РНК в мРНК остается только
часть 4 Хранение и переработка информации
334
один J-сегмент (см. рис. 25.4). Рекомбинация ДНК -
совершенно беспорядочный процесс. Таким образом,
в конечной мРНК, кодирующей L-цепь молекулы IgG,
любой из 300 V-сегментов соединяется с любым из
4 J-сегментов, что дает 1200 различных комбинаций.
Рекомбинация неточна, потому что места разреза и
повторного соединения немного различаются: это увеличивает
число вариантов L-цепи примерно до 3000.
Ген Η-цепи организован аналогично, но в его
образовании участвует гораздо большее число вариабельных
сегментов, дающих еще больше вариантов гена Н-цепи.
Формирование антиген-связывающего участка при
комбинации вариабельных частей Н- и L-цепей,
существование большого числа генов, кодирующих эти цепи,
а также многообразие различных участков связывания
объясняют способность иммунной системы
вырабатывать громадное число различных антител. Таким
образом, каждая развивающаяся В-клетка собирает наугад
гены, кодирующие одно антитело со своим спеиифичес-
ким антиген-связывающим участком.
Формирование генов, кодирующих иммуноглобулин
в диплоидных клетках, приведет к тому, что каждая
В-клетка будет продуцировать не один, а два
специфичных антиген-связывающих участка. Однако процесс
аллельной эксклюзии сопровождается экспрессией
генов легкой и тяжелой цепей иммуноглобулинов только
на одной из гомологичных хромосом.
«Изобретательность» иммунной системы
заключается и в том, что после активации В-клетки происходит
быстрая мутация в области V, приводящая к увеличению
многообразия антител. Многие из этих мутаций могут
уменьшить сродство антитела к антигену, но некоторые
будут его увеличивать. Антиген преимущественно
связывается с клетками, вырабатывающими мембраносвя-
занные антитела с более высоким сродством к нему.
Поскольку связывание антигена ведет к размножению
клеток, организм может отобрать клетки, наиболее
эффективно вырабатывающие антитела против
определенного антигена. После контакта с антигеном начинается
быстрый процесс «эволюции» - аффинное созревание,
которое совершенствует эффективность антител,
увеличивая их сродство к антигену.
Какова роль хелперных Т-клеток
в активации В-клеток для секреции
антител?
В-клетка, активированная в ходе связывания антигена с
фиксированным на ее поверхности антителом,
захватывает антиген и осуществляет его расщепление на мелкие
фрагменты, которые взаимодействуют с пептидными
рецепторами, называемыми МНС-белками. Затем
фрагменты антигена транспортируются к клеточной
поверхности и помещаются в участках связывания
МНС-белков (рис. 25.5). Молекулы МНС являются гликопроте-
инами главного комплекса гистосовместимости (МНС -
от англ. major histocompatibility complex). В данном
случае молекулы МНС принадлежат к классу II (см. ниже)
и экспрессируются теми клетками в иммунной системе,
которые функционируют в качестве
специализированных антиген-представляющих, включая В-клетки.
Подвергнутые процессингу антигены, расположенные
Виргильная В-клетка
О
Антитело, фиксированное в мембране,
выставляет антиген-связывающий
участок как рецептор
г
Встречает специфический антиген
-с
Комплекс «Рецептор-антиген»
подвергается эндоцитозу
Антиген фрагментируется на пептиды;
клетка синтезирует МНС - гликопротеины класса II
Фрагменты антигена в связанном
состоянии экспонированы на клеточной
поверхности в бороздке
молекулы МНС класса II
В-клетка находится теперь в активированном состоянии: она не
продуцирует антител до тех пор, пока не свяжется с хелперной
клеткой, которая была активирована антиген-представляющей
клеткой, несущей на своей поверхности тот же антиген. В этом
случае В-клетки созревают в секретирующие антитело
плазматические клетки и размножаются за счет выделения
цитокинов хелперной клеткой
Рис. 25.5. Процесс активации виргильной В-клетки до ее
превращения в плазматическую клетку, продуцирующую
антитела
глава 25 Иммунная система
335
в комплексе с молекулами МНС на поверхности В-кле-
ток, могут быть «узнаны» хелперными Т-клетками,
имеющими рецепторы, специфические для комплекса
МНС-антиген. Хелперные Т-клетки антител не образуют,
однако они продуцируют антиген-специфические
поверхностные рецепторы TCR(ot англ. Τ Cell Receptors),
которые располагаются на Т-клетках. TCR узнает
не фрагмент самого антигена на поверхности АРС,
не молекулу МНС, а только комплекс молекулы МНС
класса II с фрагментом антигена. Взаимодействию АРС
и хелперной Т-клетки способствует ее белок CD4. Он
связывается с константным белком клетки АРС, который
является белком МНС класса Π (рис. 25.6). Термин CD4
означает кластер дифференциации', он связан с методом,
использованным для определения специфических
маркеров дифференциации в клетке. Цифра «4» обозначает
специфический участок, найденный экспериментальным
путем. Белок CD4 - это рецептор, к которому
прикрепляется вирус СПИДа. Когда хелперная Т-клетка,
поступившая после созревания в кровоток, соединяется
с соответствующим комплексом МНС-антиген,
выставленным на циркулирующей АРС, происходит ее
активация (см. рис. 25.6). Если активированной хелперной
Т-клетке встретится В-клетка, экспонирующая тот же
комплекс МНС-антиген (что был исходно представлен
Т-клетке клеткой АРС), Т-клетка связывается с этой
В-клеткой, выделяя цитокины. Последние стимулируют
размножение В-клеток (рис. 25.7), и образовавшиеся
плазматические клетки (секретирующие В-лимфоциты)
«выплескивают» защитное антитело. В плазматических
клетках хорошо развит эндоплазматический ретикулум,
необходимый для секреции белка (см. с. 300).
Почему в рассматриваемом случае антитело секре-
тируется, а не остается зафиксированным в мембране?
Ответ заключается в различном сплайсинге интронов
мРНК генов иммуноглобулинов (см. с. 271). На З'-конце
гена иммуноглобулина есть сегмент, кодирующий
полипептидную последовательность, которая закрепляет
антитело в мембране. Начало секреции вызывается
переключением в механизме сплайсинга, в ходе которого
этот сегмент удаляется из мРНК.
Клетки памяти
При активации и пролиферации В-клеток не все клетки
клона превращаются в секретирующие антитела
плазматические клетки. Часть их становится длительно
живущими клетками памяти, которые могут
циркулировать в организме годами: они составляют основу
долговременного иммунитета от повторной инфекции
Если потом произойдет контакт с соответствующим
антигеном, иммунная система отреагирует очень быстро.
Т-клетки также участвуют в формировании
иммунологической памяти.
Рецептор
Т-клетки
МНС белки класса II, представляющие
подвергнутый процессингу антиген
Эта Т-клетка имеет рецептор, Хелперная Т-клетка
который связывается с после выделения TCR
комплексом антиген-МНС из тимуса
на АРС
CD4
МНС Π
Эта АРС захватывает антиген,
подвергает его процессингу с
АРС (антиген- образованием фрагментов и
представляющая экспонирует его в своем МНС белке,
клетка) «приглашая» хелперную Т-клетку
узнать его соответствующим
рецептором
Аутокринная стимуляция
интерлейкином-2 вызывает
пролиферацию клеток
АРС продуцирует интерлсйкин-1 и другие
цитокины. Они стимулируют хелперную
клетку к выделению интерлейкина-2.
который может индуцировать клеточную
пролиферацию
Рис. 25.6. Активация хелперной Т-клетки клеткой АРС (antigen-presenting cell)
Аутокринная стимуляция хелперной Т-клетки позволяет клеткам размножаться после того, как произошло их разделение.
CD4 - гликопротеин, расположенный на Т-клетке, он взаимодействует с белком МНС типа II клетки АРС. Это взаимодействие
необходимо для связывания рецептора Т-клетки (TCR) с комплексом «АРС-МНС-антиген». Белок CD4 является рецептором,
при помощи которого вирус СПИДа инфицирует хелперную Т-клетку
часть 4 Хранение и переработка информации
336
Активированная В-клетка
^^^^^^ Фрагмент антигена,
[ ^ /'связанный с
I ^К МНС класса II уЩ] т.ыеша
>v Рецептор /
N. Т-клетки ,/ Активированная хелперная
N. >^ Т-клетка
гррр Хелперная
Т-клетка
Секреция цитокинов
стимулирует деление клеток
В-клетка теперь активирована,
она делится и созревает в клон
секретирующих антитело В-клеток,
называемых плазматическими клетками
Рис.25.7. Активация хелперной Т-клеткой превращения
В-клетки в клон плазматических клеток, секретирующих
антитело
Цитокины - факторы роста, стимулирующие клетки к
делению. В ответ на активацию В-клетки, которая в результате
становится плазматической клеткой, различный процессингтранс-
криптов РНК приводит к элиминации участка антитела,
ответственного за заякоривание в мембране. Считается, что
цитокины, в том числе секретируемые хелперной Т-клеткой,
активируют цитотоксические клетки
Т-клетки и клеточный иммунитет
Как уже было сказано, TCR-рецепторы на хелперных
Т-клетках узнают антигены, ассоциированные с
молекулами МНС класса II, которые присутствуют на АРС
и В-клетках. Другая группа Т-клеток - цитотоксические,
или киллерные клетки - имеет рецепторы, узнающие
антигены на молекулах МНС класса I. Последние
присутствуют на поверхности большинства клеток
организма. Киллерные Т-клетки взаимодействуют только с
клетками-хозяевами, выставляющими чужеродный антиген
на своих молекулах МНС класса 1. Хотя это
утверждение в общем верно, результаты недавних исследований
указывают на то, что в определенных случаях
цитотоксические клетки могут узнавать белки МНС класса П.
Мы не хотим запутывать читателей, однако важно
представлять невероятную сложность иммунной системы,
работа которой во многом остается непонятной.
На поверхности киллерной Т-клетки расположен
гликопротеин CD8, который взаимодействует с
константным белком МНС класса I. Поскольку связывание
клеток происходит путем взаимодействия CD8-MHC,
цитотоксические клетки атакуют только клетки,
содержащие МНС класса I. Цитотоксические клетки
узнают и убивают клетки организма, ставшие
аномальными, например, при вирусной инфекции. В
инфицированной вирусом клетке вирусный белок (синтезированный
в клетке) подвергается расщеплению на пептиды,
которые экспонируются на поверхности клетки в комплексе
с молекулами МНС класса I (рис. 25.8).
Антиген-специфичная киллерная Т-клетка связывается с комплексом
антиген-МНС инфицированной клетки и разрушает ее,
вызывая перфорацию мембраны (см. рис. 25.8) или
инициируя апоптоз.
Роль цитокинов в иммунной системе
В главе 26 мы обсудим, каким образом при помощи
гормоноподобных сигнальных молекул осуществляется
связь между клетками. Важным классом таких молекул
являются факторы роста, к которым относятся белки,
подающие клеткам сигнал к размножению. Когда
хелперная Т-клетка активируется антиген-представляющей
клеткой, она секретирует цитокины, в их числе и интер-
лейкины, стимулирующие размножение и созревание
обоих типов клеток. Цитокины секретируются хелпер-
ными Т-клетками при контакте с В-клетками, и
некоторые из них участвуют в активации киллерных Т-клеток.
Одни цитокины стимулируют фагоциты, другие
вызывают переключение класса антител у В-клеток,
стимулируют деление клеток и созревание В-клеток в
плазматические клетки. Иммуносупрессор циклоспорин,
используемый при трансплантации органов, ингибиру-
ет образование интерлейкинов хелперными Т-клетками.
Почему иммунная система
человека столь неистово
отторгает чужеродные
человеческие клетки?
На первый взгляд, есть что-то озадачивающее в самом
факте существования механизма иммунной атаки на
клетки, перенесенные от одного индивида к другому. Почему
происходит отторжение трансплантанта ткани? Ведь
эволюции не был известен такой перенос. Главной
причиной отторжения чужеродного тканевого трансплантанта
глава 25 Иммунная система
337
Вирусный белок, синтезированный
в результате инфекции вирусом
Вирусный белок подвергается
расщеплению на фрагменты
Фрагменты вирусного антигена,
связанные с МНС класса I
TCR
CD8
Цитотоксическая
Т-клетка
в высоко полиморфный главный комплекс гистосовме-
стимости. Для каждого гена, кодирующего МНС белок,
в популяции существует много аллелей (вариантов).
Поэтому маловероятно, что молекулы МНС разных
индивидов будут одинаковыми. МНС-антигены - это
те же молекулы, что и антиген-пептидные рецепторы
класса I и П. Чужеродная молекула МНС распознается
киллерной Т-клеткой как молекула МНС собственного
организма, образовавшая комплекс с чужеродным
антигеном; поэтому несущая его клетка будет атакована ци-
тотоксическими Т-клетками. Напоминаем, что рецептор
киллерной Т-клетки узнает комплекс молекулы МНС
и антигена, а не каждый компонент в отдельности.
Эволюция такой вариабельности молекул МНС в популяции
могла бы выступать в качестве защиты видов в целом
от гибели вследствие инфекции. Предположим, что
случайно вирус развивается таким образом, что его
подвергнутый процессингу антиген выставляется на молекуле
МНС клетки-хозяина: в этом случае он распознается
клеточной иммунной системой как нормальный - или
не распознается совсем. Тогда вирус может
размножаться и убить своего хозяина. Впрочем, вряд ли у
следующего инфицированного индивида окажутся те же
аллели МНС, поэтому вероятность повторения событий,
описанных выше, уменьшается. Следовательно,
несмотря на гибель отдельных индивидов, болезнь вряд ли
распространится по всей популяции.
МНС ί TCR
CD8
Рис. 25.8. Последовательность развития событий в клетке
в ответ на появление чужеродного антигена
На рисунке не показано, что деление цитотоксической клетки
стимулируется цитокинами, выделяемыми при взаимодействии
хелперной Т-клетки и В-клетки. Это приводит к образованию
клона цитотоксических клеток, специфичных в отношении
определенного антигена; часть клеток клона цитотоксических
клеток развивается в клетки памяти. Инфицированная клетка
может быть убита посредством перфорации мембраны при
освобождении белка перфорина или в результате апоптоза
служат молекулы МНС на клетке. Эти молекулы
составляют семейство гликопротеинов, где класс I
и класс II - подсемейства, структурно различающиеся
между собой. Кодирующие их гены кластеризованы
Дополнительная литература
Иммунная система
Sci. Amer. 1993. Vol. 269 (3). P. 20-108.
(Прекрасно иллюстрированный полный выпуск
«Scientific American», посвященный всем аспектам иммунитета.)
Гемопоэз
и гемопоэтические факторы роста
MetcalfD. The 1991 Florey Lecture. The
colony-stimulating factors: discovery to clinical use. // Philos. Trans. Roy.
Soc. London B. 1991. Vol. 333. P. 147-173.
(Содержательный обзор, написанный
первооткрывателем этих важных белков. Хотя уровень представленного
материала носит более экспериментальный характер, чем
это нужно для студентов, он прекрасно освещает
данную тему, от открытия этих неидентифицированных
факторов до их широкого клинического использования.)
часть 4 Хранение и переработка информации
338
Kelso A. The immunophysiology of Τ cell-derived
cytokines // Today's Life Science. 1993. Vol. 5(4). P. 30-40.
(Прекрасное резюме о роли цитокинов в иммунной
системе.)
Элиминация незрелых клеток
реагирующих на аутоантигены
Collins Μ. Death by a thousand cuts // Curr. Biology. 1991.
Vol. 1. P. 140-142.
(Краткое сообщение о программируемой клеточной
смерти незрелых Т-клеток в тимусе.)
Моноклональные антитела
Yelton D., ScharffM. D. Monoclonal antibodies: a power-
full new tool in biology and medicine // Annu. Rev. Biochem.
1981. Vol. 50. P. 657-680.
(Обзор методов получения моноклональных антител и их
применения.)
Chien S., Silverstein S. С. Economic impact of
applications of monoclonal antibodies to medicine and biology//
FASEB J. 1993. Vol. 7. P. 1426-1431.
(Интересное описание этапов разработки
моноклональных антител и их практического применения.)
Представление антигенов
Engelhard V. Η. How cells process antigens // Sci. Amer.
1994. Vol.271 (2). P. 44-51.
(Сообщение о процессинге антител и демонстрация
фрагментов на белках МНС.)
Патогены и иммунная система
Goodenough U. W. Deception by pathogens // Amer. Sci.
1991. Vol. 79. P. 344-355.
(Увлекательное объяснение способов защиты бактерий
и вирусов от иммунологического выявления на
молекулярном уровне. Ясно изложены иммунные механизмы.)
Nowak Μ. Α., McMichael A. J. How HIV defeats the
immune system // Sci. Amer. 1995. Vol. 273(2). P. 42-49.
(Представляет гипотезу, согласно которой
продолжительная эволюция вируса иммунодефицита человека в
организме ведет к иммунному истощению СПИДа).
Вопросы к главе 25
1. Опишите структуру молекулы IgG.
2. Объясните, каким образом гены кодируют
множество различных антител.
3. Какие вы знаете типы лимфоцитов и каковы их
функции?
4. Что такое антиген-представляющая клетка?
5. Когда клетка-хозяин выставляет, например,
вирусный антиген на своей поверхности, на каком классе
МНС молекул он выставлен? Какой класс МНС
молекул узнает цитотоксическая Т-клетка?
6. Что такое «теория клональной селекции»?
7. В чем заключается принцип уклонения от аутоимму-
нитета, или каким образом иммунная система
становится устойчивой к аутоантигенам?
8. Образование антител после стимуляции В-клетки
(для перехода в плазматическую клетку) хелперной
Т-клеткой не изменяет антиген-связывающего
участка антитела. Но антитело должно секретироваться,
а не оставаться в мембране. Каким образом это
достигается?
9. Когда В-лимфоцит (плазматическая клетка)
начинает секретировать антитело, последнее может
обладать относительно низким сродством к своему
антигену, но этот недостаток быстро устраняется. Каким
образом?
10. Какие иммунные системы включают гликопротеин
CD4, а какие - CD8? Какова их роль в иммунной
реакции? Какое отношение имеет CD4 к вирусу
СПИДа?
глава 25 Иммунная система
339
Oft Химическая сигнализация в организме
глава ^U
В главе 12 мы уже обсуждали регуляцию метаболизма
внешними факторами, обеспечивающими сообщение
между клетками. Жизнедеятельность каждой отдельной
клетки подчинена нуждам организма как единого
целого. Животное не может существовать без сложной
системы межклеточных коммуникаций, которые
осуществляются через разнообразную систему команд.
Теперь выяснилось, что внешнее управление клетками
намного сложнее, чем это можно было представить.
Механизмы, осуществляющие отдельные виды регуляции,
известны, однако до конца неясно, как все регуляторные
факторы объединяются в единую систему в целом организме или
хотя бы в отдельной клетке. Сообщение между клетками
обеспечивают сигнальные молекулы. Они выделяются из
одних клеток и мигрируют к другим, снабженным
рецепторами, способными воспринимать эти сигналы. Это -
клетки-мишени. Связывание сигнальной молекулы с
рецептором приводит к биохимическому ответу клетки.
Сигнал может передаваться при непосредственном
контакте между двумя клетками. Как пример можно
привести активацию хелперной Т-клетки (см. главу 25)
антиген-представляющей клеткой. Наличие щелевых
контактов или регулируемых пор между соседними
клетками позволяет молекулам мигрировать от одной
клетки к другой, осуществляя прямое сообщение
между цитоплазмами и координируя клеточные функции.
В это главе мы разберем следующие вопросы.
• Какие виды клеточной активности контролируются
внешними сигналами?
• Какие типы сигнальных молекул участвуют в
химической сигнализации?
• Какие клетки выделяют сигнальные молекулы.
Каким образом контролируется их выделение и как они
доходят до своих клеток-мишеней?
• Как клетки-мишени распознают сигналы?
• Как клетка отвечает на принятый сигнал?
Какие виды клеточной
активности регулируются
внешними сигналами?
Для удобства их можно разделить на две группы.
Регуляция, не затрагивающая экспрессию гена.
Примерами служат: изменение активности ферментов,
участвующих в метаболизме жиров и углеводов
(см. главу 12); активация произвольного сокращения
поперечно-полосатой мышцы ацетилхолином (см. главу 28);
открытие лиганд-зависимых пор в мембранах нервной
клетки при включении нервного импульса (рис. 26.24).
Регуляция, затрагивающая экспрессию генов.
Большинство факторов внешнего управления действуют
именно этим способом. Поскольку регуляция
экспрессии гена влияет практически на все процессы,
протекающие в клетках, список таких процессов безграничен.
У животных контроль экспрессии генов осуществляется
главным образом на уровне инициации транскрипции
гена, и это, в свою очередь, зависит от активности
специфических факторов транскрипции (см. с. 267).
Таким образом, основная часть этой главы касается
механизмов, посредством которых факторы внешнего
управления влияют на транскрипцию специфических генов.
Большинство внешних сигнальных молекул, доставляя свои
сигналы, не входят в клетку, а взаимодействуют с
внешними доменами мембраносвязанных рецепторов. Поэтому
возникают вопросы: как сигналы, воспринимаемые
снаружи клеточной мембраны, приводят к определенным
событиям внутри клетки (трансмембранная передача сигнала)!
Как сигнал, генерируемый на цитоплазматической стороне
мембраны, передается к контролируемому участку? В
случае контроля гена это означает передачу сигнала от
цитоплазматической мембраны к ядру клетки (рис. 26.1). Кроме
того, небольшое число растворимых в липидах сигнальных
молекул могут проходить через липидный бислой внутрь
клетки, где они контактируют с внутриклеточными
рецепторами, участвующими в регуляции гена (см. рис. 26.1).
Что такое
сигнальные молекулы?
С химической точки зрения - это белки, большие и
маленькие пептиды, стероиды, эйкозаноиды, катехолами-
ны, тироксин и оксид азота. К числу первых шести групп
веществ относятся: инсулин, глюкагон, вазопрессин,
половые гормоны, простагландины и адреналин.
Биологическая классификация включает следующие
группы регуляторов:
1) гормоны;
2) факторы роста и цитокины;
3) нейромедиаторы.
часть 4 Хранение и переработка информации
340
Сигнальный лиганд
(водорастворимый)
т м
Клеточные
ответы
Цитоплазма я
Внутрикле
рецептор
на
Сигнальный лиганд
(растворимый в липидах)
Рис. 26.1. Рецептор-опосредованная сигнализация
Водорастворимые сигнальные молекулы не могут пройти
сквозь липидный бислой. Они связываются с внешними
доменами рецепторов. Растворимые в липидах сигнальные
молекулы, например стероиды и тироксин, непосредственно
поступают в клетку и связываются с внутриклеточными
рецепторами. Некоторые внутриклеточные рецепторы находятся в ядре;
стероидные рецепторы после связывания с лигандом
двигаются в ядро. В случае оксида азота внутриклеточный
рецептор вырабатывает cGMP, который может вызывать прямые
метаболические эффекты
Гормоны
Это «классические» сигнальные молекулы, большинство
из которых известно давно, вероятно потому, что в
организме они находятся в относительно больших
количествах. Гормоны образуются клетками,
специализирующимися на их продукции и собранными в железы, се-
кретирующие гормоны прямо в кровоток, а не в протоки,
сообщающиеся с внешней средой (как это происходит
при секреции панкреатических пищеварительных
ферментов в кишечник). Поэтому эндокринные железы
называют еще беспроточными железами. Гормоны
«находят» свои клетки-мишени при помощи системы
кровообращения, достигая тканей, расположенных
на значительном удалении от гормон-секретирующей
железы. Известно большое число гормонов и подробно
описаны их биологические эффекты. Основные гормоны
и оказываемое ими действие приведены в таблице 26.1.
Факторы роста
В недавно опубликованной статье было выдвинуто
предположение о том, что этому классу сигнальных молекул
больше подходит название регуляторные факторы
развития, поскольку их роль становится понятнее, если
рассматривать их в качестве регуляторов развития,
а не факторов, стимулирующих рост клеток.
Факторы роста - это регуляторные белки, выделяемые
клетками той же ткани, к которой они принадлежат,
например гепатоцитами, лимфоцитами и т. д. Факторы роста
взаимодействуют с наружным рецептором клетки. Первым
был открыт фактор роста из тромбоцитов (PDGF; от англ.
platelet-derived growth factor), который, как оказалось,
образуется и во многих других клетках. Другой
классический пример - фактор роста эпидермиса (EGF; от англ.
epidermal growth factor). Обнаружено много
разнообразных факторов. Около 20 из них участвуют в регуляции
развития гемопоэтических клеток в костном мозге. Многие
факторы роста являются колоний-стимулирующими
факторами (CSF; от англ. colony-stimulating factors), или
интерлейкинами. Свое название они получили за
стимулирование роста колоний определенных лейкоцитов
в экспериментальных условиях. Интерлейкинами они
названы «в честь» лейкоцитов, влияющих посредством
секреции на другие лейкоциты. Большинство факторов
роста были открыты благодаря их митогенному
(ростовому) влиянию на клетки, однако действие факторов роста
гораздо сложнее. При определенных ситуациях один и тот
же фактор может стимулировать или ингибировать
клеточную дифференцировку, оказывая совершенно
различное воздействие на одну и ту же клетку. Вот почему
термин фактор регуляции развития представляется более
уместным для описания этих регуляторов, но мы будем
использовать общепринятый - факторы роста.
Термин цитокин используется иммунологами для
факторов роста, участвующих в иммунном ответе.
Несмотря на разнообразие биологических эффектов,
все факторы роста взаимодействуют с внешними
рецепторами клетки и опосредованно влияют на инициацию
транскрипции специфических генов.
Чем кроме характеристик, упомянутых выше,
фактор роста отличается от гормона? Как уже отмечалось,
гормоны выделяются в систему кровообращения и
разносятся по всему организму, достигая своих
клеток-мишеней. Большинство факторов роста в своем действии
паракринны: они диффундируют на коротком
расстоянии и действуют местно только на близлежащие клетки.
Хотя есть и такие, что действуют аутокринно,
стимулируя секретирующие их клетки (например, интерлей-
кин-2 стимулирует пролиферацию Т-клеток).
глава 26 Химическая сигнализация в организме
341
Таблица. 26.1
Основные гормоны и их функции
Секретирующий орган
Гипоталамус
Передняя доля гипофиза
Задняя доля
гипофиза
Щитовидная железа
Паращитовидная
железа
Кора надпочечников
Мозговое вещество
надпочечников
Гонады
Печень
Поджелудочная
железа
Гормон
Рилизинг-факторы
Соматостатин (также
и из поджелудочной железы)
Тиреотропный гормон
Адренокортико-
тропный гормон (АКТГ)
Гонадотропины
(лютеинизирующий
гормон, LH,
и фолликулостимули-
руюший гормон, FSH)
Соматотропин
Пролактин
Антидиуретический
гормон (ADH), или
вазопрессин
Окситоцин
Тироксин (Т4)
и трииодтиронин (Т3)
Паратиреоидный гормон
Кальиитонин (также
из щитовидной железы)
Глюкокортико-
стероиды (кортизол)
Минералокортико-
стероиды (альдостерон)
Катехоламины
(адреналин,
норадреналин)
Половые гормоны
(тестостерон из яичек,
эстрадиол и прогестерон
из яичников)
Соматомедины
(инсулиноподобные
факторы роста: IGFI, IGFII)
Инсулин
Глюкагон
Ткань-мишень
Передняя доля
гипофиза
Передняя доля
гипофиза
Щитовидная железа
Кора надпочечников
Яички и яичники
Печень
Молочная железа
Почечные канальцы
Гладкая мускулатура
Печень, мышцы
Костная ткань,
почки, кишечник
Костная ткань,
почки
Многие ткани
Почки, кровь
Печень, мышцы,
сердце
Органы
воспризводства,
вторичные половые
органы
Печень, костная
ткань
Печень, мышцы
Многие ткани
Функция
Стимуляция секреции
гормонов
Тормозит выделение
соматотропина
Стимулирует выделение
тироксина (Т4)
и трииодтиронина (Т3)
Стимулирует выделение
адренокортикостероидов
Стимулирует выделение
половых гормонов и развитие
клеток
Стимулирует синтез инсулино-
подобных факторов роста IGFI
и IGFII
Необходим для лактации
Способствует реабсорбции
воды
Стимулирует сокращение
матки
Стимулирует метаболические
процессы
Поддерживает уровень
Са2+ в крови, стимулирует
реабсорбцию и захват
Са2+, поступающего
с пищей
Ингибирует реабсорбцию Са2+
Способствует глюконеогенезу
Поддерживает водно-солевой
баланс
Мобилизуют жирные кислоты
и глюкозу в кровоток
Способствуют созреванию
организма и функционированию
половых органов
Стимулируют рост
Стимулирует синтез гликогена,
липогенез, синтез белка
Стимулирует распад гликогена,
липолиз
часть 4 Хранение и переработка информации
342
Нейромедиаторы
Многие из этих сигнальных молекул участвуют в
обеспечении координированной работы нейронов. Мы
ограничимся кратким рассмотрением примеров,
непосредственно относящихся к нашей теме. Нейроны
симпатической (непроизвольной) нервной системы,
которая, например, иннервирует клетки жировых депо,
по получении нервного импульса секретируют норадре-
налин. Мотонейроны, иннервирующие произвольную
поперечно-полосатую мышцу, вызывают ее сокращение,
выделяя ацетилхолин из нервных окончаний. Таким
образом, жировая клетка может регулироваться
адреналином, образовавшимся в надпочечниках и перенесенным
кровью (см. главу 12), или норадреналином. Различие
заключается в том, что последний путь доставки более
оперативен и сигнал передается точно клетке-мишени.
Последовательность реакций синтеза катехоламинов
приведена на рис. 26.2.
Как контролируется
высвобождение сигнальных
молекул?
Регуляция секреции гормонов
Эндокринная система находится под иерархическим
контролем. Гипоталамус, представляющий вершину в цепи
команд, сам является объектом классической
регуляции по типу обратной связи (рис. 26.3). Гипоталамус -
одна из древнейших и наиболее жизненно важных
частей мозга; в нем образуются гормоны,
контролирующие высвобождение других гормонов из передней доли
гипофиза, локализованного у основания мозга. По этой
причине гипоталамические гормоны еще называют ре-
лизинг-гормонами, или релизинг- факторами.
Гормоны передней доли гипофиза побуждают
эндокринные железы-мишени организма выделять свои
гормоны. Последние являются «конечными»
гормонами в цепи; они вызывают соответствующие ответы
в тканях-мишенях, не влияя на другие железы.
Поскольку гормоны передней доли гипофиза
стимулируют образование гормонов других желез, их
называют тройными гормонами, или тропинами. Как уже
отмечалось, высвобождение гормонов передней доли
гипофиза само по себе регулируется по принципу
обратной связи, когда конечные продукты (гормоны,
действующие непосредственно на клетки-мишени)
ингибируют выделение гипоталамических гормонов;
таким образом поддерживается уровень
циркулирующих в крови гормонов. Гипоталамус получает
информацию от остальных отделов мозга, осуществляя
церебральный контроль над эндокринной системой
(см. рис. 26.3).
Не все эндокринные железы находятся под
контролем гипоталамуса. Задняя доля гипофиза регулируется
CH2-CHNH2-COOH
Тмрозмм-
гмдрожсжлаза
CHrCHNH2-COOH
ДОФА-
декарбоксилаза НО
CH2-CH2NH2
+ С02
Дигидроксифенилаланин
(ДОФА)
CHOH-CH2NH-CH3
CHOH-CH2-NH2
. Адреналин Норадреналин
Рис. 26.2. Синтез и структуры катехоламинов
глава 26 Химическая сигнализация в организме
343
иным способом; высвобождение инсулина и глюкагона
из поджелудочной железы напрямую зависит от уровня
глюкозы в крови.
Поступление
информации в мозг
\
Гипоталамус
—*- Ингибирование
Гипоталамические гормоны]
(релизинг-факторы) ι
Задняя доля гипофиза —-
Передняя доля гипофиза ι
^ Окситоцин: вазопрессин
Регуляция по ^
принципу "Ν< Циркулирующие тропные
обратной связи ^ гормоны
^^ (и пролактин)
(Щитовидная железа,
кора надпочечников,
гонады, печень
Тироксин, циркулирующие
адренокортикостероиды,
половые стероиды:
соматомедины
(инсулиноподобные
факторы роста)
\
Ткани-мишени -*--
Регуляция высвобождения
нейромедиаторов
Нервный импульс, проходящий по аксону нервной
клетки, вызывает выделение посредством экзоцитоза
нейромедиаторов из везикул нервных окончаний. Механизм
этого высвобождения будет описан позже.
Удаление сигнальных молекул
Сущность контроля заключается в его обратимости.
Выделенные сигнальные молекулы должны быть
удалены, в противном случае исходный сигнал длился бы
неопределенно долго. Классическим примером удаления
сигнальных молекул служит разрушение ацетилхолина
ацетилхолинэстеразой, присутствующей в нервных
синапсах. Удаление сигнала необходимо для
подготовки синапса к восприятию следующего нервного
импульса. Для ацетилхолина это происходит в ходе следующей
реакции:
СН
\
СН,—N+-CH2—СН2—О—СО-СНя
Н20
СН,
Ацетилхолин
Физиологические ответы
Рис. 26.3. Гипоталамической регуляция выделения гормонов
Регуляция высвобождения
факторов роста
О ней известно относительно немного, и вполне
возможно, что выделение этих факторов контролируют
многочисленные механизмы. Повреждение эпителиального
слоя кровеносных сосудов приводит к разрыву
тромбоцитов и выделению PDGF, который стимулирует
деление клеток и репарацию; но PDGF также образуется
многими другими клетками организма. В иммунной системе
активация В- и Т-клеток вызывает выделение факторов
роста (цитокинов), стимулирующих их пролиферацию
(см. главу 25). Факторы роста могут стимулировать
клетки к выделению других факторов роста. Вкратце,
факторы роста участвуют в процессах развития и репарации
клеток и в защитных механизмах, например,
иммунного ответа, который включает экстенсивный контроль
клеточной пролиферации и дифференцировки.
Ацетилхолинэстераза
СН
\
сн3—n+-ch2—сн2он + сн3—соо- + н+
СН,
Холин
Ацетат
Катехоламины, выделяемые нервными окончаниями,
захватываются соседними клетками или удаляются
посредством метаболического разрушения.
Каким образом
сигналы распознаются
клетками-мишенями?
Как уже отмечалось, для каждого сигнала
клетки-мишени имеют рецепторы на внешней поверхности или
внутри клетки. С ними сигнальные молекулы связываются,
вызывая изменения в конформации рецепторного белка
(или белков), что приводит к определенному ответу.
Несколько гормонов (стероиды, тироксин, а также оксид
часть 4 Хранение и переработка информации
344
азота) растворимы в липидах и поступают прямо в
клетку, непосредственно через липидный бислой,
связываясь с внутриклеточными рецепторами (см. рис. 26.1).
Структуры тиреоидных гормонов и двух стероидных
гормонов представлены на рис. 26.4. Остальные
сигнальные молекулы водорастворимы и связываются
с рецепторами на поверхности клеточной мембраны
(см. рис. 26.1).
Сущность регуляции - специфичность: сигнальная
молекула (называемая лигандом, или агонистом)
исключительно точно взаимодействует со своим
рецептором - так же, как субстрат со своим ферментом. Вот
почему гормон, разносимый током крови по всему
организму, влияет только на свои клетки-мишени. Если
клетка не имеет специфического рецептора, она «не видит»
этого сигнала. Точно также телеприемник без
соответствующей антены не может принимать сигнал
спутникового телевидения.
CH2-CHNH2-COOH
CH2-CHNH2-COOH
Тирозин
CHrCHNHrCOOH
Тетраиодтиронин
(Т4, тироксин)
Трииодтиронин
(Т3)
Тестостерон
Прогестерон
Рис. 26.4. Структуры гормонов
а - Структуры тирозина и тиреоидных гормонов;
б - структуры двух стероидных гормонов
Как связывание лиганда
приводит к клеточному ответу?
Внутриклеточные
рецептор-опосредованные ответы
Стероидные гормоны регулируют экспрессию
специфических генов клеток-мишеней на уровне инициации
транскрипции. К их числу относятся глюкокортикоиды,
эстроген и прогестерон (см. табл. 26.1). Эти
растворимые в липидах гормоны легко проходят сквозь
липидный бислой клетки к своим рецепторам.
Рецепторы эстрогена и прогестерона локализованы
в ядре, а глюкокортикоидный рецептор - в цитоплазме.
Все они имеют «цинковые пальцы» (см. с. 279) в
качестве ДНК-связывающего домена и существуют в
комплексе с белками теплового шока (Hsp). Как мы помним,
белки теплового шока являются шаперонами (см. с. 298),
участвующими в сворачивании и поддержании
определенной конформации ряда белков.
Наиболее хорошо исследован «цинковый палец»
глюкокортикоидного рецептора, поэтому мы опишем его
в качестве примера. В цитоплазме рецептор связан с
комплексом белков теплового шока, которые
экранируют ядерную сигнальную метку (специфическую
пептидную последовательность).
Когда глюкокортикоидный гормон связывается со
специфическим участком на рецепторе, комплекс белков
теплового шока диссоциирует от него, обнажая
сигнальную метку, и рецептор транспортируется в ядро. Там он
соединяется в форме димера с глюкокортикоид-чувст-
вительным элементом ДНК (рис. 26.5). Последний
представлен палиндромом, каждая половина которого
соединена с двумя «цинковыми пальцами» (см. рис. 21.22)
молекулы рецепторного димера.
В случае рецепторов других стероидных гормонов
ядерная сигнальная метка не экранирована комплексом
белков теплового шока, так что вся структура
транспортируется в ядро. Но до тех пор, пока Hsp не
освободится в ходе присоединения гормона, связывания с ДНК
не происходит.
Мембранные
рецептор-опосредованные ответы
Многочисленные межклеточные связи в организме
осуществляются через мембранные рецепторы. На рис. 26.6.
приведены два типа таких рецепторов. В первом случае
связывание лиганда активирует внутренний домен
рецептора, во втором - связывание лиганда вызывает диме-
ризацию рецепторов, что сопровождается активацией
глава 26 Химическая сигнализация в организме
345
Рецептор
Комплекс Hsp
Л
К
\/ Глюкокортикоид
к
В ысвобождение
комплекса Hsp
Цитоплазма
ДНК-связывающий
участок
Ядро
J
Ядерная сигнальная
метка
Ядерная мембрана
Транскрипция
активированного
гена
Участок, чувствительный к глюкокортикоиду
Рис. 26.5. Активация гена стероидными гормонами
В качестве примера взят глюкокортикоидный рецептор из над-
семейства рецепторов стероидных гормонов и тироксина,
которые образуют комплексы с белками теплового шока (Hsp) и
связываются «цинковыми пальцами» с участками ДНК
внутренних доменов. Каким образом сигнал,
генерируемый на цитоплазматической стороне мембраны,
передается к участку контроля - сложный вопрос, поэтому
мы сначала рассмотрим мембранные рецептор-опос-
редуемые ответы, а затем познакомимся с отдельными
сигнальными путями, ведущими к клеточным ответам.
Резюме по мембранным
рецептор-опосредуемым ответам
Основной предмет обсуждения этого раздела - фосфо-
рилирование белка под действием использующей АТР
протеинкиназы. Трудно преувеличить значение фосфо-
рилирования белков в процессах клеточной регуляции:
у животных фосфорилирование участвует в реализации
гормонального контроля, регуляции факторами роста,
регуляции фаз клеточного цикла циклинами и в
контроле сокращения гладкой мышцы. На рис. 26.7. показаны
Внеклеточная - .
часть ч\/
Грансмембранный |
домен
Цитоплазма- у
тический
домен
Цитоплазма
6 Внеклеточная
часть
Рецепторный Сигнал
домен
А.
Биохимический ответ
Сигнал
>
1.
Димеризация
тирозинкиназных
рецепторов
Цитоплазма Биохимический ответ
Рис. 26.6. Трансмембранная передача сигнала рецепторами
а - Связывание лиганда с рецептором вызывает аллостеричес-
кое изменение цитозольного домена рецепторной белковой
молекулы;
б - связывание лиганда приводит к димеризации мембранных
рецепторов.
Этот механизм применим к рецепторам, обладающим тирози-
нкиназной активностью (см. с. 353). Связывание двух цито-
плазматических доменов приводит к биохимическому ответу
Дефосфорилированньи
белок
Внешний Внешний
Протеинфосфатаза!
Н20
Метаболический
контроль
АТР
Фосфорилированный
белок
Изменение ферментативной
активности.
Образование активного
фактора транскрипции
Генетический контроль
Рис. 26.7. Основной принцип регуляции для большинства
внешних сигналов
Фосфорилирование белка изменяет его свойства, что
приводит к определенному клеточному ответу. Обратите внимание,
что фосфорилирование может стать причиной активации или
дезактивации различных белков. Внешние сигналы в
конечном счете влияют на активность протеинкиназы и протеин-
фосфатазы
часть 4 Хранение и переработка информации
346
основные принципы регуляции посредством фос-
форилирования. Стоит еще раз обратиться к рис. 12.8,
иллюстрирующему центральную роль фосфорилирова-
ния в проведении внешних сигналов.
Из вышесказанного со всей очевидностью следует,
что в большинстве случаев рецептор-опосредуемая
сигнализация включает активацию протеинкиназ. Дефос-
форилирование фосфопротеинов катализируют проте-
инфосфатазы, и эта стадия также может быть объектом
для контроля. Рис. 26.8. суммирует взаимоотношения
регуляторных систем, опосредуемых мембранными
рецепторами. Схема показывает также обратимость
действия сигнала путем разрушения вторичного посредника.
Сигнальная _
молекула
-► Связывается с рецептором
Отмена Удаление
сигнала
Активирует (в некоторых
случаях ингибирует)
протеинкиназу(протеинкиназы)
Активирует или \
ингибирует Фосфорилирует
активность ~* белки
ферментов
вторичного
посредника
(если он
участвует в
этом процессе)
Метаболические
эффекты
Активирует
фактор(ы)
транскрипции
Отмена сигнала
(фосфопротеинфосфатазы)
Дефосфорилирует
белки
Контролирует специфическую
транскрипцию гена
Синтез специфических
белков
Клеточные эффекты
Рис. 26.8. Упрощенное изображение главных событий
в мембранной рецептор-опосредованной сигнализации
Обратите внимание, что одна стрелка может отражать
несколько этапов данного процесса
Модуляция рецепторного ответа
на сигнал
Повторное воздействие сигнала на клетку-мишень
часто приводит к более слабому ответу. В случае одного
класса рецепторов адреналина регуляция по принципу
обратной связи сопровождается фосфорилированием
рецепторного белка и снижению чувствительности (это
называется десенситизацией) рецептора к лиганду.
Во избежание путаницы отметим, что для других типов
рецепторов, которые будут описаны позднее, фосфорили-
рование является составной частью процесса активации.
Другой тип модуляции ответа заключается в контроле
числа функциональных рецепторов в мембране. В
случае рецептора инсулина комплекс инсулин-рецептор
захватывается внутрь клетки в ходе эндоцитоза. Процесс
возвращения рецептора назад к мембране
контролируется числом рецепторов в ней. Это может
способствовать снижению ответа клетки к данному сигналу и,
таким образом, ослаблять действие регуляторной системы;
последнее известно как понижающая регуляция, или
даунрегуляция (англ. downregulation).
Описание отдельных
сигнальных путей внутри клетки,
связывающих активацию
рецептора с клеточным ответом
сАМР-опосредованные пути
Поскольку в главе 12 (см. с. 166) мы уже познакомились
с регуляцией метаболизма путем изменения активности
протеинкиназы под действием сАМР, в этом разделе мы
обсудим, как изменяется уровень самого сАМР в клетке
и как сАМР регулирует экспрессию гена.
Многие гормоны - первичные посредники,
включая адренокортикотропный гормон (АСТН),
антидиуретический гормон (ADH), гонадотропины, тироид-
стимулирующий гормон (TSH), паратиреоидный
гормон, глюкагон, катехоламины (адреналиннорадреналин)
и соматостатин (см. табл. 26.1), используют сАМР в
качестве вторичного посредника. Этот далеко не полный
список приведен только для демонстрации крайне
разнообразного влияния сАМР на различные клетки.
Система работает потому, что ответ сАМР у данной клетки
соответствует сигналу, который клетка способна уловить
своими специфическими рецепторами. Таким образом,
в клетке А сигнал X увеличивает уровень сАМР,
который обуславливает клеточные ответы, соответствующие
сигналу X. У клетки В нет рецепторов для сигнала X, но
есть для сигнала Y; они также увеличивают уровень
сАМР, но в клетке В это вызывает ответы,
соответствующие сигналу Y.
глава 26 Химическая сигнализация в организме
ЪАП
Мы обсудим два случая, связанные с проявлением
этих эффектов: 1) как связывание сигнальной молекулы
с рецептором контролирует уровень сАМР в клетке;
2) как сАМР влияет на транскрипцию гена.
Контроль уровня сАМР в клетках
сАМР образуется в клетке из АТР под действием
фермента аденилатциклазы, локализованого на
внутренней стороне клеточной мембраны (рис. 26.9).
Давайте рассмотрим типичный пример активации
аденилатциклазы адреналином, в результате которой
в печени и скелетной мышце образуется сАМР
(см. с. 167). Рецептор (в данном случае Р2-адренорецеп-
тор) - белок, полипептидная цепь которого прошивает
мембрану 7 раз (рис. 26.10). Каждый из 7
трансмембранных сегментов содержит около 19 гидрофобных
аминокислот. Последние образуют α-спиральную
структуру, достаточную для перекрывания гидрофобного липид-
ного бислоя мембраны. Адреналин «попадает» в щель
между трансмембранными спиралями.
С внутренней (цитоплазматической) поверхностью
рецепторного белка связан регуляторный G-белок,
который имеет 3 субъединицы: α, β и γ. Поскольку это 3
различных полипептида, белок называется гетеротример-
ным G-белком, или просто тримерным G-белком.
На сс-субъединице расположен участок, который может
связывать либо GTP, либо GDP. Когда он занят GDP,
не происходит ничего (рис. 26.11, а).
Когда молекула адреналина связывается с
рецептором, цитоплазматический домен последнего
претерпевает конформационные изменения. Это, в свою
очередь, вызывает конформационные изменения
связанного с ним G-белка. Последнее приводит к тому, что
G-белок (названный в данной системе Gs; «s» означает
стимуляторный) обменивает GDP на GTP G-белок не
может производить этот обмен до тех пор, пока не будет
соединен с рецептором, связанным с гормоном.
Комплекс а-субъединица-GTF отсоединяется от G-белка,
мигрирует к молекуле аденилатциклазы и активирует ее,
что приводит к образованию АТР (рис. 26.11, б, в).
NH2
9
0"-Р-0-Р-0-Р-0-СНо^
О" Ο" Аг кС^
ОН ОН
Аденилатциклаза
Аденозинтрифосфат
(АТР)
РР(
X
1Н2
JL
о-сн2 _
Нз,
Η
0=F> о ОН
Циклический аденозин-3',5'-
монофосфат (сАМР)
NH2
9
N
0=Р-^0 ОН
I
О"
н2о
Место гидролиза
Фосфодиэстераза О —Р—О—СН?*^
^ ° н^_^н
Циклический аденозин-
3',5'-монофосфат (сАМР)
ОН ОН
Аденозинмонофосфат
(AMP)
Рис. 26.9. Образование и гидролиз сАМР
а - Образование З1, 5'-циклического AMP с участием аденилатциклазы;
б - гидролиз 3\ 5-циклического AMP фосфодиэстеразой
часть 4 Хранение и переработка информации
348
Внешняя сторона
клеточной мембраны
Олигосахаридные
боковые цепи
Рецептор
А<ЛГ)
Внутренняя сторона
клеточной мембраны
С00"
Петля участвует в
активации G-белков
Рис. 26.10. Расположение Р2-адренорецептора в клеточной
мембране
Таким образом, гормон стимулирует синтез сАМР,
используя в качестве посредника G-белок (см. рис. 26.11, β).
Активация образования сАМР cc-GTP-субъединицей
должна быть ограничена во времени: в противном
случае одиночный гормональный стимул будет
действовать неопределенно долго. Если рецептор больше
не связывает гормон, образование сАМР должно
прекратиться; гидролиз с AMP в клетке фосфодиэстеразой
завершает процесс (см. рис. 26.9, б).
Ситуация подобна той, что происходит на лестнице
с ограниченным временем освещения: когда вы
нажимаете кнопку - свет зажигается, а потом кнопка
начинает медленно возвращаться в исходное положение - и
свет выключается через минуту или две. Время от
времени вы должны нажимать кнопку, чтобы свет не гас.
G-белок как раз и является «хронометражным
прибором», который ограничивает время активации аденилат-
циклазы.
сс-Субъединица G-белка в связанной с GTP форме
(которая активирует аденилатциклазу) обладает
ферментативной СТРазной активностью. Она гидролизует GTP
до GDP и Pf (рис. 26.11, г). СТРазная активность низка,
так что гидролиз GTP происходит не сразу. Как только
в ходе гидролиза образуется GDP, сс-субъединица
возвращается в исходное состояние. Она отсоединяется
от аденилатциклазы, которая становится неактивной,
и в связанном с GDP состоянии присоединяется к β-
Тримерный
G белок
ъг
Аденилатциклаза
(неактивная)
Η α -GDP
Ύ
Адреналин *
У α τρ
Гормона *·
больше нет
Аденилатциклаза
(неактивная)
Гормон
обмен GTP/GDP присутствует
GDP высвободился
Аденилатциклаза
(активная)
α
ATP
сАМР
Аденилатциклаза
(активная)
ЧБ
GTP
+ Н20
α
ATP
сАМР
GDP + Ρ/
Аденилатциклаза
(неактивная)
GDP-
Рис. 26.11. Регуляция активности аденилатциклазы
адреналином
и γ-субъединицам, образуя комплекс G-6e/zoAc-GDP, кон-
тактирущий с рецептором (рис. 26.11, д). Если
последний все еще связывает молекулу гормона, весь цикл
может начаться сначала (см. рис. 26.11, б). Место GDP
занимает GTP, сс-субъединица диссоциирует для того,
чтобы активировать молекулу аденилатциклазы. Если
рецептор больше не связывает молекулу гормона
(см. рис. 26.11, а), процесс останавливается. Таким
образом, чтобы продолжить синтез сАМР, сс-субъединица
глава 26 Химическая сигнализация в организме
349
перемещается взад и вперед от рецептора к аденилат-
циклазе. Продолжительность ее пребывания на аде-
нилатциклазе определяется временем, необходимым для
гидролиза присоединенной молекулы GTP.
Система обладает эффектом усиления: одна
связанная с рецептором молекула гормона может
активировать одну за другой молекулы G-белка, а
следовательно, одна молекула гормона, связываясь с рецептором,
способна привести к активации нескольких молекул
аденилатциклазы и образованию большого числа
молекул сАМР. Важность гидролиза GTP в процессе синтеза
АТР можно проиллюстрировать на примере холеры.
Клетки слизистой кишечника выделяют в просвет
кишечника Na+, и сАМР стимулирует этот процесс.
Холерный токсин необратимо ингибирует вТРазную
активность ос-субъединицы G-белка. Таким образом,
гормональный сигнал, активирующий аденилатциклазу,
не может быть выключен; она оказывается
«замороженной» в состоянии cc-GTP. Продолжительное образование
сАМР приводит к массированной потере ионов Na+,
за которыми следуют молекулы воды, вызывая
изнуряющий понос и возможную смерть от потери жидкости
и электролитов.
В случае, приведенном на рис.26.11, GTP-cc-субъеди-
ница стимулирует активность аденилатциклазы. Адре-
норецепторы другого типа (сс2-адренорецепторы)
действуют аналогичным образом, однако здесь
используется другая субъединица oc-GTP (Gf), которая ингибирует
активность аденилатциклазы. а- и β-Адренорецепторы
опосредуют различные эффекты катехоламинов. Как мы
уже знаем (см. с. 167), при необходимости внезапного
действия адреналин стимулирует печень и скелетные
мышцы, что реализуется через Р2-адренорецепторы,
активирующие аденилатциклазу. В отличие от них
а2-адренорецепторы ингибируют аденилатциклазу.
Еще один подтип - α,-адренорецепторы - также
имеют свои отличия: они не используют с AMP в качестве
вторичного посредника, а вызывают высвобождение
Са2+ посредством фосфоинозитидного пути (см. с. 352).
Таким образом, один гормон может оказывать
совершенно противоположное влияние, в зависимости от
типа рецептора.
Как сАМР действует
на транскрипцию генов?
В главе 12 мы описали, как сАМР активирует протеинки-
назу А (ПКА). Помимо участия в контроле активности
ферментов, ПКА является частью важного механизма
регуляции экспрессии гена. Промоторы нескольких
сАМР-индуцибельных генов содержат сАМР-чувстви-
тельные элементы (CREs). Когда ПКА фосфорилирует
CRE-связывающий белок (CREB; от англ. cAMP-respon-
sive element binding protein), он становится активным
фактором транскрипции типа «лейциновой молнии»
(рис. 26.12). Существует семейство CREB-белков,
выполняющих, по-видимому, различные функции в клетке.
Итак, мы сказали, что сАМР является вторичным
посредником (см. с. 166) в передаче внешнего сигнала
функциональным элементам как в цитоплазме, так и в ядре
клетки. Другой циклический нуклеотид - cGMP -
выполняет ту же функцию в иных регуляторных системах,
которые мы и рассмотрим.
Адреналин Аденилатциклаза
связывается р2-адрено- (активная)
.рецептор
АТР
Активирует
G-белок
САМР
Активирует
аденилатциклазу
Цитоплазма
Активирует
протеинкиназу А (ПКА)
Фосфорилирует
CREB
Рис. 26.12. Схематичное изображение, иллюстрирующее
функцию Р2-адренорецептора
Рецептор-опосредованная стимуляция аденилатциклазы и
увеличение уровня сАМР вызывают активацию сАМР-зависимой
протеинкиназы (ПКА). Фосфорилирование CREB формирует
активный центр факторов транскрипции для специфических
генов
Передача сигнала
с использованием циклического GMP
в качестве вторичного посредника
Ряд сигналов вызывает увеличение уровня cGMP
внутри клетки, что в свою очередь активирует
протеинкиназу (IlKG). Образование cGMP из GTP катализируют
часть 4 Хранение и переработка информации
350
о
л
о -р-о-р-о-р-о-сн2^
о- о- о- н^н
ОН ОН
GTP
Гуанилатциклаза
РР,
H2N
0-СН2
У
НЗД
о=р—о он
0"
cGMP
Рис. 26.13. Образование З'^'-циклического GMP, катализируемое гуанилатциклазой
мембраносвязанная и растворимая формы фермента
гуанилатциклазы (рис. 26.13). В первом случае
гуанилатциклаза является внутриклеточным доменом
мембранного рецепторного белка. Связывание гормона
с внешним рецептором приводит к активации
внутриклеточного домена и увеличению синтеза cGMP. Этот
путь отличается от механизма, при помощи которого ад-
ренорецептор запускает образование сАМР: в данном
случае внутренний домен рецептора сам является
гуанилатциклазой и активируется непосредственно в ходе
присоединения гормона (рис. 26.14). Образовавшийся
cGMP опосредует клеточный ответ, активируя особую
протеинкиназу (FIKG). Примером этого типа регуляции
может служить натрийуретический фактор предсердий,
вырабатываемый эндотелиальными клетками: он
стимулирует секрецию Na+ почками. Эффекты cGMP более
специализированы по сравнению с сАМР. Они
включают расслабление гладкой мускулатуры и воздействуют
на нервные клетки и зрение (см. ниже).
В цитозоле есть и другая, растворимая форма
гуанилатциклазы. В качестве простетической группы она
содержит молекулу гема, с которой связывается,
пожалуй, самая простая межклеточная сигнальная
молекула - N0, или оксид азота. Молекула гема
представляет чрезвычайно чувствительный детектор N0; она
передает сигнал активации ферменту. N0 образуется
в эндотелиальных клетках сосудистой системы из гуа-
нидиновой группы аргинина под действием фермента
нитроксидсинтазы. N0 диффундирует в мускулатуру
кровеносных сосудов, вызывая образование cGMP,
который, в свою очередь, способствует расслаблению
мышц и расслаблению сосудов. Ацетилхолин
стимулирует образование оксида азота, из чего следует, что
оно может опосредоваться нервной регуляцией. Оксид
азота также синтезируется в ответ на механическое
воздействие, оказываемое кровотоком на эндотелиальные
клетки выстилки кровеносных сосудов, приводя к
расширению сосудов (вазодилатации).
л
^—^ Активирует
^^образование -
сАМР
"~~— Ингибирует— -"
образование
сАМР
riiv
ATP
сАМР
Оксид азота (NO)
Плазматическая мемС ан;
Цитоплазма
Активирует
протеинкиназу
Соответствующие клеточные ответы
Рис. 26.14. Схематичное изображение, иллюстрирующее основные принципы действия циклических нуклеотидов
в качестве вторичных посредников в клеточной сигнализации
R, - рецептор а7-адренергического типа; R9 - рецептор β,-адренергического типа; R3 - участок связывания натрийуретического
пептида предсердий, один из доменов которого встроен в мембрану, а каталитический домен ориентирован в сторону. АЦ -
аденилатциклаза плазматической мембраны, ГЦ, - гуанилатциклаза плазматической мембраны, ГЦ7 - цитоплазматическая
гуанилатциклаза, активируемая оксидом азота (NO)
глава 26 Химическая сигнализация в организме
351
Тринитроглицерин - лекарство, давно
используемое для лечения стенокардии, из него медленно
образуется оксид азота, вызывая тем самым расслабление
сосудов и уменьшение нагрузки на сердце. Поскольку
за считанные секунды NO окисляется до N02 и N03, он
является сигнальной молекулой локального действия;
будучи быстро растворимым в липидах, оксид азота
легко выходит из продуцирующих его клеток и
переходит в соседние. Оксид азота является частью
сложной регуляторной системы с множественными
физиологическими эффектами. Рис. 26.14 суммирует
основные принципы действия циклических нуклеотидов
в качестве вторичных посредников в межклеточной
сигнализации.
Гормоны, передающие сигнал при
помощи различных вторичных мес-
сенджеров; фосфоинозитидный
каскад
До сих пор мы знакомились с разнообразной группой
гормонов, использующих в качестве вторичных
посредников циклические нуклеотиды - сАМР и cGMP.
Другая группа гормонов и факторов роста использует для
передачи сигналов иные внутриклеточные механизмы.
Потенциальные гормоны связываются со
специфическими рецепторами клетки, которые также
взаимодействуют с G-белками (т. е. с GTP-связывающими
белками ), но в передаче сигнала циклические нуклеотиды
не участвуют. Примерами гормонов, использующих
такой механизм, могут служить тиротропин-релизинг
гормон, гонадотропин-релизинг гормон и фактор роста
PDGF.
Фосфолипид фосфатидилинозит-4,5-дифосфа!
(Р1Р2) образуется в мембране при фосфорилировании
фосфатидилинозита, структура которого приведена в
главе 10. Еще раз отметим, что G-белок связывает рецептор-
ный стимул с сигнальным путем внутри клетки и
действует в качестве «молекулярного хрономегражного
прибора». Взаимодействие гормона с рецептором вызывает
у связанного с рецептором G-белка обмен GDP на GTP.
Комплекс G -GTP мигрирует и активирует мембрано-
связанный фермент фосфолипазу С, которая расщепляет
Р1Р2 на инозиттрифосфат (1Р3) и диацилглицерин
(ДАГ) (рис. 26.15). G-белок гидролизует GTP до GDP и Р/5
приводя к самоинактивации фермента.
1Р3 вызывает высвобождение Са2+ из просвета эн-
доплазматического ретикулума (ЭР), содержащего
высокие концентрации этого иона. 1Р3 открывает в
мембране лиганд-зависимые Са2+-каналы (см. ниже),
обеспечивая поступление иона в цитоплазму. Под действием
Н20
Фосфолипаза С
ОН
Диацилглицерин (ДАГ)
+
0"
Фосфатидилинозит-4,5-дифосфат
Рис. 26.15. Гидролиз фосфатидилинозит-4,5-дифосфата
(Р1Р2) до диацилглицерина (ДАГ) и инозиттрифосфата 1Р3
Са2+-АТРазы ионы Са2+ постоянно возвращаются в
просвет ЭР, а 1Р3 и ДАГ удаляются ферментативным путем.
Таким образом, соединение гормона или любого
другого агониста с рецептором, задействованным в фос-
фоинозитидном каскаде, приводит к увеличению
внутриклеточного ДАГ и Са2+ (рис. 26.16), а обращение
процесса происходит после диссоциации комплекса
гормон-рецептор
ДАГ является физиологическим активатором особой
протеинкиназы, отличающейся от ПКА и ПКС Для ее
максимальной активации также необходим Са2+.
Активируемая ДАГ протеинкиназа носит название ПКС;
она связана с молекулами фосфатидилсерина,
расположенными на цитозольной стороне клеточной
мембраны. ПКС участвует в регуляции многочисленных
клеточных процессов, фосфорилируя различные
белки-мишени и некоторые факторы роста. Важную роль ПКС
в контроле за делением клеток иллюстрирует онкоген-
ный эффект форболовых эфиров, являющихся так
часть 4 Хранение и переработка информации
352
.. Активный
V рецептор
GTP
Gp
GDP
1Рз
Открываемый
Са2+-канал\
Са2+
Эндоплазма-
тический
ретикулум
χ
GDP
Са*
Иротеинкиназа
(активная)
Ч-Длаз1 тическая
*·· мембрана
Р1Р2 '
\D4G--'r
.ip3
Белок
Протеинкиназа С ф'осфорилиро-
ванный белок
Кальмодулин I
Клеточные ответы
GTP
/ Са2+ Са2+-кальмодулин
Са2+-АТРаза .
Активируе-j
Активирует
ADP>
Фосфобелок *
Клеточные ответы
·» Активирует
Другие ферменты (например,
фосфодиэстераза сАМР,
02+ 4g2+-* -ϊ
АТР
Белок (например,
легкие епи миозина
Рис. 26.16. Фосфатидилинозитидный каскад: взаимосвязь
ДАГ, 1Р3 и Са2+ в качестве вторичных посредников
G -белок связывается с фосфолипазой С только в комплексе
с GTP; обращение процесса происходит при гидролизе GTP
floGDP
называемыми опухолевыми промоторами. Форболовые
эфиры - аналоги ДАГ, активирующие ПКС. Ниже
приведены структуры ДАГ и форболового эфира
(запоминать структуру последнего не обязательно).
О
II
СН2—О—С—R
I О
I и
СН—О—С—R
СН2ОН
Диацилглицерин (ДАГ) О
(R = цепь жирной кислоты)
СН2ОН
Форболовый эфир
Может показаться абсурдным, что ДАГ оказывает
такой же активирующий эффект на ПКС, что и агент,
являющийся промотором образования опухоли. Различие
их действия, по-видимому, заключается в том, что ДАГ
быстро разрушается и активирует ПКС только тогда,
когда это требуется, в то время как форболовые эфиры
«живут» дольше, пролонгируя столь нежелательный
организму сигнал.
На рис. 26.16 показано, что стимуляция описанного
в этом разделе типа рецептора приводит к образованию
двух посредников - ДАГ и Са2+, причем оба необходимы
для максимальной активации ПКС. Однако Са2+ и сам
по себе является важным вторичным посредником.
Каковы другие
мессенджерные эффекты кальция?
В реализации эффектов Са2+ как вторичного
посредника участвует кальмодулин. Этот широко
распространенный белок имеет 4 высокоаффинных участка для
ионов Са2+; их связывание вызывает изменение конфор-
мации кальмодулина. Активированный таким образом
кальмодулин изменяет активность ферментов (и других
белков), с которыми он взаимодействует. Существует ряд
Са2+-кальмодулин-активируемых протеинкиназ; из
этого следует, что реализация многих эффектов Са2+ в
клетке также включает фосфорилирование белка.
Список белков-мишеней этих киназ включает гли-
когенфосфорилазу и гликогенсинтетазу (с. 88-89).
Поскольку один тип адренорецепторов (сц) активирует
фосфоинозитидный путь, вызывая увеличение Са2+
в цитоплазме клетки, адреналин может влиять на
метаболизм гликогена как посредством этого механизма, так
и через сАМР. Другими белками-мишенями Са2+-каль-
модулин-зависимых киназ являются киназы легких
цепей миозина (см. с. 386).
Рецепторы,
ассоциированные с тирозинкиназой
Хотя в этом разделе мы все еще знакомимся с
мембранными рецепторами, теперь нам предстоит рассмотреть
очень важную область, совершенно отличную от всего
вышесказаного.
Фосфорилирование - основной этап регуляции,
однако мишенью фосфорилирования является аминокислота
тирозин самого рецепторного белка, т. е. происходит его
самофосфорилирование (аутофосфорилирование).
Все описанные до сих пор протеинкиназы фосфори-
лируют гидроксильные группы остатков серина или
треонина своих белков-мишеней. Теперь мы обсуждаем
глава 26 Химическая сигнализация в организме
353
активацию рецептора, которая обусловлена фосфори-
лированием гидроксильной группы специфических ти-
розиновых остатков белков в ответ на связывание
сигнальной молекулы (рис. 26.17, а). Само по себе это
может показаться вполне тривиальным, но связанные
с тирозинкиназой рецепторы представляют особый
тип рецепторов, активирующих специфические
сигнальные пути. Их важность подчеркивает то, что у
большинства факторов роста и некоторых гормонов,
например инсулина, есть рецепторы подобного типа.
Для иллюстрации такой сигнальной системы мы
рассмотрим фактор роста эпидермиса (EGF; от англ.
epidermal growth factor).
Связывание лиганда вызывает димеризацию
рецепторов EGF в мембране клетки (рис. 26.17, б). Это
приводит к множественному фосфорилированию остатков
тирозина в цитоплазматических доменах рецепторов.
Цитоплазматические домены являются тирозинкина-
зами, которые, возможно, фосфорилируют друг друга
в ходе димеризации. Фосфорилирование приводит
к активации сигнального пути от мембранного
рецептора к ядру клетки и завершается активацией фактора
транскрипции.
Ras-путь - распространенный
сигнальный путь от мембраны
к гену эукариот
I
NH
I
сн-сн2
I
с=о
I
I
ATP ADPNH
Уон^-
сн-сн
I
Остаток
тирозина
в белке
Тирозин- С=0
киназа |
ΟΙ
V о-Р=0
/ I
0"
Остаток
фосфотирозина
Внешняя сторона
Сигнал (EGF)
^мС ана
1.
Димеризация
рецепторов EGF
Цитоплазма
Тирозинкиназные
домены
Я зри
\к1ивац1 ··
сШОфИИЦ ι
Димеризация рецепторных
молекул и фосфорилирование
\ тирозиновых групп рецептора
Внутриклеточный
сигнальный путь
Рис. 26.17. Активация различных ферментативных и
генетических процессов при взаимодействии рецептора EGF
со специфическими лиганлами
а - Белковый уровень: фосфорилирование тирозина
тирозинкиназой;
б- клеточный уровень: суммарное влияние EGF на клетку-
мишень
Белок, известный под названием Ras-белок,
присутствует во всех клетках эукариот. Это небольшой
мономерный GTP-связывающий белок, обладающий СТРазной
активностью. Оказалось, что он является частью
главного сигнального пути от тирозинкиназы,
ассоциированной с рецепторами факторов роста, к факторам
транскрипции генов. Особенность этого пути заключается
в том, что в нем нет низкомолекулярных вторичных
посредников: все его компоненты принадлежат к белкам.
Ras-белок и другие составляющие этого
внутриклеточного сигнального пути важны для регуляции
клетки; мутации, обуславливающие появление аномальных
форм этих белков - онкогенны, они приводят к
неконтролируемому делению клеток.
Рис. 26.18 иллюстрирует эту систему. В цитоплазме
есть белок, связывающий рецептор фактора роста
(GRB, от англ. growth factor receptor-binding protein),
который связан с так называемым SOS-белком. При
помощи 8Н2-домена GRB взаимодействует с фосфори-
лированным рецептором. Этот домен найден также у
семейства цитоплазматических белков, которые, как
полагают, играют роль в связывании фосфорилированного
рецептора. Название SH2 происходит от наличия
домена, гомологичного домену 2 Src-белка, кодируемого
онкогеном вируса саркомы Рауса (см. с. 314). Отсюда
для краткости - SH2.
Соединение GRB-SOS с активированным
рецептором вызывает в Ras-белке обмен GDP на GTP
(рис. 26.19). Это в точности напоминает активацию
G-белка при образовании сАМР (см. рис. 26.11).
Ras-белок обладает низкой СТРазной активностью,
выполняющей функцию молекулярного «хронометражного
прибора».
Следующим компонентом этого пути является
Raf-белок - протеинкиназа серин-треонинового типа,
для активации которой необходим Ras-GTP. Это начало
часть 4 Хранение и переработка информации
354
Неактивный рецептор EGF
Цитоплазма
Ras
Ι
GDP Ras-белок
неактивен, когда
с ним связян GDP
Сигнальный путь
неактивен
EGFI '
Цитоплазма
GRB
SOS
GRB связывается с
фосфорилированным
рецептором через SH2 домен
Связывание EGF
с рецепторами вызывает
димеризацию последних
и фосфорилирование
остатков тирозина
их цитоплазматических
доменов
G В результате
связывания
tef EGF с рецепторами
Ras-белок
обменял GDP на GTP
Комплекс Ras-GTP
активирует Raf-белок,
который в свою
очередь активирует
протеинкиназу
(Митоген-активируемые
протеинкиназы,
или MAP)
Каскад протеинкиназ
Активация факторов транскрипции
(например, Jun, Fos), ведущих к активации
гена и клеточной пролиферации
Рис. 26.18. Активация Ras-пути EGF (упрощенная схема)
Светлым показано неактивное состояние, темным - активное
состояние.
а - Неактивное состояние без EGF;
б - активированное с EGF; GRB - белок связывающий
рецептор фактора роста;
SOS (son ofsevenless) - белок, участвующий в этом
сигнальном пути, получил свое имя «в честь» соответствующей
мутации (sevenless mutation) у дрозофилы.
Raf, Ras, Jun, и Fos получили названия от своих онкогенов.
Обратите внимание, что рецептор фосфорилируется по
остатку тирозина, но последующие киназы каскада относятся к се-
рин/треониновому типу. Механизм активации Raf-киназы
Ras-GTP до сих пор детально не выяснен
серин/треонинового протеинкиназного каскада,
заканчивающегося фосфорилированием белков-факторов
транскрипции; активация последних способствует
их присоединению к соответствующим чувствительным
элементам и обеспечивает транскрипцию
специфических генов
GTP
Для реакции
необходим
рецептор-
активируемый
белковый
комплекс
GRB-SOS
GDP
GTP
Рис. 26.19. Активация Ras-белка
Ras-путь и онкогенез
В главе 23 мы объяснили, что вызывающие рак гены,
переносимые ретровирусами, являются мутированными
формами протоонкогенов, присутствующих в
нормальных клетках. Ras- и Raf-белки - это продукты
протоонкогенов; когда соответствующие онкогены вносятся
ретровирусами в клетки, они вызывают рак. Похоже, что
онкогенные копии Ras- и Raf-белков (как и другие
компоненты Ras-пути, показанные на рис. 26.18)
постоянно находятся в «активированном» состоянии. Внутрикле-
точный сигнальный путь ведет себя так, словно
сигнальная молекула находится на рецепторе постоянно.
Таким онкогеном является \-erbB, который кодирует
аномальный рецептор EGF. У него отсутствует внешний
домен, но цитоплазматический домен находится
постоянно в фосфорилированном состоянии (рис. 26.20).
Таким образом, в присутствии любого из этих онкогенных
белков Ras-путь работает в неконтролируемом режиме.
Онкогены могут появляться не только в ходе ретрови-
русной инфекции: нормальные протоонкогены иногда
трансформируются в онкогены в результате мутации.
Превращение протоонкогенов в онкогены может
происходить не только в результате мутации одного
основания. Хромосомные перестройки способствуют
транслокации генов, участвующих в контроле
дифференциации и деления клеток. Это ведет к образованию
онкогенов по нескольким путям.
L Протоонкоген способен оказаться под контролем
другого регуляторного элемента, вызывающего
избыточную экспрессию (англ. overexpressiori)
определенного белка. Например, лимфома Бэркитта
возникает тогда, когда ген с-тус становится под контроль
промотора гена иммуноглобулина.
глава 26 Химическая сигнализация в организме
355
Внешняя сторона цИТОПлазма
клетки
а
Нет EGF, сигнальный путь неактивен
б
Ρ
EGF присоединен;
сигнальный путь активен
Ρ *- ■* Ядро
О
β
ρ
Нет EGF, аномальный
ρ рецептор активен
- - *- Ядро
..." Усеченная форма рецептора
*"*"·*· EGF с постоянно активной
тирозинкиназой
(кодируется онкогеном \-erb)
г
Нет EGF, аномальный компонент
сигнального пути активен
—τ—*" *~ Ядро
— Онкогенный компонент
Рис. 26.20. Продукты онкогенов как компоненты
сигнальных путей от мембраны к ядру: в качестве примера
использован рецептор EGF
а - Нормальный рецептор - неактивен;
б - нормальный рецептор - активен;
в - постоянно активный усеченный рецептор;
г- нормальный рецептор или компонент сигнального пути
постоянно активен
2. Транслокация может вызывать слияние генов и
приводить к образованию онкогенных гибридных
белков.
3. Амплификация способна вызвать избыточную
продукцию регуляторного белка.
Рак является результатом приобретения клеткой
множественных генетических аномалий. В дополнение
к превращению протоонкогенов в онкогены, развитие
рака часто включает потерю функционального гена-су-
прессора опухоли. Около половины всех случаев
возникновения злокачественных опухолей у человека
сопряжено с потерей гена-супрессора опухоли р53. Продукт
генар53 оказывает комплексные эффекты, включающие
часть 4 Хранение и переработка информации
356
стимуляцию механизмов репарации ДНК; однако
наиболее существенным является его способность
контролировать переход от фазы Gj к фазе S клеточного
цикла, участвуя в предотвращении неконтролируемого
деления клетки.
Замечание по терминологии
Как отмечалось ранее, онкогены называют по названию
переносящих их ретровирусов - и записывают
курсивом. Так, онкоген ras был найден у вируса саркомы крыс
(англ. Rat Sarcoma virus). Онкоген идентифицируется
с буквой «ν» (для вТфуса), а протоонкоген - с буквой «с»
(цитоплазматический, англ. cytoplasmic). Таким образом,
эти гены обозначают соответственно как v-ras и c-ras
и т.д. Белки-продукты записываются как Ras, Raf и т.д.
(курсивом не выделяются).
Белок SOS назван по мутации в гене рецепторов
у плодовых мушек (sevenless): белок-продукт этого гена
называется son of sevenless, отсюда и образовалось
название SOS. В дальнейшем оказалось, что этот белок
повсеместно присутствует у эукариот. (Не путать этот
белок с SOS-ответом Е. сой!)
Почему Ras-путь такой длинный?
Заманчиво предположить, что протеинкиназный каскад
является механизмом амплификации, аналогичным
механизмам распада гликогена и свертывания крови.
Вполне возможно, что Ras-путь взаимосвязан в
нескольких точках с другими регуляторными путями.
Накапливается все больше данных в пользу
существования обширной интерференции (англ. crosstalk),
регуляторных механизмов. Так, Ras-белок может фос-
форилироваться и активироваться ПКС, после того как
последняя будет активирована в фосфоинозитидном
пути. Разные протеинкиназы Ras-пути могут иметь
другие белки-мишени, активируя таким образом
альтернативные пути и регуляторные каскады. Кроме того,
активированный тирозинкиназный тип рецептора
может соединяться с 8Н2-белками других сигнальных
путей, так что один тип рецептора способен
активировать несколько путей (рис. 26.21). Используя аналогию
с электричеством, Ras- и другие сигнальные пути
являются электронными контурами на очень сложной
приборной панели с пересекающимися связями,
однако пока мы не можем видеть, как этот «прибор»
функционирует в качестве интегрированной регуляторной
системы. Характерной чертой сигнальных путей
является наличие белок-белковых взаимодействий,
которые, по-видимому, образуют сети такой же степени
сложности, как метаболические пути.
Активированный
димеризованный
тирозинкиназный
тип рецептора
ча
мора
с рецептором, фосфорилируют два 8Н2-белка, которые
после этого двигаются в ядро, где собираются в
активный фактор транскрипции и способствуют
транскрипции ISRE-элементов (рис. 26.22).
Цитоплазма
Ρ
Цитоплазматическая
мембрана
Различные
сигнальные
пути
♦ t ♦ t
Множественные
клеточные эффекты
\ Белки с SH2-доменами
связанные с различными
аутофосфорилированными
участками
надимерных рецепторах
Рис. 26.21. Гипотетическое изображение, показывающее,
каким образом множественные белки с БШ-доменами
могли бы играть роль связующих звеньев в интегрированной
регуляторной системе
С Ras-путем резко контрастирует другой
внутриклеточный рецептор, опосредующий относительно простой
сигнальный путь, в котором не участвует тирозинкина-
за. Рассмотрим его более подробно.
Рецепторы
интерферона ***..
Интерферон
•*.ч JAK-киназы
/ (неактивны)
>^тт"
N
Димреизованный
рецептор ""*""
.-■·· STAT-белки
""> Киназы активны
Происходит фосфорилирование
остатков тирозина рецептора
Прямой внутриклеточный
сигнальный путь от рецептора к ядру
Сигнализация интерферонами
В 1957 году было показано, что клетки позвоночных,
инфицированные вирусом, секретируют белки,
названные интерферонами, которые защищают от тех же или
других вирусов (это явление было названо
интерференцией). Антивирусная активность интерферонов до
конца не понята, но известно, что на генах есть
интерферон-стимулируемые чувствительные элементы (ISRE;
от англ. interferon-stimulated response elements), так
что присоединение интерферона к рецептору клетки
вызывает специфические ответы генов клетки.
Рецептор γ-интерферона отличается от описанного для
EGF. С ним связаны две молекулы цитоплазматической
тирозинкиназы (JAK-киназы). Связывание лиганда
вызывает димеризацию рецепторов; это приводит к
активации тирозинкиназы, в результате чего остатки
тирозина рецепторного димера фосфорилируются. Это,
в свою очередь, вызывает связывание двух цитозольных
белков с фосфорилированным рецептором. Белки
имеют те же 8Н2-домены, что и у GRB-белка Ras-пути; как
уже отмечалось, SI-12-домен найден у белков,
взаимодействующих с фосфорилированными рецепторами ти-
розинового типа. Протеинкиназы, все еще связанные
ДНК
STAT-белки связываются
с фосфорилированным рецептором
по их SI-12-домену
и фосфорилируются
Фосфорилированные STAT-белки
входят в ядро, чтобы образовать
активный фактор транскрипции
Активный
фактор
транскрипции
прикрепляется
к интерферон-
активируемому
элементу
Рис. 26.22. Сигнальный путь, при помощи которого γ-ин-
терферон активирует специфическую транскрипцию гена
STAT - передатчик сигнала и активатор транскрипции; GAS -
активируемый γ-интерфероном элемент последовательности.
Обратите внимание, что в отличие от рецептора EGF, который
сам является тирозинкиназой, в этом пути димерный
рецептор активирует цитоплазматические тирозинкиназы, или Янус-
киназы (JAK, Janus kinases), названные в честь двуликого Януса
(они имеют два киназных центра). У α-интерферона и
нескольких цитокинов есть аналогичные сигнальные пути
глава 26 Химическая сигнализация в организме
357
Насколько общим
является этот прямой путь?
Основной вопрос в том, насколько общей является эта
система, или же она специфична в отношении лишь
нескольких сигнальных молекул. Широкое
распространение системы мог бы обеспечить механизм, где
неограниченное число сигналов оказывало бы специфическое
влияние на клетки. Известно, что второй интерферон
и несколько цитокинов, участвующих в регуляции
образования красных кровяных телец, используют именно
этот путь. Можно предположить, что различные фос-
форилированные рецепторы способны связывать разные
цитозольные SI-12-белки, которые затем превращаются
в различные специфические факторы транскрипции
и обеспечивают передачу большого числа сигналов
к специфическим клеточным «органам управления».
В заключение можно привести цитату из обзора
в журнале «Nature», где говорится: «В течение
следующих нескольких лет появятся сотни и даже тысячи
статей, описывающих, как различные белки посылают свои
сигналы по внутриклеточным сигнальным путям».
Внешние сигналы,
регулирующие лиганд-зависимые
ионные каналы
Мы подошли к совершенно особому типу управления,
в котором немедленным результатом связывания лиганда
с мембранным рецептором является открытие каналов
в мембране (рис. 26.23). Для иллюстрации будет
использована нейрональная сигнализация.
Внешняя сторона
клетки Лиганд (сигнал)
<^
^γ· Рецептор
γ
Мембрана )
w ~ λ
Движение иона
Канал закрыт |
Цитоплазма
Клеточный ответ
Рис. 26.23. Лиганд-зависимый канал
Приведены каналы, специфичные для ионов Са2+, Na4 и К+
часть 4 Хранение и переработка информации
358
Каким образом
связывание ацетилхолина
с мембранным рецептором
приводит к нервному импульсу?
Как упоминалось ранее, мотонейрон, подающий
сигнал к сокращению произвольной
поперечно-полосатой мышцы, получает от пресинаптического нейрона
ацетилхолин в качестве сигнальной молекулы; он
выделяется в синапс и обеспечивает проведение нервного
импульса к другому нейрону, который побуждает
мышцу сокращаться (см. рис. 26.24).
Нейроны, как и другие животные клетки, содержат
высокую концентрацию К+ внутри клетки и низкую -
снаружи, в то время как уровень Na+ высок снаружи
и низок внутри клетки. Такое распределение ионов
является результатом действия ионного насоса Na+/K+-
АТРазы в мембране.
В состоянии покоя (когда не происходит передачи
нервных импульсов) мембрана клетки более
проницаема для К+, чем для Na+. В силу более высокой
концентрации внутри клетки К+ выходит наружу, а поскольку
мембрана непроницаема для анионов, последние
клетку не покидают. Это создает положительный заряд
снаружи и отрицательный - внутри, т. е. поляризацию
мембраны. Утечка К+- процесс
самоограничивающийся. Дальнейшей утечке препятствует
образующаяся разность зарядов. Достигается состояние
равновесия, при котором потенциал покоя клетки составляет
около -70 мВ (отрицательный заряд внутри).
Рецептор ацетилхолина представляет собой лиганд-
зависимый №+/К+-канал. При связывании
ацетилхолина канал открывается, позволяя Na+ двигаться в клетку,
а К+ - из нее (см. рис. 26.24). Однако, вследствие
гораздо более высоких концентраций, Na+ входит в клетку
быстрее, чем К+ выходит из нее, что приводит к
уменьшению отрицательного заряда внутри. Т. е. мембрана
деполяризуется, причем только в непосредственной
близости от ацетилхолиновых рецепторов синапса. Это
и есть начальный стимул.
Распространение нервного импульса вдоль
аксона происходит за счет вызываемого ацетилхолином
повторной деполяризации мембраны, однако в этом
участвует другой тип Na+- и К+-каналов, названный
потенциал-зависимым каналом, который
открывается при падении потенциала покоя (с -70 до -40 мВ).
Изменение потенциала происходит на узко ограниченном
участке мембраны и связано с открытием лишь соседних
потенциал-зависимых каналов. Каналы открыты очень
короткое время, затем они закрываются, и наступает
период восстановления. Только после восстановления
Рецептор
ацетилхолина
Ацетилхолин-зависимый
канал инициирует
деполяризацию мембраны
Потенциал-зависимые Na+-
и К+-каналы распространяют
нервный импульс по аксону
Ацетилхолиновые
везикулы
^ Ацетилхолин
Мышца
Открытие потенциал-зависимых
Са2+ каналов вызывает резкий
приток Са2+ и экзоцитоз
ацетилхолиновых везикул
в синаптическую щель
Рис. 26.24. Цепи событий, в результате которых ацетилхолиновая стимуляция мотонейронов приводит к выделению
ацетилхолина в нервно-мышечном синапсе, что сопровождается сокращением поперечно-полосатой мышцы (упрощенно)
каналы способны ответить на следующий сигнал.
После того как пройдет волна возбуждения, Na+/K+-Hacoc
восстанавливает исходную концентрацию ионов.
Подробное знакомство с механизмом нервной
проводимости выходит за рамки этого раздела и всей
книги. Важно то, что волна деполяризации мембраны
проходит по аксону до нервных окончаний. В мембране
последних есть потенциал-зависимые Са2+-каналы,
которые открываются, когда волна деполяризации их
достигнет, позволяя внеклеточному Са2+ войти в клетку.
Это приводит к слиянию везикул с ацетилхолином, с
клеточной мембраной и высвобождению их путем эк-
зоцитоза(см. рис. 26.24). Са2+-АТРаза выкачивает Са2+,
восстанавливая состояние покоя. Выделившийся
ацетилхолин связывается с рецепторами мышечных
клеток. Дальнейшая последовательность событий описана
в главе 28.
Индуцируемый Са2+ экзоцитоз везикул имеет более
общее биологическое значение, чем просто участие
в выделении нейромедиаторов. Стимулируемый Са2+
экзоцитоз везикул происходит при выделена
пищеварительных ферментов из поджелудочной железы; однако
здесь выделение Са2+ происходит по фосфоинозитидно-
му пути (см. с. 353).
Оплодотворение яйцеклетки также вызывает
индуцируемый Са2+ экзоцитоз везикул, создающий барьер
для последующего проникновения спермы.
Другим важным процессом, в котором участвует ли-
ганд-зависимое управление порами, является зрение.
Зрение: процесс, зависимый
от контроля открываемой лигандом
поры
В основе зрения лежит превращение светового сигнала
в химические изменения, приводящие к образованию
импульсов в зрительном нерве, несущем сигналы в мозг.
Свет поглощается сетчаткой позвоночных,
имеющей два типа клеток: палочки - для восприятия черно-
белого и сумеречного света - и колбочки для цветного
и яркого света. В палочках есть три сегмента. В средней
части расположен обычный комплекс органелл. Один
конец образует синапс с биполярной клеткой, которая,
соединена со зрительным нервом. Другой конец -
цилиндрический (палочкообразный) сегмент, в котором
есть «батарея» мембранных дисков (около 2000),
погруженная в цитоплазму (рис. 26.25). Диски содержат све-
товоспринимающий аппарат.
Передача светового сигнала
Детали процесса восприятия света сложны; мы
познакомимся лишь с основными принципами (рис. 26.26).
В темноте гуанилатциклаза обеспечивает в клетках
колбочек довольно высокий уровень cGMP. В клеточной
мембране есть лиганд-зависимые катионные каналы,
которые cGMP поддерживает в открытом состоянии.
Постоянный приток Na4 в темноте определяется
более низким потенциалом на клеточной мембране;
равновесие между поступающим и выходящим из клетки Na+
глава 26 Химическая сигнализация в организме
359
Цитоплазма Плазматическая Внешняя
мембрана сторона клетки
Лиганд-зависимый
катионный канал
Наружный сегмент
содержащий
пластинки
(диски) и
фоторецепторный
аппарат
! <
j С
; с
1
': <
I <
Уплощенные диски,
упакованные
...-' молекулами
родопсина
"■ ^
Приток Na+ через
~ -· —Na+ cGMP-зависимые
Na+ катионные каналы
Темнота _
Открывает
катионный
канал ~*—
cGMP cGMP
Катион
Внутренний
сегмент,
содержащий
митохондрии и
синтетический
аппарат клетки
Na+
_ к+ *"·■ Отток Na+
под действием
Na/K+-ATPa3bi
Na+
К+
Ядро
Синаптическая
терминаль
Рис. 26.25. Структура палочки
устанавливает потенциал на уровне -30 мВ (что
отличается от ситуации в нейроне, где в состоянии покоя
Ыа+-каналы закрыты и мембранный потенциал -70 мВ).
Рецептором, воспринимающим световые фотоны
в дисках палочек, является родопсин - комплекс белка
опсина и 11 -^мс-ретиналя. Свет вызывает активацию
родопсина за счет конформационного изменения
зрительного пигмента, который полностью становится
шрднс-ретиналем. Посмотрите на структуру,
приведенную на рис. 26.27, и обратите внимание на сходство
молекулы ретиналя с витамином А и β-каротином.
Активация родопсина запускает цепь реакций с
участием тримерного G-белка (и гидролиза GTP, играющего
«обычную» роль молекулярных часов). Все это в конце
концов приводит к активации фермента, гидролизую-
щего GTP - cGMP-фосфодиэстеразы. Она похожа по
действию на сАМР-фосфодиэстеразу (см. рис. 26.9, б).
Восстановление
в темноте как
указано в тексте
Свет
Свет вызывает активацию родопсина,
что приводит к обмену GDP-GTP
на G-белке и активации
cGMP-фосфодиэстеразы
Вызывает
гиперполяризацию
плазматической ■■■·
мембраны или
увеличение
отрицательного
заряда внутри
Зрительный сигнал
Рис. 26.26. Процесс зрения (упрощенное изображение)
Действие света на родопсин приводит к активации cGMP-фос-
фодиэстеразы. Снижение уровня cGMP влечет за собой
закрытие катионного канала, гиперполяризацию клеточной
мембраны и передачу импульса по зрительному нерву
Снижение уровня cGMP приводит к закрытию Na+
каналов и гиперполяризации мембраны. Таким образом
зрительный сигнал поступает в зрительный нерв.
Восстановление клетки после поглощения фотона
света - сложный процесс. Он включает: 1) инактивацию
родопсина; 2) снижение уровня Са2+ в клетке и
стимуляцию синтеза cGMP; 3) сложный кругооборот
превращений молекулы родопсина. В конечном итоге уровень
cGMP на свету падает, №+-каналы закрываются и
гиперполяризация клеточной мембраны ведет к поступлению
зрительного сигнала в зрительный нерв. При освещении
уровень cGMP восстанавливается, №+-каналы
открываются - и мы готовы к восприятию следующего фотона.
часть 4 Хранение и переработка информации
360
Н3С СН
β-Каротин
н3с сн3н сн3 н сн3
I
Η
Η Η
Витамин А
н3сч сн3у сн3 у
Н,С^ *СН
I
15СНО
11-1<мс-Ретиналь
Н3С СН3^ fH3 Η СН3
. '-Г***^'*"
Η Η
трднс-Ретиналь
Η
Рис. 26.27. Структуры β-каротина, витамина А и ретиналя
а - Структура β-каротина - основного пигмента фотосинтеза у растений и предшественника витамина А у животных;
б - структура витамина А;
в - структура 11 -z/uc-ретиналя и гараиоретиналя
Дополнительная литература
Передача сигнала
Trends Biochem. Sci. 1992. Vol. 17. P. 367-443.
(Весь выпуск посвящен различным аспектам клеточной
сигнализации, включая мембранные рецепторы,
образование и разрушение вторичных посредников, протеинки-
назы и фосфатазы и активацию факторов транскрипции.)
G белки
binder Μ, Gilman A. G. G proteins// Sci. Amer. 1992.
Vol. 267(1). P. 36-43.
(G-белки на внутренней поверхности мембраны
клетки координируют клеточные ответы для множества
сигналов.)
Рецепторы
Lincoln Т. М., Cornwell Т. L. Intracellular cGMP receptor
proteins // FASEB J. 1993. Vol. 7. P. 328-338.
(Описывает биологическую роль cGMP как вторичного
посредника.)
Pawson Т. Look at a tyrosine kinase //Nature. 1994. Vol. 372.
P. 726-727.
(Представлены данные и различные мнения о
трехмерной структуре инсулинового рецептора.)
Heldin С.-Η. Dimerisation of cell surface receptors in
signal transduction // Cell. 1995. Vol. 80. P. 213-223.
(В обзоре описаны димеризация и олигомеризация
рецепторов, которая является начальным ответом на
действие многих факторов роста и цитокинов.)
Оксид азота
Snyder S.H., Brendt D. S. Biological roles and mechanism
of action of nitric oxide// Sci. Amer. 1992. Vol. 266 (5).
P. 2^-35.
(Хорошо иллюстрированная статья о роли и механизме
действия оксида азота.)
глава 26 Химическая сигнализация в организме
361
Brendt D. S., Snyder S. H. Nitric oxide: a physiologic
messenger molecule // Annu. Rev. Biochem. 1994. Vol. 63.
P. 175-195.
(Хорошо читаемый обзор, главная ценность которого -
освещение роли оксида азота в организме в целом.)
Сигнальные пути
Ε eg L. A. The many roads that lead to Ras // Science. 1993.
Vol. 260. P. 767-769.
(Приводится обзор различных сигнальных путей,
сходящихся на Ras-белке.)
Nishizuka Υ. Protein kinase C and lipid signalling for
sustained cellular responses // FASEB J. 1994. Vol. 9. P. 484-496.
(Прекрасное резюме по сигнальным путям, в которых
используются гидролиз инозитсодержащих фосфолипи-
дов и деградация других различных липидных
компонентов мембраны.)
Karin M., Hunter Т. Transcriptional control by protein
phosphorylation: signal transmission from the cell surface to
the nucleus // Curr. Biology. 1995. Vol. 5. P. 747-757.
(Хорошо иллюстрированное сравнительное описание
Ras-пути, где происходит транслокация активированных
протеинкиназ из цитоплазмы в ядро, и пути JAK-кина-
зы, характеризующиеся транслокацией активированных
факторов транскрипции в ядро.)
Quilliam L Α., Khosravi-Far R., HuffS. Υ, Der С. J.
Guanine nucleotide exchange factors: activators of the Ras super-
family of proteins // BioEssays. 1995. Vol. 17. P. 395-404.
(Обширный, но вполне читаемый обзор взаимодействия
Ras-пути с другими сигнальными маршрутами.)
Ihle J. N. STAT and MAPKs: obligate or opportunistic
partners of signalling // BioEssays. 1996. Vol. 18. P. 95-98.
(Спекулятивное обсуждение возможного
взаимодействия сигнальных путей Ras и STAT.)
Гемопоэтические факторы роста
MetcalfD. The 1991 Florey Lecture. The colony-stimulating
factors: discovery to clinical use // Philos. Trans. Roy. Soc.
London B. 1991. Vol. 333. P. 147-173.
(Глубокая работа, сделанная первооткрывателем этих
важных белков. Несмотря на в общем исследовательский
уровень излагаемого материала, автор охватывает
проблему в целом: от обнаружения неидентифицированных
факторов до их широкого клинического применения.)
Интерфероны
Johnson Η. Μ, Bazer F. W., Szente Β. Ε., Jarpe Μ. A. How
interferons fight disease// Sci. Amer. 1994. Vol. 270 (5).
P. 4(M19.
(Описываются структура интерферонов и сигнальные
пути, при помощи которых они влияют на клетку.)
часть 4 Хранение и переработка информации
362
IhleJ. N., WithuhnB. Α., Quelle Ε W., Yamamoto К.,
Thierfelder W. E., Kreider В., Silvennoinen O. Signalling by
the cytokine receptor superfamily: JAKs and STATs // Trends
Biochem. Sci. 1994. Vol. 19. P. 222-227.
(Исчерпывающий обзор по сигнальному пути
интерферонов, интерлейкинов, колонийстимулирующих
факторов и гормона роста от клеточной мембраны до ядра.)
Гомеостаз кальция
Bootman Μ. D., Berridge M. J. The elemental principles
of calcium signalling// Cell. 1995. Vol. 83. P. 675-678.
(Миниобзор по источникам Са2+, используемым для
генерации сигналов: выделение Са2+ из
внутриклеточных депо и вход Са2+ через плазматическую мембрану.)
Monteith G. R., Roufogalis В. D. The plasma membrane
calcium pump - a physiological perspective on its
regulation // Cell Calcium. 1995. Vol. 18. P. 459^70.
(Роль Ca2+/Mg2+-3aBHCHMoft АТРазы плазматической
мембраны в клеточной сигнализации и во
внутриклеточном гомеостазе кальция.)
Онкогенез
Cavanee W. К., White R. L. The genetic basis of cancer //
Sci. Amer. 1995. Vol. 272 (3). P. 50-57.
(Описывается накопление генетических дефектов,
которые могут вызывать превращение нормальных клеток
в раковые.)
Клеточный цикл и рак
Elledge R. Μ, Lee W.-H. Life and death by p53 //
BioEssays. 1995. Vol. 17. P. 923-930.
(Суммирует многие функции продукта гена р53,
включая его роль в супрессии рака.)
Nigg Ε. A. Cyclin-dependent protein kinase: key
regulators oftheeukaryotic cell cycle//BioEssays. 1995. Vol. 17.
P. 471^80.
(Описывается роль фосфорилирования циклинзависи-
мыми протеинкиназами в регуляции клеточного цикла.)
Ионные каналы
NeherE., Sakmann В. The patch clamp technique// Sci.
Amer. 1992. Vol. 266 (3). P. 44-51.
(Дает возможность проводить исследования по лиганд-
и потенциал-зависимым ионным каналам.)
Changeux J.-R Chemical signalling in the brain // Sci Amer.
1993. Vol. 269 (5). P. 30-37.
(Описаны строение и работа ацетилхолинового рецептора.)
Catterall W. A. Structure and function of voltage-gatedion
channels //Annu. Rev. Biochem. 1995. Vol. 64. P. 493-531.
(В обзоре описаны натриевый, кальциевый и калиевый
каналы.)
Вопросы к главе 26
1. Какие классы сигнальных молекул осуществляют
сообщение между клетками?
2. Используя простую схему, сравните действие на
клетки внешних сигнальных молекул, растворимых
и нерастворимых в липидах.
3. Как регулируется образование с AMP? Опишите роль
GTP в этом процессе. Какое отношение имеет GTP
к холере?
4. Как действует сАМР в качестве вторичного
посредника?
5. Каким образом оксид азота осуществляет свое регу-
ляторное действие?
6. сАМР и cGMP - не единственные вторичные
посредники. Опишите другую подобную систему.
7. Существуют важные ген-активирующие регулятор-
ные пути, в которых низкомолекулярные вторичные
посредники не участвуют. Опишите такой путь.
8. Если белок имеет 8Н2-домен, то какова его
возможная роль?
9. Объясните, каким образом аномальные компоненты
клеточных сигнальных путей могут способствовать
развитию рака.
10. Опишите путь регулирования активности генов
интерфероном.
11. Сделайте общее сравнение механизма активации
трех типов рецепторов: А - адренорецептора,
активирующего аденилатциклазу; Б- рецептора EGF
Ras-пути; В - рецептора интерферона.
12. Что представляет собой потенциал-зависимый
Са2+-канал? Поясните на примере.
13. Что такое лиганд-зависимый канал? Приведите
пример подобного канала, участвующего в зрительном
процессе.
глава 26 Химическая сигнализация в организме
363
глава
27
Красная кровяная клетка
и функции гемоглобина
Самой важной молекулой в доставке кислорода от
легких к тканям является гемоглобин, который
представляет собой не просто белок, пассивно связывающийся
с кислородом, а сложную молекулярную машину, в
задачу которой входит захват максимального количества
кислорода в легких, доставка его к периферическим
тканям, отбор оттуда С02, и транспортировка его в легкие.
Гемоглобин - это белок глобин, простетической
группой которого является гем. В этой главе мы
обсудим несколько аспектов, связанных с транспортом газов
и начнем с синтеза гема и его регуляции. После этого мы
рассмотрим регуляцию синтеза глобина и, наконец,
обсудим физиологическую роль гемоглобина в организме.
Красные кровяные клетки
В организме человека образуется 160 млн красных
кровяных клеток, или эритроцитов, в минуту. Они
циркулируют в крови в течение 110 дней и затем разрушаются.
Зрелые эритроциты млекопитающих не имеют ядра
и представляют собой сплющенный, двояковогнутый
диск (рис. 27.1). Такая форма клетки, поддерживаемая
цитоскелетом, обеспечивает большую поверхность
по сравнению с клетками сферической формы. Это,
наряду с меньшим диффузионным расстоянием,
усиливает газообмен между клеткой и внеклеточной средой.
Общая поверхность всех эритроцитов составляет около
4000 м2.
У эмбриона эритроциты образуются в печени и
селезенке. У взрослых же этот процесс происходит в
костном мозге плоских костей, в которых кроветворные
стволовые клетки непрерывно размножаются и
продуцируют предшественников всех типов кровяных клеток,
как это показано на рис 25.1 (стволовая клетка
постоянно осуществляет самовоспроизводство, а ее
потомство способно к дифференцировке). Отдельные клетки
дифференцируются в тот или иной специализированный
тип кровяных клеток (эритроциты, различные
лейкоциты, тромбоциты). Образование разных типов клеток
крови - регулируемый процесс; обнаружены
различные регуляторные белки, способные вызывать
размножение специфических клеток-предшественников. Белок
эритропоэтин, вырабатываемый в мозговом слое
почек, стимулирует образование эритроцитов. Поскольку
скорость образования эритропоэтина обратно
пропорциональна парциальному давлению кислорода в ткани,
регуляция уровня красных кровяных клеток происходит
автоматически: при недостатке кислорода
вырабатывается больше гормона, а следовательно, и больше
красных кровяных клеток.
На ранних стадиях эритроциты содержат ядро,
однако позднее у млекопитающих оно исчезает и образуется
незрелая красная клетка ретикулоцит, которая
поступает в кровоток. Ретикулоциты содержат мРНК, и
синтез белка в них происходит до тех пор, пока клетка в
процессе созревания не превращается в эритроцит. При
окрашивании клетки рибосомы и мРНК выглядят как
темный пигментированный ретикулум. Отсюда и
произошло название этих клеток - ретикулоциты. В зрелой
клетке нет митохондрий, и для выработки энергии она
использует гликолиз с образованием лактата. В зрелом
эритроците активен пентозофосфатный путь, в ходе
которого происходит восстановление NADPH.
Рис. 27.1. Сканирующая электронная микрофотография
эритроцита
Любезно предоставлена профессором W. G. Breed, Department
of Anatomy, University of Adelaide
Гем и его синтез
Полная структура гема (рис. 27.3.), которую помнят
далеко не все биохимики, представляет собой комплекс
двухвалентного железа с протопорфирином (рис. 27.2).
часть 5 Транспорт кислорода и С02
366
Рис. 27. 2. Изображение контура молекулы гема
сн2=сн сн3
сн=сн2
сн2 сн2
I I
сн2 сн2
соо- соо-
Рис. 27. 3. Структура гема
Эта форма найдена в гемоглобине; другие гемы,
различающиеся своими боковыми цепями, обнаружены в цитохромах.
На одной стороне молекулы находятся 2 нуклеофильных
остатка пропионовой кислоты (СН 7-СН9-СОО~ ), другие боковые
цепи гидрофобны. В миоглобине гем расположен в
углублении молекулы, причем его гидрофильная часть обращена к
поверхности, а гидрофобные группы - внутрь молекулы белка
Последний образован 4 пиррольными кольцами: 4
замещенных пиррола связаны метиленовыми (=СН-)
мостиками, так что создается система сопряженных
двойных связей (т. е. при движении по кругу одинарные
и двойные связи в молекуле чередуются). Это придает
порфирину и гему тёмнокрасный цвет.
В геме 4 пиррольных атома N связаны с Fe2H
(рис. 27.4), оставляя 2 из 6 связей Fe2+ свободными и
доступными для других лигандов.
—N ; N—
Fe2+
-/!чм-
Рис. 27. 4. Связывание Fe2+ в геме
Атом железа в геме может связаться с 6 лигандами: 4 связи
заняты атомами пиррольного азота, а 2 другие расположены
над и под плоскостью листа соответственно
Синтез гема
На долю эритроцитов приходится наибольшее
количество гема, содержащегося в организме человека.
Поэтому рассмотрение вопроса о его синтезе в этой главе
вполне уместно, хотя гем простетических групп цитохромов
и других белков синтезируется и в других клетках.
Синтез гема представляется нелегкой задачей, для
решения которой животным необходимо, как ни
удивительно, только два исходных реагента: глицин
и сукцинил-СоА. С последним вы встречались при
рассмотрении цикла лимонной кислоты (см. с. 120).
Эти вещества необходимы для синтеза аминолеву-
линовой кислоты (англ. aminolevulinic acid; ALA),
реакцию образования которой катализирует фермент
аминолевулинатсинтаза (ALA-синтаза) (рис. 27.5).
CO-S-CoA
I
сн2
I
сн2
I
соон
Сукцинил-СоА
CH2(NH2)COOH
Глицин
ALA-синтаза
CO-CH2-NH2
+ С02 + CoA-SH
СН2
I
СН2
I
СООН
5-Аминолевулиновая кислота (ALA)
Рис. 27. 5. Первая стадия синтеза гема, катализируемая
5-аминолевулинатсинтазой (ALA-синтазой)
Структуры приведены в неионизированной форме
5-Аминолевулиновая кислота - единственный
предшественник синтеза порфирина. Две ее молекулы,
использующиеся для образования пиррола - порфобили-
ногена (ПБГ), подвергаются дегидратации под
действием ALA-дегидратазы (рис. 27.6). Остальные стадии
биосинтеза гема включают в себя соединение 4 молекул
ПБГ в единую структуру, модификацию групп боковых
цепей и образование хелатных комплексов с
двухвалентным атомом железа. Промежуточными тетрапиррола-
ми на стадиях между образованием ПБГ и
формированием гема являются бесцветные уро- и копропорфири-
ногены (содержащие метиленовые мостики) и красный
глава 27 Красная кровяная клетка и функции гемоглобина
367
соон соон
соон сн2 соон сн2
I I ALA- I I
СН2 СН2 деги атазг fH2 CH2
с:н2" о:=с ► с — с + \г н2о':
Г----- I И II ■___-t_J
/Сн2 :~б h2:n /CH2 n
NH2 ' J NH2
Две молекулы ALA Порфобилиноген (ПБГ)
Рис. 27.6. Синтез порфобилиногена (ПБГ) - стадия
дегидратации, катализируемая ALA-дегидратазой
Представлены неионизированные структуры.
Порфобилиноген является монопирролом; гемоглобин - тетрапиррол
протопорфирин (содержащий метеновые мостики).
Путь синтеза гема представлен на рис. 27.7.
Метаболический путь синтеза гема имеет любопытные
особенности. Первая реакция - синтез ALA - происходит внутри
митохондрий, после чего ALA поступает в цитоплазму.
Однако конечные три стадии снова протекают в
митохондриях. Почему так происходит, пока неясно.
Регуляция биосинтеза гема и доставка
железа в эритроцит
Регуляция биосинтеза гема представляет особый
интерес, поскольку она непосредственно связана с
поступлением и хранением железа в клетке.
Происходит это так.
• Активность ALA-синтазы лимитирует весь процесс
синтеза гема.
• Синтез ALA-синтазы, а следовательно, и ее уровень
регулируется количеством железа.
• Уровень железа зависит от количества
белка-рецептора трансферрина.
• Уровень железа по механизму обратной связи
регулирует синтез трансферрина.
При биосинтезе гема скорость-лимитирующей
стадией является синтез ALA. Образование самого фермента
ALA-синтазы регулируется в красных кровяных клетках на
уровне трансляции. На 5'-нетранслируемом конце (т. е. на
участке инициации трансляции) мРНК фермента имеет
шпилечную петлю, названную железо-чувствительным
элементом (IRE; англ. iron-responsive element). При
низком уровне железа IRE-связывающий белок
присоединяется к IRE и предотвращает трансляцию (рис. 27.8).
При высоком уровне железа белок высвобождается в
результате его взаимодействия с железо-серным кластером
(небольшим комплексом, состоящим из железа, цистеина
и неорганической серы ), после чего начинается
трансляция: таким образом достигается соответствие
между скоростью синтеза гема и запасом железа.
Синтез гема начинается задолго до образования ре-
тикулоцита в проэритробласте, содержащем ядро, так
что трансляция мРНК происходит в этих клетках и
продолжается на стадии ретикулоцита.
Возникает вопрос: каким образом регулируется
уровень железа в клетке? Железо транспортируется
в плазме крови в виде комплекса с белком трансфер-
рином, который образуется в печени. Этот комплекс
захватывается клетками в ходе
рецептор-опосредованного эндоцитоза. В неэритроидных клетках
захват определяется уровнем белка-рецептора
трансферрина, синтез которого регулируется уровнем железа
в клетке. Железо осуществляет контроль за
стабильностью мРНК белка-рецептора трансферрина, которая
содержит на З'-конце молекулы
железо-чувствительный элемент. При низком уровне железа в клетке
IRE-связывающий белок присоединяется к мРНК
белка-рецептора, защищая ее от деградации. В тех
случаях, когда уровень железа в клетке высок,
IRE-связывающий белок отсоединяется, что приводит к снижению
стабильности мРНК белка-рецептора трансферрина.
Таким образом, железо вызывает даунрегуляцию, или
снижение числа рецепторов трансферрина, которые,
в свою очередь, снижают потребление железа
клетками. При низком уровне железа все происходит в
обратном порядке. Пока не установлено, применим ли
этот механизм к эритроидным клеткам, однако
известно, что в эритроцитах есть рецепторы трансферрина,
число которых увеличивается в процессе созревания
клетки.
Запасы железа в эритроцитах находятся в виде
ферритина - комплекса белка апоферритина и
неорганического железа. Печень также является важным
местом хранения железа. Интересно, что синтез
апоферритина регулируется, в сущности, по такому же механизму,
как и синтез фермента ALA-синтазы: железо вызывает
присоединение IRE-связывающего белка к мРНК
апоферритина, способствуя синтезу белка.
Все клетки человека синтезируют гем тем же путем,
что и клетки крови (исключая незначительные отличия).
Гем нужен для цитохромов в цепи транспорта
электронов (см. с. 123), а также и для других ферментов.
Эритроциты более всего нуждаются в заполнении зрелой
клетки гем-содержащим белком - гемоглобином. Вслед
за красными кровяными клетками по количеству гема
идет печень, благодаря высокому содержанию в ней ци-
тохромаР450 (см. с. 210). Регуляция ALA-синтазы в
разных клетках имеет определенные отличия. В печени гем
часть 5 Транспорт кислорода и С02
368
дестабилизирует мРНК ALA-синтазы, а также
тормозит транспорт молекул фермента к «месту
назначения» - в митохондрии. мРНК фермента не имеет
железо-чувствительного элемента, который
существует в эритроидных клетках. Избыточная продукция
ALA-синтазы и высокий уровень ALA в крови при
острой перемежающейся порфирии у пациентов
коррелирует с появлением свойственных этому
заболеванию неврологических симптомов (их молекулярная
основа пока неизвестна). У таких больных частично
блокирован путь биосинтеза гема. Хлорофилл также
является тетрапирролом, однако интересно, что ALA у
растений синтезируется в ходе другой реакции.
Но вернемся к красным кровяным клеткам.
Митохондриальный
матрикс
соо-
I
СН2
СН2
СО— S— СоА ALA-синтаза
СН2- ΝΗί
I
соо-
соо-
I
сн2
сн2
► с=о
СН2-ЫНз
5-Аминолевулиновая
Рг Рг
Протопорфирин IX
\jb/\- eiH атаза
Цитоплазма
соо-
соо- сн2
I I
СН2 СН2
Порфобилиноген
NH7
- де а\ти
носн2
Гидроксиметилбилан
Рг
Уроген III синтазг
ь косинтаза
Изменение боковой
цепи и окисление
Ас
Уропорфириноген III
Рис. 27.7. Биосинтез гема
глава 27 Красная кровяная клетка и функции гемоглобина
369
Ситуация А: Концентрация железа в клетке высокая
Железо-серный кластер прикреп-
„» ленный к IRE-связывающему
белку не присоединяется к IRE,
IRE если концентрация железа высока.
Белки инициации трансляции
могут прикрепиться и мРНК
транслируется
мРНК,
Щ——ШШша^м^м Кодирующая
-Л
AUG
ALA-синтазу
Место связывания
белков инициации
трансляции
ALA-синтаза
I
ALA
I
Гем
Ситуация Б: Концентрация железа в клетке низкая
IRE-связывающий белок
присоединяется к IRE
AUG
/
Прикрепление факторов
инициации трансляции Нет образования ALA-синтазы
стерически затруднено I
i
Нет трансляции
Нет синтеза гема
Рис. 27.8. Регуляция синтеза гема изменением концентрации
железа в эритроцитах
Регуляторный механизм, по-видимому, зависит от
ALA-синтазы, имеющей короткий полупериод жизни: как только
прекращается синтез фермента, тут же останавливается и синтез
гема. Фермент, как известно, в лизатах ретикулоцитов в
условиях in vitro, а также, по-видимому, и в самих эритроцитах,
нестабилен. Вопрос о том, как контролируется уровень
железа в клетке, рассматривается отдельно. IRE -
железо-чувствительный элемент, представляющий собой шпилечную петлю
мРНК. Когда IRE-связывающий белок прикрепляется к IRE,
он, вероятно, стерически препятствует присоединению
белков инициации трансляции. Связывание железа с IRE-связы-
вающим белком снижает его сродство к IRE
Распад гема
Эритроциты разрушаются главным образом ретику-
лоэндотелиальными клетками селезенки,
лимфатических узлов, костного мозга и печени. Удаление сиаловых
кислот из гликопротеинов эритроцитарной мембраны
служит сигналом старения эритроцитов. Лишенные
сиаловых кислот углеводные компоненты связываются
с рецепторами этих клеток, что приводит к эндоцитозу
старых эритроцитов.
Фермент гемоксигеназа раскрывает тетрапиррольное
кольцо, высвобождая железо для повторного
использования и образуя линейный тетрапиррол - биливердин
(рис. 27.9). Биливердин восстанавливается в билирубин.
Последний не растворим в воде. Связываясь с
сывороточным альбумином, он переносится кровью в печень,
где после присоединения двух остатков глюкуроновой
кислоты становится гораздо более полярным, а затем эк-
скретируется с желчью в кишечник. Модификация и
частичная реабсорбция некоторых составных
компонентов желчи придает желтый цвет моче (предполагается,
что билирубин функционирует в качестве антиоксидан-
та, см. с. 213).
Как регулируется синтез глобина
Гемоглобин состоит из гема, связанного с белком
глобином. Два компонента - глобин и гем - должны
образовываться примерно в одинаковых количествах, для
чего и существует координирующая регуляторная связь.
В опытах с лизатами ретикулоцитов было показано, что
в отсутствие гема протеинкиназа фосфорилирует один
из факторов (eIF2) инициации синтеза белка. Он
обладает способностью останавливать инициацию
трансляции в красных кровяных клетках и таким образом
предотвращать синтез глобина. В присутствии гема киназа
инактивируется, а фосфатаза нормализует активность
фактора инициации. Таким образом, синтез глобина
происходит лишь в присутствии гема. Этот механизм
специфичен для эритроидных клеток.
Транспорт газов в кровь
Растворимости кислорода в обычных растворах
недостаточно, чтобы обеспечить им ткани организма.
Необходимы переносчики кислорода в крови. Аналогичным
образом С02 не может транспортироваться с
достаточной скоростью от тканей к легким в простом растворе.
Каково химическое обоснование роли
гемопротеинового комплекса
как переносчика кислорода?
Неорганический ион Fe2+, но не Fe3+, может связываться
с кислородом. Однако ионы Fe2+ спонтанно окисляются
в Fe3+, так что неорганическое железо само по себе
не является хорошим переносчиком кислорода. Fe2+
часть 5 Транспорт кислорода и С02
370
-Fe-
V KJ x
Гем
■~e Гемоксигеназа
CO (главным образом
HoO в селезенке)
NADP+
Ν Ν Ν
Биливердин
NADPH
Биливердин
ре^ктаза λ
NADP+ *S\
Ν
°^ Ν "** ^ Ν ^7C Ν ^ " Ν ""°
Η Η
Билирубин
(плохо растворим в Н20)
2-иОР-глюкуронат -^ι
UDP-глюкуронозил- ] Печень
трансфераза 2UDP ^\
соо- соо-
I I
R R
\ - V (
0^ N ^—" N ^y^ N ^^ N "^0
Η Η Η
Билирубин-диглюкуронид
(растворим в Н20)
Рис. 27.9. Реакция, катализируемая гемоксигеназой, и
последующее восстановление биливердина
Двойные связи в структурах колец не показаны. В результате
реакции одна метиленовая группа превращается в окись
углерода, и таким образом раскрывается структура кольца.
Биливердин восстанавливается в билирубин, связывается с 2
молекулами глюкуроновой кислоты и выводится из организма.
Существует однако, некоторая неясность в стехиометрии гем-
оксигеназной реакции; полагают, что в реакции участвуют
3 молекулы 07. По аналогии с цитохромом Р450 можно
предположить, что для превращения 3 атомов кислорода в Н70
требуются 3 молекулы NADPH
в составе гема может также связывать кислород, но и оно
быстро окисляется до Fe3+, образуя гематин. Поэтому
свободный гем также не является хорошим
переносчиком кислорода. Для окисления гема необходимо, чтобы
2 его молекулы взаимодействовали с 1 молекулой
кислорода. Связывание гема, расположенного в углублении
белка, предотвращает такое взаимодействие, поэтому
ионы Fe2+ в составе гема гемоглобина гораздо более
устойчивы к окислению, чем свободный гем. Важно
прояснить, почему гем в цитохромах при транспорте
электронов (см. с. 123) находится поочередно в состояниях
Fe 2+ и Fe3+, а гем в гемоглобине функционирует только
в состоянии Fe2+, на которое взаимодействие с
кислородом не влияет. Дело в том, что атом железа в
гемоглобине связан с 4 атомами азота пиррольных колец, а
пятая и шестая координационные связи расположены
непосредственно над и под плоскостью структуры гема;
одна из этих связей соединена с остатком гистидина,
а другая доступна для связывания с кислородом.
Структура миоглобина и его роль
в связывании кислорода
Миоглобин является красным пигментом мышц.
Предварительное знакомство с его структурой поможет
понять более сложную организацию гемоглобина.
Миоглобин просто снабжает мышечную клетку
кислородом. Он захватывает кислород из крови и переносит
его внутрь клетки, где тот используется митохондриями
для выработки энергии. Миоглобин - сложный белок,
состоящий из одной полипептидной цепи и связанной
с ней молекулой гема. Белок имеет сферическую форму
и содержит ряд α-спиралей (рис. 27.10), а молекула гема
расположена в углублении между двумя из них.
Насыщение миоглобина кислородом при увеличении
парциального давления последнего характеризуется
гиперболической кривой (рис. 27.11) такого же типа, как
в случае кривой зависимости активности фермента
от концентрации субстрата, описанной ранее для
«классического» фермента (см. с. 157). Миоглобин обладает
высоким сродством к кислороду и потому без труда
забирает его из оксигемоглобина крови (см. рис 27. 11,
демонстрирующий относительное сродство к
кислороду двух переносчиков). В мышцах, где концентрация
кислорода падает в процессе транспорта электронов
и образования Н20, миоглобин отдает связанный
кислород. Таким образом, он функционирует как очень
простой переносчик кислорода.
Миоглобин стал первым белком, для которого была
определена трехмерная структура, и это достижение
считается поворотным пунктом в биохимии.
глава 27 Красная кровяная клетка и функции гемоглобина
371
У а2 J
1 ρ·
Glu6
Ν
■ Glu6
щ
Рис. 27.10. Модели молекул миоглобина и гемоглобина
а - Простая компьютерная модель, демонстрирующая укладку (фолдинг) полипептидной цепи и положение гема в молекуле
миоглобина;
б - более сложная модель миоглобина;
в - модель, показывающая расположение субъединиц в молекуле гемоглобина.
В углублении в центре молекулы встраивается 2,3-дифосфоглицерат в дезоксигенированном состоянии (помечено X). При
серповидной клеточной анемии два остатка глутаминовой кислоты в положении 6 в β-цепях заменены на валины, создавая
гидрофобные участки на молекуле
Структура гемоглобина
Гемоглобин состоит из 4 белковых субъединиц (см.
рис. 27.10), каждая из которых напоминает свернутую
полипептидную структуру миоглобина и содержит гем,
способный связывать кислород. Молекула гемоглобина
взрослого человека состоит из 2 идентичных а- и 2
идентичных β-субъединиц.
Связывание кислорода с гемоглобином
Молекула гемоглобина связывает 4 молекулы
кислорода, по одной на каждую субъединицу. Насыщение
гемоглобина кислородом описывается не
гиперболической кривой, как в случае миоглобина, а сигмоидной,
причем эта кривая сдвинута вправо по отношению к
кривой, характерной для миоглобина (см. рис. 27.11).
Для достижения 50% насыщения гемоглобина
кислородом необходима более высокая концентрация
последнего, чем в случае миоглобина, что указывает на более
высокое сродство миоглобина к кислороду.
Гемоглобину необходимо забрать как можно больше кислорода из
легких и отдать его капиллярам в тканях. Наибольшая
крутизна сигмоидной кривой (свидетельствующая о
наибольшей отдаче кислорода) наблюдается при
давлении 02, соответствующем его давлению в капиллярах
часть 5 Транспорт кислорода и С02
372
s
о
et
О
α.
о
100—ι
емоглооин
0-
Приблизитсльнос давление
кислорода в капиллярах
р02
Рис. 27.11. Кривая насыщения гемоглобина кислородом
Более высокое сродство к кислороду у миоглобина, по
сравнению с гемоглобином, означает, что миоглобин в мышцах
легче забирает кислород из крови
(см. рис. 27.11). Насыщение гемоглобина кислородом
происходит при более высоком давлении кислорода в
легких.
Как появляется сигмоидная кривая
насыщения кислородом
Вы познакомились с кинетикой, описываемой сигмоид-
ной кривой, при обсуждении регуляции метаболизма
(глава 12). Там упоминалось, что аллостерические
ферменты, как правило, являются мультисубъединичными
белками, которые подвергаются конформационному
изменению при связывании с молекулой субстрата. Это
приводит к изменению сродства фермента к
последующим молекулам субстрата. То же самое применимо и
к связыванию кислорода с гемоглобином.
Несмотря на то, что молекулы гема в гемоглобине
достаточно удалены друг от друга, первоначальное
связывание кислорода с одной субъединицей ускоряет
связывание молекул кислорода с остальными
субъединицами. Это явление известно как гомотропный
положительный кооперативный эффект
{гомотропный потому, что участвует только кислород). Именно это
обусловливает сигмоидный характер кривой. Эффект
заключается в том, что первоначальное сродство дезоксиге-
моглобина к кислороду в 200 раз ниже, чем сродство на
заключительном этапе связывания. Насыщение
кислородом (оксигенация), соответствующее конечному отрезку
кривой, ограничено числом участков его связывания.
Механизм аллостерического изменения
молекулы гемоглобина
Как уже упоминалось, присоединение одной молекулы
кислорода к молекуле гемоглобина облегчает
связывание кислорода остальными субъединицами молекулы
гемоглобина. В соответствии с согласованной моделью
(см. с. 159), гемоглобин существует в двух конформаци-
онных состояниях: «напряженном», или Т-состоянии
(англ. tense) с низким сродством к кислороду, и
«расслабленном», или R-состоянии (англ. relax) с высоким
сродством к кислороду. Оба состояния находятся в
свободном равновесии. В отсутствие кислорода преобладает
Т-состояние. При связывании кислорода увеличивается
вероятность того, что все четыре субъединицы
молекулы гемоглобина будут находиться в R-состоянии
(высокого сродства); при этом равновесие Τ <-> R сдвигается
вправо.
Τ <-> R <-> R02
Поскольку исследование насыщения гемоглобина
кислородом проводится на большом числе молекул, то
возможно, чем больше кислорода связывается, тем
большее число молекул гемоглобина находится в
R-состоянии, а следовательно, увеличивается и сродство к
кислороду в растворе. Подобная ситуация характерна для
аллостерических ферментов (см. с. 159-160). С
использованием метода рентгеноструктурного анализа была
определена конформация тетрамерной структуры
гемоглобина в оксигенированном и дезоксигенирован-
ном состояниях. Как уже упоминалось ранее,
гемоглобин является тетрамером и состоит из 2 идентичных
απ 2 идентичных β-субъединиц. В тетрамере их
обозначают как α,, α2, β, и β2 По данным рентгено-структур-
ного анализа, контакты между а- и β-субъединицами
слегка различаются между собой.
Чтобы составить более ясное представление о
гемоглобине, целесообразно рассматривать его структуру
в виде двух гетеродимеров, образованных а- и
β-субъединицами: ο^β, и α2β2. Именно взаимодействие между
двумя гетеродимерами в тетрамере приводит к
перегруппировке Т<-> R. Относительное перемещение диме-
ров при переходе из Т- в R-состояние обусловленно
связыванием кислорода (рис. 27.12).
Что вызывает это аллостерическое изменение в
целой молекуле гемоглобина при связывании его с одной
или несколькими молекулами кислорода? Изменение
инициируется небольшим перемещением атома железа
при связывании кислорода с гемом, что и обусловливает
переход Τ <-> R. Хотя гем, как уже упоминалось, является
плоской молекулой, в дезоксигенированном состоянии
атом железа располагается над плоскостью молекулы,
поскольку размер атома слишком велик, чтобы
встроиться в тетрапиррольную структуру. В
дезоксигенированном гемоглобине тетрапиррольная структура не совсем
плоская (рис. 27.13).
глава 27 Красная кровяная клетка и функции гемоглобина
373
,<*ι
Оксигсппрованныи
(Χ?
Дсюксмгсниронанный
Рис. 27.12. Схематическое изображение относительного
расположения субъединиц в дезоксигенированной и окси-
генированной молекулах гемоглобина, оси которых
представлены прямыми линиями
Пары субъединиц ос,, β, и ос2, β2 должны рассматриваться как
единичный димер субъединиц. При окислении эти димеры
вращаются с наклоном около 15° относительно друг друга.
Черная и серая линии показывают относительное расположение
димеров в Т-состоянии. В оксигенированном (R) состоянии
красная линия показывает вращение α2/β2 димера по
отношению к α,/β, димеру, который изображается фиксированной
линией. Перемещение изменяет контакты ос1 β2/(Χ2 β, между ди-
мерами
Атом Fe2+ связывается с остатком гистидина одной
из α-спиралей, входящих в состав субъединицы. Эту
α-спираль обозначают как F, а гистидиновую группу -
F8 (рис. 27.13, а). Благодаря электронным изменениям,
F8 ос-спирали субъединицы глобина
—Г His I—
Тетрапиррол
Fe
Оксигенация
His
-N-Fe-N-
I
О
II
О
Рис. 27.13. Структурные изменения в геме молекулы
гемоглобина при оксигенации (схематическое изображение)
а - Молекула гема в дезоксигемоглобине с тетрапиррольной
структурой, вытянутой в слегка колоколообразную форму;
б - присоединение гема в оксигемоглобине
происходящим при связывании кислорода, атом железа
в геме существенно уменьшается в диаметре и входит
в плоскость тетрапиррола, что делает всю молекулу
более плоской. При этом перестраивается и сама
молекула белка (см. рис. 27.13).
Этот незначительный сдвиг атома железа приводит
к большим структурным изменениям где-нибудь в
другом месте молекулы белка за счет «рычагоподобного»
эффекта, влияющего на структуру полипептидной цепи.
Это «где-нибудь» является точкой, в которой а-субъе-
диница одного димера взаимодействует с β-субъеди-
ницей другого (на границе а^2 / а^). При таком
взаимодействии образуется ряд слабых связей между
аминокислотными остатками двух субъединиц. В Т-состоянии
мы имеем один набор, в R-состоянии - другой. Эти
связи удерживают димеры вместе. Таким образом,
инициированное атомом железа перемещение приводит к
замене одного набора связей между димерами на другой,
что вызывает относительную ротацию димеров (см.
рис. 27.12).
Следует отметить, что существование структур,
соответствующих Т- и R- состояниям гемоглобина полностью
доказано методами рентгеноструктурного анализа
кристаллов гемоглобина и оксигемоглобина соответственно.
Однако получить кристаллы частично окисленного
гемоглобина невозможно, и потому структура (или
структуры) последнего неизвестны. Поэтому промежуточные
стадии (если они есть вообще), при которых происходят
взаимопревращения Т- и R-форм, не установлены.
Важная роль 2,3-дифосфоглицерата
(ДФГ) в функции гемоглобина
Вы уже знакомы с промежуточным продуктом
гликолиза 1,3-дифосфоглицератом (см. рис. 8.6), однако до сих
пор мы еще не говорили о том, что в клетке также
синтезируется 2,3-фосфоглицерат (ДФГ).
О О С00"
С-0—Р—0"
0
II
-р-
I
о-
снон
о
сн2—о—р—о-
о-
1,3-Дифосфоглицерат
О
I II
сн—о—ρ—οι
о-
0
II
СН-,—О—Ρ—О-
о-
2,3-Дифосфоглицерат
ДФГ играет важную физиологическую роль в
транспорте кислорода, снижая сродство к нему гемоглобина
и, таким образом, повышая отдачу кислорода тканям.
ДФГ сдвигает кривую диссоциации оксигемоглобина
вправо. Вдоль всей тетрамерной молекулы гемоглобина
часть 5 Транспорт кислорода и С02
374
есть углубление (см. рис. 27.10). Боковые группы
положительно заряженных аминокислот проецируются
внутрь этого углубления. Молекула ДФГ, несущая при
рН крови 4 отрицательных заряда, по своей величине
и конфигурации соответствует размеру углубления и
образует ионные связи с положительно заряженными
участками белка. При этом удерживать гемоглобин вде-
зоксигенированном состоянии помогает перекрестное
сшивание β-субъединиц. В дезоксигенированном (Т)
состоянии гемоглобин может вместить молекулу ДФГ.
Однако при оксигенации в R-состоянии, в результате
конформационных изменений в белке, углубление
становится меньше и не может вместить молекулу ДФГ.
Способность ДФГ прочно связываться и
стабилизировать дезоксигенированное состояние благоприятствует
освобождению кислорода в капиллярах. В результате
имеет место следущий процесс.
НЬ-кислород <-> НЬ + кислород (1)
(расслабленное) (напряженное)
НЬ + ДФГ <-> НЬ-ДФГ (2)
(напряженное) (напряженное)
Реакция (2) с ДФГ способствует сдвигу равновесия
реакции (1) в сторону высвобождения кислорода.
Если кровь израсходовала весь запас ДФГ,
гемоглобин остается фактически насыщенным кислородом даже
при более низкой, чем в капиллярах ткани,
концентрации: вследствие этого он неспособен достаточно
эффективно отдавать кислород тканям.
Влияние ДФГ на связывание кислорода с
гемоглобином показано на рис 27. 14.
Чем выше концентрация ДФГ, тем более выражено
преобладание дезоксигенированной формы
гемоглобина. Создается регуляторная система: при низком
давлении кислорода в тканях в красных кровяных клетках
синтезируется большее количество ДФГ (посредством
100—ι ^ ^
S / /
о / /
§- /Нет ДФГ /
^ I / /
к / /
м / /
ω / /
к / /
g / / Снижение сродства
В I / к кислороду в
о / ^S присутствии ДФГ
Рис. 27.14. Кривые насыщения гемоглобина кислородом,
иллюстрирующие эффект 2,3-дифосфоглицерата (ДФГ)
регуляторного механизма, способствующего
увеличению отдачи кислорода, детали которого мы не
рассматриваем). При акклиматизации к условиям высокогорья
содержание ДФГ в красных кровяных клетках
увеличивается. Следует заметить, что хотя ДФГ и вызывает
большую отдачу кислорода тканям, сниженное сродство
гемоглобина к кислороду практически не влияет на
степень оксигенации гемоглобина в легких. Нормальная
молярная концентрация ДФГ в крови примерно
эквивалентна концентрации тетрамерного гемоглобина.
В регуляторной системе ДФГ есть еще одна
тонкость, которая показывает, как небольшие изменения
в белках могут вызвать значительные физиологические
эффекты. При снабжении плода кислородом
необходимо, чтобы гемоглобин плода забирал кислород из окси-
гемоглобина матери через плаценту. Для этого
гемоглобин плода должен иметь более высокое сродство к
кислороду, чем материнский переносчик. Это достигается
замещением β-субъединиц гемоглобина взрослого
человека γ-субъединицами плода, в каждой из которых
отсутствует один из положительных зарядов β-субъедини-
цы. Отсутствуют как раз те заряды, которые у
гемоглобина взрослого человека находятся в углублении, куда
встраивается ДФГ. Имея на 2 ионные группировки
меньше, гемоглобин плода связывает ДФГ менее прочно.
Поэтому ДФГ менее эффективно снижает сродство
гемоглобина к кислороду, что обеспечивает
гемоглобину плода более высокое сродство к нему по сравнению
с материнским гемоглобином, который легко отдает
свой кислород.
Влияние рН на связывание кислорода
с гемоглобином
Гемоглобин в дезоксигенированном состоянии имеет
более высокое сродство к протонам, чем оксигемогло-
бин. Другими словами, R-форма является более
сильной кислотой, чем Т-форма (дезоксигенированная); в
результате при связывании с кислородом происходит
диссоциация протонов от молекулы гемоглобина. Этот
феномен известен как эффект Бора:
Hb + 402^Hb(02)4 + (H+)n (1)
где η - величина порядка 2; число зависит от целого
комплекса параметров.
Протоны высвобождаются, например, из остатков
гистидина в белке. Причиной высвобождения являются
конформационные изменения, возникающие при
переходе из состояния Т- в R-состояние влияющие на
ионизацию (рКа) таких групп (о диссоциации гистидина
см. с. 40).
ιβα 27 Красная кровяная клетка и функции гемоглобина
375
Роль изменений рН в транспорте
кислорода и С02
Эффект Бора, описанный выше, имеет важное
физиологическое значение. Образующийся в тканях С02 должен
транспортироваться в легкие. Он поступает в эритроциты,
где фермент карбоангидраза превращает его в Н2СОэ,
который диссоциируется на бикарбонат-ион и протон:
Захват Η не стереохимичен
со2 + н2о<-+н2со3 + нсо3
(2)
Последний сдвигает равновесие в уравнении (1) влево,
заставляя НЬ02 отдавать свой кислород - эффект,
соответствующий физиологическим потребностями.
НСОэ пассивно продвигается через анионный канал
(см. с. 63) по градиенту концентраций в сыворотку.
Продвижение НСОэ не сопровождается перемещением Н+,
поскольку нет канала, позволяющего ему пройти через
мембрану эритроцита. Для сохранения электрического
баланса (равновесия) при выходе НСОэ из клетки, О
перемещается внутрь ее через тот же анионный канал.
Такое двойное перемещение известно как хлоридный
сдвиг (рис. 27. 15).
Растворенный НСОэ движется вместе с венозной
кровью обратно в легкие. Здесь изменения
концентрации протонов также помогают достичь желаемых
физиологических результатов. Высвобождение протона из
гемоглобина при оксигенации приводит к двум основным
результатам. Происходит образование НС03 из НС03 за
счет простого равновесного процесса:
НСО з + Н+ <-> НС03
Это позволяет карбоангидразе образовать С02. Следует
отметить, что субстратом карбоангидразы является НС03
(но не НС03):
НС03 <-> Н20 + С02
Разрушение НСО^ в красной кровяной клетке
обусловливает вхождение в нее НСОэ из сыворотки и выход
СГ~ по градиенту концентраций, так что в легких
происходит обратный хлоридный сдвиг, приводящий к
выведению С02 с выдыхаемым воздухом.
Небольшие количества С02 переносятся с кровью
в растворенном виде, однако большая часть (около 75%)
транспортируются в виде НСО^. Около 10-15% С02
переносит гемоглобин. С02 спонтанно взаимодействует
с незаряженными ЫН2-группами глобина с
образованием карбаминовых групп:
RNH2 + С02 <-> RNHCOOH <-> RNHCOO" + Н+
В этой реакции участвуют остатки лизина и
аргинина, боковые аминогруппы которых имеют высокие
- К ткани
Η
НЬ09
Н+
-^ Hb + 02
С02 + Н20 - Н2С03 - Н+ + НСОз
Анионный
канал
НСО,
С02 от ткани
Ка боангидраза С1
Красная кровяная клетка С1
(Хлоридный сдвиг)
Транспорт к легким
О из легочных
альвеол
Н+
НСО"
СГ
нь + о2—-ньо2 +вн+
НСО§ + Н+—^Н2СОз -Н20 + С02
СI Карбоанги аза
Красная кровяная клетка
С07
Выдыхаемый воздух
Рис. 27.15. Транспорт С02 в крови
а - Реакции в капиллярах тканей;
б - реакции в легких.
На рисунке не показан транспорт С07 в виде карбаминовых
групп гемоглобина
значения рКа и потому остаются преимущественно в
незаряженном состоянии. Доступные RNH2-rpynnbi - это
в основном концевые аминогруппы (боковые группы
лизина и аргинина имеют слишком высокую рКа, чтобы
быть незаряженными).
Поддержание рН в крови
Из всего вышеизложенного ясно, что значительные
изменения в концентрации иона водорода в крови
связаны с транспортом С02 и кислорода. При образовании
кислоты в тканях значение рН в красных кровяных
клетках должно снижаться. Буферные свойства НСО^,
фосфатов и самого гемоглобина играют существенную
роль в поддержании физиологического значения рН.
Эффект Бора, описанный выше, также играет роль
буфера. При высвобождении кислорода гемоглобин
принимает протоны (см. уравнение 1), что составляет примерно
часть 5 Транспорт кислорода и С02
376
половину ионов Н+, генерированных за счет С02 в
тканях, и это поддерживает рН в красных кровяных
клетках на уровне физиологических значений.
Серповидноклеточная анемия
Это заболевание обычно приводится в учебниках для
иллюстрации того, как замена в белке одной аминокислоты
на другую вызывает серьезные последствия. В норме
в β-субъединицах тетрамерной структуры гемоглобина
аминокислотой в шестом положении является глутами-
новая кислота, боковая группа которой заряжена
отрицательно и характеризуется высокой гидрофильностью
(см. рис. 27.10, в). В гемоглобине больных серповиднок-
леточной анемией глутаминовая кислота заменена
гидрофобным остатком валина. Кодонами в мРНК для глу-
таминовой кислоты являются триплеты GAA и GAG.
Замена в результате мутации центрального основания А
на U - это все, что требуется для замены аминокислоты,
поскольку триплеты GUА и GUG являются кодонами
валина. Обусловленное валином появление аномального
гидрофобного участка на субъединице глобина
становится причиной того, что дезоксигенированная
молекула гемоглобина связывается с гидрофобным карманом
на другой молекуле, приводя к образованию длинной
переплетенной жесткой структуры. Оксигенированный
гемоглобин, благодаря другой конформационной
структуре, препятствует этому. Длинная дезоксигемоглобиновая
структура нарушает нормальную форму эритроцитов,
превращая ее из двояковогнутого диска в серповидную,
которая способна блокировать капилляры. Такие
эритроциты преждевременно разрушаются, способствуя
развитию анемии. Если поражены обе гомологичные
хромосомы, заболевание может оказаться смертельным,
особенно при низком давлении кислорода в условиях
высокогорья, где дезоксигенация гемоглобина
необычайно высока. Заболевание широко распространено в
географических зонах, где наиболее часто встречается
злокачественная форма малярии. Высокий показатель
заболеваемости можно объяснить положительной
селекцией генома носителей аномальных генов. Серповидная
красная кровяная клетка «неудобна» для развития
малярийного плазмодия. Поскольку при малярийной
интоксикации смертность среди здоровых людей существенно
выше, чем среди страдающих гетерозиготной
серповидно-клеточной анемией, можно заключить, что последняя
повышает выживаемость людей, заболевших малярией.
Дополнительная литература
Гем и его синтез
StrakaJ. G., Rank J. M, Bloomer R. Porphyria and
porphyrin metabolism // Annu. Rev. Med. 1990. Vol.41.
P. 457-469.
(В обзоре рассмотрены нарушения ферментов
биосинтеза тема, которые ведут к порфирии, и использование
порфиринов в терапевтических целях.)
ChenJ.-J., London I. M. Regulation of protein synthesis
by heme-regulated eIF-2a kinase // Trends Biochem. Sci.
1995. Vol.20. P. 105-108.
(Подробный обзор, посвященный регуляции синтеза
гемоглобина с участием протеинкиназы eIF-2a.)
Гемоглобин и миоглобин
Branden С ToozeJ. Introduction to а protein structure.
Garland Publishing, 1991.
(Хорошо иллюстрированное описание всех аспектов
структуры белка, включая гемоглобин и миоглобин.)
Вопросы к главе 27
1. Опишите первые две стадии биосинтеза гема у
животных.
2. Каково медицинское значение регуляции синтеза
ALA-синтазы в печени?
3. Объясните, каким образом уровень железа
регулирует активность ALA-синтазы красных кровяных
клеток.
4. Сравните кривые диссоциации кислорода у мио-
глобина и гемоглобина.
5. Каков возможный механизм связывания кислорода
с гемоглобином, описываемый сигмоидной кривой?
6. Связывание молекулы кислорода с гемоглобином
вызывает конформационные изменения в белке.
Опишите механизм этих изменений.
7. Объясните, как гемоглобин плода может связываться
с кислородом, получаемым от материнского
переносчика.
8. Объясните значение хлоридного сдвига в красных
кровяных клетках.
27 Красная кровяная клетка и функции гемоглобина
Ъ11
QO Мышечное сокращение
глава ^О
Мышцы - высоко специализированные органы, в
которых приспособления для механического движения
развиты наиболее сильно. В этой главе мы рассмотрим
молекулярные основы мышечного сокращения, а в
следующей - двигательные системы немышечных клеток.
Напоминание о конформационных
перестройках в белках
Мышечное сокращение осуществляется благодаря
согласованным перемещениям белковых молекул,
возникающим в результате изменения их конформации. Эти
конформационные изменения обусловлены
взаимодействием белковых молекул с различными лигандами.
Для того чтобы «молекулярный двигатель»
осуществлял движение при помощи конформационных
изменений белка, необходима параллельная
противодействующая структура: если зафиксировать молекулу-
двигатель, то будет перемещаться молекула-партнер,
и наоборот. Прежде чем говорить о механизме
мышечного сокращения, отметим, что зафиксированным
«двигателем» является миозин, а противодействующей
движущейся структурой - актиновое волокно (филамент).
Типы мышечных клеток
и их энергообеспечение
Существуют два основных типа мышечных клеток -
гладкие и поперечно-полосатые. Первые обнаружены
в кишечнике и кровеносных сосудах; они сокращаются
непроизвольно и находятся под контролем вегетативной
нервной системы. Гладкие мышцы сокращаются
медленно, но могут поддерживать сокращение в течение
длительного времени. Свое название они получили потому,
что при микроскопическом исследовании у них не
обнаружено поперечной исчерченности (в отличие от
другого типа мышц). Поперечно-полосатые мышцы
сокращаются быстро и являются основой скелетной
мускулатуры, произвольные сокращения которой регулируются
двигательными нервами. Сердечной мышце также
свойственна исчерченность, однако, в отличие от скелетных
мышц, она сокращается непроизвольно, и структура
мышечных клеток миокарда имеет свои отличительные
особенности.
В произвольной поперечно-полосатой скелетной
мускулатуре различают два основных типа волокон -
быстро и медленно переключающиеся. Белые
поперечно-полосатые мышцы животных состоят из быстро
переключающихся волокон с недостаточным
кровоснабжением: АТР в них образуется в ходе гликолиза. Эти
мышцы осуществляют мгновенные реакции спасения
(бегство или борьбу), но из-за низкого выхода АТР при
гликолизе и накопления молочной кислоты в них
довольно скоро развивается утомление.
Медленно переключающиеся красные волокна
способны на более продолжительное сокращение. Они
обильно снабжаются кровью и характеризуются
высоким содержанием митохондрий и связанного с миогло-
бином кислорода. АТР в них образуется в процессе
окислительного фосфорилирования, которое более
эффективно, чем гликолиз, в случае образования АТР. Но
в критических ситуациях для доставки необходимых
количеств кислорода красным волокнам требуется время
и учащение частоты сердечных сокращений. Поэтому
ответ красных мышц (по сравнению с белыми)
происходит медленнее, зато они могут долго функционировать
без признаков утомления. У человека произвольные
поперечно-полосатые мышцы содержат оба типа
волокон, их соотношение зависит от конкретной роли
данной мышцы в организме. Мышцы спины должны
выдерживать напряжение, необходимое для поддержания
позы, в течение долгого времени, поэтому они содержат
главным образом медленно переключающиеся волокна.
Глазные мышцы, участвующие в движении глазного
яблока, - хороший пример быстро переключающихся
волокон.
Во всех типах мышц запаса АТР, от которого в
значительной степени зависит сокращение, хватает только
на очень короткий период интенсивного сокращения.
И здесь важную роль играет резервное макроэргическое
соединение креатин фосфат, свободная энергия
гидролиза которого (AG0') составляет -43,0 кДж · моль-1
(ср. с -30,5 кДж · моль1 для гидролиза АТР до ADP
и Pf.). Когда в процессе сокращения АТР превращается
в ADP и Р;, в ходе креатинкиназной реакции проио
ходит регенерация АТР за счет использования
фосфатных групп молекул креатинфосфата.
При сокращении:
АТР + Н20 -> ADP + Р,- .
часть б Механическая работа клеток
380
При регенерации ATP:
ADP + креатин-® -> ATP + креатин.
При расслаблении мышц АТР ресинтезируется в
процессе окислительного метаболизма, а регенерация креатин-
фосфата происходит в ходе той же креатинкиназной
реакции, которая в присутствии высокого уровня АТР
и низкого ADP легко обратима.
Как уже отмечалось, креатинфосфат (см. ниже) -
макроэргическое соединение, фосфатная группа
которого может обратимо передаваться на ADP с
образованием АТР. Из-за высокой реакционной способности
фосфатной группы возможно спонтанное
(некаталитическое) образование креатинина. Это соединение не
несет никаких функций и ежедневно выделяется с мочой
в количестве, пропорциональном мышечной массе.
О
II
NH-C
οι
ΝΗ—Р=
H2N=C
/
=0
I
°~
λΝ—СН2—СОО"
СН3
Креатинфосфат
HN=C
N СН2
I
СН3
Креатинин
Структура скелетной
поперечно-полосатой мышцы
Их мембрана - сарколемма - соединена через
нервно-мышечные синапсы с нервными окончаниями,
которые передают сигнал, запускающий сокращение.
Мембрана клетки окружает цитоплазму,
множественные ядра и многочисленные митохондрии. Цитоплазма
заполнена большим количеством идущих по всей
длине клетки сократительных нитей - миофибрилл,
каждая из которых окружена мембранными мешочками
саркоплазматического ретикулума.
Структура миофибриллы
Миофибрилла - сократительная нить, проходящая по
всей длине мышечной клетки. Она разделена на
сегменты, называемые саркомерами (см. рис. 28.2), которые
ограничены Z-дискам и (от нем. Zwischen -
находящийся между). При сокращении Z-диски сближаются, а
каждый саркомер - и следовательно, вся миофибрилла -
укорачивается. Это укорачивает все мышечное волокно,
обусловливая тем самым сокращение мышцы.
Ζ диск
Толстый филамент
Ζ диск
1 мкм
Тонкий филамент
Один саркомер -2,3 мкм
Мышца состоит из длинных многоядерных клеток
мышечных волокон (рис. 28.1).
Саркоплазматический ретикулум,
Большое количество окружающий каждую миофибриллу
миофибрилл в клетке в виде уплощенного мешочка, просвет
которого отделен от саркоплазмы
20Р 100 мкм
Плазматическая мембрана Цитоплазма Митохондрии
(сарколемма) (саркоплазма)
Рис. 28.1. Строение поперечно-полосатого мышечного
волокна
в θ е
• · сг · °
© О ' ~~) θ Θ ( ^ ® ©
β ζ^) © © С~2) о © ζ^ о
..© G (J^) Θ © (~^ © ©
Тонкие филаменты о © Толстые филаменты,
каждый из которых окружен
тонкими филаментами
Рис. 28.2. Строение толстых и тонких филаментов в
саркомере
а - Фаза расслабления. Поперечная исчерченность среза
миофибриллы при электронной микроскопии обусловлена
проходящими пучками белка;
б - фаза сокращения;
в - поперечное сечение.
На рисунке представлено только несколько волокон, хотя
в действительности каждый саркомер содержит большое
количество миофибрилл
глава 28 Мышечное сокращение
381
Как сокращается саркомер?
Z-диски - прочные белковые диски, расположенные
на каждом конце саркомера. К ним присоединены
тонкие филаменты, направленные к центру саркомера. Они
образованы белком актином и прочно соединены одним
из своих концов с Z-диском. Тонкие филаменты
инертны и служат «храповым механизмом», к которому
прикладывается сила, стягивающая Z-диски друг к другу.
В мышцах позвоночных тонкие филаменты имеют
гексагональную упаковку и прикреплены к двум дискам,
а каждый саркомер несет несколько таких
упорядоченных структур. Внутри каждой гексагональной «клетки»,
образованной шестью тонкими филаментами,
расположен толстый филамент. Он обладает пальцеобразными
выступами, которые совершают фактическую работу
сокращения. При помощи «храпового механизма» они
сдвигают тонкие волокна к центру, стягивая Z-диски
(см. рис. 28.2).
Такова общая картина мышечного сокращения.
Чтобы понять, как оно происходит, мы должны рассмотреть
участвующие в нем молекулярные структуры.
Структура и действие толстых и тонких
филаментов
Тонкие филаменты
Мономерный глобулярный белок актин называется
G-актином («G» означает глобулярный) (рис. 28.3).
В структуре его молекулы различают два домена,
соединенных узким перешейком, придающим молекуле
полярность. При определенных условиях в клетке
молекулы G-актина легко полимеризуются и образуют двуспи-
ральную структуру F-актина, в котором все молекулы
ориентированы в одном направлении - от головки
к хвосту, (см. рис. 28.3)
θ θ
Молекулы G-актина
Стрелки указывают полярность молекул
F-актин
Рис. 28.3. Схема строения актинов
а - G-актин глобулярный;
б-F-актин фибриллярный, образующийся при
полимеризации G-актина
Тонкие филаменты образованы двунитевыми
структурами, их концы обозначают знаками (+) и (-).
Толстые филаменты
Рассмотрим теперь белок миозин, каждая молекула
которого имеет стержень, образованный двумя
закрученными относительно друг друга α-спиралями, называемыми
также тяжелыми цепями миозина (рис. 28.4, а).
Структура каждой из спиралей такова, что регулярно
расположенные остатки гидрофобных аминокислот
участвуют в гидрофобном взаимодействии с другой α-спиралью,
образуя жесткую палочкообразную структуру. Каждая
цепь оканчивается глобулярной головкой, к которой
присоединены две небольших полипептидных цепи,
называемые легкими цепями миозина.
Толстый филамент состоит из нескольких тысяч
молекул миозина, организованных биполярно, как
показано на рис. 28.4, б. Толстые филаменты занимают
центральное положение относительно окружающих их
тонких филаментов при помощи других белков, входящих
в состав саркомера. Один из них - титин, названный
так из-за огромной (англ. titanic) молекулярной массы.
Такое центральное положение обеспечивает
оптимальное расстояние, позволяющее головкам миозина
контактировать с актиновыми филаментами.
Легкие цепи
Промежуточная зона
Головки миозина
Рис. 28.4. Организация толстого филамента
а - Строение молекулы миозина, состоящей из двух тяжелых
цепей, каждая из которых заканчивается миозиновой
головкой. К ним присоединены две различных легких цепи;
б - толстый филамент, образованный из молекул миозина,
соединенных биполярным способом
Тонкие филаменты заякорены на Z-дисках своими
(+)-концами так, что головки миозина на обоих концах
толстого филамента одинаково ориентированы по
отношению к «полюсам» тонких филаментов (рис. 28.5).
часть 6 Механическая работа клеток
382
Z-диск
Полярность тонких филаментов
Z-диск
\ \
Плюс-конец Головка
актинового миозина
филамента на Z-диске
Толстый филамент
(миозин)
Тонкий филамент
(актин)
Рис. 28.5. Организация толстых и тонких филаментов в саркомере
Стрелки указывают полярность филаментов актина. При сокращении головки миозина движутся вдоль актиновых филаментов
по направлению к их (+)-концам: при этом два Z-диска подтягиваются друг к другу
Как головка миозина
преобразует энергию гидролиза АТР
в механическую работу актиновых
филаментов?
Головка миозина обладает АТРазной активностью,
и в ходе гидролиза АТР до ADP и Р/ в ней происходят
конформационные изменения. Без соответствующего
разъяснения это наверняка вызовет замешательство.
Ведь до сих пор мы рассматривали случаи, когда
расщепление АТР использовалось для совершения работы,
и было бы естественно предположить, что сокращение
мышцы происходит за счет гидролиза АТР до ADP и РР,.
В АТР-зависимых процессах, которые были
рассмотрены ранее, шел непрямой гидролиз АТР: сначала фосфо-
рильная группа или AMP переносилась к реагирующей
молекуле, с последующим высвобождением либо РР/5
либо AMP, т. е. использовались сопряженные реакции.
При мышечном сокращении механическая работа
осуществляется под действием прямого гидролиза АТР
(без ковалентных интермедиатов). Однако фактически
«силовой удар» при сокращении происходит не при
гидролизе АТР, как этого можно было ожидать, а после
отделения ADP от белка. Именно когда ADP покидает
белок, происходит основное выделение свободной энергии,
так что «силовой удар» при мышечном сокращении
приходится не на гидролиз АТР, а на освобождение ADP от
головки миозина. В то же время в ходе превращения
комплекса белок-ATY* в комплекс белок-ADP плюс Р.,
изменение свободной энергии невелико. В растворе же
превращение свободного АТР в свободные ADP и Pf
характеризуется большим отрицательным значением
AG°\ так что общая энергетическая концепция
гидролиза АТР как движущей силы остается неизменной.
Головка миозина может находиться в напряженной
конформации (готовой к совершению усилия) и
расслабленной конформации (после совершенного
усилия). Головка присоединяется к актиновому фила-
менту в напряженной конформации и затем наносит
«силовой удар» на актин (рис. 28.6), после чего
принимает расслабленную конформацию.
Актиновый филамент,
движущийся
к центру саркомера -
Силовой
удар
Головка миозина, присоединенная
к актиновому филаменту
в «напряженной» конфигурации
Головка миозина
изменяет свою
конформацию
Рис 28.6. Сокращение скелетной мышцы (упрощенное
изображение)
Теперь мы более подробно рассмотрим этапы этого
цикла, начинающегося в точке, когда головка миозина
только что осуществила «силовой удар». Головка
присоединена к актину в расслабленной конформации. Потом
события развиваются как показано на рис. 28.7.
глава 28 Мышечное сокращение
383
Актиновый филамент
Ύσχ
©
-г*
АТР
Миозин
отделяется
Головка
миозина
\©
Φ
Н20
Миозин связывается
в «напряженной» конфигурации
ΔΤΡ '
М| г Гидролиз АТР
Головка принимает
первоначальную
конфигурацию
ji присоединяется к актину
X^V'v'VV
(D
дрр СИЛОВОЙ
удар
Сила, действующая
на актиновый филамент
Рис. 28.7. Сокращение скелетной мышцы (упрощенное изображение)
Номера соответствуют этапам, указанным в тексте. Головка миозина только что осуществила «силовой удар» по тонкому фила-
менту актина. Обратите внимание на то, что «силовой удар» связан с высвобождением ADP с головки миозина
1. АТР присоединяется к головке миозина и вызывает
ее отделение от актина.
2. АТР гидролизуется до ADP и Р,.; после
освобождения Pf. головка миозина переходит в напряженную
конформацию и присоединяется к актину.
3. Головка миозина наносит «силовой удар», связанный
с высвобождением ADP. Это вызывает конформаци-
онные изменения головки, которая поворачивается
в шарнирной области.
4. Головка миозина в расслабленной конформации
связана с актином до тех пор, пока ее не высвободит
поступающая молекула АТР, которая и обеспечивает
возврат в первоначальное состояние.
В мышечном сокращении участвует большое число
молекул миозина. Если АТР недоступен, все головки
миозина «сшиты» поперечными связями с актином
(поскольку не происходит первого этапа цикла). Это
приводит к образованию жесткой структуры мышц
(состояние трупного окоченения), которое начинается при
истощении запасов АТР. Способность головки миозина
осуществить весь рассмотренный процесс была
продемонстрирована in vitro в экспериментах с шариками
латекса, покрытыми изолированными головками миозина,
которые буквально «ходили» по иммобилизованным
волокнам актина в присутствии АТР и Са2+.
Как регулируется сокращение
произвольных поперечно-полосатых
мышц?
Сокращение скелетных мышц инициируется нервным
импульсом, вызывающим высвобождение ионов Са2+
из саркоплазматического ретикулума в миофибриллы.
Ионы Са2+ быстро удаляются из миофибриллы (см.
ниже), в результате чего сокращение прекращается. До тех
пор, пока под действием продолжающегося нервного
импульса не высвободится большое количество ионов Са2+,
сокращение не произойдет.
Как Са2+ вызывает сокращение?
Основным белком тонких филаментов является актин,
но с ними связаны и другие белковые молекулы. Одна
из них - тропомиозин - состоит из двух субъединиц.
Это довольно небольшая, вытянутая молекула, которая
расположена в бороздке, образуемой двумя актиновыми
нитями тонкого филамента. На 1 молекулу тропомиозина
приходится примерно 7 молекул актина. Молекулы двух
белков последовательно накладываются друг на друга,
часть 6 Механическая работа клеток
384
формируя непрерывную нить. Тонкий филамент имеет
две бороздки, и молекулы тропомиозина располагаются
в каждой из них (рис. 28.8 ).
Нити тропомиозина Тропониновый комплекс,
образованы из мономеров; присоединенный к концу
каждый мономер соединен каждого мономера
с семью мономерами актина тропомиозина
Актиновые филаменты
Рис. 28.8. Взаиморасположение молекул актина и
тропомиозина
Каждая молекула тропомиозина связывает 7 мономеров
актина, формируя протяженную нить в бороздках актинового фи-
ламента. К одному из концов каждой молекулы
тропомиозина присоединен тропониновый комплекс
К одному из концов молекулы тропомиозина
присоединен тропонин - комплекс, состоящий из трех
глобулярных белков. Тропонин-тропомиозиновый
комплекс чувствителен к ионам Са2+, которые
вызывают небольшое перемещение нити тропомиозина
относительно тонкого филамента, что позволяет головке
миозина присоединиться к актиновому волокну. Тропо-
миозин, как считалось, блокирует присоединение
миозина к актину, в то же время другие наблюдения
свидетельствуют о том, что Са2+ не влияет на связывание
миозина с тонким филаментом. Известно, впрочем, что
пока миозин-актиновый силовой цикл не включается,
нет ионов Са2+.
Не вдаваясь в подробности, необходимо отметить,
что ионы Са2+ связываются с тропонином, вызывая
конформационные изменения в молекуле
тропомиозина, который каким-то образом активирует
миозин-актиновый силовой цикл, приводящий к гидролизу АТР
и мышечному сокращению. Удаление ионов Са2+
обращает вспять всю последовательность событий и
прекращает сокращение: это приводит нас к механизму
регуляции выхода и удаления ионов Са2+.
Транспорт ионов Са2+ в мышце
Каждая миофибрилла мышечной клетки окружена
мембранным мешочком саркоплазматического ретикулума,
Са2+-АТРаза которого перекачивает ионы Са2+ из ци-
тозоля, окружающего миофибриллу, в просвет
ретикулума (АТР-зависимым способом против градиента
концентрации). Этот механизм поддерживает низкую
концентрацию Са2+ в миофибрилле« предотвращает
мышечное сокращение. В регуляции содержания
Са2+участвует также белок мембраны саркоплазматического
ретикулума, функционирующий в качестве канала для
пассивного транспорта ионов Са2+. Обычно он
закрыт, но при поступлении к мышечной клетке нервного
импульса открывается и впускает ионы Са2+ из
просвета ретикулума в миофибриллу, вызывая сокращение.
Для длительного мышечного сокращения все сар-
комеры миофибриллы и все миофибриллы в мышце
должны отвечать на импульс двигательного нерва
одновременно, иначе сокращение окажется
некоординированным и малоэффективным. Нервный импульс
вызывает освобождение ацетилхолина в
нервно-мышечном синапсе, а это, в свою очередь, вызывает локальную
деполяризацию плазматической мембраны, быстро
распространяющуюся по всей мембране (см. с. 358). В
процессе деполяризации происходит открытие
потенциал-зависимых Са2+-каналов саркоплазматического
ретикулума и выход иона к миофибрилле. Для обеспечения
быстрой доставки сигнала ко всему саркоплазматичес-
кому ретикулуму клетки плазматическая мембрана
имеет впячивания (инвагинации) в виде поперечных (Т)
трубочек. Последние проникают в миофибриллу в области
Z-диска и вступают в прямой контакт с мембраной
саркоплазматического ретикулума, позволяя таким образом
электрическому сигналу практически мгновенно
достигать всех сократительных единиц, управляемых этим
нервным импульсом (рис. 28.9).
Чем гладкая мышца отличается
по структуре и регуляции
от поперечно-полосатой?
Гладкие мышцы обнаружены в стенках кровеносных
сосудов, в кишечнике, мочеполовой и других системах.
Их длинные веретенообразные клетки имеют одно ядро
и объединяются, образуя мышцы, чья форма
соответствует выполняемым функциям (например,
кольцевидная в кровеносных сосудах или сетевидная в мочевом
пузыре).
Основные принципы сокращения те же, что и в
поперечно-полосатой мышце, где молекулы миозина
осуществляют силовое воздействие на актиновый
филамент, используя в качестве источника энергии гидролиз
АТР и аналогичный «силовой цикл». Однако
сократительные компоненты в гладких мышцах не так высоко
глава 28 Мышечное сокращение
385
Аксон нерва -
Сарколемма удалена
..для демонстрации
миофибрилл внутри
клетки
Миофибриллы
Нервно-мышечный
синапс
Миофибрилла ···:
Z-диск·
■ Сарколемма
Т-трубочка
Саркоплазматический ретикулум
удален для показа
окружающих его миофибрил
Рис. 28.9. Нервно-мышечный синапс
а - Плазматическая мембрана (сарколемма) мышечного волокна, образующая нервно-мышечный синапс;
б - схема поперечных (Т) трубочек, которые передают сигнал о деполяризации плазматической мембраны к саркоплазматичес-
кому ретикулуму (СР).
Считается, что деполяризация плазматической мембраны непосредственно передается на Са24-каналы СР, вызывая тем самым
быстрое высвобождение из канала Са2+
организованы. Нет миофибрилл и повторяющихся сар-
комеров, поэтому при микроскопии отсутствует
поперечная исчерченность. Вместо саркомерной
структуры волокна актина проходят по всей длине клетки
(маленькой по сравнению с клеткой
поперечно-полосатой мышцы) и одним концом прикреплены к клеточной
мембране.
Регуляция сокращения
гладкой мышцы
Хотя ионы Са2+ и отвечают за инициацию
сокращения, его регуляторный механизм не такой, как в
поперечно-полосатой мышце. Саркоплазматический
ретикулум отсутствует. Гладкая мышца сокращается гораздо
медленнее (в 50 раз), чем поперечно-полосатая (около
5 секунд). От гладкой мышцы не требуется почти
мгновенного сокращения всех структур, как в случае
поперечно-полосатой: сокращение отдельных клеток
распространяется в более медленном темпе. Нервный
импульс вегетативной нервной системы открывает
Са2+-канал в мембране гладкомышечных клеток и
вызывает вход ионов Са2+ внутрь клетки из внеклеточной
среды. Специальные соединения между клетками
позволяют нервному сигналу распространяться от клетки
к клетке по всей мышце.
Как Са2+ регулирует сокращение гладкой
мышцы?
Как уже говорилось, в головке каждой молекулы
миозина расположены два небольших полипептида - легкие
цепи миозина. В гладкой мышце одна из этих легких
цепей (р-легкая цепь) ингибирует связывание головки
миозина с актиновым филаментом и таким образом
предотвращает сокращение. Са2+ активирует киназу
легких цепей миозина, которая в присутствии АТР фос-
форилирует р-легкую цепь и снимает ингибирование,
вызывая таким образом сокращение.
часть 6 Механическая работа клеток
386
Ионы Са2+ активируют киназу опосредственно; они
соединяются с белком кальмодулином (см. с. 353),
вызывая в нем конформационные изменения, в результате
которых комплекс Съ2+-кальмодулин присоединяется
к неактивной киназе и активирует ее. При понижении
уровня ионов Са2+ процесс обращается, а фосфатаза
дефосфорилирует легкую цепь миозина, вызывая
расслабление мышцы. Общая схема регуляции
представлена на рис. 28.10.
Помимо нервной регуляции, в регуляции
сокращения гладких мышц принимают участие некоторые
гормоны, например норадреналин, вызывающий
сокращение определенных мышц кровеносных сосудов.
КМ
Кальмодулин
Са2+
-Са2+
Комплекс
Са -кальмодулин
Неактивная
КЛЦМ
Киназа легких
цепей миозина
(КЛЦМ)
Са2+
КМ
Активная
КЛЦМ
АТР
Неактивный
миозин
ADP>
Легкая цепь миозина-Гр)
Активный миозин
Сокращение
Рис. 28.10 Механизм активации сокращения гладкой
мышцы ионами Са7"
Дополнительная литература
BrayD. В. Cell movements. Garland Publishing, 1992.
(Интересная, хорошо иллюстрированная схемами и элек-
тронномикроскопическими фотографиями книга,
содержащая сообщения о всех аспектах движения в клетках.)
Мышечное сокращение
CookeR. В. The actomyosin engine// FASEB J. 1995.
Vol. 9. P. 636-642.
(В работе рассматриваются структуры головки миозина
и актиновых филаментов, а также способ генерации
механической силы в процессе мышечного сокращения.)
Сокращение гладких мышц
Small J. V. Structure-function relationships in smooth
muscle: the missing links// BioEssays. 1995. Vol. 17.
P. 785-792.
(Обсуждаются структурная организация гладкомышеч-
ной клетки и регуляция ее сокращения.)
Вопросы к главе 28
1. Каковы различия между быстро и медленно
сокращающимися мышечными волокнами? Каково их
биологическое значение?
2. Изобразите строение миофибриллы, саркоплазма-
тического ретикулума и саркомера.
3. Объясните, каким образом головка миозина
преобразует энергию гидролиза АТР в механическую
энергию.
4. Как регулируется сокращение саркомера
произвольной поперечно-полосатой мышцы?
5. Как регулируется сокращение гладкой мышцы?
глава 28 Мышечное сокращение
387
QQ Роль цитоскелета в поддержании формы
глава £ЛЭ клеток и осуществлении механической
работы
На первый взгляд кажется, что немышечные клетки
механической работы не совершают. Также легко
представить животную клетку в виде бесформенного мешка,
наполненного протоплазмой. Но и это неправильно,
поскольку у клеток есть цитоскелет (комплекс
внутренних перегородок), который принимает участие в
перемещении внутриклеточных структур, движении клеток
и поддержании их формы.
В первом приближении необходимость этих видов
механической работы не столь очевидна. Но в таком
случае животная клетка без плотной клеточной стенки
была бы маленьким аморфным образованием. Форма
большинства типов животных клеток индивидуальна,
и в ее поддержании важную роль играют
внутриклеточные структуры. Например, форма уплощенного
двояковогнутого диска эритроцита поддерживается при
помощи специальных белковых перегородок,
взаимодействующих с мембраной.
Многие клетки постоянно изменяют свою форму,
поэтому цитоскелет, определяющий ее, все время
изменяется. Такая структура цитоскелета, отличающаяся
от структуры, свойственной эритроцитам или
микроворсинкам кишечника (см. рис. 4.1), характерна для
животных клеток. Растительные и бактериальные клетки
с прочными клеточными стенками относятся к другой
категории.
Подвижность - неотъемлемое свойство животных
клеток; помимо наиболее ярких примеров амебоидного
движения макрофагов и других лейкоцитов,
мигрирующих в ткани, многие клетки передвигаются по
поверхности ткани, что важно в ходе эмбрионального
развития и при нормальном заживлении ран. Другой тип
активного (немышечного) сокращения наблюдается
в процессе деления клетки. Тогда вокруг её
экваториальной зоны образуется перетяжка, которая приводит
к разделению клетки. Еще одна форма движения -
пульсация микроворсинок, способствующая перемещению
поверхностных слоев слизи, например в дыхательных
путях, или биение жгутиков сперматозоидов.
Эукариотическая клетка - очень большой объект,
если соотносить ее размеры с низкой скоростью
диффузии веществ в растворе. Бактериальная клетка намного
меньше и поэтому «полагается» на диффузию
растворенных веществ, в то время как многие эукариотические
клетки нуждаются в сложной внутренней транспортной
системе. Пузырьки аппарата Гольджи должны
перемещаться к поверхности клетки или в другие места
назначения. Движение молекул мРНК от ядра в цитоплазму
также происходит по строго заданному маршруту.
Другим ярким примером служит аксон нерва, который
может достигать более полуметра в длину; синтез белков
и формирование везикул происходит в теле клетки, и
некоторые из этих компонентов должны быть доставлены
к окончанию аксона. При помощи диффузии такое
сделать невозможно! Гигантская клетка водоросли Nitella
настолько велика, что диффузия не смогла бы
обеспечить ее существование; поэтому клетка поддерживает
постоянные транспортные потоки веществ в
цитоплазме. Еще одним примером внутриклеточного транспорта
может служить расхождение хромосом в дочерние
клетки в процессе клеточного деления.
Суммируя все вышесказанное, можно
констатировать, что клетки имеют сложную внутреннюю
организацию, обеспечивающую поддержание формы,
процессы движения и внутриклеточного транспорта. Эта
внутренняя организация менее выражена, чем
сократительный аппарат мышечных клеток, поскольку: 1)
структуры, осуществляющие перечисленные виды работ,
выявляются только при определенном окрашивании
клеток; 2) многие из них постоянно модифицируются в
зависимости от сиюминутных потребностей клетки.
Прежде чем мы перейдем к детальному
рассмотрению роли цитоскелета в определении формы клеток
и в производстве ею механической работы, стоит
отметить ряд важных положений.
• Актиновые филаменты есть практически во всех
клетках; они участвуют в определении формы
животных клеток и в осуществлении амебоидных и
«ползающих» движений.
• Актиновые филаменты играют также роль
внутриклеточных «железнодорожных путей», по которым
интенсивно перемещаются составные части клетки.
• Μиозиноподобные молекулы существуют и в
немышечных клетках. Один тип молекул миозина
(аналогичный мышечному миозину) осуществляет
силовое воздействие на расположенные рядом актиновые
филаменты, обеспечивая движение клетки. Другие
типы молекул миозина (минимиозины),
расположенные вдоль актиновых транспортных путей,
перемещают различные внутриклеточные «грузы».
часть 6 Механическая работа клеток
388
Источником энергии служит АТР.
• Особая транспортная система существует в виде
микротрубочек, также играющих роль
транспортных путей, по которым перемещаются молекулярные
«двигатели», тянущие за собой внутриклеточный
«груз». Используя энергию АТР, такой «двигатель»
воздействует на соседние микротрубочки, производя
скользящие движения и выполняя таким образом
механическую работу, как и актомиозиновая система.
• Третий тип компонентов цитоскелета представлен
промежуточными филаментами (они имеют
промежуточные размеры по сравнению с размерами
филаментов актина и микротрубочек). Одни из них
(ламины) формируют сеть, которая является
составной частью ядерной оболочки. Промежуточные фи-
ламенты придают прочность клеткам эпидермиса,
а кератиновые промежуточные филаменты -
клеткам волос.
После этого краткого введения рассмотрим
компоненты цитоскелета более подробно.
Роль актина и миозина
в немышечных клетках
Щеточная каемка,
образованная
микроворсинками
Эпителиальная
клетка кишечника
...;.-.-- Микроворсинки
Актиновые филаменты,
проходящие вниз в клетку
Актин относится к числу простых белков, наиболее
широко представленных в большинстве эукариотических
клеток. Актины различных типов клеток сходны между
собой: это высококонсервативные белки, образующие
филаменты по типу описанных для мышечной клетки
(см. главу 28). Миозин также является универсальным
компонентом эукариотических клеток, хотя и
присутствует в них в гораздо меньших количествах.
Преобладание актина над миозином в немышечных клетках
связано с тем, что помимо участия в сокращении актин
играет также структурную роль
Структурное значение актина и его роль
в движении клеток
Наиболее наглядным примером структурной роли
актина может служить строение микроворсинок клеток
щеточной каемки, пальцевидные выступы которой
значительно увеличивают адсорбционную поверхность
кишечника (рис. 29.1). Актиновые филаменты
пронизывают большинство животных клеток. Особенно плотно
они располагаются около цитоплазматической
мембраны, в которой закреплены пучки актиновых
филаментов, участвующие в поддержании формы клетки
(рис. 29.2, а). Упорядоченность актиновых филаментов
Рис. 29.1. Строение актиновых филаментов микроворсинки
Формирование прочной структуры цитоскелета обусловлено
взаимодействием актиновых филаментов с другими белками
с образованием поперечных сшивок
и образование сетей зависят от множества других
белков, связанных с актином. Актиновые филаменты
микроворсинок - структуры постоянные, однако и они
способны к разборке и самосборке. Когда фагоцит движется
в сторону поглощаемой частицы, можно видеть, как
актиновые филаменты продолжаются в псевдоподии
(«лопастеобразные» выросты клетки, вытягивающиеся
в направлении движения).
Актиновые волокна участвуют в амебоидных и
«ползающих» движениях клеток. Это результат действия
миозина II (обычных молекул миозина мышечного
типа). Приблизительно 16 таких молекул при
необходимости собираются в небольшие биполярные
волокна, подобные толстым филаментам
поперечно-полосатой мышцы. Их действие очень похоже на действие
саркомера: биполярные волокна развивают
сократительное усилие на соседние актиновые филаменты и
вызывают натяжение мембраны клетки, к которой
прикреплены филаменты (рис. 29.3). Регуляция сокращения
глава 29 Роль цитоскелета в поддержании формы клеток и осуществлении механической работы
389
Рис. 29.2. Микрофотографии актиновых волокон и
микротрубочек
а - Актиновые волокна миобластов мыши, визуализированные
при помощи антител к актину. Волокна пронизывают клетку
насквозь и их пучки (волокна напряжения) присоединены в
фокальных точках к мембране клетки. Волокна быстро
разбираются и собираются. Фотография любезно предоставлена
P. Gunning, Children's Medical Research Institute, Sydney;
б - микротрубочки в цитоплазме клетки, исходящие из центра
организации микротрубочек или центросомы
(как и в гладкой мышце) связана с фосфорилировани-
ем легких цепей миозина (см. главу 28).
Довольно точно определена сократительная роль акто-
миозиновой системы в цитокинезе - разделении
цитоплазмы в процессе деления клетки. Во время анафазы
кольцо актиновых филаментов собирается в
экваториальной плоскости, где и происходит разделение.
Считается, что актиновые филаменты заякорены в
плазматической мембране и перекрывание этих волокон
(с противоположной полярностью) дает возможность
сокращения цитоплазматическим миозинам. По мере
развития сокращения это образование распадается,
и цитоплазма распределяется между двумя дочерними
клетками. Такая система служит превосходным
примером временной организации сократительных
комплексов, входящих в структуру немышечных клеток.
Роль актина и миозина
во внутриклеточном транспорте
веществ
Актиновые филаменты могут служить транспортными
путями, по которым АТР-зависимые молекулярные
двигатели перемещают клеточные компоненты. В главе 28
упоминалось, что если в эксперименте отдельные
головки миозина (мышечного типа) прикрепить к шарикам
латекса, то в присутствии АТР и Са2+ они будут
двигаться по иммобилизованным актиновым волокнам. «Хвост»
молекулы миозина мышечного типа устроен таким
образом, что обеспечивает самосборку миозина в
биполярный (толстый) филамент, который способствует
скольжению соседних актиновых волокон, вызывая таким
образом сокращение.
Существует семейство молекул миозина с
обычными миозиновыми головками, но вместо длинного
палочкообразного хвоста, свойственного мышечному
миозину, у них хвост маленький, отсюда и произошло их
название - минимиозины. Молекулы минимиозина
не могут формировать биполярные пучки, но за счет
энергии АТР они перемещаются по актиновому фила-
менту. Хвосты минимиозинов предназначены для
присоединения к другим структурам, например к
мембранам везикул: если такое присоединение происходит,
минимиозин будет тянуть везикулу по актиновому фила-
менту. В гигантской водоросли Nitella минимиозины
тянут эндоплазматический ретикулум по неподвижным
актиновым филаментам через всю клетку. Хвосты
минимиозина присоединены к эндоплазматическому ретику-
луму, пронизывающему цитоплазму. Они подтягивают
его, вызывая непрерывную циркуляцию всего
содержимого цитоплазмы.
Идентифицировано множество необычных
миозинов (немышечного типа). Они обнаружены как у
дрожжей, так и у позвоночных. У всех миозинов
немышечного типа есть глобулярные актин-связывающие головки
и различные хвосты, которые взаимодействуют с
определенными мембранными структурами.
часть 6 Механическая работа клеток
390
Биполярное соединение молекул
миозина II в пучок толстого филамента
Приложенная сила
Актиновый филамент
Приложенная сила
Рис. 29.3. Тянущее действие миозина II на актиновые филаменты
Энергию процесса обеспечивает гидролиз АТР. Выполнение системой полезной работы будет зависеть от соответствующего
прикрепления актинового филамента к клеточной мембране для изменения формы клетки. Каждая молекула миозина имеет
участок, способный к взаимодействию с другими молекулами, который определяет самосборку биполярного волокна. На схеме
представлено продольное сечение - биполярные комплексы имеют цилиндрическую форму. Сокращение обусловлено
скольжением актиновых филаментов относительно пучков миозина
Микротрубочки, клеточное
движение и внутриклеточный
транспорт
Что такое микротрубочки?
Микротрубочки образуются при полимеризации белка
тубулина, который является гетеродимером,
образованным субъединицами а- и β-тубулина. В процессе
полимеризации сс-тубулин одного димера контактирует с β-ту-
булином следующего с образованием протофиламентов,
13 продольных рядов которых и формируют полую
трубку. Полимеризация происходит в направлении от
головы к хвосту таким образом, что микротрубочка имеет
определенную полярность; ее концы обозначаются
соответственно как (+)- и (-)-концы. Микротрубочки
в клетке нестабильны, они могут быстро собираться
и разбираться. Нестабильность обусловлена
незащищенными концами, которые способствуют быстрому
разрушению микротрубочек путем деполимеризации на
свободные субъединицы тубулина. В клетке (-)-концы
связаны с центром организации микротрубочек
(ЦОМТ) - структурой, расположенной около ядра,
которая содержит (в животных клетках) пару маленьких
телец - центриолей - образованных из слившихся
микротрубочек. Клетка пронизана микротрубочками,
исходящими из ЦОМТ (см. рис. 29.2, б и 29.4).
Что защищает (+)-концы микротрубочек?
Как уже отмечалось, микротрубочки быстро
распадаются, если их концы не защищены. ЦОМТ защищает
(-)-конец. В клетке микротрубочки растут (и
распадаются) с (+)-концов. При достижении микротрубочкой
щент
;ство
+
риолярное^ ^ ι
*
ι " С
1
\
т rfc Микротрубочки
- \ ♦
и --...- *
- - *"%% Центриоли
Цитоплазма
+ +
Рис. 29.4. Микротрубочки, исходящие из ЦОМТ
Знаки (+) и (-) указывают полярность микротрубочек.
Центриоли - пара трубчатых структур, построенных из
соединенных микротрубочек; расхождение микротрубочек происходит
в веществе, окружающем центриоли
соответствующей мишени белки последней защищают
ее. Микротрубочки могут расти из ЦОМТ в
совершенно случайных направлениях, но лишь те из них, что
вступят в контакт с соответствующим компонентом клетки,
будут защищены и стабилизированы
белками-мишенями, в то время как остальные разрушатся. До сих пор
остается неясным, что же защищает от распада
растущую микротрубочку, прежде чем она достигнет белковой
мишени. Согласно современной модели, субъединица
тубулина связывает GTP и в виде такого комплекса
защищает (+)-конец. После ее присоединения к трубочке
происходит медленный гидролиз GTP до GDP и Р,. Таким
образом, вновь добавленные субъединицы тубулина в
течение короткого времени будут находиться в комплексе
с GTP, что временно защищает конец микротрубочки
глава 29 Роль цитоскелета в поддержании формы клеток и осуществлении механической работы
391
от разрушения. Гидролиз GTP играет роль уже
знакомого нам «молекулярного хрономегражного прибора»: если
добавление новых молекул тубулина происходит до
того, как последняя присоединенная субъединица
потеряет GTP, микротрубочка будет защищена. Если же GTP
на свободном конце гидролизуется раньше, то, согласно
этой модели, распад микротрубочки неизбежен. В
ресничках и жгутиках, где микротрубочки являются
постоянной структурой (см. ниже), после их сборки
происходит ковалентная модификация белка, препятствующая
распаду. Такова основная схема строения
микротрубочек. Теперь рассмотрим их функции.
Функции микротрубочек
Как и актиновые филаменты, микротрубочки участвуют
в поддержании формы клетки, ее движении и
внутриклеточном транспорте.
Молекулярные двигатели,
обеспечивающие движение
по микротрубочкам
Идентифицированы два типа АТР-зависимых
двигателей - кинезин и динеин. Кинезин перемещается
по микротрубочке от (-)- к (+)-концу, а динеин - в
противоположном направлении. Каждый из них имеет две
глобулярные головки, напоминающие миозиновые
(рис. 29.5, а), с легкими цепями, направленными в
противоположные стороны.
Существуют семейства молекул кинезина и динеина,
специализирующиеся на выполнении различных
функций в разных тканях. Головки, совершающие
движение по микротрубочке, вероятно, не изменяются, а вот
структура хвоста модифицируется для соединения с
различными структурами клетки (рис. 29.5, б). В качестве
примера работы молекулярных двигателей можно
рассмотреть перемещение везикулы по микротрубочке (см.
ниже). В присутствии АТР молекулы кинезина и
динеина (обладающие АТРазной активностью) будут
перемещаться по иммобилизованным на твердой поверхности
волокнам микротрубочек точно также, как головки
миозина передвигаются по иммобилизованным актиновым
филам ентам.
Роль микротрубочек в движении клеток
Микротрубочки играют важную роль в движении
ресничек и жгутиков. Клетки, выстилающие дыхательные
ходы, имеют большое количество ресничек,
пульсирующее движение которых выводит слизь и удаляет
чужеродные частицы. Сперматозоид перемещается
при помощи вращения жгутика. Реснички и жгутики
часть б Механическая работа клеток
392
а
..--'Легкие
цепи
б f ^\
I -I—- Везикула
(+Г (-)
D
Микротрубочка
Рис. 29.5. Строение и функция кинезина
а - Строение молекулы кинезина;
б - транспортировка везикулы по микротрубочке молекулой
кинезина (схематично)
по структуре похожи: они берут начало из базальных
телец, напоминающих центриоль, и микротрубочки
проходят вниз по всей их длине; к микротрубочкам
присоединены молекулы динеина, которые служат
двигателями. Используя энергию АТР, они перемещаются
по микротрубочкам по направлению к (-)-концу,
вызывая смещение последних относительно друг друга
и приводя к волнообразным движениям ресничек или
жгутиков.
Роль микротрубочек в транспорте
везикул внутри клетки
Микротрубочки участвуют во внутриклеточном
транспорте. Пузырьки, образуемые аппаратом Гольджи,
направляются в различные места клетки строго по
назначению. Детали такого запрограммированного движения
и перемещения пузырьков неизвестны, но,
по-видимому, в нем участвуют микротрубочки и АТР-зависимые
двигатели.
Как уже упоминалось, аксон нервной клетки бывает
настолько длинным, что его окончание может
находиться на значительном расстоянии от тела клетки, где
происходит образование везикул и синтез белков. Для их
транспорта по аксону вытягиваются микротрубочки,
ориентированные (+)-концом к окончанию
Молекулярный двигатель кинезин одним концом
присоединяется к мембране пузырька и передвигается по
микротрубочке в направлении к (+)-концу (см. рис. 29.5, б).
Другой двигатель - цитоплазматическийдинеин -
осуществляет транспорт в обратном направлении от
аксона к телу клетки. Наглядным примером движения
по микротрубочкам может служить транспорт
пигментных везикул (рис. 29.6).
..
Рис. 29.6 Транспорт пузырьков по микротрубочкам
Микрофотография получена при электронном сканировании
транспортируемых по микротрубочкам пузырьков,
содержащих пигмент в хроматофоре «рыбы-белки». Изменение цвета
клетки вызвано перемещением пузырьков к центру клетки
и обратно. Mts - микротрубочки; РМ - плазматическая
мембрана. (Размер шкалы = 0,5мкм)
Роль микротрубочек в митозе
Стадии митотического цикла клетки показаны на
рис. 29.7, а.
В профазе центросомы (то же, что и ЦОМТ в неде-
лящихся клетках) удваиваются и расходятся к
противоположным полюсам клетки; из них выходят
микротрубочки, образуя веретено деления. ДНК уплотняется,
формируя набор видимых в световой микроскоп
хромосом. В прометафазе ядерная мембрана исчезает,
микротрубочки веретена входят в ядерную область и
присоединяются к центромерам хроматид. Они
представляют собой «сестринские» хромосомы, сформированные
при репликации ДНК в интерфазе, которые связаны
в центральных областях, образуя Х-образные структуры
(см. рис. 19.9). После разделения каждая хроматида
станет хромосомой. Хроматиды в метафазе располагаются
в экваториальной плоскости, готовясь к анафазе. Затем
хроматиды разделяются и двигаются к полюсам
(анафаза А), а те удаляются друг от друга, расходясь в
разные стороны (анафаза В). В телофазе происходит
восстановление ядерной мембраны. Это сопровождается
глава 29 Роль цитоскелета в поддержан
разделением клетки путем расхождения цитоплазмы
{цитокинез), которое осуществляется актомиозиновой
системой. Затем в каждой дочерней клетке центросома
становится центром образования множества
микротрубочек, характерных для интерфазной клетки.
Механизм перемещения хромосом
(анафаза А)
До сих пор неизвестно, что вызывает перемещение
хромосом к полюсам клетки после их присоединения
к волокнам кинетохора (рис. 29.7, б), однако ясно, что
эту работу может совершать молекулярный двигатель
типа динеина. В процессе движения хромосом волокна
кинетохора укорачиваются. Микротрубочки не
способны к сокращению, и поэтому предполагается, что
хромосома перемещается по волокну, а микротрубочки
подвергаются деполимеризации после ее прохождения.
Механизм разделения полюсов
(анафаза В)
В этом процессе участвуют полярные волокна (см.
рис. 29.7, а), которые не присоединяются к хромосомам,
а перекрываются в экваториальной области. Поскольку
полюса перемещаются обособленно, волокна
удлиняются, чтобы сохранить это перекрывание. Его наличие
позволяет предположить, что в расхождении полюсов
участвуют молекулярные двигатели, с которыми связаны
астральные волокна, выходящие из полюсов.
Независимо от типа используемого механизма, установлено,
что разделение полюсов нуждается в АТР.
Промежуточные филаменты
Описанная выше актиновая сеть содержит микрофила-
менты диаметром 6 нм; диаметр микротрубочек 20 нм.
Третий тип сети в клетках эукариот образован филамен-
тами с диаметром в среднем 10 нм, поэтому их
называют промежуточными филаментами (ПФ).
Существует несколько типов ПФ, образованных
различными группами белков, которые характеризуются
высокой гомологичностью. Обычно они имеют
стержневой филамент, содержащий приблизительно 350
аминокислотных остатков с изменяющимися у разных типов
ПФ-концами. Это разнообразие белков контрастирует
с однотипными белковыми субъединицами актиновых
филаментов и микротрубочек.
Различные типы ПФ, обнаруженные в клетках
эукариот, экспрессируются на различных стадиях развития
и дифференцировки. Впрочем, роль сетей ПФ остается
невыясненной. Как уже упоминалось, промежуточные
филаменты придают прочность клеткам эпидермиса.
формы клеток и осуществлении механической работы
393
Центросомы с парой
перпендикулярно
..--*; расположенных
центриолей 4
Профаза
Образующееся
митотическое
веретено
Телофаза
:. Полярные
волокна
->. Астральные
волокна
Метафаза
Разделение клетки
и.. Хроматида
Волокно
кинетохора
Кинетохор
Анафаза
Области
центромера
Рис. 29.7 Митоз
а - Упрощенное изображение стадий митоза. Полярные волокна выделены цветом; предполагается, что они отвечают за
расхождение полюсов в разные стороны. Черные волокна - волокна кинетохора, ответственные за перемещение хромосом к полюсам.
Все волокна являются микротрубочками;
б - присоединение волокна кинетохора (схематично)
Белковые ламины образуют сеть, соединенную с
внутренней поверхностью внутренней ядерной мембраны;
они связаны с ядерными порами. В нервных клетках
существуют нейрофиламенты - механическая опора
длинным аксонам; филаменты десмина расположены
в Z-дисках саркомеров. Вызывает недоумение тот факт,
что недостаток промежуточных филаментов в
культивируемых клетках, вызванный мутациями или введением
определенных антител, не мешает им расти и
благополучно делиться.
часть 6 Механическая работа клеток
394
Дополнительная литература
Микротрубочки в транспорте везикул
Bray D. Cell movements. Garland Publishing, 1992.
(В монографии рассмотрены все аспекты движения
в клетках. Книга содержит простые схемы, хорошо
проиллюстрирована электронными микрофотографиями.)
Lewin В. Genes V. Oxford University Press, 1994.
Актин в немышечных клетках
Stossel Т. Ρ The machinery of cell crawling // Sci. Amer.
1994. Vol.271 (3). P. 40-47.
(В статье рассматриваются ассоциация и диссоциация
актиновых филаментов, лежащие в основе клеточного
движения, а также их регуляция, которая включает
кальций-, фосфолипид- и актинсвязывающие белки.)
Микротрубочки в клеточном движении
ByardE. Η., LangeВ. Μ. Η. Tubulin and microtubules//
Essays Biochem. 1991. Vol. 26. P. 13-25.
(Обобщающий обзор по данной теме.)
Goldstein L. S. В. В. With apologies to Scheherezade: tails
of 1001 kinesin motors // Annu. Rev. Genet. 1993. Vol. 27.
P. 319-351.
(Глубокий обзор, посвященный кинезиновым минидви-
гателям.)
Walker R. Л., Sheetz M. P. Cytoplasmic microtubule-as-
sociated motors// Annu. Rev. Biochem. 1993. Vol.62.
P. 429-451.
(Детальный обзор, посвященный минидвигателям и их
роли.)
Wallee R. В., Sheetz M. P. Targeting of motor proteins //
Science. 1996. Vol. 271. P. 1539-1544.
(Рассматривается перемещение кинезинов и динеинов
по микротрубочкам.)
McNivenM. Л., Ward J. В. В. Calcium regulation of
pigment transport in vitro// J. Cell Biol. 1988. Vol. 106.
P. 111-125.
(Экспериментальная, но легко читаемая статья о
транспорте пигментов в пузырьках.)
Микротрубочки в митозе
Glover D. Μ, Gonzalez С, Raff J. W The centrosome // Sci.
Amer. 1993. Vol. 268 (6). P. 32-39.
(Обзор, посвященный этой относительно
малоизученной структуре.)
Вопросы к главе 29
1. Какие функции выполняет актин в немышечных
клетках?
2. Что такое микротрубочки?
3. Микротрубочки с незащищенными концами
подвергаются спонтанной диссоциации. Что защищает их
концы?
4. Что такое кинезин и динеин? Проиллюстрируйте
ответ схемой.
5. В процессе деления клетки в метафазе хромосомы
движутся от экваториальной плоскости
обособленно. Микротрубочки присоединены к кинетохорам
и сокращаются по мере расхождения хромосом.
Означает ли это, что микротрубочки сокращаются?
Объясните ответ.
6. Что такое промежуточные филаменты?
7. Каковы функции промежуточных филаментов?
глава 29 Роль цитоскелета в поддержании формы клеток и осуществлении механической работы
395
Ответы на вопросы
Глава 1
1. Количество АТР, которое может быть
синтезировано при использовании 5000 кДж свободной энергии
составляет 5000/55, что соответствует 90,91 моль.
Количество образовавшейся динатриевой соли АТР
в этом случае составит 551 · 91 г в день, или 50 141 г
(72 % веса тела человека). Синтез такого количества
АТР возможен потому, что АТР в нашем организме
непрерывно распадается до ADP и Р/9 и вновь
синтезируется.
2. Значение AG0' (-55 кДж · моль-1) относится к
стандартным условиям, когда АТР, ADP и Р,
присутствуют в концентрации 1,0 М. В клетке концентрации
этих веществ значительно ниже, и фактическое
значение AG для синтеза АТР будет отличаться от AG0':
[ADP][PJ
AG = AG° + ^r-2,303 1g
[АТР]
3. АТР и ADP являются высокоэнергетическими
соединениями фосфорного ангидрида, в то время как
AMP - фосфорный эфир с низкой энергией.
Значительная экзоэргоничность гидролиза АТР и ADP
обусловлена рядом факторов. Во-первых,
высвобождение фосфата снижает напряжение, вызванное
электростатическим отталкиванием отрицательно
заряженных фосфатных групп. Высвобожденные
фосфат-ионы расходятся. Во-вторых, экзоэргоничес-
кому характеру гидролиза способствует большая, чем
у фосфатных групп, резонансная стабилизация
свободного фосфат-иона. Гидролиз AMP вызывает
незначительное увеличение резонансной стабилизации.
4. Очень незначительную, поскольку форма этой
кривой зависит от времени, выбранного для измерения
живности. При более высоких температурах
вероятность инактивации фермента возрастает; при
любой заданной температуре количество инактивиро-
ванного фермента пропорционально времени
инкубации фермента при этой температуре. При
исследовании термостабильности фермента
правильнее инкубировать фермент одинаковое время при
различных температурах и после охлаждения измерять
активность в каждом образце при нормальной
температуре инкубации.
5. А. Неполярные молекулы не могут образовывать
водородные связи с молекулами воды. Поэтому
молекулы воды, окружающие молекулу бензола,
организуются в более высокоупорядоченную структуру,
образуя водородные связи между собой. Такое
увеличение упорядоченности понижает энтропию
и увеличивает энергию системы; поэтому при
соприкосновении с водой молекулы бензола
вынуждены минимизировать поверхность взаимодействия
Бензол-Вода сначала путем формирования
сферических глобул, а затем - отдельного слоя. Результат
известен как гидрофобное взаимодействие.
Б. Полярные группы глюкозы могут образовывать
водородные связи с водой.
В. Ионы Na+ и С1~ гидратируются, и в результате
снижения ионного притяжения свободная энергия
уменьшается. Такое разделение ионов
характеризуется понижением энтропии.
6. Фермент АМР-киназа переносит фосфатную группу
от АТР на AMP в реакции АТР + AMP -^ 2ADP.
Гидролиз не происходит, поэтому существенных
изменений AG0' в реакции не наблюдается.
7. В клетке РР, будет гидролизоваться до 2Р|5
поскольку в полностью очищенном ферменте нет
неорганической пирофосфатазы.
В первом случае AG0' общей реакции соответствует
-32,2-33,4 + 10 кДж · моль1 = -55,6 кДж · моль1.
Для полностью очищенного фермента AG0' =
= -22,2 кДж · моль-1.
8. Ионные связи, водородные связи и Ван-дер-Вааль-
совы силы со средними энергиями 20, 12-29,
и 4-8 кДж · моль1 соответственно. Значение
энергии активации формирования слабых связей очень
небольшое, и поэтому такие связи могут легко
образовываться без участия катализатора и также легко раз-
руШЯГШ, ЧТОприЮДЮ К гибкости структур,
стабилизируемых слабыми связями. Установлено, что для
ассоциации молекул необходимо большое количество
слабых связей с малым радиусом действия. Такие связи
и являются основой биологической специфичности.
9. А.
ОООО
НО-Р-О НО-Р-О" НО-Р-О" -0-Р-0"
рНО АН РН4 АН рН9 А" рН14 °"
Ответы на вопросы
396
Приемлемым значением рКа будет 7,2.
рН = 7,2 + lg [Na2HP04]/[NaH2P04] = 7,2 + lg 1 = 7,2
В. Гистидин со значением рКа 6.5.
10. Значение AG0' в реакции определяет, может ли
реакция протекать, но ничего не говорит о скорости, с
которой она будет протекать (если вообще будет).
Скорость реакции определяется энергией активации
и скоростью образования переходного состояния.
11. Ферменты катализируют химические реакции по
ряду причин:
• их активные центры фиксируют переходное
состояние субстрата прочнее, чем исходный субстрат,
понижая тем самым энергию активации;
• в активных центрах формируется
предпочтительная ориентация реагирующих молекул;
• ферменты могут осуществлять общий кислотно-
основный катализ реакций;
• ферменты способствуют правильной
пространственной ориентации металлсодержащих групп, что
облегчает протекание реакций.
Глава 2
1. Это полипептидная цепь, образованная большим
количеством аминокислот, соединенных пептидными
(-CO-NH-) связями.
2. Полипептидная цепь белка свернута в
определенную, обычно компактную глобулу, точная укладка
которой (фолдинг) зависит от нековалентных и ко-
валентных (дисульфидных) связей между
боковыми группами аминокислот. При высокой
температуре эти связи разрушаются, вызывая тем самым
разупорядочивание полипептидных цепей белка и
превращение их в запутанную нерастворимую
массу, лишенную биологической активности. Это и есть
денатурация белка.
3. Все примеры даны на с. 38 и 39.
Л.Л. Около 4; Б. Около 10,5-12,5; В. Около 6,0.
5. Суммарный заряд этой молекулы зависит от
содержания аспарагиновой и глутаминовой кислот, а также
лизина, аргинина и гистидина. Карбоксильные и
аминогруппы прочих аминокислот задействованы в
образовании пептидных связей (исключая две концевые).
6. Первичная, вторичная, третичная и четвертичная (см.
рис. 2.2).
7. α-Спираль, β-складчатый слой и неупорядоченная
спираль, или область соединительной петли.
α-Спираль и β-слой удовлетворяют потенциалу водородных
связей основной полипептидной цепи (см. рис. 2.3
и 2.4). Неупорядоченная спираль не имеет
определенной структуры. Она обычно находится на
поверхности белка, где возможно образование водородных
связей с водой.
8. См. структуру волокон коллагена, приведенную на
рис. 2.8.
9. Десмозиновая группа, показанная на рис. 2.9.
Глава 3
1. Наличие двух гидрофобных хвостов вместо одного.
2. Нет.
3. Только путем экзоцитоза, эндоцитоза или при
помощи специальных транспортных механизмов (см.
главу 22).
4. Он действует в качестве буфера текучести мембран,
предотвращающего чрезмерную ассоциацию
полярных липидов.
5.
СН2—ОСО—R
СН—ОСО—R
I о
I и
сн2—о—ρ—οι
о-
6. Лецитин (холин), кефалин (этаноламин), фосфати-
дилсерин (серии).
7. См. с. 56.
8. Холин заменен на галактозу и олигосахарид
соответственно.
9. См. с. 56.
10. Образуемый i^c-ненасыщенными жипрнокислотны-
ми хвостами и фосфолипидными мембранами изгиб
предотвращает плотную упаковку хвостов жирных
кислот липидного бислоя, что делает мембрану
более текучей.
11. Из-за наличия окружающей липидные мембраны
гидратной оболочки. Ее удаление энергетически
невыгодно.
12. Для облегченной диффузии необходимы мембранные
белки, позволяющие растворам пересекать
мембрану в любом направлении согласно градиенту их
концентрации. Для этого процесса энергия не
требуется. Примером может служить транспорт анионов
в красных клетках крови, который позволяет ионам
С1~ и НС03~ проходить через мембрану в любом
направлении.
Ответы на вопросы
397
13. Если концентрацию снаружи обозначить Сх, а
концентрацию внутри обозначить С2, то:
[С2]
AG = RT 2,303 \g-~rz =
=8,315-298K-2,303 lg—=5706кДж-моль-1.
14. См. рис. 3.10.
Глава 4
1. Пепсин в виде пепсиногена, химотрипсин в виде хи-
мотрипсиногена, трипсин в виде трипсиногена, эла-
стаза в виде проэластазы, карбоксипептидаза в виде
прокарбоксипептидазы.
2. · Протеолитические ферменты потенциально
опасны тем, что они могут атаковать белки,
выстилающие протоки пищеварительных желез.
• В тонком кишечнике отсутствуют компоненты,
атакуемые амилазой, поэтому амилаза сразу
синтезируется в форме активного фермента.
• Муцины защищают клетки от протеолитической
атаки.
3. В желудке кислое значение рН вызывает конформа-
ционные перестройки (изменения) в пепсиногене.
Они активируют саморасщепление молекулы с
отщеплением от пепсиногена пептида,
поддерживающего фермент в неактивном состоянии.
Образовавшийся пепсин сразу же начинает активировать
собственный синтез из пепсиногена, т. е. происходит
автокаталитическая каскадная активация. В тонком
кишечнике энтеропептидаза (фермент,
вырабатываемый клетками кишечной стенки) активирует
переход трипсиногена в трипсин, а тот в свою очередь,
активирует остальные зимогены.
4. Панкреатит, или воспаление поджелудочной железы,
вызванное протеолитическим повреждением пакре-
атических клеток.
5. Потому что клетки кишечника способны всасывать
только мономеры (и моноацилглицерины). Липиды,
белки, полисахариды и дисахариды всасываться не
могут.
6. См. рис. 4.4
7. Молоко содержит лактозу, гидролиз которой до
глюкозы и галактозы катализирует фермент лактаза. В
зрелом возрасте многие люди теряют способность
синтезировать этот фермент. Лактоза не всасывается и
подвергается брожению в толстом кишечнике. Благодаря
Ответы на вопросы
398
осмотическому эффекту лактоза способствует
поступлению воды в кишечник, вызывая понос.
8. ТАГ, или триацилглицерин - сложный эфир
холестерина и жирных кислот.
Первичный эфир
СН20-СО—R
СНОСО—R
I
СН2ОСО— R
Первичный эфир
9. Липид (субстрат) эмульгируется движением
кишечника и при помощи моноацилглицерина и свободных
жирных кислот (образовавшихся при
первоначальном переваривании вместе с солями желчных
кислот) таким образом, чтобы липаза могла атаковать
эмульгированную поверхность субстрата. Продукты
переваривания вместе с солями желчных кислот
(свободные жирные кислоты и моноацилглицерин)
переносятся клетками кишечника в виде
дискообразных смешанных мицелл, обладающих высокой
емкостью для этих продуктов. Вероятно, на
поверхности клетки мицеллы разрушаются.
10. Свободные жирные кислоты и моноацилглицерин
ресинтезируются в триацилглицерины, которые
включаются в состав липопротеиновых частиц - хи-
ломикронов. ТАГ, холестерин и его эфиры образуют
гидрофобное ядро хиломикронов, окруженное
оболочкой из фосфолипидов и некоторых белков. Хи-
ломикроны всасываются при помощи экзоцитоза
в лимфу и в конечном счете высвобождаются в виде
эмульсии в кровь через грудной проток.
Глава 5
1. Хранению глюкозы в свободном виде мешает ее
осмотическое давление. Осмотическое давление
раствора связано с количеством растворенных частиц.
При полимеризации тысяч молекул глюкозы в
единую молекулу гликогена осмотическое давление
понижается.
2. Запасы гликогена в печени истощаются после
24-часового голодания, а запаса ТАГ обычно хватает на
несколько недель. Триглицериды являются более
мощным источником энергии, поскольку они более
восстановлены и не гидратированы. Кстати говоря,
если бы энергия, содержащаяся в липидах, запасалась
в виде гликогена, то масса тела была бы значительно
большей.
3. Да, потому, что глюкоза может превращаться в аце-
тил-СоА.
Нет, потому, что ацетил-СоА не может превратиться
в пируват.
Не существуют, поскольку в нашем организме нет
специальных запасных белков, сохраняющих
аминокислоты (исключение составляет молоко).
4. · Печень поддерживает гомеостаз глюкозы в
организме, длительное время сохраняя глюкозу в виде
гликогена. При голодании печень высвобождает
глюкозу для поддержания постоянного уровня сахара в
крови. Это необходимо для нормального
функционирования мозга.
• При длительном голодании печень синтезирует
глюкозу для энергообеспечения мозга. Печень
преобразует липиды в кетоновые тела, которые мозг
(и другие ткани) могут использовать для частичного
покрытия энергетических расходов;
• В печени синтезируются жиры, предназначенные
для экспорта в другие ткани.
5. Нет.
6. Жировые клетки сохраняют жир длительное время.
При голодании жировые клетки высвобождают
жирные кислоты.
7. Эритроциты полностью зависят от глюкозы, которую
в процессе гликолиза перерабатывают в лактат.
Вместе с потерей митохондрий при созревании красные
кровяные клетки теряют способность синтезировать
АТР иным путем, кроме гликолиза.
8. Когда содержание глюкозы в крови высоко,
выделяется инсулин. Его выделение служит сигналом для
запаса тканями питательных веществ. По мере
понижения уровня содержания глюкозы в крови
снижается уровень инсулина и повышается уровень глюка-
гона. Значение уровня глюкагона служит сигналом
для высвобождения глюкозы печенью и жирных
кислот адипоцитами. Глюкоза и жирные кислоты
используются тканями. Захват глюкозы клетками мозга не
зависит от уровня инсулина. Он может происходить
даже при голодании, когда уровень инсулина
чрезвычайно низок.
Кроме того, адреналин может вызывать интенсивную
мобилизацию глюкозы и жирных кислот с целью
преодоления экстремальной ситуации.
Глава 6
1. Глюкозо-1 -фосфат + UTP —> UDP-глюкоза + РР,,
PPi + Н20 -> 2Р,.,
UDPG + гликоген,. —> UDP + гликоген. + ^.
Гидролиз РР, делает полную реакцию сильно экзо-
эргоничной.
2. Реакция образования глюкозо-1-фосфата из
неорганического пирофосфата никогда не протекает в
клетке, потому что ее субстрат (неорганический пирофос-
фат) немедленно разрушается. Эта реакция обратна
происходящей в клетке, однако в соответствии с
правилами систематической номенклатуры, фермент
назван по прямой реакции (см. рис. 6.3).
3. Печень и в меньшей степени почки, поскольку
именно в этих тканях сосредоточен фермент глюко-
зо-6-фосфатаза.
4. Глюкоза + АТР —> глюкозо-6-фосфат + ADP.
Глюкокиназа имеет гораздо большую Км для
глюкозы, чем гексокиназа. При голодании печень
возвращает глюкозу в кровь. Это необходимо прежде всего для
снабжения глюкозой мозга и эритроцитов. Сразу
после захвата клетками мозга и печени глюкоза фосфо-
рилируется. Более низкое сродство глюкокиназы
к глюкозе (по сравнению с гексокиназой) приводит
к отсутствии конкуренции за сахар крови между
мозгом и печенью. Печень эффективно усваивает
глюкозу только при ее высоком уровне в крови.
Поскольку глюкокиназа не ингибируется глюкозо-6-фосфатом
подобно гексокиназе, фосфат может захватывать
глюкозу и синтезировать гликоген даже при высоком
уровне внутриклеточного глюкозо-6-фосфата.
5. Предотвращение детской галактоземии диетой,
лишенной галактозы, возможно потому, что
потребности таких больных в галактозе
удовлетворяются за счет обратимой эпимеризации UDP-глюкозы
в UDP-галактозу. Поэтому исключение галактозы
из диеты не предотвращает синтез гликолипидов
и гликопротеинов, содержащих галактозу.
6. В капиллярах липопротеинлипаза отщепляет жирные
кислоты из ТАГ, и они немедленно поглощаются
окружающими клетками.
7. ЛПОНП - липопротеины очень низкой плотности.
Они похожи на хиломикроны и переносят
холестерин и триацилглицерины от печени к
периферическим тканям.
8. Холестерин переносится из печени к
периферическим тканям в ЛПОНП. ЛПОНП по мере потери ТАГ
превращаются сначала в ЛППП, а потом - в ЛПНП.
Последние захватываются периферическими
тканями. Однако некоторые из ЛППП и ЛПНП
снова поступают в печень. ЛППП и ЛПНП получают
холестерин от периферических тканей от ЛПВП,
что создает обратный поток холестерина от тканей
к печени.
9. Путем превращения в соли желчных кислот в печени.
Ответы на вопросы
399
10. Фермент ЛХАТ переносит остатки жирных кислот
от лецитина на холестерин.
11. ТАГ не освобождаются; гормонзависимая липаза,
активируемая глюкагоном (или адреналином в
критических ситуациях), отщепляет свободные жирные
кислоты, которые переносятся кровью к тканям
в комплексе с сывороточным альбумином.
Глава 7
1. Гликолиз, цикл лимонной кислоты и электронтранс-
портная цепь. Эти три этапа окисления глюкозы
протекают в цитоплазме, матриксе митохондрий
и внутренней митохондриальной мембране
соответственно.
2. Структуры приведены на с. 100.
АН2 + NAD+ <-> А + NADH + Н+
В + NADH + Н+ <-> ВН2 + NAD+
3. FAD - переносчик водорода, имеющий следующее
строение: циклическая структура изоаллоксазина -
рибит-фосфат - фосфат -рибоза - аденин. FAD -
производное рибофлавина, представляющее собой
простетическую группу, ковалентно прикрепленную
к ферменту, и восстанавливающуюся до FADH2.
4. При аэробном гликолизе глюкоза расщепляется до пи-
рувата. Образующийся NADH реокисляется
митохондриями. При анаэробном гликолизе скорость
гликолиза превышает способность митохондрий к
реокислению NADH. Это может происходить,
например, при экстремальной ситуации («бегство» или
«борьба»). Если накапливающийся NADH не будет
быстро окислен, то из-за дефицита NAD+
произойдет торможение гликолиза и выработки АТР. В
критической ситуации NADH реокисляется в ходе
восстановления пирувата в лактат.
сн3сосоо-
Пируват
+ NADH + Н+
CHoCHOHCOO +NAD+
Лактатде-
ги геназа
Лактат
5. Структуры на с. 101.
AG гидролиза равна -31 кДж - моль-1 по сравнению
с 20 кДж · моль-1 для эфиров карбоксильной группы;
следовательно, тиоловые эфиры являются
высокоэнергетическими соединениями.
6. Пируват + CoASH + NAD+ ->
-> Ацетил-S-CoA + NADH+ + Н+ + С02
AG0' = -33,5 кДж · моль-1.
7. Ацетильная группа поступает в цикл лимонной
кислоты.
8. NAD и FADH2 реокисляются электронтранспортной
цепью с образованием Н20 и генерацией АТР.
9. Согласно уравнению Нернста, значения AG0' и Е^
связаны между собой следующим образом:
AG°' = -nFAE^,
где F - число Фарадея = 96,5 кДж · моль-1, а Е^ -
разница между редокс-потенциалами донора и
акцептора электронов.
В данном примере Щ = -1,035 В (-0,219 - 0,816 В).
Поэтому AG0' = -2 (96,5 кДж · моль-1); (-1,035 В ) =
= -3(-1,035) = 194,06 кДж · моль-1.
10. Расщепление жирных кислот до ацетил-СоА.
11. А Да. Глюкоза превращается в пируват, а пируват-
дегидрогеназа преобразует его до ацетил-СоА.
Последний может использоваться для синтеза липидов.
Б. Нет. Жирные кислоты расщепляются до
ацетил-СоА. Для синтеза глюкозы необходим
пируват. Реакция, катализируемая пируватдегидрогена-
зой, необратима. Поэтому у животных ацетил-СоА
(а тем более жирные кислоты) не могут
превращаться в глюкозу.
В. Да, но не путем обращения пируватдегидрогеназ-
ной реакции (см. подпись к рис. 7.11).
Глава 8
1. Превращение С6-молекулы в две С3-молекулы
происходит при альдозном расщеплении, которое
катализирует фермент альдолаза. Для этого необходима
структура типа*
R
I
с=о
I
R'—С—R"
I
н—с-он
I
R
При изомеризации глюкозо-6-фосфата во фруктозо-
6-фосфат образуется альдольная структура, которая
может подвергаться такому расщеплению
СНО
I
снон
I
снон
I
снон
I
снон
СН2ОРО§
снон
I
со
I
снон
I
снон
I
снон
СН2ОРО§
Глюкозо-6-фосфат Фруктозо-6-фосфат
Ответы на вопросы
400
2. Процесс синтеза ATP, когда на ADP переносится
высоко энергетическая фосфорильная группа, ковалентно
связанная с субстратом. Примером может служить
окисление глицеральдегид-3-фосфата (см. с. 112).
3. Пируваткиназа катализирует обратную реакцию, но
по номенклатуре киназам всегда дают названия по
реакции, в которой используется АТР. Эта реакция
необратима, потому что ее продукт, енолпируват,
спонтанно изомеризуется в кето-форму и этой
реакции отвечает большое отрицательное значение AG0'.
4. 3 и 2 молекулы соответственно. При фосфоролизе
гликогена с использованием Р, образуется глюкозо-
1-фосфат, который изомеризуется в глюкозо-6-фос-
фат. Для образования глюкозо-6-фосафата из
глюкозы требуется затратить молекулу АТР.
5. Потому, что цитоплазматический NADH не может
непосредственно проникать в митохондрии; значит,
электроны должны транспортироваться посредством
одного из двух альтернативных челночных
механизмов (шунтов). Малат-аспартатный шунт (см. рис. 8.9)
восстанавливает митохондриальный NAD+, а глице-
рофосфатный челночный механизм
восстанавливает FAD глицерофосфатдегидрогеназы внутренней
мембраны митохондрий. Редокс-потенциал первого
более отрицателен, следовательно, восстановленные
эквиваленты поступают в электронттранспортную
цепь на уровне различных дыхательных комплексов.
В случае глицерофосфатного шунта выход АТР ниже.
6. При расщеплении сукцинил-СоА из GDP и Р,
образуется молекула GTP. При этом высвобождается одна
молекула Н20, которая используется в цикле (и как
таковая не появляется), уравнивая суммарный баланс.
7. При окислении образуется β-кетокислота, которая
легко декарбоксилируется.
8. Реакция, катализируемая пируваткарбоксилазой,
пополняет запас оксалоацетата:
С00"
I
с=о
I
СН3
о
+ НС03-+ АТР-
-сосг
с-
I + ADP + Р/ + Н+.
Н2С —СОО"
9. Биотин, витамин группы В. При использовании АТР
он образует реакционноспособный карбоксибиотин,
который может передавать карбоксильную группу
субстратам.
ADP + Р,·
10. Это показано на рис. 8.19.
11. Общее в том, что оба эти вещества - подвижные
переносчики электронов. Убихинон соединяет
дыхательные комплексы I и II с комплексом III, а цитох-
ром с соединяет комплексы III и IV. Первый
локализуется в гидрофобном липидном бислое;
последний - в водной фазе на поверхности внутренней
мембраны митохондрий.
12. Для перекачивания протонов из матрикса
митохондрий через внутреннюю мембрану и генерации
электрического и протонного градиентов, которые могут
быть использованы для синтеза АТР.
13. У эукариот перенос восстановленных эквивалентов"'
от цитоплазматического NADH к электронтранспор-
тной цепи митохондрий осуществляется при помощи
челночных механизмов. В случае глицерофосфатного
шунта теряется 1 молекула АТР (из тех, что
потенциально могут образоваться при окислении NADH).
Поскольку при гликолизе из глюкозы образуются
2 молекулы NADH, это ведет к потере 2
потенциально возможных молекул АТР. У Е. coli такой проблемы
не существует. Кроме того, в отличие от клеток
эукариот, клетки Е. coli не расходуют энергию для
транспорта АТР и ADP через мембрану митохондрий.
Глава 9
1. · Свободные жирные кислоты (из жировой ткани),
переносятся сывороточным альбумином крови.
• Липопротеинлипаза высвобождает свободные
жирные кислоты и триацилглицерины из хиломик-
ронов.
• Свободные жирные кислоты могут доставляться
из ЛПОНП, образованных в печени; (их
высвобождение происходит тем же путем, что и в
предыдущем случае.
2. Клетки мозга и эритроциты (эритроциты не имеют
митохондрий). См. главу 5.
3.· Активация для образования из жирных кислот
ацил-СоА производных;
• на внешней мембране митохондрий;
• в митохондриальном матриксе;
• как производные карнитина (см. рис. 9.1).
Ответы на вопросы
401
4. Реакции, приведенные на рис. 9.2, аналогичны
реакциям: сукцинат^> фумарат^> мешать
оксалоацетат цикла лимонной кислоты (как по реакциям, так
и по используемым акцепторам электронов).
5. Из 1 молекулы пальмитиновой кислоты образуется
8 молекул ацетил-СоА и происходит восстановление
7 молекул FADH2 и NADH. При окислении NADH
и FADH2 образуется соответственно 2,5 и 1,5
молекулы АТР. Если добавить к этому выход АТР в ходе
окисления 8 молекул ацетил-СоА (10 АТР на
молекулу), то всего образуется 108 молекул АТР (считая
GTP из цикла лимонной кислоты за АТР). Из этого
количества необходимо вычесть 2 молекулы АТР,
использованные в реакции активации.
Следовательно, суммарный выход составляет 106 молекул АТР.
6. После 2 раундов β-окисления г^с-£)3-еноил-СоА изо-
меризуется в т/?днс-£>2-еноил-СоА (см. реакцию
нас. 134).
7. В случаях быстрого высвобождения жиров из
жировых клеток, как это происходит при голодании или
диабете, печень превращает ацетил-Co А в кетоновые
тела, которые поступают в кровь. Кетоновые тела
преимущественно утилизируются мышцами,
способствуя сохранению глюкозы; мозг может покрывать
приблизительно половину своих энергетических
потребностей за счет кетоновых тел.
8. Синтез ацетоацетата происходит в матриксе
митохондрий, а холестерин синтезируется в мембране эндоп-
лазматического ретикулума.
Глава 10
1. Жирные кислоты синтезируются из двухуглеродных
фрагментов, донорами которых является трехуглерод-
ная молекула - малонил-СоА. Ацетил-СоА
превращается в малонил-СоА путем АТР-зависимого
карбоксилирования. Последующее декарбоксилиро-
вание имеет большое отрицательное значение AG0'.
Другими словами, цикл карбоксилирования и декар-
боксилирования необходим, чтобы сделать
добавление двух углеродных единиц к растущей цепи
жирной кислоты необратимой реакцией.
2. Они показаны на рис. 10.1.
3. У эукариот все реакции проходят на одной белковой
молекуле, ферментные функции которой
осуществляют отдельные домены. Функциональной
единицей эукариотической синтазы жирных кислот
является димер, объединяющий две молекулы в единое
целое. У Е. coli различные реакции катализируются
отдельными ферментами. Преимущество эукариот
Ответы на вопросы
402
заключается в том, что промежуточные продукты
прямо передаются от одного активного центра
к другому. У Е. coli продукты должны
диффундировать к следующему ферменту, поэтому весь процесс
синтеза протекает медленнее.
4. См. структуры на с. 141. NAD+ используется в ката-
болических реакциях - он принимает электроны
в процессах окисления и выработки энергии. NADP+
участвует в процессах восстановительного синтеза.
Существование двух этих соединений представляет
собой форму метаболической компартментализации,
которая облегчает независимое регулирование
процессов.
5. Тканями, в которых преимущественно сосредоточен
синтез жирных кислот, являются печень, жировые
клетки и ткани молочной железы в период лактации.
6. Ацетил-СоА в митохондриях превращается в цитрат,
который транспортируется в цитозоль, где цитратли-
аза расщепляет его на ацетил-СоА и оксалоацетат.
Это АТР-зависимая реакция, что обеспечивает
полное расщепление цитрата:
Цитрат + АТР + CoA-SH + Н2 ->
-> ацетил-СоА + оксалоацетат + ADP + Р,-.
7. Оксалоацетат восстанавливается в малат NADH-зави-
симой малатдегидрогеназой митохондрий. Малат
в цитозоле окисляется и декарбоксилируется в пиру-
ват малик-ферментом в NADP4-зависимой реакции.
Такая схема позволяет эффективно переключать
восстановительные эквиваленты от NADH к NADPH.
Образовавшийся пируват возвращается в
митохондрии (см. рис. 10.4). При этом восстанавливается
только одна молекула NADPH в расчете на образованную
молекулу малонил-СоА, в то время как для реакций
восстановления в процессе синтеза жирной кислоты
необходимы две. Недостающую молекулу
предоставляет глюкозо-6-фосфатдегидрогеназная система,
описанная в главе 13.
8. Схема этого процесса показана на рис. 10.5.
9. В синтезе фосфолипидов на основе глицерина
возможны два пути. В первом из них фосфатидная
кислота присоединяется к спирту типа этаноламина
(см. рис. 10.6). Для этого спирт активируется (в
синтезе фосфолипидов активированной молекулой
всегда является CDP-спирт). Для синтеза некоторых фос-
фолипидов активируется диацилглицерин (см.
рис. 10.6). Это происходит, опять же, путем
образования комплекса CDP-диацилглицерин. Ситуация
напоминает использование UDP-глюкозы во всех
случаях, когда требуется активированный остаток глюкозы.
10. · Эйкозаноиды - биологические регуляторы, состя-
щие из 20 атомов углерода (от греч. eikosi- двадцать).
К эйкозаноидам относятся простагландины, тромбок-
саны и лейкотриены.
• Из полиненасыщенных жирных кислот.
• Простагландины вызывают болевую реакцию,
воспаление и лихорадку. Тромбоксаны участвуют
в агрегации тромбоцитов. Лейкотриены
стимулируют сокращение гладких мышц и, вызывая спазм
воздухоносных путей, способствуют развитию астмы.
• Аспирин ингибирует циклооксигеназу (фермент,
участвующий в синтезе простагландинов), поэтому
он может подавлять боль и лихорадку, а также
препятствовать свертыванию крови.
11. Мевалоновая кислота - первый метаболит,
предназначенный только для синтеза холестерина.
Структурные аналоги мевалоновой кислоты ингибируют
ГМГ-СоА-редуктазу - фермент, ответственный за
образование мевалоната.
Глава 11
1.Мозг не может использовать жирные кислоты, он
должен получать глюкозу. Это же относится к
красным клеткам крови, которые не имеют митохондрий
и могут производить энергию только путем гликолиза.
2. Потому что субстратом пируваткиназы является
енольная форма пирувата, а кето-енольное
равновесие сильно сдвинуто в сторону образования кето-
формы; следовательно, фермент не имеет субстрата.
Решение этой проблемы лежит в наличии обходного
метаболического пути, где на образование фосфое-
нолпирувата расходуются две высокоэргичные
фосфатные группы:
Пируват-
карбоксилаза
Пируват-h ATP + HCCV ►
► Оксал оацетат + ADP + Р/ + Н+;
Фосфоенолпируват-
ap6oKCl·
Оксалоацетат + GTP *+ ►
-* ► Фосфоенолируват +GDP+ С02
З.Фруктозо-1,6-дифосфатаза и глюкоза-6-фосфатаза,
катализирующие образование фруктозо-6-фосфата
и глюкозы-6-фосфата соответственно.
4. Нет. Свободная глюкоза образуется только в печени
и почках.
5. При нормальном питании (не голодании)
повышенная мышечная активность может вызвать образование
лактата при анаэробном гликолизе. Он
транспортируется кровью в печень, где преобразуется обратно
в глюкозу. Высвобождение глюкозы в кровь и ее
захват мышцами завершают этот цикл (см. рис. 11.4).
6. Глицеринкиназа необходима для синтеза глюкозы
из глицерина (см. рис. 11.5). Освобождение
глицерина происходит при голодании, когда поддержание
уровня глюкозы в крови становится жизненно
важной задачей. Поскольку глюконеогенез протекает
в печени, то имеет смысл транспортировать глицерин
именно туда, а не метаболизировать его в жировых
клетках, которые не могут высвобождать глюкозу
в кровь.
7. С помощью глиоксилатного цикла (см. рис. 11.6).
Глава 12
1. Первый - аллостерическая регуляция; второй - ко-
валентная модификация фермента, основным
механизмом которой является фосфорилирование.
2· γ μ
v max χ- ■
л I
ο * / /
« 2 /Неаллоу
/стеричесу Аллостерический
кии / ферМент
/фермент/
Концентрация субстрата [S]
3. Аллостерические эффекторы обычно действуют,
изменяя сродство фермента к субстрату.
Концентрация субстрата для такого фермента должна быть ниже
насыщающей, что, как правило, и бывает.
Положительный аллостерический эффектор сдвигает сигмо-
идную кривую зависимости скорости реакции от
концентрации субстрата влево, а отрицательный
эффектор - вправо (см. рис. 12.5). Сигмоидная
зависимость усиливает влияние аллостерических
эффекторов на скорость реакции и, таким образом,
увеличивает чувствительность регуляции (см. рис. 12.4).
4. В этих условиях они не оказывают никакого
влияния, поскольку изменение сродства фермента к
субстрату при насыщающей концентрации субстрата
не изменяет скорости реакции.
Ответы на вопросы
403
5. Согласованный и последовательный механизмы
показаны на рис. 12.7 и 12.8.
6. Поскольку структура аллостерического эффектора
отличается от строения субстрата, различные
метаболические системы могут осуществлять регулятор-
ные взаимодействия путем аллостерической
регуляции.
7. Внутриклеточная регуляция обычно аллостери-
ческая. Именно она позволяет каждой клетке
поддерживать баланс метаболических путей. Правда, алло-
стерическая регуляция не может определять
направление метаболизма в клетке в целом, в зависимости
от потребностей всего организма. Это задача
внеклеточной системы регуляции (гормонов и т. д.),
которая осуществляет управление деятельностью клеток
в соответствии с физиологическими потребностями
организма.
8. Схема приведена на рис. 12.9. AMP активирует гли-
когенфосфорилазу и фосфофруктокиназу, в то время
как АТР ингибирует ФФК. Высокое молярное
отношение ATP/ADP тормозит гликолиз, а понижение
уровня АТР (приводящее к увеличению AMP)
ускоряет его. При высоком уровне цитрата замедляется
поступление метаболитов из гликолиза в цикл
лимонной кислоты. Высокий уровень ацетил-СоА может
свидетельствовать о недостатке оксалоацетата и,
следовательно, о необходимости активации пируваткар-
боксилазы для осуществления анаплеротической
реакции. В то же время высокий уровень ацетил-СоА
указывает на «бесперебойную» поставку пирувата
гликолизом, который в данный момент уместно
затормозить на уровне ФЕП.
9. Прямая аллостерическая регуляция; фосфорилиро-
вание пируватдегидрогеназы (и ее инактивация) под
действием киназы; дефосфорилирование протеин-
фосфатазой, которая обращает эффект фосфорили-
рования (см. рис. 12.10).
10. Как показано на рис. 12.11.
11. Высокий уровень глюкозы крови стимулирует
выделение инсулина, низкий - выделение глюкагона.
12. Первичным посредником (месенджером) является
гормон типа адреналина. При взаимодействии с
рецептором клетки он вызывает увеличение
внутриклеточного содержания вторичного посредника, который
и оказывает влияние на метаболизм. Для двух
упомянутых в вопросе гормонов вторичным
месенджером является сАМР. Он аллостерически активирует
протеинкиназу А (ПКА), которая и осуществляет
разнообразные метаболические эффекты.
13. Путем мобилизации переносчиков глюкозы в
мембрану клетки (см. рис. 12.17).
Ответы на вопросы
404
14. с AMP запускает каскадную активацию, которая
начинается с активации ПКА (см. схему на с. 170).
15. Различия суммированы на рис. 12.22.
16. Разные клетки имеют рецепторы для различных
гормонов. Если клетка А имеет рецептор гормона X,
то сАМР в этой клетке оказывает эффект,
соответствующий гормону X. Клетка В не имеет рецептора
гормона X, но имеет рецептор для гормона Y,
поэтому сАМР в клетке В вызывает эффекты,
свойственные гормону Y, но не гормону X.
17. Фруктозо-2,6-дифосфат. сАМР увеличивает его
уровень (см. рис. 12.25).
18.Глюкагон, активируя синтез своего вторичного
посредника сАМР, стимулирует глюконеогенез.
Одновременно с этим сАМР активирует киназу, которая
фосфорилирует пируваткиназу, вызывая ее ингиби-
рование. В мышце адреналин увеличивает
образование вторичного посредника с AMP, который
стимулирует гликолиз для выработки энергии; поэтому
торможение пируваткиназы здесь было бы
неуместным.
19.сАМР активирует гормон-чувствительную липазу,
которая катализирует гидролиз триглицеридов.
Глава 13
1. Пентозофосфатный путь поставляет рибозо-5-фос-
фат для синтеза нуклеотидов, образующийся NADPH
используется в синтезе липидов. В пентозофосфато-
ном пути пентозы претерпевают сложные
взаимопревращения с образованием молекул, содержащих
от 3 до 7 атомов углерода.
2. В окислительной стадии происходит превращение
глюкозо-6-фосфата в рибозо-5-фосфат с
выделением С02 и восстановлением NADP+.
3. Главными являются трансальдолаза и транскетолаза
(см. рис. 13.2), но могут участвовать также
ферменты гликолиза.
4. Поскольку трансальдолаза и транскетолаза
используют в качестве донора только кетозу, часть рибо-
зо-5-фосфата преобразуется в ксилулозо-5-фосфат.
Затем происходят следующие превращения:
1. 2 С5 —> С3 + С7 (транскетолаза),
2. С7 + С3 —> С4 + С6 (трансальдолаза),
3. С5 + С4 —> С3 + С6 (транскетолаза).
2 С5 в реакции 1 - рибозо-5-фосфат и ксилулозо-
5-фосфат; конечный С3-компонент - глицераль-
дегид-3-фосфат, превращающийся в глюкозо-
6-фосфат с потерей Pf.. В конечном счете 6 молекул
рибозо-5-фосфата преобразуются в 5 молекул
глюкозо-6-фосфата и Ρ,. Таким образом, клетка
может производить NADPH без увеличения
суммарного содержания рибозо-5-фосфата.
5 В окислительной стадии глюкозо-6-фосфат
превращается в рибозо-5-фосфат с выделением С02. Если
учесть, что из 6 молекул глюкозо-6-фосфата
образуется 6 молекул рибозо-5-фосфата, из которых может
ресинтезироваться 5 молекул глюкозо-6-фосфата,
тогда в суммарном уравнении будут фигурировать
6 молекул С02. Т. е. на бумаге это выглядит как
окисление молекулы глюкозы. В действительности же
одна молекула глюкозо-6-фосфата не преобразуется
в 6 молекул С02.
6. NADPH требуется для восстановления глутатиона -
молекулы, необходимой для защиты эритроцитов.
Больные с наследственным дефицитом глюко-
зо-6-фосфатдегидрогеназы чувствительны к
антималярийному препарату памахину, вызывающему
гемолитическую анемию.
Глава 14
1. «Световые» реакции включают расщепление воды
при использовании энергии света и восстановление
NADP+ в NADPH. В ходе «темновых» реакций
NADPH используется для восстановления С02 и воды
в углеводы. Термин «темновой» подразумевает, что
для протекания таких реакций свет не требуется (это
не значит, что они происходят только в темноте).
На самом деле наиболее интенсивно темновые
реакции протекают при ярком солнечном свете.
2. Это хлорофилл, улавливающий световые фотоны.
При возбуждении фотоном один из электронов
молекулы хлорофилла переходит на более высокий
энергетический уровень. Резонансный перенос энергии
позволяет передавать это возбуждение от одной
молекулы к другой, пока его не уловят молекулы
специального реакционного центра, которые не участвуют
в резонансной передаче энергии, а направляют
электроны в электронтранспортную цепь (см. рис. 14.4).
3. · Синтез АТР по хемиосмотическому механизму
в ходе переноса электронов через ^-комплекс
фотосистемы II (см. рис. 14.6).
• Синтез АТР в условиях полного восстановления
молекул NADP+, когда электроны, транспортируемые
переносчиками фотосистемы 1, поступают на
^-комплекс, способствуя образованию большего
количества АТР (см. рис. 14.7).
4. Хлорофилл Р680+, который является реакционным
центром пигмента фотосистемы II: он возбуждается
при резонансной передаче энергии от антенной
молекулы хлорофилла и передает электрон феофитину -
первому компоненту цепи переноса электронов
фотосистемы II. Р680+ характеризуется сильными элек-
тронакцепторными свойствами, т. е. является
сильным окислителем.
5.Тилакоиды формируются при инвагинации
внутренней мембраны хлоропласта (сравните с
внутренней мембраной митохондрий), что объясняет
кажущуюся противоположную ориентацию протонного
насоса.
6. Фермент «Rubisco» (рибулозо-1,5-дифосфаткарбокси-
лаза) расщепляет рибулозо-1,5-дифосфат на две
молекулы 3-фосфоглицерата; в ходе этой реакции
происходит фиксация одной молекулы С02.
7. См. рис. 14.10.
8. Последовательность этих реакций приведена на
рис. 14.9.
9. Если С02 сначала фиксируется ферментом «Rubisco»
в молекуле 3-фосфоглицерата, то растение относят
к С3-растениям. В реакции, катализируемой этим
ферментом, кислород и С02 конкурируют друг с
другом. Однако, при высокой температуре и интенсивном
солнечном свете, у С4-растений С02 первоначально
фиксируется пируват-Р-дикиназой и ФЕП-кар-
боксилазой в оксалоацетат (см. рис. 14.11), который
восстанавливается до малага. Последний поступает
в цикл Кальвина. Декарбоксилирование малага под
действием малик-фермента приводит к
значительному увеличению отношения С02/02 (концентрация
С02 в клетках может повышаться в 10-60 раз).
Полная схема приведена на рис. 14.11. У различных
С4-растений могут встречаться незначительные
вариации этой схемы, но основная стратегия
избегания оксигенирования под действием рибулозо-5-кар-
боксилазы остается неизменной.
10. У животных пируваткиназа не может образовывать
фосфоенолпируват из пирувата. Однако у растений
есть фермент пируват-Р-дикиназа, который,
используя две фосфатные группы АТР, осуществляет такое
превращение.
Глава 15
I. Окисление приводит к образованию основания Шиф-
фа, которое гидролизуется в водной среде.
ι , Н20
I -2н Ι ν Ι
CHNH2 — C=NH ^ С=0 + NH3.
Ответы на вопросы
405
2. Глутаминовая кислота.
3. Трансдезаминирование - наиболее
распространенный механизм. Аминогруппа большинства
аминокислот переносится на α-кетоглутарат, с образованием
глутамата. Последний дезаминируется глутаматде-
гидрогеназой:
1. Алании + сс-кетоглутарат —> пируват + глутамат.
2. Глутамат + NAD++ Н20->
—> сс-кетоглутарат + NADH + NHJ.
Суммарная реакция:
Алании + NAD+ + Н20 -^ пируват + NADH + NHJ.
4. Пиридоксальфосфат (см. рис. 15.2). Механизм пере-
аминирования приведен на рис. 15.3.
5. Удалением Н20 и H2S соответственно (см. рис. 15.4).
6. Глюкогенными аминокислотами являются те, при
дезаминировании которых в конечном счете
образуется пируват (или фосфоенолпируват). Их
превращение в пируват может включать ряд промежуточных
стадий, приводящих, например, к образованию
промежуточных метаболитов цикла лимонной кислоты.
Из кетогенных аминокислот образуется ацетил-СоА.
Только лейцин и лизин являются исключительно ке-
тогенными, а некоторые аминокислоты, такие как
фенилаланин, являются смешанными. Кетоновые
тела образуются из кетогенных аминокислот только
в исключительных случаях, например, при голодании.
Во всех остальных случаях ацетил-СоА окисляется
обычным образом.
7. Фенилаланин обычно не подвергается переами-
нированию; он превращается в тирозин и после
этого метаболизируется (см. рис. 15.5). Если
превращение фенилаланина в тирозин нарушено,
фенилаланин подвергается переаминированию с
образованием фенилпирувата, который вызывает
необратимое поражение мозга у детей и ведет к ранней
смерти.
8. Тетрагидробиоптерин служит донором
восстановительных эквивалентов для образования Н20 из
одного атома молекулы кислорода, используемой в
реакции гидроксилирования.
9. Активация происходит путем образования 5-адено-
зилметионина (SAM). SAM имеет структуру иона
сульфония, существенно облегчающую отщепление
метильной группы. Образование SAM показано на
рис. 15.6.
10. См. рис. 15.8.
11. И обильное белковое питание, и голодание
характеризуются высокой скоростью дезаминирования
аминокислот (в мышцах при голодании белки
разрушаются для поддержания синтеза глюкозы), и аминный
азот должен быть преобразован в мочевину
12.· Аммоний превращается в глутамин, который
транспортируется в печень и гидролизуется.
Глютамат + аммоний ——^—^ » Глютамин,
ATP ADP + Р/
Глютамин + Н20 Глютамат + аммоний.
• Аминный азот транспортируется из мышц в виде
аланина. Аланиновый цикл показан на рис. 15.9.
Глава 16
1. Для разрушения нежелательных молекул и структур,
попавших в клетку путем эндоцитоза, а также для
завершения жизненного цикла клеточных компонентов.
2. Первичными лизосомами называются пузырьки,
образованные аппаратом Гольджи. Они содержат
гидролитические ферменты - кислые гидролазы
с оптимумом рН 4,5-5,0, который поддерживается
при помощи протонного насоса.
3. Объект-мишень находится внутри или эндоцитозного
пузырька, или аутофагосомы. Первичная лизосома
сливается с этими образованиями в единый пузырек -
вторичную лизосому, в которой и происходит
переваривание.
4. Существует большая группа наследственных лизосо-
мальных болезней накопления, обусловленных
отсутствием определенных гидролитических
ферментов, что вызывает перегрузку лизосом веществами,
от которых клетка обычно избавляется.
5. При болезни Помпе - наследственном заболевании,
характеризующемся недостатком лизосомальной
сс-1,4-гликозидазы - лизосомы перегружены
гликогеном. Однако неясно, почему гликоген удаляется
именно таким путем; лизосомы никак не
вписываются в хорошо известную схему метаболизма
гликогена.
6. Пероксисомы - мембраносвязанные пузырьки цито-
золя, содержащие оксидазы. Оксидазы окисляют
разнообразные субстраты с использованием кислорода
и образованием Н202:
R'H2 + 02 -> R' + Н202.
Пероксид затем используется для окисления других
субстратов:
RH2 + H202->R + 2H20.
Ответы на вопросы
406
В пероксисомах происходит укорочение жирных
кислот с очень длинной цепью. Предполагают, что здесь
же осуществляется окисление боковой цепи
холестерина с образованием солей желчных кислот. При
некоторых тяжелых наследственных заболеваниях
в ряде тканей обнаружено отсутствие пероксисом.
Глава 17
1. Первичный сигнал, инициирующий свертывание
крови, очень мал (в количественном отношении),
поэтому для достижения быстрого ответа
необходимо каскадное усиление, способствующее
многократной амплификации исходного сигнала. На
заключительном этапе протромбин активируется
и превращается в тромбин-активный протеолити-
ческий фермент.
2. Белок фибриноген в мономерном состоянии
защищен от спонтанной полимеризации взаимным
отталкиванием отрицательно заряженных фибринопепти-
дов (см. рис. 17.2). Удаление фибринопептидов
тромбином способствует ассоциации мономеров
фибрина (см. рис. 17.3).
3. Между мономерами, входящими в состав полимера,
формируются поперечные связи, образованные в ходе
ферментативного трансамидирования между
боковыми группами глутамина и лизина:
CONH2 + H3N+ -> CONH + NH4+.
4. Витамин К в карбоксилазной реакции является
кофактором. В ходе превращения протромбина в
тромбин происходит карбоксилирование остатка глутами-
новой кислоты и образовавшийся карбоксиглутамат
связывает Са2+.
I I
сн2 сн2
I I
сн2 + со2 нс—соо-
I I
соо- с—соо-
5. Цитохром Р450 участвует в превращении
гидрофобных ксенобиотиков в водорастворимые, катализируя
следующую реакцию:
АН + 02 + NADPH + Н+ -> АОН + Н20 + NADP+.
6. Для восстановления одного атома кислорода до Н20.
7. Остаток глюкуронида присоединяется к ОН-группе
чужеродной молекулы, делая ее более полярной (см.
рис. 17.5).
8. Это АТР-зависимая транспортная система мембран,
при помощи которой различные вещества
выводятся из клеток. Она транспортирует стероиды в секре-
тирующих клетках коры надпочечников, но все
транспортируемые вещества (включая противораковые
лекарства, используемые в химиотерапии) являются
жирорастворимыми амфипатическими веществами,
что свидетельствует о более общем значении этой
системы.
9. Нейтрофилы выделяют эластазу в слизь,
выстилающую поверхность легочной ткани, которая
разрушает эластин - структурный материал легких. Это
способствует превращению маленьких альвеол в
большие структуры со значительно меньшей
поверхностью газообмена. В норме а,-антитрипсин крови
диффундирует в легкие и ингибирует эластазу,
предотвращая повреждение альвеол. Курение приводит
к двум последствиям: 1) оно способствует
инактивации а,-антитрипсина в результате преобразования
боковой группы метионина в сульфоксид (S —> S = 0);
2) раздражение легкого привлекает нейтрофилы, что
увеличивает количество высвобождаемой эластазы.
10. Это молекула кислорода, получившая
дополнительный электрон.
02 + е~ -> 02~ ·
Супероксид - чрезвычайно реакционноспособная
молекула, вызывающая повреждение макромолекул.
11. Супероксид опасен, потому что, атакуя молекулы, он
инициирует образование других свободных
радикалов, вызывающих лавинообразную цепь реакций,
нарушающих нормальное протекание жизненно
важных процессов. Большинство клеток зукариот
содержат супероксиддисмутазу и каталазу, которые
катализируют следующие реакции:
20^ + 2Н+ —> Н202 + 02 (дисмутаза),
2Н202 —> 2Н20 + 02 (каталаза).
Кроме того, в клетках есть антиоксиданты типа
аскорбиновой кислоты и витамина Е. Антиоксиданты
являются гасящими агентами. При атаке
супероксидом из них образуется свободные радикалы,
реакционная способность которых недостаточна для
поддержания цепной реакции.
Глава 18
1.5-фосфорибозил-1-пирофосфат (ФРПФ) -
универсальный агент. Он образуется из
рибозо-5-фосфата и АТР, а пути его использования приведены
нас. 218-219.
Ответы на вопросы
407
2.Тсчрагидрофолат (FH4)
Формил-РНд :
"N^CH,
,10
Η
o=r
I
Η
3. Серии гирокси мстила за переноси! -СН2ОН -группу
серима на FH4, с выделением глицина и
образованием N\NI(,-mct илен-ГН4, который окисляется
до N\NI()-mcihiihji-FH4 в ΝΛΙ)Ρ-зависимой реакции.
1 идролиз последнею приводи! к формил-КН4.
4. Она риботилируег гуанин и гипоксантин в процессе
реутилизации пуринов.
5. У детей, больных синдромом Лсша-Нихана.
отсутствует ГГФРТ, полому реутилизации пуринов
не происходит. Однако в мозге есть прямой путь,
приводящий (благодаря повышенному уровню ΦΡΙΙΦ)
к избыточному синтезу пуриповых нуклеотидов
tie novo. У больных отмечен избыток мочевой
кислоты, однако использование аллонуринола
(тормозящею образование мочевой кислоты) не облегчает вы-
раженности неврологических симптомов, причины
которых остаются пока пеобъяснепными. У больных
подагрой уровень мочевой кислоты также повышен,
но неврологическая симптоматика отсутствует.
6. Аллопуринол превращается в аллокеантин. который
ингибирует ксант иноксидазу. Это превращение
катализирует сама ксантиноксидаза, следовательно, имеет
место суицидное иш ибирование (самоинактивация).
7. См. рис. 1К.8.
8. Hei.l имиднлатсишетаза переносит С^-грунпу от ме-
ίiiJicii-H 14 iiadlJMP и восстанавливает седо метиль-
ной группы. Донорами водорода служит FH4.
который превращаемся в I 1т\.
9. Он иш noiipyei восстановление F1I-, в 1Ί14.11оследпий
необходим для шмидилакчнпеташой реакции, и при
его дефиците тормозится образование dTMP. а
следовательно, синтез ДНК и деление клеили.
10. Витамин В12 необходим для метилирования гомоцис-
теина до мет ионика, а донором мстильиых ι руин
служит метил-КН4. Недостаток витамина Вр приводит
к тому, что FH4 оказывается «пойманным» в виде мс-
тилсодержащею соединения, которое не может быть
использовано в других фолат-зависимых реакциях.
Глава 19
I.Cm.c. 23 I.
Ответы па вопросы
408
2. Генетический материал должен бьпь максимально
химически устойчив. ДНК более устойчива, чем РНК.
г>к) обусловлено тем. что 2'-()Н-груипа рпбозы
делает возможной нукдеофильную атаку на фоефодн >-
фирпую связь, снижая тем самым устойчивость РНК
по сравнен!по с ДНК.
3. Линейная структура цепи ДНК ославляла бы ι пдро-
фобные поверхности основании обращенными в
коду. За счет шдрофобных сил основания укладьжа-
Ю!ся вместе, изгибая фосфодп )фирную свял, (см.
рис. 19.3, а).
4. В-ДНК. правозакручепная. К) пар оснований.
5. У одной цепи ДНК цепочка нуклеотидов 5'—> 3' inei
в одном направлении, а у другой в
противоположном. Таким образом, каждый конец линейной моле
кулы ДНК содержи! 5'-конен одной цени и З'-конец
другой цени.
6. Направление 5'—> 3' означает что вы двигаетесь οι
концевой 5'-()Η-ι руппы к З-ОН-группе полинуклео
тидиой цепи.
7. 11ривсдснная структура показывает одну цепь
следующего дуплекса:
5' CATAGCCG 3"
3' GTATCGGC 5'
Спаривание оснований по Уо!Сону-Крику объясняв
комплементарную последовательное!ь. Линейную
последовательность одной цени ДНК условились
записывать с 5'-концом. расположенным слева.
X. · ДНК хромосом пкариот. намотанная вокруч окта-
мера гистоновых белков в два оборота (146 нар
оснований). Нуклеоеомы последовательно сое
шпоны линкерной ДНК.
• Когда хроматин расшепляеιся Д11Казой. фра!
менты, содержащие 146 нар оснований ДНК, защищены
от действия фермента: тто наблюдение привело к οι-
крьгтию пуклеосом (см. рис. 19.7).
Глава 20
1. Фрагмент ДНК, репликация которого начинается
с одной точки начала репликации.
2. Перед рспликативпой вилкой образуклея
положительные сверхвнтки.
3. У Е. eo/i гнраза (топоизомераза ΙΠ вводит отрнца-
тсльныесверхвитки. У зукариот топоизомераза I
расслабляет положительные сверхвптки.
4. См. рис. 20.6 и 20.7.
5. При наматывании ДНК вокру! нуклеоеомы
возникают локальные отршииельные с верх витки, но
поскольку свяin при пом нс нарушаются,
суммарной отришпелыюй сверхспирализации нет.
Локальные οιрн нательные сверхвитки компенсированы
локальными положительными сверхвитками в других
местах. Они расслабляются иол действием юноню-
меразы I. оставляющей суммарные отрицательные
сверхвткп в ДНК.
6.С1ДГР. dGTP. cKTP и dTTP.
7. Цп юшн лсл ко дезамипируется с образованием ура-
пила: если бы урапил был нормальным компонентом
ДНК. ю было бы невозможно распознавать и
скорректировать мутацию, гак как спаривание Λ-U ι
очно 1акое же. как π Λ--Τ .
8. · I lei. Требуе1ся праймер. ДНК-полпмсраза не мо-
жс1 инициировав сишез новых пеней.
• Синтез пдс1 в направлении 5'—> 3',
следовательно, новая пет, ^оптируется в направлении 5'—> 3'.
π новые н\к1еотнды добавляю1ся к свободному
З'-ОН-коицч предшествующего пуклеошда.
9. Гидролиз пеоркшическою пирофосфата: спаривание
ос 11 о ва 11 и й 11 у к: ι со 111; ю в.
10. Высокая пропессивпость означает, что после
присоединения к \iaipinic ДНК полимераза III
перемещаемся \ю ней на большое расстояния без
отсоединения, синтезируя новую пень. Такой эффекд
досми аегся «кольцевым зажимом» кольцом белков,
которое собирается позади полимеразы, и цепь ДНК
проскальзывае! сквозь ие\о (см. рис. 20.14).
11. Полимераза I фермет. соединяющий фрагменты
О казак и в непрерывную отстающую цепь. Fx актив-
пост суммированы па рис. 20.16.
12.11равнльное спаривание входящих нуклеот идтрифос-
фатов с матрицей имеет первостепенную важность.
Полимера <а III /; coli шкжеможс!
пеправляιьошибки считывания. КеЗ'—» 5'-жзоиуклеазная активное!ь
удаляет последний включенный пуклеотид в случае
неправильного спаривания оснований.
13. Существует мет илзависимая система репарации
Е. coli {см. рис. 20.19). У человека также существуют
белки, выполняющие аналогичную функцию; при их
недостатке повышается риск канцерогенеза.
14.11ри облучении ДИК удырафиолеювым светом
образуются дпмеры пгмипа. Два смежных тимиповых
основания связываются ковалентно. Репарация может
осуществляйся либо прямой светочувствительной
системой, которая разрушает связи и восстанавливает
отдельные основания, либо молекула подвергается
жецнзнонпой репарации (см. рис. 20.20).
15. Как показано па рис. 20.22. после удаления З'-ирай-
мера фрагмета Оказаки осчается переплицирован-
ный участок.
16. С помощью теломериой ДНК. Схема ее синтеза
приведена на рис. 20.23.
Глава 21
1. РНК-полимераза использует рпбопуклеошды ATP
СТР. GTP и UTP в качестве субстратов; а ДНК-поли-
мераза dATP. d(TP. dGTP и dTTP. РНК-полпмера-
νά может иницинроват ь синтез новых цепей: ДНК-по-
лимеразе для пою необходим праймер. При синтеза
РНК никогда не происходи! исправления ошибоь
считывания.
2. Они приведены на рис. 21.4. Промотор iimcci άκη
Прибпова и элемент 35.
3. РНК-полимераза неспепифически присоединяете?
к ДНК. по при взаимодействии с молекулой сигма
фактора фермент связывается точно с промоюром
Нлок Прибпова и элемент 35 способствуют правил,
ной ориентации полимеразы. Фермент сиптезпруе
несколько фоефодиэфирных связей, после чего сш
ма-фактор отделяется, и полимераза продолжае
дальнейшую транскрипцию мРНК.
4. Один из них обусловлен структурой шпилечной G-1
петли, показанной па рис. 21.7. В ней внутреннее
спаривание оснований G^C в мРНК предо1вращас
связывание с матричной цепью ДНК. Последователь
ности уридиповых иуклеот идов еще более ослабля
ют связь с ДНК (пара U=A содержит только две водо
родных связи), облегчая отделение. Второй сносен
гермипацни связан с Rho-фактором хеликазой
которая раскручивает ι ибрид мРНК ДНК. На участ
кетермипапии полимераза останавливается
(возможно, благодаря повышенному содержанию G-C ικ
ттом участке ДНК); Rho-фактор догоняет се и
отделяет мРНК.
5.Точная последовательность оснований блока
Прибпова и )лемента -35, расстояние между ними, а
также основания в области от +1 до -ТО.
6. См. рис. 21.10.
7. У эукариот первичный транскрипт содержит
нитроны, которые должны быть удалены; 5'-конец
копируется; механизм терминации неизвестен: мРНК
(за редким исключением) нолиадспнлмроващ/
с 3'-кон на.
8. Механизм показан на рис. 21.13. Консенсусиая
последовательность определяет место сплайсинга,
а реакция трансперификапни сопровождается
незначительным изменением свободной энергии.
Прерывистые тепы могут облегчить эволюцию, способе ι в\ я
ιасованшо жзоиов. Дифференцированный сплайепш
Ответы па вопросы
может также приводить к тому, что один и тог же ген
определяет синтез различных белков.
9. Полимсраза III эукариот присоединяется не к ДНК,
а к белковому комплексу, который собирается на ДНК
(рис. 21.19). Кроме того, с этим комплексом может
быть связано несколько факторов транскрипции
(см. рис. 21.20 ).
10. Белки «спираль-поворот- спираль», белки «лейци-
новой молнии», белки типа «цинковых пальцев»
и белки гомеодомена.
Глава 22
1.Если бы использовались только 20 кодонов,
осталось бы 44 нскодируюших триплета. Любая мутация
в кодирующей области гена с высокой долей
вероятности вызывала бы инактивцию гена,
преждевременно внедряя стоп-кодон. При использовании 61 кодо-
на замена основания либо не вызывает изменений
(из-за вырожденности генетического кода), либо
приводит к замене одной аминокислоты на другую
(часто похожую по структуре). Три триплета
аминокислоты не кодируют и являются стоп-кодонами.
2. Потому что существует механизм качания
(неоднозначного связывания).
3. Последовательность оснований РНК всегда
записывают так, что 5'-консц расположен слева. Когда
мРНК изображается таким способом, основания ап-
тикодона τ РНК должны быть приведены антипарал-
лельно, поэтому молекулу τ РНК и записывают в
перевернутом виде с 5'-концом справа.
4. Проверка правильности присоединения
аминокислоты ктРНК. Пауза в катализируемом F.F-Tu
гидролизе GTP на стадии элонгации дает время несиарен-
ным аминоацил-тРНК покинуть рибосому.
5. Гидролиз GTP до GDP и Р#., приводит к конформа-
ционным изменениям в G-бслках. GTP участвует
в сборке инициаторного комплекса Е. с γ;//., в
доставке аминоацил-тРНК к рибосоме с помощью EF-Tu
фактора. GTP используется также на этапе
транслокации.
6. Для объяснения понадобится рис. 22.11.
Приведенный на рисунке «раскачивающийся» механизм
(модель I) имеет то преимущество, что при
перемещении от одного участка к другому тРНК никогда
полностью не отделяется. Это также означает, что
пеитидильная группа не должна перемещаться
относительно рибосомы. На рис. 22.11 приведены два
возможных механизма. Модель II объясняет, почему
рибосома имеет две субъединицы.
Ответы на вопросы
410
7. Эту несовместимость объясняем рис. 22.! 3. Механизм
нахождения инициирующего кодона AUG
предусматривает существование только одного о ар юного
участка. Как показано на рис. 22.9, инициация у
прокариот может одновременно осуществляться с
нескольких стартовых участков.
8. Шапсроны связываются с растущими
полипептидами и иредогвращают их преждевременное
неправильное сворачивание. Высвобождение шаперонов в
нужный момент облегчает правильное сворачивание,
однако полной ясности в понимании этого процесса
пока нет (подробнее см. с. 298).
9. Болезни, вызванные приопами (см. с. 298).
10. См. рис. 22.16.
Глава 23
1.При использовании механизма нормального
рецептор-опосредованного эндопитоза (см. рис. 23.1).
2. Последний должен перенести в клетку РНК-реп.ш-
казу; (+)-цспью РНК вируса является мРНК.
кодирующая РНК-репликазу.
3. Вирус коровьей оспы реплицируется в цитоплазме:
полимсраза хозяина находится в ядре.
4. РНК рстровируса копируется в двухцепочечиую
ДНК, которая внедряется в хромосому хозяина
(см. рис. 23.5).
5. Механизм показан на рис. 23.3.
6. Защитное действие антител при гриппе направлено
па белок гемагглютинин. Он постоянно мутирует
но поскольку на молекуле гемагглютинина сущее
unci большое количество эпиюпов, потеря
иммунитета происходит постепенно. Это ι антигенный дрейф
делает людей восприимчивыми к инфекции, но
частичная защита ослабляет течение заболевания.
Формирование в результате рекомбинации между двумя
различными штаммами совершенно нового re\iai-
гл юти ни на может привести к возникновению
смертельно опасной пандемии.
7. Поверхностные белки вируса специфически
связывают нейраминовую (сиаловую) кисло ι у, которая
присутствует и на конце молекулы глпкофорина >рш-
роцитов. Поэтому при смешивании с эритроцитами
вирусы связываются с ними, вызывая ai ипоттщанию.
8. Вирус связывается с кле1кой хозяина при номошп
гликопротсина, оканчивающегося нейра.миновой
кислотой. Фермент, катализирующий разрушение непра-
миновой кислоты, необходимой для проникновения
в клетку вирусу может быть нужен для: а)
облегчения выхода новых вирусных частиц, способных проч-
но связываться клеточными гл и ко протеи нам и; 6) для
разжижения слизи, содержащей нейраминовую
кислоту, что облегчает вирусу путь к поверхности
клетки. Разжижение слизи может также помогать
распространению вируса при чихании.
9. Бактериофаг может входить в логическую или лию-
генную фазы (см. рис. 23.7).
\0.Л. Маленькая, инфекционная, обнаженная молекула
РНК. лишенная оболочки.
Б. Нет. У вироидов отсутствуют длинные
кодирующие последовательности оснований, которые могли
бы содержать информацию об аминокислотной
последовательности белка.
Глава 24
1. Панкреатическая ДНКаза разрезает ДНК
беспорядочно. Фермент рестрикции разрезает ДНК в строго
определенных местах, узнавая короткие
последовательности оснований.
2. Адсниновые основания ДНК всех прилегающих
к гексамеру последовательностей R-шгамма Е. coli
метилированы, поэтому фермент не распознает их.
Чужеродная ДНК такой защиты не имеет.
3. «Липкими концами» называют концы,
образующиеся при зигзагообразных разрезах, которые
формируют большинство ферментов рестрикции.
I
- G;AAT Τ С - -G ААТТС-
>
-cttaa;g- -cttaa g -
f
Выступающие концы ДНК будут автоматически
«слипаться» в результате спаривания оснований.
4. Клон генов - клонированный участок ДНК,
идентичный последовательности ДНК в хромосоме. Клон
кДНК означает комплементарную ДНК, которая
идентична мРНК гена. Клон генов содержит интроны,
а кДНК - нет.
5. Этапы подготовки геномной библиотеки показаны на
рис. 24.1.
6. Этапы выделения приведены на рис. 24.2.
7. Нуклеотид, лишенный З'-ОН-группы. Когда ДНК-по-
лимераза присоединяет дидезоксинуклеотидтрифос-
фат к вновь синтезируемой ДНК, дальнейший рост
цепи ДНК прекращается. О применении этого метода
при секвенировании - на с. 323 -324.
8. Участок ДНК на хромосоме можно амнлифици-
ровать. Процесс включает копирование фрагмента
ДНК и многократное копирование копий. Значение
этого процесса состоит в том, что, располагая
минимальным количеством ДНК (слишком малым для
любых исследований), можно наработать необходимое
для эксперимента количество интересующего
фрагмента. Помимо фермента и субстратов требуются
праймеры для копирования фрагмента в обоих
направлениях, следовательно, должна быть известна
последовательность оснований на концах амплифи-
цируемого фрагмента.
9. Плазм и да с удобным участком для встраивания
клона кДНК определенного белка, содержащая также
соответствующие бактериальной ДНК
транскрипционные (промотор) и трансляционные сигналы. ДНК
транскрибируется, и мРНК транслируется в
бактериальной клетке. Такой плазмидный вектор может быть
использован для наработки больших количеств
интересующего белка.
10. Рестрикационным анализом называют методический
прием для выявления генных дефектов. Если ДНК
нарезается ферментами рестрикции, а полученные
фрагменты разделяются при помощи
гель-электрофореза, то, используя гибридизационные методы,
электрофоретические полосы можно
визуализировать. При мутациях картина таких полос будет
меняться.
Глава 25
1.См. рис. 25.3.
2.Образование многочисленных генов
иммуноглобулинов представлено на рис. 25.4.
3. В-лимфоциты (В-клетки) производят антитела
(после активации и созревания в плазматические
клетки). Для этого В-клеткам необходим контакт с хел-
перными Т-лимфоцитами (Т-клетками). Цитотокси-
ческие Т-клетки (или киллерные клетки) связываются
с клетками хозяина, представляющими чужеродный
антиген, вызывая их гибель посредством перфорации
мембраны или стимуляции апоптоза.
4. Антиген-представляющая клетка - тип фагоцита,
который поглощает чужеродный антиген,
подвергает его процессингу на фрагменты и выставляет
антиген в комплексе с молекулами МНС класса П. Если
неактивные хелперные Т-клетки взаимодействуют
с комплексом антиген-МНС, то происходит активация
Т-клеток (см. рис. 25.4).
5. В обоих случаях это МНС класса I.
Ответы на вопросы
411
6. Теорией клопалыюй селекции называется теория,
объясняющая процесс селекции клеток, секрети-
рующмх am и ι ела. Каждая В-клетка вырабатывает
свое антитело. Для любою антигена есть несколько
специфичных В-клеток. но только когда антиген
связывается с антителом, фиксированным на
мембране В-клетки. та пролиферпрует с образованием
клопа. Таким образом антиген автоматически
выбирает В-клетки, подвергаемые пролиферации.
7. Все антигены, которые связываются в процессе
первичного созревания В-клекж в костном мозге или
Т-клеток в тимусе, являкнея аутоаитигенами. На них
не должно быть иммунного ответа, поэтому па
дайной стадии развития клетки, реагирующие на аутоап-
тигепы, будутунич'южены. 11осле выхода из костного
мол а или тимуса связывание с антигеном активирует
клетки.
8. Выделение антитела достигается при помощи
различного сплайсинга нитронов: у З'-конна гена
иммуноглобулина существует жзон. кодирующий
последовательность якорных полипептидов. В начале
секреции переключение сплайсинга приводит к их
исчезновению.
9. Начало секреции в В-клетках сопровождается
соматической мутацией, которая модифицирует
вариабельный участок. Поскольку связывание антигена
вызывает активную быструю клеточную
пролиферацию, такая модификация служит механизмом отбора
клеток, производящих «лучшие» антитела.
10.CD4 есть на поверхности хелперных Т-клеюк. Он
связывается с константным белковым компонентом
молекул МНС класса II, полому взаимодействует
юлько с В-клетками, хелперпыми Т-клетками и ап-
тигеп-представляющими клетками, которые содержат
белки згою класса. На поверхности цитотоксичес-
ких Т-клсток есть белок CDS, который связывается
с молекулами МНС класса I: поэтому Т-киллерпые
клетки взаимодействуют только с клетками,
содержащими уют тип молекул. Белок CD4 является
рецептором, с помощью которого вирус СПИДа
инфицирует хелперпые Т-клстки.
Глава 26
1. Зпдокрипныс гормоны, факторы роста π нейро-
меднаторы.
2.Действие сигнальных молекул, растворимых и
нерастворимых влипидах. представлено на рис. 26.1.
3. Схема регуляции адепилатппклазы - фермента,
ответственною за сшпхмсЛМР изображена па рис. 26.1!.
Ответы на ({опросы
412
Гидролиз GTP играет роль молекулярных часов.
При холере токсин необратимо ннактивпруел GTPajy.
в результате чего образование сЛМΡ остается
повышенным. Ото приводит к потере Na и воды организмом.
4. сЛМР аллостеричеекп активирует протеипкиназу Λ
(ПКА), которая фосфорилпрует различные белки-мп-
шепп. включая ферменты и факторы транскрипции
Кроме того, существуют гены, содержащие еАМР-
чуветвнтельные элементы (CRKs). Из лого следил,
что с AM Р. возможно, оказывает существенное
влияние на генную регуляцию.
5. Окид азота NO активирует цптоплазматпческую г\а-
нилатциклазу. которая синтезирует cCiMP. Последний
является вторичным посредником и активпр\ст ИКС
6. Такой системой является фосфоннозшилный каскад.
Он включает рецептор-зависимую активацию мемо-
рапосвязаппой фосфолнпагы с. высвобождающей \\\ю-
зиттрифосфат IP. и диацнлглицерип ДА Г. IP,
увеличивает концентрацию цпюплазматнческого Са: .
а ДА Г активирует прочеппкпыазу ИКС. Таким
образом. IP. и ДАГ являются вторичными посредниками
(см. рис. 26.16).
7. Ras-нуть имеет рецепторы шрознпкпназною гнил
и включает фосфорилироваппе белков-посредников
(ем. рис. 26.18).
8. Такой белок можс1 связываться с фосфорплпроваппым
рецептором тирозннкнназного типа (см. рис. 26.21)
и активировать клеточные регулятор! 1ые пути.
9. Если рсцен юр или. например, компонеш Ra»-n\in
аномально постоянно акшвен. избыточная жепрсс-
сия гена, кодирующего факторы фапскрпишш.
может способствовать неконтролируемому делению
клетки (см. рис. 26.22).
10. См. рис. 26.22.
11.· Активация адрепореиснюра вызывает аллостерп-
ческое изменение циюнлазматичеекого домена,
связывающегося с гормоном.
• Активация рецептора fXiF RAS-iiyin вьпывае!
димеризацию рецептора и акшвацию аутофоефорп-
лирования остатков тирозина.
• Активация рецептора интерферона включас!
димеризацию рецептора и связывание со специальном
тирозпнкиназой. которая фосфорплируег репей юр
(см. рис. 26.6. 26.17 и 26.22).
12.Такой Са-' капал, есть, например, в мембране
периною синапса, который высвобождает аисшлходип
для инициации мышечного сокращения. Капа.ι
открывается, когда до пего доходи ι волна
деполяризации мембраны нейрона. Резкое изменение копцен-
ί рации Са: вызывает освобождение ацетнлхо.пша
(см. рис. 26.24).
13. Jm aii.HaiiiiciiMiiiM называемся канал,
открывающийся при свя зываипп со специфическим лпгаплом.
В процессе евсювоспрня ι ия капюниые каналы
мембран палочек обрываются при помощи c(iMP.
С'всι вызываем снижение копцешрации cCjMP. чю
приводи! к закрьи ию капала Возникающая ι ипер-
полярпзацпя мембран палочек преобразуется в
импульс зрительно! о нерва.
Глава 27
1. На первой стдпп 5-а.минолсвулнпа1 (ALAleiinuna ка-
кын *нр\С1 реакцию взаимодепемвпя i.niiiuiia с сукци-
пид-СоА(см.рпс. 27.5). а на втроп - М-А-дегпдрагаза
обра з\ с ι пиррол nopi|)o6n.ninoicH (см. рис. 27. 6).
2. Повышенная акмпвноемь ALA-сппга'ш в печени
обнаружена при oeipoii перемежающееся порфирии,
\01я взаимосвязь мсжд\ образованием ALA и певро-
лотческпмп спмпюмами непопягна.
3. Механизм ре ι \ляппп акцизное ι π показан на рис. 27.8.
4. Мпоглобип обладает более высоким сродством
к кнелоролу. В 01ЛИЧПС οι ешмопдпой кривой
γόμοι лобина. кривая насыщения мио1лобнна имеет вил
ι ипербо.ты (см. рис. 27. | 1).
З.Соыаспо Припяти молели. icmomoohh существует
в еосюяипях низкою (Г) и высокою (R) сролства
к кислороду Связывание кислорода смещает
равновесие в строну увеличения количества молекул,
находящихся в R-cocтяпни.
6. При связывании кпелорола с железом, входящим
13 сослав гема. железо перемешаемся в плоскость пор-
фпрнпового кольца. Для mm. чюбы но произошло,
необходимо некоюрое уплощение тераииррола.
Железо присоединяется к К8-гпстидипу белка,
вызывая иереем ройк\ белковой молекулы, чю
приводи! чплошеипю ютраииррола. Вес ή и перемешепия
ВЛИЯЮ1 на взапмодейемзпесубьедппиц, вызывая кон-
формациоппып переход тефамера из Г- в R-форму
(см. рис. 27.13).
7. Гемо1лобпн взрослою человека 13 дезокеш ениро-
ванном сосюяипи iimcci полос ι ь. в ко юрой 2.3-дп-
(|)ое(|)01Лицера1 (ДФ1 ) связываемся с положительно
заряженными ip\iina\ni белков. Связывание ДФГ
возможно юлько в дезокешенированном состоянии,
по лому оно снижаем сродемво 1емо1лобипа к
кислороду Фетальпый ι емомобпп содержи τγ-субъединицу
вмеею (^-субьединппы. характерной для взрослых.
В γ-субьедпипце нем одной из заряженных групп,
\ часпзч Ю1нп> в ei'ii ;ывапнн ДФГ. Полому ДФГ свя-
зываетя с фскиьпым ϊομοι looiтом менее прочно.
a ci о сродемво к кислороду выше, чем \ \iaiepiincKoio
1смотлобипа.
8. ">ю необходимо для ipaiicnopia СО, в виде НСО
(см. рис. 27.15).
Глава 28
1 . ИСТОЧНИКОМ AT Ρ ДЛЯ быСТрЫХ ВОЛОКОН СЛуЖИ Ι 1ЛПК0-
лиз, а для медленных окисли ιедьпый метаболизм.
Ьысмрые волокна обеспечиваю! μι повепные реакции
спасения («бел см во» пли «борьба»), но в таких
волокнах бысмро развивается уюмлеппе. Медленно
сокращающиеся волокна получаю1 АТР в ходе
окислительного метаболизма и могут функционировав
значи ic.'ibHO дольше без признаков утомления, но для
увеличения выработки щергии требуется время. У
людей имеются оба типа мышц. Мышцы спины,
поддерживающие позу в течение длшельпых периодов,
относятся в основном к тину медленно сокращающихся,
а мышны глазною яблока к быстро сокращающимся.
2. См. рис. 28.! it 28.2.
3. Ответ суммирован па рис. 28.7.
4. Стимуляция мышечного рецептора анстил холи пом
ведет к высвобождению ионов Са2' из саркоплазма-
шческого ретикулума. С актиповыми филамешами
связаны молекулы тропомиозппа. которые, в свою
очередь, содержат Са2 -чувет ви тельный тропонпно-
вый комплекс. В отеутспзие Са2 фопомиозин
блокирует взаимодействие юловки миозина с актиповым
фпламепюм. При связывании Са2 стропоппиовым
комплексом происходит конформацпоннос
изменение, способствующее связыванию миозина с топким
филаментом и развитию сокращения. Са2 -АТРаза
возвращает Са2 в саркоплазма!ичеекпй ретикулчм.
и сокращение завершается.
5. В головках миозина гладкой мышцы имеется р-лег-
кая пень, которая иигпбирус! связывание юловки
миозина с актиповым филаментом, препякмвуя
сокращению. Нейрогенпая стимуляция открываем
каналы, позволяющие Са2 входить в клетку. Са2
связывается с регул я торным белком кальмодулииом.
который активируем кппазу леч ких пеней миозина.
Фоефорилировапие легкой цени снимает пнгибптор-
пый )ффекц и происходи! сокращение.
Глава 29
1. В пемышечпых клемках актин образуем фпдамешы.
Актпновые волокна, прикрепленные к клетчиой
Ответы пи вопросы
4!3
мембране, позволяют небольшим пучкам молекул
миозина развивать тянущую силу. Помимо участия в
сокращении нсмышечных клеток актиновые филаменты
формирую! путь, по которому передвигаются молекулы
минимиозина. 11оследние имеют обычные миозиновые
головки, но длинная хвостообразная структура,
свойственная миозину мышц, заменена коротким хвостом,
к которому могут прикрепляться транспортные
пузырьки.
2. Микротрубочки - тго полые трубки, образованные
при полимеризации субъединиц белка тубулина. Они
имеют определенную полярность, и их концы
обозначаются соответственно как (+)- и (-)-консц.
3. Центр организации микротрубочек защищает (-)-ко-
нец. (+)-Концы растущих трубочек защищены
комплексом тубулин-GTP. GTP медленно гидролизуется,
снимая защиту. Если новые субъединицы
присоединяются после гидролиза GTP, происходит
деполимеризация микротрубочки. Когда микротрубочка
достигает своей «мишени», се (+)-конец становится
защищенным.
4. Кинезин и динсин - молекулярные двигатели,
которые перемещаются по трубочкам и тянут за собой
внутриклеточные «грузы». Кинезин и динеин
передвигаются по микротрубочке в противоположных
направлениях: от (-)- к (+)-концу и от (+)- к (-)-кон-
цу соответственно.
5. Микротрубочки не могут сокращаться. Укорочение
происходит вследствие деполимеризации
микротрубочек, но что вызывает движение хромосом, пока
неизвестно.
6. Промежуточными называют филаменты с диаметром
10 нм. Их размеры занимают промежуточное
положение между размерами филаментов микротрубочек
(20 нм) и актина (6 нм).
7. Промежуточные филаменты служат структурной
основой волос, придают жесткость структурам типа
нервных волокон и Z-дисков саркомеров.
Ответы на вопросы
414
Благодарности за предоставление
иллюстративного материала
Рисунок 1.8. Любезно предоставлен R. Rogers (Роджерс).
Рисунки 2.6, а- <\ Воспроизведены по соответствующим
рис. 2.10. а: 2.15. с5:4.1, а и 4.4. из книги: Branden (.,
Tooze.J. Introduction to protein structure. N.Y.: (iarland
Publishing. 19е) 1. Рис.2.10. а и 4.4 использованы с
разрешения (iarland Publishing; рис. 2.15. о и 4.4 с
разрешения.!. Richardson (Ричардсон).
Рисунок 2.7. Воспроизведен по рис. 2 в с га ι ье: Jcntoft N. //
Trends Biochcm. Sci. 1990. Vol. 15. P. 293.
Рисунок 3.1 5. Любезно нредосшвлен профессором
W. G. Breed (Ьрпд). Department of Anatomy, University
of Adelaide.
Рисунок 6.13. Любезно предоставлен доктором Т. Forte
(Форте), Lawrence Berkeley Laboratory, University of
California.
Рисунок 9.4. Воспроизведен по рис. 25.7 из учебника:
Biochemistry (4-е и здание), авторское право L. Stryer
(Страйср). С разрешения \\. 11. Freeman and Company.
Рисунок 12.17. Модифицированный вариат рис. 7. в
статье: Kamielietal. Biol. Chem. 1981. Vol. 256. P. 4772.
Рисунки 19.3,<v:<\ Адаптированные варианты
соответствующих рис. 2.4, а и 3.1 из книги: Calladine С. R.,
Drew ll. R. Understanding DNA. Academic Press, 1994.
Рисунок 19.4 Воспроизведен по рис. 2.1. из книги:
Ptashne Μ. A genetic switch. Oxford: Cell Press
and Blackwell Scientific Publications. 1987. Авторское
право Cell Press.
Рисунок 19.5. Любезно предоставлен S. Kumar(Кумар).
Рисунок 19. N. Скопирован с рис. 28.19 из книги: Lewin В.
Genes V. Oxford: Oxford University Press, 1994.
Электронную микрофотографию для книги предоставила
В. Hamkalo (Хчмкелоу).
Рисунок 19.22. Любезно предоставлен S. Kumar (Кумар).
Рисунок 20.6. Скопирован с рис. 2.24 из книги: Singer M.,
Berg P. Genes and genomes. Sausalito, California:
University Science Books, 1991.
Рисунок 20.7. Скопирован с рис. 2.26,<7 из книги: Singer
Λ/.. Berg P. Genes and genomes. Sausalito, California:
University Science Books. 1991.
Рисунок 20.14. Скопирован с рис. 3,</ в обзоре: Krishna
Т. S. R.. Kong\.-P, CaryS.. Burgers Ρ Μ., KuriyanJ. li
Cell. 1994. Vol. 79. P. 1233. Авторское право Cell Press.
Фо гографию любезно предоставил доктор J. Kuriyan
(Кераиеп).
Рисунок 20.15. Адаптированный вариант рис. 511-12
из книги: DNA Replication, Supplement by Kornberg,
1982. Авторское право W. 11. Freeman and Company.
С разрешения W. Η. Freeman and Company.
Рисунок 20.17. Скопирован с рис. 3 в обзоре: Echols //.,
Goodman Μ. Ε //Annu. Rev. Biochcm. 1991. Vol. 60.
P. 490. С разрешения Annual Reviews Inc. Оригинал
в Kennard О. //Nucleic Acids and Molecular Biology.
1987. Vol. LP 25.
Рисунок 20.24. Воспроизведен но рис. 34.1 из книги
Lewi n В. Genes V Oxford: Oxford University Press, 1994.
Рисунок 21.16. Воспроизведен по рис. 29.10 из книги
Lewi η В. Genes V. Oxford: Oxford University Press, 1994.
Рисунок 21.20, о. Скопирован с рис. 3,ί/ в статье:
Ohiendorf D. //.. Anderson U. E and Matthews В. W. .
J.Molec. Evolution. 1983. Vol. 19. P. 109. С разрешения
Springer-Verlag.
Рисунок 21.21 /). Адаптированный вариант рис. 3,г> в
обзоре: E/lenberger Т. Ε. et al. //Cell. 1992. Vol. 71. P. 1223.
Авторское право Cell Press.
Рисунок 21.23. Адаптированный вариант рис. 4 в обзоре:
Ross ./. //Microbiol. Rev. 1995. Vol. 59. P. 423.
Рисунок 21.24. Скопирован с рис. 2 в обзоре: Ross J. ■'
Microbiol. Rev. 1995. Vol. 59. P. 423.
Рисунок 22.2. Скопирован с рис. 2 в статье: Ho/brook S. R.,
Sussman J. L., Warrant R. W. and Kirn S. //. //Mol. Biol.
1978. Vol. 123. P. 631. С разрешения Springer-Verlag.
Рисунок 22.5. Скопирован с рис. 9.5 из книги: Lewi η Β.
Genes V Oxford: Oxford University Press, 1994.
Рисунок 22.11. Адаптированный вариант рис. 4 в
обзоре: NoIIerH. El/Annu. Rev. Biochem. 1991. Vol.60.
P. 193. С разрешения Annual Reviews, Inc.
Рисунок 22.12. Адаптированный вариант рис. 1 в обзоре:
NoIIer И. Ε II Annu. Rev. Biochem. 1991. Vol. 60. P. 191.
С разрешения Annual Reviews, Inc.
Рисунок 23.2. Скопирован с рис. 1.1 из книги: Ptashne M.
A genetic switch (2-е издание). Oxford: Cell Press and
Blackwell Scientific Publications, 1986. Авторское
право Cell Press.
Рисунок 23.8 Любезно предоставлен профессором
R. H. Symons (Саймоне), Department of Plant Science.
Waitc Institute, University of Adelaide.
Рисунок 24.6. Любезно предоставлен доктором Ch. Hahn
(Хан).
глава I Химия, тергия и метаоо.тзм
415
Рисунок 25.1. Воспроизведен в модифицированном виде
по оригиналу in MctcalfD. '.'' Phil. Trans. Roy. Soc.
Lond. ИЗ v3, P. 147. С разрешения автора и Royal Society
(Королевское общее ι во).
Рисунок 26. К). Воспроизведен по оригиналу в статье:
Dohlman II. G . Сигои Μ. С, and Lefkowirz R. J. /
Biochem. 1987. Vol. 26. P. 2660. С разрешения American
Chemical Society (Американское химическое
общество).
Рисунок 27.1. Любезно предоставлен профессором
W. Breed (Ьрпд), Department of Anatomy, University
of Adelaide.
Рисунки 27. К), а-с. Воспроизведены по cooibci-
ствующим рис. 2.9, ή: 2.10, а и 3.7 из киши: Brandcn С,
Toozc J. Introduction to protein structure. Ν. Υ.: Garland
Publishing, 1991. Рис. 2.9. ή нсиолмован с разрешения
Д. Lesk (Леек): рис. 2. 1().</ и 3.7 с разрешения Garland
Publishing.
Рисунок 29.2. а. Любезно прелое кжлеп доктором
P. (innning(I'aiiHiiHi ),Children's Medical Research
Institute. Sydney.
Рисунок 29.2, ή. Скопирован с рис. 2.13 π* κιιιιι π: Lew ι η Β
(ienes Υ. Oxford: Oxford University Press. 1994. Φοιο-
i рафию для книги предоставил P. Solomon (Соломин)
Рисунок 29.6. Скопирован с рис. \с в McXivcn Μ. Α..
Ward J. В. Cell Biol. 19SS. Vol. 7 (106) P. 1 1 I.С
разрешения Rockefeller Univcrsil\ Press.
часть I ВкеОсиис a \u иические реакции Kwuil··;
416
Предметный указатель
Агглютинация, 312
а гон ист, 167
адгезия, 67
аденилаткипаза, 222, 176, 348
- активация адреналином, 348
адеыиловая кислота, 27, 216
адснин, 216
аденозилгомоцисдеин, 198
S-аденозилметионин. 198
-синтез из мстионина, 198
аденозин, 217
адснозиндифосфат, 25
аденозинмонофосфат, 25
адснозинтрифосфат. 25
адреналин,85
-регуляция обмена i'jiiikoiсна. 169
- действие на клетки мышц и печени, 171
- регуляция гормон-чуведвительной липазы, 97
-стимуляция высвобождения глюкозы в кровь, 170
азотимидин, 314
аквапорин, 59
а кон hi аза, 119
активация аминокислот, 288
активация жирных кислот, 132
активация панкреатических ферментов, 74
активация иротеолитических ферментов, 74
активированный С02. 138
активные формы кислорода, 212
активный СО:, 122
активный транспорт, 63
актин, 382
аланин, 149
- участие в глюконеогенезе, 150
- транспортная форма аминного азота в крови, 202
алкаптонурия, 198
алкогольдегидрогеназа, 102
аллоксантин, 223
аллолактоза. 269
аллопуринол,223
аллостерическая регуляция ферментов, 158
- механизм, 158
-обратимость, 160
аллостерическис регуляторы, 159
аллостсричсскис ферменты. 158
аллостерическис )ффскюры. 158, 159
альдолаза, 150
альдолазнос расщепление, 110
альдольная конденсация, 110
альтернативный еплайсиш. 273
аметоитерин, 226
амилаза. 72
амилоза, 76
амилопсктин, 76
аминоакриловая кислота, 196
- в процессе превращения серима в миру ват, 196
аминоацил-тРНК-синтстаза, 288
аминокислоты, 36, 192
- алифатические, 38
- ароматические, 39
- белковые. 36. 37
- глкжогенные, 196
- дезаминированис, 194
- заменимые, 192
- заряженные, 39
- кетогенные, 195
- небелковые, 38
-незаменимые, 192, 193
- транедезаминироваиие (переаминирование), 194
аминолевулиновая кислота (ALA), 367
аминоптерии, 226
аминотрансферазы, 195
амплификация сегментов ДНК, 325
анаболизм, 21
анаболические процессы, 21
анаплеротическая реакция, 122
- в цикле лимонной кислоты. 122
анатомия пищеварительного траюа, 72
анемия, 226
анкирип,66
антибиотики, 297
- влияние на биосинтез белка, 297
антигенная изменчивость, 312
антиген-представляющис клетки, 336
антикодон. 286
антилейкемические лекарства, 226
антиоксиданты, 2 13
антипараллсльность пеней ДНК, 235
417
антипорт, 65
антитела, 329
структура, 332
- классы, 333
- функции,333
α,-антитрипсин, 212
- в легких, 212
- влияние табачного дыма, 2 12
антифолатные лекарственные препараты, 226
антифолаты, 226
апиримидинизация, 255
апобелки, 96
-ароВ 100.96
- ароВ 48, 96
-ароСП.%
- ароЕ, 96
аиолипопротеин В. 76
аиолипопротеины. 96
апоптоз. 332
апофермет, 46
аиоферригин. 368
апурииизация, 225
арахидоиовая кислота, 58
синтез простагландинов и тромбоксанов, 145
аргиназа, 200
аргинин, 39
- в цикле мочевины, 200
- механизм синтеза, 200, 201
аргининосукпинат, 201
аргининосукцинатлиаза, 201
аргининосукпинатсинтс!аза, 213
асиартатаминотрансфераза растений, 191
аспартатамиио! рансфераза, 195
аспирин,146
атеросклероз, 97
атсросклеротичсскис бляшки, 97
аттенуапия, 270
аутоантигены, 331
аутоиммунные заболевания, 329
аутокатали ι ическая активация, 74
аутофагосомы, 204
аффинное созревание, 335
ацетилг алактозам и и, 62
ацетилглкжозамин, 56, 62
ацетил-СоА, 103
ацстил-СоА-карбоксилаза. 138
- активация цитратом in vitro, 164
ингибирование СоА-производными жирных кислот.
165
ацетилхолин. 358
метаболизм, 344
роль в мышечном сокращении, 358
ацетилхолин'эстераза, 344
ацетоацетат, 85
- образование из ацетил-СоА, 135
ацидоз. 151
ацилпереносящий белок, 138
ацил-СоА-дегидрогепаза, 134
ацил-СоА-сит стаза, 132
ацилтрансфераза, 96
ALA-дсгидратаза, 367
ALA-синтаза. 367
регуляция уровня. 368
АТРаза, 64
-Н+>К+-АТРа,а. ?4
- Na+/K+-ATKna. 64
АТР-синтаза митохондрий, 126
АТР-синтаза хлороиластов, 187
- ориентация в мембране, 187
А1ЖЕ-1Юследователыюсти, 282
Бактериальная плазмида, 322
бактериофаг лямбда, 314
- строение, 314
- жизненный цикл. 314. 315
- самосборка, 315
баланс азота в организме, 192
белки, 36
- первичная структура, 37, 40
- вторичная структура, 41
-третичная структура, 43-45
- беспорядочный клубок. 43
- четвертичная структура. 41, 46
белки волос, 48
белки «лейциновой молнии». 279
белки тепловою шока, 267, 299
- фолдинг, 299
белок-активатор катаболизма, 268
белок-ингибитор фосфатазы I, 171
бслок-псрсносчик -эфиров холестерина. 97
белые мышечные волокна. 380
библиотека генов, 319, 320
биливердип, 370
билирубин,370
- антиоксидантные свойства, 213
биомембрапы, 60
биотин,122
418
бокс Прибнова, 265
болезнь
I-клеток, 302
Гоше, 57
кленового сиропа, 197
коровьего бешенства, 298
- Крюцфельда-Якоба, 298
Помпе, 205
- Тея-Сакса, 57, 205
буферные растворы, 31, 32
В-клстки, 330
в
Вакуоль. 59
ван-лер-ваальеовы силы, 28
варфарин. 209
везикулы. 52
вектор, 322. 327
взаимодействие кодом антикодоп, 287
вир! ильные В-клстки, 331
- активация, 335
вирном,308
вироиды, 3 15
вирус
- гриппа. 312, 313
- коровьей оспы. 3 11
СПИДа. 312
вирусы, 308
высвобождение из клеток, 3 10
- механизмы репликации, 311
типы генетического материала, 309
витамин F, 219
- реакция восстановления, 219
витамин Λ, 360, 361
витамин Β|Ί, 226
- кофактор метионинсинтазы. 226
витамин li, 213
- ловушка кислородных радикалов, 213
витамин К, 209
роль в свертывании крови, 210
витамин С, 213
ловушка кислородных радикалов, 213
внутренняя мембрана митохондрий, 102
водорасщепляюший центр, 186
водородная связь, 28
вторичный посредник, 166
высоко энергетический фосфат, 22
Галактоза. 56
- в церебро зидах, 56
- превращение в ипокозу, 91
- фосфорилирование галактокипазой, 91
галактоземия наследственная, 92
β-галакгозидаза, 156
синтез у Е. coli. 156
β-галактозидаза эпителиальных клеток, 77
β-галактозидпсрмсаза, 267
галактозидтрансацетилаза, 267
галактокиназа, 91
ганглиозид, 56, 57, 62
гастрин, 74
- механизм активации аденилатциклазы, 349
гексадеканоевая кислота, 53
гсксо зофосфат и зомера за, 150
гексокиназа. 91
- Кк] для глюкозы, 91
гель секвенирующий, 324
гем, 366
синтез, 367
- распад, 370
- регуляция синтеза, 368
гемап лютинин. 312
гемато-пшефалический барьер. 148
гемоглобин, 372
механизм аллостерического изменения молекулы.
373
- структура, 372
связывание кислорода, 372
влияние рН, 375
гемофилия А, 208
ген, 240
- множественные копии. 240
- 5'-конец, 265
нстранслирусмая область, 264
- терминаторная область, 265
генетический код, 285
вырожденность, 285
геномная библиотека, 320
геномное клонирование, 318
генотсрапия. 327
гены, 267
- домашнею хозяйства, 274
- конститутивно жсиресспруемыс, 267
- некодирующие белки. 283
- прерывистые, 271
- рибосомной и тРНК, 240
419
гепарин, 209
гстерохроматин, 240
гибридизация, 233
гидроксиапил-СоА, 134
гидроксиацил-СоА-дегидрогсназа, 134
β-гидроксибутират, 85
β-грдроксибутиратдсгидрогсназа, 135
гидроксилазы, 49
гпдроксилизин, 48
гидроксилированис, 48
3-гидроксиметил-З-глутарил-СоА, 135
гидроксимстилглутарил-СоА-редуктаза. 146
в регуляции синтеза холестерина, 146
гидроксипролин, 49
гидрофобное взаимодействие, 29
гиперхолсетеринемия, 97
гипогликемия, 83
гипоксантин, 217
минорное основание, 217
гипоксантин-гуанин-фосфорибозилтрансфсраза
(ГТФРТ), 222
гираза, 245
- обратная гираза термофильных бактерий, 247
гистидин, 39
гистоноподобпые белки, 240
гистоны, 237
комплекс с ДНК, 238
главный комплекс гистосовмсстимости, 335
глазок, 366
гликоген, 81, 205
расщепление в лизосомах, 205
гликогемин. 86
гликогенсинтаза, 170
- фосфорилированис протсинкиназой А, 170
- регуляторный каскад активации, 171
гликогенфосфорилаза, 162
- активация AMP, 162
взаимопревращение а и «-форм под действием
киназы и фосфатазы, 170
ингибирование АТР и глюкозо-6-фосфатом, 163
обращение активации фосфорилазы <7, 171
гликогенфосфорилаза, 89
гликозидпая связь, 76
гл π кози л и рован и е, 46
гликолиз. 99
- общая характериешка процесса, 101
аэробный, 102
аназ-рорбнмй, 102
1ермодииамически необратимые реакции, 148
ппколипиды, 57
l in ко протеины. 62
171И КО С ψ И Η ГОЛ И ГI ИДЫ, 5 7
гликофорин, 62, 66
гликохолевая кислота, 78
гл и оке плат, 152
глиоксилатный цикл, 152
глиоксисомы, 152
глицеральдегид-3-фосфатдегидрогсназа, 112, 160
кооперативные свойства, 160
глицсрип-3-фосфатдегидрогеназа, 114
глицеринкииаза, 151
роль в превращении глицерина в глюкозу, 151
глицерофосфатдегидро! епаза, 143
глицерофосфатная челночная система, 114
глицерофосфолиппд, 57
глицин, 38
глобин, 370
регуляция синтеза в )ршроидны\ клетках, 370
глутаматдегидрогеназа, 194
роль в обмене аминокислот. 194
γ-глутамил-цистсинил-глицин. 213
глутамип. 202
- транспортная форма аммиака в крови, 202
глутаминаза, 202
печени, 202
ночек, 202
глутаминсинтетаза. 202
глутатион, 213
-дефицит в эритроцитах и возникновение гемолиза,
214
глутатионпероксидаза, 214
глутатионредуктаза, 214
глюкагон, 84, 165
- активация фруктозо-1,6-дифосфатазы, 175
- регуляция активности фосфофруктокиназы и
пируваткиназы, 174, 175
- регуляция гормон-чувствительной липазы, 97
регуляция уровня глюкозы в крови, 165
глкжогенные аминокислоты, 149
-образование при голодании, 149
глюкозамин, 56
глюкозо-6-фосфатаза, 89. 150
- субклеточная локализация, 89
глюкозо-6-фосфатдегидрогсназа, 178
глюкозо-аланиновый цикл, 202
глюкозофосфатизомераза. 1 К)
глкжокиназа, 87
- Км для глюкозы, 91
глюконсогенсз, 83
- в печени, 148
- карбоксилирование пиру вата, 149
- схема реакции, 148
420
ферменты, 148, 149
головка миозина, 383
- АТРазная активность, 383
- конформационные изменения, 383
напряженная конформация. 383
- расслабленная конформация, 383
юлоданис, 84
Гольлжи комплекс, 301
гомейотические гены, 279
гомсобокс, 280
гомеодомеппые белки, 280
гормональная регуляция гликолиза и глюконеоп
175, 176
- в печени и мышцах, 175
гуапилаткиназа, 222
гуанилатцикдаза, 251
гуаниловая кислота, 217
гуанин, 230
губчатая энцефалопатия, 298
гуморальный иммунитет, 329
G-белки, 348
GRB-белок, 354
GSH, 213
GSSG. 214
д
Дау н ре гуляния, 167
двенадцатиперстная кишка, 74
девствящий фермент, 90
дегидрогеназы. 46
деградация белков, 304
дезаминированис аминокислот, 194
дезоксиадснозилкобаламин, 136
дезоксиаденозин, 217, 23 1
дезокси! уанозин, 231
дезоксинуклеозиды, 231
дезоксирибоза, 230
дезоксирибонуклеиновая кислота (ДНК), 230, 242,
- антипараллельность цепей, 235-237
большая и малая бороздки, 235
- геномная, 3 18
двойная спираль, 233
кДНК, 222
- комилементарность пар оснований, 233, 293
- геометрия комплементарных пар, 234
- липкерпая. 238
-локализация в клетке, 230
митохондриальная, 282
- пары оснований Уотсона-Крика, 233
петельная структура ДНК Е. соПщ 238, 239
- плавление, 233
- повторяющиеся последовательное ι и, 240
- полярность цепей, 235-237
- провирусная, 313
- размер молекул, 237
- релаксированная форма, 244
- репарация повреждений ДНК у Е. coli, 254
- прямая репарация, 255
- репарация с жепизией нуклеотида, 255
- АР-сайта, 256
- сателлитная, 240
- сверхенирализация, 244
- положительная, 244
- отрицательная, 244
- синтез, 249
- направление роста цени, 249
лидирующая цепь, 250
- запаздывающая цепь, 250
- стопка оснований, 234
- теломерная, 257
- синтез, 258
- упаковка в ядре, 237
- уровни организации, 238
- химическая стабильность, 232, 233
- цепь, 232
- кодирующая, 264
- кодирующая часть структуры, 232
- матричная, 264, 265
- нскодирующая, 264
- нсматричная цепь, 264, 265
- Alu-послсдовательности, 240
- А-форма, 237
- В-форма. 237
-Z-форма, 237
дезоксирибонуклеотидьг 217
- dAMP, dGMP, dCMP, dTMP(TMP), 217
дезоксирибополинуклеотид, 23 1
- структура, 23 1, 232
дезоксицитидин, 231
дскарбоксилирование. 85
денатурация, 37
дефосфорилирование, 64
диабет сахарный, 85
диацилглицерин, 56
- в синтезе фосфолипидов, 143
- вторичный посредник, 352
дш пдробиотерпп. 197
лш пдрокспбепзол (катехол). 200
лнгндрофолш, 226
лш идрофолатредуктаза, 226
дидезокси-меюд секвепирования ДНК, 323
липеии,392
дипукдеотид, 231
ли мет ил, 36
диссоциация, 23
дисульфидные свят, 45
2,3-дифосфоглицерат, 374
дифтерийный токсин. 297
ингибирование НН2-транслоказы, 297
диффузия облегченная, 67
ллинный концевой повтор, 313
ДНКазы, 204
ДНК-лигаза, 252
ДНК-полимераза, 248, 249
ДНК-полимеразы Е. coli. 248. 249
- Pol I, 248
-Pol 11,248
- Pol III, 248
ДНК-полимсразы эукариот, 256
ДНК-связывающие белки, 277
гомеодоменные белки, 278
спираль поворот спираль, 278
типа «лейциновой молнии». 279
- тина «цинковых пальцев», 279
локин! -белки, 300
дол ихол фосфат, 301
домен, 47, 48
дыхательная цепь митохондрий. 124
баланс между синтезом АТР и транспортом
электронов, 127
выкачивание прогонов, 127
комплексы переносчиков электронов. 123-125
- механизм синтеза АТР, 126
дыхательный контроль. 164
Dam-метилаза, 254
DnaB. 243
DnaA. 243
Ε
Ьдюпл-СоА-ι iupai аза. 134
енолпирува!. 1 13
еполфосфагы. 25
ж
Железо-чувствительный элемент. 281
желчные кислоты, 78
з
Зажим, 251
затравка гликогена, 86
зимогены, 72
зонд гибридизации, 320
И
Изоаллоксазин, 101
и зо цитрат, 119
изоцитратдегидрогеназа, 119
изоцитратлиаза, 152
имидазол, 40
иммунитет, 329
иммунная система, 329
- роль цитокинов, 337
иммуноглобулины. 332
- структура lg G, 332
инвертированные концевые повторы, 259
ингибитор, 21
- трипсина,75
фосфатазы I, 171
- фосфорилирование протеинкиназой А, 170
ингибиторы протсиназ, 112
в крови, 112
индукция. 268
индуцированное соответствие, 19
инициация, 243
инозин. 217
инозинмонофосфат, 218
инозиновая кислота, 218
- синтез dc novo. 220
инозиттрифосфат, 352
инсерционная последовательность, 259
инсулин, 167
- активация фосфатазы I, 1 71
-индукция глюкокиназы. 169
мобилизация переносчика глюкозы, 168
- регуляция метаболизма, 167
422
-регуляция поглощения глюкозы и жиров клепками,
165
интеграла, 313
интегральные белки, 60
интерфаза, 393
интерфазные хромосомы, 239
интерферон,357
- рецепторы, 357
- ISRE-элсменты, 357
- JAK-киназа, 357
ингроны. 273
- модель ранних нитронов, 273
- модель поздних нитронов. 273
ионные связи, 28
использование холестерина в организме, 94
IRE-связывающий белок. 282
к
Кадхерины, 67
кальмодулин. 171, 172
- в реализации 'эффектов Са2+, 353
кальций, 384, 385
- транспорт в мышце, 385
капсид, 308
карбамоилфосфат. 201
карбамоилфосфатсинтетаза, 201, 202
- аллостсрическая регуляция N-ацетил шута
202
карбоангидраза, 74
карбоксибиотин, 138
карбоксилирование пиру вата, 148
карбоксипегггидаза. 75
кардиолипин.55
карнитин-ацилтрансфераза I, 133
- локализация. 133
карнитин-ацилтрансфераза II, 133
-локализация, 133
β-каротин, 213
- антиоксидантные свойства. 213
- провитамин А, 361
каскад реакций. 207
катаболизм. 21
катал аза, 213
катализ, 18
катализатор, 18
каталитический центр. 19
катехол. 200
катехоламины, 343
- синтез и структура, 343
квашиоркор, 192
кератин, 42, 48, 50
β-кетоацил-СоА, 133
β-кетоацилеинтаза, 138
β-кетоацил-СоА-тиолаза, 133
а-кетоглутарат, 119
- в обмене аминокислот, 198
источник для синтеза глутамата, 198
а-кетоглутаратдегидрогеназа, 120
кетонсмия при диабете. 97
кетоновые тела, 85
ксфалин, 55
киллерные клетки, 329
киназа, 27, 87
- легких цепей миозина, 386
- фосфорилазы, 170
- активация ионами Са2+, 171
кинезин, 392
кинетика ферментативной реакции, 156, 157
- модель Михаэлиса-Ментен, 156, 157
кинстохор, 393
кирромицин, 297
клатрин, 205
- в механизме проникновения вируса в клетку, 308
клетки памяти, 336
клеточный иммунитет, 329
клон генов, 3 18
клональной селекции теория, 331
клонирование кДНК, 322
клонирование, 318
клофибрат, 206
кодон, 285
колбочки, 359
коллаген, 48-50
коллагенозы, 50
комплекс цитохромов bf\ 186
комплемент, 333
консенсусные последовательное! и, 265
константа Михаэлиса, 157
конститутивные гены, 267
копропорфириногены, 367
Кори цикл, 15 1
коррекция неправильного спаривания, 103
кофермент А, 103
- строение, 103
коферменты, 46
красные мышечные волокна, 380
крахмал, 76
креатин, 381
423
креашнкиназа, 380
креапшфосфат, 380
крнсш. 102
ксантиноксидаза, 213. 223
генератор перекиси водорода. 213
ксенобиотики, 210
- группы веществ, 210
накопление в организме, 210
способы модификации, 21 1
ксеродермальный пигмент*, 252
копирование, 270
л
Л акта за, 77
л акта г. 150
источник пиру вата для глюконеогенсза, 150
лактатдегидро! еназа, 101
лактоза, 77
лактозный оперой, 267
. ι а кто зо 11 ерм саза, 268
ламнпм,389
легкие цепи миозина, 382
легкий голод, 84
лейкотриены, 146
роль в организме, 146
лестница секвенирующая. 325
лецитин,55
лецитин-холестсрин-ацил-трапсфераза (ЛХАТ), 96
Леша-Нихана синдром. 222
неврологическая симптоматика, 223
лиганд-зависимые каналы, 358
лидерная последовательность. 303
лпдерный пептид, 270
лизин, 39
лизогепная клетка, 314
лизогенпый путь. 314
ли зосомные болезни, 205
лпзосомы, 204
вторичные, 205
первичные, 204, 205
слияние с аутофагосомами. фагосомами, эндо-
сомамп,205
фермешы, 204
лпзоцпм. 3 10
лимфа. 79
липаза юрмоичувсизтельпая. 78, 97
- активация в жировых клетках, 176
зависимость от физиологических потребностей
организма, 93
- сАМР-зависимое фосфорилированис, 176
липидные мембраны, 58
липидный бислой, 58 60
липкие концы, 3 19
липоевая кислота, 116
липооксигеназа, 146
липоиротсинлипаза, 93
·- в молочной железе при лактации, 156
лииопротеины, 94
ли по сомы, 60
литический путь, 314
ловастатин, 146
ловушки кислородных радикалов, 213
ЛПВП, 94-97
ЛПНП, 94-97
ЛПОНП, 94-97
ЛППП, 94-97
Μ
Макрофаги, 330
макроэргические производные фосфорной кислоты. 23
макроэргическис связи, 25
малат-аспартатная челночная система, 115
малатдегидрогеназа, 121, 142
малатсинтаза, 152
малик-фермент, 141, 190
малонил-СоА, 138
мальтаза, 77
мальтоза, 76
маныоза. 62
матрикс митохондрий, 102
мевалонат, 146
ме!алобласгичсская анемия, 226
мембранные белки, 46
мембраны митохондрий, 62
мембраны хлоропластов, 62
метаболизм аминокислот, 192
-общая схема катаболизма. 193
метаболизм жиров, 164, 165
- регуляция внеклеточными агентами, 165
метаболизм ксенобиотиков, 210
- вторичная модификация, 211
метаболические реакции, 14
424
мстафазныс хромосомы, 238
метгемоглобин, 214
метил-зависимое исправление ошибок, 254
мстилмалонил-СоА-мутаза, 136, 226
мел илмалонил-СоА-эпимсраза, 136
мстионин,198
-синтез S-аденозилметионина, 198
- перенос метильных групп, 198
метионипсинтаза, 226
метотрексат, 226, 240
механизм аллостерической регуляции, 158
механизм действия ферментов, 18
микротрубочки. 389
- функции, 391
минимиозины,390
минорные основания, 217
миопюбин, 371
- связывание кислорода, 372
- структура, 372
миозин, 382
митохондрии, 69
- строение, 102
мицелла, 53
мобилизация запасов жиров, 83
м одел ь « kj ι юч-за мок», 19
модель кооперативного связывания субстрата Моно,
Уаймена, Шанжс, 159
- напряженное Т-состояние фермента, 159
- расслабленное R-состояние фермента, 159
модель последовательная Кошланда, Нсмети, Филмора,
160
- напряженное Т-состоянис фермента, 160
- расслабленное R-состоянис фермента, 160
молочная кислота, 16
мононенасыщенные кислоты, 58
монооксигеназы, 197
моносахариды. 56
монослои, 59
мотив, 277
мочевая кислота, 213, 223
- антиоксидантные свойства, 213
- образование, 223
- роль в развитии подагры, 223
мутаротация, 76
муцины,73
мышцы поперечно-полосатые, 384
- регуляция сокращения, 384
мягкий сгусток, 208
МНС-белки, 335
Η
Налидиксовая кислота, 247
- инактивация ι иразы бактерий, 247
направленный мутагенез. 327
насосы, 66
неактивные факторы транскрипции, 276
нсгистоновые белки, 240
нейраминидаза, 312
нейромедиаторы, 66, 343
- норадреналин, 343
- регуляция гормон-чувствительной липазы, 97
- ацетилхолин, 343
нейтральные липиды, 57
нейтрофилы, 212
неоднозначное соответствие («качание»), 287, 288
неполярные липиды, 53
никотинамидадсниндикуклеотид (NAD), 100
-восстановление, 100
-структура, 100
-функции, 100
никотинамидадениндинуклеотидфосфа! (NADP), 141
- в синтезе жирных кислот, 141
нитроксидсинтаза, 351
нуклеаза, 80
нуклеиновые кислоты, 80
нуклеозид. 216
- общая структура, 216
- нумерация атомов, 216
нуклеозиддифосфаткиназа, 222
нуклеозидтрифосфаты, 219
нуклеокапсид, 308
нуклеосома, 238
нуклеосомная сердцевина, 238
нуклеотид, 216, 230
- нумерация атомов, 230
- общая структура, 216
- строение, 230
- углеводный компонент, 216
NADH, 114
- реокисление челночными системами переноса
электронов, 114
NADPH, 141, 142
О
Облегченная диффузия, 77
образование НС1 в желудке, 73
425
обратная гранскриптаза, 313
ограниченный иротеолиз, 207
Оказаки фрагменты, 250
- петельная модель синтеза, 251
окаймленная ямка, 308
окисление биологическое, 99
β-окисление жирных кислот, 134
окисление жиров и аминокислот, 107
- генерация энергии, 107
окисление ненасыщенных жиров, 134
окислительное фосфорилирование, 106, 126, 127
- хсмиосмотический механизм, 127
оксалоацетат, 1 52
- в ιлюконсо1 снезе, 152
оксигеназа смешанного действия, 210
оксид азота, 351
октадсканоевая кислота, 53
олигосахариды, 55
онкоген, 314, 356
онкогенные ретровирусы, 314
оперон, 268
- лактозный, 268
- триптофановый, 269
орнитин,200
орнитинтранскарбомоилаза, 201
оротовая кислота, 225
- в синтезе пиримидинов, 225
отжиг, 233
открытая рамка считывания, 316
отношение Р/О, 130
π
Палиндром,278
палочки, 359
пальмитиновая кислота, 58
памахин, 180
пандемия, 3 12
панкреатит, 75
панкреатические проферменты, 75
панкреатические ферменты, 75
иантотсновая кислота, 103
парная симметрия, 278
пентозофосфатный путь, 178
в эритроцитах, 180
- значение, 178
- неокислительная стадия, 178
окислительная стадия. 178
пепсин, 20, 74
426
пепсиноген, 74
пептидилпролинизомераза, 278
переваривание, 72
- белков, 73
- белков в тонком кишечнике, 74
- жиров, 78
- крахмала, 76
- углеводов, 75
перекись водорода, 213
- образование, 213
- роль в образовании гидроксильных радикалов, 213
пернициозная анемия, 226
пероксисомы, 136, 299
- роль в обмене липидов, 205, 206
печень: роль в гомеостазс глюкозы в крови, 83
пигмент, 61
пиридоксаль-5'-фосфат, 195
- в реакциях трансаминирования, 195
ниридоксальфосфал, 195
пиридоксамин,195
пиридоксаминфосфат, 195
пиримидин,217
пиримидиновыс нуклеотиды, 224
- синтез, 225
пиримидиновые основания, 217
- структура, 217
пирофосфат, 24
пирофосфатаза, 27
пиру ват, 148
- в глюконсогенезе, 148
- окислительное декарбоксилирование, 116
- транспорт в митохондрии, 116
пируватдегидрогеназа (ПДГ), 116
- ингибированис ацетил-СоА и NADH, 164
- активация CoA-SH и NAD\ 164
- регуляция киназы и фосфатазы, 164
пируватдекарбоксилаза дрожжей, 103
пируваткарбоксилаза, 141. 148
пируваткиназа, 113
плазмалоген, 57
плазматические клетки, 329
плазматические мембраны, 62
плазмида, 322
плазмин, 209
плазминоген, 209
пластогидрохинон, 186
пластохинол, 186
пластохинон, 183, 186
пластоцианин, 183, 186
подагра, 223
-лечение аллопуринолом, 223
поли-А.281
полиадснилированис. 270
поли-А-«χ воет». 281
полимеразная псиная реакция. 325
полимеризация микротрубочек, 391
иолинуклеотид. 230
- структура цепи. 232
полиовирус, 311
полипептид, 60
полисахариды. 75, 76
полисома, 295, 296
полярные л и π иды, 57
поперечно-полосатая мышца. 380
иорфобилиноген, 367
последовательность GATC, 254
последовательность Шайна-Дальгарно, 293
потенциал-зависимые каналы, 358
потенциалы окислительно-восстановительные, 104
праймаза, 249
- синтез праймера. 249
праймер, 248
ирсбелок, 303
нримахин. 214
приоиы,298
пробелок, 303
продолжительное голодание, 84. 85
промежуточные филаменты, 389
ирометафаза, 393
промотор, 264
- «сильный». 667
- «слабый», 267
нропионил-СоА, 139
пропионил-СоА-карбоксилаза. 136
простагландины, 145
- физиологические функции, 146
- синтез, 145
протсазы. 74
протеин азы, 74
протсии-дисульфидизомераза, 298
протеинкиназа, 161, 169
- инактивация белкового ингибитора фосфатазы, 171
- роль в превращении фосфорилазы в в а. 169
протеинкиназа А, 169
- фосфорилированис киназы фосфорилазы, 169
протеинкиназа С. 353
протеинкиназа сАМР-зависимая. 161
- структура и механизм аллостерической активации
сАМР, 161
- роль в регуляции активности ферментов, 161
протеинкиназа G, 350
нротсинфосфатаза I, 171
- дефосфорилпроваиие фосфорилазы а. 171
протеолиз, 73
иротсолитические ферменты, 74
протондвижущая сила, 126
протоонкоген, 314, 355
протромбин, 207
профаза, 393
проферменты, 75
процессинг, 272
проэластаза, 75
прыгающие гены, 259
псевдогены, 240
птсроилглутаминовая кислота, 219
пуриновыс нуклеотиды, 217-219
- синтез de novo, 219
- регуляция синтеза, 224
пуриновые основания, 216
-структура, 217
- реутилизация при синтезе нуклеотидов, 222
пуф хромосомный, 273
Ρ
Разветвляющий фермент, 88, 89
расщепление гликогена, 89
реакции переноса одыоуглеродных фрагментов, 218,219
реакционные центры, 184
реакция «бегства или борьбы», 169
регуляторный каскад, 170, 171
- схема активации фосфорилазы, 171
регуляция уровня глюкозы в крови гормонами, 166
регуляция активности ферментов, 155
регуляция количества ферментов, 155, 156
редактирование мРНК, 272
редактирование транскриптов, 272
редокс-пары, 104
редокс-потенциалы, 104
резонансный перенос энергии, 184
рекомбинантная плазмида, 323
рекомбинантный фаг, 319
релизинг-гормоны, 343
реннин,74
репарирующая система. 254
- компоненты, 254
- Mut H, 254
- Mut L, 254
- Mut S, 254
репликативная вилка, 242
- ферментативный комплекс Ε. coli, 250
427
устройство у эукариот, 257
репликация, 242
полу консервативная, 242
репликон, 243
репрессия, 268, 270
реирессор, 268
ресинтез нейтральных лииидов, 79
ретикулоцит, 366
ретиналь, 360
рстровирусы, 313
- в генотерапии, 314
реутилизация пуриновых оснований, 222
рефоллинг, 297
рибит, 101
рибонуклеиновая кислота (РНК), 262
- мРНК, 262
- общие свойства, 263
- редактирование, 282
- полицистронная, 263
-стабильность и структуры ее определяющие,
- роль AURE-послсдовательности, 282
-мяРНК, 272
- отличие от ДНК, 262
-рРНК, 282, 289, 290
- тРНК, 246, 247, 282
- структура, 246, 247
- инициаторная, 291
рибонуклеотидредуктаза, 224
рибосома, 270, 289
А-участок, 294
- Е-участок. 294, 295
- Р-участок, 294, 295
- структура, 289
- эукариот, 295
риботилирование, 218
рибофлавин,101
рибулозо-1,5-дифосфат. 188
- образование, 188
рибулозо-1,5-дифосфаткарбоксилаза, 187
рицин,297
РНК-иолимсраза II, 270
копирование транскрибируемой РНК, 270
РНК-нолимераза ДНК-зависимая, 262
- направление злонгации цепи. 263
- характеристика катализируемой реакции, 262
РНК-рсиликаза. 312
родопсин, 360
Ras-пугь, 354
- и онкогенсз. 355
Rubisco. 188
С
Самоинактивация ксантиноксидазы, 189
- при метаболизме аллоиуринола. 189
сарколемма, 381
саркомер, 381
саркоплазматический рстикулум, 381
Саузерн-блот-анализ, 327
свертывание крови, 207
- внешний механизм, 207
- внутренний механизм, 207
- регуляция свертывания, 208. 209
- схема, 208
сверхспираль, 244
световосиринимающий аппарат, 359
свободные жирные кислоты, 93
связывание субстрата, 158-160
- гомотропнос кооперативное, 159
1 сдвиг рамки считывания, 290
ссквенирование, 37
секвенирование ДНК, 323
- метод Максама-Гилберта, 323
- метод Сэнгера, 323, 324
секреторные белки, 46
сердцевина нуклеосомная, 238
серии.39
- дезаминирование, 195
сери нгидрокси мстил аза, 220
- перенос гидроксиметильной группы, 220
сериндегидратаза, 195
ссриновыс протсиназы, 20
серповидноклеточная анемия, 377
сиаловая кислота, 56
сигма-фактор, 266
сигма-факторы, 267
- регуляция транскрипции генов. 267
сигнал-узнающая частица, 300
сигнальная пептидаза, 300
сигнальные молекулы, 340
- биологическая классификация. 340
симвастатин, 146
сим порт, 65
- аминокислот и ионов натрия, 75
- глюкозы и ионов натрия, 77
сит азы, 88
синтез, 198
- аланина, 198
- аминокислот, 198
- аспарагиновой кислоты, 198
- белка в митохондриях. 296,297
428
- глицина, 199
- глутаминовой кислоты, 198
- глицсрофосфолипидов, 143
- глюкозы из глицерина. 151, 152
- глюкозы из II пру вата, 148
- жирных кислот, 140,
локализация в клетке. 141
-стадии восстановления, 140
кетоновых тел, 135
ненасыщенных жирных кисло!, 142
- нуклеотидов
- образование АТР и GTP из AMP и GMP, 222
превращение dUMP в dTMP, 225
- образование UTP из UMP, 224
- СТР из UTP, 224
-ОМРиАМРиз инозиновой кислоты, 221
- триглицеридов, 143
холестерина, 146
- регуляция, 146
синтстазы. 88
скользящее кольцо. 250
- стабилизация одиночных цепей ДНК в реиликатив-
ной вилке, 250
скрининг бляшек, 320
слабые связи, 27
слизи, 46
смешанная мицелла. 78
соляная кислота, 73
- механизм образования. 74
соотношение между AMP, ADP и АТР, 27
спсктрин, 66
СПИД, 311
сплайсинг. 271
альтернативный, 273
- механизм, 271
- сплайсосомы, 272
стартовая точка. 265
стеариновая кислота. 53
стероидные гормоны, 346
регуляция жсирессии генов, 345
рецепторы, 345
роль белков теплового шока. 345
стоп-кодоны, 285
стрептомицин. 297
стжинг-взаимодействия, 253
субстратное фосфорилированис. 112
- в гликолизе, 113
- в цикле лимонной кислоты, 1 15
сукцинатдегидрогеиаза, 121
сукцинил-СоА, 120, 136
- в синтезе тема, 367
при окислении жирных khcjuvi с нечетным числом
атомов, 136
сукцинил-СоА-син тстаза, 120
супероксид, 212
- мутации митохонриалыюй ДНК. 212
- образование в организме. 212
- образование при спонтанном окислении. 212
супсроксиддисмутаза, 212
сфероцитоз, 66
сфингозин, 55
сфинюмислин, 56
SOS-белок, 356
SSB-белки, 247
Τ
β-Талассемия, 272
тасование жзонов, 272
таурин, 78
таурохолсвая кислота, 78
Т-димеры, 255
теломераза, 257
телофаза, 393
темновые реакции, 183
1срминация, 266-270
1Страгидробиоптерин. 195
тстрагидрофолаг, 219
тиаминиирофосфат, 116, 178
тилакоиды, 183
- организация фотосинтстичсского аппарата, 183
тимидилатсинтетаза, 226
тимин, 217
τ иол аза, 133
типы водородных связей. 28
тиреоидные гормоны, 345
тирозин, 39
тирозинкиназа, 354
- аутофосфорилирование, 354
титин.382
тканевой активатор плазминогена, 207
Т-клетки, 330
- клеточный иммунитет, 337
а-токоферол, 213
толстые филамепты, 382
гонкие филаменты, 382
топоизомераза, 245
- биологическая роль, 245
топоизомераза I, 245
429
юиоизомераза II, 245
топоиюмераза I эукариот, 246
точка начала репликации, 242
транс-Т^-еноил-СоА, 134
трансальдолаза, 178
трансаминазы, 195
трансаминирование, 194
механизм реакции, 195
трансгенез, 327
трансгенные животные, 327
транедезаминирование, 195
транскарбамоилазы, 201
транскрипция, 263
- гена в митохондриях, 2X2
- инициация у Е. coli, 264
- распаковывание ДНК, 273
- расплетание ДНК, 266
- терминация транскрипции, 266
- фазы, 265
транслоказа, 294
транслокация, 294, 295
трансляция, 263
- инициация, 290, 291
-yE.col'u 291,292
трансметилаза, 198
транспозаза, 259
транспозиция, 272
гранспозоны, 259
транспорт белков, 302, 303
- в лизосомы, 302
- в митохондрии, 303
- в цитоплазматическис мембраны. 302, 303
- в ядро, 303
транспорт триглицеридов и холестерина, 96, 97
трансферрин, 368
треонин, 39
триглицериды, 58
- содержание в различных липопротеинах, 96, 97
тринитроглицерин,352
триозофосфатизомераза, 112
триозофосфатизомеразы, 45
трипсин, 75
трипсиноген, 75
тршнофановый оперон, 269
тромбоксаны. 145
- агрегация тромбоцитов, 146
тромбоциты,366
гронины,343
тропоколлаген, 48. 49
тропомиозин. 384
фопоиин, 384
тропонин-тропомиозиновый комплекс, 385
тубулин, 391
тупые концы, 322
тяжелые цепи миозина, 382
TAF-белки, 276
ТАТА-бокс, 275
TFIID-комплекс, 276
ТВР-белок, 276
У
Убиквитин, 304
убихинон,124
- одно- и двухэлсктронное восстановление, 124
унипорт, 66
уравнение Гендерсона-Хассельбаха, 23
уравнение Михаэлиса-Мен ген, 18
урацил,217
уреаза, 19
уридиловая кислота, 217
уридилтрансфераза, 92
уридин, 217
уридинтрифосфаг в синтезе гликогена, 87
UDP-галактоза, 92
UDP-глюкоза, 91,92
UDP-глюкуронозилтрансфераза, 211
UDP-глюкозопирофосфорилаза, 87, 88
Φ
Фа го сомы, 204
фагоцитирующие лейкоциты, 333
фагоцитирующие нейтрофилы, 212
фагоцитоз, 204
фагоциты, 213
фазы клеточного цикла, 243
фактор Rho, 266
фактор VIII, 208
фактор X, 207
фактор XII, 207
фактор активации тромбоцитов, 334
фактор инициации IF2, 291
фактор инициации IF3, 291
- связывание с 308-субчастицей, 291
фактор инициации транскрипции D для полимеразы И, 2"/
430
факторы роста, 341
-CSF, 341
-EGF, 341
-PDGF. 341
регуляция высвобождения, 344
факторы элонгации цитонлазматическис, 293
фенилаланин. 197
- метаболизм, 197
фенилалаиингидроксилаза, 197
фенилкстонурия (ФКУ), 197
фенил пиру ват. 197
фенобарбитал, 211
- индукция цитохрома Р450 и системы глюкурони-
лирования, 211
феофитин, 186
фермет, 18
ферментативный катализ, 18-21
фермент-субстратный комплекс, 156
ферменты Михазлиса-Ментсн. 156
ферменты рестрикции. 317
- биологическая функция, 317
ферредоксин. 186
фсррсдоксин-ЫАОР+-рсду ктаза, 186
ферригемоглобин, 214
ферритин, 368
феррогемоглобин. 214
фибриллы. 50
фибрин. 208
фибриновые тяжи, 208
- образование, 208
фибриноген, 208
-структура, 208
- превращение в фибрин, 208
фибринопептиды, 208
фиксация атмосферного С(Х. 188
фитостсрины, 58
флавинадениндинуклеотид (FAD), 101
- восстановленная форма, 101
- окисленная форма, 101
строение, 101
флавинмононуклеотид (FMN), 101
- восстановление, 101
- строение, 101
флип-флоп. 57
фолдинг, 297
фолиевая кислота, 219
форболовые зфиры, 353
формилтрансфераза, 219
формильная группа, 219
Nl(,^op\ULi-FH4. 219
фосфат, 24
фосфатаза, 161
фосфатидилинозит, 60. 62
фосфатидилхолин,55
фосфатидилэтаноламин. 55
фосфатидная кислота, 54
фосфоглицераткиназа, 112
фосфоглицератмутаза, 113
3-фосфоглицерат, 187, 188
-образование, 187
- образование крахмала при фоюсинтезс, 188
фосфоглицериды, 54
фосфоглюкомутаза. 87
6-фосфоглюконатдегидрогеназа, 178
фосфодиэс ι ераза, 166
фосфодиэфирная связь, 232
фосфоенол пиру ват, 25, 113, 148
- образование в глюконеогенезс, 149
фосфоенолнируваткарбоксикиназа, 149
фосфоснолпируваткарбоксилаза растений, 189
фосфоинозитидный каскад, 353
фосфолипаза С, 352
4-фосфопантотеин, 139
фосфорибозиламин, 218
5-фосфорибозил-1 -пирофосфат, 218
фосфорилаза /?, 169
- активация AMP, 162
фосфорилирование, 161
- гидроксильной группы серина, 161
- роль в регуляции ферментативной активности. 161
фосфорильные группы, 22-24
фосфоролиз гликогена, 89
фосфофруктокиназа, ПО, 163, 175
-активация фруктозо-6-фосфатом, 163
фосфоэфиры, 24
фотодыханис, 189
фотосинтез, 182
- механизм евстозависимого восстановления
NADP+, 185
- общее уравнение, 184
- схема световой фазы, 183
фотосистема I, 184
фотосистема II, 184
фотофосфорилированис, 187
ФРПФ, 218
ФРПФ-синтетаза, 218
- аллостерическое ингибированис, 224
фруктоза, 75
фруктозо-1,6-дифосфат, 163
-активация нируваткиназы, 163
фру ктозо-1,6-д и фосфатаза, 149
- ишибированис AMP, 163
431
фрукто'зо-2,6-дифосфат, 174
активация фосфофруктокиназы, 174
ингибирование фруктозо-1,6-дифосфатазы, 174
фу коза, 62
фумараза, 121
фусидовая кислота, 297
ФФК2, 174
X
Хсликаза, 243
- раскручивание двойной спирали ДНК, 250
хслпсрные Т-клетки, 330
хиломикрон, 93
- поглощение жира клетками, 93
хилом и кроны, 79
- состав, 79
химотрипсин, 75
химотриисиноген, 75
химус, 74
хитин аза, 214
хлорамфеникол, 197
хлоридный сдвиг, 376
хлоропласты, 182, 183
-строение, 183
хлорофилл, 183
- строение, 184
хлорофилл я, 184
хлорофилл я, 184
холерный токсин, 299
холестерин, 58, 78
- содержание в различных липоиротсинах, 96, 97
- использование в организме, 94
- «хороший холестерин», 97
- «плохой холестерин», 97
холецистокинин, 74
«холостые циклы», 154, 155
- на примере глюконсогенсза и гликолиза, 155
потенциальная опасность, 154
холофермент, 46
хроматиды, 393
хроматин, 237, 274
-транскрипционноактивные области, 273, 274
хромосомы, 239
Ц
Целлюлоза, 80
пет ρ организации микротрубочек, 391
церамид, 56
церсброзиды, 56
цианид, 26
цикл
- Кальвина, 188
- схема реакций, 189
- Кребса, 99
- лимонной кислоты, 115-119
- механизм реакций, 119
мочевины, 200
-Q-цикл, 127, 128
циклический аденозинмонофосфат (сАМР), 166
циклическое фотофосфорилированис, 187
циклооксигеназа, 21
циклоспорин, 298
- ингибирование образования ингерлейкинов, 337
циклофилины, 298
цистатионурия, 198
цистеин, 39, 193
цистеиндесульфураза бактерий, 196
цистин, 45
цистрон, 263
цитидиловая кислота, 217
цитидинмонофосфат, 217
цитозин, 217, 231
цитокинез, 390
цитокины, 337
цитоскелет, 66, 389
цитотоксические клетки, 329
цитохром Р450, 210,211
- в реакции гидроксилирования ксенобиотиков, 211
- индукция фенобарбиталом, 211
- синтез стероидных гормонов в митохондриях
надпочечников, 211
- участие в разрушении гема, 211
цитратлиаза, 141
цитрате и нтаза. 119
цитруллин,200
- превращение в аргинин, 200
С3-растсния, 189
С4-путь, 189
- схема реакций, 190
С4-растения, 189
сАМР, 166, 167
- аллостсричсская активация протеинкииазы, 167
- действие на транскрипцию генов, 350
- ингибирование ФФК2, 174, 175
- реакция гидролиза, 168
- регуляция уровня в клетках, 348-350
- синтез из АТР, 166
сАМР-фосфодиэстераза, 166
432
сАМР-чувствительныс элементы промоторов, 350
САР. 268
cGMP, 350, 351
- вторичный посредник, 351
cGMP-фосфодиэстераза, 360
СТР, 144
- в синтезе фосфолипидов, 144
ш
Шапероны, 298
- роль в фолдинге, 298
Шиффовы основания, 194
шпилечная петля, 266
- гистоновой РНК, 281
шпилька, 270
Эйкозаноиды, 145, 340
экзон, 271
экзонуклеаза, 252
экзопептидазы, 74
экзоцитоз, 59, 60
экспресия гена, 240, 263
экспрессирующий вектор, 322
эксцизионная репарация, 255
-у эукариот, 258
эксцизия, 255
эксцинуклеаза, 255
эластаза, 75, 212
- нейтрофилов, 212
- ингибирование а,-антитрипсином в легких, 212
эластин, 50
электронтранспортная цепь митохондрий, 99, 123-125
-железо-серные центры, 124
- иерархия переносчиков электронов, 104-106
-цитохромы, 123
-убихинон, 124
-FMN-белок, 124
элемент-35, 265
элонгация, 248, 265
- механизм, 393-395
эмульгирование жиров, 78
эмфизема легких, 212
- роль курения, 212
эндокринные гормоны, 341
- гипоталамическая регуляция выделения, 343
эндонуклеаза, 255
эндонуклеазы рестрикции, 317
эндопептидазы, 75
эндоплазматический ретикулум. 68. 300
- гликозилирование белков, 301
- транспорт белков, 299
эндосома, 205
- проникновение вируса в клетку, 308
эндоферменты, 76
эндоцитоз, 59, 205
- проникновение вируса в клетку, 308
энергия активации, 18
энергоснабжение мозга и эритроцитов, 83, 84
энзим,18
энтальпия, 14, 15
энтеропептидаза, 75
энтропия, 14, 15
энхансеры, 275
эпимераза, 91
- в окислении жирных кислот, 136
эпимеризация, 91
эпитоп, 333
эритромицин, 297
эритроциты, 148, 366
- старение, 205, 366
этанол, 59
этерификация холестерина ЛПВП, 97
эухроматин, 240
эффект Бора, 375
ю
Юношеский диабет, 97
Ядерная сигнальная метка, 345
ядерные поры, 303, 394
якорная последовательность, 303
433
Оглавление
Предисловие редакторов перевода 5
Предисловие к английскому изданию 6
Благодарности 8
Некоторые традиционные сокращенные
обозначения соединений и единиц,
используемые в данной книге 10
Часть 1. Введение в химические
реакции клетки
Глава 1. Химия, энергия и метаболизм 14
Что определяет возможность протекания химических
реакций? 14
Изменение свободной энергии и обратимость реакций 15
Роль необратимых реакций в стратегии метаболизма ... 16
Почему клетки используют такую стратегию
метаболизма? 16
Из чего складывается величина Δ07 17
Стандартная свободная энергия
и константы равновесия 17
Допустим, что AG отрицательно. От чего зависит,
будет ли протекать соответствующая реакция в клетке
с ощутимой скоростью? 17
Природа ферментативного катализа 18
Как работает фермент? 1 8
Как расщепление пищи в клетках сопряжено
с внутриклеточными реакциями, потребляющими
энергию 21
Высокоэнергстический фосфат 22
Что такое «макроургический фосфат»? 23
В чем главные структурные особенности
высокоэнсргстических фосфатов? 23
Что в клетке служит переносчиком
фосфорильной группы? 25
Как ΛΤΡ подвергается химическим превращениям? 26
Как с помощью ΛΤΡ совершаются другие виды работ? 27
Замечание о еоопюшении между AMP. ADP и АТР 27
Слабые связи и изменения свободной энергии 27
Что вызывает образование и разрыв слабых связей? .... 29
Пели слабые связи так легко рвутся,
каково их значение в биохимических системах? 29
Буферные растворы и значения рКt 30
Как связаны значения рК t и буферные растворы 3 1
Дополнительная литература 32
Вопросы к главе 1 33
Часть 2. Структура белков и мембран
Глава 2. Структура белков 36
Первичная структура белков 36
Что такое нативный белок? 37
Каковы основные факторы, определяющие
трехмерную структуру белка? 38
Структура 20 белковых аминокислот 38
Гидрофильные аминокислоты 39
Аминокислоты, используемые для особых целей 39
Ионизация аминокислот 40
Уровни структурной организации белка:
от первичной структуры к четвертичной 40
Вторичная структура белков 41
α-Спираль 41
^-Складчатый слой 42
Беспорядочный клубок.
или петли полипептидной цепи 43
Третичная структура белков 43
Как строятся белки из а-спиралей.
^-складчатых слоев и соединительных петель? 43
Какие силы поддерживают третичную структуру? 45
Какое отношение к третичной структуре
имеют ковал ситные S-S- связи? 45
Четвертичная структура белков 46
Мембранные белки 46
Белковые конъюгаты 46
Белковые модули, или домены 47
434
Чем интересны домены? 47
Белки волос и соединительных тканей 48
а-Кератины волос, шерсти, рогов и копыт 48
Структура коллагенов 48
Структура эластина 50
Тайна укладки белка 50
Дополнительная литература 50
Вопросы к главе 2 51
Глава 3. Молекулярная организация
клеточных мембран 52
Зачем клетке нужна мембрана? 52
Лииидный бислой 53
Каковы липидные компоненты клеточных мембран? .... 53
Какие полярные группы связаны в фосфолипидах
с остатком фосфатидной кислоты? 55
О номенклатуре мембранных липидов 57
Почему существует так много типов мембранных
липидов? 57
Каков жирнокислотныи состав мембранных липидов? 57
Какую роль играет холестерин в мембранах? 58
Слияние липидных бислосв 58
Проницаемость липидных бислоев 59
Мембранные белки и структура биологических
мембран 60
Что удерживает интегральные белки
в липидном бислое? 60
Заякоривание периферических белков в мембране 61
Мембранные глнкопротеины 62
Функции мембран 62
Транспорт веществ в клетку и из клетки 62
Пассивный транспорт, или облегченная диффузия 63
Активный транспорт 63
Устройство Να +/Κ+-насоса 64
Совместный перенос молекул
чера мембраны (симпорт) 64
Антипорт 65
Унипорт 66
Преобразование сигналов 66
Роль плазматической мембраны в поддержании формы
клеток и их подвижности 66
Межклеточные взаимодействия:
плотные соединения, контактные поры,
ответственные за клеточную адгенпо белки 66
Внутриклеточные мембраны 67
Дополнительная литература 69
Вопросы к главе 3 69
Часть 3. Метаболизм
Глава 4. Переваривание и всасывание пищи 72
Химический состав пищи 72
Переваривание и всасывание 72
Анатомия пищеварительного тракта 72
Что такое переваривание и всасывание
с'энергетической точки зрения 72
Почему организм сам себя не переваривает? 72
Образование проферментов зимогенов 72
Защита эпителиальных клеток с помощью слизи ... 73
Переваривание белков 73
Образование HCI в желудке 73
Пепсин - протеолитический фермент желудка 74
Переваривание белков в тонком отделе кишечника 74
Активация панкреатических проферментов 75
Перенос аминокислот из кишечника в кровяное русло 75
Переваривание углеводов 75
Переваривание крахмала 76
Переваривание и всасывание жиров 78
Что происходит с жирными кислотами
и моноглицеридами внутри эпителиальных клеток
щеточной каемки? 79
Ресинтез нейтральных липидов 79
Что представляют собой хиломикроны? 79
Переваривание других компонентов пищи 80
Дополнительная литература 80
Вопросы к главе 4 80
Глава 5. Предварительные сведения
о распределении и утилизации источников
энергии в различных тканях организма 81
Запасание пищи в теле животного 81
Как различные виды пищевых веществ хранятся
в клетках? 81
435
Глюкоза запасается в виде гликогена XI
Хранение .жиров 8 1
Запасаются :ш аминокислоты в организме? 82
Энергетический метаболизм в различных тканях 82
Роль гормонов в координации распределения
пищевых веществ 84
После всасывания пищевых веществ 84
Легкий голод 84
Продолжительное голодание 84
Диабет 85
Экстремальная ситуация 85
Вопросы к главе 5 85
Глава 6. Биохимические механизмы
транспорта, хранения и мобилизации пищи 86
Превращение глюкозы в организме 86
Механизм синтеза гликогена 86
Как синтез гликогена обеспечивается
необходимой энергией? 86
Как в печени образуется глюкоза? 89
Почему в печени работает глюкокиназа,
а в других тканях - гсксокиназа? 91
Что происходит с другими сахарами,
поглощенными в тонком кишечнике? 91
Пути превращения аминокислот в организме
(в рамках проблемы энергоснабжения) 92
Пути превращения жиров и холестерина
в организме 92
Поглощение жира клетками из хиломикронов 93
Судьба жиров и холестерина 94
Использование холестерина в организме 94
Линопротеины, участвующие
в транспорте жира и холестерина 94
Аполинопротеины 96
Механизм транспорта триглицеридов и холестерина
из печени к другим тканям и обратный перенос
холестерина в печень 96
Как жиры покидают жировые клетки? 97
Как жирные кислоты переносятся кровью? 98
Дополнительная литература 98
Вопросы к главе 6 98
Глава 7. Получение энергии из пищи.
Введение 99
Образование энергии из глюкозы 99
Основные этапы окисления глюкозы 99
Биологическое окисление
и системы переноса водорода 99
NAD" -важный переносчик электронов 99
FAD - важный переносчик электронов 101
Гликолиз -первый этап окисления глюкозы 101
Цикл лимонной кислоты - второй этап
окисления глюкозы 102
Перенос электронов на кислород
третий этап окисления глюкозы 104
Иерархия переносчиков электронов
в электротрапепоргной цепи 104
Генерация энергии при окислении жиров
и аминокислот 107
Взаимозаменяемость разных видов «топлива» 107
Вопросы к главе 7 108
Глава 8. Гликолиз, цикл лимонной кислоты,
система транспорта электронов: реакции
этих метаболических путей 108
Стадия I. Гликолиз 109
Глюкоза или гликоген? 109
Почему на начальном этапе гликолиза
расходуется АТР? I 10
Зачем глюкозо-6-фосфат превращается
во фруктозо-б-фосфат? 110
Изменения свободной энергии AG0 и AG
при альдолазном расщеплении 110
Взаимопревращение дигидроксиацетонфосфата
и глицеральдсгид-3-фосфата 112
ί липеральдегид-З-фоефатдсгидрогсназа.
Образование макроэргического фосфата 112
Заключительные стадии гликолиза 113
Баланс АТР при гликолизе 114
Реокисленис цитоплазматического NADH
челночными системами переноса электронов 114
Транспорт пирувата в митохондрии 115
Стадия 2. Цикл лимонной кислоты 115
Превращение пирувата в ацетил-СоА-
предварительный этап цикла лимонной кислоты 116
В чем истинная магия цикла? 117
Упрощенное описание цикла лимонной кислоты 118
436
Механизмы реакций цикла лимонной кислоты 119
Синтез цитрата 119
Превращение цитрата в а-кетоглутарат 119
Чет ырехугл сродные кислоты 120
Сопряжение гидролиза сукцинил-СоА
с синтезом GTP 120
Превращение сукцината в оксалоацетат 121
Что определяет общее направление реакций в цикле? 121
Стехиометрия цикла 121
Субстратное обеспечение цикла лимонной кислоты ... 121
Стадия 3. Цепь переноса электронов от NADH
и FADH2 на кислород 123
Цепь переноса электронов 123
Что представляют совой
переносчики электронов? 123
Расположение переносчиков электронов 125
Как свободная энергия, высвобождающаяся при
переносе электронов, используется для синтеза АТР? 126
Как выкачиваются прогоны? 127
Как поток протонов может привести к синтезу АТР? 129
Транспорт ADP в митохондрии и АТР из митохондрий 129
Баланс между синтезом АТР
и транспортом электронов 130
Выход АТР при окислении молекулы глюкозы
до СО, и Η,Ο
130
Используется ли потенциальная энергия
в виде протондвижущей силы для выполнения
других задач, не связанных с синтезом АТР? 130
Дополнительная литература 131
Вопросы к главе 8 131
Глава 9. Энергетическая ценность жиров 132
Механизм образования ацетил-СоА
из жирных кислот 132
Активация жирных кислот
путем синтеза их ацил-СоА-производных 132
Транспорт СоА-ироизводных жирных кислот
в митохондрии 132
Превращение ацил-СоА в ацетил-СоА
внутри митохондрии 133
Энергетический выход при окислении жирных кислот 134
Окисление ненасыщенных жиров 134
Всегда ли ацетил-СоА, образующийся при
β-окислении, поступает в цикл лимонной кислоты? 135
Как ацетоацетач образуется из ацетил-СоА? 135
Окисление жирных кислот с нечетным числом
углеродных атомов 136
Окисление жирных кислот в пероксисомах 136
Дополнительная литература 137
Вопросы к главе 9 137
Глава 10. Переход от катаболизма
к анаболизму. Синтез в организме жиров
и родственных им соединений 138
Механизм синтеза жиров 138
Общие принципы 138
Ацилпереносяший белок и β-кетоацил-синтаза 138
Механизм синтеза СоА-производных жирных кислот 139
Организация процесса синтеза жирных кислог 140
Стадии восстановления
в ходе синтеза жирных кислот 140
Что такое NADP ? 141
Где синтезируются жирные кислоты? 141
Синтез ненасыщенных жирных кислот 142
Синтез триглицеридов и мембранных липидов
из жирных кислот 143
Синтез глицерофосфолипидов 143
Место синтеза мембранных липидов 144
Синтез простагландинов
и родственных им соединений 145
Простагландины и тромбоксаны 145
Лейкотриены 146
Синтез холестерина и его регуляция 146
Дополнительная литература 147
Вопросы к главе 10 147
Глава 11. Синтез глюкозы в организме
(глюконеогенез) 148
Механизм синтеза глюкозы из пирувата 148
Откуда печень получает пируват для глюконеогенеза? 149
Синтез глюкозы из глицерина 15 I
Синтез глюкозы посредством глиоксилатного цикла... 152
Дополнительная литература 153
Вопросы к главе 11 153
437
Глава 12. Регуляция метаболизма
углеводов и жиров 154
Зачем необходима регуляция? 154
Потенциальная опасность «холостых» циклов
в метаболизме 154
Как регулируется активность ферментов? 155
Управление метаболизмом
путем изменения количества ферментов 155
Управление метаболическими процессами
путем изменения активности ферментов 156
Основные сведения о ферментативной кинетике .... 156
Гиперболическая кинетика «классическою» фермента 156
Какие ферменты метаболического
пути нужно регулировать? 157
Природа регуляторных ферментов 158
Аллостеричсская регуляция ферментов 158
Механизм аллостерической регуляции ферментов 158
Обратимость аллостерической регуляции 160
Аллостеричсская регуляция -
метаболическая концепция огромной мощности 160
Роль фосфорилировапия в регуляции
ферментативной активности 161
Регуляция отдельных метаболических путей 161
Два типа управления - внутреннее и внешнее,
или внеклеточное 161
Основные моменты внутренней регуляции
метаболизма углеводов и липидов 162
Внутриклеточная регуляция
метаболизма гликогена 162
Гликолиз и глюконеогенез 162
Внутриклеточная регуляция
пируватдегидрогеназы, цикла лимонной кислоты
и окислительного фосфорилировапия 163
Внутриклеточная регуляция процессов
окисления и синтеза .жирных кислот 164
Регуляция метаболизма углеводов и жиров
внеклеточными агентами 165
Как регулируется содержание в крови инсулина,
глюкагона и адреналина? 1 65
Как работают гормоны глюкагон. адреналин
и инсулин? 1 66
Что такое вторичный мессенджер? 166
Что служит вторичным мессенджером
для глюкагона. адреналина и норадреналина? 166
Управление числом рецепторов 167
Как инсулин управляет метаболизмом? 167
Регуляция поглощения клетками глюкозы и жиров 168
Регуляция синтеза и расщепления гликогена
внеклеточными агентами 169
Регуляция расщепления гликогена 169
Механизм регуляции синтеза
и расщепления гликогена с помощью сАМР 1 70
Обращение активации фосфори. шзы 171
Как регулируется гликогенсинташ? 172
Регуляция гликолиза и глюкопеогенеза
внеклеточными агентами 172
Как с AMP регулирует активность
фосфофруктокиназы? 1 74
Регуляция метаболизма жиров
внеклеточными агентами 175
Дополнительная литература 176
Вопросы к главе 12 176
Глава 13. Зачем нужен
альтернативный путь окисления
глюкозы - пентозофосфатный? 178
Окислительная стадия 178
Неокислительная стадия и ее значение 178
Как полностью окислить глюкозу? 179
Зачем эритроцитам пентозофосфатный путь? 180
Дополнительная литература 180
Вопросы к главе 13 180
Приложение: Реакции, участвующие в превращении
рибозо-5-фосфата в глюкозо-6-фосфат 181
Глава 14. Фотосинтез - способ передачи
энергии электронам воды 182
Общие сведения 182
Место фотосинтеза - хлоропласт 182
Фотосинтетический аппарат и его организация
в гилакоилной мембране 183
Что такое хлорофилл? 184
Механизм светозависимого восстановления NADP 185
Расщепление воды фотосистемой II 186
Как генерируется АТР? 186
Как CO., превращается в углеводы? 187
Как образуется 3-фосфоглицерат ? 187
Из чего синтезируется рибулозо-1,5-дифосфат? 1 88
На чем споткнулась эволюция? 188
438
С4-муть 189
Дополнительная литература 190
Вопросы к главе 14 191
Глава 15. Метаболизм аминокислот 192
Баланс азота в организме 192
Общий метаболизм аминокислот 193
Аспекты метаболизма аминокислот 193
Дезаминирование аминокислот 194
Меха ни sm реакций трансаминирования
(переаминирования) 195
Особые механизмы дезаминирования 195
Судьба кетокислоты, или углеродных скелетов
дезаминированных аминокислот 195
Метаболизм фспилаланина 197
Мстионин и перепое метильных групп 198
Куда переносятся метальные группы ? 198
Синтез аминокислот 198
Синтез глугаминовой кислоты 198
Синтез аспарагиновой кислоты и аланипа 198
Синтез ссрина 198
Синтез глицина 199
Синтез других соединений из аминокислот 200
Что происходит с аминогруппами после
их удаления из аминокислот? Цикл мочевины 200
Механизм синтеза аргинина 200
Превращение цитруллина в аргинин 201
Как аминный азот транспортируется в печень
для переработки в мочевину 202
Глутамин транспортная
форма аммиака в крови 202
Алании транспортная форма
аминного азота в крови 202
Дополнительная литература 203
Вопросы к главе 15 203
Глава 16. Внутриклеточные органеллы,
удаляющие отходы 204
Лизосомы 204
Как предназначенные для разрушения вещества
попадают в лизосомы? 204
О важности лизосомалыюю переваривания 205
Пероксисомы 205
Дополнительная литература 206
Вопросы к главе 16 206
Глава 17. Защитные ферментативные
механизмы организма 207
Свертывание крови 207
Что служит сигналом к образованию сгустка? 207
Как тромбин вызывает образование тромбов? 208
Регуляция свертывания крови 208
Крысиный яд, свертывание крови и витамин К 209
Защита от попавших в организм
чужеродных веществ 210
Цитохром Р450 210
Вторичная модификация: присоединение полярных
остатков к продуктам реакций, катализируемых Р450 211
Реакция печени на ксенобиотики 211
Множественная защита 211
Защита организма от воздействия
собственных протеиназ 212
Защита от активных форм кислорода 212
Витамины С и Ε как ловушки кислородных радикалов 213
Ферментативное уничтожение супероксида
супероксиддисмутазой 213
Стратегия защиты с помощью глутатиона 213
Дополнительная литература 214
Вопросы к главе 17 215
Глава 18. Метаболизм нуклеотидов 216
Структура и номенклатура нуклеотидов 216
Углеводный компонент нуклеотидов 216
Основания нуклеотидов 216
Номенклатура 216
Структура оснований 217
Присоединение оснований в нуклеотидах 218
Синтез пуриновых и пиримидиновых нуклеотидов 218
Пуриновые нуклеотиды 218
5-Фосфорибозил-\-пирофосфат (ΦΫΪΧΦ) 218
Реакции одноуглеродного переноса в синтезе
пуриновых нуклеотидов 219
439
Откуда поступает формшьная группа в фориш-¥Нл? 220
Как образуются АТР и GTP из AMP и GMP? 222
Реутилизация пуриновых оснований
при синтезе нуклеотидов 222
Какова физиологическая роль реутилизации
пуриновых оснований? 222
Образование мочевой кислоты из пуринов 223
Регуляция синтеза пуриновых нуклеотидов 224
Синтез пиримидиновых нуклеотидов 224
Как образуются дезоксирибонуклеотиды? 224
Превращение dUMP в dTMP 225
Тетрагидрофолат, витамин В12 и пернициозная
анемия 226
Дополнительная литература 227
Вопросы к главе 18 227
Часть 4. Хранение и переработка
информации
Глава 19. ДНК: структура
и локализация в клетках 230
Что такое нуклеиновые кислоты? 230
Первичная структура ДНК 230
Какие основания входят в ДНК? 230
Связь оснований с дезоксирибозой 231
Каковы физические свойства компонентов
поли нуклеотидов? 231
Структура дезоксирибополинуклеотида 231
Почему дезоксирибоза? Почему не рибоза? 232
Двойная спираль ДНК 233
Цепи ДНК антипараллельны: что это значит? 235
Каковы размеры молекулы ДНК? 237
Каким образом ДНК упакована в ядре? 237
Как структура ДНК соотносится
с компактными хромосомами эукариот,
видимыми в световой микроскоп? 239
Что такое ген в молекулярных терминах? 240
Некоторые вариации «стандартного» гена 240
Повторяющаяся ДНК 240
Где мы сейчас находимся? 241
Дополнительная литература 241
Вопросы к главе 19 241
Глава 20. Синтез и репарация ДНК 242
Общие принципы репликации ДНК 242
Регуляция инициации репликации ДНК в Е. coli 243
Инициация репликации ДНК у эукариот 243
Расплетание двойной спирали ДНК
и сверхспирализация 244
Как устраняются положительные сверхвитки
перед репликативной вилкой? 245
Какова биологическая роль топоизомераз? 246
Что такое SSB? 247
Положение на данный момент 248
Основные ферментативные реакции,
катализируемые ДНК-полимеразами 248
Проблемы и еще раз проблемы при синтезе ДНК... 249
Как инициируется новая нить? 249
Как решается проблема полярности цепей
при репликации ДНК? 249
Ферментативный комплекс
репликативной вилки Е. coli 250
Что происходит с фрагментами Оказаки? 252
Коррекция неправильного спаривания оснований
полимеразой III 252
Метил-зависимое исправление (репарация) ошибок ... 254
Репарация повреждений ДНК у Е. coli 254
Устройство репликативной вилки эукариот 256
Проблема репликации концов хромосом эукариот 257
Как синтезируется теломерная ДНК? 257
Репарация повреждений ДНК у эукариот 258
Транспозоны, или прыгающие гены 259
Является ли единственным описанный механизм,
при помощи которого синтезируется ДНК? 259
Дополнительная литература 260
Вопросы к главе 20 260
Глава 21. Транскрипция гена -
первый этап в механизме контроля
за биосинтезом белка 262
Матричная РНК 262
440
Структура РНК 262
Как синтезируется мРНК? 262
Некоторые общие свойства мРНК 263
Некоторая важная терминология 263
О чем пойдет речь дальше 264
Транскрипция гена Е. coli 264
Что определяется термином «ген» у прокариот? 264
Что мы имеем в виду, говоря о 5'-конце гена? 265
Фазы транскрипции гена 265
Инициация транскрипции в Е. coli 265
Расплетание ЦИК 266
Терминация транскрипции 266
Скорость инициации транскрипции гена у прокариот 267
Контроль транскрипции
различными сигма-факторами 267
LaoonepoH 267
Каким образом осуществляется
регуляция отдельных генов Е. colP. 267
Структура /яооперона Е. coli 268
Транскрипция генов и ее контроль у эукариот 270
Основные процессы, обусловливающие
образование мРНК у эукариот 270
Терминация транскрипции у эукариот 270
РИК-полимераза II кэпирует
транскрибируемую РНК 270
Что такое прерывистые гены? 271
Механизм сплайсинга 271
Каков биологический статус интронов? 272
Каково происхождение прерывистых генов? 273
Альтернативный сплайсинг 273
Механизм транскрипции генов эукариот
и ее контроль 273
Распаковывание ДНК для транскрипции 273
Необходимость генетического контроля
у дифференцированных эукариот 274
Структура генов эукариот II типа 275
Механизм инициации транскрипции генов эукариот 276
Стабильный комплекс инициации 276
Какова роль транскрипционных факторов? 276
Структура ДНК-связывающих белков 277
Белки с мотивом «спираль-поворот-спираль» (ИТИ) 278
Белки, содержащие «лещиновую молнию» 279
Белки с мотивом «цинковые пальцы» 279
Гомеодоменные белки и эмбриональное развитие 219
Стабильность мРНК и контроль экспрессии генов 280
Структуры, определяющие стабильность мРИК
и их роль в регуляции экспрессии 281
Транскрипция гена в митохондриях 282
Гены, не кодирующие белки 283
Дополнительная литература 283
Вопросы к главе 21 284
Глава 22. Синтез, внутриклеточный
транспорт и деградация белков 285
Основные принципы процесса биосинтеза белка ... 285
Как транслируются кодоны 286
Транспортные РНК, или тРНК 286
Механизм неоднозначного соответствия, или «качания» 287
Как аминокислоты присоединяются
к молекулам тРНК 288
Рибосомы 289
Инициация трансляции 290
Инициация трансляции у Е. coli 291
Элонгация - следующий за инициацией этап
трансляции 293
Цитоплазматические факторы элонгации 293
Механизм элонгации 293
Терминация биосинтеза белка 295
Что такое полисома? 295
Чем отличается синтез белка у эукариот? 296
Как происходит синтез белка в митохондриях? 296
Влияние антибиотиков и токсинов на синтез белка 297
Как сворачивается полипептидная цепь,
синтезированная на рибосоме? 297
Ферменты, участвующие в фолдинге белка 298
Молекулярные шапероны и фолдинг 298
Болезни, вызываемые прионами, и фолдинг белка 298
Другие функции молекулярных шаперонов 299
Как синтезированные белки доставляются
по назначению? 299
441
Что такое эндоплазматический ретикулум (ЭР)? 300
Как белки проходят через мембрану ЭР? 300
Гликозилирование белков в просвете ЭР 301
Что происходит с полипептидом,
перенесенным в просвет шероховатого ЭР? 301
Что такое «адресные сигналы» на белках,
участвующих в сортировке в аппарате Гольджи? 302
Упаковка лизосомных белков 302
Каким обра юм метятся интегральные белки
цитоплазматических мембран? 302
Является ли перенос всех белков
ко-трансляционным? 303
Деградация белков 304
Дополнительная литература 305
Вопросы к главе 22 307
Глава 23. Вирусы и вироиды 308
Жизненный цикл вируса 308
Типы генетического материала у различных вирусов 309
Как вирусы высвобождаются из клеток? 310
Механизмы репликации некоторых вирусов 311
Вирус коровьей оспы 311
Полиовирус 3 11
Вирус гриппа 312
Ретровирусы 3 13
Онкогенные ретровирусы 314
Бактериофаг лямбда 314
Что такое вироиды? 315
Дополнительная литература 316
Вопросы к главе 23 316
Глава 24. Клонирование гена, метод
рекомбинантной ДНК, генная инженерия 317
В чем заключались проблемы выделения генов? 3 17
Первый шаг: разрезание ДНК эндонуклеазами
рестрикции 317
Какова биологическая функция
ферментов рестрикции? 318
Клонирование гена, или как выделяют гены 318
Что такое клонирование? 319
Выделение клона гена человека 319
Создание библиотеки генов человека 319
Как конструируются рекомбинантные молекулы? 319
Скрининг бляшек на выбранный ген человека 320
Клонирование кДНК человека 322
Создание библиотеки кДНК человека 322
Что можно сделать с клонированной ДНК? 322
Бактериальные плазмиды 322
Определение последовательности оснований
клонированного фрагмента ДНК 323
Схема дидезокси-метода секвестрования ДНК 323
Как все это интерпретировать
в виде последовательности оснований? 324
Использование полимеразной цепной реакции (ПЦР)
для амплификации сегментов ДНК 325
Применение технологии амплификации
рекомбинантной ДНК 326
Экспрессия гена в клетках бактерий и эукариот 326
Направленный мутагенез 327
Трансгенез 327
Определение генетических аномалий анализом
рестрикции или блоттингом по Сау зерну 327
Дополнительная литература 328
Вопросы к главе 24 328
Глава 25. Иммунная система 329
У иммунной системы
существуют два защитных механизма 329
Где в организме локализована иммунная система? 330
Основная стратегия иммунной защиты 331
Механизмы иммунитета: В-клстки, хслперные
Т-клетки и образование антител 332
Структура антител 332
Каковы функции антител? 333
Классы антител 333
Как достигается разнообразие антител? 334
Какова роль хелперных Т-клеток в активации
В-клеток для секреции антител? 335
Клетки памяти 336
Т-клетки и клеточный иммунитет 337
Роль цитокинов в иммунной системе 337
442
Почему иммунная система человека столь неистово
отторгает чужеродные человеческие клетки? 337
Дополнительная литература 338
Вопросы к главе 25 339
Глава 26. Химическая сигнализация
в организме 340
Какие виды клеточной активности регулируются
внешними сигналами? 340
Что такое сигнальные молекулы? 340
Гормоны 341
Факторы роста 341
Нейромедиаюры 343
Как контролируется высвобождение сигнальных
молекул? 343
Регуляция секреции гормонов 343
Регуляция высвобождения факторов роста 344
Регуляция высвобождения нейромедиаторов 344
Удаление сигнальных молекул 344
Каким образом сигналы распознаются
клетками-мишенями? 344
Как связывание лиганда приводит к клеточному
ответу? 345
Внутриклеточные рецешор-опосредованпые ответы .. 345
Мембранные рецептор-опосредованные ответы 345
Резюме по мембранным рецептор-опосредусмым
ответам 346
Модуляция рецепторного ответа на сигнал 347
Описание отдельных сигнальных путей
внутри клетки, связывающих активацию
рецептора с клеточным ответом 347
сАМР-оносрелованные пути 347
Контроль уровня с AMP в клетках 348
Как сАМР действует па транскрипцию генов? 350
Передача сигнала с использованием
циклического GMP в качестве вторичного посредника. 350
Гормоны, передающие сигнал при помощи различных
вторичных мсссспджеров; фосфоинозитидный каскад 352
Каковы другие меесенджерные -эффекты кальция? 353
Рецепторы, ассоциированные с тирозинкиназой 353
Ras-путь - распространенный сигнальный путь
οι мембраны к гену эукариот 354
Ras-путь и онкогена 355
Замечание по терминологии 356
Почему Ras-путь такой длинный? 356
Прямой внутриклеточный сигнальный путь
от рецептора к ядру 357
Сигнализация иптерферонами 357
Насколько общим является тот прямой путь? 358
Внешние сигналы, регулирующие
лиганд-зависимые ионные каналы 358
Каким образом связывание ацетилхолина с мембран-
нымрсцептором приводит к нервному импульсу? 358
Зрение: процесс, зависимый
от контроля открываемой лигандом поры 359
Передача светового сигнала 359
Дополнительная литература 361
Вопросы к главе 26 363
Часть 5. Транспорт кислорода
и С02 (углекислого газа)
Глава 27. Красная кровяная клетка
и функции гемоглобина 366
Красные кровяные клетки 366
Гем и em синтез 366
Синтез тема 367
Регуляция биосинтеза гема
и доставка железа в эритроцит 368
Распад гема 370
Как регулируется синтез глобина 370
Транспорт газов в кровь 370
Каково химическое обоснование роли гемоиротеи-
повогокомплекса как переносчика кислорода? 370
Структура миоглобина
и его роль в связывании кислорода 371
Структура гемоглобина 372
Связывание кислорода с гемоглобином 372
Как появляется сигмоидная кривая насыщения
кислородом 373
Механизм аллостерического изменения молекулы
гемоглобина 373
Важная роль 2,3-дифосфоглицерата (ДФГ)
в функции гемоглобина 374
Влияние рН па связывание кислорода с гемоглобином 375
443
Роль изменений рН в транспорте кислорода и CO., 376
Поддержание рН в крови 376
Серповидноклеточная анемия 377
Дополнительная литература 377
Вопросы к главе 27 377
Часть 6. Механическая работа
в клетке
Глава 28. Мышечное сокращение 380
Напоминание о конформационных перестройках
в белках 380
Типы мышечных клеток и их энергообеспечение ... 380
Структура скелетной поперечно-полосатой мышцы ... 381
Структура миофибриллы 381
Как сокращается саркомер? 382
Структура и действие
толстых и топких филаментов 382
Тонкие филаменты 382
Толстые филаменты 382
Как головка миозина преобразует энергию гидролиза
АТР в механическую работу актиновых филаментов? 383
Как регулируется сокращение произвольных
поперечно-полосатых мышц? 384
Как Са2* вызывает сокращение? 384
Транспорт ионов Саг в мышце 385
Чем гладкая мышца отличается по структуре
и регуляции от поперечно-полосатой? 385
Регуляция сокращения гладкой мышцы 386
Как Са2* регулирует сокращение гладкой мышцы/ 386
Дополнительная литература 387
Вопросы к главе 28 387
Глава 29. Роль цитоскелета
в поддержании формы клеток
и осуществлении механической работы 388
Роль актина и миозина в немышечных клетках 389
Структурное значение актина и его роль в движении
клеток 389
Роль актина и миозина
во внутриклеточном транспорте веществ 390
Микротрубочки, клеточное движение
и внутриклеточный транспорт 391
Что такое микротрубочки? 391
Что защищает (+)-концы микротрубочек? 391
Функции микротрубочек 392
Молекулярные двигатели, обеспечивающие
движение по микротрубочкам 392
Роль микротрубочек в движении клеток 392
Роль микротрубочек в транспорте везикул
внутри клетки 392
Роль микротрубочек в митозе 393
Механизм перемещения хромосом (анафаза А) 393
Механизм разделения полюсов (анафаза В) 393
Промежуточные филаменты 393
Дополнительная литература 395
Вопросы к главе 29 395
Ответы на вопросы 396
Благодарности за предоставление
иллюстративного материала 415
Предметный указатель 416
444